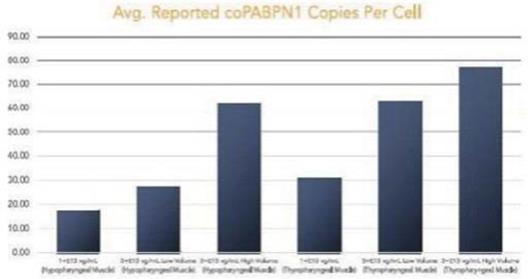
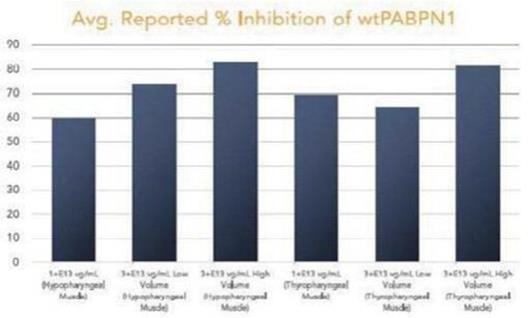
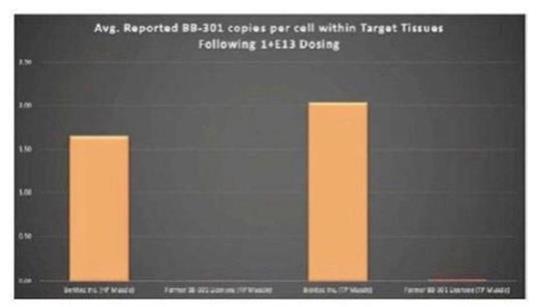
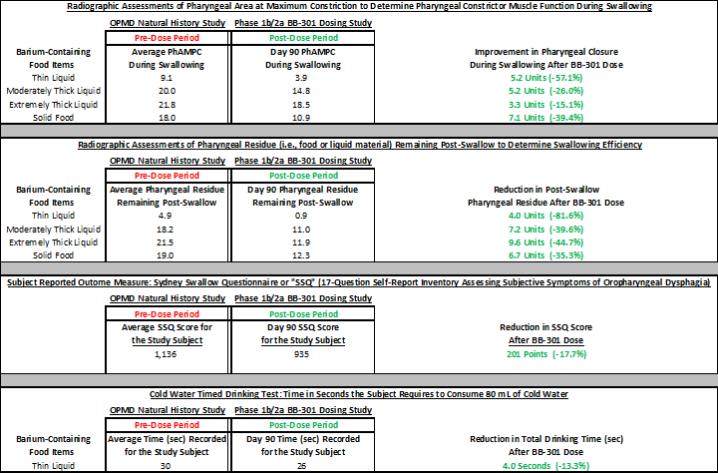
圖 1
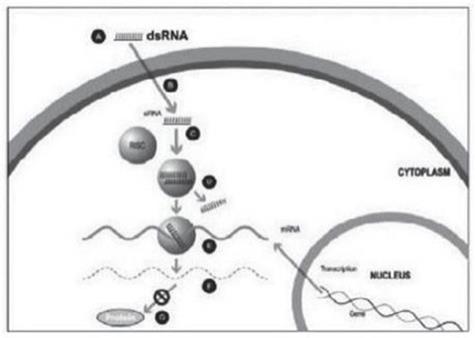
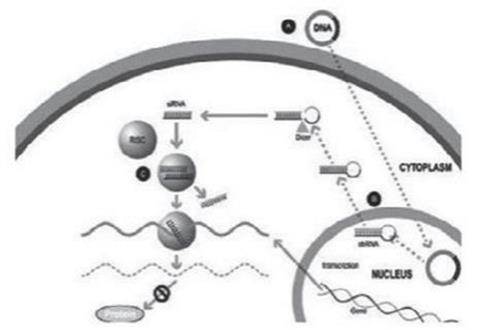
實驗室合成了一種小型雙鏈 RNA 或 dsRNA 分子(A,圖 1),包括一條稱為感覺鏈的鏈和另一條相互互補的鏈(稱為反義鏈)。這些小型 dsRNA 被稱為小型幹擾 RNA 或 siRNA。感官鏈的序列對應於靶基因 mRNA 的短區域。siRNA 被輸送到目標細胞(B,圖 1),其中一組被稱為 RNA 誘導沉默複合物(RISC)的酶處理 siRNA(C,圖 1),其中一條鏈(通常是感覺鏈)被釋放(D,圖 1)。RISC 使用反義鏈來尋找具有互補序列(E,圖 1)的 mRNA,從而導致目標 mRNA 的裂解(F,圖 1)。因此,mRNA(蛋白質產生)的輸出沒有發生(G,圖 1)。包括Alnylam Pharmicals Inc.(“Alnylam”)在內的幾家公司在其RNai候選產品中使用了這種方法。
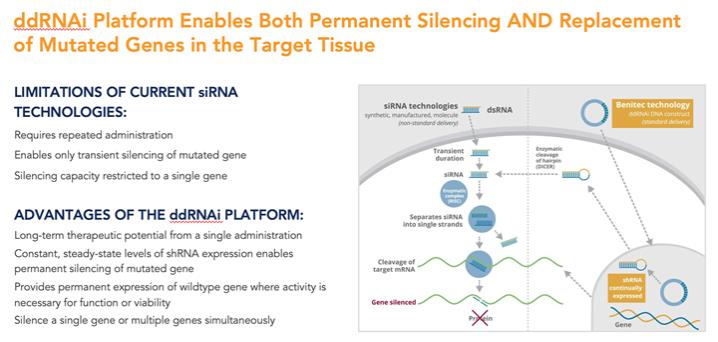
重要的是,許多遺傳疾病不適合採用圖 1 中概述的傳統基因沉默方法,因為患病細胞可能會產生感興趣的野生型蛋白質和該蛋白的致病突變變體的混合物,而潛在的基因突變可能太小,無法通過僅使用基於 siRNA 的方法來選擇性靶向該蛋白的致病變體。在這些情況下,如果不同時沉默感興趣的野生型細胞內蛋白,就很難有選擇地沉默致病蛋白,而野生型細胞內蛋白的存在對於正常細胞功能的進行至關重要。
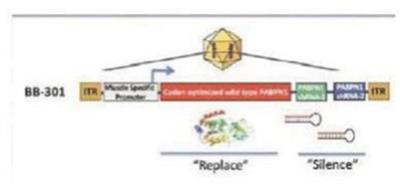
我們專有的沉默和替換技術利用了RNAi的獨特特異性和強大的基因沉默能力,同時克服了基於siRNA的疾病管理方法的許多關鍵侷限性。
在標準的RNAi方法中,雙鏈siRNA是合成產生的,然後通過對RNA進行化學修飾或替代遞送方法將其引入靶細胞。儘管使用這種方法已在多種臨牀適應症中證明瞭療效,但基於siRNA的方法仍然存在許多侷限性,包括:
| • | 臨牀管理需要重複使用基於siRNA的治療藥物進行多個週期,以保持療效; |
23