目錄表
我們的管道
我們已經組裝了一系列具有治療多種神經退行性疾病潛力的基因治療產品候選產品組合。我們的發展計劃包括:
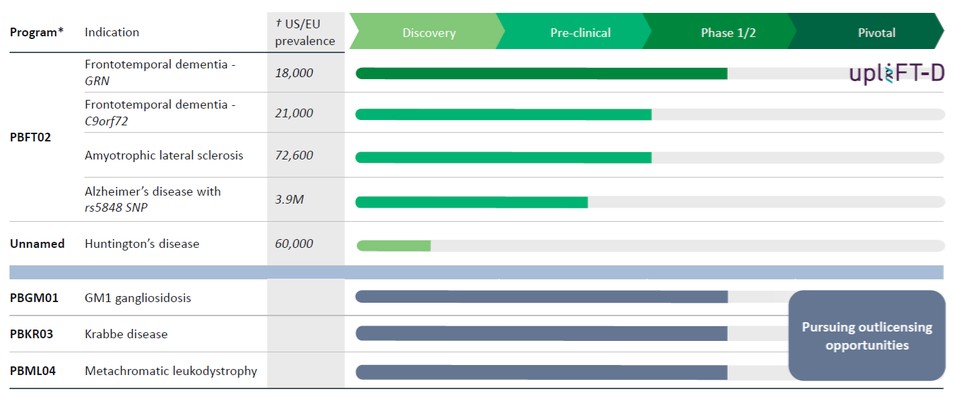
*仍有8個額外的CNS管道許可證選項;之前行使了3個許可證選項,權利隨後歸還給賓夕法尼亞大學。
第三方來源的†美國/歐盟流行率
PBFT02治療FTD-GRN
我們目前正在開發PBFT02,它利用AAV1衣殼來提供GRNPGRN編碼,用於治療FTD-GRN。FTD-GRN是一種可遺傳形式的FTD,患者在GRN基因,導致了PGRN的缺陷。PGRN是一種複雜的高度保守的蛋白質,被認為在細胞內穩態、神經發育和炎症中具有多種作用。有證據表明,FTD和其他神經退行性疾病中的PGRN缺乏可能與溶酶體功能障礙有關。
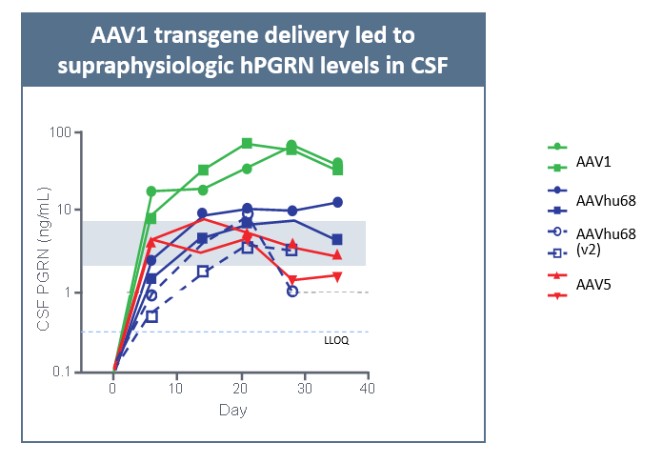
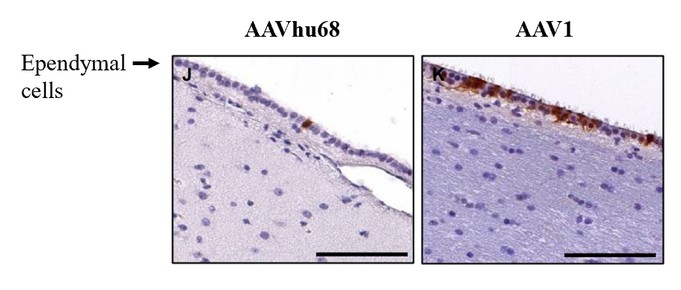
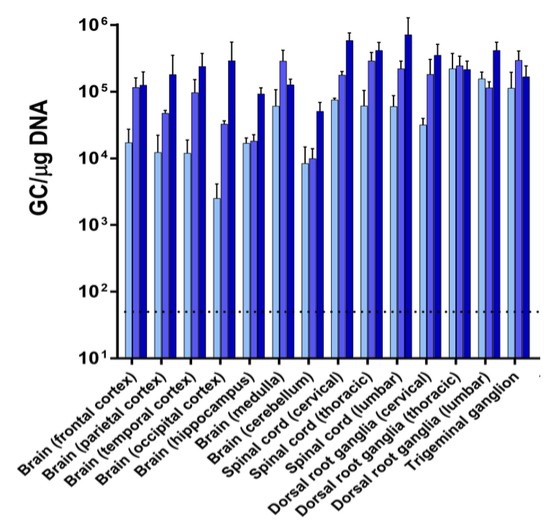
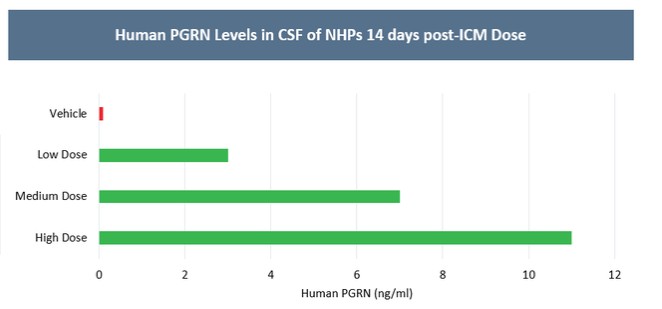
目前,還沒有被批准用於治療FTD的疾病修改療法-GRN。根據臨牀前研究的結果,我們認為PBFT02可能提供FTD-GRN預後顯著改善的患者。我們選擇AAV1衣殼和ICM給藥作為PBFT02,是因為這種方法導致了人PGRN在非人類靈長類動物(NHP)的大腦和脊髓中廣泛而強勁的表達,而且與其他被測試的血清型相比,使用AAV1獲得的腦脊液(CSF)中的PGRN水平更高。與健康受試者的腦脊液水平相比,ICM將AAV1注射到NHP導致人PGRN的腦脊液超生理水平,並且超過了AAVhu68或AAV5的NHP的水平。我們擁有來自美國食品和藥物管理局(FDA)的有效IND,並在多個國家/地區批准了PBFT02的臨牀試驗授權或CTA。我們正在進行我們的Uplift-D試驗,這是一項針對確診為有症狀的FTD患者的PBFT02的國際、多中心、開放標籤、單臂1/2期臨牀試驗。GRN.
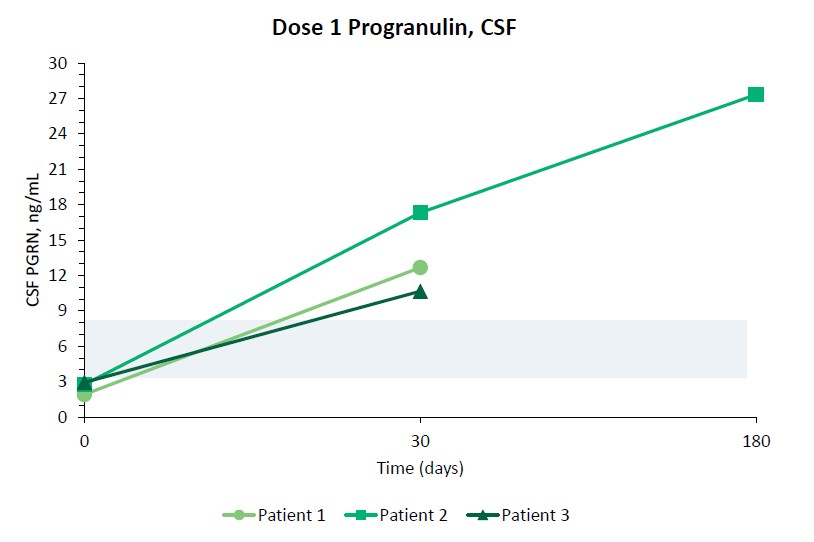
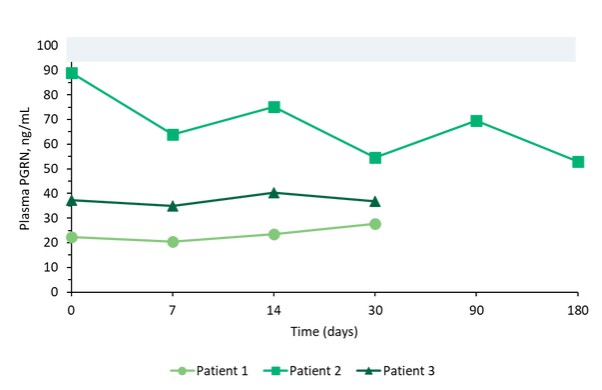
我們報告了2023年12月我們的Uplit-D試驗隊列1中的三名患者的初步安全性和生物標記物數據。在這項試驗中,劑量1的PBFT02治療後30天,腦脊液中PGRN的超生理水平從10.7到17.3 ng/m L(n=3),超過了健康成人對照組3.3到8.2 ng/m L(n=61)。在第一個在PBFT02注射後6個月達到的患者中,腦脊液PGRN保持在27.3 ng/mL的超生理水平。相比之下,在PBFT02治療後,所有三名患者的血漿PGRN水平沒有改變,在整個可用隨訪期內保持與基線濃度相似,低於健康成人對照組的水平。
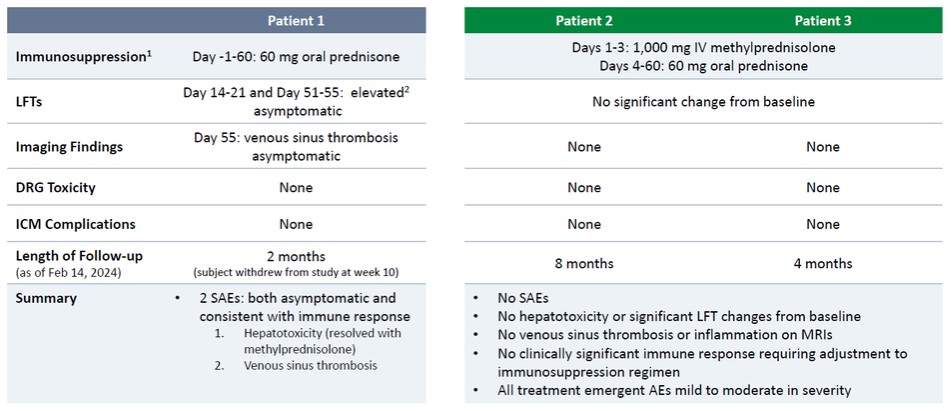
正如先前報道的,根據最初的試驗方案,患者1接受了低水平的免疫抑制(每天60毫克口服潑尼鬆),經歷了兩次嚴重的不良事件,即SAE,兩者都沒有症狀和
7