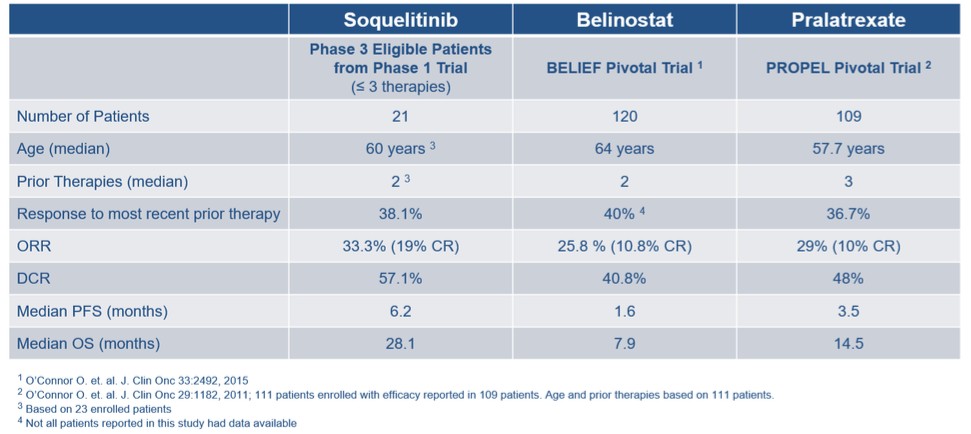
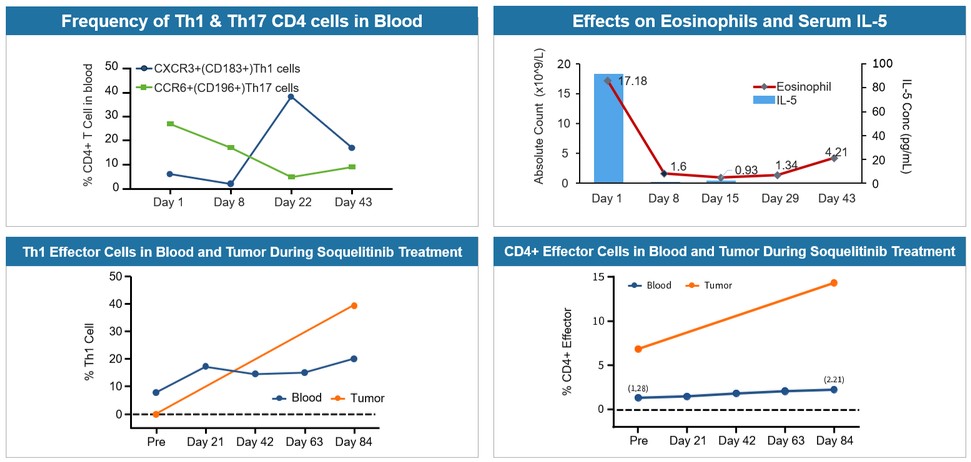
目錄表
產品線
我們的候選產品線包括以下內容:
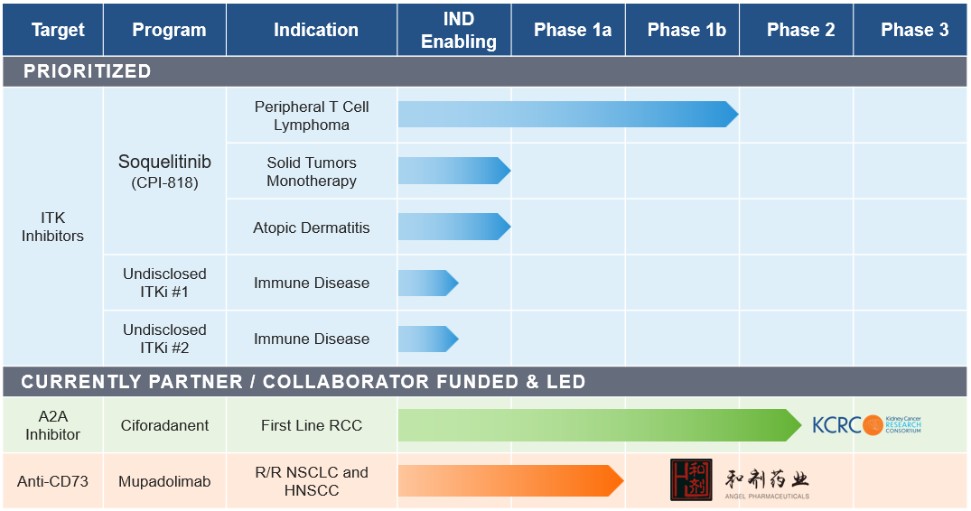
Soquelitinib(CPI—818),ITK抑制劑。Soquelitinib是一種研究性選擇性、口服生物可利用的ITK共價抑制劑。ITK是一種在T細胞信號傳導和分化中發揮作用的酶,主要在T細胞中表達,T細胞是在免疫應答中發揮重要作用的淋巴細胞。T細胞淋巴瘤是T細胞的惡性腫瘤,增殖和擴散遍及全身。這些淋巴瘤通常具有通過T細胞受體途徑的緊張性信號傳導,其中涉及ITK。抑制ITK可導致該信號通路的阻斷,並控制惡性腫瘤的生長。此外,淋巴瘤和實體瘤的關鍵生存機制之一被認為是正常T細胞的重編程,以在組織中創造一個抑制抗腫瘤免疫反應並有利於腫瘤生長的環境。我們相信這種酶的高選擇性抑制劑將促進正常T細胞抗腫瘤免疫的誘導,並可能用於治療實體瘤以及淋巴瘤。
選擇性抑制ITK可以阻斷Th2細胞的產生和功能,可能導致幼稚T細胞分化為Th1細胞,這一過程被稱為Th1偏斜。Th1細胞導致殺傷性T細胞的生成,可以消除腫瘤細胞或病毒感染細胞。Th1細胞產生幹擾素γ和腫瘤壞死因子,這是已知破壞癌細胞的細胞因子。我們認為,soquelitinib可以導致正常免疫反應的重新編程,這也可能有益於治療某些自身免疫和過敏性疾病。過度活躍的Th2細胞在自身免疫性疾病和過敏性疾病中發揮作用,通過阻斷Th2功能及其炎症細胞因子的產生,可以潛在地通過選擇性ITK抑制來改善這些疾病。
涉及ITK的T細胞信號傳導在許多T細胞淋巴瘤的發展中是必需的。ITK細胞信號通路與B細胞中發生的信號通路相似,後者由一種稱為BTK的同源酶介導,該酶是伊維替尼的靶點,已批准用於B細胞淋巴瘤和白血病患者的治療。ITK在許多T細胞淋巴瘤中表達,包括外周T細胞淋巴瘤("PTCL")、血管免疫母細胞性T細胞淋巴瘤("AITL")、皮膚T細胞淋巴瘤("CTCL")、間變性大細胞淋巴瘤("ALCL")、自然殺傷T細胞淋巴瘤("NKTCL")和其他T細胞惡性腫瘤。
在完全缺乏ITK表達的ITK基因敲除小鼠中,T細胞表現出T輔助細胞分化和細胞因子分泌的缺陷,但保留分化為分泌IL—2的細胞毒性T細胞的能力,
2