目錄表
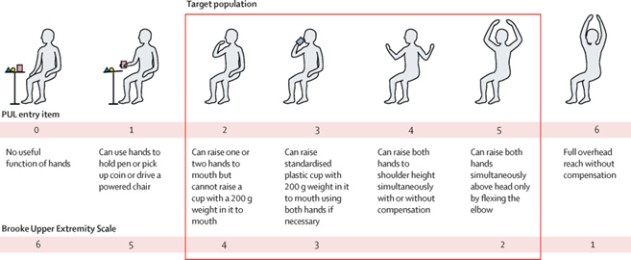
上肢性能(PUL條目) (1)
(CAP—1002當前DMD目標人羣)
| (1) | 圖片來源:HOPE—2 柳葉刀 出版物(2022年3月) |
隊列B的入組正在進行中,該隊列旨在入組約44名受試者,以1:1的比例隨機分配至CAP—1002或安慰劑組。在4次CAP—1002或安慰劑給藥後,將在第12個月對每個個體隊列進行主要療效和安全性分析。我們計劃於2024年第二季度完成隊列B的入組。隊列B使用在我們聖地亞哥工廠生產的產品。
HOPE—3研究的主要結局指標將是上肢功能("PUL")v2.0,這是一種專門設計用於評估高位(肩)、中段(肘)和遠端(腕和手)功能的經驗證的工具,其概念框架反映上肢功能的無力進展。HOPE—3還將測量各種次要終點,包括心功能評估。
根據我們的RMAT指定,在2023年第三季度,我們與FDA舉行了一次B類會議,討論了我們的生產計劃,預計可能提交BLA申請。在這次會議上,我們確認了我們的第三階段,希望—3計劃的一致性。此外,我們還討論了商業生產活動的計劃,包括效價測定和其他產品放行標準,以支持商業化。我們計劃在2024年第一季度與FDA會面,繼續討論我們的BLA途徑。在即將舉行的B類會議中,我們打算討論我們進一步的CMC商業發佈計劃(如果獲得批准),以加快我們的BLA提交途徑。我們的最終目標是提交BLA,允許使用我們聖地亞哥工廠生產的CAP—1002商業產品。
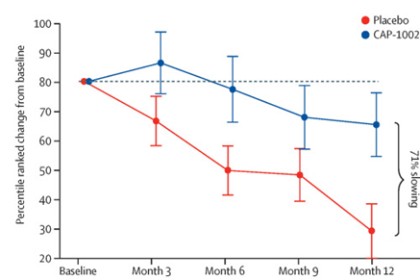
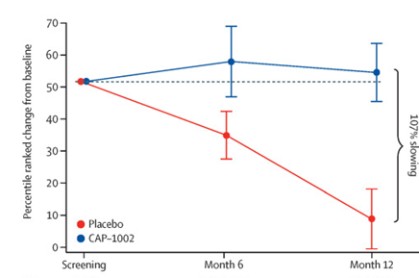
II期HOPE—2臨牀試驗: HOPE—2是一項隨機、雙盲、安慰劑對照臨牀試驗,在美國多個研究中心進行,並於2021年完成。該臨牀試驗旨在評價CAP—1002重複靜脈給藥的安全性和有效性,用於有骨骼肌損害證據的男孩和年輕男性,無論活動狀態如何。研究中約90%的患者為非活動性患者,所有患者均接受穩定的類固醇治療。兩個治療組的人口統計學和基線特徵相似。希望2的最終一年結果發表在 《柳葉刀》2022年3月,表明該試驗達到了PUL v1.2的中級維度的主要療效終點(p = 0.01)和完整PUL v2.0的其他陽性終點(p = 0.04)。雖然中級PUL v1.2是試驗確定的主要終點,但我們也使用PUL v2.0進行了分析,因為FDA建議使用更新後的PUL v2.0作為主要療效終點以支持BLA。左心室射血分數(LVEF)(心臟泵功能的總體指標)在安慰劑組隨時間推移降低,但CAP—1002組改善,顯示心臟病進展減慢107%(p = 0.002)。此外,數據表明心臟功能的整體改善,如指數容積(LVESV,LVEDV)測量。這些是心臟功能的替代指標,被認為與長期結局相關。此外,數據顯示生物標誌物CK—MB減少,CK—MB是一種只有在心肌細胞損傷時才會釋放的酶。在正常人受試者中,血液中通常沒有可測量的CK—MB。眾所周知,DMD中的持續性肌細胞損傷導致與心肌細胞損失相關的病理性高酶水平。據我們所知,這是DMD中第一個將心臟功能穩定與細胞損傷生物標誌物減少相關的臨牀研究。 除了類固醇,DMD的功能保留是罕見的。安慰劑組患者的結果與自然病史一致,但在治療組中,大多數患者在一年治療期間這些終點穩定或改善。CAP—1002在整個研究期間通常安全且耐受良好。除了過敏症
7