CCCC-2023123100016625792023財年錯誤Http://fasb.org/us-gaap/2023#CollaborativeArrangementMemberHttp://fasb.org/us-gaap/2023#CollaborativeArrangementMemberP5YP3M00016625792023-01-012023-12-3100016625792023-06-30ISO 4217:美元00016625792024-02-14Xbrli:共享00016625792023-12-3100016625792022-12-31ISO 4217:美元Xbrli:共享00016625792022-01-012022-12-310001662579美國-美國公認會計準則:普通股成員2021-12-310001662579US-GAAP:AdditionalPaidInCapitalMembers2021-12-310001662579Us-gaap:AccumulatedOtherComprehensiveIncomeMember2021-12-310001662579美國-公認會計準則:保留預付款成員2021-12-3100016625792021-12-310001662579美國-美國公認會計準則:普通股成員2022-01-012022-12-310001662579US-GAAP:AdditionalPaidInCapitalMembers2022-01-012022-12-310001662579Us-gaap:AccumulatedOtherComprehensiveIncomeMember2022-01-012022-12-310001662579美國-公認會計準則:保留預付款成員2022-01-012022-12-310001662579美國-美國公認會計準則:普通股成員2022-12-310001662579US-GAAP:AdditionalPaidInCapitalMembers2022-12-310001662579Us-gaap:AccumulatedOtherComprehensiveIncomeMember2022-12-310001662579美國-公認會計準則:保留預付款成員2022-12-310001662579美國-美國公認會計準則:普通股成員2023-01-012023-12-310001662579US-GAAP:AdditionalPaidInCapitalMembers2023-01-012023-12-310001662579Us-gaap:AccumulatedOtherComprehensiveIncomeMember2023-01-012023-12-310001662579美國-公認會計準則:保留預付款成員2023-01-012023-12-310001662579美國-美國公認會計準則:普通股成員2023-12-310001662579US-GAAP:AdditionalPaidInCapitalMembers2023-12-310001662579Us-gaap:AccumulatedOtherComprehensiveIncomeMember2023-12-310001662579美國-公認會計準則:保留預付款成員2023-12-31CCCC:細分市場0001662579CCCC:實驗室設備成員2023-12-310001662579US-GAAP:ComputerEquipmentMembers2023-12-310001662579Cccc:OfficeEquipmentFurnitureAndFixturesMember2023-12-310001662579美國公認會計準則:MoneyMarketFundsMembers2023-12-310001662579美國-公認會計準則:公允價值輸入級別1成員美國公認會計準則:MoneyMarketFundsMembers2023-12-310001662579美國公認會計準則:MoneyMarketFundsMembers美國-公認會計準則:公允價值輸入級別2成員2023-12-310001662579美國-公認會計準則:公允價值投入級別3成員美國公認會計準則:MoneyMarketFundsMembers2023-12-310001662579美國-公認會計準則:美國證券成員2023-12-310001662579美國-公認會計準則:美國證券成員美國-公認會計準則:公允價值輸入級別1成員2023-12-310001662579美國-公認會計準則:美國證券成員美國-公認會計準則:公允價值輸入級別2成員2023-12-310001662579美國-公認會計準則:美國證券成員美國-公認會計準則:公允價值投入級別3成員2023-12-310001662579美國-公認會計準則:公司債務證券成員2023-12-310001662579美國-公認會計準則:公允價值輸入級別1成員美國-公認會計準則:公司債務證券成員2023-12-310001662579美國-公認會計準則:公司債務證券成員美國-公認會計準則:公允價值輸入級別2成員2023-12-310001662579美國-公認會計準則:公允價值投入級別3成員美國-公認會計準則:公司債務證券成員2023-12-310001662579美國-公認會計準則:公司債務證券成員2023-12-310001662579美國-公認會計準則:公允價值輸入級別1成員美國-公認會計準則:公司債務證券成員2023-12-310001662579美國-公認會計準則:公允價值輸入級別2成員美國-公認會計準則:公司債務證券成員2023-12-310001662579美國-公認會計準則:公允價值投入級別3成員美國-公認會計準則:公司債務證券成員2023-12-310001662579美國-公認會計準則:美國政府債務證券成員2023-12-310001662579美國-公認會計準則:公允價值輸入級別1成員美國-公認會計準則:美國政府債務證券成員2023-12-310001662579美國-公認會計準則:美國政府債務證券成員美國-公認會計準則:公允價值輸入級別2成員2023-12-310001662579美國-公認會計準則:公允價值投入級別3成員美國-公認會計準則:美國政府債務證券成員2023-12-310001662579美國-公認會計準則:美國證券成員2023-12-310001662579美國-公認會計準則:公允價值輸入級別1成員美國-公認會計準則:美國證券成員2023-12-310001662579美國-公認會計準則:美國證券成員美國-公認會計準則:公允價值輸入級別2成員2023-12-310001662579美國-公認會計準則:公允價值投入級別3成員美國-公認會計準則:美國證券成員2023-12-310001662579美國-公認會計準則:公允價值輸入級別1成員2023-12-310001662579美國-公認會計準則:公允價值輸入級別2成員2023-12-310001662579美國-公認會計準則:公允價值投入級別3成員2023-12-310001662579美國公認會計準則:MoneyMarketFundsMembers2022-12-310001662579美國-公認會計準則:公允價值輸入級別1成員美國公認會計準則:MoneyMarketFundsMembers2022-12-310001662579美國公認會計準則:MoneyMarketFundsMembers美國-公認會計準則:公允價值輸入級別2成員2022-12-310001662579美國-公認會計準則:公允價值投入級別3成員美國公認會計準則:MoneyMarketFundsMembers2022-12-310001662579美國-公認會計準則:美國證券成員2022-12-310001662579美國-公認會計準則:美國證券成員美國-公認會計準則:公允價值輸入級別1成員2022-12-310001662579美國-公認會計準則:美國證券成員美國-公認會計準則:公允價值輸入級別2成員2022-12-310001662579美國-公認會計準則:美國證券成員美國-公認會計準則:公允價值投入級別3成員2022-12-310001662579美國-公認會計準則:公司債務證券成員2022-12-310001662579美國-公認會計準則:公允價值輸入級別1成員美國-公認會計準則:公司債務證券成員2022-12-310001662579美國-公認會計準則:公允價值輸入級別2成員美國-公認會計準則:公司債務證券成員2022-12-310001662579美國-公認會計準則:公允價值投入級別3成員美國-公認會計準則:公司債務證券成員2022-12-310001662579美國-公認會計準則:美國政府債務證券成員2022-12-310001662579美國-公認會計準則:公允價值輸入級別1成員美國-公認會計準則:美國政府債務證券成員2022-12-310001662579美國-公認會計準則:美國政府債務證券成員美國-公認會計準則:公允價值輸入級別2成員2022-12-310001662579美國-公認會計準則:公允價值投入級別3成員美國-公認會計準則:美國政府債務證券成員2022-12-310001662579美國-公認會計準則:美國證券成員2022-12-310001662579美國-公認會計準則:公允價值輸入級別1成員美國-公認會計準則:美國證券成員2022-12-310001662579美國-公認會計準則:美國證券成員美國-公認會計準則:公允價值輸入級別2成員2022-12-310001662579美國-公認會計準則:公允價值投入級別3成員美國-公認會計準則:美國證券成員2022-12-310001662579美國-公認會計準則:公允價值輸入級別1成員2022-12-310001662579美國-公認會計準則:公允價值輸入級別2成員2022-12-310001662579美國-公認會計準則:公允價值投入級別3成員2022-12-310001662579美國-公認會計準則:公司債務證券成員2023-12-31CCCC:安全0001662579美國-公認會計準則:美國政府債務證券成員2023-12-310001662579美國-公認會計準則:公司債務證券成員2022-12-310001662579美國-公認會計準則:美國政府債務證券成員2022-12-310001662579美國-公認會計準則:美國證券成員2022-12-310001662579CCCC:實驗室設備成員2022-12-310001662579Us-gaap:LeaseholdsAndLeaseholdImprovementsMember2023-12-310001662579Us-gaap:LeaseholdsAndLeaseholdImprovementsMember2022-12-310001662579美國-GAAP:傢俱和固定設備成員2023-12-310001662579美國-GAAP:傢俱和固定設備成員2022-12-310001662579美國-GAAP:OfficeEquipmentMembers2023-12-310001662579美國-GAAP:OfficeEquipmentMembers2022-12-310001662579US-GAAP:ComputerEquipmentMembers2022-12-310001662579美國-美國公認會計準則:建設正在進行成員2023-12-310001662579美國-美國公認會計準則:建設正在進行成員2022-12-310001662579CCCC:水城租賃會員CCCC:辦公室和實驗室空間成員2017-07-310001662579CCCC:水城租賃會員CCCC:辦公室和實驗室空間成員2017-07-012017-07-31CCCC:期間0001662579CCCC:水城租賃會員CCCC:辦公室和實驗室空間成員2018-04-300001662579CCCC:水城租賃會員CCCC:新租賃空間成員2022-01-012022-01-310001662579CCCC:水城租賃會員CCCC:新租賃空間成員2022-01-310001662579CCCC:水城租賃會員CCCC:新租賃空間成員2022-12-310001662579CCCC:水城租賃會員CCCC:新租賃空間成員2023-12-31CCCC:合同0001662579CCCC:水城租賃會員2022-01-31Xbrli:純0001662579CCCC:默克協議成員2023-12-112023-12-110001662579CCCC:默克協議成員CCCC:研究和開發里程碑成員2023-12-110001662579CCCC:默克協議成員2023-12-11CCCC:目標0001662579CCCC:默克協議成員2023-12-310001662579CCCC:BettaAgreement成員2023-05-292023-05-290001662579CCCC:研究和開發里程碑成員CCCC:BettaAgreement成員2023-05-290001662579CCCC:DrugApplicationApprovalMemberCCCC:BettaAgreement成員2023-05-290001662579CCCC:BettaAgreement成員2023-05-290001662579SRT:最小成員數2023-05-292023-05-290001662579CCCC:BettaAgreement成員2023-12-310001662579CCCC:BettaAgreement成員2023-01-012023-12-310001662579美國-公認會計準則:外國成員2023-01-012023-12-310001662579CCCC:RestatedRocheAgreement成員2018-12-012018-12-310001662579CCCC:RestatedRocheAgreement成員2020-11-300001662579CCCC:RestatedRocheAgreement成員2023-12-310001662579CCCC:RestatedRocheAgreement成員Cccc:LeadSeriesIdentificationAchievementMember2020-11-012020-11-300001662579CCCC:RestatedRocheAgreement成員2020-11-012020-11-300001662579CCCC:RestatedRocheAgreement成員Cccc:ResearchDevelopmentAndCommercialMilestonePaymentsMemberSRT:最大成員數2020-11-300001662579CCCC:RestatedRocheAgreement成員SRT:最大成員數CCCC:OneTimeSalesBasedPayments成員2020-11-300001662579CCCC:RestatedRocheAgreement成員SRT:最小成員數2023-01-012023-12-3100016625792018-12-31CCCC:性能禁用0001662579CCCC:RestatedRocheAgreement成員2018-12-310001662579CCCC:研究和開發目標成員2023-12-310001662579CCCC:OptionRightsMember2023-12-310001662579CCCC:生物生成研究和許可協議成員SRT:最大成員數2020-02-012020-02-29CCCC:蛋白質0001662579CCCC:生物生成研究和許可協議成員2020-02-012020-02-290001662579CCCC:生物生成研究和許可協議成員CCCC:研究和開發里程碑成員SRT:最大成員數2020-02-012020-02-290001662579CCCC:生物生成研究和許可協議成員SRT:最大成員數CCCC:OneTimeSalesBasedPayments成員2020-02-012020-02-290001662579SRT:最小成員數CCCC:生物生產許可協議成員2023-01-012023-12-310001662579CCCC:生物生產許可協議成員2023-12-31CCCC:績效_義務0001662579CCCC:研究和開發服務成員CCCC:生物生產許可協議成員2023-01-012023-12-310001662579CCCC:研究和開發服務成員CCCC:生物生產許可協議成員2023-12-310001662579CCCC:Calico許可協議成員2017-03-012017-03-310001662579CCCC:Calico許可協議成員2021-08-012021-08-310001662579CCCC:Calico許可協議成員2021-09-012021-09-300001662579CCCC:Calico許可協議成員2017-03-012020-06-300001662579CCCC:Calico許可協議成員Cccc:PotentialResearchDevelopmentAndCommercialMilestonePaymentsMemberSRT:最大成員數2017-03-012017-03-310001662579CCCC:Calico許可協議成員SRT:最大成員數CCCC:OneTimeSalesBasedPayments成員2017-03-012017-03-310001662579CCCC:研究和開發服務成員CCCC:Calico許可協議成員2023-01-012023-12-310001662579CCCC:RocheAgreement成員2023-01-012023-12-310001662579CCCC:RocheAgreement成員2022-01-012022-12-310001662579CCCC:生物基因協議成員2023-01-012023-12-310001662579CCCC:生物基因協議成員2022-01-012022-12-310001662579CCCC:CalicoAgreement成員2023-01-012023-12-310001662579CCCC:CalicoAgreement成員2022-01-012022-12-310001662579CCCC:BettaAgreement成員2022-01-012022-12-310001662579CCCC:RocheAgreement成員2023-12-310001662579CCCC:生物基因協議成員2023-12-310001662579CCCC:RocheAgreement成員2022-12-310001662579CCCC:生物基因協議成員2022-12-310001662579CCCC:CalicoAgreement成員2022-12-310001662579Cccc:RestatedRocheAgreementBiogenLicenseAgreementAndCalicoLicenseAgreementMember2023-12-310001662579Cccc:CreditAgreementWithPerceptiveLifeSciencesMasterFundLTDMemberCCCC:TermLoanMember2020-06-050001662579Cccc:CreditAgreementWithPerceptiveLifeSciencesMasterFundLTDMemberCCCC:TermLoanMemberCCCC:TrancheOneMembers2020-06-050001662579Cccc:CreditAgreementWithPerceptiveLifeSciencesMasterFundLTDMemberCCCC:TermLoanMemberCCCC:TranscheTwoMembers2020-06-050001662579Cccc:CreditAgreementWithPerceptiveLifeSciencesMasterFundLTDMemberCCCC:TermLoanMemberCCCC:TrancheOneMembers2020-06-3000016625792020-10-31CCCC:投票0001662579CCCC:AtmOfferingsMembersSRT:最大成員數Cccc:EquityDistributionAgreementWithCowenAndCompanyLLCMember2021-11-012021-11-300001662579CCCC:AtmOfferingsMembersCccc:EquityDistributionAgreementWithCowenAndCompanyLLCMember2023-01-012023-12-310001662579CCCC:AtmOfferingsMembersCccc:EquityDistributionAgreementWithCowenAndCompanyLLCMember美國公認會計準則:次要事件成員2024-01-012024-01-310001662579CCCC:AtmOfferingsMembersCccc:EquityDistributionAgreementWithCowenAndCompanyLLCMember2021-11-012023-12-310001662579CCCC:AtmOfferingsMembersCccc:EquityDistributionAgreementWithCowenAndCompanyLLCMember2023-12-310001662579CCCC:AtmOfferingsMembersCccc:EquityDistributionAgreementWithCowenAndCompanyLLCMember2022-01-012022-12-310001662579Cccc:TwoThousandFifteenIncentiveStockOptionAndGrantPlanMember2015-12-280001662579SRT:最小成員數Cccc:TwoThousandFifteenIncentiveStockOptionAndGrantPlanMember2015-12-282015-12-280001662579Cccc:TwoThousandFifteenIncentiveStockOptionAndGrantPlanMemberSRT:最大成員數2015-12-282015-12-280001662579Cccc:TwoThousandFifteenIncentiveStockOptionAndGrantPlanMember2015-12-282015-12-280001662579Cccc:TwoThousandAndTwentyStockOptionAndIncentivePlanMember2020-09-300001662579Cccc:TwoThousandAndTwentyStockOptionAndIncentivePlanMember2023-12-310001662579Cccc:TwoThousandFifteenIncentiveStockOptionAndGrantPlanMember2023-12-310001662579Cccc:TwoThousandAndTwentyStockOptionAndIncentivePlanMember2021-01-012021-01-010001662579Cccc:TwoThousandAndTwentyStockOptionAndIncentivePlanMember美國公認會計準則:次要事件成員2024-01-010001662579美國-公認會計準則:研究和開發費用成員2023-01-012023-12-310001662579美國-公認會計準則:研究和開發費用成員2022-01-012022-12-310001662579美國-公認會計準則:一般和行政費用成員2023-01-012023-12-310001662579美國-公認會計準則:一般和行政費用成員2022-01-012022-12-310001662579Cccc:TwoThousandAndTwentyStockOptionAndIncentivePlanMember2022-12-310001662579Cccc:TwoThousandAndTwentyStockOptionAndIncentivePlanMember2022-01-012022-12-310001662579Cccc:TwoThousandAndTwentyStockOptionAndIncentivePlanMember2023-01-012023-12-310001662579SRT:最小成員數Cccc:TwoThousandAndTwentyStockOptionAndIncentivePlanMember2023-01-012023-12-310001662579Cccc:TwoThousandAndTwentyStockOptionAndIncentivePlanMemberSRT:最大成員數2023-01-012023-12-310001662579SRT:最小成員數Cccc:TwoThousandAndTwentyStockOptionAndIncentivePlanMember2022-01-012022-12-310001662579Cccc:TwoThousandAndTwentyStockOptionAndIncentivePlanMemberSRT:最大成員數2022-01-012022-12-310001662579美國-GAAP:受限股票單位RSU成員2022-01-310001662579美國-GAAP:受限股票單位RSU成員2022-02-280001662579美國-GAAP:受限股票單位RSU成員2023-12-310001662579美國-GAAP:受限股票單位RSU成員2022-12-310001662579美國-GAAP:受限股票單位RSU成員2023-01-012023-12-310001662579CCCC:TimeBasedRestratedStockUnitsMember2023-02-280001662579CCCC:TimeBasedRestratedStockUnitsMember2023-01-310001662579CCCC:TimeBasedRestratedStockUnitsMember2023-12-310001662579CCCC:TimeBasedRestratedStockUnitsMember2022-12-310001662579CCCC:TimeBasedRestratedStockUnitsMember2023-01-012023-12-310001662579美國-公認會計準則:員工股票期權成員Us-gaap:ShareBasedPaymentArrangementEmployeeMember2023-07-310001662579美國-公認會計準則:員工股票期權成員Us-gaap:ShareBasedPaymentArrangementEmployeeMember2023-07-012023-07-310001662579美國-公認會計準則:員工股票期權成員Us-gaap:ShareBasedPaymentArrangementEmployeeMemberUs-gaap:ShareBasedCompensationAwardTrancheOneMember2023-07-012023-07-310001662579美國-公認會計準則:員工股票期權成員Us-gaap:ShareBasedPaymentArrangementEmployeeMember2023-12-310001662579Cccc:TwoThousandTwentyEmployeeStockPurchasePlanMember2020-09-012020-09-300001662579Cccc:TwoThousandTwentyEmployeeStockPurchasePlanMember2023-01-012023-12-310001662579Cccc:TwoThousandTwentyEmployeeStockPurchasePlanMember2023-12-310001662579Cccc:TwoThousandTwentyEmployeeStockPurchasePlanMember2021-01-012021-01-010001662579Cccc:TwoThousandTwentyEmployeeStockPurchasePlanMember2021-01-010001662579Cccc:TwoThousandTwentyEmployeeStockPurchasePlanMember2022-01-010001662579美國-公認會計準則:員工股票期權成員Us-gaap:ShareBasedPaymentArrangementEmployeeMemberUs-gaap:ShareBasedCompensationAwardTrancheTwoMember2023-07-012023-07-310001662579美國-GAAP:國內/地區成員美國-公認會計準則:研究成員2023-12-310001662579美國-GAAP:國內/地區成員美國-公認會計準則:研究成員2022-12-310001662579美國-公認會計準則:研究成員美國-公認會計準則:州和地方法律法規成員2023-12-310001662579美國-公認會計準則:研究成員美國-公認會計準則:州和地方法律法規成員2022-12-310001662579美國公認會計準則:國際收入服務IRSM成員2023-12-310001662579CCCC:購買公共股票的選項成員2023-01-012023-12-310001662579CCCC:購買公共股票的選項成員2022-01-012022-12-310001662579SRT:最大成員數2023-01-012023-12-310001662579美國公認會計準則:次要事件成員2024-01-092024-01-090001662579美國公認會計準則:次要事件成員2024-01-09 美國
美國證券交易委員會
華盛頓特區,20549
_____________________________________
表格10-K
_____________________________________
(標記一)
| | | | | |
| x | 根據1934年《證券交易法》第13或15(D)條提交的年度報告 |
截至本財政年度止12月31日, 2023
或
| | | | | |
| o | 根據1934年《證券交易法》第13或15(D)條提交的過渡報告 |
對於從日本到日本的過渡期,日本將從中國過渡到日本,中國將從日本轉向日本。
佣金文件編號001-39567
_____________________________________
C4治療公司
(註冊人的確切姓名載於其章程)
_____________________________________
| | | | | |
| 特拉華州 | 47-5617627 |
(述明或其他司法管轄權
公司或組織) | (税務局僱主 識別號碼) |
阿森納大道490號, 套房120 沃特敦, 體量 | 02472 |
| (主要執行辦公室地址) | (郵政編碼) |
註冊人的電話號碼,包括區號:(617) 231-0700
_____________________________________
根據該法第12(B)條登記的證券:
| | | | | | | | | | | | | | |
| 每個班級的標題 | | 交易
符號 | | 註冊的每個交易所的名稱 |
| 普通股,每股面值0.0001美元 | | CCCC | | 納斯達克全球精選市場 |
根據該法第12(G)條登記的證券:無
_____________________________________
如果註冊人是證券法第405條規定的知名經驗豐富的發行人,請用複選標記表示。是o 不是x
用複選標記表示註冊人是否不需要根據該法第13或15(D)條提交報告。是o 不是x
用複選標記表示註冊人是否:(1)在過去12個月內(或註冊人需要提交此類報告的較短時間內)提交了1934年《證券交易法》第13或15(D)條要求提交的所有報告,以及(2)在過去90天內一直遵守此類提交要求。是x不是o
用複選標記表示註冊人是否在過去12個月內(或在註冊人被要求提交此類文件的較短時間內)以電子方式提交了根據S-T規則第405條(本章232.405節)要求提交的每個交互數據文件。是x不是o
用複選標記表示註冊人是大型加速申報公司、加速申報公司、非加速申報公司、較小的報告公司或新興成長型公司。請參閲《交易法》第12b-2條規則中“大型加速申報公司”、“加速申報公司”、“較小申報公司”和“新興成長型公司”的定義。
| | | | | | | | | | | | | | |
| 大型加速文件服務器 | o | | 加速文件管理器 | o |
| 非加速文件服務器 | x | | 規模較小的報告公司 | x |
| 新興成長型公司 | o | | | |
如果是一家新興的成長型公司,用複選標記表示註冊人是否已選擇不使用延長的過渡期來遵守根據《交易所法》第13(A)節提供的任何新的或修訂的財務會計準則。o
用複選標記表示註冊人是否提交了一份報告,證明其管理層根據《薩班斯-奧克斯利法案》(《美國聯邦法典》第15編,第7262(B)節)第404(B)條對其財務報告的內部控制的有效性進行了評估,該評估是由編制或發佈其審計報告的註冊會計師事務所進行的。o
如果證券是根據該法第12(B)條登記的,應用複選標記表示登記人的財務報表是否反映了對以前發佈的財務報表的錯誤更正。o
用複選標記表示這些錯誤更正中是否有任何重述需要對註冊人的任何執行人員在相關恢復期間根據第240.10D-1(B)條收到的基於激勵的補償進行恢復分析。o
如果證券是根據該法第12(B)條登記的,應用複選標記表示登記人的財務報表是否反映了對以前發佈的財務報表的錯誤更正。o
用複選標記表示這些錯誤更正中是否有任何重述需要對註冊人的任何執行人員在相關恢復期間根據第240.10D-1(B)條收到的基於激勵的補償進行恢復分析。o
用複選標記表示註冊人是否是空殼公司(如《交易法》第12b-2條所定義)。是o不是x
註冊人的非關聯公司持有的有投票權和無投票權普通股的總市值,基於普通股在納斯達克股票市場2023年6月30日的收盤價,為$129,573,953.
截至2024年2月14日,註冊人已發行普通股的數量為68,600,669.
以引用方式併入的文件
註冊人將根據第14A條在截至2023年12月31日的財政年度結束後120天內提交的2023年股東年會的最終委託書的部分內容以引用的方式併入本年度報告的第三部分Form 10-K,其範圍在本文所述的範圍內。
| | | | | | | | |
審計師事務所ID:185 | 審計師姓名:畢馬威會計師事務所 | 審計師位置:美國馬薩諸塞州波士頓 |
目錄表
| | | | | | | | |
| | 頁面 |
第一部分 | | |
第1項。 | 業務 | 1 |
第1A項。 | 風險因素 | 36 |
項目1B。 | 未解決的員工意見 | 78 |
第二項。 | 屬性 | 79 |
第三項。 | 法律訴訟 | 79 |
第四項。 | 煤礦安全信息披露 | 79 |
| | |
第II部 | | |
第五項。 | 註冊人普通股市場、相關股東事項與發行人購買股權證券 | 80 |
第六項。 | [已保留] | 80 |
第7項。 | 管理層對財務狀況和經營成果的探討與分析 | 80 |
第7A項。 | 關於市場風險的定量和定性披露 | 89 |
第八項。 | 財務報表和補充數據 | 89 |
第九項。 | 會計與財務信息披露的變更與分歧 | 89 |
第9A項。 | 控制和程序 | 89 |
項目9B。 | 其他信息 | 90 |
項目9C。 | 關於妨礙檢查的外國司法管轄區的披露 | 90 |
| | |
第三部分 | | |
第10項。 | 董事、高管與公司治理 | 91 |
第11項。 | 高管薪酬 | 91 |
第12項。 | 某些實益擁有人的擔保所有權以及管理層和相關股東的事項 | 91 |
第13項。 | 某些關係和相關交易,以及董事的獨立性 | 91 |
第14項。 | 首席會計費及服務 | 91 |
| | |
第四部分 | | |
第15項。 | 展示、財務報表明細表 | 92 |
項目16 | 表格10-K摘要 | 94 |
關於前瞻性陳述的特別説明
本年度報告以Form 10-K或Form 10-K的形式,包括題為“管理層對財務狀況和經營結果的討論和分析”的章節,包含基於管理層的信念和假設以及管理層目前掌握的信息的明示或暗示的前瞻性陳述。儘管我們認為這些前瞻性陳述中反映的預期是合理的,但這些陳述涉及未來事件或我們未來的運營或財務表現,涉及已知和未知的風險、不確定性和其他因素,可能會導致我們的實際結果、業績或成就與這些前瞻性陳述明示或暗示的任何未來結果、業績或成就大不相同。本10-K表格中的前瞻性陳述可能包括但不限於關於以下方面的陳述:
•我們的研究和開發計劃以及我們當前和未來的臨牀前研究和臨牀試驗的啟動、時間、進度、結果、安全性和有效性以及成本,包括關於研究或試驗的啟動和完成時間、試驗結果將在多長時間內可用以及我們的研究和開發計劃的聲明;
•我們有能力為我們的運營獲得必要的資金,以完成我們候選產品的進一步開發、製造和商業化;
•我們有能力為當前或未來的任何候選產品獲得並保持監管部門的批准;
•預計我們現有的現金和現金等價物以及有價證券將足以支付我們的運營費用和資本支出要求的時間段;
•我們識別和開發用於治療其他疾病適應症的候選產品的能力;
•我們的候選產品的潛在屬性和優勢;
•我們可能開發的任何候選產品的市場接受度和臨牀實用性的速度和程度;
•我們候選產品的定價和報銷,如果獲得批准,包括一旦獲得批准,如果我們的產品後來根據2022年《通貨膨脹降低法案》或其他適用法律與醫療保險和醫療補助服務中心進行強制性價格談判,可能會降低產品的定價;
•競爭對我們當前或未來的任何候選產品的影響,以及我們行業當前和未來競爭對手的創新;
•執行我們的業務戰略計劃,我們可能開發的任何產品候選產品,以及我們的魚雷® (Target 或已租出P腐朽E在……裏面D漸進器OPtimizer)平臺;
•我們第三方戰略合作伙伴繼續與我們的候選產品相關的研究、開發和製造活動的能力和意願,包括我們根據與F.Hoffmann-La Roche Ltd和Hoffmann-La Roche Inc.、Roche,Biogen MA,Inc.、Biogen、Betta PharmPharmticals,Co.、Betta Pharma、Merck Sharp&Dohme LLC或Merck的現有合作協議推進計劃的能力,或其他新的合作協議;
•我們能夠為我們的候選產品建立和維護的知識產權保護範圍;
•對我們未來的支出、收入、資本需求和額外融資需求的估計;
•如果獲得批准,未來與第三方就我們的候選產品的製造和商業化達成的協議;
•我們候選產品的市場規模和增長潛力,以及我們為這些市場服務的能力;
•我們的財務業績;
•美國和其他國家的監管動態;
•我們與第三方供應商和製造商簽訂合同的能力以及他們充分履行合同的能力;
•已有或可能獲得的競爭性療法的成功;
•我們吸引和留住關鍵科學或管理人員的能力;
•與我們的競爭對手和本行業有關的發展;以及
•其他風險和不確定性,包括第一部分第1A項--本表格10-K中的風險因素中討論的風險和不確定性。
在某些情況下,前瞻性表述可以通過“將”、“可能”、“應該”、“可能”、“預期”、“打算”、“計劃”、“目標”、“預期”、“相信”、“估計”、“預測”、“潛在”、“繼續”或這些術語的否定或其他類似術語來識別,儘管並不是所有的前瞻性表述都包含這些詞語。這些聲明只是預測。你不應該過分依賴前瞻性陳述,因為它們涉及已知和未知的風險、不確定性和其他因素,這些因素在某些情況下是我們無法控制的,可能會對結果產生重大影響。可能導致實際結果與當前預期大不相同的因素包括,除其他事項外,在題為“風險因素”一節和本10-K表其他部分列出的因素。如果這些風險或不確定性中的一個或多個發生,或者如果我們的基本假設被證明是不正確的,實際事件或結果可能與前瞻性陳述中明示或暗示的大不相同。任何前瞻性陳述都不是對未來業績的承諾或保證。
本10-K表格中的前瞻性陳述代表我們截至本10-K表格發佈之日的觀點。我們預計隨後發生的事件和事態發展將導致我們的觀點發生變化。然而,雖然我們可能會選擇在未來的某個時候更新這些前瞻性陳述,但我們目前無意這樣做,除非適用法律要求這樣做。因此,您不應依賴這些前瞻性陳述來代表我們截至本10-K表格日期之後的任何日期的觀點。
本10-K表格可能包括我們從行業出版物和研究、調查以及第三方進行的研究中獲得的統計數據和其他行業和市場數據。行業出版物和第三方研究、調查和研究一般表明,他們的信息是從被認為可靠的來源獲得的,儘管它們不保證此類信息的準確性或完整性。我們沒有獨立核實這些來源中包含的信息。
風險因素摘要
我們實施業務戰略的能力受到許多風險的影響,在做出投資決定之前,您應該意識到這些風險。這些風險在第一部分第1A項--本表格10-K中的風險因素--中有更全面的描述。這些風險包括:
•我們是一家臨牀階段的生物製藥公司,運營歷史有限,自成立以來一直蒙受重大虧損。到目前為止,我們還沒有從產品銷售中獲得任何收入。我們預計,至少在未來幾年內,我們將繼續產生鉅額費用和不斷增加的運營虧損,可能永遠不會實現或保持盈利。截至2023年、2023年和2022年12月31日的年度,我們的淨虧損分別為1.325億美元和1.282億美元。
•我們將需要大量的額外資金來實現我們的業務目標並繼續我們的運營。如果我們無法在需要時籌集資金,我們可能會被要求推遲、限制、減少或終止我們的研究或產品開發計劃或未來的商業化努力。
•我們基於魚雷平臺發現和開發候選產品的方法未經驗證,這使得我們很難預測成功開發任何產品的時間、成本和可能性。
•雖然我們是一家臨牀階段的公司,並已開始對幾種候選產品進行臨牀試驗,但我們從未完成過任何一種候選產品的臨牀試驗。如果我們無法開發、獲得監管機構對我們的候選產品的批准和/或將其商業化,或者如果我們在做這些事情方面遇到重大延誤,我們的業務可能會受到損害。
•我們不能確定我們的臨牀前試驗和臨牀試驗的及時完成或結果。此外,臨牀前研究的結果可能不能預測臨牀試驗的結果,我們開始的任何早期臨牀試驗的結果也可能不能預測後期臨牀試驗的結果。
•我們的臨牀前研究和臨牀試驗可能無法充分證明我們的任何候選產品的安全性和有效性,這將阻止、推遲或需要額外的研究或分析來繼續開發、監管批准和商業化我們當前和未來的候選產品。
•我們與羅氏、Betta Pharma和默克公司有持續的合作協議,與Biogen和Calico也有合作協議,這兩家公司的研究期限分別於2023年6月30日和2023年3月13日到期。我們還可能在未來尋求與第三方進行更多合作,以開發和/或商業化我們的某些候選產品。然而,我們可能永遠無法實現這些現有的或潛在的協作安排下的全部潛在好處。
•我們面臨着激烈的競爭,這可能會導致其他人比我們更成功地發現、開發或商業化針對相同適應症和/或患者羣體的產品。
•我們依賴,並預計將繼續依賴第三方生產我們的候選產品,用於臨牀前和臨牀測試,以及用於商業生產(如果我們的任何候選產品獲得市場批准)。這種對第三方的依賴可能會增加我們無法及時或以可接受的成本或質量供應足夠數量的候選產品的風險。
•如果我們無法為我們的候選產品獲得所需的營銷批准、商業化、製造、獲得和維護專利保護或獲得市場認可,或者如果我們在這樣做方面遇到重大延誤,我們的業務將受到嚴重損害,我們從產品銷售中獲得收入的能力將受到嚴重損害。
•如果我們無法為我們的技術和產品獲得並保持專利保護,或者如果獲得的專利保護範圍不夠廣泛或不夠可執行,我們的競爭對手可能會開發和商業化與我們相似或相同的技術、候選產品和產品,我們成功將我們的技術、候選產品和產品商業化的能力可能會受到損害。
關於公司推薦人的説明
除文意另有所指外,本10-K表格中的術語“C4治療”、“公司”、“我們”、“我們”和“我們”是指C4治療公司及其合併的子公司。
關於商標的説明
我們擁有或有權使用與我們的業務運營相關的各種商標、服務標記和商品名稱,包括我們的公司名稱、C4治療、我們的徽標、我們的魚雷技術平臺的名稱以及我們的BIDAC和MONODAC蛋白質降解器候選產品的名稱。本表格10-K還可能包含第三方的商標、服務標記和商標名,這些都是其各自所有者的財產。我們在本招股説明書中使用或展示第三方的商標、服務標記、商號或產品,不是為了也不暗示我們與我們的關係,或我們的背書或贊助。僅為方便起見,本招股説明書中提及的商標、服務標記和商號可不使用®,TM或SM但省略此類引用並不意味着我們不會在最大程度上根據適用法律主張我們的權利或這些商標、服務標記和商號的適用所有者的權利。
第一部分
項目1.業務
概述
我們是一家臨牀階段的生物製藥公司,致力於實現靶向蛋白質降解(TPD)科學的承諾,以創造改變患者生活的新一代小分子藥物。通過利用我們專有的魚雷平臺,我們能夠有效地設計和優化小分子蛋白質降解器,通過利用人體破壞不需要的蛋白質的自然過程,對其預期目標高度活躍。我們相信我們的新型口腔產品候選產品有可能克服抑制劑經常出現的耐藥性,瞄準目前“無法下藥”的靶點,並改善患者的結果。到目前為止,我們已經成功地設計了幾種蛋白質降解劑,並將其應用於臨牀,覆蓋了一系列目標類別,根據我們臨牀試驗的數據,我們的候選產品已經顯示出強大的目標降解性。
我們的魚雷平臺有能力設計兩種類型的蛋白質降解器,這兩種降解器方法都反映在我們的臨牀管道中。我們將第一類降解劑稱為MonoDAC降解劑,業內一些人將其稱為“分子膠”。MonoDAC降解物通過與E3連接酶結合並在E3連接酶上創建新的表面來增強E3連接酶與靶蛋白的結合來發揮作用。我們將第二種類型的降解劑稱為BiDAC降解劑,業內一些人也將其稱為異雙功能降解劑。BiDAC降解物的設計是這樣的:分子的一端與致病目標蛋白結合,另一端與E3連接酶結合。這些降解方法中的每一種都旨在產生相同的終點:使E3連接酶離致病蛋白質足夠近,這樣E3連接酶就可以給它貼上破壞的標籤。
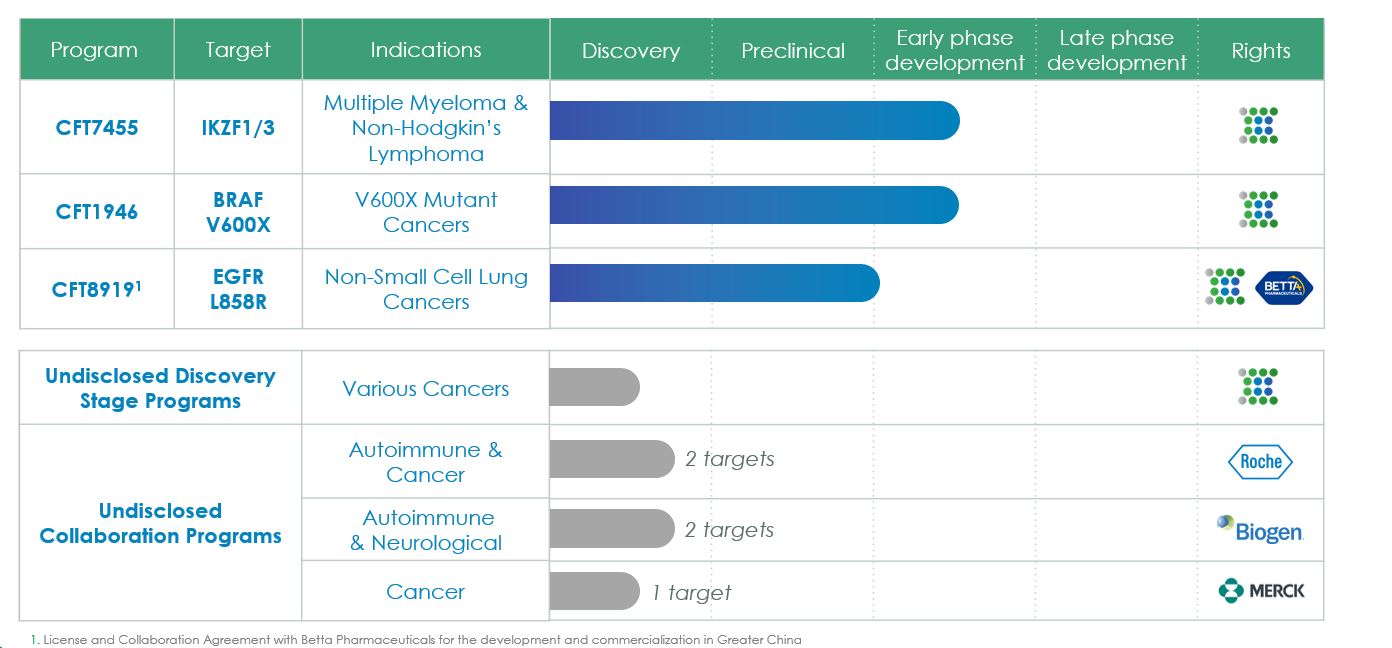
我們的臨牀前和臨牀流水線包括MonoDAC和BiDAC降解物,這些降解物有可能針對各種癌症和其他適應症的致病蛋白,用於未滿足患者需求的領域。我們全資擁有的流水線專注於腫瘤學,我們的合作戰略使我們能夠探索其他適應症。我們的整個渠道,包括夥伴關係,如下圖所示。
我們最先進的候選產品CFT7455是一種口服生物可用MonoDAC降解劑,稱為IKZF1和IKZF3,目前正處於多發性骨髓瘤(MM)和非霍奇金淋巴瘤(NHL)的臨牀開發中。以IKZF1和IKZF3為靶點有很強的機制基礎和明確的生物學定義,以一種新型降解劑為靶標有可能解決一個重要的未得到滿足的需求。在我們的臨牀前研究中,這種候選產品已經展示了具有良好藥理學特性的有效和選擇性的蛋白質降解。2021年6月,我們為該候選產品啟動了首個人類1/2期臨牀試驗。2021年8月,美國食品和藥物管理局(FDA)批准CFT7455用於治療MM。2023年12月,我們提供了CFT7455 1/2期試驗劑量遞增部分的陽性臨牀數據,作為單一療法並與地塞米松聯合治療MM。我們繼續推進MM和NHL正在進行的1/2期臨牀試驗。
我們的下一個最先進的候選產品CFT1946是一種口服生物可用BiDAC降解劑,旨在有效和選擇性地對抗BRAF V600X突變蛋白,用於治療黑色素瘤、非小細胞肺癌、結直腸癌和其他含有這種突變的惡性腫瘤。在臨牀前研究中,CFT1946在CRC和NSCLC BRAF V600X異種移植模型以及黑色素瘤患者來源的異種移植或PDX,BRAF抑制劑耐藥模型中比標準護理療法更有效。此外,CFT1946在臨牀前轉移性黑色素瘤中樞神經系統(CNS)模型中是活躍的。2023年1月,我們啟動了CFT1946的首個人類1/2期臨牀試驗,用於治療BRAF V600X突變實體腫瘤,包括非小細胞肺癌、結直腸癌和黑色素瘤。2024年1月,我們分享了正在進行的1/2階段試驗的前兩個劑量升級隊列的PK和PD數據,顯示了劑量比例暴露和口服生物利用度,這與BRAF的深度降解有關。我們繼續進行BRAF V600X癌症的1/2期臨牀試驗,包括黑色素瘤、非小細胞肺癌和結直腸癌。
此外,我們正在開發CFT 8919,這是一種在NSCLC中具有L 858 R突變的表皮生長因子受體(EGFR)的口服生物可利用、變構、多藥選擇性BiDAC降解劑。在臨牀前研究中,在Ba/F3細胞模型中,與L 858 R單突變相比,CFT 8919對EGFR抑制耐藥的EGFR突變(包括L 858 R-C797 S、L 858 R-T790 M和L 858 R-T790 M-C797 S)表現出等效的抗增殖活性 體外培養。2023年5月,我們與Betta Pharma簽訂了一項許可和合作協議,就CFT8919在內地中國、香港特別行政區、澳門特別行政區和臺灣的開發和商業化進行合作,我們保留在世界其他地區開發和商業化CFT8919的權利。此外,2023年6月,FDA批准了CFT8919的研究新藥或IND申請,2023年12月,倍他醫藥獲得了中國領導的國家醫療產品管理局對CFT8919的臨牀試驗申請或CTA批准。
除了這些最初的候選產品外,我們還針對我們自己的專利流水線和我們與默克、生物遺傳和羅氏合作開發的流水線開發了針對臨牀驗證和目前不可用藥的目標的新降解劑,從而進一步使我們的流水線多樣化。我們設計的降解物在臨牀前研究中成功地實現了血腦屏障的穿透,這是開發有可能治療腫瘤學腦轉移以及神經退行性疾病等治療領域的藥物的關鍵一步。我們還相信,在許多治療領域和適應症中,利用我們的魚雷平臺開發新型降解劑可能是有利的。
我們的戰略
我們致力於通過發現、開發和商業化破壞致病蛋白質的新療法來改變癌症和其他疾病的治療方法。
我們戰略的關鍵要素是:
•推進我們的臨牀口腔腫瘤降解劑計劃。使用我們專有的魚雷平臺,我們已經產生了用於癌症治療的新產品候選產品,我們相信這些產品有可能改善患者的預後。根據我們的臨牀試驗結果,我們將與FDA合作,討論適用於我們的候選產品的潛在加速開發和加速審批途徑。
•繼續將我們的內部渠道集中在我們認為可以從TPD方法中受益的腫瘤學目標上。通過我們的靶點識別過程,我們評估TPD在哪裏可以對患者產生超大的影響,要麼是通過它改善現有治療方式的潛力,要麼是通過選擇目前被抑制劑認為“不可用藥”的靶點,在生物學上表明,降解這些靶點可能會對疾病產生影響。在考慮為研究活動選擇什麼目標時,生物學扮演着重要的角色。我們選擇與某種疾病有明確遺傳聯繫的目標。我們還考慮降解劑如何提供比抑制劑或其他治療選擇更好的好處。
我們還將臨牀開發作為目標選擇過程的一部分。我們審查潛在的目標,以確保我們可以前瞻性地選擇患者進行臨牀試驗,並能夠確定註冊的路徑。我們還評估了可用的生物標記物分析,以幫助我們確定哪些患者可能從研究治療中受益。我們正在與我們的合作伙伴將我們目前的專有計劃集中在選定的腫瘤學適應症以及其他適應症上。
•與合作伙伴合作,實現魚雷平臺的全部價值。我們與羅氏、生物遺傳和默克公司現有戰略合作,在這些合作下,我們正在努力在多個治療領域和目標上識別和開發新的降解物,包括降解物-抗體結合物或DAC等新方法。這些夥伴關係使我們能夠推進和加快我們的研究,並使我們能夠獲得更多的能力,並擴大我們的魚雷的用途 站臺。
蛋白質降解研究綜述
蛋白質降解
蛋白質是一種大而複雜的分子,在人體內扮演着許多關鍵角色。由於它們在生物功能中的核心作用,蛋白質相互作用控制着導致健康和疾病狀態的機制。疾病通常是由突變引起的,這些突變改變了蛋白質的正常功能,進而導致蛋白質功能障礙,進而導致疾病。最近的科學進步繼續暗示特定蛋白質在多種疾病狀態中的作用。
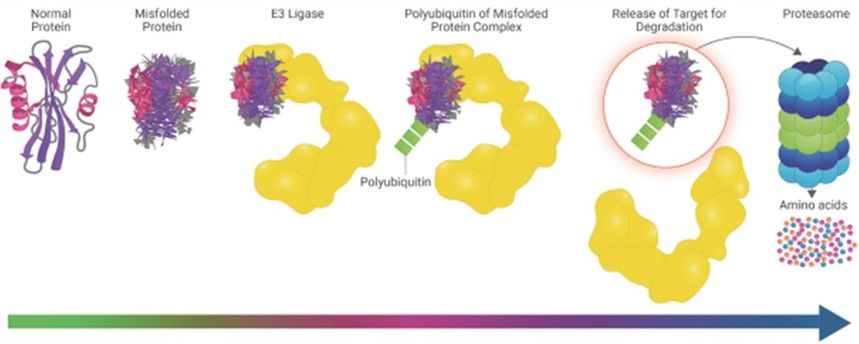
細胞內的蛋白質水平受其合成速率和降解速率之間的平衡控制。蛋白質降解提供了一種自然的機制來維持蛋白質水平的穩定平衡,去除老化或有缺陷的蛋白質,或迅速消除某些調節蛋白質對特定信號的反應。人體有一種高度保守的降解機制,稱為泛素蛋白酶體系統,簡稱UPS,可以識別蛋白質並將其分解成其組成的氨基酸。這一過程在一定程度上是由一種名為E3連接酶的蛋白質家族介導的。E3連接酶的主要作用是充當質量控制檢查員,識別陳舊、損壞、錯誤摺疊或其他被認為準備降解的蛋白質。當E3連接酶確定要降解的目標蛋白質時,它會在一個稱為泛素化的過程中附加一個名為泛素的分子標籤。這種泛素化過程通常會持續到目標蛋白被標記上多個泛素為止,這就是所謂的多泛素化。一旦目標蛋白被多泛素化,它就會被E3連接酶釋放,然後很快被蛋白酶體識別,蛋白酶體是細胞的回收工廠。蛋白酶體將多聚泛素化的蛋白質降解成其組成的氨基酸,然後這些氨基酸可以循環形成新的蛋白質或被細胞排泄出來。此過程如下圖所示。
大約5%的人類基因專門編碼泛素-蛋白酶體系統的組成部分。此外,許多具有治療意義的蛋白質通常由E3連接酶調控。總而言之,這些因素強調了E3連接酶在正常細胞功能中所起的重要作用,以及它們如何被用來對抗治療性蛋白質靶標。
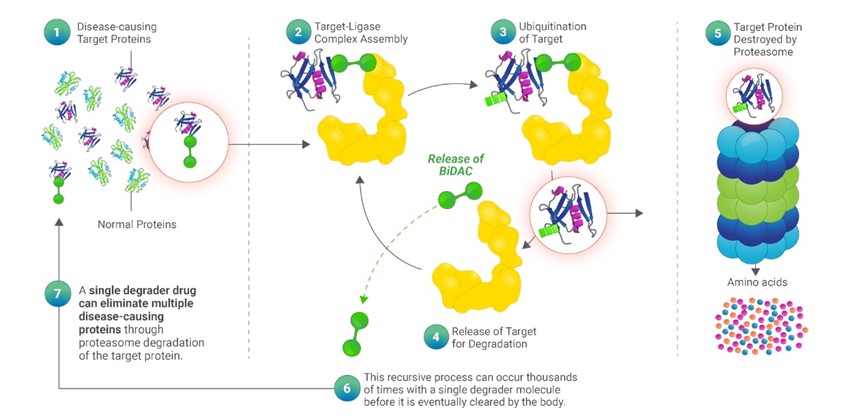
靶向蛋白降解物代表了一種新的方式,它試圖利用這種自然降解機制來破壞致病的目標蛋白,否則E3連接酶不會將其作為破壞的目標。我們的降解物介導的靶向蛋白質降解過程如下圖所示。
我們的MonoDAC降解物和BiDAC降解物遵循相同的催化過程,第一步是在天然E3連接酶、降解物和目標蛋白之間形成一個複合體,我們稱之為三元複合體。對於BiDAC降級器,這在上圖的步驟2中顯示。在步驟3中,可進行泛素化的合適的三元複合體的形成導致靶蛋白的多泛素化。一旦目標蛋白的一個分子的多泛素化過程完成,降解物被釋放,如步驟4所示,然後在步驟5中由蛋白酶體降解目標蛋白。由於降解物的釋放不變,這種遞歸過程--結合目標蛋白、與E3連接酶形成三元複合體、多泛素化和釋放以進行降解--可能與單個降解物分子一起發生數千次,直到最終被身體清除。重要的是,天然的蛋白質降解過程和我們的降解物介導的靶向蛋白質降解都發生得很快,從最初的靶標-連接酶相遇到多泛素化,再到蛋白酶體釋放降解,大約只有幾毫秒。通過這種方式,蛋白質降解物充當自然過程的催化劑,我們將這個過程稱為催化循環,這是降解物和傳統蛋白質抑制劑的關鍵區別,必須與目標蛋白質結合才能保持有效。
靶向蛋白質降解相對於傳統蛋白質抑制劑的優勢
目前的許多靶向治療都是基於抑制目標蛋白質生物功能的小分子。以抑制劑為基礎的治療的主要侷限性之一是,需要持續的目標佔有率水平才能抑制蛋白質的生物功能,從而達到療效。由於藥物的藥理作用是由藥物暴露情況決定的,藥物作用的總體時間和持續時間取決於藥物的吸收、分佈和消除。這些暴露可能很難實現,並可能增加重大偏離目標的副作用的可能性。這種方法的另一個限制是需要找到與蛋白質上特定活性部位結合的化合物,從而導致功能抑制,因為目標蛋白質上有許多小分子可以結合的部位,但對整體功能沒有影響。
我們認為,靶向蛋白質降解是一種新的方法,與傳統的小分子抑制劑方法相比,它可以提供顯著的潛在好處,包括改進和持續的效力、快速和遞歸的催化效果、高選擇性和廣闊的靶向前景。
提高並持續的效力
降解物能夠提供成倍放大的效果,因為單個降解物分子可以遞歸地在大量目標蛋白質上發揮作用。降解物分子能夠多次重複其催化循環的這種能力稱為催化放大。相比之下,傳統的蛋白質抑制劑依賴於抑制劑分子與目標蛋白質的一對一結合,只有當抑制劑結合時,蛋白質才會失活。這意味着需要更高濃度的蛋白質抑制劑藥物才能達到與蛋白質降解劑相同的治療效果。
除了需要比蛋白質抑制劑少得多的藥物外,降解劑對目標蛋白質功能的影響比傳統抑制劑更持久。這是因為一旦抑制物不再與目標蛋白結合,目標蛋白的活性就會恢復,而降解物完全消除其目標蛋白,致病活性就會被阻止,直到細胞能夠合成替代蛋白--這個過程可能需要幾個小時甚至幾天。因此,
與典型的可逆抑制劑不同,降解劑的效果在從體內清除後可以很好地保持下去。降解劑完全消除其靶標的能力是相對於傳統抑制劑提高效力的另一種機制。這是因為一種蛋白質通常具有多種功能,每一種功能都由不同的結構域介導。抑制物只能幹擾受其結合區域直接影響的蛋白質功能。在致病蛋白質中,多種功能對致病活性有貢獻(例如,酶活性異常以及形成多蛋白質複合體的能力),降解物將消除所有功能,因此與僅阻止一種功能的抑制劑相比,降解物具有更深遠的影響。所有這些因素都意味着,降解劑可能有助於實現更持久的生物效應和更好的臨牀結果。
高選擇性
蛋白質抑制的主要挑戰之一是試圖識別和開發只針對癌細胞或致病蛋白質的分子,而不對正常細胞或其他蛋白質產生有害影響,即通常所説的脱靶效應。
蛋白質降解催化循環中的每個步驟都需要靶蛋白和E3連接酶的特定定位以通過循環進行,並且這些定位要求可以用作過濾器以增加降解劑分子的選擇性,使得即使降解劑結合多種蛋白質,也只有靶蛋白最終被降解。例如,降解物不僅與靶蛋白相互作用,而且必須以靶向E3連接酶的方式進行,並呈現適合於生產性靶蛋白泛素化的三元複合物構象。因此,即使降解劑與非靶蛋白結合,所得三元複合物也可能不具有適於促進泛素化和隨後降解的構象。我們能夠利用泛素-蛋白酶體蛋白降解途徑的這些內在特性來設計對致病蛋白具有高度選擇性的降解劑。
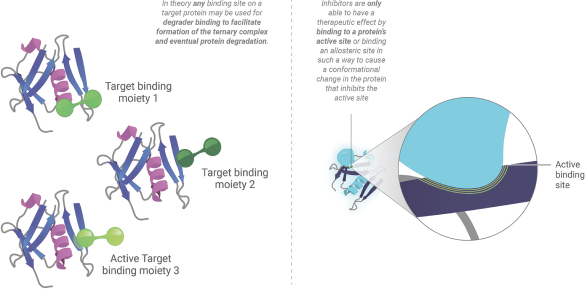
廣闊的目標市場
傳統的抑制劑只有在它們能夠與幹擾其功能的致病蛋白質上的位點緊密結合時才能產生治療效果。這需要抑制劑直接結合到蛋白質的活性位點,或以導致損害蛋白質活性的構象變化的方式結合到變構位點。這固有地限制了傳統抑制劑可尋址的可藥物化靶點的數量,因為許多潛在的結合位點不在幹擾功能的蛋白質區域中。相反,降解劑可以使用蛋白質上的任何結合位點來促進與E3連接酶形成三元複合物,從而導致其破壞。此外,儘管抑制劑需要對其結合位點的高親和力以維持佔據和阻斷功能,但降解劑可以以瞬時方式與相對較弱的親和力相互作用,並且仍然能夠實現蛋白質的泛素化和破壞。靶向蛋白質降解極大地擴展了潛在的靶池,以包括目前被認為不可用的那些的顯著比例。
我們的方法
我們採用全面的方法來發現、選擇和開發候選產品,以最大限度地發揮我們蛋白質降解劑的潛在治療效益。我們尋找可能從降解劑中獲益最多的具有高價值蛋白質靶點的指標,並將催化降解週轉率作為評估蛋白質降解的關鍵指標。為此,我們在實驗工具、計算和預測模型以及團隊專業知識方面進行了大量投資,以通過我們的TORPEDO平臺分析和優化我們降解器的催化能力。此外,我們利用我們的平臺來優化我們的降解劑的能力,以啟動泛素-蛋白酶體蛋白降解循環並預測其功能。 體內.由於我們的TORPEDO允許快速優化 平臺和我們的能力
預測降級效果的平臺體內,我們能夠快速高效地將項目從目標確定階段推進到候選開發階段。
我們的魚雷 站臺
我們專有的魚雷平臺是一組實驗方法和工具的集合,使我們能夠設計、分析和預測降解劑的性能,這使我們能夠在分子水平上設計降解劑以增強效力和選擇性。該平臺的關鍵要素包括:
•關注催化效率:我們尋求通過將我們的分析技術和預測模型集中在降解劑特性和最終蛋白質降解之間的關係來優化催化降解週轉率。我們的降解器旨在激活E3連接酶,促進目標蛋白結合和泛素化,導致整體目標快速降解。我們的降解者能夠遞歸地重複這一過程,同一個降解者分子與目標蛋白質的許多副本相互作用,使我們能夠優化我們的候選產品,以實現催化降解週轉,從而創造出具有更大治療效果的候選產品。我們的MonoDAC降解劑和BiDAC降解劑需要獲得足夠的結合親和力來啟動短暫的三元絡合物形成,但與傳統抑制劑不同的是,它們不需要實現長期穩定的結合來達到預期的生理效果。在我們的一些臨牀前研究中,我們觀察到,即使結合劑較弱,也可以產生非常有效的降解劑,因為它們可能允許更高的催化降解週轉率,這是我們優先考慮的,以實現更高的活性。
•對Cereblon的投資:我們的鉛降解者利用Cereblon作為E3連接酶。人類蛋白質組中有600多種E3連接酶,其中生物學特性最好的不超過50種。少數E3連接酶,包括Cereblon、VHL、MDM2、IAPs和ç-TRCP,已被報道適用於靶向蛋白質降解。我們選擇將Cereblon作為我們的蛋白質降解方法的E3連接酶目標,原因有幾個:
◦已批准的藥物沙利度胺、來那度胺和泊馬度胺的廣泛臨牀經驗表明,使用Cereblon可以影響人類疾病的靶向降解。這些分子的作用機制是通過使它們與Cereblon形成絡合物來降解疾病靶標,特別是IKZF1和IKZF3。來那度胺和泊馬度胺都是被批准的藥物,自2006年和2013年分別獲得批准以來,一直是MM治療的護理標準的一部分。總之,這一經驗在臨牀上證實了Cereblon是一種E3連接酶,可以用來生產有效的藥物。
◦Cereblon在組織中廣泛表達,存在於包括細胞質和細胞核在內的所有細胞室,可能允許Cereblon介導的靶向蛋白在廣泛的臨牀環境和潛在靶點中降解。
◦我們已經開發了多種獨特的專有Cereblon粘合劑,我們為改善類藥物特性而設計,例如增強口服生物利用度、溶解性、滲透性和穩定性,我們所有的候選產品和計劃都受益於我們專有Cereblon粘合劑的這些特性。我們的Cereblon活頁夾庫為降解器發現提供了一個專有且功能強大的工具包。這種Cereblon粘合劑工具包能夠以更模塊化的方法識別和優化降解劑,因為這些粘合劑類別中的每一種都編碼了不同的類似藥物的屬性,重要的是,在蛋白質降解後從Cereblon表面的獨特“退出軌跡”,這可以促進更好的目標降解週轉。
通過專注於催化效率和利用Cereblon,我們建立了TPD領域獨有的能力。通過這些功能,我們可以:
•設計、分析和預測降級器性能:我們在計算方法和工具上進行了大量投資,以增強我們合理設計降解器的能力。在硅膠中模型允許我們在我們的管道中設計具有更強的效力和選擇性的降解器。此外,我們的細胞降解分析提供了高質量的數據,我們使用基於基本酶學原理的專有和獨特的框架進行分析,這使我們能夠預測任何劑量下目標降解的深度和持續時間。體內.
◦我們已經開發了高通量的細胞降解分析,旨在產生定量數據,顯示降解劑濃度和目標蛋白質降解之間的關係。與傳統的蛋白質印跡方法相比,這種方法能夠以更高的精度和更高的吞吐量對蛋白質降解進行定量。我們的實驗數據在我們的專有模型中的應用
然後使我們能夠預測蛋白質降解動力學,並使我們能夠快速迭代和改進候選降解劑,併為優化催化降解週轉的性能進行設計。
◦我們還建立了一個評估和平衡降解物濃度、時間和目標蛋白質降解之間關係的酶學框架,以確定降解物誘導蛋白質降解的關鍵動力學參數。我們已經將這個框架擴展到專有的PK/PD模型,這些模型將這些動力學參數與新陳代謝和PK暴露情況相結合,以預測體內性能降級。我們對降級性能的預測通常通過以下方式得到驗證體內PD實驗,使用標準的蛋白質印跡分析方法測量腫瘤樣本中的靶標降解。我們發現,這些模型將細胞分析與預測聯繫起來體內性能顯著加快了我們的發現過程,我們相信這將增加從臨牀前模型成功過渡到臨牀的可能性。
•開發MonoDAC和BiDAC降解器:我們的平臺提供了靈活性,通過量身定製的方法解決不同類別的致病蛋白質。對於那些存在可配基結合位點的靶蛋白,我們可以利用我們的Cereblon工具箱開發BiDAC降解劑。對於不存在可配基結合位點或缺乏足夠特異性的靶蛋白,如轉錄因子,我們可以利用我們專有的包含7,000多種化合物的MonoDAC文庫來篩選針對靶標的匹配。一旦我們確認命中目標,我們就能夠利用我們魚雷平臺的其他元素來優化MonoDAC或BiDAC降級器。
這些功能有助於我們的平臺將重點放在創造我們認為將呈現最低生物和毒性風險的候選產品上,同時還解決了未滿足的醫療需求。
我們的候選產品-高效和選擇性的靶向蛋白質降解劑
我們目前正在進行CFT7455和CFT1946的首個人類1/2期臨牀試驗。這些方案針對的是現有療法仍未得到充分治療的目標。
CFT7455:治療多發性骨髓瘤和非霍奇金淋巴瘤的IKZF1/3降解劑
我們正在開發CFT7455,一種針對IKZF1/3的口服生物降解劑,用於治療MM和NHL。我們選擇IKZF1和IZKF3作為我們的初始降解目標,因為它們具有強大的機理基礎和明確的生物學定義。在臨牀前研究中,CFT7455在多發性骨髓瘤(MM)、外周T細胞淋巴瘤(PTCL)和套細胞淋巴瘤(MCL)皮下移植小鼠模型中顯示出強勁的活性,提供了臨牀前概念驗證。我們相信,CFT7455的差異化藥理作用,包括其高效力,可能會在我們正在進行其開發的每個適應症中轉化為臨牀結果的改善。
2023年1/2月,我們公佈了正在進行的CFT 7455單藥治療和聯合地塞米松治療MM的I/II期臨牀試驗的臨牀數據。數據顯示,14天給藥/14天停藥方案是最佳方案,截至數據截止日期,CFT 7455耐受性良好,中性粒細胞減少是預期的靶向毒性,最常見的3級或以上不良事件。此外,在既往接受過多種MM治療(包括B細胞成熟抗原或BCMA治療)的患者中,CFT 7455表現出抗骨髓瘤活性和國際骨髓瘤工作組(IMWG)反應。
IKZF 1和IZKF 3是某些血液癌症的生物學靶點
IKZF 1和IKZF 3是淋巴-骨髓多能祖細胞通過成熟免疫細胞(包括T細胞和漿細胞,如B細胞)分化的核心轉錄因子。特別地,通過阻止B細胞的成熟,在B細胞驅動的血癌(例如MM、B細胞淋巴瘤和骨髓增生異常綜合徵)中存在抗增殖作用。除了B細胞成熟對IKFZ 1/3的這些細胞內在依賴性外,第三方研究顯示IKZF 1和IKZF 3的降解導致T細胞中IL-2表達增強,這意味着IKZF 1/3降解也誘導T細胞活性並可能發揮抗癌作用。此外,IKZF 1/3先前已被驗證為臨牀實踐中的靶點,因為來那度胺和泊馬度胺主要靶向IKZF 1/3作為其作用機制。
我們的首次人體I/II期試驗
於2021年6月,我們啟動了首次針對CFT 7455的人體1/2期臨牀試驗。本試驗旨在主要研究安全性、耐受性和抗腫瘤活性。次要和探索性目的是表徵CFT 7455的PK和PD特徵。該試驗正在評估CFT 7455作為NHL患者的單藥治療,以及與地塞米松聯合治療複發性或難治性MM患者。我們繼續招募患者參加1期劑量遞增試驗的這兩個組。
多發性骨髓瘤
在美國,MM佔所有新發癌症病例的近1.8%。美國國家癌症研究所估計,2023年美國有35,730例MM新發病例,12,590例死亡。儘管MM患者的總體結局在過去幾十年中有了大幅改善,但MM患者的預後較差,根據美國國立衞生研究院(NIH)的數據,預計中位五年相對生存率僅為57.9%。因此,仍有大量需求未得到滿足。
大多數MM患者對治療有初步反應。根據熒光原位雜交(FISH)對骨髓的研究,將患者分為高風險和標準風險兩類。適合造血細胞移植的高危患者接受聯合方案的誘導治療,通常包括IKZF 1/3靶向藥物如來那度胺,以減少幹細胞收集前的腫瘤細胞數量。或者,不適合造血細胞移植的患者立即接受聯合方案,通常使用三到四類藥物,包括IKZF 1/3靶向藥物和類固醇,通常是地塞米松,直到進展或不可接受的毒性。
然而,儘管有各種治療選擇,大多數患者最終會進展和/或經歷連續復發。在我們的CFT 7455臨牀項目中,我們最初關注的是複發性或難治性MM患者,這些患者接受過至少三線指定的既往治療,包括來那度胺、泊馬度胺、蛋白酶體抑制劑和抗CD 38單克隆抗體或mAb。我們認為,正在進行的CFT 7455 I/II期試驗的劑量遞增數據有可能證明CFT 7455對患者有意義的益處。
外周t細胞淋巴瘤
PTCL是NHL的異質性和典型的侵襲性組。美國國立衞生研究院的監測、流行病學和最終結果計劃(SEER計劃)估計,2023年美國有80,540例NHL新發病例,20,180例死亡。PTCL佔美國和歐洲所有NHL診斷的約5%至15%。PTCL患者的5年總生存率為30- 50%。
PTCL是一種異質性惡性腫瘤,有許多亞型,雖然這些亞型的結局各不相同,但許多PTCL患者的結局很差。例如,在沒有定義亞型的PTCL患者中,通常稱為未另行説明的PTCL或PTCL-NOS,五年總生存率約為20%至32%。此外,血管免疫母細胞性、自然殺傷/T細胞淋巴瘤、成人T細胞白血病/淋巴瘤、肝脾、腸病型或ALK-外周T細胞淋巴瘤患者的中位5年總生存率均低於50%。雖然化療的初始總體反應率約為40%至75%,但大多數患者最終復發。化療後的中位無進展生存期或PFS為12至14個月,中位5年生存率約為20%至30%。來那度胺已在一項2期臨牀試驗中對PTCL進行了評估,並顯示總體緩解率為22%至26%。然而,Cereblon調節劑如來那度胺未被廣泛使用或批准用於治療PTCL。根據我們的臨牀前數據,我們相信如果獲得批准,CFT 7455有可能為這些患者創造有意義的益處,併成為這些患者的既定護理標準。
套細胞淋巴瘤
根據美國癌症協會的數據,MCL是一種成熟的B細胞NHL,約佔所有NHL診斷的5%。根據美國國立衞生研究院的數據,每年每20萬人中就有一個病例發生。接受標準治療的患者的中位總存活率為四到五年。除了在臨牀試驗中測試的藥物外,一線治療選擇通常包括常規化學免疫治療、利妥昔單抗和放射治療的某種組合。大多數MCL患者有連續復發的經歷,並接受各種藥物的治療,包括IKFZ1/3靶向藥物、BTK抑制劑或bcl2抑制劑ventoclax。來那度胺被批准用於在之前的兩種療法(其中一種包括波特佐米)後復發或進展的MCL患者,部分基於觀察到的總應答率約為26%。然而,來那度胺並沒有被廣泛用於治療MCL。因此,我們相信,如果獲得批准,CFT7455有可能顯著改善結果,併成為這些患者的既定護理標準。
CFT1946:降解BRAF V600X治療黑色素瘤、結直腸癌和非小細胞肺癌
我們正在開發CFT1946,一種BRAF V600X的口服生物降解劑。我們選擇BRAF V600X作為目標,是因為它強大的機械原理、良好的生物學定義和未得到滿足的需求。在美國,BRAF突變發生在大約5%的癌症中,這意味着每年約有10萬名患者被診斷為BRAF突變癌症。在所有的BRAF突變中,大約70%-90%是用V600取代另一種氨基酸。我們計劃首先在三種類型的癌症中進行CFT1946的開發:黑色素瘤,其中BRAF V600X突變發生在大約35%的晚期患者中;結直腸癌,其中BRAF V600X突變發生在大約5%到10%的患者中;以及非小細胞肺癌,其中BRAF V600X突變發生在大約1%到2%的患者中。在病人身上,有
對於那些在批准的BRAF抑制劑後復發或對其沒有反應的人來説,這仍然是一個高度未得到滿足的需求。我們正在評估CFT1946在各種BRAF V600X實體腫瘤中的應用,包括黑色素瘤、結直腸癌和非小細胞肺癌,這些特定適應症的當前護理標準BRAF抑制劑是一線設置的Dradfenib聯合曲美替尼治療黑色素瘤,無進展生存期(PFS)為11.4個月,Enorafenib聯合西妥昔單抗治療二線出租的CRC,PFS為4.2個月,以及Dradfenib聯合曲美替尼治療NSCLC,PFS為15.2個月。我們認為,突變特異性的BRAF V600X降解劑可以提供比目前可用的BRAF抑制劑顯著的機械益處,並有可能顯著改善臨牀結果。
我們的首個人類1/2期臨牀試驗
2023年1月,我們啟動了CFT1946的首個人類1/2期臨牀試驗,我們繼續招募患者參加這項正在進行的臨牀試驗。1/2期臨牀試驗將主要調查安全性、耐受性和抗腫瘤活性,次要和探索性目標是表徵CFT1946的PK和PD特徵。這項研究的第一階段的初始階段將評估CFT1946作為單一藥物治療BRAF V600X實體腫瘤患者,包括結直腸癌、黑色素瘤和非小細胞肺癌患者,在之前接受BRAF抑制劑治療後。隨着第一階段試驗的進展,我們可能會增加一支額外的ARM來評估CFT1946與曲美替尼聯合治療BRAF V600X黑色素瘤和先前BRAF抑制劑治療後的非小細胞肺癌患者,我們還可能增加另一支ARM來評估CFT1946與西妥昔單抗聯合治療BRAF V600X結直腸癌在先前BRAF抑制劑治療後的療效。
BRAF V600X是一種常見的、眾所周知的致癌突變
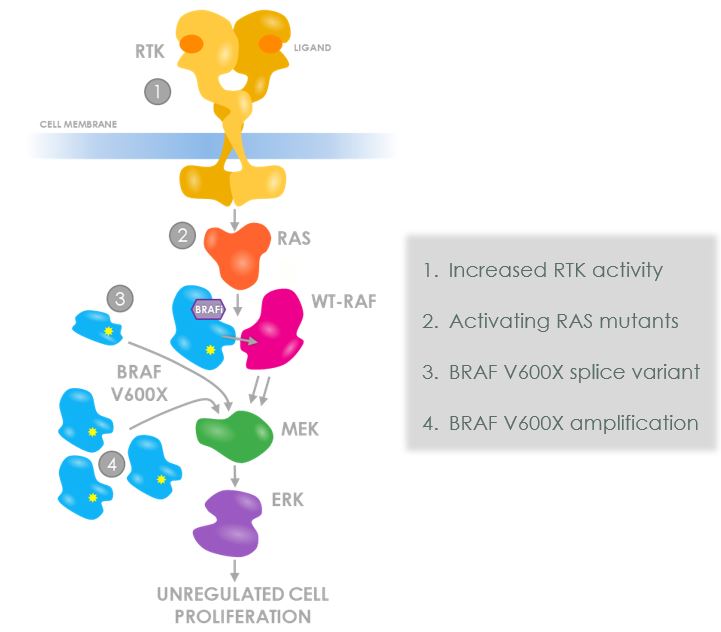
BRAF是參與向下跌發送信號以啟動細胞增殖的幾個蛋白激酶之一,該信號通路被稱為絲裂原活化蛋白激酶,或MAPK。MAPK通路將細胞外的增殖信號傳導到細胞核,發出細胞增殖的信號。許多癌症的特徵是該MAPK通路各組成部分的激活突變,包括BRAF V600X突變,它賦予MAPK通路的結構性激活,促進致癌轉化,並可導致腫瘤生長。
BRAF基因第600密碼子上氨基酸Valine的單鹼基替換,稱為V600X,被稱為I類突變,當這些V600X突變導致穀氨酸取代Valine(最常見的此類突變)時,它們被稱為V600E突變。
BRAF V600X突變體結構性地激活MAPK途徑,這意味着細胞的增殖在沒有收到正常激活該途徑所需的細胞外增殖信號的情況下被激活。之所以發生結構性激活,是因為BRAF V600X突變體能夠作為單一蛋白質或單體發出信號,而包括BRAF和CRAF在內的野生型RAF蛋白質必須形成兩種蛋白質的複合體或二聚體,才能進行下游信號傳遞。這種結構性激活導致MAPK細胞增殖途徑的過度激活,導致腫瘤細胞的增殖和腫瘤的生長。已批准的BRAF V600E小分子抑制劑-維莫拉非尼、達普拉非尼和恩可拉非尼-阻斷突變的BRAF單體對MAPK途徑的結構性激活。然而,這些分子對BRAF的抑制不可避免地會導致各種抗性機制的出現。與其他MAPK通路靶點不同,這種耐藥性不是由於ATP結合口袋的突變造成的。相反,第一代抑制劑容易受到促進BRAF V600X和其他RAF合作伙伴之間形成二聚體的抗性機制的影響,這些合作伙伴可能包括BRAF V600X本身、野生型BRAF或野生型CRAF。這是因為這些抑制劑受到一種被稱為矛盾激活的現象的影響,在RAF二聚體的背景下,藥物不能同時抑制兩種RAF蛋白。因此,BRAF V600X的抑制形式可以在促進RAF二聚體形成的細胞環境中有助於激活非藥物結合的RAF夥伴。下面是腫瘤細胞在第一代BRAF抑制劑存在的情況下利用這種脆弱性產生耐藥性的四種方式。
•RTK活動增加:增強的RTK信號可以激活RAS,從而促進藥物結合的BRAF V600X與野生型RAF蛋白之間形成二聚體。由於只有BRAF V600X被抑制劑佔據,野生型RAF伴侶同時被二聚體激活並上調MAPK途徑。
•激活RAS突變體:在缺乏上游RTK信號的情況下,RAS的突變可以激活其促進藥物結合的BRAF V600X和野生型RAF蛋白之間形成二聚體的能力。如上所述,這導致非抑制物結合的野生型RAF夥伴上調MAPK活性。
•BRAF V600X接頭變種:通過選擇性剪接產生缺乏RAF V600X亞型的RAF V600X亞型,或由外顯子3-5編碼的RBD。在沒有RBD的情況下,這些BRAF亞型即使在低水平的RAS活性存在下也會二聚化,一旦形成BRAF V600X二聚體,抑制劑只能阻止其中一個BRAF V600X夥伴,使另一個未被佔據的BRAF V600X夥伴被激活並自由上調MAPK活性。
•BRAF V600X增強版:基因改變會導致BRAF V600X基因的許多額外副本--這種現象被稱為基因放大。這反過來會導致BRAF V600X蛋白濃度升高。
在高濃度下,BRAF V600X蛋白將形成二聚體,而不需要上游RTK或RAS激活。再一次,第一代抑制劑不能同時結合和抑制兩個BRAF V600X配對,使得未被佔據的BRAF V600X二聚體配對被激活,並自由上調MAPK活性。
我們認為,BRAF V600X突變的靶向蛋白質降解提供了從根本上改善現有BRAF抑制劑的潛力,因為降解劑比一般抑制劑具有優勢,因為降解突變體BRAF消除了整合到各種基於BRAF二聚體的抗性機制中的可能性,這些機制導致MAPK途徑的矛盾激活。因此,CFT1946有可能克服因適應症而異的耐藥機制。對於黑色素瘤和非小細胞肺癌,獲得性耐藥發生在儘管BRAF V600X單體被抑制,但仍然通過如上所述的MAPK途徑激活的地方。在結直腸癌中,還有一個額外的複雜因素,即抑制MAPK信號轉導導致作為內在耐藥機制的EGFR的激活,這就是為什麼標準的護理治療方法包括BRAF抑制劑和抗EGFR抗體的組合,如西妥昔單抗。
黑色素瘤
根據國家癌症研究所的數據,2023年美國約有97,610名患者被診斷為黑色素瘤,其中約5%,即每年約4,800名患者將患有轉移性疾病。此外,約35%的晚期黑色素瘤患者攜帶BRAF V600X突變,其中約90%是BRAF V600E突變。
對於BRAF V600X突變的無法切除或轉移性黑色素瘤患者,推薦的一線治療方案是抗PD-1單藥,如pembrolizumab或nivolumab,或與BRAF抑制劑,如達普拉非尼、維莫拉非尼或Enorafenib,以及MEK抑制劑,如曲美替尼、cobimetinib或binimetinib的聯合治療。然而,相當數量的接受這種聯合治療的患者沒有足夠的反應或沒有持久的反應,因為出現了對該治療的耐藥性。
目前BRAF V600E/K黑色素瘤患者的護理治療標準是使用BRAF和MEK抑制劑的組合,達普拉非尼和曲美替尼,該方案的中位PFS約為11.4個月。
結直腸癌
根據國家癌症研究所的數據,2023年,美國約有153,020名患者被診斷為結直腸癌。在這些患者中,約20%(約3萬名患者)被診斷為轉移性疾病,約25%(約3.7萬名患者)被診斷為局部疾病,這些疾病將隨着轉移而復發。大約5%到10%的CRC患者患有BRAF V600E,或每年大約發生11,000例CRC BRAF V600E病例。
BRAF V600E突變的患者如果在以前的治療中取得進展,可以接受Enorafenib和西妥昔單抗的聯合治療,這兩種藥物都是抑制劑治療。然而,這種方案的療效有限,中位PFS約為4.2個月。
非小細胞肺癌
根據國家癌症研究所的數據,2023年,美國約有238,340名患者被診斷為非小細胞肺癌,其中約53%,即126,320名患者被診斷為轉移性疾病,與當地診斷的腫瘤相比,複發率很高。大約1-2%的非小細胞肺癌患者有BRAF V600X突變。
具有BRAF V600E突變的新診斷NSCLC患者在一線接受達普拉非尼和曲美替尼治療時,典型的中位PFS為15.2個月。在接受達普拉非尼和曲美替尼治療之前,曾接受化療或其他靶向藥物治療的患者的中位PFS僅為9個月左右。
臨牀前發展
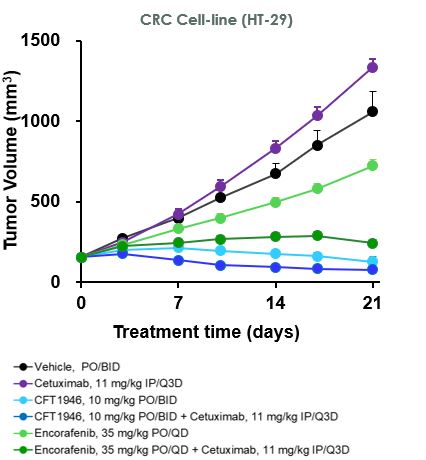
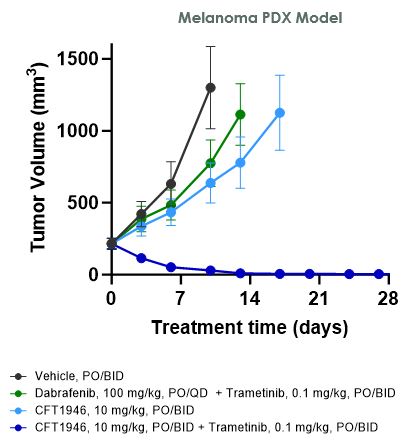
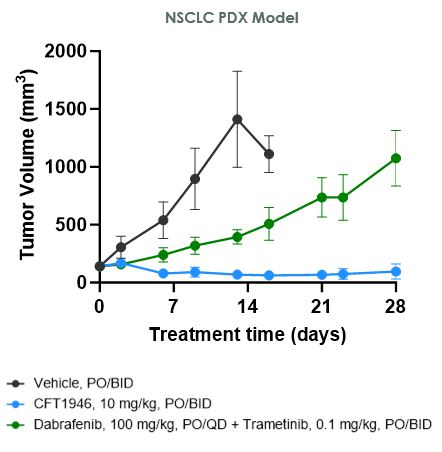
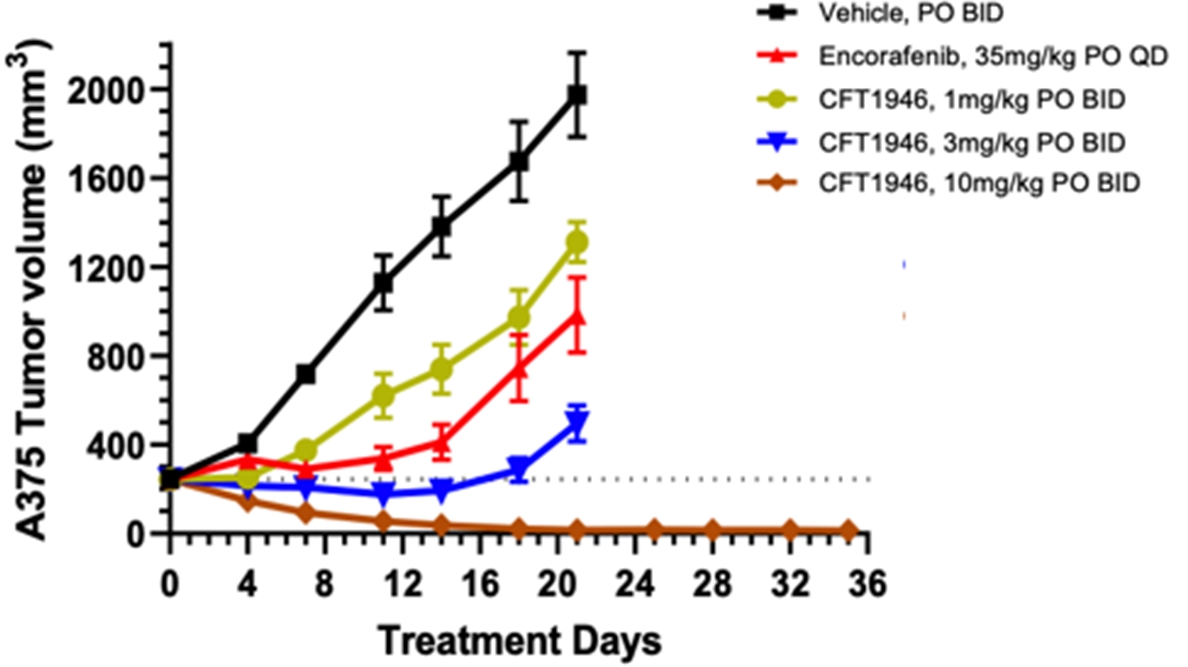
在臨牀前研究中,我們已經證明CFT1946對BRAF V600X結直腸癌、非小細胞肺癌和黑色素瘤的治療比標準治療更有效。如下圖所示,在HT-29結腸癌細胞株異種移植模型中,CFT1946作為單一療法以及與西妥昔單抗聯合使用時,顯示出比西妥昔單抗單藥、恩可拉非尼單藥和恩可拉非尼與西妥昔單抗聯合治療的療效更強。在非小細胞肺癌PDX異種移植模型中,CFT1946作為單一療法,對達普拉非尼和曲美替尼的聯合治療顯示出增強的反應。此外,在黑色素瘤PDX BRAF抑制劑耐藥模型中,與達普拉非尼聯合曲美替尼相比,CFT1946聯合曲美替尼顯示腫瘤深度消退。
在A375BRAF V600E黑色素瘤模型中,我們觀察到CFT1946對腫瘤生長具有劑量依賴性的抑制作用,並在每天兩次口服10 mg/kg的最低有效劑量下顯示出腫瘤消退。此外,CFT1946從2 mg/kg開始(口服和每天兩次)均顯示出優於FDA批准的BRAF V600E抑制劑Enorafenib的活性。此外,在下面顯示的異種移植研究中沒有觀察到不良反應(通過顯著減輕體重來評估)。
我們還證明瞭CFT1946在臨牀前轉移性黑色素瘤中樞神經系統(CNS)模型中是活躍的。此外,與恩可拉非尼相比,CFT1946延長了生存期並減輕了中樞神經系統的負擔。
CFT8919:高效、口服、變構、突變選擇性EGFR L858R降解劑
我們正在開發CFT8919,一種口服生物可用的EGFR L858R變構降解劑,用於治療非小細胞肺癌。我們之所以選擇以EGFR為靶點,是因為其明確的生物學特性,以及EGFR激酶抑制劑所面臨的侷限性,我們相信我們的降解劑方法可能能夠克服這些侷限性。我們最初的目標人羣是EGFR L858R驅動的非小細胞肺癌患者,他們在接受包括奧西美替尼在內的經批准的EGFR抑制劑治療後進展。我們相信,由於蛋白質降解的獨特優勢,CFT8919有可能克服對標準護理EGFR抑制劑的耐藥性,從而產生更深層次和更持久的反應。2023年5月,我們與Betta Pharma就CFT8919在包括內地中國、香港特別行政區、澳門特別行政區和臺灣地區的中國地區的開發和商業化訂立獨家許可及合作協議。我們相信,這一合作伙伴關係將使我們能夠在由於大中國地區EGFR L858R驅動的非小細胞肺癌高發而導致EGFR驅動耐藥突變的患者中加快CFT8919的整體開發,最大限度地增加可能從CFT8919中受益的患者數量。因此,一期臨牀研究將由鬥魚製藥在大中國地區進行。在Betta Pharma從第一階段試驗中產生結果之前,我們不打算在大中國以外的地方啟動臨牀研究。我們保留在大中國之外開發和商業化CFT8919的所有權利。
EGFR是一種具有已知耐藥機制的腫瘤靶標蛋白
EGFR是一種受體酪氨酸激酶,參與控制細胞分裂和存活的細胞信號通路。EGFR基因的突變導致某些類型的癌細胞中的EGFR蛋白信號異常,包括一組非小細胞肺癌患者。在已知的EGFR酪氨酸激酶結構域突變中,約90%發生在外顯子19的缺失或外顯子21的點突變,後者導致858密碼子(L858R)上的精氨酸取代亮氨酸。外顯子21的L858R激活突變約佔EGFR突變NSCLC的25%~45%。EGFR酪氨酸激酶抑制劑或TKI已經開發出來,並提供了顯著的臨牀益處。然而,患者最終會產生耐藥性,通常是通過獲得EGFR的第二次耐藥性突變。T790M是繼第一代和第二代EGFR TKI之後最常見的耐藥突變,包括吉非替尼、厄洛替尼、阿法替尼和達科米替尼。第三代共價EGFR抑制劑osimertinib可以克服這種耐藥機制,目前已被批准用於一線治療,但獲得性耐藥仍然是一個問題。在奧西美替尼治療後進展的患者缺乏有效的治療選擇,而EGFR C797S突變是最常見的靶向耐藥機制。
CFT8919是一種針對EGFR L858R的突變選擇性降解物,它在抗藥性次級突變(T790M和/或C797S)的設置中仍然有效。我們相信CFT8919可能有機會成為一線治療的一個組成部分,由於降解劑相對於標準蛋白抑制劑的優勢,我們希望實現更深層次和更持久的反應。此外,由於30%-40%的突變的EGFR非小細胞肺癌患者會發生腦轉移,對中樞神經系統的滲透足以推動這一區域的治療效果是我們選擇CFT8919作為開發候選者的關鍵因素。
非小細胞肺癌
根據美國國家癌症研究所的數據,2023年美國約有238,340名患者被診斷為非小細胞肺癌,其中10%至15%的患者患有突變的EGFR或mEGFR。EGFR突變在具有亞洲血統的非小細胞肺癌患者中尤其常見。在中國,大約有693,000名患者被診斷出
對於每年的NSCLC,大約50%的診斷是由EGFR突變驅動的。此外,30%到40%的EGFR突變患者會發生腦轉移。
EGFR L858R突變是第二種最常見的激活EGFR突變,在美國和中國的EGFR診斷中發現了大約40%的突變。當接受標準的EGFR抑制劑治療時,攜帶EGFR L858R突變的患者與表現出外顯子19缺失的患者相比,治療結果更差。在EGFR L858R患者的一線治療中使用奧西美替尼時,中位PFS為14.4個月,而外顯子19缺失的患者為21.4個月。在該患者羣體中,EGFR介導的對奧西美替尼耐藥最多的機制是C797S突變。這一較短的PFS中位數和耐藥性表明L858R患者羣體的醫療需求尚未得到滿足。
臨牀前發展
我們已經進行了臨牀前實驗,以表徵我們的EGFR降解劑的活性特徵,並證明CFT8919是一種有效的、高選擇性的EGFR L858R口服生物降解劑,具有廣泛的靶向耐藥突變和顱內活性。
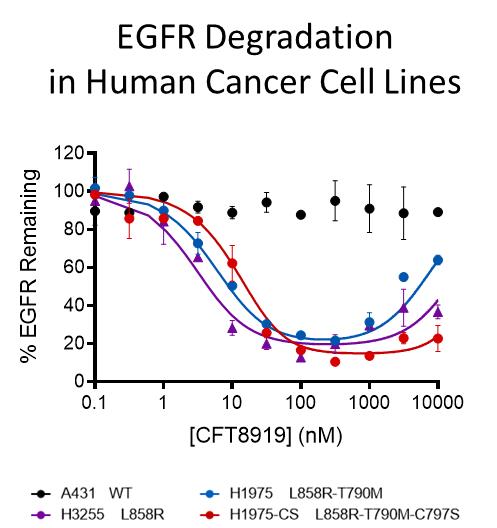
在人類癌細胞系中體外培養,我們觀察到CFT8919在低納摩爾濃度下誘導EGFR L858R的有效降解,而在10µM以下不誘導野生型降解。重要的是,CFT8919在存在T790M和T790M-C797S等二次抗性突變的情況下仍保持其活性。這反映在下圖中。
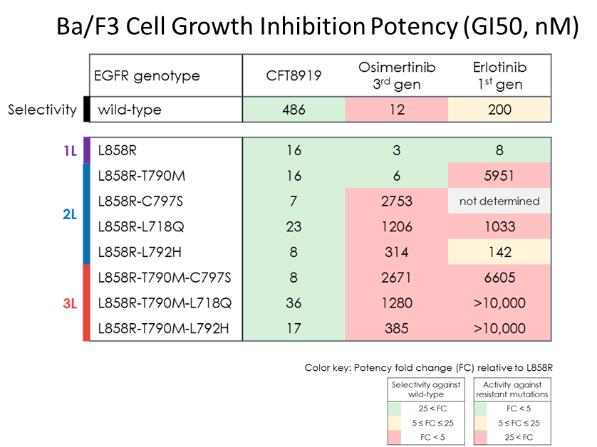
此外,我們在BA/F3細胞模型中針對一組廣泛的EGFR耐藥突變評估了CFT8919體外培養。通過檢測不同濃度的EGFR突變轉化的BA/F3細胞在72小時內的增殖情況,確定細胞生長抑制效價GI50。如下表所示,與L858R單一突變相比,CFT8919對一大組EGFR次級突變表現出同等的抗增殖活性,這些突變導致對已批准的EGFR抑制劑(如osimertinib和erlotinib)產生獲得性耐藥性。
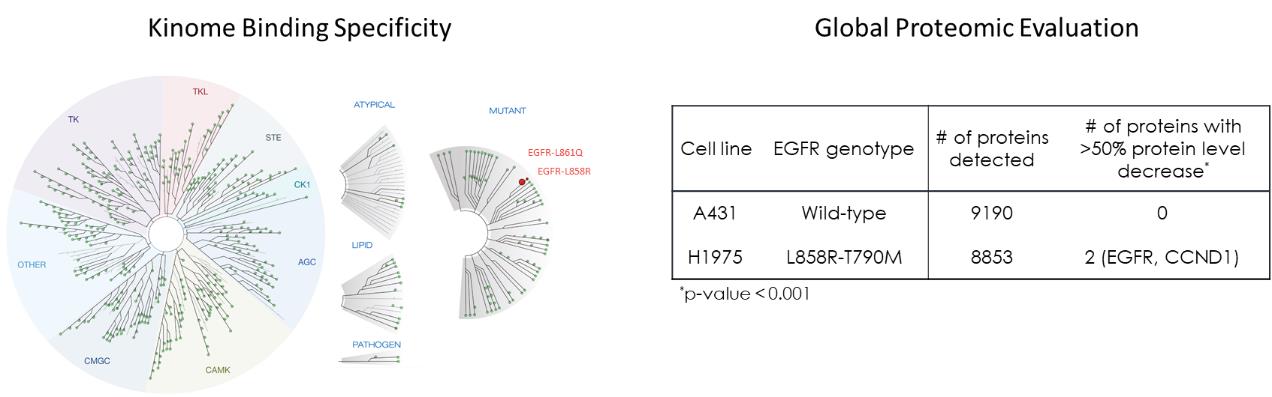
通過Kinome圖譜和全球蛋白質組學評估CFT8919的結合和降解選擇性,沒有發現CFT8919有顯著的靶外活性。Kinome選擇性通過使用
DiscoveryX KINOME掃描檢測468個激酶,如左圖所示。當CFT8919在100 nm的單一濃度下測試時,在50%的截止點上沒有野生型激酶顯示結合。CFT8919表現出顯著結合的唯一激酶是靶標上的外顯子21 EGFR激活突變體L858R和L861Q。此外,我們還評估了A431EGFR-WT和H1975 EGFR-L858R-T790M細胞系經300 nM CFT8919處理6小時後蛋白質組的降解選擇性。體外培養。如右表所示,對超過8000個蛋白質的測量表明,只有EGFR-L858R-T790M和CCND1的蛋白質水平在CFT8919處理後下降了50%以上,顯示出其高度選擇性的降解特徵。我們認為CCND1蛋白的丟失是由EGFR抑制的生物學效應引起的,而不是直接降解,因為這種變化在osimertinib EGFR抑制劑治療中也觀察到了。
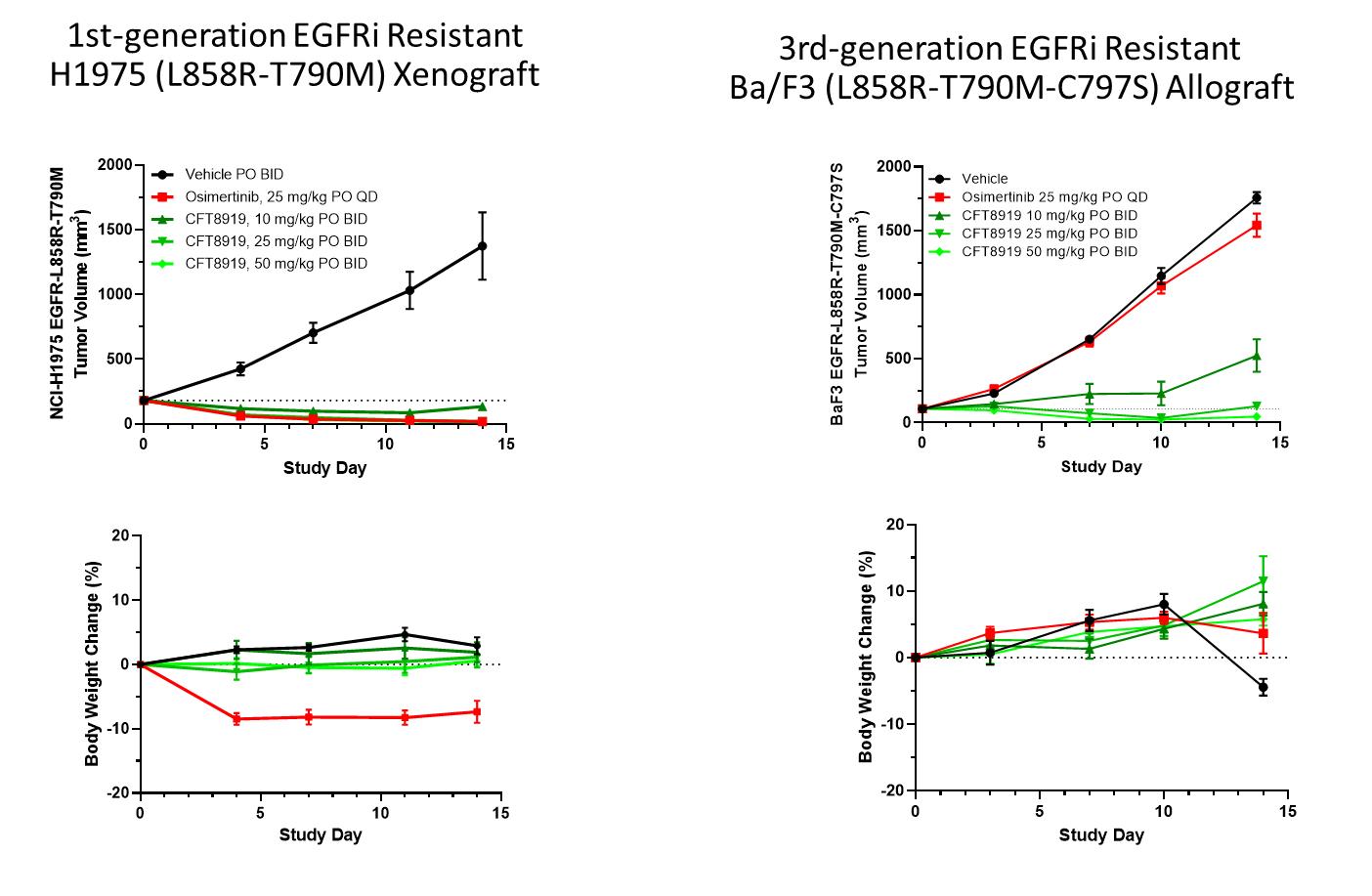
活體內在H1975 EGFR-L858R-T790M異種移植瘤中檢測CFT8919的活性(1ST耐TKI)和BaF3 EGFR-L858R-T790M-C797S(耐奧西美替尼)同種異體移植模型。在H1975異種移植模型中,每天兩次口服CFT8919(BID)顯示出劑量依賴性的活性,在低至10 mg/kg的劑量下觀察到腫瘤的消退。給藥劑量>25 mg/kg的CFT8919可使BID>25 mg/kg的CFT8919對BA/F3、EGFR-L858R-T790M-C797S同種異體移植瘤實現消退,而奧西美替尼則如預期的那樣失活。所有劑量的耐受性都很好,體重沒有明顯下降。這反映在下圖中。
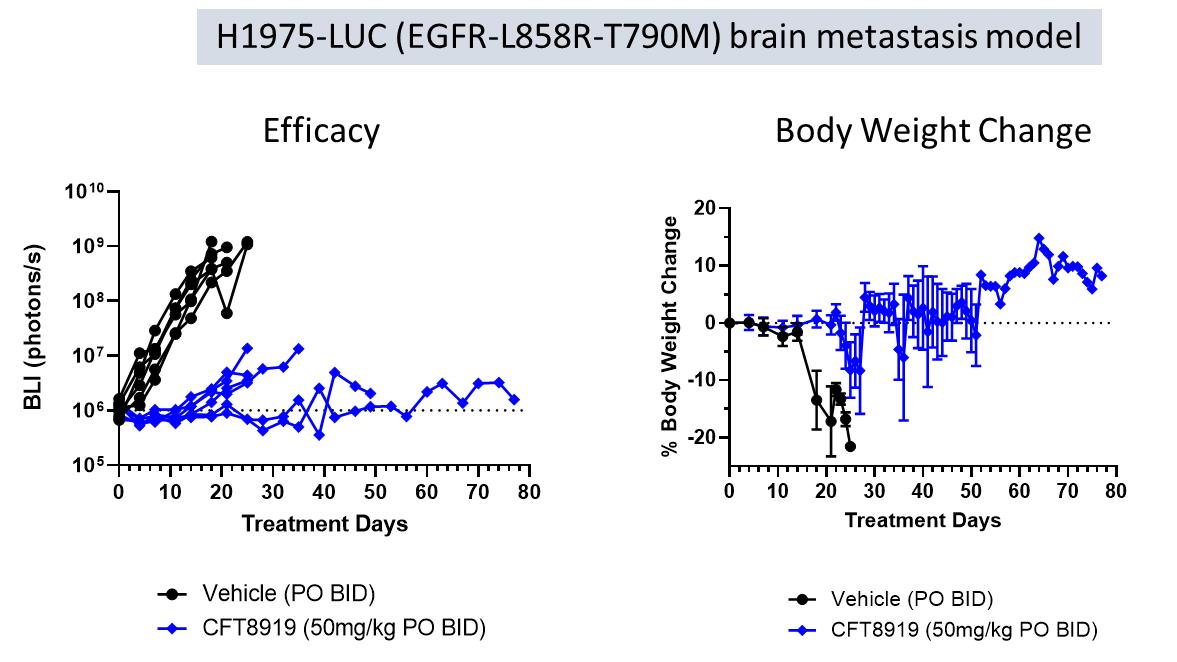
在H1975-Luc腦轉移模型中評價CFT8919的腦內活性。雌性BALB/c裸鼠頸動脈注射H1975(EGFR L858R-T790M)熒光素酶表達細胞。採用生物發光成像技術(BLI)跟蹤腫瘤生長情況。如下圖所示,
CFT8919顯示,口服給藥後,腫瘤負擔迅速顯著減少,體重減輕最小,表明其在中樞神經系統中具有活性。
我們的其他探索計劃
除了上面討論的項目外,我們還在推進其他各種發現階段的流水線項目。根據我們的戰略,我們逐個目標地評估我們可能開發的降解劑是否會提供一種令人信服的差異化方法,而不是治療同一疾病的標準或其他方法,並與我們將生物學和毒性風險降至最低的重點一致,並將重點放在高度未滿足的醫療需求上,包括罕見疾病。這些早期發現計劃包括已經在臨牀前模型中顯示出在適當情況下能夠穿越血腦屏障的化合物。我們的探索計劃是我們完全控制和擁有的內部計劃和與我們的合作伙伴合作的計劃的組合。
協作和許可協議
默克許可和協作協議
2023年12月,我們與默克公司簽署了一項許可和合作協議,或稱默克許可協議,以合作開發降解物-抗體結合物(DAC)並將其商業化,DAC是一種新興的方法,旨在選擇性地靶向和中和癌細胞中的致病蛋白。根據許可協議的條款,我們根據我們的某些知識產權授予默克公司全球獨家許可,以開發、製造和商業化針對最初未披露的腫瘤學目標的DAC。默克公司負責所有開發、監管批准、製造和商業化成本。根據默克許可協議的條款,默克已經預付了1000萬美元的現金。對於針對初始目標的DAC,我們有資格獲得總計約6億美元的里程碑式付款,外加淨銷售額的分級版税。此外,作為合作的一部分,我們授予默克公司在我們的某些知識產權下獲得全球獨家許可的選擇權,以開發、製造和商業化針對另外三個目標的DAC,每個目標都需要支付期權行使價格。如果默克公司行使這些選項,這些額外的計劃還將在默克許可協議中詳細説明的特許權使用費期限內提供額外的潛在里程碑和特許權使用費。如果默克公司行使其延長合作的所有選項,我們將有資格在整個合作中獲得高達約25億美元的潛在付款。
Betta Pharma許可和協作協議
於2023年5月,吾等與Betta Pharma或Betta Pharma許可協議訂立許可及合作協議,就CFT8919的開發及商業化進行合作,CFT8919是一種口服生物可用BiDAC降解劑,旨在有效及選擇性地對抗由內地中國、香港特別行政區、澳門特別行政區及臺灣組成的大中國地區,該地區由內地中國、香港特別行政區、澳門特別行政區及臺灣組成,我們保留在全球其他地區開發及商業化CFT8919的權利。
根據Betta Pharma許可協議的條款,我們根據我們的某些知識產權授予Betta Pharma獨家許可,以在大中國開發、製造和商業化用於人類的CFT8919。貝塔製藥負責在大中國地區的所有開發、監管審批、製造和商業化成本,但貝塔製藥作為我們在大中國地區的代理商參與由
我們。作為合作的一部分,Betta Pharma預付了1,000萬美元的現金,我們有資格獲得高達3.57億美元的里程碑付款,外加CFT8919在大中華區中國的淨銷售額的分級特許權使用費。Betta Pharma向我們支付的特許權使用費從低到中兩位數的百分比不等,但在Betta Pharma許可協議中描述的特定情況下可能會有所減少。此外,作為合作的一部分,我們同意在收到美國食品和藥物管理局批准的CFT8919新藥申請後,向Betta Pharma支付高達4,000萬美元的里程碑式付款,這筆里程碑式的金額是根據Betta Pharma納入的預期臨牀試驗中的患者百分比和批准的治療路線得出的。此外,我們已同意就本地區(除中國以外的世界其他地區)CFT8919的淨銷售額向Betta Pharma支付較低的個位數百分比範圍內的分級特許權使用費,但在某些情況下會如Betta Pharma許可協議中所述進行某些削減。Betta Pharma許可協議下所有預期專利使用費的使用費期限應在(I)該產品在該國家/地區首次商業銷售十二(12)週年、(Ii)涵蓋該產品在該國家/地區的任何法規排他期屆滿、以及(Iii)涵蓋該產品的最後一項許可專利在該國家/地區到期之日起,以逐個產品和國家/地區為基礎終止。此外,2024年1月,Betta Pharma的一家附屬公司以2500萬美元購買了我們的普通股,如下所述。
生物遺傳研究合作與許可協議
2018年12月,我們與Biogen簽訂了一份合作研究和許可協議,即Biogen協議,據此,我們同意與Biogen合作,並使用我們專有的蛋白質降解劑平臺來研究、開發和鑑定小分子蛋白質降解劑。2020年2月,我們對Biogen協議進行了修訂,進一步明確了Biogen對目標結合部分(分子的一部分)的所有權,以及針對或結合合作目標的任何相關知識產權。本修訂案進一步規定,Biogen許可我方使用這些Biogen靶向結合部分和任何相關知識產權的權利,以便開展Biogen協議項下預期的研發活動。
根據Biogen協議,我們根據我們的知識產權授予Biogen獨家許可,並有權通過多個層次進行分許可,(a)根據雙方商定的研發計劃進行候選開發活動,及(b)利用所有降解物和產品在世界上的任何用途。
根據Biogen協議的條款,我們負責根據協議中規定的目標選擇和替換程序對Biogen選擇的一些目標進行研發活動。我們必須提供開展候選開發活動所需的所有資源,以合理的謹慎和技能並根據適用法律和Biogen協議開展這些活動,並努力完成適用開發計劃中規定的活動,並向Biogen交付一定數量的降解劑,這些降解劑針對符合一系列預定標準的每個目標。我們和Biogen還負責為Biogen的目標選擇過程提供信息的研究活動,Biogen將支付自己的費用,並將償還我們的費用,最高可達一定金額。百健協議項下的研究期限於二零二三年六月結束。
在Biogen開始針對Biogen選擇的每個目標的降解劑的IND使能研究後,Biogen負責並同意使用商業上合理的努力在某些地區針對每個目標的至少一種產品進行所有進一步開發、監管事務、生產和商業化。
在執行Biogen協議後,Biogen向我們支付了4500萬美元的預付款,作為候選開發活動的預付款。在Biogen收到針對每個目標的符合預定義標準的降解劑後,我們有資格獲得每個目標200萬至500萬美元的付款。在Biogen開始針對每個目標的發展候選人的第一個IND使能研究後,Biogen需要支付800萬美元。對於每個目標,Biogen需要向我們支付(a)開發和商業化里程碑付款總額高達3500萬美元,以及(b)銷售里程碑付款總額高達2600萬美元,以實現針對該目標的所有產品的一定數量的淨銷售額,每個都受到一定的削減。如果Biogen延長合作期限並選擇其他目標,則開發、商業化和銷售里程碑付款總額將增加。此外,Biogen還必須向我們支付產品的版税,-根據每種產品的淨銷售額,按中等個位數的百分比,按產品和國家分列,但有一定的減少。
除非提前終止,否則Biogen協議將在最後一個產品和國家的專利權有效聲明到期時失效,該專利權涵蓋適用國家適用產品獲批標籤中的物質組成或使用方法。我們和Biogen各自可以終止Biogen協議(a)關於一個或多個開發候選人,產品或合作目標,或僅在Biogen的情況下,整個協議,因為另一方未解決的重大違反其義務,以及(b)整個協議
在另一方破產、資不抵債或類似程序時。為了方便起見,Biogen還可以完全終止或針對一個或多個開發候選者、產品或合作目標終止Biogen協議。
羅氏修訂和重新簽署許可協議
2016年3月,我們與羅氏簽訂了許可協議,此後多次修改,包括在2016年6月、2017年3月和2018年12月。我們將這一修訂和重述的協議稱為羅氏協議。根據羅氏協議,我們同意與羅氏合作,使用我們的專有魚雷平臺治療癌症和其他適應症,研究、開發、製造靶向約束性降解劑藥物,並將其商業化。根據羅氏協議的條款,我們負責根據協議中規定的目標選擇和替換程序,對羅氏選擇的若干目標進行臨牀前研究和開發活動。我們還負責為針對特定目標的產品進行第一階段臨牀試驗,並負責與適用研究計劃相關的製造活動,但羅氏有權在預定時間承擔製造責任。我們和羅氏各自分擔這些研究活動的費用。
根據羅氏協議,我們授予羅氏獲得獨家全球許可的獨家選擇權,並有權通過多個級別進行再許可,以開發針對每個合作目標的產品並將其商業化。在行使針對特定目標的選擇權時,羅氏負責針對該目標的產品的製造、開發和商業化,費用自負。然而,我們可以選擇共同開發針對特定目標的產品,在這種情況下,我們將負責與此類聯合開發產品相關的部分開發成本,並有資格從此類聯合開發產品的銷售中獲得更多版税。我們還可以選擇對我們已行使共同開發選擇權的產品進行聯合詳細説明。如果我們行使共同細節選擇權,我們將負責共同細節成本的一部分。我們通常有權選擇不參與這些共同開發和共同細化的活動。
2020年11月,我們簽署了羅氏協議的進一步修正案,提供了一種機制,通過該機制,我們和羅氏可以通過簽訂共同目標終止協議,在逐個目標的基礎上相互同意終止羅氏協議。在這種性質的終止後,經修訂的羅氏協議規定,支持以抑制為其行動模式的產品(稱為羅氏領域)的所有專有技術和知識產權權利將恢復到羅氏,而支持以降級為其行動模式的產品(稱為C4T領域)的所有專有技術和知識產權的所有權利將恢復到我們手中。此外,這項修正案規定,在簽訂共同目標終止協議後,羅氏將對符合羅氏領域的合作產生的任何專有技術和知識產權擁有權利和責任,我們將對符合C4T領域的合作產生的任何專有技術和知識產權擁有權利和責任。為了支持這種權利分配,根據修正案,羅氏向我們和我們提供了一份永久的、不可撤銷的、全額繳足的、獨家的(即使對於授權方也是如此)、根據相互目標終止協議分配給一方的專利的可再許可(包括多層)許可,以及根據相互終止協議可分配給一方的、永久的、不可撤銷的、非排他性的、可再許可的(包括多層的)專有技術許可。
2020年11月,通過加入這項修正案,我們和羅氏共同同意終止關於目標EGFR的羅氏協議。此外,在2021年11月、2022年7月和2023年12月,我們和羅氏共同同意終止關於BRAF的羅氏協議以及另外兩個未披露的目標。因此,羅氏現在可以自由地在羅氏領域追求這些目標,我們也可以自由地在C4T領域追求這些目標,羅氏領域與這些目標相關的所有專有技術和知識產權的權利和責任都歸還給了羅氏各方,與C4T領域的這些目標相關的所有專有技術和知識產權的權利和責任都返還給了我們,羅氏將C4T領域的專利轉讓給了我們。2023年9月,羅氏根據羅氏協議指定了一個新的未披露目標,將其添加到合作中。由於這些努力,目前仍有兩個目標是這一合作的一部分。2023年12月,我們和羅氏共同同意進一步修訂羅氏協議,以調整羅氏對剩餘兩個目標的選擇權的時間,以在羅氏收到劑量範圍發現數據包後開始。
在簽署羅氏協議後,我們從羅氏收到了4000萬美元的預付款。此外,我們從羅氏獲得每一項積極研究計劃的年度研究資金,並有資格在達到某些目標的預定研究和開發成功標準時獲得額外付款。根據修訂後的羅氏協議,如果羅氏對剩餘兩個目標中的任何一個行使選擇權,羅氏有義務支付800萬美元的行權費。對於羅氏行使的每個目標選項,我們有資格在與相應產品相關的某些研究、開發和商業里程碑實現時獲得高達2.73億美元的里程碑付款,但受基於知識產權覆蓋範圍的某些減免和排除的限制。羅氏還需要向我們支付每個目標高達1.5億美元的一次性基於銷售的里程碑付款
達到某一特定目標的產品的淨銷售額。最後,我們有資格獲得按羅氏根據其行使的選擇權銷售的產品淨銷售額的個位數中位數到十幾歲左右的百分比不等的分級特許權使用費,但須有一定的減免。對於我們行使共同開發權的產品的銷售,適用的特許權使用費將以較低的個位數百分比增加。
除非較早前終止,否則羅氏協議將於羅氏協議項下的特許權使用費或其他付款義務未到期或將到期之日終止。我們和羅氏各自可以完全或逐個目標或逐個產品地終止羅氏協議,在我們的情況下,可以逐個國家地終止羅氏協議,原因是另一方違反了羅氏協議規定的義務,或另一方破產、資不抵債或類似的程序。為了方便起見,羅氏可以逐個目標、逐個產品或逐個國家終止羅氏協議。如果我們被羅氏的競爭對手收購,羅氏有權要求我們根據羅氏協議終止我們的研究、開發和共同詳細説明活動,在此之後,我們將沒有資格收到此類終止活動的付款。
競爭
生物技術和製藥行業的特點是技術進步迅速,競爭激烈,非常重視知識產權和專有產品。雖然我們相信我們的技術、專業知識、科學知識和知識產權為我們提供了競爭優勢,但我們面臨着來自許多不同來源的潛在競爭,包括主要製藥、專業製藥、生物技術公司、學術機構、政府機構以及進行研究、尋求專利保護並建立研究、開發、製造和商業化合作安排的公共和私人研究機構。我們不僅必須與其他專注於蛋白質降解的公司競爭,而且我們成功開發和商業化的任何候選產品都將與現有療法和未來可能出現的新療法競爭。此外,我們的行業的特點是專利數量眾多,專利侵權指控頻繁。
我們的重點是使用我們的魚雷平臺發現和開發蛋白質降解療法。其他開發用於蛋白質降解的嵌合小分子的公司包括但不限於Arvinas,Inc.,BioTheryX,Inc.,Captor Treateutics,Inc.,Cullgen Inc.,Foghorn Treateutics,Inc.,Frontier Medicines Corporation,GluBioTreateutics,Inc.,Haisco製藥集團,Kymera Treateutics,Inc.,Monte Rosa Treateutics,Inc.,Nurix Treateutics,Inc.,Orum Treateutics,Inc.,PhoreMost Ltd.,Plexium,Inc.,Salarius PharmPharmticals,Inc.,Seed Treatetics,Inc.,SK,Life Science Labs,Inc.(SK生物製藥有限公司的子公司)和Vividion治療公司(拜耳股份公司的子公司)。此外,幾家大型製藥公司已經披露了在該領域的臨牀前投資,包括安進公司、阿斯特拉斯製藥公司、阿斯利康公司、百時美施貴寶公司(及其子公司Celgene公司)、葛蘭素史克公司、基因泰克公司和諾華國際公司。除了來自其他蛋白質降解療法的競爭外,我們開發的任何產品也可能面臨來自其他類型療法的競爭,如小分子、抗體、T細胞或基因療法。
我們的主要候選產品針對腫瘤適應症。最常見的治療腫瘤適應症患者的方法是手術、放射和藥物治療,包括化療、激素治療、細胞治療和靶向藥物治療。市場上銷售的癌症藥物療法多種多樣。在許多情況下,這些藥物聯合使用以提高療效。目前批准的一些藥物療法是品牌藥物,受專利保護,其他藥物是在仿製藥的基礎上提供的。這些批准的藥物中有許多是治療藥物,被醫生、患者和第三方付款人廣泛接受。總體而言,儘管過去幾十年來癌症的治療取得了長足的進步,目前市場上的治療方法也為許多患者提供了好處,但這些治療方法在一定程度上都受到其療效和不良事件發生率的限制,而且沒有一種方法能成功地治療所有患者。因此,癌症的發病率和死亡率仍然很高。
除了目前上市的藥物外,還有幾種候選產品正在進行臨牀前開發,用於治療腫瘤學適應症。這些正在開發中的產品可能會提供目前市場上的療法所不能提供的療效、安全性、便利性和其他好處。因此,它們可能會對我們獲得市場批准的任何候選產品構成重大競爭。
如果我們的任何候選產品被批准用於我們預期進行臨牀試驗的適應症,它們將與上述療法和目前上市的藥物以及任何可能正在開發的藥物競爭。我們也有可能面臨來自其他生物或藥物方法的競爭,以及來自其他類型療法的競爭。
我們的許多現有或潛在競爭對手,無論是單獨或與合作伙伴合作,在研發、製造、臨牀前測試、進行臨牀試驗、獲得監管批准和營銷批准的產品方面,都比我們擁有更多的財務資源和專業知識。這些競爭對手也在與我們競爭
招聘和留住合格的科學和管理人員,建立臨牀試驗場地,為臨牀試驗註冊患者,以及獲取與我們的計劃相輔相成或必要的技術。製藥和生物技術行業的合併和收購可能導致更多的資源集中在我們數量較少的競爭對手身上。規模較小或處於初創階段的公司也可能成為重要的競爭對手,特別是通過與大型和成熟公司的合作安排。這些競爭對手還在招聘和留住合格的科學和管理人員、建立臨牀試驗場地、臨牀試驗的患者註冊以及獲取與我們的計劃相輔相成或必要的技術方面與我們競爭。如果我們的競爭對手開發和商業化比我們可能開發的任何產品更安全、更有效、副作用更少或更少、更方便或更便宜的產品,我們的商業機會可能會減少或消失。我們的競爭對手也可能比我們更快地獲得FDA或其他監管機構對其產品的批准,這可能會導致我們的競爭對手在我們能夠進入市場之前建立強大的市場地位。此外,在許多情況下,我們的競爭能力可能會受到保險公司或其他尋求鼓勵使用仿製藥的第三方付款人的影響。目前市場上有針對我們正在尋求的某些適應症的仿製藥,預計未來幾年將有更多的仿製藥上市。如果我們的候選產品獲得批准,我們預計它們的定價將顯著高於競爭對手的仿製藥。
影響我們所有計劃成功的關鍵競爭因素,如果獲得批准,很可能是它們的有效性、安全性、便利性、價格、仿製藥競爭水平和可報銷。
製造業
我們不擁有或運營,目前也沒有建立任何製造設施的計劃。我們依賴,並預計將繼續依賴第三方合同製造組織,即CMO,用於藥物物質和成品。
我們目前以採購訂單的方式從這些製造商那裏獲得我們的供應,對於我們的候選產品和其他材料沒有長期的承諾供應安排。如果這些製造商中的任何一家因任何原因無法提供給我們,我們相信有許多潛在的替代產品,儘管我們在確定和鑑定此類替代產品時可能會出現一些延誤。欲瞭解更多信息,請參閲標題為“風險因素--與依賴第三方相關的風險--製藥產品的生產非常複雜,可能會因各種原因導致產品延誤或損失。我們與第三方簽訂合同,生產我們的候選產品,用於臨牀前測試和臨牀試驗,並預計將繼續這樣做以實現商業化。這種對第三方的依賴增加了我們將沒有足夠數量的候選產品或產品的風險,或者我們將無法以可接受的成本或質量或在合適的時間獲得我們想要或要求的數量的風險,這可能會推遲、阻止或損害我們的開發或商業化努力。”
我們所有的候選藥物都是低分子有機化合物,在生物製藥界通常被稱為小分子,但我們的BiDAC降解物往往比傳統的小分子療法更大。我們選擇這些化合物不僅是基於它們潛在的臨牀活性和耐受性,而且還基於它們的物理性質。我們的候選產品採用可靠的、可重複使用的合成工藝,以現成的原料為原料,並以易於放大的化學為基礎。我們希望繼續開發能夠在合同製造設施中以成本效益高的方式生產的候選藥物。
商業化計劃
我們還沒有建立自己的商業組織或分銷能力,因為我們的候選產品仍處於臨牀開發階段。除受我們合作協議約束的項目外,我們保留所有正在開發的項目的完全商業化權利。在我們的任何候選產品獲得上市批准之前,在適當的時候,我們需要制定一項計劃,將它們在美國和其他關鍵市場商業化。我們目前預計,我們將建立自己的專注、專業的銷售和營銷組織,以支持我們獲得營銷批准的候選產品在美國的商業化,並且這些產品可以通過這樣的能力商業化。我們希望利用與一個或多個第三方的各種類型的合作、聯合促銷、分銷和其他營銷安排,將我們的候選產品在美國以外的市場或需要更大規模的銷售和營銷組織的情況下商業化。
隨着候選產品在我們的渠道中取得進展,我們的商業計劃可能會改變。我們的一些研究計劃針對的是潛在的更大的適應症。數據、開發計劃的規模、目標市場的規模、商業基礎設施的規模以及製造需求都可能影響我們在美國、歐洲和世界其他地區的戰略。
知識產權
我們的商業成功在一定程度上取決於我們確保和維護我們的蛋白質降解技術的專利和其他專有保護的能力,包括我們的魚雷平臺、我們獨家擁有的候選產品、我們與羅氏共同擁有的候選產品以及與我們業務相關的技術訣竅。為了保護我們的核心技術和產品,我們需要成功地起訴、辯護並在必要時執行我們的知識產權,特別是我們的專利權,保護我們的商業祕密,並在不侵犯他人有效和可執行的知識產權的情況下運營。對於我們的候選產品,我們一般打算在相關情況下尋求涵蓋物質成分、藥物成分、使用方法(包括聯合療法)、製造工藝和工藝中間體的專利保護。我們在開發新技術和候選產品的同時,不斷評估和完善我們的知識產權戰略。我們目前計劃根據我們的知識產權戰略,在適當的情況下,包括我們尋求適應競爭或改善我們的商業機會的領域,提交更多的專利申請。
像我們這樣的生物製藥公司的專利地位通常是不確定的,可能涉及複雜的法律,科學和事實問題。此外,管轄知識產權保護的法律可能會隨着時間的推移而發生變化,因為發佈了新的司法判決或通過了新的法律、規則或條例。此外,專利申請中要求的覆蓋範圍在專利發佈之前可能會大大減少,即使在發佈之後,其範圍也可能會被重新解釋和質疑。因此,我們不能保證我們的任何候選產品將受到有效的、可執行的專利的保護或繼續受到保護。我們也無法預測我們目前正在尋求的專利申請是否會在任何特定司法管轄區作為專利發佈,或者任何已發佈專利的權利要求是否會提供足夠的專有保護,免受競爭對手的侵害。我們持有的任何專利都可能受到第三方的質疑、規避或無效。
我們專利的排他性條款取決於獲得這些專利的國家的法律。在我們目前提交申請的國家,專利期限為自非臨時專利申請的最早提交日期起20年。美國專利的期限可以延長,以補償獲得銷售藥品的監管批准所需的時間(稱為專利期限延長),或美國專利商標局在專利申請過程中造成的延遲(稱為專利期限調整)。例如,Hatch-Waxman法案允許FDA批准的新化學實體藥物的專利期限延長至超過涵蓋批准藥物或其使用的專利的普通到期日五年。專利期限延長的時間長短與藥物接受監管審查的時間長短以及審查過程中的勤勉程度有關。在美國,專利期限延長不能將專利期限從產品批准之日起延長超過14年,並且只有一項專利涉及批准的藥物或其使用方法。歐洲也有類似的專利延期,稱為補充保護證書。某些其他司法管轄區也可能有延長專利期限的法律框架。我們目前打算在我們擁有合格專利且可獲得延長的任何司法管轄區內,就我們的任何已頒發專利尋求產品的專利期限延長;但是,無法保證適用的監管機構(包括美國FDA)將同意我們對是否應授予此類性質的延長以及(即使授予)這些延長的長度的評估。此外,即使我們的任何專利被擴展或調整,這些專利,包括這些專利的擴展或調整部分,也可能被美國或外國的最終管轄權法院認定為無效或不可執行。
專利和專利申請
截至2023年12月31日,我們總共擁有17項已頒發的美國專利、40多項美國專利申請(包括臨時和美國實用新型申請)、7項根據《專利合作條約》(PCT)提交的專利申請,以及250多項在國外待審的專利申請。
我們的專利組合通常分為兩類:平臺專利申請,旨在涵蓋與我們專有的TORPEDO平臺相關的發明,以及蛋白質靶向特異性降解劑專利申請,其中每一類都在下文中詳細描述。
TORPEDO平臺組合
我們獨家擁有我們的平臺專利資產,該資產是使用我們專有的TORPEDO平臺設計的。截至2023年12月31日,我們的平臺專利組合包括13項已頒發的美國專利、18項待決的美國專利申請、1項PCT專利申請和超過75項待決的外國專利申請。該專利組合涵蓋了我們與Cereblon E3泛素連接酶或CRBN結合的各種配體,無論是單獨的,作為MonoDAC分子的一部分,還是作為BiDAC分子的一部分,BiDAC分子包括疾病修飾蛋白靶點的蛋白質配體。
具體地,該平臺組合由十九個專利家族組成,涵蓋TORPEDO平臺,其具有針對各種類別的CRBN配體和由其衍生的降解劑的物質組合物權利要求,以及針對以下的權利要求:
相關的使用方法、藥物組合物和製造方法。這些家族中的專利,如果發佈和維護,將在2037年至2044年之間到期,而不考慮潛在的專利期延長或調整。
產品和目標組合
我們針對特定目標降解物的專利申請,包括我們的候選產品,集中在物質的組成、藥物組成、使用方法和製造過程中,涉及旨在降解致病蛋白質的新型化合物。截至2023年12月31日,我們擁有4項已頒發的美國專利,24項未決的美國專利申請,6項PCT專利申請,以及175多項涵蓋我們的降解劑和候選產品的外國專利申請。
具體地説,截至2023年12月31日,我們擁有四個專利系列(兩項美國專利、三項美國專利申請、一項PCT專利申請和五十四項外國專利申請),嚮導致IKZF1/3蛋白質靶標降解的化合物提出物質組成和藥物組成要求,以及相關的治療癌症和製造過程的使用方法。這些專利家族中有三個專利系列包括針對物質組合物的權利要求,具體包括CFT7455,我們的主要候選產品之一和相關的使用方法,如果通過支付所有必需的費用發佈和維護,將分別於2040年、2041年和2043年到期,而不考慮任何可能的專利期延長或調整。涵蓋我們的IKZF1/3降解器的第四個專利系列針對的是與前三個系列中所涵蓋的不同的專利系列,如果通過支付所有必需的費用進行授權和維護,將於2039年到期,而不考慮任何可能的專利期延長或調整。
截至2023年12月31日,我們擁有三個專利系列(一項美國專利、四項美國專利申請、一項PCT專利申請和二十四項外國專利申請),其權利要求涉及涵蓋我們的BRD9降解器的物質組合物,包括我們的CFT8634候選產品,以及相關的藥物組合物、使用方法和製造方法。聲稱優先於這些專利申請的美國和外國專利,如果通過支付所有必需的費用來授予和維護,將分別於2041年、2042年和2044年到期,而不考慮任何可能的專利期延長或調整。
截至2023年12月31日,我們擁有四個專利系列(五項美國專利申請,一項PCT專利申請,28項外國專利申請),其權利要求涉及我們的BRAF降解器及其相關使用方法、藥物組合物和製造方法的物質組合物。其中兩個專利系列包括針對物質組成的一般和具體權利要求,涵蓋CFT1946,我們的主要候選BRAF產品,以及相關的使用方法,以及聲稱優先於這些專利申請的美國和外國專利,如果通過支付所有必需的費用授予和維護,將分別於2042年和2043年到期,而不考慮任何可能的專利期延長或調整。涵蓋我們的BRAF降解器的另外兩個專利系列旨在與前兩個系列所涵蓋的專利系列分開,聲稱優先於這些專利申請的美國和外國專利,如果通過支付所有必需的費用獲得批准和維護,將分別於2041年和2044年到期,而不考慮任何可能的專利期延長或調整。
截至2023年12月31日,我們擁有兩個專利系列(一項美國專利、兩項美國專利申請和50項外國專利申請),其權利要求涉及涵蓋我們的EGFR降解器的物質組合物,包括我們的CFT8919候選產品,以及相關的使用方法、藥物組合物和製造工藝。聲稱優先於這些專利申請的美國和外國專利,如果通過支付所有必需的費用來授予和維護,將分別於2040年和2042年到期,而不考慮任何可能的專利期延長或調整。
截至2023年12月31日,我們擁有兩個專利系列(兩個美國專利申請和九個外國專利申請),其權利要求涉及我們的RET降解劑及其相關使用方法、藥物組合物和製造工藝的物質組合物。聲稱優先於這些專利申請的美國和外國專利,如果通過支付所有必需的費用來授予和維護,將分別於2041年和2044年到期,而不考慮任何可能的專利期延長或調整。
截至2023年12月31日,我們擁有8項美國專利申請、3項PCT專利申請和12項外國專利申請,這些專利申請的權利要求涉及降解劑的組合物、未披露的目標和相關的使用方法、藥物組合物和製造方法。聲稱優先於這些專利申請的美國和外國專利,如果通過支付所有必要的費用來授予和維護,將在2040年至2044年之間到期,而不考慮任何可能的專利期延長或調整。
與羅氏共同擁有的協作專利申請
截至2023年12月31日(如上所述,羅氏在2022年7月和2023年12月向我們轉讓了與未披露目標相關的專利的每一項權利),我們沒有與羅氏共同擁有任何專利申請或專利。如果未來提交與正在進行的合作活動相關的新專利申請,我們對任何此類未來專利申請的權利將受上述羅氏協議的管轄。
目標平臺協作
我們與戰略合作伙伴合作,擴大我們的平臺潛力,包括羅氏、Calico、Biogen和默克。根據與這些合作伙伴的協議,我們一般將我們單獨或共同發明的開發候選專利權轉讓給適用的合作伙伴,以換取根據這些協議正在開發的產品的經濟利益。
商業祕密
我們還依靠商業祕密、技術訣竅和持續創新來發展和保持我們的競爭優勢。根據我們與他們簽訂的協議,我們的任何員工和顧問在任何公司擁有的專利申請中被識別出來,都會將他們在任何此類專利申請中可能擁有的任何權利轉讓給我們。我們還依賴與員工、顧問和其他顧問簽訂的保密或其他協議來保護我們的專有信息。我們的政策是要求收到重要機密信息的第三方與我們簽訂保密或其他協議,其中包含對我們的機密和商業祕密信息的適當保護。
商標
我們在美國和海外擁有各種註冊和未註冊的商標和服務商標,包括C4 Treeutics、我們的房屋標誌、我們魚雷平臺的名稱以及我們的BIDAC降解器和MONODAC降解器的名稱。
政府監管
FDA和州和地方司法管轄區以及外國的類似監管機構對藥品的臨牀開發、製造和營銷提出了實質性要求。這些機構和其他聯邦、州和地方實體管理研究和開發活動,以及我們產品的測試、製造、質量控制、安全、有效性、標籤、儲存、包裝、記錄、跟蹤、批准、進口、出口、分銷、廣告和促銷。
美國政府對藥品的監管
在美國,FDA根據聯邦食品、藥物和化粧品法案(FDCA)及其實施條例對藥品進行監管。獲得監管批准和隨後遵守適用的聯邦、州、地方和外國法規和條例的過程需要花費大量的時間和財力。在產品開發過程、批准過程或批准後的任何時間,如果申請人未能遵守適用的美國要求,可能會受到各種行政或司法制裁,例如FDA拒絕批准未決的NDA、撤回批准、實施臨牀擱置、發出警告信、產品召回、產品扣押、完全或部分暫停生產或分銷、禁令、罰款、拒絕政府合同、恢復原狀、退還、退還或民事或刑事處罰。
FDA在候選產品可以在美國上市之前所需的程序通常包括以下幾個方面:
•必須按照GLP進行的非臨牀實驗室和動物試驗;
•向FDA提交研究用新藥或IND申請,該申請必須在臨牀試驗開始前生效;
•由獨立的機構評審委員會或IRB對每個臨牀站點或在每個試驗開始之前集中批准;
•充分和良好控制的人體臨牀試驗,以確定擬議的候選產品的安全性和有效性,以供其預期用途,根據良好的臨牀實踐或GCP進行;
•向FDA提交保密協議並支付使用費;
•如果適用,令人滿意地完成了FDA諮詢委員會的審查;
•對生產設施和選定的臨牀研究人員進行批准前檢查,以瞭解他們是否符合當前良好的生產實踐或cGMP和GCP;
•令人滿意地完成FDA對臨牀試驗地點的審計,以確保符合GCP和臨牀數據的完整性;以及
•FDA審查和批准NDA,以允許特定適應症的商業營銷。
測試和審批過程需要大量的時間、精力和財力。
臨牀前研究
臨牀前研究包括對藥物化學、藥理學、毒性和藥物製劑的實驗室評估,以及評估潛在安全性和有效性的動物研究。在開始候選產品的第一次臨牀試驗之前,贊助商必須向FDA提交臨牀前試驗和臨牀前文獻的結果,以及製造信息、分析數據和任何可用的臨牀數據或文獻,以及其他必要信息,作為IND的一部分。即使在IND提交之後,一些臨牀前研究仍可能繼續。IND在FDA收到後30天自動生效,除非FDA在30天內對臨牀試驗的進行提出安全擔憂或問題並強制臨牀暫停。在這種情況下,IND贊助商和FDA必須在臨牀試驗開始之前解決任何懸而未決的問題。因此,提交IND可能不會導致FDA授權開始臨牀試驗。
臨牀試驗
臨牀試驗涉及在符合GCP要求的合格研究人員的監督下,給人類受試者服用研究用新藥。在產品開發期間進行的每一項後續臨牀試驗,以及對以前提交的臨牀試驗的修改,都必須單獨提交給現有的IND。此外,參與臨牀試驗的每個機構的獨立IRB必須在臨牀試驗在該地點開始之前審查和批准任何臨牀試驗的計劃、其知情同意書和其他與研究對象的通信。在臨牀試驗進行期間,IRB必須繼續監督它,包括研究計劃的任何變化。
監管當局、IRB或贊助商可隨時以各種理由暫停或中止臨牀試驗,包括髮現受試者面臨不可接受的健康風險、臨牀試驗沒有按照FDA或IRB的要求進行,或者如果藥物與受試者受到意想不到的嚴重傷害有關。一些研究還包括一個數據安全監測委員會,該委員會在臨牀試驗期間接受對非盲目數據的特殊訪問,如果它確定對受試者存在不可接受的安全風險或其他理由,如沒有療效證明,可能會建議贊助商停止臨牀試驗。
人體臨牀試驗通常分三個連續階段進行,這些階段可能會重疊,也可能會合並。
•第一階段-最初進行研究是為了測試該產品在健康志願者或有目標疾病或條件的受試者中的安全性、劑量耐受性、結構-活性關係、作用機制、吸收、代謝、分佈和排泄。如果可能,一期臨牀試驗也可用於獲得產品有效性的初步適應症。
•階段2-對照研究是對患有特定疾病或狀況的一組受試者進行的,以提供足夠的數據來評估初步療效、最佳劑量和給藥計劃,以及安全的擴展證據。在開始更大規模和更廣泛的3期臨牀試驗之前,可以進行多個2期臨牀試驗以獲得信息。
•階段3-這些臨牀試驗通常在更大的受試者羣體中進行,以提供具有統計學意義的有效性證據,並在多個臨牀試驗地點的擴大受試者羣體中進一步測試安全性。這些臨牀試驗旨在確定產品的總體風險/益處概況,併為產品標籤提供充分的基礎。這些臨牀試驗可以在美國以外的試驗地點進行,只要全球地點也代表美國人口,並且在全球地點進行的研究符合FDA的規定和指導,如遵守GCP。
通常,在這些開發階段,臨牀試驗是在與FDA或外國監管機構協商的情況下設計的。正在開發的適應症可能會影響在進行臨牀試驗期間採用的研究設計,例如一線癌症治療適應症,它可能需要證明臨牀優勢或非劣勢於現有治療方法的面對面的數據。一線癌症適應症開發計劃的時間表也可能比三線或更長時間尋求的適應症更長,因為監管機構希望加快那些癌症進展的人獲得更晚線治療的機會,儘管有可用的和更早的治療方法。因此,許多新的腫瘤學產品最初尋求三線治療的適應症,這是任何腫瘤學適應症中較小的可用治療人羣,以及任何後來尋求的批准
那些針對更大治療人羣的早期治療系列產品可能需要進行額外的臨牀試驗。
FDA已經實施了可能影響腫瘤學候選產品開發的舉措。例如,2021年啟動的優化項目是一項倡議,旨在改革腫瘤學藥物開發中的劑量選擇和劑量優化範式,以強調選擇不僅最大限度地提高藥物療效而且最大限度地提高其安全性和耐受性的一個或多個劑量。Optimus項目將要求腫瘤學藥物的開發商在正在進行的計劃中實施戰略,在劑量選擇中利用非臨牀和臨牀數據,包括在試驗中對一系列劑量進行隨機評估的潛在需要,並要求這些研究在開發計劃中儘早進行。
此外,FDA在2022年3月發佈了一份題為《擴展隊列:用於首個人類臨牀試驗以加快腫瘤藥物和生物製品的開發》的最終指南,其中概述了藥物開發商如何在腫瘤藥物開發的早期階段(即第一個人體臨牀試驗)利用通常被稱為無縫試驗設計的適應性試驗設計,將傳統的三個階段的試驗壓縮為一個稱為擴展隊列試驗的連續試驗。支持個人擴展隊列設計的信息包括在IND申請中,並由FDA進行評估。擴大隊列試驗可能會提高藥物開發的效率,減少開發成本和時間。
FDA可能會要求,或者公司可能會在產品獲得批准後進行額外的臨牀試驗。這些所謂的4期試驗可能會成為批准後需要滿足的條件。4期試驗的結果可以確認候選產品的有效性,並可以提供重要的安全信息。
臨牀試驗必須在符合GCP要求的合格研究人員的監督下進行,其中包括要求所有研究對象以書面形式提供他們參與任何臨牀試驗的知情同意,並由IRB審查和批准該研究。調查人員還必須向臨牀試驗贊助商提供信息,以允許贊助商向FDA披露具體的財務信息。臨牀試驗是根據詳細説明試驗目標、試驗程序、用於監測安全性的參數和要評估的療效標準以及統計分析計劃等的方案進行的。一些臨牀試驗的信息,包括試驗和試驗結果的描述,必須在特定的時間框架內提交給NIH,以便在其網站上公開傳播。詳細説明臨牀試驗結果的進展報告必須至少每年提交給FDA,如果發生嚴重的不良事件,則更頻繁地提交。
用於進行人體臨牀試驗的研究藥物的製造受cGMP要求的約束。進口到美國的研究藥物和活性藥物成分也受到FDA關於其標籤和分銷的監管。此外,美國以外的研究用藥物產品的出口須受接受國的監管要求以及美國《食品藥品管制法》下的出口要求的約束。詳細説明臨牀試驗結果的進展報告必須至少每年提交給FDA和IRB,如果發生嚴重的不良反應,則更頻繁地提交。
在臨牀試驗的同時,公司通常會完成額外的動物研究,還必須開發關於候選產品的化學和物理特性的額外信息,以及根據cGMP要求最終確定商業批量生產產品的工藝。製造過程必須能夠始終如一地生產高質量的候選產品批次,尤其是必須開發用於測試最終產品的特性、強度、質量和純度的方法。此外,必須選擇和測試適當的包裝,並進行穩定性研究,以證明候選產品在保質期內不會發生不可接受的變質。
FDA加快審查和批准計劃
FDA有各種計劃,包括快速通道指定、突破性治療指定、加速批准和優先審查,這些計劃旨在加快或簡化用於治療嚴重或危及生命的疾病或狀況的藥物的開發和FDA審查過程,並展示解決未滿足的醫療需求的潛力。這些計劃的目的是比FDA標準審查程序更早地向患者提供重要的新藥。
根據快速通道計劃,候選新藥的贊助商可以要求FDA在候選藥物IND提交的同時或之後,將特定適應症的候選藥物指定為快速通道藥物。為了有資格獲得快速通道認證,FDA必須根據贊助商的請求,確定一種產品旨在治療嚴重或危及生命的疾病或狀況,並展示出滿足未得到滿足的醫療需求的潛力。FDA將確定,如果一種產品將提供一種不存在的療法,或者提供一種基於療效或安全因素的潛在優於現有療法的療法,該產品將滿足未滿足的醫療需求。快速通道指定為與FDA審查團隊的互動提供了更多機會,並可能允許滾動審查NDA
在提交完整的申請之前,如果贊助商提供了提交保密協議部分的時間表,FDA同意接受保密協議的部分,並確定時間表是可接受的,贊助商在提交保密協議的第一部分時支付任何所需的使用費。然而,FDA審查申請的時間段目標直到NDA的最後一部分提交後才開始。如果FDA確定資格標準不再適用,它可能決定撤銷快車道指定。
此外,如果一種藥物打算單獨或與一種或多種其他藥物聯合使用,用於治療嚴重或危及生命的疾病或狀況,並且初步臨牀證據表明,該藥物可能在一個或多個臨牀重要終點顯示出比現有療法有實質性改善,例如在臨牀開發早期觀察到的實質性治療效果,贊助商可以申請突破性的治療指定。被指定為突破性療法的藥物有資格獲得FDA關於高效藥物開發計劃的密集指導,對產品開發和審查的組織承諾,包括高級管理人員的參與,以及與Fast Track產品一樣,也有資格獲得NDA的滾動審查。如果符合相關標準,快速通道和突破性治療產品都有資格獲得加速批准和/或優先審查。
根據FDA的加速審批路徑,FDA可能會批准一種針對嚴重或危及生命的疾病的藥物,該藥物為患者提供比現有治療更有意義的治療益處,其基礎是合理地可能預測臨牀益處的替代終點,或者可以比不可逆轉的發病率或死亡率更早地測量的臨牀終點,該臨牀終點合理地可能預測對不可逆轉的發病率或死亡率或其他臨牀益處的影響,同時考慮到病情的嚴重性、稀有性或流行率以及替代療法的可用性或缺乏。在此基礎上批准的候選藥物必須遵守嚴格的上市後遵從性要求,包括完成4期或批准後臨牀試驗,以確認對臨牀終點的影響。根據2022年食品和藥物綜合改革法案(FDORA),FDA現在被允許酌情要求這些試驗在批准之前或在獲得加速批准的產品批准之日後的特定時間段內進行。根據FDORA,FDA增加了加快程序的權力,例如,如果驗證性試驗未能驗證產品的預期臨牀益處,則可以撤回在加速批准下批准的藥物或適應症的批准。所有根據加速審批條例批准的候選藥物的宣傳材料都必須經過FDA的事先審查,除非FDA另行通知贊助商。
一旦提交了旨在治療嚴重疾病的產品的NDA,如果FDA確定該產品如果獲得批准,將在安全性或有效性方面提供顯著改善,FDA可能會指定優先審查指定。優先審查意味着FDA審查申請的目標是6個月,而不是根據《處方藥使用費法案》(PDUFA)指南的標準審查10個月。根據目前的PDUFA業績目標,這六個月和十個月的審查期是從新分子實體的60天提交日期而不是收到日期起計算的,這通常會使自提交之日起審查的時間線增加大約兩個月。
即使一種產品符合這些計劃中的一個或多個,FDA也可以在以後決定該產品不再符合資格條件,或者決定FDA審查或批准的時間段不會縮短。此外,治療嚴重或危及生命的疾病的研究藥物的製造商被要求提供,例如通過在其網站上張貼其關於迴應擴大准入請求的政策。此外,快速通道指定、突破性治療指定、加速批准和優先審查不會改變批准的標準,也可能最終不會加快開發或批准過程。
食品和藥物管理局提交的保密協議和審查
假設成功完成所需的臨牀和臨牀前測試,以及其他項目,產品開發的結果,包括化學、製造和控制、非臨牀研究和臨牀試驗,將與擬議的標籤一起提交給FDA,作為NDA的一部分。提交保密協議需要向FDA支付一筆可觀的使用費。這些使用費必須在第一次提交申請時提交,即使申請是滾動提交的。在某些情況下,可以免除或減少費用。免除申請費的一個依據是,如果申請人僱用的員工少於500人,包括附屬公司的員工,申請人沒有批准的產品營銷申請,該產品已被引入或交付到州際商業,並且申請人,包括其附屬公司,正在提交其第一個營銷申請。孤兒指定藥品也免收申請費。
根據《兒科研究公平法》,一種新的有效成分、適應症、劑型、給藥方案或給藥途徑的NDA或補充NDA必須包含足夠的數據,以評估該藥物在所有相關兒科亞羣中聲稱的適應症的安全性和有效性,並支持該產品對安全和有效的每個兒科亞羣的劑量和給藥。除非法規另有要求,否則PREA不適用於已被批准為孤兒藥物指定的適應症的藥物,除非PREA將向原始NDA申請新的有效成分,如果該藥物是分子上的孤兒指定的
用於治療成人癌症的靶向癌症產品,針對FDA確定與兒童癌症的生長或進展密切相關的分子靶點。FDA可以主動或應申請人的要求,批准推遲提交部分或全部兒科數據,直到批准該產品用於成人或完全或部分免除兒科數據要求之後。
FDA可能會將含有活性成分(包括活性成分的任何酯或鹽)的藥物申請提交給諮詢委員會,這些藥物之前沒有得到FDA的批准。FDA還可能將提出安全性、純度或有效性難題的藥物提交諮詢委員會。諮詢委員會通常是一個由臨牀醫生和其他專家組成的小組,他們審查、評估申請並就是否應該批准申請以及在什麼條件下提出建議。FDA不受諮詢委員會建議的約束,但它在做出決定時會仔細考慮這些建議。
FDA審查申請,以確定產品對於其預期用途是否安全有效,以及製造控制是否足以確保和保持產品的特性、強度、質量和純度。在批准保密協議之前,FDA將檢查生產該產品的一個或多個設施。FDA將不會批准申請,除非它確定製造工藝和設施,包括合同製造商和分包合同,符合cGMP要求,並足以確保產品在所要求的規格下一致生產。此外,在批准NDA之前,FDA通常會檢查一個或多個臨牀試驗地點,以確保符合GCP。
一旦FDA收到申請,在接受申請之前,它有60天的時間審查NDA,以確定它是否基本上完成了允許進行實質性審查。一旦提交的申請被接受,FDA就開始對NDA進行深入審查。根據FDA在PDUFA下達成的目標和政策,FDA對新分子實體或NME的標準NDA的審查目標是從60天的申請日期起10個月。對於優先審查申請,FDA對NME NDA的審查目標是在60天申請日期的六個月內。這樣的截止日期被稱為PDUFA日期。PDUFA日期只是一個目標,FDA並不總是達到其PDUFA日期。如果FDA要求或NDA贊助商在審查期間提供補充信息或澄清以修正原始申請,則審查過程和PDUFA日期也可延長。
一旦FDA對該申請的審查完成,FDA將發佈一份完整的回覆信或CRL或批准信。CRL表示申請的審查週期已完成,申請尚未準備好審批。CRL通常包含為了確保NDA的最終批准而必須滿足的特定條件的聲明,並且可能需要額外的臨牀或臨牀前測試或其他信息或分析,以便FDA在重新提交的NDA中重新考慮申請。即使提交了額外的信息,FDA最終也可能決定該申請不符合批准的監管標準。如果這些條件得到了FDA的滿意,FDA可能會簽發批准信。批准函授權該藥物的商業營銷,並提供特定適應症的具體處方信息。
如果不符合適用的監管標準,FDA可以推遲或拒絕批准NDA,要求額外的測試或信息,要求上市後測試和監督以監測產品的安全性或有效性和/或施加其他條件,包括分銷限制或其他風險管理機制。例如,FDA可能要求風險評估和緩解戰略,或REMS,作為批准或批准後的條件,以減輕任何已確定或懷疑的嚴重風險,並確保藥物的安全使用。FDA可以根據上市後研究或監測計劃的結果,阻止或限制產品的進一步銷售,或施加額外的上市後要求。批准後,對批准的產品的某些類型的更改,如增加新的適應症、製造更改和額外的標籤聲明,將受到進一步的測試要求、FDA通知和FDA事先審查和批准的約束。此外,如果出現新的安全信息,可能需要額外的測試、產品標籤更改或FDA通知。
如果產品獲得監管部門的批准,這種批准可能會對該產品可能上市的指定用途進行限制,或者可能包括禁忌症、盒裝警告或產品標籤中的其他警告或預防措施,從而導致盒裝警告。方框警告是FDA在藥品標籤中提出的最嚴格的警告,當有合理證據表明該藥物與嚴重危險有關時。FDA也可能不會批准將申請人尋求的所有標籤索賠納入其中。一旦獲得批准,如果沒有保持對上市前和上市後監管標準的遵守,或者如果產品進入市場後出現問題,FDA可能會撤回產品批准。此外,FDA可能要求進行第四階段上市後研究,以監測批准產品的有效性或安全性,並可能根據這些上市後研究的結果限制該產品的進一步銷售。
美國審批後要求
我們根據FDA批准製造或分銷的任何產品都將受到FDA的持續監管,包括定期報告、產品抽樣和分銷、廣告、促銷、藥品短缺報告、遵守作為批准條件施加的任何批准後要求,如4期臨牀試驗或REMS,以及記錄和報告要求,包括不良經歷。
在批准後,對批准的產品的許多更改,如增加新的適應症或其他標籤聲明,都必須事先得到FDA的審查和批准。對於批准的產品,還有持續的年度計劃費用要求,以及具有臨牀數據的補充應用程序的新申請費。藥品製造商及其分包商,以及那些提供產品、成分和成分的公司,必須向FDA和某些州機構註冊,並列出其藥品,並接受FDA和這些州機構對cGMP和其他要求的定期宣佈和突擊檢查,這些要求施加了程序和文件要求。
對製造工藝的更改受到嚴格監管,通常需要FDA事先批准或通知才能實施。FDA的規定還要求調查和糾正與cGMP和規範的任何偏差,並對贊助商和贊助商可能決定使用的任何第三方製造商提出報告和文件要求。因此,製造商必須繼續在生產和質量控制方面花費時間、金錢和精力,以保持cGMP合規性。
後來發現產品存在以前未知的問題,包括嚴重程度或頻率非預期的不良事件,或生產工藝,或不符合監管要求,可能導致撤回上市許可,強制修訂已批准的標籤以添加新的安全性信息或其他限制,強制進行上市後研究或臨牀試驗以評估新的安全性風險,或在REMS計劃下實施分配或其他限制,以及其他後果。
FDA嚴格監管藥品的營銷和推廣。公司只能做出與FDA批准的標籤一致的安全性和有效性相關的聲明。醫生根據其獨立的專業醫學判斷,可能會將合法獲得的產品用於產品標籤中未描述的用途,以及與我們測試和FDA批准的用途不同的用途。但是,禁止製造商和代表他們的第三方以與批准的標籤不一致的方式銷售或推廣藥物。FDA和其他機構執行禁止推廣標籤外使用的法律和法規,被發現不當推廣標籤外使用的公司可能要承擔重大責任。
不遵守FDA的任何要求都可能導致重大的不利執法行動。這些制裁可能包括各種行政或司法制裁,例如拒絕批准待決申請、吊銷或吊銷執照、撤回批准、實施臨牀暫停或終止臨牀試驗、警告信、無標題信件、修改宣傳材料或標籤、產品召回、產品扣押或拘留、拒絕允許進口或出口、完全或部分暫停生產或分銷、禁止、禁令、罰款、同意法令、公司誠信協議、拒絕政府合同和現有合同下的新命令、禁止參與聯邦和州醫療保健計劃、賠償、返還、返還或民事或刑事處罰,包括罰款和監禁。如果不遵守FDA關於推廣處方藥的要求,也可能會導致調查,指控他們違反了聯邦和州醫療保健欺詐和濫用等法律以及州消費者保護法。這些制裁中的任何一項都可能導致負面宣傳,以及其他不利後果。
美國營銷排他性
FDA規定了非專利監管專有期,這為批准的NDA的持有者提供了有限的保護,使其在FDA批准NDA後的三年或五年內免受其批准的藥物所代表的創新在市場上的新競爭。新的化學實體或NCE可以獲得五年的排他性。NCE是一種不含FDA在任何其他NDA中批准的活性部分的藥物。活性部分是指負責藥物物質治療活性的分子或離子,不包括使藥物成為酯、鹽、包括具有氫或配位鍵的鹽或其他不涉及原子之間共享電子對的非共價鍵的分子、衍生物,如分子的絡合物(即,由兩個化合物的化學作用形成)、螯合物(即,化合物)或籠合物(即,捕獲分子的聚合物骨架)。在排他期內,FDA可能不接受或批准由另一家公司提交的簡化新藥申請或ANDA或505(B)(2)NDA
它包含先前批准的主動部分。然而,如果提交了第四款認證,ANDA或505(B)(2)申請可以在NCE排他性到期前一年提交。
根據《孤兒藥品法》,如果一種藥物產品旨在治療一種罕見的疾病或疾病,FDA可以將該藥物產品指定為“孤兒藥物”(通常意味着,在美國,如果無法合理預期在美國開發和製造用於治療該疾病或疾病的藥物產品的成本將從該產品的銷售中收回,則該藥物產品在美國影響的人數不到20萬人或更多)。公司必須在提交保密協議之前申請孤兒藥物指定。如果這一請求被批准,FDA將披露治療劑的身份及其潛在用途。孤立產品的指定不會在監管審查和批准過程中傳達任何優勢,也不會縮短監管審查和批准過程的持續時間。
如果一種孤兒指定藥物獲得FDA對其具有此類指定的疾病或條件的第一次批准,或在其指定的罕見疾病或條件下的選定適應症或使用,該產品通常將獲得孤兒藥物排他性。孤立藥物排他性意味着FDA在七年內不得批准同一產品的同一適應症的任何其他申請,除非在某些有限的情況下。如果一種孤兒指定的藥物獲得了上市批准,其適應症的範圍比其孤兒產品申請中指定的適應症更廣泛,它可能沒有資格獲得排他性。在某些情況下,孤兒專利也不會阻礙另一種產品的批准,包括如果具有相同適應症的相同有效成分的後續產品被證明在臨牀上更好於批准的產品,因為它具有更好的療效或安全性,或者對患者護理做出了重大貢獻,或者如果擁有孤兒藥物專利的公司無法滿足市場需求。此外,FDA可以批准一種以上的產品用於同一孤兒適應症或疾病,只要這些產品含有不同的有效成分。此外,競爭對手可能會對同一產品獲得批准,但對孤立產品具有排他性的不同適應症則可能獲得批准。
此外,FDCA還鼓勵贊助商在兒科人羣中進行藥物研究。如果贊助商自願完成一項兒科研究,並且公平地迴應了FDA發佈的書面請求,則該藥物可能有資格獲得兒科獨家專利。如果被批准,兒科專有權將為所有包含被授予兒科專營權的活性部分的經批准的藥物產品的現有監管專有期和專利條款增加6個月。
美國以外的監管
關於我們的候選產品在美國以外的開發,我們將受到類似的外國法律法規的約束,包括歐盟和中國。
歐盟藥物開發
在歐盟,我們的候選產品也將受到廣泛的監管要求。與美國一樣,醫藥產品只有在獲得主管監管機構的營銷授權後才能上市,而且歐盟的臨牀前和臨牀研究的各個階段都受到嚴格的監管控制。
2014年4月,歐盟通過了歐盟第536/2014號臨牀試驗條例,該條例於2022年1月31日取代了臨牀試驗指令2001/20/EC。《臨牀試驗條例》的暫時性條款規定,到2025年1月31日,所有正在進行的臨牀試驗必須過渡到該條例。新的臨牀試驗條例的主要特點包括:通過臨牀試驗信息系統通過單一入口點簡化申請程序;為申請準備和提交一套單一文件,以及簡化臨牀試驗贊助商的報告程序;臨牀試驗申請評估的統一程序分為兩部分(第一部分包含科學和醫藥產品文件,第二部分包含國家和患者層面的文件)。第一部分通過所有歐盟成員國主管當局的協調審查進行評估,在該審查中,已提交臨牀試驗授權申請(有關成員國)參考成員國編寫的報告草案。第二部分由每個有關成員國單獨評估。已經為臨牀試驗申請的評估設定了嚴格的最後期限。相關倫理委員會在評估程序中的作用仍由相關歐盟成員國的國家法律管轄,但總體相關時間表由《臨牀試驗條例》確定。《臨牀試驗條例》自生效之日起將有三年的過渡期。
在歐盟,兒科委員會或EMA的PDCO必須在提交營銷授權申請或MAA之前批准兒科調查計劃或PIP,除非EMA已批准(1)特定產品豁免、(2)類別豁免或(3)推遲PIP中包括的一項或多項措施。PIP概述了製藥公司在兒科人羣中調查新醫藥產品的戰略。在提交MAA或修改現有的營銷授權之前,EMA評估公司是否遵守了每個相關PIP中列出的商定研究和措施。如果申請人在所有歐盟成員國獲得上市授權,或獲得歐盟委員會在集中程序中授予的上市授權,並且針對兒科人羣的研究結果包括在產品信息中,即使是否定的,該藥物也有資格額外獲得6-
通過延長任何補充保護證書或SPC的期限進行合格專利保護的一個月的期限,只要在提交產品的SPC申請的同時,或在SPC到期前的任何時候提出這一延期申請。在孤兒藥品的情況下,可以將孤兒市場的專有權延長兩年。這一兒科獎勵受到特定條件的制約,在開發和提交符合PIP的數據時不會自動獲得。
歐洲聯盟加速審查和發展
PRIME或優先醫學是EMA提供的一項計劃,旨在加強對針對未得到滿足的醫療需求的藥物開發的支持,並對代表重大創新的產品提供加速評估,其中MAA將通過集中程序進行。要獲得Prime的資格,候選產品需要早期臨牀證據,證明該療法有可能提供比現有療法更大的治療優勢,或使沒有治療選擇的患者受益。中小型企業的產品可能比大公司更早有資格加入Prime計劃。Prime的好處包括從EMA的科學委員會任命一名報告員,在MAA之前提供持續支持和幫助積累知識,在關鍵發展里程碑進行早期對話和科學建議,以及有可能在申請過程更早的時候對產品進行資格鑑定以進行加速審查。收到Prime指定不會改變審批標準,但可能會加快開發或審批過程。如果產品在開發過程中不再符合資格標準,可能會撤回Prime計劃下的支持。
歐盟藥品審查和批准
在歐盟,醫藥產品只有在獲得營銷授權或MA後才能商業化。有兩種類型的MA。由歐共體根據歐洲藥品管理局人用藥品委員會(CHMP)的意見,通過集中程序頒發的集中式MA,在整個歐盟以及EEA的其他成員國(冰島、列支敦士登和挪威)有效。對於某些類型的產品,如某些生物技術生產的藥品、被指定為孤兒藥品的產品、高級治療藥品(即基因療法、體細胞療法或組織工程藥物)以及含有用於治療艾滋病毒、艾滋病、癌症、神經退行性疾病、糖尿病、自身免疫和其他免疫功能障礙和病毒疾病的新活性物質的藥品,必須實行集中程序。對於含有歐盟尚未授權的新活性物質的產品,或構成重大治療、科學或技術創新或符合歐盟公共衞生利益的產品,集中程序是可選的。
國家MA由歐盟成員國的主管當局頒發,僅覆蓋其各自的領土,適用於不屬於集中程序強制範圍的產品。如果一種產品已被授權在歐盟成員國銷售,則該國家MA可通過互認程序在另一成員國獲得承認。如果該產品在申請時沒有在任何成員國獲得國家MA,它可以通過分散的程序在各成員國同時獲得批准。根據分權程序,向尋求MA的每個成員國的主管當局提交一份相同的卷宗,申請者選擇其中一個作為參考成員國,或RMS。RMS主管當局編制評估報告草案、產品特性概要草案或SmPC,以及標籤和包裝傳單草案,送交其他成員國(指有關成員國)批准。如果有關成員國基於對公眾健康的潛在嚴重危害,沒有對RMS提出的評估、SmPC、標籤或包裝提出異議,則該產品隨後在所有成員國(即RMS和有關成員國)獲得國家MA。
根據上述程序,在授予MA之前,EMA或歐盟成員國主管當局根據有關產品質量、安全性和有效性的科學標準,對產品的風險效益平衡進行評估。
兒科用營銷授權,或PUMA,可用於已經獲得授權的藥物,不再受SPC或符合SPC資格的專利覆蓋,並將僅用於兒童開發。PUMA是一種專門的MA,涵蓋專門為兒科人羣開發的醫藥產品的適應症和配方,如果這種開發是根據批准的PIP進行的。申請PUMA有各種激勵措施,包括進入集中程序,即使產品本來不屬於該程序的強制範圍。
歐盟孤兒稱號
在歐盟,歐盟委員會在收到EMA孤兒藥物產品委員會的意見後,如果其贊助商能夠證明:(1)該產品旨在診斷、預防或治療危及生命或慢性衰弱的疾病;(2)或(I)這種情況,歐盟委員會將就該產品授予孤兒稱號
當提出申請時,該產品在歐盟的影響不超過10,000人中的五人,或者(Ii)如果沒有孤兒身份帶來的好處,該產品不太可能在歐盟產生足夠的回報,以證明其開發所需的投資是合理的;以及(3)沒有令人滿意的診斷、預防或治療這種疾病的方法授權在歐盟銷售,或者,如果存在這種方法,該產品將對受該疾病影響的人產生重大好處。
在歐盟,孤兒指定使一方有權獲得經濟激勵,如減少費用或免除費用,並在授予MA後授予10年的市場排他性。在這一市場排他期內,歐洲藥品管理局、歐盟委員會或歐盟成員國的任何主管機構都不能接受申請,也不能批准“類似醫藥產品”的市場準入。“類似醫藥產品”的定義是含有與批准的孤兒醫藥產品中所含的一種或多種類似活性物質的醫藥產品,其目的是用於相同的治療適應症。如果在第五年結束時確定不再符合孤兒指定標準,包括證明產品的利潤足以不足以證明維持市場排他性是合理的,則這一期限可縮短至六年。在非常特殊的情況下,可向與授權孤兒產品類似的醫藥產品授予授權書,例如:(I)確定類似醫藥產品比授權產品更安全、更有效或在臨牀上更好;(Ii)授權孤兒產品的MA持有人同意授權該類似醫藥產品;或(Iii)授權孤兒產品的MA持有人不能供應足夠的孤兒醫藥產品。在提交MA申請之前,必須要求指定為孤兒。孤兒指定不會在監管審查和批准過程中傳遞任何優勢,也不會縮短監管審查和批准過程的持續時間。
中國的藥品開發
所有在中國進行的尋求上市批准的臨牀試驗必須得到中國國家醫療產品管理局的批准,並在符合GCP要求的醫院進行。除了支持開發的獨立中國試驗外,進口藥品申請者還可以將中國臨牀站點納入國際多中心試驗(IMCT)。國內生產的藥物不受外國批准的要求,與以前的做法不同,國家藥品監督管理局決定允許這些藥物也通過IMCT進行開發。
2019年修訂的《中華人民共和國藥品管理法》,現已對新藥臨牀試驗採用默示審批制度。如果在60個工作日後,申請人沒有收到藥物評價中心(CDE)的任何反對意見,試驗就可以繼續進行,而不是像以前那樣,臨牀試驗的預批准過程較長,申請人必須等待肯定的批准。通過廢除GCP認證制度和要求審判地點遵循更簡化的通知程序,審判地點還擴大了審判地點的數量。
NMPA於2021年11月敲定了以臨牀價值為導向的腫瘤學藥物研發指南,作為其政策的一部分,旨在鼓勵具有重大臨牀價值的創新腫瘤學藥物的研究和開發,並阻止重複研究和開發對患者的臨牀價值最低或沒有臨牀價值的藥物。
在中國進行的臨牀試驗必須通過藥物臨牀試驗信息平臺(http://www.chinadrugtrials.org.cn).)註冊和發佈申請人需在獲得臨牀試驗批准後一個月內對試驗信息進行預登記,以獲得試驗的唯一註冊號,並在第一個受試者登記參加試驗之前完成某些後續信息的登記。取得臨牀試驗批准後一年內未取得上述預註冊和註冊的,申請人應提交説明,三年內未完成手續的,臨牀試驗批准自動失效。
其他醫保法
醫療保健提供者、醫生和第三方付款人將在我們獲得市場批准的任何產品的推薦和處方中發揮主要作用。我們的業務運營以及目前或未來與第三方付款人、醫療保健提供者和醫生的任何安排可能會使我們面臨廣泛適用的欺詐和濫用以及其他醫療法律和法規,這些法律和法規可能會限制我們開發、營銷、銷售和分銷我們獲得上市批准的任何藥物的業務或財務安排和關係。在美國,這些法律包括但不限於州和聯邦反回扣、虛假聲明、醫生透明度以及患者數據隱私和安全法律法規,包括但不限於下述法律法規。
•聯邦反回扣法規,除其他事項外,禁止個人和實體直接或間接、公開或隱蔽地以現金或實物直接或間接、公開或祕密地索要、提供、支付、接受或提供任何報酬(包括任何回扣、新娘或某些回扣),以誘導或獎勵或回報個人轉介或購買或推薦任何商品或服務,而根據聯邦醫療保健計劃,可對其進行全部或部分付款
醫療保險和醫療補助。個人或實體不需要實際瞭解聯邦《反回扣法令》或違反該法令的具體意圖即可實施違法行為。有一些法定例外和監管避風港保護一些常見的活動不被起訴。違規行為將被處以民事和刑事罰款,並對每一次違規行為處以罰款,外加最高三倍的薪酬、監禁和被排除在政府醫療保健計劃之外;
•聯邦民事和刑事虛假索賠法律,包括民事虛假索賠法案或FCA,禁止個人或實體故意向聯邦政府提交或導致提交虛假、虛構或欺詐性的付款或批准索賠;故意做出、使用或導致做出或使用虛假陳述或記錄材料,以虛假或欺詐性的索賠或向聯邦政府支付或轉移金錢或財產的義務;或故意隱瞞或故意不正當地逃避或減少向聯邦政府支付金錢或財產的義務。根據FCA,即使製造商沒有直接向政府付款人提交索賠,如果他們被認為“導致”提交虛假或欺詐性索賠,他們也可能被追究責任。此外,政府可以斷言,就民事虛假索賠法而言,包括因違反聯邦反回扣法規而產生的物品或服務的索賠構成虛假或欺詐性索賠。FCA還允許充當“告密者”的個人代表聯邦政府提起訴訟,指控違反FCA的行為,並分享任何金錢追回;
•聯邦民事罰款法,除其他事項外,對向聯邦醫療保險或州醫療保健計劃受益人提供或轉移或支付報酬的行為處以民事罰款,如果此人知道或應該知道這可能會影響受益人對聯邦醫療保險或州醫療保健計劃可報銷服務的特定提供者、從業者或供應商的選擇,除非適用例外情況。政府還可以斷言,根據聯邦民事罰金法,包括因違反聯邦反回扣法規而產生的物品或服務的索賠構成虛假或欺詐性索賠;
•1996年《健康保險可攜性和責任法案》,或HIPAA,對明知和故意執行計劃或試圖執行計劃,詐騙任何醫療福利計劃,包括私人付款人,明知和故意挪用或竊取醫療福利計劃,故意阻礙對醫療保健違法行為的刑事調查,或偽造、隱瞞或掩蓋重大事實,或作出與提供或支付醫療福利、項目或服務有關的任何重大虛假陳述,規定刑事和民事責任。與聯邦《反回扣法令》類似,個人或實體不需要實際瞭解該法令或違反該法令的具體意圖即可實施違法行為;
•HIPAA經2009年《衞生信息技術促進經濟和臨牀健康法》及其各自的實施條例修訂,除其他外,對承保實體及其業務夥伴提出了與可單獨識別的健康信息的隱私和安全有關的具體要求,包括強制性合同條款和對此類信息的技術保障的必要實施。HITECH還創建了新的民事罰款等級,修改了HIPAA,使民事和刑事處罰在某些情況下直接適用於商業夥伴,並賦予州總檢察長新的權力,可以向聯邦法院提起民事訴訟,要求損害賠償或禁制令,以執行聯邦HIPAA法律,並尋求與提起聯邦民事訴訟相關的律師費和費用;
•《醫生支付陽光法案》是《患者保護和平價醫療法案》的一部分,經2010年《醫療保健和教育和解法案》修訂,或統稱為《醫療保險法案》,該法案對某些藥品、器械、生物製品和醫療用品製造商提出了新的年度報告要求,要求這些製造商向醫生(定義包括醫生、牙醫、驗光師、足科醫生和脊椎按摩師)、某些其他有執照的醫療從業者和教學醫院,以及這些醫生及其直系親屬持有的所有權和投資權益,支付某些款項和向醫生(包括醫生、牙醫、驗光師、足科醫生和脊椎按摩師)支付費用;以及
•類似的州和外國法律和法規,如州反回扣和虛假索賠法,可能適用於銷售或營銷安排和涉及由非政府第三方付款人(包括私人保險公司)償還的醫療項目或服務的索賠,範圍可能比聯邦同等法律更廣泛;州和外國法律要求製藥公司遵守制藥業的自願合規指南和聯邦政府頒佈的相關合規指南,或以其他方式限制向醫療保健提供者支付的款項;州和外國法律,要求藥品製造商報告與藥品定價和付款有關的信息,以及向醫生和其他保健提供者進行其他價值轉移,並限制營銷做法或要求披露營銷支出和定價信息;州和地方法律要求註冊
藥品銷售代表;在某些情況下管理健康信息隱私和安全的州和外國法律。這些數據隱私和安全法律可能在很大程度上不同,而且往往不會被HIPAA先發制人,這可能會使合規工作複雜化。
此外,製藥商還可能受到聯邦和州消費者保護以及不正當競爭法律和法規的約束,這些法律和法規對市場活動進行了廣泛的監管,可能會損害消費者。
藥品和生物製品的分銷須遵守額外的要求和條例,包括廣泛的記錄保存、許可、儲存和安全要求,以防止未經授權銷售藥品。
這些法律的全部範圍和執行都是不確定的,並受到當前醫療改革環境的快速變化的影響。聯邦和州執法機構繼續加強對醫療保健公司與醫療保健提供者之間互動的審查,這導致了醫療保健行業的一系列調查、起訴、定罪和和解。政府當局可能會得出結論,認為我們的業務做法不符合當前或未來涉及適用欺詐和濫用或其他醫療保健法律和法規的現行或未來法規、法規或判例法。如果我們的運營被發現違反了任何這些法律或任何其他可能適用於我們的相關政府法規,我們可能會面臨重大的民事、刑事和行政處罰、損害賠償、罰款、監禁、交還、被排除在政府資助的醫療保健計劃之外(如Medicare和Medicaid)、聲譽損害、額外的監督和報告義務,如果我們受到公司誠信協議或類似和解的約束,以解決有關違反這些法律的指控,並削減或重組我們的業務。如果我們預期與之有業務往來的任何醫生或其他醫療保健提供者或實體被發現不遵守適用法律,他們可能會受到類似的行動、處罰和制裁。確保商業安排符合適用的醫保法,以及對政府當局可能進行的調查做出迴應,可能會耗費時間和資源,並可能分散公司對其業務的注意力。
承保和報銷
在美國和其他國家的市場上,根據自己的病情接受處方治療的患者和提供處方服務的提供者通常依賴第三方付款人來報銷全部或部分相關的醫療費用。因此,即使候選產品獲得批准,該產品的銷售在一定程度上也將取決於第三方付款人,包括美國的政府醫療計劃,如Medicare和Medicaid,商業健康保險公司和管理型醫療保健組織,為該產品提供保險和建立足夠的補償水平的程度。在美國,第三方付款人之間沒有統一的藥品保險和報銷政策。因此,藥品的承保範圍和報銷範圍因付款人而異。確定第三方付款人是否將為產品提供保險的過程可以與設置價格或報銷率的過程分開,一旦保險獲得批准,付款人將為產品支付費用。第三方付款人越來越多地挑戰所收取的價格,審查醫療必要性,審查醫療產品和服務的成本效益,並實施控制以管理成本。第三方付款人可以將承保範圍限制在批准清單上的特定產品,也稱為配方表,該清單可能不包括特定適應症的所有批准產品。
為了確保任何可能被批准銷售的產品的保險和報銷,公司可能需要進行昂貴的藥物經濟學研究,以證明該產品的醫療必要性和成本效益,以及獲得FDA或其他類似監管批准所需的成本。此外,公司可能還需要向購買者、私人健康計劃或政府醫療計劃提供折扣。儘管如此,候選產品可能不被認為是醫學上必要的或具有成本效益的。一旦產品獲得批准,第三方付款人不承保產品的決定可能會減少醫生的使用率,並對銷售、我們的運營和財務狀況產生實質性的不利影響。此外,第三方付款人決定為產品提供保險並不意味着將批准足夠的報銷率。此外,一個付款人決定為一種產品提供保險並不能保證其他付款人也會為該產品提供保險和報銷,而且不同付款人的保險和報銷水平可能有很大不同。
控制醫療成本已成為聯邦、州和外國政府的優先事項,產品價格一直是這一努力的重點。各國政府對實施成本控制計劃表現出了極大的興趣,包括價格控制、限制報銷和要求替代仿製藥。採取價格控制和成本控制措施,以及在現有控制和措施的司法管轄區採取更嚴格的政策,可能會進一步限制一家公司從銷售任何經批准的產品中獲得的收入。承保政策和第三方付款人報銷率可能隨時發生變化。即使一家公司或其協作者獲得監管批准的一個或多個產品獲得了有利的承保和報銷狀態,未來也可能實施不太有利的承保政策和報銷費率。
醫療改革
在美國和一些外國司法管轄區,關於醫療保健系統的多項立法和監管改革以及擬議的改革,已經並可能繼續存在,旨在擴大醫療保健的可獲得性,提高醫療保健的質量,並控制或降低醫療保健的成本。例如,2010年,美國國會頒佈了ACA,其中包括改變政府醫療保健計劃下產品的覆蓋範圍和支付方式。ACA包括對我們的潛在產品候選產品非常重要的條款:
•對生產或進口指定品牌處方藥和生物製品的任何實體徵收不可扣除的年度費用,並根據這些實體在某些政府醫療保健計劃中的市場份額在這些實體之間進行分攤;
•擴大了醫療補助計劃的資格標準,從而潛在地增加了製造商的醫療補助回扣責任;
•擴大了醫療補助藥品退税計劃下製造商的退税責任;
•擴大了符合340B藥品折扣計劃的實體類型;
•後續生物製品的許可框架;
•在CMS建立醫療保險和醫療補助創新中心,以測試創新的支付和服務交付模式,以降低醫療保險和醫療補助支出,可能包括處方藥支出;
•建立了聯邦醫療保險D部分承保缺口折扣計劃,要求製造商在承保間隔期內向符合條件的受益人提供適用品牌藥品談判價格70%的銷售點折扣,作為製造商的門診藥物在聯邦醫療保險D部分承保的條件;以及
•創建了一個以患者為中心的結果研究所,以監督、確定優先事項並進行臨牀有效性比較研究,併為此類研究提供資金。
自ACA頒佈以來,美國還提出並通過了其他立法修改。2011年8月,《2011年預算控制法》除其他外,包括將向醫療保險提供者支付的醫療保險總金額每財年削減2%。這些削減於2013年4月1日生效,由於隨後對法規進行了立法修訂,這些削減將一直有效到2031年。2013年1月,《2012年美國納税人救濟法》簽署成為法律,其中包括進一步減少了向包括醫院、成像中心和癌症治療中心在內的幾家醫療服務提供者支付的醫療保險,並將政府追回向醫療服務提供者多付款項的訴訟時效從三年延長到五年。2021年3月11日,總裁·拜登簽署了《2021年美國救援計劃法案》,從2024年1月1日起,取消了針對單一來源和創新多來源藥物的法定醫療補助藥品退税上限,目前該上限為藥品平均製造商價格的100%。由於2010年的法定現收現付法案、2021年美國救援計劃法案以及後續立法導致的預算赤字估計增加,在沒有進一步立法的情況下,從2025年開始,向提供者支付的醫療保險金額將進一步減少。 這些法律和法規可能導致醫療保險和其他醫療保健資金的進一步減少,並以其他方式影響我們可能獲得監管批准的任何候選產品的價格,或開出或使用任何此類候選產品的頻率。
此外,支付方法可能會受到醫療保健立法和監管舉措的影響。例如,CMS可能會開發新的支付和交付模式,如捆綁支付模式。此外,最近政府對製造商為其商業產品定價的方式進行了更嚴格的審查,導致國會進行了幾次調查,並提出並頒佈了州和聯邦立法,旨在提高產品定價的透明度,審查定價與製造商患者計劃之間的關係,以及改革政府對藥品的計劃補償方法。例如,在聯邦一級,總裁·拜登發佈了多項旨在降低處方藥成本的行政命令。2023年2月,衞生部還發布了一項提案,以迴應總裁·拜登2022年10月發佈的一項行政命令,其中包括一項擬議的處方藥定價模型,該模型將測試有針對性的醫療保險支付調整是否足以激勵製造商完成通過 加速審批路徑批准的藥品的確證試驗。儘管其中一些措施和其他擬議中的措施可能需要通過額外的立法獲得授權才能生效,拜登政府可能會撤銷或以其他方式改變這些措施,但拜登政府和國會都表示,他們將繼續尋求新的立法措施來控制藥品成本。
2022年8月,愛爾蘭共和軍簽署成為法律。IRA包括幾項可能對我們的業務產生不同程度影響的條款,包括從2025年起將Medicare Part D受益人的自付上限降低至2,000美元,對Medicare Part D中的某些藥物施加新的製造商財務責任,允許美國政府就某些沒有仿製藥或生物相似競爭的高成本藥物和生物製品的Medicare Part B和Part D價格上限進行談判,要求公司為某些價格增長快於通脹的藥品向Medicare支付回扣,以及推遲限制藥房福利經理可以收取的費用的回扣規則。此外,根據IRA,孤兒藥物不受聯邦醫療保險藥品價格談判計劃的限制,但前提是它們有一個孤兒疾病名稱,並且唯一批准的適應症是針對該疾病或條件的。如果一種產品獲得了多個罕見疾病的指定或有多個批准的適應症,它將沒有資格獲得孤兒藥物豁免。愛爾蘭共和軍的實施目前受到正在進行的訴訟,質疑愛爾蘭共和軍的醫療保險藥品價格談判計劃的合憲性。愛爾蘭共和軍對我們的業務和整個醫療保健行業的影響尚不清楚。
美國的個別州也越來越多地通過立法並實施旨在控制藥品定價的法規,包括價格或患者報銷限制、折扣、對某些產品准入和營銷成本披露的限制以及透明度措施,在某些情況下,旨在鼓勵從其他國家進口和批量購買。例如,如果獲得批准,我們可能會在美國面臨來自對藥品實施價格管制的外國療法的開發候選藥物和研究藥物的競爭。在美國,FDA發佈了一份最終指導文件,概述了製造商為FDA批准的藥物獲得額外的國家藥品代碼的途徑,該藥物最初打算在外國銷售,但已授權在該外國銷售。目前尚不清楚最終指引的市場影響。毒品重新進口的支持者可能會試圖通過立法,在某些情況下直接允許再次進口。如果立法或法規允許重新進口藥物,可能會降低我們可能開發的任何產品的價格,並對我們未來的收入和盈利前景產生不利影響。此外,法律規定的對第三方付款人支付金額的價格控制或其他限制可能會對我們的業務前景、財務狀況和運營結果產生不利影響。此外,地區醫療當局和個別醫院越來越多地使用招標程序來確定哪些藥品和供應商將包括在他們的處方藥和其他醫療保健計劃中。這可能會減少對我們候選產品的最終需求,或者給我們的產品定價帶來壓力。
2018年5月30日,《審判權法案》簽署成為法律。除其他事項外,該法律還為某些患者提供了一個聯邦框架,允許他們獲得某些已完成第一階段臨牀試驗並正在進行調查以獲得FDA批准的研究用新藥產品。在某些情況下,符合條件的患者可以在不參加臨牀試驗和根據FDA擴大准入計劃獲得FDA許可的情況下尋求治療。根據《試用權法案》,藥品製造商沒有義務將其藥品提供給符合條件的患者。
在美國以外,確保產品的覆蓋範圍和足夠的付款也面臨挑戰。在許多國家,處方藥的定價受到政府的管制。例如,在加拿大,專利藥品的價格管制立法目前正在經歷重大變化,這可能會對在加拿大銷售產品的公司的盈利能力產生重大影響。與政府當局的定價談判可能遠遠超出產品獲得監管批准的範圍,可能需要進行臨牀試驗,將產品的成本效益與其他可用的療法進行比較。進行這樣的臨牀試驗可能代價高昂,並導致商業化延遲。
人力資本資源
截至2023年12月31日,我們擁有145名全職員工,其中61名擁有醫學博士和/或博士學位。在這些全職僱員中,105名從事研究和開發活動,44名從事一般和行政活動。2024年1月9日,我們實施了一項計劃,以降低運營成本並更好地使我們的員工隊伍與業務需求保持一致,因此,我們的員工人數減少了約30%。我們目前有9名高級領導人,其中7人擔任執行幹事。我們的員工沒有工會代表,也沒有集體談判協議的覆蓋範圍。我們認為我們與員工的關係很好。
我們相信,我們未來的成功取決於我們繼續吸引和留住高技能員工的能力。我們為員工提供有競爭力的工資和獎金、股權機會、支持持續學習和成長的發展計劃,以及促進他們生活方方面面福祉的穩健就業方案,包括醫療保健、退休計劃和帶薪休假。作為我們提升和留住人才努力的一部分,我們還對持續發展進行投資。
我們致力於工作場所的多樣性、公平性和包容性,因為我們相信,擁有包容和公平的環境的多元化勞動力是企業成功的關鍵組成部分。我們的目標是培養一種文化,在這種文化中,每一名員工都能蓬勃發展,做出貢獻,做到最好。在這一重點下,我們制定了目標,在我們的整個勞動力隊伍中推動多樣性、公平和包容性倡議,從與經理和員工合作制定戰略,瞭解他們自己的偏見和學習包容性領導技能,到促進不同背景的領導人的進步,並通過支持我們的當地社區努力彌合服務不足社區的機會差距。在2022年成立的多樣性、公平和包容性理事會的幫助下,我們繼續專注於將我們的多樣性、公平和包容性倡議納入我們的日常工作。
企業信息
我們於2015年10月根據特拉華州的法律註冊成立。我們的主要執行辦公室位於馬薩諸塞州沃特敦,02472,阿森納路490號,Suite120,我們的電話號碼是(617231-0700)。我們有一家全資子公司,C4T證券公司,一家馬薩諸塞州的公司。
可用信息
我們的網站地址是www.c4Treatutics.com。我們網站上的信息並不包含在此作為參考。我們將在以電子方式將材料存檔或提供給美國證券交易委員會後,在合理可行的範圍內儘快在我們的網站上免費提供我們的10-K表格年度報告、10-Q表格季度報告、8-K表格的當前報告,包括證物,以及根據交易法第13(A)或15(D)節提交或提供的報告的任何修訂。美國證券交易委員會擁有一個互聯網網站http://www.sec.gov,,其中包含以電子方式向美國證券交易委員會提交的報告、委託書和信息聲明以及其他有關發行人的信息。
我們的公司治理準則、商業行為和道德準則以及(I)審計委員會、(Ii)組織、領導力和薪酬委員會以及(Iii)提名和公司治理委員會章程的副本張貼在我們的網站www.c4Treateutics.com的“公司治理”標題下,任何需要副本的人都可以通過撥打電話(617231-0700)聯繫我們或寫信給C4治療公司,C4治療公司,地址:490Arsal Way,Suite120,Wattown,Massachusetts 02472,收件人:公司祕書。
我們打算使用新聞稿、我們的公司網站(包括我們的投資者關係網站)以及我們的LinkedIn和Twitter賬户(如下所列)作為披露重大非公開信息的手段,並遵守FD法規下的披露義務。
 Www.linkedin.com/Company/c4-Treatutics-Inc.
Www.linkedin.com/Company/c4-Treatutics-Inc.. Https://twitter.com/C4Therapeutics
Https://twitter.com/C4Therapeutics
第1A項。風險因素。
投資我們的普通股涉及很高的風險。在評估我們的公司時,您應仔細考慮下列風險以及本Form 10-K年度報告中的所有其他信息,包括題為“管理層對財務狀況和經營結果的討論和分析”的部分,以及本Form 10-K年度報告末尾的財務報表和相關注釋。下面以及我們提交給美國證券交易委員會的其他文件中描述的風險和不確定性,可能並不是我們面臨的唯一風險和不確定性。下列任何事件或事態的發生,如果真的發生,都可能損害我們的業務、財務狀況、經營結果和增長前景。因此,我們普通股的市場價格可能會下降,您可能會失去對我們普通股的全部或部分投資。
與我們的財務狀況和額外資本需求有關的風險
我們是一家臨牀階段的生物製藥公司,自成立以來已經發生了重大虧損。我們預計至少在未來幾年內會出現虧損,可能永遠不會實現或保持盈利。
我們是一家臨牀階段的生物製藥公司,運營歷史有限。截至2023年、2023年和2022年12月31日的年度,我們的淨虧損分別為1.325億美元和1.282億美元。截至2023年12月31日,我們的累計赤字為5.284億美元。到目前為止,我們沒有從產品銷售中獲得任何收入,主要通過出售我們的股權來為我們的運營提供資金,包括公開發行我們的普通股、我們的合作收益和債務融資。我們仍處於候選產品開發的早期階段。因此,我們預計還需要幾年時間,如果有的話,我們才能有一個候選產品供監管部門批准和商業化。我們可能永遠不會在這些活動中取得成功,即使我們成功了,也可能永遠不會產生足以實現盈利的收入。為了實現並保持盈利,我們必須成功地開發、獲得營銷批准,並將產生大量收入的產品商業化。這將要求我們在一系列具有挑戰性的活動中取得成功,包括但不限於,成功完成我們候選產品的臨牀前研究和臨牀試驗,發現更多候選產品,與第三方建立進行臨牀試驗的安排,採購臨牀和商業規模的製造,獲得候選產品的營銷批准,製造、營銷和銷售我們可能獲得營銷批准的任何產品,確定合作伙伴以開發我們確定的候選產品或現有候選產品的其他用途,以及成功完成合作夥伴候選產品的開發。
我們預計,至少在未來幾年內,我們將繼續招致鉅額費用和不斷增加的運營虧損。我們預計我們的費用將大幅增加,如果我們:
•啟動、進行併成功完成我們的候選產品的首次人類臨牀試驗和後期臨牀試驗,並擴大我們專有研究和開發組合的範圍;
•利用我們的魚雷平臺識別更多候選產品,然後將其推向臨牀前和臨牀開發;
•擴展我們魚雷平臺的能力;
•為任何成功完成臨牀試驗的候選產品尋求市場批准;
•最終建立銷售、營銷和分銷基礎設施,並擴大外部製造能力,將我們預期獲得市場批准的任何產品商業化;
•推進、擴大、維護和保護我們的知識產權組合;以及
•隨着我們推出更多候選產品和/或繼續開發現有候選產品,管理人員配置需求以滿足不斷變化的業務需求。
此外,我們預計繼續產生與上市公司運營相關的額外成本,包括重大的法律、會計、保險、投資者關係和其他費用。
如果FDA、歐洲藥品管理局或其他監管機構要求我們在目前預期的基礎上進行試驗,或者我們在為我們的任何候選產品建立適當的製造安排或完成臨牀試驗或臨牀開發方面遇到任何延誤,我們的費用可能會超出我們的預期。
由於與藥品開發相關的眾多風險和不確定性,我們無法準確預測我們將產生的增加費用的時間或金額,或者我們何時能夠實現盈利。即使我們確實實現了盈利,我們也可能無法維持或提高季度或
按年計算。如果我們不能實現並保持盈利,將降低公司的價值,並可能削弱我們籌集資金、維持研發努力、擴大業務或繼續運營的能力。我們公司的價值或我們普通股的價值下降,也可能導致您的全部或部分投資損失。
如果我們開發的一個或多個候選產品被批准用於商業銷售,我們預計將產生與這些批准的候選產品商業化相關的鉅額成本。即使我們能夠從銷售任何經批准的產品中獲得收入,我們也可能無法盈利,可能需要獲得額外的資金才能繼續運營。
我們將需要大量的 為實現我們的業務目標和繼續我們的運營提供額外資金。如果我們無法在需要時籌集資金,我們可能會被要求推遲、限制、減少或終止我們的研究或產品開發計劃或未來的商業化努力。
我們預計與我們正在進行的活動相關的費用將大幅增加,特別是當我們準備和啟動、進行和完成我們正在進行的和計劃中的候選產品的人類第一階段1/2臨牀試驗、推進我們的魚雷平臺和繼續研發活動、擴大我們的專有研發組合以及啟動和繼續我們當前和未來的臨牀前計劃的臨牀試驗,並可能尋求市場批准的時候。此外,如果我們的任何候選產品獲得市場批准,我們預計將產生與產品製造、營銷、銷售和分銷相關的鉅額商業化費用。此外,我們預計繼續產生與上市公司運營相關的額外成本。因此,我們將需要獲得與我們的持續業務有關的大量額外資金。如果我們無法在需要時或在有吸引力的條件下籌集資金,我們可能會被要求推遲、限制、減少或終止我們的研究、產品開發計劃或任何未來的商業化努力,或者授予我們開發和營銷我們本來更願意自己開發和營銷的候選產品的權利。
截至2023年12月31日,我們擁有約2.817億美元的現金、現金等價物和有價證券。我們相信,這些資金連同我們在2024年1月初收到的資金(I)結算根據我們的“按市場”發售出售的股份,(Ii)根據先前披露的股票購買協議完成向Betta Pharma的一家關聯公司出售股份,以及(Iii)根據我們2023年12月的許可和合作協議從默克獲得的預付款,以及預期從重組中節省的成本,將足以支付我們計劃到2027年的運營費用。我們基於可能被證明是錯誤的假設做出了這一估計,我們可能會比目前預期的更早耗盡現有的資本資源。我們未來的資本需求將取決於許多因素,包括:
•我們正在進行的和計劃中的候選產品的人類第一階段1/2臨牀試驗的時間、進度、成本和結果,以及這些候選產品的任何未來臨牀開發;
•臨牀開發階段計劃和我們的其他候選產品和開發計劃的範圍、進度、成本和結果;
•我們追求的其他候選產品的數量和開發要求;
•我們持續合作的成功;
•對我們的候選產品進行監管審查的成本、時間和結果;
•未來商業化活動的成本和時間,包括產品製造、營銷、銷售和分銷,以及我們獲得或預期獲得上市批准的任何產品的未來商業化活動的時間和成本;
•從我們獲得上市許可的候選產品的商業銷售中獲得的收入(如果有)以及收到任何此類收入的時間;
•我們在臨牀前研究、臨牀試驗和/或供應鏈中遇到的任何延誤或中斷,包括由於任何全球衞生流行病造成的延誤;
•準備、提交和起訴專利申請、維護和執行我們的知識產權以及為任何與知識產權有關的索賠辯護的成本和時間;以及
•我們有能力在有利的條件下與其他生物技術或製藥公司建立合作安排,以開發我們的候選產品或將其商業化,或進入我們的魚雷平臺。
我們目前的現金、現金等價物和有價證券將不足以通過監管機構的批准為我們的任何候選產品提供資金。因此,我們將需要籌集大量額外資本,以完成
我們的候選產品的開發和商業化。確定潛在的候選產品以及進行臨牀前研究和臨牀試驗是一個耗時、昂貴和不確定的過程,需要數年時間才能完成。我們可能永遠不會生成獲得市場批准和實現產品銷售所需的必要數據或結果。此外,我們的候選產品如果獲得批准,可能不會獲得商業成功。我們的商業收入,如果有的話,將來自銷售我們預計在幾年內無法投入商業使用的產品,如果根本沒有的話。在可接受的條件下,我們可能無法獲得足夠的額外資金,或者根本沒有。此外,由於有利的市場條件或戰略考慮,我們可能會尋求額外的資本,即使我們認為我們有足夠的資金來執行當前或未來的運營計劃。
如果我們開發的一個或多個候選產品被批准用於商業銷售,我們預計將產生與這些批准的候選產品商業化相關的鉅額成本。即使我們能夠從銷售任何經批准的產品中獲得收入,我們也可能無法盈利,可能需要獲得額外的資金才能繼續運營。
我們仍處於開發生命週期的早期階段,這可能會使您很難評估我們業務到目前為止的成功程度和我們未來的生存能力。
我們於2015年底開始運營,到目前為止,我們的活動僅限於組織和配備我們的公司人員、業務規劃、籌集資金、進行發現和研究活動、提交專利申請、確定潛在的候選產品、開發和推進我們的魚雷平臺、進行臨牀前研究、與第三方就製造我們的候選產品的首批數量建立安排,以及準備和進行早期臨牀試驗。雖然我們有幾個正在進行的臨牀試驗,但我們所有其他候選產品仍處於發現階段。我們還沒有證明我們有能力成功完成任何臨牀試驗,獲得市場批准,直接或通過第三方製造商業規模的產品,或進行成功的產品商業化所需的銷售、營銷和分銷活動。因此,如果我們有更長的運營歷史,或者如果我們過去已經成功地完成了一些或所有類型的活動,您對我們未來成功或生存能力的任何預測都可能不那麼準確。
此外,作為一家生物製藥公司,我們可能會遇到不可預見的費用、困難、併發症、延誤等已知和未知的挑戰。我們需要在某個時候從一家專注於研發的公司轉型為一家能夠支持商業活動的公司,而我們可能無法成功實現這一轉型。
我們預計,由於各種因素,我們的財務狀況和經營業績將繼續在每個季度和每年大幅波動,其中許多因素是我們無法控制的。因此,您不應依賴任何季度或年度的業績作為未來經營業績的指標。
籌集額外資本可能會對我們的股東造成稀釋,限制我們的運營,或者要求我們放棄對我們的技術或候選產品的權利。
在我們能夠從產品銷售中獲得可觀收入之前,我們預計將通過股權發行、債務融資、合作、戰略聯盟以及營銷、分銷或許可安排來為我們的現金需求提供資金。儘管我們可能會根據我們的合作收到潛在的未來付款,但我們目前沒有任何承諾的外部資金來源。如果我們通過出售股權或可轉換債務證券籌集額外資本,您的所有權權益將被稀釋,我們未來可能發行的任何證券的條款可能包括清算或其他優惠,對您作為普通股股東的權利產生不利影響。債務融資和優先股融資可能涉及的協議包括限制或限制我們採取具體行動的能力的契約,如招致額外債務、進行收購、資本支出或宣佈股息。
如果我們通過與第三方的合作、戰略聯盟或營銷、分銷或許可安排來籌集更多資金,我們可能不得不放棄對我們的技術、未來收入來源、研究計劃或候選產品的寶貴權利,或者以可能對我們不利的條款授予許可證。
與發現和開發我們的候選產品相關的風險
我們基於魚雷平臺發現和開發候選產品的方法未經驗證,這使得我們很難預測開發任何產品的時間、成本和成功開發的可能性。
利用靶向蛋白質降解治療疾病是一種新的治療模式。我們未來的成功取決於這種新的治療方法的成功開發。使用定向蛋白質降解的小分子候選產品,如通過我們的魚雷平臺開發的產品,很少在人體上進行測試,通過我們的魚雷平臺開發的產品候選產品都沒有在美國、歐洲或任何其他司法管轄區獲得批准。開發這些類型的治療產品的可行性背後的數據都是初步的
而且是有限的。如果目標蛋白質降解物的其他開發商做出任何不利的學習,我們候選產品的開發可能會受到實質性影響。利用泛素蛋白酶體途徑降解蛋白質靶標的小分子的發現和發展在很大程度上受到以下因素的阻礙:泛素-蛋白酶體系統特定成分的功能、生物化學和結構生物學的複雜和有限的瞭解,包括參與靶蛋白泛素化的E3連接酶及其所需的輔助蛋白,以及促進蛋白質-蛋白質相互作用的工程化合物的挑戰。
構成我們在魚雷平臺下開發降解器候選產品的基礎的科學研究正在進行中,支持開發魚雷平臺衍生療法可行性的科學證據既是初步的,也是有限的。此外,某些癌症患者對已批准的抑制致病蛋白的藥物表現出固有的初級耐藥性,而其他患者對這些抑制劑產生了繼發性耐藥性。儘管我們相信我們的候選產品可能有能力降解對目前市場上銷售的致病酶抑制劑產生耐藥性的特定突變,但患者對我們候選產品的任何固有的初級或獲得性二級耐藥性都將阻止或削弱它們的臨牀益處,如果構成我們努力基礎的科學研究被證明是矛盾的,情況就會如此。
雖然我們有幾個正在進行的臨牀試驗,但目前,我們還沒有完成任何候選產品的臨牀試驗。因此,我們才剛剛開始評估我們的主要候選產品在患者身上的安全性,我們還沒有評估我們的任何其他早期候選產品在人體上的安全性。儘管我們的一些早期候選產品在動物研究中產生了明顯的結果,但它們對動物的影響的安全性數據集有限。此外,這些候選產品可能不會在人類身上表現出相同的化學和藥理特性,並可能以不可預見的、無效的或有害的方式與人類生物系統相互作用。因此,我們目前或未來的任何候選產品的治療可能會產生我們目前無法預測的不良影響。
此外,與其他更知名或經過廣泛研究的候選產品相比,像我們這樣的新產品候選產品的監管審批過程可能更昂貴,花費的時間也更長。儘管其他公司也在開發基於靶向蛋白質降解的療法,但目前還沒有監管機構批准任何這種性質的療法。因此,我們更難預測開發我們的候選產品的時間和成本,我們也無法預測我們魚雷平臺或任何類似或競爭對手的蛋白質降解平臺的應用是否會導致開發出通過上市批准的候選產品。我們未來遇到的任何與我們的魚雷平臺或我們的任何研究計劃相關的開發問題都可能導致重大延誤或意外成本,或者可能阻礙商業上可行的產品的開發。這些因素中的任何一個都可能阻止我們完成臨牀前研究或我們可能啟動的任何臨牀試驗,以及我們可能在及時或有利可圖的基礎上開發的任何候選產品的商業化。
我們是一家臨牀階段的生物技術公司,雖然我們已經開始對我們的某些候選產品進行臨牀試驗,但我們的大多數其他候選產品仍處於發現階段。如果我們無法推進到臨牀開發、開發、獲得監管部門對我們的候選產品的批准並將其商業化,或者在這方面遇到重大延誤,我們的業務可能會受到實質性的損害。
我們是一家臨牀階段的生物技術公司,雖然我們有幾項正在進行的臨牀試驗,但我們的大多數其他候選產品目前都處於發現階段。因此,他們失敗的風險很高。我們投入了幾乎所有的精力和財力來構建我們的魚雷平臺,並確定和開展我們目前的候選產品的臨牀前開發,包括我們的領先項目。我們從產品銷售中獲得收入的能力將在很大程度上取決於我們一種或多種候選產品的成功開發和最終商業化。我們預計這種情況在幾年內都不會發生。我們候選產品的成功將取決於以下幾個因素:
•我們的財政和其他資源是否充足;
•成功啟動臨牀試驗;
•成功的患者登記,並進行和完成臨牀試驗;
•從適用的監管機構獲得上市許可的收據和相關條款;
•為我們的候選產品獲得和維護專利或商業祕密保護和法規排他性;
•與第三方製造商就我們候選產品的臨牀和商業供應做出適當安排;
•開發候選產品,以實現其預期的和適合其預期適應症的治療特性;
•建立銷售、營銷和分銷能力,並在獲得批准後單獨或與他人合作啟動我們產品的商業銷售;
•如果患者、醫學界和第三方付款人批准了我們的產品,則接受我們的產品;
•獲得並維持第三方保險和適當的補償;
•建立我們產品的持續可接受的安全狀況,並在批准後保持該狀況;
•有效地與其他療法競爭;以及
•我們的第三方協作合作伙伴在及時開發我們的候選產品(S)的市場上實現上述任何一項的技能和成功。
如果我們不能及時或根本成功地實現這些因素中的一個或多個,我們可能會遇到重大延誤或無法成功地將我們的候選產品商業化,這可能會對我們的業務造成實質性的損害。此外,如果我們沒有獲得監管部門的批准,我們可能無法繼續運營。
作為一家公司,我們在完成臨牀前研究以實現IND的歸檔、提交IND或啟動、登記和進行臨牀試驗方面的經驗相對有限。
作為一家公司,我們在完成啟用IND的臨牀前研究方面的經驗來自於我們開始四種候選產品的臨牀開發工作。雖然這項工作代表着相當大的進步,但到目前為止,作為一家公司,我們在啟動、登記和進行臨牀試驗方面的經驗仍然相對有限。部分原因是,儘管我們在這一領域繼續取得進展,但我們不能確定我們計劃的臨牀試驗是否會按時開始、登記或完成。此外,即使適用的監管機構在首次提交IND時同意我們IND中規定的臨牀試驗的設計和實施,我們也不能保證這些監管機構將來不會改變他們的要求。這些考慮因素適用於上述IND,我們未來可能提交的額外IND,以及我們可能作為現有或新IND修正案提交的新臨牀試驗。
此外,大規模臨牀試驗將需要大量額外的財務和管理資源,並依賴第三方臨牀研究人員、合同研究組織或CRO和顧問。依賴第三方臨牀研究人員、CRO和顧問可能會導致我們遇到我們無法控制的延誤,對於目前處於臨牀開發中的每個候選產品,我們都聘請了一名CRO來領導我們的首個人類1/2階段臨牀試驗。在我們的臨牀前研究或臨牀試驗中依賴第三方可能會使我們面臨這樣的風險,即他們可能無法充分遵守研究或試驗方案或遵守我們計劃提交給監管機構的任何研究或試驗所需的良好實驗室實踐或良好臨牀實踐或GCP。我們也可能無法及時或根本無法確定和聯繫足夠的研究人員、CRO和顧問,我們也可能在臨牀試驗開始後確定並在過去確定CRO有必要改變。不能保證我們能夠在沒有現有的或潛在的未來CRO的情況下,在我們的其他候選產品需要的情況下,以我們及時或完全可以接受的條款進行談判並達成適當的合同安排。
我們的臨牀前研究和臨牀試驗可能無法充分證明我們的任何候選產品的安全性和有效性,這將阻礙或推遲開發、監管批准和商業化。此外,臨牀前研究的結果可能不能預測以後的研究或試驗的未來結果,臨牀試驗的初步成功可能不能表明這些試驗完成時或在後期臨牀試驗中獲得的結果。
在獲得監管機構批准我們的任何候選產品的商業銷售之前,我們必須通過漫長、複雜和昂貴的臨牀前研究和臨牀試驗證明,我們的候選產品在每個目標適應症中都是安全有效的。這項測試費用高昂,可能需要數年時間才能完成。此外,這些活動的結果本質上是不確定的。失敗可能在臨牀開發過程中的任何時候發生,由於我們的許多候選產品處於早期開發階段,從未在人體上進行過測試,因此失敗的風險很高。此外,由於目標蛋白質降解器是一個相對較新的候選產品類別,這一類別中的其他開發商在臨牀前或臨牀測試中看到的任何失敗或不良結果都可能對我們計劃的成功產生實質性影響。我們可能永遠不會成功地開發出適銷對路的產品。
我們候選產品的臨牀前研究和早期臨牀試驗的結果也可能不能預測後期臨牀試驗的結果。臨牀前和臨牀數據往往容易受到不同解釋和分析的影響,許多公司認為他們的候選產品在
儘管如此,臨牀前研究和臨牀試驗仍未能獲得其產品的上市批准。儘管候選產品可能在臨牀前研究和早期臨牀試驗中顯示出有希望的結果,但在隨後的臨牀試驗中可能被證明不是有效或安全的。我們正在進行的和計劃中的候選產品的人類第一階段1/2臨牀試驗的劑量遞增部分的結果可能無法預測這些候選產品或任何其他候選產品的進一步臨牀試驗的結果,也可能不足以使我們能夠進行到1/2階段臨牀試驗的第二階段。在動物身上進行試驗的條件與在人類身上進行試驗的條件不同,因此,動物研究的結果可能不能準確預測人類的體驗。
通過臨牀前研究和臨牀試驗的候選產品失敗通常會導致極高的流失率。儘管通過臨牀前研究和/或初步臨牀試驗取得了成功,但臨牀試驗後期階段的候選產品可能無法顯示出所需的安全性和有效性。同樣,早期較小規模的臨牀試驗可能不能預測大規模關鍵臨牀試驗的最終安全性或有效性。特別是,在我們計劃的這些試驗設計的早期臨牀試驗中,患者數量較少,可能會使這些試驗的結果對後來的臨牀試驗的結果不那麼具有預測性。生物製藥行業的許多公司在高級臨牀試驗中遭受了重大挫折,原因是缺乏療效、療效持久性不足或不可接受的安全性問題,儘管在早期的試驗中取得了良好的結果。大多數開始臨牀前研究和臨牀試驗的候選產品從未被批准為適銷對路的產品。在我們的臨牀開發中,任何這種性質的挫折都可能對我們的業務、財務狀況、運營結果和前景造成實質性損害。
此外,我們預計我們候選產品的第一批臨牀試驗將是開放標籤研究,患者和研究人員都知道患者是否正在接受研究產品候選,或者是現有的批准藥物或安慰劑。這是我們正在進行的第一次人類臨牀試驗,也將是我們目前預計進入臨牀開發的其他候選產品的第一次人類臨牀試驗。開放標籤臨牀試驗通常只測試研究產品,有時會以不同的劑量水平進行測試。開放標籤臨牀試驗受到各種限制,這些限制可能誇大任何治療效果,因為開放標籤臨牀試驗中的患者在接受治療時是知道的。此外,開放標籤臨牀試驗可能會受到“調查者偏見”的影響,即那些評估和審查臨牀試驗的生理結果的人知道哪些患者接受了治療,並可能在瞭解這一知識的情況下更有利地解釋治療組的信息。
我們可能或已經進行的任何臨牀前研究或臨牀試驗可能不會證明獲得監管部門批准將我們的候選產品推向市場所需的安全性和有效性。如果我們正在進行的或未來的臨牀前研究或臨牀試驗的結果對於我們候選產品的安全性和有效性沒有定論,如果目標降級的證據與臨牀療效無關,如果我們沒有達到具有統計和臨牀意義的臨牀終點,或者如果我們的候選產品存在安全問題,我們可能會阻止或推遲獲得這些候選產品的上市批准。在某些情況下,由於許多因素,同一候選產品的不同臨牀前研究和臨牀試驗的安全性或有效性結果可能存在顯著差異,包括方案中規定的試驗程序的變化、患者羣體的大小和類型的差異、臨牀試驗方案的變化和遵守以及臨牀試驗參與者的退學率。
雖然我們已經開始對我們的幾個候選產品進行臨牀試驗,其中一些仍在進行中,但我們還沒有開始對我們的任何其他候選產品進行臨牀試驗。與所有藥物的情況一樣,使用我們的候選產品可能會產生與靶上毒性、靶外毒性或其他藥物毒性機制相關的副作用,包括基於化學的毒性。我們的臨牀試驗結果可能會揭示這種性質的副作用的嚴重程度和普遍性,這是不可接受的。如果觀察到不可接受的毒性水平,或者如果我們的候選產品具有其他意想不到的特徵,我們可能需要放棄它們的開發,修改我們的開發計劃,以劑量水平和/或劑量時間表或其他方式,或者將開發限制在更狹窄的用途或子羣,其中從風險-收益的角度來看,不良副作用或其他特徵不那麼普遍、不那麼嚴重或更容易接受。例如,由於觀察到的安全信號,我們先前在CFT7455正在進行的1/2期臨牀試驗中修改了劑量計劃,因為我們繼續推進這項臨牀試驗。此外,如果我們觀察到不可接受的副作用水平,或者如果其他類似靶向蛋白質降解劑的開發商發現其候選藥物的副作用嚴重或普遍無法接受,我們的試驗可能會暫停或終止,FDA或類似的外國監管機構可以命令我們停止進一步開發或拒絕批准我們的任何或所有靶向適應症的候選產品。與藥物相關的副作用也可能影響患者招募或納入患者完成正在進行的試驗的能力,或導致潛在的產品責任索賠。許多化合物最初在治療癌症的早期測試中表現出了希望,但後來發現它們會產生副作用,阻止這種化合物的進一步發展。任何這些情況都可能嚴重損害我們的業務、財務狀況和前景。
根據我們臨牀試驗中公佈或公佈的臨時頂線和初步數據得出的結論和分析可能會隨着更多的患者數據的獲得而發生變化。此外,我們提供的所有臨時數據仍須遵守審計和核查程序,這些程序可能會導致最終數據發生重大變化。
有時,我們可能會公佈臨牀試驗的中期或頂級初步數據。我們可能進行的臨牀試驗的中期數據面臨這樣的風險,即隨着患者登記的繼續和更多患者數據的獲得,一個或多個臨牀結果可能會發生實質性變化。此外,初步或最重要的數據仍需接受審計和核實程序,這可能會導致最終數據與我們先前宣佈或公佈的初步數據存在重大差異。因此,在獲得最終數據之前,應謹慎看待中期和初步數據。初步或中期數據與最終數據之間的不利差異可能會嚴重損害我們的聲譽、業務、財務狀況、運營結果和前景。
藥物開發是一個漫長而昂貴的過程,結果不確定。我們可能會在完成或最終無法完成我們候選產品的開發和商業化過程中產生意外成本或遇到延遲。
雖然我們已經開始了幾種候選產品的臨牀試驗,其中一種我們選擇關閉,並預計通過我們的合作伙伴Betta Pharma開始CFT8919的臨牀試驗,但我們所有其他候選產品目前仍處於發現階段,所有候選產品失敗的風險仍然很高。我們無法預測我們的候選產品何時或是否會在人體上證明有效或安全,或者是否會獲得上市批准。在獲得監管部門批准銷售任何候選產品之前,我們必須進行廣泛的臨牀試驗,以證明我們的候選產品在人體上的安全性和有效性。
臨牀測試費用昂貴,難以設計和實施,可能需要數年時間才能註冊和完成,並且不確定時間和結果。一個或多個臨牀試驗的失敗可能發生在該過程的任何階段。在臨牀試驗期間或作為臨牀試驗的結果,我們可能會遇到許多不可預見的事件,這可能會推遲或阻止我們獲得上市批准或將我們的候選產品商業化的能力,包括:
•延遲或未能與監管機構就臨牀試驗設計達成共識,或無法產生可接受的臨牀前結果以進入人體臨牀試驗;
•我們的候選產品或進行臨牀試驗所需的其他材料的供應或質量可能不足或不充分,包括由於我們或與我們簽訂合同執行某些功能的第三方在測試、驗證、製造和向臨牀現場交付候選產品方面的延遲;
•延遲或未能與預期試驗地點或CRO就可接受的臨牀試驗合同或臨牀試驗方案達成協議;
•監管機構或機構審查委員會或IRBs未能授權我們或我們的研究人員在預期試驗地點開始臨牀試驗或進行臨牀試驗;
•設計臨牀試驗和為尚未得到很好研究的疾病選擇終點的困難,以及對疾病的自然歷史和病程知之甚少;
•選擇某些臨牀終點,這些終點可能需要較長時間的臨牀觀察或結果數據分析;
•我們候選產品的臨牀試驗所需的患者數量可能比我們預期的要多,這些臨牀試驗的登記人數可能比我們預期的要慢,參與者退出這些臨牀試驗的比率可能高於我們的預期,或者無法回來進行治療後的跟進,或者我們可能無法招募到合適的患者參與我們的臨牀試驗;
•我們的候選產品可能有不良副作用或其他意想不到的特徵,導致我們或我們的研究人員、監管機構或IRBs暫停或終止我們的臨牀試驗;
•由於各種原因,我們可能不得不暫停或終止我們候選產品的臨牀試驗,包括髮現參與者暴露在不可接受的健康風險中;
•與我們簽訂合同的第三方可能未能及時遵守監管要求或履行其對我們的合同義務,或者根本不遵守;
•監管機構或IRBs要求我們或我們的研究人員因各種原因暫停或終止臨牀試驗,包括不符合監管要求或不可接受的安全風險;
•我們候選產品的臨牀試驗可能會產生否定或不確定的結果,我們可能決定或監管機構可能要求我們進行額外的臨牀試驗,修改我們的發展計劃,如劑量水平和/或劑量時間表或其他方面,或放棄產品開發計劃;
•我們候選產品的臨牀試驗成本可能比我們預期的要高;
•人員短缺,包括但不限於我們進行臨牀試驗的機構缺乏訓練有素或經驗豐富的臨牀研究助理或醫務人員,或參與現場承包和激活的這些機構缺乏足夠的支持人員,可能會導致延誤或對我們及時有效地進行臨牀試驗造成其他挑戰;
•由於嚴重不良事件、對某類候選產品的擔憂或對我們的臨牀試驗操作、試驗地點或製造設施的檢查,監管機構強制實施臨牀暫停;
•與候選產品相關的嚴重不良事件的發生,被認為超過了其潛在的好處;以及
•任何全球衞生流行病造成的中斷,例如最近的新冠肺炎大流行,可能會增加我們遇到此類困難的可能性,或導致我們在啟動、登記、進行或完成計劃中的臨牀試驗方面出現其他延誤。
由於美國或其他司法管轄區不斷變化的監管政策,我們在臨牀開發項目中也可能遇到挑戰。例如,2021年,FDA的腫瘤學卓越中心啟動了優化項目,這是一項改革腫瘤學藥物開發中劑量選擇的倡議,這一倡議仍在實施中。如果FDA認為我們沒有充分確定為我們的候選產品選擇的一個或多個劑量是否最大限度地提高了療效以及安全性和耐受性,FDA可能會要求我們進行額外的臨牀試驗或生成額外的劑量相關信息,這可能會顯著推遲和/或增加我們臨牀開發計劃的費用。
如果我們被要求對我們目前考慮的候選產品進行額外的臨牀試驗或其他測試,如果我們無法成功登記或完成我們候選產品的臨牀試驗或其他測試,如果這些試驗或測試的結果不呈陽性或僅為輕微陽性,或者如果存在與我們的候選產品相關的安全問題,我們可能:
•延遲獲得我們的候選產品的市場批准(如果有的話);
•獲得批准的適應症或患者羣體並不像預期或期望的那樣廣泛;
•獲得包括重大使用或分銷限制或安全警告在內的標籤的批准;
•被要求進行額外的臨牀試驗以支持上市審批;
•讓監管部門撤回或暫停批准,或以風險評估和緩解戰略(REMS)的形式對候選產品的分銷施加限制;
•接受額外的上市後測試要求或產品管理方式的更改;或
•我們的產品在獲得上市許可後將被撤下市場。
如果我們在臨牀前研究或臨牀試驗或獲得上市批准方面遇到延誤,我們的產品開發成本也將增加。雖然我們已經開始了幾種候選產品的臨牀試驗,並預計通過我們的合作伙伴貝達製藥開始CFT 8919的臨牀試驗,但我們不知道我們的任何其他臨牀試驗是否會按計劃開始,是否需要重組或是否會按計劃完成。重大的臨牀試驗延遲也可能縮短我們可能擁有將我們的候選產品商業化的獨家權利的任何時間,或可能允許我們的競爭對手在我們之前將產品推向市場,並損害我們成功將我們的候選產品商業化的能力,這可能會損害我們的業務,經營業績,財務狀況和前景。
此外,癌症治療有時被表徵為一線、二線或三線。FDA通常批准新的腫瘤治療最初僅用於三線或以後使用,這意味着在兩種或更多種其他治療失敗後使用。當癌症被足夠早地檢測到時,一線治療,通常是全身性抗癌治療(例如,化療)、手術、放射療法或這些療法的組合有時足以治癒癌症或在不治癒的情況下延長生命。當既往治療無效時,對患者進行二線和三線治療。我們正在進行的和計劃中的早期臨牀試驗將針對接受過一種或多種既往治療的患者,我們預計,我們最初將尋求監管機構批准我們的主要候選產品作為二線或二線藥物。
三線治療隨後,對於那些被證明具有足夠益處的產品(如果有),我們將尋求批准作為一線治療,但我們開發的任何候選產品,即使獲批用於二線或三線治療,也可能不會獲批用於一線治療,並且在尋求和/或獲得任何一線治療批准之前,我們可能必須進行額外的臨牀試驗。
靶向蛋白質降解是一種新的模式,繼續吸引現有和新興的生物技術和製藥公司的極大興趣。因此,我們面臨着巨大的競爭,這可能導致其他人在我們之前或比我們更成功地發現、開發或商業化用於相同適應症和/或患者人羣的產品。
生物技術和製藥業的特點是技術發展迅速、競爭激烈和高度重視專利產品。我們面臨並將繼續面臨來自使用蛋白質降解、抗體治療、抑制性核酸、免疫治療、基因編輯或基因治療開發平臺的第三方的競爭,以及來自專注於更傳統的治療方式(如小分子抑制劑)的公司的競爭。我們所面臨的和將要面臨的競爭可能來自多個方面,包括大型製藥、特種製藥和生物技術公司、學術機構、政府機構以及公共和私營研究機構。
靶向蛋白降解是一種新興的治療方式,有可能提供改善患者結局的治療。因此,一些生物技術和製藥公司已經在努力開發基於降解的療法,進入這一領域的公司數量繼續增加。我們知道幾家生物技術公司正在開發基於嵌合小分子的候選產品,用於靶向蛋白質降解,包括Arvinas,Inc.,Astellas Pharma Inc.,BioTheryX,Inc.,Captor Therapeutics,Inc.卡爾根公司,Foghorn Therapeutics,Inc. Frontier Medicines Corporation,Glubio Therapeutics,Inc., Kymera Therapeutics,Inc. Monte Rosa Therapeutics,Inc. Nurix Therapeutics,Inc.,Orum Therapeutics,Inc.公司簡介Plexium,Inc.,Salarius Pharmaceuticals,Inc.,Seed Therapeutics,Inc. SK生命科學實驗室,Ltd.(SK Biopharmaceuticals的子公司)和Vividion Therapeutics,Inc.(Bayer AG的子公司)。此外,幾家大型製藥公司和學術機構已經披露了在該領域的投資和研究,包括安進公司、阿斯利康公司、百時美施貴寶公司(及其子公司Celgene Corporation)、葛蘭素史克公司、基因泰克公司、Novartis International AG的。除了來自其他蛋白質降解療法的競爭外,我們開發的任何產品也可能面臨其他類型療法的競爭,如小分子、抗體、T細胞或基因療法。
我們的許多現有或潛在競爭對手,無論是單獨或與合作伙伴合作,在研發、製造、臨牀前測試、進行臨牀試驗、獲得監管批准和營銷批准的產品方面,都比我們擁有更多的財務資源和專業知識。這些競爭對手還在招聘和留住合格的科學和管理人員、建立臨牀試驗場地和臨牀試驗患者登記以及獲取與我們的候選產品互補或必要的技術方面與我們展開競爭。製藥和生物技術行業的合併和收購可能會導致更多的資源集中在我們數量較少的競爭對手身上,這些競爭對手的規模可能很難與之競爭。規模較小或處於初創階段的公司也可能成為重要的競爭對手,特別是通過與大型和成熟公司的合作安排。如果我們的競爭對手開發和商業化比我們可能開發的任何候選產品更安全、更有效、副作用更少或更少、更方便或更便宜的產品,我們的商業機會可能會減少或消失。我們的競爭對手也可能比我們更快地獲得FDA或其他監管機構對其候選產品的批准,這可能會導致我們的競爭對手在我們能夠進入市場之前建立強大的市場地位。此外,在許多情況下,我們的競爭能力可能會受到保險公司或其他尋求鼓勵使用仿製藥的第三方付款人的影響。目前市場上有針對我們正在尋求的某些適應症的仿製藥,預計未來幾年將有更多的仿製藥上市。如果我們的候選產品獲得批准,我們預計它們的定價將顯著高於競爭對手的仿製藥。
我們使用淨營業虧損結轉和研發税收抵免結轉的能力可能有限。
截至2023年12月31日,我們有2.149億美元的聯邦淨運營虧損結轉,3.153億美元的美國州淨運營虧損結轉,其中部分在2043年之前的不同日期到期。根據現行法律,在2017年後開始的納税年度中產生的聯邦淨營業虧損(如果有的話)不會到期,可能會無限期結轉,但我們在2020年12月31日之後開始的納税年度中扣除此類聯邦淨營業虧損的能力將僅限於淨營業虧損結轉或公司調整後應納税所得額的80%中較小的一個(受制於1986年修訂的《國税法》第382節)。目前還不確定各州是否以及在多大程度上會遵守聯邦税法。此外,在州一級,可能會有一段時間暫停使用或以其他方式限制使用淨營業虧損,這可能會加速或永久增加州應繳税款。
截至2023年12月31日,我們還有美國聯邦和州研發税收抵免結轉分別為1260萬美元和520萬美元,這些抵免將在不同的日期到期,一直持續到2043年。這些税收抵免結轉可能到期,未使用,不能用來抵消我們未來的所得税債務。
此外,根據修訂後的1986年《國內税法》第382和383條,或該法規以及州法律的相應規定,如果一家公司在三年內經歷了股權所有權按價值超過50%的“所有權變更”,該公司使用變更前淨營業虧損結轉和其他變更前税收屬性來抵消變更後收入或税收的能力可能會受到限制。2021年,我們完成了從成立到2020年12月31日的所有權變更研究,得出的結論是,我們經歷了守則第382節定義的所有權變更。然而,有限結轉或到期未使用的結轉並無淨營業虧損。我們還沒有更新這項研究,以評估在2022年至2023年期間是否發生了所有權變更。我們可能經歷了其他尚未確定的所有權變化,這可能會導致我們的淨營業虧損和税收抵免結轉到期,然後才能使用,我們可能會經歷隨後的股票所有權變化,其中一些是我們無法控制的。因此,如果我們賺取淨應納税所得額,並確定所有權發生變化,而我們利用歷史淨營業虧損和税收抵免結轉的能力受到實質性限制,這將通過有效增加我們未來的納税義務來損害我們未來的經營業績。
如果在我們可能開發的任何候選產品的開發過程中發現了嚴重的不良事件、不良副作用或意外的特徵或結果,我們可能需要修改、放棄或限制這些候選產品的進一步臨牀開發。
雖然我們已經開始了幾個候選產品的臨牀試驗,並預計通過我們的合作伙伴Betta Pharma開始CFT8919的臨牀試驗,但我們所有其他候選產品目前仍處於發現階段,這意味着我們還沒有對我們的任何其他候選產品進行人體臨牀試驗。我們無法預測我們可能開發的任何候選產品何時或是否會被證明對人類是安全的。不能保證通過我們的魚雷平臺開發的任何候選產品不會引起不良副作用,這些副作用可能在臨牀前或臨牀開發過程中的任何時候出現。
通過我們的魚雷平臺開發的候選產品或任何蛋白質降解產品候選產品的潛在風險是,健康的蛋白質或不以降解為目標的蛋白質將被降解,或者目標蛋白質本身的降解可能會導致不良事件、不良副作用或意外的特徵或結果。在使用通過我們的魚雷平臺開發的候選產品進行治療後,還存在延遲不良事件的潛在風險。
如果我們開發的任何候選產品與嚴重的不良事件或不良副作用相關,或具有其他意外的特徵或結果,我們可能會選擇或需要放棄其開發,修改我們的開發計劃,以劑量水平和/或劑量時間表或其他方式,或將開發限制在不良事件、不良副作用或其他特徵不太普遍、不太嚴重或從風險效益角度來看更容易接受的特定用途或子羣。任何此類事件的發生都會對我們的業務、財務狀況、運營結果和前景產生不利影響。許多最初在治療癌症或其他疾病的早期測試中表現出希望的候選產品後來被發現會產生副作用,阻礙候選產品的進一步臨牀開發,或者限制它們在市場上的競爭力。例如,單一的BRAF抑制劑可以導致一種稱為角化皮膚瘤的繼發性惡性腫瘤,這是一種皮膚癌,是由於BRAF與抑制劑結合時矛盾地激活而引起的。
如果我們在臨牀試驗中招募患者時遇到延誤或困難,我們提交申請和獲得必要的上市批准的時間可能會推遲,或者我們可能會被完全阻止獲得上市批准。
如果我們不能按照FDA或美國以外的類似監管機構的要求,找到並招募足夠數量的合格患者參加這些試驗,我們可能無法為我們的候選產品啟動臨牀試驗。我們已經將三種候選產品CFT7455、CFT8634和CFT1946分別於2021年6月、2022年5月和2022年12月進行了首次人體臨牀試驗,CFT7455和CFT1946的臨牀試驗目前正在進行中,計劃於CFT8919進行。雖然我們相信我們將能夠為我們正在進行和計劃中的每一項臨牀試驗招募足夠數量的患者,但我們無法確定地預測招募患者參加試驗將會有多困難,其中一些試驗是罕見的適應症。我們識別和招募符合條件的患者參加我們的候選產品臨牀試驗的能力可能會被證明是有限的,或者我們招募這些試驗的速度可能比我們預期的要慢。此外,我們的一些競爭對手正在對候選產品進行臨牀試驗,這些候選產品與我們的候選產品具有相同的適應症,因此,有資格參加我們臨牀試驗的患者可能會選擇參加
對我們競爭對手的候選產品進行臨牀試驗。患者登記參加臨牀試驗還受到其他因素的影響,包括:
•正在調查的疾病的嚴重程度;
•有關試驗的資格標準;
•臨牀試驗中提供的候選產品的已知風險和益處;
•促進及時登記參加臨牀試驗的努力;
•醫生的病人轉診做法;
•臨牀試驗地點是否有適當和足夠的工作人員;
•由於臨牀試驗方案所需程序的範圍和侵入性給患者帶來的負擔,其中一些程序可能不方便和/或不舒服;
•在治療期間和治療後充分監測患者的能力;
•為潛在患者提供臨牀試驗地點的距離和可用性;以及
•任何全球衞生流行病的影響,例如最近的新冠肺炎大流行,可能會影響臨牀試驗的進行,包括減緩潛在登記或減少臨牀試驗的合格患者數量,或幹擾患者返回臨牀試驗現場進行所需的監測、程序或後續工作的能力。
我們無法招募足夠數量的患者參加臨牀試驗,或者我們無法及時這樣做,將導致重大延誤,並可能要求我們完全放棄一項或多項臨牀試驗。我們臨牀試驗的登記延遲還可能導致我們候選產品的開發成本增加,這將導致我們公司的價值下降,並限制我們獲得額外融資的能力。
我們可能會花費有限的資源來追求特定的候選產品或適應症,而無法利用可能更有利可圖或成功可能性更大的候選產品或適應症。
由於我們的財務和管理資源有限,我們將重點放在我們確定的特定適應症的研究項目和產品上。因此,我們可能會放棄或推遲尋找其他候選產品或後來被證明具有更大商業潛力的其他跡象的機會。我們的資源分配決策可能會導致我們無法利用可行的商業產品或有利可圖的市場機會。我們在當前和未來研發計劃以及特定適應症候選產品上的支出可能不會產生任何商業上可行的產品。如果我們沒有準確評估特定候選產品的商業潛力或目標市場,我們可能會通過合作、許可或其他版税安排放棄對該候選產品有價值的權利,而在這種情況下,保留該候選產品的獨家開發權和商業化權利對我們更有利。
我們或我們的合作伙伴可能會將我們的候選產品與其他藥物結合開發。如果FDA或美國以外的類似監管機構不批准這些其他藥物,或撤銷他們對這些其他藥物的批准,或者如果我們選擇結合我們的候選產品進行評估的藥物出現安全性、有效性、製造或供應問題,我們可能無法獲得我們的候選產品的批准或將其推向市場。
根據我們許多候選產品的研究設計,一旦從我們的首個人類1/2階段臨牀試驗的劑量遞增部分確定了推薦劑量,我們通常計劃將該臨牀試驗的一部分與一種或多種其他藥物結合進行。我們沒有為任何目前批准的藥物開發或獲得市場批准,也沒有製造或銷售任何我們可能與我們的候選產品結合研究的藥物。如果FDA或美國以外的類似監管機構撤銷對我們打算與我們的候選產品組合提供的一個或多個藥物的批准,我們將無法將我們的候選產品與那些被撤銷的藥物組合在一起銷售。
如果這些藥物中的任何一種出現安全性或有效性問題,我們可能會遇到重大的監管延遲,FDA或美國以外的類似監管機構可能會要求我們重新設計或終止某些臨牀試驗。如果我們使用的藥物被替換為我們為候選產品選擇的適應症的護理標準,FDA或美國以外的類似監管機構可能會要求我們進行額外的臨牀試驗。此外,如果製造或其他問題導致我們決定與我們的候選產品結合使用的藥物供應短缺,我們可能無法在目前的時間表上完成我們候選產品的臨牀開發,甚至根本無法完成。
即使我們的候選產品獲得上市批准或商業化,以便與其他現有藥物聯合使用,我們仍將面臨FDA或美國以外的類似監管機構可能撤銷與我們的候選產品聯合使用的藥物的批准,或者這些現有藥物可能出現安全性、有效性、製造或供應問題的風險。
聯合療法通常用於癌症的治療,如果我們選擇開發我們的任何其他候選產品,與其他藥物或癌症以外的適應症聯合使用,我們將面臨類似的風險。這可能導致我們自己的產品被從市場上撤下,或者在商業上不那麼成功。
我們可能不會成功地發現或發現其他潛在的候選產品。
雖然我們目前的臨牀階段計劃專注於腫瘤學目標,但我們戰略的一個關鍵要素是應用我們的魚雷平臺開發針對廣泛目標和新治療領域的候選產品,如神經退行性變、老年病和傳染病。我們正在進行的治療發現活動可能無法成功地確定對治療癌症或其他疾病有用的候選產品。我們的研究計劃最初可能在確定潛在候選產品方面表現出希望,但由於多種原因未能產生用於臨牀開發的候選產品,包括:
•在進一步研究後,潛在的候選產品可能被證明具有有害的副作用或其他特徵,表明它們不太可能是將獲得上市批准或獲得市場接受的藥物;
•潛在的候選產品在治療其目標疾病方面可能無效;或
•潛在候選產品的目標適應症的市場規模可能會隨着時間的推移而縮小,這是由於護理標準的提高,以至於沒有必要進一步發展。
確定新產品候選產品的研究項目需要大量的技術、財力和人力資源。我們可能會選擇將我們的努力和資源集中在一個最終被證明是不成功的潛在產品候選上。如果我們無法確定適合臨牀前和臨牀開發的候選產品,我們將無法在未來時期從產品銷售中獲得收入,這可能會對我們的財務狀況造成重大損害,並對我們的股票價格造成不利影響。
如果我們不能在我們宣佈和預期的時間框架內實現預期的發展目標,我們產品的商業化可能會被推遲,因此,我們的股票價格可能會下跌。
有時,我們可能會估計各種科學、臨牀、法規和其他產品開發目標的預期完成時間,我們有時將其稱為里程碑。這些里程碑可能包括開始或完成臨牀前研究和臨牀試驗,以及提交監管文件。我們可能會不時地公開宣佈其中一些里程碑的預期時間。這些里程碑中的每一個現在和將來都是基於無數的假設。與我們的估計相比,這些里程碑的實際時間可能會有很大差異,在某些情況下,原因超出了我們的控制。如果我們沒有達到公開宣佈的這些里程碑,或者根本沒有達到,我們的收入可能會低於預期,我們產品的商業化可能會推遲或永遠不會實現,因此,我們的股票價格可能會下跌。
與依賴第三方有關的風險
我們預計將依靠第三方進行我們未來的臨牀試驗,這些第三方的表現可能不令人滿意,包括無法在截止日期前完成此類試驗。
我們目前依靠CRO進行臨牀試驗,因為我們目前沒有計劃對我們的任何候選產品進行獨立的臨牀試驗。此外,我們必須與第三方研究網站簽訂合同,進行我們的臨牀試驗。就像我們依賴貝塔醫藥高效地在大中國開發CFT8919一樣,我們未來也可能同樣依賴其他第三方合作伙伴在特定的時間期限內在不同的地區開發我們的一個或多個產品。我們與這些CRO、站點和其他第三方的協議可能會因各種原因而終止,包括第三方未能履行。如果我們需要達成替代安排,或者如果我們需要為正在進行的臨牀試驗更換CRO,就像我們過去所做的那樣,我們可能會在臨牀開發活動中遇到延誤。
我們對研究和開發活動對CRO的依賴將減少我們對這些活動的控制,但不會免除我們如何執行這些活動的責任。例如,我們將繼續負責確保我們的每一項臨牀試驗都按照適用的IND中的一般研究計劃和方案進行。此外,FDA要求遵守進行、記錄和報告臨牀試驗結果的標準,通常稱為GCP,以確保數據和報告的結果是可信和準確的,並保護試驗參與者的權利、安全和機密性。GCP符合性不僅擴展到以下項目的發起人
不僅限於臨牀研究,也包括參與臨牀研究的CRO和站點在內的第三方。同樣,世界各地的其他監管機構也要求遵守類似的標準,這些標準也適用於臨牀試驗贊助商和其他第三方,如CRO和臨牀試驗地點。
此外,這些CRO或站點可能與其他實體有關係,其中一些可能是我們的同行或競爭對手。如果與我們合作的CRO或站點不能按照法規要求或我們規定的規程成功履行其合同職責、在預期期限內完成或進行我們的臨牀試驗,我們將無法獲得或可能延遲獲得我們候選產品的營銷批准,並且我們將無法或可能延誤我們將候選產品成功商業化的努力。
藥品生產是一項複雜的工作,由於各種原因容易出現產品延誤或損失的情況。我們與第三方簽訂合同,生產我們的候選產品,用於臨牀前測試和臨牀試驗,並預計將繼續這樣做以實現商業化。這種對第三方的依賴增加了我們將沒有足夠數量的候選產品或產品的風險,或者我們將無法以可接受的成本或質量或在合適的時間獲得我們想要或要求的數量的風險,這可能會推遲、阻止或損害我們的開發或商業化努力。
我們不擁有或經營,目前也沒有建立任何製造設施的計劃。我們依賴並預計將繼續依賴CMO提供藥物物質和成品。這種對第三方的依賴可能會增加我們沒有足夠數量的候選產品或產品的風險,或者我們無法以可接受的成本或質量獲得我們想要或要求的數量的風險,這可能會推遲、阻止或損害我們的開發或商業化努力,包括需要進行審批前檢查或對製造地點進行檢查,而FDA因任何原因無法在審查期內完成這些要求的檢查。
我們可能無法與CMO達成協議,也無法以可接受的條件這樣做。即使我們能夠與CMO達成協議,依賴第三方製造商也會帶來額外的風險,包括:
•在監管、合規、質量保證和製造成功方面依賴第三方;
•第三方CMO可能違反制造協議;
•CMO可能因監管問題、財務破產、不遵守適用法律或其他原因而暫時或永久停止提供我們所需的服務或完全關閉業務的風險;
•可能盜用我們的專有信息,包括我們的商業祕密和專有技術;以及
•第三方可能在成本高昂或對我們造成不便的時候終止或不續簽協議,或者CMO無法在我們需要的時候為我們提供製造時段。
關於我們的候選產品,我們只有有限的技術轉讓協議,這些現有的安排不適用於商業供應。我們在採購訂單的基礎上獲得了許多關鍵材料。因此,我們沒有關於我們的候選產品和其他材料的長期承諾安排。如果我們預期收到或獲得任何候選產品的上市批准,我們將需要與一個或多個第三方建立或已經建立商業生產協議。
第三方製造商可能無法遵守當前良好的製造實踐或cGMP、法規或美國以外的類似監管要求。我們的一些分子是高度有效的,在沒有額外安全數據的情況下,它們會獲得高職業暴露波段,即OEB。這些指定的OeB規定了作為我們候選產品製造的一部分必須採取的遏制和其他預防措施,並且對於具有高OEB標號的分子,用於限制有資格製造我們的分子的CMO的數量。我們或我們的CMO未能遵守適用的法規,包括我們的CMO使用我們的高效材料和相關安全協議的能力,可能會導致對我們實施制裁,包括臨牀封存、罰款、禁令、民事處罰、延遲、暫停或撤回批准、吊銷許可證、扣押或召回候選產品或產品、運營限制和刑事起訴,其中任何一項都可能對我們的產品供應產生重大不利影響。
我們的候選產品和我們可能開發的任何產品都可能與其他候選產品和產品競爭製造設施。因此,我們可能無法優先使用這些設施,甚至根本無法使用。在cGMP法規下運營的製造商數量有限,能夠為我們生產的製造商數量有限,特別是在某些情況下,考慮到我們化合物的效力或OEB。
我們現有或未來製造商的任何業績失敗或業績延遲都可能推遲臨牀開發或營銷授權。例如,在過去,與我們合作的一位CMO在我們的CFT7455候選產品的生產運行中的製造步驟中出現了機械問題。雖然這個問題
如果我們最終沒有推遲提交CFT7455 IND的時間,將來我們可能會遇到製造問題,這將對我們的候選產品的開發產生實質性影響,並且這種事件的發生在很大程度上不是我們所能控制的。我們目前沒有為藥物或藥物製品的多餘供應或第二來源作出安排。如果我們現有的CMO不能按約定履行職責,我們可能需要更換他們。雖然我們已經確定了幾個可以生產部分或全部候選產品的潛在替代供應商,但在我們選擇和鑑定替代製造商時,更換供應商可能會導致顯著的額外成本和我們的運營延誤,我們可能會受到我們可以選擇的供應商的限制,特別是對於具有較高OEB標識的化合物,或者我們可能無法與替代製造商就可接受的條款達成協議。
我們目前和預期未來對他人生產我們的候選產品或產品的依賴可能會對我們未來的利潤率和我們將任何及時和有競爭力地獲得營銷批准的產品商業化的能力產生不利影響。
此外,我們目前依賴單一來源供應商為我們的臨牀前和臨牀試驗供應的供應鏈的一部分。如果我們當前或未來的供應商,無論是原材料、藥物物質還是藥品,都不能為我們的臨牀前研究和臨牀試驗提供足夠的材料,我們可能會在尋找和鑑定新的供應商或製造商時遇到開發工作的延誤。
我們所依賴的第三方製造商可能會將他們自己的專有工藝合併到我們的候選產品製造工藝中。我們對第三方專有製造過程的控制和監督有限。如果第三方製造商修改其流程,這些修改可能會對我們的製造產生負面影響,包括需要額外生產或更換製造商的產品丟失或故障,這兩種情況都可能顯著增加我們候選產品的成本並顯著推遲生產。
隨着我們的候選產品通過臨牀前研究和臨牀試驗獲得批准和商業化,我們預計產品開發和製造工藝的各個方面將不斷髮展,以優化工藝和結果。其中一些產品和製造工藝變更可能涉及使用第三方專有技術,這可能導致我們需要從第三方獲得許可。此外,這些類型的變更可能需要我們對監管申請進行修訂,這可能會進一步延遲我們任何候選產品使用改良製造工藝的時間範圍。
此外,當我們將候選產品推進到後期臨牀試驗階段並計劃候選產品的潛在商業化時,我們可能會確定有必要或適當引入其他製劑和/或原料藥供應商,這可能導致候選產品的生產工藝發生變化,並可能要求我們向監管機構提供額外信息。如果我們為候選產品引入額外的CMO,我們還可能需要證明分析可比性和/或進行額外的橋接研究或試驗,所有這些都需要額外的時間和費用。
我們與第三方有現有合作關係,據此我們從事若干候選產品的研究、開發及商業化。如果這些合作中的任何一個不成功,我們可能無法利用這些候選產品的市場潛力。此外,這些合作可能會影響我們的知識產權。
截至2023年12月31日,我們有四個正在進行的合作涉及我們的研究計劃:
•我們於2015年12月與羅氏簽訂的合作協議,我們於2018年12月對其進行了修訂和重申,並在此後定期進行進一步修訂,並就兩個目標進行了持續的合作活動;
•我們於二零一七年三月與Calico訂立的合作協議,該協議於二零二一年九月就一個項目延長,其研究年期於二零二三年三月屆滿;
•我們於2018年12月與Biogen訂立的合作協議,該協議於2020年2月修訂,根據Biogen協議的預期,有關提名目標的若干研究活動將於2023年6月研究期限結束後持續一段時間;及
•我們於2023年12月與默克達成合作協議,針對一個初始目標開發和商業化降解物-抗體偶聯物(DAC),默克可以選擇在規定的時間內添加多達三個額外的目標。
根據這些合作協議,我們通常負責根據合作伙伴選擇的目標,利用我們的TORPEDO平臺開發候選藥物。此外,這些協議規定,我們的合作伙伴
擁有為其選定和保留的目標開發降級器的專有權。因此,我們不允許單獨或與另一個合作伙伴一起追求潛在利益的目標,而該目標受到這些限制的約束。
此外,如果我們的合作沒有導致產品的成功開發和商業化,或者如果我們的合作者之一終止與我們的協議或選擇不繼續合作中的一個項目,我們可能不會收到任何未來的研究資金或里程碑或特許權使用費根據該合作或關於該終止的計劃。如果發生這種情況,我們可能會決定放棄該計劃或自行推進該計劃,這將要求我們在未來的基礎上為該計劃投入額外的資源。此外,如果我們的合作者之一一般終止與我們的協議,他們被允許在90至270天的通知下方便地這樣做,或者針對特定目標或與我們在指定時間內仍未解決的重大違反協議有關,我們可能會發現更難吸引新的合作者,我們的發展計劃可能會被推遲,或者我們在商業和金融界的形象可能會受到不利影響。所有與產品開發、市場營銷相關的風險 本報告所述的批准和商業化適用於我們合作者的活動。
我們的合作者也可能無法正確獲取、維護、執行或捍衞我們的許可程序所產生的知識產權或專有權利,或者可能以可能危及我們的專有信息或使我們的專有信息無效或使我們面臨潛在訴訟的方式使用我們的專有信息。例如,Roche、Biogen、Calico和Merck在適用的合作安排下對特定許可項目擁有強制執行和捍衞某些知識產權的優先權,儘管我們可能有權在我們的合作者不執行和捍衞這些知識產權的情況下承擔這些知識產權的強制執行和捍衞,但我們這樣做的能力可能會受到他們的行為的影響。此外,如果任何許可程序後來恢復給我們,我們保護與該程序相關的任何知識產權或其他專有權利的能力將受到我們的合作者在程序恢復之前所做的知識產權申報或採取的其他步驟的影響。此外,我們的合作者可能擁有或共同擁有我們與他們合作所產生的產品的知識產權,在適用的情況下,我們將沒有將合作知識產權商業化的專有權。
我們可能會在未來形成或尋求合作或戰略聯盟,或達成額外的許可協議,我們可能無法實現這些合作,聯盟或許可協議的好處。
2023年5月,我們與貝達藥業簽訂了貝達藥業許可協議,根據該協議,我們將與貝達藥業合作 CFT 8919在大中華區的開發和商業化,同時保留在世界其他地區開發和商業化CFT 8919的權利。在未來,W我們可能會與第三方建立或尋求戰略聯盟、創建合資企業或其他合作關係,或與第三方達成額外的許可協議,我們認為這些協議將補充或增強我們在候選產品和我們可能開發的任何未來候選產品方面的開發和商業化努力。在我們可能達成的任何其他合作安排中,我們可能的合作者包括大中型製藥公司和生物技術公司。然而,我們有可能無法訂立此性質的合作協議,或任何潛在新合作安排的條款可能並不有利。
例如,我們可能尋求達成合作安排,以推進我們的候選產品CFT 7455在MM或其他適應症中的應用,或者我們可能形成或尋求形成合作安排,以使我們能夠在指定地理區域開發和商業化候選產品,就像我們在CFT 8919以及我們與貝達製藥的合作中所做的那樣。此外,正如我們在與默克的合作協議中所做的那樣,我們可能會尋求籤訂合作協議,使其他公司能夠訪問並利用我們的TORPEDO平臺開發針對我們合作伙伴選擇的目標的藥物。任何這些關係都可能要求我們產生非經常性費用和其他費用,增加我們的近期和長期支出,發行證券稀釋我們現有的股東或擾亂我們的管理和業務。
此外,我們在尋找合適的戰略合作伙伴方面面臨着激烈的競爭,這類交易的談判過程既耗時又複雜,而且成本高昂。此外,我們為我們的候選產品建立戰略合作伙伴關係或其他替代安排的努力可能不會成功,因為它們可能被認為處於協作努力的開發階段太早,第三方可能不認為我們的候選產品具有展示安全性和有效性並獲得市場批准的必要潛力。此外,我們現有的合作伙伴可能決定收購或與其他開發目標蛋白降解劑或針對我們候選產品所針對的目標或適應症的公司合作,這可能會對我們的業務前景、財務狀況和運營結果產生不利影響。
因此,如果我們簽訂額外的協作協議和戰略合作伙伴關係或許可我們的候選產品,如果我們無法成功地將這些交易與我們現有的運營和公司文化相結合,我們可能無法實現這些交易的好處。
與我們的候選產品商業化相關的風險
即使我們的任何候選產品獲得市場批准,也可能無法獲得醫生、患者、第三方付款人和醫學界其他人為商業成功所必需的市場接受度。
如果我們的任何候選產品獲得市場批准,它可能仍無法獲得醫生、患者、第三方付款人和醫學界其他人的足夠市場接受度。例如,目前的癌症治療方法,如化療和放射治療,在醫學界已經很成熟,醫生可能會繼續依賴這些治療方法。如果我們的候選產品沒有達到足夠的接受度,我們可能無法從產品銷售中獲得可觀的收入,我們也可能無法盈利。如果我們的候選產品獲準用於商業銷售,市場對其的接受程度將取決於許多因素,包括:
•與替代療法相比的療效和潛在優勢;
•任何副作用的發生率和嚴重程度,特別是與替代療法相比;
•我們以有競爭力的價格提供我們的產品出售的能力,以及政府當局要求我們談判我們產品的定價以及這些強制性談判的時間的能力;
•與替代療法相比,給藥的方便性和簡易性;
•目標患者羣體嘗試新療法的意願,以及治療這些患者的醫生開出這些療法的意願;
•市場營銷、銷售和分銷支持的實力;
•第三方保險的覆蓋範圍和適當的報銷情況;
•與其他產品批准有關的任何上市批准的時間;
•患者權益倡導團體的支持;以及
•任何對我們的產品與其他藥物一起使用的限制。
作為一家公司,我們目前沒有營銷和銷售組織,也沒有營銷產品的經驗。如果我們無法建立營銷和銷售能力,或無法與第三方達成協議來營銷和銷售我們的候選產品,如果獲得批准,我們可能無法產生產品收入。
作為一家公司,我們目前沒有銷售、營銷或分銷能力,也沒有營銷產品的經驗。我們打算髮展一支內部營銷組織和銷售隊伍,這將需要大量的資本支出、管理資源和時間。我們將不得不與其他製藥和生物技術公司競爭,以招聘、聘用、培訓和留住營銷和銷售人員。
如果我們無法或決定不建立內部銷售、營銷和分銷能力,如果獲得批准,我們將尋求與第三方銷售、營銷和分銷合作伙伴就我們產品的銷售和營銷達成安排。然而,我們不能保證我們能夠以有利的條件建立或維持這些類型的安排,或者如果我們能夠這樣做,或者如果我們能夠這樣做的話,我們不能保證這些第三方安排將提供有效的銷售力量或營銷和分銷能力。我們獲得的任何收入都將取決於這些第三方的努力,而這可能不會成功。我們可能對這些第三方的營銷和銷售活動幾乎沒有控制權,我們從產品銷售中獲得的收入可能會低於我們自己將候選產品商業化的收入。我們在尋找第三方來幫助我們銷售和營銷我們的候選產品時也面臨着競爭。
不能保證我們能夠發展內部銷售和分銷能力,或與第三方合作伙伴建立或保持關係,以便在美國或海外將任何產品商業化。
我們的候選產品的市場機會可能相對較小,因為我們預計它們最初只會被批准用於那些沒有資格接受其他批准的治療或先前治療失敗的患者。此外,我們對目標患者人羣流行率的估計可能不準確。
我們正在開發針對癌症的候選產品,但癌症療法有時被描述為一線、二線、三線或後續線,而FDA通常最初只批准針對特定用途的新療法。當癌症被發現得足夠早時,一線治療有時足以治癒癌症或延長生命,而不需要治癒。當一線治療--通常是化療、抗體藥物、腫瘤靶向小分子、免疫治療、激素治療、放射治療、手術、其他靶向治療或這些治療的組合--被證明不成功時,可能會進行二線治療。二線療法通常包括更多
化療、放射治療、抗體藥物、腫瘤靶向小分子或它們的組合。三線治療可以包括化療、抗體藥物和小分子腫瘤靶向治療、更具侵入性的手術形式和新技術。我們預計最初將尋求批准我們的候選產品,在大多數情況下作為二線或三線療法,用於復發或難治性癌症患者。隨後,對於那些被證明足夠安全和有益的候選產品(如果有的話),我們預計將尋求批准作為二線療法和潛在的一線療法,但不能保證我們的任何候選產品,即使被批准為第二、第三或隨後的治療路線,隨後也會被批准用於較早的路線治療。此外,在我們的產品在早期系列治療中獲得任何批准之前,我們可能必須進行額外的臨牀試驗。
我們對患有我們目標癌症的人數、可能對他們的腫瘤進行基因測序的人數以及能夠接受特定治療路線並有可能從我們的候選產品治療中受益的這些癌症患者的子集進行的預測,都是基於我們的合理信念和估計。這些估計來自各種來源,包括科學文獻、診所調查、患者基金會或市場研究,可能被證明是不正確的或過時的。此外,新的治療方法可能會改變我們正在瞄準的癌症的估計發病率或流行率。因此,即使我們的候選產品被批准用於二線或三線治療,有資格使用我們的候選產品進行治療的患者數量可能會比預期的要少得多。此外,我們還沒有進行市場研究,以確定如果針對每一種腫瘤類型有不同的批准療法,治療醫生將如何預期開出一種被批准用於多種腫瘤類型的產品。
即使我們或在CFT8919的情況下,Betta Pharma獲得了我們任何候選產品的營銷批准,我們的產品也可能會受到不利的定價法規、第三方報銷做法或醫療改革舉措的影響,其中任何一項都會影響我們的業務。
管理新藥上市審批、定價、覆蓋範圍和報銷的規定因國家而異。當前和未來的立法可能會大幅改變審批要求,這可能會涉及額外的成本,並導致獲得批准的延遲。一些國家要求藥品的銷售價格獲得批准後才能上市。在許多國家,定價審查期從批准營銷或產品許可後開始。為了在一些國家獲得報銷或定價批准,我們可能需要進行一項臨牀試驗,將我們候選產品的成本效益與其他可用的療法進行比較。在一些外國市場,處方藥定價即使在獲得初步批准後,仍然受到政府的持續控制。因此,我們或在CFT8919的情況下,Betta Pharma可能會獲得某一候選產品在特定國家/地區的營銷批准,但隨後會受到價格法規的約束,這些法規會推遲產品的商業發佈,可能會有很長的時間,這將對我們在該國家/地區銷售產品所產生的收入(如果有的話)產生負面影響。因此,不利的定價限制可能會阻礙我們收回在一個或多個候選產品上的投資的能力,即使我們的候選產品獲得了市場批准。
我們的,就CFT8919而言,Betta Pharma能否成功地將任何候選產品商業化,還將在一定程度上取決於政府醫療保健計劃、私人健康保險公司和其他組織為這些產品和相關治療提供的保險範圍和足夠的補償。政府當局和第三方付款人,如私人健康保險公司和健康維護組織,決定他們將支付哪些藥物並建立報銷水平。
美國醫療保健行業和其他地方的一個主要趨勢是成本控制。聯邦醫療保險藥品價格談判計劃由CMS作為2022年通脹降低法案(通常稱為IRA)的一部分進行管理,如果我們的產品被選中進行談判,該計劃可能適用於我們的產品,如果這些產品獲得批准,這可能會大幅減少我們可以從我們的產品中獲得的收入。政府當局和第三方付款人試圖通過限制特定藥物的覆蓋範圍和報銷金額來控制成本。政府當局和第三方付款人越來越多地要求製藥公司在標價的基礎上向他們提供預定的折扣,並對醫療產品的定價提出挑戰。我們商業化的任何產品都可能無法獲得保險和報銷,即使有,報銷水平也可能不令人滿意。報銷可能會影響我們獲得市場批准的任何候選產品的需求或價格。為我們的產品獲得並維持承保範圍和適當的報銷可能很困難。我們可能被要求進行昂貴的藥物經濟學研究,以證明覆蓋範圍和報銷或相對於其他療法的報銷水平。此外,根據IRA的要求,如果獲得批准,我們可能需要與聯邦醫療保險就我們候選產品的定價進行談判,這些談判價格將在產品獲得批准九年後生效。如果沒有覆蓋範圍和足夠的報銷,或者報銷僅限於有限的級別,我們可能無法成功地將我們獲得營銷批准的任何候選產品商業化。
在獲得新批准的藥物的保險和補償方面可能會有很大的延誤。此外,覆蓋範圍可能比FDA或美國以外類似監管機構批准該藥物的目的更有限。此外,有資格獲得保險和報銷並不意味着在所有情況下都將支付藥物的費用,或支付我們的成本,包括研究、開發、知識產權、製造、銷售和分銷費用。如果適用,新藥的臨時報銷水平也可能不足以支付我們的成本,並且可能不會成為永久性的。報銷率可能會因藥物的使用和臨牀環境的不同而有所不同,可能基於已經為低成本藥物設定的報銷水平,也可能納入其他服務的現有付款中。藥品的淨價可能會通過政府醫療保健計劃或私人付款人要求的強制性折扣或回扣,以及未來任何限制從可能以低於美國價格銷售的國家進口藥品的法律的放鬆來降低。
在美國,第三方付款人對產品的承保和報銷沒有統一的政策。因此,我們產品的承保範圍和報銷範圍因付款人而異。確定付款人是否將為產品提供保險的過程可以與設置付款人將為產品支付的償還率的過程分開。一個付款人決定為一種產品提供保險並不能保證其他付款人也會為該產品提供保險和補償。第三方付款人也可以將承保範圍限制在批准的清單或配方表上的特定產品,其中可能不包括FDA批准的特定適應症的所有產品。第三方付款人在設置自己的報銷政策時通常依賴於聯邦醫療保險覆蓋政策和支付限制。我們無法迅速從政府資助和私人支付人那裏為我們開發的任何經批准的產品獲得保險和足夠的報銷率,這可能會對我們的經營業績、我們籌集產品商業化所需資金的能力以及我們的整體財務狀況產生不利影響。
針對我們的產品責任訴訟可能會導致我們承擔重大責任,並限制我們可能開發的任何產品的商業化。
我們面臨着與在人體臨牀試驗中測試我們的候選產品相關的固有產品責任風險,如果我們以商業方式銷售我們可能開發的任何產品,我們將面臨更大的風險。如果我們不能成功地針對我們的候選產品或產品造成傷害的索賠為自己辯護,我們將招致重大責任。無論是非曲直或最終結果,賠償責任可能導致:
•對任何候選產品或我們可能開發的產品的需求減少;
•終止臨牀試驗;
•撤銷已批准藥品的上市批准、產品召回、限制批准或“黑箱”警告或禁忌症;
•臨牀試驗參與者的退出;
•相關訴訟的重大辯護費用和/或增加的產品責任保險費用;
•對試驗參與者或患者給予鉅額金錢獎勵;
•收入損失;
•損害我們的聲譽和媒體的嚴重負面關注;
•減少管理層資源以推行業務策略;以及
•無法將我們可能開發的任何產品商業化。
我們目前維持產品責任保險,以支持我們的臨牀開發活動。我們可能需要購買額外的產品責任保險,因為我們擴大了我們的臨牀試驗,如果我們開始商業化我們的候選產品。保險範圍越來越貴。我們可能無法以合理的費用或足夠的金額維持保險範圍,以應付可能出現的任何責任。
與我們的知識產權有關的風險
如果我們無法為我們的技術、候選產品和產品獲得並維持專利保護,或者如果獲得的專利保護範圍不夠廣泛或可執行,我們的競爭對手可能會開發和商業化與我們類似或相同的技術、候選產品和產品,我們成功商業化我們的技術、候選產品的能力,產品可能受損,或我們可能無法在市場上有效競爭。
我們依靠專利、商業祕密保護和保密協議的組合來保護我們的知識產權,並防止他人利用我們的平臺技術、我們的管道候選藥物、我們可能開發的任何未來候選藥物及其使用或製造。
我們的商業成功在一定程度上取決於我們在美國和其他國家獲得並維護專利和其他專利保護的能力,這些專利和保護涉及我們的專有技術、候選產品和產品。我們尋求通過在美國和海外提交與我們的新技術和候選產品相關的專利申請來保護我們的專有地位。任何向第三方披露或盜用我們的機密專有信息都可能使競爭對手迅速複製或超過我們的技術成就,從而侵蝕我們在市場上的競爭地位。此外,我們擁有、共同擁有或許可的專利申請可能無法在美國或其他國家頒發專利。
專利訴訟過程既昂貴又耗時,我們可能無法以合理的成本或及時提交、起訴和維護所有必要或可取的專利申請。我們也有可能在獲得專利保護為時已晚之前,無法確定我們的研發成果中可申請專利的方面。此外,在某些情況下,我們無權控制專利申請的準備、提交和起訴,或維護專利和專利申請,包括我們從第三方許可的技術或我們許可給我們的合作者的技術。因此,不得以符合我們業務最佳利益的方式起訴和強制執行這些專利和申請。
生物製藥行業的專利地位通常高度不確定,涉及複雜的法律和事實問題,一直是許多訴訟的主題。此外,外國的法律可能不會像美國的法律那樣保護我們的權利。科學文獻中發表的發現往往落後於實際發現,美國和其他司法管轄區的專利申請通常在申請18個月後才發表,有時甚至根本不發表。因此,我們不能確切地知道,我們是否是第一個在我們擁有、共同擁有或許可的專利或未決專利申請中提出要求的發明,或者我們是第一個為這些發明申請專利保護的發明人。因此,我們或我們合作者的專利權的頒發、範圍、有效性、可執行性和商業價值都是高度不確定的。我們的未決和未來的專利申請可能不會導致頒發專利,從而保護我們的技術、候選產品或產品的全部或部分,或有效地阻止其他公司將競爭技術、候選產品和產品商業化。美國和其他國家的專利法或專利解釋或其他法律的變化可能會降低我們的專利和潛在應用的價值,縮小我們的專利保護範圍,或者導致我們被要求向第三方支付使用費。此外,如果我們的專利和專利申請提供的保護的廣度或強度受到威脅,可能會阻止公司與我們合作,授權、開發或商業化當前或未來的候選產品。
我們擁有、共同擁有和許可的專利產業主要由專利申請組成,其中許多申請處於起訴的早期階段。即使我們擁有、共同擁有和許可的專利申請以專利的形式發佈,它們的發佈形式也不會為我們提供任何有意義的保護,阻止競爭對手與我們競爭,或以其他方式為我們提供任何競爭優勢。我們的競爭對手可能能夠通過以非侵權方式開發類似或替代技術、候選產品或產品來規避我們擁有、共同擁有或許可的專利。
專利的頒發對於其發明性、範圍、有效性或可執行性並不是決定性的,我們擁有的、共同擁有的和許可的專利或我們的合作者獲得的專利可能會在美國和國外的法院或專利局受到挑戰。這些挑戰可能導致失去獨佔性或經營自由,或專利主張全部或部分縮小、無效或無法執行,這可能限制我們阻止他人使用或商業化類似或相同的技術、候選產品和產品的能力,或限制我們的技術、候選產品和產品的專利保護期限。考慮到新產品候選產品的開發、測試和監管審查所需的時間,保護我們候選藥物產品的專利可能會在商業化之前或之後不久到期。因此,我們擁有、共同擁有和許可的專利組合,或我們合作者的專利組合,可能不會為我們提供足夠的權利,以排除其他公司將與我們相似或相同的產品商業化。
專利法或專利法的改變可能會降低我們專利的整體價值或增加對我們專利的第三方挑戰,從而削弱我們保護我們候選產品的能力。
專利改革立法可能會增加圍繞我們專利申請的起訴以及我們已頒發專利的執行或辯護的不確定性和成本。2011年9月16日,《萊希-史密斯美國發明法》(Leahy-Smith America Invents Act)簽署成為法律,並對美國專利法進行了一系列重大修改。這些變化包括影響專利申請起訴方式的條款,也可能影響專利訴訟。美國專利商標局(USPTO)制定了新的法規和程序來管理Leahy-Smith法案的管理,許多與Leahy-Smith法案相關的專利法實質性變化,包括第一發明人申請條款,於2013年3月16日生效。Leahy-Smith法案及其實施可能會增加我們專利申請的起訴以及我們已頒發專利的執行或辯護的不確定性和成本,所有這些都可能對我們的業務和財務狀況產生不利影響。第一個提交的
Leahy-Smith法案的條款要求我們在從發明到提交專利申請期間迅速採取行動,因為始終存在第三方可能提交專利申請的風險,這可能會阻礙我們的專利申請。然而,即使我們打算迅速採取行動,情況可能會阻止我們迅速提交或起訴我們發明的專利申請。Leahy-Smith法案還擴大了符合現有技術的披露範圍,這可能會影響我們獲得發明專利保護的能力。
《萊希-史密斯法案》首次建立了新的程序,第三方可以根據這些程序對美國已頒發的專利提出質疑,包括授權後審查, 各方間審查和推導程序,所有這些都是在美國專利商標局進行的對抗性程序。自《萊希-史密斯法》生效以來,一些第三方一直在利用這類訴訟尋求並實現取消其競爭對手的部分或全部已頒發專利的權利要求。根據Leahy-Smith法案,對於優先權日期為2013年3月16日或之後的專利(我們所有專利申請的情況都是如此),第三方可以在專利發佈時開始的九個月窗口期內的任何時間提交授予後審查申請。此外,對於優先權日為2013年3月16日或之後的專利,第三方可以提交申請, 各方間在提交授予後審查申請的九個月期限屆滿後進行審查。批地後覆核程序可以任何質疑理由提出,而 各方間根據適用法律,美國專利商標局對這些類型的對抗性訴訟的審查標準是在不推定美國專利有效的情況下進行的,如果第三方試圖通過在美國聯邦法院提起訴訟來使專利無效,則適用這一標準。美國專利商標局於2018年11月11日發佈了一項最終規則,宣佈它現在將使用美國聯邦法院目前使用的相同權利要求解釋-這是所用詞語的普通和普通含義-來解釋這些USPTO訴訟中的專利權利要求。由於這種監管環境,如果我們的任何專利在這種性質的USPTO程序中受到第三方的質疑,則無法保證我們將成功捍衞受質疑的專利,這可能導致我們部分或全部失去受質疑專利的權利。
由於這項立法,我們或我們的合作者的專利權的頒發、範圍、有效性、可重複性和商業價值存在高度不確定性,這可能對我們的業務、財務狀況、經營業績和前景產生不利影響。
我們可能會捲入訴訟,以保護或執行我們的專利,我們的許可人的專利或其他知識產權,這可能是昂貴的,耗時的和不成功的。
競爭對手可能侵犯我們的授權專利、我們的許可方或合作方的專利或我們的其他知識產權。為了應對侵權或未經授權的使用,我們可能需要提交侵權索賠,這可能是昂貴的,耗時的和不可預測的。我們對侵權者提出的任何索賠都可能引起這些當事人對我們提出反訴,指控我們侵犯了他們的專利。此外,在專利侵權訴訟中,法院可能會裁定我們或我們的許可人或合作者的專利全部或部分無效或不可執行,嚴格限制專利的權利要求,或以我們的專利不涵蓋相關技術為由拒絕阻止另一方使用相關技術。任何訴訟程序的不利結果可能會使我們的一項或多項專利面臨被宣佈或實際上被宣佈無效、無法執行或被狹義解釋的風險。即使我們成功地維護了我們的專利權,法院也可能不會判決足以補償我們損失的補救措施。此外,我們可能沒有足夠的財務或其他資源來尋求充分執行我們的專利,以對抗被認為的侵權者,這可能對我們產品的盈利能力產生重大不利影響。
我們可能需要從第三方獲得知識產權許可,並且這種性質的許可可能無法獲得或可能無法以商業合理的條款獲得。
第三方可能持有對我們的產品或我們的合作者的產品的開發或製造非常重要或必要的知識產權,包括專利權。因此,我們可能有必要使用第三方的專利或專有技術將我們自己或我們的合作者的技術或產品商業化,在這種情況下,我們或我們的合作者將被要求從該第三方獲得許可。該知識產權的許可可能無法獲得,或可能無法以商業合理的條款獲得,這可能會對我們的業務和財務狀況產生不利影響。
第三方知識產權的許可和獲取是一種競爭性做法。那些可能比我們更成熟或擁有更多資源的公司也可能正在採取戰略,授權或獲得我們認為必要或有吸引力的第三方知識產權,以便將我們的候選產品商業化。更成熟的公司可能比我們有競爭優勢,因為他們更大的規模和現金資源,或者更強的臨牀開發和商業化能力。我們可能無法成功完成此類談判,並最終獲得圍繞我們可能尋求收購的其他候選產品的知識產權。
第三方可能會提起法律訴訟,指控我們侵犯了他們的知識產權,其結果將是不確定的,並可能對我們的業務成功產生不利影響。
我們的商業成功取決於我們和我們的合作者開發、製造、營銷和銷售我們的候選產品並使用我們的專有技術而不侵犯第三方專有權利的能力。在生物製藥行業有相當多的知識產權訴訟,以及挑戰專利的行政訴訟,包括複審、授權後審查、各方間美國專利商標局的審查、派生程序或幹預程序,以及外國司法管轄區的異議和其他類似程序。
我們可能會加入或威脅未來與我們的候選產品和技術有關的知識產權訴訟或訴訟,包括派生、複審、授權後審查、國際 零件審查或幹預美國專利商標局的訴訟程序。第三方可能會根據現有專利或未來可能授予的專利向我們提出侵權索賠。隨着生物製藥行業的擴張和專利的頒發,我們的候選產品可能會引發侵犯他人專利權的索賠的風險增加。可能存在我們目前不知道的第三方專利,即與我們候選藥物的使用或製造相關的材料、配方、製造方法或治療方法的權利要求。由於專利申請可能需要數年時間才能發佈,因此可能存在當前正在處理的專利申請,這些申請可能會導致我們的候選產品可能會侵犯已頒發的專利。此外,第三方可能會在未來獲得專利,並聲稱我們的候選產品或使用我們的技術侵犯了這些專利。
如果我們被有管轄權的法院認定侵犯了第三方的知識產權,我們可能被要求從適用的第三方知識產權持有者那裏獲得許可證,以繼續開發和營銷我們的候選產品、產品和技術。然而,我們可能無法以商業上合理的條款或根本無法獲得任何所需的許可證。即使我們能夠獲得許可,它也可能是非排他性的,從而使我們的競爭對手能夠訪問向我們許可的相同技術。我們可能會被迫停止將侵權技術或產品商業化,包括通過法院命令。此外,如果我們被發現故意侵犯專利,我們可能被判承擔金錢損害賠償責任,包括三倍損害賠償和律師費。侵權的發現可能會阻止我們將候選產品商業化,或者迫使我們停止一些業務運營,這可能會對我們的業務造成實質性損害。聲稱我們盜用了第三方的機密信息或商業祕密,可能會對我們的業務產生類似的負面影響。
許多其他公司以及大學和其他組織在與我們的產品相同的領域申請並獲得專利,這些領域是目標蛋白質降解劑或我們的平臺技術,這些專利申請可能會在未來對我們或我們的合作者提出指控,這可能會對我們的業務成功產生不利影響,如果成功,可能會導致昂貴的訴訟,可能會影響我們產品的盈利能力和/或禁止銷售或使用我們的產品。
我們的MonoDAC和BiDAC候選產品是降解特定蛋白質的小分子藥物。許多公司和機構在該一般領域中具有專利申請和頒發的專利,例如Accutar Biotechnology,Inc.,安進公司,Amphista Therapeutics,Ltd. Araxes Pharma,LLC,Arvinas,Inc.,Astellas Pharma Inc.,AstraZeneca PLC,Aurigen Discovery Technologies,Ltd.,Bayer AG(及其子公司Vividion Therapeutics,Inc.),Beigene Co. Ltd. BioTheryX,Inc.,勃林格殷格翰國際有限公司、百時美施貴寶公司(及其子公司Celgene Corporation)、Captor Therapeutics Inc.卡爾根公司,Dana-Farber癌症研究所及其蛋白質降解中心,辯證治療公司,Foghorn Therapeutics,Inc. Frontier Medicines Corporation、GlaxoSmithKline PLC、Genentech,Inc. Glubio Therapeutics,Inc.,Hinova Pharmaceuticals,Inc.,楊森生物技術公司,Kymera Therapeutics,Inc. Monte Rosa Therapeutics,Inc. Novartis International AG、Nurix Therapeutics,Inc.、Orum Therapy,Inc.,大冢製藥公司,Phoremost有限公司,Plexium,Inc.,Prelude Therapeutics,Inc. Inc.,Roche AG,Salarius Pharmaceuticals Inc.,Salarius Pharmaceuticals,Inc.,Seed Therapeutics,Inc.四川海思科製藥有限公司有限公司,SK生命科學實驗室(SK Biopharmaceuticals Co. Ltd的子公司),密歇根大學醫學院,頂點製藥公司,等人如果任何這些公司或機構或其他未列入此列表的公司或機構聲稱其專利之一被我們可能開發或使用或製造的任何候選產品或產品侵犯,我們或我們的合作者可能會捲入昂貴的訴訟,這可能會對我們的業務前景,財務狀況和運營結果產生不利影響。需要大量的時間,並導致我們的管理團隊成員和廣大員工分心。此外,如果此類性質的訴訟成功,可能會對我們產品的盈利能力產生重大不利影響或禁止其銷售。我們可能不知道目前或將來可能懸而未決的專利申請,這些專利申請可能會影響我們的業務或產品。專利申請通常在提交後6至18個月內公佈,並且在已經待決的申請中提出的新權利要求有時在一段時間內對公眾(包括我們)不可見。此外,即使在專利申請公開之後,我們可能還沒有看到該專利申請,因此可能不知道已提交和公佈的專利申請的權利要求或範圍。因此,我們無法提供任何
保證在我們的技術的一般領域內執業的第三方不會提出或尚未提出涵蓋我們的一個或多個候選產品或產品或其使用或製造方法的專利權利要求。如果發生這種情況,我們或我們的合作者(如適用)可能必須採取措施嘗試使適用的專利或申請無效,在這種性質的情況下,我們或我們的合作者可以選擇不這樣做,或者我們的嘗試可能不會成功。例如,2023年5月1日,我們向USPTO專利審判和上訴委員會(PTAB)提交了一份請願書,尋求對美國專利號11,414,416的所有權利要求進行授權後審查(稱為'416專利),並且在2023年10月23日,我們向PTAB提交了一份請願書,尋求對美國專利號11,560,381(稱為'381專利)的所有權利要求進行授權後審查。'416專利和'381專利都涉及用於治療BRD 9相關病症的化合物。由於專利所有人於2023年8月21日提交了對所有'416專利權利要求的法定免責聲明,從而放棄了他們對' 416專利的所有合法權利,因此'416專利的挑戰得到了成功解決。 由於專利所有人於2024年1月17日提交了對所有'381專利權利要求的法定免責聲明,從而放棄了他們對' 381專利的所有權利,因此'381專利的挑戰得到了成功解決。如果我們決定需要第三方專利或專利申請的許可,我們可能會發現許可可能無法以合理的條款獲得,或者根本無法獲得,這可能會阻止我們或我們的合作者銷售產品或使用我們的專有技術。
我們的候選產品如果獲得批准,將受到1984年《藥品價格競爭和專利期限恢復法》的約束,該法案在美國也被稱為《哈奇-韋克斯曼法案》,這可能會增加與試圖銷售我們產品的仿製藥公司提起訴訟的風險,並可能導致我們失去專利保護。
由於我們的臨牀候選藥物是藥物分子,將由FDA藥物評估和研究中心進行審查,商業化後,它們將在美國受到Hatch-Waxman法案的專利訴訟程序的約束,該法案允許仿製藥公司提交簡化新藥申請,或ANDA,向FDA申請批准,僅使用生物等效性數據銷售我們藥物的仿製藥。根據Hatch-Waxman法案,我們將在FDA的“具有治療等效性評價的批准藥品”綱要(有時稱為橙皮書)中列出涵蓋我們的藥品或其各自使用方法的專利。
有關於專利的詳細規則和要求,這些專利可能會提交給FDA,以便在橙書中列出。我們可能無法獲得涵蓋我們的候選產品的專利,這些候選產品包含一項或多項滿足橙皮書中上市要求的權利要求。即使我們提交了一項專利在Orange Book中上市,FDA可能會拒絕列出該專利,或者仿製藥製造商、美國聯邦貿易委員會或其他實體可能會對上市提出質疑。如果我們的候選產品之一獲得批准,並且涵蓋該候選產品的專利沒有列在橙皮書中,對於任何未列出的專利,仿製藥製造商不必提前通知我們向FDA提交的任何ANDA申請,即可獲得銷售該候選產品的仿製版本的許可。
目前,在美國,FDA可能會為新的化學實體或NCE授予五年的數據排他性,NCE是指在任何其他新藥申請或NDA中未包含FDA批准的有效部分的藥物。我們預計我們的所有產品都將符合NCE的資格;然而,FDA在審查該藥物的營銷申請之前,不會對NCE狀態進行評估。仿製藥公司可以在我們指定為NCE的任何藥物產品獲得批准四年後向FDA提交ANDA。仿製藥公司提交ANDA被認為是侵犯專利的技術行為。仿製藥公司可以證明,它將等到我們的上市專利自然到期之日才銷售我們產品的仿製藥版本,或者可以證明我們的一項或多項上市專利無效、不可強制執行或未被侵犯。如果仿製藥製造商選擇後者,我們將有45天的時間對仿製藥公司提起專利侵權訴訟。如果我們這樣做,很可能會對我們Orange Book列出的一項或多項專利發起挑戰,因為仿製藥製造商認為我們列出的專利無效、不可強制執行或未被侵犯。如果提起訴訟,FDA不得在我們收到仿製藥製造商的認證通知後30個月內,或在主審法院根據當事人的某些行為或法院的最終裁決裁定我們聲稱的專利主張無效、不可執行或未被侵犯的較短或更長時間之前,禁止FDA發佈對仿製藥的ANDA最終批准。如果我們沒有在Orange Book中正確列出我們的相關專利,或者如果我們沒有及時根據ANDA對仿製藥公司的認證提起訴訟,或者如果我們沒有在由此產生的專利訴訟中獲勝,我們可能會失去從基於專利保護的專利市場受益的能力,我們可能會發現醫生將轉向開和分配我們藥物產品的仿製藥。此外,即使我們在橙皮書中正確列出我們的相關專利,及時提起訴訟,並在訴訟中獲勝,仿製藥訴訟也可能會給我們帶來巨大的成本,無論是在律師費和員工時間方面,還是在很長一段時間內都是如此。此外,不止一家仿製藥公司試圖同時銷售一種創新者的藥物是很常見的,因此,我們可能會同時面臨來自仿製藥製造商的多起訴訟的成本和分心。我們還可能確定有必要以允許仿製藥公司進入我們的
在我們的專利到期之前或以其他方式對我們的專利的強度、有效性或可執行性產生不利影響的市場。
許多製藥公司一直是美國聯邦貿易委員會或另一個國家的相應機構嚴格審查的對象,審查的依據是它們進行或解決與製藥產品相關的專利訴訟的方式。事實上,某些審查導致了違反反壟斷的指控,有時會導致罰款或權利喪失。我們不能確定我們不會也受到這種性質的審查,或者這種性質的審查的結果對我們有利,或者任何這種性質的審查不會導致罰款或處罰。
過去幾年,美國聯邦貿易委員會(Federal Trade Commission,簡稱FTC)向聯邦法院提起了多起訴訟,以反競爭為由挑戰創新者公司和仿製藥公司之間達成的ANDA訴訟和解協議。例如,聯邦貿易委員會採取了一種激進的立場,即任何有價值的東西都是一種支付,無論支付與否。根據他們的做法,如果創新者作為專利和解的一部分,同意在授予第一家仿製藥公司挑戰涵蓋創新者藥物的Orange Book上市專利的180天期限內,不推出或推遲推出授權仿製藥,或者在沒有付款的情況下談判推遲進入,聯邦貿易委員會可能會認為這是不可接受的反向支付。製藥行業的公司辯稱,這些類型的協議是藥物創新者為應對風險而做出的理性商業決策,因此,如果和解條款在專利的排他性潛力範圍內,這些和解協議應該免受反壟斷攻擊。2013年,美國最高法院以五票贊成、三票反對的裁決FTC訴Actavis,Inc.駁回了製藥業和聯邦貿易委員會關於所謂反向支付的論點。相反,最高法院認為,涉及以延遲進入換取對價的“反向支付”和解是否應受到反競爭分析取決於五個方面的考慮:(A)對競爭產生真正不利影響的可能性;(B)支付的理由;(C)專利權人造成反競爭損害的能力;(D)支付的金額是否是專利弱點的可行替代物;以及(E)對大額不合理付款的反壟斷責任並不妨礙訴訟當事人解決其訴訟,例如,允許仿製藥在品牌藥物的專利到期之前進入市場,而專利權人不向仿製藥製造商支付費用。此外,反向支付是否合理取決於其大小、相對於專利權人預期的未來訴訟費用的規模,以及與它可能代表支付的其他服務的獨立性(如阿特維斯),以及缺乏任何其他令人信服的理由。相反,最高法院認為,反向支付和解可能違反反壟斷法,並受到標準的反壟斷理由規則分析的約束,有責任證明協議在聯邦貿易委員會上是非法的。在作出這一裁決時,最高法院將這一理由分析的結構安排留給了下級法院。
如果我們面臨藥品專利訴訟,包括與仿製藥公司的Hatch-Waxman訴訟,我們可能會面臨FTC的這種性質的挑戰,這種挑戰可能會影響我們如何或是否解決案件,即使我們強烈反對FTC的立場,我們也可能面臨鉅額費用或罰款。我們根據《哈奇-瓦克斯曼法案》與仿製藥公司達成的任何訴訟和解,也可能受到第三方付款人的挑戰,如保險公司、直接購買者或其他認為自己受到和解不利影響的人。這類後續訴訟可能是集體訴訟,費用可能很高,可能會持續多年。如果我們面臨這種性質的訴訟,我們可能無法成功地擊敗這些索賠,因此,我們可能受到鉅額付款義務的約束,我們可能無法全部或部分滿足這些義務。
我們可能無法根據美國1984年的《藥品價格競爭和專利期恢復法》獲得專利期延長,因此,我們的候選產品如果獲得批准,可能在足夠長的時間內得不到專利保護。
在美國,1984年的《藥品價格競爭和專利期限恢復法》允許每種產品的一項專利在正常到期後延長最多五年,如果涉及治療方法專利,則僅限於批准的適應症。專利期延長的長度通常計算為臨牀試驗期的一半加上FDA審查NDA期間的整個時間,減去我們在這些時間段內的任何延遲時間。專利期的延長也有限制,從藥品批准之日起不超過14年。因此,如果我們選擇並被授予最近提交和發佈的專利的專利期延長,我們可能不會從可能的專利期延長中獲得全部好處,如果有的話。我們也可能根本不被授予專利期延長,因為例如,我們未能在適用期限內申請,未能在相關專利到期前申請,或其他未能滿足眾多適用要求中的任何一項。此外,如果FDA批准的產品的有效成分在FDA批准的較早產品中受到監管審查和批准,則FDA批准的產品的監管審查期限不得作為延長專利期限的基礎。此外,適用當局,包括美國的FDA和USPTO以及其他國家/地區的任何同等監管機構,可能不同意我們對是否有這種性質的延期的評估,並可能拒絕批准我們的專利延期,或者可能批准比我們要求的更有限的延期。如果發生這種情況,我們的競爭對手可能會在我們的專利到期後,通過參考我們的臨牀和臨牀前數據獲得競爭產品的批准,並比其他情況下更早推出他們的產品。如果發生這種情況,可能會對我們創造產品收入的能力產生不利影響。
在歐洲,補充保護證書可將專利期限延長至最多五年,以補償在監管審查期間失去的專利期限,如果根據商定的兒科調查計劃獲得臨牀試驗數據,這一期限可延長至五年半。雖然歐洲所有國家都必須提供補充保護證書,但歐洲國家之間沒有統一的立法,因此,藥物開發商必須逐個國家申請補充保護證書。因此,一家公司可能需要花費大量資源在所有相關國家申請和接收這些證書,並可能在一些但不是所有國家(如果有的話)獲得它們。
專利法的弱化以及美國和外國法院的執法可能會影響我們保護市場的能力。
過去幾年,美國最高法院在專利案件中發佈了一些意見,許多人認為這些意見可能會削弱美國的專利保護,要麼縮小某些情況下可用的專利保護範圍,認為某些類型的創新不可申請專利,要麼通常以其他方式使法院更容易宣佈專利無效。此外,最近有人提議對美國和其他國家的專利法進行更多修改,如果被採納,可能會影響我們為我們的專有技術獲得專利保護的能力,或者我們執行我們專有技術的能力。根據美國國會、美國法院、美國專利商標局和其他國家相關立法機構未來的行動,管理專利的法律和法規可能會以不可預測的方式發生變化,從而削弱我們獲得新專利或執行我們現有專利和未來可能獲得的專利的能力。
一些外國司法管轄區的法律對知識產權的保護程度不如美國,許多公司在外國司法管轄區保護和捍衞這類權利方面遇到了重大困難。如果我們在保護知識產權方面遇到這樣的困難,或因其他原因而無法在外國司法管轄區有效保護我們的知識產權,我們的商業前景可能會受到嚴重損害。例如,我們可以成為外國反對程序的一方,例如在歐洲專利局,或在外國法院的專利訴訟和其他程序。如果是這樣的話,啟動和繼續此類訴訟程序所產生的不確定因素可能會對我們在市場上競爭的能力產生不利影響。外國對抗性訴訟的費用也可能很高,在許多外國司法管轄區,敗訴一方必須支付勝訴一方的律師費。
我們可能會受到第三方的索賠,聲稱我們、我們的員工、顧問或承包商挪用了適用的第三方的知識產權,或要求我們認為是我們自己的知識產權的所有權。
我們僱用的個人以前曾受僱於大學以及其他生物技術或製藥公司,包括我們的競爭對手或潛在競爭對手。我們已從合作者、潛在的被許可方和可能受合同保密和非使用義務約束的其他第三方那裏收到機密和專有信息。儘管我們努力確保我們的員工在為我們工作時不使用他人的專有信息或專有技術,但我們可能會受到指控,即這些員工或我們使用或披露了任何此類員工前僱主的知識產權,包括商業祕密或其他專有信息。我們還可能面臨前僱主或其他第三方對我們的專利擁有所有權權益的索賠。可能有必要提起訴訟來抗辯這些指控。我們可能無法成功地為這些索賠辯護,如果我們未能為任何此類索賠辯護,除了支付金錢損害賠償外,我們還可能失去寶貴的知識產權,如有價值知識產權的獨家所有權或使用權。即使我們成功了,訴訟也可能導致鉅額成本和聲譽損失,並分散我們管理層和其他員工的注意力。
此外,雖然我們的政策是要求可能參與知識產權開發的員工、顧問和承包商簽署協議,將任何由此產生的知識產權轉讓給我們,但我們可能無法與實際上開發我們視為自己的知識產權的每一方執行類似協議。這種性質的轉讓協議可能不會自動執行或可能被違反,我們可能會被迫向第三方提出索賠或為他們可能對我們提出的索賠辯護,以確定我們視為我們的知識產權的所有權。此外,員工或承包商可以創造一項發明,但不通知我們,在這種情況下,我們可能會失去發明的好處,員工或承包商可能會離開去其他地方開發發明。
知識產權訴訟可能會導致我們花費大量資源,並分散我們的人員對正常職責的注意力。
即使解決方案對我們有利,與知識產權索賠相關的訴訟或其他法律程序也可能導致我們產生鉅額費用,並可能分散我們的技術和管理人員的正常責任。此外,可能會公佈聽證會、動議或其他臨時程序或事態發展的結果,如果證券分析師或投資者認為這些結果是負面的,可能會產生重大影響。
對我們普通股價格的不利影響。這種性質的訴訟或訴訟可能大幅增加我們的運營虧損,並減少可用於開發活動或任何未來銷售、營銷或分銷活動的資源。我們可能沒有足夠的財政或其他資源來適當地進行此類訴訟或訴訟程序。我們的一些競爭對手可能比我們更有效地承擔訴訟或這類訴訟的費用,因為他們有更多的財政資源。專利訴訟或其他訴訟的發起和繼續帶來的不確定性可能會損害我們在市場上的競爭能力。
獲得和維持專利保護有賴於遵守政府專利局提出的各種程序、文件、費用支付和其他要求,如果我們不遵守這些要求,對我們專利的保護可能會減少或取消。
任何已頒發專利的定期維護費應在專利申請和任何由此產生的專利的有效期內分幾個階段支付給美國專利商標局和外國的專利局。美國專利商標局和外國專利局在專利申請過程中要求遵守許多程序、文件、費用支付等要求。雖然在許多情況下,可以根據適用規則通過支付滯納金或通過其他方式糾正疏忽失效,但在某些情況下,不遵守規定可能導致專利或專利申請被放棄或失效,從而導致相關法域的專利或專利權部分或全部喪失。可能導致專利或專利申請被放棄或失效的不合規事件包括但不限於:未能在規定的時限內對官方行動做出迴應,未支付費用,以及未能適當地使其合法化並提交正式文件。在這種情況下,我們的競爭對手可能會進入市場,這將對我們的業務產生不利影響。
如果我們不能保護我們的商業祕密的機密性,我們的商業和競爭地位將受到損害。
除了為我們的一些候選技術和產品申請專利外,我們還依靠商業祕密,包括未獲專利的訣竅、技術和其他專有信息,以保持我們的競爭地位。我們尋求保護這些商業祕密,部分是通過與有權訪問這些商業祕密的各方簽訂保密協議,例如我們的員工、公司合作者、外部科學合作者、合同製造商、顧問、顧問和其他第三方。我們還與我們的員工和顧問簽訂保密和發明或專利轉讓協議。這些協議可能不能有效阻止機密信息的披露,也不能有效地將知識產權轉讓給我們,並且在未經授權披露機密信息或其他違反協議的情況下,可能無法提供足夠的補救措施。此外,其他人可能會獨立發現我們的商業祕密和專有信息。在這種情況下,我們不能主張任何針對該第三方的商業祕密權利。儘管做出了這些努力,但任何一方都可能違反協議,泄露我們的專有信息,包括我們的商業祕密,而我們可能無法就此類違規行為獲得足夠的補救措施。執行一方當事人非法披露或挪用商業祕密的主張是困難、昂貴和耗時的,而且這種性質的爭端的結果本質上是不可預測的。我們可能需要昂貴和耗時的訴訟來尋求強制執行和確定我們的專有權利的範圍,而我們未能獲得或維持商業祕密保護可能會對我們的競爭業務地位產生不利影響。此外,美國以外的一些法院不太願意或不願意保護商業祕密。2016年的《捍衞商業祕密法》是一部美國聯邦法律,允許商業祕密的所有者在商業祕密被挪用時向聯邦法院提起訴訟。國會通過這項法律,試圖加強商業祕密擁有者的權利,這些人的寶貴資產被未經授權而被拿走。如果我們的任何商業祕密是由競爭對手合法獲取或獨立開發的,我們將無權阻止他們或他們向其傳達信息的人使用該技術或信息與我們競爭。如果我們的任何商業祕密被泄露給競爭對手或由競爭對手獨立開發,我們的競爭地位將受到損害。
對於我們的某些專利,我們只有有限的地理保護,我們可能無法在世界各地保護我們的知識產權。
申請、起訴、維護和保護涵蓋我們在世界所有國家的候選產品的專利將是令人望而卻步的昂貴。因此,我們在美國以外的一些國家的知識產權可能沒有我們在美國獲得的保護那麼廣泛。覆蓋我們在世界所有國家的候選產品的許可內專利可能同樣昂貴得令人望而卻步,如果我們真的有這些許可內機會的話。此外,即使在我們開發或商業化我們候選產品的司法管轄區內,授權或備案、起訴、維護和捍衞專利也可能昂貴得令人望而卻步或不切實際。競爭對手可以在我們沒有獲得專利保護或許可專利的司法管轄區使用我們和我們許可人的技術來開發他們自己的產品,並進一步可能向我們和我們許可人擁有專利保護的地區出口其他侵權產品,但執法力度不如美國或
歐盟。這些產品可能與我們的候選產品競爭,而我們或我們的許可方的專利或其他知識產權可能不能有效或不足以阻止他們競爭。
此外,我們可能會決定放棄國家和地區的專利申請,因為它們仍然懸而未決。每項國家或地區專利的授予程序是一個獨立的程序,可能導致相關專利局拒絕申請,而實質上類似的申請由其他機構批准的情況。例如,相對於其他國家,中國對專利性的詳細描述要求更高。此外,仿製藥製造商或其他競爭對手可能會對我們或我們的許可人專利的範圍、有效性或可執行性提出質疑,要求我們或我們的許可人進行復雜、漫長和昂貴的訴訟或其他訴訟。仿製藥製造商可以開發、尋求批准並推出我們產品的仿製藥版本。同樣常見的情況是,根據國家的不同,同一候選產品或技術的專利保護範圍可能會有所不同。
一些司法管轄區的法律對知識產權的保護程度不如美國和歐盟的法律或法規,許多公司在這些司法管轄區保護和捍衞專有權方面遇到了重大困難。此外,某些國家的法律制度,特別是某些發展中國家的法律制度,不支持專利、商業祕密或其他形式的知識產權的執行,這可能會使我們難以阻止某些司法管轄區的競爭對手以侵犯我們的專有權的方式銷售競爭產品。
在外國司法管轄區強制執行我們的專利權的訴訟,無論是否成功,都可能導致鉅額成本,並將我們的努力和注意力從我們業務的其他方面轉移,還可能使我們或我們的許可人的專利面臨被無效或狹隘解釋的風險,可能增加我們或我們的許可人的專利申請不頒發的風險,或可能引發第三方對我們提出索賠。我們可能不會在我們發起的任何訴訟中獲勝,而可能會向敵方支付損害賠償或其他補救措施,這可能具有重大的商業意義。如果我們勝訴,判給我們的損害賠償或其他補救措施(如果有的話)可能沒有商業意義。因此,我們在世界各地執行我們的知識產權的努力可能不足以從我們開發或許可的知識產權中獲得顯著的商業優勢。此外,雖然我們打算在我們預期的重要市場保護我們的知識產權,但我們不能確保我們能夠在我們可能希望營銷我們的候選產品的所有司法管轄區啟動或保持類似的努力。因此,我們在這些國家保護我們的知識產權的努力可能不夠充分,這可能會對我們在所有預期的重要國外市場成功地將我們的候選產品商業化的能力產生不利影響。如果我們或我們的許可人在這些司法管轄區遇到保護困難或無法有效保護對我們業務重要的知識產權,這些權利的價值可能會降低,我們可能會在這些司法管轄區面臨額外的競爭。
在一些司法管轄區,強制許可法迫使專利權人向第三方授予許可。此外,一些國家限制專利對政府機構或政府承包商的可執行性。在這些國家,專利權人的補救措施可能是有限的,這可能會大大降低這種專利的價值。如果我們或我們的任何許可人被迫在與我們的業務相關的專利下向第三方授予許可,或者如果我們或我們的許可人被阻止針對第三方執行專利權,我們的競爭地位可能會在這些司法管轄區受到嚴重損害。
與監管事項相關的風險
從FDA和外國監管機構獲得監管批准是漫長、耗時的,而且本質上是不可預測的,如果我們最終無法為我們的候選產品獲得營銷批准,我們的業務將受到嚴重損害。
獲得FDA和外國監管機構批准所需的時間是不可預測的,但通常需要在臨牀試驗開始後多年,並取決於許多因素,包括監管機構的相當大的酌情決定權。此外,批准標準、法規或獲得批准所需的臨牀數據的類型和數量可能會在候選產品的臨牀開發過程中發生變化,並可能因司法管轄區而異。我們尚未獲得任何候選產品的營銷批准,我們現有的候選產品或我們未來可能尋求開發的任何候選產品(獨立或與我們的合作伙伴之一)都可能永遠不會獲得營銷批准。
我們的候選產品可能會因為許多原因而無法獲得或保留營銷批准,包括以下原因:
•FDA或外國監管機構,在這裏分別稱為衞生當局,可能不同意我們臨牀試驗的設計或實施;
•我們可能無法向衞生當局證明,就其建議的適應症而言,候選產品是安全有效的,或根據衞生當局的標準,該候選產品具有足夠的強度、身份或質量;
•臨牀試驗結果可能達不到衞生部門批准的統計顯著性水平;
•我們可能無法證明候選產品的臨牀和其他益處大於其安全風險;
•衞生當局可能不同意我們對臨牀前研究或臨牀試驗數據的解釋;
•從我們候選產品的臨牀試驗中收集的數據可能不足以支持向FDA提交保密協議或向外國監管機構提交其他申請,或在美國或任何其他國家或司法管轄區獲得上市批准;
•衞生當局可能發現與我們簽訂臨牀和商業用品合同的第三方製造商的製造工藝或設施存在缺陷或不獲批准;以及
•衞生當局的批准標準、政策或法規可能會發生重大變化,導致我們的臨牀數據不足以批准。
這一漫長的藥物開發過程,以及未來臨牀試驗結果的不可預測性,可能會導致我們無法獲得監管部門的批准,從而無法銷售我們的任何候選產品,這將嚴重損害我們的業務、運營結果和前景。FDA和其他衞生當局在審批過程中擁有相當大的自由裁量權,並決定何時或是否獲得我們任何候選產品的監管批准,包括在加速審批的情況下。即使我們相信從我們的候選產品的臨牀試驗中收集的數據是有希望的,這些數據可能也不足以支持FDA或任何其他衞生當局的批准。
此外,即使我們獲得批准,監管機構也可能批准我們的任何候選產品,其適應症少於或超過我們要求的範圍,可能不批准我們打算為產品收取的價格,可能批准取決於昂貴的上市後臨牀試驗的表現,或者可能批准候選產品的標籤不包括該候選產品成功商業化所必需或可取的標籤聲明。上述任何一種情況都可能對我們的候選產品的商業前景造成實質性損害。
即使我們的任何候選產品在美國獲得了FDA的批准,我們也可能永遠不會在任何其他司法管轄區獲得批准或將其商業化,這將限制我們實現其全部市場潛力的能力。
為了在任何特定的司法管轄區銷售任何產品,我們必須在每個國家的基礎上建立和遵守關於安全性和有效性的眾多和不同的監管要求。
美國FDA的批准並不確保其他國家或司法管轄區的監管機構也批准。然而,未能在一個司法管轄區獲得批准,可能會對我們在其他地方獲得批准的能力產生負面影響。此外,在一個國家進行的臨牀試驗可能不會被其他國家的監管機構接受,一個國家的監管批准並不能保證任何其他國家的監管批准。
審批流程因國家/地區而異,可能涉及額外的產品測試和驗證,以及額外的行政審查期限。尋求外國監管機構的批准可能會給我們帶來困難和增加成本,並需要額外的臨牀前研究或臨牀試驗,這可能是昂貴和耗時的。各國的監管要求可能有很大差異,可能會推遲或阻止我們的產品在這些國家推出。我們沒有任何候選產品在任何司法管轄區獲得批准銷售,包括在國際市場,我們作為一家公司也沒有在國際市場獲得監管批准的經驗。如果我們未能遵守國際市場的監管要求,或未能獲得和保持所需的批准,或者如果國際市場的監管批准被推遲,我們的目標市場將會減少,我們開發的任何產品充分發揮市場潛力的能力將無法實現。
即使我們獲得了任何候選產品的監管批准,我們也將受到持續的監管義務和持續的監管審查的約束,這可能會導致大量額外費用,如果我們未能遵守監管要求或我們的候選產品遇到意想不到的問題,我們可能會受到處罰。
如果我們的任何候選產品獲得批准,它們將遵守持續的法規要求,包括製造、標籤、包裝、儲存、廣告、促銷、抽樣、記錄保存、進行上市後研究以及提交安全性、有效性和其他上市後信息,包括美國聯邦和州的要求以及可比的外國監管機構的要求。此外,我們可能會被要求進行
在難以進行或完成的特殊人羣中進行審批後研究。對於我們在批准後進行的任何臨牀試驗,我們還將繼續遵守cGMP和GCP要求。
製造商和製造商的設施必須遵守FDA和類似的外國監管機構的廣泛要求,包括確保質量控制和製造程序符合cGMP法規和適用的產品跟蹤和追溯要求。因此,我們和我們的CMO將接受持續的審查和檢查,以評估是否符合cGMP以及是否遵守任何NDA、其他上市申請和之前對檢查觀察結果的回覆中做出的承諾。因此,我們和我們的合作伙伴必須繼續在所有合規領域(包括製造、生產和質量控制)投入時間、金錢和精力。
我們為我們的候選產品獲得的任何監管批准可能會受到該產品可能上市的已批准指示用途的限制或批准條件的限制,或者包含可能代價高昂的上市後測試的要求,包括監控候選產品的安全性和有效性的第四階段臨牀試驗和監測。FDA還可能要求將REMS計劃作為批准我們的候選產品的條件,這可能需要對患者的長期隨訪、用藥指南、醫生溝通計劃或確保安全使用的其他要素的要求,例如限制分發方法、患者登記和其他風險最小化工具。類似的外國監管機構也可能有類似於REMS的計劃。此外,如果FDA或類似的外國監管機構批准我們的候選產品,我們將必須遵守包括提交安全和其他上市後信息以及報告和註冊在內的要求。
如果沒有遵守監管要求和標準,或者如果產品上市後出現問題,FDA可能會強制實施同意法令或撤回批准。後來發現我們的候選產品存在以前未知的問題,包括預料不到的嚴重程度或頻率的不良事件,或我們的第三方CMO或製造工藝,或未能遵守監管要求,可能會導致修訂批准的標籤以添加新的安全信息;實施上市後研究或臨牀試驗以評估新的安全風險;或根據REMS計劃實施分銷限制或其他限制。除其他外,其他潛在後果包括:
•限制我們產品的銷售或製造,將產品從市場上召回,或自願或強制召回產品;
•罰款、警告信或暫停臨牀試驗的;
•FDA拒絕批准待處理的申請或對我們提交的已批准申請的補充,或暫停或撤銷許可證批准;
•扣押或扣留產品,或拒絕允許進口或出口我們的候選產品;以及
•禁制令或施加民事或刑事處罰。
FDA對投放市場的產品的營銷、標籤、廣告和促銷進行嚴格監管。產品只能按照經批准的適應症和經批准的標籤的規定進行促銷。FDA和其他機構積極執行禁止推廣標籤外用途的法律和法規,被發現不當推廣標籤外用途的公司可能會承擔重大責任。然而,醫生可以根據其獨立的醫學判斷,為標籤外使用的合法可用產品開處方。FDA不規範醫生選擇治療的行為,但FDA確實限制了製造商在產品標籤外使用問題上的溝通。FDA和類似的外國監管機構的政策可能會改變,可能會頒佈額外的政府法規,以阻止、限制或推遲對我們候選產品的監管批准。我們無法預測美國或國外未來的立法或行政行動可能產生的政府監管的可能性、性質或程度。如果我們緩慢或無法適應現有要求的變化或採用新的要求或政策,或者如果我們無法保持監管合規性,我們可能會失去我們可能獲得的任何營銷批准,我們可能無法實現或維持盈利。
FDA指定的突破性療法,即使被授予我們的任何候選產品,也可能不會導致更快的開發或監管審查或批准過程,也不會增加我們的候選產品獲得上市批准的可能性。
我們可能會為我們當前和未來的一些或所有候選產品尋求突破性療法稱號,包括CFT7455和CFT1946。突破療法被定義為一種旨在單獨或與一種或多種其他藥物聯合治療嚴重或危及生命的疾病或狀況的藥物,初步臨牀證據表明,該藥物可能在一個或多個臨牀重要終點顯示出比現有療法顯著的改善,例如在臨牀開發早期觀察到的實質性治療效果。適用於已獲得
作為突破性療法,FDA和試驗贊助商之間的互動和溝通可以幫助確定臨牀開發的最有效途徑,同時將無效控制方案中的患者數量降至最低。被FDA指定為突破性療法的藥物也可能有資格獲得其他快速批准計劃,包括加速批准。
FDA有權將其指定為突破性療法。因此,即使我們認為我們的候選產品之一符合被指定為突破性療法的標準,FDA也可能不同意,而是決定不做出這樣的指定。無論如何,儘管突破性療法指定旨在加快獲得此類指定的藥物的開發和審查,但與根據非快速FDA審查程序考慮批准的候選產品相比,收到候選產品的突破性治療指定可能不會導致更快的開發過程、審查或批准,也不能確保FDA最終批准候選產品。此外,即使我們的一個或多個候選產品符合突破療法的條件,FDA稍後也可能決定該產品不再符合資格條件。因此,即使我們打算為我們的主要候選產品以及我們治療各種癌症的部分或全部未來候選產品尋求突破療法稱號,也不能保證我們將獲得突破療法稱號。
FDA的快速通道指定,即使授予我們的一個或所有主要候選產品,或我們當前或未來的任何其他候選產品,也可能不會導致更快的開發或監管審查或批准過程,也不會增加我們的候選產品獲得上市批准的可能性。
在各種情況下,我們可能會為我們的一個或多個候選產品尋求快速通道認證。如果一種藥物用於治療嚴重或危及生命的疾病,並且該藥物顯示出解決這種疾病未得到滿足的醫療需求的潛力,藥物贊助商可以為特定的適應症申請FDA快速通道指定。我們可能會為我們的一個或所有主要候選產品和/或某些未來候選產品尋求快速通道認證,但不能保證FDA會將這一地位授予我們建議的任何候選產品,我們可能只有在多次申請後才能成功獲得FDA對某個候選產品的快速通道認證。根據FDA提供的政策和程序,Fast Track開發產品的贊助商提交的營銷申請可能有資格優先審查,但收到Fast Track指定並不保證有任何此類資格或FDA最終批准上市。FDA擁有廣泛的自由裁量權,是否授予快速通道認證,因此,即使我們認為特定的候選產品有資格獲得該認證,也不能保證FDA會決定授予它。即使我們確實獲得了Fast Track認證,即使Fast Track認證旨在加快獲得此類認證的藥物的開發和審查,與傳統的FDA程序相比,我們可能不會經歷更快的開發過程、審查或批准,而且獲得Fast Track認證並不能保證FDA的最終批准。此外,如果FDA認為我們的臨牀開發計劃的數據不再支持快速通道指定,它可能會撤回該指定。此外,FDA可以隨時撤銷任何Fast Track的指定。
我們已經獲得了CFT7455的孤兒藥物指定,如果我們決定為任何其他當前或未來的候選產品尋求孤兒藥物指定,我們可能不會成功或可能無法保持與孤兒藥物指定相關的好處,包括補充市場排他性的潛力。
2021年8月,FDA授予CFT7455治療MM的孤兒藥物稱號。我們可能會為我們目前或未來的一個或多個候選產品尋求孤兒藥物稱號。包括美國和歐盟在內的一些司法管轄區的監管機構可能會將患者人數相對較少的藥物指定為孤兒藥物。根據《孤兒藥品法》,FDA可以將一種用於治療罕見疾病或狀況的藥物授予孤兒藥物稱號,這種疾病或狀況的定義是,在美國,患者人數低於20萬人,或在美國患者人數超過20萬人,但沒有合理的預期,在美國開發和提供藥物的成本。將從該藥物在美國的銷售中恢復。在美國,獲得孤兒藥物稱號使當事人有權獲得財政獎勵,如為臨牀試驗費用、税收優惠和用户費用減免提供贈款資金的機會。在FDA批准孤兒藥物指定後,FDA公開披露該藥物的仿製藥身份及其潛在的孤兒用途。雖然孤兒藥物指定旨在促進針對罕見疾病或疾病的藥物開發,但孤兒藥物指定不會在監管審查和批准過程中傳遞任何優勢,也不會縮短監管審查和批准過程的持續時間。此外,雖然收到孤兒藥物指定可能導致FDA放棄在兒科人羣中進行研究的任何義務,但這種豁免可能不適用於腫瘤藥物
如果一種具有孤兒藥物指定的產品隨後獲得了FDA對其具有這種指定的疾病的特定活性成分的第一次批准,該產品有權獲得孤兒產品排他性,這意味着FDA可能不會批准任何其他申請,包括NDA,在七年內銷售相同適應症的同一藥物,除非在有限的情況下,如顯示出相對於具有以下特徵的產品的臨牀優勢
孤兒藥物排他性,或FDA發現孤兒藥物排他性持有者沒有證明它可以確保獲得足夠數量的孤兒藥物,以滿足指定藥物所針對的疾病或狀況的患者的需求。因此,即使我們的一個候選產品獲得了孤兒藥物的排他性,FDA仍然可以批准其他含有不同有效成分的藥物用於治療相同的適應症或疾病。此外,如果我們無法生產足夠的產品供應,FDA可以放棄孤兒藥物的獨家經營權。
我們還可能為我們的其他主要候選產品和/或我們的部分或全部其他當前或未來候選產品在其他孤兒適應症中尋求孤兒藥名稱,其中這些候選產品的使用具有醫學合理的基礎。即使我們獲得孤兒藥認定,如果我們尋求批准的適應症比孤兒藥認定的適應症更廣泛,則在美國的獨家營銷權可能會受到限制,如果FDA後來確定認定申請存在重大缺陷,或者如果我們通過製造商,無法保證足夠數量的產品以滿足罕見疾病或病症患者的需求。此外,即使我們為其他候選產品尋求孤兒藥認定,我們也可能永遠不會獲得這些認定。例如,FDA對適用於組織不可知療法的孤兒藥認定的監管考慮表示擔憂,並且FDA可能會以限制或阻止我們獲得孤兒藥認定或孤兒藥排他性的能力的方式解釋FDCA及其孤兒藥法規,如果我們的候選產品獲得批准,我們的目標適應症。
在適用的情況下,我們可能會根據FDA的加速批准途徑尋求對我們候選產品的批准。這種途徑可能不會加快開發或監管審查或批准流程,也不會增加我們的候選產品獲得上市批准的可能性。
我們計劃尋求加速批准我們的主要候選產品,並可能尋求批准未來的候選產品,在適用的情況下,使用FDA的加速批准途徑。如果產品治療嚴重或危及生命的疾病,並且通常提供比現有療法有意義的優勢,則該產品可能有資格獲得加速批准。此外,必須證明對替代終點的影響,該替代終點有合理可能預測臨牀獲益,或對可在不可逆發病率或死亡率或IMM之前測量的臨牀終點的影響,該臨牀終點有合理可能預測對IMM或其他臨牀獲益的影響。根據《食品和藥品綜合改革法案》(通常稱為FDORA),允許FDA在適當情況下要求在批准前或批准後的規定時間內對獲得加速批准的產品進行批准後確證性研究。FDORA還要求申辦者每180天向FDA發送一次關於這些研究狀態的更新,包括招募目標的進展,FDA必須立即公開發布這些信息。FDORA還賦予FDA更大的權力,如果申辦者未能及時開展此類活動,向FDA發送必要的更新,或者如果此類批准後研究未能驗證藥物的預期臨牀益處,則可以快速撤銷加速批准;並採取行動,如罰款,對那些未能盡職盡責地進行任何批准後確證性研究或未能及時向該機構提交進展報告的公司。此外,FDA通常要求對獲得加速批准的產品的宣傳材料進行預先批准,這可能會對產品的商業發佈時間產生不利影響。因此,即使我們尋求對我們的任何候選產品使用加速審批途徑,我們也可能無法獲得加速審批,即使我們這樣做,該產品也可能不會經歷更快的開發或監管審查或審批流程。此外,獲得加速批准並不能保證產品的加速批准最終會轉換為傳統批准。
FDA可能會在書面請求中確定兒科信息對我們獲得批准的候選產品有益,並要求我們進行兒科研究。我們可能選擇不進行這些研究,或者如果我們選擇進行這些研究,我們可能無法完成這些研究,或者這些研究產生的數據可能不被FDA接受。
《食品、藥品和化粧品法案》或《FDC法案》第505(A)節為在兒童中進行藥物研究的藥品生產商提供了激勵措施。被稱為“兒科排他性條款”,這項法律為製藥商提供了額外的六個月的非專利排他性,這些製藥商對新的和目前上市的藥品進行了可接受的兒科研究,根據FDA的書面要求,兒科數據將是有益的。因此,如果我們收到FDA的兒科研究書面請求,進行兒科臨牀研究並在法定時限內提交FDA接受的報告,我們可以獲得超過所有其他類型專利和非專利的額外六個月的監管排他性,專利獨佔權則對我們所有已批准的含有兒科獨佔權的活性成分的藥品有效。但是,即使我們收到FDA對我們的一種或多種藥品進行兒科研究的書面請求,我們也可能決定不進行或無法進行符合FDC法案第505(A)條的兒科研究,或者我們可能進行研究
FDA不接受用於此目的。如果出現這種情況,我們將不會獲得額外的六個月監管排他性延長。
我們與客户、醫療保健提供者和第三方付款人的關係直接或間接地受到或將受到外國、聯邦和州醫療保健欺詐和濫用法律、虛假索賠法律、健康信息隱私和安全法律以及其他醫療保健法律法規的約束。如果我們無法遵守或沒有完全遵守這些法律,我們可能會面臨重大處罰。
美國和其他地方的醫療保健提供者和第三方付款人將在我們獲得營銷批准的任何候選產品的推薦和處方中發揮主要作用。我們目前和未來與醫療保健專業人士、主要研究者、顧問、客户和第三方付款人的安排使我們受到各種聯邦和州欺詐和濫用法律以及其他醫療保健法律的約束,這些法律可能會限制我們研究、銷售、營銷和分銷候選產品(如果我們獲得營銷批准)的業務或財務安排和關係。特別是,我們候選產品的研究,以及醫療保健項目和服務的推廣、銷售和營銷,以及醫療保健行業的某些業務安排,都受到廣泛的法律約束,旨在:(i)防止欺詐、回扣、自我交易和其他濫用行為;(ii)保證健康信息的安全和隱私;以及(iii)提高醫生、教學醫院和藥品、醫療器械和生物製品製造商之間財務關係的透明度。這些法律法規可能限制或禁止各種定價、折扣、營銷和促銷、結構和佣金、某些客户獎勵計劃以及其他業務或財務安排。見標題為“商業-其他醫療保健法律“和”商業-醫療改革“在我們的2022年年度報告中。
確保我們與第三方的業務安排和做法符合適用的醫療保健法律和法規可能會付出高昂的代價。政府機構可能會得出結論,認為我們的商業行為可能不符合當前或未來涉及適用欺詐和濫用的法令、法規或判例法,或其他醫療保健法律和法規。如果我們的運營被發現違反任何這些法律或任何其他可能適用於我們的政府法規,我們可能會受到重大的民事、刑事和行政處罰、損害賠償、罰款、沒收、監禁、被禁止參加政府資助的醫療保健計劃,如Medicare和Medicaid,如果我們受到企業誠信協議或類似協議的約束,以解決有關不遵守這些法律、合同損害、聲譽損害的指控,以及削減或重組我們的業務。不斷變化的合規環境以及構建和維護強大且可擴展的系統以符合具有不同合規或報告要求的多個司法管轄區的需求增加了醫療保健公司可能違反一項或多項要求的可能性。
如果我們預期與之開展業務的醫生或其他提供者或實體被發現不遵守適用法律,他們可能會受到重大的刑事、民事或行政制裁,包括被排除在政府資助的醫療保健計劃之外。即使以有利於我們的方式解決,與醫療保健法律法規相關的訴訟或其他法律程序也可能導致我們產生大量費用,並可能分散我們的技術和管理人員的正常職責。此外,還可以公佈審理結果、動議或其他臨時程序或事態發展。如果證券分析師或投資者認為這些結果是負面的,它可能會對我們的普通股價格產生重大不利影響。此類性質的訴訟或法律程序可能會大幅增加我們的經營虧損,並減少可用於開發、製造、銷售、營銷或分銷活動的資源。與適用的醫療保健法律和法規有關的訴訟或其他程序的啟動和持續導致的不確定性可能對我們在市場上的競爭能力產生不利影響。
我們的候選產品在美國和國外的成功商業化將部分取決於第三方支付者(包括政府機構和私人醫療保險公司)提供的覆蓋範圍和足夠的報銷水平,以及實施有利於我們候選產品的定價政策。未能獲得或維持我們的候選產品的覆蓋範圍和足夠的報銷,如果獲得批准,可能會限制我們營銷這些產品的能力,並降低我們產生收入的能力。
我們可能獲得監管批准的任何產品的承保範圍和報銷狀態存在重大不確定性。在美國和其他國家,因其病情而接受治療的患者通常依賴第三方支付者償還與其治療相關的全部或部分費用。第三方付款人(包括政府醫療保健計劃(例如,醫療保險,醫療補助或TRICARE在美國),管理式醫療服務提供者,私人健康保險公司,健康維護組織和其他組織是必不可少的大多數患者能夠負擔得起的醫療服務和醫藥產品,如我們的候選產品。第三方支付者決定他們使用哪些藥物
將支付並建立報銷水平。見標題為“”的部分承保和報銷“在我們的2022年年度報告中。
我們能否成功地將我們的候選產品商業化,在一定程度上將取決於第三方付款人為我們的產品和相關治療提供的覆蓋範圍和足夠的補償。此外,付款人決定為產品提供保險並不意味着將批准足夠的償還率。如果無法獲得保險和足夠的報銷,或僅限於有限的級別,我們可能無法成功地將我們的候選產品商業化。即使提供了保險,批准的報銷金額也可能不夠高,不足以讓我們建立或保持足夠的定價,以實現足夠的投資回報。如果第三方付款人決定不承保或不單獨報銷使用我們產品的醫療產品或治療,一旦獲得批准,可能會減少醫生對我們產品的使用。
我們不能確保美國和其他國家/地區的保險和報銷適用於我們當前或未來的候選產品,或使用我們當前或未來候選產品的任何程序,並且可能獲得的任何報銷可能不夠充分,或可能在未來減少或取消。
在美國,第三方付款人對產品的承保和報銷沒有統一的政策。因此,我們產品的承保範圍和報銷範圍因付款人而異。確定付款人是否將為產品提供保險的過程可以與設置付款人將為產品支付的償還率的過程分開。一個付款人決定為一種產品提供保險並不能保證其他付款人也會為該產品提供保險和補償。第三方付款人也可以將承保範圍限制在批准的清單或配方表上的特定產品,其中可能不包括FDA批准的特定適應症的所有產品。在美國,有關新藥報銷的主要決定通常由聯邦醫療保險和醫療補助服務中心(CMS)做出,CMS是美國衞生與公眾服務部(HHS)的一個機構。CMS將決定我們的產品是否以及在多大程度上將在聯邦醫療保險下得到覆蓋和報銷,而私人支付者往往在很大程度上遵循CMS。付款人在確定報銷時考慮的因素基於產品是否為:
•在其健康計劃下有保障的福利;
•安全、有效,並且在醫學上是必要的;
•適用於特定的患者;
•具有成本效益;以及
•既不是試驗性的,也不是調查性的。
我們不能確保我們候選產品的承保範圍和報銷範圍可用,或準確估計我們候選產品的潛在收入,也不能保證我們可能開發的任何產品都能獲得承保和報銷。
此外,美國和海外的第三方付款人加大力度限制或降低醫療成本,可能會導致付款人組織限制新批准產品的承保範圍和報銷水平,因此,它們可能無法為我們的候選產品支付或提供足夠的付款。為了確保任何可能被批准銷售的產品的承保範圍和報銷,我們可能需要進行昂貴的藥物經濟學研究,以證明我們產品的醫療必要性和成本效益,以及獲得FDA或類似監管批准所需的成本。此外,我們可能還需要向購買者、私人健康計劃或政府醫療計劃提供折扣。儘管如此,我們的候選產品可能不被認為是醫學上必要的或具有成本效益的。如果第三方付款人不認為一種產品與其他可用的療法相比具有成本效益,他們可能不會在批准後將該產品作為其計劃下的一項福利覆蓋,或者,如果他們這樣認為,支付水平可能不足以讓公司銷售其產品以盈利。我們預計,在潛在銷售我們的任何候選產品時,都會遇到來自第三方付款人的定價壓力。
最後,在一些外國國家,藥品的擬議定價必須獲得批准,才能合法上市。各國對藥品定價的要求差別很大。例如,歐盟成員國可以限制其國家健康保險制度提供報銷的醫療產品的範圍,並可以控制供人使用的醫療產品的價格。為了獲得報銷或定價批准,其中一些國家可能要求完成臨牀試驗,將特定候選產品的成本效益與目前可用的療法進行比較。歐盟成員國可以批准醫藥產品的具體價格,也可以對將醫藥產品投放市場的公司的盈利能力採取直接或間接控制的制度。歐盟成員國之間的做法正出現分歧。例如,在法國,有效的市場準入將由與醫院達成的協議支持,產品可能由社會保障基金報銷。藥品價格是與保健品經濟委員會協商的。不能保證任何對藥品實行價格管制或報銷限制的國家將允許有利的
我們的任何候選產品的報銷和定價安排。從歷史上看,在歐盟推出的產品並不遵循美國的價格結構,通常歐盟的價格往往明顯低於美國的價格。
頒佈和未來的醫療保健立法可能會增加我們推進臨牀項目、獲得候選產品的上市批准和商業化的難度和成本,並可能影響我們可能設定的價格。
在美國和其他司法管轄區,我們預計將繼續對醫療保健系統進行多項立法和監管方面的改革和擬議中的改革,這可能會影響我們未來的運營結果。特別是,美國聯邦和州一級已經並將繼續採取一些舉措,尋求降低醫療成本和提高醫療質量。美國的個別州也越來越多地通過立法並實施旨在控制藥品和生物製品定價的法規,包括價格或患者報銷限制、折扣、對某些產品准入和營銷成本披露的限制以及透明度措施,在某些情況下,旨在鼓勵從其他國家進口和批量購買。見標題為“”的部分企業-政府監管-醫療改革在我們的202年度報告中。
我們預計,未來將採取更多的州和聯邦醫療改革措施,其中任何一項都可能限制州和聯邦政府覆蓋特定醫療產品和服務的程度,並可能限制聯邦和州政府為醫療產品和服務支付的金額。這可能會導致對我們開發的任何候選產品的需求減少,或者可能導致額外的定價壓力。
在美國以外的市場,報銷和醫療保健支付制度因國家而異,許多國家對特定產品和療法設定了價格上限。美國以外的價格管制法規可能會對特定市場的盈利能力產生重大影響,如果這些法律發生變化,還會帶來進一步的不確定性。
我們無法預測美國或任何其他司法管轄區未來的立法或行政行動可能產生的政府監管的可能性、性質或程度。政府、保險公司、管理醫療組織和其他醫療服務付款人繼續努力控制或降低醫療成本和/或實施價格管制,可能會產生不利影響:
•對我們候選產品的需求,如果我們獲得監管部門的批准;
•我們有能力為我們批准的產品設定一個我們認為公平的價格;
•我們創造收入、實現或保持盈利的能力;
•我們須繳交的税項水平;及
•資金的可得性。
如果我們或我們可能接觸的任何第三方緩慢或無法適應現有要求的變化或採用新的要求或政策,或者如果我們或這些第三方無法保持監管合規性,我們的候選產品可能會失去可能已獲得的任何監管批准,我們可能無法實現或維持盈利。
如果我們從我們贊助的臨牀試驗中獲得可識別的患者健康信息,根據適用的隱私法,我們可能在美國和其他司法管轄區面臨潛在的責任。
大多數醫療保健提供者,包括我們可以從其獲得患者健康信息的某些研究機構,都受到根據1966年《健康保險可攜帶性和責任法案》(HIPAA)頒佈的隱私和安全法規的約束,該法案經《經濟和臨牀健康信息技術法案》修訂。根據事實和情況,如果我們以未經HIPAA授權或允許的方式獲取、使用或披露由HIPAA覆蓋的實體維護的可單獨識別的健康信息,我們可能會受到民事、刑事和行政處罰。此外,我們可能受到州法律的約束,要求在個人信息被泄露時通知受影響的個人和州監管機構,這是比HIPAA保護的健康信息更廣泛的信息類別。
全球數據保護格局正在迅速發展,我們可能會受到許多聯邦、州和外國法律法規以及監管指南的制約或影響,這些法規和法規管理着個人數據的收集、使用、披露、傳輸、安全和處理,例如我們收集的與臨牀試驗相關的參與者和醫療保健提供者的信息。在可預見的未來,實施標準和執法做法可能仍然不確定,這可能會給我們的業務帶來不確定性,影響我們或我們的服務提供商在某些司法管轄區運營或收集、存儲、轉移、使用和共享個人數據的能力,導致我們承擔責任或向我們施加額外的合規或其他成本。任何我們未能或被認為未能遵守聯邦、州或外國
法律或自律標準可能導致負面宣傳、轉移管理時間和精力,以及政府實體或其他人對我們提起訴訟。
在美國,2018年加州消費者隱私法,或CCPA,於2020年1月生效。CCPA為消費者提供了新的數據隱私權,併為公司提供了新的運營要求,包括對處理消費者或家庭某些個人數據的實體施加更多隱私和安全義務。這些要求可能會增加我們的合規成本和潛在的責任。CCPA賦予加州居民更大的權利,可以訪問和刪除他們的個人信息,選擇不分享某些個人信息,並接收有關他們的個人信息如何使用的詳細信息。CCPA規定了對違規行為的民事處罰,以及對數據泄露的私人訴權,預計這將增加數據泄露訴訟。雖然目前受HIPAA和臨牀試驗法規約束的受保護健康信息有例外情況,但CCPA可能會影響我們的某些業務活動。
此外,加州隱私權法案(CPRA)對CCPA進行了修訂,該法案於2023年1月1日生效。CPRA提出的修正案大大修改了CCPA,包括對涵蓋的企業施加額外義務,擴大消費者權利,並對某些敏感個人信息施加新的義務。CPRA還創建了一個新的國家機構,該機構有權實施和執行CCPA。經修訂的《反海外腐敗法》的影響可能是重大的,可能需要我們修改我們的數據收集或處理做法和政策,併產生大量成本和支出,以努力遵守並增加我們在監管執法和/或訴訟中的潛在風險。
此外,許多州已經通過了廣泛的消費者隱私法,這些法律在許多方面與CCPA相似,隨着許多其他州提出類似的法律,其他州很可能會效仿,也會通過全面的注重隱私的立法。如果通過,這類立法可能會增加複雜性、要求的進一步變化、限制和潛在的法律風險,需要在合規計劃中投入額外的資源,影響數據收集戰略和以前有用的數據的可用性,並可能導致合規成本增加和/或業務實踐和政策的變化。在美國不同的州存在全面的隱私法將使我們的合規義務更加複雜和代價高昂,並可能增加我們受到執法行動或以其他方式因不合規而承擔責任的可能性。此外,除了州一級的全面法律外,一些州一直在提出或通過針對隱私特定方面的法律。例如,華盛頓州通過了一項範圍廣泛的法律,保護HIPAA不涵蓋的醫療和健康相關信息的隱私,少數州通過了專門針對生物識別信息的法律。
隨着隱私和數據保護法的數量和複雜性的增加,以及全球法律或法規的其他變化,特別是與加強對某些類型的敏感數據的保護相關的變化,例如來自我們臨牀試驗的醫療數據或其他個人信息,可能會導致政府對我們採取執法行動和重大處罰,並可能對我們的業務、財務狀況或運營結果產生重大不利影響。
在美國之外,我們還面臨着嚴格的隱私和數據保護法律的挑戰。例如,歐洲經濟區(EEA)的立法者通過了歐盟或歐盟一般數據保護條例(EU GDPR)和歐盟GDPR,並將其轉換為聯合王國、英國GDPR,統稱為GDPR。GDPR對位於歐洲經濟區和英國的受試者的個人數據的控制器和處理器施加了更嚴格的數據保護合規要求,包括對包括健康、生物識別和遺傳信息在內的“特殊類別數據”的特殊保護,並規定了對不合規的重大處罰。此外,GDPR為EEA成員國提供了廣泛的權利來制定補充國家法律,如與健康、基因和生物識別數據處理相關的法律,這可能會進一步限制我們使用和共享此類數據的能力,或者可能導致我們的成本增加,並損害我們的業務和財務狀況。GDPR包括可能適用於我們業務的合規義務,這可能會導致我們改變業務實踐,並增加對違規行為的經濟處罰(包括對最嚴重違規行為可能處以最高2000萬歐元(英國GDPR規定為1750萬英鎊)的罰款和上一財政年度我們全球年營業額的4%,以及根據GDPR第82條任何個人要求的經濟或非經濟損害賠償的權利)。除了此類罰款外,我們還可能面臨訴訟和/或負面宣傳,這可能會對我們的聲譽和業務產生實質性的不利影響。
GDPR還賦予數據主體和消費者協會一項私人訴訟權利,可以向監管當局提出投訴,尋求司法補救,並就違反GDPR造成的損害獲得賠償。此外,GDPR還包括對跨境數據傳輸的限制。GDPR可能會增加我們在處理受GDPR約束的個人數據方面的責任和責任。在……裏面
此外,我們可能需要建立額外的機制,以確保遵守GDPR,包括個別國家實施的GDPR要求。
GDPR要求我們告知資料當事人我們如何處理他們的個人資料以及他們如何行使他們的權利,確保我們有有效的法律基礎來處理個人資料(如果這是同意的話,獲得同意的要求有更高的門檻),並在敏感的個人數據(即健康數據)被大規模處理的情況下任命數據保護官員。此外,GDPR在整個歐洲經濟區和英國引入了強制性的數據泄露通知要求,要求我們保存我們的處理活動的記錄,並在存在高風險處理的情況下記錄數據保護影響評估,在我們與服務提供商簽訂合同時對我們施加額外的義務,要求我們採取適當的技術和組織措施來保護個人數據,並要求我們採取適當的隱私治理,包括政策、程序、培訓和數據審計。我們正在採取措施,在適當和適用於我們的情況下遵守GDPR,但這是一個持續的合規過程。遵守GDPR將是一個嚴格和耗時的過程,可能會增加我們的業務成本或要求我們改變我們的業務做法。如果我們遵守GDPR或其他適用的歐洲經濟區和英國法律法規的努力不成功,或被認為不成功,可能會對我們在歐洲經濟區和/或英國的業務產生不利影響。
值得注意的是,GDPR對個人數據從歐洲經濟區和英國轉移到歐洲經濟區/英國以外的其他地區或第三國實施了嚴格的規則,在某些情況下,主管數據保護當局(包括美國)認為這些地區或第三國沒有提供“足夠”的隱私保護,除非存在克減或制定了足夠的國際轉移保障措施(例如,歐盟委員會批准的標準合同條款,或歐盟SCC,以及英國國際數據轉移協議/附錄,或英國IDTA)。在依賴歐盟SCC或英國IDTA進行數據傳輸的情況下,我們還可能被要求在個案的基礎上對根據歐盟SCC和英國IDTA進行的傳輸進行傳輸影響評估,以確保接受國的法律提供“基本上相同的”保護,以保護傳輸的個人數據,與歐洲經濟區和英國的規定相同,如果不符合這一標準,我們可能需要採取補充措施。EEA和英國數據保護制度下的國際轉移義務將需要大量的努力和成本,並可能導致我們需要對EEA和英國個人數據的位置以及我們可以利用哪些服務提供商來處理EEA和英國個人數據做出戰略考慮。任何無法按照數據保護法將個人數據從歐洲經濟區轉移到美國的行為都可能阻礙我們進行試驗的能力,並可能對我們的業務和財務狀況造成不利影響。
儘管英國被視為歐盟GDPR下的第三個國家之一,但歐盟委員會通過了一項有利於英國的充分性決定,允許數據從歐洲經濟區成員國轉移到英國,而不需要額外的保障措施。英國政府已經證實,從英國到歐洲經濟區的個人數據傳輸仍然是自由流動的。英國和歐盟在數據保護法某些方面的關係仍不清楚,也不清楚英國數據保護法律和法規在中長期將如何發展,以及進出英國的數據傳輸將如何長期受到監管。此外,英國政府還在英國立法程序中引入了數據保護和數字信息法案,或稱英國法案。英國法案的目的是改革英國退歐後的數據保護制度。如果獲得通過,英國法案的最終版本可能會進一步改變英國與歐洲經濟區數據保護制度之間的相似性,並威脅到歐盟委員會對英國充足率的決定。此外,歐洲經濟區成員國通過了國家法律,以實施可能部分偏離GDPR的GDPR。此外,歐洲經濟區成員國的主管當局對GDPR義務的解釋可能因國家不同而略有不同,因此我們預計歐洲經濟區不會在統一的法律環境中運作。歐盟GDPR和英國GDPR各自的條款和執法在未來進一步分化的可能性給我們帶來了額外的監管挑戰和不確定性。這種對未來英國法律法規及其與歐盟法律法規相互作用的缺乏清晰度,可能會增加我們處理個人數據以及我們的隱私和數據安全合規計劃的法律風險、複雜性和成本,並可能要求我們針對英國和歐洲經濟區實施不同的合規措施。
在美國和歐洲以外,我們設有CRO或以其他方式開展業務的許多司法管轄區也在考慮和/或已經制定了全面的數據保護立法。然而,我們可能會根據GDPR和適用的歐洲經濟區成員國和英國隱私法,在我們採取任何措施遵守它們的情況下招致責任、費用、成本和其他運營損失。
在我們在歐洲經濟區和英國處理個人數據的司法管轄區內,我們可能受到當地數據保護當局的監督,包括我們的業務活動涉及監控歐洲經濟區或英國的個人行為(例如,在進行臨牀試驗時)。我們依賴許多第三方提供我們的服務,其中一些第三方代表我們處理EEA和/或英國個人的個人數據。我們與每一家此類提供商訂立或打算訂立合約安排,根據這些安排,他們在合約上有義務只根據我們的指示處理個人資料,並進行或打算盡最大努力確保他們有足夠的技術和組織安全措施到位。
此外,某些健康隱私法、數據泄露通知法、消費者保護法和基因測試法可能直接適用於我們和/或我們合作者的運營,並可能對我們收集、使用和傳播個人健康信息施加限制。我們或我們的合作者可能獲得健康信息的患者,以及可能與我們共享此信息的提供者,可能擁有限制我們使用和披露信息的能力的法定或合同權利。我們可能需要花費大量資本和其他資源,以確保持續遵守適用的隱私和數據安全法律。聲稱我們侵犯了個人隱私權或違反了我們的合同義務,即使我們被發現沒有責任,辯護也可能是昂貴和耗時的,並可能導致負面宣傳,可能會損害我們的業務。
如果我們或第三方CMO、CRO或其他承包商或顧問未能遵守適用的聯邦、州/省或地方監管要求,我們可能會受到一系列監管行動的影響,這些監管行動可能會影響我們或我們的承包商開發和商業化我們的候選療法的能力,可能會損害或阻止我們能夠商業化的任何受影響療法的銷售,或者可能會大幅增加我們療法的開發、商業化和營銷的成本和支出。任何威脅或實際的政府執法行動也可能產生負面宣傳,並要求我們投入大量資源,否則這些資源可以用於我們業務的其他方面。越來越多地使用社交媒體可能會導致責任、數據安全遭到破壞或聲譽受損。
此外,我們受制於上述每項醫保法的州和國外同等法律,其中一些法律的範圍可能更廣,可能適用於無論付款人是誰。
如果我們或我們的第三方製造商和供應商未能遵守環境、健康和安全法律法規,我們可能會受到罰款或處罰,或產生可能對我們的業務成功產生不利影響的成本。
我們受到許多環境、健康和安全法律法規的約束,包括那些管理實驗室程序以及危險材料和廢物的處理、使用、儲存、處理和處置的法律和法規。我們的研究和開發活動涉及使用生物和危險材料,併產生危險廢物產品。我們通常與第三方簽訂合同,處理這些材料和廢物。我們無法消除這些材料的污染或傷害風險,這可能會導致我們的商業化努力、研發努力和業務運營中斷,環境破壞導致昂貴的清理,並根據適用的法律和法規對這些材料和指定廢物的使用、儲存、處理和處置承擔責任。儘管我們相信我們的第三方CMO在處理和處置這些材料時使用的安全程序大體上符合這些法律法規規定的標準,但我們不能保證情況確實如此,也不能消除這些材料意外污染或傷害的風險。在發生此類事件時,我們可能要對由此造成的任何損害負責,該責任可能超出我們的資源範圍,州、聯邦或其他適用機構可能會限制我們對某些材料的使用和/或中斷我們的業務運營。此外,環境法律法規復雜,變化頻繁,並有變得更加嚴格的趨勢。我們無法預測這種性質的任何變化的影響,也不能確定我們未來的合規。此外,為了遵守當前或未來的環境、健康和安全法律法規,我們可能會產生鉅額成本。這些現行或未來的法律法規可能會損害我們的研究、開發或生產努力。不遵守這些法律法規也可能導致鉅額罰款、處罰或其他制裁。
雖然我們維持工人補償保險以支付我們的成本和開支,但我們可能會因使用危險材料或其他與工作有關的傷害而導致員工受傷,但該保險可能無法為潛在的責任提供足夠的保險。我們不承保特定的生物廢物或危險廢物保險、工人補償或財產和傷亡及一般責任保險,包括因生物或危險廢物暴露或污染而產生的損害和罰款。
我們受到美國和某些外國進出口管制、制裁、禁運、反腐敗法律和反洗錢法律和法規的約束。遵守這些法律標準可能會削弱我們在國內和國際市場上的競爭能力。我們可能會因違規行為面臨刑事責任和其他嚴重後果,這可能會損害我們的業務。
我們受出口管制和進口法律法規的約束,包括美國出口管理條例、美國海關條例、由美國財政部外國資產管制辦公室實施的各種經濟和貿易制裁條例、1977年修訂的美國反海外腐敗法、美國聯邦法典第18篇第201節中包含的美國國內賄賂法規、美國旅行法、2001年美國愛國者法以及我們開展活動所在國家的其他州和國家的反賄賂和反洗錢法律。反腐敗法被廣泛解釋,禁止公司及其員工、代理人、承包商和其他合作者直接或間接地授權、承諾、提供或提供不正當的付款或任何其他有價值的東西給公共或私營部門的接受者。在未來,我們可能會在美國以外的地方聘請第三方進行臨牀試驗
一旦我們進入商業化階段和/或獲得必要的許可、許可證、專利註冊和其他監管批准,我們就有權將我們的產品銷往國外。我們還可能與政府機構或政府附屬醫院、大學和其他組織的官員和員工進行直接或間接的互動。我們可能要為員工、代理、承包商和其他合作者的腐敗或其他非法活動負責,即使我們沒有明確授權或實際瞭解這些活動。任何違反上述法律和法規的行為都可能導致重大的民事和刑事罰款和處罰、監禁、喪失進出口特權、取消資格、重新評估税收、違反合同和欺詐訴訟、名譽損害以及其他後果。
與員工事務、管理增長和運營事務相關的風險
我們高度依賴我們的關鍵人員,並預計將招聘新的關鍵人員。如果我們不能成功地吸引和留住高素質的人才,我們就可能無法成功地實施我們的商業戰略。
我們在競爭激烈的生物技術和製藥行業的競爭能力取決於我們吸引和留住高素質的管理、科學、醫療、銷售和營銷人員以及其他人員的能力。我們高度依賴我們的管理層、科學和醫務人員,包括我們的總裁和首席執行官、首席科學官、首席醫療官、首席財務官、首席法務官、首席人事官和首席業務官。失去任何高管、其他關鍵員工和其他科學和醫療顧問的服務,以及無法找到合適的替代者,都可能導致產品開發的延遲,並損害我們的業務。雖然我們預計,如果我們整合新任命的幹事和管理人員,我們將參與一個有序的過渡進程,但我們面臨與管理過渡有關的各種風險和不確定因素,包括將管理人員的注意力從業務關切上轉移,未能留住其他關鍵人員,或失去機構知識。
我們在馬薩諸塞州沃特敦的設施中開展業務。馬薩諸塞州地區是許多其他生物製藥公司以及許多學術和研究機構的總部。我們市場對技術人員的競爭非常激烈,可能會限制我們以可接受的條件聘用和留住高素質人員的能力,甚至根本不能。美國移民和工作授權法律法規的變化,包括那些限制科學和專業人才流動的法律法規,可能會受到政治力量和經濟活動水平的重大影響。如果移民或簽證法律法規的立法或行政變更損害了我們的招聘流程和目標或涉及非美國公民的項目,我們的業務可能會受到實質性的不利影響。
為了鼓勵有價值的員工留在我們公司,除了工資和現金獎勵外,我們還提供股票期權和其他股權獎勵,這些獎勵可以隨着時間的推移或基於里程碑的實現而授予。隨着時間的推移,授予我們員工的股權獎勵的價值可能會受到我們股價波動的重大影響,這些波動超出了我們的控制範圍,而且在任何時候都可能不足以抵消其他公司提供的更有利可圖的報價。對於根據里程碑成就授予的股權獎勵而言,情況可能也是如此。儘管我們努力留住有價值的員工,但我們的管理、科學和開發團隊的成員可能會在短時間內終止與我們的僱傭關係。雖然我們與我們的高管員工有僱傭協議,但這些僱傭協議規定可以隨意僱用,這意味着我們的任何高管員工都可以在任何時候離開我們的工作崗位,無論是否通知。我們的成功還有賴於我們有能力繼續吸引、留住和激勵高技能的初級、中級和高級管理人員,以及初級、中級和高級科學、醫療、一般和管理人員。
此外,我們還擁有科學和臨牀顧問,幫助我們制定發展和臨牀戰略。這些顧問不是我們的員工,可能與其他實體有承諾、諮詢或諮詢合同,這可能會限制他們對我們的可用性。此外,我們的顧問可能會與其他公司達成協議,幫助這些公司開發可能與我們競爭的產品或技術。
我們的內部計算機系統或我們的任何合作者、供應商、承包商或顧問的計算機系統可能會出現故障或遭遇安全漏洞,這可能會導致我們的產品開發計劃受到實質性破壞,並可能損害我們的聲譽或使我們承擔責任,並對我們的業務和財務業績產生不利影響。
我們的內部計算機系統以及任何合作者、供應商、承包商或顧問的計算機系統都容易受到計算機病毒、未經授權的訪問、自然災害、恐怖主義、戰爭以及電信和電氣故障的破壞。雖然到目前為止,我們還沒有經歷過任何重大系統故障、事故或安全漏洞,但如果發生此類事件並導致我們的運營中斷,可能會導致我們的開發計劃和業務運營的重大中斷,無論是由於我們的商業機密或其他專有信息的丟失,還是由於其他類似的中斷。例如,已完成或未來的臨牀試驗中的臨牀試驗數據丟失可能會導致我們的市場審批工作延遲,並顯著增加我們恢復或複製數據的成本。任何中斷或安全漏洞將導致我們的數據或應用程序丟失或損壞,或
如果不適當地披露機密或專有信息,我們可能會招致責任,我們的競爭地位可能會受到損害,我們候選產品的進一步開發和商業化可能會被推遲。此外,我們可能對我們存儲的第三方信息負有數據安全義務。未經授權訪問或使用任何此類第三方數據或信息可能會導致罰款或其他處罰,可能會影響我們與這些第三方的關係和我們的運營。
我們的平臺、系統和網絡的任何實際或感知的安全漏洞都可能損害我們的聲譽和品牌,使我們面臨訴訟和可能的責任風險,並要求我們花費大量資本和其他資源來應對和緩解安全漏洞帶來的問題。我們維持足夠的網絡犯罪和責任保險的能力可能會降低。一些司法管轄區已制定法律,要求公司在涉及某些類型的個人數據的數據安全漏洞時通知個人,我們與某些合作伙伴達成的協議要求我們在發生安全事件時通知他們。這些類型的強制性披露成本高昂,可能導致負面宣傳,並可能導致我們的合作伙伴對我們的數據安全措施的有效性失去信心。這些事件中的任何一項都可能損害我們的聲譽或使我們承擔責任,並對我們的業務和財務業績產生實質性和不利的影響。儘管我們維持網絡責任保險,但我們不能確定其承保範圍是否足以彌補實際發生的責任,或者我們是否會繼續以經濟合理的條款獲得保險,或者根本不能。
我們的員工、獨立承包商、供應商、主要調查人員、CRO和顧問可能從事不當行為或其他不當活動,包括不遵守監管標準和要求以及內幕交易法律。
我們面臨的風險是,我們的員工、獨立承包商、供應商、主要調查人員、CRO、CMO和顧問可能從事欺詐行為或其他非法活動。除其他外,這些當事人的不當行為可能包括:
•故意、魯莽或疏忽的行為或披露違反研究和試驗方案或FDA或類似外國監管機構規定的未經授權的活動;
•違反美國和國外的醫療欺詐和濫用法律法規;
•違反與我們普通股交易有關的美國聯邦證券法;以及
•未能準確報告財務信息或數據。
特別是,醫療保健行業的銷售、營銷和商業安排受到旨在防止欺詐、不當行為、回扣、自我交易和其他濫用行為的廣泛法律法規的約束。這些法律法規規範了廣泛的定價、折扣、營銷和促銷、銷售佣金、客户激勵計劃和其他業務安排。其他形式的不當行為可能涉及不當使用在臨牀試驗過程中獲得的信息,或在我們的臨牀前研究或臨牀試驗中創造虛假數據,這可能導致監管制裁,並對我們的聲譽造成嚴重損害。我們通過了適用於我們所有員工的商業行為和道德準則以及其他公司治理和合規文件、政策和章程。然而,並不總是能夠識別和阻止員工和其他第三方的不當行為。此外,我們為發現和防止此類活動而採取的預防措施可能無法有效控制未知或未管理的風險或損失,或保護我們免受因未能遵守這些法律或法規而引起的政府調查或其他行動或訴訟。此外,我們還面臨這樣的風險,即有人可能會指控欺詐或其他不當行為,即使沒有發生。如果針對我們提起任何此類訴訟,而我們未能成功地為自己辯護或維護我們的權利,這些訴訟可能會對我們的業務產生重大影響,包括施加民事、刑事和行政處罰、損害賠償、罰款、可能被排除在聯邦醫療保險、醫療補助和其他聯邦醫療保健計劃之外、合同損害、聲譽損害、利潤和未來收益減少、和/或削減我們的業務,其中任何一項都可能對我們的業務前景、財務狀況和運營結果產生不利影響。
與我們普通股相關的風險
如果我們決定在未來籌集更多資本,你的投資將受到稀釋。
我們未來可能會根據市場情況、戰略考慮和運營要求,選擇通過出售股票或其他可轉換為股票的證券來籌集額外資本。在一定程度上,我們通過這種方式籌集額外資本,我們的股東將被稀釋。未來我們普通股或其他股權證券的發行,或認為可能發生這種性質的出售,可能會對我們普通股的交易價格產生不利影響,並削弱我們通過未來發行股票或股權證券籌集資金的能力。無法預測未來普通股的出售或可供未來出售的普通股是否會對我們普通股的交易價格產生影響。
目前,我們的普通股在納斯達克全球精選市場上市。然而,在這樣的市場上可能沒有足夠的流動性來使您能夠出售您所持有的我們普通股。
目前,我們的普通股在納斯達克全球精選市場上市。如果我們股票的活躍交易市場不能持續,您可能無法快速或按市價出售您的股票。我們無法預測投資者對我們的興趣將在多大程度上導致維持一個活躍、流動性強的交易市場。此外,不活躍的市場還可能削弱我們通過出售普通股籌集資金的能力,並可能削弱我們達成戰略合作伙伴關係或以我們的普通股作為對價收購公司或產品的能力。
如果證券或行業分析師不發表或停止發表關於我們業務的研究或報告,或者如果他們對我們的股票發表不利或誤導性的意見,我們的股價和交易量可能會下降。
我們普通股的交易市場正在並將繼續受到行業或證券分析師發佈的關於我們、我們的業務或目標蛋白質降解空間的研究和報告的影響。我們無法控制這些分析師。不能保證現有的分析師將繼續提供研究報道,或者新的分析師將開始提供研究報道。儘管我們已經獲得了分析師的報道,但如果報道我們的任何分析師對我們、我們的商業模式、我們的知識產權或我們的股票表現發表了負面或誤導性的意見,或者如果我們的臨牀前研究和未來的臨牀試驗和運營結果未能滿足任何這些分析師的預期,我們的股價可能會下跌。如果其中一位或多位分析師停止對我們的報道,或未能定期發佈有關我們的報告,我們可能會失去在金融市場的可見度,這反過來可能導致我們的股價或交易量下降。
我們普通股的價格可能會波動很大,這可能會給我們普通股的購買者帶來重大損失。
我們普通股的股票交易價格一直不穩定,並可能繼續受到各種因素的廣泛波動,其中一些因素是我們無法控制的。股票市場,特別是較小的生物製藥公司的市場,經歷了極端的波動,這種波動往往與特定公司的經營業績無關。由於這種波動,你可能無法以或高於你獲得普通股的價格出售你的普通股。我們普通股的市場價格可能受到許多因素的影響,包括:
•有競爭力的產品或技術的成功程度或護理標準的改變;
•我們的候選產品或競爭對手的臨牀前研究和臨牀試驗結果;
•我們臨牀開發活動的時間和進展,以及我們發佈臨牀試驗數據的時間;
•美國和其他國家的法規或法律發展;
•與專利申請、已頒發的專利或者其他專有權利有關的開發或者糾紛;
•關鍵人員的招聘或離職;
•與我們的任何候選產品或臨牀開發計劃相關的費用水平以及我們持有的現金、現金等價物和有價證券的價值;
•我們努力發現、開發、獲取或許可其他候選技術或產品的結果;
•關於財務結果、發展時間表或證券分析師建議的估計的實際或預期變化;
•我們的財務業績或那些被認為與我們相似的公司的財務業績差異;
•改變醫療保健支付制度的結構;
•製藥和生物技術部門的市場狀況;
•公共衞生危機、流行病和流行病的影響,如最近的新冠肺炎大流行;
•一般經濟、行業和市場狀況;以及
•“風險因素”一節中描述的其他因素。
如果上述任何因素被認為可能對我們的業務、前景或運營產生負面影響,或者如果我們的經營業績低於投資者或證券分析師的預期,我們普通股的價格可能會大幅下跌。在過去,隨着一家公司證券市場價格的波動,證券
針對該公司的集體訴訟經常被提起。如果對我們提起這種性質的訴訟,可能會導致我們產生鉅額費用來為這些索賠辯護,並轉移管理層的注意力和資源,這可能會嚴重損害我們的業務、財務狀況、運營結果和前景。此外,我們的董事和高級職員責任保險成本可能會因此類訴訟而增加,在我們的保險公司被要求向我們提供任何保險之前,我們的保險免賠額可能會很大。
我們在使用我們籌集的資本方面擁有廣泛的自由裁量權,可能無法有效地使用我們的資本。
我們的管理層在運用我們之前融資的淨收益方面擁有廣泛的自由裁量權,包括我們的首次公開募股和後續公開發行,並可以將所得資金用於不會改善我們的運營業績或提高我們普通股價值的方式。如果我們的管理層未能有效地使用這些資金,可能會導致財務損失,這可能會對我們的業務產生不利影響,導致我們的普通股價格下跌,並推遲我們候選產品的開發。在使用之前,我們可能會將我們融資活動的淨收益以不產生收入或貶值的方式進行投資。
我們的高管、董事和主要股東將有能力控制或顯著影響提交給股東批准的事項。
我們的高管和董事,再加上我們的股東,他們通過向美國證券交易委員會提交的文件報告説,他們總共持有我們已發行普通股的5%以上,從而實惠地擁有我們相當大比例的股份。因此,我們的高管和董事,加上我們超過5%的股東,有能力通過這一所有權地位控制我們。這些股東如果共同行動,將繼續控制提交給我們股東批准的事項,以及我們的管理和事務。例如,如果這些人選擇一起行動,他們將控制董事的選舉和對我們所有或幾乎所有資產的任何合併、合併或出售的批准。所有權控制的這種集中可能:
•推遲、推遲或阻止控制權的變更;
•鞏固我們的管理層和董事會;或
•妨礙涉及我們的其他股東可能希望的合併、合併、接管或其他業務合併。
我們的章程文件和特拉華州法律中的反收購條款可能會推遲或阻止控制權的變更,這可能會限制我們普通股的市場價格,並可能阻止或挫敗我們的股東更換或罷免我們目前管理層的嘗試。
我們修訂和重述的公司註冊證書以及修訂和重述的章程中包含的條款可能會推遲或阻止我們公司的控制權變更或我們的股東可能認為有利的董事會變動。其中一些規定包括:
•董事會分為三個級別,交錯三年任期,其結果是不是所有董事會成員都將一次選舉產生;
•禁止股東通過書面同意採取行動,其結果是,所有股東行動都必須在我們的股東會議上採取;
•要求股東特別會議只能由董事會根據當時在任董事的多數贊成票通過的決議召集;
•股東提名和提名進入董事會的事先通知要求;
•要求我們的股東不得罷免我們的董事會成員,除非是出於法律要求的任何其他投票,並獲得當時有權在董事選舉中投票的我們有表決權股票中不少於三分之二的流通股的批准;
•要求批准不少於三分之二的有表決權股票的全部流通股,以通過股東行動修訂任何附例或修訂公司註冊證書的特定條款;以及
•董事會在未經股東批准的情況下,按照董事會決定的條款發行優先股的權力,優先股可以包括高於普通股持有人權利的權利。
此外,由於我們是在特拉華州註冊成立的,我們受特拉華州公司法第203條的規定管轄,該條款可能禁止股東擁有我們已發行有表決權股票的15%或更多的某些業務合併。這些反收購條款和我們修訂和重述的證書中的其他條款
成立公司以及修訂和重述章程可能會使股東或潛在收購者更難獲得對我們董事會的控制權,或者發起當時的董事會反對的行動,還可能推遲或阻礙涉及我們公司的合併、收購要約或代理權之爭。這些規定還可能阻止代理權競爭,並使您和其他股東更難選舉您選擇的董事或導致我們採取您希望的其他公司行動。任何延遲或阻止控制權變更、交易或董事會變動都可能導致我們普通股的市場價格下跌。
作為一家上市公司,我們將繼續產生額外的成本,我們的管理層將需要投入大量時間在合規倡議和公司治理實踐上。
作為一家上市公司,我們將繼續承擔大量的法律、會計和其他費用,而作為一傢俬人公司,我們不需要這樣做。2002年的薩班斯-奧克斯利法案,或薩班斯-奧克斯利法案,多德-弗蘭克華爾街改革和消費者保護法,納斯達克股票市場有限責任公司的上市要求和其他適用的證券規則和法規對上市公司提出了各種要求,包括建立和維持有效的披露、財務控制和公司治理做法。我們的管理層和其他人員在這些合規倡議上投入了大量時間。此外,這些規則和法規增加了我們的法律和財務合規以及保險成本,並使一些活動更加耗時和昂貴。例如,這些規章制度使我們獲得董事和高級管理人員責任保險變得更加困難和昂貴,這反過來可能會使我們更難吸引和留住合格的董事會成員。
我們不斷評估這些規章制度,不能總是預測或估計我們可能產生的額外成本金額或這些成本的時間。這些規則和條例也常常受到不同的解釋,在許多情況下是因為它們缺乏特殊性,因此,隨着監管機構和理事機構提供新的指導意見,它們在實踐中的適用可能會隨着時間的推移而演變。這可能導致關於遵守事項的持續不確定性,以及不斷修訂披露和治理做法所需的更高成本。
根據《薩班斯-奧克斯利法案》第404條或第404條,我們必須提交一份管理層關於我們財務報告的內部控制的報告。然而,作為一家“較小的報告公司”,我們將不會被要求包括由我們的獨立註冊會計師事務所出具的財務報告內部控制的認證報告,直到我們不再是一家較小的報告公司為止。在截至2023年12月31日的財政年度結束時,我們符合《1934年證券交易法》(修訂後的《證券交易法》)或《交易法》所定義的“非加速申報公司”和“較小的報告公司”的資格。我們遵守第404條的規定,就必須產生大量的會計費用,並花費大量的管理努力。
我們將需要繼續提供內部資源,可能聘請外部顧問,並通過詳細的工作計劃,以評估和記錄財務報告內部控制的充分性,繼續酌情采取步驟改進控制程序,通過測試驗證控制措施是否如文件所述發揮作用,並實施財務報告內部控制的持續報告和改進程序。儘管我們做出了努力,但我們仍有可能在規定的時間框架內或根本無法得出結論,即我們對財務報告的內部控制是有效的,符合第404條的要求。此外,我們不能向您保證,我們過去或未來採取的措施將防止未來在財務報告內部控制方面出現重大缺陷或重大缺陷。如果我們在未來發現一個或多個重大弱點,可能會導致金融市場的不良反應,並限制我們未來進入資本市場的機會,因為我們對我們的精簡綜合財務報表的可靠性失去了信心。
我們修訂和重述的章程指定特定法院作為可能由我們的股東發起的某些訴訟的獨家論壇,這可能限制我們的股東獲得有利的司法論壇處理與我們的糾紛的能力。
根據我們經修訂和重述的章程,除非我們書面同意選擇其他法院,否則特拉華州衡平法院是針對以下情況的州法律索賠的唯一和專屬法院:(i)代表我們提起的任何衍生訴訟或法律程序;(ii)任何聲稱我們的任何董事、高級職員或其他僱員違反對我們或我們的股東負有的信託責任的訴訟;(iii)因本協議或本公司服務所引起或與其有關的任何爭議應向人民法院提起訴訟,並以中華人民共和國法律為管轄法律。或(v)主張受內部事務原則所管限的申索的任何訴訟。我們在我們的修訂和重述章程中將此條款稱為特拉華州論壇條款。特拉華州法院條款不適用於根據1933年《證券法》(經修訂)、《證券法》或1934年《交易法》(經修訂)或《交易法》產生的任何訴訟事由。
我們經修訂和重述的章程進一步規定,除非我們書面同意選擇替代法院,否則美國馬薩諸塞州地區法院應為解決任何投訴的唯一和專屬法院。
根據《證券法》提出訴訟理由,因為我們的總部位於馬薩諸塞州沃特敦。我們在經修訂和重述的章程中將此條款稱為聯邦論壇條款。此外,我們修訂和重述的章程規定,任何購買或以其他方式獲得我們股本股份的任何權益的個人或實體均被視為已通知並同意特拉華州論壇條款和聯邦論壇條款。
特拉華州法院規定和聯邦法院規定在我們的修訂和重述的章程可能會對股東在追求任何此類索賠額外的訴訟費用。此外,這些法院選擇條款可能會限制我們的股東在司法法院提出索賠的能力,他們認為這有利於與我們或我們的董事,管理人員或員工的糾紛,並可能會阻止對我們和我們的董事,管理人員提起訴訟,即使訴訟,如果成功,可能有利於我們的股東。此外,儘管特拉華州最高法院於二零二零年三月裁定,根據特拉華州法律,聲稱要求根據《證券法》向聯邦法院提出申索的聯邦法院選擇條文“表面有效”,但其他法院是否會執行我們的聯邦法院條文仍存在不確定性。如果聯邦法院條款被發現無法執行,我們可能會產生與解決此類問題相關的額外費用。《聯邦法院條款》還可能對聲稱該條款不可執行或無效的股東徵收額外的訴訟費用。特拉華州衡平法院和美國馬薩諸塞州地方法院也可能做出與其他法院不同的判決或結果,包括考慮提起訴訟的股東所在地或以其他方式選擇提起訴訟的法院,此類判決可能對我們的股東更有利或更不利。
由於我們預計在可預見的未來不會對我們的股本支付任何現金股息,因此資本增值(如果有的話)將是您唯一的收益來源。
我們從未宣佈或支付過我們股本的現金股息。我們目前打算保留我們未來的所有收益,如果有的話,為我們業務的增長和發展提供資金。因此,我們普通股的資本增值(如果有的話)將是您在可預見的未來唯一的收益來源。
市況不穩定及經濟及市況下滑可能對我們的業務、財務狀況及股價造成嚴重不利後果。
我們的經營業績可能會受到全球經濟及全球金融市場的整體狀況的不利影響。例如,全球金融危機造成資本和信貸市場的極端波動和混亂。同樣,COVID-19大流行及近期地緣政治緊張局勢(包括俄羅斯與烏克蘭及以色列與哈馬斯之間的衝突)相關的重大波動,已導致資本及信貸市場出現重大不穩定及幹擾。美國和海外的全球經濟狀況繼續波動和不確定。我們的業務可能會受到市場經濟及政治變動的不利影響,包括通脹率上升、利率上升、供應鏈中斷、經濟衰退、貿易限制、關税增加或潛在新關税,以及美國實施的經濟禁運。嚴重或長期的經濟衰退可能會對我們的業務造成各種風險,包括對我們候選產品的需求減弱,也可能影響我們在需要時以可接受的條款籌集額外資金的能力。我們的整體業務策略可能會受到任何此類性質的經濟衰退、動盪的商業環境或持續不可預測及不穩定的市場狀況的不利影響。如果目前的股票和信貸市場惡化,或沒有改善,可能會使任何必要的債務或股票融資更加困難,成本更高,稀釋作用更大,或根本無法獲得。
未能及時以有利的條件獲得任何必要的融資可能會對我們的增長戰略、財務業績和股價產生不利影響,並可能要求我們推遲、修改或放棄臨牀開發計劃。此外,我們目前的一家或多家服務提供商、製造商和其他合作伙伴可能無法在經濟困難時期生存下來,這可能直接影響我們按時和按預算實現運營目標的能力。
從歷史上看,證券集體訴訟通常是在一家公司的證券市場價格下跌後對其提起的。這一風險與我們特別相關,因為生物技術和製藥公司近年來經歷了大幅的股價波動。如果我們被起訴,可能會導致鉅額成本和管理層注意力和資源的轉移,這可能會對我們的業務前景、財務狀況和運營結果產生不利影響。
影響金融服務業的不利事態發展,例如涉及流動性、違約或金融機構或交易對手方不履行義務的實際事件或擔憂,可能會對公司當前和預期的業務運營及其財務狀況和運營結果產生不利影響。
涉及流動性有限、違約、業績不佳或影響金融服務業或金融服務業其他公司的其他不利發展的實際事件,或對任何此類事件或其他類似風險的擔憂或傳言,過去和未來可能會導致整個市場的流動性問題。例如,2023年3月10日,硅谷銀行(SVB)
由加州金融保護和創新部關閉,該部指定聯邦存款保險公司或FDIC為接管人。同樣,2023年3月12日,Signature Bank和Silvergate Capital Corp.分別被捲入破產管理程序。儘管財政部、美聯儲和聯邦存款保險公司的一份聲明指出,SVB的所有儲户在關閉僅一個工作日後就可以取用他們的所有資金,包括無保險存款賬户中的資金、信貸協議下的借款人、信貸協議下的借款人、信貸協議下的信用證和某些其他金融工具、簽名銀行或FDIC接管的任何其他金融機構,但可能無法提取其中未提取的金額。雖然這些銀行倒閉都沒有給本公司帶來重大風險,但如果我們的任何貸款人或任何此類工具的交易對手被接管,我們可能無法獲得這些機構持有的資金。此外,如果我們的任何合作伙伴、供應商或與我們有業務往來的其他方無法根據此類工具或與此類金融機構的貸款安排獲得資金,這些各方向我們支付債務或達成要求向我們支付額外款項的新商業安排的能力可能會受到不利影響。在這方面,更廣泛的金融服務業對流動性的擔憂依然存在不確定性。類似的影響過去也曾發生過,例如在2008-2010年金融危機期間。
通貨膨脹和利率的快速上升導致之前發行的利率低於當前市場利率的政府債券的交易價值下降。儘管財政部、聯邦存款保險公司和聯邦儲備委員會已經宣佈了一項計劃,向以金融機構持有的某些此類政府證券為擔保的金融機構提供高達250億美元的貸款,以降低出售此類工具可能造成的潛在損失的風險,但廣泛存在的客户提款需求或金融機構對立即流動性的其他需求可能會超出此類計劃的能力。此外,不能保證美國財政部、聯邦存款保險公司和聯邦儲備委員會在未來其他銀行或金融機構關閉的情況下會提供未投保資金的渠道,或者他們會及時這樣做。
我們定期評估我們認為必要或適當的銀行和其他關係,包括確保我們在這些關係中有適當的多元化。儘管如此,我們獲得的資金來源和其他信貸安排足以為我們目前和預計的未來業務運營提供資金或資本化,可能會受到影響本公司、與本公司直接簽訂信貸協議或安排的金融機構、或整個金融服務業或整體經濟的因素的嚴重影響。除其他外,這些因素可能包括流動性緊張或失敗、履行各類金融、信貸或流動資金協議或安排下的義務的能力、金融服務業或金融市場的中斷或不穩定,或對金融服務業公司前景的擔憂或負面預期。這些因素可能涉及與本公司有財務或業務關係的金融機構或金融服務行業公司,但也可能包括涉及金融市場或一般金融服務行業的因素。
項目1B。未解決的員工評論。
不適用。
項目1C。網絡安全。
網絡風險管理與策略
在董事會審計委員會的監督下,我們實施並保持了持續的網絡安全風險管理計劃,重點是識別、評估和緩解網絡風險。我們與多家第三方供應商接洽,這些供應商提供從持續的安全諮詢服務到安全監控和響應管理的各種服務。此外,我們還有一個評估和審查第三方供應商和服務提供商網絡安全做法的程序,包括酌情使用供應商問卷和合同安全要求。除了這些努力外,我們還實施了持續的企業風險管理計劃,其中包括旨在識別、評估和應對網絡安全風險的流程。我們的網絡安全工作遵循行業標準,包括由網絡安全技術支持的定期、有針對性的風險評估,這些技術包括旨在監控、識別和應對網絡安全風險的第三方安全解決方案和監控工具。
此外,作為一家上市公司,我們受到圍繞我們的內部控制的各種監管要求的約束,包括我們對我們的信息技術系統的控制及其對我們財務報表或系統的影響。我們已聘請第三方供應商向我們提供有關遵守這些要求的建議,包括與網絡安全相關的控制措施,以及降低相關風險的策略。如果我們要找出任何構成網絡安全風險的控制缺陷,將向首席財務官和審計委員會報告,並酌情提出糾正行動計劃。
與網絡安全風險相關的治理
我們的網絡風險管理計劃及相關運營和流程由我們的董事信息技術部管理,並與法律和人力資源團隊進行協商。目前,董事的信息技術職位由擁有17年以上網絡安全、信息技術和系統工程經驗的個人擔任。信息技術部董事向首席人事官報告。
信息技術部董事定期與首席人事官會面,監測和審查我們網絡安全風險管理進程的成果,並討論和處理與網絡安全風險管理戰略相關的事項。信息技術部與首席法務官和首席人事官一起,向審計委員會提交定期報告,審計委員會負責審查和監督公司的風險管理流程,包括網絡安全風險。首席財務官、首席人事官和首席法務官和/或法律團隊的其他高級成員參加審計委員會會議,審計委員會會議通常由首席財務官領導,以及董事會全體會議。
我們的企業風險管理流程由首席法務官和首席財務官監督。在收集企業風險信息時,網絡安全被具體列為風險類別,我們的企業風險評估過程的結果,包括與網絡安全有關的風險,也會定期與審計委員會和高級管理層討論。
項目2.財產
我們目前在馬薩諸塞州沃特敦租賃了約111,611平方英尺的辦公和實驗室空間,租約將於2032年到期。根據將於2025年到期的轉租安排,我們在馬薩諸塞州沃特敦轉租了約31,039平方英尺的辦公室和實驗室空間。我們相信,在可預見的未來,我們的設施足以滿足我們目前的需求,並將在需要時提供適當的額外空間。
項目3.法律訴訟
我們目前不是任何實質性法律程序的一方。我們可能會不時地受到各種法律程序和索賠的影響,這些訴訟和索賠是在我們正常的業務活動過程中出現的。無論結果如何,由於辯護和和解成本、管理資源轉移等因素,訴訟可能會對我們產生實質性的不利影響。
第4項礦山安全信息披露
不適用。
第II部
第5項:註冊人普通股市場、相關股東事項和發行人購買股權證券。
市場信息
我們的普通股自2020年10月2日起在納斯達克全球精選市場公開交易,交易代碼為CCCC。在此之前,我們的普通股沒有公開市場。
持有者
截至2023年2月15日,我們的普通股約有60名登記持有者。這一數字不包括其股份由街道上的被提名者持有的實益所有者。
分紅
自成立以來,我們從未宣佈或支付過任何現金股利。我們打算保留未來的收益(如果有的話),為我們業務的運營和擴張提供資金,在可預見的未來不會向普通股持有者支付任何現金股息。
根據股權補償計劃獲授權發行的證券
關於我們的股權補償計劃的信息通過引用第12項併入本文,某些實益擁有人的擔保所有權以及管理層和相關股東的事項,本年度報告的表格10-K。
最近出售的未登記證券
沒有。
發行人或關聯購買人購買股權證券
於截至2023年12月31日止年度內,吾等或任何關聯買家或任何代表吾等或代表關聯買家行事的人士並無購買本公司普通股的任何股份。
第六項。[已保留]
不適用。
第七項:管理層對財務狀況和經營成果的討論和分析。
以下對我們財務狀況和經營結果的討論和分析應與我們的合併財務報表以及本年度報告中其他部分以Form 10-K格式包含的這些報表的相關附註一起閲讀。除了歷史財務信息外,以下討論和分析還包含涉及風險、不確定性和假設的前瞻性陳述。為便於列報,本文中的一些數字已進行了四捨五入。我們的實際結果可能與這些前瞻性陳述中預期的結果大不相同,這是許多因素的結果,包括本年度報告10-K表中第1A項(風險因素)下討論的那些因素。
概述
我們是一家臨牀階段的生物製藥公司,致力於兑現TPD科學的承諾,創造改變患者生活的新一代小分子藥物。通過利用我們專有的魚雷平臺,我們能夠有效地設計和優化小分子蛋白質降解器,通過利用人體破壞不需要的蛋白質的自然過程,對其預期目標高度活躍。我們相信,我們的新型口服產品候選產品有潛力克服抑制劑經常出現的耐藥性,瞄準目前“無法下藥”的靶點,並改善患者的結果。
我們的臨牀前和臨牀流水線包括MonoDAC和BiDAC降解物,這些降解物有可能針對各種癌症和其他適應症的致病蛋白,用於未滿足患者需求的領域。我們全資擁有的流水線專注於腫瘤學,我們的合作戰略使我們能夠探索其他適應症。
我們最先進的候選產品CFT7455是一種口服生物可用MonoDAC降解劑,稱為IKZF1和IKZF3,目前正處於多發性骨髓瘤(MM)和非霍奇金淋巴瘤(NHL)的臨牀開發中。以IKZF1和IKZF3為靶點有很強的機制基礎和明確的生物學定義,以一種新型降解劑為靶標有可能解決一個重要的未得到滿足的需求。在我們的臨牀前研究中,這種候選產品已經展示了具有良好藥理學特性的有效和選擇性的蛋白質降解。2021年6月,我們為該候選產品啟動了首個人類1/2期臨牀試驗。2021年8月,美國食品和藥物管理局(FDA)批准CFT7455為治療MM的孤兒藥物。2023年12月,我們
提供了CFT7455階段1/2試驗的劑量遞增部分的陽性臨牀數據,作為單一療法並聯合地塞米松治療MM。我們繼續推進正在進行的MM和NHL的1/2階段臨牀試驗。
我們的下一個最先進的候選產品CFT1946是一種口服生物可用BiDAC降解劑,旨在有效和選擇性地對抗BRAF V600X突變蛋白,用於治療黑色素瘤、非小細胞肺癌、結直腸癌和其他含有這種突變的惡性腫瘤。在臨牀前研究中,CFT1946在CRC和NSCLC BRAF V600X異種移植模型以及黑色素瘤患者來源的異種移植或PDX,BRAF抑制劑耐藥模型中比標準護理療法更有效。此外,CFT1946在臨牀前轉移性黑色素瘤中樞神經系統(CNS)模型中是活躍的。2023年1月,我們啟動了CFT1946的首個人類1/2期臨牀試驗,用於治療BRAF V600X突變實體腫瘤,包括非小細胞肺癌、結直腸癌和黑色素瘤。2024年1月,我們分享了正在進行的1/2階段試驗的前兩個劑量升級隊列的PK和PD數據,顯示了劑量比例暴露和口服生物利用度,這與BRAF的深度降解有關。我們繼續進行BRAF V600X癌症的1/2期臨牀試驗,包括黑色素瘤、非小細胞肺癌和結直腸癌。
此外,我們正在開發CFT8919,這是一種口服生物利用、變構、突變選擇性的表皮生長因子受體或EGFR的BiDAC降解劑,在非小細胞肺癌中具有L858R突變。在體外的臨牀前研究中,CFT8919對耐受EGFR抑制的突變(包括L858R-C797S、L858R-T790M和L858R-T790M-C797S)顯示出與L858R單一突變相同的抗增殖活性。2023年5月,我們與Betta Pharma簽訂了一項許可和合作協議,就CFT8919在內地中國、香港特別行政區、澳門特別行政區和臺灣的開發和商業化進行合作,我們保留在世界其他地區開發和商業化CFT8919的權利。此外,2023年6月,FDA批准了CFT8919的研究新藥或IND申請,2023年12月,倍他醫藥獲得了中國領導的國家醫療產品管理局對CFT8919的臨牀試驗申請或CTA批准。
除了這些最初的候選產品外,我們還針對我們自己的專利流水線和我們與默克、生物遺傳和羅氏合作開發的流水線開發了針對臨牀驗證和目前不可用藥的目標的新降解劑,從而進一步使我們的流水線多樣化。我們設計的降解物在臨牀前研究中成功地實現了血腦屏障的穿透,這是開發有可能治療腫瘤學腦轉移以及神經退行性疾病等治療領域的藥物的關鍵一步。我們還相信,在許多治療領域和適應症中,利用我們的魚雷平臺開發新型降解劑可能是有利的。
最新發展動態
2024年1月9日,我們宣佈了2024年要根據我們的戰略計劃執行的業務優先事項。由於這些修訂的優先事項,我們重組了我們的業務,並削減了大約30%的員工。此外,我們宣佈通過公司的市場(或自動取款機)機制,以5.42美元的平均購買價出售13,686,743股普通股,淨收益為7180萬美元。
2024年1月4日,我們宣佈完成根據先前宣佈的與Betta Pharma的全資子公司達成的股票購買協議所設想的交易。收盤時,我們以每股4.49美元的收購價出售了Betta Pharma的一家附屬公司的5,567,928股普通股,總投資額為2500萬美元。收購價格較截至2023年5月29日簽訂股票購買協議前兩個工作日的60個交易日成交量加權平均值溢價25%。
2023年12月12日,我們公佈了CFT7455作為單一療法並聯合地塞米松治療MM的1/2期試驗的劑量遞增部分的陽性臨牀數據。我們繼續推進MM和NHL正在進行的1/2期臨牀試驗。
2023年12月12日,我們宣佈與默克公司達成獨家許可和合作協議,開發降解物-抗體結合物,簡稱DAC,這是一種新興的方法,旨在選擇性地靶向和中和癌細胞中的致病蛋白。
2023年11月20日,我們宣佈任命歐文·休斯為董事會成員。
2023年11月1日,我們宣佈了一項投資組合優先級決定,不會將CFT8634推進到臨牀試驗的第一階段劑量升級部分。
經營成果的構成部分
收入
到目前為止,我們還沒有從產品銷售中獲得任何收入,在可預見的未來也不會從產品銷售中獲得任何收入。到目前為止,我們的收入來自研究合作和許可協議。我們根據每項協議確認預期業績期間的收入。我們希望我們的
未來幾年的收入將主要來自我們目前的合作協議和我們未來可能達成的任何額外合作。到目前為止,我們還沒有收到任何現有合作協議下的任何版税。
有關我們與默克、Betta Pharma、羅氏、Biogen和Calico Life Science LLC或Calico的合作協議的説明,請參閲注8,協作和許可協議在本年度報告10-K表格中的合併財務報表中。
研發費用
研究和開發費用主要包括我們的研究活動產生的成本,包括我們的發現努力和我們的候選產品開發,包括:
•從事研發職能的人員的工資、福利和其他相關費用,包括基於股票的薪酬費用;
•根據與第三方的協議發生的費用,包括合同研究組織和代表我們進行研究、臨牀前和臨牀活動的其他第三方,以及製造我們用於臨牀前和臨牀試驗的候選產品的第三方;
•外部顧問的費用,包括他們的費用和相關的旅費;
•臨牀前研究和臨牀試驗的實驗室用品和購置材料的費用;
•與CFT8634的逐步關閉活動相關的成本;
•與設施有關的費用,包括設備的直接折舊費用、設施租金和維修費及其他業務費用的分配費用;
•第三方許可費。
我們按所發生的費用來支付研究和開發費用。外部開發活動的成本是根據對完成具體任務的進度的評估,使用我們的供應商提供給我們的信息確認的。這些活動的付款是基於個別協議的條款,這些條款可能與發生的成本模式不同,並在我們的合併財務報表中作為預付或應計研究和開發費用反映。未來將收到用於研究和開發活動的貨物或服務的不可退還的預付款被記錄為預付費用,並在相關貨物交付或提供服務時支出。
我們預計,由於我們計劃的臨牀前和臨牀開發活動,我們的研發費用將繼續大幅增加。
一般和行政費用
一般和行政費用主要包括行政、財務、法律、業務發展和行政職能人員的工資和其他相關成本,包括基於股票的薪酬。一般和行政費用還包括與公司事務有關的法律費用;會計、審計、税務和諮詢服務的專業費用;保險費;差旅費用;與設施有關的費用,其中包括直接折舊成本、設施租金和維護分配費用以及其他運營成本。
我們預計未來我們的一般和行政費用將繼續增加,以支持更多的研發活動。這些增長可能包括與僱用更多人員有關的更高成本;向外部顧問、律師和會計師支付的費用;以及投資者和公關費用。
其他收入(費用)
其他收入(費用),淨額主要包括:
•長期債務的利息支出和攤銷;
•債務清償損失;以及
•從我們的現金、現金等價物和有價證券中賺取的利息和其他收入,以及有價證券的折扣增加。
行動的結果
截至2023年12月31日和2022年12月31日的年度比較
收入
我們的協作和許可協議收入包括以下內容(以千計):
| | | | | | | | | | | | | |
| 截至十二月三十一日止的年度, |
| 2023 | | 2022 | | |
| 來自協作協議的收入: | | | | | |
| 生物遺傳協議 | $ | 10,623 | | | $ | 19,214 | | | |
| 羅氏協議 | 9,063 | | | 4,939 | | | |
| | | | | |
| 印花布協議 | 1,070 | | | 6,943 | | | |
| 來自協作協議的總收入 | $ | 20,756 | | | $ | 31,096 | | | |
與截至2022年12月31日的年度相比,截至2023年12月31日的年度收入減少了1030萬美元,主要原因是:
•根據Biogen協議確認的收入減少860萬美元,主要是由於Biogen協議的研究期限將於2023年6月結束,研究期限結束後的一段時間內將對提名目標進行額外的研究活動,所有這些都是Biogen協議所預期的;以及
•根據Calico協議確認的收入減少590萬美元,原因是截至2023年3月31日,履約義務和交易價格完全滿足。
這部分被羅氏協議項下確認的410萬美元收入增加所抵消,這是由於我們在2023年完成了一項指定目標的研究活動。
研究和開發費用
下表彙總了我們的研發費用(單位:千):
| | | | | | | | | | | | | |
| 截至十二月三十一日止的年度, |
| 2023 | | 2022 | | |
| 研發費用: | | | | | |
| 人員費用 | $ | 42,706 | | | $ | 41,068 | | | |
| 臨牀前和開發費用 | 28,496 | | | 42,033 | | | |
| 臨牀費用 | 19,238 | | | 10,643 | | | |
| 設施和用品 | 13,594 | | | 13,978 | | | |
| 專業費用 | 8,605 | | | 7,047 | | | |
| 知識產權及其他費用 | 5,067 | | | 3,072 | | | |
| | | | | |
| 研發費用總額 | $ | 117,706 | | | $ | 117,841 | | | |
截至2023年12月31日止年度的研發費用較截至2022年12月31日止年度減少10萬美元,主要是由於與進展到臨牀階段的額外項目相關的臨牀前和開發成本減少1350萬美元。
這些減少額被以下因素部分抵消:
•由於正在進行的CFT 7455和CFT 1946的1/2期臨牀試驗以及CFT 8634的1期臨牀試驗,臨牀費用增加了860萬美元;
•知識產權和其他公司支出增加200萬美元;
•員工費用增加170萬美元,代表工資和福利成本,這是由於增加員工人數以支持我們的臨牀項目;以及
•專業費用增加160萬美元。
一般和行政費用
下表彙總了我們的一般和行政費用(以千為單位):
| | | | | | | | | | | | | |
| 截至十二月三十一日止的年度, |
| 2023 | | 2022 | | |
| 一般和行政費用: | | | | | |
| 人員費用 | $ | 30,244 | | | $ | 30,546 | | | |
| 專業費用 | 7,365 | | | 8,576 | | | |
| 設施和用品 | 2,076 | | | 2,371 | | | |
| 其他費用 | 2,396 | | | 1,296 | | | |
| 一般和行政費用總額 | $ | 42,081 | | | $ | 42,789 | | | |
與截至2022年12月31日的年度相比,截至2023年12月31日的年度一般和行政費用減少了70萬美元,主要是由於專業費用減少了120萬美元,人員和設施成本減少了60萬美元,但其他費用增加了110萬美元,部分抵消了這一下降。
其他收入(費用)
下表彙總了我們的其他收入(支出)(單位:千):
| | | | | | | | | | | | | |
| 截至十二月三十一日止的年度, |
| 2023 | | 2022 | | |
| 其他收入(費用),淨額 | | | | | |
| 利息和其他收入,淨額 | $ | 9,812 | | | $ | 3,575 | | | |
| 長期債務關聯方的利息支出和攤銷 | (1,373) | | | (2,216) | | | |
長期債務清償損失 | (621) | | | — | | | |
| 其他收入(費用)合計,淨額 | $ | 7,818 | | | $ | 1,359 | | | |
與截至2022年12月31日的年度相比,截至2023年12月31日的年度其他收入(支出)增加650萬美元,原因是2023年我們的投資利息增加導致利息和其他收入增加620萬美元。
所得税費用
在截至2023年12月31日的一年中,所得税支出為130萬美元,而截至2022年12月31日的一年中沒有所得税支出。這主要是由於向中國税務機關支付的1,000,000美元預扣税款與根據Betta許可協議收到的預付款有關,以及與一月份收到的Betta里程碑付款有關的預扣税款2萬美元(見附註8及附註12)。
流動資金和資本資源
流動資金來源
自成立以來,我們發生了重大的運營虧損。我們預計,在可預見的未來,隨着我們項目的繼續發展,我們將產生鉅額費用和運營虧損。我們目前沒有任何批准的產品,也從未從產品銷售中獲得任何收入。到目前為止,我們主要通過出售優先股、公開發行我們的普通股以及合作伙伴的付款來為我們的運營提供資金。自.起2023年12月31日,我們擁有約2.817億美元的現金、現金等價物和有價證券。
2021年11月,我們向美國證券交易委員會提交了自動生效的S-3表格登記説明書,登記了數量不詳的普通股、優先股、債務證券、權證和/或其任意組合的單位的發行、發行和銷售。我們同時與作為銷售代理的Cowen and Company,LLC簽訂了一項股權分配協議,根據註冊聲明和與註冊聲明或ATM計劃一起提交的相關招股説明書,我們將不時發行和銷售高達2億美元的普通股。該公司總共出售了13,686,743股普通股,平均價格為每股5.42美元,扣除佣金和費用後的收益為7180萬美元。在截至2023年12月31日的一年中,根據自動取款機計劃結算的普通股為11,186,142股,淨收益為5,770萬美元。2024年1月,2500,601股進行了結算,淨收益為1410萬美元。(見附註10)。
關於簽署Betta Pharma許可協議,本公司Betta Pharma與Betta Pharma的關聯公司(Betta Investment(Hong Kong)Limited,或Betta Investment)簽訂了一份股票購買協議,協議日期為#年。2023年5月29日,或鬥魚股份購買協議,根據該協議,鬥魚投資同意購買本公司5,567,928股普通股或股份,總購買價約為2,500萬美元,或每股4.49美元,較鬥魚股份購買協議生效日期前兩個交易日60個交易日的成交量加權平均收盤價溢價25%。《鬥魚股份購買協議》對此類協議有某些慣常的限制。根據Betta股票購買協議完成的交易發生在2024年1月(見附註8).
現金流
下表彙總了本報告所述期間我們的現金來源和用途(以千計):
| | | | | | | | | | | | | |
| 截至十二月三十一日止的年度, |
| 2023 | | 2022 | | |
| 現金、現金等價物和限制性現金的淨變化: | | | | | |
| NET在業務活動中的應用 | $ | (106,838) | | | $ | (105,939) | | | |
| 投資活動提供(用於)的現金淨額 | 158,349 | | | 58,422 | | | |
| 融資活動提供的現金淨額 | 45,489 | | | 1,147 | | | |
| 現金、現金等價物和限制性現金淨變化 | $ | 97,000 | | | $ | (46,370) | | | |
經營活動
截至2023年12月31日的年度,用於經營活動的現金淨額主要受以下因素驅動:
•淨虧損1.325億美元;
•應收賬款增加1,030萬美元;以及
•經營租賃負債減少470萬美元。
這些數額被以下項目部分抵銷:
•與股票補償費用、折舊和攤銷有關的非現金費用3530萬美元,以及我們使用權資產賬面金額的減少;
•預付費用以及流動和長期資產減少270萬美元;
•由於我們的新協作協議,遞延收入增加了380萬美元;以及
•應計費用和其他流動負債增加130萬美元。
截至2022年12月31日的年度經營活動中使用的現金淨額主要受以下因素驅動:
•淨虧損1.282億美元;
•遞延收入減少2,270萬美元,原因是根據我們的合作協議確認收入;以及
•應付賬款減少330萬美元。
這些數額被以下項目部分抵銷:
•3,760萬美元的非現金支出,與股票補償費用、折舊和使用權資產賬面金額的減少有關;
•應計費用和其他負債增加510萬美元;
•應收賬款減少420萬美元。
投資活動
截至2023年12月31日的年度,投資活動提供的1.583億美元現金淨額可歸因於可出售證券到期收益1.601億美元(扣除購買),並被購買財產和設備的170萬美元所抵消。
截至2022年12月31日的年度,投資活動提供的5840萬美元現金淨額可歸因於6,390萬美元的有價證券收益(扣除購買),並被購買財產和設備的550萬美元所抵消。
融資活動
截至2023年12月31日的年度,融資活動提供的4550萬美元現金淨額主要由我們5770萬美元的市場發行收益推動,但預付感知信用控股III,LP定期貸款的剩餘本金餘額1250萬美元抵消了這一影響。
截至2022年12月31日的一年中,融資活動提供的110萬美元現金淨額主要是由行使股票期權的80萬美元收益推動的。
資金需求
自我們成立以來,我們已經發生了重大的運營虧損,我們預計在可預見的未來,隨着我們推進候選產品的臨牀前和臨牀開發,我們將繼續招致重大支出和不斷增加的運營虧損。此外,作為一家上市公司,我們預計將繼續產生與運營相關的成本。
具體地説,我們預計未來的支出將增加,如果我們:
•繼續我們正在進行的人類第一階段1/2試驗;
•推動更多候選產品進入臨牀前和臨牀開發階段;
•繼續投資我們專有的魚雷平臺;
•推進、擴大、維護和保護我們的知識產權組合;
•隨着我們推出更多候選產品或繼續開發現有候選產品,管理人員需求以滿足不斷變化的業務需求。
•為任何成功完成臨牀試驗的候選產品尋求市場批准;以及
•最終建立銷售、營銷和分銷基礎設施,並擴大外部製造能力,將我們可能獲得營銷批准的任何產品商業化。
由於與我們候選產品的開發和商業化相關的許多風險和不確定性,我們無法估計與我們當前和預期的臨牀前和臨牀開發相關的增加的資本和運營成本。我們未來的資本需求將取決於許多因素,包括:
•我們的主要候選產品正在進行和計劃中的人類第一階段1/2試驗的進度、成本和結果,以及這些領先候選產品的任何未來臨牀開發;
•我們其他候選產品和開發計劃的臨牀前和臨牀開發的範圍、進度、成本和結果;
•我們追求的其他候選產品的數量和開發要求;
•我們現有和未來與第三方合作伙伴合作的進展和成功,包括我們是否在實現里程碑後從合作伙伴那裏獲得額外的研究支持或里程碑付款;
•對我們的候選產品進行監管審查的成本、時間和結果;
•準備、提交和起訴專利申請、維護和執行我們的知識產權以及為任何與知識產權有關的索賠辯護的成本和時間;
•我們願意並有能力以有利的條件與其他生物技術或製藥公司建立更多的合作安排,以便開發或商業化目前或更多的未來候選產品;
•對於我們獲得市場批准的任何候選產品,未來商業化活動的成本和時間,包括產品製造、營銷、銷售和分銷;以及
•從我們的候選產品的商業銷售中獲得的收入(如果有),我們獲得了營銷批准。
由於上述預期支出,我們將需要獲得大量額外資金,以支持我們的持續運營和執行我們的長期業務計劃。在我們能夠從產品銷售中獲得可觀的收入之前,我們預計將通過股票發行、債券發行、合作、戰略聯盟以及營銷、分銷或許可安排來為我們的現金需求提供資金。儘管我們可能會通過與羅氏、Biogen、Calico、Betta Pharma和默克的合作獲得潛在的未來里程碑和特許權使用費付款,但截至2023年12月31日,我們沒有任何承諾的外部資金來源。在可接受的條件下,我們可能無法獲得足夠的額外資金,或者根本沒有。如果我們無法在需要時或在有吸引力的條件下籌集資金,我們可能會被要求推遲、限制、減少或終止我們的研究、產品開發或未來的商業化努力,或者授予我們開發和營銷我們本來更願意自己開發和營銷的候選產品的權利。
如果我們通過出售股權證券籌集額外資本,每個投資者的所有權權益將被稀釋,這些證券的條款可能包括清算或其他優惠,對您作為普通股股東的權利產生不利影響。優先股融資如果可行,可能涉及的協議包括限制或限制我們採取特定行動的能力的契約,例如進行收購或資本支出或宣佈股息。
如果我們通過與第三方的合作、戰略聯盟或營銷、分銷或許可安排來籌集更多資金,我們可能不得不放棄對我們的技術、未來收入來源、研究計劃或候選產品的寶貴權利,或者以可能對我們不利的條款授予許可證。
合同義務
以下是截至2023年12月31日我們的重要合同義務摘要(單位:千):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 總計 | | 少於
1年 | | 1至3
年份 | | 4至5個
年份 | | 多過
5年 |
| | | | | | | | | |
經營租賃承諾額 (見注6) | $ | 89,046 | | | $ | 8,828 | | | $ | 18,459 | | | $ | 21,008 | | | $ | 40,751 | |
| | | | | | | | | |
| | | | | | | | | |
我們在正常業務過程中與第三方CRO簽訂臨牀試驗、臨牀前研究、製造和其他服務以及用於運營目的的產品的合同。這些合同一般規定在通知後一段時間後終止,因此我們認為我們在這些協議下的不可撤銷義務不是實質性的,它們不包括在上表中。如果里程碑或特許權使用費付款或其他合同付款義務的時間和金額未知或不確定,我們沒有將其包括在上表中。
關鍵會計估計
管理層對財務狀況和經營結果的討論和分析是以我們的綜合財務報表為基礎的,這些報表是根據美國公認的會計原則編制的。在編制我們的合併財務報表和相關披露時,我們需要作出估計和假設,這些估計和假設會影響我們綜合財務報表中資產和負債、收入、成本和費用的報告金額,以及或有資產和負債的披露。我們基於歷史經驗、已知趨勢和事件以及我們認為在當時情況下合理的各種其他因素進行估計,這些因素的結果構成了對資產和負債賬面價值的判斷的基礎,而這些資產和負債的賬面價值並不是從其他來源很容易看出的。我們在持續的基礎上評估我們的估計和假設。在不同的假設或條件下,我們的實際結果可能與這些估計不同。
雖然我們的重要會計政策在附註2中有更詳細的描述,重要會計政策摘要對於本年度報表10-K表中的綜合財務報表,我們認為以下會計政策對我們編制綜合財務報表所使用的判斷和估計最為關鍵。
合同收入
我們根據會計準則編纂或ASC,606,與客户簽訂合同的收入或ASC 606。根據ASC 606,當其客户獲得對承諾商品或服務的控制權時,實體確認收入,其金額反映了實體預期從這些商品或服務交換中獲得的對價。為了確定實體確定屬於ASC606範圍內的安排的收入確認,實體在協議開始時或在協議的實質性修改時執行以下五個步驟:(1)確定與客户的合同(S);(2)確定合同中的履約義務;(3)確定
交易價格,包括可變對價(如果有);(4)將交易價格分配給合同中的履約義務;(5)當實體履行履約義務時(或作為)確認收入。
我們認為上文第(五)步下履約債務的履行情況是一項重要的會計估計。更具體地説,確定研究和開發服務履約義務的實現程度,其滿意度模式是用迄今發生的費用與未來發生和預計發生的總費用相比較來衡量的,這是由關鍵的會計估計推動的。
在估計未來預計發生的費用時,管理層使用其最新的預算和長期計劃,並根據任何相關信息進行調整。雖然這是我們截至報告期的最佳估計,但由於研發活動的範圍和時間可能會隨着時間的推移而發生重大變化,因此未來預計將發生的成本需要管理層的判斷。我們可能會根據幾個因素調整研發活動的範圍,例如支持候選產品進步所需的額外工作或試驗中患者數量的變化。此外,研究和開發服務可能不再屬於合作協議的範圍,就像我們的某些項目一樣。預計發生研發成本的時間可能會因外部因素而改變,例如製造或供應鏈造成的延誤,或招募患者的困難;或內部因素,如計劃的優先順序。我們對研發服務的範圍和時間相對於實際範圍和時間的估計可能會對收入確認產生重大影響。
預付和應計的研究和開發費用
作為編制合併財務報表的一部分,我們需要估計我們應計的研究和開發費用。這一過程包括審查報價單和合同,確定已為我們提供的服務,並在我們尚未收到發票或以其他方式通知實際成本的情況下,估計所提供的服務水平和服務產生的相關成本。我們根據我們當時所知的事實和情況,在我們的合併財務報表中對截至每個資產負債表日期的應計費用進行估計。此外,在某些情況下,向供應商支付的款項可能會超過所提供的服務水平,並導致費用的預付款,在這種情況下,此類金額將反映為預付費用和其他流動資產。在應計服務費時,我們估計將提供服務的時間段和每段時間的努力程度。如果服務的實際執行時間或努力程度與我們的估計不同,我們會相應地調整應計費用或預付費用的金額。儘管我們預計我們的估計與實際發生的金額不會有重大差異,但我們對所提供服務的狀態和時間的理解與所提供服務的實際狀態和時間可能有所不同,並可能對報告金額產生重大影響。
將來收到用於研究和開發活動的貨物或服務的不可退還的預付款將被遞延,並以預付費用和其他流動資產的形式資本化。資本化金額在相關貨物交付或提供服務時計入費用。
經營租賃(遞增借款利率)
我們根據ASC主題842對租賃進行核算,租契,或ASC 842。根據ASC 842,在租賃安排開始或修改時,我們可能被要求重新計量我們的租賃負債和相應的使用權資產。租賃負債是通過使用遞增借款利率計算租賃安排下租賃付款的現值來計量的。遞增借款利率是指我們必須支付的利率,在抵押的基礎上,借款的金額相當於類似期限內的租賃付款,相當於類似經濟環境下的租賃期限。
由於我們的租賃中隱含的遞增借款利率不容易確定,我們使用基於開始日期可獲得的信息的估計遞增借款利率來確定用於計算租賃付款現值的貼現率。由於我們沒有近期的對外借款,增量借款利率是根據金融機構提供的價值和借款期限,並根據公司和市場特定因素進行調整後提供的關於指示借款利率的信息來確定的。這一決定需要管理層的判斷,包括在確定同行羣體、我們自己的風險概況以及根據公司和市場特定因素進行調整時。
儘管我們預期我們對增量借款利率的估計不會在合理的敏感性範圍內產生重大差異,但在選擇適當的利率時需要做出判斷,為我們的租賃選擇的利率將對綜合資產負債表中租賃負債和相應使用權資產的價值產生影響。
股票期權
我們按公允價值將授予員工和非員工的所有股票獎勵作為股票薪酬支出進行會計處理。我們以股票為基礎的支付包括股票期權、限制性股票單位和普通股的授予,包括需要歸屬的普通股。獎勵的計量日期為授予之日,基於股票的補償成本以直線方式確認為必要服務期(通常為授權期)的費用。基於股票的補償費用按提供相關服務的職能在合併經營報表和綜合損失表中分類。我們確認了已授予部分獎勵的基於股票的薪酬支出。沒收在發生時被記錄下來。每個股票期權授予的公允價值在授予之日使用Black-Scholes期權定價模型進行估計。布萊克-斯科爾斯期權定價模型包括各種假設,包括獎勵的預期期限、獎勵預期期限內的預期波動率和預期無風險利率、預期股息支付以及基於股票獎勵的普通股的公允價值。
我們認為預期波動率是一個關鍵的會計估計。由於我們沒有足夠的交易歷史,我們使用一組具有代表性的上市生物製藥公司(包括我們自己)的平均歷史波動率來計算預期波動率,以用於Black-Scholes期權定價模型。這一假設反映了我們的最佳估計,但它涉及基於市場狀況的內在不確定性,這些市場狀況通常不受我們的控制。因此,如果使用不同的波動率,基於股票的薪酬成本可能會受到實質性影響。
新會計公告
關於新會計準則的信息,見附註2,重要會計政策摘要,在本年度報告Form 10-K中加入我們的綜合財務報表。
第7A項。關於市場風險的定量和定性披露。
我們在正常的業務過程中面臨着市場風險。這些風險主要包括利率敏感性。我們的生息資產包括現金、現金等價物和有價證券。我們的利息收入對一般利率水平的變化很敏感,主要是美國利率。截至2023年12月31日,我們擁有1.551億美元的有價證券。我們的有價證券是短期證券,加權平均到期日為0.6年。因此,雖然這些賺取利息的工具帶有一定程度的利率風險,但利息收入的歷史波動對我們來説並不顯著。
項目8.財務報表和補充數據
根據本項目8要求提交的財務報表附在本年度報告的表格10-K之後。這些財務報表的索引見項目15,展品和財務報表附表,本年度報告的表格10-K。
第九項會計和財務披露方面的變更和分歧。
沒有。
項目9A.控制和程序。
信息披露控制和程序的評估
我們的管理層在我們的首席執行官和首席財務官的參與下,評估了截至本年度報告10-K表格所涵蓋的期間結束時,我們的披露控制和程序的有效性,這些控制和程序符合《交易法》第13a-15(E)和15d-15(E)條的規定。《交易法》規則13a-15(E)和15d-15(E)中定義的術語“披露控制和程序”是指公司的控制和其他程序,旨在確保公司在根據交易法提交或提交的報告中要求披露的信息在美國證券交易委員會規則和表格指定的時間段內得到記錄、處理、彙總和報告。披露控制和程序包括但不限於旨在確保公司根據《交易法》提交或提交的報告中要求披露的信息被積累並傳達給公司管理層,包括其主要高管和主要財務官,或酌情履行類似職能的人員的控制和程序,以便及時做出關於所需披露的決定。管理層認識到,任何控制和程序,無論設計和操作得多麼好,都只能為實現其目標提供合理的保證,管理部門在評估可能的控制和程序的成本-效益關係時必須運用其判斷。
根據對我們截至2023年12月31日的披露控制和程序的評估,我們的首席執行官和首席財務官得出結論,截至該日期,我們的披露控制和程序在合理保證水平下是有效的。
管理層財務報告內部控制年度報告
我們的管理層負責建立和維護對我們財務報告的充分內部控制。根據《交易法》頒佈的規則13a-15(F)和15d-15(F)中對財務報告的內部控制定義為由我們的主要高管和主要財務官設計或在其監督下,並由我們的董事會、管理層和其他人員實施的程序,以提供關於財務報告的可靠性和根據美國公認會計原則為外部目的編制財務報表的合理保證。我們對財務報告的內部控制包括以下政策和程序:
•與保存合理、詳細、準確和公平地反映我們對我們資產的交易和處置的記錄有關;
•提供合理的保證,保證交易被記錄為必要的,以便根據美國公認會計原則編制財務報表,並且我們的收入和支出僅根據我們管理層和董事的授權進行;以及
•提供合理保證,防止或及時發現可能對我們的財務報表產生重大影響的未經授權獲取、使用或處置我們的資產。
由於其固有的侷限性,財務報告的內部控制可能無法防止或發現錯誤陳述。對未來期間進行任何有效性評估的預測都有這樣的風險,即由於條件的變化,控制可能會變得不充分,或者遵守政策或程序的程度可能會惡化。
我們的管理層評估了截至2023年12月31日的財務報告內部控制的有效性。在進行這項評估時,管理層使用了特雷德韋委員會贊助組織委員會(COSO)在其2013年內部控制-綜合框架中提出的標準。根據我們的評估,我們的管理層得出結論,截至2023年12月31日,我們對財務報告的內部控制基於這些標準是有效的。
財務報告內部控制的變化
我們不斷尋求提高內部控制的效率和效力。這導致對整個公司的流程進行了改進。在截至2023年12月31日的年度內,我們對財務報告的內部控制(根據《外匯法案》第13a-15(F)和15d-15(F)條的定義)沒有發生重大影響或合理地可能對我們的財務報告內部控制產生重大影響的變化。
控制措施有效性的固有限制
任何財務報告內部控制制度的有效性,包括我們的內部控制制度,都受到內在限制,包括在設計、實施、運作和評估控制和程序時行使判斷力,以及無法完全消除不當行為。因此,任何財務報告的內部控制制度,包括我們的制度,無論設計和運作有多好,都只能提供合理的保證,而不是絕對的保證。此外,對未來期間進行任何有效性評估的預測可能會因為條件的變化而出現控制不足的風險,或者政策或程序的遵守程度可能會惡化。我們打算繼續對我們的業務需要或適當的內部控制進行監控和升級,但不能保證這些改進將足以為我們提供對財務報告的有效內部控制。見“風險因素--作為一家上市公司,我們將繼續產生額外的成本,我們的管理層將需要投入大量時間來實施新的合規舉措和公司治理做法。”
項目9B。其他信息。
沒有。
項目9C。披露妨礙檢查的外國司法管轄區。
沒有。
第三部分
項目10.董事、行政人員和公司治理
第10項所要求的信息將包括在我們向美國證券交易委員會或美國證券交易委員會提交的關於2024年股東年會的最終委託書中,該聲明將在本年度報告10-K表格涵蓋的年度結束後120天內提交,並通過引用併入本文。
第11項.行政人員薪酬
第11項所要求的信息將包括在我們提交給美國證券交易委員會的最終委託書中,該委託書將在本年度報告所涵蓋的關於2024年股東年會的10-K表格年度報告所涵蓋的年度結束後120天內提交,並通過引用併入本文。
第12項:某些實益所有人的擔保所有權和管理層及相關股東事項。
第12項所要求的資料將包括在本年度10-K表格所涵蓋的有關2024年股東周年大會的年報所涵蓋的年度報告後120天內提交給美國證券交易委員會的最終委託書中,並以引用方式併入本文。
第十三條某些關係和相關交易,以及董事的獨立性。
第13項所要求的信息將包括在我們提交給美國證券交易委員會的最終委託書中,該委託書將在本年度報告所涵蓋的關於2024年股東年會的10-K表格年度報告所涵蓋的年度結束後120天內提交,並通過引用併入本文。
第14項主要會計費用及服務
第14項所要求的信息將包括在我們提交給美國證券交易委員會的最終委託書中,該委託書將在本年度報告所涵蓋的關於2024年股東年會的10-K表格年度報告所涵蓋的年度結束後120天內提交,並通過引用併入本文。
第四部分
項目15.物證、財務報表附表
1.財務報表
有關本文所列財務報表的列表,請參閲合併財務報表索引載於本年度報告表格10-K的F-1頁,通過引用併入本項目。
2.財務報表附表
財務報表附表已被省略,因為它們要麼不是必需的,要麼不適用,或者這些信息已列入合併財務報表或其附註。
3.展品
| | | | | | | | | | | | | | | | | | | | |
展品
數 | 展品説明 | 表格 | 檔案
數 | 日期
歸檔 | 展品
數 | 已歸檔
特此聲明 |
| 3.1 | 第五份現行有效的註冊人註冊證書 | 8-K | 001–39567 | 10/06/2020 | 3.3 | |
| | | | | | |
| 3.2 | 註冊人第二次修訂和重新修訂附例的格式 | S-1 | 333–248719 | 09/10/2020 | 3.5 | |
| | | | | | |
| 4.1 | 修訂和重新簽署了註冊人、其權證持有人和若干股東之間的《投資者權利協定》,日期為2020年6月5日 | S-1 | 333–248719 | 09/10/2020 | 4.1 | |
| | | | | | |
| 4.2 | 普通股股票證書樣本格式 | S-1/A | 333–248719 | 09/28/2020 | 4.3 | |
| | | | | | |
| 4.3 | 根據經修訂的1934年《證券交易法》第12條登記的證券説明 | 10-K | 001-39567 | 03/11/2021 | 4.4 | |
| | | | | | |
| 10.1# | 經修訂的2015年股票期權和授予計劃及其授予協議的格式 | S-1 | 333–248719 | 09/10/2020 | 10.1 | |
| | | | | | |
| 10.2# | 2020年股票期權和激勵計劃及其獎勵協議的格式 | 10-K | 001-39567 | 02/23/2023 | 10.2 | |
| | | | | | |
| 10.3# | 2020年員工購股計劃 | S-1/A | 333–248719 | 09/28/2020 | 10.3 | |
| | | | | | |
| 10.4# | 高級管理人員現金獎勵獎金計劃 | S-1 | 333–248719 | 09/10/2020 | 10.4 | |
| | | | | | |
| 10.5# | 《董事賠償協議》格式 | S-1 | 333–248719 | 09/10/2020 | 10.5 | |
| | | | | | |
| 10.6# | 高級船員彌償協議的格式 | S-1 | 333–248719 | 09/10/2020 | 10.6 | |
| | | | | | |
| 10.7# | 行政人員聘用協議格式 | S-1 | 333–248719 | 09/10/2020 | 10.7 | |
| | | | | | |
| 10.8# | 登記人與安德魯·赫希之間的僱傭協議,日期為2020年9月6日 | S-1 | 333–248719 | 09/10/2020 | 10.8 | |
| | | | | | |
| 10.9† | 註冊人與Biogen MA,Inc.之間的合作研究和許可協議,日期為2018年12月28日 | S-1 | 333–248719 | 09/10/2020 | 10.10 | |
| | | | | | |
| 10.10† | 註冊人與Biogen MA,Inc.之間的合作研究和許可協議的第1號修正案,日期為2020年2月25日 | 10-K | 001-39567 | 03/11/2021 | 10.11 | |
| | | | | | |
| 10.11† | 修訂和重新簽署註冊人、F.Hoffmann-La Roche Ltd和Hoffmann-La Roche Inc.之間的許可協議,日期為2018年12月20日 | S-1 | 333–248719 | 09/10/2020 | 10.11 | |
| | | | | | |
| | | | | | | | | | | | | | | | | | | | |
展品
數 | 展品説明 | 表格 | 檔案
數 | 日期
歸檔 | 展品
數 | 已歸檔
特此聲明 |
| 10.12† | 註冊人、F.Hoffmann-La Roche Ltd和Hoffmann-La Roche Inc.之間修訂和重新簽署的許可協議的第一修正案,日期為2020年11月12日 | 10-K | 001-39567 | 03/11/2021 | 10.13 | |
| | | | | | |
| 10.13† | 註冊人與Calico生命科學有限責任公司之間的合作和許可協議,日期為2017年3月13日 | S-1 | 333–248719 | 09/10/2020 | 10.12 | |
| | | | | | |
| 10.14 | 480阿森納集團有限責任公司授予註冊人的租約,日期為2017年7月5日,經修訂 | S-1 | 333–248719 | 09/10/2020 | 10.14 | |
| | | | | | |
| 10.15 | 第三次修訂租約,日期為2021年11月24日,由C4治療公司和哥倫比亞馬薩諸塞州阿森納辦公物業公司之間的租約 | 8-K | 001-39567 | 11/30/2021 | 10.1 | |
| | | | | | |
| 10.16† | 許可和合作協議,日期為2023年5月29日,由C4治療公司和Betta製藥有限公司簽署。 | 8-K | 001-39567 | 05/30/2023 | 10.1 | |
| | | | | | |
| 10.17† | C4治療公司、Betta製藥有限公司和Betta投資(香港)有限公司之間的股票購買協議,日期為2023年5月29日。 | 8-K | 001-39567 | 05/30/2023 | 10.2 | |
| | | | | | |
| 10.18† | 許可和合作協議,日期為2023年12月11日,由C4治療公司和默克·夏普·多姆有限責任公司簽署 | | | | | X |
| | | | | | |
| 10.19# | 非限制性股票期權協議激勵獎的形式 | | | | | X |
| | | | | | |
| 21.1 | 註冊人的子公司 | | | | | X |
| | | | | | |
| 23.1 | 獨立註冊會計師事務所畢馬威會計師事務所同意 | | | | | X |
| | | | | | |
| 31.1 | 依照依照2002年《薩班斯-奧克斯利法案》第302節通過的1934年《證券交易法》第13a-14(A)和15d-14(A)條頒發的首席執行官證書 | | | | | X |
| | | | | | |
| 31.2 | 根據依照2002年《薩班斯-奧克斯利法案》第302節通過的1934年《證券交易法》第13a-14(A)和15d-14(A)條對首席財務官的證明 | | | | | X |
| | | | | | |
| 32.1* | 依據2002年《薩班斯-奧克斯利法案》第906條通過的《美國法典》第18編第1350條對主要行政官員的證明 | | | | | X |
| | | | | | |
| 32.2* | 根據2002年《薩班斯-奧克斯利法案》第906條通過的《美國法典》第18編第1350條對首席財務官的證明 | | | | | X |
| | | | | | |
| 97 | 高管薪酬追回政策 | | | | | X |
| | | | | | |
| 101.INS | 內聯XBRL實例文檔 | | | | | X |
| | | | | | |
| 101.SCH | 內聯XBRL分類擴展架構文檔 | | | | | X |
| | | | | | | | | | | | | | | | | | | | |
展品
數 | 展品説明 | 表格 | 檔案
數 | 日期
歸檔 | 展品
數 | 已歸檔
特此聲明 |
| | | | | | |
| 101.CAL | 內聯XBRL分類擴展計算鏈接庫文檔 | | | | | X |
| | | | | | |
| 101.DEF | 內聯XBRL分類擴展定義Linkbase文檔 | | | | | X |
| | | | | | |
| 101.LAB | 內聯XBRL分類擴展標籤Linkbase文檔 | | | | | X |
| | | | | | |
| 101.PRE | 內聯XBRL分類擴展演示文稿Linkbase文檔 | | | | | X |
| | | | | | |
| 104 | 封面交互數據文件(格式為內聯XBRL文檔,包含在附件101中) | | | | | |
_____________________________________
| | | | | |
| # | 指管理合同或任何補償計劃、合同或安排。 |
| † | 根據美國證券交易委員會的規定,本展品的部分內容(以星號表示)將被略去。 |
| * | 證物32.1和32.2是提供的,不應被視為為《交易法》第18條的目的而提交,或以其他方式承擔該條款的責任,也不得被視為通過引用將該證物納入根據《證券法》或《交易法》提交的任何登記聲明或其他文件中,除非該申請中另有説明。 |
項目16.表格10-K摘要
不適用。
簽名
根據經修訂的1934年《證券交易法》第13或15(D)節的要求,註冊人已正式安排由正式授權的下列簽署人代表其簽署本報告.
| | | | | | | | |
| C4治療公司 |
| | |
日期:2024年2月22日 | 發信人: | /S/安德魯·J·赫希 |
| | 安德魯·J·赫希 |
| | 總裁與首席執行官 |
根據修訂後的1934年《證券交易法》的要求,本報告已由以下注冊人以登記人的身份在指定日期簽署。
| | | | | | | | | | | | | | |
| 名字 | | 標題 | | 日期 |
| | | | |
| /S/安德魯·J·赫希 | | 首席執行官總裁和董事 | | 2024年2月22日 |
| 安德魯·J·赫希 | | (首席行政主任) | | |
/S/肯德拉·R·亞當斯 | | 首席財務官兼財務主管 | | 2024年2月22日 |
肯德拉·R·亞當斯 | | (首席財務官) | | |
撰稿S/馬克·莫斯勒 | | 首席會計官 | | 2024年2月22日 |
馬克·莫斯勒 | | (首席會計主任) | | |
| /S/布魯斯·唐尼 | | 董事長兼董事 | | 2024年2月22日 |
| 布魯斯·唐尼 | | | | |
| /S/肯尼思·C·安德森,醫學博士 | | 董事 | | 2024年2月22日 |
| 肯尼思·C·安德森醫學博士 | | | | |
| /S/勞拉·貝森,醫學博士 | | 董事 | | 2024年2月22日 |
| 勞拉·貝森醫學博士 | | | | |
| 撰稿S/格倫·杜賓 | | 董事 | | 2024年2月22日 |
| 格倫·杜賓 | | | | |
| /S/唐娜·格羅根,醫學博士 | | 董事 | | 2024年2月22日 |
| 唐娜·格羅根醫學博士 | | | | |
歐文·休斯Owen Hughes | | 董事 | | 2024年2月22日 |
歐文·休斯 | | | | |
| /S/Utpal Koppikar | | 董事 | | 2024年2月22日 |
| Utpal Koppikar | | | | |
| 撰稿S/馬爾科姆·索爾特 | | 董事 | | 2024年2月22日 |
| 馬爾科姆·索爾特 | | | | |
合併財務報表索引
| | | | | |
獨立註冊會計師事務所報告 | F-2 |
| |
合併資產負債表 | F-3 |
| |
合併經營報表和全面虧損 | F-4 |
| |
股東權益合併報表 | F-5 |
| |
合併現金流量表 | F-6 |
| |
合併財務報表附註 | F-8 |
獨立註冊會計師事務所報告
致股東和董事會
C4 Therapeutics,Inc.:
對合並財務報表的幾點看法
我們已審計隨附的C4 Therapeutics,Inc.的綜合資產負債表。本公司於2023年12月31日及2022年12月31日的綜合財務報表及附屬公司(“本公司”)截至該日及2022年12月31日的綜合財務報表、相關的合併經營及綜合虧損報表、合併股東權益報表、合併現金流量表及相關附註(統稱合併財務報表)。我們認為,綜合財務報表在所有重大方面公允地列報了貴公司於二零二三年及二零二二年十二月三十一日的財務狀況以及截至該日止年度的經營業績和現金流量,符合美國公認會計原則。
意見基礎
這些合併財務報表由公司管理層負責。我們的責任是根據我們的審計對這些合併財務報表發表意見。我們是一家在美國上市公司會計監督委員會(PCAOB)註冊的公共會計師事務所,根據美國聯邦證券法以及美國證券交易委員會和PCAOB的適用規則和法規,我們必須與公司保持獨立。
我們是按照PCAOB的標準進行審計的。這些準則要求我們計劃和執行審計,以獲得關於合併財務報表是否沒有重大錯報的合理保證,無論是由於錯誤還是舞弊。本公司並無被要求對其財務報告的內部控制進行審計,我們也沒有受聘進行審計。作為我們審計的一部分,我們被要求瞭解財務報告的內部控制,但不是為了對公司財務報告內部控制的有效性發表意見。因此,我們不表達這樣的意見。
我們的審計包括執行評估合併財務報表重大錯報風險的程序,無論是由於錯誤還是欺詐,以及執行應對這些風險的程序。這些程序包括在測試的基礎上審查關於合併財務報表中的金額和披露的證據。我們的審計還包括評價管理層使用的會計原則和作出的重大估計,以及評價合併財務報表的整體列報。我們相信,我們的審計為我們的觀點提供了合理的基礎。
關鍵審計事項
關鍵審計事項指在對合並財務報表進行當期審計時產生的、已傳達或要求傳達給審計委員會的事項:(1)涉及對綜合財務報表具有重大意義的賬目或披露;(2)涉及我們特別具有挑戰性的、主觀的或複雜的判斷。我們確定不存在關鍵的審計事項。
/s/畢馬威律師事務所
自2016年以來,我們一直擔任本公司的審計師。
波士頓,馬薩諸塞州
2024年2月22日
C4治療公司
合併資產負債表
(單位為千,不包括每股和每股金額)
| | | | | | | | | | | |
| 十二月三十一日, |
| 2023 | | 2022 |
| 資產 | | | |
| 流動資產: | | | |
| 現金和現金等價物 | $ | 126,590 | | | $ | 29,754 | |
| 流通有價證券 | 127,091 | | | 246,399 | |
| 應收賬款 | 11,799 | | | 1,473 | |
| 預付費用和其他流動資產 | 5,709 | | | 9,931 | |
| 流動資產總額 | 271,189 | | | 287,557 | |
| 非流通有價證券 | 28,008 | | | 60,962 | |
| 財產和設備,淨額 | 7,132 | | | 7,400 | |
| 使用權資產 | 63,956 | | | 70,116 | |
| 受限現金 | 3,443 | | | 3,279 | |
| 其他資產 | 2,723 | | | 1,526 | |
| 總資產 | $ | 376,451 | | | $ | 430,840 | |
| | | |
| 負債和股東權益 | | | |
| 流動負債: | | | |
| 應付帳款 | $ | 1,446 | | | $ | 1,172 | |
| 應計費用和其他流動負債 | 20,630 | | | 19,769 | |
| 遞延收入,當期 | 15,471 | | | 16,618 | |
| 經營租賃負債,流動 | 5,219 | | | 4,700 | |
| 長期債務,扣除債務折扣-關聯方,流動 | — | | | 2,287 | |
| 流動負債總額 | 42,766 | | | 44,546 | |
| 遞延收入,扣除當期 | 21,814 | | | 16,895 | |
| 經營租賃負債,當期淨額 | 65,757 | | | 70,970 | |
| 長期債務,扣除債務貼現-關聯方,扣除流動 | — | | | 9,195 | |
| 總負債 | 130,337 | | | 141,606 | |
| 承付款及或有事項(見附註6及附註9) | | | |
| 股東權益: | | | |
優先股,面值為$0.0001每股;10,000,000授權股份,以及不是分別於2023年及2022年12月31日已發行或發行在外的股份 | — | | | — | |
普通股,面值$0.0001每股;150,000,000授權股份,以及60,467,188和48,966,216截至2023年12月31日和2022年12月31日的已發行和已發行股票 | 6 | | | 5 | |
| 額外實收資本 | 774,618 | | | 689,256 | |
| 累計其他綜合損失 | (127) | | | (4,137) | |
| 累計赤字 | (528,383) | | | (395,890) | |
| 股東權益總額 | 246,114 | | | 289,234 | |
| 總負債和股東權益 | $ | 376,451 | | | $ | 430,840 | |
附註是這些合併財務報表的組成部分。
C4治療公司
合併經營報表和全面虧損
(單位為千,不包括每股和每股金額)
| | | | | | | | | | | | | |
| 截至十二月三十一日止的年度, |
| 2023 | | 2022 | | |
| R從協作協議中獲得收益 | $ | 20,756 | | | $ | 31,096 | | | |
| 運營費用: | | | | | |
| 研發 | 117,706 | | | 117,841 | | | |
| 一般和行政 | 42,081 | | | 42,789 | | | |
| 總運營費用 | 159,787 | | | 160,630 | | | |
| 運營虧損 | (139,031) | | | (129,534) | | | |
| 其他收入(費用),淨額 | | | | | |
| 長期債務關聯方的利息支出和攤銷 | (1,373) | | | (2,216) | | | |
| 提前清償債務損失 | (621) | | | — | | | |
| 利息和其他收入,淨額 | 9,812 | | | 3,575 | | | |
| | | | | |
| 其他收入(費用)合計,淨額 | 7,818 | | | 1,359 | | | |
| 所得税前虧損 | (131,213) | | | (128,175) | | | |
| 所得税費用 | (1,280) | | | — | | | |
| 淨虧損 | $ | (132,493) | | | $ | (128,175) | | | |
| 每股淨虧損--基本虧損和攤薄虧損 | $ | (2.67) | | | $ | (2.62) | | | |
| 加權平均股數-基本及攤薄 | 49,640,505 | | | 48,861,665 | | | |
| | | | | |
其他綜合收益(虧損): | | | | | |
| 有價證券的未實現收益(虧損) | 4,010 | | | (3,362) | | | |
| 綜合損失 | $ | (128,483) | | | $ | (131,537) | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
附註是這些合併財務報表的組成部分。
C4治療公司
股東權益合併報表
(單位為千,不包括份額)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 普通股 | | 其他內容
已繳費
資本 | | 累計
其他
全面
收入(虧損) | | 累計
赤字 | | 總計
股東的
權益 |
| 股票 | | 金額 | | | | |
| 截至2021年12月31日的餘額 | 48,688,875 | | | $ | 5 | | | $ | 658,091 | | | $ | (775) | | | $ | (267,715) | | | $ | 389,606 | |
| 股票期權的行使和股票單位的釋放 | 235,450 | | | — | | | 777 | | | — | | | — | | | 777 | |
| 根據2020年ESPP發行普通股 | 31,402 | | | — | | | 372 | | | — | | | — | | | 372 | |
| 基於股票的薪酬 | — | | | — | | | 29,858 | | | — | | | — | | | 29,858 | |
| 有價證券未實現虧損 | — | | | — | | | — | | | (3,362) | | | — | | | (3,362) | |
| 淨虧損 | — | | | — | | | — | | | — | | | (128,175) | | | (128,175) | |
| 其他 | 10,489 | | | — | | | 158 | | | — | | | — | | | 158 | |
| 截至2022年12月31日的餘額 | 48,966,216 | | | $ | 5 | | | $ | 689,256 | | | $ | (4,137) | | | $ | (395,890) | | | $ | 289,234 | |
| 根據市場股本方案發行普通股,淨額 | 11,186,142 | | | 1 | | | 57,672 | | | — | | | — | | | 57,673 | |
| 股票期權的行使和股票單位的釋放 | 13,096 | | | | | 58 | | | — | | | — | | | 58 | |
| 在歸屬受限股單位時發行普通股,扣除為預扣税款而回購的股份 | 117,745 | | | — | | | (110) | | | — | | | — | | | (110) | |
| 根據2020年ESPP發行普通股 | 133,784 | | | — | | | 368 | | | — | | | — | | | 368 | |
| 基於股票的薪酬 | — | | | — | | | 27,235 | | | — | | | — | | | 27,235 | |
| 有價證券的未實現收益 | — | | | — | | | — | | | 4,010 | | | — | | | 4,010 | |
| 淨虧損 | — | | | — | | | — | | | — | | | (132,493) | | | (132,493) | |
| 其他 | 50,205 | | | — | | | 139 | | | — | | | — | | | 139 | |
| 截至2023年12月31日的餘額 | 60,467,188 | | | $ | 6 | | | $ | 774,618 | | | $ | (127) | | | $ | (528,383) | | | $ | 246,114 | |
附註是這些合併財務報表的組成部分。
| | | | | | | | | | | | | |
| 截至十二月三十一日止的年度, |
| 2023 | | 2022 | | |
| 經營活動中使用的現金流: | | | | | |
| 淨虧損 | $ | (132,493) | | | $ | (128,175) | | | |
| 對淨虧損與業務活動中使用的現金進行核對的調整: | | | | | |
| 基於股票的薪酬費用 | 27,235 | | | 30,016 | | | |
| 折舊及攤銷 | 1,880 | | | 1,676 | | | |
| 減少使用權資產的賬面金額 | 6,159 | | | 5,896 | | | |
| 增加有價證券的(溢價)折扣 | (3,784) | | | 714 | | | |
| 債務貼現關聯方攤銷 | 405 | | | 713 | | | |
| 提前清償債務損失 | 621 | | | — | | | |
| 其他 | 77 | | | — | | | |
| 經營性資產和負債變動情況: | | | | | |
| 應收賬款 | (10,327) | | | 4,244 | | | |
| 預付費用及其他流動和長期資產 | 2,723 | | | 388 | | | |
| 應付帳款 | 273 | | | (3,333) | | | |
| 應計費用和其他流動負債 | 1,316 | | | 5,083 | | | |
| 經營租賃負債 | (4,695) | | | (507) | | | |
| 遞延收入 | 3,772 | | | (22,654) | | | |
| 用於經營活動的現金淨額 | (106,838) | | | (105,939) | | | |
| 投資活動提供的現金流: | | | | | |
| 有價證券到期日收益 | 289,955 | | | 283,445 | | | |
| 購買有價證券 | (129,898) | | | (219,527) | | | |
| 購置財產和設備 | (1,708) | | | (5,496) | | | |
| | | | | |
| 投資活動提供的現金淨額 | 158,349 | | | 58,422 | | | |
| 融資活動提供的現金流: | | | | | |
| 根據市場股本方案發行普通股所得收益,淨額 | 57,673 | | | — | | | |
| 支付長期債務−關聯方 | (12,500) | | | — | | | |
| 行使股票期權所得收益 | 58 | | | 777 | | | |
| 用於預扣税款的普通股回購付款 | (110) | | | — | | | |
| 其他 | 368 | | | 370 | | | |
| 融資活動提供的現金淨額 | 45,489 | | | 1,147 | | | |
| | | | | |
| 現金、現金等價物和限制性現金淨變化 | 97,000 | | | (46,370) | | | |
| 期初現金、現金等價物和限制性現金 | 33,033 | | | 79,403 | | | |
| 期末現金、現金等價物和限制性現金 | $ | 130,033 | | | $ | 33,033 | | | |
| | | | | |
| 現金、現金等價物和限制性現金的對賬: | | | | | |
| 年終現金、現金等價物和限制性現金 | $ | 130,033 | | | $ | 33,033 | | | |
| 減去:受限現金 | (3,443) | | | (3,279) | | | |
| 年終現金及現金等價物 | $ | 126,590 | | | $ | 29,754 | | | |
附註是這些合併財務報表的組成部分。
目錄表
C4治療公司
合併現金流量表--續
(單位:千)
| | | | | | | | | | | | | |
| 截至十二月三十一日止的年度, |
| 2023 | | 2022 | | |
| 補充披露現金流量信息: | | | | | |
| 應收賬款遞延收入 | $ | 10,000 | | | $ | — | | | |
| 為利息關聯方支付的現金 | $ | 990 | | | $ | 1,624 | | | |
| 繳納所得税的現金 | $ | 1,003 | | | $ | — | | | |
| | | | | |
| 非現金投資和融資活動的補充披露: | | | | | |
| 應付賬款和應計費用中的資本支出 | $ | 18 | | | $ | 473 | | | |
| 因取得使用權資產而產生的經營租賃負債 | $ | — | | | $ | 44,067 | | | |
附註是這些合併財務報表的組成部分。
注1。業務性質
C4治療公司或與其子公司本公司一起,是一家臨牀階段的生物製藥公司,致力於實現定向蛋白質降解(TPD)科學的承諾,以創造改變患者生活的新一代小分子藥物。通過利用其專有的魚雷平臺,該公司有能力有效地設計和優化小分子蛋白質降解劑,通過利用人體破壞不需要的蛋白質的自然過程,對其預期目標高度活躍。該公司相信其新型口腔產品候選產品有可能克服抑制劑經常出現的耐藥性,瞄準目前“無法下藥”的靶點,並改善患者的結果。該公司於2015年10月7日在特拉華州註冊成立,主要辦事處位於馬薩諸塞州沃特敦。
流動資金和資本資源
自成立以來,該公司的主要活動一直集中在進行研究和開發活動、建立公司的知識產權、招聘人員和籌集資金以支持這些活動。到目前為止,該公司的運營資金主要來自出售可贖回可轉換優先股、通過合作協議公開發行公司普通股以及債務融資。
該公司自成立以來發生經常性虧損,包括淨虧損#美元。132.5百萬美元和美元128.2截至2023年12月31日和2022年12月31日的年度分別為100萬美元。此外,截至2023年12月31日,公司的累計虧損為1美元。528.4百萬美元。到目前為止,該公司還沒有從產品銷售中獲得任何收入,因為它的候選產品都沒有被批准商業化。該公司預計在可預見的未來將繼續產生營業虧損。
該公司預計其現金、現金等價物和有價證券為$281.7截至2023年12月31日的100萬美元將足以為其從這些合併財務報表發佈之日起至少未來12個月的運營提供資金。因此, 綜合財務報表的編制是基於假設公司將繼續作為一家持續經營的企業,並考慮在正常業務過程中實現資產以及償還負債和承諾。
風險和不確定性
該公司在早期開發階段面臨與其他生命科學公司相同的風險,包括但不限於籌集額外資金的能力的不確定性,產品開發和商業化,競爭對手開發新技術創新的不確定性,對關鍵人員的依賴,產品的市場接受度,缺乏營銷和銷售歷史,產品責任,專有技術和知識產權的保護,以及對食品和藥物管理局(FDA)和其他政府法規的遵守。如果該公司沒有成功地將其計劃推進到人體臨牀試驗中並通過人體臨牀試驗成功地將其任何候選產品商業化,或者通過與其他公司的合作,該公司可能無法產生產品收入或實現盈利。不能保證公司的研發工作會成功,不能保證公司的知識產權會得到充分的保護,不能保證開發的任何產品都會獲得必要的政府監管批准,也不能保證任何批准的產品在商業上是可行的。即使該公司的產品開發工作取得成功,該公司何時(如果有的話)將從產品銷售中獲得可觀的收入也是不確定的。該公司在技術快速變化和來自制藥和生物技術公司的激烈競爭的環境中運營。
注2.重要會計政策摘要
陳述的基礎
本公司已按照美國公認會計原則或美國公認會計原則編制隨附的綜合財務報表。本説明中對適用指南的任何提及均指財務會計準則委員會(FASB)的會計準則編纂(ASC)和會計準則更新(ASU)中的權威美國公認會計原則。
合併原則
該公司的綜合財務報表包括C4治療公司及其全資子公司、馬薩諸塞州證券公司C4T證券公司的賬目。所有重大的公司間餘額和交易都已在合併中沖銷。
預算的使用
根據美國公認會計原則編制合併財務報表要求管理層作出估計和假設,以影響截至合併財務報表日期的資產和負債額以及或有資產和負債的披露,以及報告期內的收入和支出的報告金額。本公司根據過往經驗及其認為在當時情況下合理的各種因素作出估計及假設。這一過程可能會導致實際結果與編制綜合財務報表時使用的估計金額大不相同,如果這些結果與歷史經驗不同,或者其他假設被證明不是實質上準確的,即使該等假設在做出時是合理的。管理層使用主觀判斷的領域包括但不限於根據本公司的研發合作安排確認的收入的金額和時間、預付和應計研發費用、長期資產使用壽命的估計、長期資產減值的評估、使用權資產和租賃負債的計量、用於計量租賃負債的遞增借款率以及基於股份的薪酬支出的估值和確認。本公司按持續基準評估估計,並根據過往經驗、已知趨勢及相信合理的其他各種假設作出估計,其結果構成對資產及負債賬面值作出判斷的基礎。實際結果可能與這些估計大相徑庭。
細分市場
運營部門被定義為企業的組成部分,其獨立和離散的信息可供首席運營決策者在決定如何分配資源和評估業績時進行評估。該公司擁有一運營部門。公司首席運營決策者兼首席執行官負責綜合管理公司的運營,以分配資源為目的。該公司的所有長期資產都在美國持有。
現金等價物
本公司將所有購買時到期日在三個月或以下的高流動性投資視為現金等價物。本公司的現金等價物按公允價值經常性計量。
受限現金
限制性現金包括根據本公司馬薩諸塞州沃特敦設施租賃協議條款的要求存入單獨受限銀行賬户的現金,如附註6中進一步描述的,租契.
有價證券
公司將購買之日剩餘到期日超過三個月的有價證券歸類為可供出售證券。剩餘到期日大於一年的有價證券歸類為非流動資產。可供出售證券按公允價值列賬,未實現損益計入累計其他綜合(虧損)收入,作為股東權益的組成部分,直至實現。在購買時產生的任何溢價或折扣將在票據有效期內攤銷和/或增加利息收入和/或費用。已實現損益採用特定的確認方法確定,並計入其他收入(費用)。
信用風險集中
可能使公司面臨嚴重集中信用風險的金融工具主要包括現金、現金等價物、有價證券和受限現金。本公司在金融機構的存款可以超過政府的保險限額。本公司相信其並無重大信貸風險,因為其存款存放於管理層認為具有高信貸質素的金融機構,而本公司並無因該等存款而蒙受任何損失。此外,該公司還制定了有關信用評級和到期日的指導方針,旨在保障本金餘額和維持流動性。該公司根據其投資政策維持其基金,該政策定義了允許的投資,規定了信貸質量標準,並旨在限制對任何單一發行人的信貸風險。
金融工具的公允價值
ASC主題820,公允價值計量或ASC 820,為按公允價值計量的工具建立了公允價值層次結構,區分了基於市場數據的假設(可觀察到的投入)和公司自己的假設(不可觀察到的投入)。可觀察到的投入是市場參與者在為資產定價時使用的投入或
基於從獨立於本公司的來源獲得的市場數據的責任。不可觀察到的投入是反映公司對市場參與者將用於為資產或負債定價的投入的假設,並基於當時可獲得的最佳信息而制定。ASC 820將公允價值確定為交換價格或退出價格,代表在市場參與者之間有序交易中出售資產所收到的金額或轉移負債所支付的金額。作為在公允價值計量中考慮市場參與者假設的基礎,ASC 820建立了一個三級價值層次結構,該層次結構區分了以下各項:
•第1級--反映活躍市場中相同資產或負債的活躍市場報價的可觀察投入。
•第2級-市場中直接或間接可觀察到的第1級投入以外的投入。
•第三級-不可觀察的投入,由很少或沒有市場活動支持,並可能使用公司制定的假設的估計來制定,這些假設反映了市場參與者可能使用的假設。
在某種程度上,估值是基於在市場上較難觀察到或無法觀察到的模型或投入,公允價值的確定需要更多的判斷。因此,本公司在釐定公允價值時所作出的判斷,對分類為第3級的工具的判斷程度最高。公允價值層次內的金融工具水平是以對公允價值計量有重大意義的任何投入中的最低水平為基礎。本公司在每個報告期結束時評估不同級別之間的轉移。
財產和設備
財產和設備按成本減去累計折舊入賬。維修和保養支出在發生時計入費用。當資產被報廢或處置時,資產和相關的累計折舊從賬目中取消確認,任何由此產生的收益或損失都計入淨虧損的確定。
設備折舊在資產的估計使用年限內採用直線法計算如下:
| | | | | | | | |
| 資產類別 | | 預計使用壽命 |
| 實驗室設備 | | 5年份 |
| 計算機設備 | | 3年份 |
| 辦公設備、傢俱和固定裝置 | | 5年份 |
| 租賃權改進 | | 使用年限或剩餘租賃期較短的 |
租契
本公司根據ASC主題842對租賃進行會計處理,租契,或ASC 842。於安排開始時,本公司根據當時的特定事實及情況、已確認資產(S)的存在(如有)及本公司對已確認資產(S)的使用控制權(如適用),決定該項安排是否屬租賃或包含租賃。根據ASC 842的規定,公司選擇將租賃和非租賃組成部分合併為所有租賃的單一組成部分。經營租賃負債及其相應的使用權資產根據預期租賃期內未來租賃付款的現值入賬。對於收到的獎勵等項目,可能需要對使用權資產進行某些調整。該公司在評估租賃安排時通常只包括初始租賃期;除非有合理確定該公司將續期,否則續訂租賃的選項不包括在評估中。租賃合同中隱含的利率通常不容易確定。因此,在計算租賃負債時,本公司採用其遞增借款利率,即在類似經濟環境下以抵押方式借入相當於租賃款項的金額所產生的利率。租賃費用在預期租賃期內以直線方式確認。
長期資產減值準備
每當事件或環境變化顯示某項資產的賬面價值可能無法收回時,本公司便會評估其長期資產的減值。將持有和使用的資產的可回收性是通過將資產的賬面價值與資產預期產生的未來未貼現淨現金流量進行比較來衡量的。如該等資產被視為減值,應確認的減值以該資產的賬面價值超過該資產的公允價值的金額計量。待處置資產按賬面值或公允價值減去出售成本中較低者列報。
承付款和或有事項
因索賠、評估、訴訟、罰款和罰款及其他來源產生的或有損失的負債,在很可能已發生負債且金額可以合理估計的情況下記錄。與或有損失相關的法律費用在發生時計入費用。
收入確認
公司根據ASC主題606確認收入,與客户簽訂合同的收入或ASC 606。本標準適用於所有與客户簽訂的合同,屬於其他標準範圍的合同除外。根據ASC 606,實體在其客户獲得對承諾的商品或服務的控制權時確認收入,其金額反映了實體預期為換取這些商品或服務而獲得的對價。
該公司與戰略合作伙伴簽訂了在ASC 606範圍內的合作和許可協議,根據該協議,它可以向第三方獨家許可其候選產品的研究、開發、製造和商業化的權利。這些安排的條款通常包括向公司支付以下一項或多項費用:(1)不可退還的預付許可費;(2)某些費用的報銷;(3)額外商品或服務的客户選擇費;(4)發展里程碑付款;(5)監管和商業里程碑付款;以及(6)特許產品淨銷售額的特許權使用費。
在確定履行每項協議規定的義務時應確認的適當收入數額時,公司執行以下步驟:(1)確定合同中承諾的貨物或服務;(2)確定承諾的貨物或服務是否是履約義務,包括它們在合同中是否不同;(3)交易價格的計量,包括對可變對價的限制;(4)將交易價格分配給履約義務;(5)當公司履行每項履約義務時(或作為履行義務時)確認收入。作為該等安排的會計安排的一部分,本公司必須根據其判斷釐定:(A)根據上文第(Ii)步的釐定而產生的履約責任數目;(B)上文第(Iii)步的交易價格;(C)上文第(Iv)步的交易價格分配合同所確定的每項履約責任的獨立售價;及(D)上文第(V)步的履約責任的合約條款及履行模式。本公司使用判斷來確定除特許權使用費外的里程碑或其他可變對價是否應包括在交易價格中,如下所述。交易價格按相對獨立銷售價格分配給每一項履約義務,公司在履行合同項下的履約義務時確認收入。
為滿足收入確認標準而欠本公司的款項或根據合作協議條款應付的合同款項在本公司的綜合資產負債表中作為應收賬款入賬。在滿足收入確認標準之前收到的金額作為遞延收入記錄在公司的綜合資產負債表中。預計在資產負債表日後12個月內確認為收入的金額被歸類為當期遞延收入。在資產負債表日後12個月內未被確認為收入的金額被歸類為遞延收入,扣除當期部分。
預付許可證費
如果對公司知識產權的許可被確定有別於協議中確定的其他承諾或履行義務,當許可轉讓給客户並且客户能夠使用許可並從中受益時,公司確認分配給許可的不可退還的預付費用的收入。在評估承諾或履行義務是否有別於其他承諾時,公司會考慮以下因素:客户的研究、製造和商業化能力;公司保留任何關鍵權利;以及相關專業知識在一般市場的可用性。此外,本公司會考慮客户能否在未收到餘下承諾的情況下,就其預期目的從承諾中獲益、承諾的價值是否取決於未兑現的承諾、是否有其他供應商可提供餘下的承諾,以及該承諾是否可與剩餘承諾分開識別。對於與其他承諾相結合的許可證,公司將行使判斷,以評估合併履行義務的性質,以確定合併履行義務是隨着時間的推移還是在某個時間點得到履行,如果隨着時間的推移,則確定為確認收入而衡量進展的適當方法。本公司在每個報告期內評估進度指標,並在必要時調整業績指標和相關收入確認。
客户選項
如果一項安排被確定包含允許客户獲得額外商品或服務的客户選擇權,作為客户選擇權基礎的商品和服務在安排開始時不被視為履約義務,因為它們取決於選擇權的行使。本公司評估客户對免費或折扣獲得額外商品或服務的實質性權利或選擇權的選擇。如果客户選擇權被確定為代表一項實質性權利,則該實質性權利在安排開始時被確認為單獨的履約義務。本公司根據相對獨立銷售價格將交易價格分配給材料權利,該相對獨立銷售價格是根據確定的折扣和客户行使期權的可能性確定的。分配給重大權利的金額最早在行使選擇權之前不會確認為收入。如果期權沒有行使,目標被終止,公司將加速並確認與實質性權利履行義務相關的所有剩餘收入。
研究和開發服務
公司合作協議下的承諾可能包括公司為客户或代表客户提供的研究和開發服務。公司研發工作產生的付款或報銷被確認為按毛數提供和提供服務,因為公司是此類工作的委託人。來自客户的報銷和對客户的付款是與客户合作關係的結果,而不是客户關係,如共同開發活動,作為研究和開發費用的減少記錄。
里程碑付款
在包括髮展里程碑付款的每項安排開始時,本公司評估里程碑是否被認為有可能實現,並使用最可能金額法估計交易價格中包含的金額。如果很可能不會發生重大的收入逆轉,相關的里程碑價值將包括在交易價格中。不在公司或被許可方控制範圍內的里程碑式付款,如監管批准,在收到這些批准之前不被認為是可能實現的。該公司對科學、臨牀、監管、商業和其他必須克服的風險等因素進行評估,以實現這一評估中的特定里程碑。在確定是否可能不會發生重大收入逆轉時,需要做出相當大的判斷。在隨後的每個報告期結束時,本公司會重新評估所有受限制的里程碑的實現概率,並在必要時調整其對整體交易價格的估計。任何此類調整都以累積追趕為基礎進行記錄,這將影響調整期間的收入和收益。
有關協作收入會計處理的進一步討論,請參閲注8.協作和許可協議。
研發費用
研發成本按已發生費用計入,包括與進行臨牀前研究和臨牀試驗相關的成本,主要包括工資、基於股票的薪酬和其他員工福利支出、實驗室相關用品和與公司研發活動相關的其他運營成本,包括分配的與設施相關的支出和聘請進行研發活動的外部供應商的外部成本。
臨牀試驗費用是該公司研究和開發費用的重要組成部分。在公司候選產品的持續開發過程中,公司歷來與代表公司進行各種臨牀試驗活動的第三方簽訂合同。作為編制合併財務報表過程的一部分,本公司必須估計其應計研究和開發費用。公司根據當時已知的事實和情況在合併財務報表中對截至每個資產負債表日期的應計費用進行估計,包括審查未平倉合同和採購訂單,與其人員溝通以確定已代表公司提供的服務,並在公司尚未收到發票或以其他方式通知實際成本的情況下估計所提供的服務水平和服務產生的相關成本。如果與第三方簽訂的合同金額被修改(例如,由於臨牀試驗方案或要執行的工作範圍的變化),公司將在預期的基礎上相應地修改相關的應計項目。合同範圍的修改在引起修改的事實變得合理確定的期間計入費用。
此外,向公司供應商支付的款項可能會超過所提供的服務水平,並導致預付費用,在這種情況下,此類金額將反映為預付費用和其他費用
流動資產。在應計服務費時,本公司估計將提供服務的時間段和每段時間需要花費的努力程度。如果實際執行服務的時間或努力程度與估計不同,公司將相應調整應計費用或預付費用的金額。未來收到的用於研究和開發活動的貨物或服務的不可退還的預付款將被遞延,並以預付費用和其他流動資產的形式資本化。資本化金額在相關貨物交付或提供服務時計入費用。
基於股票的薪酬
本公司適用ASC主題718,薪酬--股票薪酬,或ASC 718,以説明其基於股票的支付。根據ASC 718,公司決定獎勵是否應被分類並計入責任獎勵或股權獎勵。公司授予員工的所有股票獎勵都被歸類為股權獎勵,並在公司的財務報表中根據授予日期的公允價值確認,公允價值採用布萊克-斯科爾斯期權定價模型計量。布萊克-斯科爾斯期權定價模型使用基於股票的獎勵的預期期限、預期波動率、無風險利率、股息率和普通股的公允價值來估計股權獎勵的公允價值。
•本公司採用“簡化”方法估計預期期限,即預期期限等於期權的歸屬期限和合同期限的算術平均值。
•由於缺乏足夠的公司特定歷史和隱含波動率數據,本公司的預期波動率計算基於一組具有代表性的上市公司的歷史波動率,這些公司具有與本公司相似的特徵,包括產品開發階段和生命科學行業重點。
•期權預期期限內的無風險利率是根據期權授予期間有效的美國國債收益率曲線計算的。
•預期股息收益率假設為零由於本公司從未派發過股息,目前亦無計劃就其普通股派發任何股息。
•以股票為基礎的獎勵的普通股的公允價值是公司普通股在授予之日的市場報價。
•公司在發生沒收行為時予以確認。
獎勵的公允價值在獎勵的必要服務期(通常是歸屬期間)內按直線原則確認為基於股票的補償支出。對於同時受績效和基於服務的歸屬條件約束的獎勵,如果按照管理層的最佳估計,績效條件被認為有可能實現,我們將確認剩餘服務期內的基於股票的薪酬支出。以股票為基礎的補償費用在綜合業務和綜合損失報表中的分類方式與獲獎者的工資成本分類或獲獎者服務付款分類的方式相同。
股權獎勵的任何條款或條件的任何變化都被視為對獎勵的修改。遞增補償成本是指修改後的裁決的公允價值超過緊接其條款被修改前的原始裁決的公允價值的部分(如有),以修改日期的裁決的公允價值和其他相關因素為基礎計算。對於既得獎勵,公司確認修改發生期間的增量補償成本。對於未歸屬的補償,本公司確認剩餘必需服務期內的費用、增量補償成本和修改日期原始補償的剩餘未確認補償成本之和。如果修改後的裁決的公允價值低於緊接修改前的原始裁決的公允價值,公司確認的最低補償成本為原始裁決的成本。
所得税
本公司採用資產負債法核算所得税,該方法要求確認已在合併財務報表或本公司納税申報表中確認的事件的預期未來税務後果的遞延税項資產和負債。根據此方法,遞延税項資產及負債乃根據綜合財務報表與資產及負債的税基之間的差額而釐定,按預期差額將撥回的年度的現行税率釐定。遞延税項資產和負債的變動計入所得税準備。本公司評估其遞延税項資產從未來應課税收入中收回的可能性,並在其根據現有證據的權重認為全部或部分遞延税項資產更有可能無法變現的情況下,建立估值撥備。
本公司使用ASC主題740的規定評估其不確定的税務狀況,所得税,或ASC 740,它規定了税務狀況在綜合財務報表中確認之前需要滿足的確認門檻,該標準使用財務報表確認和計量納税申報單中採取或預期採取的税收狀況的“更可能”標準。所得税撥備包括被認為適當的任何由此產生的税收準備金或未確認的税收優惠的影響,以及相關的淨利息和罰款。
綜合損失
綜合損失被定義為企業在一段時期內因非所有者來源的交易和其他事件和情況而發生的權益變化。綜合虧損包括淨虧損以及股東權益的其他變化,其中包括不包括在淨虧損之外的某些權益變化。該公司其他全面收益的唯一組成部分是有價證券的未實現收益和虧損。
每股淨虧損
每股基本和攤薄淨虧損採用當期已發行普通股的加權平均數計算。本公司計算每股攤薄(虧損)收益時,會考慮期內已發行的股票期權及未歸屬限制性股票的攤薄效應,除非該等證券具有反攤薄作用。
倘普通股等值股份具有反攤薄影響,則計算每股攤薄虧損淨額時不包括該等股份。於本公司錄得虧損淨額之期間,每股攤薄虧損淨額一般與每股基本虧損淨額相同,原因為倘具反攤薄影響,則不會假設已發行具攤薄影響之普通股。
持續經營的企業
在每個報告期,本公司評估是否存在對本公司在綜合財務報表發佈日期後一年內持續經營能力產生重大疑問的條件或事件。如果公司認為存在重大疑問,並且公司的計劃沒有緩解,或者當公司的計劃緩解了對公司持續經營能力的重大疑問時,公司需要做出某些額外的披露。公司的評估需要分析未來的經營預算和對公司現金需求預期的預測,並將這些需求與當前的現金、現金等價物和有價證券餘額進行比較。
近期發佈的會計準則
於二零二三年十二月,FASB發佈ASU 2023-09,所得税(主題740):所得税披露的改進。或ASU 2023-09。ASU 2023-09要求公司的年度財務報表在費率調節中包括一致的類別和更多的信息分類,並按司法管轄區分類支付所得税。ASU 2023-09於2025年12月15日之後開始的公司年度報告期間生效。採用的是前瞻性方法或完全追溯性的過渡方法。允許提前採用。本公司現正評估採納會計準則第2023-09號對其綜合財務報表的影響。
注3.公允價值計量
下表呈列有關本公司按經常性基準以公允價值計量的金融資產的資料,並顯示截至2023年12月31日用於釐定該等公允價值的公允價值層級(以千計):
| | | | | | | | | | | | | | | | | | | | | | | |
| 公允價值 | | 1級 | | 2級 | | 3級 |
| 現金等價物: | | | | | | | |
| 貨幣市場基金 | $ | 103,564 | | | $ | 103,564 | | | $ | — | | | $ | — | |
| 美國國債 | 14,972 | | | — | | | 14,972 | | | — | |
| 公司債務證券 | 7,588 | | | — | | | 7,588 | | | — | |
| 有價證券: | | | | | | | |
| 公司債務證券 | 128,705 | | | — | | | 128,705 | | | — | |
| 美國政府債務證券 | 20,428 | | | — | | | 20,428 | | | — | |
| 美國國債 | 5,966 | | | — | | | 5,966 | | | — | |
| 現金等價物和有價證券總額 | $ | 281,223 | | | $ | 103,564 | | | $ | 177,659 | | | $ | — | |
下表載列本公司於2022年12月31日按公允價值層級劃分的金融資產公允價值(單位:千元):
| | | | | | | | | | | | | | | | | | | | | | | |
| 公允價值 | | 1級 | | 2級 | | 3級 |
| 現金等價物: | | | | | | | |
| 貨幣市場基金 | $ | 28,705 | | | $ | 28,705 | | | $ | — | | | $ | — | |
| | | | | | | |
| 美國國債 | 799 | | | — | | | 799 | | | — | |
| 有價證券: | | | | | | | |
| 公司債務證券 | 234,327 | | | — | | | 234,327 | | | — | |
| 美國政府債務證券 | 47,641 | | | — | | | 47,641 | | | — | |
| 美國國債 | 25,393 | | | — | | | 25,393 | | | — | |
| 現金等價物和有價證券總額 | $ | 336,865 | | | $ | 28,705 | | | $ | 308,160 | | | $ | — | |
本公司將其貨幣市場基金分類為公平值層級內的第一級資產,該等基金根據活躍市場的市場報價估值,並無估值調整。
有價證券包括美國國債、美國政府債務證券和公司債務證券,所有這些證券均根據ASC 320分類為可供出售, 投資--債務和股權證券。有價證券被歸類於公允價值等級的第二級,這是因為定價投入不同於活躍市場的報價,於報告日期可直接或間接觀察到,而公允價值是使用模型或其他估值方法定期確定的。
注4.有價證券
截至2023年12月31日的有價證券包括以下內容(以千為單位):
| | | | | | | | | | | | | | | | | | | | | | | |
| 攤銷 成本 | | 毛收入 未實現 收益 | | 毛收入 未實現 損失 | | 公平 價值 |
| 有價證券,當前: | | | | | | | |
| 公司債務證券 | $ | 100,903 | | | $ | 16 | | | $ | (221) | | | $ | 100,698 | |
| 美國政府債務證券 | 20,457 | | | 14 | | | (43) | | | 20,428 | |
| 美國國債 | 5,965 | | | 1 | | | — | | | 5,966 | |
| 非流通有價證券 | | | | | | | |
| 公司債務證券 | 27,901 | | | 120 | | | (14) | | | 28,007 | |
| | | | | | | |
| | | | | | | |
| 流通和非流通有價證券總額 | $ | 155,226 | | | $ | 151 | | | $ | (278) | | | $ | 155,099 | |
截至2022年12月31日的有價證券包括以下內容(以千為單位):
| | | | | | | | | | | | | | | | | | | | | | | |
| 攤銷 成本 | | 毛收入 未實現 收益 | | 毛收入 未實現 損失 | | 公平 價值 |
| 有價證券,當前: | | | | | | | |
| 公司債務證券 | $ | 183,270 | | | $ | 2 | | | $ | (2,068) | | | $ | 181,204 | |
| 美國政府債務證券 | 40,986 | | | — | | | (1,184) | | | 39,802 | |
| 美國國債 | 25,650 | | | — | | | (257) | | | 25,393 | |
| 非流通有價證券 | | | | | | | |
| 公司債務證券 | 53,592 | | | 2 | | | (471) | | | 53,123 | |
| 美國政府債務證券 | 8,000 | | | — | | | (161) | | | 7,839 | |
| | | | | | | |
| 流通和非流通有價證券總額 | $ | 311,498 | | | $ | 4 | | | $ | (4,141) | | | $ | 307,361 | |
被歸類為現貨的有價證券的到期日不到一年。被歸類為非流動證券的有價證券是指:(I)到期日超過一年,以及(Ii)我們不打算在未來12個月內清算的證券,儘管這些資金可供使用,因此被歸類為可供出售。沒有可用的-
截至2023年12月31日持有的待售債務證券或2021年12月31日剩餘到期日超過五年。
截至未實現虧損頭寸的有價證券2023年12月31日由以下部分組成(單位:千,證券數量除外):
| | | | | | | | | | | | | | | | | |
| 數量
證券 | | 公平
價值 | | 毛收入
未實現
損失 |
| 連續12個月未實現虧損的有價證券: | | | | | |
| 公司債務證券 | 64 | | $ | 68,563 | | | $ | (146) | |
| 美國政府債務證券 | 9 | | 11,914 | | | (22) | |
| | | | | |
| 連續未實現虧損超過12個月的有價證券: | | | | | |
| 公司債務證券 | 18 | | 22,138 | | | (89) | |
| 美國政府債務證券 | 3 | | 5,979 | | | (21) | |
| | | | | |
| 未實現虧損的有價證券總額 | 94 | | $ | 108,594 | | | $ | (278) | |
截至未實現虧損頭寸的有價證券2022年12月31日由以下部分組成(單位:千,證券數量除外):
| | | | | | | | | | | | | | | | | |
| 數量
證券 | | 公平
價值 | | 毛收入
未實現
損失 |
| 連續12個月未實現虧損的有價證券: | | | | | |
| 公司債務證券 | 63 | | $ | 134,027 | | | $ | (1,262) | |
| 美國政府債務證券 | 6 | | 15,748 | | | (245) | |
| 美國國債 | 4 | | 5,575 | | | (11) | |
| 連續未實現虧損超過12個月的有價證券: | | | | | |
| 公司債務證券 | 36 | | 82,375 | | | (1,277) | |
| 美國政府債務證券 | 7 | | 31,892 | | | (1,100) | |
| 美國國債 | 4 | | 19,817 | | | (246) | |
| 未實現虧損的有價證券總額 | 120 | | $ | 289,434 | | | $ | (4,141) | |
基於歷史經驗、市場數據、發行人特定因素和當前經濟狀況等因素,他的公司做到了不是It‘不要在2023年12月31日記錄信貸損失準備金和2022年12月31日,與這些證券相關。
注5.財產和設備
財產和設備由以下部分組成(以千計):
| | | | | | | | | | | |
| 截至12月31日, |
| 2023 | | 2022 |
| 財產和設備 | | | |
| 實驗室設備 | $ | 8,042 | | | $ | 8,757 | |
| 租賃權改進 | 4,712 | | | 4,682 | |
| 傢俱和固定裝置 | 1,422 | | | 1,181 | |
| 辦公設備 | 621 | | | 529 | |
| 計算機設備 | 98 | | | 191 | |
| 在建工程 | — | | | 183 | |
| 總資產和設備 | 14,895 | | | 15,523 | |
| 減去:累計折舊 | (7,763) | | | (8,123) | |
| 財產和設備合計(淨額) | $ | 7,132 | | | $ | 7,400 | |
與財產和設備有關的折舊費用如下(以千計):
| | | | | | | | | | | | | |
| 截至十二月三十一日止的年度, |
| 2023 | | 2022 | | |
| 折舊費用 | $ | 1,578 | | | $ | 1,676 | | | |
注6.租契
2017年7月,該公司簽訂了馬薩諸塞州沃特敦總部辦公和實驗室空間的租賃合同,或沃特敦租賃。沃特敦租約有一個不可取消的期限:十年具有可擴展的選項一其他內容五年制在整個租期內,租金將會上升。此外,沃特敦租賃公司要求該公司提供抵押品,這些抵押品在綜合資產負債表上被記為限制性現金。水城租賃於2018年4月開始。水城租賃被歸類為經營租賃,於2018年4月開始時,本公司記錄了#美元的租賃負債。15.1100萬美元的使用權資產16.7百萬美元,其中包括$1.5百萬美元的建設成本由公司出資。在最初計算租賃負債和使用權資產時,公司沒有包括額外的五年制由於管理層不相信本公司會行使該期權,故有合理把握行使該期權。除租金外,本公司還負責按比例支付與租賃空間相關的公共區域維護、房地產税和財產保險所產生的按比例分攤的費用,這些費用被計入可變租賃成本。
於2021年11月,本公司對水城租約或經修訂租約作出修訂。修訂租賃旨在延長本公司現有租賃空間或現有租賃空間的租賃期,並提供額外的辦公和實驗室空間或新租賃空間。經修訂租賃於二零二二年一月開始生效,而本公司就新租賃空間支付租金的責任於二零二二年三月開始生效,新租金責任的金額已加入本公司就現有租賃空間支付租金的持續責任中。經修訂的租約將於2032年3月終止,即10自新租賃空間起租之日起計數年。經修訂的租約須按固定租金加租,最高可達$2.6,併為本公司提供選擇,以延長經修訂租約的租約期一其他內容五年制句號。於簽訂經修訂租約後,本公司將抵押品增加至$3.3截至2023年12月31日、2023年12月31日和2022年12月31日,這筆現金在隨附的合併資產負債表上列為限制性現金。除租金外,根據經修訂租賃條款,本公司亦負責按比例支付與租賃空間有關的公用區域維護、房地產税及財產保險所產生的按比例分攤成本,包括現有租賃空間及於2022年1月新租賃的空間,該等款項計入變動租賃成本。
經修訂租約的會計處理
由於經修訂租約延長現有租賃空間的年期並提供使用新租賃空間的通道,根據ASC 842的規定,本公司將經修訂租約入賬為二單獨的合同:1)修改現有租賃協議,以延長現有租賃空間的租賃期限,以及2)新租賃空間的使用權的新租賃協議。
由於本公司於簽訂經修訂租約後維持對現有租賃空間的控制,本公司錄得使用權資產及租賃負債增加#美元。20.1於經修訂租賃籤立後,現有租賃空間於經延長租期內的新租金支付及目前的遞增借款利率所涉及的百萬歐元。計算現有租賃空間的租賃負債和使用權資產不包括額外的五年制由於本公司並不相信有合理確定性購股權將獲行使,故本公司並無就該等購股權授出任何購股權。
如上文所述,新租賃空間的租賃於二零二二年一月開始。因此,本公司錄得使用權資產$44.4及相應的租賃負債44.1萬元,新租的空間。新租賃空間的租賃負債及使用權資產的計算並不包括額外 五年制由於本公司不相信有合理確定性,該選擇權將被行使。如上所述,修訂後的租約規定,2.6截至2022年12月31日止年度,租户改善津貼已全數動用。
於2022年5月,本公司與第三方就位於馬薩諸塞州沃特敦阿森納路490號200室的部分辦公室及實驗室空間訂立分租協議(或分租)。該轉租的租期於二零二五年六月屆滿。
租賃費用的組成部分如下(以千計):
| | | | | | | | | | | | | |
| 截至十二月三十一日止的年度, |
| 2023 | | 2022 | | |
| 租賃費: | | | | | |
| 經營租賃成本 | $ | 10,030 | | | $ | 10,031 | | | |
| 可變租賃成本 | 1,979 | | | 2,026 | | | |
| 轉租收入 | (2,577) | | | (1,481) | | | |
| 總租賃成本 | $ | 9,432 | | | $ | 10,576 | | | |
下表概述達致租賃負債時所應用之租期及增量借款利率:
| | | | | | | | | | | |
| 截至12月31日, |
| 2023 | | 2022 |
| 剩餘租期 | 8.1年份 | | 9.1年份 |
| 增量借款利率 | 5.3 | % | | 5.3 | % |
截至2023年12月31日的不可撤銷租賃下,截至2023年12月31日的每一年的未來租賃付款如下(以千為單位):
| | | | | |
| 未貼現的租賃付款: | |
| 2024 | $ | 8,828 | |
| 2025 | 9,093 | |
| 2026 | 9,366 | |
| 2027 | 9,646 | |
| 2028 | 11,362 | |
| 此後 | 40,751 | |
| 未貼現的租賃付款總額 | 89,046 | |
| 減去:推定利息 | (18,070) | |
| 截至2023年12月31日的總經營租賃負債 | $ | 70,976 | |
注7.應計費用和其他流動負債
應計費用和其他流動負債包括以下各項(以千計):
| | | | | | | | | | | |
| 截至12月31日, |
| 2023 | | 2022 |
| 應計費用和其他流動負債: | | | |
| 應計研究和開發 | $ | 11,243 | | | $ | 9,824 | |
| 應計薪酬和福利 | 7,344 | | | 6,831 | |
| | | |
| 其他 | 2,043 | | | 3,114 | |
| 應計費用和其他流動負債總額 | $ | 20,630 | | | $ | 19,769 | |
截至2023年12月31日,3.1與CFT8634的逐步結束活動有關的成本中的1.6億美元包括在應計研發餘額中。
注8.協作和許可協議
默克許可和協作協議
2023年12月11日,該公司與默克公司或默克協議簽訂了獨家許可和合作協議,開發降解物-抗體結合物(DAC),這是一種新興的方法,旨在選擇性地靶向和中和癌細胞中的致病蛋白。
根據默克協議的條款,公司收到了一份$10.0百萬2024年1月預付款。該公司和默克公司將合作開發針對最初未披露的腫瘤學目標的DAC,這是合作所獨有的。對於針對這一初始目標的DAC,公司有資格獲得總計約為$600600萬美元,以及未來銷售的分級版税。該協議還為默克公司提供了擴大合作範圍的選項,包括三合作獨有的其他目標,這可能會產生期權行權付款以及潛在的里程碑和特許權使用費。如果默克公司行使其所有延長合作的選擇權,該公司將有資格獲得最高約$2.5整個協作過程中的潛在付款金額高達2000億美元。
合作由一個聯合研究委員會或JRC管理,該委員會由默克公司和該公司的代表組成。在某些情況下,每一方還擁有各種終止權,包括但不限於監管安全中斷、專利挑戰或另一方的重大違約,但須受某些條件的限制。終止協議須視乎司法人員敍用委員會的決定。
默克協議會計
該公司在默克協議開始時確定了一項履行義務,其中包括:(1)獨家許可證和(2)最初未披露的腫瘤學目標和聯合研究計劃的研究活動。本公司認定,許可證和研究活動彼此沒有區別,因為如果沒有本公司進行的研究活動,許可證的價值有限。參與默克聯合研究中心以監督與默克協議相關的研究活動和技術轉讓被確定為在數量和質量上都無關緊要,因此被排除在履行義務之外。本公司確認分配給這一履約義務的交易價格,因為使用輸入法提供的研究和開發服務,與每個研究發展目標迄今發生的成本與為履行基本義務而發生和預計未來發生的總成本相比,是成比例的。控制權的移交發生在這一時期,在管理層看來,這是在履行履約義務方面取得進展的最佳衡量標準。
截至2023年12月31日,交易總價為$10.0百萬被分配給研究和開發服務業績義務和$10.0百萬分配的成交價的一部分仍未得到滿足。
應付本公司尚未收到的款項記為應收賬款,收到的尚未確認為收入的款項在本公司的綜合資產負債表中記為遞延收入。
Betta Pharma許可和協作協議
2023年5月29日,公司與Betta Pharma簽訂許可與合作協議,或Betta Pharma許可協議,以進行合作關於CFT8919在大中國地區的開發和商業化,
本公司由內地中國、香港特別行政區、澳門特別行政區及臺灣組成,並保留大中國或C4T地區以外的世界其他地區的CFT8919的權利。
根據Betta Pharma許可協議的條款,本公司根據本公司的若干知識產權授予Betta Pharma獨家許可,以開發、製造和商業化用於中國大區人類所有用途的CFT8919。貝塔製藥負責在大中國地區的所有開發、監管審批、製造和商業化成本,但在公司贊助的一項全球試驗中擔任公司在大中國地區的代理的情況除外。作為合作的一部分,Betta Pharma預付了1美元的現金10.0百萬美元給公司,並已同意最高可達357.0總計里程碑付款100萬英鎊,外加大中國地區淨銷售額CFT8919的分級特許權使用費。這些款項由人民Republic of China國家税務機關代扣代繳。Betta Pharma支付給公司的特許權使用費從低到中兩位數的百分比不等,在Betta Pharma許可協議中描述的某些情況下可能會有一定的減免。此外,作為合作的一部分,公司已同意向Betta Pharma支付里程碑式的款項,金額最高可達$40.0在該公司收到FDA批准的CFT8919新藥申請後,這一里程碑式的金額基於Betta Pharma公司登記的預期臨牀試驗中患者的百分比和批准的治療路線。此外,該公司還同意就C4T地區CFT8919的淨銷售額在較低的個位數百分比範圍內向Betta Pharma支付分級特許權使用費,但在Betta Pharma許可協議中描述的某些情況下可能會減少。
關於簽署Betta Pharma許可協議,本公司Betta Pharma與Betta Pharma的關聯公司(Betta Investment(Hong Kong)Limited,或Betta Investment)簽訂了一份股票購買協議,協議日期為#年。2023年5月29日,或Betta股票購買協議,以及Betta Pharma許可協議或Betta協議,根據該協議,Betta Investment同意購買5,567,928公司普通股,或股份,總收購價約為$25.0百萬美元,或美元4.49每股,這代表着一個25較鬥魚購股協議生效日期前兩個交易日的60個交易日成交量加權平均收盤價溢價%。《鬥魚股份購買協議》對此類協議有某些慣常的限制。由於鬥魚購股協議下的交易直到2024年1月4日才完成,截至2023年12月31日,鬥魚購股協議下的交易並無記錄。
協作是我的由Betta Pharma和該公司的代表組成的聯合指導委員會負責管理。在完成CFT8919第一階段試驗的劑量遞增階段後,為了方便起見,Betta Pharma可能至少在以下情況下終止Betta Pharma許可協議90提前幾天發出書面通知。在某些情況下,每一方還擁有各種終止權,包括但不限於監管安全中斷、專利挑戰或對方的實質性違約,但須受某些條件的限制。
貝塔協議會計
該公司預計將根據Betta協議確認一種類型的安排--許可協議--的收入。BETTA協議將包括以下活動:(1)知識產權許可,(2)臨牀製造供應協議,(3)製造技術轉讓,(4)商業製造供應協議。自.起2023年12月31日,目前交易總價為$12.0百萬美元,其中包括10.0百萬美元的預付現金對價和2.0根據Betta Pharma許可協議,2023年12月實現了100萬個里程碑。與Betta協議相關的收入確認預計將在根據臨牀製造供應協議交付臨牀供應時開始,該協議截至2023年12月31日。該公司已收取淨額#美元。9.0從Betta Pharma的預付款中獲得100萬美元,在支付了相關的預扣税$1.0向中國税務機關繳税(見附註12)。截至日前,尚未確認任何收入2023年12月31日,關於分配給Betta協議的交易價格。
應付本公司尚未收到的款項記為應收賬款,收到的尚未確認為收入的款項在本公司的綜合資產負債表中記為遞延收入。
羅氏協作和許可協議
2016年3月,本公司與羅氏簽訂許可協議,該協議於2016年6月修訂,並於2017年3月再次修訂。本公司和羅氏於2018年12月修訂並重述了該協議(經如此修訂)。這項修訂和重述的協議稱為羅氏協議。根據羅氏協議,該公司和羅氏公司同意合作,利用公司專有的魚雷平臺治療癌症和其他適應症,研究、開發、製造和商業化具有靶向約束力的降解藥。根據羅氏協議,公司可以選擇加入某些共同開發權,在這種情況下,公司將
從針對該目標的產品獲得更高的未來產品銷售的版税。此外,如果公司選擇某些共同詳述權,它還有權獲得某些商業化成本的補償。在簽訂羅氏協議後,公司收到額外的預付代價#美元40.0百萬美元。
2020年11月,本公司簽署了一項進一步的修正案,其效果是規定各方將發展至五潛在目標,羅氏保留針對這些目標的產品許可和商業化的選擇權。2020年11月的修訂還提供了一種機制,通過該機制,本公司和羅氏可以通過簽訂共同目標終止協議,以逐個目標的方式相互同意終止羅氏協議。一旦簽訂共同目標終止協議,羅氏協議規定,支持以抑制為其行動模式的產品的專有技術和其他知識產權的所有權利和責任歸羅氏所有,支持以降級為其行動模式的產品的專有技術和其他知識產權的所有權利和責任歸羅氏所有。為支持這一權利分配,羅氏向本公司提供永久不可撤銷的、全額繳足的、獨家的(即使對於授權方)、可再許可(包括多個層次)的專利和專有技術許可,用於根據相互目標終止協議分配給一方的專利和專有技術。隨着與羅氏的研究活動隨着時間的推移而進步和發展,現在有二各方繼續合作的目標,羅氏維持其許可和商業化針對這些目標的產品的選擇權二目標。2023年12月,本公司簽署了羅氏協議的第二次修正案,其效果是更新協議的條款,因為它與二各方繼續合作的目標。根據《羅氏協議》的第二項修正案,羅氏保留對針對這些目標的產品進行許可和商業化的選擇權,但其選擇權的時間調整為在羅氏收到劑量範圍發現數據包後開始。《羅氏協定》第二次修訂對2023年的會計沒有實質性影響。
根據經修訂的羅氏協議,公司收到年度研究計劃付款#美元1.0每一項積極的研究計劃都有100萬美元。對於二對於各方仍在合作的目標,羅氏需要向公司支付#美元的費用2.0在目標進展到領導系列確定成就階段時,將有100萬美元的資金。如果羅氏對這些目標之一行使期權,羅氏必須向公司支付#美元的期權行權費8.0百萬美元。
根據修訂後的羅氏協議,對於羅氏行使的每個目標期權,公司有資格獲得最高達#美元的里程碑付款273.0在與相應產品相關的某些開發里程碑實現後,可根據知識產權覆蓋範圍進行某些減少和排除。羅氏還被要求向公司支付高達#美元的費用。150.0在以一次性銷售為基礎的里程碑付款中,每一目標在針對該目標的產品達到指定的淨銷售額水平時支付百萬美元。最後,羅氏需要向公司支付按羅氏根據其行使的選擇權銷售的產品淨銷售額的個位數中位數至十幾歲中期不等的分級特許權使用費,但須有一定的減幅。對於公司行使共同開發權的產品的銷售,適用的特許權使用費税率將以較低的個位數百分比增加。
該合作由一個聯合研究委員會(JRC)管理。在羅氏對特定目標行使其期權之前,公司對JRC擁有控制權,此後羅氏接管JRC,羅氏可在幾種情況下逐個目標或逐個產品終止羅氏協議,至少在90提前幾天發出書面通知。
羅氏協議會計
在啟動時,該公司確定了十二《羅氏協議》中的履行義務,由六潛在的研發目標當時包括在合作中,以及羅氏對每個目標持有的選擇權六目標。使用該公司的知識產權進行研發活動以及參與JRC的非獨家免版税許可被確定為承諾的服務。然而,本公司認為,研發許可證和研發服務彼此沒有區別,對JRC的參與在數量和質量上都無關緊要。
羅氏協議的交易總價根據履約義務的相對獨立銷售價格分配給履約義務。分配的交易價格通過以下兩種方式之一確認為來自協作協議的收入:
•研究和開發目標:本公司確認分配給每項研究和開發業績義務的交易價格部分,即使用一種投入方法提供研究和開發服務時,與每個研究開發目標迄今發生的成本與為履行基本義務而發生和預計未來發生的總成本的比例
與所述研發目標相關。控制權的移交發生在這一時期,在管理層看來,這是在履行履約義務方面取得進展的最佳衡量標準。
•期權:分配給期權的交易價格被認為是實質性權利,在羅氏選擇行使或不行使期權以許可潛在研究和開發目標並將其商業化的期間確認。
下表彙總了總交易價格對該安排下確定的履約義務的分配情況,以及截至2023年12月31日未滿足的交易價格金額(以千為單位):
| | | | | | | | | | | |
| 交易記錄 價格 已分配 | | 交易記錄 價格 不滿意 |
| 履約義務: | | | |
| 研發目標 | $ | 61,149 | | | $ | 17,394 | |
| 期權權利 | 6,757 | | | 1,670 | |
| 總計 | $ | 67,906 | | | $ | 19,064 | |
應付本公司尚未收到的款項記為應收賬款,收到的尚未確認為收入的款項在本公司的綜合資產負債表中記為遞延收入。
生物遺傳研究合作與許可協議
2018年12月,該公司與生物遺傳公司簽訂了合作研究和許可協議,或生物遺傳協議。2020年2月,本公司和Biogen修訂了Biogen協議,以進一步明確Biogen對目標結合部分(分子的一部分)的所有權,以及針對合作目標或與合作目標綁定的任何相關知識產權。此項修訂進一步規定,Biogen授權本公司使用此等Biogen目標約束性部分及任何相關知識產權(視需要而定),以進行Biogen協議項下預期的研究及開發活動。根據生物遺傳基因協議的條款,該公司和生物遺傳基因公司同意合作開展研究活動,通過依賴靶向蛋白質降解(TPD)作為其作用模式的藥物,開發治療阿爾茨海默病和帕金森病等神經疾病的新療法,所有這些藥物都是使用公司的降解器技術創造的。根據生物遺傳協議的條款,該公司致力於開發利用降解劑技術的TPD療法五靶蛋白在一段時間內54幾個月,2023年6月結束。在逐個目標的基礎上,在成功完成規定的目標評估期後,生物遺傳公司承擔繼續發展每個目標的全部權利和責任。
以換取Biogen的非獨家研究許可證,以及$45.0公司已獲得許可,可開發、商業化和生產與每個目標相關的產品(這取決於不取消協議),為藥物發現提供初步研究服務,為其知識產權提供非獨家研究和商業許可,並參與聯合指導委員會或生物遺傳研究聯合委員會。該公司還有義務參與其他潛在目標的早期研究活動或沙箱活動,由生物遺傳研究公司選擇,最高金額不得超過;為這些服務進行的任何工作都由生物遺傳研究公司報銷,生物遺傳研究公司向公司報銷某些FTE費用。截至2021年8月31日,公司在沙箱活動下的義務已完成。對於任何目標,在達到開發候選標準之後,在任何支持IND的研究之前,生物遺傳公司將承擔所有費用和開支,並將擁有與生物遺傳研究協議項下正在開發的任何降解物和所有包含該降解物的產品有關的進一步活動的唯一自由裁量權和決策權。生物遺傳公司還被要求向公司支付高達$35.0發展里程碑中每個目標的百萬美元和美元26.0每個目標以一次性銷售額為基礎的付款為第一個產品,以實現一定水平的淨銷售額。此外,生物遺傳公司還需要根據全球產品淨銷售額,逐個許可產品向公司支付特許權使用費。所有里程碑式的付款和基於銷售的付款都是在公司達到該目標的聯合研究計劃中規定的標準後支付的,屆時生物遺傳公司將控制與商業化目標相關的產品;這些付款的收到取決於針對生物遺傳公司商業化目標的產品的進一步開發,而不需要公司做出任何額外的研究和開發努力。
合作由Biogen JSC管理,Biogen對該JSC擁有控制權,並且Biogen可以在幾種情況下逐個目標或逐個產品地終止Biogen協議,至少在90提前幾天發出書面通知。
生物遺傳協議會計
根據生物遺傳研究和開發服務協議,該公司確認的收入如下:
•研發服務:該公司確定了一《生物遺傳協議》開始時的履約義務,是一項綜合履約義務,包括(1)許可證,(2)面向所有人的目標評價階段的研究活動五目標和(3)每個目標的聯合研究計劃階段。本公司認定,許可證和研究活動彼此沒有區別,因為如果沒有本公司進行的研究活動,許可證的價值有限。參與生物遺傳研究中心監督研究活動和與生物遺傳許可協議相關的技術轉讓被確定為在數量和質量上都不重要,因此被排除在履行義務之外。本公司確認分配給這一履約義務的交易價格,因為使用輸入法提供的研究和開發服務,與每個研究開發目標迄今發生的成本與為履行與所述研究和開發目標相關的基本義務而發生和預計未來發生的總成本相比,是成比例的。控制權的移交發生在這一時期,在管理層看來,這是在履行履約義務方面取得進展的最佳衡量標準。
截至2023年12月31日,交易總價為美元55.0百萬美元用於研究和開發服務業績義務和#美元0.8分配的成交價中有100萬仍未得到滿足。
應付本公司尚未收到的款項記為應收賬款,收到的尚未確認為收入的款項記入本公司綜合資產負債表的遞延收入。
Calico協作和許可協議
2017年3月,本公司與Calico簽訂合作及許可協議,或Calico協議,根據該協議,本公司與Calico同意合作開發小分子蛋白質降解劑並將其商業化,用於治療包括癌症在內的老年性疾病,為期五年制截止日期為2022年3月。2021年8月,該公司向Calico提供了將某一項目的研究期限延長至多一年的選擇權一年制截止日期為2023年3月。2021年9月,Calico行使了選擇權,產生了一美元1.0向公司支付百萬延期付款。此外,Calico將根據研究階段的不同,繼續按特定的市場費率向公司償還一些FTE。截至2023年3月13日,印花布協議的研究期限已經到期,公司與該協議相關的研究活動基本完成。
根據Calico協定,Calico預付了#美元。5.0百萬元及若干年度付款合共$5.0至2020年6月30日,根據研究階段的不同,按指定的市場費率支付目標啟動費用並向公司報銷若干FTE。對於每個目標,公司有資格獲得最高$132.0潛在開發和商業里程碑付款為百萬美元,來自協作努力產生的所有產品的銷售。Calico還被要求向公司支付最高#美元65.0以一次性銷售百萬元為基礎的第一款產品實現一定水平的淨銷售額。此外,Calico還需要按授權產品按授權產品、按全球產品淨銷售額按個位數中位數的百分比向公司支付版税。所有里程碑式的付款和基於銷售的付款都是在公司達到該目標的聯合研究計劃中規定的標準之後進行的,屆時Calico將控制與商業化目標相關的產品;公司是否收到這些付款取決於Calico對商業化產品目標的進一步發展,而不需要公司需要任何額外的研究和開發努力。
截至2023年12月31日,交易總價為美元13.0向研發服務履約義務分配了100萬美元,交易價格已全部分配並得到滿足。
應付本公司尚未收到的款項在本公司的簡明綜合資產負債表上記為應收賬款。
從協作協議確認的收入彙總
來自協作協議的收入如下(以千為單位):
| | | | | | | | | | | | | |
| 截至十二月三十一日止的年度, |
| 2023 | | 2022 | | |
| 來自協作協議的收入: | | | | | |
| 羅氏協議 | $ | 9,063 | | | $ | 4,939 | | | |
| 生物遺傳協議 | 10,623 | | | 19,214 | | | |
| 印花布協議 | 1,070 | | | 6,943 | | | |
| 《貝塔協議》 | — | | | — | | | |
| 來自協作協議的總收入 | $ | 20,756 | | | $ | 31,096 | | | |
與協作和許可協議相關的補充信息包括公司截至2023年12月31日的綜合資產負債表中的以下內容(以千計):
| | | | | | | | | | | | | | | | | | | | | | | |
| 帳目 應收賬款 | | 延期 收入, 當前 | | 延期 收入, 海流淨額 | | 延期 收入, 總計 |
| 補充信息: | | | | | | | |
| 羅氏協議 | $ | — | | | $ | 2,667 | | | $ | 11,814 | | | $ | 14,481 | |
| 《貝塔協議》 | 1,799 | | | 4,000 | | | 8,000 | | | 12,000 | |
| 默克協議 | 10,000 | | | 8,000 | | | 2,000 | | | 10,000 | |
| 生物遺傳協議 | — | | | 804 | | | — | | | 804 | |
| | | | | | | |
| 總計 | $ | 11,799 | | | $ | 15,471 | | | $ | 21,814 | | | $ | 37,285 | |
與協作和許可協議相關的補充信息包括公司截至2022年12月31日的綜合資產負債表中的以下內容(以千計):
| | | | | | | | | | | | | | | | | | | | | | | |
| 帳目 應收賬款 | | 延期 收入, 當前 | | 延期 收入, 海流淨額 | | 延期 收入, 總計 |
| 補充信息: | | | | | | | |
| 羅氏協議 | $ | 417 | | | $ | 4,649 | | | $ | 16,895 | | | $ | 21,544 | |
| 生物遺傳協議 | — | | | 11,427 | | | — | | | 11,427 | |
| 印花布協議 | 1,056 | | | 542 | | | — | | | 542 | |
| 總計 | $ | 1,473 | | | $ | 16,618 | | | $ | 16,895 | | | $ | 33,513 | |
與協作和許可協議相關的補充財務信息包括(以千計):
| | | | | | | | | | | | | |
| 截至十二月三十一日止的年度, |
| 2023 | | 2022 | | |
| 在期初計入合同負債的已確認收入 | $ | 20,185 | | | $ | 25,412 | | | |
| 以前各期全部或部分履行履約義務所確認的收入 | 43 | | | 518 | | | |
截至2023年12月31日,根據羅氏協議、貝塔協議、默克協議和生物遺傳協議分配給履約義務的部分未得到滿足的交易價格總額為美元41.9百萬美元。
注9.長期債務和權證關聯方
與長期債務有關的一方由以下部分組成(以千計):
| | | | | | |
| | 十二月三十一日,
2022 |
定期貸款相關方,扣除未攤銷債務發行成本和債務貼現#美元后的淨額1,0182022年12月31日 | | $ | 11,482 | |
| 減:當前部分 | | 2,287 | |
| 長期債務關聯方總額,扣除當期和貼現 | | 9,195 | |
2020年6月5日,在完成B輪融資的同時,本公司與Perceptive Advisors LLC或Perceptive的關聯公司Perceptive Credit Holdings III,LP簽訂了一項信貸協議,規定本金借款總額最高可達$20.0百萬美元,分兩批提供,每批$12.5百萬美元和美元7.5百萬美元。基於感知在信貸協議開始時對本公司普通股的所有權,感知被視為本公司的關聯方。
2020年6月,本公司提取了第一批#美元12.5百萬美元,或定期貸款。該公司決定不提取於2021年6月30日到期的第二批資金。
於2023年5月17日,本公司訂立信貸協議修正案,據此,本公司及其貸款人同意以SOFR取代LIBOR基準,SOFR受紐約聯邦儲備銀行監管。由於金融市場行為監管局計劃於2023年6月30日逐步淘汰LIBOR,該公司修改了其信貸協議。SOFR匯率的使用自2023年7月1日起生效。
2023年7月,根據信貸協議的條款,本公司支付預付款費用的義務已被取消。2023年7月26日,公司選擇償還定期貸款的剩餘未償還本金餘額。關於償還定期貸款的未償還餘額,解除了對公司幾乎所有資產的留置權。公司發生了一筆$0.6截至2023年12月31日的年度,這筆債務的清償虧損為100萬英鎊。
截至2023年12月31日和2022年12月31日的年度的利息支出為$1.0百萬美元和美元1.5百萬美元。
注10.股東權益
普通股
2020年10月,本公司授權發行的優先股為10,000,000並將其可發行的法定普通股增加到150,000,000股票,兩者都有$0.0001每股面值。
普通股持有者有權一在所有股東會議上為每股股份投票,並以書面行動代替規定的會議。在優先股持有人有權獲得的所有優先股息全部付清的情況下,普通股持有人有權從合法可用資金中獲得股息。如果公司發生清算、解散或清盤,在支付或撥備支付公司的所有債務和負債以及優先股持有人就清算資產分配而有權獲得的所有優先金額後,普通股持有人有權按比例分享公司剩餘可供分配的資產。普通股持有人沒有優先認購權或其他認購權,也沒有關於普通股的贖回或償債基金的規定。
市場上的股票計劃
2021年11月,本公司向美國證券交易委員會提交了自動生效的S-3表格登記説明書,對發行、發行和銷售數額不詳的普通股、優先股、債務證券、權證和/或其任何組合的單位進行登記。同時,公司與作為銷售代理的Cowen and Company,LLC簽訂了一項股權分配協議,以規定公司發行和出售至多$200.0根據註冊聲明和與註冊聲明或自動櫃員機計劃一起提交的相關招股説明書,不時在“在市場”發行的普通股達數百萬股。截至2023年12月31日的年度,11,186,142淨收益為$的股份57.7100萬美元,通過自動取款機計劃結算。2024年1月,2,500,601股票結算的淨收益為#美元。14.11000萬美元。基於上述2023年12月和2024年1月的活動,本公司共出售了13,686,743其普通股的平均價格為$5.42每股收益,扣除佣金和手續費後為$71.81000萬美元。不是銷售是根據ATM計劃在截至2022年12月31日的年度內完成的。
注11.基於股票的薪酬
2015年激勵性股票期權和獎勵計劃
2015年12月28日,公司董事會通過2015年度激勵性股票期權與授予計劃,或2015年度計劃,並預留2,525,327根據本計劃發行的普通股。
2015年計劃授權董事會或董事會委員會向符合條件的公司員工、外部董事和顧問授予激勵性股票期權、非限制性股票期權和限制性股票獎勵。根據2015年計劃授予的期權通常在五或八年有一座懸崖位於一年此後的季度歸屬以及失效或被沒收的期權可以再次授予。根據2015年計劃授予的所有備選方案的合同期限為十年自授予之日起生效。
2020年股票期權和激勵計劃
2020年9月8日,公司董事會通過了C4治療公司2020年股票期權和激勵計劃,或2020年計劃,並於2020年9月30日生效。在被收養後,有6,567,144根據2020年計劃為發行保留的普通股。本公司董事會、董事會薪酬委員會,以及在某些情況下,本公司首席執行官有權根據2020年計劃授予廣泛的股票獎勵,包括股票期權、股票增值權或SARS、限制性股票獎勵或RSA、限制性股票單位或RSU、績效獎勵和股票紅利獎勵給公司的高級管理人員、員工、董事和包括顧問在內的其他關鍵人員。
自2020年計劃生效後,本公司停止發放2015年計劃下的贈款。然而,2015年計劃繼續管理根據該計劃授予的未決裁決的條款和條件。根據2015年計劃授予獎勵的普通股股票,如果在2015計劃終止後不再通過沒收或其他方式獲得此類獎勵,將可根據2020年計劃發行。截至2023年12月31日,有3,358,336根據2020年計劃,可供未來授予的股份。
2020年計劃規定每年增加,從2021年1月1日開始,在每個財政年度的第一天增加,一直持續到2020年計劃期滿,等於(I)5(二)2020年度計劃管理人(公司董事會或董事會薪酬委員會)確定的較少數量的普通股。2024年1月1日,2020計劃的年度增長導致了額外的3,023,359授權發行的股票被添加到2020年計劃中。
基於股票的薪酬支出如下(單位:千):
| | | | | | | | | | | | | |
| 截至十二月三十一日止的年度, |
| 2023 | | 2022 | | |
| 基於股票的薪酬費用: | | | | | |
| 研發 | $ | 11,826 | | | $ | 13,108 | | | |
| 一般和行政 | 15,409 | | | 16,908 | | | |
| 基於股票的薪酬總支出 | $ | 27,235 | | | $ | 30,016 | | | |
下表彙總了截至2023年12月31日的年度公司股權獎勵計劃下的股票期權活動:
| | | | | | | | | | | | | | | | | | | | | | | |
| 數量
選項 | | 加權平均
行權價格 | | 加權平均
剩餘
合同
期限(年) | | 集料
固有的
價值
(單位:千) |
| 截至2022年12月31日的未償還債務 | 6,993,052 | | | $ | 21.57 | | | 7.78 | | $ | 1,031 | |
| 授與 | 4,180,270 | | | 3.87 | | | | | |
| 已鍛鍊 | (13,096) | | | 4.46 | | | | | |
| 沒收/過期 | (1,685,217) | | | 18.48 | | | | | |
| 截至2023年12月31日的未償還債務 | 9,475,009 | | | $ | 14.34 | | | 7.61 | | $ | 8,261 | |
| 自2023年12月31日起可行使 | 3,838,707 | | | $ | 20.54 | | | 6.68 | | $ | 482 | |
| 已歸屬且預計將於2023年12月31日歸屬 | 9,475,009 | | | $ | 14.34 | | | 7.61 | | $ | 8,261 | |
與該公司的期權活動有關的其他信息如下:
| | | | | | | | | | | | | |
| 截至十二月三十一日止的年度, |
| 2023 | | 2022 | | |
| 已授予期權的加權平均公允價值 | $ | 2.84 | | | $ | 13.44 | | | |
| 已行使期權的內在價值(千) | $ | 22 | | | $ | 1,796 | | | |
截至2023年12月31日,與未償還期權相關的未確認補償成本為1美元51.6百萬美元,預計將在加權平均期間確認2.1好幾年了。
下表總結了布萊克-斯科爾斯期權定價模型中用於確定授予員工的股票期權公允價值的假設:
| | | | | | | | | | | | | |
| 截至十二月三十一日止的年度, |
| 2023 | | 2022 | | |
| 預期購股權年期(年) | 5.50 - 6.11 | | 5.50 - 6.11 | | |
| 無風險利率 | 3.45% - 4.73% | | 1.47% - 4.22% | | |
| 預期波動率 | 81.99% - 86.15% | | 81.36% - 88.75% | | |
| 預期股息收益率 | 0.00% | | 0.00% | | |
基於業績的限制性股票單位
2022年1月和2月,公司董事會授權發行563,500基於業績的限制性股票單位,或PSU,授予某些員工,包括2020年計劃下的公司領導團隊成員。PSU的歸屬取決於某些發現和臨牀里程碑的成就的確定,或特定市場條件的滿足。在授予時,每個PSU自動轉換為一公司普通股的份額。
下表彙總了截至2023年12月31日的年度,公司股權計劃下與PSU有關的活動:
| | | | | | | | | | | |
| | 股票 | | 加權平均
授予日期
公允價值 |
| 截至2022年12月31日的未償還債務 | 465,291 | | | $ | 25.09 | |
| 授與 | — | | | — | |
既得(1) | (135,301) | | | 26.52 | |
| 被沒收 | (175,990) | | | 28.91 | |
| 截至2023年12月31日的未償還債務 | 154,000 | | | $ | 19.47 | |
(1) 已授予的PSU包括17,556公司保留的股份,用於支付法定的最低預扣税。
截至2023年12月31日,與未償還PSU相關的未確認賠償成本為#美元。3.1100萬美元,預計將在加權平均期間確認1.2好幾年了。
基於時間的限制性股票單位
2023年1月和2月,公司董事會授權發行824,600根據2020年計劃,向員工提供基於時間的限制性股票單位(RSU)。RSU的歸屬取決於基於時間的歸屬條件。該等RSU於授出日以授出日相關股份的市價計值。在授予時,每個RSU自動轉換為一公司普通股的份額。
下表彙總了公司股權計劃下截至2023年12月31日的年度與RSU有關的活動:
| | | | | | | | | | | |
| | 股票 | | 加權平均
授予日期
公允價值 |
| 截至2022年12月31日的未償還債務 | — | | | $ | — | |
| 授與 | 824,600 | | | 5.37 | |
| 既得 | — | | | — | |
| 被沒收 | (74,390) | | | 5.67 | |
| 截至2023年12月31日的未償還債務 | 750,210 | | | $ | 5.35 | |
截至2023年12月31日,與未償還RSU相關的未確認賠償成本為#美元。3.5100萬美元,預計將在加權平均期間確認3.2好幾年了。
獎勵補助金
在2023年7月的兩個不同日期,公司董事會批准授予非限制性股票期權,以購買351,000將公司普通股分給兩名新員工。根據這些獎勵授予的期權在以下期限內授予四年有一座懸崖位於一年之後每季度進行一次歸屬。截至2023年12月31日,這些股票期權均未歸屬或被沒收,以及351,000在這些激勵獎下,期權仍未結清。
2020年員工購股計劃
2020年9月,公司董事會通過了C4治療公司2020年員工股票購買計劃,或2020年ESPP。符合資格的員工可授權扣除工資,最高可達15在要約期內其合格薪酬的%。公司可持有一每年或更多的招股期限,在此期間,員工將能夠根據2020年的ESPP購買股票。該公司發行了133,784截至2023年12月31日的年度內的股份。
截至2023年12月31日,公司擁有1,673,129根據2020年ESPP,可供未來發行的股票。2020年ESPP規定,從2021年1月1日起至2030年1月1日,每個財政年度的第一天將增加年度加薪,相當於(I)1前一年12月31日普通股流通股的百分比,(2)656,714股份,或(Ii)由2020年ESPP管理人確定的較少股份數量。2024年1月1日,2020年ESPP的年度增長導致了額外的604,671授權發行的股票將被添加到2020年ESPP。
注12.所得税
所得税(福利)費用由以下各項組成(以千計):
| | | | | | | | | | | | | |
| 截至十二月三十一日止的年度, |
| 2023 | | 2022 | | |
| 現行税收規定: | | | | | |
| 現行聯邦條款 | $ | — | | | $ | — | | | |
| 當前國家規定 | 76 | | | — | | | |
| 現行外國條款 | 1,204 | | | — | | | |
| 總當期撥備 | 1,280 | | | — | | | |
| 遞延税項準備: | | | | | |
| 遞延聯邦撥備 | — | | | — | | | |
| 遞延狀態準備金 | — | | | — | | | |
| 遞延外匯準備金 | — | | | — | | | |
| 總税額撥備 | $ | 1,280 | | | $ | — | | | |
税率對賬
綜合財務報表中反映的按法定聯邦税率計算的預期所得税(福利)費用與所得税的對賬如下:
| | | | | | | | | | | | | |
| 截至十二月三十一日止的年度, |
| 2023 | | 2022 | | |
| 按聯邦法定税率計算的所得税優惠 | 21.0 | % | | 21.0 | % | | |
| 州税-聯邦税網 | 4.2 | % | | 8.0 | % | | |
| 聯邦信貸 | 3.5 | % | | 1.4 | % | | |
| 國家信用 | 1.4 | % | | 0.1 | % | | |
| 其他永久性差異 | (0.2) | % | | (0.8) | % | | |
| 預提税金 | (0.9) | % | | — | % | | |
| 基於股票的薪酬 | (2.8) | % | | (3.0) | % | | |
| | | | | |
| | | | | |
| 估值免税額 | (27.2) | % | | (26.7) | % | | |
| 總計 | (1.0) | % | | — | % | | |
本公司截至2023年12月31日的有效税率受到中國預扣税義務的影響,該預扣税義務與與Betta許可協議相關的預付款和里程碑付款有關(見附註8)。這就產生了#美元的税收撥備。1.2百萬美元,有效税率為(1.0截至2023年12月31日止的百分比。
遞延税金的重要組成部分
遞延所得税反映了(A)用於財務報告的資產和負債的賬面金額與用於所得税的金額之間的暫時差異以及(B)營業損失和税收的淨税收影響。
信用結轉。公司遞延税項資產和遞延税項負債的重要組成部分如下(以千計):
| | | | | | | | | | | |
| 截至12月31日, |
| 2023 | | 2022 |
| 遞延税項資產: | | | |
| 淨營業虧損 | $ | 61,036 | | | $ | 54,300 | |
| 資本化研究和實驗支出 | 51,401 | | | 27,568 | |
| 經營租賃負債 | 20,611 | | | 21,443 | |
| 研發和投資税收抵免 | 18,049 | | | 11,519 | |
| 基於股票的薪酬 | 9,061 | | | 6,296 | |
| 遞延收入 | 3,881 | | | 8,457 | |
| 外國税收抵免 | 1,204 | | | — | |
| 資本化啟動成本 | 555 | | | 699 | |
| 未實現損益 | — | | | 1,090 | |
| 其他 | 27 | | | 354 | |
| 遞延税項總資產總額 | 165,825 | | | 131,726 | |
| 遞延税項負債: | | | |
| 使用權資產 | (18,573) | | | (19,868) | |
| 固定資產 | (1,663) | | | (1,738) | |
| | | |
| 減去:估值免税額 | (145,589) | | | (110,120) | |
| 遞延税金淨額 | $ | — | | | $ | — | |
本公司已對影響遞延税項資產變現的正面和負面證據進行了評估。截至2023年12月31日、2023年12月31日和2022年12月31日,根據本公司的歷史營業虧損,本公司得出結論,其遞延税項資產的利益很可能無法實現。因此,本公司已為截至2023年12月31日、2023年12月31日及2022年12月31日的遞延税項資產提供全額估值準備。截至2023年12月31日和2022年12月31日的遞延税項資產估值免税額為#美元。145.6百萬美元和美元110.1分別為100萬美元。估值津貼淨額增加#美元。35.5百萬美元和美元34.7在截至2023年12月31日和2022年12月31日的年度內分別為100萬美元。截至2023年12月31日止年度的變動主要是由於淨營業虧損及税項抵免結轉增加、研究及實驗開支資本化,以及年內確認的遞延收入減少所致。
截至2023年12月31日和2022年12月31日,該公司擁有214.9百萬美元和美元171.6美國聯邦淨營業虧損總額,或NOL,分別結轉,可用於抵消未來的應税收入。2017年12月頒佈的減税和就業法案(TCJA)通常允許2017年後產生的聯邦損失無限期結轉,但一般會將NOL結轉金額限制在較小者或公司應納税所得額的80%(受1986年修訂的美國國税法(IRC)第382條的約束)。此外,2017年後產生的虧損將不會結轉。2018年前產生的虧損一般可在公司的NOL結轉或公司的應納税所得額中較小的範圍內扣除,並自虧損產生之日起20年內可供扣除。出於美國聯邦所得税的目的,該公司2017年後產生的聯邦NOL為$214.9100萬美元,這些債券不會過期。本公司沒有任何在2019年前產生的可用NOL,因為這些NOL在2019年已全部使用。冠狀病毒援助、救濟和經濟安全法案,或CARE法案,暫時允許公司將2018、2019年和2020年產生的淨運營虧損結轉到前五個納税年度。此外,這些年產生的淨營業虧損可以完全抵消上一年的應税收入,而不需要80TCJA規定的應納税所得額的%。
截至2023年12月31日、2023年12月31日和2022年12月31日,該公司的美國州淨營業虧損總額為$315.3百萬美元和美元273.6分別為100萬美元,可用於抵消到2043年在不同日期到期的未來應税收入。
在2023年12月31日、2023年12月31日和2022年12月31日,公司擁有美國聯邦研究信貸結轉$12.6百萬美元和美元9.1分別為100萬美元,可用於抵消未來的聯邦所得税債務,這些債務將在2043年之前的不同日期到期。在2023年12月31日、2023年12月31日和2022年12月31日,公司擁有美國州研究信貸結轉$5.2
百萬美元和美元2.9分別為100萬美元,可用於減少到2038年在不同日期到期的未來税收負擔。
淨營業虧損和税收抵免結轉可能會受到年度限制,如果重要股東的所有權權益在三年期間的某些累積變化超過50%,分別根據IRC第382條和383條以及類似的國家規定定義。這可能會限制每年可用於抵消未來應税收入或納税義務的税收屬性的數量。年度限額的金額是根據緊接所有權變更前的公司價值確定的。隨後的所有權變更可能會進一步影響未來幾年的限制。2022年,公司完成了從成立到2020年12月31日的所有權變更研究,以評估自成立以來是否發生了所有權變更或是否發生了多次所有權變更。這項研究的結果表明,公司經歷了國税法第382條定義的所有權變更,但有不是淨營業虧損的結轉將是有限的,並將因此類所有權變更而到期而未使用。鑑於持續虧損的狀況,本公司目前尚未根據IRC第382條對2020年12月31日之後的任何活動進行分析。已就與本公司營業淨虧損及税項抵免結轉有關的遞延税項資產撥備全數估值撥備,如有需要作出調整,該項調整將由估值撥備的調整抵銷。
本公司將在所得税支出中確認與未記錄的税收優惠相關的利息和/或罰款。截至2023年、2023年和2022年12月31日,公司擁有不是與未記錄的税收優惠相關的應計利息或罰款。
該公司在美國、加利福尼亞州、佛羅裏達州和馬薩諸塞州提交所得税申報單。聯邦和州所得税申報單通常要對截至2020年12月31日的納税年度進行税務審查。在本公司具有税務屬性結轉的情況下,產生該等屬性的納税年度仍可經國税局或國家税務機關審核後調整至未來期間使用的程度。該公司目前沒有接受任何税務機關的審查。
注13.每股淨虧損
如注2所述,重要會計政策摘要,對於公司報告淨虧損的期間,潛在攤薄證券已被排除在每股攤薄淨虧損的計算之外,因為它們的影響將是反攤薄的。因此,用於計算基本每股淨虧損和稀釋後每股淨虧損的已發行普通股加權平均數是相同的。在計算所指期間的稀釋後每股淨虧損時,該公司不計入根據期末已發行金額列報的下列潛在普通股,因為計入這些股份會產生反攤薄效果:
| | | | | | | | | | | | | |
| 截至十二月三十一日止的年度, |
| 2023 | | 2022 | | |
| 購買普通股的期權 | 9,475,009 | | | 6,993,052 | | | |
| | | | | |
每股基本虧損和稀釋虧損的計算方法是將淨虧損除以已發行的加權平均普通股(單位為千股,不包括股票和每股數據):
| | | | | | | | | | | | | |
| 截至十二月三十一日止的年度, |
| 2023 | | 2022 | | |
| 分子: | | | | | |
| | | | | |
| | | | | |
| 淨虧損--基本虧損和稀釋虧損 | $ | (132,493) | | | $ | (128,175) | | | |
| 分母: | | | | | |
| 加權平均已發行普通股-基本和稀釋後普通股 | 49,640,505 | | | 48,861,665 | | | |
| 每股淨虧損--基本虧損和攤薄虧損 | $ | (2.67) | | | $ | (2.62) | | | |
注14.固定繳款計劃
公司有一個401(K)退休計劃,即401(K)計劃,根據該計劃,所有員工可以繳納最多90他們合格補償的%,最高可達美國國税局規定的最高允許金額。公司與之匹配100對401(K)計劃的繳款百分比3%,並與另一個50捐款百分比 5%,總計
潛在的匹配4每名員工的百分比。在截至2023年12月31日、2023年12月31日和2022年12月31日的每個年度內,公司貢獻了約$1.1百萬美元和美元1.0分別為401(K)計劃提供100萬美元。
注15.承付款和或有事項
法律程序
本公司目前並未參與任何重大法律程序。於每個報告日期,本公司評估潛在虧損金額或潛在虧損範圍是否根據權威性指引處理或有事項的規定而可能及合理地評估。本公司在發生與該等法律訴訟有關的費用時支出.
注16.後續事件
2024年1月9日,該公司實施了一項計劃,以降低運營成本,並使其員工隊伍更好地適應其業務需求。根據成本削減計劃,該公司裁員約30%。該公司估計,它將產生約#美元的一次性重組費用2.7百萬美元,包括員工遣散費、福利和相關解僱費用,公司預計將在2024年第一季度支付其中大部分費用。