我們的管道
下圖總結了我們當前的開發計劃:
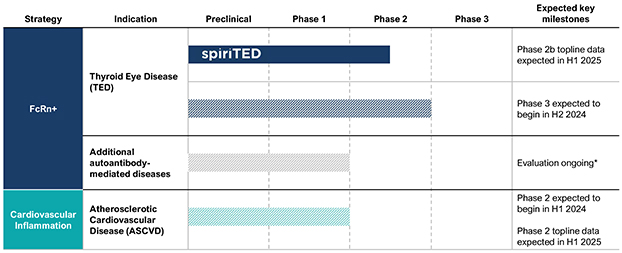
注意:影線條表示尚未開始的試驗。監管機構 提交的時間和臨牀試驗里程碑可能會發生變化,並可能與美國食品和藥物管理局進一步討論
| * | 其他適應症的臨牀開發計劃可能會在適應症選擇和與 FDA 的討論 後發生變化 |
從上圖中可以看出,我們計劃確定 TOUR006 的更多適應症機會。 此外,我們將繼續評估我們認為存在的資產的新許可和收購機會 護理標準 免疫、炎症和其他疾病患者的潛力發生變化。
我們的戰略
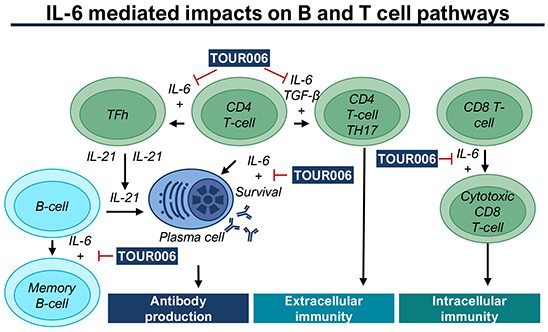
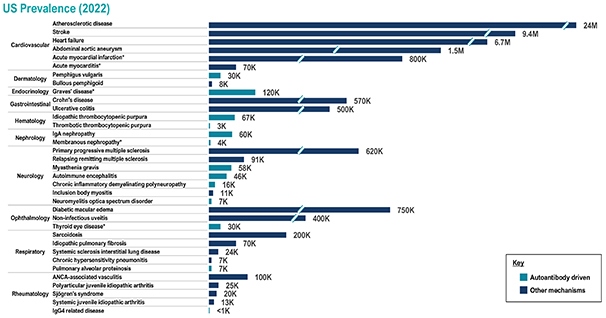
我們尋求識別和開發具有變革性的藥物,這些藥物有可能開發出新的藥物 護理標準在未得到滿足的醫療需求嚴重的地區。我們計劃採用以人類數據為中心的適應症選擇,確定儘管正規行業發展有限,但在實踐中仍成功使用 IL-6 途徑 抑制劑的疾病,以及我們認為 TOUR006 有可能對現有護理標準帶來重大改善的疾病。我們還計劃利用競爭對手 IL-6 途徑抑制劑項目的 臨牀試驗的見解,目標是迅速將 TOUR006 納入已經消除外部風險的適應症。我們 認為,這種專注於利用現有的人類數據可以使我們確定具有很高臨牀和商業成功潛力的適應症,並可以最大限度地發揮 TOUR006 的價值。
我們戰略的關鍵要素包括:
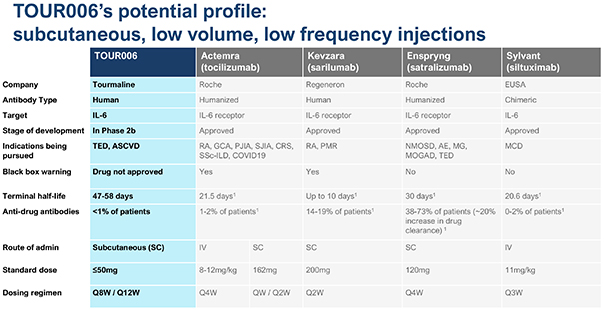
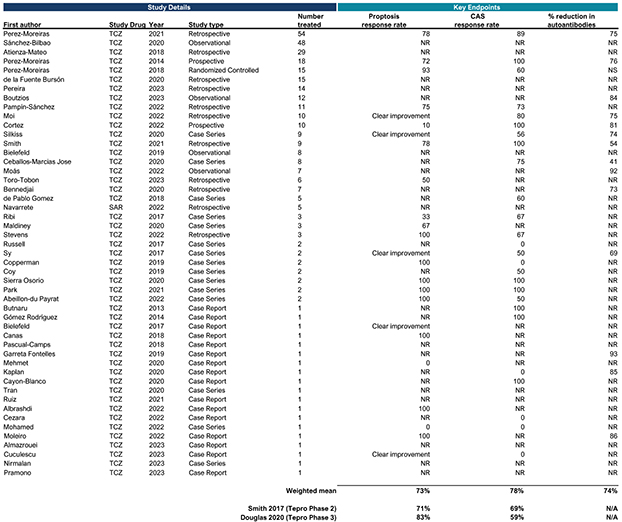
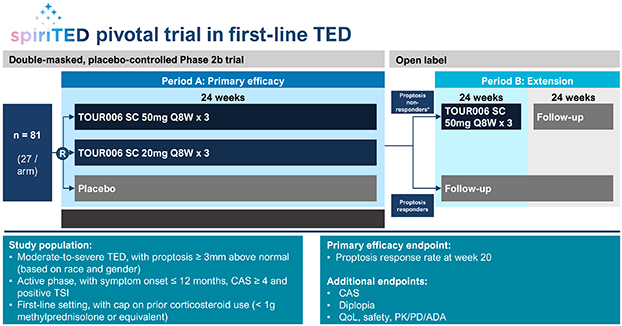
| | 在我們的 fcRN+ 戰略中,通過對以 TED 作為我們治療自身抗體驅動疾病的灘頭 適應症的患者的臨牀開發,推進 TOUR006 的發展。基於支持活性 TED 中白細胞介素-6 途徑抑制的文獻、迄今為止觀察到的 IL-6 類別良好的長期安全特徵,以及不頻繁的皮下給藥可能帶來的低管理負擔,我們的初始候選產品 TOUR006 有可能為治療 TED 提供差異化的產品特徵。2023 年 9 月,我們啟動了關鍵的 2b 期 SpiritED 試驗,以評估 TOUR006 治療 TED 的安全性和有效性,我們預計將在 2025 年上半年報告該試驗的主要結果。此外,我們預計將於 2024 年在 TED 中啟動 TOUR006 的關鍵 三期試驗。這項計劃中的3期試驗的主要數據預計將於2026年公佈。 |
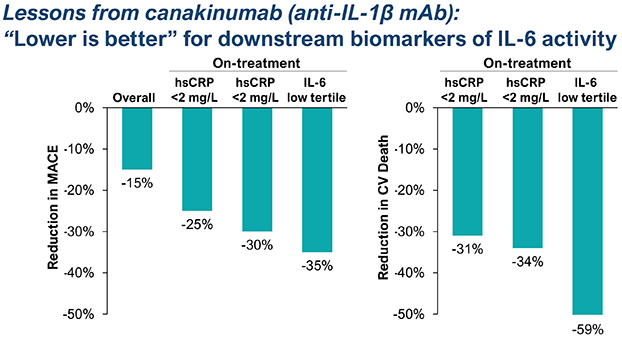
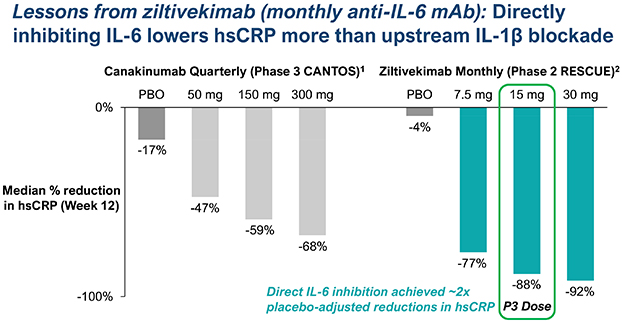
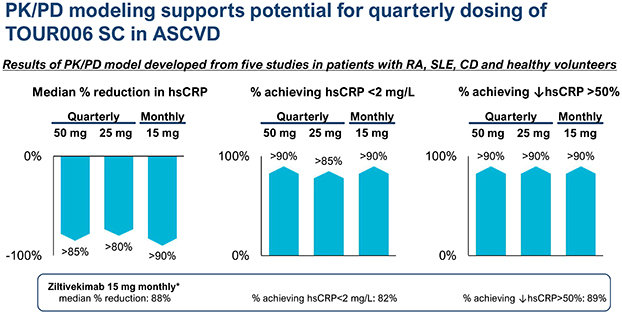
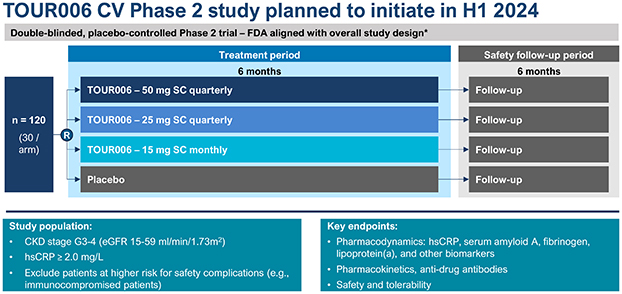
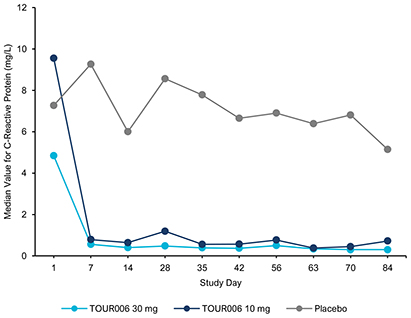
| | 在我們的心血管炎症策略中,通過對 ASCVD 患者的臨牀開發推進 TOUR006。我們認為,TOUR006 有可能為治療心血管疾病的炎症風險提供差異化的產品特徵,並有可能每三個月皮下給藥一次。我們計劃在 2024 年上半年啟動一項 2 期臨牀試驗,以評估 TOUR006 治療心血管疾病的安全性、藥代動力學 (PK) 和藥效學 (PD),預計將在 2025 年上半年公佈主要數據。 |