目錄表
我們正在籌備的所有項目都處於臨牀前開發階段,總結如下:
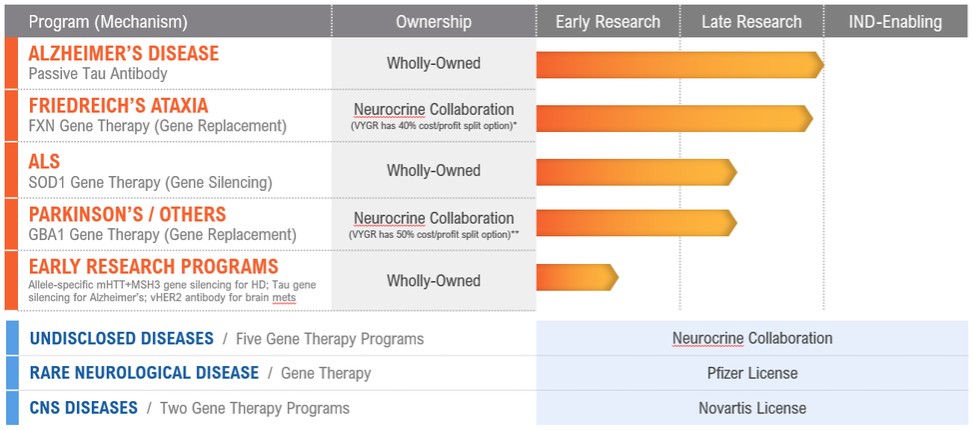
*在第一階段讀出後,旅行者公司可以選擇:(1)根據60/40的成本和利潤分享安排(Neurocrine/Voyager)與Neurocrine Biosciences在美國共同開發和共同商業化,或(2)授予Neurocrine Biosciences在美國的全部商業權,以換取基於美國銷售的里程碑式付款和特許權使用費。
*第一階段讀出後,旅行者公司可以選擇:(1)根據50/50的成本和利潤分享安排,在美國與Neurocrine Biosciences共同開發和共同商業化,或(2)授予Neurocrine Biosciences在美國的全部美國商業權,以換取基於美國銷售的里程碑式付款和特許權使用費。
抗Tau抗體方案治療阿爾茨海默病
疾病概述
我們正在開發專有抗體,選擇性地靶向並減少病理性tau的傳播,用於治療tauopathy,我們的主要適應症是阿爾茨海默病(AD)。Tau病理的傳播與AD的疾病進展和認知能力下降密切相關,在美國約有600萬人受到影響,並給社會帶來越來越大的醫療負擔。最近,抗澱粉樣蛋白抗體已被批准用於治療阿爾茨海默病,但仍有大量藥物需求未得到滿足。
我們的治療方法
長期以來,我們一直致力於開發專有的和互補的方法來幹擾tau病理的進展,據信tau病理是AD和其他tauopathy的核心。減少有毒的tau聚集體可能會減緩這些疾病的疾病進展和認知能力下降。我們正在探索被動使用我們的抗tau抗體。我們的抗tau抗體具有不同的特性,包括改進了對tau蛋白特定區域的靶向,與第一代方法相比,可以提供更好的輪廓。我們相信,我們針對C末端的抗體與其他方法有很大的不同。此外,我們相信,在IND申請獲得批准後,利用正電子發射斷層掃描(PET)對人類tau進行成像的臨牀評估,加上測量血漿和腦脊液生物標記物,有可能實現有效和加速的人類生物學證據演示。
臨牀前研究
在2022年8月舉行的阿爾茨海默病協會國際會議上,我們展示了我們專有的抗tau抗體的數據,該抗體針對的是具有高親和力的中間結構域和C末端,並在小鼠模型的臨牀前研究中顯示出良好的生物物理特性和強大的活性。在P301S播種繁殖互作中
10