在仔細審查了FDA的信息要求後,2021年12月,公司決定不按照FDA在2021年10月的反饋中要求的順序方法來處理Viaskin花生的開發計劃。該公司估計,FDA新提出的順序方法將需要至少五輪交換,這需要FDA在啟動印章之前進行比對
6個月
安全性和粘附性研究。因此,2021年12月,公司宣佈了啟動關鍵階段的計劃
3
一種改良的Viaskin花生貼片(MVP)在預期患者羣體中用於兒童的安慰劑對照療效試驗。該公司認為這種方法是最直接的,可以潛在地證明改良的Viaskin花生系統的有效性、安全性和改善的體內粘附性。FDA證實,通過口頭和書面交流,該公司的戰略變化是一致的。
2022年,公司宣佈新階段
3
改良的Viaskin花生(MVP)貼片的關鍵研究將在Young
(4-7
6歲)和對花生過敏的兒童更敏感。
2021年8月2日,該公司宣佈已從EMA收到第180天未解決問題清單,這是規定的EMA審查過程的既定部分。這是一封信,意在包括該進程那個階段的任何剩餘問題或反對意見。EMA表示,他們的許多反對意見和第120天問題清單中的主要反對意見已經得到回答。一個主要的反對意見仍然是180日。主要反對意見質疑數據的侷限性,例如,由一項關鍵研究支持的臨牀相關性和效果大小。
2021年12月20日,公司宣佈撤回對Viaskin花生的MAA,並正式通知EMA我們的決定。最初的申請得到了來自單一安慰劑對照階段的數據的支持
3
稱為PEPITES的關鍵試驗
(V712-301).
撤回的決定是基於CHMP的觀點,即到目前為止,從單一關鍵臨牀試驗獲得的數據不足以排除在審查週期的第180天出現重大異議。該公司相信,第二次Viaskin花生關鍵臨牀試驗的數據將支持Viaskin花生在歐盟獲得許可的一條更穩健的道路。該公司打算在該數據集可用時重新提交MAA。
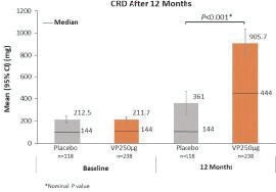
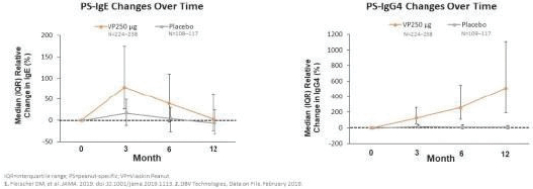
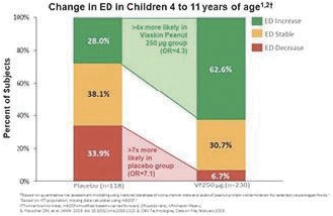
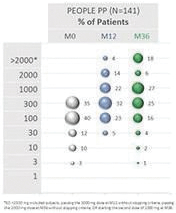
2020年6月,該公司宣佈,在A部分,兩個治療分支的患者在治療12個月後顯示出一致的治療效果,這是通過雙盲安慰劑控制的食物挑戰和生物標記物結果進行評估的。A部分的受試者沒有包括在B部分中,A部分的療效分析沒有從統計學上證明任何一種劑量相對於安慰劑的優越性。這些結果證實了這一年齡組正在進行的250微克劑量的研究,這是研究B部分正在研究的劑量。表位B部分的登記工作於2021年第一季度完成。
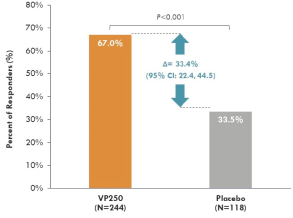
2022年6月,我們宣佈了表位B部分的陽性背線結果,招募了362名1-3歲的受試者,其中244人和118人分別在活動組和安慰劑組。在積極治療組和安慰劑治療組之間,根據年齡和基線疾病特徵進行登記。
該公司打算進一步分析表位的數據,並探索Viaskin花生在1至3歲兒童中的監管途徑,因為這一弱勢羣體的高度需求未得到滿足,而且缺乏獲得批准的治療方法。
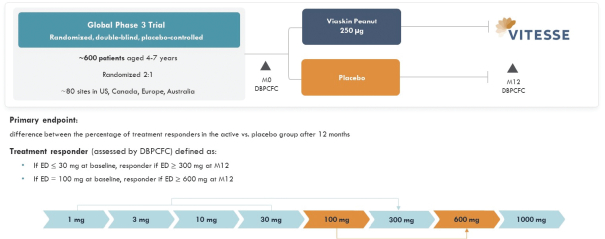
2022年9月7日,我們宣佈啟動Vitesse,一個新的階段
3
改良維亞斯金花生(MVP)貼片在兒童中的應用研究
4-7
多年來對花生過敏。我們將啟動定義為將試驗方案提交給選定的研究地點,以供隨後的機構審查委員會(IRB)/道德委員會(EC)批准。