目錄表
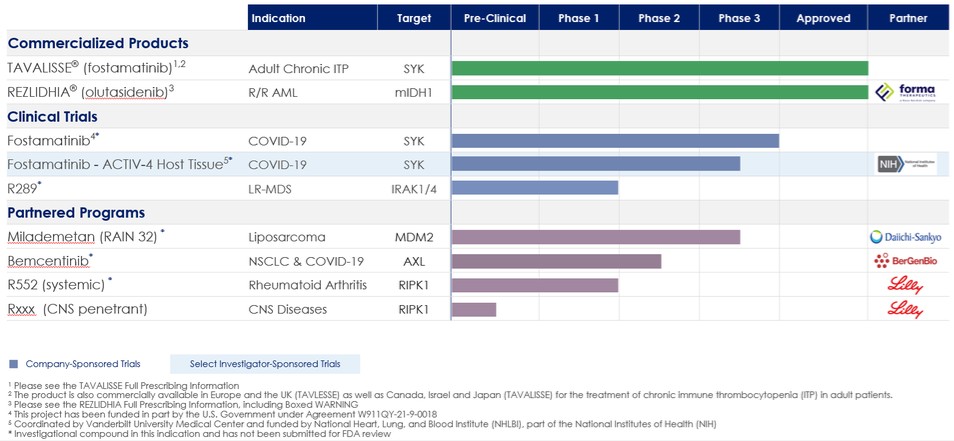
我們的產品組合
下表總結了我們的投資組合:
商業產品
他伐他利/福司他替尼在ITP中的應用
疾病背景。據估計,美國有81,300名成人患者患有慢性ITP。在ITP患者中,免疫系統攻擊並破壞人體自身的血小板,後者在血液凝結和癒合中發揮積極作用。由於血小板計數低,ITP患者可能會遭受嚴重的瘀傷、出血和疲勞。目前治療ITP的方法包括類固醇、模擬血小板生成素(TPO)的血小板生成增強劑和脾切除術。
口服福司他替尼程序。福斯塔替尼以片劑形式服用,可阻止免疫細胞內SYK的激活。ITP的典型特徵是身體產生抗體,附着在血流中的健康血小板上。免疫細胞識別這些抗體,並將其附着在它們上,從而激活免疫細胞內的SYK酶,並觸發抗體和附着的血小板的破壞。當SYK被福斯塔替尼抑制時,它會中斷這種免疫細胞功能,使血小板逃脱破壞。在我們的第二階段臨牀試驗中,16名成人慢性ITP患者口服福斯塔替尼,結果發表在血樣研究表明,福斯塔替尼顯著增加了某些ITP患者的血小板計數,包括那些目前可用的其他藥物失敗的患者。
我們的福斯塔替尼治療免疫性血小板減少症(FIT)第三階段臨牀計劃共有150名ITP患者,他們被隨機分成兩個相同的多中心、雙盲、安慰劑對照臨牀試驗。這些患者被診斷為持續性或慢性ITP,血小板計數始終低於每微升血液30,000。三分之二的受試者口服福斯塔替尼,每天兩次,每次100毫克,另三分之一的受試者接受相同時間表的安慰劑。受試者預計將繼續接受長達24周的治療。在治療的第四周,未能達到某些血小板計數和達到某些耐受性閾值的受試者,可以將他們的福斯塔替尼(或相應的安慰劑)劑量增加到150毫克,每日2次。該計劃的主要療效終點是在24周前出現穩定的血小板反應,在最後6次合格抽血中,至少有4次的血小板計數達到或超過每微升血液50,000。2016年8月,我們宣佈了第一項FIT研究的結果,報告稱福斯塔替尼達到了研究的主要療效終點。研究表明,接受福斯塔替尼治療的患者中有18%的患者實現了穩定的血小板反應,而接受安慰劑對照組的患者中沒有患者。2016年10月,我們公佈了第二項FIT研究的結果,報告了應答率(治療組為16%,安慰劑組為4%)與第一項研究一致,儘管差異不具有統計學意義。在ITP中
26