斯普林格和倫納德·I·佐恩。中國移動擁有的部分權利是獨家授權給我們的。到目前為止,我們是所有後續專利家族的唯一合法所有者。
如上所述,我們的轉化生長因子β技術的一部分被授權給揚森。這被分割為PCT/US2017/042162(出版為WO2018/013939),已被國有化。被許可人帶頭起訴這一專利家族。被許可人還擁有我們的平臺技術的非獨家許可,以使他們能夠在許可領域進行開發。
下面提供了我們的專利系列的簡要説明,以及預計的專利期限,其中不包括任何可能的專利期限調整或延長。
站臺
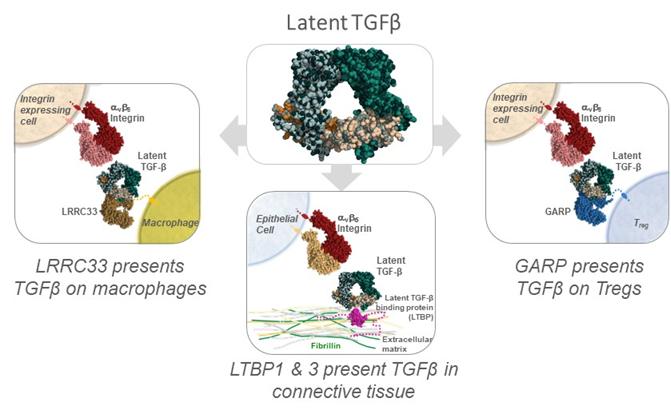
我們生產生長因子超細胞激活的選擇性調節劑的新方法廣泛體現在我們最早的兩個“平臺”專利家族--PCT/US2013/068613(發表為WO2014/074532)和PCT/US2014/036933(發表為WO2014/182676)中。這些專利家族針對調節生長因子的轉化生長因子β超家族的激活的方法和篩選特定靶向非活性形式的生長因子的單抗的方法,從而防止成熟生長因子的激活(例如釋放)。轉化生長因子β超家族是一組30多種相關的生長因子/細胞因子,調節多種生物學過程,包括轉化生長因子β1和肌肉生長抑素(又稱GDF-8)。平臺家族中已頒發的美國專利包括:美國專利號9,573,995 (issued 02/21/2017); 9,758,576 (issued 09/12/2017); 9,580,500 (issued 02/28/2017); 9,399,676 (issued 07/26/2016); 9,758,577 (issued 09/12/2017); 10,597,443 (issued 03/24/2020); and 10,981,981 (04/20/2021). There is also a granted European (“EP”) platform patent: EP2981822 (granted on 09/02/2020), which was validated in 37 states. These US and EP patents are projected to expire in 2034.
具體地説,EP2981822已經授權了針對能夠結合包括轉化生長因子β1原的重組抗原的抗體或包括轉化生長因子β1 LAP複合體的生長因子-原結構域複合體的物質組合物,以及針對製造這種抗體的方法的權利要求。EP2981822目前是歐洲專利局正在進行的反對程序的主題。
美國專利號9,573,995發佈了針對一種抗體的物質組合物權利要求,該抗體與與人轉化生長因子β1 LAP複合體相關的GARP特異性結合。
美國專利第9,758,576號已經發布了針對分離的單抗或其片段的物質組合物,其特異性地結合前/潛伏GDF-8/myostatin複合體的前域,從而防止GDF-8/myostatin前域的Arg 75和Asp 76殘基之間的蛋白水解性切割,從而抑制成熟GDF-8/myostatin生長因子從複合體中釋放。
美國專利號9,580,500已經發布了針對基於噬菌體展示文庫的抗體生產方法的權利要求,該方法用於鑑定與GARP/原轉化生長因子β1複合體結合的抗體。
美國專利號9,399,676已經發布了針對基於噬菌體展示文庫的抗體生產方法的權利要求,該方法用於鑑定與經過酶切割的前/潛伏GDF-8複合體結合的抗體。已發佈的美國專利第9,758,577號中包含了相關的產品逐個過程的權利要求。
美國專利第10,597,443號發佈的權利要求廣泛涵蓋一種藥物組合物的製造方法,該組合物含有一種抗體,該抗體與轉化生長因子β的一個大的潛在複合體結合,從而調節轉化生長因子β信號。
此外,美國專利第10,981號發佈的權利要求廣泛涵蓋一種藥物組合物的製造方法,該組合物含有一種抗體,該抗體與前/潛伏的GDF-8結合,但不與成熟的GDF-8結合,並抑制GDF-8信號。