目錄
臨牀試驗。我們預計將在2020年下半年提交IND,以支持一項初步的第一階段研究,以確定FAP-2286作為治療劑的劑量和耐受性。我們持有美國和全球對FAP-2286的權利,不包括歐洲(包括俄羅斯、土耳其和以色列),在那裏3個基點保留權利。我們還同意與3個鹼基合作開展一項發現計劃,該項目旨在為肽靶向放射性核素治療提供最多三個額外的、未公開的目標,我們將為任何結果的產品候選方獲得全球權利。
克洛維斯成立於2009年。我們已經建立了我們的組織,以支持創新的腫瘤學藥物開發,以治療癌症人羣的特定亞羣。為了實施我們的戰略,我們已經組建了一支經驗豐富的團隊,在全球臨牀和非臨牀開發、管理操作和腫瘤學商業化方面具有核心競爭力,並與專門從事配套診斷開發的公司建立了合作關係。
臨牀開發管道
我們繼續評估在選定的病人羣體中使用Rubra的情況,並在適當情況下與合作伙伴合作進行相應的診斷開發。我們對Rubra的開發策略集中在我們認為患者羣體表現出更高頻率的突變BRCA腫瘤或具有其他同源重組缺陷的腫瘤(HRD)的適應症上,PARP抑制劑已經在腫瘤中顯示出臨牀或臨牀前活性。我們也正在開發利維坦的組合,包括與魯布拉,基於鼓勵的數據,在臨牀研究的其他類似的腫瘤學化合物。FAP-2286目前是IND輔助臨牀前研究的主題,我們計劃在2020年下半年提交IND。下表概述了正在進行或計劃進行的克洛維斯或合作伙伴贊助的研究:
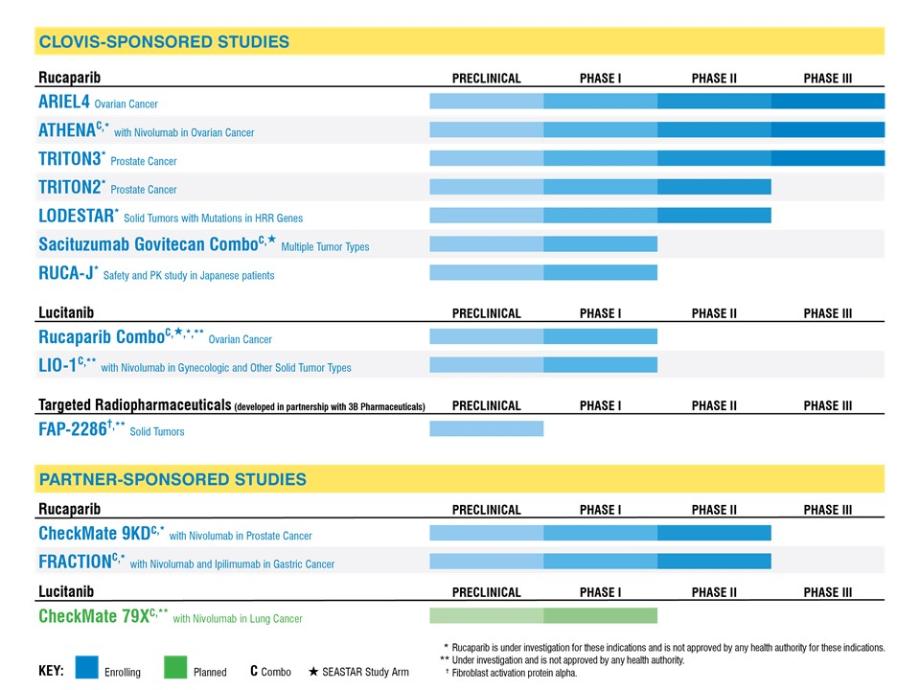
在其中某些審判中,我們或我們的合作伙伴可能定期或持續地獲得臨時數據,這些數據不會在我們獲得這些數據的同一時間框架內公開提供,或者根本不會公開提供。
5