目次
このようなタンパク質は、製造前および/または製造後に改変されます。治療用タンパク質のエンジニアリングと改変は、生物学的特性(グリコシル化、半減期、免疫原性など)を改善することで、追加の臨床的利益をもたらす可能性があります。
当社のProCellex技術には、生物学的最適化、複雑なタンパク質発現を処理する能力、効率、機能強化、迅速な水平スケールアップによる改善による柔軟な製造、製造プロセスの簡素化、哺乳類の成分によるウイルス汚染のリスクの排除、知的財産の利点など、多くの独自の利点があります。
私たちは、現代のバイオテクノロジーの不可欠な基盤である組換えタンパク質の開発、発現、製造のために、植物細胞培養技術に基づいてProCellexを開発しました。私たちは、アグロバクテリウムの本来の能力を利用して、DNA断片を植物の染色体に移し、植物細胞のゲノムが目的の特定のタンパク質をコードできるようにすることで、新しい組換え治療用タンパク質を開発しています。その後、アグロバクテリウムを介した形質転換細胞は特定のタンパク質を生成し、それを抽出して精製し、さまざまな病気の治療薬として使用できます。
当社のProCellexテクノロジーは、酵素、ホルモン、モノクローナル抗体、サイトカイン、ワクチンなど、さまざまな薬剤クラスに属する複雑な治療用タンパク質を発現するために利用できます。最初のヌクレオチドクローニングからタンパク質製品の大規模生産まで、タンパク質発現プロセス全体は、現在の適正製造基準(cGMP)に準拠した管理プロセスの下で行われています。私たちの植物細胞培養技術では、高度な遺伝子工学や化学修飾が施されたニンジンやタバコ(BY-2)細胞などの細胞を使用し、使い捨てで柔軟なバイオリアクターシステムで工業規模で増殖させます。私たちのシステムには、生産プロセスのどの時点でも、哺乳類や動物由来の成分、トランスジェニック野外で育てられた植物、または植物全体は含まれていません。
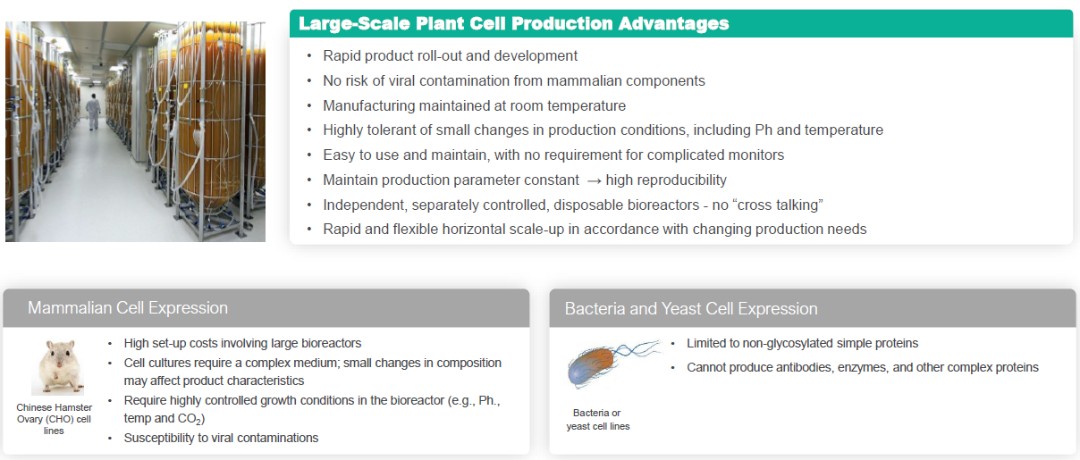
細胞バンクからのスケールアップ段階の開始から大規模生産までの細胞増殖は、柔軟で無菌のカスタム設計のポリエチレンバイオリアクターを使用してクリーンルーム環境で行われ、組換えタンパク質の生産に哺乳類ベースのシステムで一般的に使用される大型のステンレス製バイオリアクターを使用する必要はありません。ProCellexリアクターは使いやすくメンテナンスも簡単で、水平方向への迅速なスケールアップが可能で、哺乳類のウイルス汚染のリスクもありません。当社のバイオリアクターは、シンプルで安価な、化学的に定義された増殖培地を使用した植物細胞の成長に最適です。植物細胞培養用にカスタム設計され最適化されたリアクターは、初期投資が少なく、低コストで迅速に拡張できます。
植物細胞生産の利点
17

