アメリカ アメリカ
証券取引委員会
ワシントン、コロンビア特別区20549
表
(タグ 一)
| 1934年証券取引法第13又は15(D)節に基づいて提出された年次報告 |
締め切りの財政年度について
あるいは…。
| 1934年証券取引法第13条又は15条に基づいて提出された移行報告 |
に対して,_から_への過渡期.
手数料ファイル番号
(登録者の正確な名称は、その定款に規定されている名称と同じ)
(州や他の管轄区域 会社(br}や組織) |
(I.R.S.雇用主 標識 番号) | |
| (主に実行オフィスアドレス ) | (Zip コード) |
登録者の電話番号は市外局番を含んでいます
同法第12条(B)に基づいて登録された証券:
| クラスごとのタイトル | 取引 個の記号 | 登録された各取引所の名称 | ||
|
|
同法第12条(G)により登録された証券:なし
登録者が証券法規則405で定義されている有名な経験豊富な発行者であれば、再選択マークで
を示してください。はい。☐
登録者が当該法第13条又は第15条に基づいて報告書を提出する必要がない場合は,フックで
を示してください。はい。☐
再選択マークは、登録者が、(1)過去12ヶ月以内(または登録者がそのような報告の提出を要求された短い期間内)に、1934年の証券取引法第13または15(D)節に提出されたすべての報告書を提出したかどうか、および(2)
が過去90日以内にそのような提出要件に適合しているかどうかを示す
再選択マークは、登録者が過去12ヶ月以内に(または登録者がそのような文書の提出を要求されたより短い時間以内に)S−T規則(本章232.405節)405条に従って提出されることを要求するすべての対話データファイルを電子的に提出したかどうかを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな申告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
| 大型 加速ファイルサーバ | ☐ | 加速した ファイルマネージャ | ☐ | |
| ☒ | 小さな報告会社 | |||
| 新興成長型会社 |
もしbrが新興成長型会社である場合、登録者が延長された移行期間を使用しないことを選択したかどうかを再選択マークで示して、取引法第13(A)節に従って提供された任意の新しいまたは改正された財務会計基準を遵守してください
登録者が報告書を提出したか否かを再選択マークで示し、その経営陣が“サバンズ-オキシリー法案”(“米国法典”第15編7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する
証券が当該法第12(B)条に基づいて登録されている場合は、届出書類に含まれる登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かをチェックマークで示してください
これらのエラーのより真ん中に再記述があるかどうかをチェックマークで示すことは、登録者の任意の幹部が関連する回復中に受信したインセンティブベースの報酬を§240.10 D−1(B)に従って回復分析する必要があるかどうかを示す
登録者が空殻会社であるかどうかをチェックマークで表す(“取引法”第12 b-2条で定義されている)。はい、違います
2023年6月30日現在,すなわち登録者が最近完成した第2四半期の最終営業日は,ナスダック資本市場の普通株の終値に基づいて,登録者の非関連会社が保有する登録者普通株の総時価は約$である
2024年3月29日現在、登録者が発行した普通株式数は.
参照により統合されたファイル :
カタログ表
| 前向き陳述に関する警告説明 | 1 | |
| 第1部 | 2 | |
| 第 項1. | 業務.業務 | 2 |
| 1 a項目. | リスク要因 | 21 |
| 項目 1 B. | 未解決従業員意見 | 74 |
| プロジェクト 1 C. | ネットワーク·セキュリティ | 75 |
| 第 項2. | 属性 | 76 |
| 第 項3. | 法律訴訟 | 76 |
| 第 項. | 炭鉱安全情報開示 | 76 |
| 第II部 | 77 | |
| 第 項5. | 登録者普通株市場、関連株主事項及び発行者による株式証券の購入 | 77 |
| 第 項6. | [保留されている] | 77 |
| 第 項7. | 経営陣の財務状況と経営成果の検討と分析 | 77 |
| 第 7 A項。 | 市場リスクの定量的·定性的開示について | 86 |
| 第 項8. | 財務諸表と補足データ | 86 |
| 第 項9. | 会計と財務情報開示の変更と相違 | 86 |
| 第 9 A項。 | 制御とプログラム | 86 |
| 第 9 B項。 | その他の情報 | 86 |
| 第 9 C項. | 検査を妨害する外国司法管轄区域を開示する。 | 86 |
| 第三部 | 87 | |
| 第 項10. | 役員·幹部と会社の管理 | 87 |
| 第 項11. | 役員報酬 | 90 |
| 第 項12. | 特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 | 90 |
| 第 項13. | 特定の関係や関連取引、取締役の独立性 | 91 |
| 第 項14. | 最高料金とサービス | 94 |
| 第4部 | 95 | |
| 第 項15. | 展示品と財務諸表の付表 | 95 |
| 第 項16. | 表格10-Kの概要 | 97 |
| サイン | 98 | |
| i |
前向き陳述に関する警告的説明
本年度報告書には、将来の事件や私たちの将来の財務業績に関する前向きな陳述が含まれており、これらの陳述は、私たちの経営陣の現在の信念と期待を表しています。このような陳述は、多くの既知および未知のリスク、不確実性、および他の 要因に関連し、これらの要素は、私たちの未来の実際の結果、業績、または業績をもたらす可能性があり、このような前向き陳述が明示的または暗示する任意の未来の結果、業績、または業績とは大きく異なる。前向き表現は、すべての非歴史的事実の表現を含み、これらの表現は、“信じる”、“予想”、“予想”、“推定”、“計画”、“計画”、“目標”、“可能”、“できる”、“可能”、および同様の表現またはフレーズによって識別することができる。私たちのこれらの展望的な陳述は、主に経営陣の現在の期待と未来の事件と財務傾向に基づいており、これらの事件と財務傾向は、私たちの財務状況、運営結果、業務戦略、財務需要に影響を与える可能性があると考えられます。前向き表現は、以下の態様に関する明示的または暗示的表現を含むが、これらに限定されない
| ● | 私たちの現金トラックは |
| ● | 著者らの臨床前と臨床活動の開始、時間、進展と結果、Volasertibの2 a期試験と著者らの研究開発計画を含む |
| ● | どんな臨床試験データの利用可能性や時間への期待も |
| ● | 候補製品を進め臨床試験を成功させる能力を |
| ● | 私たちの将来の臨床試験の計画は |
| ● | 臨床試験のために十分な数の候補製品を生産し、適切な状況で商業化することができます |
| ● | 監督管理承認を申請するために必要なデータを含む、届出および承認の時間または可能性を規制する |
| ● | 承認されれば候補品の商業化 |
| ● | 私たちの候補製品の潜在的な利点は |
| ● | もし承認されれば、私たちの候補製品の定価と精算 |
| ● | 他の候補製品を開発し商業化する能力は |
| ● | 私たちのビジネス戦略 |
| ● | 私たちのビジネスモデル、業務戦略計画、候補製品、技術を実施します |
| ● | 私たちは私たちの候補製品と技術のために知的財産権保護範囲と期限を確立し、維持することができる |
| ● | 私たちの支出、将来の収入、資本需要、追加融資需要を推定する |
| ● | 私たちのbrは、連携の能力とこのような協力の利点を確立し、維持します |
| ● | 私たちは、私たちの寄付資金レベルを維持することができ、または追加的な贈与または他の非希釈資金源と、そのような贈与に関連する約束を得ることができます |
| ● | 私たちの競争相手と私たちの産業と関連した発展。 |
すべての展望的な陳述は危険、仮定、そして不確実な要素と関連がある。あなたは未来の事件の予測として展望的な陳述に依存してはいけない。イベントの発生および予期される結果の実装は、多くのイベントに依存し、いくつかのイベントまたは のすべてのイベントは、予測できないか、または我々の制御範囲内にある。実際の結果は予想された結果と大きく異なる可能性がある。“プロジェクト1 A. リスク要因”、“プロジェクト7.経営層の財務状況と経営成果の討論と分析” および本年度報告の他の部分を参照して、これらのリスク、仮説と不確実性、およびその他のリスクと不確実性 をより全面的に討論する。これらのリスク、仮定、および不確実性は、必ずしも実際の 結果が私たちの任意の前向き陳述で表現された結果と大きく異なるすべての重要な要素をもたらす可能性があるとは限らない。他の未知または予測不可能な要素もまた私たちの結果を損なうかもしれない。
我々が本年度報告に含まれるすべての前向き陳述は,本年度報告の発表日に得られた情報に基づいている。私たちは、新しい情報、未来のイベント、または他の理由でも、いかなる前向きなbr宣言を公開更新または修正する義務、特にいかなる義務も拒否しない。これらのリスク、不確実性、仮説を考慮して、本年度報告で議論された前向きイベントは発生しない可能性がある。
| 1 |
第 部分I
| 第 項1. | 商売人 |
2023年10月16日、先に発表された期日が2023年2月22日の合併協定および計画(“合併協定”)に基づいて、イスラエルの有名な実験室有限会社(以下、“会社”と略す)、デラウェア州の会社と同社の完全子会社Vibrant合併子会社(“合併子会社”)と、デラウェア州の以前有名な実験室会社(“重要”)と呼ばれていたデラウェア州会社が合併子会社を合併した。本年度報告では,文意が別に指摘されているほか,“当社”,“br}”および“当社”に言及する場合は,当社とその付属会社を指す.言及した“VBLT” は合併前の血管生物製薬有限会社を指す。合併についての詳細は、以下の“ビジネス-重要な背景および会社の歴史-合併”を参照されたい。
業務 の概要
注目すべきは、ある臨床分期プラットフォーム治療会社が癌患者のために正確な予測性薬物を開発したことである。その独自のbrを通じて精確な薬物プラットフォーム或いはPPMP、有名な生物シミュレーション患者の癌治療を予測する離体する(身体の外で)、単一の患者がその実際の治療に臨床的反応を生じるかどうかを正確に予測することが求められる。公認医療センターと行った4つの独立臨床検証試験では,PPMP予測応答者の精度は83%−100%であった。従来の精密薬物よりも良い医療結果を追求するために,PPMPは遺伝的あるいは他のバイオマーカーに依存しない。代わりに,PPMP産生細胞の薬物に対する生物反応の多次元測定を行い,計算アルゴリズムによりこれらのデータを統合して患者反応予測器に変換する。
PPMP は治療を開始する前に臨床反応が期待される患者 を識別し、選択することができ、この患者群において迅速な治療発展を実現する可能性があることを目的としている。そのPPMPを用いて臨床活性を示した許可と開発あるいは共同開発の研究br化合物を著しく評価し,従来の薬物開発と比較して患者の反応 の向上とこれらの化合物の開発の成功率,速度,価値の向上を目指している。
PPPPを使用して、注目すべき目標は、治療を受ける患者のサブセットの10%~30%に限定されるので、注目される臨床活動が示されているが、放棄された許可内資産であり、これは、開発成功、規制承認、br}および商業化の障害である可能性があるからである。注目すべきは、それはそのPPMPプラットフォーム上で数百項目の資産をスクリーニングと評価し、その中の多くの資産に対して、どの患者が臨床反応が出現するかを比較的に高い精度で予測できると信じている。これは、予測応答者を臨床試験に選択的に組み込む機会を提供し、より高い応答率を提供する臨床応答者において、これらの資産の発展を選択的に迅速に追跡することができる。
Br}は,そのPPMPを用いてますます多くの予測的に正確な薬物組み合わせを作成することが求められていることに留意されたい。PPMPはすでにその前の2種類の候補予測精確な薬物の選択において著しい指導作用を得ており、この2種類の臨床段階の候補薬物はプラットフォーム予測の急性骨髄性白血病(AML)応答者 に応用される。PPPP由来の主要資産はVolasertibであり,これはPolo様キナーゼ1(“PLK 1”) 阻害剤であり,多くの癌細胞の細胞周期停滞やアポトーシスを誘導できることが証明されている。注目すべきは、2024年第2四半期に、6人の患者に対する用量最適化前奏を含む成人AMLにおけるVolasertibの使用を評価する第2段階試験を開始することである。 上位6名の患者の結果は2024年第4四半期に発表される予定であり,初のPPMP予測応答者は同一四半期に を登録する予定である。また,CicloMed LLC(“CicloMed”)と共同開発し,急性骨髄性白血病患者のためにファントロピロールを治療したことに注目すべきである。この共同開発パートナーシップは,PPMPが臨床前にフォスコロピロロへの応用に成功した結果である。また、内部許可を行うためにPPMPを使用して他の注目されている資産を決定し、その開発パイプを構築する際に他の資産を迅速に追跡することに留意されたい。
患者の細分化,疾患,医療結果の予測におけるPPMPのカバー範囲を拡大していくことにより,顕著な目標は正確な医学領域を予測するリーダーとなり,患者が彼らに最大の効果を与える可能性のある治療を求め,受け入れる方法を徹底的に変えることである。
予測性 正確な薬品プラットフォーム(“PPMP”)
概要
PPPPは、患者の癌および正常細胞の治療または治療の組み合わせに対する生物学的反応を測定することによって、治療前に所与の治療または治療の組み合わせに対する単一の患者の臨床反応を予測する診断プラットフォームであることに留意されたい離体するそれは.注意すべきPPMPは内部生物学者、エンジニアとデータ科学者チームが独占的に開発したものであり、現在機能が完備しているが、効率と予測性能の角度から見ると、PPMPは依然として絶えず発展と改善している。
注目すべきPPMPは治療された細胞や細胞機能に対する癌細胞の反応を測定するため、遺伝子変異に対して設計された伝統的な精密薬物ではなく、遺伝子バイオマーカーにも制限されない。分析された各患者サンプルについて、注意すべきPPMPは、2値結果-予測応答者または予測無応答者を生成する。公認学術機関と協力した4つの独立協力研究において、注目すべきPPMPプラットフォームは臨床反応患者の識別において83%-100%の予測精度(陽性予測値(PPV)とも呼ばれる)を示した。83%−100%の予測精度は,臨床試験が予測応答者を選択的に募集すれば,83%−100%の応答率が期待できることを意味する。また,注目すべきPPMPはフォキシロピロロ2 a期試験の臨床結果を100%の正確性で予測した。
注目すべきは,他社買収により納得できる治療法を示すことに重点を置いているが,他社だけではこれ以上豊富ではなく,そのポートフォリオに適さないと考えられる10%−30%の患者 に対してのみ のさらなる開発を拒否している。これらの注目されているが放棄された資産をそのプラットフォーム上で有意に照準および試験するために、その固有のPPPPを使用して、そのPPPPがこの患者亜群を識別できることが証明された場合、これらの患者のためにこれらの化合物を選択的に許可し、残りの開発を迅速に追跡する。この戦略を用いて,最も高い医療ニーズを有する患者の予後(例えば,現在の看護基準では応答率0−50%)の特に改善が求められていることに注意されたい。
| 2 |
PPPPは、工学、デジタル技術、計算データ科学を含む生物学的力と技術的力を結合していることに注意されたい。 PPMPは、1つの遺伝経路や他の特徴に注目するのではなく、測定する離体する様々なシグナルや次元の薬物に対する癌と正常細胞の生物学的反応。その後、各患者サンプルの数十万個のデータ点が統合され、計算アルゴリズムによって、患者が実際の治療に反応する可能性があるかどうかを記述する患者反応予測値に変換される。注目すべきPPMPは、良性学習 サイクルおよび持続的最適化をサポートし、患者サンプルごとに高スループットの自動化プラットフォームとして設計されている。有名な実験室はアメリカ病理学学会(“CAP”)の認可を得た。
患者および医師にとって重要なPPMPの使用は簡単である:癌細胞および正常細胞を含む血液または骨髄サンプルは、重要な会社に運ばれ、その最適化された特許条件下で所与の1つまたは複数の薬剤と共処理される。PPMPは患者の癌細胞と正常細胞の間の差異行為と反応を測定し、単細胞レベルまで精確に計算し、 アルゴリズムはこれらの数十万個のデータ点を患者反応予測値に変換した。PPMPは、サンプルを受け取った後3~5日以内に患者の予想される反応を決定し、したがって、臨床的に操作可能な時間範囲内である。
持続的に増加しているデータバンクは、1900億行以上のデータを含み、これらのデータは、患者組織、血液サンプル、および臨床結果の分析から来ていることに留意されたい。このデータバンクは完全に内部で生成された単細胞分解能データセットから構成されている. は長年の自動化高スループットスクリーニングと良性学習周期の最適化を経ている。この専用データバンク は注目すべきデジタルバックボーンであり、そのプラットフォーム機能を疾患から疾患と追加の に拡張して医療結果を予測する戦略を推進する。
PPMPの臨床検証
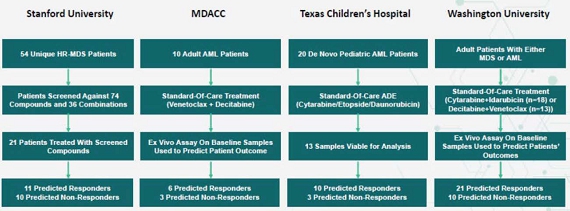
これまで,全世界で公認されている大学病院血液学領域のリーダーと協力して4つの臨床検証試験を行ってきたことに注目すべきである。これらの研究はいくつかの異なる血液癌と多種の標準治療方案におけるPPMPの表現を評価した。研究設計,患者数とテストの看護療法基準を以下の図にまとめた。
全ての研究は似たようなデザインを採用しています
| ● | 患者は治療を受ける前にすぐに研究に入った | |
| ● | 末梢血或いは骨髄のベースラインサンプルを獲得し、患者の反応を評価した離体する有名なPMPプラットフォームで発表されています | |
| ● | 次に患者 は治療を受け,その治療の結果(有効または無効)をプラットフォームから得られた予測結果と比較する。 |
感度、特異度、全体精度、陰性予測値、陽性予測値を含む、様々なパラメータを用いてPPMPなどの予測性診断テストの性能を定量化することができる。PPMPの最終的な期待用途は,所与の治療に最も反応する可能性のある患者を選択することであるため,PPVは最も関連する性能パラメータであることに注意されたい。PPVは,その実際の治療(実際の臨床応答者)に対して臨床反応を得た患者(実際の臨床応答者)のすべてのPPMP(PPMP予測応答者)患者の反応スコアが陽性である患者におけるパーセンテージで計算される。例えば,PPVが50%であることは,PPMP結果から反応が予測されるキューでは,50%の患者が実際の治療に臨床反応していることを意味する。臨床的に治療に対する反応が必ずしも疾患の長期治癒を招くとは限らないことに注意すべきである。

| 3 |
血液病研究の結果
骨髄異形成症候群(MDS)
スタンフォード大学との協力において、注目すべき臨床検証研究を行い、そのPPMPのハイリスクMDS患者の治療反応の予測における表現を評価した。この研究では,2016年9月から2019年3月までのサンプルを収集し,MDS治療の様々な薬物 療法をカバーしている。研究進行時には,5−アゾシチジンとジシタビンはFDAが承認したMDS治療薬であったが,他のすべての療法は,クロフラビンとアグリコシド,ボチゾミとデキサメタゾン,および5−アゾシトシンと万カーネーションを含めて研究されている。臨床治療効果評価は国際ワーキンググループ2006年MDS治療効果標準を採用し、治療効果を完全緩和(“CR”)と部分緩和(“PR”)と定義した。患者の末梢血液と骨髄は治療決定の前に直ちにbrを採取し,74種類もの単一治療薬と36種類もの併用治療薬の反応性をNOTICEのPPMPで評価した。治療後の臨床結果はスタンフォード大学の医師が評価し,PPMPによる予測結果と比較した。全部で21名の患者はすべての研究要求に符合し、そして全体分析に組み入れられた。全体的に注目すべきPPMPが100%のPPVを示したことは,PPMPが予測した個々の患者が医師が選択した治療に反応することを意味し,実際に治療に対する臨床反応を示している。PPPPの予測精度をより確実に推定するために、Bootstrapping(統計再サンプリング技術)を用いて患者データをシミュレーションし、92%のPPVを得た[95%CI=0.69-1.0]それは.ブートストラッピングは、複数のアナログサンプルを生成するために単一のデータセットを再サンプリングする統計的 プロセスである。この過程は,標準誤差 (サンプル平均値と可能な全体平均値との距離)と信頼度区間(CI)(95%のブートストラップシミュレーションが落ちた平均 性能範囲)の計算を可能にする.
成人急性骨髄性白血病
MD Anderson癌センターとの協力の中で、注目すべき会社は1つの臨床検証研究を行い、そのPPMPの急性骨髄性白血病患者の標準治療組み合わせベンニトクラクスガジシタビンの治療効果の予測における表現を検証した。この研究では,2018年8月から2021年2月までの間に地西他浜と万乃馨の併用治療を受けた患者のサンプルを収集した。この組み合わせは研究開始時にFDAの承認を得ていなかったが,FDAは1カ月後にAML治療の承認を得た。2017ヨーロッパ白血病ネットワーク(“ELN”)AML反応標準を用いて臨床反応を評価し、反応をCRとCRと定義し、計数回復不完全 (“CRI”)と定義した。治療前に患者の末梢血を採取し、PPMPを通じて患者の連合治療に対する反応性を評価した。患者の実際の治療後の臨床反応を評価し、PPMP予測反応と比較した。 計9名の患者はすべての研究要求に符合し、全体分析に入れた。全体的に注意すべき内部研究によると,PPMPはPPV 83%を示しており,PPMPが予測した治療反応の83%の患者が実際の治療に対して確実に臨床反応を得ていることを意味している。ブートストラップ·サンプリング技術は80%のPPVを生み出しました[95%CI=0.5-1.0].
ワシントン大学とのもう一つの協力において、ある臨床検証研究は2種類の異なる標準治療組み合わせに対するAML患者の反応を予測するPPMPの表現を検証した:ブンネデガジシタビン或いは阿糖胞配糖体ガイダビシン。この研究では,2020年3月から2022年6月までの間にアグリコシドと伊達ビシンやジシタビンと万カーネーションを受けた患者のサンプルを収集した。brという研究が行われている研究は引き続き患者を募集している。2017 ELN AML反応標準を用いて臨床反応を評価し、反応 をCR、CRIと形態無白血病状態と定義した。治療前に患者の末梢血を採取し、顕著なPPMPを通じて併用治療に対する患者の反応性を評価した。患者の実際の治療後の臨床結果を評価し,PPMP予測の反応と比較した。
ブンネデガデシタビン治療を受けた患者群では、13名の患者がすべての研究要求に適合し、全体的な分析に組み込まれている。全体的に注意すべきPPMPはPPVが100%であることを示し、これはPPMPが予測したすべての患者が彼らの治療に反応していることを意味し、br}連合治療は確かに実際の治療に対する臨床反応が得られたことを意味する。ブートストラップ·サンプリング技術は86%のPPVを生み出しました[95%CI= 0.63-1.00].
アラビシン治療を受けたグループの患者に対して、計18名の患者がすべての研究要求に符合し、全体分析に組み入れられた。全体的に、注意すべきPPMPはPPVが100%であることを示し、これはPPMPが予測したすべての患者がこの治療に反応していることを意味する。br}連合治療は確かに彼らの実際の治療に臨床反応を得た。ブートストラップサンプリング技術のPPVは100%である[95%CI =1.0-1.0].
| 4 |
小児科AML
テキサス児童病院との協力の中で、NORIGNは臨床検証研究を行い、小児科標準治療(アラビノシド、ダウノマイシンとエトロポシド(“ADE”)の三連療法)の治療効果の予測におけるPPMPの表現を検証した。この研究は2015年9月から2020年10月までの間のサンプルを収集した。研究中の患者はADEプラスまたはアトルバノンを含む通常の化学療法を受けており,臨床試験(NCT 03568994)で調査されている。1年間の無再発生存期間(RFS)或いは最小残留病(MRD)が1%未満であることによって臨床治療効果を評価する。RFSは治療後患者が無病状態を維持する持続時間,MRDは治療後に残存する少量の癌細胞を表す。急性骨髄性白血病患者。治療前に患者の末梢血と骨髄を採集し、PPMPを用いて患者の連合治療に対する反応を評価し、そして微小残留病(MRD)陰性或いは1年無再発生存率と比較した;これらの臨床結果指標は治療の持続性と関係がある。MRDと1年無再発生存率をPPMPによる予測結果と比較した。
MRD陰性を結果とし、13名の患者はすべての研究要求に符合し、全体分析に組み入れた。全体的に注目すべきPPMPのPPVは100%であり,これはすべてのPPMP予測応答者が実際の治療においてMRD陰性を確実に実現していることを意味しており, ブートストラップサンプリング技術は100%のPPVを産生している[95%CI=1.0-1.0].
1年無再発生存率の結果として,13名の患者がすべての研究要求に適合し,全体分析に組み込まれている。全体的に注意すべきPPMPはPPVが90%であることを示している[95%CI=0.9-1.0]これは,PPMP予測応答者の90%が実際の治療で1年間無再発生存を実現していることを意味している。ブートストラップ·サンプリング技術は99%のPPVを生成します[95%CI= 0.9-1.0].
補完性診断テストとセット診断テスト
どの患者および患者部分が利用可能な看護治療基準(疾患によって)に有効または無効であるかを識別および検証することによって、そのプラットフォームを液体および固形腫瘍に拡張することが計画されており、癌を超える可能性があることに注意されたい。br}単一の患者集団の持続的な増加に対する理解、彼らの利用可能なおよび研究中の治療に対する予測反応、および彼らが満足されていない医療需要(例えば、有効な治療選択がない患者)は、顕著な標的薬物許可および開発戦略を推進するために、独自のbr情報および補足診断を提供することが期待できる。
例えば、高度に満たされていない需要を有する患者集団(例えば、30%未満の患者が利用可能な看護治療基準 に反応する場合)が決定され、これらの患者のPPPPベースの研究治療の追求および許可が達成され、医療および商業的影響を最大に予測する治療によって、最も高い医療需要を有する患者にサービスすることに専念する。
このような正確な医学を予測する臨床開発計画では,NOWARYのプラットフォームが患者登録や治療を指導する患者選択の補助診断 として用いられている。
治療用製品開発
ヴォラセテブ
以上のように,注目すべき業務戦略は,治療資産の許可と開発に専念しており,これらの資産は約10%−30%の患者で納得できる臨床活動を示しており,PPMPを用いて を識別することができる。Volasertibはブリンガー-インゲルハイム社(Boehringer Inglheim,“BII”)によって開発され、固形腫瘍適応と急性骨髄性白血病(AML)において多くの臨床試験が行われている。
ヴォラセチブの研究背景と臨床経験
2021年、OnCoheros Biosciences Inc.(“OnCoheros”)からVolasertibの世界開発権と商業化権利を獲得したことに注目した。OnCoherosはボストンに本社を置くバイオテクノロジー会社であり、小児癌の新しい治療法の推進に専念している。VolasertibはPLK-1阻害剤であり、急性骨髄性白血病と他の腫瘍タイプ(固形腫瘍を含む)において活性を示し、顕著な未満足の医療需要を有する。PPMP上のVolasertib の表現に基づいて、同社はそのPPMPを利用して治療前にVolasertib反応のある患者の目標を識別と選択し、潜在的に は応答率を高め、Volasertibのこの患者群における臨床発展を迅速に追跡する。
NCT 0080485は、ヨーロッパとアメリカ血液学センターが87名のAML患者に対して行った第二段階の研究であり、Volasertibと小用量のアグリコシド(V+LDAC)治療の場合、CR+CRI率は31.0%であり、LDAC単独で治療したCR+CRI率は13.3%であり、これは小用量のアグリコシド(“LDAC”)単一治療と比べ、LDACにVolasertibを添加することは応答率を高めることができることを表明した。LDAC群と比べ、V+LDAC群の中位無事象生存期間は明らかに延長した(5.6ケ月vs 2.3ケ月;リスク比0.57;95%信頼区間0.35~0.92;P=.021);中位総生存期間はそれぞれ8.0ケ月vs 5.2ケ月であった(危険性比0.63;95%信頼区間0.40~1.00;P=0.047)。毒性において、深刻な有害事象(CTCAE分類3~5級)は以下の通りである:V+LDAC群23例(54.8%)とLDAC群7例(15.6%)は好中球減少症(白血球数の低下による発熱と/或いは疾病)が出現した。V+LDACの20名の患者(47.6%)とLDAC(2,3)の10名の患者(22.2%)が感染(細菌或いはウイルス性質)とbr}感染(寄生虫性質)を発生した。
| 5 |
NCT 01721876において、666名のAML患者に対する3期フォローアップ研究は世界各地で研究を行ったが、しかし、患者の総有効率は27.7%(V+LDAC)であり、プラセボ+LDACは17.1%(P+LDAC)であり、このことは一部の患者にVolasertib媒介のメリットが存在し、たとえこのような差が統計学的意義がなくても、このような差は統計学的意義がないことを表明した。毒性については、V+LDAC群による死亡(5級)の発生率(31.2%)がP+LDAC群(18.0%)より高く、これはV+LDAC群の比較的に高い感染と感染発生率(17.1%対6.3%)による可能性がある。毒性の面で、深刻な不良イベント(CTCAE分類3~5級)は以下の通りである:V+LDAC群258例(58.8%)、P+LLDAC群63例(28.4%)。V+LDAC群255例(58.1%)とP+LDAC群85例(38.3%)に感染と感染が発生した。
また,主に尿路上皮癌,卵巣癌,非小細胞肺癌を含む固形腫瘍に7項目の早期試験が行われており,これらの試験の結果,Volasertibは全体的に耐性が良好であることが示唆された。毒副作用では,ボルラシェブ治療患者に重篤な副作用が発生した:好中球減少55例(29.2%),貧血(赤血球減少)16例(8.5%),血小板減少19例(10.1%),白血球減少9例(4.8%)。
また,BIは2015年にVolasertib治療を受けた患者のための同情的使用計画を開始した。この計画では,2021年11月現在も7名の患者がVolasertibの長期治療を受けて反応している。
BIを利用したVolasertibデータセットと長年の臨床前と臨床開発とともに,Volasertibの 後期再開発計画を再設計し,臨床応答率と結果(Designantを用いたPPPP) の向上とVolasertibの耐性改善を目指している(BIによる特殊な後段階分析 3の結論による患者管理の強化):
臨床耐性を最大限に向上させ(骨髄抑制と感染を減少させる)ために,Volasertibの治療指数と試験を成功させるために,PPPPを用いて反応可能な患者を選択するのではなく,Volasertib臨床試験の設計を強化することに注目すべきである。注意すべきは、 計画の拡張機能には、
| - | 計量カスタマイズの投与量:急性骨髄性白血病2期と3期の臨床試験に350 mgのボルラザブ投与量を静脈注射し、患者の体重/体表を考慮しない。BIの事後分析により、比較的に軽い患者は350 mgの用量を過剰に服用し、更に高い毒性を招くことを示した。注意すべきは,使用量をカスタマイズした用量で,患者の体重/体表に応じて調整することである。 |
| - | 感染制御:適切な予防措置やプロトコルで規定されている感染予防措置を行えば,かなりの割合の致命的な感染が予防できると結論できる.これはいくつかの研究に参加するセンターがまだ到着していない可能性があり、特に世界第三段階試験である。 |
| - | AML第2期と第3期臨床試験では,VolasertibとLDACを併用した。公認された医学専門家と一致するために,有名なのは異なるVolasertibグループパートナーを選択する可能性がある。 |
| 6 |
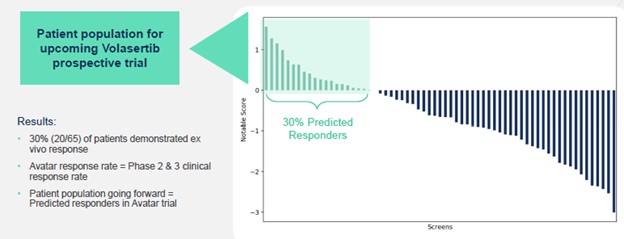
試験成績を高めるためには,そのPPMPを用いてVolasertibに臨床反応する患者を選択的に募集して試験に参加することに注意されたい。Volasertib敏感患者の選択におけるPPMPの表現を評価するために,NOTICEは1つの項目を行った前 生体そのPPMPを用いて期待患者キューで行ったアバター試験。体外アバター試験は体外でのみ薬物応答性をテストすることを含む;実際に薬物治療を受けていない患者はいない。インビトロ仮想アバタ試験は、全体的な予測検出性能を評価する上で情報を提供することができ、そのような研究において生成される任意の結果は、実際の患者試験中にさらに検証する必要があり、その後、検出を予測検出として定義することができる。この研究では,65名のAML患者から患者サンプルを収集し,ロックされたbrテスト方法を用いてPPMPテストを行い,予測応答者を階層化したことが有名である。PPMPは65名の患者のうち20名あるいは約30%の患者がVolasertibの治療に反応すると予測しており,これはBIがそのAML第二段階研究で発見した応答率を反映しており,これは有名なbrがそのPPPPプラットフォームが計画されたbr}臨床試験においてその実際のVolasertib治療に反応するAML患者を確実に識別できると信じていることと一致している。この65名の患者のPPMP結果によると、有名な会社は、このbr試験では50%から100%の参加者が彼らの実際のVolasertib治療に対して臨床反応を得る可能性があると予測している。臨床反応だけでvolasertibが臨床治療効果を証明できるとは限らないこと、あるいはその安全性がFDA或いは類似監督部門の許可を得る要求に符合することを証明することに注意すべきである。次の図は説明的であり,実際の患者テストの結果を反映していない。
注目すべきは,Volasertibの開発戦略は4つのPPPP臨床検証試験の経験と,最近2023年12月に報告されたフォックスロックス試験結果の100%正確なPPMP予測を利用していることである。
注目すべきは, は2024年の今後数カ月で再発性/難治性AML(r/r AML)の単腕開放タグVolasertib第2段階研究を開始する予定であることである。PPMP予測に焦点を当てたVolasertib応答者の成功の第2段階研究の後,登録の第3段階研究を開始する予定であることに注意されたい。規制の観点からは,PPMPを患者選択ツールとして使用し,PPMPをFDA基準の随伴診断に適合させ,volasertibに対するNDA 提出とともに市販前承認(PMA)提出が必要であることに注意した。Volasertibの発売には,対応する診断PMA提出およびVolasertib NDA提出を同時に承認する必要がある。
注目すべきは、 は2024年第1四半期にINDを提出し、2024年第2四半期に第2段階計画を開始し、2024年第4四半期に6人の患者に対する第2段階用量最適化前奏データを提供し、2024年第4四半期に第1位のPPMP選択のbr患者を募集することである。
OnCoheros 許可プロトコル
2021年10月8日、OnCoheros Biosciences Inc.(“OnCoheros”)と独占許可協定(“OnCoheros協定”)を締結し、これにより、OnCoherosはOnCoherosプロトコルによって保持されたあるタイプの血液癌およびすべての他の癌に関連する適応(通常、小児科固形腫瘍の適応を含む)(“許可されたbr}領域”)のための小分子Volasertibのグローバル独占開発および商業化権利を買収したことに注目すべきである。許可領域は:急性リンパ球性白血病;急性巨核球性白血病;急性骨髄性白血病;急性前骨髄性白血病;慢性リンパ球性白血病;慢性骨髄性白血病;慢性骨髄性白血病;青少年顆粒球性白血病;ホジキン病;白血病;リンパ腫(間変性大細胞と大B細胞);及び非ホジキンリンパ腫を含む。OnCoheros 協定については、2021年10月8日に、卓越はOnCoherosと1,500,000ドルの未来株式簡単協定(“OnCoheros Safe”) を締結し、これにより、OnCoherosのいくつかの株株を買収する権利を卓越して取得した。
| 7 |
OnCoherosとベーリングインゲルハイム国際有限公司(“BII”)が2020年4月5日及び2021年10月に改訂された2019年8月1日に締結した譲渡及び許可協定(“BII 協定”)条項によると、OnCoherosはいくつかの譲渡特許(“譲渡特許”)、及びある許可製品(“許可製品”)の開発及び商業化の権利を取得した。OnCoherosプロトコルによれば,OnCoherosプロトコルに記載されている条項や条件に基づいて,OnCoherosはOnCoherosプロトコルに記載されている条項や条件に基づいて,許可された製品を開発および商業化する権利があることに注意されたい.OnCoherosがその権利を許可,売却,譲渡または譲渡またはBIIプロトコルで付与された関連許可を終了することを決定した場合,当社は優先購入権と 優先交渉権を持つ.
“OnCoherosプロトコル”によれば、OnCoherosに特許使用料を支払わなければならないことに留意されたい。金額は、公平な取引においてOnCoheros、その関連会社または再ライセンシーにライセンス製品を販売する総金額(有名、その関連会社またはそれらのそれぞれの再被許可者間の取引に関連する販売を含まないが)、OnCoherosプロトコルに列挙された世界的な許可領域内のいくつかの費用または支出の総額(“純売上”)を減算することを意味するまた、OnCoherosに合計8ドルまでの料金を支払う必要がありますいくつかの開発マイルストーン事件が発生した時に100万ドルのマイルストーン支払いを受けた。
OnCoherosプロトコルによると,純売上高の変動割合範囲に応じて許可領域における許可製品の純売上高の特許権使用料をOnCoherosに支払う必要があり,この特許権使用料パーセンテージは1桁の中央値から10代程度であることに注意されたい.なお、ライセンス使用料の支払い義務は、ライセンス製品のライセンス分野における国/地域に基づく商業発表から始まり、(A)ライセンス特許の最後の有効な権利主張は、その国/地域で満了することを主張し、(B)適用データまたは他の規制固有権が満了し、(C)このようなライセンス製品は、その国/地域でビジネス形態で発行されてから10年後に終了することに留意されたい。
“OnCoherosプロトコル”によれば、追加的に終了しない限り、“注意すべき”使用料の支払い義務が満了するまで、割り当てられた特許の権利は、各国において を継続しなければならない:(A)“OnCoherosプロトコル”の重大な違反、または“OnCoheros”の破産によってOnCoherosによって終了されるか、または(B)“OnCoheros”が“OnCoherosプロトコル”、“Ii)”OnCoheros“破産、(Iii)”ベースBIIプロトコル“に深刻に違反し、終了する。代替的に、(Iv)いつでも、特定の国/地域全体または特定の許可製品についても、 はOnCoherosに90(90)日の書面通知を発行する。OnCoherosプロトコルを終了する場合には,BIIライセンシーとしてOnCoherosを負担する権利と義務を選択することができることに注意されたい.
製造業
注目すべきは, は第三者製造に依存していることであるヴォラセテブBrは契約製造組織やCMOと協定を締結し、彼らのために薬物物質を生産していることに気づいたヴォラセテブ. はそのすべてのCMOが現在の良好な製造仕様またはcGMP要求に従って生産活動を行うことを著しく要求する.注目すべきは,これらのCMOは臨床供給や商業規模生産を支援する能力があることであるが,現在のところ商業生産をカバーする正式な合意は何もないことに注意されたい。他のCMOと協定を締結して、薬品や完成品薬品の供給を生産することも選択できることに注意されたい。
販売 とマーケティング
注目すべき候補製品が承認された場合、単独または他社と協力して、米国でマーケティングおよび商業化を行い、国際市場を選択することに注目すべきである。癌患者は腫瘍学者、医学遺伝学者、神経学者によって管理されているため、彼らは的確な販売チームによって実現できると信じていることに注意すべきである。
| 8 |
競争Volasertib
製薬業界の特徴は技術発展が迅速で、競争が激しいことだ。その候補製品、技術、知識、経験と科学資源は優れた競争優勢を提供していると考えられているが、主要な製薬と生物技術会社、学術機関、政府機関及び公共と個人研究機関などからの競争 に直面している。注目すべき成功した開発および商業化された候補製品は、ラベル外療法および将来発売される可能性のある新しい療法を含む承認された治療レジメンと競合する。NOTIGNの有効な他の療法との競争能力に影響する重要な要素は治療効果、安全性、投与方法、コスト、販売促進活動レベル及びNOTIGN製品の知的財産権保護を含む。多くの有名な競争可能な会社は研究開発、製造、臨床前テスト、臨床試験を行い、監督管理許可とマーケティング許可を得た製品の面で有名な会社よりもっと多くの資源と資本を持っている。
FDAが承認したPLK 1阻害剤は現在のところないが,いくつかの臨床開発の異なる段階にある。Onvansertib(カーディフ腫瘍社)、GSK 461364(GSK,plc)、rigosertib(Onconova Treateutics,Inc.)Cy140(Cyclacel製薬会社)とこれらはすべてPLK 1抑制を示し、いくつかの実体と血液系悪性腫瘍に発生した。より多くの会社がこれらの患者集団の解決に適したPLK 1阻害剤計画を持っている可能性があり、これらの会社は注目すべき努力と競争力があるかもしれないが、具体的な臨床開発計画は開示されていない。小さいか早い段階にある会社は、腫瘍学に集中した治療会社を含み、重要な競争相手である可能性もあり、特に大型および成熟会社との協力を通じて手配する可能性がある。brこれらの会社はまた、合格した科学と管理者を採用し、維持し、br臨床試験サイトを設立し、患者を臨床試験に参加させ、補充或いは有名な計画に必要な技術面で有名な会社と競争する可能性がある。brが承認されれば、政府と個人支払人の精算もbr重要製品の定価と競争力に著しく影響する。注目すべき競争相手は、注目すべきライバルよりも早くFDAまたは他の規制機関のその製品に対する承認を得る可能性があり、これは、その候補製品の承認を得る可能性があり、これにより、その競争相手が著しく承認された後にその製品を商業化する前に強力な市場地位を確立することができる可能性がある。
シプロフロキサシン (CPX-POM)
2021年7月20日、CicloMed LLC(“CicloMed”)と共同開発および利益共有プロトコル(“CicloMedプロトコル”)が締結されたことに注目すべきであり、CicloMedが急性骨髄性白血病の治療のためのCicloProxプロドラッグ(“CPX-POM”)製品を研究および開発する際にCicloMedを使用する正確な腫瘍学診断試験である。CicloMed合意の条項により,双方は共同開発費用を平均的に分担するが,各方面が指定した共同指導委員会が制御する予算を遵守する必要がある。 で利益イベントが発生した場合,純収益はプロトコルの によって規定される補償シェアを双方の間で分配し,通常50/50であり,顕著な最高補償シェアは発生した総共同開発コストの中央値から1桁の倍数を超えてはならないことを前提としている.補償シェアは,(I)側の共同開発計画による総共同開発コストと(Ii)双方がその計画による総共同開発コストの和の比率 に対応するパーセンテージである.双方の間で、CicloMedはCPX-POMとCPX-POM結果のみに関連するすべての共同開発知的財産権 を持ち、顕著には顕著なテストとテスト結果 のみに関連するすべての共同開発知的財産権を持ち、それぞれが自分の背景と以前の知的財産権を保持する。
フォキシロピロロの研究背景と臨床経験
アスピリン(CPX−POM)は,静注後に活性代謝物CPXを尿路全体に選択的に輸送するカンザス大学癌センターおよび先進医学革新研究所の科学者によって発明されたトロピロールのプロドラッグである。CPXは細胞増殖,クローン形成,球体形成を抑制し,細胞周期を増加させてSおよびG 0/G 1期に停滞する。メカニズム的には,CPXはNotch信号の活性化を抑制する。分子シミュレーションと細胞熱変位分析により、シクロホスファミドはγ-分泌酵素複合蛋白である早老素1とニコルシンに結合し、この2種類の蛋白はNotch活性化に必要であることを表明した。
を作る体内にある臨床前の原理検証として,福司可羅は検証されたN−ブチル−N−(4−ヒドロキシブチル) ニトロソアミン(BBN)マウス膀胱癌モデルで試験を行った。CPX-POMは毎日1回連続4週間、投与量は235 mg/kgと470 mg/kgであり、膀胱重量(腫瘍体積の代替物)を著しく低下させ、そして腫瘍の低分期への転移を招く。同時に、拡散指数も低下した。また,CPX−POMで処理した動物膀胱組織では早老素1とHes−1の発現が減少した。
Brの最初のヒト1期試験(NCT 03348514)が完成した後、フォスコクロピロールの薬理活性は現在膀胱切除術を行う筋肉浸潤性膀胱癌患者(NCT 04608045)に対する1期拡大コホート研究(NCT 04608045)、及び経尿道膀胱腫瘍電気切除術(NCT 04525131)を計画する新しい診断と再発尿路上皮癌患者に対する2期試験(NCT 04525131)を行っている。
| 9 |
2021年7月、CicloMedプロトコルの条項に基づいて、CicloMedは再発または難治性AML成人患者におけるフォキシロップの1 b/2 a期臨床試験(NCT 04956042)を開始した。実験の一部として,この研究に参加した患者を登録し,注目すべきPPMPにおけるフォシクロビル に対する感受性を評価した。PPPP結果に基づいて試験参加患者を選択しなかった患者は,潜在的な偏見を避けるために,試験期間中にPPMP分析は実際の患者結果に対して盲目的であった。このグループの大量の前処理を経た患者の中で、臨床治療効果はAML医学研究と実践中の標準標準によって定義されている。
2023年12月には,この試験の背線結果が有名に報告された。大量の予備治療を受けた18名の患者がbr試験に参加し,そのうち9名の患者がレジメン別に応答評価を行った。推奨された第二段階投与量で投与したところ,耐性は良好であった。しかし、評価可能な9人の患者のうち1人は完全に緩和しなかった。病状は安定しており,4カ月を超え,評価可能な2名の患者に認められた。
重要なことは,PPPPは,この試験に参加したすべての患者がフォズキシロップに反応しないと予測していることである。この予測の正確性は,患者の治療に対する実際の反応によって確認された。これらのPPMPの結果は,登録された患者集団がフォスシクロビルに反応しない傾向にあることを示しており,最初にPPPP を用いて予測応答者のみを選択的に募集すれば,2 a期試験の負の臨床結果は回避されている可能性が示唆された。さらなる分析が行われている。
知的財産権
注目すべき成功は、その候補製品、製造および加工 発見および他の技術に対する独自の保護を獲得および維持し、他人の固有の権利を侵害することなく運営し、他人が注目すべき固有の権利を侵害することを防止する能力があることにある程度依存する。注目すべき計画は、ノウハウ、発明および改善に関連する現在の米国および外国特許の保護、およびより多くの米国および外国特許の起訴を含む様々な方法を使用して独自の地位を保護することであり、これらの特許は、その業務の発展および実施に非常に重要であることに留意されたい。例えば、その許可者、br、または重要な協力者が現在、その候補医薬物質からなる特許 を所有しているか、または出願しており、1つまたは複数の臨床項目の使用方法をカバーする特許保護が一般的に求められていることに留意されたい。顕著にはまた、商業秘密、商標、技術ノウハウ、持続的な技術革新、および潜在的な許可内機会に依存して、顕著な独自の地位を発展させ、維持する。
Volasertib 特許
著名な は2021年10月にOnCoheros許可協定を締結し、この協定に基づいて、62件の特許と特許出願の下の全世界独占権利を買収し、Volasertibの開発、製造と商業化を行った。OnCoheros 許可協定によると、有名な会社はVolasertibに関連する米国と多くの外国司法管轄区特許権の独占許可を持っている。OnCoherosライセンス協定によって付与された特許権は、米国における19(19)件のライセンス特許および1(1)件の特許出願と、欧州特許庁、ドイツ、フランス、イギリスおよび日本を含む他の司法管轄区で許可された40(40)件の特許および2(2)の特許出願とを含む。特許ファミリーでVolasertibをカバーするすべての3(3)項目の米国特許は2023年2月26日に満了しており、Volasertibは通常brの形態に属する物質組成物である。特許ファミリーの2つ(2)項の米国特許は、Volasertibの物質としての組成物およびその様々な結晶形態を含む2027年1月30日に満了する。現在臨床開発中のVolasertib 配合のアメリカ物質成分特許は2026年6月29日に満期になり、特許期限調整或いはいかなる特許 の延長、及び関連する外国の同業者を含まない。
取引 秘密
特許に加えて、商業秘密や技術ノウハウに依存して競争地位を発展させ、維持していることが明らかになった。 は、一般に、そのトラフィックのいくつかの態様を保護するために商業秘密によって保護されており、これらの態様は、特許 によって保護されているか、または適合していないことに留意されたい。購買力平価には注目すべき商業機密が含まれている。しかし,PCMPは現在特許保護されておらず,特許出願 もない.有名なのは、その従業員、コンサルタント、科学コンサルタント、請負業者およびパートナーと秘密協定および発明譲渡協定を確立することによって、商業秘密およびノウハウを保護する。これらのプロトコルは,一般に,個人や実体と重要人物との関係中に開発または公表されるすべての機密情報 は,関係期間と後に秘密にしなければならないことを規定している.これらのプロトコルはまた、有名またはその業務に関連する仕事のために生成されたすべての発明、および雇用または譲渡中に構想または完了した発明は、有名なbr独自財産でなければならないと一般的に規定されている。さらに、我々の独自情報が第三者に盗用されることを防止するために、物理的および技術的セキュリティ対策のような他の適切な予防措置がとられていることに留意されたい。
政府の法規
Volasertibと他の薬品のアメリカでの承認
アメリカ政府当局(“U.S.”)連邦、州と地方各級及びその他の国と司法管轄区では、欧州連合(“EU”)を含む、その他の事項を除いて、 は薬品の研究、開発、テスト、製造、品質管理、承認、ラベル、包装、貯蔵、記録保存、販売促進、広告、流通、承認後の監視及び報告、マーケティングと輸出入薬品(例えばVolasertib)に対して広範な監督管理を行う。通常、新薬が発売できる前に、大量のデータを獲得し、その品質、安全性と有効性を証明し、そして各監督管理機関特有のフォーマット に組織し、監督管理機関の審査と承認を提出しなければならない。
| 10 |
FDA承認プロセスの概要
アメリカでは、医薬品はFDAによって広く規制されている。“連邦食品、薬品と化粧品法”(“FDCA”)及びその他の連邦と州の法律法規は薬品の研究、開発、テスト、製造、貯蔵、記録保存、承認、ラベル、普及とマーケティング、流通、承認後のモニタリングと報告、サンプリング及び輸出入などを管理する。適用された米国の要求を遵守しないことは、FDAが未解決のNDAの承認を拒否し、警告または無見出し手紙、製品リコール、製品差し押さえ、生産または流通の完全または部分的な一時停止、禁止、罰金、民事罰、および刑事起訴のような様々な行政または司法制裁を受ける可能性がある。
医薬品brは、米国において、新製品または承認製品のいくつかの変更に対する製品開発は、通常、臨床前実験室および動物試験に関連し、IND(臨床試験が開始される前に施行されなければならない)、および十分なbrおよび制御された良好な臨床試験をFDAに提出して、FDAの承認を求める各適応に対する安全性および有効性 を決定する。FDA発売前の審査要求を満たすには通常長年の時間を必要とし、実際の所要時間は製品或いは疾病のタイプ、複雑性と意外性によって大きく異なる可能性がある。
臨床前試験は製品化学、調合と毒性の実験室評価、及び製品特性と潜在安全性と有効性を評価する動物試験を含む。臨床前試験の実施は必ず良好な実験室実践を含む連邦法規と要求に符合しなければならない。臨床前試験の結果はINDの一部として他の情報とともにFDAに提出され,製品化学,製造と制御に関する情報および提案された臨床試験案が含まれている。IND提出後,生殖毒性や発ガン性の動物試験など,長期的な臨床前試験を継続する可能性がある。ヒト臨床試験を開始する前に,各INDを提出してから30日間の待機期間が必要である。FDAがこの30日間INDにコメントもINDにも疑問を提起しなければ,INDで提案された臨床試験が開始される可能性がある。
Brの臨床開発段階は、試験スポンサーまたは試験スポンサーの制御下に雇われていない医師であり、良好な臨床実践(GCP)の要求に適合し、すべての研究対象に任意の臨床試験に参加することを要求するインフォームドコンセントを提供することを含む、合格した調査者の監督下で健康ボランティアまたは疾患の影響を受けた患者に研究製品を提供することを含む。臨床試験は,臨床試験目標,投与手順,被験者の選択と排除基準,および被験者の安全性および治療効果を評価するためのパラメータを詳細に説明するレジメンで行われた。すべての議定書とその議定書に対するいかなる後続の修正もINDの一部としてFDAに提出されなければならない。さらに、各臨床試験は、臨床試験に参加する個人のリスクが最小限に低下し、予期される利益に対して合理的であることを保証するために、臨床試験を行う各機関のIRBによって審査および承認されなければならない。IRBはまた、各臨床試験対象またはその法定代表者に提供されなければならないインフォームドコンセント を承認し、完了まで臨床試験を監視しなければならない。また,行っている臨床試験と完成した臨床試験結果を公的登録機関に報告することが求められている。臨床試験の多くの情報は,www.Clinicaltrials.govサイトで公表されるために,特定の時間範囲で提出されなければならない。臨床試験登録の一部として,製品,患者群,調査段階,試験地点や研究者,臨床試験の他に関する情報が公開される。brスポンサーも完成後に臨床試験の結果を検討する義務がある。これらの裁判結果の開示は、場合によっては裁判完了日以降2年に延期することができる。競争相手はこの公開情報 を用いて開発計画の進捗状況を知ることができる.人体臨床試験は通常3つの連続段階に分けて行われ、この3つの段階は重複或いは合併する可能性がある
| ● | 第1段階臨床試験は、通常、一部の健康ボランティアまたは疾患の影響を受ける患者に関するものであり、彼らは最初に単剤を使用し、その後、多剤候補製品を使用する。これらの臨床試験の主な目的は薬物の新陳代謝、薬理作用、副作用耐性と安全性を評価することである。 | |
| ● | 第二段階の臨床試験は、予想される利益を産生するために必要な投与量を決定するために、疾患の影響を受ける患者を研究することに関する。同時に安全性と更なる薬物動態学と薬効学情報を収集し、可能な副作用と安全リスクを識別し、そして初歩的な治療効果評価を行った。 | |
| ● | 第3段階臨床試験は通常、複数の地点のより多くの患者に関連し、必要なデータを提供し、期待用途における製品の有効性、使用中の安全性を証明し、そして製品の全体収益/リスク 関係を確定し、製品審査に十分な基礎を提供することを目的としている。これらの試験は、プラセボおよび/または他の対照治療との比較を含むことができる。治療の持続時間が常に延長され、製品のマーケティング期間中の実際の使用をシミュレートする。 |
登録試験は監督管理機関が候補薬物の治療効果と安全性を評価する要求を十分に満たす臨床試験であり、この薬物が承認されたことが合理的であることを証明することができる。通常,登録試験は3期試験 であるが,試験設計が臨床的利益の信頼性の高い評価を提供し,特に満たされていない医療ニーズが存在する場合には2期試験である可能性がある。
| 11 |
承認後のbr臨床試験は、時々4期臨床試験と呼ばれ、初歩的な発売許可後に行うことができる。これらの試験は期待される治療適応患者の治療から追加の経験を得るために用いられ、特に長期安全フォローアップに用いられる。場合によっては,FDAはNDAを承認する条件として4期臨床試験を強制的に実行することができる。
臨床試験結果を詳細に説明した報告は少なくとも毎年FDAに提出されなければならず,重篤な有害事象が発生すればより頻繁に提出される。FDAまたはスポンサーは、臨床試験を随時一時停止または終了することができ、またはFDAは、研究患者が受け入れられない健康リスクに直面していることを発見することを含む、様々な理由に基づいて他の制裁を適用することができる。同様に,臨床試験がIRBの要求に沿って行われていない場合,あるいは薬物が患者に意外な重篤な傷害を与えた場合,IRBはその施設の臨床試験の承認を一時停止または終了することができる。
必要な臨床試験が完了した後,秘密保持プロトコルを用意してFDAに提出する。製品が米国市場で販売される前に、FDAの承認を得る必要がある。NDAは、すべての臨床前、臨床および他の試験の結果、および製品の薬理、化学、製造および制御に関するデータの編集を含まなければならない。機密協定の準備と提出の費用は非常に高い。多くのNDAの提出には高額な申請使用料が必要であり,メーカーおよび/または承認されたNDA下のスポンサーも合格製品の年間計画費を支払う必要がある。
FDAはNDAを受信した日から60日があり,当該機関が申請が十分であるかどうか,実質的な審査が許可されているかどうかの敷居に基づいて,申請を受け入れるかどうかを決定する。提出された申請が受け入れられると、FDAは深い検討を始めた。FDAはNDAのレビューでいくつかのパフォーマンス目標を設定することに同意した。FDAの目標は,薬品の標準審査を10カ月以内に完了し,6カ月以内に優先審査を完了することである。優先審査は、FDAが治療において大きな進展を得るか、または適切な治療法がない治療法を提供する薬剤を決定するのに適用することができる。FDAは、いくつかの遅延された情報を考慮するために、または提出中に提供された情報の情報を明確にするために、標準審査および優先審査の審査プロセスを3ヶ月延長することができる。
FDAはまた、新薬製品の申請または安全性または有効性の問題を提起する医薬製品の申請を諮問委員会に提出することができる--通常、臨床医および他の専門家を含むグループであり、審査、評価を行い、申請が承認されるべきかどうかについて提案することができる。FDAは諮問委員会の提案によって制限されていないが、それは一般的にそのような提案に従っている。NDAを承認する前に、FDAは、通常、GCPに適合することを確実にするために、1つまたは複数の臨床場所を検査する。さらに、FDAは薬物を製造する1つ以上の施設を検査するだろう。FDAは、cGMPに適合しない限り、この製品を承認せず、NDAに含まれるデータは、研究された適応において安全かつ有効であることを証明する多くの証拠を提供する。
FDAが機密協定と生産施設を評価した後、それは承認状または完全な返信を発行するだろう。完全な応答文は、一般に、提出中の不足点を概説し、FDAが出願を再検討するために、大量の追加のテストまたは情報 を必要とする可能性がある。FDAがNDAを再提出する際にこれらの欠陥を満足的に処理した場合、FDAは承認書を発行する。FDAは、そのような再提出を、2ヶ月または6ヶ月以内に、含まれる情報タイプに基づいて検討することを約束した。
この薬物の商業マーケティングを許可し、特定の適応の具体的な処方情報を提供する。NDA承認の条件 として、FDAは、薬物の利益が潜在的リスクよりも大きいことを保証するために、リスク評価および緩和策またはREMSを必要とする可能性がある。REMSは、薬物ガイドライン、医療専門家のコミュニケーション計画、および安全な使用を確保する要素、またはETASUを含むことができる。ETASUは、処方または調剤のための特殊なトレーニングまたは認証、場合によっては調剤、特殊な監視、および患者登録簿の使用を含むことができるが、これらに限定されない。REMSに対する要求はこの薬物の潜在的な市場と収益力に重大な影響を与える可能性がある。また、製品承認には、薬物の安全性や有効性を監視するために、大量の承認後のテストと監督が必要となる可能性がある。承認されると、規制基準が守られていない場合や初期マーケティング後に問題が発見された場合、製品承認が撤回される可能性がある。
承認された申請で確立されたいくつかの条件を変更 し,適応,ラベルや製造プロセスや施設の変更 を含み,新たなセキュリティプロトコルまたは秘密プロトコル付録を提出し,FDAの承認後に変更を実施する必要がある.新適応のNDAサプリメントは通常,オリジナル申請と類似した臨床データが必要であり,FDAがNDAサプリメントを審査する際に使用するプログラムや行動は,NDAを審査する際に使用するプログラムや行動と同じである。
| 12 |
アメリカの独占経営権
新しい化学物質(“NCE”)がNDAの承認を得た後、この薬物は5年間の市場独占期間を得ることになり、その間FDAはこの薬物の模倣薬の承認を求める任意の簡略化新薬申請(ANDA)を受けることができない。薬物のいくつかの変更、例えばパッケージ挿入に新たな適応 を添加することは、3年間の独占期間に関連しており、その間FDAは、変更された模倣薬を含むANDA を承認することはできない。第4項の認証を提出した場合、ANDAはNCE排他性満了の1年前に提出することができる。 Orange Bookに記載されている特許がなければ、第4項の認証がない可能性があるため、ANDAは排他期間が満了する前に提出されない可能性がある。
特許延期
国家薬品監督管理局の承認後、関連薬物特許の所有者は、1つの特許出願のために最大5年間の特許延期を行うことができる。許容される特許期間 は、薬物試験段階(INDとNDA提出との間の時間)と全審査段階(NDA提出と承認との間の時間、最長5年)の半分で計算される。FDAが、出願人が職務調査を経ずに承認を求めていると判断した場合、時間を短縮することができる。展示期間後の総特許期間は承認日から14年以下である。
出願段階で満了する可能性のある特許については,特許所有者は一時特許延期を請求することができる。臨時特許延期 は特許期間を1年間延長し,最大4回延長することができる.承認ごとに臨時特許が延期され,承認後特許延期は1年減少する.米国特許商標局の取締役は,特許延期を申請している特許に含まれる薬物が承認される可能性が高いことを確認しなければならない。秘密保持契約を提出していない薬物 は一時特許延期を得ることができない。
迅速な追跡指定と承認の加速
FDAは重篤あるいは生命に危険な疾患や疾患を治療するための薬物の開発と審査の加速を促進する必要があり、これらの薬物は有効な治療方法がなく、このような疾患が満たされていないbr}医療需要を満たす潜在力を示している。Fast Track計画によると、新薬候補のスポンサーは、候補薬物のIND提出と同時にまたは後に、FDAに特定の適応の候補薬をFast Track薬として指定することを要求することができる。FDAは、スポンサーからの要請を受けてから60日以内に、その候補薬がFast Track指定を受ける資格があるかどうかを決定しなければならない。
FDAとより頻繁なインタラクションができるなどの他の利点に加え,FDAは申請完了前にFast Track薬物セキュリティプロトコルのbr部分の審査を開始することができる。申請者が余剰情報を提出するスケジュールを提供し、申請者が適用された使用料を支払うことができれば、スクロール審査を行うことができる。しかしながら、FDA審査申請の期間目標は、セキュリティプロトコルの最後の節が提出されるまで開始される。また,FDAがFast Track指定が臨床試験中に出現したデータの支持を得なくなったと考えた場合,FDAはその指定を撤回する可能性がある。
FDAの加速承認規定によれば、FDAは、臨床利益を合理的に予測する可能性のある代替終点に基づいて、または不可逆的な発病率または死亡率よりも早く測定可能な臨床終点に基づいて、不可逆的な発症率または死亡率または他の臨床的利益への影響を合理的に予測し、病状の重症度、希少性または流行度、および代替治療の利用可能性または不足を考慮して、患者に意味のある治療利益を提供することができる深刻または生命に危険な疾患に対する薬剤を承認することができる。
Br臨床試験において、代替終点は疾病或いは状況の実験室或いは臨床症状の測定であり、患者の感覚、機能或いは生存状況の直接測定の代わりになる。代替終点は通常、臨床終点よりも容易または迅速に測定 を行う。この基礎の上で承認された候補薬物は必ず厳格な発売後のコンプライアンス要求を遵守しなければならず、 の4期完成或いは承認後の臨床試験を含み、臨床終点への影響を確認する。“2022年食品·薬物総合改革法案”(FDORA)によると、FDAは、このような試験を、承認前または承認を加速させた製品が承認された日後の特定の期間内に行うことを状況に応じて要求することができる。必要な承認後研究ができなかったか,あるいは発売後の研究期間中に臨床的利益が確認されなかった場合,FDAが速やかに市場からbrを撤回することを許可する。FDAは、一般に、機関から別の通知がない限り、加速規制によって承認された候補製品のすべての販売促進材料がFDAの事前審査を通過しなければならないことを要求する。
| 13 |
画期的なbr治療指定
FDAの画期的な薬物指定はFDA高級従業員により広範な開発相談機会を提供し、薬物の承認申請のスクロール審査を許可し、深刻または生命に危険な疾患を治療するための薬物申請を提出する際に臨床データの支持を得るか、またはbr}の初歩的な臨床証拠が1つ以上の臨床重要終点で既存の治療法よりも実質的に改善されている可能性があることを示している場合、この製品は優先審査を受ける資格がある可能性があることを示している。画期的療法計画によると,新薬候補薬のスポンサーは,候補薬物のIND届出と同時にあるいはその後,特定の適応の候補薬を画期的療法として指定することをFDAに依頼することができる。FDAはスポンサーからの要請を受けて60日以内に候補薬が画期的な治療指定を受ける資格があるかどうかを決定しなければならない。
Volasertibと他の薬品のEUでの承認
概要
EUでは、注目すべき候補製品もまた広範囲な規制要求を受ける可能性がある。アメリカと同様に、医薬製品 は主管監督機関のマーケティング許可を得た後にのみ発売できる。アメリカと類似して、EUの臨床前と臨床研究の各段階は厳格な監督管理によってコントロールされている。
“臨床試験指令2001/20/EC”、“GCPに関する指令2005/28/EC”及びEU各加盟国の関連国家実施条項はEU臨床試験審査制度を管理している。この制度によると、申請者は事前に臨床試験を行うEU加盟国国家主管部門の承認を得なければならない。また,出願人 は主管倫理委員会が賛成の意見を発表した後のみ,特定の試験地点で臨床試験を開始することができる。臨床試験申請にはIMPDや汎用技術文書などの文書が添付されており,2001/20/EC号指令,2005/28/EC号指令に規定されている支援情報が添付されており,EU各加盟国の実施国規定に関連し,適用される指導文書でさらに詳細に説明されている。臨床試験期間中に発生したすべての予期せぬ深刻な副作用は,これらの反応が発生した国家主管部門と加盟国の道徳委員会に報告しなければならない。
2014年4月,新たな臨床試験条例,(EU)第536/2014号が採択された。“臨床試験条例”はすべてのEU加盟国に直接適用され、現在の臨床試験指令2001/20/ECを廃止する。EUで行われたすべての臨床試験は、新しい臨床試験条例が施行されるまで、現在の適用条項の制約を受け続ける。行われている臨床試験が“臨床試験条例”に管轄される程度は,“臨床試験条例”がいつ適用されるか,個別の臨床試験の持続時間に依存する。もし臨床試験が“臨床試験条例”の発効日から3年以上持続すれば、“臨床試験条例”はその時にこの臨床試験の適用を開始する。
新しい“臨床試験条例”はEUの臨床試験の審査プロセスを簡略化することを目的としている。この条例の主な特徴は、単一の入口点である“EUポータル”を通じて申請手続きを簡略化すること、申請のための単一の文書の準備と提出、臨床試験スポンサーの報告手続きを簡略化すること、および統一された臨床試験申請評価プログラムであり、このプログラムは2つに分けられる。第1の部分は、臨床試験許可申請が提出されたすべてのEU加盟国(関連加盟国)の主管当局によって評価される。 第2の部分は、各関連加盟国によって個別に評価される。すでに臨床試験申請の評価に厳格な締め切りを設定した。評価手続きにおける倫理委員会の役割は、EU加盟国に関する国内法律によって引き続き管轄されるだろう。しかし、全体的に関連するスケジュールは臨床試験条例によって定義されるだろう。
EUで医薬品の上場許可を得るためには、有名な会社は、いわゆる集中式または国家許可プログラムに従ってマーケティング許可申請(“MAA”)を提出することができる。
集中式 プロセス
集中化プログラムは、欧州薬品管理局(“EMA”)の賛成意見に基づいて、すべてのEU加盟国およびアイスランド、リヒテンシュタイン、ノルウェーで有効である単一マーケティング許可を付与することを規定している。特定のバイオテクノロジープロセスによって製造された医薬品、孤児医薬品として指定された製品、先進的な治療薬(例えば、遺伝子療法、体細胞療法または組織工学薬)、およびHIV/エイズ、癌、糖尿病、神経変性疾患または自己免疫疾患 および他の免疫機能障害およびウイルス疾患などの特定の疾患を治療するための新しい活性物質を含む製品については、集中手順が実行されなければならない。重大な治療、科学的または技術的革新を代表する製品、またはその許可が公衆の健康に有利になる製品については、集中化手順がオプションである。中央手続き によると、申請者が人用薬品委員会(“CHMP”)からの質問に回答した場合、環境保護局がMAAを評価する最長期限は210日(タイマーを含まない)である。特別な場合、医療製品が重大な公衆健康利益を有することが予想される場合、特に治療革新の観点から、CHMPは加速評価を承認する可能性がある。加速評価プログラムによる重大な影響評価の期限は150日であり,停止クロックは含まれていない.
| 14 |
国の認可手続き
他の2つの可能な経路はいくつかのEU諸国の医薬製品を許可することができ、これらの経路は集中プログラムの範囲に属さない研究用医薬製品に使用することができる
| ● | 分散した プログラム.分散プロセスを使用して、出願人は、1つ以上のEU諸国において、どのEU諸国でも承認されておらず、集中手続の強制範囲に属さない医薬品の承認を同時に申請することができる。 | |
| ● | 相互承認手続き。相互承認手続きでは、EU加盟国の国家手続きに基づいて、薬品がまずその国で許可される。その後、関連国が元の国のマーケティング許可の有効性を認めることに同意したプログラムにより、他のEU諸国にさらなるマーケティング許可を求めることができる。 |
上記の手順によれば、MAAを付与する前に、欧州経済区または欧州経済区(“EEA”)加盟国の主管当局は、製品の品質、安全性、有効性に関する科学的基準に基づいて、製品のリスク-利益バランスを評価しなければならない。
EU規制排他性
EUでは、マーケティングを許可した新製品(すなわち、参考製品)は、8年間のデータ独占権と追加の2年間の市場独占権を取得する資格がある。データ独占期の後発薬申請者がEUが後発薬の発売許可を申請する時に参考製品ファイルに含まれる臨床前と臨床試験データに依存して、参考製品が初めてEUで許可を得た日から8年以内である。市場独占期のbrは成功した後発薬申請者がその製品をEUで商業化し、参考製品がEUで初めて許可されるまでの10年間を禁止した。10年前の8年間に、マーケティング許可保持者が1つまたは複数の新しいbr治療適応の許可を得た場合、10年間の市場専門期間は最長11年に延長することができ、許可前の科学的評価中に、これらの適応は、既存の療法と比較して有意な臨床的利益を有すると考えられる。
薬品審査関連法規−世界の他の地域
EUや米国以外の他の国/地域,例えば東欧,ラテンアメリカまたはアジア諸国/地域では,臨床試験,製品許可,定価,精算を行う要求は管轄区によって異なる。しかし、多くの国/地域 は、米国FDAまたはEMAがパケットを審査および承認することを参照および/または参照し、これらの国/地域の承認プロセスを促進および加速する可能性がある。しかし,臨床試験はCGCP要求と適用の法規制要件および“ヘルシンキ宣言”に起源する倫理原則に基づいて行わなければならない。スポンサーが適用される外国監督管理要求を遵守できない場合、罰金、監督管理許可の一時停止または撤回、製品リコール、製品差し押さえ、経営制限、刑事起訴などの処罰を受ける可能性がある。
注意すべき予測正確医学プラットフォームテスト(PPMPテスト)規定
CLIA とCMS診断
医療保険と医療補助サービスセンター(“CMS”)はアメリカ衛生と公衆サービス部(Department Of Health And Human Services)内の一つの機関であり、 は1988年の臨床実験室改善修正案(“CLIA”)を通じてアメリカで行われたすべての臨床実験室テスト(研究を除く)に対して監督を行う。すべての人体サンプルに対して臨床実験室サービスを行い、疾病診断、予防或いは治療情報を提供する臨床実験室はすべてCLIA認証を受けなければならない。実験室 はCLIA認証を取得し,CMS検査確認のCLIA要求に適合していることを証明しなければならない。注目すべきは、2018年に初歩的なCLIA認証を取得したことだ。注目すべき実験室は2021年にCAPの追加認証を獲得し,この組織はCMSによって臨床実験室の第三者審査者として認められた。ニューヨーク州とカリフォルニア州を含むいくつかの州は、この州から標本を受け取る州外実験室から許可証を取得し、州の個別実験室法規を遵守することを要求している。
| 15 |
もし検査実験室がCLIAの要求を満たしていなければ、一時停止、制限或いは著者らのCLIA証明書の取り消しなどの処罰を受け、及び改正計画の指導、国家現場モニタリング、民事罰金、民事禁止訴訟或いは刑事罰を受ける可能性がある。したがって、MedicareおよびMedicaid受益者に提供されるサービスのための請求書を発行する資格があるように、CLIAコンプライアンスおよび認証を維持することが計画されていることに留意されたい。NORIGNのような診断テスト会社がCLIA計画に違反する要求を発見され制裁を受けると,その業務が損なわれる可能性がある。州許可法を守らないと,州許可当局に追加制裁を科す可能性がある。
FDA(Br)診断法
FDA 予測性診断テストは、現在、独立した実験室で開発されたテストまたはLDTとして提供されるPPMPのような承認または許可を必要としない。新薬や新薬適応の随伴診断として薬物の開発や製薬会社と協力して予測診断テストを発売する場合には,発売前承認またはPMAまたは510(K)許可br}を得ながらマッチング治療の新薬承認を求めることに注意する必要がある。歴史的に見ると、FDAは、予測診断試験またはLDTのような中心実験室でのみ実施される試験に対して法執行裁量権を行使する。FDAはLDTのみを提供する実験室がこの機関の医療機器に対する要求を遵守することを要求していない(例えば、機関登録、設備上場、品質システムの監督管理、上場前許可或いは上場前承認及び上場後制御)。
FDAはLDTに適用する法規を提案しているが,FDAは現在これらの法規は承認·施行されていないと考えている。2014年中、FDAは、現在市場で販売されている大多数の高価値LDTテストを最終的に510(K) 許可またはPMAを得る必要があるLDTの規制フレームワークの提案方法を記載した指導文書草案を発表した。実施すれば、この規制枠組みは、大多数の病院臨床実験室が彼らが行ったいくつかのテストを放棄するか、あるいは規制許可または承認を求めてこれらのテストを行うことができるように要求されるだろう。これらの提案は国会、病院業界、独立臨床実験室から強くボイコットされた。FDAは2016年末に、現時点で指導文書草案を最終的に決定するつもりはないと述べた。しかしながら、FDAは、LDTの潜在的な規制方法について議論し続けている。
注目すべき候補治療製品とPPMP診断テストの定価と精算
治療製品の定価と精算は、研究と開発計画によって作成された臨床概況または処方情報に記載されたその規制承認の広さを含む複数の要素に依存する。臨床資料の不確定性或いはこれらの要素の中の任意の要素は未来に販売可能な任意の重要製品のカバー範囲と精算状態に対する不確定性 に転化する。任意の重要製品の販売(または製品自体を販売することによって得られる収入)は、連邦医療保険や医療補助などの政府医療計画、商業健康保険会社、管理型医療組織または製薬会社を含む第三者支払者が製品コストを支払う程度にある程度依存する。第三者支払者が試験に保険を提供するかどうかを決定するプロセスは、薬品価格を設定するか、または支払人が薬品のために支払うべき販売率を決定するプロセスとは分離されることがある。第三者支払者は、承認リスト上の特定の試験製品 に保証範囲を制限することができ、特定の適応のすべての利用可能な試験を含まない可能性がある。
任意の製品の保険と精算を得るためには、治療製品や診断テストの医療必要性と費用効果を証明するために、強力な薬物経済学研究が必要である可能性がある。研究が成功しても,支払者は注意すべきbr製品が医学的に必要であり,採算に合わないとは思わないかもしれない。第三者支払者がテストに保険 を提供することを決定することは、十分な販売率が承認されることを意味するわけではない。また、支払者は、1つの商品に保険を提供することを決定し、他の支払者もその商品に保険及び十分な補償を提供することを保証することができない。第三者 精算は、製品開発への投資の適切なリターンを実現するために、十分に高い価格レベルを著しく維持するのに十分ではない可能性がある。
医療コストの抑制はすでに連邦、州と外国政府の優先事項になり、検査と薬品価格 はずっとこの努力の重点である。第三者支払者は、医療製品やサービスの価格に挑戦し、医療の必要性を審査し、試験製品、薬品、医療サービスの費用対効果を審査し、安全性と有効性を疑問視することが増えている。もしこれらの第三者支払者が、私たちの製品が他の利用可能な製品または治療方法と比較して費用対効果があると思わない場合、彼らは私たちの製品を保証しないかもしれない、または、もし彼らがそう思う場合、支払いレベルは有名な会社にbrの利益でその製品を販売させるのに十分ではないかもしれない。米国政府、州立法機関、外国政府はコスト制御計画の実施に大きな興味を示し、価格制御と精算制限を含む政府が支払う医療コストの増加を制限している。このような制御および措置、および既存の制御および措置を有する司法管轄区域における制限政策を採用することは、顕著な検出製品を使用する必要がある診断製品または治療の支払いを制限する可能性があり、顕著な純収入および業績に悪影響を及ぼす可能性がある。
| 16 |
定価 と精算案は国/地域によって異なる。一部の国は追加的な研究を完成させ、ある特定のテストの費用効果を現在利用可能なテストと比較することを要求するかもしれない。全体的には,医療コスト,特に処方薬や検出製品の下り圧力が大きくなっている。そのため、新製品の参入にはますます高い壁が設けられている。また、ある国/地域では、低価格市場からの国境を越えた輸入製品が競争圧力をもたらし、国/地域の価格設定を低下させる可能性がある。テスト製品に価格制御や精算制限がある国/地域では、私たちのどの製品にも割引の精算および定価手配が許可されていない可能性があります。
保証範囲br保険証、第三者精算料率、定価規則は随時変化する可能性がある。
その他 医療保険法
製品承認後の製造、販売、販売促進、その他の活動も、CMS、HHS監察長事務室、HHS民権事務室、HHSの他の部門、司法省を含むFDA以外の多くの米国規制機関によって規制されている。
医療保健提供者、医師と第三者支払人は任意の製品を推薦と処方する時に主要な役割を果たし、 注目すべき製品はすでに市場の許可を得た。現在と未来と第三者支払人、医療保健提供者と医師の手配は注目すべき広範に適用される詐欺と濫用及びその他の医療法律と法規に直面する可能性があり、これらの法律と法規は注目すべき市場、販売と流通治療と診断製品の業務或いは財務手配と関係を制限する可能性がある。米国では、これらの法律は、州および連邦反リベート、虚偽声明、医師透明性、br、および患者データプライバシーおよびセキュリティ法律法規を含むが、これらに限定されないが、これらに限定されない。
米国連邦反リベート法規(AKS)は、任意の個人または実体が知られている場合、直接または間接的に、任意の商品、施設、物品またはサービスを提供、開示または隠蔽的に提供し、任意の報酬を誘導または提供し、br}購入、レンタル、手配または推薦購入、レンタル、購入、レンタルまたは任意の商品、施設、物品またはサービスを購入または推薦し、Medicare、Medicaidまたは他の連邦医療保健計画に従って、全部または部分的に精算することができるように禁止されている。用語“報酬” は価値のあるものを含むと広く解釈されている。AKSは薬品と医療機器メーカーと処方者、購入者、処方管理人と受益者の間の手配に適していると解釈されている。いくつかの法定例外と規制安全港保護のいくつかの一般的な活動は起訴されていないが、例外と安全港の範囲は非常に狭い。特定の適用された法定例外や安全港を規制するすべての要求を満たしていないことはAKSによるものではなく、このような行為自体が不法である。逆に,そのすべての事実や状況の累積審査に基づいて,ケースベースでそのスケジュールの正当性を評価する.いくつかの裁判所は、この法規のbr意図要求を、報酬の手配に関連する任意の目的が連邦医療をカバーする業務の転換を誘導することである場合、この法規が違反されていると解釈する。また,個人やエンティティは,法規や法規違反の具体的な意図を実際に知ることなく違反を実施することができる.また、連邦民事虚偽請求法案によると、AKS違反による物品やサービスを含むクレームは、虚偽または詐欺的クレームを構成する。2020年11月20日、監察長室(“OIG”)がAKSのさらなる改正を決定した。最終規則によれば、OIGは、臨床医、提供者、および他の人との間のいくつかの調整ケアおよび価値に基づく配置を保証するために、AKSの下で安全な港保護を増加させる。 最終規則(いくつかの例外がある)は2021年1月19日に施行される。業界はこのルール がどのような影響を与えるか(あれば)を評価し続けるだろう.
さらに、“自己回診”に対する連邦法、すなわち一般的に言われている“スタック法”によれば、個人または家族によって検出されたエンティティにおいて投資または所有権の権益を有する医師が実験室サービスを含む特定の指定された医療サービスに転じることが禁止されており、これらのサービスは医療保険および医療補助計画のカバー範囲に属する。この禁止はまたStark法に違反して提出された任意のテスト費用を支払うことを含む。“スタック法”の移転禁止を回避する計画に参加した人は、このような手配や計画ごとに最高100,000ドルの罰金を科される可能性がある。また、スタック法に違反して連邦医療保険または医療補助計画にクレームを出したり、クレームを起こしたりする者は、毎回請求書を提出するたびに最高15,000ドルの民事罰金、最高請求額の3倍の評価に直面し、連邦政府支払人計画から除外される可能性がある。スタック法に違反して提出された請求書 はMedicareまたはMedicaidによって支払うことができず、このような について請求書がいかなる金額を受け取ることを禁止している人は、そのような金額を返却する義務があります。多くの州に類似した法律があり,医療保険や医療補助転転に限定されていない。
| 17 |
さらに、連邦“虚偽請求法”によれば、個人またはエンティティ(製造業者を含む)が虚偽または詐欺的なプロジェクトまたはサービスクレームを連邦計画(MedicareおよびMedicaidを含む)に意図的に提出する場合、またはMedicareおよびMedicaidを含む連邦計画(MedicareおよびMedicaidを含む)に虚偽または詐欺的な項目またはサービスクレームを提出し、クレームによって提供されていない項目またはサービスのクレーム、または医療上不必要な項目またはサービスのクレームを引き起こす場合、連邦虚偽クレーム法案に従って個人またはエンティティ(製造業者を含む)に民事処罰を加える可能性がある。有名なbr}が薬品のように支払人に直接クレームを提出しなくても適用可能である。政府は、このような製品の製造業者 が、顧客に不正確な請求書やコード情報 を提供したり、ラベル外で製品を宣伝したりすることによって、虚偽または詐欺的なクレームを“招く”と考える可能性がある。いくつかの生物製薬、医療機器、その他の医療保険会社は、顧客に製品を無料提供する疑いがあるため、連邦虚偽クレームと民事罰金法律によって起訴されており、顧客が連邦計画に製品費用を徴収することが予想されている。他の会社も起訴されているが、これらの会社のマーケティング製品が未承認(例えば、またはラベル外)の使用のために使用され、虚偽のbrクレームの提出をもたらすからである。さらに、民事罰金法規は、連邦健康計画にクレームを出すことが決定されたか、またはそれに至った任意の人に処罰を加え、そのクレームがクレームに従って提供されていない項目またはサービスbrまたは虚偽または詐欺性であることを知っているか、または知るべきである。
注意すべきbrは、報告卸売業者または私たちの製品の推定小売価格に関する将来のマーケティングと活動(承認されれば)、医療補助フィードバック情報を計算するための価格報告、および私たちの製品の連邦、州、第三者の精算に影響を与える他の情報{br]、および候補製品の販売とマーケティングは、これらの法律によって厳格に審査されている。
1996年に“連邦健康保険携行性と責任法案”(HIPAA)は追加の連邦刑法を制定し、計画を知りながら故意に実行または実行しようとし、虚偽または詐欺的な口実、陳述または約束の方法で任意の医療福祉計画(個人第三者支払人を含む)が所有または管理または保管している任意の金銭または財産を詐欺または獲得することを禁止し、brは医療福祉計画の資金または財産を故意に流用または盗んだことを知りながら、自発的に医療保健犯罪の刑事調査を阻害した。医療福祉、プロジェクトまたはサービスの交付または支払いに関連する重大な事実を隠蔽または隠蔽または隠蔽するか、または任意の重大な虚偽、架空、または詐欺的な陳述を行う。AKSと同様に,個人や実体は法規や法規違反の具体的な意図を実際に知る必要がなく違反を実施することができる.
また、最近、連邦と州政府は医師や他の医療提供者に支払う規制を強化する傾向があります。 他の事項を除いて、ACAは“医師支払い日光法案”によってカバーされたbrメーカーに対して新たな年間報告要求を実施し、医師(医師、歯医者、視光師、足科医師および脊椎マッサージ師を含むと定義されている)や教育病院に何らかの支払いと“価値移転”を提供し、医師 およびその直系親族が持つ所有権および投資権益を要求している。これらの報告義務は、2022年1月1日から、ある非医師提供者(例えば、医師アシスタントや勤務看護師)への価値移転を含む価値に拡大される。すべての支払い、価値譲渡、および所有権または投資利益のために必要な情報をタイムリーに、正確かつ完全に提出できなかったことは、民事罰金をもたらす可能性がある。保険引受メーカーは90年前に報告書を提出しなければならないこれは…。その後、各例年の日付および報告されたbr情報は、検索可能なウェブサイト上で公開されて提供される。
有名なbrはまた,連邦政府やその業務を行う州のデータプライバシーやセキュリティ法規の制約を受ける可能性がある.HITECHにより改正されたHIPAA及びそのそれぞれの実施条例は、2013年1月25日に発表されたHIPAA総合規則 を含み、保険実体及びその業務パートナーが持つ個人が健康情報を識別できるプライバシー、安全と伝送に対して具体的な要求を提出した。他の事項に加えて、HITECHは、HIPAAのセキュリティ基準 を“業務パートナー”に直接適用し、すなわち、保護された健康情報を受信、維持、または送信する保証エンティティの独立請負者またはエージェントを作成し、保証エンティティまたは代表保証エンティティにサービスを提供することに関する情報を提供し、 は、私たちの正常な業務過程において、重要人物が“業務パートナー”とみなされるかどうかを不明であるにもかかわらず、“業務パートナー”とみなされる。HITECH はまた、実体、商業パートナー、可能な他の 個人に適用される民事と刑事罰を増加させ、州総検察長に新たな権力を与え、連邦裁判所に民事訴訟を提起し、連邦HIPAA法を執行するために損害賠償または禁止令を要求し、連邦民事訴訟に関連する弁護士費と費用を求めることができる。さらに、州法 は、場合によっては健康情報のプライバシーおよびセキュリティを管理し、その多くの法律は、互いに大きく異なる であり、同じ要求がなく、コンプライアンス作業を複雑にする可能性がある。EUのデータプライバシーとセキュリティに関する議論は、以下の“欧州データ収集”を参照されたい。
| 18 |
例えば、カリフォルニア州の“消費者プライバシー法”(CCPA)は2020年1月に施行され、その後カリフォルニア州総検察長は最終法規を公布した。この法律は,カリフォルニアの消費者にその個人情報の収集と使用に関する広範な権利を与え,ある企業にデータ保護義務を課している。CCPAは、HIPAAによって制限された保護された健康情報、または研究において収集、使用または開示された個人情報には適用されないが、CCPAは依然として我々の業務活動に影響を与える可能性がある。また、2020年11月3日、カリフォルニア州有権者は投票イニシアティブの下でカリフォルニアプライバシー権法案(CPRA)を可決した。CPRAは、新しい消費者権利 および追加のデータ保護義務を含むように既存のCCPAを修正する。CPRAでの新しいデータ保護要求は,2022年1月1日以降に収集された情報に適用される.最終法規の公布に伴い、カリフォルニア州総検察長はすでにCCPAに違反した人に対して法執行行動を取り始めている。CCPAとCPRA改正案の実施をめぐる不確実性は,我々の業務が個人データや保護された健康情報に関する変化する規制環境に脆弱性があることを示している。カリフォルニア法律 はプライバシーとプロセス強化の需要をさらに拡大し、リソース投入を約束してコンプライアンスをサポートしている。また、昨年は10以上の州でCCPAやCPRAに類似した条項の法案が提出された。他の州では近い将来CCPAやCPRAのような法律が成立する可能性があり,連邦データ保護法も登場する可能性がある。
同様の州および外国詐欺および乱用法律、例えば州反リベートおよび虚偽クレーム法律は、医療プロジェクトまたはサービスに関する販売またはマーケティング配置およびクレームに適用される可能性がある。 このような法律は一般的に広く、様々な国家機関と個人的な行動によって実行される。また、Medicaidや他の州によって精算が計画されているプロジェクトやサービスを除いて、多くの州には類似した詐欺や法規や法規が乱用されており、その範囲はより広く、任意の支払者に適用される可能性がある。いくつかの州の法律は製薬会社に製薬業界の自発的コンプライアンスガイドラインと関連する連邦政府コンプライアンスガイドラインを遵守することを要求し、そして薬品メーカーに医者と他の医療保健提供者への支払いと他の方法で価値、マーケティング支出或いは薬品定価に関する情報を報告することを要求する。
製品を販売するために、有名なのはまた、薬品と生物製品のメーカーと卸売業者の登録を要求する法律を含む州法律を守らなければならない。いくつかの州では、これらのメーカーまたは流通業者が州に営業場所がなくても、製品brをその州に搬送する製造業者および流通業者を含む。一部の州はまた、メーカーと流通業者が流通チェーンの中で製品の系統を確立することを要求しており、いくつかの州はメーカーと他の人が流通チェーン中の製品の流れを追跡し、追跡できる新しい技術を採用することを要求している。いくつかの州は立法を公布し、製薬と生物技術会社にマーケティングコンプライアンス計画を確立し、州に定期報告を提出し、定期的に販売、マーケティング、定価、臨床試験およびその他の活動を公開し、brおよび/またはその販売代表を登録することを要求している。そして、薬局および他の医療エンティティが、販売およびマーケティングのための特定の医師処方データを製薬およびバイオテクノロジー会社に提供することを禁止し、いくつかの他の販売およびbrマーケティング行為を禁止する。注意すべきすべての活動は、連邦と州消費者保護と不正競争法によって制限される可能性がある。
これらの法律の範囲も実行も不確定であり,現在の医療改革環境では急速な変化の影響を受けている。 は特に適用例や法規の欠如を考慮している。連邦と州法執行機関は最近、医療保険会社と医療保健提供者の間の相互作用の審査を強化し、医療保健業界の一連の調査、起訴、有罪判決、和解を招いた。政府当局は、注目すべき業務やり方が、現在または将来、詐欺および乱用または他の医療保健法律および法規の適用に関連する現行または将来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。顕著な運営がこれらの法律のいずれかまたは任意の他の顕著な政府法規に違反していることが発見された場合、会社は、重大な民事、刑事および行政処罰、損害賠償、罰金、返却、契約損害、名声損害、利益減少および将来の収益、監禁、政府援助の医療計画(例えばMedicareおよびMedicaidなど)から医薬品を除外すること、私たちの業務を削減または再構成すること、およびこれらの法律違反に関する疑惑を解決するために、会社の誠実な合意または他の合意によって拘束された追加の報告義務および監督となる可能性がある。いずれも顕著な業務運営能力や財務業績に悪影響を及ぼす可能性がある。それと業務を展開することが明らかに予想されている任意の医師または他の医療提供者またはエンティティが、適用される法律を遵守していないことが発見された場合、彼らは、政府が援助する医療計画から除外することを含む重大な刑事、民事または行政制裁を受ける可能性がある。業務手配が適用される医療法に適合していることを確保することや,政府当局が行う可能性のある調査に対応することは,時間や資源がかかる可能性があり,会社の業務への注意を分散させる可能性がある。
| 19 |
ヨーロッパデータ収集
EU域内またはEUからの個人健康データの収集と使用は、“データ保護指令”および“一般データ保護条例”(GDPR)の規定によって管轄されている。この指令は,個人データに関する個人の同意,個人への情報提供,主管国家データ保護機関へのデータ処理義務の通知,個人データの安全と秘密についていくつかの要求を行っている。データ保護指令やGDPRは、個人データをEUから米国に移すことにも厳しいルールを実施しています。データ保護指令、GDPR、EU加盟国の関連国家データ保護法の要求を守らなければ、罰金やその他の行政処罰を受ける可能性があります。GDPRはEUに新たなデータ保護要求を導入し,データ保護規則違反に巨額の罰金を科した。GDPR条例は、臨床試験を含む、個人データに追加の責任と責任を課す可能性があり、 が新しいデータ保護ルールを遵守することを保証するために追加のメカニズムを確立する必要があるかもしれない。これは重い負担をもたらし、私たちの業務、財務状況、運営結果、将来性に悪影響を及ぼす可能性がある。
その他 法規要件
注目すべき業務は、研究開発に少量の危険材料を使用し、その予測性診断テストを正常に実行する過程で、規制された医療廃棄物 を生成する。このテーマは様々な連邦、州、そして地方環境と安全法律法規に注目すべきだ。現行の規制構造下のいくつかの規定は厳格な責任を規定し、過失や不注意を考慮することなく、当事者に潜在的な責任を負わせることを要求している。環境汚染や個人が危険物質に接触した場合,有名あるいは他の人の業務運営による損害や罰金が責任を問われる可能性がある。Br法の変化や新法規の発展がその業務運営やコンプライアンスコストにどのように影響するかは著しく予測できない。
現在と未来の立法
アメリカと他の司法管轄区では、医療保健システムに関する複数の立法と法規の変更及び提案された変更は注目すべき候補製品の発売承認を阻止或いは延期し、承認後の活動 を制限或いは規範化し、そしてその利益を影響して任意の候補製品を販売する能力に影響する可能性がある。注目すべきは、現在の法律および将来取られる可能性のある他の医療改革措置が、より厳しいカバー基準をもたらす可能性があり、注目すべきまたは任意の協力者によって得られる可能性のある価格 に追加の下振れ圧力をもたらす可能性があることである。
注目すべき背景と会社の歴史
注目すべきは、2014年にデラウェア州の会社に登録されたことだ。最初に,個々の患者にとってどの癌治療が最も有効であるかを医師が認識するための診断ツールとしてPPMPを開発したことに注意されたい。その後,同社はその使命を拡大し,そのPPMP を応用して研究化合物の同定と検証を簡略化·加速し,サービスに基づく合意に基づいて複数のバイオテクノロジー会社や製薬会社と協力した。2021年、OnCoherosプロトコルとCicloMedプロトコルを締結することによって、注目されるのは純粋な診断会社から正確な薬物を予測する総合診断と治療プラットフォーム治療会社 の設計と開発或いは共同開発に発展した。
当社は2023年12月31日現在、34,285,714株の法定普通株を有しており、そのうち9,018,261株は発行済み普通株であり、約284,437株は発行済み普通株、普通株式承認証、RSUを行使する際に保留された普通株である。
| 20 |
合併する
2023年10月16日、合併協定に基づき、当社、合併付属会社及び卓越実験室有限会社の間の合併子会社は卓越実験室会社と合併し、卓越実験室会社に編入し、卓越実験室会社は合併後も引き続き当社の存続実体及び全額付属会社とした。合併が発効した時、いかなる株主も行動しない場合、1株当たり合併前の有名な実験室会社S普通株の流通株を発行し、1株当たり額面0.001ドル(合併前S社の流通株を含む)、合併前S社の流通株奨励を含み、0.0629株会社の普通株を得る権利に変換し、1株当たり額面0.35ニューシェケルである。合併発効後、当社は直ちに発行済み会社と発行済み会社普通株に対して35株1株の逆株式分割(“逆株 分割”)を行う。合併及び合併協議で行われる取引を完了した後、(I)前著名株式有限会社 持分所有者は完全に償却した上で当社の約71.9%の既発行株式を所有し、 がすべて株式承認証を行使して94,988株会社の普通株を購入し、160,635株会社の普通株の標的オプション を含んで有名な実験室会社のS普通株を購入し、この普通株は当社が成約時及び 会社が成約時の純現金によって調整した後である。および(Ii)前血管生物製薬有限会社の株主は当社の発行済み株の約28.1%を所有している。
今回の合併は株式取引所が財務会計と報告目的のために実施した逆資本再編とみなされている。br}有名な実験室会社は会計買収者とされている。その株主は合併後に会社をコントロールしているため、血管生物遺伝有限会社は合法的な買収者であるにもかかわらず。したがって、我々の合併財務諸表に反映される資産や負債および歴史業務は、有名な実験室会社が報告会社であったように、有名な実験室会社の資産と負債および歴史業務である。普通株式,株式承認証,オプションに言及したすべての内容は,合併後,逆分割後 に基づいて提案された。
従業員 と人的資源
2024年4月1日現在,16名のフルタイム従業員を著しく有しており,その大部分が研究開発活動 に従事している。注目すべきは,従業員には現在労働組合代表もなく,集団交渉合意 もなく,従業員との関係が良いと考えていることに注意されたい。
人的資本目標の管理業務における顕著な重点はキーパーソンの誘致、発展と維持を含む。注目すべきチームは,その使命と組織の成功に重要であり,その従業員の専門的な発展を支援することに取り組んでいる。その管理チームは、その成長戦略を効果的に実施し、株主価値を推進し続けるために必要な経験を持っていることに注意されたい。報酬決定を行う際に注目すべきは、業界基準を含み、重要な人員を吸引し、維持するために、競争力のある報酬と福祉を提供し、使命と患者を中心とした安全、包容、尊重の職場を提供すると考えられることである。
| 1 a項目. | リスク要因 |
私たちの普通株に投資することは高い危険と関連がある。私たちの普通株を購入する前に、本年度報告の10-K表の他の情報に加えて、以下のリスク要因をよく考慮しなければなりません。以下に説明するリスクおよび不確実性は、我々が現在考えている重大なリスクおよび不確実性であり、これらのリスクおよび不確実性は、当社や業界に特定されていると考えられる。これらの リスクに加えて,我々の業務は我々の現在未知のリスクの影響を受ける可能性がある.実際に上記または他のいかなるリスクが発生した場合、私たちの業務は悪影響を受ける可能性があり、私たち普通株の取引価格は低下する可能性があり、あなたの投資は全部または部分的に損失する可能性があります。
| 21 |
顕著な業務·財務状況と資本要求に関するリスク
注意すべきbrは、その運営履歴が限られていることを評価することができ、小型開発会社がよく遭遇する問題、費用、困難、複雑な状況、遅延に基づいて、注目すべき成功の可能性を考慮しなければならない。
注目すべきは,臨床段階の正確な治療会社が2014年6月に設立され,運営歴史が限られていることである。設立以来、NOTICEの業務は主に知的財産権の獲得と許可、研究を展開し、NOWINGのPPMP及び現在の候補薬物VolasertibとFosiclopiroxに対して臨床前研究と臨床試験を行うことに限られている。 NOTICEはまだいかなる候補製品の監督管理許可を得ていない。したがって、注目すべき将来の成功または生存能力の予測、または注目すべき業務および将来性の任意の評価は、不正確である可能性がある。発展中の小型会社が新企業を設立する際によく遭遇する問題,費用,困難,複雑な状況や遅延,競争の激しい運営環境を考慮して,マイウェイ成功の可能性を考慮しなければならない。
注目すべき は、様々な要素 のため、その財務状況と経営業績は引き続き四半期ごとと毎年変動し、その多くの要素は制御できないと予想される。注目すべきは、同社は最終的に研究開発に専念する会社からビジネス活動ができる会社に転換する必要があることだ。注目すべきは、予見できない費用、困難、複雑な状況、遅延に遭遇する可能性があり、このような移行では成功しない可能性があるということだ。注目すべき経営歴史が限られているため、その業務が利益を上げることが保証されていないか、あるいは支出を支払い、注目すべき期待活動をサポートするのに十分な収入が発生するかどうかに注意されたい。しかも、注目すべき候補品がFDAの承認を受ける保証はない。
注目すべきは,設立以来重大な損失が発生しており,予見可能な将来も重大な損失が予想されるが,注目すべき経営陣は公認会計基準による管理職の必要な評価に基づいており,持続経営企業としての能力に大きな疑いがあると結論している。
注意すべき はすでに大部分の財務資源を研究と開発に投入し、注目すべき臨床前と臨床開発活動 を含む。これまでは,主に研究資金および持分や転換可能な証券の売却によってその運営に資金を提供してきたことに注目してきた。注目すべきは,同社の開発活動の拡大とその臨床前計画の推進に伴い,引き続き大量と増加した費用,損失,負のキャッシュフローを招くことが予想されることである。重要な候補製品が開発に成功していない場合、または資金の不足を含む商業化に成功していない場合、または重要なマーケティング承認後に十分な収入が生じていない場合、利益を達成することができず、そのビジネスが失敗する可能性がある。ADNOTYが規制部門の承認を得て候補製品を販売することに成功しても、その収入は、その候補製品が市場承認を得た任意の市場の規模と、その製品が十分な市場受容度と十分な市場シェアを得る能力に依存する。
注目すべきは, は予見可能な将来に巨額の費用と増加していく運営損失が続くと予想されることである。注目すべき純損失は四半期によって大きく変動する可能性がある。注目すべきは、 と以下の場合、その費用が大幅に増加することである
| ● | 引き続き独立し、その戦略連盟プロトコルに基づいてその候補製品に対して研究、臨床前と臨床開発を行った | |
| ● | 現在の候補薬物と追求可能な任意の未来候補薬物のためにbrの任意の他の適応の臨床前研究と臨床試験を開始する | |
| ● | より多くの候補薬剤または技術を買収することによって、その候補薬剤の組み合わせを確立し続けること | |
| ● | その知的財産権の組み合わせを開発、維持、拡大、保護し続ける | |
| ● | PPMPの開発、メンテナンス、拡張を継続する; | |
| ● | 他の候補製品を決定するために を探す; | |
| ● | 臨床試験の現在と未来の候補薬物の完成に成功するために監督管理の承認を求める |
| 22 |
| ● | 最終的に、販売、マーケティング、流通とその他の商業インフラを構築し、それが発売許可を得る可能性のある任意の候補薬物を商業化する | |
| ● | 臨床試験に成功した候補製品のために市場承認を求める | |
| ● | 追加の臨床、監督、研究、科学、行政、会計、行政者を募集する | |
| ● | 上場企業の運営として追加的な法律、会計、その他の費用が発生する | |
| ● | 注目すべき運営とその製品開発·計画の将来の商業化作業を支援するために、追加のインフラを作成する。 |
Br}を達成し、利益を維持するためには、巨大な市場潜在力を有する1つまたは複数の候補薬剤を開発し、最終的に商業化するか、またはその1つまたは複数の候補薬剤を業界パートナーに許可しなければならないことに注意されたい。一連の挑戦的なbr活動の中で成功を得るためには、ADNOTYが一連の挑戦的な活動の中で成功する必要があり、その候補薬物の臨床試験を完成し、同業者審査の出版物を通じてそのデータと研究結果を発表し、商業規模の生産プロセスを開発し、上場許可を獲得し、製造、マーケティング 及び発売許可を得る可能性のあるすべての現在と未来の候補薬物の販売、及び任意の発売後の要求 を満たす必要がある。このような活動の多くは初歩的な段階に過ぎず、場合によっては、これらの活動のいくつかの活動はまだ開始されていないことに留意されたい。注目すべきは、これらの活動のいずれでも成功しない可能性があり、成功しても、利益を達成するために十分なbr収入を生成しない可能性があることに留意されたい。
薬物開発に関連する多くのリスクと不確定要素 のため、費用の時間或いは金額を正確に予測できないこと、或いは注意すべき薬物がいつ或いは市場の許可を得てその任意の候補薬物を商業化するかどうかに注意するべきである。FDAまたはEMAのような他の規制機関(例えば、EMA)が現在予想されている基礎の上で研究および試験を行う必要がある場合、または開発またはその現在または将来の候補薬剤の任意の計画または将来の臨床前研究または臨床試験の完了に何らかの遅延が生じる場合、その費用は増加する可能性があり、利益はさらに遅れる可能性がある。
たとえbrが優れていても確実に利益を実現すれば,四半期や年度の収益性を維持または向上させることができない可能性がある。注目すべきは、利益を実現し維持することができないことは、その価値を低下させ、資金調達、研究開発努力の維持、業務の拡大、あるいは運営継続能力を弱める可能性があることである。注目すべき価値の低下は、投資家 が同社への投資の全部または一部を失う可能性もある。
したがって、経営陣は、財務諸表付記1に情報を開示しているが、注目すべき独立監査人は、2023年12月31日までの年次財務諸表報告書に、この不確実性について説明を含む。注意すべき2023年財務諸表には、このような不確実性がもたらす可能性のある資産回収性および資産分類または負債金額および分類への将来の影響を反映するための調整が含まれていないことに留意されたい。
は成立以来限られた収入しか生じておらず,永遠に利益が得られない可能性があることに注意されたい.
注目すべきは、収益を創出し、利益を達成する能力は、その単独または戦略同盟パートナーとPPMPおよび注目すべき候補製品の開発を成功させ、必要な規制承認を得て商業化する能力に依存する。予測可能な未来には、その製品の販売から相当な収入が得られないことに注意されたい。注目すべきは、将来的に製品販売から収入を得る能力は、以下の成功に大きく依存する
| ● | 候補製品の研究と臨床前開発を完成させる | |
| ● | 候補製品の臨床試験を開始し、完成し、良好な結果を得た | |
| ● | 臨床試験に成功した候補製品のために市場承認を求め、獲得し、維持する | |
| ● | 第3者との供給と製造関係の確立と維持 |
| 23 |
| ● | 連合パートナーと協力して、市場の承認を得る可能性のある候補製品を発売し、それを商業化し、独立して発売すれば、販売チーム、マーケティング、流通に成功した | |
| ● | 知的財産権の組み合わせを維持し、保護し、拡大すること | |
| ● | 合格した人材を引きつけ、採用し、引き留める。 |
予測薬や薬品開発に関わる多くのリスクや不確実性から,増加費用の時間や金額を予測できないこと,いつ利益を達成または維持できるか(あれば)であることに注意されたい。また、FDAまたは外国監督機関が現在予想されている基礎の上で研究と試験を行うことを要求する場合、 の注目すべき費用は予想を超える可能性がある。
1つまたは複数の注目すべき許可または開発候補製品が商業販売のために承認された場合であっても、任意の承認された製品の商業化に関連する巨額のコストが生じることが予想される。承認された製品を販売することから収入を得ることができても、利益が得られない可能性があり、運営を継続するために追加資金が必要かもしれない。
注意すべきことに、 は、許容可能な条項で獲得できないか、または全く得られないかもしれない追加の資本を調達する必要があると予想される。
臨床前研究や臨床試験を含めた医薬製品の開発は高価である。VolasertibとFosiclopirox および任意の他の候補薬に関する臨床試験を継続し、開発し、開始し、VolasertibとFosiclopirox および任意の他の候補薬に関する臨床試験を継続し、より多くの候補薬の決定と開発を求める;注目すべきbr}が発売される可能性のある様々な薬物を商業化するために、注目すべきbr}が市販される可能性のある様々な薬物を商業化するために、より多くの候補薬の決定と開発を求める;臨床開発のためのより多くの候補薬物の生産が要求され、商業化を実現する可能性がある;その知的財産権の組み合わせを維持、拡大、保護する;PPMPの開発、維持と拡大;臨床、品質管理と科学者のような多くの人員を雇用し、保留する;その薬物開発を支援し、上場会社の義務を履行することを助ける人員を含む運営、財務と管理情報システムと人員を増加させる;およびその研究開発計画を支援するために設備と有形インフラを増加させる。
注目すべきは,PPMP,Volasertib,Fsciclopirox,他の任意の候補薬剤の開発を進めるために多くの資金が必要であることである(S)。さらに、現在の候補薬剤の将来の開発のために、1つまたは複数のパートナー または1つまたは複数の適応のために開発される可能性のある任意の未来の候補薬剤を探すことができることに注意されたいが、適切な条項、タイムリー、またはその任意の候補薬剤とパートナー関係を確立することができないか、または許可を終了することができない可能性があることに注意されたい。いずれにしても、注目すべき既存の現金、現金等価物、および他の資本資源は、その計画によって行われるすべての努力に資金を提供するのに十分ではなく、その候補薬物の開発または他の臨床前研究の完了に資金を提供するのにも十分ではない。したがって、追加の資金は、公開または私募株式発行、債務融資、協力および許可手配、または他の 源によって得られることに注目すべきである。注目すべきは、それには何の約束もない外部資金源がないということだ。受け入れ可能なbr条項では,さらなる融資が得られない場合や,融資をまったく受けられない可能性がある.必要なときに資金を調達できなかったことは、その財務状況やその業務戦略を実施する能力にマイナス影響を与えることに注意されたい。
注目すべきは, はその研究開発費が進行中の活動により大幅に増加することが予想され,特にその が臨床試験により候補製品VolasertibとFosiclopiroxを推進している場合である。それは、その運営を支援するために追加の資本 を調達する必要がある可能性があり、受け入れ可能な条項でそのような資金を得ることができないか、またはそのような資金を全く得ることができない可能性があることに留意されたい。その計画が変化しないか、またはいかなる状況の変化も、その資本資源枯渇の速度が現在予想されているよりも速いことを保証することは著しくない。例えば、重要な前臨床試験または臨床試験は、技術または他の困難に遭遇する可能性がある。 これらのイベントのいずれも、予想を超える重要な開発コストを増加させる可能性がある。注目すべき長期計画を支援するために、新製品候補製品の臨床前または臨床試験を開始することを選択した場合、追加の資本を調達するか、または他の戦略連合を介して資金を得る必要があるかもしれない。いずれの場合も、規制部門の承認を得て、現在および将来の候補製品を商業化するために追加の資本が必要になることに注意されたい。
| 24 |
任意の追加的な拠出努力は、STNOTINGの経営陣の日常活動の注意をそらす可能性があり、これは、STNOTINGの現在および将来の候補製品を開発および商業化する能力に悪影響を及ぼす可能性がある。さらに、将来の融資 が十分な金額またはその許容可能な条項(あれば)で利用可能であることは保証されないことに注意されたい。必要な場合、または許容可能な条項で追加資本を調達できない場合、必要とされる可能性がある
| ● | 現在および将来の候補製品のすべての開発または商業化を大幅に延期、削減、または停止する | |
| ● | より早い段階で研究開発計画の戦略連合を求めることは、他の場合に望ましいのではなく、または が他の場合に得られる条件で戦略連合を求めることである;または | |
| ● | 不利な条項でbrまたは許可を放棄し、技術または任意の現在および将来の候補製品に対するその権利を放棄し、そうでなければ、それは 自身を開発または商業化することを求める。十分な金額または許容可能な条項で追加資本を調達できない場合、開発および商業化努力が阻止され、その業務、運営業績、および将来性に大きな悪影響を及ぼす。 |
注目に値する未来の資金需要は、短期的にも長期的にも、多くの要素に依存する
| ● | ボルザチブとフォスコピロロ及びその他の候補薬物の臨床前研究と臨床試験の範囲、進展、時間、コストと結果; | |
| ● | 公私パートナーシップ管理計画の維持、拡大、更新に関する費用 | |
| ● | 規制承認の費用、時間、結果を求める | |
| ● | 研究開発の拡大と潜在的な商業インフラの構築に伴い、その従業員数の増加と関連コスト | |
| ● | 市販承認された任意の候補薬物の許可または商業化活動のコストbrのようなコストは、医薬品販売、マーケティング、流通および製造能力を確立するコストおよび時間を含む任意の将来のパートナーの責任ではない | |
| ● | 任意の協力、許可協定、または他の手配を締結する能力、条項、および時間 | |
| ● | 現在および将来の候補薬剤の商業販売から得られる収入 ; | |
| ● | 特許出願を準備し、提出し、起訴し、その知的財産権を維持し、保護し、知的財産権に関するクレームについて抗弁する費用; | |
| ● | 将来の候補薬の数とその開発要件を追求しています | |
| ● | Brは、その運営の規制政策または法律の変化に影響を与える可能性がある | |
| ● | 商業努力に影響を与える可能性のある医師受容度や医学会提案の変化 ; | |
| ● | 新薬候補薬や技術を獲得するコストは | |
| ● | 購買力平価管理計画調達データに関するbrコスト; | |
| ● | ネットワークセキュリティシステムの維持と拡張に関連するコスト; | |
| ● | 上場企業としての運営コスト。 |
| 25 |
将来的に重要な株主の所有率をさらに希釈することを含む、重要な株式を売却して発行する権利、または重要な普通株を購入する権利は、重要な株主の所有権パーセンテージをさらに希釈し、その株価の下落を招く可能性がある。
未来には顕著な計画運営を継続するための追加的な資金が必要になるだろう。株式証券を発行することで追加資本を調達する著しい程度で、その株主は大幅な希釈を経験する可能性がある。普通株式、転換可能証券、または他の株式証券を1回または複数回の取引で販売することができ、価格および方法は時々顕著に決定されることができる。Brの普通株、変換可能証券、または他の株式証券が1回または複数回の取引で著しく販売されている場合、投資家はその後の販売によって大幅に希釈される可能性がある。これらの売却はまた、重要な既存株主の実質的な希釈をもたらす可能性があり、新しい投資家は重要な既存株主よりも高い権利を得る可能性がある。
注目すべきbrにも持分計画があり、注目すべき従業員、取締役と顧問に株式オプションとその他の持分に基づく奨励を付与することを規定し、株式承認証を発行した。これらのRSU、オプション、および引受権証のいずれの行使も、追加の株式発行 をもたらし、希釈効果を生じる可能性がある。これらの証券の登録に伴い、多くの証券が公開市場に転売することができる。その既存の株主は、公開市場でかなりの数の重要な普通株を売却したり、このような売却が発生する可能性があると考えられ、重要な普通株の市場価格を低下させ、追加のbr株証券を売却することで資金を調達する能力を弱める可能性がある。このような売却が普通株の現行市価に及ぼす影響は予測できない。
注意すべきbrは他の生物製薬会社からの激しい競争に直面しており,効率的な競争ができなければ注目すべき経営業績が影響を受ける。
生物製薬業界の特徴は競争が激しく、革新が迅速だということだ。注目すべき競争相手は、他の化合物や薬物を開発することができ、類似またはより良い効果を達成することができるかもしれない。有名な潜在競争相手は大型国際製薬会社、老舗生物技術会社、専門製薬会社と大学及びその他の研究機関を含む。多くの注目すべき競争相手は、より多くの研究開発者と経験豊富なマーケティングと製造組織、および成熟した販売チームのようなより多くの財務、技術、および他の資源を持っている。規模が小さいか、あるいは早い段階にある会社も重要な競争相手であることが証明される可能性があり、特にそれらが疾患を治療する新しい方法を開発した場合、注目すべき候補製品も治療に集中していることが示されている。古い製薬会社はまた、新しい治療法の発見と開発を加速させたり、注目すべき開発候補製品を時代遅れにする可能性のある新しい療法を許可するために大量の資金を投入する可能性がある。技術ビジネス適用性の進歩とこれらの業界の投資資金の増加により、競争はさらに激化する可能性がある。注目すべき競争相手は、単独またはbrパートナーとの協力にかかわらず、その候補製品よりも有効で、より安全で、商業化が容易で、またはコストが低い薬物または生物学的製品の開発、買収、またはその技術および製品を開発するために必要な特許保護を得ることに成功することが可能であることに留意されたい。注目すべきは、その候補製品の開発と商業成功に影響を与える重要な競争要素 は治療効果、安全性、耐性、信頼性、使用利便性、価格と精算である。
薬品が規制部門の承認を得た場合であっても、競争相手製品の供給や価格は、候補製品に対する需要や定価を制限する可能性がある。価格競争や医師が既存の治療法から重要な候補製品に切り替えたくない場合や,医師が他の新薬や生物学的製品に変更したり,その候補製品を限られたまま使用することを選択したりした場合,同社はその業務計画を実施できない可能性がある。
| 26 |
注目すべき候補製品VolasertibとFosiclopiroxは臨床開発の初期段階にあり、その商業可能性 は現在と未来の臨床試験、監督管理許可及び候補薬物開発に通常固有のリスクに依存する。 注目すべき候補製品が成功的に推進或いは開発できなければ、その業務は実質的な損害を受ける。
最近,重要候補製品VolasertibやFosiclopiroxの開発が成功しなければ,会社に大きな悪影響を与える可能性がある.これまで、候補製品のマーケティング、流通、または販売に成功的または商業的に販売されていないことに留意されたい。注目すべき業務の成功は主にその候補製品の臨床試験の開発を成功に推進できるかどうかに依存し、候補製品をFDA或いはその他の国/地区の監督管理機関の許可販売を獲得させ、そして最終的に候補製品を有名或いは戦略パートナーから商業化に成功させる。進行中の臨床試験の結果がその候補製品の継続開発を支持または証明することは明らかに保証されない、または それはFDAまたは他の国/地域の類似規制機関の承認を得て、その候補製品の開発 を推進する。
注目すべき候補製品は厳格な安全と治療効果の監督管理標準を満たさなければ、臨床開発を推進或いは完成することができ、或いは販売を許可することができる。これらの基準を満たすためには,高価で長い臨床試験を行い,許容可能な製造技術を開発し,規制部門の候補製品の承認を得なければならないことに注意されたい。これらの努力にもかかわらず、注目すべき候補製品 はないかもしれない
| ● | 同じ患者集団を治療するために、既存の薬剤または開発されている他の候補製品よりも多くの治療または他の医療的利点を提供すること; | |
| ● | 現在と未来の臨床試験で安全かつ有効であることが証明された | |
| ● | 期待された効果を達成する | |
| ● | 不良または予期せぬ影響がなく、適用される法規基準に適合し、商業的に適切な数量および許容可能なコストで調製および製造することができる;または | |
| ● | ITやパートナーによって商業化に成功した。 |
製薬と生物製薬業界の多くの会社は発展のすべての段階で重大な遅延、挫折と失敗を経験し、臨床試験においても奮い立つ結果を得た。さらに、注目すべき候補製品に関する前臨床研究および臨床試験から収集されたデータから良好な安全性および有効性を示しても、 のような結果は、米国FDAまたは他の司法管轄地域の他の同様の規制機関の規制承認を得るために機密協定またはBLAの提出を支持するのに十分ではない可能性があり、これは、マーケティングおよび製品の販売に必要である。
注目すべき候補製品は、さらなる臨床開発に入る前、あるいは有名または協力者によって商業化される前に、大量の追加的な研究と開発作業、大量の財政資源の承諾、br}と監督管理の承認が必要となる。注目すべきは、その候補製品が薬物開発過程で成功し、あるいは の商業的に実行可能な製品につながることを保証することはできないことである。注目すべきは、その候補製品が少なくとも数年以内に有名または協力者によって商業化されないということだ。
重要候補薬物の発見と開発に関するリスク
注意すべきは,薬物発見や薬物開発における経験が限られており,規制部門の承認が得られず,その候補薬物を市場に出すことができない可能性があることである。
候補薬物を買収する前に,参加していなくても臨床前や臨床開発を制御できないことに注意されたい。また,それから候補薬物を獲得した各当事者が適用するプロトコル,法律,法規,科学基準に基づいてこのような研究と開発を行い,適用候補薬を買収する前に行われたすべての臨床試験の結果を正確に報告し,これらの研究や試験のデータを正確に収集することに注意されたい。いずれも発生していない場合、顕著な予想される開発時間およびコストが増加する可能性があり、これは、これらの候補薬剤の発売承認およびこれらの候補薬剤から任意の将来の収入を得る見通しに悪影響を及ぼす可能性がある。
| 27 |
最近,VolasertibやFsciclopiroxの開発を推進する能力に注目すべきである.NOTICATE単独またはパートナーとVolasertibおよびFosiclopirox およびその他の候補薬剤の臨床開発を開始または完了できない場合、市場承認または商業化に成功するか、またはそのような点で重大な遅延がある場合、Designantのbr}業務は深刻な損害を受ける可能性がある。また,OnCoheros Biosciences,Inc.(“OnCoheros”)からVolasertib Indへの移行が完了するまで,米国でVolasertibの開発を継続しない可能性があることに注意されたい。このようなIND転移のいかなる遅延も有名な会社が計画したVolasertib臨床試験を延期する可能性がある。
注目すべきは、現在規制部門の承認を得ている薬剤は何もなく、適切な候補薬が開発されていない可能性があることである。注意すべきは、同社はその候補薬の発展を進めるために多くの精力と財力を投入し、PPMPを開発していることである。注目すべき将来性は、それまたは任意の未来のパートナーが開発し、市場の承認を得、1つまたは複数の疾患適応の候補薬物を商業化する能力に大きく依存する。
VolasertibとFosiclopiroxおよび有名な他の候補薬の成功は以下のいくつかの要素を含むいくつかの要素に依存する
| ● | FDAまたは任意の類似の外国監督機関にINDを提出した後、候補薬物臨床試験の許可と未来の臨床試験の提案設計を獲得した | |
| ● | その候補薬物と潜在候補薬物の臨床試験の開始、進捗、時間、コストと結果 | |
| ● | FDA或いは任意の類似の外国監督管理機関に満足させる安全性、耐性と有効性概況を確立し、上場許可を得る | |
| ● | 臨床開発および任意の商業販売のための十分なPPMPおよび原材料および医薬製品の高品質データ源の持続可能性; | |
| ● | 米国と関連する世界市場で特許、商業秘密保護、規制排他性を獲得し、維持する | |
| ● | その将来の協力者の業績(あれば); | |
| ● | 適用される規制機関に必要な上場後の承認承諾の程度 | |
| ● | 第3者原材料サプライヤーと製造業者との供給手配を確立する | |
| ● | 販売のために適切な包装された完成品薬品を得るために、第三者製造業者との手配を確立すること | |
| ● | 知的財産権を保護することです | |
| ● | 任意の市場の承認後に商業販売の開始に成功した | |
| ● | 任意の上場承認後に許容可能なセキュリティプロファイルを継続する; | |
| ● | 患者、医療界、第三者支払者の商業的受容度 | |
| ● | それは他の療法と競争する能力である。 |
これらの要素の中には、臨床試験の結果、FDAまたは任意の類似した外国の規制機関が、それが提出可能な任意の規制文書を審査するのに要する時間、その知的財産権に対する潜在的な脅威 および任意の未来のパートナーの製造、マーケティング、および販売努力を含む注目すべきではない多くの がある。NOTICATIONが単独でまたは任意の未来のパートナーと開発し、その候補薬物のマーケティング承認を得て商業化に成功することができない場合、または上記の任意の要因または他の理由で遅延が発生した場合、NOTICATIONの業務は深刻な損害を受ける可能性がある。FDAと類似外国機関の監督管理審査過程は長く、時間がかかり、高価であり、しかも本質的に予測できず、もし有名な が最終的にその候補薬物の監督管理許可を得られなければ、その業務は深刻な損害を受ける。
| 28 |
FDAや類似外国当局の承認を得るのに要する時間は予測できないが,臨床試験開始後には数年を要する可能性があり,規制機関のかなりの裁量権を含む多くの要因に依存する。重要な候補薬物の臨床前研究や早期臨床試験の結果は後期臨床試験の結果を予測できない可能性がある。臨床前研究と初歩的な臨床試験で進展を得たが、臨床試験後期段階の候補薬物は必要な安全性と有効性特徴を示すことができないかもしれない。バイオテクノロジーや製薬業界の会社が高度臨床試験で大きな挫折を経験することはまれではなく,臨床研究を行う際に非臨床結果が発見されたことや,これまで報告されていなかった有害事象を含めて臨床研究で安全性や有効性観察が行われていることが原因である。ノビート社の将来の臨床試験結果は成功しない可能性があり,早期研究には潜在的な有望な結果があるにもかかわらず,ノヴィット社が類似した挫折に直面しないことを確認することはできない。注目すべき産業では、候補薬物の歴史的失敗率が高い。また,候補薬物の臨床開発過程では,承認政策,法規あるいは承認を得るために必要な臨床データのタイプや数が変化する可能性があり,管轄区域によって異なる可能性がある。どの候補薬も規制部門の最終承認を得ておらず、その既存の候補薬や将来開発を求める可能性のある候補薬は規制部門の承認を得られない。
注目すべきbr候補薬物は多種の原因で発売許可を得られない可能性があり、以下の原因を含む
| ● | FDAまたは同様の外国の規制機関は、ゲノムまたはバイオマーカー署名を使用して、薬物治療効果に反応する可能性のある患者を識別することを含むが、これらに限定されない臨床試験の設計または実施に同意しない可能性がある | |
| ● | それはFDAまたは同様の外国の監督管理機関に候補薬物がその提案の適応に対して安全かつ有効であることを証明できない可能性がある | |
| ● | それは、その候補薬剤の臨床試験を行うために、関連するゲノムまたはバイオマーカー署名または他の指定された入選基準を有する十分な数の患者を識別および募集できない可能性がある | |
| ● | 臨床試験の結果はFDA或いは類似の外国監督管理機関が許可した統計的意義レベルに符合しない可能性がある | |
| ● | FDAまたは同様の外国の規制機関は、臨床前研究または臨床試験データの説明にSTNOTINGに同意しない可能性がある | |
| ● | その候補薬剤の臨床試験から収集されたデータは、秘密保護プロトコルの提出または他の提出をサポートするのに十分ではないか、または米国または他の場所の規制承認を得るのに十分ではない可能性がある | |
| ● | FDAまたは同様の外国の規制機関は、臨床および商業供給契約を締結する第三者製造業者の製造プロセスまたは施設に欠陥があるか、または承認されていないことを発見する可能性がある | |
| ● | FDAなどの外国の監督管理機関の承認政策や法規は重大な変化が発生する可能性があり、その臨床データが承認を得るのに十分ではない。 |
注目すべきは,br}までその候補薬剤のすべての臨床試験が完了していないことである。したがって、注意すべきは、その起動された任意の臨床試験の実行および完了を成功的に管理するために十分な人員配置を含む必要な能力を備えていない可能性があり、それによって、br}の注目すべき適時にその候補薬物のマーケティング承認を得ることができない、または全くできない。この長い審査過程及び未来の臨床試験結果の予測不可能性は注目すべき会社が監督部門によるその候補薬物の上場許可を得られなかったことを招く可能性があり、これはその業務、運営結果と将来性を深刻に損害する。
さらに、承認されても、規制機関は、その任意の候補薬剤の適応が著しく申請された適応よりも少ないか、またはそれを超えることを承認することができ、その薬剤の期待価格を承認しない可能性があり、高価な発売後の臨床試験の表現によって承認される可能性があり、候補薬剤を承認する可能性のあるラベルは、候補薬剤の商業化に必要または必要なラベル宣言を含まないか、またはその流通を制限する可能性がある。上記のいかなる制限または要求も重要な候補薬物の商業的将来性に実質的な損害を与える可能性がある。
| 29 |
注目すべきbr}は、これまでFDAまたは同様の外国当局に、任意の候補薬剤に対する秘密協定や同様の薬物承認申請を提出しておらず、その候補薬剤が臨床試験で成功したり、規制部門の承認を得たりするかどうかを決定することはできない。また、注目すべき候補薬剤は、臨床試験で成功しても、規制部門の承認を得ることができない可能性がある。有名なbrが規制部門の候補薬物の承認を得なければ,運営を継続できない可能性がある。規制部門の承認を得ることに成功し、その1種以上の候補薬剤を販売しても、その収入は、規制部門の承認を得て商業権を有する地域の市場規模にある程度依存する。注目すべき候補薬物が患者向け市場で推定されているほど大きくない場合、あるいは候補薬物に対する価格が高すぎる場合、承認されれば、このような薬物の販売から相当な収入が得られない可能性があることに注意されたい。
注目すべきは、米国とEUおよびより多くの国·地域で候補薬物の商業化を実現するために、規制部門の承認を求めることである。規制承認の範囲は他の国/地域で類似しているが、多くの他の国/地域で単独の監督管理許可 を得るためには、これらの国/地域の安全性と有効性に関する多くの異なる監督管理要求を遵守し、臨床試験と注目すべき候補薬物の商業販売、定価と流通に適用される可能性のある制限 を管理しなければならないが、これらの司法管轄区での成功を予測することはできないことに注意されたい。
注目すべきbrは、注目すべき候補薬剤の開発を継続するために、注目すべき臨床試験において特定のゲノムまたはバイオマーカー署名を有する患者を登録することに依存する可能性がある。NOTIGNがNOTICEの臨床試験で特定のゲノムやバイオマーカー署名を有する患者を募集できなければ,NOTICATEの研究,開発,商業化努力は不利な影響を受ける可能性がある。
臨床試験案により速やかに臨床試験を完了し、他の事項に加えて、顕著なゲノムやバイオマーカー署名を有する十分な数の患者を募集する能力があるか否かにも依存し、これらの患者は確定し、研究が終了するまで研究に残る。br}顕著な臨床試験の患者登録は様々な理由で困難に直面する可能性がある。患者登録は、顕著に決定された特定のゲノムまたはバイオマーカー署名を有する患者集団の大きさと性質、患者と臨床地点との近接度、試験の資格基準、臨床試験の設計、試験の主要終点を分析するために必要な患者群の規模、患者と研究地点の近接度、適切な能力と経験を有する臨床試験研究者を顕著に募集する能力、患者の同意を著しく獲得し、維持する能力、患者登録は多くの要素の影響を受ける。臨床試験に参加した患者は,完成前に試験を脱退するリスクが調査されており,注目すべき適応として承認される可能性のある任意の新薬を含む,検討中の候補薬の他の利用可能な治療法に対する潜在的な優位性に対する競争の臨床試験や臨床医や患者の見方である。注目すべきは,他の製薬会社と臨床場所,医師,腫瘍臨床試験への参加の厳しい要求を満たす限られた数の患者を争うことである。また,臨床試験の機密性により,顕著な がどれだけ条件を満たしているかを知らない患者が競争的研究に組み込まれる可能性があるため,どの患者が顕著なbrの臨床試験に参加できないか。十分な患者を募集できないため、重要な臨床試験は延期または終了される可能性があります。br}遅延や計画を満たすことができない患者登録はコスト増加や重要な臨床試験の遅延や終了を招く可能性があり、重要な薬物開発能力に有害な影響を与える可能性があります。
臨床テストの遅延 はコストの著しい増加を招き、その創造能力を遅らせる可能性がある。
FDAや他の規制機関がNotecがその候補薬物 計画や将来の試験設計を受け入れることは保証されない。注目すべきは,その臨床試験に遅延が生じる可能性があり,計画中の臨床試験が時間どおりに開始されるかどうか,再設計が必要かどうか,時間どおりに患者を募集あるいは時間どおりに完成するかどうか(あれば)であることである。臨床試験は様々な原因で遅延する可能性があり、以下の点に関連する遅延を含む
| ● | Br規制を受けることで裁判を開始することができる |
| 30 |
| ● | 予想される契約研究機関(“CRO”)と臨床試験地点とは受け入れ可能な条項について合意し、その条項は広範な交渉を行うことができ、異なるCROと試験地点の間に有意差が存在する可能性がある | |
| ● | 各場所で機関審査委員会(“IRB”)の承認を受ける | |
| ● | 適切な患者を募集して試験に参加する; | |
| ● | 試験を行うのに十分なインフラ(データ収集を含む)を有するbr個の臨床サイトを決定する | |
| ● | 臨床brサイトは試験方案から外れているか、または試験から退出している | |
| ● | 実験中に発生した患者の安全問題を解決する | |
| ● | Br名の患者は試験を完成し、或いは戻って治療後のフォローアップを行った | |
| ● | 十分な数の臨床試験場所を増やすことができます | |
| ● | 臨床試験のために十分な数量と品質の候補薬剤を生産する。 |
注意すべきことは、brはまた、臨床試験中または臨床試験のために多くの予見不可能なイベントを経験する可能性があり、これらのイベントは、上場承認またはその候補薬物を商業化する能力を延期または阻止する可能性があることである
| ● | IT は監督機関からフィードバックを受ける可能性があり、その臨床試験の設計を修正することを要求する | |
| ● | それはその臨床試験患者のための能力がない可能性があり、これらの臨床試験は特定のゲノムまたはバイオマーカー署名を必要として登録する資格がある | |
| ● | その候補薬物の臨床試験は陰性または不確定な結果をもたらす可能性があり、それは決定または監督機関が追加の臨床試験を要求するか、または薬物開発計画を放棄することを要求する可能性がある | |
| ● | 候補薬剤の臨床試験に必要な患者数は、予想よりも多い可能性があり、これらの臨床試験の登録速度は、予想よりも遅い可能性があり、または参加者がこれらの臨床試験から退出する速度は、予想よりも高い可能性がある | |
| ● | その第三者請負業者は、監督管理要求を直ちに遵守することができないか、またはその契約義務を履行することができない可能性があり、または全く遵守しない | |
| ● | 候補薬物の臨床試験コストは予想よりも高いかもしれない | |
| ● | 候補薬物の供給または品質、または候補薬物の臨床試験を行うために必要な他の材料が不足または不足している可能性がある | |
| ● | 規制機関は、その候補薬物を承認する要求を修正することができ、またはこれらの要求は、その予想とは異なる可能性がある | |
| ● | 臨床試験を行う将来の協力者はいずれも上記のいずれの問題に直面する可能性があり、彼らが自分に有利だと思っているがあまり理想的ではない方式で臨床試験を行う可能性がある。 |
Br}がその候補薬剤に対して現在予想されている以上の追加の臨床試験または他の試験を行うことに注意する必要がある場合、 その候補薬剤の臨床試験または他の試験を成功させることができないことに注意すれば、これらの試験または試験の結果が陽性でない場合、または軽度陽性である場合、または安全問題がある場合、注意すべき可能性がある
| ● | 発生計画外コスト ; |
| 31 |
| ● | 重要な候補薬物の発売承認を遅延するか、上場承認を得ていない | |
| ● | 一部の国では上場承認を得ているが、他の国では発売承認を得ていない;期待あるいは期待されていないbrの広範な適応或いは患者群については、上場承認を得ている | |
| ● | 市場の承認を得て、ブロック警告を含む重要な使用または流通制限または安全警告を含むラベルを貼り付ける | |
| ● | 追加の上場後のテスト要求を受ける | |
| ● | 発売承認を得た後,この薬物を市場に投入した。 |
また,将来的にCRO,癌研究センター,臨床試験地点に依存して臨床試験の適切かつタイムリーな進行を確保しようとしていることに注目すべきであり,その約束した活動について合意する予定であることに注意されたい。それらは要求された効果を達成していない可能性があり、あるいは他の製薬会社が行っている他の臨床試験からの競争に直面している可能性がある。
臨床試験が一時停止または終了された場合、データ安全監視委員会(“DSMB”)、そのような試験を行う機関のIRB、またはFDAまたは の他の規制機関の提案を含む遅延に遭遇する可能性があることに留意されたい。このような主管部門は一連の要素のために臨床試験を一時停止または中止する可能性があり、これらの要素は: が法規の要求或いは有名な臨床規程に従って臨床試験を行うことができなかった;FDA或いは他の監督機関が臨床試験操作或いは試験場を検査することによる臨床休止を実施することを招く;予見できない安全問題或いは不良副作用;ある種の薬物を使用するメリットを証明できなかった;政府法規或いは行政措置の変化;あるいは臨床試験を継続するのに十分な資金が不足している。
また,海外での臨床試験は,注目すべき現在と将来の候補薬がもたらす可能性があるように,余分なbrリスクをもたらし,その臨床試験の完成を遅らせる可能性がある。これらのリスクには,外国に登録された患者が医療サービスや文化的慣習の違いにより臨床合意を遵守できなかったこと,外国規制計画に関する追加行政負担の管理,およびこのような外国に関連する政治的·経済的リスクが含まれる。
注目すべき候補薬剤の任意の臨床試験の完了が遅延または終了した場合、注目すべき候補薬剤の商業的将来性は損なわれ、注目すべき候補薬剤が収入を生成する能力は遅延される。そのほか、どの遅延による注目すべき臨床試験の完成はすべて注目すべきコストを増加させ、その候補薬物の開発と承認過程を緩和し、そして薬物の販売開始と収入を創造する能力を著しく脅かす。br}のいずれの状況も顕著な業務、財務状況と将来性に重大な損害を与える可能性がある。さらに、臨床試験の開始または完了遅延をもたらす多くの因子は、最終的には 監督部門が注目すべき候補薬物の承認を拒否する可能性もある。
早期臨床試験と以前の臨床試験の結果は後の試験結果を代表しないかもしれない。
重要な候補薬物の非臨床と臨床前研究及び臨床試験のbr結果は重要な候補薬物の後期臨床試験の結果を予測できない可能性があり、臨床試験の中期結果は必ずしも最終結果を予測できるとは限らない。場合によっては,同一候補製品の異なる臨床試験間の安全性や有効性結果に有意差がある可能性があり,原因は多く,試験プログラムやレジメンに規定されているこのようなプログラムの時間の変化,患者群の大きさやタイプの違い,投与レジメンの変化,投与レジメンの遵守,臨床試験参加者の中退率などがある。例えば,2期と3期の臨床試験では,ボリンガー−インゲルハイムと小用量のアラベリンの併用による急性骨髄性白血病(“AML”)患者におけるVolasertibの治療効果を評価したが,注意すべきVolasertibのAML患者における計画臨床試験は異なる結果を招く可能性がある。Boehringerインゲルハイム委託による3期研究は主要終点試験成績結果には達していないが,BIIのVolasertibデータセットおよび長年の臨床前と臨床開発を用いてVolasertibの後期再開発計画を再設計し, 臨床応答率と結果(Boehringerを用いたPPMPによる)の向上とVolasertibの耐性改善(患者 管理を改善することにより,BIIの特殊な3期分析から得られた結論に適合する)が,結果が と異なる保証はないことに注意されたい。臨床試験後期段階の候補製品は必要な安全性と有効性を示すことができないかもしれないが、すでに非臨床研究と初歩的な臨床試験を通じて進展を得たが。臨床前と臨床活動から得られたデータ は異なる解釈を受ける可能性があり、これは監督部門の注目すべき候補製品の承認を延期、制限或いは阻止する可能性がある。
| 32 |
注目すべき候補薬物は、不良副作用を引き起こす可能性があり、あるいはその規制承認を遅延または阻止し、承認ラベルの商業イメージを制限し、または上場承認(ある場合)後の重大な負の結果をもたらす可能性がある他の特性を有する。
重要な候補薬物による不良副作用は、顕著または規制機関の中断、延期、または臨床試験の一時停止を招く可能性があり、より厳格なラベルまたはFDAまたは他の類似の外国機関の規制承認遅延または拒否を招く可能性がある。どんな注目すべき候補薬も副作用があるかもしれない。この場合、注目すべき研究を行う機関を含むdsmb、FDAまたはIRBsの推奨の下で、注目すべき臨床試験を一時停止または終了することができることに注目することができ、またはFDAまたは同様の外国の規制機関は、注目すべき任意またはすべての標的適応の候補薬剤の承認を停止または拒否するように注意すべきbrを命令することができる。治療に関連する副作用brはまた、患者の臨床試験を完成させる能力に影響を与えるか、または潜在的な製品責任クレームを引き起こす可能性がある。また、治療医療従事者は、これらの副作用を正確に識別または管理できない可能性がある。注意すべき候補薬物を使用した医療従事者は、注意すべき臨床試験および任意の注目すべき候補薬の商業化後の副作用を知るために、注意すべき候補薬を使用する医療従事者を訓練しなければならないことに注意されたい。ノビット候補薬物の潜在的副作用を識別または管理する上で訓練が不足し、患者の負傷或いは死亡を招く可能性がある。これらの状況のいずれも、brの顕著なビジネス、財務状況、および将来性を深刻に損なう可能性がある。
さらに、もし1つ以上の注目すべき候補薬物が発売許可を得た場合、注目すべき或いは他の候補薬物は後にこのような薬物による不良副作用 を発見し、いくつかの潜在的な重大な負の結果を招く可能性がある
| ● | 規制当局はこのような薬物の承認を撤回するかもしれない | |
| ● | 注意すべきは、このような薬を患者に投与する方法を変更するためには、薬を呼び戻す必要があるかもしれない | |
| ● | 特定の薬物の販売または流通または医薬またはその任意の成分の製造プロセスに追加の制限を適用することができる; | |
| ● | 規制当局は、“ブラックボックス”警告やタブーのようなラベルに警告を追加することを要求するかもしれない | |
| ● | リスク評価および緩和策(REMS)を実施するか、または患者に配布するために、そのような副作用のリスクを概説する薬物ガイドラインを作成する必要がある可能性があることに留意されたい | |
| ● | 注意すべきは、 が起訴され、患者への傷害に責任を負う可能性があることである | |
| ● | 注目すべき薬はそれほど競争力がなくなるかもしれません | |
| ● | 注目すべき 名声は影響を受ける可能性がある. |
これらのイベントのいずれも、Designantが特定の候補薬剤または特定の候補薬剤に対する市場の適応の受容度を獲得または保持することを阻止し、(承認されれば)、Designantの業務、運営結果、および将来性を深刻に損なう可能性がある。 Designantは、PPMPに基づいて候補薬剤を発見および開発する方法が革新的であり、開発の初期段階である ;Designantは、商業的価値のある薬剤を開発できるかどうか分からない。
| 33 |
注目すべきは,PPMPを用いてバイオマーカー認識と患者層を用いた候補薬物チューブ の腫瘍薬の開発を試みていることである。PPMPをすでに失敗し、放棄された或いは他の方法で臨床終点に達しなかった薬物に応用し、それから1種の精確な腫瘍学方法を開発し、作用機序、潜在的な連合薬品使用と潜在的な反応患者群を確定することは1種の強力な策略であるが、注意すべき方法は革新的な であり、開発の初期段階にもある。注目すべき方法は革新的であり、開発の初期段階にあるため、注目すべき候補薬物の開発に要するコストと時間は予測が困難であり、しかも注意すべき努力は商業的に実行可能な薬物の発見と開発に成功しないかもしれない。注目すべき候補薬が注目すべき患者群の疾患に及ぼす影響も不正確である可能性があり,注目すべき方法の実用性や注目すべき方法に対する有用性の見方を制限する可能性がある。また、注目すべき研究と治療の定義患者数の推定は予想を下回る可能性があり、これは注目すべき臨床試験を行う能力に悪影響を与える可能性があり、また注目すべき薬物の任意の市場規模に悪影響を与える可能性があるため、注意すべき薬物は商業化に成功する可能性がある。注意すべき 方法は,期待されるように時間を節約し,成功率を向上させたり,コストを下げたりしない可能性があり,そうでなければ,期待されるようにパートナーを吸引したり,迅速あるいは経済的に効率的に新薬を開発することができない可能性があることに注意されたいので,最初に予想したように の注目すべき方法を商業化できない可能性があることに注意されたい。
PPMPはより多くの潜在的な候補薬物の顕著な発見と開発を助けることができないかもしれない。
PPMPによる任意の注目すべき薬物発見または薬物開発は、商業的価値または治療効果を有する化合物の識別に成功しない可能性がある。PPMPは当初、潜在的な候補薬物の決定に希望を示す可能性があるが、様々な原因で臨床開発または商業化に利用可能な実行可能な候補薬物を生成できなかった
| ● | 新薬候補薬物を決定する研究br計画は大量の技術、財政と人的資源を必要とし、新薬候補薬物の確定には成功しない可能性があることに注意されたい。STNOTINGが臨床前および臨床開発のために適切な追加化合物を決定できない場合、STNOTING開発候補薬および将来の製品収入を得る能力は影響を受ける可能性があり、これはSTNOTINGの財務状況に重大な損害を与え、STNOTINGの株価に悪影響を及ぼす可能性がある | |
| ● | PPMPによって決定された化合物 は、その有効性、安全性または耐性を証明できない可能性がある | |
| ● | PPMPは、ゲノムまたはバイオマーカー署名を特定の癌に関連させるデータを求めることは、患者人種の影響を受ける可能性があり、NORIGN候補薬剤の治療効果を制限する可能性がある | |
| ● | 潜在的な候補薬物はさらなる研究において有害な副作用または他の特徴を有することが証明される可能性があり、それらが上場承認を得られ、市場から受け入れられる可能性が低いことを示している | |
| ● | 競合他社は、注目すべき潜在的候補薬が競争力を失ったり、吸引力を低下させたりする代替療法を開発する可能性がある | |
| ● | 潜在的候補薬は許容可能なコストで生産できない可能性がある。 |
既存の法規を遵守できなかった行為に注意しても、注意の名声と経営業績を損なう可能性がある。
注目すべき は米国連邦,州,外国政府によって広く規制され,市場ごとに,承認後にVolasertibとFosiclopiroxを販売する予定であることに注意されたい。例えば、FDAのGCP、良好な実験室仕様またはGLP、および現在のGMP要件、または適用される外国規制機関の要件を含むすべての法規要件が遵守されなければならないことに留意されたい。FDA承認前または承認後のcGMP要件を含む適用された法規が明らかに遵守されていない場合、FDAまたは他の外国規制機関は顕著な制裁を行う可能性がある。薬物がFDAによって承認されていても、規制機関は、薬物の指示用途やマーケティングに重大な制限をかけたり、高価である可能性のある発売後の研究に持続的な要求を加えたりする可能性がある。
これらの法律に違反して顕著な行動をとるいかなる行動も、それを著しく弁護することに成功しても、巨額の法的費用を著しく発生させ、顕著な業務運営に対する著しい管理層の注意を移動させ、顕著なbrの名声を損なう可能性がある。コンプライアンス作業に大量の資源がかかることに注意されたいが、このような費用は予測不可能であり、顕著な業績に悪影響を及ぼす可能性がある。
| 34 |
FDAや他の規制機関の政策は変わる可能性があり、追加の政府法規が公布される可能性があり、規制部門の注目すべき候補薬物の承認を阻止、制限、または延期する可能性がある。例えば2016年12月21日ST世紀治療法案、あるいは治療法案、署名が法律となる。その他の事項以外に、“治療法案”は薬品監督を現代化し、革新を刺激することを目的としているが、その最終的な実施状況はまだ不明である。注意すべきである場合、既存の要求の変化に適応できない場合、または新しい要求または政策を採用することができない場合、または注意すべきである場合、法規遵守性を維持することができない場合、得られ、利益を達成または維持できない可能性があるマーケティング承認を失う可能性があることに注意されたい。これは、その業務、将来性、財務状況、および運営結果に悪影響を及ぼす。
さらに、米国または海外の将来の立法または行政または行政行動によって生じる可能性のある政府規制の可能性、性質、または程度を予測できないことに注意されたい。将来の立法または行政または行政行動 がFDAが正常な過程で監督と実行活動に従事する能力に制限を加えると、重要な業務は負の影響を受ける可能性がある。さらに、既存の要求の変化に著しく適応できない場合、または新しい要求または政策を採用することができない場合、または法規コンプライアンスを著しく維持できない場合、得られ、利益を達成または維持できない可能性がある任意のマーケティング承認 を著しく失う可能性がある。
米国以外の上場承認を求めることに注意すれば,広範な法規制に制限される可能性があり,ヨーロッパや他の司法管轄区でbr薬品の上場承認を得ることができない可能性がある。
米国の法規以外に、重要又はその協力者が国際的にVolasertibとFosiclopirox 及び重要な他の候補薬物の発売を承認することを求める場合、重要又はその協力者は、臨床試験及び任意の重要薬物の商業販売及び流通を含む他の司法管轄区域の様々な法規の制約を受ける。 重要又はその協力者が適用されるFDA規制の承認及び上場承認を得るか否かにかかわらず、外国での臨床試験又は発売を開始する前に外国の規制機関の必要な承認を得なければならない。臨床試験、薬品許可、定価と精算を管理する要求と流れは国によって異なる。
必要な臨床データを得ることを前提に,注目すべき会社はパートナーとともにヨーロッパや米国以外の他の司法管轄区でVolasertibとFosiclopiroxおよび注目すべき他の候補薬の上場承認を求める予定である。EUおよび他の多くの外国司法管轄区で有名な薬物を販売し販売するためには、有名またはその潜在的な第三者パートナー は単独のマーケティング許可を得、多くの異なる法規要件を遵守しなければならない。承認プロセスは国/地域によって異なる であり,追加的なテストにつながる可能性がある.承認を得るのに要する時間は、FDAの上場承認を得るのに要する時間とは大きく異なる可能性がある。米国以外の規制承認プロセスには、通常、FDA承認の取得に関するすべてのリスクが含まれる。また,米国以外の多くの国/地域では,薬品精算を承認してから,その国/地域で販売を許可しなければならない。有名またはその潜在的な第三者協力者は、米国以外の規制機関の承認をタイムリーに得ることができないかもしれない(もしあれば)。FDAの承認は他の国または管轄区域の規制機関の承認を確保するものではなく、米国以外の1つの規制機関の承認も他の国または司法管轄区の規制機関またはFDAの承認を確保することはできない。しかし、1つの国/地域で規制承認を得ることができなかったり、遅延したりすることは、他の国/地域の規制プロセスに負の影響を与える可能性がある。NOTINGは市販承認を申請できない可能性があり,NOTIGNの薬物をどの市場でも商業化するための承認を得ることができない可能性がある。
また、2024年6月23日は英国民が離脱国民投票で離脱8周年を投票することを示し、通常は英国離脱と呼ばれる。今日、イギリスはEUから離脱し、実質的にEU規則の制約を受けない。イギリスの規制枠組みの大部分はEU指令と法規に由来し、イギリスの離脱の影響はイギリス或いはEUの重要な候補薬物の承認に関する監督管理制度に実質的な影響を与える可能性がある。イギリスの離脱または他の理由により、いかなる遅延が得られても、またはいかなるマーケティング承認も得られない場合、 は、STEPINGがその候補薬剤をイギリスおよび/またはEUで商業化することを阻止し、 STNOTINGが収入を創出し、利益を達成し、維持することを制限する。上記のいずれかの場合、重要薬物は、イギリスおよび/またはEUが規制部門にその候補薬剤の承認を求める努力を制限または延期させることを余儀なくされる可能性があり、これは、重要薬物の業務に実質的な悪影響を及ぼす可能性がある。
| 35 |
FDAと他の規制機関は非ラベル用途の普及を禁止する法律法規を積極的に実行している。
もし がその候補薬物のラベル外用途を不適切に普及させることが発見されたことに注意すれば,承認されれば重大な責任を負う可能性があることに注意する。そのような法執行はこの産業でもっと一般的になった。FDAや他の規制機関は、RIGNOTYの候補薬のような処方薬製品に関する販売促進声明を厳格に規制し、承認されれば。特に、医薬品brは、医薬品によって承認されたbrタグによって示されるように、FDAまたは他の規制機関によって承認されていない使用に使用されてはならない。NOTIGNがNOTIGN推奨適応のためのNOTING候補薬の市場承認を得た場合、医師 は依然として承認されたラベルと一致しない方法で彼らの患者にNOTIGNの薬剤を使用することができ、医師 個人が彼らの専門医学的判断を信じていれば、このように使用することができる。しかし,Attenantが任意の非ラベル用途のためのNotation‘s薬の普及が発見された場合,連邦政府は民事,刑事および/または行政処罰を行い,Attenantへの罰金を求めることができる。FDAまたは他の規制機関は、法令または会社の誠実な合意に同意することを有名にすることを要求することもでき、または特定の販売促進行為の監視、変更、または制限のための永久禁止を求めることができる。顕著な候補薬物の普及管理に成功できなければ,承認されれば,顕著に重大な責任を負う可能性があり,その業務や財務状況に重大な悪影響を及ぼす。
はFast Trackを指定する可能性があるため、より速い開発や規制審査や承認プロセスを経験しない可能性があることに注意されたい。
臨床的または臨床的データではなく、重篤な疾患の治療に使用される薬剤が、そのような疾患が満たされていない医療要件を満たす可能性があることを示す場合、薬物スポンサーは、FDA Fast Track称号を申請することができる。候補薬物のための迅速なチャネル指定を求めることに注目すべきであれば、FDAからこの称号を獲得しない可能性があることに注意されたい。しかし,Fast Track認証を卓越して取得しても,Fast Track認証 は優れたマーケティング承認や任意の特定の時間範囲での承認を確保することはできない.Fast Track指定は、従来のFDAプログラムと比較して、より速い開発や規制審査または承認プロセスを経験しない可能性があることに留意されたい。また,FDAがFast Trackの指定が重要な臨床開発計画のデータに支持されなくなったと考えると,その指定を撤回する可能性がある。高速チャネル指定自体はFDAに適合した優先審査手順を保証することはできない.
FDAが重要な候補薬物として指定した突破的治療法はもっと速い開発或いは監督審査或いは審査過程を招くことができない可能性があり、重要な候補薬物が発売承認を獲得する可能性を増加させない。
注目すべきは, がそのいくつかの候補薬物に画期的な治療指定を求める可能性があることである。画期的な治療法は、単独または1つまたは複数の他の薬剤との併用治療が深刻または生命に危険な疾患または状態を治療することを意図した薬剤として定義され、予備臨床証拠は、1つまたは複数の臨床的意義を有するbrの終点で、臨床開発早期に観察されるような既存の治療法よりも有意な改善を示す可能性があることを示す。画期的な治療法として指定された薬物と生物製品に対して、FDAと試験スポンサー間の相互作用とコミュニケーションは最も有効な臨床開発経路の決定を助けることができ、同時に無効対照方案中の患者数を最低に下げることができる。FDAで画期的な治療法に指定されている薬物も加速承認を得る資格がある。
FDAは を画期的療法に指定する権利がある。したがって,その候補薬の1つが画期的療法として指定された基準に適合していても,FDAは同意せず,このような指定を行わないことにした可能性がある。画期的な治療指定が得られても,FDA通常の手順により承認を考慮した薬剤と比較して,候補薬剤のこのような指定を受けても,より速い開発過程,審査や承認を招くことはなく,FDAの最終承認を保証することはできない。また,1つまたは複数の有名な候補薬が画期的な治療法となる資格があっても,FDAは今後これらの薬剤が資格条件を満たしていないことを決定したり,FDAの審査や承認を決定する時間帯が短縮されなくなる可能性がある。
| 36 |
有名な候補薬物の商業化に関するリスク
すべての臨床前研究や臨床試験に成功したとしても,その1つまたは複数の候補薬物の商業化に成功しない可能性がある。
Attenantが必要な臨床前研究と臨床試験を完了したとしても、上場承認プロセスは非常に高価で、br}で不確定であり、Attenantが候補薬の一部または全部の商業的承認を得ることを阻止する可能性がある。Attenantが必要な規制承認を得ることができない場合、あるいは必要な規制承認を得る上で遅延が生じた場合、Attenantはその候補薬をbrとして商業化することができず、Attenantの創造能力は深刻な損害を受ける。
注目すべき候補薬物及び開発と商業化に関連する活動は、設計、テスト、製造、安全性、有効性、記録保存、ラベル、貯蔵、承認、広告、販売促進、販売と流通、輸出と輸入を含み、すべてアメリカFDAと他の監督機関及びアメリカ以外のEMAと類似規制機関の全面的な監督管理を受けている。候補薬物が市販承認されなければ,候補薬物 を商業化することはできない。注目すべき候補薬剤は、米国または任意の他の管轄区域で申請を提出していないか、または上場承認を得ていない。
著名な は、マーケティング承認を得るために必要な申請の提出および支援に経験が限られており、この過程で第三者 CROまたは他の第三者コンサルタントまたはサプライヤーに依存して支援を提供することが予想される。上場承認を得るためには、治療適応ごとに監督機関に大量の臨床前と臨床データ及び支持情報を提出し、候補薬物の安全性と有効性を確定する必要がある。上場承認を得るためには、薬品の生産過程に関する情報を監督管理機関に提出し、監督管理機関が生産施設を検査する必要がある。NOTICEの候補薬物 は無効である可能性があり、中程度の効果しかないかもしれない、あるいは不良或いは意外な副作用、毒性或いは他の 特徴を有することが証明される可能性があり、これらの特徴はNOTICEの発売許可を阻止するか、或いは商業使用を阻止或いは制限する可能性がある。新しい抗癌薬は通常、既存の治療に反応しないまたは再発した患者集団にのみ適用される。NOTICEの候補薬剤が市販承認されれば,付随するタグはこのようにNOTIGNの薬剤の承認使用を制限する可能性があり,この はこの薬剤の販売を制限する可能性がある。
アメリカと国外で発売承認を得る過程 は高価であり,承認されれば数年の時間 を要する可能性があり,また関連する候補薬物のタイプ,複雑性と新規性 を含む様々な要素によって大きく変化する可能性がある。開発期間中の上場承認政策の変更、追加法規または法規の変更または公布、または各提出された薬品申請の監督審査の変更は、申請の承認または拒否の遅延を招く可能性がある。監督管理機関は審査過程中にかなりの自由裁量権を持っており、いかなる申請を受け入れることを拒否することができ、また注意すべきデータが承認を得るのに十分ではないことを決定することができ、追加の臨床前、臨床或いはその他の研究を行う必要がある。また、前臨床研究と臨床試験から得られたデータの異なる解釈は候補薬物の上場承認 を遅延、制限或いは阻止する可能性がある。注目すべき最終的に得られた上場承認は、制限されたり、制限されたり、承認された後の約束を受けたりして、承認された薬物が商業的に不可能になる可能性がある。
注意した薬物が市場の受け入れを得られなければ,注意の業務が影響を受け,注意は将来のbr運営を支援できない可能性があるからである。
多くの要因は、注目すべき薬物または任意の注目すべき開発または買収された製品に対する市場の受容度に影響を与える可能性がある
| ● | 他の製品に対する同じまたは同様の治療の有意な薬剤の価格; | |
| ● | 指定用途と治療の重要な薬物の有効性と安全性に対する患者、医師と衛生保健界の他のメンバーの見方; | |
| ● | 注目すべきは、その販売およびマーケティング努力に資金を提供する能力があることである | |
| ● | 注目すべき販売とマーケティング努力の有効性。 |
注目すべき薬物が市場から受け入れられなければ、注目すべきは、開発、テスト、および規制機関の新薬候補に対する承認を得ること、および注目すべき承認された薬物の販売とマーケティング努力を拡大することを含む将来の運営に資金を提供できない可能性があり、これは注目すべき業務に影響を受けることになる。
| 37 |
有名なbrは孤児薬物指定に依存していくつかの有名な候補薬物を商業化する可能性があり、孤児薬物指定が承認されても、このような指定は市場排他性或いは他の商業的優位性或いは予想される商業利益をもたらさない可能性がある。
注目すべきは, はその候補薬物の孤児薬物指定に依存する可能性があることである。米国では,孤児薬物指定は一方が臨床試験費用,税収割引,ユーザ費用減免のために贈与資金を提供する機会など,財政的インセンティブを得る権利がある。さらに、孤児の薬物名を有する薬物がその後、このようなbr}名を有する疾患に対するFDAの最初の市販承認を得た場合、この薬物は孤児薬物の独占経営権を得る権利がある。米国の孤立薬物独占経営権規定によると、FDAは完全な守秘協定を含む他の申請を承認しない可能性があり、7年以内に同じ適応の同一薬物を販売するが、brが限られた場合を除いて、適用される独占経営期間はヨーロッパで10年である。もし1つの薬物が指定された孤児薬物の基準を満たさなくなった場合、あるいはもしその薬物が十分な収益力を持っている場合、市場独占経営権はもはや合理的ではなく、ヨーロッパ独占経営期間は6年間に短縮することができる。
Brが注目に値する場合であっても、任意の将来の協力者が候補薬物の孤児薬物の称号を得ることができない場合であっても、または彼らは候補薬剤の残留薬物排他性を維持することができない可能性がある。医薬製品の開発に関する不確実性により,Togningは最初に発売承認された候補薬物ではない可能性があるが,医薬製品の開発に関する不確実性により,孤児の適応を指定する孤児薬物の称号を獲得しており,同一候補薬物の孤児薬物の称号も持つ別の会社 がDesignantに先立って同じ適応症の発売承認を得る可能性もある。これが発生した場合、競合会社の排他期間が満了するまで、注意すべき申請 は承認されない可能性がある。さらに、承認を著しく求める適応範囲が孤児指定の適応よりも大きい場合、米国における独占営業権は制限される可能性があり、またはFDAが指定された要求に重大な欠陥があると後に決定した場合、またはまれな疾患または疾患を有する患者の需要を満たすのに十分な数の薬剤が著しく保証されていない場合、独占的なマーケティング権利を失う可能性がある。さらに、著名または任意の将来の協力者が薬物の孤立した薬物排他性を獲得しても、この排他性は、異なる活性部分を有する異なる薬剤が同じ場合に許可される可能性があるので、競合から薬物を効果的に保護することができない可能性がある。孤児薬が承認された後であっても、FDAが、後者の薬剤が臨床的により安全で、より有効であると結論した場合、または患者ケアに大きな貢献をした場合、または孤児排他性を有する薬剤のメーカー が十分な薬剤数を維持できないと結論した場合、FDAはその後、同じ状況に対して同じ活性部分を有する同じ薬剤を承認することができる。孤児薬物指定は薬物の開発時間や監督審査時間を短縮することもなく、監督審査或いは審査過程中にこの薬物にいかなる優勢をもたらすこともなく、競争相手が同じ候補薬物を重要な薬物の適応として承認することを阻止することもなく、注目すべき薬物が孤児薬物指定を獲得することを承認することではない。
2017年8月3日、米議会はFDARAと略称するFDA 2017年再認可法案を可決した。FDARAは他の事項のほかに、FDAの事前に存在する監督管理解釈を編纂し、薬品スポンサーが他の方面で以前許可された同じ稀な疾病に対する薬物と同じ孤児薬物の臨床優位性を証明することを要求してこそ、孤児薬物排他性を得ることができる。立法 は以前の前例を覆し、即ち“孤児薬品法”はその臨床優勢にかかわらず、FDAに孤児排他期 を認めることを明確に要求した。また、2021年の総合支出法案では、FDARAで編纂された解釈がFDARA発行前に孤児の称号を発行する場合に適用されることが明らかになったため、国会はこの解釈をさらに変更しなかったが、製品承認はFDARA公布後に行われた。国会やFDAは“孤児薬物法案”とその法規と政策をさらに再評価するかもしれない。FDAがいつ、あるいはどのように未来に孤児の薬物法規と政策を変更する可能性があるかどうかを知らないことに注意し、いかなる変化もどのように注意の業務に影響を与える可能性があるかどうかを確定しない。FDAが孤児薬物法規や政策を変化させる可能性があることにより,NOWARYの業務は悪影響を受ける可能性がある。
| 38 |
ADNOTINGがFDAによって治療薬候補承認に関連する随伴診断の承認を要求され、ADNOTINGがFDAによる診断装置の承認を得ていない場合、またはFDA承認を得る上で遅延に直面している場合、ADNOTINGは候補薬剤を商業化することができず、ADNOTINGの創収能力は深刻な損害を受ける。
FDAのガイドラインによれば、FDAがキット診断装置を決定することが新しい治療薬または適応を安全かつ有効に使用するために重要である場合、セット診断がこの適応のためにも承認または承認されていない場合、FDAは通常、治療薬または新しい治療薬適応を承認しない。“連邦食品、薬物、化粧品法案”(FDCA)によると、診断に伴う診断は医療機器として規制されており、FDAは通常、発売前の承認またはPMAを得て診断を行うために、癌治療に反応する患者を選択することを目的とした診断に伴う診断を要求している。臨床および臨床前データを収集し、FDAに提出し、厳格な上場前審査に関連するFDAによって審査されることを含むPMAプロセスであって、その間、出願人は、設備設計、製造、およびラベルを含む装置の安全性および有効性をFDAに準備して合理的に保証しなければならない。PMAは保証されておらず,かなりの時間を要する可能性があり,FDAは最終的に申請中の欠陥 によってPMA提出の決定を“承認できない”と決定し,追加の臨床試験や他のデータを要求する可能性があり,これらのデータの生成は高価で時間がかかる可能性があり,また は承認を大きく遅らせる可能性がある。そのため、もしFDAが治療性候補薬物のセット診断許可を獲得することに注意すべきであり、FDAの診断設備に対するFDAの承認を獲得或いは遅延していないことに注意すべきであれば、適時に候補薬物を商業化できない可能性があり、或いは根本的にできず、注意すべき創収能力は深刻な影響を受ける。
上場許可を得た候補薬物はすべて発売後に制限され、或いは市場から撤退する可能性があり、もし監督管理要求を著しく遵守できなかった場合、或いは予測できない問題を顕著に経験した場合、その中のいずれかの薬物が承認された時、重大な処罰を受ける可能性がある。
明らかに発売許可を得た候補薬物は、この薬物の製造プロセス、承認後の臨床データ、ラベル、広告と販売促進活動と共に、FDAと他の監督管理機関の持続的な要求と審査を受ける。これらの要求には、安全と他の発売後の情報と報告の提出、登録とリスト要求、記録とファイルの製造、品質管理、品質保証とそれに応じたメンテナンスに関するcGMP要求 が含まれ、医師へのサンプル配布と記録保存に関する要求がある。候補薬剤が上場承認されても、承認は、REMSの実施要求を含む、当該薬剤が発売可能な指定用途の制限または承認条件の制限を受ける可能性がある。新しい抗癌薬は通常、既存の治療に反応しないまたは再発した患者集団にのみ適用される。任意のNOTIGN‘s候補薬剤が市販承認された場合、付随するタグは、このような方法でNORIGN’S薬剤の承認使用を制限する可能性があり、これは、薬剤の販売を制限する可能性がある。
FDAはまた、REMSの採用と実施を含む薬物の安全性或いは有効性を監視するために、高価な発売後の研究或いは臨床試験とモニタリングを要求する可能性がある。FDAその他の機関は、司法省、又は司法省を含み、医薬品の承認後のマーケティング及び販売促進を密接に規制し、承認の適応のみを確保し、承認されたラベルの規定による薬物の販売及び流通を確保する。FDAと米国司法省はラベル外使用に関するメーカーのコミュニケーションに厳しい制限を加えており, はその承認された適応に応じてその薬を販売していないことに注意すると,ラベル外マーケティングの法執行行動を受ける可能性があることに注意している。医薬品の普及および広告に関連する虚偽請求法案を含むFDCAおよび他の法規に違反することは、連邦および州医療詐欺および法律乱用、および州消費者保護法違反を告発する調査および法執行行動につながる可能性がある。
| 39 |
さらに、以前未知の有害事象または重要な薬物、製造業者または製造プロセスに関連する他の問題が後に発見された場合、または規制要求を遵守できなかった場合、様々な結果が生じる可能性がある
| ● | このような薬物、製造業者、または製造プロセスに対する制限br}; | |
| ● | 薬のラベルやマーケティングの制限と警告; | |
| ● | 薬品の流通や使用に対する制限 | |
| ● | 発売後の研究や臨床試験が求められている | |
| ● | 警告brの手紙またはタイトルのない手紙; | |
| ● | 薬品を市場から引き揚げる; | |
| ● | 未解決の出願の承認を拒否するか、または顕著に提出された承認された出願の追加を拒否する | |
| ● | Br薬品を呼び戻す; | |
| ● | 罰金、利益または収入の返還、 | |
| ● | 上場承認の一時停止または撤回 | |
| ● | 任意の潜在的協力者との関係を破壊する ; | |
| ● | 不利なメディア報道および有名な名声への損害; | |
| ● | ノヴィット薬の輸入や輸出を拒否しました | |
| ● | 麻薬を回収する; | |
| ● | 民事または刑事罰の命令または適用を禁止する;または | |
| ● | ノビット薬を使用した患者の訴訟に関連する。 |
注目すべきは, は競争が激しく急速に変化する業界で運営されていることである.
生物技術と製薬薬物開発競争は激しく、迅速かつ重大な技術進歩の制約を受けている。注目すべき成功は、費用効果に基づいて新薬と革新薬物を許可、獲得、開発と監督部門の許可を得る能力に大きく依存し、そしてそれを市場に投入することに成功した。このようにする過程で、有名な顔は、すでに大きな市場シェアを有する大型、全面的に統合され、成熟した製薬会社、専門製薬と生物製薬会社、学術機関、政府機関および他の民間機関、ならびに米国、EUおよび他の司法管轄区域の公共研究機関を含む様々な企業からの激しい競争に直面し続けるであろう。
多くの会社は研究開発、製造、臨床前試験、臨床試験を行い、監督管理の許可と許可を得た薬品の販売などの方面の財力と専門知識は注目すべき競争している或いは未来に競争する可能性のある会社をはるかに超えている。これらの第三者は合格した科学と管理人員を募集と維持し、臨床試験場と臨床試験患者登録を確立し、補充或いは必要な技術を獲得する面で顕著な競争となっている。製薬とバイオテクノロジー業界の合併と買収は、少数の有名な競争相手により多くの資源を集中させる可能性がある。
技術ビジネス適用性の進歩とこれらの業界投資資金の増加により、競争はさらに激化する可能性がある。注目すべき競争相手は、注目すべき開発可能な任意の候補薬剤よりも有効または安価であるかもしれない薬剤の開発に成功し、獲得することができるかもしれない。
老舗製薬やバイオテクノロジー会社は,新規化合物の発見や開発を加速させるために大挙して投資したり,新たな化合物の許可を得たりする可能性があり,注目すべき候補薬物の競争力を低下させる可能性がある。また、承認された薬物と競争するいかなる新薬も治療効果、利便性、耐性と安全性の面で納得できる優勢を示しなければならず、価格競争 を克服し、商業的に成功することができる。したがって、注目すべき競争相手は、注目すべき前に特許保護、発見、開発、FDA承認を得ることに成功するか、または薬物を商業化することに成功する可能性があり、これは注目すべき業務および運営結果に悪影響を及ぼすであろう。
| 40 |
注目すべき競争相手の薬物の供給は需要を制限する可能性があり,注目すべき価格は任意の注目すべき商業化候補薬(あれば)に課金することができる。既存またはその後に発売された薬物との競争ができないことは、顕著な業務、財務状態、および運営結果を損なうことになる。
もし満足できる販売およびマーケティング能力を発展させることができないことに注意する場合、Volasertibおよびフォスピロまたはその発売許可を得た他の任意の候補薬剤の商業化に成功しない可能性があることに注意する.
注目すべきは, にはマーケティングや薬品販売の経験がないことである。承認された場合、Volasertib およびFsciclopiroxまたは任意の他の候補薬剤の販売およびマーケティングについてはまだ合意されていないことに留意されたい。通常,製薬会社は何百人もの販売代表や関連する販売·マーケティング担当者グループを雇用し,大量の医師や病院を訪問する。注目すべきは、第三者と協力して注目すべき薬物をマーケティングすること、またはそれ自体が注目すべき薬物のマーケティングおよび販売を求める可能性があることに注意されたい。第三者との協力を著しく求める場合、顕著に許容可能な条項で協調合意に到達できるかどうかは、著しく決定できない。マイウェートが直接マーケティングとその薬品の販売を求める場合、マイウェートは熟練したマーケティングと販売者を追加的に募集する必要がある。これらのマーケティングおよび販売能力のいずれかまたはすべてを提供するために、第三者関係を取得または確立することができるかどうかは、著しく判断できない。直接販売チームや契約販売チームを構築したり、直販と契約販売チームを組み合わせて重要な薬物をマーケティングすることは高価で時間がかかり、brの任意の薬物の発表を延期する可能性がある。さらに、注目すべきは、任意の期間にわたって直接および/または契約販売チームを維持することができるか、または注目すべき販売努力が、顕著な収入を生成または増加させるのに十分であるか、または顕著な販売努力が永遠に利益をもたらすことを保証することができないことに留意されたい。
たとえNOTINGが規制部門の承認を得ても,VolasertibやFosiclopiroxやNOTINGの他の候補薬を商業化することができれば,NOTIGNの候補薬物は医師や医学界に受け入れられない可能性がある。
Volasertib、FosiclopiroxおよびNORIGNの他の候補薬、またはNOTINGによって独立して、またはパートナーとの開発に成功した任意の他の候補薬剤が、医師、病院、および他の医療機関によって受け入れられることは保証されない。VolasertibとFosiclopiroxおよび未来の注目すべき候補薬物開発は主要な製薬とバイオテクノロジー会社の生産と販売のいくつかの薬物と競争する。市場が注目すべき薬物の受容度は多くの要素に依存しています
| ● | 注目すべき他の候補薬VolasertibとFsciclopiroxの臨床治療効果と安全性の実証; | |
| ● | VolasertibおよびFosiclopiroxおよび注目すべき他の候補薬剤の発売承認および商業発売の時間; | |
| ● | ボルテブ、フォスコピロなどの候補薬の臨床適応を承認した(S) | |
| ● | 薬品ラベルと包装挿入要求; | |
| ● | 既存の抗癌薬物市場の持続的な興味と増加と比較して、注目すべき候補薬物の優勢と劣勢 ; | |
| ● | 販売、マーケティング、流通支援の実力 ; | |
| ● | 薬物brは絶対価格および代替療法に対して定価である | |
| ● | 将来の医療法、法規、医療政策の変化 | |
| ● | 精算コードとカバー範囲は,選定管轄区での可用性 ,および政府と第三者支払人精算政策の将来の変化 である. |
| 41 |
監督部門の許可を得た候補薬物のカバー範囲と精算状態に対して、著しい不確定性が存在する。米国や他の国/地域の市場では、規制部門の許可を著しく得て商業販売のための医薬品の販売は、第三者決済者が精算できるかどうかにある程度依存する。第三者支払者には、政府衛生行政部門、管理型医療サービス提供者、個人健康保険会社、その他の組織が含まれる。
医療保健改革措置は有名な候補薬物の商業成功を阻害或いは阻止する可能性がある。
米国政府や他の政府は医療改革の推進に強い興味を示している。政府が講じたいかなる改革措置も,米国や国際医療医薬品やサービスの定価や政府機関や他の第三者支払者が提供する精算金額に悪影響を及ぼす可能性がある。アメリカと外国政府、保険会社、医療組織と他の医療保健サービスの支払いを管理する人為的に医療コストをコントロール或いは低減するための持続的な努力は、注意すべき薬品の公平な価格を制定する能力及び注目すべき収入の創造、利益を実現し、維持する能力に不利な影響を与える可能性がある。
米国では,一部の州では薬品価格制御や患者訪問制限が実施されており,他の州ではその医療補助計画の実施が考えられている。最近の州立法努力は通常薬品コストの透明性を高めたり、薬品価格を制限したりすることに重点を置いている。また,大規模な管理的保健組織や処方薬福祉マネージャーの増加,後発薬代替の普遍的な存在が,処方薬の価格上昇を阻害している。継続的に厳格な公衆による薬品価格の審査に加え、政府や支払人の力に加えて、生産者や営業者が製品価値に応じて価格を設定または調整する能力を制限することが可能である。米国以外では、EU、日本、中国を含む多くの主要市場に政府の広範な参加があり、医療保健に資金を提供し、その点で薬品の定価と精算を固定している。そのため、注目すべき製品はますます多くの政府の決定と予算行動の影響を受ける。新しいまたは提案された製品を保証することができない製品は、コスト効果があるとみなされるか、または適切なリターンを達成するために、そのような製品の製造者または営業業者が適切な価格レベルを維持することができるように、十分な第三者精算を得るであろう。
医療の利用可能性、製品およびサービスの交付または支払い方法、または販売、マーケティングまたは定価に関連する新しい 法律、法規および司法判断、または既存の法律、法規および裁決の新しい解釈は、重要なbr}の潜在的収入を制限する可能性があり、その研究および開発計画を修正する必要がある可能性がある。定価と精算環境は未来に変化する可能性があり、いくつかの原因でもっと挑戦的になり、アメリカの現在の行政当局が提出した政策、新しい医療保健立法或いは政府衛生行政部門が直面している財政挑戦を含む。具体的には、アメリカと一部の外国司法管轄区では、医療システムを変更するための多くの立法と監督管理提案があり、その方式は著しい薬品販売収益能力に影響を与える可能性がある。
例えば,“医療·教育協調法案”(ACAと略す)で改正された“患者保護·平価医療法案” は,政府医療計画や民間保険会社が医療保健に資金を提供する方式を大きく変え,製薬業に大きな影響を与えている。ACAには多くの条項が含まれており,これらの条項は顕著なビジネスおよび運営 に影響を与え,顕著な将来の潜在的収入に負の影響を与える可能性が予想される。例えば,ACAは政府プロジェクトでブランド処方薬を販売する製薬メーカーや輸入業者に差し引くことのできない消費税 を徴収していることに注目すべきは,注目すべき薬物のコストを増加させることである。また、ACA条項の一部として、Medicare Part D処方薬計画に現在存在している資金不足 を埋め、有名なのは、ドーナツ穴内のある受益者の薬品に政府交渉価格に相当するbr}から50%のブランド処方薬割引を提供することを要求される(その後、 は70%に増加し、2018年両党予算法(BBA)による2019年1月1日までの現在の割引)。同様に,ACAはブランド医薬品メーカーが支払うべき医療補助フィードバックレベルを15.1%から23.1%に向上させ,医療補助管理のヘルスケア組織が支払うべき薬品に対してbr}返却点を要求する。ACAはまた、この計画に従って薬品を購入することができる合格カバーエンティティのリストを拡大することを含む340 B医薬品割引計画の重大な変更を含む。また,ACAが作成した医療保険福祉資格の拡大により,保険カバー範囲を有する患者数が増加することが予想され,これらの患者は注目すべき薬剤を受け入れる可能性がある。ACAまたは任意の将来の医療改革立法が顕著な業務に及ぼすすべての具体的な影響を予測するのは時期尚早であるが、それらは顕著な業務および財務状況に実質的な悪影響を及ぼす可能性がある。
| 42 |
国会ではACAや2003年の医療保険処方薬,改善·現代化法案やMMAなどの立法が定期的に可決され,処方薬に関する医療保険精算·引受政策が改正されている。このような法律の施行は規制と二次規制政策によって持続的に改正されるだろう。現在行われている予算交渉の一部として,国会では連邦医療保険政策をより多く修正することも考えられるが, には連邦医療保険処方薬政策が含まれている可能性がある。現在のところこのような立法の範囲は決定されていないが、将来の立法や法規が注目すべき推奨薬物のカバー範囲や価格を低下させない保証はない。他の第三者支払者は、医療製品やサービスの課金 に挑戦することが増えている。注意すべき人にとっては,連邦医療保険や個人支払者から保険や精算を求めることで時間がかかり高価になる。NOTINGの推奨薬剤は、費用対効果があるとみなされない可能性があり、カバー範囲 および精算が、NOTINGが利益に基づいてNOTINGによって推奨される薬剤を販売することができないか、または十分ではない可能性がある。さらなる連邦や州提案や医療改革がある可能性があり,注目すべき候補薬の価格 を制限し,注目すべきビジネス機会をさらに制限する可能性がある。注目すべき運営結果は、提案された医療改革、連邦医療保険処方薬カバー立法、このような現在または未来の立法が私営保険会社の支払い金額に与える可能性のある影響、および将来公布または採用される可能性のある他の医療改革の実質的な悪影響を受ける可能性がある。
2007年9月、2007年“食品と薬物管理局改訂法”を公布し、FDAにもっと強い発売後の許可を与え、 は発売後の研究と臨床試験を要求し、新しい安全情報に基づいてラベル変更を行い、FDAが許可したREMSを遵守する権力を含む。FDAがこの権力を行使することは、薬物開発、臨床試験、および規制審査期間の遅延またはコスト増加を招き、承認後の規制要件の遵守を保証し、承認された薬物の販売および/または流通を制限する可能性があるコストを増加させる可能性がある。
医療保健提供者、医師および第三者支払者との関係は、適用されるリベート、詐欺および乱用、br}虚偽声明、透明性、健康情報プライバシーと安全、および他の医療保健法律および法規の制約が適用され、違反すれば、刑事罰、民事処罰、契約損害、名声損害、行政負担および利益および将来の収入を減少させる可能性があることに注意されたい。
医療保健提供者、医師、第三者支払人は、上場承認された候補薬物を推薦と処方する過程で主要な役割を果たす。注意すべきは、医療保健提供者、医師と第三者支払人との将来の手配 は注目すべき広範に適用される詐欺と濫用及びその他の医療法律と法規に直面する可能性があり、これらの法律と法規は業務或いはbr}財務手配と関係を制限する可能性があり、これらの手配と関係を通じて、注目すべき市場、販売と流通はいかなるマーケティング許可を得た薬品 である。また、米国連邦や州政府およびそれで業務を展開している外国司法管轄区の政府は、透明性の法律や患者のプライバシー法規を遵守する可能性がある。適用される連邦と州医療法律法規の制限は
| ● | 他の事項に加えて、“連邦反バックル条例”は、個人を誘惑または奨励するために、現金または実物の形態で、現金または実物の形態で直接または間接的に報酬を請求、提供、受信または提供し、またはこれを見返りとして、個人のための推薦または購入、注文、推薦または手配、連邦医療保険および医療補助などの連邦医療計画に従って支払うことができる任意の商品またはサービスを禁止する | |
| ● | 連邦虚偽請求法案は、民事通報者または準訴訟を含む個人または実体に刑事罰を適用し、他のほかに、個人または実体が連邦医療保健計画支払いに関する虚偽または詐欺的クレームの提出を意図的に提出するか、または虚偽クレームの支払いまたは回避、連邦政府に資金を支払う虚偽陳述または記録材料を減少または隠蔽することを含む虚偽陳述または記録材料を提供することを含み、潜在的責任は、強制3倍損害賠償およびbr重大な各クレーム処罰を含む |
| 43 |
| ● | 1996年の連邦健康保険携行性および責任法案、またはHIPAAは、詐欺の任意の医療福祉計画または医療事項に関する虚偽陳述を行う計画に刑事責任および民事責任を適用する | |
| ● | HIPAAは、2009年に“衛生情報技術促進経済·臨床衛生法”及びそのそれぞれの実施条例により改正された“衛生情報技術促進経済·臨床衛生法”に加え、強制契約条項を含む個人が識別できる健康情報のプライバシー、安全、伝送を保護する義務を規定している | |
| ● | 連邦医師が陽光法案の適用を要求する保険薬品メーカーは、医師および教育病院に支払われたお金およびその他の価値移転を報告した | |
| ● | 同様の国および外国の法律、例えば、国の反リベートおよび虚偽請求の法律および透明性法規は、brの販売またはマーケティングの手配、および非政府第三者支払者(民間保険会社を含む)によって精算される医療項目またはサービスに関するクレームに適用される可能性がある。 |
いくつかの州の法律は製薬会社に製薬業界の自発的コンプライアンスガイドラインと連邦政府が公布した関連コンプライアンスガイドラインを遵守することを要求し、そして薬品メーカーに医者と他の医療保健提供者への支払いと他の価値移転或いはマーケティング支出に関する情報を報告することを要求する可能性がある。また、いくつかの州と地方の法律はこの司法管轄区に薬品販売代表を登録することを要求している。場合によっては、州法や外国法は健康情報のプライバシーやセキュリティも管轄しており、その多くの法律は互いに大きく異なり、HIPAAに奪われず、コンプライアンス作業を複雑化させることが多い。
注意すべき第三者の業務手配に適合した医療法律や法規を確保する努力 は大量のコストに及ぶ。政府当局は、注目すべき業務慣行が、現在または将来、詐欺および乱用または他の医療保健法律法規の適用に関連する現在または将来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。注目すべき業務がこれらの法律のいずれかまたは他の任意の他の注目すべき政府法規に適用される可能性があることが発見された場合、重大な民事、刑事および行政処罰、損害賠償、罰金、監禁、brの薬品を政府が援助する医療計画(例えば、MedicareおよびMedicaid)から除外し、注目すべきbr業務を削減または再編する可能性がある。それと業務を展開することを明らかに望む任意の医師または他のヘルスケア提供者またはエンティティが、brが適用される法律に適合していないことが発見された場合、彼らは、政府援助に参加する医療計画から除外されることを含む刑事、民事または行政処罰を受ける可能性がある。
最近の制定と将来の立法は、注目すべき候補薬物が上場承認され、それを商業化することの難しさとコストを増加させ、米国または外国の管轄区域で承認された任意の薬物の顕著な入手可能な価格に影響を与える可能性がある。
アメリカといくつかの外国司法管轄区では、医療保健システムに関する多数の立法と法規変更及び提案された変更 は重要な候補薬物の上場承認を阻止或いは延期し、承認後の活動を制限或いは規範化し、そしていかなる上場許可を獲得した候補薬物を顕著な利益販売する能力 に影響する可能性がある。製薬産業はこのような努力の重点であり、立法計画の大きな影響を受けてきた。現在の法律、および将来取られる可能性のある他の医療改革措置は、より厳しいカバー基準をもたらす可能性があり、FDAによって承認された任意の薬物の価格に追加の下振れ圧力を与える可能性がある。
米国では,MMAは医療保険カバーと薬品の支払い方法を変更した。この立法は高齢者が薬品を購入する医療保険のカバー範囲を拡大し、医師が管理する薬品の平均販売価格に基づく新しい精算方法を導入した。さらに、この法案は、任意の治療カテゴリがカバーする薬物の数を制限するための許可を提供する。この立法のコスト削減措置および他の条項は、任意のbrによって承認された薬物のカバー範囲および価格を低下させる可能性がある。
| 44 |
MMAは連邦医療保険受益者の薬品福祉にのみ適用されるが、個人支払者は自分の精算率を設定する際には通常連邦医療保険カバー政策と支払い制限 に従う。したがって,MMAによるどの精算減少も個人支払者の支払いのような の減少を招く可能性がある.
2010年3月、オバマ総裁は“米国弁護士協会”に署名し、法律にした。注目すべきビジネスに潜在的重要性を有するACA条項において、 は、注目すべき商業化能力および許可販売が許可される可能性のある任意の候補薬剤の価格 を含むが、以下のように含まれる
| ● | 指定されたブランドの処方薬および生物学的製剤を製造または輸入する任意のエンティティに控除できない年会費を徴収する | |
| ● | 医療補助薬品リベート計画によると、メーカーが支払わなければならない法定最低リベートが増加する | |
| ● | 民事虚偽申告法と連邦反リベート法規、新しい政府の権力調査と違反行為に対する処罰の強化を含む医療詐欺と乱用法律を拡大する | |
| ● | 新しいMedicare Part D Coverage Gap割引計画では、製造業者は、交渉価格に基づいて50%(2019年1月1日から70%)の販売時点割引を提供することに同意しなければならない | |
| ● | メーカーの医療補助税還付を延長する | |
| ● | 医療補助計画の資格基準を拡大する | |
| ● | 公衆衛生サービス薬品定価によって割引を受ける資格のある実体の拡張 ; | |
| ● | 医師や教育病院のいくつかの財務計画との新しい要求を報告する | |
| ● | 製造業者および流通業者が医師に提供する薬品サンプルの新しい要求を毎年報告する | |
| ● | 患者を中心とした新しい結果研究機関は、優先事項を監督、決定し、臨床有効性比較研究 を行い、このような研究に資金を提供する。 |
また、“ACA”が公布されて以来、他の立法改正も提案され、採択された。これらの変化には、2011年の“予算制御法案”と2012年の“米国納税者救済法”が含まれており、それ以外にも、2013年から医療保険提供者への医療保険支払い総額を前期当たり最大2%減少させ、同法規の後続立法改正により、いくつかの種類の提供者への医療保険支払いを減少させ、政府が提供者に多額の支払いを取り戻す訴訟時効期間を3年から5年まで延長することを含む2032年まで有効となる。
2022年8月、2022年に“インフレ率低減法”(略称“アイルランド共和軍”)が法律に署名した。IRAはいくつかの条項を含み、これらの条項は異なる程度で重要な業務に影響を与える可能性があり、Medicare Part D受益者のために2,000ドルの自己負担上限を作成し、Medicare Part D中のすべての薬品に新しいメーカーの財務責任を適用し、アメリカ政府がある模造薬或いは生物類似競争のない高コストの薬物と生物製品の交渉 Medicare Part BとPart D定価を許可し、会社のインフレより速い薬品価格のMedicareにリベートを支払うことを要求し、薬局福祉マネージャーを受益者に引き戻すことを要求するバックオフ規則を含む。Ireland共和軍が注目すべき業務と医療産業全体に及ぼす影響はまだ不明だ。
これらの新しい法律は、連邦医療保険および他の医療資金のさらなる減少をもたらす可能性があり、他の方法で注目すべき任意の候補薬剤が得られる可能性のある価格 の注目すべき規制の承認を得る可能性のある候補薬剤または任意のそのような候補薬剤の処方または使用頻度に影響を与える可能性がある。また、アメリカ議会は最近数回の調査を行い、州と連邦立法を提出し、薬品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、連邦医療保険下の薬品のコストを下げ、政府計画の薬品の精算方法を改革することを提案した。
| 45 |
注目すべきbrは,これらの医療改革,および将来とりうる他の医療改革措置は,医療保険や他の医療資金のさらなる減少,より厳しいカバー基準,新たな支払い方法,および注目すべき任意の承認薬の価格および/または医師が任意の承認薬物を実施することによって得られる精算レベルの追加的な下振れ圧力をもたらす可能性が予想される。精算レベルを下げることは,顕著に受け取った価格brや顕著な薬物の処方や投与頻度に悪影響を及ぼす可能性がある。連邦医療保険や他の政府が計画している精算のいかなる減少も、個人支払者の支払いの同様の減少を招く可能性がある。
処方薬のコストも米国でかなり議論されており、国会議員は新たな立法や行政措置でこのようなコスト問題を解決すると表明している。これまで、アメリカ議会は最近数回の調査を行い、そして州と連邦立法を提出し、公布し、薬品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、連邦医療保険制度下の薬品のコストを下げ、政府計画薬品精算方法を改革する。任意の提案された措置は、有効になるために追加の立法許可を通過する必要があるが、国会は、医薬品コストを制御するために、新しい立法および/または行政措置を求め続けることを示している。州レベルでは、立法機関は、価格または患者の精算制限、割引、特定の製品へのアクセスおよびマーケティングコスト開示の制限、および透明性措置を含む、医薬品および生物製品の価格設定を制御するための法規を立法および実施することによって、場合によっては、他の国からの輸入および一括購入を奨励することを目的としている。
すでに立法と監督管理の提案を提出し、承認後の要求を拡大し、薬品br製品の販売と販売促進活動を制限する。注意は、追加の立法変化が公布されるかどうか、またはFDAの法規、ガイドラインまたは解釈が変更されるかどうか、またはこれらの変化が注意候補薬物の上場承認にどのような影響を与える可能性があるかどうかを決定することができない。アメリカ議会がFDA審査手続きの審査を強化することは上場承認を大幅に延期或いは阻止する可能性があり、そして更に厳格な製品ラベルと上場後のテスト及びその他の要求の影響を明らかに受ける可能性がある。
現在または将来従事している任意の第三者製造業者または請負業者が環境、健康、および安全法律法規に準拠できなかった場合、罰金または罰金に処せられたり、顕著な業務を損なう可能性のあるコストまたは責任が生じる可能性がある。
注意すべきbrおよび現在従事している第三者製造業者および将来従事する可能性のある任意の第三者製造業者は、実験室プログラムおよび危険材料および廃棄物の処理、使用、貯蔵、処理および処理を管理する法律法規を含む多くの環境、健康および安全法律法規の制約を受けるであろう。注目すべき事業は、化学品および生物学的材料を含む危険性および可燃性材料の使用に関する第三者製造業者または請負業者による作業を含む。顕著な事業は、危険廃棄物製品も生成する。注目すべきは、一般的に第三者とこのような材料と廃棄物を処分する契約を締結したということだ。注目すべきは、このような材料が汚染や傷害の危険を除去できないということだ。危険な材料の使用による汚染または傷害の場合、NOTIGNは、それによって生じる任意の損害に責任を負う可能性があり、任意の 責任は、NOTIGNのリソース範囲を超える可能性がある。危険材料の放出や清掃を管理するいくつかの環境法によると,責任は連携しており,非を考慮せずに加えることができる。このような法律および法規を遵守できなかったため、DIGNINGはまた、br民事または刑事罰金および処罰に関連する巨額の費用を生じるか、またはDIGNING活動を制限または禁止する禁止の制約を受ける可能性がある。
危険材料の使用により従業員にダメージを与える可能性のあるコストおよび支出をカバーするために、一般責任保険および労働者補償保険を有しているが、この保険は潜在的な責任に十分な保険を提供できない可能性がある。環境責任または有毒侵害行為に保険を提供しないことに注意されたいが、これらのクレームは、注目すべき会社またはその請負業者が生物、危険または放射性材料を貯蔵または処分することに関連するクレームを対象とする可能性がある。
さらに、現在または未来の環境、健康および安全法律法規を遵守するためには、巨額のコストが生じる可能性がある。これらの現在または未来の法律および法規は、著しい研究、開発、または生産努力を損なう可能性がある。このような法律や法規を遵守できなかったことは、巨額の罰金、処罰、または他の制裁にもつながる可能性があることに注意されたい。
| 46 |
さらに、NOTINGの現在および将来の任意の第三者契約製造業者または他の請負業者の運営については、適用される環境、健康および安全な法律法規に準拠していない場合、またはNOTIGN医薬品に関連する廃棄物を適切に処理できない場合、NOTINGは、それによって生じる任意の損害、名声(Br)損害、またはNOTING候補薬剤または医薬品の生産および供給において中断された責任を負う可能性がある。また、いずれかの第三者契約メーカーが環境、健康、安全法律法規を遵守しないことにより禁止または他の制裁を受けた場合、注目すべきサプライチェーンは悪影響を受ける可能性がある。
は、PPMPの取得、開発、強化、または配備に必要な技術面で挑戦される可能性があることに注意されたい。
注目すべきは、データ収集、データ処理、クラウドに基づくプラットフォーム、分析、統計予測と予測、モバイル計算、ソーシャルメディア分析、その他のアプリケーションと技術のための複雑なコンピュータシステムとソフトウェアを必要とする。注目すべきは、業界横断技術リーダーの使用革新への依存を増加させ、注目すべき適用需要と応用に基づいてこれらを調整することによって、注目すべき技術リスクをある程度解決したことである。注目すべき業界をサポートするいくつかの技術は急速に変化しており、これらの変化には許容可能なコストでタイムリーかつ効率的な方法で適応し続けなければならないことに注意されたい。 注目すべきは、複雑な問題に明確な答えを提供しながら、使いやすい形でデータを取得し、使用し続ける必要がある。新しい技術を著しく開発、獲得または統合できる保証はなく、これらの新技術 が顕著な需要を満たすこと、あるいは顕著な期待目標を達成することを保証することはできず、競争相手のように迅速または経済的に効率的にできる保証もない 。重大な技術変革は購買力平価淘汰管理計画を時代遅れにする可能性がある。注目すべき持続的な成功は、絶えず変化する技術、増加するデータと情報に管理と処理する能力、および変化する内部および業界の需要に応答するために、プラットフォームと機能の性能、特性、信頼性を向上させる能力に依存する。注意すべき は困難に遭遇する可能性があり、PPMP高度バージョンの成功設計、開発、テストと発売を延期或いは阻止する可能性があり、それによって注目すべき新薬候補薬物を識別する能力を制限した。PPPPを利用した新しいサービスや既存サービスの強化は,注目すべき要求を十分に満たすことができない可能性がある.これらの故障のいずれも、顕著な経営業績や財務状況に重大な悪影響を及ぼす可能性がある。
注目すべき第三者依存に関するリスク
注意すべきは, は第三者による注目すべき臨床前研究と臨床試験である。これらの第三者が彼らの契約法律および規制義務を成功裏に履行できない場合、または予想される最終期限前に完了した場合、重要な薬物は規制部門の承認を得ることができないか、またはその薬物を商業化することができず、顕著な業務が深刻な損害を受ける可能性がある。
有名なbrは引き続き第三者医療機関、臨床研究者、契約実験室と他の第三者CROに依存し、進行中の臨床前と臨床プロジェクトのデータを監視と管理することを計画してきた。明らかにこれらの処方が顕著な臨床前研究と臨床試験を実行することに依存し、そしてそのbr活動のいくつかの方面のみを制御する。しかし、注目すべきは、すべての研究が適用された合意、法律、法規、科学的基準に従って行われることを保証する責任があり、CROへの依存が注目すべき規制責任を免除しないことに注意されたい。NOTICE及びそのCROはGCPを遵守しなければならず、GCPはFDA、欧州経済地域(EEA)加盟国の主管当局及び類似の外国監督機関がすべての臨床開発中のNOTIGN薬物に対して実行する法規とガイドラインである。
規制当局は,試験スポンサー,主要調査者,試験地点を定期的に検査することでこれらのGCPを実行している。有意またはそのいずれかのCROが適用されるGCPに準拠していない場合、顕著な臨床試験において生成された臨床データは、信頼できないbrと考えられる可能性があり、FDA、EMAまたは同様の外国の規制機関は、顕著な上場申請を承認する前に追加の臨床試験を行うことを必要とする可能性がある。特定の監督管理機関が検査を行った後、この監督管理機関はいかなる注目すべき臨床試験がGCP規定に符合することを確定することは明らかに保証できない。また,注意すべき臨床試験は,現行の良好な製造規範(“cGMP”)規定により生産された製品を用いて行わなければならない。注意すべきは,これらの規定を遵守できなければ,臨床試験を繰り返す必要がある可能性があり,規制部門の承認過程が遅れることである。
| 47 |
がこれらの第三者CROの任意の関係と著しく終了した場合、代替CRO と合意することができないか、または商業的に合理的な条項でそうすることができない可能性がある。また,注目すべきCROは注目すべき従業員ではなく,このようなCROとのプロトコルによって注目すべき救済措置を提供するほか,注目すべき臨床,非臨床,臨床前計画に十分な時間と資源を投入しているかどうかを制御できないことに注意されたい。CROがその契約義務または義務を成功裏に履行できなかった場合、または予期された期限内に達成できなかった場合、CROがCROを交換する必要がある場合、またはCROが獲得した臨床データの品質または正確性が重要な臨床方案、法規要求または他のbrの原因を遵守できなかったために影響を受ける場合、重要な臨床試験は延長、延期または終了される可能性があり、重要な候補薬物は監督部門の承認を得られないか、商業化に成功する可能性がある。そのため,注目すべき候補薬物の運営結果やビジネスの将来性が損なわれ,注目すべきコストが増加する可能性があり,注目すべき収入の発生能力が遅れる可能性がある。
有名な契約と締結された多くの第三者もまた、有名なbr競争相手を含む他の商業実体と関係がある可能性があり、彼らはまたこれらの競争相手のための臨床試験または他の薬物開発活動を行っている可能性があり、これは有名なbr}競争地位を損なう可能性がある。注意すべき良好な実験室規範(“GLP”)の臨床前研究または注目すべき臨床試験を行う第三者が、その契約責任または義務、作業停止、予期されるbr締め切りに達していない、注目すべきプロトコルとの終了または交換が必要である場合、または彼らが得た臨床データの品質または正確性が注目すべき臨床試験手順またはGCPまたは任意の他の原因を遵守できないことによって損害を受けた場合、注意すべきbr}は代替第三者と新しい手配を締結する必要がある可能性がある。追加のCROの交換または追加は、追加のコスト に関連し、管理時間および重点を必要とする。しかも、新しいCROが仕事を始める時、自然な過渡期がある。したがって,br}遅延が出現し,それに必要な臨床開発スケジュールを著しく満たす能力に大きな影響を与える可能性がある。注目すべきbr}は、そのCROとの関係を慎重に管理しているにもかかわらず、将来的に同様の挑戦や遅延に遭遇しないこと、またはこれらの遅延または挑戦が注目すべき業務、財務状況、および見通しに実質的な悪影響を与えないことに注意される保証はない。
注意すべき は、第三者がその候補薬物を生産する臨床供給に大きく依存し、注目すべき は、第三者が任意の承認された候補薬物を生産する商業的供給に依存することを意図している。したがって、第三者製造業者がFDAまたは同様の規制機関の承認を得られなかった場合、またはbr}NOTINGに十分な数または許容可能な価格で医薬品製品を提供することができなかった場合、その薬剤の開発を停止または延期したり、将来の薬物の商業化を延期したり、またはその利益を低下させる可能性がある。
製薬製品の製造は非常に複雑で、大量の専門知識、資本投資、プロセス制御と技術ノウハウが必要である。 製薬生産中のよく見られる困難は:原材料の調達と生産、化学 と開発活動から生産活動への技術の移転、初期生産設計の検証、製造技術の拡張、コストと生産量の向上、品質制御と安定性要求の確立と維持、汚染とオペレータのミスの除去、法規遵守要求の維持を含むかもしれない。FDAによって規定されているcGMPが内部でインフラや能力を得る計画もないことや、将来の注目すべき薬物の臨床試験および商業化の需要を満たすために十分な化合物供給を生産する計画もないことに注意されたい。薬品生産施設で検査を受けて初めてFDAは新薬製品の販売を許可し,すべての注意すべき使用しようとしているメーカーはFDAが規定しているcGMP規定を遵守しなければならない。
したがって、注目すべき は、第三者メーカーに注目すべき開発可能な候補薬物の臨床供給に依存することが予想される。これらの第三者メーカーはCGMPや他の適用される法律法規の遵守が要求される。これらの第三者がこれらの要求を遵守したり、十分な品質管理、品質保証、合格者の能力を維持したりする能力を著しく制御することはできない。FDAまたは任意の他の適用可能な規制機関が、これらの第三者が他の候補薬剤または開発に成功する可能性のある任意の薬剤を生産する施設に欠陥または不承認があることを発見した場合、またはそのような承認を撤回するか、または有名なサプライヤーまたは契約製造業者がこれ以上供給または生産しないと決定した場合、代替生産施設を探す必要がある可能性があることに注意されたい。この場合、臨床または商業供給の製造業者を許容可能な条件で決定することができない可能性が著しく、または全くできない可能性がある。これらの要素のいずれも重要な候補薬物の開発、監督許可或いはマーケティングの能力に深刻な影響を与え、重要な業務に不利な影響を与える。
| 48 |
注意すべき および/またはその第三者メーカーは、注意すべき制御範囲を超える事態の発展に悪影響を受ける可能性があり、これらの事態は、注意すべき薬物のさらなる生産を延期または阻止する可能性がある。不利な発展には、労使紛争、資源制限、br}出荷遅延、在庫不足、ロット故障、新冠肺炎疫病或いはその他の流行病或いは伝染病爆発に関連する影響、思わぬ汚染源、重要な製造技術、製造過程で使用される設備に関する訴訟、br}或いは物質組成、不安定な政治環境、テロ行為、戦争、自然災害及びその他の天災人災br}が含まれる可能性がある。NOTINGまたはその第三者製造業者が上記のいずれかの困難に遭遇した場合、または契約義務を遵守できなかった場合、NOTIGNが臨床試験または商業用途のための任意の薬剤を提供する能力が脅かされる。これは、NOTIGNの臨床試験および商業生産の完了に関連するコストを増加させる可能性がある。さらに、生産中断は、進行中の臨床試験の終了および/または新たな臨床試験の開始を招き、追加費用を支払う可能性がある。注意すべきは, は在庫を解約し,規格に適合していないか安全検査を通過した薬品に他の費用や費用 を発生させなければならない可能性があることである。もし生産困難が受け入れ可能なコスト、費用と時間範囲内で解決できない場合、顕著にその臨床開発と商業化計画を放棄させる可能性があり、これは顕著な業務、将来性、財務状況と顕著な証券の価値に重大な不利な影響を与える可能性がある。
注目すべきは、 または注目すべき第三者製造業者は、注目すべき候補薬剤 の生産を十分な品質および数量で拡大することに成功できない可能性があり、これは、注目すべき候補薬剤の開発を延期または阻止し、承認されたbr}薬剤を商業化することになる(ある場合)。
NOTICEの候補薬物に対して臨床試験を行い,任意の承認された候補薬物あるいはそのメーカーを商業化するためには, はそれらを大量に生産する必要がある。注意して、またはその製造業者は、タイムリーまたは費用便益のある方法では、または任意の注目すべき候補薬剤の製造能力を増加させることに成功しない可能性がある。また,拡張活動中に品質の問題が生じる可能性がある.NOTINGまたはその任意の製造業者がNOTIGN生産の候補薬剤 を十分な品質および数で成功的に拡大できない場合、候補薬剤の開発、試験および臨床試験は延期されるか、または不可能である可能性があり、任意の結果薬物の規制承認または商業投与が延期または入手できない可能性があり、これはNOTIGNの業務に深刻な損害を与える可能性がある。NOTINGが商業供給のためのNOTING候補薬剤 を獲得または維持できない場合、または商業的に合理的な条項でそうすることができない場合、NOTIGNはその候補薬剤の開発に成功し、商業化できない可能性がある。
第三者パートナーを見つけて薬物開発コストを協力したり分担したりすることができず,同社の業務,財務状況,経営結果に大きな被害を与える可能性がある。
その候補特許薬物の開発および商業化戦略には、第三者との協力計画 を形成することが含まれている可能性があることに留意されたい。既存と未来の協力者は、彼らが適用する仕事と資源を決定する上で大きな裁量権を持ち、その義務を期待通りに履行できない可能性がある。潜在的な第三者パートナーには、バイオ製薬、製薬、バイオテクノロジー会社、学術機関、および他の実体が含まれる。第三者協力者は、以下のような点で有名な人を支援するかもしれません
| ● | 研究、臨床前開発、臨床試験、製造を援助する | |
| ● | 規制部門の承認を求めています | |
| ● | 未来の候補薬を商業化することに成功した。 |
が著しくさらなる協力プロトコルを確立できなければ,薬物開発と商業化を著しく自費で行う必要がある可能性がある。このような承諾は,注目すべき開発可能な候補薬物の数を制限し,注目すべき資本要求を著しく増加させ,注目すべき内部資源に追加的な圧力をもたらす可能性がある。重要な失敗 は重要な業務、財務状況、運営結果に重大な損害を与える可能性がある。
さらに、許可、協力、および第三者との他のプロトコルへの依存は、多くの注目すべきリスクに直面する可能性がある。これらの協定の条項は有名な製薬会社に有利ではないかもしれないし、有名な製薬会社にその候補薬物のいくつかの権利を放棄することを要求するかもしれない。特定の分野で1人の協力者のみと協力することに著しく同意する場合、他のエンティティと著しく協力する機会が制限される可能性がある。潜在的な新しい協力者との長い交渉は,候補薬物の研究,開発あるいは商業化の遅延を招く可能性がある。注目すべき協力者は代替技術 を求めることを決定し、あるいは注目すべき権利を獲得した任意の候補薬物の開発或いは商業化に成功しなかったことは、注目すべき業務、財務状況と運営結果に重大な損害を与える可能性がある。
| 49 |
銀行機関が持っている現金残高はFDICの保証範囲を超えている。
1つまたは複数の金融機関が保有する大量の現金および現金等価物が連邦保険の限度額を超えていることに注意されたい。銀行機関が著しく保有している有利子と無利息口座は連邦預金保険会社(“FDIC”)によって保証され、最高250,000ドルに達する。ほとんどの注目すべき銀行機関が持つ現金残高 はFDICのカバー範囲を超えている.注目すべきは、これが正常な商業的危険だということだ。
候補製品の発見と開発に関するリスク
注目すべき候補製品の臨床研究は成功しないかもしれない。顕著に臨床 候補製品研究から成功した結果を得ることができない場合、あるいはその過程で重大な遅延に遭遇すれば、顕著な業務は実質的なbr}損害を受ける可能性がある。
注意すべきことは、 は商業マーケティングのための製品を承認しておらず、すべての注目すべき候補製品は臨床 テストに入るか、臨床開発段階にあることである。利益を実現し維持できるかどうかは規制部門の承認を得ることにかかっており、承認されれば、その候補製品は単独でも第三者との協力でも商業化に成功することに注意されたい。監督部門の重要候補製品の商業流通の許可を得る前に、重要或いは既存或いは未来の協力者は広範な臨床試験を行い、重要な候補製品の安全性と有効性を証明しなければならない。
注目すべき候補製品の成功は以下のいくつかの要因に依存する
| ● | 臨床試験に成功し、臨床研究を完成し、結果は良好であった | |
| ● | 規制機関に適用された上場承認を受けた | |
| ● | 現在および未来の候補製品のために特許および商業秘密保護を取得し、維持すること | |
| ● | 第三者と製造関係を確立し、維持するか、または顕著な自己製造能力を確立すること | |
| ● | 販売チーム、マーケティング、および流通インフラの構築に成功することを含む、注目すべき製品を商業化することに成功した(承認された場合)、単独でも他社との連携も含む。 |
もし が上述の1つまたは複数の要素を著しくタイムリーにまたは根本的に実現できなかった場合、重大な遅延が発生するか、またはその候補製品の開発または商業化に成功できない可能性があり、これは顕著な 業務に重大な損害を与える。
注目すべきbr候補製品の単独使用または他の承認された医薬品または研究用新薬との併用時に副作用が生じる可能性があり、これは、さらなる開発または規制承認を延期または阻止するか、または承認された後に使用を制限する可能性がある。
重要な候補製品による不良br}副作用は、顕著なまたは規制機関の中断、延期、または臨床試験 の一時停止を招く可能性があり、より厳しいラベルをもたらす可能性があり、またはFDAまたは他の規制機関の規制承認を延期または拒否する可能性がある。 の顕著な試験結果は、副作用の重症度および流行率が高く、受け入れられない可能性がある。この場合、NOTINGの 試験は一時停止または終了される可能性があり、FDAまたは同様の外国規制機関は、NOTIGNの任意またはすべての目標適応の候補製品の開発を停止または拒否するようにNOTINGに命令することができる。このような副作用はまた、患者の募集、入選患者が試験を完成する能力、或いは潜在的な製品責任クレームを招く可能性がある。上記事件のいずれも、顕著な業務、財務状況、運営結果、および見通しに重大な悪影響を及ぼす可能性がある。
| 50 |
また, はその性質に応じた臨床試験は潜在患者集団のサンプルでのみ候補製品を試験した。患者数が限られているため、このような試験で暴露される時間も限られており、より多くの患者が候補製品に接触する前に、候補製品の稀かつ深刻な副作用を発見できない可能性がある。
もし注目すべき候補製品が上場承認され、深刻な、予期せぬ或いは見たくない副作用を招く場合、多くの潜在的な重大な負の結果を招く可能性がある
| ● | 規制当局は、製品の承認を撤回、一時停止または制限するか、または修正されたリスク評価および緩和策の形態で製品の流通に制限を加えることができる | |
| ● | 規制当局は警告や禁忌症のようなラベル宣言の追加を要求するかもしれない | |
| ● | 注目すべきは、製品の投与方法を変更したり、追加の臨床試験や発売後のモニタリングを行う必要がある可能性がある | |
| ● | 注意すべきは、brが起訴され、患者への傷害に責任を負う可能性があることである | |
| ● | 注目すべき 名声は影響を受ける可能性がある. |
これらの事件のいずれも、影響を受けた製品に対する市場の受容度を著しく獲得または維持することを阻止する可能性があり、将来の製品の商業化コストを大幅に増加させ、これらの製品の商業化によって収入を生成する能力を著しく低下させる可能性がある。
たとえ が必要な臨床前研究と臨床試験を著しく完成したとしても、候補製品を商業化する監督管理許可を著しく得るかどうかは有意に予測できないため、未来の製品がいかなる収入を産生する時間 を著しく予測することはできない。
注目すべきは、適切な規制機関(例えばFDA)が候補製品 を審査して承認する前に、製品を商業化することができないことである。監督管理機関は審査過程を適時に完成できない可能性があり、あるいは注目すべきは、多くの理由で、監督部門の承認を得ることができない可能性があることである
| ● | 規制当局は注意すべき臨床試験の設計や実施に同意しない | |
| ● | このような権威機関は臨床前研究あるいは臨床試験データの有名な解釈に同意しないかもしれない | |
| ● | このような当局は、臨床機関または医療基準が米国とは異なる国で行われる可能性のある試験の臨床データを受け入れない可能性がある | |
| ● | 明らかな臨床試験の不利または不明確な結果、または結果はFDAまたは類似の外国規制機関によって承認された統計的意味レベル に適合しない可能性がある | |
| ● | 重篤なbrおよび予期しない薬物関連副作用、参加者または個人は、注目すべき候補製品に類似した薬剤 ; | |
| ● | 臨床試験で研究されている集団は、注目すべき承認を求めるすべての集団における安全性を確保するために、十分に広く、または十分な代表性を有している可能性がある | |
| ● | 注目すべきは、候補製品の臨床的および他の利益がその安全リスクよりも大きいことを証明できない可能性があることである | |
| ● | このようなbr当局は、機密協定の提出または他の提出を支援するために、重要な候補製品の臨床試験から収集されたデータが受け入れ可能であるか、または米国または他の場所の規制承認を得ることを支持することに同意しない可能性があり、そのような 当局は、追加の臨床前研究または臨床試験を要求する可能性がある |
| 51 |
| ● | これらの機関は、重要な候補製品の配合、ラベル、および/または仕様に同意しない可能性がある | |
| ● | このようなbr当局は、重要な臨床および商業用品契約に関連する重要な第三者製造業者の製造プロセスまたは施設に欠陥があることを発見するか、または政策を承認することができるかもしれない | |
| ● | このような機関の法規 は重大な変化が発生する可能性があり、重要或いは任意の重要な未来の協力者の 臨床データは承認に十分ではない。 |
FDA諮問委員会が承認を制限することや提案が承認されないことを提案した場合、追加の 遅延を招く可能性がある。また、製品開発、臨床試験、審査過程において、将来の立法または行政行動における追加の政府法規、または規制機関の政策の変化により、遅延や拒否が生じる可能性があることに注意されたい。
もし注目すべき候補製品が監督管理部門の許可を得たら、注目すべき製品は依然として広範な監督管理要求 に直面し、注目すべき製品は未来の発展と監督管理困難に直面する可能性がある。
NOTIGNが米国で規制承認された場合であっても、FDAは、NOTIGNの候補製品の指定された用途またはマーケティングに重大な制限を加えるか、またはコストの高い可能性のある承認後の研究または発売後の監視に継続的な要求を加える可能性がある。FDAはまた、注意すべき候補製品を承認する条件としてリスク評価および緩和戦略を要求する可能性があり、その中には、制限された分配方法、患者登録、および他のリスク最小化ツールのような薬品使用ガイドライン、医師コミュニケーション計画、または安全使用を保証する追加の要素の要件が含まれる可能性がある。さらに、製品の生産プロセス、パッケージ、流通、有害事象報告、ラベル、広告、販売促進および記録保存は、広く持続的なFDA法規要件、および他の適用可能な連邦および州法律の制約を受けるであろう。これらの要求 は、不良事象および他の上場後の情報および報告、登録、およびcGMP法規の継続的な遵守を含む。承認されたセキュリティ協定の保持者はまた、新しいまたは追加の申請を提出し、承認された製品、製品ラベル、または製造プロセスをいくつかの変更することができるために、FDAの承認を得なければならない。予期しない程度または頻度の不良反応、または製品生産施設の問題など、以前に未知の製品問題、または製品生産施設の問題が顕著に発見された場合、監督機関は、br}のリコールを要求するか、または市場から製品を撤回するか、または生産を一時停止することを含む、製品または製造施設に制限を加えることができる。
任意の注目すべき候補製品を承認した後、適用される監督管理要求を遵守できないことに注意すれば、監督管理機関は:
| ● | 注意すべき行為は違法だと警告状を出した | |
| ● | 禁令を求めたり、民事または刑事罰または罰金を科したりする | |
| ● | Brを一時停止するか、規制承認を撤回するか | |
| ● | 現在行われている臨床試験を一時停止します | |
| ● | 合併企業が提出した係属中の守秘協定または秘密保持協定の承認を拒否する補足合意; | |
| ● | 製品の差し押さえや製品のリコールを要求する | |
| ● | Brを拒否して、有名な会社が政府契約を含む供給契約を締結することを許可する。 |
政府は違法行為の疑いのあるどの調査にも多くの時間と資源をかけて対応する必要がある可能性があり,また は負の宣伝が生じる可能性がある.上記のいずれかのイベントまたは処罰が発生した場合、注目すべき将来の製品の商業化能力(承認された場合)を抑制し、収入を生成する可能性がある。
| 52 |
重要なbr}は、特定の研究計画または候補製品を追求するために重要な財務および人的資源を使用する可能性があり、 を利用することなく、より利益的または成功可能性の高い計画または候補製品を利用することができる。
莫大な財力と人的資源が限られているため、顕著には戦略決定を行わなければならず、どの製品 候補製品を開発するかを決定し、他の候補製品または後により大きな商業潜在力を有することが証明された他の兆候とのビジネスチャンスを放棄または延期する可能性がある。注目すべき資源配分決定は、実行可能な商業製品や利益のある市場機会を利用できないことに注目される可能性がある。注目すべき研究開発計画と特定の適応候補製品の支出はいかなる商業的に実行可能な製品も発生しない可能性がある。特定の候補製品の商業的潜在力または目標市場が正確に評価されていない場合、戦略連合、許可または他の印税配置によって候補製品に貴重な権利を放棄する可能性があることに注意されたい。この場合、候補製品の独占開発および商業化権利をより有利に保持する可能性があるか、または治療領域の候補製品 に内部資源を割り当てる可能性があり、この分野での協力計画を達成することがより有利になる可能性があることに注意されたい。
顕著な は他のバイオテクノロジーと製薬会社からの激しい競争に直面しており,顕著に有効な競争ができなければ,その経営業績は影響を受ける。
生物製薬業界の特徴は競争が激しく、革新が迅速だということだ。注目すべき競争相手は、他の化合物や薬物を開発することができ、類似またはより良い効果を達成することができるかもしれない。有名な潜在競争相手は大型国際製薬会社、老舗生物技術会社、専門製薬会社と大学及びその他の研究機関を含む。多くの注目すべき競争相手は、より多くの研究開発者と経験豊富なマーケティングと製造組織、および成熟した販売チームのようなより多くの財務、技術、および他の資源を持っている。規模が小さいか、あるいは早い段階にある会社も重要な競争相手であることが証明される可能性があり、特にそれらが疾患を治療する新しい方法を開発した場合、注目すべき候補製品も治療に集中していることが示されている。古い製薬会社はまた、新しい治療法の発見と開発を加速させたり、注目すべき開発候補製品を時代遅れにする可能性のある新しい療法を許可するために大量の資金を投入する可能性がある。バイオテクノロジーと製薬業界の合併と買収は、注目すべき競争相手により多くの資源を集中させる可能性がある。技術ビジネス適用性の進歩やこれらの業界に投資する資本の増加により、競争はさらに激化する可能性がある。注目すべき競争相手は、単独であっても、パートナーと協力しても、注目すべき候補製品よりも効率的で、安全で、商業化しやすい、またはコストの低い薬物または生物学的製品の開発、または注目すべき技術および製品開発に必要なノウハウまたは信頼性の高い特許保護の開発、取得または独占に成功する可能性がある。注目すべき候補製品の開発および商業成功に影響を与える重要な競争要素brは、治療効果、安全性、耐性、信頼性、使用利便性、価格および精算であることに注意されたい。
注意すべき薬品が規制部門の承認を得ても,注目すべき競争相手の製品の供給や価格は需要を制限する可能性があり,注目すべき価格は注目すべき候補製品に課金することができる。価格競争または医師が既存の治療方法から注目すべき候補製品に切り替えたくない場合、または医師が他の新薬または生物学的製品に変更したり、限られた場合に使用するために注目すべき候補製品を保存することを選択したりする場合、brの注目すべき候補製品の業務計画を著しく実施できない可能性がある。
重要な候補製品の商業成功は、これらの候補製品に対する医学界(医師、患者、および医療支払人を含む)の受け入れの程度に依存する。
候補製品の市場受容度は、多くの要素に依存するであろう
| ● | 他の製品と比較して臨床安全性と有効性を示した |
| 53 |
| ● | 医師、患者、医療支払者は比較的便利で、管理しやすく、受け入れやすい | |
| ● | どんな有害事象の罹患率や重症度も | |
| ● | このような製品のFDA承認ラベルに含まれる制限または警告; | |
| ● | 代替治療の可用性 ; | |
| ● | 価格設定と費用効果; | |
| ● | 注目すべきまたは任意の注目すべき協力者の販売およびマーケティング戦略の有効性; | |
| ● | 注目すべきは、 が病院または支払人の処方承認を得る能力である | |
| ● | 注目すべきは、十分な第三者保険および適切な補償を得ることができ、維持することができることである | |
| ● | 患者は第三者保険なしで費用を自己負担する意思がある。 |
製品が承認されたが、医師、患者、および医療支払者の十分な受容度に達していない場合、著しく は、製品から十分な収入を生成することができず、実質的に利益を達成または維持できない可能性がある。このような激化した競争は、価格設定の低減および割引の向上を要求する圧力がますます大きくなっていることが注目すべき製品の商業化であるため、現在および将来の候補製品の任意の将来の潜在的収入を減少させる可能性がある。
が販売およびマーケティング能力を著しく確立できない場合、または顕著な 候補製品をマーケティングおよび販売するために第三者と合意できない場合、有意にいかなる収入も生成できない可能性がある。
注意すべきことは、brは現在、薬品を販売、マーケティング、流通する組織がなく、そのような組織の構築と維持のコストがこのような費用効果を超える可能性があることである。承認され得る任意の製品をマーケティングするためには、顕著な販売、マーケティング、管理、および他の非技術的能力を確立するか、または第三者 と手配してこれらのサービスを実行しなければならない。将来の計画については、アライアンスパートナーに完全に依存して販売や マーケティングを行う可能性があることに注意されたい。さらに、米国以外の市場または顕著な資源範囲外の他の大規模市場を含む、他の候補製品を商業化するために、第三者と戦略同盟を達成する可能性があることに注意されたい。注目に値する は販売組織を構築しようとしているが、注目すべきであれば、承認を得ることができれば、米国で任意の候補製品 を販売することができるが、商業化要求が注目すべき利用可能な資源を超える場合、米国の現在および将来の候補製品 のための戦略同盟を確立することも考慮されるであろう。これはこれらの製品の販売収入 を減少させるだろう。
承認された場合、任意の将来の戦略同盟パートナーは、注目すべき候補製品の商業化に十分な資源を投入しない可能性があり、そうでなければ、著しく制御できない要因により製品商業化に失敗する可能性がある。注意する価値があればはいそれ自体のマーケティングおよび販売チームに属さない候補製品を医療専門家および地理的地域(米国を含む)の医療専門家およびbrに販売することができるように、有効な連合を確立することができない、または 重要な将来の戦略連盟パートナーが承認される可能性のある候補製品を商業化することに成功しない場合、重要な製品販売収入の能力に悪影響を及ぼすであろう。
が著しく独立していない場合、または第三者と十分な販売、マーケティング、および流通能力を確立できない場合、 は著しく十分な製品収入を生成できない可能性があり、利益を得ることができない可能性がある。注目すべきは、多くの会社と競争し、これらの会社は現在、広範で資金の豊富なマーケティングと販売業務を持っているということだ。マーケティングおよび販売機能を実行するための内部チームまたは第三者のサポートがない場合、これらのより成熟した会社との競争に成功できない可能性がある。
| 54 |
Brが著しく承認されて任意の承認された製品を米国以外の地域で商業化すれば、国際業務に関連する様々なリスクが顕著な業務に実質的な悪影響を及ぼす可能性がある。
許可された製品を米国以外の場所で商業化することに注意すべきであれば、会社は国際業務関係の構築に関連する追加のリスクに直面することが予想される
| ● | 外国の薬品審査に対する異なる監督管理要求 | |
| ● | 異なる支払人精算制度、政府支払人または患者自己支払い制度、価格制御 | |
| ● | 知的財産権の保護を減らすことです | |
| ● | 関税、貿易障壁、規制要求の意外な変化 | |
| ● | インフレ、特に外国経済と市場の政治的不安定を含む経済的疲弊 | |
| ● | 外国に住んだり旅行したりする従業員は税収、雇用、移民、労働法を遵守する | |
| ● | 源泉徴収賃金税を含む外国税 | |
| ● | 外国通貨の変動は、運営費用の増加と収入の減少、他の国での業務展開に付随する他の義務を招く可能性がある | |
| ● | 労働力の労働騒乱はアメリカよりも一般的な国の不確実性である | |
| ● | 海外の原材料供給や製造能力に影響を与えるいかなる事件による不足 | |
| ● | 業務brは、地政学的行動(戦争やテロを含む)や自然災害(地震、台風、洪水、火災を含む)による中断。 |
承認されれば,重要な候補製品は保証範囲のbrと十分な精算が得られない可能性があり,br}の重要な製品の利益販売が困難になる可能性がある。
市場 が著しく開発した任意の候補製品の受容度や販売は保険や精算政策に依存し,将来の医療改革措置の影響を受ける可能性がある。政府当局と第三者支払人、例えば個人健康保険会社、政府支払人、健康維持組織は、彼らがどの薬のために支払い、精算レベルを確立するかを決定する。注目すべきは、私たちは現在と未来の候補製品がカバー範囲と十分な精算を得ることができることを保証することができないということだ。アメリカでは、連邦医療保険と医療補助サービスセンター(“CMS”)はアメリカ衛生と公衆サービス部(“HHS”)内の一つの機関であり、連邦医療保険の下で新薬の保険を受けるかどうか及び新薬がどの程度精算されるかを決定する。個人支払者はCMSが策定したカバー範囲 精算政策に大きく従う傾向がある。CMSが新製品候補製品の精算についてどのように決定するかを予測することは困難である。精算金額不足は、注目すべき未来の製品への需要を減らしたり、その価格を下げたりする可能性があります。また、1人の支払人がある製品に保険を提供することを決定することは、他の支払人もその製品に保険 を提供する保証はありません。精算が得られない場合や限定精算のみでは,開発に成功し,承認される可能性のある候補製品 を商業化できない可能性があることに注意されたい。したがって、製品の市場への投入に著しく成功しても、 は医学的に必要または費用効果があるとは考えられない可能性があり、任意の製品の精算金額は、競争に基づいてその製品を著しく販売するのに十分ではないかもしれない。
米国や一部の外国司法管轄地域では、医療システムを変更するための立法と規制提案が多くあり、これは製品販売収益性に顕著な影響を与える可能性がある。これらの立法および/または法規の変化は承認後の薬品精算に否定的な影響を及ぼすかもしれない。大量の後発薬の提供も注目すべき将来の製品の精算可能性を大幅に低下させる可能性がある。注意すべきことは,管理型ヘルスケアの傾向,健康維持組織の影響力が大きくなっていること,追加的な立法変化により, のいずれの注目すべき開発製品の販売も定価圧力に直面することである。全体的に,特に処方薬の医療コスト下り圧力は将来的に増加し続けることが期待される。例えば、患者または医療提供者に精算する政府および個人支払者は、医薬品br製品の価格を低減するために、より大きな前期割引、追加リベート、および他の特典を求めるようになっている。注目すべき未来の製品の精算範囲を得ることに成功し、維持できなかった場合、あるいはその過程で重大な遅延が発生した場合、注目すべき未来の製品は市場の受け入れを得ることが困難になり、注目すべき業務は損害を受ける。
| 55 |
また、ある非アメリカ司法管轄区では、薬物の提案定価は必ず承認されなければならず、合法的に発売されることができる。各国の薬品定価に対する要求は千差万別である.例えば,EUはその加盟国にオプションを提供し,その国の医療保険システムが精算を提供する医療製品の範囲を制限し,人が使用する医療製品の価格を制御している。加盟国は医薬製品の具体的な価格を承認することができ、医薬製品を市場に投入する会社の収益力を直接或いは間接的に制御する制度を採用することもできる。薬品に対して価格制御或いは精算制限を実行することを保証できないいかなる国/地域でも、任意の注目すべき製品に対する優遇的な精算と定価手配が許可される。歴史的に見ると、EUで発売された製品はアメリカの価格構造に従わず、通常価格ははるかに低くなることが多い。
環境,健康,安全法律法規に注意が従わなかった場合,注意は罰金や罰 に処せられたり,注意の業務成功に重大な悪影響を与える可能性のあるコストが生じる可能性がある.
注意すべきことは、brは多くの環境、健康と安全法律法規の制約を受けており、実験室プログラムおよび危険材料と廃棄物の処理、使用、貯蔵、処理と処理を管理する法規を含む。注目すべき業務は、化学品および生物学的材料を含む危険かつ可燃性材料の使用に関するものである。注目すべき業務は危険な廃棄物製品も発生する。注意すべきは、通常、第三者とこれらの材料や廃棄物を処理する契約を締結することである。注目すべきは、このような材料による汚染や傷害のリスクを除去できないということだ。危険な材料の使用による汚染または傷害の場合、NOTING は、それによって生じる任意の損害に責任を負う可能性があり、任意の責任がNOTIGNのリソース範囲を超える可能性がある。民事や刑事罰金や処罰に関連した巨額の費用が発生する可能性もあることに注意されたい。
危険材料の使用または他の仕事に関連する傷害によって怪我をした注目すべき従業員のコストおよび支出をカバーするために労働者補償保険を維持することに注意すべきであるが、この保険は潜在的な責任に十分な保険を提供することができない可能性がある。また,現在または将来の環境,健康,安全法律法規を遵守するためには,顕著なリスクが大きなコストを生じる可能性がある。これらの現行または未来の法律法規は注目すべき研究、開発、または生産努力を損なう可能性がある。これらの法律法規を遵守しないことはまた、巨額の罰金、br処罰、または他の制裁を招く可能性がある。
注目すべき第三者依存に関するリスク
注意すべき は第三者に依存して注目すべき化合物配合、研究、臨床前研究と臨床試験のいくつかの方面を行い、 これらの第三者の表現は満足できない可能性があり、締め切り前にこのような調合、研究 或いはテストを完成できなかったことを含む。
注意すべきbr}は,注目すべき薬物発見活動,化合物調製研究,臨床前 研究と候補製品の臨床試験のすべての面を独立して行うことは望ましくない。注目すべきは、現在依存しており、引き続き第三者による注目すべき臨床前研究、臨床研究と調合開発のある方面に依存すると予想されている。
これらの第三者のいずれか一方はNOTICEとの契約をいつでも終了することができる.著しい代替手配が必要であれば, は顕著な製品開発活動を延期する。注目すべき研究開発活動のこれらの第三者への依存は、これらの活動に対する注意すべき制御を減少させるが、注意すべき責任を軽減することはない。 例えば、自分で開発·商業化に注意すべき候補製品については、引き続き を担当してINDを有効にする研究や臨床試験を試験的な研究計画と プロトコルに従って行うことに注意すべきである。
もし これらの第三者がその契約職責を成功裏に履行できず、予期される最終期限内に重要な研究 を完成または行うことができない場合、法規要件または重要な研究計画と合意に基づいて、重要なものは達成できない、 または必要な臨床前研究の完成を遅延させて、重要な選択が可能な製品候補INDを に提出するために、できない、または延期される可能性があり、これらの候補製品の開発および商業化に成功する。
| 56 |
注目すべきbrは、第三者メーカーが注目すべき臨床前候補製品の供給を生産することに依存し、注目すべきは、将来の候補製品の臨床供給に依存することであり、これらの候補製品は、臨床試験および任意の承認された候補製品の商業供給を著しく推進することになる。
注目すべき候補者が自分で製品 を製造すれば受けないリスクを含む、第三者製造商会に依存するリスク
| ● | 製品の規格や品質の要求を一貫して満たすことはできない | |
| ● | 十分な製造能力を遅延または調達できないか、または拡大することができない | |
| ● | 製造 と製造規模拡大に関する製品品質の問題; | |
| ● | 拡張に必要な新しい設備および施設のコスト および検証; | |
| ● | CGMPや類似の海外基準を満たしていません | |
| ● | 製造または供給協定を商業的に合理的な条件下で第三者と交渉することはできない | |
| ● | 第3者との製造プロトコルを終了または更新しない方法または時間は、著しい損失または損害をもたらす | |
| ● | 限られた数の原料源に依存し、場合によっては単一源の原材料に依存するので、これらの製品コンポーネントの十分な供給が確保できない場合、現在および将来の候補製品 をタイムリーに、十分な数で、または許容可能な条件下で製造および販売することができないことに注意されたい | |
| ● | 現在、単一源サプライヤーから調達した任意の原材料は合格した予備サプライヤーが不足している | |
| ● | 注目すべき第三者製造業者またはサプライヤーの運営は、製造業者またはサプライヤーの倒産を含む注目すべき業務または運営とは無関係な条件によって妨害される可能性がある | |
| ● | キャリア 制御できない中断やコスト増加;および | |
| ● | 所定のストレージ条件で製品をタイムリーに納品できませんでした。 |
これらの事件のいずれも、臨床研究の遅延または規制部門の承認を得ることができないか、または承認された場合、Designantが将来の製品の商業化に成功する能力にも影響を及ぼす可能性がある。いくつかの事件は、禁止、リコール、差し押さえ、または生産の完全な一時停止、または部分的な一時停止を含むFDAの行動の基礎となる可能性がある。
注目すべきは, 候補製品の薬物物質は限られた供給源に依存し,サプライチェーンにおけるどの中断も これらの候補製品の開発と商業化遅延を招く可能性があることである。
有名なbrはすでに限られた数量のサプライヤーと製造関係を構築し、原材料を生産し、薬物物質は を用いて注目すべき候補製品を作成した。このようなサプライヤーが注目すべき候補製品で原材料を生産する可能性は限られている可能性がある。さらに、そのようなプロセスが供給者に属さない場合、または公共分野内にある場合、各供給者は、そのような構成要素を製造するためにライセンスを必要とする場合がある。どの上場承認の一部としても、メーカーとその流れは商業化前にFDAの資格を取得しなければならない。承認された仕入先の供給が中断されると、商業供給に大きな中断が生じる可能性がある。代替サプライヤーは機密協定によって資格を補充する必要があり、これはさらなる遅延を招く可能性がある。新しいサプライヤーに依存して商業生産を行う場合、FDAや米国以外の他の規制機関も追加的な研究を行う必要がある可能性がある。サプライヤーの交換は大量のコストに関連する可能性があり、注目すべき臨床と商業スケジュールの遅延を招く可能性がある。
| 57 |
これらの要因は、臨床試験の遅延、監督提出、必要な承認または重要な候補製品の商業化を招く可能性があり、より高いコストを著しく発生させ、その製品の商業化に著しく成功することを阻止する可能性がある。また、顕著なサプライヤーが必要な商業数量の活性医薬成分を商業的に合理的な価格でタイムリーに渡すことができず、実質的に同じコストでタイムリーに生産できる1つ以上の代替サプライヤーを著しく得ることができない場合、顕著な臨床試験が延期される可能性があり、または潜在的なbr}収入を著しく損失する可能性がある。
製造 は,製品や規制承認コストを増加させたり,商業化を遅延させたりする可能性があるという問題が生じる可能性がある.
候補製品を製造し、必要な安定性テスト、製品、包装、設備とプロセスに関連する問題は の改善或いは解決が必要である可能性があり、任意の臨床試験を継続し、監督部門の商業マーケティングに対する許可を得ることができる。注目すべきbrは、明らかな不純物が発見される可能性があり、これは、規制機関の審査強化、臨床計画および規制承認の遅延、注目すべき運営費用の増加、または候補製品または任意の承認された製品の承認を得ることができないか、または維持することができない可能性がある。
注意すべき は第三者による注意すべき臨床試験,監督,監督に依存しようとしており,これらの第三者の表現が満足できなければ,注目すべき業務を損なう可能性がある。
注意すべきbr}はCROと臨床試験地点に依存して適切かつタイムリーに注意すべき臨床試験を確保する予定である。有名な は彼らの活動を管理するプロトコルがあるが,有名なものは彼らの実際の表現への影響は限られている.注目すべきCRO活動の のいくつかの態様のみを制御することに注意されたい。しかし、注意すべき責任を確保するすべての臨床試験は適用された合意、法律、法規と科学標準に従って行われ、注目すべきCROへの依存は注意すべき監督管理責任を免除しない。
有名なbrと有名なCROはFDAまたは他の監督機関のGCPを遵守し、INDを有効にする研究と臨床試験の結果を行い、記録し、報告して、データと報告の結果が信頼性と正確な であることを保証し、未来の臨床試験参加者の権利、完全性と機密性を保護することを要求される。FDAと非米国規制機関は,定期検査試験スポンサー,主要研究者,臨床試験地点によりこれらのGCPを実行している。著名なbrまたは著名な将来のCROが適用されるGCPを遵守できない場合、重要な臨床試験で生成された臨床データは信頼できないと考えられる可能性があり、FDAまたは適用される非米国規制機関は、関連する司法管轄区域の任意のマーケティング申請を承認する前に追加の臨床試験を行うことを著しく要求する可能性がある。検査後,FDAあるいは適用された非米国規制機関 はNOTIGNの将来の臨床試験がGCPに適合していないことを決定する可能性がある。また,将来の臨床試験 は潜在的な薬物製品の安全性と有効性を評価するのに十分な試験対象が必要となることに注意されたい。したがって、注目すべき将来のCROがこれらの規定を遵守できない場合、または十分な数の患者を募集できない場合、このような臨床試験を繰り返す必要がある可能性があり、これは規制承認過程を遅らせることになる。
注目すべき未来のCROは注目すべき従業員ではなく、彼らが十分な時間と資源を注目すべき未来の臨床と非臨床プロジェクトに投入するかどうかを制御できないことに注意されたい。これらのCROはまた、重要な競争相手を含む他の商業実体と関係がある可能性があり、彼らはまた、これらのエンティティのための臨床試験を行っているか、または重要な競争地位を損なう可能性のある他の薬物開発活動 を行っている可能性がある。注目すべき将来CROが契約 の職責または義務の履行に成功せず、期待された期限内に達成できなかった場合、または彼らが得た臨床データの品質または正確性が注目すべき臨床方案または法規要件または任意の他の原因を遵守できなかった場合、影響を受ける場合、注意すべきbr}臨床試験は延長、延期または終了される可能性があり、また注目すべき候補製品は監督部門の承認を得られない可能性があり、あるいはそれを商業化することに成功できない可能性がある。したがって、注目すべき財務業績とこのような 製品および注目すべき開発の任意の候補製品のビジネス見通しは損なわれ、注目すべきコストが増加する可能性があり、注目すべき創造能力が延期される可能性がある。
| 58 |
注目すべきは、他の第三者に依存して、可能な臨床試験を行うことができる任意の医薬製品を記憶および配布することを意図していることである。注目すべきディーラーの任意のパフォーマンス の任意の失敗は、注目すべき候補製品の臨床開発またはマーケティング承認を延期する可能性があり、または承認された場合、注目すべき製品の商業化を延期し、追加の損失をもたらし、注目すべき潜在的製品収入 を奪う可能性がある。
注目すべき知的財産権に関するリスク
もし が顕著に現在および未来の製品および候補製品に関連する知的財産権を獲得または保護できない場合、顕著な市場で効果的な競争ができない可能性がある。
注目すべき成功は、注目すべき候補製品のために他人の知的財産権を取得し、維持するためのbrライセンス、注目すべき候補製品を開発および製造するための方法、および注目すべき候補製品を使用する患者を治療する方法、および注目すべきビジネス秘密を保護し、第三者が注目すべき独自の権利を侵害することを防止し、他人の固有の権利を侵害することなく運営する能力を含む、注目すべき特許および他の形態の知的財産権を取得および維持する能力にある程度依存する。バイオテクノロジーや製薬分野の特許の強度は複雑な法律や科学的問題に関連しており,不確実である可能性がある。おそらく中に入るかもしれない特許出願を有することは、米国または他の国/地域をカバーする製品の特許を生成できない可能性があることに留意されたい。有名な特許および特許出願に関連するすべての潜在的に関連する既存技術が発見されたことは保証されず、このような従来技術 は、特許を無効にするか、または係属中の特許出願に基づいて特許が発行されることを阻止する可能性がある。特許が確実に発行されても、 第三者は、その有効性、実行可能性、または範囲に疑問を提起する可能性があり、これは、このような特許の範囲の縮小または無効をもたらす可能性がある。また、疑問視されていなくても、注目すべき特許および特許出願は、注目すべき知的財産権を十分に保護することができないか、または注目すべき権利要件をめぐる他の人の設計を阻止する可能性がある。
重要な計画または候補製品に関する特許出願が保有または許可されていない場合、またはそれらの保護の広さまたは強度が脅かされている場合、会社と重要な協力開発候補製品 を阻止し、重要な将来の製品の商業化能力を脅かす可能性がある。私たちは、どのような特許が発行されるかを保証することはできません。 があれば、または発行された任意の特許が無効であるかどうか、実行不可能であるかどうか、または第三者の脅威にさらされるかどうかを保証することはできません。 特許は、ライセンス後の挑戦、 の再審査、または米国特許商標局(“USPTO”)または外国特許庁に異議を提起することを含む1つまたは複数の行政手続きによって挑戦される可能性があります。成功した特許または任意の他の特許に挑戦する行為は、開発可能な任意の候補製品の商業化に成功するために必要な権利を奪う可能性がある。
米国および他のほとんどの国/地域の特許出願は、提出後しばらくは秘密であり、公開前にも秘密である特許出願もあるため、注目すべきは、候補製品に関連する特許出願を最初に提出することに注目すべきではないことに留意されたい。また、場合によっては、1つ以上の第三者が米国で特許出願を提出し、同じ主題を要求している場合には、どの出願人 がその主題の特許を取得する権利があるかを決定するために、干渉と呼ばれる行政訴訟手続きを開始することができる。このような第三者によって開始されるか、または注目すべき干渉手順が必要である可能性があり、注目すべき特許または特許出願に対する発明の優先度、または注目すべきライセンス者の特許または特許出願に対する優先度を決定するために必要である可能性がある。不利な結果は、関連技術の使用を著しく停止する必要があるか、または勝利者から許可権 を著しく得る必要がある可能性がある。もし勝利者が完全でないか、または商業的に合理的な条項で注目すべきライセンスを提供する場合、注意すべき業務は損害を受ける可能性がある。このような訴訟では特許または特許出願の弁護が成功しない可能性があり、成功しても巨額のコストを招く可能性があり、注目すべき管理職や他の従業員の注意を分散させる可能性があることに注意されたい。
しかも、特許のライフサイクルは限られている。アメリカでは、特許の自然失効期間は一般的に申請後20年だ。しかし、特許の有効期間は様々な延期がある可能性があり、それが提供する保護は限られている。製品の特許有効期限が切れると,後発薬からの競争に直面する可能性がある。また,規制承認に著しく遅れが生じた場合,特許保護された候補製品を著しく市場に出すことができる時間が短縮される可能性がある.
| 59 |
特許によって提供される保護に加えて、特許を実施することが困難なプロセス、および特許がカバーされていないノウハウ、情報または技術に関する薬物発見および開発プロセスの他の任意の要素を含む商業秘密保護および秘密保護協定に依存して特許を出願できないノウハウを保護することに留意されたい。br}注目すべき各従業員は、従業員発明協定を介して彼らの発明を注目すべきものに譲渡することに同意しているが、注目すべきすべての従業員、コンサルタント、コンサルタント、および注目すべきノウハウにアクセスできる第三者は、 秘密保持プロトコルの締結には情報や技術が必要であるが,このようなすべての プロトコルが正式に実行されていることを保証することは重要ではなく,重要なビジネス秘密や他の機密独自情報を漏洩しないことや,競合他社が重要なビジネス秘密を他の方法で取得したり,実質的に同等の 情報や技術を独立して開発したりしないことが重要である.さらに、他の人は、有名な商業秘密および固有情報を独立して発見することができるかもしれない。例えば、FDAは、その透明性イニシアティブの一部として、有名な商業秘密または他の固有情報と考えられる情報を含む、より多くの情報を定期的に公開するかどうかを検討しており、FDAの開示政策が将来どのように変化する可能性があるかは不明である(もしあれば)。
また,ある国の法律の専有権に対する保護の程度や方式は米国の法律とは異なる。そのため、米国と海外で注目すべき知的財産権を保護し、守る上で重大な問題に直面する可能性があることに注意されたい。注目すべきは、注目すべき技術に関連する非特許知的財産権の第三者への開示を阻止することができず、注目すべき任意のこのような強制的に実行可能な商業秘密によって保護される保証がない場合、注目すべき市場で競争優位性を確立または維持することができない可能性があり、これは、顕著な業務、運営結果、および財務状態に重大な悪影響を及ぼす可能性があることに留意されたい。
第三者 知的財産権侵害クレームは、注目すべき開発と商業化努力を阻害または延期する可能性がある。
注目すべき商業的成功は、第三者の特許および独自の権利の侵害を回避することにある程度依存する。アメリカ国内と国外で大量の訴訟があり、生物技術と製薬業界の特許と他の知的財産権 に関連し、特許侵害訴訟を含む。開発候補特許が求められている有名な分野には、第三者が所有する米国や外国から発行された特許 や未解決の特許出願が多く存在する。バイオテクノロジーや製薬業界の拡張やより多くの特許の発行に伴い、注目すべき製品 候補製品が第三者特許権を侵害される可能性があるというリスクが増加している。
サード·パーティ は、明らかに許可されていない場合にそのノウハウが使用されていると主張することができる。重要候補製品の使用または製造に関連する第三者特許または 特許出願が存在する可能性があり、これらの特許請求項は、材料、配合、製造方法または処理方法である。特許出願が発行されるまでに数年かかる可能性があるため、現在処理されている特許出願 が存在する可能性があり、これらの出願は、今後注目すべき候補製品が侵害される可能性のある特許をもたらす可能性がある。さらに、第三者は将来的にbr件の特許を取得する可能性があり、注目すべき技術の使用がこれらの特許を侵害していると主張する可能性がある。管轄権のある裁判所 が重要な任意の候補製品の製造プロセス、製造中に形成された任意の分子または任意の最終製品自体をカバーするために任意の第三者特許を保有している場合、任意のそのような特許の所有者は、重要な適用特許下の許可を得ない限り、またはそのような特許 が満了するまで、重要な候補製品を商業化する能力を阻止することができるかもしれない。同様に、管轄権のある裁判所が、NOTINGの処方、製造プロセス、または使用方法の様々な態様をカバーするために任意の第三者特許を保有している場合、任意のそのような特許の所有者は、NOTINGが許可またはその特許の満了を得ない限り、NOTING の開発および商業化に適用される候補製品の能力を阻止することができるかもしれない。 は、いずれの場合も、そのような許可は、商業的に合理的な条項または全く得られない可能性がある。
注目すべき製品に対してクレームを出す当事者は、禁止または他の衡平法救済を得ることができ、これは、注目すべき1つまたは複数の候補製品のさらなる開発および商業化を効果的に阻止することができるかもしれない。これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、顕著な業務の管理層または従業員資源を大量に移転させる。注意すべき侵害行為のクレームに成功した場合、意図的に侵害された3倍の損害賠償および弁護士費、特許権使用料の支払い、注目すべき侵害製品の再設計、または第三者から1つまたは複数の許可を得ることを含む大量の損害賠償を支払わなければならない可能性があり、または大量の時間およびお金の支出が必要である可能性がある。
| 60 |
がライセンスを取得できなかった場合、またはこれらのプロトコルにおける注意すべき義務を履行することに注意されたい場合、これらのプロトコルによれば、第三者から知的財産権を取得したり、他の理由で注目すべき 許可者との業務関係を中断したりすることに注意すべきであり、注目すべき業務に重要な許可権を失う可能性がある。
注意すべき は知的財産権ライセンスプロトコルの一方であり,これらのプロトコルは注目すべき業務に重要であり,将来的にはより多くの ライセンスプロトコルが締結されることが予想される.注目すべき既存ライセンスプロトコルは重要会社に様々な義務を課し,注目すべき会社は将来のライセンス契約 に様々な義務を課すことが予想される.
注目すべき は、注目すべき研究を進めたり、注目すべき候補製品の商業化 を許可するために、第三者からライセンスを取得する必要がある可能性があり、時々そうすることに注意されたい。これらのライセンスは、合理的なコストまたは合理的なbr条項で取得できない可能性があることに留意されたい。この場合、注目すべき1つまたは複数の注目すべき候補製品 をさらに開発および商業化することはできず、これは注目すべきビジネスを深刻に損なう可能性があることに留意されたい。注目すべき未来の製品に対して強制的に実行される可能性のある第三者特許 が存在しない保証はなく、注目すべき販売を禁止する禁止を招くこと、または、注目すべき販売については、第三者に印税および/または他の形態の賠償 を支払う義務があることに留意されたい。
注意すべきbr}は、注目すべき特許または注目すべき被許可者の特許の保護または強制執行に関する訴訟を含む可能性があり、これは、高価で、時間がかかり、成功しない可能性がある。
競争相手 は注目すべき特許や注目すべき被許可者の特許を侵害する可能性がある。権利侵害または不正使用と戦うために、有名なbrは、費用がかかり、時間がかかる可能性がある侵害クレームを提出する必要があるかもしれない。さらに、侵害訴訟では、裁判所は、私たちまたは有名なライセンシーの特許が無効または強制的に実行できないと判断することができ、または有名な特許が関連技術をカバーしていないことを理由に、他の 側の関連技術の使用を阻止することを拒否することができる。任意の訴訟または弁護手続きにおいて不利なbrの結果が発生した場合、有名な1つまたは複数の特許が無効または狭義の解釈が宣言されるリスク に直面する可能性があり、注目すべき特許出願を発表できないリスクに直面させる可能性がある。
注意すべきは,訴訟における弁護が失敗する可能性があり,成功しても巨額のコストを招き,注目すべき経営陣や他の従業員の注意を分散させる可能性があることである.注目すべき知的財産権は、注目すべき知的財産権の盗用を単独でまたは注目すべき許可者と共に防止することができない可能性があり、特に法律では米国のようにこれらの権利を十分に保護していない可能性がある国である。
また, は知的財産権訴訟が大量の発見を必要とするため,このような訴訟期間中には,開示により機密情報が漏洩する可能性があることに注意すべきリスクがある.公聴会、動議、または他の一時的な手続き、または事態の発展の結果も公表することができる。証券アナリストや投資家がこれらの結果がマイナスであると考える場合、注目すべき普通株の価格に実質的な悪影響を及ぼす可能性がある。
有名なbrは、有名な従業員、コンサルタント、または独立請負業者が第三者の機密情報を誤って使用または開示したとして告発される可能性がある。
注目すべきは,br雇用者は以前,他のバイオテクノロジーや製薬会社に雇われていたことである。注意すべきは、有名または注目すべき従業員、コンサルタント、または独立請負業者が無意識に、または注意すべき従業員の元雇用主または他の第三者の機密情報を使用または開示しているというクレームを受ける可能性があることに留意されたい。注意はまた、前雇用主または他の第三者が注意している特許において所有権権を有するというクレームを受ける可能性がある。訴訟を通じてこれらのクレームに対抗する必要があるかもしれません。 これらのクレームを正当化することに成功した保証はなく、著しく成功すれば、訴訟は巨額の費用を招き、顕著な経営陣や他の従業員の注意を分散させる可能性がある。
| 61 |
注目すべき業務運営と業界に関するリスク
顕著な未来の成功は重要な幹部を顕著に吸引と維持し、合格者を吸引、維持と激励する能力に依存する。
顕著な は、顕著な実行チームの主要なメンバーに高度に依存しており、彼らのサービスの任意の減少または損失は、顕著な目標の達成に悪影響を及ぼす可能性がある。注目すべき主要幹部 と雇用契約を締結していることに注意されたいが,彼らの誰もが注目すべき仕事をいつでも離れることができるが,注目すべき従業員はすべて Willの従業員であるからである。注目すべき業務募集と他の合格従業員を維持するために、科学と技術者を含むことも、注目すべき成功の鍵となる。注目すべき業界は現在熟練した管理者が不足しており, という状況が続く可能性がある。そのため、技能人材に対する競争は非常に激しく、離職率が高い可能性がある。注目すべきは,類似した技能を持つ個人に対する多くの製薬会社間の競争に鑑み,受け入れ可能な条件で人員を吸引·保持できない可能性があることである。また、臨床試験で成功できなかったことは、合格者の採用と維持をもっと挑戦的にする可能性がある。いかなる幹部や肝心な従業員を募集できない、あるいはいかなる幹部或いは肝心な従業員のサービスを失うことは、注意すべき研究、開発と商業化目標の進展を阻害する可能性がある。
注意すべき は注目すべき組織を拡張する必要がある可能性があり,このような成長を管理することが困難である可能性があり,注目すべき の運営を中断する可能性がある.
今後、注目すべき従業員基盤を拡大して、注目すべき管理、科学、運営、商業、財務、その他の資源を増加させ、より多くのコンサルタントと請負業者を招聘する可能性があることに注意すべきである。将来の成長は、より多くのbr従業員、コンサルタント、および請負業者を含む、より多くの追加責任を負うことになります。また、注意すべき経営陣は、注意すべき日常活動から過剰な注意を移し、これらの成長活動を管理するために多くの時間を投入する必要があるかもしれない。注目すべき は、注目すべき業務拡張を効率的に管理できない可能性があり、これは、注目すべき インフラが脆弱であること、または操作ミス、業務機会の喪失、従業員の流失、または余剰従業員の生産性の低下を招く可能性がある。注目すべき予想成長は、大量の資本支出を必要とする可能性があり、より多くの候補製品を開発するような他の項目から財務資源を移転する可能性がある。さらに、顕著な管理層が顕著な成長を効果的に管理できない場合、顕著な支出は予想よりも多く増加する可能性があり、顕著な収入を生成および/または増加させる能力が低下する可能性があり、顕著な業務戦略を実施できない可能性がある。注目すべき未来の財務表現と注目すべき候補製品の商業化と有効な競争の能力はある程度注目すべき が未来の成長を効果的に管理する能力に依存する。
有名なbr従業員は監督管理基準と要求を守らないこと及び インサイダー取引を含む不当な行為或いはその他の不当な活動に従事する可能性がある。
注目すべきは、 が従業員詐欺または他の不正行為のリスクに直面していることである。従業員の不正行為は、FDAおよび非米国規制機関の法規を故意にまたは意図的に遵守できず、FDAおよび非米国規制機関に正確な情報を提供すること、米国および国外の医療詐欺および乱用法律法規を遵守し、財務情報またはデータを正確に報告すること、または注意事項に不正な活動を開示することを含む可能性がある。特に、医療保健業界の販売、マーケティングと業務手配は広範な法律法規に制約され、詐欺、不当行為、リベート、自己取引とその他の濫用行為 を防止することを目的としている。これらの法律法規は、様々な価格設定、割引、マーケティングおよび販売促進、販売手数料、顧客インセンティブ計画、および他のビジネススケジュールを制限または禁止する可能性があります。
従業員の不当行為はまた臨床試験過程で得られた情報の不適切な使用に関連する可能性があり、これは監督管理 の処罰を招き、そして顕著な名声に深刻な損害を与える可能性がある。注意事項は行動基準を通過したが、従業員の不適切な行為を常に識別し、阻止することができるわけではなく、そのような行為を発見し、防止するための予防措置は、未知または未管理のリスクまたは損失を制御する上で、または政府の調査または他の行動または訴訟から注意事項を保護する上で有効ではない可能性がある。注意すべき会社に対してこのような訴訟が提起された場合、注意すべきことは、注意すべき権利を自己弁護または維持することに成功しなかったことであり、これらの訴訟は、民事、刑事および行政処罰、損害賠償、罰金、連邦医療保険、医療補助および他の政府医療保健計画から除外される可能性があり、追加の報告要件および/または監督を含む注目すべき業務に重大な影響を与える可能性があり、特に、違反、引渡し、監禁、br、および契約損害の疑いを解決するために、会社の誠実な合意または同様の合意に拘束される場合を含む。このような行動を最終的に成功的に防ぐことに成功したとしても、そのような過程で財務および管理リソースを移転することが要求される可能性があり、負の宣伝を招く可能性があり、これらのすべてが注目すべき業務を損なう可能性があることに注意されたい。
| 62 |
将来の顧客と第三者支払者との関係、および有名ないくつかの業務運営は、連邦と州の医療詐欺と乱用法律、虚偽申告法、医療情報プライバシーと安全法律の制約を直接または間接的に受ける可能性がある。有名でこれらの法律を遵守できないか、完全に遵守できない場合、有名な人は刑事罰、民事処罰、契約損害、名声損害および利益と将来の収入の減少に直面する可能性がある。
注目すべき任意の候補製品がFDAの承認を得て、米国でこれらの製品の商業化を開始した場合、注目すべき業務は、注目すべき顧客を直接または間接的に通過し、さらに様々な連邦および州詐欺および乱用法律を遵守する可能性があるが、これらに限定されないが、連邦反リベート法規および連邦虚偽クレーム法案を含む。このような法律は注目すべき販売、マーケティング、教育計画などに影響を及ぼすかもしれない。また,連邦政府および注目すべき業務に従事する米国各州と外国司法管轄区のbr患者のプライバシー法規の制約を受ける可能性があることに注意されたい。注目すべき運営能力に影響を及ぼす可能性のある医療に関する法律と法規は、
| ● | 個人およびエンティティが、インフォームドコンセントおよび意図的な場合に、個人の推奨を誘導または見返りに、または連邦医療計画(例えば、MedicareおよびMedicaid計画)に従って支払い可能な項目またはサービスを購入または推薦するために、直接的または間接的に報酬を請求、受信、提供、または支払いすることが禁止されている連邦反バックル法。報酬は価値のあるものを含むものとして広く解釈されている。多くの法定免除と規制避風港はいくつかの一般的な活動を起訴から保護しているが、免除と避風港の範囲は狭く、これらの活動が免除や避難港の資格を満たしていなければ、審査や処罰を受ける可能性がある。反リベート法違反の有罪判決は連邦医療計画への参加を強制的に排除することを要求した。この法規は、医薬品製造業者と、製品を購入することができるか、または他の人(処方医、患者、購入者、および処方マネージャーを含む)との間の配置に適用されている。また、ACAは、米国政府が主張できる“社会保障法”を改正し、連邦民事虚偽クレームについては、連邦“反リベート条例”違反による物品やサービスを含むクレームが虚偽または詐欺的クレームを構成することを規定している。これに対する処罰は以下のとおりである。 | |
| ● | 連邦民事および刑事虚偽申告法および民事金銭罰法は、連邦“虚偽清算法”を含み、この方法は、民事告発者または準訴訟を含む個人または実体に刑事または民事処罰を適用し、その理由は、虚偽または詐欺的であるか、または虚偽陳述をして回避するために、連邦政府(MedicareまたはMedicaidを含む)に提出されるクレームを引き起こすことを含む。連邦政府への資金支払い義務を減少または隠蔽する。“虚偽請求法案”の下の責任は、医療業界において潜在的に大きな意味を持っている。この法規は、各虚偽請求または陳述には、損害賠償金の3倍および5,500ドル~11,000ドルの強制罰金を支払う必要がある(2022年5月9日以降に評価された違反行為については、各虚偽請求または陳述の罰金は12,537ドル~25,076ドル )と規定されているからである。 | |
| ● | 任意の個人またはエンティティに罰を加える民事罰金法規であって、他の事項に加えて、個人またはエンティティは、連邦医療保健計画にクレームを提起することをもたらしたか、またはそのクレームがクレームに従って提供されていないプロジェクトまたはサービスまたは虚偽または詐欺的なプロジェクトまたはサービスであることを知っているか、または知るべきである。 | |
| 63 |
| ● | HIPAA,brは、支払人(例えば、公共または個人)にかかわらず、支払人(例えば、公共または個人)にかかわらず、故意に実行または実行しようとする計画を知りながら、または虚偽または詐欺的な言い訳、陳述または約束によって、任意の医療福祉計画が所有または保管または制御された任意の金銭または財産を得る行為に民事および刑事罰を適用する。医療違法行為の刑事調査を故意に妨害し、重大な事実を故意に偽造、隠蔽または隠蔽するか、または医療に関連する医療福祉、プロジェクトまたはサービスの提供または支払いに関連する任意の重大な虚偽陳述 を行う。 | |
| ● | HIPAAは、“2009年健康情報技術促進経済·臨床健康法案”(HITECHと略称する)及びその実施条例を改正した“HIPAA”を経て、特定のタイプの個人と実体、例えば医療保健提供者、健康計画と医療保健 交換所、“カバー実体”及びその“業務パートナー”と呼ばれ、実体の受信或いは個人識別可能な健康情報を取得する独立請負業者又は代理人を代表して、プライバシーに関する要求を提出した。個人は健康情報の安全と伝達を識別することができる。 |
| ● | 連邦医師支払い陽光法案“は、医薬品、器具、生物製品および医療用品のいくつかのメーカーがMedicare、Medicaidまたは児童健康保険計画の下で支払いを受けることができることを要求するが、具体的な例外を除いて、医師(医師、歯医者、検眼師、足科医師および脊医を含むと定義される)および教育病院への支払いまたは移転価値に関する情報、または医師および教育病院の要求または代表医師および教育病院によって指定された実体または個人を毎年CMSに報告しなければならない。そして、適用される製造業者と適用される共同購入組織は、医師およびその直系親族が保有する所有権および投資権益を毎年CMSに報告することを要求している。これらの報告義務は拡大され、2022年1月1日から、会社はまた、非医師提供者および他のタイプの医療保健専門家(例えば、医師アシスタントや看護師従業員)への支払いおよび移転の価値を報告しなければならない。br}がすべての保証支払い、価値および所有権移転または投資利益をタイムリーに、正確かつ完全に提出できなかった場合、民事罰金を招く可能性がある。また、多くの州では支払いや他の価値移転の報告も管理されており、その多くは互いに大きく異なり、往々にして先制者がなく、“陽光法案”よりも禁止性があり、コンプライアンス作業をさらに複雑化させる可能性がある。 | |
| ● | 多くの州と外国の法律は上述の連邦法律と同等であり、例えば、反リベートおよび虚偽請求法は、任意の第三者支払人(商業保険会社を含む)の精算の物品またはサービスに適用可能である;州法律は、製薬会社に製薬業の自発的なコンプライアンスガイドラインと連邦政府が公布した関連するコンプライアンスガイドラインを遵守することを要求する;州法律は、医薬品メーカーに医師および他の医療保健提供者またはマーケティング支出への支払いおよび他の価値のある移転に関する情報を報告することを要求する。医薬品販売は、登録された州および現地の法律を代表することを要求し、場合によっては健康情報のプライバシーおよび安全を管理する州および外国の法律であり、多くの法律は互いに大きく異なり、同じ効果を有さない可能性があり、それによってコンプライアンス作業を複雑化することができる。 |
また、EUは、95/46/EC号命令を含むが、これらに限定されない独自のデータセキュリティおよびプライバシー法的枠組みを構築している。“欧州一般データ保護条例”には、健康情報処理に特化した新しい条項、より高い制裁、および非EU会社をこの条例に組み込むことを目的とした域外措置が含まれている。
Brの顕著な経営が、上記の任意の法律または顕著な他の政府法規または法律に違反していることが発見された場合、民事、刑事および行政処罰、損害賠償、罰金、連邦医療保険、医療補助および他の政府医療保健計画から除外される可能性があり、追加の報告要件brおよび/または監督を含む可能性があり、特に、違反、引渡し、監禁、契約損害、名声損害、利益減少および将来の収益に関する告発を解決するために、会社の誠実な合意または同様の合意の制約を受けている場合、顕著な業務を削減又は再編し、いずれも顕著な業務運営能力及び顕著な運営実績に悪影響を及ぼす可能性がある。
| 64 |
最近のbrと将来の医療保険立法はさらに顕著な業務運営に影響を与える可能性がある。
米国や一部の外国司法管区は、医療システムを変更するために、注目すべき製品の収益性に影響を及ぼす可能性があるいくつかの立法および規制提案を検討しているか、または公布している。米国や他の地域の政策立案者や支払者の中で,医療システムの変革を推進し,前述したような医療コストの抑制,質の向上,参入拡大の目標を実現することに大きな興味がある。米国では、製薬業はこれらの努力の重点であり、重大な立法計画の大きな影響を受けてきた。
注目すべきは、将来取られる可能性のある医療改革措置は、より厳しいカバー基準とより低い精算をもたらす可能性があり、承認された任意の製品の価格に追加の下振れ圧力をもたらす可能性があることを予想している。連邦医療保険または他の政府援助計画の清算を減少させることは、個人支払者の支払いの同様の減少をもたらす可能性がある。
注目すべきは, が将来とりうる医療改革の取り組みを予測できないことである。連邦、州、外国の立法と規制はさらに発展する可能性があり、進行中の措置は薬品価格の圧力を増加させることに注意されたい。このような改革は、開発に成功する可能性のある候補製品の予想収入に不利な影響を与える可能性があり、顕著な全体的な財務状況および候補製品を開発する能力に影響を与える可能性がある。
有名な は潜在的な製品責任に直面しており,有名な成功クレームに対して重要であれば,大量の責任や コストを招く可能性がある。
現在と未来の臨床試験で注目すべき候補製品を使用し、いかなるマーケティング許可を得た製品を販売することは、注目すべき製品を製品責任クレームのリスクに直面させる。消費者、ヘルスケア提供者、製薬会社、または販売または他の方法で有名な製品に接触する他の人は、有名なbr製品に対して製品責任クレームを提起する可能性がある。例えば、注目すべき現在および将来の製品または候補製品の使用は、予期しない悪影響を及ぼす可能性があり、これは、潜在的な製品責任クレームをもたらす可能性がある。製品責任クレームに著しく成功できなければ、 が顕著であることは重大な責任とコストを招く可能性がある。さらに、製品責任クレームは、是非曲直や最終結果にかかわらず、以下のようになる可能性がある
| ● | 顕著な営業権の減少 ; | |
| ● | 臨床試験参加者は脱退しました | |
| ● | 関連訴訟による費用 ; | |
| ● | 主要業務に対する経営陣の関心を分散させる | |
| ● | 患者や他のクレーム者に巨額のお金の報酬を与えます | |
| ● | 注目すべき候補製品を商業化することはできません | |
| ● | 商業販売への使用が承認されると,有名候補製品への需要は減少した |
注目すべきbr}は,現在と将来の臨床試験における注目すべき治療法の使用に関する製品責任保険を獲得している。しかしながら、このような保険カバー範囲は、著しく受ける可能性のある任意の費用または損失を補償するのに十分ではない可能性がある。さらに、保険範囲はますます高価になっており、将来的には、責任による損失から著しく保護するために、合理的なbrコストまたは十分な金額で保険範囲を獲得または維持することができない可能性がある。注目すべき候補製品が市場承認を得た場合、商業製品の販売を含む注目すべき保険カバー範囲を拡大しようとすることに注意されたいが、商業的に合理的な条項または十分な金額で製品責任保険を得ることができない可能性があることに注意されたい。思わぬ悪影響を及ぼす薬物に基づく集団訴訟では,多額の判決が下されることがある。成功した製品責任クレームまたは一連のクレームは顕著な株価下落を招く可能性があり、判決が顕著な保険範囲を超えた場合、顕著な運営および業務業績に悪影響を及ぼす可能性がある。
| 65 |
ネットワークセキュリティリスクおよび注目すべきコンピュータハードウェア、ソフトウェア、インターネットアプリケーションおよび関連ツールおよび機能の機密性、完全性および利用可能性を維持できなかったことは、注目すべき名声を損なう可能性があり、および/または注意すべきbr}に費用、罰金または訴訟を負担させる可能性がある。
注目すべきビジネスは、大量のデータを処理、分析、保存する必要がある。また,注目すべき業務はグローバル企業ソフトウェアシステムに依存して運営·管理されていることに注意されたい。注目すべきは,注目すべき 従業員の個人身分情報も維持していることである.したがって、優れた業務は、優れたコンピュータハードウェア、ソフトウェア、ネットワーク、インターネットサーバおよび関連インフラの持続的、有効、信頼性および安全な動作に依存する。注目すべきハードウェアやソフトウェアの故障や内部研究者の注目すべきデータへのアクセスが中断された場合、注目すべき業務が影響を受ける可能性がある。注目すべき従業員および会社データの完全性および保護は、注目すべき業務に重要であり、従業員は、注意すべき個人情報を十分に保護することに高い期待を持っている。情報、安全、プライバシー法を管理する規制環境の要求はますます高くなり、発展し続けている。適用されるセキュリティやプライバシー法規 を遵守することは,著しい運営コストを増加させる可能性がある.注目すべきコンピュータおよび通信ハードウェアは、物理およびソフトウェアによって保護されているが、火災、嵐、洪水、停電、地震、電気通信障害、物理またはソフトウェア侵入、ソフトウェアウイルス、および同様のイベントの影響を受けやすい。このようなイベントは、不正アクセス、開示、および非公開情報の使用をもたらす可能性がある。犯罪者がコンピュータシステムを攻撃するための技術は複雑であり,変化が頻繁であり,世界で規制の少ない遠隔地に由来する可能性がある.したがって,これらの脅威 に能動的に対応したり,十分な予防措置を実施したりすることができない可能性があることに注意されたい。注意すべきコンピュータシステムが漏洩すれば、罰金、損害賠償、訴訟、法執行行動を受ける可能性があり、注意すべきは商業機密が失われる可能性があり、このような状況が発生すると、注目すべき業務を損害する可能性がある。さらに、他社が提供するインターネットアクセスの任意の継続的な中断は、注目すべき業務を損なう可能性がある。
業務中断は、将来の収入や財務状況を深刻に損なう可能性があり、大幅なコストと支出を増加させる可能性がある。
注目すべき業務、およびCROと他の請負業者とコンサルタントの業務は、地震、電力不足、電気通信故障、水不足、洪水、ハリケーン、台風、火災、極端な天気条件、医療流行病と他の自然或いは人為的災害或いは業務中断の影響を受ける可能性があり、注目すべきは主に自己保険である。このような業務中断の発生は、著しい運営および財務状況を深刻に損なう可能性があり、著しいコストおよび支出を増加させる可能性がある。
注意すべきは、本社はカリフォルニア州フォスター市にあり、野火や地震が発生しやすい地域であることだ。上記及びその他の自然災害は、当社の経営を深刻に混乱させ、当社の業務、経営業績、財務状況及び見通しに重大な悪影響を及ぼす可能性がある。自然災害、停電、または他の事件が発生し、重要なインフラの損傷や運営中断を招く場合、すべての または重要な本部の大きな部分を使用することができず、 が重要であることは難しいかもしれないし、場合によってはかなり長い期間重要な業務を継続することは不可能である。 は、深刻な災害または同様の 事件が発生した場合、重要に制定された任意の災害復旧および業務連続計画が十分ではないことが証明される可能性がある。注意すべき災害復旧と業務連続計画の限られた性質により、顕著に巨額の支出 が生じる可能性があり、これは顕著な業務に重大な悪影響を及ぼす可能性がある。
注目すべき は,注目すべき組織の規模と注目すべき外部ベンダ関係の範囲を増やす必要があり,注目すべき は成長管理に困難に直面する可能性がある.
注目すべきは、現在の管理および科学者、システム、および施設は、注目すべき将来の成長をサポートするのに十分ではない可能性があることである。顕著な運営、成長、様々なプロジェクトを効果的に管理する必要があり、顕著であることが要求される
| ● | 注意すべき臨床試験を有効に管理し、注目すべき現有と計画の臨床試験を含む |
| 66 |
| ● | 注意すべき内部開発作業を効率的に管理するとともに、注意すべきライセンス者、請負業者、その他の第三者に対する契約義務を履行する | |
| ● | 注目すべき業務、財務および管理制御、ならびに報告システムおよびプログラムの改善を継続する; | |
| ● | 十分な数の才能のある従業員を引き付け、維持し続ける。 |
注目すべきは、br}が利用しており、将来的にサプライヤーと研究パートナーまたは協力者のサービスを利用して任務を実行することが予想され、 臨床前研究と臨床試験管理、統計と分析、監督事務、医療相談、市場研究、処方 開発、化学、製造と制御活動、他の薬物開発機能、法律、監査、財務相談と投資家関係を含む。注目すべき成長戦略はまた、これらおよび他の将来のタスクを実施するために、注目すべき請負業者またはコンサルタントチーム を拡大する必要があるかもしれない。多くのコンサルタントがその業務の多くの重要な機能をアウトソーシングすることに注目すべきであるため、これらのコンサルタントを効率的に管理し、彼らがその契約 義務を成功裏に履行し、期待される期限を満たすことを確実にする必要があることに注意されたい。しかし、注意すべきアウトソーシング活動を効果的に管理できない場合、またはコンサルタントが提供するサービスの品質または正確性が任意の理由で影響を受ける場合、注目すべき臨床試験は延長、延期または終了される可能性があり、注目すべき候補薬剤は監督部門の承認を得ることができないか、あるいは他の方法で注目すべき業務を進めることができない可能性がある。注目すべき既存のコンサルタントを経済的に合理的な条件で管理できる保証はなく、または他の適任な外部請負者およびコンサルタントを見つけることができないか、または全くできない。顕著に新入社員の採用と顕著なコンサルタントや請負業者チームを拡大することで顕著な組織を効果的に拡張できない場合、顕著な は顕著な候補薬物のさらなる開発と商業化に必要な任務を成功させることができない可能性があるため、顕著な研究、開発、商業化目標を達成できない可能性がある。
将来のネットワーク攻撃やセキュリティホール、およびセキュリティを維持する効果的な情報技術システムのコストを含む重要な情報技術システムを中断することは、重要な業務や運営結果に悪影響を与える可能性がある。
注目すべき業務の効率的な運営は、注目すべき購買力平価を形成するカスタマイズされた情報技術システムを含むコンピュータハードウェアおよびソフトウェアシステムに高度に依存する。情報システムはコンピュータハッカーやネットワークテロリストの安全破壊を受けやすい。注目すべきは、注目すべき情報システム上で維持される機密 および固有の情報を安全に維持するために、業界で公認されているセキュリティ対策および技術に依存し、リスクが制御されることを保証するために、これらの システムおよびアプリケーションの維持およびアップグレードに投資し続けることに留意されたい。注目すべきは、注目すべきネットワークセキュリティシステムをどのように維持し、アップグレードしても、注目すべき侵入がないことを保証することはできず、許可されていない各方面が機密や個人情報にアクセスしないことを保証することはできず、いかなるこのような事件が適時に発見されることも保証できない。犯罪者が不正な場合に敏感なデータにアクセスするために使用される技術はしばしば変化し、一般にターゲットに対して攻撃を開始するまで識別され、これらの技術を予見できない可能性があり、または十分な予防措置が実施されている可能性があることに留意されたい。重大なデータセキュリティホールの程度を適時に発見、確定できず、それに対して適切な応答を行うことは、顕著な業務、財務状況と運営結果に重大な不利な影響を与える可能性がある。さらに、情報システムが利用できないか、またはこれらのシステムは、重大な災害またはトラフィック中断により、これらのシステムに格納されているデータにアクセスできないこと、または顕著な需要を満たすためにサポート機能を維持することができないことを含む任意の理由で予期されるように動作することができず、そのようなイベントに応答できないか、またはそのようなイベントから回復することができず、顕著なトラフィックを混乱させる可能性があり、性能低下および管理コストの増加を招き、顕著なトラフィックおよび運営結果に影響を受ける可能性がある。
さらに、 注目すべきトラフィックには、注目すべき従業員に関する個人情報 および当社および注目すべきベンダに関する独自のビジネス情報を含む敏感なデータの受信および格納が含まれる。法律要求,個人許可または適用される プライバシーポリシーが許可されている場合には,業務展開に協力するサプライヤーと情報を共有することも可能であることに注意する.
| 67 |
情報セキュリティ対策が採用されているにもかかわらず、すべての重要なITシステムまたは重要なプロバイダを決定することができないITシステム は、既知のbr}マルウェア、将来開発される可能性のあるマルウェア、または他のマルウェアの任意の未来のネットワーク攻撃またはセキュリティホールを防止、抑制、または検出することができることに留意されたい。ネットワーク攻撃は急速に発展しており、ますます複雑になっており、検出が困難になっており、したがって、これらの攻撃を予見したり、十分な予防措置を実施したりすることができない可能性がある。 さらに、不正な当事者は、詐欺、詐欺、または注目すべき従業員またはサプライヤーに関する他の形態の詐欺によって、注目すべきシステムまたはプロバイダのシステムまたは施設へのアクセスを試みる可能性がある。任意の攻撃または違反行為が情報損失、破損または盗用をもたらす場合、重要な臨床試験に参加する者、株主および他の人のクレーム、および監督機関によるコストの高い調査または法執行行動は、著しい悪影響を及ぼす可能性がある。これらのクレームや,重要なシステムをアップグレード,修復,交換するのに多大な時間と費用がかかり,重要な 運営を大きく妨害する可能性もある.顕著な臨床試験に参加した人の信頼を失う可能性もあり、顕著な名声および将来の販売に損害を与える可能性がある(あれば)。さらに、将来の技術進歩に対応するために、より厳しいプライバシーおよび情報セキュリティ法律および標準を遵守し、技術システムを開発、維持およびアップグレードするコストは巨大である可能性があり、br}は、新しいシステムまたはアップグレードされたシステムおよび技術を実施することに関連する問題に遭遇し、br}既存システムの保守または十分なサポートを中断する可能性があることに留意されたい。
マイウェートはより多くの候補薬の獲得、開発、マーケティングに成功できず、ノワールの成長能力を弱める可能性がある。
注目すべき成長戦略の一部として、より多くの候補薬剤および技術を評価、買収、許可、開発および/またはマーケティングする可能性があることに注意されたい。注目すべき内部研究能力は限られており、製薬と生物製薬会社、学術科学者、その他の研究者が候補薬物或いは技術を注目すべき会社に売却或いは許可することに依存する可能性があることに注目すべきである。この戦略の成功は、有望な候補薬物と技術を顕著に識別、選択、獲得する能力にある程度依存する。候補薬物許可証または買収を提案、交渉、実施する過程は長く複雑である。他の会社、brは、より多くの財務、マーケティング、および販売資源を持ついくつかの会社を含み、有名会社と候補薬物や技術の許可または買収を争う可能性がある。注目すべき資源は限られており,潜在的な候補薬物や技術の買収や許可を識別·実行することができず,注目すべき既存のインフラに統合されている。さらに、企業は、完成していない潜在的な買収にリソース を使用することができるかもしれないし、そのような努力の予想される収益 を達成できない可能性があることに注意されたい。また、注目すべきは、 が許容可能であると考えられる条項でより多くの候補薬剤を得る権利を得ることができないか、または全くできない可能性があることに注意されたい。
さらに、将来的に知的財産権を買収することは、多くの運営と財務リスクをもたらすかもしれない
| ● | 未知の債務にさらされるリスクは | |
| ● | 重要な業務を中断し、重要な管理職および技術者の時間と注意を移動させて、得られた候補薬物または技術を開発する | |
| ● | 買収コストを支払うために大量の債務が発生したり、証券発行を希釈したりする | |
| ● | 調達コストが予想以上に高いこと | |
| ● | 分担費用が増加しました。 |
任意の注目すべき候補薬物は、商業販売または対外許可の前に、br FDAおよび適用される外国規制機関による広範な臨床試験および承認を含む追加の開発作業を必要とする可能性がある。すべての候補薬物は監督管理機関の承認のために十分な安全と有効性が証明されない可能性がある典型的な薬物開発失敗リスクが出現しやすい。さらに、注目すべき薬物は、任意の注目すべきbrが開発または承認される可能性のある薬物が利益を得て生産されるか、または市場に承認される可能性があることを保証することはできない。
| 68 |
注目すべきは、本報告で使用される統計データ、市場データ、および他の業界データ、および予測は、市場研究、公開利用可能な情報および業界出版物からであり、これらの情報は信頼できるが、いかなる第三者によって確認されていないことに留意されたい。
本報告には,注目すべき業界,注目すべき業務および市場の推定,予測およびその他の情報 が含まれており,これらの市場の推定規模やいくつかの医療 状況に関するデータが含まれている。本報告に掲載されている業界、市場及び類似データは、すべて顕著な内部推定と研究、及び学術及び業界研究、出版物、調査及び第三者(政府機関を含む)による研究から来ている。いくつかの場合、注目すべきことは、このようなデータの出所が明確に言及されていないということだ。 推定,予測,予測,市場研究や類似手法に基づく情報は固有に不確実性の影響を受けており,実際のイベントや状況は本情報で想定しているイベントや状況とは大きく異なる可能性がある.注目すべき内部研究は信頼できるにもかかわらず,このような研究は第三者の検証は得られていない。
イスラエルへの登録に注意に関するリスク
あなたの有名な株主としての権利と責任はイスラエルの法律によって管轄され、イスラエルの法律はいくつかの点でアメリカの会社の株主の権利と責任とは異なるかもしれない。
Br注意はイスラエルの法律に基づいて登録されているため、その株主の権利と責任が注目されている組織条項とイスラエルの法律の管轄である。これらの権利および責任は、いくつかの重要な点で米国会社の株主の権利および責任とは異なる。特に、イスラエルの会社の株主は、会社や他の株主に対する権利を行使し、その義務を履行する際に、善意と慣用的な方法で行動し、会社における権力の乱用を回避する義務があり、他の事項を除いて、会社定款の改正、会社法定株式の増加、合併会社の承認、株主の承認を必要とする関連側取引のような株主総会でのいくつかの事項の採決を含む。株主も一般的に他の株主への差別を禁止する義務がある。また,持株株主や株主は,株主投票結果を決定したり,会社の高級社員を委任したり阻止する権限を持っていることを知っていれば,その等の投票や会社への委任について公平に行う責任がある.しかし、イスラエルの法律はこの公正な義務の実質的な内容を定義していない。この義務の性質やこれらの規定の影響を理解する上で,注意すべき判例法は限られている.これらの規定は、重要な株主に追加的な義務や責任を課すと解釈される可能性があるが、これらの義務や責任は通常、米国会社の株主に押しつけられない。
イスラエルの法律と注目すべき改正と再記述の会社定款 は、第三者が注目すべき買収を買収したり、注目すべきコストを増加させたりすることを難しくする可能性があり、そうしても注目すべき株主に利益を得ることができる。
イスラエルの法律は合併を監督し、指定されたハードルを超えた株式の買収を要求する際に買収要約を提出し、取締役、高級管理者または大株主に関連する取引は特別な承認を必要とし、このような取引に関連する可能性のある他の事項を規範化する。例えば、買収者が発行済み株保有者の少なくとも95%の積極的な応答を受けた場合にのみ、会社のすべての発行済み株式と流通株に対する買収要約を完成させることができる。要約買収の完了には,少なくとも98%の会社流通株 が入札されない限り,要約買収に個人的な利益がない多くの要人の承認を得る必要がある.また、株主は、買収要約の受け入れを示す株主(買収側がその買収要約に規定されていない限り、要約を受けた株主は評価権を求めることができない)を含み、買収要約が完了してから6ヶ月以内の任意の時間にイスラエル裁判所に買収の対価格変更を請求することができる。
さらに、イスラエルの税務考慮は、その有名株主またはその居住国br}がイスラエルと税金条約を締結していない一部の株主にとって潜在的な取引を望ましくない可能性があり、このような株主がイスラエルの税収から税金減免を受けることを可能にする。例えば、イスラエル税法は米国税法のように免税株取引所を認めない。合併に関して、イスラエル税法は場合によっては納税 を延期することを許可しているが、延期はいくつかの条件の満足に依存し、場合によっては、取引の日から2年間の保有期間を含み、その間に参加会社の株式の売却と処分はいくつかの制限を受けている。また,ある株式交換取引については,繰延納税の時間は限られており,この期限が満了した場合には,株式処分が発生しなくても税金を納めなければならない。
| 69 |
ある米国の株主が明らかに“統制された外国企業”と記述されていれば、不利な税収結果を受ける可能性がある
米国連邦所得税については、“制御された外国会社”(“cfc”)に分類される非米国会社の各 “10%株主”は、通常、米国連邦税収の収入にこのような 10%株主のcfcの“F分部収入”における比例シェアと米国財産への投資収益を計上することが要求され、 cfcがその株主に割り当てられていなくてもよい。10%の株主が投票権のある非米国会社のすべての種類の株の総投票権またはその会社の株式の総価値の50%以上を直接または間接的に所有している場合、非米国会社は通常、米国連邦所得税 に分類される。“十パーセントの株主”とは、その会社が投票する権利があると考えられるすべてのカテゴリーの株式総投票権を有するか、または所有すると考えられる10%以上の米国人を意味する(本準則の定義による)。クロロフルオロカーボンの地位の決定は,帰属規則を含めて複雑であり,これらの規則の適用状況は完全には決定されていない。
注意すべきは, は2022年12月31日までの納税年度にフッ素塩化炭素であるとは考えていないこと,あるいは現在フッ素塩化炭素であることに注意されたい。しかし,推定所有権規則を適用した後,米国連邦所得税の目的で,米国人とみなされる株主は十分なbr株式を直接あるいは間接的に獲得する可能性があり,10%の株主とみなされ,会社の任意の他の10%の株主とともに,米国連邦所得税にとってフッ化炭素とされていることに注意されたい。米国連邦所得税の規定によると、その一部の株主は10%の株主であることに注意されたい。保有者はCFC 10%株主の潜在的不利なアメリカ連邦所得税結果について自分の税務顧問に相談しなければならない。
注意すべきbr}は2023納税年度にPFICに分類されると予想され、その米国の株主は不利な税収結果を受ける可能性がある。
一般に、任意の課税年度において、顕著な総収入の少なくとも75%が受動的収入である場合、または有意な資産価値の少なくとも50%が、受動的収入を生成するため、または現金を含む受動的収入を製造するための資産に起因することができる場合、顕著な は、米国連邦所得税目的のPFICとして記述されるであろう。これらのテストに関して、受動的収入には、配当金、利息、および投資財産の売却または交換の収益、ならびに賃貸料および特許権使用料以外の賃貸料および特許使用料が含まれ、これらの収入は、貿易または事業を積極的に展開することに関連する関係者 から受信される。PFICとして表現されている場合、顕著な普通株を売却することによって達成される収益を資本収益ではなく、米国所有者である個人が立派な普通株から受け取るために適用される配当金に適用する割引率を失うこと、および顕著な普通株を売却することによって達成される収益を一般収入とすることを含む、米国株主が不利な税収結果を受ける可能性がある。
立派な資産投資会社の地位は、権勢会社の収入の構成とその資産の構成と価値に依存するため(一部は現像会社の普通株の時価を参考にして決定される可能性がある)、現像会社がいかなる課税年度にもプライベート資産投資会社とみなされない保証はない。注目すべきは,2021年または2022年の納税年度にPFIC はないと予想され,本納税年度のPFICであることに注意されたい。しかし、公共投資委員会が事実に基づく年次決定作業であることに注目すべきかどうかは、採用の原則や方法が不明確である場合があり、異なる解釈がある可能性がある。注目すべきプライベートエクイティ投資会社の地位はその収入の構成と資産の構成と価値( は部分的に注目すべき普通株の時価を参考にして決定することができ、その等の株式は変動する可能性がある) であるため、現在の個人資本投資会社の地位を最終的に決定することはできず、アメリカ国税局が重要会社が上述した個人資本投資会社の地位に関する決定に疑問を提起しないことを保証することはできないことに注意されたい。
注意すべきいくつかの顧問がいるイスラエル列国では政治、経済、軍事が不安定になる可能性があり、これは注目すべき業務結果に悪影響を及ぼす可能性がある。
有名なbrはイスラエルの法律登録によって成立し、その財務と会計業務に参与するいくつかの有名な顧問 はイスラエル諸国に位置する。したがって、イスラエルの政治的、経済的、軍事的条件は顕著な業務に直接影響を及ぼす。1948年にイスラエルが成立して以来、イスラエルとその隣国の間で何度も武力衝突が発生した。
イスラエルの敵対行動、またはイスラエルの経済的または財政的状況の著しい低下は、注目すべき業務に悪影響を及ぼす可能性がある。2000年10月以来、テロ暴力事件は増加している。2023年10月に開始された敵対行動、またはイスラエルの他の政治的または経済的要因を含む持続的かつ再爆発的な敵対行動は、有名な業務を損なう可能性がある。
| 70 |
課税収入が生成された場合、享受可能な税金優遇要件は、様々な条件を満たし、将来的にbrを阻止または減少させる可能性があり、これは、著しいコストおよび税金を増加させる可能性がある。
もしbrが課税収入を生み出した場合、それは1959年にイスラエルの“資本投資奨励法”(“投資法”)が“利益企業”に提供したいくつかの税金優遇を受ける資格があるだろう。“恩恵を受けた企業”の税収優遇を継続して享受する資格があるようにするためには,改正された“投資法”とその条例に規定されているいくつかの条件を満たし続ける必要がある。また、“投資法”に基づき、イスラエル税務当局は2012年を“受益企業”選挙年に選択したことに注目すべきだ。本税務条例 によって得られる利益は、この条例に規定されている条件の満足状況に依存する。しかも、未来にこのような税金優遇は減少または終了するかもしれない。これらの税金優遇が減少され、キャンセルされ、または終了された場合、注意すべきイスラエルの課税所得額は、イスラエルの正常な会社税率に適用される。イスラエル社の2018年以降の標準会社税率は23%である。 また、買収によりイスラエル以外での活動が著しく増加すれば、その拡大活動は将来のイスラエル税優遇計画に組み込む資格がない可能性がある。
注目すべき普通株保有に関するリスク
重要な普通株の市場価格は大きく変動する可能性があります。買収価格であなたの株を転売できないかもしれません。
重要な普通株は活発な取引市場がないかもしれない。重要な普通株の取引が活発でなければ、あなたは迅速にあるいは市場価格であなたの株を売ることができないかもしれません。
注目すべき普通株の市場価格はずっと変動し続ける可能性がある。注目すべき株価は、以下の要因を含む様々な要因によって大幅に変動する可能性がある
| ● | 医療産業や全体的な経済減速 | |
| ● | 注目すべき任意の製品の十分な構成要素供給を得ることができないか、または許容可能な価格で供給することができない | |
| ● | 注目すべき候補製品の開発と商業化に成功しなかった | |
| ● | 追加資金を得ることができなかった | |
| ● | 注目すべきは、彼らが法規要求を遵守する能力を含む、その製品部品の製造を含む、依存可能な第三者の業績である | |
| ● | 候補製品の現在と未来の臨床試験の結果 | |
| ● | 予期しないbrまたは任意の注目すべき製品の使用に関連する深刻なセキュリティ問題; | |
| ● | 不利な規制決定; | |
| ● | 重要なビジネスパートナー協定を含む重要な合意の締結または終了; | |
| ● | 任意の注目すべき知的財産権を強制的に執行または擁護し、または他人の知的財産権を守るために、訴訟を提起し、実質的に進展し、または訴訟を終了する | |
| ● | 有名な商業パートナーまたは競争相手によって発表された新製品または製品増強、臨床進展または不足、重大な契約、商業関係または資本約束の公告; | |
| 71 |
| ● | 従来技術および製品、または出現する可能性のある新しい技術および製品からの競争; | |
| ● | 重要な従業員が流出しました | |
| ● | 特許、訴訟事項、および特許保護を著しく得る能力(Br)の顕著な許可および所有技術を含む、係争または独占権に関連する他の発展; | |
| ● | 証券アナリスト(ある場合)の顕著な普通株の推定または提案が変化した ; | |
| ● | 公衆、立法機関、規制機関、投資界のバイオ製薬業界に対する見方 | |
| ● | 将来的に注目すべきまたはその株主は注目すべき普通株を売却する | |
| ● | 研究開発費の一般的かつ特定の業界の経済状況に影響を及ぼす可能性がある | |
| ● | 取引量が低く、関連会社の持ち株比率が高い | |
| ● | ナスダックでの重要証券の上場状態が変化する ; | |
| ● | 医療支払い制度の仕組みを変え | |
| ● | 顕著な財務業績の期間変動 |
また、株式市場は全体的に大きな変動を経験しており、通常は個別会社の経営業績とは無関係である。これらの広範な市場変動は、注目すべき普通株の取引価格にも悪影響を及ぼす可能性がある。
従来、ある会社の証券市場価格の変動に伴い、株主はこれらの会社に対して集団証券訴訟を起こすことが多かった。このような訴訟を提起すれば、巨額のコストや経営陣の注意力や資源の移転を招く可能性があり、これは顕著な収益性と名声を深刻に損なう可能性がある。
注目すべき普通株の取引量は限られてきた。
有名普通株がナスダックに上場しているにもかかわらず、これらの有名普通株の市場での流動性は限られている。これは、保有者がその有名普通株を売却することを難しくする可能性がある。重要な普通株の活発な取引市場が続く保証はない。また、株式市場は一般的に極端な価格と出来高の変動を経験しており、これらの変動は往々にして上場企業の経営業績と関係がないか比例しない。広範な市場と業界 要素はその実際の経営業績にかかわらず、有名普通株の市場価格にマイナス影響を与える可能性がある。市場で盛んになる有名な普通株の市場価格と流動性はあなたのbr支払いの価格より高いか低いかもしれないし、多くの要素の重大な影響を受ける可能性があり、その中のいくつかの要素は著しい制御範囲を超えている。
注目すべき 主要株主はそのかなりの割合の株式を所有しており,株主の承認が待たれる事項を大きく制御することができる.
2024年2月16日現在,重大事項の最適情報によると,その役員,上級管理者,5%の株主とそれぞれの関連会社実益が重要事項既発行議決権株式の約38%を有している。したがって,これらの株主はその所有権地位 によって顕著な影響を与える能力がある.これらの株主は、株主の承認を必要とするすべての事項を決定することができるかもしれない。例えば、彼らが一緒に行動すれば、これらの株主は、取締役選挙、重要な組織文書の修正、または任意の合併、資産の売却、または他の重大な会社取引を承認することができるかもしれない。これは、あなたが重要株主の一つであると考えられる重要な普通株に対する能動的な買収提案や要約を阻止または阻止する可能性があります。
| 72 |
注目すべき は、予見可能な未来に注目すべき普通株のために配当金を支払うつもりはないので、いかなる見返りもその株の価値 に制限される。
注意すべきことに、 は株の任意の現金配当金を発表または支払いしたことがない。注目すべきは、現在、将来の収益 は業務の発展、運営、拡張のために維持されることが予想され、予測可能な未来にはいかなる現金配当金 も発表または支払うことはないと予想されることである。したがって、株主へのどんな見返りもその株の増価に限定されるだろう。さらに、イスラエルの法律はAttenantが配当金を発表して支払う能力を制限し、Attenantの配当金にイスラエルの源泉徴収を課す可能性がある。また、注目すべきは、配当金(免税収入から)を支払うことは、それに対してあるイスラエル会社の所得税を徴収することに遡る可能性があるが、注意すべきは、本来これらの税金を支払う必要がないことである。
株式研究アナリストが顕著な業務に関する研究報告を発表しない場合、または彼らが不利なコメントを発表したり、顕著な普通株の格付けを引き下げたりすると、顕著な普通株の価格が下落する可能性がある。
有名普通株の取引市場は株式研究アナリストが発表した有名 及びその業務に関する研究と報告にある程度依存する。研究アナリストの報告を得ていない場合、または1つ以上の証券アナリストが有名普通株の格付けを引き下げた場合、またはこれらのアナリストが他の不利なコメントや予想を発表した場合、有名企業が有名またはその業務に関する報告を満足または停止することができない場合、有名普通株の価格は下落する可能性がある。
上場企業に影響を与える法律や法規を遵守することにより、コストや経営陣への要求が生じることに注意されたい。
注目すべきは、上場企業の報告要求に関連するコストを含む、多くの法律、会計、その他の費用が発生することに注目すべきである。注目すべきは、 サバンズ-オクスリ法案下の要求、および米国証券取引委員会とナスダックが実施する新しいルールを含む、コーポレート·ガバナンス要件に関するコストが生じることにも注意されたい。これらの規制は、顕著な法律と財務コンプライアンスコストを増加させ、いくつかの活動をより時間とコストを高くすることが予想される。例えば、注目すべき管理チームは、合併完了前に注目すべき幹部から構成され、その中には 以前に上場会社を管理·運営したことがない人もいるかもしれない。これらの幹部やその他の人員は、上場企業の運営に関する専門知識を獲得し、適用される法律法規を遵守するために多くの時間を投入する必要があるだろう。これらのルールやbrルールは,有名な役員や上級管理者が責任保険を得ることを困難かつ高価にする可能性もある.そのため、注目すべきは、適格な人を注目すべき取締役会に参加したり、注目すべき実行者に参加したりすることがより困難である可能性があり、これは投資家の注目すべき自信に悪影響を与え、注目すべき業務または株価 が影響を受ける可能性がある。また、上場企業の運営を支援するためには、より多くの経験や人員を増やす必要があるかもしれない。これらの分野の任意の既存人員の流失またはそのような拡張を効果的に達成または管理できないことは、そのインフラが弱くなる可能性があり、顕著な業務、財務状況、および運営結果が重大な悪影響を受ける可能性がある。
逆買収 注目すべき定款文書中の条項は、注目すべき会社を買収することをより困難にする可能性がある。
注意すべき会社定款や定款大綱の条項は買収を延期または阻止する可能性がある。会社法は、投票、分配、または他の事項に関するいくつかの優先権利を提供する株式 および優先購入権を有する株式を含む、普通株式とは異なる権利を有する株式を有名に創設および発行することを可能にする。重大な改訂と重述された会社定款によると、現在優先株が許可されていない。将来的に、特定の クラスの優先株を許可し、作成し、発行する価値がある場合、付随する可能性のある特定の権利に基づいて、このような株は、買収を阻止または阻止する能力があるか、またはその普通株の時価に対する潜在的なプレミアム を達成することを他の方法で阻止する能力があるかもしれない。1種類の優先株の認可と指定は、注目すべきbr改正と再記載された組織定款細則を改正する必要があり、これは事前に多数の投票権を持つ保有者の承認を得て、株主総会で注目すべき発行と流通株に投票権を与える必要がある。
| 73 |
注意すべき は、より小さい報告会社に適した情報開示とガバナンス要求低下の優位性を利用することが予想され、その普通株の投資家に対する吸引力の低下を招く可能性がある。
有名なbrは年収1億ドル未満,公開上場は7億ドル未満であるため,米国証券取引委員会規則の下で小さいbr報告会社の資格に適合し続ける。規模の小さい報告会社としては、簡略化された役員報酬開示および米国証券取引委員会文書中の財務諸表開示要求 のような低減された開示要求 を利用することができることに留意されたい。米国証券取引委員会は規模の小さい報告会社であるため、その提出された文書に開示されている情報が減少し、投資家がその運営業績と財務見通しを分析することが困難になる可能性がある。D注意は予測できず、これらの免除に依存すれば、投資家が注意する普通株吸引力の低下を発見するかどうか。一部の投資家がその普通株の吸引力が低下していると考えていれば、 その普通株の取引市場はそれほど活発ではない可能性があり、その株価はより変動する可能性がある。注意すべきは、その会社がより小さい報告会社でなくなるまで、より小さな報告会社に適した報告免除を利用することができることである。注目すべきは、(I)第2四半期の最終営業日までの非関連会社が保有する株式時価が2.5億ドル未満である場合、または(Ii)第2四半期の最終営業日までに完了した直近の会計年度の年収が1億ドル未満であり、第2四半期の最終営業日までの非関連会社が保有する株式時価がbrドル未満である場合には、引き続き規模の小さい報告会社であることに注意されたい。
注目すべきは、ナスダックの持続的な上場要求を満たしていないことが、注目すべき普通株の銘柄取得につながる可能性があることだ。
もしbrがナスダックの持続的な上場要求を著しく満たしていなければ、例えば会社の管理要求或いは最低終値要求のように、ナスダックは措置を取って、顕著な普通株を退市することができる。このような退市は、注目すべき普通株の価格にマイナス影響を与える可能性があり、希望時に注目すべき普通株を売却または購入する能力を弱める可能性がある。一旦カードを取られると、注意すべきいかなる回復も上場規定を遵守する行動が注目すべき普通株の再上場を許可し、市場価格を安定させたり、注目すべき普通株の流動性を改善したり、注目すべき普通株がナスダック最低買収入札要求を割ってしまうことを防止したり、将来ナスダックの上場要求に合わないことを防止することができることに注意すべきである。
注目すべきは、経営陣の上場企業の経験が限られているということだ。上場企業の結果として、Attenantは追加の規制コンプライアンス要求に制約され、これらの要求はAttenantが効率的に運営できるように効率的に管理されなければならない。
注目すべきは、 は上場企業として運営されておらず、大量の法律、会計、その他の費用が発生し、これらの費用は民間会社 によるものではないことである。著名な管理チームを構成する個人が上場会社を管理する経験は限られており, かつ上場企業に関する複雑かつ変化していく法律を遵守していない経験がある。注意すべき管理チームや他の人員はコンプライアンスに多くの時間を投入する必要があり、有効あるいは効率的に を管理することが上場会社への移行を管理できない可能性があることに注意されたい。
注目すべきは, が証券集団訴訟の影響を受ける可能性があることである.
従来、証券集団訴訟は、ある会社の証券市場価格が下落した後に提起されることが多かった。このリスクは特に注目すべきであり、バイオテクノロジーと製薬会社は近年著しい株価変動を経験しているからである。このような訴訟に著しく直面すれば、巨額のコストと経営陣の注意力や資源の分流を招く可能性があり、顕著な業務を損なう可能性がある。
| 項目 1 B. | 未解決 従業員意見 |
は適用されない.
| 74 |
| プロジェクト1 C。 | ネットワーク·セキュリティ |
統治する
取締役会と監査委員会監督
私たちの取締役会は、ネットワークセキュリティ、リスク開放、政策とやり方、および管理層が取った検出、brがこのようなリスクを監視し、制御し、これらの開放が私たちの業務、財務業績、運営と名声に与える潜在的な影響のステップを審査し、管理層と議論するために、監査委員会に監督責任を委託している。 監査委員会は関連するネットワークセキュリティ問題とbrリスクを解決し、幅広いテーマをカバーするためにプライバシーとデータセキュリティに関する報告とプレゼンテーションを受信した。このような報告書とプレゼンテーションは役員情報技術によって提供される。監査委員会に報告書を提出するほか、社内で何らかのセキュリティ事件を報告し、適切な場合には、速やかに監査委員会に報告するための合意を作成した。
情報技術の取締役
取締役は社内ネットワークや情報セキュリティのあらゆる面を管理しています。DIT担当:
| ● | 重要な計算環境とネットワーク環境のセキュリティ政策、基準、要求を制定する | |
| ● | Brの潜在的なセキュリティ脅威、関連ネットワークイベント、および監視是正措置の実行を監視することによって、重要な自己所有およびホスト資産およびリソースを不正アクセスから保護する; | |
| ● | ネットワークシステムとアプリケーション上で一致した方法で の注目すべきセキュリティポリシーとネットワークと情報セキュリティ計画の遵守を促進する | |
| ● | 安全分野では 安全思想リーダーを提供する. |
我々のDITはネットワークセキュリティ脅威からの重大なリスクを評価·管理する上で重要な管理役割を果たしている。DITは豊富な技術指導経験とネットワークセキュリティ専門知識を持っており、これらの経験は約20年の経験から来ており、私営と上場会社で高級職務を担当し、クラウドシステムの構築師とネットワークエンジニアを務め、専らフォーチュン500強会社のためにデータとクラウド安全の技術設計と実施を行っている。
リスク 管理と戦略
私たちの情報と私たちの顧客の情報を機密性、完全性、または可用性の不正なリスクから保護するために、合理的に設計されたネットワークおよび情報セキュリティ計画を維持します。我々の計画は、プロバイダおよびプロバイダからの第三者リスクを含むネットワークセキュリティ脅威のリスクを評価、識別および管理するための政策、プラットフォーム、プログラム、およびプロセスを含み、この計画は、通常、情報資産の損失または損害を最大限に低減し、イベント解決を促進するために、セキュリティイベントおよび脅威をタイムリーに識別して応答することを目的としている。
我々 は,重要なネットワークの継続的でリアルタイムに近いセキュリティ監視を保持し,調査,行動,ネットワークセキュリティイベント に対応する.このセキュリティ監視は、利用可能であれば、例えば、近リアルタイムデータ関連、態勢感知報告、活動イベント調査、ケース管理、傾向分析、および予測セキュリティ警報のようなツールを利用する。我々は各種のメカニズムを通じてネットワークセキュリティ脅威からのリスク を評価、識別と管理し、これらのメカニズムは不定期にデスクトップ練習を含めて、私たちの準備状況とイベント応答プロセス、業務部門評価、制御格差分析、脅威モデリング、影響分析、内部監査、外部監査、浸透テスト及び第三者を招いて私たちの情報セキュリティ計画を分析することを含む可能性がある。我々は、脆弱性 テストを行い、識別された脆弱性の深刻さ、重要な顧客および私たちの顧客への潜在的な影響、および発生する可能性を評価します。 我々は、セキュリティポリシーに基づいてその機能を維持するために、セキュリティ制御を定期的に評価します。リスク評価プロセスの一部として,公認されている掲示板,第三者,その他のソースからネットワークセキュリティ 脅威情報を取得する.また,キーインフラストラクチャエンティティとして,多くの機関と連携し,ネットワークやキーインフラの保護を支援し,さらに我々のネットワークセキュリティ脅威情報に情報を提供する.
| 75 |
イベント応答に関しては、会社は、セキュリティイベントに応答するための汎用的なフレームワークを提供するために、ネットワークセキュリティイベント応答計画およびデータプライバシーイベント応答計画(総称してDIRPと呼ぶ)を通過する。このフレームワークは、DITによってDITに識別または報告されたセキュリティイベントに識別、検証、分類、記録、および応答するプログラムを確立する。DIRPは、重要な情報および計算資産を保護する機能またはサービスを必要とするすべての重要者(請負者およびパートナーを含む)、および会社が所有または管理するすべてのデバイスおよびネットワーク サービスを実行するように適合されている。
DIRPは,上級管理職や他の重要な利害関係者に調査結果を報告し,適切な場合に状況を理解して参加させるなど,事故を調査·抑制·緩和するための協調的な多機能方法を策定した。一般的に、私たちのイベント応答の流れはNIST(国家標準と技術研究所)の枠組みに従い、準備、検出と分析、抑制、根絶と回復、および事件後の救済の4つの段階に重点を置いている。
ネットワークセキュリティリスクの影響
2023年には,我々の業務戦略,運営結果,財務状況に重大な影響を与える可能性が高いと考えられるネットワークセキュリティホールも発見されなかった.
| 第 項2. | 特性 |
私たちのbr社の本社は現在カリフォルニア州フォスター市にあり、レンタル期間は4年で、残り3年1ヶ月 です。現在のレンタル料は毎月43,894ドルで、毎年3.0%上昇している。
| 第 項3. | 法的手続き |
私たちは通常の業務過程で発生したクレームに関する訴訟や他の法的手続きに時々巻き込まれる可能性があります。私たちは現在、私たちの運営業績、財務状況、またはキャッシュフローに重大な悪影響を及ぼす可能性のある法的手続きに参加していません。
| 第 項. | 鉱山安全情報開示 |
は適用されない.
| 76 |
第 第2部分
| 第 項5. | 登録者普通株,関連株主事項及び発行者が株式証券を購入する市場 |
市場情報
私たちのbr普通株はナスダック資本市場に看板を掲げて上場して、コードは“NTBL.”です
記録保持者
私たちの譲渡代理によると、2024年3月29日現在、私たちの普通株の登録保有者は138人です。私たちは他の株主とは違う投票権を持っている株主はいない。一部の株式は“街道”名義で所有されているため、当該等の株式の実益所有者の数は知られていないか、または上記の数字に含まれている。この数の登録所有者は,その株式が他のエンティティが信託形式で保有する可能性のある株主 も含まれていない.
配当をする
私たちは株主に現金配当金を支払うことを発表したり、支払ったことがありません。私たちは現在、私たちの業務運営のために、未来のすべての収益(あれば)を維持すると予想している。さらに、イスラエルの法律によると、私たちが普通配当金を支払う能力はbrによって制限される可能性がある。
エージェントに接続する
我々の 委譲エージェントはEquiniti Trust Company,LLCである.Equiniti Trust Company,LLCの電話番号は(718)921-8124である.
最近販売されている未登録証券
ない。
| 第 項6. | [保留されている] |
| 第 項7. | 経営陣の財務状況と経営結果の検討と分析 |
以下、我々の財務状況と経営結果に関する議論は、我々の財務諸表と共に および本年度報告書の他の部分に含まれるこれらの報告書に関する注釈を読むべきである。歴史財務情報以外に、以下の討論と分析には、リスク、不確定性、仮説に関連する展望的な陳述も含まれている。“前向きな陳述に関する警告説明”を参照されたい。多くの要素、“第1 A項”で議論されている要素を含むため、選択されたイベントに対する実際の結果および時間は、これらの前向き陳述において予想されるものとは大きく異なる可能性がある。リスク要因“ および本年度報告における他の部分。
概要
注目すべきは,臨床段階のプラットフォーム療法会社が,癌患者のために正確な予測薬物を開発していることである。その独自のPredictive Precision 薬物プラットフォームやPPMPにより,有名なバイオシミュレーション患者の癌治療である離体する(インビトロ)およびbr}は、単一の患者がその実際の治療に臨床反応を生じるかどうかを正確に予測することを求めている。公認医療センターと行った4つの独立臨床検証試験では,PPMP予測応答者の精度は83%−100%であった。従来の精密薬物よりも優れた医療効果を追求するために,PPMPは遺伝や他のバイオマーカーに依存しない。代わりに,PPMP産生細胞の薬物に対する生物反応の多次元 測定を行い,これらのデータを計算アルゴリズムにより統合して患者反応予測器に変換する。
PPMPは注目すべき人が治療を開始する前に期待される臨床的に有効な患者を識別と選択でき、そして潜在的にこの患者群の中で迅速な治療発展を実現することを目的としている。注目すべきは,そのPPMPを用いて臨床活性を示した許可と開発あるいは共同開発の研究化合物 を評価し,患者の反応改善やbr}が従来の薬物開発と比較してこれらの化合物を開発する成功率,速度,価値であることに注意されたい。
PPMPを使用して、注目すべき目標brは、納得できる臨床活動を示しているが、この活動は、治療を受ける患者の10%~30%の患者サブセットに限定されるので、放棄された許可内資産である。これは、開発、規制承認、商業化に成功する障害である可能性があるからである。brは、そのPPMPプラットフォーム上で数百の資産をスクリーニングし評価することに注意し、その中の多くの資産について、どの患者が臨床反応を行うかを比較的高い精度で予測することができると信じている。これは,予測されたbr応答者を臨床試験に選択的に組み込む機会を提供し,より高い応答率を提供する臨床応答者におけるこれらの資産の発展を加速させた。
注目すべきは,そのPPMPを用いてbr}の予測的正確な薬物組み合わせを作成することが求められていることである。PPMPはその最初の2種類の候補予測正確な薬物の選択においてすでに著しい指導作用を得ており、この2種類の候補薬物は急性骨髄性白血病(AML)のプラットフォーム予測トランスポンダの開発に適している。PPMPからのリード資産はVolasertibであり、1種のPolo様キナーゼ1(“PLK 1”)阻害剤 であり、多種の癌細胞の細胞周期停滞とアポトーシスを誘導できることが証明された。注目すべきは、6人の患者に対する用量最適化前奏を含む成人AMLにおけるVolasertib の第2段階試験が2024年第2四半期に開始されることが予想されることである。これらの最初のbr 6名の患者の結果は2024年第4四半期に発表される予定であり、第1陣のPPPP予測応答者は同一四半期に登録される予定である。また,CicloMed LLC(“CicloMed”)と共同開発し,急性骨髄性白血病患者にファンスピロを提供していることに注意されたい。この共同開発パートナーシップは,PPMPが臨床前にフォス·キシロップへの応用に成功した結果である。また、内部許可を行うためにPPMPを使用して他の注目されている資産を決定し、その開発パイプを構築する際に他の資産を迅速に追跡することに留意されたい。
患者の細分化,疾患,医療結果の予測におけるPPPPのカバー範囲を拡大していくことにより,卓越した目標は正確な医学領域を予測するリーダーとなり,患者が彼らに最適な効果をもたらす可能性の最も高い治療を求めて受け入れる方式 −が患者やヘルスケアコミュニティに大きな影響を与えることである。
合併取引
2023年10月16日、合併協定により、当社、合併子会社と卓越実験室会社の間の合併子会社は卓越実験室会社と合併してbr卓越実験室会社に合併し、卓越実験室会社は合併後も引き続きbr社の存続実体と完全子会社として機能した。合併発効時に、いかなる株主も行動しない場合、合併前の有名な実験室会社のS普通株1株当たり額面0.001ドル(合併前S社の流通株を含む)の発行および流通株(合併前S社の発行済み流通株を含む)は、br}0.0629株会社普通株(“交換比率”)、1株当たり0.35新シェケル額面(“会社普通株”)に変換され、合併発効後、当社は直ちに発行済み会社と発行済み会社普通株に対して35株1株の逆株式分割(“逆株式分割”)を行う。合併及び合併協議で行われる取引を完了した後、(I)前の有名株式有限会社の株式所有者は完全な償却ベースで当社の約71.9%の発行済み株式を保有し、仮に全部承認株式証を行使して94,988株会社の普通株を購入し、160,635株会社の普通株を含み、会社が成約時及び会社の成約時の現金純分に基づいて調整した後に負担するS普通株を購入する。および(Ii)前血管生物製薬有限公司の株主は同社の約28.1%の既発行株を持っている。
合併は、財務会計や報告目的に用いられる株式取引所による逆資本再編とみなされる。有名な実験室会社は会計購入者とみなされている。その株主は合併後に会社をコントロールしたので、血管生物遺伝有限会社は合法的な買収者であるにもかかわらず。したがって、私たちの合併財務諸表に反映される資産と負債および歴史的業務は、有名な実験室会社がずっと報告会社であるかのように有名な実験室会社の資産と負債である。普通株式、株式承認証とオプションに言及したすべての内容は合併後、逆分割後の基礎の上で提出した。
財務 概要
| 77 |
収入 と収入コスト
注意すべき は、規制部門の承認を得て、その任意の治療製品が商業化されない限り、どの製品の販売からも実質的な収入を得ないことが予想される。
注目すべきは、2023年12月31日と2022年12月31日までの年間において、アウトソーシングプロバイダとして、限られた基礎の上で何らかの診断サービスを提供し続けていることに留意されたいが、このような活動は、その主要かつ持続的な中央業務を代表するものではなく、そのため、同社は実質的なbr収入を得ることはないと予想されることに留意されたい。
注目すべきは,実験室サンプルを扱う診断テストによりテスト結果を顧客に提供し,同社は診断サービスの収入を確認し,その予想される対価を反映し,顧客との契約規定の義務を履行していることである。収入記録には,顧客との契約の決定,契約中の履行義務の決定,取引価格の決定,取引価格の履行義務への分配,エンティティが契約履行義務を履行する際に収入を確認する5段階収入確認モデルを採用した。通常、 は、クライアントからの契約または購入注文を有しており、実行すべき診断サンプル数を含む必要な条項が規定されていることに留意されたい。約束履行義務のある事前支払いは何も受けていないということに注意されたい。そのため、2023年12月31日と2022年12月31日まで、繰延収入は何も記録されていない。2023年12月31日と2022年12月31日まで、2023年12月31日までの契約資産 は記録されていないことに注意されたい。顧客に請求書を発行できない契約義務は何も完了していないことに注意されたい。 サービス収入の物質コストは販売コストと非物質コストとして記録され、運営費用に記録されている。
研究と開発費
研究開発費は発生時に費用を計上する。研究開発費には、研究開発活動に関連する賃金と人員コスト、材料コスト、外部臨床薬物製品の製造コスト、外部サービスコスト、研究開発設備の修理、メンテナンスと減価償却コスト、および研究開発活動のための施設コストが含まれる。将来の研究開発活動のための貨物またはサービスを提供するための前金は、貨物が関連サービスを交付または実行する際に資本化され、費用に計上されない。重要なのは、それが貨物を送達することが予期されているか、またはサービスを提供することが予期されているかどうかを評価し、エンティティが貨物の提供またはサービスの提供をもはや予期していない場合に、資本化された前払いの任意の部分の費用を計上することである。
注目すべきは, がVolasertibとFosiclopiroxの開発と承認に必要な余剰研究を展開することにより,その研究と開発が著しく増加することである。将来の研究開発費には
| ● | 従業員関連費用、例えば、給料、ボーナスおよび福祉、コンサルタント関連費用、株式報酬、管理費用関連費用、および注目すべき研究開発者の出張関連費用; |
| ● | CROおよび上記臨床研究の実施を支援するコンサルタントとの合意による費用; |
| ● | 臨床試験に関連する製造コストと包装コスト,NDA届出支援に必要な安定性やその他の研究,商業投与のための薬品の生産; |
| ● | PPMP、Volasertib、Fsciclopiroxに関する研究開発費用、および他の注目すべき製品は開発を選択する可能性がある配合、 と |
| ● | 研究を賛助する費用は である. |
研究と開発活動は注目すべき業務計画の核心であり続ける。臨床開発後期にある製品 は通常臨床開発早期にある製品よりも高い開発コストを有しており,これは主に後期臨床試験の規模と持続時間が増加しているためである。注目すべきは,人員や報酬コストの増加に伴い,今後数年でその研究開発費が大幅に増加することであり,後期臨床研究を行い,VolasertibやFosiclopiroxおよび任意の他の未来製品の承認を求める準備をしていることに注意されたい。
| 78 |
VolasertibおよびFsciclopiroxおよび任意の他の未来製品の臨床試験持続時間、コスト、および時間は、これらに限定されない様々な要因に依存するであろう
| ● | 承認に必要な試験回数; | |
| ● | 個々の患者の実験コストは | |
| ● | 試験に参加した患者数; | |
| ● | 実験に含まれるサイト数; | |
| ● | 実験を行った国·地域 | |
| ● | 条件に適合する患者を登録するのに要する時間長; | |
| ● | 患者が受ける用量の数 | |
| ● | 患者の中退率や中途停止率 | |
| ● | 規制当局が要求する潜在的な追加的な安全監視または他の研究; | |
| ● | 患者のフォローアップ時間; | |
| ● | 規制承認のスケジュールと受信;および | |
| ● | 注目すべき候補製品の効果と安全性の概要。 |
一般料金 と管理費用
一般費用および行政費用は、主に、賃金、福祉、および株式ベースの報酬を含む従業員に関連する費用を含み、行政、財務、業務発展、施設、および行政機能のための人員のために使用される。その他の重大なコストには、研究開発費に含まれていない施設費用、特許や会社の事務に関連する法律費用、会計費用、税務、コンサルティングサービス費用が含まれています。
注目すべきは、将来的に一般的かつ管理費が増加し、その持続的な研究開発活動を支援することが予想されることである。 これらの増加には、給与や従業員関連費用、 および外部コンサルタント、弁護士、会計士の費用を含む人員募集に関する増加コストが含まれる可能性がある。また、ナスダック資本市場と米国証券取引委員会の要求、保険、投資家関係コストの遵守など、上場関連コストが増加することに注意されたい。
所得税 税
2023年12月31日と2022年12月31日までの年度では,所得税支出や福祉は確認されていない。注目すべき繰延税金資産は主に経営赤字の純繰越から構成されている。2023年12月31日現在、連邦と州の純運営赤字は約1兆208億ドルに転換し、海外純運営損失は2兆636億ドルに転換していることに注目すべきである。繰延税金資産の全額推定手当を維持することに注意すべきである。brは持続的な利益運営が実現されておらず、近いうちに課税収入がないことに注目すべきである。したがって、設立以来、所得税割引は何の記録も記録されていないことに注意されたい。
| 79 |
運営結果
2023年12月31日までと2022年12月31日までの年次比較
次の表に2023年12月31日までの年度と2022年12月31日までの年度と比較した顕著な経営実績(単位:千)を示す
| 2023 | ||||||||||||
| 12月31日までの年間 、 | 増す | |||||||||||
| 2023 | 2022 | (減少) | ||||||||||
| サービス収入 | $ | 310 | $ | 8 | $ | 302 | ||||||
| サービスコスト | 197 | - | 197 | |||||||||
| 運営費用 : | ||||||||||||
| 研究開発 | 4,706 | 7,776 | (3,070 | ) | ||||||||
| 通常 と管理 | 10,064 | 5,156 | 4,908 | |||||||||
| 運営損失 | (14,657 | ) | (12,924 | ) | (1,733 | ) | ||||||
| その他 収入(費用)、純額 | 3,393 | (1,483 | ) | 4,876 | ||||||||
| 純損失 | $ | (11,264 |
) | $ | (14,407 | ) | $ | 3,143 | ||||
サービス 収入
2022年12月31日までの年度と比較して、2023年12月31日までの年間サービス収入は000万ドルから30万ドルに増加した。この30万ドルの増加は卓越して他人にbrサービスを提供したためだ。
研究と開発費
研究·開発費は2022年12月31日までの年度と比較して、2023年12月31日現在の780万ドルから470万ドルに低下した。310万ドルの減少、または66.0%の減少は、主に以下の計画の増減によるものである
| 2023 | ||||||||||||
| 12月31日までの年間 、 | 増す | |||||||||||
| 2023 | 2022 | (減少) | ||||||||||
| 工学.工学 | $ | 829 | $ | 2,176 | $ | (1,347 | ) | |||||
| 医療事務 | 295 | 284 | 11 | |||||||||
| 操作可能な | 90 | 210 | (120 | ) | ||||||||
| 科学プロジェクト | 2,949 | 3,871 | (922 | ) | ||||||||
| ヴォラセテブ | 463 | 623 | (160 | ) | ||||||||
| 小計 | 4,626 | 7,164 | (2,538 | ) | ||||||||
| 他にも | 80 | 612 | (532 | ) | ||||||||
| 合計する | $ | 4,706 | $ | 7,776 | $ | (3,070 | ) | |||||
工事費 はPPMPプラットフォームをサポートするために必要なコストである.工事費は2022年12月31日までの年度と比較して、2023年12月31日現在の220万ドルから80万ドルに低下した。130万ドル減少、あるいは61.9%減少したのは、2022年12月31日までの年度内に、2023年12月31日までの年度内に賃金総額が増加しなかったため、賃金総額と賃金関連コストが70万ドル減少し、技術コストが10万ドル減少し、施設コストが60万ドル減少したためである。
医療費用には臨床計画費用が含まれている。医療事務費は2023年12月31日と2022年12月31日まで変わらない。
| 80 |
運営費用 は運営実験室のコストである。2022年12月31日までの年度と比較して、2023年12月31日までの年度の運営費は20万ドルから10万ドルに低下した。10万ドル、すなわち57.1%減少したのは、賃金と賃金に関する費用が10万ドル減少したためだ。
科学プロジェクトは探索的な研究と開発プロジェクトである。2023年12月31日までの1年間で、科学プロジェクト支出は90万ドル減少し、減少幅は23.8%で290万ドルに低下したが、2022年12月31日までの年度は390万ドルだった。減少の主な原因は,賃金と賃金関連費用が100万ドル減少し,Fosiclopiroxプロジェクトに関するものが50万ドル減少したが,実験室用品が10万ドル増加し,施設コストが40万ドル増加し,減価償却が10万ドル減少したことである。
Volasertib は癌に関連する小分子であり、いくつかの用途を持っている。Volasertib費用 は,2022年12月31日までの年度と比較して,2023年12月31日までの年度の60万ドルから50万ドルに低下した。10万ドル減少したり25.7%減少したのは,Volasertib関連の実験室コストが10万ドル減少したためである。
一般料金 と管理費用
2022年12月31日までの年度と比較して,2023年12月31日までの年度の一般·行政費は520万ドル から1010万ドルに増加した。490万ドルまたは95.2%増加したのは、主に合併に関連する第三者請負業者が530万ドル増加し、主に役員と上級管理者保険に関連する保険支出が30万ドル増加し、株式ベースの報酬が30万ドル増加したが、給与と給与関連支出の80万ドル減少と減価償却支出20万ドルの減少によって相殺された。
その他 収入(費用)、純額
2022年12月31日までの年度と比較して、2023年12月31日までの年度は、他の純収入(費用)が150万ドルの他費用純額から340万ドルの他収入純額に増加した。4,900,000ドルまたは328.8%増加したのは、主にDシリーズ金庫の転換損失1,700,000ドル、Cシリーズ金庫および償還可能優先株式証の派生公正価値が7,500,000ドル増加し、利息収入が1,000,000ドル増加したが、2022年までの年度購買力平価ローンに関する1,000,000ドルの債務を免除したためである。
純損失
純損失は2022年12月31日までの年度と比較して、2023年12月31日までの年度の1,440万ドルから1,130万ドルに低下した。310万元減少したのは21.8%であり、上記の要因によるものである。
流動性資本資源財務要件
概要
成立から2023年12月31日までの間に、顕著なのは主にその発展努力による運営損失と負のキャッシュフローを受け、累計損失は8,230万ドルに達した。著しくは,主に優先株と安全株の発行および2023年10月16日の合併完了後の現金流入によりその運営に資金を提供した。注目すべきは, は予見可能な未来に正のキャッシュフローはないと予想され,既存の現金資源 が今後12カ月の運営を維持するのに十分であるとも信じていないことである私たちは現在、発生した持続的なコストと費用を支払い、私たちの製品開発に資金を提供し、業務計画を実行できるように、私たちのコスト構造を支援するのに十分な収入 を生成する必要があります。もし私たちが私たちの業務計画に資金を提供するのに十分な収入を生み出すことができない場合、私たちは債務および/または株式証券を売却することでこのような融資を集めることを求めるつもりです。株式を増発すると、既存の株主の持分が希釈されます。もし私たちが必要な時に追加資金を得ることができない場合、あるいは私たちが受け入れられる条項でこのような資金を得ることができない場合、私たちは業務計画を実行したり、発生したコストや費用を支払うことができなくなり、私たちの業務、財務状況、運営結果に大きな悪影響を与えます。私たちが十分な収入を創出したり、私たちの製品を商業化するために十分な資本を調達したりしても、私たちは持続可能な経営企業として経営を継続する能力は、私たちの収入が私たちの業務運営を支援するレベルに達したときにのみ実現できます。
2024年4月1日現在、注目すべき現金と現金等価物は820万ドルである。現在の現金状況と会社計画のbr費用稼働率によると、経営陣は卓越が2024年11月までその運営に資金を提供できると信じている。この推定 に基づく仮定は、誤りであることが証明され、予想よりも早くその資本リソースが枯渇する可能性があることに留意されたい。
は少なくとも予見可能な未来には,運営損失が大幅に増加することが予想されることに注意されたい。Volasertib、Fosiclopirox、または任意の他の未来の製品の開発が成功し、規制部門の承認を得ない限り、製品収入は生じないことに注意されたい。計画された臨床試験の時間や他の研究や開発活動への支出により,NOTICEの純損失は四半期間と年度の間に大きく変動する可能性がある。注目すべきは,その費用は2024年に大幅に増加すると予想され,VolasertibとFosiclopiroxの臨床開発を進め, を上場企業として運営していることに注意されたい。注目すべきは、予見可能な未来には、運営に正のキャッシュフローが生じないことが予想されることである。注目すべき経営陣は、予見可能な未来には、研究や開発活動の拡大により、規制部門の承認を得る前に、追加的な重大な損失が生じ続けると予想している。規制部門の承認は保証されず、永遠に得られないかもしれない。そのため,これらのことは,継続的な経営を続ける企業としての持続的な経営能力を大きく疑っている。
前述の展望的情報は私たちが合理的だと思う仮説に基づいて誠実に作成された。しかし,予測の到達可能性や予測に基づく仮定の信頼性を保証することはできない. 予測は,どの試みも運営結果に固有の不確実性の影響を受け,特に新製品やサービスに関連している場合である.使用されたいくつかの仮定は必然的に現実にならず、予期せぬイベントが発生することになる。 したがって、運営の実際の結果は予測とは異なる可能性があり、このような変化は実質的である可能性があり、私たちに不利である。 したがって、このような結果が実現される保証はない。また、技術変化、新製品発表、競争圧力により、現在の計画を変更する必要があるかもしれません。
| 81 |
未来の資金需要
注目すべきbrは、注目すべき株式、債務、協力および許可スケジュール、または他のソースを公開または非公開で発行することによってさらなる資金を得る必要があると予想され、その要件は、多くの要因に依存するであろう
| ● | 注目すべき候補製品の薬物開発作業、臨床前開発活動、実験室テストと臨床試験の範囲、時間、進捗とコスト |
| ● | 注目すべき臨床項目の数と範囲は追求を決定する |
| ● | 規制当局が重要な候補製品のコスト、時間、結果を検討する準備と受け入れ |
| ● | 開発と商業製造活動の範囲とコスト |
| ● | 上場許可を得た場合、注目すべき候補製品の商業化に関するコストと時間 |
| ● | 他の候補品や技術の程度を大幅に買収することができるかもしれない |
| ● | 特許出願の準備、提出、起訴、注目すべき知的財産権、および知的財産権に関連する訴訟の費用; |
| ● | は、有利な条件で協力関係を確立し、維持することができることに留意されたい |
| ● | 注目すべきbrは、注目すべき候補製品の開発および最終的に注目すべき製品の販売を支援する人員、br}FDA承認を含む、運営システムおよび注目すべき合格者を吸引、採用、保持する能力を強化するために努力している |
| ● | 注目すべき業務、財務、管理システムの実施 |
| ● | 上場企業としての関連コスト。 |
これらまたは他の変数のいずれかは、任意の注目すべき候補製品を開発する際の結果が変化し、候補製品開発に関連するコストおよびスケジュールを著しく変更する可能性がある。また,注目すべき運営計画は将来的に変化する可能性があり,運営需要やそのような運営計画に関連する資本要求を満たすための追加資本が必要であることに注意されたい。
十分な追加資金は許容可能な条項で有名な会社に提供できないかもしれないし、全くできないかもしれない。十分な金額またはその許容可能な条項で資金を調達できない場合、VolasertibおよびFosiclopiroxまたは任意の将来の製品の開発または商業化を大幅に延期、削減または停止しなければならないか、または運営を停止しなければならない可能性がある。
株式または転換可能な債務証券を売却することによって追加の資本を調達する程度を顕著にするために、顕著な株主の所有権権益が希釈されるであろう。これらの証券の条項は、清算または一般株主の権利に悪影響を及ぼす他の特典を含む可能性がある。債務融資および優先持分融資(利用可能な場合)は、追加債務を生成すること、資本支出を行うこと、または配当を宣言することなど、特定の行動を著しくとる能力を制限または制限する能力を含む契約に関連する可能性がある。
が第三者との協力、戦略連合またはマーケティング、流通または許可スケジュールによって追加の資金を著しく調達する場合、顕著な技術、将来の収入フロー、br計画または提案された製品を研究する貴重な権利を著しく放棄すること、または著しく不利になる可能性のある条項で許可を付与することが要求される可能性がある。注意すべきである場合、必要なときに株式または債務融資によって追加の資金を調達することができない場合、注目すべき薬物開発または将来の商業化努力を延期、制限、減少または終了する必要があるか、または任意の未来の製品を開発およびマーケティングする権利が付与される可能性があり、そうでなければ、注意すべきbr}は自分自身を開発およびマーケティングすることをより望むであろう。
| 82 |
現在比較的早期の研究段階にあることに注意するため,持続可能な収入を生成できる製品や知的財産権の開発には大量の時間と資源が必要となる。したがって、注目すべき業務は、今後数年間、持続可能な運営収入を生み出すことはあまり不可能であり、決してそうしない可能性がある。また,営業収入が著しく発生できる程度では,正の収益と運営 キャッシュフローが著しく実現できる保証はない.
キャッシュフロー
2023年12月31日までと2022年12月31日までの年度比較
次の表には、以下の各時期の主要な現金源と用途(千計)を示す
| 十二月三十一日までの年度 | 増す | |||||||||||
| 2023 | 2022 | (減少) | ||||||||||
| 経営活動用の現金 | $ | (13,716 | ) | $ | (11,642 | ) | $ | (2,074 | ) | |||
| 投資活動が提供する現金 | 15,540 | 924 | 14,616 | |||||||||
| 融資活動で提供された現金 | 7,822 | 9,898 | (2,076 | ) | ||||||||
| 現金および現金等価物の純増加(減額) | $ | 9,646 | $ | (820 | ) | $ | 10,466 | |||||
経営活動に使われるキャッシュフロー
2023年12月31日および2022年12月31日までの年度まで、経営活動に用いられた現金純額はそれぞれ1,370万ドルおよび1,160万ドルで、210万ドルまたは約17.8%増加した。この増加は,主に役員や上級管理者保険に関する前払い保険が約90万ドル増加したことと,他の運営資本 が増加したためである。
投資活動のキャッシュフロー
2023年12月31日までの年度の投資活動の現金純額は1,550万ドルで、2022年12月31日までの年度の現金純額は90万ドルで、1,460万ドルまたは約1,581.8%増加した。増加の主な原因は合併に関連した現金収入だ。
融資活動のキャッシュフロー
2023年12月31日までの年度融資活動の現金純額は780万ドル、2022年12月31日までの年度の現金純額は990万ドルで、210万ドルか約21.0%減少した。2023年、この金額は転換可能な優先株と関連外管局合意を発行した純収益である。2022年、これらの額はオプションの行使、転換可能な優先株の発行、OnCoheros安全協定の発行から受け取った純収益である。
重要な会計政策と試算の使用
NOTINGの財務状況と運営結果の検討と分析NOTIGNに基づく財務諸表は、公認会計基準に基づいて作成された。これらの財務諸表を作成するには、資産、負債、収入、および費用報告金額に影響を与える推定および判断、および財務諸表における開示または資産および負債に注意する必要がある。その推定と判断を顕著に継続的に評価し、計算すべき支出、繰延税金項目の資産推定値の準備及び無形資産の推定値に関する推定と判断を含む。その推定は,歴史的経験,既知の傾向,事件,および当時合理的とされていた様々な他の要因に基づいており,これらの要因の結果は資産や負債の帳簿価値を判断する基礎を構成しているが,これらの資産や負債の帳簿価値は他の源 からは明らかに見えないことに注意されたい。異なる仮定または条件では、実際の結果は、これらの推定値とは異なる可能性がある。
| 83 |
我々の財務諸表は、経営陣が財務諸表を作成する過程で使用される会計ポリシーおよび推定および仮定の影響を受ける。 これらのポリシーの完全な要約は、本明細書の他の部分に含まれる財務諸表付記2に含まれる。我々はすでに以下のように我々の財務状況、運営結果、キャッシュフローを報告する上で特に重要な会計政策を決定しており、これらの政策は管理層が重大な判断を適用する必要がある。
株に基づく報酬費用
我々 は公正価値確認条項財務会計基準委員会会計基準編纂(“FASB ASC”)718を採用した。また、米証券取引委員会は第107号“従業員会計公報”を発表した株式支払“ (”SAB 107“)は、米国証券取引委員会の意見に基づいて補足FASB ASC 718アプリケーションガイドを提供する。FASB ASC 718によれば、確認された補償コストは、FASB ASC 718の規定に従って推定された付与日公正価値 によって付与された株式ベースの支払いのすべての補償コストを含む。
我々 はBlack-Scholesオプション定価モデルを用いてオプション公正価値を推定する.オプション定価モデルは、多くの仮定を必要とし、その中で最も重要なのは、予想株価変動、予想される付与前失敗率、および予想されるオプション期間(付与日からオプション行使または満了までの時間量)である。
すべての が非従業員に発行した株式オプションまたは他の権益ツールは、卓越して受信した商品またはサービスと引き換えに、発行された権益ツールの公正価値に従って入金される。付与時に直ちに付与されていない非従業員権益に基づく支払い は、付与期間中に料金として記入される。
公正価値計測
公正価値会計は、公正価値で財務報告書において日常的に確認または開示されるすべての金融資産および負債に適用される。現金および現金等価物,売掛金,売掛金および売掛金などの金融商品は満期日が相対的に短いため,公正価値に近い。
貸借対照表では,公正価値で恒常的に記録されている資産と負債は,その公正価値を計測するための投入に関連する判断レベルに基づいて分類される.公正価値は、計量日に市場参加者間で秩序的に取引される資産または負債の元本または最も有利な市場上で負債を移動させるために、受信された資産の交換価格または支払いを終了する価格として定義される。
株式発行によって発行された引受権証 は現金または無現金で行使される可能性があり、発行された株式数が可変である は派生負債とみなされるため、公正価値に応じて計量される。
転換可能な優先株を買い戻す引受権証
注意すべき は公正価値によって経常的にそのCシリーズ株式証負債(3級負債に分類)を計量し、総合経営報告書に記録されている公正価値変動及び総合損失計によって提出し、株式証行使、満期或いはその他の 事実及び情況を承認するまで持分証負債を権益ツールに再分類する。公正価値は オプション定価逆解法を用いて決定される.Cシリーズ権証負債2023年12月31日現在の公正価値は、Black-Scholes オプション定価モデルを用いて権証公正価値を推定して決定される。
開発コストを計算すべきだ
注目すべきは,臨床研究組織(CRO)との契約義務による費用は費用を計算することである。これらの 契約の財務条項は協議する必要があり、これらの条項は契約によって異なり、支払いの流れが材料やサービスを提供する期限 と一致しない可能性がある。適切な費用をサービスおよび努力の支出期限に適合させることにより、財務諸表に適切な試験費用を反映させることを注目すべきである。
| 84 |
償還可能な転換優先株
注意すべき は,発行日に公正価値記録に変換可能優先株を償還することができ,例外がない限り発行コスト を差し引くことである.2022年12月31日現在、償還可能な転換可能優先株は、一部の自社の制御範囲内ではない償還特徴が含まれているため、付属貸借対照表で株主赤字以外の仮株式とされている。償還可能な転換可能な優先株は一般に償還できない;しかし、制御権事項にいくつかの変動が発生し、清算、売却または譲渡の有名な制御権を含む場合、償還可能な優先株の所有者は重要な登録証明書の条項に基づいてその清算優先権を獲得する権利がある。償還可能な優先株の帳簿価値は であり、当該等の清算イベントが発生する可能性がある場合には、その清算優先順位に応じて調整される。
償還可能な転換可能優先株式株式証負債
注目すべきは、対象株式が償還可能または償還可能である場合、償還可能な転換可能優先株を購入する権利証は、公正価値によって負債に分類され、各報告期間にツールを公正価値に調整することである。償還可能な優先株を購入する引受権証は資産負債表ごとに再計量し、行使または満期になるまで再計量しなければならないが、公正価値のいかなる変動も他の収益の構成部分、総合経営報告書及び全面赤字の純額であることが確認された。著者らはBlack-Scholesオプション定価モデルを使用して、転換可能な優先株式証の公正価値を推定した。償還可能な転換可能優先持分証債務の発行に関する発売コスト は相対基準で割り当てられ、発生時に費用を計上する。合併後、このような権利証はもう普通株式に変換することができる。
収入 確認
2023年12月31日までの一年において、重要な主要な創立活動と業績義務はその独自のプラットフォームを使用して診断サービスを提供することを含み、このプラットフォームは主に主に自身の研究開発に従事する実体がbrを利用してより的確かつより効率的な薬物発見方法で治療組み合わせを識別することを含む。このような活動は主で持続的な中央業務を代表するものではない。
注目すべきは,実験室サンプルを扱う診断テストによりテスト結果を顧客に提供し,同社は診断サービスの収入を確認し,その予想される対価を反映し,顧客との契約規定の義務を履行していることである。収入記録には,顧客との契約の決定,契約中の履行義務の決定,取引価格の決定,取引価格の履行義務への分配,エンティティが契約履行義務を履行する際に収入を確認する5段階収入確認モデルを採用した。通常、 は、クライアントからの契約または購入注文を有しており、実行すべき診断サンプル数を含む必要な条項が規定されていることに留意されたい。約束履行義務のある事前支払いは何も受けていないということに注意されたい。そのため、2023年12月31日と2022年12月31日まで、繰延収入は何も記録されていない。2023年12月31日と2022年12月31日まで、契約資産は何も記録されていないことに注意されたい。 顧客に請求書を発行できない業績義務が何も完了していないことに注意すべきである。サービスの物質コスト 収入は販売コスト,サービスの非物質コストは運営費用に記入する。
FASB ASC 606によると取引先と契約した収入また,履行義務を履行する際に収入 を確認し,約束された財やサービスを顧客に譲渡することにより,これらの履行義務の履行と引き換えに注目すべき が獲得する権利のある対価格を反映している.
| 85 |
第 7 A項。 |
市場リスクの定量的·定性的開示について |
は適用されない.
| 第 項8. | 財務諸表と補足データ |
私たちの総合財務諸表は、私たちの独立公認会計士事務所の報告とともに、2023年12月31日までの年次報告書のF-1ページから始まります。
| 第 項9. | 会計·財務開示における会計担当者の変化と相違 |
ありません
| 第 9 A項。 | 制御 とプログラム |
制御とプログラムを開示する
我々は、米国証券取引委員会に開示される必要がある重大な財務および非財務情報がタイムリーに記録され、処理され、集約され、報告されることを保証するために、私たちの開示制御およびプログラムの有効性を評価している。我々の評価によると、我々の経営陣は、最高経営責任者(CEO)および最高財務責任者(CFO)を含み、本報告でカバーされた期間終了までの間、我々の開示制御および手順(1934年の証券取引法規則13 a-15(E)および15 d-15(E)で定義されているように)が有効であると結論付けた。 それにもかかわらず、私たちの開示制御とプログラムが私たちの社内人員が重要な情報を開示できなかったすべての状況を発見または暴露する保証はありません。そうでなければ、私たちの報告には他の情報を列挙しなければなりません。
経営陣財務報告内部統制年次報告書
我々の経営陣は、“取引法”に基づいて公布された規則13 a-15(F)に定義されている財務報告の十分な内部統制の確立と維持を担当している。我々の内部統制システムは、財務報告の信頼性を確保し、公認された会計原則に基づいて外部目的のために公表された財務諸表を作成·公平に掲載することを目的としている。すべての内部制御システムは, がどんなに良く設計されていても固有の限界がある.したがって、有効と判断されたシステムであっても、財務諸表の作成や列報の面で合理的な保証を提供することしかできず、誤った陳述を防止または発見することができない可能性がある。また,将来のいずれかの有効性評価を行う予測 は 条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムを遵守する程度が悪化する可能性がある.
我々の経営陣は、我々の最高経営責任者と最高財務責任者を含め、“取引法”公布の規則13 a-15(C)に基づいて、トレデビル委員会後援組織委員会が発表した“内部統制-総合枠組み”(2013)に基づいて、今年度の報告に係る期間終了までの財務報告の内部統制の有効性を評価した。評価結果によると、経営陣は、財務報告書の内部統制に対して2023年12月31日から有効であると結論した。
財務報告内部統制変更
2023 年 12 月 31 日を末日とする四半期中に、当社の財務報告に関する内部統制 ( 取引法規則 13 a—15 ( f ) および 15 d—15 ( f ) に定義される ) に重大な影響を及ぼす、または重大な影響を及ぼす合理的な可能性のある変更はありませんでした。
| 第 9 B項。 | その他 情報 |
ない。
| 第 9 C項. | 検査を阻止する外国司法管轄区に関する情報を開示する。 |
は適用されない.
| 86 |
第 第3部分
| 第 項10. | 役員·役員·会社管理 |
以下の表は、 2024 年 4 月 1 日現在の執行役員および取締役の年齢を含む特定の情報を示しています。
| 名前.名前 | 年ごろ | ポスト | ||
| 役員と役員 | ||||
| トーマス { br} ボック | 59 | 最高経営責任者兼取締役 | ||
| スコット A 。マクファーソン | 62 | 最高財務官 | ||
| ジョセフ·ワグナー | 56 | 首席科学官 | ||
| 非執行役員 | ||||
| トモ·Pバーツtsi(1) | 59 | 取締役会議長 | ||
| ピーター·ファンバーグ(1) | 63 | 役員.取締役 | ||
| ミシェル·ガレン(1) | 67 | 役員.取締役 | ||
| マイケル·ライス(1) | 59 | 役員.取締役 | ||
| 外部 取締役 | ||||
| トーマス·I·H·デュビン(1) (2) | 62 | 外部 取締役 | ||
| トーマス·グランニ(1) (2) | 59 | 外部 取締役 |
| (1) | 独立 取締役は、ナスダックのルールに管轄されています。 |
| (2) | イスラエルの法律によって定義された外部 取締役。 |
執行官
トーマス·A·バーク博士は取締役CEO兼最高経営責任者それは.Thomas Book博士は現在59歳で,2020年10月に取締役に入社し,2021年2月に臨時CEO,2021年4月に永久CEOに任命された。ボック博士は20年以上のバイオテクノロジー業界の経験と、10年間の学術医学と研究経験を持っている。2020年12月から2021年3月まで人工知能薬物設計·開発会社Ordaosの上級顧問を務めた。2020年7月から2021年1月まで、早期健康科学技術会社Tirili LLCの創業者兼CEOを務めた。配偶遺伝性BRCA癌診断にヒントを得て,ボック博士はHeritXを設立し,2015年3月から2019年2月までHeritX Inc.の最高経営責任者兼取締役を務め,HeritXは癌前ワクチン,免疫予防,遺伝子修復による癌予防の先駆者である。これまで,アレックソン製薬会社の実行管理チームで総裁医療事務上級副総裁を務め,スタートアップ企業を超まれな疾患分野のグローバルリーダーにしてきた。Alexionに加入する前に、ボック博士はノワール腫瘍部とCelgeneの全世界医療事務部を設立し、指導し、副総裁と全世界医療事務主管を務めた。Celgeneに加入する前に,Bock博士はヨーロッパに安入した血液学と腫瘍学医学取締役を務めていた。バイオ製薬業界に参入する前に、ボック博士はヨーロッパ医療センターとアメリカ国立ヒトゲノム研究所で癌、幹細胞生物学、遺伝子治療分野の医療実践と研究に従事していた。br}ボック博士はRWTH亜メダカ大学で医学博士号を取得し、コロンビア商学院で工商管理修士号を取得した。ボーク博士はコロンビア大学ビジネススクール医療諮問委員会の議長も務めた。生物製薬業界と学術医学領域における彼の広範な管理と科学経験に基づいて、ボック博士はボーク博士が取締役会に入る資格があると信じている。
ジョセフ·ワグナーは首席科学官ジョセフ·ワグナー博士は、現在56歳で、2020年6月に有名な会社に入社し、20年以上の治療学と診断学開発経験を持っている。ワグナー博士は2017年9月から2020年2月までカリフォルニア大学薬物発見連盟の執行役員を務め、2020年3月から2020年5月まで独立顧問を務め、その後有名な会社に入社した。バグナー博士は訓練された薬剤学者であり、バイオテクノロジー業界の大部分のキャリアの中でチームと組織の構築に専念し、主にベンチ研究から第2段階の臨床試験までの小分子と生物療法の開発に専念し、BriaCell Treeutics、OncoCyte、Cell Tartingなどの会社で最高経営責任者/最高技術者を務めている。ワグナー博士は腫瘍学、神経病学と心血管疾患を含む各種適応のプロジェクトを指導し、そして生物情報学の努力をリードし、新しい標的、バイオマーカーと随伴診断を開発した。ワグナー博士はロチェスター大学で神経科学学士号を取得し,デューク大学で薬理学博士号を取得した。
| 87 |
スコット·A·マクファーソンは首席財務官現在62歳のマクファーソンは2023年3月以来注目すべき首席財務官を務めている。マクファーソンさんは、Rego Payment Architecture,Inc.の最高財務責任者を務め、2015年7月から2022年8月までの間に、場外に上場した金融サービスソフトウェアプロバイダであった。マクファーソンさんは、2019年9月にUS Environmental,Inc.の兼任財務ディレクターとして招聘され、2022年3月にオーナーの死去以来、臨時CEOを務めてきた。アメリカの環境会社は主に環境廃棄物の輸送と処分に従事する環境サービス会社である。マクファーソンさんはまた、VerifyMe,Inc.のチーフ財務官を2014年12月1日から2017年7月15日まで、2012年12月から2013年10月まで務めた。VerifyMe,Inc.は上場企業であり、人員と製品認証分野でハイテク解決策を提供している。マクファーソンさんは、2015年4月から2015年7月までCannLabs,Inc.のCEOを務め、2014年6月から2015年7月までの間にCEOを務めた。これまで、マクファーソンさんは、Rego Payment Architecture,Inc.の最高財務責任者を2010年8月から2012年11月まで務めていた。マクファーソンさんは、2005年1月にMcPherson、CPA、PLLCを設立し、今でも彼によって管理されている。同事務所は、異なる業界の多くの顧客に会計、税務、訴訟支援サービスを提供している。同社は、最初にサバンズ-オクスリー法案を遵守·維持するために、小型上場企業のプログラムを作成し、実施させることに成功した。これらすべてのサービスは、マクファーソンさんの指示の下で実施されました。
外部と非執行役員
トモ·P≡tsi。現在59歳のP≡tsiはバイオテクノロジーと製薬業界で30年以上の仕事経験を持っている。P≡Tsiさんは、2022年3月以降、複数のバイオ製薬会社および投資家のコンサルタントを務めてきました。2020年7月から2022年2月までナスダック社(SGEN Inc.)で執行副社長を務め、癌に専念した米国のバイオテクノロジー会社である。2012年11月から2020年6月まで、Celgene Corp.(後に百時美施貴宝社(ニューヨーク証券取引所株式コード:BMY)に買収された)で様々な職務を担当し、ヨーロッパ、中東とアフリカ地域および世界市場の総裁、ヨーロッパと国際運営の総裁を務め、主に癌治療療法の開発と商業化に従事している。Patsiさんは、戦略的取引およびパートナーシップに参加し、Celgeneと百時美施貴宝の国際ビジネス統合を共同で主導しました。P≡tsiさんは、Celgeneに加入する前にヨーロッパのヒトゲノム科学部の副社長を務め、ヨーロッパとアメリカのアンサンブルで11年間にわたって増加してきた役割を務めています。P?TSIさんは、2023年4月からアクセンヘルスの取締役会メンバーを務めており、2023年3月以降は株式会社取締役(ロンドンAIM:FARN、ナスダックFirst North:FARON)の非執行役員を務めている。P≡tsiさんは登録薬剤師であり、ヘルシンキ大学薬学部薬理学の修士号を有しています。 P?Tsiさんは、バイオテクノロジーおよび製薬業界での広範な管理スキルと経験によって、取締役会のメンバーになる資格があることに注目してください。
トーマス·I·H·デュビンです現在62歳のデュビンは製薬会社の幹部と弁護士です。デュビン·さんは過去5年間、複数のバイオ製薬会社のコンサルタントや取締役会のメンバーを務めてきた。2001年から2013年にかけて、Alexion製薬(“Alexion”)を開発段階からSスタンダード500指数株会社に発展させた首席法務官とコア実行チームのメンバーだった。Alexionでは、ドゥビンさんは、法律、政府の事務、定価および精算、人的資源、企業コミュニケーションおよびその他の機能を担当し、オーストラリア·ストラシア地域のビジネス責任を担当します。Alexionに加入する前に、デュビン·さんは、社長副弁護士、チレックス社の総法律顧問、ワーナー·ランバート社助理の総法律顧問を務めていました。さん·デュビンのキャリアは、ニューヨーク市Cravath,Swine&Moore法律事務所から始まります。デュビンさんは現在、Cellphire治療会社のCEO、ノーウォーカー病院の会長、コネチカット州革新会社の取締役会のメンバー、エール大学公衆衛生リーダー委員会のメンバー、神話製薬会社のコンサルタント委員会のメンバーを務めています。デュビンさんは、2015年から2018年まで、BioBlast製薬会社(ナスダック·コード:ORPN)の取締役会メンバーを務め、2014年から2021年まで、米国ユダヤ人世界サービス会社の受託者を務めました。さん·デュビンは、ニューヨーク大学法学部で法学博士号、エール大学公衆衛生研究所で修士号、アーマースト大学で優秀な成績で学士号を取得しました。ドゥビンさんは、法律およびビジネススキルの豊富なだけでなく、バイオテクノロジーの分野での経験も持っているので、取締役会に勤めている資格があることに注目してください。
| 88 |
ピーター·ファンバーグです現在63歳のファンバーグさんは、金融サービス業で30年以上の経験を持つ。2020年4月以来、ファンバーグさんは、Sporos BioVentures,Inc.の創始者であり、これは私営バイオテクノロジー会社であり、腫瘍学における薬物開発の流れを変更することに専念しています。2019年6月以来、FeinbergさんはBoxcar PartnersとBoxcar PMJ LPのパートナーと創設メンバーを務めており、Boxcar PMJ LPはバイオテクノロジー投資に専念するベンチャー企業である。ファンバーグさんは、2018年10月以来、新興セキュリティ·ソリューションの共同創業者である。ファンバーグさんは、BridgeBio Pharma,Inc.(ナスダック·コード:BBIO)の共同創業者であり、遺伝子疾患に着目した上場企業であり、2014年12月に同社の設立を支援しました。ファンバーグはこれまで、奥本ハイマー社で取締役社長兼機関株取引主管を務めており、任期は1982年7月から2015年12月まで。ファンバーグさんは、2021年以降、バイオテクノロジー企業である免疫工学会社(ナスダック·コード:IMRX)の取締役会メンバーを務めており、バイオインフォマティクスを適用して新しい薬物発見手法を創出することを目指しています。ファンバーグさんは、ホイッティール大学で金融学士号を取得した。なお、ファンバーグさんは、金融やバイオテクノロジー産業で幅広いリーダーシップと経験を持っているため、取締役会に勤めている資格があります。
ミシェル·ガレンですGalenさんは現在67歳で、2016年7月から現在まで、幹部とリーダーシップ発展コーチ、バイオ製薬幹部の組織顧問を務めている。Galenさんは2015年4月から2016年6月までの間にShire PharmPharmticals PLC(現在武田)の高級副総裁兼首席通信官を務めた。これまで、Galenさんは2012年1月から2015年1月までの間にスイスのバーゼルに駐在していたノワ製薬のグローバル広報担当を務めていた。これまで、Galenさんはノワール製薬副総裁兼全世界コミュニケーションと宣伝主管総裁を務めたことがある;副総裁兼腫瘍事務グローバル主管;副総裁兼ノワール分子診断会社のコミュニケーションと対外事務グローバル主管;及びノワ製薬会社企業コミュニケーション副総裁を務めた。NPCに加入する前に、Galenさんはニューヨークの有力なグローバル広報会社の企業実践部門の取締役社長であり、コミュニケーション、問題管理と戦略慈善事業の変革に特化している。彼女は受賞歴のある記者で、“ビジネスウィーク”の法務や社会問題の編集者を務めていた。2017年1月から2021年10月まで、GalenさんはCardax,Inc.の取締役社員であり、全世界衛生理事会と全世界腫瘍学委員会のメンバーを務めている。ガレンはニューヨーク州弁護士協会のメンバーで、Stock,Stock +LavanとSkadden,Arps,Slate,Meagher&Flomで勤務していた。Galenさんはニューヨーク大学法学部で法学博士号、コロンビア大学でジャーナリズム修士号、ジョージワシントン大学で心理学学士号を取得した。有名なbrは、Galenさんは生物技術とメディア/広報業界で広範なコミュニケーション、組織変革及び商業と患者の提唱経験を持っているため、取締役会に在任する資格があると信じている注目すべきは、Galenさんがバイオテクノロジーと製薬業界で豊富な経験を持っているため、彼女は取締役会に就く資格があるということだ。
トーマス·グランニです現在59歳のGraneyさんは、企業開発、ビジネス戦略、ポートフォリオ管理、サプライチェーン管理、コミュニケーション、投資家関係など、世界的な金融経験を豊富に持っています。Graneyさんは、2021年5月から2023年12月までの間にOxurion NVのCEOを務めます。Graneyさんは、CEOに昇進する前に2020年10月からOxurion NVのCEOを務めています。グランニ·さんはこれまで、2017年9月から2019年2月までVertex製薬のチーフ財務官を務め、2014年9月から2017年9月までは鉄木製薬のチーフ財務責任者、企業戦略ディレクターの上級副社長を務めていた。グランニは鉄木製薬に参入する前にジョンソン&その子会社と20年以上協力して、ethiconグローバル最高財務官(Br)を4年間務めてきた。Graneyさんは現在、Mogrify Ltd.の取締役会のメンバーと監査委員会の会長を務めており、2017年から2023年までAC免疫SAの取締役会のメンバーを務めています。Graneyさんは、デラウェア大学から会計学の学士号を取得し、マーケティング、金融、国際ビジネスMBAの学位をニューヨーク大学レナード·N·ストーンビジネススクールから取得しました。注意すべきことは、Graneyさんは、バイオテクノロジーと製薬業界の経験だけでなく、豊富な財務スキルを持っているので、取締役会のメンバーを務める資格があります。
マイケル·ライスです現在59歳のライスは2021年7月以来VBLの取締役会メンバーを務めている。ライスさんは、ポートフォリオ管理、投資銀行、資本市場について豊富な経験を持っています。ライスは2010年以来生命科学投資家関係コンサルティング会社LifeSci Advisors LLCの創始パートナーであり、2013年以来研究駆動型投資銀行LifeSci Capital LLCの創始パートナーである。以前、ライスさんはCanaccel Adams医療投資銀行の共同責任者であり、そこでは彼は債務と株式融資に参加しました。ライスさんもまた、Think Equity Partners取締役の執行役員 は、大量の取引を実行することを含む医療資本市場の管理を担当しています。これまで、米国銀行で取締役主管を務め、大型ヘッジファンドや私募株式医療基金にサービスを提供するとともに、投資銀行と密接に協力してきた。ライスさんは、メリーランド大学を卒業し、経済学の学位を取得し、現在9メートルバイオ製薬会社(ナスダック·コード:nmtr)とナヴィディアバイオ製薬会社(ニューヨーク証券取引所コード:NAVB)の取締役会メンバーです。注目すべきは、ライスが広範な金融·業界背景を持っているため、取締役会のメンバーになる資格があると考えていることだ。
| 89 |
取締役会委員会
有名なbrには、監査委員会、報酬委員会、指名委員会の3つの常設委員会が株主に対する責任の履行に協力している。注目すべき取締役会委員会は、注目すべきbr書面定款と会社統治政策に基づいて適用される。
注意すべきbr取締役会には、注目すべき監査と財務手続きを監督し、注目すべき独立公認会計士事務所の選択を担当する監査委員会が設置されている。監査委員会のメンバーはThomas Graney (議長)、Thomas Dubin、Peter Feinberg、Michael Riceである。注意すべき取締役会は、審査委員会の各メンバーが取締役上場規則と取引所法案の下で適用される米国証券取引委員会規則が指す“独立米国証券取引委員会”であることが決定した。
取締役会には報酬委員会が設置されており、役員会が役員報酬戦略を監督し、役員報酬および従業員福祉計画の管理を審査·承認する責任があることに注意されたい。給与委員会はまた有名な役員の報酬の審査と承認を担当するだろう。以下の“報酬検討·分析”に規定されている場合を除き、執行幹事は役員または役員の報酬を確定しないが、報酬委員会の要求に応じて資料やアドバイスを提供する。報酬委員会のメンバーはトーマス·デュビン(議長)、ミシェル·ガレン、トーマス·グランニ、マイケル·ライス。注意すべきことは、取締役会は報酬委員会の各メンバーが“独立取締役”であることを決定しており、その意味は適用される“ナスダック上場規則”および“米国証券取引委員会取引委員会法案”の下の適用規則に適合している。
注目すべき取締役会には、年間株主総会で独立役員に指名を推薦する候補者が注目すべき取締役会に入る指名委員会がある。注目すべき取締役会候補を審査する際には、指名委員会と注目すべき取締役会の独立メンバーは、注目すべき取締役会が変化する需要 を考慮し、現在または予想される将来の需要を満たす候補者を探す。指名委員会のメンバーはTuomo Patsi(議長)、Peter Feinberg、Thomas Graney。注目すべきは,指名委員会のメンバーごとに“独立した取締役”であり,適用されるナスダック上場規則や取引法の適用された米国証券取引委員会規則に適合することを意味していることである。
道徳基準
我々は、我々の最高経営責任者やCEO、または同様の機能を実行する他の者を含む、我々のすべての取締役および従業員に適用されるビジネス行動および道徳的基準を採択した。商業行為と道徳規則の全文は無料で請求することができ、住所はカリフォルニア州94404フォスター市ハッジ大通り320号有名な実験室有限会社会社秘書である。もし私たちが任意の上級職員または役員の業務規則brを実質的に修正または任意の免除を与える場合、私たちは、私たちのウェブサイトまたは現在の8-Kフォーム報告書で、そのような修正または免除の性質を開示します。
取引法第16条(A)条を遵守する
取引法第 16(A)節は、当社の高級管理者及び取締役及び当社の登録カテゴリを有する株式証券が10%を超える個人(総称して報告者と呼ぶ)に、米国証券取引委員会に所有権報告及び所有権変更 を提出することを要求する。米国証券取引委員会は、通報者は、彼らが提出したすべての第16条(A)条の表のコピーを会社に提供しなければならないと規定している。
注目すべき審査で受け取ったこのような表のコピーと我々に提出された申告のみに基づいて,2023年12月31日までの年度内に,すべての上級職員,取締役および10%を超える実益株主に適用されるすべての申告要求がタイムリーに遵守されていると信じている.
| 第 項11. | 役員報酬 |
登録者が2024年2月16日に米国証券取引委員会に提出した最終依頼書 に含まれるS−K法規第402および407(E)(4)および(E)(5)項に要求される 開示は、参照によって本明細書に組み込まれる。
| 第 項12 | 安全な利益所有者の所有権と管理層と関連する株主のこと |
登録者が2024年2月16日に米国証券取引委員会に提出した最終委託書に含まれるS−K法規403項に要求される開示は、参照によって本明細書に組み込まれる。
| 90 |
株式報酬計画
次の表は、2023年12月31日までの会社のすべての持分報酬計画のいくつかの情報を提供します。
| 番目 | |||||||
| 証券 | |||||||
| 残り | |||||||
| に使用可能 | |||||||
| 未来.未来 | |||||||
| 発行する. | |||||||
| 下 株式 | |||||||
| 番目 | 補償する | ||||||
| 証券 〜 | 重み付けの- | 平面図 | |||||
| BE | 平均値 | (含まれない) | |||||
| 発行 アッポン | トレーニングをする | 証券 | |||||
| 運動 の | 価格 の | 反射する | |||||
| 卓越した | 優れた | in コラム | |||||
| オプション 株価上昇権 | オプション | (a)) | |||||
| プラン カテゴリ | (a) | (b) | (c) | ||||
| 株式証券所有者が承認した報酬計画 | 284,437 | $ | 49.67 | 305,626 | |||
| 株式所有者の承認されていない報酬計画 | - | $ | - | - | |||
| 第 項13. | いくつかの関係や関連取引、および取締役の独立性 |
以下に説明する は、2022年1月1日以降に発生するすべての取引、および現在提案されているすべての取引であり、その一方および :
| ● | 関連する金額は、120,000ドルを超えるか、または超える(または、少ない場合、2023年および2022年12月31日の平均総資産の1%である); | |
| ● | 任意の役員役員、5%以上の任意のカテゴリの発行された株式を所有する任意の人、または前述の者の任意の直系親族またはエンティティの任意のメンバーが、直接的または間接的な重大な利益を所有するか、または所有するであろうか。 |
2023年2月、LifeSci Advisorsと投資家関係サービスについて主サービス契約を締結し、毎月の求人費は約20,000ドルであることに注意されたい。プライマリサービスプロトコルにより,6(6)カ月前に書面終了通知を出し,任意の理由でLifeSci Advisorsプロトコルを終了することができることに注意されたい(LifeSci Advisorsであってもよい).有名な取締役スターのマイケル·ライスはLifeSci Advisorsの責任者です。
| 91 |
2022年2月、有名な実験室会社は簡単な将来の株式契約(“Cシリーズ金庫”)の提供を開始した。有名な実験室会社は2022年2月から2022年3月まで,Cシリーズ金庫の販売から合計約400万ドルの収入を得た。Cシリーズ金庫を売却する融資のうち、約64.9%がS前取締役会メンバーと関連がある人や関連がある人から来ている。
2023年2月21日から、成約前融資の一部として、有名な実験室会社が外管局投資家に合計約440万ドルのDシリーズ金庫を売却した。約46.0%の融資は、有名なラボ会社の元取締役会メンバーSと関連しているか、または関連している当事者から来ている。Dシリーズ金庫は注目すべきD-1シリーズ優先株に変換され、価格は投資家がDシリーズ融資で支払った価格より30%割引されている。
合併終了前の2023年には,有名な実験室会社が合計約600万ドルのD−2シリーズ優先株 を売却した。そのうちの約34.3%の融資(Dシリーズ金庫の売却金額を含まない)は、有名な実験室会社の前取締役会メンバーSと関連があるか、または関連がある方から来ている。
取締役 独立
ナスダック上場基準の要求によると、上場会社の取締役会メンバーの多くは取締役会が決定した“独立”メンバーでなければならない。著名な取締役会はすでに決定し、取締役は上場標準を適用する意味内で独立 すべきである。適用されたナスダック規則によると,トーマス·A·バークを除いて,7人の取締役のいずれも“独立した” と見なす資格がある。
外部 取締役
“会社法”によると、イスラエル国内又は海外の証券取引所に上場して取引されている株式又はその株式を公衆に発行したイスラエルの会社は、少なくとも2人の外部取締役を取締役会のメンバーに任命しなければならない。外国証券取引所(例えばナスダック)に上場する会社については、このような外部取締役はイスラエル住民である必要はない。外部 取締役は厳しい独立性基準を満たさなければならない。うちの外部役員はデュビン·さんとグランニ·さんです。
(br}“会社法”では、以下の場合、個人が外部取締役に指名·任命または任命される資格がない:(br}(I)被著名人が会社“持株株主”の親族である場合、または(Ii)著名人または著名人の親族、パートナー、雇用主、著名人の他の人が著名人の直接または間接的な部下である場合、または上記のように制御されている会社が、すでに禁止されていた従属関係または他の喪失関係があった場合(以下定義する)、(A)会社は、会社に対して支配権を有する者(すなわち、“持株株主”)またはその持株株主の親族、または任命時または任命日の2年前に会社またはその持株株主によって制御される任意の会社と;または(B)会社が持株株主または少なくとも25%の投票権を有する株主を有していない場合は、当時取締役会議長、会社行政総裁、会社発行株または投票権を5%以上保有していた者、または会社の首席財務官である。
用語“持株株主”とは,公職に就くことではなく,会社の活動を指揮する能力のある株主を指す.株主がその会社の50%以上の“制御手段”を持っている場合、その株主は同社に対して“制御権”を持っていると推定されるため、同社の持株株主とみなされる。“制御手段”の定義は,(1)一方の会社または他の会社の該当機関の株主総会で投票する権利,(2)会社の取締役または社長を任命する権利である。上記持株率を決定するために、会社の承認を提出した取引において個人利益を有する2つ以上の株主は、連名所有者とみなされる。
“会社法”によると、親族という言葉は、配偶者、兄弟姉妹、両親、祖父母または子孫、配偶者の兄弟姉妹、両親または子孫、および上記の各個人の配偶者として定義される。
| 92 |
“会社法”によると、“(禁止された)従属関係”という言葉と同様のタイプの禁止関係は、(“会社法”および会社法によって公布された条例によって無視できる関係とみなされるいくつかの例外的な場合の制約を含む):
| ● | 雇用関係; | |
| ● | 定期的または制御的に保持されている商業または専門関係(および、すでにサービスにいる外部取締役にとって−このような関係が定期的に保持されていなくても(無関係な関係を含まない)、および が、それに従って公布された会社法および法規に適合しない対価格を受け入れた場合、および) | |
| ● | 役職brを担当し,その株式初公募前にプライベート会社で取締役を務める役職は含まれておらず,このbr取締役が初公募後に外部取締役として任命されることを前提としている. |
“会社法”は、“br”の役職者を、CEO、社長、最高業務マネージャー、副社長、副社長、上記のいずれかの職務を担当する他の誰であっても、その肩書きがどうであるか、および取締役、または最高経営者または社長に直接所属するマネージャーと定義する。
さらに、誰かの職、専門または他の活動が、その人の董事人としての役割と利益衝突を引き起こす可能性がある場合、または他の方法でその人の外部取締役としての能力を妨害するか、またはその人がイスラエル証券管理局またはイスラエル証券取引所の従業員である場合、その人は“外部取締役”を務めてはならない。
また、一人が会社から直接又は間接的な補償を受けた場合、彼又は彼女が外部取締役サービスとして支払われた金を含む場合は、彼又は彼女は外部取締役を継続することができないが、“会社法”及び会社法の規定により許可されたものを除く。
外部取締役は取締役会の在任終了後、会社、その持株株主或いはその持株株主が制御するいかなる実体も、当該前の外部取締役及びその配偶者、子供に直接或いは間接的な利益を提供してはならない。これは、前外部取締役によって制御される会社を含む、会社またはその持株株主として制御される会社の責任者または取締役の採用、またはそのような任意の会社を雇用するか、またはそのような会社を直接または間接的に考慮するためにサービスを提供することを含む。元海外取締役及びその配偶者又は子供については、この制限有効期間は2年であり、元海外取締役の他の親族については、この制限期間は1年である。
外部取締役を任命する際に,会社持株株主または持株株主の親族を除くすべての取締役会メンバーが同一の性別であれば,任命された外部取締役は他の性別 でなければならない.
会社法が公布した規定によると、少なくとも1人の外部取締役は、他の監査委員会のメンバー(ナスダック証券市場規則に基づいて独立した取締役である)が“財務および会計専門知識”を有していない限り、“財務および会計専門知識”を有していなければならず、他の1人以上の外部取締役は“専門知識”を持たなければならない。
取締役外部者は再任してはならず、条件は:(1)当該取締役は“会計と財務専門長”を有し、 あるいは(2)“専門能力”を有し、再任を獲得した日に、別の外部 取締役は“会計と財務専門長”を有し、取締役会中の“会計と財務専門家”の人数は少なくとも取締役会が確定した最低人数に等しい。デュビン·さんとグランニ·さんは、会計および財務の専門知識を備えていることが確認されました(残りの取締役はいずれもプロファイリングされています)。
取締役は、以下の条件のうちの1つを満たす場合にのみ専門知識を有する:(1)取締役は、経済学、工商管理、会計、法律または公共管理専門の学位を有し、(2)取締役は、他の任意の分野の学位を有しているか、または会社の主要業務分野の他の形態の高等教育 を達成しているか、または会社の外部取締役職に関連する分野を有しているか、または(3)取締役は、少なくとも5年間の業務経験を有している。または(A)業務範囲の広い会社で高級業務管理職に就く、(B)会社の主要業務分野で高級職に就く、または(C)公共行政部門で高級職に就く、または少なくとも5年以下の2つ以上の職に在任した累積経験。
| 93 |
“会社法”によると、外部取締役は株主総会で多数票で選出され、次のいずれかがあれば
| ● | 少なくとも 非持株株主が保有する、任命中に個人利益のない株主が保有する株式の多数(株主と持株株主との関係による個人利益を含まない) は、その提案に賛成票を投じている(棄権株主が保有する株式は考慮されてはならない);または | |
| ● | 当該等株主が外部取締役の選挙に反対票を投じた株式総数は、当社の総投票権の2%以下である。 |
“会社法”では,外部役員の初期期限は3年と規定されている。その後、外部取締役は、最大2つの追加の3年間の任期であるが、以下に説明するいくつかの例外を除いて、条件は :
| (1) | 外部取締役の著名人の任期は、会社の少なくとも1%の投票権を持つ1人以上の株主が推薦し、株主総会で公正多数で可決され、その再選を支持する非持株、公正株主が保有する株式の総数が会社の総投票権の2%を超え、会社法による外部微博の有名人の従属関係に対する追加制限を受けることが条件である | |
| (2) | 彼または彼女の任期は取締役会によって推薦され、株主総会で外部取締役を初めて選挙するために必要な同様に公正な多数の承認を得る(上記のように) | |
| (3) | 前項(1)節の規定により、非常勤取締役は任期を1期延長するごとに、自発的にサービス提供を提出し、承認された。 |
ナスダック株式市場を含むある外国証券取引所で取引されているイスラエル社の外部取締役の任期を無期限に延長することができ、3年延長するごとに、外部取締役が取締役会及びその委員会の仕事に対する専門的な知識及び特別な貢献を確認することを前提として、当該追加期限(S)を再任することは会社に有利である。また、 外部取締役の再任は、初当選と同じ株主投票要求(上述したように)に適合しなければならない。 外部取締役の再選が株主総会で承認される前に、株主に、彼または彼女が以前に在任していた任期および取締役会と監査委員会がその任期延長を提案した理由を通知しなければならない。
外部取締役 は,選挙や裁判所が要求する株主数が同じ特殊多数に達した場合にのみ罷免でき,またこの2つの場合, は,外部取締役がその任命の法定資格を満たしていない場合や自社への忠誠義務に違反した場合にのみ である.取締役の外部取締役に空きが生じ、会社の外部取締役が2名以下になった場合、会社法によれば、取締役会は、その後2人の外部取締役があるように、株主総会をできるだけ早く(かつ3ヶ月以内に)開催しなければならない。
外部取締役は“会社法”が採択した規定に基づいて報酬を得ることしかできない。外部役員の報酬は任命前に決定され、いくつかの例外的な場合を除いて、その任期内に変更することはできない。
ナスダック上場規則による“独立取締役”の定義は,会社法による“外部取締役”の定義と大きく重なることから,会社法により取締役外部取締役を務めるどの取締役もナスダック上場規則の独立性に対する要求を満たすと考えられる.しかし、取締役は取締役上場規則に適合しない場合には、会社法 により“外部ナスダック”と認定される可能性があり、その逆も同様である。会社法の“外部取締役”の定義には、外部取締役の独立判断能力を損なう要因が存在しないことを確保するための基準が含まれている満たさなければならない法定基準が含まれている。ナスダック上場規則の“独立取締役”に関する定義規定は、取締役会は独立取締役の独立判断能力を弱めるいかなる要素を考慮しなければならない以外にも、類似した要求があるが、要求はやや緩い。また、“会社法”の要求によると、外部取締役の任期は3年(2年を超えない追加3年任期)となる。しかし,特殊な 多数の株主は“外部取締役”を選挙しなければならないが,“独立役員”は通常の 多数選挙で選出されることができる.
取締役会の権限を行使する各取締役会は、少なくとも1人の外部取締役を含まなければならない。br監査委員会および報酬委員会は、当時取締役会に勤務していたすべての外部取締役を含まなければならず、br監査委員会は、イスラエルの法律で独立した取締役として定義された多数から構成されなければならない。外部取締役は、報酬委員会の多数のメンバーでなければならない。監査委員会の議長と給与委員会の議長は役員外部の人たちが担当しなければならない
会社法の規定によると、外部取締役及び独立取締役のいくつかの免除及び減免について、イスラエル国外で証券を取引する会社に付与される。注目すべきは、このような免除と救済が使用されることができるということだ。
| 第 項14. | 依頼人 会計士費用とサービス |
登録者が2024年2月16日に米国証券取引委員会に提出した最終依頼書のうち,登録者を承認する独立公認会計士事務所の提案に記載されているbr}について開示し,引用により本明細書に組み込む。
| 94 |
第4部
| 第 項15. | 表と財務諸表明細書 |
| 添付ファイル 番号: | 説明する | |
| (A)(1) | 財務諸表 | |
| 有名な実験室有限会社の合併財務諸表は本報告第16項以降である。 | ||
| (A)(2) | 財務諸表明細書 | |
| ない。 | ||
| (A)(3) | 陳列品 | |
| 2.1++ | 血管生物製薬有限会社、Vibrange Merge Sub,Inc.と著名な実験室会社との間の合併協定および計画は、2023年2月22日(2023年9月5日を参照して米国証券取引委員会に提出されたS-4表登録声明の添付ファイル2.1合併である。 | |
| 3.1 | 登録者会社規約(2023年3月14日に米国証券取引委員会に提出された10−K表年次報告書添付ファイル3.1編入を参照)。 | |
| 3.2 | 登録者組織覚書(2023年3月14日に米国証券取引委員会に提出されたForm 10−K年度報告添付ファイル3.2を参照して合併)。 | |
| 3.3 | 登録者定款修正案は、期日が2023年10月16日である(添付ファイル3.1を参照して2023年10月16日に米国証券取引委員会に提出された現在の8-K表報告書に組み込まれる)。 | |
| 4.1 | 普通株式証明書フォーマット(2014年7月29日に米国証券取引委員会に提出されたF-1表登録説明書添付ファイル4.2を参照して編入)。 | |
| 10.1* | 血管生物製薬株式会社の従業員持株及び株式オプション計画(2011年)及びその合意フォーマット(2014年6月6日に米国証券取引委員会に提出されたF−1表登録声明の添付ファイル10.1を参照して組み込む)。 | |
| 10.2* | 血管生物株式有限会社従業員持株及びオプション計画(2014年)及びそれに基づいて締結した資本利得税オプション協議フォーマット(2014年6月25日にアメリカ証券取引委員会に提出されたF-1表登録説明書添付ファイル10.17)に結合した。 | |
| 10.3* | 血管生物製薬株式会社の誘導計画(2022年)およびその入札プロトコルのフォーマット(2022年2月15日に米国証券取引委員会に提出された現在の報告を参照することによって6-K表の添付ファイル99.1に組み込まれる)。 | |
| 10.4* | 血管生物製薬有限会社はその高級管理者及び取締役と締結した免除及び賠償契約書表(2014年6月25日にアメリカ証券取引委員会に提出されたF-1表登録説明書添付ファイル10.2を参照して合併)。 | |
| 10.5* | 血管生物製薬株式会社はDror Haratsと再記述した幹部採用協定で,期日は2022年1月20日である(合併内容は2023年3月14日に米国証券取引委員会に提出されたForm 10−K年度報告添付ファイル10.5参照)。 | |
| 10.6* | 血管生物製薬有限会社とGrand H Services Ltd.は2022年1月20日に再記述された“諮問とサービス協定”であり、この協定は2022年8月23日に改訂された(合併内容は2023年3月14日に米国証券取引委員会に提出されたForm 10-K年度報告の添付ファイル10.6参照)。 |
| 95 |
| 10.7* | 血管生物製薬株式会社とSam Backenrothとの間の招聘状は,2021年10月4日である(合併内容は2023年3月14日に米国証券取引委員会に提出されたForm 10−K年度報告の添付ファイル10.7参照)。 | |
| 10.8 | 事前融資株式証表(合併内容は2021年4月12日に米国証券取引委員会に提出された現在の6-K表報告書の添付ファイル4.1参照)。 | |
| 10.9+ | 著名な実験室会社およびOnCoheros Biosciences Inc.によって2021年10月1日に締結された独占許可協定(2023年9月5日に米国証券取引委員会に提出されたS-4表登録声明の添付ファイル10.9を参照して編入される)。 | |
| 10.10 | ブリンガー·インゲルハイム国際株式会社とOnCoheros Biosciences Inc.が2019年8月1日に発行した付状は、2020年4月5日に施行された改正案1改正(2023年9月5日に米国証券取引委員会に提出されたS-4表登録声明の添付ファイル10.10を参照して編入される)。 | |
| 10.11+ | 有名な実験室会社とCicloMed LLCは、2021年7月20日に署名された共同開発および利益共有協定(2023年9月5日に米国証券取引委員会に提出されたS-4表登録声明の添付ファイル10.11を参照して組み込まれる)。 | |
| 10.12 | Hatch Drive Associatesと著名な実験室会社との間の標準工業/商業単テナントリースは、2019年3月25日となっている(合併は、2023年9月5日に米国証券取引委員会に提出されたS-4表登録声明の添付ファイル10.12を参照)。 | |
| 10.13 | Hatch Drive Associates,LLCとEignant Labs,Inc.との間のリース協定第1修正案は,2023年4月27日である(2023年9月5日に米国証券取引委員会に提出されたS−4表登録声明の添付ファイル10.13を参照して編入される)。 | |
| 10.14* | 2015年の有名な実験室株式計画(2023年9月5日に米国証券取引委員会に提出されたS-4表登録説明書の添付ファイル10.17参照)。 | |
| 10.15 | 著名な実験室会社とC-1シリーズ優先株購入者が2021年6月に署名したCシリーズ株式承認協定表(添付ファイル10.18を参照して2023年9月5日に米国証券取引委員会に提出されたS-4表登録声明に組み込まれている)。 | |
| 10.16 | 著名な実験室会社とC-1シリーズ優先株購入者が2021年6月に締結した株式オプション奨励協定表(2023年9月5日に米国証券取引委員会に提出されたS-4表登録声明の添付ファイル10.19を参照して編入される)。 | |
| 10.17++ | 著名な実験室会社と投資家が2023年2月22日に署名した株式購入協定(合併して2023年9月5日に米国証券取引委員会に提出されたS-4表登録声明の添付ファイル10.20)。 | |
| 10.18* | 有名な実験室会社およびThomas Bockは、2021年4月30日に改正および再署名された雇用協定(2023年9月5日に米国証券取引委員会に提出されたS−4表登録声明の添付ファイル10.14を参照して組み込まれる)。 | |
| 10.19* | 著名な実験室会社とジョセフ·ワグナーが2020年6月15日に締結した雇用協定(2023年9月5日に米国証券取引委員会に提出されたS-4表登録声明の添付ファイル10.15を参照して組み込む)。 | |
| 10.20* | 著名な実験室会社とスコット·A·マクファーソン社が2023年3月1日に発行した婚約状(2023年9月5日に米国証券取引委員会に提出されたS-4表登録声明の添付ファイル10.16を参照して編入される)。 | |
| 10.21* | 解放および賠償協定表(2023年10月16日に米国証券取引委員会に提出された現在の8−K表報告書の添付ファイル10.4を参照して組み込まれる)。 |
| 96 |
| 10.22+ | 免疫歩行治療会社と血管生物遺伝有限会社との間の資産購入協定は、2023年10月1日である(添付ファイル10.1を参照して2023年10月2日に米国証券取引委員会に提出された現在の8-K表報告書に組み込まれる)。 | |
| 21.1 | 登録者の子会社 | |
| 23.1 | Withum Smith+Brown PCは同意します | |
| 23.2 | 徳勤法律事務所が同意した | |
| 31.1 | 2002年のサバンズ-オキシリー法第302条に基づいて最高経営責任者証明書が発行された。 | |
| 31.2 | 2002年のサバンズ-オキシリー法第302節に基づいて首席財務官証明書が発行された。 | |
| 32.1 | “米国法典”第18編1350条によると、2002年の“サバンズ-オキシリー法案”第906節で可決された最高経営責任者と最高財務官の証明による。 | |
| 97 | 払戻政策 | |
| 101.INS | 相互接続 XBRLインスタンス文書−インスタンス文書は、XBRLタグがイントラネットXBRL文書内に埋め込まれているので、対話データファイルには表示されない。 | |
| 101.書院 | 連結 XBRL分類拡張アーキテクチャ文書. | |
| 101.カール | 連結 XBRL分類拡張はリンクベース文書を計算する. | |
| 101.def | 連結 XBRL分類拡張Linkbase文書を定義する. | |
| 101.介護会 | XBRL分類拡張ラベルLinkbase文書を連結する. | |
| 101.Pre | インライン XBRL分類拡張プレゼンテーションLinkbase文書. | |
| 104 | 表紙 ページ相互データファイル。 |
| * | 管理職の報酬計画、合意、または手配を表します。 |
| + | 本展示品の 部分(で[****])登録者が、(I)漏れた情報が重要ではないと判断しているので、(Ii)漏れた情報が開示されている場合、登録者に競合障害をもたらす可能性があるから漏れている。 |
| ++ | S−K条例第601(B)(2)項によれば、いくつかの付表及び証拠物は省略されている。何か漏れたスケジュールおよび/または証拠 があれば、米国証券取引委員会に提供することが要求されるべきである。 |
| 第 項16. | 表 10-K要約 |
ない。
| 97 |
サイン
1934年の証券取引法第13または15(D)節の要求によると、登録者は、本報告が正式に許可された署名者によってその署名を代表するように正式に促進された。
| 有名なbr実験室、有限会社。 | ||
| 日付: 2024年4月11日 | 差出人: | /S/ トーマス·A·バーク |
| トーマス·A·バーク | ||
| CEO | ||
授権書
これらの陳述を通じて、以下に署名するすべての人は、Thomas A.BookとScott A.McPherson, を彼または彼女の真の合法的な事実代理人および代理人として構成し、任命し、彼または彼女の任意およびすべての身分で、彼または彼女の名義、場所および代替、テーブル10-Kの任意およびすべての身分で、本年度報告の任意およびすべての修正に署名し、その中のすべての証拠物およびそれに関連する他の文書を提出することを知っている。証券取引委員会に、上記の事実代理人及び代理人及びその各個人に十分な権力及び権力を付与し、その本人が可能又はbr本人にできるすべての意図及び目的を尽くし、それに関連するすべての必要かつ必要な事項を行い、ここで承認して確認し、これらのすべての事実代理人及び代理人、又は彼らのいずれか、又は彼ら又は彼らの代替者、brを合法的に行うことができ、又はそれによってなされたすべてのことを行うことができる。
1934年の証券取引法の要求によると、本報告は、以下の者によって登録者として指定日に署名された。
| 名前.名前 | タイトル | 日取り | ||
| /S/トーマス A.バーク | 最高経営責任者兼取締役 | 2024年4月11日 | ||
| トーマス·A·バーク | ||||
| /S/ スコット·A·マクファーソン | 最高財務官 | 2024年4月11日 | ||
| スコット A 。マクファーソン | (首席財務官と首席会計官) | |||
| /S/ トマー·パトシー | 董事局議長と役員 | 2024年4月11日 | ||
| トマー·パーシー | ||||
| /S/トーマス I.H.デュビン | 役員.取締役 | 2024年4月11日 | ||
| トーマス·I·H·デュビン | ||||
| /S/ ピーター·ファンバーグ | 役員.取締役 | 2024年4月11日 | ||
| ピーター·ファンバーグ | ||||
| /S/ ミシェル·ガレン | 役員.取締役 | 2024年4月11日 | ||
| ミシェル·ガレン | ||||
| /S/トーマス グランニ | 役員.取締役 | 2024年4月11日 | ||
| トーマス·グランニ | ||||
| /S/ マイケル·ライス | 役員.取締役 | 2024年4月11日 | ||
| マイケル·ライス |
| 98 |
連結財務諸表インデックス
| ページ | |
| 独立公認会計士事務所報告(PCAOB: |
F-2 |
| 独立公認会計士事務所報告(PCAOB:
|
F-4 |
| 合併貸借対照表 | F-5 |
| 合併経営報告書と全面赤字 | F-6 |
| 連結優先株と株主権益変動表(損失) | F-7 |
| 統合されたキャッシュフロー表 | F-8 |
| 連結財務諸表付記 | F-9 |
| F-1 |
独立公認会計士事務所報告{br
Br社の株主と取締役会へ
有名なbr Labs,Ltd.
連結財務諸表に関する意見
我々はすでに添付の卓著実験室有限会社(“当社”)が2023年12月31日に発行した総合貸借対照表及び関連する総合運営及び全面赤字報告書、償還可能転換可能優先株及び株主権益(損失)の変動、現金流量及び関連付記(総称して“総合財務諸表”と呼ぶ)を監査した。総合財務諸表は,当社の2023年12月31日までの財務状況と,2023年12月31日までの経営業績とキャッシュフローをすべての重要な面で公平に反映しており,米国公認の会計原則に適合していると考えられる。
実体の持続経営企業としての継続経営能力に大きな疑いがある
添付された連結財務諸表の作成は、当該エンティティが継続して経営を継続する企業であると仮定する。総合財務諸表付記1に記載されているように、この実体は経営により経常赤字であり、成立以来の経営活動のキャッシュフローは負であり、赤字を累積しており、その継続経営能力に大きな疑いを抱かせる。付記1は、これらの事項における経営陣の計画も説明しています。連結財務諸表には、このような不確実性の結果によって生じる可能性のあるいかなる調整も含まれていません。私たちはこの問題に対する見解を何も修正していない。
2022年財務諸表改訂版
当社の2022年12月31日までの年度の総合財務諸表は、付記1及び2で述べた株式の逆分割に関する会計変動を総合財務諸表の調整影響 に遡及適用する前に、他の計数師が審査しており、その日付が2024年4月11日の報告書は、この等の報告書に対して保留のない意見を表明している。付記1および2で述べたように、2022年財務諸表の調整を監査し、2023年10月16日に適用される逆株式分割を追跡した。私たちはこの追跡調整が適切であり、適切に適用されたと思う。しかし、トレーサビリティ調整以外に、吾らは招聘されて御社の2022年財務諸表を審査、審査或いは応用していないため、私らは2022年財務諸表全体に対して意見を発表したり、いかなる他の形式の保証をしたりしない。
意見を求める根拠
これらの合併財務諸表は会社経営陣が担当しています。私たちの責任は、私たちの監査に基づいてこれらの連結財務諸表に対して意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に基づいて監査を行いました。これらの基準は、合併財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、監査を計画し、実行することが要求されます。当社はその財務報告書の内部統制を監査する必要もありません。私たちの監査の一部として、財務報告の内部統制を知る必要がありますが、社内財務報告の内部統制の有効性について意見を述べるためではありません。したがって、私たちはそのような意見を表現しない。
我々のbr監査には,連結財務諸表の重大な誤報リスクを評価するプログラム,エラーによるものであっても詐欺であっても,これらのリスクに対応するプログラムを実行することが含まれる.これらの手続きは、連結財務諸表中の金額および開示に関する証拠をテストに基づいて検討することを含む。私たちの監査には、評価に使用される会計原則と経営陣による重大な推定と、連結財務諸表の全体列報を評価することも含まれています。私たちの監査は私たちの意見に合理的な基礎を提供していると思います。
重大監査事項
以下に述べる重要な監査事項とは、合併財務諸表を当期監査する際に生じる事項であり、 は監査委員会に伝達または要求され、(1)総合財務諸表に対して重大な意義を有する勘定または開示に関するものであり、(2)私たちが特に挑戦的で主観的または複雑な判断に関するものである。重要な監査事項のコミュニケーション は、合併財務諸表に対する私たちの全体的な意見をいかなる方法でも変えることはなく、私たち は以下の重要な監査事項を伝達することによって、重要な監査事項或いはそれに関連する勘定或いは開示について単独の意見を提供することはない。
| F-2 |
業務グループ
重要な 審査事項説明
2023年10月16日に、期日が2023年2月22日の合意と計画(“合併協定”)に基づいて、有名な実験室有限会社(前身はイスラエル会社血管生物遺伝有限会社)、デラウェア州会社及び血管生物遺伝子有限会社の完全資本付属会社Vibrant Merger Sub,Inc.及びデラウェア州会社Vibrant Merge Sub,Inc.が有名な実験室会社と合併し、合併後引き続き存続実体及び重要な実験室の完全資本付属会社とした。総合財務諸表付記1および付記3にさらに記載されているように、当社は会計基準編纂(“ASC”)805に基づいて、業務統合(ASC 805)を会計処理する。
管理層brは、取引前後の管理層および管理の構成、および会社が投票権を有する権益のパーセンテージを含むASC 805のすべての基準を評価する。評価によると、同社は、この取引は逆合併 に計上され、有名な実験室会社は会計買収側であると結論した。
私たちは、会社の確定会計買収側の評価が重要な監査事項であることを確認しました。(1)会計買収側の評価確認および(2)資本再編の会計処理および当社が財務報告の内部統制で発見した重大な弱点に触れているため、業務 合併は重要な監査事項とされている。これは会計フレームワークの適用と関連した広範囲な監査作業を必要とする。
| ● | 指示的要因の相対的重要性を単独かつ全体的に評価する際には、職位組合せ投票権、取締役会、管理職の構成を含む監査役の高度な判断力が必要である。企業合併を開始する実体もあります | |
| ● | 資本再編の計算は、すべての普通株と優先株の転換を含み、 はDシリーズ安全プロトコルと転換可能なDシリーズ優先株の転換を含み、 は複雑な過程である。 |
違う結論は合併会計上の大きな違いを招くだろう。
監査において重要な監査事項をどのように解決するか
以下は私たちがこの重要な監査問題を解決するために実行した主な手続きだ。
我々は以下の方法で会社の会計買収側と資本再編に関する結論をテストした
| ● | 合併協定や関連添付ファイルの条項の理解を記録することを含む経営陣の評価過程を審査する | |
| ● | 取締役会と経営陣の構成を検討し | |
| ● | 審査されました。公開届出書類とアメリカ証券取引委員会マージに関連してマージ後の を招く通信メッセージ, | |
| ● | 普通株と転換可能優先株の条項を審査し、普通株に転換した金額を再計算した | |
| ● | 合併に関する書類、合併協定、定款及び定款を含む会社文書、及び合併前及び合併後の両社の取締役会紀要、及び | |
| ● | 資本再編に対する管理職の計算を再計算する |
私たち は2024年以来当社の監査役を務めています。
/s/
2024年4月11日
PCAOB ID番号100
| F-3 |
独立公認会計士事務所報告
有名な実験室有限会社の株主と取締役会に。
財務諸表に対する見方
著者らはすでにbr前に付記2で述べた会計変動を2022年12月31日までの総合財務諸表、卓著実験室有限会社及びその付属会社(“当社”)の総合貸借対照表、関連総合経営報告書及び全面赤字、償還可能転換可能な優先株変動及び株主権益(損失)及び現金流量変動の調整の影響に遡及適用した。および関連付記 (総称して“財務諸表”と呼ぶ)(財務諸表付記2で述べた遡及調整影響前の2022年財務諸表はここに記載されていない)。2022年財務諸表は,付記2から で述べた会計変更の影響をたどるように調整する前に,御社の2022年12月31日までの財務状況,2022年12月31日現在の経営業績とそのキャッシュフローを米国公認の会計原則 に従って公平に列記したと考えられる。
吾らは、付記2に記載された会計変更を財務諸表 に遡及適用するために、調整を審査、審査または適用していないため、当社等は、当該等遡及調整 が適切であるか否か及び適切に適用されて意見を発表したか否か、又は任意の他の形式の保証を行うことはない。このような追跡的な調整は他の監査員によって監査される。
経営を続ける企業
添付されている2022年12月31日現在および2022年12月31日現在の財務諸表は、当社が継続経営企業として作成されることを想定して作成されています。当社は設立以来、経常的な運営損失と経営活動によるマイナス赤字を受け、br}赤字を蓄積し、当社が持続経営企業として経営を継続する能力に大きな疑いがあることを示しています。注1では、経営陣のイベントや状況の評価、経営陣のこれらの事項の計画も紹介されています。財務諸表には、このような不確実性の結果による調整は含まれていません。この問題について、私たちの意見はこれで変わらないだろう。
意見の基礎
これらの財務諸表は会社の経営陣が担当しています。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
PCAOBの基準 に基づいてレビューを行った。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査する必要はなく、私たちも招聘されて監査を行っていません。私たちの監査の一部として、財務報告の内部統制を知ることが求められていますが、社内財務報告の内部統制の有効性について意見を述べるためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には,財務諸表の重大な誤報リスクを評価するプログラム,エラーによるものであっても詐欺であっても,これらのリスクに対応するプログラム を実行するプログラムが含まれる.これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。私たちの監査には、経営陣が使用する会計原則の評価と重大な推定、財務諸表の全体的なレポートの評価も含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
/s/
2024年4月11日
当社は、 2023 年から当社の監査役として業務を開始しました。2024 年には前任監査役になりました。
| F-4 |
有名なbr実験室、有限会社。
合併貸借対照表
(千ドル単位で、1株当たりおよび1株当たりの金額は含まれていない)
| 2023年12月31日 | 2022年12月31日 | |||||||
| 資産 | ||||||||
| 現在の 資産: | ||||||||
| 現金 と現金等価物 | $ | $ | ||||||
| 前払い料金と他の流動資産 | ||||||||
| 流動資産合計 | ||||||||
| 財産と設備、純額 | ||||||||
| ファイナンス リース使用権資産、純 | ||||||||
| 運営 レンタル使用権資産 | ||||||||
| 投資 in SAFE | ||||||||
| その他 資産 | ||||||||
| 総資産 | $ | $ | ||||||
| 負債、償還可能転換優先株式、 株主資本 ( 赤字 ) | ||||||||
| 流動負債 : | ||||||||
| 売掛金 | $ | $ | ||||||
| 費用とその他の流動負債を計算しなければならない | ||||||||
| 売掛金と売掛金の関係者 | ||||||||
| 融資リース負債、流動 | ||||||||
| 営業 レンタル負債、流動 | ||||||||
| 流動負債合計 | ||||||||
| 融資リース負債、当期金額を差し引く | ||||||||
| 営業レンタル負債、当期金額を差し引く | ||||||||
| 償還可能な転換可能優先株式証債務 | ||||||||
| 総負債 | ||||||||
| 支払いを受ける とあるか | ||||||||
| 系列
は転換可能優先株、額面$を償還することができる, そして2023年12月31日と2022年12月31日までの認可株式、および
そして2023年12月31日と2022年12月31日までに発行·発行された株;総清算優先権$ | ||||||||
| Bシリーズは転換可能優先株、額面$を償還可能です,
そして2023年12月31日と2022年12月31日までに認可された株、および
と2023年12月31日と2022年12月31日までに発行および発行された株;総清算優先権は$ | ||||||||
| Cシリーズは転換可能優先株、額面$を償還可能です,
そして2023年12月31日と2022年12月31日までに認可された株、および
と2023年12月31日と2022年12月31日までに発行および発行された株;総清算優先権は$ | ||||||||
| 株主権益 | ||||||||
| 普通株、新シェケルチケットの価値は と2023年12月31日と2022年12月31日までに認可された株、および と2023年12月31日現在と2022年12月31日現在発行·未返済 | ||||||||
| 普通株 、$ 額面;そして2023年12月31日と2022年12月31日までに認可された株とそして 2023年12月31日現在と2022年12月31日までに発行された株式 | ||||||||
| 追加実収資本 | ||||||||
| 累積赤字 | ( | ) | ( | ) | ||||
| その他の総合収益を累計する | ||||||||
| 株主権益(赤字)合計 | ( | ) | ||||||
| 総負債、償還可能な転換可能優先株、 株主資本 ( 赤字 ) | $ | $ | ||||||
付記は連結財務諸表の構成要素である。
| F-5 |
有名なbr実験室、有限会社。 合併経営報告書と全面赤字 (千ドル単位で、1株当たりおよび1株当たりの金額は含まれていない)
|
| 年度まで | ||||||||
| 十二月三十一日 | ||||||||
| 2023 | 2022 | |||||||
| サービス収入 | $ | $ | ||||||
| サービスコスト | ||||||||
| 毛利 | ||||||||
| 運営費用 | ||||||||
| 研究開発 | ||||||||
| 通常 と管理 | ||||||||
| 運営費総額 | ||||||||
| 運営損失 | ( | ) | ( | ) | ||||
| その他 収入(費用) | ||||||||
金庫の公正価値変動 | ( | ) | ||||||
| 償還可能優先株式証負債の公正価値変動 | ( | ) | ||||||
| その他の収入,純額 | ||||||||
| ( | ) | |||||||
| 純損失 | ( | ) | ( | ) | ||||
その他総合収益 | ||||||||
| 外貨換算調整変動 | ||||||||
| 総合損失 | $ | ( | ) | $ | ( | ) | ||
| 1株当たり基本と希釈後の純損失 | $ | ) | $ | ) | ||||
| 加重平均 発行済み普通株式、基本普通株、希釈後普通株 | ||||||||
付記は連結財務諸表の構成要素である。
| F-6 |
有名なbr実験室、有限会社。
償還可能な転換優先株と株主権益総合変動表(損失)
(千ドル単位で、1株当たりおよび1株当たりの金額は含まれていない)
2023年12月31日と2022年12月31日までの年度
| 償還可能な転換優先株 | 普通株 株 | 普通株 株 | 追加の 個の実収 | 積算 | 累積総合 | 株主総数 権益 | ||||||||||||||||||||||||||||||||||
| 株 | 金額 | 株 | 金額 | 株 | 金額 | 資本 | 赤字.赤字 | 収入.収入 | (赤字) | |||||||||||||||||||||||||||||||
| 残高 2021年12月31日 | $ | $ | $ | $ | $ | ( | ) | $ | $ | ( | ) | |||||||||||||||||||||||||||||
|
C-1シリーズを発行して転換可能な優先株を償還し、発行コスト$を差し引く | - | |||||||||||||||||||||||||||||||||||||||
|
C-2シリーズ転換可能優先株を発行し,安全協定と引き換えに,C系列償還可能優先株に割り当てられた収益
転換可能優先株証負債を差し引いて$とする | - | - | ||||||||||||||||||||||||||||||||||||||
| Aシリーズ転換可能転換優先株発行普通株 | ( | ) | ( | ) | - | |||||||||||||||||||||||||||||||||||
| B系列転換により転換可能優先株発行普通株 | ( | ) | ( | ) | - | |||||||||||||||||||||||||||||||||||
| 普通株式オプションを行使する{br | - | - | ||||||||||||||||||||||||||||||||||||||
| 株に基づく報酬 | - | - | - | |||||||||||||||||||||||||||||||||||||
| 純損失 | - | - | - | ( | ) | ( | ) | |||||||||||||||||||||||||||||||||
| 残高 2022年12月31日 | ( | ) | ( | ) | ||||||||||||||||||||||||||||||||||||
| 血管生物製薬有限会社との逆合併は,2023年10月16日から発効する | - | - | ||||||||||||||||||||||||||||||||||||||
| 合併後の有名な実験室有限会社の普通株を発行し、交換比率は1株合併前に注目すべき実験室会社普通株 | - | ( | ) | ( | ) | ( | ) | |||||||||||||||||||||||||||||||||
| Aシリーズ発行可転換優先株普通株 | ( | ) | ( | ) | - | |||||||||||||||||||||||||||||||||||
| A,B,C,Dシリーズの償還可能株に関する普通株を発行し,発行コスト $を差し引く | - | - | ( | ) | ||||||||||||||||||||||||||||||||||||
| A,BとC系列償還可能優先株に関する逆希釈普通株 を発行し,発行コスト $を差し引く | - | - | ( | ) | ||||||||||||||||||||||||||||||||||||
| Bシリーズを発行して転換可能優先株普通株を償還できる | ( | ) | ( | ) | - | |||||||||||||||||||||||||||||||||||
| Cシリーズ発行可転換優先株普通株 | ( | ) | ( | ) | - | |||||||||||||||||||||||||||||||||||
| D-1シリーズ金庫を転換してDシリーズ優先株
を発行することにより、発行コスト#ドルを差し引く | - | - | ||||||||||||||||||||||||||||||||||||||
| D-2シリーズ金庫を転換してDシリーズ優先株
を発行することにより、発行コスト#ドルを差し引く | - | - | ||||||||||||||||||||||||||||||||||||||
| Dシリーズ優先株を発行する | - | - | ||||||||||||||||||||||||||||||||||||||
| D-1とD-2シリーズ金庫改装損失 | - | - | - | - | ||||||||||||||||||||||||||||||||||||
| 変換Dシリーズ合併前に転換可能優先株発行普通株 を償還することができる | ( | ) | ( | ) | - | |||||||||||||||||||||||||||||||||||
| 合併前の有名な実験室会社の株式オプションを行使する | - | - | ||||||||||||||||||||||||||||||||||||||
| 株に基づく報酬 | - | - | - | |||||||||||||||||||||||||||||||||||||
| 純損失 | - | - | - | ( | ) | ( | ) | |||||||||||||||||||||||||||||||||
| 総合収入 | - | - | - | - | ||||||||||||||||||||||||||||||||||||
| 残高 2023年12月31日 | $ | $ | $ | $ | $ | ( | ) | $ | $ | |||||||||||||||||||||||||||||||
付記は連結財務諸表の構成要素である。
| F-7 |
有名なbr実験室、有限会社。
統合現金フロー表
(千ドル単位で、1株当たりおよび1株当たりの金額は含まれていない)
| 12月31日までの年間で | ||||||||
| 2023 | 2022 | |||||||
| 経営活動のキャッシュフロー | ||||||||
| 純損失 | $ | ( | ) | $ | ( | ) | ||
| を調整し、純損失と経営活動で使用されている現金純額を照合する: | ||||||||
| 減価償却 | ||||||||
| 株に基づく報酬 | ||||||||
| 非現金レンタルを経営しています | ||||||||
| 固定資産処分損失 | ||||||||
| 有価証券販売損失 | ||||||||
| PPPローン減免から を得る | ( | ) | ||||||
金庫の公正価値変動 | ||||||||
| 償還可能優先株式証負債の公正価値変動 | ( | ) | ||||||
| 経営性資産と負債変化 | ||||||||
| 前払い料金と他の流動資産 | ( | ) | ||||||
| その他 資産 | ||||||||
| 売掛金 | ||||||||
| 費用とその他の流動負債を計算しなければならない | ( | ) | ( | ) | ||||
| 売掛金-関連先 | ( | ) | ||||||
| 経営的リース負債 | ( | ) | ( | ) | ||||
| 純額 経営活動で使用した現金 | ( | ) | ( | ) | ||||
| 投資活動のキャッシュフロー | ||||||||
| 財産と設備を購入する | ( | ) | ( | ) | ||||
| 有価証券を購入する | ( | ) | ||||||
| 財産と設備を処分して得られる収益 | ||||||||
| 有価証券満期日収益 | ||||||||
| 有価証券を売る収益 | ||||||||
| 企業合併現金純額 | ||||||||
| 投資活動が提供する現金純額 | ||||||||
| 融資活動のキャッシュフロー | ||||||||
| 従業員株式オプションを行使する収益 | ||||||||
| 転換可能な優先株と引受権証を発行して得られる収益 を発行し,発行コストを差し引く | ||||||||
| 融資リース債務の償還 | ( | ) | ||||||
| 外管局協定を発行して得られた収益の純額 | ||||||||
| 純融資活動から提供された現金 | ||||||||
| 現金と現金等価物の純増加(減少) | ( | ) | ||||||
| 為替レート変動が現金に与える影響 | ||||||||
| 年初現金 と現金等価物 | ||||||||
| 年末現金 と現金等価物 | $ | $ | ||||||
| 追加:非現金融資活動の開示: | ||||||||
| 融資リース使用権資産融資リース負債の支給 | $ | $ | ||||||
| 経営的リース使用権資産を発行する経営リース負債 | $ | $ | ||||||
| 普通株を発行してAシリーズを償還可能な転換優先株を償還する | $ | $ | ||||||
| 普通株を発行してBシリーズを償還可能な転換優先株を償還する | $ | $ | ||||||
| 普通株を発行してCシリーズを償還可能な転換優先株を償還する | $ | $ | ||||||
| A,B,C,Dシリーズに関連する償還可能な逆希釈株式と引き換えに普通株を発行する | ||||||||
| 転換可能優先株 | $ | $ | ||||||
| A、B、C、Dシリーズに関する償還可能奨励株普通株を発行する | ||||||||
| 転換可能優先株 | $ | $ | ||||||
| 普通株を発行して金庫を切り替える | $ | $ | ||||||
| C系列権証発行時に割り当てられた公正価値 | $ | $ | ||||||
| Cシリーズ課税費用のうち償還可能転換優先株発行コスト | $ | $ | ||||||
| AシリーズとBシリーズの償還可能な転換優先株を普通株に変換する | $ | $ | ||||||
付記は連結財務諸表の構成要素である。
| F-8 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
注 1-業務の組織と記述
有名なbr実験室有限会社は、前身は血管生物遺伝会社であり、イスラエル会社である(“有名”)。これらの連結財務諸表には、2023年12月31日現在の3つの完全子会社:顕著な実験室会社(“顕著な米国”)、VBL,Inc.(“VBL”)および顕著な治療会社(“Treateutics”)(合計“会社”)が含まれている。すべての重要な会社間取引は合併で廃止された。
注目すべきは、米国が2014年にデラウェア州の会社として登録設立されたことだ。最初,有名な米国では,どの癌治療が個々の患者に最も有効であるかを決定するための医師の診断ツールとしてPPMPが開発された。有名なアメリカはその後、その使命を拡大し、そのPPMPを応用して研究化合物の鑑定と検証を簡略化と加速し、サービスに基づく合意に基づいて複数の生物技術会社と製薬会社と協力した。2021年、OnCoherosプロトコルとCicloMedプロトコルを締結することによって、アメリカの有名な会社は純粋な診断会社から精密薬物を予測する総合診断と治療プラットフォーム治療会社 を設計と開発或いは共同開発するように発展した。
2023年10月16日に、期日が2023年2月22日の合併協議と計画(“合併協議”)に基づいて、合併付属会社は有名な実験室有限会社、合併付属会社及び著名なアメリカ会社と合併し、合併付属会社は合併後も引き続き重要な実験室有限会社の存続実体及び全額付属会社(“合併”)として存在し続ける。合併が発効したとき、いかなる株主も何の行動も取らなかった場合、1株当たり合併前に発行及び発行された顕著な普通株は、額面が$である合併前の顕著な米国流通株奨励に関する株式を含む1株(“顕著な米国普通株”)は、権利獲得に変換される有名な実験室有限会社普通株式(NIS)株式(“株式交換比率”)1株当たり額面(“会社普通株”または“顕著普通株”)。合併発効時間に続いて注目すべきは
が実現されたことである
合併の終了に伴い、ナスダック資本市場に上場した注目すべき普通株 は注目すべき実験室有限会社と注目すべき普通株と改名し、取引コードは“VBLT”であり、2023年10月17日にナスダック資本市場で取引を開始し、取引コードは“NTBL” である。
流動資金と持続的経営評価
社は設立以来運営中に損失と負キャッシュフローが出現した。2023年12月31日と2022年12月31日までの会社の累積赤字は約$である
Br社がその運営に資金を提供する能力は追加の資本を必要とし、会社は許可または協力協定を含む追加の債務または持分を発行することでこれらの資本を調達しようとしている。
これらのbr計画は、関連条件やイベントを緩和することを目的としており、これらの状況やイベントは、会社が経営を継続する能力があるかどうかを大きく疑わせるが、これらの計画は会社の制御範囲内に完全にはないため、管理層は、これらの計画が有効に実施される可能性が低いことを確認している。
これらの財務諸表は継続経営に基づいて作成されており,会社が経営を継続できない場合に資産や負債の金額や分類を調整する必要がある可能性のあるいかなる調整も含まれていない。
Br社はその薬物プラットフォームや治療法の開発を継続しており,これが会社資金の主な用途である。経営陣は、予見可能な未来には、研究開発活動の拡大により、監督管理の承認を得る前に、運営に追加的な重大な損失と負のキャッシュフローが生じ続けると予想している。規制部門の承認を受けることは保証されないし、絶対に承認されないかもしれない。
| F-9 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
会社が経営を続けるかどうかは、これらの計画を成功させ、融資源を獲得し、最終的に利益運営を実現できるかどうかにかかっている。しかしながら、そのような融資が承認されていない場合、発生していない、または適切なレベルまたは許容可能な条項で代替融資を得ることができない場合、または利益運営を達成することができない場合、会社は、運営費用を大幅に低減し、その部分開発計画を延期、縮小またはキャンセルすること、任意の候補製品の商業化権利について協力または他の同様の手配を達成すること、その候補製品に許可証 を発行し、無担保資産を販売すること、またはこれらの組み合わせを要求される可能性がある。これらの行動のいずれも、会社の業務、経営結果、財務状況、および/または所定の債務に資金を提供することができないそのタイムリーまたは根本的な能力に重大な悪影響を及ぼす可能性がある。もし同社が十分な資本を得られなければ、それは運営停止を余儀なくされる可能性がある。
注: 2-重大会計政策
(a) 陳述の基礎
添付されている総合財務諸表はすでに米国公認会計原則(“GAAP”)に基づいて作成され、当社が提出した期間の財務状況を公平に報告するために必要なすべての調整が含まれている。本説明内の適用会計指針に対するいかなる言及も、財務会計基準委員会(“財務会計基準委員会”)がアメリカ証券取引委員会(“アメリカ証券取引委員会”)の規則制度に基づいて公布した“会計基準編纂”及び“会計基準更新”に掲載された権威公認会計原則を指す。
注意すべきことは
影響を受けた
(b) 合併原則
連結財務諸表は有名な実験室有限会社及び完全子会社の口座を含み、これらの口座はすべてドル建てである。すべての会社間の残高と取引はすでに合併中に販売されている。
(c) 予算と判決の使用
公認会計原則に基づいて総合財務諸表を作成することは、通常、管理層にいくつかの推定および仮定を要求し、これらの推定および仮定は、総合財務諸表および付記に報告された金額に影響を与える。当社は、資産及び負債に関する推定及び仮定、並びに連結財務諸表日の又は有資産及び負債の開示及び報告期間内に報告された費用金額を定期的に評価する。経営陣が主観的判断を使用する分野には、リース負債や使用権資産の計量、長期資産の減価、株式ベースの報酬、研究開発コスト、償還可能な転換可能優先株式証負債 が含まれるが、これらに限定されない。管理職は過去の経験や他の様々な当時の状況で部下が合理的と考えている仮説に基づいて推定されているが,これらの仮定の結果は資産や負債の帳簿価値を判断する基礎となっており,そのような資産や負債の帳簿価値は他の出所から容易に見られるわけではない。異なる仮定や条件では,実際の結果はこれらの推定値と大きく異なる可能性がある .
| F-10 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
(d) 本位貨幣と列報貨幣
1) 本位貨幣と列貨幣種
ドルは当社が業務を展開している主な経済環境の通貨です。 そのため、当社とその米国子会社の機能通貨と届出通貨はドルです。
2) 取引と残高
最初にドル建ての取引と残高はその元の金額に記載されています。非ドル通貨残高はそれぞれ非通貨残高と通貨残高の過去の為替レートと現在の為替レートを使用してドルに換算される。非ドル取引および統合レポートの他の項目について運営と総合損失(以下に示す)は、以下のレートを使用する:(I)取引-取引日のレートまたは平均レート、(Ii)他の項目(減価償却や償却などの非貨幣資産負債表項目から)-履歴レート。
すべての為替損益は運営と総合損失.
(e) 信用リスクその他のリスクと不確定要因が集中している
会社を深刻な集中信用リスクに直面させる可能性のある金融商品は主に現金と現金等価物
を含む。同社は金融機関に大量の現金残高を持ち、年間を通じて連邦保険の$#上限を常に超えている
会社は他の早期生物製薬会社と類似したリスクに直面しているが、これらに限定されない:十分な追加資金を得る必要があり、現在或いは未来の臨床前研究或いは臨床試験は失敗する可能性があり、第三者に依存して臨床試験を行い、その候補製品のために監督とマーケティングの許可を得る必要があり、競争相手の新しい技術革新を開発する必要があり、会社の候補製品を商業化し、市場の受け入れを獲得し、そのノウハウを保護する必要があり、及び第三者との適切な製造手配を確保し、維持する必要がある。このような努力は多くの追加資本、十分な人員インフラ、そして広範囲なコンプライアンスと報告書を必要とするだろう。当社の候補製品はまだ開発中であり、これまで当社の候補製品 は販売が許可されていないため、当社は製品販売から何の収入も得ていません。Br社の研究開発が成功することを保証することはできず、会社の知的財産権が十分な保護を得るか、あるいは十分な保護を維持することは保証されず、開発されたいかなる製品も必要な政府監督管理の許可を得ることが保証されず、いかなる承認された製品も商業実行可能性があることを保証することはできない。同社の製品開発事業が成功しても、その会社がいつ(あれば)製品販売から収入を得ることができるかは定かではない。同社の経営環境は急速な技術変革と他の製薬やバイオテクノロジー会社からの激しい競争である。また、会社は従業員、コンサルタント、その他の第三者のサービスに依存している。
重要な
顧客代表
| F-11 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
(f) 細分化市場
会社はその業務を報告可能な運営部門として運営·管理しており,様々な形態の癌を治療する予測精度薬を開発する業務である。会社の最高経営責任者は経営意思決定者であり、まとめた上で財務情報を審査し、資源を割り当て、財務業績を評価する。当社のすべての長期資産はアメリカ合衆国に保存されており、すべての収入と損失はアメリカ合衆国によるものです。
(g) 現金と現金等価物
当社はすべての高流動性投資を現金および現金等価物とし、引き出し日や用途制限を受けない短期銀行預金(預金日から最大3カ月)を含む会社を重大な信用リスクに直面させる可能性のある金融商品には、主に現金と現金等価物が含まれる。当社は連邦保険の金融機関に銀行預金を保持しており、これらの預金は連邦保険の限度額を超える可能性がある。会社の現金と現金等価物を持つ金融機関が違約すれば、会社は信用リスクに直面し、その程度は貸借対照表に記録されるsそれは.同社の現金と現金等価物預金には何の損失もありません。
(h) 繰延発売コスト
Br社は、このような融資が完了するまで、進行中の株式融資に直接関連するいくつかの法律、専門会計、その他の第三者費用を繰延発売コストとする。株式融資完了後、これらのコスト
は株主権益(損失)に計上され、発行による追加実収資本の減少となる。
進行中の株式融資を放棄すれば、繰延発行コストは直ちに合併経営報告書に営業費用と全面赤字を計上する
(i) 財産と設備、純額
財産と設備は減価償却累計額を差し引いたコストで申告する。減価償却は直線法を用いて耐用年数内の減価償却を記録し,資産投入時から計算する。各資産の推定耐用年数は以下のとおりである
| コンピュータ 装置 | |
| 実験室装置 | |
| 家具と事務設備 | |
| レンタル権改善 |
資産が売却または廃棄された場合、コストおよび関連減価償却は総合貸借対照表から除外され、それによって生じる損益は総合経営報告書と全面赤字に反映される。メンテナンス·メンテナンス は発生時に費用を計上し,重大な交換や改善のコストを資本に計上する。
| F-12 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
(j) 長期資産減価準備
イベントまたは状況変化が資産が回収できない可能性があることを示す場合、
社は、その長期資産(例えば、財産および設備)の帳簿価値を評価する。保有および使用される資産の回収可能性は、帳簿金額を資産または資産グループによって予想される推定未割引将来のキャッシュフローと比較することによって評価される。帳簿金額が推定された未割引将来のキャッシュフローを超えた場合、資産帳簿価値が公正価値を超えた部分について減値損失を確認する。あったことがある
(k) 収入確認
Br社は、2023年12月31日と2022年12月31日までの年度内に、アウトソーシングプロバイダとして限られた基礎の上で何らかの診断サービスを提供するが、これらの活動は、その主要で持続的な中央業務を代表するものではない。
会社が診断サービス収入を確認する金額は、会社が顧客と締結した契約規定の義務を履行する際に、実験室サンプルの診断テストを処理し、テスト結果を顧客に提供することで期待される対価格を反映している。収入記録は、顧客との契約の決定、契約中の履行義務の決定、取引価格の決定、取引価格の履行義務への割り当て、およびエンティティが履行義務を履行する際または履行義務として収入を確認することを含む5段階収入確認モデルを採用する。当社は、実行すべき診断
サンプル数を含む指定された条項を有する契約または顧客からの調達注文を通常有する。当社は残りの履行義務がある前払いを受けていません。
そのため、
過去の経験や経営陣が不良債権を見積もる際に確認すべき他の要因を考慮し、必要に応じて不良債権準備を構築する。これらの要因には、売掛金の増加と構成、不良債権準備と売掛金の関係、および現在の経済状況が含まれる。対応金額の回収可能性を決定するには、会社が将来の事件や傾向を判断する必要がある。不良債権準備は、会社の単一顧客と全体ポートフォリオの評価に基づいて決定される。この流れには、履歴入金経験、顧客口座の現在の帳簿状態、および会社の顧客の財務状況の審査が含まれる。これらの要因の審査に基づいて、会社は特定の顧客と売掛金の組み合わせに対する全体的な準備を確立または調整する。はい2023年12月31日と2022年12月31日の間に、すべての売掛金が回収可能とされているため、不良債権準備 を計上する必要はないと考えられる。
(l) 賃貸借証書
ASC 842では賃貸借証書会社は、そのスケジュールに存在する事実および状況に基づいて、そのスケジュールがテナントであるか、またはテナント を含むかどうかを決定する。そしてレンタル開始日にレンタル分類、確認、計量を決定します。
会社は,リース開始日にリースが融資や経営リースの分類基準に適合しているかどうかを決定する際に, (1)リースがリース期限終了時に対象資産の所有権をテナントに移転するか否か,(2)リースがテナント購入テナントに付与されて行使する対象資産の選択権を合理的に決定するか否か,(3)リース期限が対象資産の残存経済寿命の主要部分であるか否か,を考慮する。(4)リース支払いとテナント担保の残存価値の和の現在値が対象資産の全公正価値に等しいか、または実質的に超えているか否か、および(5)対象資産に専門性があるか否かは、リース満了時にレンタル者に他の用途がないと予想される。同社の賃貸対象は、2023年12月31日と2022年12月31日まで、不動産と実験室設備を含み、そのうちの1つを除いて経営的賃貸に分類されている。当社には2023年12月31日現在、融資リースがあり、2022年12月31日現在、当社は融資リースを行っていません。
Br社は融資リースの形でいくつかのデバイスをレンタルします。リースの経済的実質は設備購入の融資取引 である。そこで、今回リースした使用権資産は、総合貸借対照表に融資リース、減価償却累計控除後の使用権(“ROU”)資産を計上し、該当金額は融資リース債務の当期部分または融資リース債務の非流動部分(場合によって決まる)に計上する。融資リース資産は直線法でリース年限で償却し、短い場合は直線で償却し、減価償却費用を計上する。融資リース義務に関する 利子を利息支出に計上する。
賃貸と非賃貸構成要素を含む不動産賃貸プロトコル は単一の賃貸構成要素として入金される。当社は、ラボ設備レンタルと非レンタル部分を統合してレンタルしないことを選択しました。取消不能期限が12ヶ月未満の賃貸契約は、当社の総合貸借対照表 には計上されません。当該等短期賃貸に関するレンタル料金は直線法でレンタル期間別に確認します。
| F-13 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
経営的リース使用権資産と経営的リース負債はリース開始日にリース期間内のリース支払いの現在値により確認される。ROU資産代表会社がリース期間内に対象資産を使用する権利は,経営的リース負債代表会社がリースによるリース金の支払いを義務付けている。レンタル支払いの現在値を決定する際に、レンタルに隠れている金利が容易に確定できない場合、当社はレンタル開始日の情報に基づいて逓増借款金利を使用します。当社は、賃貸開始日に得られる情報に基づいて増額借入金利を決定し、この情報は、類似期間内に担保方式で借金することによる内部開発金利 を表し、金額は類似経済環境下での賃貸支払いに相当する。同じ事実や状況に異なる 判断を適用すると見積り金額が異なる可能性がある.
(m) 転換可能優先株を償還する
例外の場合を除いて、当社は発行日に公正価値記録に従って転換可能優先株を償還し、発行コストを差し引くことができます。償還可能な転換可能な優先株はすでに添付の総合貸借対照表に株主権益(損失)以外の一時的権益とされており、このような株式にはいくつかの完全に当社がコントロールしていない償還特徴が含まれているからである。償還可能な転換可能優先株は一般的に償還できない;しかし、清算、売却または当社の制御権の譲渡を含む制御権の変動が発生した場合、償還可能な優先株の所有者は会社登録証明書の条項に従ってその清算優先権を享受することができる。このような清算イベントが発生する可能性がある場合、転換可能な優先株を償還可能な帳簿価値は、その清算選好に基づいて調整される。すべての償還可能な転換可能優先株は合併時に転換された。
(n) 転換可能優先株承認持分負債を償還することができる
対象株式brが償還可能または償還がある場合、当社は転換可能な優先株を購入する引受権証を公正価値で計算された負債に分類し、各報告期間に関連ツールを公正価値に調整する。転換可能な優先株を購入する引受権証(Br)は、行使または満期まで資産負債表ごとに再計量する必要があり、公正価値のいかなる変動も他の収入(支出)の構成要素として確認され、総合経営報告書および全面赤字で純額であることが確認された。 の発売と転換可能な優先持分証債務の発行に関するコストは相対基準で分配され、発生時に費用を計上する。
(o) 研究と開発費
研究開発費は発生時に費用を計上する。研究開発費には、研究開発活動に関連する賃金と人員コスト、材料コスト、外部臨床薬物製品の製造コスト、外部サービスコスト、研究開発設備の修理、メンテナンスと減価償却コスト、および研究開発活動のための施設コストが含まれる。将来の研究開発活動のための貨物またはサービスを提供するための前金は、貨物が関連サービスを交付または実行する際に資本化され、費用に計上されない。当社は、貨物の納入またはサービスの提供が期待されているかどうかを評価し続け、エンティティが貨物の納入またはサービスの提供を望まなくなった場合には、資本化された前払いの任意の部分を費用に計上する。
(p) 研究と開発費用を計算すべきである
Br社は,研究,臨床前研究,臨床試験,製造開発の見積コストの計上費用を研究開発費の重要な構成要素である課税費用と他の流動負債に記録している。会社のいくつかの持続的な研究開発活動は、第三者サービスプロバイダ、契約研究機関(“CRO”)と契約開発·製造機関(“CDMO”)によって行われる。これらの契約の財務条項はbr交渉の影響を受け、これらの条項は契約によって異なり、支払いの流れがこのような契約に基づいて会社に材料やサービスを提供する期限と一致しない可能性がある。当社は,それぞれのプロトコルによる実工程見積り数に基づいて,これらの第三者とのプロトコル項で発生するコストを提案している.当社は,内部人員や外部サービスプロバイダとサービスの進捗,完了段階または実際のスケジュール(開始日と終了日)およびそのようなサービスのために支払う取り決め費用について検討することで,見積りコストを決定する.
| F-14 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
サービスの実際の実行時間または作業レベルが推定値と異なる場合、会社はそれに応じて研究開発費に影響を与える計算費用または前払い費用を調整する。実際に発生した金額と実質的な差はないと予想されているが、提供されたサービスの実態や時間に対する会社の提供状態や時間の理解が異なる可能性があり、報告された金額が任意の特定の時期に高すぎたり、低すぎたりする可能性がある。これまで、会社は研究や開発費の以前の見積もりに大きな調整はなかった。
会社は従業員、コンサルタント、取締役への長期的なインセンティブとして株式インセンティブ計画を維持している。この計画は、奨励的株式オプション(“ISO”)、非法定株式オプション(“NSO”)および制限株式奨励を発行することを許可する。
Br社は、付与日株式報酬の推定公正価値を計量し、必要なサービス期間内のこれらの報酬の補償費用を確認し、このサービス期間は、通常、対応する報酬の帰属期間である。会社は直線法を用いてサービスから与えられた報酬に基づく費用 を記録している.当社は発生した没収行為を計算します。業績ベースの奨励については、業績基準に達する可能性がある場合、会社は加速帰因法を用いて、必要なサービス期間内の株式ベースの報酬支出を確認する。
各株式奨励の公正価値は、付与された株式数と付与日の普通株式価値とに基づいて決定される。合併後、会社は活発な普通株市場を持ち、ブラック-スコアーズオプション定価モデルを採用し、このモデルは一連の複雑な主観仮説を使用する必要があり、普通株の推定公正価値、期待変動率、無リスク金利、期待配当率とオプションの期待期限を含む。
合併前、米国の重要な普通株と制限株は活発な市場が不足しており、取締役会(“取締役会”)は経営陣と独立第三者評価会社の協力の下でその普通株と制限株の公正価値を決定し、株式奨励brを付与する必要がある。各株式オプション報酬の公正価値は、付与された日にブラック·スコアーズオプション定価モデルを用いて推定される。有名なアメリカ会社はずっと個人会社であり、会社の特定の歴史と隠れた公正価値情報が不足しているため、有名なアメリカ会社の普通株と制限性株の最適な推定公正価値を確定するには重大な判断が必要である。取締役会は毎回奨励を承認する会議で多くの客観と主観的要素を考慮して、重要なアメリカ普通株式オプションの公正価値 を確定する。考慮要因は、(I)米国の有名な普通株の同期独立第三者推定値の結果と、米国の有名な償還可能な転換可能優先株のその普通株に対する価格、権利、優遇および特権、(Ii)米国の有名な普通株の市場性の欠如、(Iii)実際の経営および財務業績、(Iv)米国の有名な発展段階に関連する現在の業務状況および予測、を含むが、これらに限定されない。(V) 当時の市場状況を考慮して、流動性イベントを実現する可能性、例えば、初公募株または有名な米国株の売却、 (Vi)重要な米国株の前例取引、および(Vii)重大なマイルストーンおよび研究開発作業の進展 に関連する。
合併後、当社は当社の普通株の加重平均履歴変動率を用いて予想される普通株変動率を決定した。合併前に、当社は、発行条項がほぼ類似したオプションの指導会社の履歴変動率の加重平均値を用いて予想株価変動率を決定し、当社が自身の取引株価変動性に関する十分な履歴データを有するまで、 を継続する予定である。当社の株式オプションの期待期限は簡略化方法で決定されました。無リスク金利は、奨励付与時に発効した米国債収益率曲線を参考にして決定され、期間は奨励の予想期限にほぼ等しい。当社はまだ支払っておらず、普通株の現金配当金の支払いも期待されていません。そのため、期待配当率はゼロと仮定しています。
社がその合併経営報告書において株式による補償費用と全面損失を分類する方式は,受賞者の現金補償コストを分類する方式と同様である.
| F-15 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
当社が付与日を決定する際に使用する仮説、付与された株式奨励の公正価値および2023年12月31日現在の当社株式インセンティブ計画における株式奨励活動の概要については、付記12を参照されたい。
(r) 公正価値計量
公正価値会計は、公正価値に応じて連結財務諸表において日常的に確認または開示されるすべての金融資産および負債に適用される。現金や現金等価物、売掛金や売掛金などの金融商品は満期日が相対的に短いため、公正価値に近い。
総合貸借対照表に公正価値で経常的原則で入金された資産および負債は、その公正価値を計量するための投入に関する判断レベルに基づいて分類される。公正価値は、計量日に資産のために元本または最も有利な市場で負債を移転するか、または市場参加者間の秩序ある取引において負債を転送するために支払われる交換価格または退出価格として定義される。
会社は公正価値レベルを使用して金融資産と負債の公正価値を決定し、公正価値レベルは公正価値を計量するために使用できる3つのレベルの投入を以下のように記述した
| レベル 1- | 観察可能な 計量日と同じ資産や負債の活発な市場での未調整見積などの観察可能な投入。 |
| レベル 2- | 資産または負債については、投入 を直接または間接的に観察することができる(第1レベルに含まれる見積は除く)。これらのオファーは、アクティブ市場における同様の資産または負債の見積もりと、非アクティブ市場における同じまたは同様の資産または負債の見積もり とを含む。 |
| 第 レベル3- | 観察できないbr}は、管理層が市場参加者が計量日にどのような資産または負債を価格設定するのに最適な推定値を使用するかの入力を反映する。推定技術に固有のリスクとモデル投入に固有のリスクを考慮する。 |
(s) 所得税
会社はバランスシート法を用いて所得税を計算し、繰延税金資産と負債口座残高は、資産と負債の財務報告と税ベースとの差に基づいて決定され、制定された税率と予想差が逆転した場合に発効する法律を用いて計量される。当社の繰延税金資産の一部または全部が現金化できない可能性が高い場合、当社は評価準備を提供します。
Br社は福祉確認モデルを用いて所得税や有事項を計算する。監査時に税務頭寸がbrよりも持続できない可能性があると考えられる場合、その頭寸の技術的価値のみに基づいて、この利点を確認する。同社は以下の金額を決定することで収益を測定している
同社はイスラエル、アメリカ連邦司法管轄区、各州司法管轄区で税金を払わなければならない。当社の損失のため、当社は設立以来主管部門の所得税審査を受けてきました。当社の政策
は、所得税に関する利息支出と罰金を所得税支出として確認する構成要素である。2023年12月31日までに
基本 普通株株主が1株当たり純損失を占めるべき計算方法は、普通株株主は純損失を除いて期間内に発行された普通株の加重平均株式数を占め、潜在的に薄い証券を計上しないことである。
| F-16 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
普通株株主が1株当たり純損失を占めるべき計算方法は、普通株株主が純損失を 当期に発行された普通株と潜在的希薄化証券の加重平均で割るべきである。希釈後の1株当たり純損失を計算する際に、転換可能な優先株、株式オプション、償還可能な優先株を購入できる引受権証は潜在的な希薄化証券とみなされる。
当社は証券参加定義に適合した株式を発行しているため、当社は2段階法を用いて1株当たりの基本および償却純損失を計算している。2段階法は,参加証券 を一般株主が本来獲得可能な収益を持つ権利と見なす収益分配式である.当社の参加証券は契約上当該等株式保有者に配当に参加する権利を与えているが、契約上は当該等株式の保有者に当社の損失を分担することは要求されていない。したがって,会社が純損失を報告している間,このような損失はこのような参加証券 に計上されない。
したがって,当社が純損失を報告している間は,1株当たり純損失は1株当たりほぼ純損失と同様であり,希釈性普通株 はその効果が逆償却であれば発行されていないとみなされないからである。
(u) 引受金とその他の事項
クレーム、評価、訴訟、罰金および罰金、および他のソースによって引き起こされるまたは損失のある負債 および が負債が発生している可能性が高く、金額が合理的に推定できる場合、その負債を記録する。損失の有無に関する法的費用は発生時に費用を計上する。
(v) 最近の会計公告
新しい会計声明は、財務会計基準委員会または他の基準策定機関によって時々発表され、指定された発効日から当社が採択される。最近発表された会計声明 経営陣は、会社の財務状況、経営業績やキャッシュフローに実質的な影響を与えないと考えている。
(w) 最近採用された会計公告
2023年12月31日現在、新たに発表された会計基準が採用されていないことは、当社の連結財務諸表に大きな影響を与える。
注: 3-業務合併
合併·合併協定で予想される取引が完了した後、顕著に発表される 逆方向株は交換比率で普通株を合併前の有名な実験室会社の前利益関係者に分割する。合併および合併協議で行われる取引が完了した後、合併前の有名な実験室会社の前利害関係者は約を持っている% 完全に希釈した上で重要な発行済み普通株 を販売することを含む 普通株式入札オプションと 普通株関連株式権証は合併前の有名な実験室会社の株を購入します。合併前の有名な実験室会社の普通株の保有者は約合併後に発行された株式の% は注目に値する。
ASC 805によれば、会社はこの取引を有名な実験室有限会社(前身は血管生物製薬株式会社)との逆合併として会計処理を行う。合法的な買収者と合併前の有名なLabsとして,Inc.は会計購入者としている.取引の結果として、同社は今回の合併を株式資本再編と見なしている。同社は約$を受け取りました
所持約% 合併後の普通株式はロックプロトコルに制限されており、この合意によれば、これらの株主は、合併発効後60日以内に、関連、売却、交換、質権、または他の方法で任意の会社の株式の選択権を譲渡、授出してはならないことに同意している。
| F-17 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
注: 4-公正価値計量
以下の表は会社の財務負債を示し、公正価値レベルごとに公正価値の計量(千単位)を行う
| 2023年12月31日まで | ||||||||||||||||
| レベル 1 | レベル 2 | 第 レベル3 | 合計 公正価値 | |||||||||||||
| 負債.負債 | ||||||||||||||||
| 優先持分証責任 | $ | $ | $ | $ | ||||||||||||
| 2022年12月31日まで | ||||||||||||||||
| レベル 1 | レベル 2 | 第 レベル3 | 合計 公正価値 | |||||||||||||
| 負債.負債 | ||||||||||||||||
| 優先持分証責任 | $ | $ | $ | $ | ||||||||||||
2023年12月31日と2022年12月31日までの年度では,第1級,第2級または第3級の間に移行は発生しなかった。また他にも
権証の価値は,2023年12月31日まで(付記11)のブラック·スコアモデルが推定した権証価値に基づいて計算される。
合併協定については、当社はいくつかの投資家と将来の株式(SAFE)簡易協定
を締結し、当社は$を獲得しました
2023年6月28日、著名な投資家はある投資家とD-2安全協定を締結し、投資家はDシリーズ購入協定に基づいてD-2シリーズ優先株を購入することを承諾した。D-2外部管理局は、割引なしにD-2シリーズ優先株に変換し、ドル対ドルに基づいて、Dシリーズ購入プロトコル項目における各投資家が不足している購入価格を低下させる。金庫所持者が金庫を交換した合併時のD-2系列優先株の株式。
以下は、当社の2023年12月31日までの安全株式証明責任および安全手形活動の概要である
| 償還可能な転換可能優先株式証債務 | 安全なbr手形 | |||||||
| 2021年12月31日現在の残高 | $ | $ | ||||||
| 権証発行時の公正価値 | ||||||||
| 価値変動を公平に承諾する | ||||||||
| 2022年12月31日までの残高 | ||||||||
| 安全手形発行時の公正価値 | ||||||||
| 公正価値 を変更 | ( |
) | ( |
) | ||||
| マージ時の変換 | ( |
) | ||||||
| 2023年12月31日までの残高 | $ | $ | ||||||
| F-18 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
償還可能な優先株式証負債の公正価値変動 は、権利証が2023年12月31日に公平な市場推定値に基づいて、1部当たりの株式証の価値が減少したためである。減収は主に関連株の価格がこれまでの予想を大幅に下回ったためである(付記2(R))。
安全手形の発行時の公正価値には$が含まれている
2023年12月31日及び2022年12月31日現在、金融商品(現金及び現金等価物、短期銀行預金、制限された銀行預金、その他の流動資産及び売掛金)の公正価値は帳簿価値とほぼ同じである。
注: 5-貸借対照表の構成要素
前払い料金と他の流動資産
次の表は、2023年12月31日、2023年12月31日、2022年12月31日までの前払い費用と他の流動資産の構成要素(単位:千)を示している
| 十二月三十一日 | ||||||||
| 2023 | 2022 | |||||||
| 売掛金 | $ | $ | ||||||
| 従業員 留任ポイント | ||||||||
| 費用を前払いする | ||||||||
| 前払い給付 | ||||||||
| 臨床費用を前払いする | ||||||||
| 前払い費用とその他の流動資産を合計する | $ | $ | ||||||
当社は、2020年度と2021年度に、米国政府が“コロナウイルス援助、救済、経済安全法”(以下、“CARE法案”と略す)に基づいて新冠肺炎に提供する救済条項から利益を得る。CARE法案は、2021年度までに従業員に支払われるいくつかの適格なbr賃金に基づいて、ある就業税に対して提供される払戻可能な税収控除である従業員留任控除(“従業員留任相殺”)を提供する。“CARE法案”によると、会社は税金控除を受ける資格があり、2021年末までの合格賃金追加減免条項に基づいて、引き続き追加税金控除を受ける資格がある。2023年12月31日と2022年までに
財産と設備、純額
次の表は、2023年12月31日と2022年12月31日までの財産と設備の純額構成要素(千計)を示しています
| 十二月三十一日 | ||||||||
| 2023 | 2022 | |||||||
| コンピュータ 装置 | $ | $ | ||||||
| 実験室装置 | ||||||||
| 家具と事務設備 | ||||||||
| レンタル権改善 | ||||||||
| 減算: 減価償却累計 | ( | ) | ( | ) | ||||
| 財産と設備合計 純額 | $ | $ | ||||||
減価償却費用は約$
| F-19 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
外管局投資
2021年10月、当社は未来株式(“OnCoheros Safe”)について簡単な合意を締結し、金額は$とした
費用とその他の流動負債を計算しなければならない
次の表は、2023年12月31日と2022年12月31日までの課税費用とその他の流動負債の構成要素(単位:千)を示しています
| 十二月三十一日 | ||||||||
| 2023 | 2022 | |||||||
| 課税費用 | $ | $ | ||||||
| 従業員費用を計算する | ||||||||
| 累計ボーナス | ||||||||
| 計算すべき費用とその他の流動負債総額 | $ | $ | ||||||
注: 6-売掛金と売掛金に関係する当事者
当社は、2023年12月31日、2023年12月31日、2022年12月31日まで、関連先に以下の金額(千計)を不足しています
| 2023年12月31日 | 2022年12月31日 | |||||||||||||||||||||||
| 勘定.勘定 | 応策 | 勘定.勘定 | 応策 | |||||||||||||||||||||
| 対処する | 費用.費用 | 合計する | 対処する | 費用.費用 | 合計する | |||||||||||||||||||
| 前 会長 | $ | $ | $ | $ | $ | $ | ||||||||||||||||||
| 取締役会 メンバー | $ | $ | $ | $ | $ | $ | ||||||||||||||||||
前取締役会長とのコンサルティングサービスについては、会社が記録した一般的かつ行政費用は#ドルであった
取締役会メンバーとのコンサルティングサービスについて、会社が記録した一般的かつ行政的費用は#ドルです
| F-20 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
注: 7-共同開発と許可協定
OnCoheros プロトコル
2021年9月、当社はOnCoherosと独占許可協定(“OnCoheros協定”)を締結し、これにより、当社は成人のいくつかのタイプの癌に関連する用途のために、小分子volasertibのグローバル独占開発権と商業化権利を取得した。OnCoheros協定の条項によると、OnCoherosは、会社の許可を得ていない癌治療薬volasertibを開発および商業化する権利を保持する。
協定条項によると、同社は追加の臨床と規制マイルストーンの支払いを義務しており、総額は最高で
$です
同社は2021年10月にOnCoherosと外管局と合意し、金額は$となった
CicloMed プロトコル
2021年7月,当社はCicloMed LLC(“CicloMed”)と共同開発·利益共有プロトコル(以下,“CicloMedプロトコル”と呼ぶ)を締結し,CicloMedが急性骨髄性白血病を治療するCicloProx製品を開発する際に当社を用いた精密腫瘍診断テストを内容とした。共同開発プロトコルの条項によると、CicloMedは臨床試験操作を実行する主要な責任を持っているが、主に顕著な予測精度
医学プラットフォームの最適化に顕著に集中している。発効日
の後,双方は行っている臨床試験に関する費用を平均的に分担する。CicloProx製品が商業化されて開発·販売されれば、双方は純収益を共有するだろう。会社は
$を記録した
注: 8-賃貸借証書
2023年2月、会社は#ドルの設備融資リース契約を締結した
2023年4月、同社はカリフォルニア州フォスターシティにある施設の賃貸契約を延長した。レンタル期間は2023年6月から2027年5月に延長されます。当社は、4ヶ月の通知brおよび通知年度の4ヶ月の基本レンタル料を早期終了契約費用として提供した後、2025年3月に発効した賃貸契約を終了する権利があります。加重平均増額借入金金利は
次の表は、2023年12月31日と2022年12月31日までの年間レンタル総費用(千単位)をまとめています
| 2023年12月31日 | 2022年12月31日 | |||||||
| ROU資産の償却-融資リース | $ | $ | ||||||
| リース負債利息(Br)-融資リース | $ | $ | ||||||
| 融資リース負債を支払う現金 | $ | $ | ||||||
| レンタル負債経営のための現金 | $ | $ | ||||||
| 運営 レンタル料 | $ | $ | ||||||
| 可変 レンタル料 | $ | $ | ||||||
| 短期レンタル料金 | $ | $ | ||||||
| F-21 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
次の表は、2023年12月31日までの賃貸負債満期日と賃貸負債の入金状況(単位:千):をまとめています
| レンタル義務 | ||||||||
| 融資リース | 施設レンタル | |||||||
| 2024 | $ | $ | ||||||
| 2025 | ||||||||
| 2026 | ||||||||
| 2027 | 87 |
240 | ||||||
| 2028年以降 | ||||||||
| 将来割引されていないレンタル支払いを合計する | ||||||||
| 差し引く: 利子を計上する | ( |
) | ( |
) | ||||
| 賃貸負債合計 | $ | $ | ||||||
会社ROU資産と関連賃貸負債に関する情報 は以下のとおりである(残存期間と割引率を除いて千計):
| 2023年12月31日 | ||||||||
| 融資リース | 施設レンタル | |||||||
| 流動融資リース負債 | $ | $ | ||||||
| 非流動融資リース負債 | $ | $ | ||||||
| 当期賃貸負債を経営する | $ | $ | ||||||
| 非流動経営リース負債 | $ | $ | ||||||
| 重み 平均残存賃貸年数(年) | ||||||||
| 重み 平均割引率 | % | % | ||||||
| 2022年12月31日 | ||||||||
施設 レンタルする |
装備 賃貸借証書 |
|||||||
| 当期賃貸負債を経営する | $ | $ | ||||||
| 非流動経営リース負債 | $ | $ | ||||||
| 重み 平均残存賃貸年数(年) | ||||||||
| 重み 平均割引率 | % | % | ||||||
注: 9-賃金保障計画ローン
2021年2月、会社はPaycheck保護計画に従って本チケットを申請し、#ドルの承認を得た
| F-22 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
注: 10-資本構造
普通株 株
2023年10月16日つまり合併が終わる前に注意すべきことは文章に対する修正案を提出した協会のメンバーイスラエル社の登録は逆株式分割(新しいシェケルの追加を含む)の額面を反映している
普通株1株当たり)、配当金を増加させる(会社に所有させる普通株式と新シェケルの許可
2022年12月31日までに有名なアメリカのライセンスが発表されました$の株額面普通株。取締役会が任意の転換可能な優先配当金をすべて支払った後、普通株主 は配当を得る権利があると発表した場合、1株当たりの普通株の所有者は1票を獲得する権利がある。2022年12月31日現在、配当は発表されていない。
簡単な未来持分プロトコル
2022年1月から5月までの間に、有名なアメリカ会社はある投資家と未来の株式(2022年金庫)について簡単な合意に達し、$を獲得した
流動性イベントや解散事件が発生した場合、2022金庫の所持者は自動的に購入金額の一部を得る権利がある。2022年金庫は発行時に負債として記録され、報告日ごとに再計量され、価値変動は他の収入(費用)、総合経営報告書の純額、全面損失に記録されています。
有名なアメリカが2022年6月からC-1シリーズの償還可能な転換可能優先株を発行することについて、発行価格は$C-1シリーズ株については、2022年金庫の保有者が株式融資に参加することができる。Br}金庫はC-2シリーズ株に変換することで決済され、発行価格は$です1株当たり,合計するC-2系列は優先株
を償還可能である.C-1シリーズとC-2シリーズの償還可能な転換可能な優先株を総称してCシリーズの償還可能な転換可能優先株と呼ぶ。純収益$
| F-23 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
償還可能な転換優先株
2022年12月31日までに有名なアメリカのライセンスが発表されました $の株額面Aシリーズ、BシリーズとCシリーズの償還可能な転換可能優先株(総称して“償還可能転換可能優先株”、“優先株”または“償還可能転換優先株”と呼ぶ)。
2022年6月から2022年7月まで、アメリカで有名なC-1シリーズの償還可能な転換優先株を投資家に売却する1株当たりの総収益は$
発行された各Cシリーズの償還可能な転換可能な優先株に対して、有名なアメリカ会社はまた引受権証を発行し、Cシリーズの償還可能な優先株(“Cシリーズ株式承認証”)を購入する。約$
2022年6月、有名なアメリカは会社登録証明書を改訂し、すべての既存の償還可能な転換可能優先株株主にCシリーズ優先株発行に参加することを要求する特別強制転換条項を加えた。C系列優先株発行に参加できなかったことは、保有者の優先株 を自動的に普通株に変換することになる。2022年7月Aシリーズ償還可能な転換可能優先株和C系列優先株の発行に関与していないため、B系列償還可能優先株の株式は普通株に変換され、Bシリーズ償還可能優先株総数は減少する。
2023年12月31日までの1年間であるが、合併前に、有名な米国会社はある投資家と安全協定を締結し、合併により、有名なアメリカ会社が獲得した
償還可能な変換可能優先株は、以下の権利および特権を有する:
オプション 変換
1株当たり転換可能優先株を償還可能であり、任意の時間に所有者によって普通株に変換することができ、方法は、元の発行価格を変換時の有効変換価格で割ることである。2022年12月31日現在、転換可能優先株を償還可能な初期1株当たりの変換価格は元の発行価格に相当するため、任意の調整を行う前に 1対1のベースで変換を行う。
各適用株式交換価格は、将来の任意の株式分割または株式組み合わせ、再分類または類似株の交換、有名な米国会社の再編、合併または合併、または有名な米国会社が普通株を発行または売却する費用が適用株式交換価格よりも低いことに応じて調整される。
強制 変換
1株当たり償還可能な転換可能優先株は、より早い変換率から決定される普通株式数に自動的に変換される
| F-24 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
清算 優先
任意の自動または非自発的清算、解散または清算の場合、発行された償還可能な優先株の保有者は、転換後および同等の割合でその株主に割り当てられた有名な米国の資産から比例して支払う権利があり、普通株式所有者に任意の金を支払う前に、1株当たりの金額は、(I)このようなbrシリーズ優先株の適用元発行価格に、申告されているが支払われていない配当金のいずれかの大きい者に等しい。または(Ii)当該等清盤、解散、清盤または清盤事件の直前とみなされるすべてのbr株償還可能転換可能優先株がすべて普通株に変換されたときに支払われるべき1株当たりの金額。
償還可能な優先株式所有者に割り当てられた有意なドル資産が均等所有者に支払うのに十分でない場合、任意の合法的に割り当て可能な有意なドル資産は、償還可能な変換可能な優先株式所有者の間で比例的に割り当てられ、割合は、各均等所有者が他の方法で獲得する権利がある優先順位の金額である。
上記で指定されたすべての優先金額を償還可能な転換可能な優先株保有者に支払った後、任意の残りの株主に割り当てることができる顕著な米国資産は、普通株式株主に比例して割り当てられる。
配当をする
転換可能な優先株を償還可能なbr所有者は、任意の合法的に利用可能な資産から配当を得る権利があり、普通株式の任意の配当金を発表または支払いする前に、普通株の任意の配当金を発表または支払いする前に、著名な米国取締役会によって発表された場合にのみ、配当を得る権利がある。この配当は累積的ではない。2023年12月31日と2022年12月31日までに、累積配当金や延滞はない。
投票する.
転換可能優先株を償還可能な保有者1人当たり、普通株総株式数に相当する投票権 を得る権利があり、このような転換可能優先株を償還可能な株式は、記録日から普通株に変換することができる。転換可能な優先株を償還可能な保有者は、普通株保有者と1つのカテゴリとして投票する。
Aシリーズは転換可能な優先株を償還可能なbr保有者が独占的に保有し、普通株に変換した上で単独カテゴリとして一緒に投票し、取締役を1人選出して有名な米国取締役会に入る権利がある。Bシリーズは償還可能な優先株の保有者が独占的に保有し、普通株に変換した上で単独カテゴリとして一緒に投票し、1人の取締役を選出して米国の有名な取締役会に入る権利がある。
次輪希釈防止保護
有名なアメリカ会社が無代償または各株の償還可能な転換可能な優先株の有効な転換価格を下回る場合、このシリーズの転換価格は を引き下げて、一連の転換可能な優先株を転換可能な普通株の数を増加させる。
合併時には、すべての償還可能優先株株式が顕著な普通株に変換される。償還可能な転換可能優先株契約に基づいて、Aシリーズの償還可能な転換可能優先株株主は受け取りました普通株、Bシリーズ償還可能転換優先株保有者が受け取りました普通株、Cシリーズ償還可能転換優先株保有者が受け取りました普通株とDシリーズ償還可能転換優先株保有者が受け取りました 普通株式。また、償還可能な転換可能優先株株主合計は受け取りました 逆希釈普通株式と 奨励的普通株。
注: 11-転換可能な優先株を買い戻す引受権証
2021年4月、注目すべき発表
Cシリーズの償還可能な転換可能な優先株を発行する上で、アメリカの有名な引受権証はCシリーズの償還可能な転換可能な優先株を購入した(“Cシリーズ株式承認証”、総称して“Cシリーズ株式承認証”と呼ばれる)。br}Cシリーズ株式承認証所有者は最も多く購入する権利がある
| F-25 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
Br社は公正価値によってそのCシリーズ株式証負債(3級負債に分類)を計量し、総合経営報告書内の公正価値変動及び全面損失を計上し、株式証が行使され、満期或いはその他の事実が行使されるまで 及び状況により持分証負債を権益ツールに再分類した。公正価値は オプション定価逆解法を用いて決定される.Cシリーズ株式証負債の2022年12月31日までの公正価値は、br確率を用いて予想リターン方法を加重することによって決定され、この方案では、有名なアメリカは上場企業との合併を完了し、brのシナリオでは、有名なアメリカはその後撤退するまで運営を継続し、これはオプション定価方法を用いて推定される。Cシリーズ株式証負債2023年12月31日までの公正価値はBlack-Scholesオプション定価モデル を用いて決定された。
権利証の公正価値を推定する際には、以下の仮定を採用した
2023年12月31日まで :
| 無リスク金利 | % | |||
| 期待寿命(年) | ||||
| 期待変動 | % | |||
| 年間配当収益率 | % |
2022年7月までの発行時点:
| 無リスク金利 | % | |||
| 期待寿命(年) | ||||
| 期待変動 | % | |||
| 年間配当収益率 | % |
2022年12月31日まで :
| 無リスク金利 | % | |||
| 期待寿命(年) | ||||
| 期待変動 | % | |||
| 年間配当収益率 | % |
以下は、会社が2023年12月31日と2022年12月31日までの年度の償還可能優先株式証債務活動の概要を示す
| 償還可能な転換可能優先株式証債務 | ||||
| 2021年12月31日までの残高 | $ | |||
| 権証発行時の公正価値 | ||||
| 公正価値 を変更 | ||||
| 2022年12月31日までの残高 | ||||
| 公正価値 を変更 | ( | ) | ||
| 2023年12月31日までの残高 | $ | |||
| F-26 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
2000年計画
2000年2月、注目すべき取締役会は、2008年に改訂されたオプション計画(“2000計画”)を承認した。 は2000計画により、会社は保留したNISの普通株従業員と非従業員に割り当てられた会社額面
2011年計画
2011年4月、注目すべき取締役会は新たなオプション計画(“2011計画”)を承認した。2011年の計画によると、 会社は最大で保留しました 普通株式(ただし普通株式は、2000計画に保持されている未割り当てプールから抽出されなければならない)を従業員および非従業員に割り当てる
2014持分インセンティブ計画
注目すべき株主は2014年9月、従業員の持株および株式購入計画(2014) (“2014計画”)の採択を承認し、公開発売終了時から発効した。2014年の計画によると、注目すべきは保留されていることです 普通株式(ただし(Br)普通株式は、2011年に予定されている未分配資金プールから抽出されなければならない)。購入株式を行使する際に発行される普通株は、配布時に直ちに他の普通株と同じ権利が付与される
2015年株式インセンティブ計画
注目すべきは、米国は2015年8月に2015年株式インセンティブ計画(“2015計画”)を採択し、従業員、取締役、コンサルタントにISO、NSO、 と制限株を付与することを規定していることだ。2015年計画共同許可将来の発行に備えて株式 を予約する.2015年計画の改正案によると,別の項目がある2017年の株式 、2019年の株 ,および2022年の株 は将来の発行に備えて保留されることが許可されている。2023年12月31日までに株式 は、2015年計画に基づいて将来のために予約された普通株を発行し、合併によって有名に採択されている。
| オプション 未完了 | ||||||||||||||||
| 未完了のオプションの総数は である | 加重平均 行の重み | 加重平均 残り契約期間 | 内在的価値を集約する | |||||||||||||
| ( 年内に) | (単位:千) | |||||||||||||||
| 2022年12月31日までの未返済債務 | $ | $ | ||||||||||||||
| マージにより 重要なオプションを仮定する | ||||||||||||||||
| 授与する | $ | |||||||||||||||
| 鍛えられた | ( | ) | $ | |||||||||||||
| キャンセルします | ( | ) | $ | |||||||||||||
| 2023年12月31日までの未返済債務 | $ | $ | ||||||||||||||
| 2023年12月31日から行使可能 | $ | $ | ||||||||||||||
| に帰属しており,2023年12月31日に帰属する予定である | $ | $ | ||||||||||||||
| F-27 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
行使された株式オプションの内的価値の合計は$である 1,000ドル 2023年12月31日までと2022年12月31日まではそれぞれ千元である.あったことがある 2023年と2022年12月31日までの2015年計画での限定株式活動(RSA)。
重要なアメリカ株式オプションの仮定
合併協定の条項によれば、合併前に合併発効時間直前に行使されていない重要な米国の株式購入項目の下のすべての米国の権利および責任が顕著であると仮定することに留意されたいが、各株式購入(帰属の有無にかかわらず)は、発効直前に発効する条項と実質的に同じ条項で、顕著な普通株を購入する権利を代表する購入権に変換されるが、br}発行可能な顕著な普通株数およびその等購入株の1株当たりの行使価格は、 逆株分譲によって調整される。
株に基づく報酬費用
あります 2023年12月31日までに年度内に付与されたオプション。加重平均付与日2022年12月31日までの年度内に付与されたオプションの公正価値は$1株あたり 。有名な米国は、株式の報酬に基づく公正な価値を決定するために、高い主観的仮定を使用することを要求するブラック·スコアーズオプション定価モデルを使用して株式オプションの公正価値を推定する。従業員と非従業員株式オプションの公正価値は、奨励に必要なサービス期間内に直線原則で費用として確認される。これらの仮定 は:
| ● | 無リスク金利 ·無リスク金利は、付与時に有効な米国財務省ゼロ金利債券 に基づいており、期限は期待オプション期限に対応する。 | |
| ● | 期待変動 ·当社は個人持株であり、その普通株は取引履歴がないので、予想変動率は、株式オプション付与の予想期間と等しい期間における上場バイオテクノロジー会社の平均変動率に基づいて推定される。比較可能な会社は,ライフサイクルや専門分野における類似規模,段階 に基づいて選択される.上場企業になって以来、公開市場で取引される普通株の変動性を利用する。 | |
| ● | 所期の 期限·予想期間は、株式ベースの報酬が突出すると予想される期間を表す。オプション付与の期待期限 は簡略化方法を用いて決定される.簡略化方法は,期限を株式報酬の帰属時間と契約期限の中点 とする.歴史的なトレーニングデータが不足しているため、当社はこの方法を使用しています。 | |
| ● | 期待配当率 ·会社は、その普通株に配当金を支払ったこともなく、その普通株に対して配当金を支払う計画もない。したがって、同社が使用している期待配当収益率はゼロとなる。 |
| 12月31日までの年間 、 | ||||||||
| 2023 | 2022 | |||||||
| 所期の 期限(年単位) | - | |||||||
| 無リスク金利 | % | % | ||||||
| 期待配当率 | % | % | ||||||
| 期待変動 | % | % | ||||||
| F-28 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
| 12月31日までの年間 、 | ||||||||
| 2023 | 2022 | |||||||
| 研究開発 | $ | $ | ||||||
| 通常 と管理 | ||||||||
| 合計する | $ | $ | ||||||
2023年12月31日現在、未確認株式奨励に関する株式報酬支出総額は$百万,重み付き平均残り時間で を確認する何年もです。
注: 13-引受金とその他の事項
従業員福祉計画
Br社はその従業員のために401(K)で定義された支払い計画を開始した。この計画はすべての従業員に繰延納税賃金減額を提供する。従業員の支払いは自発的だ。従業員は最高で年間給与の100%を本計画に使用することができるが、米国国税局が決定した年間最高限度額に制限されている。同社はその401(K)計画の下でそれに貢献していない。
事件があったり
当社は通常業務の過程で起こる法的訴訟に時々巻き込まれる可能性があります。2023年12月31日または2022年12月31日までの年間で、当社にはいかなる重大な法律手続きもなく、現在もいかなる重大な法律手続きの懸案や脅威もない。
賠償する
正常な業務過程において、有名な会社は協定を締結し、 は賠償条項を含む可能性がある。イスラエルの法律で許可されている場合、役人または役員がこのような職務を担当しているか、またはそのような職務中に発生したいくつかの事件または事件がある場合、その高官および取締役に対する賠償が注目される。場合によっては、補償は合意の終了後に継続されるだろう。これらの規定により、将来支払う必要がある可能性のある最大潜在金額は を決定できないことに注意されたい。これらの賠償条項に関連する訴訟を弁護したり、クレームを解決したりするために物質コストが発生したことはないことに注意されたい。 現在いかなる賠償要求も知られていないことに注意すべきである。したがって,2023年12月31日と2022年12月31日まで,これらの賠償権利や合意に関する責任は何も記録されていないことに注意されたい。
注: 14-所得税
連邦法定所得税と会社の2023年12月31日と2022年12月31日までの実際の所得税支出の入金は以下の通り
| 12月31日までの年間 、 | ||||||||
| 2023 | 2022 | |||||||
| 連邦法定所得税 | % | % | ||||||
| 州所得税 | ||||||||
| 外国所得税 | ||||||||
| PPP(Br)ローン減免 | ||||||||
| 研究開発単位 | ||||||||
| ASC 740-10予約 | ( | ) | ( | ) | ||||
| 外管局は責任を重算する | ( | ) | ||||||
| 転換可能債務損失 | ( | ) | ||||||
| 調整を延期する | ( | ) | ||||||
| 合併によって得られた繰延税金資産 | ||||||||
| 為替レートは実際に上ります | ||||||||
| 他にも | ( | ) | ( | ) | ||||
| 評価免税額の変更 | ( | ) | ( | ) | ||||
| 所得税引当総額 | % | % | ||||||
| F-29 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
繰延所得税は、財務報告目的のために記録された金額と税務目的のための金額 との一時的な違いによる純税収の影響を反映している。繰延所得税は以下の部分からなる(千計)
| 十二月三十一日 | ||||||||
| 2023 | 2022 | |||||||
| 繰延納税資産: | ||||||||
| 純営業損失繰越 | $ | $ | ||||||
| 税金の繰越免除 | ||||||||
| 財産 と設備 | ||||||||
| 資本化研究と実験コスト | ||||||||
| 株の報酬 | ||||||||
| 他にも | ||||||||
| 小計 | ||||||||
| 推定手当(Br) | ( | ) | ( | ) | ||||
| 純繰延税金資産(負債) | $ | $ | ||||||
繰延税金資産が現金化できない可能性が高い場合、評価準備が提供される。将来の課税収入を通じて繰延税金資産の不確定性
について、当社はすでに全額推定値を提供してbrを準備しているため、繰越その他の繰延税金資産の純営業損失についていかなる利益も確認していない。
推定免税額が増加した$
2023年12月31日現在、同社の連邦、州、海外純営業損失(NOL)は約br}$に転換している
2023年12月31日まで、同社の連邦とカリフォルニア研究開発(R&D)クレジットは約brドルに転換した
会社が将来純営業損失を利用する能力は、“国税法”第382節と同様の州税法で定義されているように、過去の や将来の所有権変更の場合に大きな制限を受ける可能性がある。会社 に所有権変更(定義のような)が発生すると,その純営業損失繰越と信用の使用は にかなりの年次制限を受ける可能性がある.年間限度額は純営業損失や使用前の信用満期になる可能性があります。
会社は、財務諸表において確認、計量、列報、および開示されたか、または納税申告書上で採用されると予想される任意の不確定税収ヘッドの総合モデルが規定されているASC 740−10“所得税における不確実性会計”を遵守する。当社は、最初にFASB解釈第48号として発表されたFASB ASCテーマ740-10に規定されている条項を採用した所得税における不確実性会計それは.この声明は、税収が確定していないことを確認する税収優遇に“より可能性がある”という基準を設定している。
| F-30 |
有名なbr実験室、有限会社。
連結財務諸表付記
2023年12月31日
不確定な 税務頭寸は以下のように構成される(千計)
| 十二月三十一日 | ||||||||
| 2023 | 2022 | |||||||
| 期初残高 | $ | $ | ||||||
| 今年度の新規税額 | ||||||||
| 期末 残高 | $ | $ | ||||||
先に述べた未確認の税金優遇については
同社はアメリカ内のいくつかの州に連邦所得税申告書と所得税申告書を提出した。当社は現在連邦または州司法管轄区域所得税機関の審査範囲内にありません。任意のNOL使用日から、すべての納税申告書はそれぞれ3年と4年以内に連邦と州当局の審査のために開放される。
| 12月31日までの年間 、 | ||||||||
| 2023 | 2022 | |||||||
| 分子: | ||||||||
| 純損失 | $ | ( | ) | $ | ( | ) | ||
| 分母: | ||||||||
| 加重平均 普通株流通株は、1株当たりの基本と希釈後の純損失の計算に用いられる | ||||||||
| 1株当たり純損失、基本損失と希釈後の損失: | $ | ) | $ | ) | ||||
会社の潜在的希薄化証券は、その影響が逆薄になるため、1株当たりの純損失の計算から除外されている。したがって,基本と希釈後の1株当たり純損失を計算するための加重平均普通株流通株数は同じである。各償却計算に含まれていない潜在的希薄化証券は以下の通りである
| 12月31日までの年間 、 | ||||||||
| 2023 | 2022 | |||||||
| 系列A-1償還可能転換優先株 | ||||||||
| 系列A-2償還可能転換優先株 | ||||||||
| 系列A-3償還可能転換優先株 | ||||||||
| 系列A-4償還可能転換優先株 | ||||||||
| 系列A-5償還可能転換優先株 | ||||||||
| 系列A-6償還可能転換優先株 | ||||||||
| B-1シリーズ償還可能転換優先株 | ||||||||
| B-2シリーズ償還可能転換優先株 | ||||||||
| C-1シリーズ償還可能転換優先株 | ||||||||
| C-2シリーズ償還可能転換優先株 | ||||||||
| 転換可能な優先株を買い戻す引受権証 | ||||||||
| 株式 オプション、発行済みと未償還 | ||||||||
| 合計する | ||||||||
注: 16-後続事件
Br社は、2024年4月1日まで(連結財務諸表が発行可能な日)までのすべてのイベントを評価しており、その間、以下に開示するイベントを除いて、正常な業務運営プロセス以外に開示すべきイベントは何も発生していない。
2024年1月24日会社発表1人から5人の取締役会のメンバーと関連側の取締役会のメンバーに選択権が付与される。オプションの公正価値はBlack-Scholesオプション定価モデルを用いて計算され,公正価値は約 $であるそれは.公正価値は帰属期限になる何年もです。
2023年3月22日、株主は会社員の持株とオプション計画(2024年)を承認した。
| F-31 |