CK 0001824293-202312310001824293誤り2023会計年度0.03330.142900.14290P 15 D0.142900018242932023-01-012023-12-3100018242932023-06-30ISO 4217:ドル00018242932024-03-20Xbrli:共有00018242932023-10-012023-12-310001824293Ck 0001824293:AmendedAndRestated 2018 EquityIncentivePlanMember2023-04-212023-04-210001824293Ck 0001824293:AmendedAndRestated 2018 EquityIncentivePlanMember2023-04-2100018242932019-12-31ISO 4217:ドルXbrli:共有0001824293アメリカ公認会計基準:オプションメンバーを呼ぶ2015-12-3100018242932022-07-3100018242932022-10-3100018242932023-12-3100018242932022-12-3100018242932022-01-012022-12-3100018242932021-12-310001824293アメリカ-アメリカ公認会計基準:普通株式メンバー2021-12-310001824293US-GAAP:AdditionalPaidInCapitalMembers2021-12-310001824293アメリカ-公認会計基準:前払いメンバーを保留2021-12-310001824293US-GAAP:AdditionalPaidInCapitalMembers2022-01-012022-12-310001824293US-GAAP:変換可能ノードPayableMemberUS-GAAP:AdditionalPaidInCapitalMembers2022-01-012022-12-310001824293US-GAAP:変換可能ノードPayableMember2022-01-012022-12-310001824293アメリカ-アメリカ公認会計基準:普通株式メンバー2022-01-012022-12-310001824293US-GAAP:AdditionalPaidInCapitalMembersUS-GAAP:BridgeLoanMembers2022-01-012022-12-310001824293US-GAAP:BridgeLoanMembers2022-01-012022-12-310001824293アメリカ-公認会計基準:前払いメンバーを保留2022-01-012022-12-310001824293アメリカ-アメリカ公認会計基準:普通株式メンバー2022-12-310001824293US-GAAP:AdditionalPaidInCapitalMembers2022-12-310001824293アメリカ-公認会計基準:前払いメンバーを保留2022-12-310001824293US-GAAP:AdditionalPaidInCapitalMembers2023-01-012023-12-310001824293アメリカ-アメリカ公認会計基準:普通株式メンバー2023-01-012023-12-310001824293アメリカ-公認会計基準:前払いメンバーを保留2023-01-012023-12-310001824293アメリカ-アメリカ公認会計基準:普通株式メンバー2023-12-310001824293US-GAAP:AdditionalPaidInCapitalMembers2023-12-310001824293アメリカ-公認会計基準:前払いメンバーを保留2023-12-3100018242932023-04-212023-04-21Xbrli:純0001824293アメリカ公認会計基準:副次的事件メンバー2024-01-292024-01-290001824293アメリカ公認会計基準:副次的事件メンバー2024-01-302024-01-300001824293アメリカ-GAAP:投資家のメンバー2023-04-212023-04-210001824293アメリカ-GAAP:投資家のメンバー2023-04-2100018242932023-04-210001824293アメリカ-アメリカ公認会計基準:普通株式メンバー2023-04-212023-04-210001824293アメリカ公認会計基準:副次的事件メンバーCK 0001824293:PublicOfferingMember2024-02-012024-02-010001824293アメリカ公認会計基準:副次的事件メンバーCk 0001824293:PreFundeWarrantsメンバーCK 0001824293:PublicOfferingMember2024-02-010001824293アメリカ公認会計基準:副次的事件メンバーCK 0001824293:シリーズB 1汎用保証メンバーCK 0001824293:PublicOfferingMember2024-02-010001824293アメリカ公認会計基準:副次的事件メンバーCK 0001824293:シリーズB 2汎用保証メンバーCK 0001824293:PublicOfferingMember2024-02-010001824293アメリカ公認会計基準:副次的事件メンバーCK 0001824293:PublicOfferingMember2024-02-010001824293アメリカ公認会計基準:副次的事件メンバーCk 0001824293:PreFundeWarrantsメンバー2024-02-010001824293アメリカ公認会計基準:副次的事件メンバーCK 0001824293:シリーズB 1汎用保証メンバー2024-02-010001824293アメリカ公認会計基準:副次的事件メンバーCK 0001824293:シリーズB 2汎用保証メンバー2024-02-010001824293アメリカ公認会計基準:副次的事件メンバーCK 0001824293:シリーズA 1質保証メンバー2024-02-010001824293SRT:重み平均メンバCK 0001824293:シリーズT質保険メンバー2023-09-300001824293CK 0001824293:周辺機器のメンバー2023-12-310001824293US-GAAP:ComputerEquipmentMembers2023-12-310001824293米国-公認会計基準:従業員株式オプションメンバー2023-01-012023-12-310001824293米国-公認会計基準:従業員株式オプションメンバー2022-01-012022-12-310001824293アメリカ公認会計基準:保証メンバー2023-01-012023-12-310001824293アメリカ公認会計基準:保証メンバー2022-01-012022-12-310001824293米国-公認会計基準:制限された株式メンバー2023-01-012023-12-310001824293米国-公認会計基準:制限された株式メンバー2022-01-012022-12-310001824293CK 0001824293:GRIOperationsMember2023-04-210001824293CK 0001824293:為替保証メンバー2023-04-210001824293CK 0001824293:GRIOperationsMember2023-04-2000018242932023-04-2000018242932023-04-220001824293アメリカ-公認会計基準:公正価値入力レベル1メンバー2023-12-310001824293アメリカ-公認会計基準:公正価値入力レベル2メンバー2023-12-310001824293アメリカ-公認会計基準:公正価値投入レベル3メンバー2023-12-310001824293CK 0001824293:保証責任メンバー2022-12-310001824293CK 0001824293:保証責任メンバー2023-04-210001824293CK 0001824293:保証責任メンバー2023-04-222023-12-310001824293CK 0001824293:保証責任メンバー2023-12-310001824293アメリカ公認会計原則:投入価格を測るメンバーSRT:重み平均メンバ2023-12-310001824293SRT:重み平均メンバUS-GAAP:入力期待タームメンバーの測定2023-12-310001824293アメリカ-公認会計基準:投入予想分割率を評価するメンバーSRT:重み平均メンバ2023-12-310001824293アメリカ-公認会計基準:投入リスクを測定する自由金利メンバーSRT:重み平均メンバ2023-12-310001824293US-GAAP:ComputerEquipmentMembers2022-12-310001824293アメリカ-GAAP:家具と固定機器のメンバー2023-12-310001824293アメリカ-GAAP:家具と固定機器のメンバー2022-12-310001824293アメリカ-GAAP:投資家のメンバーCK 0001824293:BridgeSecuritiesPurcheプロトコルメンバー2022-12-130001824293アメリカ-GAAP:投資家のメンバーCK 0001824293:BridgeSecuritiesPurcheプロトコルメンバー2022-12-132022-12-13CK 0001824293:オフ0001824293アメリカ-GAAP:投資家のメンバーCK 0001824293:BridgeSecuritiesPurcheプロトコルメンバー2022-12-140001824293アメリカ-GAAP:投資家のメンバーCK 0001824293:BridgeSecuritiesPurcheプロトコルメンバー2022-12-142022-12-140001824293アメリカ-GAAP:投資家のメンバーCK 0001824293:BridgeSecuritiesPurcheプロトコルメンバー2023-03-090001824293アメリカ-GAAP:投資家のメンバーCK 0001824293:BridgeSecuritiesPurcheプロトコルメンバー2023-03-092023-03-090001824293CK 0001824293:ブリッジ保証メンバーアメリカ-GAAP:投資家のメンバー2023-03-090001824293CK 0001824293:ブリッジ保証メンバーアメリカ-GAAP:投資家のメンバー2022-12-142022-12-140001824293CK 0001824293:ブリッジ保証メンバーアメリカ-GAAP:投資家のメンバー2023-03-092023-03-090001824293CK 0001824293:為替保証メンバーアメリカ-GAAP:投資家のメンバー2023-04-210001824293CK 0001824293:BridgeSecuritiesPurcheプロトコルメンバー2022-12-310001824293CK 0001824293:BridgeSecuritiesPurcheプロトコルメンバー2023-12-310001824293CK 0001824293:TEPNoteMemberUS-GAAP:変換可能ノードPayableMember2018-11-3000018242932018-11-300001824293CK 0001824293:TEPNoteMemberUS-GAAP:変換可能ノードPayableMember2018-11-012018-11-300001824293CK 0001824293:TEPNoteMemberUS-GAAP:変換可能ノードPayableMember2019-12-310001824293CK 0001824293:TEPNoteMemberUS-GAAP:変換可能ノードPayableMember2019-12-012019-12-310001824293CK 0001824293:TEPNoteMemberUS-GAAP:変換可能ノードPayableMember2021-05-012021-05-310001824293CK 0001824293:TEPNoteMemberUS-GAAP:変換可能ノードPayableMember2022-07-012022-07-310001824293CK 0001824293:TEPNoteMemberUS-GAAP:変換可能ノードPayableMember2022-10-012022-10-310001824293CK 0001824293:TEPNoteMemberUS-GAAP:変換可能ノードPayableMember2022-10-310001824293CK 0001824293:TEPNoteMemberUS-GAAP:変換可能ノードPayableMember2022-12-012022-12-310001824293CK 0001824293:TEPNoteMemberUS-GAAP:変換可能ノードPayableMember2022-01-012022-12-310001824293米国-GAAP:PutOptionMembers2018-11-300001824293米国-GAAP:PutOptionMembers2018-11-012018-11-3000018242932022-12-012022-12-310001824293アメリカ-GAAP:投資家のメンバーCK 0001824293:シリーズA 1質保証メンバー2023-05-080001824293アメリカ-GAAP:投資家のメンバーCK 0001824293:株式権証シリーズA 1メンバー2023-05-080001824293CK 0001824293:シリーズA 2保証メンバーアメリカ-GAAP:投資家のメンバー2023-05-080001824293アメリカ-GAAP:投資家のメンバーCK 0001824293:株式権証シリーズA 2メンバー2023-05-080001824293アメリカ-GAAP:投資家のメンバーCK 0001824293:シリーズT質保険メンバー2023-05-080001824293CK 0001824293:持分保証メンバーアメリカ-GAAP:投資家のメンバーUS-GAAP:AdditionalPaidInCapitalMembers2023-12-310001824293CK 0001824293:ブリッジ保証メンバーアメリカ-GAAP:投資家のメンバー2023-05-080001824293CK 0001824293:為替保証メンバーアメリカ-GAAP:投資家のメンバー2023-05-080001824293CK 0001824293:ブリッジ保証メンバーアメリカ-GAAP:投資家のメンバーUS-GAAP:AdditionalPaidInCapitalMembers2023-12-310001824293CK 0001824293:銀行家保証メンバー2023-05-080001824293CK 0001824293:銀行家保証メンバーアメリカ-GAAP:投資家のメンバーUS-GAAP:AdditionalPaidInCapitalMembers2023-12-310001824293アメリカ公認会計原則:投入価格を測るメンバーSRT:重み平均メンバCK 0001824293:株式保証取引所保証および銀行家保証メンバー2023-12-310001824293SRT:重み平均メンバCK 0001824293:株式保証取引所保証および銀行家保証メンバーUS-GAAP:入力期待タームメンバーの測定2023-12-310001824293アメリカ-公認会計基準:投入予想分割率を評価するメンバーSRT:重み平均メンバCK 0001824293:株式保証取引所保証および銀行家保証メンバー2023-12-310001824293アメリカ-公認会計基準:投入リスクを測定する自由金利メンバーSRT:重み平均メンバCK 0001824293:株式保証取引所保証および銀行家保証メンバー2023-12-310001824293CK 0001824293:VallonMembers2022-05-310001824293CK 0001824293:保証人保証メンバー2021-02-280001824293CK 0001824293:保証期間2025年12月まで2023-12-310001824293CK 0001824293:保証期間2026年2月まで2023-12-310001824293CK 0001824293:保証期間2027年5月2023-12-310001824293CK 0001824293:保証期間2027年7月まで2023-12-310001824293CK 0001824293:保証期間2028年4月2023-12-310001824293CK 0001824293:保証期間2028年12月まで2023-12-310001824293SRT:最大メンバ数米国-公認会計基準:従業員株式オプションメンバーCK 0001824293:GRIOperationsPlanMember2023-01-012023-12-310001824293CK 0001824293:GRIOperationsPlanMember2023-04-210001824293SRT:最大メンバ数Ck 0001824293:AmendedAndRestated 2018 EquityIncentivePlanMember米国-公認会計基準:従業員株式オプションメンバー2023-01-012023-12-310001824293Ck 0001824293:AmendedAndRestated 2018 EquityIncentivePlanMember2023-12-310001824293米国-公認会計基準:研究·開発費メンバー2023-01-012023-12-310001824293米国-公認会計基準:研究·開発費メンバー2022-01-012022-12-310001824293アメリカ-公認会計基準:一般と行政費用メンバー2023-01-012023-12-310001824293アメリカ-公認会計基準:一般と行政費用メンバー2022-01-012022-12-310001824293米国-公認会計基準:従業員株式オプションメンバー2023-01-012023-12-310001824293米国-公認会計基準:従業員株式オプションメンバー2022-01-012022-12-310001824293米国-公認会計基準:従業員株式オプションメンバー2023-12-310001824293米国-公認会計基準:従業員株式オプションメンバー2022-12-310001824293CK 0001824293:AardvarkTreeuticsIncMember2023-08-222023-08-220001824293CK 0001824293:FormerChiefExecutiveOfficerMember2023-04-212023-04-210001824293米国-GAAP:国内/地域メンバー2023-12-310001824293CK 0001824293:StateTaxAuthorityMember2023-12-310001824293CK 0001824293:LocalTaxAuthorityMember2023-12-310001824293アメリカ-公認会計基準:研究メンバー米国-GAAP:国内/地域メンバー2023-12-310001824293CK 0001824293:StateTaxAuthorityMemberアメリカ-公認会計基準:研究メンバー2023-12-31 アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
________________________________
表:10-K
________________________________
(マーク·オネル)
| | | | | |
| x | 1934年証券取引法第13項又は15(D)項に基づいて提出された年次報告 |
本年度までの十二月三十一日, 2023
あるいは…。
| | | | | |
| ¨ | 1934年証券取引法第13項又は15(D)項に基づいて提出された移行報告 |
日本から日本への過渡期において、日本から日本への移行期、日本と日本との間の過渡期
手数料書類番号001-40034
Gri Bio,Inc.
(登録者の正確な氏名はその定款に記載)
| | | | | | | | | | | | | | |
| デラウェア州 | | 82-4369909 | |
| | | | |
| (明またはその他の司法管轄権
法団および組織として設立する) | | (国際税務局雇用主身分証明書番号) | |
| | | | | |
2223プラヤ通り, #208, ラホア, カルシウム.カルシウム 92037
| (619) 400-1170
|
| (主な執行機関の住所、郵便番号を含む) | (登録者の電話番号、市外局番を含む) |
同法第12(B)項に基づいて登録された証券:
| | | | | | | | | | | | | | |
| クラスごとのタイトル | | 取引コード | | 登録された各取引所の名称 |
| 普通株は、1株当たり0.0001ドルの価値があります | | グレイ | | ナスダック資本市場 |
同法第12(G)条により登録された証券:なし
登録者が証券法規則第405条で定義されている有名な経験豊富な発行者であるかどうかをチェックマークで示す。答えは肯定的で、違います¨ 違います。 x
登録者が当該法第13節又は第15節(D)節に基づいて報告書を提出する必要がない場合は,複選マークで示してください。答えはイエスです¨ 違います。 x
再選択マークは、登録者が、(1)過去12ヶ月以内(または登録者がそのような報告の提出を要求された短い期間内)に、1934年の証券取引法第13節または15(D)節に提出されたすべての報告書を提出したかどうか、および(2)過去90日以内にそのような提出要件に適合しているかどうかを示す。これは大きな問題ですはい、そうです x*¨
再選択マークは、登録者が過去12ヶ月以内に(または登録者がそのような文書の提出を要求されたより短い時間以内に)S−T規則405条(本章232.405節)に従って提出されることを要求した各対話データファイルを電子的に提出したかどうかを示す。彼は言いましたはい、そうです x 違います¨
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小報告会社”、“新興成長型会社”の定義を参照されたい。
| | | | | | | | | | | |
大型加速ファイルサーバ | ¨ | ファイルマネージャを加速する | ¨ |
非加速ファイルサーバ | x | 規模の小さい報告会社 | x |
| | 新興成長型会社 | x |
新興成長型企業であれば、登録者が、取引法第13(A)節に提供された任意の新しいまたは改正された財務会計基準を遵守するために、延長された移行期間を使用しないことを選択するか否かを再選択マークで示す¨
登録者が報告書を提出したかどうかを再選マークで示し、その経営陣が“サバンズ·オクスリ法案”(“米国法典”第15編7262(B)節)第404(B)条に基づいて、その監査報告書を作成または発表する公認会計士事務所の財務報告内部統制の有効性を評価した¨
証券が同法第12条(B)に基づいて登録されている場合は,登録者が届出中の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示す¨
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に第240.10 D−1(B)条に従って受信されたインセンティブベースの補償に従って回復分析を行う必要があるかどうかを再選択マークで示す¨
登録者が空殻会社であるか否かをチェックマークで示す(同法第12 b-2条で定義されている)。*はい、違います¨*x
2023年6月30日まで(登録者が最近完成した第2四半期の最終営業日)現在,非関連会社が保有している登録者普通株の総時価は約$である9.5登録者普通株2023年6月30日のナスダック資本市場における最後の報告販売価格に基づいて計算される。
2024年3月20日までに3,196,488登録者の普通株が発行された
引用で編入された書類
ない。
カタログ
Gri Bio,Inc.
| | | | | | | | |
| | 第…ページ,第 |
| 前向き陳述に関する説明 | |
| リスク要因の概要 | |
| 市場、業界、その他のデータ | |
| 第1部: | 1 |
第1項。 | 商売人 | 1 |
プロジェクト1 A | リスク要因 | 67 |
項目1 B。 | 未解決従業員意見 | 67 |
プロジェクト1 C。 | ネットワーク·セキュリティ | 67 |
第二項です。 | 特性 | 68 |
第三項です。 | 法律手続き | 68 |
第四項です。 | 炭鉱安全情報開示 | 68 |
| | |
| 第II部 | 69 |
第5項。 | 登録者普通株式市場、関連株主事項及び株式証券発行購入 | 69 |
第6項。 | [保留されている] | 69 |
第七項。 | 経営陣の財務状況と経営成果の検討と分析 | 69 |
プロジェクト7 A。 | 市場リスクの定量的·定性的開示について | 78 |
第八項です。 | 財務諸表と補足データ | 78 |
第9項。 | 会計·財務開示面の変化と会計士との相違 | 79 |
プロジェクト9 Aです。 | 制御とプログラム | 79 |
プロジェクト9 B。 | その他の情報 | 80 |
プロジェクト9 Cです。 | 検査妨害に関する外国司法管区の開示 | 80 |
| | |
| 第三部 | 81 |
第10項。 | 役員、行政、会社の管理 | 81 |
第十一項。 | 役員報酬 | 86 |
第十二項。 | ある実益所有者の担保所有権及び経営陣及び株主に関する事項 | 92 |
十三項。 | 特定の関係と関連取引と取締役の独立性 | 94 |
14項です。 | チーフ会計士費用とサービス | 96 |
| | |
| 第4部 | 97 |
第十五項。 | 展示品と財務諸表の付表 | 97 |
第十六項。 | 表格10-Kの概要 | 99 |
| | 署名ページ | 100 |
前向き陳述に関する警告説明
この2023年12月31日までの財政年度Form 10−K年度報告(年次報告)には、経営陣の信念や仮定、経営陣が現在把握している情報に基づく前向きな陳述が含まれている。タイトルは“プロジェクト1.業務”,“プロジェクト1 A”の各節のいくつかの陳述である.リスク要因“、”項目7.経営層の財務状況と経営結果の検討と分析“及び本年度報告の他の部分には前向き陳述が含まれている。場合によっては、“予想”、“信じる”、“考慮”、“継続”、“可能”、“推定”、“予想”、“可能”、“可能”、“計画”、“潜在”、“予測”、“プロジェクト”、“求める”、“すべき”、“目標”、“将”、“将”などの言葉の否定または他の同様の用語によって、前向きな陳述を識別することができる。これらの前向きな陳述は、以下の態様に関する陳述を含むが、これらに限定されない
•私たちの損失の歴史と追加資本の需要は私たちの運営に資金を提供して、私たちは受け入れ可能な条件で追加資本を得ることができない、あるいは持続的に経営する企業として続ける能力がありません。もし私たちが十分な資金を得ることができなければ、私たちは清算が必要で、これは私たちの株主の投資に価値がないかもしれません
•私たちはナスダック資本市場に上場し続けることができます。特に私たちは現在ナスダック1.00ドルの入札価格規則とナスダックが現在適用されている株主権益要求を遵守していないことを考慮して、
•私たちの限られた経営の歴史と発展中の小さな会社が直面している困難は
•これらの支出の時間および金額を含む再構成に関連する予想される現金支出
•計画中の臨床試験を開始する時間
•計画された研究新薬(IND)または新薬申請(NDA)の時間;
•現在と未来の候補製品の研究、開発、商業化が計画されている
•新しい協力を行い、そのような協力協定に規定された義務を履行する能力がある
•候補製品の臨床的効果、潜在的利益、および市場受容度
•商業化、マーケティング、製造能力、戦略
•他の製品または重大なビジネス潜在力を有する候補製品を識別する能力;
•会社の競争相手とその業界に関する発展と予測
•政府の法律法規の影響
•会社が知的財産権の地位を保護する能力
•将来の収入、支出、資本需要、および追加融資需要に関する推定
•2024年1月29日に施行される普通株式(普通株式)の逆分割(2024年1月29日発効)が私たちの普通株式価格または取引に及ぼす影響または潜在的な影響に関するいかなる陳述、またはナスダック資本市場での普通株の上場能力を維持する能力に関する任意の声明;
•信仰宣言と前述の仮定のいずれかの宣言。
これらの展望的陳述は、“項目1 A”に記載されたリスク、不確定要素、および仮説を含むいくつかのリスク、不確実性、および仮説の影響を受ける。リスク要因“と本年度報告書の他の部分。また、私たちは競争が非常に激しく、変化が迅速な環境で運営されており、新たなリスクが時々発生している。私たちの経営陣はすべてのリスクを予測することはできませんし、すべての要素が私たちの業務に与える影響を評価することもできません。あるいは任意の要素や要素の組み合わせは、実際の結果が私たちが行う可能性のある任意の前向きな陳述に含まれる結果と大きく異なる程度をもたらす可能性があります。これらのリスク、不確定性と仮定を考慮して、本年度報告で議論された前向き事件と状況は発生しない可能性があり、実際の結果は展望性陳述中の予想或いは示唆の結果と大きく異なる可能性がある
あなたは未来の事件の予測として前向きな陳述に依存してはいけない。私たちは展望性陳述に反映された予想は合理的であると考えているが、私たちは展望性陳述に反映された未来の結果、活動レベル、業績或いは事件と状況が実現或いは発生することを保証できない。本年度報告書の発行日後に、法的要件がない限り、これらの陳述が新しい情報、実際の結果、または私たちが予想する変化と一致するように、任意の理由で任意の前向き陳述を公開更新する義務はありません
あなたは、私たちの将来の実際の結果、活動レベル、業績、および事件および状況が私たちの予想と大きく異なる可能性があることを理解するために、本年度報告書および本年度報告書で引用され、年間報告書添付ファイルとして米国証券取引委員会(米国証券取引委員会)に提出された文書を読まなければなりません。
本年度報告では,文意が別に指摘されているほか,“会社”,“GRI Bio”,“我々”,“我々”および“我々”はいずれもGRI Bio,Inc.を指す.
リスク要因の概要
以下は,本年度報告表格10−K第I部第1 A項“リスク要因”に記載されている主なリスクの概要である。“リスク要因”の節で述べたリスクは投資家にとって重要であると信じているが,現在知られていないことや現在重要でないと考えている他の要因も我々に悪影響を与える可能性がある.以下の要約は,我々が直面している重大なリスクの詳細な要約と見なすべきではなく,“リスク要因”の節と本年度報告10-K表に記載されている他の情報と合わせて読むべきである
私たちの財務状況と追加資本需要に関連するリスク
•成立以来、重大な純損失が発生しており、予測可能な未来に重大な純損失が続くことが予想される。私たちは決して利益を上げないかもしれないし、決して利益を上げないかもしれない。
•私たちは多くの追加資本が必要になるだろう。必要に応じて、または許容可能な条件下でそのような資金を集めることができない場合、私たちは、私たちの1つまたは複数の研究および薬物開発計画、将来の商業化努力、または他の操作の延期、減少、および/またはキャンセルを余儀なくされる可能性がある。
•私たちの監査役は、継続経営企業としての持続的な経営能力に大きな疑いを示しており、追加融資を受けることができなければ、継続的に経営している企業として経営を続けることができないかもしれません。
研究開発と製薬業に関連するリスク
•私たちのビジネスは、私たちの主要な候補製品GRI-0621の成功と、私たちが臨床開発に進める可能性のある任意の他の候補製品に高度に依存しています。私たちのすべての候補製品は大量の追加開発が必要で、それから規制部門の承認を求めて製品を商業使用に投入することができます。
•臨床開発は長く、複雑で高価な過程に関連し、結果は不確定である。また,我々の候補製品の臨床前研究や早期臨床試験の結果は後期臨床試験の結果を予測できない可能性がある。
私たちの候補製品の商業化に関するリスクは
•承認された候補製品のために十分な精算や保険を得ることができない場合、これらの候補製品をマーケティングする能力を制限し、収入を創出する能力を低下させる可能性があります。
•いずれの候補製品も米国食品医薬品局(FDA)の承認を得ても、米国以外で承認されたり、これらの候補製品が商業化されたりすることは決してなく、すべての市場潜在力を実現する能力を制限する可能性がある。
•私たちは現在マーケティングや販売組織もなく、製品を商業化した経験もない。私たちはこのような能力を発展させるために多くの資源を投入しなければならないだろう。もし私たちがマーケティングと販売能力を確立できない場合、あるいは第三者と合意して私たちの製品をマーケティングして販売することができなければ、私たちは私たちの任意の承認を得る可能性のある候補製品から製品収入を生成することができないかもしれない。
•医療提供者、医師、処方医、購入者、第三者支払者、慈善組織および患者との関係は、刑事制裁、民事処罰、契約損害、名声損害、利益および将来の収入の減少に直面する可能性があるリベート、詐欺および乱用、および他の医療保健法律および法規との関係が適用されるだろう。
•行われている医療立法や規制改革措置は、私たちの業務や運営結果に実質的な悪影響を及ぼす可能性がある。
•米国食品医薬品局、米国証券取引委員会および/または他の政府機関の資金不足は、彼らが重要な指導部や他の人員を採用し、維持する能力を阻害し、新製品やサービスの適時な開発や商業化を阻止するか、あるいはこれらの機関が私たちの業務運営に依存する可能性のある正常な業務機能を履行することを阻止する可能性があり、これは私たちの業務に負の影響を与える可能性がある。
•もし私たちが環境、健康、安全の法律法規を守らなければ、罰金や罰金を科されたり、私たちの業務、財務状況、または運営結果に重大な悪影響を及ぼす可能性のあるコストが発生する可能性があります。
私たちの知的財産権に関するリスクは
•私たちの成功は私たちの知的財産権を保護する能力にある程度かかっている。私たちの固有の権利と技術を保護することは難しくて高価で、私たちはそれらの保護を保障できないかもしれない。
•私たちは未来に許可や他の協力協定を締結するかもしれないし、これらの協定は私たちにいくつかの義務を加えるかもしれない。第三者と締結されたこのような未来の合意に規定された義務を履行できなければ、将来の業務に非常に重要である可能性のある許可権を失う可能性があります。
•第三者の知的財産権侵害に対するクレームはコストが高く、時間がかかる可能性があり、私たちの製品の発見、開発と商業化努力を阻害または延期する可能性がある。
私たちの第三者への依存に関するリスクは
•私たちは第三者に依存して臨床試験を行い、私たちの候補製品を生産し、他のサービスを提供します。もしこれらの第三者が要求通りに契約責任の履行に成功し、期待時間を満たすか、あるいは他の方法で試験または実行を行い、規制要求を遵守できない場合、私たちは期待通りにあるいは臨床開発を成功させることができず、監督管理の承認を得ることができないか、あるいは私たちの候補製品を商業化することができず、私たちの業務は実質的な損害を受ける可能性がある。
•私たちは第三者の製造と供給供給者に依存するため、私たちの研究開発、臨床前と臨床開発材料の供給は限られたり中断したりする可能性があり、あるいは数量あるいは品質は満足できないかもしれない。
•私たちは将来的に第三者との協力を求めて、私たちの候補製品を開発して商業化することができるかもしれません。私たちの将来の協力は私たちの業務に非常に重要になります。もし私たちが協力できない場合、あるいはこれらの協力が成功しなければ、私たちの業務は不利な影響を受ける可能性がある。
管理業務と運営に関するリスク
•もし私たちが重要な管理職を失った場合、あるいは私たちがより多くの高技能者を募集できない場合、私たちは既存製品候補製品を開発したり、新製品候補製品を決定して開発する能力が損なわれ、市場シェアや市場シェアを失ってしまい、私たちの競争力を低下させる可能性があります。
•私たちは私たちの情報システムをネットワーク攻撃から十分に保護できない可能性があり、サイバー攻撃は個人データを含む機密や独自の情報が漏洩し、私たちの名声を損なう可能性があり、私たちを重大な財務と法的リスクに直面させる可能性がある。
融資、私たちの普通株式、資本要求に関するリスク
•私たちは私たちの普通株の株価が非常に不安定になると予想する。
•将来的に私たちの証券を売却して発行することは、私たちの株主の持ち株比率をさらに希釈し、私たちの株価を下落させる可能性がある。
•私たちの普通株の上場は現在ナスダック資本市場や他のナスダック市場の規則に適合していません。私たちの普通株がナスダックから撤退した場合、私たちは巨額の経済的処罰を受ける可能性があり、および/または株式証券を公開または私的に売却することで追加資本を調達する能力と、私たちの投資家が私たちの普通株式を処分したり、正確な一般株式市場値の見積もりを得る能力に悪影響を及ぼす可能性がある。
•2024年1月の逆株式分割は、分割前に対する我々の株価の価値を低下させ、普通株の流動性を低下させる可能性がある。
市場、業界、その他のデータ
この年間報告書は、これらの市場の推定規模および特定の医療条件の発生率に関するデータを含む、私たちの業界、私たちの業務、および私たちの候補薬物市場の推定、予測、およびその他の情報を含む。私たちは、我々の内部推定と研究および独立した業界出版物、政府出版物、市場研究会社の報告、または信頼できると考えられる他の独立したソースから、本年度報告に列挙された業界、市場、および同様のデータを取得した。いくつかの場合、私たちはこのようなデータの出所を明確に言及しなかった。業界出版物および第三者研究、調査および研究は、一般に、それらの情報は、そのような情報の正確性または完全性を保証しないにもかかわらず、信頼できるソースから得られることを示している。私たちの内部データと推定は、貿易·商業組織および私たちの業界の他の連絡先から得られた情報と、私たちの経営陣の業界状況の理解に基づいています。私たちは私たちの内部研究が信頼できると信じているが、それはまだ独立したメッセージ源によって確認されていない。見積り,予測,予測,市場研究や類似手法に基づく情報自体が不確定要因の影響を受け,実際のイベントや状況は本情報で想定しているイベントや状況とは大きく異なる可能性がある.私たちは本年度報告に含まれるすべての開示に責任があり、私たちはこれらの業界の出版物と第三者研究、調査、研究が信頼できると信じている。本年度報告に記載されている任意の第三者情報に関するいかなる誤った記述も知られていないが,その推定,特に予測に関する推定は,多くの仮定に関連しており,リスクや不確定要因の影響を受け,本年度報告“リスク要因”の節や本年度報告の他の部分で議論された要因を含む様々な要因によって変動する可能性がある。
第I部
第1項:商業銀行業務
概要
我々は臨床段階の生物製薬会社であり,炎症,線維化,自己免疫疾患を引き起こす免疫反応失調に関連する重篤な疾患に対する発見,開発,商業化革新療法に専念している。これらの疾患を治療する療法の開発において業界の先頭となり,このような疾患患者の生活を改善することを目標としている。
我々の主要候補製品GRI−0621は1型ナチュラルキラーT細胞の経口阻害剤である。GRI−0621もタザロチンの経口製剤であり、タザロチンは合成RAR−βおよびガンマ選択性アゴニストであり、米国で乾癬およびざ瘡の局所治療のために許可されている。2023年12月31日現在、1700人以上の患者に経口製品として52週間にわたる評価が行われている。われわれは特発性肺線維化(IPF)のような重篤な線維化肺疾患の治療にGRI−0621を開発しており,生命を脅かす進行性肺線維化疾患であり,米国では約140,000人が影響を受け,米国では毎年40,000例もの新症例がある。一部の人々はIPFが世界300万人に影響を与えていると推定している。現在2つの承認された肺線維化治療法があるが,いずれも総生存率の改善には相関しておらず,両方法とも著明な副作用に関連しており,治療コンプライアンスが悪い。これまでのGRI−0621とタザロチン経口投与の早期試験の予備データでは,GRI−0621耐性が良好であり,被験者のiNKT細胞活性を抑制することが観察された。活性化iNKTはIPF,原発性硬化性胆管炎(PSC),非アルコール性脂肪性肝炎(NASH),アルコール性肝疾患(ALD),全身性エリテマトーデス(SLE),多発性硬化症(MS),潰瘍性大腸炎(UC)患者および他の適応患者で上昇することが証明されている。これらの患者では,活性化されたiNKT細胞がより重篤な疾患に関与している。米国食品医薬品局(FDA)はIPF治療のためのGRI−0621のIND申請を承認しており,我々はランダム,二重盲検,マルチセンター2 a期バイオマーカー研究でGRI−0621を評価する予定であり,2023年12月にこの研究への登録を開始した。この試験のTOPLINE結果は2024年下半期に発表されると予想される。
私たちの候補製品の組み合わせはまた、GRI−0803および500種以上の化合物の専用ライブラリを含む。GRI−0803はライブラリーからスクリーニングされた先導分子であり,新規な2型ナチュラルキラーT(NKT)細胞の経口アゴニストである。我々は自己免疫疾患の治療にGRI−0803を開発しており,SLEや狼瘡や多発性硬化症の大部分の臨床前作業において,免疫系は誤って自分の健康組織,特に関節や皮膚を攻撃しているが,身体のほぼすべての器官や組織に影響を与える。この場合は致命的である可能性があり,虚弱な疲労や疼痛発作をきたすことが多く,成人患者の半分近くが仕事ができなくなる。米国では狼瘡は16万から20万人の患者に影響しており,米国では約8万から10万人の患者が腎炎を有しており,SLEの最も深刻な表現の一つであり,通常確定診断後5年以内である。狼瘡は治癒できないが,医療介入やライフスタイルの変化はそれをコントロールするのに役立つ。全身性エリテマトーデスの治療は主に免疫系活動を抑制する免疫抑制薬を含む。過去50年間に2種類の薬剤のみがループスに承認され,新たな治療法が早急に必要とされてきた。INDの承認により,最初のSLEに対する1 aと1 b段階試験でGRI−0803を評価する予定である。我々は2024年上半期に1 aと1 b段階試験に関するINDを提出する予定である。適応を評価し続け,この計画のさらなる発展に最適な適応を選択するが,最初の重点は狼瘡であった。
私たちのパイプは
私たちはすべての候補製品の世界的開発権と商業化権利を保持している。以下のグラフは我々の計画に関する重要な情報をまとめたものである.われわれはまたいくつかの臨床前と臨床資産を進展させており,これらの資産は疾患に関連する臨床前モデルにおいて希望を示している。
__________________
図1.GRIのパイプライン−GRI−0621およびGRI−0803
我々の最初のポイントはIPF治療のためのGRI−0621の開発であった。GRI−0621はタザロチンの経口製剤であり、タザロチンは合成レチノイン酸受容体(RAR)−βおよびガンマ選択性アゴニストであり、米国で乾癬およびざ瘡の局所治療のために許可されている。GRI−0621は,IPF患者や他の間質性肺疾患患者に集積することが証明されているiNKT細胞の活性を阻害する。活性化されたiNKT細胞はIPF,肝臓,その他の線維化条件下で過剰発現し,進行疾患と有意に関与していることが他の人と証明されている。GRI−0621は,他の肺線維化疾患,NASH,ALD,腎線維化,急性慢性肝不全,薬物性肝障害(DILI)や他の急性適応を含む多様な線維化や関連疾患を治療する潜在力があると信じられている。大量の臨床前研究において、iNKT細胞の活性を抑制することは炎症、マクロファージ群の活性化、トランスフォーミング増殖因子(TGF)-βと繊維化を著しく減少させた。INKT細胞に特化した治療薬はない。
われわれは14名の肝臓障害慢性肝疾患患者において2 a期試験を行い,GRI−0621を評価した。この研究は当初60名の患者を評価する予定であったが,募集困難とNASH臨床研究設計に関するFDAの更新ガイドラインのため,14名の患者を募集した後に研究を中止する行政決定を行った。この限られた数の患者ではGRI−0621耐性が良好であることが認められたが,統計学的に有意な終点には至らなかった。2023年12月、2 a段階試験の登録を開始し、この試験のTOPLINE結果が2024年下半期に発表されることが予想されます。
著者らはまたGRI-0803、新型の2型NKT細胞活性化剤を開発しており、著者らは多種の自己免疫モデルにおける治療メリットを観察した。GRI−0803はSLEや関連腎炎,多発性硬化症,自己免疫性肝炎,その他の自己免疫疾患の治療の可能性があると信じている。
また,Jado Technologies GmbHから得られた500種類以上の新規化合物のコーパスを持つ.このライブラリーはGRI-0124(ミチフォキシン)の構造と機能をシミュレートすることを目的としており、GRI-0124は有効な2型NKT細胞活性化剤である。GRI−0803は、ライブラリから選択された主要候補製品である。
著者らは数十年間にナチュラルキラーT細胞(NKT)活性及び健康と疾病における作用を研究した経験に基づいている。わが社は三人の免疫学者によって創立され、その中に1人の国際公認のNKT細胞研究の先頭者を含み、彼はNKT亜群の初歩的な表現に貢献し、1型と2型NKT細胞がそれぞれのリガンドのT細胞受容体と結合することを表現し、そして炎症、繊維化と自己免疫性疾患における1型と2型NKT細胞の作用を同定と表現した。
われわれの創始者と経営陣の経験は,NKT細胞の活動と慢性炎症,線維化,自己免疫疾患における役割に独自の知見を提供していると信じている。私たちはW.Marc Hertz博士が指導し、私たちの社長とCEOはバイオテクノロジー幹部で、Pharmexa社とMultimmer BioTreateutics社の最高経営責任者を務め、Pharmexa A/Sの上級管理職の一部でもあります。アルバート·アグロー博士は私たちの医療責任者で、バイオテクノロジーや製薬業界で豊富な経験を持ち、グローバル臨床開発会社バーリンガー·インゲルハイム国際有限会社やバイエル社で高級職を務め、Cynapsus治療会社で上級職(医療責任者)を務めています。VTV治療有限会社(ベテラン副総裁発展)と崇高治療有限会社(最高経営責任者)。アグロー博士はマクマスター大学の病理学·分子医学系で教職を務めている。私たちの首席科学官Vipin Kumar Chaturvedi博士はNKT細胞研究の国際公認のリーダーである。GRIの技術は,NKT細胞亜群と炎症,線維化と自己免疫疾患における異なる作用を認識することに基づいている。クマール博士は医学教授であり、カリフォルニア大学サンディエゴ校免疫調節実験室の責任者でもある。私たちは取締役会の支持を得た
同社はこれまで,TEP Biotech,LLC(TEP),AcquiPharma Holdings Ltd,Altium Healthcare Inc.を含む家族理財室と有力な生命科学投資家によって援助されてきた。
私たちの戦略
我々の目標は,重大な需要を満たしていない疾患に対する治療薬の開発と商業化の先駆者となることである。我々の最初の重点は,NKT細胞の活性と脱調免疫反応の駆動における役割に対する候補製品の開発であった。私たちの戦略は以下の重要な構成要素に重点を置いている
•特発性肺間質線維化におけるGRI−0621の臨床応用の有効な推進それは.約36名のIPF患者の中でランダム、二重盲検、プラセボ対照の2 a期試験を行う予定であり、2024年下半期にTOPLINEデータがあると予想される。この孤児疾患は治療に十分な治療が得られず,GRI−0621はこれらの患者の最初の真の疾患修正療法になる可能性があると信じている。この試験の結果が肯定的であると仮定すると,欧州連合(EU)でGRI−0621の申請が条件付きで承認され,米国での登録試験とみなされる可能性がある2 b段階試験を開始する予定である
•SLEに対する最初の1 a/1 b段階研究によるGRI−0803研究の推進それは.INDの承認により,最初のSLEに対する1 aと1 b段階試験でGRI−0803を評価する予定である。私たちは2024年上半期にこの裁判についてINDに提出される予定だ。
•疾患におけるiNKTと2型NKT細胞の理解を用いて,GRI−0621,GRI−0803,後続適応における他の候補品の評価を継続したそれは.失調免疫反応を駆動する生物過程に対する治療法の開発に取り組んでいる会社として,我々のリーダーシップを拡大する予定である。私たちはまた、私たちの製品の組み合わせや支援技術を拡大するために、業務発展の機会を選択的に求めるつもりです。
•炎症性線維症自己免疫疾患を含む患者を中心とした会社を設立し続けていますそれは.患者を中心とする会社を構築し、患者の需要を満たす過程において、著者らは臨床医師、患者権益団体、卓越医学センターと医学肝心なオピニオンリーダーと協力し、これらの疾病の症状と結果をもっとよく理解し、できるだけ早く患者にもっと良い治療方法を開発と提供し、そしてこれらの疾病に対する認識を高める。
•私たちの候補製品のビジネス価値を最大化するそれは.私たちはすべての候補製品の世界的な開発権と商業権を維持した。私たちは限られたターゲットを絞ったビジネスチームを通じて、私たちのポートフォリオの中にはまれな状況で米国とEUの監督管理によって承認された任意の製品を商業化するつもりだ。また、戦略的協力の柔軟性を評価し、パートナーを探して私たちの製品を他の地域で商業化し、商業インフラの建設に大量の投資が必要な場合に私たちの製品を商業化するつもりです。
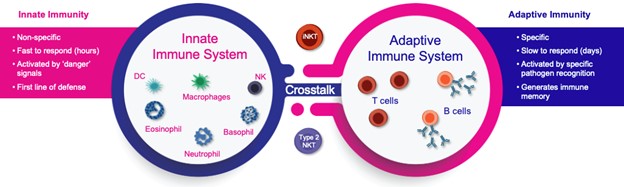
NKT細胞と免疫系
著者らの方法は、NKT細胞が天然免疫系と後天性免疫系の間の機能的絆であり、NKT細胞の活動を調節することによって、失調した免疫反応をリセットすることができ、それによって一連の急性と慢性病を潜在的に治療することができるという発見に基づいている。
__________________
図2.NKT細胞は適応免疫系と先天性免疫系を連結した先天性T細胞である。
NKT細胞は適応性免疫系と先天性免疫系を連結する先天性T細胞である(図2参照)。それらはNKとT細胞の共通特性を有し、重要なサイトカイン/ケモカインの発現を制御し、免疫反応の重要な調節因子である
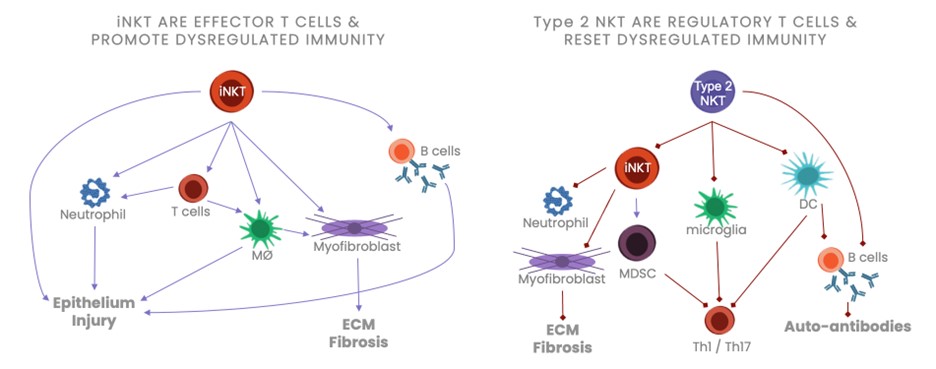
INKT細胞は効果性T細胞であり、肺、肝と自己免疫徴候に発病作用を発揮することができる;2型NKT細胞は制御性T細胞であり、それはiNKT細胞及び他のタイプの細胞の活性を抑制し、そして抗炎症反応を支持する。2型NKT細胞は、いくつかの線維症および自己免疫疾患の炎症反応による障害を最小限に低減するために、破壊的な炎症促進環境および細胞毒性環境から抗炎症および保護環境に反応を転移させることができる(図3参照)
__________________
図3.iNKTと2型NKT細胞は炎症制御に逆の役割を果たしている(左図中の矢印は活性化を示し,右図中の矢印は抑制を示している)。
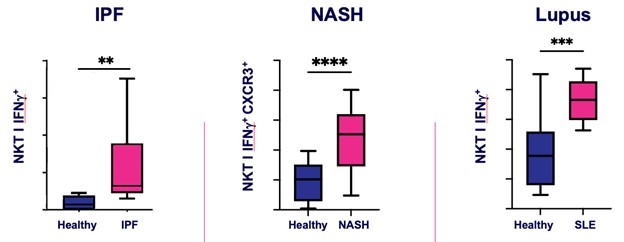
INKT細胞の反復活性化は慢性肺疾患を招き、そして患者の体内で上昇することができる。INKT細胞活性を調節することはすでにIPF動物モデルにおいて治療作用を有することが観察され、活性化されたiNKT細胞はIPF、NASHとSLE患者の肺、その他の慢性炎症性、繊維化と自己免疫性疾患群に集まる。
__________________
図4.IPF,NASH,SLE患者のPBMC試料では健常人と比較して活性化されたiNKT細胞が増加している。
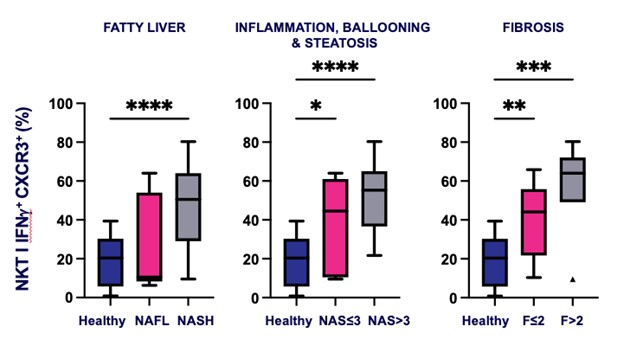
現在のIPF療法は肺機能の低下を緩和しているが,全体の生存率を向上させることはできない。INKT細胞の活性を調節し、マクロファージの分極、形質転換増殖因子-βの産生と筋線維芽細胞の活性化を促進する能力は、それらが繊維化の進展を緩和し、IPFの生存結果を改善する可能性があることを表明した。活性化されたiNKT細胞はIPF患者において著しく上昇し、これらの患者の重要な薬効学的バイオマーカーになる可能性がある。健常個体から軽度の非アルコール性脂肪肝や進行NASHへの進展に伴いNASH患者で活性化したiNKT細胞の増加が認められ,iNKTはIPF患者の類似バイオマーカーである可能性が信じられた(図5参照)。
__________________
図5.疾患の健康,軽度から晩期への進展に伴い,NASH患者ではCXCR 3+インターフェロン−γ+活性化したiNKT細胞が増加している。
肺、腎臓と肝繊維化モデルの中で-IPF、SLE、NASH、ALD、DILIと自己免疫性肝炎-iNKT細胞を含み、組織損傷を媒介する中で重要な発病作用を発揮し、それは迅速な蓄積、活性化とサイトカインとケモカインの分泌を通じて炎症低下を誘導し、その中にIL-1β炎症体と好中球募集を活性化し、繊維化を促進する筋線維芽細胞/肝星状細胞の分化と活性化、コラーゲン沈着と繊維化を含む。
GRIはまた、シス-テトラ炭素アルケニル硫黄脂質(硫黄脂質)、あるリン脂質とGRI-0124を含むいくつかの2型NKT細胞活性の調節剤を同定した。GRI−0803およびGRIの500以上の化合物ライブラリーは、GRI−0124に構造的に関連している。GRI−0803とGRI−0124のインビボ応用は2型NKT細胞を活性化し,活性化したiNKT細胞の増殖を抑制する。つまり,これらのデータはiNKT阻害剤(例えばGRI−0621)と2型NKT調節剤(例えばGRI−0803)およびGRI−0124とGRI−0729がともに炎症免疫反応のバランスに作用するモデルを支持していると考えられる。
肺疾患
IPFはまれな生命を脅かす疾患であり,進行性線維化と異常瘢痕が特徴であり,時間の経過とともに酸素の血流への進入を阻止することで肺の構造や機能を破壊し,肺の悪化や破壊を招く。IPFで最もよく見られる症状は呼吸急と乾性咳である
私たちの候補製品の組み合わせ
特発性肺線維化に対するGRI−0621
GRI−0621はFDAが承認した皮膚科外用製品であるタザロチン(エチル6−エチル)の経口ゲルカプセル製剤である[2−(4,4−ジメチルチオ−6−イル)アセチレン]ニコチン)、合成RAR−β及びガンマ選択性アゴニスト及びiNKT細胞の有効な阻害剤。タザロチンは乾癬およびざ瘡のための局所製剤が承認され、1700人以上の患者において経口製品として52週間にわたって被験者に服用されている。同社はIPF治療のためのGRI−0621を開発している。
IPF背景と市場チャンス
IPFは進行性肺繊維化の最もよく見られると最も深刻な形式であり、アメリカでは約140,000名の患者が影響を受けている。米国では毎年確定診断された新症例は4万例に達し,主に65歳から70歳までの人々に影響を与え,人口高齢化に伴い米国の流行率は上昇すると予想される。確定診断後の生存期間は2年から3年であり,IPFと診断された患者の平均期待寿命は3年から5年であった
特発性肺線維化の治療現状とその限界
軽度または中等度の症状を有するIPF患者の一部は、Boehringer Inglheim製薬会社が販売しているOfevというinetedanibまたはGenentech USAによって販売されているEsbrietというピルフェニドンで治療することができる。これら2つの薬剤は、IPFに関連する肺機能低下の進行および肺機能の悪化を緩和することが証明されているが、いずれも総生存率の改善には関係なく、有意な副作用に関連している。9 tedanibを服用した患者では,60%を超える患者に下痢症状があり,約14%の患者で肝酵素レベルが上昇すると推定されている。ピルフェニドンで治療した患者では,約30%に皮疹が出現し,約9%に感光性反応が出現し,両者とも投与量の減少や中止を招く可能性がある。この2種類の薬物はより深刻な疾病を有する患者に対して一定の治療効果があるが、これらの虚弱患者は不良事件で使用を中止する比率が高く、それらの使用を制限している。2017年に第三者によって発表された290人の医師に対する調査では、IPF患者の半数以上がこの2つの薬剤のいずれも使用しておらず、医師が臨床的利益に十分な自信を持っていないこと、安全性への懸念など、様々な原因があることが分かった。メオ診療所研究者が2019年に提出した処方記録の遡及性コホート分析では,IPF患者のピルフェニドンと9 tedanibの割合は治療法ごとの約10%であり,早期の観察結果,すなわち多くのIPF患者が積極的に治療されていないことが支持された。それにもかかわらず、2019年のピルフェニドンとナチダニの世界総売上高はそれぞれ12億ドルと16億ドルを超えた
私たちの解決策-GRI-0621
IPF患者を治療する経口ゲルカプセル製剤としてGRI−0621を開発している。GRI−0621は、調節失調に対する免疫反応下流の疾患症状ではなく、iNKT細胞の活動を抑制することによって、疾患の免疫反応失調をリセットすることを意図しているため、現在のIPF療法とは異なる。GRI-0621は経口製剤として約1700名の乾癬、ざ瘡、肝疾患患者において評価されており、これらの患者群と研究において、この分子はビタミンA増加症に関連する典型的な有害事象(頭痛、背部痛、足痛、唇炎、高血糖、関節痛、筋肉痛、関節障害、鼻乾、皮膚乾燥、皮疹と皮膚炎)に対して良好な耐性を有している
臨床前研究では,iNKT細胞を欠く動物はIPF,NASH,ALD,自己免疫性肝疾患,DILIモデルで線維化からの影響を認めた。同様に,iNKT細胞の活性を抑制することは,動物を線維化から保護および/または治療することができる。繊維化は複雑な動態過程であり、異なる組織中の複数のシグナル分子、分化経路と多種の細胞タイプに関連する。そのため,創部修復機序が慢性炎症/損傷により誤った場合,組織の瘢痕化,硬直を招き,最終的に機能障害が出現する。それは非常に複雑であるが、科学文献により、共通の生物学的機序が肺、肝臓と腎臓などの異なる組織の繊維化を推進することが示唆された
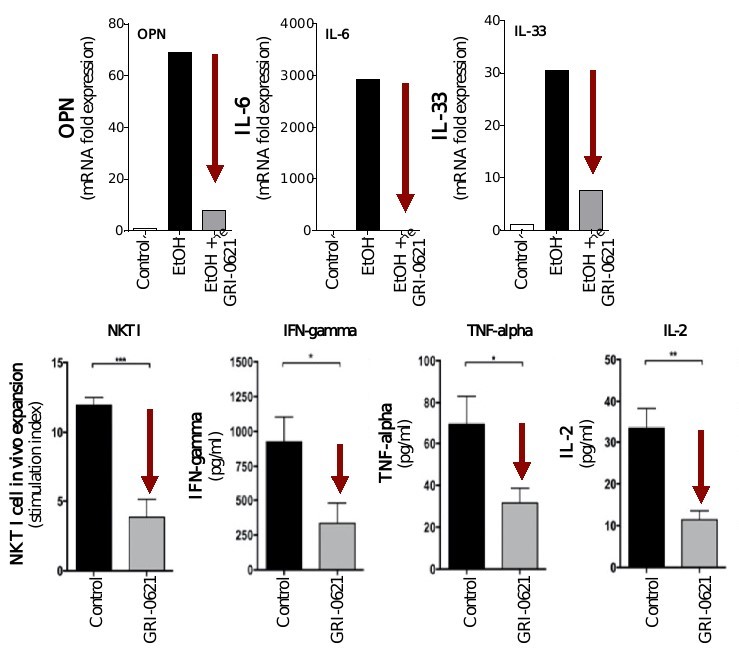
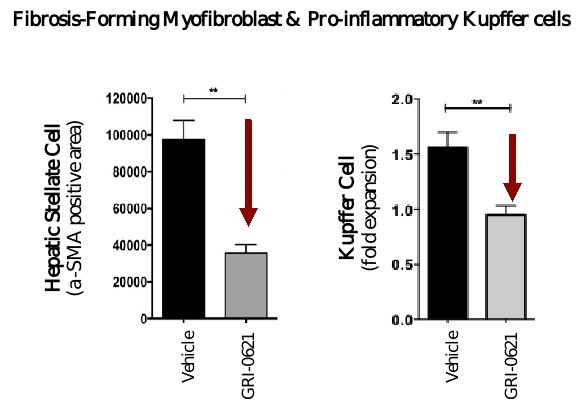
われわれの臨床前研究では,肝線維化動物モデルにGRI−0621を用いることでiNKT細胞(図6参照)から分泌される炎症促進サイトカインや,炎症を促進するKupffer細胞と線維化を促進する筋線維芽細胞/肝星状細胞の成熟と活性化が観察された(図7と10参照)。
__________________
図6.線維化動物モデルで観察されたGRI−0621は,iNKT細胞の体内拡張および活性化を抑制し,炎症性サイトカインを抑制する。
__________________
図7.観察されたGRI−0621は、クッパー細胞および筋線維芽細胞/肝星細胞の活性化および成熟を阻害する。
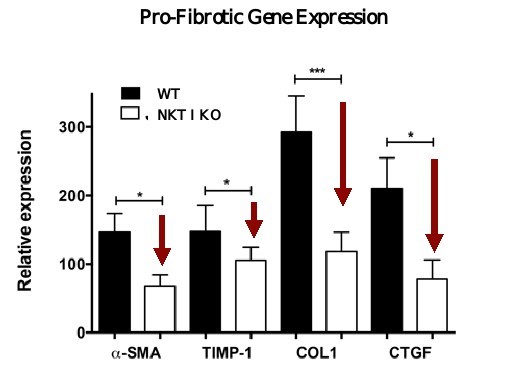
従来,iNKT細胞を欠くiNKTノックアウト(KO)動物は線維化モデルにおいて野生型動物(WT)に関連する線維化促進遺伝子を上方制御できなかったことが観察されてきた(図8参照)。
__________________
図8.iNKT欠乏線維化動物モデルで観察されたCTGFを含むキー線維化遺伝子の抑制。
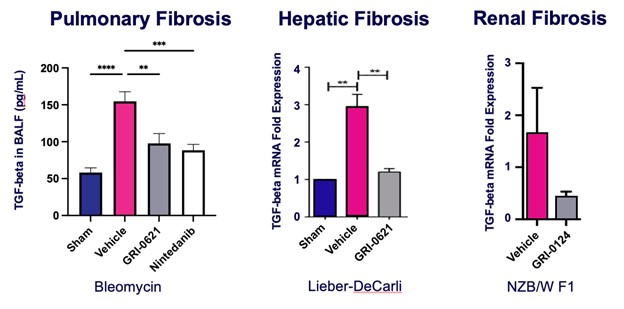
転化成長因子-βは繊維化形成を推進する最も重要なシグナル分子の一つである。われわれの肺、肝臓、腎臓繊維化モデルにおいて、iNKT阻害剤或いは2型NKT細胞活性化剤を用いてiNKT細胞機能を不活化させ、この繊維化の重要なメディエーターの顕著な抑制を招いた(図9参照)
__________________
図9.iNKT細胞を抑制することは、肺と肝繊維化モデルにおける形質転換増殖因子-βを著しく低下させる。
著者らの臨床前研究において、炎症促進サイトカイン、クープファー細胞、活性化筋線維芽細胞、繊維化促進遺伝子発現と繊維化の重要な可溶性メディエーター転化増殖因子-βの減少は肝と肺繊維化モデル中のコラーゲン沈着と繊維化の減少を招く(図10、11と12参照)。
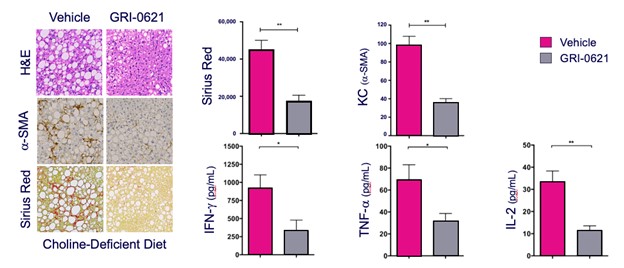
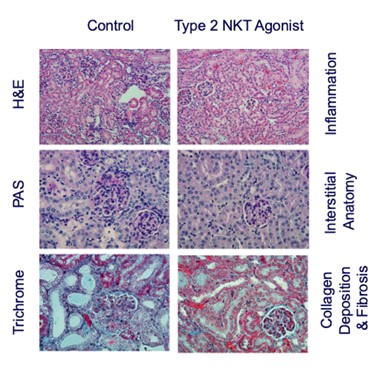
__________________
図10.コリン欠乏Lで定義されるNASHモデルでは,肝臓炎症および脂肪変性(H&E),筋線維芽細胞活性化(抗SMA)および線維化(シリウスレッド)が抑制される(左側組織パネルおよび上部棒グラフ),ならびにインターフェロン−γ,腫瘍壊死因子−αおよびIL−2(底棒グラフ)。
__________________
図11.ブレオマイシン肺線維化モデルにおいて観察される炎症、炎症性サイトカイン、および形質転換増殖因子−βを予防することができるiNKT阻害剤。
__________________
図12.ブレオマイシン肺線維化モデルでは,GRI−0621が肺炎症(H&E)および線維化(Mason‘s trichrome)を抑制することが観察された。
肝疾患対象におけるGRI−0621試験段階2 aの使用
著者らは2 a期試験において、肝機能障害の慢性肝疾患患者におけるGRI-0621の応用を評価した。この研究は当初60名の患者を評価する予定であったが,募集困難とNASH臨床研究設計に関するFDAの更新ガイドラインのため,14名の患者を募集した後に研究を中止する行政決定を行った。この限られた数の患者ではGRI−0621耐性が良好であり,肝機能テスト,血清CK−18,iNKT細胞活性の改善を示したが,統計学的有意な終点には至らなかった。副作用は一般に軽度であり,RARb g興奮症と一致した(下表参照)。
| | | | | | | | | | | | | | | | | | | | |
| 全因 | | プラセボ(n=4) | | GRI−0621 4.5 mg(n=4) | | GRI−0621 6.0 mg(n=5) |
| シリアスTEAE | | 0 | | 0 | | 0 |
| TEAEレベル1 | | 0 | | 0 | | 0 |
| 2級特級英語試験 | | 0 | | 0 | | 0 |
| TEAEレベル3/4/5 | | 0 | | 0 | | 0 |
| 治療と関係がある | | | | | | |
| Chelitis | | 0 | | 0 | | 0 |
| Naseau | | 0 | | 0 | | 0 |
| 肌が乾燥している | | 0 | | 0 | | 0 |
| プリティス | | 0 | | 0 | | 0 |
| 頭が痛い | | 0 | | 0 | | 0 |
| マラジア | | 0 | | 0 | | 0 |
| 高血圧.高血圧 | | 0 | | 0 | | 1* |
| 胃腸炎 | | 0 | | 0 | | 0 |
| 扁桃腺炎 | | 0 | | 0 | | 1* |
| クレアチンホスホキナーゼ | | 0 | | 0 | | 0 |
| 乳酸脱水素酵素 | | 0 | | 0 | | 0 |
| カリウム | | 0 | | 0 | | 0 |
__________________
*二次治療緊急有害事象(TEAE)
GRI−0621製造
我々は第三者契約メーカーに依存して前臨床研究および臨床試験のためのGRI−0621を生産し、前臨床研究または臨床試験製品供給の製造施設は何も生産していない。著者らは限られた数量の薬品供給者に依存し、臨床研究のためのGRI-0621調合薬品を生産するメーカーを招聘し、これは早いから中期までの臨床開発の標準業界実践である。もしこれらのサプライヤーが私たちが要求した数量で私たちに供給することができない場合、あるいは私たちに供給することができない、あるいは他の方法で彼らの私たちへの供給義務を滞納する場合、私たちは許容可能な条件で適時または根本的に他のサプライヤーから代替供給を得ることができないかもしれません。また、何らかの理由でメーカーの交換を要求された場合、新しいメーカーの施設やプログラムが品質基準およびすべての適用される法規やガイドラインに適合しているかどうかを確認することを要求されます。我々はまた、例えば比較可能な研究を製造することによって、任意の新しい製造業者または製造プロセスが、以前にFDAまたは他の規制機関に提出された仕様に基づいて、私たちの候補製品を生産することを検証する必要がある。臨床用品の比較可能性の証明には成功しない可能性があり,追加の臨床試験が必要かもしれない。新メーカーの検証に関する遅延は、タイムリーまたは予算内で候補製品を開発する能力に悪影響を及ぼす可能性があります。
特発性肺線維化患者に対するGRI−0621 2 a期試験
2023年12月,2 a段階の試験の登録を開始した。この試験は約36名のIPF患者の中で12週間の多中心、多国、無作為、プラセボ対照試験を行う。背景療法でIPFと診断された被験者では,4.5ミリグラム量で12週間の治療でプラセボと比較した。被験者はスクリーニング訪問を完了し,病歴,現状,実験室評価,合併症,それに伴う薬物を評価する。これらの知見によれば、対象は、2つの治療群のうちの1つ:4.5ミリグラムGRI−0621またはプラセボのうちの1つにランダムに配置され、2:1でランダムに配置される。週12週間の検査では、GRI-0621の安全性、薬物動態および治療効果/作用機序が評価され、6週および12週の血液中のiNKT細胞および12週における気管支肺胞洗浄液中のiNKT細胞の活性化によって評価される。副次的な終点として、様々なバイオマーカーも支持するように評価される
GRI−0621の作用機序。被験者は服薬完了後少なくとも2週間追跡される。この試験は,必要な数の被験者を募集するのに約6カ月を要し,1回目の被験者の初回訪問後約10カ月以内に完了したはずである。この試験の裏線データは2024年下半期に利用可能でなければならない。この試験の最終結果は、用量、安全サンプル量、臨床関連終点、および後続登録計画を設計する際のFDAとのコミュニケーションの持続時間を決定するために使用される
GRI−0803全身性エリテマトーデス関連性ループス腎炎の治療
全身性エリテマトーデス背景
SLEは最もよく見られる狼瘡タイプであり,米国では16万から20万人の患者に影響し,米国では毎年2.4万人もの人がこの疾患と診断されている。全身性エリテマトーデスは主に女性に影響を与え,通常15歳から44歳の間に開始される。全身性エリテマトーデスは1種の自己免疫性疾患であり、免疫システムは自分の組織を攻撃し、影響を受ける器官の広範な炎症と組織損傷を招く。それは関節、皮膚、脳、肺、腎臓と血管に影響を与える。狼瘡は治癒できないが,医療介入やライフスタイルの変化はそれをコントロールするのに役立つ。すべての人種の人がこの病気を患っている可能性があるが,アフリカ系アメリカ人女性の新症例数は非スペイン系白人女性の3倍である。アフリカ系アメリカ人女性はよくアフリカ系白人女性より若くてこの病気にかかり、更に深刻で生命に危害を及ぼす合併症が出現する。それはスペイン系、アジア系、アメリカ先住民の子孫の女性にもよく見られる。治療方案を堅持することはしばしば問題であり、特に出産適齢期の若い女性の中で。SLEの治療には重篤な副作用が生じる可能性のある強い免疫抑制薬が必要である可能性があるため,女性患者は妊娠前や妊娠中に薬物の服用を中止し,未出生児を保護しなければならない。
狼瘡性腎炎と全身性エリテマトーデスの治療現状とその限界
SLEの治療と管理は疾病の重症度と疾病の表現に依存する。ヒドロキシクロロヒドリンはSLEの長期治療において核心的な役割を果たしており、SLE治療の基礎である。コルチコステロイド、非ステロイド抗炎症薬、および免疫抑制剤(例えば、アザチオプリン、シクロホスファミド、シクロスポリン、メトトレキサートおよびミコフェノール酸エステル)もSLEの治療および管理に使用されている。これらの治療法は適度に有効であるだけであり,長期使用では安全性や/あるいは免疫抑制が問題となる。B細胞枯渇抗体リツキシマブはSLE治療に承認されていないが,ある患者亜群に有益であるようである。
FDAは過去50年間に全身性エリテマトーデスに対する2つの標的療法,belimumab,anifrom Lumabを承認した。2011年、FDAはBelimumab(Benlysta)を承認し、Bリンパ球刺激因子に対する抗体であり、標準治療と組み合わせて、軽~中度全身性エリテマトーデスの治療に使用し、B細胞標的による自己免疫疾患の治療効果に追加の臨床検証を提供した。しかしながら、Benlystaの適度な治療利益および遅延した疾患介入は、過剰に活性化したB細胞を抑制するための追加の治療戦略が必要であることを示している。2021年、10年間この疾患を治療してきた最初の新薬であり、標準治療を受けている中重度疾患を有する成人のために許可された1型インターフェロン受容体抗体anifrom Lumabに随一である。
狼瘡性腎炎は全身性エリテマトーデスのよく見られる表現であり、不可逆的な腎臓損害を招くことができる。この疾患は複雑、異質性であり、多種の細胞タイプ及び免疫と非免疫機序に関連する。疾患進展の特徴は糸球体損傷、炎症、細胞浸潤と繊維化である。免疫複合体の沈着は炎症小体やI型インターフェロンを介した経路を招き,内皮機能障害を招き,原因抗体による補体を介した損傷を伴う。
私たちの解決策-GRI-0803
科学研究により、iNKTは急性腎損傷、虚血再灌流損傷と狼瘡性腎炎などの腎臓疾患において重要な発病作用を果たしている。したがって,ループス患者(上図4参照)や自発性ループスモデルではiNKT細胞が活性化される。注目すべきは,2型NKTの活性化による樹状細胞を介したiNKT細胞の抑制である。われわれの臨床前研究では,1種の2型NKT活性化分子GRI−0803がマウスもヒトのiNKT細胞も抑制作用が認められた。GRI-0803の経口投与、1種の2型NKT活性化分子が観察され、狼瘡性腎炎を抑制でき、全体の生存率を著しく向上させた
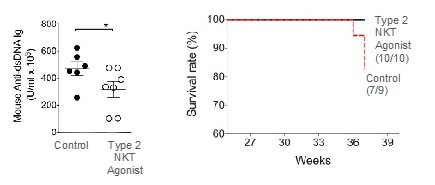
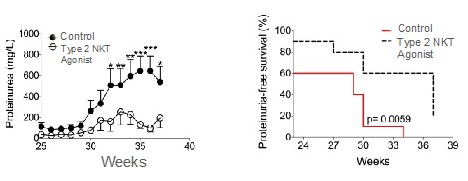
ループス腎炎自発モデルでGRI−0803を毎週経口投与したところ,IL−17とIL−6を含む炎症性サイトカインの有意な抑制が認められた(図12参照)。他の線維化を引き起こす分子は,形質転換成長因子−βを含めて抑制され,コラーゲン沈着や腎臓線維化を引き起こすことも観察されている(図13参照)。これは,細胞(B細胞やT細胞を含む)の腎臓や糸球体病理への侵襲を伴うことが観察された。また,GRI−0803投与後,病原性抗二本鎖DNA抗体と尿卵白尿の有意な抑制が認められた(図13と14参照)。また,GRI−0803は血漿細胞様樹状細胞や腎障害に関与するI型インターフェロンシグナル経路遺伝子の活性化を遮断することも観察された。腎臓疾患の抑制は無蛋白尿動物の全体生存率の改善に反映されている。
Lipocalin 2(Lcn 2)は多種の免疫細胞から分泌される糖タンパク質であり、自己免疫性疾患において炎症促進免疫反応を促進し、狼瘡性腎炎の重症度の指標とすることができる。興味深いことに,他の炎症遺伝子では,対照群と比較してGRI−0803を経口投与した動物腎臓におけるLcn 2の発現が有意に抑制された(図12参照)。
__________________
図12.GRI−0803の経口投与後に観察された自発性ループスモデルにおけるいくつかの重要な炎症促進、線維化、および腎臓疾患促進遺伝子の阻害。
__________________
図13.自発性ループスモデルでは,GRI−0803投与は炎症細胞浸潤(H&E),糸球体病理(PAS),腎線維化(三色)を抑制することが観察された
__________________
図14.GRI−0803を用いた治療後のループスモデルでは,血清中の抗dsDNA抗体の抑制と総生存率の増加が観察された。
__________________
図15.GRI−0803経口投与動物において、尿蛋白尿および自発性狼瘡性腎炎の有意な阻害が観察された
GRI−0803製造
我々は第三者契約メーカーに依存して臨床前研究のためのGRI−0803を生産し,臨床前研究製品供給の製造施設は何も生産していない。著者らは単一或いは限られた数量の薬品供給者に依存し、単一メーカーを招いて臨床研究のためのGRI-0803調合薬物製品を生産し、これは早いから中期までの臨床開発の標準業界実践である。もしこれらのサプライヤーが私たちが要求した数量で私たちに供給することができない場合、あるいは私たちに供給することができない、あるいは他の方法で彼らの私たちへの供給義務を滞納する場合、私たちは許容可能な条件で適時または根本的に他のサプライヤーから代替供給を得ることができないかもしれません。また、何らかの理由でメーカーの交換を要求された場合、新しいメーカーの施設やプログラムが品質基準およびすべての適用される法規やガイドラインに適合しているかどうかを確認することを要求されます。我々はまた、例えば比較可能な研究を製造することによって、任意の新しい製造業者または製造プロセスが、以前にFDAまたは他の規制機関に提出された仕様に基づいて、私たちの候補製品を生産することを検証する必要がある。臨床用品の比較可能性の証明には成功しない可能性があり,追加の臨床試験が必要かもしれない。新メーカーの検証に関する遅延は、タイムリーまたは予算内で候補製品を開発する能力に悪影響を及ぼす可能性があります。
GRI-0803フェーズ1実験
我々はGRI−0803毒理学計画完了後に第1段階試験を開始する予定である。結果が肯定的であると仮定して、私たちは2024年上半期にIND申請を提出する予定だ。単回漸増用量(SAD)試験は健康ボランティアで行われる。6剤までが12人の被験者で評価され,そのうち10人がGRI−0803用量の治療を受け,2人がプラセボ治療を受けた。各キューの安全性は、独立安全審査委員会(ISRB)によってGRI臨床管理と共に評価される。第1のキューが完了した後、後続のキューは、前のキューに投与された後2週間以内に開始される。薬物動態学と安全性はSAD試験の主要な終点である。この試験の完了には約3カ月を要するはずであり,最初のキュー投与から始まる
ISRBの提案によると、多重漸増用量(MAD)試験はSAD試験中の用量3が完了した後に開始される。MAD試験は、SADの結果に依存する4つの用量のGRI−0803を検査する。各キューに10名の被験者:8名がGRI−0803,2名がプラセボを服用していた。コホート投与量は4週間持続し、そして薬を飲んだ後に2週間の安全追跡を行い、前の2群は健康被験者であり、両群の最高投与量はSLE患者で完成する。安全性と多用量薬物動態学はこの薬の主要な終点になるだろう
狂った裁判。探索的結果は,第3および第4のキューで検査され,いくつかのバイオマーカー(例えば,サイトカイン)およびNKT細胞活性化マーカーが含まれる。MAD試験は完成まで約5カ月かかり,背線試験結果は2024年第4四半期末に発表される
競争構造
生物技術と生物製薬業界の特徴は技術が迅速に進歩し、競争が激しく、独自製品を高度に重視していることである。著者らは著者らの技術、管理チームの専門知識、臨床能力、研究開発経験と科学知識が私たちに競争優勢を提供したと信じているが、著者らは生物技術と生物製薬会社、学術機関、政府機関及び公共と個人研究機関を含む多くの異なる源からの日々激しい競争に直面している。我々が開発と商業化に成功した任意の候補製品は,既存の療法や将来出現する可能性のある新しい療法と競争するであろう
現在いくつかの大型バイオテクノロジーやバイオ製薬会社が疾患治療製品を開発しており,GRIの目標もIPF,SLE,MS,UC,PSC,NASHを含めて将来的に照準されている可能性がある。我々の知る限り,臨床開発においてNKT細胞を上記のいずれかの疾患を治療する方法としている会社はないが,これらの疾患の治療を目指している会社には,多くの財源を持つ大手会社が含まれていることが知られている
IPF-アスリーカン、ブリンガー-インゲルハイム国際有限公司、百時美施貴宝、およびノワ製薬。他の大量の資源を持つ小さい会社はAvalyn Pharma Inc.,Bellerophon Treateutics,Inc.,Endeavor Biomedicines,Inc.,Horizon Treateutics Public Limited,Pplant Treateutics,蘇州ゼルゲン生物製薬有限会社,共同治療会社とVicore Pharma Holding ABである。
全身性エリテマトーデス-アステラス製薬会社、アスリコン、オリニア製薬会社、生物遺伝会社、グラクソ·スミスクライン、ジョンソン、ナイクタ治療会社、ロ氏ホールディングス、およびサイノフィアンバンテ。他の大量の資源を持つ小さな会社は,Anthera製薬会社,aurinia製薬会社,ImmuPharma PLC,Kezar生命科学社,Vera治療会社,Viela生物会社である。
PSC.PSC-Albireo Pharma,Inc.,Avolynt Inc.,Calliditas Treateutics AB,ダウン製薬会社,Chemomab Treateutics,CymaBay Treateutics,Inc.,Dr.Falk Pharma GmbH,Galmed PharmPharmticals Ltd.,Gattair Pharma Co.,Genfit Corp.,Gilead Sciences,Inc.,HighTide Treeutics Inc.,免疫会社,Invea Treeutics,Inc.,LISCure Biosciences Inc.,Mirum Mireutics,Inleoscy,Inoldc.
ナッシュ-アスリコン、礼来、ジリッド科学、メルク、ノボノルド/S、ノワ製薬、ファイザー、およびローホールディングス。他の大量の資源を持つ小さな会社は,Enanta製薬会社,Ionis製薬会社,NGMバイオ製薬会社,Pplant治療会社,Terns製薬会社,89 bio社である
ミリ秒-生物遺伝会社、百時美施貴宝社、EMD Serono社、ジョンソン、メルク社、ノワ製薬、セノフィ、Teva製薬工業有限公司、および羅氏ホールディングス
カリフォルニア大学--エバーヴィ社、アスリコン、百時美施貴宝社、礼来社、ジリッド科学会社、ヤンソンバイオテクノロジー会社、ジョンソン、ファイザー、メルク社、ミレニアム製薬会社、主役治療会社、羅氏ホールディングス、武田製薬有限会社。
私たちの候補製品の成功に影響を与える重要な競争要素は有効性、安全性、コストと利便性かもしれない。私たちの多くの競争相手は、単独でも彼らとのパートナーでも、私たちより多くの資源を持っていて、市場で足場を固めて、研究開発、製造、臨床前と臨床テストの方面で専門知識を持って、監督管理の許可及び清算とマーケティングの許可を得た製品を獲得します。これらの競争相手はまた、合格した科学、販売、マーケティングと管理者を募集し、維持し、臨床試験場と臨床試験の患者登録を確立し、そして私たちの計画と相補的或いは必要な技術を獲得する上で私たちと競争している。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。より多くの合併と買収は私たちの競争相手により多くの資源を集中させるかもしれない。
知的財産権
私たちは私たちの業務に商業的または戦略的重要性を持つノウハウと情報を保護するために努力している。私たちは、私たちの候補療法または私たちの候補療法を生産するための技術、私たちの候補療法の物質成分、およびその使用および製造方法をカバーすることを目的とした特許権の獲得と維持を求めている
私たちの業務に重要な他の発明もあります米国や他の管轄地域で戦略的または商業的価値のある特許権を取得することも求められている。
私たちは、使用方法のような、私たちの独自技術および現在の独自製品および関連方法をカバーするために、6つの特許家族を代表する特許出願を提出した。2024年2月15日現在、私たちの特許資産には、12件の許可された米国特許、米国で係属している2つの非臨時特許出願、65件の許可された外国特許、および16件の現在各外国司法管轄区で係属している外国特許出願が含まれている
具体的には、GRI-0621、および炎症性疾患のような疾患の関連方法を治療するための特許シリーズを有する。この家族は、3つの米国特許と20件の外国特許(オーストラリア、ブラジル、カナダ、中国、ヨーロッパ(9カ国/地域で有効)、香港、日本、韓国、メキシコ、ロシア)を取得した。この家族の特許出願は、例えば、欧州特許機関、中国、日本、韓国を含む複数の司法管轄区域で決定されている。この特許シリーズの特許は2032年に満了する予定であり,いかなる特許期限の調整や延長も行われない
我々はまた、GRI−0803およびそれを用いた疾患の治療に関連する方法のための特許ファミリーを有している。この家族は3つのアメリカ特許と9つの外国特許(カナダ、ヨーロッパ(7カ国·地域で有効)と香港を取得した。この一連の特許出願はアメリカとヨーロッパ特許機関で審査されている。この特許家族の特許は2032年に満了する予定であり、いかなる特許期限の調整や延長も行われない
さらに、GRI−0729に関連する特許系列と、この特許を用いて疾患を治療する関連方法とを有する。この家族は、4つの米国特許および13件の外国特許(カナダ、ヨーロッパ(11カ国/地域で検証)および香港)を付与している。この特許家族の特許は2032年に満了する予定であり、いかなる特許期限の調整や延長も行われない
私たちはまた、GRI-0124およびそれを使用して疾患を治療する関連方法のための特許ファミリーを有している。この家族は14件の外国特許(台湾、オーストラリア、中国、欧州(7カ国で有効)、香港、イスラエル、メキシコ、ロシア)を取得した。例えば、米国、アラブ首長国連邦、ブラジル、中国、日本、ロシア、カナダ、香港、韓国では、この一連の特許出願が審理されている。この特許家族の特許は2035年に満了する予定であり、いかなる特許期限の調整や延長も行われない
著者らは新しい技術と候補治療薬を開発すると同時に、絶えず私たちの知的財産権戦略を評価と完備している。私たちの業務の発展に伴い、私たちは競争に適応したり、潜在的な機会をつかむために、私たちの知的財産戦略を追求するために、より多くの特許出願を提出するかもしれない
個別特許の期限は、このような特許を取得した国の法律にかかっている。私たちが出願したほとんどの国では,特許期間は非臨時特許出願が最初に提出された日から20年である。しかし,FDA要求を遵守したり,米国特許商標局(USPTO)が起訴中に遭遇した遅延による遅延により,米国特許の有効期限を延長することができる。例えば、ハッジ·ワックスマン法は、FDAが承認した薬物の特許期間を特許満了後最大5年間延長することを許可している。特許期間の延長の長さは,薬物が規制審査を受ける時間の長さと関係がある。特許延期は、製品承認日から14年を超えることができず、承認された薬物に適用される特許を延長することしかできない。ヨーロッパおよび他の管轄区域にも同様の規定があり、承認された薬物をカバーする特許の期限を延長する。将来、私たちの候補治療薬がFDAの承認を得たら、私たちはこれらの候補治療薬をカバーする特許期間の延長を申請する予定だ。私たちは、これらの管轄区域があれば、条件に適合する特許もありますが、米国特許商標局やFDAを含む適用当局が、このような延長を承認すべきかどうか、そのような延長の長さを承認すべきかどうかの評価に同意する保証はありません
また,データ独占性,市場独占性,特許期限調整,特許期限延長(利用可能であれば)に依存することが予想される
政府規制と製品審査
その他の事項以外に、アメリカ連邦、州と地方の各レベル及びその他の国の政府当局は薬品と私たちが開発している薬品と候補製品の研究、開発、臨床試験、テスト、製造(任意の製造変更を含む)、許可、薬物警戒、不良事件報告、リコール、包装、貯蔵、記録、ラベル、広告、販売促進、流通、マーケティング、輸出入などの方面に対して広範な監督管理を行った。米国や外国で規制の承認を受ける手続きや、その後適用される法規や条例の遵守には、多くの時間と財力が必要だ。
アメリカ政府の規制
米国では,FDAは“連邦食品,薬物と化粧品法”(FDCA)及びその実施条例に基づいて薬品を規制している。製品開発プロセス、承認プロセス、または承認後の任意の時間において、出願人が適用された米国の要求を遵守できなかった場合、FDAおよび司法省(DoJ)または他の政府エンティティによって提起された様々な行政または司法制裁、例えば、FDAが係属中のNDAの承認を拒否すること、承認の撤回、臨床棚上げの実施、警告状の発行、製品リコール、製品差し押さえ、生産または流通の完全または部分的停止、禁止、罰金、政府契約の拒否、原状回復、返品または民事および/または刑事罰を受ける可能性がある
FDAが新薬が米国で発売される前に必要なプログラムには、一般に以下のような態様が含まれる
•実験室テスト、可能な動物研究と調合研究などの非臨床と臨床前研究を完成し、FDA良好な実験室規範(GLP)とその他の適用法規に符合する
•ヒト臨床試験が開始される前に有効でなければならないINDをFDAに提出する
•試験を開始する前に、各臨床場所をカバーするIRBの承認を得ることができる
•良好な臨床実践(GCP)に基づいて十分かつ良好に制御された人体臨床試験を行い、各適応に対する提案薬物製品の安全性と有効性を確定する
•セキュリティプロトコルをFDAに提出し、適用される場合、適用される場合の申請使用料を支払い、このセキュリティプロトコルに対するFDAの受け入れ;
•適用されれば、FDA諮問委員会の審査が満足的に完了する
•現在の良好な製造仕様(CGMP)の遵守状況を評価し、施設、方法、および薬物の特性、強度、品質、および純度を維持するのに十分な施設、方法、および制御を保証するために、FDAの生産製品の1つまたは複数の製造施設の承認前検査を満足的に完了させる
•GCP要件および臨床データの完全性を保証するために、FDAの臨床試験会場に対する監査を満足的に完了させる
•NDAに対するFDAの審査と承認。
臨床前研究
臨床前或いは非臨床研究は製品の化学、毒性と調合に対する実験室評価、及び潜在安全性と有効性を評価する潜在動物研究を含む。2022年12月29日に法律となった2023年総合支出法案(P.L.117−328)に署名し、薬物の非臨床試験は生体動物試験を含むことができるが、必要ではないと規定したFDCAが改正された。改訂された言語によれば、スポンサーは、様々なインビトロ分析(例えば、細胞ベースの分析、器官チップまたは微生理システム)、コンピュータ研究(すなわち、コンピュータモデリング)、他のヒトまたは非ヒト生物学に基づく試験(例えば、生物印刷)または体内動物試験を完了することによって、非臨床試験要件を満たすことができる。
候補製品の安全性を支援するためにFDAに提出される前臨床試験は、GLP法規および米国農務省の動物福祉法に適合しなければならない。薬品スポンサーはFDAに臨床前試験結果、及び生産情報、分析データと任意の利用可能な前アメリカ臨床データ或いは関連文献をINDの一部として提出しなければならない。IND提出後も,いくつかの非臨床試験が継続される可能性がある。INDはFDAが受信した30日後に自動的に発効し、それ以前にFDAが1つまたは複数の提案された臨床試験に対して懸念または問題を提起しなければ、臨床試験を保留する。この場合,INDスポンサーやFDAは臨床試験開始前に未解決の問題を解決しなければならない。したがって,INDの提出はFDAが臨床試験の開始を許可しない可能性がある。臨床休止はIND中の任意の時間に発生することができ、1つまたは複数の特定の研究またはINDによるすべての研究に影響を与える可能性がある。
また、FDAやスポンサーは、研究対象が受け入れられない健康リスクに直面していることを発見することを含む、随時様々な理由で臨床試験を一時停止または終了することができる。同様に、臨床試験がIRBの要求に従って行われない場合、または候補製品が患者に予期せぬ深刻なダメージを受けることに関連している場合、IRBは臨床試験の承認を一時停止または終了することができる。
臨床試験
臨床試験は、GCP要求に適合する合格した研究者の監督の下でヒト被験者にINDを投与することに関連し、すべての研究対象に要求することを含む
書面では、IRBが同意要求を免除されない限り、任意の臨床試験に参加することを要求し、臨床試験から報告されたデータおよび結果が信頼性かつ正確であることを保証することを要求する。臨床試験は,試験目標を詳細に説明し,被験者の資格を決定する基準,用量計画,安全性をモニタリングするためのパラメータ,有害事象をタイムリーに報告するプログラム,および評価する有効性基準のシナリオの下で行った。INDの一部として,各臨床試験の案と任意の後続の案修正案をFDAに提出しなければならない。さらに、IRBは任意の臨床試験が開始される前にこの計画を審査して承認しなければならない。
いくつかの臨床試験および臨床試験結果に関する情報は、ClinicalTrials.gov登録上で公開されるために、特定の時間範囲で国家衛生研究院に提出されなければならない。対象の臨床研究や法律規定の研究結果を適時に登録できなかったことは民事罰金を招く可能性があり、また違反側が連邦政府の将来の支出を獲得することを阻止する。政府はすでにこれらの要求を遵守できなかった臨床試験スポンサーに対して法執行行動をとっている。
人体臨床試験は通常3つの連続段階に分けて行われ、この3つの段階は重なる可能性があり、合併する可能性もある
第一段階:この候補製品は、最初に健康なヒト対象または標的疾患または状態を有する患者に導入され、安全性、用量耐性、吸収、代謝、分布、排泄が試験され、可能であれば、その有効性の早期兆候が得られる。第一段階臨床試験期間中に、薬物の薬物動態学と薬理作用に関する十分な情報を得ることができ、良好かつ科学的に有効な第二段階臨床試験の設計制御を可能にする。
第二段階:候補製品は、可能な副作用と安全リスクを決定するために、より大きいが依然として限られた患者集団に使用され、特定の標的適応に対するこの製品の治療効果を初歩的に評価し、用量耐性および最適用量を決定する。第二段階の臨床試験は通常良好な制御と密接なモニタリングを受けている。
第3段階:候補製品は、制御された良好な臨床試験においてより多くの患者集団のために使用され、通常は地理的に分散された臨床試験地点であり、承認された製品の有効性および安全性を統計的に評価するのに十分なデータを生成し、製品の全体的なリスク-利益プロファイルを確立し、製品のラベルに十分な情報を提供する。第二段階臨床試験と比較して、第三段階臨床試験は通常より多くの参加者に関連する
承認後試験は,“4期”臨床試験と呼ばれることがあり,最初の上場承認後に行われる可能性がある。これらの試験は,治療適応が予想される患者の治療から追加的な経験を得るために用いられている。場合によっては、FDAは“4期”の臨床試験を強制的に実行する可能性がある。
ヒト臨床試験は本質的に不確定であり、第一段階、第二段階と第三段階の臨床試験はいかなる指定された期間でも成功しないかもしれない、あるいは全く成功しないかもしれない。さらに、所与の臨床試験は複数の段階の要素を組み合わせることができるが、会社がある臨床試験を特定の段階に指定することは、この研究がこの段階に対するFDAの要求を満たすのに十分であるとは限らない。なぜなら、プログラムおよびデータを提出および審査する前に、この決定を下すことができないからである
重要な試験は1種の臨床試験であり、FDAの候補製品の安全性と有効性に対する評価要求を満たすと考えられ、規制承認を支持するために、単独で使用できるか、または他の重要な試験または非肝心な試験と一緒に使用することができる。一般的に、重要な試験は3期試験であるが、設計が臨床利益の良好な制御と信頼できる評価を提供すれば、特に医療需要を満たしていない領域では、それらは2期試験である可能性がある。近年、FDAは、新薬承認に適した法的基準である“有効な実質的な証拠”の証明を支持するために、データ提出のタイプ、数量、および時間を決定する際に規制柔軟性を行使することを望むようになってきており、以下ではさらに議論する
国会は最近FDCAを改正し、3期臨床試験のスポンサーや新薬の他の“肝心な研究”にマーケティング許可を支持し、このような臨床試験のための多様な行動計画の設計と提出を要求した。行動計画には,スポンサーの多様な学生募集目標と,目標の理由やスポンサーがこれらの目標をどのように達成するかの記述が含まれなければならない。スポンサーはスポンサーが関連する臨床試験案をFDA審査に提出する前にFDAに多様な行動計画を提出しなければならない。FDAは多様な行動計画の一部またはすべての要求を免除することができる。FDAがスポンサーの多様性行動計画に反対したり,重大な変更を要求したりすれば,関連臨床試験の開始を遅らせる可能性がある。
臨床開発計画中のFDAとの相互作用
INDが承認され臨床試験を開始した後,スポンサーはFDAとの相互作用を継続する。臨床試験結果を詳細に説明する進捗報告は少なくとも毎年FDAに提出しなければならず,深刻な有害事象が発生すればより頻繁に提出される。さらに、以下のいずれかのIND安全報告書は、深刻かつ予期しない疑わしい副作用、他の研究または動物またはインビトロ試験の結果のうちの1つにFDAに提出されなければならない
この製品に暴露された人類は重大なリスクがあることを示唆した;方案或いは研究者マニュアルに列挙された情況と比べ、臨床上深刻な副作用の疑いの発生はいかなる重要な増加がある
また,スポンサーは臨床開発計画のあるときにFDAと会う機会がある。具体的には,スポンサーはIND提出前(IND前会議),第2段階臨床試験終了時(EP 2会議),NDA提出前(NDA前会議)にFDAと会うことができる。他の時間に会議を開催することを要求することもできる。これらの会議はスポンサーにこれまで収集してきたデータの情報をFDAと共有する機会を提供し,FDAに次の段階の開発に関する提案を提供した。例えば,EOP 2では,スポンサーはその第2段階の臨床結果を検討し,その鍵となる第3段階臨床試験計画(S)を提案し,新製品の承認を支援すると考えられる。このような会議は自ら行うことができ,電話会議/ビデオ会議や書面回答により,スポンサーからFDAへの質問とその機関への回答を記録するだけでよい.FDAは、議事録および諮問書簡で伝達された応答は、スポンサーに対する提案および/または提案のみを構成するため、スポンサーは、そのような提案および/または提案の制約を受けないと述べている。しかし,実践的には,スポンサーがFDAの提案に沿って臨床計画を設計していないことは,その計画を大きな失敗リスクに直面させる可能性がある。
新発展区を受け入れる
必要な臨床試験,臨床前研究および臨床試験の結果,および製品の化学,製造,制御,安全更新,特許情報,乱用情報および提案されたラベルに関する情報が,出願の一部としてFDAに提出され,候補製品を1つまたは複数の適応に押し出すことの承認を要求すると仮定する。データは、製品使用の安全性および有効性を試験するために、または研究者によって開始された研究を含む多くの代替源からの臨床試験からのものである可能性がある。上場承認を支援するためには,提出されたデータは品質と数量で薬物製品の安全性と有効性を十分に決定しなければならない。“処方薬使用者費用法案”(PDUFA)による申請提出と審査に要する費用は高く,承認された申請の発起人は合格処方薬製品評価に基づく年間計画費用を納付する必要がある。これらの費用は、一般に年に1回調整され、場合によっては免除および免除がある可能性があり、例えば、公衆の健康を保護するために免除が必要であり、費用は革新に大きな障害となるか、または申請者は小企業であり、審査のために最初の人間治療申請を提出する。
FDAは、すべての出願を受信してから60日以内にすべての出願を予備審査し、実質的な審査のためにスポンサー申請が十分に完全であるかどうかをその時または前に通知しなければならない。関連部分では、FDAの法規は、FDAがすべての関連情報およびデータを受信する前に、申請は提出されたとみなされてはならないと規定している。FDAが出願がこの基準を満たしていないと判断した場合、出願人に提出拒否(RTF)決定を発行する。一般に、RTFの根拠は、情報または必要な情報を明らかに見落としている部分のような行政的不完全さであり、安全性および有効性の評価を見落としたり、説明を適切に使用するために必要なキーデータ、情報または分析を提供したり、または情報の内容、提示または組織が不十分であったりして、実質的かつ有意義な検討を行うことができない。FDAは申請を受け入れるのではなく、より多くの情報を提供することを要求するかもしれない。この場合、申請は追加情報と共に再提出されなければならない。再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。
新発展区を検討する
出願が受け入れられた後、FDAは出願の深い実質的な審査を開始した。FDAは、提案された製品がその予期される用途に対して安全に有効であるかどうか、許容可能な純度プロファイルを有するかどうか、および製品がcGMPに従って製造されているかどうかを決定するために出願を審査する。
FDAがPDUFAで合意した目標と政策によると、FDAは10ヶ月間、新しい分子実体としての標準出願の予備審査を完了し、“優先審査”を有する出願については6ヶ月の期間がある。FDAは、新しい情報を考慮するために、審査プロセスをさらに3ヶ月延長することができ、または出願人が明確な説明を提供する場合には、FDAが最初の提出後に発見した係属中の欠陥を解決することができる。これらの審査目標にもかかわらず,NDA審査過程は非常に長い可能性があり,FDAによる申請の審査がPDUFA目標行動日まで延長されることは珍しくない。多くの革新的薬物製品(生物製品を除く)は、米国食品薬品監督管理局第505条(B)(1)条に提出された機密協定に基づいてFDAの上場承認を得ており、一般に伝統的または“完全守秘協定”と呼ばれている。1984年、“ハッジ-ワックスマン法案”の採択に伴い、短い規制案が確立され、FDAが革新者または“参考”製品に基づく模倣薬を承認することを許可し、国会はまたFDCA第505(B)(2)条を公布し、伝統的なNDAと後発薬応用を組み合わせた混合経路を提供した。第505条(B)(2)条は、出願人が、その適用を支援するために、以前のFDAの既存製品の安全性および有効性データの発見、または出版された文献に部分的に依存することを可能にする。第505条(B)(2)NDAは、FDAが新規または改善した処方または以前に承認された製品の新しい使用を承認するための代替方法を提供することができる
これは安全性や有効性を証明するための新しい臨床データを必要とするだろう。第505条(B)(2)条は、出願人が薬物が安全であるか又は有効であるかを証明するために行われた研究における情報に少なくとも部分的に依存する機密協定の提出を許可し、これらの情報は、出願人によって行われたものでもなく、出願人のために行われたものでもなく、出願人も参照権又は使用権を得ていない。第505条(B)(2)の出願人は、以前に承認された製品に対する研究の依存が科学的に適切であると判断できる場合、いくつかの臨床前または臨床研究を行う必要性を除去または減少させることができる。FDAはまた、承認された製品からの変更を支援するために、非臨床研究および臨床研究を含む追加の研究または測定を企業に要求する可能性がある。新製品の安全性および/または有効性を決定するために必要な研究タイプおよびデータ範囲、例えば、薬物の投与経路を局所から経口に変更する効果は、科学的な方法で駆動され、ケースに基づいて決定される。次に、FDAは、参照製品のラベル適応のすべてまたは一部が承認された新製品候補と、第505条(B)(2)条のNDA出願人がデータを提出した任意の新しい適応とを承認することができる。
出願を審査する際には,FDAは通常,申請者に情報要求を提出し,回答の最終期限を設定する.FDAはまた,新製品の製造施設の承認前検査を行い,製造プロセスや施設がcGMPに適合しているかどうかを確認する。FDAは、製造プロセスおよび施設がcGMPに適合していることを決定し、要求された仕様の下で製品が一貫して生産されることを保証するのに十分でない限り、この製品を承認しないであろう。
FDAはまた、INDおよびGCP要件に適合することを保証し、FDAに提出された臨床データの完全性を保証するために、スポンサーおよび1つまたは複数の臨床試験場所を検査することができる。その従業員や第三者引受業者が“一般公共サービス計画”や“一般公共サービス規則”を遵守することを確保するために,申請者は訓練,記録保存,作成,品質制御などに多大な時間,お金,労力を費やす可能性がある.試験がIND下で行われる場合,FDAは通常外国からの臨床試験のデータを受け,NDAを支持する。外国の臨床試験がINDによるものでなければ,研究がGCPによって行われ,FDAが現場検査によりデータを検証することができれば(必要であれば),FDAはNDAを支持するデータを受け入れることができる。FDAは通常、上場申請に国内臨床試験のいくつかのデータ支持が要求されているが、以下の場合、FDAは上場承認の唯一の根拠として外国データを受け入れることができる:(1)外国データは米国人口とアメリカの医療実践に適用される、(2)研究は公認能力を有する臨床研究者が行うこと、および(3)データは有効であると考えられ、現場検査を必要としない、あるいはFDAが検査を行う必要があると考えられる場合、FDAは現場検査や他の適切な手段でデータを検証することができる。
さらに、FDAは、安全性または有効性の問題を提起する新製品候補出願を含む申請を、審査、評価、および提案を行って、申請を承認すべきかどうか、およびどのような条件で承認すべきかを決定するために諮問委員会に提出することができる。通常,諮問委員会は臨床医や他の科学専門家を含む独立した専門家からなるグループであり,申請を審査,評価し,申請を承認すべきかどうか,どのような条件でアドバイスを提供すべきかを担当する。FDAは諮問委員会の提案に制限されていないが、最終承認決定を下す際にこれらの提案を考慮する。
臨床試験のデータは常に決定的ではなく,FDAやその諮問委員会は異なる方法でデータを解釈する可能性があり,スポンサーが同じデータを解釈する可能性がある。FDAはまた、臨床試験データを再分析する可能性があり、これは、FDAおよび出願人が審査中に広範な議論を行うこと、または規制部門の承認を延期、制限、または阻止することをもたらす可能性がある。FDAはタイムリーに承認されないかもしれないし、全く承認されないかもしれない。
FDAはまた、医薬品の利益がそのリスクよりも大きいことを保証し、製品の安全な使用を保証するために、リスク評価および緩和戦略(REMS)を提出する必要があると判断した場合、リスク評価および緩和戦略(REMS)の提出を要求する可能性がある。REMSは、制限された分配方法、患者登録、または他のリスク最小化ツールのような薬物ガイドライン、医師コミュニケーション計画、評価計画、および/または安全使用を保証する要素を含むことができる。FDAは具体的な状況に応じてREMSに対する要求および具体的なREMS条項を決定する。FDAがREMSが必要であると考えている場合、申請されたスポンサーは提案されたREMSを提出しなければならず、FDAはREMSのない出願を承認しないであろう。
さらに、改正および再許可された2003年の“小児科研究公平法”によれば、いくつかの新薬または新薬補充剤は、すべての関連する小児科亜群において主張される適応の安全性および有効性を評価し、安全で有効な各小児科亜群に対するこの製品の投与および投与をサポートするのに十分なデータを含まなければならない。FDAは、成人のために使用されるか、または小児科データの要求を完全にまたは部分的に免除するために製品が使用されることが承認されるまで、申請者の要求に応じて、または小児科データの一部または全部の提出を延期することができる。法規が別途要求されない限り、小児科データ要求は孤児の称号を有する製品には適用されない。
新発展区に関する決定
FDAは、その製品が安全であるかどうか、およびその予期される用途に有効であるかどうかを決定するために出願人を審査し(S)、後者の決定は、大量の証拠に基づく。米国食品薬品監督管理局の規定によると、“実質的な証拠”という言葉は、“科学的訓練と経験を経た専門家が臨床調査を含み、関連製品の有効性の証拠を評価するための十分かつ良好な調査を行い、その上で、これらの専門家は、そのラベルまたは提案のラベルに規定、推薦または提案の使用条件下でその主張または表示の効果を有すると公平かつ責任を持って結論を出すことができる”と定義されている
FDAのこのエビデンス基準の解釈は,新製品の有効性を確認するためには,少なくとも2回の十分かつ良好な制御の臨床調査が必要である。しかしながら、場合によっては、FDAは、いくつかの特徴および追加情報を有する単一の実験がこの基準を満たす可能性があることを示している。このやり方は1998年に国会で認められました立法は関連部分で規定されています[アメリカ食品医薬品局は]関連する科学的決定に基づいて、良好な臨床調査からのデータおよび確認性証拠(調査の前または後に得られる)が有効性を決定するのに十分である場合、FDAは、そのようなデータおよび証拠を実質的な証拠と見なすことができる。法のこの改正は,FDAが十分かつ良好に制御された臨床調査を発見する可能性があることを認識しており,対照試験外の支持性データを含めて有効性を確立するのに十分である。2019年12月、FDAはガイドライン草案を発表し、有効性の実質的な証拠を確立するために必要な研究をさらに説明した。2023年9月、この機関は2019年の“有効性実質性証拠”ガイドライン草案中の提案を補充と拡張し、“十分かつ制御された良好な臨床調査と検証性証拠に基づいて有効性を証明する実質的な証拠”と題する二番目のガイドライン草案を作成した。第2の文書は、十分かつ良好に制御された臨床調査の結果をサポートするために、1つまたは複数のソースからのデータ(例えば、臨床データ、機械的データ、動物データ)の使用に関するより多くの詳細を提供し、確認証拠と見なすことができるデータタイプの例を提供する第1の文書を補足する。このような確定されたケースの性質のため、FDAは引き続き、スポンサーが十分かつ制御された臨床調査と確認性証拠を加えることによって有効な実質的な証拠を構築しようとするなら、彼らは早期にこの機関と接触する必要があると強調した。
申請およびすべての関連情報を評価した後、諮問委員会のアドバイス(ある場合)および製造施設および臨床試験地点の検査報告を含めて、FDAは完全な返信(CRL)または承認書を発行する。この結論を達成するために、FDAは、この薬剤が有効であることを決定しなければならず、その期待利益は、患者に対する潜在的リスクよりも大きい。この“利益−リスク”評価は、この製品の安全性および有効性に関するNDAにおける多くの証拠によって提供される。この評価はまた他の要素の影響を受け、潜在疾病の深刻性及び現有の治療法がどの程度患者の医療需要を満たしているか;発売前の臨床試験証拠はどのようにこの製品の発売後の環境における実際の使用状況の不確定性を推定するか;及びリスク管理ツールが特定のリスクを管理する必要があるかどうかを含む。この評価に関連して,FDA審査チームはすべての個別審査と他の文書を1つの“行動パッケージ”にまとめ,FDA審査の記録となっている。審査チームはその後、FDAの高官が決定を下すための提案を発表した。
CRLは,申請の審査周期が完了したことを示しており,申請は現在の形で承認されない.CRLは、通常、提出中の不足点を列挙し、FDAが申請を再検討するために、大量の追加のテストまたは情報を必要とする可能性がある。CRLは、追加の臨床または他のデータ、追加の重要な第3段階臨床試験(S)、および/または臨床試験、臨床前研究または生産に関連する他の重要で時間のかかる要件を必要とする可能性がある。CRLが発行された場合、出願人は、FDAが決定した欠陥に1年間応答することができ、FDAは、出願が撤回されたと考えるか、または申請者が6ヶ月の応答期間を追加的に延長することを適宜承認することができる。FDAは、発行されたCRLの再提出を2ヶ月または6ヶ月以内に検討することを約束しており、具体的には、含まれる情報のタイプに依存する。しかしながら、この補足情報を提出しても、FDAは最終的に、その申請が承認された規制基準を満たしていないと決定する可能性がある
一方,その製品の商業マーケティングを承認し,特定の適応に関する具体的な処方情報を提供する。すなわち、承認は、FDA承認のラベルに記述された使用条件(例えば、患者数、適応)に限定される。さらに、解決すべき特定のリスク(S)に応じて、FDAは、承認後に製品の安全性をさらに評価するための承認後試験(4期臨床試験を含む)を要求するために、製品ラベルに禁忌症、警告または予防措置を含むことを要求することができ、試験および監視計画は、製品の商業化後に製品を監視すること、または販売および使用制限、または製品の潜在的な市場および利益に大きな影響を与える可能性のあるREMS下の他のリスク管理メカニズムを含む他の条件を適用することを要求することができる。FDAは発売後の試験或いはモニタリング計画の結果に基づいて、製品の更なるマーケティングを阻止或いは制限することができる。承認後、新たな適応の追加、製造変更、および追加のラベル宣言など、承認された製品のいくつかのタイプの変更は、さらなるテスト要件およびFDAの審査および承認を受けることになる。
FDA特別迅速審査計画
FDAは特定の製品を指定して加速開発或いは審査を行う権利があり、もしこれらの製品が深刻な或いは生命に危害を及ぼす疾病或いは状況の治療において満たされていない医療需要を解決することを目的としている場合。これらの計画には、高速チャネル指定、画期的な治療指定、優先審査指定が含まれる。これらの計画の目的は,FDA標準審査プログラムよりも早く患者に重要な新薬を提供することである。
迅速なチャンネル認証を取得する資格があるため、FDAはスポンサーの要求に基づいて、深刻な或いは生命に危害を及ぼす疾病或いは状況を治療することを目的とした製品を確定し、満足されていない医療需要を満たす潜在力を示しなければならない。FDAは、製品が存在しない療法を提供するか、または治療効果または安全要因に基づく潜在的に既存の療法よりも優れた治療法を提供する場合、満たされていない医療需要を満たすことを決定するであろう。高速チャネル指定は、FDA審査チームと相互作用するより多くの機会を提供し、完了した申請を提出する前にNDAコンポーネントのスクロール審査を可能にすることができ、スポンサーがNDA部分を提出するスケジュールを提供した場合、FDAはNDAの部分を受け入れることに同意し、スケジュールが許容可能であると判断し、スポンサーはNDAの第1の部分を提出する際に任意の必要な使用料を支払うことができる。また,迅速チャネルの指定が臨床試験中に出現したデータの支持を得なくなった場合,スポンサーがその指定を撤回したり,FDAが指定を撤回したりする可能性がある
また,2012年のFDA安全·革新法案(FDASIA)の公布に伴い,INDスポンサーの要求に応じて,国会でFDAが“画期的療法”に指定された治療候補薬のための新たな規制計画が作成された。画期的な治療法は、1つまたは複数の他の薬剤と単独でまたは1つまたは複数の他の薬剤と組み合わせて重篤または生命を脅かす疾患または状態を治療することを意図した薬剤として定義され、初歩的な臨床証拠は、1つまたは複数の臨床的に重要な終点において、臨床開発早期に観察される実質的な治療効果のような既存の療法よりも有意な改善を示す可能性があることを示す。FDAは画期的な治療法の承認申請の開発と審査を加速するために、適時に製品スポンサーと会議を行い、それに提案を提供するなど、突破的な治療法に対して何らかの行動を取らなければならない。
最後に、薬物が重篤な疾患を治療し、承認された場合、安全性または有効性の面で有意な改善を提供する場合、FDAは、製品を優先的に検討するように指定する可能性がある。FDAがマーケティング申請を提出する際に,具体的な状況から他の既存療法と比較して,提案薬が疾患の治療,予防または診断における有意な改善を表すかどうかを決定する。顕著な改善は,ある疾患の治療の有効性の向上,治療を制限する薬物反応の除去あるいは大幅な減少,記録されている患者のコンプライアンスの向上,重篤な結果の改善,あるいは新亜群の安全性と有効性の証拠に現れる可能性がある。優先審査指定の目的は、全体的な注意とリソースをこのような申請の評価に誘導し、FDAがマーケティング申請に行動する目標を10ヶ月から6ヶ月に短縮すること、すなわち申請の日から新しい分子実体のNDAに行動することである。
1つの製品がこれらの計画のうちの1つまたは複数に適合していても、FDAは、製品がもはや資格条件に適合していないことを後で決定することができ、またはFDAの審査または承認を決定する期間が短縮されないことができる。また,迅速チャネル指定,画期的治療指定,優先審査は承認の基準を変更することはなく,最終的に開発や承認過程を加速させない可能性もある。
承認ルートを加速する
さらに、深刻または生命を脅かす疾患の治療における研究された安全性および有効性、および既存の治療よりも意味のある治療利益を提供する製品は、承認を加速させることができ、これは、(I)十分かつ良好に制御された臨床試験に基づいて、医薬製品が臨床的利益を合理的に予測する可能性のある代替終点に効果があることを決定することができること、または(Ii)中間臨床終点において、不可逆的な発症率または死亡率(IMM)よりも早く測定することができ、深刻性を考慮して、IMMまたは他の臨床的利益への影響を合理的に予測することができることを意味する。このような状況の希少または流行と代替治療が利用可能か不足している。承認の条件として、FDAは、迅速な薬剤停止手順を必要とする可能性があるIMMまたは他の臨床終点に対する期待される効果を検証および説明するために、承認を加速させた薬物のスポンサーの発売後の研究を要求することができる。加速的な承認を得た薬品は伝統的に承認された薬品と同じ安全と有効性法定基準に適合しなければならない。加速承認計画によって検討·承認されている薬剤製品の宣伝材料は,FDAの事前審査を経なければならない。
承認を加速するために、代替終点は、例えば実験室測定、放射画像、バイタルサイン、または他の臨床的利益を予測することができると考えられるが、それ自体は臨床的利益の測定基準ではない標識である。代替終点は通常、臨床終点よりも容易または迅速に測定される。中間臨床終点は治療効果の測定であり、1種の薬物の臨床利益、例えばIMMに対する効果を合理的に予測することが可能であると考えられる。FDAの中間臨床終点による加速承認の経験は限られているが,
終点自体は臨床利益と伝統的な承認の基礎ではなく、基礎があれば結論を出し、治療効果は合理的に1種の薬物の最終長期臨床利益を予測する可能性がある。
加速承認経路は病気経過が長く、薬物の期待される臨床利益を測定するために比較的に長い時間を必要とする環境に最もよく用いられ、代用或いは中間臨床終点への影響が非常に速く発生した。例えば、加速承認は、様々な癌を治療するための薬剤の開発および承認に広く使用されており、治療の目標は、通常、生存率を向上させること、または発症率を低下させることであり、典型的な病気経過の持続時間は、臨床または生存上の利点を証明するために長い、場合によっては大型の臨床試験を必要とする。
承認を加速する方法は、一般に、薬物の臨床的利益を検証および説明するために、勤勉な方法で追加的な承認後の検証的研究を行うことにスポンサーが同意することに依存する。そのため、この基礎の上で承認された候補薬物は必ず厳格な発売後のコンプライアンス要求を遵守し、4期或いは承認後の臨床試験を完成し、臨床終点への影響を確認することを含む。また、2023年の総合支出法案の一部として、国会は、マーケティングを継続する前に加速的に承認された無効薬の患者への潜在的リスクを軽減するために、FDAに追加の法定権力を提供した。FDCAのこれらの修正案によると、機関は、加速的な承認を得た製品のスポンサーに、承認前に検証試験を行うことを要求することができる。スポンサーはまた、試験が完了するまで、検証性試験の進捗報告を6ヶ月ごとに提出しなければならず、これらの報告はFDAのウェブサイトで発表される。必要な承認後研究を行わない場合、あるいは発売後の研究期間中にこの製品の期待される臨床的利益が確認できなければ、FDAがこの薬剤の承認を撤回することを許可する。国会では最近、FDAがスポンサーの検証性試験が製品主張の臨床的利益を検証できない場合に、迅速なプログラムを用いて製品承認を撤回することを選択することを可能にする法律も改正された。加速承認計画によって検討·承認されている薬剤製品の宣伝材料は,FDAの事前審査を経なければならない。国会で最近可決された法定改正案の前に,いくつかの腫瘍学スポンサーが加速承認により発売された薬物製品の特定適応を自発的に撤回した。最近、FDAは2024年2月に、この薬物の検証的研究が臨床効果を確認できなかった後、この法律改正されたプログラムを使用して承認を加速することを初めて発表した。承認経路の見直しを加速させることは、今後数年間継続する可能性があり、今後さらなる立法および/または行政改革を招く可能性がある。
承認後に要求する
FDAによって生産または流通を許可された薬品はFDAの普遍的かつ持続的な監督管理を受けなければならず、その中には記録保存、定期報告、製品サンプリングと流通、広告と販売促進、および製品の不良反応の報告に関連する要求が含まれている。承認後、承認された製品の大多数の変更は、新たな適応または他のラベル宣言を追加するなど、FDAの事前審査および承認を経なければならない。適応または製造プロセスまたは施設の変化を含む製品のいくつかの修正は、FDAへの提出を支援するために、出願人が追加のデータを開発するか、または追加の臨床前研究および臨床試験を行う必要がある可能性がある。前述したように,いずれの上場製品にも継続的な年間使用料要件と,臨床データを持つ補充アプリケーションの新規出願料がある
FDAはNDAを承認するための条件として、いくつかの承認後の要求を加えるかもしれない。例えば、FDAは、第4段階の臨床試験を含む上場後試験を要求し、製品の商業化後の安全性と有効性をさらに評価し、監視するために監視を行う可能性がある
また,FDAの規定では,製品は特定の承認施設で生産され,cGMPに適合しなければならないことが求められている。CGMPは、人員、建物および施設の組織、装置、構成要素および薬品容器および閉鎖的な制御、生産およびプロセス制御、包装およびラベル制御、保有および分配、実験室制御、記録および報告、ならびに返品または回収された製品に関する要件を含む。医薬品メーカーや他の生産·流通承認薬品に参加する実体は、食品·薬物管理局といくつかの州機関にその機関を登録し、食品·薬物管理局の定期的な抜き打ち検査を受けて、cGMPと他の法律を遵守しているかどうかを検査しなければならない。製造プロセスの変更は厳しく規制されており,変更の重要性により,FDAが事前に承認して実施する必要がある可能性がある。FDAの規定では、cGMPから外れた状況を調査·是正し、スポンサーや任意の第三者メーカーに報告や文書要求を行うことも求められている。そのため、メーカーは、cGMPおよび品質管理と品質保証に適合する他の側面を維持するために、生産と品質管理に時間、お金、精力をかけ続けなければならない
FDAは発売薬品のマーケティング、ラベル、広告と販売促進活動を厳格に監督する。製品は承認されるまで商業的に普及することはできないが,承認された薬物は一般にその承認の適応や製品承認のラベルに記載されている患者群に基づいてしか普及できない。販促声明はまた、安全性および有効性に関する声明を含む、製品のFDA承認のラベルと一致しなければならない。政府は、消費者向け広告、業界など、処方薬の特定の背景での普及を密接に検討している−
科学や教育活動やインターネットやソーシャルメディアを利用したキャンペーンを後援しています医師はラベル外の用途のために合法的な製品を処方する可能性があるが、メーカーはこのような用途を販売したり普及させたりしてはならない。FDAは最近、薬品メーカーがどのように医療保健提供者と許可されていない用途に関する真実、科学的合理と臨床関連情報の現代化提案を共有するかについて概説したガイドライン草案を発表した
その後、製品には、予期されない深刻度または頻度の不良事象、または生産プロセス、または法規要件を遵守できなかったことを含む、以前に未知の問題が存在することが発見され、新しいセキュリティ情報を追加するために承認されたラベルの強制改訂をもたらす可能性がある;新しい安全リスクを評価するために発売後研究または臨床試験を実施すること、またはREMS計画に従って流通または他の制限を実施することが可能である。規制を遵守しない他の潜在的な結果は以下のことを含む
•製品の販売または製造を制限または一時停止し、製品を完全に市場から撤回またはリコールすること;
•製造施設または製造ラインを閉鎖すること、または新しい製造要件を課すことを含む生産プロセス中断;
•承認された臨床試験には、罰金、警告状、または他の実行書または臨床保留が科される
•宣伝材料とラベルを強制的に修正し、訂正情報を発表する
•FDAが承認すべきNDAまたは承認されたNDAの補充剤の承認を拒否するか、または製品承認を一時停止または撤回すること;
•製品の差し押さえ、差し押さえ、あるいは製品の輸出入を許可しないことを拒否した
•民事または刑事罰の命令または適用を禁止する;または
•法令、企業誠実協定、資格取り消し、または連邦医療計画から除外されたことに同意する
そのほか、処方薬製品の流通は“処方薬販売法”(PDMA)の制約を受け、この法は連邦一級の薬品と薬品サンプルの流通を規定し、各州の薬品流通業者に対する登録と監督管理に最低基準を設定した。PDMA,州法ともに処方薬製品サンプルの配布を制限し,配布中の責任の確保を求めている。最近、米国で流通されているいくつかの処方薬を識別し、追跡するための電子システムを構築することを目的とした“医薬品サプライチェーン安全法”(DSCSA)が公布された。DSCSAは薬品メーカー,卸,流通業者に10年間の段階的と資源集約型の義務を要求し,最終的に2023年11月に終了する。しかし、FDAは、医薬品サプライチェーン内の貿易パートナーに必要な追加時間を収容するために、包装レベルで電子薬物追跡に対するDSCSAの要求を全面的に実施するために、1年の“安定期”から2024年11月までを発表した。
時々、新しい立法と法規が施行される可能性があり、これらの法規はFDA規制製品の承認、製造、マーケティングの法定条項を著しく変える可能性がある。例えば、FDAは、2022年2月に、各州が医薬品卸売業者に許可証を発行する国家基準を改訂するための提案された法規を発表し、州政府が第三者物流業者に許可証を発行するための新しい最低基準を確立し、州計画なしに使用するための連邦システムを作成し、各計画はDSCSAによって強制的に実行される。さらなる立法または規制の変化が公布されるかどうか、あるいはFDAの法規、ガイドライン、または解釈が変わるかどうか、またはこれらの変化の影響が(あれば)何になるかは予測できない。
排他性と後続製品の承認を規制する
ハッジ·ワックスマン排他性
FDCAに対するHatch−Waxman修正案の一部としてFDCA第505(B)(2)条が制定されたほか、国会は、FDAが以前NDAによって承認された薬剤と同じ有効成分を含み、生物学的同等性を有することが証明された後発薬を承認するための短い規制計画を確立した。後発薬の承認を得るためには,出願人は短い新薬申請(ANDA)を当該機関に提出しなければならない。ANDAは統合文書であり,その中には他の事項のほかに,有効薬物成分,生物学的同等性,薬品調製,後発薬の規格と安定性および分析方法,製造過程検証データと品質制御プログラムに関するデータと情報が含まれている。ANDAは安全性と有効性を証明する臨床前および臨床データを含むことができないため、“略語”である。逆に、このような申請を支持するためには、イミテーション製薬メーカーは、以前に機密協定によって承認された薬物製品(参考上場薬物(RLD)と呼ばれる)に以前に行われた臨床前と臨床テストに依存しなければならない。
具体的には,ANDAを承認するためには,FDAは後発薬が有効成分,投与経路,剤形,薬物強度,薬物使用条件においてRLDと同様であることを発見しなければならない。同時に、FDAはこの模造製薬と革新薬が生物学的同等性を有することを確定しなければならない。この法規によると、模倣薬の吸収速度と程度が市販薬物の吸収速度と程度と有意差がなければ、模倣薬は生物的にRLDに等しい。第505(B)(2)NDA経路と異なり、NDA経路は後続申請者が追加の臨床試験或いは非臨床研究からのデータを行って提出することを許可し、参考製品に対する提案変更(S)を支持し、ANDA規制経路は申請者が生物利用度或いは生物学的同等性データ以外の新しい臨床データを提出することを許可しない
ANDAが承認されると,FDAはその出版物“治療同等性評価を有する承認された薬物製品”(“オレンジブック”とも呼ばれる)において,この後発薬がRLDと“治療同等性”を有するかどうかを指摘する。内科医や薬剤師は,治療上等価な後発薬がRLDを完全に代替できると考えている。さらに、いくつかの州の法律および多くの医療保険計画の実施のため、FDA指定の治療同等性は、処方医または患者が知らない場合、またはその同意を得ない場合に、後発薬の代替をもたらすことが多い。このようなオレンジブック指定の薬学的実践の重要性を考慮して、国会は最近、申請者が評価を要求した場合、承認後6ヶ月以内にいくつかの505(B)(2)の薬物の治療同等性評価を行うようにFDAに指示した。
NDA審査および承認プロセスの一部として、出願人は、出願人の製品または治療使用方法をカバーすると主張する各特許をFDAに列挙することを要求される。新薬が承認されると、その薬物出願に記載されている各特許はオレンジブックに発表される。逆に、オレンジブックに記載されている薬剤は、ANDAまたは505(B)(2)NDAの承認をサポートするために、潜在的な後続のライバルによって参照されることができる。このプロセスにおけるFDAの役割は、純粋に“閣僚級”であり、それらが医薬製品またはその承認の使用方法をカバーするかどうかを決定するために、各特許における特許の請求項を審査または評価しない。FDCAおよびFDA実施条例で定義されていないNDA保有者が列挙する必要がある範囲外の特許は、競争相手や他の利害関係者から定期的に挑戦されるか、FDAの行政的挑戦手順によって、裁判所システムにおいて反競争または不公平とみなされる可能性がある。特に、連邦貿易委員会は2023年9月に、このような上場がより安い模造薬からの競争を損ない、人為的にブランド価格を上昇させる可能性があるため、オレンジマニュアルに“不正”を提出することを審査することを表明した政策声明を発表した。連邦貿易委員会は2023年11月にこの行動をとり,大手製薬会社10社の100件以上の“不正”特許リストを公開し,これらの特許に対するFDAの行政手続きを開始した。連邦貿易委員会、他の政府機関、製薬業者あるいは他の利益関係者が“不正”特許リストの政策問題を優先的に処理し続けるかどうか、及びこの分野で重大な訴訟を発展させるかどうかはまだ観察が必要である
ANDA申請者がFDAに出願する場合、FDAオレンジブックに記載されている参照製品の任意の特許をFDAに証明する必要がある。具体的には、出願人は、(I)要求された特許情報がまだ提出されていないこと、(Ii)に記載されている特許が満了していること、(Iii)に記載されている特許が満了していないが、特定の日に満了し、特許が満了した後に承認を求めること、または(Iv)に記載された特許が無効であるか、または新製品の侵害を受けないことを証明しなければならない。また,第505条(B)(2)条のNDA出願人は,承認された製品の検討に依存するため,出願人は,オレンジマニュアルに記載されているNDAが承認した製品の任意の特許をFDAに証明しなければならず,ANDA出願人と同程度である
後続の出願人がイノベーティブにリストされた特許に挑戦していない場合、FDAは、すべての要件参照製品のリスト特許が満了するまで、ANDAまたは505(B)(2)出願を承認しないであろう。新製品が承認された製品の上場特許又はそのような特許を侵害しない無効な認証を第4項認証と呼ぶ。後続の出願人が第4項の認証をFDAに提供した場合、FDAがANDA届出を受けると、出願人はまた、NDA及び特許所有者に第4項の認証の通知を送信しなければならない。そして、NDA及び特許所有者は、第4項の認証の通知に対して特許侵害訴訟を提起することができる。第四項の認証を受けてから45日以内に特許侵害訴訟を提起することは、より早い30ヶ月、特許満了、訴訟和解または侵害事件においてANDAまたは505(B)(2)出願人に有利な裁決まで、FDAがANDAまたは505(B)(2)NDAを承認することを自動的に阻止する
ANDAまたは505(B)(2)の出願も、オレンジブックに記載された参照製品の任意の適用非特許排他性満了まで承認されないであろう。FDCAに対するHatch−Waxmanの修正案は、新しい化学物質(NCE)NDAの許可を得た最初の出願人に、米国内の5年間の非特許データ排他期間を提供する。本条項の場合、NCEは、FDAが以前に任意の他のNDAで承認された活性部分を含まない薬剤を意味する。活性部分は薬物物質の生理的あるいは薬理作用を担う分子またはイオンである。このようなNCE排他性が付与されている場合、ANDAまたは505(B)(2)NDAは、提出された書類に第4項の証明が添付されていない限り、5年の満了前にFDAに提出することができず、この場合、出願人は、原製品の承認後4年以内に出願を提出することができる。
FDCAはまた,NDAまたはNDA付録が申請者が行ったり賛助したりする1つまたは複数の新たな臨床研究(バイオアベイラビリティや生物学的同等性研究を除く)の報告を含む場合,FDAはこれらの研究が承認申請に重要であると考え,FDCAは3年間のデータ排他性も規定している。この3年間の専門期間は、通常、新しい適応、剤形、投与経路または成分の組み合わせなど、以前に承認された薬物製品の変化を保護する。新たな臨床研究を行う法定要求を満たしていれば,先に承認された活性部分を含む医薬製品は3年間の独占経営権を得ることになる。5年間のNCE排他性とは異なり、3年間の排他的裁決は、FDAがANDAまたは505(B)(2)NDAを受け入れることを阻止せず、元の薬物製品が承認された日にその薬物の模倣薬を承認することを求め、逆に、この3年間の排他性は、新しい臨床研究に関連する使用条件のみをカバーし、一般的な事項として、FDAが元の活性成分を含む薬物の後続申請を承認することを禁止しない
5年および3年の排他性も、FDCA第505条(B)(1)条に従って提出された従来のNDAの提出または承認を遅延させることはないが、従来のNDAを提出する出願人は、安全性および有効性を証明するために必要なすべての臨床前研究および十分かつ制御された臨床試験の参照権を行うか、または得ることを要求されるであろう
孤児薬の指定と排他性
孤児医薬品法によれば、FDAは、一般に疾患または状態であり、(I)米国で200,000人未満、または(Ii)米国で200,000人を超え、そのようなタイプの疾患または状態を治療する薬剤を米国で開発および提供することに合理的な期待がなく、米国での販売から回収される稀な疾患または状態を治療するための薬剤のための孤児薬剤名を付与することができる。国会は、製品候補者が孤児指定の第2の選択を得ることができる立法提案、いわゆる“コスト回収”経路を定期的に審議している
秘密保持協定を提出する前に、指定孤児薬を申請しなければならない。FDAが孤児薬物指定を承認した後、FDAは、治療薬の識別情報およびその潜在的な孤児用途を開示するであろう。この投稿は、薬物が孤児薬としてもはや指定されていないかどうかを示す。最近の法廷事件は,FDAが孤児薬物の排他的範囲を決定する方法に挑戦しているが,現在,同機関は管理条例の長期的な解釈を適用し続けており,孤児薬物施行条例を変更するつもりはないことを示している。議会はまた未来のある時点でこの分野の法律を修正するために行動するかもしれない。
1つ以上の候補製品は同一の適応の孤児薬物指定を得ることができ、同一の候補製品は1つ以上の合格孤児適応を指定することができる。孤児薬を指定するメリットは、税収控除の開発とFDA処方薬使用料の免除を含む。残留孤児薬物指定は、候補製品の機密協定が提出された場合や、規制審査および承認過程においていかなる利点も伝達されず、規制審査および承認過程の継続時間を短縮することもない。
孤児薬物の称号を有する製品がその後、その称号を有する適応に対するFDAの最初の承認を得た場合、この製品は、孤児製品の排他性を得る権利があり、これは、7年以内に、FDAが、以下にさらに説明する限られた場合を除いて、同じ薬物の同じ適応の任意の他のマーケティング申請を承認することができないことを意味する。孤児の排他性は異なる薬物の同じ稀な疾病或いは疾病に対する承認を妨げることはなく、同じ薬物の異なる疾病に対する承認を阻止することもない。したがって、FDAは依然として異なる薬物を同じ適応または疾患の治療に使用することを許可することができる。また,孤児製品に指定された薬物が市販承認された場合,その適応範囲は指定されたものよりも広く,孤児薬物排他性を得る資格がない可能性がある。
場合によっては、同じ薬物を使用する別の製品は、同じ薬物を使用して同じ疾患を治療する後続製品が、承認された製品よりも臨床的に有効または安全であることが証明された場合、または患者ケアに重大な貢献をした場合、または孤児薬物排他性を有する会社が、指定された薬剤によって対象とされる疾患または状態の患者の需要を満たすのに十分な量の薬剤を保証することができない場合を含む、同じ場合には許可されない。FDAは現在、臨床優勢を証明した上で、1種の薬物が孤児製品の独占特許を獲得する資格がある時、臨床優勢発見要約を発表することを要求されている。
さらに、FDAは、一般的な疾患の小児科亜群の製品に追加の孤児薬物名を付与しないことが予想されることを示すガイドラインを決定した。しかし、FDAは、(I)まれな小児科亜群を含む稀な疾患、(Ii)有効な孤児亜群を構成する小児科亜群、または(Iii)有効な孤児亜群を構成する稀な疾患、または(Iii)実際に小児科群の中で成人群とは異なる疾患である場合に、すべての他の指定基準に適合する薬物を孤児薬物指定に付与することを意図している。
特許期間を延長する
FDCAによれば、FDAの承認を得た処方薬の特許は、特定の法定および規制要件を満たす場合に、製品開発およびFDA規制審査中に失われた特許期間を5年間にわたって回復することを可能にする限られた特許期間を延長する資格がある可能性があると主張されている。特許期間延長の長さは,特許発効期間中に薬物が規制審査を受ける時間の長さと関係がある。FDA規制をカバーする新医療製品の特許付与の回復期は、通常、人体臨床研究を開始する日と製品発売前承認申請提出日との間の半分の時間であり、製品承認申請提出日と最終承認日との間の時間を加える。特許期間回復は特許の残存期間の延長には利用できず,製品承認日から合計14年を超える。承認された薬品に適用される特許は1つのみ延期する資格があり,延期出願は関連特許が満了する前に提出しなければならない。複数の製品をカバーする特許は、そのうちの1つが上場承認された場合にのみ延期される。米国特許商標局は,FDAと協議した後,任意の特許期間の延長または回復の出願を審査·承認する。
小児科排他性
小児科専門権は、米国が提供する別の非特許マーケティング専門権であり、承認された場合、任意の既存の規制専用権または上場特許の期間に追加の6ヶ月の市場保護を追加することを規定する。これは特許期間の延長ではないが、FDAが別の出願を承認できない規制期間を効果的に延長する。
“小児ベストプラクティス法”(BPCA)によれば、スポンサーがFDAによって要求された候補製品の活性部分の子供における使用に関する情報を提出した場合、いくつかの候補治療薬は、追加6ヶ月の排他性を得ることができる。これらのデータは,この製品が研究されている小児科群で有効であることを示す必要はなく,逆に小児科臨床試験がFDAの書面要求に公平な対応をしていると考えられていれば,追加的な保護を与える。FDAは、適応の承認または承認されていない研究に関する書面請求を発行することができるが、小児科集団または一部の小児科集団における候補製品の使用に関連する情報がその集団に健康利益をもたらす可能性があると判断した場合にのみ、そうすることができる。書面出願の発出は主催者に述べた裁判を要求しない。
アメリカの他の医療法律法規
FDA以外にも、製品承認後の製造、販売、販売促進、その他の活動も米国の他の規制機関によって規制される可能性がある。製品の性質によると、これらの機関は、医療保険および医療補助サービスセンター(CMS)、衛生·公衆サービス部(HHS)の他の部門、米国司法省、連邦貿易委員会(FTC)、薬品監督管理局、職業安全·健康管理局、および州および地方政府を含むことができる。
例えば、米国では、処方薬バイオ製薬製品の販売やマーケティングは州や連邦の詐欺や乱用法に従わなければならない。これらの法律には、処方薬製造業者(またはそれを代表する側)を含む任意の人が、インフォームドコンセントおよび意図的な場合には、連邦医療保険または医療補助などの連邦医療計画に基づいて支払われる可能性がある代替薬の購入、推薦、注文または処方を含む任意の報酬を請求、受け入れ、提供、または支払いすることが規定されている連邦反リベート法規が含まれている。この法律に違反した行為は、監禁、刑事罰金、行政民事罰金、連邦医療計画から除外された罰を受けるだろう。また,“患者保護と平価医療法案”(ACA)などは連邦“反リベート法規”(AKS)の意図要求と,“健康保険流通と責任法案”(HIPAA)により制定された5つの医療詐欺刑事法規の2部を改正した。個人または実体は、法規内のこの2つの規定またはそれらの具体的な意図を実際に理解する必要はなく、特に、任意の医療福祉計画の金銭または財産を詐欺的または詐欺的に獲得しようとする計画または詐欺の実行を禁止または実行しようとする計画またはトリックに関して、個人が医療補助を受ける資格があるように資産を処分することを禁止する。また,政府は現在,“虚偽請求法”により,連邦AKS違反による物品やサービスのクレームが虚偽や詐欺的クレームを構成していると断言できる。
定価と返却計画は,米国の1990年の“総合予算調節法”の医療補助帰点要求およびACAの最近の要求に適合しなければならない。総務省連邦供給スケジュールの許可されたユーザに製品を提供する場合は、他の法律および要求が適用される。“医師支払い陽光法案”にも連邦透明性要件があり、連邦医療保険または医療補助カバーのFDA承認を要求する薬物、設備、生物製品、および医療用品の製造業者は、医師、教育病院およびいくつかの高度な非医師医療保健への支払いおよび他の価値移転に関する情報をCMSに毎年報告する
事業者と医師の所有権と投資権益。処方薬製品はまたアメリカの“毒物予防包装法”に適用される児童保護包装要求に符合しなければならない
製造、販売、販売促進、その他の活動はまた、連邦と州消費者保護および不正競争法によって制限される可能性がある。いくつかの州の法律は、製薬会社が製薬業の自発的なコンプライアンスガイドライン、または連邦政府が公布した関連するコンプライアンスガイドラインを遵守することを要求し、また、これらの法律が適用される要求が“医師が日光を支払う法案”よりも厳しい場合、製薬業者に医師および他の医療保健提供者への支払いまたはマーケティング支出に関する情報を報告することを要求する。州、連邦、外国の法律は、“自由貿易法”を含み、場合によっては健康情報のプライバシーと安全を管理し、その多くは互いに大きく異なり、HIPAAに占領されず、コンプライアンス作業を複雑化させることが多い。
これらの法律または規制要件のいずれかを守らない場合、会社は可能な法律または規制行動に直面するだろう。状況に応じて、適用される規制要件に適合しないことは、刑事起訴、罰金またはその他の処罰、禁止、リコールの要求、製品の差し押さえ、生産の完全または部分的な一時停止、製品の承認の拒否または撤回、または政府契約を含む会社の供給契約の締結の許可を拒否する可能性がある。
アメリカ以外の政府規制
米国の法規に加えて、臨床試験および私たちの製品の任意の商業販売と流通を含む様々な外国法規の制約を受け、承認されれば、直接または流通パートナーを通過することができる。FDAの候補製品の承認を得るか否かにかかわらず、これらの国や地域で臨床試験や製品の販売を開始することができる前に、外国や経済地域(EU、カナダ、イギリスなど)の規制機関の必要な承認を受けなければならない。外国規制承認プロセスには、上述したFDA承認に関連するすべてのリスクが含まれており、他の国や管轄区域で承認を得るのに要する時間は、FDA承認を得るのに要する時間とは異なり、FDA承認を得るのに要する時間よりも長い可能性がある。一部の外国司法管轄区の薬物製品の審査手続きはアメリカと類似しており、臨床研究を開始する前に臨床試験申請を提出することを要求し、INDと非常に類似している。例えば,ヨーロッパでは,臨床試験申請(CTA)は各国の国家衛生当局と独立した倫理委員会に提出されなければならず,FDAやIRBのようなものである。CTAが一国の要求に応じて承認されると,臨床試験開発は継続可能である。EU規制制度によると、候補医薬製品の規制承認を得るためには、NDAと類似したマーケティング許可申請(MAA)の提出が求められるが、他の事項に加えて、特定の国に対する文書要求がある点で異なる。EU以外の国,例えば東欧,ラテンアメリカやアジアの国,最近のイギリスでは,臨床試験,製品承認,定価,精算の要求は国によって異なる。1つの国または管轄区域で規制承認を得ることは、他の国または管轄区域で規制承認を得ることを保証することはできないが、1つの国または管轄区域で規制承認を得ることができなかったか、または遅延して監督管理許可を得ることは、他の国または司法管轄区の規制手続きに悪影響を及ぼす可能性がある。また、一部の国は、彼らの国の承認を支持するために、米国の承認を得るための臨床研究を受け入れないか、あるいは彼らの国の地元の人に追加的な研究を要求する可能性がある。また、ある海外市場では、薬品の定価は政府によってコントロールされており、場合によっては、不足していることや精算が不足している可能性がある。もし私たちが適用された外国監督管理要求を遵守できなかった場合、私たちは罰金、規制許可の一時停止または撤回、製品のリコール、製品の差し押さえ、経営制限、刑事起訴などの処罰を受ける可能性がある
2020年1月31日から、イギリスはもはやEU加盟国ではないため、イギリスで医薬製品を販売するには単独のマーケティング許可申請と承認が必要である。薬品と保健製品監督機関(MHRA)はイギリスの独立した薬品監督機関である。
ヨーロッパの臨床試験と薬品規制
アメリカと同じように、医薬製品は監督管理機関のマーケティング許可を得た後にのみEUで販売することができる。アメリカと類似して、EUの臨床前と臨床研究の各段階は重要な監督管理によって制御されている
欧州臨床試験指令によると、EUはすでに加盟国の国家立法を通じて臨床試験承認制度を実施した。この制度によれば,申請者は臨床試験を行うEU加盟国の主管国当局の承認を得なければならない。また,出願人は主管倫理委員会が賛成の意見を発表した後にのみ臨床試験を開始することができる。臨床試験申請はヨーロッパ臨床試験指令と加盟国の相応国家法律で規定されている支持情報を持つ研究用薬品ファイルを添付し、適用された指導文書の中でさらに詳細に説明しなければならない。2014年4月,新たな臨床試験条例(EU)第536/2014号(“臨床試験条例”)が可決され,2022年1月31日に施行された。臨床試験条例はすべてのEU加盟国に直接適用され、廃止された
先の臨床試験指令2001/20/EC。現在行われている臨床試験は“臨床試験規例”の管限を受ける程度は、個別の臨床試験の持続時間に依存する;1つの臨床試験が“臨床試験規則例”の適用日から3年以上持続すれば、その時“臨床試験規則例”はこの臨床試験に適用される。また,2023年2月1日から臨床試験情報システム(CTIS)により実施された新たなEU範囲内の申請プログラムは,新たな臨床試験申請を提出する強制的なプログラムとなっている。
新しい臨床試験条例はEUの臨床試験の審査を簡略化と簡素化することを目的としている。この条例の主な特徴は、単一入口点による申請手続きの簡略化、申請のための単一文書の準備と提出、および臨床試験発起人の報告手続きの簡略化、臨床試験申請を評価する統一的な手続きを含む
EUでの薬物の発売承認を得るためには,申請者は集中的または分散した手順でMAAを提出しなければならない。集中化手続きは、すべてのEU加盟国、アイスランド、リヒテンシュタイン、ノルウェーに対する有効な単一マーケティング許可を欧州委員会によって付与することを規定している。特定の製品には、特定のバイオテクノロジーによって生産された医薬品、孤児医薬品として指定された製品、高度な治療製品(例えば、遺伝子療法、体細胞療法または組織工学薬)、および特定の疾患を治療するための新しい活性物質を含む製品が含まれ、集中手順は強制的である。特定の疾患を治療するための新しい活性物質を含む製品、および患者の利益に有利な高度な革新性または集中処理を有する製品については、集中処理がオプションである可能性がある。中央手続きによると、欧州薬品管理局(EMA)がMAAを評価する最長期限は210日であり、タイマーを含まず、申請者は人用薬品委員会(CHMP)からの質問に答えるために追加の書面または口頭情報を提供する。CHMPは特殊な状況下で加速評価を承認することができ、特に治療革新の角度から見ると、1種の医薬製品が重大な公衆衛生利益を有することが予想される場合。加速評価プログラムによる重大な影響評価の期限は150日であり,停止クロックは含まれていない.
特定のEU加盟国で製品を販売することを希望する申請者は、分散されたプログラムを使用することができ、これらの国の製品は、以前にどのEU加盟国でもマーケティング承認を受けたことがない。分散手続きは,出願人が1つの加盟国に評価申請(加盟国参照)を申請し,承認を希望する他の加盟国(関連加盟国)を具体的に列挙することができると規定している
EUでは、マーケティングの許可を得た製品だけが販売促進を行うことができる。マーケティング許可は原則として5年間有効であり、マーケティング許可は5年後に、EMAまたはライセンス加盟国の主管当局によるリスク-収益バランスの再評価に基づいて更新することができる。そのため、上場授権書所有者は上場授権書が失効する少なくとも6ヶ月前に、マーケティング授権書を付与してから導入されたすべての変化を含むEMA或いは主管当局に品質、安全性と有効性に関する文書の総合バージョンを提供しなければならない。一旦更新されると、上場許可の有効期限は無期限であり、欧州委員会または主管当局が薬物警戒に関する正当な理由に基づいて5年間の継続を決定しない限り、5年間の継続が決定される。いかなる認可後も、認可が失効した後3年以内に薬品は実際にEU市場(集中手続きであれば)または許可会員国の市場に投入されていない(いわゆる日没条項)。
また、EUでの販売が許可されても、処方薬は一般公衆ではなく、医療専門家にしか普及できない。すべての販促活動は、製品特性の要約に記載されている詳細に従って行われなければなりません。宣伝材料はまた、EUの製薬業界機関が制定した様々な法律および行為規則に適合しなければならない。これらの法律および行為規則管理(他の事項を除く)の販売者の訓練、販売促進主張およびその理由、比較広告、誤った広告、裏書き、および(許可された場合)公衆広告。このような要求を守らないことはEU加盟国の主管当局に処罰を与える可能性がある。処罰には、警告、その薬品の宣伝停止、宣伝材料の没収、罰金、可能な監禁が含まれる可能性がある。
2023年4月、欧州委員会は既存の一般薬品立法を改正し、代替する提案を発表した。現在の提案に従って採択され、実施されれば、これらの改正はEUの薬物開発と承認のいくつかの側面を大きく変えるだろう。
イギリスの新薬に対する規制
英国は2020年1月31日にEU(通称“離脱”)を離脱し、過渡期は2020年12月31日に満了する。英国とEUは2021年1月1日に発効する“貿易·協力協定”という貿易協定を締結した。我々は現在,貿易·協力協定が我々の業務に与える潜在的な影響と,英国MHRAがこれまでに発表してきたイギリスでの医薬品の許可やマーケティング要求に関する指導意見を評価している
イギリスの医薬製品に対する監督管理枠組みは医薬製品の品質、安全性と有効性、臨床試験、マーケティング許可、商業販売と流通をカバーしているため、EUからの指令と法規により、イギリスの離脱は将来この種類の製品に適用される監督管理制度とイギリスが候補製品の承認に重大な影響を与える可能性がある。このような結果は、私たちがヨーロッパで業務を展開することをより困難で高価にし、私たちの臨床、製造、規制戦略を複雑化させ、私たちが規制承認を得て維持する能力を弱めることと、承認されれば、私たちの製品や候補製品がヨーロッパで商業化する能力を弱めるかもしれない。
最近、2023年3月、イギリス政府と欧州委員会は北アイルランド議定書、すなわちウィンザー枠組みの代わりに規制枠組みについて合意した。ウィンザー枠組みは2025年1月1日から適用される予定であり、英国の薬品規制を含む北アイルランド議定書下の既存制度を変更する。具体的には,MHRAはイギリス(すなわち大ブリテンおよび北アイルランド連合王国)で販売しようとしているすべての薬剤を承認することを担当し,EMAは北アイルランドで販売しようとしている薬剤の承認には参加しなくなる。
カナダの医薬製品の規制
カナダ衛生部はカナダ連邦当局であり、カナダ人が獲得できる薬物とその他の治療製品の安全性、有効性と品質を監督、評価、監視する。カナダ衛生部の製品の審査、承認、監督管理の規制手続きはFDAが行った規制手続きと類似している。カナダのヒト被験者で候補製品の臨床試験を開始するためには,カナダ衛生部に提出し,CTAを承認しなければならない。しかも、すべての連邦規制の実験は研究倫理委員会の承認と監視を受けなければならない。審査委員会は研究に関連する文書を研究·承認し、試験データを監視する。
薬品市場の許可を得る前に、メーカーは“食品と薬品法”(カナダ)及びその関連法規(“食品と薬物条例”を含む)の要求に基づいて、製品の安全性、有効性と品質の実質的な科学的証拠を提出しなければならない。これらの情報は通常新薬提出(NDS)の形で提出される。カナダ保健省は提出された情報を審査し,外部コンサルタントや諮問委員会を用いて薬物の潜在的なメリットやリスクを評価することがある。審査終了後に患者の利益が薬物関連のリスクを超えていると結論した場合、この薬物に薬品識別番号(DIN)を発行し、その後、市場許可保持者(すなわちNOCとDIN所持者)がカナダでこの薬物を販売することを許可するコンプライアンス通知(NOC)を発行する。国家薬品監督管理局に授与された薬品は追加の発売後のモニタリングと報告要求を受ける必要があるかもしれない。
製造、包装/ラベル、輸入、流通と卸売薬品及び薬品に関連する検査実験室を経営するすべての機関は、“食品と薬物条例”が明確に免除されない限り、“薬品経営許可証”を持っていなければ1つ以上の許可活動に従事することができない。薬品経営許可証を発行する基礎は,この施設が“食品·薬品条例”に規定されているcGMPに適合することを確保し,カナダ衛生部がcGMP検査を行うことである。外国で生産された薬品の輸入業者は,外国地点がcGMPに適合していることを証明できなければならず,これらの外国地点は輸入業者の薬品経営許可証に含まれている。
薬品が最初に市場の承認を得た後、規制義務と監督は続いている。例えば、各市場許可保持者は、カナダで発生した深刻な薬物副作用およびカナダ国外で発生した任意の深刻な意外薬物副作用をタイムリーに報告することを含む、受信した任意の薬物副作用に関する新しい情報を報告しなければならない。発売許可保持者はまた,製品発売後に発見された任意の新たな安全性と有効性をカナダ衛生部に通報しなければならない。
薬品のカバー、定価と精算と医療改革
我々の製品の販売は、上場が許可されれば、連邦医療保険と医療補助、商業保険と管理の医療保健組織を含む政府医療計画のような第三者支払者のカバー範囲と精算範囲にある程度依存する。これらの第三者決済者は価格に挑戦し、医療製品やサービスのカバー範囲や精算金額を制限することが増えている。承認された製品の保険や精算に重大な遅延がある可能性があり、保険範囲はFDAや他の国·地域規制機関がこの製品を承認する目的よりも限られている可能性がある。第三者決済者に補償を求めるのは時間がかかって高い。さらに、精算を受ける資格があるということは、どの製品もすべての場合に支払われることを意味するわけではありません。または支払いの費用率は、研究、開発、製造、販売、流通を含む私たちのコストをカバーします。新製品の仮払い(適用すれば)も私たちのコストを支払うのに十分ではない可能性があり、恒久的な支払いにならない可能性があります。支払率は、製品の使用や臨床環境によって異なる可能性があり、精算された低コスト製品によって許容される支払いに基づく可能性があり、他のサービスの既存の支払いに組み込まれる可能性もある。製品の正味価格は第三者支払者が要求する強制的な割引或いはリベートと未来の法律のいかなる緩和によって低下する可能性があります
現在、米国より低い価格で販売される可能性のある国からの製品の輸入を制限している。米国では、第三者支払者が自分の精算政策を設定する際には通常Medicare保証政策と支払い制限に依存しているが、Medicare保証範囲と精算確定以外にも、彼らは独自の方法と承認の流れを持っている。したがって、第三者支払者は、1つの商品に保険を提供することを決定し、他の支払者もその商品に保険を提供することを保証することはできない。
また,医療コストの抑制は連邦と州政府の優先順位となっており,薬品価格はこの努力の重点となってきた。アメリカ政府、州立法機関と外国政府はコスト制御計画の実施に深い興味を示し、これらの計画は価格制御、カバー範囲と精算に対する制限及び模造薬代替に対する要求を含む。価格制御とコスト制御措置、および既存の制御·措置を講じている司法管轄区域でより制限的な政策をとることで、我々の純収入と業績をさらに制限することができる。私たちの候補製品の第三者精算減少または第三者支払者は、私たちの候補製品をカバーしないことを決定することは、医師の候補製品への使用を減少させ、私たちの販売、運営結果、財務状況に実質的な悪影響を与える可能性があります。また、政府はメーカーがその販売する製品に価格を設定する方式に対してより厳格な審査を行い、国会で数回の調査を行い、連邦と州立法を提出し、製品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、政府の薬品の精算方法を改革することを提出し、公布した。米国の個別州も、価格または患者の精算制限、割引、ある製品への参入の制限、マーケティングコストの開示および透明性措置を含む立法および実施により、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。2020年12月、アメリカ最高裁判所は、連邦法律は各州の監督薬局福祉マネージャー(PBM)と医療保健と薬品サプライチェーンの他のメンバーの能力を妨害しないと一致し、この重要な決定は各州がこの領域で更なるかつ積極的な努力をすることを招いた。連邦貿易委員会は2022年中にPBM業界のやり方に対して全面的な調査を展開し、これはこのような実体の運営、薬局ネットワーク或いは財務手配に対するより多くの連邦と州立法或いは監督管理提案を招く可能性がある。実際、米国議会や州立法機関は、業界を検討し、様々な公共政策の懸念を解決するための新たな規制方法を提案している。例えば、今国会の会議期間中、衆参両院は多くのPBM改革を考慮している;その中にはリベートの廃止などの様々な立法提案が含まれている;サービス料を薬品、割引またはリベートの価格から分離する;価格差の定価を禁止する;行政費用を制限する;PBMに処方配置理由を報告することを要求する;透明性を促進する。アメリカの既存のPBM業界を変える重大な努力は薬品サプライチェーンと他の利益関係者の業務に影響を与える可能性があり、私たちのような生物製薬製品開発業者を含む。
また、2022年8月、総裁·バイデンは“2022年インフレ降下法案”(IRA)に署名し、法律にした。他の点では、アイルランド共和軍には複数の条項があり、連邦医療保険計画や米国全体に販売されている薬品の価格に影響を与える可能性がある。連邦医療保険BやDの部分にカバーされている薬品メーカーは現在連邦政府にリベートを支払わなければならず,その薬品の価格上昇速度がインフレ率よりも速い場合。この計算は薬品に基づいて行われており、連邦政府に不足している税金還付額は、連邦医療保険B部分またはD部分で支払われている薬物製品の数に直接依存する。また、CMSは2026支払い年度から毎年、模造薬や生物類似競争を含まずに、選択された数量の単一源D部分薬物について薬品価格交渉を行う。CMSはまた,選定数のB部分薬の薬品価格を2028年から交渉する。CMSが1つの薬物を選択して交渉すれば,このような薬物による収入は減少すると予想される。CMSはこれらの新たなライセンスの実施を開始し,2023年10月に製薬業者と合意し,価格交渉を行っている。しかし、この計画のアメリカ生物製薬業界への影響はまだ確定しておらず、一部の原因は複数の大手製薬会社と他の利害関係者(例えばアメリカ商会)がCMSに対して連邦訴訟を提起しており、この計画は様々な原因で違憲、その他の苦情であると述べている。このような訴訟は現在も進行中だ。
また、一部の外国の国では、薬品の提案価格は必ず承認されなければならず、合法的に発売されることができる。各国の薬品定価に対する要求は大きく異なる。例えば、EUでは、EUレベルで薬品の定価と精算を管理する唯一の法的文書は、理事会第89/105/EEC号指令(価格透明性指令)である。価格透明性指令の目的は、EU加盟国で確立された価格設定と補償メカニズムが透明で客観的であり、EU医薬製品の自由な流動と貿易を阻害せず、市場競争を阻害、防止、または歪曲しないことを確保することである。“価格透明性指令”は、個別のEU加盟国の価格設定および補償決定に基づく具体的な基準についていかなる指導も提供しておらず、個別EU加盟国の定価や補償レベルに直接的な影響を与えていない。EU加盟国は、その国の健康保険制度が精算を提供する医療製品の範囲を自由に制限し、価格および/または
人用薬品の精算水準。EU加盟国は、医薬製品の具体的な価格または精算レベルを承認することができ、あるいは医薬製品を市場に投入する責任を負う会社の収益力に対して、数量に基づく手配、上限、参考定価メカニズムを含む直接または間接的な制御制度をとることができる
いくつかのEU加盟国では、フランス、ドイツ、アイルランド、イタリア、スウェーデンを含め、医療製品の衛生技術評価(HTA)が定価や精算手続きでますます一般的な部分となっている。EU加盟国の技術援助手続きはこのような国の国家法律によって管轄されている。HTAは,特定の医療製品が個別の国の国家ヘルスケアシステムで使用されている公共健康影響,治療影響および経済的·社会的影響を評価するプログラムである。HTAは通常、個別医薬製品の臨床治療効果および有効性、安全性、コストおよび費用効果、および医療システムに対するそれらの潜在的な影響に重点を置いている。医療製品のこれらの要素を市場で提供されている他の治療案と比較した。特定の医薬製品に関するHTAの結果は、EU個別加盟国主管当局がこれらの医薬製品の価格設定と補償地位を与えることに影響を与えることが多い。EU加盟国間の具体的な医薬製品のHTAが定価と補償決定に与える影響の程度はそれぞれ異なる。例えば、HTA機構が制定されていないEU加盟国は、ある特定の医薬製品の定価および補償に関する決定によってHTAフレームワークが発達した国が行うHTAにある程度依存することができる
コスト抑制の努力に加えて、米国や一部の外国司法管轄地域でも、医療システムに関するいくつかの立法·規制改革、提案中の改革が継続されており、これらの改革は、候補製品の上場承認を阻止または延期したり、承認後の活動を制限したりする可能性がある。例えば、2023年4月、欧州委員会は既存の一般薬品立法を改正し、代替する新しい指令と新法規に関する提案を発表した。現在の提案に従って採択され、実施されれば、これらの改正はEUの薬物開発と承認のいくつかの側面を大きく変えるだろう。FDAおよび他の規制機関の政策は変わる可能性があり、私たちの現在または未来の候補製品に対する規制承認を阻止、制限、または延期するために追加の政府法規が公布されるかもしれない
データプライバシーと個人情報保護
私たちは、データプライバシーを管理し、個人情報(健康情報を含む)を保護する法律および法規によって制限されている。プライバシーとデータ保護の立法と規制構造は発展し続け、プライバシーやデータ保護問題にますます注目されており、これらの問題は引き続き私たちの業務に影響を与える。米国では、私たちは州セキュリティホール通知法、健康および個人情報のプライバシーを保護する州法律、ならびに個人情報の収集、使用、開示、および伝送を規制する連邦および州消費者保護法の制約を受ける可能性がある。これらの法律は互いに重なり、往々にして衝突し、すべての法律は裁判所と政府機関の異なる解釈を受け、複雑なコンプライアンス問題をもたらしている。もし私たちが適用された法律と法規を守らなければ、私たちは刑事罰を含めて処罰や制裁を受けるかもしれない。当社のお客様と研究パートナーは、HIPAAや州健康情報プライバシー法など、健康情報のプライバシーやセキュリティを管理する法律を遵守しなければなりません。もし私たちが知らずにHIPAAによって保護された健康情報を取得することを“保護された健康情報”と呼ぶ場合、私たちの顧客や研究協力者は強制的に実行される可能性があり、私たちは保護された健康情報を不正に受信したり、協力したり、HIPAA違反をそそのかしたりすることで直接責任を負う可能性がある。
健康と個人情報を保護する州法はますます厳しくなっている。例えば、“カリフォルニア医療情報秘密法”は健康情報と他の個人身分情報の使用と開示に対して制限的な要求を提出した。カリフォルニア消費者プライバシー法(CCPA)は、以下に説明する一般的なデータ保護条例(GDPR)のいくつかの重要な条項を反映する。CCPAは個人情報の定義を拡大することによって、カリフォルニア州の消費者のための新しいデータプライバシー権を構築し、未成年者から消費者データを収集するための特殊な規則を実施し、CCPAに違反し、合理的なセキュリティ手続きとやり方を実施できなかった企業のために新しい、深刻な可能性のある法定損害賠償枠組みを作成し、それによってカバーする企業のために新しいプライバシー枠組みを構築する。カリフォルニア消費者権益法案(CPRA)は2023年1月1日に発効し、CCPAの内容を強化した。CCPAが採択されて以来、他のいくつかの州(例えば、コネチカット州、コロラド州、バージニア州、デラウェア州、フロリダ州、アイオワ州、モンタナ州、オレゴン州、テネシー州、テキサス州、ユタ州)もカリフォルニア州の法律との大きな違いを含む包括的な消費者プライバシー法を公布し、業界や他の利益関係者のコンプライアンスをさらに複雑化させている。米国の他の州でもCCPAのようなプライバシー法が考えられている。
欧州では,GDPRが2018年5月に施行され,臨床試験データを含むEUデータ保護法の範囲を拡大し,EU域内個人に関する個人データを処理または制御処理する非EUエンティティが実施された。GDPRは守らなければならない要求を規定している
EUに設置された資料当事者の個人資料を処理する時、これらの資料は:より多くのその個人資料がどのように使用されるかに関する開示資料を提供する;組織はそれが有効な同意を得たか、あるいはすでに他の法律根拠がその資料処理活動の合理性を証明したことをより高い基準で証明しなければならない;ある場合、資料保護者を委任する義務がある;新しい個人の“忘れられた”権利と資料携帯権、及び既存の権利(例えば閲覧要求)を強化しなければならない;責任の原則を強化し、政策、プログラム、訓練と監査を通じてコンプライアンスを証明する;及び新しい強制資料漏洩制度を含む。特に,医療や健康データ,遺伝子データ,バイオメトリクスデータは1人を一意に識別するために用いられ,これらのデータはいずれもGDPR下の“特殊クラス”データに分類され,より大きな保護を受けており,追加的なコンプライアンス義務が必要である。さらに、EU加盟国は制限を含むこのようなデータカテゴリに追加的な条件を適用する広い権利を持っている。これは、GDPRが、EU加盟国が主に特定の処理状況(特殊なカテゴリデータおよび科学的または統計的目的の処理を含む)においてGDPRの要求を欠陥させることを可能にするためである。EU諸国がGDPRと協調するために自国の立法を再制定することに伴い、GDPRの削減の導入が許可された場合を含め、EU加盟国のすべての法律·法規の遵守状況を監視する必要がある。GDPRはまた,EUからEU以外の国への個人データの移行を禁止しており,欧州委員会が十分なデータプライバシー法を持つと考えている国への移行や,承認されたデータ転送機構による移行を行わない限りである.2020年7月16日、欧州連合裁判所(CJEU)は、Schrems IIというマークシミリアン·シュレムス氏がFacebook(C-311/18)を訴えた事件に対して、Schrems IIという記念碑的な意見を発表した。この判決は、(A)通常、EU加盟国と米国との間のデータ転送メカニズム(例えば、標準契約条項)に依存し、(B)EU-米国のプライバシー盾を無効にし、多くの会社がこのメカニズムに依存してこのようなデータをEUから米国に移したことがある
2023年7月10日、欧州委員会は、EUから米国にデータを移転する新しいメカニズムであるEU-米国データプライバシー枠組み(略称フレームワーク)に関する十分性決定を採択した。このフレームワークは、EUの個人に、そのデータにアクセスする権利を取得する権利、または誤ったまたは不正に処理されたデータの訂正または削除を取得する権利を含むいくつかの新しい権利を提供する。十分性決定は,Schrems II決定で提案されたいくつかの点を解決するために,新たな拘束力のある保障措置を導入した行政命令に署名した後に行われる.新たな義務は、米国の情報機関が必要かつ適切な範囲でしかデータを取得できないことを確保し、国家安全目的のためのデータ収集に関する欧州人の苦情を処理するための独立かつ公正な救済メカニズムを構築することを目的としていることに留意されたい。欧州委員会は十分な決定を下しながら、米国の事態を検討していくだろう。事態が適用法域の保護レベルに影響すれば,十分な決定を調整あるいは撤回することができる.連合データ保護当局の未来の行動は予測が難しい。一部の顧客または他のサービスプロバイダは、これらの変化する法律および法規に応答して、私たちができない、またはしたくないいくつかのプライバシーまたはデータに関連する契約約束をすることを要求するかもしれません。これは既存または潜在的な顧客または他のビジネス関係を失うことになる可能性がある。
これに関連して,イギリスの離脱とイギリスの離脱移行期間が2020年12月31日に終了することに伴い,EU GDPRはイギリス(イギリスGDPRと略す)で実施されている。英国GDPRは2018年の英国データ保護法と並んで、EU GDPRのいくつかの削減措置を英国法に盛り込んでいる。イギリスGDPRによると、イギリスでは設立されていないが、イギリスで個人への商品やサービスの提供に関する個人データを処理したり、その行動を監視したりする会社は、イギリスのGDPRの制約を受ける--その要求(現在)はGDPRでの要求とほぼ一致するため、類似のコンプライアンスおよび運営コストを招き、潜在的な罰金は1,750万ポンドまたは世界売上高の4%に達する可能性がある。
アメリカの“海外腐敗防止法”
全体的に、1977年に改正された“反海外腐敗法”(FCPA)は、外国人官僚の公的な身分で行われた任意の行為または決定に影響を与えるため、またはいかなる人または誰との業務を維持し、または誰または誰との業務を維持し、または業務を誰に導くために、任意の他の不正な利益を確保するために、外国人役人への支払い、支払い、約束または許可のための金銭または任意の価値のあるものの支払いを禁止する。これらの禁止は、“任意の外国人官僚”に支払われる金だけでなく、“任意の外国政党またはその役人”、“任意の外国政治職候補者”または任意の人に支払われる金にも適用され、上記のいずれかのカテゴリの誰にも提供、与え、または約束されることを知っている。“海外腐敗防止法”によると、“外国役人”には、外国政府部門、機関又は機関の役人又は従業員が含まれる。“ツール性”という言葉は広く、国有または国家統制の実体を含むことができる。重要なのは、米国当局は、公共医療および/または公共教育システムを有する国では、医療専門家の多くと外国の病院、診療所、研究機関、医学院の他の従業員が“海外腐敗防止法”の下の“外国人役人”であると考えている。私たちが海外で私たちの製品をテストして販売する時、私たちが外国の医療専門家や研究者と交流する時、もし私たちの任意の候補製品が将来外国の監督機関の許可を得たら、私たちと私たちを代表する代理人が私たちの製品やサービスを販売したり、必要な許可と承認を得ることに関する過度または贅沢な飲食、旅行、娯楽を含む任意の賄賂、プレゼントやチップを提供することを防止するために十分な政策と手続きを制定しなければならない。FCPAはまた、その証券がアメリカに上場する会社に会計条項を遵守することを要求して、すべての取引の帳簿と記録を正確かつ公平に反映することを要求します
管理会社は、国際子会社を含み、国際業務のために適切な内部会計制御制度を策定·維持する。
環境、健康、安全規制
私たちは多くの連邦、州と地方の環境、健康と安全(EHS)の法律法規の制約を受けて、これらの法律法規は安全作業条件、製品管理、環境保護と製品の処理或いは処分に関連し、私たちの共同研究実験室で処理する可能性のある危険または潜在的な危険材料、医療廃棄物と伝染病材料の発生、貯蔵、処理、使用、輸送、放出と処置を管理することを含む。その中のいくつかの法律はまた私たちが私たちの業務を行うために許可または許可を得ることを要求する。このような法律を遵守できなかったり、適用された許可証を取得して遵守できなかった場合、巨額の罰金に直面したり、許可証が取り消される可能性があり、あるいは私たちが業務を展開する能力が制限される可能性があります。私たちのいくつかの開発と製造活動は時々危険材料の使用に関連するかもしれません。私たちは適用される環境法律、法規、許可証、許可証を遵守していると信じています。しかし、私たちはEHS債務が未来に発展しないことを確実にすることができない。EHS法律法規は複雑で変化が頻繁であり,時間の経過とともにより厳しくなることが多い。適用される法律·法規を遵守するコストは重要ではないにもかかわらず、新たな法律や改正された法律や法規が私たちの業務に与える影響、あるいは既存および将来の法律·法規の解釈や実行方法のいかなる変化も予測できず、必要なライセンスや許可を取得または維持できることを保証することはできない。
人的資本資源
GRIは2024年3月1日現在4人の従業員を持ち,いずれも常勤社員である。私たちは、私たちの現在と未来の従業員とコンサルタントの知的資本が私たちの業務の重要な推進力であり、私たちの将来の見通しの鍵でもあると信じている。
GRIの企業情報
Vallon PharmPharmticals,Inc.(Vallon)は2018年1月にデラウェア州法律に基づいて成立し、2018年6月に組織、創立と初期資本化活動を完成した。GRI Bio Operations,Inc.(前身はGRI Bio,Inc.)は2009年5月にデラウェア州法律に基づいて登録成立し、名称はGlycoregity,Inc.であり、そして2015年7月29日に会社登録証明書を修正し、GRI Bio,Inc.と改名した。
2023年4月21日、2023年2月17日に改訂された2022年12月13日に発効した合併協定と計画(合併協定)に基づき、Vallon、GRI OperationsとVallon Merge Sub,Inc.(当社のデラウェア州会社と完全子会社Vallon Merge Sub,Inc.)の間で合併し、Merge SubはGRIと合併してGRI(合併)に組み込まれ、GRIは自社の完全子会社として合併後も存在する。合併については、合併発効時期(発効時期)に先立ち、当社は30株1株の割合で会社普通株を逆株式分割(4月の逆分割)を行った。また,合併の終了に伴い,同社は会社登録証明書と定款を修正し,その名称を“Vallon PharmPharmticals,Inc.”から“Vallon PharmPharmticals,Inc.”に変更した。GRI Bio,Inc.へ
私たちの主な実行事務所はカリフォルニア州ラホア92037ラホア208号Avenida de La Playa 2223にあります
利用可能な情報
我々は,改正された1934年証券取引法(取引法)に基づき,我々の年間報告書(Form 10−K),四半期報告(Form 10−Q),現在のForm 8−K報告,依頼書と情報声明,その他の情報を米国証券取引委員会に提出した。アメリカ証券取引委員会のウェブサイトで私たちのアメリカ証券取引委員会の届出書類を読むことができます。
米国証券取引委員会には、米国証券取引委員会に電子的に提出された報告書、依頼書及び情報声明、その他の発行者に関する情報が含まれた相互接続サイトが設けられているHttp://www.sec.gov。
私たちのサイトの住所はWwwn.GriBio.comそれは.本年度報告に含まれる情報および本サイトを介してアクセス可能な情報は本年度報告に含まれず,本年度報告の一部にも属さない
新興成長型会社の地位
私たちは“新興成長型会社”で、2012年のJumpStart Our Business Startups Act(JOBS法案)の定義によると、5年に及ぶ間も新興成長型会社である可能性がある。私たちがまだ新興成長型企業である限り、私たちは、新興成長型企業に適用されず、他の上場企業に適用される特定の開示要求の免除に依存することを許可され、意図されている。これらの免除には
•私たちの役員報酬の開示を減らします
•役員報酬や黄金パラシュート手配について拘束力のない株主相談投票は行われなかった
•財務報告書に対する私たちの内部統制を評価する際に、監査人の認証要求を免除する。
私たちはこの報告書で削減された報告書の要求を利用して、私たちがもはや新興成長型会社ではなくなるまでそうし続けることができる。我々は、(A)本年度の総収入が12.35億ドル以上となるまで“新興成長型企業”とし、(B)2026年12月31日、IPO完了5周年後の本年度の最終日、(C)過去3年間で10億ドルを超える転換不能債券を発行した日、または(D)米国証券取引委員会規則に基づいて申告を加速した大手企業とみなす。JOBS法案第107条は,新興成長型会社は延長された過渡期間を利用して新たなまたは改正された会計基準を遵守することができると規定している
プロジェクト1 A.様々なリスク要因の評価
私たちの業務を評価する際には、以下に説明するリスクと、本年度報告書および私たちの他の公開文書に含まれる他の情報とをよく考慮しなければなりません。もし実際に以下のいかなるリスクが発生すれば、私たちの業務、財務状況、経営業績、未来の成長見通しは重大な不利な影響を受ける可能性がある。この場合、私たちの普通株の市場価格は下落するかもしれない。
私たちの財務状況と追加資本需要に関連するリスク
成立以来、重大な純損失が発生しており、予測可能な未来に重大な純損失が続くことが予想される。私たちは決して利益を上げないかもしれないし、決して利益を上げないかもしれない。
設立以来、私たちは重大な純損失が発生し、主に株式と債務融資を通じて私たちの運営に資金を提供してきた。私たちは私たちの持続的な運営に関連した巨額の研究開発と他の費用を発生させ続けている。2023年12月31日と2022年12月31日までの年度の純損失はそれぞれ1300万ドルと320万ドルだった。2023年12月31日現在、私たちの累計赤字は3150万ドルです。私たちはほとんどの資源と努力を研究と開発に投入しており、製品販売から収入を得るのにあと数年かかると予想される。1つ以上の候補製品のマーケティング承認を得て商業化しても、より多くの潜在的な候補製品を開発して販売するためには、大量の研究開発や他の費用が発生し続けると予想される。
私たちは予測可能な未来に大きな損失を被ると予想していますもし私たちの費用が大幅に増加すると予想しています
•私たちの主要候補製品GRI−0621と他の候補製品の臨床開発を推進し、成功すれば後期臨床試験も行われる
•新しい候補品を発見し開発しました
•私たちの臨床前開発計画を臨床開発に進めました
•さらに製造プロセスを開発し候補品を製造します
•流行病、サプライチェーンと労働力不足、労働スト、停止またはボイコット、自然災害と地政学的衝突(例えばウクライナと中東の紛争)、臨床前研究、臨床試験、私たちが依存している第三者サービスプロバイダから得たサービス、または私たちのサプライチェーンの遅延または中断
•臨床試験に成功した候補製品のために監督部門の承認を求める
•GRI−0621、我々の他の候補製品、および任意の将来の候補製品(承認された場合)を商業化すること;
•研究開発活動の数を増やして候補製品を決定し開発します
•追加の臨床開発、品質管理、科学、管理者を招聘する
•私たちの臨床開発と製造努力、上場企業としての私たちの運営を支援する人員を含む、私たちの運営、財務、管理システムを拡大し、人員を増加させる
•販売、マーケティング、医療事務、流通インフラを構築し、マーケティングの承認を得ることができ、単独または第三者と連携して商業化しようとしている任意の製品を商業化する
•私たちの知的財産権の組み合わせを維持し、拡大し、保護する
•他の技術または製品候補への投資または許可;
•以下にさらに説明するように、私たちは私たちの登録権協定の義務を履行することができない
•このような活動に従事するために私たちの組織を拡大し続けている。
利益を実現し、維持するためには、巨大な市場潜在力を持つ製品を開発し、最終的に商業化しなければならない。これは私たちが一連の挑戦的な活動の中で成功することを要求します。臨床前の研究と臨床試験を完成して、候補製品のマーケティング許可を得て、必要かもしれない製品を製造、マーケティングと販売することを含みます
上場承認を得て、任意の発売後の要求を満たす。私たちは決してこのような活動やすべての活動で成功しないかもしれないし、たとえ私たちが成功しても、私たちは利益を達成するのに十分な収入を得られないかもしれない。もし私たちが確実に利益を達成したら、私たちは四半期や年度の収益性を維持したり向上させることができないかもしれない。もし私たちが実現して利益を維持できなければ、会社の価値を下げ、資金を調達し、研究開発努力を維持し、業務を拡大し、あるいは運営を継続する能力を弱める可能性がある。
私たちは多くの追加資本が必要になるだろう。必要に応じて、または許容可能な条件下でそのような資金を集めることができない場合、私たちは、私たちの1つまたは複数の研究および薬物開発計画、将来の商業化努力、または他の操作の延期、減少、および/またはキャンセルを余儀なくされる可能性がある。
生物技術と生物製薬製品を開発することは、臨床前研究と臨床試験を含む、非常に時間がかかり、高価と不確定な過程であり、完成するのに数年かかる。設立以来、私たちの業務は大量の現金を消費した。我々が行っている活動に関連する費用が増加することが予想され,特にGRI−0621,GRI−0803,任意の他の候補製品を計画した臨床試験では,規制部門の承認を開発または求め,承認されれば発売され商業化される可能性がある。特に,追加資金を調達せずに臨床試験や開発を継続することはできないと予想される。私たちはまた上場企業の運営に関する追加コストが発生すると予想している。したがって、私たちは私たちの持続的な業務を維持するために多くの追加資金を得る必要があるだろう。もし私たちが必要な時や受け入れ可能な条件下で資金を集めることができなければ、私たちは私たちの1つまたは複数の研究および薬物開発計画または将来の商業化努力を延期、減少または廃止することを余儀なくされるかもしれない。
2023年12月31日現在、約180万ドルの現金と現金等価物を保有しており、累計赤字は約3150万ドル。より多くの資金を獲得すれば,特にGRI−0621とGRI−0803の臨床試験を行い,我々の発見計画を推進し,我々の製品開発努力を継続する際に,我々の計画活動に大量の財務資源を投入する予定である。吾等はまた、吾等に登録声明の取得の効力、及び転売取引所承認株式証(定義は後述)及び株式証(定義は後述)に関連する株式の登録声明の効力を維持することを要求する登録権協定の契約者である。もし私たちがこれらの義務を履行できなかったら、私たちは巨額の現金罰金を受けます。今年度の報告日まで、このようなお金を支払う資源がなく、私たちの業務を継続することができない可能性が高いです
本所は2023年10月13日にS-3表“引受権証と権利証標的株式要約返送登録書”を提出し、2023年12月4日に提出された“S-1表S-3号事前発効改正案”により改正され、2023年12月15日に発効を宣言した。2024年2月1日、吾らは署名ページに記載されている各買い手と証券購入協定(購入契約)を締結し、これにより、吾らは公開発売(I)330,450株普通株(株式)、(Ii)4,669,550株の合計4,669,550株の普通株の予備資本権証(予備資本権証)を行使することに同意し、(Iii)5,000,000株B-1シリーズ普通権証(B-1シリーズ普通権証)を行使し、合計5,000,000株の普通株と交換することができる。(Iv)5,000,000株B-2シリーズ普通権証(B-2シリーズ普通権証,およびB-1シリーズ普通権証とともに発行された普通権証)と,行使可能な普通株総数は5,000,000株である.一般権証と前払い資金権証は本年度報告では“権証”と呼ばれる。これらの証券の発売組み合わせは、(A)1株または1部の事前資本権証、および(B)B-1シリーズ一般権証とB-2シリーズ普通権証であり、総購入価格は1.1ドル(1部の事前資本金権証から0.0001ドル減算)である。
また、上場企業の運営に関連した追加コストが引き続き発生すると予想される。私たちの現在の運営計画によると、私たちの既存の現金と現金等価物は、2024年下半期までの運営費用と資本支出需要を支払うのに十分であると信じています。2022年12月13日に当社,GRI運営会社とAltium Growth Fund L.P.(Altium)によりこの特定証券購入プロトコルにより発行されたT系列株式承認証は現在強制行使可能である。私たちが基づいているこのような推定は間違っていることが証明されるかもしれないし、私たちは私たちが予想していたよりも早く私たちが利用できる資本資源を枯渇させるかもしれない。私たちの将来の資本需要と既存資源が私たちの運営を支援する時間は私たちが予想していたのとは大きく違うかもしれませんが、私たちは私たちの候補製品を再開発するために追加の資金が必要になります。私たちの支出水準は新しい開発と進行中の開発と会社活動によって異なります。我々の候補製品開発に関連する時間や活動の長さは非常に不確定であるため、開発にどれだけの実際の資金が必要かを推定し、承認、マーケティング、商業化活動を仮定することはできない
我々は,2024年2月に完成した発行で調達した資金に加え,短期的に大量の資金を調達し,ナスダックが現在適用されている株主権益要求を再遵守し,以下に述べるように運営を継続する必要がある.私たちは短期的にもっと多くの資金を集めるつもりだ。しかし、受け入れ可能な条件では、もしあれば追加的な資金を得ることができないかもしれない。私たちが私たちの現金需要を満たすのに十分な収入を生み出すことができる前に、私たちは公開または私募株式発行、債務融資、協力、戦略連合、許可手配、および他のマーケティングまたは流通手配を通じて、将来の現金需要に資金を提供する予定です。もし私たちが
公開または私募株式発行によってより多くの資金を調達するためには、これらの証券の条項は、清算または他の特典を含む可能性があり、私たち普通株主の権利に悪影響を及ぼす。さらに、普通株または転換可能または普通株に交換可能な証券を売却することで追加資本を調達する場合、私たちの株主の所有権権益は希釈される。さらに、いかなる債務融資も、追加債務を招く、資本支出を行う、または配当を発表するなど、私たちが具体的な行動を取る能力を制限または制限する固定支払義務と契約を負担させる可能性がある。もし私たちがマーケティングと流通手配、または第三者との協力、戦略連合、または許可手配によってより多くの資本を調達する場合、私たちは、いくつかの価値のある知的財産権または他の権利が私たちに与える候補製品、技術、将来の収入流、または研究計画を放棄しなければならないかもしれない。私たちはまた、任意の候補製品の初期段階で私たちの任意の候補製品のためのパートナーを探すこと、または候補製品または技術に対する私たちの権利を放棄することを要求されるかもしれません。そうでなければ、私たちは自分自身の開発または商業化を求めます。インフレ、流行病、地政学的事件、または他の金融市場要因による市場変動は、必要に応じて資本を得る能力にも悪影響を及ぼす可能性がある。もし私たちが十分な追加資金を得ることができない場合、私たちは私たちの運営計画を再評価し、支出の削減を余儀なくされる可能性があり、サプライヤーとの支払い期限を延長し、可能な場合に資産を清算し、私たちの開発計画の一部または全部を延期、削減、または廃止し、私たちが本来選択した優遇条件よりも低いために私たちの技術に対する権利を放棄したり、運営を完全に停止したりする必要があるだろう。これらの行動は私たちの業務、経営結果、私たちの将来の見通し、私たちの普通株の価値に大きな影響を与えるかもしれないので、私たちの株主は彼らの投資から何の価値も得られないかもしれない。また、追加融資を獲得しようとすると、経営陣の時間と注意力を日常活動から移し、発見や製品開発への関心を分散させている
私たちの監査役は、継続経営企業としての持続的な経営能力に大きな疑いを示しており、追加融資を受けることができなければ、継続的に経営している企業として経営を続けることができないかもしれません。
設立以来、私たちは赤字を出してきましたが、これまで株式や債務証券を発行することで運営に資金を提供してきました。予測可能な未来には,我々が候補薬を開発し続けるにつれて,損失を受け続け,負の運営キャッシュフローが生じ,計画された運営活動を支援するための追加資金が必要となると予想される。我々の独立公認会計士事務所は、2023年12月31日までの年次財務諸表に関する報告書には説明段落が含まれており、継続的に経営している企業として経営を継続する能力に大きな疑問があることを示しています。相当な製品収入を生み出すことができるまで、株式発行、債務融資、または他の資本源(戦略的許可、協力、または他の同様の合意を含む)を通じて、私たちの運営および資本融資需要に資金を提供し続ける可能性が予想されます。上述したように、私たちが十分な追加資金を得ることができない場合、私たちは私たちの運営計画を再評価し、支出の削減を余儀なくされる可能性があり、サプライヤーとの支払い条件を延長し、可能な限り資産を清算し、私たちの開発計画の一部または全部を延期、削減、または廃止するか、またはそれが選択したよりも不利な条件で私たちの技術に対する権利を放棄する必要があるだろう。これらの行動は、私たちの業務、経営結果、私たちの将来の見通し、普通株の価値に大きな影響を与えるかもしれないので、私たちの株主は彼らの投資から何の価値も得られないかもしれない。
研究開発と製薬業に関連するリスク
私たちのビジネスは、私たちの主要な候補製品GRI-0621の成功と、私たちが臨床開発に進める可能性のある任意の他の候補製品に高度に依存しています。私たちのすべての候補製品は大量の追加開発が必要で、それから規制部門の承認を求めて製品を商業使用に投入することができます。
私たちは現在商業販売が許可されていない製品で、決して適切な製品を開発できないかもしれません。GRI−0621は我々の主要な候補製品であるため、GRI−0621が安全または治療効果の問題、開発遅延、規制問題、または他の問題に遭遇した場合、我々の開発計画および業務は深刻な損害を受けるであろう。私たちの主要候補製品GRI−0621、GRI−0803、または私たちの任意の他の候補製品の販売において任意の収入を生成する前に、私たちは、1つまたは複数の司法管轄区域で追加の臨床開発、規制審査、および承認を行わなければならない。このような努力は多くの投資を必要とし、私たちは私たちの候補製品を開発し続ける財力がないかもしれない。
私たちはいくつかの挫折を経験するかもしれません。これらの挫折は、規制部門の候補製品の承認、規制保護の程度、または私たちがそれを商業化する能力を延期または阻止する可能性があります
•我々と類似した候補製品の臨床試験に対する我々の前研究または臨床試験または他の人の陰性または非決定的な結果は、追加の臨床前試験または臨床試験または計画放棄の決定または要求をもたらす
•私たちの臨床試験中の被験者または私たちの候補製品に似た薬物または療法を使用した個人が経験した製品に関連する副作用;
•INDまたは同様の外国出願の提出を遅延させるか、または臨床試験を開始するために規制機関の必要な承認を得ることができなかったか、または臨床試験が開始されると一時停止または終了した
•FDAまたは同様の外国当局は、製品開発中に市場許可のいくつかの結果を測定または支援する任意の規制要件を含む、我々の臨床試験の範囲または設計に適用される条件を含む
•流行病、労働力不足、または他の地政学的事件による被験者の臨床試験への参加を延期する
•臨床試験の被験者の中退率は高い
•臨床試験に必要な候補製品または他の材料の供給または品質不足
•規制要件に適合するようにコスト効果のある方法で候補製品に挑戦しています
•臨床試験費用は予想以上だった
•他の治療法とは競争できません
•特定の管轄区域で孤児の指定を確保または維持することができなかった
•私たちの候補製品は臨床試験期間中の治療効果が良くなかった
•FDAや他の監督機関の臨床試験場の検査と審査に不利である
•私たちの第三者請負業者または調査者は、監督要求をタイムリーにまたは根本的に遵守しなかったか、またはその契約義務を履行しなかった
•一般的な臨床試験または特に私たちの技術に追加の規制監視を適用することを含む、規制要件、政策、およびガイドラインの遅延および変更;または
•FDAと似たような外国の規制機関のデータの違いに対する解釈。
私たちは、臨床開発および規制提出プロセスのいくつかの態様、私たちの知的財産権および私たちの製造、マーケティング、流通および販売努力、または任意の未来のパートナーへの潜在的な脅威を含む多くの要素を完全に制御することができません。規制承認の遅延や私たちが規制承認を得られなかったことは、私たちの業務、将来性、そして運営結果を損なうだろう。
臨床開発は長く、複雑で高価な過程に関連し、結果は不確定である。また,我々の候補製品の臨床前研究や早期臨床試験の結果は後期臨床試験の結果を予測できない可能性がある。
任意の候補製品を商業化するために必要な規制承認を得るためには、広範な臨床前研究と臨床試験により、我々の候補製品の人体上の期待用途が安全かつ有効であることを証明しなければならない(S)。臨床試験費用は高価であり,完成まで数年かかる可能性があり,その結果自体も確定していない。特に,FDAが新薬を承認する一般的な方法は,関連患者群における関連薬物の2つの良好な制御の第3段階臨床試験から陽性データを得ることである。3期の臨床試験は通常数百人の患者に関連し、コストが高く、完成するまで数年かかる。
候補製品は試験のどの段階でも失敗する可能性があり,早期の臨床前研究や臨床試験で有望な活性シグナルが観察された後でも同様である。著者らの候補製品の臨床前研究と早期臨床試験の結果は後期臨床試験の結果を予測できないかもしれない。そのほか、臨床試験の初歩的な成功はこれらの試験が完成した後に得られた結果を代表しないかもしれない。通常,候補製品が臨床試験で失敗することによる自然流出率は極めて高い。臨床前研究と初歩的な臨床試験で進展を得たが、臨床試験後期段階の候補製品は必要な安全性と有効性を示すことができないかもしれない。生物製薬業界のいくつかの会社は治療効果の不足或いは受け入れられない安全性の問題のため、高度な臨床試験で重大な挫折を受け、早期の試験で良好な結果を得たにもかかわらず。一般に、臨床試験を開始する候補製品の多くは製品として承認されておらず、将来の任意の臨床試験が最終的にGRI−0621、GRI−0803、または我々の任意の他の候補製品のさらなる臨床開発に成功またはサポートされることは保証されない。
開発の初期段階で有望そうな候補製品は、いくつかの理由で市場に参入できない可能性がある
•前臨床研究または臨床試験は、候補製品の有効性が予想より低い(例えば、臨床試験がその主要な終点(S))に到達できない可能性があるか、または許容できない副作用または毒性を有することを示す可能性がある
•適用監督機関が臨床意義があると考えられる臨床終点を確立できなかった
•同じ病気の状態で競争製品を開発し
•製造コスト、配合問題、定価または精算問題、または候補製品を経済的にしない他の要因;
•他の会社とその競争製品と技術の独占権は、これらの製品と技術は私たちの候補製品の商業化を阻止するかもしれない。
国会は最近FDCAを改正し,3期臨床試験や新薬の他の“キー研究”のスポンサーにこのような臨床試験のための多様な行動計画を設計·提出し,マーケティング許可を支援することを求めている。行動計画は,学生募集の適切な多様性目標と,これらの目標の理由やスポンサーがこれらの目標をどのように達成するかの記述を記述しなければならない。我々の候補製品はいずれも臨床開発の第3段階に達していないにもかかわらず,多様な行動計画の一部またはすべての要求の免除が得られない限り,第3段階試験または重要な研究案をFDA審査に提出する前にFDAに多様な行動計画を提出しなければならない。多様性行動計画が私たちの候補製品の未来の第三段階試験の計画と時間スケジュールにどのように影響するかはまだ不明であり、FDAがこのような計画の中でどのような具体的な情報を期待するかも不明である。しかしながら、FDAが私たちの候補製品の将来のために行われる任意の第3段階実験のために提案された多様な行動計画に反対する場合、このような試験の開始は延期される可能性があり、承認された任意の多様な行動計画の要求を満たすことを試みる際に、多様な患者集団を募集することが困難になる可能性がある。
また、FDAや同様の外国規制機関が我々の候補製品を規制する際に使用する基準は判断が必要であり、変化する可能性があり、これらの基準がどのように適用されるかを確定的に予測することは困難である。著者らは臨床前と臨床活動データに対するいかなる分析も監督管理機関の確認と解釈を得る必要があり、これは監督部門の承認を延期、制限或いは阻止する可能性がある。新しい政府規定のせいで、私たちはまた予期しない遅延や費用増加に遭遇する可能性がある。このような規制の例は、将来の立法または行政行動、または製品開発およびFDA規制審査中のFDA政策の変化を含む。私たちは立法の変化が公布されるかどうか、あるいはFDAや外国の法規、指導や解釈が変わるかどうか、あるいはこれらの変化の影響が何か可能性があれば予測できない。例えば、2023年4月、欧州委員会は既存の一般薬品立法を改正し、代替する新しい指令と新法規の提案を発表した。現在の提案に従って採択され、実施されれば、これらの改正はEUの薬物開発と承認のいくつかの側面を大きく変えるだろう。
FDAはまた、諮問委員会と呼ばれる専門家グループを要求し、承認を支援する安全性および有効性データの十分性を審議することができる。諮問委員会の意見はFDAに拘束力はないが,FDAの意思決定過程や我々が開発した任意の候補製品の承認を得る能力に大きな影響を与える可能性がある。
もし私たちが外国で臨床試験を行うか、あるいは外国の司法管轄区で上場許可を求めるならば、私たちは臨床試験の進行、製造とマーケティング許可、定価と第三者精算を含む多くの外国の監督管理要求を守らなければならない。外国監督審査手続きは国/地域によって異なり、上述したFDA承認に関連するすべてのリスクと、外国司法管轄区の現地法規を満たすことによるリスクが含まれる可能性がある。さらに、承認を得るのに要する時間は、FDA承認を得るのに要する時間とは異なる可能性がある。FDAの承認は米国以外の規制機関の承認を確保しておらず、その逆も同様である。私たちの競争相手も私たちよりも早く彼らの候補製品のためにFDAや規制機関の承認を得ることができ、これは私たちの競争相手が私たちが市場に入る前に強力な市場地位を確立することができ、あるいは私たちの開発をより複雑にする可能性がある。
われわれが臨床試験で患者を募集する際に困難に遭遇すると,われわれの臨床開発活動は延期されたり,他の悪影響を受けたりする可能性がある。
患者を確定し、それを著者らの候補製品の臨床研究に参与させることは著者らの成功に非常に重要である。著者らの臨床研究の完成時間はある程度著者らが患者を募集して著者らの候補製品のテストに参加する速度に依存し、もし著者らが登録時に困難に遭遇すれば、著者らは臨床試験中に遅延に遭遇する可能性がある。FDAが要求するこれらの試験に十分な数の条件を満たした患者を見つけて募集できなければ,候補製品の臨床試験を開始または継続できない可能性がある。
様々な理由から,臨床試験では患者登録の困難に遭遇する可能性がある。臨床試験方案に基づいて適時に臨床試験を完成し、他の事項以外に、試験が終了するまで十分な数の患者を募集する能力があるかどうかに依存する。患者の登録は多くの要素に依存している
•協定に規定されている患者資格と排除基準
•試験の主な終点と患者の過程を決定するために必要な患者数を分析した
•患者が私たちの実験に参加したいかどうか
•患者と試験場所の距離
•実験の設計
•私たちは適切な能力と経験を持つ臨床試験研究者を募集することができます
•研究中の候補製品の他の利用可能な治療法に対する潜在的な利点およびリスクに対する臨床医および患者の見方は、私たちが調査中の適応のために承認される可能性のある任意の新製品を含む
•市場で入手可能な競争療法および他の競争候補製品の臨床試験の利用可能性;
•患者の同意を得て維持する能力を持っています
•臨床試験に参加した患者が試験完了前に試験を終了するリスク。
例えば,肝障害慢性肝疾患患者のパイロット段階2 a試験でGRI−0621を評価した場合,この研究では当初60名の患者を評価する予定であったが,NASHの臨床研究設計に関する挑戦やFDAの更新ガイドラインを募集したため,14名の患者を募集した後に研究を中止する行政決定を行った
また,我々はIPFの治療のためのGRI−0621を初歩的に開発しており,孤立した適応である。したがって,GRI−0621の臨床試験では被験者募集が困難になる可能性があり,一部は患者群の規模が小さいこと,あるいは臨床レジメンに含まれる安全実験室の負担などが原因である。さらに、私たちの臨床試験は、私たちの試験に参加することを選択する可能性のある患者の中には、私たちの競争相手による試験に参加することを選択する可能性があるので、他の臨床試験と同じ治療分野での製品の候補を争奪するであろう。合格した臨床研究者の数は限られているため、著者らはいくつかの競争相手が使用している同じ臨床試験地点で著者らのいくつかの臨床試験を行い、このような臨床試験地点で臨床試験を行うことができる患者数を減少させることが予想される。
そのほか、臨床試験の適時登録は臨床試験地点に依存し、これらの場所は流行病、供給と労働力不足及び地政学事件などを含む全世界の健康問題の不利な影響を受ける可能性がある。これらの遅延および潜在的な開発スケジュール遅延は、当社の業務、見通し、および運営結果に悪影響を及ぼす可能性があります。
著者らが時々発表或いは公表した臨床試験の中期、主要と初歩データは更に多くの患者データの出現に従って変化する可能性があり、そして監査と検証プログラムの制限を受け、これは最終データの重大な変化を招く可能性がある。
私たちは時々私たちの臨床研究の中期、初歩または主要なデータを公開し、これらのデータは当時利用可能なデータの初歩的な分析に基づいており、結果および関連する発見と結論は特定の研究または試験に関連するデータをより全面的に検討した後に変化する可能性がある。私たちはまた、私たちのデータ分析の一部として、すべてのデータを全面的かつ詳細に評価する機会がないか、または受け取る機会がないかもしれないという仮説、推定、計算、および結論を出した。したがって,我々が報告した臨床試験の中期,背線,あるいは予備結果は,これらの研究報告の最終結果とは異なる可能性があり,あるいは追加のデータを受信して全面的な評価を行うと,異なる結論や考慮要因がこれらの結果を合格させる可能性がある。バックラインデータはまだ監査と確認手続きを受ける必要があり、これは最終データが私たちが以前に発表した予備データと大きく異なる可能性がある。したがって、最終的な完全なデータを得る前に、バックラインデータを慎重に見るべきである。
臨床試験の中期データは患者登録の継続とより多くの患者データの獲得に伴い、1つ或いは複数の臨床結果が実質的に変化する可能性があるというリスクに直面している。初期または中期データと最終データとの間の不利な違いは、私たちのビジネスの将来性を深刻に損なう可能性があります。臨床試験完了時に有利な中期分析が有利な最終結果を生む保証はない。
同様に,われわれの2 a期肝障害患者試験におけるGRI−0621の評価は,最初に60名の患者を評価することを目的としており,募集挑戦およびNASH臨床研究設計に関するFDAの最新ガイドラインにより,14名の患者を募集した後に本研究を中止する行政決定を行ったことから,GRI−0621は耐性が良好であり,肝機能テスト,血清CK−18,iNKT細胞活性ともに改善したことが明らかになったが,この研究の動力不足により統計学的意義のある終点には至らなかったため,本研究を中止する行政決定を行った。著者らがこの2 a期試験から観察した結果は、未来のいかなる潜在的な臨床前研究或いは臨床試験の結果を代表しないかもしれない。
著者らの臨床前研究と候補製品の臨床研究期間中に、法規要求、FDA指導或いは意外事件の変化が発生する可能性があり、これは臨床前或いは臨床研究方案の変化、或いは追加の臨床前或いは臨床研究要求を招く可能性があり、これは私たちのコスト増加を招き、そして著者らの開発スケジュールを延期する可能性がある。
著者らの臨床前研究と臨床研究期間中、法規要求、FDA指導或いは意外事件の変化は著者らに臨床前研究と臨床研究方案の修正を迫る可能性がある。FDAあるいは同様の外国の監督管理機関も追加の臨床前研究と臨床研究要求を強制的に実施する可能性がある。我々の臨床研究プログラムの修正または変更は、終点の変更を含み、FDAまたは同様の外国の規制機関およびIRBsに再提出して審査および承認する必要があり、これは、コストを増加させたり、臨床研究の時間を遅らせたり、成功したりする可能性がある。同様に、臨床前研究の修正はコストを増加させたり、それらの前研究の時間を遅らせたり、成功したりする可能性がある。もし私たちが任意の臨床前または臨床研究の完了を遅延または終了した場合、あるいは追加の臨床前または臨床研究を要求された場合、私たちの候補製品の商業的将来性が損なわれる可能性があり、製品収入の能力が延期されることを確認します。
私たちに製品責任訴訟を提起すれば、私たちは大量の財務または他の責任を負うことができ、私たちの候補製品の商業化を制限することが要求されるかもしれない。
臨床試験でGRI−0621、GRI−0803、および他の任意の候補製品を試験するため、我々は固有の製品責任リスクに直面し、任意の製品を商業化すれば、より大きなリスクに直面する。例えば、私たちの候補製品が臨床試験、製造、マーケティング、または販売中に傷害をもたらすと思われるか、または他の態様では不適切であることが発見された場合、私たちは起訴されるかもしれない。GRI−0621は、以前FDAによってのみ局所投与が許可されていた経口製剤であり、特に、GRI−0621は、慢性使用適応および新しい患者集団における安全性/有効性を評価するために、より多くの研究対象に適用されるので、新たな重篤な有害事象を識別する必要がある可能性がある。このような製品責任クレームは、製造欠陥、設計欠陥、製品固有の危険について警告、不注意、厳格な責任、または保証違反の告発を含む可能性がある。州消費者保護法によると、クレームも主張することができる。もし私たちが製品責任クレームで自分自身を弁護することに成功できなければ、私たちは重大な責任を招いたり、私たちの候補製品の商業化を制限することを要求されるかもしれません。このようなクレームを正当化することに成功しても、多くの財政と管理資源が必要だ。事件がどうであっても最終的な結果がどうであろうと、賠償責任は次のようになるかもしれない
•候補品を市場に出すことはできません
•私たちの製品への需要が減少しました
•私たちの名声を損なう
•臨床試験参加者は脱退し、臨床試験を継続することができない
•規制当局が調査を開始しました
•罰金、禁止、刑事罰
•関連訴訟の弁護費用
•経営陣の時間と資源を移転する
•裁判参加者に多額の報酬を提供し
•製品のリコール、撤回またはラベル付け、マーケティング、または販売促進制限;
•収入損失
•利用可能なすべての保険と私たちの資本資源を枯渇させる
•承認された場合、候補製品を商業化することはできない
•私たちの株価は下落した。
潜在的な製品責任クレームを防止するために、許容可能なコストで十分な製品責任保険を得ることができず、私たちが開発した製品の商業化を阻止または阻害する可能性がある。私たちは臨床試験保険を受けることができないかもしれないし、不利な条項で私たちの任意の臨床試験の任意の責任をカバーするのに十分な金額の臨床試験保険を得ることができないかもしれない。私たちの保険証書にも様々な例外があるかもしれません。私たちは製品責任クレームの影響を受けるかもしれません。私たちは保険範囲を持っていません。私たちは私たちの保険範囲の制限を超えたり、私たちの保険カバー範囲内でない裁判所の裁決または和解合意で達成された任意の金額を支払う必要があるかもしれません。私たちはこれらの金額を支払うために十分な資本を持っていないか、または得ることができません。私たちが未来の会社のパートナーと合意しても、私たちは損害賠償を受ける権利があります。何かクレームがあれば、この賠償は利用できないか十分かもしれません。
FDAの505番目(B)(2)の経路を私たちの主要な候補製品に使用することを希望します。もしこの経路が利用できなければ、私たちの候補製品の開発は現在予想されているより長い時間、より多くのコスト、およびより大きな複雑性と危険を必要とするかもしれませんし、どうしても成功しないかもしれません。
FDAの505(B)(2)経路に基づいてGRI−0621の承認と,我々が開発可能な他の候補薬を開発して求める予定である。FDAが私たちがこの規制経路を使用できないと判断した場合、私たちはFDCA第505条(B)(1)条に従って“完全”または“独立した”秘密協定を通じて規制承認を求める必要があるだろう。これは、追加の臨床試験を行い、追加の安全性および有効性データおよび他の情報を提供し、非臨床データを含む可能性がある規制によって承認された追加の基準を満たすことを要求するだろう。このような状況が発生すると、FDA承認を得るのに要する時間および財政資源、およびこれらのプロジェクトに関連する開発複雑性およびリスクが大幅に増加する可能性があり、これは私たちの業務や財務状況に実質的な悪影響を及ぼす可能性がある。
1984年の薬品価格競争と特許期限回復法は,非公式にハジ−ワックスマン法と呼ばれ,FDCAに505(B)(2)条が追加された。第505条(B)(2)条は、承認に必要な情報の少なくとも一部が、出願人又は出願人のための研究及び情報からではなく、出願人が参照権を得ていない場合に秘密協定を提出することを許可する。第505条(B)(2)条は,FDCAにより我々に適用される場合である。これにより、我々がFDAに提出したNDA部分は、GRI−0621の開発計画を加速させる可能性がある共通分野のデータまたはFDAが承認された化合物の安全性および有効性に関する以前の結論に依存することを可能にするであろう。また、505(B)(2)申請者は彼らが提出可能な研究タイプ、データと情報の面で大きな柔軟性があり、NDA承認の要求を支持し、新薬製品のために有利な利益-リスク概況を構築し、新薬の期待用途に対する有効性を証明した(S)にもかかわらず、申請者はその薬物製品と申請者が依存しようとしているすべての上場薬物との間に科学的な架け橋を構築する責任を負い、その提案による研究は科学的で合理的である。FDAが出願人が提案した後続の薬物製品の開発計画に同意しない場合、承認された製品からの変更を支援するために、非臨床的および臨床的研究を含む追加の研究または測定を企業に要求する可能性がある。FDAはまた、スポンサー提案よりも多くの臨床終点を検討することを要求または要求することができる。そのため、新製品の安全性及び/或いは有効性を確定するために必要なデータ範囲、例えば薬物を局部投与経路から経口投与経路に変更する効果は、科学的に駆動され、そしてケースの基礎の上で確定される。IPFを治療するためにFDAに提案されたGRI−0621の安全性および有効性の研究および臨床試験を保証することはできないし、505(B)(2)NDA経路を用いて開発された任意の将来の候補薬剤を使用することが可能であり、候補製品と関連する上場薬との間のすべての差を支持するのに十分であると考えられる。例えば、FDAが以前に局所投与のための活性成分のみを承認した経口製剤の承認を得るために、我々が予想していたよりも多くのセキュリティデータを収集することが要求される可能性がある。
FDAが505(B)(2)条の解釈に成功して挑戦された場合、または国会が現在利用可能な規制経路を変更するために法規を修正した場合、FDAは505(B)(2)条の政策およびやり方を変更する可能性があり、これは、FDAが505(B)(2)条に従って提出した任意のNDAを承認することを延期または阻止する可能性がある。また、製薬業は競争が激しく、第505条(B)(2)項の国家薬品監督管理局は、第505条(2)で言及された先に承認された薬品の発起人の特許権を保護するために特別な要求を遵守しなければならない。私たちの1人以上の候補者に対して505(B)(2)条の規制経路を使用することができても、これが最終的により速い製品開発またはより早い承認につながることは保証されない。
さらに、FDAの505(B)(2)経路に従うことができないことによるいかなる遅延も、我々のGRI−0621候補製品よりも新しい競争製品の市場進出をもたらす可能性があり、これは、私たちの競争地位および将来性に実質的な悪影響を及ぼす可能性がある。FDAの505(B)(2)の経路に従うことが許可されても、GRI-0621または私たちの将来の任意の候補製品が商業化に必要な承認を得ることを保証することはできません。
規制部門が私たちの候補製品を承認することに関するリスク
私たちは1つ以上の候補製品のための迅速なチャネル認証を求めるかもしれませんが、このような認証を取得しなくても、そのような認証を取得しても、そのような認証は、実際にはより速い開発や規制審査や承認プロセスをもたらさない可能性があります。
候補製品が重篤な疾患の治療に使用され、非臨床または臨床データが、このような疾患が満たされていない医療需要を満たす可能性があることを示す場合、製品スポンサーはFDA Fast Track称号を申請することができる。もし私たちが候補製品のために高速チャネル認証を求めると、FDAの認証を受けることができないかもしれない。しかし、Fast Track認証を取得しても、Fast Track認証は、マーケティング承認または任意の特定の時間範囲で承認されることを保証することはできません。従来のFDAプログラムと比較して、高速チャネル指定のより速い開発または規制審査または承認プロセスを経験しない可能性があります。また,我々の臨床開発プロジェクトのデータが高速チャネルの指定をサポートしなくなった場合,FDAはその指定を破棄する可能性がある。高速チャネル指定だけではFDA優先審査プログラムに適合する資格は保証されない。
私たちがどんな候補製品の規制承認を得ても、私たちは持続的な規制義務と持続的な規制審査の制約を受けるだろう。持続的な規制要件を遵守することは私たちに大きな追加費用をもたらすかもしれないが、このようなコンプライアンスを維持できなかったいかなる行為も私たちを処罰させ、私たちの業務に影響を与える可能性がある。
もし私たちの任意の候補製品が承認されたら、私たちは製造、ラベル、包装、貯蔵、広告、販売促進、サンプリング、記録保存、発売後研究及び安全、治療効果とその他の発売後情報を提出することを含む持続的な法規要求を遵守し、アメリカ連邦と州の要求及び比較可能な外国監督機関の要求を含む。また,われわれが承認後に行ったいずれの臨床試験についてもcGMPとGCPを遵守していく。
メーカーおよびその工場は、品質管理と製造プロセスがcGMPおよび適用される追跡および追跡要求に適合することを確保することを含む、広範なFDAおよび類似の外国規制機関の要求を遵守しなければならない。したがって、私たちと契約製造業者は、cGMPの遵守状況、任意のマーケティング申請で行われた約束の遵守状況、および以前の検査意見への対応を評価するために、持続的な審査および検査を受ける。したがって、私たちと私たちと協力している他の人たちは、製造、生産、品質管理を含む規制コンプライアンスのすべての分野に時間、お金、エネルギーをかけ続けなければならない。
私たちの候補製品のために得られた任意の規制承認は、その製品が発売される可能性のある承認指示用途の制限または承認条件によって制限される可能性があり、または候補製品の安全性および有効性を監視する第4段階の臨床試験および監視を含む、コストの高い上場後試験の要求を含む可能性がある。FDAはまた、REMS計画を私たちの候補製品を承認する条件として要求することができ、これは、患者の長期フォローアップ、薬物使用ガイドライン、医師コミュニケーション計画、または安全な使用を確保する他の要素の要求、例えば、分配方法、患者登録、および他のリスク最小化ツールを制限する必要があるかもしれない。また、FDAや同様の外国規制機関が私たちの候補製品を承認した場合、安全および他の上場後の情報の提出、報告および登録を含む要求を遵守しなければなりません。もし私たちが最初に候補製品の発売許可を加速するルートを通じて獲得したならば、著者らは成功した発売後の臨床試験を行い、著者らの製品の臨床利益を確認する必要があるかもしれない。発売後の臨床試験は成功しない或いはこのような試験を完成できなかったことは上場承認の撤回を招く可能性がある。
私たちが市場の承認を得たい任意の候補製品については、広告や販売促進に関する要求も守らなければならない。FDAは市場に投入された製品のマーケティング、ラベル、広告、販売促進を厳格に規制している。製品は承認された適応と承認されたラベルの規定でしか販売促進できません。しかし、会社はラベルと一致しない真実で誤解されない情報を共有する可能性があり、FDAは最近、製薬業者が承認されていない用途に関する科学的で合理的な臨床関連情報を医療保健提供者とどのように共有するかを提案するガイドライン草案を発表した。FDAや他の機関はラベル外用途の普及を禁止する法律や法規を積極的に実行しており,ラベル外用途の普及が不適切であることが発見された会社は重大な責任を負う可能性がある。
規制要件や基準が守られていない場合、あるいは製品発売後に問題が発生した場合、FDAは同意法令を強制的に実施したり、承認を撤回したりする可能性がある。その後、私たちの候補製品には、意外な深刻さや頻度の不良事件、または当社の第三者製造業者または製造プロセスを含む、以前に未知の問題が存在し、または法規要件を遵守できなかったことが発見され、承認されたラベルの修正につながる可能性があります
新しい安全情報を追加する;発売後の研究或いは臨床試験を実施して新しい安全リスクを評価する;あるいはREMS計画に従って流通制限或いはその他の制限を実施する。他の他の潜在的な結果には
•私たちの製品の販売や製造を制限し、製品を市場からリコールするか、または自発的または強制的に製品をリコールする
•発売後の研究あるいは臨床試験の新しい要求を行う
•罰金、警告状、または臨床試験を一時停止する者
•FDAは、処理すべき出願または私たちが提出した承認された出願の追加を拒否する
•薬の承認を一時停止したり撤回したり
•自発的または強制的な製品リコールと関連する宣伝要求;
•生産を停止しています
•製品を差し押さえたり抑留したり、私たちの候補製品の輸入または輸出を許可することを拒否したり;
•禁止令、同意法令、または民事または刑事処罰を適用する。
政府が違法の疑いのあるいかなる調査にも対応するのに時間と資源がかかり、否定的な宣伝が生じる可能性が予想される。現行の規制要件を遵守しないいかなる行為も、我々が製品を開発·商業化する能力に重大な悪影響を及ぼす可能性があり、私たちの価値や経営業績も悪影響を受けるであろう。さらに、FDAおよび他の規制機関の政策は変わる可能性があり、私たちの候補製品に対する規制承認を阻止、制限、または延期するための追加の政府法規が公布されるかもしれない。米国や海外の将来の立法や行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することはできない。もし私たちが既存の要求の変化に緩やかに適応できない場合、あるいは新しい要求や政策を採用することができない場合、または規制適合性を維持できない場合、私たちは私たちが得る可能性のあるマーケティング承認を失う可能性があり、利益を達成したり維持することができないかもしれない。
私たちの候補製品の商業化に関するリスクは
我々が開発した候補製品が市場承認を得ても,医師,患者,第三者支払人,医学界の他の人がビジネス成功に必要な市場受容度を達成できない可能性がある。
我々が開発したGRI−0621、GRI−0803、または任意の他の候補製品が市場承認を得ても、医師、患者、および第三者支払者(例えば、MedicareおよびMedicaid計画および管理看護組織)および医学界の他の人の十分な市場受容度を得ることができない可能性がある。また,第三者支払者が提供する保険は,既存および将来の医療コスト低減のための医療改革措置の影響を受ける可能性がある。もし私たちが開発した候補製品が十分な受容度に達していなければ、著しい製品収入が生じないかもしれませんし、利益を上げることができないかもしれません。
すべての候補製品が商業販売のために承認された場合、市場の受け入れ度は多くの要素に依存する
•代替療法と比較した治療効果と潜在的優位性
•私たちの製品を提供することができ、承認されたら競争力のある価格で販売することができる
•代替療法よりも便利で投与しやすい
•対象患者群が新たな療法を試みる意欲と,医師がこれらの治療法を処方する意欲
•私たちと私たちの候補製品に適した様々な科学組織が発表したガイドラインの私たちの候補製品に関する提案
•有力なマーケティングと流通支援
•十分な第三者保険と適切な補償を得ることができる
•任意の副作用の流行率および重症度、ならびに任意のラベル警告(ブロック警告を含む)、予防措置または禁忌症の言語および範囲。
.
医療製品の販売は医師が治療処方を出す意思にも依存し、これはこれらの医師に基づいて製品が安全で、治療が有効であり、費用効果があることを確定した可能性が高い。また,異なる医師団体による治療ガイドラインや影響力のある医師の観点から製品を取り入れたり排除したりすることは,他の医師が治療処方を処方する意思に影響する可能性がある。医師、医師組織、病院、他の医療保健提供者、政府機関または個人保険会社が競合療法と比較してOUT製品が安全で、治療効果が高く、コスト効果が高いかどうかを予測することはできない。任意の候補製品が承認されたが、このような各当事者の十分な受容度に達していない場合、候補製品から十分な収入を生成または得ることができず、利益を達成または維持することができない可能性がある。政府や他の第三者支払者が商業化されたどの製品にも保険と十分な補償レベルを提供しなければ、市場受容度や商業成功は低下するだろう。
承認された候補製品のために十分な精算や保険を得ることができない場合、これらの候補製品をマーケティングする能力を制限し、収入を創出する能力を低下させる可能性があります。
私たちが承認した製品の定価、保証範囲、精算(ある場合)は、私たちのビジネス努力や他の開発計画を支援するのに十分でなければならず、第三者支払者(政府および個人保険会社を含む)の保険および精算の可用性および十分性は、ほとんどの患者が医療費を負担できるようにするために重要である。私たちが承認した製品(ある場合)の販売は、私たちが承認した製品のコスト(ある場合)がどの程度健康維持、管理医療、薬局福祉および同様の医療管理組織または政府支払人および個人支払者によってどの程度支払われるかに大きく依存する。保険や精算を受けることができない場合、あるいは限られた金額だけでは、無料で補助金を提供したり、製品を商業化することに成功しないかもしれません。
また,新たに承認された製品の保険カバー範囲や精算に関する不確実性が大きい。アメリカでは、新薬の保証と精算に関する主な決定は通常HHS内のCMSによって行われ、CMSは新薬の決定及びどの程度連邦医療保険の下で保険と精算を受けるかを決定する。個人支払者はCMSが確立した保険精算政策に大きく従う傾向がある。CMSが私たちのような新製品候補者の精算がどのような決定を下すのか、承認されれば、私たちの製品候補者はどのような精算コードを受け取るかもしれないと予測することは難しい。
米国以外では、国際業務は通常、広範な政府価格制御や他の価格制限法規によって制約されており、欧州、カナダ、その他の国のコスト制御措置はますます重視され、処方薬の定価と使用に圧力を与え続けると考えられる。多くの国では、国家衛生システムの一部として、薬品価格は異なる価格制御メカニズムの制約を受けている。価格制御や定価規制の他の変化は、私たちが製品に受け取ることができる費用を制限するかもしれません(もしあれば)。したがって、米国以外の市場では、潜在収入は商業的に合理的な収入と利益を生み出すのに十分ではない可能性がある。
また,米国や海外の政府や個人支払者が医療コストの制限や低減に力を入れており,新薬のカバー範囲や精算レベルが制限されている可能性があるため,あれば,我々の製品に十分な支払いや十分な支払いを提供できない可能性がある。医療機関の影響力が大きくなっていることや,追加的な立法変化を含む管理型医療の傾向が大きくなっているため,薬品関連の価格設定圧力に直面することが予想される。全体的な医療コスト,特に処方薬の下振れ圧力はすでに増加しており,今後も増加し続けると予想される。したがって、私たちの製品の中で規制部門の承認を得た製品があっても、その収益性はより困難になる可能性がある。
たとえ私たちの候補製品がFDAの承認を得ても、私たちは決してアメリカ以外で承認されたり、これらの候補製品を商業化したりすることはできません。これは、私たちがそのすべての市場潜在力を達成する能力を制限するかもしれません。
米国以外の市場で任意の製品を販売するためには、安全性と有効性に関する他国の多くの規制要件を確立し、遵守しなければならない。一国で行われる臨床試験は、他の国の規制部門に受け入れられない可能性があり、一国の規制承認は、他のどの国でも規制承認を受けることを意味するものではない。承認手続きは国によって異なり、追加の製品テストと検証、および追加の行政審査期限が含まれる可能性があります。外国の監督管理機関の承認を求めることは著者らの重大な遅延、困難とコストを招く可能性があり、追加の臨床前研究或いは臨床試験を必要とする可能性があり、これは高価で時間がかかるだろう。各国の規制要求には大きな違いがある可能性があり、私たちの製品がこれらの国で発売されることを延期または阻止する可能性がある。これらと他の規制要件を満たすことは高価で、時間がかかり、不確定であり、予期しない遅延が生じる可能性がある。また、私たちはどの国でも規制承認を得ることができず、他の国の規制承認過程を延期したり、マイナス影響を与える可能性がある。私たちは国際市場を含めてどの司法管轄区域での販売も承認されていませんし、規制機関の経験も得ていません
国際市場で承認された。もし私たちが国際市場の規制要求を守らない場合、あるいは必要な承認を得て維持できなければ、私たちが製品全体の市場潜在力を実現する能力は損なわれるだろう。
私たちは現在マーケティングや販売組織もなく、製品を商業化した経験もない。私たちはこのような能力を発展させるために多くの資源を投入しなければならないだろう。もし私たちがマーケティングと販売能力を確立できない場合、あるいは第三者と合意して私たちの製品をマーケティングして販売することができなければ、私たちは私たちの任意の承認を得る可能性のある候補製品から製品収入を生成することができないかもしれない。
私たちは内部販売、マーケティング、または流通能力を持っていない。ある会社として、私たちは以前医薬製品のマーケティング、販売と流通の方面で経験がなく、販売組織の構築と管理は重大なリスクに関連し、私たちは私たちの雇用、維持と激励、十分な販売手がかりを生成し、販売とマーケティング人員に十分な訓練を提供し、異なる地理的な位置に分散した販売とマーケティングチームを有効に管理する能力を含む。販売、マーケティング、および流通能力の発展における私たちのどんな失敗や遅延も、承認される可能性のある候補製品の商業化に悪影響を及ぼすだろう。直接販売チームや流通システムを構築する第三者と協力して、自分たちの販売チームや流通システムを強化したり、自分たちの販売チームや流通システムの代わりにしたりすることも選択できます。私たちは、受け入れ可能な財務条項で協力することができないか、またはコンサルタントまたは外部サービスプロバイダを招いて、私たちの販売、マーケティング、および流通機能を支援することができないかもしれません。また、私たちが第三者に依存してこれらの機能を実現すれば、私たちの製品の収入と収益力(あれば)は、私たちが開発したどの製品のマーケティング、販売、流通よりも低くなる可能性があります。私たちはこれらの第三者に対して支配権がほとんどない可能性が高く、彼らのいずれも必要な資源と注意力を投入して、私たちの製品を効果的に販売し、マーケティングすることができないかもしれません。もし私たち自身がまたは1つ以上の第三者との手配によって、私たちが所有する可能性のある任意の承認された候補製品を商業化することに成功できなかった場合、私たちは将来どんな製品収入も発生できないかもしれないので、私たちは重大な追加損失を招くだろう。
医療提供者、医師、処方医、購入者、第三者支払者、慈善組織および患者との関係は、刑事制裁、民事処罰、契約損害、名声損害、利益および将来の収入の減少に直面する可能性があるリベート、詐欺および乱用、および他の医療保健法律および法規との関係が適用されるだろう。
米国や他の地域の医療提供者,医師,第三者支払者は薬品の推奨と処方に主な役割を果たしている。第三者支払者および顧客との合意は、AKSおよび連邦虚偽クレーム法案を含むが、AKSおよび連邦虚偽クレーム法案を含むが、これらの法律および法規は、このような会社の販売、マーケティングおよび流通薬品の業務または財務配置および関係を制限する可能性がある幅広い適用される詐欺および乱用、ならびに他の医療法律および法規に直面する可能性がある。特に、私たちの候補製品の研究、および医療製品やサービスの普及、販売とマーケティング、および医療業界のいくつかのビジネス配置は、詐欺、リベート、自己取引、および他の乱用を防止するための広範な法的制約を受けている。これらの法律法規は、幅広い価格設定、割引、マーケティングおよび販売促進、構造および手数料(S)、特定の顧客インセンティブ計画、および他のビジネススケジュールを制限または禁止する可能性があります。これらの法的拘束を受けた活動は,患者を募集して臨床試験を行う過程で得られた情報の不正使用に関するものである。“プロジェクト1.企業-政府法規および製品承認-他の米国のヘルスケア法律および法規”というタイトルの章を参照してください
これらの法律の範囲も執行も不確定であり,現在の医療改革環境の急速な変化の影響を受けている。商業手配が適用される医療保険法に適合することを確保することや,政府当局が行う可能性のある調査に対応することは,時間や資源がかかる可能性があり,会社の業務その他への注意を分散させる可能性がある。
政府および法執行当局は、私たちの業務実践が、詐欺および乱用または他の医療保健法律および法規を適用する現在または未来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。もし私たちにこのような訴訟を提起した場合、私たちは私たちの権利を弁護したり、維持することに成功しませんでした。これらの行動は、重大な民事、刑事および行政処罰、損害賠償、罰金、返還、監禁、名誉被害、連邦および州政府が援助する医療保健計画から除外される可能性があり、契約損害、削減または制限、およびこれらの法律違反に関する告発、および追加の報告義務および監督を解決するために、私たちの業務に重大な影響を与える可能性があります。さらに、私たちがそれと業務を展開することを期待している任意の医師または他のヘルスケア提供者またはエンティティが、適用された法律に適合していないことが発見された場合、彼らは、政府が援助する医療計画から除外されることを含む重大な刑事、民事または行政制裁を受ける可能性がある。これらの法律に違反するいかなる行為も、成功して弁護されても、巨額の法的費用を招き、経営陣の企業運営への注意をそらす可能性がある。今後発売される製品の販売を禁止または制限または撤回することは、不利な方法で業務に大きな影響を与える可能性がある
行われている医療立法や規制改革措置は、私たちの業務や運営結果に実質的な悪影響を及ぼす可能性がある。
規制、法規、または既存の規制の解釈の変化は、私たちの製造スケジュールの変更を要求すること、製品ラベルを追加または修正すること、私たちの製品をリコールまたは停止すること、または追加の記録保存要件を要求することなど、私たちの将来の業務に影響を与える可能性があります。このような変化を強制的に実施すれば、私たちの業務運営に悪影響を及ぼす可能性がある。もし私たちが既存の要求の変化に適応できない場合、あるいは新しい要求や政策を採用することができない場合、あるいは規制適合性を維持できない場合、私たちは私たちが得る可能性のあるマーケティング承認を失う可能性があり、私たちは利益を達成したり維持することができないかもしれない。“プロジェクト1.企業--政府規則と製品審査--薬品保険、定価、精算と医療改革”と題する章を見た
また、米国や海外(カナダやヨーロッパを含む)の政府や第三者支払者が医療コストの制限や低減に力を入れており、これらの組織が新承認製品の保証範囲や精算レベルを制限する可能性があるため、候補製品をカバーしたり、十分な支払いを提供することができない可能性がある。アメリカでは、特殊薬品の価格設定に関する立法と法執行の関心が増加している。具体的には、米国議会は最近数回の調査を行い、薬品定価の透明性を高め、連邦医療保険下の処方薬のコストを下げ、定価とメーカー患者計画との関係を審査し、政府計画の薬品精算方法を改革するための連邦と州立法を提出し、公布した。最近では2022年8月に総裁·バイデンが複数の条項を含む“アイルランド共和法”に署名したことで,これらの条項は連邦医療保険計画や米国全体に販売されている薬品の価格に影響を与える可能性がある。
他の点では、アイルランド共和軍には複数の条項があり、連邦医療保険計画や米国全体に販売されている薬品の価格に影響を与える可能性がある。連邦医療保険BやDの部分に覆われた薬品や生物製品のメーカーは現在,連邦政府にリベートを支払わなければならず,その薬品の価格上昇速度がインフレ率よりも速い場合。この計算は薬品に基づいて行われており、連邦政府に不足している税金還付額は、連邦医療保険B部分またはD部分で支払われている薬物製品の数に直接依存する。また、CMSは2026支払い年度から毎年、模造薬や生物類似競争を含まずに、選択された数量の単一源D部分薬物について薬品価格交渉を行う。CMSはまた,選定数のB部分薬の薬品価格を2028年から交渉する。CMSが1つの薬物を選択して交渉すれば,このような薬物による収入は減少すると予想される。CMSはこれらの新しい許可の実施を開始し、2023年10月に製薬業者と第1セットの合意を締結し、価格交渉を行った。しかし,アイルランド共和軍の米国バイオ製薬業界への影響はまだ不確定であり,一部の原因は複数の大手製薬会社や他の利害関係者(例えば米国商会)がCMSに対して様々な理由で違憲,その他の苦情を訴えていることである。このような訴訟は現在も進行中だ。
州レベルでは、立法機関は、価格または患者の精算制限、割引、特定の製品参入およびマーケティングコスト開示の制限、および透明性措置を含む薬品の価格設定を制御するための法規を立法および実施することによって、場合によっては、他の国からの輸入および大量購入を奨励することを目的としている。法律で規定されている第三者支払者の支払金額の価格制御またはその他の制限は、私たちの業務、財務状況、運営結果、将来性を損なう可能性があります。また,地域医療当局や個別病院では,どの薬品やサプライヤーが処方薬や他の医療計画に含まれるかを決定するために入札プログラムが使用されるようになってきている。これは私たちの製品の最終需要を減らしたり、私たちの製品の価格設定に圧力を与える可能性があります。これは私たちの業務、財務状況、運営結果、将来性にマイナスの影響を与えるかもしれません。また、アメリカ最高裁判所は2020年12月に一致して、連邦法律は先制せず、各州がPBMと医療保健と薬品サプライチェーンの他のメンバーを監督する能力を剥奪し、この重要な決定は各州がこの分野で更なるかつ積極的な努力をすることを招いた。
連邦貿易委員会は2022年中にPBM業界のやり方に対して全面的な調査を展開し、これはこのような実体の運営、薬局ネットワーク或いは財務手配に対するより多くの連邦と州立法或いは監督管理提案を招く可能性がある。米国議会と州立法機関は、ますます多くの業界を検討し、公共政策の懸念とされている様々な問題を解決するための新しい規制方法を提案している。米国の既存のPBM業界を変える重大な努力は、私たちのようなバイオ製薬製品開発業者を含む医薬品サプライチェーン全体および他の利害関係者の業務に影響を与える可能性がある。また、2023年9月、連邦貿易委員会は政策声明を発表し、医薬品開発者がFDAのオレンジブックに列挙したいくつかの“不正”特許は不公平な貿易慣行を代表し、業界はその分析に基づいて潜在的な法執行行動の準備をすべきであることを明らかにした。連邦貿易委員会は2023年11月にこの行動をとり,大手製薬会社10社の100件以上の“不正”特許リストを公開し,これらの特許に対するFDAの行政手続きを開始した。連邦貿易委員会、他の政府機関、製薬業者、または他の利害関係者が引き続き優先しているかどうか
“不正”特許上場の政策問題と、この分野で重大な訴訟が発展するかどうか。そのため、生物製薬業界の商業実践に対する規制と政府の興味は引き続き拡大し、不確定なリスクとなっている。
これらの法律および将来の州および連邦医療改革措置は将来的に採択される可能性があり、いずれも連邦医療保険および他の医療保険資金のさらなる減少をもたらし、他の方法で規制の承認を得る可能性のある任意の候補製品の価格、またはそのような候補製品の処方または使用頻度に影響を与える可能性がある。また、管理型ヘルスケアの傾向、健康維持組織のますます増加する影響力、コスト制御措置、追加の立法変化により、今後の任意の承認された候補製品の販売において価格設定圧力に直面することが予想される。
米国食品医薬品局、米国証券取引委員会および/または他の政府機関の資金不足は、彼らが重要な指導部や他の人員を採用し、維持する能力を阻害し、新製品やサービスの適時な開発や商業化を阻止するか、あるいはこれらの機関が私たちの業務運営に依存する可能性のある正常な業務機能を履行することを阻止する可能性があり、これは私たちの業務に負の影響を与える可能性がある。
FDAが新製品を審査·承認する能力は、政府予算と資金レベル、肝心な人員の雇用と維持及びユーザー費用の支払いを受ける能力、及び法律、法規と政策の変化を含む様々な要素の影響を受ける可能性がある。したがって、その機関の平均検討時間は近年変動している。また、政府が米国証券取引委員会や我々の業務に依存する可能性のある他の政府機関に提供する資金は、研究開発活動に資金を提供する機関を含め、政治プロセスの影響を受けており、政治プロセス自体が不安定で予測不可能である
FDAや他の機関の中断も、新薬が必要な政府機関によって審査および/または承認されるのに要する時間を遅らせる可能性があり、これは私たちの業務に悪影響を及ぼすだろう。例えば、過去数年間、2018年12月22日から2019年1月25日まで、米国政府は何度も閉店しており、食品·医薬品局や米国証券取引委員会などの規制機関は、食品·医薬品局、米国証券取引委員会、その他の政府従業員を休暇させ、重要な活動を停止しなければならない。政府が長期的に停止したり減速したりすれば、FDAが提出した規制文書をタイムリーに審査して処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。また、将来的に政府の閉店は、適切な資本化と事業継続のために、私たちが公開市場に進出し、必要な資本を得る能力に影響を与える可能性がある。歴史的に見ると、米国政府は停止したことがあり、最近の政府の停止も脅かされていた。政府の停止がどのくらい続くか、それが私たちの様々な行動に管轄権を持つ連邦機関にどのような影響を与えるかはよくわからない
私たちはアメリカと外国のいくつかの反腐敗、反マネーロンダリング、輸出規制、制裁、その他の貿易法律と法規の制約を受けている。私たちは違反によって深刻な結果に直面するかもしれない。
他の問題ではアメリカです外国の反腐敗、反マネーロンダリング、輸出規制、制裁およびその他の貿易法律および法規(総称して貿易法と呼ばれる)は、会社およびその従業員、代理人、臨床研究組織、法律顧問、会計士、コンサルタント、請負業者および他のパートナーの許可、承諾、提供、公的または民間部門の受取人の腐敗または不当な支払いまたは任意の他の価値のあるものを直接または間接的に受け入れることを禁止する。貿易法違反は、大量の刑事罰金と民事処罰、監禁、貿易特権の喪失、資格取り消し、税収の再評価、契約違反と詐欺訴訟、名声損害、その他の結果を招く可能性がある。私たちは政府機関や政府付属病院、大学、その他の組織の役人や従業員と直接または間接的な相互作用を持っている。私たちはまた、アメリカ以外での私たちの活動がタイムリーに増加すると予想している。我々は、第三者を招いて臨床試験および/または必要な許可、許可、特許登録、および他の規制承認を得ることを計画しており、私たちは、このような活動を明確に許可していなくても、または事前にそのような活動を知っていなくても、私たちの人員、代理またはパートナーの腐敗または他の不正活動のために責任を負わなければならないかもしれない。
もし私たちが環境、健康、安全の法律法規を守らなければ、罰金や罰金を科されたり、私たちの業務、財務状況、または運営結果に重大な悪影響を及ぼす可能性のあるコストが発生する可能性があります。
私たちの研究開発活動と私たちの第三者メーカーとサプライヤーの活動は、私たちの候補製品の成分と他の危険化合物を含む危険材料の制御された貯蔵、使用と処分に関連している。私たちと私たちの製造業者とサプライヤーはこのような危険な材料の使用、製造、貯蔵、処理、処分に関する法律法規を遵守しなければならない。場合によっては、これらの危険材料とそれらを使用して発生した様々な廃棄物は、私たちと私たちの製造業者の施設に貯蔵され、使用と処分を待っている。私たちは汚染リスクを除去することができません。これは私たちの商業化努力、研究開発努力、業務運営の中断、環境破壊による高価な整理、およびこれらの材料と特定の廃棄物製品の使用、貯蔵、処理、処理を管理する適用法律と法規に規定された責任を招く可能性があります。私たちと私たちの第三者製造業者がこれらの材料を処理して処分する際に使用するセキュリティプログラムは全体的にこれらの法律と法規に規定された基準に適合していると信じていますが、状況がそうであることを保証することはできません
このような材料の意外な汚染や傷害の危険を除去する。この場合、私たちはそれによって生じるいかなる損害に対しても責任を負うことができ、このような責任は私たちの資源範囲を超える可能性があり、州、連邦、または他の適用機関は、特定の材料の使用を減少させ、および/または私たちの業務運営を中断するかもしれない。また,環境法律法規は複雑で変化が頻繁であり,より厳しくなる傾向にある。私たちはこのような変化の影響を予測することもできないし、私たちの未来のコンプライアンス状況を決定することもできない。私たちは現在危険廃棄物保険を受けていません。
私たちの知的財産権に関するリスクは
私たちの成功は私たちの知的財産権を保護する能力にある程度かかっている。私たちの固有の権利と技術を保護することは難しくて高価で、私たちはそれらの保護を保障できないかもしれない。
私たちの業務は、私たちの特許技術および私たちの候補製品、それらのそれぞれの構成要素、合成中間体、製剤、共同療法、それらを製造する方法および治療方法のための特許、商標および商業秘密保護の獲得および維持に大きく依存し、これらの特許を第三者の挑戦から保護することに成功している。私たちが許可されていない第三者が私たちの候補製品を製造、使用、販売、提供、または輸入することを阻止する能力は、これらの活動をカバーする有効かつ実行可能な特許の下で私たちが所有する権利の程度、および裁判所が禁止救済措置を発表するかどうかに依存する。もし私たちが開発したいかなる製品や技術のために特許保護を獲得して維持することができない場合、あるいは獲得した特許保護範囲が十分でなければ、私たちの競争相手は私たちと類似または同じ製品や技術を開発し、商業化する可能性があり、私たちが開発する可能性のある任意の候補製品の商業化の能力は不利な影響を受ける可能性がある
特許出願プロセスは高価で時間がかかり、私たちはすべての必要または望ましい特許出願を合理的なコストまたはタイムリーに提出して起訴することができないかもしれない。特許を申請する過程には多くのリスクがあり、私たちが私たちが申請した特許を成功的に獲得することは保証されない。しかも、私たちはすべての関連市場で特許保護を追求、獲得、または維持しないかもしれない。我々は,特許保護を得るのが遅くなるまで,我々の研究開発成果で特許を申請できることを確認できない可能性もある.さらに、場合によっては、私たちは、第三者から許可を得て、第三者に与えられ、私たちのライセンシーまたはライセンシーの技術に依存することができるかもしれないことを含む、特許出願の準備、提出および起訴を制御する権利がないかもしれない。
バイオテクノロジーやバイオ製薬分野の特許の強度は複雑な法律や科学的問題に関連しており,不確実である可能性がある。私たちが所有しているまたは許可されている特許出願は、発行された特許を生成することができない可能性があり、その声明は、私たちの候補製品または米国または他の国/地域におけるその使用をカバーする。特許が確実に発行されたとしても、第三者はその有効性、実行可能性、または範囲に疑問を提起する可能性があり、これは、このような特許が縮小され、無効にされ、または強制的に実行されない可能性がある。また、それらが挑戦されていなくても、私たちの特許と特許出願は、私たちの候補製品を含めて、私たちの技術を十分に保護できないかもしれません。あるいは、他の人が私たちの声明を中心に設計することを阻止します。もし私たちが持っている特許出願が私たちの候補製品に提供する保護の広さや強度が脅かされれば、会社が私たちと協力して私たちの候補製品を開発することを阻止し、私たちがそれを商業化する能力を脅かすかもしれない。また、臨床試験で遅延に遭遇すれば、特許保護の下で候補製品を販売できる時間が短縮されるだろう。
私たちは私たちの技術(私たちの候補製品を含む)に関連する特許出願を最初に提出した会社であることは確認できません。そうでなければ、私たちは私たちの技術(私たちの候補製品を含む)の特許保護から除外される可能性があります。
私たちは私たちが最初の発明未解決特許出願でカバーされた発明者であることを確認することができません。もし私たちがそうでなければ、私たちは優先権紛争の影響を受けるかもしれません。さらに、すべての請求項が2013年3月16日までに優先日を得る権利がある米国出願については、誰が私たちが出願した特許権利要件がカバーする任意の主題を最初に発明したかを決定するために、第三者が介入手続きを開始するか、または米国特許商標局によって訴訟を提起することができる。同様に、特許請求の範囲の少なくとも1つが2013年3月16日までに優先日を有する権利を有していない米国出願については、特許請求の範囲の標的が以前の発明者の開示から由来しているか否かを決定するために、派生プログラムを提起することができる。
私たちは、特定の特許の一部または全期間または特定の特許出願の全期間を放棄することを要求されるかもしれない。私たちが知らない従来技術は、特許または特許請求の範囲の有効性または実行可能性に影響を及ぼす可能性があるかもしれない。我々が知っているかもしれないが,クレームの有効性や実行可能性に影響を与えるとは考えられない既存技術があるが,最終的にはクレームの有効性や実行可能性に影響を与えることが発見される可能性がある。もし挑戦されれば、私たちの特許は裁判所によって有効または強制執行可能であると宣言されるか、または有効かつ強制的な執行が発見されても、私たちの候補製品が十分に保護されるか、または裁判所によって競争相手の技術または製品によって侵害されると判断されることを保証することはできない。私たちは、私たちの活動に関連すると考えられる競争相手の特許または特許出願を分析し、私たちの候補製品に対して自由に運営できると考えているかもしれませんが、私たちの競争相手は、私たちが関係ないと思う特許を含む発表されたクレームを得るかもしれません。これは、私たちの努力を阻害したり、私たちの製品候補や私たちの活動がこのようなクレームを侵害したりする可能性があります。他社が開発した製品は我々の製品と同様の効果がある可能性がある
私たちの特許または他の知的財産権を侵害しない独立製品、または私たちの製品をカバーする可能性のある特許の請求項を中心に設計されます。
法的手段は限られた保護しか提供できず、私たちの権利を十分に保護できないか、あるいは私たちの競争優位性を獲得または維持することができるかもしれないので、未来の私たちの所有権の保護の程度は不確定だ。
私たちは未来に許可や他の協力協定を締結するかもしれないし、これらの協定は私たちにいくつかの義務を加えるかもしれない。第三者と締結されたこのような未来の合意に規定された義務を履行できなければ、将来の業務に非常に重要である可能性のある許可権を失う可能性があります。
私たちの候補製品ルートの拡大に努力するために、私たちはいくつかの許可または他の候補製品許可に関する他の協力協定を締結するかもしれない。このような合意は、様々な職務調査、マイルストーン支払い、特許権使用料、保険、または他の義務を私たちに強要するかもしれない。もし私たちがこれらの義務を履行できなかった場合、私たちの許可者またはパートナーは関連合意を終了する権利があるかもしれません。この場合、このようなライセンス知的財産権がカバーする製品を開発またはマーケティングすることができません。
さらに、ライセンス契約によると、知的財産権に関する紛争が生じる可能性があります
•ライセンス契約に従って付与された権利範囲および解釈に関連する他の問題;
•私たちの候補製品、技術、およびプロセスは、ライセンス契約に拘束されていないライセンス側の知的財産権をどの程度侵害しているか
•私たちの協力開発関係に基づいて、特許と他の権利を再許可する
•私たちのライセンス契約の下での義務と、どのような活動がこれらの職務義務を満たしていますか
•私たちのライセンス者、私たちおよびパートナーが知的財産権を共同で創造または使用することによって生成された発明およびノウハウの発明および所有権;
•特許技術発明の優先権。
また,我々が現在第三者から知的財産権や技術許可を得ているプロトコルは複雑であり,このようなプロトコルのいくつかは様々な解釈の影響を受ける可能性がある.可能性のある契約解釈の相違の解決は、関連する知的財産権または技術に対する私たちの権利と考えられる範囲を縮小するか、または関連協定の下での私たちの財務または他の義務を増加させる可能性があり、いずれも、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。さらに、我々が許可している知的財産権をめぐる紛争が、商業的に許容可能な条項で現在の許可手配の能力を維持していることを阻害または損害すれば、影響を受けた候補製品の開発および商業化に成功することができない可能性があり、これは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
さらに、私たちは、これらの許可内の特許および特許出願の維持および起訴、または私たちの許可内の知的財産権に関連する任意の他の知的財産権の維持および起訴について限られた支配権を有する可能性がある。例えば、私たちは、任意の未来のライセンシーのそのような活動が適用された法律および法規に準拠しているか、または効果的かつ強制的に実行可能な特許および他の知的財産権を生成するだろうと判断することはできない。私たちは、ライセンシーが知的財産権の第三者侵害者に対して侵害訴訟を提起したり、私たちに許可されたいくつかの知的財産権を弁護する方法に限られた支配権を持っています。ライセンシーの権利侵害訴訟や弁護活動は私たち自身が行うほど激しくないかもしれない。
第三者の知的財産権侵害に対するクレームはコストが高く、時間がかかる可能性があり、私たちの製品の発見、開発と商業化努力を阻害または延期する可能性がある。
私たちのビジネス成功は、私たちの候補製品を開発、製造、マーケティング、販売し、第三者の固有の権利を侵害することなく、当社の独自技術を使用する能力にある程度依存します。バイオテクノロジーおよび生物製薬業界には、干渉、派生、当事者間の審査、許可後の再審、および米国特許商標局における再審手続き、または外国法ドメインにおける異議および他の同様の手続きを含む特許および他の知的財産権に関する訴訟、および特許に挑戦する行政訴訟が多い。私たちは、私たちの候補製品および/またはノウハウが彼らの知的財産権を侵害していると主張する特許または他の知的財産権を持つ第三者の将来の訴訟の脅威に直面するか、または受ける可能性がある。我々が我々の候補製品を開発している分野には,第三者が所有する米国や外国から発行された特許や係属中の特許出願が多く存在する.バイオテクノロジーやバイオ製薬業界の拡張や特許の発行に伴い,我々の候補製品は他人の特許権侵害のクレームを引き起こすリスクが増加する可能性がある。さらにそれは
私たちを含む業界参加者は、どの特許が様々なタイプの薬物、製品、またはその使用または製造方法をカバーしているのかを常に明確にしているわけではない。したがって,我々の分野で発行される特許や提出された特許出願数が多いため,第三者は我々の候補製品,技術または方法を含む特許権を持っていると主張する可能性がある.
さらに、公聴会、動議、または他の一時的な手続きまたは事態の発展の結果を公開発表する可能性があり、証券アナリストまたは投資家がこれらの結果がマイナスであると考える場合、私たちの普通株式価格に大きな悪影響を及ぼす可能性がある。このような種類の訴訟や訴訟は、私たちの運営損失を大幅に増加させ、開発活動に利用できる資源を減少させる可能性がある。私たちはそのような訴訟や法的手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らがより多くの資源を持っているので、私たちよりも複雑な特許訴訟の費用を効果的に受けるかもしれない。さらに、いかなる訴訟の開始および継続に生じるいかなる不確実性も、運営を継続するために必要な資金を調達する能力に重大な悪影響を及ぼす可能性があり、または私たちの業務、運営結果、財務状況、および見通しに重大な悪影響を及ぼす可能性がある。さらに、知的財産権訴訟や行政訴訟に関連する大量の開示要求により、私たちのいくつかの機密情報が開示によって漏洩される可能性がある。
第三者は私たちが許可されていない状況で彼らのノウハウを使用していると主張するかもしれない。
我々の候補製品の組成、使用または製造された物質、材料、配合、製造方法、または処理方法を含む、私たちが現在知らない第三者特許の請求項が存在する可能性がある。私たちが現在知らない現在処理されている特許出願が存在する可能性があり、これらの出願は、私たちの候補製品またはその使用または製造が発行された特許を侵害する可能性がある。さらに、第三者は将来的に特許を取得し、私たちの技術を使ってこれらの特許を侵害したと主張するかもしれない。管轄権のある裁判所が、私たちの候補製品、私たちの候補製品または材料を製造するための中間体、私たちの処方または製造または使用方法の様々な態様をカバーするために、任意の第三者特許を保有している場合、任意のそのような特許の所有者は、許可を得ない限り、またはその特許が満了するまで、または最終的に無効または実行不可能であると判定されない限り、候補製品を開発および商業化する能力を阻止することができる。いずれの場合も、そのような許可は商業的に合理的な条項や全く存在しないかもしれない。もし私たちが商業的に合理的な条項で第三者特許の必要な許可を得ることができない場合、あるいは根本的にできなければ、候補製品を商業化する能力が損なわれたり、延期されたりする可能性があり、逆に私たちの業務を深刻に損なう可能性がある。我々が許可を得ても,我々の競争相手が我々に許可された同じ技術にアクセスできるように非排他的である可能性がある.さらに、私たちの特許および特許出願によって提供される保護の広さまたは強度が脅かされている場合、会社が私たちと協力し、現在または将来の候補製品を許可、開発、または商業化することを阻止するかもしれない。
私たちにクレームを出した当事者は、禁止令や他の公平な救済を求めて獲得する可能性があり、これは、私たちの候補製品のさらなる開発と商業化を効果的に阻止することができるかもしれない。これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、当社の業務における従業員資源を大量に移転することになる。もし私たちに対する侵害クレームが成功した場合、私たちは故意に侵害した3倍の損害賠償金と弁護士費、第三者から1つ以上の許可証を取得し、印税を支払うか、または私たちの侵害製品を再設計することを含む大量の損害賠償を支払わなければならないかもしれないし、大量の時間とお金の支出が必要かもしれない。私たちはそのようなライセンスがあるかどうか、あるいは商業的に合理的な条項で提供されるかどうかを予測できない。また、訴訟がない場合であっても、第三者からライセンスを取得して、私たちの研究を進めたり、候補製品の商業化を許可したりする必要があるかもしれません。私たちはもしあれば、合理的な費用または合理的な条項でこのような許可証のいずれかを得ることができないかもしれない。この場合、私たちは私たちの候補製品をさらに開発して商業化することができなくなり、これは私たちの業務を深刻に損なう可能性があります。
第三者は、我々の従業員やコンサルタントが機密情報を誤って使用または漏洩したり、商業秘密を流用したりしたと主張する可能性がある。
バイオテクノロジーやバイオ製薬業界でよく見られるように、私たちが雇った個人は以前、私たちの競争相手や潜在的な競争相手を含む大学や他のバイオテクノロジーやバイオ製薬会社に雇われていた。現在、私たちに対するクレームが解決されていないにもかかわらず、私たちの従業員およびコンサルタントが、私たちのために働いているときに他人の固有情報またはノウハウを使用しないことを確実にするために努力しているにもかかわらず、私たちまたは私たちの従業員、コンサルタントまたは独立請負業者は、商業秘密または他の独自情報を含む、以前の雇用主または他の第三者の知的財産権を不注意または他の方法で使用または開示する可能性がある。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭的損害賠償を支払う以外に、私たちは貴重な知的財産権や人員を失う可能性がある。このようなクレームに対抗することに成功しても、知的財産権クレームに関連する訴訟や他の法的手続きは、巨額の費用を発生させる可能性があり、私たちの技術や管理者の正常な責任を分散させる可能性があります。さらに、証券アナリストまたは投資家がこれらを考える場合、公聴会、動議または他の一時的な手続きまたは事態の発展の結果が公表される可能性がある
もし結果が否定的なら、それは私たちの普通株価格に実質的な悪影響を及ぼすかもしれない。このような種類の訴訟や訴訟は、私たちの運営損失を大幅に増加させ、開発活動に利用できる資源を減少させる可能性がある。私たちはそのような訴訟や法的手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らの財政資源がはるかに大きいので、私たちよりもこのような訴訟や訴訟の費用を効果的に負担するかもしれない。特許訴訟または他の知的財産権関連訴訟の開始および継続によって生じる不確実性は、市場での競争能力に悪影響を及ぼす可能性がある。
他の人たちは私たちの知的財産権の所有権を持っていると主張するかもしれないが、これは私たちを訴訟に直面させ、私たちの将来性に大きな悪影響を及ぼすかもしれない
第三者は、私たちまたは私たちのライセンシーの1つまたは複数の特許または他の独自または知的財産権の所有権を要求することができる。第三者は、影響を受けた製品または製品の臨床試験、製造、およびマーケティングを禁止するために、金銭賠償および/または禁止を求める法的訴訟を提起することができる。私たちは現在、第三者が私たちの特許や他の知的財産権について提起したいかなるクレームや主張も知らないが、第三者がこのような特許や知的財産権のクレームや利益を主張しない保証はない。もし私たちがどんな訴訟に巻き込まれたら、私たちの資源の大部分を消費し、私たちの技術と管理者の多くの心配を招くかもしれない。これらの行動のいずれかが成功すれば、任意の潜在的損害賠償責任に加えて、影響を受けた製品の製造または販売を継続するためのライセンス取得を要求される可能性があり、この場合、巨額の印税を支払うか、または私たちに特許交差許可を付与することを要求される可能性がある。もしあれば、そのような許可が商業的に受け入れられる条項で提供されるかどうかは予測できない。最終的に、私たちは特許侵害や他の知的財産権侵害の疑いで候補製品の商業化を阻止されたり、いくつかの側面の業務運営を停止させられたりする可能性がある。また、知的財産権訴訟の結果は不確定要素の影響を受け、これらの不確定要素は証人の振る舞いや信頼性、いかなる敵の身分も含めて事前に十分に定量化することができない。知的財産権事件では特に、これらの事件は専門家が合理的に同意しない可能性のある技術的事実に関する専門家の証言に依存するかもしれない。
私たちは許容可能な条項で任意の未来の候補製品を開発するために必要な権利を獲得または維持することができないかもしれない。
私たちの計画は他の候補製品に関連する可能性があり、第三者が保有する独占権を使用する必要があるかもしれないので、私たちの業務の成長は、私たちがこれらの独占権を獲得、許可、または使用する能力にある程度依存するかもしれない。
私たちの候補製品はまた、効率的かつ効率的に動作するために特定の処方が必要である可能性があり、これらの権利は他の人によって保持されている可能性がある。私たちは私たちの化合物と既存のバイオテクノロジーとバイオ製薬化合物を含む製品を開発するかもしれない。私たちは、当社の業務運営に必要または重要であると考えられる任意の成分、使用方法、プロセス、または他の第三者知的財産権を第三者から得ることができないかもしれません。私たちは合理的な費用または合理的な条項でこれらのライセンスのいずれかを得ることができないかもしれないが、もしあれば、これは私たちの業務を損なうだろう。私たちは、このような第三者知的財産権がカバーする成分や方法の使用を停止する必要があるかもしれませんし、このような知的財産権を侵害しない代替方法の開発を求める必要があるかもしれません。たとえこのような不可能な代替方法を開発することができても、追加のコストと開発遅延を招く可能性があります。我々が許可を得ることができても,非排他的である可能性があり,我々の競争相手が我々に許可された同じ技術にアクセスできるようにする.この場合、私たちは代替技術を開発または許可するために多くの時間と資源を必要とするかもしれない。
第三者知的財産権の許可や買収は競争分野であり、私たちよりも成熟しているかもしれないし、より多くの資源を持っている会社も、私たちの候補製品を商業化するために必要または魅力的だと思う第三者知的財産権許可または買収戦略を実施しているかもしれません。より成熟した会社は私たちより競争優位にあるかもしれません。それらの規模、現金資源、そしてより強い臨床開発と商業化能力のためです。このような交渉を成功させ、最終的に買収を求める可能性のある他の候補製品をめぐる知的財産権を得ることができる保証はない。
私たちは、私たちの特許が人かもしれない特許を保護または強制する訴訟に巻き込まれたり、他人の特許権に挑戦したりすることができます。これは高価で、時間がかかり、成功しないかもしれません。
化学品や試薬サプライヤーのような競争相手または他の第三者は、私たちの特許または私たちの現在または未来の許可者の特許を侵害する可能性がある。権利侵害や不正使用に対抗するために、私たちは費用が高く時間がかかるかもしれない侵害請求を要求されるかもしれない。さらに、侵害訴訟では、裁判所は、私たちの1つまたは複数の特許が無効または強制的に実行できないと判断するか、または私たちの特許が関連技術または他の理由をカバーしないことを理由に、他方の関連技術の使用を阻止することを拒否することができる。任意の訴訟または弁護手続きにおける不利な結果は、私たちの1つまたは複数の特許が無効であることが宣言されるか、強制的に実行できないか、または狭義の解釈に直面するリスクに直面する可能性があり、私たちの特許出願を発行できないリスクに直面させる可能性がある。これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、当社の業務における従業員資源を大量に移転することになる。
私たちは、米国特許商標局に一方的な再審、当事者間再審または許可後の再審手続において特許主張を審査することを要求することによって、第三者の米国特許における特許請求の可能性に挑戦することを選択することができる。このような手続きは高価で、私たちの時間や他の資源を消費するかもしれない。欧州特許庁(EPO)または他の外国特許庁の特許反対手続において第三者の特許に挑戦することを選択することができる。このような訴訟に反対する費用は巨大かもしれないし、私たちの時間や他の資源を消費するかもしれない。もし私たちが米国特許商標局、欧州特許庁、または他の特許庁で有利な結果を得ることができなかった場合、私たちは私たちの候補製品または独自技術が私たちの特許を侵害している可能性があると非難する第三者の訴訟に直面する可能性がある。
さらに、米国のいくつかの特許出願は、特許発行前に秘密にされている可能性があるため、米国および多くの外国司法管区の特許出願は、通常、提出されてから18ヶ月後に公表され、科学文献の出版物は、実際の発見よりも遅れていることが多く、他の人が、私たちが所有し、許可されている特許または私たちが審理している出願に含まれている技術について特許出願を提出していないか、または適用されている場合、最初の発明または最初にその技術に関連する特許出願を提出した最初の会社であることができるかもしれない。私たちの競争相手はすでに提出されている可能性があり、将来的に特許出願を提出することができ、私たちと類似した製品または技術をカバーすることができる。このような任意の特許出願は、私たちが所有している特許出願または特許よりも優先される可能性があり、これは、そのような技術をカバーする発行された特許を取得する権利を要求することができる。他方が私たちによって所有されているか、または許可されている発明に米国特許出願を提出したような場合、米国における発明の優先度を決定するために、USPTOが発表した干渉または派生プログラムに参加しなければならないかもしれない。もし私たちまたは私たちの許可者のうちの1つが妨害または派生プログラムの一方であれば、私たちが所有している発明または私たちに許可された発明に対する米国特許出願に関連して、私たちが成功しても、私たちは巨額のコストを招き、管理職を移転する時間と他の資源を費やす可能性がある。
我々の特許または特許出願または我々の許可者の発明に関連する発明の優先権を決定するために、第三者によって引き起こされるか、または米国特許商標局によって発表された干渉または派生プログラムが必要である可能性がある。不利な結果は、現在の特許権の喪失を招く可能性があり、関連技術の使用を停止したり、勝利者から許可権を得ようとしたりすることが要求される可能性があります。もし勝利者が商業的に合理的な条項で私たちに許可を提供しない場合、あるいは私たちに許可を全く提供しない、あるいは非独占的な許可を提供し、私たちの競争相手が同じ技術を獲得した場合、私たちの業務は損害を受ける可能性がある。訴訟や介入手続きは、私たちの利益に不利な決定を招く可能性があり、私たちが成功しても、巨額のコストを招き、私たちの経営陣や他の従業員の注意を分散させる可能性があります。私たちは私たちの商業秘密や機密情報の盗用を単独でまたは私たちの許可者と一緒に防ぐことができないかもしれません。特に法律ではアメリカのようにこれらの権利を十分に保護していないかもしれません。
さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。さらに、公聴会、動議、または他の一時的な手続き、または事態の発展の結果が発表される可能性がある。もし証券アナリストや投資家がこれらの結果がマイナスだと思っていれば、私たちの普通株の価格に大きな悪影響を及ぼす可能性がある。
私たちの特許保護の獲得と維持は、政府特許機関によって提出された様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
私たちが所有しライセンスしている特許および特許出願の定期維持費、継続費、年会費、および様々な他の政府費用は、特許有効期間内に米国特許商標局および外国特許代理機関に段階的に支払われる。米国特許商標局および各種外国政府特許機関は、特許出願中および特許発行後に、いくつかのプログラム、文書、費用支払い、およびその他の規定を遵守することを要求する。多くの場合、適用規則に従って滞納金を支払うか、または他の方法で不注意を是正することによって失効を是正することができるが、場合によっては、規定を遵守しないことは、特許または特許出願が放棄または失効され、関連法ドメインの特許権の一部または全部の喪失をもたらす可能性がある。特許または特許出願の放棄または失効をもたらす可能性のある不正事件には、規定された期限内に公式行動に応答できなかったこと、費用が支払われていなかったこと、および適切に合法化され、正式な文書が提出されなかったことが含まれるが、これらに限定されない。場合によっては、意図しない不正事件であっても、特許権を永久的かつ取り返しのつかないように危険にさらす可能性がある。この場合、私たちの競争相手が市場に参入する可能性があり、これは私たちの業務に実質的な悪影響を及ぼすだろう。
私たちの候補製品をカバーする特許は、法廷または米国特許商標局(または外国特許庁)で疑問が提起された場合、無効または実行不可能であることが発見される可能性がある。
もし私たちまたは私たちの許可者のうちの1つが第三者に対して訴訟を起こして、私たちの候補製品のうちの1つをカバーする特許を強制的に執行する場合、被告は私たちの候補製品をカバーする特許が無効であり、および/または実行不可能であることを反訴することができる。米国の特許訴訟では、被告が特許の無効および/または強制執行不可能と主張する反訴が一般的であり、第三者は様々な理由に基づいて特許が無効または強制的に実行できないと断言することができる。第三者も米国や海外の行政機関に類似したクレームを出すことができ、訴訟範囲外でも同様である。このようなメカニズムには、再審査、当事者間の審査、贈与後審査、および外国法ドメインにおける同等の手続き(例えば、反対手続き)が含まれる。このような訴訟は私たちの特許がこれ以上私たちの候補製品をカバーしないように撤回したり修正したりするかもしれない。法的に無効と実行不可能と断言された後の結果は予測できない。例えば、有効性の問題について、私たちは無効な以前の技術がないことを確認することはできないが、私たち、私たちの特許弁護士、特許審査員は起訴中にこれを知らない。もし被告が無効および/または強制できない法的主張で勝った場合、または私たちが他の方法で私たちの権利を十分に保護できなかった場合、私たちは私たちの候補製品の少なくとも一部またはすべての特許保護を失うだろう。このような特許保護の喪失は、私たちの業務および私たちの技術および候補製品を商業化または許可を得る能力に実質的な悪影響を及ぼす可能性がある。
私たちの最初の特許は私たちの最初の製品がアメリカや外国の管轄区域で発売承認される前か近いうちに満期になるかもしれません。私たちの既存の特許が満期になると、私たちは他の人たちを排除してこのような発明を実施する権利を失うかもしれない。これらの特許の満期はまた、私たちの業務、運営結果、財務状況、および将来性に類似した大きな悪影響を及ぼす可能性があります。私たちは、特許として発行された場合、可能な特許期限の調整や延長を考慮することなく、2032年から2035年までの期限が予想される独自技術または候補製品を対象とした特許出願を有している。しかし、米国特許商標局、欧州特許庁、または他の関連外国特許機関がいずれかの特許出願を承認することは保証されない。
米国と外国の管轄区域の特許法の変化は特許の全体的な価値を低下させ、製品を保護する能力を弱める可能性がある。
米国においても外国司法管区においても,特許法または特許法解釈の変化は,特許出願をめぐる起訴および発行された特許の実行または弁護の不確実性およびコストを増加させる可能性がある。他の特許性要件を満たしていると仮定すると,2013年3月16日までは,米国では,最初に発明して保護された発明を要求した者が特許を有しているが,米国以外では,最初に特許出願を提出した者が特許を有している。2013年3月16日、“ライシー·スミス米国発明法”(“米国発明法”)によれば、米国は第1の発明者から出願制に移行し、このような制度の下で、他の特許性要件を満たすと仮定して、第1の特許出願を提出した発明者は、第三者が第1の発明によって要求された発明であるか否かにかかわらず、発明の特許を取得する権利がある。2013年3月16日以降に米国特許商標局に特許出願が提出されたが、我々の前の第三者は、その第三者が発明を行う前に発明をしていても、我々の発明をカバーする特許を付与することができる。これは私たちが発明から特許出願までの提出時間を認識することを要求するだろう。米国およびほとんどの他の国/地域の特許出願は、提出後または発行前の一定期間秘密であるため、私たちまたは私たちのライセンス者が、私たちの候補製品に関連する任意の特許出願または(Ii)発明または私たちのライセンシーの特許または特許出願に要求される任意の発明を最初に提出することを決定することはできない。
米国の発明法にはいくつかの重大な変化も含まれており、これらの変化は特許出願の起訴方法に影響を与え、特許訴訟に影響を与える可能性もある。これらの措置は、第3者が特許訴訟中に米国特許商標局に以前の技術を提出することを可能にすることと、米国特許商標局によって管理される許可後のプログラム(ライセンス後審査、当事者間の審査および派生プログラムを含む)が特許有効性を攻撃する追加の手続きとを含む。USPTO訴訟における証拠基準は、米国連邦裁判所が特許請求の無効を宣言するために必要な証拠基準よりも低いため、第三者は、USPTO手続きにおいて、USPTOが権利要求を無効とするのに十分な証拠を提供する可能性があり、同じ証拠が最初に地域裁判所訴訟で提出された場合には、権利請求を無効にするのに十分ではない。したがって,第三者は米国特許商標局の手続きを用いて我々の特許主張の無効を宣言しようとする可能性があり,第三者が地域裁判所訴訟で最初に被告として疑問を提起すれば,我々の特許主張は無効ではない.したがって、米国発明法およびその実施は、私たちの所有または許可をめぐる特許出願の起訴および私たちが所有または許可した発行された特許の実行または弁護の不確実性およびコストを増加させる可能性があり、これらのすべては、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
また,バイオテクノロジーやバイオ製薬の開発や商業化における会社の特許地位は特に不確定である。米国最高裁判所の最近の裁決は、場合によっては入手可能な特許保護範囲を縮小し、場合によっては特許所有者の権利を弱める。この一連の事件は特許取得後の有効性と実行可能性の面で不確実性をもたらしている。未来の行動による
米国議会、連邦裁判所、米国特許商標局と同様に、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、私たちの既存の特許の組み合わせおよび私たちの将来の知的財産権の保護と実行能力に重大な悪影響を及ぼす可能性がある。
私たちは限られた外国の知的財産権を持っていて、世界各地で私たちの知的財産権を保護して実行できないかもしれない。
私たちのアメリカ以外の知的財産権は限られている。世界のすべての国で候補製品の申請、起訴、特許保護の費用は目を引くほど高く、私たちのアメリカ以外のいくつかの国での知的財産権はアメリカほど広くないかもしれない。また、いくつかの外国の法律は知的財産権の保護の程度はアメリカの連邦や州法律に及ばない。したがって、私たちは、米国以外のすべての国/地域で第三者が私たちの発明を実施したり、米国または他の管轄区域で私たちの発明を使用して製造された製品を販売または輸入することを阻止することができないかもしれない。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの技術を使用して自分の製品を開発することができ、また、他の侵害製品を私たちが特許保護を持っているが法執行力がアメリカに及ばない地域に輸出することもできる。これらの製品は、私たちが発行した特許を持っていない司法管轄区域で私たちの製品と競争するかもしれませんが、私たちの特許主張や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。
多くの会社は外国の管轄区域の知的財産権の保護と保護に重大な問題に直面している。特定の国の法律制度、特にある発展途上国の法律制度は、特許、商業秘密、および他の知的財産権保護、特にバイオテクノロジーおよび生物製薬製品に関連する特許、商業秘密および他の知的財産権保護の強制執行に賛成しない可能性があり、これは、私たちの特許の侵害や第三者に対する競争製品の販売行為を阻止することを困難にする可能性があり、これらの行為は全体的に私たちの独占権を侵害する。第三者が外国の管轄区域で訴訟を起こし、私たちの特許権の範囲または有効性に挑戦し、巨額のコストを招く可能性があり、私たちの努力と注意を私たちの業務の他の側面から移すことができます。外国の管轄区域で私たちの特許権を強制的に執行する訴訟手続きは、巨額のコストを招き、私たちの努力と注意を私たちの業務の他の側面から移転させる可能性があり、私たちの特許が無効または狭義に解釈されるリスクに直面する可能性があり、私たちの特許出願を発表できないリスクに直面させ、第三者からのクレームを引き起こす可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。したがって、私たちが世界各地で私たちの知的財産権を実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネスメリットを得るのに十分ではないかもしれません。
もし私たちが特許期間延長やデータ排他性を得ていない場合、または同様の非米国立法が保護期間を延長し、私たちが開発する可能性のある任意の候補製品をカバーしていれば、私たちの業務は実質的な損害を受ける可能性がある。
新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちが持っている特許と許可された特許の組み合わせは、他社が私たちと似ているか同じ製品を商業化することを排除するために十分な権利を提供してくれないかもしれない。FDAが私たちが開発する可能性のある任意の候補製品の上場承認の時間、期限、詳細によると、私たちの1つ以上の米国特許は、“ハッジ·ワックスマン法案”によって限られた特許期間を延長する資格がある可能性がある。ハッジ·ワックスマン法は、FDA規制審査中に失われた特許期間の補償として、特許期間を最大5年間延長することを許可している。1つの特許期間の延長は、製品が承認された日から計14年の期間を超えてはならず、1つの特許を延長することしかできず、承認された薬物、その使用方法又はその製造方法に関する請求項を延長することしかできない。しかし,テスト段階や規制審査中に職務調査が行われていないこと,適用の最終期限内に出願できなかったこと,関連特許が満了する前に出願できなかったこと,適用要求を満たしていなかったことなどの理由で延期が得られなかった可能性がある。しかも、特許保護の適用期間や範囲は私たちが要求しているものよりも短いかもしれない。また,EU内では,データ独占性,マーケティング保護,孤児適応の市場独占性や小児科延期など,医療製品に提供される規制保護が,現在審査中であり,今後数年で減少する可能性がある。もし私たちが特許期間の延長を得ることができない場合、あるいはどのような延長の期限が私たちが要求しているのよりも短い場合、あるいはデータ独占性や他の規制保護が減少した場合、私たちの競争相手は私たちの特許が満了した後に競争製品の承認を受ける可能性があり、私たちの業務、財務状況、運営結果、および見通しは実質的に損なわれる可能性がある。
私たちの第三者への依存に関するリスクは
私たちは第三者に依存して臨床試験を行い、私たちの候補製品を生産し、他のサービスを提供します。もしこれらの第三者が要求通りに契約責任の履行に成功し、期待時間を満たすか、あるいは他の方法で試験または実行を行い、規制要求を遵守できない場合、私たちは期待通りにあるいは臨床開発を成功させることができず、監督管理の承認を得ることができないか、あるいは私たちの候補製品を商業化することができず、私たちの業務は実質的な損害を受ける可能性がある。
私たちは引き続き第三者CROに依存して、私たちの臨床プロジェクトを実施、監視、管理することに依存し、計画してきた。私たちはこのような当事者たちに依存して臨床試験を実行し、私たちは彼らの活動のいくつかの側面だけを管理して制御している。私たちはまだ私たちのすべての実験が適用された方案、法律、法規、そして科学的基準に従って行われ、私たちのCROへの依存が私たちの規制責任を免除しないことを確実にする責任がある。私たちと私たちのCROおよび他のサプライヤーは、FDAおよび同様の外国の規制機関が私たちの臨床開発におけるすべての候補製品に対する要求を含む、すべての適用された法律、法規、およびガイドラインを遵守しなければならない。もし私たちまたは私たちの任意のCROまたはサプライヤーが適用された法律、法規またはガイドラインを遵守できなかった場合、私たちの臨床試験によって生じる結果は信頼できないと考えられるかもしれませんが、FDAまたは同様の外国の規制機関は、私たちの上場申請を承認する前に追加の臨床試験を行うことを要求するかもしれません。私たちのCROや他のサプライヤーがこれらの要求を満たすことを保証することはできません。あるいは任意の規制機関が検査を行った後、この規制機関は、私たちの任意の臨床試験を含む努力が適用される要求に適合すると判断します。私たちがこれらの法律、法規、あるいはガイドラインを守らないことは、私たちが臨床試験を繰り返す必要があるかもしれない。これは高価であり、規制承認過程を遅延させるだろう。
もし私たちがこれらの第三者CROとの任意の関係が終了した場合、または彼らが隔離され、その場の配置令、閉鎖、または他の制限を受け、彼らの業務を意外に削減しなければならない場合、私たちはCROの代替との合意をタイムリーに達成できないか、または商業的に合理的な条項でそうすることができないかもしれない。また、他の顧客に対して、私たちのCROは私たちの臨床試験を優先しない可能性があり、CRO人員のいかなる変動やCRO従業員の分配の遅延は私たちの臨床試験に負の影響を与える可能性がある。CROがその契約義務または義務を成功裏に履行できなかった場合、あるいは予想された最終期限内に達成できなかった場合、私たちの臨床試験は延期または終了される可能性があり、私たちは現在の候補製品に関する私たちの計画を満たすことができないかもしれない。CROはまた、予想よりも高いコストを伴う可能性があり、これは私たちの財務状況と運営に否定的な影響を及ぼすかもしれない
また、私たちは第三者メーカーに依存して私たちの臨床段階候補製品を生産し、彼らの責任は通常、第三者サプライヤーから必要な材料を購入して、臨床試験や規制承認のための候補製品を生産することを含む。私たちの候補製品を生産するためのいくつかの原材料のサプライヤーの数は限られており、代替サプライヤーを見つけることができない可能性があり、私たちの臨床試験候補製品の生産が中断される可能性があり、承認されれば、最終的に商業販売に使用されることになると予想される
私たちは一般に臨床試験を開始することを望んでいませんが、私たちが試験を完成するのに十分な候補製品供給があると信じない限り、候補製品の供給のいかなる重大な遅延や中断、あるいは候補製品の生産過程における原材料や他の材料部品は、私たちの臨床試験の完成を延期する可能性があり、規制機関が私たちの候補製品を承認する時間を遅らせる可能性があり、これは私たちの業務と運営結果を損なう可能性があります。私たちはまだ候補製品の商業製造コストを確実に見積もるための十分な情報を持っていませんが、私たちは現在候補製品を製造するコストは商業的に不可能かもしれません。したがって、私たちは商業的に実行可能な製品を開発することができないかもしれない。
さらに、私たちの第三者製造業者への依存は、私たちを以下の追加のリスクに直面させます
•私たちは、合格した潜在的なメーカーの数が限られているので、許容可能な条件で、または私たちの候補製品を生産することができないメーカーを見つけることができないかもしれない。NDA承認後、生産場所の変更にはFDAの追加承認が必要となる可能性がある。この承認には新しいテストとコンプライアンス検査が必要になるだろう
•私たちの第三者製造業者は、もしあれば、私たちの製品または生産に必要な数量と品質を適時に制定して製造することができないかもしれません
•労働力不足、インフレ、自然災害、または地政学的衝突のため、私たちの第三者メーカーは運営の削減または中止を余儀なくされる可能性があり、これは候補製品に対して行われているおよび将来の臨床試験の能力を損なう可能性がある
•私たちの未来の第三者製造業者は、約束通りに契約製造業務を履行できないかもしれないし、契約製造業務において、私たちの臨床試験または私たちの候補製品の生産、保存、および流通に要する時間を継続的に提供しないかもしれません
•医薬品メーカーはFDAと州機関の定期的な抜き打ち検査を受け続け、cGMPと他の政府法規および相応の外国基準を厳格に遵守することを保証し、第三者メーカーがこれらの法規と基準を遵守するかどうかを制御することができない
•もし任意の第三者メーカーが私たちの製品の製造過程を改善した場合、私たちは所有していないかもしれないし、許可することができないかもしれないし、あるいは私たちの候補製品の製造過程で私たちの第三者メーカーが行った任意の改善された知的財産権を共有しなければならないかもしれない
•私たちの第三者製造業者は私たちとの合意を違反または終了するかもしれない。
これらのリスクの各々は、私たちの臨床試験、私たちの候補製品の承認(あれば)を延期したり、私たちの候補製品の商業化を延期したり、より高いコストをもたらしたり、私たちの潜在的な製品収入を奪ったりする可能性がある。また,我々は第三者に依存して候補製品の放出試験を行い,臨床試験の被験者に渡した。これらの試験が適切に行われていない場合、試験データは信頼できず、私たちの臨床試験中の対象または私たちの候補製品を使用して治療された患者(将来的に任意の候補製品が承認された場合)は、深刻な損害のリスクに直面する可能性があり、これは製品責任訴訟を引き起こす可能性がある。
私たちの従業員、独立請負業者、主要な調査者、CRO、コンサルタントまたはサプライヤーは、法規基準と要求を遵守しないことを含む不適切な行為または他の不適切な活動に従事する可能性があります
私たちは、従業員、独立請負業者、主要な調査者、CRO、コンサルタント、またはサプライヤーが詐欺または他の不正活動に従事する可能性があるリスクに直面しています。これらの当事者の不正行為は、意図的、無謀、および/または不注意な行為、または以下の規定に違反する不正な活動を開示することを含む可能性がある:FDA法規は、FDAへの真の、完全かつ正確な情報の報告を要求する法律、製造基準、連邦および州医療詐欺および法律法規の乱用、または財務情報またはデータを真に、完全かつ正確に報告することを要求する法律を含む。これらの法律法規は、広範な価格設定、割引、マーケティングおよび販売促進、販売手数料、顧客インセンティブ計画、および他のビジネス計画を制限または禁止する可能性があります。これらの法的制約を受けた活動はまた、臨床試験中に得られた情報の不適切な使用または虚偽陳述、あるいは著者らの臨床前研究または臨床試験において虚偽データを作成することに関連し、これは規制制裁と私たちの名声に深刻な損害をもたらす可能性がある。
私たちは、私たちの従業員や他の第三者の不正行為を常に識別し、阻止することができるわけではありません。このような活動を発見し、防止するための予防措置は、未知または未管理のリスクや損失を効果的に制御することができないか、またはそのような法律や法規に準拠できないことによる政府の調査または他の行動や訴訟から私たちを保護することができない可能性があります。また、私たちは、起きていなくても、このような詐欺や他の不正行為を告発する可能性があるというリスクに直面している。もし私たちにこのような訴訟を提起し、私たちが私たちの権利を弁護または維持することに成功しなかった場合、これらの訴訟は、民事、刑事および行政処罰、損害賠償、罰金、連邦医療保険、医療補助および他の連邦医療保健計画から除外される可能性があり、契約損害、名声損害、潜在的利益および将来の収益の減少、および私たちの業務の削減を含む、私たちの業務に大きな影響を与える可能性があり、いずれも私たちの業務、財務状況、運営結果、または見通しに悪影響を及ぼす可能性がある。
私たちは第三者の製造と供給供給者に依存するため、私たちの研究開発、臨床前と臨床開発材料の供給は限られたり中断したりする可能性があり、あるいは数量あるいは品質は満足できないかもしれない。
私たちは第三者契約メーカーに依存して、臨床前研究と臨床試験のための候補製品を生産します。私たちは臨床試験製品を生産する製造施設を持っていない。著者らの臨床前と臨床開発製品の供給が制限或いは中断されないことを保証することができず、またそれらが満足できる品質を持つことを保証することができず、或いは引き続き受け入れ可能な価格で供給することができない。候補製品の製造過程はFDAと外国規制機関の審査を受けなければならない。サプライヤーと製造業者は、適用された製造要求を満たし、cGMPのような規制基準に適合するために、監督機関の要求された厳格な施設とプロセス検証テストを受けなければならない。もし私たちのどの製造業者もこれらの要求を遵守できなかった場合、または品質、時間、または他の側面での私たちの義務を履行できなかった場合、または私たちの部品や他の材料の供給が他の理由で限られたり中断されたりした場合、私たちは自分で材料を製造することを余儀なくされるかもしれませんが、私たちは現在能力や資源を持っていない、あるいは他の第三者と合意していますが、私たちは合理的な条項でそうすることができないかもしれません。いずれの場合も,代替供給源の確立に伴い,われわれの臨床試験は著しく延期される可能性がある。場合によっては、私たちの候補製品を製造するために必要な技術的スキルまたは技術は、元の製造業者固有または独自である可能性があり、困難に遭遇する可能性があり、またはそのようなスキルまたは技術を予備または代替サプライヤーに譲渡することを禁止する契約制限が存在する可能性があり、またはそのようなスキルまたは技術を根本的に譲渡できない可能性がある。また、メーカーは、当該メーカーが独立して所有する我々の候補製品の製造に関する技術を持つことができる。これらの要素は、私たちの製造業者への依存を増加させ、あるいは他の第三者が私たちの候補製品を生産するために、その製造業者のライセンスを取得することを要求するだろう
GRI−0621、GRI−0803、および我々の他の候補製品の製造は、独占サプライヤーに依存するか、または場合によっては、限られた数のサプライヤーに依存する。もしこれらの供給者が私たちに必要な数量を供給できない場合、あるいは供給が全くできない場合、あるいは他の方法で私たちへの供給義務を滞納した場合、私たちは受け入れ可能な条件でタイムリーまたは根本的に他の供給者から代替供給を得ることができないかもしれない。さらに、これらのサプライヤーのいずれかが私たちとの契約に違反した場合、私たちがこのような違約に関連する法的救済は、私たちが受ける可能性のあるいかなる損害も補償するのに十分ではないかもしれません。また、何らかの理由でメーカーの交換を要求された場合、新しいメーカーの施設やプログラムが品質基準およびすべての適用された法規やガイドラインに適合しているかどうかを確認するように要求されます。我々はまた、例えば比較可能な研究を製造することによって、任意の新しい製造業者または製造プロセスが、以前にFDAまたは他の規制機関に提出された仕様に基づいて、私たちの候補製品を生産することを検証する必要がある。臨床用品の比較可能性の証明には成功しない可能性があり,追加の臨床試験が必要かもしれない。新メーカーの検証に関する遅延は、タイムリーまたは予算内で候補製品を開発する能力に悪影響を及ぼす可能性があります
GRI−0621、GRI−0803、または任意の他の候補製品の規制承認を得た場合、私たちは引き続き第三者メーカーに依存することが予想される。私たちが第三者とすでにまたは将来製造手配を達成した範囲内で、私たちはこれらの第三者に依存して、品質管理と保証に関する要求を含む契約と監督管理の要求に適合する方法でその義務を適時に履行する。もし私たちが候補製品の第三者製造を獲得したり維持したり、商業的に合理的な条項でそうすることができなければ、私たちは候補製品の開発と商業化に成功できないかもしれない。
私たちは将来的に第三者との協力を求めて、私たちの候補製品を開発して商業化することができるかもしれません。私たちの将来の協力は私たちの業務に非常に重要になります。もし私たちが協力できない場合、あるいはこれらの協力が成功しなければ、私たちの業務は不利な影響を受ける可能性がある
我々の戦略の一部は、パートナーが重大なビジネスおよび/または開発能力を増加させることができると考えられる標識および地理的位置におけるパートナーシップを考慮することである。しかも、私たちはまだ商業的な能力を持っていない。したがって、私たちは将来的に他の会社と協力して、私たちの計画や技術に重要な技術と資金を提供することができます。私たちが未来に行う任意の協力は、協力者が彼らが適用する仕事と資源を決定する上で大きな裁量権を持っていることを含む多くのリスクをもたらす可能性があり、予想通りに彼らの義務を履行できない可能性があります。協力者は、任意の未来の許可協定に基づいて、開発進捗や活動に関するタイムリーで正確な情報を提供できない可能性があり、これは、私たちが投資家に進捗状況を報告し、他の方法で候補製品を計画する能力に悪影響を与える可能性があります。私たちは、特許権、契約解釈、または第一選択開発過程の相違を含む協力者と意見が分かれている可能性があります。候補製品の研究、開発、または商業化の遅延または終了を招く可能性があります。または訴訟または仲裁につながる可能性があり、いずれも時間がかかり高価になり、協力が協力者によって終了する可能性があり、終了すれば、適用可能な候補製品をさらに開発または商業化するために追加の資金を調達する必要があるかもしれない。
私たちは私たちの候補製品のために適切なパートナーを探す上で激しい競争に直面しており、交渉過程は時間がかかり複雑である。私たちが私たちの1つまたは複数の候補製品との協力関係を成功させるためには、潜在的な協力者は、私たちが求めている条項や他の会社が許可できる製品だから、これらの候補製品を彼らが魅力的だと思う市場で経済的価値を持っているとみなさなければならない。協力の交渉と記録は複雑で時間がかかる。もし私たちが適時に、受け入れ可能な条項によって、または適切なパートナーと合意できない場合、私たちは候補製品の開発を削減し、その開発計画または私たちの1つまたは複数の他の開発計画を減らしたり、その潜在的な商業化を延期したり、いかなる販売またはマーケティング活動の範囲を縮小したり、または私たちの支出を増加させ、自費で開発または商業化活動を行わなければならないかもしれない。もし私たちが私たちの支出を増やし、私たち自身の開発や商業化活動に資金を提供することを選択すれば、私たちはより多くの専門知識と追加の資本を得る必要があるかもしれないが、これらは私たちが受け入れられない条件であるか、あるいは全く得られないかもしれない。もし私たちが将来の協力を達成できなかったり、必要な開発および商業化活動を展開するのに十分な資金や専門知識がなければ、私たちの候補製品をさらに開発し、それらを市場に出し、薬品販売から収入を生み出したり、私たちの技術を開発し続けたりすることができない可能性があり、私たちの業務は実質的な悪影響を受ける可能性がある。私たちが新たな戦略協力を成功させたとしても、私たちが合意した条項は私たちに不利になる可能性があり、例えば、候補製品の開発や承認が延期または承認された製品販売に失望した場合、私たちはこのような戦略的協力を維持できないかもしれない。私たちの候補製品に関連する任意の新しい戦略協力協定の任意の遅延は、私たちの候補製品の開発と商業化を延期し、市場に進出してもそれらの競争力を低下させる可能性がある。
管理業務と運営に関するリスク
もし私たちが重要な管理職を失った場合、あるいは私たちがより多くの高技能者を募集できない場合、私たちは既存製品候補製品を開発したり、新製品候補製品を決定して開発する能力が損なわれ、市場シェアや市場シェアを失ってしまい、私たちの競争力を低下させる可能性があります。
競争の激しい生物技術と生物製薬業界における私たちの競争能力は私たちの高い素質管理、科学と医療人員を引き付ける能力にかかっている。私たちはW.Marc Hertz、私たちの社長とCEO、私たちの最高科学者Vipin Kumar Chaturvedi、そして私たちの最高医療責任者Albert Agoを含む私たちの管理、科学、そして医療者に非常に依存している。私たちの幹部、他の重要な従業員、他の科学と医療コンサルタントのサービスを失って、私たちは適切な代替者を見つけることができなくて、製品開発の遅延を招き、私たちの業務を損なう可能性があります。
私たちはカリフォルニア州ラホアの工場で事業を展開しています。この地域は多くの他のバイオテクノロジー会社、生物製薬会社、研究機関の本部だ。私たちの市場は技術者に対する競争が非常に激しく、私たちが受け入れられる条件で高い素質の人員を採用し、維持する能力を制限する可能性があり、甚だしきに至っては根本的にはできない
価値のある従業員をわが社に引き付けるために、給料や現金インセンティブのほか、時間の経過とともに付与された持分奨励を提供しています。時間が経つにつれて、従業員に付与された株式奨励の価値は、私たちの株価変動の大きな影響を受ける可能性があり、これらの変動は私たちの制御範囲を超えており、いつでも他社が提出したより利益のある見積もりを相殺するのに十分ではないかもしれない。私たちは価値のある従業員を引き留めるために努力しているにもかかわらず、私たちの管理、科学、開発チームのメンバーは短時間で私たちとの雇用関係を終了するかもしれない。私たちの重要な従業員は好きなような従業員であり、これは私たちのどの従業員も通知または通知せずにいつでも離職できることを意味します。私たちは私たちが持っているか加入可能な任意の“キーパーソン”保険証書が私たちのすべての重要な従業員の損失を十分に補償することができるという保証はない。私たちの成功はまた私たちが引き続き高技能の初級、中級と高級科学と医療人員を吸引、維持、激励できるかどうかにかかっている。
経営陣や他のキーパーソンを引き付けることができなければ、私たちの候補製品の開発や商業化に成功したり、他の方法で私たちのビジネス計画を実施することができないかもしれません。
近年、バイオテクノロジー産業は高い流出率を経験している。競争の激しい生物製薬業界における著者らの競争能力は、科学、医療、監督、製造と管理技能と経験を持つ高技能と経験者の能力を吸引、維持、激励することに依存する。私たちは将来合格者を引き付けることができないかもしれません。一部の原因は生物製薬会社間の限られた数の合格者に対する激しい競争です。私たちはそれと競争する多くの他の生物製薬会社がより多くの財務と他の資源、異なるリスク状況、そしてその業界でのより長い歴史を持つだろう。私たちの競争相手は、より高い報酬、より多様な機会、および/またはより良い職業昇進の機会を提供するかもしれない。これらすべての競争要因は、私たちが高素質の人材を誘致し、維持し続ける能力を制限する可能性があり、これは、私たちの候補製品の開発と商業化に成功し、現在の想定に沿って私たちの業務と運営を発展させる能力にマイナスの影響を与えるかもしれない。
私たちは私たちの情報システムをネットワーク攻撃から十分に保護できない可能性があり、サイバー攻撃は個人データを含む機密や独自の情報が漏洩し、私たちの名声を損なう可能性があり、私たちを重大な財務と法的リスクに直面させる可能性がある
我々の内部コンピュータシステムおよび任意の将来の協力者および他の請負業者またはコンサルタントのコンピュータシステムは、コンピュータウイルス、ネットワーク釣り、または他の許可されていないアクセス、自然災害、テロ、戦争、電気通信、および電気故障の破壊を受けやすい。このような事件が発生し、当社の運営中断を招く場合、私たちの開発計画および業務運営中断、財務損失、当社のビジネス機密または他の固有情報の損失、および当社の名声被害およびその他の負の影響をもたらす可能性があります。例えば、未来の臨床試験における臨床試験データの紛失は著者らの監督管理の承認作業の遅延を招く可能性があり、そして著者らのデータの回復或いは複製のコストを著しく増加させる。もしどんな中断やセキュリティホールが私たちのデータやアプリケーションを紛失したり、破損したり、機密または独自の情報を適切に開示しない場合、私たちは責任を招く可能性があり、私たちの競争地位が損なわれる可能性があり、私たちの候補製品のさらなる開発と商業化は延期される可能性がある
私たちの日常的な運営では、私たちは、私たちまたは私たちの第三者プロバイダが運営する情報技術システムに依存して、電子情報を処理、送信、保存します。私たちの製品発見作業では、名前、郵送住所、電子メールアドレス、電話番号、臨床試験情報など、様々な個人データを収集して使用することができます。成功したネットワーク攻撃は、知的財産権、データ、または他の流用資産の盗難または破壊、または他の方法で私たちの機密または独自の情報を危険にさらし、私たちの運営を混乱させる可能性があります。ネットワーク攻撃の頻度,複雑さ,強度が増加しており,発見されにくくなってきている.サイバー攻撃には不法行為が含まれている可能性があります
外国政府に敵対する行為、工業スパイ活動、電気通信詐欺およびその他の形態のネットワーク詐欺、有害マルウェアの配備、サービス拒否、社会工学詐欺、または他のデータセキュリティ、機密性、完全性、および可用性を脅かす手段。成功したサイバー攻撃は、運営中断を含むが、運営中断に限定されず、金融情報、商業秘密、財務損失、会社戦略計画の開示を含む機密商業情報が流用されることを含む深刻な負の結果をもたらす可能性がある。私たちは私たちの情報システムを保護するために資源を投入しているにもかかわらず、私たちはネットワーク攻撃が脅威であることを認識しており、私たちの努力が情報セキュリティホールを防止することを保証することはできません。これらの抜け穴は、私たちの業務、法律、財務、または名声を損なう、あるいは私たちの運営結果と財務状況に実質的な悪影響を及ぼすことになります。セキュリティホールや不正アクセスを防止または軽減し、個人データ(研究参加者の個人データを含む)を使用または開示できなかった行為は、州(州違反通知法)、連邦(例えば、連邦貿易委員会法案第5条)、国際法(例えばGDPR)による重大な責任を負い、私たちの名声に重大な悪影響を与え、私たちの研究能力に影響を与え、私たちの業務を混乱させる可能性がある
私たちは、私たちの第三者プロバイダに有効なセキュリティ措置を実施し、そのような任意の故障、欠陥、または違反を識別して修正することに依存します。私たちまたは私たちの第三者プロバイダが、私たちの情報技術システムおよびデータ完全性を効果的に維持または保護できなかった場合、または私たちの情報技術システムの重大な中断を予見、計画または管理できなかった場合、私たちまたは第三者プロバイダは、このようなネットワーク攻撃を予防、検出および制御することが困難である可能性があり、そのような攻撃は、上述した損失および医師、患者および私たちのパートナーとの紛争、規制制裁または処罰、運営費用、支出または収入損失の増加、民事責任または他の不利な結果をもたらす可能性があり、いずれも、私たちの業務、運営結果、財務状況、将来性、およびキャッシュフローに実質的な悪影響を及ぼす可能性がある。このような第三者がセキュリティホールを防止または軽減できなかったか、またはそのような情報を取得または開示することができなかったことは、私たちに同様の不利な結果をもたらす可能性がある。このようなセキュリティやデータプライバシー漏洩の影響を防止または軽減できなければ、訴訟や政府調査に直面する可能性があり、業務の潜在的な中断を招く可能性があります。例えば、2020年1月1日に発効したCCPAは、カリフォルニアの消費者のためのプライバシー権を創出し、ある個人データを処理するエンティティのプライバシーおよびセキュリティ義務を増加させる。CCPAは,違反行為に対する民事処罰と,データ漏洩訴訟の可能性を増加させたデータ漏洩に対する個人訴権を規定している。“包括的平和協定”は2023年1月1日に発効し、“包括的平和協定”の内容を強化する。HIPAAや臨床試験法規に制約された情報には例外があるが,CCPA(適用すれば)は依然として我々の業務に影響を与え,我々のコンプライアンスコストや潜在的責任を増加させる可能性があり,多くの類似した法律が連邦レベルで提出され,他の州で実際に実施されている。たとえば,データプライバシーやセキュリティにおける外国法律法規では,GDPRが2018年5月にEUで発効し,EUデータ主体の個人データ処理に厳しい要求が出されている.GDPRを遵守しなければならない企業は、より強力なデータ保護要件の規制法執行、および不適合であれば2000万ユーロまたは不適合会社の世界年収の4%までの罰金が科される可能性があるコンプライアンス義務およびリスクに直面しなければならない。
私たちの現在の業務は1つの場所に集中しており、私たちまたは私たちが依存している第三者は地震や他の自然災害の悪影響を受ける可能性があり、私たちの業務の連続性や災害復旧計画は、地震、病気の発生、または他の自然災害を含む深刻な災害の影響から私たちを十分に保護できないかもしれません。
私たちの現在の業務はカリフォルニア州ラホアの施設にある。洪水、火災、爆発、地震、極端な天気条件、医療流行病、電力不足、電気通信故障、または他の自然または人為的事故または事件のような計画外の事件は、私たちの施設や私たちの第三者契約製造業者の製造施設を十分に利用できず、特に日常生活において、私たちの財務および運営状況に大きなマイナス影響を与える可能性がある。その中のいくつかの自然事件は気候変化によって悪化するかもしれない。これらの施設を使用できないとコスト増加、候補製品の開発遅延、私たちの業務運営が中断する可能性があります。地震やその他の自然災害は、私たちの運営をさらに混乱させ、私たちの業務、財務状況、運営結果、見通しに実質的な悪影響を及ぼす可能性がある。さらに、世界的な気候変化は、いくつかのタイプの自然災害がより頻繁に発生したり、影響が大きくなったりする可能性がある。自然災害、停電、または他の事件が発生した場合、本社の全部または大部分を使用することができなくなり、私たちの研究施設や私たちの第三者契約メーカーの製造施設のような重要なインフラを破損したり、他の方法で運営を中断したりすることは難しいかもしれません。場合によっては、かなり長い間私たちの業務を継続することはできません。
深刻な災害や同様の事件が発生した場合、我々の既存の災害復旧および業務連続計画は十分ではないことが証明される可能性がある。私たちの災害復旧と業務連続計画の限られた性質のため、私たちは大量の費用を発生する可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性があります。私たちのリスク管理政策の一部として、私たちは私たちの業務に適していると思うレベルに保険範囲を維持します。しかし、これらの施設で事故や事件が発生した場合、保険金額がどんな損害や損失を補うのに十分な保証はできません。もし私たちの工場や私たちの第三者契約メーカーの製造工場が以下のような理由で稼働できなければ
事故や事件または任意の他の理由により、短い時間であっても、私たちの任意またはすべての研究開発プロジェクトは損害を受ける可能性がある。
我々の業務は,健康大流行やそれに依存する第三者が重要な製造施設,臨床試験地点集中,あるいは他の業務運営地域を持つ流行病の影響を受ける可能性がある
我々の業務は健康大流行や流行病の影響を受ける可能性があり,その臨床試験地点や他の業務運営が集中している地域では,それに依存する第三者メーカーやCROの運営に大きな中断を与える可能性がある。このような流行病或いは流行病は生産力に負の影響を与え、業務を混乱させ、臨床計画とスケジュールを遅延させる可能性があり、その重症度は任意のこのような流行病或いは流行病が私たちが正常な過程で業務を展開する能力に加えた任意の制限及び他の制限の長さと重症度に部分的に依存する。これらや類似した運営中断は、私たちの業務、運営業績、財務状況に悪影響を及ぼす可能性があります。
隔離、自宅滞在、および同様の政府命令、またはこのようなコマンド、閉鎖または他の業務運営行為の制限が発生する可能性があるとの見方は、米国および他の国の第三者製造施設の人員、または材料の利用可能性またはコストに影響を与える可能性があり、これは私たちのサプライチェーンを乱す可能性がある。これらの材料の多くは1つ以上のサプライヤーによって獲得される可能性があるが、任意の大流行による制限は、私たちのサプライチェーンを混乱させたり、私たちの候補製品のために十分な材料を得る能力を制限したりする可能性がある。
私たちの保険は私たちの財務状況に悪影響を及ぼす可能性のあるクレームに十分な保証レベルを提供しないかもしれません。
私たちは私たちがこのような規模とタイプの業務を満たすのに十分だと思う保険を維持する。しかし、私たちはいくつかの種類の損失が経済的に合理的に保険に加入できない、あるいは保険に加入できないと思う。
私たちは将来的に潜在的な集団訴訟や株主派生訴訟を含む証券訴訟の影響を受けるかもしれない。私たちが取締役やある上級職員と締結した賠償協定や、デラウェア州会社法(DGCL)は、取締役や上級職員としての身分やサービスとして生じる可能性のある責任について賠償することを求めているかもしれません。もしD&O保険がなければ、私たちの上級管理者と取締役が彼らが提供してくれたサービスによって法的訴訟を受けた場合、私たちはこのような訴訟のために抗弁あるいは賠償金額を支払います。これは私たちの財務状況、運営業績、流動性に重大な悪影響を与える可能性があります。
私たちの普通株、融資、資本要求に関連するリスク
私たちは私たちの普通株の株価が非常に不安定になると予想する。
私たちの普通株の市場価格はずっと大きな変動の影響を受け続ける可能性がある。歴史的には,バイオテクノロジーや他の生命科学会社証券の市場価格は特に不安定であり,毎日大幅に変動している場合でも同様である。私たちの普通株式市場の価格変動を引き起こす可能性のある要素には、これらに限定されない
•私たちは将来の候補製品のための規制承認をタイムリーに得る能力と、そのような承認を得るための遅延または失敗
•私たちはナスダック資本市場の上場要求と、私たちの普通株の任意の退市や潜在的な退市を守ることができます
•候補製品が承認された場合、ビジネス成功は得られなかった
•未来の候補製品を作る問題は
•現在と未来のどんな臨床試験の結果も
•パートナーは、キービジネスパートナー合意を含む重要な合意を締結、終了、または違反する
•任意の知的財産権を強制的に実行または保護するため、または他人の知的財産権を守るために提起された、実質的な進展、または任意の訴訟の終了;
•希釈性株式融資と重大株式証券発行公告
•ビジネスパートナーまたは競合他社は、新しい商業製品、臨床進展または不足、重大な契約、ビジネス関係または資本約束を発表する
•意味のある株式アナリストの報道や、アナリストが私たちの株の格付けを下げることができませんでした
•登録権協定に基づいて義務を履行する能力;
•重要な従業員の流失。
また、バイオテクノロジー業界の株式市場は全体的に大きな変動を経験しており、この変動は往々にして個別会社或いはある業界の経営業績とは無関係である。このような広範囲な市場変動はまた私たちの普通株の取引価格に悪影響を及ぼすかもしれない。
従来、ある会社の証券市場価格が変動した後、株主はこれらの会社に対して集団証券訴訟を起こすことが多かった。このような訴訟を起こすと、巨額のコストや経営陣の注意力や資源の移転を招く可能性があり、収益性や名声を深刻に損なう可能性がある。さらに、このような証券訴訟は、逆方向M&Aまたは私たちが従事している他のタイプのM&A活動の後に発生することが多い。もしそのような訴訟が提起されれば、私たちの業務に否定的な影響を及ぼすかもしれない。
将来的に私たちの証券を売却して発行することは、私たちの株主の持ち株比率をさらに希釈し、私たちの株価を下落させる可能性がある。
将来的には、研究開発、マーケティングの増加、新しい人員の募集、私たちの製品の商業化、運営中の上場企業として活動を継続するなど、私たちの計画中の業務を継続するために多くの追加資本が必要になると予想されています。ある程度、私たちは株式証券を発行することで追加資本を調達し、私たちの株主は深刻な希釈を経験するかもしれない。私たちは1回または複数回の取引で、私たちの普通株、転換可能証券、または他の株式証券の株を時々決定された価格と方法で売ることができる。もし私たちが1回以上の取引で普通株、転換可能証券、または他の株式証券を販売する場合、投資家はその後の売却によって深刻に希釈される可能性がある。このような売却は私たちの既存株主の実質的な希釈を招く可能性もあり、新しい投資家は私たちの既存株主よりも優れた権利を得ることができるかもしれない
2023年12月31日まで、全部で(I)530,375株の普通株の直接或いは間接関連株式承認証((I)116,353株の普通株関連A-1シリーズの株式承認証を含み、A-1シリーズの株式承認証はTシリーズの株式承認証を行使する時に発行することができ、Tシリーズの株式承認証はすでに現金で本店の使用価格を支払い、及び(Ii)116,353株の普通株関連A-2シリーズの株式証は普通株株式を購入することができ、A-2シリーズの株式承認証はTシリーズ株式証の行使時に発行することができる。T系列株式承認証が現金で本店権価格を支払うことによりすべて行使されたと仮定すると,(Iii)4,576株普通株は,普通株を購入する他の発行された株式承認証の基礎とし,(Iv)4,318株普通株は,改訂·改訂されたGRI Bio,Inc.2018年持分インセンティブ計画(前身はVallon PharmPharmticals,Inc.2018持分インセンティブ計画)の下で未償還オプションがある場合に発行することができ,および(V)追加の28,324株普通株は,その日まで帰属制限されたオプションを行使する場合に発行可能であり,GRI BRI,未来に発行可能なオプションがない場合に発行することができると仮定する.Inc.2015年持分インセンティブ計画(GRI運営計画)またはA&R 2018計画。購入契約に基づいて証券を発行する場合、A-1シリーズ株式証の条項によると、A-1シリーズ株式承認証の行価格は額面、即ち1株当たり0.0001ドルに低下する。
2023年12月31日現在,交換権証行使時に合計60,227株の普通株が発行され,A−2シリーズ株式承認証行使時に合計163,185株の普通株が発行され,いずれの場合もキャッシュなしで発行されており,会社は何の収益も受けていない。私たちは無現金ベースで行使された引受証から何の収益も得ないだろう。これらの証券の所有者またはその関連会社は、過去および将来に私たちの普通株の取引価格を低くし、私たちの株主に深刻な希釈をもたらす可能性がある公開市場またはひそかに協議された取引で私たちの普通株を大量に販売する可能性がある。また、このような大量の普通株の登録と上場取引は、私たちの株価の変動性を増加させたり、私たちの株価に大きな下振れ圧力を与えたりする可能性がある
私たちは予測可能な未来に何の配当金も支払わないと予想している。
私たちの現在の期待は、私たちが未来の収益を維持し、私たちの業務の発展と成長に資金を提供することだ。したがって、予測可能な未来には、私たち普通株の資本付加価値(あれば)が私たちの株主の唯一の収益源になるだろう。
上場企業としては、大幅なコスト増加を招き続け、当社の経営陣はコンプライアンスイニシアティブに多くの時間を投入することを求められます。また、適切かつ効率的な内部統制を維持できなければ、正確な財務諸表をタイムリーに作成する能力が損なわれる可能性がある。
上場企業として、私たちは“証券取引所条例”、改正された“2022年サバンズ-オキシリー法案”(SOX)や他の適用される証券規則や法規に基づいて、大量の法律、会計、その他の費用を発生させている。さらに、私たちはナスダック株式市場有限責任会社(ナスダック)とナスダック資本市場の規則を守らなければならない。
これらの規則は上場企業に様々な要求を加え、有効な情報開示と財務制御、適切な会社管理やり方の確立と維持を要求している。私たちの経営陣と他の人たちはこのようなコンプライアンス計画に多くの時間を投入し続けるだろう。しかも、このような規則と規制は私たちの法律と財務コンプライアンスコストを増加させ、いくつかの活動をより時間的で高価にする。例えば、これらの規則および条例は、取締役および高級管理者責任保険をより難しく、より高価にすることを可能にし、同じまたは同様の保険を得るために、低減された保険限度額および保険範囲を受け入れることを要求されるかもしれない。また、ナスダック資本市場の上場要求は、取締役の独立性、株主会議、承認と投票、依頼書の募集、利益衝突と行為準則に関連するある会社の管理要求を満たすことを要求している。私たちの経営陣と他の人たちは私たちがこのすべての要求を遵守することを確実にするために多くの時間を投入しなければならない。したがって、私たちは合格者を私たちの取締役会、私たちの取締役会委員会、あるいは執行役員に引き付けることが難しいかもしれません。
SOXは、他の事項に加えて、財務報告および開示制御および手続きに対して効率的な内部統制を維持することを要求する。したがって、経営陣がSOX第404条(第404条)の要求に応じてこれらの制御の有効性を報告できるように、財務報告の内部統制を定期的に評価することが要求される。また、私たちの独立監査人は、同様の評価を要求される可能性があり、財務報告書の内部統制の有効性について報告することができる。第404条と関連条例を遵守するこれらの努力は、大量の財政と管理資源を必要とする約束を継続するだろう。財務報告書および第404条の他のすべての側面に対する内部統制の完全性を維持することが予想されるが、将来的に我々の制御システムの有効性をテストする際に大きな弱点が発見されないことは確認できない。重大な欠陥が発見されれば、私たちは米国証券取引委員会や他の規制機関の制裁や調査を受ける可能性があり、これは追加の財務と管理資源、コストの高い訴訟、あるいは大衆が私たちの内部統制に自信を失っていることが必要であり、これは私たちの株式の市場価格に悪影響を及ぼす可能性がある
私たちは現在、より小さな報告会社に適した開示と管理要件を利用しており、これにより、私たちの普通株の投資家に対する吸引力が低下する可能性がある。
我々の公開流通株は2.5億ドル未満であるため,米国証券取引委員会規則により,規模の小さい報告会社になる資格がある。規模の小さい報告会社として、簡略化された役員報酬開示及び低減された財務諸表開示要求などの低減された開示要求を利用することができ、米国証券取引委員会に提出された文書に提出することができる。私たちは比較的規模の小さい報告会社であるため、米国証券取引委員会に提出された文書に開示されている情報が減少し、投資家が私たちの運営結果や財務見通しを分析することを難しくする可能性がある。私たちはもし私たちがこのような免除に依存すれば、投資家が私たちの普通株の吸引力の低下を発見するかどうか予測できない。したがって、一部の投資家が私たちの普通株の吸引力が低下していることを発見すれば、私たちの普通株取引市場はそれほど活発ではなくなり、私たちの株価はもっと変動するかもしれない。私たちは、私たちがもう小さな報告会社ではなく、私たちの公衆流通株が2.5億ドルを超えると、この地位が終了するまで、より小さな報告会社に適用される報告免除を利用するかもしれない。この場合、もし私たちの年収が1.00億ドルを下回って、そして私たちの上場流通株が7.00億ドルを下回ったら、私たちは依然として規模の小さい報告会社になることができます。
新興成長型企業の減少に適した開示やガバナンス要件も利用しており、投資家に対する私たちの普通株の吸引力の低下を招く可能性がある。
“雇用法案”の定義によると、私たちは“新興成長型会社”です。新興成長型企業は、これらの基準が民間企業に適用されるまで、新たなまたは改正された会計基準の採用を延期することができる。新興成長型企業としては、第404条の監査人認証要求を遵守することは要求されておらず、定期報告及び依頼書で役員報酬に関する開示義務を削減し、役員報酬について拘束力のない諮問投票を行うことや、株主が以前に承認されていない金パラシュート支払いを承認するという要求を受けない。また、新興成長型企業として、これらの基準が民間企業に適用されるまで、上場企業と民間企業に対して異なる発効日を有する新規または改訂された会計基準の採用を延期することを選択した。したがって、私たちの財務諸表は、上場企業の発効日に該当する会社と比較できない可能性があります。私たちは投資家が私たちがこのような条項に依存して私たちの株の魅力が低下していることを発見する可能性があるかどうかを予測できない。したがって、一部の投資家が私たちの株の吸引力が低下していることを発見すれば、私たちの株式取引市場はそれほど活発ではなくなり、私たちの株価はもっと変動するかもしれない。
また、雇用法第102条(B)(1)条免除新興成長型企業は、民間企業(すなわち、施行が宣言されていない証券法登録声明又は“取引法”に基づいて登録されていない証券種別)が、新たな又は改正された財務会計基準の遵守を要求されるまで、新たな又は改正された財務会計基準を遵守する。JOBS法案では,会社は延長からの移行期間を選択し,非新興成長型会社に適用される要求を遵守することができると規定しているが,いずれの選択脱退も撤回できない。私たちは、この延長された移行期間を採用しないことを選択しており、これは、基準が発表または改訂された場合、その基準が上場企業または民間企業に対して異なる適用日があれば、私たちは新しい成長型企業として、民間企業が新しい基準または改正基準を採用することが要求されるまで、新しい基準または改正基準を採用しないことを意味する。これは、私たちの財務諸表を別の上場企業と比較させる可能性があります。別の上場企業は新興成長型会社でも新興成長型会社でもありませんので、使用する会計基準の潜在的な違いにより、延長された過渡期間を使用しないことを選択することは困難または不可能です。
(I)第2四半期末までに非関連会社が保有する普通株式時価が7億ドルを超える事業年度が終了するまで新興成長型会社とし、(Ii)当該事業年度の総収入が12.35億ドルを超える事業年度が終了するまで、(Iii)3年以内に10億ドルを超える転換不能債券を発行する期日、又は(Iv)証券法に基づいて提出された有効登録声明は、普通株を初めて売却した日から5周年後の財政年度が終了する。
私たちの普通株の上場は現在ナスダック資本市場や他のナスダック市場の規則に適合していません。私たちの普通株がナスダックから撤退することは、株式証券を公開または非公開で売却することで追加資本を調達する能力と、私たちの投資家が私たちの普通株式を処分したり、正確な一般株式市場値の見積もりを得る能力に悪影響を及ぼす可能性があります。
ナスダック資本市場ルールはまた、普通株の終値を1株当たり少なくとも1.00ドル(最低入札価格ルール)に維持することを求めている。2024年1月5日、私たちはナスダック従業員から手紙を受け取り、私たちは普通株の終値が30営業日連続で1.00ドルを下回ったので、ナスダック上場規則第5550(A)(2)条に規定されている最低入札価格規則に適合しなくなったことを指摘した。この手紙は通知(以下のように定義する)に対する補足である.この手紙は私たちが引き続きナスダック資本市場に上場することに影響を与え、直ちに影響を与えないだろう。ナスダック上場規則第5810(C)(3)(A)条によると、最低入札価格規則を再遵守するために、180暦の期限、または2024年7月3日(コンプライアンス日)に期限があります。私たちの普通株が180日間の期間内に10営業日連続の最低終値が1株当たり少なくとも1.00ドルである場合、ナスダックがその裁量権を行使しない限り、最低入札価格規則に適合する。もし私たちがコンプライアンス日までにコンプライアンスを回復できなければ、私たちは適用されるナスダック上場規則の条件を満たすことを前提として、180暦の追加期限を得る資格があるかもしれない。コンプライアンス日までに、私たちの普通株の連続10取引日の終値が1株当たり0.10ドル以下であれば、従業員はナスダック上場規則第5810条に基づいて、私たちの普通株について従業員の退市決定を発行します。2024年1月29日、2024年1月の逆株式分割を実施し、最低入札価格ルールを再遵守しようとする憲章改正案を提出した。ナスダックはその後、私たちの普通株は180日間の連続10取引日の終値が1株1.00ドルを超えたが、ナスダックは裁量権を行使し、10日の期限を延長し、最低見積規則を再遵守したとは思わないと報告した。コンプライアンスを再獲得できる保証はありませんし、普通株の入札価格が分割後の任意のナスダック監視期間や他の期間に要求される最低入札価格を1ドル以上維持する保証もありません。私たちは最低入札価格規則を再遵守するために、私たちの普通株を追加的な逆株式分割を行う必要があるかもしれないが、このような分割が承認または完了する可能性があるという保証はない
2023年11月22日、私たちはナスダック(ナスダック)上場資産部(従業員)から手紙を受け取り、2023年9月30日までの10-Q表四半期報告書で提供された情報に基づいて、ナスダック資本市場に引き続き上場する最低株主権益要求を遵守していないことを通知した。ナスダック上場規則第5550(B)(1)条の要求によると、ナスダック資本市場に上場する会社は、その上場証券の時価が3,500万ドルを下回り、年間純収益が50万ドル未満であり、少なくとも250万ドルの株主権益(株主権益要求)を維持しなければならない。ナスダックの規定によると、私たちは2024年1月8日までに株主権益要求を再遵守する計画(コンプライアンス計画)を提出することを要求されている。この通知は当社が引き続きナスダック資本市場に上場することに即時的な影響はありませんが、当社が他の継続上場規定を遵守しているかどうかに依存しなければなりません。2024年1月22日、従業員は、株主権益要求を再遵守するために、2024年5月20日まで期限を延長することを許可した。従業員2024年1月22日の書簡によると、私たちは株式発行を完了して、少なくとも600万ドルの毛収入(株式発行)を調達し、2024年5月24日までに公開利用可能な報告を提出し、従業員とナスダックに株主の持分要求を遵守する証拠を提供しなければならない。私たちは株式発売を完了しましたが、得られたお金は私たちが再び株主の権益要求に符合するのに十分ではありません
近いうちに。2024年6月30日までの四半期報告で株主権益要求に適合していることが証明できなければ、私たちはカードを外される可能性がある。私たちが再び遵守を得ることができるという保証はない。
もし私たちの普通株がナスダックによって取引された場合、私たちの普通株は場外取引市場または他の場外取引市場で取引する資格があるかもしれませんが、もし私たちがブランドを外したり、市場から撤退したり、これらの市場で取引された場合、私たちは株式証券を公開または個人的に売却することで追加資本を調達し、投資家に私たちの普通株を処分したり、正確な一般株式市場価格の見積もりを得させたりする可能性が高いです。しかも、私たちの普通株がこのような代替取引所や市場で取引する資格があることは保証されない。
我々の普通株がナスダック資本市場のような国家証券取引所に上場しない限り、我々の普通株も“細価格株”取引に関する規制および制限を受ける可能性があり、“細価格株”とは、1株当たり取引価格が5ドル未満の証券を指し、適用法規に規定されている他の免除により、細価格株の定義を免除することはできない。これらの要求と条例は、関連するコンプライアンス活動に従事したいブローカーや取引業者が減少する可能性があるため、二次市場における証券の流動性を深刻に制限する可能性がある。われわれの普通株が国家証券取引所に上場していない場合、細価格株取引に関する規則や制限は、投資家が第三者に株式を売却する能力を制限する可能性があり、二級市場での取引活動は減少する可能性がある。
また、吾等が適用されるナスダック上場要求を遵守できなかったことも、吾等がTシリーズ株式承認証の行使を要求する株式条件を満たしていないことを招き、吾等が各投資家合意に基づいて罰金責任を負わなければならない可能性がある。上記のいずれも、我々の業務、経営結果、見通し、および普通株の価値に悪影響を及ぼす可能性がある。
このような理由やその他の理由で退市することは、私たちの資金調達能力に大きな影響を与え、私たちの業務と私たちの普通株の価格に悪影響を及ぼす可能性があります。
2024年1月の逆株式分割は、分割前に対する我々の株価の価値を低下させ、普通株の流動性を減少させる可能性がある。
我々は2024年1月29日に法に基づいて2024年1月の逆株式分割を実施し、2024年1月30日に、私たちの普通株は分割後の基礎で取引を開始した。2024年1月の逆株式分割がなくても、私たちが発行した普通株の実際の価値が低下することは保証されません。2023年4月の逆株式分割後、私たちの株式の取引量は異なり、2023年4月の逆株式分割は、私たちの普通株の株式流動性を低下させる可能性があります。2024年1月の逆株式分割後の流通株数が減少したことを考慮すると、2024年1月の逆株式分割の悪影響を受ける可能性があり、特に2024年1月の逆株式分割により我々普通株の市場価格が増加しなければ。また、2024年1月の逆株分割は、我々の普通株を持つ奇数ロット(100株未満)の株主数を増加させる可能性があり、これらの株主がその株を売却するコストの増加と、このような売却を実現することをより困難にする可能性がある。現在の入札価格が最低入札価格ルールに適合していないことから、普通株の追加的な逆株式分割を完了する必要があると予想されるが、このような分割が承認または完了する保証はない。
株式の逆分割後、私たちの普通株の市場価格は新しい投資家を引き付けることができない可能性があり、機関投資家を含め、これらの投資家の投資要求を満たすことができない可能性がある。したがって、私たちの普通株の取引流動性は改善されないかもしれない。
私たちの普通株の高い市場価格は、より大きな投資家の興味を引き出すのに役立つかもしれないが、2024年1月の逆株式分割を含む逆方向株式分割は、機関投資家を含む新しい投資家の株価を引き付けることにつながると信じている。しかも、私たちの普通株の市場価格がこのような投資家の投資要求を満たすという保証はない。したがって、私たちの普通株の取引流動性は必ずしも改善されるとは限らない。2024年1月に逆株式分割を行う主な目的は、ナスダック上場要求を満たす最低入札価格ルールを支援するために、私たちの普通株の価格を向上させることですが、2024年1月の逆株式分割は、私たちの普通株価格が最低入札価格ルールに達していません。したがって、私たちは私たちの普通株を追加的に逆方向株式分割する必要があるかもしれないが、これは承認されたり完了したりしないかもしれない。逆株式分割が私たちの普通株市場価格の持続的な割合を上昇させることは保証されない。これは多くの要素に依存して、私たちの業務と財務表現、一般市場状況と未来の成功の見通しを含むが、これらの要素は私たちが発行した普通株の数量とは関係がない。実際、2024年1月の逆株式分割は、株式発行とその後の引受権行使の希釈効果が原因の一部であり、私たち普通株の市場価格が比例的に上昇し続けていない。株式の逆分割や上場後しばらくの間、会社普通株の市場価格が下落することは珍しくない。
税法の変化は私たちの業務に悪影響を及ぼすかもしれない。
アメリカ国税局、アメリカ財務省、その他の政府機関はアメリカ連邦、州、地方所得税に関する規則を絶えず審査している。税法の変化(これらの変化は追跡力を持つ可能性がある)は、私たちまたは私たちの普通株の所有者に悪影響を及ぼすかもしれない。近年、このような変化は多く発生しており、未来も変化し続けるかもしれない。将来の税法の変化は、私たちの業務、キャッシュフロー、財務状況、または運営結果に実質的な悪影響を及ぼす可能性があります。
私たちの普通株の活発な取引市場は発展しないかもしれません。私たちの株主は彼らの普通株を転売して利益を稼ぐことができないかもしれません。もしあれば。
私たちの普通株の活発な取引市場は永遠に発展したり持続しないかもしれない。もし私たちの普通株の活発な市場が発展したり持続したりしなければ、私たちの株主は彼らの株を魅力的な価格で売ることが難しいかもしれない。
株式研究アナリストが我々、我々の業務又は我々の市場に関する研究又は報告を発表しない場合、又は不利な研究又は報告を発表しなければ、我々の株価及び取引量は低下する可能性がある。
私たちの普通株の取引市場は株式研究アナリストが発表した私たちと私たちの業務に関する研究と報告書の影響を受けるだろう。株式研究アナリストは今のところなく、私たちの普通株の研究報告を提供することを選択しない可能性もあり、このような不足研究報告は私たちの普通株の市場価格に不利な影響を与える可能性がある。もし私たちが株研究アナリストの報告書を持っていたら、私たちはアナリストや彼らの報告書に含まれている内容と意見を制御できないだろう。1つ以上の株式研究アナリストが私たちの株式格付けを引き下げたり、他の不利なコメントや研究を発表したりすれば、私たちの普通株の価格は下落する可能性がある。1つ以上の株式研究アナリストが私たちの報告書を停止したり、私たちの報告書を定期的に発表できなかったりすれば、私たちの普通株に対する需要が減少する可能性があり、これは逆に私たちの株価や取引量を低下させる可能性がある。
私たちは1つまたは複数の株主派生訴訟または集団訴訟の被告になる可能性があり、将来のどのような訴訟も、私たちの業務、財務状況、運営結果、およびキャッシュフローに悪影響を及ぼす可能性がある。
私たちと私たちのいくつかの上級管理者および役員は、将来の1つまたは複数の株主派生訴訟または他の集団訴訟の被告になる可能性がある。これらの訴訟は、私たちの経営陣の正常な業務運営に対する関心と資源を移し、その弁護に関連する巨額の費用(大量の弁護士費や他の専門顧問費用に限定されないが、このような訴訟当事者になっているか、またはそのような前任の上級管理者や取締役への賠償の潜在的な義務を含む)を生じる可能性がある。もしこれらの訴訟が本当に発生した場合、私たちは実質的な損害賠償の支払いを要求されるかもしれないし、未来の行動の禁止および/または他の罰、救済、または制裁に同意することに同意するかもしれない。また、将来的にはどのような株主訴訟も、私たちの名声、私たちの候補製品を開発し続ける能力に悪影響を与え、収入を創出する能力を損なう可能性があります。したがって、これらの問題の最終的な解決は私たちの業務、財務状況、経営結果、キャッシュフローに重大な悪影響を与える可能性があり、それによって私たちの普通株の取引価格に負の影響を与える可能性がある。
私たちの定款とデラウェア州法律は株主が有利だと思う買収を阻止するかもしれない。
当社の会社登録証明書(憲章)および当社の改正·再記述された定款(定款)のいくつかの条項およびデラウェア州法律の適用条項は、株主がその株式からプレミアム取引を得る可能性があること、または私たちの株主がその最適な利益に適合すると考える取引を含む、実際または潜在的な支配権変更または管理層変更に関連する取引を遅延または阻止する可能性がある。私たちの憲章と付例の規定:
•誰が株主総会を開催できるかを制限する
•累積投票権は規定されていない
•定員に満たなくても、当時在任していた取締役の大多数が賛成票を投じてこそ埋めることができることになっている
•デラウェア州衡平裁判所はある法律の請求の独占的な裁判所になると規定されている
•アメリカ合衆国連邦地域裁判所は“証券法”に基づいて法律クレームを行う独占的なフォーラムになると規定されている。
さらに、デラウェア州会社法第203条は、特定の条件が満たされない限り、議決権のある株式を発行した15%以上の人と業務統合を行う能力を有する実益を制限することができる。これが
株式を買収した後、制限期間は3年となる。これらの規定は、我々の管理チームを強化し、現行価格よりも高い割増でその株を潜在的な買収者に売却する機会を株主に奪う可能性がある。このような制御権プレミアムが得られない可能性がある場合は、私たちの普通株の価格を下げるかもしれない
さらに、私たちの憲章は、私たちが書面で代替裁判所を選択することに同意しない限り、デラウェア州衡平裁判所は、(I)私たちを代表して提起された任意の派生訴訟または訴訟、(Ii)私たちの任意の取締役、上級管理職および従業員が私たちまたは私たちの株主に対して受託責任を負っていると主張する任意の訴訟、(Iii)DGCL、私たちの憲章または私たちの定款の任意の規定に基づいてクレームを提起する任意の訴訟、または(Iv)内部事務原則によって管轄されるクレームを主張する任意の訴訟に関連する、と規定されている。いずれの事件においても、衡平裁判所に管轄され、その中で被告に指名された不可欠な当事者に対して属人管轄権を持っている。これらの条項は,首相と裁判官をデラウェア州法と連邦証券法の適用においてより一致させ,特に会社紛争の解決に経験が豊富であり,他のフォーラムに比べてより速いスケジュールで事件を効率的に管理し,多裁判所訴訟からの負担から保護すると信じている。しかし、これらの規定は私たちの役員や上級職員に対する訴訟を阻止する役割を果たすかもしれない。他社の会社登録証明書に類似した裁判所選択地条項の実行可能性が法的手続きで疑問視されており、我々に提起された任意の適用訴訟において、裁判所は、憲章に記載されている選択地条項がこのような訴訟において適用されないか、または実行できないことを発見する可能性がある。この裁判所条項の選択は、取引法によって提起された任意の訴訟に対する連邦政府の排他的管轄権範囲を排除または制限しない。取引法第27条は,連邦政府が取引法又はその下の規則及び条例を実行するために生じた任意の義務又は責任に対して提起されたすべての訴訟に対して排他的連邦管轄権を有すると規定されている。したがって,排他的裁判所条項は,取引法で規定されているいかなる義務や責任を実行するための訴訟にも適用されず,連邦裁判所が排他的管轄権を持つ他のクレームにも適用されず,排他的裁判所条項を“取引法”クレームに適用するつもりもない.証券法第22条には,連邦裁判所及び州裁判所は,証券法又はその下の規則及び条例で規定されている任意の義務又は責任を執行するために提起されたすべての訴訟に対して同時管轄権を有すると規定されている。そのため、裁判所が証券法に基づくクレームに関する書面選択裁判所の規定を実行するかどうかには、まだ不確実性がある。さらに、この裁判所条項の選択は、デラウェア州衡平裁判所に対象物管轄権がないというクレームには適用されないだろう。憲章のフォーラム条項を選択することは、私たちの株主が連邦証券法とその規則と条例を遵守する義務を放棄することを招くことはない。
項目1 B.未解決の作業者意見
ない。
項目1 C:ネットワークセキュリティ問題について
ネットワーク·セキュリティ
私たちは、患者、業務パートナー、従業員の私たちの業務に対する信頼と信頼を維持することが重要であることを認識し、私たちの業務運営とシステムの機密性、完全性、可用性を保護するために努力している。私たちの取締役会は私たちのリスク管理活動を監督することに積極的に参加して、ネットワークセキュリティは私たち全体のリスク管理方法の重要な構成要素です。全体的に、我々は、ネットワークセキュリティ脅威を識別、予防、緩和し、ネットワークセキュリティイベントが発生したときに効率的に対応することに重点を置いて、収集および格納された情報の機密性、セキュリティ、および可用性を維持するために、包括的な方法によってネットワークセキュリティリスクに対応することを求めている。
ネットワークセキュリティリスク管理と戦略
不正アクセス、ネットワークセキュリティ攻撃、およびハッカーによって実装されたセキュリティイベント、およびハードウェアおよびソフトウェアシステムへの意図しない破損または中断、データ損失、および機密情報が流用されるなど、ネットワークセキュリティに関連するリスクに直面しています。ネットワークセキュリティ脅威からの重大なリスクを識別して評価するために、我々のシステムが有効であることを保証し、我々の内部および外部脅威を定期的に監視するセキュリティ監視計画を含む情報セキュリティリスクに備えたネットワークセキュリティ計画を維持し、私たちの情報資産の機密性および完全性を確保する。我々は,ネットワークセキュリティ脅威のリスクを他社リスクとともに考え,我々全体のリスク評価プロセスの一部としている.以下の“サイバーセキュリティ管理”でより詳細に議論されているように、我々の取締役会は、我々のネットワークセキュリティリスク管理および戦略プロセスを監視しており、これらのプロセスは、私たちの首席財務官がリードしている。
キーデータとシステムの可用性を提供し、監督管理のコンプライアンスを維持し、ネットワークセキュリティの脅威を受ける重大なリスクを管理し、ネットワークセキュリティ事件を防止と対応するために、私たちは以下の活動を行った
•新しいデータ保護法を監視し、これらの法律の要求に適合するように私たちの流れを修正します
•私たちの政策、やり方、契約(例えば、適用される)を通じて、従業員と私たちのサービスを提供する第三者に機密情報とデータに慎重に対応することを要求します
•ファイアウォール、侵入予防·検出システム、アンチマルウェア機能、およびアクセス制御など、我々の情報システムをネットワークセキュリティ脅威から保護するための技術保障措置を採用する
•私たちの従業員にネットワークセキュリティ脅威に関する訓練を提供し、1つの手段として、彼らに有効なツールを把握させてネットワークセキュリティ脅威に対応し、そして私たちが絶えず変化している情報セキュリティ政策、標準、プロセスと実践を伝達する。
私たちのイベント応答計画は、イベントの分類、評価の深刻さ、報告、抑制、調査と修復の流れ、および潜在的に適用される法的義務を遵守し、私たちの業務と名声に対する損害を軽減することを含む、私たちが準備、検出、応答、回復するために取った活動を調整する。
我々のプロセスはまた、第三者サービスプロバイダの使用に関連するネットワークセキュリティ脅威リスクを解決し、これらの第三者サービスプロバイダは、従業員データまたは我々のシステムにアクセスすることができる。また、ネットワークセキュリティ面の考慮は、第三者サービスプロバイダの選択と監視にも影響を与える。
我々は、タイトル下の項目1 Aに、従来の任意のネットワークセキュリティイベントのリスク、私たちに重大な影響を与えたかどうか、または私たちの業務戦略、経営成果、または財務状況を含む、決定されたネットワークセキュリティ脅威のリスクを説明した私たちは私たちの情報システムをネットワーク攻撃から十分に保護できないかもしれません。サイバー攻撃は、個人データを含め、機密や独自の情報の漏洩を招き、私たちの名声を損ない、重大な財務的、法的リスクに直面させる可能性がありますこれらの開示は、参照によって本明細書に組み込まれる。
過去3年間、私たちは何の重大なサイバーセキュリティ事件も経験しておらず、私たちがサイバーセキュリティ事件による費用も取るに足らない。これには処罰と和解が含まれているが、どんな処罰も和解もない。
サイバーセキュリティ?ガバナンス
ネットワークセキュリティは私たちのリスク管理プロセスの重要な構成部分であり、私たちの取締役会と経営陣が注目している分野でもある。私たちの取締役会はサイバーセキュリティ脅威からの危険を監視する責任がある
ネットワークセキュリティ脅威を検出した後、我々の取締役会は、我々のネットワークセキュリティ脅威リスク管理と戦略プロセスに関する管理層の最新の情報と、このようなリスクに対応するための管理層のステップを受け取る。私たちの取締役会はまた、確立報告のハードルに適合したネットワークセキュリティイベントに関するタイムリーな情報と、それが解決されるまで、そのようなイベントの継続的な更新についての情報を受け取ります
我々のネットワークセキュリティリスク管理と戦略プロセスは,我々の首席財務官が管理し,情報セキュリティの管理,ネットワークセキュリティ戦略の策定,必要に応じて効率的な情報やネットワークセキュリティ計画を実施する上で経験豊富なコンサルタントによって支援されている.上述したように、首席財務官は私たちの取締役会に重大なネットワークセキュリティ脅威リスクを報告した。
第2項:すべての財産を管理する
私たちの行政事務室はカリフォルニア州ラホア92037ラホア208号Avenida de La Playa#2223にあります。2024年3月に満期になったレンタルによって、私たちはここで約1,100平方フィートのオフィス空間を借りました。私たちは私たちの施設が私たちの現在の需要を満たすのに十分だと信じている。
3つ目:法的訴訟を継続する
私たちは現在、いかなる重大な法的手続きの当事者でもなく、私たちのいかなる未解決または脅威に対する法的手続きも知らず、私たちの業務、経営業績、または財務状況に悪影響を及ぼす可能性があると思います
4つ目:炭鉱の安全情報開示
適用されません。
第II部
第五項:登録者普通株、関連株主事項及び発行者が株式証券を購入する市場
私たちの普通株は現在ナスダック資本市場に上場しており、コードはGRIです。2024年3月1日現在、登録株主約17人が3196,488株の普通株と未発行の優先株を保有している
株式補償計画に基づいて発行された証券
第3部、第12項を参照。“ある実益所有者の保証所有権及び管理層及び関連株主事項”は、我々の持分補償計画に関する情報を知る。
最近売られている未登録証券
ない。
発行者および関連購入者が株式証券を購入する
ない。
配当政策
私たちは私たちの普通株について現金や他の配当金を発表したり、支払ったりしていないし、私たちは予測可能な未来にも現金や他の配当金を発表したり支払うことはないと予想される。
第6項。以下の内容:[保留されている]
プロジェクト七、会社経営陣の財務状況と経営成果の検討と分析
あなたは私たちの財務状況と経営結果に関する以下の議論と分析、並びに私たちの財務諸表と関連する注釈を読んで、本年度報告のF-1ページから始めるべきです。本議論および分析に含まれるまたは本年度報告の他の部分に記載されている情報は、リスクおよび不確実性要因に関する前向きな陳述を含む、我々の業務および関連融資の計画および戦略に関する情報を含む。多くの要因のため、“項目1 A”と題する節に列挙された要素が含まれる。本年度報告における“リスク要因”と“前向き陳述に関する特別な説明”によれば,我々の実際の結果は,本稿に含まれる前向き陳述に記述または示唆された結果とは大きく異なる可能性がある。
概要
我々は臨床段階の生物製薬会社であり,炎症,線維化,自己免疫疾患を引き起こす免疫反応失調に関連する重篤な疾患に対する発見,開発,商業化革新療法に専念している。これらの疾患を治療する療法の開発において業界の先頭となり,このような疾患患者の生活を改善することを目標としている。
我々の主要候補製品GRI−0621はiNKT細胞の経口阻害剤である。GRI−0621もタザロチンの経口製剤であり、タザロチンは合成RAR−βおよびガンマ選択性アゴニストであり、米国で乾癬およびざ瘡の局所治療のために許可されている。2023年12月31日現在、1700人以上の患者に経口製品として52週間にわたる評価が行われている。我々はIPFなどの重篤な線維性肺疾患の治療にGRI−0621を開発しており,生命を脅かす進行性肺線維化疾患であり,米国では約140,000人が影響を受けており,米国では毎年40,000例もの新しい症例があり,一部の人はIPFが全世界300万人に影響していると推定している。現在2つの承認された肺線維化治療法があるが,いずれも総生存率の改善には相関しておらず,両方法とも著明な副作用に関連しており,治療コンプライアンスが悪い。これまでのGRI−0621とタザロチン経口投与の早期試験の予備データでは,GRI−0621耐性が良好であり,被験者のiNKT細胞活性を抑制することが観察された。活性化したiNKTはIPF,PSC,NASH,ALD,SLE,MS,UCおよび他の適応患者で上昇することが他の人と証明されている。これらの患者では,活性化されたiNKT細胞がより重篤な疾患に関与している。2023年12月、2 a段階試験の登録を開始し、この試験のTOPLINE結果が2024年下半期に発表されることが予想されます。
私たちの候補製品の組み合わせはまた、GRI−0803および500種以上の化合物の専用ライブラリを含む。GRI−0803はライブラリーからスクリーニングされた先導分子であり,新規な2型NKT細胞経口アゴニストである。我々は自己免疫疾患の治療にGRI−0803を開発しており,SLEや狼瘡や多発性硬化症の大部分の臨床前作業において,免疫系は誤って自分の健康組織,特に関節や皮膚を攻撃しているが,身体のほぼすべての器官や組織に影響を与える。この場合は致命的である可能性があり,虚弱な疲労や疼痛発作をきたすことが多く,成人患者の半分近くが仕事ができなくなる。米国では狼瘡は16万から20万人の患者に影響しており,米国では約8万から10万人の患者が腎炎を有しており,SLEの最も深刻な表現の一つであり,通常確定診断後5年以内である。狼瘡は治癒できないが,医療介入やライフスタイルの変化はそれをコントロールするのに役立つ。全身性エリテマトーデスの治療は主に免疫系活動を抑制する免疫抑制薬を含む。過去50年間に2種類の薬剤のみがループスに承認され,新たな治療法が早急に必要とされてきた。INDの承認により,最初のSLEに対する1 aと1 b段階試験でGRI−0803を評価する予定である。我々は2024年上半期に1 aと1 b段階試験に関するINDを提出する予定である。適応を評価し続け,この計画のさらなる発展に最適な適応を選択するが,最初の重点は狼瘡であった。
最新の発展動向
証券購入協定
2024年2月1日に,吾等は買収合意)を締結し,これにより,(I)330,450株の普通株を公開発売および売却すること,(Ii)4,669,550株が合計4,669,550株の普通株を行使できる予備資本権証,(Iii)総額5,000,000株の普通株を行使できる5,000,000株B−1シリーズ普通権証,および(Iv)行使可能な5,000,000株B−2シリーズ普通権証,総収益は550万ドルであることに同意した。これらの証券の発売組み合わせは、(A)1株または1部の事前資本権証、および(B)B-1シリーズ一般権証とB-2シリーズ普通権証であり、総購入価格は1.1ドル(1部の事前資本金権証から0.0001ドル減算)である。
ある所有権制限の場合、株式承認証は発行時に行使することができる。各前払い資金株式承認証は1株当たり0.0001ドルの価格で普通株を行使することができ、しかも満期にならない。各B-1シリーズ普通権証は2024年2月6日に発行されてから5年以内に、1株1.10ドルの価格で1株普通株に転換することができる。各B-2シリーズ普通株式承認証は2024年2月6日に発行されてから18ヶ月以内に、1株1.10ドルの価格で1株普通株に転換することができる。購入契約に基づいて証券を発行する場合、A-1シリーズ株式証の条項によると、A-1シリーズ株式承認証の行価格は額面、即ち1株当たり0.0001ドルに低下する
2024年1月逆方向株式分割
2024年1月19日、私たちの株主は2024年1月の逆株式分割を承認した。私たちの取締役会は2024年1月の逆株式分割比率、すなわち7対1を承認した。承認された後、私たちは2024年1月の逆株式分割を午後4:01から実施するために、デラウェア州国務長官に憲章の改正案を提出した。東部時間2024年1月29日。我々の普通株の株式は2024年1月30日に分割後のベースで取引を開始した。2024年1月の逆株式分割のため、デラウェア州国務長官に提出された憲章改正案の提出と発効前に、私たちは普通株7株当たり、発行されたものも発行されたものも、自動的に合併して転換します(これ以上の行動は必要ありません)。2024年1月の逆株分割に関する断片的な株は発行されていない。そうでなければ、普通株式の断片的な株式を取得した株主は現金で代替され、その価格は株主が本来獲得する権利のある断片的な株式に等しい。2024年1月の逆方向株式分割は、普通株の流通株総数を逆分割前の4,520,233株から逆分割後の総流通株645,738株に減少させる。
他に説明がある以外に、本年報に掲載されているすべての財務資料、株式番号、オプション番号、株式承認証番号、その他の派生証券番号及び行権価格はすでに調整され、2024年1月の逆株式分割を実施する
ナスダックコンプライアンス-持分欠損
2023年11月22日、私たちはナスダック従業員から手紙を受け取り、2023年9月30日までの10-Q表シーズン報で提供された情報に基づいて、ナスダック資本市場に引き続き上場する最低株主権益要求に適合しないことをGRIに通知した。ナスダックの規定によると、私たちは2024年1月8日までに私たちのコンプライアンス計画を提出して、株主権益要求を再遵守することを要求された。2024年1月22日、従業員は、株主権益要求を再遵守するために、2024年5月20日まで期限を延長することを許可した。従業員2024年1月22日の手紙によると、私たちは株式発行を完成して提供しなければなりません
従業員とナスダックが株主権益要件を遵守していることを証明するために、2024年5月24日までに公開利用可能な報告書を提出する。株式発売を完了しましたが、得られたお金は株主の権益要求に再適合させるのに十分ではありませんので、短期的に大量の追加資金を集める必要があります。2024年6月30日までの四半期報告で株主権益要求に適合していることが証明できなければ、私たちはカードを外される可能性がある。
ナスダックコンプライアンス-入札価格不足
2024年1月5日、私たちはナスダック従業員から手紙を受け取り、私たちは普通株の終値が30営業日連続で1.00ドルを下回ったので、ナスダック上場規則第5550(A)(2)条に規定されている最低入札価格規則に適合しなくなったことを指摘した。この手紙は上記の通知に対する補足です。この手紙は私たちがナスダック資本市場に上場し続けることに効果がありません。ナスダック上場規則第5810(C)(3)(A)条によると、最低入札価格規則を再遵守するために、2024年7月3日までの180暦の期限がある。私たちの普通株が180日間連続して10営業日の最低終値が1株当たり少なくとも1.00ドルであれば、ナスダックがその裁量権を行使してこの10日間の期間を延長しない限り、最低入札価格規則に適合する。もし私たちがコンプライアンス日までにコンプライアンスを回復できなければ、私たちは適用されるナスダック上場規則の条件を満たすことを前提として、180暦の追加期限を得る資格があるかもしれない。規則日の前に、私たちの普通株の連続十取引日の終値が1株当たり0.10ドル以下であれば、従業員はナスダック上場規則第5810条に基づいて、私たちの普通株に対して従業員の退職決定を発行します。2024年1月29日、2024年1月の逆株式分割を実施し、最低入札価格ルールを再遵守しようとする憲章改正案を提出したが、再遵守できる保証はない。私たちの普通株の株価が分割後のいかなるナスダック監視期間または他の期間に要求される最低購入価格1.00ドル以上を維持することは保証されない。
Vallon製薬会社と合併した
2023年4月21日,我々は統合プロトコルに従ってGRI運営との統合を完了した.解決のために,会社登録証明書と定款を修正し,我々の名称を“Vallon PharmPharmticals,Inc.”から“Vallon PharmPharmticals,Inc.”に変更した。GRI Bio,Inc.へ
発効時期には
有効期間直前に発行された1株当たりGRI運営普通株(GRI運営普通株)は、株式融資(以下に定義する)に従って発行されたGRI運営普通株の任意の株式を含み、0.0374(交換比率)に相当する数の普通株を得る権利に自動的に変換される。
(A)GRI運営普通株株式を購入する各オプション(それぞれ,GRI運営オプション)は,GRI運営計画(GRI運営計画,帰属の有無にかかわらず)の発効時間直前に行使されず,普通株式株式を購入するオプションに変換され,吾らはGRI運営計画の条項(想定オプション)に基づいてGRI運営計画とその各GRI運営オプションを仮定している.各仮説引受権に制約された普通株式数は、(I)有効時間の直前に有効なGRI運営オプションに制約されたGRI運営普通株式数に(Ii)株式交換比率を乗じ、得られた数字を最も近い普通株式整数に丸め込むことによって決定される。各仮説株式購入時に普通株を発行できる1株当たりの権価は以下の方法で決定される:(A)発効直前に発効したこの仮説株式購入の1株当たりの権価を(B)両替比率で割って、得られた1株当たりの権価を最も近い整数仙に丸める。任意の仮説株式購入権を行使するいかなる制限も引き続き十分な効力を有しているが、このような仮説株式購入の期限、使用可能性、帰属スケジュール、および任意の他の条文は他の点で変わらない。
(B)GRI運営普通株株式を購入する1部当たりの株式承認証(GRI運営株式承認証)は、発効直前に当社が普通株を購入する権利証(株式承認証)に引受して変換し、その後(I)1部当たりの引受権証は普通株にのみ適用される。(Ii)仮説株式証明書に制約された普通株式数は、(A)有効期間直前に有効なGRI運営承認権証に制約された普通株式数に(B)交換比率を乗じ、得られた数字を普通株式に最も近い整数に丸めて決定され、(Iii)各仮想株式証を行使する際に発行可能な普通株の1行当たりの権価は、(A)即時発効GRI運営株式証制約を受けたGRI運営普通株の1株当たり権価によって決定される
発効時間の前に、(B)レートを両替し、それによって生成された行権価格を最も近い整数セントに丸める。
(C)過橋承認株式証(定義は以下参照)は、合計60,227株の自社普通株を購入するために、株式承認証(取引所承認株式証)と交換された。取引所株式証はブリッジ株式証と実質的に類似した条項を含み、初期行使価格は1株103.11ドルに相当する。それ以来、取引所株式証は現金なしで全面的に行使された。
(D)GRI Operations制限株式報酬に関連するすべての権利は、当社が負担し、会社限定株式報酬に変換し、報酬の株式数に株式交換比率を乗じ、得られた数字を最も近い普通株式整数に切り捨てる。GRIトラフィック制限株式報酬の期限、実行可能性、ホームスケジュール、および他の条項は、他の態様では不変のままである。
証券購入協定(ブリッジ融資)
合併協定の締結について,GRI運営会社はAltiumと2022年12月13日に発効する証券購入協定(Bridge SPA)を締結し,この協定によると,他の事項を除いて売却株主は元金総額約330万ドルの高級保証手形を購入し,GRI運営会社は合計約250万ドルの購入価格(ブリッジ手形)と引き換えに元金総額約330万ドルの優先保証手形を発行した。また,GRI運営はAltiumに引受権証を発行し,合計357,854株のGRI運営普通株(ブリッジ株式証)を購入した。合併の結果,発効期間中にブリッジ株式証は取引所承認株式証と交換される.取引所株式証はブリッジ株式証と実質的に類似した条項を含み、初期行使価格は1株103.11ドルに相当する。取引所株式証の行使価格は分割及び類似した資本再編事件によって調整される可能性がある。
証券購入協定(株式融資)
Bridge SPAを除いて、合併協定の締結について、当社、GRI運営とAltiumは2022年12月13日の証券購入協定(株式SPA)を締結した。株式SPAによると,GRI運営は取引終了直前にAltiumに969,602株のGRI運営普通株(初期株)を発行し,3,878,411株のGRI運営普通株(追加株)をホストエージェントに信託した.合併完了時には、合併により初期株式が合計36,263株普通株に変換され、追加株式が合計145,052株普通株に変換される。2023年5月8日、当社とAltiumは、株式SPAの条項に基づいて、実益所有権制限に適合した場合、追加株式を交換するために発行されたすべての普通株式(信託株式)をAltiumに支払うことを許可する。
株式特別行動計画によると、私は2023年5月8日にAltiumに(I)A-1シリーズ株式承認証を発行し、1株94.57ドルの初期行権価格で181,316株の普通株を購入した;(Ii)A-2シリーズ株式承認証は、1株103.18ドルの初期行権価格で163,185株の普通株を購入した(これ以降すべて行使した);及び(Iii)Tシリーズ株式承認証は、1株85.96ドルの行権価格で購入(X)116,353株普通株を購入し、及び(Y)Tシリーズ株式承認証は現金で総取引価格を支払い、全数行使した。追加額のA-1シリーズ株式承認証とA-2シリーズ株式承認証は、それぞれの使用価格で116,353株の普通株(総称して権証と呼ぶ)を購入する。ある持分条件を満たす場合にのみ、私たちはTシリーズ株式承認証を強制的に行使することができる。Tシリーズ株式承認証は現在、吾などの強制行使の権益条件の制限を受けていないが、Tシリーズ株式承認証が指す期間の普通株式価値の加重平均価格が少なくとも1株64.47ドルの要求を満たしていないためである。購入契約に基づいて証券を発行する場合、A-1シリーズ株式証の条項に基づいて、A-1シリーズ株式証の行使は額面、即ち1株当たり0.0001ドルに減少する。
肝心な会計政策
我々の財務諸表は米国公認会計原則(GAAP)に基づいて作成されている。これらの財務諸表を作成するには、財務諸表に報告されている資産、負債、収入および費用、または資産および負債の開示に影響を与える推定および判断を行う必要があります。我々は過去の経験,既知の傾向や事件,および様々な当時の状況に属すると考えられる合理的な要素から推定しているが,これらの要因の結果は資産や負債の帳簿価値を判断する基礎を構成しており,当該などの資産や負債の帳簿価値は他の出所から容易に見られるものではない。異なる仮定または条件では、実際の結果は、これらの推定値とは異なる可能性がある。
私たちの主な会計政策は、私たちの財務諸表付記により詳細に説明されていますが、以下の会計政策は、財務諸表作成に使用される判断と推定に最も重要であると考えられます。
株に基づく報酬
我々はASCテーマ718に基づいて従業員と非従業員の株式報酬の費用を確認した報酬--株式報酬それは.ASC主題718は、公正な価値に基づく方法を使用して、このような取引を計算することを必要とする。オプションの推定公正価値は、オプション付与日の公正価値に基づいて帰属中に償却され、Black-Scholesオプション定価モデルを使用して計算される。私たちは発生した罰金を計算に入れた
従業員の株式購入計画によって発行されたオプション株の公正価値を推定するには、著者らの普通株の推定公正価値、オプションの期待寿命、株価変動性、無リスク金利と期待配当を含む主観的仮定を入力する必要がある。ブラック·スコアーズオプション定価モデルで使用される仮説は、本質的に主観的であるため、多くの変数、不確実性および仮説、および管理層判断の適用に関連する管理職の最適な推定を代表する。もしどんな仮定が変化すれば、私たちの株に基づく報酬支出は未来に大きく違うかもしれない。
ブラック·スコアーズオプション定価モデルで使用されているこれらの仮定は以下のように推定される
•所期期限それは.十分な企業固有の履歴データが不足しているため、従業員オプションの予想期限は、米国証券取引委員会従業員会計公告第107号に規定される“簡略化”方法に従って決定される、すなわち、期待期間は、オプションの既得期限と元の契約期間との算術平均値に等しい。非従業員オプションの期待期間は契約期間に等しい
•予想変動率それは.予想変動率は我々の業界内の類似エンティティの履歴変動率に基づいており,これらの変動率はSAB第107号で述べた期待期限仮定に見合っている
•無リスク金利E.無リスク金利は,付与時に有効な米国債の対応金利に基づいており,期限は想定した期待期限に見合っている
•配当を期待するそれは.予想配当収益率は0%であり、私たちは歴史的に支払ったことがなく、予測可能な未来にも私たちの普通株の配当金を支払わないからだ。
賃貸借証書
私たちは2016-02年度会計基準更新(ASU)に基づいてレンタルを会計処理しますレンタル(テーマ842)アメリカとアリゾナ州立大学2018-10年度テーマ842“レンタル”の編纂改善ASU 2018-11とレンタル(テーマ842):的確な改善両方とも、ASU 2016-02で行われたいくつかの修正を明確にして強化した。ASUSは貸借対照表ですべての賃貸資産と賃貸負債を確認し、賃貸手配に関する重要な情報を開示することで、実体間の透明性と比較性を向上させた。オフィススペースの賃貸契約を締結しました。経営的な賃貸契約であることを確認しました。
財務運営の概要
研究と開発費
研究·開発費用には,研究·開発活動に関連する人員コストがあり,第三者請負業者による研究,臨床試験および薬物供給と材料の製造が含まれている
我々の研究開発費には,主に我々の主要候補製品GRI−0621の開発計画に関するコストが含まれている。これらの費用には
•従業員に関連する支出、例えば、賃金、花紅および福祉、顧問に関連する支出、例えば顧問費および花紅、株式ベースの報酬、管理支出に関連する支出、および研究者および開発者に関連する出張支出;
•CRO,CMO,研究実験室と我々の臨床前開発,プロセス開発,製造と臨床開発活動,およびわれわれの臨床·非臨床研究を実施するコンサルタントの合意を支援することによる費用。
私たちの直接研究開発費用は候補製品によって追跡されていますが、従業員コストと私たちの発見仕事、実験室用品、施設に関連するコスト(他の間接コストを含む)を特定のものに分配しません
候補製品は、このような費用が複数の計画に配置されているからだ。私たちが候補製品のための計画中の臨床と臨床前活動を展開することに伴い、私たちの研究開発費は今後数年で増加すると予想される。
一般と行政費用
一般及び行政支出は主に行政人員及びその他の行政人員の給与及びコンサルティング関連支出、専門費用及びその他の会社の支出を含み、法律及び会計費用、出張費用、施設に関連する支出及び会社事務に関するコンサルティングサービスを含む。
私たちは、ナスダック資本市場とアメリカ証券取引委員会が要求したサービス費用、役員と上級管理者の保険、法律と会計費用、投資家関係コスト、私たちがより多くの人員を雇用することによって増加した人件費を含む上場企業に関連するコストが発生するため、私たちの一般的かつ行政費用が大幅に増加することを予想します。
株式証法的責任
2022年5月、バロンは証券購入協定について株式承認証を発行した。VallonはASC 815-40に基づいて株式承認証を評価しデリバティブとヘッジ−実体自己資本の契約(ASC 815-40)、株式承認証のうち、場合によっては行使価格の低下に関連する条項は、株式承認証が株式構成要素に計上される可能性を排除すると結論付けられる。そこで,権利証は発行時の公正価値に応じてBlack-Scholes推定モデルを用いて計測し,貸借対照表に負債として入金する.株式承認証の公正価値は報告日ごとに計量し,公正価値の変動は変動期の総合経営報告書で確認した
その他の収入
2023年8月22日、Aardvark Treateutics,Inc.(Aardvark)と資産購入協定(Aardvark協定)を締結し、この合意に基づいて、Aardvarkは、Medice Arzneimittel P≡Tter GmbH&Co.kgとのライセンス契約に同意し、日付は2020年1月6日、(Ii)Adair候補製品に関連するいくつかの特許、および(Iii)注意欠陥/多動障害および傾眠症の治療のためのIND 133072号Adairに記載されている処方の文書(契約製造およびFDA通信)。協定の条項によると、私たちは30万ドルの前払い現金を受け取り、このお金は他の収入として確認された。私たちには、Aardvarkがいくつかの未来のAdair法規と販売マイルストーンを実現することに依存する潜在的な追加マイルストーン支払いを得る資格がある。前金に加えて、私たちは近いうちにAardvarkのマイルストーン支払いを受けないと予想され、これらの潜在的なマイルストーン支払いは将来実現、支払い、または受信されないかもしれない。
債務の弁済
債務返済には、TEPが保有するいくつかの部分変換可能チケット(TEPチケット)を修正することによって確認された費用が含まれる。改訂された債務の公正価値が原始債務の帳簿価値を超え、弁済損失をもたらした場合は弁済とみなされる。
利子収入,純額
利息支出には、債務割引償却、債務発行コスト、TEP手形と橋手形に関する利息支出が含まれる。利息収入には私たちの現金と機関銀行が持っている現金等価物から稼いだ利息が含まれています。
最近発表された会計公告
私たちはすべてのASUSの適用性と影響力を考慮する。以下では議論されていないASUSが評価された後、財務諸表への不適用または予想の影響はわずかであると判断された。
FASBは2023年12月にASU 2023-09を発表しました所得税(話題740):所得税開示の改善ASU 2023-09の改正は、所得税開示を改善することによって、所得税開示の透明性および決定有用性を向上させることを目的としており、主に税率調整および支払われた所得税情報に関するものである。ASU 2023-09は、公共エンティティに対して2024年12月15日以降の年間期間に有効であり、早期採用を許可している。私たちは現在この更新が私たちの財務諸表に及ぼす影響を評価している。
2023年10月、FASBはASU 2023-06を発表した開示改善:米国証券取引委員会の開示更新と簡略化イニシアティブに応答して編纂改訂を行う中の修正案 ASU 2023-06は、編纂中の様々なテーマの開示および提示要件を明確にまたは改善し、これらの要件を米国証券取引委員会の規定と一致させるためのいくつかの変化を代表する。米国証券取引委員会の既存の開示要求に制約されているエンティティについては、各改正の発効日は、米国証券取引委員会がS-X条例またはS-K条例から関連開示を削除する発効日となる
早期養子縁組を禁止する。私たちは現在、この更新の影響とその発効日を評価していますが、更新は私たちの財務諸表に実質的な影響を与えないと予想されます。
2022年1月1日にASU 2020-06を通過しました債務--転換可能な債務および他のオプション(主題470-20)と派生ツールおよびヘッジ--エンティティ自身の権益の契約(主題815-40)それは.ASU 2020-06は、いくつかの負債および持分の特徴を有する金融商品にGAAPを適用することによって決定された問題を処理する。改訂は、変換可能なツールに関する指導意見およびエンティティ自身の株式契約の派生商品範囲の例外に関する指導意見の改訂に重点を置いている。この基準を採用することは私たちの財務諸表に実質的な影響を与えない
比較的小さな報告会社の状態
我々はS-K条例第10(F)(1)項で定義された“より小さい報告会社”である。規模の小さい報告会社は、2年間の監査済み財務諸表のみを提供することを含む、いくつかの減少した開示義務を利用する可能性がある。いずれの会計年度の最終日までにも、(1)非関連会社が保有する我々の普通株の時価が2.5億ドル以上でない限り、または(2)完成した会計年度内の年収が1.00億ドル以下であり、前の6月30日までに非関連会社が保有している我々の普通株の時価が7.00億ドル以下であれば、比較的規模の小さい報告会社となる。私たちが削減された開示義務を利用する場合、これは私たちの財務諸表を難しくするか、または他の上場企業と比較することができないかもしれません。
新興成長型会社の地位
雇用法案の定義によると、私たちは“新興成長型企業”であり、5年に及ぶ間も新興成長型企業である可能性がある。私たちがまだ新興成長型企業である限り、私たちは、新興成長型企業に適用されず、他の上場企業に適用される特定の開示要求の免除に依存することを許可され、意図されている。これらの免除には
•私たちの役員報酬の開示を減らします
•役員報酬や黄金パラシュート手配について拘束力のない株主相談投票は行われなかった
•財務報告書に対する私たちの内部統制を評価する際に、監査人の認証要求を免除する。
私たちはこの報告書で削減された報告書の要求を利用して、私たちがもはや新興成長型会社ではなくなるまでそうし続けることができる。我々は、(A)本年度の総収入が12.35億ドル以上となるまで“新興成長型企業”とし、(B)2026年12月31日、IPO完了5周年後の本年度の最終日、(C)過去3年間で10億ドルを超える転換不能債券を発行した日、または(D)米国証券取引委員会規則に基づいて申告を加速した大手企業とみなす。JOBS法案第107条は,新興成長型会社は延長された過渡期間を利用して新たなまたは改正された会計基準を遵守することができると規定している
経営成果
2023年まで、2023年と2022年12月31日までの年度比較
次の表に2023年12月31日までの年度と2022年12月31日までの年度の経営実績(単位:千)を示す
| | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 |
| | | |
| 運営費用: | | | |
| 研究開発 | $ | 3,232 | | | $ | 242 | |
| 一般と行政 | 8,155 | | | 1,997 | |
| 総運営費 | 11,387 | | | 2,239 | |
| 運営損失 | (11,387) | | | (2,239) | |
| その他の収入 | 250 | | | — | |
| 株式証負債の公正価値変動を認める | 182 | | | — | |
| 債務返済損失 | — | | | (325) | |
| 利子支出,純額 | (2,082) | | | (653) | |
| 純損失 | $ | (13,037) | | | $ | (3,217) | |
研究と開発費
2023年12月31日と2022年12月31日までの年間研究開発費はそれぞれ320万ドルと20万ドル。研究開発費が300万ドル増加したのは,主にGRI−0621登録発展計画に関する費用が190万ドル増加し,人員費用が40万ドル増加し,相談費が60万ドル増加し,知的財産権費用が10万ドル増加したためである。
一般と行政費用
2023年12月31日と2022年12月31日までの年度の一般·行政費はそれぞれ820万ドルと200万ドル。610万ドルの増加は、主に合併と上場コストにより会計、法律、投資銀行、その他の費用が420万ドル増加し、人員コストが150万ドル増加し、保険費用が30万ドル増加し、コンサルティングやその他の一般·行政費用が20万ドル増加したためである
その他の収入
2023年12月31日までの3カ月間,2023年8月に締結されたAardvark合意条項により受け取った支払いにより,その他の収入は30万ドルであった。
権証責任の公正価値変動
公正価値変動20万ドル代表2023年12月31日までの年度内の未弁済株式証の公正価値は減少した。
債務の弁済
債務の返済は30万ドルこの年度までに2022年12月31日以下の理由によりTEP注釈のいくつかの部分の修正。改訂された債務の公正価値が原始債務の帳簿価値を超え、弁済損失をもたらした場合は弁済とみなされる。
利子支出,純額
2023年12月31日と2022年12月31日までの年間の純利息支出はそれぞれ210万ドルと70万ドルで、本チケットの未返済と関係がある。利息支出純額の増加は橋手形に関する利息によるものである
流動性と資本資源
設立以来、私たちはすでに赤字が発生しており、予測可能な未来にも赤字が続くと予想されている。2023年12月31日、2023年12月31日、2022年12月31日までの年度まで、それぞれ1300万ドルと320万ドルの純損失を出しています。2023年12月31日現在、私たちの累計赤字は3150万ドルです。
我々はこれまで,普通株式,変換可能手形,短期本券,Paycheck Protection計画本票を発行することで運営資金需要を満たしてきた.2023年12月31日現在、私たちは180万ドルの現金と現金等価物を持っている
次の表は、示す期間のキャッシュフロー(千単位)をまとめた
| | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 |
| 提供された現金純額(使用): | | | |
| 経営活動 | $ | (8,990) | | | $ | (1,085) | |
| 投資活動 | (8) | | | (3) | |
| 融資活動 | 10,797 | | | 1,007 | |
| 現金と現金等価物の純増加 | $ | 1,799 | | | $ | (81) | |
経営活動のキャッシュフロー
2023年と2022年12月31日までの年度では、それぞれ900万ドルと110万ドルが経営活動に使われている。790万ドルの増加は、主に私たちの純損失が980万ドル増加し、前払いおよびその他の費用および売掛金のための現金が110万ドル増加したが、非現金調整純増加180万ドルによって相殺され、非現金調整には、株式による補償費用、債務割引、債務発行コストの償却および債務清算損失、および支払いすべき帳簿を支払うための現金が130万ドル減少した
投資活動によるキャッシュフロー
投資活動が提供する現金純額は、2023年12月31日と2022年12月31日までにそれぞれ8,000ドルと3,000ドルであり、コンピュータ設備の購入に関連している。
融資活動によるキャッシュフロー
2023年12月31日までの年間で,融資活動が提供する現金純額は1080万ドルであり,これは株式SPA(以下の定義)の1230万ドルの収益,橋梁債券の第2弾融資の130万ドルの収益,および合併に関する90万ドルの現金によるものである。これらの収益は、合併に関連する300万ドルのコスト、Bridge Notesに関連する50万ドルの債務発行コスト、Equity SPAに関する20万ドルの株式発行コストによって相殺される。2022年12月31日までの年度、融資活動が提供する現金純額は100万ドルで、主に約束手形収益が含まれている
株式証券購入協定
合併契約に署名する際には、Vallon、GRI Operations、および売却株主が株式SPAを締結し、これにより、売却株主は、合併完了直前にGRIが運営する普通株の株式を発行するために、Bridge Notesの未償還元金および利息を現金で12,250ドル投資し、Bridge Notesの任意の未償還元金および課税利息を解約することに同意する。株式SPAによると,GRI運営部は取引終了直前に売却株主に初期株を発行し,546ドルの発売費用を差し引いた後,追加の株式をホストエージェントに預け,純収益は11,704ドルであった.
取引完了時には、合併事項により、初期株式は合計36,263株の私たちの普通株に変換され、追加株式は合計145,052株の我々の普通株に変換されます。2023年5月8日、株式SPAの条項に基づいて、売却株主と共に、実益所有権制限に適合した場合、追加株式と引き換えに発行されたすべての普通株を売却株主に支払うことを許可する
将来の資金需要
2023年12月31日と2022年12月31日までの年度の純損失はそれぞれ1300万ドルと320万ドルだった。2023年12月31日現在、私たちは180万ドルの現金を持っており、累計赤字は3150万ドルです。私たちは計画された活動に大量の財源を投入したいと思っています特に私たちが計画した臨床試験を準備し起動して行う際には
GRI−0621とGRI−0803は,我々の発見計画を進め,我々の製品開発努力を継続している。また、上場企業の運営に関する追加コストも発生すると予想されています
2024年2月1日に、吾等は購入合意を締結し、これにより、吾等は、(I)330,450株普通株、(Ii)4,669,550株を公開発売で発行·売却することに同意し、総株式4,669,550株普通株、(Iii)5,000,000株B-1シリーズ普通権証合計5,000,000株普通株および(Iv)5,000,000株B-2シリーズ普通株を行使できる合計5,000,000株普通株を行使でき、総収益は550万ドルであることに同意した。これらの証券の発売組み合わせは、(A)1株または1部の事前資本権証、および(B)B-1シリーズ一般権証とB-2シリーズ普通権証であり、総購入価格は1.1ドル(1部の事前資本金権証から0.0001ドル減算)である。
ある所有権制限の場合、株式承認証は発行時に行使することができる。各前払い資金株式承認証は1株当たり0.0001ドルの価格で普通株を行使することができ、しかも満期にならない。B-1シリーズ普通株式承認証は2024年2月6日に発行されてから5年以内に、1株1.10ドルの価格で1株普通株に転換することができる。2024年2月6日以降の18ヶ月以内に、各B-2シリーズ普通株式承認証は1株1.10ドルの価格で1株普通株に変換することができる。購入契約に基づいて証券を発行する場合、A-1シリーズ株式証の条項によると、A-1シリーズ株式承認証の行価格は額面、即ち1株当たり0.0001ドルに低下する。
私たちの現在の運営計画によると、私たちの既存の現金と現金等価物は、2024年下半期までの運営費用と資本支出需要を支払うのに十分であると信じています
したがって、私たちは私たちの持続的な業務と関連した多くの追加資金を得る必要があるだろう。合併に関連して発行されたTシリーズ株式証は現在会社の強制行使の制約を受けていない。それらの強制行使の持分条件がTシリーズ株式承認証が規定している期間の私たちの普通株の価値加重平均価格は1株当たり少なくとも64.47ドルの要求を満たしていないからである。私たちは株式証券および/または短期または長期債務手配を増発することで資金を調達しようとしているが、私たちの研究や開発努力が成功しても、必要に応じてこのような融資がある保証はない。もし私たちが十分な追加資金を得ることができない場合、私たちは私たちの運営計画を再評価し、支出の削減を余儀なくされる可能性があり、仕入先との支払い条件を延長し、可能な場合に資産を清算し、私たちの開発計画の一部または全部を延期、削減または廃止するか、あるいは私たちの技術の権利を放棄する必要があり、条件は私たちが選択した優遇に及ばない、あるいは運営を完全に停止する。これらの行動は私たちの業務、経営結果、未来の見通し、そして私たちの普通株の価値に大きな影響を与えるかもしれない。また、追加融資を得ようとすることは、経営陣の時間や注意力を日常活動から移し、発見や製品開発への関心を分散させる可能性がある。そのため、私たちが経営を続けている企業として継続する能力に大きな疑いが生じています。私たちは、少なくとも予測可能な未来に、私たちは重大で増加していく運営損失を受け続けると予想する。私たちが開発を成功させ、規制部門の承認を得て、私たちの現在または未来の候補製品の商業化に成功しない限り、製品収入は生じないと予想されます。
第1 A項を参照。本年度報告書の“リスク要因”は、我々の巨額の資本要求に関連する追加リスクを知るためのものである。
契約義務その他の約束
著者らは正常業務過程において第三者契約組織と臨床試験、臨床前研究、製造とその他の運営目的のサービスと製品について契約を締結した。これらの契約は一般に通知後の一定期間で契約を終了することが規定されているため,これらの合意の下での撤回不可能な義務は実質的ではないと考えられる.
表外手配
私たちは表外取引には何も参加しません。通常の業務運営で生じる保証または義務を除いて、私たちはいかなる保証や義務も負いません。
第7 A項には、市場リスクに関する定量的かつ定性的な開示が含まれる
適用されません。
第8項:財務諸表及び補足データ
本プロジェクトの要求に基づいて作成された財務諸表はここで参考にして,項目15における適用情報から抜粋した.本年度報告の“展示品·財務諸表明細書”は,F−1ページから示した。
項目9.報告会計·財務開示面の変化と会計担当者との相違
ない。
項目9 A:管理制御とプログラム
情報開示制御とプログラムの評価
取引法規則13 a-15(E)および15 d-15(E)に定義されている開示制御およびプログラムという言葉は、取引法に従って提出または提出された報告書において会社が開示を要求する情報が、米国証券取引委員会規則および表によって指定された期間内に記録、処理、集約および報告されることを確実にするための制御および手順を意味する。開示制御およびプログラムは、そのような情報の蓄積を確実にし、その主要幹部および主要財務官を含む会社管理層に適宜伝達することを目的とした制御および手順に限定されないが、開示要求に関する決定をタイムリーに行うために、これらに限定されない。
我々の開示制御およびプログラムを設計·評価する際に、管理層は、任意の制御およびプログラムが、どんなにアイデアや操作が完全であっても、絶対的な保証ではなく、合理的な保証を提供することしかできず、開示制御およびプログラムの目標を達成することを確保することができることを認識している。また、開示制御及びプログラムを設計する際には、我々の管理層は、可能な開示制御とプログラムの費用対効果関係を評価する際にその判断を適用することを要求される。任意の制御システムの設計も、将来のイベント可能性のいくつかの仮定に部分的に基づいており、任意の設計がすべての潜在的な将来の条件でその目標を成功的に達成する保証はなく、時間の経過とともに、制御が条件の変化によって不十分になる可能性があり、またはポリシーまたはプログラムを遵守する程度が悪化する可能性がある。制御システム固有の制限により、エラーや詐欺によるエラー陳述が発生し、検出されない可能性がある。
我々の最高経営責任者(我々の最高経営責任者)と我々の最高財務官(我々の最高財務官)は、本年度報告書の期間終了までの間、取引所法案下の規則13 a 15(E)および15 d 15(E)で定義された開示制御およびプログラムの有効性を評価した。この評価に基づき、我々の最高経営責任者および最高財務責任者は、2023年12月31日までに、我々の開示統制および手続きが有効であると結論した。
財務報告の内部統制に関する経営陣の報告
私たちの経営陣は財務報告書の十分な内部統制を確立して維持する責任がある。財務報告の内部統制は公認会計基準に基づいて財務報告の信頼性と外部報告目的の財務諸表の作成に合理的な保証を提供することを目的とした過程である。
財務報告の内部統制は、(1)合理的で詳細かつ正確かつ公平に資産取引および処置を反映した記録を維持すること、(2)公認会計基準に基づいて財務諸表を作成し、その管理層および取締役の許可のみに基づいて収支を行うために必要な取引を記録するための合理的な保証を提供すること、および(3)その財務諸表に重大な影響を与える可能性のある不正な買収、使用または処分を防止またはタイムリーに発見するための合理的な保証を提供する、という政策および手順を含む。
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来の期間の有効性をどのように評価するかの予測には,条件の変化により制御措置が不十分になる可能性がある,あるいはそのような制御措置に含まれる政策やプログラムの遵守度が悪化する可能性がある.
私たちの経営陣は、2023年12月31日までの財務報告書の内部統制に対する我々の有効性を評価した。この評価を行う際には、経営陣は、“内部統制--総合枠組み(2013)”で決定された基準をテレデビル委員会協賛組織委員会(COSO)が使用した。これらの基準は,制御環境,リスク評価,制御活動,情報や通信,モニタリングなどの分野に関連している。経営陣の評価には、文書の作成、評価、その財務報告の内部統制の設計と運営効果のテストが含まれている。
以上のように、経営陣の流れと評価によると、経営陣は、2023年12月31日現在、財務報告の内部統制に有効であると結論している。
公認会計士事務所認証報告
雇用法案による“新興成長型会社”の免除により、本年度報告にはわが公認会計士事務所の認証報告は含まれていません
財務報告の内部統制の変化
2023年12月31日までの四半期内に、財務報告の内部統制には何の変化もなく、これらの変化は、私たちの財務報告の内部統制に大きな影響を与えたり、合理的になったりする可能性があります。
プロジェクト9 B:他の資料の提供
ルール10 b 5-1取引スケジュール
2023年12月31日までの3ヶ月以内に、私たちの役員または上級管理者通過するあるいは…終了しました“ルール10 b 5−1取引スケジュール”または“非ルール10 b 5−1取引スケジュール”は、各用語がS−K条例408項で定義されている。
第9項C.検査妨害の開示を禁止する外国司法管区
ない。
第III部
プロジェクト10.役員、役員、およびコーポレートガバナンス管理職を含む
次の表に私たちの役員、取締役被著名人、そして私たちの役員に関するいくつかの情報を示します。
| | | | | | | | | | | | | | |
| 名前.名前 | | 年ごろ | | ポスト |
| 行政員 | | | | |
| マーク·ヘルツ博士です。 | | 54 | | 取締役最高経営責任者総裁 (首席行政主任) |
| リンエン·ケリー | | 47 | | 首席財務官 (首席財務会計官) |
| Vipin Kumar Chaturvedi博士 | | 64 | | 首席科学官 |
| アルバート·アグロー博士 | | 59 | | 首席医療官 |
| | | | |
| 非従業員役員と取締役が指名する | | | | |
デヴィッド·スクルス(1)(2) | | 50 | | 取締役、取締役会長 |
| デヴィッドパン屋 | | 59 | | 役員.取締役 |
カミラ·V·シンプソン理系修士(1)(2)(3) | | 52 | | 役員.取締役 |
ロロフ·ユンゲン(1)(3) | | 58 | | 役員.取締役 |
(1)監査委員会のメンバー。
(2)報酬委員会のメンバー。
(3)会社管理委員会のメンバーを指名して指名します。
行政員
マーク·ヘルツ博士です。2023年4月以降、私たちの総裁兼最高経営責任者、取締役会のメンバーを務めてきました。2009年にGRI運営会社を共同設立し、会社設立以来最高経営責任者や取締役会長を務めてきた。管理職以外に、ヘルツ博士は2005年から2009年までGemVax、2014年から2018年までEvozym Biologics Inc.及び2008年以来多重合生物治療会社の取締役会メンバーを務めたことがある。1998年以来、ヘルツ博士は生物技術業界の会社でいくつかの高級職を務めている。ヘルツ博士はボデン学院で生物学学士号を取得し、コロラド大学医学院で免疫学と微生物学博士号を取得した。ヘルツ博士はGRI運営会社の共同創業者兼最高経営責任者としてのサービスと、バイオテクノロジー業界での豊富な経験から、私たちの取締役会のメンバーになる資格があると信じている。
リンエン·ケリー2023年4月の合併が完了して以来、私たちの最高財務官を務めてきました。彼女は20年以上の経験を持ち、生命科学、技術、電子商取引分野の民間と上場企業をリードし、公共会計面の基礎を持っている。2021年5月から合併完了まで、彼女はバロンの首席財務官を務めた。2016年から2021年までOptiNose,Inc.で取締役グローバル財務報告総監兼役員を務め,OptiNose,Inc.は数百万ドルの収入を得た専門製薬会社である。彼女のキャリアの中で、彼女はFlower Orthopedics、Iroko PharmPharmticals、LLC、Genaera Corporationなどの私営や上場企業で財務総監兼最高財務官上級副総裁を務めたことがある。ケリーのキャリアはビマーウェイ会計士事務所から始まりました。このような職務を担当している間、ケリーの仕事は数百万ドルの融資、M&A調査、支援を含む。彼女はまた財務監督、内部と外部財務報告、予測と財務分析、そして投資家と公共関係について豊富な経験を持っている。ケリーさんはリハーイ大学で商業経済学の学士号を取得し、会計学を専攻し、ペンシルバニア州の公認会計士(非勤務状態)である。
Vipin Kumar Chaturvedi博士2023年4月から私たちの首席科学官を務めてきた。2009年にGRI運営会社を共同設立し、同社設立以来、取締役会メンバーや科学顧問委員会の議長を務めてきた。Chaturvedi博士は2009年から2017年まで、2022年から2023年4月までGRI運営部首席科学官を務めた。2015年4月以来、チャトゥルヴィディ博士はカリフォルニア大学サンディエゴ校免疫調節実験室医学教授を務めてきた。2015年、チャトゥルヴェディ博士は他人と共同でイギリスのシム経済学シミュレーションソフトウェア会社を設立し、2015年から2022年7月まで取締役の非執行役員を務めた。また、チャトゥルヴィディ博士は2009年以降、コンサルティング会社Vidur Discovery、LLCの取締役会に勤務してきた。Chaturvedi博士はインドカンプル大学で生物学学士号を取得し、インド医学教育·研究研究所で生化学、分子生物学、免疫学修士号を取得し、インド科学研究所で生化学博士号を取得した。
アルバート·アグローです博士号。2023年4月から同社のコンサルタントを務め、首席医療官という肩書を持っている。2009年にGRI運営会社を共同設立し、2017年8月から2023年4月までGRI運営会社の顧問を務め、首席医療官の肩書を務めた。アグロー博士は2021年4月からコロンビア治療会社の総裁兼最高経営責任者を務め、バイオテクノロジーや製薬業界で20年以上の経験を持ち、2018年3月から2021年4月まで崇高な治療会社の最高経営責任者を務め、2012年6月から2016年9月までCynapsusの首席医療官を務めた。また,アグロー博士は現在マクマスター大学病理学·分子医学部のアシスタント教授を務めている。アグロー博士はマクマスター大学医学部で免疫学博士号を取得しました
非従業員取締役
デヴィッド·スクルス2023年4月以来、当社の取締役会メンバーを務めてきました。彼は全世界の生命科学業界で20年以上の経験を持ち、金融と商業発展幹部、取引協力者、法律顧問と取締役会のメンバーを務めた。Szekeresさんは、2016年3月にHeron Treateutics,Inc.に加入し、2023年8月まで最高経営責任者兼財務責任者を務めました。これまで、2014~2016年の間にRegulus治療会社の首席商務官、首席財務、会計官、総法律顧問を務めていた。Szekeresは2008年からLife Technologies CorporationのM&A担当も務め、同社は2014年2月にThermo Fisher Scienceに買収された。Szekeresさんは現在、Sanford Burnham Prebys、AureMatch、Animantisの取締役会メンバーであり、Leossal Biosciencesの執行顧問取締役会メンバーでもある。2014年3月からEdico Genome Inc.の取締役会メンバーを務め、2018年にIlluminaに買収され、2014年10月からPatara Pharmaを務め、2018年までRoivant Sciencesに買収された。Szekeresさんは、カリフォルニア大学オーウェン校で犯罪、法律、社会学士号、デューク大学法学部で法学博士号を取得しています。我々は、Szekeresさんは、バイオテクノロジーおよび生物学的治療の他の業界の役員としての経験が豊富であることを信じて、彼が私たちの取締役会に在籍する資格を持っていると信じています。
デヴィッドパン屋2019年1月15日から2019年8月23日まで私たちの取締役会メンバーを務め、2021年2月12日に私たちの普通株初公募が完了した後、再び取締役の取締役に任命されました。これまで、2019年1月15日から2023年4月12日まで華竜の総裁兼最高経営責任者を務めてきた。2018年1月15日からバロンに任命された。彼は以前、Alcobra Ltd.(現在はArcturusと呼ぶ)の臨時最高経営責任者とCEOを務めており、そこでAdairの発展を監督していた。Alcobra株式会社に入社する前、神経科学事業部ビジネス戦略や新業務副社長を務めるなど、シャルル製薬会社で10年間働いていた。この職に就いている間、Bakerさんは、神経科学的許可機会のビジネス評価、パイプライン中枢神経系(CNS)製品の管理のビジネス努力を指導し、長期的な戦略計画プロセスを主導しました。これまで、シャルルVyvanseのグローバル社長を務めてきたが、そこでVyvanseの発売をリードし、日本でのパートナーシップ構築に成功し、カナダとブラジルで発売するなど、世界拡張努力をリードしてきた。以前、ベイカーさんは、シャルル社のすべてのADHD製品マーケティング部副社長を務めていました。1990年から2004年までの間に、ベックさんは、マーケティング、販売、マーケティング、研究、ビジネス開発においてますます多くの責任を担ってきたメルク社で働いています。中枢神経系薬の知識や経験に加えて、ベックさんの専門職はまた、骨粗鬆症、片頭痛、高脂血症の治療を含んでいます。彼は5種類の薬のマーケティングに直接参加し、どの薬の年間売上高も10億ドルを超えた。ベイカーさんは、デューク大学を卒業し、経済学、コンピュータ科学の学士号を取得した優れた成績で卒業しました。彼はデューク大学福庫商学院の工商管理修士号を取得した。ベイカーさんは、私営医療広告会社Benchworks,Inc.の取締役会にも勤めています。私たちは、ベイカーさんは、私たちの社長やCEOとしてサービスを提供し、また、バイオテクノロジーの分野で彼の幅広い専門知識を持っており、彼は私たちの取締役会のメンバーとしての資格を持っていると信じています。
ロロフ·ユンゲン2023年4月以来、当社の取締役会メンバーを務めてきました。彼は連続創業者、会社創設者、研究開発/商業開発リーダーであり、多くの治療分野と機能分野で豊富な経験を持っている。栄根さんは2022年7月以来遺伝子治療会社Adolore BioTreateuticsのCEOを務め、2019年6月以来革新分子会社の創業者/CEOを務め、2018年9月以来AsteRx Pharma Consultingの管理パートナーを務めている。2012年、Matinas BioPharma(omega-3と脂質結晶ナノ薬物送達)を設立し、2018年3月に退職するまで上場企業(ニューヨーク証券取引所コード:MTNB)に発展した。HumiraやLovazaなどの製品の開発と商業化には、栄根さんが不可欠です。Matinas BioPharmaの創設に先立ち、榮恩さんは2010年から2012年までTrygg Pharmaで執行副社長を務め、ノルウェーのAker Groupが処方薬omega-3事業に入るのを手伝って、最終的にFMCに売却した。Akerに加入する前に、Rongenさんは、Relant PharmPharmticals(グラクソ·スミスクラインによって買収)された知的財産とポートフォリオ管理副社長であり、そこでLOVAZAの許可を得て、開発と投入前の活動をリードしていました。彼のキャリアの初期には、栄根さんはバズフ製薬会社(アボット/エバービーに買収された)でHUMIRAや他の免疫学的プロジェクトのグローバル製品の取締役を務めていた。Rongenさんのキャリアは、Arthur D.Little‘s Technology Innovation Management Practiceの管理コンサルタントとWilkerson Group(IBMに買収された)のバイオテクノロジー/製薬コンサルタントから始まりました。Rongenさんは、オランダのワフニンゲン大学で分子科学(バイオテクノロジー/バイオテクノロジー)工学の修士号と修士号を取得しています
西北大学ケロッグ商学院商工管理学博士。私たちは、栄根さんは生物製薬業界の経験から、彼は私たちの取締役会に在籍する資格があると信じています。
カミラ·V·シンプソンM.Sc2023年4月以来、当社の取締役会メンバーを務めてきました。2017年10月以来、彼女はSpruce Biosciences取締役会のメンバーを務めてきた。2021年4月以来、シンプソンはZehna Treeuticsの最高経営責任者を務めており、Zehna Treeuticsはクリーブランド診療所が剥離した初期段階のバイオテクノロジー会社である。2019年4月以来、シンプソンさんはRare Strategic、LLCの管理メンバーと社長を務め、バイオテクノロジー会社に戦略アドバイスとコンサルティングサービスを提供してきた。シンプソンさんは2020年12月にDyve Biosciencesの取締役会に加入した。2017年4月から2019年4月まで、SimpsonさんはBioMarin高級副総裁兼製品組合せ開発主管を務め、会社と研究開発管理、プロジェクト指導者、プロジェクト管理、競争情報、ポートフォリオ戦略と商業分析を担当した。2014年10月から2017年4月まで、SimpsonさんはBioMarinグローバル監督事務グループの副総裁を務め、2014年3月から2014年10月まで、SimpsonさんはBioMarinグローバル監督事務副総裁を務めた。彼女はまたシャルルで12年間働き、そこでますます多くの職責を担当し、最終的に副総裁監督事務の早期発展と業務発展のポストを務めた。Simpsonさんはアイルランドゴルヴェ大学の理科学士号、英キングストン大学の理科(栄誉)学士号、英国ロンドン大学の優秀な理科修士号を持っている。私たちは、シンプソンさんがバイオテクノロジー業界で幹部、役員、顧問を務める豊富な経験をして、彼女が私たちの取締役会のメンバーになる資格があると信じている。
家族関係
役員、役員、または役員役員や役員に指名された人の間には家族関係はありません。
取締役会構成
私たちの定款と定款は、私たちの取締役会の取締役数は時々取締役会または私たちの株主決議によって決定されなければなりませんが、私たちの取締役会の現在の規模は5人のメンバーです。
私たちの付例はまた、私たちの役員が取締役選挙で投票する権利のある大多数の株式保有者が投票する権利がある場合、または理由がない場合に免職することができると規定している。
私たちの現在と未来の幹部と重要な職員たちは私たちの取締役会によって適宜決定される。私たちの取締役会はまた報酬委員会と監査委員会のような特定の委員会を設立することを選択することができる。
私たちの取締役会は3種類に分類され、各任期は3年間交錯している。年次株主総会では、任期満了の取締役が再選挙され、再選後の第3回年次会議まで在任する。したがって、我々の株主年次会議では、1つのレベルの取締役のみが選択され、他のレベルの取締役はそれぞれ3年間の任期の残り時間内に存在し続けることになる。私たちの取締役は以下の3つに分類されます
•第一種取締役はデヴィッド?ベックで、2024年に開催される株主総会で任期が満了した
•第二種取締役は、2025年に開催される年次株主総会で任期が満了するRoelof RongenとCamilla V.Simpson,M.Sc.である
•三番目の役員はW.Marc Hertz博士とDavid Szekeresであり、彼らの任期は2026年に開催される年間株主総会で満了する。
私たちの定款と付例規定は、取締役会規模の増加による取締役会の空きを埋めることができるのは、私たちの取締役会のみである。査定役員数を増やすことで増加した取締役職は、各カテゴリが可能な限り査定役員数の3分の1で構成されるように、この3つのカテゴリに置かれる。
役員は自主独立している
ナスダック資本市場の上場要求によると、独立取締役は上場日から12ヶ月以内に上場会社の取締役会で多数を占めなければならない。また、指定された例外を除いて、上場企業の監査、報酬と指名委員会及び企業管理委員会のメンバーは上場日から12ヶ月以内に独立しなければならない。監査委員会のメンバーはまた、取引法規則10 A-3に規定されている基準を含む他の独立性基準を満たさなければならず、報酬委員会メンバーはまた、取引法規10 C-1に規定されている独立性基準を満たさなければならない。取締役は、同社取締役会が当該人の関係が取締役がその職責を履行することを妨害しないと判断した場合にのみ、独立判断を行使する場合にのみ、その人材が“独立した取締役”となる資格がある。上場企業監査委員会のメンバーは、取引法第10 A-3条の規定により独立とみなされ、次の場合を除いてはならない
監査委員会、取締役会または他の取締役会メンバーとしてのアイデンティティ:(1)上場企業またはその任意の子会社の任意の相談、相談または他の補償費用を直接または間接的に受け入れること、取締役会サービス報酬を除く;または(2)上場企業またはその任意の子会社の関連者。規則10 C-1については、独立とみなされるためには、上場企業報酬委員会の各メンバーにとって、取締役が当該会社と関係があるか否かを決定することに関連するすべての具体的な要因を考慮しなければならず、これは、取締役が報酬委員会メンバーの職責において管理層から独立した能力を含むことに重要であるが、これらに限定されない:取締役の報酬源は、その会社が取締役に支払う任意の相談または他の補償料、および取締役がその会社またはその任意の子会社または関連会社に関連しているかどうかを含む。
当社の取締役会は、W.Marc Hertz博士とDavid·ベックを除いて、すべての取締役会のメンバーが独立取締役であり、ナスダック資本市場規則とアメリカ証券取引委員会についても含めて独立取締役であることを決定した。当該等の独立性決定を行う際には、当社取締役会は、各非従業員取締役と吾等との関係と、取締役会が彼等の独立性の決定に関連すると考えている他のすべての事実及び状況を考慮しており、非従業員取締役1人当たりの自社株の実益所有権を含む。私たちの取締役会と各委員会の構成と運営はナスダック資本市場のすべての適用要件とアメリカ証券取引委員会の規則に適合しています。
取締役会各委員会
私たちの取締役会は、監査委員会、報酬委員会、指名·コーポレートガバナンス委員会を設立し、私たちの業務管理を促進するために他の委員会を設置することが可能です。会員たちは辞任や取締役会が別の決定をするまで、この委員会に勤めている。私たちの取締役会とその委員会は年間の会議スケジュールを作成し、状況に応じて時々特別会議を開催し、書面で同意した方法で行動することができる。
私たちの取締役会は以下に述べるように、様々な役割と権力を委員会に委譲することを期待している。各委員会は定期的に取締役会全体にその活動と行動を報告する。取締役資本市場の上場基準によると、私たちの取締役会の各委員会のメンバーは独立したナスダックになる資格があります。私たちの取締役会の各委員会は取締役会の承認を受けた書面規定を持っている。
全ての規約のコピーはウェブサイトwww.griBio.comに掲示されています“投資家“部分。我々のサイトに含まれる情報は,本Form 10-K年次報告に引用的に組み込まれることはない.私たちは、非アクティブなテキストとしてのみ、私たちのサイトアドレスをここに登録しています。
監査委員会
私たちの監査委員会のメンバーはRoelof Rongen、Camilla V.Simpson、M.Scです。監査委員会議長のDavid·スゼクルスです
私たちの監査委員会は取締役会に協力して私たちの財務諸表の完全性を監督します。私たちは法律と法規の要求を遵守します。独立公認会計士事務所の資格、独立性と業績、私たちの財務リスク評価とリスク管理の設計と実施。他の事項を除いて、私たちの監査委員会は、私たちの経営陣と私たちの開示制御と手続きの十分性と有効性を検討し、議論する責任があります。我々の監査委員会はまた、我々の経営陣及び独立公認会計士事務所と年次監査計画及び監査活動の範囲、我々財務諸表の年次監査の範囲及び時間、監査の結果、財務諸表の四半期審査を検討し、適切な場合に当社の財務事務のいくつかの面について調査を開始する。
我々の監査委員会は、会計、内部会計制御又は監査事項に関する任意の苦情、並びに我々の従業員が問題のある会計又は監査事項について提出した秘密及び匿名苦情を受信、保留及び処理するための手続きの構築及び監視を担当する。しかも、私たちの監査委員会は私たちの独立公認会計士事務所の任命、報酬、保留、監督を直接担当しています。私たちの監査委員会は、独立公認会計士事務所の採用と解任、すべての監査採用条項と費用、および独立監査師との非監査採用を許可するすべての資格を独占的に承認する権利があります。私たちの監査委員会は私たちの政策と手続きに基づいてすべての関係者たちの取引を検討して監視する。
私たちの監査委員会のすべてのメンバーは独立しており、アメリカ証券取引委員会の規則と監査委員会のメンバーに適用されるナスダック資本市場上場基準の制約を受けている。当社の取締役会は、David司徒克思は“アメリカ証券取引委員会”の規定が指す監査委員会の財務専門家資格に符合し、そして“ナスダック資本市場上場基準”の財務複雑度に対する要求に符合すると認定した。この決定を下したとき、当社取締役会は、Szekeresさんの過去の経験、ビジネス洞察力、および独立性を考慮しています。私たちの独立公認会計士事務所と経営陣は定期的に私たちの監査委員会と私的に会います。
私たちは、私たちの監査委員会の構成と運営がサバンズ法案404節のすべての適用要件と、すべての適用されたアメリカ証券取引委員会とナスダック資本市場規則と規定に適合すると信じている。私たちはこのような要求が私たちに適用される限り、未来の要求事項を守るつもりだ。
報酬委員会
私たちの報酬委員会のメンバーはDavid·スケックスと報酬委員会のカミラ·V·シンプソン会長だ。
私たちの給与委員会の各メンバーはアメリカ証券取引委員会の規則と報酬委員会のメンバーに適用されるナスダック資本市場上場基準の下で独立しています。当社の報酬委員会は、役員(取引所法案第16条に基づいて報告された役員を含む)の報酬形態及び金額を監督し、従業員及び他のサービスプロバイダに対する当社の株式及び非持分インセンティブ計画、並びに当社の報酬計画に関連するいくつかの他の事項を管理するように協力しています。他の職責に加えて、私たちの報酬委員会は、私たちの最高経営責任者のパフォーマンスを評価し、彼と協議し、私たちの他の幹部(取引所法案第16条に基づいて報告された役人を含む)のパフォーマンスを評価します。私たちの報酬委員会はまたGRI運営計画と私たちのA&R 2018計画を管理しています。報酬委員会は、私たちのCEOの報酬を決定し、CEOが不在の場合にその問題について決定する責任があります。
給与委員会は、役員報酬と役員報酬の審議と決定に以下の手順と手順を採用した
•役員および役員の報酬スケジュール、計画、政策、および計画を評価、推薦、承認、審査し、
•現金と持分に基づく報酬計画を管理する
•取締役会が役員報酬に関連する任意の他の職責について取締役会に提案する。
指名と会社管理委員会
私たちが指名して会社管理委員会のメンバーはCamilla V.Simpson、M.Scだ。ロロフ·ユンゲン会長を指名して会社統治委員会議長に任命しました
私たちの指名と会社管理委員会の各メンバーは独立しており、アメリカ証券取引委員会の規則とナスダック資本市場の上場基準に符合し、指名と会社管理委員会のメンバーに適用される。私たちの指名とコーポレートガバナンス委員会の義務は以下の通りです
•取締役会とその各委員会の構成、組織と管理について取締役会全員に評価し、提案した
•役員候補者の評価と推薦を行い
•現在の取締役会のメンバーのパフォーマンスを評価し
•必要に応じて取締役のための継続教育計画を立て、
•取締役会とその委員会に情報を伝える過程を監督します
•毎年自分の業績と指名と会社統治委員会の規約を審査します
•行政総裁及びその他の行政者の後任計画を監督する手続き、及び
•会社のためにガバナンスガイドラインを策定·推薦する。
リスク監督における取締役会の指導構造と取締役会の役割
取締役会は会社の統制と指導を担当している。現在、取締役会は会長と最高経営責任者の職を分離することを決定した。ヘルツ博士は会社の最高経営責任者と取締役会のメンバーを務めるだろう。Szekeresさんは取締役会の議長を務めます。取締役会では、ヘルツ博士を取締役会メンバーとしての経営陣と、Szekeresさんを非執行役員として、非執行役員をはじめとする非執行役員との連絡が、この仕組みが当社にとって有利になると信じています。
私たちの取締役会の主な機能の一つは私たちのリスク管理過程を知ることだ。取締役会は常設のリスク管理委員会ではなく、取締役会全体および取締役会の各常設委員会管理という監督機能を直接通過し、これらの委員会はそれぞれの監督分野の固有のリスクを処理する。特に、我々の取締役会は戦略的リスクの監視·評価を担当しており、我々の監査委員会は、当社が直面している主要な財務リスクの開放を考慮して検討する責任があり、これらの開口を監視·制御するための経営陣のステップは、リスク評価や管理を行うプログラムを管理するガイドラインや政策を含む。監査委員会はまた法律と規制要求の遵守状況を監視する。私たちの指名とコーポレートガバナンス委員会は、不正または不正な責任創造行為を防止することに成功したかどうかを含む、当社のコーポレート·ガバナンス慣行の有効性を監督します。私たちの給与委員会は私たちの任意の報酬政策と計画が過度な冒険を奨励する可能性があるかどうかを評価して監視する。
報酬委員会は内部の人と連動して参加する
私たちの給与委員会のメンバーは私たちの役員や従業員を務めたことがない。私たちの上級職員は現在、任意の他の実体の取締役会、報酬委員会、または他の1人以上の上級職員が私たちの取締役会または報酬委員会のメンバーと同等の機能を担当する委員会を務めておらず、前の財政年度に在任したこともない。
ビジネス行為と道徳的基準
私たちは、私たちすべての従業員、役員、役員に適用される書面商業行為と道徳基準(“行動基準”)を採択しました。“行動基準”は、正確な会計記録と財務報告、利益衝突の回避、私たちの財産と情報の保護と使用、法律と規制要件の遵守など、基本的な道徳とコンプライアンスに関する原則とやり方をカバーしている。私たちの行動基準は私たちのウェブサイトwww.GriBio.comの“投資家-会社管理”の部分で入手でき、要求に応じて株主に無料で提供され、書面は会社秘書、住所:2223 Avenida de la Playa、Suite 208、La Jolla、CA 92037。
私たちの指名と会社管理委員会は私たちの行動基準を監督する責任があり、従業員、幹部、または役員が行動基準に対するいかなる免除も承認しなければならない。私たちは、ウェブサイトがそのような修正または免除のプレスリリースをその後、ナスダック規則によって許可されない限り、放棄または修正後の4営業日以内に、現在のForm 8-K報告書において、私たちの行動基準の任意の将来の修正または免除を開示するつもりである。
インサイダー取引政策
私たちはインサイダー取引政策を維持し、他の事項を除いて、一般的に公衆に知られていない、または入手していない当社に関する重大な情報を知っているすべての人は、私たちの高級社員、役員、および従業員を含め、インサイダー取引政策に基づいて記載されていないプログラムに従って事前に取引決済を取得する前に、当社の株式に係る取引に従事することを禁止しています。これには空売り、株式のヘッジ、そして私たちの株に関連する派生証券の取引が含まれる
第11項:役員報酬の増加
報酬総額表
新興成長型企業として、私たちが任命した役員が過去2つの完全な財政年度に稼いだり、彼らに支払った報酬を開示しなければならない。
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 名称と主要ポスト | | 年.年 | | 給料(元) | | 株奨励(ドル)(1) | | 選択権 受賞額(ドル)(2) | | 非持分 激励する (ドルを)補償する(3) | | 他のすべての 補償する ($) | | 合計(ドル) |
マーク·ヘルツ博士です。 | | 2023 | | 404,447 | | | | — | | | — | | | 437,500 | | | 67 | | | | 842,014 | |
| 社長と最高経営責任者 | | 2022 | | 343,750 | | (4)(5) | | — | | | — | | | — | | | — | | | | 343,750 | |
| リンエン·M·ケリー | | 2023 | | 304,063 | | | | — | | | 114,347 | | | 237,500 | | | 9,367 | | (6) | | 665,277 | |
首席財務官 | | 2022 | | 283,893 | | | | 20,820 | | | 210,428 | | | 150,000 | | | 8,400 | | (6) | | 673,541 | |
アルバート·アグロー博士 | | 2023 | | 162,500 | | | | — | | | — | | | 113,750 | | | 130,736 | | (7) | | 406,986 | |
首席医療官 | | 2022 | | — | | | | — | | | — | | | — | | | — | |
| | — | |
デヴィッドパン屋(9) | | 2023 | | 144,577 | | | | — | | | 89,264 | | | 75,000 | | | 988,211 | | (8)(10) | | 1,297,051 | |
元総裁兼最高経営責任者 | | 2022 | | 417,266 | | | | 20,820 | | | 251,494 | | | 210,000 | | | 19,983 | | (10) | | 919,564 | |
(1)この欄の金額は、FASB ASCトピック718から計算された制限株式単位(RSU)の総付与日公正価値を表す。これらの額は,必ずしも実行者が実現可能なオプション報酬に関する実際の価値に対応するとは限らない.本欄に報告されたオプション報酬を推定する際に作成した仮定は、Vallonの既監査財務諸表に記載されている(付記3)重要な会計政策の概要--株式報酬11を付記します株に基づく報酬)は、2023年12月31日現在のForm 10-K年次報告に含まれている。米国証券取引委員会の規定によると、履行条件に制約された任意の奨励の付与日公正価値は、履行条件に基づく可能な結果である。ASCテーマ718項では、2022年12月31日までの1年間に補償費用が何も確認されていないため、付与日までに実現不可能と考えられる業績条件を有するRSU奨励は本欄には含まれていない。2022年12月、ベックとケリーに授与されたRSU賞は廃止された。ASC特集718項で付与日に等しい補償費用が確認され,キャンセル日にキャンセルされた賠償金の公正価値が付与される予定である
(2)FASB ASCトピック718により計算された会計年度内に付与された株式オプションの総付与日公正価値を反映する。これらの額は,必ずしも実行者が実現可能なオプション報酬に関する実際の価値に対応するとは限らない.GRIが審査した総合財務諸表(付記3)は,本欄報告のオプション報酬を推定する際の仮定を述べている重要な会計政策の概要-株に基づく報酬11を付記します株に基づく報酬)は、2023年12月31日現在の10-Kフォーム年次報告書に含まれ、このフォームは、米国証券取引委員会に提出されている。
(3)この欄の額は,指定された実行幹事があらかじめ定められた業績目標達成状況に応じて年度に稼いだ業績ボーナスである。参照してください“役員報酬−報酬要素−ボーナスと非持分インセンティブ計画報酬“下だ。
(4)2022年12月7日、ヘルツ博士はGRI運営会社の普通株の369,003株制限株を獲得し、そのうち275,250株は2022年1月1日から2022年9月30日まで獲得した275,250ドルの給料を置換し、ヘルツ博士当選後に放棄した。ヘルツはまた6.85万ドルの現金給料を受け取った。2022年12月7日に発行された余剰限定株は、2021年に稼いだ賃金と関係がある。
(5)制限株式報酬に含まれる金額は、FASB ASC主題718から計算されたこのような株式報酬の総付与日公正価値を表す。合併完了後に帰属する株式。
(6)これらの金額は、私たちの簡単なアイルランド共和軍計画の下で指定された実行幹事口座への等額入金を反映している。
(7)これらの額は、アグロー博士が2023年7月1日までに彼に支払った相談費を反映しており、この日は同社に雇われた日だった。
(8)これらの額は、Vallonの簡単な個人退職口座計画の下で指定実行幹事口座へのマッチング入金、自動車手当、および指定執行幹事の利益のために支払われた短期および長期障害および生命保険金額を反映している。これらの金額は2023年と2022年にそれぞれ11,400ドルと10,200ドルの簡単な個人退職口座マッチング入金を反映している。2023年と2022年にそれぞれ1,875ドルと6,000ドルの自動車手当と2023年と2022年にそれぞれ1,991ドルと7,983ドルの保険福祉を反映している。
(9)合併が2023年4月21日に完了すると、ベックさんは2023年4月21日から当社を辞任した。
(10)ベイカーさんとの現在の雇用契約および彼の現在の雇用契約の条項に基づいて支払われている945,000ドルの散財料(うち603,750ドルが2023年に支払われた)と、ベイカーさんが当社の取締役会で取締役サービスを担当している補償27,945ドルを含みます。
補償要素
2023年基本給
2023年4月20日、ヘルツ博士は会社の最高経営責任者に任命され、ケリーさんはCEOに任命され、発効時期から発効した。ヘルツとケリーと締結された雇用協定はそれぞれ375,000ドルと312,500ドルの年間基本給を規定している。2023年7月1日、私たちの首席医療官アグロー博士は、基本賃金325,000ドルを規定する雇用協定に調印しました
ボーナスと非持分インセンティブ計画の報酬
2023年4月20日、ヘルツ博士は会社の最高経営責任者に任命され、ケリーさんは会社の最高財務責任者に任命され、発効時期から発効した。彼の雇用協定によると、ヘルツ博士は自由に支配可能な年間業績ボーナスを得る資格があり、目標ボーナスは当時の年間基本給の50%に相当する。彼女の雇用協定によると、ケリーは適宜の年間業績ボーナスを得る資格があり、目標ボーナスは就任1年目の現在の年間基本給の20%に相当し、その後彼女は当時の年間基本給の35%、契約ボーナス10万ドル、留任ボーナス5万ドルは、取引終了1周年に支払われる。彼の雇用協定によると、アグロー博士は自由に支配可能な年間業績ボーナスを得る資格があり、目標ボーナスは当時の年間基本給の35%に相当する。
年間業績ボーナスのほか、2022年12月、ケリーさんは7.5万ドルのボーナスを獲得し、このボーナスは合併完了時に支払われた。
合併前に,ヘルツ博士のボーナスはGRI運営取締役会が適宜決定した。2021年3月、ヘルツ博士は合併完了時に支払われた25万ドルの賞金を獲得した。
2023年期間に付与されたオプション奨励
2023年9月26日、彼女とGRIの雇用協定に基づき、ケリーさんは10.64ドルの取引価格で11,904株の普通株を購入する選択権を獲得した。
条件に合った退職計画
私たちは退職、繰延補償、年金、または利益共有計画を維持しない。
雇用協定
私たちは私たちが任命したすべての幹部たちと雇用協定を締結した。雇用協定では、行政者は基本給を取得し、年間現金ボーナスを取得する資格があり、具体的には会社の特定のマイルストーンおよび/または個人目標の実現にかかっている。雇用協定によると、私たちの給与委員会または取締役会は、各役員の基本給と目標ボーナスを定期的に審査する。雇用協定には特定の解雇給付も規定されており、以下のタイトルは“終了または制御権変更時の潜在的支払い."
私たちが指定した幹部も私たちのすべての退職や団体福祉計画に参加する権利がありますが、このような計画に適用される条項と条件を守らなければなりません。また,指名された実行幹事ごとの雇用協定には,機密情報を開示しない,互いに卑下したり譲渡したりしないことに関する限定的な契約が含まれている.ヘルツ博士のほか、指名された幹部一人一人と締結された雇用協定には、競業禁止や非招待状条項も含まれている。
終了または制御権変更時の潜在的支払い
以下に説明する潜在的な支払いに加えて、指定された実行幹事のサービスがどのような方法で終了しても、その指定された実行幹事は、未払い賃金および適用される他の課税給付を含む、そのサービス中に稼いだ補償金を得る権利がある。また,任命された幹部一人ひとりが,会社が理由なくその雇用関係を終了した場合に何らかの福祉を得る権利があるか,あるいは退職する十分な理由がある。
マーク·ヘルツ博士です。
彼と私たちの雇用合意によると、ヘルツ博士の雇用が理由なく中止されたり、ヘルツ博士が十分な理由で終了したりすれば、いずれの場合も制御権変更と関係がない場合、ヘルツ博士は以下の解散費福祉を受ける権利がある
•通年の実績結果に基づいて、12ヶ月の基本給を連続して支払い、退職当時の割合で計算されるボーナスを加え、当該人員がその年に少なくとも6ヶ月雇用されることを条件とする
•コブラは12ヶ月間の補助金を受け続けている(あるいは彼が代替保険を受けた早い日)。
彼と私たちの雇用協議によると、ヘルツ博士の雇用が私たちに理由なく中止されたり、ヘルツ博士が十分な理由で終了したりすれば、制御権取引変更後の一年以内に、いずれの場合も、ヘルツ博士は以下の解散費を得る権利がある
•18ヶ月の基本給を連続して支払い、退職会計年度目標ボーナス150%に相当する一括払いを加えたが、割合では計算しない
•コブラは18ヶ月の補助金(またはそれ以上の日に代替保険を受けさせる)を継続した
•終了日までに保有しているすべての株式ベースの未償還奨励金の付与を加速し、いかなる業績奨励も“目標”の業績水準を満たすとみなされ、どの株式オプションもその任期中に返済されていない。
リンエン·ケリー
ケリーさんと私たちの雇用契約によると、ケリーさんの雇用が理由なく中止されたり、ケリーさんに正当な理由で中止されたり、いずれの場合も支配権の変更に関係なく、ケリーさんは以下の解散費給付を受ける権利があるだろう
•通年の実績結果によると、9カ月連続で基本給を支払い、離職当時の割合で計算されたボーナスは、その年に少なくとも六ヶ月雇用されることが条件となっている
•コブラは9ヶ月の補助金(またはそれ以上の日に代替保険を受けるために保険を受け続ける)。
ケリーさんと私たちの雇用契約によると、ケリーさんの雇用が理由なく中止されたり、ケリーさんが十分な理由で終了したりすれば、支配権変更取引後の一年以内に、ケリーさんは以下の解散費給付を受ける権利があるだろう
•12ヶ月連続の基本給には、会計年度目標ボーナス100%を終了することに相当する一括払いが加えられ、比例して計算されない
•コブラは12ヶ月の補助金(または彼女が代替保険を受けた早い日)を継続して保証している
•終了日までに保有しているすべての株式ベースの未償還奨励金の付与を加速し、いかなる業績奨励も“目標”の業績水準を満たすとみなされ、どの株式オプションもその任期中に返済されていない。
アルバート·アグロー博士
彼と私たちの雇用合意によると、アグロー博士の雇用が理由なく中止されたり、アグロー博士が正当な理由で中止されたり、いずれの場合も支配権の変更に関係なく、アグロー博士は以下の解散費を得る権利がある
•通年の実績結果に基づいて、9ヶ月の基本給を連続して支払い、退職当時の割合で計算されるボーナスを加え、その人員がその年に少なくとも6ヶ月雇用されることを条件とする
•コブラは9ヶ月の補助金(または早い日に代替保険を受けるために)を継続して保証している。
アグロー博士との雇用契約によると、アグロー博士の雇用が理由なく中止されたり、アグロー博士が十分な理由で終了したりすれば、支配権取引変更後の一年以内に、アグロー博士は以下の解散費を得る権利がある
•12ヶ月連続の基本給には、会計年度目標ボーナスの終了に相当する100%の一括払いが加えられ、比例して計算されない
•12ヶ月間のコブラ継続保険の補助金(または彼が代替保険を受けるより早い日)
•終了日までに保有しているすべての株式ベースの未償還奨励金の付与を加速し、いかなる業績奨励も“目標”の業績水準を満たすとみなされ、どの株式オプションもその任期中に返済されていない。
財政年度終了時の優秀株奨励
株式オプション賞
次の表は、2023年12月31日現在、私たちの指定役員が個別奨励に基づいて保有している未償還株式オプション奨励を示し、各株式オプション奨励の背後にある株式総数を示しており、これらの株式は、(I)行使可能であるが行使されていない、(Ii)行使不能および未行使、および(Iii)各奨励の総金額を含む。Leanne KellyとDavid Bakerに2023年12月31日までの年間のすべての持分報酬を授与することは、A&R 2018計画に基づいて行われます。その条項によると、合併直前に発行された普通株を購入した既得、未満期、未行使の引受権は有効時間経過後も行使されていない
合併直前に発行された普通株式の各未帰属,未満期および未行使の引受権は発効時に取り消され,掛け値は何もない
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | オプション大賞(3) | |
| 名前.名前 | | 行使されていない証券標的数,行使可能なオプション(#) | | 未行使の証券の標的数、未行使のオプション(#) | | 株式インセンティブ計画賞:未満期オプションを行使していない証券対象数(#) | | オプション取引権価格(ドル) | | オプション期限 | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
リンエン·ケリー | | 145 | | — | | | — | | | 768.60 | | | 5/14/2031 | |
首席財務官 | | 59 | | — | | | — | | | 1,182.30 | | | 2/15/2032 | |
| | | | 11,904 | (1) | | | | | 10.64 | | | 9/22/2033 | |
デヴィッドパン屋 | | 222 | | — | | | — | | | 386.40 | | | 10/1/2028 | |
| 元CEO | | 291 | | — | | | — | | | 462.00 | | | 2/5/2029 | |
| | 208 | | — | | | — | | | 768.60 | | | 5/14/2031 | |
| | 72 | | — | | | — | | | 1,182.30 | | | 2/15/2032 | |
| | 410 | | 4,516 | (2) | | — | | | 20.30 | | | 8/10/2023 | |
__________________
(1)株式オプション奨励は、帰属開始日の1周年(2023年9月22日)に25%が付与され、役員が引き続き私たちに雇用された後、毎月2.083%(このような株式の1/48)が付与される。
(2)株式オプション奨励金は、授権日(2023年8月10日)に8.33%、Bakerさんが全四半期ごとに取締役会に残り、8.33%(株式等の1/12)を授与されます。
(3)2023年12月31日現在、ヘルツ博士とアグロー博士は未返済のオプションや株式奨励を持っていないため、本表には含まれていない。
役員報酬及び補償表
私たちの役員報酬計画は、私たちが高い素質のある役員を引き付ける能力を強化し、彼らの利益を私たちの株主の長期的な利益と一致させることを目的としています。この計画は通常、現金部分と株式部分を含み、前者は非従業員取締役会でのサービスを補償することを目的とし、後者は非従業員取締役と株主の利益を調整することを目的としている。当社の従業員である取締役は当社取締役会でのサービスにより追加報酬を得ません。
給与委員会は毎年、非従業員取締役に支払われた報酬を審査し、取締役会全員に適切な調整提案を行う。この年度審査の一部として、給与委員会は、各非従業員取締役が取締役会とその各委員会に在任するのに要する大量の時間と技能レベルを考慮する。報酬委員会は、取締役報酬計画の市場競争力を維持することを求め、当社の役員報酬計画と同業者チームが維持する報酬計画とを比較します。
2023年8月10日から、非従業員役員報酬計画を改訂し、再説明すると、非従業員取締役会メンバー1人当たり以下の報酬が得られることが規定されています
•取締役会に在任している年間現金前払金は40,000ドル、監査委員会に在任している年間現金前払金は7,500ドル、給与委員会での年間現金前払金は6,000ドル、指名·会社統治委員会での年間現金前払金は5,000ドルであり、非従業員取締役は任意の年度予備招聘金を受け入れることを選択し、現金の代わりに株式オプションを付与することができる。
•取締役会のメンバーに初めて任命または当選された非従業員取締役は、一定数の私たちの普通株を購入するための初期株式オプションを獲得し、その商数は100,000ドルに等しい。取締役が初めて選ばれた日または任命された日の終値で割ると、通常3年以内に四半期ごとに分割して付与される。
•非従業員董事は、(I)2023年8月10日以降の任意の株主周年総会日に取締役会に在任し、この会議が開催された日に非従業員取締役を少なくとも6ヶ月務めており、(Ii)はその会議の直後に非従業員取締役を継続し、獲得した商数に相当する普通株を購入するための購入権を自動的に付与する
普通株の年間会議日の終値で50,000ドルを割ることで、普通株は通常1年以内に四半期ごとに分割払いになります。
受け取った他の考慮事項を除いて、私たちが改訂して再説明した非従業員役員報酬計画の規定によると、会長を務める非従業員取締役会のメンバーは以下の追加的な考慮を受ける
•監査委員会の議長は毎年15000ドルの雇用費を追加的に受けるだろう。
•給与委員会の議長は毎年12,000ドルの雇用金を追加するだろう。
•指名と会社統治委員会の議長は毎年1万ドルの雇用費を追加するだろう。
•取締役会の議長は毎年30000ドルの雇用費を追加的に受けるだろう。
逆に、非従業員取締役は、現金ではなく株式オプションを獲得しているため、会長を務めることで年間招聘金を獲得することを選択する可能性がある。
役員報酬
次の表は、2023年に非従業員役員に支払われる報酬情報を提供します
| | | | | | | | | | | | | | | | | | | | |
| 現金で稼ぐか支払うかの費用(ドル) | | オプション奨励(ドル)(1)(3) | | 合計(ドル) |
デヴィッドパン屋 | 27,945 | | | 89,264 | | (2) | | 117,209 | |
ロロフ·ユンゲン | 40,171 | | | 89,264 | | (2) | | 129,435 | |
カミラ·V·シンプソンM.Sc | 45,062 | | | 89,264 | | (2) | | 134,325 | |
デヴィッド·スクルス | 63,575 | | | 89,264 | | (2) | | 152,839 | |
________________
(1)FASB ASCトピック718により計算された会計年度内に付与された株式オプションの総付与日公正価値を反映する。これらの額は,必ずしも実行者が実現可能なオプション報酬に関する実際の価値に対応するとは限らない.本欄に記載されているオプション報酬の推定値は、当社が財務諸表の審査を経て掲載されているものとする(付記3参照)重要な会計政策の概要-株に基づく報酬11を付記します株に基づく報酬)は、本年度報告に含まれます。
(2)4,926株普通株を購入するオプションは2023年8月10日に授与され、授出日に8.33%に帰属し、取締役はその後取締役会の全四半期ごとに保持されている8.33%(同等株式の1/12)である。
(3)次の表は、2023年12月31日現在、私たちの非従業員取締役が持っている普通株に関する発行済みオプションと株式奨励の流通株総数を示しています
| | | | | | | | | | | |
| 名前.名前 | | 傑出オプション賞 | |
| デヴィッドパン屋 | | 4,926 | |
| ロロフ·ユンゲン | | 4,926 | |
| カミラ·V·シンプソンM.Sc. | | 4,926 | |
| デヴィッド·スクルス | | 4,926 | |
バロン役員の報酬
次の表は、ワロン非従業員取締役が合併完了後に辞任する前に2023年に支払われた報酬情報を提供します
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 現金で稼ぐか支払うかの費用(ドル) | | 株奨励(ドル) | | オプション奨励(ドル) | | 合計(ドル) |
| リチャード·アンマー | 9,123 | | | — | | | | — | | | | 9,123 | |
| メイヌ·カーソン | 15,205 | | | — | | | | — | | | | 15,205 | |
| ジョセフ·ペイン | 12,164 | | | — | | | | — | | | | 12,164 | |
| マレイラ·ソレル | 37,640 | | | — | | | | — | | | | 37,640 | |
第十二項:特定の実益所有者及び経営陣の保証所有権及び関連株主事項の決定
次の表は、私たちが知っている2023年12月31日までの株式の実益所有権のいくつかの情報を示しています
•私たちが知っているすべての人または関係者のグループは私たちの株式の5%以上の実益所有者です
•私たちのすべての任命された執行官は
•私たちのすべての役員と私たちの役員が指名しました
•私たちのすべての役員、役員、役員指名者は一つの団体としています。
私たちは米国証券取引委員会の規則と規定に基づいて実益所有権を決定しており、これらの情報は必ずしも実益所有権が他の目的のために使用されているとは限らない。これらの規則によれば、利益所有権は、個人またはエンティティが投票権または投資権を個別にまたは共有する任意の株式を有し、個人またはエンティティが2023年12月31日から31日後60日以内に株式オプションの行使などによって取得する権利を有する証券を含む。脚注に明記され、適用された場合に共同体財産法を遵守しない限り、私たちは、私たちに提供された情報に基づいて、次の表に記載された個人および実体が、その実益に対して所有するすべての普通株に対して唯一の投票権および投資権を有すると信じている。
次の表の実益所有権パーセンテージは、2023年12月31日現在発行された645,738株普通株式に基づいている。
| | | | | | | | | | | | | | |
| | 実益所有普通株 |
| 実益所有者の氏名又は名称及び住所 | | 実益所有持分 | | 総普通株の割合を占める |
役員および指名された行政員(1) | | | | |
マーク·ヘルツ博士です。(2) | | 55,072 | | 8.53 | % |
リンエン·ケリー(3) | | 233 | | * |
デヴィッドパン屋(4) | | 2,067 | | * |
ロロフ·ユンゲン(5) | | 1,232 | | * |
カミラ·V·シンプソンM.Sc(5) | | 1,232 | | * |
デヴィッド·スクルス(5) | | 1,232 | | * |
全役員と執行幹事(8人)(6) | | 104,339 | | 16.92 | % |
*実益を代表して私たちが発行した普通株式の1%未満の株式を持っています。
(1)別の説明がない限り、受益者の住所はC/o GRI Bio、Inc.2223 Avenida de la Playa、Suite 208、La Jolla、CA 92037である。
(2)55,072株普通株で構成されている。
(3)(I)29株普通株式および(Ii)204株普通株を含み、2024年1月15日から60日以内に行使可能な株式オプションにより発行することができる。
(4)(I)42株の普通株式と(Ii)2,025株が2024年1月15日から60日以内に行使可能な株式オプションに従って発行可能な普通株を含む。
(5)1,232株普通株からなり、2024年1月15日から60日以内に行使可能な株式オプションにより発行可能。
(6)(1)上記脚注(2)~(5)で述べた普通株、(2)Vipin Kumar Chaturvedi博士が保有する24,598株の普通株、および(3)アルバート·アグロー博士が保有する24,598株の普通株を含む。
株式報酬計画情報
次の表は、2023年12月31日までの株式報酬計画の情報を示しています
| | | | | | | | | | | | | | | | | | | | | | | |
| | 未償還オプションを行使する際に発行すべき証券数 | | 未償還オプションの加重平均行権価格 | | 株式補償計画に基づいて将来発行可能な証券の数((A)欄に反映されている証券を除く) |
| 計画種別 | | (a) | | (b) | | (c) |
証券保有者が承認した持分補償計画(1) | | 32,642 | | | $ | 169.84 | | | 30,952 | | (2) |
| 証券保有者の許可を得ていない持分補償計画 | | — | | | — | | | — | | |
合計する | | 32,642 | | | $ | 169.84 | | | 30,952 | | |
__________________
(1)A&R 2018計画により認可された我々の普通株の株式数は、A&R 2018計画が満期になるまで毎年1月1日に自動的に増加し、金額は前年の12月31日に発行された普通株総数の4%に相当し、当社の取締役会または報酬委員会がこの年度に少ない数の株式を増加させることを決定したことによる。
(2)合併については、2023年4月20日、会社株主は、この計画下の奨励として、24,129株の普通株総数から30,952株の普通株を増やすことを含むA&R 2018計画を承認した。
(1)A&R 2018計画の普通株式が含まれています。この計画の説明については、付記11を参照されたい株に基づく報酬“本年度報告書に含まれる財務諸表に含まれる。
GRI Bio,Inc.2018年持分インセンティブ計画の改訂と再起動説明
2023年4月21日、会社株主はA&R 2018計画を承認した。A&R 2018は2023年4月21日に発効する予定で,株主はA&R 2018計画(I)に対する修正案を承認し,株式総数を増加させる24,129共有する30,952A&R 2018計画に基づいて奨励として発行される普通株は、(Ii)A&R 2018計画下の奨励的株式オプションの行使により発行可能な普通株の総最高数を380,952株に増加させ、(Iii)A&R 2018計画の期限を2033年1月1日に延長し、(Iv)会社株主のさらなる承認を経ずに奨励とされる“再定価”行動を禁止し、(V)A&R 2018計画に基づいて任意の非従業員取締役に付与される株式の付与日の公正価値と、任意の例年に任意の非従業員取締役に支払われる任意の他の現金補償の合計が75万ドルを超えてはならないこと、および(V)改正非従業員取締役への奨励限度額。このような非従業員役員が最初に取締役会に参加した当時、この数字は1,000,000ドルに増加した。
A&R 2018計画は、当社の従業員、取締役、コンサルタントに奨励株式オプション、非法定株式オプション、制限株式、業績単位、業績株、RSU、その他の株式ベースの奨励を付与することを規定しています。A&R 2018計画の目的は、重大な責任を担うポストの最適な利用可能な人員を吸引し、維持し、私たちの従業員、取締役、コンサルタントに追加の激励を提供し、私たちの業務の成功を促進することである。A&R 2018計画では、2024年1月1日から2033年1月1日(当該日を含む)までの各カレンダー年度の初日に毎年増加し、前のカレンダー年度最終日の流通株式総数の(X)4%以上、および(Y)取締役会が決定した比較的少ない数の株式に相当することが規定されている。A&R 2018は、行使価格および計画に基づいて付与されたいくつかの報酬の条項を修正することを管理者にさらに許可する計画である。
GRI Bio,Inc.2015年持分インセンティブ計画の説明
GRI運営計画は2015年7月10日にGRI運営株主の承認を得た。合併協定によると、当社は2023年4月21日に、GRI運営計画とその発効時期に付与された未完了奨励を担当した。GRI運営計画は,取締役会または取締役会が指定した委員会によって管理される.GRI運営計画は、管理者が行使価格を修正し、その計画に基づいて何らかの契約を付与する条項をさらに許可することをさらに許可する。GRI運営計画によると、新たな奨励を支給することはできません。2023年12月31日現在,GRI運営計画の下で未解決の賞はない。
第十三項:特定の関係及び関連取引の確立、並びに取締役の独立性
以下は、2022年1月1日以来、私たちが参加している各取引または一連の同様の取引の概要です
•総額が12万ドルを超えるか、または2023年12月31日と2022年12月31日までの私たちの総資産の1%を超えるか、および
•当社の任意の取締役又は行政者、当社の5%の株式を保有している者又はその直系親族は、直接又は間接的な重大な利益を有することがあります。
TEPはチケットを変更できます
2018年11月、GRI運営およびTEPは、TEP手形および株式承認証購入契約を締結し、合意に基づいて、TEPは、TEP手形および引受権証と交換して、1株0.01ドルの行使用価格で最大96,428株のGRI運営普通株をGRI運営に最大500万ドルの資金を提供することに同意した。TEP手形はGRI運営会社の資産を担保とし,年利12%の金利で元金残高を返済していない場合は簡単な利息とすべきである。未償還元金と受取利息残高総額は,最初にGRIが運営する次の融資(TEP手形の定義)と2020年5月2日(満期日)の早い日に満期となる。
TEP説明の改訂
2019年12月、GRI運営とTEPはTEP説明を改訂しました。TEPが第2の250万ドルに資金を提供する代わりに、TEPはGRIに最初の追加の50万ドルの前払いを提供し、変換可能なチケット1枚と交換して、最大で購入することができる2,467GRIは普通株を運営し、行権価格は$0.011株あたり,および特定のコールオプションプロトコルに従ってGRIが運営する権利を譲渡する.このコールオプション協定は2015年に締結され、GRI運営部門に最大の買い戻しを提供した5,674取引相手が持つGRIは普通株を運営しており、価格は#ドルです187.182025年4月1日までのいつでも。
2020年7月,TEP手形満期日を2020年8月31日まで延長し,2021年3月,TEPはGRI運営が2021年10月31日までに期限超過元金と課税利息残高を支払うことができなかったため救済措置を行使する既存の権利を放棄することに同意した。
2021年5月、GRI運営およびTEPは、別個の変更された変換オプションを有する変換可能なチケットと交換するために、GRI運営に第2の50万ドルの追加の前払いを提供することに同意したTEPチケットをさらに修正した。
2022年7月、GRI運営およびTEPはさらにTEPチケットを改訂し、TEPは、購入のために変換可能なチケットと引受権証明書とを交換するために、GRI運営に第3の追加の125,000ドルの前金を提供することに同意した167GRIは普通株を運営し、行権価格は$0.01一株ずつです。
TEPチケットの変換
2022年12月に合併協定の署名に伴いTEPチケットは全文に変換されました22,172GRIとTEPが署名した変換プロトコルにより,GRIは普通株の株式を運営する.変換後,TEPはGRI運営普通株の5%を超える実益所有者となる.
株式融資
2022年12月13日から2023年5月8日までの間に、私たちはAltiumと証券購入協定、高級担保手形、権証と取引所株式証を締結した。備考4を参照。“バロンと合併”と注11。本年度報告F−1ページからの連結財務諸表における“株主権益(赤字)”である。
雇用協定
私たちは私たちの特定の執行官たちと雇用協定を締結した。“プロジェクト11--行政職報酬”を参照。
持分補助金
私たちは私たちの特定の幹部と取締役会の会員たちに株式オプションを付与した。“プロジェクト11--行政職報酬”を参照
賠償と責任制限
デラウェア州一般会社法第145条授権法団は、その役員及び高級職員が、ある会社の役員又は高級職員を務めているか、又はその一方の訴訟、訴訟及び法律手続きによる法的責任となることを保障している。賠償は、そのような行動、訴訟、または訴訟によって実際かつ合理的に生じる費用(弁護士費を含む)、罰金、および和解金額を含む判決、罰金、および役員または官僚をカバーすることができる。第百四十五条会社は、このような訴訟、訴訟又は法律手続を最終的に処分する前に、取締役及び上級管理者が発生した費用(弁護士費を含む)を支払うことを許可する。また、第145節では、会社は、その役員及び上級職員を代表して保険を購入·維持する権利があり、彼らが取締役又は高級職員として負ういかなる責任、又は彼らの身分によるいかなる責任も負う権利があり、会社が第145条に基づいて取締役又は高級職員のこのような責任について取締役又は上級職員に賠償する権利があるか否かにかかわらず、そのような責任を負うことができる。
吾らはすでに改訂及び重述された会社登録証明書及び改訂及び再記載された会社定款に条文を採用して、取締役の個人責任を制限又は除去し、当該等の条文はDGCLによって許可されており、その現在の存在又は将来が改訂される可能性がある。したがって、取締役は、私たちまたは私たちの株主が取締役としての受託責任に違反することによって、私たちまたは私たちの株主に個人的な責任を負うことはありませんが、以下の責任は除外します
•取締役の私たちや私たちの株主に対する忠誠義務に違反します
•善意でない行為やしないこと、または故意の不正行為に関連しているか、または違法であることを知っている
•配当金または株式の不法購入、償還または他の分配に関連する任意の不正支払い;または
•取締役は不当な個人利益の取引をむさぼる。
これらの責任制限は連邦証券法下の取締役の責任を変えることはなく、禁止令や撤回などの衡平法救済措置の可用性にも影響を与えない。
また、私たちの付例は以下のように規定している
•DGCLが現在存在している場合や将来改訂される可能性がある場合には、取締役会の適宜決定の下で、私たちの役員、上級管理職、および特定の従業員に対して最大限の賠償を行います
•限られた例外を除いて、私たちの役員に弁護士費を含む合理的な費用を前払いし、私たちの取締役会の適宜の決定の下で、私たちの上級職員とある従業員に、彼らが私たちを代表してくれたり、私たちを代表してサービスすることに関する法律手続きに関連する費用を前払いします。
私たちは私たちのすべての役員と賠償協定を締結し、私たちの役員とこのような合意に到達しようとしています。これらの協定は、私たちはデラウェア州の法律で許可されたすべての役員、私たちの幹部、時々彼らの付属会社も含めて、私たちのすべての役員、私たちの幹部を最大限賠償することを規定している。私たちは、弁護士費(判決、罰金、和解金額は含まれていませんが)を含む前払い費用を各保障された役員、役員、または関連会社に提供し、賠償を受けることができる任意の訴訟に関連して、その人を取締役または役員として私たちを代表する、または私たちの権利を促進するために提起された任意の訴訟または訴訟を賠償します。さらに、私たちのいくつかの役員または上級管理者は、その関連会社または他の第三者によって提供されるいくつかの賠償、前借り費用または保険を得る権利がある可能性があり、これらの賠償は、本明細書で指す取締役と同じである取締役または上級管理者のサービスに起因する訴訟に適用される可能性がある。それにもかかわらず、私たちは賠償協定で同意しており、私たちはそのような役員または高級管理者に対する責任が主であり、当該等連合会社または他の第三者は、そのような取締役が発生した支出または責任について賠償を提供するいかなる責任も副次的である。
役員は自主独立している
第10項を参照。役員,役員,コーポレートガバナンス管理−取締役の独立性−
わが社の取締役会各委員会
当社の取締役会はすでに監査委員会、報酬委員会及び指名及び会社管理委員会を設立し、各委員会はすべて当社の取締役会が採択した書面定款に基づいて運営されている。10項を参照。“役員,役員,会社管理−取締役会委員会−.”
14項目目:総会計士費用とサービス料
独立公認会計士事務所はSadler,Gibb&Associates LLC, ユタ州ドレッパー監査役事務所ID:3627.
次の表は,2023年12月31日,2023年12月31日,2022年12月31日までの年間でSadler,Gibb&Associates LLCサービスで発生した費用総額を示している。
| | | | | | | | | | | |
| 十二月三十一日 |
| 2023 | | 2022 |
料金を審査する (1) | $ | 156,323 | | | $ | 204,878 | |
監査関連費用 (2) | — | | | — | |
税金.税金(3) | — | | | — | |
他のすべての費用 (4) | — | | | — | |
| 合計する | $ | 156,323 | | | $ | 204,878 | |
(1)料金を審査する私たちの独立公認会計士事務所が当社の年次財務諸表を監査し、当社の四半期報告書に含まれる財務諸表を審査するため、または通常提供されるこれらの財政年度の法定および規制書類または約束に関するサービス、および米国証券取引委員会に提出された登録声明文書および証券発行に関する慰問状について提供される専門サービスの総費用を指します。
(2)監査関連費用私たちの独立公認会計士事務所が提供する保証及び関連専門サービスから徴収される費用総額を指し、このような費用は私たちの財務諸表の審査或いは審査の表現と合理的に関連しており、“審査費用”の項に記載されていない。
(3)税金.税金私たちの独立公認会計士事務所が税務コンプライアンス、税務提案及び税務計画サービスが提供する専門サービスについて徴収する総費用のことです。
(4)他のすべての費用独立公認会計士事務所が提供する他のすべての製品とサービスの課金総額を指しますが、他のカテゴリのサービスは含まれていません
監査委員会は、独立性、資格、業績(例えば、適用される)に基づいて、独立公認会計士事務所がすべての監査サービスおよび非監査サービスを提供する採用および費用を事前に承認する。監査委員会は、監査委員会を構成することができ、監査委員会の1つ以上のメンバーからなるグループ委員会は、監査及び許可された非監査サービスに対して予め承認された権限を与えることができるが、特定の金額を超えてはならない。Sadler,Gibb&Associates LLCが本報告の間に提供したすべての監査サービスは、私たちの取締役会の承認を得ました。
監査および非監査サービスの事前承認
私たちの監査委員会は、すべての監査および非監査サービスを承認するための政策および手続きを採択し、これらのサービスは私たちの独立した公認会計士事務所が行います。これらの政策及び手続は、一般に、監査又は非監査サービスを提供する公認会計士事務所を招聘することはなく、当該サービスが予め我々の監査委員会の特別承認を受けているか、又は後述するいずれかの事前承認手続に従って行われなければならない。
私たちの監査委員会は時々私たちの独立公認会計士事務所が今後12ヶ月以内に私たちに提供される特定のタイプのサービスを期待することを事前に承認するかもしれません。いずれの事前承認も、提供される特定のサービスまたはサービスタイプに関する詳細な説明であり、通常、最高額に制限される。
アメリカ証券取引委員会とアメリカ上場会社会計監督委員会の監査師の独立性に関する要求によると、私たちの監査委員会は独立した公認会計士事務所の任命、報酬、監督を担当しています。この責任を認識するために、独立公認会計士事務所により提供されるすべての監査及び許可された非監査サービスを、監査委員会又は議長(監査委員会会議間でそのような承認が必要な場合)に予め承認しておく。これらのサービスは、監査サービス、監査関連サービス、税務サービス、および他のサービスを含むことができる。
第IV部
項目15.すべての展示品、財務諸表明細書
(a)以下の書類は本報告の一部として提出される:
(1)財務諸表それは.当社の財務諸表は、独立公認会計士事務所Sadler、Gibb&Associates LLCの財務諸表とともに本年度報告書に含まれ、F-1ページから始まります。
(2)財務諸表明細書それは.その中に記載されている資料が財務諸表または付記に適用または記載されていないことが要求されるので、すべての付表は省略される。
(3)展示品です下記(B)項を参照。
(b)陳列品
以下の“展示品索引”に記載されている展示品は、本年度報告の一部としてアーカイブまたは格納されている。
| | | | | | | | | | | | | | | | | | | | |
| | | 引用で編入する |
| 展示品番号: | | 説明する | 同封アーカイブ | 表 | 日取り | 書類番号 |
| 2.1△ | | 会社,GRI Bio,Inc.とVallon Merge Sub,Inc.の間の合併プロトコルと計画は,2022年12月13日である. | | 8-K | 12/13/22 | 001-40034 |
| 2.2 | | 当社,GRI Bio,Inc.とVallon Merge Sub,Inc.の間の合意と合併計画修正案は,2023年2月17日とした。 | | S-4/A | 02/24/23 | 333-268977 |
3.1 |
| 改訂および再予約された会社登録証明書 | X | | | |
3.2 |
| 付例を改訂および再制定する | | 8-K/A | 05/26/23 | 001-40034 |
4.1 |
| 普通株式証明書サンプル。 | | S-1 | 10/23/20 | 333-249636 |
4.2△ |
| 2022年12月13日、Vallon製薬会社と投資先が署名した登録権協定。 | | 8-K | 12/31/22 | 001-40034 |
| 4.3 | | ブリッジ授権書表。 | | 8-K | 12/13/22 | 001-40034 |
| 4.4 | | 普通株式引受権証第1号修正案フォーマット。 | | 8-K | 07/26/22 | 001-40034 |
| 4.5 | | 株式承認株式証形式。 | | 8-K | 12/13/22 | 001-40034 |
| 4.6 | | 株式証明書を両替します。 | | 8-K | 12/13/22 | 001-40034 |
| 4.7△ | | GRIバイオ運営会社は手形のフォーマットを高度に保証する。 | | 8-K | 12/13/22 | 001-40034 |
| 4.8 | | TEP Biotech,LLCへの引受権証は,2018年11月2日に発行された。 | | S-4 | 12/23/22 | 333-268977 |
| 4.9 | | TEP Biotech,LLCに発行された株式引受権証は,2019年12月3日である. | | S-4 | 12/23/22 | 333-268977 |
| 4.10 | | TEP Biotech,LLCへの引受権証は,2022年7月7日に発行された。 | | S-4 | 12/23/22 | 333-268977 |
| 4.11 | | 欧宝·グレイブに発行された株式引受権証は、2022年7月7日。 | | S-4 | 12/23/22 | 333-268977 |
| 4.12 | | 欧宝·グレイブに発行された株式引受権証は、2020年11月4日となっている。 | | S-4 | 12/23/22 | 333-268977 |
| 4.13 | | Dinver LLCに発行された株式引受権証は、日付は2020年11月4日。 | | S-4 | 12/23/22 | 333-268977 |
| 4.14 | | Eric Reiterに発行された株式引受権証は、2020年11月4日となっている。 | | S-4 | 12/23/22 | 333-268977 |
| 4.15 | | FT 618 Investments,LLCに発行された引受権証は,2020年11月9日とした。 | | S-4 | 12/23/22 | 333-268977 |
| 4.16 | | FT 618 Investments,LLCに発行された引受権証は,2020年12月28日である。 | | S-4 | 12/23/22 | 333-268977 |
| 4.17 | | “2020年株式証改訂表”。 | | S-4/A | 01/27/23 | 333-268977 |
| 4.18 | | “2022年株式証改訂表”。 | | S-4/A | 01/27/23 | 333-268977 |
| 4.19△ | | GRI Bio Operations,Inc.とAltium Growth Fund,LP間の証券購入プロトコルは,2022年12月13日である. | | 8-K | 12/13/22 | 001-40034 |
| 4.20△ | | 当社,GRI Bio Operations,Inc.とAltium Growth Fund,LP間の証券購入プロトコルは,2022年12月13日とした。 | | 8-K | 12/13/22 | 001-40034 |
| 4.21△ | | 当社,GRI Bio Operations,Inc.とAltium Growth Fund,LP間の証券購入プロトコルの総合修正案は,期日は2023年2月17日である。 | | S-4 | 03/06/23 | 333-268977 |
| 4.22 | | 投資家権利協定は、当社とSalmon Pharma GmbHによって署名され、日付は2019年7月25日です。 | | S-1 | 10/23/20 | 333-249636 |
| 4.23△ | | 会社とその特定の株主との間の投票合意は、2020年12月30日となる。 | | S-1 | 10/23/20 | 333-249636 |
| 4.24 | | 資金を前払いして株式証明書を発行する. | | S-1/A | 01/31/24 | 333-276025 |
| | | | | | | | | | | | | | | | | | | | |
| 4.25 | | B-1系列一般権証の表. | | S-1/A | 01/31/24 | 333-276025 |
| 4.26 | | B-2シリーズ通常権証の表. | | S-1/A | 01/31/24 | 333-276025 |
| 4.27 | | 証券説明。 | X | | | |
10.1# |
| A&R 2018持分インセンティブ計画。 | | 10-Q | 05/15/23 | 001-40034 |
10.2# |
| 会社の2018年株式インセンティブ計画下の非限定株式オプション協定フォーマット。 | | S-1 | 10/23/20 | 333-249636 |
10.3# |
| 会社の2018年株式インセンティブ計画下のインセンティブ株式オプションプロトコルフォーマット。 | | S-1 | 10/23/20 | 333-249636 |
10.4# |
| Gri Bio Operations,Inc.2015年持分インセンティブ計画。 | | S-4 | 12/23/22 | 333-268977 |
10.5 |
| ライセンス契約は,会社とMedice Arzneimittel Putter GmbH&Co.KGが署名し,日付は2020年1月6日である. | | S-1 | 10/23/20 | 333-249636 |
10.6 |
| 会社サポートプロトコルのフォーマットは、GRI Bio Operations,Inc.と各プロトコルで指定された各当事者との間で署名され、日付は2022年12月13日である。 | | 8-K | 12/13/22 | 001-40034 |
10.7 |
| GRI Bio Operations,Inc.とプロトコルで指定された各当事者との間のサポートプロトコルフォーマットは,2022年12月13日である. | | 8-K | 12/13/22 | 001-40034 |
10.8 |
| 配給代理契約は、会社とラデンブルクタルマン社が締結し、期日は2022年5月13日。 | | 8-K | 05/13/22 | 001-40034 |
10.9△ |
| 証券購入協議フォーマット | | 8-K | 05/13/22 | 001-40034 |
10.10△ |
| 修正案番号:1証券購入協定は、当社と署名ページで決定された各購入者との間で署名され、期日は2022年7月25日である。 | | 8-K | 07/26/22 | 001-40034 |
10.11# |
| GRI生物運営会社とショーン·エドワーズとの間の奨励的株式オプション協定は,2016年11月4日である。 | | S-4 | 12/23/22 | 333-268977 |
10.12# |
| GRI Bio Operations,Inc.とRoHit Loombaとの間の非限定的株式オプションプロトコルは、2016年11月4日である。 | | S-4 | 12/23/22 | 333-268977 |
10.13# |
| GRI生物運営会社とGerald Yakatanとの間の非限定的株式オプション協定は、2016年11月4日である。 | | S-4 | 12/23/22 | 333-268977 |
10.14△ |
| GRI生物運営会社とアルバート農業会社が署名した制限株式購入協定は,2009年11月1日であった。 | | S-4 | 12/23/22 | 333-268977 |
10.15# |
| GRI生物運営会社とアルバート農業会社との間の制限株式奨励協定は、日付が2015年4月2日であり、GRI生物運営会社とアルバート農業会社との間の制限株式奨励協定改正案により改正され、期日は2022年12月7日である。 | | S-4 | 12/23/22 | 333-268977 |
10.16#△ |
| GRI Bio Operations,Inc.およびVipin Kumar Chaturvediが署名した制限株式購入協定は、2009年3月28日である。 | | S-4 | 12/23/22 | 333-268977 |
10.17# |
| GRI Bio Operations,Inc.とVipin Kumar Chaturvediとの間の制限株式奨励協定は、日付が2015年4月2日、GRI Bio Operations、Inc.とVipin Kumar Chaturvediとの間の制限株式奨励プロトコル修正案によって改正され、日付は2022年12月7日である。 | | S-4 | 12/23/22 | 333-268977 |
10.18# |
| GRI生物運営会社とショーン·エドワーズとの間の制限的株式奨励協定は,2021年3月31日である。 | | S-4 | 12/23/22 | 333-268977 |
| 10.19# | | GRI生物運営会社とショーン·エドワーズとの間の制限的株式奨励協定は,2022年12月7日である。 | | S-4 | 12/23/22 | 333-268977 |
| 10.20#△ | | GRI生物運営会社とW.Marc Hertzが署名した制限株式購入協定は、2009年3月28日である。 | | S-4 | 12/23/22 | 333-268977 |
| 10.21# | | GRI Bio Operations,Inc.とW.Marc Hertzとの間の制限株式報酬プロトコルは、日付が2015年4月2日であり、日付は、2022年12月7日であるGRI Bio Operations,Inc.とW.Marc Hertzとの間の制限株式奨励プロトコル修正案によって修正される。 | | S-4 | 12/23/22 | 333-268977 |
| 10.22# | | GRI生物運営会社とW.Marc Hertzが署名した制限株式奨励協定は、2021年3月31日である。 | | S-4 | 12/23/22 | 333-268977 |
| 10.23# | | GRI生物運営会社とW.Marc Hertzが署名した制限株式奨励協定は、2022年12月7日である。 | | S-4 | 12/23/22 | 333-268977 |
| 10.24# | | 諮問·臨床諮問委員会協定は,GRI生物運営会社とRohit Loomba,M.D.が署名し,日付は2016年6月3日であった。 | | S-4 | 12/23/22 | 333-268977 |
| 10.25# | | 諮問·科学諮問委員会協定は、GRI Bio Operations,Inc.とVipin Kumar Chaturvediが自らまたはVidur Discovery LLCを介して署名され、日付は2018年10月31日である。 | | S-4 | 12/23/22 | 333-268977 |
| 10.26△ | | La Jolla ShorePlaza、LLCとGRI Bio Operations、Inc.の間の賃貸契約は、2018年3月2日であり、この物件はカリフォルニア州ラホア208号Avenida de la Playa 2223、Suite 208、92037、2021年2月16日、2024年2月20日に改訂された。 | X | | | |
| 10.27# | | 非従業員役員報酬計画を修正して再作成した。 | | S-1/A | 12/04/23 | 333-274972 |
| 10.28# | | 賠償協議形式。 | | 8-K | 04/21/23 | 001-40034 |
| 10.29# | | GRI生物運営会社とマーク·ヘルツ博士が署名した雇用協定は,2023年2月20日である。 | | S-4/A | 02/24/23 | 333-268977 |
| | | | | | | | | | | | | | | | | | | | |
| 10.30# | | GRI生物運営会社とLeanne M.Kellyとの雇用契約は,2023年2月20日である。 | | S-4/A | 02/24/23 | 333-268977 |
| 10.31# | | GRI Bio Operations,Inc.とVipin Kumar Chaturvediとの間の雇用契約は,2023年2月20日である。 | | S-4/A | 02/24/23 | 333-268977 |
| 10.32# | | GRI生物運営会社とアルバート·アグロー博士が署名した諮問協定は,2023年1月9日である。 | | 8-K | 04/21/23 | 001-40034 |
| 10.33# | | 当社とDavid·ベックとの別居協定は、2023年4月21日となっています。 | | 8-K | 04/21/23 | 001-40034 |
| 10.34# | | 当社がアルバート·アグロー博士と締結した雇用契約は、2023年7月1日となっています。 | | 10-Q | 08/14/23 | 001-40034 |
| 10.35△ | | 資産購入協定は,会社とAardvark治療会社が署名し,期日は2023年8月22日である。 | | 8-K | 08/23/23 | 001-40034 |
| 10.36△ | | 証券購入協議フォーマット。 | | S-1/A | 01/31/24 | 333-276025 |
| 10.37 | | 代理契約書のフォーマットを配置する。 | | S-1/A | 01/31/24 | 333-276025 |
21.1 |
| 付属会社 | | S-1/A | 12/04/23 | 333-274972 |
| 23.1 | | 独立公認会計士事務所の同意 | X | | | |
24.1 |
| 役員及びある行政員の授権書(署名ページに掲載) | | | | |
31.1 | | 最高経営責任者は、取引法第13 a-14(A)または15 a-14(A)条に基づいて認証される。 | X | | | |
31.2 | | 取引法第13 a-14条(A)又は15 a-14(A)条に基づいて首席財務官を証明する | X | | | |
| 32.1+ | | 2002年にサバンズ·オクスリ法案第906条で可決された“米国法典”第18編1350条による主要行政官の証明 | X | | | |
| 32.2+ | | 2002年サバンズ·オキシリー法第906条で可決された“米国法典”第18編1350条による首席財務官の証明 | X | | | |
| 97 | | 政策を取り戻す。 | | 10-Q | 11/14/23 | 001-40034 |
| 101インチ | | XBRLインスタンスドキュメント | | | | |
| 101 SCH | | XBRL分類拡張アーキテクチャリンクライブラリ文書 | | | | |
| 101キャリブレーション | | XBRL分類拡張計算リンクライブラリ文書 | | | | |
| 101 DEF | | XBRL分類拡張Linkbase文書を定義する | | | | |
| 101実験 | | XBRL分類拡張タグLinkbaseドキュメント | | | | |
| 101プレミアム版 | | XBRL分類拡張プレゼンテーションLinkbaseドキュメント | | | | |
| 104 | | 表紙対話データファイル(添付ファイル101に含まれるイントラネットXBRL形式) | | | | |
他の説明がない限り、証拠品はすべて手紙に保存されている。
△ S-K条例第601(A)(5)項によると、付表と証拠物は省略されている。米国証券取引委員会の要求に応じて、会社は任意の漏れたスケジュールの補充コピーを提供することを約束した。
#Bは、管理契約または任意の補償計画、契約、またはスケジュールを表します。
+:本10-Kフォーム年次報告添付ファイル32.1に添付されている認証は、米国証券取引委員会に届出されたものとはみなされず、参照によって登録者が1933年“証券法”(改訂本)または1934年“証券取引法”(改訂本)に従って提出された任意の文書に組み込まれてはならず、このような文書に含まれる任意の一般的な登録言語にかかわらず、本表の10-K年度報告日の前または後に行われてはならない。
サイン
1934年“証券取引法”第13節又は第15節(D)節の要求に基づいて、登録者は、正式に許可された以下の署名者が代表して本年度報告に署名することを正式に手配した。
| | | | | | | | |
| Gri Bio,Inc. |
| | |
| 日付:2024年3月28日 | 差出人: | /S/W.Marc Hertz |
| | 名前:W.Marc Hertz博士 |
| | 役職:総裁と最高経営責任者 |
署名と授権書
このような陳述を通じて、以下の署名のすべての人が、David·ベックを彼または彼女の真の合法的な事実代理人および代理人として構成し、指定することを知っている。誰もが、彼または彼女の名義、場所、代理、任意およびすべての身分で、本Form 10-K年次報告書の任意およびすべての修正に署名し、その中のすべての証拠物およびすべての関連文書を米国証券取引委員会に提出し、上記の事実代理人と代理人、およびそれらそれぞれを付与する。関連する場所内および周囲で必要とされるすべてのことおよびしなければならないすべてのことを行い、実行する権利が完全にあり、その可能性または自ら行うことができるすべての意図および目的を尽くし、本明細書でそのようなすべての事実代理人および代理人またはそのうちのいずれか1人、またはその1人以上の代替者を承認および確認することは、本条例に従って任意の行為および事柄を合法的に行うか、または手配することができる。
1934年の証券取引法の要求に基づき、本年度報告は、指定日に登録者として以下の者代表登録者によって署名された。
| | | | | | | | | | | | | | |
| サイン | | タイトル | | 日取り |
| | | | |
| タイトル/著者Marc Hertz,Ph.D. | | 取締役最高経営責任者総裁 (首席行政主任) | | 2024年3月28日 |
| マーク·ヘルツ博士です。 | | | |
| | | | |
| /S/リンエン·ケリー | | 首席財務官 (首席財務会計官) | | 2024年3月28日 |
| リンエン·ケリー | | | |
| | | | |
| 寄稿S/David査読者スクールス | | 取締役、取締役会長 | | 2024年3月28日 |
| デヴィッド·スクルス | | | | |
| | | | |
| 寄稿S/David·ベック | | 役員.取締役 | | 2024年3月28日 |
| デヴィッドパン屋 | | | | |
| | | | |
| /投稿S/ロロフ·ユン | | 役員.取締役 | | 2024年3月28日 |
| ロロフ·ユンゲン | | | | |
| | | | |
| 題名/責任者:M.Sc./S/Camilla V.Simpson,M.Sc. | | 役員.取締役 | | 2024年3月28日 |
| カミラ·V·シンプソンM.Sc | | | | |
連結財務諸表索引
| | | | | |
独立公認会計士事務所報告 | F-2 |
2023年12月31日と2022年12月31日までの連結貸借対照表 | F-3 |
2023年12月31日と2022年12月31日までの連結業務報告書 | F-4 |
2023年12月31日までと2022年12月31日まで年度株主権益(赤字)総合変動表 | F-5 |
2023年12月31日と2022年12月31日までの統合現金フロー表 | F-6 |
連結財務諸表付記 | F-7 |
独立公認会計士事務所報告
GRI Bio,Inc.取締役会と株主へ:
財務諸表のいくつかの見方
GRI Bio,Inc.(“当社”)2023年12月31日までと2022年12月31日までの連結貸借対照表,2023年12月31日までの2年間の各年度に関する総合経営報告書,株主権益(損失)とキャッシュフローの変化および関連付記(総称して“財務諸表”と呼ぶ)を監査した。上記財務諸表は、すべての重要な点において、当社の2023年12月31日と2022年12月31日までの財務状況、および2023年12月31日までの2年間の各年度の経営結果とキャッシュフローを公平に反映しており、米国公認の会計原則に適合していると考えられる。
持続経営に関する解釈的段落
添付財務諸表の作成は、同社が引き続き経営を継続する企業であると仮定している。財務諸表付記2で述べたように、当社は経営により常に赤字を計上しており、純資本不足が発生しており、その継続経営能力に大きな疑いを抱かせている。付記2は、これらの事項における経営陣の計画も説明しています。財務諸表には、このような不確実性の結果に起因する可能性のある調整は含まれていません。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査することを求められておらず、私たちも招聘されて監査を行っていません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性について意見を述べるためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている
/S/サドラー,Gibb&Associates,LLC
2022年以来、私たちは会社の監査役を務めてきた。
ドレッパーテキサス州
2024年3月28日
Gri Bio,Inc.
合併貸借対照表
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
| | | | | | | | | | | |
| 十二月三十一日 |
| 2023 | | 2022 |
| 資産 | | | |
| 流動資産: | | | |
| 現金と現金等価物 | $ | 1,808 | | | $ | 9 | |
| | | |
| 前払い費用と他の流動資産 | 1,126 | | | 303 | |
| 流動資産総額 | 2,934 | | | 312 | |
| 財産と設備、純額 | 8 | | | 4 | |
| 経営的リース使用権資産 | 14 | | | 67 | |
| 総資産 | $ | 2,956 | | | $ | 383 | |
| | | |
| 負債と株主権益(赤字) | | | |
| 流動負債: | | | |
| 売掛金 | $ | 1,410 | | | $ | 1,294 | |
| 費用を計算する | 1,270 | | | 36 | |
| 従業員からの前払い | — | | | 5 |
| 株式証法的責任 | 3 | | | — |
| 橋はこのチケットを受け取ります。純額です | — | | | 602 | |
| 賃貸負債を経営し、流動 | 14 | | | 57 | |
| 流動負債総額 | 2,697 | | | 1,994 | |
| | | |
| 非流動経営賃貸負債 | — | | | 14 | |
| 総負債 | 2,697 | | | 2,008 | |
| | | |
| 引受金及び又は有事項(付記13) | | | |
| | | |
| 株主権益(赤字): | | | |
普通株、$0.0001額面価値250,000,000ライセンス株;645,738そして142,8202023年12月31日と2022年12月31日までの発行済株式 | — | | | — | |
| 実収資本を追加する | 31,792 | | | 16,871 | |
| | | |
| 赤字を累計する | (31,533) | | | (18,496) | |
| 株主権益合計 | 259 | | | (1,625) | |
| 総負債と株主権益(赤字) | $ | 2,956 | | | $ | 383 | |
連結財務諸表の付記を参照。
Gri Bio,Inc.
連結業務報告書
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
| | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 |
| | | |
| 運営費用: | | | |
| 研究開発 | $ | 3,232 | | | $ | 242 | |
| 一般と行政 | 8,155 | | | 1,997 | |
| 総運営費 | 11,387 | | | 2,239 | |
| 運営損失 | (11,387) | | | (2,239) | |
| その他の収入 | 250 | | | — | |
| 株式証負債の公正価値変動を認める | 182 | | | — | |
| 債務返済損失 | — | | | (325) | |
| 利子支出,純額 | (2,082) | | | (653) | |
| 純損失 | $ | (13,037) | | | $ | (3,217) | |
| 普通株は基本的に1株当たり純損失と希釈して1株当たり純損失 | $ | (28.25) | | | $ | (24.95) | |
| 加重平均発行済み普通株式、基本普通株式、希釈後普通株 | 461,566 | | | 128,994 | |
連結財務諸表の付記を参照。
Gri Bio,Inc.
合併株主権益変動表
(千単位で、株を除く)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | 普通株を償還できる | | | 普通株 | | その他の内容
実収資本 | | 積算
赤字.赤字 | | 株主の
権益(赤字) |
| | 株 | | 金額 | | | 株 | | 金額 | | | |
| バランス、2021年12月31日 | | 1,116 | | | $ | 124 | | | | 121,631 | | | $ | — | | | $ | 10,431 | | | $ | (15,279) | | | $ | (4,848) | |
| 株に基づく報酬費用 | | — | | | — | | | | — | | | — | | | 25 | | | — | | | 25 | |
| 転換可能な本チケットに関する引受権証及び非又は利益転換特徴を発行する | | — | | | — | | | | — | | | — | | | 60 | | | — | | | 60 | |
| 転換不可本票に関する引受権証を発行する | | — | | | — | | | | — | | | — | | | 30 | | | — | | | 30 | |
| 本チケットの両替ができます | | — | | | — | | | | 22,172 | | | — | | | 5,337 | | | — | | | 5,337 | |
| 報酬を支払うために発行された制限的な株式奨励 | | — | | | — | | | | — | | | — | | | 417 | | | — | | | 417 | |
| 橋本票の発行に関する引受権証を発行する | | — | | | — | | | | — | | | — | | | 571 | | | — | | | 571 | |
| 普通株を償還することができる | | (1,116) | | | (124) | | | | (1,116) | | | — | | | — | | | — | | | — | |
| 制限株の帰属 | | — | | | — | | | | 133 | | | — | | | — | | | — | | | — | |
| 純損失 | | — | | | — | | | | — | | | — | | | — | | | (3,217) | | | (3,217) | |
| バランス、2022年12月31日 | | — | | | $ | — | | | | 142,820 | | | $ | — | | | $ | 16,871 | | | $ | (18,496) | | | $ | (1,625) | |
| 株に基づく報酬費用 | | — | | | — | | | | — | | | — | | | 388 | | | — | | | 388 | |
| 制限株帰属 | | — | | | — | | | | 23,501 | | | — | | | — | | | — | | | — | |
| 株式証明書発行 | | — | | | — | | | | — | | | — | | | 532 | | | — | | | 532 | |
| 授権証行使 | | — | | | — | | | | 229,645 | | | — | | | 12 | | | — | | | 12 | |
| 決算前融資における普通株の発行 | | — | | | — | | | | 173,558 | | | — | | | 11,721 | | | — | | | 11,721 | |
| 普通株式を発行して橋越し手形を決済する | | — | | | — | | | | 7,756 | | | — | | | 3,333 | | | — | | | 3,333 | |
| 普通株を発行して逆資本再編費用に用いる | | — | | | — | | | | 4,363 | | | — | | | 1,875 | | | — | | | 1,875 | |
| 逆資本再編でワロン株主に普通株を発行する | | — | | | — | | | | 64,095 | | | — | | | (2,940) | | | — | | | (2,940) | |
| 純損失 | | — | | | — | | | | — | | | — | | | — | | | (13,037) | | | (13,037) | |
| バランス、2023年12月31日 | | — | | | $ | — | | | | 645,738 | | | $ | — | | | $ | 31,792 | | | $ | (31,533) | | | $ | 259 | |
連結財務諸表の付記を参照。
Gri Bio,Inc.
統合現金フロー表
(単位:千)
| | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 |
| 経営活動のキャッシュフロー: | | | |
| 純損失 | $ | (13,037) | | | $ | (3,217) | |
| 純損失と業務活動で使用されている現金の照合の調整: | | | |
| 減価償却費用 | 4 | | | 3 | |
| 債務償却と発行コスト | 2,104 | | | 217 | |
| 株に基づく報酬費用 | 388 | | | 25 | |
| 債務返済損失 | — | | | 325 | |
| 株式証負債の公正価値変動を認める | (182) | | | — | |
| 経営性リース使用権資産を減らす | 53 | | | 47 | |
| 営業資産と負債の変動: | | | |
| 前払い費用と他の流動資産 | (547) | | | (35) | |
| 売掛金 | 2,159 | | | 897 | |
| 費用を計算する | 125 | | | 696 | |
| リース負債を経営する | (57) | | | (43) | |
| 経営活動用の現金 | (8,990) | | | (1,085) | |
| | | |
| 投資活動: | | | |
| | | |
| | | |
| 財産と設備を購入する | (8) | | | (3) | |
| 投資活動用の現金 | (8) | | | (3) | |
| | | |
| 融資活動: | | | |
| 従業員からの前払い | 190 | | | 35 | |
| 従業員の立て替えを返済する | (195) | | | (30) | |
| 本券を発行して得た金を転換できない | — | | | 125 | |
| 両替できない本チケットを返済します | — | | | (125) | |
| 転換本券を発行して得た金 | — | | | 125 | |
| 本チケットの返済に転換可能です | — | | | (125) | |
| 橋を渡る本券を発行して得た金 | 1,250 | | | 1,250 | |
| 決算前融資で普通株で得た金を発行する | 12,250 | | | — | |
| 株式承認証を行使して得られた収益 | 12 | | | — | |
| 逆資本再編成で得られた現金 | 941 | | | — | |
| 逆資本再編成コストを支払う | (2,984) | | | — | |
| *繰延株式発行コストの支払い | (517) | | | (111) | |
| *債務発行コストの支払い | (150) | | | (13) | |
| *普通株式の償還が可能 | — | | | (124) | |
| 融資活動で提供された現金 | 10,797 | | | 1,007 | |
| | | |
| 現金および現金等価物の純増加(減額) | 1,799 | | | (81) | |
| 期初現金及び現金等価物 | 9 | | | 90 | |
| 期末現金および現金等価物 | $ | 1,808 | | | $ | 9 | |
| | | |
| キャッシュフロー情報の追加開示: | | | |
| 利子を支払う現金 | $ | — | | | $ | 33 | |
| 追加開示または非現金活動: | | | |
| 橋越しローンの返済用の株を発行する | $ | 3,333 | | | $ | — | |
| 変換可能なチケットの非または収益変換特徴があります | $ | — | | | $ | 60 | |
| 報酬を支払うために発行された制限的な株式奨励 | $ | — | | | $ | 417 | |
| 本票に関連して発行された引受権証の債務割引及び追加実収資本を認定する | $ | 532 | | | $ | 601 | |
| 当票の割引 | $ | — | | | $ | 5,337 | |
| 逆資本再編で得られた純負債 | $ | 3,881 | | | $ | — | |
| 債務と繰延株式発行コストは売掛金と売掛金に計上される | $ | 226 | | | $ | 340 | |
| 逆資本再編コストを支払うために株を発行する | $ | 1,875 | | | $ | — | |
| 株式発行コストを支払うために株式承認証を発行する | $ | 18 | | | $ | — | |
連結財務諸表の付記を参照
カタログ表
Gri Bio,Inc.
連結財務諸表付記
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
1. 業務の組織と記述
Gri Bio,Inc.(GRIまたは会社)は、カリフォルニア州ラホヤに本社を置き、2009年5月にデラウェア州に登録設立された。
GRIは臨床段階の生物製薬会社であり,炎症,線維化や自己免疫疾患を引き起こす免疫反応失調に関連する重篤な疾患に対する発見,開発,商業化革新療法に専念している。同社の目標は,これらの疾患を治療する治療法の開発において業界の先頭となり,これらの疾患患者の生活を改善することである。同社の主要候補製品GRI−0621は1型ナチュラルキラーT細胞(INKT I)の経口阻害剤であり,特発性肺線維化(IPF)などの重篤な線維化肺疾患の治療に開発されている。同社の候補製品の組み合わせはまた、GRI−0803および500種類以上の化合物の専用ライブラリを含む。GRI−0803はライブラリーから選ばれた先導分子であり,新規な2型ナチュラルキラーT細胞(NKT II)経口アゴニストであり,自己免疫疾患の治療に開発されており,その大部分の臨床前作業は全身性エリテマトーデス(SLE)や狼瘡や多発性硬化症(MS)で行われている。
バロン製薬会社との逆合併。
2023年4月21日に、当社、GRI Bio Operations、Inc.(前身はGRI Bio,Inc.)およびVallon Merge Sub,Inc.(当社の完全子会社)が2023年2月17日に改訂された2022年12月13日に改訂された合併協定および計画に基づき、Merge SubはGRI Operationsと合併してGRI Operations(合併)に組み込まれ、GRI Operationsは自社の完全子会社として存在し続ける。合併に関連して、合併発効時間(発効期間)の直前に、当社はその普通株を1:30の割合で逆株式分割(2023年4月の逆株式分割)を行った。当社は2024年1月29日、その普通株を7対1の割合で逆株式分割(2024年1月の逆株式分割および2023年4月の逆株式分割)を行った。別の説明がない限り、これらの財務諸表に記載されているすべての株式および1株当たりの金額は、逆株式分割を反映している。また,合併の終了に伴い,会社は“Vallon PharmPharmticals,Inc.”と改名した.GRI Bio,Inc.へ
陳述の基礎
付記4で述べたように、合併は逆資本再編成で入金され、逆資本再編により、合併前の自社の歴史財務諸表は会計取得側GRI業務の履歴財務諸表である。合併前の総合財務諸表及び付注内に記載されているすべての普通株、1株当たり及び関連資料はすでに適用範囲内で遡及調整し、すべての列報期間の株式交換比率(定義は後述)及び逆株式分割を反映する。
逆株分割
2023年4月21日、合併に関連し、発効時期に先立ち、会社は2023年4月の逆株式分割を実施した。2024年1月30日、会社は2024年1月の逆株式分割を実施した。株主資本および付属財務諸表に記載されているすべての株式および1株当たりの金額は、すべての列報期間の1:30逆方向株式分割および7:7逆方向株式分割を反映するように遡及調整されている。
2. 流動性
これらの財務諸表を作成する根拠は、当社は継続的に経営している会社であり、他の事項を除いて、同社は正常な業務過程で資産の実現と負債の返済を考慮していることである。同社は設立以来運営から何の重大な収入も生じておらず、予見可能な未来にもこのような収入は生じないと予想される。同社は2009年の設立以来営業損失を出しており、$が発生している31,5332023年12月31日までの累計赤字。同社はこれまで、株式や債務証券を発行することでその運営資金需要に資金を提供してきた。2023年12月31日まで、会社の現金は約ドルです1,808.
合併協定の締結については,当社,GRI運営とAltium Growth Fund,LP(Altium)は2022年12月13日に証券購入協定(株式SPA)を締結し,この合意によりAltiumは投資$に同意した12,250そして、合併完了直前にGRI運営普通株の株式を発行すると引き換えに、ブリッジ手形(定義は後述)の任意の未償還元本および当算利息を解約する。株式SPAによると
カタログ表
Gri Bio,Inc.
連結財務諸表付記
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
終値直前にGRI運営部門が発表しました969,602GRI運営普通株(初期株)をAltiumと3,878,411GRIが普通株式を運営する株式(追加株式)はホストエージェントがホストし,純収益は#ドルである11,704、発売費用を差し引く$546.
終値時には、合併により、最初の株式を合算に両替します36,263当社普通株、額面$0.00011株(普通株)と余分株式を合計に換算する145,052普通株株。2023年5月8日、当社とAltiumは、株式SPAの条項に基づいて、実益所有権制限に適合した場合、追加株式を交換するために発行されたすべての普通株をAltiumに支払うことを許可する
当社は2024年2月1日に証券購入協定(購入契約)を締結し、この合意に基づき、当社は公開発売方式での発行及び販売に同意した:(I)330,450普通株式、(二)4,669,550予融資権証(予融資権証)が行使可能な総金額は4,669,550普通株式、(三)5,000,000B-1シリーズ普通権証(B-1シリーズ普通権証)が行使可能な総金額は5,000,000普通株式(Iv)5,000,000B-2シリーズ普通権証(B-2シリーズ普通権証,およびB-1シリーズ普通権証,普通権証)が行使可能な総金額は5,000,000普通株式、総収益は$5,500それは.2023年12月31日までの10−K表年次報告(年報)では,一般権証と前払い資金の権証を“権証”と呼ぶ。これらの証券は(A)項の組み合わせで発売されている1つは共有したり1つはあらかじめ出資した引受権証は,(B)とともに1つはB-1シリーズ一般権証と1つはB-2シリーズ一般権証、総購入価格は$1.10($を差し引く0.0001あらかじめ出資した引受証ごとに)。
ある所有権制限の場合、株式承認証は発行時に行使することができる。すべての前払い資金株式承認証は行使することができる1つは普通株、1株当たり価格は$です0.0001そして期限が切れません。B-1シリーズの一般権証はすべて行使できます1つは普通株、1株当たり価格は$です1.10一つの上に5年制発行日は2024年2月6日以降です。B-2シリーズの一般権証はすべて行使できます1つは普通株、1株当たり価格は$です1.10そのために18-発行日2024年2月6日以降1ヶ月。購入契約に基づいて証券を発行することについては、A-1シリーズ株式証の発行権価格は額面、すなわち#ドルに低下する0.0001A-1シリーズ株式証の条項に基づいて、1株当たり。
当社の現在の運営計画によると、当社はその既存の現金及び現金等価物が2024年下半期までの運営支出及び資本支出需要を支払うのに十分であると信じている
同社が経営を継続する能力があるかどうかは、その研究·開発計画を含むより多くの資本を調達してその業務活動を支援する能力があるかどうかにかかっている。合併に関連して発行されたTシリーズ株式承認証は現在会社の強制行使の制約を受けていないが,強制行使の持分条件には(その他の事項を除く)普通株の価値加重平均価格が少なくとも$であるためである64.47Tシリーズ株式承認証で指定された期限内に、1株当たり満足されていない。同社は株式証券および/または短期または長期債務手配を増発することで資金を調達しようとしているが、たとえ会社の研究や開発努力が成功しても、必要に応じてこのような融資がある保証はない。同社が受け入れ可能な条項で追加融資を受けることができず、将来の運営ニーズを十分に満たすために必要な金額を得ることができない場合には、運営の減少または完全な停止を余儀なくされる可能性がある。そのため、これらの財務諸表が発表された日から、会社が経営を続ける企業として1年間経営を続ける能力があるかどうかは、大きな疑問がある。これらの財務諸表は、記録された1つまたは複数の資産額の回収可能性および分類、ならびにこのような不確実性に起因する可能性のある負債分類に関連するいかなる調整も含まない。
3. 重要会計政策の概要
本年度報告が指す“権威性指針”系とは、財務会計基準委員会(FASB)の会計基準編纂(ASC)と会計基準更新(ASU)における米国公認の会計原則を指す。
合併原則
連結財務諸表には、GRI Bio,Inc.およびその完全子会社GRI Bio Operations,Inc.のアカウントが含まれています。すべての会社間残高および取引はログアウトされました。
カタログ表
Gri Bio,Inc.
連結財務諸表付記
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
予算の使用
公認会計基準に従って財務諸表を作成することは、報告中に報告された資産および負債額、財務諸表の開示日または資産および負債、および報告の費用額に影響を与えるために、管理層に推定および仮定を要求する。推定と仮定は主に株式購入推定値、転換可能な手形内に派生ツール、株式証発行及びその後の再評価、繰延税項資産に関連する推定免税額、収入確認、計算支出及び融資リースの逓増借款金利を推定することについて作成する。実際の結果が会社の見積もりと異なる場合、あるいはこれらの見積もりが今後の間に調整された場合、会社の経営業績は、どのような見積もりの変化からも利益を得る可能性があり、このような変化の悪影響を受ける可能性もある。
現金と現金等価物
現金等価物とは、購入時にいつでも元の満期日が3ヶ月以下の現金に変換できる高流動性投資であり、2023年12月31日現在、2023年12月31日現在、通貨市場基金への投資を含む。銀行のアメリカ銀行の預金保険は最高$に達しています250連邦預金保険会社が提供します。同社の未保険現金残高は#ドルです1,2242023年12月31日。会社は2022年12月31日までの現金残高を全額保険に加入している。
公正価値計量
当社はASC 820に従い、公正価値計量と開示(ASC 820)財務諸表の公正価値およびその金融商品の公正価値の開示を計量するために。ASC 820は、GAAPにおいて公正価値を計量する枠組みを確立し、公正価値計量に関する開示を拡大する。公正価値は、計量日の市場参加者間の秩序ある取引において売却資産から得られた価格または支払いされた移転負債の価格として定義される。公正価値計量と関連開示の一致性と比較可能性を高めるために、ASC 820は公正価値等級を確立し、公正価値を計量するための推定技術の投入を3つの大体のレベルに分けた。ASC 820によって定義される公正価値レベルの3つのレベルは以下のとおりである
レベル1: 報告日まで、活発な市場での同じ資産または負債の見積もり
レベル2:第1レベルに含まれるアクティブ市場オファー以外の定価投入は、報告日までに直接または間接的に観察されることができる
レベル3:投入に価格を設定し、これらの投入は通常観察できない投入であり、市場データの実証もない
2023年12月31日現在、会社の金融商品には、現金、現金等価物、前払い費用および他の流動資産、支払すべき勘定、計算費用、およびいくつかの負債分類株式証が含まれている。貸借対照表に報告されている現金,現金等価物,前払い費用および他の流動資産,売掛金および売掛金の帳簿価値は,これらのツールの短期満期日に応じてその公正価値に近い。当社は、事件発生日や移転状況が変化した場合に価値階層間の移転を公平にすることを確認しています。2023年12月31日に、負債分類株式証以外に、公正価値によって経常的な基礎によって計量された金融資産或いは負債はなかった。
2022年5月、バロン製薬会社(Vallon)は証券購入協定に関する引受権証を発行した。VallonはASC 815-40に基づいて株式承認証を評価しデリバティブとヘッジ−実体自己資本の契約(ASC 815-40)、株式承認証のうち、場合によっては行使価格の低下に関連する条項は、株式承認証が株式構成要素に計上される可能性を排除すると結論付けられる。そのため、株式承認証は貸借対照表に負債として入金される。VallonはBlack-Scholes推定モデルを用いて権利証の発行時の公正価値を記録した
当社は報告日ごとに株式証の価値を再推定し、その経営報告書に任意の公平値変動を記録する必要がある。評価には公正価値計量に重大な意義があり観察できない仮定を用いる必要があるため、権利証の推定値は公正価値レベルの第三級とされている。第3級株式証券負債の公正価値変動は、2023年12月31日現在の年度経営報告書に反映されている。
繰延株式発行コスト
繰延株式発行コストとは、証券発行予定による直接発生できる逓増法律コストである。この等コストは成約時にそれぞれ発売された総収益から差し引かれる。
カタログ表
Gri Bio,Inc.
連結財務諸表付記
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
財産と設備
財産と設備はコストに応じて列記する.会社は資産投入時に減価償却を始めました。コンピュータと周辺機器は使用年数内に直線的に減価償却する3年.
賃貸借証書
当社は、契約が一定期間内に確定した財産、工場または設備の使用権を譲渡または制御して対価格と交換するかどうかを決定することで、契約開始時のレンタルであるかどうかを決定する。レンタルは融資リースや経営的賃貸に分けられます。付属貸借対照表で確認されたリース使用権(ROU)資産とリース負債は,それぞれリース期間内に対象資産を使用する権利とリースによるリース金の支払い義務を表す。
以下の1つまたは複数の基準に適合する場合、当社は、リースを融資リースに分類する:(I)リースは、リース期間終了時に対象資産の所有権を自社に譲渡すること、(Ii)リースは、自社が行使する対象資産を合理的に決定することを購入する選択権を付与すること、(Iii)リース期間が対象資産の残存使用年数の大部分であること、(Iv)リース支払総額の現在値が対象資産を実質的に超える全ての公正価値に等しいか、あるいは(V)標的資産には専門性があり,レンタル期間終了時にレンタル者の他の用途がないことが予想される.同社は2023年12月31日と2022年12月31日まで、融資リースを何もしていない。上記のいずれの基準を満たしていないレンタルは経営的レンタルに分類される。
レンタル開始日には、当社はレンタルを経営するROU資産とリース負債を確認していますが、オリジナルレンタル期間が12ヶ月以下の短期経営リースは除外しています。ROU資産は最初にコストで計量され、主にレンタル負債の初期金額に任意のレンタル前金を含む。賃貸負債は、最初に支払われていない賃貸支払いの現在値で計量され、当社が類似した経済環境下で基礎賃貸に類似した金額や条項の担保融資の逓増借款金利推定を用いて割引を行う。この割引率を使用したのは、会社の賃貸契約に隠されている金利が通常確定しにくいからだ。
既存の賃貸資産に追加の期間(すなわち、元の賃貸契約に含まれていない期間)の使用権を付与する賃貸修正は、別個の契約として入金されないが、これらの修正を実行する際には、契約満了に応じた残りの対価格のレンタル期間、分類、割引率、および計量が再評価される。
経営的リースのリース費用はレンタル期間内に直線法で確認し、営業費用に計上する。
債務割引
本票項の元金立て替えに関連する引受権証、普通株及び引受オプション譲渡の相対公正価値、転換可能本票修正に関する埋め込み転換オプションの公正価値の増加、及び非利益転換機能の内在価値は債務割引として記録され、実際の利子法に従って手形推定条項で追加利息支出として償却される。
起債コスト
債務発行コストとは、債務発行によって直接発生することができる増分法的コストとその他のコストである。この等コストは負債帳額面に関する直接減少額に計上され、実際の利息法により債務推定年間内に追加利息支出として償却される。
株式証法的責任
当社はASC 815-40に基づいて、2022年5月の登録直接融資(付記10)に関する発行権証を評価し、デリバティブとヘッジ−実体自己資本の契約(ASC 815-40)、株式承認証のうち、場合によっては行使価格の低下に関連する条項は、株式承認証が株式構成要素に計上される可能性を排除すると結論付けられる。この等承認持分証はASC 815が想定する派生ツールの定義に符合するため、この等承認持分証は添付の総合貸借対照表に派生負債として入金され、ASC 820に従って初期及び各報告日に公正価値で計量される公正価値計量公正価値変動は変動期間内に付随する総合経営報告書で確認される。派生負債は、最終的に株式証行使時に普通株式に変換されるか、または株式承認証の期限が満了したときに終了する。行使時の株式の内在的価値
カタログ表
Gri Bio,Inc.
連結財務諸表付記
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
発行された株は株主権益に移される。発行済み株の内在価値と株式承認証の公正価値との差額は、行使期間内に付随する総合経営報告書に取引所損益と表記される。
研究と開発
研究·開発コストは発生時に費用を計上する。研究·開発費用には,研究·開発活動に関連する人員コストがあり,第三者請負業者による研究,臨床試験および薬物供給と材料の製造が含まれている。当社は,提供するサービスの見積もりと発生したコストに基づいて,外部サービスプロバイダ(契約研究組織や臨床研究者を含む)が発生する費用を計算しなければならない。
株に基づく報酬
会社はASCテーマ718に基づいて従業員と非従業員の株式報酬の費用を確認した株に基づく報酬(ASC 718)。ASC 718は、公正な価値に基づく方法を使用して、そのような取引を会計処理することを要求する。オプションの推定公正価値は、オプション付与日の公正価値に基づいて帰属中に償却され、Black-Scholesオプション定価モデルを使用して計算される。同社は発生した没収を計算した。当社がオプションを付与する際の対象株式の公正価値を考慮する際には、当社は、市場取引によって決定される公正価値を含むいくつかの要因を考慮している。株式オプションに基づく報酬には、株式オプションがいつ行使されるかと株価変動の推定および判断が含まれる。株式購入の時間は当社がコントロールできるものではなく、複数の要素に依存し、当社の時価及び株式購入所有者の財務目標を含む。これらは株式報酬支出に実質的な影響を与える可能性があるが、キャッシュフローに影響を与えることはない。最終的に付与された株式ベースの報酬の推定は、実際の結果または更新された推定が元の推定と異なる場合、例えば判断する必要がある推定数を訂正している間は,額を累積調整数と記す同社は、発表された従業員とコンサルタントオプションの両方に対して、契約条項ではなく、予想期限を使用している。
所得税
所得税は貸借対照法で計算される。当社は繰延税金資産及び負債と当社の資産及び負債の財務報告基準と課税基準との一時的な差異、及び純営業損失の繰越予想収益を確認した。一時的差額予想決済期間に適用される税率及び法的変動が繰延税項(ある場合)に及ぼす影響は、公布期間中の当社の財務諸表に反映されている。証拠の分量によって、繰延税金資産の一部または全部が現金化できない可能性が高い場合、繰延税金資産の計量は必要に応じて減少する。同社は2023年12月31日、2023年12月31日、2022年12月31日までに、すべての繰延税項目の純資産に全額推定準備が必要であると結論した。当社は添付されている総合財務諸表に不確定な税務状況、利息または罰金の金額を記録していません。
普通株1株当たり純損失
普通株1株当たりの基本純損失は、毎年発行されている普通株の加重平均株式数から計算される。ASC 260によれば、1株当たりの普通株式償却純損失は、毎年発行されている普通株の加重平均株式数に、毎年発行されているオプションとみなされる希釈効果を加えて計算される1株当たりの収益当社は2023年まで、2023年および2022年12月31日までに純損失を記録したため、1株当たりの普通株の純損失は期内の普通株1株当たりの純損失とほぼ同じであり、潜在的な希薄化証券の影響は逆薄であるからである。
カタログ表
Gri Bio,Inc.
連結財務諸表付記
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
普通株1株当たりの純損失を計上しない普通株等価物は以下のとおりである
| | | | | | | | | | | |
| 十二月三十一日 |
| 2023 | | 2022 |
| 株式オプション | 32,642 | | | 12,781 | |
| 株式承認証 | 298,553 | | | 8,130 | |
| 権利の制限株を買い戻す | — | | | 23,500 | |
| 331,195 | | | 44,411 | |
最近の会計公告
当社はすべてのASUSの適用性と影響力を考慮しています。以下では議論されていない成果が評価され、財務諸表への不適用または予想の影響が最小となるように決定された
FASBは2023年12月にASU 2023-09を発表しました所得税(特集740):所得税開示の改善(ASU 2023-09)。ASU 2023-09の改正は、所得税開示を改善することによって、所得税開示の透明性および決定有用性を向上させることを目的としており、主に税率調整および支払われた所得税情報に関するものである。ASU 2023-09は、公共エンティティに対して2024年12月15日以降の年間期間に有効であり、早期採用を許可している。経営陣は現在、この更新が会社の財務諸表に与える影響を評価している。
2023年10月、FASBはASU 2023-06を発表した開示改善:米国証券取引委員会の開示更新と簡略化イニシアティブに応じた編纂改訂(ASU 2023-06). 中の修正案 ASU 2023-06は、編纂中の様々なテーマの開示および提示要件を明確にまたは改善し、これらの要件を米国証券取引委員会の規定と一致させるためのいくつかの変化を代表する。米国証券取引委員会(米国証券取引委員会)の既存の開示要求に制約されている実体については、毎回改正された発効日は、米国証券取引委員会がS-X法規またはS-K法規から関連開示の発効日を削除し、早期採用を禁止することとなる 経営陣は現在、この更新とその発効日の影響を評価しているが、更新は会社の財務諸表に実質的な影響を与えないと予想される。
2022年1月1日、会社はASU 2020-06を通じて債務--転換可能な債務および他のオプション(主題470-20)と派生ツールおよびヘッジ--エンティティ自身の権益の契約(主題815-40)(ASU 2020-06)。ASU 2020-06は、いくつかの負債および持分の特徴を有する金融商品にGAAPを適用することによって決定された問題を処理する。改訂は、変換可能なツールに関する指導意見およびエンティティ自身の株式契約の派生商品範囲の例外に関する指導意見の改訂に重点を置いている。この基準を採用することは会社の財務諸表に実質的な影響を与えていない。
4. Vallonと合併する
2023年4月21日、合併協定により、合併子会社はPrivate GRIと合併してPrivate GRIに組み込まれ、Private GRIは当社の完全子会社として存続する。取引の終了に伴い、同社は会社登録証明書と定款を修正し、その名称を“Vallon PharmPharmticals,Inc.”から“Vallon PharmPharmticals,Inc.”に変更した。GRI Bio,Inc.へ
発効時期には
(a)有効期間直前に発行されるGRI運営普通株(GRI運営普通株)1株当たり、株式融資(以下に定義する)に従って発行されたGRI運営普通株のいずれかを含み、以下に定義する数に相当する数の会社普通株を取得する権利に自動的に変換される0.0374(為替レート)。
(b)GRI運営普通株株式を購入する各オプション(各,GRI運営オプション)は,GRI運営計画(GRI運営計画,帰属の有無にかかわらず)の発効時間直前に行使されず,普通株株式を購入するオプションに変換され,当社はGRI運営計画(想定オプション)の条項に基づいてGRI運営計画とその各GRI運営オプションを負担する.各仮説引受権に制約された普通株式数は、(I)有効時間の直前に有効なGRI運営オプションに制約されたGRI運営普通株式数に(Ii)株式交換比率を乗じ、得られた数字を最も近い普通株式整数に丸め込むことによって決定される。1株あたりの引受権を行使する場合,普通株を発行できる1株当たりの権益は
カタログ表
Gri Bio,Inc.
連結財務諸表付記
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
その整理方法は、(A)発効時間直前に発効した仮説株式購入に関する1株当たりの権価を(B)両替比率で割って、得られた1株当たりの権価を最も近い整数仙に丸めることである。任意の仮説株式購入権を行使するいかなる制限も引き続き十分な効力を有しているが、このような仮説株式購入の期限、使用可能性、帰属スケジュール、および任意の他の条文は他の点で変わらない。
(c)GRI運営普通株式を購入した株式毎の株式承認証(GRI運営権証)は、発効直前に当社が負担し、普通株を購入する権利証(仮設権証)に変換し、その後(I)各仮定権証は普通株式にのみ適用される。(Ii)各仮定株式証に制約された普通株式数は、(A)有効期間直前に有効なGRI運営株式証に制約された普通株式数に(B)株式交換比率を乗じ、結果を最も近い普通株式整数に丸め込むことによって決定される。(Iii)各仮説株式証を行使する際に普通株を発行可能な1株当たりの権価は、以下の方法で決定される:(A)発効直前に発効するGRI業務株式証規約の制限を受けたGRI業務普通株の1株当たりの権価を(B)両替比率で除算し、得られた行権価格を最も近い整数分に上方丸める。
(d)橋を渡る株式証(注9)はすでに株式承認証(取引所株式承認証)と交換され、合算して購入する60,227会社普通株の株です。取引所株式証には過橋株式証と実質的に類似した条項が含まれており、初期行権価格はドルに相当する103.11一株ずつです。取引所株式証はすでにキャッシュレスに基づいて全面的に行使されている。
(e)GRI運営制限株式報酬に関するすべての権利は、当社が負担し、会社限定株式報酬に変換し、奨励された株式数に交換比率を乗じ、得られた数字を会社普通株の最も近い整数に切り捨てる。GRIトラフィック制限株式報酬の期限、実行可能性、ホームスケジュール、および他の条項は、他の態様では不変のままである。
公認会計原則によると、今回の合併は逆資本再編とされているが、ワロンの主要資産は現金と現金等価物であるからである。会計目的に関しては、GRI運営は、合併条項および他の要因に基づいて会計買収側として決定されており、これらの要因は、(I)合併直前にGRIが運営する持分所有者が合計で買収権利約を所有または保有することを含む85当社普通株式流通株及び合併直前に当社株主が所有する約15当社の普通株式流通株の2%(Ii)GRI運営部門は合併後の会社の多数(5個中4個)の取締役会席を保有しており、(Iii)GRI運営部門管理部門は合併後の会社の経営陣の中で大部分の要職を持っている。合併して間もなく422,333当社は普通株式の株式を発行しました。
次の表は、合併で負担する純負債を示しています
| | | | | |
| 2023年4月21日 |
| 現金と現金等価物 | $ | 941 | |
| 前払い資産とその他の資産 | 310 | |
| 売掛金と売掛金 | (4,190) | |
| 負担した純負債総額 | (2,939) | |
| また:取引コスト | (2,984) | |
| 総純負債に取引コストを負担する | $ | (5,923) | |
上記の取引コストを除いて、有効時間には、4,363普通株はGRI運営会社の財務コンサルタントに発行され、合併に関するサービスを提供する。
5. 公正価値計量
当社は、ASC 820におけるガイドラインを用いて、経常的に計量された金融資産と負債を会計処理する。公正価値とは、秩序のある市場取引において資産を売却するか、または負債を移転させることによって徴収される価格である
カタログ表
Gri Bio,Inc.
連結財務諸表付記
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
日付を測定する参加者。したがって、公正価値は市場に基づく計量であり、資産または負債の定価のために市場参加者が使用する仮定に基づいて決定される。
当社は公正価値階層構造を採用し,市場データに基づく仮説(観察可能な投入)と実体自身の仮説(観察不可能な投入)を区別している。指導意見は公正価値計量を以下の3種類の1つで分類と開示することを要求した
レベル1:同じ、制限されていない資産または負債が、計量日に利用可能なアクティブ市場の未調整オファー。
レベル2:非アクティブな市場でのオファー、または資産または負債の全期間内に直接または間接的に観察可能な投入。
レベル3:価格または推定技術は公正な価値計量に重大な意義があるが観察できない投入が必要である(すなわち、市場活動の支持が少ないか、あるいはない)。
資産または負債がどの種類の資産または負債に属するかを決定するには、重大な判断が必要である。会社はすべての報告期間でその階層開示状況を評価するだろう。2023年12月31日までの年次では,1級,2級,3級の間には何の移行もなかった。
以下の表は、ASC 820要求の各公正価値レベルを示し、2023年12月31日まで、会社が公正価値によって日常的に計量した負債を示している
| | | | | | | | | | | | | | | | | | | | |
| | 市場オファー(1級)を活発にする | | 重要な他に観察可能な投入(第2レベル) | | 重要な他に観察できない入力(レベル3) |
| | | | | | |
| | | | | | |
| 負債: | | | | | | |
| 株式証法的責任 | | $ | — | | | $ | — | | | $ | 3 | |
2022年12月31日現在、会社は公正な価値で恒常的に計量された資産や負債を持っていない
次の表に示す変動は、レベル3負債の公正価値である
| | | | | |
| 株式証法的責任 |
| 2022年12月31日までの公正価値 | $ | — |
| 公正価値2023年4月21日(合併日) | 185 |
| 推定値変動 | (182) |
| 2023年12月31日現在の残高 | $ | 3 |
ブラック·スコアーズ推定値モデルは、株式証明書の公正価値を推定するために使用され、その仮定は以下のとおりである
| | | | | | | | |
| | |
| | 2023年12月31日 |
| 波動率 | | 171.0 % |
| 予想期限(年単位) | | 2.5 |
| 配当率 | | 0.0 % |
| 無リスク金利 | | 4.12 % |
カタログ表
Gri Bio,Inc.
連結財務諸表付記
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
6. 財産と設備
| | | | | | | | | | | |
| 十二月三十一日 |
| 2023 | | 2022 |
| コンピュータ装置 | $ | 21 | | | $ | 13 | |
| 家具と固定装置 | 13 | | | 13 | |
| 34 | | | 26 | |
| 減価償却累計 | (26) | | | (22) | |
| $ | 8 | | | $ | 4 | |
財産や設備に関する減価償却費用は#ドルです4そして$32023年12月31日まで,2023年12月31日と2022年12月31日までである。
7. 賃貸借証書
同社は経営賃貸契約に基づいて事務施設をレンタルしている。レンタル契約は、毎月の固定レンタル料の支払いと、レンタル期間全体にわたって毎月可変の光熱費と運営費用を支払うことを要求しています。当社はリース開始時に継続期間権を評価し続け、賃貸を分類し、賃貸負債を測定する際に、その予想される賃貸期間内に行使する継続権を合理的に決定することを計上している。レンタルプロトコルは、通常、実質的な可変レンタル支払い、残存価値保証または制限契約を必要としない。
以下の表に、会社合併貸借対照表で確認された経営リース資産と負債を示す
| | | | | | | | | | | | | | | | | |
| | | 十二月三十一日 |
| 貸借対照表行プロジェクト | | 2023 | | 2022 |
| 非流動経営リース資産 | その他の資産 | | $ | 14 | | | $ | 67 | |
| 賃貸負債を経営する: | | | | | |
| *現在の営業賃貸負債 | その他流動負債 | | 14 | | | 57 | |
| *非流動経営リース負債 | その他負債 | | — | | | 14 | |
| --営業賃貸総負債 | | | $ | 14 | | | $ | 71 | |
将来の最低賃貸支払いは以下の通りです
| | | | | | | | |
| | 2023年12月31日 |
| 2024 | | 14 | |
| 合計する | | 14 | |
| 差し引く:推定利息 | | — | |
| リース負債現在価値を経営する | | $ | 14 | |
運営リースコストは$592023年12月31日まで、2023年12月31日、2022年12月31日までの毎年。経営リースコストは総合経営報告書の販売、一般および行政費用に計上される。
レンタル負債を計上した金額に支払う現金は#ドルです65そして$542023年12月31日まで,2023年12月31日と2022年12月31日までである。この金額は統合現金フロー表の経営活動に含まれている。
カタログ表
Gri Bio,Inc.
連結財務諸表付記
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
8. 費用を計算する
計算すべき費用は以下の通りです
| | | | | | | | | | | |
| 十二月三十一日 |
| 2023 | | 2022 |
| 課税費用: | | | |
| 研究と開発を担当する | $ | 93 | | $ | — | |
| 総務部と行政部 | 441 | | — | |
| *給与総額および関連 | 736 | | 33 | |
| 他の人は | — | | 3 | |
| *費用の合計額 | $ | 1,270 | | | $ | 36 | |
9. 本票
橋越し融資
合併協定の締結については,GRI運営はAltiumと2022年12月13日(Bridge SPA)に署名した証券購入協定を締結し,この合意により,GRI運営は元金総額#ドルの優先保証元票(Bridge Note)を発行した3,333総買い取り価格と引き換えに$2,500.
大橋手形は#年に発行された二つ成約:(I)初めて成約#ドル1,667元金総額(買い換え総価格#ドル)1,250)は2022年12月14日に成約し、および。(Ii)2回目の成約1,667元金総額(買い換え総価格#ドル)1,250)は2023年3月9日に閉鎖された。橋手形は会社のすべての資産に対する留置権を担保としている
また、各資金が到着した後、Altiumは購入合計を受け取りました178,927普通株式(大橋株式証)の株式。橋梁株式証の発行権価格は1ドルである9.31適用発行日以降の任意の時間に行使することができ、その期限は60株式証明書を承認した日から数ケ月、すべての関連株式証株式は自由に流通することができる
これは1ドルです1,250承諾日の相対公正価値に基づいて、初めて成約して得られた金の一部は過橋手形と過橋株式証に分配され、分配金額は#ドルである679そして$571それぞれ,である.これは1ドルです1,250約束日までの相対公正価値に基づいて、二回目に成約して得られた金の一部は過橋手形と過橋株式証明書に割り当てられ、分配金額は#ドルである718そして$532それぞれ,である
Bridge SPAを除いて、合併協定に署名した場合、当社、GRI運営およびAltiumは株式SPA(付記10)を締結し、これによりAltiumは投資$に同意します12,250そして、合併完了直前にGRIが運営する普通株の株式と交換するために、ブリッジ手形の未償還元本および当算利息を解約する
2023年4月21日に、当社は合併を完了し、橋式手形の未返済元金及び受取利息を解約し、ブリッジ株式証で取引所株式証を交換した。取引所株式証には過橋株式証と実質的に類似した条項が含まれており、初期行権価格はドルに相当する103.111株当たりの収益は分割と資本再編事件の調整の影響を受ける。
橋梁手形はASC 835-30会計指針により株式決済債務として入金されるため,初期帳簿純値は実利子法により償還金額に増加する。会社が発生する債務発行コストは#ドルだ2052022年12月31日までの年度と902023年12月31日までの年間で、Bridge SPAによる債務発行に関連している。未償却債務割引と債務発行コストの合計は#ドルである1,0652022年12月31日まで。債務償却と発行コストによる利息支出は#ドルである2,1042023年12月31日までの年度。
Tep備考
2018年11月、GRI運営会社はTEP Biotech,LLC(TEP)と変換可能な手形と株式証明書購入協定を締結し、協定に基づいて、TEPは最大ドルの資金を提供することに同意した5,000GRI運営部門に転換可能なチケット(TEPチケット)と引受権証を交換して、最大購入します3,606GRI運営会社普通株、行権価格は$0.01一株ずつです。TEP手形は,GRI運営会社の資産と未償還元金残高の応算単純利息を担保とし,金利は12年利率です
カタログ表
Gri Bio,Inc.
連結財務諸表付記
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
未償還元金と課税利息残高総額は、最初にGRIが運営する次の融資(早い者と定義)と2020年5月2日までに満期となる。最初の$2,500TEP手形下の資金の一部は2018年11月の合意調印時に資金を獲得した
2019年12月、GRI運営とTEPはTEP説明を改訂しました。TEPの代わりに2つ目の$を援助します2,500TEPは最初に#ドルを前払いしました500GRI運営部で転換可能な本チケットと交換して、株式証明書を最大購入します2,467GRI運営会社普通株、行権価格は$0.011株あたり,および特定のコールオプションプロトコルに従ってGRIが運営する権利を譲渡する.このコールオプション協定は2015年に締結され、GRI運営部門に最大の買い戻しを提供した5,674取引相手が持つGRI運営会社の普通株は、価格は#ドル187.182025年4月1日までのいつでも
2020年7月,TEP手形満期日を2020年8月31日まで延長し,2021年3月,TEPはGRI運営が2021年10月31日までに期限超過元金と課税利息残高を支払うことができなかったため救済措置を行使する既存の権利を放棄することに同意した。
2021年5月、GRI運営およびTEPはTEP説明を修正し、TEPは2回目の追加前払い#ドルに同意した500変更された変換オプションを個別に有する変換可能なチケットは、GRIオペレータによって交換される
2022年7月、GRI運営とTEPはさらにTEPチケットを修正し、TEPは3回目の追加前払い#ドルに同意した125GRI運営部門に転換可能な本チケットと引受権証を交換して、最大購入します167GRI運営会社普通株、行権価格は$0.01一株ずつです。
2022年10月、GRI運営とTEPは転換協定を締結し、この合意に基づき、合併協定(付記4)の全面署名後に発効し、$3,500特恵債券の未償還元金と$650関連課税利息の自動変換22,172GRIが運営する普通株は、価格を$に変換します187.18一株ずつです。また,最初の橋手形決済後,GRI運営部門は現金で$を返済する125TEP手形の3つ目の追加前払いと$15関連課税利息。発している22,172株式交換および支払い$140元金と受取利息残高は,GRI業務はTEP手形項の下でのすべての義務を完全に履行する.
2022年12月,合併協定の全面的な署名と第1陣のブリッジ手形の完了後,GRI業務は発行された22,172株式を変換して$を支払う140転換協議の条項により、元金と受取利息残高を計算する。付記9“TEP手形”節内の株式番号及び行使又は転換価格をさかのぼって為替レートを反映する。
変換の一部として$4,150元金と応算利息とドルを換算する863転換日までの関連没収計上すべき利息のうちの一部を株主損失に計上する。TEP手形で確認された支払利息は$3522022年12月31日までの年度。
10. 株主権益
普通株
合併協定の締結については,会社,GRI運営会社,Altiumが株式SPAに進出し,このSPAによりAltiumは$投資に同意した12,250そして、合併完了直前にGRIが運営する普通株の株式と交換するために、ブリッジ手形の未償還元本および当算利息を解約する。株式SPAによると、終値直前にGRI運営部門が発表しました969,602GRIが運営する普通株(初期株)をAltiumと3,878,411GRIが運営する普通株式(追加株式)はホストエージェントがホストし,純収益は#ドルである11,704、発売費用を差し引く$546.
終値時には、合併により、最初の株式を合算に両替します36,263普通株式および合計に換算すると145,052普通株株。2023年5月8日、当社とAltiumは、株式SPAの条項に基づいて、実益所有権制限に適合した場合、追加株式を交換するために発行されたすべての普通株をAltiumに支払うことを許可する。
カタログ表
Gri Bio,Inc.
連結財務諸表付記
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
普通株を償還できる
2018年11月,GRI運営は株主と合意し,この合意により,株主はGRI運営にすべてまたは一部の購入を要求する権利がある1,116株主が#ドルで保有しているGRI運営会社普通株111.161株当たり(下落オプションを見る)。番外オプションの行使期限は:(一)従30日プライベートGRIが株式または債務融資を完了して終了する前日15歳その後の平日、または(Ii)プライベートGRIがプロトコルに違反した後の任意の時間。
2022年12月、株主は権利を行使し、GRI運営部は償還する1,116GRIが運営する普通株価格は$124 ($111.161株当たり)。償還した株はGRI運営会社が解約します。付記10“普通株償還可能”節の株式番号及び行使又は転換価格は、株式交換比率を遡及的に反映する。
普通株式引受証
株式SPAによると、当社は2023年5月8日にAltium(I)シリーズA-1に株式承認証を発行し、購入する181,316普通株は,発行価格は$である94.57、(Ii)A-2シリーズ株式承認証163,185普通株は,発行価格は$である103.18,および(Iii)Tシリーズ購入株式承認証(X)116,353普通株は,発行価格は$である85.96および(Y)T系列株式承認証を行使する際には,116,353追加のA-1シリーズ権証とA-2シリーズ権証は、それぞれ購入しなければなりません116,353普通株は,発行価格は$である94.57そして$103.18,(総称して権証と呼ぶ)
A-1シリーズ株式承認証の有効期限は60発行日から数ヶ月、A-1シリーズ株式承認証のすべての株は自由に取引することができる。A-2株式承認証は一つあります2年制任期は2025年6月まで。Tシリーズ株式承認証の有効期限は24Tシリーズ株式証を承認した日から数ヶ月、すべての関連株は自由に取引することができる。付記2で述べたとおりである。流動性ある持分条件を満たす場合、会社はTシリーズ株式承認証を強制的に行使することができる。権利証は、いくつかの現金行使特徴または無現金行使特徴を含み、資産分配および基本取引に関連するいくつかの他の権利を含む。A-1シリーズ株式証の行使価格はある希釈発行を調整する可能性があり、すべての権利証は標準的な逆希釈調整を行う必要がある。2023年12月31日現在、A-2権証はすべて行使され、A-1権証とT権証はすべて決済されていない。株式承認証は権益と分配の公正価値#ドルに分類される5,675追加実収資本を計上する。
橋梁SPAによると、橋手形の一部ごとに資金を獲得した後、Altiumは橋梁株式証明書を受け取った。橋梁株式証の発行権価格は1ドルである9.31適用発行日以降の任意の時間に行使することができ、その期限は60株式証明書を承認した日から数ケ月、すべての関連株式証株式は自由に流通することができる。合併が完了した後,橋承認株式証は取引所承認株式証と交換され,合算を購入する60,227普通株株。取引所株式証には過橋株式証と実質的に類似した条項が含まれており、初期行権価格はドルに相当する103.111株当たりの収益は分割と資本再編事件の調整の影響を受ける。2023年12月31日まで、すべての大橋株式証はすでに行使された。過橋株式証は株式に分類され、分配の公正価値は#ドルである2,860追加実収資本を計上する。
取引終了について、GRI運営会社は、GRI運営会社の普通株の株式を購入するために、その財務顧問株式承認証(コンサルタント株式承認証)を付与し、発効時にこれらの普通株が行使可能な総金額とする343普通株は,発行価格は$である429.73一株ずつです。顧問株式証明書は5年制学期です。2023年12月31日まで、すべてのAdvisor株式承認証は決済されていない。コンサルタント株式承認証は権益と公正価値#ドルに分類される18追加実収資本を計上する
ブラック·スコルスオプション定価モデルは、権利証、取引所権証、および顧問権証の公正価値を推定するために使用され、その加重平均は以下のように仮定される
| | | | | |
| 波動率 | 167.6 % |
| 予想期限(年単位) | 1.69 |
| 配当率 | 0.0 % |
| 無リスク金利 | 4.37 % |
2022年5月、バロンは引受権証を発行して購入しました17,619普通株、行使価格は$197.05証券購入協定に関連した1株当たり収益。これらの株式承認証には1つの項目がある5年制学期です。これらの株式承認証は
カタログ表
Gri Bio,Inc.
連結財務諸表付記
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
負債は、各貸借対照表の日に再評価される。公正価値$32023年12月31日現在、添付されている総合貸借対照表(付記5)の権証負債にこれが反映されている。
Vallonの2021年2月の初公開について,Vallonは引受業者に引受権証(引受業者株式承認証)を付与し,合計を購入した542普通株、行使価格は$2,100.00一株ずつです。引受業者の引受権証は5年制学期です。
同社は2023年12月31日現在、以下の未償還株式権証を持って普通株を購入することができる
| | | | | | | | | | | | | | |
| 株式数 | | 1株当たりの権益 | | 期日まで |
| 116,353 | | $85.96 | | 2025年12月 |
| 542 | | $2,100.00 | | 2026年2月 |
| 3,524 | | $197.05 | | 2027年5月 |
| 167 | | $0.01 | | 2027年7月 |
| 343 | | $429.73 | | 2028年4月 |
| 181,316 | | $94.57 | | 2028年12月 |
11. 株に基づく報酬
2015年株式インセンティブ計画
GRI運営は,改訂されたGRI Bio,Inc.2015持分インセンティブ計画(GRI運営計画)を採用し,GRI運営が従業員,取締役,コンサルタントに株式オプション,制限株式奨励,その他の持分に基づく奨励を付与できるようにした。GRI運営計画により付与される株式オプションの契約期間は一般に10何年もです。合併完了後、会社はGRI運営計画と12,781その発行された未行使と未行使のオプションにより,GRI運営計画による報酬の付与を停止する
2018年の株式インセンティブ計画の改訂と再確認
2023年4月21日、株主は改訂されたGRI Bio,Inc.2018年持分インセンティブ計画を承認し、前身はワロン製薬会社2018年持分インセンティブ計画(A&R 2018計画)である。A&R 2018計画はこれまでに会社の取締役会の承認を得ているが、株主の承認が待たれている。A&R 2018は2023年4月21日に発効する予定で、株主は(I)株式総数を増加させることを含むA&R 2018計画の修正案を承認した24,129共有する30,952A&R 2018計画に基づいて普通株を奨励として発行すること、(Ii)A&R 2018計画の期限を2033年1月1日に延長すること、(Iii)会社の株主のさらなる承認を受けずに報酬とみなされる“再定価”行動を禁止すること、および(Iv)非従業員取締役への奨励制限を改正すること。
A&R 2018は、会社が従業員、取締役、コンサルタントに株式オプション、制限株式、およびその他の持分ベースの奨励を付与することを可能にする計画です。当社がA&R 2018計画に基づき付与した株式オプションの契約期間は一般的に10何年もです。2023年12月31日現在、A&R 2018計画によって付与された奨励代表購入権または権利は、最大で獲得することができます32,642普通株が発行され30,952普通株式はA&R 2018計画に基づいて予約して発行します。A&R 2018計画に基づいて発行される株式数を予約することにより、A&R 2018計画の“常青樹”条項に基づいて、2024年1月1日から2033年1月1日(2033年1月1日を含む)までの例年の初日に、数量を超えない株式を増加させることができる4前年の最後の日に発行された普通株式総数のパーセンテージ。
同社は、GRI運営計画とA&R 2018計画に基づいて発行された株式オプションに関する株式報酬を、2023年12月31日、2023年12月31日、2022年12月31日までの年度運営報告書の以下の費用種別に記録している
カタログ表
Gri Bio,Inc.
連結財務諸表付記
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
| | | | | | | | | | | |
| 12月31日までの年度 |
| 2023 | | 2022 |
| 研究開発 | $ | — | | $ | — |
| 一般と行政 | 388 | | 25 |
| 合計する | $ | 388 | | $ | 25 |
当社は従業員及び非従業員に付与された公平な価値に基づいて、従業員及び非従業員の持分奨励を計量付与し、当該等の奨励が必要なサービス期間又は表現に基づく期間(一般に奨励に関する帰属期間)内の補償支出を確認する。サービスに基づく持分報酬の計量日は付与日であり、株式に基づく報酬コストは、必要なサービス期間内の費用であることが確認され、これは、いくつかの業績ベースの奨励の獲得期間である。もし会社が業績条件に達する可能性が高いと結論した場合、業績に基づく奨励費用を記録する
次の表は、2023年12月31日までの年間従業員と非従業員の株式オプション活動状況を示しています
| | | | | | | | | | | | | | | | | |
| オプション数 | | 加重平均行権値 | | 加重平均残契約期間(年) |
| 2022年12月31日に返済されていません | 16,081 | | $ | 278.17 | | 4.71 |
| 授与する | 31,608 | | $ | 16.66 | | |
| 鍛えられた | — | | — | | |
| 没収される | (15,047) | | $ | 251.13 | | |
| 2023年12月31日現在の未返済債務 | 32,642 | | $ | 37.41 | | 9.55 |
| 2023年12月31日に行使できます | 4,318 | | $ | 176.27 | | 8.81 |
| すでに帰属しており、2023年12月31日に帰属する予定です | 32,642 | | $ | 37.41 | | 9.55 |
2023年12月31日現在、未返済と行使可能な株式オプションがすべて切れているため、違います。内在的価値。2023年12月31日現在,付与予定の未帰属株式オプションに関する未確認補償コストは$である424それは.この未確認の補償は加重平均償却期間中に確認される予定である2.9何年もです
ブラック·スコアーズオプション定価モデルは、付与日が付与されたときの各株式オプション付与の公正価値を推定するために使用され、以下の加重平均仮定を採用する
| | | | | | | | | | | |
| 12月31日までの年度 |
| 2023 | | 2022 |
| 波動率 | 129.54 | % | | 90.39 | % |
| 予想期限(年単位) | 5.84 | | 5.98 |
| 配当率 | 0.00 | % | | 0.00 | % |
| 無リスク金利 | 4.34 | % | | 2.00 | % |
| 日普通株の公正価値を付与する | $ | 14.91 | | $ | 27.02 |
オプション推定方法には,ブラック-スコアズ手法が含まれており,主観的仮定を入力する必要があり,以下で議論する.
•オプションの期待期間は、米国証券取引委員会第107号上場企業会計基準第107号“株式支払”(上場企業会計基準第107号)に規定されている“簡略化”方法に従って決定される、すなわち、企業が十分な履歴データを欠いているため、期待寿命はオプションの帰属期限と元の契約期間との算術平均値に等しい
•予想変動率は、SAB第107号で述べた予想条項の仮定に見合った当社の歴史的変動率と当社業界内の類似エンティティの変動率との加重平均値に基づいている。
•無リスク金利は,付与時に有効な米国国庫券の支払金利をもとに,期限は想定した期待期限に見合っている。
カタログ表
Gri Bio,Inc.
連結財務諸表付記
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
•予想配当収益率は0%であり、同社は歴史的に支払われていないため、予測可能な未来にもその普通株の配当は支払われない。
12. 資産購入協定
2023年8月22日、会社はAardvark Treateutics,Inc.(Aardvark)と資産購入協定(Aardvark協定)を締結し、これにより、Aardvarkは、(I)社とMedice Arzneimittel P≡Tter GmbH&Co.kgが2020年1月6日に署名した許可協定、(Ii)会社Adair候補製品に関するいくつかの特許、および(Iii)米国食品·薬物管理局に提出されたIND番号133072、Adair治療多動症および発作性睡眠病のADAIRに記載された処方の文書(契約製造およびFDA箱件)を購入することに同意した。Aardvark協定の条項によると、会社は前払い現金#ドルを受け取った250これは、他の収入として確認されている。同社には、Aardvarkが将来的にいくつかのAdair規制と販売マイルストーンを実現することに依存する潜在的な追加マイルストーン支払いを得る資格がある。前金に加えて、当社は近いうちにAardvarkのマイルストーン支払いを受けないと予想され、これらの潜在的なマイルストーン支払いは将来的に実現、支払い、または受信されない可能性がある。
13. 引受金とその他の事項
雇用協定
会社はその高級管理者と雇用契約を締結し,会社が理由なく雇用を中止したり,従業員が十分な理由で雇用を中止したりした場合には,解散費と福祉を与えることを規定している。また、支配権変更後に雇用を終了する場合には、一部の持分報酬の帰属が加速する可能性がある。
別居と釈放協定
当社のDavid David元行政総裁が合併により辞任したことについて、当社とベイカーさんは2023年4月21日に別居および離職協議(別居協議)を締結しました。別居協議と雇用契約の条項によると、ベイカーさんは現在の給料とコブラの一部の福祉を受け取り続けます18会社の給与慣行によると、支払わなければならない月数。ベイカーさんも同じように150%の目標ボーナスと、将来のいくつかのマイルストーン支払いの支払金額を減らすことに同意します。
14. 所得税
米国連邦法定所得税率で計算された所得税支出(福祉)と財務諸表中の所得税支出の入金は以下のとおりである
| | | | | | | | | | | |
| 十二月三十一日 |
| 2023 | | 2022 |
| 連邦法定税率で計算される所期所得税割引 | 21.0 | % | | 21.0 | % |
| 連邦福祉を差し引いた州税と地方税 | 8.4 | | | 7.7 | |
| 控除できない項目やその他の項目 | (12.1) | | | (11.0) | |
| 研究開発単位 | 1.1 | | | — | |
| 評価免除額を変更する | (18.4) | | | (17.7) | |
| 合計する | — | % | | — | % |
繰延所得税は、財務報告目的のための資産および負債の帳簿価値と所得税目的のための額との間の一時的な差異の純影響を反映する。
カタログ表
Gri Bio,Inc.
連結財務諸表付記
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
会社の繰延税金資産と負債の主な構成要素は以下の通りである
| | | | | | | | | | | |
| 十二月三十一日 |
| 2023 | | 2022 |
| 繰延税金資産: | | | |
| 連邦と州の営業純損失が繰り越す | $ | 12,848 | | | $ | 3,211 | |
| シェアに基づく報酬 | 166 | | | 177 | |
| 応算項目その他 | 255 | | | 16 | |
| 賃貸負債 | — | | | 20 | |
| 資本化研究開発コスト | 1,099 | | | 91 | |
| 研究開発税収控除 | 153 | | | — | |
| 繰延税項目総資産 | 14,521 | | | 3,515 | |
| 差し引く:繰延税金負債 | — | | | (19) | |
| 減算:推定免税額 | (14,521) | | | (3,496) | |
| 繰延税項目純資産 | $ | — | | | $ | — | |
会社の赤字履歴によると、会社は2023年12月31日、2023年12月31日、2022年12月31日までの繰延税金資産に全額推定準備金を記録している。同社はその推定手当を約#ドル増加させた11,0252023年12月31日までの事業年度。当社はこの手当の撤回を支持する十分な証拠があるまで、推定手当を維持しようとしている
同社の連邦、州、地方の純営業損失は2023年12月31日現在、ドルに転換した45,185, $29,552、と$17,772別々にする39,061連邦純営業損失の中で、繰越期限が切れず、残りのドル6,1242029年に満期になります。州政府の損失も2029年に満期になるだろう。現地の純営業損失は2024年に満期になる。2023年12月31日現在、会社は連邦と州の研究開発税収控除控除額を$としています136そして$17それぞれ,である.連邦信用繰越は2043年から満期になり、州信用繰り越しは満期にならない。改正(IRC)された1986年の国税法第382及び383条の規定によると、IRCで定義されたこのような経営損失純額、貸方繰越及びその他の税務属性によると、過去の所有権の重大な変化及び未来の会社所有権の重大な変化によって制限される可能性がある。
IRC第382及び383節の規定によれば、当社の所有権のいくつかの重大な変動は、将来の所得税の純営業損失およびクレジット繰越金額を減少させるために制限または将来的に使用される可能性がある(IRCで定義された当社の所有権に重大な変動が発生した)。将来の所有者や持分移転は純営業損失や信用繰り越しが制限される可能性がある。
同社はアメリカ連邦司法管轄区とカリフォルニア州、ペンシルベニア州とフィラデルフィアで所得税申告書を提出した。2020年から2023年度までの納税年度は、当社の課税対象の主要司法管区の審査を受けることができます。以前に発生した税務属性のため、正常な訴訟時効を超えた財政年度は依然として税務機関の監査に供することができ、これらの属性はすでに繰り越しており、使用時にその後の年度に監査を行う可能性がある。
当社は、確認すべき税務倉位をより可能な確認敷居を用いて評価しているが、確認資格に適合する税務倉位は、すべての関連情報を完全に知っている税務機関と効率的な決済を行った後に実現される可能性が50%を超える最大税収割引額として計量される。2023年、2023年、2022年12月31日まで、会社は違います。未確認所得税割引は、確認すれば会社の有効税率に影響を与えます。会社は課税利息と所得税支出で収益が確認されていないことに関する罰金を確認します。当社がまだ確定していない不確定税務状況は、関連税務機関がまだ審査すべき年度に関係する。当社は赤字繰越状況にあるため、当社は通常、赤字繰越のあるすべての納税年度に米国連邦、州、地方所得税当局の審査を受けなければならない。
カタログ表
Gri Bio,Inc.
連結財務諸表付記
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
15. 後続事件
資本再編
2024年1月30日、会社は2024年1月の逆株式分割を実施した。株主権益および添付財務諸表に記載されているすべての株式および1株当たりの金額は、すべての列報期間中の7株1株の逆株式分割を反映するように遡及調整されている。
証券購入協定
2024年2月1日に当社は購入契約を締結し、これにより、当社は発売中の発行および販売に同意しました:(I)330,450普通株式、(二)4,669,550行使可能な事前出資株式証合計4,669,550普通株式、(三)5,000,000B-1シリーズ一般権証が行使可能な総金額は5,000,000普通株式(Iv)5,000,000B-2シリーズ一般権証が行使可能な総金額は5,000,000普通株式、総収益は$5,500それは.これらの証券は(A)項の組み合わせで発売されている1つは共有したり1つはあらかじめ出資した引受権証は,(B)とともに1つはB-1シリーズ一般権証と1つはB-2シリーズ一般権証、総購入価格は$1.10($を差し引く0.0001あらかじめ出資した引受証ごとに)。
いくつかの所有権制限の規定の下で、株式承認証は発行時に行使することができる。すべての前払い資金株式承認証は行使することができる1つは普通株、1株当たり価格は$です0.0001そして期限が切れません。B-1シリーズの一般権証はすべて行使できます1つは普通株、1株当たり価格は$です1.10一つの上に5年制発行日は2024年2月6日以降です。B-2シリーズの一般権証はすべて行使できます1つは普通株、1株当たり価格は$です1.10そのために18-発行日2024年2月6日以降1ヶ月。購入契約に基づいて証券を発行する場合、A-1シリーズ株式証の発行価格は額面、すなわち#ドルに低下する0.0001A-1シリーズ株式証の条項に基づいて、1株当たり。