アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
本財政年度末まで
あるいは…。
_から_への過渡期
依頼書類番号:
(登録者の正確な氏名はその定款に記載)
| (登録設立又は組織の州又はその他の管轄区域) | (税務署の雇用主 識別子) |
| (主にオフィスアドレスを実行) | (郵便番号) |
登録者の電話番号は、
市外局番を含む:
同法第12条(B)に基づいて登録された証券:
| クラスごとのタイトル | 取引コード | 登録された各取引所の名称 | ||
同法第12(G)条により登録された証券{br
株式権証明書は、1株当たり普通株の半分を行使でき、額面は0.0001ドル、行使価格は1株11.5ドルである。
登録者が証券法規則第405条で定義されている有名な経験豊富な発行者であれば、再選択マークで示してください。はい☐
登録者が“取引所法案”第13節又は第15(D)節に基づいて報告書を提出する必要がない場合は、複選マークで示してください。はい☐
再選択マークは、登録者が(1)過去12ヶ月以内に(または登録者がそのような報告の提出を要求されたより短い時間以内に)1934年の証券取引法第13節または15(D)節に要求されたすべての報告書を提出したかどうか、および(2)過去90日以内にそのような提出要件に適合しているかどうかを示す
再選択マークは、登録者が最初の12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間)に、S−T法規(本章232.405節)第405条の規定に従って提出しなければならない各相互作用データファイルを電子的に提出したか否かを示す
再選挙マークで登録者が大型加速申請者,加速申請者,非加速申請者,小さい報告会社か新興成長型会社かを示している。取引法12 b-2条のルールにおける定義 の“大型加速申告会社”、“加速申告会社”、“小報告会社”、“新興成長型会社”を参照されたい
| 大型加速ファイルサーバ | ☐ | ファイルマネージャを加速する | ☐ |
| ☒ | 規模の小さい報告会社 | ||
| 新興成長型会社 |
新興成長型会社である場合は、登録者 が延長された移行期間を使用しないことを選択したか否かをチェック番号で示して、“取引所法案”第13(A)節に提供される任意の新しいまたは改正された財務会計基準を遵守してください☐
登録者がbrに関する報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オキシリー法案”(“米国法典”第15編7262(B)節)404(B)節による財務報告に対する内部統制の有効性の評価を証明する。
証券がこの法(Br)12(B)節に基づいて登録されている場合は、届出文書に含まれる登録者の財務諸表が以前に発表された財務諸表の誤りを反映して訂正されたか否かをチェックマークで示してください
これらのエラーのより真ん中に再記述 があるかどうかをチェックマークで示すことは、登録者の任意の役員が、関連回復期間内に§240.10 D−1(B)によって受信されたインセンティブベースの報酬に基づいて回復分析を行うことを要求する☐
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条に規定するように)☐
ない
2023年6月30日,すなわち登録者が最近第2四半期の最終日を完了し,登録者の非関連会社が保有している登録者普通株の総時価は$である
登録者の普通株流通株数2024年3月28日現在の株は
BIOMX Inc.
2023年12月31日までの年間Form 10−K年報
| 第1部 | ||
| 1つ目:ビジネス活動 | 1 | |
| プロジェクト1 A.リスク要因 | 28 | |
| 項目1 B:未解決スタッフの意見 | 70 | |
| プロジェクト1 C。ネットワーク·セキュリティ | 70 | |
| 項目2.財産 | 70 | |
| 項目3.法的手続き | 70 | |
| プロジェクト4.炭鉱の安全状況の開示 | 70 | |
| 第II部 | ||
| 項目5.登録者普通株、関連株主事項及び発行者が株式証券を購入する市場 | 71 | |
| 第六項です[保留されている] | 71 | |
| プロジェクト7.経営陣の財務状況と経営成果の検討と分析 | 72 | |
| プロジェクト7 A.市場リスクに関する定量的かつ定性的開示 | 83 | |
| 項目8.財務諸表と補足データ | 83 | |
| 項目9.会計·財務開示における会計担当者との変更と相違 | 83 | |
| プロジェクト9 A.制御とプログラム | 83 | |
| プロジェクト9 B.その他の資料 | 84 | |
| プロジェクト9 Cです。検査妨害に関する外国司法機関の開示 | 84 | |
| 第三部 | ||
| プロジェクト10.役員、役員、および企業管理 | 85 | |
| プロジェクト11.役員報酬 | 90 | |
| プロジェクト12.特定の実益所有者および経営陣の保証所有権および株主に関する事項 | 95 | |
| 第13項:特定の関係及び関連取引、並びに取締役独立性 | 97 | |
| プロジェクト14.総会計士費用とサービス料 | 98 | |
| 第4部 | ||
| プロジェクト15.各種証拠品および財務諸表の添付表 | 100 | |
| 項目16.表格10-Kの概要 | 101 | |
i
2024年3月15日、BiomX Inc.は、デラウェア州の適応バクテリオファージ治療会社(APT)を買収し、BiomX Inc.,APT、デラウェア州のBTX Merger Sub I,Inc.とデラウェア州の有限責任会社BTX Merger Sub IIとの間の合併プロトコルおよび計画または合併プロトコルに従って買収した。ただし、本年度報告で言及されている“会社”、“BiomX”、“私たち”、“我々”または“我々”は、2023年12月31日および2022年12月31日までのすべての財務情報を含むBiomX Inc.およびその連結子会社(APTを含む)を意味し、2023年12月31日および2022年12月31日までの財務情報、および2024年3月15日までの他のbr情報を含むが、本年度報告または年次報告で言及されている内容にはAPTは含まれていない。本年度報告で言及されたBiomX Ltd.とは、我々のイスラエルの完全子会社BiomX Ltd.を指す。ここでの会社記述は買収後の会社であり、APTの業務統合を反映している。本年報の他の部分で述べたように、2019年10月28日、特殊目的買収会社Chardan Healthcare Acquisition Corp.はBiomX Ltd.と合併し(以下のように定義する)、BiomX Inc.と改名した
前向き陳述に関する警告声明
本年度報告には、改正された1933年“証券法”第27 A節又は“証券法”と、改正された1934年“証券取引法”第21 E節又は“取引法”が指す前向き陳述が含まれている。本年度報告書に含まれる非純歴史的陳述はすべて前向き陳述である。前向きな陳述は、私たちの期待、信念、計画、目標、意図、仮説、および他の非歴史的事実に関する陳述を含む。“予想”、“信じる”、“継続”、“推定”、“予想”、“予定”、“可能”、“進行中”、“br}”、“計画”、“潜在”、“予測”、“プロジェクト”、“将”または同様の言葉またはフレーズ、 またはこれらの言葉またはフレーズの否定または否定などの言葉またはフレーズは、前向き陳述を識別することができるが、これらの言葉が必ずしも展望性を有さないことを意味するとは限らない。本年度報告における展望的陳述は,われわれの運営,キャッシュフロー,財務状況の開示,およびわれわれの臨床前と臨床発展計画,われわれのバクテリオファージ療法の安全性,耐性と有効性,およびその臨床前と臨床研究の進行,設計,目標とタイミングおよび発表結果の陳述を含むが,これらに限定されない。
前向きな陳述は、本年度報告の複数の位置に現れ、“経営陣の財務状況および経営結果の議論および分析”および“業務”を含むが、これらに限定されない
| ● | 収入を創出し、運営資金要求を満たすのに十分な資金を調達することができる |
| ● | APTの業務を会社に統合する |
| ● | 買収と関連する民間投資取引に関するいくつかの提案に対する私たちの株主の承認を受けた |
| ● | バクテリオファージ技術を用いて候補製品を開発する方法に関連する予測不可能な時間およびコスト |
| ● | ロシアのウクライナ侵攻や世界のロシア、ベラルーシおよび関係者への制裁、テロ、ハリケーン、火災、洪水、汚染、地震などの自然災害や他の悲劇的な事件を含むが、政治的および経済的に不安定である |
| ● | 米国食品医薬品局(FDA)の候補製品の任意の非米国臨床試験の受け入れを獲得した |
| ● | 私たちは患者を募集して臨床試験に参加し、予想された時に予想された発展マイルストーンを実現することができる |
| ● | 新しい製品の機会と買収を求め、効率的に開発し、これらの製品の機会と買収から価値を得る能力 |
| ● | 候補製品に予期せぬ問題が発生し、ラベルおよび他の制限を遵守できなかったことによる処罰および市場撤退 |
| ● | 全体的な経済状況、私たちの現在の低株価と他の私たちの運営に影響を与える要素、私たちの業務の連続性、私たちの臨床前と臨床試験、そして私たちの追加資本を調達する能力を含む | |
| ● | 持続的な規制義務の遵守と成功した持続的な規制審査に関する費用 |
| ● | 私たちの候補製品の市場受容度と他の候補製品を識別または発見する能力 |
II
| ● | 臨床前と臨床試験に必要な特定のバクテリオファージカクテルの高力価を得ることができます |
| ● | 特殊な原材料の獲得可能性とグローバル·サプライチェーンの挑戦 |
| ● | 私たちの候補製品は薬品の安全性と有効性、あるいは生物製品の安全性、純度と効力を証明する能力があり、悪影響を与えることがない |
| ● | 私たちの候補製品の将来の高度な臨床試験の成功を期待しています |
| ● | 私たちには必要な規制の承認を得ることができます |
| ● | 私たちの候補製品のための製造プロセスの開発の遅れ |
| ● | 同様の技術からの競争は、私たちの候補製品よりも効率的で、より安全で、または私たちの候補製品よりも早く市場の承認を得た製品である |
| ● | 不利な価格設定法規、第三者精算やり方、医療改革の措置が候補製品の販売や療法の収益性に与える影響 |
| ● | 私たちの知的財産権を保護し、第三者と現在と未来のライセンスの条項と条件を守る |
| ● | 第三者の知的財産権及び職務発明権譲渡の報酬又は使用料を侵害する |
| ● | 私たちは、第三者が保有する候補製品または将来の候補製品の開発に必要な独自の権利を取得、許可、または使用することができます |
| ● | 合成生物学や遺伝子工学の倫理、法律、社会的懸念は、私たちの候補製品の市場受容度に悪影響を及ぼす可能性がある |
| ● | 第三者協力者に依存して |
| ● | イスラエル国民の政治、経済、軍事的不安定、特に10月7日の襲撃後のガザの戦争、他の中東諸国とのより多くの潜在的な衝突、イスラエル政府が提案し続けた司法とその他の立法改革 |
| ● | 私たちは重要な従業員を引き付けたり、従業員との競争禁止条項を実行する能力を引き付けたりします |
| ● | 薬品生産コンプライアンス以外の適用法律、法規を遵守していない |
| ● | ネットワークセキュリティイベントを含む潜在的なセキュリティホール; |
| ● | 本報告では,29ページから“リスク要因”と題する節で議論した他の要因について述べた。 |
前向き表現は既知と未知のリスクと不確実性の影響を受け、私たちの経営陣が不正確である可能性のある仮定に基づいて、これらの仮定は実際の 結果と展望性表現中の予想或いは暗示の結果とは大きく異なる可能性がある。これらの陳述は、本年度報告書提出日までに提供された情報に基づいており、これらの情報は、このような陳述の合理的な基礎を構成していると考えられるが、このような情報は、限られているか、または不完全である可能性があり、私たちの陳述は、すべての潜在的に利用可能な関連情報について詳細な調査または検討が行われていることを示すように解釈されてはならない。このような陳述は本質的に不確実であり、投資家はこのような陳述に過度に依存しないように注意されている。多くの原因により、実際の結果は展望性陳述で予想された結果と大きく異なる可能性があり、本年度報告の“リスク要因”と題する節で議論した要素を含む。法律が適用されてbrが要求される可能性がない限り、私たちは、本年度の報告日以降の状況またはイベントを反映するために、または予期しない事象の発生を反映するために、任意の前向きな陳述を公開的に修正する義務がない。しかし、あなたは、本年度報告日の後、時々米国証券取引委員会または米国証券取引委員会に提出された報告書に記載されている要因およびリスクを検討すべきである
三、三、
リスク要因の概要
以下の要約は、プロジェクト1 Aにおいて、より詳細なリスク議論を見つけることができる企業が直面する多くのリスクについて概説する。“リスク要因”は以下のとおりである.あなたは私たちの証券に投資する時、このようなリスクと不確実性を慎重に考慮しなければならない。私たちの業務に影響を与える主なリスクと不確実性は含まれているが、これらに限定されない
| ● | 私たちは臨床段階の会社で、運営の歴史は限られており、設立以来赤字が続いている。私たちは、引き続き巨額の費用を招き、予測可能な未来に、私たちは引き続き重大な損失を招くと予想している。 |
| ● | 私たちは将来的に私たちの業務を支援するためにより多くの資本を調達する必要があります。これらの業務は私たちに有利な条項で提供されない可能性があり、私たちの株主が私たちの第三者への債務を大幅に希釈したり増加させたりする可能性があります。 |
| ● | 私たちの財務諸表には、持続的な経営を続ける企業としての能力があるかどうかを示す説明段落が含まれており、これは、合理的な条項や新たな融資を受けることができないことを阻止するかもしれません。 |
| ● | APTに対する私たちの買収が株主価値を増加させる保証はない。 | |
| ● | バクテリオファージ技術を用いた候補製品の開発が求められており,潜在的な成功や開発の時間やコストを予測することは困難である。我々の知る限り,これまで米国やEUで薬物として承認されたバクテリオファージは何もない。 |
| ● |
吾らは最大の努力を尽くして株主が交換可能な優先株株式(定義は以下参照)及び買収(定義は以下参照)及び2024年3月パイプ(定義は以下参照)で発行された引受権証の転換を獲得しなければならない。もし私たちが転換可能優先株の初回発行後150日以内にこのような承認を得なければ、転換可能優先株の現金決済が必要になるかもしれない。 |
| ● | 私たちの候補製品は臨床試験を経なければならず、薬品に必要な安全性と有効性、あるいは生物製品の安全性、純度と効力を証明できない可能性があり、私たちのどの候補製品も悪影響を招く可能性があり、これは規制部門の承認および/または商業化を大幅に延期または阻止するだろう。 | |
| ● |
私たちはまだ私たちの候補製品の構成開発を終えていない。 |
| ● |
私たちは他の候補製品 の識別や発見に成功できないかもしれない。 |
| ● |
ボルト独自製品プラットフォーム に依存して我々のバクテリオファージ療法を開発していく予定である。もし私たちの競争相手が似たようなプラットフォームを開発して競争相手の候補製品を開発すれば、私たちの競争地位は実質的に損なわれる可能性がある。 | |
| ● |
私たちの限られた運営の歴史は、私たちの業務のこれまでの成功状況を評価し、私たちの将来の生存能力を評価することを難しくするかもしれません。 |
| ● | 私たちは製品販売から何の収入も得たことがなく、永遠に利益を上げないかもしれないし、実現しても収益力を維持できないかもしれない。 | |
| ● |
著者らの候補製品の臨床前研究結果は臨床試験或いは後期臨床開発の結果を予測できない可能性がある。 |
| ● | 私たちの候補製品は厳格な規制承認要求を受けています。これは私たちのマーケティングを延期し、阻止したり、制限したり、私たちの候補製品を開発する能力を制限するかもしれません。 |
| ● | 私たちと医療保健提供者、医師、第三者支払者との関係は、適用される反リベート、詐欺、乱用、および他の医療法律法規の制約を受けることになり、これは私たちを刑事制裁、民事処罰、契約損害、名声損害、そして他の結果に直面させるかもしれない。 |
四
| ● | たとえ治療適応のための任意の候補製品の規制承認を得ても、持続的な規制コンプライアンス義務と持続的な規制審査の制約を受けることになり、多くの追加費用を招く可能性がある。また、私たちのどの候補製品も、承認されれば、ラベルや他の制限や市場撤退を受ける可能性があり、規制要求を遵守できなかったり、予期しない製品問題に遭遇したりすれば、処罰を受ける可能性があります。 | |
| ● |
私たちが開発する可能性のあるどの製品も、不利な価格設定法規、第三者精算やり方、あるいは医療改革措置の制約を受ける可能性があり、どんな候補製品や治療法を販売して利益を得ることが困難になるかもしれません。 | |
| ● |
行われている医療立法や規制改革措置は,我々の業務や運営結果に実質的な悪影響を及ぼす可能性がある。 |
| ● | 私たちが維持しているライセンス契約は、2015年のライセンス契約(以下、定義)を含めて、私たちの業務に非常に重要です。もし私たちまたは私たちの許可協定の他の当事者が許可協定を十分に履行できなかった場合、または私たちまたは彼らが許可協定を終了した場合、私たちのバクテリオファージに基づく候補治療製品の開発、テスト、製造、生産、および販売は延期または終了され、私たちの業務は悪影響を受けるだろう。 |
| ● | 私たちは第三者の許可の知的財産権に強く依存しており、これらの許可のいずれかを終了または制限することは、重大な権利の損失を招き、私たちの業務に実質的な損害を与える可能性がある。 |
| ● | 私たちは特許と独自技術に依存している。もし私たちがこの知的財産権を十分に保護できなければ、あるいは製品マーケティングの独占経営権がなければ、私たちの製品商業化能力は影響を受ける可能性がある。 | |
| ● |
もし私たちが第三者の権利を侵害した場合、私たちは製品の販売を禁止され、損害賠償および/または印税の支払いを余儀なくされ、抗弁訴訟を余儀なくされる可能性がある。 | |
| ● |
我々はバクテリオファージに基づく療法 を特定するために独自の製品プラットフォームに依存している。もし私たちの競争相手が似たようなプラットフォームを開発して競争相手の候補製品を開発すれば、私たちの競争地位は実質的に損なわれる可能性がある。 | |
| ● |
私たちは商業秘密と他の形態の非特許知的財産権 財産保護に依存している。もし私たちが私たちのビジネス秘密を守ることができなければ、他の会社は私たちともっと効果的に競争できるかもしれない。 | |
| ● |
もし私たちが第三者の知的財産権侵害で起訴された場合、または私たちが介入手続きを余儀なくされた場合、これは高価で時間がかかり、この訴訟または介入の不利な結果は私たちの業務に大きな悪影響を及ぼすだろう。 | |
| ● |
第三者関係は私たちの業務に非常に重要だ。我々 が我々の連携を維持したり,新たな関係を構築できなかったり,これらの関係が成功しなければ,我々の業務は に悪影響を受ける可能性がある. |
| ● | 私たちの本部、研究開発、そして他の重要な行動はイスラエルに設置されているので、私たちの成果は、最近ハマスとガザ地区からの他のテロ組織との戦争を含むイスラエルの政治的、経済的、軍事的不安定の悪影響を受ける可能性がある |
v
| ● |
私たちが受け取った研究と開発のためのイスラエル政府の支出は、私たちがイスラエル国外で製品を製造し、技術を譲渡する能力を制限し、特定のbr条件を満たすことを要求した。もし私たちがこれらの条件を満たしていなければ、前に受け取った贈与と利息と罰金を返金する必要があるかもしれません。 |
| ● | ドル、新イスラエルシェケル、ユーロと他の外貨との為替レート変動は、私たちの将来の収入と支出にマイナスの影響を与える可能性がある。 | |
| ● |
私たちまたはイスラエルまたはアメリカにいる私たちの上級職員や役員に対するアメリカの判決を実行することは難しいかもしれませんし、イスラエルでのアメリカ証券法のクレームを主張したり、私たちの上級職員や役員に訴訟手続きを送達したりすることは難しいかもしれません。 | |
| ● |
私たちの候補製品は特殊な原材料の利用可能性に依存しており、私たちは受け入れ可能な条項やこれらの原材料を全く入手できないかもしれません。 |
| ● | 私たちの普通株式の中に相当の数の株式が発行された引受権証とオプションを行使したり、私たちの転換可能な優先株を転換したりする時に発行する必要があり、一度行使したり転換したりすると、私たちの証券保有者の株式希釈を招く可能性があります。 | |
| ● |
私たちは私たちの普通株に配当金を支払ったことがなく、私たちは予測可能な未来に、私たちは私たちの普通株に現金配当金を支払わないと予想している。 | |
| ● |
私たちの公共株式証明書(以下の定義)はすでに取得されており、私たちは未来に引き続き私たちの証券を上場させることができないかもしれない。 | |
| ● |
私たち普通株や他の証券の市場価格は変動して大幅に変動する可能性があり、これは私たち普通株の購入者に大きな損失をもたらす可能性があります。 | |
| ● |
“小さな報告会社”として、比較的大きな上場企業のより少ない開示を提供することが許可されており、これにより、私たちの普通株の投資家への吸引力が低下する可能性がある。 | |
| ● |
私たちの成功はある程度私たちが肝心な幹部を維持する能力、及び合格した人材を吸引、維持と激励する能力にかかっている。 | |
| ● |
環境、社会、およびガバナンス(ESG) プロジェクトに関する予想は、追加コストを増加させ、新たなリスクに直面させる可能性がある。 | |
| ● |
コンピュータシステムに障害、ネットワーク攻撃、または私たちのネットワークセキュリティに欠陥があれば、私たちの業務と運営は影響を受けるだろう。 |
VI
第1部
1つ目:ビジネス活動
概要
我々は臨床段階の製品発見会社brが天然や工学バクテリオファージ技術を用いた製品を開発しており,嚢胞性線維化や糖尿病性足骨髄炎などの慢性疾患に関連する特定の有害細菌を標的かつ死滅させることを目的としている。バクテリオファージやバクテリオファージは細菌、種特異性、菌株の限られたウイルスであり、標的細菌に感染、増幅、殺すことは、哺乳動物細胞の不活性ウイルスと考えられている。自然産生バクテリオファージの特許の組み合わせと合成生物学を用いた新たなバクテリオファージの創出により,我々はバクテリオファージに基づく療法を開発し,大市場と孤児疾患の解決を目指した。
感染治療の緊急性(急性であっても慢性であっても)、標的細菌のバクテリオファージに対する感受性(例えば、複数の細菌に対するバクテリオファージカクテルを識別する能力)および他の考慮要因に基づいて、バクテリオファージベースの2つの製品タイプを提供する
| (1) | 固定カクテル療法−この方法では、幅広い細菌株をカバーし、同じ製品を用いて広範な患者集団の治療を可能にするために、選択されたバクテリオファージを固定数含む単一製品が開発される。固定式カクテルは我々独自のBoltプラットフォームを用いて開発され,その中で高スループットスクリーニング,配向進化,バイオインフォマティクス法 を用いて最適なファージカクテルを生産している。 | |
| (2) | 個人化治療-この方法では、特定の患者を治療するために単一の最適バクテリオファージが個人的に適合された大型ファージライブラリーが開発される。最適バクテリオファージと患者とのマッチングは、固有のバクテリオファージ感受性試験またはPSTを使用して行われ、複数の考慮要因を同時に分析することは、より短い回転時間を維持しながら、バクテリオファージライブラリーを効率的にスクリーニングすることを可能にする。 |
我々の治療計画では,バクテリオファージ療法を用いて疾患に関連する特定の病原菌菌株を標的とすることに集中している。我々のバクテリオファージに基づく候補製品 は,我々ボルト独自の研究開発プラットフォームを用いて開発した。Boltプラットフォームは唯一無二であり、学際的な先端方法と能力を採用し、計算生物学、微生物学、バクテリオファージ合成工程及びbr細菌宿主の生産、生物分析テスト開発、製造と調合を含み、柔軟かつ効率的に天然或いはbr工学バクテリオファージの組み合わせ或いはカクテルを開発することを実現する。このカクテルは機能的に相補的なバクテリオファージを含み、広範な標的宿主範囲、薬剤耐性を防止する能力、生物被膜浸透、安定性、および製造しやすいなど、多種の特性に対して最適化されている。
我々の目標は,バクテリオファージに基づいて有害細菌を正確に特定する能力と,自然産生と合成工程を用いて創出された様々なバクテリオファージを含む様々なバクテリオファージをスクリーニング,識別,結合する能力に基づいて,これらの治療法を開発することである。
私たちの製品ライン
次のグラフ は,我々の候補製品のチャネル,彼らの現在の状態,および来るマイルストーンの予想時間を決定する.私たちは承認または販売可能な製品は何もありません。私たちの候補製品はまだ臨床前と臨床開発段階にあり、私たちは製品販売から何の収入も得ていません。
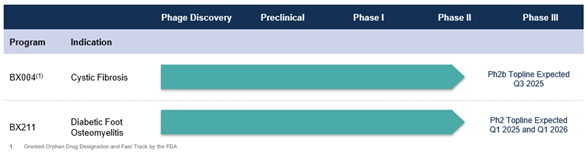
進行中の計画
BX 004−嚢胞性線維症の治療
BX 004は我々が開発している以下の原因による慢性肺感染に対する候補治療バクテリオファージ製品である緑膿菌や緑膿菌CF患者の発病率と死亡率の主要な貢献者である。抗生物質に対する耐性増強、特に慢性閉塞性肺疾患患者において、これは広範に使用されている薬物brが長時間と重複する広域抗生物質治療コースを含み、通常児童時期から始まり、そして多剤耐性菌株の出現を招くためである。臨床前段階で体外培養BX 004は抗生物質耐性菌株brに対して活性であることが明らかになった緑膿菌生物膜を透過する能力を示し,生物膜は細胞外ポリマーに包まれた表面関連微生物細胞の集合であり,抗生物質耐性の要因の一つでもある。
1
慢性気管支炎患者の慢性呼吸器感染のための1 b/2 a期試験緑膿菌。二つの部分から成っています。この研究は嚢胞性繊維化治療発展ネットワークに基づく提案を設計した。
2023年2月末,BX 004の1 b/2 a段階試験の第1部分を評価する積極的な結果 を発表した。第1部では,BX 004の安全性,耐性,薬物動態またはPK,および慢性骨髄性白血病患者9名(BX 004服用7名,プラセボ服用2名)の7日間の漸増治療期間中の微生物学的活性を評価した緑膿菌Br肺部感染は単回漸増投与量と複数回投与設計を採用した。
1 b/2 a段階試験の第1の部分の結果は、BX 004治療の使用に関連する安全事象が発生していないことと、平均緑膿菌コロニー形成単位、すなわちCFUが、15日目(ベースラインと比較して):-1.42対数(BX 004)および-0.28対数(プラセボ)であることとを含む。この減少は抗生物質吸入の標準ケアの上に出現し,服薬中にBX 004治療を受けたすべての患者でバクテリオファージが検出され,15日目(治療終了後)の数名の患者に含まれ,プラセボを受けた患者ではバクテリオファージが検出されず,プラセボと比較して治療期間または治療後にbr}BX 004に治療関連耐性が生じた証拠はなく,治療持続時間が短いため,1秒以内に予測された力呼気量やFEV 1にはbrの影響が検出されなかった。
2023年11月、BX 004の1 b/2 a段階試験の第2部分を評価する積極的な結果を発表した。1 b/2 a期試験の第2の部分の目標は、BX 004の安全性および耐性を評価して、BX 004のより多くのCF患者における安全性および耐性を評価することであり、より長い治療時間が第1の部分よりも大きな効果をもたらす可能性が予想される。第2の部分では、34人のCF患者が2:1の割合でBX 004治療をランダムに受け、23人のCF患者がBX 004治療を受け、11人の患者が霧化プラセボによる10日間を吸入した。
1 b/2 a段階試験の第2部分の主な結果は、以下の 調査結果を含む
| ● | 薬物の安全かつ耐性が良好で、関連するSAE(深刻な有害事象)或いは関連する猿類(急性肺加重)を研究して薬物を研究した。 |
| ● | BX 004群では,ベースラインCFU定量患者21名(14.3%)中3名(14.3%)が痰培養陰性となった緑膿菌治療10日後(4日後の患者2名を含む)では,プラセボ群10人中0人(0%)であった。 |
| ● | BX 004はプラセボと比較してベースライン肺機能低下(FEV 1)の所定の亜群患者で積極的な臨床効果を示した |
| ● | 全群でBX 004はプラセボと比較した緑膿菌痰中のレベル の変化が大きいのは,薬物管理の検討開始が看護抗生物質治療案の基準 の起動と一致しているためかもしれない。連続レジメン吸入抗生物質看護基準の予め指定された亜群患者では、BX 004とプラセボは10日目に痰中の緑膿菌のレベルを減少させた:両群間のベースラインとの変化の差-2.8 log 10 cfu/g痰(ベースラインとの変化-2.91比-0.11)は、第1部分の結果を超えた。 |
| ● | 交替/循環背景抗生物質レジメンは糖尿病の変動と関係がある可能性が高い緑膿菌これらのレベルはこの亜群における緑膿菌感染の減少を観察する能力を混同する可能性がある。 |
| ● | 研究期間中,現在利用可能なデータによると,BX 004を服用した患者ではプラセボと比較して治療に関連するバクテリオファージ耐性の証拠は認められなかった。 |
2023年8月、FDAはBX 004 Fast Track 治療を許可し、以下の原因による慢性気道感染の治療を許可した緑膿菌CF.患者の細菌菌株br}また,2023年12月,BX 004はFDAの孤児薬物指定を獲得した。
BiomXは慢性うっ血性心不全患者のための無作為、二重盲検、プラセボ対照のマルチセンター2 b期研究を開始する予定である緑膿菌2024年第4四半期の肺感染。この研究は,約60名の患者を募集し,BX 004またはプラセボと2:1の割合でランダムに比較することを目的としている。治療は1日2回吸入し,8週間持続する予定である。この研究はBX 004の安全性と耐性を監視し、微生物の減少を証明することを目的としている緑膿菌FEV 1測定の肺機能と患者報告の結果のような臨床パラメータに対する負担と評価の影響。研究結果は2025年第3四半期に発表される予定だ
2
BX 211−糖尿病性足骨髄炎の治療
BX 211は個人化されたバクテリオファージ療法であり黄色ブドウ球菌あるいは…黄色ブドウ球菌それは.個人化されたバクテリオファージ療法は、各患者から生検および単離された黄色ブドウ球菌の特定の菌株から、固有バクテリオファージバンクから特定のバクテリオファージを選択する。DFOは細菌性骨格感染であり、通常感染した足潰瘍から発展し、糖尿病患者の切断の主な原因である。科学文献は、動物モデルにおけるバクテリオファージを用いた骨髄炎治療の潜在的な利点を証明していると信じており、バクテリオファージ療法によるDFO患者の治療に大量に成功した慈悲例は、バクテリオファージ療法を用いたDFO治療方法を支持している。
進行中の無作為,二重盲検,プラセボ対照のマルチセンター第二段階研究では,黄色ブドウ球菌関連DFO患者に対するBX 211の安全性,耐性,有効性を調査し,約45名の被験者を募集する予定であり,割合は2:1,BX 211またはプラセボに対する割合は2:1である。BX 211またはプラセボは、毎週局所および静脈投与されるか、または1週目に静注により投与され、2~12週目にのみ局所経路で投与されるように設計されている。12週間の治療期間中、すべての対象は、場合によっては抗生物質治療を含む看護基準に従って治療を継続すると予想される。13週目には初めて背線研究結果を読み出し,骨髄炎に関連する創部の癒合状況を評価し,その後52週目に2回目の読み取りを行い,X線,臨床評価, と確立されたバイオマーカー(赤血球沈降率とC反応蛋白)から切断率と骨髄炎の解決を評価する予定である。これらの読み取り値はそれぞれ2025年第1四半期と2026年第1四半期に発表される見通しだ。
アメリカ国立衛生研究院の嚢胞性線維化の研究
米国国立衛生研究院と抗菌薬耐性リーダーグループによる研究を支持しています緑膿菌FDAで新薬やEIND手当下のCF患者の感染を緊急研究している。この1 b/2期マルチセンター,無作為,二重盲検,プラセボ対照試験は,嚢胞性線維症患者における単剤IVバクテリオファージ療法の安全性と微生物学的活性を評価している緑膿菌.
保留の番組
BX 005−アトピー性皮膚炎の治療
BX 005は私たちがbrに対する人気のバクテリオファージ製品の候補品です黄色ブドウ球菌あるいは…黄色ブドウ球菌アトピー性皮膚炎炎症の進行や増悪に関与する細菌黄色ブドウ球菌アトピー性皮膚炎患者の皮膚は健康な人の皮膚よりもっと豊富で、皮膚損傷皮膚は非皮膚皮膚よりもっと豊富である。紅斑が出現するとその数も増加し,優占細菌となる。brは細菌の負荷を減少させることである黄色ブドウ球菌BX 005の設計目的は、皮膚微生物群の組成を“爆発前” 状態に変換し、臨床的利益を提供することである。臨床前段階で体外培養研究により、BX 005は薬剤耐性菌株を含む90%を超える菌株を根絶できる黄色ブドウ球菌菌株(米国とヨーロッパの被験者の皮膚から120株分離)。FDAは2022年4月8日、同社BX 005のIND申請を承認した。
本年度報告の日まで,我々のCFとDFO計画に資源を優先しているため, BX 005の開発を休止しており,その開発再開に関する指導 を提供することはできない。
プロテーゼ関節感染、PJIと略称される
我々はPJIを治療するための個人化バクテリオファージ療法は、黄色ブドウ球菌、表皮ブドウ球菌、糞腸球菌など様々な細菌を対象としている。この治療は2020年7月にFDAから孤児薬物の称号を与えられた。本年度報告日までに,我々のCFとDFO計画に資源を優先的に割り当てるため,この計画の開発作業 を一時停止し,その開発再開に関する指導を提供することができなかった.
3
私たちの戦略
我々の目標は,バクテリオファージに基づいて有害細菌を正確に特定する能力と,自然産生と合成工程を用いて生成されたbrを含む様々な製品を開発し,これらの治療法を開発する能力と,異なるバクテリオファージをスクリーニング,識別·最適化する能力に基づいている。私たちは続けるつもりです
| ● | バクテリオファージを基礎とした鉛系製品によるCFOとDFO治療の臨床安全性と有効性を研究した |
| ● | 私たちの既存の適応と可能な新しい適応のためにバクテリオファージ療法が対象とする新しい病原細菌を決定し |
| ● | 我々の独自のXMarkerプラットフォームに基づき,微生物群に基づくバイオマーカー試験を開発·協力し,疾患診断や補助診断として利用することができる。 |
バクテリオファージ発見プラットフォームは
著者らの方法はいくつかの要素の集合である:バクテリオファージに対する理解は迅速に増加し、バクテリオファージ行為とそのゲノムとの間の関連を含む;ますます多くの証拠により、特定の有害細菌の存在は慢性疾患、例えばCFに影響を与える可能性があり、それらを原則的にバクテリオファージによる治療 に適合させる;及び異なる学術センターからのますます多くの逸話報告は、バクテリオファージ治療が他の治療法に無効な重症患者に同情することに成功した。我々のバクテリオファージ治療製品候補製品は、健康な微生物領域元素を破壊することなく、病原性細菌を正確に位置決めすることによって、疾患および疾患を治療する潜在力を有すると信じている。
我々はバクテリオファージの候補製品に基づいて,固定ファージカクテルも個人化されたバクテリオファージ治療も,Bolt とPSTという独自の研究開発プラットフォームを用いて開発した。Bolt、プラットフォームは唯一無二であり、学際的な先端方法と能力を採用し、計算生物学、微生物学、バクテリオファージ及びその生産細菌宿主の合成工程、生物分析テスト開発、brと調合を製造し、柔軟かつ効率的な天然或いは工程バクテリオファージの組み合わせ或いはカクテルの開発を実現する。
PSTプラットフォームは、固有アッセイbrを使用して、所与の患者から単離された特定の標的細菌を治療する最適なバクテリオファージを探すために、広範なファージライブラリーをスクリーニングすることを可能にする。
Boltは最適化されたファージカクテルの迅速な開発を目指している。これらのカクテルは自然に生産されたものや人工的に合成されたバクテリオファージからなる可能性がある。カクテル は機能相補的なバクテリオファージを含み、広範な標的宿主範囲、br耐性を防止する能力、生物被膜浸透、安定性、製造が容易など、多種の特性に対して最適化されている。最適化されたファージカクテルの臨床前開発には1−2年を要すると予想される。
私たちは様々な技術を組み合わせて、これらの技術は私たちの科学創始者の実験室から来て、私たちの内部で開発したものです。我々の科学創始者によって開発された技術は有力な科学誌に記述されている。我々の科学創始者の一人であるRotem Sorek教授はヴァイズマン科学研究所分子遺伝系の教授であり、バクテリオファージゲノム学と細菌防御機構の世界の先頭者である。br}もう一人の科学創始者であり、ウィスコンシン大学免疫学系教授Eran Elinav教授は微生物群と人類の健康と疾病との関連を研究する専門家である。私たちの3人目の科学創始者であるティモシー·K·Lu教授は、遺伝子回路とバクテリオファージを設計するための合成生物学的方法の世界リーダーであり、マサチューセッツ工科大学電気工学とコンピュータ科学系合成生物学グループと生物工学系をリードしている。 また、2017年にイスラエルの私有株会社RondinX Ltd.を買収することにより、著者らはウィスコンシン大学コンピュータ科学と応用数学系のリーディング計算生物学者エラン·シーゲル教授が開発した高スループットbr}ゲノム解析技術を獲得した。これらの先駆者のそれぞれの分野における技術と専門知識の結合は,微生物群を正確に操作することで,複雑なヒト疾患や疾患の治療に専念できるようにするために重要である
4
また,ロバストかつ高スループットなPSTテストを実現するための独自検出·スクリーニング技術を開発した。PSTプラットフォームは最先端の自動化と先進的な微生物測定 を結合した。結果は多種の要素に基づく最適バクテリオファージマッチングの再現性の決定的な決定であり、バクテリオファージ感染の成功、薬剤耐性突然変異の抑制と抗生物被膜の活性を含む。
製造業
私たちは最先端の工業方法を利用して私たちの候補製品を製造する製造プロセスを開発した。これらの過程は現在の良好な製造規範或いはcGMPに符合し、そして適切な規模を持ち、著者らの臨床研究需要を満たし、そして監督管理機構の人体研究に対する要求を満たすことを目的としている。
2021年2月、私たちは私たちのアメリカの良い製造実践(GMP)、製造、テスト、開発をゲザスバーグ工場の6100平方フィートの空間に統合し、2021年3月、私たちはイスラエルナイスジオナ本社にある6500平方フィートの新しい製造工場に引っ越した。両施設とも臨床ロットのわれわれの早期臨床開発に必要な候補製品の生産を目指しており,この開発段階のコンプライアンス に適合してEINDを支持している。
Ness Ziona工場は、液体、クリーム、半固体または乾燥形態の外用、経口、吸入および注射されたバクテリオファージ製品を支持するための2つの薬物バクテリオファージ製造/開発のための2つのスイートと、製剤および最終医薬製品生産チャンバとを含む。
ゲザスバーグ工場は、上流種子バンクのための3つの生産工場からなり、1つは薬物物質バクテリオファージ生産のためのものであり、1つは最終薬物製品の処方および充填のためのものである。この施設はまた、液体形態で発表された注射可能なバクテリオファージ製品brをサポートするための内部品質管理試験実験室を備えている。プロセス開発のために追加の実験室空間が割り当てられ、将来のGMP拡張に再使用することができる利用可能な実験室およびオフィス空間 がある。
我々が現在運営している製造モデル は,インターワークフロー開発,製造とテストと,必要に応じて第三者開発,製造,テスト,物流組織にアウトソーシングする柔軟性を組み合わせている.私たちは複数のメーカー、テスト実験室、および製品候補流通のための第三者物流倉庫維持サービス協定と合意した。これらのサービスプロトコルは通常短期的であり, は延期または更新することができる.そこで,BX 004については,我々のインターワークフロー開発活動を補完する第三者を招聘した. 我々は,その組織の経験,能力,能力,規制地位に基づいてその組織を選択した.製造·開発プロジェクト は内部従業員チームが管理し,製造プロセスに応じた技術面と法規要求 を確保する。
我々がゲザスバーグに位置する内部GMP工場では他のファージバンク候補製品 を生産し,総称してBX 211と呼ぶ。このような候補製品は内部 従業員がcGMPに従って生産·発表する。私たちは内部で多くの放出テストをリリーステストし、テストを合格した実験室にもアウトソーシングします。また、第三者物流倉庫を利用して製品を保存し、臨床サイトに配布します
5
将来の需要に応じて2つのGMP サイトを1つのサイトに統合することを考えている.現在はビジネス規模の製造能力は必要ありませんが、適切な時期には、私たちの業務の拡大が含まれている可能性がある大規模なcGMP内部製造能力を評価する予定です。
知的財産権
私たちは、米国および国際的に私たちの候補製品や発見プラットフォームのために特許保護を求め、維持することを含む、私たちの業務に重要であると考えられるノウハウを保護するために努力しています。私たちはまた、商標、商業秘密、技術ノウハウ、著作権、持続的な技術革新、ライセンス内の機会に依存して、私たちの独自の地位を発展させ、維持しています。私たちの知的財産権に関連するリスクに関するより多くの情報は、参照してください“危険要素-私たちが許可して共有された知的財産権に関連する危険。”
我々は,我々が行っている候補製品開発のためのレシピ,関連処理方法,製造方法または同定の特許出願を提出し,我々の独自製品プラットフォームの発見に基づいて,我々の知的財産権 を拡大していく予定である.私たちの成功は、私たちが私たちの業務に関連する重要なビジネス技術、発明およびノウハウの特許および他の独自保護を取得し、維持する能力があるかどうかに依存し、私たちが入手可能な任意の特許を保護し、私たちの商業秘密およびノウハウの機密性を保護し、第三者が効果的かつ強制的に実行可能な特許および独自の権利を侵害することなく運営されるであろう。
米国およびいくつかの他の司法管轄区域の特許出願は18ヶ月以上秘密にされており、科学または特許文献に開示されている発見は、実際の発見および特許出願よりも遅れていることが多いため、係属中の特許出願によってカバーされる発明の 優先権を決定することができない。したがって、私たちは、最初の発明のいくつかの特許出願に開示された主題のbrでもなく、そのような主題をカバーする特許出願を最初に提出した人でもなく、私たちは、発明の優先度を決定するために、米国特許商標局またはUSPTOが発表した妨害訴訟または派生訴訟に参加しなければならないかもしれない。
特許組合
私たちの特許の組み合わせは、自己特許出願、 および授権特許出願および共有特許出願(特許出願でもある)を含む。参照してください“危険要素-私たちが許可して共有された知的財産権に関連する危険。いくつかの申請については、起訴はまだ開始されておらず、他のいくつかの申請は米国と米国以外の選定された司法管轄区域で起訴の初期段階にある。私たちは4つの特許家族しか持っていない。私たちは日本の慶応大学または慶応義烏大学と共同で米国特許家族を持ち、業達研究開発有限会社と共同で国際特許家族(アメリカ、オーストラリア、カナダ、欧州特許庁国家届出)を所有し、WIS技術移転事務室と共同で国際特許家族(アメリカ、ヨーロッパ)を所有し、慶応義烏と業達と共に国際特許家族を所有している。このような共同所有特許出願について、私たちはYedaとKeioからの独占ライセンスを持っている。私たちはYedaまたはKeioがその製品の組み合わせにおける残りの特許と特許出願に対する独占的な許可 を持っている。
私たちの製品の組み合わせの大部分は、私たちの候補製品、特にCFとアトピー性皮膚炎、および炎症性腸疾患(IBD)、原発性硬化性胆管炎および結腸直腸癌、ならびに私たちの細菌標的発現とバクテリオファージ発見技術プラットフォームなどの開発を中止したプロジェクトに関連する候補製品である。私たちの候補製品をカバーするほとんどの未解決特許出願はまだ起訴され始めていない。起訴は長い過程であり、その間、最初に米国特許商標局によって提出された審査請求の範囲は、それらが本当に発行されれば、発行時に大きく縮小されることが多い。我々のライセンス特許出願と共通所有の特許出願については,以下のように となると予想される.
6
今回の買収では、我々の知的財産権の組み合わせをさらに強化し、発行または許可された7つの特許、19件の特許シリーズ(米国、ヨーロッパ、オーストラリア、カナダ、中国、インド、日本、韓国、イスラエル、ブラジル、南アフリカの出願を含む)を含むAPTの組み合わせを追加した。APTの特許および特許出願は、2037年6月から2043年10月までの有効期間を有する医薬組成物およびそのような組成物の治療方法、製造方法に関連する特許および特許出願を含む。
CF
我々は、慢性肺炎患者の特に一般的なシュードモナス肺感染を治療するバクテリオファージの組み合わせ、これらのバクテリオファージの組み合わせの使用方法、およびこれらのバクテリオファージの組み合わせに反応する患者を識別する方法を含む医薬組成物に対する請求項を含む1つの特許ファミリー(米国、オーストラリア、カナダ、欧州特許庁、日本および中国)のみを有する。この計画から我々の主要バクテリオファージ組合せの係属中の出願から発行された任意の米国特許は,発行されれば2042年に満了する予定である。特許期限の調整または特許期間の延長は、より遅い満期日をもたらす可能性がある。
アトピー性皮膚炎
我々は、皮膚感染(特にアトピー性皮膚炎患者によく見られる)を治療するためのバクテリオファージの組み合わせ、これらのバクテリオファージの組み合わせの使用方法、およびこれらのバクテリオファージの組み合わせに反応する患者を識別する方法を含む医薬組成物に対する請求項を含む1つの特許シリーズ(米国、オーストラリア、カナダ、欧州特許庁および日本)のみを有する。本計画における主要バクテリオファージの組み合わせをカバーする任意の米国特許が処理中のbr出願から発行され,発行されれば2042年に満了する予定である。特許期限の調整や特許期限の延長は、より遅い満了日をもたらす可能性がある。
特許期間
個別特許の期限は,特許を取得した国·地域の特許の法的期限に依存する。私たちが特許出願を提出したほとんどの国/地域では、br米国を含み、基本期限は、特許要求優先権の最初に提出された非臨時特許出願の提出日から20年である。米国特許の期限は、特許期限調整によって延長することができ、これは、米国特許商標局の行政遅延による特許所有者の損失を補償する。場合によっては,米国特許の期限は免責声明を終了することにより を短縮し,その期限をより早く満期された特許に短縮する.1984年の“薬品価格競争および特許期限回復法”(略称“ハッジ-ワックスマン法”)によると、米国特許の有効期限は、特許付与後に薬物が開発および規制審査段階にある少なくとも 部分時間を示すために、資格延長資格がある可能性がある。FDA 承認が有効成分の最初の発売を許可する薬剤の場合、“ハッジ-ワックスマン法案”は、そのようなFDA承認薬物の物質組成、FDA承認薬物治療方法、および/またはFDA承認薬物の製造方法をカバーする少なくとも1つの請求項を含む米国特許の有効期限を延長することを可能にする。延長された特許期間は、特許未延長満了後のより短い5年またはFDAが薬物を承認した日から14年を超えることができず、1つの特許は1回以上の製品を延長することができない。延期期間中、許可が得られた場合、専有権の範囲は、承認された使用のための承認された製品に限定される。欧州や日本を含むいくつかの外国司法管区にも同様の特許期限延長条項があり、適用される外国規制機関が承認した薬物の特許期間の延長を許可している
将来、私たちの候補製品 がFDAによって承認された場合、私たちは、これらの候補製品、その使用方法、および/または製造方法のための特許を適切な場合に延長することを予想する。しかし、米国のFDAを含む適用当局が、このような延期を承認すべきかどうかの評価と、承認されれば、このような延期の期限を承認することに同意する保証はありません。
7
ビジネスの秘密と技術ノウハウ
特許に加えて、私たちは商業秘密と技術ノウハウに依存して、私たちの競争地位を発展させ、維持しています。私たちは通常、私たちの業務における を特許保護から保護するために商業秘密に依存しています。または特許保護には適していないと考えています。私たちは、私たちの従業員、コンサルタント、科学コンサルタント、請負業者、および協力者と秘密協定および発明譲渡プロトコルを確立することによって、商業秘密および技術的ノウハウを保護します。これらの合意は、個人または実体と私たちとの関係期間または後に開発または漏洩されたすべての機密情報は、関係期間および後に秘密にしなければならないと規定しています。これらのプロトコルはまた、私たちのために達成された仕事または私たちの業務に関連する仕事から生成されたすべての発明が、適用される場合、雇用または割り当て中に構想または完了された場合、私たちの固有財産となることを規定する。また、物理的および技術的セキュリティ対策など、その独自の情報が第三者に盗用されることを防止するための他の適切な予防策も講じている。
私たちは、私たちの従業員およびコンサルタントと契約を締結することを含む、私たちの固有の情報およびビジネス秘密を保護する措置を取っていますが、第三者は、実質的に同じ固有の情報および技術を独立して開発したり、他の方法で私たちのビジネス秘密にアクセスしたり、私たちのbr技術を開示したりすることができます。したがって、私たちは私たちの商業秘密を意味的に保護し、その独占的な使用から利益を得ることができないかもしれない。私たちの知的財産権に関連するリスクに関するより多くの情報は、参照してください“リスク要因−我々の許可と共同所有する知的財産権に関するリスク .”
競争
バイオテクノロジーと製薬業界の特徴は技術の進歩が迅速で、競争が激しく、独自製品を強調することである。私たちの技術、知識、経験は私たちに競争優位を提供してくれると信じていますが、私たちはより多くの資源を持つ大きな製薬会社を含む多くの異なる源からの激しい競争に直面しています。専門バイオテクノロジー会社、学術研究機関、政府機関及び公共及び民間機関も競争力のある製品と技術の潜在的な源である。私たちのすべての候補製品の成功に影響を与える重要な競争要素は有効性、安全性、発売時間、 コスト、販売促進活動レベルと知的財産権保護を含むと考えられる。
私たちは多くのバイオテクノロジー会社が疾患治療のためのバクテリオファージ製品を開発していることを知っている。いくつかのバイオテクノロジー会社,例えばLocus Biosciences,Inc.,Armata PharmPharmticals,Inc.およびSNIPR Biome,および学術機関には,自然産生バクテリオファージや合成生物学的手法を用いた発見段階や臨床計画があることが知られている。また、いくつかの研究と発売製品が、私たちの候補製品に対する指標を処理するために使用できることが知られていますが、これらに限定されません
| ● | Cf:トラカフタ、サイドコ、Pulmozyme、トブマイシン、アンモニア曲南 |
| ● | DFO:TP−102 Phaxiamにより開発されたバクテリオファージベース製品Technphage開発 |
私たちの多くの競争相手は、単独でも彼らのbr戦略パートナーとも、私たちよりも多くの財力、技術、人的資源を持っており、候補製品の発見と開発、FDAと他の規制機関の製品の承認、およびこれらの製品の商業化の面でより多くの経験を持っている。したがって、私たちの競争相手は私たちよりも候補製品の発見に成功し、このような候補製品の承認を得て、広範な市場で受け入れられるかもしれない。私たちの競争相手の製品は、私たちが商業化する可能性のある任意の製品よりも効果的であるか、またはより効率的にマーケティングおよび販売され、私たちの候補製品を時代遅れにしたり、競争力を失ったりする可能性があり、その後、任意の候補製品の開発および商業化の費用を回収することができる。新薬の市場進出と先進技術の出現に伴い、私たちは激しく日々激しい競争に直面すると予想している。
これらの第三者は合格した科学研究、臨床、製造、販売、マーケティングと管理人員を募集と維持し、臨床試験場所と臨床試験患者登録を確立し、そして著者らの計画と相補或いは必要な技術を獲得する方面で著者らと競争する。
8
販売とマーケティング
我々は,内部販売やマーケティング能力を確立したり,他社との協力により候補薬物製品の商業化を実現したりする予定である。
2021年10月、私たちはMaruhoの子会社と株式購入協定を締結し、Maruhoは日本をリードする皮膚病薬会社のbrであり、この協定によると、私たちはMaruhoに1株8.00ドルで375,000株の普通株を発行し、総収益は300万ドルである。br}私たちはまたMaruhoに優先的な約束権を与え、Maruhoが日本で私たちのBX 005候補製品を使用してアトピー性皮膚炎を治療することを許可した。現在保留されている第1/2段階研究結果が利用可能になった後、最初の オファーを得る権利が開始される。
政府の監督管理
アメリカとその他の国/地区の政府当局は薬品と生物製品の研究、開発、テスト、製造、品質管理、審査、ラベル、包装、保存、記録保存、販売促進、広告、流通、承認後の監視と報告、マーケティングと輸出入 などを監督する。通常、新薬或いは生物が人体臨床試験で研究或いは発売できる前に、必ず大量のデータを獲得し、その品質、安全性、有効性、純度及び/或いは効力を証明し、各監督管理機関の特定のフォーマットに組織し、審査を提出し、製品が研究或いは発売予定の監督管理機関によって許可される。
アメリカの生物製品開発プロセス
アメリカでは、FDAは“連邦食品、薬物と化粧品法”(FDCA)とその実施条例、“公衆衛生サービス法”(PHSA)及びその実施条例に基づいて薬品を監督する。医薬品と生物製品もまた他の連邦、州、そして地方法規によって制限されている。規制の承認を得て、その後適切な連邦、州、地方法規を遵守する過程には多くの時間と財力が必要だ。製品開発,承認または上場後の過程において,出願人が適用された米国の要求をいつでも遵守できなかった場合,行政または司法制裁を受ける可能性がある。他の行動に加えて、これらの制裁は、FDAが未解決の申請の承認の拒否、承認またはライセンスの撤回、臨床封印、無タイトルまたは警告状、製品のリコールまたは市場撤回、製品の差し押さえ、生産または流通の完全または部分的な一時停止、禁止、罰金、政府契約の拒否、原状回復、返還、返還、および民事または刑事処罰を含むことができる。どんな機関や司法法執行行動も私たちに実質的な悪影響を及ぼすかもしれない。
私たちの現在のいくつかの候補製品と未来の候補製品 は生物製品許可証申請またはBLAプロセスを通じてFDAの承認を得なければならず、その後アメリカで合法的に発売されることができる。このプロセスは、一般に以下のことを含む
| ● | 適用法規に基づいて広範な臨床前研究を完成し、必要であればGLP要求による研究を含む |
| ● | ヒト臨床試験が開始される前に有効でなければならないINDをFDAに提出する |
| ● | 各臨床試験が開始される前に、各臨床試験場所で機関審査委員会またはIRBによって承認される |
| ● | 適用されたIND法規、良好な臨床実践或いはGCP要求と他の臨床試験関連法規に基づいて、十分かつ制御された人体臨床試験を行い、各提案適応の研究製品の安全性、純度、効力と有効性を確定する |
| ● | 食品医薬品局にBLAを提出しました |
| ● | FDAはBLAを受信してから60日以内に再審申請を受けることを決定した |
| ● | 施設、方法、および生物の特性、強度、品質、および純度を維持するのに十分な制御を保証するために、cGMP要件に適合する状況を評価するために、生物を生産する1つまたは複数の製造施設の承認前検査を良好に完了させることが好ましい |
9
| ● | FDAは、BLAを支持するデータを生成する臨床試験場所を監査することができる |
| ● | FDAがBLAの使用料を審査する(適用費用免除が適用されない限り) |
| ● | 米国で任意の商業マーケティングまたは生物販売を行う前に、FDAは、任意のFDA顧問委員会の意見を考慮することを含むBLAの審査および承認を含む。 |
臨床前研究とIND
臨床前研究には製品化学と処方の実験室評価があります体外培養動物研究と、治療使用の理論的基礎を確立し、場合によっては有害事象が発生する可能性を評価する。臨床前研究の実施は連邦法規と要求の制約を受け、場合によっては安全/毒理学研究に対するGLP法規を含む。INDスポンサーは,臨床前試験の結果を生産情報,分析データ,任意の利用可能な臨床データや文献,臨床試験計画などとともにFDAに提出し,INDの一部としなければならない。INDはFDAがヒトに研究製品の使用を許可する要求であり,ヒト臨床試験が開始される前に発効しなければならない。IND提出後,いくつかの長期的な臨床前テストを継続する可能性がある。INDはFDAが受領してから30日後に自動的に発効し、それ以前に、FDAは1つまたは複数の提案された臨床試験に対して懸念または問題を提起し、試験を保留する。この場合,INDスポンサーやFDAは臨床試験開始前に未解決の問題を解決しなければならない。したがって,INDの提出はFDAが臨床試験の開始を許可しない可能性がある。
臨床試験
臨床試験は、合格した調査員の監督の下で、候補薬物やバイオ製品を健康ボランティアや病気の影響を受けた患者に使用することに関連している。br医師は通常、試験スポンサーの雇用や制御を受けない。臨床試験は,臨床試験目標,投与手順,被験者選択と排除基準,および被験者の安全性と有効性を監視するためのパラメータ を詳細に説明するレジメンに基づいて行われ,何らかの有害事象が発生した場合に臨床試験を終了する停止ルールを確保することを含む。各スキームおよびスキームの任意の修正は、INDの一部としてFDAに提出されなければならない。臨床試験 はFDAがGCP要求を含む規定に従って行わなければならず,すべての研究対象にインフォームドコンセントを要求することを含む。また,いずれの臨床試験も臨床試験を行う機関のIRBが審査·承認し,サービスを提供しなければならない。IRBは研究参加者の福祉と権利 の保護を担当し,臨床試験に参加する個人のリスクが最低に低下したかどうか,期待収益に対して合理的であるかどうかを考慮している。IRBはまた、各臨床試験対象またはその法律代表によって署名されなければならないインフォームドコンセントの形態および内容を承認し、完成まで臨床試験を監視しなければならない。また,行っている臨床試験や完成した臨床試験結果を公的登録機関に報告する要求もある。臨床試験結果を含むいくつかの臨床試験に関する情報は、www.Clinicaltrials.govサイト上で発表するために、特定の時間範囲で提出されなければならない。
臨床試験は通常3つの連続段階で行われ,第1段階,第2段階と第3段階と呼ばれ,重なる可能性がある。
| ● | 第1段階の臨床試験は、一般に、一部の健康ボランティアまたは疾患の影響を受ける患者に関連し、彼らは最初に単剤に接触し、その後、多剤候補製品に接触する。これらの臨床試験の主な目的は候補製品の新陳代謝、薬理作用、副作用耐性と安全性を評価することである。 |
10
| ● | 第2段階臨床試験は、一般に、概念証明を評価するため、および/または後続研究のための用量レジメン(S)を決定するために、疾患の影響を受ける患者の研究を含む。同時に、安全性と時々更なる薬物動態学と薬効学情報を収集し、可能な副作用と安全リスクを識別し、そして初歩的な治療効果の評価を行った。 |
| ● | 第三段階臨床試験は通常、複数の場所の大量の患者に関連し、必要なデータを提供し、この製品の期待用途に対する有効性、使用中の安全性を証明し、そしてこの製品の全体的な利益/リスク関係を確立し、新薬ラベルに十分な基礎を提供することを目的としている。 |
承認後の試験は、4期臨床試験とも呼ばれ、初歩的な発売承認後に行うことができる。これらの試験を行ったのは,期待される治療適応患者の治療からより多くの経験を得るためである。場合によっては,FDAはBLAを承認する条件として4期臨床試験を強制的に実行することができる。
その他の情報を除いて、臨床試験結果を詳細に説明する進捗報告は少なくとも毎年FDAに提出されなければならず、書面のIND安全報告は、深刻かつ意外な疑わしい有害事象、他の研究または動物を発見するためにFDAおよび調査者に提出されなければならない体外培養測定 は人類被験者に重大なリスクがあることを表明し、方案或いは研究者マニュアルに記載されているのと比較して、深刻な副作用の疑いの発生率は臨床上どのような重要な増加がある。
第1段階、第2段階、第3段階、および の他のタイプの臨床試験は、指定された時間内に成功できない可能性がある(あれば)。FDAまたはスポンサーは、患者が許容できない健康リスクに曝露されていることを発見することを含む、いつでも様々な理由で臨床試験を一時停止または終了することができる。同様に、臨床試験がIRBの要求に従って行われていない場合、または生物試験が患者に意外な深刻なダメージを受けた場合、IRBは、その機関の臨床試験の承認を一時停止または終了することができる。また、いくつかの臨床試験は、臨床試験スポンサーまたはデータ安全監視委員会によって組織された独立した合格専門家グループによって監視される。このグループは,実験のあるデータへのアクセスにより,テストが指定されたチェックポイントで できるかどうかを許可する.
臨床試験と同時に、会社はより多くの動物研究を完成することができ、生物化学と物理特性に関するより多くの情報を開発し、cGMP要求に基づいて最終的に商業量産製品のプロセスを決定しなければならない。製造プロセスは一貫して高品質の製品ロットを生産できる必要があり,また,会社は最終製品の特性,強度,品質,純度をテストする方法を開発しなければならない。また,適切な包装を選択·テストし,候補製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
11
FDA審査プログラム
臨床試験が完了した後、研究製品が提案された1つ以上の指定用途に対して安全かつ有効であるかどうかを評価し、 の効力と純度の法規要件を満たすために、データ を分析する。臨床前研究および臨床試験の結果は、その後、BLAの一部としてFDA に提出され、製品の品質および他の関連データを確保するために提案されたラベル、化学および製造情報である。 BLAは、1つまたは複数の指定された適応を指定する生物の上場を承認する要求であり、安全性、純度、および効力の証明を含まなければならない。申請には,臨床前研究や臨床試験の陰性とファジィ結果,陽性のbr}結果が含まれる可能性がある。データは、製品使用の安全性および有効性を試験するために、または研究者によって開始された研究を含む多くの代替源からの臨床試験からのものである可能性がある。上場審査を支持するために、提出したデータは品質と数量で十分でなければならず、研究製品の期待適応、純度と効力の安全性と有効性を確定し、FDAを満足させる。生物製剤が米国で発売される前に,FDAの承認を得なければならない。改正された“処方薬使用者費用法”によると,いずれの生物製剤にも使用料が付与されなければならない。FDAは毎年PDUFAユーザ料金を調整する。場合によっては、小規模企業が初めて申請した費用を免除することを含む費用を免除または減免することができる。また,孤児薬として指定された製品については,孤児適応も含まれていない限りBLASで使用料を評価することはない。
FDAは、届出を受ける前に提出されたすべてのBLAを審査し、BLAの届出を受け入れるのではなく、より多くの情報を提供することを要求する可能性がある。FDAは受信後60日以内にBLA届出を受け入れるかどうかの決定を行わなければならず,このような決定にはFDAが届出を拒否することが含まれる可能性がある。提出された申請が受け入れられると,FDAはBLAの深い審査を開始する.FDAがPDUFAで合意した目標および政策によれば、FDAは、元のBLAの予備審査を完了し、出願人に応答し、優先審査のために指定された元のBLAの提出日から6ヶ月間、FDAが10ヶ月間有する。FDAは常にそのPDUFA標準と優先BLASの目標日を満たすわけではなく、審査過程はFDAがより多くの情報を提供することを要求したり、明確にしたりすることによって延長されることが多い。
BLAを承認する前に、FDAは、それらがcGMP要件に適合しているかどうかを決定するために、新製品の製造施設を承認前に検査する。FDAは、製造プロセスおよび施設がcGMP要件 に適合していることを決定し、製品が要求された仕様内で一貫して生産されることを保証するのに十分でない限り、製品を承認しないであろう。FDAはまた,GCP要求に適合することを確保するために臨床試験のデータを審査することができる。さらに、FDAは、通常、臨床医および他の専門家を含むグループであり、申請が承認されるべきかどうか、およびどのような条件で(あれば)承認すべきかを検討、評価、および提案するために、新製品または製品の申請を諮問委員会に提出することができる。FDAは諮問委員会の提案に制限されていないが、承認決定を下す際にこれらの提案を考慮する。FDAは臨床試験データを再分析する可能性があり,FDAや出願人の審査過程で広く議論される可能性がある。
FDAはBLAを評価した後,承認状または完全な返信を発行する.この生物学的薬物の商業マーケティングを許可し、特定の適応の具体的な処方情報を提供する。完全な返信は、申請の審査期間が終了し、現在の申請が承認されないことを示している。完全な応答文は、一般に、FDAによって決定されたBLA内のすべての特定の欠陥を記述する。完全な返信は、追加の臨床データおよび/または臨床試験、臨床前研究または生産に関連する他の重要かつ時間の要求を必要とする可能性がある。完全な返信が発行された場合、出願人 は、BLAを再提出し、手紙で発見されたすべての不足点を解決するか、または申請を撤回することができる。このようなデータや情報を提出しても,FDAはBLAが承認基準を満たしていないと認定する可能性がある.臨床試験から得られたデータは常に決定的ではなく,FDAのデータ解釈はスポンサーの同じデータに対する解釈とは異なる可能性がある。
孤児薬名
1983年の“孤児薬品法”または“孤児薬品法”によれば、FDAは、米国では一般に200,000人未満、または米国では200,000人を超える影響を与え、そのような疾患または疾患の開発および提供に対する米国での製品の開発および提供コストが製品の販売から回収されることを合理的に予想することができない稀な疾患または疾患の治療のための医薬または生物製品に孤児の称号を付与することができる。BLAを提出する前に,生物 の孤児薬物指定を申請しなければならない。FDAが孤児薬物指定を承認した後、FDAは、治療薬の識別およびその潜在的な孤児の使用を開示するであろう。孤児薬物指定は規制承認過程でいかなる優位性を伝達したり、持続時間 を短縮したりしない。
12
孤児薬物指定は,一方が臨床試験コスト,税収割引,ユーザ費用減免のために贈与資金を提供する機会など,財政的インセンティブを得る権利がある。孤児指定を有する製品がその後、そのような指定された疾患または状況に対するFDAの最初の承認を得た場合、br製品は、孤児薬物排他性を得る権利があり、これは、FDAが、限定された場合、例えば、より効率的、より安全、または患者ケアに重大な貢献をする方法によって、または医薬品供給問題において、排他的孤児よりも優れた臨床症状を有する製品を示すことによって、限定された場合を除いて、承認日から7年以内に同じ適応に対して同一の薬物を販売する任意の他の出願を許可しないことを意味する。しかし,競合相手は 同じ適応に対する異なる製品の承認,あるいは異なる適応に対する同じ製品の承認を得る可能性があるが,これらの製品はラベル外で孤立した適応に用いることができる.孤立薬br}競争相手がFDAによって定義された同じ製品が承認される前に承認されるか、または承認された同じ適応を求めている場合、または私たちの製品が競合他社の製品範囲に含まれていると決定された場合、排他性は7年以内に私たちの製品が承認されることを阻止する可能性もある。もし私たちの孤児薬に指定された製品が発売承認され、その適応範囲が指定された適応よりも大きければ、孤児薬物の独占経営権を得る権利がない可能性がある。br}2023年12月、BX 004はFDAの孤児薬物称号を獲得した。
開発と審査計画を加速する
FDAは特定の標準に符合する新薬と生物製品の審査過程を加速或いは促進することを目的とした迅速なチャンネル計画を持っている。具体的には、新薬および生物製品が深刻または生命に危険な疾患の治療に使用され、臨床前または臨床データが、この疾患が満たされていない医療需要を満たす可能性があることを示す場合、迅速なチャネル指定を受ける資格がある。高速チャネル指定は 製品と検討中の特定の適応の組合せに適している.迅速チャネル計画を含むFDAがマーケティングのために提出した製品は、迅速なチャネル計画を含むFDAが開発および審査を加速するために意図された他のタイプの計画の資格に適合する可能性があり、例えば、優先的な審査および承認の加速が可能である。製品が深刻または生命に危険な疾患を治療する場合、優先審査を受ける資格があり、承認されれば、既存の療法と比較して安全性と有効性の面で有意な改善を提供する。FDAは、指定された優先審査の新薬または生物学的出願を評価するために、追加のbrリソースを使用することを試み、br}審査を促進するために努力する。
もし製品が深刻な或いは生命に危害を及ぼす疾患を治療し、代替終点に対して臨床利益を合理的に予測する可能性がある影響を示し、あるいは臨床終点に対して不可逆的な発病率或いは死亡率或いはIMMよりも早く測定する可能性があり、そしてbr}が合理的にIMM或いは他の臨床利益への影響を予測する可能性がある場合、この製品も加速承認を得る資格がある。承認の一つの条件として,FDAは通常,承認を加速させる薬物や生物のスポンサーに十分かつ良好に制御された上場後臨床試験を要求する。このような臨床試験が予期される臨床的利益を検証できなかった場合、またはスポンサーがこのような試験をタイムリーに行うことができなかった場合、加速的な承認を得た製品は、迅速な撤回手順を実行する必要がある可能性がある。
さらに、1つの医薬または生物学的製剤が単独でまたは1つまたは複数の他の薬剤または生物学的製品との併用を計画している場合、深刻または生命に危険な疾患の治療のためのbr}が使用され、初期臨床証拠は、製品が現在承認されている治療法よりも実質的に改善されている可能性があることを1つまたは複数の臨床的重要終点で示す可能性がある場合、医薬または生物学的製剤が突破的療法として指定される資格がある可能性があることを示す。画期的な治療指定の利点には,迅速チャネル指定と同様の利点と,有効な薬物開発計画を確保するためのFDAの密な指導がある。
製品がこれらの計画のうちの1つまたは複数に適合していても、FDAは、製品がもはや資格条件を満たしていないと後で決定することができ、またはFDAが審査または承認する期間が短縮されない可能性がある。さらに、迅速チャネル指定、優先審査、加速承認、および画期的な治療指定は、承認の基準を変更することはないが、開発または承認プロセスを加速させる可能性がある。
13
小児科情報
2003年の小児科研究公平法或いはPREAによると、BLA或いはBLAの補充はすべての関連小児科亜群で主張されている適応の生物安全性と有効性を評価し、製品の安全に有効な各小児科亜群の投与量と投与を支持するためのデータを含まなければならない。FDAは小児科データの提出を延期することを許可するか、またはすべてまたは部分的な免除を与える可能性がある。新しい活性成分、新適応、新剤形、新投与レジメンまたは新投与経路を含む薬物マーケティング申請を提出する計画のスポンサーは、第2段階会議終了後60日以内に予備小児科研究計画またはPSPを提出しなければならず、そのような会議がない場合には、第3段階または第2/3段階研究が開始される前に可能な限り早く予備小児科研究計画を提出しなければならない。初期PSPは、研究目標および設計、年齢グループ、関連する終点および統計方法、またはそのような詳細な情報を含まない理由、および小児科研究データの提供を延期または完全または一部免除する要求および支援情報の要求を含むbr}スポンサー計画によって行われる1つまたは複数の小児科研究の大綱を含まなければならない。br}FDAおよびスポンサーはPSPについて合意しなければならない。臨床前研究,早期臨床試験および/または他の臨床開発計画から収集したデータから小児科計画の変更を考慮する必要があれば,スポンサーは合意した初期PSPに対する修正案 を随時提出することができる。
発売後要求
新製品が承認された後、製造業者および承認された製品は、br活動の監視および記録保存、不良体験の報告、販売促進および広告要件の遵守を含むFDAの規制を継続し、許可されていない用途または患者集団(“非ラベル使用”と呼ばれる)への製品の使用を制限すること、および業界スポンサーの科学的および教育活動を制限することを含む。医師はラベル外の用途のために合法的な製品を開く可能性があるが、メーカーはこのような用途を販売したり普及させたりしてはならない。処方薬と生物宣伝材料は初回使用時にFDAに提出されなければならない。さらに、適応、ラベルまたは製造プロセスまたは施設の変化を含む生物学的修正がある場合、出願人は、新しいBLAまたはBLAサプリメントの承認を得るために提出および提出を要求される可能性があり、これは、追加のデータまたは臨床前研究および臨床試験を開発する必要があるかもしれない。
FDAはまた、製品の安全な使用を保証するために、リスク評価および緩和策を要求すること、またはREMSを含む、承認時に他の条件を追加することができる。FDAがREMSを必要とすると結論した場合,BLAのスポンサーは提案したREMSを提出しなければならない。必要に応じて、FDAは、承認されていないREMSなしにBLAを承認することはない。 REMSは、投与ガイドライン、医師コミュニケーション計画、または配布方法、患者登録、および他のリスク最小化ツールを制限するなどの安全な使用を確保する要素を含むことができる。承認またはマーケティングに関するこれらの制限は、製品の商業販売促進、流通、処方、または配布を制限する可能性があります。新たに発見または開発された安全性または有効性データは、新たな警告や禁忌症の追加を含む製品承認のラベルを変更する必要がある可能性があり、REMSの実施や発売後の研究を含めて、新たに発見された安全問題 を評価することを含む他のリスク管理措置を実施する必要がある可能性がある。製品承認は、規制基準を満たしていないことや、初期マーケティング後に問題が発生したことで撤回される可能性があります。
FDA法規は、製品が特定の承認された施設で生産され、cGMP法規に適合することを要求し、その中で、品質管理および品質保証、記録および文書の維持、およびcGMPとの任意の偏差を調査および是正する義務が要求される。製造業者および他の承認された医薬品または生物製品の製造および流通に関連するエンティティは、FDAおよび特定の州機関にその機関を登録し、brがcGMP要件および他の法律を遵守することを確実にするために、FDAおよび特定の州機関の定期的な抜き打ち検査を受けなければならない。そのため、メーカーはcGMPコンプライアンスを維持するために、生産と品質管理に時間、お金、精力を投入し続けなければならない。CGMP規定に適合しないことを含む違反が発見され、法執行行動を引き起こす可能性があり、承認後に製品問題が発見されると、リコールを含む製品、製造業者、またはBLAを承認した所有者の制限を招く可能性がある。
14
生物模倣薬と排他性
2009年の“バイオ製品価格競争および革新法案”は、FDA許可の基準生物製品と生物学的に類似しているか、または交換可能であることが証明された生物製品のための簡略化された承認経路を作成した。PHSAのこの修正案は繰り返し検出を最小限にしようとある程度努力している。生物類似性は分析研究、動物研究と1つ或いは複数の臨床試験を通じて証明することができ、それは生物製品が参考製品と高度に類似していることを要求し、臨床では活性成分の微小な差異がないにもかかわらず、しかも製品と参考製品は安全性、純度と効力の面で臨床上意義のある差異がない。
互換性は、生物製品が参照製品生物と類似していることを必要とし、この製品は、参照製品と同じ臨床結果を生じることが予想され、複数回投与された個人の場合、製品および参照製品は、参照生物製品の独占的使用に関連する安全リスクを増加させることなく、または相対的に治療効果を低下させるリスクを増加させることなく、以前の投与後に交互または交換することができる。
参照生物製品は初めて許可を得た日から12年以内にデータ独占権を取得し、FDAは参照生物製品に基づく生物類似または交換可能な製品の申請を受け入れず、参照製品が初めて許可を得た日から4年になる。“初許可”とは、一般に、米国で特定の製品が許可された初期日を意味する。最初に許可された日brは、生物製品の許可日を含まず(かつ、新しい固有期間は適用されない)、生物製品の補充のために許可された場合、または生物製品の同じ発起人または製造業者(おそらく、利益担い手または他の関連エンティティ)のための後続の出願が、新たな適応、投与経路、投与スケジュール、剤形、送達システム、送達デバイスまたはbrの強度をもたらす変更を行うために行われる(生物学的br製品構造の修正を含まない)。または生物製品の構造を安全性、純度または効力の変化を引き起こさない修飾を行う。
小児科排他性はアメリカのもう一つの監督管理の市場排他性 であり、“児童最適薬品法”により生物製品価格競争と革新法を通じてそれを生物製品 に応用する。もし小児科専門権を付与すれば、現有の監督管理 専門期間に基づいて6ケ月増加し、この期限は到着しなければ小児科専門権を適用することができない。この6ヶ月間の専門権は、FDAがスポンサーの要請に応じてそのような書面請求を行うことができるが、FDAによって発行されたこのような試験の“書面請求”に基づいて小児科試験を自発的に完了することに基づいて付与することができる。
セット診断
私たちの臨床試験中、および私たちが開発中または将来開発可能な製品の商業化に関連する可能性がある場合、私たちは、診断に伴うbrを使用して、私たちの個人化バクテリオファージ治療下で特定の患者を治療するのに最適なバクテリオファージを識別することができ、私たちのファージカクテルに敏感な患者 をより正確に識別するのを助けることができる。同時診断は、特定の治療製品から利益を得る可能性が最も高い患者を識別することができ、特定の治療製品を使用することによって治療によって深刻な副作用のリスクを増加させる可能性のある患者を識別すること、またはより高い安全性または有効性を達成するために治療を調整するために、特定の治療製品に対する治療に対する反応を監視することができる。診断に伴い医療機器としてFDAの規制を受けているため,商業化前に承認または承認が必要である。リスクレベルはリスク低減に応用できる制御措置と結合し、セット診断設備が発売前承認が必要かどうか或いは510(K)上場前通知プロセスを通じて承認を得るかどうかを決定した。新しい治療製品のためには、そのセット診断装置は、製品の安全かつ有効な使用に不可欠であり、 キット診断装置は、治療と同時に開発および承認されるべきか、または510(K)を通過しなければならない。セット診断装置の使用は治療製品のラベルに規定される。
15
アメリカ以外の政府規制
アメリカの法規以外に、私たちは他の管轄区域の各種法規を遵守して、その中には薬物製品の臨床試験と私たちの候補製品の承認、製造と流通を含む。生物由来の原材料は独特の汚染リスクを受けるため、それらの使用はいくつかの国で制限される可能性がある。FDAの候補製品の承認を得るか否かにかかわらず,臨床試験や製品のこれらの国/地域での販売を開始する前に,外国の規制機関の必要な承認を得なければならない。もし私たちが適用された外国監督管理要求を遵守できなかった場合、私たちは罰金、規制許可の一時停止または撤回、製品のリコール、製品の差し押さえ、経営制限、刑事起訴などに処せられるかもしれない。
臨床試験
米国以外のある国/地域の規制プロセスは米国のプロセスと類似しており,ヒト臨床試験開始前に臨床試験申請を提出することが求められており,INDと非常に類似している。例えば,EUでは,各臨床試験の臨床試験申請やCTAは関連する国家衛生当局と各国の独立倫理委員会に提出されなければならず,各国では,試験は単一のEUポータルサイトで統一的に評価され,FDAやIRBのようになる。CTAは に臨床試験指令(および加盟国対応国法律)に規定された支援情報を添付した研究薬品アーカイブを添付し,適用された指導文書でさらに詳細に説明しなければならない。CTAが一国の要求に応じて承認されると,臨床試験は継続することができる。イスラエルでは、臨床試験を開始するにはEUのために説明されたような手続きが必要だ。臨床試験を管理する要求と流れは国によって異なる。すべての場合,臨床試験はGCPと適用される法規制の要求および“ヘルシンキ宣言”に起源する倫理原則に基づいて行われなければならない。
審査の流れ
私たちの製品を市場に出すためには、各製品のマーケティング許可を得て、多くの異なる法規要求を守らなければなりません。承認手続きは国/地域によって異なり、米国の承認を得るためのテストと比較して、追加のテストに関連する可能性がある。海外で承認を得るのに要する時間は,FDA承認を得るのに要する時間とは大きく異なる可能性がある。一国で行われた臨床試験は他の国の規制機関に受け入れられないかもしれない。米国以外の規制承認プロセスは、通常、FDA承認の取得に関するすべてのリスクの影響を受ける。また,米国以外の多くの国/地域では,製品の精算を許可してから,その国/地域で販売を許可する必要がある。
EUの監督管理制度の下で医薬製品の発売許可を得るためには、申請者は集中式或いは分散式プログラムに従ってマーケティング許可申請或いはMAAを提出しなければならない。分散したプログラムは、申請者が選択した加盟国間の連携に基づいている。 実質的に、申請者はMAAを科学的に評価し、製品情報を審査する“先頭”加盟国を選択する。他の会員国はこのような評価と審査の結果を認めなければならないが、“公衆衛生に深刻な潜在リスクがある”場合は除外される。分散されたプログラムは、選択された各国/地域で全国マーケティング許可を付与することをもたらす。この手続きは集中手続きの強制範囲に属さない限り、すべての医療製品に適用される。実際、それは高度に革新的な製品、模倣薬ではなく、非処方薬に使用され、生物模倣薬に使用されるようになってきている。
集中化手続きは、すべてのEU加盟国に有効な単一マーケティング許可を欧州委員会によって付与することを規定している。特定の医薬製品の場合、いくつかのバイオテクノロジーによって生産された医薬製品、孤児医薬製品、高度な治療薬またはATMPに指定された製品、および新しい活性物質を含み、特定の疾患を治療するための製品が表示され、集中プログラムが実行されなければならない。新規活性物質を含み、他の疾患の治療のために指定された製品、高度に革新的な製品、または患者の利益に適合した集中処理製品については、集中処理が任意である。
16
中央プログラムによると、人用薬品委員会(CHMP)はヨーロッパ薬品管理局(EMA)に設立された主要な科学委員会であり、未来の医薬製品の科学評価を担当している。CHMPはまた、既存のマーケティング許可の修正または拡張を評価するなど、いくつかの許可された後の および保守活動を担当する。評価MAAの最長時間は210日であり,クロックポーズは含まれていない.欧州委員会はマーケティング許可を承認または拒否し、 は会員国代表に関連する手続きに従う。ごく少数の場合を除いて、欧州委員会の決定はCHMPの科学的評価に適合している。
条例(EC)1394/2007により、具体的な規則はATMPに適用され、このカテゴリは遺伝子治療医療製品、体細胞治療医療製品と組織工学医療製品を含む。これらのルールは,一般法規要求をATMPの具体的な特徴に適応させるために,生産,臨床試験,薬物警戒に関するガイドラインの通過を引き起こしている。条例(EC)1394/2007では“病院免除”が導入され,認可病院は販売許可を得ずに内部使用のためのATMPを開発し,欧州連合の薬剤法を遵守している。病院免除は本質的に複合ATMPであり、すでにすべての加盟国で交換されており、時々、病院免除下のATMPはマーケティング許可を有するATMPの競争的代替品である。国家病院の病院免除の広範な使用は、ATMPの一般的な法律制度を破壊することなく、欧州委員会と加盟国との間で病院免除をより合理的に適用することを検討した。
マーケティング許可は原則として5年間の有効期間であり、マーケティング許可は5年後にEMAまたは許可加盟国の主管当局によるリスクと収益バランスの再評価に基づいて更新することができる。このため、マーケティング許可所有者は、マーケティング許可が無効になる少なくとも6ヶ月前に、マーケティング許可が付与されてから導入されたすべての変化を含む、品質、セキュリティ、および有効性に関するファイルの統合バージョンをEMAまたは主管当局に提供しなければならない。更新後,上場許可の有効期限は無期限であり,欧州委員会や国家主管当局 が薬物警戒に関する正当な理由に基づいてもう一度更新することを決定しない限り。いかなる許可も、認可終了後3年以内に薬品をEU市場(集中手続きである場合)または認可加盟国の市場に実際に投入されていない場合は無効である(いわゆる日没条項)。
孤児の称号
米国を除く他国は、まれな疾患を治療する薬剤や生物製品の開発·マーケティングを支援するための具体的な法制度を採択している。
例えば、欧州連合では、欧州経済地域(EU、アイスランド、リヒテンシュタインおよびノルウェーを加えた)または欧州経済地域(薬剤の開発が合理的であることを証明するために十分な見返りを得ることができない場合)で10,000人以下の5人以下の生命または慢性衰弱を脅かす疾患の診断、予防または治療を目的とした孤児薬物指定の承認が組織されている。その製品は影響を受けた人たちに顕著な利益をもたらすだろう。EMAの孤児薬品委員会(COMP)は孤児基準が満たされているかどうかを検査し、これについて意見を提出し、孤児の身分は欧州委員会からbr}を授与した。医療製品を承認する際、委員会は孤児指定基準の適合状況を再審査する予定であり、これは通常孤児指定承認数年後に発生する。もし当時指定孤児の基準 を満たさなくなった場合、欧州委員会は孤児の身分を撤回する。
EUでは、孤児薬物指定はスポンサーが費用を下げたり費用を免除したり、薬品が承認されてから10年間の市場排他性を獲得するなど、スポンサーに経済的奨励を受ける権利がある。市場排他性は、EMAまたは国家規制機関が別のMAAを検証することができず、欧州委員会または国家規制機関も、この期間内に同じまたは同様の医薬製品および同じ治療適応について別のマーケティング許可を付与することができない。孤児薬物指定基準を満たさなくなり、製品の利益が十分に高く、市場排他性を維持するのが合理的であることを証明するのに十分でない場合、この10年間の期限は6年に短縮される可能性がある。製造業者が患者の需要を満たすのに十分な数の医薬製品を保証できない場合、またはその他の製品がbrによって承認された孤児製品よりも臨床的に優れていることが証明された場合、別の医薬製品に対する孤児の排他性を失う可能性がある。もし1種の薬物がより安全で、より有効で、或いは患者の看護に重大な貢献があれば、それは臨床的に優れている。MAAを提出する前に,孤児薬物指定を申請しなければならない。孤児薬物指定は監督審査と審査過程でいかなる優勢を伝えることもなく、監督管理審査プロセスの持続時間を短縮することもなく、しかも上場許可を得る前に、いかなる規制排他性も負担しない。
17
開発と承認を速める
多くの司法管轄区域は機序を確立し、brは事前にこの薬物を承認することを許可し、それが医療需要を満たしていない患者の手にもっと早く到達するようにした。例えば、欧州連合は、集中手続きに特化した2つのメカニズムを含むいくつかの迅速な承認メカニズムを確立している
| ● | 加速承認:公衆衛生の観点から、特に治療革新の観点から、将来の医薬製品が重大な意義を有する場合、EMAはMAAの最長評価期間を210日から150日に短縮することができる。 |
| ● | 条件付きマーケティング許可:そのマーケティング許可プロセスの一部として、欧州委員会は、通常要求されるデータではなく、不完全なデータに基づいてマーケティング許可を付与する可能性がある。 |
CHMPが発見された場合、この医薬製品の安全性と有効性に関する全面的な臨床データが提供されていないにもかかわらず、以下のすべての要求を満たす場合、条件付きマーケティング許可を付与することができる
| ● | 医療製品のリスク/利益バランスは正である |
| ● | 申請者は全面的な臨床データを提供することができる可能性が高い |
| ● | 満たされていない医療ニーズは解決されます |
| ● | 医薬品の市場への即時発売に関する公衆健康へのメリットは,依然としてデータを補充する必要があるという事実に固有のリスクを超えている。 |
条件付きマーケティング許可の承認は、通常、申請された臨床部分が完全に完了していない場合に限定される。しかし,不完全な臨床前 や品質データが受け入れられる可能性があり,十分な理由があれば,製品が緊急時に公衆衛生上の脅威に対応しようとしている場合にのみ である。
条件付きマーケティング許可の有効期限は1年で、更新できます。承認の条件は、通常、保有者が行っているbr試験の完了や新たな試験を行い、収益-リスクバランスを確認し、薬物警戒データを収集することを要求します。 は上場許可の条件を満たすと、条件付きマーケティング許可は を通常のマーケティング許可に変換します。しかしながら、EMAが設定した時間範囲内で条件が満たされていない場合、条件マーケティング許可は更新を停止する。
EMAはまた,いわゆる“良質”(優先薬品)の地位を実施し,複雑な革新医療製品の開発と加速承認を支援し,満たされていない医療需要を満たしている。良質な地位はEMAが関連する科学委員会と早期対話を行うことができ、そしていくつかの支払人と と対話する可能性があり、それによってEMAの科学と監督支持を強化した。MAAを第一の地位とする加速評価も開かれており、 は通常、加速評価から利益を得る可能性のある薬品、すなわち公共健康の観点から、特に治療革新の観点から大きな意義を有する薬物に保持されている。
最後に、“特殊な場合”には、すべての医薬製品(すなわち、分散されたプログラムおよび集中されたプログラム)がMAから利益を得ることができる。このマーケティング許可は、深刻な疾患または満たされていない医療需要のための許可を必要とする医薬品のために保持されているので、条件付きマーケティング許可に近いが、出願人は、マーケティング許可を付与するために必要な法定完全データセットを有していない。しかし、条件付きマーケティングライセンスとは異なり、申請者は紛失したデータを提供する必要もなく、提供する必要もありません。毎年この医薬製品のリスク-利益について検討されている。したがって,“特殊な場合”はMAの最終承認を得たにもかかわらず, 医薬製品のリスク−収益バランスは毎年審査され,リスク−収益比が有利でなければ上場認可 を撤回する。
18
小児科学
ますます多くの司法管轄区域は小児科群の中で強制的な検査を要求している。EUはアメリカとスイスを含む他の司法管轄区域を刺激する複雑で非常に厳格な制度を公布した。任意の出願が、(I)新しい活性物質を含む医薬品のbrまたは(Ii)認可された医薬品の新しい治療適応、医薬形態または投与経路の任意の出願を承認し、例えば、br}が依然として補足保護証明書またはSPCによって保護された活性物質を含むか、またはSPC保護を受ける資格がある特許を含み、 は小児科データを含まなければならない。そうでなければ、監督管理当局はその申請を承認しないだろう。これらの場合,成人使用に関連しても小児科データを提出しなければならない。EMAが小児科開発の全部または一部の免除をそれぞれ承認した場合、小児科データの提出を要求しない、または完全に要求する。さらに、EMAが、成人人口の提出を遅延させないようにMAAの提出を延期することを許可した場合、提出を延期することができる。
小児科データは小児科調査計画やPIPを実施することで生成され,この計画は会社が成人PK研究が完了した後に提出され,EMAが に同意し,通常いくつかの修正を経ている。PIPは,将来の医薬品を児童に使用する際の安全性と有効性を証明するために,行うすべての研究ととるべき措置を示している。EMAは会社の要求に応じてPIPの修正に同意することができる。 PIPの範囲は成人治療適応や成人応用がその一部である条件,さらには活性物質の作用機序であり,EMA準適宜によって決定される。この非常に広範な裁量権は,EMAが成人適応とは異なる小児適応の開発を会社に求められるようになった。
PIPを完成させて会社に小児科奨励を受ける資格を持たせることは,SPC期間を6カ月延長することができ,孤児薬品であれば追加 年の市場排他性である。他の条件に加えて、報酬は、PIPが完全に完了したこと、小児科医療製品がすべての加盟国で承認されたこと、および何らかの方法(例えば、小児科適応を承認すること)で、製品特性要約において小児科研究結果に言及する条件に依存する。
発売後要求
多くの国は米国と類似した発売後の要求、特に安全モニタリング或いは薬物警戒を実施している。EUでは,薬物警戒データは,非ラベル使用に関する を含む規制当局による承認後の安全性や有効性研究の基礎である。このような要求を守らないことは、重大な経済的処罰とマーケティング許可の一時停止または撤回につながる可能性がある。
保護証明書の追加と規制の排除
米国以外のいくつかの国/地域では、私たちのいくつかの特許は、規制部門が私たちの候補製品および任意の未来の候補製品の承認の時間、期限、および具体的な詳細に依存する限られた特許期間延長を得る資格がある可能性がある。さらに、許可された薬物および生物製品は、規制の排他性(特許による特許保護を除いて)から利益を得ることができる。
欧州連合では、(EC)469/2009条例でSPCが規定されている。SPCは、法律の要求 が医薬製品を市場に投入する前に安全性および有効性試験を行い、マーケティング許可を得て失われた特許保護を補償するための特許期限の延長である。SPC は、“基礎特許”(特許所有者によって選択された特許であってもよく、製品、プロセスまたは応用特許)によって保護された任意の活性物質を申請することができ、EU製薬法による販売許可を得るまで医薬製品として市場に投入されていない。SPCの有効期間は最長5年であり、欧州経済区の初のマーケティング許可日から、特許とSPCの総合保護は15年を超えてはならない。SPC権利は、基礎特許およびマーケティング許可によって同時に制限される、すなわち、SPCは、基礎特許 によって付与された権利と同じ権利を付与するが、マーケティング許可によってカバーされる活性物質(および後に承認される任意の医薬製品の使用)に限定される。
19
SPCはヨーロッパレベルで規制されているが,それらは各国特許庁によって付与されている。SPCの付与には、国家特許庁が付与した基礎特許及び 上場許可が必要であり、これは、医薬製品として国内での最初のマーケティング許可である。 また、活性物質はSPCを取得していなければならず、SPCの出願は、最も遅いものを基準として、欧州特許庁の第1回マーケティング許可又は基礎特許付与後6ヶ月以内に国家特許庁に提出されなければならない。
将来、私たちは、臨床試験の予想期間および関連MAAの提出に関連する他の要因に依存して、現在の満期日後の特許寿命を延長するために、現在所有または許可されている1つまたは複数のヨーロッパ特許のためにSPCを出願するかもしれない。
さらに、EUでは、薬品 は以下の法規排他性から利益を得ることができる:データ排他性、市場保護、市場排他性、および小児科奨励。
新活性br物質(参考薬品)を含む医薬製品には8年間のデータ独占権が付与され,その後2年間の市場保護が与えられた。データ独占性brは他社が参考薬物br製品のマーケティング許可ファイル中の非臨床と臨床データを参考に模倣薬MAA目的を提出することを防止し、市場保護は他社が後発薬を市場に出すことを防止する。グローバルマーケティング許可の概念によれば、マーケティング許可所有者による医薬製品の任意のさらなる開発(例えば、新しい適応、新しい形態、br}を活性物質に変更する)は、いかなる新しいまたは追加の保護も引き起こさない。 任意の新しい開発プロジェクトの許可は、規制保護において最初のマーケティング許可に属するとみなされ、 したがって、新しい開発プロジェクトは、許可された後にのみ残りの規制保護から利益を得ることができる。唯一の例外は新たな治療適応であり,既存療法と比較して有意な臨床的メリットをもたらしていると考えられる。この新しい は、許可前の8年以内(すなわち、データ独占期間内)に許可を得ることを前提として、グローバルマーケティング許可のために1年間の市場保護を増加させることを示す。また、“有効物質”の新治療適応は1年間のデータ排他性から利益を得ているが、新適応を支持する非臨床·臨床データに限られている。brは欧州医薬品局で少なくとも10年間のどの活性物質も有効物質の資格を満たしていることを承認している。
生物模倣薬は8年間のデータ独占期間が終わった後に簡略化された審査ルートを通じて承認を得ることができ、そして10年或いは11年の市場保護期間後に発売することができる。生物模倣薬の承認は、申請者が生物模倣薬と生物医薬製品との間の類似性を証明し、EMA定義の非臨床および臨床データを提出することを要求する。生物類似法制度は,主に環境保全局が生物活性物質種別に適用する科学ガイドラインにより制定されている。米国と異なり、互換性 は加盟国ごとに規制されている。
市場排他性は孤児の地位を持つ医薬製品に対して提供される唯一の規制保護である。市場排他性はEMAまたは国家監督管理機関が承認後の10年以内に(上記参照)別のMAAを検証することができないようにし、欧州委員会または国家監督管理機関は同じ または類似の医薬製品と同じ治療適応に対して別のマーケティング許可を与えることができない。
小児科奨励は別の規制排他性である。 PIPが完了した後、会社は小児科奨励を受ける資格があり、これはSPCの期間を6ヶ月延長するか、または孤児薬品の場合、2年間の市場排他性を追加することとすることができる(上記参照)。PIPが自発的にbrに基づいて達成された場合、すなわち、SPCまたは基礎特許によって保護されていないか、またはSPCまたは基礎特許によって保護されなくなった承認された医薬製品の場合、小児科報酬は、“小児科用途マーケティング許可”またはPUMAの形態を採用する。この特殊なライセンスは、グローバルマーケティングのライセンスではないので、8年間のデータ独占と2、3年の市場保護から利益を得る。
20
他のアメリカの医療保険法やコンプライアンスの要求は
医薬品マーケティングに対するFDAの制限に加えて、私たちは医療業界の詐欺や乱用のための様々な連邦や州法律の制約を受ける可能性があります。これらの法律は、私たちの業務や財務の手配、そして私たちがマーケティング、販売、流通、私たちが承認された製品(あれば)の関係に影響を与える可能性があります。私たちの運営能力に影響を与える可能性のある法律には
| ● | 他の事項に加えて、現金または実物を直接または間接的に、公開的または間接的に現金または実物で直接または間接的に要求し、受信し、提供し、提供するか、または任意の報酬(任意のリベート、賄賂またはリベートを含む)を支払い、誘導または交換として、個人を紹介するか、または購入、レンタル、注文または推薦することを禁止し、連邦医療保険および医療補助計画のような連邦医療保険および医療補助計画に従って支払い可能な任意の商品、施設、物品またはサービスを提供すること;個人や実体は連邦反リベート法規を実際に知る必要はなく、この法規に違反する具体的な意図を持つ必要もなく違反を実施することができる。また、政府は、連邦反リベート法規違反による物品またはサービスのクレームを含み、連邦虚偽クレーム法案または連邦民事金銭処罰法規については、虚偽または詐欺的クレームを構成していると断言することができる |
| ● | FCAのような連邦民事および刑事虚偽申告法および民事金銭罰法は、個人または実体に刑事および民事処罰を適用し、民事告発者または準訴訟を許可する以外に、故意に連邦政府に虚偽または詐欺的な支払いクレームを提出または提出させること、連邦政府への金銭または財産の支払いまたは移転の虚偽または詐欺的クレームまたは義務を行うこと、使用または使用を招くこと、または虚偽陳述または記録材料の使用を招くこと、または連邦政府に金銭または財産を支払う義務を故意に隠蔽または故意に不当に回避または減少させること、または連邦政府に金銭または財産を支払う義務を回避または減少させることを含む |
| ● | 民事罰金法は、他に加えて、医療保険または医療補助受益者に無料または公平な市場価値よりも低い任意の方法で物品またはサービスを譲渡すること(限られた例外)を含むが、これらの物品またはサービスが、連邦または州政府案によって精算可能な物品またはサービスを選択する特定のサプライヤーに影響を与える可能性があることを知っているか、または知るべきであるが、これらの物品またはサービスが受益者に影響を与える可能性があることを知っているか、または知っているべきである |
| ● | 1996年の“健康保険携帯及び責任法案”、またはHIPAAは、支払人(例えば、公共または個人)にかかわらず、支払人(例えば、公共または個人)にかかわらず、任意の医療福祉計画が所有または保管または制御されている任意の金銭または財産を、虚偽または詐欺的な言い訳、陳述または約束によって、故意に、または故意に偽造、隠蔽、または隠蔽し、または任意の重大な虚偽陳述を行うことを禁止する新しい連邦刑法を制定し、医療に関連するプロジェクトやサービス連邦反リベート法規と同様に、個人または実体は、この法規または法規違反の具体的な意図を実際に理解する必要がなく、違反を実施することができる |
| ● | 連邦“平価医療法案”(Affordable Care Act、ACAと略称する)の透明性要件は、連邦医療保険、医療補助または児童健康保険計画に基づいて支払われる薬品、設備、生物製品および医療用品のメーカーが毎年アメリカの衛生·公衆サービス部に医師(医師、歯科医師、視光師、足科医師および脊医を含むと定義される)、ある非医師従事者(医師アシスタント、勤務看護師、臨床看護師専門家、麻酔師アシスタント、登録看護師および登録看護師助産師)と教育病院支払いまたは他の方法で価値を移転することに関する情報を米国の衛生·公衆サービス部に報告することを含む、連邦“平価医療法案”(Affordable Care Act、略称ACA)の透明性要件を含む。これらの医師とその直系親族が所有している所有権と投資権益 |
| ● | 連邦政府価格報告法は、複雑な価格指標を政府計画に正確かつタイムリーに計算し、報告することを要求している |
| ● | 連邦消費者保護·不正競争法は、市場活動や消費者を損なう可能性のある活動を広く規制している。 |
21
さらに、他にも、上記の各医療保険法の州および海外等価物の制約を受けており、いくつかの法律の範囲は、支払者が誰であるかにかかわらず、より広い可能性がある。アメリカの多くの州は連邦反リベート法規のような法律を通過しており、その中のいくつかの法律は政府支払人だけでなく、個人保険会社を含む任意の出所から精算された医療サービスを得るのに適している。そのほか、いくつかの州はすでに法律を通過し、製薬会社に2003年4月の総監察長コンプライアンス計画 製薬メーカーガイドライン及び/或いはアメリカの製薬研究とメーカーが医療保健専門家と相互作用する基準 を遵守することを要求した。いくつかの州ではまた、他のマーケティング制限を実施したり、製薬会社に州政府へのマーケティングや価格開示を要求したりしている。このような州の要求を守るために何が必要なのかは曖昧で、もし私たちが適用された州の法律要求を遵守できなければ、私たちは処罰を受けるかもしれない。最後に,健康情報のプライバシーや安全を管理する州や外国の法律もあり,その多くの法律は互いに大きく異なり,HIPAA に先制されず,コンプライアンス作業を複雑化することが多い。
これらの法律の広さおよび法定例外および安全港が利用可能な狭いbrのために、私たちのいくつかのビジネス活動は、1つまたは複数のそのような法律の挑戦を受ける可能性がある。
詐欺および法律違反行為は、懲罰、罰金、監禁および/または連邦および州医療保健計画(例えば、MedicareおよびMedicaid)の排除または一時停止、および米国政府との契約の禁止を含む刑事および/または民事制裁を受ける可能性がある。また,個人は連邦FCAおよびいくつかの州の虚偽クレーム法に基づいて米国政府を代表して訴訟を起こす能力がある。
法執行部門は詐欺と法の乱用をますます重視しており、私たちのいくつかの接近はこのような法律の挑戦を受けるかもしれない。私たちの現在と将来の第三者との業務配置と、私たちの業務が全体的に適用される医療法令に適合するようにする努力は、多くのコストに関連しています。政府当局は、私たちと医師および他の医療保健提供者との手配を含む我々の業務慣行を結論する可能性があり、その中の一部の人は、サービスを提供する補償として株式オプションを取得し、現在または将来的に詐欺および乱用の適用に関連する法律、法規、機関指導または判例法(Br)または他の医療保健法律および法規に適合しない可能性がある。もし私たちにこのような訴訟を提起した場合、私たちは私たちの権利を弁護したり、維持することに成功しませんでした。これらの訴訟は、民事、刑事および行政処罰、損害賠償、返還、罰金、監禁、Medicare、Medicaidおよび他の連邦医療保健計画から除外される可能性があり、契約損害、名声損害、利益および将来の収益の減少、および私たちの業務を削減することを含み、これらはいずれも私たちの業務運営能力および運営結果に悪影響を及ぼす可能性があります。また,我々の任意の候補製品の米国国外での承認と商業化は,上記の医療法の外国等価物 や他の外国法律の制約を受ける可能性もある。
もし私たちがそれと業務を展開する任意の医師や他の医療提供者や実体が法律に適合していないことが発見された場合、彼らは政府の援助された医療計画から除外されることを含む刑事、民事または行政制裁を受ける可能性があり、これはまた私たちの業務に悪影響を及ぼす可能性がある。
米国連邦反リベート法規と非常に類似しており、EUも医師に医療製品の購入、供給、注文または使用を誘導または奨励するために、医師に福祉または利点を提供することを禁止している。医師に福祉または優遇を提供することは、主に加盟国の国家反賄賂法によって管轄されており、例えばイギリスの2010年の“反賄賂法”や国の反賄賂条項(フランス、ベルギーなど)を受けている。このような法律に違反することは巨額の罰金と監禁につながるかもしれない。いくつかの会員国では、医者に支払われる費用は公開されなければならない。さらに、医師との合意は、通常、医師の雇用主、その主管する専門組織、および/または個別加盟国の規制機関によって事前に通知され、承認されなければならない。これらの要求は、加盟国に適用される国家法律、業界規範、または専門行動基準 に規定されている。これらの要求を守らないことは、名声リスク、公開非難、br行政処罰、罰金または監禁につながる可能性がある。
22
法規を付加する
これらの規定に加えて,環境保全や有害物質に関する州や連邦法は,“職業安全と健康法”,“資源節約と回収法”,“有毒物質制御法”を含めて,我々の業務に影響を与える。これらの法律と他の法律は私たちの様々な生物、化学、放射性物質の使用、処理、処分を規範化しており、これらの物質は私たちの業務で使用され、廃棄物を発生させる。もし私たちの運営が環境汚染を招いたり、個人を危険物質に暴露したりすれば、私たちは損害賠償と政府罰金の責任を負う可能性があります。私たちは私たちが適用される環境法律を実質的に遵守し、これらの法律を遵守し続けることは、私たちの業務に実質的な悪影響を与えないと信じています。しかし、私たちはこのような法律の変化が私たちの未来の運営にどのように影響するか予測できない。
アメリカの“海外腐敗防止法”
米国の“反海外腐敗法”は、会社や個人が何らかの活動に従事して業務を取得または保留したり、公的な身分で働いている人に影響を与えたりすることを禁止している。外国の政府関係者、政府職員、政党または政治候補者に支払いを提案し、任意の価値のあるものを支払うことを提案し、業務を獲得または保留しようとするか、または公的身分で働く人に他の方法で影響を与えることは、不法である。同様の規則はフランスのような世界の多くの他の国にも適用される“意向書 薩平“)またはイギリス(イギリス“反収賄法”)。
アメリカの医療改革
アメリカの医療業界と他の地域の主な傾向の一つはコストを抑えることだ。政府当局や他の第三者支払者は,カバー範囲や特定医療製品の精算金額を制限することでコストを抑制しようとしている。例えば、2010年3月、ACAが公布し、他の事項以外に、 は医療補助薬品還付計画の下で大多数のメーカーが支払うべき最低医療補助税金還付を高めた;この方法に基づいて、吸入、輸液、点滴、インプラントまたは注射の薬品計算メーカーが医療補助薬品還付計画の下で不足している払い戻しを導入し、医療補助薬品還付計画を医療補助管理に参加する看護計画を使用する個人処方に拡大し、ある連邦医療保険D部分受益者に対して強制割引を行い、メーカーが連邦医療保険D部分で外来診察を受ける薬物の条件として;製薬会社の連邦医療保健計画における販売シェアに基づいて、薬品メーカーに新しい年会費を支払うことを要求した;新しい患者を中心とした結果研究所を創立し、監督、優先事項を確定し、そして の臨床有効性比較研究を行い、及びこのような研究に資金を提供した;そしてCMSでMedicare&Medicaid革新センターを創立し、革新の支払いとサービス交付モードをテストし、MedicareとMedicaidの支出を下げる。
公布以来,ACAは多くの の重大な変化が発生した。2021年6月17日、米国最高裁はACAに対する最新の司法挑戦を却下したが、ACAの合憲性を具体的に裁くことはなかった。米国の最高裁が裁決を下す前に,総裁·バイデンはACA市場によるbr}医療保険の取得を目的とした2021年2月15日から2021年8月15日までの特別保険期間を開始する行政命令を発表した。行政命令はまた、特定の政府機関に、医療の獲得を制限する既存の政策や規則を審査し、再考するよう指示した。最近、総裁·バイデンは2021年3月11日に“2021年米国救援計画法案”に署名し、2024年1月1日から法定の医療補助薬品還付上限を廃止し、現在この上限は薬品メーカーの平均価格の100%である
23
また、2011年の“予算制御法案”と2015年の“両党予算法案”は、各年度に医療保険提供者に支払う医療保険支出総額を2%減少させ、この削減は追加の国会行動を取らない限り2030年まで有効となる。また、2013年1月2日、米国納税者救済法 が法律に署名し、病院、画像センター、癌治療センターなどいくつかの提供者に支払う医療保険を減らし、政府が提供者に多額の支払いを取り戻す訴訟時効を3年から5年に延長した。最近,政府はメーカーがその市場製品に価格を設定する方式をより厳しく審査し,最近のいくつかの国会調査や提案された法案を招き,製品定価の透明性を高め,定価とメーカー患者計画との関係を審査し,br政府計画薬品精算方法を改革することを目的としている。2022年8月、“インフレ率低減法案”はMedicare がある高支出、単一源のMedicare B部分或いはD部分の薬物交渉薬品価格を許可した。アメリカ各州もますます積極的に立法と法規を実施して、価格或いは患者の精算制限、割引、ある製品への参入とマーケティングコスト開示の制限、透明性措置を含む薬品の定価を制御し、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。
私たちは将来、より多くの外国、連邦、州医療改革措置を取ることが予想され、そのいずれも連邦と州政府が医療製品とサービスのために支払う金額を制限する可能性があり、これは限られたカバー範囲と精算、そして私たちの製品に対する需要の減少を招く可能性があり、br}が承認されると、あるいは追加の価格設定圧力になる可能性がある。
保証と精算を請け負う
私たちが規制部門の許可を得たどの製品の保証範囲も清算状態には重大な不確実性がある。アメリカでは、化粧品は通常保険と精算資格を満たしていないため、化粧品として販売されている製品は保険を受けたり精算したりすることはありません。米国や他の国·地域の市場では、規制部門の承認を得て商業販売のための任意の製品の販売は、保証範囲の可用性および第三者支払者の精算にある程度依存する。第三者支払者には、政府当局、管理されている医療サービス提供者、個人健康保険会社、その他の組織が含まれる。支払者が製品に保険を提供するかどうかを決定するプロセスは、支払者が製品のために支払う支払率を設定するプロセスと分離される可能性がある。第三者支払者(Br)は、承認されたリストまたは処方表上の特定の製品に保証範囲を制限することができ、特定の適応のためのすべてのFDAによって承認された製品を含まない可能性がある。もし第三者支払人が私たちの製品を保証しないと決定したら、承認されると、医者の私たちの製品に対する使用を減らし、私たちの販売、運営結果と財務状況に重大な悪影響を与える可能性があります。また、支払人が製品に保険を提供することを決定することは、十分な販売率を承認することを意味するものではない。製品開発投資の適切なリターンを実現するために、十分な価格レベルを維持することができるように、十分な第三者精算が得られない可能性がある。
また,製品 の保証範囲と精算範囲は支払人によって異なる。第三者支払者は、特定の医療製品またはサービスを保証することを決定し、他の支払者もその医療製品またはサービスに保険を提供するか、または適切な販売率で保険 を提供することを保証することができない。
したがって,保証範囲決定プロセス は,支払者ごとに我々の製品を用いた科学的かつ臨床的支援を提供することが求められ,時間のかかるbrの流れとなる。
第三者支払者は、安全性と有効性に加え、価格や医療製品やサービスの医療必要性と費用対効果にますます多くの挑戦をしています。 任意の製品の保険や精算を獲得し維持するためには,その製品の医療必要性と費用効果を証明し,規制承認を得るのに必要な費用を証明するために高価な臨床試験を行う必要があるかもしれない。第三者支払者が、ある製品が他の利用可能な療法と比較して費用対効果がないと考えている場合、彼らは、その製品をその計画下の福祉brとしてカバーしないかもしれない、または、支払いレベルが、会社がその製品を販売するのに十分ではないと考える場合がある。
24
米国以外の多くの国/地域では,薬品brの定価が政府によって規制されている。例えば、EUでは、価格設定と補償プログラムは会員国によって異なる。一部の国·地域では、価格を精算することを合意した後にのみ、製品を販売することができると規定されている。一部の国は追加の研究を完成し、特定の治療法の費用効果を現在利用可能な治療法またはいわゆる衛生技術評価と比較して、精算または定価の承認を得ることを要求するかもしれない。他のbr国/地域は会社が自ら製品価格を決定することを許可する可能性があるが,製品数を監視し,医師にガイドラインを発表して処方数を制限する。各国が医療支出を管理しようとしていることに伴い,薬品や医療機器の価格や使用を抑える努力が継続する可能性がある。
データプライバシーとセキュリティ法
多くの州、連邦と外国の法律は、消費者保護法律と法規、個人情報の収集、伝播、使用、アクセス、秘密と安全を管理し、健康に関連する情報を含む。米国では、データbr}違反通知法、健康情報プライバシーおよびセキュリティ法律(HIPAAを含む)および連邦および州消費者保護法(例えば、連邦貿易委員会法案第5条)を含む健康および他の個人情報の収集、使用、開示および保護を管理する多くの連邦および州法律法規が、私たちの運営または私たちのパートナーの運営に適用される可能性がある。また、“カリフォルニア消費者保護法”、“カリフォルニアプライバシー法”、“一般データ保護条例”(GDPRと略す)のような州および非米国の法律は、場合によっては健康関連情報を含む個人情報のプライバシーやセキュリティを管理しており、その中のいくつかはHIPAAよりも厳しく、その多くは互いに大きく異なり、同じ効果を生じず、コンプライアンス作業を複雑化させる可能性がある。これらの法律を遵守しない場合は、重大な民事及び/又は刑事罰及び私的訴訟に処せられる可能性がある。プライバシーやセキュリティ法律、法規、その他の義務は絶えず変化し、互いに衝突し、コンプライアンス作業を複雑化させ、調査、訴訟または行動を招き、それによって重大な民事および/または刑事罰やデータ処理制限を招く可能性がある。
材料協定
許可協定
業達との許可協定
2015年6月22日、BiomX Ltd.は、Yedaと研究および許可協定、または修正されたYeda 2015ライセンス協定を締結し、BiomX Ltd.は、微生物グループに基づく候補治療製品の開発、試験、製造、生産および販売に関連するいくつかの独自技術および研究情報のグローバル独占許可brを取得し、プロトコルで指定された候補製品、我々のバクテリオファージ発見プラットフォーム、および特許のための候補製品を含む。コンサルタントの仕事によるバクテリオファージ候補製品の研究と他の権利はプロトコルでbrを決定し,BiomX株式会社が援助したWISでさらなる研究を行った。
本ライセンスについては,BiomX Ltd.は年間10,000ドルの払戻不可能な許可料を支払うことに同意した。また,BiomX株式会社は2015年のライセンス契約で合意された研究予算に合計約200万ドル を貢献した。BiomX Ltdはまた、Yeda 2015ライセンスプロトコルによってカバーされる製品および診断キットの純売上に、より低い1桁のbr}ビットの階層印税を支払い、許可プロトコルに記載された減額で支払うことを要求される。brライセンスプロトコルに含まれる製品および診断キットは、CFに対する製品およびバクテリオファージベースの療法および関連技術プラットフォームによる治療の可能性のある任意の他の適応を含む。BiomX株式会社がYEDA 2015ライセンス契約に従ってその権利を再許可する場合、BiomX株式会社は、受信したプロトコルに記載された再許可受領書の割合で表される10代から25歳までの範囲で、YEDAに追加の再許可使用料を支払う義務があるであろう。BiomX Ltd.は、YEDA 2015ライセンス契約に従って許可された特許の出願および維持費用 を支払う義務がある。2015年のライセンス契約について、BiomX Ltd.もいくつかの普通株を発行し、これらの普通株はその後、業務合併の一部として193,406株に転換された。もし私たちがいくつかの合併と買収に参加した場合、私たちは取引によって受け取った対価格の約1%に相当する金額を業界に支払う義務がある。
いずれか一方が早期に終了しない限り、付与されたライセンスは、その製品が国/地域の最後のライセンス特許(2039年の満了予定)の満了が遅くなるまで、各国/地域およびライセンスに従って開発された各製品において有効に維持され、製品が国/地域で初めて商業販売される日から11年となる。YEDA 2015ライセンス契約は、プロトコルがカバーする最後の特許の満了と、どの国/地域でも任意の製品の最初の商業販売が連続して15年以内に行われない期間(遅い者を基準とする)で終了する。BiomX Ltd.がYEDA 2015年ライセンス契約に記載されているいくつかの職務調査および開発要件およびマイルストーンを遵守できなかった場合、YEDAもプロトコルを終了する可能性がある。BiomX株式会社またはYEDAは、通知期限後、または他方の清算、破産、債務不履行、解散、または他の同様の業務の終了後に、YEDA 2015年のライセンス契約を終了することができる。“Yeda 2015ライセンスプロトコル”の終了後、BiomX Ltd.は、時間の経過に加えて、Yeda 2015ライセンスプロトコルに記載された独自技術および研究成果に対する我々の権利に関するYedaに、Yedaがその後、私たちの権利の使用を許可する第三者に付与される場合、BiomX Ltd.は、Yedaがその後、私たちの権利を使用する第三者に許可を与える場合、BiomX Ltd.は、Yedaがこの許可によって実際に受け取った純利益を共有する権利を有することを前提としている。BiomXと業界達2015ライセンスプロトコルに基づく開発費用の上限 に制限されている。
25
BiomX株式会社は特許の起訴と維持決定についてYedaと相談した。YEDAは主にライセンス情報 (ライセンス中の定義のような)に関連する起訴および保守を担当し、私たちは後続の結果(例えば、br}ライセンスの定義)に関連する起訴および保守を担当する。BiomX Ltd.とYedaは相談権を得る権利がある。BiomXはすべての特許と出願の起訴と維持に関連した費用を担当している。
YEDAの承認を経て,BiomX株式会社はライセンス項下の特許権を行使する権利がある。Yedaは訴訟に参加することを選択することができますが、私たちは訴訟に関連するすべての費用を担当しています。 私たちがYedaに可能な訴訟を最初に通知した後、私たちがYedaに通知しなければ、私たちは権利を強制的に執行したり、訴訟を提起しようとしています。Yedaは自分で訴訟を提起する権利を保持しています。
アメリカ海軍独占許可証
2017年3月16日、APTはアメリカ合衆国と独占許可(2019年1月10日改正、またはUSN許可協定、海軍またはUSN長官代表)を締結し、この協定によると、APTは、“特定細菌に対するバクテリオファージ組成物および成分選択方法”またはUSN許可特許、および約350個のバクテリオファージ(またはUSNライセンス特許、USN材料と総称する)を含む関連材料を含む米国、カナダ、ヨーロッパを含む領土全体の独占許可を取得した。工業的または医療的使用を含む、すべての用途の治療および/または除去のための多剤耐性細菌。USNライセンスプロトコルによれば、APTは、USNライセンス特許が、2022年12月31日までに商業開発計画に規定されたマイルストーンに一致するライセンス発明を実用化するために、USNライセンス特許が発明において要求または開示されるかもしれない発明を開発およびマーケティングするために、商業開発計画または商業開発計画を実行することに同意し、その後、USNライセンス契約の残りの時間内に、ライセンス発明の利点を合理的に公衆に提供し続ける。ライセンス期間内に、任意のライセンス発明またはライセンス発明を使用して米国で使用または販売することによって製造された製品は、実質的に米国で製造されなければならない。同社はUSNライセンスプロトコルに関連するbrバクテリオファージをそのバクテリオファージ治療のために開発した潜在的バクテリオファージ源として用いている。
USNライセンスプロトコルについては,APT はUSNに5,000ドルのライセンス実行使用料を支払った.また、(I)USNライセンス特許の任意の請求項によって定義される、またはUSNライセンス特許の任意の請求項によって定義される組成物、(Ii)ライセンス発明において要求される方法によって製造された、(Iii)USN材料に基づく、または(Iv)USNライセンス特許に見つからないUSNによって作成された情報br}または(Iv)DNA配列データを含むUSNライセンス特許に基づいて生成された情報br}または(Iv)ベース、または(Iv)DNA配列データを含む製品または印税含有製品の正味売上高に1桁単位の使用料を支払う必要がある。ライセンス発明に関連する臨床試験データおよび詳細な実験室方法。
APTは2018年から2020年まで毎年最低5,000ドルの印税を支払うことに同意し、その後20,000ドルを支払うことに同意した。APTはまた、FDA承認を受けて180日以内に(A)許可された特許権使用料を支払い、特許権使用料を有する製品を販売し、(B)ある収入の閾値に達したときに、100,000ドル以下の記念碑的特許権使用料 を支払うことにも同意する。また,任意の印税製品から再許可されたすべての収入の割合を25%程度の割合で表すことに同意した.
私たちはUSNライセンス特許を統制して勤勉に起訴し、アメリカと外国の管轄地域でUSNライセンス特許の起訴と維持に関連するすべての費用を支払う責任がある。2021年1月1日までに年次進捗報告を提出することに同意し,許可発明の実用化のための努力を説明し,その後このような実用を受けるまでである。
(I)USNが“商業開発計画”を実行していないと判断した場合、(Ii)USNが“USNライセンス契約”を終了することが必要であると判断した場合、(I)USNライセンス契約の日後に発行され、合理的に満たされていない公共使用要件を満たすために、(Ii)USNライセンス契約の終了後120日後に“USNライセンス契約”を終了することが必要であると判断した場合、(Iii)“USNライセンスプロトコル”の申請またはそれによって要求された任意の報告において、意図的に重大な虚偽の陳述や重大な事実を見落とした場合、 または(Iv)私たちはUSN許可協定に深刻に違反し、書面通知後30日以内に修復されませんでした。
26
ウォルター·ヨシ陸軍研究院と許可協定を締結しました
APTは、2021年8月24日、Walterヨシ陸軍研究所(WRAIR)と生体材料許可協定(または2022年8月31日に修正されたWRAIR許可協定)を締結し、この協定によれば、APTは、治療/予防のためのバクテリオファージ製品を開発および商業化するために、約100個のバクテリオファージまたはWRAIR材料を含む特定の材料および情報の非独占的な世界的許可を得た緑膿菌, バウマンアシネトバクター, 黄色ブドウ球菌, 肺炎クレブシエラ外傷や尿路感染大腸菌.大腸菌そして膣腸桿菌 細菌が感染する。同社は、そのバクテリオファージ治療法の潜在的な源として、WRAIR許可プロトコルで提供されたバクテリオファージを使用している。
WRAIR許可プロトコルについては,APTはWRAIRに数千ドル程度の初期実行費を支払い,毎年数千ドル程度の保守費 を支払うことに同意した.WRAIR材料またはWRAIRライセンス製品を含む製品の純売上高には、より低い桁数パーセントで表される印税を支払う必要があるが、WRAIRライセンスプロトコルの説明に従って減少する必要がある。また、WRAIR許可プロトコルに従って私たちの権利を再許可する場合、任意の再許可使用料から取得した再許可領収書の10分の1の割合で表すために、追加の再許可使用料をWRAIRに支払う義務があります。また、追加の印税 は、期限を過ぎて支払われた印税に基づいて評価することもできる。
WRAIRに年次進捗報告 を提出する義務があり、WRAIR許可プロトコルによって許可された任意の発明を実用化するための努力、 およびWRAIRが要求するまたは開発計画の予想または要求された任意の他の情報を詳細に説明する。WRAIRライセンスプロトコルでのbr表現の一部として,WRAIRライセンスプロトコルの発効日から4年間にWRAIRライセンス製品を用いて臨床試験中の1人目の患者に用量 を行うことに同意した。
本ライセンス契約の一部として、WRAIRがWRAIR材料および/またはその使用に関連する非一時的特許出願を提出した場合、WRAIRは、特許許可の必要性および/または取得可能性を評価することを我々およびWRAIRに通知する義務がある。この場合,非排他性や排他的許可を交渉する優先拒否権 を持つ.
WRAIRライセンスプロトコルは、各WRAIR材料について、その条項に従って早期に終了しない限り、WRAIRライセンスプロトコルにこのようなWRAIR材料を追加した日から10年以内に満了する。私たちは60日間の書面通知後にWRAIR許可協定を終了することができ、もし私たちが約束を違反し、書面で違約を通知してから90日以内にその違約が救済されなかった場合、WRAIRは終了する可能性があります。
従業員
2023年12月31日現在、58人のフルタイム従業員と13人のアルバイト従業員がいます。私たちの21人の従業員は博士または医学博士号を持っており、53人の従業員は現在研究開発と臨床活動に従事している。私たちの従業員たちは労働組合代表もなく、集団交渉合意の範囲 もない。私たちは私たちと従業員との関係がしっかりしていると思う。
企業情報:
私たちの最高経営責任者事務室の郵送先はイスラエル7414003ネスツィオナ4階アインシュタイン通り22番地で、電話番号は(972)72-394-2377です。私たちの会社のサイトの住所はwww.bibix.comです。当ウェブサイトの内容は、本年度報告または私たちが提出した任意の他の報告または文書に引用的に組み込むことを意図しておらず、これらのウェブサイトへの任意の参照は、非アクティブなテキスト参照としてのみ使用される。
27
第1 A項。リスク要因
私たちの証券に投資する前に、以下に説明するリスクと不確実性、本年度報告書の他の情報をよく考慮しなければなりません。上記のいずれかのリスクが発生した場合、私たちの業務、財務状況、運営結果または見通しは実質的な悪影響を受ける可能性がありますので、私たち証券の市場価格は下落する可能性があり、あなたはすべてまたは一部の投資を損失する可能性があります。本年度報告書にはまた、リスクと不確定要素に関する前向きな陳述が含まれている。“前向き陳述に関する警告声明”を参照してください。 いくつかの 要因のため、以下の陳述を含むため、私たちの実際の結果は、これらの前向き陳述で予想される結果とは大きく異なる可能性があります。
私たちのビジネス、技術、業界に関連するリスク
我々は臨床段階の会社であり,運営歴史は限られており,設立以来赤字が続いている。私たちは引き続き巨額の費用を招くことを予想し、予測可能な未来には引き続き重大な損失を招くだろう。
私たちは臨床段階の生物製薬会社で、運営の歴史は限られている。2015年にBiomX株式会社Sが設立されて以来、私たちは毎年赤字を出しています。2023年12月31日現在、私たちの累計赤字は1.63億ドルで、予測可能な未来にますます大きな損失が出ることが予想されます。臨床前開発と臨床br試験と活動コストが高い。私たちはすでに予測可能な未来にほとんどの資源 を投入して、私たちの候補製品の研究開発と臨床試験を行っている。私たちは短期的に私たちの候補製品の商業販売から何の収入も得られないと予想しています。また、今回の買収により、我々の将来の業務、見通し、財務状況、経営業績は、歴史的時期やわが経営陣が予測している状況と大きく異なる可能性があります。
2023年12月31日と2022年12月31日までの年度、私たちの運営損失はそれぞれ2530万ドルと2720万ドルです。最近APTを買収したため、私たちの費用レベルは増加すると予想されています
| ● | 未来の候補製品のために研究、臨床前と臨床開発を開始し、継続する |
| ● | より多くの候補製品を発見し開発し、私たちの臨床製品ラインをさらに拡大することを求めた |
| ● | 臨床試験に成功した候補製品のためにマーケティングと監督管理の承認を求める |
| ● | より多くの臨床開発候補製品の生産が要求され、商業化が可能である |
| ● | 私たちの知的財産権の組み合わせを維持し、拡大し、保護する |
| ● | 臨床、品質管理、科学者のようなより多くの人員の募集と保留を含む、私たちの研究開発インフラを拡大する |
| ● | 将来的に販売、マーケティング、流通、その他の商業インフラを構築して、上場許可を得た製品を商業化する |
| ● | 我々の製品開発·商業化を支援する者を含め、運営、財務、管理情報システム及び人員を増加させ、上場企業としての義務を履行するのを支援する。 |
28
私たちは将来的に私たちの業務を支援するためにより多くの資本を集める必要があります。これらの業務は私たちに有利な条項では得られない可能性があり、私たちの株主が大幅に希釈したり、第三者への債務を増加させたりする可能性があります。
2023年12月31日まで、私たちは現金、現金等価物、制限現金1,590万ドルを持っています。設立以来、私たちは運営の経常赤字と負の運営キャッシュフローが存在しています。将来、私たちは私たちの運営と製品開発活動を支援するためにより多くの資金を集める必要があります。短期的には、私たちが持っている現金、政府、その他の贈与、将来の株式や債務融資から、私たちの運営や他の候補製品に関連する開発活動に資金を提供し続ける予定です。また,2023年12月7日にH.C.ウェインwright&Co.,LLCやウェインライトと管理人として“市場発売プロトコル”や“ATMプロトコル”を締結し,この合意により,総発行価格750万ドルに達する普通株を随時Wainwrightで発行·販売することができる.ATM協定によると、私たちは普通株を売る義務がありません。2023年5月4日、会社の株主の許可を得て、会社は2023年2月のパイプの2回目の閉鎖を完了し、600万ドルの毛収入を追加した。2023年12月7日、2024年1月2日に米国証券取引委員会によって発効が宣言されたS-3表の棚上げ登録声明を提出した。また、買収を完了すると同時に、私たちは2024年3月15日に1つの私募、即ち2024年3月のパイプを完成し、証券法の免除登録要求とある投資家 に基づいて、このような投資家は私たちのXシリーズ無投票権転換可能優先株を購入し、1株当たりの額面価値は0.0001ドル、あるいは転換可能な優先株、あるいは転換可能な優先株、あるいは株式承認証、あるいは私募株式権証を購入して、合計108,208,500株会社の普通株を購入し、総収益は約5,000万ドルである。1株当たり転換可能優先株 は合計1,000株の普通株に変換できる。
2024年3月のPIPEの制限を受けて、私たちはATMプロトコルに従って株を売却し続け、他の方法で私たちの保留登録声明を使用して、時々追加のbr資金を調達することができます。私たちはまた2023年2月と2024年3月のパイプラインプロジェクトで私たちがしたように、個人的に資金を調達するかもしれない。私たちはまた、協力者や他の人との手配を通じて資金を求めることができ、これらの手配は、候補製品の権利を放棄することを要求するかもしれません。そうでなければ、私たちは独立した開発または商業化を求めることができます。
もし私たちが以前の開発段階で1つ以上の現在または未来の候補製品の協力に参加した場合、このような協力の条項は、私たちがより後の段階で協力に参加するよりも、または製品を独立して商業化する場合には及ばないかもしれない。もし私たちが株式発行によって追加資金を調達した場合、これらの証券の条項は清算または他の特典を含み、私たちの株主の権利に悪影響を与え、あるいは私たちの株主に深刻な希釈をもたらす可能性がある。もし私たちが債務融資を通じて追加資本を調達する場合、それは固定支払義務の制約を受け、追加債務を招く、資本支出を招く、配当金を発表する、または買収することができるかもしれない知的財産権のような特定の行動をとる能力を制限または制限する契約の制約を受ける可能性がある。
薬物の開発や臨床試験の費用が高い。私たちの未来の支出需要は多くの要素に依存するだろう
| ● | 私たちの研究開発と臨床活動のコスト、時間、進行度 |
| ● | 私たちのターゲットバクテリオファージ、治療戦略、その他の研究開発活動に関連する製造コスト |
| ● | 私たちが構築することができる任意の協力、許可、買収、または他の計画の条項と時間 |
| ● | 従業員に関する費用、外部コンサルタントに支払う費用などの外部費用 |
| ● | 規制承認を求めるコストと時間と、規制要件の遵守に関連するコストと時間; |
| ● | 任意の特許出願、請求項、特許、および他の知的財産権の提出、起訴、弁護、および実行費用。 |
国内·国際経済状況と懸念に基づいて、国内·国際株式·債務市場 は高度な変動と動揺を経験し続けている可能性がある。これらの経済状況や懸念が持続または悪化し、市場が変動し続けたり、米国の株式市場がその後熊市や衰退したりした場合、市場はイスラエル-ハマス戦争、ロシアのウクライナ侵攻、それによる世界のロシア、ベラルーシ、関係者への制裁や他の地政学的不確実性や不安定源などのマイナスの影響を受け、私たちの経営業績や流動性は、必要に応じて資金を調達しにくくし、株価が下落する可能性があるなど、多くの面で不利な影響を受ける可能性がある。
29
必要なときや受け入れ可能な条項(あれば)で十分な資金 を提供することは保証されない.私たちがもっと多くの資金を得ることができないことは、私たちの業務、財務状況、そして運営結果に大きな悪影響を及ぼすかもしれない。また、もし私たちがbrに基づいて適時に追加資金を得ることができなければ、私たちが経営を続けている企業として経営を継続する能力は大きく疑われ、倒産リスクが増加し、私たちの株主の投資は完全に損失するだろう。
私たちの財務諸表には、持続的な経営企業としての私たちの持続的な経営能力に対する深刻な疑いに関する説明段落が含まれており、これは、合理的なbr条項や新たな融資を得ることができないことを阻止するかもしれません。
私たちの財務諸表には、持続的な経営企業としての継続的な経営能力に対する深刻な疑いを説明する解釈段落が含まれています。我々が継続的に経営している企業として継続できるかどうかには大きな疑問があると結論した。設立以来、私たちは1.63億ドルの赤字を蓄積してきました。これまで、私たちはまだ私たちの運営から収入を生み出していません。今後12ヶ月以内に製品販売から顕著な収入 は発生しないと予想されています。予測可能な未来に、私たちの現金需要は増加するかもしれない。2023年12月31日現在、私たちは1,590万ドルの現金 と現金等価物を持っています。
私たちの手元の現金と現金等価物および短期預金は、少なくとも12ヶ月の運営資本と資本支出要件を満たすのに十分だと信じています。しかし、私たちの株主が2024年3月のパイプラインや買収に関連する転換可能な優先株の転換を承認しないリスクがあるため、現金決済転換可能優先株を要求される可能性があり、2024年4月3日から少なくとも12ヶ月間継続的に経営し続けることができるかどうかには大きな疑問がある。私たちの持続的な経営企業としての持続的な経営は多くの要素に依存しており、私たちは5ヶ月以内に株主の承認を得て転換可能な優先株を転換する能力、追加資金を調達する能力、私たちのCF臨床試験の成功、APT業務の統合に成功する能力、および満期時に債務を返済する能力を含む。私たちは私たちが未来のいかなる資金も得ることができるかどうかを確認することはできません。私たちが獲得する可能性のあるこのような資金は私たちの運営に資金を提供するのに十分ではないかもしれません。もし私たちが十分な資金を得ることができなければ、私たちは経営を続けることができないかもしれない。
APTに対する私たちの買収が株主価値を増加させる保証はない。
2024年3月、私たちは買収でAPTを買収しました。 買収や関連取引の実施が株主価値を損なわないことや、他の方法で私たちの業務に悪影響を与えないことを保証することはできません。今回の買収は、私たちの業務と管理チームの間の統合挑戦を招く可能性があり、これはbrの管理と業務中断を招く可能性があり、そのいずれも私たちの運営結果と業務の将来性に影響を与え、このような買収の私たちの株主に対する価値を損なう可能性がある。
私たちは株主が買収で発行した転換可能な優先株と引受権証および2024年3月のパイプラインの転換を承認するために合理的な最善を尽くす必要がある。もし私たちが転換可能優先株の初回発行後150日以内に承認されなければ、私たち は現金決済転換可能優先株を要求される可能性があります。
合併協議によると、吾等は株主総会(“株主総会”)を開催し、株主承認を取得するために(I)“ニューヨーク証券取引所米国証券市場規則”に基づいて、交換可能優先株転換及び行使承認証(以下、定義を参照)を普通株式株式(普通株既発行株式の19.9%を超える)、(Ii)新株 奨励計画又は改訂当社現行の株式奨励計画(“2024奨励計画”)を採択し、及び(Iii)必要があれば、交換可能株の転換及び株式承認証の行使のために、十分な追加の普通株式を保有することを許可するために、当社の会社登録証明書を改訂する。株主の承認を受けていない場合には、承認されるまで少なくとも90日ごとに追加株主会議を開催する必要があり、大量のコスト を招き、経営陣の注意を分散させる可能性がある。また、転換可能優先株の初回発行後150日以内に転換可能優先株を承認していない場合には、転換可能優先株保有者の70%の書面請求を保有すべきであり、転換可能優先株指定証明書に記載されているように、転換可能優先株保有者毎に、変換可能優先株の公正価値に相当する現金金額を支払うように要求される。私たちは必要であれば、大量の転換可能な優先株を決済するのに十分な流動資金を持っていないと予想している。現金和解は私たちのコントロール下ではなく、私たちが経営を続けている企業として継続する能力に大きな疑いを抱かせた。
30
バクテリオファージ技術を用いた候補製品の開発が求められており,開発時間やコストを予測することは困難な方法である。本年度報告日までに,これまで米国やEUで薬物として承認されているバクテリオファージはないことが知られている。
私たちはバクテリオファージ技術を使って私たちの候補製品を開発している。私たちはいません。私たちが知っている限りでは、この方法に基づく製品候補に対するFDAや同様の外国の規制機関の規制承認を得ている他の会社もありません。そして当を受ける体外培養そして体内にある細胞培養や動物モデルにおけるバクテリオファージの行動が特徴化されており,ヒトにおけるバクテリオファージ療法の使用に関する多くの文献があり,ヒトにおけるバクテリオファージ療法の安全性と有効性は良好に制御された現代臨床試験では広く研究されていない。以前のバクテリオファージ療法の研究の多くは第二次世界大戦前と第二次世界大戦後の旧ソ連で行われており、適切な対照群の設計が不足しているか、あるいは対照群が全くない。また,これらの研究が行われて以来,次の数十年間で看護基準が大きく変化し,治癒率向上に関する先の説との関連性が弱まってきた。著者らが開発した任意の候補製品brは、実験室および他の臨床前研究においてそれらの治療特性を患者に示すことができない可能性があり、それらは予測不可能、無効、さらには有害な方法でヒト生物システムと相互作用する可能性がある。私たちの方法 が承認可能または販売可能な製品の開発につながることは確認できません。また,時間の経過とともにバクテリオファージの細菌標的が候補製品に耐性を持つ可能性があり,新たなファージカクテルを開発することで克服できない可能性もあり,あるいは標的病原体世界を十分にカバーしたカクテルを構築できない可能性がある。
我々の候補製品が規制部門の承認を得ているが,医師,医療支払者,患者の十分な承認が得られていなければ,利益を達成するのに十分な製品収入が生じない可能性がある。私たちの成功は、私たちが薬物の候補製品として私たちが対象とする疾患を専門的に治療する医師に依存し、彼らがよりよく知っており、より多くの臨床データが得られる可能性のある既存の治療レジメンを代替または補充するために、私たちの候補製品を使用することに関する潜在的治療レジメンを開発するであろう。私たちの成功はまた消費者の私たちの商業化製品の受け入れと採用にかかっている。我々の候補製品の臨床前研究および臨床試験における有害事象、または類似製品を開発する他の臨床試験における有害事象、およびそれによる宣伝、およびバクテリオファージ療法分野の任意の他の有害事象は、我々が開発する可能性のある任意の製品に対する需要の減少を招く可能性がある。承認された製品に対する市場の受け入れ度は、複数の要因に依存する
| ● | 製品の有効性 |
| ● | 副作用の流行率や重症度は |
| ● | 代替療法と比較して潜在的な優位性や劣勢 |
| ● | 相手が便利で管理しやすい |
| ● | 有力なマーケティングと流通支援 |
| ● | 製品の絶対価格と代替療法に対する価格; |
| ● | 十分な第三者が保険を受けたり精算したりします。 |
ビジネス規模で私たちの候補製品を開発するには大量の技術、財力、人材が必要になるだろう。私たちと第三者パートナーは、私たちの候補製品のための製造能力の開発に遅延がある可能性があり、効率的な臨床試験を行うために必要な規模に達しない可能性があり、規制機関の私たちの候補製品の承認を得たり、私たちの製品を生産するビジネス の数を得ることができません(承認された場合、または他の方法で発売が許可されている場合)。
31
私たちの候補製品は臨床試験を経なければならず、brは薬品に必要な安全性と有効性、あるいは生物製品の安全性、純度、効力を証明できない可能性があり、私たちの任意の候補製品は悪影響を招く可能性があり、これは規制部門の承認および/または商業化を大幅に延期または阻止するだろう。
私たちが規制機関の候補製品に対する承認を得るか、あるいは他の方法で製品を薬物或いは生物製品として発売することを許可する証拠を得る前に、私たちは安全性と有効性を証明するために、広範な臨床前のbrと人体臨床テストを行わなければならない、あるいは生物製品、安全性、純度と効力の場合、FDA或いは他の監督管理機関を満足させる必要がある。候補製品の臨床試験は監督管理機関の上場許可を得るのに十分であり、あるいは発売前に安全であることを証明し、費用が高く、完成するのに数年かかる。また,これらの臨床試験の結果は,我々の候補製品の安全性や有効性がさらなる開発を承認または保証するのに十分であることを示していない可能性がある。著者らの方法は、特定の病原菌菌株に対して、微生物群の組成を変化させ、患者に潜在的な治療或いは美容的利益を提供するために、バクテリオファージ組み合わせ或いはカクテルを設計することを目的としている。しかし,根絶選定の目標が潜在疾患に臨床的に意義のある影響を与える保証はなく,例えば疾患の病理 が不明な場合である。さらに、我々が対象とする細菌は、疾患に関連している可能性があるが、疾患の病因または原因因子ではない可能性があり、または候補製品が対象としていない他の細菌が存在する可能性があり、それらは潜在的な疾患のより意味のある駆動因子である。さらに、私たちの候補製品は、標的臓器または組織に到達するために有効な送達ツールを使用する必要があり、私たちの予期される送達システムが、私たちの候補製品が患者の所望の位置 に到達することを可能にすることを保証することはできない。安全性はまず臨床前試験と早期臨床試験を通じて確定しなければならず、それから治療効果を評価と確定することができ、それによってFDA或いはその他の監督管理機関の上場許可を得ることができる。私たちの臨床試験は不良な副作用または否定的または不確定な結果をもたらす可能性があり、私たちは決定または監督機関が追加の臨床および/または臨床前のbrテストまたは計画の放棄を要求するかもしれない。
持続的な地政学的不安定はbrに悪影響を与え,われわれの臨床試験を含めてわれわれの業務に悪影響を与え続ける可能性がある。
イスラエル-ハマス戦争やロシア-ウクライナ紛争のような地政学的不確実性と不安定さを含む世界的な経済、政治、人口、ビジネス状況は、私たちのサプライチェーンを間接的に中断し、私たちが受け入れられる条件で資金を調達する能力、その他の影響を損なう可能性がある。私たちはまた他の中断に直面するかもしれません。これは私たちの業務、臨床前研究、臨床試験に深刻な影響を与えるかもしれません
| ● | 患者を臨床試験に参加させるのは遅延したり困難です |
| ● | 臨床サイト起動の遅延または困難は、臨床サイト調査員と臨床サイトスタッフを募集する上での困難を含む |
| ● | 人員不足、生産減速または停止、および交付システムの中断のため、私たちの契約製造組織から私たちの候補製品の供給を受けることを中断または遅延した |
| ● | 私たちの元の発見と臨床活動を中断したり遅延させたりする。 |
32
治療適応のための候補製品を得るために必要な規制承認を得ることができない場合、候補製品を商業化することができない、または商業化の過程で遅延することになり、私たちの将来の創造能力は深刻な被害を受けるだろう。
我々の候補製品及びその治療適応の開発と商業化に関する活動は、その設計、テスト、製造、安全性、有効性、記録保存、ラベル、貯蔵、承認、広告、販売促進、販売、流通、輸出入を含み、すべてアメリカFDAと他の監督機関及び同等の外国監督管理機関によって監督されている。私たちが私たちの任意の治療適応製品を商業化することができる前に、私たちは市場の承認を得なければならない。私たちはまだどの司法管轄区の規制機関からもどの候補製品の発売の承認も得ていません。私たちの候補製品や私たちが将来開発を求める可能性のあるどの製品も規制部門の承認を得られないかもしれません。
米国や他の国/地域で治療適応の規制承認を得る過程は非常に高価であり,追加の臨床試験が必要であれば数年を要する可能性があり,様々な要因によって大きく異なる可能性があり,これらの要因は候補製品のタイプ,複雑性,新規性 を含む。開発中の市場承認政策の変更、追加法規または法規の変更または公布、または各提出されたINDまたは同等の申請タイプの規制審査の変更は、承認の遅延または申請 の拒否をもたらす可能性があります。FDAと同等の外国の監督管理機関は承認過程においてかなりの自由裁量権 を持っており、いかなる申請も拒否することができ、私たちのデータが承認を得るのに十分ではなく、追加の臨床前、臨床あるいは他の研究を行う必要があると決定することもできる。様々な理由で、私たちの候補製品は規制部門の承認を得るのが遅れたり、承認されなかったりする可能性があります。 には以下の理由が含まれています
| ● | FDAまたは同等の外国の規制機関は、人々、用量レベル、用量方案および生物分析分析方法、または私たちの臨床試験の実施を含む設計に同意しない可能性がある |
| ● | 候補薬がその提案の適応に対して安全かつ有効であること、または関連する随伴診断が適切な患者群を識別するのに適していることをFDAまたは同等の外国規制機関に証明することはできないかもしれない |
| ● | 臨床試験の結果は、FDAまたは同等の外国規制機関が承認を要求する統計的意義レベル、例えば私たちのざ瘡候補製品に適合しない可能性がある |
| ● | 候補製品の臨床的および他の利益がその安全リスクよりも大きいことは証明できないかもしれない |
| ● | FDAまたは同等の外国の規制機関は、前臨床研究または臨床試験データの解釈に同意しないかもしれない |
| ● | 私たちの候補製品の臨床試験から収集されたデータは、マーケティング申請または他の提出をサポートするのに不十分であるか、または米国または他の場所の規制承認を得るのに十分ではない可能性がある |
| ● | FDAまたは同等の外国規制機関は、私たちと臨床および商業用品契約を締結する第三者メーカーの製造プロセスまたは施設を承認できない可能性がある |
| ● | FDAまたは同等の外国規制機関の承認政策や法規は大きく変化する可能性があり、私たちの臨床データは承認を得るのに十分ではない。 |
大量に開発されている薬物のうち,一部のみがFDAや同等の外国規制承認手続きに成功し,商業化されている。長い承認過程および未来の臨床試験結果の予測不可能性は、監督部門の承認を得られず、その候補製品を市場に投入することができず、これは私たちの業務、運営結果、将来性を深刻に損なう可能性がある。
33
FDAはまた、諮問委員会と呼ばれる専門家グループが必要である可能性があり、安全性と有効性データが治療適応の承認を支持するのに十分であるかどうかを審議する。諮問委員会の意見は拘束力はないが、完成した臨床試験によって開発された任意の候補製品が承認される能力に大きな影響を与える可能性がある。EUでは,EMAは高級治療薬資格に適合する任意の候補製品の安全性と有効性データをEMA高度治療委員会で審査しなければならないと考えており,EMA委員会は高度治療薬専門家グループである。
また,米国のPREAとEUの小児科法規によると,FDAあるいは同等の外国規制機関は小児科人口での強制的な検出を要求することができる。米国またはEUで承認された出願は、すべての関連する小児科亜集団において生物学的製剤が主張する適応の安全性および有効性を評価し、製品が安全で有効な各小児科亜群の用量および投与をサポートするためのデータを含まなければならない。FDAまたは同等の外国の規制機関は、小児科対象者へのデータの全部または一部の免除または延期を適宜承認することができる。FDAが小児科患者のデータを必要とする場合、より多くの資金を投入して強制的な小児科臨床試験と研究を行わなければならないが、成人医療製品の承認は通常影響を受けるべきではない。もしこのような小児科研究の結果が肯定的でなければ、私たちの候補製品は子供のために承認されないだろう。
さらに、私たちが承認されても、br規制機関は、私たちの要求された治療適応以下またはそれ以上の治療適応のために、私たちの任意の候補製品を承認することができ、適切な患者集団の使用制限または禁忌症を含む可能性があり、私たちの製品のために徴収しようとしている価格を承認しないかもしれないし、高価な発売後の臨床試験の表現に依存するかもしれないし、または製品の商業化に成功するために必要または所望のラベル宣言の候補製品が含まれていない可能性がある。上記のいずれの場合も、私たちの候補製品のビジネス見通しに実質的な損害を与える可能性があります。
もし私たちが承認を得る上で遅延に遭遇した場合、あるいは私たちが候補製品の承認を得られなかったら、私たちの候補製品のビジネスの将来性が損なわれる可能性があり、私たちの将来の創造能力は深刻な損害を受けるだろう。
私たちは製品販売から何の収入も得たことがありません。br}は永遠に利益を上げないかもしれません。あるいは利益を達成しても、利益を持続できないかもしれません。
私たちが相当な収入を生み出し、利益を達成できるかどうかは、私たちおよび私たちが協力する可能性のある第三者が開発を成功させ、規制要求を満たす能力にかかっており、これらに限定されないが、任意の必要な規制承認を得て、私たちの候補製品を商業化することを含む。私たちは現在、規制要件に適合していないか、または必要な承認がなく、私たちの候補製品 を販売しており、これらの要求を決して満たしたり、獲得したりしないかもしれない。予測可能な未来に、私たちは製品販売から収入を得られないことが予想されます。もしあれば。もし私たちの任意の候補製品が臨床試験で失敗した場合、あるいは私たちの任意の候補製品が規制要求に適合していない場合、必要な時に監督管理の承認を得ることを含むか、または私たちの任意の候補製品が発売後に市場の承認を得られなかった場合、私たちは決して利益を上げないかもしれない。私たちが未来に利益を達成しても、私たちは後続の時期に収益性を維持することができないかもしれません。私たちが将来製品販売から収入を得る能力は、私たちの以下の成功に大きく依存しています
| ● | 私たちの候補製品の研究、臨床前、臨床開発を完成させる |
| ● | 臨床試験を終えた候補製品のために規制とマーケティングの承認を求めています |
| ● | 製品マーケティングの規制要件を満たす |
| ● | 私たちの候補製品のための持続可能で拡張可能で反復可能で譲渡可能な製造プロセスを開発すること; |
34
| ● | 販売チーム、マーケティング、流通インフラを構築すること、またはパートナーと協力することによって、私たちが監督とマーケティングの許可を得たり、他の方法で発売が許可された候補製品の発表と商業化を行う |
| ● | 承認された製品の市場受容度 |
| ● | 競争的な技術や市場の発展に対応しています |
| ● | 必要に応じてより多くの内部システムとインフラを実施する |
| ● | 新製品候補を識別し、検証する |
| ● | 私たちが参加する可能性のある任意の協力、許可、または他の手配で有利な条件を交渉する |
| ● | 特許、商業秘密、および専門知識を含む、私たちの知的財産権の組み合わせを維持、保護、拡大し、 |
| ● | 人材を引きつけ、採用し、引き留める。 |
我々が開発した1つ以上の候補製品が商業販売を許可されたり、他の方法で発売が許可されたりしても、任意の承認された製品の商業化に関連する巨額のコストが生じることが予想される。FDA、EMA、 または他の同等の外国規制機関が現在の予想に基づいて臨床試験および他の研究を行うことを要求した場合、私たちの費用は予想を超える可能性があります。 たとえ承認された製品の販売から収入を得ることができても、利益を得ることができなくても、運営を継続するために追加の資金が必要になるかもしれません。もし私たちが利益を上げることができなければ、あるいは私たちが持続的な損失を補うことができなければ、私たちの業務、財務状況、運営結果は実質的な悪影響を受けるかもしれない。
我々は,ある細菌の存在に関する医療状況を治療するための候補製品の開発を求めている。我々の成功は市場の広範な受容度に大きく依存し、薬物製品、医師の採用と使用において、これは商業成功に必要である。
私たちの候補薬品がFDAや外国規制機関の許可を得ても、私たちの候補製品の商業成功は消費者の私たちの商業化製品の受け入れと採用に依存する。私たちの候補製品の臨床前研究や臨床試験または他の類似製品を開発する臨床試験における有害事象、およびそれによって生じる宣伝は、私たちが開発する可能性のある任意の製品に対する需要を減少させる可能性がある。
さらに、私たちの候補薬物の商業的成功は、承認された治療的適応および承認された任意の他の適応に対する小児科医および他の医師の広範な使用および使用に大きく依存するであろう。私たちの方法が承認または販売可能な製品の開発につながることは確認できません。
著者らの臨床前と臨床テストに必要な特定のバクテリオファージカクテルのために高力価を得ることは困難であり、時間がかかる可能性がある。
私たちの候補製品は特定の特性を満たすために設計されたファージカクテルです。私たちの契約製造業者と複数のバクテリオファージのカクテルを生産し、私たちの臨床前および臨床試験を満たすのに十分な高力価またはレベルのバクテリオファージを得ることは困難または時間がかかる可能性がある。場合によっては、臨床試験に必要な数を得るために、製品を複数回実行する必要があるかもしれません。これは私たちの臨床試験スケジュールの遅延を招き、生産コストと関連費用を増加させる可能性がある。また,br生産過程を複製することは困難である可能性があり,我々の候補製品が臨床開発過程で進展するにつれて,より多くの数が必要となるからである。
35
著者らの候補製品の臨床前研究結果は臨床試験或いは後期臨床開発の結果を予測できない可能性がある。
我々の候補製品(例えばBX 004とBX 005)の臨床前研究は、動物疾患モデル研究を含み、候補製品の安全性を正確に予測できない可能性があるため、さらなる人体臨床試験を許可する。特に、有望な臨床前テストは、プロトファージ製品の潜在的な効果が、これらの製品がヒトの臨床環境において疾患に対応する能力を予測できない可能性があることを示している。例えば、バクテリオファージ活性を検討した場合体外培養そして体内にある私たちのバクテリオファージカクテルがヒト被験者に投与された場合、これらの結果は重複しないかもしれない。いずれの臨床前研究においても有望なデータがあるにもかかわらず,われわれのバクテリオファージ技術は臨床試験では有効ではない可能性がある。
FDAまたは同等の外国規制承認基準を満たすためには,十分かつ良好に制御された臨床試験において,候補薬物製品がその期待用途に対して安全かつ有効であることを証明しなければならない。前臨床試験と早期臨床試験の成功は今後の臨床試験も成功することは確保できない。著者らの前臨床試験の初歩的な結果も後の分析或いはその後のより大きな臨床試験によって実証されない可能性がある。製薬業界の多くの会社は高度臨床試験で大きな挫折を経験し,早期の臨床試験でエキサイティングな結果を得ても,臨床試験を開始した候補製品の多くはbr商業販売が許可されていない。
われわれが患者を募集してわれわれの臨床試験に参加することが困難であれば,われわれの臨床開発活動は延期されたり,他の悪影響を受けたりする可能性がある。
臨床試験の完了は、私たちが十分な数の患者を募集する能力に依存し、これは多くの要素の関数である
| ● | 評価のための治療の終点を選択し |
| ● | 議定書に規定されている資格基準 |
| ● | 研究を受けた製品候補製品の期待収益; |
| ● | 臨床試験治療の終点に必要な患者群の大きさを分析した |
| ● | 適切な能力と経験を持つ臨床試験研究者やサイトを募集することができます |
| ● | 患者の同意を得て維持する能力は |
| ● | 他の治療の臨床試験から患者を奪い合う。 |
われわれは臨床試験で患者を募集する際に困難に遭遇し続ける可能性があり,コストを増加させたり,これらの臨床試験の時間や結果に影響したりする可能性がある。患者数が比較的少ない疾患では,特にそうである。また,われわれが試験した潜在患者 は,われわれの目標疾患として十分に診断あるいは識別されていない可能性があるか,あるいはわれわれの研究の入選基準 に適合していない可能性がある。
FDAや同様の外国規制機関が要求する臨床試験に参加するのに十分な数の条件を満たす患者が見つからなければ,臨床試験を開始または継続できない可能性がある。また,患者を発見し診断する過程は高価であることが証明される可能性がある。私たちは私たちの任意の臨床試験のために十分な数の患者を募集することができません。これは重大な遅延を招きます。あるいは私たちは1つ以上の臨床試験を放棄する必要があるかもしれません。
36
著者らの臨床試験の遅延は予想されたbrの予想された開発マイルストーンを達成できず、コストを増加させ、私たちの候補製品が監督部門の承認と商業化を得る能力を遅延させる可能性がある。
著者らの臨床試験の遅延は著者らのbrが期待した臨床マイルストーンを達成できず、そして著者らの製品開発コストに重大な影響を与える可能性があり、そして監督管理部門の著者らの候補製品に対する承認を延期する可能性がある。計画中の臨床試験は予定通りに開始されたり完成したりしないかもしれないし,まったくできないかもしれない。
臨床試験は様々な理由で延期されるかもしれません
| ● | 臨床試験規模で一貫した生産を可能にするために、私たちの候補製品の製造能力の開発を遅延させる |
| ● | 私たちの内部製造作業は失敗し、臨床試験を支援するのに十分な量のバクテリオファージを持続的かつタイムリーに生産することができませんでした |
| ● | 私たちが計画した臨床試験を開始し完成させるために財政資源の獲得可能性があります |
| ● | 遅延と臨床研究者は研究設計について合意した |
| ● | 遅延と監督機関は試験設計について合意したり、監督機関の許可を得て試験開始を遅延させたりする |
| ● | 臨床材料の入手が遅れています |
| ● | 臨床試験に参加した患者の募集速度は予想より遅い |
| ● | 規制制限または禁止(例えば、ネットワークセキュリティおよびデータプライバシー法を遵守しない場合、規制当局からの制限または禁止) |
| ● | 臨床試験場所、他の第三者、または私たちは臨床試験プロトコルおよび/または試験方案を遵守できなかった |
| ● | 受け入れ可能な臨床試験合意条項に対する遅延と意図された場所との合意、またはIRBまたは独立倫理委員会の承認を遅延させること; |
| ● | 私たちの臨床試験中に発生した有害な安全事件。 |
もし私たちが予定通りに臨床試験を開始したり、完成できなければ、私たちの証券価格は下落するかもしれない。重大な臨床前または臨床試験遅延は、候補製品を商業化する独占的な権利を有する私たちの任意の期限を短縮するか、または私たちの競争相手が私たちよりも先に製品を市場に出すことを可能にし、候補製品を商業化することに成功する能力を弱める可能性があり、私たちの業務brと運営結果を損なう可能性がある。
37
著者らの現在或いは未来の候補製品は不良な影響 をもたらす可能性があり、その臨床開発を停止し、その承認或いは発売を阻止し、その商業潜在力を制限し、或いは重大な負の結果を招く可能性がある。
悪影響を及ぼす可能性があり、私たちまたは監督当局の臨床試験の中断、延期、または一時停止を招き、より厳しいラベルやFDAあるいは同等の外国の監督管理機関の遅延または上場拒否を招く可能性がある。我々の実験結果は,重篤で受け入れられない重症度,副作用や意外な特徴の盛行率を示す可能性がある。
私たちの候補製品を開発する際に悪影響が発生した場合、私たち、FDAまたは同等の外国規制機関、IRBs、または私たちの研究を行う機関の独立道徳委員会、またはデータ安全監視委員会は、私たちの臨床試験を一時停止または終了することができ、またはFDAまたは同等の外国規制機関は、私たちの任意またはすべての目標適応の候補製品の承認を拒否することができる。
我々の候補製品brの安全性と耐性は,引き続き第1段階臨床試験の形で評価していく予定である。我々の現在および未来の候補製品は、規制機関と議論された条件下で、可能な範囲内で、適用可能な場合に安全性試験を行うが、すべての薬物の副作用 が予測可能または予想されるわけではない。予見できない悪影響は臨床開発過程で発生する可能性があり、あるいは、もしこのような不良影響が比較的に希であれば、著者らの製品が監督管理機関の許可を得て発売された後に発生する可能性があり、br}がより多くの患者に暴露されることを招く。例えば,バクテリオファージをスクリーニングして安全問題を最小限にしようとしているが,バクテリオファージに毒性遺伝子,抗生物質耐性遺伝子,溶解源遺伝子,インテグラーゼ遺伝子あるいは他の有毒遺伝子が出現するリスク,あるいは患者免疫系のバクテリオファージに対する副作用を除去する保証はない。これまでbrは証明されておらず,進行中や将来の臨床試験がいずれの候補製品が人体にとって安全であるかどうかを証明するかどうかも予測できない。また,われわれの候補製品の臨床試験は丁寧に定義された患者群で行われ,これらの患者は臨床試験に入ることに同意した。したがって,われわれの臨床試験では,候補製品 の有意な正面効果が実際の正面効果よりも大きい(あれば),あるいは不良副作用を認識できない可能性がある。
最終的に、私たちの一部またはすべての候補製品 は人間の使用に安全ではないことが証明されるかもしれない。また,任意のボランティアや患者がわれわれの臨床試験に参加することにより,あるいは悪影響を受けているようであれば,重大な責任を負う可能性がある。これらの事件のいずれも、私たちの候補製品に対する市場の受け入れ度を達成または維持することを阻止し、商業化コストを大幅に増加させる可能性がある。
我々は候補製品の組成開発 を完了していない.
私たちの候補製品の開発には、この候補製品に必要な細菌を複数のバクテリオファージを分離、選択、最適化、組み合わせる必要がある。我々の任意の候補製品のためのバクテリオファージを選択することは、選択されたbrバクテリオファージの組み合わせが標的細菌を成功裏に死滅させる能力、単一のバクテリオファージと同じbr部分の細菌標的との交差反応の程度、バクテリオファージを組み合わせて法規的要件を満たす能力、十分な数のバクテリオファージを生産する能力、第三者の知的財産権、および他の要因を含む様々な要因に基づいているが、これらに限定されない。BX 004の初期処方 を選択したが、承認されれば、この初期処方が製品候補製品の最終製剤 となる保証はない。もし私たちが予想される時間範囲で候補製品の調合開発を完了できなければ、 私たちの製品開発スケジュールと候補製品の規制承認が延期される可能性があります。
38
私たちは引き続き私たちの候補製品のための製造プロセスを開発しなければなりません。どんな遅延や私たちがそうできなくても は私たちの臨床試験を遅延させます。
我々の候補製品の製造プロセスや臨床試験におけるこのようなプロセスの拡大が挑戦をもたらす可能性があり, はこの作業をタイムリーに達成できる保証はない(あれば)。これらの製造プロセスの開発や拡大のいずれの遅延も臨床試験の開始を延期し、私たちの業務を損なう可能性がある。私たちの製造能力を拡大するためには、追加の内部製造能力を確立し、1つまたは複数のパートナーと契約を締結するか、または両者を両立させる必要がある。我々の技術 や設備やツールの生産プロセスは複雑であり,我々の 候補製品を生産する際には思わぬ困難に遭遇する可能性がある.例えば、バクテリオファージを産生するための製造宿主は、それらのゲノムに1つ以上の統合バクテリオファージを含む可能性があり、これらの統合バクテリオファージを除去できない場合、生産されたバクテリオファージの生産に挑戦をもたらす可能性がある。私たちが内部で製造能力を確立し続けることができるか、または1つまたは複数の適切なパートナーを見つけることができるか、または両方を組み合わせて、必要な数量および品質要件を満たすことができるという保証はない。私たちの生産規模の拡大に伴い、製造と製品品質の問題が発生する可能性があります。 私たちの製造能力を確立したり拡大したりする上で、いかなる遅延も実現できず、候補製品を開発する能力を弱める可能性があります。
2019年第3四半期、私たちはイスラエルのナイスジオナの本社に独自の製造工場を設立し、初の人体臨床研究を行った。APTは2021年2月、GMP生産、テスト、開発をゲザスバーグ工場に統合した。2021年3月、私たちはイスラエルのネチオナ本部にある新しい製造工場に引っ越した。我々の新施設は,cGMP要求に基づいて第1段階と第2段階の臨床研究の正確な製造を検証するための持続的な内部検査を受けている。もしこれらの施設が私たちの候補製品生産のcGMP規格に適合していない場合、私たちは製造プロセスの追加的な修正に資金を提供し、追加の検証研究を行ったり、代替製造施設を探す必要があるかもしれません。これらはいずれも私たちに大きなコストをもたらし、そのような製品候補製品の承認を得る時間を数年遅延させることになります。
もし私たちが工場で生産された任意の候補製品にマーケティング申請を提出した場合、製造工場は、欧州、FDA、およびcGMP法規に適合することを確実にするために、持続的な定期検査を受けるであろう。これらの法規やbrを遵守する基準は複雑でコストが高く,必ず遵守できる保証はない。適用された法規を遵守しないいかなる行為も、制裁(罰金、禁止、民事処罰を含む)、規制機関が私たちの候補製品の上場、遅延、一時停止または承認の撤回、許可証の取り消し、候補製品または製品の差し押さえまたはリコール、運営制限、刑事起訴を承認できなかった可能性がある。
もし私たちの競争相手が私たちよりも効果的で、安全で、あるいは私たちの前にマーケティング許可を得ることができれば、私たちのビジネス機会は制限されるかもしれません。
バイオテクノロジーと製薬産業の競争は非常に激しく、まだ激化し続けている。より規模が大きく,我々よりもはるかに多くの資源を持つ会社では,従来の療法brや新たな作用機序を有する療法など,我々が求めている適応の開発計画を積極的に求めている。また,他社は治療および非治療用途のためのバクテリオファージベースの製品を開発しており,バクテリオファージ開発および製造における専門的な知識を利用して,我々の製品と競合する製品の開発を試みることが可能である。
我々brはまた,学術機関,政府機関,薬物や療法の発見と開発に従事する民間や公的研究機関からの潜在的な競争に直面している。私たちの多くの競争相手は研究開発、臨床前テスト、臨床試験を行い、監督管理の許可を得て、製造、販売とマーケティングの方面で私たちよりずっと多くの財力と専門知識を持っている。規模が小さいか早い段階にある会社も重要な競争相手となる可能性があり、特に大手や老舗製薬会社との協力で手配されている。
EUでは,潜在的な競争は病院や薬剤師が販売許可なしに生産·使用する医薬製剤からも由来しており,一般に“複方”と呼ばれている。いくつかの会員国では、国家当局は一般的に医療費を減らすために復薬を普及させる。
私たちの競争相手は、私たちの候補製品よりも有効で、副作用が少なく、より安全で、より手頃な製品の開発に成功するかもしれません。これは、私たちの候補製品の競争力を低下させるか、または競争力を欠くことを可能にし、孤立製品の称号を付与または保持することを阻止するでしょう。これらの競争相手はまた、私たちと競争し、合格した科学と管理者を募集し、保留し、臨床試験場と臨床試験患者登録を確立し、そして私たちの計画と相互補完あるいは私たちの業務に有利な技術と技術許可証を獲得する。また,特許保護を得ることができる競争相手は,我々が以前に規制部門の承認を得て製品の商業販売を開始していたが,すでにそうしているライバルは顕著な競争優位性を有している可能性がある.
39
私たち は他の候補製品を決定または発見することで成功しないかもしれない。
我々が現在開発している候補製品以外の治療機会を我々の技術を用いて評価する予定であるが,様々な理由から,他の候補製品を臨床開発に用いることができない可能性がある。例えば、私たちの研究br方法は、潜在的な候補製品の識別に成功しない可能性があり、または私たちが決定した製品が、販売できないか、または規制部門の承認を得ることができないように、有害な副作用または他の特徴を有することが証明される可能性がある。また,調達困難,例えば多様性の欠如,タイムリーあるいは全く得られない試料,あるいは試料中の汚染など,根絶対象細菌のバクテリオファージを決定することができない可能性がある。我々は,研究療法の要求に応じたファージカクテルを設計する際にも困難に遭遇する可能性があり,細菌の我々のバクテリオファージに対する抵抗力の増強,我々のバクテリオファージの影響を受ける宿主細菌の範囲,異なる細菌の生育状態に対する活性の多様性,我々のバクテリオファージ中の毒性問題br,および我々の候補製品の安定性,頑強性,製造が容易である。さらに、設計合成された工学的バクテリオファージは、所望の特性または挙動を有するバクテリオファージを開発することができない可能性があり、これらの特性または挙動は、実行可能な治療法として使用するのに適しているか、または免疫原性、毒性、および他の安全問題などの不良特性を含むバクテリオファージを引き起こす可能性がある。
私たちの戦略の重要な部分は私たちのスクリーニング技術を利用して臨床開発で追求すべき候補製品を決定することである。もし私たちが他の潜在的な候補製品を発見し開発できなかったら、私たちは私たちの業務を発展させることができないかもしれません。私たちの運営結果は深刻な損害を受ける可能性があります。このような候補製品は、商業化販売の前に、臨床前研究、臨床試験およびFDAおよび/または適用される外国の監督管理機関の承認を含む追加的で時間のかかる開発作業が必要となる。すべての候補製品 は薬品開発固有の失敗リスクが発生しやすい。
合成生物学や遺伝子工学に関する法的要求や倫理や社会的懸念は,我々の技術使用を制限または阻止し,我々の収入を制限する可能性がある。
私たちの技術は合成生物学と遺伝子工学を使用することを含むかもしれない。いくつかの国では、遺伝子組換え生物を使用して生産された薬物は、より厳しい法律制度によって制限される可能性があり、これは複雑で非常に挑戦的であることが証明される可能性があり、特に小型生命科学会社にとっては。例えば、EUでは、医薬製品や化粧品に関する一般的なルールに加えて、遺伝子組換え生物に関するルールが適用される。高級治療薬に関する規定も適用される可能性がある。
また,合成生物学や遺伝子工学の安全と環境被害に対する公衆の見方やその倫理への関心は,公衆の我々の技術,候補製品やプロセスに対する受容度に影響を与える可能性がある。私たちと私たちの協力者が合成生物学や遺伝子工学に関する法的挑戦や倫理や社会的懸念を克服できなければ、私たちの技術、候補製品、プロセスは受け入れられないかもしれない。これらの挑戦と懸念は、費用の増加、規制審査、規制の強化、私たちの候補製品の輸入に対する貿易制限、私たちの計画の遅延または他の障害、または私たちの製品の一般的な受け入れと商業化をもたらす可能性がある。私たちが設計および生産した候補製品は、制御された実験室で自然に生成された生物または酵素と類似しているか、またはより良い特性を有しているが、このような生物 を非制御環境に放出することは、予期しない結果をもたらす可能性がある。このような放出によって生じる任意の悪影響は、私たちの業務、財務状態、または運営結果に実質的な悪影響を及ぼす可能性があり、私たちは、それによって生じる任意の損害に責任を負う可能性がある。
我々 は、特定の候補製品または指示を追求するために限られたリソースを費やす可能性があり、より利益またはより成功する可能性の高い候補製品または指示 を利用することができない。
我々は財務や管理資源が限られているため, が最も成功する可能性があると考えられる特定の適応に焦点を当てて候補製品を開発する予定であり,市場承認においても商業化においても。したがって、私たち は、他の候補製品を求めることを放棄または延期するか、またはより大きな商業的潜在力を有することが証明される可能性のある他の指示 を放棄または延期する可能性がある。例えば、私たちはBX 001を開発するのに多くの時間と資源を費やしましたが、私たちはBX 001と、私たちのBX 005候補製品とCRC開発を停止して、私たちはこの仕事を無期限に一時停止しました。
私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。現在と未来の研究開発計画および特定の適応候補製品への支出は、いかなる商業的に実行可能な候補製品も生じないかもしれない。もし私たちが特定の候補製品の商業的潜在力や目標市場を正確に評価できなければ、候補製品の独占的な開発と商業化の権利を保持することが私たちにより有利な場合、私たちは協力、許可、または他の印税配置を通じて候補製品の貴重な権利を放棄するかもしれない。
我々は,我々のBolt独自製品プラットフォームに依存して我々のバクテリオファージ療法を開発していく予定である。もし私たちの競争相手が似たようなプラットフォームと競争相手の候補製品を開発すれば、私たちの競争地位は実質的に損なわれる可能性がある。
著者らのbr Boltプラットフォームは私たちが特定の病原菌brに対してバクテリオファージ治療候補薬物を迅速に開発、製造と調合でき、そして著者らの過去6年間の経験と技術改善と技術進歩の実施を結合することができるようにした。与えられた適応については,プラットフォームは通常,プロジェクト開始後約12−18カ月以内に患者の臨床概念検証研究,すなわち第2段階結果を完成させることを許可しているが,ある適応では,適応,目標細菌のアイデンティティ,募集速度,キューの大きさ,その他の要因により,br概念の臨床検証時間がより長くなる可能性があり,このスケジュール上で臨床概念検証を実現できないか,あるいは全く実現できない可能性がある。われわれの最初の目標は,われわれのCFプロジェクト開始後約12−18カ月以内に患者の臨床概念検証研究を完成させることである。我々がBolt プラットフォームを用いた経験は限られており,我々が期待している利点を実現できない可能性がある.私たちが私たちの資源を利用して私たちのBoltプラットフォームをさらに発展させる限り、私たちはそれの成功にもっと依存するかもしれない。
40
私たちの業務には、製品責任クレームの重大なリスクがあります。もし私たちが十分な責任保険を受けていなければ、製品責任クレームは私たちに対する重大な責任を招くかもしれない。
私たちの業務は私たちを人類治療製品の開発、製造、マーケティングに固有の重大な潜在製品責任リスク に直面させた。是非曲直や最終結果にかかわらず、製品責任クレームは次のようになるかもしれない
| ● | 私たちの臨床試験を遅延または完了できなかった; |
| ● | 臨床試験を脱退した参加者; |
| ● | 私たちの製品への需要を減らす候補製品; |
| ● | 私たちの名声を損なう |
| ● | 訴訟費用; |
| ● | 私たちの巨額のお金に対する報酬;そして |
| ● | 管理職や他の資源を私たちの運営の重要な側面から移す。 |
製品のマーケティングに成功すれば、製品責任クレームは、FDAまたは同等の外国規制機関に、私たちの製品、私たちの製造プロセス、および施設、または私たちのマーケティング計画の安全性または有効性を調査することにつながる可能性があります。このような調査はまた、私たちの製品をリコールすること、またはより深刻な法執行行動をとること、またはこれらの製品が使用可能な適応を制限すること、または承認を一時停止または撤回することをもたらす可能性がある。
著者らのbrは現在限られた臨床試験保険証書しかなく、ある地区の臨床試験をカバーしている。もし私たちの候補製品や私たちが開発する可能性のある他の化合物が市場の承認を得たら、私たちは私たちの保険カバー範囲を拡大し、商業製品の販売を含めるつもりです。しかしながら、保険範囲は高価であり、私たちは保険範囲を合理的なコストまたは根本的に維持できない可能性があり、私たちが所有または獲得した保険範囲は、潜在的なクレームまたは損失をカバーするのに十分ではない可能性がある。
41
当社の従業員、独立請負業者、コンサルタント、ビジネスパートナー、およびサプライヤーは、規制基準および要件を遵守しないことを含む、不適切な行為または他の不適切な活動に従事する可能性があります。
私たちbrは、従業員、独立請負業者、コンサルタント、ビジネスパートナー、サプライヤーが従業員詐欺や他の不正活動を行うリスクに直面しています。これらの当事者の不正行為には、故意、無謀、および/または不注意な行為が含まれている可能性があり、FDAおよび他の同様の外国規制機関の法律を遵守できず、FDAおよび他の類似外国規制機関に真実、完全かつ正確な情報を提供し、私たちが制定した製造基準を遵守し、米国の医療詐欺および法の乱用および類似の外国詐欺的不正行為法律を遵守するか、または財務情報またはデータを正確に報告するか、または許可されていない活動を私たちに開示する。もし私たちの候補製品がFDAの承認を得て、アメリカでこれらの製品を商業化し始めたら、私たちのこのような法律の下での潜在的なリスクは著しく増加し、私たちはこのような法律を遵守することに関連するコストも増加する可能性がある。これらの法律は私たちの現在の主要な研究者と患者の活動、提案と未来の販売、マーケティングと教育計画に影響を与える可能性がある。
私たちの限られた運営の歴史は、これまでの業務の成功を評価することが難しくなり、私たちの将来の生存能力を評価することも困難になるかもしれない。
2015年に設立されて以来、BiomX Ltd.はその臨床前計画を通じて、そのほとんどの資源をバクテリオファージ技術を有する候補製品の開発に投入し、その知的財産権の組合せを構築し、サプライチェーンを開発し、その業務を計画し、資金を調達し、そしてこれらの運営に一般と行政支持を提供した。私たちは、私たちがどんな臨床研究や他の重要な臨床試験を成功させ、監督管理の許可を得て、商業規模の製品を製造し、あるいは第三者代表が私たちにそうすることを手配したり、成功した製品の商業化に必要な販売とマーケティング活動を行うことができることを証明していません。したがって、もし私たちがもっと長い運営歴史があれば、私たちの未来の成功または生存能力に対するいかなる予測もそんなに正確ではないかもしれません。
また,スタートアップ企業としては,予見できない費用,困難,合併症,遅延などの既知と未知のbrに遭遇する可能性がある。私たちが私たちの候補製品を進めるにつれて、研究に集中している会社から、臨床開発やビジネス活動(成功すれば)を支援できる会社に移行する必要があります。そのような移行で、私たちは成功しないかもしれない。
私たち は私たちの組織の規模を拡大する必要があるかもしれないし、このような成長を管理する際に困難に直面する可能性がある。
私たちの研究、開発、製造、商業化計画と戦略のために、追加の管理、運営、販売、マーケティング、財務、その他の人員が必要かもしれません。将来の成長は経営陣のメンバーに大きな追加的な責任をもたらす
| ● | 他の従業員の決定、採用、補償、統合、維持、および激励 |
| ● | 臨床候補を決定し、私たちの製造プロセスを拡張し、私たちの候補製品をナビゲーションする臨床とFDA審査の流れを含む、私たちの内部研究と開発を効果的に管理する |
| ● | 私たちの運営、財政、管理制御、報告システム、そして手続きを改善する。 |
私たちの将来の財務業績と候補製品を商業化する能力は、私たちが将来の成長を効果的に管理する能力にある程度依存し、私たちの経営陣はまた、これらの成長活動を管理するために多くの時間を投入するために、不比例な注意を日常活動から移さなければならないかもしれない。
もし私たちがより多くの従業員を雇用し、私たちのコンサルタントや請負業者チームを拡大することで私たちの組織を効果的に拡大することができなければ、私たちは私たちの候補製品のさらなる開発と商業化に必要な任務を実行することに成功しないかもしれないので、 は私たちの研究、開発、商業化の目標を達成できないかもしれない。
42
政府規制に関するリスク
私たちの候補製品は厳格な規制承認要求を受けており、これは私たちのマーケティング を遅延、阻止、あるいは制限したり、私たちの候補製品を開発する能力を制限したりする可能性があります。
著者らの研究開発活動、臨床前研究、臨床試験及び著者らの候補薬物の期待製造とマーケティング はすべてアメリカFDAとその他の監督管理機関及びヨーロッパと他の地方の類似機関の広範な監督管理を受けている。FDAまたは同等の外国規制承認基準を満たすためには,十分なbrと良好な制御の臨床試験において,我々の候補薬物製品がその期待用途に対して安全かつ有効であることを証明しなければならない。規制承認プロセスは高価で時間がかかり、監督管理承認を受ける時間を予測することは困難である。バクテリオファージ療法の不確実性を考慮して、私たちの候補製品は、規制部門の承認を得るために、予想よりも長い時間が必要になるかもしれないし、決して承認されないかもしれない。私たちは多くの時間と財力がかかった後であっても、私たちのすべての候補製品のために規制部門の許可を得ることができない。規制承認の延期や拒否は、私たちが製品収入を創出し、収益性を達成することを延期または阻止するかもしれない。
我々の候補製品開発に対する規制 要求は不確実で変化している.これらの法律を変更したり、現在の解釈を変更したり、これらの法律を適用することは、私たちの候補製品を開発し、商業化する能力に重大な悪影響を及ぼす。EUを含む多くの国/地域では、バクテリオファージ療法の法律および規制の地位はまだ不明である。我々の任意の候補製品の開発中に、規制承認政策の変更、追加法規または法規の変更または公布、または提出された製品申請の規制審査実践の変更は、承認の遅延を招く可能性があり、または規制承認申請が拒否される可能性がある。
規制 の承認を受けると,我々の販売製品の指定用途の制限や,製品承認の ラベルを受ける可能性がある.このような制限は私たちの潜在的な製品収入に悪影響を及ぼすかもしれない。規制部門の承認はまた高価な発売後の後続研究を条件とする可能性がある。さらに、製品に関連するラベル、パッケージ、有害事象報告、貯蔵、広告、販売促進および記録保存は、広範な継続的な法規要件の制約を受けるであろう。また、どのような上場製品についても、当社のメーカーおよび当社の製造施設は、FDAまたは他の規制機関の登録および上場要件および継続的な審査および定期検査を受けることになります。適用される規制要求を遵守しないことは、罰金、監督管理審査の一時停止、製品のリコール、製品の差し押さえ、運営制限、刑事起訴を招く可能性がある。
突破的なFDA承認の治療指定または迅速チャネル指定は、私たちがbr適応を治療するために開発した任意の候補製品が承認されても、より速い開発、規制審査または承認過程を招くことはなく、私たちのいかなる候補製品も米国で発売承認される可能性を増加させることはない。
アメリカでは、BX 004または他の開発されている候補製品を含む画期的な治療法の称号を私たちのいくつかの候補製品に求めるかもしれない。画期的な治療法は、1つまたは複数の他の療法と共に、重篤または生命に危険な疾患または状態を治療することを意図した療法として定義され、予備的な臨床的証拠は、1つまたは複数の臨床的に重要な終点において、例えば、臨床開発早期に観察される有意な治療効果のような既存の治療法よりも有意な改善を示す可能性があることを示している。画期的な治療法として指定された療法では,FDAと試験スポンサーとのインタラクションやコミュニケーションは,臨床開発の最も有効な経路 の決定を助けるとともに,無効対照レジメンにおける患者数を最低にすることができる。画期的な指定はまた、スポンサーに転がり審査BLAの潜在力を提供した。画期的療法に指定されたのはFDAの裁量権である。
EUでは、主要(優先薬物)の地位は突破的な治療指定に似ている。EMAは主要な地位を実施し、複雑、革新的な医療製品の開発と加速を支持し、満足されていない医療需要を満たす。良質な地位はEMAが早期に関連する科学委員会と対話を行うことができ、そしていくつかの支払人と対話することが可能になり、それによってEMAの科学と監督管理支持を強化した。良質な地位はEMAから適宜授与され、重点は加速評価を行う資格のある上場許可の医薬製品(公共衛生の角度から、特に治療革新の角度から見ると、重大な意義を持つ医薬製品)である。
したがって, 我々の候補製品の1つが画期的療法や良質な状態に指定された基準に適合していると信じていても, FDAやEMAはそれぞれ同意せず,このような指定を行わないことに決定する可能性がある。いずれの場合も、従来の手順に従って承認を考慮する療法と比較して、画期的な治療指定または候補製品を得る良質な状態は、実際には、より速い開発プロセス、審査または承認をもたらすことはなく、最終的に承認されることも保証されない可能性がある。さらに、 が、我々の1つまたは複数の候補製品が突破的療法の資格に適合しているか、またはFDAまたはEMAの良質な地位をそれぞれ獲得している場合であっても、 以降、これらの候補製品がもはや資格条件を満たしていないか、または審査または承認の期間 を短縮しないことを決定する可能性がある。
アメリカでは、私たちのいくつかの治療適応候補製品のために高速チャネル認証を求めるかもしれません。治療の目的が重篤または生命に危険な疾患を治療することであり、そのような疾患の満たされていない医療需要を満たす可能性がある場合、治療スポンサーは迅速なチャネル認証を申請することができる。FDAは広範な裁量権を持ってこの認証を付与するかどうかを決定するため,特定の候補製品がその認証を取得する資格があると考えても,FDAがその認証を付与することを保証することはできない.2023年8月、私たちはアメリカでBX 004の高速チャネルの称号を取得した。Fast Track認証を取得したにもかかわらず、従来のFDAプログラムと比較して、より速い開発プロセス、審査、承認を経験しない可能性があります。FDAが我々の臨床開発計画のデータがこの認証をサポートしていないと考えると、Fast Track認証を撤回する可能性があります。高速チャネル指定だけではFDA優先審査プログラムに適合する資格は保証されない。
43
他の国では、患者にとって特に重要な薬物の承認を加速させるための計画がとられているかもしれない。例えば、欧州連合では、公衆衛生の観点から、特に治療革新の観点から、欧州医薬品局は、主に注目されている医薬製品の加速評価(210日ではなく150日)に同意することができる。また、主管監督機関は、“特殊な場合”に、すべての必要な安全性と有効性データを収集していない場合には、需要を満たしていないために設計された薬品又は孤児薬品に対して市場許可を与えることができる。特別な場合のマーケティング許可は決定されているが、医薬品br製品のリスク−収益バランスを毎年検討し、販売許可が負である場合には、マーケティング許可を撤回しなければならない。また、集中化されたプログラムによれば、欧州委員会は、必要なすべてのセキュリティおよび有効性データを取得していない場合に“条件付きマーケティング許可”を付与することができる。条件付きマーケティング許可は、失われたデータを生成すること、またはセキュリティ対策を増加させるために必要な条件を保証することに依存する。有効期間は1年で、すべての条件が満たされるまで年に1回更新しなければならない。EMAで設定された時間範囲内で条件が満たされていない場合、マーケティング許可は の更新を停止する。迅速チャネル指定と同様に、EUの主管規制機関は、広範な自由裁量権を有し、このような加速的な評価または承認を承認するかどうかを決定し、そのような評価または承認を承認しても、従来のプログラムよりも速い開発過程、審査、または承認を経験しない可能性がある。
私たちの現在と未来の候補治療製品について、私たちはFDAまたは同等の外国の規制機関から孤立した薬物指定を獲得し、維持することができないかもしれない。
米国では、孤児医薬品法によれば、FDAは、米国で200,000未満の患者または米国で200,000を超える患者に発生するbrを定義することができるが、薬物または生物を開発するコストは、米国では販売から回収されることはなく、まれなbr疾患または疾患の治療に使用される薬物または生物に孤児薬物の称号を与えることができる。2023年12月,我々は米国でBX 004の孤児薬物名 を獲得した。米国では,孤児薬物指定は一方が経済的インセンティブを得る権利があり,例えば臨床試験費用,税収割引,ユーザ費用減免に贈与資金を提供する機会 である。さらに、孤児の薬物名を有する製品がその後、FDAによる名称を有する疾患の最初の承認を得た場合、この製品は、孤児薬物独占性を得る権利があり、これは、FDAがNDAを含む7年以内に、同じ適応で同じ薬物または生物学的製剤 を販売する他の申請を承認することができないことを意味し、限られた場合、例えば、孤児薬物に対する独占性を有する製品に対する臨床的利点を示すか、または元の製造業者が十分な製品数を保証することができないことを意味する。
さらに、孤児が指定した適応よりも広い適応の承認を求める場合、米国での独占営業権が制限される可能性があり、またはFDAが後に指定要求に重大な欠陥があると判断した場合、または孤児指定疾患または条件を満たすのに十分な数の製品を確保できない患者の需要がある場合、独占営業権を失う可能性がある。また,我々が製品の孤立薬物排他性を獲得しても,この排他性は競合からの保護に有効ではない可能性があり,異なる活性部分を有する異なる薬物が同じ条件で承認される可能性があるからである。孤児指定製品brが承認された後であっても、FDAが 後の薬剤がより安全で、より有効であることが証明された場合、または患者ケアに大きな貢献があると結論した場合、FDAはその後、同じ状況に対して同じ活性部分を有する比較的新しい薬剤を承認することができる。孤児の薬物指定は薬物の開発時間或いは監督審査時間を短縮することもなく、監督管理 の審査或いは審査過程中に薬物にいかなる優勢をもたらすこともない。しかも、私たちは私たちの候補製品のために孤児薬物の称号を申請するかもしれないが、私たちは決してそのような称号を得ないかもしれない。
連合にはまた孤立した薬品法制度が存在する。EMAの孤児薬物製品委員会(COMP)は、欧州経済地域(EU、アイスランド、リヒテンシュタイン、ノルウェーを含む)における10,000人当たり5人以下の生命または慢性衰弱に影響を及ぼす疾患を診断、予防または治療するために、孤児薬物名の開発について決定した。または(Ii)生命を脅かす深刻な虚弱または深刻な慢性疾患であり、インセンティブがない場合、欧州経済地域における薬剤の販売は、医薬または生物学的製品の開発に投資する必要があることを証明するのに十分ではない可能性が高い。孤児に与えられる称号は、満足できる診断、予防または治療法がないことを要求するか、または、そのような方法が存在する場合、将来の薬剤がそのような疾患の影響を受ける人に大きな利点を有することが要求される。将来の製品は、外科手術、許可された医薬製品、および複合製剤(いくつかの条件に依存する)を含む、このようなまれな状況のすべての既存の治療方法と比較しなければならないので、後者の場合の試験は厳しい。市場許可時には、孤児状態の維持を考慮して、会社は孤児指定を再審査する。もしこれ以上指定された基準を満たさなければ、欧州委員会は孤児の指定を撤回するだろう。発売時に孤児の称号を保持することは、称号が付与されてから許可されたすべての薬物/生物製品が、満足できる治療または顕著な利益の不足を決定することと関係があることを意味する。
Brを獲得すれば,孤立薬物指定は,費用の低減や費用の免除,10年間の市場排他性 の獲得などの財務的インセンティブを得る権利がある。市場排他性のため、欧州医薬品管理局または国家主管部門は販売許可申請を承認できず、欧州委員会または国家主管部門も同じまたは類似したbr薬物/生物および治療適応の販売許可を承認することができない。孤児指定基準brがもはや満たされていない場合、製品が市場独占性を維持することが合理的であることを証明するのに十分な利益がないことを示すことを含む場合、10年の期限が6年に短縮される可能性がある。もし、製造業者が患者の需要を満たすのに十分な数の薬剤が保証されない場合、またはその製品が承認された孤児製品よりも臨床的に優れていることが証明されている場合、別の薬剤/生物製品に対する孤児の排他性を失う可能性もある。もし1種の薬物/生物薬がより安全で、より効果的で、或いは患者看護に重大な貢献をすれば、それは臨床的に優れている。
44
健康およびデータ保護法律法規を遵守できないことは、クレーム、政府の法執行行動(br民事または刑事罰を含む可能性がある)、規制行動、個人訴訟および/または負の宣伝をもたらす可能性があり、私たちの運営結果および業務に負の影響を与える可能性がある。
私たちは連邦、州、外国データ保護法律法規(すなわちプライバシーと安全に関連する法律法規)によって制約される可能性があります。米国では、連邦健康情報プライバシー法、州消費者プライバシー法、州データ漏洩通知法、州健康情報プライバシー法、連邦および州消費者保護法(例えば、連邦貿易委員会法第5条)を含む多くの連邦および州法律法規が、私たちの業務または私たちの協力者の業務に適用される可能性がある。また,我々は第三者(臨床試験データを取得した研究機関を含む)からHIPAAのプライバシーやセキュリティ要求に拘束された第三者(臨床試験データを取得する研究機関を含む)を取得する可能性があり,HIPAAは2009年に“経済的·臨床的健康の健康情報技術”を改訂した。事実および状況によると、HIPAAによってカバーされているエンティティがHIPAAによって許可されていないまたは許可されていない方法で維持されている個々の識別可能な健康情報を意図的に取得、使用、または開示する場合、私たちは刑事罰を受ける可能性がある。
国際データ保護法はまた追加的な 要求を加える可能性がある。この場合、GDPR第2016/679号条例(多くの他の国際データ保護法を除く)は、EUに位置する個人データを収集および/または処理する際の業務に影響を与える可能性がある。GDPRは2018年5月25日から適用(従来適用されていたデータ保護フレームワークの代わりに) を適用し,ドメイン外カバー範囲を持つ.GDPRは、加盟国が特定の分野に対して特別なカテゴリのデータを処理することを含む特定の要求を行うことを可能にし、このような国の枠組みの下で、私たちはさらに制限および不適切なリスクに直面する可能性がある。私たちは、その活動がGDPRによって発見される可能性があるかどうかを評価していない。
私たちが収集して処理したデータタイプは、健康、バイオメトリクス、および遺伝データに関連する可能性があるので、これらのタイプのデータは、特殊なクラスのデータとみなされ、より高い保護が与えられるので、GDPRルールに準拠しない高いリスク(または異なるEU加盟国におけるGDPRルールの局所的な低下)に直面する可能性がある。EUの臨床試験における個人データ処理に関するルール,要求や枠組みの違い (処理データを選択する法的根拠,収集した個人データの可能な用途など)を考慮すると,リスクはさらに増加する。他のフレームワークとの相互作用もありますGDPRはEUに厳しいデータ保護要求, および違反会社への潜在的罰金を導入し,最高2000万ユーロあるいは世界の年商の4%に達する。我々の加工活動がGDPR(あるいは 現地低下)に適合していないと考えられれば,規制機関も我々の加工活動を制限する能力があり,我々の活動のあり方に大きな影響を与える可能性がある。GDPRは、個人データの収集、使用、開示に多くの要求を出しており、有効な高い基準と、その個人データのどのような使用に関する具体的な情報を個人に提供するか、規制機関に通知し、影響を受けた個人に個人データ違反行為、広範な新しい内部プライバシー管理要求、および個人 がその強化されたプライバシー権を行使することを許可する義務(例えば、その個人データをアクセス、訂正、削除する権利、その同意を撤回することなど)を含む多くの要求を行っている。サービスプロバイダー、CROなどの第三者と契約を締結する際の権利と義務。また、GDPRには欧州経済圏以外のデータ転送の制限も含まれている。GDPRが規定しているこのような個人データを移転する実際のメカニズムはより厳格な監督管理と司法審査を受けている。もし私たちが既存のメカニズムに依存してヨーロッパ経済区、イギリス、br、あるいは他の管轄区域から個人データを転送できなければ、私たちはこれらの地域で個人データを転送できないかもしれない。また、イギリスの投票はEU離脱を支持し、一般的に“イギリス離脱”と呼ばれ、イギリスのデータ保護立法がGDPRから離脱するかどうか、イギリスに出入りするデータ転送をどのように規制するかに不確実性をもたらしている。
45
アメリカと国際データ保護法律法規を遵守することは、私たちがいくつかの司法管轄区域で運営する能力に影響を与えることを制限し、私たちがデータを収集、使用、開示する能力を制限する契約において、より重い義務を負うことを要求することができるかもしれない。このような法律法規は、個人または他のデータを使用して共有する能力を制限し、私たちのコストを増加させ、私たちの業務および財務状況を損なう可能性があります。米国および国際データ保護の法律および法規を遵守しないことは、クレーム、政府の法執行行動(民事または刑事罰を含む可能性がある)、規制行動、個人訴訟および/または負の宣伝を招き、私たちの経営業績および業務に負の影響を与える可能性がある。また,我々または我々の潜在的協力者 が情報を取得する臨床試験対象と,この情報を共有する提供者は,契約的に情報の使用や開示能力を制限する可能性がある。私たちがプライバシー権を侵害していると主張し、データ保護法を遵守できなかったり、契約義務に違反したりして、私たちが責任を負わないと認定されても、弁護の費用が高く時間がかかり、不利な宣伝を招く可能性があり、私たちの業務を損なう可能性があります。最後に、私たちは政府機関、法執行機関、情報機関の要求に基づいて個人データの開示を要求されるかもしれない。この開示は、データプライバシーの法律、規則、および法規を遵守できなかったか、または遵守できなかったと考えられる可能性があり、同じまたは他の管轄地域で私たちに訴訟または訴訟を提起することをもたらす可能性があり、私たちの名声およびブランドに悪影響を及ぼす可能性がある。
医療提供者、医師、第三者支払者との関係は、刑事制裁、民事処罰、契約損害、名声br損害、利益および将来の収入減少に直面する可能性がある逆控除、詐欺、乱用、および他の医療保健法律法規の制約を受けることになります。
医療 米国や他の地域の医療提供者,医師,第三者支払者は薬品の推薦と処方に主な役割を果たしている。第三者支払者および顧客との合意は、連邦反リベート法規およびFCAを含むが、これらに限定されないが、そのような会社の販売、マーケティングおよび流通薬品の業務または財務配置および関係を制限することができる外国同等の法律を含む、製薬業者に広く適用される詐欺および乱用および他の医療保健法律法規に直面する可能性がある。特に、医療製品およびサービスの普及、販売およびマーケティング、ならびに医療業界におけるいくつかの商業的配置は、詐欺、リベート、自己取引、および他の乱用行為を防止するための広範な法律によって制限されている。これらの法律法規は、幅広い価格設定、割引、マーケティングおよび販売促進、構造および手数料、特定の顧客インセンティブ計画、および他のビジネススケジュールを制限または禁止する可能性があります。これらの法律に拘束されている活動は、患者を募集して臨床試験を行う過程での不適切な使用に関する情報も含まれています。私たちの運営能力に影響を与える可能性のある適用連邦、州、外国のヘルスケア法律法規は含まれていますが、これらに限定されません
| ● | 連邦反リベート法規は、他の事項に加えて、任意の報酬(任意のリベート、賄賂またはリベートを含む)を知りながら故意に請求、受け取り、提供または支払うことを禁止し、直接または間接的に現金または実物の形態で誘導または見返りとして、個人を推薦し、または購入、レンタル、注文または推薦し、任意の商品、施設、物品またはサービスを推薦し、brは連邦医療保険と医療補助計画などの連邦医療計画に基づいて、すべてまたは部分的に支払う。法規に違反する具体的な意図が実際に理解されていない場合には、個人又は実体は法規違反と判断される可能性がある。また,FCAについては,連邦反リベート法規違反によるbr物品やサービスを含むクレームが虚偽や詐欺的クレームを構成している.逆リベート法規は、医薬品メーカーと処方者、購入者、および処方管理者との間の配置に適用されると解釈される。いくつかの法定例外と規制避難港はいくつかの一般的な活動を起訴されないように保護する | |
| ● | 連邦医療保険法を含む連邦民事·刑事虚偽精算法であって、個人または実体が故意に虚偽または詐欺的精算を提出することを禁止し、連邦医療保険、医療補助または他の連邦医療保険計画に支払いまたは承認し、虚偽記録または報告書材料を作成、使用または使用し、虚偽または詐欺的クレームまたは連邦政府に資金を支払うか移転する義務を負うことを禁止する連邦民事および刑事虚偽精算法。故意に隠したり、故意に不正に逃げたり、減少したり、連邦政府に資金を支払う義務を隠したりする。メーカーが虚偽や詐欺的なクレームを“招く”と考えられていれば,政府支払者に直接クレームを提出しなくても,FCAによって責任を負うことができる。FCAはまた、FCA違反行為を告発し、いかなる金銭的回収も共有する“密告者”としての個人代表が連邦政府を代表して訴訟を起こすことを許可した |
46
| ● | HIPAAは、支払人(例えば、公共または個人)にかかわらず、支払人(例えば、公共または個人)にかかわらず、任意の医療福祉計画が所有または管理されている任意の金銭または財産を取得し、または詐欺または詐欺的な言い訳、陳述または約束によって、支払人(例えば、公共または個人)にかかわらず、任意の医療福祉計画が所有または管理または制御された任意の金銭または財産を取得することを禁止する新しい連邦刑法を制定する。医療事務の提供または支払いに関連する医療福祉、プロジェクトまたはサービスに関連する重大な事実を任意のトリックまたは手段で隠蔽または隠蔽するか、または任意の重大な虚偽陳述を行う。連邦の“反リベート法規”と同様に、一人または実体は、実際に法規を理解していない場合、または具体的な意図が法規に違反していない場合に、“反リベート法規”に違反すると判断することができる | |
| ● | 連邦医師支払陽光法案は、Medicare、Medicaidまたは児童健康保険計画(いくつかの例外を除く)に基づいて支払いを受けることができる医薬品、設備、生物製剤および医療用品の製造業者が毎年米国衛生·公衆サービス部に医師(医師、歯科、検眼士、足科医および脊医を含む)、brのいくつかの非医師従事者(医師アシスタント、医師補佐官、医師アシスタント、医師補佐官、医師補佐官、勤務看護師、臨床専門看護師、麻酔科医アシスタント、登録看護師麻酔科医と登録助産師)と教育病院、および医師とその直系親族が持つ所有権と投資権益 | |
| ● | 連邦消費者保護と不正競争法は、市場活動を広く規制し、消費者の活動を損なう可能性がある | |
| ● | 州反リベートおよび虚偽請求法律のような同様の州および外国の法律は、医療に関する販売またはマーケティング手配およびクレーム(Br)が連邦同等の法律よりも広い可能性がある非政府第三者支払者(個人保険会社を含む)によって精算されるプロジェクトまたはサービスに適用可能であり、州と外国の法律は、製薬会社が製薬業界の自発的コンプライアンスガイドラインおよび連邦政府が公布した関連コンプライアンスガイドラインを遵守することを要求するか、または他の方法で医療保健提供者への支払いを制限することを要求する。医薬品製造業者に、医師および他の医療提供者への支払いまたは他の方法での価値またはマーケティング支出に関する情報の報告を要求する州および外国の法律; | |
| ● | EUと他の外国条項。 |
薬品流通は他の要求と法規の制約を受け、広範な記録保存、許可、保存、無許可販売を防止するための安全要求、及びEU国家を含むいくつかの外国国家で強制的に実施された偽造防止機能を含む。
47
これらの法律の範囲も実行も不確定であり,現在の医療改革環境では急速な変化の影響を受けている。 は特に適用例や法規の欠如を考慮している。連邦と州法執行機関は最近、医療保険会社と医療保健提供者の間の相互作用の審査を強化し、医療保健業界の一連の調査、起訴、有罪判決、和解を招いた。業務手配が適用される医療法に適合していることを確保し,政府当局が行う可能性のある調査に対応することは,時間や資源がかかり,会社の注意力を分散させる可能性がある。
従業員の不正行為を常に識別し、阻止できるわけではなく、不正行為を発見し、防止するための予防措置 は、未知または未管理のリスクや損失を効果的に制御できない可能性があり、あるいはこのような法律や法規を遵守しないことによる政府調査や他のbr行動や訴訟から私たちを保護することができるかもしれない。我々の業務手配 が適用される医療法に適合することを確保する努力は巨額のコストに及ぶ可能性がある。政府と法執行当局は結論を出すかもしれないが、私たちの業務実践は、解釈が適用される詐欺や乱用、または他の医療保健法律および法規の現在または未来の法規、法規または判例法に適合していない可能性がある。このような法律や法規の要求を守らなければ、私たちは法律や法規の行動に直面するかもしれない。状況に応じて、適用される規制要件を満たすことができないことは、民事、刑事および行政処罰、損害賠償、罰金、返還、個人監禁、連邦および州政府が援助する医療計画から除外される可能性があり、契約損害および私たちの業務の削減または制限を招く可能性があり、もし私たちが会社の誠実な合意または他の合意の制約を受けた場合、これらの法律違反に関する告発を解決するために追加の報告義務と監督を負う必要がある。これらの法律に違反するいかなる行為も、弁護に成功しても、製薬メーカーに巨額の法的費用が発生し、経営陣の業務運営への注意をそらす可能性がある。 将来の上場製品の販売や撤回の禁止または制限は、不利な方法で業務に大きな影響を与える可能性がある。
また,我々の任意の候補製品の米国国外での承認と商業化は,上記の医療保健法や他の外国法の外国等価物の制約を受ける可能性もある。
FDAは他と同等の外国規制機関が微生物群に作用する製品の開発や商業化に追加的な法規や規制を実施する可能性があり,予測が困難である可能性がある。
FDAと他の国の類似した外国の規制機関は、ヒト微生物グループに作用する製品のようなバイオテクノロジー製品および候補製品をさらに規制する興味を示している。米国連邦や州レベルの機関や米国議会委員会や他の政府や管理機関もバイオテクノロジー業界をさらに規制する興味を示している。そのような行動は私たちの候補製品の一部またはすべての商業化を延期または阻止するかもしれない。他の人が行った非INDヒト臨床研究或いは微生物グループ製品の臨床試験の不良進展 はFDA或いは他の監督機関が著者らの任意の候補製品に対する審査要求を変更することを招く可能性がある。これらの規制審査機関と委員会とその公表された新しい要求やガイドラインは、規制審査の流れを延長し、追加の研究や試験を行うことを要求し、私たちの開発コストを増加させ、規制の立場と解釈の変化を招き、私たちの候補製品の承認とbr}の商業化を延期または阻止し、あるいは承認後の重大な制限や制限を招く可能性がある。私たちが私たちの候補製品を推薦する時、私たちはこれらの規制機関に相談し、適用される要求とガイドラインを遵守するように要求されるだろう。もし私たちがこれをできなかったら、私たちはこのような候補製品の開発を延期または停止することを要求されるかもしれない。これらの追加的な流れは、私たちが予想していたよりも長い審査と承認の流れをもたらす可能性があります。規制承認プロセスを増加または延長し、あるいは私たちの候補製品開発をさらに制限することによる遅延はコストが高い可能性があり、私たちの臨床試験を適時に完成し、私たちの現在と未来の候補製品を商業化する能力にマイナス影響を与える可能性がある。
たとえbrが治療適応のための任意の候補製品の規制承認を得たとしても、持続的な規制コンプライアンス義務および持続的な規制審査の制約を受けることになり、多くの追加費用を招く可能性がある。また、私たちのどの候補製品も、承認されれば、ラベルや他の制限や市場撤退を受ける可能性があり、規制要求を遵守できなかったり、意外な製品候補問題に遭遇した場合、処罰される可能性があります。
もし私たちの任意の候補製品が治療適応のために承認された場合、私たちは製造、ラベル、包装、貯蔵、流通、広告、販売促進、サンプリング、記録保存、輸出、輸入、発売後の研究と提出、安全性、有効性およびその他の上場後情報を含む持続的な法規要件を遵守し、アメリカの連邦と州要求および同等の外国監督機関の要求を含む。また,われわれが承認後に行ったいずれの臨床試験についても,cGMP とGCP要求を遵守していく。
48
メーカー とメーカーの工場は、品質管理と製造プロセスがcGMP法規に適合することを確保することを含むFDAと同等の外国規制機関の広範な要求を遵守しなければならない。したがって、私たちと私たちの契約メーカー は、cGMPのコンプライアンスを評価し、任意のセキュリティプロトコル、他の マーケティング申請、および以前の検査意見への応答で行われた約束を遵守するために、持続的な審査および検査を受ける。したがって、私たちと私たちのパートナーは、製造、生産、および品質管理を含むすべてのコンプライアンス分野に時間、お金、エネルギーを投入し続けなければならない。
FDA或いは同等の外国監督管理機関は重要な上場後権力を持っており、例えば、新しい安全情報に基づいてラベルを変更することを要求する権利があり、そして発売後の研究或いは臨床試験に薬物使用に関連する深刻な安全リスクを評価することを要求する。私たちの候補製品については、私たちが得た任意の規制承認は、候補製品の安全性および有効性を監視するための第4段階の臨床試験および監視を含む可能性の高い上場後試験の要件を含むかもしれない。br}FDAまたは同等の外国規制機関は、REMS計画を私たちの候補製品を承認する条件として要求することも可能であり、これは、患者への長期フォローアップ、薬物使用ガイドライン、医師のコミュニケーション計画、または安全使用を確保するための追加要素brを要求することができる。例えば、制限された配信方法、患者登録表、および他のリスク最小化ツール。また、FDAまたは同等の外国規制機関が私たちの候補製品を承認した場合、安全および他の上場後の情報の提出、報告および登録を含む要求を遵守しなければなりません。
規制要件や基準を遵守していない場合、あるいは製品発売後に問題が発生した場合、FDAまたは同等の外国規制機関は、同意法令を強制的に実施したり、承認を撤回したりすることができる。もし我々の候補製品が後に以前の未知の問題 を発見した場合、予想されない深刻度または頻度の不良事象、または私たちの第三者製造業者またはbr}製造プロセスを含むか、または規制要件を遵守できない場合、新しいbr}セキュリティ情報を追加するために承認されたラベルの改訂を招き、発売後の研究または臨床試験を実施して新しい安全リスクを評価するか、またはREMS計画に従って流通制限または他の制限を実施する可能性がある。他の他の潜在的な結果には
| ● | 私たちの製品の販売を制限したり、市場から製品をリコールしたり、自発的または強制的に製品をリコールしたりします |
| ● | 罰金、警告、または無タイトルの強制実行手紙、または臨床試験の一時停止; |
| ● | FDAまたは同等の外国規制機関は、私たちが提出した係属中の出願または承認された出願を承認することを拒否するか、または許可証の承認を一時停止または取り消し; |
| ● | 製品が私たちの候補製品の輸入または輸出を許可することを差し押さえたり差し押さえたり拒否したり; |
| ● | 禁止または民事または刑事処罰を適用する。 |
FDAまたは同等の外国規制機関は、市場に投入された薬品のマーケティング、ラベル、広告、販売促進を厳格に管理する。製品は承認された適応と承認されたbrラベルや他の規制マーケティング経路の規定に基づいて販売促進しかできません。FDAと同等の外国規制機関はラベル外用途の普及を禁止する法律法規 を積極的に実行しており,ラベル外用途の普及が不適切であることが発見された会社は重大な責任を負う可能性がある。FDAまたは同等の外国規制機関の政策は変化する可能性があり、追加の政府法規 を公布して、私たちの候補製品に対する規制承認を阻止、制限、または延期する可能性がある。私たちが既存の要求の変更や新しい要求や政策の採用にゆっくりまたは適応できない場合、またはコンプライアンスを維持できない場合、 私たちは私たちが得る可能性のあるマーケティング承認を失う可能性があり、これは私たちの業務、潜在的な顧客、および収益性を達成または維持する能力に悪影響を及ぼすだろう。
49
FDAまたは同等の外国規制機関の政策が変わる可能性があり、追加の政府法規が公布される可能性があり、 は私たちの候補製品に対する規制承認を阻止、制限、または延期する可能性がある。米国や海外の将来の立法や行政や行政行動によって生じる可能性のある政府規制の可能性、性質、程度も予測できない。もし私たちがゆっくりしているか、既存の要求の変化や新しい要求や政策の採用に適応できない場合、あるいは私たちが規制適合性を維持できない場合、私たちは法執行行動の影響を受ける可能性があり、私たちは利益を達成したり維持することができないかもしれない。
もし私たちまたは任意の未来のパートナーが安全監視または薬物警戒要求を含む法規要件を遵守しなければ、重大な経済的処罰につながる可能性もある。
われわれは米国以外の地域で我々の候補製品の臨床試験を行う可能性があり,FDAはこのような試験のデータを受け入れない可能性がある。
私たちはすでにアメリカ以外の候補製品のためにいくつかの臨床試験や一部の臨床試験を継続している可能性があります。FDAや同様の外国の規制機関がアメリカ国外や他の司法管轄区で行われている臨床試験を受け入れる研究データは、ある条件によって制限される可能性があり、全く受け入れられないかもしれません。外国の臨床試験のデータが米国での上場承認の唯一の根拠となることを意図している場合、FDAは通常、(I)データがアメリカの人口とアメリカの医療実践に適用されない限り、外国のデータのみに基づいて申請を許可しない。(Ii)試験は公認能力を有する臨床研究者によって行われ、GCP規定に適合する。(Iii)データは、FDAによる現場検査を必要とせずに有効であると考えることができ、またはFDAがこのような検査を行う必要があると考えられる場合、FDAは、現場検査または他の適切な方法でデータを検証することができる。また,海外の研究データが承認の唯一の根拠としようとしなくても,FDAはこれらのデータを上場承認申請の支援として受け入れず, 研究がGCP要求に応じて良好な設計·実施を行わない限り,FDAは必要と考えた場合に現場検査により研究データ を検証することができる。多くの外国の規制機関もまた似たような承認要求を持っている。また、このような外国裁判は、裁判を行う外国司法管轄区域に適用される現地法によって管轄される。FDAまたは同様の外国規制機関が米国または適用司法管轄区域外で行われた試験データを受け入れることは保証されない。FDAまたは任意の類似した外国の規制機関がこのようなデータを受け入れない場合、追加の試験が必要となることになり、これは高価で時間がかかる可能性があり、私たちが開発する可能性のある現在または将来の候補製品brが適用司法管轄区域で商業化承認を得ることができない可能性がある。
私たちが開発する可能性のあるどの製品も、不利な価格設定法規、第三者精算やり方、または医療改革措置の制約を受ける可能性があり、これは、任意の候補製品や治療法を販売することを困難にするかもしれない。
新医療製品の定価を管理する法規は国によって異なる。したがって、特定の国/地域で規制部門の許可を得る可能性があるが、その後、その国/地域の価格設定法規の制約を受け、これらの法規は、製品の商業発表時間 を遅延させ、その製品をその国/地域で販売することによって生じる収入に悪影響を及ぼす。また、私たちが任意の承認された製品の商業化に成功できるかどうかは、これらの製品が政府衛生行政部門、個人健康保険会社、その他の組織から精算できる程度にある程度依存するだろう。たとえ私たちが1つ以上の治療製品を市場に投入することに成功しても、これらの製品は費用効果があると思われない可能性があり、どの製品の精算金額も競争に基づいて販売させるのに十分ではないかもしれない。もし私たちの開発と他のコストに基づいて、治療用製品に対して十分に高い価格を取ることができなければ、私たちの将来の収益性は不利な影響を受けるかもしれない。
行われている医療立法や規制改革措置は、私たちの業務や運営結果に実質的な悪影響を及ぼす可能性がある。
法規、法規、または既存の法規の解釈の変更は、例えば、 (I)私たちの製造スケジュールの変更、(Ii)製品ラベルの追加または修正、(Iii)私たちの製品 のリコールまたは生産停止、または(Iv)追加の記録保存要件を要求する、私たちの将来のビジネスに影響を与える可能性があります。このような変更が実施されれば、私たちの業務の運営に悪影響を及ぼす可能性があります。
アメリカでは、医療費を制御するための多くの立法計画があり、継続されるだろう。例えば,2010年3月,ACAが採択され,政府や民間保険会社が医療保健に資金を提供する方式 を大きく変え,米国製薬業に大きな影響を与えた。その他の事項以外に、ACAは生物製品を比較的に低コストの生物模倣薬の潜在競争に直面させた;吸入、輸液、点滴、移植或いは注射の薬物に対して、メーカーがMedicaid薬品返却計画の下で獲得すべきリベートを計算した;メーカーがMedicaid薬品リベート計画の下で最低医療補助バックル を高めた;そしてリベート計画をMedicaid管理の看護組織に登録した個人に拡大した。それはまたあるブランドの処方薬メーカーに対する年間費用と税収を確立し、そして新しい Medicare Part Dカバーギャップ割引計画を作成し、メーカーは現在そのカバー間隔期間内に条件を満たす受益者にブランド薬品交渉価格の50%の販売時点割引を提供することに同意しなければならず、メーカーの 外来薬物がMedicare Part Dでカバーする条件とした。
50
ACAのいくつかの側面は公布以来、司法、行政、そして国会の挑戦を受けてきた。2021年6月17日、米国最高裁はACAに対する最新の司法挑戦を却下したが、ACAの合憲性を具体的に裁くことはなかった。最高裁が裁決を下す前に,総裁·バイデンはACA市場による医療保険の取得を目的とした2021年2月15日から2021年8月15日までの特殊保険期間を開始する行政命令を発表した。行政命令はまた、ある政府機関に医療の取得を制限する既存の政策とルールを審査·再考するよう指示した。最近、総裁·バイデンは2021年3月11日に2021年の米国救援計画法案に署名し、2024年1月1日から法定の医療補助薬品のリベート上限を廃止し、現在は薬品平均メーカー価格の100%である。バイデン政府の他の医療改革措置(あれば)が我々の業務にどのように影響するかは不明である。
これらの法律および将来の州および連邦医療改革措置は将来的に採択される可能性があり、いずれも連邦医療保険および他の医療資金の追加的な削減をもたらす可能性があり、規制によって承認される可能性のある任意の候補製品の価格、またはそのような候補製品の処方または使用頻度に影響を与える可能性がある。
連合国でも似たような運動が観察された。定価と精算基準は国によって異なり,国の医療保険システム予算に対する消費を削減するために定期的に改訂·引き締めされている。また,参考定価制度(一国の価格は他の価格が通常低い国の価格から計算される) は従来高値を与えていた国の価格を低下させた。
FDAや他の政府機関の資金不足や世界的な健康問題による中断(Br)は、重要な指導部および他の人員の採用、保留または配置の能力を阻害する可能性があり、あるいは新しい製品や修正された製品のタイムリーまたは開発、承認または商業化を阻止することは、私たちの業務に負の影響を与える可能性がある。
FDAが新製品を審査および/または承認する能力は、政府予算および資金レベル、法律、法規および政策の変化、FDAのキーパーソンの雇用および保留、およびユーザ費用支払いを受け入れる能力、およびFDAが通常の機能を履行する能力に影響を与える可能性のある他のイベントを含む様々な要因の影響を受ける可能性がある。したがって、FDAの平均審査時間は近年変動している。また,研究や開発活動を援助する他の政府機関への政府の援助は政治プロセスの影響を受けており,政治プロセス自体が不安定で予測不可能である.FDAおよび他の規制機関の中断も、必要な規制機関による新薬および生物製品の審査および/または承認に要する時間を遅らせる可能性があり、これは私たちの業務に悪影響を及ぼすだろう。例えば、ここ数年間、米国政府は何度か閉鎖されており、FDAのようないくつかの規制機関は、FDAのキー従業員を休暇にし、キー活動を停止しなければならない。
私たちはアメリカと外国のいくつかの反腐敗、反マネーロンダリング、輸出規制、制裁、その他の貿易法律法規の制約を受けています。もし違反すれば、私たちは深刻な結果に直面する可能性があります。
その他の事項のうち、米国と外国の反腐敗、反マネーロンダリング、輸出規制、制裁および他の貿易法律法規は、総称して貿易法と呼ばれ、会社およびその従業員、代理人、臨床研究組織、法律顧問、コンサルタント、請負業者および他のパートナーの許可、約束、提供、提供、誘致、直接または間接的に公共または民間部門の受給者に腐敗または不正なお金または任意の他の価値のあるものを支払うことを禁止する。貿易法違反は、巨額の刑事罰金および民事処罰、監禁、監禁を招くことができる。貿易特権の喪失、除名、税務再評価、違約と詐欺訴訟、名声損害、その他の結果。私たちは政府機関や政府付属病院、大学、その他の組織の役人や従業員と直接または間接的な相互作用を持っています。私たちはまた、私たちの非アメリカ業務が時間の経過とともに増加すると予想しています。我々は、第三者を招いて臨床試験および/または必要なbr許可、許可証、特許登録、および他の規制承認を得ることを計画しており、私たちは、このような活動を明確に許可していなくても、または事前にそのような活動を知っていなくても、私たちの人員、代理またはパートナーの腐敗または他の不正活動のために責任を負わなければならないかもしれない。
51
我々が許可したものと共通して所有する知的財産権に関するリスク
Yeda 2015ライセンス契約を含むYedaと維持されているライセンス契約は、私たちの業務に非常に重要です。もし私たちまたは私たちの許可プロトコルの他のbr側が許可プロトコルを十分に履行できなかった場合、または私たちまたは彼らが許可プロトコルを終了した場合、私たちのバクテリオファージに基づく候補治療製品の開発、テスト、製造、生産、および販売は延期または終了され、私たちの業務は悪影響を受けるだろう。
Br}Yeda 2015ライセンスプロトコルは、プロトコルで指定された候補製品、私たちのバクテリオファージ発見プラットフォームのための候補バクテリオファージ製品、ならびにbrプロトコルで決定されたコンサルタントの仕事および私たちが支援したさらなる研究によって生成された候補バクテリオファージ製品の特許、研究および他の権利を含むバクテリオファージベースの候補治療製品の開発、試験、製造、製造および販売に関連する特定の技術および研究情報のグローバル独占許可を規定する。YEDA 2015ライセンスプロトコルは、YEDA 2015ライセンスプロトコルによってカバーされた最後の特許が満了し、15年連続してどの製品もどの国/地域で初めて商業販売されなかったときに を終了するかを終了する。YEDA 2015ライセンス契約に記載されているいくつかの勤勉さおよび開発要件およびマイルストーンを遵守できない場合、YEDAはまた、合意 を終了することができます。 私たちまたはYEDAは、通知期間後、または他方の清算、破産、債務不履行、解散、または他の同様の業務終了後に合意を終了することができます。契約終了時には、時間の経過に加えて、Yeda 2015ライセンス協定に記載されている私たちのノウハウおよび研究成果に関する権利をYedaに付与しなければならない。もしYedaがその後、私たちの権利を使用する第三者に許可を与えるならば、Yedaがその許可によって実際に受け取った純収益を共有する権利があることを前提として、Yedaに排他的で撤回できない、永久的、全額支払い可能な再許可の世界的許可を付与しなければならない。私たちが2015年のライセンス契約に関連して発生した開発費用の上限に制限されています。YEDA 2015ライセンス契約の詳細については、“を参照されたい”業務−材料プロトコル−ライセンスbr}プロトコル−業達とのライセンスプロトコル。”
私たちのライセンス契約を終了することは、私たちの製品および商業化の大きな遅延をもたらす可能性があり、これは、私たちの内部能力を最初に拡張することなく、または第三者と他の合意を達成することなく、私たちの候補製品(バクテリオファージベースの治療製品を含む)を商業化することを阻止するかもしれない。どんな代替協力やライセンスも私たちに不利になるかもしれません。
我々の は第三者の許可の知的財産権に高度に依存しており、これらの許可のいずれかを終了または制限することは、 が重大な権利の損失を招き、私たちの業務に実質的な損害を与える可能性がある。
私たちは現在、第三者協力者によって提供された許可に依存しています。私たちの技術のいくつかの側面と私たちの既存のいくつかのプロジェクトのために使用されています。特に、私たちはYedaを含む第三者が持っているいくつかの特許の独占的で印税の許可を得ています。YEDA 2015ライセンスプロトコルは、我々のバクテリオファージ発見プラットフォームで使用されるバクテリオファージベースの候補治療製品の開発、試験、製造、生産および販売に関連するいくつかの技術および研究情報の許可、ならびにプロトコルで決定されたコンサルタントの仕事および私たちが支援したさらなる研究によって生成された候補バクテリオファージ製品の特許、研究、およびbr}の他の権利を提供する。
もし私たちが支払い条項を含むライセンス契約下の義務を履行できなかった場合、私たちのライセンス側は、私たちのライセンス契約を終了する権利がある可能性があり、この場合、私たちは、これらのライセンスプロトコルに含まれる製品 を開発、製造、マーケティング、または販売することができない可能性がある。もし私たちが契約義務を履行しなければ、私たちはまた許可協定による他の処罰に直面するかもしれない。このようなイベント は,このようなライセンスプロトコルに基づいて我々が開発した製品の価値に大きな悪影響を与える可能性がある.私たちの1つまたは複数のライセンスプロトコルを終了するか、またはこれらのライセンスプロトコルの下での私たちの権利を減少またはキャンセルすることは、同じ特典条項で提供できないか、または全く得られない可能性がある新しいまたは回復されたライセンスプロトコルを交渉しなければならないことをもたらす可能性があり、これは、影響を受けた候補製品を商業化できないことを意味するかもしれない。
将来的には、第三者からの特定の特許権およびノウハウの追加許可に依存する可能性があり、これらの特許権およびノウハウは、私たちの候補製品および独自製品プラットフォームの開発に非常に重要である。我々が将来的に許可を得る特許権 は、1つ以上の第三者の権利によって保持される可能性がある。したがって、このような第三者はそのような知的財産権のいくつかの権利を持っている可能性がある。
52
さらに、このようなライセンス契約の条項によれば、私たちは、第三者から許可を得た特許および特許出願の準備、届出、起訴および維持を制御する権利がなく、特許および特許出願の実行および弁護を制御する権利がない可能性がある。私たちは、私たちの許可者によって制御される私たちの許可内の特許出願(およびそこから発行される任意の特許)が、私たちの業務の最良の利益に合った方法で準備、提出、起訴、維持、強制執行、および弁護されるかどうかを決定することはできません。私たちの許可者がこのような特許権を起訴、維持、強制的に執行し、擁護することができなかった場合、またはこれらの特許出願(またはそれによって発行された任意の特許)の権利を失う場合、私たちが許可を得る権利は減少または廃止される可能性があり、私たちは、このような許可権に制約された任意の候補製品および独自製品プラットフォーム技術を開発および商業化する権利が悪影響を受ける可能性があり、競争相手の競合製品の製造、使用および販売を阻止することができない可能性がある。私たちの将来の潜在的許可者たちのこのような活動が適用された法律法規に適合しているかどうか、あるいは効果的かつ強制的に実行可能な特許や他の知的財産権が生じるかどうかを判断することはできません。さらに、私たちが第三者または第三者から許可される可能性のある特許および特許出願への起訴を制御する権利がある可能性があっても、私たちbr}は、特許起訴を制御する日前に発生するので、私たちの潜在的な将来の許可者、ライセンシーおよびその弁護士の行動または非作為によって悪影響または損害を受ける可能性がある。
バイオ製薬会社(我々と我々の許可者を含む)の特許地位は通常不確実であり,複雑な法律や事実に関連して考慮されているため,その有効性や実行可能性を確実に予測することはできない。私たちの許可と共同所有の知的財産権は疑問視され、実行不可能、無効、または回避されるかもしれない。私たちと私たちの許可者は、これらの権利(およびそれがカバーする製品およびサービス)が有効かつ強制的に実行可能な特許、著作権または商標によって保護されているか、または商業秘密として効果的に維持されていることを前提として、第三者の無許可使用から私たちの知的財産権を保護することができるだろう。
我々のライセンシーまたは私たちによって取得された任意の特許は、再審査されるか、または他の方法で無効または最終的に発見されて実行できない可能性がある。特許出願プロセスおよび特許紛争を管理するプロセスは、時間がかかり高価である可能性がある。私たちまたは私たちの許可者のうちの1つが第三者に対して当社の製品に関連する特許を強制的に執行するために法的訴訟を提起した場合、そのような訴訟における被告は、主張された特許を無効および/または強制的に実行することができない。米国の特許訴訟では,被告 が無効または強制執行不可能と主張する反訴が一般的であり,被告が主題特許や関連特許に対して米国特許商標局に提出した有効性挑戦も一般的である。有効性疑問の理由は、新規性の欠如、明らかな、実施できない、書面記述要件を満たしていない、不確実性、 および/または特許可能な主題を主張できなかったことを含む、いくつかの法定特許性要件 のいずれかを満たすことができなかったと言われている可能性がある。主張を実行できない理由は,係争特許/Sの起訴に関係している人が,米国特許商標局に重要な情報を故意に隠したり,起訴中に誤った陳述をしたりしている可能性があるからである.実行不可能な主張の他の理由には,特許権の濫用や反競争使用に関する告発, および欺瞞的意図を持つ不正解リストに関する告発がある.第三者は米国特許商標局に類似したクレームを提起することもでき、訴訟範囲外でも同様である。無効および/または実行不可能な断言に関するいかなる結果も予測不可能である.被告または第三者が無効および/または強制執行できない法的主張で勝訴した場合、私たちおよび私たちの許可者は、挑戦された特許/Sの少なくとも一部、さらには全ての権利要件を失うであろう。このような特許保護の喪失は、私たちのbr}業務に実質的な悪影響を及ぼす可能性がある。
私たち は特許とノウハウに依存している。もし私たちがこの知的財産権を十分に保護できなければ、あるいは私たちがそうしなければ、 は私たちの製品のマーケティングに独占経営権がなく、私たちが製品を商業化する能力が影響を受ける可能性がある。
私たちのビジネス成功は、他の人が私たちの候補製品を販売することを防止し、これらの特許を侵害から保護し、実行し、他人の固有の権利を侵害することなく運営するために、私たちが十分な特許保護を得ることができるかどうかにある程度依存するであろう。私たちの候補製品を第三者から不正に使用しないように保護することは、私たちの候補製品またはその製造または使用をカバーする有効かつ強制的に実行可能な特許を持っているかどうか、または効果的な商業秘密保護を持っているかどうかにかかっている。もし私たちの特許出願が発行された特許が生成されていない場合、または私たちの特許が無効であることが発見された場合、私たちは、その中で主張されている発明を製造、使用、または販売する能力を失う。私たちは限られた数の特許と出願されている特許を持っている。
53
バイオテクノロジー会社の特許地位は複雑な法律と事実の問題に関連して不確実である可能性がある。これは,これまでバイオテクノロジー特許の政策応用と政策変化が世界的に審査·実行されてきたことが一致しなかったためである。一部の国の法律は知的財産権の保護程度は特許制度が完備されている国の法律に及ばない可能性があり、これらの国は私たちの知的財産権を保護するための十分な規則と手続きが不足している可能性がある。また,特許法や特許法解釈の変化 は我々の知的財産権の価値を低下させる可能性がある。私たちは私たちのすべての特許出願が特許の発行につながることを保証することはできません。私たちの特許出願や私たちが許可する可能性のある他の特許出願で許容される可能性のあるクレームの広さを予測することもできません。
Leahy-Smith America発明法は、第三者がUSPTO特許裁判および控訴委員会の前で発行された特許の有効性に疑問を提起することができるように、当事者間審査、知的財産権または認可後審査のような特許付与後審査手続きに関連するプログラムを規定している。各プロセスには異なる資格基準と提示可能な異なる特許性挑戦がある。知的財産権 は、誰(特許に対して1年以上訴訟を提起した当事者を除く)が、既存技術の期待または明らかな理由で特許の有効性に疑問を提起することを可能にする。医薬品をカバーする特許は知的財産権の面で後発薬会社とヘッジファンドの攻撃を受けている。疑問視された特許が発行されてから9ヶ月以内である場合、第三者は、米国特許商標局に許可後審査を申請することができ、これは、任意の無効理由に基づくことができ、従来技術特許または印刷出版物に限定されるものではない。
発行後の手続きでは、米国特許商標局の規則および条例は、通常、特許所有者ではなく特許挑戦者をひいきする傾向がある。例えば、地域裁判所訴訟とは異なり、発行後の手続きにおいて疑問視される権利要件は、最も広範な合理的な意味を与えられ、これは、特許要件が従来技術によって無効と宣言される可能性があるか、または特許仕様においてサポートが不足している可能性を増加させる。別の例として、地域裁判所訴訟とは異なり、発行された特許は有効性推定がないため、挑戦者が無効を証明する責任は、より高い明確かつ納得できる証拠基準ではなく、証拠の優位性によって証明される。これらのルールや他のルールにより,米国特許商標局が公表した統計データによると,発行後のプログラムでは,高い割合の債権が無効と宣言されている.また、ごく少数の例外を除いて、米国特許商標局に締約国間審査や贈与後審査を申請する長期的な要求はない。言い換えれば、侵害されていない、または特許標的に対して商業的利益が不足していると告発されていない会社は、依然として発行された特許の審査を米国特許商標局に申請することができる。したがって、たとえ私たちが特許を発行しても、これらの特許の下での私たちの権利は挑戦される可能性があり、 は最終的に競争製品やプロセスに対して十分な保護を提供することができない。
将来の私たちの所有権に対する保護の程度は不確定であり、法的手段は限られた保護しか提供できないため、私たちの権利を十分に保護できないかもしれないし、私たちの競争優位性を獲得したり維持したりすることができるかもしれない。例えば:
| ● | 私たちは私たちの発明のために特許出願を提出した最初の会社ではないかもしれない |
| ● | 他の会社は、私たちの特許範囲内ではなく、私たちの任意の候補製品と類似または代替の候補製品を独立して開発することができる |
| ● | 私たちが処理している特許出願は、発行されたbr件の特許を生成しないかもしれない |
| ● | 私たちが発行した特許は、商業的に実行可能な製品に基礎を提供しないかもしれないし、競争優位性を提供してくれないかもしれないし、第三者の挑戦を受ける可能性がある |
| ● | 他の人たちは私たちの特許範囲を超えた競争力のある製品を生産するために、私たちの特許声明を中心に設計されるかもしれない |
| ● | 私たちは私たちの候補製品に関連する他のbr可特許技術を開発しないかもしれない |
| ● | 私たちは国家司法管轄区の指定代理人の勤勉さに依存しています。彼らは私たちを代表して行動し、未解決の国内及び外国特許出願の起訴を制御し、付与された国内及び外国特許を維持します。 |
54
発行された特許は、私たちが特許技術を実践したり、特許製品を商業化する権利があることを保証しない。第三者 は、私たちの特許製品を商業化し、私たちの特許技術を実践することを阻止する阻止特許を持っている可能性があります。 私たちが発行した特許と将来発行される可能性のある特許は、挑戦、無効、または回避される可能性があり、これは、競争相手が同じまたは関連する候補製品を販売することを阻止する能力を制限するか、または私たちの候補製品の特許保護期間を制限する可能性があります。さらに、潜在的な製品の開発、テスト、および規制審査には多くの時間がかかるため、私たちの任意の候補製品が商業化できる前に、どの関連特許も商業化後の短い期間にわたって失効または有効に維持され、それによって、その特許の任意の利点を弱める可能性がある。これらの特許は 特許期間を延長できないかもしれない。
私たちが私たちの候補製品および独自製品プラットフォームを開発し、商業化する権利は、他の人が私たちに現在および未来のライセンスを付与する条項および条件によってある程度制限される可能性がある。
私たちのいくつかの許可権利は私たちに製品とサービスの様々な側面に対する運営自由を提供することができる。私たちは他の会社から追加のbrライセンスを取得して、私たちの研究、開発、商業化活動を推進する必要があるかもしれない。
ライセンス契約により、私たちとライセンシーとの間で知的財産権紛争が発生する可能性があります
| ● | ライセンス契約に従って付与された権利範囲および解釈に関連する他の問題; |
| ● | 私たちの製品、サービス、技術、プロセス、およびライセンス契約に拘束されていないライセンス側の知的財産権がどの程度侵害されているかどうか |
| ● | 私たちは協力開発関係の下で特許と他の権利を第三者に再許可する権利; |
| ● | 私たちのライセンス契約の下での義務と、どのような活動がこれらの職務義務を満たしていますか |
| ● | 私たちの許可者と私たちおよびその協力者が共同で知的財産権を創造または使用することによって生成される発明およびノウハウの在庫および所有権 および |
| ● | 特許技術の発明優先権。 |
もし私たちがこのような紛争に勝つことができなければ、私たちはこのような許可協定の下での権利の一部または全部を失うかもしれない。
また,我々は現在,第三者の知的財産権や技術を許可するプロトコルは複雑であり,このようなプロトコルのいくつかの 条項は様々な解釈の影響を受ける可能性がある.起こりうる任意の契約解釈分岐の解決 は、関連する知的財産権または技術に対する私たちの権利と考えられる範囲を縮小することができ、または可能な を増加させることは、関連プロトコルにおける私たちの財務または他の義務であると考えられ、これらのいずれも、私たちの業務、財務状況、運営結果、および見通しに大きな悪影響を及ぼす可能性がある。さらに、我々が許可している知的財産権をめぐる紛争が、商業的に許容可能な条項で現在の許可スケジュールを維持する能力を阻害または弱めると、影響を受けた製品やサービスの開発に成功し、商業化することができない可能性があり、これは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
ライセンス契約がなければ,これらのプロトコルに基づいて特許を侵害する可能性があり,ライセンスプロトコルが終了すれば,許可側から訴訟を受ける可能性がある.訴訟は私たちに巨額の費用をもたらし、私たちの経営陣の注意を分散させるかもしれない。もし私たちが勝訴しなければ、私たちは三倍の損害賠償金、弁護士費、費用と費用、印税を含む損害賠償金の支払いを要求されるかもしれません。私たちはまた私たちの製品やサービスの販売を禁止される可能性があります。これは私たちが製品やサービスを提供する能力、私たちが運営を続ける能力、そして私たちの財務状況に悪影響を及ぼすかもしれません。
55
もし私たちが第三者の権利を侵害した場合、私たちは製品の販売を阻止され、損害賠償および/または印税の支払いを余儀なくされ、brに抗弁訴訟を強要される可能性がある。
我々 は,我々が現在開発している製品がどの第三者の権利を侵害しているか,あるいは第三者 に侵害されているとは思わない.しかし,我々の技術が将来他人の権利侵害や他人に侵害されることが発見されないことは保証されない.さらに、場合によっては、特許出願は特許発行前に秘密にされている。科学的または特許文献で発見された発行時間は、通常、基礎発見および特許出願の提出日よりもはるかに遅い。特許の発行には数年を要する可能性があるため、我々の知らない現在承認されている出願が存在する可能性があり、これは、私たちの製品または候補製品が発行された特許を侵害する可能性がある。例えば、サポートを提供する処理中のbr特許出願が存在する可能性があり、または、我々の1つまたは複数の製品によって発行された特許を侵害するクレームを支援するように修正することができる。この場合、他の人は私たちに侵害請求をするかもしれません。もし私たちがこれらの特許を侵害したり、彼らの知的財産権を不正に使用していることが発見されたら、もし私たちがこれらの第三者の特許権を故意に侵害していることが発見されたら、私たちはbrの3倍の損害賠償金を含めて損害賠償金の支払いを余儀なくされるかもしれません。
私たちが支払わなければならない可能性のある任意の損害賠償に加えて、私たちはその知的財産権の所有者から許可を得ること、印税協定を締結すること、または私たちの製品を再設計して、この知的財産権の使用を避けることを要求されるかもしれない。このような処罰のすべては経済的でないか不可能であることが証明されるかもしれない。私たちは商業的に合理的な条項でこのような許可書や知的財産権 を得ることができないかもしれない。たとえ我々が許可を得ることができても,我々の競争相手が同じ許可技術にアクセスできるように非排他的である可能性がある. この場合,代替技術の開発や許可に時間と資源がかかる可能性がある.もし私たちがそうできなければ、私たちは影響を受けた製品を開発したり、商業化することができないかもしれません。これは私たちの業務に実質的な損害を与える可能性があります。逆に,我々 は我々の許可や共有技術を侵害する第三者にクレームを付けることができない可能性がある.したがって、私たちが許可を得て共有している技術は、競争相手に対して十分な保護を提供できないかもしれない。
製薬業界の特徴は特許と他の知的財産権に関する訴訟範囲が広いことだ。さらに, は,解決策が我々に有利であっても,我々が許可しているおよび/または共有知的財産権に関する任意の訴訟や他の手続きは,我々にとって大きなコストとなる可能性がある.このような訴訟はすべて私たちの管理努力を分散させ、私たちはこのような訴訟を成功させるために十分な資源 を持っていないかもしれない。任意の訴訟の開始と継続による不確実性は、私たちが運営を継続する能力を制限するかもしれない。
また, は,我々の開発プロセスが他の開発プロジェクト候補に及ぶ可能性があり,第三者が持つ独占権を使用する必要がある可能性があるため,我々の業務の成長は,これらの独占権を取得,許可,または使用する能力にある程度依存する可能性がある.さらに、我々の候補開発者は、効率的かつ効率的に動作するために特定の処方を必要とする可能性があり、これらの権利は、他の人によって保持される可能性がある。 私たちは、私たちが決定した第三者から任意の成分、使用方法、プロセス、または他の第三者知的財産権 を取得または付与することができない可能性がある。第三者知的財産権の許可·買収は競争分野であり、多くのより成熟した企業も許可や買収を求めており、魅力的と考えられる第三者知的財産権の戦略を求めている可能性がある。これらの老舗会社は私たちより競争優位を持っているかもしれません。それらの規模、現金資源、br及びもっと強い臨床開発と商業化能力のためです。
例えば,米国や外国の学術機関と協力し,これらの機関との書面合意に基づいて我々の臨床前研究や開発を加速させることがある。一般に、これらの機関は、協働によって得られた任意の技術的権利のライセンスを交渉するためのオプションを提供してくれる。知的財産権のこのような最初の交渉権にかかわらず、私たちは指定された時間範囲内で、または私たちが受け入れられる条項の下でライセンスを交渉することができないかもしれない。もし私たち がそれができなければ、その機関は知的財産権を他の側に提供する可能性があり、これは私たちの計画を継続することを阻止するかもしれない。
しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。私たちはまた、私たちの投資が適切なリターンを得るための条項で許可したり、第三者知的財産権を取得することができないかもしれません。もし私たちが第三者知的財産権を要求する権利を得ることができなければ、私たちの業務、財務状況、成長の見通しは影響を受ける可能性があります。
56
私たち は、買収、許可、または他の方法で、私たちの候補製品、独自の 製品プラットフォーム技術、または他の技術に必要な権利を得ることができないかもしれません。
我々 は現在、第三者からの許可によっていくつかの知的財産権を有する権利を有しており、私たちの候補製品および独自の 製品プラットフォーム技術を開発している。いくつかの医療会社および学術機関は、バクテリオファージに基づく療法分野で、特許を有している可能性があり、および/または提出されており、我々の業務に関連する可能性のある特許出願を提出している可能性があるバクテリオファージに基づく治療分野で競合している。これらの第三者特許の侵害を回避するためには,このような第三者知的財産権所有者からそのような特許の許可を得ることが必要または慎重であることが分かる可能性がある.私たちはまた、第三者からいくつかの技術のライセンスを取得する必要があるかもしれません。私たちは、現在または未来の候補製品のために、これらの技術を評価しているかもしれません。しかし、私たちは、合理的なコストまたは合理的な条項で第三者からそのような許可を得ることができないか、または他の方法で取得または許可することができないかもしれません 私たちの現在または未来の候補製品および私たちの独自製品プラットフォームにとって必要な任意の成分、使用方法、プロセス、または他の知的財産権です。 第三者知的財産権の許可または取得は競争分野であり、より多くの老舗企業 は、魅力的または必要と考えられる第三者知的財産権許可または取得戦略をとる可能性があります。これらの老舗会社は私たちより競争優位を持っているかもしれません。それらの規模、資本資源及びもっと強い臨床開発と商業化能力のためです。しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。私たちはまた私たちが投資から適切な見返りを得ることを可能にする条項に従って許可したり、第三者知的財産権を取得することができないかもしれない。
もし私たちが必要な第三者知的財産権の権利を得ようとしたが最終的に失敗した場合、私たちは私たちの技術、候補製品、またはそれらを製造する方法、または代替技術を開発または許可するのに多くの時間と資源をかけて再設計する必要があるかもしれません。これらはすべて技術的または商業的に不可能かもしれません。もし私たちがそうできなければ、私たちは影響を受けた候補製品を開発したり、それを商業化することができず、私たちの既存の独自のbr製品プラットフォーム技術を使用し続けることができないかもしれません。これは、私たちの業務、財務状況、運営結果、および将来性を深刻に損なう可能性があります。
我々は我々の独自の製品プラットフォームに依存してバクテリオファージに基づく療法を識別する。もし私たちの競争相手が似たようなプラットフォームを開発して競争相手の候補製品を開発すれば、私たちの競争地位は実質的に損なわれる可能性がある。
私たちは、独自の技術、発明、および他の固有の情報に依存して、私たちの競争地位を強化する。私たちの独自製品プラットフォームのために、私たちはノウハウが私たちの主要な知的財産だと思う。私たちの臨床試験は臨床データを収集することを可能にして、私たちはこれらのデータをフィードバックサイクルとして使用して、私たちの独自の製品プラットフォームを改善します。特に,この独自製品プラットフォームを尊重した場合,時間の経過とともに,これらのデータは独立して開発され,この方法や技術者の流れを記述した定期刊行物文章が発表されることにより業界内に伝播する可能性が予想される。
私たち は、私たちの任意の候補製品と競合する可能性のある製品を識別して開発するために、私たちの競争相手が必要な知識を持っているか、または獲得した可能性があることを排除して、私たちの既知のbrデータと類似したデータを分析して表現することはできません。今まで、私たちの競争相手はもっと多くの財務、製品開発、技術、人的資源を持っているかもしれない。また,我々の競争相手 は,翻訳科学的手法を用いて候補製品を認識·開発する上でより豊富な経験を持つ可能性がある.
私たちの競争相手は、私たちの独自製品 プラットフォームと同じまたは同様の技術または方法を使用して、彼ら自身の候補製品を開発することを禁止できないかもしれない。もし私たちの競争相手が共同療法を開発すれば、私たちが将来性のある製品や候補製品を開発·マーケティングする能力は大幅に低下する可能性があり、これは私たちの業務、財務状況、見通し、運営結果に実質的な悪影響を及ぼす可能性がある。
57
私たちは商業秘密と他の形態の非特許知的財産権保護に依存している。もし私たちが私たちの商業機密を保護できなければ、他の会社は私たちともっと効果的に競争するかもしれない。
私たちのビジネス秘密に依存して、私たちの技術のいくつかの態様は、私たちのバクテリオファージの製造と精製の独自のプロセスを含む。ビジネス秘密は保護が困難であり,特に製薬業界では,規制承認過程において,製品に関する多くの情報が公開されなければならない。私たちは私たちのビジネス秘密を保護するために合理的に努力しているにもかかわらず、私たちの従業員、コンサルタント、請負業者、外部科学協力者、および他のコンサルタントは、私たちの情報を競争相手に意図的にあるいは意図的に漏らしてしまうかもしれない。第三者が私たちの商業秘密情報を不正に取得して使用することを強制することは高価で時間がかかり、結果は予測できない。さらに、米国以外の裁判所は商業機密を保護しないことをあまり望まないかもしれない。また,我々の競争相手は同等の知識,方法,ノウハウを自主的に開発することができる.
もし私たちが第三者の知的財産権侵害で起訴された場合、または私たちが介入手続きを余儀なくされた場合、費用がかかり、その訴訟または介入における不利な結果は、私たちの業務に大きな悪影響を及ぼすだろう。
私たちが候補製品を商業化する能力は、第三者の独占権を侵害することなく候補製品を開発、製造、マーケティング、販売する能力に依存する。第三者が所有する多くの米国および外国特許および特許出願は、抗感染製品の一般的な分野または我々の候補製品に関連する可能性のある他の分野に存在する。もし私たちが特許を侵害していることが証明された場合、特許 が無効であることが証明できなければ、私たちは要求された発明の使用または販売を禁止される可能性がある。さらに、特許出願が発行されるまでに数年かかる可能性があるため、現在処理されている私たちの未知の特許出願が存在する可能性があり、これらの出願は、今後、私たちの候補製品が発行された特許を侵害する可能性があるか、または私たちが所有または許可している特許または出願に関する妨害訴訟手続き を引き起こす可能性がある。私たちが知らない既存の特許が存在する可能性もあり、私たちの候補製品は無意識に私たちの特許を侵害しているかもしれないし、または妨害手続きに巻き込まれている可能性がある。
生物技術と製薬業界の特徴は大量の特許が存在し、特許侵害疑惑で頻繁に訴訟を提起することである。私たちの候補製品が臨床試験段階にある限り、私たちの臨床活動 はアメリカ“アメリカ法典”第271(E)条に規定されている免除範囲に属し、この条項は開発とFDAへの情報の合理的な提出に関する特許侵害責任を免除していると信じている。著者らの臨床研究候補薬物製品の商業化に伴い、私たちに対する特許侵害クレームの可能性が増加した。私たちは私たちの活性臨床研究薬と私たちがそれらを製造するための方法と、私たちが普及しようとしている方法 が他の側の特許や他の独占権を侵害しないことを確認しようと努力しているが、私たちはそれらができないことを確認することはできません。競争相手や他の当事者はいかなる場合でも私たちが彼らの独占権を侵害していると主張する可能性があります。
私たちは私たちの候補製品、私たちがそれらを製造するための方法、または私たちがそれらの用途を広めるつもりだと主張して、他の人の知的財産権を侵害しているため、未来の訴訟に直面するかもしれない。私たちが私たちの候補製品を製造して商業化する能力があるかどうかは、私たちが採用した製造プロセスと私たちの候補製品の使用が第三者特許を侵害していないことを証明する能力があるかどうかにかかっているかもしれません。第三者特許が私たちの候補製品またはその使用または製造をカバーしていることが発見された場合、私たちは損害賠償金の支払いを要求されるか、または禁止される可能性があるので、br許可証を取得しない限り、私たちの候補製品を商業化することができない。私たちは許容可能な条項で許可証を得ることができないかもしれない。
我々 は,譲渡された職務発明権に対する従業員の報酬や使用料のクレームを受ける可能性があり,これは 訴訟を招き,我々の業務に悪影響を与える可能性がある.
私たちの知的財産権の大部分は私たちの職員たちが私たちのために働く過程で開発されたものだ。イスラエルの“特許法”(第5727-1967号)又は“特許法”によれば、従業員が在任中に発想した発明は、1つの会社に雇用された範囲の一部として、“職務発明”とみなされ、雇用主に属し、従業員と雇用主との間に従業員に職務発明権を与えない具体的な合意である。特許法はまた,雇用主と従業員の間にこのような合意がない場合,イスラエル補償·使用料委員会は,特許法に基づいて構成された機関である従業員がその発明によって報酬を得る権利があるかどうかを決定しなければならないと規定している。私たちは通常、従業員と発明協定を締結し、この合意に基づいて、従業員は、その雇用または採用の範囲内で創造された任意の発明のすべての権利を私たちに譲渡する。私たちの従業員は私たちの職務発明権の譲渡に同意したにもかかわらず、譲渡された発明に対する報酬の支払いを要求するクレームに直面する可能性がある。このようなクレームのため、私たちは、私たちの現職または前任者従業員に追加の報酬または特許使用料を支払うことを要求されるか、または訴訟を提起されることを余儀なくされる可能性があり、これは、私たちの業務に負の影響を与える可能性がある。
58
我々の第三者への依存に関するリスク
我々の は第三者に依存して我々の臨床試験を継続しており,これらの第三者の表現は満足できない可能性があり, は締め切りまでにこのような試験を完了できないことを含む。
我々は引き続き契約研究組織やCROと臨床研究者などの第三者に依存して、私たちの臨床試験を行い、管理している。
私たちの研究開発活動におけるこれらの第三者への依存は、これらの活動に対する私たちの統制を減少させるだろうが、私たちの責任を軽減することはない。例えば、私たちは私たちのすべての臨床試験が試験の全体的な研究計画と方案に従って行われていることを確実にする責任がある。さらに、FDAは、データおよび報告の結果が信頼性および正確であることを保証し、試験参加者の権利、安全、および福祉を保護するために、GCPに従って臨床試験結果を行い、記録し、報告することを要求する。他の国の規制機関もまた私たちが守らなければならない臨床試験に要求がある。実施中の臨床試験を指定された時間範囲で登録し,完成したbr臨床試験結果を政府後援のデータベースClinicaltrials.govに発表する必要がある。そうしないと罰金、否定的な宣伝、民事と刑事制裁につながる可能性があります。
さらに, これらの第三者は他のエンティティと関係がある可能性があり,その中のいくつかは我々の競争相手である可能性がある.これらの第三者がその契約義務の履行に成功しなかった場合、予想される締め切り前に仕事を完了できなかった場合、停止に遭遇し、私たちとの合意を終了するか、または交換する必要があるか、または法規の要求または私たちが宣言した方案に従って私たちの臨床試験を行わなかった場合、私たちは代替第三者と新しい計画を達成する必要があるかもしれません。これは困難、高価または不可能である可能性があり、私たちの臨床試験は延長、遅延、終了、または重複する必要があります。上記のいずれかが発生した場合、候補製品のマーケティング承認を得ることができない場合や、候補製品の商業化に成功する努力を延期することができない可能性もあります。
私たちbrはまた、他の第三者に依存して、私たちの臨床試験の貯蔵と配布薬品の供給に依存しています。私たちのディーラーはもしどんな業績ミスが発生したら、私たちの候補製品の臨床開発或いはマーケティング審査或いは製品の商業化を延期し、追加の損失をもたらし、私たちの潜在的な製品収入を奪うかもしれません。
第三者関係 は我々の業務に非常に重要である.もし私たちが私たちの協力を維持したり、新しい関係を作ることができない場合、あるいはこれらの 関係が成功しなければ、私たちの業務は悪影響を受ける可能性があります。
我々の製品開発能力は限られており,まだ販売,マーケティングあるいは流通能力を備えていない.したがって,我々は他社や学術機関と関係を築き,重要な技術を提供してくれており,将来的にはこれらの協力や他の協力を通じてより多くの技術や資金を得ることができるかもしれない。私たちが築いた関係は多くのリスクをもたらすかもしれません
| ● | 第三者は彼らが適用する努力と資源を決定する上で大きな裁量権を持っており、将来の第三者協力者も大きな裁量権を持っているかもしれない |
| ● | 現在および未来の第三者 は、予想通りにその義務を履行できない可能性がある |
| ● | 現在および将来の第三者 は、規制部門の承認を得た任意の候補製品を開発および商業化してはならないか、または臨床試験結果、第三者戦略重点の変化または利用可能な資金または外部要因(リソースを移転する可能性があるか、または競争優先度を作成する戦略的取引を作成する可能性がある)に基づいて、開発または商業化計画を継続しないことを選択してはならない |
59
| ● | 第三者は臨床試験を延期し、臨床試験計画に資金不足を提供し、臨床試験を停止或いは候補製品を放棄し、新しい臨床試験を繰り返し或いは行うことができ、或いは新しい調合の候補製品に臨床試験を行うことを要求することができる |
| ● | 第三者が競争力のある製品の開発に成功する可能性があると考え、または私たちよりも経済的に魅力的な条項で商業化できる場合、現在および将来の第三者は、第三者と共に直接または間接的に私たちの製品および候補製品と競合する製品を独自に開発または間接的に開発することができる |
| ● | 私たちと協力して発見された候補製品は、私たちの現在または未来の第三者によって、彼ら自身の候補製品または製品と競争するとみなされる可能性があり、これは、これらの第三者が私たちの候補製品の商業化のための資源を投入することを停止させる可能性がある |
| ● | 現在および将来の第三者は、候補製品または製品の開発、製造、流通、またはマーケティングに関する適用法規要件を遵守しない可能性がある |
| ● | 現在および未来には、規制部門の承認を得た場合、そのような製品または製品をマーケティングおよび流通するために十分なリソースが投入されていない可能性がある1つまたは複数の候補製品に対してマーケティングおよび流通権利を有する第三者が存在する |
| ● | 特許権、契約解釈、または第一選択開発プロセスにおける分岐を含む現在または将来の第三者との分岐は、候補製品の研究、開発または商業化の遅延または終了をもたらす可能性があり、候補製品に対して追加の責任を負うことになる可能性があり、または訴訟または仲裁を引き起こす可能性があり、いずれも時間がかかり高価になる可能性がある |
| ● | 現在および未来の第三者は、私たちの知的財産権を正確に維持したり守ったりすることができないかもしれないし、私たちの固有の情報を何らかの方法で使用して、私たちの知的財産権や固有の情報を危険にさらしたり、破壊したり、潜在的な訴訟に直面させたりするために訴訟を招く可能性がある |
| ● | 現在と未来の第三者は他人の知的財産権を侵害する可能性があり、これは私たちを訴訟と潜在的な責任に直面させるかもしれない |
| ● | 現在および将来の第三者は、規制フレームワーク(例えば、ネットワークセキュリティおよび/またはプライバシーフレームワークに限定されないが)に違反する可能性があり、これは、私たちを訴訟および潜在的な責任に直面させたり、要求したり、彼らとの関係を終了させたりする可能性がある |
| ● | 現在または将来の第三者が業務合併に参加する場合、協力者は、私たちが許可した任意の候補製品の開発または商業化を淡水化または終了する可能性がある |
| ● | パートナーは現在と未来の協力関係を終了することができ、終了すれば、適用可能な候補製品をさらに開発するために、または商業化するために、より多くの資金を集める必要があるかもしれない。 |
もし私たちの関係が製品の発見、開発、商業化に成功しなかった場合、または私たちの第三者協力者の一人が私たちとの合意を終了した場合、私たちは協力項目の下で未来の研究資金、マイルストーン、または印税支払いを受けないかもしれない。これらの合意に基づいて予想される資金が得られなければ、私たちの技術と候補製品の開発が遅れる可能性があり、候補製品と私たちの技術を開発するために追加のbr資源が必要かもしれません。さらに、もし私たちの現在または未来の任意の第三者協力者が私たちとの合意 を終了した場合、私たちは新しい協力者を引き付けることがもっと難しいことを発見するかもしれません。商業および金融界での私たちの名声は不利な影響を受ける可能性があります。
60
交渉と記録関係は複雑で時間がかかる.また,最近では大手製薬会社間の業務合併数が大きく,将来の潜在的パートナー数の減少を招いている。私たちは適切な協力者を探す上で激しい競争に直面しています。 私たちが協力について最終的な合意を達成できるかどうかは、協力者の資源と専門知識の評価、提案された協力の条項と条件、提案された協力者の多くの要素の評価に依存します。
私たちは協力関係の維持や構築に成功しないかもしれません これは私たちの開発能力に悪影響を及ぼす可能性があり、必要な規制の承認を得られれば、私たちの候補製品を商業化するかもしれません。
将来,我々の臨床 開発を進めるために,あるいは任意の潜在的候補製品や技術の外部許可に関連して,協力 合意を求めることが可能である。さらに、私たちは、米国内または海外で私たちの候補製品を開発、販売、マーケティング、および/または流通するために、医療技術、製薬またはバイオテクノロジー会社のbrとの協力計画を考慮し、および/またはマーケティングパートナーと戦略的関係を確立することを求めるかもしれない。もし私たちが潜在的なパートナーと合意できない場合、 私たちは影響を受けた候補製品や計画の業務目標を満たすことができない可能性がある。連携スケジュールは複雑であり,交渉,記録,実施に時間がかかり,連携 や他の代替手配の確立や実施に成功しない可能性がある.私たちが設立した任意の協力または他の手配の条項は私たちに不利かもしれないが、このような任意の協力の成功は私たちの協力者の努力と活動に大きく依存するだろう。さらに、私たちの協調 プロトコルは、コストが高い場合、または損害をもたらした場合に、第三者によって終了または更新されない可能性がある。成功しなかったパートナーのbrを引き付けることは、私たちの製品開発および/または商業化作業の遅延を招く可能性があり、これは、私たちの財務状況および運営業績を損なう可能性があります。
イスラエルでの私たちのビジネスに関わるリスクは
私たちの本部、研究開発、そして他の重要な業務はイスラエルに設置されているので、私たちの業績は、最近ハマスとガザ地区からの他のテロ組織との戦争を含むイスラエルの政治、経済、軍事的不安定の悪影響を受けるかもしれない。
私たちの実行事務室と研究開発施設はイスラエルに設置されている。しかも、私たちのほとんどの重要な職員たちとすべての職員たちはイスラエルの住民だ。したがって、イスラエルの政治、地政学、経済、そして軍事条件は私たちの業務に直接影響を及ぼすかもしれない。1948年のイスラエル建国以来、イスラエルとその隣国アラブ諸国、ハマス(ガザ地区を支配するイスラムテロ民兵と政治団体)、ヒズボラ(レバノンを拠点としたイスラムテロ民兵や政治団体)、地域で活躍している他のテロ組織との間でいくつかの武力衝突が発生した。これらの紛争は、ミサイル攻撃、敵意の浸透、イスラエル各地の民間人の目標に対するテロに関連しており、これらはイスラエルのビジネス条件に負の影響を与えている。イスラエルに関連するいかなる敵対行動やイスラエルとその貿易パートナーとの間の貿易中断または中断は、イスラエルの全体的なビジネス条件、特に私たちの業務に負の影響を与え、私たちの製品開発、運営、および運営結果に悪影響を及ぼす可能性がある。
2023年10月、ハマステロリストはガザ地区からイスラエル南部の境界に浸透し、民間人と軍事目標に対して一連の攻撃を発動した。ハマスはまた、イスラエルとガザ地区の境界沿線やイスラエル列国内の他地域のイスラエル人口と工業センターに対して広範なロケット弾攻撃を発動した。このような攻撃は多くの民間人たちと兵士たちの死傷者と誘拐をもたらした。襲撃後、イスラエルの安全内閣はハマスに宣戦布告し、これらのテロ組織がロケット弾やテロを継続するとともに、これらのテロ組織に対する軍事行動を開始した。また、これらの事件が始まって以来、イスラエル北部とレバノンとの境界(ヒズボラテロ組織と国境を接する)と南部の境界(イエメンフーセ運動と国境を接し、以下に述べる)は敵対行動を続けている。レバノンヒズボラとの敵対行動はエスカレートする可能性があり、他のテロ組織、ヨルダン西岸のパレスチナ軍事組織、イランのような他の敵対国も敵対行動に参加する。このような葛藤は未来にもっと大きな地域紛争にエスカレートするかもしれない。
イスラエルの安全内閣がハマスに対する戦争を宣言し、他の組織と敵対行動を起こす可能性があることを考慮して、数十万のイスラエルの予備役が徴発されて直ちに兵役に服し、その中には9人の従業員が含まれており、いずれも管理職や肝心な従業員ではなく、予備役に徴用され、そのうち4人は常勤の職場に復帰し、戦前の軍事予備役を履行していた。戦争が続く限り,我々の人員 は予備役に徴用される可能性があり,長期の予備役でも短期の周期的予備役でも.兵役召集により、人員の長時間欠勤は私たちの業務、将来性、財務状況、運営結果に実質的な悪影響を及ぼす可能性がある。
61
2023年10月7日の戦争勃発以来、私たちの運営はこのような状況の悪影響を受けておらず、私たちの業務運営も中断されていません。そのため、私たちの製品や業務開発活動は軌道上にあります。しかし、イスラエルの現在のハマスに対する戦争の強度と持続時間は現段階では予測が困難であり、この戦争が私たちの商業と業務およびイスラエル全体の経済に与える影響も同様である。もし戦争が長く続いたり、レバノン、シリア、そしてヨルダン川西岸のような他の戦線に拡大したりすれば、私たちの行動は不利な影響を受けるかもしれない。
この地域のいかなる武力衝突、テロ、または政治的不安定はビジネス環境に悪影響を及ぼす可能性があり、私たちの運営結果と私たちの普通株の市場価格を損なう可能性があり、資金調達を難しくする可能性がある。
私たちの商業保険は中東安全情勢に関連する事件によって起こりうる損失を保証していません。イスラエル政府は過去にテロや戦争によるいくつかの損害の回復価値を保証していたが、この政府保険は不変であるか、あるいは維持すれば、私たちの損害を全額賠償するのに十分であることを保証することはできません。私たちによるいかなる損失や損害も、私たちの業務に実質的な悪影響を及ぼす可能性があります。
最後に、イスラエル国内の政治的状況は私たちの行動に影響を及ぼすかもしれない。イスラエルは2019年から2022年の間に5回の総選挙を行い、2023年10月までにイスラエル政府はイスラエルの司法制度の広範な改革を求め、広範な政治討論や動乱を引き起こした。今まで、このような計画は基本的に保留された。イスラエルの実際または考えられる政治的不安定または政治環境のいかなる負の変化も、個別または全体的にイスラエル経済に不利な影響を与える可能性があり、更に私たちの業務、財務状況、経営結果、および成長の見通しに悪影響を与える可能性がある。
私たちの行動は管理職やキーパーソンが兵役義務を履行するために中断されるかもしれない。
本稿の発表日までに、私たちは現在99人の常勤従業員を持っており、そのうち64人はイスラエルに位置し、5人の上級管理職を含んでいます。私たちイスラエルのある従業員と顧問は、私たちの上級管理者を含めて、彼らが40歳になるまで軍事予備役を履行する義務があるかもしれません(イスラエル武装部隊予備役で特定の職務を担当している将校や他の市民については、40歳以上)、軍事衝突が発生した場合、現役を召集される可能性があります。テロ活動の増加に対応するために、しばらく軍事予備役が大量に徴兵された。未来には似たような大規模な軍事予備役召集が発生する可能性がある。私たちの運営は、大量の管理者、役員、従業員、コンサルタントの不在によって中断される可能性があります。このような中断は、私たちの業務や運営に実質的な悪影響を及ぼす可能性があります。
私たちが受け取った研究と開発支出のためのイスラエル政府の支出は、イスラエル国外での製品の製造と技術移転の能力を制限し、brが指定した条件を満たすことを要求している。もし私たちがこれらの条件を満たしていなければ、私たちは前に受け取った贈与と利息と罰金の払い戻しを要求されるかもしれない。
私たちの研究と開発は資金を得ており、その一部はイスラエル革新局(IIA)から提供された寄付金から来ている。したがって、私たちはイスラエルの“工業研究と発展を奨励する法”または“研究法”の要求を守らなければならない。2023年と2022年12月31日までの年間で、国際投資協定から得られた贈与総額はそれぞれ100万ドルと110万ドルだった。支出はそれぞれ2023年12月31日と2022年12月31日までの年間研究開発総支出の7.3%と6.1%を占めている。
研究法によると、これらの贈与を用いて開発された各製品の主要部分をイスラエルで生産したり、他の方法で特別な承認を申請しなければなりません。私たちは提案された製造活動の移転に必要な承認を得ることができないかもしれません。政府が開発した製品をイスラエル国外で生産することを許可されても、印税税率が向上する可能性があり、最高達成金額の300% の支払いを要求される可能性があり、利息を加えて、具体的にはイスラエル国外で行われる生産量に依存する。この制限は私たちがこれらの製品や技術をアウトソーシングしたり、私たち自身の製造操作に従事する能力を弱めるかもしれない。
62
また、研究法によると、限られた場合であり、IIA研究委員会が承認した場合にのみ、許可された方法での譲渡を含めて、IIAが援助した技術および関連知的財産権および独自技術をイスラエル列国以外に譲渡することが禁止されている。私たちは、提案された譲渡に必要な承認を受けないかもしれませんが、受け取っても、IIAが自ら決定した部分をIIAに支払うことを要求される可能性があり、IIAがこのような取引を承認した後、このような技術を非イスラエルエンティティに売却または発行する際に受け取った対価格またはマイルストーンおよび使用料の支払いを考慮すると、最大600% 金額および利息を支払うことができます。
これらの制限は、私たちが私たちの技術資産を売却したり、イスラエル国外で生産を実行またはアウトソーシングしたり、他の方法で私たちの技術ノウハウをイスラエル国外に移す能力を弱める可能性があり、いくつかの行動および取引についてIIAの承認を得て、IIAに追加の印税および他の金額 を支払うことを要求するかもしれない。また,我々の普通株式の任意の支配権変更や所有権変更は,非イスラエル市民やbr住民を“研究法”で定義された“利害関係者”にするためには,あらかじめIIAに書面で通知する必要があるが,我々はこの要求を遵守できず,場合によっては刑事責任を招く可能性がある.
このような制限は私たちが付与された印税を全額返済した後でも適用されるだろう。
私たちは私たちの研究と開発活動に資金を提供するために、イスラエル政府から寄付を受け続けているかもしれない。もし私たちがこれらの研究開発支出の資金を失ったら、私たちは未来の研究開発プロジェクトの資金と技術改善を実施する上で困難に直面する可能性があり、これは私たちの経営業績を損なうだろう。
2023年12月31日までに、IIAから合計800万ドルの贈与を受けました。BiomX株式会社はFutuRxインキュベーターの一部として設立されたインキュベーター会社であり,2017年前に我々の資金の大部分はIIAの贈与とインキュベーターからの資金であり,後者はIIAの支持を得た。インキュベーターを離れた後、私たちは引き続きIIAの贈与を申請し、受け入れます。このような援助の要求と制限は研究法で見つけることができる。研究法によると、これらのIIAを用いて開発された製品またはサービスの全部または一部を贈与して販売して得られた収入の3%~3.5%の特許使用料はイスラエル政府に支払われなければならない。私たちはこの2つのプラットフォーム技術を開発し、少なくとも一部はこれらの寄付金の資金を使っているので、私たちは私たちの任意の候補製品の販売にこれらの印税を支払い、規制部門の承認を得る義務があります。私たちの候補製品の生産がイスラエルで行われ、IIAの助成金によって援助された技術がない限り、非イスラエルエンティティに販売または許可されている限り、支払われる最高許可使用料は、通常、私たちに発行された贈与の100%を超えず、ドル預金に適用される12ヶ月の保証隔夜融資金利またはSOFRに相当する年利を加えて、各カレンダー年度の第1取引日に公表される。2023年12月31日現在、IIAに対する将来の支払い約束の元金と利息残高は約790万ドルである。私たちの現在と計画されている製品開発活動に資金を提供する一部として、追加の支出を得るために後続の支出申請を提出する可能性があります。
これらの贈与は、私たちの一部の人員、下請け業者との開発活動、その他の研究開発コストと支出に資金を提供した。しかしながら、これらの賞が全ての援助を受けていない場合、またはIIAの予算制限や政府政策決定などの理由で、将来的に追加的な贈与が付与されない場合、将来の研究開発および技術改善の実施に資金を提供する能力が損なわれ、候補製品を開発する能力に負の影響を与えることになる。
63
ドル、新イスラエルシェケル、ユーロと他の外貨との為替レート変動は私たちの将来の支出にマイナス影響を与える可能性がある。
私たちが証券を売る収益は通常ドルで計算されます。私たちの本部はイスラエルに置かれていて、そこでは、私たちの一般的で行政費用の大部分と研究と開発コストは新イスラエルシェケル(NIS)で発生している。将来の費用はユーロやポンドなどの外貨で計算されるかもしれない。したがって、私たちの財務業績は各国の通貨為替レートの変動の影響を受ける可能性があります。例えば、2020年の間、新シェケルのドルに対する平均為替レートが強くなっていることが見られ、これはイスラエルの支出のドル価値を増加させています。NISが2020年と2021年のようにドルに強くなれば、主に人員や施設に関連する費用である我々イスラエルの支出のドル価値が増加するだろう。我々は外貨契約(主にオプションと長期契約)を用いて貸借対照表項目の為替リスクをヘッジする。しかしながら、これらの外国為替契約は、会計目的のためにヘッジツールとして指定されているわけではないため、無効である可能性がある。これまで、為替レート変動のリスクの開放はまだ我々の業務に重大な悪影響を与えていないにもかかわらず、将来の変動が我々の経営業績や財務状況に重大な悪影響を与えない保証はない。
適用された雇用法によると、私たちは競争条約を施行できないかもしれない。
私たちは一般的に従業員たちと競争禁止協定を締結する。これらの協定は私たちの従業員が限られた時間内に私たちと直接競争したり、私たちの競争相手や顧客のために働くことを禁止します。私たちは従業員が働いている管轄区の法律に基づいてこれらの合意を実行することができないかもしれませんし、競争相手が私たちの以前の従業員やコンサルタントが私たちの仕事中に培ってくれた専門知識を制限することは難しいかもしれません。例えば、イスラエル労働裁判所は、会社の商業秘密または他の知的財産権を保護するように、裁判所が認めた雇用主の限られた数の物質的利益のうちの1つを損なうことを証明しなければならない元従業員の競業禁止約束を強制的に実行する雇用主を求めなければならない。
私たちが課税収入を生成する時、私たちが享受できる税金優遇要件は、様々な条件を満たし、将来的には阻止または減少される可能性があり、これは私たちのコストと税金を増加させるかもしれない。
そして、課税収入が発生した場合、1959年の資本投資法、同法およびその改正条例で定義された“技術優先企業”および/または“優先企業”に提供されたいくつかの税金優遇を受ける資格があるので、法律で規定されている収入減収会社税率(範囲7.5%~16%)を享受することができる。もし私たちがこのような身分を得る資格がなければ、私たちのイスラエルでの課税収入はイスラエルの正常な会社税率を適用します。イスラエル社の標準会社税率は23%です。法律とその条例によって私たちに提供される福祉 は法律と条例で規定されている条件を満たさなければならない。さらに、将来的にこのような税金優遇は減少または終了するかもしれない。
私たちまたはイスラエルまたはアメリカの上級職員や役員に対する米国の判決を実行することは難しいかもしれませんし、イスラエルにおける米国証券法のクレームを主張したり、私たちの上級職員や役員に訴訟手続きを送達したりすることは難しいかもしれません。
私たちのすべての役員や管理者がアメリカ住民であるわけではありません。彼らと私たちの資産の大部分はアメリカ以外にあります。アメリカ内では私たちや私たちの非アメリカ常駐役員や上級職員に法的手続き書類を届けることは難しいかもしれません。イスラエルの裁判所は、イスラエルがこのようなクレームを提起する最適なbrフォーラムではないかもしれないので、私たちまたは私たちの非アメリカの官僚および取締役のためのアメリカ証券法違反に基づくクレームの審理を拒否する可能性がある。また、イスラエルの裁判所がクレームの審理に同意しても、それがクレームに適用されるのはイスラエルの法律であり、アメリカの法律ではないと判断することができる。米国の法律の適用が発見された場合,適用される米国の法律の内容が事実であることを証明しなければならず, これは時間がかかり高価な過程である可能性がある。特定の手続き事項はまたイスラエルの法律によって管轄されるだろう。イスラエルはこのような事項に対する拘束力のある判例法をほとんど言及していない。さらに、イスラエルの裁判所は、米国で得られた私たちまたは私たちの非米国役員や幹部に対する判決を実行しない可能性があり、これは、私たちまたは私たちの非米国人や役員に対する判決を収集することを困難にするかもしれない。
64
また、一方の非イスラエルの判決がその法律がイスラエルの裁判所の判決を実行することを規定していない国で行われた場合、その判決の実行がイスラエルの国々の主権または安全を損なう可能性がある場合、その判決が詐欺によって得られた場合、または正当な手続きがない場合、その判決が同じ当事者との間で同じ事項について下した別の有効な判決と一致しない場合、イスラエルの裁判所はその判決を実行しない。 又は外国訴訟を提起する際には,同一当事者間の同一事項の訴訟がイスラエルの裁判所又は法廷で議決されている。
製造と供給に関するリスク
私たちは第三者に依存して私たちの臨床供給の候補製品を生産し、承認されれば、第三者に依存して私たちの製品を生産して加工するつもりです。
私たちは現在、実験室設備のような原材料と他の重要な構成要素を提供する外部供給者に依存している。私たちはまだどの候補製品も生産や商業規模加工を行っていないので、私たちのどの候補製品に対してもこの操作を実行できないかもしれません。必要に応じて変更を行い,我々の候補製品の製造プロセスを で最適化し,プロセス中のわずかな変更でも安全で有効な療法をもたらすことは確認できない。
我々の製品を生産するための施設br候補者はFDAまたは同等外国規制機関の承認を得なければならず,検査はFDAまたは同等外国規制機関にマーケティング申請を提出した後に行われる。また、非治療用途のための候補製品を生産するためのどの施設も、FDAおよび外国規制機関の検査を受ける。私たちは現在、生産過程のすべての側面を制御することができず、現在、主に私たちの契約製造パートナーが法規要件、すなわちcGMP要求を遵守して、私たちの候補製品を生産することに依存している。もし私たちの製造施設が運行を開始すれば、私たちはcGMP要求を守る責任があるだろう。もし私たちまたは私たちの契約メーカー が私たちの規範とFDAまたは他の規制機関の厳格な規制要求に従って生産に成功しなければ、私たちと彼らはその製造施設の規制許可を確保および/または維持することができないだろう。しかも、私たちの契約製造業者が十分な品質管理、品質保証、合格者の能力を維持することを制御することはできません。FDAまたは同等の外国規制機関が私たちの候補製品を生産するためにこれらの施設を承認しない場合、または将来的にこのような承認を撤回する場合、私たちは承認されれば、私たちが規制機関の承認を得たり、私たちの候補製品をマーケティングする能力に深刻な影響を与えるbr代替製造施設を探す必要があるかもしれない。
著者らは製品を生産する経験が限られており、臨床試験、治療適応或いは非治療性臨床研究或いは試験の候補製品に用いられている。私たちは2019年にイスラエルのネスジオナの本社に自分の製造工場を開設した。私たちは製品が商業的に実行できるようにするのに必要なコストや数量で、規定に従って私たちの候補製品を生産することができることを保証できません。
私たちの候補製品は専門的なbr原材料の可用性に依存しており、これらの原材料は受け入れ可能な条項で提供できないか、あるいは全く得られないかもしれない。
私たちの候補製品は特定の専門的なbr原材料が必要で、その中のいくつかは資源と経験の限られた小さな会社から得られたもので、商業製品をサポートすることができません。これらの第三者サプライヤーは、特にFDA検査や医療危機、例えば広範な汚染のような非通常の場合には、私たちの需要を満たすことができないかもしれない。私たちは現在、すべてのサプライヤーと任意の時点で必要かもしれない契約を締結していません。必要であれば、受け入れ可能な条項で、あるいは彼らと契約を結ぶことができないかもしれません。したがって、私たちは臨床や商業生産を支援する重要な原材料の受け入れに遅延があるかもしれない。
65
私たちの普通株に関するリスクは
当社の相当数の普通株は、発行された株式承認証及びオプションを行使したり、自社の優先株を転換したりする場合に発行され、当該等の行使又は転換(誰が適用されるかによります)は、当社の証券保有者の権益を薄くする可能性があります。
2023年12月31日まで、私たちは全部で25,363,688株の未発行の引受権証があり、合計20,926,189株の普通株を購入することができ、加重平均行権価格は2.60ドルであり、その中のいくつかまたは単位権証は私たちの発行された単位に含まれ、それぞれ1株の普通株とbr}普通株または単位の半分で行使可能な権利証を含み、その中のいくつかは私募方式で発行されたか、または私募株式権証で発行され、いくつかまたは事前出資の株式証明は2023年2月に発行された。あるいは株式証を公開し、以前はニューヨーク証券取引所米国証券取引所で取引されていたが、コードは“PHGE.WS”、現在は場外取引市場で、取引コードは“PHGEW”である。単位株式承認証、私募株式承認証、事前融資権証と公開株式権証は、総称或いは未償還権証であり、それぞれの場合に調整を行うことができる。この等未償還引受権証を行使する範囲では,普通株 を増発し,普通株当時の保有者の希釈を招き,公開市場で転売する資格のある株式数 を増加させる。このような株を公開市場で大量に販売することは、私たち普通株の市場価格に悪影響を及ぼす可能性があります。
また、2023年12月31日現在、私たちは5,280,711株の普通株を購入する未帰属および未帰属オプションを持っている。いずれかのオプションを行使すれば、一般に公開市場で転売する資格がある(証券法の規則 144による関連会社の株式の制限を受ける)一般的にbr}普通株が発行され、これにより、私たちの証券保有者の株式が希釈される。
また,(I)買収については,普通株の発行に加えて,(A)合計40,470株の転換優先株を発行し,我々の株主が株主総会で転換可能優先株の転換を承認すれば,最大40,470,000株の普通株に変換することができ,および(B)株式承認証または合併権証に変換でき,我々の株主が株主総会で合併引受権証の行使を承認すれば,合計2,416,497株の普通株,すなわち行使可能な株式権証を行使することができる. と(Ii)は2024年3月のパイプラインに関連して,(X)合計216,417株の転換可能優先株を発行し,我々の株主が株主総会で転換可能優先株の転換を承認すれば,これらの優先株は合計216,417,000株の普通株,および(Y)私募株式権証に変換可能であり,我々の株主が株主総会で私募株式権証の行使を承認すれば,合計108,208,500株の普通株 を行使することができる.また,2024年3月パイプラインの配給代理に対する部分補償として,引受権証または配給代理権証を発行し,合併権証と私募株式証とともに,我々の株主 が株主総会で配給代理権証の転換を許可した場合,これらの株式承認証は合計9,523,809株の普通株を行使することができる。任意の転換可能な優先株が変換されるか、または任意の株式承認証が行使される限り、追加の普通株 が発行され、適用される証券法によれば、これらの普通株は、通常、公開市場で を転売する資格がある(証券法第144条の関連会社が保有する株式に制限される)。このような株を公開市場で大量に売ることは私たちの普通株の市場価格に悪影響を及ぼす可能性があります。
株主総会での承認に応じて追加オプションを付与する予定であり、将来的に追加権証や優先株を発行する可能性がある。また、適用されれば、当該等の証券を行使する際に当社普通株を増発することは、当時自社普通株を保有していた既存保有者の希釈を招き、当社普通株の市場価格に悪影響を及ぼす可能性がある。
私たちは私たちの普通株に配当金を支払ったことがなく、私たちは予測可能な未来にも私たちの普通株に現金配当金を支払わないと予想している。
私たちは普通株の現金配当金を発表したり支払ったりしたことがない。私たちは予測可能な未来に私たちの普通株に現金配当金を支払わないと予想している。私たちは現在、すべての利用可能な資金と未来の任意の収益を維持し、私たちの業務の発展と成長に資金を提供するつもりです。したがって、予測可能な未来には、私たち普通株の資本付加価値(あれば)が私たちの株主の唯一の収益源となるだろう。
私たちの公共株式証明書はすでに取得されて、私たちは未来に私たちの証券の上場を維持できないかもしれません。
私たちの普通株と単位はニューヨーク証券取引所アメリカ証券取引所で取引されています。私たちの公共株式証は以前ニューヨーク証券取引所アメリカ証券取引所で取引されていましたが、2023年6月に退市し、その後は場外取引プラットフォームPinkで取引されています。著者らの公開株式証が退市したため、その所有者の市場オファーは限られており、しかもその公開株式証の流動性は減少した。もし私たちの普通株式または単位がその後取られたら、私たちは重大なbrの重大な不利な結果に直面する可能性があります
| ● | 私たちの証券の市場オファーは限られています |
| ● | 証券の流動性が減少しています |
| ● | 私たちの株が“細価格株”であることを確認することは、私たちの証券を取引するブローカーに、より厳しい規則を遵守することを要求し、私たちの証券二級取引市場の取引活動を減少させる可能性がある |
| ● | 限られた数のニュースとアナリストの会社に対する報道; |
| ● | 将来的により多くの証券を発行したり、より多くの融資を得る能力が低下する。 |
66
私たちの普通株と他の証券の市場価格はbrが大きく変動する可能性があり、これは私たち普通株の購入者に大きな損失をもたらす可能性があります。
一般的な株式市場とバイオテクノロジー株式市場は極端な変動を経験している。我々のような小さな会社の普通株市場の特徴は,国家証券取引所で取引され大量の公開流通株を持つ比較的大きく成熟した会社の株に比べて価格変動が大きい であり,我々の株価は不確実な未来にはこれらの大きく成熟した会社の株よりも不安定であることである.
“リスク要因”の節で議論されている要因に加えて、我々の普通株(他の証券と)の価格下落も、一般市場や経済状況 や様々な他の要因によるものである可能性がある
| ● | 私たちの臨床試験は不良な結果や遅延があります | |
| ● | 規制当局は私たちの候補製品、臨床試験、または私たちの候補製品の製造プロセスに対する不利な行動を取った | |
| ● | 競争相手の技術革新、特許、または新製品を発表する | |
| ● | アメリカや他の国の規制動向は | |
| ● | 私たちや私たちの候補製品に関するいかなる訴訟も | |
| ● | 私たちの競争相手、あるいはバイオテクノロジーや製薬産業全体に関する公告 | |
| ● | 私たちが達成する可能性のある戦略連合や買収に関する事態の発展 | |
| ● | 私たちの経営業績の実際や予想の変化 | |
| ● | 証券アナリストが提案した変更またはアナリストカバー面の欠如; | |
| ● | 私たちの経営業績はアナリストの見積もりとずれています | |
| ● | 私たちには能力がない、あるいは投資家は私たちの普通株がニューヨーク証券取引所アメリカ証券取引所に上場し続けるすべての適用要件を満たし続けることができないと思って、私たちの普通株が取得される可能性があります | |
| ● | 私たちの役員、役員、主要株主は私たちの普通株を売却したり、大量の普通株を売却したりします | |
| ● | 私たちの重要な科学や管理者を失ってしまいました |
しかも、バイオテクノロジー会社の証券市場価格は従来から大きく変動してきた。いずれの会社の経営業績にも関係ないため、これらの証券の市場はしばしば重大な価格や出来高変動を経験している。さらに、私たちの業務は、大流行や他の健康危機に関連するリスクまたはリスクに対する公衆の見方の悪影響を受ける可能性があります。例えば、新冠肺炎、またはイスラエル-ハマス戦争やロシアのウクライナ侵攻、それによるロシア、ベラルーシ、関係者への世界制裁の結果です。br}感染症の深刻な爆発は、広範囲の健康危機を招き、多くの国の経済や金融市場に悪影響を与え、経済低迷を招く可能性があります。
過去には、特定会社証券の市場価格が変動した後、同社を提訴することが多かった。このような訴訟は、リソース、管理時間、および注意力を消費する可能性があり、これは私たちの業務に悪影響を及ぼす可能性があります。
67
“小さな報告会社”として、より大きな上場企業よりも少ない情報を開示することが許可されており、これは、私たちの普通株の投資家への魅力を低下させる可能性がある。
我々は現在、取引法第12 b-2条に規定されている“小さな報告会社” である。小さな報告会社として、他の上場企業に適した各種報告要求のいくつかの免除を利用する資格がある。そのため、投資家が私たちの運営結果と財務見通しを分析することはもっと挑戦的である可能性があり、これは投資家の自信低下を招く可能性がある。私たちの報告会社の地位が小さいため、投資家は私たちの普通株吸引力が低下していることを発見するかもしれません。一部の投資家が私たちの普通株の吸引力が低下していることを発見すれば、私たちの普通株取引市場はそれほど活発ではなくなり、私たちの株価はもっと変動するかもしれない。
一般的なリスク要因:
私たちの成功はある程度私たちが肝心な幹部を維持する能力、及び合格した人材を吸引、維持と激励する能力にかかっている。
私たちは最高経営責任者ジョナサン·ソロモンと私たちの管理、科学、臨床チームの他の主要なメンバーに非常に依存している。私たちは私たちの幹部と雇用協定を締結したが、彼らの誰もが私たちとの雇用関係をいつでも終わらせることができる。私たちは私たちの役員や他の職員たちにbr“キーパーソン”保険を提供しない。私たちの幹部、他の重要な従業員、他の科学と医療コンサルタントのサービスを失って、私たちは適切な代替者を見つけることができなくて、製品開発の遅延を招き、私たちの業務を損なう可能性があります。また、我々の最近のAPTの買収及び会社業務との統合は、予想可能な将来的に従業員が離職する可能性を増加させる可能性がある。
著者らは高素質の管理、臨床と科学者の能力、及び著者らがリードする学術機構、臨床医師と科学者の発展と重要な関係を維持する能力を持続的に吸引、維持し、激励する能力は、著者らの成功に重要である。バイオテクノロジー分野では、特に私たちの本部があるイスラエルでは、合格した人材に対する競争が非常に激しい。我々は,他のバイオテクノロジー,製薬会社,大学,公共·民間研究機関,その他の組織からの人員の競争に直面している。私たちはまた、他の資金が豊富で、より成熟した企業からの競争 に直面しており、仕事の安定性の観点からも、私たちはよりリスクの高い選択として扱われる可能性があります。私たちの地位は歴史の長いバイオテクノロジーや製薬会社よりも比較的新しいからです。合格人材に対する競争を考慮すると、私たちは受け入れ可能な条件でこれらの人材を誘致し、維持することができないかもしれない。もし私たちの維持、激励、採用努力が失敗したら、私たちは私たちの業務戦略を実行できないかもしれない。
環境、社会、ガバナンス (ESG)プロジェクトに関する予想は、コストを増加させ、新たなリスクに直面させる可能性がある。
一部の投資家および他の主要な利益関係者は、企業責任、特に環境、社会および管理またはESG要因に関連する責任にますます注目している。そのため、ますます企業責任の格付けを重視し、多くの第三者は企業に関する報告を提供し、企業の責任業績を評価している。また、企業の企業責任実践を評価するためのESG要因が変化する可能性があり、これにより、我々への期待が高くなり、これらの新しい基準を満たすためにコストの高い計画をとることになる可能性がある。あるいは、私たちがこれらの新しい基準を満たすことができなければ、投資家は 私たちの企業責任に関する政策が不十分だと結論するかもしれない。もし私たちの企業責任手続きや基準が各顧客が制定した基準を満たしていなければ、私たちのブランドと名声を損なうかもしれません。私たちはESGに関連する事項に投資する必要があるかもしれませんが、これは重大であり、私たちの運営結果に悪影響を及ぼすかもしれません。また,我々の競争相手の企業責任表現が我々よりも高いと考えられれば,潜在的あるいは既存の投資家は我々の 競争相手とともに投資することを選択する可能性がある.さらに、ESG事項について何らかの計画および目標を伝達する場合、そのような計画または目標を達成する上で失敗したり、失敗したりする可能性があり、または、そのような計画または目標の範囲によって批判される可能性がある。もし私たちが投資家や他の主要な利害関係者の期待を満たしていない場合、あるいは私たちの計画が計画通りに実行されていない場合、私たちの名声や財務業績は実質的な悪影響を受ける可能性がある。
68
もし私たちが将来の買収や戦略的パートナーシップに参加すれば、これは私たちの資本要求を増加させ、私たちの株主を希釈し、私たちに債務を発生させたり、負債を負担したり、他のリスクに直面させたりする可能性がある。
2024年3月15日にAPTを買収しました我々は、相補的製品、知的財産権、技術またはビジネスの許可または買収を含む様々な他の買収機会および戦略的パートナーシップを評価することができる。潜在的な買収や戦略的パートナーシップは、多くのリスクをもたらす可能性がある
| ● | 業務費と現金需要が増加した |
| ● | 追加の債務や負債を負担しています |
| ● | 株式証券を発行し |
| ● | 買収された会社の業務、知的財産権、製品を吸収し、新しい人員の統合に関する困難を含む |
| ● | 私たちの経営陣の関心を既存の製品計画と計画からこのような戦略的合併や買収を求めることに移しました |
| ● | 重要な従業員の保留、キーパーソンの流出、そして私たちがキー業務関係を維持する能力の不確実性 |
| ● | そのような取引の他方に関連するリスクおよび不確実性は、その当事者およびその既存製品または候補製品の将来性および販売承認を含む; |
| ● | 私たちは買収の目標を達成するために、買収された技術および/または製品から十分な収入を得ることができず、関連する買収や維持コストを相殺することさえできない。 |
コンピュータシステムに障害、ネットワーク攻撃、または私たちのネットワークセキュリティに欠陥があれば、私たちの業務と運営は影響を受けるだろう
セキュリティ対策が実施されているにもかかわらず、我々の内部コンピュータシステムおよび私たちが依存する第三者コンピュータシステムは、コンピュータウイルス、マルウェア、自然災害、テロ、戦争、電気通信および電気故障、インターネット上のネットワーク攻撃またはネットワーク侵入、電子メール添付ファイル、組織内部者、または組織内部システムにアクセスする権利を有する者によって破壊されやすい。セキュリティホールや中断のリスク、特にコンピュータハッカー、外国政府、ネットワークテロリストを含むネットワーク攻撃やネットワーク侵入を介して、 は通常、世界各地からの未遂攻撃や侵入の数、強度、複雑性の増加とともに増加する。このような事件が発生し、私たちの運営が中断されると、私たちの製品開発計画が実質的に妨害される可能性がある。例えば、完了した、行われている、または計画されている臨床試験における臨床試験データの損失は、われわれの臨床試験の作業遅延を招き、データの回復または複製のコストを著しく増加させる可能性がある。もし任意の中断またはセキュリティホールが私たちのデータやアプリケーションを紛失したり、破損したり、あるいは機密または独自の情報を適切に開示しない場合、私たちは重大な法的クレームおよび責任を招き、私たちの名声を損なう可能性があり、私たちの製品のさらなる開発は候補製品を延期する可能性があります。我々はまた,一般に重要な非公開情報を持ち,ネットワークセキュリティホールに関連する場合の取引に対する制限の潜在的な適用性を解決するためのコンプライアンス計画を維持する.しかし、私たちのネットワークセキュリティ環境の既存の制御とプログラムの故障 は、私たちが適時にネットワークイベント を発見、報告、あるいは応答することを阻止し、私たちの財務状況と株式価値に重大な悪影響を与える可能性がある。彼は言いました
上場企業として、私たちの運営コストは高い。
上場企業として、私たちは取締役や上級管理者保険に大きなコスト を発生させ、サービスプロバイダの法律や会計などの費用、その他の費用を支払います。私たちは取引法の報告に制約されています。その中で、私たちの業務と財務状況に関する年間報告、br}四半期報告、現在の報告をアメリカ証券取引委員会に提出することを要求しています。また、“サバンズ-オクスリ法案”および米国証券取引委員会とニューヨーク証券取引所米国証券取引所は、“サバンズ-オクスリー法案”、“ドッド·フランクウォール·ストリート改革·消費者保護法”または“ドッド·フランク法案”および上場企業会計監督委員会が“サバンズ-オックススリー法案”、“ドッド-フランクウォール街改革法案”および“消費者保護法案”および上場企業会計監督委員会がその後可決した規則を実施するために、上場企業に重大な要求を提出し、有効な情報開示と財務制御の確立と維持、企業統治実践における変化brを含む。“サバンズ·オクスリー法案”“ドッド·フランク法案”“米国証券取引委員会規則·条例”によるコーポレート·ガバナンスや情報開示への追加要求により、将来的にはこれらの費用が増加する可能性があり、特に“より小さな報告会社”でなければ。
上場企業に適用される規則や法規は、大量の法律や財務コンプライアンスコストを発生させ続けている。これらのコストは、私たちの純損失を増加させ、あるいは任意の純収入を減少させ、他の業務分野のコストを下げることが要求されるかもしれません。
69
項目1 B:未解決スタッフの意見
適用されません。
プロジェクト1 C:ネットワークセキュリティ
私たちは、私たちの情報システムを保護し、私たちのデータの機密性、完全性、br、および可用性を保護するために、ネットワークセキュリティ対策を策定、実施、維持することの重要性を認識している。我々は,内部計算機システム上でセキュリティ対策を実施し,第三者と業務パートナーが同様の措置を実施することを確保することで,ネットワークセキュリティリスクに対応する.これらのセキュリティ対策には,ファイアウォール,侵入防御·検出システム,アンチマルウェア機能,アクセス制御があり,我々の外部ITコンサルタントが評価し,バグ評価とネットワークセキュリティ脅威情報による改善を行っている.
我々の運営上級取締役はネットワークセキュリティ脅威リスクの日常評価と管理を担当し、ネットワークセキュリティイベントの予防、緩和、検出と修復を含む。
監査委員会は、第三者および業務パートナーからのリスクを含む、ネットワークセキュリティリスクおよび関連または負債に関する私たちの政策、および会社に重大な影響を及ぼす可能性のあるリスクを検討する責任があります。監査委員会は四半期ごとにネットワークセキュリティ脅威リスクに関する管理職の最新情報を受け取った。このような更新は、会社の情報技術セキュリティ計画をカバーし、その現在の状態、能力、前四半期の変化、目標と計画、そして絶えず変化するネットワークセキュリティ脅威情勢を含む。
これまで,ネットワークセキュリティ脅威からのリスク は我々に実質的な影響を与えておらず,ネットワークセキュリティ脅威からのいかなるリスクも我々の業務戦略,運営結果,財務状況を含めて合理的に会社に影響を与えるとは考えていない.詳細については“をご覧ください”リスク要因-コンピュータシステムの故障、ネットワーク攻撃、またはネットワークセキュリティ欠陥が発生すれば、私たちの業務と運営は影響を受ける。“ は本年度報告第1 A項に記載されている。私たちはネットワーク責任保険証書を維持する。しかし、私たちのネットワーク責任保険リストは、私たちに対するすべてのクレームをカバーできない可能性があり、勝訴するかどうかにかかわらず、訴訟を弁護する費用が高く、経営陣の私たちの業務と運営に対する関心を分散させる可能性があります。
項目2.財産
私たちの会社はイスラエルのナイスジオナに本社を置いています。2021年第2四半期に、私たちは新しい6,500平方フィートの製造施設を含む28,610平方フィートの新しいオフィスと実験室施設に引っ越しました。レンタル契約は2025年11月に満期となり、レンタル期間を5年延長することが選択できます。この施設はすでに臨床大量の臨床候補製品を生産でき、臨床開発に必要な需要を満たすように設計されている。2022年8月、BiomXイスラエル社はイスラエルのネチオナにあるオフィス空間の一部について転貸協定を締結した。協定の有効期間は2年で、2022年8月15日から発効する。
私たちはイスラエルのオフィスのほかに、メリーランド州ゲザスバーグで6,100平方フィートの製造施設を含む25,894平方フィートのオフィスと実験室施設をレンタルしました。brレンタル協定は2034年7月に満期になり、2029年2月に終了する権利がありますが、12ヶ月前に通知して、br費用を早期に終了する必要があります。
私たちは私たちの施設が私たちの現在の需要を満たすのに十分だと信じている。
項目3.法的手続き
私たちは時々法的手続き、調査 と私たちの業務展開に関するクレームを受けるかもしれない。私たちは現在私たちの重大な訴訟や他の重大な法的訴訟に対するいかなる当事者でもない。
プロジェクト4.炭鉱の安全状況の開示
適用されません。
70
第 第2部分
項目5.登録者普通株式市場、関連株主事項、発行者による株式証券の購入
我々の普通株と単位の株はニューヨーク証券取引所アメリカ証券取引所で取引され,コードはそれぞれPHGEとPHGE.Uである.私たちの公共株式証は場外ピンク市場で“PHGEW”のコードでオファーされました。
記録保持者
2024年3月28日までに、55,220,707株の発行済み普通株と流通株が75人の登録株主が保有している。記録保持者の数は,我々の譲渡エージェントの記録 によって決定され,普通株の受益者は含まれておらず,その株は各種証券仲介人,取引業者,登録決済機関の名義で保有されている.
配当をする
今まで、私たちは普通株に対して何の現金配当金も支払っていませんし、現金配当金を支払うつもりもありません。将来の現金配当金の支払いは私たちの収入と収益(もしあれば)、資本要求、そして全体的な財務状況に依存するだろう。その際、任意の現金配当金の支払いは当社取締役会が自ら決定します。さらに、もし私たちが債務を発生させた場合、私たちが配当金を発表する能力は、私たちが同意する可能性のある制限されたbr契約によってさらに制限されるかもしれない。
第六項です[保留します。]
71
プロジェクト7.経営陣の財務状況と経営成果の検討と分析
以下では、我々の財務状況と経営結果の検討と分析を、我々の財務諸表と本年度報告に含まれる他の部分に含まれる付記 に合わせて読むべきである。財務状況や経営結果の分析は、2023年12月31日以降に得られたためAPTは含まれていない。以下の議論および分析に含まれるいくつかの情報は、リスクおよび不確実性に関する前向き陳述を含む。様々な要素の影響により、私たちの実際の結果は、本年度報告で“展望性陳述に関する警告声明”と“リスク要因”の節で述べた要素を含む任意の前向き陳述で議論された結果と大きく異なる可能性がある。
概要
著者らは臨床段階の製品発見会社であり、天然と工学バクテリオファージ技術を用いて製品を開発し、CFとDFOなどの慢性疾患に関連する特定の有害細菌 を対象とし、殺すことを目的としている。バクテリオファージやバクテリオファージは細菌,種特異性,菌株が制限されたウイルスであり,標的細菌に感染,増幅して死滅させ,哺乳動物細胞に対して不活性であると考えられている。自然産生バクテリオファージの特許の組み合わせと合成生物学を用いた新たなバクテリオファージの創出により,大市場や孤児疾患を解決するためのバクテリオファージベース療法を開発した。
BiomX Ltd.Sが2015年に設立されて以来、私たち はほとんどの資源を組織と会社のために人員を配置し、資金を調達し、候補製品の権利を獲得或いは発見し、私たちの技術プラットフォームを開発し、関連する知的財産権を保護し、及び私たちの候補製品のために発見、研究及び開発と臨床活動を行う。私たちは販売を許可された製品もなく、私たちは製品販売から何の収入も得ていない。私たちが私たちの候補製品を発売するにつれて、私たちの費用はまだ高いと予想されています。これまで、私たちは普通株、優先株、株式承認証、政府支出、協力協定、債務を売却することで、私たちの運営に資金を提供してきました。2023年12月31日現在、証券販売から約1.54億ドルの毛収入を得ています。また、2023年12月31日現在、協力協定から200万ドルを獲得し、研究開発費から220万ドル削減しました。残りの20万ドルは2024年1月に受信された。また、2023年12月31日現在、国際投資局から合計800万ドルの贈与を受けており、2023年12月31日現在110万ドルを受け取っています。
しかも、私たちは深刻な運営損失を発生させた。私たちが製品販売から十分な収入を得て利益を達成できるかどうかは、私たちの1つ以上の候補製品の開発成功、規制部門の承認、最終的な商業化にかかっている。2023年12月31日と2022年12月31日までの純損失はそれぞれ約2,620万ドルと2,830万ドルであった。2023年12月31日現在,我々のbrの累計赤字は1.63億ドルであり,予測可能な未来には,我々の候補製品を発見段階から臨床前開発と臨床試験段階に進め,規制部門に我々のbr候補製品の承認を求めているため,引き続き巨額の支出が生じると予想される。また、いずれかの候補製品が規制部門の承認を得た場合、製品製造、マーケティング、販売、流通に関連した巨額の商業化費用が発生すると予想される。許可内 や他の候補製品の取得に関する費用も発生する可能性がある.
製品開発に関連する多くのリスクや不確実性 のため、費用を増加させる時間や金額、またはいつまたは が利益を達成または維持できるかどうかを予測することはできない。たとえ私たちが製品販売を作ることができても、私たちは利益を上げることができないかもしれない。もし私たちがbrの利益を達成できない場合、または持続的に利益を上げることができなければ、私たちは計画レベルで運営を継続できず、運営を減少または終了させることができないかもしれない。私たちは、修正、延期、制限、減少、または私たちの1つまたは複数の計画または進行中または計画中の候補製品臨床試験を含むことができるコスト低減戦略を実施することができる。2022年5月には、わが社再編計画(“会社再編”)の一部として、50%削減を含む運営コストを削減するとともに、我々が行っているCF計画を優先することを発表しました。
2023年12月31日、私たちは現金、現金等価物、制限された現金1,590万ドルを持っています。私たちの財務諸表には、次のように“-流動性と資本資源”でさらに議論されているように、私たちが持続的に経営を続ける能力がある企業が少なくとも1年から2025年4月3日までの深刻な疑いに関する解釈段落が含まれている。
72
2024年3月6日、APTと一部の他の当事者と合併協定を締結し、APTは当社の完全子会社となり、2024年3月15日から発効します。今回の買収の構造は株式交換取引であり、一回の合併において、APTのすべての発行済み株式 は合計9,164,968株のBiomX普通株、40,470株Xシリーズ優先株 に交換され、株主の承認を経て40,470,000株のBiomX普通株と、行使可能な持分証または合併権証 が行使可能なBiomX普通株2,166,497株に変換できる。買収完了後、APTの利益相続人はBiomXの完全子会社となる。合併承認株式証は、BiomX株主の承認を受けた日からいつでも1株5.00ドルの行使価格で行使され、2027年1月28日に満期となる。
買収を完了すると同時に、BiomXは既存と新しい投資家と私募融資、すなわち2024年3月のパイプを完成し、合計約5,000万ドルの総収益を発生させ、その中で投資家は(I)合計216,417株Xシリーズ優先株を購入し、株主の承認により合計216,417,000株のBiomX普通株と(Ii) 承認株式証、あるいは私募株式承認証、合計108,208,500株BiomX普通株に変換することができる。Xシリーズ優先株1株231.10ドルの合計購入価格と付随する私募株式権証でBiomX普通株500株を購入した。私募株式承認証は、BiomX株主の承認を受けた日以降の任意の時間に1株当たり0.2311ドルの使用価格で行使され、初期行使日の24ヶ月以内に満期となる。
買収事項brに続いて、2024年3月に発行された交換可能株優先株に計上しなければ、すべての交換可能株が普通株に転換すると仮定し、当社の買収前株主(事前資金援助権証所有者を含む、br}と定義する)は当社の約55%の株式を所有しているが、APTは買収前株主に当社の約45%の株式を所有している。
私たちの総合運営結果の構成要素は
収入.収入
これまで、私たちは製品販売から何の収入も得ておらず、近いうちに製品販売から何の収入も得られないと予想されています。もし私たちの製品の開発作業が候補者が成功し、任意の必要な規制承認を得たり、あるいは他の方法で任意の商業化製品または第三者との追加の 許可協定をもたらした場合、私たちは将来的に製品販売または第三者との協力または許可協定支払いから収入 を得ることができる。
運営費
研究と開発費、純額
研究開発費には主に我々の候補製品の発見と開発に関するコストが含まれている。私たちは発生した研究と開発コストを支出し、IIAの贈与によって相殺され、研究と開発協力協定の収入をより小さい程度で相殺する。これらの費用 には:
| ● | 独自のプラットフォームを開発し運営しています | |
|
|
● | CROや契約製造組織のような第三者との合意、科学開発サービスを提供するコンサルタント、下請け業者、および重要なオピニオンリーダーなど、私たちの候補製品の臨床前および臨床開発に関連する費用 |
| ● | 増幅費用と臨床前臨床試験材料の取得と製造のコスト; | |
| ● | 様々なライセンス契約に関連するライセンス維持費とマイルストーン費用 | |
| ● | 研究開発機能に従事する従業員の賃金、関連福祉、出張および株式報酬費用、およびこのような活動に従事する外部コンサルタントに支払われる費用などの外部費用を含む従業員関連費用 | |
| ● | 法規の遵守に関する費用及び特許に関する法律の費用; | |
| ● | 減価償却その他の費用。 |
我々は,我々のサービスプロバイダが提供する情報を用いて特定のタスクを達成する進捗を評価することにより,外部開発コストを確認する.
これらのコストは、複数の計画に配置されているため、個別に分類されることはないので、従業員コストや施設費用(減価償却または他の間接コストを含む)を特定の計画に割り当てることはない。著者らは主に内部資源を用いて研究と発見及び著者らの臨床前開発、技術開発、製造と臨床開発活動を監督及び管理する。これらの従業員は複数のbr計画で働いているため、私たちは計画通りに彼らのコストを追跡しない
73
次の表は私たちの研究開発費の計画支出をまとめています
| 十二月三十一日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| 千単位のドル | ||||||||
| BX 004 | 8,853 | 3,499 | ||||||
| BX 005 | 81 | 1,011 | ||||||
| 賃金と関連給付(株式報酬を含む) | 6,004 | 9,130 | ||||||
| 減価償却 | 782 | 909 | ||||||
| 賃料および関連支出 | 905 | 1,101 | ||||||
| インフラやその他の未分配や研究開発費 | 2,410 | 2,017 | ||||||
| 国際投資局からの支出と協力協定からの考慮は少ない | (2,337 | ) | (1,423 | ) | ||||
| 研究開発費総額(純額) | 16,698 | 16,244 | ||||||
研究開発活動は私たちの業務の核心だ。臨床開発後期段階にある候補製品は通常,臨床開発早期段階の候補品よりも高い開発コストを有しており,これは主に後期臨床試験の規模と持続時間が増加しているためである。我々の研究開発費は主に停止または棚上げされたプロジェクトおよび新たな開発プロジェクトを反映している。したがって,今後数年で我々の研究開発費は大幅に増加することが予想され,特に候補製品の開発を進め続けるにつれて,株ベースの報酬,請負業者コスト,施設コストを含む人員コストが増加している.私たちはまた、第三者へのマイルストーンと特許権使用料の支払いに関する追加費用を発生させる可能性があり、私たちは候補製品の権利を得るためにライセンス契約を締結しました。
一般と行政費用
一般及び行政支出は主に行政、財務、会社、業務発展及び行政機能者の賃金、関連福祉及び株式給与支出を含む。一般および行政費用には、会社および証券事務に関連する法律費用、会計、税務および監査サービスの専門費用、保険料、出張費用、施設関連費用(レンタル料を含む)、および運営関連コストも含まれる。
我々がAPT運営を統合して継続的な研究活動や候補製品の開発を支援することにより,我々の一般的かつ行政費用 は将来的に増加する可能性があると考えられる。また、上場企業に関連する巨額の会計、監査、法律、規制、コンプライアンス、役員と役員保険費用、投資家と広報費用が引き続き発生すると予想しています。私たちはこのようなサービスの追加費用が私たちの未来の一般的で行政的費用を増加させると予想する。また,候補製品が規制部門の承認を得る可能性があると考えられれば,ビジネス運営に備えているため,特に候補製品の販売やマーケティングに関する準備作業により,賃金や費用が増加すると予想される。
無形資産の償却
無形資産には進行中の研究·開発が含まれており、償却期間は3年で、2020年1月1日から2022年12月31日まで。
その他の収入
その他の収入には、2022年8月から、イスラエルのネチオナにあるオフィススペースの一部を分譲する収益が含まれている。
利子支出
支払利息には、Hercules Loan プロトコルによる利息が含まれています(以下のように定義されます。私たちはHercules Capital、Inc.またはHerculesとリスク債務融資またはHercules融資協定について融資と保証協定を締結した。Hercules融資協定によると、Herculesは当社に総額3,000万ドルまでの定期融資、または定期融資融資を提供する。2024年3月19日、当社は定期融資に基づいてすべての残りのローンを前払いし、総額は10,428,000ドルである。前金には98.3千ドルの期末費用と6.9万ドルの課税利息が含まれています。
財務費用、純額
財務費用は、純額は主に収入 或いは外貨再評価に関連する費用及び中国の銀行預金と通貨市場基金の利息収入を含む
74
経営成果
2023年12月31日までと2022年12月31日までの年次比較
次の表は、2023年12月31日と2022年12月31日までの年間総合経営実績をまとめたものです
| 十二月三十一日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| 千単位のドル | ||||||||
| R&D費用、純額 | 16,698 | 16,244 | ||||||
| 無形資産の償却 | - | 1,519 | ||||||
| 一般と行政費用 | 8,650 | 9,456 | ||||||
| 営業損失 | 25,348 | 27,219 | ||||||
| 利子支出 | 2,404 | 2,069 | ||||||
| 財務収入、純額 | (1,249 | ) | (902 | ) | ||||
| その他の収入 | (357 | ) | (134 | ) | ||||
| 税金支出 | 23 | 65 | ||||||
| 純損失 | 26,169 | 28,317 | ||||||
2023年12月31日までの1年間の研究開発費純額(IIAから受け取った贈与と研究協力の対価格を差し引く)は1,670万ドルであるのに対し、2022年12月31日までの年間は1,620万ドルである。2023年12月31日までの1年間で、前年に比べて50万ドル増加し、3%増となった要因は以下の通り
| ● | 私たちのCF候補BX 004の臨床試験に関連する費用が増加したため、530万ドル増加した |
| ● | 賃金および関連費用と株式報酬費用が310万ドル減少したのは、主に会社再編によるリストラと、ドル対新シェケル高により、われわれイスラエル子会社の賃金と関連費用が減少した |
| ● | アルツハイマー病治療の候補製品BX 005の開発が中断されたため、90万ドル減少した |
| ● | 100万ドル減少した理由は研究協力の考慮が増加して費用が減少したからです |
2023年12月31日と2022年12月31日までの毎年,IIAからの合計110万ドルの贈与を記録した。
無形資産の償却は2022年12月31日に終了した。無形資産はすべて償却されたからだ。
2023年12月31日までの年間の一般·行政費は870万ドルであるのに対し、2022年12月31日までの年間は950万ドルである。80万ドル、または8%減少したのは、主に会社役員や上級管理職の保険料が90万ドル減少したためだ。
2023年12月31日までの年度の利息支出は240万ドルであるのに対し、2022年12月31日までの年度の利息支出は210万ドルである。30万ドル、または14%増加したのは、米国の最優遇金利の引き上げによるものであり、Hercules融資協定下での利息支払いの増加を招いた。
財務収入純額は2023年12月31日現在で120万ドルであるが、2022年12月31日現在の年度純収益は90万ドルである。30万ドルの増加、すなわち33%の増加は、主に金利上昇により、わが銀行預金の利息収入が増加したためである。この成長は、ドル対新シェケル高によるbrの減少によって部分的に相殺され、より高いレート支出を招いている。
2023年12月31日までの年度は、その他の収入は40万ドルだったが、2022年12月31日までの年度は10万ドルだった。30万ドル、または300%増加したのは、わが社の再編後2022年8月に締結されたイスラエルのネチオナのオフィススペースの一部で分譲契約収益を受けたためである。
75
流動資金と資本資源:
流動資金源
私たちは私たちの製品を販売することから何の収入も得たことがなく、私たちの運営は深刻な運営損失と負のキャッシュフローを生み出した。これまで、私たちの運営資金は、主に普通株、優先株と引受権証、リスク債務、IIA贈与と協力協定の資金、および特別目的買収会社Chardan Healthcare Acquisition Corp.とBiomX Ltd.との業務統合から、Chardan Healthcare Acquisition Corp.はBiomX Inc.と改称された。2023年12月31日現在、普通株と優先株の売却から約1.54億ドルの総現金収益を得ている。2021年8月、私たちはHerculesローン協定に基づいて1,500万ドルを借りた。また,2023年12月31日と2022年12月31日までの毎年,我々の協力協定とIIAから提供された贈与から約190万ドルを獲得した
即時需要を超える現金に投資するのは主に流動性と保証のためだ。
2020年12月4日、米国証券取引委員会によって2020年12月11日に発効を宣言したS-3表の棚上げ登録 声明を提出した。また、2020年12月4日に公開市場販売協定を締結しましたSMまたはJefferies LLC Jefferiesと締結された販売契約によれば、私たちは時々Jefferiesを介して5,000万ドルまでの総発行価格の普通株を発行して販売することができる。販売契約によると、私たちはいかなる普通株も売却する義務はありません。2023年12月31日までに,売却合意により合計983,384株の普通株 を売却し,総収益は580万ドルであった。私たちは2023年12月7日に販売協定を終了した。
2021年8月16日、私たちはHerculesとリスク債務融資に関するHercules 融資協定を締結した。Hercules融資協定によると、Herculesは米国に総額3,000万ドルに達する定期融資を提供し、3ロットに分けて提供され、ある条項とbr条件の制限を受けている。第1弾の1,500万ドルは、Hercules融資協定に署名した当日に前払いされています。第2回と第3回のマイルストーンはまだ達成されておらず、期限が切れています。したがって、私たちはHercules ローン協定によって追加金額を得たことがない。私たちは2023年3月1日までに利息のみの支払いを要求され、2025年9月1日までに元金残高brと利息を月分割払いで返済することを要求されました。2024年3月19日、私たちは力神ローン協定の下の未返済金額を自発的に前払いし、この協定は満期になった。
2023年2月22日、私たちは証券法で規定されている登録免除規定に基づいて、私募発行と売却によって合計15,997,448株の私たちの普通株と14,610,714株の予資金権証、あるいは予資権証、および証券を締結し、価格はそれぞれ1株0.245ドルと1部当たりの資本権証0.244ドルである。発行コストを差し引くまで、2023年2月のパイプラインの総収益は約750万ドル。今回の発行は2部に分かれて終了しました。1回目の成約は2023年2月27日に発生し、3,199,491株の普通株と2,776,428株の予融資権証を発行し、総収益は150万ドルであった。このような事前出資の引受権証は2023年2月27日から行使可能であり、行使価格は1株当たり0.001ドルであり、満期日はない。1回目の取引では,発行コスト 20万ドルを差し引いて130万ドルの純収益を調達した。2023年4月24日、ニューヨーク証券取引所米国規則に基づき、我々の株主は最大24,632,243株の普通株の発行を許可し、その中にbr株に関する事前出資株式承認証を含む。2023年5月4日、私たちは2023年2月のパイプの第2回閉鎖を完了し、合計12,797,957株の普通株と11,834,286部の予融資権証を発行した。このような事前資金権証brは2023年5月4日から普通株1株当たり0.001ドルの行使が可能であり,満期日はない。2回目の終値では、発行コスト10万ドルを差し引いて590万ドルの純収益を集めた。2023年12月31日現在、事前資金の引受権証は行使されていない。
2023年12月7日、2024年1月2日に米国証券取引委員会によって発効が宣言されたS-3表の棚上げ登録 声明を提出した。さらに、2023年12月7日に、私たちはウェインライトとATM協定を締結し、この協定によると、私たちは時々ウェインライトを通じて750万ドルまでの総発行価格の普通株を発行して販売することができる。ATM プロトコルにより、普通株を販売する義務はありません。2024年1月1日から2024年3月26日まで、ATM協定により75,179株の普通株を発行し、総収益は19,000ドルであった。
76
2024年3月15日、買収に関連して、2024年3月のパイプラインを完成させ、このパイプにより、合計216,417株の転換可能優先株と私募株式権証を売却し、合計108,208,500株の普通株を購入し、総収益は約br}5,000万ドルであった。
私たちの財務諸表には説明的なbr段落が含まれており、私たちが経営を続けている企業として少なくとも1年から2025年4月3日まで経営を続ける能力に大きな疑問を投げかけています。将来的に、私たちは、私たちの運営費用や資本要求を支援するために、または買収のような他の目的のために、またはより多くの資金を得ることを必要とするか、または、現在ATMプロトコルに対して行われているように、私たちがHerculesローンプロトコルに対して行っているように、公的または私募株式または債務融資または協力を通じて、または他のソースからこのような追加資金を調達することを求めるかもしれない。イスラエル-ハマス戦争やイスラエルの政治的不安定などによるいくつかの中断が続いて深まった場合、私たちは追加の資本を得ることができないかもしれません。これは、将来的に運営費用やbrの資本需要を支援する能力に悪影響を与えたり、買収のような他の目的に投資したりする可能性があります。
私たちは追加的なbr融資の他の約束を得ていないし、追加的な融資を完全に得ることができると保証することもできないし、もしあれば、そのような融資は私たちに有利な条項で得られ、希釈されないだろう。私たちの将来の流動性と現金需要は、br新製品の発売と、私たちの運営支出を制御し続ける能力を含む多くの要素に依存するだろう。
キャッシュフロー
次の表は に示す各時期のキャッシュフローをまとめたものである
| 十二月三十一日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| 千単位のドル | ||||||||
| 経営活動のための現金純額 | (21,286 | ) | (29,092 | ) | ||||
| 投資活動提供の現金純額 | 1,951 | (2,107 | ) | |||||
| 融資活動が提供する現金純額 | 2,899 | 292 | ||||||
| 現金および現金等価物と限定的現金に及ぼす為替レート変動の影響 | 6 | 106 | ||||||
| 現金および現金等価物の純増加(減額) | (16,430 | ) | (30,801 | ) | ||||
77
経営活動
2023年12月31日までの1年間に,経営活動には2,130万ドルの現金純額が使用されており,純損失2,620万ドルと,我々の経営資産や負債変動に使用した現金純額250万ドルと非現金費用240万ドルが主な原因である。2023年12月31日までの年度の非現金費用には、主に株式ベースの給与支出100万ドル、減価償却と償却90万ドル、債務償却発行コスト60万ドルが含まれている。当社の運営資産および負債の純変動は、2023年12月31日までに、主に貿易勘定の60万ドルの増加およびその他の売掛金の120万ドルの増加を含むが、他の流動資産が80万ドル減少した部分から相殺される。
2022年12月31日までの年間で,運営活動には2,910万ドルの現金純額が使用されており,純損失2,830万ドルと,運営資産や負債変動に使用されている現金純額440万ドルと非現金費用370万ドルが主な原因である。2022年12月31日までの年度の非現金費用には、主に150万ドルの株式給与支出と250万ドルの減価償却と償却が含まれる。2022年12月31日現在、当社の運営資産および負債の純変動は、主に貿易勘定の200万ドルの減少およびその他の売掛金の330万ドルの減少を含むが、他の流動資産によって100万ドル減少した部分で相殺される。
投資活動
2023年12月31日までの年間で、投資活動に使用された現金純額は200万ドル、短期預金を引き出した収益は200万ドルだった。
2022年12月31日までの年間で、投資活動に使用される現金純額は210万ドルで、主に1,350万ドルに投資する短期預金を含み、一部は1,150万ドルを引き出した短期預金の収益によって相殺される。彼は言いました
私たちは私たちの投資政策に基づいて既存の現金を短期投資に投資し、投資を続けることを計画している。これらの投資には、通貨市場基金や米国債からなる投資証券、および会社や政府が支援する企業の高品質で販売可能な債務ツールが含まれる可能性がある。我々は外貨契約(主にオプションと長期契約)を用いて貸借対照表項目の通貨リスクを穴あけする。会計目的で、これらの外国為替契約はヘッジツールとして指定されていない。これらの外貨契約について、私たちが確認した収益や損失は貸借対照表項目の再評価を相殺しており、これらの項目も財務費用の純額に記録されています。2023年12月31日現在、約410万ドルの未返済外国為替契約 を持っており、公正価値資産は30万ドルです。2022年12月31日現在、私たちは約450万ドルの未返済外国為替契約を持っており、その公正価値負債は55,000ドルです。
融資活動
融資活動は2023年12月31日までの年間で300万ドルの現金純額を提供しており、主に2023年2月のパイプラインによる普通株発行による720万ドルが含まれており、発行コストを差し引いた部分は、大力神ローン協定項下の430万ドルの長期債務返済によって相殺されている。
融資活動は、2022年12月31日までの年間で、販売協定による普通株発行による30万ドルを主に含む30万ドルの現金純額を提供している
契約義務、約束、または事項がある
私たちの契約義務と約束は主に私たちが正常な業務過程で各種の研究開発組織とサプライヤーと締結した協定項目の下での力神ローン協定、経営レンタルと撤回できない調達義務と関係があります。2020年9月、私たちはイスラエルのネチオナで新しい事務と実験室空間のレンタル協定を締結した
正常な業務過程において、私たちが締結した契約と協定は様々な陳述と保証を含み、一般的な 賠償を規定している。私たちのこれらの合意の下でのリスクは未知であり、それは未来に私たちに提起されるかもしれないが、まだ提起されていないクレームに関連しているからだ。私たちは今まで、私たちの賠償義務に関連するいかなる訴訟を弁護するために請求されたり、要求されたいかなる訴訟も支払わなかった。しかし、このような賠償義務のため、私たちは未来に費用を記録するかもしれない。
私たちの会社の登録証明書と定款及び契約賠償協定によると、私たちは私たちの高級管理者と取締役に対して潜在的な賠償義務を負っています。彼らが私たちの要求に応じてこのような身分でサービスする時、彼らは一定の制限を受ける可能性があります。今まで何のクレームもありません。取締役と役人保険があります。将来可能なクレームのために支払われた任意の金額の一部を回収させることができます。
78
政府補助金と関連印税
イスラエル政府は国際投資協会を通じて助成金を提供して研究と開発プロジェクトを奨励しています。私たちは国際投資協会から援助を受けることができ、金額は研究·開発費の20%から50%まで様々です。2023年12月31日まで、国際投資協会から合計800万ドルの寄付を受けました。BiomX株式会社は、2017年までFutuRxインキュベーターの一部として設立されたインキュベーター会社であり、その資金の大部分はIIAからの贈与およびインキュベーターの資金であり、このインキュベーターはIIAによって支持されている。インキュベーターを離れた後、私たちは引き続きIIAの贈与を申請し、受け入れます。このような援助の要求と制限は研究法で見つけることができる。研究法によると、これらのIIAを用いて開発された製品またはサービスの全部または一部を贈与して販売して得られた収入の3%~3.5%の特許使用料はイスラエル政府に支払われなければならない。私たちはこの2つのプラットフォーム技術を開発し、少なくとも一部はこれらの寄付金の資金を使っているので、私たちは私たちの任意の候補製品の販売にこれらの印税を支払い、規制部門の承認を得る義務があります。
以下は、研究法に基づいてIIAから得られた贈与が負う義務について説明する
現地製造義務
私たちの候補製品の生産がイスラエルで行われ、IIAによって助成された技術が非イスラエルエンティティに販売または許可されていない限り、支払われる最高合計br印税は、一般に、例年の最初の営業日に発表されるように、一般に、私たちに提供される贈与金の100%を超えず、brドル預金に適用される12ヶ月のSOFRに相当する年利率を超える。
研究法の条項によると、IIAの事前承認を得た場合にのみ、イスラエル以外の場所で製品 を生産することができる(移転合計10%までの製造能力はこのような承認を必要とせず、この場合、IIAに通知を出さなければならず、IIAは通知が出されてから30日以内に反対してはならない)。
ノウハウ譲渡制限
研究法は、IIAが援助した技術ノウハウをイスラエル国外に移す能力を制限している。国際投資機関が援助したノウハウをイスラエル以外に移すには、事前に国際投資機関の承認を得る必要があり、研究法に規定されている式に基づいて計算すると、国際投資機関への支払いが必要になる可能性がある。償還費の上限は国際保険業協会の贈与総額の6倍であり、加えて計上すべき利息(すなわち国際保険業協会に対する総負債は、課税利息を含み、×6)である。IIAが助成したノウハウを譲渡することを望む場合、承認された条項は、取引の性質と、この譲渡に関連する支払いが私たちに支払う対価格に基づいて決定される。
受け入れ者が研究法及び関連条例の規定を遵守し、イスラエル国外へのノウハウの譲渡及び製造権利の制限を含む場合にのみ、IIAが援助したノウハウを他のイスラエル会社に譲渡することを許可することができる。
統制権の変更
その他の事項を除いて、(I)私たちの5%以上の株式または投票権の所有者になるような非イスラエル市民、住民または実体、(Ii)私たちの取締役または私たちのCEOを任命する権利があり、または(Iii)私たちの取締役の1つまたは私たちの最高経営責任者(この直接所有者に25%以上の投票権、持分または指名取締役を含む権利を含む)を任命し、IIAに通知し、IIAに贈与計画に適用される規則および規定を遵守しなければならない。上記の振込制限 を含む.
要請があれば、イスラエル国外で製品を生産することを承認したり、国際投資機関が援助するノウハウの譲渡に同意したりすることが国際投資機関の裁量である。さらに、IIAは、IIAが援助したノウハウまたは製造技術をイスラエルに移すことを可能にする任意の手配にいくつかの条件を課すことができるかもしれない。
将来的にIIA資金によって開発されたノウハウをイスラエル以外の取引に移すことに関連して、我々の株主が獲得可能な対価格(例えば、合併や同様の取引)は、IIAに支払う必要がある任意の金額を減少させる可能性がある。
2023年12月31日現在、何の販売も生じていません。IIAに対する将来の支払い約束の元金と利息残高は約790万ドルですが、2022年12月31日現在の元金と利息残高は660万ドルです。私たちが現在計画している製品開発活動に資金を提供する一部として、私たちは新しい支出を得るために後続の支出申請を提出する可能性があります。
79
展望
私たちは持続的な活動と関連した費用がほぼ同じ水準に維持されると予想する。私たちの支出は相当なものを維持し、増加するかもしれない
| ● | 私たちの候補製品を開発し続けています |
| ● | INDを支援する活動を完了し、私たちの候補製品のためのbr臨床試験を開始する準備ができている |
| ● | APTのビジネス統合に取り組んでいます |
| ● | 私たちの候補製品のために追加の臨床試験と臨床前研究を開始した |
| ● | 他の候補製品および技術の識別および開発、許可または取得を求める; |
| ● | 私たちが臨床試験に成功した候補製品のために規制の承認を求めます |
| ● | 販売、マーケティング、流通インフラを構築し、規制の承認を受ける可能性のある任意の候補製品を商業化する | |
| ● | 臨床、品質管理、商業、科学者のようなより多くの人員を採用し、保留する | |
| ● | 私たちの研究開発を支援するために、設備や実物インフラを増やすことを含む、私たちの増加する従業員基盤に適応するために、私たちのインフラや施設を拡大します。 |
私たちの財務諸表には説明的なbr段落が含まれており、私たちが経営を続けている企業として少なくとも1年から2025年4月3日まで経営を続ける能力に大きな疑問を投げかけています。私たち は、間違っていることが証明される可能性があるという仮定に基づいてこれらの推定を行い、利用可能な資本資源を予想よりも早く利用することができます。 私たちの候補製品が規制部門の承認を得たら、製品の製造、販売、マーケティング、流通に関連した巨額の商業化費用 が発生することが予想されます。具体的には、私たちがどこで商業化するかにかかっています。
もしあれば、利益を達成するのに十分な製品収入を生成することができる前に、ATMプロトコル、融資、マイルストーン支払い、IIAまたは他の政府または非営利機関、または他の外部資金源から得られる可能性のある追加贈与を含む、私たちの株式を公開または個人的に販売することによって、私たちの現金需要を満たす予定です。私たちが株式や債務市場で追加資本を調達する能力は、イスラエル-ハマス戦争、ウクライナなどの他の武力衝突や他の破壊による市場変動、私たちの証券に対する市場需要、市場が私たちの証券の需要自体に多くの発展と業務リスクと不確実性の影響を受けること、および私たちが会社に有利な価格や条項でこのような追加資本を調達できるかどうかの不確実性を含む多くの要素に依存しています。私たちが株式を売却したり、債務証券を転換して追加資本を調達できるかどうかについては、私たち株主の所有権権益は大幅に希釈される可能性があり、このような証券の条項は清算または他の 特典を含む可能性があり、彼らの普通株主としての権利に悪影響を及ぼす。債務融資および優先持分融資(利用可能であれば)に関連するプロトコルは、追加債務を生成すること、資本支出を行うこと、または配当を宣言することなど、特定の行動をとる能力を制限または制限する契約を含む。もし私たちが政府と他の第三者資金、協力br協定、戦略連合、許可手配、またはマーケティングおよび流通手配を通じて追加資金を調達した場合、私たちは私たちの技術、将来の収入フロー、研究計画、または候補製品に対する貴重なbr権利を放棄しなければならないか、または私たちに不利になる可能性のある条項でライセンスを付与しなければならないかもしれない。もし私たちが必要な時に株式や債務融資を通じてより多くの資金を調達できない場合、私たちは私たちの製品開発や将来の商業化努力を延期、制限、減少または終了することを要求されるかもしれません。あるいは私たちが自分で開発し、マーケティングすることをより望んでいた製品または候補製品の権利を与えることができます。私たちの展望に関連するリスクに関するより多くの情報は、“を参照してください”リスク要因-私たちの業務、技術、そして産業に関連するリスク
80
外国為替契約
我々は,賃金と関連費用の支払いおよび新シェケル建ての他の費用による将来のキャッシュフロー全体の変化のリスクをヘッジする長期和オプション契約を締結した。2023年12月31日と2022年12月31日まで、私たちの未平倉外国為替契約の名目金額はそれぞれ約410万ドルと450万ドルです。
キー会計見積もり数は
私たちの連結財務諸表はアメリカ公認会計基準に基づいて作成されました。我々の連結財務諸表および関連開示を作成する際には、資産、負債、収入、コストおよび支出報告金額に影響を与える推定および判断を行い、財務諸表に開示または資産および負債がある必要がある。我々の見積りは,歴史的経験,既知の傾向や事件,および当時の場合には合理的であると考えられる様々な他のbr要因に基づいており,これらの要因の結果は資産や負債の帳簿価値を判断する基礎を構成しているが,これらの資産や負債の帳簿価値は他の源からは明らかではないように見える。私たちは私たちの推定と仮定 を評価し続けている。異なる仮定や条件の下で、私たちの実際の結果はこれらの推定とは異なる可能性がある。
我々の主な会計政策は、我々の総合財務諸表付記2により詳細に記載されているが、以下の会計政策は、我々が総合財務諸表を作成する際に使用する判断と推定に最も重要なものであると考えられる。
研究と開発費用を計算すべきである
連結財務諸表作成過程の一部として、計算すべき研究開発費を見積もる必要がある。この流れは、未締結のbr契約および購入注文を審査し、私たちの適用者とコミュニケーションして、私たちを代表して実行されたサービス を決定し、領収書を受け取っていない場合、または他の方法で実際のコストを通知している場合に、実行されたサービスレベルおよびサービスによって生じる関連コストを推定することを含む。私たちのほとんどのサービスプロバイダは、あらかじめ決められたスケジュールに従って、あるいは契約マイルストーンに達したときに借金の領収書を発行してくれますが、前金が必要なものもあります。私たちは、当時私たちが知っている事実と状況に基づいて、連結財務諸表において、各貸借対照表の日付までの課税費用を推定します。我々は定期的にサービスプロバイダとこれらの推定の正確性を確認し,必要に応じて調整する.計算すべき研究および開発費用の推定例には、支払いが含まれている
| ● | 臨床前開発活動に参加したサプライヤー |
| ● | 前臨床試験と臨床試験に関するCROと研究場所; |
| ● | 臨床前と臨床試験に用いられる材料の生産に関する下請け業者。 |
我々は、複数のCROと下請け業者とのオファーと契約に基づいて、受信したサービスと費用の推定に基づいて確認費用を測定し、これらのCROと下請け業者は私たちを代表して、臨床前研究、ヒト臨床研究と臨床試験を実施し、管理している。これらの合意の財務条項 は協議することができ、契約によって異なり、支払いの不均衡を招く可能性がある。場合によっては、私たちのサプライヤーに支払われるお金は、提供されたサービスレベルを超え、料金の前払いをもたらす可能性があります。その中のいくつかの契約下の支払いは患者の成功登録といくつかのマイルストーンの完成などの要素に依存する。サービス料金を計算する際には,サービスを実行する時間帯と各時間内の作業支出レベルを見積もる.サービス実行の実際の時間または作業レベルが推定値と異なる場合、それに応じて計算すべき費用または前払い費用の金額を調整する。実際に発生した金額と実質的に異なることはないと予想されるが,実行されたサービスの実態や時間に対する実行サービスの状態や時間の理解が異なる可能性があり,任意の特定の期間で確認された金額の増加や減少の推定値が変化する可能性がある.これまで,我々は研究や開発費の先行見積もりに応じて何の大きな調整も行っていない.
株に基づく報酬
我々は、従業員および取締役に支給されたすべての株式ベースの支払い報酬の報酬支出を計量および確認することが要求されるASC 718-10“株式ベースの支払い” を適用し、推定された公正価値に基づいて従業員の株式オプションを我々の株式計画に含める。
ASC 718-10は、オプション定価モデルを使用して、日株権支払い報酬を付与する公正価値を推定することを要求する。報酬の公正価値は,我々の総合運営報告書で必要なサービス期間の費用 であることが確認された.我々は,適用ペナルティ率ではなく,株式報酬ペナルティが発生していない場合に確認した.
81
各報酬の必要なサービス期間内に非従業員報酬の公平 価値の補償費用を確認した。
我々はBlack-Scholesオプション定価モデルを用いて、株式奨励として付与された株式オプションの公正価値を推定する。オプション定価モデルは多くの仮定を必要とし,その中で最も重要なのは株価,期待変動率,期待オプション期限(付与日からオプション行使または満期までの時間)である.私たちは私たちの最近の株式販売状況を考慮することで対象株の1株当たりの公正価値を決定する。BiomX Ltd.歴史上ずっと1つの私営会社であり、その株の特定の歴史と隠れた変動率情報 が不足している。我々が用いた歴史的平均株価変動率は,我々の歴史的平均変動率とバイオテクノロジーや製薬業界で選定された上場企業同業グループの歴史的平均変動率と比較可能な総合加重平均値に基づいており,これらの会社は将来の株価傾向を代表していると考えられており,我々の普通株には十分な歴史的取引履歴がないためである。我々は,我々の株価変動に関する十分な数の履歴情報が利用できるまで,この過程を適用し続ける.私たちは以前から配当金を派遣していなかったし、配当金を発行する予測可能な計画もなかった。無リスク金利は同値期限の国債利回りに基づいている。すべての株式オプション が付与する期待オプション期限は“簡略化”方法を用いて計算される.各投入の決定の変化は、付与されたオプションの公正価値と私たちの運営結果に影響を及ぼす可能性がある。
無形資産
業務合併で買収されている研究·開発は,買収日に公正価値で確認され,その後無期限無形資産として入金され,関連研究や開発作業が完了または放棄されるまで入金される。
私たちは買収会計方法を用いて買収RondinX Ltd.に対して会計計算を行い、これは私たちに買収資産と負担した負債の公正な価値を推定することを要求した。このbr}には買収中の研究開発とあるいは価格があることが含まれている。対価格公正価値の調整 を収益に計上する.2020年1月1日、進行中の研究開発が完了した。当社は研究開発資産の耐用年数を3年と決定し、財務諸表に応じてこれらの資産を償却し始めた。2022年12月31日までの年間で、150万ドルの償却費用を記録した。この無形資産は2022年12月31日までにすべて償却された。
82
プロジェクト7 A.市場リスクに関する定量的かつ定性的開示
小さな報告会社として、本項の下で開示する必要はありません
項目8.財務諸表と補足データ
我々の財務諸表とその付記 は本年度報告のF−1ページから始まる。
項目9.会計·財務開示における変更と分岐
ない。
プロジェクト9 A.制御とプログラム
情報開示制御とプログラムの評価
我々の経営陣は、最高経営責任者および臨時最高経営責任者(それぞれ最高経営者および最高財務責任者)の参加の下、2023年12月31日現在の開示制御および手順(“取引所法案”規則13 a-15(E)および15 d-15(E)で定義されている)の有効性を評価した。上記の評価に基づき、我々の経営陣は、2023年12月31日現在、我々の開示制御及び手続が合理的な保証レベルで有効であると結論した。
経営陣の年度財務内部統制報告書
私たちの経営陣は財務報告書の十分な内部統制を確立して維持する責任がある。私たちの財務報告に対する内部統制はアメリカが公認している会計原則に基づいて財務報告の信頼性と外部目的の財務諸表の作成に合理的な保証を提供することを目的としている。
私たちの財務報告に対する内部統制は、財務報告書の作成を可能にするために米国公認の会計原則に基づいて取引を記録し、私たちの管理層と取締役の許可のみに基づいて収支 を行うことを可能にするための合理的な保証を提供することと、私たちの経営陣および取締役の許可のみに基づいて収支 を行うことと、私たちの財務報告書に重大な影響を与える可能性のある不正な買収、使用、または私たちの資産をタイムリーに発見することを防止するための合理的な保証を提供することとを含む。
その固有の限界により,財務報告の内部制御 は誤った陳述を防止あるいは発見できない可能性がある。したがって,有効と判断されたシステムであっても,財務諸表の作成や列報の面で合理的な保証を提供することしかできない.将来的にどのような有効性評価を行うかの予測は,条件の変化により制御措置が不足したり, が政策やプログラムを遵守している程度が悪化する可能性がある.
83
経営陣は2023年12月31日に財務報告書の内部統制に対する私たちの有効性を評価した。この評価を行う際には,管理層はトレデビル委員会2013年枠組み協賛組織委員会 が#年に提出した基準を用いた内部制御--統合フレームワークそれは.これらの基準の下での評価に基づき、経営陣は、2023年12月31日現在、財務報告の内部統制に有効であると判断した。
非加速申請者として“取引所法案”に基づいているので、財務報告の内部統制に関する独立公認会計士事務所の証明報告書を提供する必要はありません。現在の財務報告の内部統制の変化時間
2023年度第4四半期において、我々は、財務報告の内部制御に大きな影響を与えないか、または財務報告の内部制御に大きな影響を与える可能性がある変化を生じていない(この用語は、“取引法”の下のルール13 a−15(F)および15 d−15(F)で定義されている)。
プロジェクト9 B.その他の資料
貿易手配
2023年12月31日までの3ヶ月以内に、私たちの役員または上級管理者
株式発行を承認する
2024年4月2日、我々の取締役会は、デラウェア州会社法第204条による買収完了に基づいて発行された普通株の発行、または承認を承認する決議を採択した。本年度報告書添付ファイル99.1には、デラウェア州会社法第204条に基づいて承認を要求する情報が記載された当社取締役会が採択した決議書のコピーが記載されている。いかなるクレームであっても、欠陥のある会社法または承認に基づいて承認された推定株式は、決議に規定された認可失敗のために無効または撤回可能であるか、またはデラウェア州衡平裁判所は、デラウェア州会社法第204条による承認が無効であるか、またはいくつかの条件の下でのみ有効であり、本通知が発行されてから120日以内に提出されなければならない(本年度報告が米国証券取引委員会に提出された日から120日以内とみなされる)。
プロジェクト9 Cです。検査阻止に関する外国法規の開示
適用されません。
84
第 第3部分
プロジェクト10.役員、役員、および企業管理
以下に、2024年4月3日現在、執行役員および取締役会のメンバーを務める各人の名前、年齢、役職を示す。
| 名前.名前 | 年ごろ | ポスト | ||
| 行政員 | ||||
| ジョナサン·ソロモン | 47 | 取締役CEO兼最高経営責任者 | ||
| アサフ·オロン | 49 | 首席商務官 | ||
| マリナ·ウォルフソン | 39 | 首席財務官 | ||
| エイブラハム·ガブリエル | 39 | 臨時首席財務官 | ||
| メラフ·バサン博士 | 58 | 首席発展官 | ||
| 非従業員取締役 | ||||
| ラッセル·グレッグ博士(1)(2)(3) | 71 | 取締役と取締役会長 | ||
| ジョナサン·ライヴ(2) | 55 | 役員.取締役 | ||
| エレン·モーゼ博士(2) | 74 | 役員.取締役 | ||
| グレゴリー·メリル(3) | 58 | 役員.取締役 | ||
| エドワード·ウィリアムズ(1) | 67 | 役員.取締役 | ||
| ジェシー·グッドマン博士(3) | 72 | 役員.取締役 |
| (1) | 監査委員会のメンバー |
| (2) | 報酬委員会のメンバー |
| (3) | 委員会と会社管理委員会のメンバーを指名する |
行政員
ジョナサン·ソロモン彼は2019年10月から当社の最高経営責任者兼取締役を務めている。ソロモンさんは2016年2月にBiomX株式会社の取締役会長を務め、2017年2月から2019年10月までの間にCEOを務めた。 2007年7月から2015年12月までの間、ソロモンさんはProClara Biosciences Inc.(前身はNeuroPhage PharmPharmticals Inc.)の共同創業者兼CEOであり、ProClara Biosciences Inc.は神経変性疾患の治療方法を開発するバイオテクノロジー会社である。ProClaraに加入する前に、彼はイスラエル国防軍の機密軍事部門に10年間勤務した。ソロモンさんはB.Scを保有した。ヘブライ大学の物理学と数学修士号を優秀な成績で取得した。テルアビブ大学電気工学専攻を優秀な成績で卒業し、優秀な成績でハーバードビジネススクールMBAを取得した。
我々は、私たちの取締役会でのさんソロモンの資格は、バイオテクノロジー産業の取締役会および管理経験を豊富に含んでいると信じています。
アサフ·オロン 2019年10月から当社の首席商務官を務めます。オロンさんは、2017年1月から2019年10月までBiomX Ltd.の最高経営責任者を務めました。これまで、2006年3月から2016年12月までの間に、農業バイオテクノロジー会社Evogene Ltd.(ナスダック·コード:EVGN)で様々な職務を担当していた。同社は、戦略·事業発展部執行副社長、企業発展部執行副社長を含む独自の統合された技術インフラを利用して作物生産性の潜在的種子性状を向上させる。Evogeneに加入する前に、Oronさんは軟骨サイト株式会社のCEOを務めていた。オーレンさんは、バイオテクノロジー会社であり、整形外科分野において人工組織製品を開発し、かつ、イスラエル管理コンサルティング会社POC株式会社で高級プロジェクトマネージャー及び戦略顧問を務める。生物学(バイオインフォマティクス)と理科学士号。テルアビブ大学の化学と経済学を専攻しています
マリナ·ウォルフソン 2022年4月から会社の首席財務官を務め、現在産休中。ウォルフソンさんは2019年12月から2022年3月までの間に当社で複数の財務·運営職を務めています。ウォルフソンの経験には,大手製薬会社,ハイテク会社およびベンチャーファンドとの協力がある。当社に入社する前に、ウォルフソンさんは2010年から2019年までBioView Ltd.(トロント証券取引所コード:BIOV)で財務副総裁 を務め、2007年から2010年まで安永会計士事務所で高級監査役を務めた。ウォルフソンさんはイスラエルの公認会計士で、ベン-グリアン大学経済学と会計学学士号(栄誉)とMBA学位(栄誉、金融を専攻) を持っている。
85
Avrahamギャビー2023年11月に会社の首席財務官であるウォルフソンさんが産休を開始して以来、彼は会社の臨時首席財務官を務め、ウォルフソンさんの産休中にこのポストを担当する。任命される前に、ギャビーは2021年から2023年までOravax Inc.の首席財務官を務めており、経口ワクチンの開発に専念しているバイオテクノロジー会社である。これまで、ガベさんは、2019年から2021年までの間に、ナスダック社(Oramed PharmPharmticals Inc.)の最高財務責任者であり、同社は、経口インスリンに焦点を当てたタンパク質経口送達プラットフォームを開発しています。2015年から2019年まで、ギャビーさんは、視覚障害者、視覚障害者、読書、または他の障害者のためにウェアラブル支援技術デバイスを開発、製造、販売している会社のディレクターを務めています。Gornitzky&Co法律事務所の税務部門で働くGabayさんは、Gornitzky&Co法律事務所の税務部門で働いている。また、Gabayさんは、Nala Digital Ltd.の取締役会の役員でもある。Nala Digital Ltd.は、テルアビブ証券取引所に株式を上場している会社である。Gabayさんは、イスラエルの公認会計士とイスラエルの弁護士協会のメンバーで、テルアビブ大学の法律および会計の学士号を持っています。
メラフ·バサン博士 2019年10月から当社の首席発展官を務めます。これまで、彼女は2005年から2019年までTeva製薬工業有限会社で各種開発職務を担当し、2017年から2019年まで臨床専門研究開発翻訳科学部副主管総裁を務め、2015年から2017年まで疼痛と全世界内科副主管総裁を務め、プロジェクト指導者、革新製品開発、 2015年から2015年まで取締役グローバル革新製品開発部の高級プロジェクトチャンピオンを務めた。バサン博士は理科学士号を持っています。生物学の修士号です。彼女はテルアビブ大学で人類遺伝学博士号と神経生物学博士号を取得し、ハーバード大学医学院で神経科学博士後奨学金を取得した。
役員.取締役
ソロモンさんの伝記は上記の“執行幹事”の称号の下に掲載された。私たちの非従業員役員の履歴は以下の通りです
ラッセル·グリガー博士2019年10月から取締役および当社取締役会長を務めています。Greig博士は製薬業界で44年以上の経験を持ち、研究開発、業務開発、商業運営に関する知識と専門知識を持っている。彼はグラクソ·スミスクラインでキャリアの大部分を過ごし、そこで2003年から2008年までグラクソ·スミスクライン国際製薬部総裁と上級副総裁グローバル業務発展部を含む複数のポストを務めた。2008年から2010年にかけて、グラクソ·スミスクライン博士はグラクソ·スミスクライン企業リスク投資グループSR Oneの総裁を兼任した。また、グレッグ博士はスペインSanifit社(Vifor Pharma AGに買収)、Tigix N.V.(武田製薬有限会社に買収)、Ablynx N.V.(フランスのセノフィに買収された)、Merus N.V.(ナスダック:MRUS)の取締役会メンバーを務めたことがある。彼はSynaxin Ltd(イギリス)(益プソンに買収)、Novagali Pharma S.A.(フランス)(Santen製薬有限会社に買収)、Isconova AB(スウェーデン)(ノバ社(ナスダックコード:NVAX)に買収された)の会長を務めたことがある。彼は遺伝子生物科学会社(ナスダックコード:GNCA)とIsconova AB社の代理CEOを一時的に務めたことがある。彼はスコットランド科学諮問委員会のメンバーでもあり、スコットランドの首席長官に仕事を報告した。
我々の取締役会におけるグレッグ博士の資格には、業務発展や製薬業界の薬物研究·開発における彼の豊富な取締役会と指導経験が含まれていると信じている。
86
ジョナサン·レフ2024年3月以来、会社の取締役を務めてきました。レーフさんは、Deerfield Management Company、L.P.またはDeerfieldのパートナーとDeerfield Instituteの議長です。彼は2013年にDeerfieldに入社し、バイオテクノロジーや製薬分野のリスク投資と構造的投資に集中した。これまで、レフさんは2000年から2012年まで華平投資有限責任公司で取締役社長を務め、バイオテクノロジーや製薬分野での同社の投資を指導してきた。ライフさんはまた、NVCA(NVCA)の取締役会実行委員会のメンバーを務め、NVCA医療革新と競争力連盟の会長としてNVCAの生命科学業界の努力をリードしています。彼はバイオテクノロジー産業組織の新興会社分会にも勤めていた。ライフさんは、脊柱筋萎縮症財団の取締役会のメンバーとコロンビア大学医学センターの顧問委員会のメンバーを含むいくつかの非営利団体のガバナンスに参加しました。彼は現在上場バイオテクノロジー会社Larimar Treateutics,Inc.の取締役会に勤めている。ライヴさんはまた、バイオテクノロジーおよび製薬会社の他のいくつかの取締役会に勤務しており、2022年~2023年のARS製薬会社、2017~2019年のProteon治療会社、2014~2017年のAveXis社、2014~2016年のnivalis治療会社を勤めています。現在は、いくつかの私営生物製薬会社の取締役会に勤めており、他の私有株のバイオ製薬会社の取締役会に勤めています。ライヴさんはハーバード大学で学士号、スタンフォード大学ビジネススクールで工商管理修士号、ジョン·ホプキンス大学でバイオテクノロジーの修士号を取得した。
我々は、製薬およびバイオ産業の幅広い取締役会およびリーダーシップの経験を含む当社の取締役会でのレフさんの資格は、資本市場およびバイオ技術産業の幅広い分野で含まれると信じています。
エレン·モーゼス博士2020年10月から会社の役員を務めています。モーゼス博士は2021年3月以来、ナスダック治療有限会社(CHEMOMA:CMMB)の取締役会メンバーである。2013年から2018年までの退職前、モーゼス博士はノボノルドA/Sグローバル首席医療官を務めた。これまで,2004年からノ·ノドA/Sで様々な職務を担当しており,米国医療事務補佐副総裁から始まった。彼のキャリア全体の中で,モーゼ博士は糖尿病の新しい治療法や診断法の開発に取り組んできた。彼はベスイスラエル女性執事-ハーバード医学院-マサチューセッツ工科大学で共同で臨床研究者訓練計画を創立し、指導した。1998年から2004年まで、モーゼス博士brはジョスリン糖尿病センターで高級副総裁と首席医療官を務め、専門的にジョスリン診療所を担当した。彼は2021年12月からジョスリン糖尿病センター取締役会のメンバーを務めた。彼は非営利誹謗財団の取締役会長と青少年糖尿病研究基金会大新イングランド分会の取締役会のメンバーも務めている。モーセ博士はセントルイスのワシントン大学医学院で医学博士号を取得し、国家衛生研究院で3年間働き、タフツ新イングランド医学センターで臨床内分泌/糖尿病訓練を完成し、ハーバード商学院で衛生保健戦略を学んだ。
私たちの取締役会におけるモーセ博士の資格には、製薬業の臨床開発における豊富な指導経験が含まれていると信じている。
グレゴリー·メリル は2024年3月以来当社の取締役を務めています。メリルは2016年10月にAPTを創設し、最高経営責任者を2023年10月まで務め、2024年3月まで取締役会に在任した。現在、彼は自分の専門知識を様々なスタートアップ企業に応用し、コンサルタントから役員まで様々なポストを担当している。メリルは2015年8月から2017年12月までの間にYost Labsの最高経営責任者を務め、物理リハビリテーションやドローンナビゲーションなどの分野で慣性運動センサを開発している。2011年から2015年8月にかけて、サッカー、ホッケー、ホッケーなどのスポーツで外傷性脳損傷を引き起こす可能性のある頭部衝突を検出するためのウェアラブルセンサの開発に取り組んだBrain Sentryを設立し、リードした。2009年10月から2011年2月まで、米国海軍とミサイル防衛局の技術調達と配備支援を提供するDecision Technologiesの首席運営官を務めた。これまで,2002年3月から2009年10月までの間に,インタラクティブ実験室の創設最高経営責任者や議長として,Merrilはビデオゲームや軍事シミュレーションにおける身体活動を強化するために特許 および製品に取り組んできた.これまで彼はHT医療システム会社の創設最高経営責任者であり,同社は外科訓練シミュレータに専念し,2000年7月に没入型会社(ナスダック:IMMR)と合併した。メリルは発明家として知られ、22件の特許を持ち、マクダニエル学院精神生物学の学士号を持っている。
我々は、製薬産業における彼の薬物研究開発の経験を含む取締役会での彼の資格をさんすることができると信じています。
87
エドワード“エディ”ウィリアムズ2023年10月以来当社の取締役を務めております。ウィリアムズさんは、2021年12月以来、BioAtla,Inc.(ナスダックコード:BCAB)の取締役会メンバーを務めており、同社は腫瘍学に専念する上場バイオテクノロジー企業です。2018年1月から2022年12月まで、上場バイオ製薬会社Catalyst Biosciences Inc.(ナスダック:CBIO、現在GYRE)の取締役会メンバーを務めた。彼は現在もボン記念健康会社と革新血液学会社の非営利医療保健委員会の役員メンバーを務めている
2020年3月から2022年9月まで、WilliamsさんWilliamsはAscendis製薬会社(ナスダック·コード:ASND)のCEO、特別顧問兼臨時CEOを務めています。Ascendisに参加する前に、2006年から2017年1月まで、ウィリアムズさんは、多国籍製薬会社とバイオテクノロジー会社ノとノド(ニューヨーク証券取引所コード:NVO)で上級副社長と米国バイオ製薬社長を務めました。Novoに加入する前、2003年から2006年にかけて、ウィリアムズさんは、ノワ製薬会社の呼吸器系業務および皮膚科業務において販売副社長を務めた。さんbr}ウィリアムズは1981年にエイプジョンでキャリアを開始し、その後、2001年7月~2001年7月に販売副総裁、2001年7月から2003年5月まで東北地方販売副総裁を務めた。ウィリアムズさんはワシントン州ハンティントンのマーシャル大学とロサンゼルス州立大学の生化学学士号を有している。
我々は、製薬業界の新規化合物の上場前および商業化における彼の成功経験を含む、取締役会におけるウィリアムさんの資格は、彼の豊富な取締役会およびリーダーシップの経験を含むと信じています。
ジェシー·グッドマン博士 は2024年3月以来当社の取締役を務めています。グッドマン博士は2014年3月以来、ジョージシティ大学医療製品取得、安全·管理センターの取締役主任、医学教授、感染症主治医を務めてきた。グッドマン博士はワシントンD.C.退役軍人事務センターやWalter·リード医学センターの感染症内科医でもある。2016年に多国籍製薬会社グラクソ·スミスクラインに入社し、2023年初めまで取締役会科学委員会の議長を務めた。2018年10月以降、バイオテクノロジー会社Intellia Treeutics,Inc.の取締役会に勤務してきた。グッドマン博士は合併合意に達する前にAPTの取締役会に勤めていた。総裁博士(2015年から2020年)や米国薬局方条約会社の取締役会メンバー(2015年から現在まで)も務めている。2009年から2014年2月まで、グッドマン博士はFDA首席科学者を務めている。グッドマン博士は2009年から2012年までFDAの科学·公衆衛生担当副局長も務めた。これまで,グッドマン博士は2003年から2009年までFDA生物製品評価と研究センターの取締役 を務め,1998年から2000年までFDA専門員の上級顧問を務めていた。政府に奉仕する前に,グッドマン博士はミネソタ大学の医学教授と感染症主任であった。グッドマン博士は疾病管理センター、国家衛生研究院、世界保健機関と防疫革新連盟を含む多くの国家と国際医療保健組織の顧問委員会と委員会に勤めていた。グッドマン博士はハーバード大学生物学学士号、ミネソタ大学公衆衛生修士号、アルバート·アインシュタイン医学院医学博士号を持ち、ペンシルバニア大学病院とカリフォルニア大学ロサンゼルス校で医学、伝染病、腫瘍学の入院医師と研究員研修を受け、同校の首席入院医師でもある。彼はすでにアメリカ国立科学院医学研究所に選ばれた。
私たちの取締役会におけるグッドマン博士の資格は、製薬業界と法規の面での臨床開発における豊富な取締役会と指導経験を含むと信じている。
ビジネス行為と道徳的基準
私たちはすべての役員、上級管理職、従業員に適用されるビジネス行動と道徳基準を採択した。ビジネス行動と道徳基準は私たちのサイトwww.bibix.comで見つけることができます。もし私たちが“商業行為と道徳的規則”を実質的に修正したり、任意の役員または幹部に“規則”条項の任意の免除を付与した場合、私たちは直ちに私たちのウェブサイトで修正または放棄の性質を開示します。
88
取締役会委員会と会社管理
取締役会構成と指導構造
取締役会 は2024年4月3日まで7人のメンバーで構成されている。取締役会は取締役会議長と最高経営責任者職の合併や分離に柔軟な政策を持っている。現在、ラッセル·グレッグは私たちの独立した会長を務め、ジョナサン·さんは私たちのCEOを務めています。取締役会は、異なる役割を通じて、行政総裁は当社の日常業務と事務に集中できると信じているが、議長は重要な戦略問題、取締役会のリーダーシップ、コミュニケーションに集中できる。取締役会はこの指導構造が現在最も会社とその株主の利益に合致していると考えているが、取締役会も将来の状況がこれらの役割を合併する可能性があることを認識している。
取締役会委員会
取締役会はすでに三つの常設委員会を設置した:監査委員会、報酬委員会及び指名及び企業管理委員会、各委員会はすべて独立取締役から構成され、詳細は以下の通りである。審査委員会、報酬委員会及び指名及び企業管理委員会はすべて書面定款に基づいて運営し、各委員会はその定款の十分性を審査及び評価し、そしてその定款を取締役会に提出して承認する。監査委員会、給与委員会、指名委員会、会社管理委員会の規約はすべて私たちのウェブサイトwww.bibix.comで調べることができます。
監査委員会
私たちの監査委員会は、会社の独立会計士を招聘します:彼らの独立性と業績を審査し、会社の会計と財務報告の流れとその財務諸表の完全性を審査し、会社の財務諸表の監査と会社の独立監査士の任命、報酬、資格、独立性と業績を審査し、会社の法律と監督要求を遵守する状況を審査し、社内監査機能と財務報告の内部統制の表現を審査します。
監査委員会のメンバーはラッセル·グレッグ博士とエドワード·ウィリアムズ博士で、ニューヨーク証券取引所米国上場基準によると、いずれも独立取締役会社であり、取引所法案第10 A-3条の追加独立性要求に適合している。ラッセル·グレッグ博士は監査委員会の議長だ。監査委員会には現在、米国証券取引委員会規則で定義されている“監査委員会財務専門家” は設置されていない。
報酬委員会
私たちの報酬委員会は、会社の会社の業績目標と最高経営責任者の報酬に関する目標を毎年審査し、これらの目標と目的に基づいてCEOの業績を評価し、この評価に基づいてCEOオフィスの報酬レベルを決定し、承認し、承認しない、承認しない、修正または終了する従業員福祉計画について取締役会に提案し、CEOおよび取締役以外の私たちの役員および取締役の報酬について取締役会に提案する。会社の奨励的報酬計画や株式計画、会社の回収政策を管理している。報酬委員会は、それが適切だと思う場合、その任意の職責をグループ委員会に転任することを自ら決定する権利がある。会社の最高経営責任者brは、その報酬に関する報酬委員会の投票や審議に出席してはならない。その会社の幹部は彼ら自身の給料に参加しないことを提案した。
報酬委員会のメンバーは、エレン·モーゼ博士、ジョナサン·レフさん、ラッセル·グレッグ博士で、ニューヨーク証券取引所米国上場基準に基づいており、いずれも独立した取締役です。アラン·モーゼス博士は賠償委員会の議長だ。
89
給与委員会 は怡安ソリューションイギリス有限会社または独立給与コンサルタント怡安を招聘し、それぞれChardan Healthcare Acquisition Corp.2019年株式激励計画(または2019年計画)と会社 2015年従業員株式オプション計画(または2015計画)下のオプション交換と再定価について提案を提供する。怡安の2023年12月31日までの財政年度の主な責任は、再定価とオプション交換の方法を決定し、このような再定価とオプション交換を決定する際に検討された要因の中でこれらの提案を考慮する報酬委員会に提案を提供することを含む。
指名と統治委員会
私たちの指名と会社管理委員会は取締役会の指名人選の選考を監督します。具体的には、指名·会社管理委員会は取締役会の規模と構成について取締役会に提案し、取締役指名手続き を構築し、候補者を取締役会に選出·推薦する。指名と会社管理委員会は毎年取締役会メンバーに必要ないくつかの資格と特徴を取締役会に推薦し、取締役会の承認に供する。また、指名と会社管理委員会は取締役会全体とその個別メンバーに対する年間業績評価を確立し、監督する。指名と会社管理委員会は個人の取締役会メンバー資格を評価する際に、管理と指導経験、背景、誠実と専門精神に関連する複数の資格 を考慮する。指名と会社管理委員会は指名人選の多様性を決定するための正式な政策を考慮していないにもかかわらず、指名と会社管理委員会はいくつかの技能或いは属性、例えば財務或いは会計経験を必要とし、取締役会に時々出現する特定の需要を満たし、そしてそのメンバーの全体的な経験と構成を考慮して、広範かつ多様な取締役会メンバーの組み合わせを獲得する。指名とコーポレートガバナンス委員会は、株主と他の人が推薦した被命名者とを区別しない。
ニューヨーク証券取引所米国上場基準に従って、ジェシー·グッドマン博士と、ニューヨーク証券取引所米国上場基準に従って、委員会およびコーポレートガバナンス委員会のメンバーを指名しました。ラッセル·グレッグ博士は指名と会社統治委員会の議長だ。
プロジェクト11.役員報酬
報酬総額表
次の表は、過去2会計年度の支払または計上された報酬総額を示しており、(I)我々の最高経営責任者、(Ii)私たちの他の2人の報酬が最も高いbr役員であり、彼らは2023年12月31日までの会計年度で1人当たり100,000ドルを超え、その日までに役員を担当する。
| 名称と主要ポスト | 年.年 | 賃金.賃金 ($)(1) | ボーナス.ボーナス ($)(1) | オプション大賞(2) ($)(2) | 他のすべての 補償する ($)(1)(3) | 合計する ($)(1) | ||||||||||||||||
| ジョナサン·ソロモン | 2023 | 412,135 | 201,234 | 404,174 | 100,998 | 1,118,541 | ||||||||||||||||
| 最高経営責任者 | 2022 | 424,581 | - | 512,974 | 103,987 | 1,041,542 | ||||||||||||||||
| マリナ·ウォルフソン | 2023 | 214,727 | 76,209 | 90,742 | 46,578 | 428,256 | ||||||||||||||||
| 首席財務官 | 2022 | 231,414 | - | 116,827 | 48,002 | 396,243 | ||||||||||||||||
| メラフ·バサン博士 | 2023 | 264,105 | 101,145 | 153,218 | 72,463 | 590,931 | ||||||||||||||||
| 首席発展官 | 2022 | 280,213 | - | 206,332 | 76,373 | 562,948 | ||||||||||||||||
| (1) | すべての支払いは最初に新シェケルで支払い、各年度のドル/新シェケル年間平均為替レートをドルに換算した。 |
| (2) | この欄の金額は、ASC 718から計算された日オプション報酬を付与する公正価値を表し、サービスベースのホーム条件に関連するいかなる没収推定値も含まれない。2023年12月31日現在の年度連結財務諸表付記12.Bを参照して、当社が2023年12月31日現在と2022年12月31日までの財政年度オプション奨励付与日公正価値を決定する際の仮定を検討した。本欄で報告した金額は、これらの株式オプションの会計コストを反映しており、非従業員取締役が株式オプション帰属、株式オプションの行使、またはそのような株式オプションの普通株を売却する際に実現可能な実際の経済的価値を反映していないことに注意されたい。 |
| (3) | この欄の額は、イスラエルの従業員に福祉、障害保険、および他の習慣的または強制的な社会福祉の追加支払いを支払うことである。 |
90
報酬集計表の叙述的開示 表
オプション大賞
業務統合の前に、 は2015年計画に基づいて私たちが指定した役員にオプション奨励を付与します。業務合併終了後に私たちが指定した役員に付与されたオプション奨励 は2019年計画に従って付与されます。それぞれの場合、4分の1のオプション帰属および は、付与日1周年に行使することができ、残りのオプションは、任命された実行幹事が継続して雇用されることが条件である12等分の分割払いで行使されることができる。指定された行政者が制御権変更(定義は適用付与プロトコル参照)が発生してから12(12)ヶ月以内に理由なく非自発的終了(定義は適用付与プロトコル参照)または十分な理由で自発的に終了した場合(定義は適用付与プロトコル参照)によって終了される場合、その等オプションは付与されて行使可能である。任意の雇用契約の条項によれば、これらの報酬のうち行使されていない部分は、通常、その雇用終了日に参加者によって死亡または障害以外の理由で没収される。死亡または障害の場合、オプションは完全に行使可能であり、適用される報酬プロトコルで指定された期間内に行使可能である。
賞金賞
年に1回の会社と個人目標設定 があり、私たちが任命した役員を審査することが、潜在的な年間ボーナスを決定する基礎となっています。取締役会または給与委員会の許可を得て、私たちが任命した各幹部はその給料の特定のパーセンテージまでの年間業績ボーナスを得る資格があり、ボーナス範囲は40%から50%までだ。業績ボーナスは取締役会の審査と承認された特定の会社および/または個人目標と目標(例えば臨床と開発マイルストーン、予算と戦略目標の実現)とリンクし、著者らは毎年1回業績評価を行い、これらの目標と目標の実現状況を確定する。私たちの経営陣は主にこのような審査過程に基づいて取締役会にボーナス奨励を提案する可能性があります。報酬委員会は、具体的な企業や戦略目標の実現状況およびこのようなボーナスの資格要求と金額を最終的に決定し、取締役会にボーナス支給を承認することを提案する。2023年度には、ボーナスは、brや開発計画を推進し、いくつかの候補製品開発マイルストーンと戦略目標を満たすことによって蓄積される。
雇用協定
以下は我々が指定幹部と締結した雇用協定の説明である。
ジョナサン·ソロモン
BiomXイスラエル社とソロモンさんによって2016年2月1日に締結された雇用契約によれば、ソロモンはBiomXイスラエル社のCEOとして毎月64,000新しいシェケルの基本給、すなわち約19 500ドルを獲得する権利を有し、通常の営業時間および通常の営業日を除いて毎月最大40時間働く追加総収入16,000ニューシェケル、すなわち約4 900ドル(基本給とソロモンさんの賃金と一緒)を得る権利がある。2023年4月1日から、ソロモンさんの基本給は月100,000新シェケル(約27,778ドル、残業代は月25,000新シェケル、約6,944ドル)となった。
BiomXイスラエル社はまた、慣例に従い、ソロモンさんを代表して選出されたときにソロモンさんを代表して養老基金または経理人保険会社に入金し、その賃金の8.33%をソロモンさんソロモンが経済人保険証券を通じて保険にかけた場合、ソロモンさんが年金基金を通じて保険を加入した場合ソロモンさんが年金基金を介して保険をかけた場合、ソロモンさんの給与の6.50%であった場合、このお金は積立金または年金計画に分配されなければならない。ソロモンさんがその年金支払をマネージャー保険証書(年金基金ではなく)に割り当てることを選択した場合、当社はまたソロモンさん100%の賃金に要する料率で労働障害保険証書に従って彼に保険を提供しなければならず、そのための保険証および/または保険会社はソロモンさんの障害保険保険にその保険証書が保持している賃金までの2.50%の金額を提供する必要がある。第5723-1963号“解散料支払法”または“解散料支払法”によると、これらのお金はソロモンさんがBiomXイスラエル社から受け取る権利があった法的解散料の代わりになることを目的としている。BiomXイスラエルはまたソロモンさんの月給の7.50%を公認教育基金に寄付した。BiomXイスラエル会社はまた、ソロモンさん2000ニューシェケルの自動車修理費と交通費、すなわち毎月556ドルを返済しました。もしソロモンさんがすべてのクレームを放棄し、その雇用契約の他の条項を遵守し続ける場合、ソロモンさんはまた、(I)辞任 または(Ii)無理が解雇(良い理由と理由は両方で定義され、私たちの過去の慣行と一致する)の場合に12ヶ月間の非法的解散料(社会福祉を含む)を得る十分な理由があるとする権利を持っている。
91
マリナ·ウォルフソン
BiomXイスラエル社とウォルフソンさんが2019年12月1日に締結した雇用契約によると、彼女は私たちの首席財務官を務めている。ウォルフソンさんは毎月39,600新シェケルの基本給、約11,400ドル、毎月通常の営業時間と通常の平日を除いて最大40時間働く追加月給7,400新シェケル、約2,130ドル(ウォルフソンさんの基本給と一緒)を得る権利がある。2020年5月1日から、ウォルフソンさんの基本給は月40,000新シェケル(約11,458ドル)と、毎月10,000新シェケル(約2,865ドル)の追加毛払いとなった。2023年4月1日からウォルフソンさんの基本給は月54,080新シェケル(約15,022ドル)で、毛収入13,520新シェケル(約3,756ドル)を加えた。
ウォルフソンさんが当選した時、BiomXイスラエル会社はまた慣例に従ってウォルフソンさんを代表して養老基金或いはマネージャー保険会社に支払い、金額はウォルフソンさんの給料の8.33%に相当し、解散費基金に割り当てられ、もしウォルフソンさんがマネージャー保険リストを通じて保険に加入した場合、ウォルフソンさんの給料の5.00%に相当する追加金額を追加し、もしウォルフソンさんが年金基金を通じて保険をかけた場合、ウォルフソンさんの給料の6.50%である。その金は積立金や退職金計画に振り込まなければならない。もしWolfsonさんが彼女の年金をマネージャー保険証書(年金基金ではなく)に分配することを選択した場合、会社はまた仕事の障害保険証書に基づいて彼女に保険を提供しなければならない。保険料率はWolfsonさんの給料の75%であり、そのために保険証書および/または保険会社でWolfsonさんの障害保険証書にWolfsonさんの給料の2.50%までの金額を提供しなければならない。このようなお金はウォルフソンさんが“解散費法”に基づいてBiomXイスラエル社から得た法定解散費を代替する権利がある。BiomXイスラエル社はまた、ウォルフソンさんの月給の7.50%(15,712新シェケル、約4,364ドル)を公認教育基金に寄付した。同社は毎月Wolfsonさんに2500新シェケル(約694ドル)の自動車整備費と交通費を精算している。ウォルフソンさんにも、ウォルフソンさんがすべてのクレームを放棄し、彼の雇用合意の他の条項を守り続ける限り、ウォルフソンさんがすべてのクレームを放棄し、彼の雇用合意の他の条項を遵守し続ける限り、ウォルフソンさんがすべてのクレームを放棄し、彼の雇用合意の他の条項を遵守し続ける限り、(I)十分な理由の辞任、または(Ii)無理な解雇がある(良い理由と理由は双方で定義され、私たちの過去のやり方と一致する)。
メラフ·バサン博士
BiomXイスラエル社とBassan博士が2019年8月26日に締結した雇用協定によると、BiomXイスラエル社の首席開発官として、Bassan博士は毎月56,000新シェケルの基本給、約17,230ドル、および毎月正常営業時間と通常営業日以外で最大40時間働く追加総収入14,000新シェケル、または約4,307ドル(基本給、すなわちBassan博士の給料と一緒)を得る権利がある。2023年4月1日から、バサン博士は毎月62,800新シェケル(約17,444ドル)の基本給と、15,700新シェケル(約4,361ドル)の追加月給を受け取る権利がある。
BiomXイスラエル会社はまた慣例に従って、巴桑博士が選択した場合、巴桑博士を代表して養老基金或いはマネージャー保険会社に支払い、金額 は巴桑博士の給料の8.33%に相当し、解散費基金に分配され、もし巴桑博士がマネージャー保険証書を通じて保険を加入すれば、巴桑博士の給料の7.30%に相当し、もし巴桑博士が養老基金を通じて保険を加入すれば、巴桑博士の給料の6.50%である。その金は積立金や退職金計画に振り込まなければならない。もしBassan博士が彼女の年金支払いをマネージャー保険証書(年金基金ではなく)に分配することを選択した場合、会社はまたBassan博士の給料の75%の保険料率で彼女に仕事の障害保険を提供しなければならず、そのために保険証書および/または保険会社で障害保険にその保険証書の保険料の2.50%までの金額を納めなければならない。このお金はBassan博士がSeverance LawによってBiomXイスラエル社から得た法定解散費を代替する権利がある。BiomXイスラエル社はまたBassan博士の月給の7.50%を公認された教育基金に貢献した。同社は毎月Bassan博士に2500新シェケル(約694ドル)の自動車メンテナンスと交通費を精算している。Bassan博士はまた、非法定の9ヶ月の解散料(社会福祉を含む)を得る権利があり、Bassan博士がすべてのクレームを放棄し、その雇用合意の他の条項を遵守し続ける限り、彼女がすべてのクレームを放棄し、その雇用合意の他の条項を遵守し続ける限り、(I)辞任する十分な理由があるか、または(Ii)原因なしに解雇される(良い原因と原因は双方で定義され、私たちの過去のやり方と一致する)。
92
2023年度末未償還株式賞
次の表は、2023年12月31日現在、任命された役員が保有する未償還持分報酬に関する情報を提供しています
| オプション大賞 | ||||||||||||||||
| 名前.名前 | 授与日 | 未行使オプションを行使可能な証券数(1)(#) | 行使できない未行使オプションの証券数(1)(#) | オプション取引権価格(ドル) | オプション期限 | |||||||||||
| ジョナサン·ソロモン | 11/13/2016 | 167,434 | - | 0.54 | 01/07/2027 | |||||||||||
| 03/26/2017 (2) | 182,133 | - | 0.275 | 03/26/2027 | ||||||||||||
| 05/22/2018 (2) | 201,718 | - | 0.275 | 05/21/2028 | ||||||||||||
| 03/29/2019 (2) | 284,701 | - | 0.275 | 03/29/2029 | ||||||||||||
| 03/25/2020 (3) | 35,527 | 2,368 | 0.275 | 03/25/2030 | ||||||||||||
| 03/30/2021 (3) | 27,500 | 12,500 | 0.275 | 03/30/2031 | ||||||||||||
| 03/29/2022 (3) | 64,063 | 82,366 | 0.275 | 03/29/2032 | ||||||||||||
| 08/22/2022 | 31,250 | 68,750 | 0.66 | 08/22/2032 | ||||||||||||
| 03/01/2023 | 410,000 | 0.4 | 03/01/2033 | |||||||||||||
| メラフ·バサン博士 | 10/10/2019 (2) | 189,997 | - | 0.275 | 10/10/2029 | |||||||||||
| 03/30/2021 (3) | 8,593 | 3,907 | 0.275 | 03/30/2031 | ||||||||||||
| 03/29/2022 (3) | 31,250 | 40,179 | 0.275 | 03/29/2032 | ||||||||||||
| 08/22/2022 | 23,438 | 51,562 | 0.66 | 08/22/2032 | ||||||||||||
| 03/01/2023 | - | 100,000 | 0.4 | 03/01/2033 | ||||||||||||
| マリナ·ウォルフソン | 03/25/2020 (3) | 8,882 | 592 | 0.275 | 03/25/230 | |||||||||||
| 03/30/2021 (3) | 6,017 | 2,733 | 0.275 | 03/30/2031 | ||||||||||||
| 03/29/2022 (3) | 15,625 | 20,090 | 0.275 | 03/29/2032 | ||||||||||||
| 08/22/2022 | 23,438 | 51,562 | 0.66 | 08/22/2032 | ||||||||||||
| 03/01/2023 | - | 100,000 | 0.4 | 03/01/2033 | ||||||||||||
| 29/10/2023 | - | 59,800 | 0.275 | 10/29/2033 | ||||||||||||
| (1) | 別の説明がない限り、購入持分の帰属および行使は以下の通りである:“帰属開始日”(適用される購入権付与通知を参照)1周年の場合、引受権の帰属および行使可能な割合は25%であり、その後12回に分けて平均四半期分割、毎期6.25%である。 |
| (2) | 2023年10月29日、取締役会は、2015年計画で付与された1株当たり0.69ドル以上の元の行権価格に基づいて、現在当社従業員が保有している当社の普通株式の1株当たり未償還オプションを購入することを許可し、1株当たり0.275ドルに引き下げた。行使価格を除いて、再定価されたオプションの他の付与条項は変更されていないが、このオプションは、再定価日の1年後に行使することができる。 |
| (3) | 2023年11月9日、同社は、従業員にいくつかの条件に適合するオプションを提供する使い捨て自発的株式オプション交換または2019年計画に基づいて付与されたオプション取引所の条項および条件を定義する入札要約声明を米国証券取引委員会に提出した。当社は、取引価格の低い普通株式と交換するために、1.4~3.8の交換比率で、いくつかの現金外株式オプションで新しい株式オプションを交換し、オプションを放棄して行使可能な新オプションと交換することを提案している。オプション取引所完了日2023年12月11日に、株式オプションは条件を満たす従業員が入札し、会社は0.275ドルの行権価格で新たなオプションを付与する。 |
役員の報酬
私たちは非従業員役員報酬政策を維持し、この政策により、非従業員取締役1人当たり毎年35,000ドルの採用金を得ることができる。また、私たちの非従業員取締役は、以下の取締役会サービスの現金報酬を得ることができます(場合によっては)
| ● | 取締役会長の年間採用費は100,000ドル(年間委員会の議長やメンバーを含む) |
93
| ● | 議長を除いて、私たちの監査、報酬、指名、および企業統治委員会の各メンバーは毎年それぞれ7,500ドル、5,000ドル、4,000ドルの追加雇用費を得ている |
| ● | 私たちの監査、報酬、指名、およびコーポレートガバナンス委員会の各議長は毎年それぞれ15,000ドル、10,000ドル、8,000ドルの追加求人料を得ています。 |
私たちは四半期ごとに分割払いにします。取締役会や委員会会議への出席による各取締役の合理的な旅費、宿泊費、その他の自己負担費用も補償します。
各非従業員取締役はまた、私たちの普通株を購入する年間オプション奨励を受けます。各年度オプション奨励の4分の1は授出日の第1周年 に帰属し、残りの年間オプション奨励は12四半期分割払いで帰属するが、この取締役 は取締役会でのサービスを継続することに制限されなければならない。同社の政策は、報酬コンサルタントの提案などに応じてオプションを付与することだ。2023年、会社は非従業員取締役1人に41,000件のオプションを付与し、取締役会議長に82,000件のオプションを付与する。
次の表は、2023年12月31日までの年間で、当社の独立非従業員取締役が取締役会でのサービスのために計上または支払うべき報酬情報を示しています。ジョナサン·さんは取締役の一員であり、また私たちの従業員であり、彼は役員としてのサービスは追加の報酬を得ていませんでした。次の表にはリストされていません
| 名前.名前 | 現金で稼ぐか支払う費用 ($) | 選択権 賞.賞(2)(3) | 他のすべての補償 | 合計する ($) | ||||||||||||
| ラッセル·グリガー博士 | 100,500 | 61,077 | - | 161,577 | ||||||||||||
| マイケル·ダンバッハ(1) | 27,205 | 2,449 | - | 29,654 | ||||||||||||
| ジェイソン·マックス(1) | 25,605 | 2,449 | - | 28,054 | ||||||||||||
| エレン·モーゼス博士 | 47,560 | 33,873 | - | 81,433 | ||||||||||||
| エドワード·ウィリアムズ | 7,704 | 1,085 | - | 8,789 | ||||||||||||
| リン·シャリヴァン(1) | 54,000 | 30,538 | - | 84,538 | ||||||||||||
| 262,574 | 131,471 | - | 394,045 | |||||||||||||
| (1) | 2024年3月15日から取締役が辞任し、取締役会のメンバーを務めなくなった |
| (2) | この欄の金額は、サービスベースのホーム条件に関連するいかなる没収推定値も含まれないASC 718から計算されたオプション報酬付与日の公正価値を表す。当社の2022年と2023年12月31日までの財政年度オプション奨励付与日の公正価値を決定する仮定の検討については、2023年12月31日現在の年次報告書に連結財務諸表付記12.Bを参照されたい。この欄に報告されている金額は、非従業員取締役が株式オプション帰属、株式オプションの行使、またはそのような株式オプション関連普通株を売却する際に実現可能な実際の経済的価値を反映していないこれらの株式オプションの会計コストを反映していることに注意されたい。 |
| (3) | 2023年12月31日現在、非執行役員に付与された未返済金は合計493,800件であり、そのうち134,675件は行使可能または付与されており(場合によっては)、具体的には以下の通りである |
| 名前.名前 | 選択肢の総数 承認された | 選択肢の総数 操作可能な和 既得 | ||||||
| ラッセル·グレッグ | 185,400 | 68,839 | ||||||
| マイケル·ダンバッハ | 41,000 | - | ||||||
| ジェイソン·マックス | 41,000 | - | ||||||
| エレン·モーゼス博士 | 92,700 | 31,418 | ||||||
| エドワード·ウィリアムズ | 41,000 | - | ||||||
| リン·シャリヴァン | 92,700 | 34,418 | ||||||
| 合計する | 493,800 | 134,675 | ||||||
94
プロジェクト12.特定の利益を受けるすべての人および管理職の保証所有権および関連株主事項
株式補償計画に基づいて発行された証券
私たちは2つの持分インセンティブ計画、2015年計画と2019年計画を持っている。2015年計画によると、将来発行できる普通株はありませんが、2015年にはこの計画に基づいて付与された未償還奨励を管理し続ける予定です。2023年12月31日現在、2015年計画により、私たちの普通株2,055,836株を購入したオプションはまだ返済されていません。
2019年は取締役会によって採択され、私たちの株主が業務合併に関連することを承認する予定です。2023年12月31日まで、2019年の計画により、私たちの普通株は1,011,104株が発行できます。2019年計画によると発行可能な普通株式総数 は毎年1月1日に自動的に増加し、10年を超えず、2020年1月1日から2029年1月1日(この日を含む)まで、金額は前年の12月31日に発行された普通株式総数の4%に相当する。そこで、2024年1月1日、2019年計画により、私たちの普通株は1,839,197株 を追加発行しました。
2023年12月31日現在の2015年計画および2019年計画の詳細については、第2部-第8項-財務諸表および補足データである 合併財務諸表付記-付記12 B-株式ベースの報酬を参照されたい。
| 株式報酬計画情報 | ||||||||||||
| 2023年12月31日 | ||||||||||||
| 計画種別 | 証券数量 私たちは 発表日: 演習をする 卓越した 選択肢と 制限される 在庫品 (a) | 重み付けの- 平均値 トレーニングをする 値段 卓越した 選択肢と 制限 さ れる 在庫品 (b) | 量 証券 残り 適用することができます 未来.未来 発行する. 権益の下で 補償する 平面図 (含まれない) 証券 反映されています (A)欄) (c) | |||||||||
| 証券保有者が承認した持分補償計画 | 3,224,871 | 0.68 | 1,011,104 | |||||||||
| 証券保有者の許可を得ていない持分補償計画 | 2,055,840 | 0.32 | - | |||||||||
| 合計する | 5,280,711 | 0.54 | 1,011,104 | |||||||||
ある利益所有者と管理層の安全所有権
次の表は、2024年3月28日までの私たちの普通株式の実益所有権(他に説明がない限り)に関するbr}情報を示しており、この情報は、以下の人々から得られた私たちの普通株の実益所有権に関する情報に基づいています:(I)私たちが知っているすべての人は、私たちが発行した普通株の実益所有者であり、(Ii)私たちの各役員と取締役、 と(Iii)私たちのすべての役員と取締役はグループとして。利益所有権に関する情報は、私たちが発行した普通株式の5%以上を保有する各取締役、役員または株主が提供してくれる情報と、各利益所有者が2024年3月28日から60日以内に買収する権利がある我が普通株を含む、米国証券取引委員会に提出されたスケジュール13 Gまたは13 D(場合によって決定される)に基づいている。他に説明がある以外に、私らは表に記載されているすべての人々がその実益を持つすべての普通株に対して独占投票権と投資権を持っていると信じている。我々は2024年3月28日までの55,220,077株発行普通株に基づいて利益所有権 を計算した。
95
| 実益所有者の氏名又は名称及び住所(1) | 金額と 性質 有益 所有権 |
パーセント 類 |
||||||
| OrbiMedイスラエルGP株式会社です(2)イスラエルヘズリアE号館89メディナート·ハエフディム通り4614001イスラエル | 12,577,821 | 19.9 | % | |||||
| 嚢胞性線維化財団(3) モンゴメリー大通り4550号です。メリーランド州ベセスダ1100 Nスイート、郵便番号:20814 |
9,330,580 | 15.6 | % | |||||
|
柔軟リスク投資有限責任会社(4) カリフォルニア州サンフランシスコ4900軒A棟ライトマン通り1号、郵便番号:94129(2) |
4,598,189 | 8.3 | % | |||||
|
ディルフィールド医療革新基金II L.P(5) ニューヨーク公園大通り南345号、12階、郵便番号:10010 |
3,055,049 | 5.5 | % | |||||
|
Deerfield Private Design Fund V,L.P(6) ニューヨーク公園大通り南345号、12階、郵便番号:10010 |
3,055,049 | 5.5 | % | |||||
|
AMR行動基金L.P(7) マサチューセッツ州ボストン、フランクリン通り225号、1750部屋、郵便番号:02110 |
3,054,870 | 5.5 | % | |||||
|
Telmina Limited(8) スイスジュネーブ12号郵便ポスト393番地L通り34番地 |
2,839,714 | 5.1 | % | |||||
| 役員および指名された行政員 | ||||||||
| ジョナサン·ソロモン(9) | 1,167,096 | 2.1 | % | |||||
| マリナ·ウォルフソン(10) | 98,272 | * | ||||||
| メラフ·バサン博士(11) | 292,900 | * | ||||||
| ラッセル·グリガー博士(12) | 102,365 | * | ||||||
| ジェシー·グッドマン博士 | - | - | ||||||
| ジョナサン·レフ | - | - | ||||||
| グレゴリー·メリル | - | - | ||||||
| アラン·モーゼス先生(13) | 54,650 | * | ||||||
| エドワード·ウィリアムズ | - | - | ||||||
| 全役員と執行幹事(11名) | 2,027,633 | 3.7 | % | |||||
| * | 1%未満です |
| (1) | 別の説明がない限り、各人の営業住所はc/o BiomX Inc.,22 Einstein St.,4であるこれは…。ネス·ツィオナ7414003イスラエル |
| (2) | 当該株主及びその関連会社及び任意の他のグループとして株主又は株主の任意の関連会社と共に行動する者は、OrbiMedイスラエル生物基金GP有限組合会社、カール·L·ゴードン、エレズ·チモヴィッツを含み、実益は4,517,589株の普通株と予め出資した引受権証を有し、最大8,060,232株の普通株を買収する。(X)4,327シリーズX非投票権変換可能優先株、(Y)290,781件の株式承認証、および(Y)1,220,176件の事前計画資本権証、および(Z)2,538,500部の株式承認証を含まない。この等株式承認証及び事前出資株式証明書には発行制限が記載されており、所有者が当該等株式承認証又は事前出資株式承認証を行使することを禁止し、行使後に当該等の発行が発効した後、所有者(所有者の共同経営会社及び任意の他の者と共に所有者又は所有者の任意の連絡者とともに、OrbiMedイスラエル生物基金GP Limited Partnership、Carl L.Gordon及びErez Chimovits)が19.9%を超える既発行株式を保有することが条件となる所有権制限を受けるそれは.発行者株主の承認を得た後、Xシリーズ優先株の1株は自動的に1,000株に変換できるが、利益所有権制限の制限を受けている。2024年3月19日に米国証券取引委員会に提出された付表13 D/Aに含まれる情報と会社の記録による。 |
| (3) | (I)4,552,315株普通株および(Ii)4,778,265株を含み、60日以内に株式承認証を行使して発行可能な普通株。(I)21,635株Xシリーズ非投票権転換可能優先株、及び(Ii)10,817,500株を含まない場合、引受権証を行使する際に発行される普通株を含むことができる。Xシリーズ優先株及び当該等株式権証は、当社の株主の承認後に両替又は行使が必要である(いずれが適用されるかに応じて決定される)。会社の株主の許可を得た後、Xシリーズ優先株は1株当たり1,000株の普通株に転換することができるが、実益所有権の制限を受けなければならない。2024年3月26日に米国証券取引委員会に提出された付表13 Gに含まれる情報と会社の記録に完全に基づく。 |
| (4) | (I)4,550,000株の普通株及び(Ii)最大552,041株の普通株を買収する引受権証を含み、この等株式証は、発行制限を掲載し、保有者が事前出資の引受証の行使を禁止することを含む。ただし、当該等株式証の行使後に発効した後、所有者(所有者との連属会社及び他の所有者又は所有者との任意の連絡者が団体として行動する者)は、9.99%を超える既発行普通株株式を保有し、当該株式等は、株式証行使時に普通株式を発行した直後に発効する。ジョン·H·バーバンク3世はNimble Venturesの制御者であり,この身分は,間接実益がNimble Venturesが直接実益所有する株式を所有していると見なすことができる。2023年6月23日に米国証券取引委員会に提出された付表13 Gに含まれる情報と会社の記録による。 |
| (5) | (I)合計53,840,000株関連普通株Xシリーズ優先株53,840株を含まず、ある条件が出現すると、普通株(実益所有権制限を受ける)、または(Ii)合計20,897,175株普通株関連引受権証に変換することができ、ある条件が出現すると、普通株を行使することができる(実益所有権制限を受ける)。2024年3月22日に米国証券取引委員会に提出された付表13 Dに含まれる情報と会社の記録に完全に基づく。 |
| (6) | (I)合計53,840,000株関連普通株Xシリーズ優先株53,840株を含まず、ある条件が出現すると、普通株(実益所有権制限を受ける)、または(Ii)合計20,897,175株普通株関連引受権証に変換することができ、ある条件が出現すると、普通株を行使することができる(実益所有権制限を受ける)。2024年3月22日に米国証券取引委員会に提出された付表13 Dに含まれる情報と会社の記録に完全に基づく。 |
96
| (7) | が(I)合計42,337株関連普通株Xシリーズ優先株42,337株を含まないかどうか, がある条件が出現すると、普通株(実益所有権制限を受ける)、または(Ii)合計15,145,647株の普通株関連引受権証に変換することができ、ある条件が発生したときに 普通株を行使することができる(実益所有権制限を受ける)。2024年3月25日に米国証券取引委員会に提出された付表13 Gに含まれる情報と会社記録のみに基づいている。 |
| (8) | 2,839,714株の普通株からなる。2023年9月29日に米国証券取引委員会に提出された付表 13 Gに含まれる情報と会社記録のみに基づく。 |
| (9) | 25,000株普通株、25,000株承認株式証(所有者に最大18,750株普通株を獲得する権利を有する)、1,105,444株行使可能なオプション、および17,902部が2024年3月28日から60日以内に行使可能な追加オプションを含む。 |
| (10) | 3,750株普通株、3,750株株式承認証(保有者に最大2,813株普通株を獲得する権利を有する)、84,242株の行使可能なオプション、および7,467株が2024年3月28日から60日以内に行使される追加オプションを含む。 |
| (11) | 282,966個の行使可能なオプションと、2024年3月28日から60日以内に行使可能な9934個の追加オプションが含まれる。 |
| (12) | 3,750株普通株、3,750株株式承認証(保有者に最大2,813株普通株を購入する権利を有する)、91,339株行使可能なオプション、および4,463株が2024年3月28日から60日以内に行使可能な追加オプションを含む。 |
| (13) | 5,000株普通株式、5,000部の株式承認証(所有者に最大3,750株普通株を取得させる)、42,668件の行使可能なオプション、および2024年3月28日から60日間以内に行使可能な追加オプションを含む。 |
第13項.ある関係と関連取引、および取締役の独立性
役員は自主独立している
ニューヨーク証券取引所アメリカ証券取引所は、取締役会の多くのメンバーが“独立取締役”から構成されなければならないことを要求し、“独立取締役”は、通常、会社またはその子会社の幹部または従業員ではなく、取締役会がその客観的判断の行使を妨害すると認定する他の任意の個人でもなく、ニューヨーク証券取引所米国証券取引所と米国証券取引委員会の適用規則および法規の確立に必要な独立基準に適合するものと定義される。
ラッセル·グレッグ博士、エレン·モーゼ博士、エドワード·L·ウィリアムズさん、ジョナサン·ライヴさん、ジェシー·グッドマン博士、グレゴリー·メリルさんは私たちの独立取締役です。
取締役会は、少なくとも毎年、私たちと各取締役との間のすべての関係を評価し、関連する事実および状況を考慮して、潜在的利益衝突が存在する可能性があるかどうか、または他の方法で取締役が独立取締役としての責任を果たす能力を妨害する重大な関係があるかどうかを決定する。この評価によると、我々の取締役会は毎年、各取締役がニューヨーク証券取引所米国証券取引所と米国証券取引委員会の独立性基準の範囲内で独立しているかどうかを決定する。
関係者と取引を行う政策と手順
我々の関連者取引ポリシーは、取締役会(または監査委員会)によって承認されない限り、実際または潜在的な利益衝突を引き起こす可能性のあるすべての関連者取引を可能な限り回避することを要求する。当社が“取引法”第12 b-2条の規則で定義されている“より小さい申告会社”の資格に適合する限り、関連者取引は、私たちの関連者取引ポリシーの下で、任意の関連者(または任意の一連の類似した取引、手配または関係)と定義される取引、手配または関係(または任意の類似した取引、手配または関係)と定義され、その取引、手配または関係において、私たちbrおよび任意の関連者(政策で定義されているような)は、かつて、または参加者であり、関連する金額は、12万ドルまたは最近の2つの完全会計年度年末総資産平均の1%を超える。そして,任意の関係者が直接または間接的に重大な利益を持つ を持つか,または持つことになる.当社がもはや小さな申告会社でない場合、関連者取引は、当社と任意の関連者が現在、または参加者になる取引、手配または関係(または任意の一連の類似した取引、手配または関係)として定義され、関連金額が120,000ドルを超え、関連者が直接または間接的に重大な利益を有することがある。本政策によれば,我々が従業員,コンサルタントまたは取締役として提供するサービスを補償する取引に関する取引は関連者取引とはみなされない.
97
もし当社のbrが関係者の取引を締結或いは重大な改訂を提案した場合、当社の管理層はこのような関係者の取引を監査委員会の審査、審議及び承認或いは承認に提出しなければならない。陳述書は、(A)すべての当事者と、(B)任意の関係者(S)監査委員会がこれらの利益を十分に評価できるように取引における直接的または間接的利益と、(C)取引の目的と、(D)提案された関連者取引のすべての重大な事実と、取引の提案された総価値を含むすべての重大な事実と、債務が発生した場合、関連する元本金額を説明することと、を合理的な範囲内に含まなければならない。(E)提案された関連者取引の当社へのメリット、(Br)(F)他の比較可能な製品またはサービスの出所(例えば、適用)、(G)提案された関連者取引の条項が(状況に応じて)無関係な第三者が入手可能または由来の条項に匹敵するかどうかを評価し、(Br)管理層が提案された 関連者取引に関する提案を行い、管理層がその事項が潜在的または実際的な衝突を生じることを知りながら成果を得る。監査委員会が進行中の関連者取引を承認するか否かを考慮するように要求された場合、上記の情報 に加えて、陳述材料は、(I)取引が完了し、完了すべき作業範囲に関する説明、(Ii)取引終了の潜在的リスクおよびコストの評価、および(Iii)適切な場合に、取引修正の可能性を含む必要がある。
委員会は、提案された関連者取引を承認又は拒否する際に、(A)会社のリスク、コスト及び収益を含むが、(A)会社のリスク、コスト及び収益を含むが、(B)関連者が取締役の直系親族、取締役の直系親族又は取締役に関連する実体の直系親族である場合、(C)取引の条項及び時間、(D)他の出所の類似したサービス又は製品があるか否か、を含むが、提案された関連者の取引を承認又は拒否する際に、委員会が関連して委員会が使用することができるすべての関連事実及び状況を考慮する。(E) は、関係のない第三者に提供または提供される条項は、状況に応じて、および(F)関連者取引がどのように実現され、関連者取引政策の要求に応じて監査委員会に伝達される。審査委員会が審査委員会のみを承認してその適宜決定権を誠実に行使する場合には,既知の状況に応じて,当社とその株主の最適な利益に適合または違反しない関係者取引を行う。
その他 補償、終了、制御権変更およびその他の手配は、第11項-補償および第12項の実行-いくつかの実益所有者の保証所有権と管理層および関連株主事項に記載されており、私たちの2023年1月1日以来の唯一の関係者取引は、(I)我々は2023年2月22日に認可された非米国投資家と締結した証券購入契約を含み、嚢胞性繊維財団、OrbiMedイスラエルGP有限会社またはOrbimedとNimbleリスク投資有限責任会社を含み、各株主は私たちが発行した普通株の5%以上を持っている。私募は合計15,997,448株の私たちの普通株と14,610,714株の資本金権証に関連しており、買収価格は1株0.245ドル、1部の資本金権証1部当たり0.244ドルである。発行コストを差し引くまで、今回発行された総収益は約740万ドルだった。予備資金権証は2023年5月4日から行使可能で、行使価格は1株当たり0.001ドルで、満期日はない。これらの収益のうち、合計3,385,000株の普通株と4,778,265件の予備資本権証がCFに売却され、総収益は200万ドルであり、合計1,740,000株の普通株と 9,280,408部の予備金権証がOrbimedに売却され、総収益は270万ドルであり、合計4,550,000株の普通株と552,041部の事前資本金権証がNimble Venture LLCに売却され、総収益は125万ドルであった。(Ii)私たちは2024年3月6日にCFF、Orbimed、Telmina、Telmina{bited、Telmina{mina}またはTelmina{mina}を含むいくつかの投資家と証券購入契約を締結した。私たちの株主は一人当たり5%以上の発行済み普通株を持っています。これにより、合計216,417株の転換可能優先株と私募株式権証を売却して、合計108,208,500株の普通株を購入し、総購入価格は1株Xシリーズ優先株231.10ドルで、br私募株式証を添付します。今回発行された総収益は約5000万ドル。株主が発行済みと未発行のXシリーズ優先株の転換を許可し、ニューヨーク証券取引所米国上場規則に基づいてすべての私募株式証を行使した後、いつでも私募株式証明書を行使することができ、2024年8月12日までに株式承認証を株主投票に提出することが義務付けられ、発行価格は0.2311ドルであり、初めて引受証を行使した日から24ヶ月以内に満期となる。私募株式証の発行権価格は株式配当、株式分割、再分類などの通常調整の影響を受ける。これらの収益のうち,合計21,635株の転換可能優先株と10,817,500件の私募株式証明書がCFFに売却され,総収益は500万ドル,合計4,327株転換可能優先株と2,163,500件の私募株式証はOrbimed に売却され,総収益は100万ドル,合計2,596株の転換可能優先株と1,298,000件の私募株式証はTelminaに売却され,総収益は60万ドルであった。
プロジェクト14.チーフ会計士費用とサービス
以下にKesselman&Kesselman会計士事務所(ISR)に受け取った費用の概要と説明を示す.2023年12月31日と2022年12月31日までの財政年度。
| 12月31日までの会計年度は 2023 | 財政年度 12月31日まで 2022 | |||||||
| 料金を審査する(1) | $ | 126,000 | $ | 126,000 | ||||
| 監査関連費用(2) | $ | 97,000 | $ | 24,969 | ||||
| 税金.税金(3) | $ | 3,393 | $ | - | ||||
| 他のすべての費用 | $ | - | $ | - | ||||
| 総費用 | $ | 226,393 | $ | 150,969 | ||||
| (1) | 監査費用には、中期総合財務諸表の四半期審査および年度監査のために提供される専門サービス費用が含まれています。 |
| (2) | 監査関連費用には、本財政年度総合財務諸表監査業績に関する合理的なサービス費用が含まれていますが、監査費用は除外されています。例えば、販売契約、私たちの2023年2月のパイプライン、株主がある普通株を転売するために提出された登録報告書に関するサービス費用です。 |
| (3) | 税金には税務コンプライアンス費用と税務相談費用が含まれています。 |
98
承認前の政策と手順
監査委員会は、私たちの独立公認会計士事務所が提供するすべての監査と予め承認されたすべての非監査サービスを承認し、その後、私たちはそれを招いて非監査サービスを提供することができます。このようなサービスは監査関連サービス、税務サービス、および他のサービスを含むことができる。
以下の場合、上記の事前承認要求は、非監査サービスには適用されない
| ● | このようなすべてのサービスは、サービスを提供する財政年度内に、私たちが独立公認会計士事務所に支払う総費用の5%を超えません |
| ● | このようなサービスは、関連採用時に非監査サービス とみなされない |
| ● | このようなサービスは、年度監査が完了する前に、直ちに監査委員会(またはその代表)に注意を促し、監査委員会(またはその代表)の承認を受ける。 |
承認前の政策と手順
監査委員会は、私たちの独立公認会計士事務所が提供するすべての監査と予め承認されたすべての非監査サービスを承認し、その後、私たちはそれを招いて非監査サービスを提供することができます。このようなサービスは監査関連サービス、税務サービス、および他のサービスを含むことができる。
以下の場合、上記の事前承認要求は、非監査サービスには適用されない
| ● | このようなすべてのサービスは、サービスを提供する財政年度内に、私たちが独立公認会計士事務所に支払う総費用の5%を超えません |
| ● | このようなサービスは、関連採用時に非監査サービス とみなされない |
| ● | このようなサービスは、年度監査が完了する前に、直ちに監査委員会(またはその代表)に注意を促し、監査委員会(またはその代表)の承認を受ける。 |
99
第4部
プロジェクト15.各種証拠品および財務諸表の添付表
| (a) | 以下は本年度報告書とともに提出されたものである |
| (1) | 財務諸表目録に記載されている財務諸表 | |
| (2) | 適用されない |
| (b) | 陳列品 |
以下の証拠品は、本年度報告のアーカイブの一部として、または参照によって組み込まれる。
展示品索引:
| 展示品 | 説明する | |
| 2.1* | BiomX Inc.,BTX Merge Sub I,Inc.,BTX Merge Sub II,LLCとAdaptive Phage Treateutics,Inc.の間の合併協定と計画は,期日は2024年3月6日(合併内容は,会社が2024年3月6日に提出した現在の8-K表報告の添付ファイル2.1参照) | |
| 3.1 | 2018年12月11日から施行され、これまでに改訂された“会社登録証明書”のコピー。(2022年11月9日に会社が提出した会社の四半期報告書10-Q表の添付ファイル3.1を参照) | |
| 3.2 | 定款の改正と再制定は、2019年10月28日から施行される(会社が2019年11月1日に提出した会社現在の8-K表報告書の添付ファイル3.3を参照して設立) | |
| 3.3 | Xシリーズ優先株指定証明書フォーマット(会社が2024年3月6日に提出した会社現行8-Kレポート添付ファイル3.1合併参照) | |
| 4.1*** | 1934年改正証券取引法第12節に基づいて登録された証券説明 | |
| 4.2 | 単位証明書サンプル(会社2018年12月4日提出のS-1表登録書添付ファイル4.1登録成立参照) | |
| 4.3 | 普通株式サンプル(会社2018年12月4日提出のS-1表登録説明書添付ファイル4.2参照) | |
| 4.4 | 承認株式証サンプル(会社2018年12月4日提出のS-1表登録説明書添付ファイル4.3参照) | |
| 4.5 | 大陸株式譲渡信託会社と当社が2018年12月13日に署名した引受権証協定(当社が2018年12月18日に提出した現在の8-K表報告添付ファイル4.1登録により設立) | |
| 4.6 | 授権書表。(当社が2021年7月26日に提出した現行8-K表報告書の添付ファイル4.1参照) | |
| 4.7 | 資金を前払いして株式証明書を発行する.(当社が2023年2月27日に提出した現行8-K表報告書の添付ファイル4.1参照) | |
| 4.8 | 合併株式証表(当社が2024年3月6日に提出した当社現行8-K表報告添付ファイル4.1を参照して設立) | |
| 4.9 | 私募株式証表(当社が2024年3月6日に提出した当社現行8-K表報告添付ファイル4.2を参照して法団として設立) | |
| 4.10 | 配給代理人授権書表(当社が2024年3月6日に提出した当社現行8-K表報告添付ファイル4.3参照) | |
| 4.11 | 株式承認証表(当社が2024年3月18日に提出した当社現行8-K表報告添付ファイル4.1を参照して設立) | |
| 10.1** | 改訂されたチャールダン医療買収会社2019年総合長期インセンティブ計画(合併時参考会社が2023年7月28日に提出した付表14 Aに関する最終依頼書添付ファイルA) | |
| 10.2 | 2019年10月28日の登録権協定(会社が2019年11月1日に提出した会社現在8-K表報告の添付ファイル10.1を参照して会社として設立) | |
| 10.3**, *** | 合意の形式を達成する | |
| 10.4* | BiomX株式会社と業達研究開発有限会社が2015年6月22日に締結した改訂された研究·許可協定(会社が2019年11月1日に提出した会社の現在の8-K表報告書の添付ファイル10.5を参考に合併することにより) | |
| 10.8** | 改訂後の2015年従業員株式オプション計画(会社が2020年1月2日に提出したS-8表登録説明書添付ファイル99.1を参照) | |
| 10.9 | 会社と初期株主とChardan Capital Markets,LLCとの間の登録権協定は,2018年12月13日である。(2018年12月18日に提出された会社の現在の8-Kレポートの添付ファイル10.4参照) | |
| 10.10** | 非限定株式オプション協定表(米国は非執行役員に付与)(当社が2020年3月26日に提出した10-K表年次報告添付ファイル10.19登録成立参照) | |
| 10.11** | 非限定株式オプション協定表(米国の上級管理者への奨励)(会社が2020年3月26日に提出した10-K表年次報告添付ファイル10.20設立参照) | |
| 10.12** | オプション契約表(イスラエル賞)(当社が2020年3月26日に提出した10-K表年次報告添付ファイル10.21合併参照) | |
| 10.13* | AFI Assets Ltd.,AF-SHAR Ltd.,WISとBiomX Ltd.の間で2020年9月7日に締結された日付は2017年5月25日の賃貸契約付録である(会社が2021年3月31日に提出したForm 10-K年度報告添付ファイル10.14合併により) |
100
| 10.14* | AFI Assets Ltd.,AF-SHAR Ltd.,Wis,Nova測定システム有限会社とBiomX Ltd.の間で2020年9月7日に締結された賃貸契約(会社が2021年3月31日に提出したForm 10-K年報添付ファイル10.15合併による) | |
| 10.15 | 公開市場販売協定SM当社とJefferies LLCが2020年12月4日に締結した“S-3表登録説明書”添付ファイル1.2(当社が2020年12月4日に提出した“会社登録説明書”添付ファイル1.2)。 | |
| 10.17** | BiomX株式会社(前MBcuve Ltd.)間の雇用契約は、2016年2月1日となっているジョナサン·ソロモン(2022年5月2日に提出された改訂された10-K/A表年次報告添付ファイル10.1を参照して法団として設立) | |
| 10.18** | BiomX株式会社とMerav Bassan間の雇用契約は、2019年8月26日(会社が2022年5月2日に提出した改訂された10-K/A表年次報告添付ファイル10.2合併による) | |
| 10.19** | BiomX株式会社(前身はMBCURE Ltd.)が2017年1月1日に締結した雇用契約アサフ·オロンと(添付ファイル10.3編入会社が2022年5月2日に提出した改訂された10-K/A表年次報告を参照) | |
| 10.20 | 2023年2月22日の証券購入契約表(当社が2023年2月22日に提出した当社現行8-Kレポートの添付ファイル10.1参照) | |
| 10.21 | 登録権プロトコル表(2023年2月22日に会社が提出した現在の8-K表報告書の添付ファイル10.2参照) | |
| 10.22*,*** | 適応バクテリオファージ治療会社とアメリカ合衆国との間の独占ライセンスは、海軍長官に代表され、日付は2017年3月16日 | |
| 10.23*,*** | 第一修正案は、2019年1月10日に、海軍長官代表により、適応バクテリオファージ治療会社とアメリカ合衆国との間の独占的許可を付与する | |
| 10.24*,*** | 適応バクテリオファージ治療会社とWalter芦徳陸軍研究所との間の非独占的許可協定は,2021年8月24日である | |
| 10.25*** | ライセンス修正1,期日は2022年8月31日,適応バクテリオファージ治療会社とWalter芦徳陸軍研究所が締結した非独占ライセンス契約 | |
| 10.26 | BiomX Inc.は、添付ファイルAに記載されている購入者毎に締結された、期日が2024年3月6日の証券購入契約(会社が2024年3月6日に提出した現在の8-Kレポート添付ファイル10.1に統合されている) | |
| 10.27 | 会社とある購入者の間の登録権協定形式で、日付は2024年3月6日です(当社が2024年3月6日に提出した現行8-K表報告書の添付ファイル10.2参照) | |
| 10.28*,*** | リース契約は,2019年8月9日にARE−708 Quince Orchard,LLCと適応バクテリオファージ治療会社が締結した。 | |
| 10.29*,*** | 改正案第1号は,2020年10月28日にARE−708 Quince Orchard,LLCと適合性バクテリオファージ治療会社との間のリース契約を締結した。 | |
| 10.30*,*** | 改正案第2号は,2021年7月8日にARE−708 Quince Orchard,LLCと適応バクテリオファージ治療会社との間のリース契約を締結した。 | |
| 10.31*,*** | 期日は2021年7月15日の改正案第3号であり,ARE−708 Quince Orchard,LLCと適応バクテリオファージ治療会社によるリース契約が締結されている。 | |
| 10.32*,*** | 改正案第4号は,2022年9月27日にARE−708 Quince Orchard,LLCと適応バクテリオファージ治療会社によるリース契約が締結された。 | |
| 10.33*,*** | 改正案第5号は,2023年2月2日にARE−708 Quince Orchard,LLCと適応バクテリオファージ治療会社との間のリース契約を締結した。 | |
| 10.34*,*** | 期日は2024年3月5日の改正案第6号であり,ARE−708 Quince Orchard,LLCと適応バクテリオファージ治療会社によるリース契約が締結されている。 | |
| 21.1*** | 会社の付属会社 | |
| 23.1*** | 普華永道国際有限公司のメンバー事務所Kesselman&Kesselman会計士事務所(Isr.)の同意 | |
| 31.1*** | チーフ実行幹事は、ルール13 a~14およびルール15 d−14(A)に従って認証される。 | |
| 31.2*** | 細則13 a−14及び細則15 d−14(A)に従って首席財務幹事を確認する。 | |
| 32.1**** | 米国法第18編第1350条に基づいて最高経営責任者及び最高財務官証明書を発行する。 | |
| 97.1*** | 払戻政策 | |
| 99.1*** | 株式発行を承認する取締役会決議 | |
| 101.INS | XBRLインスタンスドキュメントを連結する | |
| 101.書院 | イントラネットXBRL分類拡張アーキテクチャ文書 | |
| 101.カール | インラインXBRL分類拡張計算リンクライブラリ文書 | |
| 101.def | インラインXBRL分類拡張Linkbase文書を定義する | |
| 101.介護会 | XBRL分類拡張ラベルLinkbase文書を連結する | |
| 101.Pre | インラインXBRL分類拡張プレゼンテーションLinkbaseドキュメント | |
| 104 | 表紙相互データファイル(添付ファイル101に含まれるイントラネットXBRLのフォーマット) |
| * | S−K法規第601(B)(10)条によれば、本展示品の一部の内容は省略されている。漏れた情報は実質的ではなく、公開開示すれば会社に競争被害を与える可能性がある。 |
| ** | 契約または補償計画または合意を管理することを指す。 |
| *** |
本局に提出します。 |
| **** | 手紙で提供する。 |
項目16.表格10-Kの概要
ない。
101
サイン
1934年の“取引法”第13又は15(D)節の要求に基づいて、登録者は、本年度報告を以下の署名者が代表して署名し、正式な許可を得るように促す。
| BIOMX Inc. | ||
| 日付:2024年4月3日 | 差出人: | /S/ジョナサン·ソロモン |
| 名前: | ジョナサン·ソロモン | |
| タイトル: | 最高経営責任者 | |
1934年の証券取引法の要求に基づき、本報告は以下の者が会社名で指定日に署名した。
| サイン | タイトル | 日取り | ||
| /S/ジョナサン·ソロモン | 最高経営責任者 | 2024年4月3日 | ||
| ジョナサン·ソロモン | (CEO)と役員 | |||
| /S/Avraham Gabay | 臨時首席財務官 | 2024年4月3日 | ||
| エイブラハム·ガブリエル | (首席財務官と首席会計官 | |||
| /投稿S/ラッセル·グレッグ | 取締役会議長 | 2024年4月3日 | ||
| ラッセル·グリガー博士 | ||||
| /S/ジェシー·グッドマン | 役員.取締役 | 2024年4月3日 | ||
| ジェシー·グッドマン博士 | ||||
| 寄稿S/ジョナサン·レフ | 役員.取締役 | 2024年4月3日 | ||
| ジョナサン·レフ | ||||
| /S/グレゴリー·メリル | 役員.取締役 | 2024年4月3日 | ||
| グレゴリー·メリル | ||||
| /S/アラン·モーゼ | 役員.取締役 | 2024年4月3日 | ||
| エレン·モーゼス博士 | ||||
| /S/エディ·ウィリアムズ | 役員.取締役 | 2024年4月3日 | ||
| エディ·ウィリアムズ |
102
BIOMX Inc.
連結財務諸表
2023年12月31日
カタログ
| ページ | ||
| 独立公認会計士事務所レポート(PCAOB名:Kesselman&Kesselman C.P.A.,PCAOB ID: | F-2 | |
| 連結財務諸表: | ||
| 合併貸借対照表 | F-3-F-4 | |
| 連結業務報告書 | F-5 | |
| 合併株主権益変動表 | F-6 | |
| 統合現金フロー表 | F-7-F-8 | |
| 連結財務諸表付記 | F-9-F-34 |
F-1
独立公認会計士事務所報告
![]()
BiomX Inc.取締役会と株主へ。
財務諸表のいくつかの見方
添付BiomX Inc.およびその付属会社(“当社”)の2023年12月31日および2022年12月31日の総合貸借対照表と、2023年12月31日までの2年間の各年度の関連総合運営報告書、株主権益変動およびキャッシュフローを審査し、関連付記(総称して“総合財務諸表”と呼ぶ)を含む。総合財務諸表は,米国公認の会計原則に従って,当社の2023年12月31日,2023年および2022年12月31日の財務状況,および2023年12月31日までの各年度の経営実績およびキャッシュフローをすべての重要な面で公平に反映していると考えられる。
その会社が継続的な企業として経営を続ける能力があるのではないかと疑っている
添付されている総合財務諸表の作成仮説は、当社は引き続き経営を継続する企業である。総合財務諸表付記1 Cで述べたように、当社は運営により重大な損失や負のキャッシュフローが発生しており、累積損失が発生しており、このような事件や状況は、当社の持続経営企業としての持続的な経営能力に大きな疑いがあることを示している。管理部門のこれらの事項に関する計画も付記1 Cに掲載されている。連結財務諸表には、このような不確実性の結果がもたらす可能性のあるいかなる調整も含まれていない。
意見の基礎
これらの連結財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の総合財務諸表 に意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従ってこれらの連結財務諸表を監査した。これらの基準は、合併財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、監査を計画し、実行することが要求されます。当社はその財務報告書の内部統制を監査する必要もありません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性に対する意見を表明するためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、連結財務諸表の重大な誤報リスクを評価するプログラム(エラーによるものであっても詐欺によるものであっても)、これらのリスクに対応するプログラム を実行することが含まれる。これらの手続きは、連結財務諸表中の金額および開示に関する証拠をテストに基づいて検討することを含む。私たちの監査には、経営陣が使用する会計原則の評価と重大な推定、合併財務諸表の全体列報の評価も含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
重要な監査事項とは、連結財務諸表を当期監査する際に生じる、監査委員会に伝達または要求された事項であり、(I)総合財務諸表に対して大きな意味を有する勘定または開示に関連し、(Ii)は、私たちが特に挑戦的、主観的、または複雑な判断に関するものである。私たちは重要な監査事項が存在しないと確信する。
/s/
公認会計士(Isr.)
普華永道国際有限公司のメンバー
2024年4月3日
2021年以来、私たちは会社の監査役を務めてきた。
F-2
BIOMX Inc.
合併貸借対照表
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 12月31日まで | ||||||||
| 2023 | 2022 | |||||||
| 資産 | ||||||||
| 流動資産 | ||||||||
| 現金と現金等価物 | ||||||||
| 制限現金 | ||||||||
| 短期預金 | ||||||||
| その他流動資産 | ||||||||
| 流動資産総額 | ||||||||
| 非流動資産 | ||||||||
| 経営的リース使用権資産 | ||||||||
| 財産と設備、純額 | ||||||||
| 非流動資産総額 | ||||||||
付記は連結財務諸表の構成要素である。
F-3
BIOMX Inc.
合併貸借対照表
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 12月31日まで | ||||||||
| 2023 | 2022 | |||||||
| 負債と株主権益 | ||||||||
| 流動負債 | ||||||||
| 売掛金 | ||||||||
| 賃貸負債の流動部分 | ||||||||
| その他の売掛金 | ||||||||
| 長期債務の当期部分 | ||||||||
| 流動負債総額 | ||||||||
| 非流動負債 | ||||||||
| 契約責任 | ||||||||
| 長期債務,当期分を差し引く | ||||||||
| 賃貸負債を経営し,当期分を差し引く | ||||||||
| その他負債 | ||||||||
| 非流動負債総額 | ||||||||
| 引受金及び又は有事項(付記10) | ||||||||
| 株主権益 | ||||||||
| 優先株、$ | ||||||||
| 普通株、$ | ||||||||
| 追加実収資本 | ||||||||
| 赤字を累計する | ( | ) | ( | ) | ||||
| 株主権益総額 | ||||||||
付記は連結財務諸表の構成要素である。
F-4
BIOMX Inc.
連結業務報告書
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 十二月三十一日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| 研究と開発(“R&D”)費用、純額 | ||||||||
| 無形資産の償却 | ||||||||
| 一般と行政費用 | ||||||||
| 営業損失 | ||||||||
| その他の収入 | ( | ) | ( | ) | ||||
| 利子支出 | ||||||||
| 財務収入、純額 | ( | ) | ( | ) | ||||
| 税引き前損失 | ||||||||
| 税金支出 | ||||||||
| 純損失 | ||||||||
付記は連結財務諸表の構成要素である。
F-5
BIOMX Inc.
合併株主権益変動表
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 普通株 | その他支払い済み | 積算 | 合計する 株主の | |||||||||||||||||
| 株 | 金額 | 資本 | 赤字.赤字 | 株権 | ||||||||||||||||
| 2021年12月31日現在の残高 | ( | ) | ||||||||||||||||||
| 公開市場販売協定項における普通株の発行純額は$ | ||||||||||||||||||||
| 株に基づく報酬費用 | - | |||||||||||||||||||
| 株式収益(*) | ||||||||||||||||||||
| 純損失 | - | ( | ) | ( | ) | |||||||||||||||
| 2022年12月31日現在の残高 | ( | ) | ||||||||||||||||||
| 公共株式個人投資項目で普通株式と引受権証(“PIPE”)を発行し、純額は#ドル | ||||||||||||||||||||
| 在庫再発行(*) | ||||||||||||||||||||
| 株に基づく報酬費用 | - | |||||||||||||||||||
| 公開市場販売協定により普通株式を発行(**) | ||||||||||||||||||||
| 純損失 | - | ( | ) | ( | ) | |||||||||||||||
| 2023年12月31日現在の残高 | ( | ) | ||||||||||||||||||
| (*) |
| (**) | |
| (***) |
付記は連結財務諸表の構成要素である。
F-6
BIOMX Inc.
統合現金フロー表
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 十二月三十一日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| キャッシュフロー--経営活動 | ||||||||
| 純損失 | ( | ) | ( | ) | ||||
| 純損失と経営活動で使用されているキャッシュフローとの照合に必要な調整 | ||||||||
| 減価償却および償却 | ||||||||
| 株に基づく報酬 | ||||||||
| 債務発行原価償却 | ||||||||
| 財務収入、純額 | ( | ) | ( | ) | ||||
| その他負債の変動 | ( | ) | ( | ) | ||||
| 資本損失,純額 | ||||||||
| 経営性資産と負債変動状況: | ||||||||
| その他流動資産 | ||||||||
| 売掛金 | ( | ) | ||||||
| その他の売掛金 | ( | ) | ||||||
| 経営賃貸純変動 | ( | ) | ( | ) | ||||
| 経営活動のための現金純額 | ( | ) | ( | ) | ||||
| キャッシュフロー--投資活動 | ||||||||
| 短期預金投資 | ( | ) | ||||||
| 短期預金収益 | ||||||||
| 財産と設備を購入する | ( | ) | ( | ) | ||||
| 財産と設備を売却して得た収益 | ||||||||
| 投資活動提供の現金純額 | ( | ) | ||||||
| キャッシュフロー--融資活動 | ||||||||
| 公開市場販売協定項における普通株の発行は,発行コストを差し引く | ||||||||
| PIPEによる普通株式と引受権証 | ||||||||
| パイプからの発行コスト | ( | ) | ||||||
| 長期債務を償還する | ( | ) | ||||||
| 株式で取得した収益 | ||||||||
| 融資活動が提供する現金純額 | ||||||||
| 現金と現金等価物および制限現金の減少 | ( | ) | ( | ) | ||||
| 現金および現金等価物と限定的現金に及ぼす為替レート変動の影響 | ||||||||
| 年初現金および現金等価物と制限現金 | ||||||||
| 年末現金および現金等価物と制限現金 | ||||||||
付記は連結財務諸表の構成要素である。
F-7
BIOMX Inc.
統合現金フロー表
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 2013年12月31日までの1年間 | ||||||||
| 2023 | 2022 | |||||||
| 連結貸借対照表上の金額を照合する: | ||||||||
| 現金と現金等価物 | ||||||||
| 制限現金 | ||||||||
| 現金と現金等価物および限定的な現金総額 | ||||||||
| キャッシュフロー情報の追加開示: | ||||||||
| 利子を支払う現金 | ||||||||
| 納めた税金 | ||||||||
| 非現金投資活動の追加開示: | ||||||||
| 売掛金に含まれる財産と設備購入 | ||||||||
付記は連結財務諸表の構成要素である。
F-8
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 注1- | 一般情報 |
| A. | 一般情報: |
BiomX Inc.(単独かつその子会社BiomX Ltd.とRondinX Ltd.,略称“会社”または“BiomX”)
2019年10月29日、当社はBiomXイスラエル社と合併し、後者は合併で生き残り、BiomX社の完全子会社となった。当社はBiomXイスラエル社の全流通株を買収した。代わりにBiomXイスラエル社の株主は
2020年2月6日には、同社の普通株もテルアビブ証券取引所で取引を開始した。2022年7月6日、同社は自発的にその普通株brをテルアビブ証券取引所から退市し、2022年10月6日に発効すると発表した。
BiomXは慢性疾患中の有害細菌を対象とした天然と改造されたバクテリオファージカクテル を開発しており,現在嚢胞性線維化に集中しており,アトピー性皮膚炎を比較的小さい程度治療している。BiomXは固有細菌標的を発見し検証し,これらの標的に対してバクテリオファージ成分をカスタマイズした。その会社はイスラエルのネスジオナに本部を置いている。
2024年3月6日、当社は米国デラウェア州のある会社と適応バクテリオファージ治療会社(“APT”)及びいくつかの他の方と協議及び合併計画(“合併協議”)を締結し、APTを当社の全額付属会社(“買収事項”)とした。付記1 Dを参照 今回の買収に関するより多くの情報は、 をお願いします。
| B. | イスラエル·ハマス戦争 |
2023年10月7日、ハマステロ組織のテロリストはガザ地区とイスラエル内の他の地域からイスラエル南部の境界に浸透し、イスラエルに前例のない攻撃を発動し、民間人と軍事目標を攻撃するとともに、イスラエル民衆に大規模なロケット弾攻撃を発動した。このような攻撃は多くの民間人たちと兵士たちの死傷者と誘拐をもたらした。これに応じて、イスラエル国民安全内閣はハマスに宣戦布告し、ハマスはロケット弾やテロを継続するとともに、これらのテロ組織に対する軍事行動を開始した。また、レバノン南部の大部分を支配するイスラムテロ組織ヒズボラがイスラエル北部の軍事·民間目標を襲撃し、イスラエルはこれに対応した。
これまでイスラエル国民はハマスと戦い続けてきましたヒズボラとの武力衝突.
BiomX本部と主要な事務所とその大部分の業務はイスラエル諸国に位置している。しかも、すべての重要な職員たちと職員たちはイスラエルの住民だ。したがって、イスラエルと周辺地域の政治、経済、軍事状況はその業務に直接影響する可能性がある。
同社の数名の従業員がイスラエル国防軍予備役に召喚されたが,開始以来,ハマスとの継続戦争はBiomXの業務や運営に実質的な影響を与えていない。また、BiomXその 計画はこれにより遅延しないと予想される.しかし、現在のところ、イスラエルがハマスと戦う戦争の強度や持続時間を予測することはできず、この戦争が最終的にBiomXの業務と運営やイスラエル全体の経済にどのように影響するかを予測することもできない。
F-9
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 注1- | 普通(続) |
| C. | 経営を続ける企業 |
会社は重大な損失を出し、運営キャッシュフローはマイナスで、累計損失は#ドル
| D. | 合併協定 |
2024年3月6日、当社はデラウェア州の全資付属会社BTX Merge Sub I,Inc.(“第一合併付属会社”)、デラウェア州の有限責任会社及び当社の全資付属会社BTX Merge Sub II,LLC(“第二合併付属会社”)と合併協定を締結した。合併協議によると、第一合併付属会社はアジア太平洋電気通信と合併してアジア太平洋電気通信に編入し、アジア太平洋電気通信はまだ残っている法団であり、当社の完全子会社となる(“初合併”)。第1の合併後、APTは直ちに合併子会社と第2の連結子会社に合併し、当該連結子会社によれば、第2の連結子会社は存続する実体である(第1の合併、すなわち“買収”とともに)。今回の買収は、米国連邦所得税の免税再編資格を満たすことを目的としている。彼は言いました
2024年3月15日、つまり買収発効時期、APTの前株主は全部で獲得した
買収を完了すると同時に、
会社はいくつかの投資家と1つの証券購入協定を締結し、この合意に基づいて、同等の投資家は共にbrを購入した
買収に続いて,パイプライン優先株や私募株式証を考慮せずに,会社買収前の株主が約brを所有している
F-10
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 注1- | 普通(続) |
| D. | 合併協定(継続) |
買収は会計基準に従ってテーマ805“企業合併”を編纂し、会計の買収方法を用いて会計計算を行う。以下の事実と状況の評価によると、当社は会計購入者として決定された
| ● | 合併協議によると、当社の買収後の取締役会は7人の取締役で構成されており、その中で当社は4つの取締役会席を指定し、買収前に当社の取締役会長は引き続きそのポスト、すなわち取引完了後の取締役会の大部分は当社が指定している。 |
| ● | 最高経営責任者や管理職の多くは買収前に会社と関連のある個人が担当する。 |
| 注2- | 重大会計政策 |
新しい会計基準を採用する以外に、一致した基礎の上で財務諸表を作成する際に採用する主要な会計政策は以下の通りである
| A. | 列報根拠と合併原則 |
添付されている総合財務諸表は、当社とその完全子会社BiomXイスラエルとRondinX株式会社の口座を含む米国公認会計原則(“GAAP”)に基づいて作成されています。すべての会社間口座と取引は合併で抹消されています。
| B. | 財務諸表を作成する際に推定数を用いる |
公認会計原則に基づいて財務諸表を作成する際には、管理層は、報告された資産及び負債額に影響を与え、財務諸表中の又は資産及び負債及び報告年度の費用金額を開示するために推定及び仮定を行う必要がある。会社の財務諸表の中で最も重要なbrは、研究開発費に関する計算項目と株式に基づく報酬報酬の推定値を推定する。このような推定および仮定は現在の事実、未来予想及びその時の状況で が合理的と思われる他の様々な要素に基づいており、その結果は資産と負債の額面を判断する基礎を構成し、他の源から知覚しにくい支出を記録する。実際の結果はこれらの推定値と大きく異なる可能性があり, とは逆である.
イスラエル-ハマス戦争が会社の業務、運営結果、財務状況に直接または間接的に影響を与える可能性のある全面的な程度は、不確定な未来の事態の発展、および現地、地域、国、国際市場への経済的影響に依存する。
| C. | 本位貨幣と外貨換算 |
当社の本位貨幣はドル (“ドル”)であり、ドルは当社の経営が置かれている主要な経済環境の通貨であるため、 は予見可能な未来に経営を継続することが予想される。最初にドル建ての取引と残高をその元の 金額に並べて示す.非ドル通貨残高はそれぞれ非通貨残高と通貨残高の過去の為替レートと現在の為替レートを使用してドルに換算される。非ドル取引および損益表の他の項目(以下に示す)については、(I)取引--取引日の為替レートまたは平均為替レート、(Ii)他の項目(減価償却や償却などの非貨幣貸借対照表項目から生じる)−履歴レートが使用される。貨幣取引収益と損失は状況に応じて財務収入純額に記載されている。
| D. | 現金および現金等価物および限定現金 |
当社は現金等価物をすべての短期·高流動性投資と見なし、引き出しや使用制限を受けない通貨市場基金と、購入日から最初の満期日が3ヶ月以下であり、引き出しや使用制限を受けず、既知の金額の現金に容易に両替できる短期銀行預金を含む。制限された現金には、短期外国為替契約を返済していないクレジット限度額に制限された契約上の資金と、リース契約によって提供される銀行保証とが含まれる。当社は総合貸借対照表に限定的な現金を現金と現金等価物と分けて列記しています。当社は、連結現金フロー表に表示されている期初と期末総額を調整する際に、現金と現金等価物を含む制限銀行預金 を調整します。
F-11
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 注2- | 重大会計政策(継続) |
| E. | 信用リスクが集中する |
私たちを信用リスクに直面させる可能性のある金融商品は主に現金、現金等価物、および短期預金を含む。このような金額は時々連邦保険の限度額を超えるかもしれない。私たちはこのような口座で何の信用損失も経験していないし、私たちがこのような資金に重大な信用リスクがあるとは思わない。同社の現金と現金等価物および銀行預金の大部分は米国とイスラエルの主要銀行に投資されている。経営陣は、当社の現金と現金等価物および銀行預金を持つ金融機関の信用リスクは低いと考えている。付記2 Jを参照されたい。
| F. | 財産と設備 |
財産と設備はコストから減価償却累計を引いて申告する
| 使用可能寿命を見積もる | ||
| 実験室装置 | ||
| コンピュータとソフトウェア | ||
| 設備と家具 | ||
| 賃借権改善 |
| G. | 長寿資産 |
米国会計基準360-10“長期資産の減値および処分”によると、事件や環境変化が資産の帳簿価値を示すたびに、推定された将来の未割引現金流量に基づいて回収できない可能性がある場合、管理層は長期資産の減値を審査する。表示されていれば,資産の額面とその公正価値との差額について減値損失を確認する.12月31日まで,2023年および2022年12月31日までは,減値支出は記録されていない。
| H. | 所得税 |
当社は貸借対照法を用いて所得税を計算します。繰延税金資産と負債は、資産と負債の財務諸表と税項との差額と、これらの差額が予想沖販売された場合の実際の税率に基づいて入金される。既存の証拠の重みに基づいて、繰延税金資産の一部または全部が現金化できない可能性が高い場合、繰延税金資産は減価される。会社は2023年12月31日、2023年12月31日、2022年12月31日まで、繰延税金資産に対して全額評価準備をしている。
当社は、ASC 740-10-25“所得税”(以下、“ASC 740”と略す)の規定を遵守しなければならない。ASC 740は、税務状態を決定しない財務諸表確認のためのより可能な閾値を規定する。ASC 740は、納税申告書において採用されるまたは予期される納税状態を確認および計量するために、最低確認閾値および計量br属性を規定することによって、所得税の会計処理を明確にする。当社は毎年、所得税課税項目が不確定税務状況に関するASC 740の指針に適合しているかどうかを評価している。当社は2023年12月31日及び2022年12月31日までの年度の不確定税務状況について何の負債も記録していません。当社は未確認の税務優遇を繰延税金資産の減値としていますが、司法管轄区の税法を適用することにより、純営業損失、類似の税務損失又は 税務相殺繰越は、決済税務状況による追加所得税を相殺するために使用することができます。
| I. | 派生活動 |
同社は通貨リスクを回避するために、外国為替契約(オプション契約と長期契約)を使用して金流を現金化している。これらの外国為替契約は、会計目的のためにヘッジツール
として指定されない。当該等外国為替契約については、当社が確認した収益や損失は、総合経営報告書の財務支出(収益)の項にも記入されている現金流量のリスコアリングを相殺している。当社は2023年12月31日現在、ドル対新シェケルの未償還短期外国為替契約を保有しており、金額は約brドルです
F-12
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 注2- | 重大会計政策(継続) |
| J. | 金融商品の公正価値 |
当社は、ASC 820“公正価値計量·開示”(“ASC 820”)に基づいて金融商品を会計処理する。ASC 820は、公正価値階層構造 を構築し、公正価値を計量するための推定技術の入力を優先順位付けする。この階層構造は,同じ資産や負債の未調整 アクティブな市場オファーを最高優先度(1レベル評価),最低優先度を観察できない投入 (3レベル尺度)に与える.ASC 820によって規定される公正価値階層構造の3つの階層は以下のとおりである
第1レベル-アクティブ市場の未調整見積 計量日に得られる同じ、制限されていない資産または負債の見積もり。
第2レベル-資産や負債のような非アクティブ市場またはアクティブ市場のオファー、オファー以外の観察可能な投入、および直接観察できないが観察可能な市場データが確認された投入。
第3級−公正価値計測に重大な意義を持ち観察できない投入の価格や推定値 が必要である。
2023年12月31日および2022年12月31日までの年間で,公正価値階層レベルに変動はなかった。
| 2023年12月31日 | ||||||||||||||||
| レベル1 | レベル2 | レベル3 | 公正価値 | |||||||||||||
| 資産: | ||||||||||||||||
| 現金等価物: | ||||||||||||||||
| 貨幣市場基金 | ||||||||||||||||
| 受取外国為替契約 | ||||||||||||||||
| 負債: | ||||||||||||||||
| 値段が合うかもしれない | ||||||||||||||||
| 2022年12月31日 | ||||||||||||||||
| レベル1 | レベル2 | レベル3 | 公正価値 | |||||||||||||
| 資産: | ||||||||||||||||
| 現金等価物: | ||||||||||||||||
| 貨幣市場基金 | ||||||||||||||||
| 負債: | ||||||||||||||||
| 含まれていないか、または割引があります | ||||||||||||||||
| 為替契約に対処する | ||||||||||||||||
F-13
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 注2- | 重大会計政策(継続) |
| J. | 金融商品公正価値(継続) |
帳簿価値が公正価値に近い金融商品は、現金及び現金等価物、制限された現金、短期預金、その他の流動資産、貿易帳簿及びその他の流動負債を含み、短期的な性質である。
当社は確率割引キャッシュフロー分析に基づいて対価格負債の公正価値を決定または有します。この公正価値計量は市場では観察できない重大な投入に基づいているため、公正価値等級中の第三級計量を代表する。あるいは考慮された公正価値はいくつかの要素、例えば:原発性硬化性胆管炎の候補製品の治療に関連する未来の臨床、開発、監督管理、商業と戦略マイルストーンの実現状況に基づいている。適用される割引率範囲は
| K. | 固定払込計画 |
イスラエル雇用法によると、BiomXイスラエル社の従業員は1963年の解散費補償法第14節(“第14節”)に基づいて賃金の一部を支給している。第14条によると、これらの従業員は、当社が彼らを代表して保険会社に入金した毎月の預金を得る権利がある。
第14条に基づいて支払われた金は、会社が将来これらの従業員に関連する任意の解散費(1963年イスラエル解散費補償法に基づく)の支払いを免れるようにする。上記br預金は資産として会社の貸借対照表に記録されておらず、いかなる負債も記録されていません。会社には将来的に追加金を支払う義務がないからです。会社員がサービスを提供する場合、会社の固定払込計画に対する貢献は総合経営報告書に計上される。これらの寄付に関する総費用
は$
米国人従業員に対して、会社は“米国国税法”第401(K)節に基づいて固定納付貯蓄計画を策定した。この計画は、BiomX Inc.の米国における最低年齢およびサービス要件に適合するすべての従業員をカバーし、参加者が税引前に年間給与の一部の支払いを延期することを可能にする。
会社はまだ従業員たちのどんな延期計画と一致するかを選択していない。2023年12月31日と2022年12月31日までの年間で、会社は401(K)Match納付の費用を何も記録していない。
| L. | 金融商品 |
当社が独立手形を発行する場合、まず、ASC 480の“負債と権益を区別する”(“ASC 480”)の規定を分析して、その手形が負債に分類されるべきかどうかを決定し、各期間の業務連結報告書においてその後の公正価値変動を確認する。ツールがASC 480の範囲内にない場合、当社は、ツールがエンティティ自身の株式にリンクされているとみなされているかどうかを決定し、br権益で分類する資格があるかどうかを決定するために、ASC 815-10の規定をさらに分析するであろう。当社が発行したすべての引受権証は、株主権益内で“追加実収資本”に分類されています。 株式証が自社自身の株式をインデックスとし、ASC 815-40“実体自己権益契約”(“ASC 815-40”)の株主権益分類要求に適合した場合、権益分類を行うことができます。
F-14
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 注2- | 重大会計政策(継続) |
| M. | 研究開発コスト |
研究·開発コストは発生時に運営報告書 を計上する。IIAが提供する特許権使用料を負担する贈与は,当社がこのような贈与を得る権利がある場合に確認し,その基礎が発生したコストであり,研究·開発費の控除とする。
| N. | 1株当たりの基本損失と赤字 |
1株当たり基本損失の算出方法は,純損失を本年度発行済み普通株の加重平均,会社普通株の無行使価格の全額株式承認証と会社普通株の全額既存予備資本権証,行権価格$で割った
| O. | 株式補償計画 |
当社は、推定された公正価値に基づいて、会社の株式計画に基づいて従業員および取締役に支給されるすべての株式ベースの支払い報酬の報酬支出を従業員株式オプションを含む株式ベースの支払い報酬のすべての報酬支出を計量および確認することを要求するASC 718-10、“株式ベース支払い”(ASC 718-10)を適用する。
ASC 718-10は、付与された日における従業員および非従業員に付与された株式報酬の公正価値を推定するために、オプション定価モデルを使用することを会社に要求する。報酬の公正価値 は,会社運営報告書において必要なサービス期間の費用であることを確認し, 階層帰属方法を採用する.当社は株式ベースの報酬報酬を株式奨励に分類している。当社では、適用ペナルティ率ではなく、株による報酬ペナルティが発生していないことを確認しました。
すべての非従業員に株式オプション或いは他の権益ツールを発行して当社が受信した商品或いはサービスの代価として、発行済み権益ツールの公正価値に従って入金する。
F-15
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 注2- | 重大会計政策(継続) |
| O. | 株式報酬計画 |
当社はブラック·スコアーズオプション定価モデルを用いて株式奨励として付与された株式オプションの公正価値を試算しています。オプション定価モデルは多くの仮定を必要とし,その中で最も重要なのは株価,期待変動率,期待オプション期限(付与日からオプション行使または満期までの時間)である.当社が採用した平均歴史株価変動率は,当社の歴史平均変動率とバイオテクノロジーや製薬業界で選定された上場企業同業グループと比較可能な総合加重平均変動率に基づいており,この等変動率は将来の株価傾向を代表すると考えられており,当社自身の普通株には十分な歴史取引 歴史がないためである。会社は、自身の株価変動に関する十分な数の履歴情報があるまで、この過程を適用し続ける。当社はこれまで配当金を派遣せず、予見可能な配当計画 もなかった。無リスク金利は同期間の国債ゼロ金利収益率に基づいている。“簡略化”方法を用いて、すべての株式オプション付与の期待オプション期間を計算する。個々の投入の査定 の変動は、付与されたオプションの公正価値および当社の経営結果に影響する可能性があります。
| P. | 賃貸借証書 |
会計基準の更新によると、“レンタル” (“ASC 842”)、会社は、開始時にレンタルであるか否かを決定する。初歩的な確認の際には、当社はリース期限内に支払われたリース支払いの現在値で負債を確認するとともに、負債金額と同じ使用権資産を確認し、任意の前払いまたは未払いで調整した後、リースに関する初期直接コストを加える。当社は開始日に得られた資料に基づいて、逓増借款金利を採用して賃貸支払いの現在値を決定します。その後の計量はリースが融資リースに分類されるかbr}経営的賃貸に分類されるかに依存する。報告期間中、当社は運営賃貸契約のみとなります。レンタル条項には、会社が選択権を行使することを合理的に確定した場合にレンタル期間を延長する選択権が含まれています。レンタル経営のレンタル料金はレンタル期間内に直線br法で確認します。
当社はすでに政策選択を行い、期限が12ヶ月を超えない賃貸契約を資本化しています
米国会計基準第360-10条によると、事件や環境変化がある資産の帳簿金額を回収できない可能性があることを示した場合、管理層は、推定された将来の未割引現金流量に基づいて、経営中の賃貸資産計の減値を審査する。表示されていれば,資産の額面とその公正価値との差額について減値損失を確認する.
| Q. | 在庫株 |
在庫株は持株を減額する形で列報し,費用は会社が負担する.
F-16
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 注2- | 重大会計政策(継続) |
| R. | 新会計公告 |
最近採用された会計公告
2016年6月、財務会計基準委員会(“FASB”) は、2016−13年度会計基準更新(“ASU”)、“金融商品−信用損失−金融商品信用損失の計測 ”を発表した。本ガイドラインは期待信用損失を反映する方法 を用いて現在発生した損失減値方法を置換し、更に広範な合理的かつ支持可能な情報を考慮してbr}信用損失推定を通知することを要求する。このガイドラインは、2023年1月1日に開始された次年度(年内の移行期間を含む)の規模の小さい報告会社に適用される(1934年の証券取引法改正規則に基づいて定義される)。当社は2023年1月1日にこの指針を採択し、この指針の採択は連結財務諸表に実質的な影響を与えないと結論した。
FASBは2021年10月、取引先との契約に基づいて契約資産および契約負債を会計処理するASU 2021−08“業務 組合せ(主題805)を発表し、業務組合で買収された契約資産および契約負債は、買収側がASC 606に従って買収日に確認および計量しなければならないことを要求した。この指導意見は、買収側が被買収側が記録した同じ金額で契約資産と契約負債を確認することにつながる。このガイドラインは、発効日または後に発生した買収に前向きに適用されなければならない。この指導意見は、2022年12月15日以降の財政年度に適用され、当該財政年度内の過渡期を含む。 は2023年1月1日から、同社はその合併財務諸表に実質的な影響を与えないと結論した。
2022年12月、FASBはASU 2022-06を発表し、 金利改革(テーマ848)を参考に:テーマ848の日没日を延期した。本ASUは、債務、リース、デリバティブ、その他の契約の金利改革関連活動に影響を与える臨時オプション実践方便を2024年12月31日まで延長する。当社は直ちにこの指針を採択し、この指針を採択することはその総合財務諸表に大きな影響を与えないと判断した。
最近発表された会計声明はまだ採用されていない
FASBは2023年11月、報告可能な支部に開示された改善“(”ASU 2023-07“)をASU 2023-07”支部報告“を発表した。本ガイドラインは、公共エンティティの部分開示を拡大し、主に、首席運営決定者に定期的に提供される重大な分部費用の開示を要求し、各報告の支部損益測定に含まれ、他の支部プロジェクトの金額および構成説明、および現在毎年要求されている報告可能な支部の損益および資産の中期開示を含む。単一の報告可能な支部を有する公的br}エンティティは、新たな開示を提供しなければならず、ASC 280、br}支部報告に要求されるすべての開示を提供しなければならない。本ガイドラインは、2023年12月15日以降の会計年度に有効である。2024年12月15日以降のbr年度内の移行期間は,早期採用が認められた。改訂は実体財務諸表に記載されているすべての 個の前期をさかのぼって適用しなければならない.当社は現在、ASU 2023-07の採用がその連結財務諸表に及ぼす可能性のある影響を評価しています。
FASBは2023年12月、“所得税開示の改善”(“ASU 2023-09”)をASU 2023-09“所得税(主題740)”を発表した。本ガイドラインは所得税開示の透明性と意思決定の有用性を向上させることを目的としている。ASU 2023-09の修正案は、主に米国および外国の管轄区域で支払われる税率調整および所得税の開示を変更することによって、所得税の強化に対する投資家の要求を満たす。ASU 2023-09は、2024年12月15日以降の会計年度に有効であり、この基準を遡及適用することを選択することができます。早期養子縁組を許可する。当社は現在、その連結財務諸表開示に影響を及ぼす可能性があることを確認するために、このガイドラインを評価している。
| 注3- | 短期預金 |
短期預金とは、br銀行に預けられている3ヶ月以上ですが、1年以下の定期預金のことです。当社が短期預金を持っている年度内に、稼いだ利息を財務収入純額 と表記します。
2023年12月31日までに会社は保証金です。2022年12月31日までの会社のドル預金は
F-17
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 注4- | その他流動資産 |
| 12月31日まで | ||||||||
| 2023 | 2022 | |||||||
| 政府機関 | ||||||||
| 前払い保険 | ||||||||
| その他前払い費用 | ||||||||
| 売掛金 | ||||||||
| 他にも | ||||||||
| 注5- | 財産と設備、純額 |
| 12月31日まで | ||||||||
| 2023 | 2022 | |||||||
| コンピュータとソフトウェア | ||||||||
| 実験室装置 | ||||||||
| 設備と家具 | ||||||||
| 賃借権改善 | ||||||||
| 減価償却累計 | ( | ) | ( | ) | ||||
その会社のほとんどの非流動資産はイスラエルに集中している。
減価償却費用は$
| 注6- | 子会社を買収する |
2017年11月、BiomXイスラエル社はRondinX株式会社の株主と株式購入協定に調印した。株式購入協定に基づき、BiomXイスラエル社は買収した
または公定価値で入金 (第3級)がある.2023年12月31日および2022年12月31日までの年間で、公正価値階層レベルに変動はありません。 付記2 Jを参照してください。
2023年12月31日現在、2022年12月31日現在の連結財務諸表には、本協定に関連する負債#ドルが含まれています
RondinX Ltd.を買収した時に獲得した無形資産はすでに2022年12月31日に全額償却された。2022年12月31日現在、業務連結報告書に記録されている償却費用は#ドルである
F-18
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 注7- | 賃貸借証書 |
2020年9月、BiomXイスラエル社はイスラエルのネチオナにあるオフィス空間賃貸協定を締結し、レンタル期間は2020年9月1日から5年間、2030年11月30日まで延長することができる。賃貸契約の下の毎月のレンタル料は約$である
2020年10月1日、当社は米コネチカット州ブランフォードオフィスビル賃貸契約を締結し、レンタル期間は2020年10月5日から25ヶ月間となった。プロトコルにより,毎月のレンタル料は約$
である
2022年8月、BiomXイスラエル社はイスラエルのネチオナにあるオフィス空間の一部について転貸協定を締結した。この協定の有効期間は2年で、2022年8月15日から始まる。プロトコルによると,
の毎月のレンタル料は約$である
Br業務統合レポートに記録されているリース費用は
| 2013年12月31日までの年度 2023 | 現在までの年度 十二月三十一日 2022 | |||||||
| レンタルの現金支払いを営む | ||||||||
| 運営中です 賃貸借証書 | ||||
| 2024 | ||||
| 2025 | ||||
| 2026 | ||||
| 2027 | ||||
| 2028 | ||||
| 2029 | ||||
| 2030 | ||||
| リース支払総額を経営する | ||||
| 計上された利息を差し引く | ( | ) | ||
| 総経営賃貸負債残高 | ||||
| 注8- | その他の売掛金 |
| 12月31日まで | ||||||||
| 2023 | 2022 | |||||||
| 従業員と関係機関 | ||||||||
| 費用を計算する | ||||||||
| 政府機関 | ||||||||
| 協商協議繰延費用と前払い分譲収入 | ||||||||
| 他にも | - | |||||||
F-19
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 注9- | 関係者との取引 |
| A. | 2019年10月、BiomXイスラエル社は#ドルの融資協定を締結した |
| B. | 付記12 Aを参照して、機関投資家、当社全取締役及び若干の行政者と締結した証券購入協定について。 |
| C. | 関連先に付与された株式オプションについては、付記12 Bを参照されたい。 |
| 付記10- | 引受金とその他の事項 |
| A. | 2021年3月、IIAは当社の嚢胞性繊維化製品候補製品に関する2つの新しい申請を承認し、総予算は新シェケルである |
2021年8月、IIAはバージョンアップ会社の製造能力を支援する申請を承認し、予算総額は新シェケルである
2022年3月、IIAは総額brの新シェケル予算の申請を承認した
2023年3月、IIAは総額brの新シェケル予算の申請を承認した
IIAとの合意によるとBiomXイスラエル社は
2023年12月31日現在,IIAが承認した贈与総額は約$である
| B. |
F-20
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 付記10- | 引受金及び又は有事項(継続) |
2019年7月、当社は業達研究開発有限公司(“業達”)と2015年に締結した研究及び許可協定(“ライセンス契約”)を改訂した。改訂によると、資本再編取引が完了した後、業達許可協定中の脱退費に関する条項はすでに改訂され、当社は業界達に改訂で述べた一次金を支払う責任があり、金額は返品費ではなく、当社のいかなる合併或いは買収に関連して徴収された代価の1%を超えず、そして各許可協定について を支払う。付記1 Dに記載された統合プロトコルは、修正案によって定義された合併または買収には適用されない。
| C. | RondinX株式会社の権益継承者として、BiomXイスラエル社は2016年3月20日にYedaと署名した許可協定の一方であり、同協定によると、同社はYedaの会社のメタゲノム標的発見プラットフォームに関する技術ノウハウ、情報、特許のグローバル独占許可を持っている。ライセンスの掛け値として,同社は毎年#ドルのライセンス料を支払う義務がある |
| D. | 2017年12月、BiomXイスラエルは日本の慶応義烏大学とJSR Corporationと特許許可協定に調印した。協定によると,BiomXイスラエル社は炎症性腸疾患(IBD)に関連するある特許権の独占特許許可を得ており,その見返りに年間ドルの許可料を支払う |
2019年4月、BiomXイスラエルは日本の慶應義塾大学とJSR Corporationと追加の特許許可協定
に署名した。合意により,BiomXイスラエル社はJSRの独占的再許可を得,原発性硬化性胆管炎の治療に関連するいくつかの特許権を取得した。その見返りに、会社は(I)ライセンス
発行料$を支払う必要があります
F-21
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 付記10- | 引受金及び又は有事項(継続) |
| F. | 2022年6月23日(“発効日”)、BiomXイスラエル社はブリンガー·インゲルハイム国際株式会社(“BI”)と研究協力協定を締結し、協力してIBDのバイオマーカーを決定した。協定によると、BiomXイスラエル社は合計#ドルの費用を得る資格がある |
| G. | 当社の賃貸負債に関する資料は、付記7を参照されたい。 |
| 注11- | 長期債務 |
2021年8月16日、当社はHercules Capital,Inc.(“Hercules”)とリスク債務融資について融資及び担保協定(“融資合意”)を締結した。融資協定によると、Herculesは会社に元金総額最大#ドルまでの定期融資を提供した
当社はいつでもローン契約に基づいて前払金の全部または一部を前払いすることができます。前払い費用は以下の通りです
定期ローンの利息は年利で計算して、利率は
に等しい
F-22
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 注11- | 長期債務(継続) |
2023年12月31日現在、定期ローンの帳簿価値には$が含まれている
連結経営報告書に利息支出が計上されている定期融資に関する利息支出は$である
融資協定の条項により、当社は当社のほとんどの知的財産権の優先留置権と担保権益を付与し、その義務の担保として
を付与します。同社はまた、Herculesが任意の後続の広範な市場融資の任意の成約に適宜参加することを許可し、その定義の最高総額は$を超えない
| 2023年12月31日 | ||||
| 2024 | ||||
| 2025 | ||||
| 元金支払総額 | ||||
| 未償却割引、債務発行コスト、期末費用の増加 | ||||
| 将来元金支払総額 | $ | |||
| 長期債務の当期部分 | ( | ) | ||
| 長期債務、純額 | $ | |||
| 付記12- | 株主権益 |
| A. | 株本: |
普通株:
2022年8月24日、会社株主は普通株式法定株式数を
国庫株:
注9 Aを参照してください。
初公募株:中国
2018年12月18日、当社は初公開(IPO)を完了しました
初公募を完成させるとともに,当社は合計を完成させた
F-23
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 付記12- | 株主権益(継続) |
| A. | 株本:(続) |
証券取引所:
付記1で述べたように、2019年10月28日の資本再編取引の一部として、当社は発行しました
また、会社は株主に以下の総数の普通株 を比例的に増発することに同意し、会社は資本再編取引後に以下の規定の条件を満たしていることを前提としている(これらはすべて会社がニューヨーク証券取引所米国取引所で取引する普通株に関するものである)
| A. |
| B. |
公募株式の個人投資:
2023年2月22日、当社は証券購入協定を締結し、発行及び販売を合わせている
未弁済資金権証の行使は、以下の利益所有権によって制限される
F-24
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 付記12- | 株主権益(継続) |
| A. | 株本:(続) |
市場販売協定:
2020年12月、証券取引委員会が2020年12月11日に発効を発表したS-3表登録声明によると、当社はJefferies LLCと公開市場
発行販売協定(“ATMプロトコル”)を締結した。(“Jefferies”)この条項は、条項
に基づいて、ATMプロトコルにおける条件および制限によって、総発行価格が最大$に達する普通株式
を時々選択して発売および販売することができると規定している
2023年12月、米国証券取引委員会が2024年1月2日に発効を発表したS-3表登録声明によると、当社はウィンライト株式会社(“ウェインライト”)と公開市場発売協定を締結し、この合意により、当社は総発行価格が最大$1に達する普通株を発行·販売することができる
丸紅プロトコル:
当社は2021年10月、日本の皮膚科を中心としたリーディング製薬会社丸紅株式会社(“丸紅”)の付属会社と株購入協定を締結し、これにより、当社は丸紅に株式を発行した
F-25
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 付記12- | 株主権益(継続) |
| A. | 株本:(続) |
CFFプロトコル:サポート
2021年12月,同社はCF財団と証券購入協定を締結し,従来から嚢胞性線維症(CF)患者の革新療法の開発を支援する役割を果たしてきた。合意条項によると、会社は最高
$を獲得します
優先株:
当社は発行を許可されている
株式承認証:
| 資本再編取引が完了した後、公募株式証を行使することができる。公開株式証を行使する際には、断片的な株式は発行されない。したがって、公共株式承認証は2つの株式承認証の倍数で行使されなければならない。株式公開承認証は満期になります
当社は以下の公開株式証明書を償還することができる |
| ● | 一部ではなく全てです | |
| ● | 販売価格は$ | |
| ● | トレーニング中のいつでも | |
| ● | 最低30日前に書面償還通知を出します | |
| ● | 会社の普通株の最後の販売価格が$以上になると | |
| ● | また、償還時にのみ、当該株式等承認証に関連し、償還時に有効な普通株式登録宣言がある |
もし会社が公共株式証の償還を要求した場合、管理層は公共株式証の行使を希望するすべての所有者に、株式承認契約に記載されている“キャッシュレス基礎”に従って公共持分証を行使することを要求する権利がある。株式承認証の行使時に普通株を発行できる使用価格と株式数は、株式配当、資本再編、再編、合併または合併を含む場合によって調整される可能性がある。しかし、株式承認証は普通株の発行価格がその行使価格を下回ることで調整されることはない。また、いずれの場合も、当社は現金純額で株式承認証を決済することを要求されません。
F-26
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 付記12- | 株主権益(継続) |
| A. | 株本:(続) | |
| 捜査命令 | 発行日 | 期日まで | トレーニングをする 値段 1株当たり | 量 の株 普通株 潜在的な 株式承認証 | ||||||||
| 株式証を公開する | ||||||||||||
| 2021年登録直接株式発行承認証 | ||||||||||||
| あらかじめ出資して株式証明書を発行する | ||||||||||||
| あらかじめ出資して株式証明書を発行する | ||||||||||||
| B. | 株式ベースの報酬: |
持分インセンティブ計画:
2015年、BiomXイスラエル取締役会は、従業員、サービスプロバイダ、および官僚にオプションを割り当てる計画(“2015計画”)を承認した。これらのオプションはBiomXイスラエル社の普通株を購入する権利を代表して、使用価格を支払う代償として使用される。また、当該等オプションは、イスラエル所得税条例第102条及び第3(I)条及び米国国税法第409 a条下の“資本収益路線”に基づいて付与され、2019年10月28日に資本再編取引を行った後に技術調整を行う。
2023年12月31日現在、2015年計画により、残りの普通株 は発行できません。
2019年、会社は新たなインセンティブ計画(“2019年計画”)によって与えられます
2019年計画によると交付可能な普通株式総数は毎年1月1日に自動的に増加し、2020年1月1日から2029年1月1日まで、金額は4%に相当する
上記の規定にもかかわらず、取締役会は、特定年度の1月1日までに行動することができ、当該年度の1月1日が増加しないこと、又は当該年度の普通株式増加の数が本稿で規定する数よりも少なくなることを規定している。
2023年12月31日までに
F-27
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 付記12- | 株主権益(継続) |
| B. | 株ベースの報酬:(継続) | |
株式オプション:
2022年3月29日、取締役会は
の付与を許可した
2022年6月21日、取締役会は
の授与を許可した
2022年8月22日、取締役会は
の授与を許可した
2022年9月30日、取締役会は
の授与を許可した
2023年3月1日、取締役会は
の付与を許可した
2023年8月21日取締役会が承認しました
2023年10月19日取締役会が承認しました
2023年10月29日取締役会が承認しました
2023年10月29日、取締役会は、BiomX従業員が現在保有している会社の普通株式を購入する各未償還オプションの行権価格(再定価)をbrに引き下げることを許可し、元の行権価格は$より高い
2023年11月9日、同社は、従業員にいくつかの条件に適合するオプションを提供する一度の自発的株式オプション交換(“オプション取引所”)の条項および条件を規定する入札要約声明を証券·取引委員会に提出した。会社は現物外株の一部を新たな株式オプションと交換することを提案し,交換割合は
| 2023 | 2022 | |||||||
| 普通株の潜在価値(ドル) | ||||||||
| 相場(ドル) | ||||||||
| 期待変動率(%) | ||||||||
| オプションの予想条項(年) | ||||||||
| 無リスク金利(%) | ||||||||
F-28
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 付記12- | 株主権益(継続) |
| B. | 株ベースの報酬:(継続) |
株式オプション:(続)
付与日に2023年と2022年に付与されたオプションが示す公正価値総額は#ドルと推定される
2023年12月31日現在,未確認の補償コスト
はすべての未帰属株式分類株式オプションに関連しており,金額は$である
| 2023年12月31日までの年度 | ||||||||||||
| 量 オプション | 重みをつける 平均値 トレーニングをする 値段 | 骨材 固有の 価値がある | ||||||||||
| 期初未返済債務 | $ | $ | ||||||||||
| 授与する | ||||||||||||
| 没収/キャンセルされる | ( | ) | ||||||||||
| 代替選択権が付与されました | ||||||||||||
| 期限が切れる | ( | ) | ||||||||||
| 鍛えられた | $ | |||||||||||
| 期末未済債務 | $ | |||||||||||
| 期限終了時に行使できます | ||||||||||||
| 加重平均残存契約寿命−2023年12月31日まで− | ||||||||||||
株式承認証:
| 捜査命令 | 発行日 | 満期になる 日取り | 1回の演習 値段 一人当たり 情報を共有する: | --数量 の株 ごく普通である 在庫品 潜在的な 株式証書を承認する | ||||||||
| 科学創業者に発行された私株権証(以下参照) | | |||||||||||
F-29
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 付記12- | 株主権益(継続) |
| B. | 株ベースの報酬:(継続) |
株式承認証:(続)
| 2017年11月BiomXイスラエルが発表 |
| 十二月三十一日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| 研究と開発費、純額 | ||||||||
| 一般と行政 | ||||||||
会社は会社役員に付与されたオプションに関する株による報酬支出
,金額は$であることを確認した
F-30
BIOMX Inc.
連結財務諸表付記
1株当たりのデータは含まれていません
| 注13- | 研究と開発費、純額 |
| 十二月三十一日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| 専門サービスと下請け業者 | ||||||||
| 賃金および関連支出 | ||||||||
| 株に基づく報酬 | ||||||||
| 減価償却 | ||||||||
| 材料と用品 | ||||||||
| 賃料および関連支出 | ||||||||
| 他にも | ||||||||
| 負債の変動が少ない(付記10 D参照) | ( | ) | - | |||||
| 連携プロトコルからの収入減少(付記10 F参照) | ( | ) | ( | ) | ||||
| 国際投資協定からの支出削減(付記10 A参照) | ( | ) | ( | ) | ||||
| 付記14- | 一般と行政費用 |
| 十二月三十一日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| 賃金および関連支出 | ||||||||
| 株に基づく報酬 | ||||||||
| 専門サービス | ||||||||
| 出張費用 | ||||||||
| 賃料および関連支出 | ||||||||
| 保険料 | ||||||||
| 他にも | ||||||||
| 付記15- | 財務支出,純額 |
| 十二月三十一日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| 為替レートの違い | ( | ) | ( | ) | ||||
| 銀行預金利子収入 | ( | ) | ( | ) | ||||
| 銀行手数料その他 | ||||||||
| 外国為替契約損失 | ( | ) | ||||||
| ( | ) | ( | ) | |||||
F-31
BIOMX Inc.
連結財務諸表付記
(ドルは千単位で、1株当たりおよび1株当たりのデータは含まれていない)
| 付記16- | 所得税 |
| A. | 同社は米国連邦司法管轄区および州と地方司法管轄区に所得税申告書を提出し、各税務機関の審査を受けている。当社の2020年以降の所得税申告書は依然として開放され、審査されています。アメリカの法定連邦所得税率は | |
| B. | BiomX株式会社とRondinX株式会社はイスラエルで所得税申告書を提出します。彼らの2017年までの納税評価は最終的だと考えられている。イスラエルの法定所得税率は |
| C. | 2023年12月31日と2022年12月31日までBiomXイスラエル繰り越しイスラエルでの損失は約$です |
| D. | 経営陣は、会社の設立以来の累計純損失の歴史と、設立以来いかなる製品を商業化していないか、製品販売から何の収入も生じていない場合を考慮し、会社は繰延税金資産のメリットを実現できない可能性があると結論した。そのため、2023年12月31日までと2022年12月31日までの繰延税金資産について全額推定準備が設けられている。経営陣は各報告期間に肯定的で否定的な証拠を再評価する。 | |
| E. | 当社の政策は、所得税支出に不確定な税収状況に関する推定利息と罰金を記録することです。2023年12月31日と2022年12月31日まで、会社には確認されていない税務頭寸、課税利息または罰金の金額記録がありません。 |
| 12月31日まで | ||||||||
| 2023 | 2022 | |||||||
| アメリカの法定連邦所得税率 | ( | )% | ( | )% | ||||
| アメリカと外国の税率の違い | ( | ) | ( | ) | ||||
| 繰延税金資産推定値変動準備 | ||||||||
| 実際の税率 | % | % | ||||||
| 12月31日まで | ||||||||
| 2023 | 2022 | |||||||
| アメリカです | ||||||||
| イスラエル | ||||||||
| 12月31日まで | ||||||||
| 2023 | 2022 | |||||||
| 繰延税金資産: | ||||||||
| 純営業損失が繰り越す | ||||||||
| 研究と開発費、純額 | ||||||||
| リース責任 | ||||||||
| 他にも | ||||||||
| 繰延税金資産総額 | ||||||||
| 繰延税金負債: | ||||||||
| 使用権資産 | ( | ) | ( | ) | ||||
| 固定資産 | ( | ) | ( | ) | ||||
| 繰延税金負債総額 | ( | ) | ( | ) | ||||
| 推定免税額 | ( | ) | ( | ) | ||||
| 繰延税項目純資産 | ||||||||
F-32
BIOMX Inc.
連結財務諸表付記1株当たりのデータは含まれていません
| 付記17- | 1株当たり基本損失 |
| 12月31日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| 純損失 | ||||||||
1株当たり基本損失の計算方法は,当期純損失
を当期発行普通株の加重平均株式数,当社普通株無行使価格の全額承認株式証と当社普通株の全額既所得資本権証
,行権価格$で割った
2023年12月31日現在、基本1株当たり損失計算には
加重平均が含まれている
2023年12月31日現在の1株当たりの赤字は含まれていないと計算
2022年12月31日までの1株当たりの赤字は含まれていないと計算
| 付記18- | 企業改制 |
2022年5月24日,会社は会社の資本資源を拡大するとともに,会社が行っている嚢胞性線維化計画を優先し,br社のアトピー性皮膚炎計画を延期する会社再編を発表した
F-33
BIOMX Inc.
連結財務諸表付記1株当たりのデータは含まれていません
| 付記19- | 後続事件 |
| A. | 2024年1月18日、会社は最後の金#ドルを受け取った |
| B. | 2024年3月6日、当社はAPT及びその他の各方面と合併協定を締結し、APTは当社の完全子会社となった。より多くの 情報については、注釈1 Dを参照されたい。会計基準編纂特別テーマ805“企業合併”の開示要求に基づいて、会社は企業合併の影響に関する情報を提供しなければならない。以下の制限により、財務諸表を発行する際に、企業合併の初期会計計算が完全ではないため、当社 には、ASC 805−10−50−4およびASC 805−30−50−3が許可する上記情報は含まれていない。 |
| a. | 買収は2024年3月15日に完了したが、Form 10-K年報における年度財務諸表の提出日は2024年4月3日である。 |
| b. | 会社年度財務諸表10-Kレポート提出日まで、会社はAPTのすべてと最終財務データ を取得していません。 |
| c. | 当社はASC 805項目に必要な購入価格配分作業を完了していません。 |
| C. | 2024年3月6日、買収完了と同時に、当社はいくつかの投資家と証券購入協定を締結し、総収益は$とした
| |
| D. | 2024年3月19日、当社は定期ローンツール下のすべての定期ローンを前払いし、総額は$とした
| |
| E. | 2024年3月21日、RondinXはイスラエルの税務当局と2018-2022年の分担に関する合意に調印した。協定の結論は、RondinXの知的財産権と従業員が買収の日にBiomXイスラエル社に移転されたということだ。したがって,RondinXの資本収益はその繰越損失1ドルに相当する |
F-34