
アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
本財政年度末まで
あるいは…。
手数料書類番号
(登録者の正確な氏名はその定款に記載)
|
|
|
(明またはその他の司法管轄権 会社や組織) |
|
(税務署の雇用主 識別番号) |
|
||
(主にオフィスアドレスを実行) |
|
(郵便番号) |
登録者の電話番号、市外局番を含む:(
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
|
取引 記号 |
|
登録された各取引所の名称 |
|
|
♪the the the |
同法第12条(G)により登録された証券:なし
登録者が証券法規則405で定義されている経験豊富な発行者である場合は、再選択マークで示してください。はい。☐
登録者が当該法第13条又は第15条(D)に従って報告書を提出する必要がないか否かを、再選択マークで示す。はい。☐
登録者が(1)過去12ヶ月以内(または登録者がそのような報告の提出を要求されたより短い期間)に、1934年の証券取引法第13または15(D)節に提出されたすべての報告を提出したかどうか、および(2)このような提出要求を過去90日以内に遵守してきたかどうかを、再選択マークで示す
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
大型加速ファイルサーバ |
☐ |
|
ファイルマネージャを加速する |
☐ |
☒ |
|
規模の小さい報告会社 |
||
|
|
|
*新興成長型企業 |
|
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に第240.10 D−1(B)条に従って受信されたインセンティブベースの補償に従って回復分析を行う必要があるかどうかを再選択マークで示す
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)。はい、違います
2023年6月30日まで(登録者が最近完成した第2財期の最終営業日),登録者の非関連会社が保有する投票権と無投票権普通株の総時価は約$である
2024年3月20日までに
引用で編入された書類
このForm 10−K年次報告の第3部は,登録者が2024年年度株主総会に提出する最終依頼書のいくつかの情報を引用しており,登録者は,登録者が2023年12月31日の財政年度終了後120日以内に第14 A条に基づいて証券取引委員会にこの声明を提出しようとしている。引用により本10-Kテーブルに明示的に含まれる情報を除いて,依頼書は本10-Kテーブルの一部として提出されるとはみなされない.
監査役事務所ID: |
監査役の名前: |
監査役位置: |
カタログ表
|
|
ページ |
|
|
|
第1部 |
|
6 |
第1項。 |
業務.業務 |
6 |
第1 A項。 |
リスク要因 |
39 |
項目1 B。 |
未解決従業員意見 |
92 |
プロジェクト1 C。 |
ネットワーク·セキュリティ |
92 |
第二項です。 |
属性 |
93 |
第三項です。 |
法律訴訟 |
93 |
第四項です。 |
炭鉱安全情報開示 |
93 |
|
|
|
第II部 |
|
94 |
五番目です。 |
登録者普通株市場、関連株主事項及び発行者による株式証券の購入 |
94 |
第六項です。 |
[保留されている] |
94 |
第七項。 |
経営陣の財務状況と経営成果の検討と分析 |
95 |
第七A項。 |
市場リスクの定量的·定性的開示について |
107 |
第八項です。 |
財務諸表と補足データ |
107 |
第九項です。 |
会計と財務情報開示の変更と相違 |
107 |
第9条。 |
制御とプログラム |
107 |
プロジェクト9 B。 |
その他の情報 |
107 |
プロジェクト9 Cです。 |
検査を妨害する外国司法管轄区域を開示する。 |
108 |
|
|
|
第三部 |
|
109 |
第10項。 |
役員·幹部と会社の管理 |
109 |
第十一項。 |
役員報酬 |
109 |
第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
109 |
十三項。 |
特定の関係や関連取引、取締役の独立性 |
109 |
14項です。 |
チーフ会計士費用とサービス |
109 |
|
|
|
第4部 |
|
110 |
第十五項。 |
展示品と財務諸表の付表 |
110 |
第十六項。 |
表格10-Kの概要 |
112 |
|
|
|
サイン |
|
113 |
2
前向き陳述に関する特別説明
このForm 10-K年間報告書は前向きな陳述を含んでいる。展望性陳述は主に“リスク要素”、“経営層の財務状況と経営結果に対する討論と分析”と“業務”と題する部分に含まれる。これらの陳述は未来の事件或いは私たちの未来の財務表現と関係があり、既知と未知のリスク、不確定要素とその他の要素に関連し、これらのリスク、不確定要素とその他の要素は私たちの実際の結果、表現或いは成果は展望性陳述と明示或いは暗示する未来の結果、表現或いは成果と大きく異なることを招く可能性がある。前向きな陳述は、以下の態様に関する陳述を含むが、これらに限定されない
場合によっては、これらの陳述は、“予想”、“信じ”、“可能”、“推定”、“予想”、“意図”、“可能”、“計画”、“潜在”、“予測”、“プロジェクト”、“すべき”、“将”、“将”またはこれらの用語の否定、および未来のイベントまたは結果に対する不確実性を表す同様の表現によって識別することができる。これらの展望的陳述は、我々の経営陣の未来の事件に対する信念、意見、見方を反映しており、本年度報告10-K表までの日に私たちに提供された推定、仮説、情報に基づいており、これらの情報はこのような陳述の合理的な基礎を構成していると考えられるが、このような情報は限られているか不完全である可能性があり、私たちの陳述は、私たちがすべての潜在的に利用可能な関連情報について詳細な調査または検討を行ったことを示すものと解釈されてはならない。このような陳述は本質的に不確実であり、危険と不確実性の影響を受ける。私たちはその中の多くのリスクについて“リスク要因”のタイトルでもっと詳しく議論するつもりだ。しかも、私たちの運営環境は競争が激しく、変化が迅速だ。新機能
3
リスクが時々出てくる。私たちの経営陣はすべてのリスクを予測することはできませんし、すべての要素が私たちの業務に与える影響を評価することもできません。あるいは任意の要素や要素の組み合わせは、実際の結果が私たちが行う可能性のある任意の前向きな陳述に含まれる結果と大きく異なる程度をもたらす可能性があります。このような不確実性を考慮して、あなたはこのような前向きな陳述に過度に依存してはいけない。
あなたはこのForm 10-K年間報告書と私たちがここで引用して証拠品としてForm 10-K年間報告書に完全に提出した文書をよく読んで、私たちの未来の実際の結果が私たちが予想していたものと大きく異なる可能性があることを理解しなければなりません。我々は,これらの警告的声明により,本年度報告の10-K表中のすべての前向き陳述を限定した.
法的要件がない限り、私たちは、新しい情報、未来のイベント、または他の理由でも、新しい情報、未来のイベント、または他の理由でも、これらの前向きな陳述を公開的に更新する義務がない、または実際の結果を更新することは、任意の前向きな陳述において予期される結果と大きく異なる可能性がある。
リスク要因の概要
本節で“リスク要因”のタイトルでより全面的に説明されているように、私たちは多くのリスクと不確実な要素に直面している。以下にそのいくつかの危険と不確実性をまとめた。以下の要約には、あなたにとって重要である可能性のあるすべての情報は含まれていません。この要約および“リスク要因”に含まれるこれらのリスクおよび不確実性のより詳細な議論を読むべきです
4
5
部分 I
プロジェクト1.バス純粋である。
概要
著者らは臨床段階の生物技術会社であり、免疫生物学に対する深い理解を利用して新しい治療法を開発し、深刻な自己免疫と炎症性疾患、或いは免疫炎症性疾患を治療し、高度に満足されていない医療需要を持っている。私たちの戦略の重点は潜在的にもっと多くの適応を求め、新しい候補製品とプラットフォームを獲得して、私たちのルートを拡大することを含む、私たちの候補製品の臨床開発を推進することである。私たちは独立したり、パートナーを通じて私たちの候補製品を商業化したり、戦略取引を通じて他の方法で私たちのチャネルを貨幣化するつもりです。
著者らの現在の臨床段階の候補製品はEQ 101とitolizumab(EQ 001)を含む。EQ 101は一流の選択性三特異性合成ペプチドであり、IL-2、IL-9とIL-15を抑制するために設計され、これらは疾病を引き起こす重要なサイトカイン標的であり、一連の免疫炎症適応の中で満たされていない需要を解決することを目的としている。Itolizumab(EQ 001)は1種の一流のモノクロナル抗体であり、それは選択的に免疫チェックポイント受容体CD 6、CD 6に対して効果性T細胞或いはT細胞の調節に中心的な役割を果たすEFF細胞、活動と人身売買は、多くの治療領域でいくつかの免疫性炎症性疾患を推進した。
我々はまた、様々なサイトカインに対して選択的にEQ 302の臨床前開発を進めている他のポリペプチドベースの候補製品の発見および最適化に取り組んでおり、EQ 302は一流の経口IL-15およびIL-21二重特異性阻害剤である。著者らの一流の免疫資産の斬新と差別化パイプラインは皮膚科、胃腸科、リューマチ科、血液科、移植科学、腫瘍科と肺科を含む多くの領域の満たされていない医療需要を満たす可能性がある。
我々は,多くの重篤な免疫炎症疾患を治療する潜在的最適疾患修正療法として,EQ 101,EQ 302,itolizumab(EQ 001)の開発に焦点を当てている。以下の図に示すように,われわれは現在EQ 101とitolizumab(EQ 001)の積極的な臨床開発計画があり,EQ 302の臨床前開発を進めている。 われわれの現在の個々の臨床計画の詳細は,以下の“われわれの現在の目標適応”の節で概説する。
6
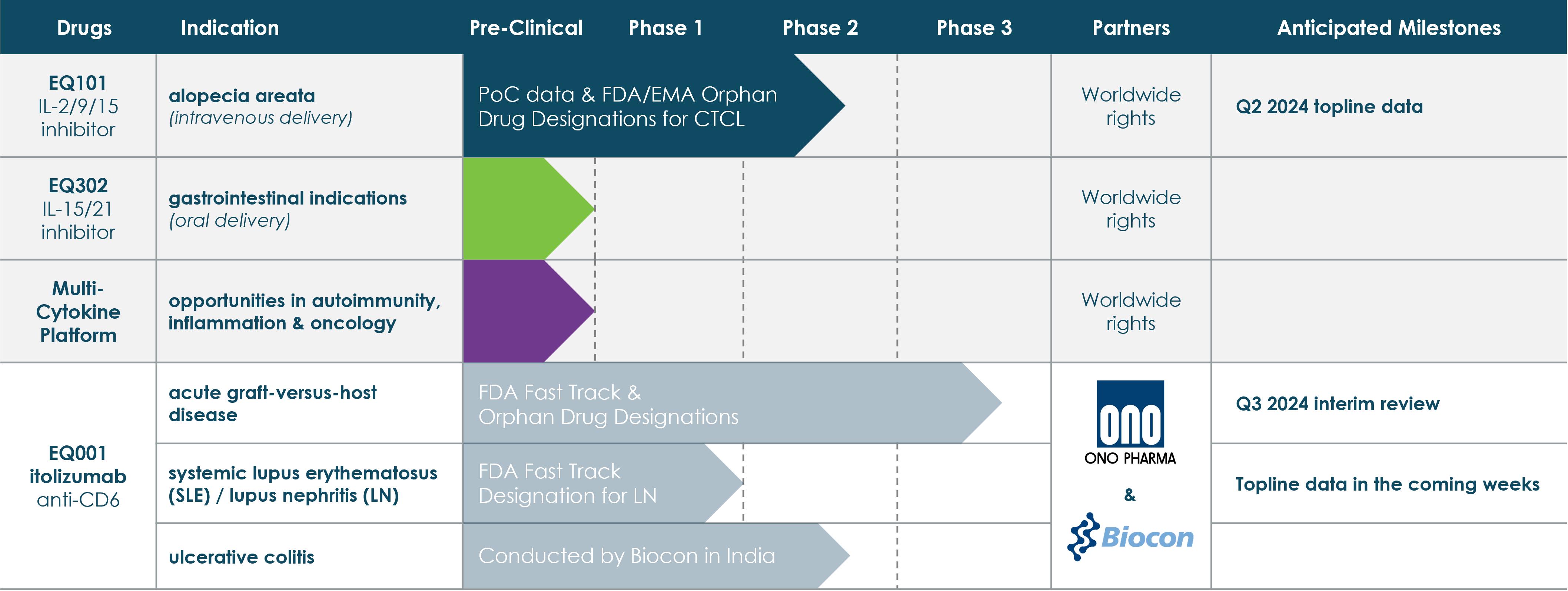
戦略.戦略
我々の目標は,先進的で完全に統合されたバイオテクノロジー会社となり,重篤な免疫炎症疾患の治療に専念することである。私たちの目標を達成するためには
採掘する
7
2022年2月、我々は、Bioniz Treateutics,Inc.,またはBionizを買収することによって、EQ 101のグローバル独占権利を取得し、EQ 302のような追加的で新規な多サイトカイン候補製品を発見する独自のプラットフォームを取得した。その買収は私たちの免疫学ルートを拡大し、一連の開発段階で一流の免疫炎症候補製品を発売した。今回の買収の詳細については、本年度報告書に添付されている財務諸表6のForm 10-Kを参照されたい。
仲間関係
小野とのオプション契約
2022年12月5日、小野と資産購入協定を締結し、この合意に基づいて小野に独占的権利を付与したが、itolizumab(EQ 001)に対する私たちの権利、またはオプションを得る義務はない。これらの権利は、すべての治療適応と、米国、カナダ、オーストラリア、およびニュージーランドでitolizumab(EQ 001)を商業化する権利を含む。オプションの交換として、小野は35億円相当の前金である2640万ドルを一度に支払ってくれた。
小野が選択権を行使すれば、小野は50億円相当の金額を一度に支払うか、三菱UFG銀行による2024年3月21日の為替レートは約3310万ドルとなる。私たちは小野が2024年下半期にオプション行使決定を下すと予想している。私たちはいくつかの開発と商業化のマイルストーンを達成する際に1.014億ドルの資金を得る資格がある。
我々はitolizumab(EQ 001)のすべての研究と開発を担当し,2022年7月1日から選択期間まで小野社が四半期ごとに資金を提供している。早期終了しない限り,選択期間はLNのEqualise臨床研究の背線データとaGVHDの赤道第3期臨床研究の中期データ交付後3カ月で満了する。
小野はいつでも書面通知で“資産購入協定”を終了することができるが、限られた場合、小野は終了後しばらくイトカマブ(EQ 001)の研究開発費と支出を返済し続ける義務がある。小野がその選択権を行使しなければ,資産購入プロトコルと選択権は自動的に終了する.資産購入協定には、双方が重大な違約時の常習停止権と、いずれか一方が2025年12月31日までに取引が完了していない場合に資産購入契約を終了することを許可する外部日(有限調整可能)も含まれている。
資産購入プロトコルにはEquilliumとOnoに関する慣用的な陳述と保証が含まれている。さらに、私たちは、itolizumab(EQ 001)の開発および採掘に適した当社の業務に関連する肯定および消極的な経営契約を含む習慣的な義務および契約の制約を受け、直接的または間接的な販売、許可、または他の方法で私たちのitolizumab(EQ 001)計画または資産購入協定に従って購入される任意の資産の直接または間接販売、許可または他の処置を求める排他的義務を禁止し、限られた場合でなければ、慣例的な上限および免責額の制限を受ける。資産購入プロトコルのさらなる詳細については、本年報10-K表に掲載されている総合財務諸表付記9を参照されたい。
Bioconとの協力と許可協定
Biocon SA(後にBiocon Limitedに譲渡、または共同譲渡、Biocon)との協力および許可協定に基づいて、私たちは2017年5月にitolizumab(EQ 001)の権利を獲得した。このプロトコルおよびその後の修正またはBioconライセンスによれば、Bioconは、米国、カナダ、オーストラリアおよびニュージーランドまたはEquillium地域での開発、製造、製造、使用、販売、販売、要約、販売、輸入、または他の方法でitolizumab(EQ 001)およびitolizumab(EQ 001)を含むまたは含む任意の医薬組成物または製剤を使用することを可能にする独占的許可を付与する。Bioconとの私たちの協力は、EQ 001臨床および商業薬物製品の独占的供給プロトコル、または臨床供給プロトコルを含む。Bioconは現在インドの工場でitolizumab(EQ 001)を商業規模で生産しており,この工場は米国食品医薬品局(FDA)の規制を受けている。また,インドBiocon社が行っている潰瘍性大腸炎患者におけるitolizumabの第二段階臨床研究を共同で援助することにも同意した。
Bioconが私たちに与えた権利の代価として、私たちはBioconに2,316,134株の普通株を発行した。また、ある規制の承認を得た後、Bioconに合計3000万ドルの規制マイルストーン支払いを支払い、製品の初の商業販売と特定レベルの製品販売を実現する際に、Bioconに合計5.65億ドルの販売マイルストーン支払いを支払う義務がある。私たちはまた、私たち、私たちの関連会社、そしてアメリカとカナダでの許可者が毎年純販売しているBiocon製品に特許使用料を支払うことを要求されました
8
私たちと私たちの付属会社(ただし私たちの許可者ではありません)のオーストラリアとニュージーランドのBiocon製品の年間純売上高のパーセンテージは1桁の中央値から10代以下の2桁まで、それぞれの場合に応じて特定の状況に応じて調整されます。この地域の承認が私たちのいくつかの臨床研究のデータを含むか、または引用した場合、場合によっては調整される可能性があり、Bioconはまた、Equillium地域以外でのitolizumabの販売のために可能な割合で印税を支払う必要がある。Onoがitolizumab(EQ 001)に対する私たちの権利の選択権を得る権利を行使する場合、上記はBioconの記念碑的支払いおよび特許使用料を欠く可能性があり、Onoの責任となり、Equillium分野以外でitolizumab(EQ 001)を販売する潜在的な特許権使用料はOnoの権利となるであろう。Bioconライセンスおよび臨床供給プロトコルの詳細については、本年度報告にForm 10−K形式で含まれる合併財務諸表付記9を参照されたい。
私たちの方法の基礎を理解する:多サイトカイン抑制
特許発見プラットフォームを用いた新規化合物標的抑制疾患関連γCサイトカイン
著者らの独自の多サイトカインプラットフォームは合理的に設計された複合ペプチドを産生し、共有受容体レベルで選択的に重要なサイトカインを遮断し、病原性サイトカインの冗長性と協同作用を標的とし、同時に非病原性シグナルを保留する。
この方法は受容体レベルで多種のサイトカイン抑制を提供し、Janusキナーゼ或いはJAK阻害剤の広範な免疫抑制と標的外安全責任を回避することが期待できる。多くの免疫介在性疾患は同じ失調サイトカインの組み合わせによって駆動され、著者らはこれらの疾病を識別する重要なサイトカインは多種の自己免疫疾患に対して個性化された治療策略を制定できると信じている。
多くのモノクロナル抗体或いはモノクロナル抗体は単一のサイトカイン或いはサイトカイン受容体を対象としており、疾病過程中に多種のサイトカインに関連する場合、疾病の病理問題を完全に解決できない可能性がある。もう一つの治療方法は、疾患に関与しているか否かにかかわらず、多くのサイトカインが利用するシグナル経路を抑制するため、Janusキナーゼ/シグナル伝達および転写活性化因子、またはJAK/STATを抑制することである。したがって、このような化合物はしばしば深刻な副作用と関連がある。また,JAK阻害剤はJAK/STATシグナル経路のみを抑制するが,われわれのポリペプチドはPI 3 KやERKを含む他の経路を抑制することを目的としている。したがって,我々のポリペプチドは重要な下流シグナル経路をより選択的に完全に抑制する可能性があり,より納得できる治療レジメンに変換される可能性があると信じている。我々の特許ポリペプチドは、他の家族の正常な機能によって健康な免疫バランスを維持しながら、複数の疾患駆動サイトカインを選択的に阻止する。参照してください図1.
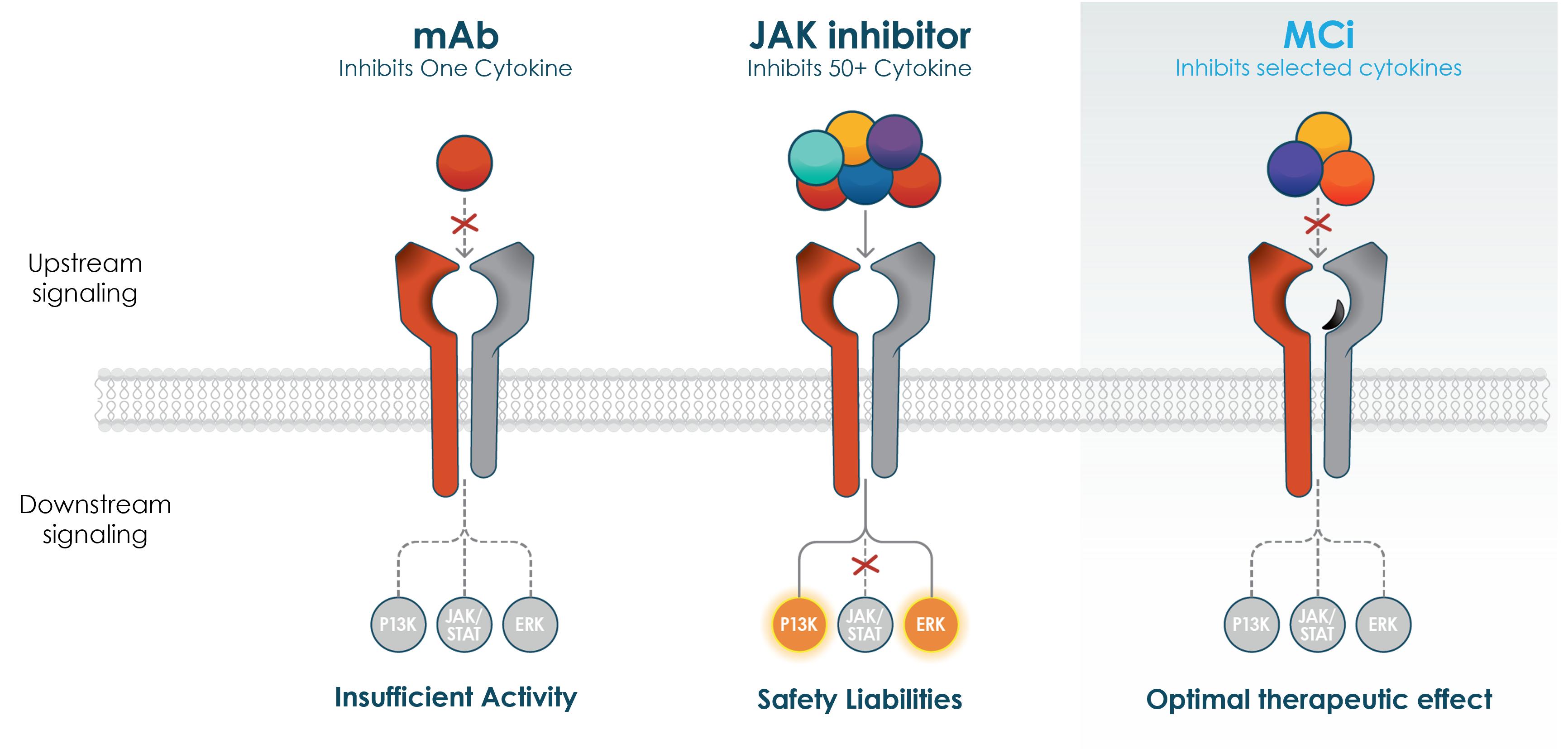
図1:モノクロナル抗体やJAK阻害剤と比較して、多種のサイトカインを合理的に設計、選択的に抑制することは最適な治療法であると考えられている。
9
Equilliumの複数のサイトカイン阻害剤は,ポケットを細胞内にバンドルすることにより,あるサイトカインの活性を選択的に遮断するgC共同受容体
EQ 101は一流の三特異性IL-2、IL-9とIL-15の阻害剤であり、この3種類の炎症性サイトカインは多種の疾患と関連している。それはこの3つの重要な病原性サイトカインを選択的に遮断し、同時に別の関連する非病原性シグナルを保持するgC サイトカインファミリーメンバーIL-4、IL-7およびIL-21(図2)である皮膚T細胞リンパ腫(CTCL)の1/2期臨床研究を通じて、EQ 101は新型の三特異性サイトカイン阻害剤であることを証明し、これは皮膚腫瘍学的指標である。この研究は安全性と耐性の主要な目標を実現し、修正後の重症度加重評価ツール或いはSWAT採点が臨床的に意義のある改善を示した。その研究では,良好な耐性と良好な安全性が証明され,薬物に関連する重篤な有害事象やSAEはなく,用量制限毒性もなく,臨床的に顕著な実験室異常もなかった。
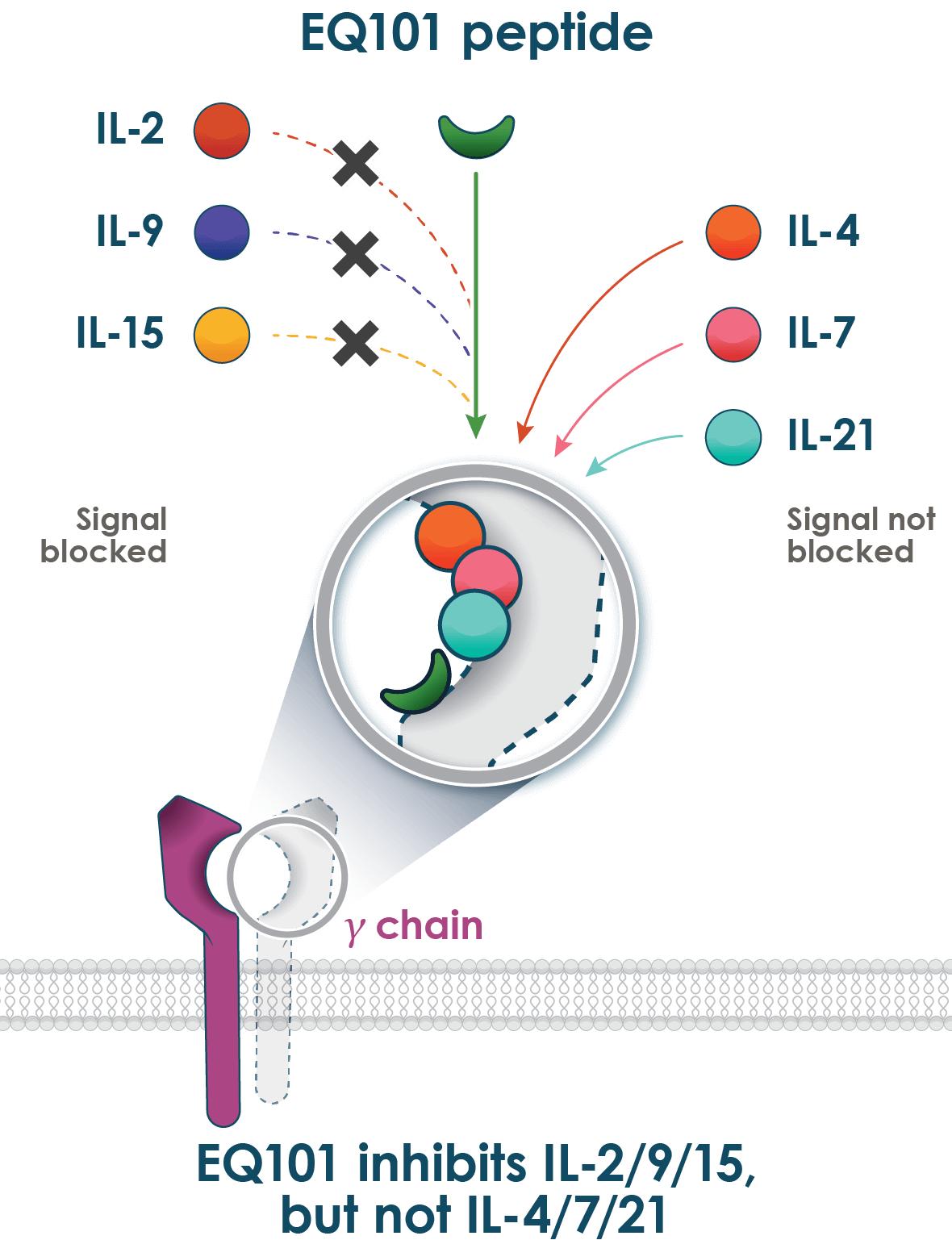
図2:EQ 101はIL−2,IL−9,IL−15を抑制するが,IL−4,IL−7,IL−21は抑制しない
EQ 302は、経口IL-15およびIL-21選択的阻害剤の一流である(図3)である。翻訳と臨床前データは乳糜潟、グルテン暴露に関連する免疫疾患を含む各種胃腸疾患を治療する潜在的な用途として支持されている。IL-15とIL-21抑制に対する高度選択性はこの2種類のサイトカインの重要な参与とよく結合し、この2種類のサイトカインの協同作用は乳糜潟と他の炎症性腸管と肝臓疾患の病理を推進した。
10
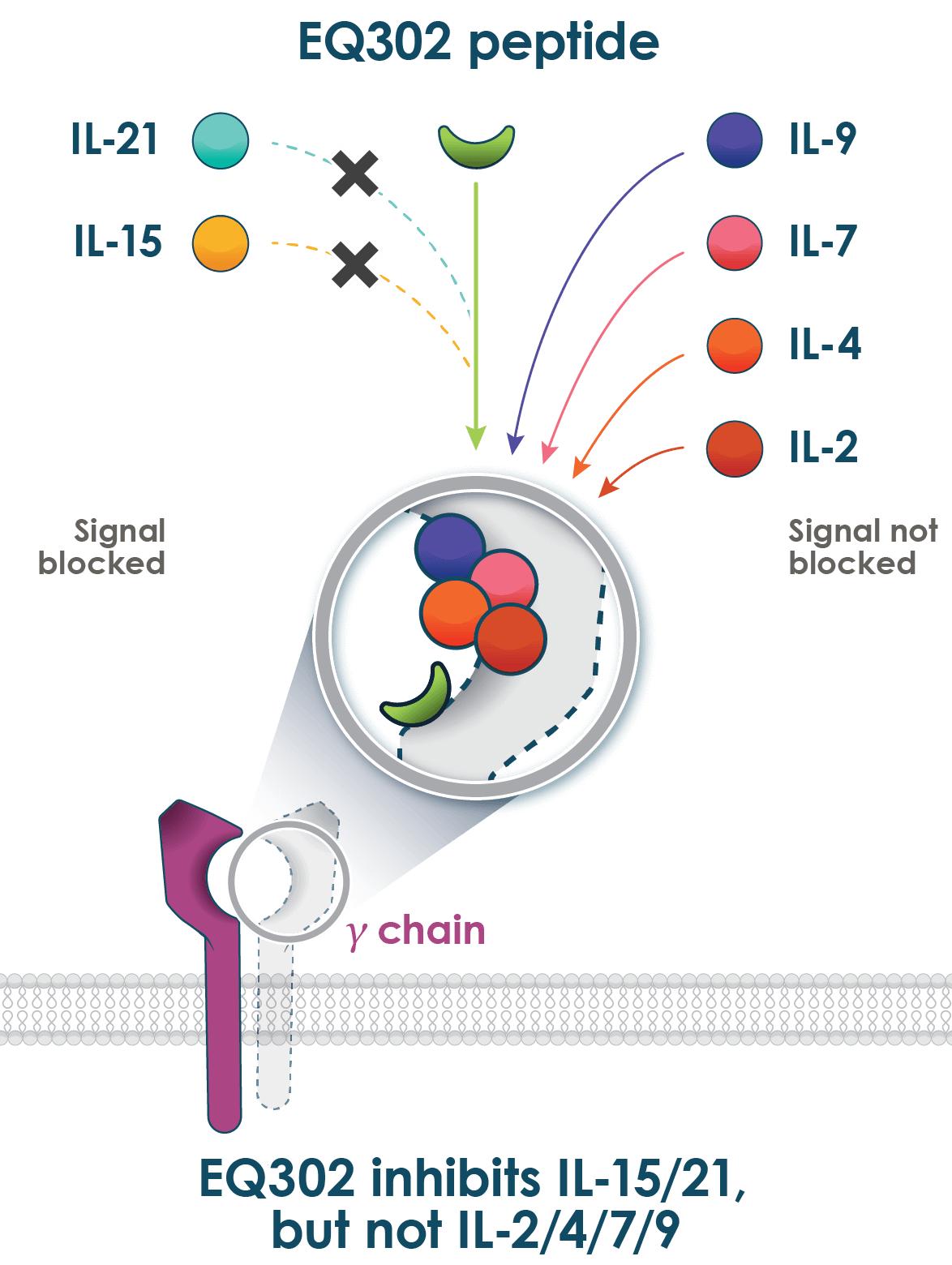
図3:EQ 302はIL−15およびIL−21を抑制するが、IL−2、IL−4、IL−7またはIL−9を抑制しない
EQ 101およびEQ 302の製品開発
EQ 101
EQ 101は合成ペプチドであり、部位特異的にポリエチレングリコールまたはポリエチレングリコール分子に共有結合している。この薬物は現在米国に位置する契約製造組織またはCMOによって良好な製造規範またはGMPに従って生産されている。EQ 101薬物製品は現在1種の凍結乾燥粉であり、静脈注射に用いられ、無菌注射用水(SWFI)と再調合した後である。われわれはEQ 101の皮下あるいはSC製剤を開発しており,後続の臨床研究に備えていく予定である。
EQ 101の臨床前概念検証研究はすでに免疫を介した脱毛動物モデルを用いて行われた。非肥満糖尿病、またはNOD、重症免疫不全、またはSCID、γc(CD 132)-/-(Nsg)マウスリンパ球欠損は、他の実験室で使用されているヒト化免疫介在性脱毛マウスモデルを確立するために使用されている(Sonntag 2015)。ヒト末梢血単核細胞を移植したヒト化マウスは重篤な全身性脱毛や脱毛などの症状を示した。重篤な脱毛の進展に伴い,移植後約45週間で動物は治療の研究を開始した。生存期間、体重、血清サイトカインレベル(すなわちIL-2、IL-6、IL-15、腫瘍壊死因子)を用いて治療反応を評価したaインターフェロンγ)、毛髪再生。
干与研究の治療効果の結果により、EQ 101によるIL-2、IL-9とIL-15シグナルの遮断は免疫介入型再障害マウスの毛髪の顕著な再生を招くことを発見した。また,EQ 101は炎症性サイトカインIL−6やインターフェロンγの循環レベルを低下させ,再生不良性貧血患者の免疫炎症過程の一部である。このマウスモデルでは,EQ 101は抗IL−2モノクロナル抗体,抗IL−15モノクロナル抗体,JAK 1/2阻害剤ruxolitinibよりも有効であるようである。これらの結果は,EQ 101が再生不良性貧血を治療する新たな潜在療法となることを示唆している。
Bionizはすでに3つの完成したヒト臨床研究においてEQ 101を評価した。一つは健康ボランティアの単一増加用量或いはSAD臨床研究であり、もう一つは健康ボランティアのクレイジーな臨床研究である。SAD研究ではEQ 101静注を受け,単回投与量は0.2,0.4,0.8,1.6,3.2または6.4 mg/kgであった。MAD研究では,被験者は0.5,1または1.5 mg/kgのEQ 101静注を週4回,または1週間に2または3 mg/kgのEQ 101を週4回受けた。この2つの研究では,43名の健常被験者が少なくとも1剤のEQ 101を受けた。EQ 101は耐性が良好と考えられ、死亡、深刻或いは深刻な治療-緊急有害事象、輸液反応或いは用量制限毒性、或いはDLTはない。
Bionizはまた大顆粒リンパ球性白血病(LGLL)或いは難治性皮膚T細胞リンパ腫(RCTCL)患者の開放ラベル1/2期用量範囲の臨床研究においてEQ 101を評価し、EQ 101の安全性、耐性、臨床治療効果とPK/PDを表現した。この研究では,50名の被験者を募集し,そのうち30名がrCTCL,20名がLGLLを有していた。4つのアップグレード
11
投与量はそれぞれ0.5 mg/kg,1.0 mg/kg,2.0 mg/kgと4.0 mg/kgであり,毎週静脈点滴し,計74週間であった。EQ 101耐性は良好で,DLTなし,輸液反応なし,死亡せず,不良事象で参加を中止した被験者は1名のみであった。EQ 101治療を受けた被験者は,IL−2およびIL−15依存細胞および炎症の減少,皮膚障害の改善および良好な全体応答率を示した。FDAと欧州薬品管理局(EMA)はそれぞれ2019年7月と2021年4月にEQ 101がCTCLを治療する孤児薬物名を授与した。
EQ 302
EQ 302は、検証された炭化水素製本技術を使用して、その特異性を維持し、魅力的な医薬製品プロファイルを達成しながら、ポリペプチドを安定化させる綴じポリペプチドである。経口投与が可能で腸内に安定して浸透しています著者らは現在EQ 302の臨床前薬理と調合開発を行い、候補製品を更に特徴づけ、最適化している。
我々の方法の基礎を理解する:CD 6-alcam経路抑制
自己免疫におけるCD 6の役割
免疫系の役割は,癌細胞を含む外来生物や細胞から身体を保護することであり,このようにする過程では,自己と非自己実体を正確に区別しなければならないという過程を耐受と呼ぶ。自己免疫は身体自己健康細胞と組織に対する免疫反応であり、多くの炎症性疾患の基本的な過程である。自己免疫はTエフェクター細胞あるいはT細胞間の関係不均衡による耐性喪失によるものであるEFF制御性T細胞やT細胞レジー細胞です。参照してください図4.

図4:自己免疫はバランス行動であるTレジー細胞はT細胞死の予防に重要な役割を果たしているEFF自己抗原を標的とする細胞は、自己免疫と組織破壊を招くことができる。
免疫検査点は免疫活性化経路の重要な調節因子であり、共刺激(活性化)であっても、共抑制(抑制)であってもよく、免疫バランスの維持と自己免疫の予防に重要である。共刺激検査点は免疫炎症性疾患を治療する魅力的な薬物標的であると考えられ,最近では免疫炎症性疾患の発展の焦点となっている。
CD 6は共刺激受容体であり、T細胞の活性化、増殖、分化と輸送過程を調節する上で不可欠な役割を果たしている。CD 6はT細胞を調節する重要なチェックポイントであるEFF自己免疫反応に重要な細胞です臨床前と臨床研究により、CD 6共刺激を遮断することは発病T細胞を選択的に抑制できることを表明したEFF細胞活動と人身売買、同時にT細胞の重要な調節機能を保留するレジー細胞です。これらの研究と潜在生物学に対する新しい知見は、CD 6が多種の免疫性炎症性疾患を治療する新しい標的であることを示唆している。
活性化された白血球接着分子,あるいはALCAMは,CD 6のリガンドであり,抗原提示細胞などの造血組織に発現し,これらの組織において,免疫シナプスの形成と最適な共刺激に重要である。AlcamとCD 6ドメイン3の結合はT細胞の活性化、増殖、分化と生存に関連するいくつかのマイトジェン活性化プロテインキナーゼ経路の下流活性化を招く。参照してください図5.
12
ALCAMは血管内皮細胞、血液脳関門、皮膚、肺、腎臓と腸管などの非造血組織にも発現し、CD 6を発現するT細胞の輸送を選択的に促進する。
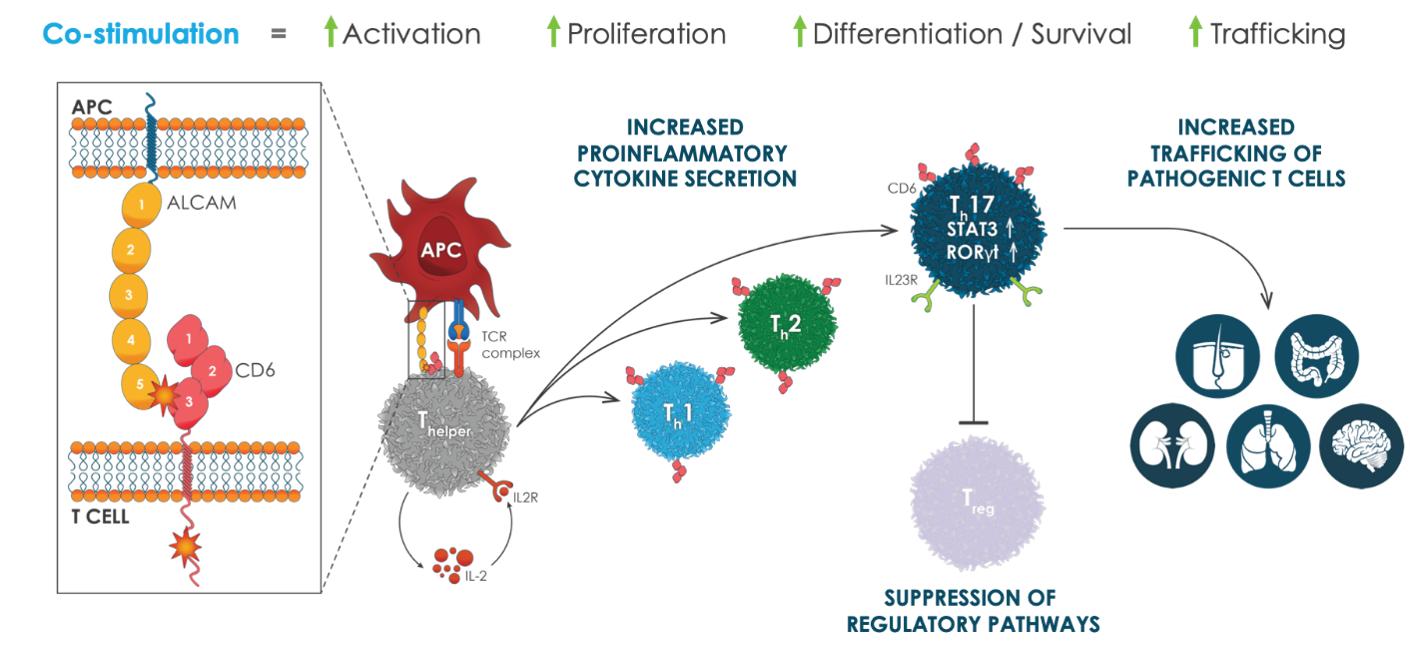
図5:CD 6共刺激駆動病原T細胞の発育と活性それは.共刺激はalcamとCD 6のドメイン3の結合により発生し,協同活性化を招き,IL−2受容体を介したT細胞の5倍に増加するEFF細胞が増殖する。共刺激はpSTAT 3とRoRγtを活性化し、IL-23 R発現の増加といくつかのT細胞の発病分泌を含む炎症促進反応を促進するEFF炎症性サイトカイン。AlCaMは皮膚、肺、腸管、血液脳関門と腎臓などの組織に発現し、選択的にT細胞の輸送を促進するEFFCD 6を発現する細胞。注目すべきはTh17細胞(ステロイド非感受性)および関連サイトカイン阻害T細胞レジー細胞活動は高Tを引き起こすh17:Tレジー慢性自己免疫疾患の比率特徴。
Tの変調EFFイトリズマブ(EQ 001)の細胞活性
Itolizumab(EQ 001)はヒトCD 6に選択的に結合し、CD 6とそのリガンドAlcamとの相互作用を抑制し、共刺激を防止し、Tを低下させるヒト化抗体であるEFF細胞活動と人身売買ですイトリズマブ(EQ 001)の臨床前研究により、CD 6の遮断はT細胞の減少を招くことが示唆されたEFFT細胞増殖を促進するいくつかの重要な経路の下方制御EFFTh 1,Th 2,Th 17などの細胞は発育している。重要なのは,CD 6を遮断して炎症を制御する重要な細胞経路の低下であり,STAT 3やRoRγtを含む。これらの経路の低下は炎症促進Tの分泌減少に伴うEFFサイトカインインターフェロン-γ、腫瘍壊死因子-α、インターロイキン6とインターロイキン17。
13
また,alcamとCD 6との結合を抑制することにより,itolizumabはリンパ球輸送を調節し,T細胞を減少させるEFF細胞は炎症組織に浸透する。その広範なマルチモード機序に基づいて、著者らはitolizumab(EQ 001)は既存の治療法に対する薬剤耐性或いは難治性疾患を含む多種の免疫性炎症性疾患を治療する潜在力を有すると信じている。参照してください図6.
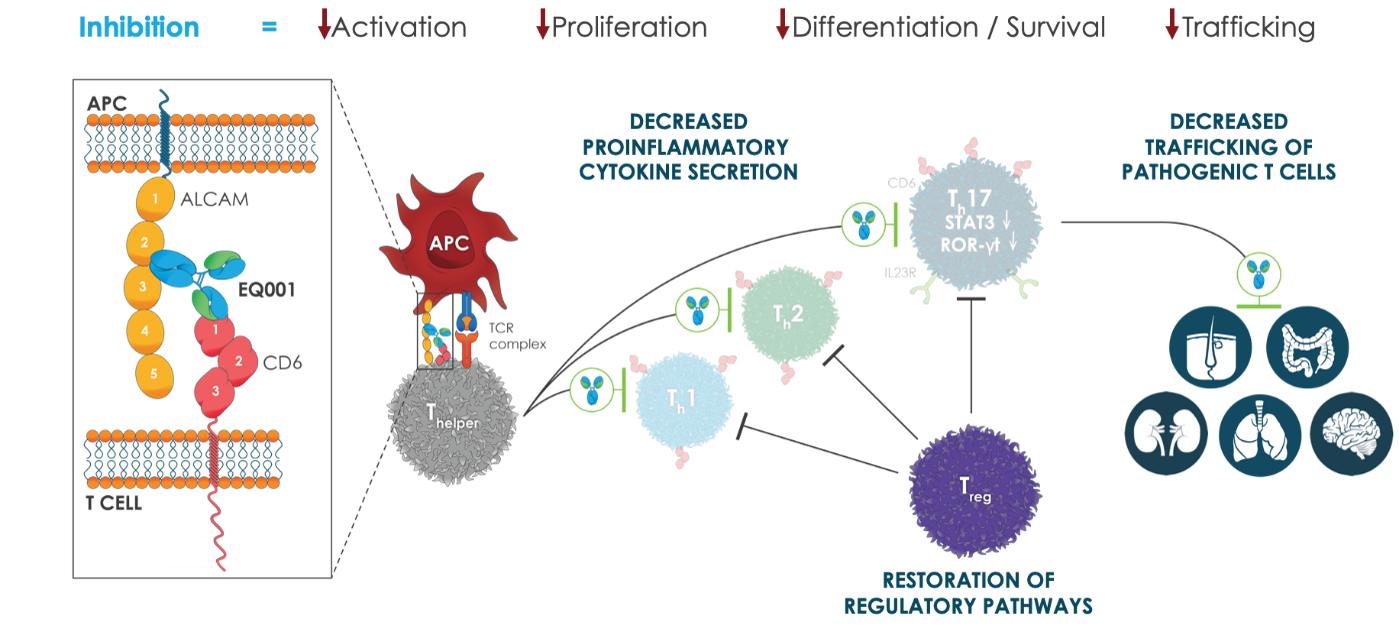
図6:イトリズマブ(EQ 001)CD 6阻害T細胞抑制EFF細胞が活性化·増殖する,差別化と販売。Itolizumab(EQ 001)はCD 6のドメイン1に選択的に結合し,alcamの相互作用を抑制し,共刺激を防止し,Tを低下させるEFF細胞が増殖する。CD 6のpSTATとRoRγtの低下を遮断し、IL-23 R発現と炎症性T細胞分泌の減少を招くEFFサイトカインインターフェロン-γ、腫瘍壊死因子-α、インターロイキン6とインターロイキン17。また,alcamとCD 6の結合を抑制し,リンパ球の皮膚,肺,腸管,血液脳関門や腎臓などの炎症組織への輸送を減少させる。T細胞数と活性の減少h17細胞がT細胞を抑制レジー細胞は免疫バランスを回復し、免疫寛容を促進する。
Biocon社のItolizumab(EQ 001)製品開発
イトーズマブはすでに臨床活性を示しており,関節リウマチ,乾癬,新冠肺炎に関連する急性呼吸窮迫症候群(ARDS)患者に対するBIOCONの臨床研究では耐性が良好であった。Itolizumabはインドで重度斑塊型乾癬の治療に許可されており,最初はBioconがアルツハイマー一次抗体のブランドで発売された。アルツハイマー病はNS 0細胞系で産生され,従来は静脈製剤でしか使用できなかった。Itolizumab(EQ 001)は中国ハムスター卵巣或いはCHO細胞株で産生された同じモノクロナル抗体配列を含み、静脈或いはSCで投与することができる。CHO細胞系は業界標準の抗体治療生産システムである。2020年9月、インド薬品監督管理局はインドBiocon社が生産したCHO細胞株製itolizumabのBioconを斑塊型乾癬の治療に使用することを許可し、同社がインドで販売している商標はアルツェモノ抗-L、あるいはアルツェモノ抗凍結乾燥化であり、そしてアルツェモノ抗-Lの緊急使用による新冠肺炎中の重度急性呼吸窮迫症候群患者のサイトカイン放出症候群の治療を許可した。アルツムモノクロナルはBiocon社によって生産されなくなり、商業販売にも使用されなくなった。Bioconは彼らのマーケティングと商業化努力をアルツェマブからアルツェマブ-Lに移行した。イトーズマブ(EQ 001)とアルツモノクロナル抗体-Lは同じ製剤の異なる薬物製品名である。
Itolizumab(EQ 001)は,BioconがインドFDAで規制されている製造工場で商業規模で生産されている。Bioconは業界標準の物理化学と生物機能表現方法を用いてデータを生成し、itolizumab(EQ 001)/アルツェマブ-Lとアルツェマブの分析生物比較性を証明した。
Bioconによって行われ、2017年第4四半期に完成したアルツェマブとイトリス抗体(EQ 001)に関する第1段階の研究において、健康ボランティアの中でIVとSCを同時に使用し、いくつかの被験者の中で一時的、可逆的な2~3級リンパ球計数の低下が観察され、臨床結果がなく、この研究は早期に終了した。臨床化学,血液学,尿分析パラメータについてはSAEは報告されておらず,他に臨床的意義のある異常や傾向は認められなかった。アルツハイマー患者のリンパ球数の類似低下はこれまで報告されていないが,従来の臨床研究では,血液学的評価の時間は十分早い時点では出現していない可能性があり,このような瞬時反応は検出できなかった。また,アルツハイマー病は従来,健常被験者ではなく活動期自己免疫疾患患者にのみ使用されていた。重要なことは,イトリズマブ(EQ 001)IVとアルツェマブのリンパ球減少幅は動力学と類似しているが,イトリズマブ(EQ 001)SCのリンパ球数の減少はSCとIV製剤の異なるPK特性に基づいて予想される。またアルズムモノクロナルは
14
インドで完成した関節リウマチと慢性斑塊型乾癬患者に対する臨床研究により、この薬の耐性は良好であり、4年間の投与量は0.2 mg/kgから1.6 mg/kgまで様々であることが示された。したがって,健常被験者の第一段階臨床研究で見られるリンパ球数の一過性低下はitolizumab(EQ 001)とアルツェマブのPD特性を代表しており,将来的にモニタリングされると考えられ,第一段階臨床研究の結果はitolizumab(EQ 001)SCとIVの炎症性疾患患者におけるさらなる臨床開発を支持している。
Bioconは現在インドで潰瘍性大腸炎患者に対するアルツハイマー−Lの第二段階臨床研究を行っており,この研究はEquilliumが共同で援助している。この研究は2022年11月に始まり、ランダム、二重盲検、プラセボ対照の臨床研究から構成され、90名の被験者に関連し、アルツハイマー-L治療中の重度潰瘍性大腸炎の安全性と有効性を評価する。
現在の目標適応は
円形脱毛症市場概要
AAは炎症性で瘢痕のない自己免疫を介した疾患であり,脱毛をきたし,重篤な疾患を有する人には治療選択が限られている。免疫系が身体の任意の毛髪成長領域の毛包を攻撃すると,このことが発生し,最も一般的なのは頭部と顔面である。全世界の再生不良性貧血の生涯発病率は約2%と推定され、罹患率は0.1%から0.2%と推定されている。再生不良性貧血はすべての人種や民族の男性と女性に影響を与える。小児や青少年では罹患率が高く,40%の症例が20歳までに発生し,80%の症例が40歳前に発生している。再生障害性疾患の発病機序はまだ完全に解明されていないが、一般的にはインターフェロンγ促進の一連のトリガーイベント(例えばストレス、感染、創傷)による毛包の免疫免除権を失い、それによって炎症性サイトカインシグナルの上昇を招き、それによって自己免疫を介した脱毛を招くと考えられている。再生不良性貧血は他の免疫介在性或いは自己免疫性疾患と関係があり、例えば甲状腺炎、白斑症とアトピー性疾患である。約50%の患者は12カ月以上持続する慢性再発と緩解性疾患を有し,約10%から35%の患者は最終的に頭皮毛髪の完全脱落(全脱毛)や頭皮と体毛の完全脱毛(全般性脱毛)を経験した。再生不良性貧血は1種の心理社会的負担があり、健康に関連する生活の質に重大な負の影響を与える可能性があり、そして抑うつと焦慮と関係がある。
EQ 101円形脱毛症治療の理論的基礎
IL-2、IL-9とIL-15は再生不良性貧血動物モデルとヒト再生不良性貧血病変組織における発現上昇が実証されている。研究により、CD 8+NKG 2 D+T細胞はAAマウスモデルの発病の必要条件と十分な条件であり、IL-2はCD 8+細胞の数を増加させ、IL-15は毛包上皮細胞のNKG 2 Dの発現とインターフェロンγの産生を増加させ、毛包上皮細胞はこれらのT細胞を異常に毛包を攻撃する細胞毒性T細胞に転化する。樹状細胞および/または毛包上皮細胞から放出されるIL−15はインターフェロンγ産生を促進する開始事象と考えられ,毛包の最初の免疫免除権を失い,IL−2を産生してT細胞増殖を引き起こす。マウスとヒト再障害皮膚に対する全世界転写スペクトルにより、遺伝子発現特徴は細胞毒性T細胞の浸透とIL-2とIL-15の上昇を表明し、すでにインターフェロンγのCD 8+NKG 2 D+効果T細胞の活性化と生存を促進することが証明された。また,IL−9はTh 2サイトカイン(IL−5,IL−6およびIL−13)の産生と分泌と増殖因子を形質転換する肥満細胞の病的活性化に関与しているbこれらの細胞毒性T細胞への毛包の抗原提示を促進することは,再生不良性貧血に関与している。
すでにいくつかの臨床研究が行われており,JAK阻害剤を用いて再障害を治療している。当初,tofacitinibとruxolitinibを用いた研究は,本来標準看護治療に無効であった少数の被験者で中等応答率を認めた。最近,いくつかの業界スポンサーの研究では,JAK阻害剤によるバリーチニブ,リライシチニブ,ドルソリーチニブによる再生不良性貧血の治療効果が観察された。ブラジルのチニブとリライシチニブはそれぞれ2022年6月と2023年6月にFDAによって再生不良性貧血の治療に許可された。
これらの臨床研究の結果とバリシチニブおよびリライシチニブの承認にもかかわらず、JAK阻害剤の治療効果はそれらのPK特性によって制限される可能性があり、これは、異なる用量間のJAKシグナル伝達の十分な遮断、およびJAKシグナル伝達に関連する追加のシグナル経路(PI 3 K、AKT、mTORおよびMAPK/ERK)をもたらす可能性があるgJAK阻害剤によって阻害されていないCサイトカインは、病的サイトカインシグナルの不完全遮断をもたらす可能性がある。また,JAK阻害剤は非疾患に関連するサイトカインシグナルを広く抑制するため,安全性に問題があり,その実用性や医師や患者に受け入れられることを制限する可能性がある。EQ 101はより選択的かつ有効なIL−2,IL−9およびIL−15(すなわち病原性サイトカイン)の阻害剤であり,JAK阻害剤よりも安全であり,AAの治療に新たな治療法を提供する可能性があると考えられる。
15
円形脱毛症の発展計画
AAとCTCLのEQ 101に2つの開放INDを提供した。2022年11月に第2段階の臨床研究を開始しました(図7)オーストラリアとニュージーランド再生不良性貧血患者のEQ 101。2023年12月,我々はこのマルチセンター,オープンラベル,概念検証臨床研究の36名の患者の募集を完了し,18歳から60歳の成人被験者であり,頭皮脱毛の少なくとも35%が再生不良性貧血によるものであることを発表した。被験者は毎週1回EQ 101を静脈注射し、投与量レベルは2 mg/kgであり、24週間連続し、その後4週間追跡した。この研究の主な目標は,EQ 101の24週間の治療期間中から重度再生不良性貧血患者に対する安全性と耐性を評価することである。第二の目標は、薬物治療効果、PK/PD特性を評価し、患者のバイオマーカーの変化を評価することである。私たちは2024年第2四半期にこの研究の裏線データを発表する予定だ。我々はEQ 101のSCレシピを開発しており,後続の臨床研究で使用する予定である。
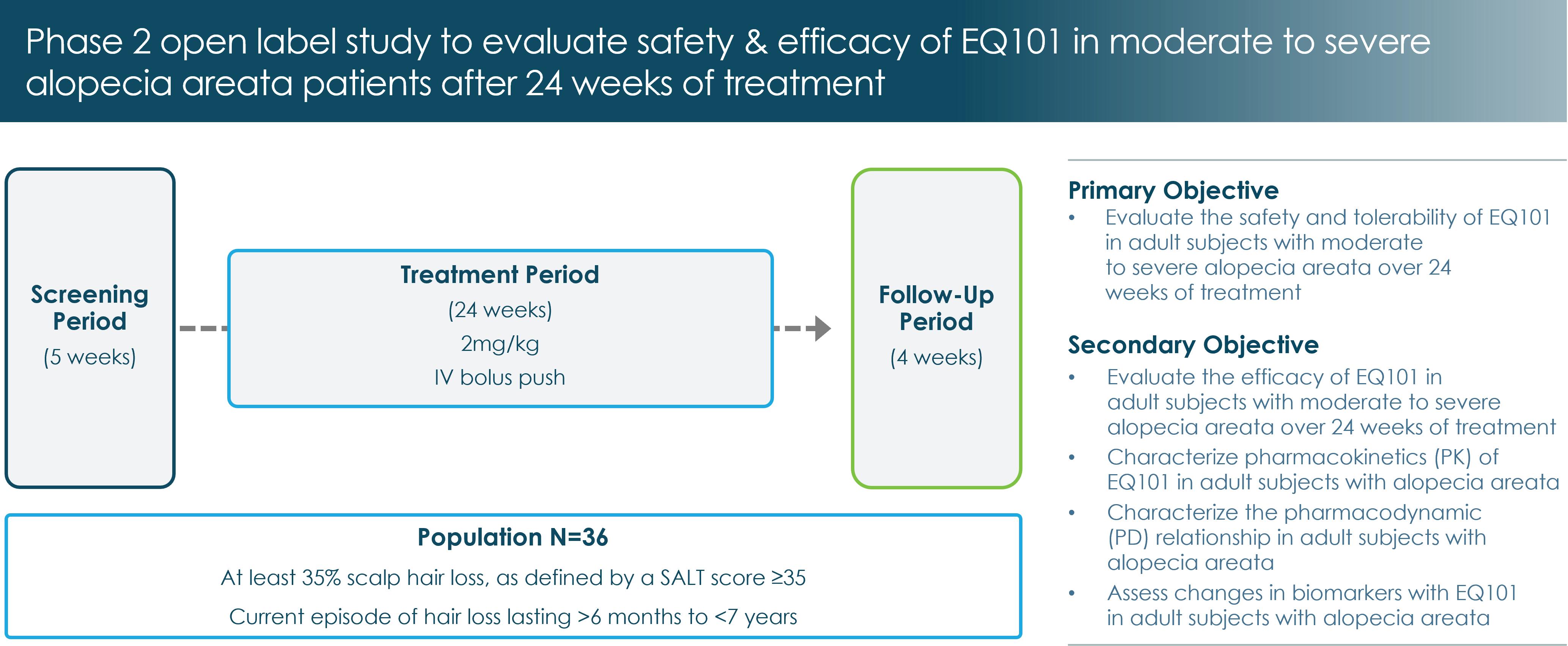
図7:EQ 101円形脱毛症治療の2期開放マーカー臨床研究
乳糜潟市場の概要
乳糜潟は1種の慢性炎症性腸管疾患であり、遺伝感受性個体の食事グルテンに対する不適切な細胞と体液免疫反応によって引き起こされる。乳糜潟は最もよく見られる自己免疫性疾患の一つであり、その罹患率は全世界人口の0.5%から1%であり、アメリカの約230万人に影響することが報告されている。過去50年間で乳糜潟の罹患率は上昇し,過去20年間で診断率も上昇している。乳糜潟は任意の年齢で発生することができるが、最もよく見られる発病は生命の最初の2年あるいは生命の第2あるいは第3の10年に発生する。乳糜潟はヒト白血球抗原(HL A)−DQ 2またはHL A−DQ 8遺伝子を発現する個体に選択的に発生する。乳糜潟では,腸上皮の粘膜炎症反応により絨毛萎縮や陰窩細胞が増殖する。乳糜潟では,粘膜完全性の喪失は,過剰な腸管や腸外疾患所見による高疾患負担に関与している。現在、まだ承認されていない乳糜潟の治療製品は、グルテンフリー飲食を厳格に遵守することが乳糜潟患者の現在疾病を管理する唯一の方法である。小児乳糜潟患者は通常完全な回復が観察されるが、40%を超える成人乳糜潟患者は飲食グルテンを完全に除去した後も組織学的異常--例えば絨毛構造損傷が存在する。グルテンフリー治療戦略をさらに複雑化させたのは,成人患者の5%が難治性の乳糜潟に進展する可能性があり,重篤な絨毛萎縮と異常上皮内リンパ球の存在が特徴であり,腸疾患関連T細胞リンパ腫の早期段階と考えられ,腸管上皮細胞に限らない潜在的致命的疾患である。
EQ 302腹腔疾患治療の理論的基礎
IL-15はすでに乳糜潟発症の重要な駆動因子と考えられている。IL−15は腸管固有層と上皮細胞で慢性的に上昇し,粘膜損傷の重症度と直接関連している。この炎症促進サイトカインは異なるタイプの細胞に作用し、多種の免疫機序の故障を招く。腸管固有層におけるIL−15の上昇は耐性樹状細胞の表現型変化を促進し,制御性T細胞やT細胞の産生が阻害されることが証明されたルール自己抗原に対する耐性を維持し、無害な食事抗原に対する耐性を促進するために重要であるリンパ球亜群。Tの約化を誘導するルール食抗原への依存は潜在的に向上しています
16
グルテン摂取に対する腸管炎症免疫反応。IL-15の発現はまた、細胞毒性リンパ細胞上で活性化されたNKG 2 D受容体及びそれに関連する細胞毒経路、及び上皮細胞表面に相補的な主要な組織適合性複合体I類鎖関連(MIC)リガンド(即ちMICAとMICB)の上昇と関係があり、それによって細胞毒性リンパ細胞のその後の殺傷を誘発する。
もう一つの乳糜潟に関連するγ−cサイトカインはIL−21である。IL−21は乳糜潟と強い遺伝子相関を示した。IL−21は腸管疾患におけるグルテン特異的CD 4 T細胞から産生され,腸管上皮内細胞毒性Tリンパ球(IE−CTL)細胞の溶解を促進する能力を有する。IL−21は活動性乳糜潟(腸管組織破壊を有する患者)にのみ過剰発現し,潜在的な乳糜潟(抗体反応のみで腸管組織損傷のない患者)では過剰発現はなかった。このことは,IL−21がIL−15とともに活動性乳糜潟で組織損傷を引き起こす可能性を示唆している。また,IL−21はB細胞分化や形質細胞産生の重要なサイトカインと考えられ,抗体反応を引き起こす。乳糜潟の1つの重要な特徴はトランスグルタミナーゼ2(TG 2)の自己抗体が存在することであり、この抗体はTG 2特異的なB細胞から産生される。小腸固有層へのIE−CTLの浸透に加え,固有層にも形質細胞増殖症の証拠があり,自己抗体産生の乳糜潟における潜在的な原因作用を強調している可能性がある。最近の研究では,腸管組織に対する細胞傷害免疫攻撃の活性化におけるCD 4 T細胞とB細胞間のクロストークの重要性が強調されている。IL−21は主要なBサイトカインとして,乳糜潟の抗体や細胞毒反応に重要な役割を果たしている可能性がある。IL−21を抑制することは,これらの患者の自己抗体産生を制御することができる。
さらにこの2種類のγ−cサイトカインの協同作用を支持する証拠は,IL−15が活動性乳糜潟患者からのIL−21分泌を促進する証拠を支持している。EQ 302はIL-15とIL-21の活性を特異的に抑制するが、家族中の残りのγcサイトカイン(IL-2、-4、-7或いは-9)を抑制せず、それによって乳糜潟中の重要な病原性サイトカインを標的とし、同時に他の不連続なγcサイトカインによって機能免疫系を保護する。EQ 302は独特な利点を有し、IL-15とIL-21の協同作用を抑制することによって、特定の二重管斉下の方法を提供することによって、乳糜潟におけるIELsの細胞毒活性を低下させることができると考えられる。EQ 302はBやT細胞アームに高度に選択的に作用することにより,アルコールタンパク質を介した炎症効果を制御する可能性が考えられる。
腹部疾患の発展計画
著者らは現在、体内薬理と調合開発を含むEQ 302の臨床前開発を行っており、候補製品を更に表現し、最適化している。積極的な結果を待つ前に,GMP製造や毒理学研究を含むEQ 302をより多くの臨床前開発に進める予定であり,これらの研究は潜在的なIND申請やヒト第一臨床研究への進展を支援することができる。
移植片対宿主病市場の概要
移植片対宿主病(GVHD)は1種の多系統疾患であり、同種造血幹細胞移植(allo-HSCT)のよく見られる合併症であり、移植された免疫系(より具体的にはT細胞)によって引き起こされるEFF細胞は受容者の身体を認識し攻撃します同種造血幹細胞移植を受けた患者では,移植片対宿主病が無再発死亡の要因である。GVHDのリスクはallo−HSCTを受ける患者の数やタイプを制限しており,GVHDリスクを低下させる治療法がallo−HSCT条件に適合した患者群を有意に拡大することができると信じられている。
国際血液·骨髄移植研究センターやその他の報告によると,2021年に米国では約8300例のallo−HSCTが行われ,2016年から2021年までの5年間で手術数は平均年間約1%のペースで増加している。新冠肺炎が大流行する前の2019年には,約8,600例の異遺伝子同種異体移植が行われた。Allo-HSCT受容者の約30~70%が移植片対宿主病を発症した。一線ホルモン治療に反応した患者の5年生存率は53%と低かったが,ステロイド難治性aGVHD患者では総5年生存率は5%と低かったことが報告されている。2021年のaGVHDの発病率は約4200名の患者であり、GVHDの総罹患率は約16000名の患者であると推定した。著者らは2030年までに、aGVHDの年間発病率は約4500名の患者に達する可能性があり、GVHDの総罹患率は約2万名の患者に達する可能性があると推定した。
イトーズマブ(EQ 001)による移植片対宿主病治療の理論的基礎
Itolizumab(EQ 001)GVHD発症機序を選択的に標的とする
安全、有効と的確なGVHD治療に対して非常に高い満足されていない医療需要がある。Tolizumab(EQ 001)はGVHDを標的とする潜在生物学的にGVHDを治療する最適な薬になる可能性があると信じている
17
高い選択性を持っていますまた,GVHD予防や慢性GVHD治療の将来を考えることにより,この方法も有望である。
既存の研究により、pSTAT 3シグナル駆動のTh 17細胞はaGVHDの発病機序において重要な役割を果たし、研究によりGVHD患者T細胞におけるpSTAT 3の発現は明らかに増加することが示唆された。AGVHDでは,aGVHDのないHSCT患者と比較して発症時のTh 17細胞やIL−17血清レベルが有意に上昇することがより多く報告されている。疾患の進展に伴い,Th 17細胞は末梢血からGVHD標的組織に入り,そこで障害を引き起こす。そのほか、aGVHD早期Th 17細胞の増幅はcGVHDへの移行過程において一定の役割を果たしている。GVHD患者ではTh 17:Tの上昇が示唆されているレジー耐性を失った割合を示す.循環中のTh 17細胞数の増加はT細胞の低下に伴うことに注意されたいレジー細胞はT細胞が失われることを示唆していますEFF細胞調節。この調節機序は同種異体反応性T細胞活性を除去するために重要であり、それによってGVHDの持続的な自己免疫反応と組織破壊を防止する。
Itolizumab(EQ 001)はaGVHDの潜在的発症機序を選択的に標的とする素子:a)T細胞を抑制すると考えられるEFF細胞増殖,b)原因Th 17細胞の進展に関連するSTAT 3経路を下方制御し,GVHDの発症を駆動する,c)T細胞の輸送を抑制するEFF細胞はGVHD標的組織に入り、更なる炎症と器官損傷を防止する;及びd)Th 17:Tを減少させるレジーGVHDの進行に関連する比率は,耐性を促進する。
CD 6を標的としたGVHD治療の第三者臨床経験
Itolizumab(EQ 001)の使用によるGVHDの治療を支持する臨床証拠は、以前に報告された第三者の臨床経験、すなわち血液系悪性腫瘍によって骨髄移植を受けた患者においてCD 6発現のT細胞が枯渇し、抗CD 6モノクロナル抗体を用いてドナー骨髄またはリンパ球注入中のT細胞を枯渇させることがaGVHDを予防する潜在力を有することが証明された。Soifferらは,ある研究において,ドナー異遺伝子骨髄にCD 6を発現するT細胞を選択的にインビトロで除去するT細胞の臨床効果を抗CD 6とウサギ補体のモノクロナル抗体を用いて評価した。報告によると、体外で抗CD 6モノクロナル抗体を用いてT細胞を除去することは有効に異遺伝子骨髄移植後の急性と慢性移植片対宿主病(GVHD)の発生率を下げることができ、移植に影響を与えない。
その後の研究はさらに、ヒト白血球抗原相合を受けた親族と非親族ドナーの異遺伝子骨髄移植患者におけるCD 6発現のT細胞除去の可能性を実証した。これらの研究において,CD 6を発現するドナー幹細胞製品の枯渇はGVHDを予防する唯一の方法である。抗CD 6モノクロナル抗体治療を受けた異遺伝子骨髄患者において、aGVHDの発生率は比較的に低く、これは混合リンパ球反応分析において、早期にCD 3を発現するTリンパ細胞が出現し、そのCD 6陰性表現型の同種異体刺激に対する反応性が低下したためである。これらの方法はCD 6をインビトロで発現するT細胞が枯渇する方法の一つであるが,さらにaGVHD発症機序におけるCD 6を発現するT細胞の役割を支持していると考え,CD 6をGVHDを調節治療する潜在的に重要な標的として検証した。
AGVHDにおける発展計画
我々とFDAのaGVHD INDは2018年7月に受け入れられた。FDAは、aGVHDの治療のためのTolizumab(EQ 001)の迅速チャネル指定を2018年12月に承認し、aGVHDの予防および治療のための孤児薬指定を2019年2月に承認した。
2019年3月に私たちはEquateを始めました, イトリズマブ(EQ 001)は第一線の連合ホルモンとしてaGVHDを治療する1 b期開放臨床研究である。EQUATE臨床研究において、著者らは完全寛解(CR)率、総有効率(OOR)、生存率とステロイド減量を含む安全性、PK、PDといくつかの臨床結果を評価した。
2023年2月,米国移植·細胞治療学会および国際血液·骨髄移植研究センターの直列会議でEQUATE臨床研究の最終安全性と有効性結果を公表した。計30名の被験者にイトリムマブ(EQ 001)を0.4、0.8または1.6 mg/kgの用量で服用した。Itolizumab(EQ 001)と全身コルチコステロイド治療の併用は迅速かつ持続的な高総臨床反応率と関係があり、その中の29日目の反応は1年以内の無進展生存期間の改善と関係がある。また,応答者は29日目にステロイドを70%,169日目に99%減少させることができた。イトーズマブ(EQ 001)は重症aGVHD患者における耐性は良好であった。これらの発見およびFDAと造血幹細胞移植領域のリーディング内科医からのフィードバックに基づき,われわれは2022年3月にEquatorを開始し,一線aGVHDの3期の重要な臨床研究である。
18
Equatorは無作為二重盲検臨床研究であり、itolizumab(EQ 001)とプラセボをaGVHDとコルチコステロイドの併用治療とする第一線療法の有効性と安全性を評価する。この研究の主な目標は早期疾患反応の実現であり,重要な副次的目標は反応の持続性,コルチコステロイドの使用,生存結果,cGVHD発生率の評価である。主要終点評価は29日目のCR率であり,キーサブエンドポイントは29日目の全体応答率またはOOR,および29日目から99日目までのCR率の持続性である。
赤道研究はitolizumab(EQ 001)とプラセボ(ランダム1:1)の静脈注射を第一線の治療の有効性と安全性と比較し、これらの患者は200名もの胃腸障害が比較的に低いIII-IV級aGVHD或いはII級aGVHDを有する成人と青少年患者を比較し、現在の治療標準-大量のコルチコステロイドと結合する。研究案によると,患者は初回大量コルチコステロイド投与後3日以内にitolizumab(EQ 001)治療を受けなければならず,治療期間は1−99日,フォローアップ期間は100−365日であった。条件に適合した被験者は,1日目にメチルプレドニゾロン2 mg/kgまたは同用量の治療を受け,A群:itolizumab(EQ 001),第1剤1.6 mg/kg,その後2週間に1回,6剤0.8 mg/kg(Q 2 W),全身コルチコステロイド(100例),またはB群:プラセボ,7剤Q 2 W,全身コルチコステロイド(100例)の2群にランダムに分けた。独立したデータ監視委員会は、安全データを定期的に審査し、約100人の被験者が29日目の評価を完了した後に中間分析を行い、治療の安全性および有効性を評価する予定である。私たちは中間検討が2024年第3四半期に行われると予想している。赤道研究の設計概要は#を参照図8.
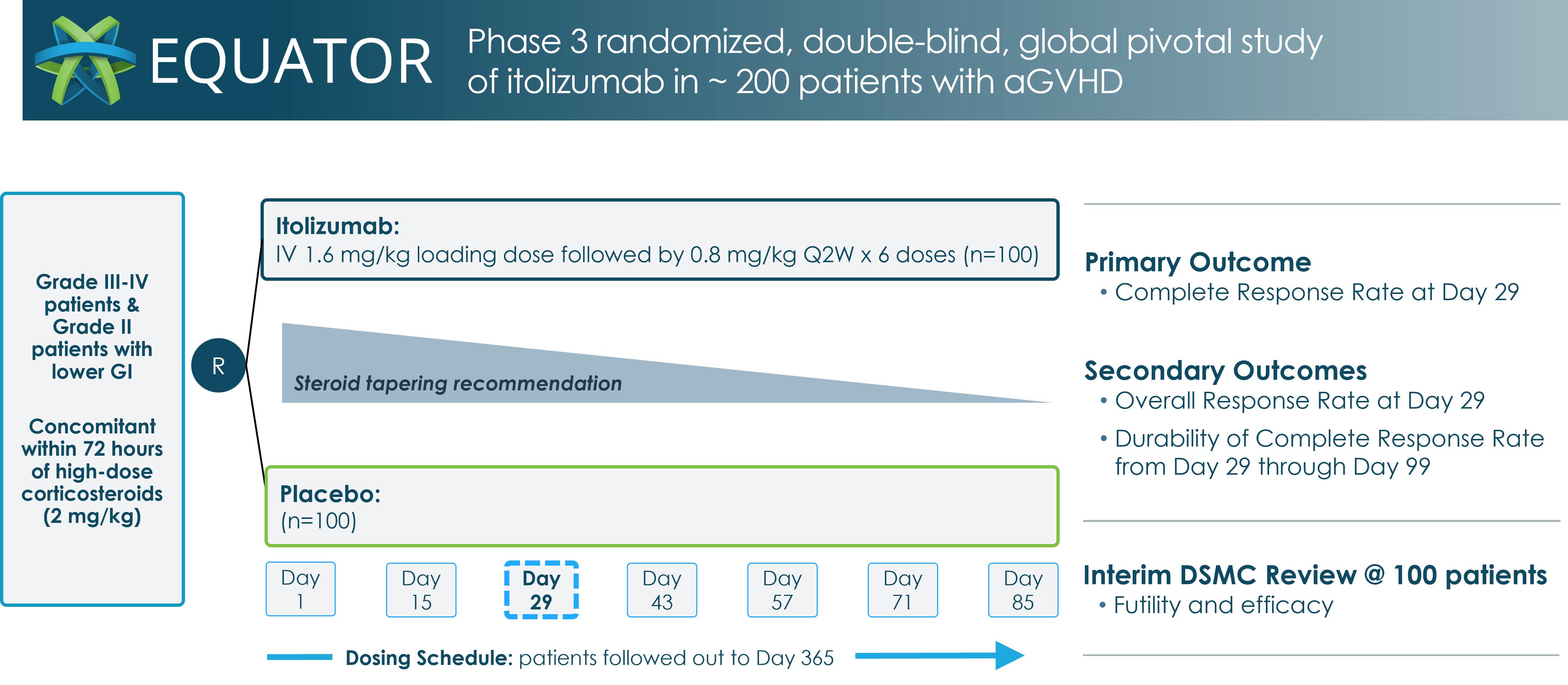
図8:赤道臨床研究設計概要
ループス市場の概要
全身性エリテマトーデスは異質性、多系統の自己免疫性疾患であり、その特徴は多種の自己抗体の存在と免疫複合体の異なる組織における沈着である。公開された出所によると,全身性エリテマトーデスは米国では25万から32.2万人に影響していると推定されている。
LNはSLEの最もよく見られる、最も深刻な表現であり、30%-60%に達するSLE患者に発生する。アメリカには10万人を超えるLN患者がいると推定されている;影響を受けた人数は非常に多いにもかかわらず、現在2種類のFDAが許可した薬物しかこの疾病を治療していない。
ループス腎炎の治療現状とその限界
現在、最も侵襲性のあるLNに対する標準治療方法は増殖性LN或いはIII或いはIV類LNと呼ばれ、広範な免疫抑制薬物、例えばプレドニゾロン、ミコフェノール酸エステル或いはMMF、及びシクロホスファミドを含み、これらの薬物は顕著な毒性を有する。LNは主に若い女性の1種の疾病であり、これらの薬物はいくつかの毒性を持ち、この人群にとって特に問題があり、体重増加、浮腫、月亮顔、感染リスク、糖尿病と不妊を含む。
これらの療法はLN患者の5年生存率を向上させたが,50%−75%までの患者は治療に無効であり,反応した患者は5年以内に再発する可能性がある。難治性や再発した患者では
19
誘導療法の予備治療には,どのような治療が有効である可能性があるかを支持するコンセンサスや強力な証拠はない。増殖性LN患者の予後は依然として悪く、40%に達する患者は末期腎臓疾患、或いはESRDに進展し、透析或いは腎臓移植が必要である。全体的に、LN患者の利用可能な選択はかなり限られており、特に標準誘導治療に無効あるいは再発した患者である。現在,LN治療のために2つの療法が承認されている。グラクソ·スミスクラインのBenlystaは2020年12月にLNへの使用が許可され,BLySに対して自己反応性B細胞の刺激を抑制する。Auinia PharmPharmticals,Inc.のS Lupkynisは2021年1月に承認され、1種のカルモジュリン阻害剤であり、IL-2の発現を遮断し、自己反応性T細胞を抑制することができる。これらの承認された製品にもかかわらず,より効果的で持続的な反応を維持し,より安全性の高い新しい療法が必要である。
エトーズマブ(EQ 001)ループス腎炎治療の理論的基礎
Itolizumab(EQ 001)選択的ターゲットTEFF狼瘡性腎炎発症機序に中心的な役割を果たす細胞
SLEやLNには自己抗体の形成や炎症性サイトカインが存在するにもかかわらず,B細胞ガイドと単一サイトカイン標的治療は臨床開発にほとんど失敗した。最近の証拠によるとTはEFF細胞はSLEとLNの発病機序において核心的な役割を果たしており、それらは組織損傷を媒介し、そしてB細胞の分化、増殖と成熟を促進することによって自己抗体の産生を増強する。複数のTEFF細胞/サイトカイン、例えばTh 1/インターフェロン-γ、Th 2/IL-4とTh 17/IL-17は、すべて全身性エリテマトーデスとLNの免疫発病機序に参与し、疾病の複雑性を強調した。しかし、Th 17細胞は重要な標的になりつつあり、既存の研究により、LN患者が免疫抑制治療を受けた後、高レベルのIL-17は不良な組織病理学的結果を示唆している。Th 17細胞レベルの上昇に伴うT細胞の減少レジー細胞は、このような機能免疫バランスの喪失がSLE患者の腎臓損害の発病機序に参与している可能性が示唆された。そこで,Tを目指してEFF細胞やT細胞を調節する分子EFFT細胞を保持しながら細胞活性をレジーSLEとLN患者に対して、ACTIVEは成功した治療策略であることが証明されている可能性がある。
Itolizumab(EQ 001)の独特な作用機序はLNの潜在的発症機序の素子を選択的に標的とすることができると考えられる:a)多種の病原性T細胞を抑制するEFF細胞とサイトカインの分泌;b)T細胞の輸送抑制EFF細胞が腎組織に入る;およびc)Th 17:Tの減少レジーLNに関する比率.
LNではItolizumab(EQ 001)の翻訳研究をサポートしている
Tの中心的な役割を考慮してEFF細胞はSLEやLNの免疫発症機序に作用し,itolizumab(EQ 001)はCD 6−alcam経路を遮断し,T細胞の活性や輸送を抑制すると考えられるEFF細胞は組織に変換され,この疾患を治療する有望な方法を代表している。この仮説を支持するために,SLEと糸球体腎炎動物モデルからの臨床前実験データは,抗CD 6モノクロナル抗体治療が炎症性サイトカインを低下させ,疾患活動性,蛋白尿,腎機能を改善したことを示している。また,LNにおけるitolizumab(EQ 001)を用いたCD 6-alcamパスウェイの標的化の有効性は,ヒト組織翻訳研究結果に支持され,発表されている臨床研究雑誌2022年1月に。この原稿は,CD 6−alcam経路が活性化したT細胞のLN発生における役割を証明するデータを強調し,LNを治療する潜在的な新しい療法としてitolizumab(EQ 001)を検討することを支持している。
ヒューストン大学が行った研究は、狼瘡研究連盟の目標識別支出によって支持され、活動期LN患者の尿液alcamレベルは明らかに上昇し、尿液alcamレベルは疾病活動と密接に関連していることを表明した。参照してください図9それは.これらのデータは更にLN患者におけるCD 6-alcam経路の潜在的な重要な発病作用を強調した。
20
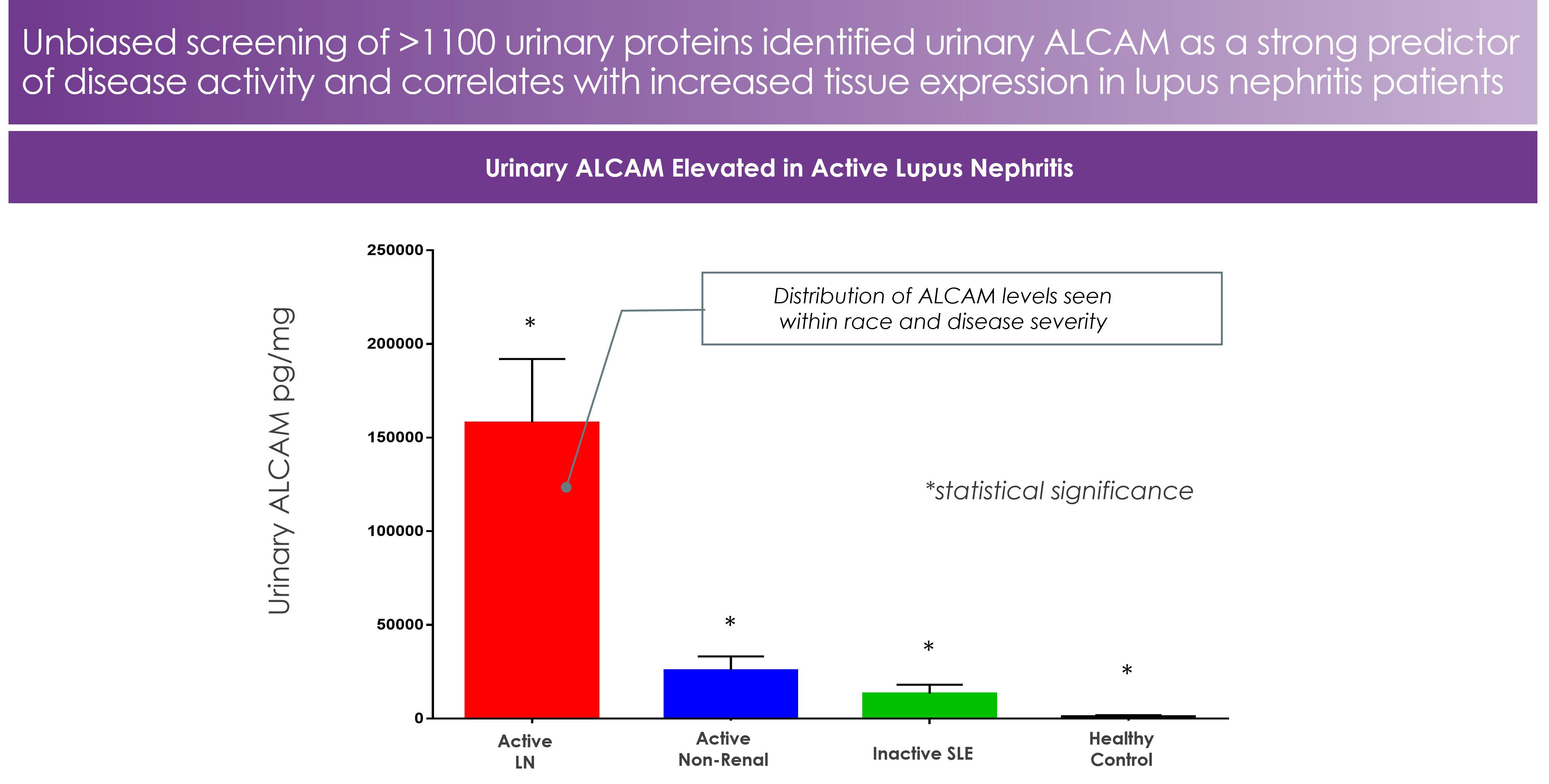
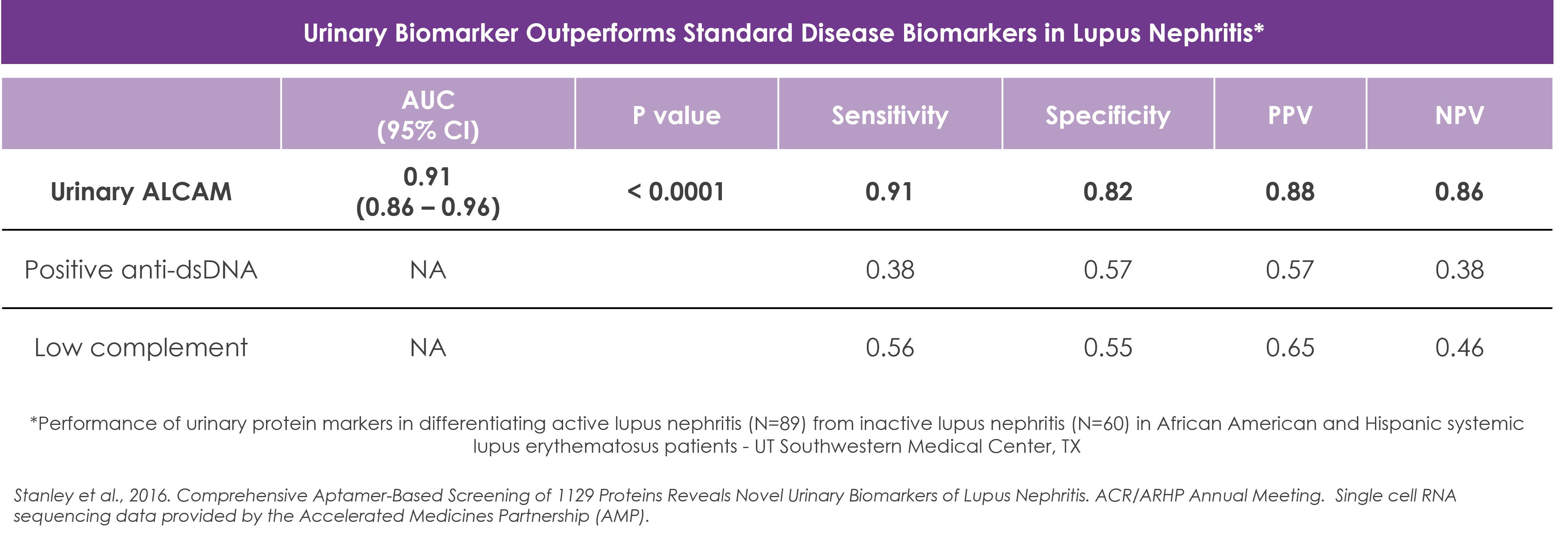
図9:ALCAMは活動期LN患者の予測バイオマーカーであるそれは.このグラフは活動期LN,活動期(非腎臓)SLE,非活動期SLEと健康対照群の尿中Alcamのレベルを描いている。ALCAMは活動期LN患者の中で最も高く、SLE患者は健康対照群より高かった。アフリカ系アメリカ人とスペイン系全身性エリテマトーデス患者における尿蛋白マーカーの活動期LN(N=89)と非活動期LN(N=60)の区別における発現を表に比較した。 |
標的検証に加えて,この尿バイオマーカーに関する研究は,我々がLNにおいてどのようにitolizumab(EQ 001)を開発するかに重要な意義を持つかもしれない。腎臓として尿を使用した非侵襲性液体生検の簡便性と拡張性は、LN患者を識別し治療する方法を潜在的に変化させる機会を提供した。CD 6-alcam経路のリアルタイム尿検出を用いて適切な患者を決定し、治療を指導し、疾患を監視するバイオマーカー誘導治療方法であって、標的治療を薬物承認まで進める機会を増加させ、患者ケアを著しく改善する可能性があるバイオマーカー誘導治療方法。具体的には、可溶性alcamやCD 6のような尿バイオマーカーの上昇は、itolizumab(EQ 001)に最も反応する可能性の高い患者を識別するために使用することができる。これらのバイオマーカーの評価は開発計画の重要な部分であり、itolizumab(EQ 001)を用いて個人化薬物バイオマーカーを探索する戦略の初歩的な基礎を形成する。
LNの発展計画
2019年7月、我々がループス/LNを治療したINDはFDAに受け入れられ、2019年12月にFDAはitolizumab(EQ 001)高速チャネルがLN治療のために指定されたことを承認した。2019年9月、著者らはSLEとLN患者のitolizumab(EQ 001)に対する1 b段階概念検証臨床研究であるEqualiseを開始した。A型部分はMAD研究であり、35名のSLE患者に関連し、0.4 mg/kgから3.2 mg/kg q 2 wまでのSC用量itolizumab(EQ 001)の安全性、耐性、PK、PDと臨床活性を評価する。この研究のB型部分はオープンタグで17個の新しいものに
21
確定診断或いは難治性LN患者はitolizumab(EQ 001)を用いて治療し、投与量は1.6 mg/kg SC Q 2 Wであり、治療コースは24週間に達した。1.6 mg/kg用量の選択は、A型研究部分の安全性、耐性、およびPK/PDデータの全体に基づいており、このデータは、1.6 mg/kg用量以上でCD 6細胞表面発現の低下がプラットフォーム期にあることを示している。A類研究と同じ目標以外に、B類研究は蛋白尿レベルとSLEDAI-2 K採点によりLN患者におけるitolizumab(EQ 001)の潜在的臨床活性を評価した。均衡化研究の設計概要を参照[図10].
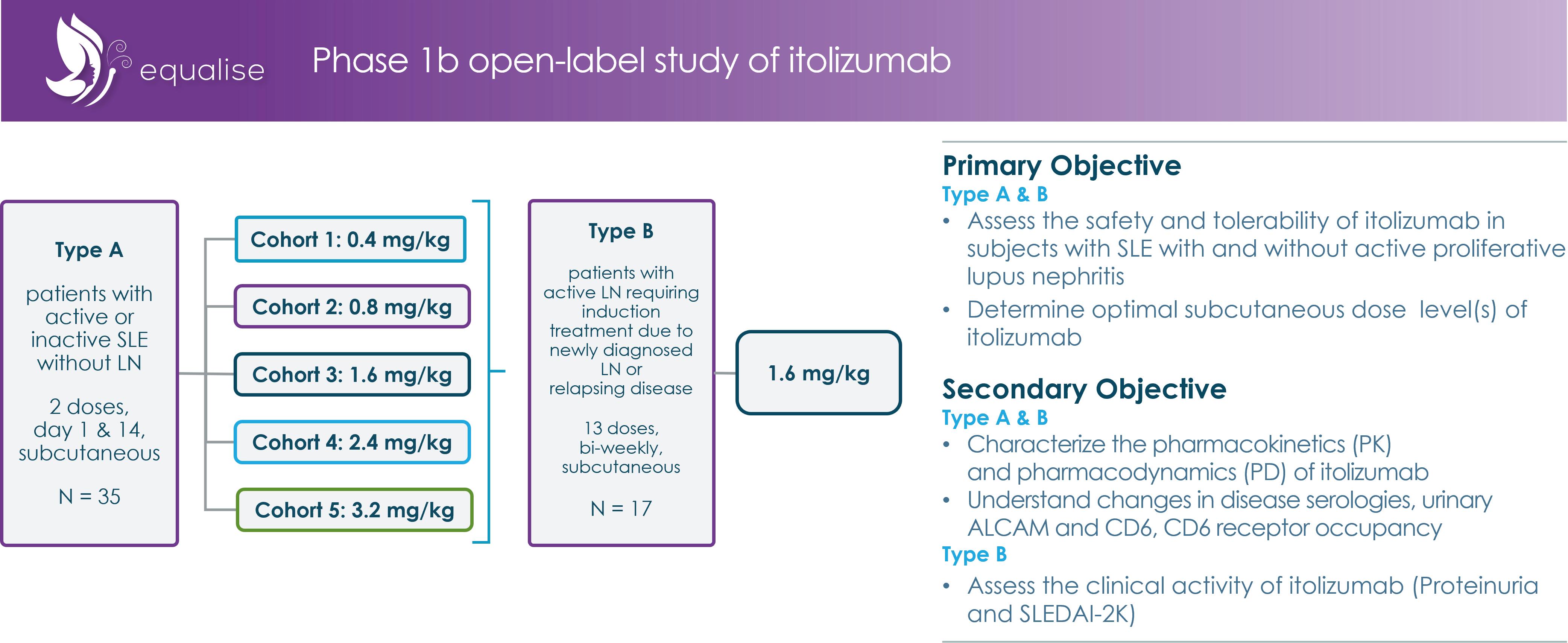
図10:EQUIZE臨床研究設計概要
2021年3月,全身性エリテマトーデス患者において平等な研究を行ったA型群の有利な背線データが報告され,イトリズマブ(EQ 001)耐性が良好であることが示された。また,itolizumab(EQ 001)は用量依存的にエフェクターT細胞表面CD 6の発現を減少させることを示し,その作用機序と一致している。
B群の研究登録が完了した後,2023年11月に米国リウマチ学会と米国腎臓病学会年次総会で公表されたこの研究部分のデータを公表した。これらのデータは,追跡期間中に最後の患者を除くすべての患者と,今後数週間で小野に提出される予定の背線データのプレビューを表している。データは,イベリズマブ(EQ 001)をMMFとコルチコステロイドとともに使用した場合,高い完全および部分応答率が生じ,高蛋白尿症被験者の尿蛋白クレアチニン比(UPCR)が平均4.9 g/gの場合,尿蛋白クレアチニン比率(UPCR)が急速かつ深さ低下したことを強調した。具体的には,28週になると73%の被験者のUPCR値が50%を超え,その40%が完全応答率であり,UPCR値は0.7 g/g以下に低下した。また,UPCR値の経時的低下と一致し,被験者は検討中に全身コルチコステロイドホルモンを徐々に減少させることができた。イトーズマブ(EQ 001)治療はリンパ球絶対数或いはALCの減少と関係があり、これは既知の薬効学的効果である。観察されたALCの低下は感染率の増加や他の不良臨床シグナルとは無関係であった。2人の被験者は少なくとも1つの深刻な有害事象を有し、調査者は、治療緊急有害事象の77%が軽度(レベル1)または中等度(レベル2)であると評価した。
知的財産権
私たちの知的財産権は私たちの業務に重要であり、私たちはそれを保護するために努力して、アメリカと国際的に私たちの候補製品、発見プラットフォーム、新しい生物発見、エピトープ、新しい治療方法と潜在的な適応、および私たちの業務のために重要な他の発明が特許保護を獲得し、維持することを含む。私たちの候補製品については、通常、私たちは物質組成と使用方法をカバーする特許保護を最初に求めるつもりだ。私たちの候補製品の開発過程全体において、私たちは、新たに発見された成分の改善、設計方法、治療方法、および他の治療目標に対するクレームを含む商業的成功を増強する可能性がある特許保護を得る他の方法を決定するつもりである。
2024年3月15日現在、私たちのitolizumabに関連する特許の組み合わせには、米国、オーストラリア、カナダ、ニュージーランドのBioconから独占的に許可された発表された特許および係属中の特許出願、および私たちが所有する特許協力条約(PCT)に基づいて提出された未解決の国際および国家段階特許出願が含まれている。Bioconライセンスの条項は、上の“業務-パートナーシップ-Bioconとの協力と許可協定”で議論されている。
22
具体的には、2024年3月15日現在、Bioconから取得したitolizumabに関する許可権には、米国で発行された9件の特許、オーストラリアで発行された6件の特許、カナダで発表された特許5件、ニュージーランドで発行されている特許6件、米国、オーストラリア、カナダ、ニュージーランドで出願されている特許が含まれている。我々が発行した5つの米国特許は、2028年に満了することが予想され(規制遅延のために特許期間を延長することはない)、itolizumab抗体配列に対する権利主張と、移植片対宿主病および移植片拒絶反応を含む様々なT細胞媒介性疾患および障害を単独または他の薬剤と共に製剤および使用して治療する方法とを含む。我々が発行する2つの米国特許は、特定のT細胞数が増加した患者に対してitolizumabを使用して多発性硬化症または炎症性腸疾患を治療する主張を含む2034年に満了することが予想される(規制遅延により特許期間を延長する場合はない)h17細胞であって、いくつかの方法がIL-23 Rの発現を監視することを含む、細胞。私たちが発行したオーストラリア、カナダ、ニュージーランドの特許は2027年から2034年の間に満期になる予定です。Bioconから得られた許可権利は、itolizumabを用いて狼瘡を治療する方法に関連する出願中の特許シリーズを含み、発行された米国特許と、米国、オーストラリア、カナダ、およびニュージーランドで出願されている特許とを含む。Bioconから得られた許可権は、米国およびカナダにおける係属中の出願およびオーストラリアおよびニュージーランドで予想される出願を含む、itolizumabを用いた高侵襲性医療プログラムの超炎症反応による細胞および臓器損傷を予防する方法に関連する係属中の特許出願シリーズをさらに含む。我々の係属中のライセンス内で特許出願が発行される可能性のある特許は,2028年から2042年の間に満了する予定であり,いかなる特許期限の調整や延長も行われない。
さらに、我々は、米国、オーストラリア、カナダ、およびニュージーランドで承認を待っているitolizumabを使用して重篤な喘息を治療する方法に関連する特許出願系列を有する。承認された場合、その特許家族によって発行される任意の特許は、2039年に満了することが予想され、いかなる特許期限の調整や延長もない。米国、オーストラリア、カナダ、ニュージーランドで承認を待っているitolizumab用量レジメン、バイオマーカー、インビトロ試験バッチ分析、および体外移植療法に関連する特許シリーズも持っている。承認された場合、その特許家族によって発行される任意の特許は、2041年に満了することが予想され、いかなる特許期限の調整や延長もない。
私たちはまた、Tolizumabを用いたLN治療の診断方法に関する特許出願中の家族をヒューストン大学システムと共同で有し、現在、米国、オーストラリア、カナダ、およびニュージーランドで出願を待っている。承認された場合,その特許家族が発行するどの特許も2040年に満了することが予想され,いかなる特許期限の調整や延長もない。
2024年3月15日現在,Bionizの買収により,複合ペプチド拮抗薬に対する特許組み合わせを完全に有している。我々のこの分枝は、IL-2、IL-9、IL-15ポリペプチドアンタゴニストEQ 101、IL-15およびIL-21ポリペプチドアンタゴニストEQ 302、他のペプチド配列、および主にγc-サイトカインファミリー空間においてポリサイトカインシグナルをペプチド調節する他の関連技術に関連する特許出願シリーズを含む他の6つの特許出願系列を含む。
これら6つの複合ペプチド特許出願ファミリーにおいて、第1のファミリーは、EQ 101をカバーする複合ペプチドに対する現在の請求項、そのようなペプチドを設計する方法、およびそのようなペプチドを使用して様々なT細胞媒介性疾患および障害(RA、免疫媒介性脱毛および筋炎を含むがこれらに限定されない)を治療する方法を含む。この家族は現在、9つの米国特許、3つのオーストラリア特許、1つのカナダ特許、1つのブラジル特許、1つの中国特許、38の欧州国家特許、および2つの日本特許を含む。米国、中国、欧州、香港、日本の申請も待っている。承認された場合、この特許家族内のどの特許も2032年に満了することが予想され、いかなる特許期限の調整や延長もない。
我々の複合ペプチド製品の組み合わせにおける第2の特許出願シリーズは、他のマルチサイトカインファミリーポリペプチドアンタゴニストに対する現在の請求項、およびそれらの製造方法を含む。このシリーズは、発行された3つの米国特許と、出願中の米国特許を含む。承認された場合、この特許家族内のどの特許も2034年に満了することが予想され、いかなる特許期限の調整や延長もない。
第3の特許出願シリーズは、IL-15およびIL-21ペプチドアンタゴニストをカバーする現在の複合ペプチド、ならびに様々なT細胞媒介性疾患および障害(乳糜潟および炎症性腸疾患を含むがこれらに限定されない)を治療するための方法を含む、請求項1~3のいずれか一項に記載の方法。この一連の特許は、3つの米国特許、2つのオーストラリア特許、24の欧州国家特許、香港特許、1つの日本特許、1つのインド特許、および韓国特許を含む。米国、オーストラリア、カナダ、中国、欧州、香港、インド、日本、韓国の申請も待っている。承認された場合、この特許家族内のどの特許も2036年に満了することが予想され、いかなる特許期限の調整や延長もない。
第4特許出願シリーズは、EQ 302をカバーする複合ペプチドと、乳糜潟および炎症性腸疾患を含むがこれらに限定されない様々なT細胞媒介性疾患および障害を治療するための現在の方法とを含む、請求項1~4のいずれか一項に記載の方法。この一連には発行されたオーストラリア特許が含まれており、米国、オーストラリア、カナダ、中国、欧州、香港、インド、日本、韓国に未解決の出願がある。承認された場合、この特許家族内のどの特許も2038年に満了することが予想され、いかなる特許期限の調整や延長もない。
23
第5および第6の特許出願ファミリーは、円形脱毛症、サイトカイン放出症候群、およびそれに関連する疾患を含むが、これらに限定されない様々な治療標的を治療するためのEQ 101に対する現在の請求項を含む。全体的に、これらの家庭には、アメリカ、オーストラリア、ブラジル、カナダ、中国、ヨーロッパ、香港、インド、日本、韓国の保留申請が含まれている。承認された場合、この特許シリーズのいずれの特許も2040年または2041年に満了することが予想され、いかなる特許期限の調整や延長もない。
我々は,米国仮特許出願及び以前に提出された仮出願の優先日利益を有することを要求する米国非仮出願及びPCT出願(適用される場合)を提出する。仮特許出願は、米国でより低コストの初特許出願を提供するためのものである。該当する非臨時特許出願は,仮出願日の後12ヶ月以内に提出されなければならない。対応する非仮出願の利点は、特許出願の優先日(S)が早い仮出願日(S)であり、最終的に発行される特許の特許期間が遅い非仮出願日から計算されることである。この制度は,優先権日を早期に取得することができ,優先権年度内に特許出願(S)に材料を増加させ,特許期間の開始を延期し,起訴費用を延期することができ,出願を審査しないことを決定した場合に有用である可能性がある。PCT制度は、特許出願の最初の優先日から12ヶ月以内に単一出願を提出し、153個のPCT加盟国を指定することを可能にし、これらの国は後でPCTによって提出された国際特許出願に基づいてこれらの国で特許を出願することができる。PCT検索機関は、特許性検索を実行し、申請料を生成する前に外国国出願の成功機会を評価するために使用することができる拘束力のない特許性意見を発表する。PCT出願は特許として発行されていないが、出願人が国家段階出願を介して任意の加盟国で保護を求めることを可能にしている。特許出願の第1の優先権の日から2年半の期限が終了したとき、PCTのどの加盟国も、直接の国出願を通過することができ、または場合によっては、欧州特許機関のような地域特許機関を介して単独の特許出願を出願することができる。PCTシステムは費用を遅延させ、国/地域特許出願の成功機会の限られた評価を可能にし、出願が出願の最初の2年半以内に放棄された場合に大量の節約を実現した。
私たちは私たちの所有と許可中の未解決の出願を起訴し、私たちが重大な製品販売が発生する可能性が予想される重要な商業市場で特許発行と保護を求めるつもりです。
2009年の“生物製品価格競争と革新法”(BPCIA)の公布に伴い、生物類似と交換可能な生物製品を承認するために短い道を開いた。簡略化された規制経路は、FDAのために生物類似生物製品を審査および承認する法的権威を確立し、既存の参照製品との類似性に基づいて、生物類似体を交換可能なものとして指定することを含む。BPCIAによれば、生物類似製品の申請は、生ブランド製品が生物製品許可証申請(BLA)によって承認されてから12年後にのみFDAの承認を得ることができる。
私たちのようなバイオテクノロジー会社の特許地位は通常不確実であり、複雑な法律、科学、および事実の問題に関連している。特許保護を得る能力およびこのような保護の程度は、従来技術の範囲、発明の新規性および非顕著性、および特許法実施要件を満たす能力を含む多くの要因に依存することを認識している。また,特許出願において要求されるカバー範囲は,特許発行前に大幅に縮小することができ,その範囲は発行後に再解釈することができる.したがって、私たちは私たちの任意の候補製品のために十分な特許保護を獲得したり維持することができないかもしれない。私たちが現在求めている特許出願が任意の特定の司法管轄区域で特許として発行されるかどうか、または任意の発行された特許の権利主張が競争相手の影響を受けないように十分な特許保護を提供するかどうかを予測することはできない。私たちが持っているどんな特許も第三者によって挑戦され、回避され、無効に発表される可能性がある。
私たちの商業的成功はまた第三者の所有権を侵害しないことにある程度依存するだろう。また、当社は、当社の製品やサービスの特定の側面を開発、製造、商業化することができる第三者独自技術のライセンス権を持っています。いかなる第三者特許を発行することが、私たちの開発または商業戦略を変更し、私たちのプロセスを変更し、許可証を取得したり、いくつかの活動を停止することを要求するかどうかはまだ確定されていません。第三者が許可した特許または特許出願が満了したか、または任意のライセンス契約に違反したり、将来の技術を開発または商業化するために必要な専有権許可を得ることができなかったりすることは、私たちに大きな悪影響を及ぼす可能性がある。もし第三者が米国で準備して提出した特許出願も私たちが権利を持つ技術を持っていると主張した場合、私たちは発明の優先権を決定するために、米国特許商標局(USPTO)の介入手続きに参加しなければならないかもしれない。私たちの知的財産権に関するリスクのより全面的な議論については、“リスク要因-知的財産権に関連するリスク”を参照されたい
個別特許の期限は特許を取得した国の法的期限に依存する。私たちが出願したほとんどの国では,特許期間は,その特許に関連する非臨時特許出願が提出された最初の日から20年である。場合によっては、特許期限調整、すなわちPTAを米国特許に提供することもできる
24
米国特許商標局から特許を取得した遅延を補償する.場合によっては、そのような特許協定は、米国特許期間を、米国特許に関連する非臨時特許出願を提出する最初の日から20年以上延長させる可能性がある。また、米国では、FDAが承認した薬物をカバーする米国特許の期限を延長する資格がある可能性もあり、これにより、FDA規制審査中に失われた特許期間の補償として特許期限の回復が可能となる。ハッジ-ワックスマン法は特許期間を特許満了後最大5年間延長することを許可している。特許期間の延長の長さは,薬物が規制審査を受ける時間の長さと関係がある。特許期間の延長は、製品承認日から14年を超えることができず、承認された薬物に適用される特許を延長することしかできない。欧州や他の外国司法管区にも同様の規定があり、承認された薬物をカバーする特許の期限を延長する。将来、私たちの製品がFDAの承認を得たら、私たちはこれらの製品の特許出願のために特許期間を延長する予定です。私たちは、特許を取得できる任意の管轄区域で、私たちが発行した任意の特許のために特許期間の延長を求める予定ですが、米国のFDAを含む適用当局が保証することはできません。このような延長を承認すべきかどうか、承認されれば、そのような延長の長さの評価に同意します。
私たちはまた、候補製品に関連する商業秘密に依存し、当社の業務において特許保護から保護されているか、または特許保護に適していないと考えられる側面を保護し、独自の情報の機密性を保護し、維持することを求めている。私たちは、従業員やコンサルタントとの契約を結ぶことを含む、当社の固有情報およびビジネス秘密を保護する措置をとっていますが、第三者は、当社の従業員やコンサルタントとのこのような合意に違反することを含む、実質的に同じ独自の情報および技術を独立して開発したり、他の方法で私たちのビジネス秘密を取得したりすることができます。したがって、私たちは私たちの商業秘密を意味的に保護することができないかもしれない。私たちの政策は、私たちの従業員、コンサルタント、外部科学パートナー、賛助された研究者、および他のコンサルタントに、私たちとの雇用や相談関係を開始したときに秘密協定を実行することを要求します。これらの合意は、個人と私たちとの関係中に開発または開示された当社の業務または財務に関するすべての機密情報は、特定の場合を除いて第三者に開示されてはならないことを規定している。私たちと従業員の合意はまた、従業員が私たちに雇われた過程で構想されたすべての発明または従業員が私たちの機密情報を使用することによって生成されたすべての発明が私たちの固有財産であることを規定している。
競争
バイオテクノロジーと製薬産業の特徴は技術が絶えず進歩し、競争が激しいことだ。私たちの候補製品、技術、知識、経験、科学資源は私たちに競争優位を提供してくれると信じていますが、私たちは主要な製薬とバイオテクノロジー会社、学術機関、政府機関、公共と個人研究機関などからの競争に直面しています。我々が開発と商業化に成功した任意の候補製品は,既存の療法や将来出現する可能性のある新しい療法と競争するであろう。私たちが他の療法と効果的に競争する能力に影響を与える重要な製品機能には、私たちの製品の有効性、安全性、利便性があります。後発薬競争の程度や政府と他の第三者支払者が提供する補償もまた私たちの製品の定価と競争力に著しく影響するだろう。私たちの競争相手も私たちよりも早くFDAや他の規制機関のその製品の承認を得ることができ、これは私たちの競争相手が私たちが市場に入る前に強力な市場地位を確立することをもたらすかもしれない。
私たちと比べて、私たちが競争する可能性のある多くの会社は研究開発、製造、臨床前テスト、臨床研究を行い、監督管理の許可とマーケティング承認製品を獲得する方面でより多くの財務資源と専門知識を持っている。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。これらの競争相手はまた合格した科学と管理人員を募集と維持し、臨床研究サイトと臨床研究患者登録を確立し、補充或いは必要な技術を獲得する上で著者らと競争している。
さらに、いくつかの会社が、私たちの候補製品と同じ適応および/または疾患の治療法のために承認される可能性があることをマーケティングまたは開発している。
円形脱毛症
現在の再生不良性貧血の治療には,様々な局所的,局所的,全身的な薬剤の使用,設備があるが,治療に対する反応は大きく異なる。第一線の治療は通常局所あるいは病巣内のコルチコステロイドに依存し,病態の重い患者では全身コルチコステロイドに依存する。難治性疾患に対しては,メトトレキサート,アザチオプリン,シクロスポリンなどの免疫抑制剤を用いることができる。現在、2種類の製品のみがFDAによって再生不良性貧血の治療に許可されている:バリシチニブ、JAK阻害剤、商標名OLumant;及びritlecitinib、JAK阻害剤、商標名はLitfuloである。臨床研究ではJAK阻害剤やS 1 P調節剤などが検討されている。円形脱毛症の薬物開発に参加した会社は,Arcutis BioTreateutics,Inc.,Aslan PharmPharmticals Limited,Bristol−Myers Squibb Company,Concert PharmPharmticals,Inc.(日製薬工業株式会社に買収),Forte Biosciences,Inc.,Horizon Treeutics Plc(安進に買収),Inmagene BiopPharmtics Co.Ltd.,Legacy Healthcare,Nektar
25
治療会社,Ornovio,Inc.,ファイザー,Q 32 Bio Inc.,Reistone Biophma,Zelgen BiopPharmticals Co.,Ltd.,Zura Bio Limited。
乳糜潟
唯一利用可能な乳糜潟治療方法は一生厳格なグルテンフリー飲食を堅持することである。多くの患者はこのような食事の維持が困難であり,グルテンの使用を避けるにもかかわらず症状が持続している患者が多く,完全に反応していない。FDAが承認した乳糜潟の治療方法は現在のところない。われわれの知る限り,多くの会社がこの疾患に対する開発計画を持っており,安進(前Proventive Bioの資産),Anokion SA,Calypso Biotech BV(ノワ製薬に買収),中外製薬株式会社,IgY免疫技術と生命科学会社,免疫会社,免疫遺伝子X社,Protegant Treeutics社,武田製薬会社,Teva製薬会社,Topas治療会社,Zedira GmbH社を含む。
AGVHD
コルチコステロイド,あるいはステロイドは,依然としてGVHDの第一線治療の標準的な看護である。現在のところFDAが承認していない療法はaGVHDの第一線の治療法に指定されている。二次治療は、2019年にステロイド難治性aGVHDの治療のために承認されたタグ外免疫阻害剤、その治療利益がまだ決定されていない、Incell Corporationのruxolitinibを含む。
また、一部の会社は、AltruBio,Inc.,ASC Treeutics,CSL Behring LLC,Cynata Treateutics Limited,ElsaLys Biotech,Evive Biotech(一帆製薬株式会社の子会社)、Humanigen,Inc.,Maat Pharma SA,Medac GmbH,Mesoblast Limited,深センXbiome Biotech,Co.,TR 1 X Bio,Vecmo Bio Holding AG(Ironwood Pharmarmartics,InZarMtics,InZarPhc.,InZarmoltics,InZarPhc.)を開発計画していることを知っている。
狼瘡性腎炎
最も深刻なLN患者に対する標準看護誘導治療は増殖性LNあるいはIIIあるいはIV類LNと呼ばれ,通常メチルプレドニゾロンを静脈内投与し,プレドニゾロンを経口投与しMMFやシクロホスファミドを添加する。維持治療の標準的なケアは,通常コルチコステロイドとMMFあるいはカルモジュリン阻害剤の組み合わせである。現在、承認された2つのLNを治療する方法がある。1つはグラクソ·スミスクラインのBenlysta(Belimumab)で、2020年に承認され、もう1つはLupkynis(Voclosporin)で、2021年1月に承認され、aurinia PharmPharmticals Inc.によって販売されている。
我々の知る限り,多くの会社がLNに対する開発計画を持っており,アスリーカン社,Corestem株式会社,CSL Behring LLC,遺伝子テーク社,ジョンソンのヤンソン製薬会社,コサ生命科学社,Nkarta社,ノワ製薬社,Omeros社,Vera治療会社を含む。
販売とマーケティング
私たちの発展段階を考慮して、私たちはまだ商業組織や流通能力を確立していない。私たちは、内部リソースまたは第三者関係によって、販売、マーケティング、患者訪問、および配布を管理することを望んでいる。大量の財務·管理資源をビジネス活動に投入する可能性がありますが、1つ以上の製薬会社と協力して、私たちのビジネス能力を強化することも考えます。Onoがその選択権を行使してitolizumab(EQ 001)に対する私たちの権利を獲得する場合、itolizumab(EQ 001)の商業化はOnが担当するであろう。
製造業
私たちは私たちの候補製品を生産する製造施設を所有したり運営したりしませんし、予測可能な未来に私たち自身の製造業務を発展させる計画もありません。我々はCMOによりEQ 101とEQ 302を生産した.BioconライセンスとBiocon供給プロトコルによると、私たちは私たちの契約メーカーBioconに依存して、私たちの臨床前研究、臨床研究、およびitolizumab(EQ 001)の商業供給に必要なすべての原材料、薬物物質、そして薬物製品を提供する。Bioconはインドのバンガロールに位置するFDAによって規制されている施設でitolizumab(EQ 001)を商業規模で生産している。
任意の未来の候補製品について、私たちは契約メーカーに依存して、私たちの臨床前研究、臨床研究、商業供給に必要なすべての原材料、薬物物質、薬物製品の需要を提供することが予想される。
政府規制と製品審査
FDAと連邦、州と地方の各レベル及び外国の他の監督機関は、その他の以外に、研究、開発、テスト、製造、品質管理、輸入、輸出、安全、
26
生物製剤の有効性、ラベル、包装、貯蔵、流通、記録保存、承認、広告、販売促進、マーケティング、承認後の監視と承認後の報告。私たちは、第三者請負業者と共に、私たちが研究したい、または承認を求めることができるかもしれない国/地域規制機関の様々な臨床前、臨床、および商業承認要件を満たすことが要求されるだろう。
米国では,FDAは連邦食品,薬物と化粧品法案(FDCA)と公衆衛生サービス法案(PHSA)とその実施条例に基づいて生物製品を規制している。FDAがバイオ製品候補製品が米国で発売される前に必要とされるプログラムは、一般に以下のような態様を含む
臨床前と臨床発展
米国で最初の候補製品の臨床研究が始まる前に、INDをFDAに提出しなければならない。INDはFDAが人類の研究を許可した新薬製品の要求である。IND提出の中心焦点は臨床研究の全体的な研究計画と方案(S)である。INDには動物や体外培養候補製品の毒理学、PK、薬理学およびPD特性を評価する研究;化学、製造および制御情報;および研究製品の使用を支援するための任意の利用可能なヒトデータまたは文献。INDはヒトの臨床研究が始まる前に施行されなければならない。INDはFDAが30日以内に提案された臨床研究に対して安全懸念または問題を提起しない限り、FDA受信後30日以内に自動的に発効する。この場合、INDは臨床的に保留される可能性があり、INDスポンサーおよびFDAは、臨床研究が開始される前に、任意の未解決の問題または問題を解決しなければならない。したがって,INDの提出はFDA認可による臨床研究の開始につながる可能性があり,そうでない可能性もある。
臨床研究は、GCPによって合格した研究者の監督の下でヒト被験者に研究製品を服用することに関連し、その中には、すべての研究被験者に任意の臨床研究への参加についてインフォームドコンセントを提供することを含む。臨床研究はプロトコルに基づいて行われており,その中で研究の目標,安全性をモニタリングするためのパラメータ,評価すべき有効性基準を詳細に説明した。製品開発中に行われるすべての後続の臨床研究および後続の任意の案修正は、既存のINDに個別に提出されなければならない。また,臨床研究を推奨する各地点の独立IRBは,その場所で臨床研究を開始する前に任意の臨床研究の計画およびそのインフォームドコンセントを審査·承認しなければならず,完成まで研究を監視しなければならない。監督管理機関、内部審査局、または保証人
27
被験者が受け入れられない健康リスクに直面していることを発見したり、研究がその規定の目標を達成する可能性が低いことを含む、いつでも様々な理由で臨床研究を一時停止することができる。いくつかの研究は、データ安全監視委員会と呼ばれる臨床研究スポンサーによって組織された独立した合格専門家グループの監視をさらに含み、委員会は、研究のいくつかのデータへのアクセスに基づいて、研究が指定されたチェックポイントで行うことができるかどうかを許可し、被験者が受け入れられない安全リスクまたは他の理由があると判断した場合、治療効果を示さない場合、臨床研究を一時停止する可能性がある。行われている臨床研究結果を公的登録機関に報告することに関する要求もある。
BLAまたはNDA承認の目的では、ヒト臨床研究は、通常、重複する可能性のある3つの連続段階で行われる。
場合によっては、FDAは、製品が承認された後に追加の臨床研究を自発的に行うことを要求することができ、または製品に関するより多くの情報を得るために自発的に行う可能性がある。これらのいわゆる4期臨床研究はBLAやNDA承認の条件となる可能性がある。臨床研究と同時に、会社は追加の動物研究を完成させ、候補製品の生物学的特徴に関する追加情報を開発することができ、cGMP要求に基づいて最終的に商業量産製品の過程を決定しなければならない。製造過程は高品質の候補製品ロットを持続的に生産できる必要があり、特に最終製品の特性、強度、品質と純度をテストする方法を開発しなければならない、あるいは生物製品に対しては、安全、純度、効力の試験方法を開発しなければならない。また,適切な包装を選択·テストし,候補製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
BLAまたはNDAの提出、審査、承認
すべての適用された法規要件に基づいてすべての要求されたテストが成功したと仮定すると、製品開発、非臨床研究、および臨床研究の結果は、BLAまたはNDAの一部としてFDAに提出され、製品を1つまたは複数の適応の市場に使用することの承認を要求する。BLAまたはNDAは、否定または曖昧な結果および積極的な発見、および製品の化学、製造、制御およびアドバイスのラベルなどに関する詳細な情報を含む、関連する臨床前および臨床研究から得られたすべての関連データを含まなければならない。BLAまたはNDAを提出するには、免除または免除が適用されない限り、FDAに大量のアプリケーション使用料を支払う必要がある。
BLAまたはNDAが提出されると、FDAは、出願を受けてから10ヶ月以内に審査基準申請を行うか、または申請が優先審査資格を満たしている場合には、FDAが提出申請を受けてから6ヶ月以内に審査基準申請を行うことを目標としている。標準審査および優先審査では、FDAがより多くの情報を提供することを要求するか、または明確にすることを要求する要件は、しばしば審査プロセスを大幅に延長する。FDAは、製品が安全で純粋かつ有効であるかどうか、およびその製造、加工、包装、または保持されている施設が、製品の持続的な安全、純度および効力を保証するための基準に適合しているかどうかを決定するために、BLAまたはNDAを検討する。FDAは諮問委員会を招集し,審査申請について臨床的知見を提供する可能性がある。BLAまたはNDAを承認する前に、FDAは、通常、製品を製造する1つまたは複数の施設を検査する。FDAは、製造プロセスおよび施設がcGMP要件に適合していることを決定し、要求された仕様の下で製品が一貫して生産されることを保証するのに十分でない限り、申請を承認しないであろう。さらに、FDAは、BLAまたはNDAを承認する前に、通常、GCPに適合することを確実にするために、1つまたは複数の臨床場所を検査する。FDAが申請、製造プロセス、または製造施設が受け入れられないと判断した場合、それは、提出された文書に不足点を列挙し、追加の試験または情報の提供を要求することが多い。任意の要求された補足情報が提出されたにもかかわらず、FDAは最終的に、その申請が承認された規制基準を満たしていないと決定する可能性がある。
28
FDAがBLAまたはNDAを評価し、研究製品および/またはその薬物を生産する製造施設を検査した後、FDAは承認状または完全な返信を発行することができる。承認書は、製品の商業マーケティングを許可し、特定の適応に関する具体的な処方情報を提供する。
完全な返信は、BLAまたはNDAにおいてFDAによって発見されたすべての欠陥を記述するが、FDAが申請をサポートするデータが承認をサポートするのに不十分であると判断した場合、FDAは、必要な検査、試験提出された製品バッチ、および/または提案されたラベルを最初に検討することなく、完全な返信を発行する可能性がある。完全な返信を発行するとき、FDAは、BLAまたはNDAがより多くの情報を提供することを要求すること、または明確にすることを含む、BLAまたはNDAが承認された条件下で、より多くの情報を提供するか、または明確にすることを含む、出願人がとる可能性のある行動を提案する可能性がある。適用される規制基準を満たしていない場合、FDAは、製品の安全性または有効性を監視するために、追加の試験または情報を要求するBLAまたはNDAの承認を延期または拒否することができ、および/または製品の安全性または有効性を監視するために発売後の試験および監視を要求することができる。
1つの製品が規制部門の承認を受けた場合、このような承認は特定の適応が付与され、製品が発売される可能性のある指定用途の制限をもたらす可能性がある。例えば、FDAは、製品の利点がそのリスクよりも大きいことを確実にするために、リスク評価および緩和戦略(REMS)を有するBLAまたはNDAを承認する可能性がある。REMSは、製品に関連する既知または潜在的に深刻なリスクを管理し、そのような薬剤の安全な使用を管理することによって、患者がこれらの薬剤を継続的に得ることを可能にするための安全戦略であり、制限された分配方法、患者登録、および他のリスク最小化ツールのような薬物ガイドライン、医師のコミュニケーション計画、または安全使用を保証する要素を含むことができる。FDAはまた,提案されたラベルを変更したり,適切な制御や仕様を作成したりすることを条件に承認することも可能である.承認されると、発売前と上場後の要求に対する遵守が保たれていない場合、あるいは製品が市場に進出した後に問題が発生した場合、FDAは製品承認を撤回する可能性がある。FDAは、製品の商業化後の安全性と有効性をさらに評価し、監視するために、1つまたは複数の第4段階上場後の研究および監視を要求することができ、これらの発売後の研究の結果に基づいて製品のさらなる販売を制限する可能性がある。
開発と審査計画を加速する
FDA承認を提出する新しい治療製品のマーケティング申請は、優先審査、迅速チャネル指定、画期的な治療、および承認の加速など、FDAの審査および承認プロセスを加速することを目的とするFDAの計画の資格に適合する可能性がある。
製品が、満足できる代替療法なしに安全で効果的な治療を提供する可能性がある場合、または市販されている製品と比較して、重篤な疾患または状態の治療、診断または予防において有意に改善された場合、優先審査を受ける資格がある。新分子実体を含む製品について、優先審査指定は、FDAが60日申請後6ヶ月以内に上場申請に行動することを目標としていることを意味する(標準審査は10カ月)。
迅速なチャネル認証を取得する資格があるために、FDAはスポンサーの要求に基づいて、深刻なまたは生命に危害を及ぼす疾患または状態を治療することを目的とした製品を決定し、存在しない治療法または治療効果または安全要素に基づく既存の治療法よりも優れている可能性があることを証明することによって、未満足の医療需要を満たす可能性があることを証明した。高速チャネル指定は、製品の開発と審査を加速させるために、FDA審査チームと頻繁に相互作用する機会を提供する。FDAはまた、完全な出願を提出する前に、スポンサーおよびFDAが出願部分のスケジュールについて合意し、BLAまたはNDAの第1の部分を提出する際に任意の必要な使用料を支払うことを前提として、高速チャネル製品を審査するBLAまたはNDA部分をスクロールすることができる。
また,スポンサーは候補品を“画期的な療法”に指定することを要求することができる。画期的な治療法は、1つまたは複数の他の薬剤または生物学的製品と単独でまたは1つまたは複数の他の薬剤または生物学的製品と組み合わせて深刻または生命に危険な疾患または状態を治療することを目的とした医薬または生物学的製剤として定義され、初歩的な臨床証拠は、1つまたは複数の臨床的に重要な終点において、臨床開発早期に観察される実質的な治療効果のような既存の治療法よりも実質的に改善された効果を示す可能性があることを示す。画期的な治療法に指定されている薬物や生物製品も加速承認を得る資格がある。FDAは、画期的な治療法の承認申請の開発と審査を加速するために、会議を適時に開催し、提案を提供するなど、何らかの行動を取らなければならない。
さらに、深刻または生命を脅かす疾患または状態の治療における安全性および有効性について研究された製品は、臨床的利益を合理的に予測する可能性のある代替終点に有効であるか、または不可逆的な発病率または死亡率よりも早く測定することができる臨床終点に有効であると判断され、病状の重症度、希少性または流行率、および代替治療を利用可能または不足し、不可逆的な発症率または死亡率または他の臨床的利益への影響を合理的に予測することを考慮すると、加速承認を得ることができる。加速承認の条件として、fdaは通常スポンサーに十分かつ良好な制御を行う上場後の臨床研究を要求し、不可逆的な発病率或いは死亡率或いはその他の臨床効果に対する期待効果を検証と記述する
29
利益。また、FDAは現在、承認を加速させる条件として宣伝材料を事前承認することを求めており、製品商業発売の時期に悪影響を及ぼす可能性がある。
1つの製品が1つまたは複数のこれらの計画の条件に適合していても、FDAは、製品がもはや資格条件を満たしていないと判断するか、またはFDAの審査および承認を決定する期間が短縮されない可能性がある。さらに、優先審査、迅速チャネル指定、画期的な治療指定、および承認の加速は、承認の基準を変更することはないが、開発または承認プロセスを加速させる可能性がある。
孤児薬名
孤児医薬品法によれば、FDAは、米国では20万人未満に影響を与えるか、または米国では20万人を超える影響を与える稀な疾患または疾患を治療するための薬剤または生物に孤児の称号を付与することができるが、米国では、そのような疾患または疾患を治療する薬剤または生物薬剤を米国で開発および提供するコストが、米国での薬剤または生物学的薬剤の販売から回収されることを合理的に予想することができない。BLAまたはNDAを提出する前に、孤児の指定を要請しなければならない。FDAが孤児の称号を付与した後、FDAは、治療剤の汎用的な識別情報およびその潜在的な孤児の使用を開示する。孤児薬物の指定は、規制審査または承認過程においていかなる利点も伝達されず、規制審査または承認過程の持続時間を短縮することもない。
孤児として指定された製品がその後、このような指定された疾患を有するFDAの最初の承認を得た場合、この製品は、孤児の排他性を得る権利があり、これは、FDAが完全なBLAまたはNDAを含む他の出願を承認しない可能性があり、限られた場合、例えば孤児薬物に対して排他的な製品に対する臨床的利点を示さない限り、7年以内に同じ適応の同じ製品を販売することを意味する。孤児排他性は、FDAが同じ疾患または状態に対して異なる薬剤または生物を承認すること、または異なる疾患または状態に対して同じ薬剤または生物を承認することを阻止しない。孤児薬物を指定する他の利点は、いくつかの研究の税金免除、およびBLAまたはNDA申請料の免除を含む。
指定された孤児製品が孤児指定の指示を得るよりも広い用途に使用されることが承認された場合、孤児の排他性を得ることはできない。さらに、FDAが指定された要求に重大な欠陥があると後に判断した場合、または製造業者がこのような稀な疾患または疾患患者の需要を満たすのに十分な数の製品を保証できない場合、米国での独占営業権を失う可能性がある。
承認後に要求する
我々は、FDAによって製造または流通を許可されたどの製品も、品質管理および品質保証、記録保存、不良イベント報告、定期報告、製品サンプルおよび流通、ならびに製品広告および販売促進に関する要求を含むFDAによって普遍的かつ持続的に規制されている。承認後、承認された製品の大多数の変更は、新たな適応または他のラベル宣言を追加するなど、FDAの事前審査および承認を経なければならない。継続的なユーザ料金要求もあり,この要求に基づいて,FDAは承認されたBLAまたはNDAで決定された製品ごとの年間計画費用を評価する.生物製造業者とその下請け業者は、FDAとある州機関に彼らの機関を登録し、FDAと特定の州機関の定期的な抜き打ち検査を受けて、cGMPのコンプライアンスを理解しなければならない。これは、私たちと私たちの第三者製造業者にいくつかの手続きと文書要求を加えている。製造プロセスの変更は厳しく規制されており,変更の重要性により,FDAが事前に承認して実施する必要がある可能性がある。FDAの規定はまた、cGMPから外れた状況を調査·是正し、私たちと私たちが使用を決定する可能性のある任意の第三者メーカーに報告することを要求している。そのため、メーカーは生産と品質管理の分野で時間、お金、精力をかけ続け、cGMPやその他の法規遵守性を維持しなければならない。
規制要件や基準が守られていない場合、あるいは製品発売後に問題が発生した場合、FDAは承認を撤回する可能性がある。その後、予想されていなかった重症度または頻度の副作用、または生産プロセス、または規制要件を遵守できなかったことを含む、以前に未知の製品問題が発見され、新しいセキュリティ情報を追加するために承認されたラベルの改訂をもたらす可能性がある;新しい安全リスクを評価するために発売後研究または臨床研究を実施すること、またはREMS計画に従って流通制限または他の制限を実施することが可能である。他の他の潜在的な結果には
30
FDAは生物製品のマーケティング、ラベル、広告、販売促進を厳格に規制している。1社はFDAが承認したラベルの規定に基づいて、安全性と有効性、純度、効力に関する声明しか提出できない。しかしながら、会社は、FDAによって承認された製品ラベルと一致する真で誤解されない情報を共有するかもしれない。FDAと他の機関は非ラベル用途の普及を禁止する法律法規を積極的に施行している。これらの要求を守らないことは、否定的な宣伝、警告状、改正広告、および潜在的な民事と刑事罰を招く可能性がある。医師は、製品ラベルに記載されていない使用のための合法的に入手された製品の処方、および我々が試験およびFDAによって承認された用途とは異なる使用を行うかもしれない。このようなラベル外の使用は医学専門科でよく見られる。医師は,異なる場合,このような非ラベル使用が多くの患者の最適な治療法であると考えるかもしれない。FDAは医者が治療を選択する時の行動を規範化しない。しかし、FDAは製品ラベルの外使用問題に対する製造業者のコミュニケーションを制限した。
診断性テストの規管
CD 6−alcam経路に関連する診断マーカーと他の尿バイオマーカーとの随伴診断を共同開発·検証し,itolizumab(EQ 001)に最も反応する可能性のあるLN患者を決定し,任意の診断協力者とFDAの設備法規を遵守させる。適用免除が適用されない限り、診断テストは商業流通の前に市場許可またはFDAの承認を得る必要がある。医療機器に適用される2つの主要なタイプのFDAマーケティング許可は、510(K)承認、および発売前承認、またはPMA承認とも呼ばれる上場前通知である。Itolizumab(EQ 001)に対して開発されたどのセット診断にもPMA経路が利用されることが予想される。
PMA申請は有効な科学的証拠支持が必要であり、これは通常、技術、臨床前、臨床と製造データを含む大量のデータを必要とし、この装置の安全性と有効性を証明し、FDAを満足させる。診断テストの場合、PMAアプリケーションは、一般に、分析および臨床検証研究に関するデータを含む。PMA審査の一部として、FDAは、製造業者が設計、テスト、制御、文書、および他の品質保証手順に従うことを要求する品質システム法規に適合することを保証するために、1つまたは複数の製造施設を承認前に検査する。FDAは2014年8月6日、“体外随伴診断装置”の開発と承認手順を述べた最終指導文書を発表した。このガイドラインによれば、キット診断装置およびその対応する薬剤は、製品ラベルにおいて示される治療のためのFDAの承認または承認を同時に得るべきである。
生体模倣薬と参考製品の排他性
2010年に法律となった“患者保護および平価医療法案”に署名されたか、またはBPCIAを含む“平価医療法案”と総称され、FDAによって承認された参照生物製品と類似しているか、または交換可能な生物製品のための短い承認経路が作成された。これまで、いくつかの生物模倣薬はBPCIAによって許可され、多くの生物模倣薬はすでにヨーロッパで承認された。FDAはすでにいくつかの指導文書を発表し,生体模倣薬の審査と承認方法について概説した。
生物類似性は分析研究、動物研究と臨床研究を通じて証明でき、即ち生物製品と参照製品は安全性、純度と効力の面で臨床的に意義のある差異がないことを証明することができる。互換性は、製品が基準製品生物と類似していることを必要とし、この製品は、任意の所与の患者において、参照製品と同じ臨床結果を生成することが期待できることを証明しなければならず、複数回投与された製品の場合、以前の投与後、生物および参照生物は、安全リスクを増加させることなく、または参照生物の独占的使用と比較して治療効果のリスクを低下させることなく、交互にまたは交換することができる。生物製品がもっと大きく、よくもっと複雑な構造に関連する複雑性、及びこのような製品を製造する技術は、FDAがまだ制定している簡略化審査経路の実施に対して重大な障害を構成した。
BPCIAによると,生物類似製品の申請は,参考製品が初めてFDA許可を得た4年後にFDAに提出されなければならない。また,FDAによる生物類似製品の承認は,参考製品が初めて許可された日から12年後に発効する可能性がある。この12年間の排他的期間内に、別の会社は依然として参照製品の競争バージョンを販売することができます。FDAが承認すれば
31
競合製品の完全なBLAは、その製品の安全性、純度および効力を証明するために、申請者自身の臨床前データと、十分かつ良好に制御された臨床研究からのデータとを含む。BPCIAはまた、交換可能な製品として承認された生物模倣薬のためのいくつかの排他的期限を設けている。この節では,FDAが“交換可能”と考えている製品が本当に州薬剤法に管轄されている薬局に取って代わられるかどうかは不明である。
BPCIAは複雑であり、FDAによって解釈され、実施され続けている。また、政府の最近の提案は12年間の参考製品専門期間を短縮しようとしている。BPCIAの他の面では,そのいくつかがBPCIAの排他的条項に影響を与える可能性があり,最近の訴訟のテーマでもある.そのため,BPCIAの最終的な影響,実施,影響は重大な不確実性の影響を受ける。
他のアメリカの医療保険法やコンプライアンスの要求は
米国では、FDAに加えて、現在および将来の業務は、連邦医療保険·医療補助サービスセンター(CMS)、米国衛生·公衆サービス部(HHS)の他の部門(例えば、監察長事務室、民権事務室と衛生資源·サービス管理局)、米国司法省(DoJ)と司法省内の個別連邦検事室、州と地方政府を含む様々な連邦、州、地方当局によって規制されている。例えば、私たちの臨床研究、販売、マーケティング、科学/教育援助計画は、“社会保障法”、“虚偽請求法”、“健康保険携行性及び責任法”(HIPAA)のプライバシー及び安全条項、並びに改正された同様の州法の反詐欺及び乱用条項を遵守しなければならない可能性がある。
他の事項に加えて、連邦反リベート法規は、任意の個人またはエンティティが、購入、レンタル、注文または手配、購入、注文または手配購入、レンタルまたは注文として、Medicare、Medicaid、または他の連邦医療計画に従って全部または部分的に精算することができる任意の物品またはサービスの見返りとして、故意に、または故意に現金または実物で直接または間接的に、公開または隠蔽的に提供、支払い、請求または任意の報酬を受けることを禁止する。報酬という単語は価値のあるものを含むと広く解釈されている。連邦反リベート法規は、治療製品製造業者と処方者、購入者、および処方マネージャーとの間の配置に適用されると解釈される。いくつかの法的例外と規制避難所がいくつかの一般的な活動を保護することは起訴されない。例外や安全港の範囲は狭く,処方,購入または推奨の報酬を誘導するために告発される可能性があるやり方に関連しており,例外や安全港の資格を満たさなければ審査される可能性がある。特定の適用された法定例外や安全港を規制するすべての要求を満たすことができなかったことは、連邦反リベート法規に基づいて、このような行為自体が不法であることを意味するわけではない。代わりに、そのすべての事実と状況の累積審査に基づいて、この手配の合法性を逐案的に評価する。私たちの接近はすべての場合、法定例外や安全港保護を規制するすべての基準を満たしていないかもしれない。
また、連邦“反リベート法規”下の意図基準は“平価医療法案”によってより厳格な基準に修正され、個人或いは実体が法規或いは法規違反の具体的な意図を実際に理解する必要がなくなり、違反を実施することができる。また,“平価医療法”は連邦“虚偽申告法”(FCA)の規定に基づき,連邦“反リベート条例”違反による物品やサービスのクレームが虚偽や詐欺的クレームを構成する判例法を編纂している。
連邦虚偽請求法は、“連邦医療保険法”および民事罰金法を含み、一般市民が政府を代表して民事訴訟によって強制的に執行することができ、任意の個人または実体が知っている場合に連邦政府に虚偽または詐欺的な支払いまたは承認請求を提出または提出することを禁止し、連邦医療保険および医療補助計画を含む、虚偽記録または陳述の作成、使用または使用を招き、虚偽または詐欺的なクレーム材料を連邦政府に提供するか、または故意に虚偽陳述をして不正な方法で回避することができる。連邦政府にお金を支払う義務を減らしたり隠したりする。クレームには、米国政府に提出された金銭または財産に対する“任意の請求または要求”が含まれている。例えば、歴史的には、製薬や他の医療会社は、顧客に製品を無料で提供する疑いがあるため、これらの法律に基づいて起訴されており、顧客が製品の連邦計画に課金することを期待している。他の会社も起訴されました。これらの会社のマーケティング製品は未承認、ラベル外の用途に使用されているため、通常は精算されず、虚偽の声明を提出することになりました。
HIPAAは、他の事項に加えて、虚偽または詐欺的な口実、陳述または約束の方法で任意の医療福祉計画(プライベート第三者支払者を含む)が所有または管理または保管する任意の金銭または財産を詐欺または取得する計画を故意に実行または実行しようと意図的に実行または実行しようとする追加の連邦民事および刑事法規を制定し、医療保健違法行為の刑事調査を意図的に阻害し、悪巧み、計画または装置、重大な事実、または医療福祉、プロジェクトまたはサービスの提供または支払いに関連する方法で任意の重大な虚偽、架空、または詐欺的陳述を偽造、隠蔽または隠蔽することを目的とする。連邦反リベート法案と同様に、“平価医療法案”も特定の意向基準を改正した
32
HIPAA下の医療詐欺法規の規定は、個人或いは実体が実際に法規或いは法規違反の具体的な意図を理解する必要がなく、違反を実施することができる。
さらに、多くの州は、連邦医療補助および他の州計画に従って精算されるプロジェクトやサービス、またはいくつかの州では、支払者が誰であるかにかかわらず、類似した通常より禁止された詐欺や法律または法規の乱用を持っている。
私たちは連邦政府と私たちが業務を展開している州のデータプライバシーと安全規制によって制限されるかもしれない。“経済と臨床衛生情報技術法案”及びその実施条例の改正を経たHIPAAは保証実体、業務パートナー及びその保証下請け業者に対して個人が健康情報を識別できるプライバシー、安全と伝送方面の要求を提出した。他の事項に加えて、HITECHは、特定の医療提供者、健康計画、および医療チケット交換所を含む商業パートナー、独立請負業者、またはカバーエンティティのエージェントに、カバーエンティティを代表してサービス提供に関連する保護された健康情報を受信または取得することを表すHIPAAのプライバシーおよびセキュリティ基準を直接適用する。HITECHはまた4つの新しい民事罰金等級を作成し、HIPAAを改訂し、民事と刑事処罰を商業パートナーに直接適用し、州総検察長に新しい権力を与え、連邦裁判所に民事訴訟を提起し、損害賠償または禁制令を要求してHIPAAを実行し、連邦民事訴訟の提起に関連する弁護士費と費用を求めることができる。また,多くの州の法律は特定の場合に健康情報のプライバシーやセキュリティを管理しており,その多くの法律は互いに大きく異なり,HIPAAに先制されておらず,HIPAAよりも禁止的な効果があり,コンプライアンス作業を複雑化している可能性がある.
私たちは承認されたら医者が管理できる製品を開発するかもしれない。現在適用されているアメリカの法律によると、いくつかの通常自己投与されていない製品(注射薬を含む)は、Medicare B部分を介してMedicareの保険を受ける資格がある可能性がある。Medicare B部分は元のMedicareの一部であり、元のMedicareは高齢者と障害者に医療福祉を提供する連邦医療保健計画であり、外来サービスと用品をカバーし、いくつかの生物製薬製品を含み、これらのサービスおよび用品は治療受益者の健康状態に医学的に必要である。メーカーの合格薬品が連邦医療保険B部分精算を獲得する条件の一つとして、メーカーは医療補助薬品返却計画と340 B薬品定価計画を含む他の政府医療保健計画に参加しなければならない。医療補助薬品還付計画は製薬業者が衛生と公衆サービス部部長と締結し、全国的な税金還付協定を発効することを要求し、各州が連邦マッチング資金を獲得する条件として、メーカーが医療補助患者に提供する外来薬物に使用する。340 B薬品定価計画によると、製造業者は割引をこの計画に参加するエンティティに拡大しなければならない。
また、多くの製薬業者は平均販売価格(ASP)と最適価格のようないくつかの価格報告指標を計算し、政府に報告しなければならない。場合によっては、これらの指標が正確かつタイムリーに提出されていない場合には、処罰が適用される可能性がある。また、これらの薬品の価格は、政府の医療計画や個人支払者が要求する強制的な割引やリベート、および将来的には米国よりも販売価格が低い国からの薬品の輸入を制限している法律を緩和することによって低下する可能性がある。
さらに、“平価医療法案”およびその実施条例における“連邦医師支払い陽光法案”または“陽光法案”は、いくつかの薬品、器具、生物および医療用品の製造業者が、そのような法律で規定されている医師、他の医療専門家(例えば、医師アシスタントおよび看護師従事者のような)、教育病院、またはその要求またはその指定された実体または個人の支払いまたは分配を表すいくつかの支払いまたは他の価値移転に関する情報をCMSに毎年報告することを要求する。医者と教育病院は、毎年医者及び直系親族が持っているいくつかの所有権と投資権益を報告する。タイムリーに正確に報告できなかったことは処罰につながるかもしれない。また、多くの州では支払いや他の価値移転の報告も管理されており、その多くは互いに大きく異なり、往々にして先制されておらず、“陽光法案”よりも尻込み的な効果が生じ、遵守努力をさらに複雑化させる可能性がある。
製品を商業流通するためには、州の薬品および生物製品の製造業者および卸売業者に登録を要求する州の法律を遵守する必要があり、いくつかの州で製品を州のメーカーおよびディーラーに輸送することを含む、これらのメーカーまたはディーラーがその州に営業場所を持っていなくても。一部の州はまた、メーカーと流通業者が流通チェーン中で製品の系統を確立することを要求しており、いくつかの州はメーカーと他の州に流通チェーン中の製品の流れを追跡し追跡できる新しい技術を採用することを要求している。いくつかの州はすでに立法を公布し、製薬と生物技術会社にマーケティングコンプライアンス計画を確立し、州政府に定期報告を提出し、販売、マーケティング、定価、臨床研究およびその他の活動を定期的に公開開示し、および/またはその販売と医療代表を登録し、薬局と他の保健実体が製薬と生物技術会社にいくつかの医師処方データを提供し、販売とマーケティングのために使用することを禁止し、そして禁止した
33
いくつかの他の販売とマーケティング実践。私たちのすべての活動は連邦と州消費者保護と不正競争法によって制限されるかもしれない。
第三者との業務配置が適用される医療法律や法規に適合することを確保することは、費用の高い努力である。もし私たちの業務が上記の任意の連邦および州医療保健法律または任意の他の現在または将来私たちに適用される政府法規に違反していることが発見された場合、私たちは民事、刑事および/または行政処罰、損害賠償、罰金、返還、監禁、MedicareとMedicaid、禁止、個人通報者によって政府名義で提起された個人訴訟、または政府契約、契約損害、名声損害、行政負担、利益減少、および将来の収入のような政府計画から除外されることを含む重大な処罰を受ける可能性があります。私たちがこれらの法律を遵守していないという疑惑や、私たちの業務の縮小または再編を解決するために、会社の誠実な合意または他の合意の制約を受けている場合、追加の報告義務および監督を負う必要があり、これらはいずれも、私たちの業務運営能力および私たちの運営結果に悪影響を及ぼす可能性があります。
保証範囲·定価·精算
私たちが規制部門の承認を得る可能性のある任意の候補製品のカバー範囲と精算状態には、重大な不確実性がある。米国や海外市場では、規制部門の承認を得て商業販売を行う任意の製品の販売は、第三者支払者がこのような製品に保険を提供する程度にある程度依存し、十分な補償レベルを確立する。米国では,第三者支払者には連邦や州医療計画,個人管理のヘルスケア提供者,医療保険会社,その他の組織が含まれている。カバー範囲および政府医療計画(例えば、米国のMedicareおよびMedicaid)および商業支払者からの十分な補償は、新製品の受容度に重要である。
私たちがすべての製品を商業化することに成功した能力はまた、政府衛生行政当局、個人健康保険会社、その他の組織がこれらの製品と関連治療に保険と補償を提供する程度にある程度依存する。政府当局や他の第三者支払人、例えば個人健康保険会社や健康維持組織は、どのような治療費を支払うかを決定し、精算レベルを確立する。第三者支払人の治療性薬物の使用に対する決定を含む第三者支払人の保証範囲と精算は多くの要素に依存する可能性がある
私たちは商業化されたどの製品にも保険があることを確実にすることができません。もし保険があれば、清算水準はいくらですか。カバー範囲はまた、FDAや同様の外国規制機関が製品を承認する目的よりも限られている可能性がある。清算は私たちが規制部門の承認を受けた任意の製品の需要や価格に影響を及ぼすかもしれない。
第三者決済者は、価格に挑戦し、医療の必要性を審査し、医療製品、療法、サービスの費用対効果を審査するとともに、それらの安全性と有効性を疑問視するようになっている。私たちの製品のための精算は特に難しいかもしれません。ブランド薬と医者の監督下で使用される薬物は往々にして価格が高いからです。私たちは、私たちの製品の医療の必要性と費用効果、FDAの承認を得るのに必要なコストを証明するために、高価な薬物経済学的研究を行う必要があるかもしれない。私たちの候補製品は医学的に必要で費用効果があると思われないかもしれない。政府または他の第三者支払人から製品の保証と精算承認を得ることは時間がかかり、高価なプロセスであり、これは、各支払人に科学的、臨床的、費用効果的なデータを提供して、個々の支払人に基づいて私たちの製品を使用するために必要かもしれないが、保険と十分な精算を得ることは保証されないかもしれない。第三者支払者が製品に保険を提供することを決定することは、十分な返済率を承認することを意味するものではない。また、第三者支払者が1つの商品に保険を提供することを決定し、他の支払者もその商品に保険を提供することを保証することはできない。製品開発への投資の適切な見返りを実現するために、十分な価格レベルを維持することができる十分な第三者精算がないかもしれない。精算が得られない場合や限られたレベルの精算のみが提供されなければ、私たちが開発に成功した任意の候補製品を商業化することに成功できないかもしれません。さらに私たちや私たちの協力者は
34
候補製品です。セット診断試験はそのセット薬品或いは生物製品の保証と精算以外に、単独で保険と精算を受けることが要求されている。医薬製品の獲得保険や精算に適用される類似の挑戦は随伴診断にも適用される。
他の国もまた違う価格設定と精算プログラムを持っている。EUでは,各国政府はその定価と精算規則や国家医療保健システムの制御によりバイオ製薬製品の価格に影響を与え,これらのシステムは消費者にバイオ製薬製品の大部分のコストを支払っている。いくつかの法域はプラスリストとネガティブリスト制度を実行し、補償価格を合意した後にのみ、製品を販売することができる。精算或いは定価の承認を得るために、その中のいくつかの国は臨床研究の完成を要求する可能性があり、特定の候補製品のコスト効果を現在利用可能な治療法と比較する。他の会員国たちは会社が自分の薬品価格を固定することを許可するが、会社の利益を監視する。医療コストの下振れ圧力が大きくなる。そのため、新製品の参入にはますます高い壁が設けられている。また、一部の国では、低価格市場からの国境を越えた輸入が一国国内の定価に商業圧力をかけている。
政府や第三者支払者が保険と十分な補償を提供できない場合、規制部門の承認を得て商業販売を行う任意の候補製品の適正性が影響を受ける可能性がある。また,管理式医療への重視,健康維持組織の日増しに増加している影響力,米国の追加立法変化は医療定価の圧力を増加させており,この圧力は増加し続けることが予想される。2022年8月に可決された“2022年インフレ低減法案”(IRA)には、薬品定価改革が含まれており、これらの改革は、候補製品を商業化することに成功した能力に悪影響を及ぼす可能性があり、私たちの候補製品の実際的または知覚的価値を低下させる可能性があり、これは私たちの業務に負の影響を与えるだろう。医療費は一般的に上昇しており,特に処方薬,医療機器,外科手術プログラムなどの治療費上昇の下振れ圧力は非常に大きくなっている。保証政策と第三者支払人の販売率はいつでも変化する可能性があります。規制部門の承認を得た1つまたは複数の製品が有利な引受·精算状態を獲得しても、将来的にはあまり有利ではない引受政策や精算料率が実施される可能性がある。
医療改革
米国や一部の外国司法管轄区では、医療保健システムに関するいくつかの立法と規制の変化、提案された変化が継続され、これらの変化は候補製品の上場承認を阻止または延期し、承認後の活動を制限または規範化し、マーケティングの許可を得た候補製品を収益的に販売する能力に影響を与える可能性がある。米国や他の地方の政策立案者や支払者の中で,医療システムの改革を推進することに大きな興味があり,医療コストの抑制,質の向上および/または参入拡大を既定の目標としている。米国では、製薬業はこれらの努力の重点であり、重大な立法計画の大きな影響を受けてきた。
“平価医療法案”は、政府と民間保険会社の医療融資と提供方式を大きく変えた。例えば、“平価医療法案”は、ブランド薬品メーカーが支払うべき医療補助税金還付の最低レベルを15.1%から23.1%に引き上げ、医療補助管理を要求する医療機関に対して支払うべき薬品の還付を徴収し、指定された連邦政府がある“ブランド処方薬”を販売する予定の製薬業者や輸入業者に控除できない年会費を徴収し、吸入、注入、点滴、移植または注射を要求した薬品に対してメーカーの医療補助薬品還付計画下でのリベートを計算し、医療補助計画の資格基準を拡大した。患者を中心とした結果研究所を創立し、監督、優先事項を確定し、臨床治療効果の比較研究を行い、そしてこのような研究に資金を提供した;そしてCMSに医療保険と医療補助革新センター(CMMI)を設立し、革新的な支払いとサービス交付モードをテストして、連邦医療保険と医療補助支出を下げ、処方薬支出を含む可能性がある。“平価医療法案”のいくつかの側面は法律と政治的挑戦を受けている。例えば、2021年6月17日、米国最高裁は、“個人強制令”が国会で廃止されたため、“平価医療法案”全体が違憲だと弁明する手続き理由に基づく挑戦を却下した。米国の最高裁が裁決を下す前に、バイデン総裁は2021年1月28日に行政命令を発表し、平価医療法案市場で医療保険を獲得することを目的とした特殊な加入期間を開始した。行政命令はまた、特定の政府機関に、医療補助モデルプロジェクトおよび免除計画の再検討、医療補助または“平価医療法案”による医療保険獲得に不必要な障害をもたらす政策を含む医療補助モデルプロジェクトおよび免除計画の再審査および見直し、医療補助または“平価医療法案”の見直しを指示する。また、2022年8月16日、バイデン総裁は、平価医療法案市場で医療保険を購入した個人への増強補助金を2025年計画年に延長することを含むアイルランド共和軍法案に署名した。2025年からアイルランド共和軍は,受益者の最大自己負担コストの著しい低減と新たなメーカー割引計画の作成により,連邦医療保険D部分計画下の“ドーナツ脆弱性”を解消した。“平価医療法案”は将来的に司法や国会の挑戦を受ける可能性がある。これらの挑戦やバイデン政府の医療改革措置が“平価医療法案”や我々の業務に影響を与えるかどうかは不明である。
35
“平価医療法案”のいくつかの側面は法律と政治的挑戦を受けている。トランプ政権は2017年1月以降、“平価医療法案”に規定されているいくつかの要求を延期、回避、または緩和するためのいくつかの行政命令やその他の指令に署名した。
私たちは、“平価医療法案”が引き続き承認された製品の保証範囲と価格に追加の下振れ圧力を与え、私たちの業務を深刻に損なう可能性があると予想している。医療保険や他の政府が計画している精算のどの減少も、個人支払者の支払いのような減少を招く可能性がある。コスト抑制措置や他の医療改革を実施することは、私たちの収入の創出、利益の実現、あるいは私たちの製品の商業化を阻止するかもしれない。これらの改革は、私たちが開発に成功し、規制承認を得る可能性のある候補製品の予想収入に悪影響を及ぼす可能性があり、私たちの全体的な財務状況や候補製品を開発する能力に影響を及ぼす可能性がある。
さらなる立法や規制によって、私たちの業務、財務状況、そして運営結果を損なう可能性があります。“平価医療法”が公布されて以来,他の立法改正も提案され,採択された。例えば、2011年8月、オバマ総裁は赤字削減合同特別委員会を作成し、国会に支出削減提案を提案する“2011年予算制御法案”に署名した。赤字削減合同特別委員会は、2012年度から2021年度までの少なくとも1.2兆ドルの赤字削減目標を実現せず、いくつかの政府プロジェクトの自動削減を触発した。これには,各年度にプロバイダに支払われる連邦医療保険総額が2%に達し,2013年4月1日から施行され,追加の国会行動がとられない限り2032年まで有効であることが含まれている。また、総裁·バイデンは2021年3月11日に“2021年米国救援計画法案”に署名し、2024年1月1日から単一源と革新多源薬に対する法定医療補助薬品還付上限を廃止し、現在この上限は薬品メーカー平均価格の100%である。2013年1月、“2012年米国納税者救済法”が法律に署名され、病院、画像形成センター、がん治療センターを含むいくつかの提供者への医療保険支払いがさらに減少し、政府が提供者に多額の支払いを取り戻す訴訟時効が3年から5年に延長された。
また、特殊薬品の価格設定実践におけるアメリカの立法と法執行の興味もますます大きくなっている。具体的には,米国議会は最近いくつかの調査と連邦立法を行い,薬品定価の透明性の向上,連邦医療保険下の処方薬のコスト低減,定価とメーカー患者計画との関係の審査,政府計画の薬品精算方法の改革を目指している。連邦レベルでは、2021年7月、バイデン政府は“米国経済における競争を促進する”という行政命令を発表し、その中には処方薬に対する条項が複数ある。バイデン行政命令への対応として,2021年9月9日,HHSは高薬価に対応した総合計画を発表し,その中で薬品定価改革の原則を概説し,国会がとりうる様々な潜在立法政策と,HHSがとりうる潜在行政行動がこれらの原則を推進することを示した。また,アイルランド共和軍(IRA)は他の事項を除いて,(1)HHSに連邦医療保険(Medicare)で覆われたある単一由来薬物と生物製品の価格について交渉するよう指示し,(2)連邦医療保険B部分とD部分にリベートを徴収し,インフレを超える価格上昇を懲罰するよう指示した。このような規定は2023年度から段階的に施行される。 2023年8月29日、HHSは、連邦医療保険薬品価格交渉計画が現在法的挑戦を受けているにもかかわらず、価格交渉を受ける上位10種類の薬物のリストを発表した。アイルランド共和軍がどのように実施されるかは不明であるが,製薬業に大きな影響を与える可能性がある。また、バイデン政府の2022年10月の行政命令に応えるために、衛生·公衆サービス部は2023年2月14日に報告を発表し、CMMIテストの3種類の新しいモデルを概説し、これらのモデルはそれらの薬品コストを低減し、可獲得性を促進し、医療の質を高める能力に基づいて評価を行う。これらのモデルが将来の任意の医療改革措置で使用されるかどうかは不明である。また、2023年12月7日、バイデン政府は“ベハ·ドール法案”下の参入権を使用することで処方薬の価格を制御するイニシアチブを発表した。2023年12月8日、米国国家標準·技術研究所は、権限行使を考慮した機関間指導枠組み草案を発表し、その中で初めて製品価格を機関が進行権を行使する際に使用できることを決定する要因とした。これまでデモの権利を行使したことはなかったが、新たな枠組みの下で、この権利が継続するかどうかは定かではない。米国の個別州も、価格や患者の精算制限、割引、ある製品への参入の制限、マーケティングコストの開示と透明性措置を含む、バイオ製薬製品の価格設定を制御するための法規を立法と実施することをますます積極的に実施しており、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。
“反海外腐敗法”
“反海外腐敗法”(FCPA)は、個人または企業が業務を獲得または保持するのを助けるために、任意の米国人または企業が、任意の外国人官僚、政党または候補者に直接的または間接的に支払い、提供または許可支払い、または任意の価値のあるものを提供することを禁止し、個人または企業が業務を獲得または保持することを目的とする。“海外腐敗防止法”は米国に上場する証券会社にも遵守を求めている
36
会計規定は、企業(国際子会社を含む)のすべての取引の帳簿及び記録を正確かつ公平に反映し、国際業務のために適切な内部会計制御システムを設計·維持することを要求する。
法規を付加する
これらの規定に加えて,環境保全や有害物質に関する州や連邦法は,“職業安全と健康法”,“資源節約と回収法”,“有毒物質制御法”を含めて,我々の業務に影響を与える。これらの法律と他の法律は私たちの様々な生物、化学、放射性物質の使用、処理と処理、これらの物質は私たちの行動、そして私たちの行動によって生成された廃棄物を規範化している。もし私たちの運営が環境汚染を招いたり、個人を危険物質に曝露させたりすれば、損害賠償と政府罰金の責任を負う可能性があります。私たちは、私たちが適用される環境法律を実質的に遵守し、これらの法律を遵守し続けることは、私たちの業務に実質的な悪影響を与えないと信じている。しかし、私たちはこのような法律の変化が私たちの未来の運営にどのように影響するか予測できない。
経済制裁に関する政府法規
様々な法律、法規、行政命令に基づいて、米国財務省外国資産制御弁公室(OFAC)は、特定の外交政策や国家安全上の理由で国家、制裁された実体、制裁された個人の特定の活動を禁止または制限する経済·貿易制裁を管理·実行する。制裁の範囲は大きく異なるが、制裁を受けた司法管轄区、実体または個人、および制裁を受けた司法管轄区、実体または個人を代表する制裁されていない個人および実体の輸出入、投資、および外国取引の円滑化に関する全面的な制限が含まれている可能性がある。
その中の一つの規制はCACRと略称されるキューバ資産統制条例だ。CACRは、キューバ政府またはキューバ国民の財産に関するほぼすべての取引に米国人が参加すること、またはキューバ政府または任意のキューバ国民が1963年7月8日以降の任意の時間に任意の性質の利益を直接または間接的に所有する財産に参加することを禁止する。CACRが活動を禁止する場合、そのような活動に従事することは、OFACによって発行される一般的または特定のライセンスの許可を取得しなければならない。イトリス抗体(EQ 001)とアルツハイマー抗体の抗体配列はいずれもキューバ国民によって独自に開発された。著者らは現在CACR中のキューバ原産薬品に関する汎用許可証に依存して輸入とitolizumab(EQ 001)に関する臨床研究を行っている。
2019年11月、OFACは、FDAに問い合わせ、OFACが“キューバ原産薬”の定義に属することを決定することを含む慎重な考慮を経て、CACR 515.547(B)および(C)節の一般的なライセンス許可は、FDAの承認を求めることを目的としたitolizumabの臨床研究を行うことを通知した。したがって,現在我々が行っているおよび計画中のitolizumab臨床研究はOFACのさらなる認可を得る必要はない。
その他の規則
私たちはまた多くの連邦、州、地方法律の制約を受けており、これらの法律は安全作業条件、製造実践、環境保護、火災危険制御と危険或いは潜在的危険物質の処分に関連している。私たちが今または未来にこのような法律と法規を遵守することは大きな費用をもたらすかもしれない。
従業員
2023年12月31日現在、私たちは44人の従業員を雇用しており、いずれもフルタイムで、研究開発活動、運営、財務、業務発展や管理に従事している。私たちはまた必要に応じて臨時職員とコンサルタントを雇う。
企業情報
我々は最初に2017年3月にデラウェア州で減衰型バイオ製薬会社に登録し,その後2017年5月にEquillium,Inc.と改称した。私たちの主な実行事務室はカリフォルニア州ラホア92037号105号スイートルーム2223 Avenida de la Playaにあります。私たちは二つの完全子会社があります。一つはデラウェア州のBioniz治療会社で、もう一つはオーストラリアの独自有限会社Equillium Australia Pty Ltdです。私たちの電話番号は(858)240-1200です。私たちのサイトの住所はWwwl.equilliumBio.comそれは.我々のウェブサイトやウェブサイトに含まれている,あるいはサイトを介してアクセス可能な情報は,参照によって本Form 10-K年次報告に組み込まれているとはみなされず,Form 10-K年次報告の一部ともみなされない.我々の10-Kフォーム年次報告、10-Qフォーム四半期報告、8-Kフォーム現在の報告、および1934年の証券取引法(改正)第13(A)および15(D)節または取引法に基づいて提出または提出されたこのようなレポートの修正案は、これらの材料を電子的にアーカイブまたは米国証券取引委員会に提供した後、合理的で実行可能な場合にできるだけ早く私たちのサイトで無料で提供される。
37
我々も取引法で定義されている“小さな報告会社”であり、より小さい報告会社が得ることができるいくつかの規模開示の利点を利用することを選択している。
本年度報告でForm 10−K形式で出現したすべてのブランド名または商標は,それぞれの所有者の財産である。我々は、本年度報告において、他の当事者の商標、商業外観または製品をForm 10−Kの形態で使用または展示し、商標または商業外観所有者と私たちとの関係、または私たちへの裏書きまたはスポンサーを示唆するつもりもない。
38
第1 A項。国際ロータリーSK因子です。
リスク要因
私たちの普通株を購入するか、保有するか、または売却するかどうかを決定する前に、以下のリスク要因および本報告書の他の情報をよく考慮しなければなりません。以下のいかなるリスクが発生しても、私たちの業務、財務状況、運営結果、および/または成長の見通しを損なう可能性があり、または、私たちの実際の結果は、本報告書で行われた私たちの展望的な陳述および私たちが時々行う可能性のある展望的な陳述に含まれる結果とは大きく異なる可能性がある。私たちの業務を評価する際には、私たちの財務諸表および関連する付記の他の情報、ならびに“経営陣の財務状況および運営結果の検討および分析”について、すべての記述要因を考慮しなければなりません。もし実際に以下のいかなるリスクが発生すれば、私たちの業務、財務状況、経営業績、未来の成長見通しは重大な不利な影響を受ける可能性がある。この場合、私たちの普通株の市場価格は下落する可能性があり、あなたは投資の全部または一部を失うかもしれない。私たちは今知らないか、あるいは私たちが現在どうでもいいと思っている他のリスクと不確実性はまた私たちの業務運営を損なう可能性がある。
私たちの財務状況と追加資本需要に関連するリスク
私たちは設立以来大きな損失を受けており、予測可能な未来に重大な損失を受け、永遠に利益を実現したり維持したりすることはないかもしれない。
我々は、2017年3月に設立された臨床段階のバイオテクノロジー会社であり、これまで、当社の人員、業務計画、資本調達、itolizumab(EQ 001)の許可権、EQ 302の初期臨床前開発を含む非臨床研究を行い、3つのINDを提出し、EQ 101、EQ 102およびitolizumab(EQ 001)の臨床開発を行い、2022年2月のBioniz買収、2022年12月のOnoとの資産購入契約、上場企業に関連する一般的かつ行政活動などの業務開発活動を行ってきた。私たちはどんな候補製品の開発から発売承認までを完了したことがなく、私たちは承認された製品の販売から何の収入も得たことがありません。そのため、私たちの業務を評価する意味のある業務はありません。バイオ製薬製品の開発に成功し、それを商業化した歴史があれば、私たちの将来の成功や生存能力の予測はそれほど正確ではないかもしれません。
生物製薬製品開発への投資は非常に投機性があり、それは大量の前期資本支出を必要とし、候補製品が監督管理の許可を得られないか、あるいは商業上実行可能な重大なリスクがあるからである。私たちは販売許可された製品から何の収入も得たことがなく、私たちは私たちの未来の損失を正確に見積もることができません。2023年12月31日と2022年12月31日までの年度の純損失はそれぞれ1330万ドルと6240万ドルだった。2023年12月31日までの累計赤字は1兆857億ドルだった。我々は研究開発活動を実行し、EQ 101とitolizumab(EQ 001)の臨床開発を推進し、EQ 302と他の候補臨床前製品の臨床前研究と潜在臨床開発を行い、発見研究を行い、候補製品の配合と設備開発を行い、候補製品の臨床開発の適応を拡大し、新製品および/または候補製品を買収または開発する可能性があり、規制部門が任意の承認された製品を承認し、それを商業化し、より多くの人を募集し、維持し、規制要求を遵守し、私たちの知的財産権を保護し、私たちの業務の行政面を管理する可能性があるため、予測可能な未来に運営損失が予想される。また,Bionizを買収する過程で,我々の生産ラインを1つの候補製品から複数の候補製品に拡張し,これらすべてが異なる開発段階にある.私たちのパイプラインの拡大は、規制機関がこれらの候補製品を承認することを求めるコストが生じるため、私たちの運営損失の増加速度を加速させるかもしれません。また、いずれかの候補製品が規制部門の承認を得た場合、いくつかの投資は承認前に行われる可能性があるより多くの販売およびマーケティング費用が発生すると予想されます。したがって,予測可能な未来には,重大な運営損失と負のキャッシュフローが継続することが予想される。このような損失はすでに私たちの財務状況と運営資本に悪影響を与え続けるだろう。
39
利益を実現し、維持するためには、巨大な市場潜在力を持つ製品を開発または買収し、最終的に商業化しなければならない。これは、私たちの候補製品の臨床前研究と臨床研究を完成させ、私たちの候補製品のマーケティング許可を得て、もし私たちがマーケティング許可を得たら、私たちの候補製品を製造、マーケティング、販売し、上場後の要求(あれば)を満たすことを含む一連の挑戦的な活動で成功することを要求するだろう。私たちはこのような活動で決して成功しないかもしれません。候補製品の承認を得て商業化することに成功しても、私たちは利益を達成するのに十分な収入を生むことができません。さらに、私たちは予測できない費用、困難、複雑な状況、遅延、および他の既知および未知の挑戦に直面するかもしれない。また,バイオ製薬製品開発に関連する多くのリスクや不確実性により,費用増加の時間や金額,あるいはいつ,あるいは利益を達成できるかどうかを正確に予測することはできない。もし私たちが確実に利益を達成すれば、私たちは四半期や年度の収益性を維持または向上させることができないかもしれません。私たちはより多くの候補製品を開発し、マーケティングするために、大量の研究開発や他の支出を生み出し続けるかもしれません。もし私たちが実現して利益を維持できなければ、会社の価値を下げ、資金を調達し、研究開発努力を維持し、業務を拡大し、あるいは運営を継続する能力を弱める可能性がある。わが社の価値の低下はあなたの投資損失の全部または一部を招く可能性もあります。
私たちは、小野がその選択権、itolizumab(EQ 001)、および任意の将来の候補製品を行使しない場合、EQ 101およびEQ 302の開発および商業化を継続して完了するために多くの追加資金を必要とするだろう。もし私たちが必要な時にこの資金を集めることができなければ、私たちは私たちの研究開発計画や他の業務を延期、減少、またはキャンセルさせることを余儀なくされるかもしれない。
私たちは今後数年間私たちの支出が大幅に増加すると予想している。候補生物技術製品の開発は資本集約型だ。著者らは著者らの候補製品に対して非臨床研究と臨床開発を行うことに伴い、著者らは発見と非臨床研究、臨床開発、法規事務、製品開発、製品品質保証と薬物警戒を含む様々な領域の能力を維持と拡大するために大量の追加資金を必要とする。また、私たちの任意の候補製品が市場の承認を得たら、マーケティング、販売、製造、流通に巨額の商業化費用が発生すると予想されます。その中のいくつかの商業化投資は承認される前に危険があるかもしれない。
2023年12月31日現在、私たちは4090万ドルの現金、現金等価物、短期投資を持っています。我々の株式買い戻し計画にこれ以上の買い戻しがないと仮定すると、2023年12月31日現在の既存の現金、現金等価物、短期投資は、2025年下半期の運営に資金を提供できるようになると予想される。しかし、変化する環境や私たちの不正確な推定は、私たちの資本使用速度が私たちの現在の予想よりも大きく速くなる可能性があり、そして私たちがコントロールできない状況のため、私たちは現在の予想よりも多くの資金を必要とするかもしれない。例えば、私たちが行っている候補製品と将来の臨床研究は、技術、登録、あるいは他の問題に直面する可能性があり、これは私たちの開発コストを私たちが予想していたよりも多く増加させる可能性があります。2023年12月31日現在、私たちは株式買い戻し計画に基づいて298,385株の普通株を買い戻し、総金額は約30万ドルです。2023年12月31日以降,本年度報告がForm 10−K形式で提出される日まで,株式買い戻し計画により,我々の普通株は買い戻しが行われていない。さらに買い戻しの時間と金額(あれば)は、私たちの普通株の価格、代替投資機会、私たちの現金資源、私たちの任意の合意下の制限、会社と監督管理要求、および市場状況を含む様々な要素に依存します。
EQ 101の臨床開発を完了するのに十分な資金がなく,小野がその選択権を行使しなければ,我々の現在の適応の規制により,itolizumab(EQ 001)が承認された。私たちは、私たちの株式買い戻し計画に基づいて任意の普通株買い戻しを行う場合、各候補製品の開発と商業化を達成するために、より多くの追加資本を調達する必要があり、これらの追加資本は、私たちの普通株または他の証券を売却することによって、あるいは代替戦略取引を締結することによって調達される可能性があり、その条項は、小野との資産購入協定のような1つ以上の候補製品の剥離を要求したり、株主が大きな希釈を受けたりすることを要求する可能性がある。
将来の資本要求は多くの要素に依存するだろう
40
2023年10月、私たちはJefferiesと2023年の現金自動支払機メカニズムを締結しました。このメカニズムによると、私たちは時々Jefferiesを通じて私たちの販売代理として私たちの普通株の株を発売し、販売することができます。総発行価格は2195万ドルに達します。本年度報告書Form 10−Kを提出した時点で、2023年の現金自動支払機融資メカニズムにおけるいかなる株も販売していません。
私たちの商業収入は、もしあれば、主に製品の販売から来ると予想されていますが、これは今後12ヶ月以内に発生する可能性はあまりありません。小野と締結された資産購入契約によると、35億円の一括払い、または約2640万ドルを受け取り、(I)小野がその独占選択権を行使してitolizumabを買収する権利があれば、50億円の一括払い、または約3310万ドル(三菱日連銀行2024年3月21日の為替レートによる)を得る権利があり、(Ii)は一定のマイルストーンを達成した際に最高1.014億ドルの収入を得る資格がある。しかし、小野がその選択権を行使する保証はなく、記念碑的な支払いを受ける保証もない。また、外国為替レートに関するリスクにより、小野が選択権を行使すれば、50億円の一括払いはドル価値が予想を著しく下回ってしまう可能性がある。したがって、私たちは私たちの持続的な業務と関連した多くの追加資金を得る必要があるだろう。私たちは受け入れ可能な条項で十分な追加融資を受けることができないかもしれないし、根本的にできないかもしれない。我々のより多くの資本を調達する能力は、潜在的な世界経済状況の悪化や米国と世界各地の信用と金融市場の中断と変動の悪影響を受ける可能性があり、これらの要素は公衆衛生流行病や疫病、銀行倒産、ロシアとウクライナ間の衝突、中東紛争、および連邦機関が日々激化するインフレ圧力に対応するために金利を高める通貨政策の変化を含む。もしこのような妨害が続いて深まったら、私たちは追加的な資本を得ることができない状況に直面するかもしれない。また、有利な市場条件や戦略的考慮により、現在または将来の運営計画のために十分な資金があると考えても、追加の資本を求めることができる。もし私たちが必要な時や魅力的な条項で資金を調達できない場合、私たちは私たちの研究開発計画や他の業務を延期、減少または廃止したり、パートナー関係を構築したり、他の方法で戦略的取引によって私たちのパイプを貨幣化することを余儀なくされます。これらの条項は、私たち自身が開発したり商業化したりする候補製品ほど有利ではないかもしれません。また、市場状況のため、既存の現金、現金等価物、および投資の一部を得ることができない可能性がある。例えば、2023年3月10日、連邦預金保険会社(FDIC)はSVBを引き継ぎ、SVBの係に任命された。連邦預金保険会社が接収した時、私たちはSVBの清掃口座に約820万ドルの資産を持っていた。私たちは2023年3月13日にこのような資金に対する完全なアクセス権限を獲得した。他の銀行や金融機関が将来、銀行システムや金融市場の金融状況に影響を与えて破産手続きや破産に入った場合、既存の現金、現金等価物、投資を取得する能力が脅かされ、私たちの業務や財務状況に大きな悪影響を及ぼす可能性がある。
41
我々の業務に関するリスクと我々の候補製品の開発と規制承認に関するリスク
私たちは現在の候補製品EQ 101、EQ 302、およびitolizumab(EQ 001)の成功した開発に強く依存しており、私たちは私たちが開発する予定の任意の適応で規制やマーケティングの承認を得ることができないか、またはこれらの候補製品の商業化に成功することができないかもしれない。
私たちの将来の成功は、EQ 101、EQ 302およびitolizumab(EQ 001)の開発に成功した能力にほぼ完全に依存し、次いでEQ 101、EQ 302およびitolizumab(EQ 001)の商業化に成功し、EQ 101を用いた再生障害性貧血の治療、EQ 302を用いた乳糜潟または他の胃腸疾患の治療、またはitolizumab(EQ 001)を用いてaGVHDおよびLNを治療することを含む、現在開発されている任意の適応のために使用されることは決して起こらないかもしれない。私たちは現在バイオ製薬製品の販売収入がありません。私たちは永遠に適切なバイオ製薬製品を開発したり商業化することができないかもしれません。
米国で任意の候補製品をマーケティング·販売することができる前に、研究·開発活動を管理し、臨床研究を開始·完了し、必要なFDA規制の承認を得、ビジネス組織を構築したり、第三者とのマーケティング協力などを行う必要がある。必要な臨床研究を成功させ、および/または規制部門の承認を得て、私たちの任意の候補製品のために十分な商業能力を開発することができることを保証することはできません。私たちはまだ候補製品のBLAやNDAをFDAに提出しておらず、米国以外の他の規制機関にも承認を申請していない。また,我々の候補製品は臨床研究で成功しても,規制部門の承認を得られない可能性がある。もし私たちが規制部門の承認を得なければ、私たちの業務、見通し、財務状況、そして経営結果は不利な影響を受けるだろう。私たちが規制部門の承認を得ても、私たちはどんな製品のいかなる商業販売からも相当な収入を得られないかもしれない。もし私たちの候補製品が承認されて、私たちがそれを商業化することに成功しなかったら、私たちは私たちの業務を維持して発展させるのに十分な収入を生むことができないかもしれません。私たちの業務、将来性、財務状況、運営結果は不利な影響を受けるでしょう。
私たちは将来的に戦略的取引を通じてパートナーシップや同様の手配を達成したり、他の方法で私たちのチャネルを貨幣化したりする可能性があり、これは私たちの投資リターンを達成する能力を損なう可能性があり、外部資金に対する私たちの需要を増加させる可能性がある。
私たちは、パートナー関係や同様の手配を達成したり、戦略的取引によって私たちのチャネルを貨幣化して、追加資本を調達し、私たちの利用可能な資金や他の資源を私たちの他のまたは未来の候補製品を開発して商業化することができます。例えば、2022年12月、小野と資産購入協定を締結し、この合意に基づいて、小野に独自の選択権を付与して、itolizumab(EQ 001)に対する権利を得る。私たちは努力しているにもかかわらず、私たちは未来のパートナー関係を構築することができないかもしれないし、他の方法で優遇条件で第三者と戦略的取引を行うことで、私たちのチャネル通貨化を実現することができないかもしれない。第三者による職務調査活動を支援し、戦略的手配の財務および他の条項について交渉することは長く、高価で複雑な過程であり、結果は不確定であり、これらの活動からいかなる財務的利益も得ることができないかもしれない。私たちの1つまたは複数の候補製品のための戦略的パートナーを探すためのどんな努力も、私たちの経営陣の時間と注意を彼らの日常活動からそらすことができ、これは、私たちが開発と商業化を継続しようとしている現在の候補製品の発見と開発に悪影響を及ぼすかもしれない。さらに、潜在的な戦略的パートナーは、代替製品を自ら開発したり、代替技術を求めたりする可能性があり、これは、任意のこのような手配の下で、将来のマイルストーンや特許使用料支払いを得ることができない可能性がある。私たちは私たちの1つ以上の候補製品を戦略的に取引するかもしれません。これらの候補製品は、私たちが開発と商業化を続けることを決定した候補製品よりも成功していることが証明されています。したがって、私たちの財務状況と研究開発活動で達成された見返りはマイナスの影響を受ける可能性があり、私たちは株式発行、債務融資、または他の資本源を通じて追加資金を求めて私たちの運営を支援する必要があるかもしれません。これは私たちの既存の株主の大量の希釈を招き、私たちの普通株の価格を下落させる可能性があります。上記のいずれも、我々の競争地位、業務見通し、財務状況、経営業績に重大な悪影響を及ぼす可能性がある。
私たちは、内部許可を通じて将来の資産の権利を得ることを望んでいるか、または私たちの現在または未来の候補製品との協力を試みているかもしれませんが、それができないかもしれません。これは、私たちの開発や商業化計画を変更したり、延期したりする可能性があります。
私たちの候補製品の開発と潜在的な商業化は費用を支払うために多くの追加資本を必要とするだろう。将来的には、バイオテクノロジーや製薬会社と協力して、候補製品を開発し、小野との資産購入協定のような潜在的な商業化を行うことになるかもしれません。適切な協力者を探すことで、私たちは激しい競争に直面するだろう。私たちはもう一つを作ることに成功しないかもしれません
42
任意の候補製品の戦略的パートナーシップまたは代替配置は、協力努力の開発段階が早すぎると考えられる可能性があるので、潜在的な締約国は、これらの候補製品が安全性および有効性を示すために必要な潜在力を有するとは考えない可能性がある。Tolizumab(EQ 001)以外の候補製品を協力して開発し、商業化すれば、候補製品の将来成功する制御権の一部または全部をパートナーに譲ることが予想される。我々が協力について最終的な合意を達成できるかどうかは,他の事項に加えて,協力者の資源や専門知識の評価,協調の条項や条件,提案された協力者のいくつかの要因の評価に依存する.これらの要素には以下のような要因が含まれている可能性がある
協力者はまた、同様の協力可能な指示を得るための代替候補製品または技術を考慮することができ、そのような連携が、私たちと私たちとの連携よりも私たちの候補製品に魅力的であるかどうかを考慮することができる。いかなる許可協定によれば、私たちはまた制限される可能性があり、特定の条項や潜在的な協力者と協定を締結することはできない。協力の交渉と記録は複雑で時間がかかる。また,最近では大手製薬会社間の大量の業務合併により将来の潜在パートナー数が減少し,合併後の会社の戦略も変化している。したがって、私たちはタイムリーで受け入れ可能な条件で協力を交渉することができず、交渉することさえできないかもしれない。もし私たちがそれができない場合、私たちは候補製品の開発を減らし、私たちの1つ以上の他の開発計画を減少または延期し、候補製品の潜在的な商業化を延期したり、任意の計画の販売やマーケティング活動の範囲を縮小したり、私たちの支出を増加させ、自費で開発、製造、または商業化活動を行わなければならないかもしれない。もし私たちが私たちの支出を増やし、私たち自身の開発、製造、商業化活動に資金を提供することを選択すれば、私たちは追加の資本を得る必要があるかもしれないし、これらの資本は受け入れ可能な条件で私たちに提供できないかもしれないし、全く得られないかもしれない。もし私たちが十分な資金がなければ、私たちは私たちの未来の候補製品をさらに開発したり、それらを市場に出して製品収入を作ることができないかもしれない。私たちがこのような協力を成功させたとしても、私たちが合意した条項は私たちに不利になるかもしれません。例えば、候補製品の開発承認が延期され、候補製品の安全性が問われたり、承認された候補製品の販売が満足できない場合、私たちはこのような協力を維持できないかもしれません。
著者らは臨床開発における経験が限られており、まだ後期臨床研究を成功させておらず、いかなる候補製品の監督管理許可も得ていない。
我々は2019年第1四半期に我々の最初の臨床研究を開始し、これはitolizumab(EQ 001)によるaGVHD治療の第1段階の臨床研究である。その時から、著者らはまた3つのitolizumab(EQ 001)の臨床研究を開始し、その中の2つは未制御喘息と狼瘡/狼瘡/LNに対する第一段階の臨床研究であり、一つはaGVHDに対する第三段階の臨床研究である。Itolizumab(EQ 001)の第1段階研究は完了しているが,aGVHDの第3段階研究は現在行われている。われわれは最近オーストラリアの健康ボランティアでEQ 102の第一段階ヒト臨床研究を完成させ,現在オーストラリアとニュージーランドのAA患者でEQ 101の第二段階臨床研究を行っている。我々は現在2つの有効なINDがFDAでitolizumab(EQ 001)を用いてaGVHDとLNを治療している。Bionizを買収することにより,FDAとINDSを達成し,EQ 101をHTLV−I関連脊髄症/熱帯痙攣対麻痺,皮膚T細胞リンパ腫(CTCL)と再障害の治療に用いた。FDAとの相互作用は限られているため、将来の相互作用の前に、FDAが要求する可能性のある情報やデータを知ることができないかもしれない。一部は我々の限られたインフラ,会社としての臨床研究の経験,規制機関の相互作用により,我々が行っていることと将来の臨床研究が時間どおりに完了するかどうかも確認できず,できれば,我々の計画の臨床研究が時間どおりに開始されるかどうか,あるいは我々の計画の開発計画がFDAに受け入れられるかどうかを決定することはできない。
著者らは非臨床研究或いは臨床研究を行う時、不良な安全性と毒理学結果が出現する可能性がある。そのほか、早期臨床研究の成功は後の臨床研究も成功することを意味するわけではなく、後期臨床研究はより広範な患者群で行われる可能性があり、異なる研究設計に関連する可能性がある。例えばitolizumabは
43
(EQ 001)およびアルツハイマー病は、異なる細胞株で生産された同じ一次モノマブ配列を有し、したがって、異なるバイオ製薬製品と考えることができる。したがって,Bioconによるアルツェルモノクロナル臨床研究で見られた結果は,われわれのitolizumab(EQ 001)の臨床研究結果を予測できない可能性がある。また,われわれの将来の臨床研究は,FDAの承認を得るために,より大きな患者集団で十分な安全性と有効性を証明する必要がある。会社は高度臨床研究でしばしば大きな挫折を経験しており,早期の臨床研究が有望な結果を示した後であっても,類似した挫折に直面しないとは確信できない。そのほか、臨床前と臨床データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品が臨床前研究と臨床研究で満足できると考えているが、しかし依然としてその製品のマーケティング許可を得られなかった。また,開発中の候補製品のうち,一部のみがBLAやNDAをFDAに提出しており,より少ない製品が商業化承認されている.
私たちが製品収入を作る能力は、私たちが上記の活動を成功させる能力と、私たちの候補製品の成功開発と最終商業化に必要な他の活動に大きく依存します。少なくとも今後数年以内にはこのような状況は起こらないと予想されます。私たちの候補製品の成功はさらに次の要素に依存するだろう
もし私たちがこれらの要素のうちの1つまたは複数をタイムリーに達成できなければ、私たちは重大な遅延に直面したり、マーケティングの承認を得ることに成功し、私たちの候補製品を商業化することができなくなり、これは私たちの業務に実質的な損害を与えるだろう。
Itolizumab(EQ 001)はCD 6を選択的に標的とするモノクロナル抗体であり,現在FDAが承認していないCD 6に対する治療法である。これにより,itolizumab(EQ 001)の臨床開発時間やコストを予測することは困難である。CD 6を狙う方法がビジネス価値のある製品を開発させるかどうかはわかりません。
標的CD 6は1種の治療方法であり、著者らが現在研究と開発している重要な構成部分を代表し、このような著者らが治療している疾病に対する治療方法の成功開発は、著者らの未来の成功に重要な役割を果たしている。これまでFDAが承認していなかったCD 6に対する薬剤は,いくつかの独立した研究でCD 6が標的であることが臨床的に証明されているが,我々のパートナーであるBioconを除いて,CD 6は伝統的に他の生物製薬会社を標的とする経路ではない。他のもっと有名あるいはもっと広く研究されている治療方法と比べ、イトリズマブ(EQ 001)などの新しい候補製品の監督管理審査過程はもっと高価で、時間がもっと長いかもしれない。Itolizumab(EQ 001)を市場に投入するために必要な規制承認の遅延または失敗、または規制承認を得るための予期しないコストは、業務を維持するために十分な収入を創出する能力を低下させる可能性がある。
44
また,itolizumab(EQ 001)と併用したキット診断試験も開発可能である。私たちまたは私たちの協力者は、これらのテストに対するFDAの承認または承認、および私たちのitolizumab(EQ 001)のために求められた承認および保証および清算を除いて、単独の保証および清算を要求されるだろう。私たちはセットの診断開発者と協力することができず、私たちの業務、財務状況、運営結果、見通しに実質的な悪影響を及ぼす可能性があります。
私たちはアメリカ、カナダ、オーストラリア、ニュージーランドでitolizumabの権利を許可した。Bioconまたは他の司法管轄区域の第三者によるitolizumabの任意の研究、臨床または商業使用中に発生するいかなる不利な発展も、私たちが規制部門の承認または成功してitolizumab(EQ 001)を商業化する能力、または他の方法で私たちの業務に悪影響を及ぼす可能性がある。
Biocon、そのキューバパートナーCIMAB S.A.およびそのライセンス保有者(私たちは制御できない)は、世界的にitolizumabを開発し、Equillium領土(以下、定義する)以外の地域でitolizumabを商業化する権利がある。Itolizumabはインドで重度斑塊型乾癬の治療に許可され,Bioconによってアルズムモノクロナル抗体として販売されている。インド薬品監督管理局はまた、インド新冠肺炎中の重度ARDS患者のサイトカイン放出症候群(CRS)を治療するために、Biocon有限緊急にitolizumabを使用することを許可した。2020年9月、DCGIは中国ハムスター卵巣(CHO)細胞株から生産されたitolizumabを慢性斑塊型乾癬の治療に使用することを許可し、中重度ARDSを有する新冠肺炎患者のCRSの緊急使用許可を制限した。この細胞系はインドで発売され、ブランド名はアルツェマブ-L、あるいはアルツェモノクロナル抗凍結乾燥化である。アルツェマブおよびアルツェマブ−Lはすでにインドで応用されており,アルツェマブ−Lはインドで同情的に使用され,ラベル外で使用され,および/または研究者による研究で使用され続けている可能性が知られている。
私たちはキューバが現在活発で進行中のイトリス抗臨床研究を持っているということを知らない。免疫分子センターはイトリス抗体の緊急使用許可を得て、キューバ重症新冠肺炎患者に応用した。イトリズマブも中国で臨床試験が行われ,急性呼吸窮迫症候群や皮膚筋炎患者に用いられていることが分かった。イトリズマブのキューバや中国での使用はNS 0細胞系で産生されたイトリズマブに限られているのに対し,イトリズマブ(EQ 001)はCHO細胞系で産生されていると考えられる。
Bioconまたは第三者によるitolizumabの臨床研究結果およびBioconまたは第三者によって支持されるitolizumabの臨床または商業使用に関連する持続的有害事象報告は、我々の開発計画およびitolizumab(EQ 001)の潜在的なビジネスの将来性に影響を及ぼす可能性がある。また,我々は制御できず,Bioconや第三者によって報告された研究結果を検証することもできない.Bioconまたは第三者報告書のデータおよび公開開示における任意のミスまたは漏れは、私たちの株価および業務計画に重大な悪影響を及ぼす可能性がある。
患者が承認された治療方法としてitolizumabを使用する場合、またはBioconまたは第三者が行ったりサポートしたりする任意の臨床研究、探索的研究または他の臨床使用中に深刻な有害事象が発生し、FDAを含む規制機関がitolizumab(EQ 001)の承認を延期、制限または拒否する可能性があり、私たちのitolizumab(EQ 001)の臨床開発を一時停止するか、または上場承認の条件として追加の臨床研究を要求することは、私たちのコストを増加させ、私たちの業務に悪影響を及ぼすだろう。私たちがitolizumab(EQ 001)の規制機関の承認を得、アルツモノマブ-Lの商業的使用またはBioconまたは第三者による臨床研究、探索的研究、または他の臨床用途で新たな深刻な安全問題が発見された場合、規制機関は製品の承認を撤回するか、または他の方法で私たちのマーケティングおよび販売itolizumabの能力を制限するかもしれない。また,このような有害事象が懸念されるため,治療医は我々の製品をあまり使用したくない可能性があり,itolizumab(EQ 001)を商業化する能力を制限し,itolizumab(EQ 001)の臨床開発を行う能力に悪影響を及ぼす可能性がある。
もし私たちが他の候補製品や製品を開発または買収できなければ、私たちの業務と見通しは制限されるだろう。
我々の戦略の1つの要素は,業務や候補製品買収により我々のチャネルを拡大し,Bionizを買収することなどで他の候補製品組合せを買収することである.この戦略の成功は、私たちの規制、開発と商業能力と専門知識の結合、および候補治療適応を識別、選択、獲得する能力に大きく依存し、これらの適応は、私たちの既存のチャネルを補充または増強することができ、または私たちが受け入れられる条項で私たちの発展または戦略計画に適合することができる。将来性のある候補製品を確定、選別、獲得するには大量の技術、財政と人的資源の専門知識が必要である。このような努力は、可能な特定の候補製品を実際に獲得することを招くことはなく、何のメリットも生じることなく、私たちの管理職の時間と資源支出の分流をもたらす可能性がある。もし私たちが第三者から適切な候補製品を探し、選択し、買収することができない場合、あるいは私たちが受け入れられる推定値や他の条項で事業を買収することができない場合、あるいは事業や新製品候補製品の買収に必要な資金を調達できなければ、私たちの業務と見通しは制限されるだろう
45
また、業務または新製品候補製品を買収したり、他の候補製品の開発を進めたりすることができるように、1つまたは複数の候補製品を剥離することを要求することができる。
さらに、我々が得た任意の候補製品は、商業販売または他の適応に拡張する前に、臨床前研究(適用されれば)、FDAおよび関連外国規制機関の広範な臨床試験および承認を含む追加的、時間のかかる開発または規制努力が必要となる可能性がある。すべての候補製品は薬品開発固有の失敗リスクに直面しやすく、候補製品は十分な安全及び/又は有効性が証明されない可能性があり、監督管理機関の許可を得られない可能性がある。さらに、承認されたどのような製品も経済的に製造または生産され、商業化に成功したり、市場に広く受け入れられたり、他の商業的に利用可能な代替製品よりも効果的または人気があるという保証はない。
また、私たちの候補製品を商業化してさらに開発することに成功しなければ、私たちの既存の候補製品に倣って、他の候補製品を買収して既存の製品の組み合わせを拡大することができないかもしれません。私たちの業務と将来性は損なわれます。
潜在的な自然災害は、その中のいくつかは気候変化の日々深刻な影響と関係がある可能性があり、臨床研究場所、著者らのオフィス空間、実験室及び/又は倉庫を損傷、破壊或いは混乱させる可能性があり、これは私たちの運営に重大なマイナス影響を与える可能性がある。
私たちは気候変化と他の自然災害の深刻化の影響を受けやすい。極端な暑さや寒さを含む天候条件の不安定な変化は、野火、洪水、吹雪、ハリケーン、その他の天候に関連する災害のリスクを増加させる可能性がある。このような極端な天気イベントや他の自然災害(例えば、地震)は、停電やネットワーク中断を招く可能性があり、運営中断を招く可能性があり、臨床研究を継続または完了する能力に影響を与える可能性があり、これは、私たちの運営に負の影響を与え、候補製品の商業化計画を延期することになる。それらはまた,我々の臨床研究場所に重大な被害や破壊をもたらし,これらの施設の一時的あるいは長期的な閉鎖を招く可能性がある。このような災害はまた、オフィスビル、実験室、従業員および/または患者の住所、その国の他の地域への移転、または臨床研究場所に行きたくない従業員および/または患者の損失または損傷、および重要な従業員を募集することができず、および/または患者を募集することができない可能性がある。これは、既存の労働力および/または患者サンプルへの悪影響、材料および/またはデータの損傷または破壊、または臨床研究および新しいデータを提供することができない可能性がある。
私たちは独占許可協定に従ってBioconからitolizumabの許可を得た。この許可の条件は、私たちがitolizumabの開発、規制承認、商業化に関連するいくつかの職務義務を履行し、規制承認と商業マイルストーン、および特許権使用料支払いに関して重大なマイルストーン支払いを支払うことである。
私たちはBioconと独占ライセンス契約を締結し、この協定によると、私たちは最初に米国とカナダで開発、製造、製造、使用、販売、販売、要約、輸入、および他の方法でitolizumabを使用し、itolizumabを含むまたは含む任意の医薬成分または製剤の独占的許可を得て、その後、オーストラリアとニュージーランド、または共同でEquillium分野での同じ独占許可を与えるために修正された。本プロトコルによれば、すべての許可権利を保持するために、所定の時間枠内で特定の開発マイルストーンを実現する義務があります。その中のいくつかのマイルストーンは私たちの統制範囲内に大きくない。ビジネス上の合理的な努力を用いて規制部門の承認を求め,規制部門の承認を得た場合には,Equillium分野でitolizumabを商業化し,2つ以上の適応でitolizumabの開発に資金を提供することを保証する義務がある。さらに、私たちは特定の規制承認と商業マイルストーンを終えた後にBioconに特定の現金マイルストーン支払いを支払う義務があり、承認された場合、私たちはまたBioconにitolizumab純売上の特許権使用料を支払わなければならない。私たちの業務計画によると、特許権使用料とマイルストーン支払いは合理的だと思いますが、私たちはこれらの義務を履行するために多くの資金が必要です。私たちの手元に現金支払いがない場合、私たちは記念碑的な支払いを支払う義務があるかもしれません。これは、臨床研究の延期、私たちの運営の削減、私たちの商業化とマーケティング努力の削減、あるいは私たちに不利な条項の履行のための資金を求める必要があるかもしれません。さらに、もし私たちが満期時に何のお金も支払うことができない場合、または許可協定の要求された時間枠内で開発マイルストーンを達成できなかった場合、または商業的に合理的な努力でitolizumabを開発、登録、商業化し、2つ以上の適応においてitolizumabの開発に資金を提供することを保証できなかった場合、Bioconは私たちの許可範囲を制限したり、合意を終了し、私たちがitolizumabを開発および商業化するすべての権利を制限する権利があるかもしれない。
我々はONOがitolizumab(EQ 001)の臨床開発と商業化に資金を提供することにさらに依存している可能性がある。小野が我々の資産購入プロトコルを終了すれば,その選択権を行使しない,あるいは実現していない
46
資産購入協定に規定されたマイルストーン、私たちの業務、そして財務状況は不利な影響を受けるだろう。
2022年12月に,吾らはOnoと資産購入協定を締結し,これにより,吾らはLNのEqualise臨床研究の背線データおよびaGVHDの赤道第3期臨床研究の中期データ交付後3カ月で満了する吾らのitolizumab(EQ 001)に対する権利を取得するためにOnoに独占選択権を付与した。オプション期間中は,itolizumab(EQ 001)のすべての研究·開発を担当し,2022年7月1日から小野社が四半期ごとに資金を提供している。ONOがこのような資金を提供できない場合、私たちの財務状況とイトキシマブ(EQ 001)の研究開発を継続する能力は不利な影響を受けるだろう。
Onoがその選択権を行使してitolizumab(EQ 001)に対する私たちの権利を獲得すれば、私たちはitolizumab(EQ 001)の臨床開発および潜在的な商業化を制御しなくなるだろう。資産購入プロトコルによると、小野の当選により、小野を代表して何らかの活動を行い、補償を得ることができるが、itolizumab(EQ 001)活動を制御することはない。小野は、将来的にFDAまたは他の規制機関にitolizumab(EQ 001)を承認する申請を提出し、FDAまたは他の規制機関によって発行される任意のitolizumab(EQ 001)の上場承認の所有者となることを担当する。FDAまたは他の規制機関がitolizumab(EQ 001)を承認した場合、小野はまた、最終製品の発売、マーケティング、および販売を担当するだろう。しかし,小野がitolizumab(EQ 001)の臨床開発に十分な関心や資源を投入しているかどうか,あるいは迅速に行われるかどうかを制御することはできない。FDAまたは他の規制機関がitolizumab(EQ 001)を承認しても、小野は1つ以上の国で最終製品の商業化を行わないことを選択することができる。これらまたは任意の他の理由で、itolizumab(EQ 001)の開発が進んでいない場合、特定の開発および商業化マイルストーンを含むitolizumab(EQ 001)からさらなる収入を得ることができず、他の方法でそのような取引の利点を達成することもできず、これは私たちの業務を損なう可能性がある。
バイオ製薬製品の開発と商業化は広く規制されており、私たちは私たちが開発を計画しているいかなる適応においても、私たちの候補製品または未来の候補製品の承認を得ることができないかもしれない。
臨床開発、製造、ラベル、包装、保存、記録保存、広告、販売促進、輸出、輸入、マーケティング、流通、不良事件報告、安全および他の発売後の情報と報告の提出、および私たちの現在の候補製品と私たちが将来開発する可能性のある任意の他の候補製品に関連する他の可能な活動は、広範な監督管理を受けている。米国では、新治療製品の発売承認はNDAまたはBLAをFDAに提出する必要があり、FDAの承認を得るまで、米国での候補製品の販売は許可されていない。NDAやBLAは広範な臨床と臨床前データおよび薬理学,化学,製造と制御に関する大量の情報支持が必要である。治療製品が発売される前に、類似した提出書類は米国以外の他の地域の関連規制機関の承認を得る必要がある。
FDAや他の適用可能な規制機関の承認は保証されず,審査·承認プロセスは高価で不確実なプロセスであり,数年かかる可能性がある。FDAのような規制機関は、承認過程においても大きな裁量権を持っている。承認を必要とする臨床前研究および臨床研究の数量およびタイプは、候補製品、疾患または候補製品治療の条件および任意の特定の候補製品に適用される法規によって異なる。臨床前研究や臨床研究に関連する時間や費用は高いにもかかわらず,失敗はどの段階でも発生する可能性がある。著者らの候補製品の臨床前と早期臨床研究結果は著者らの後期臨床研究の結果を予測できないかもしれない。
臨床研究失敗は多種の要素によるものである可能性があり、研究設計、用量選択、プラセボ効果、患者登録標準及び良好な安全性或いは有効性特徴を証明できず、臨床研究失敗は任意の段階で発生する可能性がある。生物製薬業界の会社はよく治療効果或いは不良安全性の不足による臨床研究の進展の中で挫折し、早期の研究で人を奮い立たせる結果を得たにもかかわらず。陰性または不確定の結果に基づいて、私たちは決定することができますか、あるいは監督機関は私たちに追加の臨床研究または臨床前研究を要求するかもしれません。また、臨床研究から得られたデータは異なる解釈の影響を受けやすく、監督管理機関は著者らのように著者らのデータを有利に解釈しない可能性があり、これは更に上場承認を延期、制限或いは阻止する可能性がある。
FDAおよび他の適用可能な規制機関は、様々な理由で承認候補製品を延期、制限、または拒否する可能性がある
47
一般的に、バイオ製薬製品の安全性に対する公衆の懸念は、規制部門の承認を得る能力を延期または制限する可能性があり、私たちのラベルに不利な情報が含まれているか、あるいは追加コストが発生する可能性のある他の活動が要求される可能性がある。私たちはFDAや他の適用可能な規制機関からどんな製品の承認も受けていない。このような経験の不足は、FDAまたは任意の他の適用可能な規制機関による私たちの候補製品の承認をタイムリーに得ることを阻害するかもしれない。
もし私たちが承認を得る上で遅延や私たちの任意の候補製品が承認されなかった場合、私たちのビジネスの見通しは損なわれ、私たちの収入を作る能力は重大な損害を受け、これは私たちの業務、見通し、財務状況、そして運営結果に悪影響を及ぼすだろう。
私たちが行っている、計画されている、または未来の臨床研究の任意の開始または完了の遅延、または終了または一時停止は、私たちのコスト増加を招き、資金を調達したり、収入を創出する能力を延期したり制限したりし、私たちのビジネスの見通しに悪影響を及ぼす可能性がある。
米国で任意の明らかな適応の候補製品の臨床研究を開始することができる前に、INDまたは同様の規制文書の一部として、それらの化学、製造および制御、および我々が提案する臨床研究案に関する情報を含む臨床前研究の結果および他の情報をFDAに提出しなければならない。これまで,EQ 001によるaGVHD,LN,新冠肺炎の臨床研究のためのINDのみを提出してきた。また,HTLV−I関連脊髄症/熱帯痙攣対麻痺,CTCL,AAには,EQ 101の開放INDがあり,これらのINDは最初にBionizによってEQ 101資産を買収する前に提出された。
FDAまたは米国以外の任意の他の適用規制機関から任意の適応候補製品の発売承認を得る前に、これらの候補製品の安全性と有効性を証明するために、広範な臨床研究を行わなければならない。臨床テスト費用が高く、時間がかかり、結果は確定しなかった。また、私たちは、私たちのパートナーBioconおよび契約研究組織(CRO)および他の契約者が、私たちの候補製品のために監督管理のために提出された臨床前、臨床、および品質データを部分的に依存する予定だ。私たちはこれらの契約者のサービスについて合意したりしていますが、彼らの実際の表現に対する影響力は限られています。これらの締約国が私たちにデータを提供しない場合、または適用される場合、私たちと彼らとの合意に基づいて、直ちに規制報告書を提出すると、私たちの開発計画は著しく遅延する可能性があり、私たちは独立して追加的な研究を行ったり、追加のデータを収集したりする必要があるかもしれません。いずれの場合も、私たちの開発コストは増加します。
FDAおよび他の適用可能な規制機関は、既存または任意の未来の候補製品の追加の臨床前研究を要求し、その後、臨床研究を開始することを可能にするかもしれません。これは、追加の遅延を招き、私たちの臨床前開発計画のコストを増加させる可能性があります。私たちが行っている、計画中、または未来の臨床研究の開始または完成過程のいずれのこのような遅延も、私たちの製品開発コストに著しく影響する可能性があります。私たちが行っている研究と未来の研究が予定通りに完成するかどうかはわかりませんが、できれば、あるいは私たちの研究が時間通りに始まるかどうか、できれば。臨床研究の開始と完成は様々な原因で遅延する可能性があり、以下の方面に関連する遅延を含む
48
臨床研究が我々,そのような研究を行っている機関のIRBs,そのような研究のデータ安全監視委員会,FDA,あるいはその研究を行っている国で管轄権を持つ他の規制機関や衛生当局によって修正,一時停止または終了されれば,我々も遅延に遭遇する可能性がある。このような主管部門は様々な要素によって著者らの研究方案を一時停止、終了または修正する可能性があり、これらの要素は、法規の要求または著者らの臨床方案に従って臨床研究を行うことができなかったこと、FDAまたは他の法規機関の臨床研究操作または研究場所の検査の実施による臨床一時停止、予見できない安全問題または副作用、薬物使用の利益を証明できなかったこと、政府法規または行政措置の変化、または十分な資金が不足して臨床研究を継続することを含む。また、
49
規制要求と政策は変化する可能性があり、私たちはこれらの変化に適応するために臨床研究方案を修正する必要があるかもしれない。修正案は,臨床研究案をIRBsに再提出して再検査することを要求する可能性があり,臨床研究のコスト,時間,あるいは成功に影響を及ぼす可能性がある。
私たちのいくつかの科学顧問やコンサルタントは私たちから報酬を得て、私たちの未来の臨床研究の調査者になる可能性が高い。場合によっては、私たちはFDAまたは他の規制機関にいくつかの関係を報告することを要求されるかもしれない。FDAまたは他の規制機関は結論を出すかもしれないが、私たちと主要な研究者との間の財務関係は利益の衝突をもたらし、あるいは他の方法で臨床研究の解釈に影響を与えた。したがって,FDAや他の適用規制機関は,適用された臨床研究地点で生じるデータの完全性を疑問視する可能性があり,臨床研究自体の効用が脅かされる可能性がある。これは、FDAまたは他の規制機関が私たちの上場申請の承認を遅延または拒否することを招き、最終的には、私たちの候補製品が1つまたは複数の適応で上場承認を拒否される可能性がある。もし私たちが私たちの候補製品に対する任意の臨床研究の完了を遅延または終了すれば、その候補製品の商業的将来性は損なわれ、私たちが製品収入を作る能力は延期されるだろう。また、臨床研究を完成するいかなる遅延も私たちのコストを増加させ、私たちの開発と審査過程を緩和し、そして私たちの製品販売の開始と製品販売から収入を創造する能力を危険にさらし、これは私たちの業務、財務状況、運営結果と将来性を深刻に損なう可能性がある。
進行中または計画中の臨床研究で患者を募集する際に遅延や困難に遭遇した場合、必要な規制承認を受けることは延期または阻止される可能性がある。
FDAや他の適用可能な規制機関の要求に応じて、これらの研究に十分な数の合格者を決定し、募集することができなければ、私たちが行っている、あるいは将来私たちの候補製品に対する臨床研究を継続することができないかもしれない。多種の要素は著者らの臨床研究学生募集が公共衛生流行病或いは疫病と関連する影響を含むこのような挑戦に直面する可能性があり、これらの影響は以前著者らの臨床研究学生募集に不利な影響を与えたことがある。また,我々の競争相手のいくつかは候補製品の臨床研究を行っている可能性があり,これらの候補製品は我々の候補製品治療と同様の適応となるが,本来我々の臨床研究に参加する資格がある患者は競争相手の候補製品の臨床研究に参加する可能性がある。患者の入選は他の要素の影響を受けています
私たちの臨床研究のために十分な数の患者を募集し、保留することはできません。これは重大な遅延を招きます。あるいは1つ以上の臨床研究を完全に放棄する必要があるかもしれません。我々の臨床研究の登録遅延は開発コストの増加を招く可能性があり、これはわが社の価値を低下させ、追加融資を受ける能力を制限する。
我々の候補製品に関連する不良副作用または他の安全リスクは、承認を延期または阻止する可能性があり、臨床研究の一時停止または停止、さらなる開発の放棄、承認されたラベルの商業イメージの制限、または上場承認後の重大な負の結果をもたらす可能性がある(ある場合)。
50
医薬品の一般的な状況のように,我々が行っている研究や将来の臨床研究,およびitolizumabが商業化可能な司法管轄区の臨床研究,研究者による研究や商業使用では,我々の候補製品に関連する副作用や有害事象が出現する可能性が高い。
Bioniz以前に完成した研究では,EQ 101の耐性は良好であり,健康ボランティア,大顆粒リンパ球性白血病,CTCLの被験者を含む用量制限毒性や輸液反応の報告はなかった。再生不良性貧血患者に対するEQ 101の第二段階臨床研究が行われている。
著者らが現在itolizumab(EQ 001)を使用している限られた臨床経験によると、予想される副作用は、リンパ球減少、注射部位反応、輸液/注射関連反応(発熱と頭痛を含む)、および皮疹、蕁麻疹、紅斑および掻痒を含む他の全身アレルギー反応を含む。
Itolizumab(EQ 001)臨床レジメンから発見される最も一般的な副作用は、注射部位反応(決定されたリスクとして指定される)およびリンパ球減少(重要な決定されたリスクとして指定される)である。しかも、感染は重要な潜在的危険として指定されている。リンパ細胞減少事件はイトリズマブ(EQ 001)研究で報告されたよく見られる治療緊急不良事件である。リンパ球計数の減少はイトリズマブ(EQ 001)の既知の薬効学的マーカーである。これらのイベントは通常1回目の服薬後は短く,服薬継続に伴い減少することはなく,イトキシマブ(EQ 001)の治療中止後に消失する。そのほか、リンパ細胞計数の低下は感染或いはその他の臨床後遺症と関係がない。
Bioconはまた、彼ら自身が後援する臨床研究、非ラベル使用、研究者によって開始された研究、または私たちが制御できない第三者スポンサーの研究におけるアルツェマブ-Lの使用を支持し続ける可能性がある。例えば,Bioconは潰瘍性大腸炎のTolizumab治療を検討しており,これはインドで行われている第二段階臨床研究の一部であり,Equilliumは協力して共同援助している。Bioconまたは第三者のitolizumabの持続的な使用を考慮すると、有害事象は著者らの臨床開発とitolizumab(EQ 001)の商業化に成功する能力に影響する可能性がある。また,このような有害事象が正しく報告されていないリスクがあり,我々の業務にも悪影響を及ぼす可能性がある.
Itolizumab(EQ 001)とアルツェマブは同じ初期モノマブ配列を有するが,異なる細胞系で生産されていることから,異なるバイオ製薬製品と考えられる。そのため、アルツェマブの臨床結果はイトリーモノクロナル抗体(EQ 001)の結果と関係なく、副作用を含む可能性がある。本年度報告書が10−K表に提出された日まで,itolizumabのリスク収益状況に大きな変化は認められなかった。
著者らの臨床研究結果は副作用或いは予期せぬ特徴の高さと受け入れられない深刻さと普遍性を掲示する可能性がある。著者らの候補製品による不良副作用は著者ら、FDA或いは他の適用規制機関が多種の原因で臨床研究を延期、一時停止或いは中止する可能性がある。また,われわれのaGVHD臨床研究では,大きな割合の患者がこの疾患で死亡する可能性があり,itolizumab(EQ 001)による可能性があり,itolizumab(EQ 001)の進行に影響する可能性がある。もし私たちが任意の臨床研究を延期、一時停止、または終了することを選択または要求された場合、私たちの候補製品の商業的将来性は損なわれ、候補製品から製品収入を生成する能力は延期またはキャンセルされるだろう。臨床研究で観察された深刻な不良事件は市場が私たちの候補製品を受け入れることを阻害或いは阻止する可能性がある。これらのすべての状況は私たちの業務、将来性、財務状況、そして経営結果に重大な損害を与える可能性がある。
さらに、もし私たちの任意の候補製品が臨床研究において不良副作用または予期しない特徴に関連している場合、私たちは、その開発をより狭い用途または集団に放棄または制限することを選択することができ、リスク効果の観点から、副作用または他の特徴は、それほど一般的ではなく、それほど深刻ではない、またはより容易に受け入れられ、これは、承認されれば、候補製品の商業的期待を制限するかもしれない。私たちはまた私たちの臨床研究結果に基づいて私たちの研究計画を修正することを要求されるかもしれない。早期テストで最初に希望を示した候補製品の多くは後に発見され副作用が生じ,さらなる開発を阻害する。さらに、規制当局は異なる結論を出すか、またはこれらの決定を確認するための追加的なテストを要求するかもしれない。
私たちがより大きく、より長く、より広範な臨床研究で私たちの候補製品をテストする時、異なる用量レジメンの使用、または任意の規制承認後、私たちの候補製品の使用がより広くなり、患者は早期研究で観察された疾患、傷害、不快感、および他の有害事象、および以前の研究で発生しなかったか、または検出されなかったことを報告するかもしれない。これらの副作用が開発後期または承認された後に知られた場合、これらの発見は、私たちの業務、財務状況、運営結果、および将来性に大きな損害を与える可能性があります。
51
さらに、もし私たちの任意の候補製品が発売承認され、私たちまたは他の人が後にその承認された製品または任意の関連製品による不良副作用を発見した場合、多くの潜在的な重大な負の結果を招く可能性がある
これらの事件のいずれも、私たちの任意の候補製品に対する市場の受け入れ度を達成または維持することを阻止することができ、承認されれば、私たちの業務、財務状況、運営結果、および将来性を深刻に損なう可能性がある。
著者らが時々発表或いは公表した臨床研究の中期、主要或いは初歩的なデータはより多くの患者データの獲得に従って変化する可能性があり、そして監査と検証プログラムの影響を受け、これは最終データの重大な変化を招く可能性がある。
私たちは時々私たちの臨床前と臨床研究の初歩的或いは主要なデータを公開するかもしれないが、これらのデータは当時利用可能なデータの初歩的な分析に基づいて、結果及び関連する発見と結論は特定の研究に関連するデータをより全面的に審査した後に変化する可能性がある。私たちはまた、私たちのデータ分析の一部として、すべてのデータを全面的かつ詳細に評価する機会がないか、または受け取る機会がないかもしれないという仮説、推定、計算、および結論を出した。したがって,我々が報告したTOPLINE結果は,同じ臨床前および臨床研究の将来の結果と異なる可能性があり,あるいは追加のデータを受信して十分な評価を行うと,異なる結論や考慮要因がこれらの結果を合格させる可能性がある。バックラインデータはまだ監査と確認手続きを受ける必要があり、これは最終データが私たちが以前に発表した予備データと大きく異なる可能性がある。したがって、最終データが利用可能になる前に、バックラインデータは慎重に表示されなければならない。私たちはまた時々私たちの研究の中間データを開示するかもしれない。私たちが完成可能な研究の中期データは、患者登録の継続とより多くの患者データの獲得に伴い、1つまたは複数の臨床結果が実質的に変化する可能性があるというリスクに直面している。初期または中期データと最終データとの間の不利な違いは、私たちのビジネスの将来性を深刻に損なう可能性があります。しかも、私たちまたは私たちの競争相手が中間データを開示することは私たちの普通株の価格変動を招くかもしれない。
さらに、他の人は、規制機関を含み、私たちの仮定、推定、計算、結論または分析を受け入れないか、またはデータの重要性を異なる方法で解釈またはトレードオフする可能性があり、これは、特定の計画の価値、特定の候補製品または製品の承認または商業化、およびわが社の全体的な状況に影響を与える可能性がある。さらに、私たちが開示する特定の研究または臨床研究に関する開示を選択する情報は、一般に広範な情報に基づいており、あなたまたは他の人は、私たちが決定した重大な情報または他の適切な情報が私たちの開示に含まれていることに同意しない可能性があり、私たちが開示しないことを決定したいかなる情報も、最終的には、特定の生物製薬製品、生物製薬製品候補製品または私たちの業務に関する将来の決定、結論、観点、活動、または他の側面に対して大きな意味を持つと考えられるかもしれない。もし私たちが報告したバックラインデータが実際の結果と異なる場合、あるいは規制機関を含む他の人が結論に同意しない場合、私たちが承認を得て私たちの候補製品を商業化する能力が損なわれる可能性があり、これは私たちの業務、経営業績、見通し、または財務状況を損なう可能性がある。
過去,米国以外でitolizumab(EQ 001)の臨床研究が行われており,今後も米国以外の場所でEQ 101とitolizumab(EQ 001)の臨床研究が継続されている可能性があり,aGVHDにおけるitolizumab(EQ 001)の第3段階の重要な臨床研究や,aGVHDにおけるitolizumab(EQ 001)の第3段階の重要な臨床研究が可能である
52
他の候補製品もあります。FDAはこのような研究のデータを受け入れない可能性があり,この場合,我々の開発計画が延期され,我々の業務に実質的な被害を与える可能性がある.
2017年第4四半期、Bioconはオーストラリアの健康被験者においてitolizumab(EQ 001)の臨床研究を完了し、SC版itolizumab(EQ 001)の安全性と耐性を評価した。この研究はまた、itolizumab(EQ 001)の静脈内投与とアルツモノクロナル抗体の薬物動態を比較し、SC itolizumab(EQ 001)の絶対バイオアベイラビリティを決定するための別個の段階を含むが、最初のリンパ球計数の減少と一過性リンパ球減少の発生により、この段階は早期に終了する。これらのデータをFDAに提出し,我々INDが提出したaGVHD,LN,新冠肺炎治療のための臨床研究の一部とした。しかしながら、FDAは、異なる患者集団の他の適応を有する任意の将来のIND提出に関連する臨床研究を継続することを許可しない可能性があり、高価で時間がかかる追加の第1段階の臨床研究を要求される可能性があり、私たちの開発計画のいくつかの態様を遅らせることは、私たちの業務を損なう可能性がある。
オーストラリアとニュージーランドのサイトを用いてitolizumab(EQ 001)治療がコントロールされていない中~重度喘息の1 b期臨床研究を行い,インドのサイトを用いてitolizumab(EQ 001)による狼瘡とLN治療の1 b期臨床研究を行った。また,米国以外の複数の国からの地点を用いて重要なitolizumab(EQ 001)のaGVHDにおける第3段階臨床研究をヨーロッパ,アジア,他の場所で行っている。われわれが再生不良性貧血患者で行ったEQ 101第二段階臨床研究はオーストラリアとニュージーランドで行われている。FDAはIND下で行われるのではなく、完全にアメリカ国外で行われた臨床研究のデータを受け入れることができるが、このような臨床研究データの受け入れは通常いくつかの条件の制約を受ける。例えば,FDAは臨床研究をGCPに沿って行わなければならないことが求められており,FDAが現場検査が必要であると考えていれば,現場検査で臨床研究のデータを検証できる必要がある。また,臨床研究が米国以外の場所でのみ行われている場合,FDAは通常研究の臨床案をあらかじめコメントしていないため,FDAが非米国臨床研究の研究設計や案が不十分であることを決定する可能性があり,追加の臨床研究が必要である可能性がある。アメリカ国外で臨床研究を行うことも、以下の方面と関連するリスクを含むより多くのリスクに直面させる
私たちは未来に私たちの候補製品をテストするために、より多くの兆候を決定することで、私たちのルートを拡大することに成功しないかもしれない。私たちは、ある候補製品の特定の適応を追求するために限られた資源を費やす可能性があり、他の候補製品を利用することができないか、またはより有利または成功する可能性の高い適応を利用することができる。
私たちの転化生物学計画は、最初に私たちの候補製品が治療的利益を有する可能性のある他の適応を決定する上で希望を示す可能性があるが、様々な理由で、これは、さらなる研究後、私たちの候補製品が有害な副作用を有することが証明される可能性があり、治療効果がないことに限定されるか、または他の特徴は、これらの追加の適応において上場承認および市場で受け入れられる可能性が低いことを示す可能性があることを示している。私たちの候補製品のためにもっと多くの適応を確定する研究計画は大量の技術、財力と人力資源を必要とする。
私たちの財政と管理資源が限られているため、私たちの研究計画を優先しなければならず、私たちの開発努力をいくつかの限られた適応の潜在的な治療に集中させる必要がある。したがって、私たちは、他の適応または任意の将来の候補製品の機会の追求を放棄または延期するか、または後により大きな商業的潜在力を有することが証明された候補製品を剥離することができる。私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。特定の適応候補製品を開発するための支出は、承認された製品や商業的に実行可能な製品を生成しない可能性がある。私たちが候補製品の商業的潜在力や目標市場を正確に評価していなければ、魅力の低い指示を求め、協力、許可、または他の印税手配によって候補製品に貴重な権利を放棄する可能性があり、この場合、私たちは候補製品の独占的な開発と商業化の権利を維持することがより有利になるかもしれない。
私たちの候補製品が規制機関の承認を得ても、私たちは持続的な規制義務と持続的な規制審査の制約を受けることになり、これは多くの追加費用を招く可能性がある。また私たちの誰もが
53
候補製品が承認されれば、ラベルや他の制限や市場撤退を受ける可能性があり、規制要求を守らなかったり、私たちの製品が予期しない問題に遭遇したりすれば、処罰を受けるかもしれません。
我々の候補製品に対する任意の規制承認は、製品上場の承認指示用途の制限または承認条件によって制限される可能性があり、または第4段階の臨床研究、および候補製品の安全性および有効性を監視するモニタリングを含む可能性の高い上場後試験要件を含む可能性がある。さらに、FDAが任意の候補製品を承認する場合、製品の製造プロセス、ラベル、包装、流通、有害事象報告、貯蔵、広告、販売促進、輸入、輸出、および記録保存は、高価で時間がかかる可能性がある広範かつ持続的な規制要件を受けるであろう。これらの要求は、承認後に行われた任意の臨床研究のための安全および他の上場後の情報および報告、登録を提出すること、およびcGMPおよびGCPを継続的に遵守することを含む。私たちは巨額の費用を負担して、このような複雑な規定を遵守することを保障するために時間と労力をかけなければならない。その後、製品には、予期しない重症度や頻度の不良事件、製品による不良副作用、私たちの契約製造業者または製造プロセスが遭遇した問題、または製品承認前または後に規制要求を遵守できなかったことを含む以前の未知の問題が存在することが発見され、以下の状況を招く可能性がある
さらに、任意の候補製品が市場承認された場合、FDAは、その治療法の利点がそのリスクよりも大きいことを確実にするためにREMSを採用することを要求することができ、患者に配布されるリスクを概説する薬物ガイドラインと、医療従事者に発表されるコミュニケーション計画とが含まれる可能性がある。これらの事件のいずれも、関連製品または特定の候補製品に対する市場の受容度を達成または維持することを阻止または維持することができ、私たちの業務、将来性、財務状況、および運営結果を深刻に損なう可能性がある。
また、もし私たちが承認された候補製品があれば、私たちの製品ラベル、広告、販売促進は規制要求と持続的な規制審査を受けるだろう。FDAは生物製薬製品を販売促進する可能性のある声明を厳格に規制している。特に、製品は、当該製品が承認されたラベルに反映されるように、FDA承認されていない用途に使用されてはならない。もし私たちが候補製品のマーケティング承認を得たら、医者は承認されたラベルと一致しない方法で患者に処方するかもしれない。もし私たちがこのようなラベル外の使用を普及させることを発見されたら、私たちは重大な責任を負うかもしれない。FDAや他の機関はラベル外用途の普及を禁止する法律や法規を積極的に実行しており,ラベル外用途の普及が不適切であることが発見された会社は重大な制裁を受ける可能性がある。連邦政府は不正販売促進の疑いのある会社に巨額の民事と刑事罰金を科し、いくつかの会社がラベル外販売促進に従事することを禁止している。FDAはまた、企業に同意法令または永久禁止を締結し、これらの法令または永久禁止に基づいて、特定の販売促進行為を変更または制限することを要求する。
FDAの政策は変わる可能性があり、私たちの候補製品に対する規制承認を阻止、制限、または延期するための追加の政府法規が公布されるかもしれない。米国や海外の将来の立法や行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することはできない。もし私たちが既存の需要の変化に適応できない場合、あるいは新しい需要や戦略を採用することができなければ、あるいは私たちが維持できなければ
54
法規を遵守した場合、私たちは私たちが得る可能性のあるマーケティング承認を失う可能性があり、私たちは利益を達成したり維持することができないかもしれません。これは私たちの業務、将来性、財務状況、運営結果に悪影響を及ぼすでしょう。
たとえ私たちの候補製品がどのような状況でも市場の承認を得ても、それらは医師、患者、病院、医療保険支払人と医学界の他の人が商業成功に必要な市場受容度を達成できないかもしれない。
もし私たちの任意の候補製品が任意の1つまたは複数の適応でマーケティング許可を得た場合、それはまだ医師、患者、第三者支払人、および医学界の他の人の十分な市場受容度を得ることができないかもしれない。もし彼らが十分な受容度に達していなければ、私たちは著しい製品収入を生むことができないかもしれません。私たちは利益を上げることができないかもしれません。市場の受容度は、いかなる兆候でも商業販売のために承認されれば、複数の要因に依存する
私たちは現在マーケティングや販売組織がなく、会社としても製品を商業化した経験がなく、これらの能力を開発するために大量の資源を投入しなければならないかもしれません。もし私たちがマーケティングと販売能力を確立できない場合、あるいは契約第三者と契約を締結して、私たちが許可した任意の製品をマーケティングして販売することができなければ、製品収入を生み出すことができないかもしれません。
私たちは内部販売、マーケティング、流通能力もなく、製品を商業化していない。もし私たちの候補製品が最終的に規制部門の承認を得たら、私たちはそれを効果的にマーケティングして流通することができないかもしれない。私たちは、内部販売、流通、およびマーケティング能力を発展させるためにパートナーを探すか、または大量の財務および管理リソースを投入しなければならないかもしれないが、いくつかは、私たちの任意の候補製品が承認されることを確認する前に約束されるだろう。私たちは、受け入れ可能な財務条項で協力することができないか、またはコンサルタントまたは外部サービスプロバイダを招いて、私たちの販売、マーケティング、および流通機能を支援することができないかもしれません。また、もし私たちが契約者に依存してこれらの機能を履行すれば、私たちの製品の収入と収益性(あれば)は、私たち自身のマーケティング、販売、流通製品の状況を下回るかもしれません。私たちはこのような契約者に対する制御が限られているかもしれません。彼らのいずれも必要な資源と注意力を投入して、私たちの製品を効果的に販売し、マーケティングすることができないかもしれません。私たちが自分で販売、マーケティング、流通機能を実行することを決定しても、私たちはいくつかの追加的なリスクに直面する可能性があります
私たちは激しい競争に直面しています。これは他の人が私たちよりも早く製品を発見し、開発したり、商業化したり、あるいは私たちよりも製品のマーケティングに成功する可能性があります。もし彼らの候補製品が私たちのより安全で効果的であることが証明されたら、私たちのビジネス機会は減少または消滅するだろう。
55
新製品の開発と商業化競争は激しい。我々は製薬,バイオテクノロジー,その他の関連市場で競争を展開しており,これらの市場では免疫性炎症性疾患の治療のための薬物や生物製品が開発されている。もし私たちの競争相手が開発し商業化した製品が私たちが開発する可能性のあるいかなる製品よりも安全で、より効果的で、副作用が少なく、より便利で、あるいは私たちが開発する可能性のあるいかなる製品も時代遅れまたは競争力がなければ、私たちのビジネス機会は減少または消滅するかもしれない。私たちの競争相手も私たちよりも早く製品の発売承認を得るかもしれません。これは私たちの競争相手が市場に入る前に強力な市場地位を築くことにつながるかもしれません。
EQ 101,EQ 302およびitolizumab(EQ 001)と同じ適応を有する他の製品が開発されており,そのいくつかが承認されていることが知られている。AAの治療には,礼来社がFDAから承認されたOLumant,ファイザー社も最近Litfuloに対するFDAの承認を得ている。AA薬物開発に参加する他の民間および上場企業は、Arcutis BioTreateutics、Aslan製薬有限会社、百時美施貴宝社、Concert製薬会社(太陽製薬工業有限会社に買収)、フォード生物科学会社、Horizon治療会社(安進に買収された)、Inmagene生物製薬有限会社、Legacy Healthcare、Nektar治療会社、Ornovi社、輝瑞、q 32生物会社、Reistone Biopma、Zelgen生物製薬有限会社、Zura Bio有限会社を含む。まだ承認されていない乳糜潟製品。乳糜潟に対する開発プロジェクトを有する私営·上場企業には,安進社,Anokion SA社,Calypso Biotech BV社(ノワ製薬に買収),中外製薬会社,IgY免疫技術と生命科学会社,免疫会社,免疫遺伝子X社,主役治療会社,武田製薬会社,Teva製薬会社,Topas治療会社,ジディラ社がある。現在のところFDAが承認していない療法はaGVHDの第一線の治療法に指定されている。二次治療は、2019年にステロイド難治性aGVHDの治療のために承認されたタグ外免疫阻害剤、その治療利益がまだ決定されていない、Incell Corporationのruxolitinibを含む。その他の第一線とステロイド難治性aGVHDの治療に開発計画がある私営と上場企業は、AltruBio,Inc.,ASC Treeutics,CSL Behring LLC,Cynata Treeutics Limited,ElsaLys Biotech,Evive Biotech(一帆製薬有限会社の子会社),Humanigen,Inc.,Maat Pharma SA,Medac GmbH,Mesoblast Limited,深センXbiome Biotech,Co.TR 1 X Inc.,VectivBio Holding AG(Ironwood Pharmarmartics,InZarencs,InVarticc.が承認された。2021年1月に承認された。LN薬物開発に関与する他の私営と上場企業には,アスリコン社,コリス社,CSL Behring有限責任会社,遺伝子技術会社,ジョンソンのヤンソン製薬会社,コザ生命科学社,Nkarta社,ノワ製薬社,Omeros社,ベラ治療会社がある。
私たちの多くの競争相手、例えばファイザー会社と礼来会社などの大型製薬と生物技術会社は、研究開発、製造、臨床前研究、臨床研究を行い、監督管理の許可とマーケティング許可を得た製品の面で私たちよりもっと多くの財力と専門知識を持っている。これらの競争相手はまた合格した科学と管理人員を募集と維持し、臨床研究サイトと臨床研究患者登録を確立し、補充或いは必要な技術を獲得する上で著者らと競争している。さらに、これらの大きな企業は、そのより大きな市場力を利用して、第三者とより有利な流通および販売に関する合意を達成することができ、これにより、我々に対する競争優位性を得ることができるかもしれない。
さらに、特定のカテゴリーの生物製薬製品の中のより多くの候補製品が臨床開発を通じて監督管理審査と承認に入ることに伴い、監督管理機関が必要とする可能性のある臨床データの数量とタイプは増加または変化する可能性がある。したがって、これらのカテゴリの候補製品の臨床研究結果は、市場承認または(承認された場合)商業化に有利な製品ラベルを得るために、これらの製品および候補製品と競合するか、またはより有利なリスク利益プロファイルを表示する必要があるかもしれない。リスク収益プロファイルがこれらの製品または候補製品と競争力がない場合、私たちが開発した製品は商業的に実行できない可能性があり、利益を上げることができない場合、または割引された価格設定や精算を実現できない可能性があります。このような状況で、私たちの未来の製品収入と財政状況は実質的な悪影響を受けるだろう。
製薬とバイオテクノロジー産業の合併と買収は、私たちの数の少ない競争相手により多くの資源を集中させる可能性がある。規模の小さい会社や他のスタートアップ会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配されている。これらの第三者は、合格した科学および管理者を募集し、維持し、臨床研究場所および臨床研究の被験者登録を確立し、EQ 101、EQ 302、itolizumab(EQ 001)または任意の未来計画の補充または必要な技術を取得する上で私たちと競争する。
56
私たちのすべての候補製品の成功に影響を与える重要な競争要素は、それらの有効性、安全性、利便性と精算の可用性である可能性がある。もし私たちが開発、商業化に成功し、競争相手よりも高い精算レベルを実現できなければ、私たちは彼らと競争できなくなり、私たちの業務は実質的に損害を受けるだろう。
私たちの現在の候補製品と私たちは生物製品としての将来の候補製品の承認を求めるつもりですが、予想よりも早く競争に直面するかもしれません。
2010年の患者保護および平価医療法案は、2010年の医療·教育調整法案によって改正され、または2009年の生物製品価格競争および革新法案と呼ばれる副題を含む平価医療法案と総称され、またはBPCIAは、FDA許可の参考生物製品生物と類似または交換可能な生物製品のための短い承認経路を作成する。BPCIAによると,生物類似製品の申請は,参考製品が初めてFDA許可を得た4年後にFDAに提出されなければならない。また,FDAによる生物類似製品の承認は,参考製品が初めて許可された日から12年後に発効する可能性がある。この12年間の独占期間内に、FDAが競合製品の完全なBLAを承認した場合、スポンサー自身の臨床前データおよび十分かつ良好に制御された臨床研究からのデータを含み、その製品の安全性、純度および有効性を証明するために、別の会社は依然としてこの参照製品の競合バージョンを販売する可能性がある。この法律は複雑であり、FDAはまだ説明して施行している。したがって、その最終的な影響、実施、そして意味には不確実性がある。FDAがいつこれらのBPCIAを実施するためのプロセスを完全に採用できるかどうかは不明であるが、どのようなプロセスも私たちの生物製品の将来のビジネスの将来性に重大な悪影響を与える可能性がある。
BLAによりバイオ製品として承認されたいずれの我々の候補製品も12年の専門期間を得る資格があるべきであると考えられる。しかしながら、国会の行動または他の理由により、このような排他性は短縮される可能性があり、またはFDAは、我々の製品候補製品を競合製品の参考製品とみなさないかもしれないが、これは、予想よりも早く生物類似競争のための機会を創出する可能性がある。BPCIAの他の面では,そのいくつかがBPCIAの排他的条項に影響を与える可能性があり,最近の訴訟のテーマでもある.さらに、承認されると、生物類似体が私たちのいずれかの参考製品をどの程度置換するかは、非生物製品の伝統的な模造薬代替と類似しており、これはまだ発展中のいくつかの市場および規制要因に依存するかどうかは不明である。
もし私たちの候補製品の市場機会が私たちが思っているより小さいなら、私たちの潜在収入は不利な影響を受けるかもしれません。私たちの業務は影響を受けるかもしれません。
われわれはEquillium領域のitolizumab(EQ 001)の権利のみを有しており,自己免疫や炎症性疾患のためのitolizumab(EQ 001)の開発に専念し,aGVHDやLNの治療のための患者の開発を計画している。我々はEQ 101とEQ 302の世界的な権利を有しており,AAと胃腸疾患(例えば乳糜潟)のための候補製品をそれぞれ開発する予定である。我々の候補製品治療から利益を得る可能性のあるアドレス可能な患者集団の予測は推定に基づいており,誤りであることが証明されている可能性がある。もし私たちのいかなる推定も正確でなければ、私たちの候補製品の市場機会は著しく減少し、私たちの業務に実質的な影響を与えるかもしれない。
私たちは最終的に孤児薬をEQ 101またはitolizumab(EQ 001)として指定する潜在的な利点を認識しないかもしれない。
EQ 101はすでにFDAとヨーロッパ薬品管理局からCTCL孤児薬物の称号を授与され、itolizumab(EQ 001)はすでにFDAからaGVHDの予防と治療の孤児薬物称号を授与された。FDAは,米国で患者が20万人未満のまれな疾患を治療する薬剤,あるいは20万人を超えるが治療薬の開発·マーケティングコストを回収できないと予想される薬剤に孤児を指定している。孤児薬物はマーケティング申請中に処方薬使用料を受け取る必要がなく、薬物開発スポンサーに何らかの税金免除を受ける資格があり、7年間の市場排出期(例外的な場合もある)を得る資格がある可能性がある。しかし、孤児薬物を指定することは、候補製品の開発時間や監督審査時間を短縮することも、監督審査や承認過程において候補者にいかなる利点をもたらすこともない。市場独占経営権を獲得しても、FDAが後続薬がより安全で、より効率的で、または患者看護に重大な貢献をしていると判断した場合、それは依然として同じ有効成分を含み、同じ孤児適応に使用される別の薬物を承認することができ、孤児薬物製造業者が稀な疾患または疾患を有する患者の需要を満たすために十分な数の孤児薬を確保できない場合、孤児の独占経営権が失われる可能性がある。FDAが最初の指定要求に実質的な欠陥があると後に決定すれば,孤児薬の排他性も失われる可能性がある。また,孤児薬の排他性は,FDAが異なる活性成分を含む同じまたは類似した適応の競合薬を承認することを阻止しない。孤児薬の排他性を失ったら私たちは成功できません
57
私たちの合格候補製品をカバーする任意の残りの特許を強制執行することは、私たちが予想よりも早く生物類似競争の影響を受けるかもしれない。さらに、その後の薬物がEQ 101またはitolizumab(EQ 001)と同じまたは同様の適応で発売されることが承認された場合、孤児薬の排他性を考慮することなく、より激しい競争に直面し、市場シェアを失う可能性がある。
FDAの高速チャネル指定は、実際には、より速い開発や規制審査または承認プロセスをもたらすことはないかもしれません。
AGVHDおよびLNを治療するためのitolizumab(EQ 001)の高速チャネル指定が得られている。1つの製品が深刻または生命に危険な疾患を治療するために使用され、そのような疾患が満たされていない医療需要を解決する可能性を示す場合、製品スポンサーはFDA迅速チャネル認証を申請することができる。高速チャネル指定があっても、従来のFDAプログラムと比較して、より速い開発過程、審査、または承認を経験しない可能性があります。FDAが我々の臨床開発計画のデータが高速チャネル指定をサポートしなくなったと考える場合,その指定を撤回する可能性がある。
マーケティングの承認を得ても、不利な定価規制や第三者保険·精算政策により、承認されたどの製品も商業化することに成功できない可能性があり、承認された任意の製品を利益的に販売することが困難になる可能性があります。
政府または他の第三者支払人から製品の保証範囲および十分な補償承認を得ることは、時間がかかり、高価なプロセスであり、支払人に、私たちが承認した製品の使用を支援する科学的、臨床的、および費用便益データを提供する必要があるかもしれない。新たに承認された製品については,このような保険や精算を得る上で大きな遅延が生じる可能性があり,保険範囲はFDAが承認したものよりも目的が限られている可能性がある。さらに、保険および精算を受ける資格があることは、製品がすべての場合に支払いを受けることを意味するのではなく、研究、開発、知的財産権、製造、販売、および流通費用を含む、私たちのコストをカバーするレートで支払うことを意味するわけではない。新製品の臨時精算水準(適用すれば)も私たちのコストを支払うのに十分ではない可能性があり、恒久的にならない可能性があります。販売率は製品の使用や臨床環境によって異なる可能性があり,すでに低コスト製品のために設定された精算レベルに基づいている可能性があり,他のサービスの既存支払いにも組み込まれている可能性がある。製品の正味価格は、政府医療計画または個人支払者が要求する強制的な割引またはリベート、将来の任意の薬品価格を制限する法律、および将来的に米国価格よりも低い価格で製品が販売される国からの輸入を制限する法律の任意の緩和によって低下する可能性がある。
新たに承認された製品の保険カバーと精算に関する不確実性が大きい。第三者支払者は精算戦略を設定する際には通常Medicare保証政策と支払い制限に依存するが、Medicare保証と精算確定以外にも、彼ら自身の方法と承認の流れがある。私どもが開発した任意の候補製品のカバー範囲と精算金額についての決定は支払者ごとに行います。第三者支払者は、1つの薬剤に保険を提供することを決定し、他の支払者もその薬剤に保険及び適切な補償を提供することを保証することはできない。また、第三者支払者が治療に保険を提供することを決定することは、十分な販売率を承認することを意味するものではない。第三者決済者は、価格に挑戦し、医療の必要性を審査し、医療製品、療法、サービスの費用対効果を審査するとともに、それらの安全性と有効性を疑問視するようになっている。
第三者支払者の製品使用状況の決定を含む第三者支払人の保証範囲と精算は多くの要素に依存する可能性がある
私たちが商業化しているどの製品にも保険や精算があることを確実にすることはできません。もし保険と精算があれば、清算レベルはいくらですか。ブランド療法や医師の監督下で実施される療法は高い価格に関連することが多いため,我々の製品のために十分な精算を得ることは特に困難である可能性がある。同様に,我々の候補製品は医師が管理する注射剤であるため,製品自体が単独で精算される可能性もあるし,できない可能性もある。逆に,管理医は我々の製品を用いた治療やプログラムを提供することで精算される可能性がある。政府の資金援助と個人支払者から承認された製品の保険と十分な返済率を迅速に得ることはできません
58
私たちの開発は、私たちの経営業績、製品の商業化に必要な資金を調達する能力、そして私たちの全体的な財務状況に実質的な悪影響を及ぼすかもしれません。
精算は私たちが市場で承認されたどんな製品の需要と価格に影響を及ぼすかもしれない。第三者支払者が特定の製品のために保険を受けていると仮定すると,それによる精算支払率が十分に高くない可能性があり,あるいは患者が受け入れられないと考えて受け入れられないほど高い共同支払いが必要となる可能性がある。各第三者支払者は,治療に保険を提供するかどうか,治療のためにメーカーにどの程度の金額を支払うか,その保険薬品リストまたは処方のどのレベルに置かれるかを決定する。第三者支払者の処方上の場合は,通常,患者が治療に要する共同費用を得ることが決定され,患者や医師のこのような治療に強い影響を与える可能性がある。自分の病態を治療するために処方薬を服用している患者とその処方を行う医師は,通常第三者支払者に依存してこれらの薬物に関する費用の全部または一部を精算する。患者は保険を提供しなければ、私たちの製品を使用することはあまりできません。私たちの製品の全部または大部分のコストを支払うのに十分な費用を精算するのに十分です。したがって、カバー面と十分な精算は新製品の受容度に必須的だ。カバー範囲の決定は臨床および経済基準に依存する可能性があり、より成熟またはより低コストの治療代替製品が利用可能になった場合、またはその後に利用可能である場合、これらの基準は新製品に不利である。また、私たちまたは私たちの協力者が私たちの候補製品のためのセット診断テストを開発すれば、このようなテストは単独の保証と精算プロセスによって制限され、候補製品のために求めた保証と精算以外に制限される。
管理的ヘルスケアの傾向,ヘルスケア組織の日増しに増加する影響力,追加的な立法変化により,我々の候補製品の販売に関する価格設定圧力に直面することが予想される。全体的に,医療コストの下り圧力は非常に大きくなり,特に処方薬,医療機器,外科手術プログラム,その他の治療が行われている。そのため、新製品の商業化成功にますます高い壁が設けられている。
製造に関するリスクと第三者への依存は
製薬製品、特に生物製品の製造は複雑で、私たちは私たちの候補製品を生産、流通、配布する時に困難に直面するかもしれない。Bioconを含むCMOの場合、我々のitolizumab(EQ 001)の独占CMOがこのような困難に遭遇した場合、私たちは臨床研究に候補製品を提供する能力、市場承認を得る能力、または私たちが商業的に私たちの製品を供給する能力を獲得し、承認されれば延期または停止される可能性がある。
私たちは生物製造の経験もなく、製品製造、貯蔵、流通やテストの施設も持ったり運営したりすることもなく、これらの施設を所有したり運営したりすることも望んでいません。私たちは私たちの候補製品の臨床と商業供給を満たすために第三者CMOに完全に依存している。しかし、製薬製品、特に生物製品の製造過程は複雑で、高度な監督管理を受け、そして多重リスクの影響を受けている。汚染、設備故障、設備設置或いは操作が不適切であるため、サプライヤー或いはオペレータの誤り、生産量の不一致、製品特性の変化及び生産過程の拡張が困難であるため、このような製造は製品損失の影響を受けやすい。正常製造プロセスとのわずかなばらつきでも生産量の低下,製品欠陥やその他の供給中断,コスト上昇を招く可能性がある。もし私たちのメーカーの施設で微生物、ウイルス、あるいは他の汚染が発見された場合、これらの施設は汚染を調査·修復するために長い時間閉鎖する必要があるかもしれないが、これは臨床研究を遅延させ、薬品コストの上昇を招き、私たちの業務に悪影響を及ぼす可能性がある。また,我々のメーカーの工場が米国以外にあれば,現在のitolizumab(EQ 001)のように,薬品の生産,流通,交付も国家法律法規によって制約されている。他の国の法律法規のいかなる変化、あるいは地政学問題或いは健康流行病に関連する生産或いはサプライチェーンの中断は、臨床研究を延期し、薬品コストの上昇を招き、著者らの業務に不利な影響を与える可能性がある。さらに、FDAが、我々の製造業者がcGMPを管理する法律および法規を含むFDAの法律および法規に適合していないと判断した場合、FDAは、欠陥が是正されるまでBLAの承認を拒否するか、またはBLA内の製造業者を法規に適合する製造業者に置き換える可能性がある。
そのほか、大規模生産は臨床研究或いは商業規模のリスクに用いられ、その他の以外に、コスト超過、技術拡大の潜在問題、技術再現性、安定性問題、cGMP遵守、ロット一致性及び原材料の適時供給と交付を含む。私たちの候補製品または未来の候補製品の規制承認を得ても、私たちのCMOがFDAや他の規制機関が許容できる規格で承認された製品を生産し、製品が発売される可能性のある要求を満たすために十分な数の製品を生産することができるか、または将来の潜在的な需要を満たすことができる保証はありません。また、資源制限や自然災害、労使紛争、不安定な政治環境や公衆衛生流行病により、私たちの契約メーカーは製造や輸送困難に遭遇する可能性がある。もし私たちのメーカーが
59
臨床研究や商業化のために十分な数の薬物を生産すれば、商業化の努力が損なわれ、これは私たちの業務、財務状況、運営結果、および成長の将来性に不利な影響を与える。
薬品生産プロセス、特に生物プロセスとポリペプチド合成を拡大することは、困難かつ不確定な任務であり、私たちのCMOは生産をさらに拡大し、生産を他の場所に移したり、その生産能力を管理して、私たちが供給したEQ 101、EQ 302、itolizumab(EQ 001)または他の未来の候補製品(他の生物製品を含む)または製品の需要を満たす実施と開発過程を達成するために必要な能力がないかもしれない。
2017年5月、私たちはBioconと独占的な臨床供給協定を締結し、将来的にBioconと独占的な商業供給協定を締結することに同意した。Bioconはインドバンガロールに位置するFDA規制施設でitolizumab(EQ 001)を生産している。私たちのBioconへの依存は、私たちが臨床と商業供給itolizumab(EQ 001)を実現する能力に関連するさらなるリスクと不確実性に直面させた。例えば,2020年3月には,コロナウイルスの伝播により,インド政府は26種類の活性薬物成分とその製造薬品の輸出を制限している。このような輸出制限は無期限であり、修正されたり拡大されるかもしれない。輸出制限がitolizumab(EQ 001)を含むように拡大された場合、私たちのitolizumab(EQ 001)の供給は無期限中断、遅延、または停止する可能性があり、私たちが行っている臨床研究を含むitolizumab(EQ 001)の能力の開発を継続し、重大な影響を受ける可能性があり、薬品コストの上昇を招き、私たちの業務に悪影響を及ぼす可能性がある。Bioconが我々の製造要求(輸出制限や他の理由による)を満たすことができない場合、製造を第三者にアウトソーシングする権利があり、共同指導委員会は製造を第三者に移すことを決定する可能性がある。しかしながら、生物学的製品の生産を新しい契約製造業者に移すことは、itolizumab(EQ 001)に関連するものであっても、私たちの現在または未来の任意の候補製品であっても、長い可能性があり、著しい追加コストを伴う可能性がある。契約メーカーとの製造過程を十分に検証して拡大することができても、契約メーカーと商業供給協定を交渉する必要があり、受け入れられる条項について合意できるかどうかは定かではありません。さらに、Bioconは、Biocon不足後に第三者と交渉した後であっても、itolizumab(EQ 001)の独占製造権を再獲得するいくつかの権利を有しており、これは、Biocon以外の任意の第三者製造業者がitolizumab(EQ 001)と交渉することを困難にし、高価にする可能性がある。
著者らは引き続きCROに依存して臨床研究を行い、いくつかの研究と臨床前研究を行う予定である。これらの第三者がその契約義務を満足に履行できず、適用された規制要件を遵守できなかった場合、または予想される期限までに完了した場合、私たちの開発計画は延期されたり、コストが増加したりする可能性があり、または私たちは規制部門の承認を得ることができない可能性があり、これらはすべて私たちの業務、財務状況、運営結果、および見通しに悪影響を及ぼす可能性がある。
私たち自身は臨床前テストや臨床研究のすべての側面を独立して行うことができない。したがって、私たちは第三者に依存して、私たちが行っている未来のEQ 101、EQ 302およびitolizumab(EQ 001)の臨床前研究および臨床研究、ならびに任意の他の候補製品の未来の臨床前研究および臨床研究を行う。したがって,これらの研究の起動と完了時間はこれらの第三者によって部分的に制御され,我々の開発計画が遅延する可能性がある.
具体的には,CRO,臨床研究者,コンサルタントがこれらの研究の進行とその後のデータ収集と分析に重要な役割を果たすことが望まれる。しかし、私たちは彼らの活動のすべての側面を統制できないだろう。しかし、私たちはすべての臨床研究が適用された方案と法律、法規と科学基準に従って行われることを保証する責任があり、私たちのCROと他の第三者への依存は私たちの監督責任を免除しない。もし私たちのCROが不道徳、不法、または不規則な活動に従事している場合、このような行為は私たちの業務に悪影響を及ぼす可能性があります。また、もし著者らがこのような不正行為のためにCROとの契約関係を終了すれば、異なるCROへの移行は遅延、妨害或いは他の方法で臨床研究の進展に不利な影響を与える可能性がある。我々と我々のCROはGCP要求を遵守することを要求されており,これらの要求はFDAが臨床開発における候補製品に対して実行する法規とガイドラインである。監督管理機関は定期的に研究スポンサー、臨床研究調査者、臨床研究場所を検査することによって、これらのGCP要求を実行する。もし私たちまたは私たちの任意のCROまたは臨床研究サイトが適用されたGCP要求を遵守できなかった場合、私たちの臨床研究で生成されたデータは信頼できないと考えられる可能性があり、FDAは私たちのマーケティング申請を承認する前に追加の臨床研究を行うことを要求するかもしれない。また,われわれの臨床研究はcGMP法規下で生産された製品を用いて行わなければならない。もし私たちがこれらの規定を守らなければ、私たちに臨床研究を停止および/または繰り返すことを要求するかもしれません。これは上場承認過程を延期します。
私たちが依存しているどのようなCRO、臨床研究調査者、または他の第三者が、私たちの開発活動に十分な時間および資源を投入するか、または契約の要求に従ってタスクを実行することは保証されません。もしこれらの第三者のいずれかが予想される期限内に完成できなかった場合、私たちの臨床方案を遵守したり、法規の要求を満たしたり、他の方法で目標を達成しなかったり、私たちとの協力を終了したりすることができなければ、私たちの開発計画の期間は延長されるかもしれません
60
遅延や私たちの開発活動は一時停止または終了される可能性がある。私たちの臨床研究サイトが何らかの理由で終了した場合、これらの被験者を別の合格した臨床研究サイトに移すことができない限り、このような臨床研究に参加する被験者の後続情報を失う可能性があり、これは困難であるか不可能である可能性がある。また、私たちの臨床研究の臨床研究調査員は時々私たちの科学顧問や顧問を担当し、このようなサービスのために現金や株式補償を受けるかもしれない。これらの関係と任意の関連する賠償が知覚または実際の利益の衝突をもたらし、またはFDAが財務関係が研究の解釈に影響を及ぼす可能性があると結論した場合、適用される臨床研究場所で生成されるデータの完全性が問われる可能性があり、臨床研究自体の効用が脅かされる可能性があり、これは、私たちが提出した任意のFDAによって提出されたマーケティング申請が延期または拒否される可能性がある。そのような遅延または拒否のいずれも、EQ 101、itolizumab(EQ 001)、または任意の将来の候補製品を商業化することを阻止することができる。
さらに、これらの第三者も他のエンティティと関係がある可能性があり、その中のいくつかは私たちの競争相手である可能性があり、彼らはまたこれらの競争相手のための臨床研究や他の生物製薬製品開発活動を行っている可能性があり、これは私たちの競争地位を損なう可能性がある。これらの第三者が規制要件または私たちが規定した規程に従って契約責任を成功的に履行し、予想される期限内に私たちの臨床研究を完了または行うことができない場合、私たちはEQ 101、itolizumab(EQ 001)、または任意の将来の候補製品の発売承認を得ることができないか、または延期することができず、私たちの製品の成功商業化の努力を得ることができないか、または延期することができないだろう。
私たちの契約者への依存は、私たちのビジネス秘密を共有することを要求し、これは、競争相手がこれらの秘密または私たちの商業秘密が流用または開示されていることを発見する可能性を増加させる。
私たちは契約者に依存して私たちの候補製品を研究、開発、製造するので、私たちは彼らとビジネス秘密を共有しなければならない。商業秘密および他の機密情報を共有する必要は、そのような商業秘密が我々の競争相手に知られ、意図せずに他人の技術に組み込まれるか、または開示されるか、または秘密協定に違反して使用されるリスクを増加させる。私たちの独自の地位が私たちのノウハウおよびビジネス秘密にある程度基づいていることを考慮すると、競争相手は、私たちのビジネス秘密または他の許可されていない使用または開示を独立して発見することは、私たちの競争地位を損なうことになり、私たちのビジネスに大きな悪影響を及ぼす可能性があります。
私たちのコンサルタント、従業員、請負業者、およびコンサルタントとの合意には、いくつかの限られた出版権が含まれている可能性がある。例えば、私たちが協力する可能性のある任意の学術機関は、そのような協力によって生成されたデータを発表する権利を付与されるかもしれません。任意の共同研究開発プロジェクトは、私たちの研究開発または同様の合意の条項に従って商業秘密を共有することを要求するかもしれません。私たちは私たちのビジネス秘密を保護しようと努力しているにもかかわらず、私たちの競争相手は、私たちの合意に違反したり、独立した開発をしたり、私たちの任意の協力者によって情報を発表することによって、私たちのビジネス秘密を発見するかもしれない。競争相手は私たちのビジネス秘密が私たちの競争地位を損なうことを発見し、私たちの業務に悪影響を及ぼすだろう。
知的財産権に関するリスク
もし私たちが私たちの候補製品をカバーする知的財産権を獲得したり保護することができない場合、あるいは知的財産権保護の範囲が十分でなければ、私たちの競争相手は私たちと似ているまたは同じ製品を開発し、商業化する可能性があり、私たちは私たちの市場で効果的に競争することができないかもしれない。
私たちの成功は、私たちおよびitolizumab(EQ 001)について、Bioconが、EQ 101、EQ 302およびitolizumab(EQ 001)を含む当社の独自技術、研究計画、および候補製品に関連する特許および他の知的財産権を確立、維持、保護する能力があり、他人の知的財産権を侵害することなく運営されることに大きく依存する。特許訴訟プロセスは高価で時間がかかり、私たちと私たちの現在または未来のライセンシー、ライセンシー、またはパートナーは、すべての必要または望ましい特許出願を合理的なコストまたはタイムリーに準備、提出、起訴することができないかもしれない。私たちまたは私たちの現在および未来のライセンシー、ライセンシー、またはパートナーも、特許保護を得る前に、開発および商業化活動中に私たちが行った研究または発明の特許可能な態様を識別できない可能性がある。私たちは、私たちの研究開発計画にアクセスする権利のある特許に関する当事者と秘密保持協定を締結していますが、例えば、私たちの従業員、会社協力者、外部科学協力者、CRO、契約製造業者、コンサルタント、独立請負業者、コンサルタント、および他の第三者は、これらの当事者のいずれかがこれらの合意に違反し、特許出願を提出する前にこのような結果を開示し、私たちの研究計画に関連する技術を求める特許保護の能力を危うくする可能性があります。さらに、場合によっては、私たちは、第三者から許可を得ることができ、私たちの許可者、被許可者、またはパートナーの技術に依存することができるかもしれない技術を含む、特許出願の準備、提出および起訴を制御する権利がない場合がある。したがって、このような特許および出願は、私たちの業務の最適な利益に合った方法で起訴され、強制されてはならない。私たちの現在または未来のライセンス者、ライセンシー、またはパートナーが、そのような特許および他の知的財産権を確立、維持、または保護できない場合、そのような権利は減少する可能性があるまたは
61
淘汰された。もし私たちのライセンシー、ライセンシー、またはパートナーがいかなる特許権の起訴、維持または実行に関して完全に協力しないか、または私たちの意見に同意しない場合、これらの特許権は損害を受ける可能性があります。全世界の健康問題に関する公共政策として、アメリカ政府と国際政府機関は巨大な圧力に直面する可能性があり、アメリカ国内と国外の成功が証明された疾病治療方法に対する特許保護範囲を制限することが求められている。
バイオテクノロジー会社の特許地位は通常高度に不確定であり、複雑な法律や事実問題に関連しており、近年多くの訴訟のテーマとなっており、最高裁の裁決を含む裁判所の裁決を招き、将来の特許権を執行する能力の不確実性を増加させている。また、外国の法律は米国の法律のように私たちの権利を保護し、外国の競争相手により良い機会を与え、競争相手の候補製品を創造、開発、販売することができない可能性があり、その逆も同様である。私たちは、EQ 101、EQ 302およびitolizumab(EQ 001)、および私たちの研究計画に関連する技術のような、私たちの保留特許出願における私たちの候補製品に対する権利要件を決定することができず、米国特許商標局または米国特許商標局または外国特許庁によって特許を申請可能とみなされる。さらに、それらが挑戦されていなくても、私たちの製品の組み合わせにおける特許は、第三者が関連技術を実践することを排除したり、私たちの権利要件をめぐる他の人の設計を阻止するのに十分ではない可能性がある。したがって、私たちと現在または未来のライセンシー、ライセンシーまたはパートナーの特許権の発行、範囲、有効性、実行可能性、および商業的価値は非常に不確実である。私たちおよび私たちのライセンシー、ライセンシーまたはパートナーの係属中および将来の特許出願は、私たちの技術または製品の全部または一部、またはそれらの予期される用途、製造方法または処方を保護する特許の発行を招くことなく、または他社が競合技術および製品を商業化することを効果的に阻止する可能性がある。特許審査プロセスは、私たちまたは私たちのライセンシー、ライセンシーまたはパートナーが、私たちまたは私たちのライセンシー、ライセンシーまたはパートナーの係争および将来の特許出願の特許請求範囲を縮小することを要求することができ、これは、入手可能な特許保護範囲を制限する可能性がある。過去、私たちはいつも私たちの特許出願において最初に求められた全面的な特許保護を得ることができるわけではなく、上述したように、多くのバイオテクノロジー特許訴訟の典型的な状況のように、私たちは特許訴訟過程の一部として特許請求を縮小または除去することを要求されてきた。さらに、業務および/または法的戦略の変化により、私たちまたは私たちのライセンシーはこれらの特許出願を放棄しているので、私たちまたは私たちのライセンシーが提出したいくつかの特許出願は特許を生成していない。
私たちの特許および特許出願に関連する可能性のあるすべての既存技術が、保護を要求する発明の優先日の前に、関連分野の技術者によって、または入手可能と考えられていた情報が発見されたことを保証することはできない。そのような従来技術が存在する場合、特許を無効にするか、または係属中の特許出願から特許を発行することを阻止することができ、第三者が既存技術の発行前に米国特許商標局に提出する必要があるかもしれない。たとえ特許が確実に発行されても、これらの特許が私たちの候補製品をカバーしていても、第三者は法廷または特許庁で訴訟または反対、妨害、再審査、許可後の審査、当事者間の審査、廃止または派生訴訟、または同様の訴訟を提起することができ、そのような特許の有効性、実行可能性または範囲を疑問視することができ、これは特許主張の縮小または無効を招く可能性があり、第三者が私たちの候補製品を商業化し、私たちに支払うことなく、または私たちの技術および製品の特許保護期間を制限することを可能にするかもしれない。このような訴訟を開始する法的敷居が低い可能性があるため,勝訴確率の低い訴訟であっても起動可能である.さらに、私たちの特許および特許出願によって提供される保護の広さまたは強度が脅かされている場合、結果にかかわらず、会社が現在または将来の候補製品を許可、開発、または商業化することを阻止することができる。私たちおよび私たちのライセンシー、ライセンシー、またはパートナーの特許出願は、特許が他の出願から発行されるまで、かつ、発行された特許要件が技術の範囲をカバーしない限り、これらの出願で要求される技術を実施する第三者に対して強制的に実行することはできない。
米国および他のほとんどの国/地域の特許出願は、出願後しばらくは秘密であり、一部の特許出願は発行前に秘密であるため、私たちまたは私たちの許可者が、私たちの研究計画および候補製品(EQ 101、EQ 302およびitolizumab(EQ 001))に関連する特許出願を最初に提出した会社であることを確認することはできない。私たちが効果的で強制的に実行可能な特許を持っている場合であっても、他の当事者が彼らが私たちの出願日前にその発明を商業に使用していることを証明することができれば、または他方が強制許可から利益を得ることができれば、他の人が私たちの発明を実践することを排除することはできない。しかも、特許の寿命は限られている。アメリカでは、すべての維持費が適時に支払われれば、特許の自然失効時間は通常、米国で最初の出願日から20年である。様々な延期があるかもしれないが、特許の有効期限とその提供される保護は限られている。私たちの候補製品をカバーする特許を取得しても、製品の特許有効期限が満了すると、生物学的類似薬や模倣薬を含む競争薬物からの競争に直面する可能性がある。
“ハッジ·ワックスマン法案”に基づいて米国および外国で同様の法律に基づいて特許期間を延長することができなければ、EQ 101、EQ 302、itolizumab(EQ 001)、または私たちが決定する可能性のある任意の他の候補製品に対する独占的なマーケティング期間を延長することができなければ、私たちの業務は実質的に損害を受ける可能性がある。
62
新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちが持っている特許と許可された特許の組み合わせは、他社が私たちと似ているか同じ製品を商業化することを排除するために十分な権利を提供してくれないかもしれない。私たちは私たちが特許を起訴したどの国/地域でも特許期間の延長を受けることを望んでいる。これには、米国における1984年の薬品価格競争および特許期限回復法案、または特許期限が特許満了後最大5年間延長されることを可能にするハッジ-ワックスマン法案が含まれる。ハッジ·ワックスマン法は、FDA規制審査中に失われた特許期間の補償として、各FDAが承認した製品に対して最大1つの特許を延長することを可能にする。特許期間の延長は、製品承認日から計14年の期間を超えてはならず、当該承認された薬品、その使用方法又はその製造方法に関連する請求項のみが延長することができる。私たちの候補製品が規制部門の承認を得られれば、特定の国/地域でも特許期間を延長することができる。しかし、米国の適用当局は、FDAとUSPTO、およびどのような同等の外国規制機関も含めて、このような延期が利用可能かどうかの評価に同意しない可能性があり、私たちの特許延期の承認を拒否するか、または私たちが要求したよりも限られた延期を承認する可能性があるかもしれない。このような状況が発生すれば、私たちの競争相手は私たちの開発と臨床研究への投資を利用して、私たちの臨床と臨床前データを参考にして、他の場合よりも早く彼らの製品を発売するかもしれない。
私たちの所有権の未来の保護度は不確定で、私たちは予測できません
私たちはBioconから許可を得た知的財産権に依存して、私たちの許可を終了することは重大な権利の損失を招く可能性があり、これは私たちの業務を損なうだろう。
私たちは現在Bioconから私たちの業務に非常に重要ないくつかの知的財産権の許可を得ており、将来私たちは追加の協定を締結し、貴重な知的財産権や技術の許可を提供してくれるかもしれません。私たちはBioconにある程度依存して特許出願を提出し、他の方法で私たちが許可した知的財産権を彼らの影響を受けないように保護する。私たちはこのような活動や私たちの許可内の知的財産権に関連する可能性のある他の知的財産権の制御が限られている。例えば、私たちはBioconのこのような活動が適用された法律および法規に準拠しているか、または効果的かつ強制的に実行可能な特許および他の知的財産権を生成するだろうと決定することはできない。私たちはBioconが知的財産権の第三者侵害者に対して侵害訴訟を提起したり、私たちに許可されたいくつかの知的財産権を弁護する方法に限られた支配権を持っている。私たちの許可者たちの権利侵害訴訟や弁護活動は私たち自身が行ったほど激しくないかもしれない。
さらに、ライセンス内特許は、1つまたは複数の第三者の権利によって保持される可能性がある。さらに、私たちはBioconの既存のライセンスと規定されており、未来の合意はまた私たちが様々な勤勉さ、マイルストーン支払い、特許権使用料、保険、そして他の義務を負担することを規定するかもしれない。もし私たちがこれらの義務を履行できなかった場合、私たちは損害賠償金の支払いを要求される可能性があり、私たちの許可側は許可を終了する権利がある可能性があり、この場合、私たちはこのような許可知的財産権がカバーする製品を開発またはマーケティングすることができず、競争相手や他の第三者は私たちと同じ技術や製品を得ることができるかもしれない。もし現在又は将来のライセンスが終了した場合,ライセンス者がライセンス条項を遵守しない場合,ライセンシーが侵害第三者に対してライセンスの特許を強制的に執行できなかった場合,我々の業務は影響を受けるであろう
63
許可された特許または他の権利が無効または強制的に実行できないことが発見された場合、または許容可能な条項で必要な許可を締結することができない場合、双方に許可を提供する。さらに、現在または将来のライセンスが終了した場合、または基礎特許が予期される排他性を提供できなかった場合、競争相手または他の第三者は、規制部門の承認を求め、私たちと同じ製品を販売する自由を得ることができる。さらに、私たちの許可者は私たちに許可されていない知的財産権を所有したりコントロールしたりする可能性がありますので、その是非にかかわらず、私たちは私たちが許可者の権利を侵害しているか、または他の方法で侵害しているというクレームを受ける可能性があります。ライセンス契約によると、以下の事項に関する紛争を含む知的財産権紛争が発生する可能性があります
さらに、知的財産権または技術ライセンスプロトコルは、私たちの既存のプロトコルを含めて複雑であり、このようなプロトコルのいくつかの条項は、様々な解釈の影響を受ける可能性がある。可能性のある契約解釈の相違の解決は、関連する知的財産権または技術に対する私たちの権利と考えられる範囲を縮小するか、または関連協定の下での私たちの財務または他の義務を増加させる可能性があり、いずれも、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。私たちが許可している知的財産権をめぐる紛争が許容可能な条項で現在の許可手配の能力を維持していることを阻害したり、損害を与えたりすれば、影響を受けた候補製品の開発に成功し、商業化することができない可能性がある。私たちは通常、私たちが許可した知的財産権保護に関するすべてのリスクに直面しており、これらのリスクは以下のとおりである。もし私たちまたは私たちの許可者がこの知的財産権を十分に保護できなければ、私たちの製品商業化能力は影響を受ける可能性がある。
私たちの手続きは第三者が持っている独占権を使用する必要があるかもしれないので、私たちの業務の成長は、私たちがこれらの独占権を取得、許可、または使用する能力にある程度依存するかもしれない。私たちは、私たちの候補製品にとって必要と考えられる任意の成分、使用方法、プロセス、または他の第三者知的財産権を第三者から得ることができないかもしれません。第三者知的財産権の許可·買収は競争分野であり、一部のより成熟した企業も魅力的と考えられる第三者知的財産権許可または買収戦略を求めている。これらの老舗会社は私たちより競争優位を持っているかもしれません。それらの規模、現金資源及びより強い臨床開発と商業化能力のためです。
しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。私たちはまた私たちの投資が適切な見返りを得るための条項の許可や第三者知的財産権を得ることができないかもしれない。また、将来の製品の販売にどのくらいの印税義務が支払われるかは確定できませんが、金額は大きいかもしれません。私たちの将来の印税義務の金額は、開発や商業化に成功した製品で使用される技術や知的財産権にかかっています(あれば)。したがって、私たちが製品を開発して商業化することに成功しても、私たちは利益を達成したり維持することができないかもしれない。
将来的には、私たちは第三者技術の追加許可を得る必要があるかもしれません。これらの許可は私たちに提供できないかもしれないし、商業的に不合理な条項でしか得られないかもしれません。これは、私たちがより高価で不利な方法で私たちの業務を運営することにつながるかもしれません。
時々、私たちは、我々の候補製品をさらに開発するために、またはEQ 101、EQ 302、itolizumab(EQ 001)、および/または他のようなEQ 101、EQ 302、itolizumab(EQ 001)および/または他の製品をさらに開発するために、他の第三者から我々の治療研究計画に関連する技術的許可を得る必要があるかもしれない。もし私たちが私たちの候補製品を製造、使用、または販売するために必要な任意のこのような特許を含む任意の第三者技術の許可を要求された場合、私たちは商業的に合理的な条項でそのような許可を得ることができないか、またはそのような許可を得ることができないかもしれない。私たちの任意の候補製品を開発または商業化するために必要ないかなる第三者ライセンスも取得できないことは、私たちの業務と運営を深刻に損なう可能性があり、関連する努力を放棄する可能性があります。
私たちが将来達成する可能性のあるどんな協力計画も成功しない可能性があり、これは私たちが製品を開発し、それを商業化する能力に悪影響を及ぼすかもしれない。
64
私たちが未来に行ったどんな協力も成功しないかもしれない。私たちの協力計画の成功は私たちの協力者の努力と活動に大きく依存するだろう。協力は多くのリスクに直面しています
関連する第三者特許を識別できないか、または第三者特許の関連性、範囲、または満了時間を誤って解釈することができない可能性があり、これは、私たちが製品を開発およびマーケティングする能力に悪影響を及ぼす可能性がある。
私たちはあなたに私たちの業務が既存または未来の特許を侵害しないか、または未来に侵害しないということを保証することはできません。私たちは、関連特許の識別、特許請求の範囲、または関連特許の満了を含めて、私たちのいかなる特許検索または分析も保証することはできず、私たちは、米国および海外が私たちの治療研究計画に関連しているか、または私たちの候補製品の商業化に必要な各第三者特許および未定出願、例えばEQ 101、EQ 302、itolizumab(EQ 001)および/または任意の司法管轄区域の他の特許を識別していることを確実にすることはできない。
我々の市場には、第三者が所有する米国および外国特許および処理すべき特許出願が多く存在し、我々の製品および/または候補製品の使用または製造に関連する第三者特許または特許出願を識別することができ、これらの特許または特許出願は、我々の製品および/または候補製品の使用または製造を要求する可能性がある。私たちはアメリカと海外の競争相手で、その多くはより多くの資源を持っていて、特許の組合せと競争技術に大量の投資を行って、彼らはすでに特許を申請したり、獲得したり、あるいは将来出願して特許を獲得する可能性があり、これらの特許は私たちの製品の製造、使用、販売の能力を阻止、制限、または妨害するだろう。私たちはいつも第三者が未解決の特許出願と特許を独立して審査するわけではない。米国および他の地方の特許出願は、通常、優先権を要求する最初の出願の約18ヶ月後に発行され、このような最も早い出願日は、一般に優先権日と呼ばれる。アメリカ国外で提出されないいくつかのアメリカ出願は特許が発行される前に秘密にすることができる。さらに、米国および他の地方の特許出願は、発表前に長年待つことができ、または意図せずに放棄した特許または出願を再起動することができる。さらに、開示された係属中の特許出願は、いくつかの制限された場合に、私たちの技術、私たちの製品、または私たちの製品の使用をカバーするために、後で修正することができる。したがって、待っているか最近回復している他の特許の出願があるかもしれませんが、私たちはこれらの特許を知らないかもしれません
65
EQ 101、EQ 302、itolizumab(EQ 001)および他、またはそれらの予期される用途のような、我々の研究計画および候補製品に関連する。これらの出願は、発行された特許、または以前に放棄された特許の復興をもたらす可能性があり、これは、私たちの製品を製造、使用、または販売する能力を阻止、制限、または他の方法で妨害するだろう。
特許請求の範囲は、法律の解釈、特許における書面開示、および特許の起訴履歴に依存する。特許または保留出願の関連性または範囲の解釈は正しくない可能性があり、これは私たちの製品を販売する能力に悪影響を及ぼす可能性がある。私たちの製品が第三者特許のカバー範囲内にないか、または第三者の保留出願が関連範囲のクレームを出すかどうかを誤って予測する可能性がある。私たちが関連する特許の満期日の決定は、アメリカまたは海外のどのようなものでも正しくないかもしれません。これは、私たちの候補製品を開発し、マーケティングする能力に悪影響を及ぼす可能性があります。私たちは関連特許を識別して正確に解釈することができず、私たちが製品を開発し、マーケティングする能力に悪影響を及ぼすかもしれない。
私たちは、私たちの現在の技術(私たちの研究計画、候補製品(EQ 101、EQ 302、itolizumab(EQ 001)などを含む)と、それらのそれぞれの使用、製造および処方に対して実施される可能性のある第三者特許が存在しない保証はなく、私たちの製造または将来の販売の禁止、または私たちの将来の販売について、第三者に印税および/または他の形態の賠償を支払う義務があるかもしれません。
もし私たちが第三者の知的財産権侵害を起訴されれば、このような訴訟は費用が高く時間がかかり、私たちの候補製品の開発や商業化を阻止または延期する可能性がある。
私たちのビジネス成功は、第三者の知的財産権および他の固有の権利を侵害することなく、EQ 101、EQ 302およびitolizumab(EQ 001)、ならびに他の潜在的な将来の候補製品を開発、製造、マーケティングおよび販売する能力にある程度依存する。第三者は私たちが彼らの知的財産権を侵害したり流用したと主張するかもしれない。正当な理由の有無にかかわらず、知的財産権クレームに関連する訴訟や他の法的手続きは予測不可能であり、通常コストが高く、時間がかかり、解決策が私たちに有利であっても、私たちの技術や管理者の正常な役割を分散させることを含む、私たちのコア業務から大量の資源を移転する可能性がある。また、公聴会、動議、または他の一時的な手続きや事態の発展の結果が公表される可能性があり、証券アナリストや投資家がこれらの結果がマイナスだと思う場合、私たち普通株の市場価格に大きな悪影響を及ぼす可能性がある。このような訴訟または訴訟は、私たちの運営損失を大幅に増加させ、開発活動または任意の将来の販売、マーケティング、または流通活動に使用することができるリソースを減少させる可能性がある。私たちはそのような訴訟や法的手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らがより多くの財政資源とより成熟して発展した知的財産権の組み合わせを持っているので、このような訴訟や法的手続きの費用を私たちよりも効率的に負担するかもしれない。特許訴訟または他の訴訟の開始と継続によって生じる不確実性は、市場での競争能力に重大な悪影響を及ぼす可能性がある。私たちはあなたに私たちの業務が既存または未来の特許を侵害しないか、または未来に侵害しないということを保証することはできません。
バイオテクノロジーや製薬業界には大量の知的財産権訴訟があり、私たちは私たちの候補製品に関連する知的財産権訴訟や他の対抗性訴訟の当事者になったり、これらの訴訟や訴訟の脅威にさらされたりする可能性がある。私たちは米国と海外の競争相手であり、その多くはより多くの資源を持っており、特許の組み合わせおよび競争技術に大量の投資を行っており、それらはすでに特許を出願または獲得しているかもしれないし、将来的に特許を申請して獲得する可能性があり、これらの特許は、私たちの候補製品の製造、使用、販売の能力を阻止、制限、または他の方法で妨害するだろう。第三者は既存または未来の知的財産権に基づいて私たちに権利侵害を請求するかもしれない。製薬とバイオテクノロジー産業は大量の特許を生み出しており、私たちを含む業界参加者は、どの特許が様々なタイプの製品や使用方法をカバーしているのかを常に明らかにしていないかもしれない。特許のカバー面は裁判所の解釈にかかっており、解釈は常に統一されているわけではない。もし私たちが特許侵害で起訴された場合、私たちは私たちの候補製品、製品、または方法が関連特許の特許主張を侵害していないか、または特許主張が無効または実行不可能であることを証明する必要があり、私たちはそれができないかもしれない。その無効性を証明することは難しいかもしれない。例えば、米国では、法廷で無効を証明するには、発行された特許の有効性の推定を覆すために、明確で納得できる証拠を提示する必要がある。私たちがこれらの訴訟で勝訴しても、私たちは巨額のコストを生む可能性があり、私たちの経営陣や科学者の時間と注意力はこれらの訴訟に移される可能性があり、これは私たちの業務や運営に実質的な悪影響を及ぼす可能性がある。しかも、私たちはこのような行動を円満に達成するための十分な資源がないかもしれない。さらに、知的財産権訴訟や行政訴訟に関連する大量の開示要求により、私たちのいくつかの機密情報が開示によって漏洩される可能性がある。
66
もし私たちが第三者の知的財産権を侵害していることが発見された場合、裁判所の命令を含めて、権利侵害の候補製品や製品の開発、製造、または商業化を停止させられる可能性がある。代替的に、権利侵害技術を使用し、権利侵害候補製品の開発、製造、またはマーケティングを継続するために、第三者から許可を得る必要があるかもしれない。しかし、私たちは商業的に合理的な条項や必要な許可証を得ることができないかもしれない。我々が許可を得ることができても,非排他的である可能性があり,我々の競争相手が我々に許可された同じ技術にアクセスできるようにする.さらに、もし私たちが故意に特許を侵害したことが発見された場合、私たちは3倍の損害賠償と弁護士費を含む金銭損害賠償責任を負われる可能性がある。権利侵害の発見は、候補製品を商業化したり、一部の業務運営を停止させたりすることを阻止する可能性があり、私たちの技術者や経営陣の時間と注意を移し、開発遅延を招く可能性があり、および/または、費用効果に基づいて私たちの業務に実質的な損害を与える可能性があります。我々が第三者の機密情報や商業秘密を盗用したと主張することは,我々の業務に類似した負の影響を与える可能性がある.
私たちは私たちの特許や他の知的財産権を保護したり強制したりする訴訟に巻き込まれるかもしれないが、これは高価で時間がかかり、成功しないかもしれない。
競争相手は、EQ 101、EQ 302、itolizumab(EQ 001)など、それらのそれぞれの使用、製造、および処方を含む、私たちの特許、商標、著作権、または私たちの現在および将来の候補製品に関連する他の知的財産権を侵害する可能性がある。権利侵害や不正使用に対抗するために、私たちまたは私たちの許可者は、費用がかかり、時間がかかる可能性があり、私たちの管理者や科学者の時間と注意力を分散させる権利侵害請求を要求されるかもしれません。私たちまたは私たちの許可者は、侵害者によって提起された任意のクレームに対して、私たちの特許が無効であるか、強制的に実行できないと主張することに加えて、これらの当事者に、私たちが彼らの特許を侵害したことを告発するように、私たちに反訴させる可能性がある。米国の特許訴訟では、被告が無効および/または実行不可能と主張する反訴は日常的であり、法的に無効および実行不可能と主張された後の結果は予測できない。いずれの特許侵害訴訟においても、裁判所は、私たちが所有または許可している特許の全部または一部が無効または強制的に実行できないと判断する可能性があり、私たちは、反対側が論争のある発明を使用することを阻止する権利がない。もう一つのリスクは、これらの特許の有効性が支持されても、裁判所がその特許の権利要件を偏狭に解釈するか、または私たちの特許権利要件がこの発明を含まないことを理由に、私たちが他方の論争のある発明の使用を阻止する権利がないと判断することである。我々の特許に関連する訴訟または訴訟における不利な結果は、これらの当事者または他の競争相手に対して私たちの特許を主張する能力を制限し、第三者が類似または競合製品を製造および販売する能力を制限または排除する可能性がある。
第三者によって引き起こされるか、または我々によって提起されるか、または米国特許商標局によって発表される干渉または派生プログラムは、我々の特許または特許出願に関連する発明の優先権を決定するために必要である可能性がある。不利な結果は、私たちが関連技術の使用を停止することを要求するか、または勝利者から許可を得ようとすることを要求するかもしれない。例えば、不利な結果は、関連技術の使用を停止することを要求するか、または勝利者から許可を得ようと試みることを要求することができる。もし勝利者が商業的に合理的な条項で私たちに許可を提供しない場合、あるいは私たちに許可を全く提供しない、あるいは非独占的な許可を提供し、私たちの競争相手が同じ技術を獲得した場合、私たちの業務は損害を受ける可能性がある。私たちの訴訟、介入、または派生訴訟の弁護は失敗する可能性があり、成功しても巨額のコストを招き、私たちの経営陣や他の従業員の注意を分散させる可能性がある。さらに、訴訟に関連する不確実性は、私たちの臨床研究を継続し、私たちの研究計画を継続し、第三者から必要な技術的許可を得るため、またはパートナー関係を開発する能力に重大な悪影響を及ぼす可能性があり、これらのパートナーシップは、EQ 101、EQ 302、itolizumab(EQ 001)、または私たちが決定する可能性のある他の候補製品を市場に出すのを助ける。これらのいずれも、私たちの競争業務の地位、経営結果、業務の将来性、および財務状況に悪影響を及ぼす可能性があります。同様に、私たちが商標侵害クレームを主張する場合、裁判所は、私たちが主張する商標が無効または強制執行できないと判断するか、または商標侵害を主張する側が関連商標に対する優先権を有することを主張するかもしれない。この場合、私たちは最終的にこのような商標の使用を中止することを余儀なくされるかもしれない。
侵害行為が成立したと認定しても,裁判所はさらなる侵害活動に禁止令を付与せず,金銭賠償のみを決定する可能性があり,十分な救済措置ではない可能性もある。さらに、知的財産権訴訟は大量の開示を必要とするため、私たちのいくつかの機密情報は訴訟中に開示によって漏洩される可能性がある。聴聞、動議、または他の一時的手続き、または事態発展の結果を公開することもできる。もし証券アナリストや投資家がこれらの結果がマイナスだと思っていれば、私たち普通株の株価に実質的な悪影響を及ぼす可能性がある。しかも、私たちはこのような侵害請求を提起して追跡するのに十分な財政的または他の資源を持っていることを保証することはできません。これらのクレームは通常数年続いています。たとえ私たちが最終的に勝訴しても、このような訴訟の金銭的代価と私たちの経営陣と科学者の注意の移動は、私たちが訴訟から得たいかなる利益を超えるかもしれない。
67
訴訟の費用と不確実性のため、私たちは第三者に対して私たちの知的財産権を強制的に実行できないかもしれない。
訴訟の費用と不確実性のため、第三者が私たちが発表した研究プロジェクトや候補製品に関連する特許、私たちの未解決または将来の特許出願または他の知的財産権によって発行される可能性のある任意の特許を侵害しても、そのようなクレームや訴訟を提起して実行するリスク調整コストが高すぎる可能性があり、またはわが社または私たちの株主の最適な利益に適合しない可能性があると結論するかもしれない。この場合、私たちは、状況を簡単に監視するか、または他の非訴訟の行動または解決策を開始または求めることで、より慎重なやり方を決定するかもしれない。
私たちは、私たちの従業員、コンサルタント、または独立請負業者が第三者の機密情報を誤って使用または開示することに関するクレームを受けるかもしれない。
私たちは以前他社と協力した人を、私たちの競争相手や潜在的な競争相手を含めて雇用した。将来的に、私たちは以下のような告発を受けるかもしれない:私たちまたは私たちの従業員、コンサルタント、または独立請負業者は、現在または前任雇用主または競争相手の商業秘密または他の機密情報を無意識にまたは他の方法で使用または漏洩した。私たちは、私たちの従業員、コンサルタント、および独立請負業者が、私たちのために働いているときに他人の知的財産権、独自の情報、技術的ノウハウ、または商業秘密を使用しないことを保証するために努力しているが、私たちは、個人がその競争禁止協定または競業禁止協定の条項に違反しているか、または私たちまたはこれらの個人が、現または前任雇用主または競争相手のいわゆる商業秘密または他の固有情報を意図的にまたは他の方法で使用または開示しているという疑惑を受ける可能性がある。
私たちは訴訟を通じて自分を弁護するかもしれませんが、私たちが勝訴しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性があります。これらのクレームに対する私たちの抗弁が失敗した場合、裁判所は、金銭的損害賠償の支払いを要求することに加えて、EQ 101、EQ 302またはitolizumab(EQ 001)を含む、当社の候補製品に重要な技術または機能の使用を禁止することができ、これらの技術または機能が、現在または前任雇用者の商業秘密または他の固有情報を含むか、またはそれから発見されることを前提とすることができる。さらに、このような訴訟またはその脅威は、私たちの名声、戦略連合を形成したり、協力者に私たちの権利を譲渡したり、科学コンサルタントと接触したり、従業員やコンサルタントを雇う能力に悪影響を及ぼす可能性があり、すべては私たちの業務、運営結果、および財務状況に悪影響を及ぼすだろう。
私たちは世界各地で私たちの知的財産権を保護できないかもしれない。
世界のすべての国で現在と未来のすべての特許を申請、起訴、保護することは目を引くほど高価であり、私たちのアメリカ以外のいくつかの国での知的財産権はアメリカほど広くないかもしれない。また、いくつかの外国の法律は知的財産権の保護の程度はアメリカの連邦や州法律に及ばない。したがって、私たちは、米国以外のすべての国/地域で第三者が私たちの発明を実施したり、米国または他の管轄区域で私たちの発明を使用して製造された製品を販売または輸入することを阻止することができないかもしれない。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの技術を使用して自分の製品を開発することができ、また、他の侵害製品を私たちが特許保護を持っているが法執行力がアメリカに及ばない地域に輸出することもできる。これらの製品は私たちの候補製品と競争するかもしれませんが、私たちの特許や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。
多くの国の法制度は、特許や他の知的財産権保護の強制執行を支持しておらず、これは、私たちの特許の侵害を阻止したり、私たちの独自の権利を侵害する方法で競合製品をマーケティングすることを困難にする可能性がある。例えば、一部の外国国には強制許可法があり、これらの法律によると、特許権者は第三者に許可を付与しなければならない。また、一部の国は、政府機関または政府請負業者を含む第三者に対する特許の実行可能性を制限している。このような国では、特許は限られた利点を提供するかもしれないし、利益さえないかもしれない。また,近年,欧州特許法の複雑さと不確実性が増加している。欧州では,2023年6月に発効した新たな単一特許制度は,このような制度導入前に付与された特許を含む欧州特許に大きな影響を与えるであろう。単一特許制度の下で,欧州の出願は特許付与後に単一特許裁判所(UPC)によって管轄される単一特許となる権利がある。UPCは新しい裁判所制度であるため、裁判所には前例がなく、いかなる訴訟の不確実性も増加している。UPC実施前に付与された特許は,UPCの管轄から脱退することを選択し,UPC国の国家特許として保持することができる。まだUPC管内にある特許は,UPCによる単一撤回挑戦を受けやすい可能性があり,成功すれば,UPC署名国のすべての国の特許を無効にする可能性がある.私たちはどんな潜在的な変化の長期的な影響も確実に予測できない。外国の管轄区域で私たちの特許権を強制的に執行する訴訟手続きは巨額のコストを招く可能性があり、私たちの努力と注意を私たちの業務の他の方面から移すことは、私たちの特許が無効または狭義に解釈されるリスクに直面する可能性があり、私たちの特許出願は直面する可能性がある
68
第三者が私たちにクレームをつける可能性があります。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。したがって、私たちが世界各地で私たちの知的財産権を実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネスメリットを得るのに十分ではないかもしれません。
特許保護の獲得と維持は、政府特許機関が提出した様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要件に適合しない場合、私たちの特許保護は減少またはキャンセルされる可能性がある。
特許および/または特許出願の有効期間内に、定期維持費、継続費、年会費、および特許および/または特許出願に関する様々な他の政府費用を、いくつかの段階に分けて米国特許商標局および外国特許代理機関に支払わなければならない。米国特許商標局および各種外国政府特許機関はまた、特許出願中にいくつかのプログラム、文書、費用支払い、およびその他の同様の規定を遵守することを要求している。多くの場合、適用規則に従って滞納金を支払うか、または他の方法で不注意を是正することによって失効を是正することができるが、場合によっては、規定を遵守しないことは、特許または特許出願が放棄または失効され、関連法ドメインの特許権の一部または全部の喪失をもたらす可能性がある。特許または特許出願の放棄または失効をもたらす可能性のある規定を遵守しないイベントは、規定された期限内に公式行動に応答できなかったこと、費用を支払わなかったこと、および適切に合法化されず、正式な文書を提出することができなかったことを含むが、これらに限定されない。EQ 101、EQ 302、itolizumab(EQ 001)および他の製品の特許および特許出願、ならびにそれらのそれぞれの使用、製造、および処方のような我々の研究プロジェクトおよび候補製品を維持できない場合、私たちの競争地位は、例えば、競争相手が他の場合よりも早く市場に参入する可能性がある。
私たちは私たちの商業秘密の機密性を保護できなければ、私たちの商業と競争の地位が損なわれるかもしれない追跡と実行が困難な商業秘密とノウハウに依存するかもしれない。
私たちのいくつかの候補技術および製品のための特許を申請することに加えて、私たちは、研究計画および候補製品における私たちの競争地位を維持するために、非特許の技術的ノウハウ、技術および他の独自の情報を含む商業秘密に依存することができる。私たちの候補製品の要素は、その製造および製造プロセスを含み、特許がカバーされていない独自技術、情報または技術に関連する可能性があり、したがって、これらの態様については、商業秘密および技術を私たちの主要な知的財産権と見なすことができる。私たちの従業員、私たちの施設を共有する第三者従業員、または研究、臨床研究または製造活動を行う第三者コンサルタントおよびサプライヤーの任意の意図的または意図的な開示、または第三者による私たちのビジネス秘密または独自の情報の流用(例えば、ネットワークセキュリティホールを介して)は、競争相手が私たちの技術成果をコピーまたは超えることを可能にし、それによって市場における私たちの競争地位を侵食する可能性がある。
ビジネス秘密と技術的ノウハウを保護することは難しいかもしれない。私たちは従業員に秘密条項を含む書面雇用協定を締結し、彼らが雇用中に発生した任意の発明を私たちに譲渡する義務があることを要求します。私たちは、双方の財産、潜在的な商業秘密、ノウハウ、情報を保護するための守秘義務および知的財産義務を含む、私たちの共有施設の任意の第三者と書面協定を締結します。私たちはさらに、当社の協力者、外部科学協力者、CRO、契約製造業者、コンサルタント、コンサルタント、および他の第三者のような、アクセス権限を取得した当事者と秘密および秘密協定を締結することで、私たちの潜在的なビジネス秘密、ノウハウ、および情報を保護することを求めています。私たちのコンサルタント、請負業者、および外部科学協力者の場合、これらの合意は、一般に発明譲渡義務を含む。しかし、私たちはすべての関係者とこのような合意を締結したことを確認することはできず、私たちの商業秘密や他の機密固有情報が開示されないか、あるいは競争相手が私たちの商業秘密を取得したり、実質的に同等の情報や技術を独立して開発しないかを決定することもできない。さらに、このような努力にもかかわらず、どちらも合意に違反し、私たちのビジネス秘密を含む私たちの固有の情報を漏洩する可能性があり、私たちはこのような違反について十分な救済措置を得ることができないかもしれない。許可されていない使用と開示を監視することは困難であり、私たちはまた私たちのノウハウを保護するために私たちが取った段階が有効かどうか分からない。許可されていない当事者たちはまた、複製や逆工事をしようとするかもしれないし、私たちは独自の製品のいくつかの側面だと思う。強制執行側が商業秘密を不正に開示したり流用したりする主張は困難であり、高価で時間がかかり、結果は予測できない。しかも、米国国内外のいくつかの裁判所は商業秘密をあまり望んでいないか、または保護したくない。
商業秘密は他の人たちによって独立して開発されるかもしれないし、私たちの法律の追跡を阻止するかもしれない。時間の経過に伴い、商業秘密は独立して定期刊行物文章を開発、発表し、熟練した芸術人員を1つの会社から別の会社に移転すること、あるいは学術界から業界科学職に移転することによって業界内に伝播する。それにもかかわらず
69
私たちの第三者との合意は、通常、私たちのコンサルタント、従業員、協力者、ライセンシー、サプライヤー、第三者請負業者、およびコンサルタントが、私たちのビジネス秘密に関連する可能性のあるデータを発行する能力を制限しますが、私たちの合意は、いくつかの限られた発行権を含む可能性があります。私たちは製品の開発、製造と流通、そして私たちのサービスを提供することを時々第三者に依存したいので、私たちは時々彼らと商業秘密を共有しなければならない。上述した契約および他の安全予防措置がとられているにもかかわらず、商業秘密を共有する必要は、そのような商業秘密が私たちの競争相手に知られ、意図せずに他の人の技術に組み込まれるか、または開示されるか、またはこれらの合意に違反するリスクを増加させる。もし私たちの任意の商業秘密が競争相手または他の第三者によって合法的に取得または独立して開発された場合、私たちは彼らがその技術や情報を使用して私たちと競争することを阻止する権利がないだろう。もし私たちの任意の商業秘密が競争相手または他の第三者に漏洩された場合、または競争相手または他の第三者によって独立して開発された場合、私たちの競争地位は損なわれるだろう。
私たちは私たちの特許と他の知的財産権の発明権または所有権のクレームに挑戦する対象になるかもしれない。
私たちまたは私たちの許可者は、元従業員、コンサルタント、独立請負業者、協力者または他の第三者を所有者、共同所有者、発明者、または共同発明者が私たちの特許または他の知的財産権において権利を有するというクレームを受ける可能性がある。特許出願上適切な発明者が示されていないことは、その上で発行された特許が強制的に実行できない可能性がある。発明権紛争は、発明者として指定された異なる個人の貢献に関する相互矛盾した意見、外国国民が特許標的開発に参加する外国法律の影響、我々の候補製品の開発に参加する第三者の義務衝突、または潜在的な共同発明の共同所有権に関する問題によるものである可能性がある。さらに、私たちの政策は、私たちの従業員、コンサルタント、コンサルタント、請負業者、および知的財産権のアイデアや開発に参加する可能性のある他の第三者が合意に署名し、そのような知的財産権を私たちに譲渡することを要求していますが、私たちまたは私たちの許可者は、実際に私たちが自分の知的財産権と見なしているすべての当事者とこのような合意を実行することに成功できないかもしれません。知的財産権の譲渡は自動的に実行されない可能性があり、あるいは範囲が十分でない、あるいは譲渡協定に違反する可能性がある。さらに、私たちと協定に署名した個人は、第三者(例えば、学術機関)に対して予め存在または相互競争する義務を負う可能性があり、したがって、私たちまたは私たちのライセンシーと達成された合意は、個人が開発した発明の所有権を改善する上で無効になる可能性がある。訴訟は、これらおよび他の挑戦在庫および/または所有権のクレームを解決するために必要である可能性がある。代替または追加として、私たちはこのような知的財産権上の私たちの権利範囲を明確にするために協定を締結することができる。もし私たちがこのようなクレームを弁護することができなければ、金銭損害賠償を支払うことに加えて、貴重な知的財産権、例えば貴重な知的財産権の独占所有権や使用権を失う可能性がある。このような結果は私たちの業務に実質的な悪影響を及ぼすかもしれない。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。
特許条項は候補製品に対する私たちの競争地位を十分に長く保護するのに十分ではないかもしれない。
特許権の期限は限られています。米国では、特許の自然失効は、通常、その最初の有効な非臨時出願日から20年後である。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。私たちの候補製品をカバーする特許を取得しても、製品の特許有効期限が切れると、私たちは生物類似薬や模倣薬からの競争に直面する可能性がある。米国は規制遅延に基づく特許期限の延長を持つかもしれない。しかし、各上場承認は1つの特許しか延長できず、1つの特許は単一製品に対して1回しか延長できない。また,特許期間延長期間の保護範囲は,特許請求の範囲のすべてまで延長されるのではなく,承認された製品範囲に限定される。外国司法管轄区域が特許期間延長のような法律の違いを管理することは大きく異なり、1つの特許家族から複数の特許を取得する能力を管理する法律も同様である。また,適用の最終期限内に出願を提出できなかった場合,関連特許の満了前に出願を提出できなかった場合,または適用の要求を満たしていなかった場合には,延期が得られない可能性がある.また,米国の最近の司法判断は,関連特許が特許期限調整なしに発行された家庭特許の期限調整(PTA)裁決に疑問を投げかけている。したがって,将来PTAをどのように見るか,特許満期日が影響を受ける可能性があるかどうかは肯定的ではない.もし私たちが特許期間の延長や回復を得ることができない場合、あるいはこのような延長の期限が私たちが要求したものよりも短くなれば、私たちが独占的に販売する権利がある期間は短縮され、私たちの競争相手は私たちの特許が満期になった後に競争製品の承認を受ける可能性があり、私たちの収入は大幅に減少するかもしれない。
もし私たちの商標と商号が十分に保護されていなければ、私たちは興味のある市場で知名度を作ることができないかもしれません。私たちの業務は悪影響を受けるかもしれません。
70
私たちは現在2つのEQUILLIUM商標登録があります。それぞれ5種類と42種類、1つのカナダ商標登録、5種類と42種類をカバーしています。私たちの現在または未来の商標または商号は、他の商標を侵害するための汎用または記述商標として疑問、侵害、回避、または宣言される可能性がある。私たちはこれらの商標や商品名に対する私たちの権利を保護できないかもしれないし、これらの名前の使用を停止させることができないかもしれません。私たちは関心のある市場で潜在的なパートナーや顧客の名前の承認を得るために必要です。商標登録過程で、私たちは拒否を受けるかもしれない。私たちはこのような拒否に答える機会があるだろうが、私たちはこの拒否を克服できないかもしれない。また,米国特許商標局や多くの外国司法管轄区の類似機関では,第三者は係属中の商標出願に反対し,登録商標の取り消しを求める機会がある。私たちの商標に反対またはキャンセル訴訟を提起するかもしれないが、私たちの商標は継続できないかもしれない。もし私たちが私たちの商標と商号に基づいて名称を確立することができなければ、私たちは効果的に競争できない可能性があり、私たちの業務は不利な影響を受けるかもしれない。私たちは私たちの商標と商品名を流通業者のような第三者に許可するかもしれない。これらのライセンス契約は、私たちの商標や商号をどのように使用するかに指針を提供する可能性がありますが、ライセンシーは、これらの合意に違反したり、私たちの商標およびビジネス番号を乱用したりすることで、私たちの権利を危険にさらしたり、私たちの商標や商号に関連する商標の名誉を弱める可能性があります。
知的財産権は必ずしも私たちの競争優位に対するすべての潜在的な脅威を解決できるとは限らない。
私たちの知的財産権が提供する未来の保護の程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できないか、あるいは競争優位性を維持することができるかもしれない。例えば:
このような事件が発生した場合、私たちの業務、財務状況、運営結果、将来性を深刻に損なう可能性があります。
従業員、私たちの成長、その他の法務に関するリスクを管理する
私たちは私たちのキーパーソンのサービスに強く依存している。
私たちは私たちのキーパーソンのサービスに強く依存していますブルース·D·スティールは私たちの総裁兼最高経営責任者を務めスティーブン·コーネリー博士は私たちの最高科学者を務めています私たちは彼らと雇用協定を締結したが、彼らには特定の任期がなく、彼らの誰もが私たちとの雇用関係をいつでも終わらせることができるが、私たちはまだこの人たちの中で誰かが私たちを離れようとしていることを知らないにもかかわらず。
私たちは私たちの開発、規制、運営能力を拡大することが予想されますので、私たちは管理成長に困難に直面する可能性があり、これは私たちの運営を混乱させるかもしれません。
2023年12月31日現在、私たちは44人のフルタイム従業員がいる。EQ 101とitolizumab(EQ 001)の臨床開発や、潜在的な他の候補製品の推進に伴い、私たちは私たちの
71
著者らの業務範囲は多くの領域をカバーし、非臨床研究、臨床開発、品質、法規事務、薬物警戒、製造とサプライチェーン、及び一般と行政機能を含む。EQ 101、EQ 302、itolizumab(EQ 001)、または任意の将来の候補製品が発売承認された場合、販売、マーケティング、および流通部門の従業員が増加すると予想される。私たちが予想していた未来の成長を管理するためには
私たちの将来の財務業績および私たちの候補製品を開発、製造、商業化する能力は、将来のいかなる成長の能力を効果的に管理するかにある程度依存し、私たちの経営陣はまた、これらの成長活動を大量の時間を投入して管理するために、財務および他の資源および比例しない多くの注意を日常活動から移行させなければならないかもしれない。
現在、予測可能な未来において、私たちは、私たちが行っているおよび未来の臨床研究、ならびにEQ 101、EQ 302、itolizumab(EQ 001)、および任意の未来の候補製品の製造に重大な責任を負うことを含む、いくつかのCRO、CMO、他の契約サービスプロバイダ、コンサルタント、およびコンサルタントに大きく依存し続けるであろう。必要に応じて、このような契約サービスプロバイダ、コンサルタント、コンサルタントのサービスがタイムリーに提供されていくか、または合格した代替者を見つけることができることを保証することはできません。さらに、私たちのアウトソーシング活動を効果的に管理できない場合、あるいは私たちのサプライヤーやコンサルタントが提供するサービスの品質や正確性が何らかの理由で影響を受ける場合、私たちの臨床研究は延長、延期、または終了する可能性があり、候補製品の市場承認や他の方法で私たちの業務を促進することができないかもしれません。私たちは、私たちの既存のサプライヤーやコンサルタントを経済的に合理的な条件で適切に管理することができるか、または他の適任な外部サプライヤーとコンサルタントを見つけることができるか、または全くできないことを保証することはできません。
私たちがより多くの施設をレンタルし、新入社員を雇用し、私たちのコンサルタントや請負業者チームを拡大することで、私たちの組織を効果的に拡大することができなければ、私たちの候補製品をさらに開発し、商業化するために必要な任務を実行することができない可能性があり、私たちの研究、開発、商業化の目標を達成できないかもしれません。
私たちの未来の成功は私たちが肝心な従業員、顧問と顧問を維持する能力、及び合格者を吸引、維持、激励する能力にかかっている。
近年、私たちの産業は高い流出率を経験している。競争の激しい生物製薬業界における著者らの競争能力は、科学、医療、監督、製造と管理技能と経験を持つ高技能と経験者の能力を吸引、維持、激励することに依存する。著者らは主に大サンディエゴ地区で業務を展開し、この地区は多くの他の生物製薬会社及び多くの学術と研究機構の所在地であり、合格人材に対する激しい競争を招いた。生物製薬会社の限られた数の合格人材に対する激しい競争のため、私たちは未来に合格した人材を引き付けることができないかもしれない。私たちと競争している多くの他の生物製薬会社はより多くの財務と他の資源、異なるリスク状況、そして私たちよりも長い業界の歴史を持っている。私たちの競争相手は、より高い報酬、より多様な機会、および/またはより良い職業昇進の機会を提供するかもしれない。これらすべての競争要因は、私たちが高素質の人材を誘致し、維持し続ける能力を制限する可能性があり、これは、私たちの候補製品の開発と商業化に成功し、現在の想定に沿って私たちの業務と運営を発展させる能力にマイナスの影響を与えるかもしれない。
環境、社会、ガバナンス要因に関する第三者の予想はコストを増加させ、新たなリスクに直面させる可能性がある。
近年、ある投資家、従業員、および他の利益関係者は、企業責任、特に環境、社会および管理またはESG要因に関連する責任にますます注目している。ESG評価および会社報告書の第三者プロバイダの数が増加し、基準がそれぞれ異なり、場合によっては一致しない。評価中に考慮するテーマは気候変動と人権方面の会社の努力と影響、道徳と法律の遵守、及び会社の取締役会が各種の持続可能な発展問題を監督する上での役割を含む。
72
一部の投資家は、第三者のESG格付けおよび報告を使用して彼らの投資戦略を指導するかもしれないが、場合によっては、彼らが私たちのESG実践が十分でないと思う場合、彼らは私たちに投資しないことを選択するかもしれない。会社のESG実践を評価する基準が進化しており、これは、私たちへの期待がより高くなり、これらの新しい基準を満たすためにコストの高い措置をとることになる可能性がある。あるいは、私たちが投資しないこと、または新しい基準を満たすことができないこと、または特定の第三者プロバイダの基準を満たしていないことを選択した場合、一部の投資家は、ESGに関する政策が十分ではなく、私たちに投資しないことを選択したと結論するかもしれない。
私たちのESG実践が投資家または他の利害関係者の絶えず変化する期待および基準を満たすことができない場合、私たちの名声、従業員を引き付けるまたは維持する能力、および投資またはビジネスパートナーとしての私たちの入手可能性は否定的な影響を受ける可能性がある。同様に、私たちは、私たちが発表した期間内に私たちの目標と目的を十分に追求または達成できなかったと考えられたり、様々な報告基準を満たしていなかったり、追加の規制、社会的または他の審査に直面したり、予期しないコストを適用したり、私たちの名声を損なう可能性があります。これは、逆に私たちの業務、財務状況、キャッシュフロー、および運営結果に大きな悪影響を与え、私たちの普通株の市場価値を低下させる可能性があります。
私たちの従業員、臨床研究調査者、CRO、コンサルタント、サプライヤー、および任意の潜在的なビジネスパートナーは、規制基準と要求およびインサイダー取引を遵守しないことを含む不正行為または他の不正活動に従事する可能性があります。
私たちは、従業員、臨床研究者、CRO、コンサルタント、サプライヤー、および任意の潜在的なビジネスパートナーの詐欺または他の不適切な行為のリスクに直面しています。これらの当事者の不正行為は、(I)FDA法律および法規、または真、完全かつ正確な情報の報告を要求する法律、(Ii)製造基準、(Iii)連邦および州の健康およびデータプライバシー、セキュリティ、詐欺および乱用、政府価格報告、透明性報告要件、ならびに米国および海外の他の医療保健法律法規、(Iv)セクハラおよび他の職場での不正行為、または(V)財務情報またはデータを真実、完全かつ正確に報告することを要求する法律を含む、故意、無謀、および/または不注意な行為、または許可されていない活動を開示することを含む可能性がある。このような不正行為はまた臨床研究過程で得られた情報を不当に使用する可能性があり、これは規制制裁を招き、著者らの名声に深刻な損害を与える可能性がある。我々は、すべての従業員に適用される行動基準および開示計画および他の適用可能な政策および手順を通過したが、常に従業員の不適切な行為を識別し、阻止することができるわけではなく、このような行為を発見し、防止するための予防措置は、未知または未管理のリスクまたは損失を効果的に制御することができないか、またはこれらの法律や法規を遵守できないことによる政府の調査または他の行動または訴訟から私たちを保護することができないかもしれない。もし私たちにこのような訴訟を提起し、私たちが私たちの権利を弁護したり、維持することに成功しなかった場合、これらの行動は、Medicare、Medicaidおよび他の連邦医療保健計画、契約損害、名声損害、利益および将来の収益の減少、追加の誠実な報告と監督義務、および私たちの業務を削減または再構成することを含む、私たちの業務に重大な影響を与える可能性があり、いずれも私たちの業務の運営能力および運営結果に悪影響を及ぼす可能性がある。
もし私たちの情報技術システム、または私たちが依存しているCROまたは他の第三者のシステム、または私たちのデータが漏洩された場合、私たちは規制調査や行動を含むが、制限されないが、訴訟、罰金と処罰、私たちの業務運営中断、名声損害、収入または利益損失、および他の不利な結果を含むこのような損害によって生じる不利な結果を経験する可能性がある。
私たちの通常のビジネスプロセスでは、独自および機密の商業データ、私たちが収集した臨床研究に関連する試験参加者データ、敏感な第三者データ、商業計画、取引、財務情報、知的財産権および商業秘密(総称して敏感な情報)を含む個人データおよび他の敏感な情報を収集、受信、格納、処理、生成、使用、送信、開示、提供、保護、処置、送信および共有(総称して敏感な情報)する。
したがって、私たちと私たちが依存している第三の側面は様々な変化する脅威に直面しており、これらの脅威は安全事件を招く可能性がある。ネットワーク攻撃、インターネットベースの悪意のある活動、オンラインおよびオフライン詐欺、および他の同様の活動は、私たちの敏感なデータおよび情報技術システム、ならびに私たちが依存する第三者システムの機密性、完全性、および可用性を脅かす。このような脅威は一般的であり、上昇し続けており、検出がますます困難になっており、伝統的なコンピュータ“ハッカー”、脅威行為者、“ハッカー活動家”、組織的犯罪脅威行為者、人員(例えば、窃盗または乱用によって)、複雑な民族国家および民族国家によって支持される行為者を含む様々なソースから来ている。
73
一部の行為者は、地政学的理由で軍事衝突と防御活動を組み合わせた国家行為者を含むが、これらに限定されないネットワーク攻撃に従事し、継続することが予想される。戦争と他の重大な衝突の間、私たちと私たちが依存している第三者は、報復的なネットワーク攻撃を含むこれらの攻撃のリスクを増加させやすいかもしれません。これは、私たちのシステムと運営、サプライチェーン、ならびに私たちの商品やサービスを生産、販売、流通する能力を実質的に乱す可能性があります。
我々と私たちが依存する第三者は、社会工学攻撃(深さ偽装およびネットワーク釣りによる攻撃を含む)、悪意コード(例えば、ウイルスおよびワーム)、マルウェア(高度な持続的脅威侵入の結果を含む)、サービス拒否攻撃、証拠充填、証拠取得、人員不正行為またはエラー、恐喝ソフトウェア攻撃、サプライチェーン攻撃、ソフトウェア脆弱性、サーバ障害、ソフトウェアまたはハードウェア障害、データまたは他の情報技術資産の損失、広告ソフトウェア、人工知能強化または促進の攻撃、電気通信障害、地震、火災、洪水、そして他の似たような脅威。
特に、深刻な恐喝ソフトウェア攻撃が一般的になっており、私たちの運営が深刻に中断されている可能性がある製品やサービスを提供することができます敏感な情報と収入の損失、名声被害、そして資金流用。恐喝支払いは恐喝ソフトウェア攻撃の否定的な影響を軽減するかもしれないが、例えば、適用された法律または法規によってそのような支払いが禁止されているため、私たちはそのような支払いを望んでいないか、または支払うことができないかもしれない。
ますます多くの従業員が私たちのオフィスやネットワークの外でネットワーク接続、コンピュータと設備を使用して、家、途中と公共の場所で仕事をすることを含めて、遠隔仕事はますます一般的になり、私たちの情報技術システムとデータのリスクを増加させた。さらに、将来的または過去のビジネス取引(例えば、買収または統合)は、我々のシステムがエンティティのシステムおよび技術に存在する脆弱性を買収または統合する負の影響を受ける可能性があるので、より多くのネットワークセキュリティリスクおよび脆弱性に直面する可能性がある。また,これらの買収や統合されたエンティティを職務調査する際に発見されなかったセキュリティ問題が発見される可能性があり,会社を我々の情報技術環境やセキュリティ計画に統合することは困難である可能性がある.
また、第三者サービスプロバイダへの依存は、サプライチェーン攻撃や他の脅威を含む、当社の業務運営に新たなネットワークセキュリティリスクや脆弱性をもたらす可能性があります。 我々は、クラウドベースのインフラ、データセンター施設、暗号化および認証技術、従業員電子メール、人的資本管理、文書管理、臨床前研究、臨床研究(データ管理、生体統計および安全報告を含む)、医薬品製造および他の機能を含む、様々な環境において敏感なデータを処理するために、第三者サービスプロバイダおよび技術に依存してキー業務システムを実行する。私たちはまた、第三者サービスプロバイダが他の製品、サービス、部品、または他の方法を提供して私たちの業務を運営することに依存します。これらの第三者の情報セキュリティアプローチを監視する能力は限られており、これらの第三者には十分な情報セキュリティ対策がない可能性がある。もし私たちの第三者サービス提供者がセキュリティ事件や他の中断に遭遇したら、私たちは悪い結果に直面するかもしれない。第三者サービス提供者がプライバシーや安全に関する義務を履行できなかった場合、損害賠償を受ける権利があるかもしれませんが、どの賠償も私たちの損害を補うのに十分ではないかもしれません。あるいはそのような賠償を取り戻すことができないかもしれません。 また、サプライチェーン攻撃の頻度と深刻さが増加しており、私たちのサプライチェーンにおける第三者のインフラや私たちの第三者パートナーのサプライチェーンが損なわれていないという保証はありません。
我々はすでにセキュリティ事件を防ぐためのセキュリティ対策を実施しているが,これらの措置が有効である保証はない.ハードウェアおよび/またはソフトウェアのような情報システムにおける脆弱性を検出、緩和、および修復するためのステップをとる。しかし、私たちはこのようなすべての抜け穴をタイムリーに検出して修復しないかもしれない。また,我々は,認識された脆弱性を解決するための修復策やパッチの開発と配備に遅延が生じる可能性がある.抜け穴は利用されてセキュリティ事件を招く可能性がある。
以前に決定されたまたは同様の脅威は、無許可、不法または意外に取得、修正、廃棄、損失、変更、暗号化、開示、または私たちの敏感な情報、または私たちの情報技術システム、または私たちが依存する第三者の情報にアクセスすることをもたらす可能性があるセキュリティイベントまたは他の中断をもたらす可能性がある。セキュリティイベントや他の中断は、私たち(および私たちが依存する第三者)が私たちの製品やサービスを提供する能力を破壊する可能性があります。
私たちは大量の資源を費やしたり、私たちの業務活動(私たちの臨床研究活動を含む)を修正したりして、安全事故を防止しようとするかもしれない。さらに、いくつかのデータプライバシーおよびセキュリティ義務は、特定のセキュリティ対策の実施および維持、または私たちの情報技術システムおよび敏感な情報を保護するための業界標準または合理的なセキュリティ対策を要求する可能性がある。
74
適用されるデータプライバシーおよびセキュリティ義務は、影響を受けた個人、顧客、規制機関、投資家を含むセキュリティイベントを関連利害関係者に通知することを要求する可能性があります。このような開示は費用が高く、そのような要求を開示または遵守しないことは悪い結果をもたらすかもしれない。もし私たち(または私たちが依存する第三者)がセキュリティ事件を経験した場合、またはセキュリティ事件を経験したと考えられる場合、私たちは政府の法執行行動(例えば、調査、罰金、処罰、監査、検査)、追加の報告要件および/または監督、敏感な情報の処理に対する制限(個人データを含む)、訴訟(類クレームを含む)、賠償義務、負の宣伝、名声損害、通貨資金移動、管理層の注意の移動、私たちの運営中断(私たちの候補製品の開発および商業化遅延を含む)、財務損失、および他の同様の損害に遭遇する可能性がある。私たちまたは私たちの第三者プロバイダが遭遇する可能性のあるセキュリティイベントとそれに伴う結果は、お客様が私たちの製品やサービスの使用を阻止または停止させ、新しい顧客が私たちの製品やサービスを使用することを阻止し、私たちの業務成長と運営能力にマイナスの影響を与える可能性があります。
私たちの契約には責任制限が含まれていないかもしれませんが、あっても、私たちの契約における責任制限は、私たちのデータプライバシーとセキュリティ義務に関連する責任、損害、またはクレームから私たちを保護するのに十分である保証はありません。私たちの保険範囲が私たちのプライバシーと安全慣行によって生じる責任から私たちを保護または軽減するのに十分か、または私たちを保護するのに十分かどうかは確認できません。私たちはこのような保険が商業的に合理的な条項や根本的に存在しない、あるいはそのような保険が未来のクレームを支払うと判断することはできません。
セキュリティイベントを経験することに加えて、第三者は、公共ソース、データ仲介人、または他の方法から、私たちに関する敏感なデータを収集、収集、または推定することができ、これらの情報は、私たちの組織に関する競争の敏感な詳細を漏洩し、私たちの競争優位性または市場地位を破壊するために使用される可能性がある。さらに、当社の敏感なデータは、私たちの従業員、人員、またはサプライヤーが生成的人工知能技術を使用しているか、またはそれに関連しているため、漏洩、開示、または漏洩する可能性がある。
私たちは厳格で変化するアメリカと外国の法律、法規と規則、契約義務、業界標準、政策、その他に支配されています データのプライバシーとセキュリティに関する義務。私たちが実際にまたはそのような義務を履行できなかったと考えられることは、規制調査または行動、訴訟、および大規模な仲裁要求、罰金および処罰、私たちの業務運営中断、名声損害、収入または利益損失、顧客または販売損失、および他の不利な業務結果をもたらす可能性がある。
我々のデータ処理活動は、研究参加者から情報を取得して処理し、様々な法律、法規、指導、業界基準、外部および内部プライバシーおよびセキュリティポリシー、契約要件、およびデータプライバシーおよびセキュリティに関連する他の義務など、多くのデータプライバシーおよびセキュリティ義務の制約を受けることを含む。
アメリカでは、連邦、州、地方政府は、データ漏洩通知法、個人データプライバシー法、消費者保護法(例えば、第5条)を含む多くのデータプライバシーとセキュリティ法律を公布している
連邦貿易委員会法と他の似たような法律(例えば盗聴法)。さらに、連邦1996年の“健康保険携帯性·責任法案”(HIPAA)(2009年の“健康情報技術促進経済·臨床健康法案”(HITECH)改正)のプライバシーおよびセキュリティ要件に制約された第三者(臨床研究データを取得した研究機関を含む)から健康情報を取得することができる。事実および状況によると、もし私たちがHIPAAによって許可されていない、または許可されていない方法でHIPAAによってカバーされているエンティティによって維持されている個別に識別可能な健康情報を取得、使用、または開示する場合、私たちは刑事罰を含む罰を受ける可能性がある。
過去数年間、米国の多くの州-カリフォルニア州、バージニア州、コロラド州、コネチカット州、ユタ州を含めて全面的なプライバシー法が公布され、プライバシー通知に具体的な開示を提供し、その個人データに関するいくつかの権利を住民に提供することを含む、カバーされた企業にいくつかの義務が加えられた。適用可能であれば、そのような権利は、特定の個人データにアクセス、訂正または削除する権利、および指向性広告、分析、および自動決定のような特定のデータ処理活動から退出する権利を選択する権利を含むことができる。このような権利の行使は私たちの業務と製品とサービスを提供する能力に影響を及ぼすかもしれない。いくつかの州はまた、敏感な情報を含むいくつかの個人データを処理することに対して、データプライバシー影響評価を行うなど、より厳しい要求を提出している。この州の法律は規定を守らない行為に法的罰金を科すことを許可している。例えば、2020年にカリフォルニアプライバシー権法案が改正された2018年カリフォルニア消費者プライバシー法は、総称してCCPAと呼ばれ、カリフォルニア州住民に属する消費者、商業代表、および従業員の個人データに適用され、企業にプライバシー通知において具体的な開示を提供し、そのような個人が特定のプライバシー権を行使する要求を尊重することを要求する。CCPAは、故意違反のたびに最高7500ドルの罰金を科すことができ、あるデータ漏洩の影響を受けた個人訴訟当事者が巨額の法定損害賠償を取り戻すことを許可すると規定している。CCPAはいくつかの臨床試験で処理されたデータを免除したが、CCPAはコンプライアンスコストを増加させ、著者らが維持しているカリフォルニア住民の他の個人データに関する潜在的な責任を増加させた。似たような法律も考慮されている
75
他のいくつかの州、そして連邦と地方レベルで、私たちは未来に似たような法律を採択するもっと多くの州があると予想している。CCPAのように,これらの州でも臨床試験を背景に処理されたデータが免除される可能性があるが,これらの事態はコンプライアンス作業をさらに複雑化させ,我々と我々が依存する第三者の法的リスクやコンプライアンスコストを増加させる可能性がある。
また、一部の州と地域および外国司法管轄区では、生物識別情報の収集を禁止または制限する法規が公布されている。私たちが使用しているアイデンティティ検証技術は私たちをバイオメトリクスによって制約されるかもしれない。例えば、イリノイ州生体特徴情報プライバシー法案、またはBIPAは、生体情報の収集、使用、保護、および記憶を管理する。BIPAは実質的な懲罰と法定損害賠償を規定し,大量の集団訴訟活動を引き起こしているが,我々はBIPAや同様の法律に違反した任意のクレームの訴訟や和解コストが高い可能性がある。訴訟に加えて、連邦貿易委員会(FTC)などの規制機関は、生体認証技術(顔認識技術を含む)の使用が追加的に審査される可能性があると述べている。
我々の従業員や人員は、生成的人工知能(AI)技術を使用して彼らの仕事を実行することができ、生成的AI技術における個人データの開示と使用は、様々なプライバシー法や他のプライバシー義務によって制約されることができる。各国政府は生成的人工知能をより多くの法律によって規範化することができ、より多くの法律によって規範化されるかもしれない。私たちがこの技術を使用することは追加的なコンプライアンス費用、規制調査と行動、そして訴訟につながるかもしれない。もし私たちが生成的人工知能を使用できなければ、それは私たちの業務効率を低下させ、競争の劣勢を招くかもしれない。
米国以外では、ますます多くの法律、法規、業界基準がデータプライバシーとセキュリティを管理する可能性がある。例えば,EUの一般データ保護条例,あるいはEUのGDPR,イギリスのGDPR,あるいはイギリスのGDPR,インドの情報技術法案や補完ルール,およびオーストラリアのプライバシー法案は,個人データの処理に厳しい要求をしている.
例えば、GDPRによれば、会社は一時的または最終的なデータ処理禁止および他の是正行動に直面する可能性があり、EU GDPRによると、会社は最高2000万ユーロの罰金に直面する可能性があり、イギリスGDPRによると、会社は最高1750万ポンドの罰金に直面する可能性があり、またはそれぞれの場合、会社は個人データの処理に関連する個人訴訟に直面する可能性があり、これらの罰金は、法的許可によってその利益を表す様々なデータ主体または消費者保護組織によって提起された、または世界の年収の4%を基準としている。
また、データの現地化要求や国境を越えたデータの流れの制限により、個人データをヨーロッパや他の司法管轄区域から米国や他の国に移すことができない可能性がある。ヨーロッパと他の司法管轄区は、データの現地化または他の国への個人データの移転を制限するための法律を公布した。特に,欧州経済圏(EEA)やイギリス(UK)は,米国や他のプライバシー法が不足していると一般的に考えられている国への個人データの移転を大きく制限している。他の司法管轄区域はそのデータ現地化と国境を越えたデータ転送法に対して類似の厳格な解釈を行う可能性がある。
現在、欧州経済地域の標準契約条項、イギリスの国際データ転送プロトコル/付録およびEU-米国データプライバシー枠組みおよびそのイギリス拡張(自己認証コンプライアンスへの移行を許可し、この枠組みに参加する関連米国組織)には、法律によって個人データを欧州経済地域およびイギリスから米国に移転するための様々なメカニズムがあるが、これらのメカニズムは法的挑戦を受けており、これらの措置を満たすか、依存して合法的に米国に個人データを転送することができる保証はない。もし私たちが合法的な方法で個人データをヨーロッパ経済区、イギリスまたは他の司法管轄区域からアメリカに移すことができない場合、あるいは合法的な移転の要求が煩雑すぎる場合、私たちの業務中断または降格、巨額の費用で私たちの業務または全部の業務またはデータ処理活動を他の管轄区域(例えば、ヨーロッパ)に移す必要があり、より多くの規制行動に直面し、巨額の罰金と処罰、データの転送ができないこと、パートナー、サプライヤーおよび他の第三者との協力、私たちの業務に必要な個人データの処理または移転を禁止することを含む重大な不利な結果に直面する可能性がある。また,個人データをヨーロッパ経済区やイギリスから他の司法管轄区,特に米国に移転した会社は,規制機関,個人訴訟者,活動団体のより厳しい審査を受けることになる。一部の欧州規制機関は、これらの会社がGDPRの国境を越えたデータ転送制限に違反している疑いがあるため、いくつかの企業に特定の個人データの欧州への移転を一時停止または永久停止するよう命じている。
データプライバシーやセキュリティ法律に加えて、契約上は業界組織が採用している業界基準の制約を受けており、将来的にはこのような義務に制約される可能性があります。私たちはまた、データプライバシーやセキュリティに関する他の契約義務の制約を受けており、これらの義務を守る努力は成功しないかもしれません。
我々は、データプライバシーおよびセキュリティに関連するいくつかの認証または自律原則を遵守するなど、プライバシーポリシー、マーケティング材料、および他の声明を発表します。これらの政策や材料や声明に欠陥があることが発見されれば
76
もし私たちの接近が透明性、詐欺性、不正性、または非現実的な陳述が足りなければ、私たちは調査、規制機関の法執行行動、または他の不利な結果を受けるかもしれない。
データプライバシーやセキュリティ(および消費者のデータプライバシー期待)に関する義務は急速に変化しており,ますます厳しくなり,不確実性をもたらしている.さらに、これらの義務は、法ドメイン間で不一致または衝突する可能性がある異なる適用および解釈される可能性がある。これらの義務を準備して遵守するには、私たちが大量の資源を投入する必要があり、私たちのサービス、情報技術、システム、やり方、および個人データを処理する任意の第三者を代表するサービス、情報技術、システム、およびやり方を変更する必要があるかもしれません。
私たちは、私たちのデータプライバシーやセキュリティ義務を守る努力に失敗することがある(または失敗したと考えられる)。また、私たちが努力しているにもかかわらず、私たちが依存している人や第三者がこのような義務を履行できない可能性があり、これは私たちの業務運営に悪影響を及ぼす可能性があります。もし私たちまたは私たちが依存している第三者が適用されたデータプライバシーおよびセキュリティ義務を解決または遵守できなかったとみなされた場合、私たちは、政府の法執行行動(例えば、調査、罰金、監査、検査、および同様の行動)、訴訟(集団訴訟クレームを含む)および大規模な仲裁要求、追加の報告要件および/または監督、個人データの処理の禁止、個人データの廃棄または使用の禁止、および監禁会社の管理者を含む重大な結果に直面する可能性がある。特に,原告は集団クレームや大規模仲裁要求を含むプライバシーに関するクレームを積極的に会社に提起してきた.その中のいくつかのクレームは毎回違反した上で法定損害賠償を取り戻すことを許可し、実行可能であれば、巨大な法定損害賠償をもたらす可能性があり、具体的にはデータ量と違反数量に依存する。
このような事件は、顧客流出、関連する臨床研究を含むが、これらに限定されない、私たちの名声、業務、または財務状況に悪影響を及ぼす可能性があります。個人データを処理できない、またはいくつかの司法管轄区域で運営されていること、私たちの製品を開発すること、またはそれを商業化する能力が限られていること、任意のクレームまたは調査のために時間と資源をかけて弁護すること、否定的な宣伝、または私たちのビジネスモデルまたは運営の大きな変化。
私たちの純営業損失の繰越といくつかの他の税務属性を使用する能力は限られている可能性があるので、将来の税務負担が増加するかもしれません。
2023年12月31日現在、我々が繰り越した米国連邦純営業損失総額は約7690万ドル。米国現行の連邦所得税法によると、2017年12月31日以降の納税年度に発生する米国連邦NOLは無期限に繰り越すことができるが、このような米国連邦NOLの控除額は一般的に課税収入の80%に限られている。各州がどの程度米国の現行の連邦所得税法を遵守するかどうかは定かではない。
また、改正された1986年の国内税法(IRC)第382条および383条および州法の該当条項によると、ある会社が“所有権変更”を経験した場合、これは通常、その持分所有権が3年間で累計50ポイントを超える(価値計算)と定義され、同社は変更前のNOL繰り越しや他の変更前の税収属性(例えば、税収控除を検討する)を使用して、変更後の収入や税収を相殺する能力が制限される可能性がある。私たちは2023年6月30日までに1回または複数回の所有権変更を経験したと確信している。しかし,2023年6月30日までの所有権変更は,会社がそのNOLや他の税収属性を利用する能力に大きな影響を与えないと予想される。私たちの株式所有権はその後変化したので、私たちはまた未来に所有権の変化を経験するかもしれない。その中のいくつかは私たちがコントロールできるものではないかもしれない。したがって、私たちは、将来の米国連邦課税収入(課税純収入を稼いだ場合)や他の所有権前に税収属性を変更する能力が制限される可能性があり、将来の納税義務を増加させる可能性がある所有権前変更NOL繰り越しを使用しています。さらに、州レベルでは、NOLの使用を一時停止または制限する時期がある可能性があり、これは州の課税税を加速または永久的に増加させる可能性がある。
私たちは私たちのオーストラリアの完全子会社を通じて重要な業務を展開している。もし私たちがオーストラリアで運営する能力を失った場合、あるいは私たちの子会社がオーストラリア法規によって許可された研究開発税の免除を受けることができなければ、私たちの業務と運営結果は影響を受けるだろう。
2019年1月、著者らはオーストラリアの完全子会社Equillium Australia Pty Ltdを設立し、オーストラリアとニュージーランドで暴走喘息の治療に用いられるitolizumab(EQ 001)の臨床開発を初歩的に行った。当付属会社はEQ 102の第一段階研究を行っており,この研究は完成しており,現在EQ 101の臨床研究も行われており,将来さらなる臨床研究が行われている可能性がある。地理的距離が遠いため、現在オーストラリアでは従業員が不足していることと、私たちはオーストラリアでの運営経験が不足しているため、臨床研究を含むオーストラリアとニュージーランドでの私たちの候補製品のモニタリング、開発あるいは商業化に有効あるいは成功できないかもしれない。しかも製品のための臨床研究の結果は保証できません
77
オーストラリアとニュージーランドの候補者はFDAまたは他の外国規制機関によって開発と商業化のための承認を受けるだろう。
また、オーストラリアの現行の税収法規は払い戻し可能な研究開発税の免除を規定している。もし私たちがオーストラリアでEquillium Australia Pty Ltdを運営する能力を失った場合、資格がない場合、あるいは研究開発税の免除を得ることができず、得られた返金が私たちの予想を大幅に下回った場合、あるいはオーストラリア政府が税金免除を大幅に減少またはキャンセルした場合、または監査結果に基づいて、オーストラリア税務局が以前の申告を無効と判断し、以前の返金金額の返済を要求した場合、私たちの財務予測は正しくない可能性があり、私たちの業務と運営結果は不利な影響を受けるだろう。
もし私たちが米国の輸出規制と経済制裁を守らなければ、私たちの業務、財務状況、見通しは実質的な悪影響を受ける可能性がある。
我々の業務·製品は、米国財務省外国資産制御弁公室(OFAC)によって施行された“米国輸出管理条例”および“経済貿易制裁条例”を含む米国輸出規制法律法規によって制約されている。わが社はこのような法律法規を守らなければならない。Itolizumab(EQ 001)の抗体配列はキューバの知的財産権に由来するため,キューバ原産の薬物であり,itolizumab(EQ 001)の輸入,開発,商業化がこれらの法律,制裁,条例によって拘束されると考えられる。著者らは現在、キューバ資産規制条例(CACR)によって発行されたキューバ原産薬品に関連する汎用許可証にOFACに依存しており、輸入とitolizumab(EQ 001)に関する臨床研究を行っている。OFAC汎用ライセンスがない場合、CACRによれば、itolizumab(EQ 001)のすべての開発および潜在的な商業化活動が禁止され、そのような活動を許可する特定のライセンスをOFACに申請することが要求されるが、OFACはこれを否定することができる。
OFACには、一般ライセンスのitolizumab(EQ 001)への適用性を確認するか、または汎用ライセンスがない場合には、itolizumab(EQ 001)の商業化に関連する活動を許可する特定のライセンスライセンスをOFACに提出するか、または提出された解釈指導要求をOFACに提出する。我々は同時に、OFACが共通ライセンスがitolizumab(EQ 001)に適用されることに関する決定が間違っていると結論を出した場合、提出された材料を自発的に開示することを要求する。
2019年11月,OFACはFDAに問い合わせ,OFACはitolizumab(EQ 001)が“キューバ原産薬”の定義に属することを決定し,したがって,CACR 515.547(B)および(C)節の一般ライセンス認可は,FDAの承認を求めることを目的として,Tolizumab(EQ 001)の臨床研究を決定することを含めて詳細に検討することを通知した。したがって、私たちが行っているおよび未来のitolizumab(EQ 001)の臨床研究は現在、OFACのさらなる許可を必要としない。
OFACは、キューバ原産医薬品の汎用ライセンスがitolizumab(EQ 001)に適用されると結論したが、OFACが将来汎用ライセンスを撤回または修正しないことを保証することはできず、私たちが汎用ライセンスまたは他の輸出法律法規を遵守し続けることを保証することもできない。OFACが汎用ライセンスを撤回または修正する場合、または他の方法で汎用ライセンスがitolizumab(EQ 001)に適用されないと判断し、OFACがその後、私たちの特定のライセンス要求を拒否するか、または特定のライセンスの発行を延期する場合、私たちは取引できない、または他の方法でitolizumab(EQ 001)を商業化する。この場合、Tolizumab(EQ 001)に関連するビジネスの停止を要求されることになり、私たちの財務状況やビジネスの見通しに大きな悪影響を与えることになります。さらに、一般的または特定のライセンスがない場合には、私たちの証券の譲渡、販売および/または購入が禁止される可能性があり、私たちの証券の所有権または占有権は、凍結された財産に関するOFACの肯定的報告要件によって制約される可能性がある。CACRまたは他の適用される輸出規制および制裁法に違反するいかなる行為も、私たちと私たちの特定の従業員を重大な民事または刑事罰を受ける可能性がある。
医療法や施行条例の変化、医療政策の変化は、現在予測できない方法で私たちの業務に影響を与える可能性があり、私たちの業務や運営結果に大きな悪影響を及ぼす可能性があります。
ヘルスケアシステムについては、多くの立法や規制面の変化や提案中の変化が続いており、これらの変化は、候補製品の上場承認を阻止または延期し、承認後の活動を制限または規制し、マーケティング承認を得た任意の候補製品を収益的に販売する能力に影響を与える可能性がある。米国の政策立案者や支払者の中で,医療システムの改革を推進することに大きな興味があり,その既定目標は医療コストの抑制,質の向上および/または参入拡大であり,製薬業はこれらの努力の特別な重点であり,重大な立法イニシアティブの大きな影響を受けてきた。
78
“平価医療法案”は、政府と民間保険会社が医療保健に資金を提供する方式を大きく変え、米国の製薬業に大きな影響を与えた。例えば、“平価医療法案”は、ブランド薬品メーカーが支払うべき医療補助税金還付の最低レベルを15.1%から23.1%に引き上げ、医療補助管理を要求する医療機関に対して支払うべき薬品の還付を徴収し、指定された連邦政府がある“ブランド処方薬”を販売する予定の製薬業者や輸入業者に控除できない年会費を徴収し、吸入、注入、点滴、移植または注射を要求した薬品に対してメーカーの医療補助薬品還付計画下でのリベートを計算し、医療補助計画の資格基準を拡大した。患者を中心とした結果研究所を創立し、監督、優先事項を確定し、臨床治療効果の比較研究を行い、そしてこのような研究に資金を提供する;そして連邦医療保険と医療補助サービスセンター(CMS)に連邦医療保険と医療補助革新センター(CMMI)を設立し、革新的な支払いとサービス交付モードをテストして、連邦医療保険と医療補助支出を下げるために、処方薬支出を含む可能性がある。
“平価医療法案”のいくつかの面は司法、国会、行政面で挑戦されている。例えば、2021年6月17日、米国最高裁は、“個人強制令”が国会で廃止されたため、“平価医療法案”全体が違憲だと弁明する手続き理由に基づく挑戦を却下した。また,米国最高裁判所が裁決を下す前に,総裁·バイデンは2021年1月28日に行政命令を発表し,平価医療法案市場による医療保険の取得を目的とした特殊な加入期間を開始した。行政命令はまた、特定の政府機関に、医療補助モデルプロジェクトおよび免除計画の再検討、医療補助または“平価医療法案”による医療保険獲得に不必要な障害をもたらす政策を含む医療補助モデルプロジェクトおよび免除計画の再審査および見直し、医療補助または“平価医療法案”の見直しを指示する。また、2022年8月16日、総裁·バイデンは2022年インフレ削減法案に署名し、個人が平価医療法案市場で医療保険を購入する増強補助金を2025年計画年に延長した。2025年からアイルランド共和軍は,受益者の最大自己負担コストの著しい低減と新たなメーカー割引計画の作成により,連邦医療保険D部分計画下の“ドーナツ脆弱性”を解消した。“平価医療法案”は将来的に司法や国会の挑戦を受ける可能性がある。このような挑戦やバイデン政府の医療改革措置が“平価医療法案”にどのように影響するかは不明である。私たちは平価医療法案のどんな変化にも注目し続けており、これらの変化は逆に私たちの将来の業務に影響を与える可能性がある。
“平価医療法”が公布されて以来,他の立法改正も提案され,採択された。これらの変化には、2011年の予算制御法とその後の法律に基づいて、医療保険提供者に支払われる総金額を2%/年度削減することが含まれており、この法案は2013年に始まり、国会がさらに行動しない限り、2032年まで有効となる。また、総裁·バイデンは2021年3月11日に“2021年米国救援計画法案”に署名し、2024年1月1日から単一源と革新多源薬に対する法定医療補助薬品還付上限を廃止し、現在この上限は薬品メーカー平均価格の100%である。2013年1月、“2012年米国納税者救済法”が法律に署名され、病院、画像形成センター、がん治療センターを含むいくつかの提供者への医療保険支払いがさらに減少し、政府が提供者に多額の支払いを取り戻す訴訟時効が3年から5年に延長された。新しい法律は連邦医療保険や他の医療資金のさらなる減少を招く可能性があり、これは顧客が私たちの製品の需要と負担能力、そして私たちの財務運営結果に実質的な悪影響を及ぼす可能性がある。
また,最近政府は製薬会社がその上場製品に価格を設定する方式をより厳しく審査し,いくつかの国会調査を招き,連邦立法,州政府の努力を提案し,製品定価の透明性の向上,連邦医療保険下の処方薬のコスト低減,定価とメーカー患者計画との関係の審査,政府計画の薬品精算方法の改革を目指している。連邦レベルでは、2021年7月、バイデン政府は“米国経済における競争を促進する”という行政命令を発表し、その中には処方薬に対する条項が複数ある。バイデンの行政命令に応えるために、2021年9月9日、衛生·公衆サービス部(HHS)は高薬価に対応する総合計画を発表し、その中で薬品定価改革の原則を概説し、国会が取ることができる各種の潜在立法政策、HHSが取ることができる潜在行政行動を挙げてこれらの原則を推進した。また,アイルランド共和軍(IRA)は他の事項を除いて,(1)HHSに連邦医療保険(Medicare)で覆われたある単一由来薬物と生物製品の価格について交渉するよう指示し,(2)連邦医療保険B部分とD部分にリベートを徴収し,インフレを超える価格上昇を懲罰するよう指示した。このような規定は2023年度から段階的に施行される。2023年8月29日、HHSは、連邦医療保険薬品価格交渉計画が現在法的挑戦を受けているにもかかわらず、価格交渉を受ける上位10種類の薬物のリストを発表した。アイルランド共和軍は衛生と公共サービス部が最初の数年に規制ではなく指導によってその多くの規定を実施することを許可した。これらの計画の実施に伴い,HHSは指導意見を発表·更新し続けている。アイルランド共和軍がどのように実施されるかは不明であるが,製薬業に大きな影響を与える可能性がある。また,バイデン政府の2022年10月の行政命令に応えるために,HHSは2023年2月14日にCMMIテストの3つの新しいモデルを概説し,それらに基づいて薬品コストを低減し,促進する報告書を発表した
79
獲得可能であり、そして看護の質を高める。これらのモデルが将来の任意の医療改革措置で使用されるかどうかは不明である。また、2023年12月7日、バイデン政府は“ベハ·ドール法案”下の参入権を使用することで処方薬の価格を制御するイニシアチブを発表した。2023年12月8日、米国国家標準·技術研究所は、権限行使を考慮した機関間指導枠組み草案を発表し、その中で初めて製品価格を機関が進行権を行使する際に使用できることを決定する要因とした。これまでデモの権利を行使したことはなかったが、新たな枠組みの下で、この権利が継続するかどうかは定かではない。州レベルでは、アメリカの各州はますます積極的に薬品と生物製品の定価を制御するための法規を立法と実施し、価格或いは患者の精算制限、割引、ある製品への参入の制限、マーケティングコストの開示と透明性措置を含み、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。アイルランド共和軍の薬品価格改革は、私たちの候補製品を商業化することに成功した能力に悪影響を及ぼす可能性があり、私たちの候補製品の実際または期待価値を低下させる可能性があり、これは私たちの業務に否定的な影響を与えるだろう。
これらの措置や将来講じられる可能性のある他の医療改革措置は,より厳しいカバー基準とより低い精算を招き,我々が受け取った任意の承認製品の価格に追加の下振れ圧力をもたらす可能性が予想される。連邦医療保険や他の政府援助計画のいかなる精算減少も、個人支払者の支払いの同様の減少を招く可能性がある。上場承認を得ると、コスト制御措置や他の医療改革を実施することは、私たちの収入の創出、利益の実現、あるいは私たちの薬物の商業化を阻止するかもしれない。
もし私たちのすべてのサービスプロバイダが従業員として説明されたら、私たちは雇用と源泉徴収責任と他の追加コストの影響を受けるだろう。
私たちは独立請負業者に依存して私たちに特定のサービスを提供してくれる。我々とこれらの外部サービスプロバイダとの関係構築方式は,従業員関係ではなく,独立した請負者関係が生じると考えられる.独立請負者と従業員の違いは、一般的に彼または彼女のサービス提供に関する自主権と独立性にある。高度自治と独立は通常独立した請負者関係を表し,高度制御は通常雇用関係を表す.税務または他の規制機関は、既存の法律と法規、および未来に採択された法律と法規に基づいてサービスプロバイダを独立請負業者と同定するという説に疑問を提起するかもしれない。カリフォルニア州立法機関が採択したカリフォルニア議会法案5、カリフォルニア州知事のガヴィン·ニューザムが2019年9月に法律に署名したこと、またはAB 5、議会法案2257、またはAB 2257が2020年9月に施行され、AB 5の一部が改訂されたことを含むいくつかの司法判断および立法提案が、労働者の分類方法に大きな変化をもたらす可能性があることが分かっている。AB 5とAB 2257は一般にAB 5と総称されている。AB 5はカリフォルニア州最高裁判所のDynamex Operations West,Inc.がロサンゼルス高等裁判所の一致裁決を訴えることを目的としている。労働者分類を決定するための新たなテストを導入し、従業員関係の範囲を拡大し、独立請負業者関係の範囲を縮小したと広く考えられている。AB 5は医師や心理学者を含む資格のある医療専門家を免除しているが、私たちのすべての独立請負業者が免除職業で働いているわけではない。AB 5の実行を担当する規制機関はほとんど指導を提供しておらず,その応用には大きな不確実性がある。また,AB 5は広く全国的な議論のテーマであり,他の管轄区域でも類似した法律が制定される可能性がある.そのため、州、連邦と外国人労働者の分類監督構造は今後数年でどのようになるかに重大な不確定性が存在する。現在の経済気候は予測可能な未来に労働者分類に関する討論が続くということを見せてくれる。そのような規制機関や州、連邦、または外国裁判所が、私たちのサービス提供者が独立請負業者ではなく従業員であると判断した場合、私たちは所得税の源泉徴収、社会保障、医療保険および同様の税金の源泉徴収と支払い、失業および他の関連賃金税の支払い、特定の従業員福祉の提供を要求されるだろう。私たちはまた過去に未納された税金と他の費用に責任を負い、処罰されるかもしれない。したがって、私たちを独立請負業者として説明するサービスプロバイダを従業員として分類するいかなる決定も、私たちの業務、財務状況、および運営結果に悪影響を及ぼす可能性があります。
私たちは適用される外国、連邦、州詐欺と乱用、透明性、政府価格報告書、および他の医療法規によって制限されるかもしれない。もし私たちがこのような法律を遵守できないか、または完全に遵守できなければ、私たちは巨額の処罰に直面するかもしれない。
医療提供者と第三者支払者は、市場の承認を得た任意の将来の候補製品を推薦し、処方する上で主な役割を果たすだろう。第三者支払者や顧客との手配は、広範に適用される詐欺や乱用、その他の医療法律や法規に直面する可能性があり、これらの法律と法規は、私たちの研究やマーケティング、販売、流通の私たちの製品の業務または財務配置と関係に影響を与える可能性があります。医療サービスの転換やMedicareやMedicaidへの直接の移行はコントロールできません
80
他の第三者支払者、連邦、州医療保健法律法規と詐欺と乱用、および患者の権利に関する法律と法規は現在も将来も私たちの業務に適用されます。私たちの運営能力に影響を与える可能性のある法律は含まれているが、これらに限定されない
81
私たちは、EQ 101、EQ 302、itolizumab(EQ 001)、および任意の将来の候補製品の使用に影響を与える可能性がある(承認された場合)を含む、医師および他のヘルスケア提供者と相談および科学的諮問委員会の手配を達成した。これらの法律の複雑かつ深遠な性質のため、規制機関はこれらの取引を再編成または終了しなければならない禁止措置と見なすかもしれない。そうでなければ、私たちは他の重大な処罰を受けるかもしれない。規制当局が私たちとサプライヤーとの財務関係を適用法違反と解釈した場合、これらのサプライヤーはEQ 101、EQ 302、itolizumab(EQ 001)、または任意の将来の候補製品の注文および使用に影響を及ぼす可能性があり、私たちは悪影響を受ける可能性がある。
これらの法律の範囲も執行も不確定であり,現在の医療改革環境の急速な変化の影響を受けている。連邦と州法執行機関は最近、医療保険会社、医療保健提供者、慈善基金を含む他の第三者間の相互作用の審査を強化し、医療業界の一連の調査、起訴、有罪判決、和解を招いた。政府当局は結論を出すかもしれないが、私たちの業務のやり方は、私たちと医者の相談手配を含み、その中の一部の医者は提供されたサービスの補償として株式オプションを獲得し、現在或いは未来に医療保険法の適用に関する法規、法規、機関指導或いは判例法に適合していない。調査への対応に時間と資源がかかる可能性があり、経営陣の業務への関心を分散させる可能性がある。このような調査や和解は、私たちのコストを増加させるか、または他の方法で私たちの業務に悪影響を及ぼす可能性がある。
私たちが第三者の業務手配と適用される医療法律や法規に適合することを確保することは、コストが高くなる可能性があります。もし私たちの運営がこれらの法律のいずれかまたは任意の他の現在または将来、私たちの政府の法律および法規に適用される可能性があることが発見された場合、私たちは重大な民事、刑事および行政処罰、損害賠償、罰金、返還、監禁、連邦医療保険や医療補助、契約損害、名声損害、利益減少および将来の収益減少、追加の報告義務および監督(これらの法律違反に関する告発を解決するために当社の誠実な合意または他の合意に拘束された場合)、および私たちの業務の削減または再編は、いずれも私たちの運営を深刻に混乱させる可能性がある。もし私たちがそれと業務を展開することを期待している任意の医師または他の医療提供者または実体が適用されない法律に適合していないことが発見された場合、彼らは政府の援助された医療計画から除外されることを含む重大な刑事、民事または行政制裁を受ける可能性がある。
私たちはアメリカと外国のいくつかの反腐敗、反マネーロンダリング、輸出規制、制裁、その他の貿易法律と法規の制約を受けている。私たちは違反によって深刻な結果に直面するかもしれない。
米国と外国の反腐敗、反マネーロンダリング、輸出規制、制裁およびその他の貿易法律法規、または総称して貿易法、会社とその従業員、代理人、CRO、法律顧問、
82
会計士、コンサルタント、請負業者、および他のパートナーは、直接または間接的な許可、承諾、提供、提供、誘致、または公共または民間部門の受取人の腐敗または不当な支払い、または任意の他の価値のあるものを受け入れてはならない。貿易法違反は、大量の刑事罰金と民事処罰、監禁、貿易特権の喪失、資格取り消し、税収の再評価、契約違反と詐欺訴訟、名声損害、その他の結果を招く可能性がある。私たちは政府機関や政府付属病院、大学、その他の組織の役人や従業員と直接または間接的な相互作用を持っている。私たちはまた、時間が経つにつれて、アメリカ以外での私たちの活動が増加すると予想している。契約サービス提供者による研究、臨床前研究および臨床研究、および/または必要な許可、許可証、特許登録、および他の市場承認を得ることが予想される。私たちは、私たちがそのような活動を明確に許可していなくても、私たちの人員、代理、またはパートナーの腐敗や他の不法活動に責任を負うことを要求されるかもしれない。上記の法律および条例に違反するいかなる行為も、重大な民事と刑事罰金と処罰、監禁、輸出入特権の喪失、資格取り消し、税額の再評価、契約違反および詐欺訴訟、名誉損害、およびその他の結果を招く可能性がある。
私たちの普通株式所有権に関連するリスク
私たちの経営業績にかかわらず、私たちの普通株の株価は変動したり下落したりする可能性があり、あなたはすべてまたは一部の投資を損失するかもしれません。
私たちの普通株の市場価格は多くの要素によって大幅に変動する可能性があります。その中の多くの要素は私たちがコントロールできないことです
また、株式市場は、新冠肺炎の大流行、銀行の倒産、ロシアとウクライナの間の衝突、中東の衝突を含む極端な価格と出来高の変動を経験し、これらはすべて影響を与えた
83
多くの生命科学会社の株式証券の市場価格に影響を与え続ける可能性がある。多くのバイオ製薬会社の株価変動はこれらの会社の経営業績に関係なく、あるいは比例しない。過去、市場変動期間中、株式市場は証券集団訴訟を起こした。私たちが証券訴訟に巻き込まれれば、私たちに巨額の費用を負担させ、資源や経営陣の私たちの業務への関心を移転させ、私たちの業務に悪影響を及ぼすかもしれません。
追加資本の調達は私たちの株主に希釈し、私たちの運営を制限したり、私たちの技術や候補製品に対する権利を放棄することを要求するかもしれません。
これまで、相当な製品収入を生み出すことができれば、株式発行、債務融資、および協力および許可協定の組み合わせによって、小野との資産購入協定のような現金需要を満たすことが予想されています。私たちが株式または転換可能な債務証券を売却することによって追加資本を調達する場合、あなたの所有権資本は希釈され、これらの証券の条項は清算または他の特典を含む可能性があり、普通株主としての権利に悪影響を及ぼす可能性がある。2023年10月、私たちはJefferiesと2023年の現金自動支払機メカニズムを締結しました。このメカニズムによると、私たちは時々Jefferiesを通じて私たちの販売代理として私たちの普通株の株を発売し、販売することができます。総発行価格は2195万ドルに達します。本年度報告書Form 10−Kを提出した時点で、2023年の現金自動支払機融資メカニズムにおけるいかなる株も販売していません。
債務融資が可能であれば、追加債務を招く、資本支出を行う、または配当を宣言するなど、私たちが具体的な行動をとる能力を制限または制限する契約を含むいくつかの合意が含まれる可能性がある。
もし私たちが小野との資産購入協定のような第三者との協力および許可協定によって資金を調達すれば、私たちは、私たちの技術、将来の収入流、研究計画、または候補製品に対する貴重な権利を放棄しなければならないか、または私たちに不利になる可能性のある条項で許可を与えなければならないかもしれない。もし私たちが必要な時に株式や債務融資を通じてより多くの資金を調達できない場合、私たちは私たちの製品開発や将来の商業化努力を延期、制限、減少または終了することを要求されるかもしれません。あるいは私たちは自分で開発とマーケティングをより望んでいた候補製品の権利を与えます。
金融サービス業の不利な発展に影響を与えることは、私たちの現在および予想されている業務運営および私たちの財務状況や運営結果に悪影響を及ぼす可能性がある。
金融機関の不利な事態の発展に影響を与え、例えば噂や実際に発生した流動性に関連した事件は、過去及び未来に銀行倒産や市場全体の流動性問題を招く可能性がある。例えば、2023年3月10日、SVBはカリフォルニア州金融保護·革新部によって閉鎖され、後者はFDICを担当者に任命した。同様に,2023年3月12日,Signature BankとSilvergate Capital Corp.はそれぞれ破産管理プログラムに巻き込まれた.また、2023年5月1日、連邦預金保険会社は第1共和銀行を引き継ぎ、その資産をモルガン·チェースに売却した。米国財務省、連邦預金保険会社、連邦準備委員会は、金融機関が保有するいくつかのこのような政府証券を担保とした金融機関に250億ドルまでの融資を提供し、このようなツールの売却による潜在的損失リスクを低減する計画を実施しているが、金融機関の顧客引き出しや他の流動性需要に対する広範な需要はこのような計画の能力を超える可能性があるが、このような計画が十分であるかどうかは保証されていない。また、米国財務省、FDIC、連邦準備委員会が将来、他の銀行や金融機関が倒産した場合に未保険資金のルートを提供するかどうか、あるいはタイムリーにそうするかどうかは定かではない。
SVB、Signature Bank、Silvergate Capital CorpとFirst Republic Bankの関連事項によって、私たちの流動資金や私たちの現在および予想されている業務運営、財務状況、あるいは経営結果に何の悪影響も与えていないが、金融サービス業全体の流動資金問題には不確実性が存在し、私たちの業務、私たちの業務パートナー、あるいは業界全体は現在予測できない悪影響を受ける可能性がある。
私たちの銀行関係を必要または適切に評価する必要があると思いますが、現在、将来の業務運営に資金や資本化を提供するのに十分な現金額を得ていますが、私たちと銀行関係にある金融機関に影響を与える要因は、私たちが現金を得る機会を大きく損なう可能性があります。他にも、これらの要因には、流動性の緊張または失敗、様々な金融、信用または流動資金協定または手配された義務を履行する能力、金融サービス業または金融市場の中断または不安定、または金融サービス業会社の将来性に対する懸念または否定的な予想が含まれる可能性がある。これらの要素はまた、金融市場や一般金融サービス業に関連する要素を含む可能性がある。1つまたは複数のこれらの要因に関連するイベントまたは懸念の結果には、現在および予想されているビジネス運営、ならびに私たちの財務状況および運営結果に生じる様々な重大かつ悪影響が含まれている可能性がある。これらは遅延に限定されません
84
預金または他の金融資産または未加入預金または他の金融資産の損失を取得するか、または現金管理スケジュールによって制限された資金の取得または実際の損失の取得を遅延させる、および/または現金管理スケジュールに制限された資金を終了する。
さらに、米国または国際金融システムに対する投資家の一般的な懸念は、より高い金利またはコスト、より厳しい財務および運営契約、または信用および流動性源の獲得に対する体系的な制限を含むあまり有利ではない商業融資条項を招く可能性があり、それによって、私たちはより受け入れ可能な条項が融資を受けにくく、さらには融資を得ることができなくなるかもしれない。他のリスクに加えて、利用可能な資金または現金および流動資金源の減少は、運営費用、財務義務、または他の義務を履行する能力に悪影響を及ぼす可能性があり、私たちの財務および/または契約義務に違反したり、連邦または州賃金および労働時間法違反を招いたりする可能性がある。上記のいずれかの影響、または上記の要因または他の関連または同様の要因に起因する任意の他の影響は、我々の流動資金、私たちの現在および/または予想される業務運営、ならびに財務状態および運営結果に重大な悪影響を及ぼす可能性がある。
もし私たちの普通株が大量に売却されたら、私たちの普通株の価格は下がるかもしれない。
もし私たちの普通株が大量に売却されれば、特に私たちの役員、役員、大株主の売却、あるいは私たちの普通株が大量に売却できる株があれば、市場は売却が発生すると考えて、私たちの普通株の価格は下がる可能性があります。2024年3月20日現在、私たちは35,254,752株の普通株流通株を持っている。改正された1933年証券法第144条によると、役員、役員、その他の関連会社が保有する株は出来高に制限される。私たちはすでに発行した普通株を登録して、私たちの従業員の株式激励計画に従って発行することができます。これらの株は発行後に公開市場で自由に売ることができます。既存の株主が私たちの普通株を売却することは、将来的に合理的または適切な時間と価格で株式または株式に関連する証券を売却することをより困難にし、他の株主が私たちの普通株の株を売却しにくくするかもしれない。
公開市場で大量の普通株を売却したり、大量の普通株の保有者が彼らの株を売却しようとしていると市場が考えているため、我々の普通株の市場価格は低下する可能性がある。私たちは売却が私たちの普通株の現在の市場価格に及ぼす影響を予測できない。
私どもの持分集中は、取締役選挙結果や他の株主承認を必要とする事項に影響を与える能力を含む会社の事務に影響を与える能力を制限する可能性があります。
私たちの役員、役員、そして発行された普通株の保有者の5%を超える実益は、私たちの普通株のかなりの割合を持っています。したがって、これらの株主が共同で行動することは、取締役選挙や重大な会社取引の承認を含む、我々の株主の承認を必要とするすべての事項に大きな影響を与えることになる。他の株主が反対しても、会社は行動するかもしれない。このような所有権の集中は、他の株主が有益と思われる可能性のある会社の制御権の変更を遅延または阻止する可能性もある。
私たちの株式買い戻し計画がさらに改善されたり、株主価値が向上したりする保証はありません。株式買い戻しは私たちの普通株の価格に影響を与える可能性があります。
2023年7月、我々の取締役会は、2024年12月31日までに最大750万ドルの普通株を買い戻すことができる株式買い戻し計画を承認した。株式買い戻し計画によれば、株式買い戻し計画中に公開市場取引または当社取締役会またはその指定委員会が時々承認する他の取引を介して普通株を買い戻すことができる。2023年12月31日現在、私たちは株式買い戻し計画に基づいて298,385株の普通株を買い戻し、総金額は30万ドルです。2023年12月31日以降,本年度報告がForm 10−K形式で提出される日まで,株式買い戻し計画により,我々の普通株は買い戻しが行われていない。私たちが未来にこれ以上の株の買い戻しをするという保証はない。
公開市場買い戻しは、適用される連邦証券法に従って行われ、取引法規則10 b-18の定価と数量要求範囲内で行われる。私たちはまた時々規則10 b 5-1計画を作成して、この許可の下で私たちの普通株を買い戻すことができます。買い戻しの時間と金額(あれば)は、私たちの普通株の価格、代替投資機会、私たちの現金資源、私たちの任意の合意下の制限、会社と監督管理要求、および市場状況を含む様々な要素に依存します。
85
普通株の買い戻しは私たちの普通株の市場価格に影響を与え、その変動性を増加させ、あるいは私たちの現金備蓄を減少させる可能性があり、これは私たちの未来の運営の融資能力に影響を与える可能性がある。我々の株式買い戻し計画は長期株主価値の向上を目指しているが、それができる保証はなく、短期株価変動がその計画の有効性を低下させる可能性がある。
さらに、将来のどの株の買い戻しも、私たちの“公衆流通株”(すなわち、私たちの普通株のうち非関連株主が所有し、証券市場で取引可能な株式数)を減少させる可能性がある。私たちの公開流通株を減らすことは、私たちの普通株の取引量を減少させ、流動性の減少を招く可能性があり、両方の場合、私たちの業績とは関係なく、私たちの普通株の取引価格の変動を招く可能性がある。
デラウェア州の法律と私たちが改訂して再記載した会社証明書および改正と再記述の定款中の条項は合併、要約買収あるいは代理権競争を困難にし、それによって私たちの普通株の取引価格を下げる可能性があります。
当社の改訂及び再記載された会社登録証明書及び改訂及び重述された付例の条文は、株主がその株式によって割増を得る可能性のある取引、又は当社株主がその最適な利益に適合すると考えられる取引を含む、当社の制御権又は管理層の実際又は潜在的な変更に関する取引を遅延又は阻止する可能性がある。したがって、このような規定は私たちの普通株の価格に悪影響を及ぼすかもしれない。その他の事項を除いて、当社は会社登録証明書の改訂と再記載の定款を改訂し、再記載します
これらの条項の改正は、私たちの取締役会が優先株株を発行し、任意の権利、優遇、特権を指定する能力を除いて、私たちが当時発行した普通株の少なくとも66-2/3%の保有者の承認を得る必要がある。さらに、デラウェア州会社として、私たちはデラウェア州会社法第203条の制約を受けている。これらの規定は、大株主、特に私たちが発行した議決権株の15%以上を有する株主が、一定期間内に私たちと合併または合併することを禁止することができる。デラウェア州
86
会社は、その元の会社登録証明書に明文で規定されているか、またはその登録証明書またはその株主の承認を受けた定款を修正することによって、脱退を選択することができる。しかし、私たちはこの条項から脱退することを選択しなかった。
当社の会社登録証明書の改訂、改訂、再記述された会社定款およびデラウェア州法律のこれらおよび他の条項は、株主または潜在的な買収者が私たちの取締役会の支配権を獲得することを困難にするか、またはわが社に関連する合併、買収要約または代理権競争を延期または阻害することを含む、当時の取締役会の反対行動を開始する可能性があります。これらの条項の存在は、私たちの普通株の価格に悪影響を与え、会社の取引で価値を実現する機会を制限する可能性があります。
私たちが改正して再記載した会社登録証明書は、デラウェア州衡平裁判所とアメリカ合衆国連邦地域裁判所は、私たちと私たちの株主とのほとんどの紛争の独占法廷となり、これは、私たちまたは私たちの役員、幹部、または従業員との紛争を処理するために、私たちの株主が有利な司法フォーラムを得ることを制限するかもしれない。
私たちが改訂して再説明した会社登録証明書の規定によると、デラウェア州衡平裁判所はデラウェア州成文法または普通法に基づいて提起された以下のタイプの訴訟または手続きの独占裁判所である
この規定は、取引法で規定されている義務または責任を執行するための訴訟には適用されない。また、証券法第22条は、連邦裁判所と州裁判所は、このようなすべての“証券法”訴訟に対して同時に管轄権を持っていると規定している。したがって、州裁判所と連邦裁判所はこのようなクレームを受理する管轄権を持っている。複数の司法管轄区でクレームを提訴せざるを得ないことや、異なる裁判所が不一致や逆の裁決を下す脅威などの考慮要因を回避するために、我々が改訂·再記述した会社登録証明書は、アメリカ合衆国連邦地域裁判所が証券法に基づいて提起された任意の訴因を解決するための独占的なフォーラムとなることをさらに規定している。デラウェア州裁判所はこのような選択の裁判所条項が表面的に有効であることを確定しているが、いくつかの州研究裁判所はこれらの条項を実行し、連邦裁判所に証券法のクレームを主張する訴訟を要求しているが、控訴裁判所がこれらの条項の執行可能性を確認する保証はなく、株主は依然として専属裁判所条項が指定された場所以外の場所でクレームを提出することを求めることができる。このような状況の中で、私たちは、私たちが改訂して再記載した会社登録証明書の独占フォーラム条項の有効性と実行可能性を強く主張する予定です。これは、他の法ドメインでこのような訴訟を解決することに関連する多くの追加費用を必要とする可能性があり、これらの規定がこれらの他の法ドメインの裁判所によって実行されることを保証することはできない。もし裁判所が私たちが改正して再記載した会社登録証明書のいずれかの独占的な裁判所条項が訴訟で適用されないか、実行できないことを発見した場合、私たちは州裁判所または州と連邦裁判所訴訟証券法のクレームに関連するさらなる重大な追加費用を発生させる可能性があり、これは私たちの業務、財務状況、運営結果、将来性を深刻に損なう可能性がある。
これらの排他的フォーラム条項は、司法フォーラムにおいて、私たちまたは私たちの役員、役員、または他の従業員との紛争に有利であると考える株主のクレームを提出する能力を制限する可能性があり、これは、私たちと私たちの役員、役員、および他の従業員に対する訴訟を阻止するかもしれない。もし裁判所が私たちが改正して再記載した会社登録証明書のいずれかの独占法廷条項が訴訟で適用されないか、実行できないことが発見された場合、私たちは他の管轄区域での紛争解決に関連するさらなる重大な追加費用を発生する可能性があり、これらすべては私たちの業務を深刻に損なう可能性がある。
一般リスク因子
アメリカの上場企業として、私たちは大量の法律と財務コンプライアンスコストを負担しており、私たちはサバンズ-オキシリー法案の制約を受けている。私たちは私たちが未来のいつでも私たちが財務報告書の内部統制に効果的だということを報告できるという保証はない。
米国証券取引委員会に報告書を提出した会社は、2002年サバンズ·オキシリー法案第404条の要求を遵守しなければならない。第404条経営者は、財務報告内部統制制度の確立及び維持を要求し、取引法に基づいて提出されたForm 10−K年次報告には、経営陣が会社の財務報告の内部統制の有効性を評価する報告書を含まなければならない。十分な内部資金と
87
正確な財務諸表を適時に作成するために実施された会計制御とプログラムは依然として高価で時間のかかる仕事であり、常に再評価する必要がある。もし私たちが有効な内部財務と会計統制を持っていなければ、私たちの財務報告が信頼できなくなり、私たちの業務、経営業績、財務状況に実質的な悪影響を与え、私たちの株価を下落させる可能性があります。
私たちが財務報告書を効果的に内部統制していると結論できなければ、あるいは私たちの独立公認会計士事務所が財務報告の内部統制の有効性について保留されていない意見を提供できない場合、投資家は私たちの財務諸表の信頼性に自信を失うかもしれません。報告書の要求を遵守できなかったことは、米国証券取引委員会、ナスダック資本市場、または他の規制機関の制裁および/または調査を受ける可能性もある。
また、株主急進主義、現在の政治環境、および現在の高度な政府介入と規制改革は、大量の新しい法規や開示義務をもたらす可能性があり、これは追加のコンプライアンスコストを招き、現在予見できない方法で業務を運営する方法に影響を与える可能性がある。私たちの経営陣と他の人たちはこのようなコンプライアンス計画を実施するために多くの時間を投入する必要があるだろう。さらに、どんな新しい規制や開示義務も、私たちの法律と財務コンプライアンスコストを増加させ、いくつかの活動をより時間的で高価にするかもしれない。
私たちまたは私たちが依存している当事者は、公衆衛生流行病や疫病を含む地震、火災、他の自然災害、または他の突発的、予見不可能で深刻な有害事象の悪影響を受ける可能性があり、私たちの業務連続性および災害復旧計画は、深刻な災害から私たちを十分に保護できないかもしれない。
私たちの本部と主要な研究機関は大サンディエゴ地区にあり、この地域は過去に深刻な地震と火災を経験したことがある。もし私たちの制御範囲を超えたこれらの地震、火災、他の自然災害、テロ、および似たような予見不可能な事件が、私たちの本社や研究施設の全部または大部分を使用できない場合、私たちは難しいかもしれないし、場合によっては、長い間私たちの業務を継続することはできない。災害復旧や事業継続計画がなく、特に地震保険が不足している場合には、当社の内部または第三者サービスプロバイダの災害復旧や業務連続性計画の欠落や性質に巨額の費用が発生する可能性があり、当社の業務に大きな悪影響を及ぼす可能性があります。また、私たちのサプライチェーンに不可欠な各当事者は単一の場所で運営されており、自然災害や他の突発的、予見不可能で深刻な有害事象(私たちの業務に影響を及ぼす可能性のある公衆衛生流行病や疫病を含む)の前での脆弱性を増加させている。このような事件が我々のサプライチェーンに影響を与えると,我々の臨床研究,開発計画,業務を行う能力に実質的な悪影響を及ぼす可能性がある。例えば,2020年3月には,コロナウイルスの伝播により,インド政府は26種類の活性薬物成分とその製造薬品の輸出を制限している。このような輸出制限は無期限であり、修正されたり拡大されるかもしれない。輸出制限がitolizumab(EQ 001)を含むように拡大された場合、私たちのitolizumab(EQ 001)の供給は無期限中断、遅延、または停止する可能性があり、私たちが行っている臨床研究を含むitolizumab(EQ 001)の能力の開発を継続し、重大な影響を受ける可能性があり、薬品コストの上昇を招き、私たちの業務に悪影響を及ぼす可能性がある。
米国や他の管轄区特許法の変化は、特許の全体的な価値を低下させ、私たちの候補製品を保護する能力を弱める可能性がある。
他の生物製薬会社と同様に、私たちの成功は知的財産権、特に私たちの研究計画や候補製品に関連する特許に大きく依存している。生物製薬業界で特許を獲得と実施することは技術上の複雑性と法律上の複雑性にも関連するため、コストが高く、時間がかかり、しかも内在的な不確定性を持っている。特許法または米国特許法解釈またはUSPTO規則および条例の変化は、不確実性とコストを増加させる可能性がある。2011年9月16日に法律となった“Leahy-Smith America発明法”や“Leahy-Smith Act”に署名することを含む米国と他の国の最近の特許改革立法は、私たちの特許出願をめぐる起訴や、私たちが発表した特許の実行または保護の不確実性とコストを増加させる可能性がある。“ライシー·スミス法案”は米国特許法を多くの重大な改正を行った。これらの条項は、特許出願起訴方式に影響を与える条項を含み、既存技術を再定義し、競争相手に特許の有効性に挑戦するために、より効果的かつ費用効果的な方法を提供する。これらの措置には、第三者が特許訴訟中に米国特許商標局に以前の技術を提出することを可能にすることと、特許の有効性を攻撃するために、ライセンス後審査を含む米国特許商標局によって管理される認可後手続きとが含まれる各方面間審査と派生手続き。2013年3月以降、“ライシー·スミス法案”によれば、米国は第1の発明者から出願制に移行し、このような制度の下で、他の法定要件が満たされたと仮定すると、第1の特許出願を提出した発明者は、第三者が第1の発明によって要求された発明であるか否かにかかわらず、発明の特許を取得する権利がある。しかし、“ライシー·スミス法案”とその実施は、私たちの特許出願をめぐる起訴の不確実性とコストを増加させ、私たちの将来の獲得能力を増加させる可能性がある
88
特許、および私たちが発行した特許の実施または保護は、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。
近年、米国最高裁判所はいくつかの特許事件に対して裁決を下し、場合によっては利用可能な特許保護範囲を縮小するか、場合によっては特許所有者の権利を弱めるかを決定している。米国議会、米国裁判所、米国特許商標局、および他の国の関連立法機関の将来の行動によれば、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、それによって、私たちが新しい特許を獲得したり、私たちの既存の特許と将来獲得可能な特許を実行する能力を弱める可能性がある。
私たちに対する製品責任訴訟は、私たちが重大な責任を負うことを招き、私たちが開発する可能性のある任意の候補製品の商業化を制限する可能性があります。
私たちはEQ 101、itolizumab(EQ 001)、人間の臨床研究における任意の未来の候補製品のテストに関連する固有の製品責任リスクに直面しており、私たちが開発可能な任意の製品を商業的に販売すれば、私たちはより大きなリスクに直面するだろう。もし私たちがEQ 101、itolizumab(EQ 001)、または任意の未来の候補製品または製品にダメージを与えるクレームを自己弁護することに成功できなければ、私たちは重大な責任を招くかもしれない。是非曲直や最終結果にかかわらず、製品責任クレームは次のようになるかもしれない
私たちは今製品責任保険を持っています。しかし、保険金額は私たちが発生する可能性のあるすべての責任を支払うのに十分ではないかもしれない。EQ 101,EQ 302,itolizumab(EQ 001)および任意の将来の候補製品が臨床研究によって進展するにつれて,任意の製品の商業化に成功すれば,保険カバー範囲を増やす必要があると予想される。保険範囲はますます高くなっています。私たちは可能などんな責任にも対応するために、合理的な費用や十分な金額で保険範囲を維持することができないかもしれない。
私たちまたは私たちの顧客に不利な税金法律または法規の変化は、私たちの業務、キャッシュフロー、財務状況、または経営結果に重大な悪影響を及ぼす可能性があります。
新しい収入、販売、使用、あるいは他の税金法律、法規、規則、法規あるいは法令はいつでも公布される可能性があり、これは私たちの国内と国外の収入の税収処理に影響を与える可能性がある。どんな新しい税金も、私たちの国内と国際業務運営、そして私たちの業務と財務業績に悪影響を及ぼす可能性があります。さらに、既存の税金法律、法規、規則、法規または条例は、私たちに解釈、変更、修正、または適用される可能性がある。例えば、2017年12月22日、米国連邦所得税立法署名が法律(H.R.1、“同時に発表された2018年度予算決議第2章と第5章に基づいて和解を規定する法案”)となり、非公式名称は“減税と雇用法案”であり、IRCは重大な改正が行われた。米国国税局や他の税務機関が将来的に減税や雇用法案についての指導を提供することは私たちに影響を与える可能性があり、減税や雇用法案のいくつかの側面は将来の立法で廃止または改正される可能性がある。
2022年1月1日から減税·雇用法案はIRC 174を改正し、納税者に米国と非米国での研究·実験(R&E)支出をそれぞれ5年または15年以内に資本化·償却するよう求めた。減税及び雇用法案改正案の前に、第174条は、納税者が支払又は発生した年度のR&E支出を直ちに控除すること、又は少なくとも60ヶ月の期間内に資本化及び償却を選択することを許可する。米国財務省が法規を発表しない限り、この条項の適用範囲を私たちの研究開発費のより小さいサブセットに縮小するか、または国会がこの条項を延期、修正、または廃止しない限り、それは可能性がある
89
私たちの将来の納税義務を効果的に増加させ、私たちの未来の経営業績を損なう。この規定の実際の影響は、私たちが発生する研究と開発費用の金額、私たちがこれらの減額を活用するのに十分な収入を得ているかどうか、私たちがアメリカ国内であるか海外で私たちの研究と開発活動を行っているかなど、様々な要素に依存するだろう。
2020年3月27日に公布された“コロナウイルス援助、救済、経済安全法案”または“CARE法案”と題する立法は、“減税と雇用法案”のいくつかの条項を改正した。また、最近公布されたアイルランド共和法には、ある大企業の帳簿収入に対する最低税の徴収や、会社がこれらの株を買い戻したある会社の株の買い戻しに消費税を徴収することなど、米国連邦会社の所得税に影響を与える条項が含まれている。各州が減税や雇用法案、CARE法案、アイルランド共和軍をどの程度遵守するかは定かではない。私たちは減税と雇用法案やCARE法案が私たちが現在予測している最近の最低現金税に実質的な影響を与えないと予想している。しかし、私たちは減税と雇用法案、CARE法案、アイルランド共和軍が私たちの業務に長期的な影響を及ぼす可能性があることを検討し続けている。私たちは潜在的な投資家がこの立法と私たちの普通株に投資したり、私たちの普通株を持っている潜在的な税金結果について彼らの法律と税務顧問に相談することを促す。
私たちは複数の管轄区域で税金と関連した危険に直面している。
私たちはアメリカと各州の管轄区とオーストラリアで所得税を払わなければなりません。これらの所得税申告書を作成する際には、これらの管轄区域で有効な適用税法や法規を説明する必要があり、これは支払われた税額に影響を与える可能性がある。私たちの所得税申告書は計算と仮定に基づいています。これらの計算と仮定はアメリカ国税局と他の税務機関の審査を受けています。また、私たちの税務責任を計算する際には、複雑な税務規則の適用における不透明な要素を処理することに関連する。私たちは私たちが申告表上の立場を適切に支持すると信じているが、私たちは私たちの所得税の準備が十分かどうかを決定するために、税務機関の審査の潜在的な結果を定期的に評価するつもりだ。私たちは、潜在的な改訂の可能性と金額を定期的に評価し、必要に応じて改正につながる事実が知られている間の所得税の支出、所得税、繰延税金を調整する。所得税申告表で採用されたか、または予想されて採用された任意の不確定な税務ヘッドについて、任意の追加の税金負債(利息および罰金を含む)の推定があれば、累算しなければならない。所得税の計上を決定する際には、現行の税収法律や法規の解釈に基づいて重大な判断を下す必要がある。我々の所得税支出は、異なる法定税率を有する税務管区収益の組み合わせの変化、繰延税金資産と負債推定値の変化、既存の税収政策、法律、法規または税率の変化、控除できない費用レベル(株に基づく報酬を含む)の変化、事業場所、将来の研究開発支出レベルの変化、合併と買収、または各税務機関の審査結果を含む様々な要素の悪影響を受ける可能性がある。私たちの納税推定は合理的だと信じていますが、国税局や他の税務機関が納税申告書上の私たちの立場に同意しなければ、利息と罰金を含む追加の納税義務を負うかもしれません。実質的であれば、任意の紛争最終裁決時にこれらの追加金額を支払うことは、私たちの運営結果や財務状況に実質的な影響を与える可能性があります。
もし私たちが環境、健康、安全の法律法規を守らなければ、私たちは罰金や罰金を科されたり、コストを発生したりする可能性があり、これは私たちの業務の成功に実質的な悪影響を及ぼすかもしれない。
私たちおよび私たちと施設を共有する第三者は、実験室の手続きおよび処理、使用、貯蔵、処理、危険材料および廃棄物の管理を含む多くの環境、健康および安全な法律と法規を遵守しなければならない。私たちのすべての行動は化学品、生物学的、そして放射性材料を含む危険で燃えやすい材料の使用に関するものだ。私たちのすべての業務は危険な廃棄物製品を生成するだろう。私たちは一般的に第三者と契約を結び、このような材料と廃棄物を処理する。私たちはこのような材料が汚染や傷害をもたらす危険を除去することができない。もし私たちが私たちと施設を共有している第三者が危険材料を使用して汚染や損傷をもたらした場合、私たちはそれによるいかなる損害に責任を負うかもしれません。どんな責任も私たちの資源範囲を超えている可能性があります。私たちはまた民事や刑事罰金と処罰に関連した巨額の費用を発生させるかもしれない。
危険材料の使用による従業員の負傷により生じる可能性のあるコストや支出を支払うために労働者補償保険を維持しているが、潜在的な責任を支払うのに十分ではない可能性がある。私たちは私たちが生物、危険または放射性物質を貯蔵したり処分したりすることによって、私たちが提起した環境責任や有毒侵害に対して保険を維持することはできません。
90
また、現在または未来の環境、健康、安全法律法規を遵守するためには、巨額のコストが生じる可能性がある。このような現行または未来の法律法規は私たちの研究と開発を損なうかもしれない。このような法律法規を遵守しないことはまた巨額の罰金、処罰、または他の制裁につながる可能性がある。
証券や業界アナリストが我々の業務に関する研究報告を発表しない場合、あるいは不正確または不利な研究報告を発表しなければ、私たちの株価や取引量は低下する可能性がある。
私たちの普通株の取引市場は、証券や業界アナリストが発表した私たちまたは私たちの業務に関する研究と報告にある程度依存するだろう。私たちの一人以上のアナリストが私たちの普通株格付けを引き下げたり、私たちの業務に関する不正確または不利な研究報告を発表したりすれば、私たちの普通株価格は下落するかもしれない。これらのアナリストのうちの1人以上が私たちの報告書を停止したり、私たちの報告書を定期的に発表できなかった場合、私たちの普通株に対する需要が減少する可能性があり、これは私たちの普通株価格と取引量を低下させる可能性がある。
私たちの開示統制と手続きはすべてのミスや詐欺を阻止したり検出できないかもしれない。
私たちは“取引所法案”の定期報告書の要求事項を守らなければならない。我々の開示制御および手続きは、取引所法案に基づいて提出または提出された報告書に開示されなければならない情報が蓄積され、管理層に伝達され、米国証券取引委員会規則および表に指定された期間内に記録、処理、まとめ、報告されなければならないことを合理的に確保するために設計されている。
任意の開示制御およびプログラムまたは内部制御およびプログラムは、発想および動作がどのように完全であっても、絶対的な保証ではなく、合理的な保証を提供することしかできず、制御システムの目標が達成されることを確保することしかできないと信じている。
これらの固有の限界は,意思決定過程における判断が誤りである可能性があり,簡単な誤りや誤りによって故障が発生する可能性があるという現実を含む.例えば、私たちの役員または役員は、意図せずに新しい関係または手配を開示できなかった可能性があり、関連するいかなる取引も開示できなかった。さらに、ある人の個人的な行動、2人以上の結託、または無許可超越制御は、制御を回避することができる。したがって,我々の制御システム固有の制限により,誤りや詐欺による誤り陳述が発生し,発見されない可能性がある.また、他の分野が私たちの業務にもたらすリスクを識別して解決するためのリスク管理計画やプロセスやプロセスはありません。
私たちは“小さな申告会社”と“非加速申告会社”であり、より小さい申告会社または非加速申告会社に適用されるいくつかの減少した申告および開示要求のみを遵守することを決定すれば、私たちの普通株の投資家に対する魅力を低下させる可能性がある。
我々は取引法で定義されている“小さな申告会社”と“非加速申告会社”であり、“小さい申告会社”や“非加速申告会社”であり続ける限り、他の上場企業に適用されるが“小さい申告会社”や“非加速申告会社”には適用されない各種申告要求の免除を利用することを選択することができるが、これらに限定されない。第404条監査に基づいて当社の独立公認会計士事務所に財務報告の内部統制を要求せず(私たちが“非加速申告会社”である限り)、定期報告及び委託書における役員報酬に関する開示義務を削減した(私たちが“より小さい報告会社”である限り)。2024年までには“規模の小さい報告会社”になりつつ、“非加速申告会社”になると予想しています。私たちがこれらの免除に依存することを選択すれば、投資家は私たちの普通株の吸引力が低下していることを発見するかどうか予測できない。もし一部の投資家が私たちの普通株が未来の情報開示を減らす選択によって吸引力が低下していることを発見すれば、私たちの普通株はそれほど活発ではない取引市場が出現する可能性があり、私たちの株価はもっと変動するかもしれない。
予測可能な未来に、私たちは配当金を支払うつもりはない。
私たちは私たちの株の現金配当金を発表したり支払ったりしたことがない。私たちは現在、将来のどんな収益も残して、私たちの業務の運営と拡張に資金を提供するつもりで、私たちは予測可能な未来にいかなる配当金も発表したり支払うことはないと予想しています。しかも、未来のどんな債務協定の条項も私たちが配当金を支払うことを阻止するかもしれない。したがって、株主は、将来の収益に投資する唯一の方法として、価格上昇後に彼らの普通株を売却することに依存しなければならない。
私たちは証券集団訴訟の影響を受けるかもしれない。
従来、証券集団訴訟は、ある会社の証券市場価格が下落した後に提起されることが多かった。このリスクは私たちに特に関連しています生物製薬会社は
91
近年の株価の変動。私たちがこのような訴訟に直面すれば、巨額のコストを招き、経営陣の注意と資源を移転させる可能性があり、これは私たちの業務を損なう可能性があります。
項目1 B。未解決従業員コメント。
ない。
プロジェクト1 C。CybErSecurity
リスク管理と戦略
我々は、知的財産権、独自、戦略または競争性を有する機密情報、および私たちの臨床研究および従業員に関連するデータ、または情報システムおよびデータを含む、ネットワークセキュリティ脅威の重要なコンピュータネットワーク、第三者ホストサービス、通信システム、実験室装置、ハードウェアおよびソフトウェア、および私たちのキーデータの重大なリスクを識別、評価および管理するための様々な情報セキュリティプロセスを実施し、維持した。
我々は、会社のネットワークセキュリティ脅威とリスクの識別、評価、管理を支援するために、情報技術コンサルタントの外部担当者を会社と協力させ、首席運営官、首席財務官、幹部指導チームを招聘した。このグループは、自動化ツール、ネットワークセキュリティ脅威を識別する報告およびサービスの加入、脅威および参加者の報告の分析、および我々に報告された脅威の評価を含む、様々な方法を使用して、我々の脅威環境を監視および評価することによって、ネットワークセキュリティ脅威からのリスクを識別および評価する。
環境に応じて、我々は、イベント応答計画および/またはイベント応答ポリシー、データおよび情報保護計画、いくつかのシステムのネットワークセキュリティおよびアクセス制御、データ暗号化、システム監視、ネットワーク保険、および従業員訓練など、様々な技術、物理的および組織的措置、プロセス、基準、およびポリシーを実施し、維持し、維持するために、ネットワークセキュリティおよびデータの脅威によって生じる情報システムおよびデータの重大なリスクを管理および低減することを目的としている。
ネットワークセキュリティ脅威の重大なリスクの評価と管理は、会社全体のリスク管理プロセスに統合されている。たとえば,情報技術担当者は管理層と連携し,リスク管理プロセスの優先順位を決定し,我々の業務に実質的な影響を与える可能性のあるネットワークセキュリティ脅威を緩和する.
我々は、専門サービス会社(法律顧問を含む)および脅威情報サービスプロバイダを含むネットワークセキュリティ脅威からの重大なリスクを識別、評価、管理するために、第三者サービスプロバイダを使用することができるかもしれない.
私たちの業務を運営するために、私たちは、アウトソーシング業務のキー機能、臨床研究、専門サービス、ソフトウェアすなわちサービス(SaaS)プラットフォーム、ホストサービス、クラウドベースのインフラ、暗号化および認証技術、企業生産性サービス、および他の機能のようないくつかの第三者サービスプロバイダを使用して様々な機能を実行する。私たちは、これらのプロバイダを使用することに関連するネットワークセキュリティリスクの管理を支援するための特定のプロバイダ管理プロセスを持っています。提供されたサービスの性質、処理された情報の感度および数、ならびにサービスプロバイダの識別情報に基づいて、当社のプロバイダ管理プロセスは、そのようなプロバイダのネットワークセキュリティアプローチを検討すること、契約に従って提供されるサービスおよび/またはその処理された情報に関連する義務をプロバイダに適用すること、そのサービスおよびデータ処理アプローチに関する書面アンケートを記入し、その参加中に定期的に再評価することを要求することを含むことができる。
会社に実質的な影響を与える可能性のあるネットワークセキュリティ脅威のリスクとこれらのリスクをどのように実現するかについては,“リスク要因である従業員,我々の成長管理,その他の法務に関するリスク”を参照されたい
統治する
我々の取締役会は、その一般的な監督機能の一部として、会社のネットワークセキュリティリスク管理を担当しています。取締役会監査委員会は、ネットワークセキュリティ脅威リスクの評価及び緩和に関するガイドライン及び政策を含む、リスク評価及びリスク管理における会社のガイドライン及び政策を審査する。
92
私たちのネットワークセキュリティリスク評価と管理プロセスは、当社のIT担当者を含むいくつかの会社経営陣によって実施·維持されています。彼はマイクロソフト認証と、当社の首席運営官を持ち、コーポレートガバナンス研究所から取締役ネットワークセキュリティ証明書を取得しました。
経営陣はまた、適切な人員を採用し、ネットワークセキュリティリスク考慮要因を会社全体のリスク管理戦略に取り入れ、関係者に重要な優先事項を伝えることを支援している。IT担当者と首席運営官は、ネットワークセキュリティ事件への対応準備を進め、ネットワークセキュリティプロセスを承認し、セキュリティ評価やその他のセキュリティに関する報告を審査することを支援する。
我々のネットワークセキュリティイベント応答プロセスは、状況および指定されたリスクレベルに応じて、最高財務官、CEO、および/または私たちの開示制御プログラムおよびプログラムに参加する幹部リーダーチーム全体を含むいくつかのネットワークセキュリティイベントを管理層メンバーに報告することを目的としている。IT担当者と最高経営責任者は、会社のイベント応答チームと協力し、通知を受けたネットワークセキュリティイベントの緩和と修復を支援します。また、同社のイベント対応プログラムには、取締役会監査委員会に何らかのサイバーセキュリティ事件を報告することが含まれている。
監査委員会は,会社の重大なネットワークセキュリティ脅威とリスク,これらの脅威やリスクに対応するために会社が実施しているプログラムに関するIT担当者と首席運営官から定期的に報告を受けている。監査委員会はまた、同社の情報システムおよびデータ、ならびにネットワークセキュリティ脅威、リスクおよび緩和に関する要約または陳述を受信する。
項目2.財産
2027年2月に満期になる賃貸契約によると、現在カリフォルニア州ラホヤにある本社のために約1,750平方フィートの空間を借りています。カリフォルニア州ラホヤで追加のオフィスと実験室スペースをレンタルしました。レンタルの期限はそれぞれ異なります。最初のものは2025年2月に満期になり、最新のものは2027年2月に延長されます。私たちはずっとカリフォルニア州サンフランシスコ南部でオフィススペースを借りています。この空間は2023年2月末に期限が切れて、更新していません。
既存の施設は現在の需要に対応するのに十分であると信じていますが、後日も商業的に合理的な条項で、適切な追加用地を提供します。
項目3.法律法律手続き。
ない。
プロジェクト4.地雷安全安全に開示する。
適用されません。
93
部分第2部:
項目5.登録者普通株·関連株の市場保有者は重要であり,発行者は株式証券を購入する.
市場情報
我々の普通株は2018年10月12日にナスダック世界市場で取引を開始し、コードはEQであり、2023年9月15日にナスダック資本市場に譲渡された。2018年10月12日まで、私たちの普通株は公開取引市場を持っていません。
記録保持者
2024年3月20日現在、我々の普通株には約50名の登録株主がいる。実際の株主数はこの記録保持者の人数を超えており、受益者である株主を含むが、その株式は仲介人や他の被命名者が街頭名義で保有している。この数の登録所有者は、その株式が信託形態で保有または他のエンティティが保有する可能性のある株主も含まれていない。
配当政策
私たちは私たちの株の現金配当金を発表したり支払ったりしたことがない。私たちは現在、すべての利用可能な資金と任意の将来の収益を維持し、私たちの業務運営のために、予測可能な未来に私たちの普通株にいかなる配当も支払わないと予想している。将来配当を発表する任意の決定は、私たちの取締役会が適宜決定し、私たちの財務状況、経営業績、資本要求、契約制限、一般業務条件、および私たちの取締役会が関連する他の要素に依存する可能性があります。
私たちの株式補償計画に基づいて発行された証券
私たちの株式報酬計画に関する情報は、私たちが米国証券取引委員会に提出した2024年年次総会の株主に関する最終依頼書に含まれ、引用によって本明細書に組み込まれます。
最近売られている未登録証券
ない。
発行者および関連購入者が株式証券を購入する
2023年7月、我々の取締役会は、2024年12月31日までに最大750万ドルの普通株を買い戻すことができる株式買い戻し計画を承認した。2023年12月31日現在、我々の株式買い戻し計画によると、約720万ドルが将来の買い戻しに利用できる。
第六項です[R保存された]
94
項目7.経営陣の以下の問題の議論と分析財務状況と経営実績。
私たちの財務状況と経営結果の議論と分析、本年度報告書にForm 10-K形式で出現した“財務データ精選”というタイトルの部分と、私たちの総合財務諸表と関連付記を読むべきです。本議論および分析は、リスクおよび不確実性に関連する現在の予想に基づく前向きな陳述を含む。様々な要因により、“リスク要因”の節と本年度報告10-K表の他の部分で議論された要因を含むため、我々の実際の結果は、これらの前向き陳述で予想される結果とは大きく異なる可能性がある。また“前向きな陳述に関する特別な説明”と題する節を参照されたい
概要
著者らは臨床段階の生物技術会社であり、免疫生物学に対する深い理解を利用して新しい治療法を開発し、深刻な自己免疫と炎症性疾患、或いは免疫炎症性疾患を治療し、高度に満足されていない医療需要を持っている。私たちの戦略の重点は潜在的にもっと多くの適応を求め、新しい候補製品とプラットフォームを獲得して、私たちのルートを拡大することを含む、私たちの候補製品の臨床開発を推進することである。私たちは独立したり、パートナーを通じて私たちの候補製品を商業化したり、戦略取引を通じて他の方法で私たちのチャネルを貨幣化するつもりです。
著者らの現在の臨床段階の候補製品はEQ 101とitolizumab(EQ 001)を含む。EQ 101は一流の選択性三特異性合成ペプチドであり、IL-2、IL-9とIL-15を抑制するために設計され、これらは疾病を引き起こす重要なサイトカイン標的であり、一連の免疫炎症適応の中で満たされていない需要を解決することを目的としている。Itolizumab(EQ 001)は1種の一流のモノクロナル抗体であり、それは選択的に免疫チェックポイント受容体CD 6、CD 6に対して効果性T細胞或いはT細胞の調節に中心的な役割を果たすEFF細胞、活動と人身売買は、多くの治療領域でいくつかの免疫性炎症性疾患を推進した。我々はまた、様々なサイトカインに対して選択的にEQ 302の臨床前開発を進めている他のポリペプチドベースの候補製品の発見および最適化に取り組んでおり、EQ 302は一流の経口IL-15およびIL-21二重特異性阻害剤である。著者らの一流の免疫資産の斬新と差別化パイプラインは皮膚科、胃腸科、リューマチ科、血液科、移植科学、腫瘍科と肺科を含む多くの領域の満たされていない医療需要を満たす可能性がある。我々は,多くの重篤な免疫炎症疾患を治療する潜在的最適疾患修正療法として,EQ 101,EQ 302,itolizumab(EQ 001)の開発に焦点を当てている。
2022年2月、我々は、Bioniz Treateutics,Inc.,またはBionizを買収することによって、EQ 101のグローバル独占権利を取得し、EQ 302のような追加的で新規な多サイトカイン候補製品を発見する独自のプラットフォームを取得した。EQ 101とEQ 302は合成ペプチドであり、重要な疾病駆動を抑制し、臨床検証するサイトカイン標的に特化し、いくつかの免疫炎症適応中に満たされていない需要を解決することを目的としている。2022年11月、著者らはオーストラリアとニュージーランドでEQ 101静脈注射或いはEQ 101静脈注射の第二段階概念検証臨床研究を開始し、研究対象は中から重度の円形脱毛症或いはAAである。この研究の登録作業は完了し、2024年第2四半期に背線データを発表する予定だ。著者らは現在、体内薬理と調合開発を含むEQ 302の臨床前開発を行っており、候補製品を更に表現し、最適化している。積極的な結果を待つ前に,GMP製造や毒理学研究を含め,潜在的なIND申請を支援し,最初のヒト臨床研究に入ることができるEQ 302をより多くの臨床前開発に進める予定である。
著者らは最近、オーストラリアの健康ボランティアにおいて、別の多サイトカイン標的ポリペプチドEQ 102の第一段階ヒト臨床研究を完成させた。EQ 102はIL−15とIL−21の二重特異性阻害剤であり,後者もBioniz買収の一部として買収されている。第一段階研究では、EQ 102の全体的な耐性は良好であり、薬効学的活性を示したが、初期処方のバイオアベイラビリティは予想より低かった。臨床前および翻訳データは、EQ 102と比較して、EQ 302はより高い効力を有し、腸管において安定かつ透過性を有し、最適な全身または腸管制限活動を達成するためにさらに修飾することができることを示した。2023年12月、我々は、EQ 302の最新の進展およびEQ 102に対する優れた製品構成を考慮して、胃腸および皮膚疾患患者を潜在的に治療するために、EQ 102のさらなる開発からEQ 302を臨床に移行したことを発表した。
私たちは2017年5月にBiocon SA(後にBiocon Limitedまたは一緒にBioconに譲渡された)との協力および許可協定に従って、後に修正された、またはBioconライセンスを取得するitolizumab(EQ 001)に対する私たちの権利を獲得した。2022年3月、著者らは200名の急性移植片対宿主病(AGVHD)患者の中でイトキシマブ(EQ 001)の全世界3期の肝心な臨床研究Equatorを開始した。Equator研究を開始する決定は,我々が完成したaGVHD 1 b期臨床研究(Equateと呼ぶ)の発見と,米国食品·薬物管理局(FDA)と造血幹細胞移植領域のリーディング医師のフィードバックに基づいている。楽しみにしています
95
データ安全監視委員会は2024年第3四半期に上位100人の被験者の赤道データを中間審査する。著者らは最近、EQ 001による全身性エリテマトーデス(SLE)と狼瘡性腎炎(LN)患者の1 b期概念検証臨床研究EQ 001を完成した。2023年11月,我々は米国リウマチ学会と米国腎臓学会の年次総会でこの研究のB型LN部分のデータを公表した。これらのデータはフォローアップ期間中に最後の患者を除くすべての患者を代表し,itolizumab(EQ 001)耐性が良好であり,高蛋白尿被験者に臨床的に有意な反応が生じたことを示している。今後数週間以内に小野製薬有限会社や小野製薬会社にEqualiseのB型LN部分からの背線データを提供する予定である。
著者らはまたBioconと協力し、潰瘍性大腸炎患者におけるitolizumabの第二段階臨床研究を共同で援助した。この研究はインドのBiocon社が行い、2022年11月から無作為、二重盲検、プラセボ対照の臨床研究であり、90名もの被験者で行い、中から重度潰瘍性大腸炎患者に対するitolizumabの安全性と有効性を評価する。
2022年12月5日に、吾らはOnoと資産購入協定を締結し、これにより吾等はOnoに独占選択権を付与して、吾等のitolizumab(EQ 001)又はその選択権を取得する権利を取得する。これらの権利は、すべての治療適応と、米国、カナダ、オーストラリア、およびニュージーランドでitolizumab(EQ 001)を商業化する権利を含む。オプションの交換として、小野は35億円相当の前金である2640万ドルを一度に支払ってくれた。
小野が選択権を行使すれば、小野は50億円相当の金額を一度に支払うか、三菱UFG銀行による2024年3月21日の為替レートは約3310万ドルとなる。私たちは小野が2024年下半期にオプション行使決定を下すと予想している。私たちはいくつかの開発と商業化のマイルストーンを達成する際に1.014億ドルの資金を得る資格がある。
資産購入プロトコルにより,我々はitolizumab(EQ 001)のすべての研究·開発を担当し,2022年7月1日からオプション期間中,小野が四半期ごとに資金を提供する。早期終了しない限り,選択期間はLNのEqualise臨床研究の背線データとaGVHDの赤道第3期臨床研究の中期データ交付後3カ月で満了する。
小野はいつでも書面通知で“資産購入協定”を終了することができるが、限られた場合、小野は終了後しばらくイトカマブ(EQ 001)の研究開発費と支出を返済し続ける義務がある。小野がその選択権を行使しなければ,資産購入プロトコルと選択権は自動的に終了する.資産購入協定には、双方が重大な違約時の常習停止権と、いずれか一方が2025年12月31日までに取引が完了していない場合に資産購入契約を終了することを許可する外部日(有限調整可能)も含まれている。
我々は独自の製品発見プラットフォームを持ち、それを利用して多様なサイトカインを標的と阻害するために新しいポリペプチドを設計することができ、これらのサイトカインは検証された生物学的および疾患経路に関与している。例えば,我々は最近EQ 302の臨床前データを重点的に紹介し,EQ 302は開発中のIL−15およびIL−21に対する第2世代経口多サイトカイン阻害剤である。我々の候補製品が特定の疾患の発症機序に重要な役割を果たしていると考えられる他の適応の治療効果を評価するための翻訳生物学的計画も行われている。私たちが現在と未来の適応を選択するのは、私たちの候補製品のさらなる開発を推進する科学、翻訳、臨床、ビジネス理由の分析に基づいている。
我々の設立以来,我々のすべての努力は,基本的には組織会社と会社のための人員,業務計画,資金調達,itolizumab(EQ 001)の許可権を獲得し,非臨床研究を行い,3つの調査性新薬申請やINDを提出し,候補製品の臨床開発を行い,Bionizの買収,Onoとの資産購入プロトコルや他の未完成の取引,株式買い戻し計画の開始,上場企業の運営に関連する一般的かつ行政活動の展開に集中している。また,Bionizを買収する過程で,我々の生産ラインを1つの候補製品から複数の候補製品に拡張し,これらすべてが異なる開発段階にある.この拡張は、さらなる開発コストが発生し、規制機関によるこれらの候補製品の承認を求めるため、私たちの運営損失の増加速度を加速させる可能性がある。私たちは、オプションおよび小野社が提供するitolizumab(EQ 001)開発資金と引き換えに、資産購入契約から小野社の一括払いに関する収入を得た。私たちは製品販売、マイルストーン支払い、または版税から何の収入も得ていない。設立以来、私たちは主に債務と株式融資、資産購入協定による収入を通じて私たちの運営に資金を提供してきた。
私たちは最初から損失を被った。2023年12月31日と2022年12月31日までの年度の純損失はそれぞれ1330万ドルと6240万ドルだった。2023年12月31日までの累計赤字は1兆857億ドルだった。
96
我々のほとんどの運営損失は,我々の研究·開発活動,非臨床·臨床活動,買収の進行中の研究·開発に関する費用および我々の運営に関する一般的·行政コストに起因している。
私たちは予測可能な未来に巨額の費用と増加した損失を招き続けると予想する。私たちの研究開発活動の推進に伴い、EQ 101、EQ 302、itolizumab(EQ 001)の現在と将来の臨床開発を含め、私たちの費用は大幅に増加し、候補製品の臨床開発への適応を拡大する可能性があり、私たちの多サイトカイン標的薬物発見プラットフォームによって決定された臨床前候補薬を含む新たな候補製品を獲得または開発する可能性があり、規制部門が任意の承認された候補製品の承認を求めて商業化する可能性があり、より多くの人を募集し、私たちの知的財産権を保護し、一般会社コストを生成することが予想される。我々の株式買い戻し計画にこれ以上の買い戻しがないと仮定すると、2023年12月31日現在の既存の現金、現金等価物、短期投資は、2025年下半期の運営に資金を提供できるようになると予想される。
EQ 101、EQ 302、または将来の候補製品の開発を成功させ、規制部門の承認を得ない限り、製品販売から何の収入も得られないことが予想され、今後12ヶ月以内には起こり得ない。また,小野との資産購入プロトコルによると,itolizumab(EQ 001)に関する収入は,受信した前払いオプション費用,オプション期間中のitolizumab(EQ 001)開発コストの精算,および潜在的オプション費用と潜在的マイルストーン支払いに限られている.小野がその選択権を行使しなければ、私たちは開発に成功し、規制部門のitolizumab(EQ 001)の承認を得ない限り、itolizumab(EQ 001)の製品販売から何の収入も得られないと予想されるが、これは永遠に起こらなければ今後12ヶ月以内に起こる可能性は低い。したがって、候補品を販売することで大量の収入を生み出すことができる前に、株式発行、債務融資、協力および許可協定(小野との資産購入協定など)の組み合わせで現金需要を満たすことが予想される。しかし、私たちは、もしあれば、タイムリーに、または優遇条件で追加資金を得たり、そのような他の計画を達成することができないかもしれない。ロシアとウクライナ間の衝突、中東紛争、銀行倒産、経済的インフレ圧力、政府機関の通貨政策反応、およびその他のマクロ経済要素により、世界の信用と金融市場は極端な変動を経験し、銀行倒産、流動性と信用供給の減少、消費者自信の低下、経済成長の低下と経済安定の不確実性を含む。株式や信用市場が悪化した場合、任意の必要な債務や株式融資をより難しくし、コストが高く、および/またはより希釈作用がある可能性がある。私たちは必要な時に資金を調達したり、このような他の手配を達成することができず、私たちの財務状況に負の影響を与え、私たちの研究開発計画や他の業務を延期、減少、または終了させるか、あるいは私たちが自分で開発とマーケティングをより望んでいた候補製品を開発し、マーケティングする権利を与える可能性があります。
財務概要
収入.収入
私たちは今まで治療製品の販売、開発マイルストーン、あるいは版税から何の収入も得ていません。2022年と2023年に、私たちの収入は資産購入協定の下での前払いと小野の開発資金から来ています。将来的には、開発資金や潜在的オプション行使、資産購入協定のマイルストーン支払い、および承認された製品の任意の製品販売など、さらなる収入を含む、私たちの候補製品と締結される可能性のある協力または許可協定から収入を得ることができ、これらの製品が今後12ヶ月以内に承認される可能性は低い(あれば)。私たちが製品収入を創出する能力は、小野がその選択権、および任意の未来の候補製品を行使しなければ、EQ 101、EQ 302、itolizumab(EQ 001)の成功と最終商業化に依存するだろう。EQ 101、EQ 302、itolizumab(EQ 001)または任意の未来の候補製品の開発がタイムリーに完了できなかった場合、または規制機関の私たちの候補製品の承認を得ることができなかった場合、私たちの将来の収入を創出する能力および私たちの運営業績および財務状況は大きな悪影響を受けるだろう。
小野薬業有限公司と締結した資産購入契約。
2022年12月5日、私たちは資産購入協定を締結し、この合意に基づいて、35億円または2640万ドル相当の一次前払いと引き換えにOno選択権を付与した。これらの権利は、すべての治療適応と、米国、カナダ、オーストラリア、およびニュージーランドでitolizumabを商業化する権利を含む。
小野が選択権を行使すれば、50億円、または約3310万ドルを獲得し、三菱UFG銀行2024年3月21日の為替レートに基づいて計算される。私たちはいくつかの開発と商業化のマイルストーンを達成する際に1.014億ドルの資金を得る資格がある。
我々はitolizumabのすべての研究と開発を担当し,2022年7月1日から小野社が選択期間まで四半期ごとに資金を提供している。事前に終了しない限り、オプション期間は3ヶ月になるだろう
97
LNのEqualise臨床研究のTOPLINEデータとaGVHDの赤道3期臨床研究の中期データを渡した後である。
2023年12月31日までの年度中に、小野との資産購入協定に基づいて、2,700万ドルの開発資金と910万ドルのプリペイド償却に関する収入を含む3,610万ドルの収入を確認した。
2023年12月31日現在、資産購入契約に関する繰延収入総額は1,570万ドル。
研究と開発費
研究と開発費用には主に我々の非臨床研究や候補製品の臨床開発に関するコストが含まれている。私たちの研究開発費は
私たちは発生した費用に応じて研究と開発費用を支払う。サービスが完了または貨物を受け取った場合、将来の研究開発活動で費用として使用される貨物およびサービスの前払いは払い戻しできません。
私たちの直接研究開発費用は主に外部コスト、例えば私たちの非臨床研究と臨床開発に関連するCROとコンサルタントに支払われる費用を含む。
私たちはオーストラリアの研究開発税インセンティブ、あるいは税金インセンティブは、研究開発費の減少であることを認識している。金額は私たちの合格した研究と開発支出によって確定されたもので、払い戻しはできません。申告実体は精算申請を提出した納税年度内に2000万豪ドル以下の収入がなければ、所得税免除実体によってコントロールできなくて、税金優遇を受ける資格があることを前提としています。税金優遇を受けることが合理的に保証され、関連支出が発生し、金額が確実に測定または信頼できる推定ができる場合、税収インセンティブを確認する。
我々は,小野がその選択権を行使しなければ,EQ 101,EQ 302,itolizumab(EQ 001)の開発を進め,これらの候補製品を開発している適応数を拡大し,新たな候補製品を獲得または開発する可能性があるため,予測可能な将来に我々の研究開発費を大幅に増加させる予定である。EQ 101、EQ 302とEQ 001の開発成功は非常に高い不確実性を持っている。現在,臨床前と臨床開発自体の予測不可能性により,我々の候補製品の余剰開発を完了するために必要な努力の性質,時間やコスト,あるいは我々の候補製品販売による大量の現金純流入が開始可能な時期(あれば)を合理的に見積もることはできない。臨床開発スケジュール、成功の確率と開発コストは期待と大きく異なる可能性がある。
98
臨床研究を完成するには数年或いはそれ以上の時間を要するかもしれないが、時間の長さは通常候補製品のタイプ、複雑性、意外性と期待用途によって異なる。臨床開発過程で生じる差により、臨床研究のコストはプロジェクトのライフサイクル全体で大きく異なる可能性がある
買収している研究開発費は
買収された研究開発費には、買収された候補製品が将来代替用途がないとみなされるため、Bioniz買収に関連する新候補製品開発権を得るコストが含まれる。
一般と行政費用
一般および行政費用には、株式ベースの給与と福祉、行政、人的資源、投資家関係、財務および会計機能に基づく相談費が含まれる主に賃金およびその他の関連費用が含まれる。その他の重大なコストには、特許および会社の事務に関連する法律費用、保険、出張、取締役会費用、施設コスト、および税金が含まれる。
将来的には,我々の一般的かつ行政的費用が増加し,インフラの拡大,上場企業に関する法律,監査,税務,その他の専門費用の増加,証券取引所上場や米国証券取引委員会の要求,取締役や上場企業に関連する上級管理者保険料,会計や投資家関係コストの遵守を維持することが予想される。また、規制機関の任意の候補製品の承認を得た場合、インフラの構築やそのような製品を商業化する能力に関する費用が発生すると予想される。しかし、このような承認の期間は非常に不確定であり、私たちはこのような規制の承認を得るために数年かかるかもしれない。
利子支出
利息支出には前期ローンの利息と償却割引が含まれています。
利子収入
利子収入には、主に現金、現金等価物、短期投資による利息収入が含まれ、稼ぎ時に確認される。
その他の収入,純額
99
その他(費用)収入は、純額には主にわがオーストラリア子会社に関する外貨取引収益と損失が含まれています。
所得税費用
所得税支出は連邦所得税支出と州所得税支出からなる。
経営成果
2023年12月31日までと2022年12月31日までの年度比較
次の表に2023年12月31日までと2022年12月31日までの年度の経営成果(単位:千)を示す
|
|
現在までの年度 |
|
|
|
|
||||||
|
|
2023 |
|
|
2022 |
|
|
変わる |
|
|||
収入.収入 |
|
$ |
36,084 |
|
|
$ |
15,759 |
|
|
$ |
20,325 |
|
研究開発 |
|
|
37,039 |
|
|
|
37,547 |
|
|
|
(508 |
) |
現在行われている研究と開発を買収する |
|
|
- |
|
|
|
23,049 |
|
|
|
(23,049 |
) |
一般と行政 |
|
|
13,567 |
|
|
|
17,239 |
|
|
|
(3,672 |
) |
利子支出 |
|
|
(491 |
) |
|
|
(1,053 |
) |
|
|
562 |
|
利子収入 |
|
|
2,334 |
|
|
|
420 |
|
|
|
1,914 |
|
その他の収入,純額 |
|
|
(76 |
) |
|
|
281 |
|
|
|
(357 |
) |
所得税費用 |
|
|
580 |
|
|
|
- |
|
|
|
580 |
|
収入.収入
2023年12月31日までの年間で、小野との資産購入協定に基づいて3610万ドルの収入を確認した。2023年12月31日までの1年間で、発展資金は2700万ドル、前払い償却は910万ドルだった。2022年12月31日までの年間で、小野との資産購入協定に基づいて1580万ドルの収入を確認した。前金の償却金額は400万ドル、2022年7月1日から2022年12月31日までの発展資金は1180万ドル。
研究と開発費
2023年12月31日までの年間研究開発費は3700万ドルだが、2022年12月31日までの年間は3750万ドル。研究開発費の減少には主に以下の点の変化がある
買収している研究開発費は
2023年12月31日までの1年間、買収が行われている研究開発費はないが、2022年12月31日までの年間では、このような費用は2300万ドルである。2022年12月31日までの年度に,買収の検討·開発費用は,買収の候補製品が将来代替用途がないことを決定したため,Bioniz買収を資産買収とした結果である。購入された有形純負債を超える対価格は支出された。
100
一般と行政費用
2023年12月31日までの1年間は、一般·行政費は1360万ドルだったが、2022年12月31日までの年間は1720万ドルだった。一般と行政費用の減少には主に以下の変化が含まれる
利子支出
2023年12月31日までの1年間の利息支出は50万ドルだったが、2022年までの年間110万ドルだった。利息支出には私たちの以前の期限払い手形の利息が含まれています。
利子収入
2023年12月31日までの1年間の利息収入は230万ドルだったが、2022年12月31日までの1年間の利息収入は40万ドルだった。利息収入の増加は主に2023年の平均金利が2022年よりも高いためだ。
その他の収入,純額
2023年12月31日までの1年間、その他の支出純額は10万ドルだったが、2022年12月31日までの1年間、その他の収入純額は30万ドルだった。2023年12月31日までの年度、その他の費用純額には、主に私たちのオーストラリア子会社に関する実現済み外貨取引損失純額が含まれています。2022年12月31日までの年間で、他の収入純額には小野の達成した外貨収益60万ドル、前払35億円が含まれているが、これは領収書発行と現金受領期間中に円が強くなったが、オーストラリア子会社に関連した未実現外貨取引損失純額はこの純収益を部分的に相殺したためである。
所得税費用
所得税支出は2023年12月31日までの1年間で60万ドル。私たちの2023年の所得税支出は、主に特定の項目の帳簿と税収処理の違いによる国内現金税費支出に起因する。私たちは私たちの繰延税金資産を相殺する準備があるので、繰延税の支出を計上しません。2022年12月31日までの年度には所得税支出はない。
流動性と資本資源
設立から2023年12月31日まで、私たちは主に株式と債務証券を売却することで私たちの運営に資金を提供します。また,小野との資産購入プロトコルから収益が生じているので,詳細は以下の流動資金源部分を参照されたい.2023年12月31日現在、私たちの累計赤字は1兆857億ドルで、予測可能な未来に純損失が続くと予想されています。2023年12月31日現在、私たちは2320万ドルの現金と現金等価物、1770万ドルの短期投資を持っています。
流動資金源
2023年ATM施設
2023年10月、私たちはJefferies LLCまたはJefferiesと市場での手配を締結し、この計画によると、私たちは時々Jefferiesを私たちの販売代理または2023年の現金自動支払機として、2195万ドルまでの総発行価格の普通株を発売し、販売することができる。本年度報告書Form 10−Kを提出した時点で、2023年の現金自動支払機融資メカニズムにおけるいかなる株も販売していません。
101
小野との資産購入契約
2022年12月5日、小野と資産購入協定を締結し、この協定に基づいて、小野にitolizumabに対する権利を得る権利を付与したが、義務ではなかった。これらの権利は、すべての治療適応と、米国、カナダ、オーストラリア、およびニュージーランドでitolizumabを商業化する権利を含む。オプションの交換として、小野は35億円相当の前金である2640万ドルを一度に支払ってくれた。
小野が選択権を行使すれば、小野は50億円相当の金額を一度に支払うか、三菱UFG銀行による2024年3月21日の為替レートは約3310万ドルとなる。
私たちはいくつかの開発と商業化のマイルストーンを達成する際に1.014億ドルの資金を得る資格がある。2023年12月31日現在、私たちはオプション行使支払いやマイルストーン支払いを受けていません。
我々はitolizumabのすべての研究と開発を担当し,2022年7月1日から選択期間まで小野から四半期ごとに資金を提供している。選択期間はLNのEqualise研究の背線データとaGVHDの赤道第三段階臨床研究の中期データ交付後3カ月で満期となる。
2023年12月31日現在、小野社から3800万ドルの開発資金支払いを受けている。
資金需要
我々が行っている将来の活動に関連する費用は大幅に増加することが予想され,特にEQ 101とitolizumab(EQ 001)の臨床開発を進めて拡大する際には,Onoが潜在的な新たな適応を含めてその選択権を行使しなければ,EQ 302と我々の多サイトカイン標的薬発現プラットフォームにより決定された他の新規臨床前候補薬の臨床前研究を進めることが可能である。私たちの資本の主な用途は、臨床開発サービス、非臨床研究、製造と製品供給、潜在的な新製品買収、私たちの株式買い戻し計画に従って私たちの普通株の株を買い戻す可能性があり、法律と他の規制コンプライアンス費用、従業員の報酬と関連費用、保険料、運営資本、その他の一般管理費用であると予想される。
2023年7月、我々の取締役会は、2024年12月31日までに最大750万ドルの普通株を買い戻すことができる株式買い戻し計画を承認した。この計画によれば、計画中に公開市場取引または当社取締役会またはその指定委員会が時々承認した他の取引を介して普通株を買い戻すことができる。買い戻しの時間と金額(あれば)は、私たちの普通株の価格、代替投資機会、私たちの現金資源、私たちの任意の合意下の制限、会社と監督管理要求、および市場状況を含む様々な要素に依存します。2023年12月31日まで、私たちは株式買い戻し計画に基づいて298,385株の普通株を買い戻し、総金額は30万ドルだった。2023年12月31日以降,本年度報告がForm 10−K形式で提出される日まで,株式買い戻し計画により,我々の普通株は買い戻しが行われていない。この計画によると、将来の任意の普通株買い戻しに既存の現金と現金等価物を用いて資金を提供する予定だ。
私たちの株式買い戻し計画にこれ以上の買い戻しがないと仮定すると、2023年12月31日までの既存の現金、現金等価物、短期投資は、現在計画されている2025年下半期の運営に資金を提供できるようになると予想される。私たちは間違っていることが証明される可能性があるという仮定に基づいてこの推定をして、私たちは予想よりも早く私たちの資本資源を使用することができる。しかも、私たちの運営計画は変わるかもしれないし、私たちは計画よりも早い追加資金が必要かもしれない。また,臨床研究では候補製品をテストする過程コストが高く,これらの研究の進展時間も不確定である。これらの努力の結果は不確定であるため、EQ 101、EQ 302およびitolizumab(EQ 001)または他の任意の候補製品の開発および商業化に成功した実際の金額を推定することはできず、私たちがいつ利益を達成することが可能かどうかを推定することもできない。
私たちの将来の資本需要は多くの要素に依存します
102
私たちが相当な製品収入を生み出すことができる前に、もしあれば、株式発行、債務融資、協力と許可協定の組み合わせによって、例えば小野との資産購入協定のような私たちの現金需要を満たす予定です。追加の株式または変換可能な債券を販売することは、私たちの株主に追加的な希釈をもたらす可能性があり、これらの証券の条項は、清算または他の特典を含む可能性があり、私たちの既存の普通株主の権利に悪影響を及ぼす。債務融資は債務超過義務を招き、管理文書には私たちの運営を制限する運営や融資契約が含まれる可能性がある。ロシアとウクライナの間の紛争、中東紛争、銀行倒産、経済的インフレ圧力、政府機関が取った通貨政策およびその他のマクロ経済要素により、世界の信用と金融市場は、流動性と信用供給の減少、消費者自信の低下、経済成長の低下、経済安定の不確実性を含む極端な変動を経験した。信用と金融市場のさらなる悪化と経済状況への自信が起こらない保証はない。株式や信用市場が悪化した場合、任意の必要な債務や株式融資をより難しくし、コストが高く、および/またはより希釈作用がある可能性がある。もし私たちが協力または許可協定を通じてより多くの資金を調達するならば、私たちは私たちの技術、将来の収入源、研究プロジェクト、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利である可能性があり、および/または私たちの普通株の価値を低下させる可能性のある条項で許可を付与しなければならないかもしれない。もし私たちが必要な時や魅力的な条件下で資金を調達できなければ、私たちは私たちの研究開発計画や他の業務を延期、減少、またはキャンセルさせることを余儀なくされるだろう。このような行動のいずれも私たちの業務、財務状況、そして運営結果に実質的な影響を及ぼす可能性がある。私たちの設立以来、私たちの経営活動は純損失と負のキャッシュフローを経験しており、予測可能な未来に純損失が続くことが予想される。2023年12月31日までの累計赤字は1兆857億ドルだった。小野がその選択権を行使しなければ,EQ 101,EQ 302およびitolizumab(EQ 001)の開発に関連するコスト,および我々の任意の他の候補製品が生じるため,少なくとも今後数年以内に運営損失と負のキャッシュフローが続くと予想される。
材料現金需要
103
私たちが予想する重大な現金需要には、私たちの経営リース項目の満期金額を含む契約規定の支出が含まれています。レンタルに関する詳細は、本年度報告書Form 10-Kの連結財務諸表付記7および12を参照されたい。私たちは通常キャンセル可能な調達注文に基づいて契約を締結しているので、サービス提供者と実質的にキャンセルできない調達約束を持っていません。吾等が予想する重大な現金需要は、吾等がBionizを買収する合併協定条項に基づいて支払わなければならない可能性のある規制及び商業マイルストーン事項の潜在的又は有支払を含まず、吾等が複数のエンティティと締結又は締結可能な許可協定に基づいて支払うべき規制及び商業マイルストーン事項の潜在的又は有支払又は使用料支払いも含まれておらず、当該等の許可合意に基づいて、吾等は特定の知的財産権(Biocon許可を含む)に対して内部許可を行っている。Bionizの買収に関連し、Bioconライセンスに関連する潜在的または支払いの詳細については、本年度報告書Form 10-Kに含まれる連結財務諸表付記6および9を参照されたい。
キャッシュフロー
次の表は、以下の各期間の主要な現金源および用途(千計)を示しています
|
|
現在までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
提供された現金純額(使用): |
|
|
|
|
|
|
||
経営活動 |
|
$ |
(21,783 |
) |
|
$ |
(8,733 |
) |
投資活動 |
|
|
(4,762 |
) |
|
|
18,684 |
|
融資活動 |
|
|
(9,228 |
) |
|
|
(1,215 |
) |
為替レート変動が現金に与える影響 |
|
|
(118 |
) |
|
|
5 |
|
現金および現金等価物の純増加 |
|
$ |
(35,891 |
) |
|
$ |
8,741 |
|
経営活動
2023年12月31日までの年間では,経営活動に用いられる現金純額は2180万ドルであったが,2022年12月31日までの年度は870万ドルであった。業務活動に用いられた現金純額が2,640万ドル減少した要因は,小野社から受け取った2022年12月の資産購入契約に関する一括払いが2,640万ドル減少したが,小野社の開発資金の1,370万ドル増加分で相殺されたためである。
投資活動
2023年12月31日までの年間で、投資活動のための現金純額は480万ドル。私たちが購入した短期投資総額は5,470万ドルですが、同期合計5,000万ドルの短期投資満期日によって相殺されます。2023年12月31日までの1年間の不動産·設備の購入総額は10万ドルだった。
2022年12月31日までの年間で、投資活動が提供する現金純額は1870万ドル。私たちの短期投資満期総額は3320万ドルですが、購入した合計1490万ドルの短期投資によって相殺されます。2022年12月31日までの1年間で、不動産や設備の購入総額は30万ドルだった。Bionizを買収した結果、私たちは全部で70万ドルの現金を得た。
融資活動
2023年12月31日までの年間で、融資活動のための現金純額は合計920万ドルであり、これは主に、オックスフォード金融有限責任会社とSVBとの間で合意されていた融資と保証協定や融資合意に関連した支払総額910万ドルと、30万ドルの株式買い戻しと、我々の従業員の株式購入計画に関連した20万ドルの現金と相殺されるためである。
2023年5月25日、私たちはローン契約を終了し、すべての未返済金額を全額前払いしました。2023年12月31日までの1年間に支払われた総金額は910万ドルで、(I)2022年12月31日までの元金、合計860万ドル、(Ii)前払い費用約6.2万ドル、および(Iii)最終支払い費用約50万ドルを含む。2023年12月31日まで、私たちは融資協定の下でこれ以上の義務がない。
104
2022年12月31日までの1年間、融資活動に用いられた純現金総額は120万ドル。私たちは2022年10月から未返済の支払手形を元金で支払い、総額は140万ドルです。これらの支払いは、私たちの従業員株式購入計画に関連する従業員株購入から受け取った20万ドルの現金で相殺されます。
表外手配
報告書を提出している間は、私たちはありませんし、現在もありません。アメリカ証券取引委員会規則の定義によると、私たちは何の表外手配もありません。同様に、私たちは可変利益実体にどの株式も持っていません。私たちは確かに私たちのBioconライセンスと私たちのBionizとの合併協定に含まれているいくつかのマイルストーン支払い形式のまたは対価格負債を持っていて、これらの債務は私たちの貸借対照表に反映されていない。しかし、私たちの現在の運営計画と私たちのこのような支払いの可能性と潜在的なタイミングの評価によると、これらの支払いは遠いと思い、今後12ヶ月以内に期限が切れる可能性は極めて高いと思います。
重要な会計政策と試算
私たちの経営陣の財務状況と経営結果の討論と分析は、アメリカで公認されている会計原則に基づいて作成された私たちの総合財務諸表に基づいています。総合財務諸表を作成する際には、総合財務諸表および付記に報告された資産、負債および費用金額、または資産および負債の開示に影響を与えるための推定および仮定を行う必要がある。私たちはこのような推定数値を継続的に評価するつもりだ。我々は過去の経験、既知の傾向及び事件、財務モデル及び各種の当時の状況の下で合理的な他の要素を推定し、その結果は資産及び負債の帳簿価値を判断する基礎を構成しているが、このような資産や負債の帳簿価値は他の出所から容易に見られるものではない。異なる仮定または条件では、実際の結果は、これらの推定値とは異なる可能性がある。
我々の重要な会計政策は、本年度報告10-K表の他の部分の総合財務諸表付記2により包括的に記述されているが、以下の会計政策は、我々の財務状況や経営結果を全面的に理解し評価するために最も重要であると考えられる。
収入確認
収入を確認する方法は、製品やサービスの制御権を顧客に転送することを説明し、このような製品やサービスから得られる対価格金額を反映しています。そのため,(1)顧客との契約を決定する,(Ii)契約中の履行義務を決定する,(Iii)取引価格を決定する,(Iv)取引価格を履行義務に割り当てる,(V)顧客が製品やサービス制御権を取得したときに収入を確認する,の5つのステップに従う.収入確認基準を適用する際には、契約条項およびすべての関連事実と状況を考慮します。私たちは、任意の実際的な便宜策を使用して、類似した特徴および類似した状況を持つ契約に一貫して適用されることを含む収入確認基準を含む。
お客様は私たちと契約を結ぶ側で、契約の目的は私たちの日常活動の製品やサービスを獲得して、対価格と交換することです。契約とみなされるためには、(1)契約は承認されなければならない(書面、口頭または他のビジネス慣行に基づいて)、(2)譲渡する製品またはサービスに対する各当事者の権利を決定することができ、(3)譲渡する製品またはサービスの支払い条件を決定することができ、(4)契約は商業的実質を持たなければならない(すなわち、将来のキャッシュフローのリスク、時間または金額は契約によって変化すると予想される)。そして(V)我々が獲得する権利のある製品やサービス譲渡に必要なほぼすべての対価格を受け取る可能性が高い.
履行義務は、製品やサービスを顧客に譲渡する約束と定義される。我々は、製品またはサービスのセット(または製品またはサービスのセット、または実質的に同じ譲渡パターンを有する一連の製品およびサービス)を譲渡する各コミットメントを異なるコミットメントとして識別する。(I)顧客が単独で、または顧客がいつでも利用可能な他のリソースと共に製品またはサービスから利益を得ることができ、(Ii)製品またはサービスを顧客に譲渡する約束が、契約内の他の承諾とは別に識別することができる場合、製品またはサービスは異なる。譲渡製品またはサービスのすべての異なる約束は、収入確認の会計単位である。製品またはサービスを譲渡する約束が契約中の他の約束とは別に識別できない場合、これらの約束は単一の履行義務に統合されなければならない。
取引価格は、製品やサービスの制御権を顧客に譲渡することと引き換えに、私たちが獲得する権利がある対価格金額です。取引価格を決定する際には、任意の重要な融資成分の存在、任意の可変要因の影響、非現金考慮要因、および顧客への対価格を考慮します。重要な融資成分が存在すれば、取引価格は通貨の時間価値に応じて調整される。可変性の要素があれば、私たちが受け取る予定の対価格を推定し、その金額を確認の基礎として使用しなければなりません
105
製品やサービスが顧客の手に移ったときの収入。可変対価金額を決定するには,(1)期待値方法,すなわち一連の対価可能金額の確率重み付き金額の和,(2)最も可能な金額法,一連の可能な対価格のうち単一の可能な金額を決定する方法がある.
1つの契約に複数の履行義務がある場合、取引価格を各異なる履行義務に割り当て、その金額は、それぞれの異なる履行義務を履行するために、私たちが獲得する権利のある対価格を反映している。項目ごとに異なる履行義務については、収入は、当該履行義務に適用される製品又はサービスに対する制御権を我々が移転したときに確認される。
私たちがその履行義務を履行する前に最初に対価格を受けた場合、私たちはその履行義務を履行するまで、このような対価格を繰延収入に分類する。私たちが対価を受け取る前にまず義務を履行した場合、対価格は売掛金として記録されます。
確認された資産の予想償却期間が1年以下である場合、または資産の金額が重要でない場合には、発生時に契約の取得および履行の増分コストを支出する。そうでなければ、このようなコストが契約の増分であり、基礎契約の収入確認に比例する費用で償却された場合、そのようなコストは契約資産に資本化される。
研究と開発費用を計算すべきである
私たちは、サプライヤー、コンサルタント、契約研究組織との契約に基づいて負担する研究や開発活動に関する費用を見積もることを要求されています。これらの契約の財務条項は交渉が必要であり、異なる契約の交渉が異なり、支払流量がこれらの契約に基づいて材料またはサービスを提供する期限と一致しない可能性がある。私たちは研究開発費をサービスと努力支出の期間と一致させることで、私たちの連結財務諸表にこれらの費用を反映します。著者らは、前臨床研究または臨床研究の進展に基づいて、研究または関連活動の各方面の時間によってこれらの費用を測定する。私たちは、関連契約を検討し、財務モデルを準備し、研究や他の主要者との研究進展や進行中の他のサービスに関する議論を考慮して、評価すべきカウントを決定する。研究や臨床研究において,実際の結果が我々の見積りと異なれば,費用認知率を調整する.
実際に発生した金額と実質的に差がないと予想されているにもかかわらず、提供されたサービスの状態および時間の推定が提供されたサービスの実際の状態および時間と異なる場合、これは、任意の特定の時期に報告された金額が高すぎたり、低すぎたりする可能性がある。これまで、このような費用の見積もりと実際に発生した金額との間に実質的な差はありませんでした。
株に基づく報酬費用
私たちは、付与日公正価値で従業員と非従業員の株式ベースの奨励を付与し、株式オプションと株式購入権を含み、帰属期間中の報酬支出を直線的に記録した。私たちはブラック-スコアーズオプション価格モデルを使用して私たちの株式オプション報酬を評価する。株式オプション奨励の公正価値を推定するには、管理層がいくつかの仮定に対して判断と推定を行う必要があり、私たちの普通株の変動性、私たちの株式オプションの期待期限、および計量日の期待配当収益率を含む。したがって、要因が変化すれば、経営陣は異なる仮定を使用し、株式に基づく報酬支出は将来の報酬に対して実質的に異なる可能性がある。
所得税
連結財務諸表の将来の税務結果に計上された繰延税金資産と負債を確認することを要求する貸借対照法に基づいて所得税を計算する。この方法によると、繰延税項資産及び負債は、総合財務諸表と資産及び負債の税ベースとの差額に基づいて、予想差額を用いて返送される年度の現行税率を決定する。既存の証拠に基づいて、繰延税金資産が現金化される可能性が高い場合を除いて、繰延税金資産の純額計について全額推定値を用意します。
我々は、(I)管理層が税務倉庫位の技術的優位性に基づいて、税務倉位をより維持する可能性があるかどうかを2段階で行われた手続き記録に基づいて決定し、(Ii)確認可能な閾値に適合する税務倉位について、管理層は、最終的に関連税務機関と和解したときに達成可能な最大税収割引額の50%を超えることを確認する。もし所得税支出が発生した場合、私たちは利息と罰金を確認するつもりだ。
106
最近の会計公告
最近の会計声明に関する情報は、本年度報告に他の場所のForm 10−Kに含まれる連結財務諸表の付記1を参照されたい。
第七A項。定量と合格市場リスクの開示について。
小さな報告会社は必要ありません。
プロジェクト8.財務諸表Sと補足データ。
本プロジェクトで要求される財務諸表と補足データはF-1ページから始まり,本年度報告の10-K表署名ページの後に列挙される.
項目9.Accouとの変更と分岐会計と財務情報開示の専門。
ない。
第9条。制御するプログラムがあります
情報開示制御とプログラムの評価
我々は、米国証券取引委員会に提出された定期的かつ現在の報告で開示すべき情報が、米国証券取引委員会規則および表で指定された期間内に記録、処理、集約および報告されることを保証し、必要に応じて開示すべき情報をタイムリーに決定するために、必要に応じて私たちの管理層に伝達されることを保証するために、開示制御および手続きを維持する。開示制御およびプログラムを設計および評価する際、管理層は、任意の制御およびプログラムは、設計および動作がどんなに良好であっても、予想される制御目標を達成するために絶対的な保証ではなく合理的な保証を提供することしかできないことを認識している。合理的な保証レベルを達成するためには、管理層は必然的にその判断を用いて可能な制御とプログラムのコスト-利益関係を評価する必要がある。さらに、任意の制御システムの設計も、将来のイベント可能性のいくつかの仮定にある程度基づいており、どの設計もすべての潜在的な未来の条件でその目標を成功的に達成することを保証することはできない。時間の経過とともに,制御が条件の変化により不十分になったり,政策やプログラムへの遵守度が悪化したりする可能性がある.費用対効果を有する制御システムの固有の制限により、エラーまたは詐欺によるエラー陳述が発生し、発見されない可能性がある。
我々は、2023年12月31日現在、最高経営者及び最高財務官を含む経営陣の監督の下、最高財務官を含む経営陣の参加の下で、1934年に改正された証券取引法又は“取引法”下の規則13 a−15(E)及び15 d−15(E)に基づいて定義されている開示制御及びプログラムの設計及び操作の有効性を評価している。この評価に基づき、我々の最高経営責任者および最高財務責任者は、2023年12月31日までに、我々の開示統制および手続きが合理的な保証レベルで有効であると結論した。
財務報告の内部統制に関する経営陣の報告
我々の経営陣は、取引規制13 a-15(F)および15 d-15(F)に定義されている財務報告の十分な内部統制の確立と維持を担当している。財務報告の内部統制は、米国で一般的に受け入れられている会計原則に基づいて財務報告の信頼性と外部目的の財務諸表作成のための合理的な保証を提供するために、我々の経営陣(我々の最高経営責任者、最高財務官、首席会計官を含む)の監督と参加の下で設計されたプログラムである。
2023年12月31日現在、我々の経営陣は、トレデビル委員会後援組織委員会が#年に提案した基準を用いて、財務報告書の内部統制に対する我々の有効性を評価した内部統制--統合フレームワーク(2013年フレームワーク)この評価に基づき、我々の経営陣は、2023年12月31日現在、財務報告に対する内部統制がこれらの基準に基づいて有効であると結論した。
財務報告の内部統制の変化
2023年12月31日までの四半期内に、財務報告の内部統制に大きな影響を与えたり、合理的に財務報告の内部統制に大きな影響を与える可能性のある変化はありません。
プロジェクト9 B。他にも情報です。
107
貿易手配
2023年12月31日までの3ヶ月間、私たちの役員の一人は、次の表に示すように、私たちの証券を秩序よく処分する取引計画を採択した
|
|
|
取引手配の種別 |
総株式数 |
|
|
氏名と職位 |
行くぞ |
養子縁組/終了 |
ルール |
規則ではない |
待ち株 |
期日まで |
養子縁組 |
|
|||||
(1)契約、指示または書面計画は、取引法規則10 b 5-1(C)に規定されている積極的防御条件を満たすことを目的としている。
(2) “
プロジェクト9 Cです 外国会社の情報開示について妨害検査の管轄区域
適用されません。
108
第三部
プロジェクト10.役員·役員職ICERSと会社管理。
以下に述べる以外に、本プロジェクトに要求される情報は、2023年12月31日までの財政年度終了後120日以内に“取締役選挙”、“取締役会情報と会社管理”、“取締役会情報”、“取締役情報”、“役員”、“延滞の第16(A)条報告”というタイトルの米国証券取引委員会に提出された最終依頼書または依頼書に含まれ、本年度報告Form 10−Kに参照されて組み込まれる。
我々は、我々の最高経営責任者、最高財務官、最高会計官または財務総監、または同様の機能を実行する者を含む、すべての上級管理者、取締役および従業員に適用されるビジネス行動および道徳的基準を採択した。“ビジネス行動と道徳基準”は私たちのサイトで見ることができますWwwl.equilliumBio.comそれは.我々のサイト上の情報は,本Form 10-K年次報告に引用的に組み込まれていない.もし私たちが“商業行為および道徳的規則”を実質的に修正したり、任意の役員または役員に任意の免除を付与したりすれば、私たちは直ちに私たちのウェブサイトまたは現在のForm 8-K報告書に修正または免除の性質を開示する。
プロジェクト11.行政官イー補償します。
本プロジェクトに必要な資料は,依頼書“役員および役員報酬”の節に掲載され,本年度報告Form 10−Kに参考に組み込まれる。
プロジェクト12.特定の利益所有者の保証所有権所有者と経営陣と関連する株主について。
本プロジェクトに必要な資料は委託書に“いくつかの実益所有者及び管理層の担保所有権”及び“株式補償計画によって許可されて発行された証券”と題する2節に掲載され、参考方式で本年報の10-K表に組み込まれる。
本プロジェクトに必要な資料は,依頼書に“関係者との取引および賠償”および“取締役会および会社管理に関する資料”と題した2節に掲載し,本年度報告のForm 10−Kを参考に組み込む。
第14項.元金口座弁護士代とサービス料です。
本プロジェクトに必要な資料は,依頼書“首席会計士費用およびサービス”の節に掲載され,本年度報告の10−K表に参考に組み込まれる。
109
部分IV.IV
プロジェクト15.展示品と資金エーアールアイチェックリストです。
(a)(1) 連結財務諸表:
“Equillium,Inc.総合財務諸表”と“独立公認会計士事務所報告”は,本年度報告の10-Kフォーム署名ページを含めた後,F-1ページから始まる.
(a)(2) 財務諸表付表:
これらの付表が省略されているのは、財務諸表や付記に要求された資料が記載されているため、またはこれらの資料が適用されていないか、または必要ではないためである。
(a)(3) 陳列品
展示品索引
展示品 番号をつける |
|
説明する |
|
|
|
2.1*
|
|
登録者、Bioniz治療会社、Project JetFuel Merge Sub,Inc.およびKevin Greenは、登録者、Bioniz治療会社、Project JetFuel Merge Sub,Inc.との間で2022年2月14日に署名された合併協定および計画のみで署名され、登録者が2022年2月16日に証券取引委員会の8-K表に現在報告されている添付ファイル2.1を参照することによって署名される。 |
|
|
|
3.1 |
|
改訂および再予約された登録者登録証明書は、参照登録者によって2018年10月16日に提出された現在の8−Kフォーム報告の添付ファイル3.1によって組み込まれる。 |
|
|
|
3.2 |
|
登録者規約を改訂及び再作成し,登録者が2018年10月16日に提出した現在の表格8−K報告の添付ファイル3.2を参照して編入する。 |
|
|
|
4.1 |
|
登録者普通株式証明書フォーマットは、S-1登録者登録説明書添付ファイル4.1(書類第333-227387号)を参照して合併し、改訂され、最初に2018年9月17日に米国証券取引委員会に提出される。 |
|
|
|
4.2 |
|
普通株式証の購入日は2019年9月30日で、オックスフォード谷金融有限責任会社に発行され、登録者を引用して2019年11月12日に米国証券取引委員会に提出された10-Q表四半期報告の添付ファイル4.2を合併した。 |
|
|
|
4.3 |
|
普通株購入権証は、日付を2019年9月30日にシリコンバレー銀行に発行し、登録者を引用して2019年11月12日に米国証券取引委員会に提出した10-Q表四半期報告の添付ファイル4.3を統合したものである。 |
|
|
|
4.4 |
|
普通株説明は,登録者が2020年3月26日に証券取引委員会に提出した10−K表年次報告書の添付ファイル4.4を引用する。 |
|
|
|
4.5 |
|
2021年2月5日に発行された引受権証用紙は、登録者を参照することにより、2021年2月4日に米国証券取引委員会に提出された8-K表の現在の報告書の添付ファイル4.1に組み込まれる。 |
|
|
|
10.1+ |
|
登録者とその役員と上級管理者との間の賠償協定表は、登録者登録説明書S−1表(文書番号333−227387)の添付ファイル10.1を参照して組み込まれ、この表は、2018年9月17日に米国証券取引委員会に最初に提出された。 |
|
|
|
10.2+ |
|
Equillium,Inc.2017年株式インセンティブ計画およびその下でのオプション付与通知、オプション合意および行使通知のフォーマットは、2018年9月17日に最初に米国証券取引委員会に提出された登録者S-1登録説明書添付ファイル10.2(文書番号333-227387)を参照して組み込まれる。 |
|
|
|
10.3+ |
|
Equillium 2018年株式インセンティブ計画及びその下の株式オプション付与通知、オプション協定及び行使通知のフォーマットは、登録者を参照して2018年10月16日に米国証券取引委員会に提出されたS-8登録説明書(文書番号333-227859)第99.2号添付ファイルに組み込まれる。 |
|
|
|
10.4+ |
|
Equillium,Inc.2018年従業員株購入計画は,2018年10月16日に米国証券取引委員会に提出された登録者S-8表(ファイル番号333-227859)の99.3番添付ファイルを参照して組み込まれている. |
|
|
|
10.5 |
|
登録者とBiocon SAとの間の協力及び許可協定は、参照により編入された2017年5月22日(後に2018年3月からBiocon Limitedに譲渡される)である |
110
|
|
登録者が2023年8月9日に米国証券取引委員会に提出した10-Q表四半期報告書の添付ファイル10.1。 |
|
|
|
10.6 |
|
登録者とBiocon SAとの間で2017年5月22日に締結された臨床供給協定(この協定は、その後、2018年3月にBiocon Limitedに譲渡される)であり、この協定は、登録者が2023年8月9日に米国証券取引委員会に提出された10-Q表四半期報告書の添付ファイル10.2を参照することによって組み込まれる。 |
|
|
|
10.7 |
|
標準オフィスリースは,2018年2月1日に施行され,登録者とLa Jolla ShorePlaza,LLCの間のリースにより,参照登録者が表S−1の登録説明書(書類番号333−227387)添付ファイル10.8で登録成立し,最初に2018年9月17日に米国証券取引委員会に提出された。 |
|
|
|
10.8+ |
|
登録者とDaniel M.Bradburyとの間で2018年6月1日に発行された招待状は,登録者登録声明の添付ファイル10.9を引用して当社のS−1表(文書333−227387号)に組み込まれ,この文書は最初に2018年9月17日に米国証券取引委員会に提出された。 |
|
|
|
10.9+ |
|
登録者とJason A.Kyesの間で2018年3月19日に発行された招待状は,登録者のレジストリS−1(文書番号333−227387)添付ファイル10.10を引用して統合され,2018年9月17日に米国証券取引委員会に提出された。 |
|
|
|
10.10+ |
|
登録者とBruce D.Steelの間で2018年6月1日に発行された招待状は,登録者のレジストリS−1(文書番号333−227387)添付ファイル10.11を引用して統合され,2018年9月17日に米国証券取引委員会に最初に提出された。 |
|
|
|
10.11+ |
|
登録者とスティーヴン·コネリー博士との間で2018年6月7日に改訂·再予約された招待状は,登録者のS-1表登録声明(書類番号333-227387)添付ファイル10.12を引用して統合され,2018年9月17日に米国証券取引委員会に最初に提出された. |
|
|
|
10.12 |
|
登録者とBiocon Limitedとの連携·許可協定第1改正案は,2018年9月28日に施行され,登録者とBiocon Limitedは,登録者の登録説明書S−1表(文書番号333−227387)添付ファイル10.14を引用して統合され,この書類は最初に2018年10月2日に米国証券取引委員会に提出された。 |
|
|
|
10.13 |
|
登録者とBiocon Limitedが2019年4月22日に署名した協力·許可協定第2修正案では、登録者とBiocon Limitedは、登録者を引用して2020年3月26日に米国証券取引委員会に提出された10−K表年次報告書の添付ファイル10.15を統合した。 |
|
|
|
10.14 |
|
登録者とBiocon Limitedとの間の協力·許可協定第3改正案は,2019年12月10日とし,登録者が2020年3月26日に米国証券取引委員会に提出した10−K表年次報告書の添付ファイル10.18を引用することにより統合される。 |
|
|
|
10.15+ |
|
登録者とChristine Zeelmayerとの間の招待状は,日付は2018年1月19日であり,登録者が2020年3月26日に米国証券取引委員会に提出した10−K表年次報告の添付ファイル10.19を引用して編入される。 |
|
|
|
10.16+ |
|
第1改正案は,登録者とDaniel·M·ブラッドベリーの間で発行され,2020年1月1日に施行され,登録者が2020年3月26日に米国証券取引委員会に提出された10−K表年次報告書の添付ファイル10.20を引用して統合された。 |
|
|
|
10.17+ |
|
登録者とBruce D.Steelの間で発行された要件書の第1改正案は,2020年1月1日に施行され,登録者が2020年3月26日に証券取引委員会に提出された10−K表年次報告書の添付ファイル10.22を引用して統合された。 |
|
|
|
10.18+ |
|
第1改正案は,登録者とクリスティーナ·セデルメルの間で発行され,2020年1月1日に施行され,登録者を引用して2020年3月26日に米国証券取引委員会に提出された10−K表年次報告書の添付ファイル10.23に組み込まれる。 |
|
|
|
10.19 |
|
公開市場販売協定は、登録者とJefferies LLCとの間で署名され、日付は2023年10月5日であり、登録者が2023年10月5日に米国証券取引委員会に提出された8-K表の現在の報告書の添付ファイル1.1を参照することによって組み込まれる。 |
|
|
|
111
10.20+ |
|
Equillium,Inc.非従業員役員報酬政策は改訂され、登録者を引用して2022年3月23日に米国証券取引委員会に提出された10-K表年次報告書の添付ファイル10.25に組み込まれる。 |
|
|
|
10.21 |
|
登録者とBiocon Limitedとの間の協力及び許可協定の第4の修正案は、日付が2021年4月14日であり、登録者を引用して2021年5月13日に証券取引委員会に提出された10-Q表四半期報告書の添付ファイル10.3に組み込まれる。 |
|
|
|
10.22 |
|
登録者とBiocon Limitedとの間の協力及び許可協定の第5の修正案は、2022年11月18日とし、登録者が2023年3月23日に米国証券取引委員会に提出された10−K表年次報告書の添付ファイル10.29を引用して編入する。 |
|
|
|
10.23 |
|
登録者と小野製薬有限会社が締結した資産購入契約は,期日は2022年12月5日であり,登録者と小野製薬有限会社は登録者を引用して2023年3月23日に証券取引委員会に提出した10−K表年次報告添付ファイル10.31を合併した。 |
|
|
|
10.24+ |
|
Equillium,Inc.2024年インセンティブ計画および株式オプション付与通知、オプション協定およびその下の行使通知の形態は、登録者を参照して2024年3月8日に証券取引委員会に提出された8−K表の現在報告されている添付ファイル99.1を本明細書に組み込む。 |
|
|
|
21.1 |
|
Equillium,Inc.の子会社 |
|
|
|
23.1 |
|
独立公認会計士事務所が同意します。 |
|
|
|
24.1 |
|
授権書。ここのサインページを参考にしてください。 |
|
|
|
31.1 |
|
改正証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて首席執行幹事証明書が発行される。 |
|
|
|
31.2 |
|
第13 a-14条の改正証券取引法(A)及び15 d-14(A)条に基づいて首席財務長官を認証する。 |
|
|
|
32.1** |
|
改正証券取引法第13 a−14(B)又は15 d−14(B)条及び米国法第18編第1350条に基づいて最高経営責任者及び最高財務官の認証を行う。 |
|
|
|
97.1 |
|
奨励的補償政策。 |
|
|
|
101.INS |
|
連結されたXBRLインスタンス文書-インスタンス文書は、そのXBRLタグがイントラネットXBRL文書に埋め込まれているので、対話データファイルには表示されない |
|
|
|
101.書院 |
|
イントラネットXBRL分類拡張アーキテクチャ文書 |
|
|
|
101.カール |
|
インラインXBRL分類拡張計算リンクライブラリ文書 |
|
|
|
101.def |
|
インラインXBRL分類拡張Linkbase文書を定義する |
|
|
|
101.介護会 |
|
XBRL分類拡張ラベルLinkbase文書を連結する |
|
|
|
101.Pre |
|
インラインXBRL分類拡張プレゼンテーションLinkbaseドキュメント |
|
|
|
104 |
|
表紙対話データファイル(添付ファイル101に含まれるイントラネットXBRLのフォーマット)。 |
*S-K規制第601(A)(5)項によれば、プロトコルの添付表および証拠品は省略されています。任意の漏れたスケジュールおよび/または証拠品のコピーは、米国証券取引委員会に提供することを要求しなければならない。
**改正された1934年“証券取引法”(以下、“取引法”と略す)第18節の規定によると、本証明書は提出されたものとみなされず、この条項の責任の制約も受けない。このような証明は、参照によってそのような出願が明示的に組み込まれない限り、改正された1933年の証券法または“取引法”に基づいて提出された任意の出願に引用によって組み込まれているとはみなされない。
+管理契約または補償計画を示します。
本展示品のいくつかの部分に対して、秘密保護待遇が与えられた。見落とした部分は米国証券取引委員会(Securities And Exchange Commission)に単独で提出されている。
S-K規則601項によれば、本添付ファイルのいくつかの情報は省略されている。
項目16.表10-Kの概要。
ない。
112
標札すきま
改正された1934年“証券取引法”第13又は15(D)節の要求に基づいて、登録者は、本10-K表年次報告を次の署名者によって署名することを正式に促し、正式な許可を得た.
|
|
EQUILLIUM,Inc. |
|
|
|
|
|
日付:2024年3月25日 |
|
差出人: |
/S/ブルース·D·スティール |
|
|
|
ブルース·D·スティール |
|
|
|
社長と最高経営責任者 (首席行政主任) |
権力があるのです弁護士
このような陳述を通じて、以下の署名のすべての人が、Bruce D.SteelとJason A.Kyesを構成して任命し、彼らの各々が、彼または彼女の真および合法的な事実代理人および代理人として、任意およびすべての身分で、彼または彼女の名義、職または代替として、本Form 10-K年次報告書の任意およびすべての改訂に署名し、それを証拠物およびこれに関連する他の文書と共に証券取引委員会に提出し、上記の事実代理人と代理人、およびそれらそれぞれを付与することを知っている。その場所およびその周囲で必要かつ行われなければならないすべての行為および事柄を完全に行い、実行する権利があり、その可能性または自ら行うことができるすべての意図および目的を尽くし、本明細書で上述したすべての事実代理人および代理人、またはそれらの1人または複数の代理人を承認し、確認することは、本条例に従って合法的にまたは手配されたすべての行為および事柄を行うことができる。
本報告書は、1934年に改正された証券取引法の要求に基づいて、次の者によって指定された身分及び日付で署名された。
サイン |
|
タイトル |
|
日取り |
|
|
|
|
|
/S/ブルース·D·スティール |
|
社長と |
|
2024年3月25日 |
ブルース·D·スティール |
|
最高経営責任者 (首席行政主任) |
|
|
|
|
|
|
|
/S/ジェイソン·A·ケス |
|
首席財務官 |
|
2024年3月25日 |
ジェイソン·A·ケス |
|
(首席財務官) |
|
|
|
|
|
|
|
/S/ペニートム |
|
上級副社長、金融学 |
|
2024年3月25日 |
ペニー·トム |
|
(首席会計主任) |
|
|
|
|
|
|
|
/S/Daniel M.ブラッドベリー |
|
取締役会議長 |
|
2024年3月25日 |
ダニエル·M·ブラッドベリー |
|
|
|
|
|
|
|
|
|
/S/スティーヴン·コネリー、博士 |
|
取締役会のメンバー |
|
2024年3月25日 |
スティーヴン·コネリー博士です |
|
|
|
|
|
|
|
|
|
/S/マーサ·J·デムスキー |
|
取締役会のメンバー |
|
2024年3月25日 |
マーサ·J·デムスキー |
|
|
|
|
|
|
|
|
|
/S/バラ·S·マニアン博士 |
|
取締役会のメンバー |
|
2024年3月25日 |
Bala S.Manian博士 |
|
|
|
|
|
|
|
|
|
/S/チャールズ·マクドモット |
|
取締役会のメンバー |
|
2024年3月25日 |
チャールズ·マクドモット |
|
|
|
|
|
|
|
|
|
/S/マーク·プルザンスキー医学博士 |
|
取締役会のメンバー |
|
2024年3月25日 |
マーク·プルザンスキー医学博士 |
|
|
|
|
|
|
|
|
|
/S/バーバラ·トルビン医学博士 |
|
取締役会のメンバー |
|
2024年3月25日 |
バーバラ·トルピン医学博士 |
|
|
|
|
|
|
|
|
|
S/Y.キャサリン·徐博士 |
|
取締役会のメンバー |
|
2024年3月25日 |
*Y.Katherine Xu博士 |
|
|
|
|
113
114
連結財務諸表索引
EQUILLIUM,Inc.
独立公認会計士事務所レポート(PCAOB ID:185) |
F-2 |
合併貸借対照表 |
F-3 |
合併経営報告書と全面赤字 |
F-4 |
株主権益合併報告書 |
F-5 |
統合現金フロー表 |
F-6 |
連結財務諸表付記 |
F-7 |
F-1
“独立報告書”REGISTERED会計士事務所
株主や取締役会に
Equillium,Inc.:
連結財務諸表に対するいくつかの見方
Equillium社とその子会社(当社)が2023年12月31日までと2022年12月31日までの総合貸借対照表,同年度までの関連総合経営表と全面赤字,株主権益と現金流量,および関連付記(総称して総合財務諸表と呼ぶ)を監査した。総合財務諸表は、すべての重要な面で、会社の2023年12月31日と2022年12月31日までの財務状況、および2023年12月31日と2022年12月31日までの年度の経営結果とキャッシュフローを公平に反映しており、米国公認会計原則に適合していると考えられる。
意見の基礎
これらの連結財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいてこのような連結財務諸表に意見を発表することだ。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、連結財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査することを求められておらず、私たちも招聘されて監査を行っていません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性について意見を述べるためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、連結財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであっても詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれる。これらの手続きは、連結財務諸表中の金額および開示に関する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、合併財務諸表の全体列報の評価も含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
/s/
2018年以来、当社の監査役を務めてきました。
カリフォルニア州サンディエゴ
2024年3月25日
F-2
Equillium,Inc.
合併残高シーツ
(千単位、株式及び額面データを除く)
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
短期投資 |
|
|
|
|
|
|
||
売掛金 |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
流動資産総額 |
|
|
|
|
|
|
||
経営的リース使用権資産 |
|
|
|
|
|
|
||
財産と設備、純額 |
|
|
|
|
|
|
||
その他の資産 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
負債と株主権益 |
|
|
|
|
|
|
||
流動負債: |
|
|
|
|
|
|
||
売掛金 |
|
$ |
|
|
$ |
|
||
費用を計算する |
|
|
|
|
|
|
||
繰延収入の当期分 |
|
|
|
|
|
|
||
支払手形の当期分 |
|
|
- |
|
|
|
|
|
賃貸負債の当期部分を経営する |
|
|
|
|
|
|
||
流動負債総額 |
|
|
|
|
|
|
||
長期支払手形 |
|
|
- |
|
|
|
|
|
長期繰延収入 |
|
|
- |
|
|
|
|
|
長期経営賃貸負債 |
|
|
|
|
|
|
||
総負債 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
株主権益: |
|
|
|
|
|
|
||
普通株、$ |
|
|
|
|
|
|
||
追加実収資本 |
|
|
|
|
|
|
||
その他の総合収益を累計する |
|
|
|
|
|
|
||
赤字を累計する |
|
|
( |
) |
|
|
( |
) |
株主権益総額 |
|
|
|
|
|
|
||
総負債と株主権益 |
|
$ |
|
|
$ |
|
||
添付の説明を参照してください。
F-3
Equillium,Inc.
連結業務報告書完全な損失を出しています
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
|
|
現在までの年度 |
|
|
|
現在までの年度 |
|
||
|
|
2023 |
|
|
|
2022 |
|
||
収入.収入 |
|
$ |
|
|
|
$ |
|
||
運営費用: |
|
|
|
|
|
|
|
||
研究開発 |
|
|
|
|
|
|
|
||
現在行われている研究と開発を買収する |
|
|
|
|
|
|
|
||
一般と行政 |
|
|
|
|
|
|
|
||
総運営費 |
|
|
|
|
|
|
|
||
運営損失 |
|
|
( |
) |
|
|
|
( |
) |
その他の収入(費用)、純額: |
|
|
|
|
|
|
|
||
利子支出 |
|
|
( |
) |
|
|
|
( |
) |
利子収入 |
|
|
|
|
|
|
|
||
その他の収入,純額 |
|
|
( |
) |
|
|
|
|
|
その他の収入を合計して純額 |
|
|
|
|
|
|
( |
) |
|
所得税費用前純損失 |
|
|
( |
) |
|
|
|
( |
) |
所得税費用 |
|
|
|
|
|
|
|
||
純損失 |
|
$ |
( |
) |
|
|
$ |
( |
) |
他の全面的な収入、純額: |
|
|
|
|
|
|
|
||
証券売却可能な未実現収益(赤字),純額 |
|
|
|
|
|
|
( |
) |
|
外貨換算収益 |
|
|
( |
) |
|
|
|
|
|
その他の全面収入合計,純額 |
|
|
|
|
|
|
|
||
総合損失 |
|
$ |
( |
) |
|
|
$ |
( |
) |
1株当たり基本と希釈して純損失 |
|
$ |
( |
) |
|
|
$ |
( |
) |
加重平均発行された普通株式数 |
|
|
|
|
|
|
|
||
添付の説明を参照してください。
F-4
Equillium,Inc.
合併株式報告書商工業者の権益
(単位:千、共有データを除く)
|
|
|
|
|
|
|
|
その他の内容 |
|
|
積算 |
|
|
|
|
|
合計する |
|
||||||
|
|
普通株 |
|
|
支払い済み |
|
|
全面的に |
|
|
積算 |
|
|
株主の |
|
|||||||||
|
|
株 |
|
|
金額 |
|
|
資本 |
|
|
収入(損) |
|
|
赤字.赤字 |
|
|
権益 |
|
||||||
2021年12月31日の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
Bioniz買収のために普通株を発行する |
|
|
|
|
|
|
|
|
|
|
|
- |
|
|
|
- |
|
|
|
|
||||
従業員株購入計画による普通株の発行 |
|
|
|
|
|
- |
|
|
|
|
|
|
- |
|
|
|
- |
|
|
|
|
|||
株式責任の帰属を制限する |
|
|
- |
|
|
|
- |
|
|
|
|
|
|
- |
|
|
|
- |
|
|
|
|
||
株に基づく報酬費用 |
|
|
- |
|
|
|
- |
|
|
|
|
|
|
- |
|
|
|
- |
|
|
|
|
||
総合収益 |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
|
|
|
- |
|
|
|
|
||
純損失 |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
( |
) |
|
|
( |
) |
2022年12月31日の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||||
Bioniz買収のために普通株を発行する |
|
|
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
従業員株購入計画による普通株の発行 |
|
|
|
|
|
- |
|
|
|
|
|
|
- |
|
|
|
- |
|
|
|
|
|||
普通株を買い戻す |
|
|
( |
) |
|
|
- |
|
|
|
( |
) |
|
|
- |
|
|
|
- |
|
|
|
( |
) |
株に基づく報酬費用 |
|
|
- |
|
|
|
- |
|
|
|
|
|
|
- |
|
|
|
- |
|
|
|
|
||
総合収益 |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
|
|
|
- |
|
|
|
|
||
純損失 |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
( |
) |
|
|
( |
) |
2023年12月31日の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||||
添付の説明を参照してください。
F-5
Equillium,Inc.
統合報告書またはFキャッシュフロー
(単位:千)
|
|
現在までの年度 |
|
|
現在までの年度 |
|
||
|
|
2023 |
|
|
2022 |
|
||
経営活動: |
|
|
|
|
|
|
||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
純損失と業務活動で使用されている現金の照合の調整: |
|
|
|
|
|
|
||
現在行われている研究と開発を買収する |
|
|
- |
|
|
|
|
|
減価償却 |
|
|
|
|
|
|
||
株に基づく報酬 |
|
|
|
|
|
|
||
外貨取引は純損失を実現していない |
|
|
|
|
|
|
||
定期融資割引償却と発行コスト |
|
|
|
|
|
|
||
割増償却と投資割引の増加 |
|
|
( |
) |
|
|
|
|
収入を繰り越す |
|
|
( |
) |
|
|
|
|
経営性資産と負債変動状況: |
|
|
|
|
|
|
||
売掛金 |
|
|
( |
) |
|
|
( |
) |
前払い費用と他の流動資産 |
|
|
( |
) |
|
|
( |
) |
売掛金 |
|
|
|
|
|
|
||
費用を計算する |
|
|
( |
) |
|
|
|
|
使用権資産と賃貸負債純額 |
|
|
( |
) |
|
|
|
|
経営活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
投資活動: |
|
|
|
|
|
|
||
財産と設備を購入する |
|
|
( |
) |
|
|
( |
) |
短期投資を購入する |
|
|
( |
) |
|
|
( |
) |
短期投資満期日 |
|
|
|
|
|
|
||
Bionizを買収して得た現金 |
|
|
- |
|
|
|
|
|
投資活動が提供する現金純額 |
|
|
( |
) |
|
|
|
|
融資活動: |
|
|
|
|
|
|
||
支払手形の償還 |
|
|
( |
) |
|
|
( |
) |
普通株を買い戻す |
|
|
( |
) |
|
|
- |
|
従業員の株式購入計画に基づいて普通株を発行して得た金 |
|
|
|
|
|
|
||
融資活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
現金および現金等価物に及ぼす為替レート変動の影響 |
|
|
( |
) |
|
|
|
|
現金および現金等価物の純増加 |
|
|
( |
) |
|
|
|
|
期初現金及び現金等価物 |
|
|
|
|
|
|
||
期末現金および現金等価物 |
|
$ |
|
|
$ |
|
||
キャッシュフロー情報の追加: |
|
|
|
|
|
|
||
利子を支払う現金 |
|
$ |
|
|
$ |
|
||
所得税の現金を納める |
|
$ |
|
|
$ |
- |
|
|
|
|
|
|
|
|
|
||
買収されたBioniz資産の公正価値 |
|
$ |
- |
|
|
$ |
|
|
Bioniz買収のために普通株を発行する |
|
|
- |
|
|
|
( |
) |
ビオネッツが負担した純負債は |
|
$ |
- |
|
|
$ |
|
|
添付の説明を参照してください。
F-6
連結財務諸表付記
1.組織および会計公告
業務説明
Equillium,Inc.(当社)は
同社の現在の臨床段階の候補製品はEQ 101とitolizumab(EQ 001)を含む。EQ 101は1種の随一の選択性三特異性合成ペプチドであり、IL-2、IL-9とIL-15を抑制するために設計され、これらは疾病を引き起こす重要なサイトカイン標的であり、一連の免疫炎症適応の中で満足されていない需要を解決することを目的としている。Itolizumab(EQ 001)は1種の一流のモノクロナル抗体であり、それは免疫チェックポイント受容体CD 6を選択的に標的とし、CD 6は効果性T細胞(T)を調節する過程において核心的な役割を果たしているEFF細胞)活動と人身売買は,複数の治療領域でいくつかの免疫性炎症性疾患を推進している。同社はまた、様々なサイトカインに対する他の選択的なポリペプチド候補製品の発見と最適化に取り組んでおり、EQ 302の臨床前開発が進められており、EQ 302は潜在的な一流の経口IL-15およびIL-21二重特異性阻害剤である。同社は,多種の重篤な免疫炎症疾患の潜在的同種最適疾患改良療法としてEQ 101,EQ 302とitolizumab(EQ 001)の開発に専念している。
設立から2023年12月31日まで,同社はほとんどの努力を投じて会社員を組織·配備し,業務計画を策定し,資金を調達し,itolizumab(EQ 001)許可権を付与し,非臨床研究を行い,3つの調査性新薬申請(IND)を提出し,会社候補製品の臨床開発を行い,Bioniz治療会社(Bioniz)の買収,小野製薬有限会社(Ono)との資産購入協定やその他の未完了の取引,株式買い戻し計画の開始,上場会社運営に関する一般的かつ行政活動を行っている。また、同社は製品販売、マイルストーン支払い、特許権使用料から収入を得ておらず、その業務の販売と収入の潜在力は確認されていない。
流動性とビジネスリスク
2023年12月31日現在、同社は
F-7
本10-Kフォーム年次報告書が米国証券取引委員会(米国証券取引委員会)に提出された日から、2023年31日には、少なくとも今後12ヶ月の運営に資金を提供するのに十分である。
陳述の基礎
添付されている連結財務諸表は、米国公認会計原則と米国証券取引委員会の規則と規定に基づいて作成された。本付記で言及された任意の適用指針は、財務会計基準委員会(FASB)が公布した“会計基準アセンブリ”(ASC)および“会計基準更新”(ASU)内のGAAPを意味する。
すでに前年の額に対していくつかの再分類を行い、今期の列報に符合するようにした。
合併原則
添付されている総合財務諸表には、当社及びその完全子会社の勘定が含まれている。すべての会社間取引と残高は合併で販売された。
外貨換算
同社はオーストラリアの完全子会社でその機能通貨として現地通貨を使用している。資産と負債は四半期末の為替レートでドルに換算され、収入と支出は年初から現在までの平均為替レートに換算される。報告期間の外貨換算調整は会社総合全面損失表の累積他の全面収益に計上され、累積影響は会社総合貸借対照表の株主権益部分に計上される。
最近発表され最近採用された会計公告
FASBは2021年10月にASU 2021-08を発表した業務組合(テーマ 805):顧客との契約から契約資産および契約負債を計算するこれは、購入日に公正価値に調整するのではなく、購入者が顧客との契約収入(主題606)に基づいて企業合併で購入した契約資産および負債を確認および計量することを要求する。今回の会計基準更新は、2024年度第1四半期から会社に対して発効する。同社は今回の会計基準の更新がその連結財務諸表に実質的な影響を与えないと予想している。
FASBは2023年12月にASU 2023-09を発表しました所得税(話題740):所得税開示の改善ASU 2023-09は、レート調整中の特定のカテゴリを毎年開示し、量子化しきい値に達した調整項目に追加情報を提供し、支払われた所得税を分類し、返金を差し引くことを要求する。ASU 2023-09はまた、不確定な税金状態および確認されていない繰延税金負債に関連するいくつかの既存の開示要件をキャンセルする。ASU 2023−09は,2025年12月31日までの年次報告Form 10−Kから発効した。早期養子縁組を許可する。ASU 2023-09は前向きな応用価値を持つべきである。トレーサビリティのある採用を許可する。会社は現在、この基準が会社の連結財務諸表に及ぼす影響を評価している。
2.主な会計政策の概要
予算の使用
当社の総合財務諸表を作成する際には、総合財務諸表及び付記中の資産、負債及び費用の報告金額及び又は有資産及び負債の開示に影響を与えるために、推定及び仮定を行う必要がある。会社合併財務諸表の重大な見積もりは、研究と開発費用、収入確認と株式奨励の推定値に関連している。経営陣はその見積もり数を評価し続けている。同社の歴史的経験,現在の事件の理解,将来可能な行動に基づいていると考えられるが,実際の結果は最終的にはこれらの推定や仮定とは大きく異なる可能性がある。
信用リスクと表外リスク集中
金融会社を深刻な集中信用リスクに直面させる可能性のあるツールには、現金と現金等価物、短期投資が含まれる。同社は連邦保険の金融機関で#年に預金を維持している
F-8
過剰になる連邦保険限度額。当社は当該等の口座に何の損失も生じておらず、経営陣は、当該等の預金を保有する預金機関の財務状況により、当社は重大な信用リスクに直面しないと信じている。当社の投資政策には、関連機関や金融商品の品質に対するガイドラインが含まれており、当社が投資可能な投資許可を定義しており、当社はこれらの投資が信用リスク集中のリスクを最小限に抑えることができると考えています。
総合収益(赤字)
♪the the the会社は確認期間中に連結財務諸表に純損失を含む全面赤字のすべての構成要素を報告しなければならない。全面損失は、一定期間内に非所有者源の取引とその他の事件と状況によって発生する権益変化と定義され、投資の未実現収益と損失及び外貨両替収益と損失を含む。その他の全面収益は、純額には短期投資の未実現収益または損失および外貨換算収益または損失が含まれる。
現金と現金等価物
現金および現金等価物は、いつでも利用可能な小切手および貯蓄口座内の現金、および通貨市場基金を含む。当社は購入日から原始満期日が三ヶ月以下のすべての高流動性投資を現金等価物と見なしています。会社の現金と現金等価物は、2023年12月31日、2023年12月31日、2022年12月31日まで、主に通貨市場基金で構成されている。
短期投資
販売可能な証券公正価値に基づいて帳簿を作成し,損益を実現せずに総合損失の中に列記する.債務証券を売却可能な償却コストは、割増償却と満期時に増加する割引に応じて調整される。このような償却と付加価値は利息収入に計上される.売却可能な証券の実現損益と非一時的と判断された価値低下(あれば)は他の収入や費用に計上される。証券売却のコストは具体的な識別方法によって決定される。証券を売却可能な利息と配当金に分類されて利息収入に計上される。
売掛金
売掛金には、小野資産購入協定からの貿易売掛金が含まれる(付記9参照)。貸借対照表日現在も領収書を発行していない返済可能費用は、未開債権と記入している。2023年12月31日と2022年12月31日まで、同社の未開請求書の売掛金総額は
前払い費用と他の流動資産
前払い料金および他の流動資産には、以下のものが含まれている(千計)
|
十二月三十一日 |
|
|||||
|
2023 |
|
|
2022 |
|
||
オーストラリアの研究開発税の割引 |
$ |
|
|
$ |
|
||
前払い臨床開発 |
|
|
|
|
|
||
前払い保険 |
|
|
|
|
|
||
その他売掛金 |
|
|
|
|
|
||
前払いその他 |
|
|
|
|
|
||
その他流動資産 |
|
|
|
|
|
||
前払い費用とその他の流動資産総額 |
$ |
|
|
$ |
|
||
F-9
財産と設備
賃貸借証書
会社は最初から賃貸契約かどうかを確定しています。リース使用権資産代表会社がリース期間内に対象資産を使用する権利は、リース負債代表会社がリースにより発生したリース金を支払う義務がある。初期期間が12ヶ月以上の経営リースについては、当社は開始日レンタル期間内のレンタル支払い現在値に基づいて経営リース使用権資産と経営リース負債を確認します。経営的賃貸使用権資産は、賃貸負債に支払いを加えた任意のレンタル金からなり、レンタル奨励は含まれていません。賃貸条項には、当社が継続選択権が行使されると合理的に判断した場合、または終了選択権が行使されないと合理的に判断した場合、契約を更新または終了するオプションが含まれる。当社の経営リースについては、将来の賃貸支払いの現在値を決定するための金利が容易に確定できなければ、当社はその増額借入金金利をレンタルの割引率と推定している。同社の逓増借款金利は、類似条項と支払い、類似経済環境下の担保に基づく金利近似値と推定されている。レンタル支払いのレンタル料金はレンタル期間内に直線法で確認します。当社は実際の便宜策を選択しており、レンタルと非レンタル構成要素を分離しません。
長期資産減価準備
長期資産は主に財産と設備で構成されている。イベントや状況が資産の減価可能性を示し、当該等資産推定による未割引現金流量が当該等資産の帳簿金額よりも少ない場合には、減値損失を計上する。当社の現在および過去の経営損失および負キャッシュフローはいずれも減値指標であるが、管理層は将来受け取るキャッシュフローが長期資産の帳簿価値を支持すると信じているため、設立以来何の減価損失も確認されていない。
研究と開発費用を計算すべきである
同社は,サプライヤー,コンサルタント,契約研究組織と締結された契約に基づいて負担する研究·開発活動に関する費用を推定しなければならない。これらの契約の財務条項は交渉が必要であり、異なる契約の交渉が異なり、支払流量がこれらの契約に基づいて材料またはサービスを提供する期限と一致しない可能性がある。同社は、研究·開発費用をサービスや努力支出の期間に合わせることで、その連結財務諸表にこれらの費用を反映している。同社は臨床前研究あるいは臨床研究の進展に基づいて、研究或いは関連活動の各方面の時間によって測定し、これらの費用を計算する。同社は,関連契約の審査や財務モデルの準備により見積を決定するとともに,研究や開発者と研究進展や進行中の他のサービスについての検討を考慮している。研究過程で、実際の結果がその見積もりの結果と異なれば、会社はその費用確認比率を調整する。同社は計算すべき研究·開発費用の見積もりを添付の総合貸借対照表に記載されている計算費用に分類している。
オーストラリアの研究開発税の割引
オーストラリアが開発した税収インセンティブ計画または税収インセンティブ計画によると、同社はオーストラリア税務局から条件に合った研究開発支出の現金払い戻しを受ける資格がある。申請実体の収入はオーストラリアドル以下でなければ資格がない
収入確認
会社が収入を確認する方法は、製品またはサービスの支配権を顧客に移転することを記載し、会社がその製品やサービスと交換する権利があることを反映した対価格金額を反映している。この場合,会社は,(I)顧客との契約を決定する,(Ii)契約中の履行義務を決定する,(Iii)取引価格を決定する,(Iv)取引価格を履行に割り当てる,の5ステップ法に従う
F-10
(V)顧客が製品またはサービスの制御権を取得した場合に収入を確認する。収入確認基準を適用する際には、会社は契約条項およびすべての関連事実と状況を考慮します。同社は、任意の実際的な便宜策を使用することを含む収入確認基準を、類似した特徴や類似した状況を持つ契約に一貫して適用する。
顧客は会社と契約を結ぶ側であり、契約の目的は会社が正常に活動している製品やサービスを獲得し、対価格と交換することである。契約とみなされるためには、(1)契約は承認されなければならない(書面、口頭または他のビジネス慣行に基づいて)、(2)譲渡する製品またはサービスに対する各当事者の権利を決定することができ、(3)譲渡する製品またはサービスの支払い条件を決定することができ、(4)契約は商業的実質を持たなければならない(すなわち、将来のキャッシュフローのリスク、時間または金額は契約によって変化すると予想される)。および(V)当社は,その獲得権のある製品やサービス譲渡のすべての対価格を受け取る可能性が高い.
履行義務は、製品やサービスを顧客に譲渡する約束と定義される。当社は、製品またはサービス(または1つの製品またはサービス、または実質的に同一であり、同じ譲渡モードを有する一連の製品およびサービス)を譲渡することを決定する各約束は異なる。(I)顧客が単独で、または顧客がいつでも利用可能な他のリソースと共に製品またはサービスから利益を得ることができ、(Ii)会社が製品またはサービスを顧客に譲渡する約束が契約内の他の承諾とは別に識別することができる場合、製品またはサービスは異なる。譲渡製品またはサービスのすべての異なる約束は、収入確認の会計単位である。製品またはサービスを譲渡するコミットメントが契約中の他のコミットメントとは別に識別できない場合、そのようなコミットメントは、単一の履行義務に統合されるべきである。
取引価格は、製品やサービスの制御権を顧客に譲渡することと引き換えに、会社が獲得する権利がある対価格金額である。取引価格を決定する際には,会社は重要な融資成分,任意の可変要因の影響,非現金考慮要因,顧客への対価格が存在するかどうかを考慮する。重要な融資成分が存在すれば、取引価格は通貨の時間価値に応じて調整される。可変性要因が存在する場合、会社は、その予想される対価格を推定し、その金額を、製品やサービスを顧客に移転する際の収入を確認するための基礎としなければならない。可変対価金額を決定するには,(1)期待値方法,すなわち一連の対価可能金額における確率重み付き金額の総和,(2)最も可能な金額法,一連の可能な対価格のうち単一の可能な金額を決定する方法がある.
1つの契約に複数の履行義務がある場合、会社は、各異なる履行義務を履行するために、会社が獲得する権利のある対価格を反映した取引価格をそれぞれ異なる履行義務に割り当てる。各項目別の履行義務については、収入は、会社が当該履行義務に適用される製品又はサービスの制御権を移転する際に(又は)確認する。
会社が契約履行義務を履行する前に初めて対価格を受けた場合、会社は(または)会社が履行義務を履行するまで、このような対価格を繰延収入に分類する。会社が対価格を受け取る前にまずその履行義務を履行した場合、対価格は売掛金として記録される。
確認された資産の予想償却期間が1年以上である場合、または資産の金額が重要でない場合、会社は、発生時に契約の取得および履行の増分コストを支払う。そうでなければ、このようなコストが契約の増分であり、基礎契約の収入確認に比例する費用で償却された場合、そのようなコストは契約資産に資本化される。
契約資産
会社には実質的な契約資産がなく、収入確認が貨物統制権の移転又はサービスの提供である。少量の研究や開発サービスは一定期間発生する可能性があるが,この期間は通常短い.任意の生成可能な契約資産は、会社総合貸借対照表の売掛金から信用損失準備を差し引いて売掛金に計上される。当社の契約資産には、小野資産購入契約からの貿易売掛金が含まれています(付記9参照)。貸借対照表日現在も領収書を発行していない返済可能費用は、未開債権と記入している。2023年12月31日と2022年12月31日まで、会社の未開請求書は売掛金総額$です
F-11
契約責任
その会社の契約負債には前払いと繰延収入が含まれている。同社は予想される収入確認時間に応じて前払いと繰延収入を当期収入と非当期収入に分類している。一般的に、すべての契約負債は1年以内に確認され、会社総合貸借対照表の繰延収入に計上される予定だ。繰延収入の非当期部分が含まれ、会社の総合貸借対照表に個別に開示される。
買収している研究開発費は
同社は新製品候補製品を開発する権利を獲得し続けている可能性がある。新製品候補製品の買収の支払いおよび資産買収に関連する将来のマイルストーン支払い(またはその中で解決された支払い)は、候補製品がマーケティング監督管理の承認を得ておらず、承認されていない場合には、将来的に代替用途がないことを前提とした買収進行中の研究·開発支出となる。
研究と開発
研究·開発費用には、賃金及び関連間接費用、非現金株式補償費用、第三者との手配に応じて発生する外部研究·開発費用、コンサルタント·契約研究機関がサービスを提供するコスト、及びINDの作成及び提出に関するコストを含む規制コストが含まれる。研究·開発コストは発生時に費用を計上する。
特許費用
株に基づく報酬
同社は、株式オプションと購入権を含む従業員と非従業員の株式奨励を付与日の公正価値で測定し、帰属期間中の報酬支出を直線的に記録している。同社はブラック·スコアーズオプション定価モデルを用いてその株式オプション報酬を評価した。株式オプション奨励の公正価値を推定するには、管理層がある仮定に対して判断と推定を行う必要があり、会社普通株の変動性、会社株式オプションの期待期限、期待配当率と会社普通株の計量日における公正価値を含む。したがって、要因が変化すれば、経営陣は異なる仮定を使用し、株式に基づく報酬支出は将来の報酬に対して実質的に異なる可能性がある。
所得税
当社は、連結財務諸表に含まれる事件の将来の税務結果を予想した繰延税金資産と負債を確認することを要求する貸借対照法に基づいて所得税を計算する。この方法によると、繰延税項資産及び負債は、総合財務諸表と資産及び負債の税ベースとの差額に基づいて、予想差額を用いて返送される年度の現行税率を決定する。税率変動が繰延税金資産や負債に及ぼす影響は、公布日を含む期間の収入で確認されている。
当社が繰延税金資産の範囲を確認したのは、当社はこれらの資産がより現金化する可能性があると考えています。このような決定を下す際には、管理層は、既存の課税の一時的な差の将来逆転、将来の課税収入の予想、税務計画戦略、最近の経営の結果を含む、利用可能なすべてのプラスおよび負の証拠を考慮する。経営陣が当社が将来その記録純額を超える繰延税金資産を実現できると判断した場合、管理層は繰延税金資産の推定値を調整し、所得税の支出を減らすことになる。
根拠は改正された1986年国内税法(IRC)によると、特に第382条及び383条によると、会社所有権の累積変動が超えていれば
F-12
課税税今後数年間の所得税と所得税支出はBionizによって得られた支出を含めて大きく制限されたり廃止される可能性がある。また,IRC第382条の規定により,所有権変更が実現されると,会社がこのような税収属性に関連する繰延税金資産が大幅に減少または廃止される可能性がある。廃止された場合、関連資産は繰延税金資産表から削除され、それに応じて推定免税額が減少する。また、会社税務属性繰り越しの使用制限は、確認された課税収入や当期所得税費用を増加させる可能性があります。推定免税額の存在により、所有権変更制限は顕著ではなく、会社の実際の税率に影響を与えない可能性がある。
2017年の減税·雇用法案は、2021年12月31日以降の納税年度に支払われるまたは発生した金額の研究·実験(R&E)支出の即時費用を廃止するためにIRC第174条を改正した。改訂されたIRC第174条規則は、納税者にそのR&E支出とソフトウェア開発コスト(総称してR&E支出と呼ぶ)を資本口座に計上することを要求する。資本化のコストは償却する必要があるあるいは…
当社は、(1)管理職が税務倉位の技術的価値に基づいて、その税務倉位を維持する可能性が高いかどうかを決定する2段階に分けた手順に基づいて不確定な税務倉位を記録し、(2)確認のハードルを達成する可能性の高い税務倉位について、管理層が確認した最大税収割引額を超える
1株当たり純損失
1株当たり基本純損失の計算方法は,純損失を当期発行普通株の加重平均で割ったものである。1株当たり純損失の計算方法は,純損失を当期発行済み普通株と普通株等価物の加重平均で割る。普通株式等価物は、その影響が希釈である場合にのみ含まれる。当社の潜在的希薄化証券には、当社の持分激励計画下の未償還オプションと普通株を購入する未償還引受権証が含まれており、1株当たりの純損失に対して逆償却作用があるため、計算希釈後の1株当たりの純損失には含まれていない。列報のすべての期間において、会社の純損失状況により、基本と希釈後の流通株を計算するための株式数に差はなかった。
普通株株主の1株当たり償却純損失計算に含まれない潜在的希薄化証券は以下のようになる(普通株等値株式では)、これは逆薄になるからである
|
|
現在までの年度 |
|
||||||
|
|
2023 |
|
|
|
2022 |
|
||
普通株式オプション |
|
|
|
|
|
|
|
||
普通株式引受証 |
|
|
|
|
|
|
|
||
合計する |
|
|
|
|
|
|
|
||
F-13
3.金融商品の公正価値
会計指針は公正価値を定義し、公正価値を計量するために一致した枠組みを構築し、公正価値の経常性或いは非日常性に基づいて計量する各主要資産と負債カテゴリの開示範囲を拡大した。公正価値は、市場参加者間の秩序ある取引において資産を売却して受信した金額または移転負債によって支払われた金額を表す脱退価格として定義される。したがって、公正価値は市場に基づく計量であり、資産または負債の定価のために市場参加者が使用する仮定に基づいて決定されるべきである。このような仮定を考慮する基礎として、会計基準は三級公正価値レベルを確立し、公正価値を計量する際に使用する投入の優先順位は以下の通りである
レベル1アクティブ市場の未調整オファーは、計量日に同じ制限されない資産または負債のオファーを得ることができる。
レベル2−アクティブ市場における同様の資産および負債のオファー、非アクティブ市場におけるオファー、または資産または負債の全期間にわたって直接または間接的に観察可能な投入。
レベル3−価格または推定技術は、公正な価値計量に重大な意味を有するが、観察できない投入(すなわち、市場活動のサポートが少ないか、またはない)を必要とする。
公正な価値に応じて恒常的に計量される金融資産には、会社の現金等価物および短期投資が含まれる。現金等価物には、通貨市場基金と米国国債と預金からなる短期投資が含まれる。同社は、その投資マネージャーから定価情報を取得し、一般に、報告された取引、仲介人/取引業者のオファー、および入札および/または要約を含む、標準的な観察可能な入力を使用して投資証券の公正価値を決定する。
以下の表は、会社が経常的な公正価値に応じて計量する必要がある資産と、公正価値レベルに基づくそれぞれの投入レベル(千単位)をまとめている
|
|
|
|
|
公正価値計量使用 |
|
||||||||||
|
|
|
|
|
見積もりはありますか |
|
|
意味が重大である |
|
|
意味が重大である |
|
||||
|
|
|
|
|
活発な市場 |
|
|
他にも |
|
|
見えない |
|
||||
|
|
十二月三十一日 |
|
|
同じ上の |
|
|
観察できるのは |
|
|
入力量 |
|
||||
|
|
2023 |
|
|
資産(レベル1) |
|
|
入力(レベル2) |
|
|
(レベル3) |
|
||||
短期投資: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
$ |
|
|
$ |
|
|
$ |
- |
|
|
$ |
- |
|
||
合計する |
|
$ |
|
|
$ |
|
|
$ |
- |
|
|
$ |
- |
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
公正価値計量使用 |
|
||||||||||
|
|
|
|
|
見積もりはありますか |
|
|
意味が重大である |
|
|
意味が重大である |
|
||||
|
|
|
|
|
活発な市場 |
|
|
他にも |
|
|
見えない |
|
||||
|
|
十二月三十一日 |
|
|
同じ上の |
|
|
観察できるのは |
|
|
入力量 |
|
||||
|
|
2022 |
|
|
資産(レベル1) |
|
|
入力(レベル2) |
|
|
(レベル3) |
|
||||
短期投資: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
$ |
|
|
$ |
|
|
$ |
- |
|
|
$ |
- |
|
||
合計する |
|
$ |
|
|
$ |
|
|
$ |
- |
|
|
$ |
- |
|
||
アメリカの国庫券と預金は一級投入を使って評価します。第1級証券は、同じ、制限されていない資産または負債で計算されるアクティブ市場の調整されていない見積で推定される。二次投入によって確定された公正価値は、見積もり、金利、収益率曲線などの観察可能なデータ点を利用して、判断と使用推定を行う必要があり、見積もりが変化すれば、会社の財務状況と運営結果に重大な影響を与える可能性がある。機関証券への投資は第2レベル投入を用いて推定される。二級証券は最初に取引価格で評価され、その後、直接或いは間接的に観察できる見積もり以外の投入を利用して評価と報告を行い、例えば第三者定価サプライヤーからのオファーを行う。
当社の金融商品の帳簿には、現金及び現金等価物、前払い支出及びその他の流動資産、支払すべき帳簿及び売掛金が含まれており、満期日が短いため、公正価値と比較している。2022年12月31日、当社が支払うべき手形の帳簿金額は$です
F-14
“会社”ができた
4.短期投資
次の表は、同社の短期投資(単位:千):
|
|
成熟性 |
|
償却する |
|
|
実現していない |
|
|
実現していない |
|
|
推定数 |
|
||||
|
|
(単位:年) |
|
コスト |
|
|
収益.収益 |
|
|
損 |
|
|
公正価値 |
|
||||
2023年12月31日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
|
$ |
|
|
$ |
|
|
$ |
- |
|
|
$ |
|
||||
合計する |
|
|
|
$ |
|
|
$ |
|
|
$ |
- |
|
|
$ |
|
|||
2022年12月31日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
|
$ |
|
|
|
- |
|
|
|
( |
) |
|
$ |
|
|||
合計する |
|
|
|
$ |
|
|
$ |
- |
|
|
$ |
( |
) |
|
$ |
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
会社のすべての販売可能な証券は会社が現在の業務で使用することができます。したがって、会社はこれらのすべての証券を流動資産に分類し、いくつかの個別証券の声明満期日であっても、
いくつありますか
5.財産と設備
財産と設備は以下の部分から構成される(千計)
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
家具と固定装置 |
|
$ |
|
|
$ |
|
||
機械と実験室装置 |
|
|
|
|
|
|
||
コンピュータ装置 |
|
|
|
|
|
|
||
賃借権改善 |
|
|
|
|
|
|
||
減価償却累計と償却を差し引く |
|
|
( |
) |
|
|
( |
) |
財産と設備、純額 |
|
$ |
|
|
$ |
|
||
財産や設備に関する減価償却費用は約#ドルである
6.買収
2022年2月14日、会社はBioniz証券所有者(証券所有者代表)としてのみ会社の完全子会社、デラウェア州会社Project JetFuel Merge Sub,Inc.と合併協定と計画を締結した。 Bioniz買収の対価格として,会社は(A)が最も多く発行されることに同意した
終値時に会社は譲渡代理に納入した
F-15
ビオニーズ
同社は今回の買収を決定した 買収された総資産の公正価値のほぼすべてが1組の類似した識別可能な資産に集中しているため、この買収は企業とはみなされない。会社がASC 805に従って取引を資産買収として記録しているため、または支払いが実現されたときに確認され、進行中の研究および開発に使用される。取引コストは約$
調達価格割当ての概要は以下のとおりである(単位:千):
|
|
金額 |
|
|
買収した資産: |
|
|
|
|
現金 |
|
$ |
|
|
前払い費用と他の流動資産 |
|
|
|
|
固定資産 |
|
|
|
|
買収した総資産 |
|
|
|
|
負債を抱えています |
|
|
|
|
売掛金 |
|
|
|
|
費用を計算する |
|
|
|
|
負担総負債 |
|
|
|
|
購入純負債 |
|
$ |
|
|
Bioniz買収のために普通株を発行する |
|
|
|
|
現在行われている研究と開発を買収する |
|
$ |
|
|
7.賃貸証書
同社の賃貸契約は主にカリフォルニア州ラホアにあるオフィスと実験室施設、以前はカリフォルニア州サンフランシスコ南部に位置していた施設に関連している。同社のサンフランシスコ南部のオフィススペースレンタルは#年で満期になります
会社は使用権資産を他の資産(長期)に計上し、経営リース負債を他の流動負債と長期負債に計上する。
F-16
当社の2023年12月31日現在及び同年度までの賃貸借に関するその他の資料は以下の通り(単位:千、レンタル期間及び割引率を除く)
|
|
2023年12月31日 |
|
|
貸借対照表情報 |
|
|
|
|
使用権資産 |
|
$ |
|
|
賃貸負債、流動 |
|
$ |
|
|
非流動賃貸負債 |
|
|
|
|
リース総負債 |
|
$ |
|
|
その他の情報 |
|
|
|
|
加重平均残余レンタル期間 |
|
|
||
加重平均割引率 |
|
|
% |
|
キャッシュフロー情報を補充する |
|
|
|
|
経営的リースの経営的現金流出 |
|
$ |
|
|
賃貸義務と引き換えに使用権資産 |
|
$ |
|
|
2023年12月31日までの賃貸負債満期日は以下の通り(千計)
十二月三十一日までの年度 |
|
|
|
|
2024 |
|
$ |
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 |
|
|
|
|
未割引賃貸支払総額 |
|
|
|
|
差し引く:推定利息 |
|
|
( |
) |
リース総負債 |
|
$ |
|
|
当社は2023年12月31日現在、重大な権利及び義務を生じ始めていない賃貸契約は何もありません。
8.課税料金
計算すべき費用には、以下の項目が含まれる
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
賃金とその他の従業員の福祉を計算しなければならない |
|
$ |
|
|
$ |
|
||
臨床発展 |
|
|
|
|
|
|
||
非臨床研究 |
|
|
|
|
|
|
||
その他の課税項目 |
|
|
|
|
|
|
||
応算利息 |
|
|
- |
|
|
|
|
|
費用総額を計算する |
|
$ |
|
|
$ |
|
||
9.パートナーシップ
小野薬業有限公司と締結した資産購入契約。
当社は2022年12月5日に、日本Kabushiki KaishaのOnoと資産購入協定を締結し、これにより、当社はOno独占権利(ただし義務なし)を付与して、当社のitolizumabに対する権利(株式購入)を買収する。これらの権利は、すべての治療適応と、米国、カナダ、オーストラリア、およびニュージーランドでitolizumabを商業化する権利を含む。小野はオプションの交換として会社に一括払いをした円並みになる
小野が選択権を行使すれば、小野は会社に円相当の金額を一度に支払います
F-17
同社はitolizumabのすべての研究·開発を担当しており,2022年7月1日から小野社が選択期間まで四半期ごとに資金を提供している。早期に終了しない限り
小野はいつでも書面通知で資産購入契約を終了することができるが、限られた場合、小野は終了後しばらく自社の研究開発費とitolizumabの支出を返済し続ける責任がある。小野がその選択権を行使しなければ,資産購入プロトコルと選択権は自動的に終了する.資産購入協定には、双方が重大な違約時の常習停止権と、いずれか一方が2025年12月31日までに取引が完了していない場合に資産購入契約を終了することを許可する外部日(有限調整可能)も含まれている。
資産購入協定には、当社と小野に関する慣行陳述と保証が含まれています。さらに、会社は、会社の業務の肯定および消極的な経営契約の開発および開発に適した排他的義務、会社の直接的または間接的な販売、許可または他の方法で会社のすべてまたは任意の部分のitolizumab計画または資産購入協定に従って購入された任意の資産を処理することを禁止する排他的義務を含む慣例的な義務および契約の制約を受け、限られた場合には、売却会社に関連する義務、および賠償義務が含まれない限り、慣行の上限および免責額の制限を受ける。
ASC 808を採用しています協力して計画し資産購入協定は、その協定がこのような指示に適用されることを決定する。会社は、小野は顧客を代表し、ASC 606の関連指導を応用していると結論した収入確認資産購入プロトコルの適切な会計処理を評価するために(ASC 606)。このガイドラインに基づき,同社は小野に許可を付与し,ある条件下で何らかの知的財産権を付与し,研究·開発サービスを行うことを含む義務を履行することを決定した。当社は,ある条件を満たした場合,小野にある知的財産権を付与する許可は他の履行義務と変わらないと認定しているが,このような付与は研究·開発サービスの行為や結果に依存するからである。そこで,当社は,すべての履行義務を合併履行義務として入金すべきであり,合併履行義務は研究·開発サービスを行う所期内に移行すべきであることを決定した。
同社は円の前金と貸切不可の支払いについても評価した
当社も資産購入協定項の任意の可変要素の影響を評価します。この評価は、(I)オプション費用および(Ii)様々な臨床、規制、および商業マイルストーン支払いを受ける可能性を評価する。その評価によると,同社はこれらの可変成分が発生する可能性から,取引価格に重大な可変要因はないと結論した。そのため、資産購入協定項における任意のオプション費用やマイルストーン支払いの実現には大きな不確実性があることから、当社は取引価格を設定していない。
ASC 606によれば、当社は、資産購入プロトコルの下での初期取引価格が#ドルに等しいと判断した
同社が確認した収入は#ドル
F-18
2023年12月31日までに、会社はすでに受け取りました
Biocon連携とライセンス契約
2017年5月、当社はBiocon SA(後にBiocon LimitedまたはとともにBioconに譲渡)と協力·許可協定(2018年9月、2019年4月、2019年12月、2021年4月、2022年11月に改正)、臨床供給協定、投資家権利協定、普通株購入協定(総称してライセンス契約)を締結した。ライセンス契約によれば、Bioconは、米国、カナダ、オーストラリアおよびニュージーランド(総称してEquillium分野と呼ばれる)における開発、製造、製造、使用、販売、販売、要約販売、輸入、およびitolizumabおよびitolizumabを含むまたは含む任意の医薬組成物または製剤(総称してBiocon製品と呼ぶ)を使用することを可能にする独占的許可を会社に付与する。当社はまた、このような再許可がライセンス契約の条項に適合することを前提として、複数のレベルで第三者に再許可を発行する権利があり、当社は署名後30日以内に各再ライセンス契約のコピーをBioconに提供する。会社がオーストラリアまたはニュージーランドでBiocon製品を開発および販売する権利を第三者に付与した場合、会社は、その分許可者から受信した任意の事前支払いの2桁の数百分の比をBioconに支払うことを要求され、その分許可者がBiocon製品の純売上に対する印税支払いを含むが、これらに限定されない。ライセンス契約により,同社はBioconに許可を発行し,Equillium地域以外のある国でitolizumabやBiocon製品に関する技術やノウハウの使用を許可した。ライセンス契約によると、Bioconは同社のitolizumab臨床薬物製品の独占的なサプライヤーになることに同意した。会社が初めてアメリカの規制機関の許可を得る前に、Bioconは最大3つの同時に出現した孤児の適応に臨床薬物製品を無料で提供し、Bioconの費用で他のすべての臨床薬物製品を提供する。また,同社はインドBiocon社が行っている潰瘍性大腸炎被験者に対するitolizumabの第二段階臨床研究を共同で支援することに同意した。
Bioconが会社に与えた権利の代償として,会社はBioconに発行した
また、同社はBioconに合計#ドルを支払う義務があります
10それは.支払手形
開ける2019年9月30日(発効日)に、当社は2名の貸手(貸手)と融資及び担保協定(融資協議)を締結し、これにより当社は$を借り入れます
♪the the the定期ローンは
F-19
2021年4月23日(I)最終支払率を
2022年2月、当社は融資の保証者としてBionizを追加した融資協定第3改正案(第3改正案)を締結した。
融資契約によると,その会社は最後の金を支払わなければならない
融資契約の締結については,当社は貸金人に以下の権利を行使できる引受権証を発行する
2023年5月25日、当社は全額前払いローン契約項目の下のすべての満期と借金を前払いし、融資協定を終了した。ローン契約の前金と終了について、会社は合計約#ドルを支払った
定期ローンの帳簿総額は以下の部分からなる(千計)
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
元金 |
|
$ |
|
|
$ |
|
||
追加:最終支払い費用の累積責任 |
|
|
|
|
|
|
||
差し引く:未償却割引 |
|
|
|
|
|
( |
) |
|
合計する |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
11.株主権益
2023年12月31日現在、会社の法定配当金は
その会社は所有している
2023年ATM施設
2023年10月、私たちはJefferies LLCまたはJefferiesと市場での手配を締結し、このスケジュールによると、私たちは私たちの普通株の株を発行して販売することができて、総発行価格は最高$に達することができます
F-20
株式買い戻し計画の認可
2023年7月、会社の取締役会は株式買い戻し計画を承認し、この計画によると、会社は最高$を買い戻すことができる
未償還オプションの再定価
2023年8月7日、会社取締役会はオプション再定価を承認し、2023年8月14日(発効日)に発効した。再定価は、発効日に当社従業員、高級社員及びいくつかの非従業員取締役が保有する当社普通株株式の未償還オプション(未償還オプション)を購入する場合に適用され、当該等の未償還オプションの行使価格は、当社普通株の発効日の終値よりも高く、当社2017年株式インセンティブ計画又は2018年持分インセンティブ計画(2018年計画)に基づいて付与される。発効の日から
再定価のオプションが保留期間終了日(以下に定義する)前に行使される場合、または保留期間終了日前の場合、購入持分所有者の雇用またはサービスが終了した場合、購入持分所有者は、再定価オプションに相当する各株元の使用価格の割増を要求されるであろう。保留期間終了日“とは、(I)発効日後18ヶ月の日付、(Ii)制御権変更(定義は2018年計画参照)及び(Iii)株式購入所有者が死亡、障害又は何らかの他の理由なく終了する(定義は2018計画参照)ために連続サービスを終了する(定義は2018計画参照)中で最も早い項目である。
再定価のオプションの行使価格を改定することを除いて、以前奨励的株式オプションに属していた再定価のオプションは、非法定株式オプションに改訂された(それぞれ2018年計画で定義されている)。いくつありますか
再定価の影響により、株式ベースの非現金報酬支出総額が#ドル増加した
2023年12月31日までの年度内に、会社は株式ベースの増額報酬支出総額を$と確認した
2018年株式インセンティブ計画
2018年10月、会社は既存の2017年度持分インセンティブ計画(“2017年度計画”)に代わって“2018年度持分インセンティブ計画”(“2018年度計画”)を採択した。“2018計画”では、奨励的株式オプション、非法定株式オプション、株式付加価値権、制限性株式奨励、制限株式単位奨励、業績株式奨励、業績現金奨励など様々な形式の株式奨励を付与することが規定されている。2023年12月31日現在、2018年計画は最大
F-21
2018年計画により付与されたオプションは,付与時に決定された異なる日に行使することができ,有効期限を超えない
株式オプション
以下に2023年12月31日までの年間株式オプション活動の概要を示す
|
|
未平倉オプション |
|
|
重み付けの- |
|
|
重みをつける |
|
|
骨材 |
|
||||
2022年12月31日現在の残高 |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
授与する |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
鍛えられた |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
没収とキャンセル |
|
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
2023年12月31日現在の残高(b) |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2023年12月31日までに行使可能なオプション(b) |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
いくつありますか
2023年12月31日までおよび2022年12月31日まで年度帰属株式オプションの公正価値は$
2023年12月31日現在、未帰属株式オプションに関する未確認報酬支出は$
2018年従業員株購入計画
2018年10月、当社は2018年を採択しました
当社は2023年12月31日までに発行しました
F-22
株に基づく報酬費用
連結業務報告書で確認されたすべての株式奨励と購入権の非現金株補償支出は、発生時に確認された没収を差し引くと、総額は以下の通り(千計)
|
|
現在までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
研究開発 |
|
$ |
|
|
$ |
|
||
一般と行政 |
|
|
|
|
|
|
||
合計する |
|
$ |
|
|
$ |
|
||
ブラック·スコアーズオプション定価モデルにおける従業員および非従業員株式オプション付与の公正価値を決定するための加重平均は、以下のように仮定される
|
|
現在までの年度 |
||
|
|
2023 |
|
2022 |
無リスク金利 |
|
|
||
予想変動率 |
|
|
||
予想期限(年単位) |
|
|
||
期待配当収益率 |
|
|
||
無リスク金利です無リスク金利は米国債に基づいていると仮定しており、その条項は会社株オプションの期待期限と一致している。
予想される波動性当社の経営歴史は限られており、民間会社として会社に特定された歴史や隠れた変動率が不足しているため、予想変動率は株価が公開されている業界同業者の歴史変動率を研究することで決定されると仮定している。
期限を見込む株式オプションの予想期間は、株式オプション期待未償還の加重平均期間を表す。当社はアメリカ証券取引委員会が提供する簡略化方法を用いて期待期間を推定しています。簡略化された方法は、期待期限をオプションの帰属時間および契約期間の平均値として計算する。
配当収益率を期待する期待配当仮定は、会社の歴史と配当金の予想に基づいている。その会社はまだ支払いをしておらず,進行中である
没収するそれは.会社は期間内に実際に没収された株ベースの補償費用を減らした。
未来発行の普通株を確保する
将来の発行のために予約された普通株式には以下が含まれる2023年12月31日および2022年12月31日:
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
発行済みおよび未償還株式オプション |
|
|
|
|
|
|
||
普通株式引受証 |
|
|
|
|
|
|
||
2018年の持分インセンティブ計画によると得られる報酬 |
|
|
|
|
|
|
||
従業員株購入計画 |
|
|
|
|
|
|
||
合計する |
|
|
|
|
|
|
||
12.支払いの引受および事項
賃貸借その他の負担
当社は2023年12月31日まで、2025年2月期の実験室スペースレンタルと2027年2月期のオフィススペースレンタルを含むカリフォルニア州ラホアのオフィススペースと実験室スペースを撤回できない経営リースに基づいてレンタルしています。同社はこれまでカリフォルニア州サンフランシスコ南部でオフィススペースをレンタルしており、賃貸契約は2023年2月に満期となった。
当社は通常業務中に賠償条項付きサービス契約を締結しています。これらの条項によると、当社は賠償、弁護、無害を維持し、補償された側の損失を賠償することに同意します
F-23
補償を受けた方の サービス表現。当社はこれらの賠償条項に基づいて訴訟を弁護する費用を発生させていません。
訴訟を起こす
2023年12月31日までに
13.所得税
2023年12月31日と2022年12月31日までの年度の所得税前損失準備金(福祉)部分には、以下のものが含まれています(千計)
|
|
現在までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
アメリカです。 |
|
|
( |
) |
|
|
( |
) |
外国.外国 |
|
|
( |
) |
|
|
( |
) |
|
|
$ |
( |
) |
|
$ |
( |
) |
2023年12月31日までの年間で、会社は当期連邦税務支出を$と記録した
以下は予想される連邦法定所得税と我々の年度までの実際の所得税の計上との比較である2023年12月31日および2022年12月31日(単位:千):
|
|
現在までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
法定税率で所得税を徴収する |
|
$ |
( |
) |
|
$ |
( |
) |
州所得税、連邦福祉を差し引いた純額 |
|
|
|
|
|
( |
) |
|
株に基づく報酬 |
|
|
( |
) |
|
|
|
|
上級乗組員の報酬 |
|
|
|
|
|
- |
|
|
永久品 |
|
|
|
|
|
|
||
研究と孤児薬物相殺 |
|
|
( |
) |
|
|
( |
) |
外貨利回り |
|
|
|
|
|
|
||
現在行われている研究と開発を買収する |
|
|
- |
|
|
|
|
|
評価免除額を変更する |
|
|
|
|
|
|
||
|
|
$ |
|
|
$ |
|
||
繰延税金資産の大部分と繰延税金負債の一時的な差額を生じる税務影響2023年12月31日と2022年12月31日の状況は以下の通り(単位:千)
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
繰延税金資産: |
|
|
|
|
|
|
||
純営業損失が繰り越す |
|
$ |
|
|
$ |
|
||
単位 |
|
|
|
|
|
|
||
資本化研究支出 |
|
|
|
|
|
|
||
収入を繰り越す |
|
|
|
|
|
- |
|
|
持分補償 |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
繰延税金資産総額 |
|
|
|
|
|
|
||
推定免税額 |
|
|
( |
) |
|
|
( |
) |
繰延税金資産総額,控除免除額 |
|
$ |
|
|
$ |
|
||
繰延税金負債: |
|
|
|
|
|
|
||
経営的リース使用権資産 |
|
|
( |
) |
|
|
( |
) |
収入を繰り越す |
|
|
- |
|
|
|
( |
) |
他にも |
|
|
( |
) |
|
|
( |
) |
繰延税金負債総額 |
|
$ |
( |
) |
|
$ |
( |
) |
F-24
このような資産現金化の不確実性のため、当社はその繰延税項純資産について推定値を設定して準備した。当社は繰延税金資産の回収可能性を定期的に評価している。繰延資産がより顕在化する可能性があることが確定した場合、推定免税額は減少する。同社は全額推定手当#ドルを記録した
2023年12月31日同社の連邦とカリフォルニア州の税収損失は約$に転換しました
2023年12月31日この会社には連邦と州の税収控除金があります
“によると改正された“1986年国内収入法”(IRC)、特に第382条及び383条は、会社の所有権の累積変動が超える場合
以下の表は、2023年12月31日と2022年12月31日までの年間で税収割引活動が確認されていない入金状況(単位:千)をまとめたものである
|
|
現在までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
未確認の税金割引を開始します |
|
$ |
|
|
$ |
|
||
前年度の税収状況は |
|
|
|
|
|
- |
|
|
毛減--前期税務頭寸 |
|
|
- |
|
|
|
- |
|
総増加-当期納税状況 |
|
|
|
|
|
|
||
総減少額-当期納税状況 |
|
|
- |
|
|
|
- |
|
集まって落ち合う |
|
|
- |
|
|
|
- |
|
訴訟の時効が失効する |
|
|
- |
|
|
|
- |
|
未確認の税金割引-終了 |
|
$ |
|
|
$ |
|
||
未確認の税収割引額は、会社繰延税金資産の決定に反映される。これらすべての金額は、繰延税金資産推定値準備の同等の調整によって相殺されるため、当社の実際の税率に影響を与えないことが確認された。同社は税収割引を確定していない負債は今後12カ月以内に大きな変化はないと予想している。
当社の政策は、所得税支出において所得税事項に関する利息及び/又は罰金を確認することです。2023年12月31日現在、会社の総合貸借対照表に利息や罰金がない課税項目、および
F-25
2023年12月31日までの年度総合経営報告書では利息および/または罰金は確認されていません。
連邦と州の目的のためのすべての納税年度は依然として開放され、税務司法管轄区の審査を受けている。同社はアメリカ、アメリカの各州の管轄区とオーストラリアで納税する必要がある。
2017年の税改正法案はIRCを改正し、2021年12月31日以降に開始された納税年度支払いまたは発生した金額を発効させ、研究と実験(R&E)支出の即時支出を解消し、そのR&E支出とソフトウェア開発コスト(総称してR&E支出と呼ぶ)を資本口座に計上することを納税者に要求した。資本化のコストは償却する必要がある
所得税支出は#ドルです
14.退職計画
同社は、賃金延期の条件を満たしたIRC第401(K)条に基づく従業員貯蓄計画を開始した。計画に参加した従業員はアメリカ国税局の年間納付限度額を守ることができる。“会社”ができた
15.後続の活動
2024年3月6日、取締役会報酬委員会の提案に基づき、取締役会は、2024年インセンティブ計画(以下、インセンティブ計画と略す)を採択し、承認した
F-26