
アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
本財政年度末まで
あるいは…。
1934年証券取引法第13条又は15(D)条に基づいて提出された |
手数料書類番号
(登録者の正確な氏名はその定款に記載)
(明またはその他の司法管轄権 会社や組織) |
(税務署の雇用主 識別番号) |
501軒の部屋 |
|
(主にオフィスアドレスを実行) |
(郵便番号) |
登録者の電話番号、市外局番を含む:(
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
|
取引 記号 |
|
登録された各取引所の名称 |
|
|
同法第12条(G)に基づいて登録された証券:ありません
登録者が証券法第405条に規定する有名な経験豊富な発行者である場合は、再選択マークで表示してください。はい、そうです☐
登録者がこの法第13又は15(D)条に従って報告書を提出する必要がないか否かを、再選択マークで示す。はい、そうです☐
登録者が(1)過去12ヶ月以内(または登録者がそのような報告を提出する必要があるより短い期間)に1934年の証券取引法第13条または15(D)条に提出されたすべての報告書を再選択マークで示すかどうか、および(2)このような提出要求を過去90日以内に遵守してきた
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
大型加速ファイルサーバ |
|
☐ |
|
ファイルマネージャを加速する |
|
☐ |
|
☒ |
|
規模の小さい報告会社 |
|
||
|
|
|
|
新興成長型会社 |
|
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に第240.10 D−1(B)条に従って受信されたインセンティブベースの補償に従って回復分析を行う必要があるかどうかを再選択マークで示す☐
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)。はい、そうです
i
2023年6月30日まで,すなわち登録者が最近完成した第2財期の最終営業日であり,登録者の非関連会社が保有する投票権と無投票権普通株の総時価は約#ドルである
2024年3月21日現在、登録者が発行した普通株の数は
引用で編入された書類
登録者の2024年株主総会の最終委託書に関する部分は、2023年12月31日までの財政年度終了後120日以内に証券取引委員会に提出され、ここで引用により第3部に組み込まれる。
II
カタログ表
|
|
ページ |
第1部 |
|
|
第1項。 |
業務.業務 |
7 |
第1 A項。 |
リスク要因 |
48 |
項目1 B。 |
未解決従業員意見 |
111 |
プロジェクト1 C。 |
ネットワーク·セキュリティ |
111 |
第二項です。 |
属性 |
112 |
第三項です。 |
法律訴訟 |
112 |
第四項です。 |
炭鉱安全情報開示 |
112 |
|
|
|
第II部 |
|
|
五番目です。 |
登録者普通株市場、関連株主事項及び発行者による株式証券の購入 |
116 |
第六項です。 |
[保留されている] |
116 |
第七項。 |
経営陣の財務状況と経営成果の検討と分析 |
117 |
第七A項。 |
市場リスクの定量的·定性的開示について |
128 |
第八項です。 |
財務諸表と補足データ |
128 |
第九項です。 |
会計と財務情報開示の変更と相違 |
129 |
第9条。 |
制御とプログラム |
129 |
プロジェクト9 B。 |
その他の情報 |
129 |
プロジェクト9 Cです。 |
検査妨害に関する外国司法管区の開示 |
129 |
|
|
|
第三部 |
|
|
第10項。 |
役員·幹部と会社の管理 |
131 |
第十一項。 |
役員報酬 |
131 |
第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
132 |
十三項。 |
特定の関係や関連取引、取締役の独立性 |
132 |
14項です。 |
最高料金とサービス |
133 |
|
|
|
第4部 |
|
|
第十五項。 |
展示·財務諸表明細書 |
134 |
第十六項。 |
表格10-Kの概要 |
137 |
3
前向き陳述に関する特別説明
本年度報告は,Form 10−Kまたは年次報告形式で前向き陳述を含む。改正後の1933年証券法第27 A節(“証券法”)と改正後1934年証券取引法第21 E節(“取引法”)に含まれる前向き陳述の安全港条項にこれらの前向き陳述を組み込む予定である。本年度報告に含まれる歴史的事実に関する陳述を除いて、本年度報告に含まれるすべての陳述は、私たちの将来の経営業績と財務状況、私たちの現金、現金等価物および有価証券が私たちの運営費用と資本支出要件を満たすのに十分かどうか、私たちの持続的な経営企業としての能力、業務戦略、候補製品開発、予想製品、候補製品の承認、研究開発活動とコスト、未来の収入、私たちの業務計画の成功時間と可能性、管理計画と目標、臨床試験の将来の結果とタイミング、私たちの候補製品の治療潜在力、および私たちの候補製品の市場潜在力に関する陳述を含み、すべて前向きな陳述である。これらの陳述は既知と未知のリスク、不確定性とその他の重要な要素に関連し、私たちの実際の結果、表現或いは成果は展望性陳述と明示或いは暗示の任意の未来の結果、表現或いは成果とは大きく異なる可能性がある
場合によっては、“可能”、“会議”、“すべき”、“予想”、“計画”、“予想”、“可能”、“意図”、“目標”、“プロジェクト”、“考慮”、“信じ”、“推定”、“予測”、“潜在”、“将”または“継続”またはこれらの用語の否定または他の同様の表現によって、前向きな陳述を識別することができる。すべての展望的声明書がこのような単語を含んでいるわけではないにもかかわらず。本年度報告書の展望的陳述は予測のみであり、主に私たちの現在の未来の事件と財務傾向の予想と予測に基づいており、これらの事件と財務傾向は私たちの業務、財務状況、運営結果に影響を与える可能性があると考えられる。これらの展望的陳述は、本年度報告の日までの状況のみを代表し、第1の部分1 A項で説明されるリスク、不確実性、および仮説を含む多くの既知および未知のリスク、不確実性および仮説の影響を受ける可能性がある。本年度報告書の“リスク要因”。展望性陳述自体はリスクと不確実性の影響を受けるため、その中のいくつかのリスクと不確定性は予測できないか定量化されており、いくつかは私たちが制御できないので、あなたは未来の事件の予測としてこれらの展望的陳述に依存してはならない。著者らの展望性陳述に反映された事件と状況は実現できない或いは発生できない可能性があり、実際の結果は展望性陳述中の予測結果と大きく異なる可能性がある。しかも、私たちは持続的な環境で運営している。新たなリスク要因や不確定要因が時々出現する可能性があり、管理職がすべてのリスク要因や不確定要因を予測することは不可能である。法的要件が適用されない限り、私たちは、任意の新しい情報、未来のイベント、状況の変化、または他の理由による、本明細書に含まれる任意の前向きな陳述を公開または修正するつもりはありません。
本年度報告書で使用されるように、他の説明または文脈に別の規定がない限り、言及された“オメガ”、“オメガ治療”、“会社”、“私たち”は、オメガ治療会社およびその子会社を意味する。
4
リスク要因をまとめる
私たちの業務は、第1部1 A項で述べたリスクと不確実性を含む多くのリスクと不確実性に直面している。本年度報告表格10−Kにおける“リスク要因”。私たちの普通株に投資する時、あなたはこのような危険と不確実性を慎重に考慮しなければならない。私たちの業務に影響を与える主なリスクと不確定要素は以下の通りです
5
6
第1部
プロジェクト1.ビジネス.
概要
オメガ治療会社は臨床段階の生物技術会社であり、新型プログラム可能なエピゲノムmRNA薬物を開発した。著者らのomegaプラットフォームはエピジェネティクスの力と著者らのゲノム構造に対する深い理解を利用して、正確に定位し、転写前レベルの遺伝子発現を制御し、疾病を治療或いは治療することができる。ヒトゲノムの3次元構造を解読しました遺伝子とそれに伴う制御因子は異なる進化的に保存された構造に組織され,絶縁ゲノムドメイン,あるいはIGDと呼ばれる。IGDは遺伝子制御と細胞分化の基本構造と機能単位であり、自然界の遺伝子発現に対する天然制御システムである。多くの疾患はIGD変化による遺伝子発現異常によるものである。Omegaプラットフォームは、数千個の新しいDNA配列に基づくエピゲノム“郵便番号”を系統的に識別し、検証することができ、これらの“郵便番号”はIGD中の単一の制御要素に関連する。これらのエピゲノム標的をEpiZipsと呼ぶ。著者らはEpiZipsに対して正確なエピゲノム制御を行うために、私たちのメッセンジャーリボ核酸療法を理性的に設計し、設計した。これにより,遺伝子を必要な発現レベルに正確に調節し,発現の持続時間を制御することができる。この方法を通じて、omegaプラットフォームは一連の疾病と疾病の中で広範な潜在的適用性があり、従来薬物治療ができず、治愈しにくく、治療が困難であった目標を含むと信じている。著者らが現在準備しているプロジェクトは腫瘍学、再生医学と多遺伝子疾患を含み、免疫学と心臓新陳代謝疾患を含む。
私たちは、omegaプラットフォームが提供する精確なエピゲノム制御は広範な治療適用性と転化潜在力を持っていると信じている
7
私たちのパイプは
私たちが現在準備しているプロジェクトは以下の項目を含む
知的財産権と製造能力
私たち自身の開発活動とマサチューセッツ工科大学ホワイトウッド研究所またはワイトヘイト研究所のライセンスを通じて、omegaプラットフォームと私たちのECsをカバーする重要な知的財産権を強化しました。我々はまた、自社工場を設立する計画を評価することを含む内部·外部製造能力を発展させており、当社のECの開発·商業化を支援する適切な規模·品質を提供している。
私たちの戦略
我々の目標は,omegaプラットフォームを用いてECsを設計,開発,製造,商業化することにより,最先端のプログラマブルエピゲノム薬物会社となることである。我々のビジョンは,ヒトゲノムを選択的かつ安全に誘導し,天然DNA配列を変化させることなく遺伝子発現をあらかじめ転写制御することで,重篤な疾患を治療·治癒することである。
私たちの戦略には
8
絶縁ゲノムドメイン(IGD)の背景
エピジェネティクスは生体生命のあらゆる面を系統的に制御する機序であり、細胞成長と分化から細胞死亡までである。著者らのチームはすでにエピジェネティクスの汎用オペレーティングシステムを理解し、omegaプラットフォームを構築して自然の遺伝子制御方法を複製し、治療効果を得た。IGDはこのオペレーティングシステムの組織構造を理解する鍵であり、遺伝子制御と細胞分化の基本構造と機能単位である。約15,000個のIGDがあり、私たち23個の染色体に分布する約20,000個の遺伝子をカバーしています。それらはすべての細胞に普遍的に存在し,進化的には種内と大部分の種の間で保存されている。
細胞中の遺伝子発現は通常高度に多様な制御要素、例えばエンハンサー、抑制子とプロモーターによって制御される。これらの調節エレメントは比較的短いDNA断片であり,タンパク質転写因子の結合部位として機能し,これらの転写因子は逆に他のタンパク質を募集して標的遺伝子の転写を活性化する。現在の研究により、遺伝子及びその関連する調節素子はモジュール化された方式でIGDに存在することが明らかになった。IGDの染色体ループ構造は遺伝子とその制御要素との間の相互作用が隣接するIGDと外部制御因子から遮断されることを確保し、これは正常な細胞特異性遺伝子制御を確保するために重要である。CCCTC結合因子、CTCFとアドヘシン複合体はIGD構造の形成と維持に重要な役割を果たしている。接着素はモーターです
9
これはIGDループを圧迫および拡大し、CTCFはラミニンをさらに押出し、アンカーとして機能させ、それによってIGD間の境界を強化することを阻止する。
IGDの図形表現
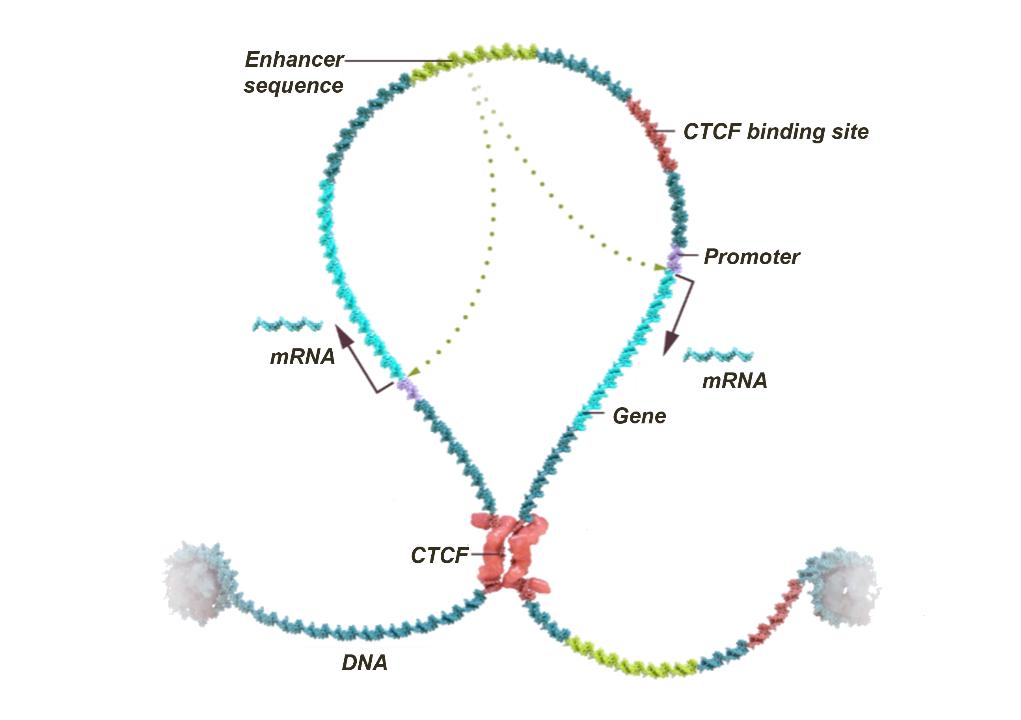
IGDはタンパク質をコードする遺伝子とその制御エレメントを含む。単一のIGDは通常1~10個の遺伝子を含み,中央値は3つの遺伝子である。エピゲノムコントローラは、1つまたは複数のIGD成分(EpiZip)を正確に制御することによって遺伝子発現を制御し、それによって特定のIGD内の遺伝子発現に影響を与えるように設計されている。コントローラはまた、複数のIGDのために多重化されてもよい。
IGDまたはその境界の任意の摂動は、その内部の1つまたはすべての遺伝子の失調を引き起こし、それによって一連の疾患状態を引き起こす可能性がある。IGDの変化は、構造的であってもよく、アンカー-CTCF結合配列、遺伝子プロモーター、およびエンハンサー領域(スーパーエンハンサーを含む)の変異または切断を含む機能的であってもよい。 例えば,乳癌,前立腺癌,腎癌を含む様々な固形腫瘍および白血病にCTCFやアドヘシンコード配列の変異が認められた。IGD境界変化は、調節要素または遺伝子の異常を含むか、または排除することを含むことができる。例えば、いくつかの癌では、IGD境界の破壊は、癌遺伝子を上方制御するために、スーパーエンハンサーと呼ばれる強力な活性化制御要素を含むようにループ相互作用を再接続することができる。遺伝子反転や転座の場合にも,類似した活性化が認められる。IGD境界のエピゲノム変化、例えばDNAメチル化異常は、CTCF結合を変化させることができ、遺伝子拒絶或いはIGD内の遺伝子を外部制御要素に暴露させることを引き起こす。この破壊の病理証拠はすでに癌(例えばグリオーマ)と遺伝性ヒト疾患(例えば脆性X症候群)で発見された。
オメガプラットフォーム
Omegaプラットフォームはこれまでにない方法を代表し,天然DNA配列を変化させることなく遺伝子発現を正確に制御することにより,疾患を治療するエピジェネティクス基礎の治療法を開発していると考えられる。著者らは、著者らのECsは精確に定位し、制御可能、調整可能と持続的な効果を提供でき、多種の疾病を治療する潜在力があると信じている。
Omegaプラットフォームは4つの基本コンポーネントの上に構築されています
1. 新薬標的としてのIGDSとEpiZipの特許データベース
我々は,生物学的優先の方法を用いて,興味のある疾患の兆候に関連する有効な遺伝子標的から標的認識を行った。独自のアルゴリズムと機械学習ツールを使って私たち自身の和を発掘します
10
公共データベースは、開発目標IGDの全面的な概況を用いて、罹患州でどのように失調を調節しているかを理解する。著者らはこれらの情報を総合して肝心な治療介入点を確定し、EpiZips、それはECsの標的であり、期待される遺伝子発現効果を達成する。この過程により,我々はすでに数千のEpiZipとIGDの膨大なライブラリーを構築し,潜在的な治療標的としている。
2. 疾患に対してオーダーメイドされたプログラマブルエピゲノムmRNA薬
著者らはプログラム可能なエピゲノムメッセンジャーリボ核酸薬物ECsの高効率と知能設計のためにモジュール基礎を創造した。これらの前向きな工学化研究薬物は著者らが精緻な特異性、制御可能な調節と作用持続時間で多数の遺伝子を調節できるようにした。われわれの内皮細胞は,2成分からなる融合タンパク質であるDNA結合ドメインとエピゲノムエフェクター蛋白を発現するプログラム可能なメッセンジャーリボ核酸療法である。DNA結合ドメインは特定のEpiZipを標的とし,精緻な特異性を有するように設計されている。エピゲノム効果蛋白は、遺伝子発現を上昇或いは下方制御し、作用持続時間を制御するために、ヒストンと転写因子のような細胞核内のDNA或いはDNA関連蛋白と相互作用することを目的としている。我々は特許アルゴリズムを用いて,DNA結合ドメインのプログラミングと最適なエピゲノム効果タンパク質の選択を含むECsを設計した。これらの計算ツールは大量の潜在的なECsを効率的に生成でき、特定の目標を治療するためにECsを設計する能力を高めることができる。
エピジェネティック制御因子(EC)*

*メッセンジャーリボ核酸薬は細胞核内でタンパク質として発現される
我々は現在特許の亜鉛フィンガー蛋白と他のDNA結合ドメインを開発している。エピゲノムエフェクタードメインの場合、我々は、DNA修飾因子、ヒストン修飾因子、および他のクロマチン再構築因子の自然発生および独自の工学的変異体を含む100個以上の単機能および多機能エピゲノムエフェクタードメインを含むライブラリーを確立し、確立し続けている。
IGDS、EpiZipの初歩的な同定及び特定の標的遺伝子に対するECsの作用機序はエピゲノムコントローラスクリーニングを利用して迅速に検証することができる。著者らのモジュール化設計方法は著者らが発見過程を加速し、遺伝子標的を識別し、そしてわずか数週間以内に初歩的な主導ECsを産生してそれらを調節することができる。
3. 標的組織と細胞にカスタマイズされた工学薬を提供する
適切な細胞と組織への輸送は私たちの技術の成功的な応用に重要だ。私たちは多様な配送方法を模索して革新している。
11
脂質ナノ粒子またはLNP送達技術を選択し,第三者臨床試験で検証され,われわれの初期計画に用いた。現在,LNPsは承認され開発されている製品に用いられている。我々はレシピを渡す上で深い専門知識を持ち,技術進歩と既定の規制前例を利用して我々自身のLNPsを開発している.我々の内皮細胞をメッセンジャーリボ核酸としてDNA結合ドメインとエピゲノムエフェクター蛋白をコードし,LNPに封入している。我々のLNPsは通常,mRNAのような核酸を被覆し,体内の器官や組織を保護·輸送し,細胞への取り込みを促進する4成分または5成分の分子からなる。私たちのLNPsは再利用可能な非ウイルス性を提供すると信じています体内にある肝、肺、中枢神経系、免疫細胞、関節、および他の細胞および組織に送達される。細胞に摂取されると,LNPはmRNA貨物を細胞質に放出し,そこではECに翻訳され,ECは核に輸送され特定のIGDにおける標的EpiZipに結合する。我々は現在,様々な内部および外部由来の一連の陽イオンとイオン化可能なLNPを探索しており,特定かつ有効であることを示す独自のLNP製剤を開発している体内にある臨床前研究中の機能交付。LNP配信技術以外の分野を拡張し,将来のECプロジェクトのための他の配信方式を評価している.
4. 業界をリードする計算、生物、ゲノムの専門知識
我々は,我々のリーディングプランから収集した編纂された学習と知見を用いて我々のプラットフォームを最適化し続け,後続の候補製品の発見と開発に情報を提供する.我々はまた,我々のEpiZipとECs知識ベースを構築し,追加し続けている.著者らは合理的かつ簡略化した方法を用いてプログラム可能なエピゲノム薬物を開発し、穏健かつ高効率な標的認識、検証、候補製品の設計と最適化を通じて、潜在的により速い臨床経路を提供する。著者らはまた、我々のEpiZipと新しいおよび独自のDNA結合ドメインとエピゲノム効果タンパク質のディレクトリを絶えず拡大し、計算方法を用いて標的上と潜在的な脱標的結合と活性を評価し、遺伝子発現の無意識的な変化を最大限に減少させる。
計算基礎
Omegaプラットフォームは新しい生物学とエピジェネティクスを利用して、私たちの重要な計算能力を通じて、治療的に遺伝子発現と細胞状態を制御し、プログラミングする。ヒトゲノムを解読するルールは、数十億個のヌクレオチド、数万個の遺伝子、および100万個までの調節配列からなる規則であり、これらすべてが三次元空間で相互作用する可能性がある--先進的な独自のアルゴリズムおよび統計データ解析技術を創造する必要がある。我々の先端計算ツールは,様々な独自アルゴリズムや深さ学習技術のライブラリ上に構築されており,IGDの位置,構造,機能を解釈し予測することができる.オメガプラットフォームが提供した重要な科学的知見は人々が治療領域と適応にまたがってEpiZipsを識別できるようにした。こんなに深いですねシリコン片理解と予測可能性はまたECsの設計と迅速な工学に直接影響し、私たちは精緻な特異性、制御可能な調節と作用持続時間で単一或いは複数の遺伝子を調節することができる。
我々は,計算とデータ優先の手法を広く応用することにより,我々の計算技術を薬物開発の連続体全体に応用した。広範な系統生物学と機能ゲノミクス手法を用いて関連するバイオマーカーを識別した。著者らは重要な翻訳モデルを用いて作用機序を検証し、臨床翻訳のリスクを加速し、潜在的に低減する。組合せ最適化技術と新しい発見努力は交付と調合設計を加速した。これは私たちが品質とコストを向上させながら、プロジェクトと製造規模を迅速に拡大することができるようにする。システムのデータ捕捉と自動化はリアルタイムでデータ駆動の意思決定を実現し,さらに複数のプログラムを並列に加速させる能力を推進している.
私たちは高いスキルの計算チームを持ち、深い専門知識と幅広い経験を持ち、omegaプラットフォームをサポートしています。このチームはIGD生物学の複雑さ、私たちのECSの設計と交付、および会社全体の計算とデータ優先の理念の統合を解決するために必要なツール、機能と専門方法を開発した。私たちはプログラム可能なエピジェネティック薬物、製造と私たちのデジタル基礎の発見と開発方面の革新を推進するために、私たちの計算チームと能力を発展させている。
12
我々の発展計画は
著者らの現在の戦略重点は腫瘍学、再生医学と多遺伝子疾患(免疫学と心臓新陳代謝疾患を含む)の発展プロジェクトである。Omegaプラットフォームは疾病過程において広範な適用性を有し、腫瘍、代謝失調、繊維化過程、免疫機能障害、血管病理と組織退化を含む。私たちはすでに生まれましたシリコン片, 体外培養そして体内にある複数の遺伝子標的のデータを自らあるいは協力することで臨床に進めることが可能である。
腫瘍学
肝細胞癌用OTX−2002
我々はOTX−2002を開発してc−Mycの発現を低下させ,c−Mycは癌遺伝子であり,50%を超えるヒト癌では発現失調があり,常に不良予後に関与しており,進行肝癌患者の潜在的治療法としている。C-Mycは肝腫瘍細胞増殖に重要な役割を果たし、しかも大多数の肝細胞癌で発現が上昇することが証明された。C−Mycに対する直接の薬物開発は挑戦的であり,その発現が厳密に制御されているため,小分子結合の特定の活性部位に乏しいタンパク質であることが明らかになった。これは,いずれの方法も転写レベルでの潜在的失調を解決できないため,標的c−Myc mRNAやタンパク質が有効である可能性が低いことを意味する。他のより二元的な遺伝子発現を下方制御する方法と異なり、内皮細胞はc-Mycの発現を正確に調節することができ、高度に増幅したMYC増幅の癌細胞を殺し、腫瘍の退化を推進するのに十分であり、健康な周囲細胞を必要とせず、低レベルのMYCだけで正常に機能することができる。
肝細胞癌は慢性肝疾患環境中で発展してきた原発性肝臓悪性腫瘍である。それは通常病気経過が遅い時に診断され、確定診断後の平均生存期間は約6~20ケ月である。2017年、米国では89,950人が肝癌と肝関連癌を患っていると推定されている。確定診断された疾患段階によれば、現在の治療方案は手術切除、チロシンキナーゼ阻害剤(TKI)、例えばソラフィニとランワチニブ、原位肝移植或いは高周波アブレーション、より末期の患者に対して、免疫検査点と抗血管内皮細胞増殖因子の併用治療、或いは経カテーテル動脈化学療法或いは放射線塞栓術、立体定位放射線治療或いは全身化学療法などの緩和治療を含む。
1組の肝癌細胞系でテストを行った時、OTX-2002はゲノム中に精確に定位した領域にエピジェネティック修飾を行い、MYC mRNAと蛋白質レベルの低下を招き、そして抗増殖作用を推進した。皮下と原位マウス異種移植肝癌モデルを用いた臨床前研究では,OTX−2002はマウスの腫瘍成長を有意に抑制したが,マウスの体重変化に有意な影響はなかった。抗腫瘍活性は腫瘍中のMYC蛋白レベルの低下と一致した。また、体外と体内研究において、OTX-2002は現在肝癌を治療する標準薬物lenvatinib或いはsorafenibと結合し、肝癌腫瘍モデル中の腫瘍成長の増強抑制に関与している。
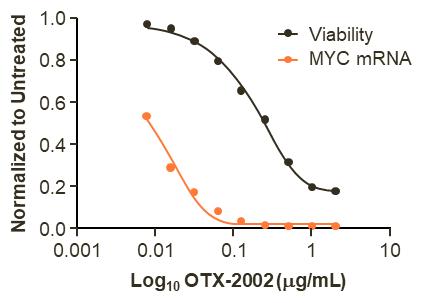
OTX−2002の異なる肝癌細胞系における臨床前研究では,OTX−2002はc−Mycの発現を低下させ,肝細胞癌サブタイプを標的とした細胞活性喪失を認め,15日間の影響を観察した。次の図に示すように欧州委員会は50肝癌細胞系ではベースラインと最大反応の間に50%の反応を与える薬物濃度を測定した。OTX−2002 c−Myc遺伝子発現ECの誘導50平均値は0.013μg/mlであり、濃度が0.147μg/mlの場合、細胞活力は50%低下した。
13
OTX−2002は発現および活性の用量反応に関連している(体外)
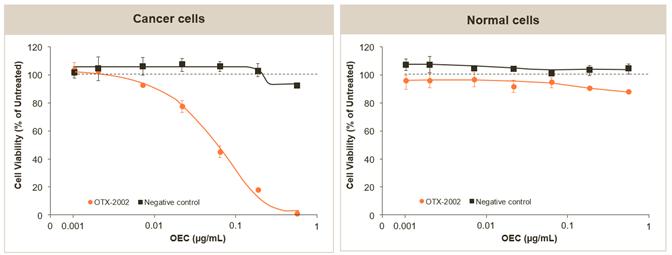
もう一つのOTX−2002単独の肝癌細胞系(Hep 3 B)の臨床前研究では,OTX−2002の癌細胞活力に対する選択的効果が証明された。以下の図に示すように,0.001から500 ng/mLの濃度範囲では,癌細胞をOTX−2002で処理するとこれらの細胞の活力は有意に低下したが,逆に,正常細胞(健康な初代ヒト肝細胞)をOTX−2002で処理した場合,細胞活力への有意な影響は認められなかった。
OTX−2002は肝癌細胞の活力を低下させたが、健康なヒト肝細胞を低下させることはなかった(体外培養)
体内に調製したLNPsを介して送達されるOTX−2002は,ヒト肝癌移植腫瘍を含むマウスの腫瘍負担を低下させた。この臨床前研究では、著者らはマウス皮下腫瘍モデル或いは小分子対照群に5日ごとに3 mg/kgのOTX-2002を投与した。以下の図に示すように,OTX−2002を用いた治療は腫瘍成長の統計的有意な抑制に関与しており,陰性対照と比較して13日目に腫瘍成長は78%抑制された。OTX−2002を用いた治療は小分子比較器を用いた治療に相当する。OTX−2002を服用したマウスの体重は有意に低下しなかった。この研究ではOTX−2002耐性は良好であり,副作用は認められなかった。
14
OTX−2002肝癌皮下移植腫瘍モデルで観察された抗腫瘍活性は,体重に有意な影響はなかった(生体内)
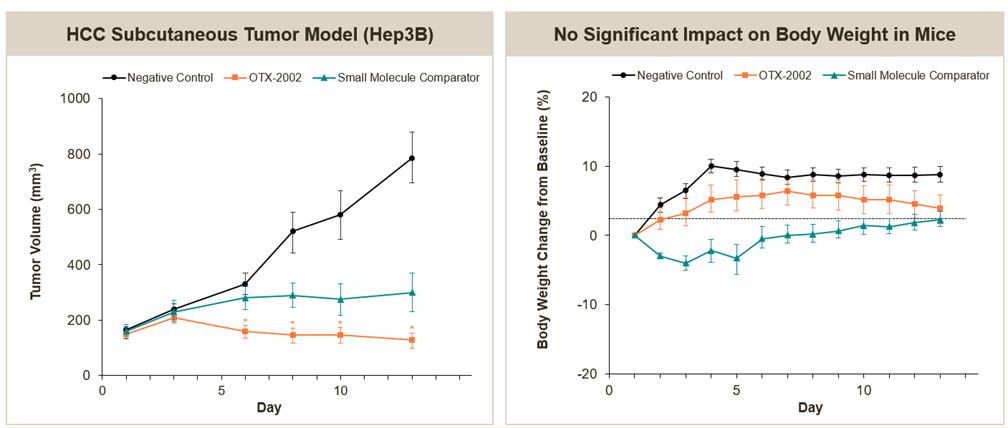
*統計学的有意差は陰性対照と比較して、t検定p.
また,OTX−2002はソラフィニと比較してヒト肝癌異種移植腫を含むマウスの腫瘍成長に同等の影響を認めた。OTX−2002は5日ごとに3 mg/kgまたはソラフェニール50 mg/kgを1日1回胃投与した。生物発光イメージング技術を用いて腫瘍の成長状況を測定した。以下の図に示すように,OTX−2002を用いた治療による発光低下は,ソラフィニを用いた治療に相当する。OTX−2002を服用したマウスの体重は有意に低下しなかった。ソラフェニを服用したマウスは体重が減少し続けた。この研究ではOTX−2002耐性は良好であり,副作用は認められなかった。
15
OTX-2002ヒト肝癌原位移植モデル体内の抗腫瘍活性及び体重変化の観察)

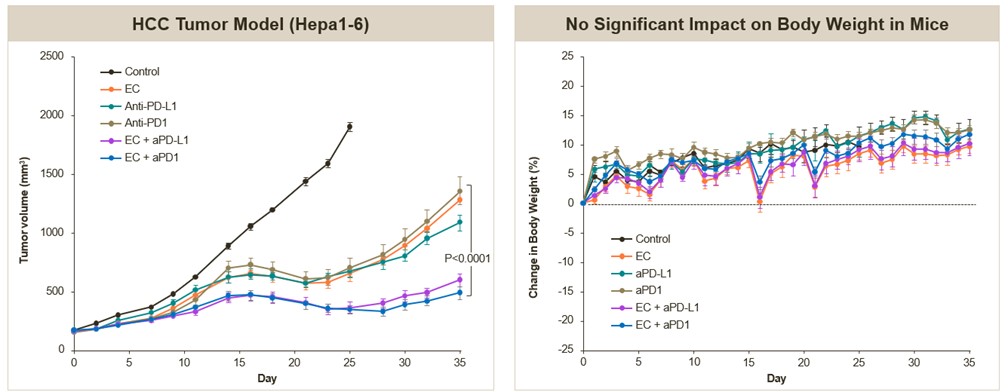
著者らはまた、検査点阻害剤、抗PD-1と抗PD-L 1薬物の併用治療効果は統計学的意義があることを観察した。皮下肝癌モデル(Hepa 1-6)においてOTX-2002のマウス代替物ECを評価する時、免疫能力群マウスは5日ごとに1 mg/kg ECを投与し、毎週10 mg/kgの抗PD 1或いは抗PD-L 1を投与し、ECと抗PD 1或いは抗PD-L 1の組み合わせ、或いは陰性対照を投与した。以下の図に示すように、陰性対照と比較して、連合治療は腫瘍成長の抑制に対して統計学的有意な意義があり、また、すべての単一治療群と比較して、腫瘍成長の抑制も統計学的に有意な意義がある。両組み合わせとも腕耐性は良好であり,研究期間中に体重への有意な影響は認められなかった。
16
ECと抗PD 1あるいは抗PDL 1の併用によるヒト肝癌移植腫瘍モデルの抗腫瘍活性と体重への影響(生体内)
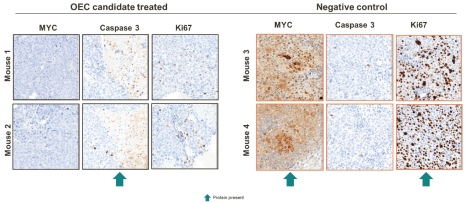
生体内OTX−2002は,調製したLNPsによりマウス皮下肝癌モデルにおいて5日ごとに3 mg/kgの用量で治療し,腫瘍負担の減少を招き,腫瘍におけるc−Myc発現と関連臨床バイオマーカーの相関変化を細胞レベルで示した。以下の図に示すように,EC候補治療と陰性対照腫瘍の組織学的切片の免疫組織化学分析を行った体内にある上述の研究により、c-Myc蛋白は腫瘍において明らかに低下し(褐色染色消失で表される)、予想Ki 67(腫瘍細胞増殖バイオマーカー)は低下し、Caspase 3(アポトーシスの1種のバイオマーカー、プログラム細胞死)は上昇することが示唆された。
肝細胞癌移植腫瘍モデルにおける臨床バイオマーカーの変化
2022年7月、著者らはアメリカ食品·薬物管理局(FDA)が著者らの研究新薬(IND)の申請を許可し、OTX-2002の1/2期臨床試験を開始し、OTX-2002による肝癌治療の初のヒト臨床試験であることを発表した。
2022年10月、MYCHELANGELOの1/2期の開始を発表TM私の臨床試験です。このグローバルな研究はOTX-2002の安全性、耐性、薬物動態学、薬効学と初歩的な抗腫瘍活性を評価しており、単一療法(第1部分)として標準看護療法(第2部分)と結合し、再発または難治性肝細胞癌および他のMYC癌遺伝子に関連することが知られている固形腫瘍タイプの治療に用いられている。
17
2022年11月、OTX-2002がFDAによって肝癌治療の孤児薬として承認されたことを発表した。
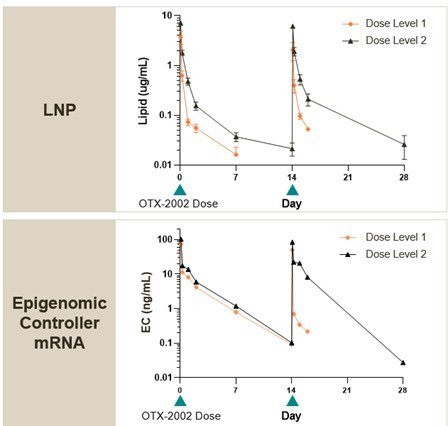
2023年9月、米国およびアジアの臨床地点で現在行われている第1/2期MYCHELANGELO I試験(NCT 05497453)の最初の2用量列の予備臨床データを発表した。2023年9月18日までのデータ締め切りは、8名の患者が2週間ごとに0.02 mg/kg(用量レベル1、n=4)あるいは0.05 mg/kg(用量レベル2、n=4)OTX-2002を静脈内投与した。細胞内遊離DNAと細胞外体mRNAレベルを測定し、MYC細胞DNAメチル化とmRNAレベルの変化を分析した。
データ遮断点まで、試験前の2つの用量レベルの初歩的な臨床データの主要なハイライトは:
安全性とフォールトトレランス:
薬物動態:
予測可能な薬物動態、薬物製品の迅速な除去が観察された
翻訳:
18
MYC IGDにおける標的遺伝子座の高度に特異的な標的関与と予想されるエピジェネティック状態変化が観察された
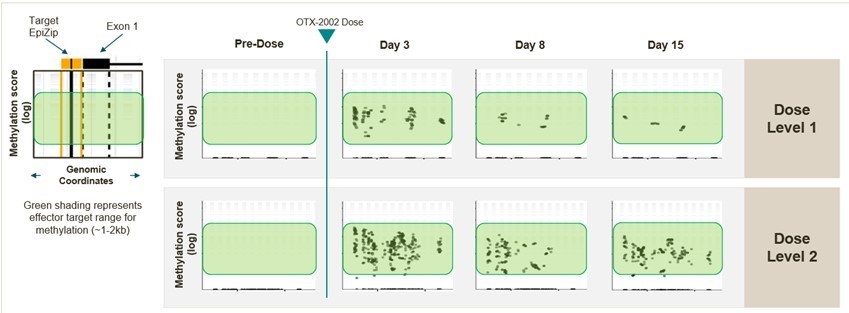
MYC mRNAの迅速かつ強力で持続的な低下が観察された

2024年1月、3人の肝癌患者において、用量レベル3(0.06 mg/kg)の用量制限毒性(DLT)ウィンドウを完成させることを発表した。2024年3月24日までのデータ締め切りは,11名の患者がOTX−2002 0.02 mg/kg(用量レベル1,n=4),0.05 mg/kg(用量レベル2,4)または0.06 mg/kg(用量レベル3,n=3)の静脈治療を受け,2週間に1回であった。
データカット日までの最初の3つのキューの中間データは、
19
OTX−2002:中期単一療法全体反応概要(2024年3月24日現在)
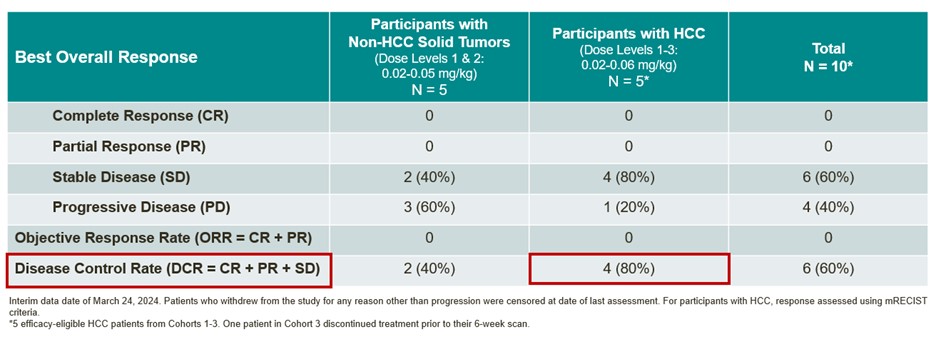
著者らは、これらの中期データはOmegaに臨床プラットフォーム検証を構築し、プログラム可能な候補mRNAを利用して制御されたエピゲノム制御の潜在力を強調し、そしてエピゲノムコントローラを次の革新療法として支持する潜在力を信じている。
これらの鼓舞的な中期データに基づいて、OTX-2002は単一治療用量のアップグレードにおいて引き続き進展を得ており、同社は現在0.12 mg/kgの用量レベルで第4グループの肝癌患者を評価している。キュー4は2024年3月初めに28日間の用量制限毒性窓口を完成した。同社では,2024年に単一療法用量増加に関する最新の臨床データをより多く報告する予定である。推奨投与量を決定した後,同社は2024年に単一療法と併用治療環境に拡張する予定である。
非小細胞肺癌治療のOTX−2101
2022年10月、非小細胞肺癌治療のINDイネーブル研究に入るための開発候補薬としてOTX-2101を選択することを発表しました. NSCLC腫瘍の約50%がc-Mycを過剰発現している。我々は,c-Mycを下方制御し,この過剰表現を減少させるためにOTX-2101を開発している.非小細胞肺癌は最もよく見られる肺癌タイプであり、すべての肺癌診断の84%を占め、2020年にアメリカでは約192,200例の新症例がある。非小細胞肺癌の5年生存率は24%であった。診断時の疾患段階によれば、現在の治療レジメンは、手術切除、光力学的治療(PDT)、レーザー治療または近距離放射線治療、化学療法、放射線治療、標的治療(例えば、TKIs)および免疫治療を他の治療と組み合わせることを含む。
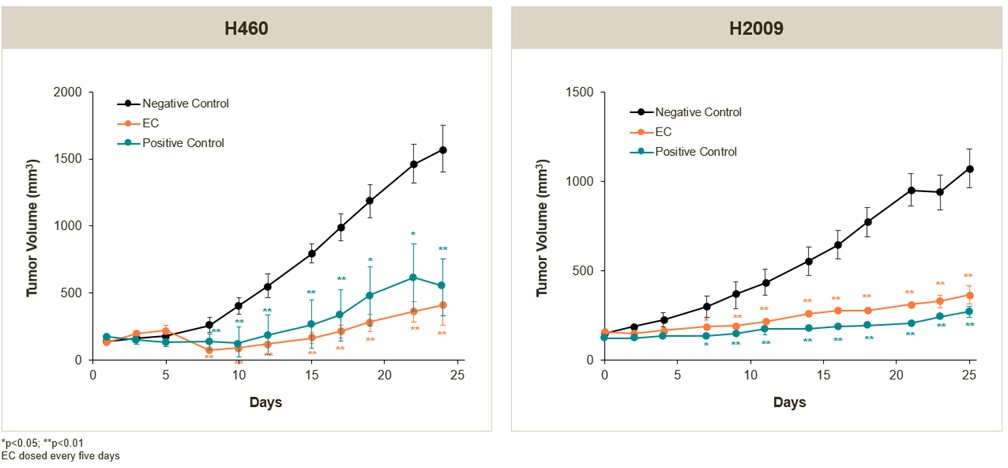
臨床前研究では,体外で一連の非小細胞肺癌細胞株に対して活性を示すEC候補が決定され,c−Myc低下に伴う細胞活力の喪失を示した。重要なことは,体外臨床前研究において,これらの候補ECの正常初代肺上皮細胞,線維芽細胞,内皮細胞に対する最小抗増殖作用が観察されたことである。また,ある臨床前研究では,臨床に関連するTKI併用治療の場合,EC候補患者は細胞増殖に対する相乗効果を示した(データは示されていない)。著者らはまた、2種類の非小細胞肺癌皮下移植モデルに対して臨床前研究を行った。これらの研究では,EC候補の3 mg/kgを5日ごとにマウスに投与した。われわれのMYC標的EC候補治療では,検討した用量段階で腫瘍サイズは統計的に有意に減少し,治療マウスの体重減少は認められなかった。この2つの研究で私たちのEC治療は
20
候補の腫瘍体積への影響は、以下の図に示すように、治療基準(陽性対照)、いくつかの癌を治療するための化学療法薬と同じである。
H 460およびH 2009非小細胞肺癌皮下移植モデルにおけるEC候補細胞の抗腫瘍活性(生体内)
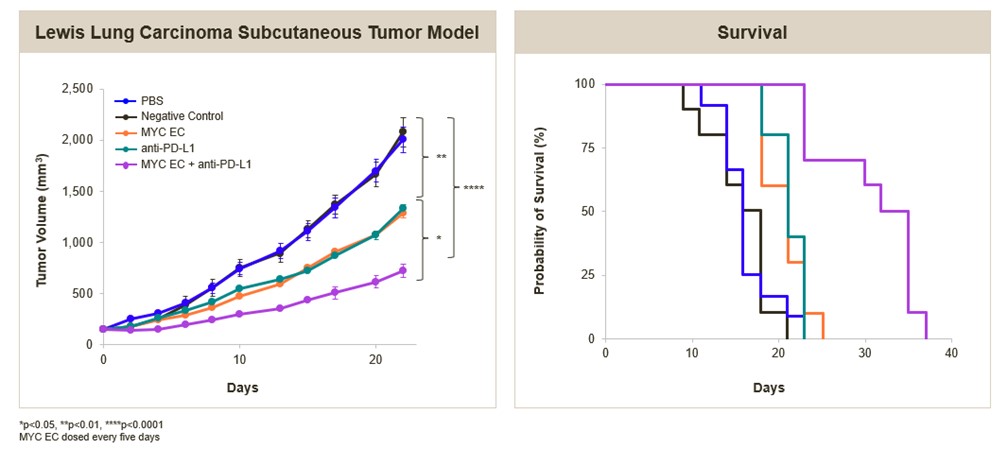
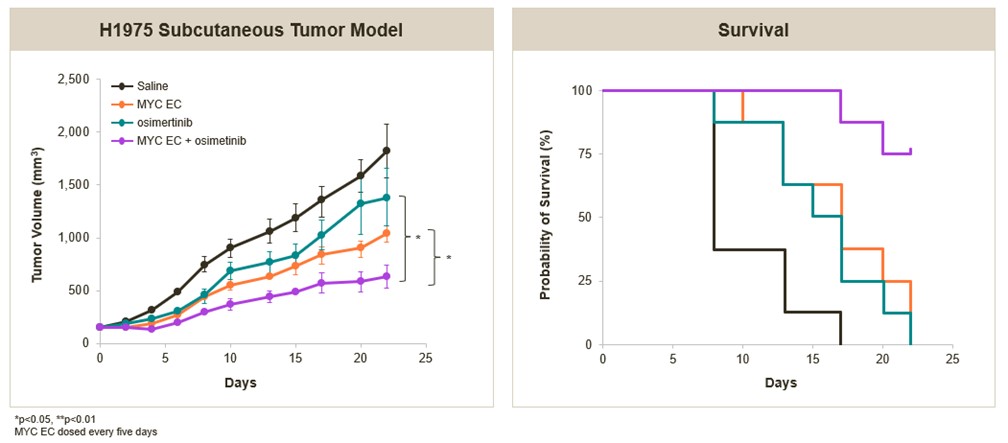
著者らはさらに、著者らのMYC標的ECの臨床前に免疫チェックポイント或いはEGFR阻害剤との併用設定を評価した。NSCLCをMYC−ECプラス抗PD−L 1抗体で治療した同遺伝子マウスモデルの抗腫瘍活性増強を以下に示す。また、非小細胞肺癌ヒト異種移植モデルにおいて、MYC-ECはEGFR阻害剤osimertinibと併用し、体外で協同して腫瘍細胞の活力を低下させ、腫瘍成長の抑制を著しく増強した。
MYC標的EC併用免疫検査点は同遺伝子マウスNSCLCモデルの抗腫瘍活性を顕著に増強した
21
MYC標的ECとEGFR阻害剤の顕著な結合
ヒト非小細胞肺癌移植腫瘍モデルの抗腫瘍活性の増強
全体的に、これらの発見はOTX-2101の末期非小細胞肺癌治療の潜在力をさらに支持した 単一療法や標準看護療法と組み合わせた環境とした。
免疫学を含む多遺伝子疾患
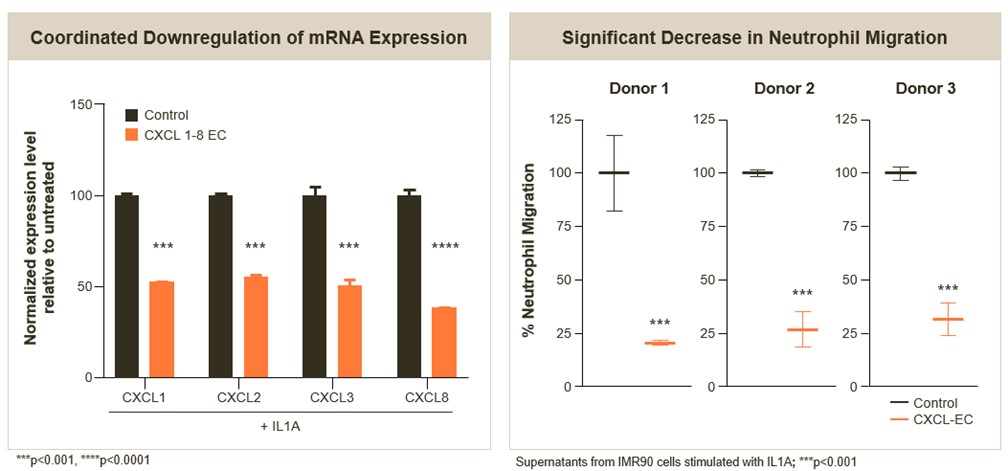
我々は、好中球喘息および急性呼吸窮迫症候群(ARDS)、皮膚疾患およびリウマチ適応、および腫瘍学などの炎症性肺疾患を含む、CXCL 1、2および3およびIL-8遺伝子クラスターの様々な潜在的適応における発現を減少させるためのEC候補レジメンを評価している。CXCL遺伝子クラスターの過剰発現はケモカインを産生し,好中球を吸引し,局所炎症を促進する。ARDSの症例では,炎症細胞を肺に募集するケモカインが疾患発症機序に重要な役割を果たしており,ARDS患者の肺細胞におけるCXCL 1,2,3とIL−8遺伝子クラスターの発現が増加している。ARDSは壊滅的な症候群であり、アメリカでの発病率は約20万で、死亡率は40%に近い。
ヒト単球CXCL 1−8候補ECに対する前研究では,投与24時間後にCXCL 1の遺伝子発現が65%低下し,CXCL 1,CXCL 2,CXCL 3,CXCL 8の遺伝子発現が対照群と比較して45%−60%の低下が認められた。体外培養したヒト肺線維芽細胞をCXCL−ECで処理したところ,培養上清は好中球遊走の有意な減少を示した。
22
ケモカイン遺伝子の多遺伝子IGD標的により好中球遊走が著しく減少した(体外)
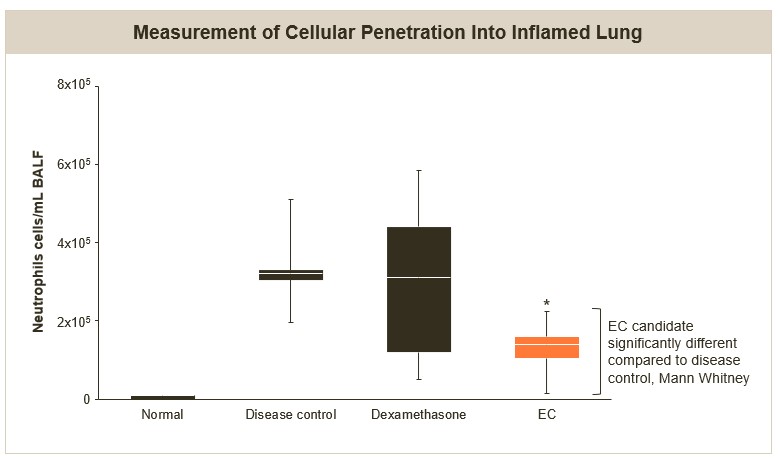
ARDS動物モデルの臨床前研究では,EC候補薬による治療後,肺好中球浸潤の著明な減少が認められた。炎症前2時間と炎症発生後8時間にそれぞれ3 mg/kgの候補EC起炎または10 mg/kgのデキサメタゾンを陽性対照とした。以下の図に示すように,疾患対照群と比較してEC候補治療72時間後にマウス気管支肺胞洗浄液(下図ではBALFと表記)中の好中球浸潤の56%の減少が認められ,炎症反応の重症度を測る指標である。
ARDSモデル好中球浸潤減少(体内にある)
* p
23
再生医学
肝再生
HNF 4の表現を増やすためにEC候補を開発しています転写主調節因子は、潜在的な方法として深刻な肝機能障害患者の肝細胞機能を回復する。HNF 4正常肝機能に重要な蛋白質、例えばビリルビン、アルブミンと代謝酵素の発現を制御することによって、肝細胞と肝臓中の他の細胞タイプの発育、分化と動態バランスを制御する。慢性肝疾患ではHNF 4低下しており肝不全の病態を引き起こしていますHNF 4の発現が増加していることがわかりました一部の肝細胞でも健康な肝機能を回復することができる。
2020年には慢性肝疾患と肝硬変が米国の主要な死亡原因であり,死亡者数は5万人を超えている。疾患の病因に応じて,治療選択にはコルチコステロイド,抗ウイルス薬,あるいは他の薬剤が含まれる可能性があり,最後の選択は肝移植である。2019年には,米国では1.3万人を超える人が肝臓移植を待っており,約12%が移植を受ける前に死亡している。
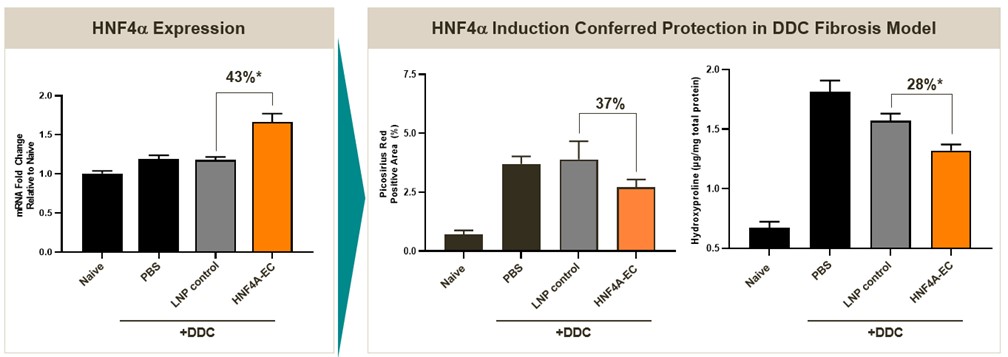
一次健康人肝細胞の臨床前研究では、単用量で著者らのEC候補薬物を使用することはHNF 4の持続的な増加を誘導することができる10日間と長く,CLDとESLD患者の肝細胞を機能状態に回復させ,肝機能を回復させるのに十分である可能性が考えられる。ECを介したhNF 4α発現上昇は,以下の図に示すように,臨床関連線維化遺伝子の体外での発現減少に関与していることが観察された。これらのデータは私たちのEC候補が提案した治療作用機序を支持する。
HNF 4α標的ECはHNF 4α発現を有意に上昇させ、
臨床前モデルにおける線維化減少の重要な指標
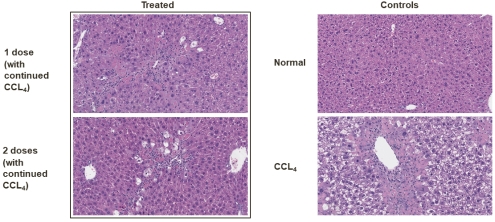
図に示すように体内にある臨床前マウス肝繊維化モデル、四塩化炭素処理を用いて肝細胞変性を誘導する(CCL標識4以下の図に示す).構築の代わりにEC候補マウスを用いた治療では,31日目と38日目に週1回投与しても2回投与しても肝細胞変性の発生率が有意に低下した。
24
EC候補マウスエージェント構築は肝臓組織学を改善した(体内にある)
私たちは現在追加の体外培養そして体内にある著者らはECの薬理、処方の最適化、治療効果と初歩的な安全性研究を候補とした。
データを翻訳する
著者らのEC候補の臨床翻訳の重要な要素の1つは、IGDに対して種を越えた遺伝子発現を調節できるEC候補を設計することである。前臨床研究ではHNF 4の変化を評価した非ヒト霊長類動物および移植され、我々のEC候補で処理されたマウス(下図ではFRG Mouseとラベル付けされている)のヒト肝組織および相同マウス標的配列に対するEC候補で処理された健康なマウスに発現される。以下の図に示すように,HNF 4の治療上の上昇が認められた対照群と比較して,マウスは246%,非ヒト霊長類は68%,FRGマウスは31%増加した。私たちは、私たちの行動メカニズムのこのような翻訳忠実度が、私たちのEC候補者と計画を引き続き発展させることを支持すると信じている。
EC候補は臨床前研究でHNF 4αの発現を増加させた(体内にある)

* p
25
データを渡す
わが社はメッセンジャーリボ核酸薬物の調合、送達、開発において豊富な内部専門知識を持っています。我々は現在,様々な内部および外部由来の一連のLNPと非LNPレシピを探索しており,具体的かつ有効であることを示す独自のレシピを開発している体内にある臨床前研究では,われわれのEC候補は次の図に示すように,多くの治療に関連する細胞や組織タイプに機能を提供している。我々の現在の送達製剤ライブラリは、肝臓(例えば、肝細胞、星細胞、Kupffer細胞)および肺(例えば、内皮、肺胞、上皮)を含む様々な組織および細胞タイプにアクセスすることができる。配信の勢いを利用して、私たちはまた、局所関節(例えば、滑膜層、軟骨細胞、免疫細胞)、中枢神経系(例えば、脊髄、脳)、および腫瘍(例えば、皮下、原位)を覆うために、私たちの能力を向上させている。このような伝達能力は一緒に私たちが私たちのチャンネルを発展させて拡大できるようにする。
内部開発独自LNPsの開発に努め,他の組織に拡張する
製造業
私たちは製造能力、生産能力と制御の発展は私たちの全体的な成功に重要であり、特に私たちが開発スケジュールを満たし、運営コストを制御し、私たちのプラットフォーム技術と候補製品のために知的財産権を生成し、保護する能力に重要であると考えている。だからこそ,臨床検証された製造·交付技術を選択し,ワクチン開発や遺伝子編集分野の様々な応用のために開発されている技術と類似した深い内部専門知識を持っている。したがって、私たちは私たち自身の経験と、以前と現在と同じモデルを使用して作られた技術改善と規制前例を利用することができる。
著者らの内部プロセスと分析開発組織はすでに十分な規模の製造プロセスを構築し、著者らの薬物物質と薬物製品の研究と早期臨床前開発需要を満たした。また,我々の薬物物質や薬品の製造に豊富な経験を持つ高技能第三者契約開発·製造組織(CDMO)を招聘し,現在の良好な製造規範(CGMP)に基づいて我々の製造プロセスを大規模に実施した。著者らはすでに第三者CDMOと製造サービスプロトコルを構築し、薬物物質と薬物製品を供給し、著者らの臨床前研究、IND毒理学研究と臨床試験に対する需要を満たす。今後数年は引き続き第三者CDMOに依存して薬品、薬品、製造品を供給することが予想される。
我々のすべての治療計画について,我々は外部協力と我々内部のLNP研究開発プラットフォームから最適なLNP交付オプションを評価した。例えば,我々の主導計画OTX−2002については,Acuitas Treateutics,Inc.やAcuitasからLNP技術の許可を得ている。Acuitasは広範なLNP知的財産権を有する会社であり,臨床用LNPの協力と開発に良好な記録がある。私たちは外部パートナーとの協力が重要なレシピと製造専門知識を提供すると信じています
26
CGMP標準下でのLNPメッセンジャーリボ核酸の策定過程のCDMOへの移行を促進する。私たちはまた私たちの候補製品を生産するためにより多くの経験豊富なCDMOを招いている。
我々の第三者CDMOと既存の内部施設を通じて、私たちは現在の研究、臨床前、臨床材料の需要を満たすのに十分な製造能力があると信じている。私たちは、現在外部に構築されている製造能力と、内部能力に加えて、私たちの今後数年間の期待需要を満たすことができると信じている。私たちは薬品と薬品生産の生産能力を監視し、CDMOとの供給協定及び新材料供給の納期は私たちの期待した需要を満たすために追加の生産能力を得ることができると信じています。また、私たちの製品は規模化生産ができ、生産と調達効率があり、これは商業競争力のあるコストにつながると信じています。
競争
スタートアップ段階のバイオテクノロジー会社として、製薬やバイオテクノロジー業界から様々な会社の競争に直面しています。この競争には小さな会社も含まれており、私たち自身よりも多くの財務や技術資源、より長い運営歴史を持つ大企業も含まれている。また、学術、政府、民間研究機関の知的財産権、技術、製品開発と競争するように努力しています。
私たちの競争相手は私たちよりもっと多くの財務資源、成熟した市場地位、研究開発、製造、臨床前と臨床テスト、監督管理の承認と精算及びマーケティング許可を得た製品に関する専門知識を持っているかもしれない。これらの競争相手はまた、合格した科学、販売、マーケティングと管理者を募集し、維持し、臨床試験場と臨床試験の患者登録を確立し、そして私たちの計画と相補的或いは必要な技術を獲得する上で私たちと競争している。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大企業と協力して手配すれば。
私たちが開発したすべての製品の成功に影響を与える重要な競争要素は、承認されれば、それらの有効性、安全性、利便性、価格、政府と他の第三者決済者が精算できるかどうかである可能性が高い。もし私たちの競争相手が私たちが開発する可能性のある任意の製品よりも効果的で、副作用が少なく、より深刻ではなく、より便利で、あるいはより安い製品を開発し、商業化すれば、私たちの任意の候補製品に対するビジネス機会は減少または消失する可能性がある。私たちの競争相手も私たちよりも早く彼らの製品のためにFDAや他の規制部門の承認を得て、私たちよりも早く製品を商業化することができるかもしれない。
CRISPR遺伝子編集,遺伝子療法,非コードRNA療法,小分子エピジェネティクスなど,遺伝子発現制御に焦点を当てた技術を開発する会社と競争したい。これらの会社には,Alnylam製薬会社,ビム治療会社,生物遺伝会社,CRISPR治療会社,Editas製薬会社,Ionis製薬会社,Intellia治療会社,ヤンソン製薬会社,ファイザー,Sangamo治療会社がある。
また,他の社ではエピゲノムコントローラや事前転写制御遺伝子発現を開発して肝癌や非小細胞肺癌を治療していることは知られていないが,アンチセンス阻害剤を開発しているIonis PharmPharmticals Inc.,アスリコン,Alnylam PharmPharmticals Inc./ゲーリー製薬やBio−Path Holdings,Inc.siRNA阻害剤のNitto Denko CorporationやSimaEconomics,Inc.がマイクロRNAシミュレーション療法のIntera TechnologB.V.を開発しており,マイクロRNAシミュレーション療法のIntera Technologies B.V.が開発されている。彼らは遺伝子治療法とMina治療有限会社を開発しており,同社は小型活性化RNA療法を開発している。
これらの技術、および小分子および生物学的製剤のような他の形態は、治療候補薬剤を開発するために使用される可能性があり、これらの候補薬剤は、私たちの現在および潜在的な将来の候補製品と競合するであろう。また,我々が開発したどのECsもその目標適応で既存の治療法と競合することが予想される。
知的財産権
27
私たちは私たちの知的財産権が戦略的資産であり、私たちに競争優位を提供する可能性があると信じている。我々は、内部開発でも第三者から許可を得ても、特許権の追求、保守、擁護、保全を含む、当社の業務に重要なビジネス的意義を有するノウハウ、発明、および改善に努めている。私たちの政策とやり方は、米国および米国以外の司法管轄区域に当社の独自技術(例えば、omegaプラットフォーム、ECs、交付および製造技術)、発明、改善、および候補製品に関連する特許出願を提出することを含む様々な方法で私たちの独自の地位を保護することであり、これらは私たちの業務の発展と実施に非常に重要である。私たちはまた、私たちのノウハウや候補製品に関するビジネス秘密やノウハウに依存しています。私たちはエピジェネティック薬物分野における私たちの特許地位を発展、強化、維持するために、革新と許可内の機会を求め続けている。また,データ独占性,市場独占性,特許期間の延長(あれば)に依存し,孤児薬物指定による規制保護を求め,依存することを計画している。私たちのビジネス成功は、私たちの技術、発明、および改善のために特許および他の固有保護を取得し、維持する能力があるかどうか、私たちの商業秘密を秘密にすること、第三者が所有する知的財産権を使用する私たちのライセンスを維持すること、私たちの特許を含む私たちの固有の権利を擁護し実行すること、および効果的かつ実行可能な特許および第三者の他の独自の権利を侵害することなく運営される可能性がある。
私たちの完全所有と許可された特許の組み合わせは、製造、交付、ECS、および私たちの治療計画を含むomegaプラットフォームの様々な側面をカバーしている。私たちの特許組合には私たちが開発している候補製品も含まれている。2023年12月31日まで、私たちの特許組合は42個の特許家族から構成されている。これらの家族のうち4つのアメリカ特許、2つのヨーロッパと日本での外国特許があり、これらの特許は2037年または2038年に満期になる。これらの特許シリーズはまた、オーストラリア、ブラジル、カナダ、中国、ユーラシア大陸、ヨーロッパ、香港、インド、日本、韓国、メキシコ、ニュージーランド、シンガポール、南アフリカおよび台湾における係属中の38件の米国特許出願(仮および非仮出願を含む)、94件の未解決外国特許出願、および国家段階に入っていない7つの所有または認可中の特許協力条約出願を含む。我々の特許組合せにおける係属中の特許出願によって発行されるか、または優先権を有すると主張する任意の米国または外国特許は、可能な特許期間の調整または延長を考慮することなく、2037年から2044年の間に満了し、すべての適切な保守、更新、年金、および他の政府費用が支払われると仮定する。私たちの目標は、私たちの独自技術(omegaプラットフォーム、ECs、交付および製造技術を含む)、発明、改善、および現在および未来の候補製品を保護するために、私たちの特許の組み合わせを拡大し続けることです。私たちの特許の組み合わせは現在、少なくとも私たちの候補製品をカバーするライセンス特許を含む。
私たちの知的財産権の組み合わせがカバーする製品と技術分野のより多くの詳細は以下の通りだ。
オメガプラットフォームに関する知的財産権
私たちの知的財産権の組み合わせには、ワイトヘード生物医学研究所(WEBR)と旗艦パイオニア革新会社または旗艦会社によって独占的または共同で独占的に許可されたomegaプラットフォームおよび交付技術のためのノウハウおよび特許権が含まれている。
我々omegaプラットフォーム技術の知的財産権の組み合わせは、ECsを使用する組成物および方法に対する特許権、IGDを標的とすることによって遺伝子発現を上方制御または下方制御する方法および組成物、エピジェネティック効果、物理的干渉物および遺伝修飾剤によるIGD調節遺伝子発現を標的とする組成物、およびIGDを識別および問い合わせる方法を含む。この製品の組み合わせは、私たちの既存の候補製品と、私たちが将来開発する可能性のある製品、そして私たちの目標または未来の可能な目標の兆候に広く関連しています。私たちのomegaプラットフォームに関連する知的財産権は私たちが持っている特許出願を含む。2023年12月31日現在、我々は、omegaプラットフォームに関連する標的遺伝子発現をエピジェネティックに調節するいくつかの方法を開示している2つの一時的な米国特許出願を有している。これらの係属中の出願(ある場合)が発行されるか、または優先権を有すると主張される特許は、特許期限の調整または延長を含まずに2044年に満了すると予想される。
私たちはまた旗艦製品から私たちのomegaプラットフォームに関連する特許と特許出願許可を取得した。2023年12月31日現在、オーストラリア、ブラジル、カナダ、中国、ユーラシア大陸、ヨーロッパ、香港、インド、日本、韓国、メキシコ、ニュージーランド、シンガポール、韓国、オーストラリア、ブラジル、カナダ、中国、ヨーロッパ、香港、インド、日本、韓国、メキシコ、ニュージーランド、シンガポール、韓国、オーストラリア、ブラジル、カナダ、中国、欧州、香港、インド、日本、韓国、メキシコ、ニュージーランド、シンガポール、韓国、メキシコ、ニュージーランド、シンガポール、韓国、オーストラリア、ブラジル、カナダ、中国、ヨーロッパ、香港、インド、日本、韓国、メキシコ、ニュージーランド、シンガポール、韓国
28
アフリカと台湾はomegaプラットフォームと関連がある。これらの係属中の出願(ある場合)に発行された特許またはそれに優先権を有すると主張する特許は、特許期間の調整または延長を含まず、2037年から2041年の間に満了すると予想される。
また,WIBRから我々のomegaプラットフォームに関する特許や特許出願許可を取得した.2023年12月31日現在、WIBRから米国特許、欧州および日本における2つの外国特許、4つの非一時的な米国特許出願、およびカナダ、中国、欧州、香港、日本およびメキシコにおける11件の外国特許出願を取得し、omegaプラットフォームに関連する標的遺伝子メチル化調節のいくつかの方法および組成を開示した。これらの係属中の出願(ある場合)が発行されるか、または優先権を有すると主張される特許は、特許期間の調整または延長を含まず、2037年または2038年に満了すると予想される。
上記の我々の特許権の記載には、非排他的ライセンス協定に従ってOmegaに付与された特許および特許アプリケーションのAcuitasが所有する権利、または日東電工社(Nitto Denko Corporation、またはNitto)によって所有され、候補薬剤の共同開発に限定される独占的ライセンス契約に従って我々に付与される特許および特許適用の権利は含まれない。
交付に関連する知的財産権
我々が提供する技術の特許的組み合わせは、LNP製剤、脂質分子および細胞透過性ポリペプチド組成物およびそれらの使用に対する特許出願を含む。私たちは、私たちの交付技術に関連するいくつかの特許出願を持っており、旗艦会社からの特定の特許出願の許可を持っている。2023年12月31日現在,我々は7つの米国仮特許出願,2つの非仮米国特許出願,10件のオーストラリア,カナダ,中国,ヨーロッパ,日本,韓国からの外国特許出願,および技術交付に関する2つのPCT特許出願を有している。これらの係属中の出願(ある場合)に発行された特許またはそれに優先権を有すると主張する特許は、特許期間の調整または延長を含まず、2041年から2044年の間に満了すると予想される。また、2023年12月31日現在、旗艦特許Oneから発行された米国特許と、技術交付に関連する非一時的な米国特許出願のライセンスを取得した。これらの係属中の出願(ある場合)が発行されるか、または優先権を有すると主張される特許は、特許期限の調整または延長を含まずに2037年に満了すると予想される。
疾患関連知的財産権
私たちの知的財産権の組み合わせにおける疾病に関連する特許権は、ある疾病と関連疾病状態に特化したECSにカバー範囲を提供した。私たちのLead計画の疾病関連特許出願は以下に記載されたものを含む。以下に説明する疾患に関連するすべての特許出願は、私たちが完全に所有するか、またはWIBRまたは旗艦会社によって独占的または共同独占的に許可される。
MYC
我々のOTX−2002計画はc−Mycファミリー癌遺伝子を対象としている。C−Mycを下方制御する内皮細胞を開発し,肝癌の治療に用いた。非小細胞肺癌の治療のためにc-Myc発現を減少させる計画も設計された。2023年12月31日現在,c−Myc関連癌の治療方法に関する3つの仮米国特許出願を有している。これらの係属中の特許出願よりも優先される特許が2044年に満了することを要求し,いかなる特許期間の調整や延長も含まないことを予想している。2023年12月31日現在、旗艦会社から米国仮特許出願、オーストラリア、ブラジル、カナダ、中国、ユーラシア大陸、ヨーロッパ、インド、日本、メキシコ、ニュージーランド、シンガポール、南アフリカ、台湾の17件の外国特許出願、およびEC物質の組成、c−Myc関連癌の治療法およびc−Myc発現の調節方法に関する3つのPCT出願を取得した。これらの未解決特許出願(ある場合)に発行された特許またはそれに優先権を有すると主張する特許は、特許期間の調整または延長を含まず、2041年から2044年の間に満了すると予想される。
CXCL 1、2、3、IL-8
CXCL 1,2,3,IL−8遺伝子クラスターの発現を減少させるためのEC候補を開発している。この計画は関節リウマチ、痛風、好中球喘息とARDSを含む広範な炎症性疾患においてケモカインの発現を減少させることを目的としている。2023年12月31日まで、台湾でフラッグシップPCT特許出願と外国出願の許可を得た
29
CXCL 1−3/IL−8免疫グロブリンに対するEC組成物、及び関節リウマチを含む炎症性疾患の治療方法。これらの未解決の特許出願から優先権を有する特許を取得または主張することを期待している 2043年までには、特許期間の調整や延長は含まれていない。
HNF 4a
私たちの肝臓再生計画は主な転写制御因子HNF 4を対象としていますaそれは.HNF 4の発現を増加させるEC候補遺伝子を開発しましたa重症肝機能障害患者の肝細胞機能を回復する。2023年12月31日現在、私たちはオーストラリア、カナダ、中国、ヨーロッパ、香港と日本に1つのアメリカ非臨時特許出願と6つの外国特許出願を持っており、EC物質成分と肝臓病の治療方法に関連している。これらの未解決特許出願(ある場合)に発行された特許またはそれに優先権を有すると主張する特許は、特許期限の調整または延長を含まずに2040年に満了すると予想される。
他の疫病地区
我々は、上述した疾患計画に加えて、遺伝子発現の上昇または下方制御から利益を得る新たなEC成分および追加疾患の治療のための特許出願を有している。
2023年12月31日現在、我々はオーストラリア、カナダ、中国、ヨーロッパ、香港、および日本に非一時的な米国特許出願および6つの外国特許出願を有しており、神経疾患の組成物および治療方法に関する。これらの係属中の出願(ある場合)が発行されるか、または優先権を有すると主張される特許は、特許期限の調整または延長を含まずに2040年に満了すると予想される。
2023年12月31日現在、私たちはオーストラリア、カナダ、中国、ヨーロッパ、香港、日本に非一時的なアメリカ特許出願と6つの外国特許出願を持っており、代謝障害の成分と治療法に関連している。これらの係属中の出願(ある場合)が発行されるか、または優先権を有すると主張される特許は、特許期限の調整または延長を含まずに2040年に満了すると予想される。
2023年12月31日現在、我々は、標的遺伝子の発現を調節することによって代謝障害を治療する成分および方法に関する一時的な米国特許出願を有している。この保留出願から発行または優先権を有する特許がある場合(ある場合)は,特許期限の調整や延長を含まず2044年に満了すると予想される。
2023年12月31日現在、我々は、標的遺伝子の発現を調節することによる癌の治療成分および方法に関する3つの臨時米国特許出願を有している。私たちは、これらの保留出願に優先すると主張する任意の特許が2044年に満了し、いかなる特許期間の調整または延長も含まないと予想する。
我々は、2023年12月31日現在、オーストラリア、カナダ、中国、ヨーロッパ、香港、および日本において、炎症性疾患の組成物および治療方法に関する非一時的な米国特許出願および6つの外国特許出願を有している。これらの係属中の出願(ある場合)が発行されるか、または優先権を有すると主張される特許は、特許期限の調整または延長を含まずに2041年に満了すると予想される。
2023年12月31日現在、脱毛組成物および治療法に関するPCT特許出願がある。私たちは、この保留出願よりも優先的であると主張する特許があれば、いかなる特許期限の調整や延長も含まず、2042年に満了すると予想する。
2023年12月31日まで、私たちは肝疾患治療組成物と方法に対する臨時アメリカ特許出願を持っている。私たちは、この保留出願よりも優先的であると主張する特許があれば、いかなる特許期限の調整や延長も含まず、2044年に満了すると予想する。
我々は,新たなプラットフォーム技術や候補製品を開発するとともに,我々の知的財産権戦略を評価·整備し,より多くの特許出願を提出する予定である.
30
許可協定
私たちはライセンス契約の側であり、これらの合意に基づいて、私たちは第三者から特許、特許出願、その他の知的財産権を許可します。許可された知的財産権は、IGDを標的とすることによって遺伝子発現を調節する方法および組成物を少なくとも部分的にカバーする。このような許可証は私たちに様々な勤勉さと財政的支払い義務を課している。私たちは未来にこのような種類の許可協定を締結し続けると予想する。私たちは次の許可協定が私たちの業務に必須的だと思う。
旗艦製品とライセンス契約を結ぶ
2019年3月に、吾らは旗艦会社と合意または旗艦協定を締結し、これにより、吾ら(I)は“当社設立”前に構想したいくつかの基礎知的財産権のすべての権利、所有権及び権益を旗艦会社に撤回及び無条件に履行することができず、吾等のBシリーズ融資或いは吾などの最高経営責任者(当該等の基礎知的財産権)が初日に雇用された(より早い者を基準とする)及び(Ii)このような基礎知的財産権の項目の下で旗艦会社から独占、グローバル、印税、再許可及び譲渡可能な許可を取得し、開発、製造及び商業化の任意の製品又はプロセス又はそのコンポーネント、開発、製造及び商業化のいかなる製品又は商業化プロセス、開発、商業化及びその製品又は商業化、いかなる製品、商業化、いかなる製品又は商業化、いかなる製品、商業化、製品又は商業化、いかなる製品、商業化、製品又は商業化、いかなる製品、商業化、製造及び商業化、いかなる製品又は商業化、製造及び商業化、いかなる製品又は商業化、製品又は商業化その製造および商業化は、フラッグシップ協定に従って治療分野で付与された許可ではなく、フラッグシッププロトコルの有効期間内に少なくとも1つの基礎知的財産権の有効な主張を侵害するであろう。さらに、旗艦会社は、以下の発明の特許を有すると主張するすべての権利、所有権および権益を撤回および無条件に私たちに譲渡することができない:(I)旗艦パイオニア会社または旗艦管理会社によって単独で構想されるか、または旗艦管理会社が私たちと共同で構想すること、(Ii)“当社設立後”、および(Iii)旗艦管理会社とのいくつかの管理協定または管理協定に基づいて行われる活動、または旗艦管理会社が私たちの事務に他の参加をするが、基礎知的財産権は含まれていない。基礎的IPは、特にomegaプラットフォームのためのものであり、標的IGDによって遺伝子発現を調節する一般的な方法および組成物(例えば、EC)と、MYC、CXCL 1、CXCL 2、CXCL 3およびIL-8のうちの1つまたは複数に関連する疾患などの様々な疾患を治療するための特定の標的に対する特定の組成物および方法とを含む。MYC、CXCL 1、CXCL 2、CXCL 3、およびIL-8のうちの1つまたは複数を調節するための治療計画を含む、我々のomegaプラットフォームおよび私たちの候補治療製品において旗艦プロトコルを使用して付与された権利。基本IPは2023年12月31日までに2037年から2043年までの間に満了する予定です。基礎知的財産権を付与する許可は私たちが旗艦協定の下で義務を遵守することにかかっている。旗艦協定の下での義務には、ライセンス製品の純売上高の特許権使用料を含む、ライセンス製品の開発と商業化に必要な支払いを商業的に合理的に利用することが含まれている。フラッグシップ協定によると、ライセンス製品と司法管轄エリアでライセンス製品と司法管轄エリアにライセンス製品の純売上高の1桁パーセントで計算された使用料をフラッグシップ製品に支払う責任があります。我々は単独で基礎IP開発に基づく任意の候補製品の臨床開発を担当している。フラッグシップ協定によると、旗艦会社は非商業研究と開発目的だけで治療学領域で基礎的知的財産権を実施する権利を保留し、管理プロトコルの下での職責を履行する。
フラッグシッププロトコルは、ライセンス製品の任意の基礎知的財産権をカバーする最後の有効クレームが満了したときに終了する最後の満了許可使用料期限で終了し、この期限は、各ライセンス製品および各司法管轄区域のライセンス製品に基づいて終了する。任意の管轄区域の許可製品の許可使用料の期限が満了し、フラッグシップ協定項の下で当該許可製品のすべての借金を全額支払った後、私たちに付与された許可は、自動的に当該司法管轄区域内の当該許可製品の非独占的全額支払い許可に変換される。便宜上、私たちは60日の書面通知後にフラッグシップ協定のすべての内容を終了する権利があります。いずれも書面通知を受けてから30日以内に是正されなかった重大な違約行為は、旗艦協定を終了することができる。また、旗艦協定で付与された権利に関連する業務の経営を停止した場合、フラッグシップは、(I)30日以内に書面通知を出して終了することができ、(Ii)破産事件に遭遇した場合は、書面通知の下で終了するか、または(Iii)任意の基礎知的財産権の有効性、特許性または実行可能性または参加可能性に疑問がある場合は、書面通知後直ちに終了することができる。旗艦が許可領域内の特固定子分野で許可製品を開発して商業化するために商業的に合理的な努力をしていないと判断した場合、フラッグシップは、事前に書面で通知された場合には、そのサブ分野における許可製品の許可を終了する権利がある。しかし、この場合、旗艦会社が書面による開発·商業化計画を承認した場合には、このようなライセンス製品やサブ分野の許可を保留することができる。
31
WIBRと独占と共同独占許可協定を締結する
2019年5月、WIBRと独占ライセンス契約、またはWIBR独占契約を締結しました。WIBR独占協定によれば、WIBRが所有または制御するいくつかの特許権の下で、独占的、世界的に、印税あり、再許可可能な許可を取得し、研究、製造、製造、使用、販売、販売、レンタルおよび輸入製品の提供、および人間および動物治療および診断の分野で行われ、許可されたプロセスを研究することができる。WIBR独占プロトコル下のライセンス特許は、主にIGDにおける遺伝子発現を調節する方法および組成物を対象としている。
2019年5月には、WIBRと共同独占ライセンス契約、すなわちWIBR共同独占合意も締結した。WIBR共同独占協定によれば、WIBRが所有または制御するいくつかの特許権の項下のグローバル共同独占、印税、再許可可能な許可を取得し、研究、製造、製造、使用、販売、販売、レンタルおよび輸入製品、および人間および動物治療および診断の分野で行われ、許可されたプロセスを取得することができる。WIBRと共通独占ライセンス者との間の共通独占ライセンスプロトコルが任意の理由でいつでも終了すれば,WIBR共通独占プロトコルでの共通独占権利は独占となる.WIBR共通独占プロトコル下のライセンス特許は、主に、IGDを標的とすることによって遺伝子発現を調節する方法および組成物を対象としている。WIBR独占プロトコルとWEBR共通独占プロトコルを総称してWEBRプロトコルと呼ぶ.
WIBRプロトコルによれば、WEBRは、研究、教育、および他の教育目的(第三者支援のための研究を含む)のために許可特許を使用する権利を保持し、非商業研究、教育、および他の教育目的のみのための非独占的許可を他の学術および非営利研究機関に付与する。
WIBR協定に基づいて付与されたライセンスは、米国政府が保有しているいくつかの以前に存在する権利によって制限される。適用法によると、米国政府は連邦研究助成によって生成されたライセンス特許に関するいくつかの権利を保持している。WIBRプロトコルに従って我々に付与される許可は、WIBRと特定のスポンサー研究契約またはSRAを締結した特定の第三者が所有するいくつかの以前に存在する権利にも制限される。SRAによれば、SRAの下で生成されたいくつかの発明またはSRA発明がライセンス特許によって支配されている場合、WIBRは第三者を起訴しないことを約束しているので、SRA発明をカバーするWIBRから許可を得た第三者の特許権主張から除外される。さらに、“WIBR独占協定”が発効してから5年後、WIBRまたは我々が第三者がライセンス特許権に基づいて提出した再許可請求を受けた場合、製造、製造、使用、販売、要約販売または輸入と当時約販売される許可製品または許可プロセスまたは私たちまたはその代表による誠実な研究または開発が直接競争しない製品またはプロセスを提供する場合には、(I)第三者が提案した分野および提案された製品に限定された非排他的二次許可を付与するために誠意を持たなければならない、または(Ii)私たちが選択したとき、WIBRが提案した製品の開発を承認するための計画を提出し、この計画の承認は無理に差し押さえてはならない。
WIBR独占プロトコルにより,中間5桁の年間ライセンス保守費をWEBRに支払う必要がある.WIBRはまた、上位3種類の許可された製品(バックアップ製品を含まない)のために合計170万ドルに達する潜在的な臨床と規制マイルストーンを獲得する権利がある。WIBRは,我々,我々の付属会社または我々の分被許可者が販売しているライセンス製品について,各国特許権が満期または放棄されるまで,ライセンス製品の純売上高から1桁パーセントの印税を得る権利がある.特定の国/地域の許可製品について、私たちはこれらの印税のいくつかの慣行の減免と補償を受ける権利がある。WIBR独占合意に基づいて我々が製品を開発または商業化する権利を再許可する権利があれば,WIBRには我々の再許可から我々の再許可から得られた一定割合の非印税支払いを得る権利があり,ゼロから低い2桁まで様々であり,この再許可を実行する際に製品の開発段階を許可することに依存する.
WIBR独占合意は、事前に終了しない限り、すべてのライセンス特許権が満了または放棄されるまで有効である。WIBRに書面で通知した後、私たちの都合の良い時にWIBR独占プロトコルを終了することができます。どちらか一方が治癒していない実質的な違約によりWIBR独占合意を終了することができる。オメガが事業を停止すれば,WEBRもWIBR独占合意を終了する可能性がある。WIBR独占協定によると,最終満期の特許が発表されれば,2038年に満期になる予定である。
32
WIBR共同独占プロトコルにより,WIBRに中5桁まで低い年間ライセンス保守費を支払う必要がある.WIBRはまた、上位3種類の許可製品(バックアップ製品を含まない)のために合計190万ドルに達する潜在的な臨床、監督管理、再許可マイルストーンを獲得する権利がある。WIBRは、当社、当社の付属会社、または当社の分割ライセンシーが販売しているライセンス製品について、ライセンス製品の純売上高に応じて一桁パーセント未満の印税を徴収し、各国特許権が満期または放棄されるまで、ライセンスサービス収入からより低い一桁パーセントの印税を得る権利があります。特定の国/地域の許可製品について、私たちはこれらの印税のいくつかの慣行の減免と補償を受ける権利がある。WIBR共同独占プロトコルに従って我々が開発または商業化したライセンス製品の権利を再許可する場合,WIBRは再許可者にライセンス特許下の権利を付与する権利を付与する権利を有しており,そのような再ライセンスプロトコルの各々は5桁の年俸中桁を獲得する権利がある.
WIBR共通独占合意は、事前に終了しない限り、すべてのライセンス特許権が満了または放棄されるまで有効である。WIBRに書面で通知した後,我々の都合の良いときにWEBR共通独占プロトコルを終了することができる.どちらか一方が治癒していない実質的な違約によりWEBR共通排他性プロトコルを終了することができる.我々が我々の業務を停止すれば,WEBRもWIBR共同独占プロトコルを終了することができる.WIBR共同独占協定によると,最終満期の特許が発表されれば,2037年に満期になる予定である。
2023年12月31日と2022年12月31日までの年間で、ライセンス維持費とマイルストーン支払いを含め、それぞれ20万ドルと20万ドル未満の費用が発生した。
Acuitasとの合意
開発と選択プロトコル
2020年10月、私たちはAcuitasと開発とオプション協定、またはAcuitasオプション合意に到達した。Acuitasオプションプロトコルにより,われわれの遺伝子調節療法とAcuitasのLNPsを組み合わせたいくつかの製品を共同開発することに同意した。双方はそのノウハウの下で他方に共同研究を行うグローバル非排他性·免版税許可を付与した。我々はAcuitasオプションプロトコルでの作業計画に基づいてAcuitasの人員費用と研究所で発生した外部費用を支払う.Acuitasオプションプロトコルによれば、Acuitasは、LNP技術またはAcuitas LNP技術に関連する特許権および独自技術に基づいて、2つの指定された標的(例えば、EC構築物)または標的を保持することに関連する非排他的、世界的、再許可可能な許可を得て、保持標的のmRNAをコードすることを含む1つまたは複数の治療製品を開発および商業化するために、選択権を付与する。各オプションおよび予約ターゲットについて、年間の技術的アクセス料およびターゲット予約および保守費を支払う義務があり、合計金額は6桁であり、これらの予約ターゲットが予約ターゲットリストから削除されるまで、または予約ターゲットに対して選択権を行使するまで、または予約ターゲットに対して選択権を行使する。第1のオプションを行使する際には、最初の非排他的ライセンスを実行した後に150万ドルのオプション行権料を支払う必要がある。第2のオプションを行使する際には、第2の非排他的ライセンスを実行した後に175万ドルのオプション行権料を支払う必要がある。2023年及び2022年12月31日までの年度内に、吾らはAcuitasオプション協定によりそれぞれ総支出40万ドル及び190万ドルを発生し、技術アクセス費、目標予約及び維持費、Acuitasサービス提供コスト、材料コスト及び償還可能コストを含む。
以前に終了しない限り、Acuitasオプション協定は、(1)2つのオプションが行使され、(2)発効日から3年になるまで有効であるが、私らは、3年間の期間をさらに2年間延長することを選択することができる。一方は、他方の治癒されていない重大な違約によって、または他方が破産または同様の事件の場合にAcuitasオプション合意を終了することができる。書面でAcuitasに通知した後、私たちは私たちの都合の良い時にAcuitasオプション合意を終わらせることができる。Acuitasオプション協定によると,最終満期の特許が発行されれば,2041年に満了する予定である。
許可協定
2021年3月、私たちはAcuitasオプション協定の下で最初のオプションを行使し、Acuitasと非独占ライセンス契約、またはAcuitasライセンス契約を締結しました。Acuitasライセンス協定に署名することと関連して、私たちは150万ドルのオプション行使費用を発生させた。AcuitasはAcuitas LNP技術に基づいて,我々のOTX−2002遺伝子調節からなる製品の研究,開発,製造,商業開発のための非独占的で世界的に許可可能なライセンスを付与してくれた
33
治療学と鍼灸のLNPs。Acuitasライセンス協定によると,最終満期の特許が発行されれば,2041年に満了する予定である。Acuitasライセンス協定によると、私たちは特定の発展マイルストーンに達するまで、Acuitasに6桁までの年間ライセンス維持費を支払う必要がある。Acuitasは合計1800万ドルに達する潜在的な臨床、規制、商業マイルストーン支払いを得る権利がある。Acuitasは、当社、私たちの付属会社、または当社の再許可者が販売する各ライセンス製品について、特定の国/地域におけるライセンス製品の純売上高に応じて、その国/地域で以下の最後の状況が発生するまで、低いビット数パーセントの印税を請求する権利がある:(I)ライセンス製品に関するすべてのライセンス特許権の満了または放棄、(Ii)ライセンス製品の任意の規制排他的満了、または(Iii)ライセンス製品が初めて商業販売されてから10年、使用料期限が可能かもしれない。ライセンス特許カバーライセンス製品がない場合、またはLNP技術に関連する第三者特許を取得することが要求された場合、所与の国/地域の各ライセンス製品に対して一定の印税減免および補償を受ける権利がある。
事前に終了しない限り、Acuitasライセンス契約は最後に満了した印税期限が満了するまで有効になるだろう。どちらか一方が破産したり、類似した事件が発生した場合には、他方が治癒していない重大な違約行為によりAcuitas許可プロトコルを終了することができる。書面でAcuitasに通知した後、私たちは簡単な時にAcuitas許可協定を終了することができる。
日立との連携とライセンス契約
2022年10月,吾らはNittoと協力及び許可協定(“Nittoプロトコル”)を締結し,この合意(その中を含む)により,NittoはそのLNP交付技術に関するNittoが所有または制御するすべての知的財産権(“NittoライセンスIP”)下の独占,グローバル,印税,完全譲渡および完全再許可可能な許可を吾らに付与した。
日東協定の条項によると、私たちは日東に100万ドルの前払い現金を支払った。具体的な開発、規制、販売マイルストーンの実現状況に応じて、将来的に8300万ドルまで日東に支払う必要がある。ライセンス製品の純売上高に応じて,特定の場合には国/地域ごとにNittoに1桁の等級,1桁パーセントの印税を支払う義務がある。
私たちが事前に終了しない限り、製品がその国の日東にさらなる印税を支払うことを許可していない場合、日東協定は国ごとに満期になる。国·地域ライセンス製品の適用印税期限が満了した後,そのライセンスは,当該国/地域のライセンス製品の全額,印税免除,永久的かつ取消不可の許可となる。いずれか一方が治癒せずに日東協定に実質的に違反した場合に日東協定を終了し、他方の破産、債務不履行、または何らかの類似の事件が発生した場合、吾等はいつでもいかなる理由でもいかなる理由でも日東協定を終了することができる。2023年12月31日までの年間で、材料コスト、日東提供サービスのコスト、精算可能コストを含む90万ドルの研究開発費を記録した。
ノボノルド社と研究協力合意に達しました
2023年12月31日,我々はノとノッドA/S(“ノとノッド”),パイオニア薬業08,Inc.(“PM SpinCo”)およびパイオニア薬業(NN),有限責任会社(“株主”)およびPM(NN)探索会社(“PMCo NN”およびPM SpinCoおよび株主,“PM実体”)と研究協力協定(“Novo RCA”)を締結し,新研究協力協定に掲載されているいくつかの条項について旗艦先鋒薬業(“旗艦”)の関連会社パイオニア薬業(NN),有限責任会社(“株主”)およびPM(NN)探索会社(“PM実体”)と連携協定を締結した。Novo RCAの条項によると、私たちはノとノドに独占的、印税あり、譲渡可能な許可を付与し、PM実体と一致した研究開発計画の下で研究と開発活動を行う権利があり、この計画は製品候補または計画目標に関連し、糖尿病を含む世界各地の人類の心臓代謝性疾患の予防、治療または制御のために使用される。Novo RCAの実行では,Novo Nordiskから510万ドルの払戻不能前金を受け取り,2027年までに約2160万ドルのコスト補償を受け,関連する研究開発活動を支援する予定である。ノおよびノドは、ライセンス製品および国/地域におけるライセンス使用料の支払い義務は、ライセンス製品が国/地域で初めて商業販売され、ライセンス製品に適用されるいくつかの有効な特許権利が、国/地域の最後の満了、およびその国/地域の規制の排他的満了後遅くとも10年以内に失効するが、以下の条件によって制限される
34
Novo RCAに規定されている特定の特許権使用料が低減され、段階的に減少する条項。詳しくは、本年度報告書の最後の総合財務諸表付記内の付記11--協力協定を参照されたい。
政府の監督管理
私たちは広範囲な規制を受けている。私たちは私たちの候補製品が生物製品として規制されると予想する。生物製品は“連邦食品、薬品と化粧品法”、“公衆衛生サービス法”、“PHS法”及びその他の連邦、州、地方と外国法規の監督管理を受けている。その他の事項以外に、“食品と薬物規制法”と“小霊通法案”及びその相応の法規はすべて生物製品の検査、製造、安全、効果、ラベル、包装、貯蔵、記録保存、流通、報告、広告及びその他の販売促進活動を管理する。
アメリカの生物製品開発プロセス
FDAが米国でバイオ医薬品を発売する前に必要なプログラムには、一般に以下のような態様が含まれる
人体で任意の候補生物製品をテストする前に、候補製品は臨床前テスト段階に入る。臨床前試験は、非臨床研究とも呼ばれ、製品の化学、毒性と調合に対する実験室評価、及び候補製品の潜在的安全性と活性を評価する動物研究を含む。臨床前試験の進行はGLPを含む連邦法規と要求に適合しなければならない。
臨床研究スポンサーは臨床前試験の結果を生産情報、分析データ、任意の利用可能な臨床データ或いは文献及び提案された臨床方案と共にFDAに提出し、INDの一部としなければならない。IND提出後も,いくつかの臨床前試験が継続される可能性がある。INDはFDAが30日以内に臨床研究を保留しない限り、FDAが受領してから30日後に自動的に有効になる。この場合,INDスポンサーやFDAは臨床研究が開始される前に未解決の問題を解決しなければならない。FDAはまた、臨床試験の前または期間のいつでも、安全考慮または規定に適合しない理由で、候補生物製品に臨床的制限を加えることができる。FDAが臨床一時停止を強制した場合、試験はFDA許可なしに再開されず、その後、FDA許可の条件下でのみ再開される可能性がある。
IND提出プログラムのほかに、ヒト遺伝子転移研究を含む組換えまたは合成核酸分子を含む細胞臨床試験のスポンサーも評価されなければならない
35
機構生物安全委員会またはIBCによって評価され、IBCは地方機関委員会であり、米国国立衛生研究院が組換えまたは合成核酸分子に関する研究ガイドラインまたはNIHガイドラインに基づいて、組換えまたは合成核酸分子を使用した研究を審査および監視する。IBCは、研究の安全性を評価し、公衆の健康または環境に対する任意の潜在的リスクを決定し、このような審査は、臨床試験開始前のいくつかの遅延をもたらす可能性がある。NIHガイドラインは強制的ではないが,関連研究がNIH組換えや合成核酸分子研究助成を受けた機関で行われているか,あるいはその助成によって行われていない限り,多くの会社や他のNIHガイドラインに拘束されていない機関は自発的にこれらのガイドラインに従っている。
臨床試験は,合格調査者の監督の下で,候補生物製品を健康ボランティアや患者に服用することに関連しており,これらの調査者は通常研究スポンサーに雇用されたりコントロールされていない医師である。臨床試験は,いくつかの有害事象が発生したときに臨床研究が停止されることを確保する停止ルールを含む,臨床研究の目標,投与手順,被験者の選択と排除基準,および被験者の安全性を監視するためのパラメータを詳細に説明するプロトコルで行われる。各スキームおよびスキームの任意の修正は、INDの一部としてFDAに提出されなければならない。臨床試験は,すべての研究対象にインフォームドコンセントを要求することを含むFDAのGCP要求を含む規定に基づいて行われなければならない。さらに、各臨床研究は、臨床研究を行う各機関に位置するか、またはサービスする独立した機関審査委員会またはIRBによって審査および承認されなければならない。IRBは研究参加者の福祉や権利の保障を担当し,臨床試験に参加する個人のリスクが最低に低下するかどうか,期待利益と比較して合理的かどうかなどの項目を考慮している。IRBはまた、各臨床研究対象またはその法律代表によって署名されなければならないインフォームドコンセントの形態および内容を承認し、完成まで臨床研究を監視しなければならない。
人体臨床試験は通常3つの連続段階に分けて行われ、これらの段階は重複或いは合併する可能性がある
承認後の臨床試験は,4期臨床試験と呼ばれることがあり,最初の上場承認後に行われる可能性がある。これらの臨床試験は,治療適応が予想される患者の治療から追加的な経験を得るためのものであり,特に長期安全なフォローアップのためである。
臨床開発のすべての段階において、監督管理機関はすべての臨床活動、臨床データと臨床研究調査人員に対して広範なモニタリングと監査を行うことを要求している。臨床試験結果を詳細に説明する年次進展報告はFDAに提出しなければならない。書面のIND安全報告書は、深刻かつ意外な有害事象、他の試験の任意の発見、実験室動物試験または体外培養実験により、人類被験者に対して重大なリスクがあり、或いは方案或いは研究者マニュアルに記載された試験と比べ、深刻な不良反応の発生率は臨床上重要な意義がある。スポンサーは15日以内にINDセキュリティ報告書を提出し,スポンサーがその情報有資格報告を確定した後でなければならない。スポンサーはまた、スポンサーが初めて情報を受け取ってから7日以内に、任意の意外、致命的、あるいは生命に危害を及ぼす疑いのある副作用をFDAに通知しなければならない。FDAまたはスポンサーまたはそのデータ安全監視委員会は、研究対象または患者が受け入れられない健康リスクに直面していることを発見することを含む、随時様々な理由で臨床研究を一時停止することができる。同様に、1つの臨床研究がIRBの要求に従って行われない場合、または候補生物製品が患者の予期しない深刻な傷害に関連している場合、IRBは、その機関の臨床研究の承認を一時停止または終了することができる。
36
行っている臨床試験や完成した臨床試験結果を公的登録機関に報告することも求められている。FDA規制製品(生物製品を含む)の臨床試験スポンサーは、www.Clinicaltrials.gov上で公開して得ることができるいくつかの臨床試験情報を登録し、開示しなければならない。製品、患者集団、調査段階、研究場所と研究者、および臨床試験の他の方面に関する情報は、その後、登録の一部として公開される。スポンサーも完成後に彼らの臨床試験結果を検討する義務がある。これらの試験結果の開示は,研究中の新製品や新適応が承認された後に延期することができる。
臨床試験と同時に、会社は通常追加の動物試験を完成し、候補生物製品の物理的特徴に関する追加情報を開発し、GMP要求に基づいて商業大量生産製品のプロセスを最終的に決定しなければならない。PHS法案では,生物製品を用いた外来製剤導入のリスク低減を支援するために,属性が正確に定義できない製品の製造制御の重要性を強調している。製造過程は一貫して高品質の候補製品ロットを生産することができなければならず、他の以外に、スポンサーは最終生物製品の特性、強度、品質、効力と純度をテストする方法を開発しなければならない。また,適切な包装を選択·試験し,候補生物製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
アメリカの審査と承認の流れ
候補生物製品の臨床試験が完了した後,生物製品の商業マーケティングの前に,FDAのBLAの承認を得なければならない。BLAには、製品開発、実験室と動物試験、人体試験、製品製造と成分の情報、提案されたラベル、その他の関連情報が含まれなければならない。さらに、小児科研究公平法またはPREAによれば、BLAまたはBLAのサプリメントは、すべての関連小児科亜群で主張される適応候補生物製品の安全性および有効性を評価し、安全で有効な各小児科亜群に対する製品の用量および投与をサポートするためのデータを含まなければならない。食品医薬品局安全·革新法案(FDASIA)は、薬物または生物製品のマーケティング申請のスポンサーを提出することを計画し、医薬または生物製品が新しい有効成分、新しい適応、新しい剤形、新しい投与レジメンまたは新しい投与経路を含む場合、第2段階会議の終了後60日以内に、またはスポンサーとFDAとの間の合意に従って予備小児科研究計画またはPSPを提出しなければならないことを要求する。
改正された処方薬使用料法案(PDUFA)によると,BLAごとに使用料を伴わなければならない。FDAは毎年PDUFAユーザ料金を調整する。場合によっては、小企業が初めて出願した出願料を免除することを含む、費用を免除または減免することができる。また,孤児薬として指定された製品については,この製品が孤児適応も含まれていない限り,BLASに対して使用料を評価しない。
出願提出後60日以内に、FDAは、機関が提出を受け入れる前に実質的に完了したかどうかを決定するために、提出されたBLAを審査する。FDAは、それが不完全であるか、または提出時に適切に審査できないと考えられる任意のBLAの提出を拒否することができ、より多くの情報の提供を要求することができる。この場合,BLAおよび付加情報を再提出しなければならない.再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。提出された申請が受け入れられると,FDAはBLAの深い実質的な審査を開始する。FDAは、提案された製品がその予期される用途に対して安全かつ有効であるかどうか、許容可能な純度プロファイルを有するかどうか、および製品が製品の特性、安全性、強度、品質、効力、および純度を確保および維持するためにcGMP要求に従って生産されるかどうかを決定するためにBLAを審査する。FDAは、新規な生物製品または安全性または有効性の問題を提起する生物製品の申請を諮問委員会に提出することができ、一般に、申請を承認すべきかどうか、およびどのような条件下で承認すべきかを審査、評価および提案するための臨床医および他の専門家を含むグループである。FDAは諮問委員会の提案に制限されていないが、それは決定を下す時にこれらの提案を慎重に考慮するだろう。バイオ製品の承認過程において、FDAはまた、候補生物製品の安全な使用を確保するために、リスク評価および緩和戦略、またはREMSを策定する必要があるかどうかを決定する。FDAがREMSが必要であると結論した場合,BLAのスポンサーは提案したREMSを提出しなければならず,必要であればFDAはREMSのないBLAを承認しないであろう。
37
BLAを承認する前に、FDAはこの製品を生産する施設を検査する。FDAは、製造プロセスおよび施設がGMP要件に適合していると判断しなければ、製品が要求された仕様内で一貫して生産されることを保証するのに十分であることを決定しない限り、この製品を承認しない。さらに、BLAを承認する前に、FDAは通常、IND研究要求およびGCP要求に従って臨床試験が行われることを確実にするために、1つまたは複数の臨床場所を検査する。
関連データおよび情報が提出されたにもかかわらず、FDAは、BLAがその承認の規制基準を満たしていないことを最終的に決定し、承認を拒否する可能性がある。臨床試験から得られたデータは常に決定的ではなく,FDAは同じデータを解釈するのではなく,異なる方法でデータを解釈する可能性がある。FDAが現在の形態のBLAを承認しないと決定した場合、FDAは、FDAによって決定されたBLA内のすべての特定の欠陥を一般的に記載する完全な返信を発行するであろう。決定された欠陥は微小である可能性があり、例えば、ラベル変更が必要であるか、または重大であり、例えば、追加の臨床試験が必要である。さらに、完全な返信状は、出願人がとり得る、申請を承認条件に置くための提案行動を含むことができる。完全な返信が発行された場合、出願人は、BLAを再提出し、手紙で決定されたすべての不足点を解決するか、または出願を撤回することができる。
1つの製品が規制部門の承認を得た場合、この承認は、特定の疾患および用量に明らかに限定される可能性があり、または使用の適応が制限される可能性があり、これは、製品の商業的価値を制限する可能性がある。さらに、FDAは、いくつかの禁忌症、警告、または予防措置を製品ラベルに含めることを要求する可能性がある。FDAは、REMSの形態で製品の流通、処方または調剤に制限および条件を適用することができ、または他の方法で任意の承認範囲を制限することができる。そのほか、FDAは発売後の臨床試験を要求する可能性があり、時々第四段階の臨床試験と呼ばれ、生物製品の安全性と有効性を更に評価することを目的とし、そして商業化された承認製品の安全性を監視するためにテストと監督計画を要求する。
FDAがPDUFAにより合意した業績目標の1つは,申請提出日から10カ月以内に90%の標準BLASを審査し,提出日から6カ月以内に90%の優先BLASを審査し,審査決定を行うことである。FDAは常にそのPDUFA標準や優先BLASの目標日を達成するわけではなく,その審査目標は時々変更される可能性がある。FDAがPDUFA目標日の前の最後の3ヶ月以内に提出材料で提供された情報に関する追加の情報または明確化を要求またはBLAスポンサーが要求する場合、審査プロセスおよびPDUFA目標日を3ヶ月延長することができる。
孤児薬名
孤児医薬品法によれば、FDAは、米国では20万人未満に影響を与えるか、または米国では20万人を超える影響を与える稀な疾患または疾患を治療するための薬剤または生物に孤児の称号を付与することができるが、米国では、そのような疾患または疾患を治療する薬剤または生物薬剤を米国で開発および提供するコストが、米国での薬剤または生物学的薬剤の販売から回収されることを合理的に予想することができない。NDAやBLAを提出する前に,指定孤児薬を申請しなければならない。FDAが孤児薬物指定を承認した後、FDAは、治療薬の模倣薬識別情報およびその潜在的孤児の使用を開示する。孤児薬物の指定は、規制審査または承認過程においていかなる利点も伝達されず、規制審査または承認過程の持続時間を短縮することもない。
孤児薬物指定を有する製品がその後、そのような指定された疾患に対するFDAの最初の承認を得た場合、この製品は、孤児薬の独占的承認(または排他性)を得る権利があり、これは、FDAが、限られた場合を除いて、7年以内に同じ疾患または状態について同じ薬物または生物学的薬剤を販売するために、完全なNDAまたはBLAを含む他の出願を承認しない可能性があることを意味する。例えば、孤児薬物排他性を有する製品に対する臨床的利点を示すか、またはFDAは、孤児薬物排他性保持者が、指定された薬物または生物学的薬物が対象とする疾患または状態の患者の需要を満たすために十分な数の孤児薬を得ることができることを証明していないことを発見する。孤児薬物の排他性は、FDAが同じ疾患または条件のために異なる薬剤または生物学的薬剤を承認することを阻止しないか、または異なる疾患または条件のための同じ薬剤または生物学的薬剤を使用することを阻止しない。孤児薬を指定する他の利点は、いくつかの研究の税金免除、およびNDAまたはBLA申請使用料の免除を含む。
指定された孤児薬物が孤児が指定された適応よりも広い用途で承認された場合,孤児薬物の排他性を得ることはできない。またアメリカでの独占営業権は
38
FDAが、指定された要求に重大な欠陥があると後に判断した場合、または製造業者が、このようなまれな疾患または疾患を有する患者の需要を満たすのに十分な数の製品を保証できない場合、国を失う可能性がある。
開発と審査計画を加速する
FDAは合格した候補製品に一連の迅速な開発と審査計画を提供した。例えば、Fast Track計画は、特定の基準に適合する新製品の審査プロセスを加速または促進することを目的としている。具体的には、候補製品が深刻または生命に危険な疾患または状態を治療することを意図し、その疾患または状況が満たされていない医療需要を満たす潜在力を示す場合、迅速なチャネル認証を取得する資格がある。高速チャネルは,候補製品と研究中の特定の適応に適した組合せを指定する.Fast Track候補製品のスポンサーは,製品開発期間中に審査チームとより頻繁なインタラクションを行う機会があり,セキュリティプロトコルやBLAが提出されると,優先審査を受ける資格がある可能性がある.高速チャネル候補製品もスクロール審査を行う資格がある可能性があり、この場合、FDAは、完全な出願を提出する前に、NDAまたはBLAの審査部分をスクロール考慮することができ、スポンサーがNDAまたはBLA部分を提出するスケジュールを提供した場合、FDAは、NDAまたはBLAの部分を受け入れることに同意し、スケジュールが許容可能であると判断し、スポンサーは、NDAまたはBLAの第1の部分を提出する際に任意の必要なユーザ料金を支払うことができる。
重篤または生命に危険な疾患や状況を治療しようとする候補品も,その開発や審査を加速するための画期的な療法指定を受ける資格がある可能性がある。初歩的な臨床証拠が、1つの製品が単独でまたは1つまたは複数の他の薬剤または生物製品と組み合わせて使用される場合、1つまたは複数の臨床的重要終点において、例えば臨床開発早期に観察される実質的な治療効果が既存の治療法よりも実質的に改善される可能性があることを示す場合、製品は画期的な治療指定を得ることができる。この指定には、Fast Track計画のすべての機能と、第1段階で開始されたより密集したFDA相互作用および指導と、高度な管理者の参加を含む候補製品開発および審査を加速する組織約束とが含まれる。
迅速なチャネル指定および/または画期的な治療指定を有する候補製品を含むFDA承認を提出する任意の医薬またはバイオマーケティング申請は、優先審査および加速承認のようなFDAの審査および承認プロセスを加速することを意図した他のタイプの計画の資格に適合する可能性がある。候補製品が深刻な疾患または状態の治療、診断または予防において著しい改善を提供する可能性がある場合、製品を優先的に審査する資格がある。新しい分子実体NDAおよび元のBLASについて、優先審査指定は、FDAの目標が、提出日から6ヶ月以内に上場申請に行動することであることを意味する(標準審査では10カ月)。
さらに、深刻または生命を脅かす疾患または状態の治療における安全性および有効性を研究する候補製品については、候補製品が臨床利益を合理的に予測することができる代替終点、または不可逆発病率または死亡率よりも早く測定することができ、不可逆発病率または死亡率または他の臨床利益を合理的に予測することができる臨床終点に対して有効であることを決定する際に、病状の重症度、希少性または流行率、および代替治療を利用可能または不足することを考慮しながら、承認を加速することができる。承認を加速する条件として、FDAは通常スポンサーに十分かつ良好な制御を行う上場後の臨床研究を要求し、不可逆的な発病率或いは死亡率或いは他の臨床利益に対する期待影響を検証と記述する。スポンサーが必要な上場後研究やこのような研究が予測の臨床的利益を検証できなかった場合、加速的な承認を得た製品は迅速な脱退プログラムの影響を受ける可能性がある。また、FDAは現在、承認を加速させる条件として宣伝材料を事前承認することを求めており、製品商業発売の時期に悪影響を及ぼす可能性がある。
2017年,FDAは21世紀の治療法の実施の一部として新たな再生医学高度療法,あるいはRMATを確立した。RMAT指定計画は、FDAが、細胞療法、治療用組織工学製品、ヒト細胞および組織製品、またはそのような治療法または製品を使用する任意の組み合わせ製品として定義されるrmatの資格に適合する薬物または生物の有効な開発計画を促進し、審査を加速させることを目的としている21世紀の治療法の要件を満たすことを意図している。(Ii)薬物または生物は、生命に深刻なまたは危険な疾患または状態を治療、修正、逆転または治癒することを意図している、および(Iii)予備臨床臨床
39
このような薬物や生物製剤が,このような疾患や状況が満たされていない医療ニーズを解決する可能性が示唆されている。RMAT指定は、FDAとより頻繁に会議を行い、候補製品の開発計画およびスクロール審査および優先審査の資格を検討することを含む画期的な治療指定のすべての利点を提供する。RMAT資格が付与された候補製品は、合理的に長期的な臨床的利益を予測する可能性のある代替物または中間終点に基づいて、または試験をより多くの場所に拡張することによって加速承認を得ることを含む、大量の臨床試験地点から得られるデータに依存する資格がある可能性もある。
迅速チャネル指定、画期的な治療指定、優先審査、加速承認、およびRMAT指定は、承認の基準を変更することはありませんが、開発または承認プロセスを加速する可能性があります。1つの製品がこれらの計画のうちの1つまたは複数に適合していても、FDAは、製品がもはや資格条件に適合していないことを後で決定することができ、またはFDAの審査または承認を決定する期間が短縮されないことができる。私たちは私たちの候補製品のためのいくつかの機会を適切に探索するかもしれない。
承認後に要求する
生物製品はFDAの普遍的かつ持続的な規制を受けており、その中には記録保存、副作用報告、定期報告、製品サンプリングと流通、および製品の広告と販売促進に関連する要求が含まれている。承認後、承認された製品の大多数の変更は、新たな適応または他のラベル宣言を追加するなど、FDAの事前審査および承認を経なければならない。どんな発売された製品についても、継続的な年間計画費用があります。バイオメーカーおよびその下請け業者は、FDAおよびある州機関に彼らの工場を登録し、FDAおよび特定の州機関の定期的な抜き打ち検査を受けて、cGMPの適合性を確保するように要求されており、これらの機関はcGMPにいくつかの手続きおよびファイル要件を適用している。製造プロセスの変更は厳しく規制されており,変更の重要性により,FDAが事前に承認して実施する必要がある可能性がある。FDAの規定では,cGMPから外れた状況を調査·是正し,報告要求を行うことも求められている。そのため、メーカーは生産と品質管理の分野で時間、お金、精力をかけ続け、cGMPやその他の法規遵守性を維持しなければならない。
規制要件や基準が守られていない場合、あるいは製品発売後に問題が発生した場合、FDAは承認を撤回する可能性がある。その後、製品には、予期されない深刻度または頻度の不良事象、または生産プロセス、または規制要件を遵守できなかったことを含む、以前に未知の問題が存在することが発見され、新しいセキュリティ情報を追加するために承認されたラベルの改訂を招く可能性がある;新しい安全リスクを評価するために発売後研究または臨床研究を実施すること、またはREMS計画に従って流通制限または他の制限を実施することが可能である。他の他の潜在的な結果には
FDAは生物製品のマーケティング、ラベル、広告、販売促進を厳格に規制している。1社はFDAが承認したラベルの規定に基づいて、安全性と有効性、純度、効力に関する声明しか提出できない。FDAと他の機関は非ラベル用途の普及を禁止する法律法規を積極的に施行している。これらの要求を守らないと
40
その中には不良宣伝、警告状、訂正性広告、および可能な民事と刑事罰が含まれている。医師は、製品ラベルに記載されていない使用のための合法的に入手された製品の処方、および我々が試験およびFDAによって承認された用途とは異なる使用を行うかもしれない。このようなラベル外の使用は医学専門科でよく見られる。医師は,異なる場合,このような非ラベル使用が多くの患者の最適な治療法であると考えるかもしれない。FDAは医者が治療を選択する時の行動を規範化しない。しかし、FDAは製品ラベルの外使用問題に対する製造業者のコミュニケーションを制限した。
生物模倣薬と排他性
2009年の“生物製品価格競争と革新法”(BPCIA)は、高度に類似している、またはFDAによって承認された参考生物製品と交換された生物製品のための短い承認の道を開いた。FDAはすでにいくつかの指導文書を発表し,生体模倣薬の審査·承認方法について概説した。生物類似性は生物製品と参照製品が安全性、純度と効力の面で臨床的に意義のある差異が存在しないことであり、通常分析研究、動物研究と臨床研究によって示される。互換性は、製品が基準製品生物と類似していることを必要とし、この製品は、任意の所与の患者において、参照製品と同じ臨床結果を生成することが期待できることを証明しなければならず、複数回投与された製品の場合、以前の投与後、生物および参照生物は、安全リスクを増加させることなく、または参照生物の独占的使用と比較して治療効果のリスクを低下させることなく、交互にまたは交換することができる。FDAによって承認された参照生物製品と類似しているか、または交換可能であることが証明された製品は、FDAが以前に承認された参照製品の安全性および有効性の決定に部分的に依存する可能性があり、これは、承認された製品を市場に投入するのに要するコストおよび時間を減少させる可能性がある。
BPCIAによると,生物類似製品の申請は,参考製品が初めてFDA許可を得た4年後にFDAに提出されなければならない。また,FDAによる生物類似製品の承認は,参考製品が初めて許可された日から12年後に発効する可能性がある。この12年間の独占期間内に、FDAが競合製品の完全なBLAを承認した場合、出願人自身の臨床前データと、その製品の安全性、純度および有効性を証明するために、十分かつ良好に制御された臨床試験からのデータとを含み、別の会社は、参照製品の競合バージョンを販売する可能性がある。BPCIAはまた、交換可能な製品として承認された生物模倣薬のためのいくつかの排他的期限を設けている。この節では,FDAが“交換可能”と考えている製品が本当に州薬剤法に管轄されている薬局に取って代わられるかどうかは不明である。
生物製品は米国でも小児科市場の排他性を得ることができる。小児科専有権が付与された場合、既存の専有期間と特許条項を6ヶ月増加させる。この6カ月間の排他性は,他の排他的保護や特許期間終了時から,FDAが発表したこのような研究の“書面請求”によって小児科研究を自発的に完成させることができる。
アメリカ以外の政府規制
私たちの候補製品は、例えば、臨床試験、マーケティング許可、上場後の要件(安全監視、詐欺および乱用法律を含む)、企業コンプライアンス計画の実施、および医療保健専門家への支払いまたは他の価値移転を含む、米国以外の司法管轄区域、特にEUまたはEUによって実施される同様の法律および法規によって制限されるであろう。生物由来の原材料は独特の汚染リスクに直面しているため、それらの使用はいくつかの国で制限される可能性がある。また,遺伝子編集技術,遺伝子治療,遺伝子テスト,遺伝子研究の倫理,社会および法律面の懸念は,追加の法規制や使用可能性の禁止過程を招く可能性がある。
私たちがFDAの候補製品の承認を得るかどうかにかかわらず、これらの国が臨床試験や候補製品の販売を開始する前に、外国の規制機関の必要な承認を得なければならない。臨床試験、製品許可、定価と精算を指導する要求と手続きは国によって異なる。適用される外国の監督管理要求を守らなければ、他のほかに、罰金、監督管理の一時停止または撤回、製品のリコール、製品の差し押さえ、経営制限、刑事起訴などの処罰を受ける可能性がある。
41
非臨床研究と臨床試験
アメリカと類似して、EUの非臨床と臨床研究の各段階は重要な監督管理によって制御されている。
非臨床研究を行うのは,新たな化学や生物物質の健康や環境安全性を証明するためである。非臨床(薬物-毒性)研究は、EU指令2004/10/ECに規定されている良好な実験室慣行またはGLPの原則を遵守しなければならない(特定の医薬製品、例えば放射性ラベル目的のための放射性薬物前駆体については、別の正当な理由がない限り)。特に非臨床研究は両者が体外培養そして体内にあるGLP原則に従って計画、実行、監視、記録、報告とアーカイブを行わなければならず、GLP原則は組織過程の品質体系と非臨床研究の条件として一連の規則と標準を規定した。このようなプロス基準は経済協力と開発組織の要求を反映する。
EUでは、医療製品の臨床試験はEUと国家法規、国際協調会議(ICH)、良好な臨床実践ガイドライン(GCP)及び“ヘルシンキ宣言”からの適用法規の要求と倫理原則に符合しなければならない。欧州委員会からの追加GCPガイドラインは、特にトレーサビリティを重視し、高級治療薬物製品(ATMP)の臨床試験に適している。臨床試験の発起人がEU内で成立していない場合、それはEU実体をその法定代表者として指定しなければならない。発起人は臨床試験保険証書を購入しなければならず、大多数のEU諸国では、発起人は臨床試験で負傷したいかなる研究対象にも“非のない”賠償を提供する責任がある。
EUの臨床試験に関連する規制構造は最近変化した。EU臨床試験条例,あるいはCTRと呼ばれ,2014年4月に採択され,EU臨床試験指令が廃止され,2022年1月31日に施行された。指示とは異なり、CTRはすべてのEU加盟国に直接適用され、加盟国がそれをさらに国家法律として実施する必要はない。CTRは臨床試験情報システムを通じてEU全体の臨床試験の評価と監督過程を著しく調整し、このシステムは集中したEU門戸とデータベースを含む。
臨床試験指令は,臨床試験を行う各加盟国で主管する国家衛生当局と独立した倫理委員会に単独の臨床試験申請(CTA)を提出することを要求しているが,FDAやIRBのように,CTRは集中的な手続きを導入し,多センター試験の単一申請の提出のみを要求している。CTRは、スポンサーが各会員国の主管当局と道徳委員会に文書を提出することを可能にし、各会員国が決定を下すことを可能にする。他の事項以外に、CTAは試験方案のコピーと被調査薬品の生産と品質情報を含む調査薬品ファイルを含まなければならない。CTAの評価手続きも統一されており、すべての関連加盟国による共同評価を含み、道徳基準を含む各加盟国が個別にその領土に関する具体的な要求を評価する。各会員国の決定は集中されたEUポータルサイトを通じてスポンサーに伝達される。CTAが承認されると,臨床研究開発は継続可能である。
CTRは3年間の過渡期が予想される。進行中の臨床試験と新たな臨床試験がCTRによってどの程度制御されるかはそれぞれ異なる。(I)2022年1月31日までに“臨床試験指令”に基づいて申請を提出した臨床試験、または(Ii)2022年1月31日から2023年1月31日までの間、かつスポンサーが“臨床試験指令”を適用する臨床試験を選択し、2025年1月31日までこの指令によって管轄されている。この日以降,すべての臨床試験(行われている臨床試験を含む)はCTR条項に拘束される。
臨床試験で使用される薬物は良好な製造規範やGMPに従って生産されなければならない。他の国と連合の範囲の規制要件も適用される可能性がある。
マーケティング許可
私たちの未来の候補製品をEUと他の多くの外国司法管轄区に推進するために、私たちは単独の規制承認を受けなければならない。より具体的にはEUでは医薬品候補は
42
マーケティング許可またはMAを取得した後に商業化される。EUの監督管理制度によると、研究用化学或いは生物製品の監督管理許可を得るためには、マーケティング許可申請、すなわちMAAを提出しなければならない。このようにする過程は,他を除いて医薬製品の性質に依存する。2つのタイプのMAがあります
上記の手順により、EMA又はEU加盟国主管当局は、製品の品質、安全性及び有効性に関する科学的基準に基づいて、製品のリスク−利益バランスを評価する。
中央プログラムによると,環境評価評価の最長時限は210日であり,クロックポーズは含まれていない。特別な場合、CHMPは、150日以下(クロックポーズを含まない)内でMAAの加速審査を行う可能性がある。満たされていない医療需要に対して、公衆健康に大きな影響を与えることが期待される革新製品は、米国の画期的な治療指定と同様のインセンティブを提供するPrime計画のような一連の迅速な開発および審査計画を得る資格がある可能性がある。2016年3月、EMAは、未満足の医療需要に対する薬物開発に対するEMAの支援を強化するための自発的な計画である優先薬物計画、またはPrime計画を開始した。その基礎は、有望な薬剤を開発している会社との相互作用と早期対話を増加させ、彼らの製品開発計画を最適化し、より早期に患者に接触するのを助けるために、彼らの評価を加速させることである。Prime指定を受けた製品開発者は加速評価を受ける資格が期待されるが,これは保証ではない。Primeの称号を持つ候補製品のスポンサーは多くのメリットを得ることができ、しかし限らず、早期にEMAと積極的な監督対話を行い、臨床試験設計とその他の開発計画要素を頻繁に討論し、及びファイルを提出した後にMAA評価を加速する。重要なのはCHMPの専任連絡先と調査委員が総理の早期に任命されたということです
43
EMA委員会レベルで製品の理解を促進する計画。最初の会議はこれらの関係を開始し、EMAの多学科専門家チームを含み、全体的な発展と監督戦略に関する指導を提供した。
また,EUでは,必要なすべての安全性や有効性データが得られていない場合には,“条件付き”MAが付与される可能性がある.条件付きMAは,失われたデータの生成やセキュリティ対策の増加を確保する条件を満たさなければならない.有効期間は1年で、すべての条件が満たされるまで年に1回更新しなければならない。未完成の研究が提供されると、“標準”のMAとなることができる。しかし,EMAが設定した時間範囲でこれらの条件を満たしていなければ,MAは更新を停止する.また、“特別な場合”では、出願人が、製品が許可され、特定の手順に従った後であっても、正常な使用条件下での有効性および安全性に関する包括的なデータを提供できないことを証明することができれば、MAも承認することができる。特に期待される適応は非常にまれであり,現在の科学的知識状態では網羅的な情報を提供することが不可能である場合や,データを生成することが一般的に受け入れられている倫理原則に違反する可能性がある場合がある.このMAは、重篤な疾患または満たされていない医療需要のために承認されるべき医薬製品を保持し、出願人は、MAに付与するために必要な合法的な完全データセットを有さないので、条件付きMAに近い。しかし,条件付きMAと異なり,申請者は失われたデータを提供する必要もなく,提供する必要もない.“特殊な場合”のMAは最終的に承認されているが,毎年薬品のリスク−収益バランスが審査されており,リスク−収益比が有利でなければMAは撤回される。
上記の手順により、MAを付与するために、欧州市場管理局又はEU加盟国主管当局は、製品の品質、安全性及び有効性に関する科学的基準に基づいて、製品のリスク効果バランスを評価する。MAの初期期限は5年である.この5年後、許可はリスク-収益バランスを再評価した上で無期限に更新することができる。
データとマーケティングは排他的です。
EUでは、マーケティングが許可された新製品候補製品または参照製品候補製品は、通常、MAで8年間のデータ独占権および追加2年間の市場独占権を取得する。承認された場合、データ固有期間は、模倣薬または生物類似薬の申請者がEUで模倣薬または生物類似薬を申請することを防止することになり、参照製品がEUで初めて許可された日から8年以内に、参照製品プロファイルに含まれる臨床前および臨床試験データに依存する。市場排他期は、参考製品がEUで最初の許可を得た10年後まで、成功した模倣薬または生物類似申請者がその製品をEUで商業化することを禁止している。MA保持者が10年の最初の8年間に1つまたは複数の新しい治療適応の許可を得た場合、10年間の市場専門期間は最大11年に延長される可能性があり、これらの新しい治療適応は、認可前の科学的評価中に既存の治療法と比較して有意な臨床的利益をもたらすことができると考えられる。しかし,製品がEU規制機関によって新たな化学/生物実体とみなされる保証はなく,製品にはデータ排他性を得る資格がない可能性がある。
生体模倣薬、すなわち参考医薬製品と類似しているが、模倣薬の定義に適合しない生物医薬製品については、例えば、原材料または製造技術の違いによる特殊な制度がある。このような製品については,適切な臨床前または臨床試験の結果を提供しなければならず,EMAのガイドラインは異なるタイプの生物製品に提供される補足データのタイプを詳細に説明している。遺伝子や細胞療法医薬製品のような複雑な生物製品に対しては,このようなガイドラインがないため,これらの製品の生体模倣薬がEUで承認される可能性は低い。しかし,EMAのガイドラインは,将来的には当時得られた科学的知識や規制経験に基づいてこれらの提案を考慮することを指摘している。
小児科発展
EUでは、許可されていない新しい薬剤候補のMAAは、EMAの小児科委員会またはPDCOと合意された小児科調査計画またはPIPに適合する小児科集団で行われた研究結果を含まなければならない。PIPは,市販認可が求められている薬物の小児科適応を支援するためのデータ生成の時間とアドバイスを規定している。PDCOは、十分なデータがあるまで、PIPの実施義務の一部または全部を延期することを許可することができる
44
成人におけるこの製品の有効性と安全性を証明する。さらに、小児臨床試験データを必要としないか、または提供するのに適していない場合、PDCOは、子供に無効または安全でない可能性があるので、これらのデータを提供する義務を免除することができ、この製品は、治療のために使用される疾患または状態が成人集団でのみ発生することが予想される場合、または小児科患者の既存の治療に対して有意な治療利益がない場合。すべてのEU加盟国でMAを取得し、研究結果を製品情報に含めると、否定的な場合であっても、6ヶ月間の補充保護証明書を取得して延期する資格があるか、または、孤児薬品については、孤児市場独占権を2年間延長することが許可される。
孤児医薬製品
EUの“孤児薬品”の認定基準は原則的にアメリカと似ている。EUにおいて、スポンサーが、(1)生命または慢性衰弱にかかわる疾患の診断、予防または治療を目的としている場合、(2)または(A)申請時に、EUにおけるこのような疾患の影響が10,000人中5人以下であること、または(B)孤児の身分によるメリットがなければ、EUで十分な見返りを生じることなく、投資が合理的であることを証明することができれば、製品は孤児として指定することができる。(3)EUがこのような疾患を満足できる診断、予防または治療する方法を許可していないか、または、そのような方法があれば、製品は、疾患の影響を受けている人に大きな利点を有するであろう。
EUでは、孤児製品に指定された申請はMAAの前に提出されなければならない。孤児薬物指定は、一方の当事者が費用を削減したり、費用を免除したり、集中プログラムを使用する権利があるなど、財政的奨励を受ける権利がある。MAを承認した後,孤児医療製品が承認された治療適応の10年間の市場専門期間を得る権利があることは,その間,規制当局が同一の適応の類似医療製品の別のMAAを受け入れることができないか,MAを承認するか,MAを延長する申請を受けることができないことを意味する。合意されたPIPにも適合する孤児薬品については,市場専門期間を2年間延長した。いかなる補充保護証明書も孤児の症状に関する小児科研究によって延期してはならない。孤児薬物の指定は、監督審査と承認過程においていかなる利点も伝達されず、規制審査と承認過程の持続時間を短縮することもない。
しかし,5年目の終了時にその製品が孤児の薬物目的地を獲得する基準に適合していないことが決定された場合,この疾患の流行率が敷居を超えていること,あるいはその製品の利益が十分に高いと判定され,市場排他性を維持することが合理的であることを証明するのに不十分であれば,10年間の市場排他性を6年に短縮することができる。次の場合、(I)第2の出願人は、その製品が認可製品と類似しているが、より安全で、より効率的で、または臨床的により良いことを証明することができ、(Ii)出願人は、十分な数の孤児医薬製品を供給することができない、または(Iii)出願人は、第2の孤児医薬製品出願に同意することができる。ある会社は自発的に一つの製品を孤児登録簿から削除することができる。
上述のEU規則はヨーロッパ経済区、すなわちEEAに一般的に適用され、27のEU加盟国にノルウェー、リヒテンシュタイン、アイスランドを加えて構成されている。
私たちまたは私たちの任意の第三者パートナーは、サプライヤー、製造業者、および流通業者を含み、MAを付与する前および後に、臨床試験、生産承認、医薬製品のMAおよびそのような製品のマーケティングに適したEUおよび加盟国の法律を遵守することができなかったか、またはMA、医薬製品の製造、法定医療保険、賄賂および反腐敗または他の適用可能な法規要件を遵守することができず、行政、民事または刑事罰をもたらす可能性がある。これらの処罰は、臨床試験またはMAの承認の遅延または拒否、製品の撤回およびリコール、製品の差し押さえ、一時停止、マーケティング許可の撤回または変更、生産の完全または部分的な一時停止、流通、製造または臨床試験、経営制限、禁止、免許取り消し、罰金、および刑事罰を含むことができる。
イギリスの離脱とイギリスの規制枠組み
イギリスは2020年1月31日にEUを離脱し、その後、EU-イギリス離脱協定の条項に基づいて、過渡期内に、既存のEU医薬製品立法は引き続きイギリスに適用される。貿易·協力協定(TCA)は英国とEUが達成したものである
45
2021年1月1日から施行される。TCAには薬品に関する具体的な条項が含まれており,薬品生産施設のGMP検査と発表されたGMP文書の相互承認が含まれているが,イギリスとEUの薬品法規が大規模に相互承認されることは予想されていない。
二次立法によりイギリス法律に転換したEU法律は引き続き“保留EU法律”として適用される。しかし、連合CTRのような新しい法案は適用されないだろう。英国政府は、医療製品や医療機器分野の既存の法規を改正または補完するために、国務大臣または適切な機関に権限を付与する新しい“2021年薬品·医療機器法”を可決した。これにより,人間の薬物,臨床試験,医療機器分野の規制格差や将来の変化を解決する上で柔軟性を許すことを目的として,今後二次立法で新たなルールを導入することが可能となる。
2021年1月1日から、薬品と医療製品規制機関(MHRA)はイギリスの独立した薬品と医療機器監督機関である。北アイルランド議定書の結果として、北アイルランドはイギリスやGBを含むイングランド、ウェールズ、スコットランドとは異なる規則が適用されるだろう;全体的に、北アイルランドはEUの規制制度に従っているが、その国家主管機関はMHRAであるだろう。2023年2月27日、イギリス政府と欧州委員会は、北アイルランド議定書、すなわち“ウィンザー枠組み”と呼ばれる新たな手配の代わりに、原則的な政治的合意を発表した。この新しい枠組みはイギリスの医薬製品に対する規制を含む北アイルランド議定書下の既存制度を根本的に変えた。特に,MHRAはイギリス市場(すなわちイギリスと北アイルランド)に輸送されたすべての医薬製品を承認することを担当するが,EMAは北アイルランドへの医薬製品の輸送を承認する上で何の役割も果たすことはできない。MHRAは,イギリスで販売されているすべての医薬製品に単一のイギリス範囲のMAを付与し,製品をイギリス各地で単一パッケージと単一ライセンスで販売できるようにする。2023年3月24日、EU·イギリス合同委員会はウィンザー枠組みを承認したため、イギリス政府とEUは立法措置を制定し、法律にする。2023年6月9日,MHRAはウィンザーフレームワークの薬品について2025年1月1日から適用すると発表した。
MHRAは、150日間の評価およびスクロール審査プログラムを含む、患者に利益を得る新薬を優先的に得るプログラムを含む、国家許可プログラムを変更している。中央許可製品のためのすべての既存のEU MAは、MA所有者が脱退を選択しない限り、GBでのみ有効であり、無料で、2021年1月1日にイギリスMAに自動的に変換またはキャンセルされる。集中プログラムを用いて欧州経済区全体で効果的なM&Aを獲得するためには,欧州経済区に会社を設立しなければならない。したがって,イギリスが離脱して以来,イギリスで設立された会社はEU集中化プログラムを使用することはできず,ヨーロッパ経済区実体はいかなる集中式MAを持たなければならない。2024年1月1日までに、MHRAは、新しいGB MAをより迅速に承認するために、新しい(集中プログラム中のMA)の承認に関する欧州委員会の決定に依存する可能性がある。新たな国際認可枠組みは2024年1月1日から実施され,この枠組みにより,MHRAは新たなGB MAの申請を決定する際に,EMAとある他の規制機関によるMAの承認決定を考慮する。
イギリスでは、MA前の孤児の称号はない。逆に,MHRAは対応するMA申請を審査しながら孤児指定申請を審査する.これらの基準は基本的に同じであるが,すべて市場のためにオーダーメイドされている,すなわちEUではなくイギリスのこのような疾患の流行率は万分の5を超えてはならない。孤児の称号が付与された場合、期限または市場独占権は、その製品が初めて承認された日からGB単位で設定される。
他の医療保険法
製薬会社は、連邦政府およびそれらが業務を展開している州と外国の司法管轄区域当局の追加的な医療監督管理と法執行を受けており、私たちが研究、販売、マーケティング、流通し、マーケティングの許可を得た任意の製品の財務配置や関係を制限する可能性がある。このような法律には、連邦および州のリベート、詐欺および乱用、虚偽声明、ならびに薬品の価格設定および支払い、ならびに医師および他の医療提供者への他の価値移転に関連する透明性のある法律および法規が含まれているが、これらに限定されない。このような法律または任意の他の適用される政府条例に違反することは、行政、民事および刑事罰、損害賠償、罰金、返還、削減、またはこれらに限定されない重大な処罰をもたらす可能性があります
46
規制を遵守せず、連邦や州医療計画から除外され、監禁された疑いを解決するために、業務、誠実な監督、報告義務を再編する。
保証と精算を請け負う
任意の製品の販売は、連邦、州と外国政府医療保健計画、商業保険とホスト医療組織、および第三者支払人のこの製品に対する清算レベルなど、第三者支払者のこの製品に対する保証範囲にある程度依存する。提供されるべき補償範囲と金額に関する決定は個々の計画に基づいて行われる。これらの第三者決済者は医療製品、薬品、サービスの保証と精算をますます減少させている。医師の監督下で管理されている製品については、このような薬物が高い価格に関連することが多いため、保険および適切な補償を得ることは特に困難である可能性がある。また,製品自体やその製品を使用した治療やプログラムは単独では精算できない可能性があり,医師の使用に影響を与える可能性がある。
米国政府、州立法機関、外国政府も引き続きコスト制御計画を実施し、価格制御、カバー範囲と精算に対する制限及び模造薬代替の要求を含む。価格制御及びコスト制御措置、並びに既存の制御及び措置を講じている司法管区において、より限定的な政策をとることにより、任意の製品の販売をさらに制限することが可能となる。いかなる製品の第三者精算または第三者支払者が製品を保証しないことを決定することは、医師の使用量や患者の製品に対する需要を減少させ、販売に実質的な悪影響を及ぼす可能性がある。
医療改革
米国では、2010年3月に“医療·教育和解法案”によって改正された“患者保護·平価医療法案”が公布され、総称してACAと呼ばれ、政府や民間保険会社が医療保健に資金を提供する方式を大きく変え、製薬業に大きな影響を与えた。ACAには、連邦医療計画の登録、補償調整、および詐欺および乱用の法律の改正を含むいくつかの条項が含まれている。例えば、ACA:
ACAのいくつかの側面は公布以来、司法、行政、そして国会の挑戦を受けてきた。2021年6月17日、米国最高裁はいくつかの州がACAに対して提出した最新の司法挑戦を却下したが、ACAの合憲性を具体的に裁くことはなかった。最高裁が裁決を下す前に,総裁·バイデンはACA市場による医療保険の取得を目的とした2021年2月15日から2021年8月15日までの特殊保険期間を開始する行政命令を発表した。行政命令はまた、作業要求を含む医療補助モデル項目および免除計画の再検討、医療補助またはACAによる医療保険カバー範囲の獲得による不必要な障害をもたらす政策の再検討、医療補助またはACAによる医療保険の取得を制限する既存の政策および規則の見直しを指示する。
ACAが公布されて以来、また、会計年度ごとに提供者に支払う医療保険総額が2%減少することを含む他の立法変化が提案され、採択され、この規定は2020年5月1日から2022年3月31日までの間停止された。
47
また,政府は最近,メーカーが販売する製品に価格を設定する方式をより厳しく審査し,国会で数回の調査を行い,製品定価の透明性を高めるための立法,定価とメーカー患者計画との関係を審査し,政府計画の薬品に対する補償方法を改革することを提案·公布した。政府はまた新冠肺炎の流行に対応するためにもっと多くの行動をとる可能性がある。アメリカの個別州もますます積極的に薬品の価格を制御するための法規を実施しており、価格或いは患者の精算制限、割引、ある製品への参入とマーケティングコスト開示の制限及び透明性措置を含み、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。
将来的には,より多くの州と連邦医療改革措置がとられ,いずれも連邦や州政府および他の第三者支払者が医療製品やサービスに支払う金額に影響を与える可能性が予想される。
データのプライバシーとセキュリティ
多くの州、連邦と外国の法律は個人情報の収集、伝播、使用、取得、秘密と安全を規範化し、健康に関する情報を含む。私たちの業務と業務の増加に伴い、私たちはアメリカ連邦と州の法律法規の制約または影響を受ける可能性があり、これらの法律法規は健康に関連する個人情報や他の個人情報の収集、使用、開示、保護を管理する。また、いくつかの非米国法は、健康関連データを含む個人データのプライバシーやセキュリティを管理しており、その多くの法律は互いに大きく異なり、同じ効果が生じず、コンプライアンス作業を複雑化させる可能性がある。適用される場合には、これらの法律を遵守しない場合には、重大な民事及び/又は刑事罰及び私的訴訟を引き起こす可能性がある。プライバシーとセキュリティ法律、法規、その他の義務は絶えず変化し、互いに衝突し、コンプライアンス作業を複雑化させ、調査、訴訟あるいは行動を招き、重大な民事および/または刑事罰およびデータ処理の制限を招く可能性がある。
従業員
2023年12月31日現在、93人のフルタイム従業員と1人のアルバイト従業員がいます。
企業情報
私たちは2016年7月にデラウェア州法律に基づいて登録成立し、名称はVL 42,Inc.
第1 A項。RISK因子です。
わが社を評価する際には、当社の連結財務諸表と本10-K年度報告書の末尾の関連付記を含む、以下に説明するリスクおよび不確実性、および当社の10-K年次報告書に含まれる他のすべての情報をよく考慮しなければなりません。以下のいずれかの事件や事態が発生した場合、私たちの業務、見通し、経営業績、財務状況は重大な影響を受ける可能性があり、私たちの普通株の取引価格は低下する可能性があります。以下に説明するリスクと不確実性は私たちが直面している唯一の危険と不確実性ではない。私たちは今知らないか、あるいは私たちが現在どうでもいいと思っている他のリスクと不確実性はまた私たちの業務に悪影響を及ぼすかもしれない。
私たちの財務状況と資本要求に関連するリスク
私たちの経営の歴史は限られており、承認された候補製品の開発や商業化に成功したり商業化されていない歴史は、私たちの業務のこれまでの成功度と将来の生存能力の将来性を評価することが困難になるかもしれません。
私たちは臨床段階の生物製薬会社です。これまで、私たちの業務は、会社に資金と人員を提供し、私たちの技術を開発し、私たちの候補製品を決定し、開発することに限られています。我々の将来性は,生物製薬会社が運営初期によく遭遇する不確実性,リスク,費用,困難を考慮しなければならない。私たちはまだ能力を証明していません
48
任意の臨床試験を行ったり完了したり、市場の承認を得たり、商業規模の製品を生産したり、製品の商業化に成功するために必要な販売及びマーケティング活動を行う。したがって、私たちがより長い運営歴史や開発に成功し、市場の承認を得て候補製品を商業化した歴史があれば、私たちの将来の成功や生存能力の予測はあるべきほど正確ではないかもしれない。また、私たちは予期せぬ費用、困難、合併症、遅延などの障害に直面する可能性がある。
私たちが業務を継続するにつれて、様々な要素によって、私たちの財務状況と経営業績は四半期ごとと毎年大幅に変動し、その多くの要素はコントロールできないと予想しています。したがって、将来の経営業績の指標として、特定の四半期や年度の結果に依存してはいけません。
設立以来、私たちはすでに重大な損失が発生しており、予測可能な未来にも重大な追加損失が予想される。
成立以来、2023年12月31日現在と2022年12月31日現在の年度純損失を含む重大な純損失が発生し、それぞれ9740万ドルと1.027億ドルとなった。2023年12月31日までの累計赤字は3兆346億ドルだった。しかも、私たちはどんな製品も商業化していないし、製品販売から何の収入も得たことがない。我々のほとんどの財政資源は,我々の臨床前開発活動や,我々の候補製品の臨床試験を準備·実施することを含めて研究·開発に投入されている。
予測可能な未来には,臨床開発による候補製品の推進,臨床前開発の継続,我々の研究·開発活動の拡大,新たな候補製品の開発,臨床前研究と臨床試験の完了,規制承認を求め,規制承認を得た場合に我々の製品を商業化することが求められ,引き続き重大な追加純損失が生じることが予想される。FDA承認を得て米国で任意の候補製品を販売するためには、バイオ製品ライセンス申請、またはBLAをFDAに提出し、FDAがその予期される用途に対して安全かつ有効であることを満足的に証明することを証明しなければならない(S)。外国の規制機関もまた似たような要求を実施した。この論証には大量の研究と動物試験からの広範なデータが必要であり、これらの試験は非臨床或いは臨床前研究、及び人体試験と呼ばれ、臨床試験と呼ばれる。また,時間の経過とともに候補製品を後続の臨床段階に進めるコストが大幅に増加することが多い。司法管轄区域であっても、私たちのすべての候補製品を市場に承認する総コストは巨大であり、正確に予測することは難しいだろう。医薬品開発に関連する多くのリスクおよび不確実性のため、費用を増加させる時間や金額を正確に予測することができないか、または製品の商業化から収入を生み出すことができるかどうか、または利益を達成または維持することができるかどうかを正確に予測することができない。私たちの支出も大幅に増加するでしょう
49
未来の損失の額と私たちがいつ利益を達成するかは不確実だ。私たちはビジネス段階の製品がなく、私たちが1つ以上の候補製品を開発することに成功するまで、製品の商業販売から収入を得ることはなく、製品販売から収入を得ることができないかもしれない。私たちは予測可能な未来に営業損失と負のキャッシュフローが続くと予想しています。これらの経営損失と負のキャッシュフローはすでにわれわれの株主権益や運営資本に悪影響を与え続けている。
私たちは受け入れ可能な条項で得ることができないか、または全く得られないかもしれない大量の追加資金が必要だ。必要な時に必要な資本を得ることができなければ、私たちの製品開発を延期、制限、減少、または中止させる可能性があります。
設立以来、私たちの運営は多くの費用を発生させた。臨床前開発,起動,候補製品の臨床開発を継続し,新たな候補製品の決定を継続して巨額の費用が発生することが予想される。
IPOと2023年2月に直接発売された収益を登録する以外に、私たちは引き続き追加の資金を必要とし、私たちが計画した臨床前開発と臨床試験に資金を提供し、新しい候補製品を開発し、株式発行、債務融資、マーケティングと流通手配、その他の協力、戦略連盟と許可手配、あるいはその他の資金源を通じて資金を調達することができる。他の融資源は割引の条件で提供されない可能性があり、もしあれば。もし私たちが受け入れ可能な条件下でより多くの資金を集めることができない場合、私たちは私たちの任意の候補製品の臨床試験を開始したり、完成させたり、FDAやいかなる外国規制機関の規制承認を求めたりすることができず、製品開発の中止を余儀なくされる可能性がある。また、より多くの資金を得ようとすることは、私たちの経営陣の日常活動に対する時間と注意力を移し、私たちの発展努力を損なう可能性がある。
2023年12月31日現在、私たちの既存の現金、現金等価物、および有価証券は、私たちが計画しているすべての努力が資金を提供するために不足するだろう。私たちの現在の運営計画によると、2023年12月31日まで、私たちの現金、現金等価物、有価証券は、2025年第1四半期までの運営費用と資本支出要求を満たすのに十分であると信じている。この推定は、正しくないことが証明される可能性があるという仮定に基づいており、私たちは現在予想されているよりも早く私たちが利用できる資本資源を使用することができる。また,以下に述べるように,我々が継続的に経営している企業として存在し続ける能力に関する条件やイベントを決定した.私たちは私たちの現在と未来の候補製品を発売して商業化するために多くの追加資金を必要とするだろう。また,我々の開発努力の過程で他の予期しないコストが発生する可能性がある.著者らの多くの候補製品は臨床前開発段階にあるため、著者らはまだいかなる臨床試験を行っていないため、著者らは候補製品の開発と商業化に成功するために必要な実際の数量を合理的に見積もることができない。また、我々は主要金融機関の口座に現金と現金等価物の大部分を保持しており、これらの機関での預金は保険限度額を超えている。市場状況はこのような機関の生存能力に影響を及ぼすかもしれない。もし私たちが現金と現金同等物を維持しているどの金融機関が倒産すれば、私たちが未保険の資金をタイムリーにまたは根本的に得ることができる保証はない。このような資金を得ることができない場合や遅延されたどんな状況も、私たちの業務と財務状況に悪影響を及ぼす可能性がある。
私たちの将来の資本需要は多くの要素に依存しています
50
私たちは許容可能な条件で追加資金を提供するかどうか、あるいは根本的にできないかどうかを確認することができない。もし私たちが受け入れられる条項や十分な追加資本をタイムリーに調達できなければ、私たちは候補製品の開発や商業化、または他の研究開発計画を大幅に延期、削減、停止しなければならないかもしれない。
もし私たちがタイムリーに資金を得ることができなければ、私たちは私たちの1つ以上の研究開発計画や任意の候補製品の商業化を大幅に削減、延期または停止することを要求されるかもしれないし、必要に応じて私たちの業務を拡大したり、他の方法で私たちのビジネスチャンスを利用することができなくなり、これは私たちの業務、財務状況、運営結果に大きな影響を与えるかもしれない。上記のどの事件も、私たちの業務、将来性、財務状況と経営結果を深刻に損害し、私たちの普通株価格の下落を招く可能性があります。
私たちの相次ぐ運営赤字は私たちが経営を続けている企業として経営を続ける能力を大きく疑っています。
設立以来、私たちは大きな損失を受けて、製品販売から収入や利益を得ることはなく、製品販売から収入や利益を得ることができないかもしれない。2023年12月31日まで、私たちは7340万ドルの現金と現金等価物と有価証券を持っています。私たちの現在の運営計画によると、2025年第1四半期まで履行するのに十分な資金があると信じています。しかし、私たちは私たちの将来の運営を支援し、継続的に経営する企業として追加の資本を集める必要があるだろう。株式発行、債務融資、マーケティングおよび流通スケジュール、ならびに他の協力、戦略連合および許可スケジュールの組み合わせ、または許容可能な条件で他のソース(あれば)を得ることを含む追加の資金を得ることができる保証はない。今後の株式発行により追加資本を調達する程度では、普通株主の所有権権益は希釈され、深刻な希釈となる可能性がある。私たちは私たちが任意のまたは十分な追加資金を得ることができるという保証がないし、もしあれば、このような資金は私たちが満足する条件で得られるだろう。もし私たちがいかなるあるいは十分な追加資金を得ることができなければ、私たちが経営を続けることができることを保証することができなければ、私たちは私たちの製品開発計画を延期、減少あるいは停止させ、あるいは他の様々な戦略的選択を考慮することを余儀なくされます.
また,これらの要因は,我々が継続的に経営している企業として継続する能力を大きく疑わせている。われわれの持続的な経営能力への大きな疑いは価格に実質的な悪影響を及ぼす可能性がある
51
普通株ごとに、私たちは融資を受けるのがもっと難しいかもしれない。もし既存または潜在的な協力者がこのような懸念のために私たちとのビジネスを拒否したり、潜在的な投資家が将来のいかなる融資にも参加することを拒否した場合、私たちが現金を増加させる能力は制限されるかもしれない。私たちが経営を続けられないかもしれないという見方は、他の人が私たちの契約義務を果たす能力を心配して私たちと付き合わないことを選択してしまう可能性があると思います。
我々は,持続経営をもとに総合財務諸表を作成し,正常業務過程における資産現金化および負債と承諾の弁済状況を考慮した。当社の年次報告書Form 10-Kに含まれる総合財務諸表には、会社が当該等の財務諸表発表後1年以内に経営を継続できない可能性があることを反映するための調整は含まれていません。もし私たちが経営を続けることができなければ、あなたはわが社への投資の全部または一部を失うかもしれません。
追加資本の調達は私たちの株主に追加的な希釈をもたらし、私たちの運営を制限し、私たちの技術や候補製品に対する権利を放棄することを要求し、私たちの株価を下落させる可能性があります。
これまで、製品販売から相当な収入を得ることができれば、株式発行、債務融資、マーケティング、流通手配、その他の協力、戦略連合と許可手配、または他のソースの組み合わせによって、私たちの現金需要を満たすことができるかもしれません。また、有利な市場条件や戦略的考慮により、現在または将来の運営計画のために十分な資金があると考えても、追加の資本を求めることができる。
私たちが株式または転換可能な債務証券を売却することによって追加資本を調達する場合、あなたの所有権資本は希釈され、これらの証券の条項は清算または他の特典を含む可能性があり、普通株主としての権利に悪影響を及ぼす可能性がある。債務融資および優先株融資に関連する可能性のある協定には、追加債務を招く、資本支出を行う、配当金を発表する、私たちの株を償還する、特定の投資を行う、特定の合併、合併または資産売却取引に従事するなどの特定の行動をとる能力が含まれている。もし私たちが第三者との協力、戦略連合またはマーケティング、流通または許可手配によってより多くの資金を調達する場合、私たちは、私たちの技術、将来の収入流、または候補製品の貴重な権利を放棄するか、または私たちに不利になる可能性のある条項でライセンスを付与することを要求されるかもしれない。もし私たちが必要な時に株式や債務融資を通じてより多くの資金を調達できなければ、私たちは私たちの製品開発や将来の商業化努力を延期、制限、減少または終了することを要求されるかもしれません。あるいは私たちが自分で開発し、マーケティングすることをより望んでいた候補製品の権利を与えることができます。
私たちの既存と未来のどんな債務も私たちの業務運営能力に悪影響を及ぼすかもしれない。
2023年12月31日現在、改正された融資と担保協定である太平洋西部銀行との融資協定によると、1900万ドルの未返済借金がある。ローン協議の満期日は2027年9月30日で、さらに2028年9月30日に延期することができる。このローンは2023年9月30日から返済され、毎月元金30万ドルと利息が支払われ、満期日が2028年9月30日まで延長されなければ、2027年9月30日に400万ドルの決済も支払われる。融資協議項下の未返済残高は変動年利で利上げされ、金利は(I)当時の最優遇金利0.50%および(Ii)5.50%(2021年12月20日以降最初の月から月単位満期)の両者の中で大きい者に相当する。私たちの未返済債務は、私たちが工務局から借金した以外のいかなる追加債務も含めて、私たちの他の財務義務と契約約束を加えて、重大な不利な結果をもたらす可能性があります
52
私たちは私たちの現在と未来の債務超過義務を私たちの当時の現金と現金同等物で返済するつもりだ。しかし、私たちは十分な資金を持っていないかもしれないし、追加の融資を手配することができないかもしれないし、融資協定や任意の他の債務ツールの下で満期になった金額を支払うことができないかもしれない。ローン契約やこのような他の債務文書に基づいてお金を支払わない、あるいは他の契約を守らないと、違約や満期金額を加速させる可能性があります。例えば、私たちの融資協定下の肯定的な契約には、私たち(および私たちの子会社に)に私たちの合法的な存在と政府の承認を維持すること、いくつかの財務報告および通知を提出すること、適切な記録および帳簿を保存すること、納税申告書をタイムリーに提出し、支払いすること、在庫と保険範囲を維持すること、PWB(例外的な場合)、および制御されたプロトコルによって制限された口座(例外を受けた場合)に現金を維持することを含む。ローン協定によると、合理的に予想されるように吾などの業務、運営、資産或いは状況に重大な悪影響を与える事件が発生すれば、すなわち違約事件である。もし違約事件が発生し、PWBが満期金額を加速した場合、私たちは支払いを加速できない可能性があり、融資者は担保債務を強制的に執行する担保権益を求める可能性がある。また、融資協定下の契約、私たちの資産を担保として、そして私たちの知的財産権の負質抵当は、私たちが追加債務融資を受ける能力を制限するかもしれない.
私たちはどんな製品収入も発生しておらず、永遠に利益を上げないかもしれない。
私たちの収益性は私たちが製品収入を作る能力にかかっている。これまで、私たちは臨床開発に成功し、規制部門の承認を得て、候補製品の商業化に成功しなければ、顕著な製品収入は生じませんでした。私たちの多くの候補製品は臨床前開発段階にあり、より多くの臨床前研究と臨床開発、監督審査と承認、安全な製造供給、成熟した商業化販売能力、大量の投資と十分な資金、及び重大なマーケティング努力が必要であり、製品販売から任意の収入を得ることができる。私たちが製品収入を作る能力は多くの要素に依存しています
53
上に挙げられた多くの要素は私たちがコントロールできないことであり、私たちが重大な遅延に遭遇したり、規制部門の許可を得たり、私たちの候補製品を商業化することを阻止したりする可能性がある。私たちの候補製品を商業化できても、製品販売が発生した後すぐに利益を達成することはできないかもしれません。もしあれば。もし私たちの候補製品を販売することで十分な収入を得ることができなければ、私たちは持続的な資金なしで運営を続けることができないかもしれない。
我々の候補製品の発見,開発,臨床前と臨床試験および規制承認に関するリスク
我々の候補製品は新しい技術に基づいており、これにより、臨床前および臨床開発、およびその後に規制承認を得る時間およびコスト(あれば)を予測することは困難である。
私たちの成功はomegaプラットフォーム技術に依存し、これは新しい技術だ。そのため、著者らの計画と候補製品が製品発見或いは鑑定、臨床前研究と臨床試験を行う時に遭遇する可能性のある臨床前と臨床開発挑戦を正確に予測することは困難である。また,最近我々のパイプライン候補製品の臨床試験を開始したため,我々の技術の人体上の安全性や有効性を評価することはできず,我々が開発した任意の候補製品の治療は現在予測できない短期的あるいは長期的な影響を与える可能性がある。さらに、私たちが計画の中で追求しているいくつかの疾患を選択するためには、動物モデルは存在しないかもしれない。著者らの技術プラットフォームの新規性を考慮して、臨床前仕事の持続時間、臨床開発、FDA或いは類似の外国監督機関は著者らの候補製品の安全性と有効性、純度と効力の患者数を確定するために臨床試験に参加することを要求するかもしれないし、これらの臨床試験が産生したデータがFDA或いは類似の外国監督機関によって受け入れられ、上場承認を支持することも保証できない。FDAおよび同様の規制機関は、私たちが提出した任意の生物製品ライセンス申請またはBLAまたは外国マーケティング申請を決定するために、通常よりも長い時間を必要とする可能性があり、承認を支持するために私たちの候補製品が十分なデータ、情報、または経験を持っていないことを最終的に決定する可能性がある。FDAなどの外国規制機関は、我々の候補製品とのより多くの経験を得るまで、リスク評価や緩和戦略やREMSなどのリスク管理計画の追加的な上場後の研究または実施を要求することも可能である。これらの要素の各々は、私たちの予想される開発コストを増加させ、私たちの候補製品の任意の商業化範囲を遅延、阻止、または制限することができる。検証プロセスには時間と資源が必要であり、独立した第三者分析が必要となる可能性があり、FDAおよび同様の外国規制機関によって受け入れまたは承認されない可能性がある。私たちは私たちの方法が単独または他の治療法と組み合わせて承認または適切な製品の開発につながると確信できない。
また,我々が計画した臨床試験からデータを得ても,我々の計画に適用されているomegaプラットフォーム技術は新たであり,外部検証を経ていないため,我々のデータは複製および/または我々や他の人から誤解されにくい可能性がある。エピゲノムコントローラは1種の新しい薬物類別を代表し、まだ臨床試験で評価を行っておらず、監督部門の許可も得られていない。したがって,臨床データのための新たな評価方法や指標を開発する必要がある可能性があり,データを分析することが困難になる可能性があり,あるいは我々にとっては,我々のECの開発にはより多くの時間やコストが必要となる可能性があり,同じ適応では他の療法よりもECsの開発が高価である可能性がある。これらの要因により、製品候補開発の時間とコストを予測することは困難であり、omegaプラットフォーム技術や任意の類似または競争的エピジェネティック技術の応用が任意の製品の識別、開発、規制承認を招くかどうかを予測することもできない。我々が将来遭遇するomegaプラットフォーム技術や我々の任意の研究プロジェクトに関する開発挑戦が重大な遅延や意外なコストを招くことは保証されず,このような開発問題が解決される保証はない.これらの要因のいずれも、前臨床研究または開始可能な任意の臨床試験を完了することを阻止するか、またはタイムリーまたは利益的に開発される可能性のある任意の候補製品を商業化することができる。
そのほか、FDAと他の監督機関の臨床試験要求及びこれらの監督管理機関は候補製品の安全性と有効性を決定するための標準は、潜在製品のタイプ、複雑性、意外性、期待用途と市場によって大きく異なる。他のより有名あるいは広範に研究された治療方式と方法と比べ、著者らのような新製品候補製品の監督管理審査過程はもっと高価で、時間がかかるかもしれない。さらに私たちは新しいものを開発しています
54
薬物治療の進展に伴い、FDA或いは類似の外国の監督管理機関は臨床試験の終点を考慮して臨床上意義のある結果を提供するリスクが増加する可能性があり、それによって産生された臨床データと結果は更に分析しにくいかもしれない。これまで、遺伝子治療製品がFDAや同様の外国規制機関の承認を得ることは少なく、これにより、私たちの候補製品が米国、EU、またはEUまたは他の管轄区域で規制承認を得るのにどのくらい時間がかかるか、またはいくらかかるかを決定することは困難であった。しかも、一つの規制機関の承認は他の規制機関が何を承認する必要があるかを代表しないかもしれない。
プログラム可能なエピジェネティック薬物を管理する規制要求はすでに変化し、未来に変化し続ける可能性がある。例えば、FDAはその生物製剤評価·研究センター(CBER)内に組織と高度治療オフィスを設置し、遺伝子治療と関連製品の審査を統合し、その審査についてCBERにアドバイスを提供するために細胞、組織、遺伝子治療諮問委員会を設立した。FDAの監督とIRBsの監督に加え、米国国立衛生研究院(National Institutes of Health、NIHと略称する)が発表したガイドラインによると、遺伝子治療臨床試験は機関生物安全委員会(IBC)の審査と監督を受けなければならない。IBCは地域機関委員会であり、当該機関の組換えや合成核酸分子を用いた研究の審査と監督を担当している。どの機関が臨床研究を開始する前に、この機関のIRBおよびそのIBCは、研究の安全性を評価し、公衆衛生または環境に対する任意の潜在的リスクを決定する。NIHガイドラインは強制的ではないが,関連研究がNIH組換えや合成核酸分子研究助成を受けた機関で行われているか,あるいはその助成によって行われていない限り,多くの会社や他のNIHガイドラインに拘束されていない機関は自発的にこれらのガイドラインに従っている。さらに、候補遺伝子治療製品の臨床試験において他の人が発生する深刻な有害事象または進展は、FDAまたは他の規制機関が私たちの臨床試験を一時停止し始めるか、または他の方法で私たちの任意の候補製品に対する承認要求を変更する可能性がある。FDAは個別遺伝子治療プログラムを継続できるかどうかを決定しているにもかかわらず,他の審査機関の審査過程や決定は臨床試験の開始を阻害または延期する可能性があり,たとえFDAがこの試験を審査して起動を許可したとしても。これらおよび他の規制審査機関、委員会および諮問グループおよびその公表された要求およびガイドラインは、追加の臨床前研究または臨床試験を行うことを要求し、私たちの開発コストを増加させ、規制の立場や解釈の変化を招き、これらの候補治療薬の承認と商業化を延期または阻止し、あるいは重大な承認後の制限または制限を招く可能性がある。似たような要求は連合にも適用される。ヨーロッパ薬品管理局(EMA)には高級療法委員会(CAT)があり、高級療法医薬製品の品質、安全性と有効性の評価を担当している(S)。ATMPは遺伝子治療薬、体細胞治療薬と組織工学薬物を含む。CATの役割は、ATMP候補者のマーケティング許可申請に関する意見草案を準備することであり、この草案はEMAに提出される。EUでは、ATMPの制定と評価はEU関連基準を背景に審議されなければならない。EMAは遺伝子治療薬製品の開発とマーケティング許可に関する新しいガイドラインを発表し、これらの新しいガイドラインを遵守することを要求する可能性がある。同様に、他の管轄区域にも複雑な規制環境が存在し、これらの環境では、私たちの候補製品のための規制承認を求め、規制環境をさらに複雑化させることが考えられるかもしれない。
規制指針の適用の変化は、規制審査過程を延長する可能性があり、追加的な研究や試験を行うことを要求し、私たちの開発コストを増加させ、規制の立場や解釈の変化を招き、私たちの候補製品の承認と商業化を延期または阻止し、あるいは重大な承認後の制限または制限を招くことができる。私たちが私たちの候補製品を推薦する時、私たちは規制機関に相談し、適用されたガイドラインを遵守するように要求されるだろう。もし私たちがこれをできなかったら、私たちはこのような候補製品の開発を延期または停止することを要求されるかもしれない。このような追加的な手続きは私たちが予想していたより長く検討と承認過程を招くかもしれない。規制審査手続きの増加或いは延長或いは私たちの候補製品開発の更なる制限による遅延はコストが高い可能性があり、そして私たちが適時に臨床試験を完成し、私たちの現在と未来の候補製品を商業化する能力にマイナス影響を与える可能性がある。
この潜在的な新しい薬物クラスでは、見かけの遺伝子コントローラ薬は承認されておらず、他の人または私たちの努力のために承認されないかもしれない。このような新薬の新規性と前例のない性質のため、リボ核酸薬物開発は重大な開発と監督管理リスクを持っている。
潜在的な新しい薬物カテゴリーとして,これまでエピゲノム制御薬はFDAや他の規制機関の承認を得ていない。私たちまたは私たちの戦略パートナーがエピゲノム制御薬の発見と開発に成功したことは非常に不確実で、多くの要素に依存しています
55
その中のいくつかは私たちや彼らがコントロールできるものではない。私たちは、メッセンジャーリボ核酸技術、交付技術、および製造プロセスに関連する決定を含む、一連の商業的決定を継続し、一定のリスクを負担して、私たち、私たちの戦略パートナー、または他の人のさらなる作業によって、これらの決定が正しくないことが証明される可能性がある。
著者らは早期開発段階で有望そうな薬物は推進できない可能性があり、臨床前段階或いは臨床段階で遅延を経験し、臨床保留を経験し、或いは多くの原因で発売できないかもしれない
我々の研究薬物は現在LNPで調製·管理されている。これらのLNPはLNPの成分に関連する系統的な副作用を引き起こす可能性があり,その中のいくつかはヒトでテストされていない可能性がある。LNPsの公認されている限界の1つは,単回投与と繰り返し投与時に炎症反応が発生する可能性があり,耐性や治療指数に影響する可能性がある。したがって,我々の特許と内部開発した独自LNPシステムの設計は高い耐性を有し,体内再投与によりLNP車両に関連する毒性を最低に低下させた。我々は我々のLNPsを最適化し続けているが,我々のLNPsが悪影響を与えない保証はない.著者らは薬物のいくつかの態様はmRNA或いは脂質からの免疫反応、及び生物経路内の副作用、或いはmRNA或いはLNPの分解を引き起こす可能性があり、その中のいずれも著者らの1つ以上の臨床前或いは臨床研究における重大な有害事象を引き起こす可能性がある。我々のLNPは、免疫反応、輸液反応、補体反応、オプソニン反応、抗体反応、IgA、IgM、IgEまたはIgGまたはそれらの何らかの組み合わせ、またはLNPに関連するいくつかの脂質またはポリエチレングリコール成分のポリエチレングリコールまたはポリエチレングリコールに対する反応のうちの1つまたは複数を全体的にまたは部分的に促進することができる。これらのタイプの副作用の多くはLNPsで広く観察されている。このような有害事象の根本的な原因については,不確実性が生じる可能性があり,将来の臨床試験で副作用を正確に予測することは困難であり,われわれの計画の著しい遅延を招くであろう.
臨床前開発は不確定であり、特にエピゲノムコントローラなどの新しい薬物に対して、したがって、私たちの臨床前計画または開発候補項目は延期、終了、または永遠に臨床に入らない可能性があり、その中のいずれも私たちのプラットフォームまたは私たちの業務に実質的な悪影響を与える可能性がある。
私たちのほとんどのプロジェクトは臨床前開発段階にある。候補薬物開発のための臨床試験を開始する前に、INDを支持する良好な実験室実践、あるいはGLP、およびアメリカ以外の同等の要求、毒理学試験を含む広範な臨床前研究を完成しなければならない。使用された疾患モデルの可変性を含む臨床前発展は不確定である。さらに臨床前研究所に進めるために必要な治療活性や安全性の特徴を有する開発候補薬,あるいは最初に有望な開発候補薬の臨床前研究結果がさらなる試験を支持していない可能性がある。私たちはまた、化学、製造および制御に関する広範な作業、またはCMC、活動(収率、純度、および安定性データを含む)が任意のINDまたは同様の外国文書に含まれるであろう。エピゲノムコントローラなどの新規薬物のCMC活動には広範な製造プロセスや分析開発が必要であり,不確定で長い。私たちは私たちの臨床前テストと研究の適時な完成或いは結果を確定することができず、FDA或いは他の監督機関が私たちの臨床前テスト或いは私たちが提案した臨床計画の結果、あるいは私たちの臨床前テスト、研究と研究の結果を受け入れるかどうかを予測することもできない
56
CMC活動は最終的に私たちのプロジェクトのさらなる発展を支持するだろう。したがって,我々が期待しているスケジュール上でINDや同様の臨床前計画申請を提出できることを保証することはできず,INDや同様の申請の提出がFDAや他の規制機関が臨床試験の開始を許可することを保証することもできない。
OTX−2002の臨床開発は延期または終了される可能性があり、私たちはOTX−2002に対する規制部門の承認を得ることができないかもしれないが、これは私たちのプラットフォームまたは私たちの業務に実質的な悪影響を及ぼすかもしれない。しかも、臨床開発には大量の資本投資が必要であり、これは私たちが支持できないかもしれない。私たちはOTX-2002と私たちの他の候補製品の開発と商業化を完了する過程で予測不可能なコストが発生したり、遅延が発生したり、最終的には達成できない可能性がある。
FDAや他の同様の外国規制機関が私たちの候補製品の販売を許可する前に、私たちの候補製品の安全性と有効性を証明するために、臨床前開発と広範な臨床試験を完了しなければならない。臨床試験費用は高く、時間がかかり、しかも不確定性が存在する。1つまたは複数の臨床試験の失敗はこの過程の任意の段階で発生する可能性があり、臨床前研究および早期臨床試験の結果は後の臨床試験の成功を予測できない可能性がある。そのほか、臨床前と臨床データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品が臨床前研究と臨床試験で満足できると考えているが、しかし依然としてその薬物の発売許可を得られなかった。
2022年7月,我々はFDAがわれわれのIND申請を承認し,OTX−2002による肝癌治療の1/2段階臨床試験を開始したことを発表し,MYCHELANGELO臨床計画により開始された初めてのヒト臨床試験である。私たちはまだ私たちの候補製品のために他のどんな臨床試験も開始したり完了していない。私たちは私たちのどんな臨床試験も計画通りに起動したり、計画通りに完成することを保証できません。将来のINDまたは同様の出願を提出することが、適用される場合、FDAまたは他の規制機関が将来の臨床試験をタイムリーに開始することを可能にするかどうかも決定できません。また,これらの試験が開始されても,規制当局のこのような臨床試験の一時停止や終了を招く可能性がある。1つまたは複数の臨床試験の失敗は試験の任意の段階で発生する可能性があり、我々の臨床試験は成功しない可能性がある。成功またはタイムリーな臨床試験の開始または完了を妨げる可能性のあるイベントは、
57
また,新冠肺炎の大流行による中断は,開始,登録,計画中や進行中の臨床試験を開始する際に困難や遅延に遭遇する可能性が増加する可能性がある。臨床試験の開始や完了に成功できない場合は、私たちの追加コストをもたらしたり、製品販売から収入を創出する能力を弱める可能性があります。臨床試験の遅延は、承認された製品が特許保護を受ける時間を短縮することも可能であり、私たちの競争相手が私たちよりも先に製品を市場に出すことが可能であり、候補製品を商業化する能力を弱める可能性があり、私たちの業務を深刻に損なう可能性がある。
臨床試験はFDAや他の適用規制機関の法的要求,法規やガイドラインに基づいて行われ,これらの政府機関や臨床試験を行う医療機関の倫理委員会やIRBsの監督を受けなければならない。臨床試験が、私たち、データ安全監視委員会またはデータ安全監視委員会またはFDAまたは任意の他の規制機関によって一時停止または終了された場合、またはそのような試験を行った機関のIRBsが、その臨床研究者およびその審査を受けた場所の参加を一時停止または終了する場合、遅延に遭遇する可能性もある。このような機関は、様々な要素のために臨床試験を一時停止または終了する可能性があり、これらの要素は、監督管理要求または著者らの臨床規程に従って臨床試験を行うことができなかったこと、FDAまたは他の監督機関の臨床試験操作または試験場所の検査の実施による臨床休止、予見できない安全問題または副作用、候補製品の使用のメリット、政府法規または行政措置の変化を証明できなかったこと、または臨床試験を継続するのに十分な資金が不足していることを含む。
また,われわれの臨床試験の首席研究員は時々私たちの科学コンサルタントやコンサルタントを務め,このようなサービスに関する報酬を得る可能性がある。場合によっては、私たちはFDAまたは同様の外国規制機関にいくつかの関係を報告することを要求されるかもしれない。FDAや同様の外国の規制機関は結論を出す可能性があり、私たちと主要な研究者との財務関係は利益の衝突をもたらしたり、他の方法でこの研究の解釈に影響を与えたりする。したがって,FDAや同様の外国の規制機関は,適用された臨床試験地点で発生するデータの完全性を疑問視する可能性があり,臨床試験自体の効用が脅かされる可能性がある。これは、fdaや同様の外国の規制機関が、私たちのマーケティング申請の承認を遅延させたり、拒否したりする可能性があります
58
このような状況は、最終的に私たちの1つまたは複数の候補製品の上場承認を拒否することにつながる可能性もある。
私たちの候補製品のすべての臨床試験の完成を遅延することは私たちのコストを増加させ、私たちの候補製品の開発と承認過程を遅くし、そして私たちの製品販売と製品収入を創造する能力を遅延或いは危険にさらす可能性がある。さらに、臨床試験の開始または完了遅延をもたらす多くの要因は、最終的には、私たちの候補製品が規制部門の承認を得ることを拒否される可能性もある。したがって、私たちの臨床試験に生じるどんな遅延も、候補製品を商業化する独占的な権利を持つ任意の期限を短縮することができ、私たちの競争相手は私たちの前に製品を市場に出すかもしれません。これは私たちの候補製品の商業的可能性を著しく低下させるかもしれません。これらの状況のいずれも、私たちの業務、財務状況、経営結果、および将来性を深刻に損なう可能性がある。
また,FDAや他の規制機関の臨床試験に関する政策が変わる可能性があり,追加の政府法規が公布される可能性がある。例えば,EUの臨床試験に関する規制構造が最近変化している。EU臨床試験条例,あるいはCTRと呼ばれ,2014年4月に採択され,EU臨床試験指令が廃止され,2022年1月31日に施行された。“臨床試験指令”は,臨床試験を行う各加盟国で主管する国家衛生当局と独立した倫理委員会に単独の臨床試験申請(CTA)を提出することを要求しているが,CTRは集中的な手続きを導入し,多センター試験の申請の提出のみを要求している。CTRは、スポンサーが各会員国の主管当局と道徳委員会に文書を提出することを可能にし、各会員国が決定を下すことを可能にする。CTAの評価手続きも統一されており、すべての関連加盟国による共同評価を含み、道徳基準を含む各加盟国が個別にその領土に関する具体的な要求を評価する。各会員国の決定は集中されたEUポータルサイトを通じてスポンサーに伝達される。CTAが承認されると,臨床試験開発は継続可能である。CTRは3年間の過渡期が予想される。進行中の臨床試験と新たな臨床試験がCTRによってどの程度制御されるかはそれぞれ異なる。(I)2022年1月31日までに“臨床試験指令”に基づいて申請を提出した臨床試験、または(Ii)2022年1月31日から2023年1月31日までの間、かつスポンサーが“臨床試験指令”を適用する臨床試験を選択し、2025年1月31日までこの指令によって管轄されている。この日以降,すべての臨床試験(行われている臨床試験を含む)はCTR条項に拘束される。我々と我々の第三者サービスプロバイダ(例えばCRO)がCTR要求を遵守することは、我々の開発計画に影響を与える可能性がある。
イギリスがどの程度その規制をEUと統合することを求めているのかは不明だ。臨床試験に関するイギリスの規制枠組みは、既存のEU立法(二次立法によってイギリス法律に定着された)に由来する。2022年1月17日、イギリスの薬品と保健製品監督機構(MHRA)は8週間のコンサルティングを開始し、内容はイギリスの臨床試験立法を再構築し、具体的な目標は臨床試験の審査を簡略化し、革新を支持し、臨床試験の透明性を高め、より大きなリスク割合を実現し、そして患者と公衆の臨床試験への参加を促進することである。MHRAは2023年3月21日に諮問結果を公表し、既存の立法を更新することを確認した。これによる立法変化は公表されておらず,最終的にイギリス法規と(EU)CTRの一致度が決定される。イギリスはその法規をEUが採用している新しい方法と緊密に結合しないことを決定し,他の国ではなく,イギリスでの臨床試験のコストに影響を与える可能性がある。
もし私たちが既存の要求の変化にゆっくりあるいは適応できない場合、あるいは新しい要求を採用したり、臨床試験を管理する政策を採用すれば、私たちの発展計画も影響を受ける可能性がある。
FDAと類似の外国規制機関の規制承認過程は長く、高価で、時間がかかり、本質的に予測できない。もし私たちが最終的に規制部門の候補製品の承認を得ることができなければ、私たちは製品収入を生むことができなくなり、私たちの業務は深刻な損害を受けるだろう。
FDAの承認なしに、私たちはアメリカで商業化、マーケティング、普及、またはどんな候補製品も販売してはいけない。外国の規制機関もまた似たような要求を実施した。FDAと類似の外国監督管理機関の承認を得るのに要する時間は予測できないが、通常は臨床試験開始後数年後に必要であり、監督機関のかなり大きな適宜決定権を含む多くの要素に依存する。また、承認政策、法規、あるいは承認を得るために必要な臨床データのタイプと数量は候補製品の臨床開発過程で変化する可能性があり、司法管轄区域によって異なる可能性がある。私たちはまだ規制部門の許可を得ていない
59
米国または任意の他の管轄区域の候補製品であっても、私たちが将来開発を求める可能性のある任意の候補製品は、規制部門の承認を得られないかもしれない。
米国や他の場所で候補製品を商業化する承認を得る前に、厳格に制御された試験によって多くの証拠を提供し、これらの候補製品が安全で有効で純粋で予期される用途であることをFDAまたは他の規制機関に満足させなければならない。私たちの候補製品の非臨床的または臨床的データが有望だと信じていても、これらのデータはFDAまたは他の規制機関の承認を支持するのに十分ではないかもしれない。FDAまたは他の規制機関はまた、承認前または承認後に私たちの候補製品に対して追加の臨床前研究または臨床試験を行うことを要求することができ、または私たちの臨床開発計画の要素に反対する可能性がある。
FDAまたは任意の外国規制機関は、私たちの候補製品の承認を遅延、制限、または拒否することができ、または追加の非臨床または臨床試験を行うことを要求することができ、または以下の理由を含むが、これらに限定されない多くの理由で計画を放棄することができる
さらに、FDAと外国規制機関は彼らの承認政策を変更し、新しい規定を制定する可能性がある。例えば、欧州委員会が2020年11月に開始した欧州薬品戦略イニシアティブを背景に、EU薬品立法は現在全面的な審査が行われている。欧州委員会は医薬製品に関するいくつかの立法文書の改正に関する提案(規制排他性期限の短縮、迅速通路の資格改正など)を可能にしている。2023年4月26日に出版された。提案された改正はまだ欧州議会と欧州理事会の同意と採択を待たなければならない。これらの提案は可決までに重大な修正が行われる可能性があり、2025年初めまではないと予想される。しかし、長期的には、これらの改正は製薬業と私たちの業務に大きな影響を与える可能性がある。
この長い承認過程と,臨床試験結果の予測不可能性は,規制部門の承認を得ることができず,任意の候補製品を市場に出すことができず,業務を大きく損なう可能性がある。
60
我々が最終的に臨床試験を完了し、我々の候補製品のBLAまたは海外マーケティング申請の承認を得たとしても、FDAまたは同様の外国規制機関は、第4段階の臨床試験を含む高価な追加試験のパフォーマンスに応じて承認される可能性があり、および/またはREMSまたは同様のリスク管理措置を実施することは、承認された後の薬剤の利益がそのリスクよりも大きいことを確実にするために必要である可能性がある。FDAまたは同様の外国の規制機関もまた、適応または患者数が私たちが最初に要求したものよりも限られた候補製品を承認する可能性がある。適用可能な規制承認を得るか得られないかのいずれの遅延も、候補製品の商業化を延期または阻止し、私たちの業務や将来性に大きな悪影響を及ぼすだろう。
著者らの候補製品は深刻な不良事件、不良副作用或いはその臨床開発を阻止し、その監督管理の承認を阻止し、その商業潜在力を制限し、或いは重大な負の結果を招く可能性のある他の特性と関係があるかもしれない。
私たちの候補製品によって引き起こされる有害事象または他の副作用は、私たち、試験の任意のDSMBまたは規制機関の臨床試験の中断、延期、または停止を招く可能性があり、より厳しいラベルやFDAまたは他の同様の外国規制機関の規制承認遅延または拒否を招く可能性がある。我々の実験結果は副作用の重症度と一般性を示す可能性があり,これは受け入れられない。この場合、私たちの実験は一時停止または終了される可能性があり、FDAまたは同様の外国規制機関は、私たちの任意またはすべての目標適応を承認する候補製品の開発を停止または拒否するように命令することができます。薬物に関連する副作用は、患者の募集または患者の試験完了能力に影響を与える可能性があり、あるいは潜在的な製品責任クレームを招く可能性がある。これらの状況のいずれも、私たちの業務、財務状況、経営結果、および将来性を深刻に損なう可能性がある。
臨床試験を行っている間、患者は彼らの研究医に疾病、傷害、不快感を含む彼らの健康変化を報告した。一般に、研究されている候補製品がこれらの状況をもたらしているかどうかを決定することは不可能だ。私たちがより大きく、より長く、より広範な臨床試験で私たちの候補製品を試験するとき、またはこれらの候補製品の使用がより広くなるにつれて(規制部門の承認を得た場合)、患者は、以前の試験で観察された疾患、傷害、不快感、および他の有害事象、および以前の試験で発生しなかったか、または検出されなかったことを報告するかもしれない。多くの場合,研究製品が大規模臨床試験で試験を行った後,あるいは承認後に患者にビジネス規模の製品を提供した後にのみ,副作用を検出することができる場合がある。
もし臨床開発過程中にいかなる深刻な不良事件が発生した場合、私たちが開発した任意の候補製品或いは製品の臨床試験は一時停止或いは終了される可能性があり、私たちの業務は深刻な損害を受ける可能性がある。治療に関連する副作用はまた、患者の募集と患者の試験完了或いは潜在的責任クレームを招く能力に影響を与える可能性がある。規制当局は私たちに任意のまたはすべての目標適応を承認する任意の候補製品の開発を停止または拒否するように命令することができる。もし私たちが任意の臨床試験を延期、一時停止、または終了することを要求された場合、これらの候補製品の商業的将来性が損なわれる可能性があり、それらまたは私たちが開発した他の候補製品から製品収入を創出する能力が延期またはキャンセルされる可能性がある。
さらに、もし私たちの1つまたは複数の候補製品が発売承認され、私たちまたは他の人が後にこのような製品によって引き起こされる不良副作用または有害事象を発見した場合、多くの潜在的な重大な負の結果をもたらす可能性があるが、これらに限定されない
61
これらの事件のいずれも、特定の候補製品に対する市場受容度を達成または維持することを阻止することができ、承認されれば、私たちの業務を深刻に損なう可能性がある.
わが社は候補製品を商業化したことがなく、現在と将来の候補製品の規制承認を得る際に遅延や予期せぬ困難に遭遇する可能性がある。
私たちは規制部門からどんな候補製品も承認されたこともなく、商業化されたこともない。FDAは、私たちが計画したBLASの任意または全部の実質的な審査を拒否するか、または私たちのデータを審査した後、私たちの申請がいかなる候補製品の規制承認を得るのに十分ではないと結論を出すかもしれない。FDAが私たちの計画中のどのBLASも承認しなければ、追加的に高価な臨床試験、臨床前研究、またはCMC研究を要求し、その後、私たちの申請を再検討することができるかもしれない。これらまたはFDAによって要求される任意の他の研究の範囲によれば、私たちが提出した任意のBLAまたは他の出願の承認は大幅に延期される可能性があり、数年遅れるかもしれないし、利用可能なリソースよりも多くのリソースが必要になるかもしれない。規制の承認を得る上でのどんな失敗や遅延も、候補製品を商業化し、収入を創出し、利益を達成し、維持することを阻止するだろう。FDAは、追加の研究を行って完了すれば、私たちが提出したBLAまたは他の申請を承認するのに十分ではないかもしれないと考える可能性もある。似たような危険は外国の管轄区域に存在するかもしれない。これらの結果のいずれかが発生すれば、私たちは私たちの候補製品の開発を放棄することを余儀なくされる可能性があり、これは私たちの業務に実質的な悪影響を与え、運営を停止させる可能性があります。外国の管轄区域での私たちの申請もまた似たような危険に直面している。
もし私たちが患者を募集して任意の臨床試験に参加する時に困難に遭遇すれば、私たちの臨床開発活動は延期されたり、他の悪影響を受けたりする可能性がある。
様々な理由から,臨床試験では患者登録の困難に遭遇する可能性がある。臨床試験方案に基づいて適時に臨床試験を完成し、他の事項以外に、試験が終了するまで十分な数の患者を募集する能力があるかどうかに依存する。患者の登録は多くの要素に依存している
さらに、我々が計画している臨床試験は、私たちの試験に参加することを選択する可能性のある患者の中には、競争相手のうちの1つによる試験に参加することを選択する可能性があるので、他の臨床試験と同じ治療領域または同様の領域を有する製品を他の臨床試験と争奪するであろう。合格臨床研究者の数が限られているため、著者らはいくつかの競争相手が使用した同じ臨床試験地点で著者らのいくつかの臨床試験を行うことが予想され、これは著者らがこれらの臨床試験地点で臨床試験を行うことができる患者数を減少させる。
患者登録の遅延はコスト増加を招く可能性があり,あるいは計画中の臨床試験の進行時間や結果に影響を与える可能性があり,これらの試験の完了や開始を阻止し,候補製品開発を進める能力に悪影響を及ぼす可能性がある。
62
著者らは時々発表或いは公表した臨床試験の一時、“主要”と初歩的なデータはより多くの患者データの獲得に従って変化する可能性があり、そして監査と検証手続きの制限を受け、これは最終データの重大な変化を招く可能性がある。
私たちは時々私たちの臨床前研究と臨床試験の初歩的或いは主要なデータを公開する可能性があり、これらのデータは当時利用可能なデータの初歩的な分析に基づいて、結果及び関連する発見と結論は特定の研究或いは試験の関連データをより全面的に審査した後に変化する可能性がある。データ分析の一部として、私たちはまた仮定、推定、計算、および結論を下すだろうが、私たちはすべてのデータを全面的かつ詳細に評価する機会がないか、または機会がないかもしれない。したがって、より多くのデータが受信され、十分に評価されると、私たちの報告の主要または予備結果は、同じ研究の将来の結果とは異なる可能性があり、または異なる結論または考慮要因が、これらの結果を合格させる可能性がある。トップラインデータと初歩データは依然として監査とチェック手続きを受けなければならず、これは最終データが以前公表されたトップラインデータ或いは初歩データと大きく異なる可能性がある。したがって、最終データが利用可能になる前に、トップラインおよび予備データは慎重に見られなければならない。
私たちはまた時々私たちの臨床前研究と臨床試験の中期データを開示することができる。私たちが完成する可能性のある臨床試験の中期データは、患者登録の継続とより多くの患者データの獲得に伴い、あるいは私たちの臨床試験の患者が彼らの疾患の他の治療を継続するにつれて、1つまたは複数の臨床結果が実質的に変化する可能性があるというリスクを受ける可能性がある。初期または中期データと最終データとの間の不利な違いは、私たちのビジネスの将来性を深刻に損なう可能性があります。しかも、私たちまたは私たちの競争相手が中間データを開示することは私たちの普通株の価格変動を招くかもしれない。
さらに、規制機関を含む他の人は、私たちの仮定、推定、計算、結論または分析を受け入れないか、またはデータの重要性を異なる方法で解釈またはトレードオフする可能性があり、これは、特定の計画の価値、特定の候補製品または製品の承認または商業化、およびわが社の全体的な状況に影響を与える可能性がある。さらに、開示された特定の研究または臨床試験に関する情報を選択することは、通常、広範な情報に基づいており、あなたまたは他の人は、私たちが決定した重要な情報または他の適切な情報が私たちの開示に含まれることに同意しない可能性がある。もし私たちが報告した中期、トップライン、または予備データが実際の結果と異なる場合、または規制機関を含む他の人が結論に同意しない場合、私たちが承認を得て私たちの候補製品を商業化する能力が損なわれる可能性があり、これは私たちの業務、経営業績、将来性、または財務状況を損なう可能性がある。
私たちは他の候補製品を決定して成功させる努力で成功しないかもしれない。
私たちの戦略の一部は新しい候補製品を決定することを含む。Omegaプラットフォームは臨床開発のための候補製品を生成できない可能性があり,これらのリスク要因で議論されている原因,および:
63
もし私たちが他の適切な候補製品を決定し、商業化に成功しなければ、これは私たちの業務戦略や財務状況に悪影響を及ぼすだろう。
我々はFDAから肝癌治療のためのOTX−2002の孤児薬物指定を取得しており,将来的にはより多くの候補製品のための孤児薬物指定を求める可能性があるが,市場排他性を含めてこのような指定や孤児薬物指定に関する利点を維持することができない可能性があり,製品収入(あれば)を減少させる可能性がある。
米国を含むいくつかの司法管轄区域の規制機関は、比較的少ない患者集団の治療を目的とした薬物または生物製品を孤児医薬製品として指定する可能性がある。孤児医薬品法によれば、薬物または生物学的薬物がまれな疾患または疾患の治療を目的としている場合、FDAは孤児薬として指定することができる。米国では、まれな疾患または疾患の定義は、通常、患者数が20万人未満であること、または米国では患者数が20万人を超えることを意味するが、米国では、米国の販売から薬剤開発コストを回収することができる合理的な期待はない。
米国では,孤児薬物指定は,税収割引やユーザ料金減免などの経済的インセンティブを当事者に与える権利がある。まれな疾患を治療するための医薬または生物製品の臨床試験は、これらの薬剤または生物製品が孤児として使用されるか否かにかかわらず、資金を贈与する機会を得ることも可能である。さらに、孤児薬として指定された医薬または生物学的製剤が、その後、そのような指定された疾患または状態を有する最初の市販承認を得た場合、製品は、限定された場合を除いて、FDAがその間に同じ疾患または状態の同じ薬物に対する別のマーケティング申請を承認することができないように、7年間の市場排他的期間を有する権利がある。もし私たちの競争相手が私たちの前に孤立した薬物の独占特許権を得ることができれば、私たちの候補製品と“同じ薬物”を構成し、同じ病気や病気を治療する製品については、長い間競争相手の製品を適用された規制機関の承認を得ることができないかもしれない。
われわれはFDAから肝癌治療のOTX−2002の孤児薬物名を取得した。私たちは私たちの未来の特定の候補製品のために孤児の称号を求めるかもしれない。しかし,これらの薬物の孤児薬物指定を成功させることはできず,孤児薬物指定に関する利点を保つことができない可能性がある。私たちが私たちの任意の候補製品のために孤児薬物排他性を獲得したとしても、この排他性は、異なる薬剤が同じ疾患または疾患のために承認されることができるので、これらの候補製品を競合から効果的に保護することができない可能性があり、孤児薬物の排他性は、FDAが別の疾患または疾患のために同じまたは異なる薬剤を承認することを阻止しない。孤児薬が孤児に独占特許を付与され、承認された後であっても、FDAが、大部分の標的集団においてより安全で、より効果的であることが証明されているので、FDAが臨床的に良いと結論した場合、FDAはその後、同じ疾患に対するその後の同じ薬物の出願を承認することができる。また,指定された孤児薬物が孤児指定の適応を得るよりも広い用途で許可されていれば,孤児薬物排他性を得ることはできない。さらに、FDAが後に指定要求に重大な欠陥があると判断した場合、またはこのようなまれな疾患または疾患患者の需要を満たすのに十分な数の製品を生産できない場合、米国における孤児薬の独占営業権を失う可能性がある。孤児薬物を指定することは薬物の開発時間や監督審査時間を短縮することもなく、監督審査或いは承認過程において薬物にいかなる利点をもたらすこともない。
私たちはすでに投資しており、omegaプラットフォームの研究開発努力をさらに強化することに引き続き投資する予定だ。このような投資は私たちの経営業績に影響を与えるかもしれません。もしこれらの投資のリターンが私たちの期待や発展速度よりも遅くなれば、私たちの収入と経営業績は影響を受けるかもしれません。
我々の技術力を利用して新たな候補製品を発見し,我々が設立して以来,omegaプラットフォームをさらに強化する研究·開発に投資していく予定である。これらの投資は、多くの時間、リスク、および不確実性に関連する可能性があり、これらの投資に関連する費用が私たちの利益率および経営業績に影響を及ぼす可能性があり、そのような投資は、市場の他の選択に対する十分な技術的優位性のリスクを生じない可能性があり、これは、逆に負担された債務およびこれらの新しい投資に関連する費用を相殺するために収入に影響を与えるであろう。バイオテクノロジー業界は技術と製品の発展に伴い迅速に変化し、これは私たちのプラットフォームが候補製品を識別と開発する能力を他の技術とプラットフォームよりも低くする可能性がある。私たちは私たちの競争地位を維持して向上させるために、omegaプラットフォームに大量の時間と資源を投入し続けなければならないと思う。このような予想された利点を達成できなければ
64
もしこれらの利点の実現が延期された場合、あるいは私たちの技術が私たちが予想していたように迅速に薬物発見プロセスを加速できなければ、私たちの収入と経営業績は不利な影響を受けるかもしれない。
私たちは迅速かつ重大な技術変革に適応し、競争力を維持するために、競争相手が発売した新製品や新技術に反応しなければならない。
私たちのプラットフォームを利用して私たち自身の候補製品を発見·開発するほか、他の生物製薬や製薬会社と協力して私たちのECsを発見·開発します。人工知能と精確な薬物設計をめぐる技術構造の特徴は著しく増強と発展する業界標準である。したがって、私たちと私たちの協力者の需要は急速に発展している。もし私たちが適切に革新して新しい技術に投資しなければ、私たちのプラットフォームはそれほど競争力がなくなる可能性があり、私たちの協力者は私たちの競争相手が提供する新しい技術、あるいは自分で薬物発見に従事することになるかもしれない。我々の多くのパートナーは,我々と協力するかどうかを決定するために初期時間投資を必要とするため,競争相手とパートナーシップや提携合意を達成すれば,このようなパートナーとのビジネス関係を再獲得することは困難である可能性があると考えられる.新しい解決策や技術改善をタイムリーに発売しなければ、私たちの製品は時間の経過とともに競争力を失う可能性があり、この場合、私たちの競争地位と経営業績が影響を受ける可能性がある。そこで,新たな技術や市場の開発と決定に多くの努力と資源を集中させ,薬物発見·開発における私たちの能力や専門知識をさらに拡大·深化させる。例えば、新たな革新的な技術や解決策をタイムリーに導入できなければ、パートナーのニーズを十分に予測できない、あるいは必要な市場受容度を得ることができなければ、私たちの業務は影響を受ける可能性があり、私たちの経営業績は不利な影響を受ける可能性がある。
私たちの候補製品の潜在的な市場機会は、私たちが予想していたより小さいかもしれない、または以前の治療を受ける資格がない患者または以前の治療を通過できない患者に限られる可能性があり、ターゲット患者集団の流行率の推定は正確ではないかもしれない。
著者らの現在と未来の目標患者集団は著者らの候補製品が解決する可能性のあるタイプの癌の発病率或いは流行率に対する信念と推定に基づいており、これらの推定は各種の源から来ており、科学文献と臨床調査を含む。私たちの予測は間違っていることが証明される可能性があり、潜在的な患者の数は予想を下回るかもしれない。私たちの候補製品がかなりの市場シェアを獲得しても、潜在的なターゲット層が少ない可能性があるので、規制部門の他の適応に対する承認を得なければ、私たちの候補製品を一線治療と二線治療に使用することを含めて、私たちは決して利益を達成しないかもしれない。
癌治療の特徴は時々第一線の治療(一線、二線、三線など)であり、FDAは通常最初に1種或いはいくつかの特定の治療方法しか承認しない。癌が十分に早く発見された時、第一線の治療は治愈を必要とすることなく、癌を治癒したり、生命を延長するのに十分であることがある。第一線の治療が成功しないことが証明された場合、通常は化学療法、抗体薬物、腫瘍標的小分子、ホルモン治療、放射線治療、手術あるいはこれらの療法の組み合わせであり、二次治療が実施される可能性がある。二線治療は、一般に、より多くの化学療法、放射線、抗体薬物、腫瘍標的小分子、またはこれらの薬剤の組み合わせを含む。三線治療は化学療法、抗体薬物と小分子腫瘍標的治療、より侵襲性のある手術形式と新しい技術を含むことができる。私たちは最初に、他の承認療法に失敗した患者の治療のための第2または第3のライン療法として、いくつかの候補製品の承認を求める予定である。その後,十分有益であることが証明された候補品については,あれば二次療法や潜在的な一次療法としての承認を求めたいが,我々の候補薬が三次療法として承認されても,二次療法や一次療法として承認される保証はない。また、二線または一線治療の承認を得る前に、追加の臨床試験を行わなければならないかもしれない。
私たちは成功しない可能性のある潜在的な候補製品に集中する可能性があり、私たちはより成功する可能性のある他の候補製品を開発する機会を放棄しなければならないかもしれない。
私たちは、最終的に成功しないことが証明された潜在的な製品候補に、私たちの努力と資源を集中させるか、または私たちの財務的予想に合わないマーケティング製品を許可または購入することを選択することができます。したがって、可能な商業製品または利益のある市場機会を利用することができず、他の候補製品または後により大きな商業可能性が証明される可能性のある他の疾患との機会を放棄または延期すること、または協力によってこれらの候補製品に対する貴重な権利を放棄することが要求される可能性がある
65
独占開発と商業化の権利を保持することが私たちに有利な場合、許可または他の特許権使用料の手配。もし私たちが他の適切な候補製品を決定し、商業化に成功しなければ、これは私たちの業務戦略や財務状況に悪影響を及ぼすだろう。
また、私たちの財力と人的資源は限られており、私たちの主要な候補製品の開発に重点を置いているため、私たちは他の未来の候補製品を追求する機会を放棄または延期する可能性があり、これらの製品は後により大きなビジネス潜在力を持っていることが証明された。私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。現在および将来の研究開発計画および他の特定の適応の将来の候補製品への支出は、いかなる商業的に実行可能な将来の候補製品も生じない可能性がある。もし私たちが特定の未来の候補製品の商業的潜在力や目標市場を正確に評価しなければ、私たちは協力、許可、または他の印税手配を通じて、これらの未来の候補製品に価値のある権利を放棄するかもしれないが、この場合、私たちはこれらの未来の候補製品の独占開発権と商業化権利を維持した方が有利になるだろう。
FDAの高速車線,突破性,再生医学高度治療指定が求められるかもしれない。これらの指定は、実際には、より速い開発または規制審査または承認プロセスをもたらすことができず、FDAが私たちが開発する可能性のある任意の候補製品を承認することを保証することもできない。
FDAの快速チャンネル、突破性と再生医学高級療法或いはRMAT計画はある合格製品の開発を加速することを目的とし、これらの製品は深刻な疾病と状況の治療を目的としている。候補製品が深刻または生命に危険な疾患の治療に使用され、臨床前または臨床データが、この製品がこの疾患が満たされていない医療需要を解決する潜在力があることを示す場合、スポンサーはFDA迅速チャネル認証を取得する資格がある可能性がある。候補製品が深刻または生命に危険な疾患の治療を意図しており、予備臨床証拠が、候補製品が1つまたは複数の臨床的重要終点において既存の療法の実質的な改善を示す可能性があることを示す場合、候補製品は突破的療法として指定することができる。もし製品候補が治療、修正、逆転、または深刻な生命を脅かす疾患を治療、修正、または治癒するための再生医学療法であり、初歩的な臨床証拠が、候補製品がこのような疾患が満たされていない医療需要を解決する潜在力を有することを示す場合、候補製品はRMAT称号を得ることができる。高速チャネル、突破、および/またはRMAT認証を求めることができるが、このような認証に成功する保証はない。たとえ私たちがそのような認証を受けたとしても、私たちは従来のFDAプログラムよりも速い開発過程、審査、または承認を経験しないかもしれない。高速チャネル、画期的な認証、またはRMAT認証は、候補製品が市場承認を得るか、または任意の特定の時間範囲で承認されることを保証することができない。また,FDAが我々の臨床開発プロジェクトのデータが高速チャネル,画期的またはRMAT指定をサポートしなくなったと考えた場合,その指定を取り消す可能性がある。高速チャネル、突破性、および/またはRMAT指定自体は、FDAの優先審査手順に適合することを保証することはできない。
たとえ私たちの候補製品がFDAの承認を得ても、私たちは決してアメリカ以外で承認されたり、そのような製品を商業化したりすることはありません。これは、私たちがそのすべての市場潜在力を達成する能力を制限するだろう。
米国以外の市場で任意の製品を販売するためには、安全性と有効性に関する他国の多くの規制要件を確立し、遵守しなければならない。一国で行われる臨床試験は、他の国の規制部門に受け入れられない可能性があり、一国の規制承認は、他のどの国でも規制承認を受けることを意味するものではない。承認手続きは国によって異なり、追加の製品テストと検証、および追加の行政審査期限が含まれる可能性があります。外国の監督管理機関の承認を求めることは著者らの重大な遅延、困難とコストを招く可能性があり、追加の臨床前研究或いは臨床試験を必要とする可能性があり、これは高価で時間がかかるだろう。各国の規制要求には大きな違いがある可能性があり、私たちの製品がこれらの国で発売されることを延期または阻止する可能性がある。これらおよび他の規制要件を満たすことは高価で、時間がかかり、不確実であり、予期しない遅延が生じる可能性がある。また、私たちはどの国でも規制承認を得ることができず、他の国の規制承認過程を延期したり、マイナス影響を与える可能性がある。私たちは国際市場を含めてどの司法管轄区でも候補製品の販売を承認しておらず、国際市場で規制承認を受けた経験もありません。もし私たちが国際市場の規制要求を守らない場合、あるいは必要な承認を得て維持できなければ、私たちが製品全体の市場潜在力を実現する能力は損なわれるだろう。
66
現在或いは未来の候補製品が市場の承認を得ても、それは医者、患者、第三者支払人と医学界の他の人が商業成功を獲得するために必要な市場受容度に達することができないかもしれない。
もし私たちが開発した任意の現在または未来の候補製品が市場の承認を得たら、それは依然として医師、患者、第三者支払人、および医学界の他の人の十分な市場受容度を得ることができないかもしれない。もし私たちが開発した候補製品が十分な受容度に達していなければ、著しい製品収入が生じないかもしれませんし、利益を上げることができないかもしれません。すべての候補製品が商業販売のために承認された場合、市場の受け入れ度は多くの要素に依存する
資金不足や世界的な健康問題によるFDAや他の政府機関の中断は、重要な指導部や他の人員の採用、保留、配置の能力を阻害する可能性があり、あるいは新製品や修正された製品のタイムリーまたは開発、承認、商業化を他の方法で阻止することは、私たちの業務にマイナスの影響を与える可能性がある。
FDAおよび外国規制機関が新製品を審査または承認する能力は、政府予算および資金レベル、法定、規制および政策変化、FDAまたは外国規制機関のキーパーソンの雇用と保留、およびユーザー費用支払いを受け入れる能力、およびFDAまたは外国監督機関が通常の機能を履行する能力に影響を与える可能性のある他の事件を含む様々な要素の影響を受ける可能性がある。したがって、FDAと外国規制機関の平均審査時間は近年変動している。また,他の研究開発活動を援助する政府機関への政府の援助は政治過程の影響を受けており,この過程は本質的に不安定で予測不可能である。EMAのようなFDAや他の機関は、アムステルダムへの移転や関連再編(人員変動を含む)後に中断が発生し、必要な政府機関の新薬の審査および/または承認に要する時間を遅らせる可能性もあり、これは我々の業務に悪影響を及ぼす。例えば、ここ数年間、米国政府は何度か閉鎖されており、FDAのようないくつかの規制機関は、FDAのキー従業員を休暇にし、キー活動を停止しなければならない。
また、新冠肺炎の流行に対応するため、アメリカ食品薬品監督管理局は国内外の異なる場所の製造施設の大部分の検査を延期した。FDAはすでに実行可能な情況下で国内施設に対する標準検査操作を回復したが、FDAは依然としてその検査活動の変化を監視と実施し、その従業員及び監督会社の安全を確保し、新冠肺炎の発展に適応し、いかなるウイルスの灰再発或いは新変種の出現は更なる検査遅延を招く可能性がある。米国以外の規制機関は、新冠肺炎の流行に対応するために似たような制限や他の政策措置をとっており、彼らの規制活動に遅延が生じる可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。政府が長期的に停止している場合、または世界的な健康問題がFDAや他の規制機関が定期的な検査、審査、または他の規制活動を阻害または阻止し続けている場合、FDAまたは他の監督管理機関が私たちの規制提出を適時に審査し、処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に大きな悪影響を及ぼす可能性がある。
私たちの保険証書は高くて、いくつかの商業リスクから私たちだけを保護して、これは私たちを重大な未保険債務に直面させます。
67
私たちは私たちの業務が直面する可能性のあるすべての種類のリスクに保険をかけない。私たちが現在維持しているいくつかの保険書には、一般責任、財産、自動車、雇用行為、労働者補償、環境責任、役員と高級管理者保険が含まれている。
私たちが将来得た任意の追加の製品責任保険は、私たちが受ける可能性のあるいかなる費用や損失を補償するのに十分ではないかもしれない。また、保険範囲はますます高くなっており、将来的には、責任による損失から私たちを保障するために、合理的なコストや十分な金額で保険範囲を維持することができないかもしれない。もし私たちの候補製品が市場の承認を得たら、私たちは商業製品販売を含む保険を購入するつもりです;しかし、私たちは商業的に合理的な条項や十分な金額で製品責任保険を得ることができないかもしれません。成功した製品責任クレームや一連の私たちに対するクレームは、私たちの株価を下落させる可能性があり、私たちの保険範囲を超えていると判断すれば、私たちが開発した任意の候補製品の開発と商業化を阻止または制限することを含む、私たちの運営および業務結果に悪影響を及ぼす可能性がある。私たちの環境責任保険は生物或いは危険材料の放出によるクレームに一定の保証範囲を提供しているが、私たちの財産、意外と一般責任保険は生物或いは危険廃棄物の暴露或いは汚染による損害と罰金は明確に含まれていない。したがって、汚染や傷害が発生した場合、損害賠償責任を請求されたり、私たちの資源を超えた罰金が科されたりする可能性があり、私たちの臨床試験や規制承認は一時停止される可能性があります。
上場企業として、取締役や上級管理者責任保険をより難しく、より高価にすることを難しくしており、同じまたは同様の保証範囲を得るために、低減された保険限度額および保証範囲を受け入れることが要求される可能性があります。したがって、私たちは合格した人を私たちの取締役会、取締役会委員会に参加したり、役員にしたりすることがもっと難しいかもしれない。しかし、私たちは私たちが既存の保険を維持し、十分な保険を提供できるかどうか分からない。いかなる重大な未保険債務も私たちが大量の金額を支払う必要があるかもしれません。これは私たちの現金と現金等価物の状況と運営結果に悪影響を及ぼすでしょう。
医療保健法やその他の法律コンプライアンス事項に関するリスク
私たちは広範囲で費用の高い政府によって規制されるだろう。
私たちの候補製品はFDA、医療保険と医療補助サービスセンター(CMS)、アメリカ衛生と公衆サービス部の他の部門、アメリカ司法省、州と地方政府及びアメリカ海外の相応機関の監督管理を含む広範かつ厳格な国内政府の監督管理を受ける。FDAは薬品の研究、開発、臨床前と臨床試験、製造、安全性、有効性、記録保存、報告、ラベル、包装、貯蔵、承認、広告、販売促進、販売、流通、輸入と輸出に対して監督管理を行う。私たちの製品が海外で販売されていれば、それらは特定の製品とその用途に対するFDAの承認を得たかどうかにかかわらず、外国政府によって広く規制されるだろう。このような外国規制はアメリカの相応の規制と同じように厳しいかもっと厳しいかもしれない。
政府の規制は私たちの製品の研究、開発、製造、販売のコストとリスクを大幅に増加させた。各候補製品の臨床前試験と臨床試験を含む審査と承認過程を監督することは、長く、高価で不確定である。私たちは臨床前研究と臨床試験を行う規制の許可を得て維持しなければならない。私たちが販売しようとしているすべての製品は規制部門の許可を得なければなりません。製品が使用する製造施設は検査され、法律の要求に適合しなければなりません。監督部門の承認を得るためには、各期待用途に対する製品の安全性と有効性、効力および純度を決定するために、各提案された治療適応のために、広範な臨床前および臨床データおよび他の支持情報を提出する必要がある。開発と承認過程には長年の時間がかかり、大量の資源が必要であり、製品の承認を招くことは決してないかもしれない。
特定の製品に対する規制承認を得ることができても、承認はその製品の指定された医療用途を制限する可能性があり、そうでなければ、製品を普及、販売、流通する能力を制限する可能性があり、コストの高い発売後監督を要求することができ、および/または継続的な発売後研究を要求する可能性がある。承認された製品の材料変更、例えば変更または修正されたラベルの製造には、さらなる規制審査および承認が必要となる可能性がある。承認されると、任意の承認が撤回される可能性があり、例えば、製品が以前未知のセキュリティ問題のような以前に未知の問題が存在することが後に発見された場合。
68
もし、私たち、私たちのコンサルタント、CDMO、CRO、または他の供給者が規制プロセスの任意の段階で適用される規制要件を遵守できなかった場合、このような不遵守が、承認申請または承認された申請の補充の遅延を招く可能性がある;FDAまたは他の規制機関を含む規制機関は、未解決の市場承認申請または承認された申請の追加の審査を拒否する;警告状、罰金、輸入および/または輸出制限、製品リコールまたは差し押さえ、禁止、完全または部分的な生産停止、民事処罰、以前に承認されたマーケティング申請または許可証の撤回、FDAまたは他の規制機関の政府契約に対する提案、および/または刑事起訴.
今後の医療法や政策の公布は、候補製品のマーケティング承認を得て商業化することの難しさやコストを増加させ、私たちの業務に悪影響を及ぼす可能性があります。
米国、EU、その他の司法管轄地域では、医療システムは、複数の立法や法規の変化、提案された変化が引き続き発生することが予想され、これらの変化は、私たちが開発している製品の発売承認を阻止または延期し、マーケティングの承認を得た任意の候補製品の承認後の活動を制限または規制し、定価と精算に影響を与え、私たちがこのような製品を販売する利益能力に影響を与える可能性がある。特に,米国連邦や州レベルでは,医療コストの低減と医療の質の向上を図る取り組みが継続されている。また,新たな条例や既存の医療法規や条例の解釈もしばしば採択されている。
2010年3月、“患者保護と平価医療法案”(Patient Protection and Affordable Care Act、ACAと略称する)が公布され、政府と私営保険会社が医療保健に融資する方式を大きく変えた。ACAの条項の中で、製薬とバイオテクノロジー産業にとって最も重要な条項は:
ACAのいくつかの側面は公布以来、司法、国会、行政面の挑戦を受けてきた。2021年6月17日、米国最高裁はいくつかの州がACAに対して提出した最新の司法挑戦を却下したが、ACAの合憲性を具体的に裁くことはなかった。最高裁が裁決を下す前に,総裁·バイデンはACA市場による医療保険の取得を目的とした2021年2月15日から2021年8月15日までの特殊保険期間を開始する行政命令を発表した。行政命令はまた、ある政府機関にそれを審査し、再検討するように指示した
69
医療保健の獲得を制限する既存の政策および規則は、作業要求を含む医療補助モデルプロジェクトおよび免除計画の再検討、医療補助またはACAによる医療保険カバー範囲の獲得が不必要な障害をもたらす政策を含む。他の医療改革措置が我々の業務にどのように影響するかは不明である。
また、ACAが公布されて以来、米国は他の立法改正を提案し、採択した。2011年8月、2011年の予算統制法により、医療保険提供者への医療保険支払総額が減少し、この法案は2013年4月に施行され、国会がさらなる行動を取らない限り、本報告の日から2031年まで有効となる。また、2013年1月、2012年に米国納税者救済法が署名され、病院、画像センター、癌治療センターを含むいくつかのタイプの提供者に支払う医療保険をさらに削減し、政府が提供者に多額の支払いを取り戻す訴訟時効を3年から5年に延長した。さらに、より広範な税金改革の一部として、孤児医薬品税控除が減少した。これらの新しい法律または将来導入される任意の他の同様の法律は、連邦医療保険および他の医療保険資金のさらなる減少をもたらす可能性があり、これは私たちの顧客および私たちの財務運営に負の影響を与える可能性がある。
また、支払い方法は医療立法と規制措置の影響を受ける可能性がある。例えば、協力医療は、結果に基づく精算など、新たな支払い·交付モデルを開発することができる。また、最近政府はメーカーが製品を販売するための価格設定方式の審査を強化し、アメリカ議会がいくつかの調査を行い、薬品定価の透明性を高め、連邦医療保険下の処方薬コストの低減及び定価とメーカー患者計画との関係を審査するための連邦立法を提出し、公布した。最近は2022年8月16日に“2022年インフレ削減法案”(IRA)が署名されて法律となっている。他の事項を除いて、アイルランド共和軍はある薬品のメーカーに連邦医療保険との価格交渉(2026年から)を要求し、価格は交渉できるが上限があり、連邦医療保険B部分と連邦医療保険D部分に基づいてリベートを実施し、インフレを超える価格上昇を処罰し(2023年に初めて満了)、D部分のカバーギャップ割引計画の代わりに新しい割引計画を用いる(2025年から)。アイルランド共和軍は,衛生·公衆サービス部(HHS)秘書が最初の数年間,規制ではなく指導によってその多くの規定を実施することを許可した。これらの計画の実施に伴い,HHSは指導意見を発表·更新し続けている。2023年8月29日、HHSは、連邦医療保険薬品価格交渉計画が現在法的挑戦を受けているにもかかわらず、価格交渉を受ける上位10種類の薬物のリストを発表した。このような理由と他の理由で、アイルランド共和軍がどのように実施されるのかは不明だ。また,バイデン政府の2022年10月の行政命令に応えるために,HHSは2023年2月14日に報告を発表し,医療保険と医療補助サービスセンター(CMS)革新センターでテストされた3種類の新しいモデルを概説し,これらのモデルは薬品コストを低減し,可獲得性を促進し,医療の質を向上させる能力を評価する。これらのモデルが将来の任意の医療改革措置で使用されるかどうかは不明である。
米国の個別州も、価格または患者の精算制限、割引、ある製品への参入の制限、マーケティングコストの開示および透明性措置を含む、薬品および生物製品の定価を制御するための法規を立法し、実施することが増えており、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。法律で規定されている第三者支払者の支払金額の価格制御またはその他の制限は、私たちの業務、運営結果、財務状況、見通しを損なう可能性があります。また,地域医療当局や個別病院では,どの薬品やサプライヤーが処方薬や他の医療計画に含まれるかを決定するために入札プログラムが使用されるようになってきている。これは私たちの候補製品に対する最終的な需要を減らすか、あるいは私たちの製品の価格設定に圧力を与えるかもしれない。
EUでは、承認されれば、同様の政治、経済、規制発展が候補製品を利益的に商業化する能力に影響を与える可能性がある。価格およびコスト制御措置に対する持続的な圧力に加えて、EUまたは加盟国レベルの立法発展は、著しい追加的な要求や障害を招く可能性があり、これは私たちの運営コストを増加させる可能性がある。EUで医療サービスを提供することは、医療サービスの確立と運営、薬品の定価と精算を含み、ほぼ完全に国家の法律と政策の問題であり、EUの法律や政策ではない。この点で,各国政府や保健サービス提供者は,保健サービスの提供や製品の定価や精算に異なる優先順位や方法を持っている。EU加盟国はその国家医療保険制度が精算を提供する薬品の範囲を自由に制限し、人が使用する薬品の価格と精算レベルを制御することができる。一部の司法管轄区域はプラスリストとネガティブリスト制度を実施しており、この制度の下で、製品は政府が価格を精算することに同意した後にのみ販売することができる。
70
EU加盟国は、薬品の具体的な価格または精算レベルを承認することができ、または薬品を市場に投入する責任を負う会社の収益力を直接または間接的に制御する制度をとることができ、数量に基づく手配、上限、および参考定価メカニズムを含む。いくつかのEU加盟国で精算や定価の承認を得るためには、私たちの候補製品の費用対効果を現地の看護標準と考えられる他の療法と比較する研究が必要かもしれない。他のEU加盟国は会社が自分の薬品価格を固定することを許可したが、会社の利益を監視した。全体的には,医療コスト,特に処方薬の下り圧力が非常に大きくなっている。全体的に言えば、大多数のEU加盟国の医療予算制限は関連医療サービスプロバイダが薬品の定価と精算を制限することを招く。これに加えて、EUや国が製品の開発やマーケティングを希望する人に増加している規制負担は、候補製品の上場承認を阻止または延期し、承認後の活動を制限したり、規制したりし、候補製品を商業化する能力(承認されれば)に影響を与える可能性がある。
2021年12月13日,第2011/24/EU号指令の改正第2021/2282号衛生技術評価条例(HTA)が可決された。この条例は2022年1月に施行されるが,2025年1月から適用され,その間に実施に関する準備と手順がとられる。この規定が適用されると、関連製品に基づいて段階的に実施される。この規定は、新医薬製品を含む衛生技術の評価におけるEU加盟国の協力を促進し、これらの分野で共同臨床評価を行うEUレベルの協力に基礎を提供することを目的としている。この法規は、EU加盟国がEU範囲内で汎用的なHTAツール、方法、およびプログラムを使用することを可能にし、患者に最大の潜在的影響を有する革新的な衛生技術の共同臨床評価を含む4つの主要分野で協力することを可能にし、科学コンサルティングを連合し、開発者はHTA当局にアドバイスを求め、新興衛生技術および将来性のある技術を決定し、他の分野で自発的な協力を継続することができる。個別EU加盟国は、衛生技術の非臨床(例えば、経済、社会、倫理)の評価を引き続き担当し、定価と精算について決定する。
米国やEU以外の市場では,精算や医療保険支払いシステムは国によって異なり,多くの国で特定製品や療法に価格上限が設定されている。
また、米国では、承認後の要求を拡大し、薬品の販売や販売促進活動を制限するための立法·規制提案がなされている。私たちは、より多くの立法変化が公布されるかどうか、あるいはFDAの法規、ガイドライン、解釈が変わるかどうか、あるいはこれらの変化が私たちの候補製品の上場承認にどのような影響を与える可能性があるかどうかを決定することはできない。また、FDA承認過程に対する国会のより厳格な審査は、上場承認を著しく延期または阻止し、より厳しい製品ラベルと上場後のテストとその他の要求の制約を受ける可能性がある。
私たちはアメリカ、EU、または任意の他の司法管轄区域の将来の立法または行政行動によって生じる可能性のある政府規制の可能性、性質、または程度を予測することができない。もし私たちまたは私たちが接触する可能性のある任意の第三者が既存の要求の変化に緩やかに適応できない場合、または新しい要求や政策を採用することができない場合、または私たちまたはそのような第三者が規制適合性を維持できない場合、私たちの候補製品は得られた可能性のあるいかなる規制承認も失う可能性があり、私たちは利益を達成したり維持することができないかもしれない。
もし私たちの候補製品が規制部門の承認を得たら、私たちと彼らは持続的な監督審査と重大な上場後の監督要求と監督を受けるだろう。
FDAまたは他の規制機関が私たちの任意の候補製品を承認した場合、私たちの候補製品の製造過程、ラベル、包装、流通、有害事象報告、貯蔵、広告、販売促進、輸入、輸出、記録保存は広範で持続的な規制要件を受けるだろう。これらの要求には,安全性や他の上場後の情報や報告,登録の提出,および承認後に行われた任意の臨床試験においてcGMPや類似の外国要求やGCPを継続的に遵守することが含まれている。また、バイオ製品メーカー及びその施設は、cGMP法規や同様の外国要求に適合することを確保するために、FDAや他の規制機関の持続的な審査と定期的な抜き打ち検査を受けている。私たちまたは規制機関が、予想されていない深刻度または頻度の不良事象、または製品の製造施設に問題があるような以前に未知の問題があることを発見した場合、規制機関は、製品のリコールを要求するか、または市場から製品を撤回するか、または生産を一時停止することを含む、製品、製造施設、または私たちに制限を加えることができる。しかも、私たちはどんな規制の承認を得ることができます
71
候補製品は、特定の年齢層の使用制限、警告、予防措置または禁忌症に関連する重大な制限を含むことができ、重い承認後の研究またはリスク管理要件を含む可能性がある。例えば、FDAは、私たちの候補製品を承認するためにREMSを必要とすることができ、これは、制限された分配方法、患者登録、および他のリスク最小化ツールのような薬物ガイドライン、医師トレーニングおよびコミュニケーション計画、または安全な使用を保証する他の要素を必要とする可能性がある。
適用された法規の要求を守らなければ、私たちは行政または司法制裁を受けるかもしれない
このような事件の発生は、候補製品を商業化し、収入を創出する能力を抑制する可能性があり、対応するのに時間と資源がかかり、負の宣伝が生じる可能性があるかもしれない。
さらに、FDAおよび他の規制機関の政策は変化する可能性があり、私たちの候補製品に対する規制承認を阻止、制限、または延期するための追加の政府法規が公布される可能性がある。米国や海外の将来の立法や行政または行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することはできない。
もし私たちが既存の要求の変化に緩やかに適応できない場合、あるいは新しい要求や政策を採用することができない場合、または規制適合性を維持できない場合、私たちは私たちが得る可能性のあるマーケティング承認を失う可能性があり、利益を達成したり維持することができないかもしれない。
米国の“ハッジ·ワックスマン法案”は,我々の各製品のために選択された特許の特許期間の延長を求める機会があり,特許期間の長さを延長することは,米国特許商標局または米国特許商標局およびFDAの審査·承認に依存すると規定されている。
米国では、“ハッジ·ワックスマン法”は、各製品の特許期間を1つの特許が正常に満了した後に最大5年間延長することを許可し、治療方法特許である場合は、承認された適応(または延長期間内に承認された任意の他の適応)に限定される。特許期間の延長の長さは、通常、臨床試験期間の半分にFDA審査BLA期間の全時間を加えて、これらの期間内の任意の遅延時間を減算する。特許期間の延長にも制限があり,薬品承認日から14年を超えない。したがって、もし私たちが最近提出·発表された特許の特許期間延長を選択して付与された場合、私たちは可能な特許期間延長からすべてのメリットを得ることができないかもしれない。例えば、適用期間内に出願できなかったか、関連特許が満了する前に出願できなかったか、または多くの適用要件のいずれかを満たすことができなかったため、特許期間の延長が全く付与されない可能性もある。また、適用当局は、米国のFDAおよびUSPTO、および他の国/地域の任意の同等の規制機関を含み、このような延期が利用可能かどうかの評価に同意しない可能性があり、私たちの特許延期の承認を拒否するか、または私たちが要求したよりも限られた延期を承認する可能性がある。このような状況が発生すれば、私たちの競争相手は私たちの特許が満期になった後、私たちの臨床と臨床前データを参考にすることで競争製品の承認を得て、他の場合よりも早く彼らの製品を発売するかもしれない。このような状況が発生すれば、私たちが製品収入を創出する能力に実質的な悪影響を及ぼす可能性がある。
72
1997年、食品と薬物管理局現代化法案(FDAMA)の一部として、国会は法律を公布し、児童薬物研究を行う薬品メーカーに激励を提供した。この法律は小児科研究と引き換えに6カ月間の排他性を規定しており,小児科排他性条項と呼ばれている。FDAMAに適合した臨床研究を行った場合、任意の法規データ独占期間と私たちの特許期間延長期間(受領すれば)で6ヶ月の期限を追加的に得ることができるかもしれない。しかし,FDAMAに適合した小児科研究を行わないことや,FDAに受け入れられないことを選択した場合,我々のデータ排他性や特許期間延長の追加6カ月の排他的延長は得られないであろう。
EUでは、追加保護証明書またはSPCは、規制審査中に失われた特許期間を補償するために特許期間を最大5年に延長するために使用することができ、合意された小児科調査計画に従って臨床試験データを取得すれば、さらに6ヶ月延長することができる(承認時に有効であれば)。すべてのEU加盟国はSPCを提供しなければならないにもかかわらず、SPCはすべての国に基づいて申請と承認されなければならない。これは、国によって異なるか、または全く付与されていない可能性があるこれらの証明書の申請と受信のコストが高い可能性があります.
我々の業務運営および調査者,医療専門家,コンサルタント,第三者支払者,患者組織と顧客との現在と将来の関係は,適用される医療規制法の制約を受け,処罰される可能性がある。
私たちの業務運営および調査者、医療専門家、コンサルタント、第三者支払人、患者組織と顧客との現在および将来の手配は、広範に適用される詐欺と乱用、および他の医療に関する法律と法規に直面する可能性があります。これらの法律は、私たちがどのように私たちの候補製品を研究、マーケティング、販売、流通するかを含めて、私たちが業務を展開する業務または財務的手配と関係を制約するかもしれない。これらの法律には
73
我々の内部運営と将来の第三者の業務配置が適用される医療法律や法規に適合することを確保することは、多くのコストに及ぶ。政府当局は、私たちのビジネス行為は、私たちと医師や他のヘルスケア提供者との関係を含むかもしれませんが、その中の一部の人は、株式または株式オプションの形で私たちに提供されたサービスが補償され、承認されれば、私たちの候補製品の注文や使用に影響を与える可能性があり、現在または未来に適用される詐欺および乱用または他の医療保健法律および法規に関する現行または将来の法規、法規、機関指導または判例法に適合しない可能性があります。私たちの業務が上記の任意の法律または私たちに適用される可能性のある他の任意の政府の法律および法規に違反していることが発見された場合、私たちは、MedicareおよびMedicaidまたは他の国または司法管轄区域の同様の計画、誠実な監督、および違反、返還、監禁、契約損害、名声損害、利益減少、および私たちの業務の削減または再構成の疑惑を解決する報告義務など、民事、刑事と行政処罰、損害賠償、罰金、政府援助の医療計画から除外される可能性がある重大な処罰を受ける可能性があります。もし私たちがそれと業務を行うことを期待している任意の医師や他の提供者や実体が適用されない法律を遵守していないことが発見された場合、彼らは政府の援助された医療計画や監禁から除外されることを含む刑事、民事または行政制裁を受ける可能性があり、これは私たちの業務運営能力に影響を及ぼす可能性がある。また、このような行為を防御するには費用がかかる可能性がある
74
これは時間がかかり、多くの人的資源が必要かもしれない。したがって、私たちが私たちに提起される可能性のあるいかなる訴訟も防ぐことに成功しても、私たちの業務は損害を受ける可能性があります.
私たちは政府の規制や他の法的義務の制約、特にプライバシー、データ保護、情報セキュリティに関する義務を受けている。実際または予想されるように、適用されるデータ保護、プライバシーおよびセキュリティ法律、法規、基準、およびその他の要求を遵守できないことは、私たちの業務、運営結果、および財務状況に悪影響を及ぼす可能性があります。
私たちの運営と業務の増加に伴い、私たちは新しいまたは追加のデータ保護法律と法規の制約または影響を受け、規制機関のより厳しい審査や関心に直面する可能性がある。米国では、2009年に“経済·臨床健康情報技術法案”によって改正された1996年の“健康保険携帯性·責任法案”およびその公布された条例、または総称してHIPAAと呼ばれ、他を除いて、個人が健康情報を識別できるプライバシー、安全、送信、違反報告に関するいくつかの基準が規定されている。多くの医療提供者は,我々がそれから患者の健康情報を取得する研究機関を含め,HIPAAが公布したプライバシーと安全法規の制約を受けている。我々は現在,HIPAA下の保証エンティティや業務パートナーに分類されているとは考えられないため,HIPAAの要求や処罰を直接受けることはない.しかしながら、事実および状況に基づいて、私たちが知らずにHIPAAによってカバーされているヘルスケア提供者または研究機関から個人識別可能な健康情報を受信し、その医療提供者または研究機関が個人識別可能な健康情報の開示に関するHIPAAの要求を満たしていない場合、重大な刑事罰に直面する可能性がある。
一部の州では、同様のプライバシーおよびセキュリティ法律法規が採択され、健康に関する個人情報や他の個人情報のプライバシー、処理、保護が管理されている。例えば、2018年6月28日、カリフォルニア州はカリフォルニア消費者プライバシー法を公布し、CCPAと略称し、2020年1月1日に施行された。CCPAはカリフォルニアの消費者にプライバシー権を創出し、ある個人情報を処理するエンティティのプライバシーとセキュリティ義務を増加させ、カリフォルニアの個人に特定の情報を開示することを要求し、これらの個人に特定の個人情報を販売しないことを選択するための新たな能力を与え、違反行為に対する民事処罰と、データ漏洩訴訟の可能性および関連リスクを増加させたデータ漏洩に対する個人訴訟権利を規定する。また,カリフォルニアプライバシー権法案(CPRA)は2023年1月1日に施行され,CCPAの重大な改正が行われた。それは、カバーされた企業に、追加の消費者権利手続き、データ使用の制限、より高いリスクデータの新しい監査要件、および敏感なデータからの退出を選択するいくつかの用途を含む追加のデータ保護義務を課す。それはまた、実質的な法規の発行を許可し、プライバシーと情報セキュリティ法執行の強化につながる可能性がある新しいカリフォルニア州データ保護機関を作成した。追加的なコンプライアンス投資と潜在的なビジネスプロセスの変更が必要になる可能性があります。これらの多く、複雑でよく変化する法規を遵守することは高価で困難であり、いかなるプライバシー法やデータセキュリティ法にも従わない、または流用、紛失または他の無許可処理、敏感または機密患者、消費者または他の個人情報を使用または開示するセキュリティ事件または違反行為は、私たち、私たちのCROの1つまたは他の第三者にかかわらず、調査コスト、重大な罰金と処罰、補償性、特殊、懲罰性および法定損害賠償;訴訟;私たちのプライバシーおよび安全実践に関する同意を含むが、これらに限定されない。影響を受けた個人に通知、信用監視サービスおよび/または信用回復サービスまたは他の関連サービスを提供することを要求し、私たちの営業許可証に不利な行動をとること、名声損害、および禁止救済を要求する。他の州でも同様の法律が可決され、州や連邦レベルで提案され続けており、これは米国がより厳しいプライバシー立法に傾いている傾向を反映している。このような法律の公布は相互衝突の要求があり、コンプライアンスを挑戦的にする可能性がある。もし私たちがHIPAA、CCPA、CPRA、または他の国内プライバシーおよびデータ保護法律の制約または影響を受けた場合、これらの法律の要求を遵守できなかったために負う任意の責任は、私たちの財務状況に悪影響を及ぼす可能性があります。
私たちのアメリカ以外の活動は追加的なコンプライアンス要求に制約され、規則に合わないために追加的な強制執行リスクが生じる可能性がある。例えば,2018年5月25日に一般データ保護条例(GDPR)が発効し,欧州経済域内の個人データの処理に厳しい要求が出された。例えば、GDPRは治外法権に適用され、データ当事者に詳細な開示を要求し、個人データを処理することができる法的根拠を開示して、個人データ(臨床試験データを含む)の収集および処理の有効な同意を取得し、敏感な個人データ(例えば、健康データ)を大規模に処理する際にデータ保護主任を任命し、データ当事者に強力な権利を提供し、政策、プログラム、訓練、およびデータ監査を含む適切なプライバシー管理をとることができる。それはまた、サービス提供者との契約時にデータ漏洩通知といくつかの義務を要求することを強制する。GDPRはヨーロッパ経済圏諸国は自分の法律と法規を制定することができると規定している
75
遺伝子、生体特徴、または健康データを含む個人データの処理を制限することは、個人データを使用および共有する能力を制限するか、またはコストを増加させる可能性がある。GDPRを遵守しなければならない企業は、より強力なデータ保護要件の規制法執行、および不適合であれば2000万ユーロまたは不適合会社の世界年収の4%までの罰金が科される可能性があるコンプライアンス義務およびリスクに直面しなければならない。罰金に加えて、GDPR違反は、規制調査、名声被害、データ処理活動の停止/変更の命令、執行通知、評価通知(強制監査のための)、および/または民事クレーム(集団訴訟を含む)を引き起こす可能性がある。他の要件では、GDPR規制は、GDPRによって制約された個人データを、このような個人データに対して十分な保護を提供することが発見されていない第3国に移すことで、米国、欧州経済地域と米国との間の既存の移動機構の有効性および持続性はまだ不確定である。欧州連合裁判所(CJEU)の判例法では,標準契約条項のみに依存する欧州委員会が承認した適切な個人データ転送機構としての標準契約形式は,必ずしもすべての場合で十分であるとは限らない可能性があり,移転はケースベースで評価されなければならない。2022年10月7日、バイデン総裁は米国の情報活動の保障措置の強化に関する行政命令に署名し、欧州経済圏から米国へのデータ移転に対するCJEUの懸念を解決するための新たな救済メカニズムと拘束力のある保障措置を導入し、2022年12月13日に発表された新たなEU·米国データプライバシー枠組み(DPF)の基礎を構成した。欧州委員会は2023年7月10日にDPFに関する十分性決定を採択し,DPFをDPF自己認証による米国実体のGDPR移行機構と効率的にした。DPFはまた、CJEU前の判決における重要な問題を解決し、標準契約条項による移転が将来的に挑戦される可能性が低いことを意味する可能性があるEU市民のための新たな救済メカニズムを導入した。我々は現在,EU標準契約条項とEU標準契約条項のイギリス付録,およびイギリス国際データ転送プロトコルとDPFに依存しており,グループ内と第三者移行において,個人データをヨーロッパ経済圏以外とイギリス(米国を含む)に移転している。私たちは、国際個人データ移転に関する既存の法律の複雑さと不確実性が引き続き存在すると予想する。特に,DPFの十分性決定が挑戦され,米国やより広い他の管轄地域への国際移転は規制機関の強化審査を受け続けることが予想される。したがって、いくつかの業務上の変更を行わなければならない可能性があり、規定された期限内に既存のデータ転送のために改訂された標準契約条項および他の関連文書を実行しなければならない。
さらに、2021年1月以降、私たちはGDPRとイギリスのGDPRを遵守しなければならず、各制度は違反行為に対して最高2000万ユーロ(1750万ポンド)または世界売上高4%の罰金を科すことができる。2023年10月12日,イギリスによるDPFの延期が発効し(イギリス政府の承認を得た),イギリスGDPRとしてイギリスからDPFに延期された自己認証を行う米国エンティティにデータを転送する仕組みである。
私たちのCDMO、CRO、または他の第三者サービスプロバイダが、私たちまたは私たちの顧客、サプライヤー、試験患者および従業員の個人識別情報および他の敏感なまたは機密情報にアクセスできることを保証することはできません。私たちはこれに責任がありますか、または彼らは私たちが適用した契約義務に違反しないか、または彼らはデータセキュリティの抜け穴や試みに遭遇しません。これは、プライバシー法律法規の下での私たちの義務および/または逆に私たちの業務、運営結果、および財務状況に悪影響を及ぼす可能性があります。私たちの契約措置と私たち自身のプライバシーと安全保障に関する措置が、第三者の処理、使用、保存、転送に関連するリスクから私たちを保護することを保証することはできません。さらに、私たちまたは私たちの協力者が健康情報を取得した患者、および私たちとこの情報を共有する提供者は、私たちが情報を使用して開示する能力を制限する法定または契約権利を持っている可能性がある。私たちは適用されるプライバシーとデータセキュリティ法律を持続的に遵守することを保証するために、多くの資本と他の資源を費やす必要があるかもしれない。私たちがプライバシー権を侵害したり、私たちの契約義務に違反していると主張して、私たちが責任がないことが発見されても、弁護は高価で時間がかかる可能性があり、否定的な宣伝を招く可能性があり、私たちの業務を損なう可能性があります。もし私たちまたは第三者CDMO、CROまたは他の請負業者またはコンサルタントが適用される連邦、州、または地方規制プライバシー要件を遵守できない場合、私たちは一連の規制行動の影響を受ける可能性があり、これらの行動は、私たちまたは私たちの請負業者が私たちの候補製品を開発および商業化する能力に影響を与える可能性があり、商業化できる任意の影響を受ける製品の販売を損害または阻止する可能性があり、あるいは私たちの製品の開発、商業化、およびマーケティングのコストおよび支出を大幅に増加させる可能性がある。いかなる脅威や実際の政府の法執行行動も否定的な宣伝を生む可能性があり、そうでなければ、これらの資源は私たちの業務の他の側面に使用できるように大量の資源を投入することが要求される。ソーシャルメディアをますます使用することは、責任、データセキュリティが破壊され、名声を損なう可能性があります
76
壊れています。上記のいずれも、我々の業務、財務状況、経営結果及び見通しに実質的な悪影響を及ぼす可能性がある。
私たちは環境、健康、安全法律法規の制約を受けており、私たちは環境コンプライアンスや救済活動に関する責任と巨額の費用を負担するかもしれない.
私たちの業務は、私たちの開発、テスト、製造活動を含めて、多くの環境、健康と安全法律法規の制約を受けています。他の事項に加えて、これらの法律および法規は、危険材料および生物材料の制御された使用、処理、放出および処置および登録、例えば化学溶媒、ヒト細胞、発癌化合物、変異原性化合物、および生殖、実験室操作および血液伝播病原体に毒性を有する化合物を管理する。もし私たちがこのような法律法規を守らなければ、私たちは罰金や他の制裁を受けるかもしれない。
我々と同様の活動をしている他社と同様に,危険や生体材料の放出や接触に関する責任を含む現在および歴史的活動に固有の環境責任リスクに直面している。環境、健康、そして安全の法律法規はもっと厳しくなっている。私たちは将来の環境コンプライアンスや救済活動に多くの費用を発生させることを要求される可能性があり、この場合、私たちの第三者メーカーの生産努力や私たちの開発努力は中断または遅延する可能性があります.
私たちと私たちの職員たちは内部と外部のコミュニケーションの手段としてソーシャルメディアツールをますます利用している。
変化するソーシャルメディア伝播ガイドを監視し、適用されるルールを遵守しようと努力していますが、私たちまたは私たちの従業員がソーシャルメディアを使用して私たちの候補製品やビジネスを伝播することは、適用要件に違反していることが発見される可能性があります。さらに、私たちの従業員は、法律法規、私たちの政策、および他の法律または契約要件に適合しない方法でソーシャルメディアを意図的または意図的に使用する可能性があり、これは、規制法執行行動、責任、商業秘密または他の知的財産権の損失を招き、または私たちの従業員、臨床試験患者、顧客、および他の人の個人情報公開をもたらす可能性がある。さらに、ソーシャルメディア上の私たちまたは私たちの候補製品に関する否定的な投稿やコメントは、私たちの名声、ブランドイメージ、および名声を深刻に損なう可能性があります。これらの事件のいずれも、私たちの業務、見通し、財務状況、および経営結果に実質的な悪影響を及ぼす可能性があり、私たちの普通株の価格に悪影響を及ぼす可能性がある。
商業化に関連するリスク
私たちの開発はまだ非常に早い段階にある。著者らの候補製品の多くは臨床前開発或いは発見段階にあり、著者らはOTX-2002のIND申請に対するFDAの承認を得て、関連する臨床試験を開始した。私たちは候補製品を商業化するために何年も必要だ。私たちの候補製品を臨床開発に進めることができず、規制部門の承認を得ることができず、最終的に私たちの候補製品を商業化したり、その過程で重大な遅延に遭遇したりすれば、私たちの業務は実質的な損害を受けるだろう。
我々の開発は非常に早期の段階にあり,これまで我々の研究と開発はomegaプラットフォームの開発,我々の最初の目標疾患適応の決定,我々の最初のECsの設計に集中してきた。著者らは著者らのいくつかの項目に対して体内前臨床研究を行っただけであり、著者らが他の項目に対して臨床前体内研究を行うことを保証することはできない。私たちが製品収入を作る能力は、私たちの候補製品の成功した臨床開発と最終商業化に大きく依存し、これは永遠に起こらないかもしれない。
アメリカで臨床試験を開始することはFDAがIND或いは外国の監督管理機関が類似の申請を受けることに依存し、FDAと他の監督機関との討論に基づいて最終的に試験設計を確定する。FDAや外国の規制機関が追加の臨床前研究の完成を要求している場合、あるいはFDAや外国の規制機関の他の要求を満たすことが要求された場合、私たちの臨床試験の開始は遅れるかもしれません。私たちがこれらの規制機関から指導を受けて導入された後も
77
当局、FDA、または他の規制機関は、私たちの臨床試験を開始するために、または私たちの試験設計または選択された臨床終点に対する受容可能な立場を変更するために、私たちが彼らの要求を満たしていることに同意しないかもしれません。これは、追加の臨床前研究または臨床試験を完成させること、または現在予想されているよりも厳しい承認条件を適用することを要求するかもしれません。
私たちの候補製品の商業化には追加の臨床前と臨床開発と規制とマーケティング承認が必要になるだろう。著者らが開発或いは上場許可を得る能力は著者らの財政とその他の資源が十分であるかどうかに依存し、必要な臨床前研究を完成し、INDの研究或いは類似研究、臨床試験及び成功登録と臨床試験を完成する。
もし私たちがこれらの活動のうちの1つまたは複数をタイムリーにまたは完全に成功させることができなければ、私たちは私たちが開発する可能性のある任意の候補製品を商業化することに成功したり、私たちの業務に実質的な損害を与えるかもしれない重大な遅延に遭遇するかもしれない。もし私たちの候補製品が規制部門の承認を得られなければ、私たちは運営を続けることができないかもしれない。
競争相手の発展は私たちの製品や技術を時代遅れにしたり、競争力に欠けたり、私たちの市場規模を縮小させるかもしれません。
私たちの業界はずっと広範な研究開発努力、迅速な技術発展、激しい競争と独自製品に対する高度な重視を特徴としている。新製品の関連市場への参入と先進技術の出現に伴い、私たちの候補製品は激しい競争とますます激しい競争に直面すると予想される。私たちは製薬、バイオテクノロジー、専門製薬会社を含む多くの異なる源からの潜在的な競争に直面している。学術研究機関、政府機関および公共·民間機関も競争力のある製品と技術の潜在的な源である。私たちの競争相手はすでに先進的な技術や方法を持っているか、または開発する可能性があり、これは彼らに競争優位を提供するかもしれない。これらの競争相手の多くはまた、私たちの候補製品において対象とされている治療カテゴリにおいて承認または開発されている化合物である可能性がある。また、これらの競争相手の多くは、単独でも協力者と一緒にも、より大きな研究開発プロジェクトを運営したり、私たちよりも多くの財力を持っていたり、より多くの経験を持っているかもしれない
もしこれらの競争相手が私たちの前に市場に進出し、より安全で、より効果的で、より安価な治療法を使用すれば、私たちの候補製品が商業化が承認されれば、利益にならないかもしれないし、開発を続ける価値もないかもしれない。製薬産業の技術は急速で大きな変化を経験しており、私たちはそれが引き続きそうすることを予想している。私たちがその開発に関連したいかなる費用を回収する前に、私たちが開発したどんな化合物、製品、またはプロセスは時代遅れになったり、経済的になったりする可能性がある。私たちの候補製品の成功は製品の効果、安全性、信頼性、獲得性、タイミング、監督管理の承認範囲、受容度と価格などの要素に依存する。著者らが成功した他の重要な要素は候補製品の開発速度、臨床開発と実験室テストの完成、監督管理の許可と製造及び大量の潜在製品の販売を含む。
私たちはエピジェネティック医学分野の新しい参入者からの競争に直面するかもしれない。他の技術を使った多くの会社と競争していますこれらの会社は現在求めているのと同じ適応を目指しています私たちの候補製品は、Alnylam製薬会社、Beam治療会社、Biogen社、CRISPR治療株式会社、Editas Medicine,Inc.,Ionis製薬会社、Janssen製薬会社、ファイザー、Sangamo治療会社を含む様々な技術(CRISPR遺伝子編集、遺伝子療法、非コードRNA療法、小分子エピジェネティクス)を開発する会社と競合することが予想されます。承認されて商業化されても、私たちの候補製品は病院、医師、患者の市場で受け入れられない可能性があります。病院、医者、あるいは患者は、私たちの製品はあまり安全ではない、あるいは効果があると結論するかもしれません
78
魅力は既存の薬に及ばない。もし私たちの候補製品がどんな理由でも市場の承認を得られなければ、私たちの収入潜在力は減少し、これは私たちの収益性に実質的な悪影響を及ぼすだろう。
私たちの多くの競争相手は私たちよりずっと多くの資本資源、強力な候補製品ルート、成熟した市場地位、及び研究開発、製造、臨床前と臨床テスト、監督管理の承認と精算及びマーケティング許可を得た製品に関する専門知識を持っている。したがって、私たちの競争相手は私たちよりも早く製品の商業化や特許や他の知的財産権保護を達成するかもしれない。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。これらの競争相手はまた合格した臨床、監督、科学、販売、マーケティングと管理人員を募集と維持し、臨床試験場と臨床試験患者登録を確立し、そして著者らの計画と相補的或いは必要な技術を獲得する上で著者らと競争している。もし私たちの競争相手が開発し商業化した製品が私たちが開発する可能性のあるどんな製品よりも安全で、より効果的で、副作用が少なく、より深刻ではなく、より便利で、あるいは私たちが開発する可能性のあるいかなる製品も時代遅れになったり、競争力がなくなったりすれば、私たちのビジネス機会は減少または消滅するかもしれない。
私たちの候補製品は予想以上に早く競争に直面するかもしれない。
2009年の“生物製品価格競争と革新法”(BPCIA)は、FDAによって許可された参考生物製品の生物学的に類似したまたは交換可能な生物製品のための短い承認経路を作成した。BPCIAによると,生物類似製品の申請は,参考製品が初めてFDA許可を得た4年後にFDAに提出されなければならない。また,FDAによる生物類似製品の承認は,この参考製品が初めて許可された日から12年後に発効する可能性がある。この12年間の独占期間内に、FDAが競合製品の完全なBLAを承認した場合、スポンサー自身の臨床前データおよび十分かつ良好に制御された臨床試験からのデータを含み、その製品の安全性、純度および効力を証明するために、別の会社は依然としてこの参照製品の競合バージョンを販売する可能性がある。
私たちは将来、BLAによって生物製品として承認された候補製品は、12年の独占経営期を得る資格があると信じている。しかしながら、国会の行動や他の理由により、この排他性は短縮される可能性があり、またはFDAは、私たちの候補製品を競合製品の参考製品とみなさず、予想よりも早く競争機会を創出する可能性がある。BPCIAの他の面では,そのいくつかはBPCIAの排他的規定に影響を与える可能性があり,訴訟のテーマでもある.アメリカ以外に、各司法管轄区は監督管理許可と以前に許可した参考製品と生物類似性を有する生物製品のために簡略化の道を確立した。例えば、2006年以来、EUは生物模倣薬に対する規制方法を確立した。さらに、承認されると、生物類似体がどの程度私たちのいずれかの参考製品を代替することができ、その方法は、非生物製品の伝統的な模造薬代替に類似しており、いくつかのまだ発展中の市場および規制要因に依存する。
私たちの候補製品の商業化の成功は、政府当局と健康保険会社が設立した保険範囲、十分な補償レベル、価格設定政策にある程度依存するだろう。もし私たちの候補製品が保証範囲を獲得または維持し、十分な補償を得ることができなければ、承認されれば、これらの製品を販売する能力を制限し、製品収入を創出する能力を低下させる可能性がある。
新たに承認された製品の保険カバーと精算に関する不確実性が大きい。米国では,連邦医療保険や医療補助計画のような第三者支払者,個人や政府支払者を含め,新薬や生物製品のカバー範囲を決定する上で重要な役割を果たしている。私たちが私たちの候補製品を商業化することに成功できるかどうかは、政府衛生行政部門、個人健康保険会社、その他の組織がこれらの製品と関連治療に提供する保険範囲と十分な補償にある程度依存する。政府当局や他の第三者支払人、例えば個人健康保険会社や健康維持組織は、どのような薬剤を支払うかを決定し、精算レベルを確立する。政府や個人支払者が提供する保険範囲と精算範囲は,多くの患者が治療費を負担できる鍵となっている。
第三者支払者は、医薬品およびサービスの課金に挑戦するようになっており、多くの第三者支払者は、同等の模倣薬、生物類似薬、またはより安価な治療法が利用可能であるため、特定の薬物および生物製品に保険および精算を提供することを拒否する可能性がある。1つかもしれません
79
第三者支払者は私たちの製品が代替可能であり、価格の低い製品のみを患者に精算するかもしれない。医師の監督下で管理されている製品については、このような薬物が高い価格に関連することが多いため、保険および適切な補償を得ることは特に困難である可能性がある。私たちの候補製品に対してより良い治療効果あるいはより良い管理利便性を示しても、既存の第三者療法の価格は私たちの候補製品に対する費用を制限するかもしれません。これらの支払者は、特定の製品の精算状態を拒否または撤回したり、新製品や既存市場製品の価格を低すぎるレベルに設定したりして、候補製品への投資から適切なリターンを実現できなくなる可能性がある。精算が得られない場合や限定精算だけでは、候補製品の商業化に成功できない可能性があり、候補製品に満足な財務リターンを得ることができない可能性もある。
米国では,連邦医療保険や医療補助計画のような第三者支払者,個人や政府支払者を含め,新薬や生物製品の保険や精算範囲を決定する上で重要な役割を果たしている。連邦医療保険計画は、個人や他の政府支払人がどのように新薬保険と精算政策を制定するかのモデルとしてますます多く使われている。しかし、米国の第三者支払者では、製品のカバーや精算に統一された政策はない。そのため、製品の保証範囲と精算範囲は支払人によって異なる。したがって、保証範囲の決定プロセスは通常、時間がかかり、高価なプロセスであり、私たちの候補製品を使用するために単独で各支払人に科学的かつ臨床的な支援を提供する必要があるが、保証範囲と十分な補償が一致するか、または最初に得られる保証はない。一部の第三者支払者は,新たなあるいは革新的な薬物療法の保証範囲をあらかじめ承認しておく必要がある可能性があり,このような治療法を用いた医療提供者に精算する。また、精算に関する規則や条例は常に変化しており、場合によっては短時間で通知されており、これらの規則や条例が変わる可能性があると考えられる。第三者決済者が私たちの候補製品の保証範囲と精算についてどのように決定するか予測できません。
アメリカ以外では、国際運営は通常広範な政府価格制御と他の市場監督管理を受けており、EUと他の司法管轄区のコスト制御措置に対する日々の重視はすでに私たちの候補製品の価格設定と使用に圧力を与え続けると信じている。多くの国では、国家衛生システムの一部として、医療製品の価格は異なる価格制御メカニズムの制約を受けている。他の国は会社が自ら医療製品を価格設定することを許可しているが、会社の利益を監督してコントロールしている。追加の外国価格規制や定価規制の他の変化は、私たちの候補製品に受け取ることができる費用を制限するかもしれません。そのため、米国以外の市場では、米国に比べて候補製品の精算が減少する可能性があり、商業的に合理的な収入や利益を生み出すには不十分である可能性がある。
また、米国や海外の政府や第三者支払者が医療コストの制限や低減に力を入れていることは、これらの組織が新承認製品のカバー範囲や精算レベルを制限する可能性があるため、私たちの候補製品に十分な支払いを提供できない可能性がある。管理的ヘルスケアの傾向,ヘルスケア組織の日増しに増加する影響力,追加的な立法変化により,我々の候補製品の販売に関する価格設定圧力に直面することが予想される。全体的に,医療コストの低下圧力は大きくなり,特に処方薬や生物製品,外科手術やその他の治療が行われている。そのため、新製品の参入にはますます高い壁が設けられている。
もし私たちが単独でまたは第三者と協力して販売、マーケティング、流通能力を確立できなければ、私たちは私たちの任意の候補製品を商業化することに成功できないかもしれません。承認されれば、どんな製品収入も発生できないかもしれません。
私たちは製品を販売、マーケティング、流通する人員やインフラが限られていて、会社として候補製品を商業化した経験がありません。そのような組織を設立して維持する費用はそうする費用効果を超えるかもしれない。
私たちは、アメリカや世界各地の他の市場で私たちの候補製品をマーケティングするために、自分の重点販売、流通、マーケティングインフラを構築することができるかもしれません(承認されれば)。私たち自身の販売、マーケティングと流通能力を確立することは、私たちが雇用し、維持し、適切に合格者を激励すること、十分な販売手がかりを生成すること、販売とマーケティング人員に十分な訓練を提供し、異なる地理的位置に分散した販売とマーケティングチームを効果的に管理する能力を含む大量の費用とリスクに関連する。私たちの内部販売、マーケティング、流通能力の発展のいかなる失敗や遅延もいかなる製品の発売を延期する可能性があります。これは私たちの製品の商業化に不利な影響を与えます
80
候補者、もし承認されたら。また、販売チームを募集してマーケティング能力を確立する候補製品のビジネス発表が何らかの理由で延期または発生していない場合には、これらの商業化費用を早期または不必要に発生させる。これは費用が高いかもしれません。もし私たちが私たちの販売とマーケティング担当者を維持したり再配置できなければ、私たちの投資は損失します。
候補製品の商業化を阻害する可能性がある要素は
内部販売、マーケティング、流通能力を確立しないか、または特定の国/地域に内部販売、マーケティング、および流通能力を確立しないことを決定することができない場合、協力手配を求めることができるかもしれません。もし私たちが協力手配を求めれば、私たちの販売は協力者の製品に対する戦略的興味と、そのような協力者が製品のマーケティングと販売に成功する能力に大きく依存するだろう。
もし私たちが自分の販売チームを構築したり、協力関係を構築して私たちの任意の候補製品を商業化することができなければ、候補製品の潜在的な商業化を延期したり、これらの候補製品に対する販売やマーケティング活動の範囲を縮小させられたりする可能性があります。もし私たちが私たちの支出を増やして商業化活動に資金を提供することを選択すれば、私たちは追加的な資本を得る必要があり、これらの資本は受け入れ可能な条件で私たちに提供できないかもしれないし、全く得られないかもしれない。私たちは理想的な場合よりも早い段階でパートナーと手配することができ、私たちは私たちの任意の候補製品に対する権利を放棄すること、または他の方法で私たちに不利な条項に同意することを要求される可能性があり、いずれも私たちの業務、運営結果、および見通しに悪影響を及ぼす可能性がある。
もし私たちが十分な販売、マーケティング、流通能力を確立できなければ、私たち自身も第三者と協力しても、私たちの他の候補製品を商業化することに成功できず、利益を上げることができず、重大な追加損失を招く可能性がある。私たちは現在広範で資金的に十分なマーケティングと販売業務を持っている多くの会社と競争するだろう。マーケティングおよび販売機能を実行するための内部チームまたは第三者の支援がなければ、これらのより成熟した会社との競争に成功することができないかもしれない。
また、十分な販売、マーケティング、流通能力が確立されていても、定価モデル、サプライチェーン、配信メカニズムなどにおける業界全体の傾向の進展は、私たちの予想から外れる可能性がある。もしこれらや他の業界の傾向が予想されていない方法で変化した場合、私たちは私たちの候補製品を商業化したり、利益を達成することに成功できないかもしれません。
私たちの将来の成長は私たちが外国市場に進出する能力にある程度依存するかもしれないが、そこでは追加の規制負担と他のリスクと不確定要素の影響を受けるだろう。
私たちの将来の収益性は、私たちの候補製品をEUを含む海外市場で商業化する能力があるかどうかにある程度かかっているかもしれませんが、私たちは第三者との協力に依存するかもしれません。外国市場関連規制機関の規制承認を得るまで、私たちは私たちの候補製品のマーケティングや普及は許可されておらず、私たちはいかなる候補製品の規制承認も得られないかもしれない。他の国で単独の規制承認を得るためには、これらの国/地域の私たちの候補製品の安全性と有効性に関する多くの異なる規制要求、および私たちの候補製品の臨床試験や商業販売、定価、流通などの規制要求を遵守する必要があり、これらの管轄区域で成功するかどうかを予測することはできない。もし私たちが承認されれば
81
もし私たちの候補製品が最終的に海外市場で商業化されれば、私たちは追加のリスクと不確実性に直面するだろう
私たちの候補製品の海外販売は、政府規制、政治的·経済的不安定、貿易制限、関税変化の悪影響を受ける可能性もある。
もし私たちの候補製品が商業化されることが承認されれば、私たちは第三者と選択的に協力して、アメリカ以外のいくつかの司法管轄区域でマーケティングを行うことができるかもしれない。私たちは国際製薬業務に関連する追加的なリスクに直面すると予想しています
私たちは以前このような分野で経験がなかった。しかも、EUと多くのEU加盟国は複雑な規制、税金、労働、その他の法的要求を提出し、私たちはこれらの要求を遵守しなければならない。多くのアメリカに本部を置くバイオテクノロジー会社は、EUで自分の製品をマーケティングする過程は非常に挑戦的であることを発見した。
特定の法律と政治的危険もまた外国の行動に固有のものだ。私たちが事業を展開する可能性のある国では、外国政府が民間企業を国有化する可能性があり、これにはリスクがある。特定の国や地域では、テロ活動、政治動乱、戦争、およびこれらの活動に対する反応は、米国よりも我々の行動に大きな脅威となる可能性がある。特定の国/地域の社会および文化規範は、実質的な法律および法規の遵守を要求する政策を含む、わが社の政策を遵守することを支持しない可能性がある。また、我々が業務を展開することが可能な国の全体的な経済·政治状況の変化は、私たちの財務業績や将来の成長にリスクとなる。また、米国国外で商業化された財務的および商業的に強力なパートナーを決定する必要があり、これらのパートナーは高度な製造と
82
私たちが要求する法律と規制コンプライアンス基準は私たちの財務業績に危険になる。私たちが世界で業務を経営するにつれて、私たちの成功は私たちがこれらのリスクと他の関連リスクを予測し、効果的に管理する能力にある程度依存するだろう。これらの要素や他の国際業務に関連する要因の結果が、私たちの業務、財務状況、または経営結果に悪影響を与えないことは保証されません。
いくつかの国、特にEUでは、処方薬の価格設定は政府によって統制されている。これらの国では,薬品発売許可を受けた後,政府当局との定価交渉にはかなりの時間がかかる可能性がある。一部の国で精算や定価の承認を得るためには、臨床試験を行う必要があるかもしれません。私たちの候補製品のコスト効果を他の利用可能な治療法と比較します。もし私たちの製品が精算を受けられない場合、あるいは範囲や金額が制限されている場合、あるいは定価が満足できないレベルに設定されている場合、私たちの業務は損害を受ける可能性があり、実質的かもしれません。
私たちの潜在的な製品に対する責任訴訟は、私たちが重大な責任を招き、私たちが開発する可能性のある任意の製品の商業化を制限するかもしれません。
臨床試験で私たちの候補製品を使用して市場の承認を得たどの製品を販売しても製品責任クレームのリスクに直面させます。消費者、医療提供者、製薬会社、または他の販売または他の方法で私たちの製品に接触した人は、私たちに製品責任を請求するかもしれません。予期せぬ悪影響を持つ製品に基づく集団訴訟では,多額の判決が下されることがある.もし私たちが製品責任クレームを正当化することに成功できなければ、私たちは大量の責任とコストを発生するかもしれないが、これらは保険範囲内ではないかもしれない。さらに、製品責任クレームは、是非曲直や最終結果にかかわらず、以下のようになる可能性がある
潜在的な製品責任クレームを防止するために、許容可能なコストで十分な製品責任保険を獲得または保持することができず、私たち単独または会社パートナーと開発した製品の商業化を阻止または阻害する可能性がある。私たちは臨床試験保険を受ける予定ですが、私たちの保険証書は各種の排除があるかもしれません。私たちは製品責任クレームの影響を受けるかもしれませんが、私たちは保険範囲を受けていません。私たちは私たちの保険範囲の制限を超えたり、私たちの保険カバー範囲内でない裁判所の裁決または和解合意で達成された任意の金額を支払う必要があるかもしれません。私たちはこれらの金額を支払うために十分な資本を持っていないか、または得ることができません。私たちが未来の会社のパートナーと合意しても、私たちは損害賠償を受ける権利があります。何かクレームがあれば、この賠償は利用できないか十分かもしれません。
私たちの第三者や製造業への依存に関するリスク
臨床前または臨床的に供給されるEC候補薬を生産する能力が制限される可能性があり,特にmRNAやLNPによる治療薬の製造需要が増加した場合には,われわれの発展計画に悪影響を及ぼす可能性がある。
83
我々は、第三者CDMOのメッセンジャーリボ核酸療法および脂質賦形剤、薬物または他の活性物質の担体または媒体として使用される脂質に依存して、我々の臨床前および臨床供給のEC候補を産生する。新冠肺炎を治療するためのワクチンは、メッセンジャーリボ核酸ワクチンおよび脂肪賦形剤を利用するワクチンを含む。アメリカ食品医薬品局はいくつかの新冠肺炎ワクチンを承認した。したがって,これらのCDMOに対する新冠肺炎ワクチンの生産需要はこれまでにないが,m RNAやlnPによる療法の生産能力は限られており,製造施設や材料の潜在力によってさらに制限される可能性があり,これらの施設や材料は1950年の“国防生産法”や同等の外国立法によって徴用され,我々の計画中の臨床試験に必要な製品は材料や製造槽を得ることが困難になる可能性がある。我々は期待した臨床前と臨床開発のために十分なECs供給を得るために努力しているが、メーカーが新冠肺炎ワクチンの供給をECsの上に置くため、供給制限と中断に遭遇する可能性がある。もし私たちが合理的な価格で適時または所望の数で必要な供給を得ることができなければ、私たちは私たちのEC候補者の開発を完了し、あるいは私たちが監督部門の私たちのEC候補者の承認を得たら、彼らを商業化する能力は実質的な悪影響を受ける可能性がある。
我々の候補ECは新技術に基づいており,複雑で製造が困難である可能性がある.私たちは製造、製品発表、賞味期限、テスト、貯蔵、サプライチェーン管理、あるいは輸送に困難に直面するかもしれません。
私たちの技術の新規性とより大規模な生産における限られた経験のため、私たちは製造、製品の発表、賞味期限、テスト、保存と供給チェーン管理或いは輸送の面で困難に直面する可能性があります。これらの困難は多くの原因によるものである可能性があり、より大規模な生産の複雑さ、設備故障、原材料と補助材料の選択と品質、分析テスト技術と製品の不安定を含むが、これらに限定されない。したがって、私たちの候補ECの臨床前または臨床開発は実質的に延期される可能性があり、あるいは新しい製剤医薬製品を使用するための新しい研究または試験を開始することが要求される可能性がある。
LNPsに包まれたmRNAコードのEC候補を産生する過程は複雑であり,良好な制御条件下で開発·製造しなければ薬理活性に悪影響を及ぼす可能性がある。しかも、私たちはまだビジネス規模で私たちのECを作っていない。私たちは生産を拡大する過程で困難に直面する可能性があり、それによって潜在的に臨床と商業供給に影響を与える。
私たちが引き続き私たちの薬品と薬品のために生産技術を開発することに伴い、私たちの生産技術の変更は逆に薬品の規格と安定性に影響する可能性がある。著者らの生産技術の変化は大量不合格を招く可能性があり、これは著者らの臨床前研究或いは任意の臨床試験の大幅な遅延を招く可能性がある。我々のEC候補は安定性があることが証明され,最終的に承認されたECの賞味期限が予想を下回ってしまう可能性がある(あれば).これは供給需要、在庫の浪費、そしてより高い商品コストのリスクをもたらす。
私たちの製品と製品の中間体は温度に非常に敏感で、私たちのいかなる製品あるいはすべての製品の安定性は予想に及ばないことを知ることができるかもしれません。私たちはまた輸送条件が製品の品質に否定的な影響を及ぼすことを発見するかもしれない。これは、私たちの1つまたは複数の候補ECの処方または製造プロセスを変更し、臨床的または商業的供給の遅延または中断をもたらす必要があるかもしれない。また,このような輸送サービスに関連するコストや限られたサプライヤーグループも供給中断のリスクを増加させる可能性がある.
私たちの革新率は高く、これはすでに高度な技術変革を招き続け、これは臨床開発期間と後に製品の比較可能性に負の影響を与える可能性がある。また、技術変革は、新しい製造インフラの変更、修正、または調達の需要を推進する可能性があります.
予測可能な未来に、私たちは第三者に依存して、私たちの研究計画、臨床前研究と臨床試験のために材料を製造と供給し、私たちはその中の多くの会社と長期契約をしていません。このような第三者への依存は、併用療法、候補製品、または私たちが開発および商業化する可能性のある任意の療法のための薬物供給を含む十分な量のそのような材料を含まないか、または許容可能なコストでそのような供給を得ることができず、私たちの開発または商業化努力を延期、阻止または損害する可能性があるというリスクを増加させる。
我々は自分たちの製造施設を開発する計画を評価し続けているが,少なくとも今後数年以内には,第三者が計画してくれた臨床試験および臨床前と臨床開発生産材料に依存することが予想される。私たちは第三者に部分的に依存して商業生産を行いたいです
84
私たちの候補製品は市場の承認を得た。私たちは現在臨床前と臨床材料を提供するために使用されているいかなる第三者メーカーと長期合意を締結していません。私たちは注文した方法で任意の必要な材料を購入します。その中のあるメーカーは私たちの生産に重要であり、これらのメーカーが私たちの競争相手に奪われているかどうかにかかわらず、あるいは受け入れられるコスト或いは品質で数量を得ることができなくても、著者らの臨床前研究或いは臨床試験を適時に行う能力を遅延、阻止或いは弱める可能性があり、そして私たちの開発と商業化努力に実質的かつ不利な影響を与えるであろう。
予測可能な未来には、引き続き第三者メーカーに部分的に依存し、マーケティング承認を得た任意の候補製品にビジネス供給を提供していくと予想される。私たちは第三者製造業者と必要な合意を維持または確立することができないかもしれないし、受け入れ可能な条項でそうすることができないかもしれない。たとえ第三者製造業者と合意できても、第三者メーカーに依存することは、追加的なリスクをもたらす
私たちは私たちの契約製造パートナーの製造過程のすべての側面を完全にコントロールすることができず、私たちの契約製造パートナーがcGMPや同様の海外法規を遵守して私たちの候補製品を製造することに依存する。第三者メーカーは、cGMP法規や米国以外の類似した法規要件を遵守できない可能性がある。もし私たちの契約製造業者が私たちの規格およびFDAまたは他の機関の厳格な規制要件に適合した材料を生産することに成功しなければ、彼らはその製造施設の許可を確保および/または維持することができないだろう。しかも、私たちの契約製造業者が十分な品質管理、品質保証、合格者の能力を維持することを制御することはできません。FDAまたは同様の外国規制機関が、これらの施設が私たちの候補製品を生産するために許可されていない場合、または将来的にそのような許可を撤回する場合、私たちは代替製造施設を探す必要があるかもしれません。これは、私たちの候補製品を開発、獲得、またはマーケティングする能力に深刻な影響を与えます。私たちまたは私たちの第三者製造業者が適用された法規を遵守できなかったことは、罰金、禁止、民事処罰、遅延、一時停止または承認撤回、許可証の取り消し、候補製品または薬物の差し押さえまたはリコール、操作制限、刑事起訴を含む制裁を実施する可能性があり、いずれも候補製品や薬物の供給に重大な悪影響を与え、私たちの業務および運営結果を損なう可能性があります.
私たちはCDMOに依存して私たちの候補製品を製造するために、私たち自身の製造施設やインフラを買収し、構築する計画を評価し続けています。私たち自身の製造施設とインフラを構築する計画は高価で時間がかかるだろうし、私たちは成功しないかもしれない。
85
私たちは、臨床前と臨床で必要な薬物物質を生産するために、CDMOへの依存を代替または補充するために、工場を借りて製造施設を建設することにしたかもしれない。もしレンタル契約を締結すれば、私たちは私たちが使用するために生産施設を改修してカスタマイズする予定です。著者らは、私たち自身の製造施設を建設することは、私たちにより良い臨床前研究と臨床試験材料の供給制御を提供し、技術変更をより迅速に実施でき、より良い長期利益率を許可することを予想している。しかし、会社として、私たちは製造施設を建設した経験がなく、私たち自身の製造施設や能力を構築することに成功しないかもしれません。そのため、我々の運営や施設を管理し、候補製品の研究開発、製造、最終商業化を継続するために必要なインフラを開発するために、より多くの人員を雇用する必要がある(承認されれば)。会社として、私たちは製造施設を設立、建設、あるいは最終的に管理した経験がありません。もし私たちが正しい位置を選択していない場合、あるいは賃貸契約を締結していない場合、あるいは計画のリフォームやカスタマイズを効果的に完了していない場合、あるいは必要な人員を募集し、全体的に効果的に私たちの成長を管理していなければ、私たちの候補製品の開発と生産は削減または延期される可能性がある。たとえ私たちが成功しても、私たちの製造能力はコスト超過、意外な遅延、設備故障、労働力不足、自然災害、電力故障と多くの他の要素の影響を受ける可能性があり、これらの要素は私たちが製造戦略を実現する期待的なメリットを阻止し、私たちの業務に実質的な悪影響を与えるかもしれない。
さらに、FDAおよび他の外国規制機関は、任意のロットの承認された製品のサンプルをいつでも提出し、適用試験結果を示す合意を要求することができるかもしれない。場合によっては、FDAや他の外国規制機関は、関連機関が発行を許可するまで大量の製品を配布しないように要求する可能性があります。製造過程中の微小な偏差は、品質属性や安定性に影響を与える偏差を含み、製品に許容できない変化を招き、大量故障や製品リコールを招く可能性がある。大量失敗や製品リコールは臨床試験や製品発表を延期する可能性があり、これは私たちに高い代価を払わせるかもしれません。そうでなければ、私たちの業務、財務状況、運営結果と将来性を損なうことになります。私たちの製造過程の問題は私たちの製品に対する臨床と市場の需要を満たす能力を制限するかもしれません。
私たちはまた、私たちの製造プロセスを操作するために必要な経験豊富な科学、品質管理、製造人員を雇用と維持する問題に直面する可能性があり、これは生産が遅れたり、適用された法規の要求を維持することが困難になる可能性がある。
私たちの製造プロセスや施設のどんな問題も、潜在的なパートナー(大きな製薬会社や学術研究機関を含む)の魅力の低いパートナーになる可能性があり、これは、より多くの魅力的な開発プロジェクトを獲得することを制限するかもしれません.
製造施設を管理する会社として、私たちは経験がありません。
私たち自身の製造施設の運営には大量の資源が必要になりますが、私たちは会社として製造施設を管理した経験がありません。一部は経験不足のため,我々の製造計画が時間どおりに完了するかどうか(あれば),あるいは自分たちの製造工場が計画してくれた臨床試験製造候補製品が開始または時間どおりに完成するかどうかを決定することはできない(もしあれば)。一部は私たちの経験が足りないため、私たちの製品の品質成功率と良率は受け入れられない或いは一致しないかもしれません。私たちは十分な品質管理、品質保証と合格者を維持できないかもしれません。さらに、もし私たちが将来1つ以上の候補製品のために現在のCDMOから私たち自身の製造施設に切り替える場合、私たちは修正された候補製品をより早いバージョンに接続するために追加の臨床前研究を行う必要があるかもしれない。私たちが計画した製造施設の獲得と運営に成功しなければ、私たちの候補製品の商業可能性に悪影響を及ぼす可能性があります。
私たちまたは私たちの第三者メーカーは十分な品質と数量で私たちの候補製品の生産規模を拡大することに成功できないかもしれません。これは私たちの候補製品の臨床進歩と商業化に影響を与えるかもしれません。
私たちの候補製品の臨床試験を行い、承認された任意の候補製品を商業化するためには、私たちと製造パートナーはこれらの製品を大量に生産する必要がある。しかし、私たちまたは彼らは私たちの任意の候補製品の製造能力をタイムリーにまたは費用効果的に向上させることに成功できないかもしれないし、全くできないかもしれない。また,上述したように,活動拡大期間中に品質の問題が生じる可能性がある.もし私たちまたは任意の製造パートナーが私たちの候補製品の生産規模を十分な品質と数量で成功的に拡大することができなければ、これらの候補製品の開発、テスト、および臨床試験は延期または実行不可能であり、それによって生じるいかなる規制承認または商業発売も延期される可能性がある
86
製品が遅延したり、入手できない可能性があります。これは私たちの業務を深刻に損なう可能性があります。供給源は時々中断する可能性があり,中断すると,合理的な期限内に許容可能な費用で供給を再開できるかどうか(一部または全部)供給できるかどうか,あるいは供給をまったく回復できないかどうかは決定できない。もし私たちが第三者が私たちの候補製品を商業的に供給するために製造したり、商業的に合理的な条項でそうすることができなければ、私たちの候補製品の開発と商業化に成功できないかもしれない。
私たちの候補製品で使用される脂肪補助材料のサプライヤーの数は限られていて、私たちのいくつかのサプライヤーは私たちの生産に重要です。もし私たちがキーサプライヤーを失ったら、私たちが候補製品開発を完成させる能力に実質的な悪影響を及ぼすかもしれない。もし私たちの候補製品が規制部門の承認を受けたら、私たちはそれらを商業化するために脂肪補助剤の供給を拡大する必要があるだろう。
私たちの候補製品中の脂質補剤成分の供給者の数は限られている。私たちはまた私たちのすべての油脂供給者たちと長期供給協定に到達しなかった。私たちは私たちの候補製品の脂質補剤成分のための追加の供給源を確立できないかもしれないし、許容可能な条件でそうすることができないかもしれない。
私たちの候補製品に脂肪補助剤を提供するサプライヤーの数は限られている。必要であれば代替サプライヤーから脂質補剤を獲得することが必要であれば,本当にあれば商業的に合理的な条件でそれらを得ることはできないかもしれない。他の会社と協力するために私たちの製造プロセスを再設計するには多くの時間と費用が必要かもしれませんが、再設計プロセスは比較可能な研究や接続研究のようなより多くの研究の必要性を引き起こす可能性があります。さらに、私たちのいくつかのサプライヤーは私たちの生産に重要であり、もしこれらのサプライヤーが私たちの競争相手に奪われたり、他の方法で流失したりすれば、私たちの開発と商業化努力に実質的な悪影響を及ぼすだろう。
著者らは依存し、引き続き第三者に依存して著者らの臨床前研究のいくつかの方面を行い、第三者に依存して著者らの計画した臨床試験を行う予定である。もし第三者がGCPによって計画された臨床試験を適時に行うことができない場合、候補製品のために監督管理の許可を求めたり、商業化する能力を求めたり阻害したりする可能性がある。
著者らはずっと第三者に依存して著者らの臨床前研究のいくつかの方面に依存し、そして第三者に依存して著者らの計画中の臨床試験を行い、そして著者らが行っている臨床前と計画中の臨床計画のモニタリングと管理データを提供する。著者らはこれらの側に依存して著者らの臨床前研究を実行し、そしてこれらの側に依存して著者らの計画した臨床試験を実行し、その活動のいくつかの方面のみを制御する。しかし、私たちは私たちのすべての研究と実験が適用された合意と法律、法規、そして科学的基準に従って行われ、私たちのCROと他の第三者への依存が私たちの規制責任を免除しないことを確実にする責任がある。私たちと私たちのCROは、FDAと同様の外国規制機関が臨床開発における候補製品に対して実行する法規およびガイドラインであるGCP要求を遵守することを要求される。規制機関は,試験スポンサー,主要調査者,試験地点を定期的に検査することでこれらのGCPを実行する。もし私たちまたは私たちの任意のCROまたは試験サイトが適用されたGCPに従わなかった場合、私たちの臨床試験で生成された臨床データは信頼できないと考えられるかもしれませんが、FDAまたは同様の外国の規制機関は、私たちのマーケティング申請を承認する前に追加の臨床試験を行うことを要求するかもしれません。特定の規制機関が検査を行った後、この監督機関は私たちのいかなる臨床試験がGCP規定に適合しているかどうかを確認することを保証することはできません。また,われわれの臨床試験はcGMP法規や米国以外の類似外国法規により生産された製品を用いて行わなければならない。私たちがこれらの規定を遵守しないことは、私たちが臨床試験を繰り返す必要があるかもしれないが、これは規制部門の承認過程を遅らせるだろう。
我々の計画中の臨床試験や臨床前研究を行ういかなる第三者も我々の従業員ではなく,また,我々がそのような第三者と合意して我々に提供してくれた救済措置を除いて,このようなCRO,研究者,他の第三者がそのような試験に十分な時間と資源を投入したり,契約要求に従って実行することは保証されない。もしこれらの第三者のいずれかが予想される最終期限内に達成できなかった場合、私たちの臨床方案を遵守したり、規制要求を満たしたり、あるいは他の方法で不合格を示した場合、私たちが計画した臨床試験は延長、延期、または終了される可能性がある。また、私たちと契約した多くの第三者も、私たちの競争相手を含む他の商業実体と関係があるかもしれません。彼らはまた、これらのエンティティのための臨床試験や他の製品開発活動を行っているかもしれません。これは、私たちの競争地位を損なう可能性があります。さらに、私たちが計画している臨床試験の首席研究者は、時々私たちの科学顧問またはコンサルタントを担当し、そのようなサービスに関連する現金および現金等価物または株式補償を得ることができるかもしれない。もしこれらの関係や関連した補償が
87
もし私たちが実際に利益の衝突があると考えたり、FDAなどの外国の規制機関が財務関係が試験の解釈に影響を与える可能性があると結論した場合、適用される臨床試験の場所で生成されたデータの完全性が問われる可能性があり、臨床試験自体の効用が脅かされる可能性があり、これは、FDAに提出された任意のBLAまたは私たちが外国の規制機関に提出した任意の同様の外国規制申請が遅延または拒否される可能性がある。そのような遅延や拒否は私たちが候補製品を商業化することを防ぐことができる。
もし私たちがこのような第三者との任意の関係が終わったら、私たちは他の第三者と合意したり、商業的に合理的な条項でそうすることができないかもしれない。追加のCRO、調査者、および他の第三者を交換または追加することは、追加のコストに関連し、管理時間および重点を必要とします。しかも、新しいCROが仕事を始める時、自然な過渡期がある。したがって,遅延が生じ,必要な臨床前と臨床開発スケジュールを満たす能力に実質的な影響を与える可能性がある。CRO、調査者、その他の第三者との関係を慎重に処理しているにもかかわらず、将来的に挑戦や遅延に遭遇しない保証はなく、これらの遅延や挑戦が私たちの業務、財務状況、および見通しに実質的な悪影響を与えないことを保証することはできない。
私たちは第三者と協力して私たちの候補製品を開発し、それを商業化する。我々は既存の連携関係の維持や将来の連携関係の構築と維持に成功できない可能性があり,我々の候補製品の開発と商業化に成功する能力を大きく制限する可能性がある.
私たちはすでに協力協定を締結しており、将来的には候補製品の開発と商業化のために他の協力関係を求めることができるかもしれない。例えば、2021年11月、私たちはPM(CF)Explorations,Inc.と5年間の協力協定を締結した;2022年10月、私たちは日東電工会社と協力と許可協定を締結した;2023年12月、私たちはノボノルドA/S、パイオニア医薬08会社と他の各方面と研究と協力協定を締結した。これらの類似した未来と任意の第三者との配置によると、私たちは、彼らと共同開発を求める可能性のある任意の候補製品の開発または潜在的に商業化されたリソースの数および時間に、私たちの協力者を共有または限定的に制御することができ、または制限されている。私たちが商業実体とのこれらの計画から製品収入を生成する能力は、私たちの協力者がこれらの手配の中で彼らに割り当てられた機能を成功的に履行する能力に依存するだろう。私たちは私たちが参加したどんな協力も成功するかどうかを予測できない。私たちの候補製品に関する協力は私たちに次のようなリスクを与えてくれる
88
より多くの財務、マーケティング、販売、技術、または他の業務資源を持つ他の会社との適切な協力を求めている場合、激しい競争に直面する可能性があります。バイオテクノロジーと製薬会社間の業務統合はまた潜在的な協力者の数の減少を招く。また、交渉過程は時間も複雑であり、私たちはタイムリーで、受け入れ可能な条件で協力を交渉することができず、交渉することさえできないかもしれない。それができなければ、私たちが協力を求めている候補製品の開発を減らしたり、潜在的な商業化を延期したり、販売やマーケティング活動の範囲を縮小したり、支出を増やしたり、自費で開発や商業化活動を行ったりしなければならないかもしれません。もし私たちが私たちの支出を増やし、私たち自身の開発や商業化活動に資金を提供することを選択すれば、私たちは追加の資本を得る必要があるかもしれないし、これらの資本は受け入れ可能な条件で私たちに提供できないかもしれないし、全く得られないかもしれない。もし私たちが十分な資金がなければ、私たちは候補製品をさらに開発したり、それらを市場に出して製品収入を生成することができないかもしれない。
私たちまたは私たちの協力者が、合意によって付与された権利を行使しないことを選択した場合、または私たちまたは私たちの協力者が候補製品を既存の運営に成功的に統合することができない場合、既存または将来の協力の利点を達成することができない可能性がある。さらに、もし私たちと私たちの任意の協力者との合意が終了すれば、私たちの協力者が私たちに権限を与えた技術と知的財産権へのアクセスが制限されたり、完全に終了したりする可能性があり、これは、私たちが協力者の技術や知的財産権を利用して私たちの候補製品を開発し続けることを延期するか、あるいはこれらの候補製品の開発を完全に停止することを要求するかもしれない。私たちはまた、適切な代替協力者を見つけたり、新しい協力者を誘致することが難しいことを発見するかもしれませんし、私たちの開発計画が延期されるかもしれませんし、商業·金融界での私たちのイメージが悪影響を受ける可能性があります。いずれの協力者も、製品開発、規制承認、商業化に関連する多くのリスクに直面する可能性があり、これらのリスクは本“リスク要因”の節で説明されているが、私たちの協力者にどんな負の影響も私たちに悪影響を及ぼす可能性がある。
89
私たちの従業員と独立請負業者は、主要な調査者、CDMO、CRO、コンサルタント、サプライヤー、および私たちが従事する可能性のある研究、開発、監督、製造、品質保証および他の薬品機能および商業化に関連する任意の第三者を含み、規制基準および要求を遵守しないことを含む不適切な行為または他の不適切な活動に従事する可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。
主要な調査者、CDMO、CRO、コンサルタント、サプライヤー、および研究、開発、監督、製造、品質保証および他の医薬品機能および商業化に関連する任意の第三者の不正行為を含む可能性のある研究、開発、規制、および他の医薬品機能および商業化に関連する任意の第三者の不正行為を含むことができ、これらの行為または不正な活動は、(I)FDAおよび他の同様の規制機関の法律、および外国司法管轄区域の同様の医療保健法律法規を含み、そのような機関に真実、完全かつ正確な情報を報告することを要求する法律、(Ii)製造基準、および(Ii)製造基準;(Iii)データプライバシー、セキュリティ、詐欺および乱用、および他の医療に関する法律、または(Iv)真の、完全かつ正確な財務情報およびデータを報告することを要求する法律。具体的には、医療業界の販売、マーケティング、および商業配置は、詐欺、不正行為、リベート、自己取引、および他の乱用行為を防止するための広範な法律法規によって制限されている。これらの法律法規は、幅広い価格設定、割引、マーケティングおよび販売促進、販売手数料、顧客インセンティブ計画、および他のビジネススケジュールを制限または禁止する可能性があります。これらの法律制約を受けた活動はまた、臨床前研究或いは臨床試験過程で得られた情報の不適切な使用或いは虚偽陳述に関連する可能性があり、臨床前研究或いは臨床試験中に詐欺性データを作成し、或いは薬品を不法に流用し、これは規制制裁を招き、そして著者らの名声に深刻な損害を与える可能性がある。従業員や他の第三者の不正行為を常に識別し、阻止することができるわけではなく、このような活動を発見し、防止するための予防措置は、未知または管理不可能なリスクや損失を効果的に制御することができないか、またはそのような法律や法規を遵守できないことによる政府調査または他の行動または訴訟から私たちを保護することができない可能性がある。さらに、誰かや政府がこのような詐欺や他の不正行為を告発する可能性があり、起こらなくてもリスクに直面している。もし私たちにこのような訴訟を提起した場合、私たちは私たちの権利を弁護したり、維持することに成功しませんでした。これらの訴訟は、重大な民事、刑事および行政処罰、損害賠償、金銭的罰金、引き渡し、Medicare、Medicaid、他の米国連邦医療計画または他の司法管轄区の医療計画に参加することから除外される可能性があり、誠実な監督および違反疑惑の解決、個人監禁、他の制裁、契約損害、名声損害、利益減少および将来の収益の減少、および私たちの業務を削減する報告義務を含むかもしれません。
もし私たちのCDMOが傷害や適用法違反の方法で危険や生体材料を使用すれば、損害賠償責任を負う可能性があります。
我々の研究·開発活動は,化学や生体材料を含む潜在的危険物質の制御使用に関するものである。私たちのメーカーはアメリカとその運営のある国の連邦、州と地方の法律法規の制約を受けて、医療と危険材料の使用、製造、貯蔵、運搬と処置を管理しています。メーカーがこれらの材料を使用,処理,貯蔵,処分する手順は法律で規定されている基準に適合していると信じているが,医療や危険材料による汚染や傷害リスクを完全に除去することはできない。このような汚染や傷害のため、私たちは責任を負うかもしれないし、地方、都市、州、または連邦当局はこれらの材料の使用を制限し、私たちの業務運営を中断するかもしれない。事故が発生すると、私たちは損害賠償責任や罰金を要求されるかもしれません。責任は私たちの資源範囲を超えるかもしれません。一般的に、私たちは医療や危険材料による責任に保険をかけない。適用される環境法律法規の遵守はコストが高く、現在または未来の環境法規は私たちの研究、開発、生産努力を損なう可能性があり、それによって私たちの業務、将来性、財務状況、または運営結果を損なう可能性がある。
知的財産権に関するリスク
もし私たちの技術や候補製品について私たちの知的財産権を獲得、維持、実行、十分に保護できない場合、あるいは取得した特許や他の知的財産権保護の範囲が広くなければ、私たちの競争相手は私たちと似ているか同じ技術や製品を開発し、商業化する可能性があり、私たちの技術や候補製品の開発と商業化に成功する能力は不利な影響を受ける可能性がある。
90
私たちは、特許、商業秘密保護、および秘密協定の組み合わせによって私たちの知的財産権を保護し、他の人が私たちの候補パイプライン製品をコピーすることを防止するか、またはそれらの使用または製造、または任意の未来の候補製品に依存して、私たちの成功は、私たちがこのような候補製品に対する米国および他の国の特許保護を獲得し、維持する能力があるかどうかに大きく依存する。
特許訴訟過程は高価で、時間がかかり、複雑であり、私たちは合理的なコストまたはタイムリーな提出、起訴、維持、強制執行、またはすべての必要または望ましい特許出願を許可することができないかもしれない。私たちは、私たちの従業員、CRO、コンサルタント、科学コンサルタント、および他の請負業者などと、私たちの研究開発成果の秘密または特許可能な当事者と秘密協定を締結する権利がありますが、これらの当事者のいずれかは、合意に違反し、特許出願を提出する前にこのような成果を開示し、特許保護を求める能力を危険にさらす可能性があります。また、科学文献における発見発表は、実際の発見よりも遅れがちであり、米国および他の管轄地域の特許出願は、通常、出願18ヶ月後に発表され、発行前にそうである特許出願もある。したがって、私たちは、私たちが私たちの特許または係属中の特許出願で主張された最初の発明であるか、または発明または候補製品に関連する任意の特許出願を最初に提出することを決定することができない。また、規制承認に遅延が生じた場合、特許保護の下で候補製品を販売する期間が短縮される可能性があります。
バイオテクノロジーや製薬分野の特許実力は複雑な法律,事実,科学的問題に関連しており,不確実である可能性がある。特許保護を受ける前に、私たちの研究開発成果で特許を申請できることを確認できない可能性があります。私たちが持っている特許出願は、発行された特許を生成できないかもしれません。その権利要件は、アメリカまたは他の外国での候補製品をカバーしています。我々の特許および特許出願に関連するすべての潜在的に関連する既存技術が発見されていることは保証されず、これらの技術は、特許を無効にするか、または係属中の特許出願から特許が発行されることを阻止する可能性がある。特許が確実に発行されても、これらの特許が私たちの候補製品をカバーしていても、第三者は、そのような特許の発明性、所有権、有効性、実行可能性、または範囲に疑問を提起する可能性があり、これは、そのような特許を縮小または失効させるか、または実行不可能と認定される可能性がある。私たちの未解決および将来の特許出願は、私たちの技術または候補製品を保護する特許の発行、または他社が競争技術および候補製品を商業化することを効果的に阻止する特許をもたらすことができないかもしれません。また、私たちが提出したいかなる米国臨時特許出願も、関連する臨時特許出願を提出してから12ヶ月以内に非臨時特許出願を提出しない限り、発行された特許になる資格がない。もし私たちがいかなる非臨時特許出願を直ちに提出しない場合、私たちは臨時特許出願の優先日と、臨時特許出願に開示された発明に関する任意の特許保護を失う可能性がある。
また,それらが挑戦されていなくても,我々の特許や特許出願は,我々の知的財産権を十分に保護し,我々の候補製品に排他性を提供したり,他の人が我々の主張をめぐる設計を阻止したりすることができない可能性がある.さらに、第三者が我々の特許を侵害することなく、類似または代替製品または方法を作成して同様の結果を達成しないことは保証されない。これらの結果のいずれも、第三者の競争を阻止する能力を弱める可能性があり、第三者競争は私たちの業務に悪影響を及ぼす可能性がある。
もし私たちが持っている私たちの計画または候補製品に関連する特許出願が発表されなかった場合、もし私たちが現在または未来に発表された特許の保護の広さや強度が脅かされている場合、またはそれらが私たちの候補製品に意味のある排他性を提供できなかった場合、会社が私たちと協力して候補製品を開発することを阻止したり、現在または未来の候補製品を商業化する能力を脅かすかもしれない。私たちは最近私たちの候補製品に関するいくつかの特許出願を提出した。私たちは、もしあれば、発行された特許、任意のそのような特許の広さ、または発行された特許が無効であることが発見されるかどうか、強制的に実行できないか、または第三者によって脅かされるかどうかをもたらす保証を提供することはできない。これらの特許または私たちが持っている任意の他の特許への成功に対するいかなる反対も、私たちが開発する可能性のある任意の候補製品を商業化することに成功するために必要な権利を奪う可能性がある。
特許の発行は、その発明性、所有権、範囲、有効性、または実行可能性に対して決定的ではなく、私たちの特許は、米国および海外の裁判所または特許庁で挑戦される可能性がある。さらに、特許の発行は、第三者が(承認された場合)私たちの候補製品の販売を阻止するか、または私たち自身の特許技術を実践することを阻止する可能性があるので、特許発明を実践する権利を与えてくれない。
2011年の“ライシー·スミス米国発明法”や“ライシー·スミス法案”を含む米国範囲の幅広い特許改革立法は、我々の知的財産権の強度や実行可能性の不確実性を増加させ、それを保護するコストを増加させる可能性がある。Leahy-Smith法案は米国特許のいくつかの重大な修正を含む
91
特許出願起訴方式及び特許訴訟に影響を与える条項を含む法律。“ライシー·スミス法案”によると、米国は“先発明”から“先出願”制度に転換し、同一発明を要求する異なる当事者が2つ以上の特許出願を提出する際に、どちらが特許を付与されるべきかを決定するために用いられる。これは、私たちが発明から特許出願を提出するまでの間に迅速に進み、勤勉に特許出願を提出することを要求するだろうが、場合によっては、私たちが発明に関する特許出願を迅速に提出または起訴することを阻止する可能性がある。ライシー·スミス法案はまた、既存技術に適合した開示範囲を拡大した。さらに、第三者が2013年3月16日に“Leahy-Smith Act”適用条項が発効する前に特許出願を提出した場合、第三者は、それが私たちの特許請求の範囲に含まれる任意の主題の第1の発明であるかどうかを決定するために、米国で介入手続きを開始することができる。私たちはまた、第三者が事前に発行した既存技術を米国特許商標局に提出する必要があるかもしれない。
Leahy-Smith Act“は、付与後審査、当事者間の審査、および派生プログラムを含む米国によって発行された特許に挑戦するための新しいプログラムを初めて作成し、これらのプログラムは、米国特許商標局で行われた対抗性プログラムであり、一部の第三者は、競争相手が発行した特許の一部または全ての権利要件をキャンセルするためにこれらのプログラムを利用してきた。優先日が2013年3月16日以降の特許については,我々のすべての特許出願があり,第三者は特許発表後9ヶ月の窓口でライセンス後審査出願を提出することができる。特許が2013年3月16日までに提出された場合は,特許発表後ただちに当事者間の審査出願を提出することができる。優先権日が2013年3月16日以降の特許については、ライセンス提出後の審査出願の9ヶ月の期限が満了した後に当事者間審査出願を提出することができる。贈与後の再審手続きは任意の疑問理由に基づいて提起することができるが、当事者間の再審手続きは公表された既存技術に基づいて疑問を提起することしかできない。USPTOのこれらの対抗性訴訟には,米国特許の有効性を推定することなく,米国連邦裁判所の訴訟で特許主張を審査することが含まれている。USPTOは2018年11月13日に施行された最終規則を発表し、現在使用されている米国連邦裁判所が現在使用している同じ請求項構築基準を使用して、USPTO訴訟における特許請求書を解釈することを発表した。これは、使用される言葉の簡単で一般的な意味である。もし私たちの任意の特許がこのようなUSPTO訴訟で第三者の挑戦を受けた場合、私たちがその特許を成功的に守ることが保証されないことは、排他性を失うことを含む挑戦された特許権を失うことになり、あるいは特許主張が縮小され、無効または実行不可能であることは、他人が他人の使用または商業化または同じ技術および製品を使用することを阻止する能力を制限するか、または私たちの技術および候補製品の特許保護期間を制限する可能性がある。最終的な結果が私たちに有利であっても、このような手続きは大量のコストを招く可能性があり、私たちの科学者と経営陣に多くの時間がかかる必要がある。
以上のような理由により、我々の特許権の発行、範囲、有効性、実行可能性、商業価値には大きな不確実性があり、これは私たちの業務、財務状況、運営結果、見通しに実質的な悪影響を及ぼす可能性がある。
第三者は、我々の技術または製品開発の知的財産権を阻害または制限する可能性がある知的財産権を獲得または制御する可能性がある。知的財産権侵害、流用、または他の侵害行為に対する第三者のクレームは、大量のコストを招いたり、私たちの開発と商業化努力を阻害したり、延期したりする可能性がある。
私たちのビジネスの成功は、私たちが実際の侵害、流用、または他の方法で第三者の特許および他の独占権を侵害することを避けることにある程度かかっている。米国国内外では、バイオテクノロジーおよび製薬業界には、特許侵害訴訟、介入、反対、再審、許可後および当事者間の審査手続き、米国特許商標局および外国司法管轄区における同様の訴訟、例えば欧州特許庁または欧州特許庁の反対など、特許および他の知的財産権に関連する訴訟が多く存在する。我々が開発候補を求めている分野には,第三者が所有する米国や外国から発行された特許や係属中の特許出願が多く存在する.製薬業を含む知的財産権に依存する業界の多くは、競争相手に対する優位性を得るための手段として知的財産権訴訟を利用している。バイオテクノロジーや製薬業界の拡張や特許の発行に伴い、上場企業としてより高い知名度と市場露出率を獲得することにより、当社の候補製品が第三者特許権侵害の疑いを受けるリスクが高まる可能性がある。一部のクレーム者は私たちよりもはるかに多くの資源を持っている可能性があり、私たちよりも複雑な知的財産訴訟費用をより大きく長時間受けることができるかもしれない。また,特許権の強制執行による特許料の抽出と和解にのみ重点を置いた特許持株会社が我々を対象とする可能性がある。
92
第三者は私たちが許可されていない状況で彼らのノウハウを使用していると主張するかもしれない。物質の組成、薬物送達、製造方法、または治療方法に関する当社の候補製品の使用または製造に関連する第三者特許または特許出願が存在する可能性がある。私たちは、私たちの技術、製品、成分、およびその使用が侵害されないか、流用しないか、または他の方法で第三者特許または他の知的財産権を侵害することを保証することはできません。特許出願が発行されるまでに数年かかる可能性があるため、現在処理されている特許出願が存在する可能性があり、これらの出願は、我々の候補製品が発行された特許を侵害する可能性がある。さらに、第三者は将来的に特許を取得し、私たちの技術を使ってこれらの特許を侵害したと主張するかもしれない。公表された未定特許出願は、私たちの候補製品または私たちの候補製品の使用をカバーするために、いくつかの制限された場合に後で修正することができる。特許発行後、特許請求の範囲は、依然として法律解釈、特許における書面開示、および特許の起訴履歴に依存する。特許または保留出願の関連性または範囲の説明は正しくないかもしれませんが、これは私たちの候補製品をマーケティングする能力に悪影響を及ぼすかもしれません。連邦裁判所で米国特許の有効性に挑戦することに成功するためには,有効性推定を克服する必要がある。この負担が重いため,このような米国特許主張の無効について明確で納得できる証拠を提出することが求められているため,管轄権のある裁判所がこのような米国特許の主張の無効を宣言する保証はない.管轄権のある裁判所が任意の第三者特許を持ち、私たちの任意の候補製品の組成、任意の候補製品の製造プロセス、または任意の候補製品の使用方法をカバーする場合、任意のそのような特許の所有者は、適用特許に基づいてライセンスを取得し、これらの特許が商業的に合理的な条項で提供されないか、またはその特許が満了する前に、その候補製品を商業化する能力を阻止することができるかもしれない。
提訴や論争のある訴訟手続きの法的敷居が低いため,勝訴確率の低い訴訟や訴訟手続きが提起される可能性があり,弁護には大量の資源が必要である.訴訟と論争のある訴訟手続きはまた高価で時間がかかるかもしれないが、私たちのこのような訴訟手続きの中の相手は私たちよりも多くの資源を投入してこのような法的行動を起訴する能力があるかもしれない。私たちの製品候補が商業化に近づいていれば、上場企業に関するより大きな知名度を得るにつれて、このような訴訟や訴訟に巻き込まれるリスクが高まる可能性があります。第三者は、そのようなクレームの是非を考慮することなく、既存の特許または将来付与される可能性のある特許に基づいて、侵害クレームを私たちに提示することができる。我々は、我々の技術および製品候補およびその用途に関連する可能性のあるすべての知的財産権を知らないかもしれないし、第三者知的財産権が無効であると誤って結論を出すことができるか、または私たちの活動および製品候補が侵害されていない、流用、または他の方法でそのような知的財産権を侵害している可能性がある。したがって、私たちは私たちの技術と製品候補、あるいは私たちの開発と商業化を確実に知ることができなくて、侵害、流用、あるいは他の方法でいかなる第三者の知的財産権を侵害することもありません。
我々にクレームをつけた当事者は、禁止または他の公平な救済を受ける可能性があり、これは、私たちの1つまたは複数の候補製品のさらなる開発および商業化を効果的に阻止することができ、および/または私たちの名声および財務業績を損なうことができる。これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、経営陣や従業員資源を当社の業務から大量に移転する可能性がある。また、公聴会、動議、または他の一時的な手続きまたは事態の発展の結果が公表される可能性があり、証券アナリストや投資家がこれらの結果がマイナスだと思う場合、私たちの普通株の価格に大きな悪影響を及ぼす可能性がある。もし私たちに対する侵害クレームが成功した場合、私たちは故意に侵害した3倍の損害賠償金と弁護士費、特許権使用料の支払い、私たちの侵害製品の再設計、登録商標に関するクレームで私たちの製品候補製品を再命名すること、または第三者から1つ以上のライセンスを取得することを含む巨額の損害賠償を支払わなければならないかもしれない。これらは大量の時間とお金の支出を必要とする可能性があり、不可能であるか、技術的に不可能である可能性がある。しかも、私たちは商業的に合理的な条項や必要な許可証を得ることができないかもしれない。たとえ私たちが許可を得ることができても、それは私たちの競争相手と他の第三者が私たちに許可された同じ技術にアクセスできるように非排他的である可能性がある;または、また、それは商業市場での私たちの成功的な競争の能力を阻害または破壊する条項を含むことができる。上記のいずれも、我々の業務、財務状況、経営結果及び見通しに実質的な悪影響を及ぼす可能性がある。
特許条項は製品に対する私たちの競争地位を十分な時間で保護するのに十分ではないかもしれない。
任意の個別特許の期限は、その特許が付与された国の適用法に依存する。米国では、すべての維持費が直ちに支払われる限り、特許の有効期限は、通常、その出願提出日から20年または最初に要求される非一時的出願日である。場合によっては拡張を提供する可能性があります
93
この場合、特許の有効期限は限られているので、それが提供する保護も限られている。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。特許期間を延長する資格のある特許については、米国や他の国/地域(ある場合)に特許期間の延長を求めることが予想されるが、我々が求めている任意の特許期間の延長、またはそのような特許期間の延長がいかなる競争優位性を提供するかは保証されない。
もし私たちが“ハッジ·ワックスマン法案”に基づいてアメリカと外国で類似した法律によって特許期間を延長しなければ、私たちの候補製品のマーケティング排他的期間を延長する可能性があり、私たちの業務は損なわれる可能性があります。
米国では,FDAが承認した薬物または生物をカバーする特許には,FDAによる上場前の規制審査中に失われた特許期間を回復する資格延長資格がある可能性がある。私たちの候補製品のFDA上場承認の時間、持続時間、条件によると、私たちの1つ以上の米国特許は、1984年の“薬品価格競争および特許期限回復法”または“ハッジ-ワックスマン法案”(Hatch-Waxman Act)に基づいて、製品開発およびFDA規制審査中に失われた有効な特許期間の補償として、承認された製品の特許を最大5年間延長することを可能にする限られた特許期間延長を受ける資格がある可能性がある。特許期間の延長は,製品が承認された日から14年間の余剰特許期間を超えてはならず,承認された薬品,使用方法又は製造方法の請求項のみを延長することができる。EUでは、私たちの候補製品は似たような法律に従って期限を延長する資格があるかもしれない。しかし、いずれの管轄区域においても、適用の最終期限内に出願を提出できなかった場合、関連特許の満了前に出願を提出できなかった場合、または適用の要件を満たすことができなかった場合、延期が得られない可能性がある。たとえ私たちが延期を許可されたとしても、延期の期限は私たちが要求したより短いかもしれない。もし私たちが特許期間の延長を得ることができない場合、あるいはこのような延長の期限が私たちの要求よりも少ない場合、私たちはその製品に対して特許権を行使する期限が実際に短縮されることができ、私たちの競争相手は市場競争製品の承認をより早く得ることができるかもしれない。これによる適用製品の長年の収入の減少は大きな可能性がある.
私たちは他の人たちから許可を得たノウハウに依存している。もし私たちが既存の許可証を失ったら、私たちは私たちの候補製品を開発し続けることができないかもしれない。
私たちは特許、ノウハウ、そして独自技術に依存しており、私たち自身のものもあれば、他の人が許可しているものもある。
私たちは、これらの合意によって付与されたライセンスを含む、旗艦パイオニア革新V会社または旗艦会社Whitehead Institute for Biomedical Research(WEBR)、Acuitas Treateutics,Inc.(Acuitas Treateutics,Inc.)およびNitto Denko Corporation(Nitto)との合意に大きく依存している。このようなライセンスはいくつかの条件で終了するかもしれない。これらのライセンスのいかなる終了も重大な権利の喪失を招く可能性があり、候補製品を商業化する能力を損なう可能性がある。
私たちはまた、将来的に他の当事者と許可協定を締結し、勤勉、開発と商業化スケジュール、マイルストーン支払い、印税、保険、その他の義務を私たちに課すことができるかもしれない。私たちは規定された時間内に私たちが使用している分野の許可製品についていくつかの開発マイルストーンを実現する義務があります。旗艦、WEBR、Acuitas、Nitto、または私たちの現在または未来の任意の他の許可者またはパートナーに対する私たちの義務を履行できない場合、私たちの取引相手は、これらの合意を終了する権利がある可能性があり、この場合、これらのプロトコルによってカバーされる任意の候補製品を開発、製造、またはマーケティングすることができない可能性があり、これは、任意のプロトコルに従って開発された候補製品の価値に悪影響を及ぼす可能性がある。これらの合意を終了したり、これらの合意の下での私たちの権利を減少またはキャンセルすることは、私たちが新しいまたは回復された条項の悪い合意を交渉しなければならないことを招き、または重要な知的財産権や技術に対する私たちの権利を含む、これらの合意の下の権利を失うことになるかもしれない。
私たちは旗艦、WIBR、Acuitas、Nittoによって特許出願を提出し、起訴し、特許を維持し、他の方法で私たちが許可した知的財産権を彼らの影響を受けないように保護する。私たちは彼らの活動または私たちの許可内の知的財産権に関連する可能性のある他の知的財産権の使用を制限することができるかもしれない。例えば、これらのライセンシーのこのような活動が適用された法律および法規に適合しているかどうか、または効果的かつ強制的に実行可能な特許および他の知的財産権が生じるかどうかを判断することはできない。
94
もし私たちが商業的に合理的な条項や第三者からライセンスを得ることができない場合、あるいはこのような合意の下での私たちの義務を履行できない場合、私たちの業務は損なわれる可能性があります。
私たちは第三者の特許や他のノウハウを使用して私たちの製品を商業化する必要がある。もし私たちがこのような技術を許可できない場合、あるいは私たちが不利な条項でそのような技術を許可することを余儀なくされた場合、私たちの業務は実質的な損害を受ける可能性がある。もし私たちが将来必要な許可を得ることができない場合、私たちは影響を受けた候補製品を開発したり、商業化することができなくなり、これは私たちの業務に実質的な損害を与える可能性があり、このような知的財産権を所有または他の方法で制御する第三者は、私たちの販売禁止を禁止すること、または私たち側が印税および/または他の形態の賠償を支払う義務を求めることができる。たとえ我々が許可を得ることができても,我々の競争相手や他の第三者が我々に許可されている同じ技術にアクセスできるように非排他的である可能性がある.第三者知的財産権の許可または買収は競争分野であり、より多くの老舗企業は、魅力的または必要と考えられる第三者知的財産権許可または買収戦略をとることができる。これらの老舗会社はその規模、資本資源及び更に強い臨床開発と商業化能力のため、私たちより競争優位を持っているかもしれない。しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。
もし私たちが必要な第三者知的財産権を得ることができない場合、あるいは私たちの既存の知的財産権を維持することができなければ、私たちの技術、候補製品、または製造方法を再設計するのに時間と資源をかけて、あるいは代替技術を開発または許可することができないかもしれません。これらはすべて技術的にも商業的にも不可能かもしれません。もし私たちがそれができなければ、影響を受けた技術や候補製品を開発または商業化することができないかもしれません。これは、私たちの業務、財務状況、運営結果、将来性を深刻に損なう可能性があります。
さらに、私たちが将来の任意の許可プロトコルの義務を履行できない場合、私たちの取引相手は、これらのプロトコルを終了する権利があるかもしれません。この場合、私たちは、これらのプロトコルによってカバーされる任意の製品の開発、製造、またはマーケティングを停止させることができないか、またはそのようなプロトコルの下での他の処罰に直面する可能性があります。このような状況は、任意のこのようなプロトコルに従って開発された候補製品の価値に実質的な悪影響を及ぼす可能性がある。これらの合意を終了するか、またはこれらの合意の下で私たちの権利を減少またはキャンセルするか、または私たちの業務の利益に適合した場合に、私たちのこのような合意の下での私たちの権利を自由に譲渡または再許可することを制限することは、私たちが重要な知的財産権または技術に対する私たちの権利、またはそのような合意に依存する1つまたは複数の候補製品のさらなる開発または商業化を阻害、遅延、または禁止することを含む、新たなまたは回復された合意をあまり有利ではない条項で交渉しなければならない可能性がある。
私たちは現在何の関連訴訟にも巻き込まれていないにもかかわらず、私たちは私たちの特許や他の知的財産権を保護または強制する訴訟に巻き込まれるかもしれないが、これは高価で時間がかかり、成功しないかもしれない。
競争相手および他の第三者は、私たちまたは将来の許可者の特許、商標、著作権、または他の知的財産権を侵害、流用、または他の方法で侵害する可能性があります。したがって、私たちは第三者に侵害、流用、または他の知的財産権に関するクレームを提起する必要があるかもしれない。権利侵害や他の許可されていない使用と戦うために、私たちは国/地域にクレームをつけることを要求されるかもしれません。これは高価で時間がかかり、私たちの管理者と科学者の時間と注意力を分散させているかもしれません。私たちはこのようなクレームを提出し、追及するのに十分な財政あるいは他の資源があることを保証することはできません。これらのクレームは往々にして数年続けなければなりません。
私たちのライセンス契約は第三者侵害者に対してライセンス特許を強制的に執行する能力に制限があります。たとえば,WIBRとのライセンスプロトコルについては,WiBRとこれまでスポンサー研究協定を締結していた特定の第三者に対して,このようなスポンサー研究プロトコルによって生成された発明についてライセンス特許を強制的に実行することはできない.また,WIBR共同独占合意については,WIBR特許権は共同独占許可されており,他方の第三者に与えられている.したがって、共同独占ライセンスを主張する特許権は、共同独占ライセンシーに対抗することは許されない。
私たちが第三者に提起したいかなるクレームも、私たちが侵害、流用、あるいは他の方法で彼らの知的財産権を侵害すると主張するように、これらの当事者に反クレームを促す可能性がある。さらに、特許侵害訴訟では、このような当事者は、私たちが主張している特許が無効または実行不可能であるか、またはその両方を反訴することができる。米国の特許訴訟では,被告が無効または実行不可能と主張する反訴が一般的である。有効性疑問の理由は,以下のいずれかを満たさなかったということである可能性がある
95
いくつかの法定要件は、新規性の欠如、明確性、または使用不可能性を含む。主張を実行できない理由は,特許起訴に関連する者が起訴中に米国特許商標局に関連情報を隠蔽したり,誤った声明をしたりしたためかもしれない.第三者は、訴訟範囲外であっても、米国や海外の行政機関にこのようなクレームを提起することができる。このようなメカニズムには、再審査、付与後再審、当事当事者再審、介入手続き、派生手続き、および外国法ドメインにおける同等の手続き(例えば、反対手続き)が含まれる。法的に無効と実行不可能と断言された後の結果は予測できない。
どのような訴訟においても、裁判所は、私たちの特許または私たちが許可した特許が無効であること、強制的に実行できないこと、および/または侵害されていないことを判断することができ、またはそのような特許の権利要件を狭く解釈することができ、または私たちの特許が関連技術をカバーしないことを理由に、他方の関連技術の使用を阻止することを拒否することができる。任意の訴訟または弁護手続における不利な結果は、私たちの1つまたは複数の特許を全部または部分的に無効、狭義の解釈または実行できないリスクに直面させる可能性があり、私たちの特許出願を発行できないリスクに直面させる可能性があり、これらの当事者または他の競争相手に対してこれらの特許を主張する能力を制限し、第三者が類似または競合製品を製造および販売する能力を制限または排除する可能性がある。同様に、私たちが商標侵害クレームを主張する場合、裁判所は、私たちが主張する商標が無効または強制執行できないと判断するか、または商標侵害を主張する側が関連商標に対する優先権を有することを主張するかもしれない。この場合、私たちは最終的にこのような商標の使用を中止することを余儀なくされる可能性があり、これは私たちの業務に実質的な損害を与え、私たちの市場地位に悪影響を及ぼす可能性がある。
私たちが知的財産権の侵害、流用、あるいは他の侵害を確定しても、裁判所はこれ以上のこのような活動に対して禁止令を発行せず、金銭損害賠償金だけを判決する可能性があり、これは十分な救済措置ではないかもしれない。さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。聴聞、動議、または他の一時的手続き、または事態発展の結果を公開することもできる。もし証券アナリストや投資家がこれらの結果がマイナスだと思っていれば、私たちの普通株の価格に実質的な悪影響を及ぼす可能性がある。上記のいずれも、我々の業務、財務状況、経営結果及び見通しに実質的な悪影響を及ぼす可能性がある。
米国や他の司法管轄地域では,特許法の弱化や裁判所や他の機関の法執行は,我々の特許保護能力に影響を与える可能性がある。
過去数年間、米国最高裁判所は特許事件においていくつかの意見を発表し、多くの人はこれらの意見が米国の特許保護を弱める可能性があると考えているか、ある場合に利用可能な特許保護範囲を縮小し、いくつかのタイプの革新は特許を申請できないと考えているか、または通常は他の方法で裁判所が特許を無効にしやすいようにしている。また,最近では米国や他の国の特許法をより多く改正することが提案されており,採択されれば,我々のノウハウのために特許保護を受ける能力,あるいは我々のノウハウを実行する能力に影響を与える可能性がある。米国議会、米国裁判所、USPTOおよび他の国の関連立法機関および他の機関の将来の行動によれば、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、それによって、私たちが新しい特許を獲得したり、私たちの既存の特許と私たちが将来獲得する可能性のある特許を強制的に執行して保護する能力を弱めることができる。
一部の外国司法管轄区の法律は知的財産権の保護程度はアメリカに及ばず、多くの会社は外国司法管轄区の保護とこのような権利を守る面で重大な困難に直面している。もし私たちが知的財産権を保護する上でこのような困難に遭遇したり、他の理由で外国の管轄区域で私たちの知的財産権を効果的に保護できない場合、私たちのビジネスの見通しは深刻な損害を受ける可能性があります。例えば、私たちは、欧州特許庁、または外国裁判所での特許訴訟や他の手続のような外国の手続に反対する側になることができる。そうであれば,このような訴訟手続きの開始と継続による不確実な要因は,我々が市場で競争する能力に大きな悪影響を与える可能性がある.外国対抗訴訟の費用も高い可能性があり、多くの外国司法管轄区では、敗訴側は勝訴側の弁護士費を支払わなければならない。
私たちの特許保護の獲得と維持は、政府特許機関によって提出された様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
米国特許商標局、欧州特許庁、および他の特許機関は、特許出願中にいくつかのプログラム、文書、費用支払い、および他の同様の条項を遵守することを要求する。周期性の
96
維持費、継続費、年会費、および特許および/または出願に関連する様々な他の政府費用は、特許および/または出願の有効期間内にいくつかの段階で米国特許商標局および米国以外の様々な政府特許機関に支払われる。私たちはこれらの費用を支払うようにシステム的に注意して、私たちは外部会社を招聘し、私たちの外部弁護士に依存してアメリカではない特許代理機関にこのような費用を支払う。多くの場合、適用される規則に基づいて、滞納金を支払うことによって、または他の方法で過失失効を修正することができるが、場合によっては、規則を遵守しないことは、特許または特許出願が放棄または失効され、関連する法域特許権の一部または全部を喪失させる可能性がある。この場合、私たちの競争相手または他の第三者は、同様のまたは同じ製品または技術で市場に参入する可能性があり、これは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
私たちは世界各地で私たちの知的財産権を強制的に施行できないかもしれない。
世界中で私たちの候補製品の出願、起訴、特許保護の費用は目を引くほど高くなり、私たちが自分の知的財産権の保護を求めている国でも、この保護はアメリカほど広くないかもしれない。ある国、特に発展途上国では、特許性に対する要求が異なる可能性があり、許容される特許請求の範囲の広さが一致しない可能性がある。また、いくつかの外国の法律は知的財産権の保護の程度はアメリカの連邦や州法律に及ばない。このような機会があれば、私たちが世界のすべての国の候補製品をカバーする許可内で特許は同じように尻込みするほど高いかもしれない。そして、私たちが候補製品を開発したり商業化したりする司法管轄区域内でも、特許の許可や届出、起訴、擁護は尻込み的または非現実的に高価になる可能性がある。競争相手は、私たちが特許保護または特許を許可していない司法管轄区域で、私たちと私たちの許可側の技術を使用して彼ら自身の製品を開発することができ、また、他の侵害製品を私たちと私たちの許可者が特許保護を持っているが、アメリカやEUの地域よりも法執行力が低い地域に輸出することもできる。これらの製品は私たちの候補製品と競争する可能性があり、私たちまたは私たちの許可者の特許または他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。
しかも、私たちはまだ未解決のままであるので、国と地域の特許出願を放棄することを決定するかもしれない。各国又は地域特許の付与手続は独立した手続であり,関連特許庁が出願を拒否し,実質的に類似した出願が他の機関によって承認されている場合を招く可能性がある。例えば,他の国よりも中国の方が特許性に対する要求が高く,具体的には主張されている薬物の医療用途について詳細に説明することが求められている。さらに、模倣薬製造業者または他の競争相手は、複雑で長く、高価な訴訟または他の手続きに参加することを要求するために、私たちまたは私たちのライセンシー特許の範囲、有効性、または実行可能性に挑戦する可能性がある。後発薬メーカーは私たちの製品の模造薬バージョンを開発し、承認を求めて発売することができる。同様に一般的な場合は、国によって同じ候補製品または技術の特許保護範囲が異なる可能性があることである。
一部の司法管轄区の法律は米国やEUの法律や法規のように知的財産権を保護しておらず、多くの会社はこれらの管轄区の専有権の保護と擁護に重大な困難に直面している。さらに、特定の国の法律制度、特に特定の発展途上国の法律制度は、特許、商業秘密または他の形態の知的財産権の強制執行を支持しておらず、特にバイオテクノロジー製品に関連する特許、商業秘密、または他の形態の知的財産権は、特定の司法管轄区域の競争相手が私たちの独占権を侵害する方法で競争製品を販売することを阻止することを困難にする可能性がある。外国司法管轄区域で我々の特許権の訴訟を強制的に執行することは、成功するか否かにかかわらず、巨額のコストを招き、私たちの努力と注意を私たちの業務の他の側面から移転させる可能性があり、また、私たちまたは私たちのライセンシーの特許が無効または狭義に解釈されるリスクに直面させる可能性があり、私たちまたは私たちのライセンシーの特許出願が発行されないリスクを増加させるか、または第三者からのクレームを引き起こす可能性がある。私たちは私たちが起こしたいかなる訴訟にも勝てないかもしれないし、敵に損害賠償や他の救済措置を支払うかもしれないが、これは大きなビジネス的意義を持つかもしれない。もし私たちが勝訴すれば、私たちに与えられた損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれません。したがって、私たちが世界各地で私たちの知的財産権を実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネスメリットを得るのに十分ではないかもしれません。また、私たちが予想している重要な市場で私たちの知的財産権を保護するつもりですが、候補製品をマーケティングしたいかもしれないすべての司法管轄区域で同様の努力を開始したり維持したりできることを保証することはできません。したがって、これらの国で私たちの知的財産権を保護するための努力が不十分である可能性があり、これは、すべての予想される重要な海外市場で私たちの候補製品を商業化することに成功した能力に悪影響を及ぼす可能性がある。もし私たちまたは私たちの許可者が知的財産権の保護に困難に直面したり、知的財産権を効果的に保護することから排除されたりすれば
97
もしこのような権利がこのような司法管轄地域での私たちの業務に非常に重要であれば、このような権利の価値が低下する可能性があり、私たちはこのような司法管轄区で追加的な競争に直面するかもしれない。
EU諸国を含むいくつかの司法管轄区では、強制ライセンス法は、特許所有者に第三者に許可を付与することを強制する。さらに、いくつかの国は、政府機関または政府請負業者に対する特許の実行可能性を制限している。これらの国では,特許権者の救済措置は限られている可能性があり,このような特許の価値を大幅に低下させる可能性がある。もし私たちまたは私たちの任意の許可者が私たちの業務に関連する特許の下で第三者に許可を付与することを余儀なくされた場合、または私たちまたは私たちの許可者が第三者に対して特許権を執行することを阻止された場合、私たちの競争地位はこれらの管轄区域で深刻な損害を受ける可能性があります。
私たちは私たちの能力に頼って私たちの特許を強制的に執行することで他人の競争を阻止しますが、いくつかの管轄区域は私たちに第三者に許可を与えることを要求するかもしれません。このような強制許可は私たちを含むいくつかの候補製品に拡張することができ、これは私たちの潜在的な収入機会を制限するかもしれない。
多くの外国国には強制許可法があり,これらの法律によると,特許権者は場合によっては第三者に許可を付与しなければならない。例えば、直接立法や国際計画によって、救命製品や高価な製品の強制許可や強制許可の脅威が発展途上国でますます一般的になっている。強制許可は私たちを含むいくつかの候補製品に拡張することができ、もし彼らがマーケティング承認を得たら、これは私たちの潜在的な収入機会を制限するかもしれない。したがって、私たちはアメリカとヨーロッパ以外のいくつかの国で第三者が私たちの発明を実施することを阻止できないかもしれない。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの技術を使用して自分の製品を開発することもできます。また、もし私たちが特許を実行して侵害活動を阻止する能力が不足していれば、競争相手は他の侵害製品を私たちが特許保護を持っている地域に輸出するかもしれません。これらの製品は私たちの製品と競争するかもしれませんが、私たちの特許や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。外国司法管区で私たちの特許権の訴訟を強制的に執行することは、成否にかかわらず、巨額のコストを招き、私たちの努力と資源を私たちの業務の他の側面から移転させる可能性があります。また、このような特許権が存在する主要市場で私たちの製品の知的財産権を保護しようとしていますが、私たちの製品を販売したいすべての司法管轄区域で同様の努力を開始または維持できることを保証することはできません。したがって、このような国で知的財産権を保護するための私たちの努力は十分ではないかもしれない。
さらに、いくつかの国は、政府機関または政府請負業者に対する特許の実行可能性を制限している。これらの国では,特許所有者は金銭救済に限られている可能性があり,政府が侵害者であれば侵害を禁止できない可能性があり,特許の価値を大幅に低下させる可能性がある。
私たちの知的財産権のいくつかは政府が援助したプロジェクトによって発見されたので、“入場”権利、いくつかの報告要件、およびアメリカ会社の選好など、連邦法規の制約を受ける可能性があり、これらの法規を遵守することは、私たちの独占権と非アメリカ製造業者との契約能力を制限するかもしれない。
“ベハ·ドール法案”によると、米国連邦政府はその財政援助の下で生成された発明に対していくつかの権利を保持している。連邦政府は自身の利益のために“非排他的、譲渡不可能、撤回不可能、支払済み許可証”を保持している。ベッハ-ドール法案はまた連邦機関に“デモの権利”を提供した。特定の場合、政府が、(1)発明を商業化し、政府援助を実現する技術の実用化に十分なステップを講じていないと判断した場合、(2)政府が公衆衛生または安全需要を満たすように行動すること、(3)連邦法規の公共使用に対する要求を満たすために政府の行動が必要である場合、政府が特定の場合に請負者または特許所有権相続人に“責任を負う1つまたは複数の出願人に”非排他性、部分排他性または排他的許可“を付与することを可能にする。もし特許所有者がそうすることを拒否した場合、政府は自分で許可を与えることができる。私たちのいくつかの許可特許は“ベハ-ドール法案”の条項によって制限されている。もし私たちの許可者が“ベハ-ドール法案”の規定を遵守できなかった場合、彼らはこのような規定によって制限された任意の特許の所有権を失う可能性があり、これは私たちの特許下の許可権に影響を与え、私たちは他人が類似したまたは同じ技術と製品を使用したり、それを商業化する能力を阻止したり、私たちの技術および製品の特許保護を制限するかもしれない。
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの商業と競争の地位は損なわれるだろう。
98
特許提供の保護に加えて、我々は、特許を出願できない独自技術、特許を実施しにくいプロセス、および私たちの候補製品発見および開発中に特許がカバーされていないノウハウ、情報または技術に関する任意の他の要素を保護するために、商業秘密保護および秘密保護プロトコルに依存して保護する。しかし、商業秘密は保護するのが難しいかもしれない。私たちは、当社の従業員、CRO、コンサルタント、科学コンサルタント、および他の請負業者のような、これらの技術およびプロセスにアクセスする権利のある当事者と秘密協定を締結することによって、私たちのノウハウおよびプロセスを保護することを求めています。私たちは私たちが可能であるか、または私たちの商業秘密またはノウハウとプロセスに接触したすべての当事者とこのような合意に到達したことを保証することができない。また、私たちは私たちのビルの実体安全と、私たちの情報科学技術システムの実体と電子安全を維持して、私たちの資料と商業秘密の完全性とセキュリティを維持することに力を入れています。私たちはこれらの個人、組織、そしてシステムに自信がありますが、合意やセキュリティ措置は違反される可能性があり、私たちの商業機密は漏洩する可能性があり、私たちはこのような違反に対応するための十分な救済措置がないかもしれません。強制執行側が商業秘密を不正に開示したり流用したりする主張は困難であり、高価で時間がかかり、結果は予測できない。しかも、米国国内外のいくつかの裁判所は商業秘密をあまり望んでいないか、または保護したくない。私たちの商業秘密または他の機密固有情報を盗用または無許可に開示することは、私たちの商業秘密の保護を失い、私たちの競争的地位を損ない、私たちの業務に実質的な悪影響を及ぼす可能性がある。私たちは、将来のビジネスパートナー、協力者、請負業者、およびビジネス秘密が盗まれるリスクの高い他の国に位置する他の他の人と、個人または外国の行為者を介した直接侵入、および国家行為者に関連しているか、または国家行為者によって支配されている人を含む商業秘密を含む私たちの独自の情報を共有する必要があるかもしれない。さらに、私たちの商業秘密または他の機密固有情報を維持するためのステップが不十分であると考えられる場合、第三者が商業秘密または他の機密固有情報を流用するのに十分な追跡権がない可能性がある。
さらに、競争相手または他の第三者が、我々のビジネス秘密および他の機密固有情報を他の方法で取得しないこと、または実質的に同じ技術およびプロセスを独立して発見または開発することを保証することはできない。私たちの候補製品や技術に関連する商業秘密や他の非特許知的財産権を第三者に漏洩することを阻止できない場合、私たちがこのような強制的に実行可能な商業秘密保護を持つことは保証されません。私たちは私たちの市場で競争優位性を確立または維持することができないかもしれません。これは、私たちの業務、運営結果、および財務状況に大きな悪影響を及ぼす可能性があります。
私たちは、私たちの従業員、コンサルタント、または独立請負業者が第三者の機密情報を誤って使用または開示し、私たちの従業員が彼らの元雇用主のいわゆる商業秘密を誤って使用または開示した、または私たちが私たち自身の知的財産権を持っていると主張するという告発を受けるかもしれない。
私たちは将来的に、私たちの競争相手や潜在的な競争相手を含む、以前に大学や他のバイオテクノロジーや製薬会社に雇われていた個人を雇うことができるようになった。私たちは、私たちの従業員、コンサルタント、および独立請負業者が、私たちのために働いているときに他人の固有情報またはノウハウを使用しないことを保証するために努力しているが、私たちは、商業秘密または他の固有情報を含む、私たちまたは私たちの従業員、コンサルタント、または独立請負業者が、これらの個人の任意の元雇用主または他の第三者の知的財産権を無意識にまたは他の方法で使用または開示しているというクレームを受ける可能性がある。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。もし私たちがこのようなクレームを弁護できなければ、金銭損害賠償の支払いに加えて、貴重な知的財産権や人員、あるいは私たちの雇用者の能力を失う可能性があり、上記のいずれの場合も、これは私たちの業務に悪影響を及ぼす可能性があります。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。
私たちの政策は、私たちのすべての従業員およびコンサルタントが彼らの発明を私たちに譲渡することを要求することであるが、従業員またはコンサルタントが私たちのために働いているときに他人が所有している知的財産権を使用する場合、関連するまたはそれによって生じるノウハウおよび発明の権利に関する論争が生じる可能性がある。私たちはまた、実際に私たちが自分の知的財産権と見なしているすべての当事者とこのような合意を構想したり開発したりすることに成功しないかもしれない。知的財産権の譲渡は自動的に実行されない可能性があり、あるいは譲渡協定が違反される可能性があり、私たちは第三者にクレームをつけさせられたり、私たちが私たちの知的財産権の所有権だと思うことを決定するために、私たちが提起する可能性のあるクレームを弁護したりすることができます。このようなクレームは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。
99
私たちの独自の権利は私たちの技術と候補製品を保護するのに十分ではないかもしれませんが、知的財産権は私たちの競争優位が直面しているすべての潜在的な脅威を解決できるとは限りません。
私たちの知的財産権が提供する将来の保護の程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できないか、あるいは競争優位性を維持することができるかもしれない。以下の例は例示的である
これらの事件のいずれかが発生した場合、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
従業員の事務、管理成長に関するリスク、そして私たちの業務に関連する他のリスク
私たちは私たちの組織を拡大する必要があり、私たちはこのような成長を管理することに困難に直面するかもしれないし、これは私たちの運営を混乱させるかもしれない。
時間の経過とともに、私たちの従業員数と業務範囲は大幅に増加し、特に製品候補開発、規制と臨床事務、医療事務、法律、財務、販売、マーケティング、流通の分野で大幅に増加すると予想される。私たちの成長活動を管理するためには、私たちの管理、運営、財務システムを継続して実施し、改善し、より多くの合格者を募集し、訓練し続けなければならない。私たちの財務資源が限られていることと、私たちの管理チームがこのような成長を期待している会社を管理する上での経験が限られているため、私たちの業務の拡張を効果的に管理したり、より多くの合格者を募集したりすることができないかもしれません。私たちが拡大するにつれて
100
組織内で、私たちは新しい職員たちを確定し、採用し、統合することが難しいかもしれない。今後の成長は私たちの経営陣に大きな追加責任をもたらすだろう
また、私たちの経営陣は、私たちの日常活動から不比例な注意を移し、これらの成長活動を大量に管理する必要があるかもしれない。私たちは私たちの業務の拡張を効果的に管理できないかもしれません。これは私たちのインフラが弱く、操作ミス、ビジネス機会の喪失、従業員の流失、残りの従業員の生産性の低下を招く可能性があります。私たちの予想成長は大量の資本支出を必要とし、開発候補製品のような他のプロジェクトから財政資源を移すことができるかもしれない。私たちの経営陣が私たちの成長を効果的に管理できなければ、私たちの費用は予想以上に増加する可能性があり、私たちは製品収入を創出および/または増加させる能力が低下する可能性があり、私たちは私たちの業務戦略を実施できないかもしれない。私たちの将来の財務業績と、私たちの候補製品を開発し、商業化し、効果的に競争する能力は、将来の任意の成長を効果的に管理する能力にある程度依存するだろう。
現在、予測可能な未来に、著者らは臨床前と臨床開発活動と製造を含むいくつかの独立組織、顧問とコンサルタントに大きく依存していくつかのサービスを提供する。必要な場合には、独立した組織、コンサルタント、コンサルタントのサービスがタイムリーに提供される保証はなく、合格した代替者を見つけることも保証されない。さらに、私たちのアウトソーシング活動を効果的に管理できない場合、あるいはコンサルタントが提供するサービスの品質や正確性が何らかの理由で影響を受ける場合、私たちが計画している臨床試験は延長、延期、または終了する可能性があり、私たちは規制機関の候補製品の承認や他の方法で私たちの業務を進めることができないかもしれません。私たちは私たちが既存のコンサルタントを経済的に合理的な条件で管理したり、他の適任な外部請負業者やコンサルタントを見つけることができるか、あるいは全くできないという保証はない。新入社員を雇用したり、コンサルタントや請負業者チームを拡大したりすることで、私たちの組織を効果的に拡張できない場合や、新しい施設を効率的に建設してこの拡張に適応できなければ、私たちの候補製品をさらに開発して商業化するために必要な任務を成功させることができない可能性がありますので、私たちの研究、開発、商業化の目標を達成できないかもしれません。
私たちと適格な人材やコンサルタントを競争する多くのバイオテクノロジーや製薬会社は、私たちよりも多くの財務や他の資源、異なるリスク状況、そしてより長い業界の歴史を持っています。もし私たちが高い素質の人員やコンサルタントを引き付け、維持することができなければ、候補製品を発見し、開発し、私たちの業務を経営する速度と成功は限られるだろう。
もし私たちが役員を失って、合格した幹部や他のキーパーソンを募集できなければ、私たちの業務は実質的な影響を受ける可能性がある。
私たちは私たちの最高経営責任者Mahesh Karande、私たちの最高科学者Thomas McCauley、私たちの最高財務官Joshua Reed、そして私たちの最高医療責任者厳モルを含む私たちの経営陣に高く依存している。私たちのすべての幹部は私たちの候補製品と私たちの運営に専門知識を持っているため、私たちのどの幹部のサービス損失も私たちの候補製品の開発を延期したり、私たちの業務運営に悪影響を及ぼす可能性があります。私たちは私たちのどんな幹部にもキーパーソン生命保険をかけません。一般的に、私たちと行政職員たちの採用計画は彼らがいつでも雇用を終了することを妨げるものではない。
さらに、私たちの将来の成功と成長は、私たちの従業員と管理者の持続的なサービス、そして私たちがより多くの人員を識別、採用、維持する能力にある程度依存するだろう。重要な従業員や管理者を交換することは困難またはコストが高い可能性があり、私たちの業界では開発に成功し、監督部門の承認を得て候補製品を商業化するために必要な技能と経験を持っている個人数が限られているため、時間がかかるかもしれない。この限られた人材バンクから募集する競争は激しく、多くの製薬と生物技術会社の間の競争を考慮すると、私たちは受け入れ可能な条件でキーパーソンを募集、訓練、維持或いは有効に激励することができないかもしれない
101
似たような人。大学や研究機関からの科学や臨床人材の競争も経験した。また、私たちは、科学と臨床コンサルタントを含むコンサルタントとコンサルタントに依存して、私たちの研究開発と商業化戦略の制定を助けてくれます。私たちのコンサルタントとコンサルタントは、私たち以外のエンティティによって採用される可能性があり、他のエンティティとの相談やコンサルティング契約に基づいて約束することができ、これは、彼らの私たちに対する利用可能性を制限するかもしれません。もし私たちが引き続き素質の高い人材を誘致し、維持することができなければ、候補製品を開発し、商業化する能力は制限されるだろう。
私たちの多くの従業員たちは私たちの大量の普通株式または多くの普通株式オプションになるか、またはこれから私たちの大量の普通株式または多くの普通株式オプションになるだろう。もし私たちの従業員が持っている株が株の元の購入価格に対して大幅に上昇した場合、あるいは彼らが持っているオプションの行権価格が私たちの普通株の市場価格よりも著しく低い場合、私たちの従業員は私たちを離れる可能性が高いかもしれない。
私たちは買収や戦略的協力を行う可能性があり、これは私たちの業務を混乱させ、株主の株式希釈を招き、私たちの財務資源を減少させ、私たちに債務を発生させたり、負債を負担したり、他のリスクに直面させたりする可能性がある。
将来的には、他の業務、製品、または技術を得るために取引を行うことができ、または許可を含む戦略的協力を達成することができる。適切な買収や協力を確実に決定すれば、有利な条件でこのような買収や協力を完成させることができず、完成することさえできないかもしれない。私たちが行ったいかなる買収や協力も、私たちの競争地位を強化しないかもしれませんし、私たちはこのような買収や協力の期待的な利益を決して実現しないかもしれません。私たちは、買収に関連する債務を発生させるか、または被買収会社の株主に私たちの普通株式または他の株式証券を発行することを決定することができ、これは私たちの既存株主の所有権パーセンテージを減少させるだろう。私たちは買収された業務や協力が発見されなかった債務によって損失を受ける可能性があり、これらの債務は私たちが売り手や私たちのパートナーから受ける可能性のある賠償範囲内ではない。さらに、私たちは、得られた任意の人員、技術、および運営を、効率的、タイムリーかつ中断のない方法で既存の業務に統合することに成功することができないかもしれません。買収や協力は、管理職の日常的な役割への関心を分散させ、キーパーソンの流失を招き、私たちの費用を増加させ、運営や他の用途に利用可能な現金や現金等価物を減らすことも可能である。将来の買収や協力の数、時間や規模を予測することもできず、どのような取引が私たちの経営業績に影響を与える可能性も予測できない。
私たちに対する訴訟は高価で時間のかかる弁護になるかもしれないし、追加的な責任を招くかもしれない。
私たちは時々法的手続きや通常の業務過程または他の側面で発生したクレーム、例えば第三者が商業紛争について提起したクレーム、および私たちの現職または元従業員が提出した雇用クレームの影響を受ける可能性がある。クレームは、政府機関、患者、または株主を含む様々な他の当事者または他の当事者によって提起されてもよい。
私たちに関連する訴訟は、巨額のコストを招き、運営上私たちの業務を制限し、経営陣の注意と資源を分散させる可能性があり、これは私たちの業務、全体の財務状況、運営結果を深刻に損なう可能性があります。保険は既存または未来のクレームを含まない可能性があり、私たちの1つ以上のそのようなクレームを完全に賠償するのに十分ではないか、または私たちが受け入れられる条項で保険を提供し続けることができる。私たちが提出した未保険や保険不足のクレームは思わぬコストを招き、私たちの運営結果に悪影響を及ぼす可能性があります。
私たちの普通株に関するリスクは
私たちの普通株の市場価格は大きく変動するかもしれない。
私たちの株価は変動するかもしれません。この変動のせいで、普通株を売って利益を得ることができないかもしれません。私たちの普通株の市場価格は多くの要素の影響を受けるかもしれません
102
103
また、株式市場全体、特にナスダック世界の精選市場、特にバイオテクノロジー会社は、極端な価格と出来高の変動を経験しており、これらの変動は往々にしてこれらの会社の経営業績と関係がないか比例しない。私たちの実際の経営業績にかかわらず、広範な市場と業界要素は私たちの普通株の市場価格にマイナス影響を与える可能性がある。従来、証券集団訴訟は、会社証券の市場価格が変動した後に会社に提起されることが多かった。このような訴訟を提起すれば、巨額の費用と経営陣の関心と資源の分流を招く可能性があり、これは私たちの業務、財務状況、運営結果を損なうことになる。
我々の役員、取締役、主要株主は、彼らが共同行動を選択すれば、株主に承認されたすべての事項を制御する能力があるだろう。
2023年12月31日現在の発行済み普通株数によると、我々の役員、取締役、私たち発行済み普通株を5%以上保有する株主と、それぞれの関連会社は合計約71%の発行済み議決権株を保有しています。したがって、これらの株主が共同行動を選択すれば、彼らは私たちの株主に提出されたすべての事項と、私たちの管理と事務を制御または顕著に影響することができるだろう。例えば、これらの株主は、取締役の選挙、私たちの経営陣の構成、および私たちのすべてまたはほとんどの資産の任意の合併、合併、または売却の承認を制御するだろう。これは、私たちの経営陣の変動を阻止したり、私たちの株主の一人としての最適な利益に合っていると考えられる私たちの普通株への能動的な買収提案や要約を阻止することができます。
私たちの総流通株の大部分は近い将来に市場に売る資格があります。これは私たちの業務が良好であっても、私たちの普通株の市場価格を大幅に低下させる可能性があります。
公開市場で私たちの普通株の大量株を売ったり、市場で大量の株を持っていると思っている人が株を売却しようとしたりすると、私たちの普通株の市場価格が下がる可能性があります。また、合計24,284,625株を保有する自社普通株の所有者は、特定の条件に適合する場合には、その株式に関する登録声明の提出、又はその株式を自己又は他の株主に提出可能な登録声明に、当該等の株式が規則144により制限されずに売却できるまで、又は権利が吾等と当該等の株主との間の株主合意条項によって終了するまで、吾等に提出する権利を要求する。私たちの株式補償計画によって発行可能なすべての普通株も登録しました。これらの株は発行後に公開市場で自由に販売することができますが、関連会社に適用される数量制限を受けています。
私たちは“新興成長型会社”であり、新興成長型会社に適用される開示要求を下げることは、私たちの普通株の投資家への魅力を低下させる可能性があります。
“2012年創業法案”または“雇用法案”の定義によると、私たちは“新興成長型企業”であり、(1)本年度の最終日まで、(A)IPO終了5周年後まで、(B)私たちの年間総収入が少なくとも12.35億ドル(インフレ調整済み)、または(C)大規模加速申告会社とみなされている。これは、私たちの非関連会社が保有する普通株の時価が、私たちが最近完成した第2四半期の最終営業日に7億ドルを超え、(2)私たちが前の3年間に10億ドルを超える転換不可能債券を発行した日を要求する。私たちがまだ新興成長型企業である限り、私たちは、他の非新興成長型企業に適用される上場企業の特定の開示要求の免除に依存することを許可され、意図されている。これらの免除には
104
我々は,本年度報告で減少した報告書負担を利用し,将来的にこのように継続する予定である。特に、私たちが新興成長型会社でなければ、私たちは役員報酬に関するすべての情報を含んでいません。私たちはもし私たちがこのような免除に依存すれば、投資家が私たちの普通株の吸引力の低下を発見するかどうか予測できない。また、雇用法案は、新興成長型会社は、延長された過渡期を利用して新たなまたは改正された会計基準を遵守することができると規定している。これにより、新興成長型企業は、それらが本来民間会社に適用されるまで、これらの会計基準の採用を延期することができる。また、新興成長型企業になる資格がなくなっても、“小さな報告会社”になる資格がある可能性があり、これにより、定期報告や依頼書で役員報酬に関する義務を減らすことを含め、多くの同じ免除開示要件を利用することができるようになります。したがって、一部の投資家が私たちの普通株の吸引力が低下していることを発見すれば、私たちの普通株取引市場はそれほど活発ではなくなり、私たちの株価は低下したり、もっと変動したりする可能性がある。
私たちは“小さな報告会社”であり、より小さい報告会社に適用される情報開示要求が低下し、私たちの普通株の投資家への吸引力を低下させる可能性がある。
私たちは“規模の小さい報告会社”と考えられている。したがって、私たちがまだ規模の小さい報告会社である限り、私たちは、3年間監査された財務諸表および役員報酬情報の提供を免除するなど、いくつかの減少した開示要求に依存する権利がある。もし私たちが小さな報告会社になる資格があれば、小さな報告会社の定義の下で収入制限を満たしているので、私たちは“低収入の報告会社”になるだろう。2002年サバンズ-オキシリー法第404条または第404条によれば、低収入の報告会社は、その財務報告書の内部統制の有効性を外部監査する必要はない。このような免除と減少の開示は投資家が私たちの運営結果と財政的見通しを分析するのを難しくするかもしれない。私たちは投資家が私たちがこのような免除に依存する可能性があるために私たちの普通株の吸引力が低下していることを発見するかどうか予測できない。したがって、一部の投資家が私たちの普通株の吸引力が低下していることを発見すれば、私たちの普通株はそれほど活発ではない取引市場がある可能性があり、私たちの株価はもっと変動するかもしれない。
上場企業として、私たちの運営コストは増加し続けており、私たちの経営陣は、新しいコンプライアンス措置やコーポレートガバナンス実践を実施するために多くの時間を投入する必要があります。
上場企業として、特に新興成長型企業ではなくなった後、多くの法律、会計、その他の費用が発生し続けており、これらの費用は上場企業になるまでは発生していません。2002年サバンズ-オクスリ法案、ドッド-フランクウォール街改革と消費者保護法、ナスダック世界精選市場の上場要求及びその他の適用された証券規則と法規は上場企業に対して各種の要求を提出し、有効な開示、財務制御と会社管理やり方を確立と維持することを含む。私たちの経営陣と他の人たちはこのようなコンプライアンス計画に多くの時間を投じている。しかも、このような規則と規制は私たちの法律と財務コンプライアンスコストを増加させ、いくつかの活動をより時間的で高価にするだろう。例えば、これらの規制は、私たちが取締役や上級管理職責任保険を獲得することをより困難かつ高価にし、逆に合格した取締役会メンバーを引き付け、維持することを困難にする可能性がある。
私たちはこのような規則と規定を評価しており、私たちが発生する可能性のある追加コスト金額やそのようなコストの時間を予測または推定することができない。これらの規則や条例は往々にして異なる解釈を持ち,多くの場合特殊性に欠けるため,規制機関や理事機関が新たな指導意見を提供するにつれて,実践における適用は時間とともに変化する可能性がある。これは遵守事項に関する持続的な不確実性と、開示と統治慣行を絶えず修正するために必要なより高いコストをもたらす可能性がある。
2002年サバンズ-オキシリー法404条または404条によると、私たちは財務報告書の内部統制に関する管理職報告書を提出しなければならない。しかし、我々は依然として新興成長型企業または低収入の比較的小さい報告会社であるにもかかわらず、我々の独立公認会計士事務所が発行する財務報告書の内部統制に関する認証報告書を含むことは要求されないであろう。規定された期間内に第404条を遵守するために、私たちは高価で挑戦的な財務報告書の内部統制の過程を記録して評価している。この点では,内部資源を提供し続け,外部コンサルタントを招聘し,詳細な作業計画によって財務報告の内部統制の十分性を評価·記録し,制御プログラムを改善する手順を継続する必要がある
105
適切な場合には、このような制御が文書規定に従って実行されているかどうかをテストにより検証し、財務報告内部制御の継続的な報告と改善の流れを実施する。私たちは努力したにもかかわらず、私たちまたは私たちの独立公認会計士事務所は、規定された時間枠内で、または根本的に結論を出すことができません。すなわち、財務報告の内部統制に有効であり、第404条の要求に適合しています。私たちは私たちの財務報告書の内部統制に重大な欠陥や重大な欠陥があることを発見するかもしれないし、私たちはタイムリーではないかもしれないし、根本的に成功的に救済できないかもしれない。私たちが発見した任意の重大な欠陥や重大な弱点を是正できなかったか、または必要な新しいまたは改善された制御措置を実施できなかったか、または実行中に困難に遭遇したことは、私たちの報告義務を履行できなかったり、私たちの財務諸表に重大な誤報を招いたりする可能性がある。1つまたは複数の重大な弱点を発見すれば、財務諸表の信頼性に対する自信を失った金融市場の不良反応を招く可能性がある。
財務報告の効率的な内部統制と効率的な開示制御プログラムを保持できなかった場合、財務結果をタイムリーに正確に報告したり、詐欺を防止したりすることができず、投資家がわが社の信頼に悪影響を及ぼす可能性がある。
我々は、米国証券取引委員会が2002年サバンズ-オキシリー法案第302および404節を実施した規則を遵守しなければならない。この2節は、経営者に、我々の四半期および年間報告書において財務および他の情報を認証し、財務報告の内部統制の有効性に関する年間管理報告を提供することを要求する。新興成長型企業と低収入の報告会社として、我々の独立公認会計士事務所は、第404条に基づいて財務報告書の内部統制の有効性を証明することを要求されなくなり、新興成長型企業または低収入の報告会社ではなくなる。このとき、私たちの財務報告内部統制に重大な欠陥が発見された場合、私たちの独立公認会計士事務所は不利な報告書を発表する可能性があります。
上場企業の要求に適合するためには、新たな内部統制やプログラムの実施、会計や内部監査者の招聘など、より多くの行動をとる必要がある。内部統制のテストと維持は、私たちの経営陣の注意を、私たちの業務運営に重要な他の問題から移すことができます。また、財務報告書の内部統制を評価する際には、404条を遵守するために設定された適用期間を満たすために、タイムリーに救済できない可能性のある重大な弱点を見つけることができるかもしれない。我々の財務報告内部統制に重大な弱点があることが発見された場合、または404条の要求を直ちに遵守できない場合、または財務報告内部統制が有効であると断言したり、独立公認会計士事務所が新興成長型企業でなくなると、財務報告内部統制の有効性について意見を述べることができない場合、投資家は私たちの財務報告の正確性と完全性に自信を失う可能性がある。したがって、私たちの普通株の市場価格は実質的な悪影響を受けるかもしれない。
私たちの開示統制と手続きはすべてのミスや詐欺を阻止したり検出できないかもしれない。
我々は、1934年に改正された証券取引法または“取引法”の定期報告要求を遵守しなければならない。私たちは、取引所法案に基づいて提出または提出された報告書で開示されなければならない情報が蓄積され、管理層に伝達され、米国証券取引委員会規則および表で指定された期間内に記録、処理、まとめ、報告されなければならないことを保証するために、合理的な保証を提供するために、開示制御および手続きを継続している。いかなる開示規制と手続きも、構想や運営がどのように詳細であっても、絶対的な保証ではなく合理的な保証しか提供できず、規制制度の目標が達成されることを確保するしかないと信じている。
これらの固有の限界は,意思決定過程における判断が誤りである可能性があり,簡単な誤りや誤りによって故障が発生する可能性があるという現実を含む.さらに、ある人の個人的な行動、2人以上の結託、または無許可超越制御は、制御を回避することができる。したがって,我々の制御システム固有の制限により,誤りや詐欺による誤り陳述が発生し,発見されない可能性がある.
証券や業界アナリストが私たちの業務に関する研究や報告を発表しない場合、あるいは彼らが私たちの株に不利または誤った意見を発表すれば、私たちの業務が良好であっても、私たちの株価や取引量が低下する可能性がある。
106
私たちの普通株の取引市場は、業界または証券アナリストが発表した私たちまたは私たちの業務に関する研究と報告の影響を受けるだろう。私たちは現在証券と産業アナリストに対する研究報告書が限られている。もし私たちのどのアナリストが私たちの普通株の格付けを引き下げたか、あるいは私たち、私たちのビジネスモデル、私たちの知的財産権、あるいは私たちの株式表現に不利または誤った意見を発表した場合、あるいは私たちの目標の臨床前研究や臨床試験と運営結果がアナリストの期待に達しなかった場合、私たちの株価は下落する可能性がある。これらのアナリストのうちの1人以上が私たちへの報告を停止したり、私たちに関する報告書を定期的に発表できなかったりすれば、私たちは金融市場で可視性を失う可能性があり、これは逆に私たちの株価や取引量を低下させる可能性がある。
私たちの会社登録証明書の改訂と再記述の会社定款やデラウェア州法律の条項は、私たちの会社を買収することをより困難にする可能性があり、これは私たちの株主に有利になる可能性があり、私たちの株主が現在の経営陣を交換または更迭しようとすることを阻止するかもしれません。
当社の改正および再記載された会社登録証明書および当社の改正および再記載された定款の条項は、あなたの株式から割増取引を得る可能性があることを含む、株主が有利と考える可能性のある会社の合併、買収、または他の支配権変更を阻止、延期または阻止する可能性があります。これらの条項はまた、投資家が将来私たちの普通株に支払いたいかもしれない価格を制限し、それによって私たちの普通株の市場価格を下げる可能性がある。また、我々の取締役会が責任を持って我々の管理チームのメンバーに命じているため、これらの規定は、株主が取締役会のメンバーを交換する難しさを増やすことで、現在の経営陣の任意の試みを交換または罷免することを阻害または阻止する可能性がある。他の事項を除いて、これらの規定には規定が含まれている
また、私たちはデラウェア州に登録して設立されたので、私たちはデラウェア州会社法第203条の規定によって管轄されています。この条項は、規定された方法で合併または合併が承認されない限り、私たちが発行した議決権のある株を15%以上保有している人が取引日後3年以内に私たちと合併または合併することを禁止しています。
107
私たちが修正して再記述した会社登録証明書は、特定の裁判所が私たちの株主が起こしうるいくつかの訴訟の独占フォーラムとして指定され、これは、私たちの株主が有利な司法フォーラムを得ることが私たちとの紛争を処理する能力を制限するかもしれない。
我々が書面で代替裁判所を選択することに同意しない限り、我々が書面で代替裁判所を選択することに同意しない限り、デラウェア州衡平裁判所は、株主が私たちに提起したクレームに関連する大多数の法的訴訟の唯一かつ独占的な裁判所であるが、“取引法”に規定されている任意の責任または義務または連邦裁判所が排他的管轄権を有する任意の他のクレームを強制的に執行するための訴訟を除外し、デラウェア州衡平裁判所が標的管轄権の欠如によって却下された任意の訴訟をデラウェア州の別の州または連邦裁判所で提起する可能性がある。私たちが改正して再記載した会社登録証明書は、私たちが書面で代替裁判所を選択することに同意しない限り、アメリカ連邦地域裁判所は、改正された1933年の証券法または証券法によって提起された任意の訴えを解決するための独占的なフォーラムとなる。任意の者又は実体が自社株の株式を購入又はその他の方法で取得する任意の権益は、当社の上述した改正及び再記載された会社登録証明書の規定に了承され、同意されたものとみなされる。
この条項は、会社紛争の解決に特に経験のある総理や証券法紛争の解決に経験のある連邦裁判官のデラウェア州法律の適用における一貫性を向上させ、他のフォーラムよりも速いスケジュールで効率的に事件を管理し、多裁判所訴訟からの負担を保護するため、私たちに利益を与えると信じている。しかしながら、この条項は、司法裁判所において、任意の株主が私たちまたは私たちの取締役、上級管理者、従業員または代理とトラブルを発生させることに有利であると考える株主のクレームを提起する能力を制限し、株主がクレームを出すコストを増加させる可能性があるので、私たちの役員、上級管理者、従業員、および代理人に対する訴訟を阻止する可能性がある。他の会社の会社登録証明書における同様の選択裁判所条項の実行可能性が法的手続きで疑問視されており、裁判所は、私たちが提起した任意の適用訴訟において、私たちが改訂および再記載した会社登録証明書に含まれる選択裁判所条項が、このような訴訟では適用されないか、または実行できないことを発見する可能性がある。もし裁判所が私たちが改正して再記載した会社登録証明書に含まれる裁判所条項の選択が訴訟で適用されないか、または実行できないことが発見された場合、私たちは他の管轄区域でこのような訴訟の解決に関連する追加費用を発生する可能性があり、これは私たちの業務、財務状況、または運営結果に悪影響を及ぼす可能性がある。
私たちは純営業損失の繰越や他の税務属性を利用して将来の課税収入を相殺する能力が一定の制限を受ける可能性があります。
2023年12月31日現在、私たちのアメリカ連邦と州の純営業損失繰越(NOL)はそれぞれ1.874億ドルと1.804億ドルであり、もしあれば、未来の課税収入を相殺することができる。2023年12月31日まで、私たちは連邦と州の研究開発信用の繰り越しがあり、それぞれ1240万ドルと490万ドルです。一般的に、改正後の1986年の“国内税法”第382条と383条または同法規によると、会社は“所有権変更”を経験し、通常3年間のスクロール期間中に、その持分所有権は価値によって計算された変化が50ポイントを超え、その利用変更前のNOLとその研究開発信用は将来の課税所得額を相殺する能力が制限されていると定義されている。著者らの現有のNOLと研究開発信用の転換は以前の所有権変更による制限を受ける可能性があり、もし私たちが所有権の変更が発生すれば、私たちがNOLと研究開発信用の転換を利用する能力は規則第382と383節の更なる制限を受ける可能性がある。また、規則382及び383条の規定によれば、将来、私たちの株式の変化、その中のいくつかは私たちの制御範囲を超える可能性があり、所有権の変化を招く可能性があります。これらの理由で、利益を達成しても、NOLや研究開発信用の大きな一部を利用できないかもしれません.
一般リスク
情報技術システムの故障,欠陥や侵入が発生すると,我々の業務や運営が影響を受ける可能性があり,我々の業績に大きな影響を与える可能性がある.
コンピュータウイルスおよび他のマルウェア(例えば、恐喝ソフトウェア)、不正アクセスまたは他のネットワークセキュリティ攻撃、自然災害(ハリケーンを含む)、テロ、戦争、火災、ならびに電気通信または電気故障の故障または破損を受けやすい、我々の情報技術システム、および私たちのCROおよび他の請負業者およびコンサルタントのシステム。私たちの正常なビジネスプロセスでは直接または間接的に収集し
108
我々の臨床試験対象および従業員の知的財産権、機密情報、臨床前および臨床試験情報、独自の業務情報、個人情報、および健康関連情報を含む、我々のデータセンター、私たちのネットワーク、または第三者のデータセンター、およびネットワークにおいて敏感な情報を格納および送信する。このような情報の安全な処理、維持、そして伝達は私たちの運営に必須的だ。私たちはセキュリティ措置を取っているにもかかわらず、私たちの情報技術およびインフラは、ハッカーまたは内部非行者の攻撃を受けやすく、または人為的ミス(例えば、社会工学、ネットワーク釣り)、技術的脆弱性、汚職、または他の中断によって破られる可能性がある。世界各地からの未遂攻撃と侵入の数、強度と複雑性の増加に伴い、セキュリティホールや破壊のリスクは普遍的に増加し、特にコンピュータハッカー、外国政府とネットワークテロリストを含むネットワーク攻撃或いはネットワーク侵入を介している。私たちはまた、インターネット技術への依存と私たちの遠隔作業の従業員の数によって、サイバー犯罪者の抜け穴を利用するためのより多くの機会を作るかもしれないので、より多くのネットワークセキュリティリスクに直面する可能性がある。私たちはすべての種類の安全脅威を予見できないかもしれないし、これらすべての安全脅威に対して効果的な予防措置を取ることができないかもしれない。サイバー犯罪者が使用する技術はしばしば変化し,起動前に識別されない可能性があり,外部サービスプロバイダ,組織犯罪分岐機関,テロ組織や敵対する外国政府や機関などの外部団体を含む様々なソースから来る可能性がある.たとえ私たちが安全事件を決定したとしても、攻撃者が制御を回避し、法医学的証拠を検出および除去または混同するためのツールおよび技術をますます多く使用しているので、事件または違反行為を十分に調査または修復することができないかもしれない。私たちのデータ保護努力と情報技術への私たちの投資は、私たちのシステムまたは私たちのCDMO、CRO、および他の請負業者、コンサルタントのシステムに重大な故障、データ漏洩、または脆弱性を防止することを保証することはできません。
私たちと私たちの特定のサービス提供者たちは時々ネットワーク攻撃とセキュリティ事件の影響を受ける。これまで、重大なシステム故障、事故、セキュリティホールを経験したことはないと思いますが、このような事件が発生して、私たちの運営や私たちの重要な第三者の運営が中断されれば、私たちの候補製品開発計画、私たちの運営、そして最終的に私たちの財務業績に大きな中断を招く可能性があります。例えば、完成した、進行中または計画中の研究または試験中の臨床前研究または臨床試験データの損失は、我々の規制承認作業を遅延させる可能性があり、データを回復または複製するコストを著しく増加させる可能性がある。任意の中断またはセキュリティホールが、私たちのデータやアプリケーションを紛失したり、破損したり、個人、機密、または独自の情報を適切に開示しない場合、私たちは責任を招く可能性があり、私たちの候補製品のさらなる開発が延期される可能性があります。このような重大なセキュリティホールは、私たちの情報技術システムに危険を及ぼす可能性があり、そこに格納された情報がアクセスされ、公開され、開示され、失われ、または盗まれる可能性がある。
このような情報のアクセス、開示、または他の損失は、法的クレームや訴訟、個人情報のプライバシーを保護する法的責任と重大な規制処罰を招く可能性があり、このような事件は私たちの運営を乱し、私たちの名声を損なう可能性があり、人々が私たちと私たちの臨床試験を行う能力に自信を失わせる可能性があり、これは私たちの名声に悪影響を与え、私たちの候補製品の臨床開発を延期する可能性がある。私たちはネットワーク責任保険を維持する;しかし、この保険は、私たちのシステムの中断または破壊によって引き起こされる可能性のある財務、法律、商業、または名声損失をカバーするのに十分ではないかもしれない。
私たちまたは私たちが依存している第三者は自然災害や流行病の悪影響を受ける可能性があり、私たちの業務の連続性や災害復旧計画は深刻な災害から私たちを十分に保護できないかもしれません。
自然災害や流行病は私たちの運営を深刻に混乱させ、私たちの業務、運営結果、財務状況、将来性に重大な悪影響を及ぼす可能性がある。自然災害、停電、大流行、または他の事件が発生した場合、本部の全部または大部分を使用することができず、私たちが依存している製造施設のような重要なインフラを損傷させたり、他の方法で運営を中断したりすることは難しいかもしれません。場合によっては、長い間私たちの業務を継続することはできません。深刻な災害や同様の事件が発生した場合、我々の既存の災害復旧および業務連続計画は十分ではないことが証明される可能性がある。私たちの災害復旧と業務連続計画の限られた性質のため、私たちは大量の費用を発生する可能性があり、これは私たちの業務に実質的な悪影響を与える可能性があります.
不安定な市場や経済状況は、私たちの業務、財務状況、株価に深刻な悪影響を及ぼす可能性がある。
109
信用と金融市場を含む世界経済は最近、流動性と信用供給の深刻な減少、金利とインフレ率の上昇、銀行と金融機関に関連する危機、消費者自信の低下、経済成長の低下、失業率の上昇、経済安定の不確定など、極端な変動と破壊を経験している。株式市場や信用市場が悪化し続けている場合、あるいは米国が衰退に入った場合、任意の必要な債務や株式融資を適時または有利な条件で得ることが難しくなり、コストが高く、または希釈作用がある可能性がある。さらに、我々の1つまたは複数のCRO、プロバイダ、CDMO、または他の第三者プロバイダは、経済低迷または衰退の中で生き残ることができない可能性がある。したがって、私たちの業務、経営業績、普通株価格は不利な影響を受ける可能性があります。
環境持続可能性と社会的イニシアティブへの日々の関心は、私たちのコストを増加させ、私たちの名声を損ない、私たちの財務業績に悪影響を及ぼす可能性がある。
投資家、患者、環境保護活動家、メディア、政府と非政府組織は、様々な環境、社会、その他の持続可能な発展問題に対する国民の関心が高まっている。私たちは圧力に直面する可能性があり、持続可能性に関連する具体的なリスク緩和戦略の設計と実施を含む、私たちの持続可能性に影響を与える事項に関する約束を行うことが求められる。私たちの業務に影響を与える環境、社会、その他の持続可能な開発問題を効果的に解決したり、関連する持続可能な開発目標を策定して実現できなければ、私たちの名声や財務業績は影響を受ける可能性があります。また、私たちがこれらの懸念を効果的に解決しても、私たちの持続可能な開発目標を実行することによってコストが増加する可能性があり、これらのコストは私たちの名声のいかなるメリットによっても相殺できない可能性があり、これは私たちの業務や財務状況に悪影響を及ぼす可能性がある。
また,環境,社会,その他の持続可能な問題への重視は,新たな報告要件を含む新たな法律や条例の採択につながる可能性がある。もし私たちが新しい法律、法規、または報告要件を遵守できなければ、私たちの名声と業務は不利な影響を受けるかもしれない。
私たちは予測可能な未来に私たちの普通株に現金配当金を支払わないと予想されるので、資本付加価値(あれば)があなたの唯一の収益源になるだろう。
私たちは普通株式に対するどんな現金配当金も発表したり支払ったりしたことがない。私たちは現在、私たちの業務の発展、運営、拡張のために、すべての利用可能な資金と将来の収益を維持し、予測可能な未来に現金配当金を発表または支払うことはないと予想している。したがって、予測可能な未来に、私たちの普通株の資本付加価値はあなたが私たちの普通株に投資する唯一の収益源になるだろう。
私たちは証券集団訴訟の影響を受けるかもしれない。
従来、証券集団訴訟は、ある会社の証券市場価格が下落した後に提起されることが多かった。このリスクは、バイオテクノロジー会社が近年大幅な株価変動を経験しているため、私たちと特に関連している。私たちがこのような訴訟に直面すれば、巨額のコストを招き、経営陣の注意と資源を移転させる可能性があり、これは私たちの業務を損なう可能性があります。
110
項目1 B。未解決従業員コメント。
適用されません。
プロジェクト1 Cネットワーク·セキュリティ.
ネットワークセキュリティリスク管理と戦略
我々は,我々のキーシステムと情報の機密性,完全性,可用性を保護するためのネットワークセキュリティリスク管理計画を策定し実施した.私たちのサイバーセキュリティリスク管理計画にはサイバーセキュリティ事件対応計画が含まれている。
著者らは国家標準と技術研究院ネットワークセキュリティフレームワーク(NIST CSF)などの業界標準フレームワークに基づいて、私たちの計画を設計と評価した。これは、特定の技術基準、規範、または要求を満たすことを意味するわけではありませんが、ガイドラインとしてNIST CSFを使用して、私たちの業務に関連するネットワークセキュリティリスクの識別、評価、管理を支援しています。
我々のネットワークセキュリティリスク管理計画は、私たちの全体企業リスク管理計画に統合され、企業リスク管理計画全体に適用される汎用的な方法、報告ルート、管理プロセスを共有し、他の法律、コンプライアンス、戦略、運営、金融リスク分野に適用される。
私たちのネットワークセキュリティリスク管理プログラムは
私たちは、私たちの運営、業務戦略、運営結果、または財務状況を含む、既知のネットワークセキュリティ脅威(これまでの任意のネットワークセキュリティイベントを含む)から、私たちに重大な影響を与えるか、または合理的に私たちに重大な影響を与える可能性があるリスクを決定していません。詳細については,“リスク要因--情報技術システムの故障,欠陥や侵入が発生すると,我々の業績に大きな影響を与える可能性がある場合,我々の業務や運営が影響を受ける可能性がある”というタイトルの節を参照されたい
サイバーセキュリティ·ガバナンス
当社取締役会は、ネットワークセキュリティリスクをそのリスク監督機能の一部とし、監査委員会(“委員会”)にネットワークセキュリティや他の情報技術リスクの監督を依頼している。その委員会は管理職たちが私たちのネットワークセキュリティリスク管理計画を実施することを監督する。
委員会は毎年私たちのネットワークセキュリティリスクに関する管理職の報告書を受け取っている。また,管理層は,必要に応じて任意の重大なネットワークセキュリティイベントおよび任意の影響の小さいイベントの最新状況を委員会に通報する.
委員会は、ネットワークセキュリティに関する活動を含む取締役会全員にその活動を報告した。取締役会の全メンバーはまた、我々のネットワークリスク管理計画に関する経営陣のブリーフィングを聴取した。取締役会メンバーは、上場企業に影響を与えるテーマに関する取締役会の継続教育の一部である内部セキュリティスタッフ又は外部専門家からネットワークセキュリティテーマの紹介を聴取する。
111
我々の管理チームは、最高財務官とデータ·情報技術副社長を含み、ネットワークセキュリティ脅威からの私たちの重大なリスクの評価と管理を担当しています。このチームは、私たちの全体ネットワークセキュリティリスク管理計画に主な責任があり、私たちの内部ネットワークセキュリティ担当者と私たちが招聘した外部ネットワークセキュリティコンサルタントを監視します。著者らの管理チームの集合経験は10年以上の指導ネットワークセキュリティ監督と事件応答情景のデスクトップ演習を含む。
我々の管理チームは、内部セキュリティ担当者のプレゼンテーション、政府、公共またはプライベートソース(私たちが招聘した外部コンサルタントを含む)から得られた脅威情報および他の情報、ならびに情報技術環境に配置されたセキュリティツールによって生成された警報および報告を含むことができる様々な手段を介して、ネットワークセキュリティリスクおよびイベントを予防、発見、緩和および修復する作業を監視する。
項目2.財産.
2021年11月、私たちはAre-MA Region No 94,LLCと賃貸契約を締結し、マサチューセッツ州ケンブリッジ市チャールズ公園1号にあるオフィスと実験室の総面積は約89,246平方フィートです。第1段階レンタルは2023年5月に開始し、第2段階レンタルは2023年8月に開始する。各段階のレンタル期間は各段階のレンタル開始日から15年後に終了しますが、いくつかの延長権によって制限されなければなりません。私たちの施設は私たちの現在の需要を満たすのに十分で、必要な時に適切な追加空間を提供すると信じている。
プロジェクト3.法律専門家割譲する。
私たちは実質的な法的手続きの制約を受けないだろう。
プロジェクト4.鉱山安全開示する。
適用されません。
私たちの執行担当者と役員に関する情報
次の表は、本年度報告10-K表までの各執行役員と取締役の名前、年齢、ポストを示しています
名前.名前 |
|
年ごろ |
|
ポスト |
行政員 |
|
|
|
|
マッハシュ·カランダー |
|
51 |
|
取締役最高経営責任者総裁 |
ジョシュア·リード |
|
51 |
|
首席財務官 |
トーマス·マッコーリー博士 |
|
55 |
|
首席科学官 |
厳モル、医学博士。 |
|
57 |
|
首席医療官 |
凌増する |
|
55 |
|
首席法律と行政官 |
非従業員取締役 |
|
|
|
|
クリスティアン·S·シャド |
|
62 |
|
取締役会議長 |
レナ·ボム |
|
63 |
|
役員.取締役 |
ルーク·M·ベシャール |
|
65 |
|
役員.取締役 |
エリオット·M·リヴィ医学博士 |
|
65 |
|
役員.取締役 |
ジョン·メンデリン博士J.D. |
|
64 |
|
役員.取締役 |
ミシェル·C·ウォーナー |
|
48 |
|
役員.取締役 |
メアリー·T·セラ |
|
60 |
|
役員.取締役 |
リチャード·A·ヤン博士です |
|
69 |
|
役員.取締役 |
行政員
112
マッハシュ·カランダー2019年6月以降、総裁兼最高経営責任者と取締役会のメンバーを務めてきました。カランダーさんは、2018年4月から2019年3月までの間に、製薬会社の社長(その後、ゼカニー製薬会社)のCEO兼CEOを務めました。カランダーさんは、2010年3月から2017年4月までの間に、米国腫瘍学副社長兼フランチャイズ主管、ノバアフリカ会社社長、ノファ社エジプト支社総裁を含む、ノルウェイ社の上級指導者を務めました。カランダーはペンシルバニア大学ウォートンビジネススクールの工商管理修士号を持っています。ジョージア工科大学とムンバイ大学を卒業し、そこで工学修士号を取得し、そこで工学学部課程を修了した。我々は、カランダーさんの豊富な生命科学とリーダーシップの経験が彼が私たちの取締役会に在籍する資格を持っていると信じています。
ジョシュア·リード2022年5月以来最高財務官を務めてきた。Reedさんは、オメガに加入する前に、2018年7月~2022年5月にAldeyra Treeuticsの最高財務責任者を務め、財務、ビジネスの発展、投資家関係、コンプライアンス、人的資源、情報技術を担当しました。Aldeyra在任中、リードは何度も融資を指導し、会社と既存と潜在投資家との相互作用を監督し、アメリカ証券取引委員会に提出された四半期と年間報告を含む会社の財務プロセスのあらゆる面を管理した。Reedさんは、Aldeyraに参加する前に、百時美施貴宝でますます重要な財務責任を負い、2016年6月から2018年7月までの間に財務副社長と米国プエルトリコ財務運営責任者を務めていました。リードさんは、百時美施貴宝の在任中にも財務計画と分析を担当し、様々な買収、資産剥離、同盟、協力協定の作業に参加しました。彼のキャリアの初期、リードはモルガン大通、スイス信用第一ボストン銀行と大通マンハッタン銀行に勤めていた。彼は現在も上場バイオテクノロジー会社Scholar Rockの取締役会メンバーと監査委員会の議長を務めている。リード·さんは、ローッグス大学で金融学の学士号を取得し、ミシガン大学ロスビジネス大学で工商管理の修士号を取得した。
トーマス·マッコーリー博士2019年7月より弊社首席科学官を務めております。2018年9月から2019年7月まで、マッコーリー博士はMacrolide PharmPharmticals(その後のZikani治療会社)の首席科学官を務め、2016年9月から2018年4月まで翻訳生物会社(元Rana治療会社)の首席科学官を務めた。2010年4月から2016年8月まで、マッコーリー博士はシャルル製薬副総裁兼全世界非臨床開発主管を務め、期間中に彼はファブリ病の治療のReplagal、高謝病を治療するVprv、ハント症候群を治療するElapraase、遺伝性血管性水腫を治療するFirazyrとドライアイを治療するXIIDRAを含むシャルルの多くの製品の開発と全世界の承認に参与した。マッコーリー博士はアラバマ大学バーミンガム校の博士号と、理学士と工学修士号を持っている。コーネル大学から来ました。
厳モル、医学博士。2022年1月より弊社首席医療官を務めております。2018年9月から2021年12月まで、ムーア博士は益プソン製薬腫瘍治療区の責任者の高級副総裁を務めた。ムーア博士は2016年9月から2018年9月まで、アンキアーノ治療会社(前身はBioCancell治療会社)の臨床開発·研究開発部門の首席医療官と上級副総裁を務めた。彼のキャリアの初期、ムーア博士はAriad、サイノフィ、グラクソ·スミスクラインと百時美施貴宝で多くのポストを担当し、全世界の医療事務と臨床開発を担当した。ムーア博士は臨床医としてサピール医療センター,メル病院,エディス·ウォルフソン医療センターで働いていた。ムーア博士はテルアビブ大学ザックラー医学院で医学学位と医学学士号を取得し、ドレクセル大学レボ商学院で工商管理修士号を取得し、ハーバード商学院で高級管理課程を修了した。
凌増する2022年3月以来、わが社の首席法律·行政官を務めてきました。2020年9月から2022年3月まで、曽さんは最近Dicerna PharmPharmticalsで首席法務官兼秘書を務め、そこで彼女は他の幹部、取締役会とその委員会と共に会社戦略、政策、コンプライアンスと管理活動を制定し、実施した。2017年8月から2020年9月まで、曽さんはノワ製薬でグループM&A法律部の副主管を務め、ノバグループの全世界取引を担当し、すべての業務部門と地域を含む。これまで、ボッシュヘルス、Penwest製薬、バルラボで様々な責任が増してきた法律行政を務めてきた。かつてはCleary、Gottlieb、Steen、Hamiltonから始まり、これまでAlkermes Inc.とLeukosite Inc.で研究員を務めていた。女性は北京大学で物理学理学学士号を取得し、ブランディス大学で生物物理学理学修士号を取得し、ジョージタウン大学で法学博士号を取得した。
113
非従業員取締役
クリスティアン·S·シャド2023年7月から当社の取締役会メンバーを務め、2023年8月から当社の取締役会長を務めています。シャダーさんは、いくつかの役員職でリーダーシップを証明することを含む、30年以上の経験を持つ民間企業や上場バイオ製薬会社での経験豊富な幹部です。2023年1月に旗艦先鋒に入社し、成長パートナーを務めた。2016年7月から2022年8月まで、総裁はアプレア治療会社の最高経営責任者。シャードは2016年からAprea治療会社の取締役会メンバーを務め、2020年9月から2023年8月まで取締役会議長を務めている。Apreaに加入する前に,Novira,Omthera製薬会社,Medarexで指導職を務めていた。Schadeさんは、業界の専門知識のほか、投資銀行から幅広い企業融資と資本市場経験をもたらし、米林とモルガン·チェースで勤務していた。2006年以降、INCELA LifeSciences,Inc.(ナスダック:IAT)の取締役会に在籍している。彼はペンシルバニア大学ウォートンビジネススクールの商工管理修士号とプリンストン大学の文学学士号を持っています。私たちは、Schadeさんはバイオテクノロジーと投資銀行の産業の豊富な指導経験で、彼が私たちの取締役会に就く資格があると信じている。
レナ·ボム2022年9月以来、私たちの取締役会のメンバーを務めてきた。ボムさんはオメガに30年以上の臨床と管理経験をもたらした。1988年から2017年にかけて、ノワ製薬株式会社とその前身であるCIBA−Geigyで複数の高級管理職を務め、最近は首席商業·医療事務官を務めている。彼はノバ腫瘍学会社の設立に成功したキーパーソンだ。彼は全世界の異なる地区の多くの爆発的なブランドの発売とライフサイクル管理を監督し、その中に腫瘍学領域のFemara、ZometaとGlivec、及びCosentyxとEntresto及び免疫学と心血管疾患領域を含む。ノワール社に入社する前に、ドイツのズビファルガーの精神病院で部門の責任者を務めていた。ボームさんは現在、Cellectis SA(ナスダック市場コード:CLLS)、Humanigen Inc.(ナスダック市場コード:HGEN)の取締役会に勤めており、2018年7月から2022年4月まで北欧Nanovector S.A.の取締役会に在籍していた。彼はドイツのウルム大学の医学学位とフランスのストラスブールキャンパスのシラー大学の工商管理修士号を持っている。最近、スイスのバーゼル/ベルン/チューリッヒ大学で公衆衛生修士課程を専攻するようになった。我々は、ボームさんは製薬業界の重要なリーダーシップ経験によって、彼が私たちの取締役会に在任する資格があると信じています。
ルーク·M·ベシャール2021年5月から私たちの取締役会のメンバーを務めてきた。ベルシャールさんは、上場企業および私株会社を中心に30年以上の経験を持つ行政指導者および最高財務責任者の職に就いています。ベシャールは2020年1月以来ずっと取締役会主席を務め、2018年10月から上場免疫腫瘍学会社Protara Treeuticsの取締役会メンバーを務めている。2014年3月から2021年11月まで、ファイザー社によって買収され、当社に上場しているベシャールさん社の臨床段階免疫腫瘍学会社Trillium Treateutics Inc.が取締役を務めています。2015年5月から2021年9月まで、ベシャールは発売されたリード臨床段階遺伝子治療会社Regenxbio,Inc.の取締役会メンバーを務めた。これまで、ベシャールは2007年から2015年2月までの間にNPS PharmPharmticals,Inc.首席財務長総裁を務めていた。NPS PharmPharmticalsは稀な疾患の治療に専念する全世界の上場生物製薬会社である。NPS製薬会社に入社する前に、ベシャールさんは2002年から2007年まで、上場企業Cambrex Corporationの最高財務責任者(CEO)を務めていましたが、同社は上場ブランドおよびジェネリック医薬品活性成分メーカーおよび関連サービス提供者です。ベシャールのキャリアは安達信会計士事務所から始まり、公認会計士だった。ベシャーはミシガン州立大学で会計と財務管理学士号を取得し、バージニア大学ダットンビジネススクール幹部課程を卒業した。我々は、ベシャールさんは製薬業界の豊富なリーダーシップ経験によって、彼が私たちの取締役会に就く資格があると信じています。
エリオット·M·リヴィ医学博士2021年3月以来、私たちの取締役会に勤めてきた。リーヴィ博士は2021年6月以来、ボストンコンサルティンググループで上級顧問を務めてきた。リヴィ博士は2014年9月から2020年6月まで安進全世界発展部高級副総裁を務め、2020年6月から2021年5月まで研究開発戦略及び運営部高級副総裁を務めた。また、2021年11月からNuCana plcの取締役会メンバーを務め、2023年4月以来Editas Medicineの取締役会メンバーを務めている。リヴィ博士はエール大学で医学博士号を取得し、エール大学で学士号を取得した。私たちはリーヴィ博士のこの業界での豊富な経験が彼が私たちの取締役会に就く資格があると信じている。
114
ジョン·メンデリン博士J.D.2020年1月以来、私たちの取締役会のメンバーを務めてきた。メンデリン博士は現在旗艦パイオニア社の執行パートナーである。メンドライアン博士は、2018年1月から2019年2月まで、Moderna社の企業·製品戦略の総裁を務めた。1996年から2017年にかけて、メンデライアン博士は、Affdium製薬会社(Debiamm Groupに買収)、Adnexus Treeutics(BMSに買収)、aTyr Pharma、Inc.またはATyr、Aurora Biosciences、Fate Treateutics、Inc.を含む複数のバイオテクノロジー会社で異なる上級管理·取締役を務めてきた。2011年から2017年にかけて、aTyrのCEOも務めた。彼のバイオテクノロジー人生はスミス-クラインとフランキー(現在はグラクソ·スミスクライン)から始まった。彼は現在、Fate社の取締役会副議長を務めており、これまでModerna、Mongraph、ATyr、Editas Medicine,Inc.の公開取締役会に勤務していた。メンデリン博士はカリフォルニア大学ロサンゼルス校の生理学と生物物理学博士号、カリフォルニア大学ヘイスティングス法学部の法学博士号、マイアミ大学の生物学学士号を有している。私たちは、メンデリン博士のバイオテクノロジー業界での広範な科学経験と経験が、彼が私たちの取締役会に就く資格があると信じている。
メアリー·T·セラ2019年6月から私たちの取締役会のメンバーを務めてきました。セラさんは現在個人が所有している免疫腫瘍学会社TriSalus生命科学会社(前身はSurefire Medical,Inc.)のCEOと総裁である。2016年1月から2016年11月まで、セイラさんはAegarie製薬会社の最高経営責任者と取締役を務めた。2016年11月、Aegarie製薬会社はQLT社と合併し、Novelion治療会社を設立した。Szelaさんは2017年11月までNovelion Treateutics Inc.のCEOと取締役会のメンバーを務めてきた。Szelaさんは2013年4月から2015年8月まで抗生物質開発会社Melinta Treateutics,Inc.のCEOと取締役会のメンバーを務めた。セラさんは1987年から2012年までアボットの高級管理職を務め、2008年1月から2010年12月まで同社の米国製薬業務の総裁を務めた。Szelaさんは以下の上場会社の取締役会メンバーを務めていた:Kura Oncology,Inc.2018年11月以来、Prometheus Biosciencesは2021年3月から2023年6月まで、Coherus Biosciencesは2014年7月から2021年8月まで、Alimera Sciences Inc.は2018年6月から2021年3月までである。セラさんはイリノイ大学の工商管理修士号と看護学士号を取得しました。私たちはSzelaさんの製薬業界での豊富な指導経験が彼女を私たちの取締役会に任命する資格があると信じている。
リチャード·A·ヤン博士です2017年8月以来、私たちの取締役会に勤めています。1984年以来、彼はホワイトウッド研究所のメンバーであり、マサチューセッツ工科大学の生物学教授であった。楊博士は2011年11月からSyros PharmPharmticals,Inc.の取締役会メンバーを務めてきた。2012年5月、米国国家科学院院士に選出され、2019年10月に米国国家医学科学院院士に選出された。楊博士はエール大学で分子生物物理学と生化学博士号を取得した。私たちはヤン博士が科学的に専門的なので、私たちの取締役会に勤務する資格があると信じている。
ミシェル·C·ウォーナー2023年8月以来、私たちの取締役会のメンバーを務めてきました。彼女は20年以上の生物製薬経験を持ち、商業と研究開発の職責をカバーし、2022年4月以来ずっとバイオテクノロジー会社Alltrnaの最高経営責任者と旗艦パイオニア会社の最高経営責任者兼パートナーを務めている。現在の職務を担当する前に、Wernerさんは2020年7月から2022年4月までノ華腫瘍学で全世界の特許経営主管、固形腫瘍主管を担当し、多種の腫瘍にまたがる疾病領域戦略を提供し、そして業務発展を指導する。ノワールに加入する前に、彼女はアスリーカンの高級指導者であり、2016年から2020年7月までの間の全世界血液学フランチャイズ主管とアメリカ腫瘍学主管を含む多くのポストを務めたことがある。Wernerさんは以前百時美施貴宝で10年間働いて、アメリカとイギリスの販売、マーケティングと市場参入方面で各種の職務を担当したことがあり、ヨーロッパと全世界の販売、マーケティングと市場参入の面ではほとんど腫瘍学方面の仕事にしか従事していない。私たちはWernerさんの製薬業界での豊富な指導経験が彼女を私たちの取締役会に任命する資格があると信じている。
115
第II部
項目5.登録者の市場普通株、関連株主事項、発行者が株式証券を購入する。
市場情報
我々の普通株は2021年7月30日からナスダック全世界精選市場で公開取引され、取引コードはOMGAである。その前に、私たちの普通株は市場を公開しなかった。
所持者
2024年3月21日現在、私たちの普通株は約79人の登録保有者がいます。実株主の数は受益者である株主を含む記録保持者の数よりも大きいが,その株式は仲介人や他の被命名者が街頭名義で保有している.登録されている株主数には、その株式が他の実体が信託形式で保有する可能性のある株主も含まれていない。
配当政策
設立以来、私たちは普通株の任意の現金配当金を発表したり支払ったりしたことがない。私たちは現在、すべての利用可能な資金と任意の将来の収益を維持し、私たちの業務の発展と拡張に資金を提供するつもりで、私たちは予測可能な未来に現金配当金を支払わないと予想しています。将来の配当金の発表と支払いの任意の決定は、私たちの取締役会が適宜決定し、私たちの運営結果、財務状況、契約制限、資本要求、業務見通し、および私たちの取締役会が関連すると考えられる他の要素を含む当時の条件に依存するだろう。
最近売られている未登録証券
2023年12月31日までの年間で、未登録証券は何も販売していません。
証券登録所得収益の使用
2021年8月3日、初公募(IPO)を完了した。初めて公開された株式は、S 1号表(番号333-257794)の登録声明に基づき、証券法に基づいて登録され、2021年7月29日に施行されることが発表された。
私たちの初公募株の純収益は約1億281億ドルで、利息貯蓄口座、短期·中期投資級証券、利下げツール、米国政府債券を含む保本投資に投資されている。当社が初めて公募した所得金の期待用途に関する資料は、2021年8月2日にルール424(B)(4)により米国証券取引委員会に提出された最終入札説明書の“所得使用”節に含まれているが、目論見書に記載されている初公開入札所得純額残高の計画用途は大きな変化はない。
第六項です[保留されている]
116
プロジェクト7.人AGEMENTの財務状況と経営成果の検討と分析。
以下、当社の財務状況と経営結果の検討と分析は、本年度報告10-K表末尾の総合財務諸表と関連付記とともに読まなければならない。本議論および分析に含まれるいくつかの情報または本Form 10−K年次報告における他の情報は、リスクおよび不確実性に関する前向きな陳述を含む、我々の業務計画および戦略に関する情報を含む。本年度報告10-K表の“リスク要因”の一部に列挙された要素を含む多くの要因の影響により、我々の実際の結果は、以下の議論および分析に含まれる前向き陳述に記載されているか、または示唆された結果とは大きく異なる可能性がある。なお,本年度報告のForm 10−Kにおける“前向き陳述に関する特別説明”の部分を参照されたい。
概要
オメガ治療会社は臨床段階の生物技術会社であり、新型プログラム可能なエピゲノムmRNA薬物を開発した。著者らのomegaプラットフォームはエピジェネティクスの力と著者らのゲノム構造に対する深い理解を利用して、正確に定位し、転写前レベルの遺伝子発現を制御し、疾病を治療或いは治療することができる。ヒトゲノムの3次元構造を解読しました遺伝子とそれに伴う制御因子は異なる進化的に保存された構造に組織され,絶縁ゲノムドメイン,あるいはIGDと呼ばれる。IGDは遺伝子制御と細胞分化の基本構造と機能単位であり、自然界の遺伝子発現に対する天然制御システムである。多くの疾患はIGD変化による遺伝子発現異常によるものである。Omegaプラットフォームは、数千個の新しいDNA配列に基づくエピゲノム“郵便番号”を系統的に識別し、検証することができ、これらの“郵便番号”はIGD中の単一の制御要素に関連する。これらのエピゲノム標的をEpiZipsと呼ぶ。著者らはEpiZipsに対して正確なエピゲノム制御を行うために、私たちのメッセンジャーリボ核酸療法を理性的に設計し、設計した。これにより,遺伝子を必要な発現レベルに正確に調節し,発現の持続時間を制御することができる。この方法を通じて、omegaプラットフォームは一連の疾病と疾病の中で広範な潜在的適用性があり、従来薬物治療ができず、治愈しにくく、治療が困難であった目標を含むと信じている。著者らが現在準備しているプロジェクトは腫瘍学、再生医学と多遺伝子疾患を含み、免疫学と心臓新陳代謝疾患を含む。
設立以来、私たちは重大な運営損失を受けた。私たちはどんな製品も商業化していないし、製品販売から何の収入も得られなかった。我々の臨床前開発活動や,我々の候補製品の臨床試験の準備と開始を含め,ほとんどの財政資源を研究·開発に投入した。これまで、私たちの運営資金は主に株式証券の売却収益と私たちの融資と担保合意下の借金から来ています。
2023年12月31日現在、私たちは7340万ドルの現金、現金等価物、有価証券を持っています。2021年8月に初公開(IPO)を完了し、これにより、引受業者が引受権を行使し、1株17.00ドルの公開発行価格で追加株式を購入し、総収益は1.411億ドルとなったため、8,300,976株の普通株を発行·売却した。保証割引と手数料と私たちが支払うべき他の発売費用を差し引いた後、約1.281億ドルの純収益を受け取りました。2023年2月には、普通株の登録直接発売を完了し、これにより、1株5.78ドルの購入価格で6920,415株の普通株を発行·売却し、推定発売費用を差し引いて約3970万ドルの純収益を得た。私たちは2023年8月にJefferies LLC(“Jefferies”)とJefferies LLC(“Jefferies”)を販売代理とする公開市場販売協定(“販売協定”)を締結し、この合意によると、私らは2022年11月8日に米国証券取引委員会のS-3表登録声明(文書番号333-268254)に時々提出することができ、“市場で”またはATM機総生産が6,000,000ドルに達する普通株を発売し、2022年11月18日に発効すると発表した。2023年12月31日までの年間で、吾らは販売契約に基づいて普通株を売却していない。
私たちが製品収入を作る能力は、私たちの1つ以上の候補製品の成功的な開発、規制承認、最終商業化にかかっているだろう。これまで、相当な製品収入を生み出すことができれば、株式発行、債務融資、マーケティング、流通手配、その他の協力、戦略連合と許可手配、または他のソースを通じて私たちの運営に資金を提供する予定です。もしあれば、私たちは割引された条件で追加的な融資源を得られないかもしれない。必要な時に株式や債務融資でより多くの資金を集めることができなければ
117
私たちの製品開発または将来の商業化努力を遅延、制限、減少、または終了することが要求されるか、または私たちが自分で開発およびマーケティングしたい候補製品を開発およびマーケティングする権利が付与されています。
予測可能な未来には,臨床開発による候補製品の推進,臨床前開発の継続,我々の研究開発活動の拡大,新たな候補製品の開発,臨床前研究や臨床試験の完了,規制部門の承認を求め,規制部門の承認を得た場合に製品を商業化することが求められ,重大な追加運営損失が生じ続けることが予想される。私たちの支出も大幅に増加するでしょう
最新の発展動向
2023年12月31日,我々はノとノッドA/S(“ノとノッド”),パイオニア薬業08,Inc.(“PM SpinCo”)およびパイオニア薬業(NN),有限責任会社(“株主”)およびPM(NN)探索会社(“PMCo NN”およびPM SpinCoおよび株主,“PM実体”)と研究協力協定(“Novo RCA”)を締結し,新研究協力協定に掲載されているいくつかの条項について旗艦先鋒薬業(“旗艦”)の関連会社パイオニア薬業(NN),有限責任会社(“株主”)およびPM(NN)探索会社(“PM実体”)と連携協定を締結した。Novo RCAの条項によると、私たちはノとノドに独占的、印税あり、譲渡可能な許可を付与し、PM実体と一致した研究開発計画の下で研究と開発活動を行う権利があり、この計画は製品候補または計画目標に関連し、糖尿病を含む世界各地の人類の心臓代謝性疾患の予防、治療または制御のために使用される。Novo RCAを実行する際には,ノドは1000万ドルの現金を前払いすることに同意し,将来の開発·販売マイルストーン支払いでは最大5.22億ドルと,ライセンス製品の純売上高の中高1桁から2桁低い数百分の印税を支払うことに同意した。このような支払いは私たちと株主の間でほぼ平均的に分配されるだろう。2024年1月、私たちは510万ドルの前払い現金を受け取った。ノとノドのライセンス製品および国/地域に対する印税の支払い義務は、ライセンス製品が国/地域で初めて商業販売され、その国/地域でそのライセンス製品に適用されるいくつかの有効な特許権利要件の最終期間の満了、およびライセンス製品の国家/地域の規制排他性満了後の遅くとも10年以内に満了するが、Novo RCAに規定されているいくつかの許可使用料の低下および段階的脱退条項によって制限される。詳しくは、本年度報告書の最後の総合財務諸表付記内の付記11--協力協定を参照されたい。
118
発展計画
OTX-2002
2022年7月,我々は米国食品医薬品局(FDA)がわれわれの研究新薬(IND)申請を承認し,OTX−2002による肝細胞癌治療の1/2期臨床試験を開始したことを発表し,ヒト初の臨床試験である。
10月には 2022年にMYCHELANGELO I臨床試験における最初の患者を発表した。MYCHELANGELO I期試験は、OTX-2002の安全性、耐性、薬物動態学、薬効学および初歩的な抗腫瘍活性を評価することを目的としている。OTX-2002は単一療法(第1部分)であり、標準看護療法(第2部分)と組み合わせて、再発または難治性肝細胞癌およびMYC癌遺伝子に関連することが知られている他の固形腫瘍タイプを治療するために使用される。この研究は米国,アジア,ヨーロッパの臨床試験地点で患者を募集する予定である。
2022年11月、OTX-2002がFDAによって肝癌治療の孤児薬として承認されたことを発表した。
2023年3月、著者らは羅氏会社と臨床供給協定を締結し、著者らの1/2期MYCHELANGELO I臨床試験の一部として、OTX-2002と羅氏社の抗PD-L 1療法atezolizumabと併用して末期MYC駆動の肝細胞癌患者を評価した。この協定の条項によると,羅氏社はアトゾールズマブを提供し,オメガ社は試験全体実施の一部として合併を評価する。
2023年9月、実施中のMYCHELANGELO I試験の第1の部分の最初の2つの用量レベルキュー(n=8)の予備安全性、耐性、薬物動態、および翻訳データを発表した。両用量レベルの全8名の患者において,高度に特異的な標的接触と標的ゲノム位置での期待エピジェネティクス変化が認められ,OTX−2002服用後,無細胞DNA MYCメチル化シグナルの強い増加が証明された。このようなMYCに対するエピジェネティック調節はすべての8名の患者のMYC発現の迅速、強力かつ持続的な低下に転化し、OTX-2002を服用した7日後、2種類の用量レベルの平均低下幅は約55%であった。この2種類の用量レベルで、OTX-2002の全体的な耐性は良好であり、用量制限毒性がない。これらの初歩的なデータに基づいて、OTX-2002は単一治療用量の増加において引き続き進展を得た。われわれは2024年に単一療法用量上昇の最新の臨床データを報告する予定である。また,2024年に単一療法と併用療法に拡張する予定である。
OTX-2101
2022年10月、我々は、非小細胞肺癌(NSCLC)治療のためのINDイネーブル研究に入るために、第2のEC開発候補としてOTX-2101を選択することを発表した.
他のヨーロッパ共同体プロジェクトは
肝細胞癌と非小細胞肺癌を除いて,われわれは引き続き臨床前研究によりオメガプラットフォーム上の他の内皮細胞を推進している。
マクロ経済状況に関する重大なリスクと不確実性
信用や金融市場を含む世界経済は最近、流動性や信用供給の深刻な減少、金利やインフレ率の上昇、銀行·金融機関に関する危機、消費者自信の低下、経済成長の低下、経済安定の不確実性など、極端な変動と破壊を経験している。不安定な市場と経済状況、そして疫病や国際政治の動揺、戦争、テロによるさらなる破壊は、私たちの業務、財務状況、運営結果に深刻な悪影響を及ぼす可能性がある。
119
私たちの運営結果の構成要素は
収入.収入
これまで、私たちは製品販売から何の収入も得ておらず、予測可能な未来にも製品販売から何の収入も得られないだろう。2023年12月31日現在,我々が確認した収入はPM(CF)Explorations,Inc.またはPMCoとの連携協定により発生しており,PMCoは旗艦会社の付属会社であり,2021年11月に締結され,この協定により,我々が行っている研究活動に関する費用補償を得る権利がある。
運営費
研究開発費
研究·開発費用には、主に研究·開発活動を展開することによるコストが含まれている
私たちは発生した費用に応じて研究と開発費用を支払う。具体的なタスク達成の進捗状況の評価により,研究や開発活動の費用を確認する.これらの活動の支払いは、発生したコストモデルとは異なる個別合意の条項に基づいており、私たちの財務諸表には、前払いまたは計算された研究および開発費用として反映される可能性がある。私たちが将来受け取る研究開発活動のための商品またはサービスのために支払われる払戻不可能な前払いは前払い費用として記録され、関連商品が提供またはサービスを提供する際に支出される。
これらのコストは、複数の計画およびomegaプラットフォームに配置されているので、私たちの発見された仕事、実験室用品、および施設に関連するコスト(減価償却または他の間接コストを含む)を特定の計画に割り当てることはありません。我々は主に内部資源を用いて我々の研究と発見活動を行い,我々の臨床前開発,プロセス開発,製造と臨床開発活動を管理している。これらの従業員たちは複数の計画と私たちの技術プラットフォームで働いているので、私たちは計画通りにこれらのコストを追跡しません。
研究開発費には,一般相談のある関連側費用や,旗艦関連会社との施設コストや分譲収入も計上している。
我々は,現在の発見と研究計画を継続し,新たな研究計画を開始し,候補製品の臨床前開発を継続し,OTX−2002や他の任意の候補製品の臨床試験を行い,我々の研究·開発費用が増加することを予想している。
一般と行政費用
一般及び行政支出は主に行政、財務、法律、人力資源、会社の業務発展及び行政機能者の賃金及びその他の関連コスト、例えばボーナスと福祉を含み、株式給与を含む。一般および行政費用には、直接減価償却コストおよび施設賃貸料および維持の分配費用、ならびに他の運営コストが含まれる法律、特許、会計、情報技術、監査、税務、コンサルティングサービス、保険、および施設に関連する費用の専門的費用も含まれる。
旗艦会社に支払う一般相談費用と、私たちを代表して生成されたソフトウェア許可に関するいくつかの関連費用と一般管理費用も含まれています。また、私たちは旗艦付属会社との施設コストと転貸収入も含む。
120
私たちは、私たちの研究開発と候補製品の潜在的な商業化を支持し続けるにつれて、私たちの一般的かつ管理費用は将来的に増加すると予想しています。また、上場企業に関連する費用は、会計、監査、法律、規制、税務コンプライアンスサービスのコスト、取締役や上級管理者の責任保険コスト、投資家や広報コストなど、引き続き増加すると予想される。
その他の収入,純額
利子収入,純額
利息支出には主に利息支払いと私たちのローンと保証協定に関する債務割引の償却が含まれています。利息収入には私たちの有価証券と通貨市場口座から稼いだ利息が含まれています。
その他の収入,純額
その他の収入(費用),純額には,主に請求書を支払った為替損益と,改訂された融資や担保プロトコルに関する成功手数料義務の公正価値変化に関する再計量損益が含まれる.決済前に、私たちの成功費用義務の公正価値の変動は、各報告期間の再計量に基づいている。
行動の結果
2023年12月31日までと2022年12月31日までの年度比較
次の表は,2023年12月31日まで,2023年12月31日と2022年12月31日までの年間業務結果,およびこれらの項目の変化(千ドルとパーセンテージで示す)をまとめたものである。
|
|
十二月三十一日までの年度 |
|
|
割増価格/ドル |
|
|
|
|
|||||||
|
|
2023 |
|
|
2022 |
|
|
(減少) |
|
|
変更率 |
|
||||
関係者からの協力収入 |
|
$ |
3,094 |
|
|
$ |
2,073 |
|
|
$ |
1,021 |
|
|
|
49 |
% |
運営費用: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
研究開発 |
|
|
77,169 |
|
|
|
81,167 |
|
|
|
(3,998 |
) |
|
|
(5 |
)% |
一般と行政 |
|
|
26,186 |
|
|
|
23,672 |
|
|
|
2,514 |
|
|
|
11 |
% |
総運営費 |
|
|
103,355 |
|
|
|
104,839 |
|
|
|
(1,484 |
) |
|
|
(1 |
)% |
運営損失 |
|
|
(100,261 |
) |
|
|
(102,766 |
) |
|
|
(2,505 |
) |
|
|
(2 |
)% |
その他の収入(費用)、純額: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
利子収入,純額 |
|
|
2,810 |
|
|
|
222 |
|
|
|
2,588 |
|
|
NM |
|
|
その他の収入,純額 |
|
|
23 |
|
|
|
(157 |
) |
|
|
180 |
|
|
NM |
|
|
その他の収入合計,純額 |
|
|
2,833 |
|
|
|
65 |
|
|
|
2,768 |
|
|
NM |
|
|
純損失 |
|
$ |
(97,428 |
) |
|
$ |
(102,701 |
) |
|
$ |
(5,273 |
) |
|
|
|
|
意味がない
収入.収入
2023年12月31日までの年間収入は310万ドル,2022年12月31日までの年間収入は210万ドルであり,2021年11月にPMCOと締結された協力協定に関する研究費の精算が含まれている。
121
研究開発費
2023年12月31日までの1年間で、研究開発費は2022年12月31日現在の8120万ドルから7720万ドルに減少し、400万ドル減少した。400万ドルの減少は、主に外部研究コストが920万ドル減少し、外部製造コストが550万ドル減少し、コンサルティング·専門費用が80万ドル減少し、実験室費用が60万ドル減少したが、人員関連費用は400万ドル増加し、その中には、業務増加を支援する株式報酬、および施設およびその他のコスト690万ドルが含まれており、この部分は減少した費用を相殺した。
一般と行政費用
2023年12月31日までの1年間で、一般·行政費は250万ドル増加し、2022年12月31日現在の2370万ドルから2620万ドルに増加した。250万ドルの成長は主にから専門家と相談費は140万ドル増加し、施設やその他の行政費用は110万ドル増加した。
利子収入
2023年12月31日までの年度の利息収入は280万ドル、2022年12月31日までの年度の利息収入は20万ドル。利子収入純額は260万ドル増加し、2023年12月31日までの1年間、金利上昇により有価証券や通貨市場口座の利息収入が増加したためである。
その他の費用、純額
その他の支出は、2023年12月31日までの年間純収益は10万ドル未満で、2022年12月31日までの年間支出は20万ドル。2023年12月31日までの1年間で,他の純収入の増加は主に固定資産の処分による収益であった。
流動資金と資本資源
流動資金源
設立以来、私たちは重大な運営損失を受けた。私たちは、予見可能な未来に、私たちの持続的な研究活動と私たちの計画とプラットフォームの開発を支持するため、巨額の費用と運営損失が生じると予想しています。私たちはまだ何の製品も商業化していません。もしあれば、数年以内に製品収入が生じないと予想しています。これまで、私たちの運営資金は、主に株式証券の売却収益、私たちの初公募株と登録直接発行、私たちの融資と保証契約下の借金を含む。
2021年8月、私たちは初公募株を完成させ、これにより、引受業者が1株17.00ドルの公開発行価格で追加株式を購入する選択権を行使したため、900,976株を含む8,300,976株の普通株を発行·売却した。総収益は1.411億ドルだった。保証割引と手数料と私たちが支払うべき他の発売費用を差し引いた後、約1.281億ドルの純収益を受け取りました。2023年2月には、普通株の登録直接発売を完了し、これにより、1株5.78ドルの購入価格で6920,415株の普通株を発行·売却し、推定発売費用を差し引いて約3970万ドルの純収益を得た。
2023年8月に、吾らはジェフリを販売代理とする販売契約を締結し、これにより、吾らは時々、我々が2022年11月8日に米国証券取引委員会に提出したS-3表登録声明(文書番号333-268254)に基づいて6,000,000ドルの普通株を発行および販売することができ、2022年11月8日に米国証券取引委員会の“市場で”やATM機に普通株を発売し、2022年11月18日に発効を宣言することができる。販売協定によれば、普通株の販売は、ナスダック全世界精選市場または我々普通株の任意の他の既存取引市場での販売、または市商への販売を含む、証券法第415条(A)条に定義された“市場別発売”の販売において行うことができる。2023年12月31日までの年間で、吾らは販売契約に基づいて普通株を売却していない。
122
キャッシュフロー
次の表は、各期間の現金源と用途(千単位)をまとめています
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
経営活動のための現金純額 |
|
$ |
(91,510 |
) |
|
$ |
(98,515 |
) |
投資活動提供の現金純額 |
|
|
47,515 |
|
|
|
(18,060 |
) |
融資活動が提供する現金純額 |
|
|
41,823 |
|
|
|
708 |
|
現金、現金等価物および限定的現金の純変化 |
|
$ |
(2,172 |
) |
|
$ |
(115,867 |
) |
経営活動
2023年12月31日までの12カ月間、経営活動に用いられた純現金総額は9150万ドルだったが、2022年12月31日までの12カ月間で、経営活動に用いられた純現金は9850万ドルだった。営業現金流出が700万ドル減少したのは、主に2023年12月31日までの1年間に確認された純損失が530万ドル減少したためだ。
投資活動
2023年12月31日までの1年間に、投資活動が提供した純現金総額は4750万ドルだったが、2022年12月31日までの1年間、投資活動で使用された現金純額は1810万ドルだった。投資活動が提供する現金が増加したのは,主に有価証券の満期日収益の増加により2,880万ドルであったのに対し,有価証券購入量は3,760万ドルと減少した。
融資活動
2023年12月31日までの年度、融資活動が提供する現金純額には、主に2023年2月に登録された直接発売による3970万ドル、発行コストと賃貸融資取引所を差し引いた260万ドルが含まれています。2022年12月31日までの1年間に、融資活動が提供する純現金には、株式オプションの行使を含む70万ドルの収益が含まれている。販売-リターンファイナンス取引に関するより多くの情報は、本年度報告の末尾の総合財務諸表付記9-承諾およびまたは事項における保温家具販売-リベートを参照してください。
融資と保証協定
2018年3月9日、太平洋西部銀行(“PWB”)と800万ドルの融資協定を締結し、2019年9月30日(“第1修正案”)、2020年1月22日(“第2修正案”)、2020年12月30日(“第3修正案”)、2021年12月20日(“第4修正案”)をさらに改正する。
2023年9月22日に、吾らは別の融資合意修正案(“第5修正案”)を締結し、この修正案によると、普華永道は融資期限を2027年9月30日まで延長し、ただ吾は2024年12月31日またはそれ以前にその株式証券を売却して得られた少なくとも5,000万ドルの現金収益および/または返却できない前期戦略パートナーシップ収益を受け取った後、融資期限日を2028年9月30日までさらに延長することができる。このローンは2023年9月30日から返済され、毎月元金30万ドルと利息が支払われ、満期日が2028年9月30日まで延長されなければ、2027年9月30日に400万ドルの決済も支払われる。利息は引き続き変動年率によって決定され、(I)当時の最優遇金利より0.50%と(Ii)5.50%の両者の中で大きいものを基準とする。我々は15,000ドルの債務発行コストを発生させ、このコストは追加定期融資の直接減少と記録され、実金利法を用いて関連定期融資の期限内に利息支出の構成要素として償却を行う。第5改正案の条項によると、私たちは第5改正案に基づいて10万ドルの成功費を支払わなければならず、また、第4改正案によると、20万ドルの成功費義務を支払わなければならない。成功費用は特定の流動性イベントが達成されたかどうかにかかっている。私たちは、成功費用債務は独立した金融商品であり、私たちの総合貸借対照表では負債に分類され、最初に記録されていることを確認した
123
公正価値は、報告期間ごとの公正価値変動は他の費用、総合経営報告書の純額と全面赤字で確認された。このような債務の公正な価値は、債務が返済されるまで、各報告期間の終了時に再計量される。
また、第5修正案によると、PWBとはいつでも少なくとも500万ドルの無制限現金残高を維持することに同意しているが、合計少なくとも500万ドルの未返済融資または融資元金残高が1000万ドルを下回った場合には終了する。
改正された融資協定下の借金は、私たちのほとんどの個人財産(私たちの知的財産を除く)を担保にしています。修正された“ローン協定”には何の金融契約もない;しかし、私たちはいくつかの肯定的で否定的な契約を守らなければならず、私たちは期限が切れるまでこれらの契約を守らなければならない.
2023年12月31日現在、私たちは7340万ドルの現金、現金等価物、有価証券を持っています。われわれが行っている活動に関する費用は大幅に増加することが予想され,特にわれわれが臨床前活動を進め,OTX−2002や他の開発中の候補製品の臨床試験を行った場合である。また、上場企業として、運営に関連した追加コストを発生させていきます。私たちの経営と資本支出の時間と金額は大きく依存するだろう
私たちの既存の現金、現金等価物、有価証券は、2025年第1四半期の運営費用と資本支出需要に資金を提供できると信じている。しかし,我々は不正確であることが証明される可能性があるという仮定に基づいてこの推定を行い,利用可能な資本資源を期待よりも早く利用することができる.成立以来,経常赤字が存在しており,予想される将来,運営損失を継続し,運営に現金を使用している。私たちは、株式発行、債務融資、マーケティングと流通手配、その他の協力、戦略連合と許可手配、または他のソースの組み合わせによって、私たちの将来の現金需要に資金を提供する予定です。資本市場の変動と米国の全体的な経済状況は必要な資金調達の重大な障害となる可能性がある
124
したがって、私たちは受け入れ可能な条件で必要な資金を得ることができないかもしれない。これは私たちが経営を続ける企業として継続する能力に大きな疑いを抱かせた。
契約義務
著者らは正常な業務過程においてCRO、CDMOとその他の第三者と臨床前研究、テストと製造サービスについて契約を締結した。このような契約は一般的に最低購入約束を含まず、一般的に私たちは書面で通知された後にキャンセルすることができる。キャンセル時に支払われるべき金額には、キャンセル日までに提供されるサービスの支払いまたは発生した費用が含まれており、当サービスプロバイダーのキャンセル不可能な義務を含み、CROおよびCDMOと何らかの手配を達成した場合には、キャンセル不可能な費用が含まれている可能性がある。契約がキャンセルされるまで、支払いをキャンセルする金額と時間がわかりません。
私たちはまた旗艦革新V,Inc.,ワイトヘード生物医学研究所、Acuitas、Nitto Denko Corporationと許可協定を締結し、これらの協定に基づいて、潜在的なマイルストーン支払い、特許権使用料、あるいは両方を支払う義務がある。このような支払いは、合意許可を使用した知的財産権の製品の開発に依存し、将来のイベントの発生に依存し、したがって、このような潜在的義務の時間および可能性は正確には知ることができない。
先に述べたように、私たちは改訂された融資協定に基づいて元金総額2,000万ドルを借り入れました。改正された融資協定条項によると、2023年9月30日から毎月30万ドルの元金と利息を返済する義務があり、満期日を2028年9月30日に延長しなければ、2027年9月30日に400万ドルの決済金を支払う義務がある。元本残高は2023年12月31日現在1,900万ドル。
私たちは2017年に締結されたキャンセル不可賃貸契約に基づいて締結されたオフィスと実験室を有しており、2024年9月に満了します。残りのレンタル期間中、私たちの毎月のレンタル料は約10万ドルです。2020年9月、この空間は他の2つの政党に完全に転貸され、この2つの政党は旗艦ブランドの付属会社である。そのうちの1つの転貸協定は2022年5月に終了し、もう1つの転貸協定は2024年9月に期限が切れる。
2021年11月、私たちはAre-MA Region No 94,LLCと賃貸契約を締結し、マサチューセッツ州ケンブリッジ市第一街140番地にあるオフィスと実験室空間をレンタルし、総面積は約89,246平方フィート、郵便番号:02142。レンタル期間は2023年5月(第1期)及び2023年8月(第2期)に開始され、レンタル期間はリース開始後15年で満了するが、いくつかの継続権利規約の制限を受けなければならない。賃貸ビル面の基本賃貸料は1平方フィート116.00元で、初めて賃貸料を支払って1周年から、毎年当時の賃貸料の3%を上方調整することができ、私たちがあるテナントを使用して手当を改善する状況に応じて、他の可能な調整を行うことができる。2023年7月、私たちは旗艦会社の付属会社と共有空間の手配を達成し、マサチューセッツ州ケンブリッジ市第一街140号にあるオフィスと実験室空間を転用して、私たちの成長計画を補充します。
重要な会計政策と試算
私たちの経営陣の財務状況と経営結果の討論と分析は私たちの合併財務諸表に基づいています。これらの報告書はアメリカで公認されている会計原則やGAAPに基づいて作成されています。これらの連結財務諸表を作成する際には、資産、負債、コスト、費用報告金額に影響を与える見積もりと判断を行う必要があります。継続的に基づいて、以下に説明するものを含む、これらの推定および判断を評価する。私たちの見積もりは歴史的経験とこのような状況で合理的だと思う様々な他の仮定に基づいています。これらの見積りと仮定は資産や負債の帳簿価値を判断する基礎を構成しており,これらの資産や負債の帳簿価値は他のソースからは明らかに見えない.実際の結果と経験はこれらの推定とは大きく異なるかもしれない。
我々の主な会計政策は、本年度報告書末の付記2である総合財務諸表付記における重要会計政策要約により包括的に記述されているが、以下の会計政策は、我々が総合財務諸表を作成するために使用される判断と推定に最も重要であると考えられる。
研究と開発費用を計算すべきである
125
連結財務諸表作成過程の一部として、我々が計上すべき研究·開発費用を見積もる必要がある。このプロセスは、未締結契約および調達注文を審査し、私たちの適用者とコミュニケーションして、私たちを代表して実行されるサービスを決定し、インボイスを受け取っていない場合、または他の方法で実際のコストを通知している場合に、実行されたサービスレベルおよびサービスによって生じる関連コストを推定することを含む。私たちのほとんどのサービスプロバイダは予定のスケジュールに従って、あるいは契約マイルストーンに達した時に私たちに借金の領収書を発行しますが、前金が必要なものもあります。私たちは、当時私たちが知っている事実と状況に基づいて、連結財務諸表において、各貸借対照表の日付までの課税費用を推定します。我々は定期的にサービスプロバイダとこれらの推定の正確性を確認し,必要に応じて調整する.検討·開発費用を算定すべき例としては,サプライヤーへの臨床前·臨床開発活動に関する費用,臨床開発·研究活動に関するCROおよび研究材料生産に関するCDMOが予想される。
我々は、第三者サービスプロバイダ(私たちに代わって臨床前および臨床研究を提供、実施および管理するCROおよびCDMOを含む)とのオファーおよび契約に基づいて、受信したサービスおよび費用に対する私たちの努力の推定に基づいて、研究開発費用を計算すべきであると推定する。これらの合意の財務条項は交渉が必要であり、契約によって異なり、支払いの不均衡を招く可能性がある。場合によっては、私たちのサプライヤーに支払われるお金は、提供されたサービスレベルを超え、料金の前払いをもたらす可能性があり、この場合、達成されると予想される時間に基づいて、料金の現在または長期分類評価が行われる。サービス料を計算する際に、サービスを提供する時間帯と各時間の努力度を見積もります。サービスの実行時間や努力の程度が推定値と異なる場合には,それに応じて計算すべき費用や前払い費用の金額を調整する.実際に発生した金額と実質的に異なることはないと予想されるが,提供されたサービスの実態や時間に対する提供サービスの状態や時間の理解が異なる可能性があり,任意の特定の時期に確認された増加または減少金額の推定値が変化する可能性がある.これまで,我々が計算した研究や開発費の先行推定には何の大きな調整もなかった.
最近発表された会計公告
吾らは、最近発表されたすべての会計声明を検討し、本年報末尾の総合財務諸表付記に開示されている付記2--主要会計政策要約が開示されていることを除いて、当該等の基準は、吾等の財務諸表に重大な影響を与えないか、又は吾等の現在の業務に適用されないことを決定した。
新興成長型企業と小さな報告会社の地位
私たちは“2012年創業始動法案”(JumpStart Our Business Startups Act Of 2012)や“雇用法案”(JOBS Act)で定義されている“新興成長型会社”の資格を満たしています。したがって、私たちは特定の削減開示と他の上場企業に適用される他の報告要件を利用することができる。特に、“雇用法案”は、新興成長型会社は、延長された過渡期を利用して新たなまたは改正された会計基準を遵守することができると規定している。私たちは、“脱退”延長を選択しない移行期間を選択したが、これは、基準が発表または改正された場合、上場企業または民間企業に対して異なる適用日があれば、民間会社が新たな基準または改正基準を採用する際に新たな基準または改訂基準を採用することができ、我々(I)が“選択脱退”延長の移行期間を撤回できなくなるまで、または(Ii)新興成長型企業の資格を持たなくなるまでそうすることができることを意味する。
証券法や取引法の定義によると、私たちも“小さな報告会社”です。(I)非関連会社が保有する我々の普通株の時価が2.5億ドル以下である限り、または(Ii)最近終了した会計年度の年収が1億ドル未満であり、非関連会社が保有する我が普通株の時価が7億ドル未満であれば、規模の小さい報告会社になり続けることができる。もし私たちが小さな報告会社であれば、私たちがもう新興成長型会社ではない場合、私たちはより小さな報告会社が得ることができるいくつかの開示要求の免除に依存し続けるかもしれない。具体的には、規模の小さい報告会社として、私たちのForm 10-K年報で最近の2つの財政年度の監査財務諸表のみを公表し、役員報酬に関する開示義務を削減することを選択するかもしれません
126
より小さい申告会社は、上記(Ii)項の規定により、当社の独立公認会計士事務所が発行する財務報告の内部統制に関する認証報告を取得する必要はない。
127
第七A項。量と質市場リスクの開示について。
我々は取引法ルール12 b-2で定義されている小さな報告会社であり,本プロジェクト7 Aに要求される情報を提供する必要はない.
プロジェクト8.財務諸表データを補完しています
項目8に要求される財務資料は本報告F−1ページから始まる。
128
項目9.との変更会計と財務開示問題で会計士の意見とは食い違っている。
ない。
第9条。コントロールとプログラムです。
制御とプログラムの有効性の制限
我々の開示制御およびプログラムを設計·評価する際に、管理層は、任意の制御およびプログラムは、設計および動作がどんなに良好であっても、予想される制御目標を達成するために合理的な保証を提供することしかできないことを認識している。また、開示制御およびプログラムの設計は、管理層に、そのコストに対する可能な制御およびプログラムの利点を評価する際に判断することが要求されるリソース制限が存在することを反映しなければならない。
開示制御とプログラムの評価
我々の経営陣は、2023年12月31日現在、最高経営責任者及び最高財務責任者の参加の下、我々の開示制御及び手順(“取引法”第13 a−15(E)及び15 d−15(E)条に定義されている)の有効性を評価している。評価によると、我々の最高経営責任者および最高財務責任者は、2023年12月31日まで、その日までの開示制御および手続きが合理的な保証レベルで有効であると結論した。
財務報告の内部統制に関する経営陣の年次報告
我々の経営陣は、“取引法”の下のルール13 a-15(F)で定義されている財務報告の十分な内部統制の確立と維持を担当している。我々の経営陣は、トレデビル委員会後援組織委員会が発表した“内部統制-総合枠組み(2013)”に規定されている基準に基づいて、財務報告書の内部統制の有効性を評価した。この評価に基づき、我々の経営陣は、2023年12月31日現在、財務報告の内部統制に有効であると結論した。
“米国証券取引委員会”規則は“新興成長型会社”に移行期間を設定しているため、今年度の10−K表には独立公認会計士事務所の認証報告は含まれていない。
財務報告の内部統制の変化
2023年12月31日までの四半期内に、財務報告の内部統制(“外国為替法案”規則13 a-15(F)および15 d-15(F)の定義に基づく)に大きな影響を与えなかったか、または合理的に財務報告の内部統制に大きな影響を与える可能性のある変化はなかった。
プロジェクト9 B。その他の情報.
(A)十分な資源を確保してリード計画を進め、そのプラットフォームを最大限に利用して短期的かつ長期的な価値機会を創出するために、同社は2024年3月に戦略優先順位を発表した。この計画の一部として、同社は組織を簡素化し、研究開発作業とコスト構造を最適化し、その現金滑走路を2025年第1四半期まで延長した。
(B)。最近、同社はある投資家と公募株の可能性について秘密裏に議論している。現在、現在の市場状況を踏まえて、当社はこのような融資を継続しないことを決定しています。同社は現金需要や事業見通しを評価し続け、市場状況が有利だと判断すれば、将来的に追加融資を求める可能性がある。
(C).2023年第4四半期に、役員または取引法第16 a-1(F)条に定義された役人
プロジェクト9 Cです。Discloこれは検査を阻止する外国司法管轄区について肯定的だ。
129
適用されません。
130
第三部
プロジェクト10.役員、執行幹事会社を管理しています
ビジネス行為と道徳的基準
私たちは、私たちの最高経営責任者、最高財務官、最高会計官、または財務総監、または同様の機能を実行する人を含む、私たちのすべての役員、高級管理者、および従業員に適用される書面による商業行為および道徳的規則を採択した。規則の最新バージョンは、当社のサイトir.omegaTreateutics.comの投資家部分で入手できます。私たちは私たちのウェブサイトでアメリカ証券取引委員会またはナスダック規則に基づいて開示する必要がある私たちのコードの任意の修正または免除を開示するつもりです. 当社のサイトアドレスへの引用は、当社のサイトに掲載されていることや本サイトを通じて提供される情報への引用ではありません。本Form 10-K年度報告の一部と見なすべきではありません。
行政員および役員
要求された私たちの執行者と役員に関する情報 本年度報告表格10−K第I部分の末尾には,本項目10は“当社行政者及び取締役に関する資料”の見出しに掲載されている。
第10項の要件の残りの情報は、2024年年次総会に関する我々が米国証券取引委員会または米国証券取引委員会に提出した最終委託書に含まれ、“会社統治”、“延滞第16条(A)報告書”(適用される場合)、および“取締役会委員会”と題され、参照によって本明細書に組み込まれる。
プロジェクト11.実行補償します。
第11条に要求される情報は、我々が米国証券取引委員会に提出した2024年株主総会に関する最終委託書に含まれ、“役員及び役員報酬”及び“報酬委員会連動及び内部者参加”(適用される場合)と題され、引用により本明細書に組み込まれる。
131
プロジェクト12.所有権を保証するいくつかの実益所有者と経営陣と関連株主の件。
株式補償計画に基づいて発行された証券(2023年12月31日現在)
計画種別 |
|
未償還オプション,株式承認証及び権利を行使する際に発行される証券数 |
|
|
未償還オプション、権証および権利の加重平均行権価格 |
|
|
持分補償計画の下で将来発行可能な証券の数(第1欄の証券を除く) |
|
|||
証券保有者が承認した持分補償計画 (1) |
|
|
8,780,737 |
(2) |
|
$ |
5.69 |
(3) |
|
|
4,019,697 |
(4) |
証券保有者の許可を得ていない持分補償計画 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
合計する |
|
|
8,780,737 |
|
|
|
|
|
|
4,019,697 |
|
|
この第12項の要件の残りの情報は、私たちが米国証券取引委員会に提出した2024年株主年次総会に関する最終委託書に含まれ、“いくつかの利益所有者および管理層の保証所有権”と題され、参照によって本明細書に組み込まれる。
プロジェクト13.特定の関係関連取引、そして役員の独立性。
第13条に要求される情報は、2024年年次総会に関する我々が米国証券取引委員会に提出した最終委託書に含まれ、“会社のガバナンス”および“特定の関係および関連者取引”と題され、参照によって本明細書に組み込まれる。
132
第14項.主体会計料金とサービス料です。
第14項の要件の情報は、2024年の株主総会に関する我々が米国証券取引委員会に提出した最終委託書に含まれ、“独立公認会計士事務所費用及びその他の事項”と題され、引用により本明細書に組み込まれる。
パー?パーT IV
133
項目15.物証、財務諸表付表
(A)(1)本報告に記載されている財務諸表一覧表は、本年度報告F-1ページ表10-Kの総合財務諸表インデックスを参照して本項目に組み込まれる。
(A)(2)財務諸表が必要でないか、適用されないか、または資料が連結財務諸表またはその付記に含まれているため、財務諸表添付表は省略されている。
(A)(3)展示品:
|
|
|
引用で編入する |
提出日 |
同封のアーカイブ/提供 |
||
展示品 番号をつける |
|
説明する |
表 |
書類番号. |
展示品 |
||
3.1 |
|
再記載した会社登録証明書。 |
8-K |
001-40657 |
3.1 |
08/03/2021 |
|
|
|
|
|
|
|
|
|
3.2 |
|
添付例を改訂及び再編成する。 |
8-K |
001-40657 |
3.1 |
12/11/2023 |
|
|
|
|
|
|
|
|
|
4.1 |
|
普通株式証明書サンプル。 |
S-1/A |
333-257794 |
4.2 |
07/26/2021 |
|
|
|
|
|
|
|
|
|
4.2 |
|
投資家権利協定の改正と再署名は2021年3月4日。 |
S-1/A |
333-257794 |
4.1 |
07/26/2021 |
|
|
|
|
|
|
|
|
|
4.3 |
|
2019年9月30日にPacWest Bankcorpに発行された株式引受権証を改訂·再発行し、Aシリーズ優先株を購入した。 |
S-1/A |
333-257794 |
4.3 |
07/26/2021 |
|
|
|
|
|
|
|
|
|
4.4 |
|
株本で説明する。 |
10-K |
001-40657 |
4.4 |
03/10/2022 |
|
|
|
|
|
|
|
|
|
10.1# |
|
オメガ治療会社とその役員と上級管理者との間の賠償協定フォーマット。 |
S-1/A |
333-257794 |
10.8 |
7/26/2021 |
|
|
|
|
|
|
|
|
|
10.2# |
|
2021年奨励計画とその下の合意形態。 |
S-1/A |
333-257794 |
10.2 |
7/26/2021 |
|
|
|
|
|
|
|
|
|
10.3# |
|
2021年従業員株購入計画。 |
S-1/A |
333-257794 |
10.3 |
7/26/2021 |
|
|
|
|
|
|
|
|
|
10.4# |
|
非従業員役員報酬計画。 |
S-1/A |
333-257794 |
10.4 |
7/26/2021 |
|
|
|
|
|
|
|
|
|
10.5# |
|
Mahesh Karandeと登録者の間の雇用協定は、2021年7月25日である。 |
S-1/A |
333-257794 |
10.17 |
7/26/2021 |
|
|
|
|
|
|
|
|
|
10.6# |
|
トーマス·マッコーリーと登録者の間の雇用協定は、2021年7月24日となっている。 |
S-1/A |
333-257794 |
10.18 |
7/26/2021 |
|
|
|
|
|
|
|
|
|
10.7# |
|
厳モルと登録者の間の雇用協定は,期日は2021年12月12日である。 |
10-K |
001-40657 |
10.8 |
3/10/2022 |
|
|
|
|
|
|
|
|
|
10.8# |
|
凌増と登録者との間の雇用協定は、期日は2022年3月18日。 |
10-Q |
001-40657 |
10.3 |
05/04/2022 |
|
|
|
|
|
|
|
|
|
10.9# |
|
ジョシュア·リードと登録者の間の雇用協定は、2022年4月28日となっている。 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
10.10# |
|
リチャード·A·ヤンと登録者との間の諮問協定は、2016年11月7日。 |
10-K |
001-40657 |
10.9 |
03/10/2022 |
|
|
|
|
|
|
|
|
|
10.11# |
|
リチャード·A·ヤンと登録者との間の諮問協定改正案は,期日は2021年10月29日である。 |
10-K |
001-40657 |
10.1 |
03/10/2022 |
|
|
|
|
|
|
|
|
|
134
10.12# |
|
リチャード·A·ヤンと登録者との間の諮問協定改正案は,期日は2022年10月5日である。 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
10.13# |
|
リチャード·A·ヤンと登録者との間の諮問協定改正案は,期日は2023年10月6日である。 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
10.14 |
|
太平洋西部銀行(n/k/a PacWest Bancorp)と登録者との間の融資·担保協定は、2018年3月9日、2019年9月30日、2020年1月22日、2020年12月30日に改訂された。 |
S-1/A |
333-257794 |
10.1 |
07/26/2021 |
|
|
|
|
|
|
|
|
|
10.15 |
|
“融資·担保協定第4改正案”は、2021年12月20日としている。 |
8-K |
001-40657 |
10.1 |
12/21/2021 |
|
|
|
|
|
|
|
|
|
10.16 |
|
融資·担保協定第5改正案は、2023年9月22日となっている。 |
8-K |
001-40657 |
10.1 |
09/22/2023 |
|
|
|
|
|
|
|
|
|
10.17 |
|
フラッグシップパイオニア革新V,Inc.と登録者との間の許可協定は、2019年3月12日である。 |
S-1 |
333-257794 |
10.1 |
07/09/2021 |
|
|
|
|
|
|
|
|
|
10.18 |
|
フラッグシップパイオニア革新V,Inc.登録者が2023年12月31日に締結した2019年3月12日のライセンス契約に関する書面協定 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
10.19 |
|
ワイトヘード生物医学研究所と登録者との間の独占許可協定は,2019年5月22日である。 |
S-1 |
333-257794 |
10.1 |
07/09/2021 |
|
|
|
|
|
|
|
|
|
10.20 |
|
2019年5月22日,ワイトヘード生物医学研究所と登録者との間で研究協力に関する放棄,確認,合意があり,発効日は2019年5月22日である。 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
10.21 |
|
ワイトヘード生物医学研究所と登録者との共同独占許可協定は,2019年5月22日である。 |
S-1 |
333-257794 |
10.1 |
07/09/2021 |
|
|
|
|
|
|
|
|
|
10.22 |
|
2019年5月22日,ワイトヘード生物医学研究所と登録者との間で研究協力に関する放棄,確認,合意があり,発効日は2019年5月22日である。 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
10.23 |
|
Acuitas Treateutics,Inc.と登録者との間の開発とオプション合意は,期日は2020年10月5日であり,改訂された。 |
S-1 |
333-257794 |
10.2 |
07/09/2021 |
|
|
|
|
|
|
|
|
|
10.24 |
|
Acuitas治療会社と登録者との間の非独占的許可協定は,2021年3月22日である。 |
S-1 |
333-257794 |
10.2 |
07/09/2021 |
|
|
|
|
|
|
|
|
|
10.25 |
|
日東電工会社と登録者との協力と許可協定は,期日は2022年10月12日である。 |
10-K |
001-40657 |
10.20 |
03/01/2023 |
|
|
|
|
|
|
|
|
|
10.26 |
|
BMR-325 Vassar Street LLCと登録者との間の賃貸契約は、2017年11月30日となっています。 |
S-1/A |
333-257794 |
10.1 |
07/26/2021 |
|
|
|
|
|
|
|
|
|
135
10.27 |
|
オメガ治療会社とARE-MA地域第94号、有限責任会社との間の賃貸契約。 |
10-K |
001-40657 |
10.1 |
03/10/2022 |
|
|
|
|
|
|
|
|
|
10.28 |
|
オメガ治療会社とARE-MA地域有限責任会社の間でレンタルされる第1修正案は、2023年5月3日 |
10-Q |
001-40657 |
10.1 |
08/03/2023 |
|
|
|
|
|
|
|
|
|
10.29 |
|
オメガ治療会社とセンダ生物科学社は空間協定改正案を共有し,期日は2022年1月31日である。 |
10-K |
001-40657 |
10.1 |
03/10/2022 |
|
|
|
|
|
|
|
|
|
10.30 |
|
共有空間協定は,期日は2023年7月12日であり,オメガ治療会社とメタファーバイオテクノロジー社が署名した。 |
8-K |
001-40657 |
10.1 |
07/13/2023 |
|
|
|
|
|
|
|
|
|
10.31 |
|
共有空間協定は,2023年7月11日にオメガ治療会社とアビオリー生物社によって署名された。 |
8-K |
001-40657 |
10.2 |
07/13/2023 |
|
|
|
|
|
|
|
|
|
10.32 |
|
共有空間協定は、日付は2023年7月12日で、オメガ治療会社とフラッグシップラボ89社によって署名された。 |
8-K |
001-40657 |
10.3 |
07/13/2023 |
|
|
|
|
|
|
|
|
|
10.33 |
|
ノボノルドA/S、パイオニア医薬08社、オメガ治療会社とその他の各方面との研究協力協定は、2023年12月31日である |
|
|
|
|
* |
|
|
|
|
|
|
|
|
10.34 |
|
オメガ治療会社とジェフリー有限責任会社が2023年8月3日に署名した公開市場販売協定 |
8-K |
001-40657 |
10.1 |
08/03/2023 |
|
|
|
|
|
|
|
|
|
10.35 |
|
オメガ治療会社とその中で指定された購入者との間の証券購入契約、日付は2023年2月22日 |
8-K |
001-40657 |
10.1 |
02/02/2023 |
|
|
|
|
|
|
|
|
|
21.1 |
|
登録者の子会社。 |
10-K |
001-40657 |
21.1 |
03/10/2022 |
|
|
|
|
|
|
|
|
|
23.1 |
|
独立公認会計士事務所徳勤会計士事務所が同意します。 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
31.1 |
|
ルール13 a−14(A)/15 d−14(A)に従って首席実行幹事証明書が発行される。 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
31.2 |
|
財務チーフ幹事は、細則13 a~14(A)/15 d−14(A)に従って認証される。 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
32.1 |
|
“米国法典”第18編第1350条に基づく最高経営責任者の証明。 |
|
|
|
|
** |
|
|
|
|
|
|
|
|
32.2 |
|
“米国法典”第18編第1350条に基づく首席財務官の証明。 |
|
|
|
|
** |
|
|
|
|
|
|
|
|
97 |
|
誤って判決された賠償に関する政策 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
101.INS |
|
連結されたXBRLインスタンス文書−インスタンス文書は、XBRLタグがイントラネットXBRL文書に埋め込まれているので、相互作用データファイルには表示されない。 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
101.書院 |
|
イントラネットXBRL分類拡張アーキテクチャ文書 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
101.カール |
|
インラインXBRL分類拡張計算リンクライブラリ文書 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
136
101.def |
|
インラインXBRL分類拡張Linkbase文書を定義する |
|
|
|
|
* |
|
|
|
|
|
|
|
|
101.介護会 |
|
XBRL分類拡張ラベルLinkbase文書を連結する |
|
|
|
|
* |
|
|
|
|
|
|
|
|
101.Pre |
|
インラインXBRL分類拡張プレゼンテーションLinkbaseドキュメント |
|
|
|
|
* |
|
|
|
|
|
|
|
|
104 |
|
表紙インタラクションデータファイル(イントラネットXBRL文書に埋め込む) |
|
|
|
|
* |
*アーカイブをお送りします。
**関数で提供されます。
#は、管理契約または補償計画を示します。
S法規第601(B)(10)(Iv)条の規定により、本展示品の一部の内容(星番号で示す)が編集されている。
項目16.表格10-Kの概要
ない。
137
サイン
改正された1934年証券取引法第13又は15(D)節の要求に基づいて、登録者は、正式に許可された次の署名者がその代表を代表して本報告書に署名することを正式に手配した.
|
|
オメガ治療会社は |
|
|
|
|
|
日付:2024年3月28日 |
|
差出人: |
/S/Mahesh Karande |
|
|
|
マッハシュ·カランダー |
|
|
|
社長と最高経営責任者 |
本報告書は、改正された1934年の証券取引法の要求に基づき、以下の登録者によって登録者として指定日に署名された。
名前.名前 |
|
タイトル |
|
日取り |
|
|
|
|
|
/S/Mahesh Karande |
|
取締役CEO社長(最高経営責任者) |
|
2024年3月28日 |
マッハシュ·カランダー |
|
|
|
|
|
|
|
|
|
/S/ジョシュア·リード |
|
首席財務官(首席財務官と首席会計官) |
|
2024年3月28日 |
ジョシュア·リード |
|
|
|
|
|
|
|
|
|
/S/クリスティアン·S·シャド |
|
取締役会議長 |
|
2024年3月28日 |
クリスティアン·S·シャド |
|
|
|
|
|
|
|
|
|
/S/Rainer Boehm |
|
役員.取締役 |
|
2024年3月28日 |
レナ·ボム |
|
|
|
|
|
|
|
|
|
/投稿S/ルーク·M·ベシャール |
|
役員.取締役 |
|
2024年3月28日 |
ルーク·M·ベシャール |
|
|
|
|
|
|
|
|
|
/S/エリオット·M·リヴィ |
|
役員.取締役 |
|
2024年3月28日 |
エリオット·M·リヴィ医学博士 |
|
|
|
|
|
|
|
|
|
寄稿S/ジョン·メンデラン |
|
役員.取締役 |
|
2024年3月28日 |
ジョン·メンデリン博士J.D. |
|
|
|
|
|
|
|
|
|
/S/メアリー·T·セラ |
|
役員.取締役 |
|
2024年3月28日 |
メアリー·T·セラ |
|
|
|
|
|
|
|
|
|
/投稿S/ミシェル·C·ヴォーナ |
|
役員.取締役 |
|
2024年3月28日 |
ミシェル·C·ウォーナー |
|
|
|
|
|
|
|
|
|
/S/リチャード·A·ヤン |
|
役員.取締役 |
|
2024年3月28日 |
リチャード·A·ヤン博士です |
|
|
|
|
138
連結財務諸表索引
独立公認会計士事務所報告 (PCAOB ID番号 |
F-2 |
合併貸借対照表 |
F-3 |
合併経営報告書と全面赤字 |
F-4 |
合併株主権益報告書 |
F-5 |
統合現金フロー表 |
F-6 |
連結財務諸表付記 |
F-7 |
F-1
独立登録者の報告公認会計士事務所
オメガ治療会社の株主と取締役会に。
財務諸表のいくつかの見方
オメガ治療会社とその子会社(“当社”)の2023年12月31日と2022年12月31日までの連結貸借対照表、2023年12月31日までの2年度の関連総合経営報告書と全面赤字、株主権益と現金流量および関連付記(総称して“財務諸表”と呼ぶ)を監査した。これらの財務諸表は,すべての重要な点で当社の2023年12月31日と2022年12月31日までの財務状況,および2023年12月31日までの2年度の経営結果とキャッシュフローを公平に反映しており,米国公認の会計原則に適合していると考えられる。
経営を続ける企業
添付財務諸表の作成は、同社が引き続き経営を継続する企業であると仮定している。財務諸表付記1で述べたように、当社の設立以来の経営による経常赤字、予想される将来の持続的な経営損失、および追加資本を調達して将来の経営に資金を提供する必要があり、持続経営企業としての持続的な経営能力に大きな疑いを抱かせる。付記1は、これらの事項における経営陣の計画も説明しています。財務諸表には、このような不確実性の結果に起因する可能性のある調整は含まれていません。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても詐欺によるものであっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査することを求められておらず、私たちも招聘されて監査を行っていません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性について意見を述べるためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
/s/
3月28, 2024
2020年以来、当社の監査役を務めてきました。
F-2
オメガ治療会社は
強固な基礎スプレー銃のシーツ
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
|
|
2023 |
|
|
2022 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
有価証券 |
|
|
|
|
|
|
||
関係者の売掛金 |
|
|
|
|
|
|
||
売掛金 |
|
|
|
|
|
— |
|
|
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
流動資産総額 |
|
|
|
|
|
|
||
財産と設備、純額 |
|
|
|
|
|
|
||
経営的リース使用権資産純額 |
|
|
|
|
|
|
||
制限現金 |
|
|
|
|
|
|
||
その他の資産 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
負債と株主権益 |
|
|
|
|
|
|||
流動負債: |
|
|
|
|
|
|
||
売掛金 |
|
$ |
|
|
$ |
|
||
費用を計算する |
|
|
|
|
|
|
||
その他流動負債 |
|
|
|
|
|
|
||
賃貸負債、流動 |
|
|
|
|
|
|
||
長期債務、流動部分 |
|
|
|
|
|
|
||
流動負債総額 |
|
|
|
|
|
|
||
非流動賃貸負債 |
|
|
|
|
|
|
||
長期債務、純額 |
|
|
|
|
|
|
||
その他負債 |
|
|
|
|
|
|
||
総負債 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
株主権益: |
|
|
|
|
|
|
||
優先株、$ |
|
|
|
|
|
|
||
普通株、$ |
|
|
|
|
|
|
||
追加実収資本 |
|
|
|
|
|
|
||
その他の総合損失を累計する |
|
|
( |
) |
|
|
( |
) |
赤字を累計する |
|
|
( |
) |
|
|
( |
) |
株主権益総額 |
|
|
|
|
|
|
||
総負債と株主権益 |
|
$ |
|
|
$ |
|
||
付記はこれらの連結財務諸表の構成要素である。
F-3
オメガ治療会社は
合併運営報告書イオンと総合損失
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
十二月三十一日までの年度 |
|
|||||
|
2023 |
|
|
2022 |
|
||
関係者からの協力収入 |
$ |
|
|
$ |
|
||
運営費用: |
|
|
|
|
|
||
研究開発 |
|
|
|
|
|
||
一般と行政 |
|
|
|
|
|
||
総運営費 |
|
|
|
|
|
||
運営損失 |
|
( |
) |
|
|
( |
) |
その他の収入(費用)、純額: |
|
|
|
|
|
||
利子収入,純額 |
|
|
|
|
|
||
その他の収入,純額 |
|
|
|
|
( |
) |
|
その他の収入合計,純額 |
|
|
|
|
|
||
純損失 |
$ |
( |
) |
|
$ |
( |
) |
普通株株主は1株当たり普通株純損失を占め,基本損失と赤字を計上すべきである |
$ |
( |
) |
|
$ |
( |
) |
加重平均普通株、普通株株主は1株当たり純損失、基本損失と希釈損失に用いる |
|
|
|
|
|
||
総合的な損失: |
|
|
|
|
|
||
純損失 |
$ |
( |
) |
|
$ |
( |
) |
その他の全面収益(損失): |
|
|
|
|
|
||
有価証券の未実現収益 |
|
|
|
|
( |
) |
|
総合損失 |
$ |
( |
) |
|
$ |
( |
) |
付記はこれらの連結財務諸表の構成要素である。
F-4
オメガ治療会社は
合併報告書Sトカゲ人権益
(単位は千で、シェアは含まれていない)
|
|
普通株 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
株式.株 |
|
|
パー?パー |
|
|
追加料金を払いました |
|
|
他の総合損益を累計する |
|
|
積算 |
|
|
合計して |
|
||||||
2022年1月1日まで |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
普通株式を発行して行使のオプションと交換する |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
その他総合損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
株に基づく報酬 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2022年12月31日まで |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
直接発行を登録するための普通株を発行し、発行コストを差し引く |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
普通株式を発行して行使のオプションと交換する |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
その他総合収益 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
株に基づく報酬 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2023年12月31日まで |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
付記はこれらの連結財務諸表の構成要素である。
F-5
オメガ治療会社は
総合政治家キャッシュストリームのTS
(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
経営活動 |
|
|
|
|
|
|
||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
純損失と経営活動で使用される現金純額の調整: |
|
|
|
|
|
|
||
減価償却 |
|
|
|
|
|
|
||
債務発行原価償却と債務割引 |
|
|
|
|
|
|
||
経営的リース使用権資産の償却 |
|
|
|
|
|
|
||
有価証券割引の増加 |
|
|
( |
) |
|
|
|
|
成功費用債務の公正価値変動 |
|
|
|
|
|
|
||
固定資産収益を処分する |
|
|
( |
) |
|
|
— |
|
株に基づく報酬費用 |
|
|
|
|
|
|
||
経営性資産と負債変動状況: |
|
|
|
|
|
|
||
関係者の売掛金 |
|
|
( |
) |
|
|
( |
) |
売掛金 |
|
|
( |
) |
|
|
|
|
前払い費用と他の流動資産 |
|
|
|
|
|
( |
) |
|
その他の資産 |
|
|
|
|
|
( |
) |
|
売掛金 |
|
|
( |
) |
|
|
|
|
費用とその他の流動負債を計算しなければならない |
|
|
( |
) |
|
|
|
|
その他負債 |
|
|
|
|
|
( |
) |
|
経営活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
投資活動 |
|
|
|
|
|
|
||
財産と設備を購入する |
|
|
( |
) |
|
|
( |
) |
財産と設備を売却して得た収益 |
|
|
|
|
|
|
||
有価証券を購入する |
|
|
( |
) |
|
|
( |
) |
有価証券満期日収益 |
|
|
|
|
|
|
||
投資活動提供の現金純額 |
|
|
|
|
|
( |
) |
|
融資活動 |
|
|
|
|
|
|
||
株式発行で得られた収益 |
|
|
|
|
|
— |
|
|
持分発行費用を支払う |
|
|
( |
) |
|
|
— |
|
リース融資収益 |
|
|
|
|
|
— |
|
|
債務を返済する |
|
|
( |
) |
|
|
— |
|
融資費用の支払い |
|
|
( |
) |
|
|
— |
|
持分激励計画に基づいて普通株所得金を発行する |
|
|
|
|
|
|
||
融資活動が提供する現金純額 |
|
|
|
|
|
|
||
現金、現金等価物、および限定的な現金純変化 |
|
|
( |
) |
|
|
( |
) |
現金、現金等価物、制限現金--期初 |
|
|
|
|
|
|
||
現金、現金等価物、制限された現金--期末 |
|
$ |
|
|
$ |
|
||
現金、現金等価物、および限定現金の入金 |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
制限現金 |
|
|
|
|
|
|
||
現金、現金等価物、および限定現金 |
|
$ |
|
|
$ |
|
||
キャッシュフロー情報の補足開示 |
|
|
|
|
|
|
||
利子を支払う現金 |
|
$ |
|
|
$ |
|
||
非現金投資·融資活動の追加開示 |
|
|
|
|
|
|
||
売掛金及び売掛金に掲げる財産及び設備購入額 |
|
$ |
|
|
$ |
|
||
付記はこれらの連結財務諸表の構成要素である。
F-6
オメガ治療会社は
連結財務諸表付記
1.ビジネスの性質とビジネス基盤プレゼンテーションのSIS
組織する
オメガ治療会社(“会社”あるいは“オメガ”)は臨床段階の生物技術会社であり、そのオメガプラットフォームを利用することによって、率先して新型プログラマブルエピゲノムメッセンジャーリボ核酸薬物を開発した。Omegaプラットフォームはエピジェネティクスの力を利用して、これは遺伝子発現と生体生活のあらゆる面を制御する機序であり、細胞の発生、成長と分化から細胞死までである。Omegaプラットフォームは基本的なエピジェネティック過程を制御し、異常遺伝子発現を正常範囲に戻すことで、天然核酸配列を変えることなく疾病の根本的な原因を是正することができる。当社は2016年7月にデラウェア州会社として設立され、その事務所はマサチューセッツ州ケンブリッジ市にある。
流動資金と持続経営
設立以来、同社はそのほとんどの資源をそのプラットフォームの構築とそのプロジェクトグループの発展の推進、その知的財産権の確立と保護、研究と開発活動、組織と会社員の配備、業務計画、資金調達、およびこれらの業務に一般的かつ行政的な支援を提供することに投入した。同社は生物技術業界の早期臨床段階会社によく見られるリスクと不確定要素の影響を受け、候補製品の成功研究、開発と製造に関連する技術リスク、候補製品の臨床開発に関連するリスク、新技術革新の競争相手の開発、キー人員への依存、ノウハウの保護、政府法規の遵守及び追加資本を獲得して運営を援助する能力を含むが、これらに限定されない。現在と未来のプロジェクトは広範な臨床前と臨床テスト及び商業化前の監督管理承認を含む大量の研究と開発努力が必要となる。このような努力は多くの追加資本、十分な人員、そしてインフラを必要とする。同社の薬物開発努力が成功しても,同社がいつ(あれば)製品販売から相当な収入を得るかは定かではない。
2021年8月に、当社は初の公募(IPO)を完了し、それに基づいて発行·販売した
同社は現金、現金等価物、有価証券を#ドルと予想している
添付の連結財務諸表は、これらの不確実性の結果に起因する可能性のあるいかなる調整も含まない。そこで,総合財務諸表の作成は,当社が継続的に経営している企業であると仮定し,正常業務過程で資産を現金化し,負債や約束の返済を考慮したものである。
F-7
マクロ経済状況に関する重大なリスクと不確実性
信用や金融市場を含む世界経済は最近、流動性や信用供給の深刻な減少、金利やインフレ率の上昇、銀行·金融機関に関する危機、消費者自信の低下、経済成長の低下、経済安定の不確実性など、極端な変動と破壊を経験している。また、不安定な市場や経済状況、ならびに疫病や国際政治の動揺、戦争やテロによるさらなる破壊は、私たちの業務、財務状況、経営業績に深刻な悪影響を及ぼす可能性がある。
陳述の基礎
添付されている総合財務諸表は、米国公認会計原則(“公認会計原則”)に基づいて作成される。本付記における適用指針に対するいかなる言及も財務会計基準委員会(“FASB”)の“会計基準編纂”(“ASC”)と“会計基準更新”(“ASU”)中の権威公認会計原則を指す。別の説明がない限り、本契約のすべての金額はドル(“ドル”)で表される。
2.主な会計政策の概要
合併原則
添付されている連結財務諸表には、オメガ治療会社とその完全子会社オメガ治療安全会社の勘定が含まれています。オメガ治療安全会社はマサチューセッツ州の子会社です。すべての会社間取引と残高は合併で販売された。
再分類する
会社は前年度の関連側費用を研究·開発および総合経営報告書と全面赤字における一般·行政費用に再分類し,本年度に適合した列報方式である。
予算の使用
公認会計基準に従って連結財務諸表を作成することは、報告期間及び報告期間中の資産、負債及び費用の報告金額、又は有資産及び負債の開示に影響を与えるために、管理層に推定と仮定を要求する。当社は過去の経験とその当時の状況で合理的と考えられる様々な要素に基づいて見積もりと仮説を立てています。
これらの総合財務諸表に反映される重大な推定および仮定は、物件および設備使用年数の選択、成功費用負債の公正価値、賃貸負債を計算するための増分借入率、研究開発費、収入確認、および株に基づく報酬に関するいくつかの判断を含むが、これらに限定されない。実際の結果はこれらの推定とは異なる可能性がある。推定数の変化は既知期間の報告結果に反映されている。
現金と現金等価物
現金は、いつでも利用可能な小切手口座内の現金を含み、現金等価物は、通貨市場口座と、購入日から元の満期日が3ヶ月以下のすべての高流動性投資とを含む。現金および現金等価物はコストごとに入金され,コストは公平価値とほぼ同じである.
有価証券
2023年12月31日現在、同社の有価証券は会社の債務証券からなり、販売可能に分類され、公正価値で報告されている。債務証券を売却できる未実現収益と損失は、実現前に株主権益として他の全面的損失の構成要素として報告される。購入時に生成される任意の割増または割引は、手形の有効期間内に償却され、および/または利息収入および/または費用が増加される。実現した損益は特定の確認方法で決定し,構成部分として他の費用純額を計上した。
♪the the the同社が有価証券を評価した場合,一時的な減値に加えて未実現損失が発生する。有価証券の非一時的な価値低下を評価する際には,価値低下が原始コストに占める割合がどの程度であるか,市場持続時間がどの程度であるかを考慮する
F-8
価値がある投資総額はその元のコストより低いため、当社は投資を一定期間保留し、公正価値と全体の市況を十分な予想を回復させる能力と意向がある。公正価値の任意の調整が当社が“非一時的”と考えている投資価値の低下を反映していれば、当社は総合経営報告書と全面赤字に費用を計上することで、投資を公正価値に減少させる。
制限現金
限定現金とは、会社のオフィス賃貸に関係する保証金として発行された信用状のために提供される担保のことである。
信用リスクが集中する
信用リスクの高度な集中の影響を受ける可能性のある金融商品は主に現金、現金等価物、および有価証券を含む。同社は、高格付けの金融機関と協力することで、自社で定義されている広範かつ多様な金融商品に投資する有価証券に関連するリスクを最小限にしようとしている。同社は信用格付けと満期日に関するガイドラインを策定し、元金残高の保障と流動性の維持を目指している。同社はその投資政策に基づいてその基金を維持し、この政策は許可された投資を定義し、信用品質基準を規定し、任意の単一発行者に対する信用リスクを制限することを目的としている。
保証と補償
デラウェア州の法律で許可されている場合、会社が会社との関係又は会社が担当している職によって発生した何らかの事件又は事件は、会社はその高級管理者、取締役、コンサルタント及び従業員に対して賠償を行う。同社は2023年12月31日と2022年12月31日まで
財産と設備
財産と設備はコストから減価償却累計を引いて申告する.減価償却費用は、資産ごとの推定耐用年数で直線法で確認され、具体的には以下のようになる
資産種別
|
使用寿命を見込む
|
コンピュータ装置及びソフトウェア |
|
実験室設備と事務家具 |
|
賃借権改善 |
廃棄又は販売時には、資産を処分するコスト及び関連する減価償却は勘定から差し引かれ、それによって生じる収益又は損失はいずれも運営損失に計上される。修理費と維持費は発生時に料金を記入します。
研究と開発費用を計算すべきである
会社は、会社の供給、臨床前および臨床研究を実施し、管理する契約研究組織(“CRO”)および契約開発および製造組織(“CDMO”)を含む第三者サービスプロバイダとのオファーおよび契約に基づいて研究および開発費用を推定する。これらの合意の財務条項は交渉が必要であり、契約によって異なり、支払いの不均衡を招く可能性がある。場合によっては、供給者に支払われるお金は、提供されたサービスレベルを超えて、費用の前払いをもたらす可能性があり、この場合、費用は、予期される達成された時間に応じて現在または長期的に分類評価されるであろう。サービス料を計算する際には、当社はサービスを提供する時間帯と時間ごとにかかる努力度を見積もる。実際にサービスを実行する時間や努力の程度が見積もりと異なる場合、会社はそれに応じて費用または前払い費用の金額を調整する。
F-9
起債コスト
当社の長期債務の発行に関するコストは債務の直接減値として記録されており、実際の利子法を用いて関連債務の寿命内で利息支出の構成要素として償却を行う。
成功費義務
改訂された普華永道との融資協定は、改訂された融資協定に記載されている指定流動資金事件が発生した場合に成功費用(“成功費用責任”)を支払うことを当社に要求する。当社は、この債務が独立した派生ツールであると認定した。したがって,成功費用債務は会社の総合貸借対照表で負債に分類され,最初に公正価値で記録され,報告期間ごとの公正価値変動は他の費用,総合経営報告書純額,全面赤字で確認された。このような債務の公正な価値は、債務が返済されるまで、各報告期間の終了時に再計量される。
株式発行コスト
同社は、このような融資が完了するまで、進行中の株式融資に直接関連するいくつかの法律、専門会計、その他の第三者費用を繰延発売コストとしている。株式融資完了後、これらのコストは発行による収益の減少として株主権益に計上される。計画された株式融資を放棄すれば、繰延発売コストは直ちに総合経営報告書における運営費用として支出される。いくつありますか
長期資産減価準備
当社はその長期資産を評価し、主に物件や設備および経営賃貸使用権減価資産を含み、事件や状況が変化して当該などの資産の額面が回収できない可能性があることを示すたびに、その資産の評価を行う。保有·使用する資産の回収可能性は,資産の帳簿価値と資産予想による将来の未割引現金流量を比較することで測定した。当該等の資産は減値とみなされ、確認すべき減値は、当該資産の帳簿価値が当該資産の公正価値を超える金額で計量される。いくつありますか
賃貸借証書
同社はマサチューセッツ州ケンブリッジ市にある会社のオフィスと実験室空間の不動産賃貸契約を持っている。それは契約の開始時にレンタルが含まれているかどうかを確認する。経営リース資産および負債はリース開始日にリース期間内にリース支払いの現在値を確認する。レンタル費用は一般的に固定されており、時間の経過とともに徐々に上昇している。浮動支払いは、会社がレンタル者の対象資産に関する運営コストを使用または共有することに関連し、発生したことを確認する。レンタル期間を決定する際には、会社は、その選択権を行使すると合理的に判断した場合に、リース契約を延長または終了する選択権を含む。隠れ金利が確定しにくい場合、会社はその増分借入金利を使用してレンタル負債を計算する。レンタル料金はレンタル期間内に直線法で確認します。
公正価値計量
公正価値は、計量日市場参加者間の秩序ある取引において、資産または負債が元金または最も有利な市場で負債を移転するために徴収または支払いされる交換価格として定義される。公正価値を計量するための推定技術は,観察可能な投入を最大限に利用し,観察不可能な投入を最大限に減少させなければならない。公正価値別に列挙された金融資産および負債は、公正価値レベルの以下の3つのレベルのうちの1つで分類および開示されなければならず、そのうちの最初の2つのレベルは可視とみなされ、最後のレベルは見えないとみなされる
F-10
収入確認
2023年12月31日までに確認された収入は、PM(CF)Explorations,Inc.またはPMCoとの連携プロトコルのみからである。当社はASU 2014-09に基づいて収入を確認しました顧客との契約の収入(主題606)それらに関連する修正案、または総称してASC 606と呼ばれる。
開始時に、会社は、契約がASC 606または他の主題の範囲内にあるかどうかを判断する。ASC 606の範囲内で決定された契約の場合、収入は、顧客が約束された商品またはサービスの制御権を取得したときに確認される。確認された収入金額は、これらの商品やサービスと交換するために、会社が獲得する権利があると予想されている対価格を反映している。この核心原則を実現するために,会社は,(I)顧客との契約を決定する,(Ii)契約中の履行義務を決定する,(Iii)取引価格を決定する,(Iv)取引価格を契約に割り当てる履行義務,および(V)契約履行義務を履行する際に収入を確認する,の5つのステップを採用する.会社が顧客の意図と支払い承諾の対価格の能力に基づいて、譲渡された商品およびサービスの実質的にすべての対価格を受け取る可能性があると決定した場合にのみ、会社は5段階モデルを契約に適用する。
契約で約束された履行義務は,顧客に移行する貨物やサービスによって決定され,これらの貨物とサービスは区別できるとともに,契約範囲内で異なる.1つの契約に複数の約束された貨物およびサービスが含まれている場合、会社は、契約の背景に区別があり、区別できるかどうかを決定するために判断を運用する。これらの基準を満たしていなければ、約束された貨物とサービスは総合履行義務として入金されるだろう。
取引価格は、商品やサービスを顧客に移転することと引き換えに、会社が獲得する権利のある対価格によって決定される。取引価格に可変対価が含まれている場合、当社は可変対価の性質に基づいて、期待値法または可能金額法を用いて取引価格に含まれるべき可変対価金額を推定する。経営陣の判断により、契約項下の累積収入が将来的に大きく逆転しない可能性が高い場合には、可変対価格が取引価格に含まれる。可変考慮に対する制約の影響を含む任意の推定は、どのような変化があるかどうかを決定するために、各報告期間において評価される。取引価格を決定するには重大な判断が必要だ。
契約が単一履行義務を含む場合、取引価格全体をその単一履行義務に割り当てる。複数の履行義務を含む契約要求は、取引価格が可変でない限り、相対的に独立した販売価格に基づいて取引価格を個々の履行義務に割り当て、かつ、履行義務または単一履行義務の一部を構成する異なるサービスに完全に割り当てられる基準に適合する。受信した対価格を相対的に独立した販売価格に応じて単独の履行義務の間に割り当てる.
その会社は一定期間、またはある時点で契約履行義務を履行する。以下の場合、収入は、時間とともに確認される:(I)顧客がエンティティ業績によって提供される利益を同時に受け取り、消費すること、(Ii)エンティティ業績が資産作成または強化時に顧客によって制御される資産を創造または強化するか、または(Iii)エンティティ業績は代替エンティティ用途の資産を生成しておらず、エンティティは、これまでに完了した業績支払いを強制的に実行する権利がある。エンティティが一定期間義務を履行していない場合、ある時点で、約束された貨物またはサービスの制御権を顧客に移すことによって、関連する履行義務を履行する。
会社の知的財産権の許可が手配で決定された他の履行義務とは異なると判定された場合、会社は、許可が顧客に譲渡され、顧客が許可を使用して利益を得ることができるときに許可に割り当てられた対価格収入を確認する。他の承諾と合併するライセンスについて、会社は、合併義務が一定期間内に履行されているか、またはある時点で履行されているかを決定するために、合併履行義務の性質を評価するために判断を利用する
F-11
時間の経過とともに、収入を確認するための進行を測る適切な方法。当社は報告期間ごとに進捗指標を評価し、必要に応じて業績指標と関連収入確認を調整します。
マイルストーン支払いを含む各手配の開始時には、会社はマイルストーンに達する可能性を評価し、最も可能な金額法を用いて取引価格に含まれる金額を推定する。将来大きな収入逆転が起こらない可能性が高い場合、関連するマイルストーン価値は取引価格に含まれるだろう。当社または被許可者の制御範囲内でないマイルストーン支払いは実現不可能とされているため、確認された収入が制限されており、経営陣は収入が逆転不可能であると断言できないからである。そして、取引価格は相対的に独立した販売価格で個々の履行義務に割り当てられ、会社は契約項目の履行義務を履行する際に収入を確認する。その後の各報告期間が終了した時点で、当社は、このようなマイルストーンを実現する可能性や任意の関連制限を再評価し、必要に応じて全体の取引価格の推定を調整します。いずれの調整も累積追跡をもとに記録されており,調整期間中の収入や収益に影響を与える.現在まで、同社は何の記念碑的な収入も確認していない。
繰延収入は、利益プロセスがピークに達する前に受け取った額から得られ、契約履行義務を履行する際に将来の期間の収入として確認される。今後12ヶ月以内に確認されると予想される繰延収入は流動負債に分類される。
研究開発費
研究·開発費は発生時に費用を計上する。研究開発支出には、賃金とボーナス、株式給与、従業員福祉、施設コスト、実験室用品、減価償却、相談費、技術許可コスト、マイルストーン支払い、外部契約研究開発と製造費用が含まれる研究開発活動を行うことによって生じるコストが含まれる。いくつかの研究及び発展活動のコストは個別手配の条項によって確認され、すでに発生したコストモデルと異なる可能性があり、総合財務諸表に前払い或いは研究及び発展コストを反映すべきである。将来的に研究および開発活動のために受信される貨物またはサービスの払戻不可能な前払いは、前払い費用として記録され、関連貨物が提供またはサービスを提供する際に支出される。
株に基づく報酬
同社の株式報酬計画は、従業員、取締役、コンサルタントに奨励的株式オプション、非制限株式オプション、株式付加価値権、制限株式奨励、制限株式単位、およびその他の株式ベースの奨励を付与することを可能にする。
当社はその公正価値に基づいて、総合経営報告書において、従業員と非従業員に与えられたすべての株式補償を費用と全面赤字と確認した。株式オプション奨励について、会社はブラック·スコアーズオプション定価モデルを用いて公正価値を推定した。会社普通株の公正価値は制限株式奨励の公正価値を決定するために使用される。
ブラック·スコアーズオプション価格設定モデルは、(I)予想株価変動、(Ii)予想報酬期間、(Iii)無リスク金利、および(Iv)期待配当を含む、いくつかの主観的仮定に基づく入力を必要とする。初公開前に当社の普通株は公開市場が不足し、及び十分な特定会社の歴史と隠れた変動率データが不足しているため、当社は1組の代表的な上場会社の歴史変動率から予想変動率を計算し、これらの上場会社は自社と似た特徴を持っており、製品開発段階と生命科学業界の重点を含む。履歴変動率は,期待期限の仮定に見合った一定期間に基づいて計算される.当社が採用している簡略化方法は
F-12
アメリカ証券取引委員会職員会計公告第107号株式支払したがって、従業員および非従業員に付与されたオプションの期待期間が計算され、したがって、十分な履歴データが不足しているため、期待期間は、オプションの帰属期限および元の契約期間の算術平均値に等しい。無リスク金利は、満期日に関連奨励の期待期限に見合った米国債に基づく。予想配当収益率はゼロと仮定しているが、同社は配当金を支払ったことがないため、現在はその普通株に何の配当も支払う計画もない。
特許費用
特許出願の提出及び起訴に関するすべての特許に関する費用は、特許が将来代替用途がないため、発生した費用として計上される。
所得税
当社は、繰延税金資産と負債が、当社の総合財務諸表および納税申告書に含まれている事件の予想される将来の税務結果であることを確認します。繰延税項資産と負債は財務諸表帳簿額面と現有資産及び負債及び赤字及び貸記繰越の税基との差額に基づいて決定し、予想差額を採用してその年に発効した制定税率を振り戻す。繰延税金資産が現金化できない可能性が高い場合は、推定準備金を差し引く。2023年12月31日まで2022年までに、当社はその繰延税金資産入金全額推定値について準備しています。当社は審査の結果、税務状況をより維持する可能性があるかどうかを決定します。より可能性がある可能性がない場合、ハンドヘルドデバイスのいかなる利点に起因することができるかは確認されない。確認された税金割引は、より確認可能な閾値に適合する任意の税務ヘッドについて、事項解決後に達成される可能性が50%を超える最大金額として計算される。所得税引当金の一部として、同社は不確定な税収状況に関連した利息及び罰金を計上する。
総合損失
総合損失とは、企業株主権益が一定期間内に非所有者源の取引及びその他の事件と状況によって発生した変化である。総合損失には純損失及び株主権益の他の変化が含まれ、その中には純損失以外に含まれないいくつかの権益変化が含まれている。同社の他の全面赤字の唯一の要素はその有価証券の未実現損益である。
1株当たり純損失
普通株株主が1株当たり基本純損失を占めるべき計算方法は、普通株株主が純損失を当期発行普通株の加重平均で割るべきである。普通株主が償却純損失を占めるべきであることは、普通株株主の純損失を調整して、希釈証券の潜在的な影響に基づいて未分配収益を再分配することによって計算される。普通株株主が希釈1株当たり純損失を占めるべき計算方法は、普通株株主は希釈純損失を当期発行普通株で割った加重平均を占めるべきであり、その中に普通株等価物の希釈影響を仮定する潜在希釈性普通株を含む。
会社が普通株株主が純損失を占めるべき期間を報告する時、普通株株主は希釈1株当たり純損失と普通株株主は基本的に1株当たり純損失を占めるべきであり、希釈性普通株の影響が反希薄であれば、希釈性普通株を発行したと仮定しないからである。同社は2023年12月31日と2022年12月31日までの年度普通株主が純損失を占めるべきだと報告した。
市場と地理情報を細分化する
業務部門はエンティティの構成要素として定義され、そのエンティティに関する離散情報は、首席業務意思決定者または意思決定グループがリソースをどのように割り当てるかを決定し、業績を評価する際に評価することができる。CODMは会社の最高経営責任者です。CODMは,その業務を米国で運営するための運営部門と見なし,その業務を管理している。
F-13
最近可決された会計公告
2023年1月1日、当社は会計基準を採択して2016-13号を更新した金融商品·信用損失:金融商品信用損失の測定(“ASU 2016-13”)。ASU 2016-13は、金融資産の予想される信用損失の測定と確認を必要としている。2019年4月、財務会計基準委員会(FASB)はASU 2019-04年度内にASU 2016-13に明らかにした主題326(金融商品--信用損失)、主題815(派生商品およびヘッジ)、および主題825(金融商品)の編纂改善それは.この指導意見は2022年12月15日以降の会計年度に適用される。この基準を採用することは、添付された連結財務諸表に関係がない。
最近採用されていない会計公告
2023年11月、FASBはASU 2023-07を発表した分部報告(主題280), 報告可能な部分に開示された改善それは.新基準は、支部情報と重大支部費用の開示を強化することを要求している。それは公共実体がその経営部門を決定する方法を変えないだろうASU 2023-07は、2023年12月15日以降に開始される会計年度と、2024年12月15日以降に開始される会計年度内の移行期間に適用され、早期採用が許可されています。新しい基準は、財務諸表に記載されている以前のすべての期間に適用されるべきである。同社は現在、その連結財務諸表への影響を評価している。
FASBは2023-09を発表しました所得税(主題740)、所得税開示の改善それは.新基準は、公共企業エンティティに、納付された所得税の情報、税率調整中の特定のカテゴリ、および量子化しきい値に適合する調整項目の追加情報を開示することを要求する。その指針は予想に基づいて適用されなければならない。公共事業体については、ASU 2023-08は2024年12月15日以降の会計年度に発効し、早期採用が許可されている。この基準は、他のすべてのエンティティについて、2025年12月15日以降の年間期間に有効である同社は現在、その連結財務諸表への影響を評価している。
3.有価証券
次の表は、同社の有価証券(単位:千):
|
2023年12月31日 |
|
|||||||||
|
原価を償却する |
|
|
未実現損失総額 |
|
|
公正価値 |
|
|||
会社債務証券 |
$ |
|
|
$ |
( |
) |
|
$ |
|
||
|
|
|
|
|
|
|
|
|
|||
|
2022年12月31日 |
|
|||||||||
|
原価を償却する |
|
|
未実現損失総額 |
|
|
公正価値 |
|
|||
会社債務証券 |
$ |
|
|
$ |
( |
) |
|
$ |
|
||
有価証券の償却コストは割増償却と満期割引増加に応じて調整される。2023年12月31日その他の総合損失を累計した残高は有価証券関連の活動のみを含む。いくつありますか
2023年12月31日まで償却コストベース回収まで、当社は赤字状態の債務証券を売却するつもりはなく、売却する必要がない可能性も高い。その結果、同社はそれができたことを確認しました
F-14
4.前払い費用と他の流動資産
前払い料金および他の流動資産には、以下のものが含まれている(千計)
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
臨床発展 |
|
$ |
|
|
$ |
|
||
研究開発 |
|
|
|
|
|
|
||
施設 |
|
|
|
|
|
|
||
その他売掛金 |
|
|
|
|
|
|
||
保険 |
|
|
|
|
|
|
||
ソフトウェア |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
$ |
|
|
$ |
|
||
5.財産と設備、純額
財産と設備、純額は以下の部分からなる(千計)
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
実験室装置 |
|
$ |
|
|
$ |
|
||
家具と固定装置 |
|
|
|
|
|
|
||
賃借権改善 |
|
|
|
|
|
|
||
コンピュータ装置 |
|
|
|
|
|
|
||
建設中の工事 |
|
|
|
|
|
|
||
総資産と設備 |
|
|
|
|
|
|
||
減価償却累計を差し引く |
|
|
( |
) |
|
|
( |
) |
財産と設備、純額 |
|
$ |
|
|
$ |
|
||
2023年12月31日までと2022年12月31日までの年度減価償却費用w$として
6. 費用を計算する
計算すべき費用には、以下の項目が含まれる
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
従業員関連費用 |
|
$ |
|
|
$ |
|
||
研究コスト |
|
|
|
|
|
|
||
専門と相談料 |
|
|
|
|
|
|
||
製造コスト |
|
|
|
|
|
|
||
利子 |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
合計する |
|
$ |
|
|
$ |
|
||
7.定期ローン
2018年3月9日、当社は太平洋西部銀行(“西太平洋銀行”)と融資協定を締結し、初歩的な借金$を締結した
2023年9月22日、当社は融資期限を延長する融資協定のもう1つの改正(“第5改訂”)を締結した
F-15
百万その持分証券を売却するための現金収益および/または払戻不可能な前払い戦略パートナーシップ収益。ローンは#年に返済を開始します
また、“第5修正案”によると、同社はPWBといつでも少なくとも#ドルの残高を維持することに同意した
改訂された融資合意項の下の借入は、当社の知的財産を除くほとんどの個人財産を担保としています。改訂された融資協定には関連財務的な約束はない;しかし、当社は期限が切れるまでいくつかの肯定と消極的な承諾を守らなければならない。
自分から2023年12月31日$
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
元金 |
|
$ |
|
|
$ |
|
||
未償却債務割引 |
|
|
( |
) |
|
|
( |
) |
帳簿純額 |
|
$ |
|
|
$ |
|
||
会社は債務の期待寿命が定期ローンの期限に等しいと確定した。負債部分の実質金利はm
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
契約利子支出 |
|
$ |
|
|
$ |
|
||
債務発行原価償却と債務割引 |
|
|
|
|
|
|
||
利子支出総額 |
|
$ |
|
|
$ |
|
||
2023年12月31日及び2022年12月31日に、定期ローンの受取利息は$
当社は以下の定期ローンに関する元金(千計)を返済しなければならない
2024 |
|
$ |
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 |
|
|
|
|
合計する |
|
$ |
|
F-16
8.金融商品の公正価値
会社の現金及び現金等価物と制限性現金の公正価値はオファー市場価格によって計量される;会社の有価証券の公正価値は活発な市場オファー以外の定価投入によって決定され、これらの価格は報告日に直接或いは間接的に見られる。その他の流動資産、売掛金、売掛金、負債はその短期的な性質のため、2023年12月31日現在、2023年と2022年までの公正価値に近い。変動金利が市場金利に近いため、当社債務の帳簿価値はその公正価値に近い。改訂された融資契約に関する成功費用責任には観察できない情報が含まれており,これらの情報は当社自身の仮定,すなわち計量日に市場活動が少ない(あれば)ことを反映しているため,当社の成功費用義務は観察不可能な情報を用いて公正価値に応じて恒常的に計測される。
会社の金融商品の公正価値を以下の表にまとめる(千単位)
|
|
2023年12月31日 |
|
|||||||||||||
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
|
合計する |
|
||||
金融資産 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
会社債務証券 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
合計する |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
金融負債 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
成功費義務 |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
||
|
|
2022年12月31日 |
|
|||||||||||||
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
|
合計する |
|
||||
金融資産 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
会社債務証券 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
合計する |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
金融負債 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
成功費義務 |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
||
工務局と締結された融資協定の第4及び第5項の改正によると、同社は共済$を支払わなければならない
2023年12月31日現在、同社は
|
|
2023年12月31日 |
|
|
2022年12月31日 |
|
||
割引率 |
|
|
% |
|
|
% |
||
流動性イベントの実現が期待される期間(年) |
|
|
|
|
|
|||
流動性イベントを実現する確率 |
|
|
|
|
||||
次の表は、会社の成功費用義務の公正価値の前転を提供し、公正価値は第三級投入によって決定される(千単位)
F-17
|
|
成功費 |
|
|
成功費 |
|
||
2023年1月1日の公正価値 |
|
$ |
|
|
$ |
— |
|
|
成功費債務の初期公正価値 |
|
|
— |
|
|
|
|
|
価値変動を公平に承諾する |
|
|
|
|
|
|
||
2023年12月31日の公正価値 |
|
$ |
|
|
$ |
|
||
9.支払いの引受およびその他の事項
賃貸借証書
同社はマサチューセッツ州ケンブリッジ市にある会社のオフィスと実験室空間のために以下の運営賃貸契約を締結した.
ヴァサ通り325番地
当社は2017年、マサチューセッツ州ケンブリッジ市ワサ街325番地にあるオフィススペースをレンタルする撤回不可能な運営リース契約を締結したそれは.会社は財産税、保険料、正常維持費を支払う必要がある。経営リースには、レンタル期間内に予約された固定最低レンタル料のアップグレードが含まれています。二零一年及び二零二年、当社と
ドングリ公園通り20番地
2020年7月13日,当社はSail Biomedicines,Inc.(LaRonde,Inc.と合併する前に,Sail Bio,Inc.,前身はSenda Biosciences,Inc.とKintai Treateutics,Inc.)と共有空間手配(以下,手配と略す)を締結した.至れり尽くせり
140 First Street(One Charles Park)
2021年11月4日、当社はARE-MA地域第94号有限責任会社と賃貸契約を締結し、レンタル総額は約
同社は2023年5月3日、一部の物件の交付日を延期し、初期基本賃貸料を$増加させることを含む賃貸借契約の第1回改訂を行った
F-18
2023年7月11日、当社は締結しましたPriori Bio,Inc.と空間配置を共有し,2023年7月12日に
2023年12月31日現在、経営性賃貸使用権の純資産額は$
使用権資産は当社がリース期間内に対象資産を使用する権利を代表し、関連リース負債は当社のリースによるリース金の支払い義務を代表する。使用権資産と当該負債はいずれもレンタル開始日にレンタル期間内にレンタル支払いの現在値によって確認されます。当社の借款は暗黙的な金利を提供していないため、当社は改訂された定期ローンの金利(このローンは完全に担保されている)と期限に合わせた保証市場金利に基づいて借入金金利を増加させると推定している。
下表は年末までのレンタル料金の構成要素をまとめています2023年12月31日と2022年12月31日(単位:千)。
|
|
現在までの年度 |
|
|||||
|
|
2023年12月31日 |
|
|
2022年12月31日 |
|
||
レンタル費用を経営する |
|
$ |
|
|
$ |
|
||
可変レンタル費用 |
|
|
|
|
|
|
||
レンタル総費用 |
|
$ |
|
|
$ |
|
||
可変レンタル費用には一般的に公共地域維持費、光熱費、財産税が含まれています。2023年12月31日までの年間 $
当社の賃貸に関する加重平均残存期間と割引率は以下のとおりである
|
|
自分から |
|
|||||
|
|
2023年12月31日 |
|
|
2022年12月31日 |
|
||
加重平均残存賃貸年限(年) |
|
|
|
|
|
|
||
加重平均割引率 |
|
|
% |
|
|
% |
||
F-19
同社の2023年12月31日までと2022年12月31日までの賃貸契約に関する補足キャッシュフロー情報は以下の通り(単位:千)
|
|
現在までの年度 |
|
|||||
|
|
2023年12月31日 |
|
|
2022年12月31日 |
|
||
賃貸負債の金額を計上するための現金 |
|
$ |
|
|
$ |
|
||
リース負債と引き換えに経営的リース資産 |
|
$ |
|
|
$ |
|
||
2023年12月31日まで、12月31日までの毎年、第1街140号とワサ325号の推定最低賃貸料は以下の通り(千単位)
2024 |
|
$ |
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 |
|
|
|
|
2028 |
|
|
|
|
その後… |
|
|
|
|
最低賃貸支払総額 |
|
|
|
|
差し引く:推定利息 |
|
|
( |
) |
リース負債現在価値を経営する |
|
$ |
|
保温家具の販売-レンタル
10.ライセンスプロトコル
旗艦パイオニア革新V,Inc.
2019年3月、当社はフラッグシップパイオニア革新V,Inc.と独占ライセンス契約を締結し、この合意に基づき、当社は指定特許権の下のグローバル独占、特許権使用料、再許可及び譲渡可能な許可を付与し、開発、製造及び商業化特許製品(“旗艦許可”)を取得した。旗艦ライセンスの条項によると、当社は当社のライセンス製品の純売上高について低い1桁パーセントの印税を支払う義務があります。特許権使用料は、その国/地域が特許製品をカバーする最後の有効特許主張が満了または放棄されるまで、会社によって国/地域によって支払われなければならない。同社には旗艦会社に特許訴訟費用の返済が義務付けられている。
印税支払いは旗艦許可下の許可製品の販売にかかっている。したがって、当該等料金が販売開始時に可能かつ推定可能であると考えられる場合には、会社は、その負担すべき金額に特許使用料費用を販売コストに計上する。
ワイトヘード生物医学研究所
はい2019年5月、当社はワイトヘード生物医学研究所(“WEBR”)(当社の取締役会メンバー1名の連属機関)と独占ライセンス契約を締結し、指定特許権により、当社はグローバル独占、特許権使用料及び再許可可能な許可を付与し、研究、製造、製造、使用、販売、要約、リース及び輸入製品、及び許可を行った工程(“WIBR独占許可”)を取得した。WIBRの独占許可の条項によると
F-20
会社$未満の払い戻し不可の前払い料金をお支払いいただきました
2019年5月には、当社もWIBRと共同独占ライセンス契約を締結し、指定特許権に基づき、当社はグローバル独占、特許料負担、再許可可能な許可を付与し、研究、製造、製造、使用、販売、要約販売、リース及び輸入製品、及び許可されたプログラム(“WEBR共同独占許可”)を行う。WIBR共同独占許可の条項によると、同社は$未満の払い戻し不可の前払いを支払った
2023年12月31日まで及び2022年12月31日まで年度会社確認費用は#ドルです
年間維持費は,適用年度の規定額に基づいて年次費用と記す。記念碑的な支払いが発生する可能性があると判断されると、支払うべき金額は研究開発費として記録される。最後に、特許権使用料支払い及び再許可非特許権使用料支払いは、ライセンス製品の販売又はWEBR独占及び共同独占ライセンス下での従属ライセンス契約の実行に依存する。したがって、販売開始または再許可契約の実行時に、当該等の費用が可能かつ試算可能であると考えられる場合には、当社は、特許権使用料費用を計算し、適用状況に応じて非特許権使用料を再許可して、当社が負担すべき金額を支払う。
Acuitas治療会社は
2020年10月,当社はAcuitas Treateutics,Inc.(“Acuitas”)と開発·オプション協定(“開発·オプション協定”)を締結した。開発とオプション合意の条項により,双方は会社の遺伝子調節療法とAcuitasの脂質ナノ粒子を結合したいくつかの製品を共同開発することに同意した。さらに、開発およびオプション協定によれば、同社は、Acuitas脂質ナノ粒子技術(“Acuitas LNP技術”)に関連する特許および独自技術に基づいて、これらの目標に関連する1つまたは複数の治療製品を開発および商業化するために、2つの指定されたターゲット(例えば、EC構築物)(“保持ターゲット”)に関連する非独占的で世界的に再許可可能な許可を得る権利がある。オプションおよび予約ターゲットごとに、当社は、その予約ターゲットが予約ターゲットリストから削除されるまで、または当社がその予約ターゲットについて選択権を行使するまで、技術的アクセス料およびターゲット予約および保守費を毎年支払う義務がある。会社がオプションを行使すれば,会社は$を支払うだろう
はい2021年3月に、当社は開発及びオプション協定の下で第1項の承認権を行使し、Acuitasと非独占許可協定(“Acuitas許可協定”)を締結し、この合意に基づいて、当社はAcuitas LNP技術項下の世界的に排他的で再許可可能な許可を付与し、当社の遺伝子調節療法及びAcuitas脂肪ナノ粒子を含む製品を研究、開発、製造及び商業開発する。株購入権の行使に関係して当社は一筆を招く
F-21
費用.費用オプション行権料$
当社は2023年12月31日及び2022年12月31日まで年度を収録しています共$
開発およびオプション合意項下のオプション執行費は,当社が2021年3月に第1オプションを行使した際に研究および発展支出とした。また,Acuitasによる技術アクセス費,目標予約費と維持費,FTE資金義務に関する費用および開発と材料コストの精算は,発生時に研究と開発費用を計上した。年間維持費は,適用年度の規定額に基づいて年次費用と記す。記念碑的な支払いが発生する可能性があると判断されると、支払うべき金額は研究開発費として記録される2022年12月31日までの年間維持費は$
日東電工会社
2022年10月12日、会社は日東電工会社(“日東”)と協力·許可協定(“日東協定”)を締結し、この協定によると、日東は日東がその脂質ナノ粒子輸送技術に関するすべての知的財産権の下で、独占的、世界的、特許権使用料のある、完全に譲渡可能かつ完全に再許可可能な許可を会社に付与した。
日東協定の条項によると、同社は現金#ドルを前払いしました
当社は2023年12月31日までに合算した$
11.連携プロトコル
PMCO
2021年11月に当社が締結しました
F-22
許可を得た製品です。双方の書面による同意により,研究計画経費を調整することができる.また、PMCoが買収または売却された場合、会社はこのような取引収益の一部を獲得する権利があるが、合意の規定に従って様々な減免や他の金額の支払いを行う必要がある.
当社はASC 606に基づいてこの手配を評価し、契約相手側PMCOは顧客であると結論した。当社は,研究活動と連携協定によって付与された独占許可が単一履行義務とみなされるため,取引価格は単一履行義務に完全に割り当てられることを決定した。基本サービスの履行及び/又は外部コストの発生に伴い、会社は時間の経過とともに単一履行義務に関する収入を確認する。
♪the the the2023年12月31日の取引総価格は$に決定された
その会社は資金援助の研究と協力収入を$と確認した
協定によると、PMCoが買収または販売された場合、同社は一部の販売収益を得る権利がある。各報告期間が終わると、会社はこのような取引が発生する可能性を評価するだろう。同社は2023年12月31日現在、同取引の収益が確認されることは不可能だと判断した。
日東電工会社
2022年10月12日、会社は日東会社と協力と許可協定を締結し、この協定によると、日東会社は日東社が所有または制御しているその脂質ナノ粒子輸送技術に関連するすべての知的財産権に基づいて、独占的、世界的範囲の、特許権使用料を負担する、完全に譲渡可能かつ完全に再許可可能な許可を会社に付与した。付記10の更なる議論を参照してください許可協定.
ノドとノド
2023年12月31日、会社はノとノッドA/S(“ノとノッド”)およびFLAGINGの関連会社パイオニア薬品08社(および合意に規定されているいくつかの条項、パイオニア薬品(NN)、有限責任会社、PM(NN)探索会社)と研究協力協定を締結した。同協定の条項によると、同社は、糖尿病を含む心臓代謝性疾患の予防、治療または制御に関連する製品候補または計画目標の合意された研究および開発計画に基づいて研究および開発活動を行うために、排他的で印税のある譲渡可能なライセンスをノボノードに付与する権利がある。
2024年1月、会社は払い戻しできない前金を受け取りました#
同社はASC 606に基づいてこの手配を評価し、契約相手の方ノとノッドが顧客であると結論した。その会社は研究活動と
F-23
連携プロトコルによって付与された独占的な許可は,区別できないため単一の組合せで義務を果たすとみなされる.時間の経過とともに,会社は入力法を用いて単一履行義務に関する収入を確認する。入力法によると,達成進捗はこれまでに発生した費用と義務履行時の推定費用総額の比率に基づいて測定されており,会社は合併履行義務履行における進捗状況を測る最適な方式であると考えている。コストに基づく収入確認入力法は,経営陣に業績義務達成に要するコストを見積もることを要求する。このような見積りを行う際には,コスト見積りに関する仮定を評価する必要がある.
2023年12月31日現在、取引総価格はbと決定されましたe $
12.優先株と普通株
2021年、会社取締役会と株主は、会社が改正·再発行した会社登録証明書を承認し、その中で規定されている
2023年2月、同社は登録を完了して普通株を直接発行し、それに基づいて普通株を発行·売却した
普通株式保有者は普通株を保有するごとに、1票の投票権を有する権利がある。優先株保有者が獲得権のあるすべての優先配当金を全額支払う場合、普通株式保有者は合法的に利用可能な資金から配当を得る権利がある。もし当社に任意の自動または非自発的な清算、解散または清算が発生した場合、当社のすべての債務と負債および優先株保有者が清算資産の分配について獲得する権利のあるすべての優先金額を支払いまたは支出した後、普通株式保有者は会社の残りの分配可能な資産を比例的に共有する権利があるべきである。
2023年12月31日まで会社は合共を予約しました
13.持分インセンティブ計画
2017持分インセンティブ計画
2017年6月、会社取締役会は“2017年度計画”を採択し、会社従業員および非従業員が会社普通株株を発行または購入することに合格奨励性株式オプションおよび非制限株式オプション、制限株式またはその他の奨励を付与することを規定した。2023年12月31日までいくつありますか
2017計画は、当社の取締役会またはその委員会によって管理されていますが、当社の取締役会が2017計画に基づいて権限または権限を付与したことを限度としています。行使価格、帰属及びその他の制限は取締役会が適宜決定するが、株式オプションの1株当たりの行使価格は下回ってはならない
F-24
日取りもちろんグラントです。2017年計画により付与された株式オプションが満期になります
2021インセンティブ·プラン
2021年7月、会社取締役会は2021年計画を採択し、会社株主はこの計画を承認した。2021年計画では、奨励的株式オプション、非法定株式オプション、制限株式奨励、制限株式単位、株式付加権などの株式ベースの奨励を付与することが規定されており、IPO後、すべての株式ベースの奨励は2021計画に基づいて付与される。その会社は最初に保留した
会社は総合経営報告書と総合損失報告書に株ベースの報酬費用を研究開発費と一般および行政費用に計上しており、総合損失状況は以下の通り(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
研究開発 |
|
$ |
|
|
$ |
|
||
一般と行政 |
|
|
|
|
|
|
||
株式に基づく報酬総支出 |
|
$ |
|
|
$ |
|
||
株式オプション
ブラック·スコアーズオプション定価モデルで用いられるストックオプション定価モデルの仮定は以下のとおりである:
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
予想変動率 |
|
|
|
|
||||
加重平均無リスク金利 |
|
|
|
|
||||
期待配当収益率 |
|
|
|
|
||||
|
|
|
|
|
|
|||
F-25
2023年12月31日までの年間における会社持分インセンティブ計画下のオプション活動の概要は以下のとおりである
|
|
|
|
|
重みをつける |
|
|
重みをつける |
|
|
骨材 |
|
||||
|
|
番号をつける |
|
|
平均値 |
|
|
契約書 |
|
|
価値がある (1) |
|
||||
2023年1月1日現在の未返済金 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
授与する |
|
|
|
|
|
|
|
|
|
|
|
|
||||
鍛えられた |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
没収される |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
期限が切れる |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
2023年12月31日現在の未返済債務 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
帰属しており、2023年12月31日に帰属する予定です |
|
|
|
|
|
|
|
|
|
|
|
|
||||
2023年12月31日から行使可能 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
(1) 総内在価値とは、関連オプションの行使価格と2023年12月31日現在の普通株の推定公正価値との差額である.
2023年12月31日および2022年12月31日までに年度内に付与された加重平均授出日の1株当たり株式購入公正価値は$
2023年12月31日までに$
2021年従業員株購入計画
会社取締役会は2021年7月に“従業員株購入計画”(以下“2021年従業員持株計画”と略す)を採択し、会社株主の承認を得た2021年のESPPの目的は、条件を満たした従業員に累積拠出金で会社の普通株を購入する機会を提供することです。2021年にESPPは、参加者が支払い(通常は賃金控除の形態で)によって普通株式を購入することを可能にし、最高金額は管理人によって決定された合格報酬を決定する。いくつかの他の制限を受けているか、または管理者が別の決定がない限り、参加者は最大で購入することができる
2021年ESPPは,2022年1月1日から2031年1月1日(2031年1月1日を含む)までの例年の初日まで,その計画により発行可能な株式数を年に1回増加させ,増加した金額は(I)に相当すると規定している
当社は2023年12月31日現在、2021年の特別引出権計画に基づいて発売期限を完了していません。2023年12月31日までに会社は
F-26
14.普通株主の1株当たり純損失
当社が普通株主が純損失を占めるべき期間を報告した場合、潜在的希薄化証券は、それらの影響が逆薄になるため、1株当たりの純損失の計算から除外されている。したがって、普通株株主が1株当たり基本純損失を占めるべきであることと希釈後の1株当たり純損失を計算するための発行済み普通株加重平均株式数は同じである。
以下の表では、会社の普通株株主が1株当たり基本純損失と償却純損失を占める計算方法(株と1株当たり金額を除く)をまとめた
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
分子: |
|
|
|
|
|
|
||
普通株主は純損失を占めなければならない |
|
$ |
( |
) |
|
$ |
( |
) |
分母: |
|
|
|
|
|
|
||
基本普通株と希釈普通株の加重平均 |
|
|
|
|
|
|
||
普通株株主は1株当たり普通株純損失を占め,基本損失と赤字を計上すべきである |
|
$ |
( |
) |
|
$ |
( |
) |
普通株株主が希釈後の1株当たり純損失を占めるべきであると計算する場合、同社は、これらの株に計上すると逆希釈効果が生じるため、期末既発行金額に基づいて申告された潜在普通株は含まれていない
|
|
12月31日まで |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
普通株購入の未償還オプション |
|
|
|
|
|
|
||
15.所得税
米国連邦法定所得税率と会社の有効所得税率の入金は以下の通りである
|
十二月三十一日までの年度 |
|
|||||
|
2023 |
|
|
2022 |
|
||
アメリカ連邦法定所得税率 |
|
% |
|
|
% |
||
連邦福祉を差し引いた州所得税 |
|
|
|
||||
研究開発税収控除 |
|
|
|
||||
控除不可/課税不可永久項目 |
|
( |
) |
|
|
( |
) |
評価免除額を変更する |
|
( |
) |
|
|
( |
) |
他にも |
|
|
|
|
|
||
有効所得税率 |
|
% |
|
|
% |
||
F-27
同社の繰延税金の構成要素は以下の通り(千計)
|
十二月三十一日までの年度 |
|
|||||
|
2023 |
|
|
2022 |
|
||
繰延税金資産: |
|
|
|
|
|
||
純営業損失が繰り越す |
$ |
|
|
$ |
|
||
研究開発信用繰り越し |
|
|
|
|
|
||
費用を計算する |
|
|
|
|
|
||
株に基づく報酬 |
|
|
|
|
|
||
無形資産 |
|
|
|
||||
IRC 174研究開発資本化 |
|
|
|
|
|
||
ASC 842リース責任 |
|
|
|
|
|||
未実現損益 |
|
— |
|
|
|
||
繰延税金資産総額 |
|
|
|
|
|
||
減算:推定免税額 |
|
( |
) |
|
|
( |
) |
税金資産を繰延し,純額 |
|
|
|
|
|
||
繰延税金負債: |
|
|
|
|
|
||
減価償却 |
|
( |
) |
|
|
( |
) |
使用権資産 |
|
( |
) |
|
|
( |
) |
未実現損益 |
|
( |
) |
|
|
— |
|
繰延税金負債総額 |
|
( |
) |
|
|
( |
) |
繰延税金純額 |
$ |
— |
|
|
$ |
— |
|
その会社は所有している
将来の税収優遇の実現は、会社が純営業損失の繰越期間内に課税収入を発生させる能力を含む多くの要素に依存する。国内税法の規定によると、当社の所有権のいくつかの重大な変化は、当社を売却するか、株式を売却することによる所有権の重大な変化を含み、将来繰り越し可能な純営業損失金額を制限または制限する可能性があり、この金額は毎年将来の課税収入の相殺に使用することができる。当社は制御権が変更されたかどうかを評価する研究を完了していないか,あるいは当社が設立されて以来,このような研究に関する重大な複雑さやコスト,将来的に追加的な制御権変更により複数回の制御権変更が発生する可能性があるかどうかを評価している.そのため、当社の将来の純営業損失や研究開発信用繰越の能力を利用した制御権変更の影響を見積もることはできません。
2023年12月31日現在、同社は
2023年12月31日と2022年12月31日まで、会社には不確定な税務頭寸はありません。同社は、未確認の税収優遇に関する利息と罰金を所得税支出の構成要素として確認する。設立以来、当社は確認されていない税務特典についていかなる利息や罰金も記録していません。
同社の設立以来のすべての納税年度はアメリカとマサチューセッツ州で所得税申告書を提出しました。すべての課税年度は依然としてこれらの管轄区で審査して、繰越とすることができます
F-28
過去数年間に生成された属性は将来的に調整される可能性がある.その会社は現在アメリカ国税局あるいは他の税務機関の審査を受けていませんアズ。
16.関連先取引
関連側取引は,2023年12月31日までに,主に賃貸料支払いおよび償還可能支出を含み,関連側から受け取った分譲収入で相殺される。
その会社の多数の株式は旗艦会社が保有しており,その保有株式の約は
2023年7月、当社は締結しました
はい2020年9月、同社はヴァサ街325号工場の全空間を転貸した
F-29
二零二年七月、当社はセル生物と共有空間手配を締結した
2019年9月、当社は約を転貸しました
付記9で述べた他の関連者取引を指す引受金とその他の事項10を付記して許可協定そして付記11協力協定.
17.従業員福祉
2018年、会社は国税法第401(K)節または401(K)計画に基づいて固定払込計画を構築した。401(K)計画は、規定された最低年齢およびサービス要件に適合するすべての従業員をカバーし、参加者が税引前に年間給与の一部の支払いを延期することを可能にする。2022年5月1日
F-30