♪the the the
アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
この年度までに
あるいは…。
1934年証券取引法第13条又は15条に基づいて提出された移行報告 |
移行期になります 至れり尽くせり
依頼書類番号:
(登録者の正確な氏名はその定款に記載)
(明またはその他の司法管轄権 会社や組織) |
(税務署の雇用主 識別番号) |
(主にオフィスアドレスを実行) |
(郵便番号) |
(
(登録者の電話番号、市外局番を含む)
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
取引コード |
登録された各取引所の名称 |
♪the the the |
同法第12条(G)により登録された証券:なし
登録者が証券法規則405で定義されている経験豊富な発行者である場合は、再選択マークで示してください。はい、そうです☐
登録者が証券取引法第13節又は第15節(D)節に基づいて報告を提出する必要がない場合は,複選マークで示してください。はい☐
再選択マークは、登録者が(1)過去12ヶ月以内(または登録者がそのような報告の提出を要求されたより短い期間)に、1934年の証券取引法第13条または15(D)節に提出されたすべての報告書を提出したかどうか、および(2)過去90日以内にそのような提出要件に適合しているかどうかを示す
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T法規第405条(本章232.405節)に従って提出を要求した各対話データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
大型加速ファイルサーバ |
☐ |
ファイルマネージャを加速する |
☐ |
|
|
|
|
☒ |
規模の小さい報告会社 |
||
|
|
|
|
|
|
新興成長型会社 |
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する☐
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する。
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に第240.10 D−1(B)条に従って受信されたインセンティブベースの補償に従って回復分析を行う必要があるかどうかを再選択マークで示す。 ☐
登録者が空殻会社であるか否かをチェックマークで示す(同法第12 b-2条で定義される)。はい
登録者の非関連会社が保有する議決権を有する普通株の総時価(当該株のナスダック資本市場における終値に基づいて、2023年6月30日、登録者が最近完成した第2四半期の最終営業日)は約$
2024年3月19日まで、登録者普通株の流通株数は、額面0.001ドル一株当たり、はい
引用で編入された書類
本報告第III部が要求する情報(本報告に記載されていない範囲内)は、本報告に関連する財政年度終了後120日以内に米国証券取引委員会に提出されなければならない2024年に開催される株主年次総会に関する最終委託書(“委託書”)を参照することにより登録者に提出される。
GYRE治療会社
表格10-Kの年報
カタログ
|
|
ページ |
第1部 |
||
第1項。 |
業務.業務 |
5 |
第1 A項。 |
リスク要因 |
62 |
項目1 B。 |
未解決従業員意見 |
123 |
プロジェクト1 C。 |
ネットワーク·セキュリティ |
123 |
第二項です。 |
属性 |
124 |
第三項です。 |
法律訴訟 |
124 |
第四項です。 |
炭鉱安全情報開示 |
124 |
|
||
第II部 |
||
五番目です。 |
登録者普通株市場、関連株主事項及び発行者による株式証券の購入 |
125 |
第六項です。 |
[保留されている] |
125 |
第七項。 |
経営陣の財務状況と経営成果の検討と分析 |
126 |
第七A項。 |
市場リスクの定量的·定性的開示について |
139 |
第八項です。 |
財務諸表と補足データ |
140 |
第九項です。 |
会計と財務情報開示の変更と相違 |
141 |
第9条。 |
制御とプログラム |
141 |
プロジェクト9 B。 |
その他の情報 |
141 |
プロジェクト9 Cです。 |
検査妨害に関する外国司法管区の開示 |
142 |
|
||
第三部 |
||
第10項。 |
役員·幹部と会社の管理 |
143 |
第十一項。 |
役員報酬 |
143 |
第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
143 |
十三項。 |
特定の関係や関連取引と取締役の独立性 |
143 |
14項です。 |
チーフ会計士費用とサービス |
143 |
|
||
第4部 |
||
第十五項。 |
展示と財務諸表明細書 |
144 |
第十六項。 |
表格10-Kの概要 |
145 |
|
|
|
展示品リスト |
146 |
|
|
|
|
サイン |
150 |
|
1
部分 I
前向きな陳述と市場データ
本年度報告における10-K表(“年次報告”)及び本文で引用された文書は、1933年証券法(“証券法”)第27 A条及び1934年“証券取引法”(“取引法”)第21 E条の意味に適合する前向き陳述を含む。歴史的事実に関する陳述を除いて、本年度報告に含まれるまたは引用された我々の戦略、将来の経営結果、将来の財務状況、将来の収入、予想コスト、見通し、計画、意図および管理目標、およびこれらの陳述に基づいている仮定に関するすべての陳述は前向きな陳述である。これらの展望的陳述は、これらの陳述に反映されたイベントまたは状況が達成または発生することを保証することができないので、未来のイベントの予測とみなされてはならない。前向き表現は、すべての前向き表現がこれらの識別語を含むわけではないが、“信じる”、“予想”、“可能”、“将”、“すべき”、“求める”、“意図”、“計画”、“形式”、“推定”または“予想”などの語、またはこれらの語およびフレーズの否定、またはこれらの語およびフレーズの他の変形または同様の用語を使用して識別される。このような展望性陳述は、管理職が現在把握している情報、管理職の経験及び歴史的傾向、現在の状況、未来の発展を期待する見方及び管理層が適切と思う他の要素に基づいて行った仮説と評価に基づいている。
未来の予想を議論し、将来の運営結果や財務状況の予測を含む、または他の“前向き”情報を述べているので、これらの声明をよく読むべきだ。これらの陳述は、私たちの計画、目標、期待、意図、および財政的表現、およびこれらの陳述が根拠とする仮定と関連がある。例えば、前向きな陳述は、以下の態様に関する任意の陳述を含む
2
3
どのような展望性陳述も未来の業績の保証ではなく、あるリスクと不確定要素の影響を受ける可能性があり、これらのリスクと不確定性は実際の結果とこのような展望性陳述中の予想或いは予想の結果とは大きく異なる可能性がある。このような差をもたらす可能性のある要因には、第1部1 A項に記載されたリスクを含む、本年度報告に記載されているリスクおよび不確定要因が含まれるが、これらに限定されないリスク要因“第2部第7項:”管理する’財務状況と経営成果に関する検討と分析本年度報告に記載されているリスクおよび不確実性は、第1部1 A項を含む、現在、どうでもよいと考えられているか、または予想されていない他のリスクおよび不確実性である可能性があるリスク要因“第2部第7項:”経営陣の財務状況と経営成果の検討と分析独占的ではなく、わが社やわが業務に関するさらなる情報は、当社の経営業績や財務状況に重大な影響を与える可能性のある要素を含めて、時々現れる可能性があります。すべての前向きな陳述は、私たちの経営陣の信念と仮定と、私たちの経営陣が現在把握している情報に基づいています。本年度報告書のすべての陳述と同様に、これらの陳述は締め切りのみを説明し、これらの陳述を更新または修正するために将来の発展を考慮する義務はない。我々の業務や財務パフォーマンスは、重大なリスクや不確定要因の影響を受けていることを投資家に想起させ、時々米国証券取引委員会(“米国証券取引委員会”)に提出された他の報告書や文書に記載されている要因を慎重に考慮すべきである。
本年度報告はまた、私たちの業界、私たちの業務とある薬品市場の推定、予測とその他の情報を含み、これらの市場の推定規模、予測成長率とある医療条件の発生率を含む。推定、予測、予測、または同様の方法に基づく情報は、不確定要因の影響を固有に受けており、実際のイベントまたは状況は、そのような情報が反映するイベントおよび状況とは大きく異なる可能性がある。他に明確な説明がない限り、私たちは、報告、研究調査、研究、およびサードパーティによって準備された類似データ、業界、医療および一般出版物、政府データ、および同様のソースから、これらの業界、商業、市場、および他のデータを取得する。いくつかの場合、私たちはこのようなデータの出所を明確に言及しなかった。この点で、私たちがどの段落でもこのようなデータの1つまたは複数のソースを言及した場合、他の平文規定または文脈に別の要求がない限り、同じ段落に出現するこのような他のデータは、同じソースからのものであると仮定すべきである。
4
プロジェクト1.BU無邪気ですね。
本節では、他の説明がない限り、言及された“私たち”、“私たち”、“私たち”および“私たちの会社”は、Gyre治療会社と私たちの多数の間接的に所有する子会社北京大陸医薬有限会社(d/b/a Gyre PharmPharmticals Co.,Ltd.)を指す。(“Gyre製薬”)。
要約.要約
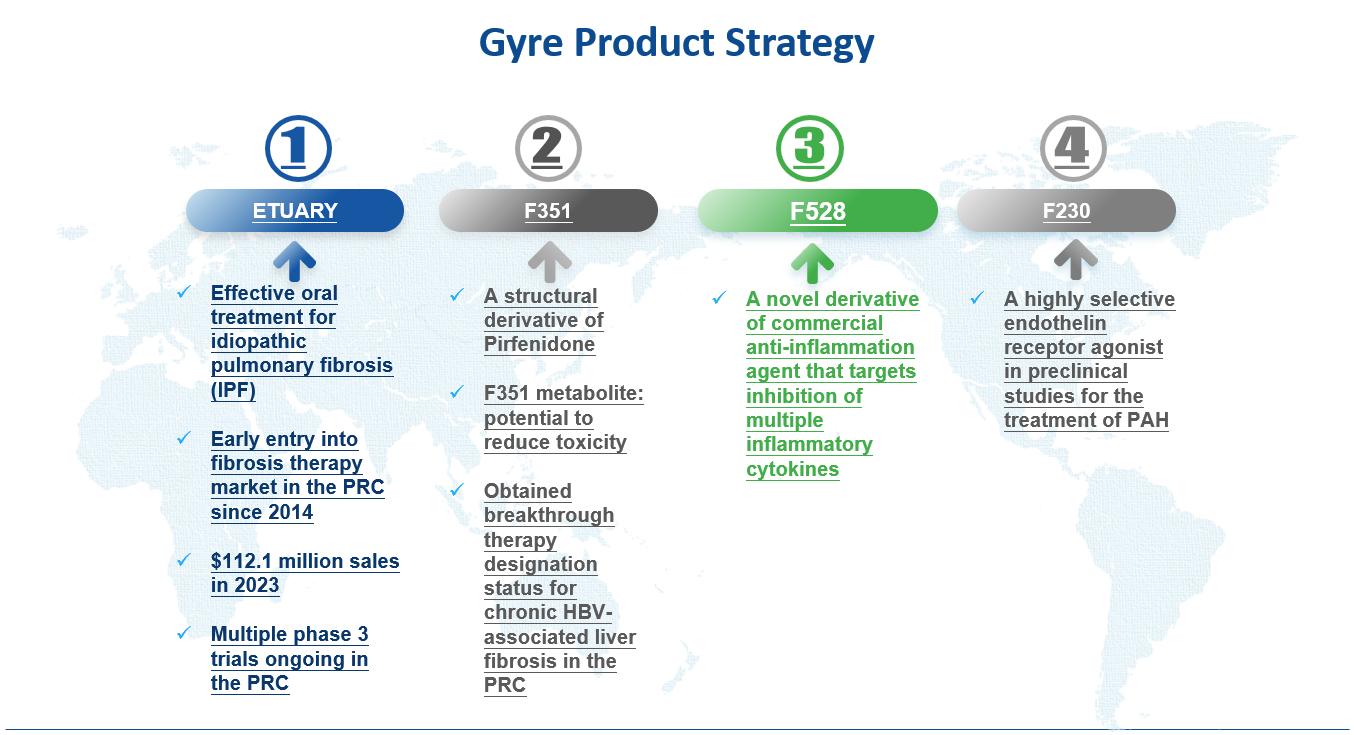
私たちは財務的に持続可能な製薬会社であり、財務成功の記録があり、臓器疾患に対する小分子抗炎症と抗繊維化薬物の開発と商業化、特に臓器繊維化に集中している。線維性疾患とは,満たされていない医療ニーズを多く有する患者である。繊維化は複雑で多段階の過程であり、複数の経路がある。多くの潜在的な抗繊維化治療標的があるが、すでに確立されているものもあれば、出現しているものもあり、単一の分子経路を解決することは繊維化を予防、阻止或いは逆転するのに十分ではないかもしれない。私たちの戦略はETUARYの開発成功と商業化の経験を利用することです®(ピルフェニドン)を新しい適応に拡張し、同様の候補薬剤を開発する。
ピルフェニドンは日本、EU、アメリカと中国が特発性肺繊維化に使用することを許可した第一種の抗繊維化薬物であり、1種の小分子薬物であり、腫瘍成長転化因子-1、腫瘍壊死因子-αと他の繊維化と炎症調節剤の合成を抑制できる。我々はすでに中国でIPF用ETUARY(ピルフェニドン)の承認を得た。
GYRE製薬会社はPirfenidoneの研究開発(“R&D”)から中国治療IPFの商業化への推進に成功した。ETUARYの年間売上高は毎年増加し続けており,2023年には1億121億ドルに達している。IPFを除くPirfenidoneは,間質性肺疾患に関連する結合組織病(“CTD−ILD”)の3つの研究を行っており,その適応と市場:SSC−ILD,DM−ILDと塵肺(“PD”)を拡大している。
F 351は,我々の主な開発候補であり,ETUARY(ピルフェニドン)の構造誘導体である。それは1種の新しい経口化学実体であり、抗繊維化、転化成長因子-1標的作用機序を有し、著者らは主要な市場で特許を持っている。研究により、F 351及びその主要な代謝物の薬物相互作用リスクは最も小さいことが分かった。IPFに潜在的な治療効果があるにもかかわらず、巨大な潜在市場と重大な満たされていない需要のため、私たちは肝線維化の治療にF 351を優先している。
5
GYRE製薬会社はすでにF 351が中国で慢性B型肝炎関連性肝繊維化を治療する第二段階試験を完成した。2期試験では,F 351の耐性は良好であり,明らかな毒性はなく,治療を受けた患者は統計的に有意な肝線維化改善を示し,270 mg/日の治療効果が最適であった。これらの結果から,中国では検証的な第3段階試験が行われている。検証性3期試験の248名の患者の募集は完了し,最後の患者は2024年に退院する予定であり,臨床結果は2025年初めに発表される予定である。
アメリカでは健康ボランティアにおけるF 351の第1段階臨床試験を完了しました私たちはIND申請を準備しており、2024年末に提出される予定だ。CHB関連肝線維化PRC第3段階試験の結果からINDの承認を待っており,2025年に2 a段階試験を開始し,F 351治療にNASHに関連する肝線維化を評価する予定である。

業務合併
2022年12月26日、Catalystと売り手が2022年12月26日に締結したいくつかの資産購入協定(“F 351合意”)に基づき、デラウェア州にあるCatalyst Biosciences,Inc.はGNI Group Ltd.(日本の法律登録に基づいて設立された有限責任会社(“GNI Japan”)およびGNI Hong Kong Limited(香港法に基づいて設立された有限責任会社(“GNI Hong Kong”,GNI Japan,“売り手”)買収(“F 351買収”)F 351資産とともに設立された。F 351プロトコルによると、Catalystは主に売り手独自F 351化合物に関連するすべての資産および知的財産権(総称して“F 351資産”と呼ぶ)を買収したが、中国に位置するこのような資産や知的財産権は除外されている。F 351プロトコルの条項およびF 351プロトコルによって予期される取引が発効したとき、Catalystは、以下の形態で売り手に35,000,000ドルを支払う:6,266,521 Catalyst普通株、1株当たり額面0.001ドル(“普通株”)、および12,340株のCatalyst X系列変換可能優先株、1株当たり0.001ドル(“転換可能優先株”)である。
同じく2022年12月26日、米国デラウェア州会社(“GNI USA”)、GNI日本、GNI香港、上海ゲノム学有限公司(中国の法律に基づいて設立された会社)、当該商業合併協定(改正された“商業合併協定”)付属書A内の個別者(個別に“少数株主”および総称して“少数株主”と呼ぶ)およびケイマン諸島株式会社(“CPI”)が締結した業務合併協議によると、他の事項を除いて、CATALYSTは、中国の法律に基づいて設立された北京大陸医薬有限公司の間接持株権を買収する(現在の業務名はGyre製薬有限会社)。(“BC”または“Gyre PharmPharmticals”)は、以下の取引に従って、(A)GNI USAは、CPI資本中のすべての普通株式を、1株当たり額面0.0001ドルをCatalyst(“CPI貢献”)に貢献し、(B)GNI USAは、法律に基づいて設立され、存在するさらなる挑戦者国際有限公司における権益に貢献する
6
(C)少数株主毎に、それが保有するそれぞれのエンティティ(業務合併プロトコルを参照することを定義する)の権益(消費者物価指数の供給および機能界別の供給、すなわち“供給”)を触媒に100%供給する。
Catalystは2023年10月27日、GNI USAと私募について証券購入協定(“証券購入協定”)(“私募”)を締結した。証券購入協定によると、GNI USAは合計8,110,000株単位(“単位”)を購入することに同意し、(I)811株交換可能優先株および(Ii)最大811株交換可能株の引受権証(“株式承認証”)に相当する。単位あたりの買い取り価格は0.6165ドル,総購入価格は約500万ドルであった.私募は2023年10月30日に終了します。この等株式承認証は即時に転換可能な優先株1株当たり4,915.00ドルの行使価格で行使することができるが、株式証条項に従って調整し、2033年10月30日に満期にしなければならない。この段落で言及した株式および1株当たりの金額は、逆株式分割を反映していない(以下の定義参照)。
有効時間は午前12時01分です米国東部時間2023年10月30日(“締め切り”)、Catalyst(I)は普通株の認可株式数を100,000,000株から400,000,000株に増加させ,(Ii)15株中1株の逆株式分割(“逆株分割”),(3)を“Gyre Treateutics,Inc.”と改称した
完了日にも、Gyreは、いくつかの業務合併協定に従って以前に公表された業務統合を完了し、逆株式分割を実施した後、(I)GNI USAは、CPI株式内のすべての普通株式を45,923,340株の一般株式と交換し、(Ii)GNI USAは、合計10,463,627株の普通株式と交換するために、FCにおける権益をGyreに与え、(Iii)各少数株主は、合計10,463,627株の普通株と交換するために、それぞれのエンティティに保有する100%の資本を提供する。出資直後には,F 351プロトコルで発行された交換可能株優先株および私募発行の交換可能優先株について転換すると仮定し,出資直前にCatalystの株主はGyre約2.5%の流通株,GNI USAはGyre約85.3%の流通株,少数株主はGyre約12.3%の流通株を持つと仮定した.
寄付が完了した後、登録者が展開する業務は主にGyreが展開し、Gyreは生物製薬会社であり、器官繊維化革新薬物の研究、開発、製造と商業化に取り組んでいる。
概要
私たちはより良い治療法臓器線維化患者の革新を通じて。私たちは私たちの使命と目標を達成するために以下の戦略を実施することを求めている
我々の中国における資源と専門知識を利用して、費用効果と効率性を持つ臨床前研究と早期臨床開発を通じて、迅速な概念検証を実現し、それを世界の他の場所に応用する
Gyre PharmPharmticalsは2002年に設立され,2023年に業務合併取引によりわが社の一部となった。中国ではIPFの開発と商業化治療に成功したETUARY(ピルフェニドンカプセル)により,強力な市場シェアを確立しており,かなりの市場シェアを占めている。ETUARYの成功はGyre PharmPharmticalsが各種の臨床試験を行う強力かつコスト効果のある研究開発資源、製造能力及び中国での広範な全国販売とマーケティングネットワークを含む全面的な製薬能力を発展させることができる。また、中国生物技術プロジェクトに対する政府の支持はGyre製薬会社が臨床前と臨床開発過程を有効かつ経済的に行う能力を増強した。Gyre PharmPharmticalsが中国の研究で獲得した知識や技術を利用して,臨床研究を行う基礎として,世界の他地域の臓器線維化において満たされていない医療ニーズを解決することが期待される。
繊維化疾患の治療における私たちの地位を確立し、製品の組み合わせを発展させ、適応の拡張を探索します
7
IPF治療のためのETUARYの中国での商業化に成功しており,ETUARYの使用を研究·開発していく予定であるはい。著者らの市場地位を確立する他の適応は、中国で行われているSSc-ILD、DM-ILDとPDを治療する第三段階の臨床試験を含む。著者らはまた、ETUARYによる糖尿病腎症(“DKD”)治療計画の第一段階臨床試験を成功した。
ETUARYの適応拡大に加え,われわれの臨床段階計画には,CHB関連肝線維化の治療のための主要候補品F 351が含まれており,Frost&Sullivanのデータによると,2022年の中国CHB関連肝線維化の罹患率は6360万患者,米国のNASH関連肝線維化の治療は,2022年1月から中国で患者を募集し,2023年10月に248名の患者の研究に全面的に組み込まれている。われわれはすでに米国でF 351の第1段階臨床試験を完了し,2024年末にINDを提出する予定である。我々の臨床段階パイプラインは、急性/急性慢性肝不全(“ALF/ACLF”)を治療するためのF 573も含む。著者らは2023年3月に中国でF 573の第二段階臨床試験を開始した。
著者らの臨床前段階製品ラインはCOPDを治療するためのF 528とPAHを治療するためのF 230を含む。著者らはF 528はCOPDに第一線の治療を提供し、そして長期の肺機能退化を減少できると考えている。2024年3月13日、私たちは中華人民共和国国家医療製品管理センターにF 230のIND申請を提出した。
学術宣伝をいっそう強化し,販売ネットワークを拡大する
学術普及を強化することは私たちの主要な販売方式の一つだ。著者らは学術組織と密接かつ長期的な協力を維持し、専門家の共通認識を促進し、国際と国内の学術会議に出席し、肝心なオピニオンリーダー(“KOL”)と密接にコミュニケーションし、著者らのブランド認知度を高めることに力を入れた。私たちはオンラインとオフラインの学術活動に積極的に参加し、学術会議を開催して、市場教育を促進し、私たちのブランド認知度を高め、私たちの製品の臨床使用を増加させる。
私たちはすでに中国で全面的な販売ネットワークを構築しており、候補製品が承認されれば、迅速に販売を実現することができる。著者らは著者らの販売チームに在職訓練を提供し、彼らに最新の研究と臨床実践を理解させ、そして著者らの製品の組み合わせの臨床利益を深く理解させた。製品販売の地域被覆面を拡大し,臨床需要を正確に位置づけ,市場浸透率を向上させるために,販売チームと資源を病院に配置し,中国のより多くの中小都市に触角を伸ばした。
付加価値業務開発と戦略連携による我々の製品組合せの慎重な構築
私たちの内部の研究開発努力と相補的なのは、私たちは買収、内部許可或いは協力を通じて、私たちの発展戦略と研究開発の原則に符合する製品と技術を導入し、引き続き業界の先端技術と製品発展と同期を維持することである。私たちは、ますます増加する業務発展ニーズを支援するために、敬業と経験豊富な業務開発チームを設立し、付加価値の機会を探したいと考えています。
私たちは将来性のある許可内や買収機会を能動的かつ慎重に探し、識別し、実行する。我々はGNI Japanからいくつかの候補薬物の知的財産権,特に中国以外のF 351の知的財産権を獲得している。私たちの医療と製薬ネットワークを利用して、私たちは引き続き国内と国際業界の先頭者と協力して、私たちのパイプライン構造を最適化し、私たちの製品の組み合わせの臨床と商業価値を最大限に高めます。
私たちは引き続き高品質の製品に集中しています。これらの製品は私たちの既存の製品ラインと相乗効果があります。例えば,我々の肝臓製品ラインを豊かにするために,慢性肝疾患患者の血小板減少症を治療する有望な製品が得られている。同時に、私たちの研究開発とビジネス経験を利用して、多発性硬化症のような他の疾患分野も探索しています。
生産能力の向上と生産コストの抑制のために私たちの施設を拡大·アップグレードする
製品ラインの研究開発を促進し、日々増加する市場需要を満たし、私たちの販売拡張計画を実現するために、著者らは北京順義に革新薬物研究開発と生産センターの建設を開始した
8
2022年6月。私たちは2024年までにさらに5億カプセルの生産能力を増加させ、私たちの販売と市場の私たちの製品と候補製品に対する需要に基づいて生産能力を拡大し続ける予定です。
また、私たちの製品の開発と商業化に伴い、私たちの生産能力を向上させたいと思います。合理的な価格安定と十分な高品質活性薬物成分(“原料薬”)の供給を確保するため、著者らは中国滄州に原料薬生産センターを設立し、技術アップグレードを通じて生産能力を拡大した。私たちはすでに今回のアップグレードを完成しました。滄州工場の年間生産能力は50トンの原料薬です。私たちの原料薬の生産能力は上流の原材料サプライヤーへの依存を減少させ、私たちのコスト管理を改善し、定価の優勢を提供してくれた。
高い素質の人材を絶えず吸引し、育成し、維持していく
利益と革新的な製薬会社として、私たちの製品ラインの特徴と私たちの発展需要に適応するために、簡素で才能のあるチームを維持することは私たちの成功の鍵である。人材を誘致し、維持するために、私たちは結束力と活力を持つ企業文化を持続的に発展させ、各従業員の訓練と発展を高度に重視している。
私たちの経験豊富な販売とマーケティングチームは私たちの持続的な成長に必須的だ。私たちの全国ネットワークを拡大し、中国の中小都市に浸透させるために、この分野の従業員を増やしていくことで、私たちの販売やマーケティング能力をさらに強化していきたいと思います。革新的な会社として、私たちは中国とアメリカの関連分野で経験豊富で熟練した生産技術者と研究開発人材を募集しています。また、私たちの戦略協力計画を調整し、実施するために、私たちの業務開発チームを設立する予定です。
私たちは次のような利点が私たちの戦略を実行できると信じています
長期的に臓器繊維化の治療に力を入れ、ETUARYは世界で初めてIPFの治療に許可された3種類の薬物の一つである
著者らのルートと2023年と2022年のETUARYはそれぞれ1.121億ドルと9920万ドルの収入であり、臓器繊維化治療において堅固な基礎を築いた。ETUARYは中国で初めてIPFを治療する薬物であり、世界で初めてIPFの治療に許可された3種類の薬物の一つでもある。
また,一連の候補薬物があり,肺,肝,腎を含む様々な臓器の線維化の治療に集中している。このうちF 351は慢性B型肝炎とNASH関連肝線維化の治療に開発されている。私たちはまた私たちの治療適応を様々な病気に拡大している。
F 351は中国初の慢性B型肝炎関連肝線維化の治療に承認された薬剤として開発され,米国ではNASH関連肝線維化治療の候補薬剤が開発されている。
肺繊維化疾患に対する最近20年間の集中研究と器官繊維化治療に蓄積した深い知識により、更に繊維化の発生は異なる器官で類似した発病機序を持っており、著者らはすでに肺繊維化から肝繊維化の治療に拡張し、未満足の臨床需要と絶えず増加する市場を満たす。著者らの革新小分子薬物F 351はすでに臨床試験において慢性B型肝炎関連性肝繊維化を治療する良好な効果を示した。その目的は肝星細胞の増殖を抑制し、同時に転化成長因子-1シグナル経路を遮断することによって肝繊維化を逆転することであり、この2つのシグナル経路は慢性B型肝炎関連性肝繊維化において重要な役割を果たしている。疾病の重症度とF 351が現有の治療方法と比較した臨床試験の進展のため、2021年3月、中国国家薬品監督管理局薬物評価センター(CDE)はF 351突破性治療称号を授与し、これは早期証拠により、この薬物が現有の治療方法より実質的に改善される可能性がある薬物の審査を加速することに役立つ。われわれは2022年1月から患者を募集してわれわれの3期臨床試験に参加した。2023年10月までに248科目の全面募集を完了し、2024年に最後の患者が退院し、2025年初めに中国でNMPA F 351の申請を提出する予定だ。
米国では2024年末にIND申請を提出する準備を積極的に行い,F 351によるNASH関連肝線維化の治療効果を評価する第二段階臨床試験を行っている。
9
専門的なマーケティングチームと全国に広がる販売ネットワーク
患者が希少であるため、孤児薬物を商業化するハードルは極めて高い。ETUARYの商業化の過程で、私たちは専門的な販売とマーケティングチームと広範な販売ネットワークを構築し、これはETUARYの強力な販売記録と主導的な市場地位を証明した。現在、私たちの販売ネットワークは中国の30の省、自治区、直轄市をカバーしている。私たちの販売ネットワークは著しく増加し、2022年と2023年にはそれぞれ2901軒と35512軒の病院と薬局をカバーした。
2023年12月31日まで、私たちの販売·マーケティングチームには391人の従業員がいて、平均9年間の仕事経験を持っています。その中で,我々が販売を担当する副総裁は25年を超える多国籍製薬会社と中国革新型製薬会社の経験を持ち,我々のコア地域マネージャーは平均17年間の業界経験と11年の管理経験を持っている。私たちの販売チームの他のメンバーの3分の1は国際製薬会社で働いており、4分の1は生物、医学、あるいは薬学学士以上の学位を持っている。私たちが統合した販売ネットワークと蓄積された管理と販売経験は、引き続き中国IPF市場の主導企業になり、新製品の発売時に迅速な販売を実現すると信じている。
大型内部製造施設と厳格な品質管理
我々の内部で原料薬や薬品を生産する能力は、サプライチェーンの厳格な制御を提供してくれ、コスト効果のある生産を維持し、予測できないサプライチェーン中断のリスクを低減することができる。私たちの2つの製造センター、製造能力、プロセスの詳細については、“を参照してください”財産-Gyre製薬会社の財産“と”-生産と品質管理-内部製造施設。”
経験豊富な上級管理チームは実行力が強い
私たちは経験豊富な管理チームを持ち、強力な実行能力を持ち、アメリカと中国で平均20年以上の業界経験を持っている。
知的財産権
私たちの成功は私たちが核心技術と知的財産権を保護する能力にある程度かかっている。私たちの知的財産権は私たちの業務に重要であり、私たちは米国と国際的に私たちの候補製品、新しい目標、適応、応用、および私たちの業務に重要な他の発明の特許保護を獲得し、維持することを含む様々な方法でそれを保護するために努力している。私たちの候補製品に対して、私たちは通常、物質成分、製造方法、使用方法をカバーする特許保護を追求しています。私たちが私たちの候補製品をさらに開発するにつれて、私たちは、新しい治療適応に対する権利主張を求めることを含む、ビジネス成功を向上させる可能性のある特許保護の新しい候補をより多く決定する予定です。私たちは製薬会社や他の業界参加者と協力協定や他の関係を締結して、私たちの知的財産権を利用したり、他人の知的財産権を獲得したりします。
特許
Gyreは,本年度報告日までに,世界で15件のライセンス特許,11件の中国での係属特許出願,および8件の特許協力条約(“PCT”)特許出願を有している。 本年度報告日まで、私たちは私たちの業務に大きな意味を持つすべての特許と特許出願の所有者です。
個別特許の期限は,特許が付与された司法管区の特許法律用語に依存する。ほとんどの法ドメインにおいて,発明の特許期間は,適用法ドメイン内の非仮特許出願の最初の要求の提出日から20年である。特許によって提供される実際の保護は、請求項および国によって異なり、特許のタイプ、そのカバー範囲、任意の特許期間の延長または調整の有無、ある特定の国に法的救済方法があるかどうか、および特許の有効性および実行可能性を含む多くの要因に依存する。
ETUARY(Pirfenidone)の物質特許は1993年8月に満期になったにもかかわらず、Gyreは現在2038年満期のプロセス特許と2024年満期のPirfenidoneに関する用量および投与特許を持っている。また、
10
GYREは、ヨーロッパ、日本、中国、カナダ、および米国で付与された特許を含む、Pirfenidone組成物を使用していくつかの細胞毒性または放射線による損傷(例えば、肺炎)を治療する一連の方法特許を有する。
我々のF 351特許組み合わせは、現在、異なる国およびPCTにおける特許および/または特許出願を含む6つの特許シリーズを含む。第1の特許シリーズは、その医薬組成物に関する方法であり、F 351を調製または使用して線維化を治療する方法であり、米国特許の1つが2024年9月22日に満了する。第2の特許シリーズは、中国における1つのライセンス特許が2028年に満了し、日本で許可された2つの特許を含むF 351の製造方法に関するものであり、両方の特許は2037年に満了し、中国における2つのPCT特許出願および6つの係属中特許出願を含む。第3の特許シリーズは、2041年に満了する中国で付与された特許からなるF 351結晶形態の製造方法に関するものである。第4の特許シリーズは、PCT特許出願および中国、カナダおよびオーストラリアでそれぞれ出願されている特許出願を含む、F 351を用いて慢性B型肝炎合併肝線維症を治療および/または予防する方法に関する。第5の特許シリーズは、中国で出願されている特許出願およびPCT特許出願を含む、肺線維症の治療または緩和または肺線維症症状の改善のための医薬組成物またはキットを製造することに関する。第6の特許シリーズは、PCT特許出願と、中国、オーストラリア、カナダ、ヨーロッパ、イスラエル、日本、韓国、メキシコ、ニュージーランド、シンガポールおよび米国でそれぞれ出願されている特許とを含む、F 351を用いた肝線維化、肝硬変、末期B型肝炎およびNASHの治療に関するF 351組成物および方法である。
F 573については、Gyreは中国で2つのライセンス特許を有しており、これら2つの特許はいずれも2031年に満了し、1つの中国での特許出願、1つのPCTの特許出願を有する。F 528の場合、Gyreは、中国、日本、ヨーロッパ、および米国でそれぞれ1つのPCT特許出願および1つの特許出願を有している。
他の適応を治療する方法、およびF 351および他の候補薬剤を製造するための新しい形態、処方、および方法をカバーするために、特許出願を引き続き提出する予定である。
商業秘密
場合によっては、私たちは、私たちの技術の様々な態様を保護するために、商業秘密および/または機密情報に依存する可能性がある。私たちは、コンサルタント、科学コンサルタント、請負業者と秘密保持協定を締結することで、私たちのノウハウやプロセスをある程度保護することを求めています。私たちは、私たちの上級管理職、研究開発チームの重要なメンバー、および私たちの業務のビジネス秘密や機密情報に触れることができる他の従業員と秘密協定やeスポーツ禁止協定を締結しました。
また、私たちは私たちのビルの実体安全と、私たちの情報科学技術システムの実体と電子安全を維持して、私たちの資料と商業秘密の完全性とセキュリティを維持することに力を入れています。これに関連するリスクの詳細については、参照のこと-リスク要因-私たちの知的財産権に関するリスク”.
商標とドメイン名
私たちは“大陸”や“大陸”のブランドで事業を展開している “と。2023年12月31日現在、我々は中国で4つの登録芸術品著作権、23件の登録ソフトウェア著作権、35の登録商標を持っている。我々は香港に7つの登録商標,1つの国際商標“ETUARY”,および“ETUARY”商標の米国,EU,日本など7カ国·地域における商標出願を有している。同じ日までに、私たちは28個のアクティブドメイン名を持っている。
“と。2023年12月31日現在、我々は中国で4つの登録芸術品著作権、23件の登録ソフトウェア著作権、35の登録商標を持っている。我々は香港に7つの登録商標,1つの国際商標“ETUARY”,および“ETUARY”商標の米国,EU,日本など7カ国·地域における商標出願を有している。同じ日までに、私たちは28個のアクティブドメイン名を持っている。
私たちのアメリカでの業務は
本節では,“私たち”,“私たちの会社”とはGyre治療会社を指す。
われわれの米国業務はカリフォルニア州サンディエゴに本部を置き,主にNASH関連肝線維化を治療するF 351(Hydronidone)の開発と商業化に焦点を当てている。F 351のNASH関連肝線維化における開発戦略はNASHげっ歯動物モデルで得られた機序研究結果およびGyre治療会社が中国でF 351による慢性B型肝炎誘導肝線維化の第2段階臨床試験を評価した結果に基づいており,安全性と有効性の主要な終点を達成し,ブレークスルーを得た
11
アメリカ国家薬品監督管理局が指定した治療方法。我々はすでに米国でF 351の第1段階臨床試験を完了し,2024年第4四半期にわれわれの第2段階臨床試験にINDを提出する予定である。
疾患概要−NASH
非アルコール性脂肪性肝炎(NASH)は深刻な非アルコール性脂肪性肝疾患(NAFLD)であり、代謝機能障害関連性脂肪変性肝疾患(MASLD)とも呼ばれ、肝臓中の余分な脂肪蓄積による一連の疾患であり、非アルコール摂取によるものであり、組織学的特徴は炎症と肝細胞損傷が付加的に存在し、もし見られる風船が膨張し、予後は明らかに悪く、肝繊維化、肝硬変或いは肝細胞癌(肝細胞癌)に進展する可能性がある。
ナッシュはアメリカと世界的に巨大で急速に増加する問題を代表している。確定診断例は上昇しており,今後10年で大幅な増加が予想される。NAFLDとNASHの流行は主に全世界の肥満流行によって推進され、NAFLDは全世界の約25%の人口に影響し、NASHは約20%から30%のNAFLD患者の中で発展する。そのため,ここ数十年来NASHの罹患率は著しく増加し,肥満,インスリン抵抗性,2型糖尿病の罹患率と類似している。これらの疾患の罹患率は,不健康な栄養習慣,例えば高フルクトース,ショ糖,飽和脂肪を食べる食事や長時間座って動かない行動により,さらに増加することが予想される。
NASHの発生と発展の重要な病理生理機序は透明脂質代謝能力の低下、インシュリン抵抗性の増加、肝細胞損傷と肝細胞損傷による肝繊維化を含む。NASH患者の肝臓脂肪の過剰蓄積は,主にカロリー摂取がエネルギー需要を超えているためである。健康な肝臓には5%未満の脂肪が含まれているが,NASHを有する人の肝臓には20%以上の脂肪が含まれている。この異常な肝臓脂肪によるNASHの進展は,NASHは肝臓壊死性炎症状態であり,瘢痕形成をきたす可能性があり,線維化とも呼ばれ,一部の人では肝硬変や肝不全−約20%から45%の患者が肝硬変に進展する可能性がある。肝硬変が非代償性肝硬変に進展する場合があり,恒久的肝障害をきたし,肝不全をきたす。また,進行線維化患者の8%が肝細胞癌に進展すると推定されている。NASHは複雑で多面的な疾患であり,肝臓だけでなく。NASH患者はよく他の重要な代謝合併症、例えば肥満、高血糖、血脂異常と全身性高血圧(一連の通常代謝症候群と呼ばれる)があり、これらは更に心血管疾患のリスクを増加させる。
NASHの病因学
NASHをきたす病理生理機序の解明は近年進展しているが,まだ不明である。過剰な熱過負荷,代謝失調,心臓代謝共通病や遺伝リスク因子はNASH発生の可能性を増加させ,多くの潜在的な機序因子が病理生理学に貢献している。NASHでは,肝臓処理は主にエネルギー基質炭水化物や脂肪酸を代謝する能力が重荷である。過剰な遊離脂肪酸が肝臓に沈着したり,肝臓からの処置が損なわれたりすると,このようなことが起こる。余分な遊離脂肪酸の蓄積は有毒脂質の形成を招く。これらの有毒な脂質はその後小胞体ストレス、酸化ストレスと炎症反応を誘導し、それによって肝細胞損傷と死亡を招く。これにより線維化やゲノムが不安定になる可能性があり,時間の経過とともに肝硬変と肝癌に悪化する可能性がある。NASHの発生と発展の重要な病理生理機序は,(1)脂質を処理する能力の低下,(2)インスリン抵抗性の増加,(3)肝細胞損傷,(4)肝細胞損傷による肝繊維化の発生と発展を含む。
診断学
NASH患者の多くは症状がなく,彼らの疾患は通常肝臓イメージングプログラム(例えば超音波)後に偶然発見され,これらのプログラムは他の理由で処方されているか,あるいは肝酵素上昇調査の一部として行われている。NASHが臨床的に疑われると肝生検でNASHを明確に診断する必要があり,NASHには脂肪変性,気腫,小葉炎を併用する必要がある。病理学的に確認されると、NAFLDおよびNASHの重症度は、組織学的に検証されたNASを用いて決定され、NASは、疾患活動性を0~8のスコアを行う。NASは、脂肪変性(0~3)、小葉炎(0~3)、肝細胞バルーン(0~2)の単一スコアの合計であるが、線維化スコアは含まれていない。線維化分期(F 0−F 4)はKleiner分類に依存する
12
(F 0=線維化なし;F 1=肝洞周囲または門脈周囲線維化(両者があるわけではない);F 2=肝洞周囲および門脈周囲線維化;F 3=橋性線維化;F 4=肝硬変)。
組織学的診断はNASHと線維化を評価するゴールドスタンダードのままである。しかし、肝生検は疼痛、出血とその他の発病率のリスク及び高いコストと関連していることから、このプログラムは一般患者のスクリーニングには適していない。さらに、組織学的診断は、1つの大きな異種器官の小片を評価することは、器官全体を代表しない可能性があり、読者間と読者内の可変性を含むスライドを読む時の顕著な可変性によって混同される。臨床リスクスコア,血清マーカー,イメージング技術などいくつかの非侵襲的ツールがNASH患者の評価にますます多く用いられている。線維スキャン−ASTスコア,Fibrosis−4指数,増強肝線維化スコア,振動制御瞬時弾性イメージングなどの非侵襲性テスト(NITS)が検証され,ますます使用されている。これらのNITSは晩期(F 3)線維化の検出に優れた陰性予測値と許容可能な陽性予測値を有し,臨床環境で使用されるようになってきている。また、核磁気共鳴プロトン密度脂肪分率測定の脂肪変性の減少とALT 17 U/Lの減少と肝臓生検の組織学的改善との間に相関があることがますます多くの証拠により示されている。彼らのガイドライン草案では、FDAはスポンサーが生化学的または非侵襲的画像バイオマーカーを識別することを奨励し、FDAがこれらのマーカーを決定し、同意すると、臨床試験において肝臓生検の代わりに患者の選択と治療効果評価に使用することができる。
これらのNITSの検証とその後の採用により将来のNASHの診断と治療率の増加が予想される。
私たちの臨床段階製品候補:f 351
F 351の概要
F 351は著者らの第一段階の臨床候補薬物であり、NASH関連性肝繊維化を治療する潜在力がある。2024年に米国でIND申請を提出し,NASH関連肝線維化治療の2 a期臨床試験を開始する予定である。
F 351は承認された抗線維化(肺線維化)薬物ETUARYの構造類似体であるが,第1段階代謝に関連する安全責任を潜在的に減少させるために修正された薬物化学が使用されている。F 351はSmad 7を介した形質転換成長因子β分解を通じて肝星細胞(“HSC”)の活性化を抑制し、そして繊維化関連遺伝子の発現を減少させ、それによって肝繊維化を治療する。形質転換成長因子は 組織中の線維形成の中枢性メディエーター。肝星細胞の活性化は肝繊維化進展過程中の中心イベントと考えられ、転化成長因子はその中の重要なメディエーターの一つである。
F 351は抑制できることが証明されています体外培養P 38γキナーゼ活性および形質転換成長因子-β1によって誘導されるHSC過剰合成コラーゲン。これは更に肝臓中のHSCsに対する抗増殖作用の支持を得た離体するF 351の抗線維化作用もいくつかの確立された研究で実証されている体内にある四塩化炭素(CCL)のような肝繊維化げっ歯動物モデル4)誘導マウス肝線維化モデル、DMN誘導ラット肝線維化モデル、HSA誘導ラット肝線維化モデルおよびNASH線維化(CCL)マウスモデル4+西部[高脂肪]食事)。NASHマウスモデルでは,F 351は線維化の重症度を有意に軽減し,ヒドロキシプロリン含有量と肝酵素(ALT),アスパラギン酸(AST)の著明な低下,肝臓脂肪変性の減少,およびいくつかの炎症性サイトカインレベルの低下(3−10 mg/kg/日),CCLのNASスコアの低下を含む肝組織の機能,生化学および組織病理学的属性の改善を示した4WD誘導線維化と細胞膨張NASHモデルは,15−50 mg/kg Bid(HEDs 144−480 mg)であり,ヒト曝露に関与している。したがって,肝線維化動物モデルにおけるF 351‘Sの分子作用機序の重要な属性はNASHに関連する様々な病因を含む肝線維化における治療効果の潜在力を支持している。
ステップ1
米国におけるF 351の臨床開発は、米国の健康ボランティアにおける単剤と多剤F 351の安全性、耐性およびPKを評価し、中国の健康ボランティアが獲得したデータに関連するデータを収集した完成した第1段階の臨床試験を含む。F 351の臨床試験は
13
INDの基礎は2016年に申請したF 351であり、抗繊維化薬物として、一連の慢性肝疾患と関連する肝繊維化に重点を置いている。
試験第1部でF 351 30 mgまたは120 mgを単回経口投与したところ,F 351は速やかに吸収し,線形PKパターンを呈し,平均消失半減期は5−6時間,M 3およびM 4代謝物は5−7時間であった。試験の第二部分でF 351 30 mgまたは120ミリグラムTIDを7日間繰り返し経口投与した後、F 351カプセルは迅速に吸収し、暴露モードと半減期は単剤F 351と類似した。TID 30 mgまたは120 mgを繰り返したところ,F 351,M 3およびM 4の適度な蓄積が認められた(1.5倍未満)。投与量正規化F 351 C最大値男性と女性のAUC値は類似していた。
全体的に、30 mgまたは120 mgを単回経口投与し、30 mgまたは120 mgのTIDを7日間繰り返し経口投与した場合、F 351の耐性は良好であった。この実験では,有害事象(AEs)による早期終了はなく,重篤な有害事象(SAEs)もなく,死亡報告もなかった。試験の第1の部分では、単剤投与後に報告される緊急治療副作用には、鼻溢音放出および分散、孤立、可逆的な実験室異常が含まれる。試験の第2部反復投与後に報告された緊急治療副作用は,頭痛(25.0%),便秘(16.7%),傾眠(12.5%)であった。腹部不快感と腹部膨満感も胃腸不良反応各1名の被験者と報告されている。試験薬物製品の安全実験室テストは全体的に臨床上の重大な変化がなく、いかなる重大な薬物による肝損傷があることを示す証拠がないこと、或いは試験薬物製品のバイタルサイン、心電パラメータ或いは体格検査の臨床上の重大な全体的な変化に起因することができる。
第二段階
NASH関連肝線維化におけるF 351の2 a期臨床開発を開始する予定である。提案された2 a期臨床試験の目標は,NASH関連肝線維化患者におけるF 351の早期概念検証(PoC)を得て,より包括的な2/3期臨床計画への拡張の基礎とすることである。この試験は新たなIND下で行われ,NASH患者群においてこの薬剤が持つ可能性のある独自のリスク−収益プロファイルを反映している。INDの申請と提案された2 a期臨床試験の開始を支援するために,米国で現在活発なINDで行われている試験で得られたすべての非臨床および臨床データ,およびこれまでに中国で完成したデータを交差引用する予定である。これらの非臨床·臨床研究で得られた結果は,この薬剤の現在の臨床リスク/利益プロファイルに関する十分な情報を提供し,提案されたF 351によるNASH関連肝線維化治療の2 a期臨床試験の安全な開始を可能にしていると考えられる。F 351の買収について、GNIはINDの所有権譲渡を開始した。
F 351計画に関する合意
F 351資産購入契約
2022年12月26日、GNI日本とGNI香港からF 351資産を買収した。F 351協定によると、吾等は主にGNI日本及びGNI香港の独自F 351化合物に関するすべての資産及び知的財産権を買収しているが、中国に位置する当該等資産及び知的財産権は除外している。F 351資産には、中国国外での15件の発行済みまたは未解決特許と特許出願が含まれており、最後の発行済み特許は2037年8月に満了する予定だ。F 351プロトコルの条項およびF 351プロトコルで予想される取引発効時間に基づいて、吾らはGNI JapanおよびGNI Hong Kongに35,000,000ドル:6,266,521株普通株および12,340株交換可能優先株を以下のような形で支払う。
競争
生物製薬業界の競争は激しく、迅速な革新と重大な技術進歩の影響を受けている。F 351と任意の未来候補製品の開発と商業成功に影響する重要な競争要素は治療効果、安全性と耐性、信頼性、投与の利便性、価格、模造薬の競争レベルと精算であると信じている。私たちの競争相手は多国籍製薬会社、専門バイオテクノロジー会社、大学、そして他の研究機関を含む。多くのバイオテクノロジーと製薬会社が私たちが対象としている同じ病気のための薬を開発またはマーケティングしている。規模が小さいか早い段階にある会社も重要な競争相手になる可能性があり、特に大手老舗会社との協力で手配されている。高値を考慮すると
14
NASHの発生に伴い,肝臓や心臓代謝疾患の治療を求める製品や療法の開発を求める会社数が増加する可能性がある。
F 351がNASH関連肝線維化の治療に承認されれば,将来の競争は5種類の主要な薬剤に由来する可能性もあり,ファニー系X受容体アゴニスト,線維芽細胞増殖因子21,甲状腺ホルモン受容体−βアゴニスト,グルカゴン様ペプチド1アゴニスト,PPARアゴニストの現在のNASH市場への進出を目指しているが,他の薬剤もある。Madrigal製薬会社のβ−甲状腺ホルモン受容体アゴニストResmetiromは,中末期肝線維化を有する成人非肝硬化性NASHの治療にFDAによって初めて承認された薬剤となり,食事や運動とともに使用可能であり,その他の候補薬剤はViking治療社のβ−甲状腺ホルモン受容体アゴニストVK 2809,NGMバイオ製薬社のFGF 19類似体Aldafermin,Sagimet Biosciences社の新規脂肪酸合成酵素(FASN)阻害剤Denifanstat,メルク社のFGFR 1 c/Kager抗体MK−3655;Akero治療社のF 21融合蛋白GFferminである。89 bio,Inc.のFGF 21融合タンパク質Pegozafermin;Galectin Treateutics Inc.のGalectin-3阻害剤Belapectin;Galmed製薬有限会社のコール酸とアラキドン酸の合成結合体Aramchol;Novo Nordisk A/SのGLP-1受容体アゴニストSemagluide;AltimmuneのGLP-1/GLP-1二重アゴニストPemviduide/ALT-801;Eli Lilly and CompanyのGIP/GLP-1二重受容体アゴニストTirzepatide;Invenva社のPPARα/Delta/Gamma Lanibanror 004 Ndisk Ndisvo-19 NdisckおよびBFKB 8488 A、Genentech由来のFGFR 1/KLBアゴニスト抗体;およびpegozafermin、89 io,Inc.からの特殊な工程による線維芽細胞増殖因子21のグリコシル化類似体。
製造と供給
NASH関連肝線維化2 a期臨床試験を支持するために必要なF 351活性医薬成分(“原料薬”)と薬品供給の製造は中国無錫広州市で完了した。原料薬と薬物製品は現在良好な製造規範(“cGMP”)レベルの品質を有し、ロット放出と安定性研究は適用する法規要求に符合する。
最近発表された“生物安全法”が、連邦機関が関連バイオテクノロジー会社のバイオテクノロジー設備またはサービスを使用するエンティティと調達契約を締結することを禁止していることを考慮して、私たちは、薬明生物会社(“薬明生物”)または私たちの他のメーカーが影響を受けないように、私たちのサプライチェーンを強化するためのいくつかの措置を講じた。薬明生物をめぐる状況を注意深く監視·評価し、米国ではF 351臨床試験のために異なるCDMOに変更するなど、様々な選択を考慮している。地政学的リスクを注視し、必要に応じて追加的な緩和措置やサプライチェーン冗長性を実施していきたい。リスク要因は“大規模な薬品製造は高度に厳しく複雑であり、私たちと私たちの第三者メーカーはこの過程で問題に直面する可能性がある”と見ている
アメリカの政府規制
その他の事項以外に、アメリカ連邦、州と地方各級及びEUを含む他の国と司法管轄区の政府当局は薬品の研究、開発、テスト、製品承認、製造、品質管理、製造変更、包装、貯蔵、記録、ラベル、販売促進、広告、販売、流通、マーケティングと輸出入などの方面に対して広範な監督管理を行った。私たちの現在の候補製品は薬品として規制される予定だ。アメリカと他の国と司法管轄区で監督管理許可を得る流れ、および商業化前後に適用される法規と法規及び他の監督管理機関の遵守状況は、私たちの製品の生産とマーケティング及び私たちの研究開発活動の重要な要素であり、大量の時間、人的資本と財力を必要とする。
薬品の審査と審査
アメリカでは、FDAと他の政府実体は連邦食品、薬物と化粧品法案(“FDCA”)及び公布された法規及び他の連邦と州法規に基づいて薬品を監督する。製品開発過程、承認過程、あるいは承認後のいつでもアメリカに適用される法律と法規の要求を守ることができなくて、私たちは様々な影響を受けるかもしれません
15
FDAの承認遅延または承認保留申請の拒否、承認撤回、臨床試験の遅延または一時停止、警告状および他のタイプの規制状の発行、製品リコール、製品差し押さえ、生産または流通の完全または部分的停止、禁止、罰金、民事罰金、政府契約の拒否またはキャンセル、連邦医療保健計画の排除、賠償、利益返還、FDA、米国司法省、州総検察長および/または他の機関の民事または刑事調査、虚偽クレーム法案訴訟および/または他の訴訟、および/または刑事起訴などの行政または司法制裁。
米国での新薬の販売·流通の承認を求める出願人は、通常、以下の義務を履行しなければならない
臨床前研究とIND
INDを提出するためには,組織は臨床試験のための実際の薬物物質と薬物製品のロットを提供しなければならない。このロットの薬物物質と薬物製品は放出前に分析特徴づけが必要であり,安定した状態に置かれている。提出時には、少なくとも1ヶ月の安定性データがINDに含まれなければならない。
前臨床研究では体外培養動物研究と、有害事象の可能性を評価し、場合によっては、治療使用の理由を確立する。臨床前研究の進行はGLP法規を含む連邦法規と要求の制約を受けている。その他の研究は実験室で製造された原料薬或いは原料薬の純度、安定性と物理形態、及び調合薬物或いは薬物製品の物理的性質、安定性と再現性を評価することを含む。INDスポンサーは,臨床前試験の結果を生産情報,分析データ,任意の利用可能な臨床データや文献,臨床研究計画などとともにFDAに提出し,INDの一部として提案された臨床研究が安全に行えることを証明しなければならない。いくつかの臨床前テスト、例えば長期毒性テスト、生殖不良事件と発ガン性の動物テストは、IND提出後も引き続き行われる可能性がある。INDはFDAが受領してから30日後に自動的に発効し,それ以前にFDAが提案された臨床試験に懸念や問題を提起しない限り,試験は棚上げにした。この場合,INDスポンサーやFDAは臨床試験開始前に未解決の問題を解決しなければならない。したがって,INDの提出はFDAが臨床試験の開始を許可しない可能性がある。
INDでの臨床試験開始後,FDAはこの試験を一時停止する可能性がある。臨床保留は,FDAがスポンサーに提案した臨床研究の延期や進行中の臨床研究の一時停止の命令である
16
調査します。一部の臨床保留はIND要求の一部の臨床仕事を遅延或いは一時停止することである。例えば、特定のプロトコルまたはプロトコルの一部が継続されることは許可されず、他のプロトコルはそうすることができる。臨床保留或いは一部の臨床保留を実施した後30日を超えない後、FDAはスポンサーに棚上げ根拠に関する書面解釈を提供する。臨床棚上げや一部の臨床放置を発表した後,FDAがスポンサー調査が継続可能であることを通知した後にのみ,調査を回復することができる。FDAは、スポンサーによって提供された情報に基づいて、上述した欠陥が修正されたかどうかを決定するか、またはFDAを満足させる、すなわち調査を継続することができる。
NDAを支持するヒト臨床研究
臨床試験は、GCP要求に従って合格した研究者の監督の下でヒト対象に研究製品を服用することを含み、すべての研究対象に任意の臨床試験に参加する前に書面でインフォームドコンセントを提供することを含む。臨床試験は書面による研究案に基づいて行い,その中で研究の目標,安全性をモニタリングするためのパラメータ,評価する有効性基準を詳細に説明した。INDの一部として,各臨床試験の案と任意の後続の案修正案をFDAに提出しなければならない。また,臨床試験に参加する各機関を代表する内部審査委員会は,その機関が任意の臨床試験の計画を開始する前にその計画を審査·承認しなければならず,この委員会は少なくとも年に1回継続的な審査と再承認を行わなければならない。他の事項以外にも,IRBは研究対象に提供される研究案とインフォームドコンセント情報を審査·承認しなければならない。IRBの運営はFDAの規定に適合しなければならない。
いくつかの臨床試験に関する情報は,そのClinicalTrials.govサイト上で公開伝播するために,特定の時間枠内でNIHに提出されなければならない。
人体臨床試験は通常3つの連続段階に分けて行われ、この3つの段階は重なる可能性があり、合併する可能性もある
第1段階:候補製品は、最初に健康なヒト対象または標的疾患または状態を有する患者に導入され、安全性、用量耐性、吸収、代謝、分布、排泄が試験され、可能であれば、その有効性の早期兆候が得られる。
第二段階:候補製品を限られた患者集団に使用して、可能な副作用および安全リスクを決定し、特定の標的疾患に対するこの製品の治療効果を初歩的に評価し、用量耐性および最適用量を決定する。
第三段階:良好に制御された臨床試験において、候補製品をより多くの患者集団に適用し、通常、地理的に分散された臨床試験場所であり、承認のために製品の有効性および安全性を統計的に評価するための十分なデータを生成し、製品の全体的なリスク-利益プロファイルを確立し、製品のラベルに十分な情報を提供する。
臨床試験結果を詳細に説明する進捗報告は少なくとも毎年FDAに提出しなければならず,深刻な有害事象が発生すればより頻繁に提出される。第1段階、第2段階、および第3段階の臨床試験は、任意の指定された時間内に成功しないか、または全く成功しない可能性がある。また、FDAやスポンサーは、研究対象が受け入れられない健康リスクに直面していることを発見することを含む、随時様々な理由で臨床試験を一時停止または終了することができる。同様に、臨床試験がIRBの要求に従って行われない場合、または薬物が患者の意外な深刻な傷害に関連している場合、IRBは、その機関またはその代表機関の臨床試験の承認を一時停止または終了することができる。FDAは通常、GCPおよび提出された臨床データの完全性を保証するために、後期臨床試験において1つまたは複数の臨床場所を検査する。
スポンサーは選択可能であるが,必要ではなく,IND下で海外臨床研究を行っている。米国では、ある外国の臨床研究がINDの下で行われる場合、放棄しない限り、すべてのIND要求を満たさなければならず、FDAの決定はいくつかの要求を放棄する可能性がある。海外の臨床研究がINDの下で行われていない場合、スポンサーは、独立した倫理委員会による審査および承認、および適切なプログラムを使用して被験者からインフォームドコンセントを得ることを含むGCPに従って行われることを含む、研究がINDまたは上場承認またはライセンス申請の支援として使用されるように、FDAが適用される法規要件に適合することを保証しなければならない
17
FDAが必要と判断すれば,現場検査で検討する。GCPは臨床研究の倫理とデータ完全性基準を含むことが要求される。
アメリカ食品医薬品局に秘密保持協定を提出します
成功に必要な臨床試験や他の要求,臨床前と臨床研究の結果,製品の化学,製造,制御,提案されたラベルなどに関する詳細な情報を仮定し,NDAの一部としてFDAに提出し,その医薬製品を1つまたは複数の適応の市場に使用することの承認を要請する。連邦法によると,臨床データを必要とする申請については,多くのNDAの提出には申請使用料が必要であり,現在は約2024年度の400万ドルであるが,承認されたNDAのスポンサーも年度計画費を納付する必要があり,現在は約2024年度の40万ドルである。これらの費用は毎年調整されます。
場合によっては、FDAは、小企業が審査に提出した最初のヒト薬物出願の申請料を免除する。小企業の定義は、付属会社の従業員を含む従業員数が500人未満の会社である。1つの商業エンティティが別の商業エンティティを制御するか、または第三者が2つのエンティティを制御するか、または制御する権利がある場合、関連企業は、第2の商業エンティティに関係する商業エンティティとして定義される。さらに、孤児として指定された処方薬製品を販売する申請は、治療薬のための珍しい疾患または疾患を指定する以外の指示が含まれない限り、処方薬使用料の制限を受けない。孤児医薬品法によれば、FDAは、米国で20万人未満の疾患または疾患を治療することを目的とした薬剤として指定することができ、または米国の売上高が開発および生産コストを回収するのに十分であるという合理的な期待がない。
FDAは、NDAを受信してから60日以内に予備審査を行い、FDAが提出書類を受信した74日目にスポンサーに通知して、申請が十分に完全であるかどうかを決定し、実質的な審査を行うことができる。FDAは秘密協定の申請を受け入れるのではなく、より多くの情報を提供することを要求するかもしれない。この場合、申請は追加情報と共に再提出されなければならない。再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。提出された申請が受け入れられると、FDAは深い実質的な審査を開始する。FDAはNDAの審査過程で具体的な業績目標を設定することに同意した。このような申請の多くは提出日から10ヶ月以内に審査され、多くの“優先審査”製品の申請は提出後6ヶ月以内に審査される。FDAは、最初の提出後にFDAによって発見された顕著な欠陥を解決するために、出願人によって提供された新しい情報を考慮して、または明確にするために、審査プロセスをさらに3ヶ月延長することができる。
秘密協定を承認する前に、FDAは通常、製品を生産する1つまたは複数の施設を検査する。FDAは、製造プロセスおよび施設がGMP要件に適合していることを決定し、要求された仕様の下で製品が一貫して生産されることを保証するのに十分でない限り、申請を承認しない。さらに、NDAを承認する前に、FDAは、GCPに適合することを確実にするために、通常、1つまたは複数の臨床場所を検査する。
FDAはまた、決定されたまたは疑われる任意の深刻なリスクを軽減するために、REMS計画の提出を要求する可能性がある。REMS計画は、制限された分配方法、患者登録、または他のリスク最小化ツールのような薬物ガイドライン、医師コミュニケーション計画、評価計画、および安全使用を確保する要素を含むことができる。
FDAは新薬の申請を諮問委員会に提出したり,なぜこのような推薦がないのかを説明するように求められている。通常,諮問委員会は臨床医や他の科学専門家を含む独立した専門家からなるグループであり,申請を審査,評価し,申請を承認すべきかどうか,どのような条件でアドバイスを提供すべきかを担当する。FDAは諮問委員会の提案に制限されていないが、それは決定を下す時にこれらの提案を慎重に考慮するだろう。
FDAの秘密保持協定に関する決定
FDAによるNDAの評価および付帯情報によると、製造施設の検査結果を含めて、FDAは承認状または完全な返信を発行する可能性がある。商品の商業マーケティングを承認し、特定の処方情報を提供する
18
兆候があります。完全な応答文は、一般に、提出文書の不足点を概説し、FDAが出願を再検討するために、多くの追加のテストまたは情報を必要とする可能性がある。これらの欠陥がNDAの再提出時にFDAによって満足的に解決された場合、FDAは承認書を発行するであろう。FDAは、含まれる情報タイプに応じて、そのような再提出された出願を2ヶ月または6ヶ月以内に検討することを約束している。この補足情報を提出しても、FDAは最終的にその申請が承認された規制基準を満たしていないと決定する可能性がある。
FDAが製品を承認する場合、製品の承認適応を制限する可能性があり、製品ラベルに禁忌症、警告または予防措置を含むことを要求し、承認後の薬物安全性をさらに評価するための承認後の研究を要求し、製品の商業化後に製品を監視するための試験および監視計画を要求するか、またはREMSを含む流通制限または他のリスク管理メカニズムを含む他の条件を適用することは、製品の潜在的な市場および収益性に大きな影響を与える可能性がある。承認後、FDAは上場後の研究あるいはモニタリング計画の結果に基づいて、製品のさらなるマーケティングを阻止または制限することを求める可能性がある。承認された製品のいくつかのタイプの変更、例えば、新しい適応の追加、製造変更、および追加のラベル宣言など、さらなるテスト要件およびFDAの審査および承認が必要です。
迅速チャネル指定、加速承認、優先審査、孤児薬指定、突破的治療計画
快速通路
FDAにはいくつかのプロジェクトがあり、ある基準に合った新薬の開発を促進することを目的としている。具体的には,新薬が重篤あるいは生命に危険な疾患を治療することを目的としており,このような疾患が満たされていない医療ニーズを解決する可能性があることが示されていれば,高速車線指定を受ける資格がある。高速チャネル指定は,製品と研究中の特定の適応の組合せに適している.新薬の臨床開発期間中,新薬のスポンサーは随時FDAにこの薬物を迅速追跡製品として指定することを要求することができる。高速チャネル指定の下で、FDAは、完全な出願を提出する前に、マーケティング申請の部分を検討することをスクロールすることができ、スポンサーが出願部分のスケジュールを提供した場合、FDAは、申請の部分を受け入れることに同意し、スケジュールが許容可能であると判断し、スポンサーは、申請の第1の部分を提出する際に任意の必要な使用料を支払うことができる。
優先審査
製品が重篤な疾患を治療するための医薬であり、承認された場合、市場で販売されている製品と比較して、疾患の治療、診断または予防の安全性または有効性の面で有意な改善を提供する場合、優先審査を受ける資格がある。FDAは、審査を促進するために、優先審査として指定された新薬出願を評価するために追加のリソースを使用しようと試みるだろう。
承認を加速する
研究中の製品は、深刻または生命を脅かす疾患の治療における安全性および有効性、および既存の治療方法よりも意味のある利点を提供する製品が加速的に承認される可能性があり、これは、十分かつ制御された臨床試験に基づいて、臨床的利益を合理的に予測する可能性のある代替終点に有効であるか、または不可逆的な発症率または死亡率への影響を早期かつ合理的に予測することができる中間臨床終点の効果に基づいて承認される可能性があることを意味する。承認の一つの条件として,FDAは承認を加速させた薬物のスポンサーに十分かつ良好に制御された上場後臨床試験を要求する可能性がある。2022年食品·薬物総合改革法案によれば、FDAは、承認前または承認が加速された製品の承認日後の特定の期間内に、このような研究を適宜要求する可能性がある。FDAはプログラムを加速する権限も増加しており,スポンサーが必要な上場後研究を行うことができなかった場合,あるいはそのような研究が予測の臨床的利益を検証できなかった場合には,承認下で承認された製品や適応の承認を撤回することができる。さらに、FDAは現在、販売促進材料の事前承認を承認を加速する条件とすることを要求しており、製品の商業発売時間に悪影響を及ぼす可能性がある。
19
孤児薬名
孤児医薬品法によれば、FDAは、米国では20万人未満の疾患または疾患に一般的に影響を与える疾患または疾患である稀な疾患または疾患を治療するための薬剤を孤児薬として指定することができる。秘密保持協定を提出する前に、指定孤児薬を申請しなければならない。FDAが孤児薬物指定を承認した後、FDAは、治療薬の識別およびその潜在的な孤児の使用を開示する。指定孤児薬物は、規制審査および承認過程においていかなる利点も伝達されず、規制審査および承認過程の持続時間を短縮することもない。孤児薬物の称号を有する製品がその後、孤児薬物称号を有する疾患に対するFDAの承認を得た場合、この製品は、孤児製品の排他性を得る権利があり、これは、FDAが限られた場合を除いて、同じ疾患のための他の同じ薬物の販売申請を7年以内に承認することができないことを意味する。これらの状況は、十分な数の薬剤を提供できないか、または薬物の新しい処方がより良い安全性または有効性を示すか、または患者のケアに大きく貢献することである。しかし,競合相手があらかじめ同じ薬物の同じ適応を承認していれば,この排他性は7年以内にその製品の承認を阻止する可能性もある。
孤児薬物の排他的範囲に対するFDAの解釈は変わるかもしれない。FDAの“孤児薬物法案”の長期解釈は,排他性はその薬物が実際に承認された孤児の適応に限られている。そのため,排他的な範囲は狭く,より広い“病気や状況”からの競争ではなく,同じ“使用や適応”からの競争から保護されてきた.2021年9月の事件ではCATALYST製薬会社はFDAを訴えたある連邦巡回裁判所は、孤児薬物の排他性が、薬剤がより狭い用途のためにのみ承認されているかどうかにかかわらず、孤児によって指定された疾患または疾患のすべての範囲をカバーすると判断するFDAの狭い解釈を棚上げした。この決定は、amifamprdine、Lambert-Eaton筋無力症候群(LEMS)の治療のための医薬に関する。FDAが本件後にどのように実施するかという決定により,排他的な薬物が得られることが制限される可能性がある。
突破的治療指定
一つの製品はまた画期的な治療法の称号を得る資格がある。画期的な治療法の指定は、1つまたは複数の他の薬剤と単独でまたは1つまたは複数の他の薬剤と組み合わせて重篤または生命に危険な疾患を治療することを目的としたFDAの潜在的新薬の審査を加速することを目的としており、“初歩的な臨床証拠は、薬物が1つまたは複数の臨床的重要な終点で既存の治療法よりも著しく改善されている可能性があることを示しており、例えば、臨床開発早期に観察された実質的な治療効果を示す”ことを目的としている。1つの薬物を画期的な治療法に指定することは,迅速チャネル計画下で提供されるのと同様の利点と,この製品開発計画に対するFDAの密な指導を提供している。迅速チャネル指定、優先審査、加速承認、および画期的な治療指定は、承認の基準を変更することはありませんが、開発または承認プロセスを加速させる可能性があります。
承認後に要求する
FDAによって生産または流通を許可された薬品はFDAの普遍的かつ持続的な監督管理を受けなければならず、その中には記録保存、定期報告、製品サンプリングと流通、広告と販売促進、および製品の不良反応の報告に関連する要求が含まれている。承認後、承認された製品の大多数の変更は、新たな適応または他のラベル宣言を追加するなど、FDAの事前審査および承認を経なければならない。どのような上場製品やそのような製品を製造する機関に対しても,継続的な年間使用料要件と,臨床データ補充応用に対する新たな出願料がある。
また,医薬品メーカーや他の承認された薬品の生産·流通に参加するエンティティは,FDAや州機関にその機関を登録し,FDAやこれらの州機関の定期的な抜き打ち検査を受け,cGMP要求を遵守することを保証しなければならない。製造プロセスの変更は厳しく規制されており,通常FDAが事前に承認して実施する必要がある。
FDAの規定はまた、cGMPとのいかなる偏差も調査·是正し、スポンサーやスポンサーが使用を決定する可能性のある任意の第三者メーカーに報告や文書要求を行うことを要求している。
20
そのため、メーカーはcGMPコンプライアンスを維持するために、生産と品質管理に時間、お金、労力をかけ続けなければならない。
承認された場合、規制要求や基準の遵守が維持されていない場合、または製品発売後に問題が発生した場合、FDAは承認を撤回する可能性がある。その後、製品に以前未知の問題が存在することを発見し、不良事件或いは意外な深刻性或いは頻度の製造技術問題、或いは法規要求を遵守できなかったことは、新しい安全情報を追加するために承認されたラベルの改訂を招く可能性がある;発売後の研究或いは臨床試験を実施して新しい安全リスクを評価する;あるいはREMS計画に従って流通或いはその他の制限を実施する。他の他の潜在的な結果には
FDAは市場に投入された製品のマーケティング、ラベル、広告、販売促進を厳格に規制している。承認の適応と承認のラベルの規定に基づいてのみ薬物を普及させることができる。FDAや他の機関はラベル外用途の普及を禁止する法律法規を積極的に実行しており,ラベル外用途の普及が不適切であることが発見された会社は重大な刑事·民事責任に直面している可能性がある。
そのほか、処方薬製品の流通は“処方薬販売法”(“PDMA”)の制約を受け、この法案は連邦一級の薬品と薬品サンプルの流通を規範化し、各州の薬品流通業者に対する登録と監督管理に最低基準を設定した。PDMA,州法ともに処方薬製品サンプルの配布を制限し,配布中の責任の確保を求めている。
Hatch-Waxman特許認証と30ヶ月の有効期限
1984年、1984年の“医薬品価格競争および特許期間回復法”(通称“ハッジ·ワックスマン改正案”)の採択に伴い、国会はFDAが法規のNDA条項に従って以前に承認した薬物と同じ模倣薬を承認することを許可した。模造薬の承認を得るためには,出願人は短い新薬申請(“ANDA”)を当該機関に提出しなければならない。このような申請を支持するために、イミテーション製薬メーカーは、以前に機密協定によって承認された薬物製品(参考上場薬物(RLD)と呼ばれる)に以前に行われた臨床前および臨床試験に依存することができる。しかし,これらの情報を参考にするためには,ANDA出願人は,FDAは実際に複製を主張するRLDの作用様式と同じであると結論しなければならない。具体的には,ANDAを承認するためには,FDAは後発薬が有効成分,投与経路,剤形,薬物強度においてRLDと同様であることを発見しなければならない。しかしながら、出願人は、投与経路、剤形または強度がRLDとは異なる薬剤として短い出願を提出するか、または固定組合せ医薬製品に異なる有効成分を含む薬剤であることをFDAに事前に承認するようにANDA適合性請願書を提出することができる(即複数の有効成分を含有する医薬品)。
同時に、FDAはこの模造製薬と革新薬が生物学的同等性を有することを確定しなければならない。この法規によると,後発薬の吸収速度と程度がRLDの吸収速度や程度と有意差がなければ,後発薬は生物的にRLDと同等である。ANDAが承認された後、FDAは、この模倣薬がRLDと治療上“等価”であることを示し、その出版物“承認された治療同等性評価を有する医薬製品”(“オレンジブック”とも呼ばれる)で承認された後発薬のための治療同等性格付けを指定している。医者や薬剤師はこの治療法は
21
同等性評価は後発薬がRLDを完全に代替できることを意味する。さらに、いくつかの州法および多くの医療保険計画の実施のため、FDA指定された治療同等性の評価は、通常、処方医または患者が知らない場合、またはその同意を得ない場合に、後発薬の代替をもたらす。
NDAまたはその付録が承認された後、NDAスポンサーは、各特許をFDAに列挙し、出願人の製品または製品の使用方法をカバーすることを要求しなければならない。NDAスポンサーにリストされたすべての特許はオレンジ色の本に発表されている。ANDA出願人がFDAに出願を提出する場合,出願人はオレンジマニュアルに記載されている参照製品の任意の特許をFDAに証明しなければならないが,ANDA出願人が承認を求めていない使用方法の特許は除外される。
具体的には、出願人は、各特許証明について:
新製品が承認された製品の上場特許又はそのような特許を侵害しないか、又は強制的に実行できない認証を第4項認証と呼ぶ。出願人が列挙された特許に異議を唱えていない場合、または特許使用方法の承認を求めていないことを示す場合、ANDA出願は、参照製品を必要とするすべての特許が満了するまで承認されないであろう。ANDA出願人が第4段落の認証をFDAに提供している場合、FDAがANDAの届出を受けると、出願人はまた、NDAおよび特許所有者に第4段落の認証の通知を送信しなければならない。そして、NDA及び特許所有者は、第4項の認証の通知に対して特許侵害訴訟を提起することができる。第4項の認証を受けてから45日以内に特許侵害訴訟を提起すると、特許満了、訴訟和解または侵害事件におけるANDA申請者に有利な裁決の30ヶ月前まで、FDAによるANDAの承認が自動的に阻止される。
以前のFDAによって承認された製品の処方または用途をNDAに従って修正することをFDAが承認する代替経路として、出願人は、FDCA第505(B)(2)条に従ってNDAを提出することができる。第505条(B)(2)条は、“ハッジ·ワックスマン改正案”の一部として制定されており、承認に必要な情報の少なくとも一部が出願人によって行われていないか、又は出願人のための研究からのものであり、出願人が参考になる権利を得ていない場合に機密協定を提出することを許可している。505(B)(2)出願人がFDAに依存する以前の安全および有効性発見が科学的および法的に適切であることを証明することができる場合、新製品のいくつかの臨床前研究または臨床試験の必要性を除去することができる。FDAはまた、以前に承認された参照薬剤との変化を支援するための臨床試験を含む追加の架橋研究または測定を企業に要求する可能性がある。次いで、FDAは、参照薬物のすべてまたは一部が承認されたラベル適応および505(B)(2)出願人が求めた任意の新しい適応のために新薬候補を承認することができる。
第505条(B)(2)の出願人が承認された製品の検討に依存する場合,出願人は,オレンジマニュアルに承認された製品として記載されている任意の特許をFDAに証明しなければならず,その程度はANDA出願人と同じである。したがって、505(B)(2)セキュリティ協定の承認は、オレンジブックに記載されている参照製品に関する任意の非特許専有権、例えば、新しい化学物質の承認を得る排他性が満了し、第4段落の認証およびその後の特許侵害訴訟の場合、より早い30ヶ月まで、訴訟または侵害事件において505(B)(2)条の出願人に有利な裁決まで、参照製品を要求するすべての特許が満了するまで放置することができる。
特許期限の回復と延長
ハッジ·ワックスマン改正案によると,新薬製品の特許は限られた特許期間延長を得る資格がある可能性がある。これらの修正案は,製品期間中に失われた特許期間を最長5年間の特許回復を可能にする
22
開発とFDA規制審査。承認の回復期は、通常、IND発効日とNDA提出日との間の時間の半分であり、NDA提出日と最終承認日との間の時間を加える。特許期間回復は特許の残存期間の延長には利用できず,製品承認日から合計14年を超える。承認された薬品に適用される特許は1つのみ延期する資格があり,延期出願は関連特許が満了する前に提出しなければならない。米国特許商標局は、FDAと協議した後、任意の特許期限の延長または回復の出願を審査および承認する。
臨床試験情報の開示
FDA規制製品の臨床試験スポンサーはいくつかの臨床試験情報を登録し、開示しなければならない。そして,登録の一部として,製品,患者群,調査段階,試験地点と調査者および臨床試験の他に関する情報が公開されている。スポンサーも完成後に彼らの臨床試験結果を検討する義務がある。場合によっては、これらの裁判結果の開示は、裁判が完了した日から最大2年に延期されることができる。競争相手はこれらの公開された情報を用いて開発計画の進捗状況を知ることができる.
他のアメリカの医療保険法やコンプライアンスの要求は
アメリカでは、FDA以外に、私たちの活動は、医療保険や医療補助サービスセンター(CMS)、アメリカ衛生·公衆サービス部を含むが、これらに限定されない様々な連邦、州、地方当局によって規制される可能性がある例えば:監察長室、米司法省(“司法省”)、司法省内の各連邦検事室、州と地方政府。例えば、販売、マーケティング、科学/教育補助計画は、“社会保障法”、“虚偽請求法”、“健康保険携行性及び責任法案”(HIPAA)のプライバシー条項、改正された類似州法の反詐欺及び乱用条項に適合しなければならない。
他の事項に加えて、連邦反リベート法規は、任意の個人またはエンティティが、購入、レンタル、注文または購入の手配、レンタルまたは購入の手配、レンタルまたは注文として、Medicare、Medicaid、または他の連邦医療計画に従って精算可能な任意の物品またはサービスの見返りとして、故意に、または故意に現金または実物で直接または間接的に、公開または隠蔽的に提供、支払い、請求または任意の報酬を受けることを禁止する。報酬という単語は価値のあるものを含むと広く解釈されている。逆リベート法規は、医薬品メーカーと処方者、購入者、および処方管理人との間の手配に適用されると解釈される。法的な例外と規制された避難所があり、いくつかの一般的な活動を起訴されないように保護する。例外や安全港の範囲は狭く,処方,購入または推奨の報酬を誘導するために告発される可能性があるやり方に関連しており,例外や安全港の資格を満たさなければ審査される可能性がある。特定の適用された法定例外や安全港を規制するすべての要求を満たしていないことは、このような行為自体が“反リベート条例”に規定された不法行為であることを意味するわけではない。代わりに、そのすべての事実と状況の累積審査に基づいて、この手配の合法性を逐案的に評価する。私たちの接近はすべての場合、法定例外や規制避難港保護のすべての基準を満たしていないかもしれない。
また、“平価医療法案”は“反リベート法規”下の意図標準を改正し、更に厳格な標準にし、個人或いは実体がこの法規或いはこの法規に違反する具体的な意図を実際に理解する必要がなく、違反を実施することができる。また,ACAは連邦虚偽申告法(以下議論)に基づき,連邦反リベート法規違反による物品やサービスのクレームを含めて虚偽や詐欺的クレームを構成する判例法を編纂している。
民事罰金法規は、任意の個人またはエンティティに処罰を加え、その個人またはエンティティは、連邦健康計画へのクレームを出したか、または結果として判断され、そのクレームがクレームに従って提供されていないプロジェクトまたはサービス、または虚偽または詐欺的であることを知っているか、または知るべきである。
他の事項に加えて、連邦虚偽申告法は、連邦政府の支払いまたは承認を要求するために、または虚偽または詐欺的クレームに関連する虚偽記録または陳述の作成、使用、または作成または使用を引き起こすために、任意の個人または実体が連邦政府に意図的に提出するか、または虚偽請求の提出を引き起こすことを禁止する。2009年の“詐欺法執行·追跡法”の改正により、クレームには
23
米国政府に提出された金銭や財産に対する“任意の請求または要求”。最近,いくつかの製薬や他のヘルスケア会社がこれらの法律により起訴されており,顧客に無料製品を提供し,顧客がその製品の連邦計画に課金することを期待しているといわれている。他の会社も起訴され、これらの会社はこの製品を未承認のため、精算できない用途にマーケティングし、虚偽の声明を提出することになった。HIPAAは、計画を故意かつ故意に実行または実行しようとすることを禁止し、虚偽または詐欺的な言い訳、陳述または承諾の方法で任意の医療福祉計画(個人第三者支払者を含む)が所有または制御または保管している任意の金銭または財産を詐欺または取得し、悪巧み、計画または装置、重大な事実、または任意の重大な虚偽、架空または詐欺的陳述によって、医療福祉、プロジェクトまたはサービスの提供または支払いに関連する情報を故意に偽造、隠蔽または隠蔽する新しい連邦刑法を制定する。
また、多くの州で同様の詐欺や法律や法規の乱用があり、医療補助や他の州が計画して精算するプロジェクトやサービスに適用されたり、いくつかの州では支払者にかかわらず適用されている。
私たちは連邦政府と私たちが業務を展開している州のデータプライバシーと安全規制によって制限されるかもしれない。HIPAAは“経済と臨床健康を促進する健康情報技術法”(“HITECH”)及びその実施条例の改正により、個人が健康情報を識別できるプライバシー、安全と伝送に対して要求を提出した。他の事項に加えて、HITECHは、保護された健康情報を受信または取得するために、カバーエンティティに代わってサービスを提供する商業パートナー、独立請負業者、またはエージェントに、HIPAAのプライバシーおよびセキュリティ基準を直接適用させる。HITECHはまた4つの新しい民事罰金等級を作成し、HIPAAを改訂し、民事と刑事処罰を商業パートナーに直接適用し、州総検察長に新しい権力を与え、連邦裁判所に民事訴訟を提起し、損害賠償または禁止令を要求して連邦HIPAA法を執行し、連邦民事訴訟に関連する弁護士費と費用を求めることができる。また,州法は特定の場合に健康情報のプライバシーやセキュリティを管理しており,その多くの法律は互いに大きく異なり,同様の効果が生じず,コンプライアンス作業を複雑にする可能性がある。
米国には、2020年にカリフォルニアプライバシー権法案(CPRA)によって改正された2018年カリフォルニア消費者プライバシー法(CCPA)など、いくつかの州プライバシー法があり、個人情報のプライバシーやセキュリティを管理する場合もある。CCPA/CPRAは、消費者、企業代表、および従業員の個人データに適用され、プライバシー通知において特定の開示を提供すること、およびカリフォルニア住民にその個人情報を提供する権利を含むカリフォルニアでビジネスをしているいくつかの企業に義務を課す。健康情報は、CCPA/CPRAの個人情報の定義に属し、この場合、特定の消費者または家庭に関連することができるか、またはHIPAAによって制約されない限り、新しい個人情報カテゴリである“敏感な個人情報”に識別、記載、または合理的に関連することができ、より大きく保護されている。いくつかの法律·法規は異なる要求を加えており、場合によってはHIPAAよりも要求が厳しい場合がある。これらの法律および法規を遵守しないことは、重大な民事および/または刑事罰、および個人訴訟に直面する可能性があり、これらのすべては、財務的および名声のリスクを招く可能性がある。
さらに、ACAの範囲内の連邦医師は、Medicare、Medicaidまたは児童健康保険計画(いくつかの例外)に従って支払うことができるいくつかの薬品、器具、生物および医療用品メーカーに、様々な医療専門家(医師、医師アシスタント、看護師従事者、臨床看護師専門家、登録看護師麻酔科医、登録助産師および教育病院を含む)へのいくつかの支払いまたは他の価値移転に関する情報を報告するか、または以下の要求またはその名義で指定された実体または個人に報告しなければならない。医者と教育病院は、毎年医者及び直系親族が持っているいくつかの所有権と投資権益を報告する。
製品を商業的に流通させるためには、州の法律を遵守し、一部の州で製品をその州のメーカーおよびディーラーに輸送することを含む州の薬品製造業者および卸売業者の登録を要求しなければならない。一部の州はまた、メーカーと流通業者が流通チェーン中で製品の系統を確立することを要求しており、いくつかの州はメーカーと他の州に流通チェーン中の製品の流れを追跡し追跡できる新しい技術を採用することを要求している。いくつかの州はすでに立法を公布し、製薬とバイオテクノロジー会社にマーケティングコンプライアンス計画を構築することを要求している
24
国は、販売、マーケティング、定価、臨床試験および他の活動および/またはその販売代表を定期的に公開し、および/またはその販売代表を登録し、薬局および他の医療保健エンティティが製薬および生物技術会社にいくつかの医師処方データを販売およびマーケティングのために提供することを禁止し、いくつかの他の販売およびマーケティング行為を禁止することを規定している。私たちのすべての活動は連邦と州消費者保護と不正競争法によって制限されるかもしれない。
もし私たちの運営が上記のいずれかの連邦および州医療保健法律または私たちに適用される任意の他の政府法規に違反していることが発見された場合、私たちは、MedicareとMedicaid、禁止、個人通報者が政府の名義で提起した個人訴訟、または政府契約、契約損害、名声損害、名声損害、行政負担、利益減少と将来の収益、ならびに私たちの業務の削減または再編を含む、民事、刑事および/または行政処罰、損害賠償、罰金、返還、MedicareおよびMedicaid、禁止、個人通報者が政府名義で提起された個人訴訟を含むが、処罰を受ける可能性がある。いずれも私たちの業務運営能力と運営結果に悪影響を及ぼす可能性があります。
保証範囲·定価·精算
私たちが規制部門の承認を得た任意の候補製品のカバー範囲と精算状態には、重大な不確実性がある。米国や他の国·地域の市場では、規制部門の承認を得て商業販売を行う任意の製品の販売は、第三者支払者がそのような製品に保険を提供する程度に依存し、十分な補償レベルを確立することになる。米国では,第三者支払者には連邦や州医療計画,個人管理のヘルスケア提供者,医療保険会社,その他の組織が含まれている。第三者支払者が製品に保険を提供するかどうかを決定するプロセスは、製品価格を決定するプロセス、または第三者支払者が製品のために支払うべき支払率を決定するプロセスから分離することができる。第三者支払者は、承認リスト上の特定の製品に保証範囲を制限することができ、処方表とも呼ばれ、FDAによって承認された特定の適応のすべての製品を含まない可能性がある。第三者決済者は、価格に挑戦し、医療の必要性を審査し、医療製品、療法、サービスの費用対効果を審査するとともに、それらの安全性と有効性を疑問視するようになっている。私たちは、私たちの製品の医療の必要性と費用効果、FDAの承認を得るのに必要なコストを証明するために、高価な薬物経済学的研究を行う必要があるかもしれない。私たちの候補製品は医学的に必要で費用効果があると思われないかもしれない。支払者が製品に保険を提供することを決定することは、十分な返済率を承認することを意味するものではない。また、支払者が1つの商品に保険を提供することを決定し、他の支払者もその商品に保険を提供することを保証することはできない。連邦医療保険精算も同様であり、異なるサプライヤーが支払いを処理するため、1つのサプライヤーの保険は他のすべてのサプライヤーが保険を提供することを保証することはできない。製品開発への投資の適切な見返りを実現するために、十分な価格レベルを維持することができる十分な第三者精算がないかもしれない。また、アメリカ連邦政府の薬品定価関連問題における立場は変化しており、不確定であり、いかなる変化もアメリカの薬品定価に実質的な影響を与える可能性があり、承認されれば、私たちの候補製品に実質的な影響を与える可能性もある。最近、米国政府はインフレ低減法案を可決し、米国衛生·公衆サービス部と連邦医療保健計画に参加したメーカーとある薬品の価格交渉を許可した。
他の国もまた違う価格設定と精算プログラムを持っている。EUでは,各国政府はその定価と精算規則及び国家医療システムの制御により医薬製品の価格に影響を与え,これらのシステムは消費者にこれらの製品の大部分のコストを支払っている。いくつかの法域はプラスリストとネガティブリスト制度を実行し、補償価格を合意した後にのみ、製品を販売することができる。精算または定価の承認を得るために、その中のいくつかの国は臨床試験の完成を要求する可能性があり、特定の候補製品のコスト効果を現在利用可能な治療法と比較する。他の会員国たちは会社が自分の薬品価格を固定することを許可するが、会社の利益を監視する。イギリスの国家健康·看護卓越研究所(NICE)も費用便益分析を考慮する必要がある。医療コストの下振れ圧力は非常に大きくなっている。そのため、新製品の参入にはますます高い壁が設けられている。また、一部の国では、低価格市場からの国境を越えた輸入が一国国内の定価に商業圧力をかけている。
政府や第三者支払者が十分な保険や補償を提供できない場合、規制部門の承認を得て商業販売を行う任意の候補製品の適正性が影響を受ける可能性がある。また,米国では管理型医療への重視度が増加しており,医療定価の圧力が増加し続けることが予想される。保証政策と第三者精算料率は随時変化する可能性がある。有利な被覆面であっても
25
規制部門の承認を得た1つ以上の製品が精算資格を取得すれば、将来的にはあまり優遇されない保証政策や精算料率が実施される可能性がある。
“反海外腐敗法”
反海外腐敗法(FCPA)は、いかなる米国人または企業が、いかなる外国人官僚、政党または候補者にも直接的または間接的に支払い、提供または許可支払い、または任意の価値のあるものを提供することを禁止し、その個人または企業が業務を獲得または保持することを支援するために、外国の実体の任意の行為または決定に影響を与えることを目的としている。“海外腐敗防止法”はまた、証券が米国に上場している会社に会計規定を遵守し、会社(国際子会社を含む)のすべての取引の帳簿や記録を正確かつ公平に反映し、国際業務のために適切な内部会計制御制度を制定·維持することを要求している。
法規を付加する
これらの規定に加えて,環境保全や有害物質に関する州や連邦法は,“職業安全と健康法”,“資源節約と回収法”,“有毒物質制御法”を含めて,我々の業務に影響を与える。これらの法律と他の法律は私たちの行動で様々な生物、化学、放射性物質、それによって生成された廃棄物を使用、処理、処分することを規定している。もし私たちの運営が環境汚染を招いたり、個人を危険物質に曝露させたりすれば、損害賠償と政府罰金の責任を負う可能性があります。私たちは、私たちが適用される環境法律を実質的に遵守し、これらの法律を遵守し続けることは、私たちの業務に実質的な悪影響を与えないと信じている。しかし、私たちはこのような法律の変化が私たちの未来の運営にどのように影響するか予測できない。
アメリカ以外の政府規制
アメリカの法規のほかに、臨床試験と私たちの製品の任意の商業販売と流通を含む他の管轄区の様々な法規の制約を受けます。FDAによる製品の承認を得るか否かにかかわらず、これらの国での臨床試験や販売を開始する前に、外国の規制機関の必要な承認を得なければならない。米国以外のある国にも類似したプログラムがあり,ヒト臨床試験開始前に臨床試験申請を提出することが求められており,INDに似ている。例えば,EUでは,臨床試験申請は各国の国家衛生当局と独立した倫理委員会に提出されなければならず,FDAやIRBのようなものである。臨床試験申請が国の要求に応じて承認されると,臨床試験開発が可能となる。生物由来の原材料は独特の汚染リスクに直面しているため、それらの使用はいくつかの国で制限される可能性がある。
臨床試験、製品許可、定価と精算を指導する要求と手続きは国によって異なる。いずれの場合も,臨床試験はGCPおよび“ヘルシンキ宣言”からの適用法規要求と倫理原則に基づいて行われた。
EUの監督管理制度によると、研究薬物の監督管理承認を得るためには、上場許可申請を提出しなければならない。
EU以外の他の国,例えば東欧,ラテンアメリカ,アジアの国では,臨床試験,製品許可,定価,精算を行う要求は国によって異なる。繰り返しますが,すべての場合,臨床試験はGCPおよび“ヘルシンキ宣言”からの適用法規要求と倫理原則に基づいて行われています。
もし私たちまたは私たちの潜在的な協力者が適用された外国の規制要求を遵守できなかった場合、私たちは罰金、規制の一時停止または撤回、製品のリコール、製品の差し押さえ、運営制限、刑事起訴などに直面する可能性がある。
従業員 アメリカでは
26
私たちは私たちが職員たちを募集し、維持し、奨励する能力が私たちの成功に必須的だと思う。私たちは機会均等な雇用主であり、従業員を尊重し尊重する労働環境の創出と維持に根本的に取り組んでいる。私たちは、個人の技能、知識、能力、仕事の表現、および他の合法的な基準に基づいて、人種、肌色、宗教、性別、性指向、性別表現またはアイデンティティ、民族系、民族血統、血統、年齢、知的または身体障害、遺伝情報、任意の退役軍人身分、任意の軍事身分または兵役申請、または法的保護を適用する任意の他のカテゴリのメンバーの身分を考慮せずに、雇用、昇進、補償、福祉および解雇に関連するすべての人的資源政策、やり方、および行動を管理するために、平等な雇用機会の原則に従うように努力している。
2023年12月31日現在、私たちはアメリカに4人の常勤従業員がいます。私たちは従業員と集団交渉をしていませんし、何の休職も経験していません。私たちは私たちが従業員と仲がいいと思う。
従業員に競争力のある報酬と福祉を提供し、良好な生活の質を実現し、未来を計画することを目標としています。私たちの福祉は、地域規範および市場選好に基づいているが、現地の法律要件を含むすべての賃金および社会福祉(有給休暇および病気休暇を含む)、および法律の要求を超える多くの追加的な福祉に基づいている。
アメリカの商業団体は
私たちは2002年に運営を始めましたデラウェア州の会社です2015年8月20日、私たちはTargacept,Inc.と合併し、2023年10月30日に寄付を完了し、Gyre治療会社になりました。私たちの会社はカリフォルニア州サンディエゴに本社を置いています。私たちは主にカリフォルニア州サンディエゴで私たちの研究開発活動と一般と行政機能を行っています。
利用可能な情報
我々のForm 10-K年次報告、Form 10-Q四半期報告、Form 8-K現在の報告、およびこれらの報告の改訂は、米国証券取引委員会に電子的に提出または提出した後、合理的に実行可能な場合にできるだけ早くwww.gyretx.comで無料で取得することができる。これらはアメリカ証券取引委員会のサイトwww.sec.govで無料で入手することもできます。米国証券取引委員会及び当サイトにおける情報又は当サイトを通じて取得された情報は、本出願に組み込まれることもなく、本出願の一部ともみなされない。
私たちの中国での業務:GYRE製薬
本節では、“私たち”、“私たち”、“私たち”と“わが社”は北京大陸医薬有限会社(d/b/a Gyre PharmPharmticals Co.,Ltd.)を指す。(“Gyre製薬”)。
GYRE製薬会社は2002年に設立され、中国で臓器繊維化疾患の治療に取り組む革新型薬物開発企業であり、研究開発、生産と商業化を一体にしている。Gyre製薬会社はGyre治療会社の間接持株子会社である。
私たちの中国での業務概要
著者らは商業段階の生物製薬会社であり、器官繊維化革新薬物の研究、開発、製造と商業化に取り組んでいる。著者らは最初にIPFの治療に集中し、次第に著者らの治療領域と研究開発努力を他の器官繊維化領域に拡大した。我々の旗艦製品ETUARYは2011年に中国で承認され、世界で初めてIPFに使用される3種類の薬物の一つである。その後、著者らは他の革新候補薬物F 351、F 528、F 230とF 573のパイプラインを開発し、ETUARYを10年間商業化することに成功した。
中国で初めて承認された特発性肺不全治療薬として、ETUARYは2017年から中国国家精算薬品目録(NRDL)に登録された。最初に承認されたIPF治療法として、ETUARYは中国の空白を埋め、急速に発展し、中国で主導的な市場シェアを維持した。2022年、中国IPF治療の総市場規模は1兆274億ドルと推定され、2031年には6.986億ドルに増加すると予想される
27
フロストとサリヴァンです。そのほか、異なる器官繊維化疾患の発病機序と繊維化過程が類似しているため、著者らは他の肺繊維化疾患、例えばILDと塵肺疾患、及び腎臓繊維化を引き起こす疾患、例えばDKDを含むETUARYの使用範囲を拡大することを求めている。IPF薬物市場におけるETUARYの成功は著者らの研究開発と登録戦略に基礎を築き、著者らは更にこのような薬物の使用範囲を拡大し、患者数の多い適応に適用する。
内部研究開発努力とGNI Japanとの連携により,ETUARYに加えて,F 351,F 528,F 230,F 573を含む臨床開発の異なる段階の候補薬を開発した。具体的には,肝線維化はわれわれが注目している分野であり,この分野での重要な候補品はF 351である。F 351は現在第3段階臨床試験にあり,慢性B型肝炎に関連する肝線維化の治療に世界で初めて承認された薬剤となる可能性がある。Frost&Sullivanのデータによると、2022年に中国の肝繊維化患者の数は1.403億人に達し、その中の約45.3%、即ち6360万人は慢性B型肝炎による肝繊維化である。われわれのF 351第二段階臨床試験では,線維化過程の逆転に積極的な結果が得られ,F 351は2021年3月にCDEに画期的な治療指定を与えられた。著者らは2022年1月から患者を募集して3期臨床試験に参加し、2023年第4四半期に募集を完了した。
著者らの長年の器官繊維化に対する研究に基づいて、著者らはまた著者らの研究開発範囲を拡大し、COPD、PAHとALF/ACLFの潜在的な治療方法を含む:
F528それは.COPD治療の臨床前研究におけるF 528の役割を評価している。F 528は新型抗炎症薬であり、多種の炎症性サイトカインを抑制することを標的とし、低毒性でCOPD進展を改善する潜在力がある体内にあるそれは.Frost&Sullivanのデータによると,2022年の中国COPD患者数は1.064億人に達し,2031年には1.101億人に達すると予想されている。現在の看護標準は主に症状を緩和し、疾病の悪化の頻度と重症度を減少し、そして心臓持久力を高めるために用いられている。著者らはF 528はCOPDに第一線の治療を提供し、そして長期の肺機能退化を減少できると予想した。
F230それは.PAH治療の前臨床研究における選択的エンドセリン受容体アンタゴニストF 230の役割を評価している。PAHは進行性で、生命を脅かす心血管疾患である。Frost&Sullivanのデータによると,2022年の中国PAH患者数は57,882人,2031年には70,279人と予想されている。2024年3月13日、私たちは中国でF 230のIND申請を提出した。
F573それは.われわれはF 573によるALF/ACLF治療の第2段階臨床試験を評価している。Frost&Sullivanのデータによると,2022年の中国ALF/ACLF患者数は39,247人に達した。ALF/ACLFの主要な治療方案は総合内科治療、非生物人工肝支持治療と肝移植を含む。しかし、ALF/ACLFを治療するための小分子または生物学的薬物は現在承認されていない。著者らは2022年1月に1人目の被験者を第1段階臨床試験に募集し、2023年3月に著者らの第2段階臨床試験を開始した。
我々のパイプライン製品の研究開発を推進すると同時に、私たちは中国にしかない臓器繊維化薬物に集中しているいくつかの生物製薬会社の一つであり、製造と商業化能力を有し、既定の記録がある。私たちの2つの製造センター、製造能力、プロセスの詳細については、“を参照してください”属性—GYRE製薬会社の性質“と”-生産と品質管理-内部製造施設。当社の専門的な販売チームと包括的な販売ネットワークの詳細については、ご覧ください販売、マーケティング、流通部門。”
私たちも中国の数少ないバイオ製薬会社の一つであり、これらの会社は発展段階の会社から持続的な利益を実現する会社に成長している。この成長は主に市場のETUARYに対する需要増加によるものであり、ETUARYは中国で発売された初めてのIPF薬物である。私たちはIPF薬物市場で限られた競争に直面して、私たちは私たちのマーケティング資源を医者にETUARYを採用することを奨励するために使用した。
私たちの製品と製品ライン
ETUARY:2011年に承認された国家1.1類IPF新薬
概要
28
ETUARY(ピルフェニドンカプセル)は2011年に国家1.1類新薬として承認され、稀な疾患IPFの治療に用いられた。中国がまだ承認されていないIPF待遇を受けて、ETUARYは2017年にNRDLに組み入れられ、その後ずっと市場シェアを占めてきた。臨床研究により、ETUARYは有効に肺機能低下とIPF疾患の進展を遅らせることができる。そのほか、異なる器官繊維化の発病機序と繊維化過程が類似していることから、著者らは現在ETUARYの治療適応を他の肺繊維化疾患、例えばSSc-ILD、DM-ILDと塵肺、及び腎臓繊維化を招く疾患、例えばDKDに拡大することに努力している。
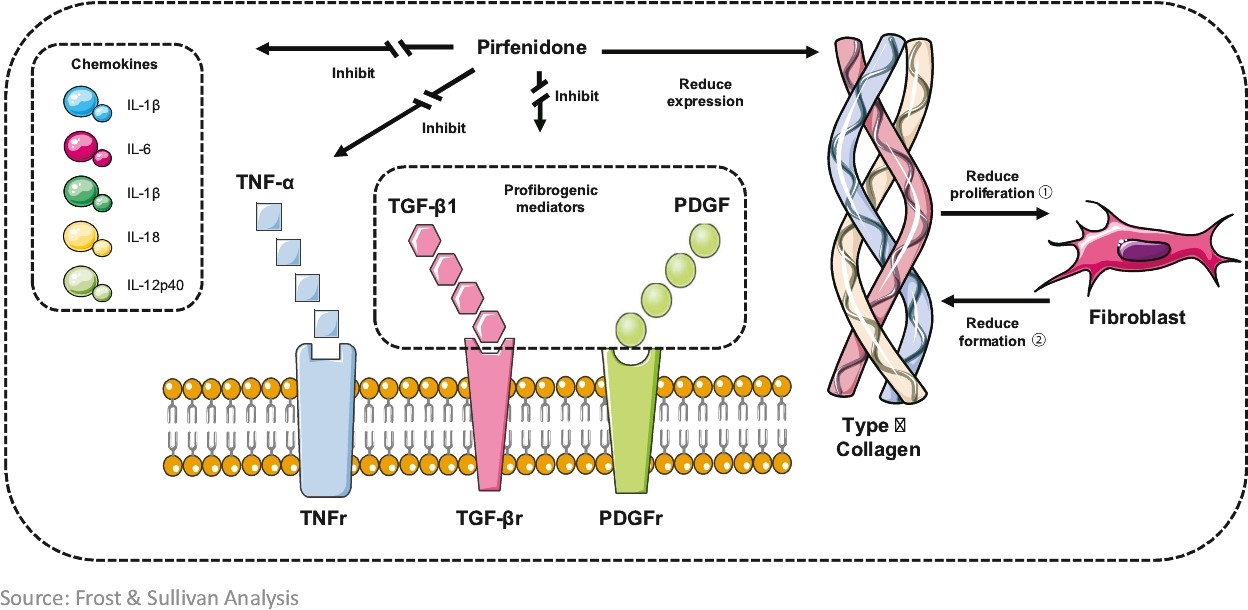
行動メカニズム
肺繊維化は肺上皮損傷後に肺胞細胞が活性化し、一連の炎症促進サイトカインを分泌し、線維芽細胞の増殖と筋線維芽細胞の分化を活性化し、細胞アポトーシス率を低下させることによるものである。ETUARYは,形質転換増殖因子1,血小板由来増殖因子と線維芽細胞増殖因子の発現を含む線維化促進メディエーターを抑制することにより,I型コラーゲンの発現を減少させ,最終的に線維芽細胞の増殖と膠原線維の合成を減少させ,細胞外基質の蓄積を減少させる。また、腫瘍壊死因子-α、インターロイキン1などの炎症メディエーターを抑制し、炎症反応を軽減する。
以下の図にピルフェニドンの作用機序を説明した。
市場のチャンスと競争
IPF
IPFはまれな疾患であり,肺原因不明の慢性進行性線維性間質性肺炎と定義され,主に高齢者に発生する。呼吸困難と肺機能の進行性悪化が特徴であり,予後不良に関与している。IPF患者の平均5年生存率は32%,平均10年生存率は16%に低下した。Frost&Sullivanのデータによると,IPFの中国における罹患率は2017年の83,002人から2022年の131,654人に増加し,複合年平均成長率は9.7%であり,2027年には214,664人,2022年から2027年の複合年平均成長率は10.3%,2031年には320,677人に増加し,複合年平均成長率は10.6%と予想されている。IPFの中国における総市場規模は二零一七年の1,360万元から二零二年の127.4元に増加し、複合年平均成長率は56.3%であり、二零二七年および二零三年はそれぞれ三億四千四百九十万元及び六億九千八百六十万元に達し、複合年平均成長率は二零二七年から二零二七年までの22.0%及び二零二七年から二零三年までの19.3%と予想される。
29
肺組織の瘢痕は不可逆的である。しかし、適切な治療は繊維化の速度を緩和し、患者の生存率を高め、患者の症状を緩和し、患者の生活の質を高める可能性がある。現在、中国で許可されているIPF薬物は2種類がある:ピルフェニドンと九チダニ。臨床により、それらはすべてIPF患者の肺瘢痕組織の形成を遅らせることができ、中国で唯一臓器繊維化を有効に治療すると考えられる薬物である。アメリカ胸科学会、ヨーロッパ呼吸学会、日本呼吸学会とラテンアメリカ胸科協会が発表した最新のIPF治療ガイドラインによると、ピルフェニドンと9 tedanibは条件付きで推薦された中等質の証拠のあるIPF薬物しかない。
中国で商業化されて以来、ETUARYはIPF薬物市場の主導者であった。ETUARYの売上高は増加を続け、2022年の9920万ドルから2023年には1億121億ドルに増加し、私たちの売上高は引き続き増加すると予想されています。まだ主導者であるにもかかわらず,イーストビィの市場シェアが最近低下しているのは,2022年の売上高1,350万ドルの安博司と2022年の売上高6,700万ドルのオフェキシブ(Ofev)を含む新薬の市場シェアが上昇しているためである。2022年、中国では、非正規医療費精算前に、中国の患者1人当たりのETUARY、安博司、OFEVへの年間支出はそれぞれ13,106ドルから13,378ドル、10,553ドルと15,132ドルである。最近IPF市場におけるETUARYの市場シェアはある程度低下したが、IPF罹患率の持続的な上昇、及びETUARYの未来にILD、塵肺疾患とその他の疾病の治療への適応を拡大するため、ETUARYの販売表現は強いと予想される。また、潜在的な市場参入者には様々な参入障壁がある。例えば、器官繊維化薬物の販売とマーケティング策略は病因治療薬物の販売とマーケティング策略と大きく異なるため、新規参入者は短期内に経験豊富と専門化された販売とマーケティングチームを構築することは困難であり、KOLと病院との長期安定協力は製品の組み合わせの開発と最適化、有効な教育と市場浸透及び患者の募集に対して臨床試験を行うことは非常に重要である。
SSC−ILD及びDM−ILD
CTD−ILDは非特発性間質性肺炎である。結合組織病(CTD)は自己免疫性疾患であり、血管と結合組織の慢性炎症を基礎とし、全身の各種器官に損害を与える。間質性肺疾患(ILD)は最も深刻な肺合併症の一つであり、CTDと関連する時に顕著な発病率と死亡率を招くことができる。
強皮症は退行性微小血管現象と免疫系活性化を特徴とするCTDであり、皮膚と内臓繊維化を招く。ILDはSScの影響を受ける患者に非常によく見られ、罹患率は約50%であり、SSc関連死亡の主要な原因である。皮膚筋炎(DM)の特徴は近位骨格筋無力と筋炎症である。糖尿病患者の中で、ILDは発病率と死亡率の主要な原因である。診断法によって糖尿病患者におけるILDの発生率は5%から45%と報告されている。
Frost&Sullivanのデータによると、中国のCTD−ILDの罹患率は2017年の約230万人から2022年の240万人に増加し、2027年には250万人、2031年には260万人に達すると予測されている。2022年のCTD−ILD患者のうち,約8.4%がSSC−ILDとDM−ILD患者である。2022年にはSSC−ILD/DM−ILD患者の抗線維化薬の市場規模は910万ドル,2027年には5310万ドル,2031年には1兆176億ドル,2022年から2027年の複合年間成長率は42.4%,2027年から2031年の複合年間成長率は22.0%と予想される。
SSc-ILDおよびDM-ILDは、特定の曝露または自己免疫疾患(例えば、強皮症および関節リウマチ)を含む既知の要素によって引き起こされる。症状は慢性咳,喀痰,喀血,進行性呼吸困難と間欠性発熱である。CTD-ILDの治療(SSc-ILDとDM-ILDを含む)はCTDの免疫抑制治療とILDの抗繊維化治療の結合であり、ILD病変の進展を有効に阻止し、患者の肺機能を保護することができる。推奨される免疫薬としては,シクロホスファミド,ミコフェノール酸エステル,アザチオプリンがある。CTD関連ILDの異なるタイプの抗繊維化治療方法はタイミング、薬物選択、投与量と治療コースなどの面である程度異なる。推奨される抗線維化薬は、ピルフェニドンおよびナチダニを含む。
ピルフェニドンは、炎症性サイトカインの抑制および炎症細胞増殖の抑制を含む抗炎症特性を有する抗線維化薬である。彼らの臨床所見は違いますがIPFは
30
SSc-ILDとDM-ILDは同じ発病機序を有し、構造細胞損傷、線維芽細胞活性化、筋線維芽細胞凝集、線維芽サイトカインと増殖因子の発現及び進展性ILDを含む。ピルフェニドンの臨床前研究結果から,著者らは第三段階臨床試験を行い,SSc−ILDとDM−ILD患者に対する治療効果を評価している。現在、9 tedanibはSSc-ILD患者の抗繊維化の治療に許可されている。
塵肺病
塵肺疾患はミネラル粉塵の吸入による一連の肺疾患であり、通常ある職業によるものである。主要な病理特徴は慢性肺炎症と進行性肺繊維化を含み、最終的に呼吸と/或いは心不全死亡を招くことができる。塵肺疾患は全世界的に普遍的に存在し、深刻な全世界の公衆衛生問題である。その高発病率と高死亡率は職業保護が不適切であり、早期診断方法と有効な治療が不足しているためである。
Frost&Sullivanのデータによると,中国では塵肺疾患の罹患率は2017年の850,299人から2022年には926,769人に増加し,2027年には962,562人,2031年には980,917人に増加すると予想されている。2027年までに塵肺抗線維化薬物の市場規模は1210万ドル,2031年には市場規模は6410万ドル,2027年から2031年の複合年間成長率は51.7%と予想される。
これまで、中国では、2種類のピルフェニドン製品が異なる臨床段階の塵肺疾患の治療に応用できる。著者らは2022年6月に著者らのETUARYによる塵肺治療の3期臨床試験の中で1人目の患者を募集し、ETUARYを中国の塵肺治療の臨床最先端の抗繊維化薬物にした。2023年12月31日現在、中国は塵肺の治療のための小分子或いは生物抗繊維化製品を許可していない。
シリカ誘導ラット肺繊維化の実験研究により、ピルフェニドンの連続投与14日と28日は肺上皮細胞の間葉系細胞への転化を遅らせることができることを表明した。これらの治療はビメンチンの顕著な低下とE-カルアドヘシンの上昇と関係があり、ピルフェニドンはシリカ誘導ラット上皮-間葉系転化を抑制できることを示した。
DKD
DKDは糖尿病による慢性腎臓疾患(CKD)である。糖尿病腎症(DKD)は糖尿病患者の腎臓の特殊な病理構造と機能変化である。そのほか、DKDはすでにCKDが末期腎臓疾患に進展する主要な原因になり、腎臓繊維化を招く主要な疾患の一つでもある。DKDは中国の糖尿病の深刻な合併症の一つであり、罹患率が高く、認識率が低く、治療率が低く、コントロール率が低いという特徴がある。
Frost&Sullivanのデータによると,DKDの中国における罹患率は2017年の4540万人から2022年の5320万人に増加し,2031年には6150万人に増加すると予想されている。中国のDKD市場は2017年の242億ドルから2022年には372億ドルに増加し、2027年には515億ドル、2031年には603億ドルに拡大すると予想される。
DKDに対する看護標準はずっと血糖コントロール、血圧コントロールと血液コントロールである。しかし、現在の治療策略は完全に有効ではなく、現在まだ有効な治療方法がDKDの予防に成功していないため、多くの患者は依然として末期腎臓疾患に進展している。現在、DKDを治療する薬物は主に血糖降下薬、降圧薬と降血脂薬がある。2023年12月31日現在、世界の多くの地域でDKD治療薬が承認されており、フェナレノン(KERENDIAの名称で発売されている)。
ピルフェニドンはその独特な作用機序でDKDに対して積極的な治療作用がある。腎臓局部に産生されるいくつかの増殖因子或いはサイトカインは進展性DKDの細胞外基質蓄積、炎症と瘢痕形成と関係があるようである。形質転換増殖因子-1システムは活性化され、1型と2型糖尿病動物モデルのDKDにおいて発病作用を果たす。また、1型や2型糖尿病患者におけるいくつかの研究では、形質転換増殖因子-β1の腎臓産生が増加していることが明らかにされています。最近では、患者の循環血液レベルや腎臓中の遺伝子発現によって、腫瘍壊死因子-α系もヒト糖尿病腎症につながっています
31
ディーケーディーです。腎臓疾患の実験動物モデルでは、ピルフェニドンは形質転換成長因子-1の産生とその後の基質沈着を抑制することが発見された。動物モデルや細胞培養研究においても,ピルフェニドンは腫瘍壊死因子−αの産生を減少させた。先の研究でも,確立されたDKD発症後,db/dbマウスにピルフェニドンを経口投与することが糸球体硬化の軽減に有効であることが示唆された。
臨床結果まとめ
2023年12月31日まで、Gyre製薬会社と上海遺伝子会社(“SG”)はすでに10項目以上の臨床試験を行い、ピルフェニドンの中国における臨床利益を探索した。IPFへの中国初の承認された薬物として,ETUARYは2 a期臨床試験を完了した後に承認された。以下はETUARYの現在の主要な臨床試験である。
ピルフェニドン治療特発性肺線維化2 a期登録臨床試験
本試験はランダム、二重盲検、多用量、平行対照、多中心2 a期の臨床試験であり、ピルフェニドンと基礎治療の併用による特発性肺繊維化に対する治療効果を観察した。目的:特発性肺繊維化患者の肺機能(動脈血ガス分析を含む)、6分間歩行試験(6 MWT)、生存時間、生活の質と高解像度CTスキャンに対するピルフェニドンカプセルの治療効果を観察し、その安全性と有効性を評価し、そして最適な臨床治療量を確定する。治療群は2つの用量群、400 mg/tid治療群と600 mg/tid治療群に分けた。各グループの患者24例。プラセボ群も2群に分けられ、4カプセル/tid群と6カプセル/tid群、各群12名の患者であった。治療群およびプラセボ群は2:2:1:1の割合で分配され、患者は層化され、ランダムに分配されてピルフェニドンまたはプラセボを受けた。主な観察指標は肺機能パラメータ、6 MWT結果と生存率である。
この試験は完了し、合計72人の患者が入選した。
試験の治療効果の結果は以下の通りである
治療効果 |
|
|
基準 |
|
|
結果(Fas、治療12カ月後) |
肺機能 |
|
|
一酸化炭素拡散能%(“DLCO%”) |
|
|
3群間のDLCO%変化は統計学的有意差があり(P=0.0306)、600 mg治療群の平均変化-2.79±9.34%、プラセボ群の平均変化-14.92±16.40%(P=0.0014)。 |
|
|
|
|
|
|
|
|
|
|
一酸化炭素拡散能力(“DLCO”) |
|
|
3群間のDLCO変化は統計学的有意差があり(P=0.0049)、600 mg治療群の平均変化-0.42±3.45%、プラセボ群の平均変化-3.14±4.44%(P=0.0016)。 |
|
|
|
|
|
|
|
|
|
|
動脈血酸素飽和度(SaO 2) |
|
|
3群間の動脈血酸素飽和度変化は統計学的有意差があり(P=0.0145)、プラセボ群と400 mg群の平均変化はそれぞれ-3.83±4.02%と-0.30±3.05%(P=0.0055)であった。 |
|
|
|
|
|
|
|
6 MWT |
|
|
脈拍血酸素飽和度(SpO 2) |
|
|
6 MWT後の3群のSpO 2変化は統計学的有意差があり(P=0.0168)、プラセボ群の平均変化-9.08±10.66%、400 mg群の平均変化0.22±7.30%(両群P=0.0062)。 |
|
|
|
|
|
|
|
32
生存率は |
|
|
適用されない |
|
|
プラセボ群、400 mg群と600 mg群の死亡率はそれぞれ20.83%、21.74%と16.67%であり、統計学的有意差がなかった。 |
17例の患者はSAEが発生したが、薬物と関係がある例は一例もなかった。プラセボ群、400 mg群と600 mg群の薬物副作用の発生率はそれぞれ41.67%、29.17%と45.83%であり、3群間に統計学的有意差がなかった。治療群の皮疹の発生率はプラセボ群と統計学的有意差があり、すべての600 mg治療群の皮疹の発生率はすべて20.83%であった。よく見られる副作用は吐き気(400 mg治療群と600 mg治療群はそれぞれ12.5%)、感光性反応(400 mg治療群は4.17%と600 mg治療群は12.5%)と傾眠(600 mg治療群は8.33%)を含むが、プラセボ群と比較して統計学的有意差がなかった。プラセボ群、400 mg群と600 mg群の副作用の発生率はそれぞれ70.83%、66.67%と66.67%であり、3群間に統計学的有意差がなかった。プラセボ群、400 mg群と600 mg群のSAEsの平均発生率は54.17%であり、3群間に統計学的差はなかった。プラセボ群、400 mg群と600 mg群のSAEs発生率(死亡率と入院率を含む)はそれぞれ29.17%、20.83%と20.83%であり、3群間に統計学的有意差がなかった。
治療12ケ月後、ピルフェニドンは有効に6 MWT直後のDLCO%、DLCO、SaO 2とSpO 2の低下を遅延させることができた。薬物に関連する副作用は認められず,皮疹や嘔気は最もよく見られる副作用であった。その結果、ピルフェニドンは特発性肺繊維化を治療する潜在力があることが分かった。
ピルフェニドン治療SSc−ILDの3期臨床試験
著者らはピルフェニドン治療SSc-ILDの有効性と安全性を評価するために、ランダム、二重盲検、プラセボ対照の多中心3期臨床試験を行っている。主な終点は治療52週におけるベースラインと比較したFVC%の変化である。144名の患者を募集して試験に参加する予定で、その中で治療群108人、対照群36人であった。
この試験は2018年6月に1人目の患者を募集した。新冠肺炎の発生と条件を満たす患者の希少性のため、この試験はまだ患者を募集しているため、現在のところ臨床結果は分析されていない。
ピルフェニドン治療DM−ILDの3期臨床試験
著者らはピルフェニドンによるDM-ILD治療の有効性と安全性を評価するために、ランダム、二重盲検、プラセボ対照の多中心3期臨床試験を行っている。主な終点は治療52週におけるベースラインと比較したFVC%の変化である。152名の患者が試験に参加し、その中に治療群114人、対照群38人であった。
この試験は2018年6月に1人目の患者を募集した。新冠肺炎の発生と条件を満たす患者の希少性のため、この試験は依然として患者を募集する過程にあり、現在まだ臨床結果が分析されていない。
ピルフェニドンによる塵肺治療の3期臨床試験
著者らはピルフェニドンによる塵肺治療の治療効果と安全性を評価するために、ランダム、二重盲検、プラセボ対照、多中心の3期臨床試験を行っている。主な終点は治療52週時のベースラインと比較した力肺活量の変化である。272名の患者が試験に参加し、治療群136人、対照群136人であった。
我々は2022年1月に倫理委員会の承認を得,2022年6月に1人目の患者を募集した。
私たちのDKD計画におけるPirfenidoneの1期臨床試験
著者らは開放ラベル、平行対照、単中心の臨床試験を行い、目的は単用量ピルフェニドンカプセルのCKD G 2とG 3 a期患者に対する安全性とPKを評価することである。入選被験者24例、その中に腎不全患者12例、健康ボランティア12例であった。
33
第一段階の臨床試験は2022年3月に完成した。本試験では,慢性腎臓病G 2とG 3 a患者のピルフェニドン使用は耐性であり,健康対照群と比較して主要な薬物動態学的パラメータに有意な変化はなく,用量調整も不要であった。
我々の臨床段階製品−F 351:CHB関連肝線維化を逆転させた1種類の新薬
概要
F 351は著者らの第三段階の臨床候補製品であり、慢性B型肝炎関連肝繊維化の治療が許可された最初の薬物になる可能性がある。Frost&Sullivanのデータによると、慢性B型肝炎は中国の肝繊維化の第一の原因であり、2022年に中国の肝繊維化患者の数は約1.403億人であり、その中の約45.3%は慢性B型肝炎によるものである。これまで,肝線維化を治療する有効な臨床治療法はなく,特効的な治療薬も全世界的に承認されていない。F 351はその第二段階臨床試験において線維化過程を逆転させる積極的な結果を示した。F 351は肝星細胞の増殖と転化増殖因子-β1シグナル経路を抑制することによって肝繊維化を逆転する可能性があり、この2つのシグナル経路は慢性B型肝炎関連性肝繊維化において重要な役割を果たしている。CHB誘導肝線維化の第二段階臨床試験の結果と,最初に発表された肝線維化治療薬の一つとして,F 351は2021年3月にCDEの画期的な治療指定を得たため,2022年1月から第三段階臨床試験への患者募集を開始した。我々は2023年第4四半期に登録を完了し,2024年に最後の患者が退院し,2025年初めに中国にNMPA F 351の申請を提出する予定である。
行動メカニズム
損傷が発生し、上皮及び/又は内皮細胞が損傷した時、炎症促進サイトカインは凝血から放出され、免疫細胞募集に用いられ、主に好中球とマクロファージである。これらの募集された免疫細胞はスカベンジャーの役割を果たし,組織断片や死亡細胞を除去し,急性炎症をきたす。同時に,免疫細胞自体はケモカインやサイトカインなどの因子を放出して炎症反応を増幅する。次に、形質転換増殖因子1、血小板由来増殖因子、インターロイキン13およびインターロイキン4のような放出因子は、限られた筋線維芽細胞の活性化および増殖を誘導する。F 351はHSCの増殖と形質転換増殖因子-1シグナル経路を抑制することによって、慢性B型ウイルス性肝炎の肝繊維化を治療と逆転することが期待される。
以下の図はF 351の作用機序を説明した
34

市場のチャンスと競争
アジアでは、CHBは肝臓の発病率と死亡率の主要な原因である。慢性B型肝炎ウイルスに感染した患者はよく肝繊維化が出現し、そして末期肝疾患、例えば非代償性肝硬変と肝癌に発展する可能性があり、干与を行わない。中国では、肝硬変の約70%はB型肝炎ウイルス感染によるものであり、これは慢性B型肝炎関連性肝繊維化治療に対する巨大な需要を反映している。
Frost&Sullivanのデータによると、全世界の慢性B型肝炎関連肝繊維化の罹患率は2017年の2.211億患者から2022年の2.578億患者に増加した。2017年から2022年まで、中国慢性B型肝炎関連肝繊維化の罹患率は6,360万から6,640万患者まで様々であり、今後10年間安定を維持することが予想される。中国の抗肝繊維化薬物市場は2017年の1.38億ドルから2022年の1.627億ドルに増加し、2027年と2031年の市場規模はそれぞれ3.38億ドルと8.012億ドルに増加し、複合年平均成長率はそれぞれ15.8%と24.1%になると予想されている。
病因治療は現在肝繊維化を治療する最もよく見られる方法である。慢性B型肝炎に関連する肝繊維化に対して、抗ウィルス治療はウィルス感染しか抑制できないが、繊維化の進展を予防、緩和或いは逆転できず、これは有効な抗肝繊維化治療に対する需要がまだ満たされていないことを表明した。中末期肝繊維化と早期肝硬変の治療は抗肝繊維化治療を提案する。2023年12月31日現在、肝線維化を治療するための化学的または生物学的薬剤は、世界的または中国で承認されていない。全世界範囲内で、現在一連の肝繊維化を治療する薬物は末期(2期或いはそれ以上)の臨床試験段階にある。これらの臨床段階の薬物の中で、F 351は中国臨床上最も先進的な候補製品であり、繊維化過程を有効に逆転する可能性がある。
われわれの臨床試験では,F 351は肝線維化を逆転させる上で有望な結果を示した。著者らは慢性B型肝炎合併肝繊維化患者の第二期臨床結果により、Ishak分期の病理採点を主要な予後指標とし、治療52週間後、治療群は肝繊維化を逆転する方面の効果はプラセボ群より優れていることを表明した。特に,270ミリグラム群では約56.1%の患者が>1の線維化消退を達成した。著者らは2022年1月に3期臨床試験の患者登録を開始し、2023年10月に登録を完了した。
臨床結果まとめ
35
2023年12月31日まで、著者らまたはSGが後援した5つ以上の臨床試験はF 351の臨床リスク/利益を探索するために使用された。以下にF 351精選キー臨床試験の結果を示す。
F 351中国における慢性B型肝炎関連肝線維化治療の2期臨床試験
著者らは1つの2期ランダム、二重盲検、プラセボ対照、エンテカビル(慢性B型肝炎ウイルス感染を治療する第一線の薬物)、多中心、用量増加試験を行い、F 351による中国慢性B型肝炎関連性肝繊維化患者の安全性と有効性を評価した。第二段階試験はランダムに240名の患者を4つの用量増加群(プラセボ、180 mg/日、270 mg/日と360 mg/日)に分け、主な終点はF 351とエンテカビル服用後の肝繊維化採点(Ishak採点システム)が一級以上低下することである。
この試験は,プラセボと比較して52週の治療で肝線維化スコアが統計的に有意に改善したという主要な終点に達した(p=0.0245)。肝繊維化消退>1者はそれぞれ25.58%(プラセボ群)、40.48%(180 mg/d)、56.10%(270 mg/d)と43.90%(360 mg/d)であった。したがって,270 mg/日の治療群が主要終点に到達できる患者の割合が最も高いことを示した。
この実験では,F 351はプラセボよりも良好な安全性の結果を示した。プラセボ群、180 mg群、270 mg群、360 mg群のSAEs発生率はそれぞれ4.65%、2.38%、2.38%、7.32%であり、統計学的有意差がなかった。全試験過程において、7名の被験者(4.17%)は7回のSAE:プラセボ群2人(4.6%)、180ミリグラム群1人(2.38%)、270ミリグラム群1人(2.38%)、360ミリグラム群3人(7.32%)を経験し、統計学的有意差はなかった。実験室検査異常、トランスアミナーゼ上昇、塞栓性脳梗塞、粉砕性骨折、骨粗鬆症、意外妊娠と高血圧。誰も死ななかった。
F 351中国における慢性B型肝炎関連肝線維化治療の3期臨床試験
著者らは3期無作為、二重盲検、プラセボ対照、エンテカビルに基づく多中心試験を行い、F 351による中国慢性B型肝炎関連性肝繊維化患者の安全性と有効性を評価している。第三段階の臨床試験は248名の患者の中でランダムに行われるように設計され、これらの患者の主要な終点はF 351とエンテカビル服用後の肝繊維化採点(Ishak採点システム)が少なくとも1つのレベルを低下させることである。
われわれは2022年1月からF 351の3期臨床試験に患者を募集し,2023年10月に募集を完了した。現在のところ分析できる臨床結果はない。私たちは2025年初めまで最高の結果があると予想している。
F 573:ALF/ACLFを治療する潜在的な1種類の新薬
概要
F 573は1種のシステインアスパラギン酸アミノトランスフェラーゼ阻害剤であり、ALF/ACLFを治療する1種類の新薬になる可能性がある。Frost&Sullivanのデータによると,2022年の中国ALF/ACLF患者数は39,247人に達した。ALF/ACLFの主要な治療方案は総合内科治療、非生物人工肝支持治療と肝移植を含む。現在,ALF/ACLFに特化した薬剤はない。著者らは2022年1月に第一段階臨床試験に参加する第一段階の被験者を募集し、2023年3月に第二段階の臨床試験を開始した。
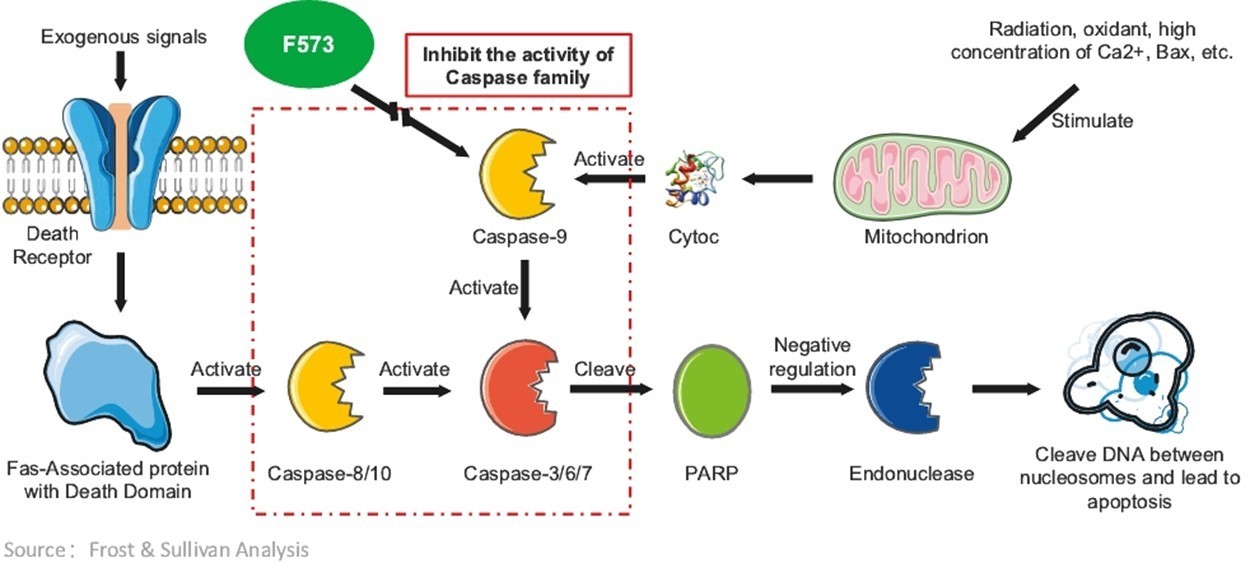
行動メカニズム
炎症反応と免疫機能障害は大量の肝細胞の死亡を招くことができ、それによってALF/ACLFを招く。そのため、正常肝細胞のアポトーシス過程を抑制することはALF/ACLFの進展を遅らせることに役立つ。肝細胞アポトーシスの主要な経路はミトコンドリア経路と死亡受容体経路を含み、その中でcaspaseファミリーは主要な実行分子として重要な役割を果たしている。F 573の主要な作用機序はcaspaseファミリーを抑制することであり、caspase 1、3、6、7、8と9の活性を含み、ポリADPリボポリメラーゼに対する切断作用を減少させ、それによって内因性或いは外因性シグナルを介した細胞アポトーシス過程を遮断する。したがって,F 573は肝不全の緩和が期待できる。
36
以下の図にF 573の作用機序を説明した
市場のチャンスと競争
ALF/ACLFは多種の要素による深刻な肝障害であり、合成解毒、代謝と生物転化機能の深刻な損傷或いは喪失を招く。ALF/ACLFは黄疸、凝血機能障害、肝腎症候群、肝性脳症と腹水などの症状を伴うことができる。ALF/ACLFの病因は非常に複雑で、肝炎ウイルス(特にB型肝炎ウイルス)と他のウイルス、薬物、肝毒性物質(特にB型肝炎ウイルス)を含む例えば:アルコールと化学剤)、細菌、寄生虫。中国では、B型肝炎ウイルス、薬物と肝毒性物質はALF/ACLFの最もよく見られる原因である。
Frost&Sullivanのデータによると,2017年の中国ALF/ACLFの罹患率は43,123名,2022年は39,247名,2027年は34,969名,2031年は31,485名と予想されている。2017年、ALF/ACLFの中国市場規模は2.786億ドル、2022年は2兆535億ドル、2027年は2.259億ドル、2031年は2.033億ドルと予想されている。
肝不全の主要な治療選択は総合内科治療、非生物人工肝支持治療と肝移植を含む。内科治療は主に一般支持治療、対症治療、病因治療と合併症治療を含む。
F 573の治療効果はすでに4つの臨床前研究で実証された。はい体外培養研究により、F 573は多種の細胞のアポトーシスに対して明らかな抑制作用がある。具体的には,F 573はHeLa細胞,ヒト正常肝細胞L 02とJurkat細胞に対して保護作用を有するとともに,Caspase−3酵素活性を抑制し,基質AC−DEVD−AMCを切断する能力を低下させる。F 573はD-GalN/リポ多糖誘導ラット爆発性肝不全による肝機能を改善でき、肝損傷を軽減し、肝細胞の壊死とアポトーシスを顕著に抑制し、急性と深刻な肝損傷に対して予防と治療作用がある。D-ガラクトース/リポ多糖によるマウス急性肝損傷の薬効学実験において、F 573はD-ガラクトースとリポ多糖によるKMマウス急性肝損傷に対して保護作用があり、そしてKMマウスの生存時間を延長することができる。F 573はConA誘導BALB/cマウス急性肝不全に対して肝機能改善と肝障害軽減作用がある。F 573は肝細胞壊死と細胞アポトーシスを顕著に抑制でき、急性と深刻な肝損傷に対して予防と治療作用がある。
臨床発展計画
F 573 ALF/ACLF治療の1期臨床試験
37
2022年1月、我々は、単剤および多剤F 573の耐性およびPKを評価するために、第1段階臨床試験のための第1の被験者を募集した。われわれは100名の健常者を募集してこの試験に参加し,2022年7月に耐性とPKの第1段階臨床観察を完了した。F 573のCmaxは0.5~2.0 mg/kgの範囲内で用量依存性がなく、AUC 0-tとAUC 0-は線形薬物動態学規則に符合する。F 573の吸収速度には性差が認められた。F 573は1日1回投与し,7日間連続して人体内に蓄積しなかった。
F 573 ALF/ACLF治療の2期臨床試験
私たちは2023年3月に第二段階臨床試験を開始した。第二段階の臨床試験は無作為、二重盲検、プラセボ対照の臨床試験である。この試験の主な目的は注射用F 573による肝障害/不全治療の有効性と安全性を評価することである。第2段階実験は3つの段階に分けられた
被験者の同意を得た後,この試験では,第1段階と第2段階のCHB患者のために薬物動態血液試料を採取した。
スクリーニング条件に適合する48人の被験者は、試験薬またはプラセボを3:1の割合で1日1回、28日間継続する予定である。被験者は同時に薬物アセチルシステイン注射を受けた。投与中止後、被験者は90日間の安全追跡を受ける。
臨床前段階の候補品は
F 230:PAHに対するEPAの選択的アンタゴニスト
F 230は選択的エンドセリンA受容体拮抗薬であり、PAHの治療に用いられる。PAHはまれな疾患であり,進行性で生命を脅かす疾患であり,心臓から肺に血液を輸送する肺動脈圧が増加することを特徴とする。PAHの1つの共通の病理特徴は血管再構築であり、肺動脈硬化と肥厚を含む。これは肺を通る血液の流れを制限し,肺動脈高圧を招き,心臓が肺に血液を輸送しにくくなる。PAHの正確な原因は不明であり,既知の治療法もない。PAHは重篤な疾患であり,治療しなければ期待寿命は短い。PAHの治療は予後不良であり,死亡率が高く,標準治療が乏しい場合には3年未満生存している。
Frost&Sullivanのデータによると,中国のPAH罹患率は2017年の49,004名から2022年の57,882名に増加し,2027年には67,682名,2031年には70,279名に増加すると予想されている。PAHの中国における市場規模は2017年の2.9億ドルから2022年の3.7億ドルに増加し、2027年には4.7億ドル、2031年には5.2億ドルに増加すると予想される。
低酸素誘導ラット肺動脈高圧の検討では,F 230は平均肺動脈圧,右室収縮圧,右室/左室加室間隔,肺動脈壁厚を有意に低下あるいは低下させる傾向にあった。最小有効量の下でも、治療群とPAH群の間のこれらの指標の差は統計学的有意差があった。
F 528:COPDの潜在的な第一線治療
38
F 528はCOPDを治療するために開発された抗炎症小分子候補薬物である。F 528は新型抗炎症薬であり、多種の炎症性サイトカインを抑制することを標的とし、極めて低い毒性でCOPDの進展を変化させる体内にあるそれは.COPDは1種の慢性炎症性肺疾患であり、肺気流閉塞を招く。それは三つの異なる疾病から構成されている:肺気腫、慢性気管支炎と慢性閉塞性喘息。慢性閉塞性肺疾患は肺内肺胞間のバリアを破壊し,気道が腫脹し粘液で閉塞される。多くの場合,COPDの進行は非常に遅く,診断される前に長年の症状が出現する可能性がある。
Frost&Sullivanのデータによると,中国のCOPD罹患率は2017年の1.027億人から2022年の1.064億人に増加し,2027年と2031年にはそれぞれ1.086億人と1.101億人に増加すると予想される。中国の慢性閉塞性肺疾患市場は2017年の9億ドルから2022年の11億ドルに増加した。2027年までにこの市場は13億ドル、2031年には15億ドルに拡大すると予想される。
COPDの薬物治療は主に症状を緩和し、疾病の悪化の頻度と重症度を減少させ、心臓の持久力と健康を高めるために用いられる。現在、確実な臨床試験証拠がなく、現有の薬物は肺機能の長期低下を緩和できることを表明した。末期COPD患者に対して、現在利用可能な治療方案による治療効果は限られている。外部からの臨床研究の結果,2%の患者は24カ月の標準薬物治療後に運動能力が改善し,健康に関する生活の質の改善が得られたと報告されていない。そのため、慢性閉塞性肺疾患患者の臨床需要は非常に満足されていない。
著者らはF 528がCOPDの第一線の治療方法になる可能性があると信じている。煙暴露とリポ多糖の気管注射によるCOPDラットの中で、F 528はその影響に対する臨床前研究により、F 528治療後のラットの肺指数、肺胞隙間と肺損傷採点は明らかに低下した。
他の候補薬
私たちの候補製品ラインを補充し、豊富にするために、慢性閉塞性肺疾患関連血小板減少の治療のためのマレイン酸アトラモプ、多発性硬化症の治療のための塩酸フェントモート、重度のざ瘡の治療のためのミノサイクリン泡、および粘液過多の呼吸器疾患を治療するためのアセチルシステイン注射剤を含むいくつかの模倣薬の営業権を獲得した。
職業、健康、安全、環境問題
私たちは様々な健康、安全、社会、環境法律法規に制約されており、私たちの運営は現地政府当局の定期検査を受けている。私たちは社会的責任を負うために努力し、環境、社会、そしてガバナンスは私たちの持続的な発展に重要であると考え、私たちは適用される健康、安全、社会、環境保護法規の遵守を促進するのに十分な政策があると信じている。
上級管理職の監督の下で、私たちは環境、社会、気候に関連するリスクが私たちの業務、戦略と財務業績に与える実際と潜在的な影響を積極的に識別し、監視し、これらの問題に対する考慮を私たちの業務、戦略と財務計画に組み入れ、特に従業員の責任、環境責任、公共責任などの分野に注目する。企業社会的責任は私たちの核心的な成長理念の一部とされ、株主のための持続可能な価値を創出する鍵でもある。
また、私たちは環境、健康、安全に関する私たちの業務の遵守状況を監視して実行します。この責任は、戦略、政策、基準、指標を訓練、制定、実施し、コーディネーターチームを通じて環境、健康と安全政策と手順、環境、健康と安全監査、およびイベント応答計画と実施を伝達することによって履行される。私たちの経営陣の監督の下で、私たちの品質管理チームはこのようなリスク発生の可能性と任意の潜在的な影響の推定程度を評価します。
ライセンスとその他の承認
39
2023年12月31日現在、業務運営に必要なすべての材料の許可と承認を取得しました。2023年12月31日現在、私たちの完全子会社(北京大陸生物医薬科学技術有限会社、中華人民共和国の法律に基づいて設立された会社)は営業許可証を取得していますが、経営活動はありません。以下の表に材料ライセンス、ライセンス、および承認の詳細を示します
免許·許可証 |
|
|
有効期限 |
|
|
権威.権威 |
薬品生産許可証 |
|
|
2022年9月から2025年9月まで |
|
|
北京市医薬品監督管理局 |
情報サービス資質証明書 |
|
|
2021年1月-2026年1月 |
|
|
北京市医薬品監督管理局 |
中関村ハイテク企業 |
|
|
2022年12月-2024年12月 |
|
|
中関村科学技術園管理委員会 |
ハイテク企業証明書 |
|
|
2022年11月-2025年11月 |
|
|
北京市科学技術委員会は 北京市財政局、北京市税務総局 |
薬品登録承認(ピルフェニドン) |
|
|
2028年8月まで有効です |
|
|
北京市医薬品監督管理局 |
薬品登録承認(ピルフェニドンカプセル) |
|
|
2028年8月まで有効です |
|
|
北京市医薬品監督管理局 |
医薬品GMP証明書(ピルフェニドン原料薬) |
|
|
2019年7月-2024年7月 |
|
|
北京市医薬品監督管理局 |
対外貿易経営者登録表 |
|
|
2022年2月から |
|
|
北京市商務委員会 |
私たちの研究と開発は
私たちは長期的な成長を達成するために、一貫して資源を投入して研究開発を行っている。内部研究開発と外部協力を通じて製品ラインの多様化と拡張を実現することは、私たちの長期的な競争力と成功に重要だと信じている。
2023年12月31日現在、私たちは中国に85人の従業員からなる集中的な内部研究開発チームを持っている。我々の研究開発部門は以下の部門から構成されている:薬物発見、化学、製造と制御(CMC)、臨床開発、医療事務と監督管理事務。我々の研究開発者は分子生物学、化学法規事務と臨床開発において豊富な専門知識を持っている。機能を越えた協力を通じて、著者らの研究開発組織は新薬製品を開発し、満足されていない臨床需要を満たすことができるようにした。
著者らは臨床需要ガイドと市場駆動の方法を用いて研究開発を行った。著者らはまず適切な薬物開発目標を確定し、著者らの発展戦略に基づいてプロジェクト評価と全体プロジェクト設計を行い、それから異なる実験プラットフォームを協調することによって実験方法論を探索と構築した。我々は,個々の候補薬物のビジネス潜在力とその開発成功の可能性,潜在的競争と最終的な市場規模をバランスさせることで,薬物開発計画を慎重に選択した。
薬物発見
著者らの分子スクリーニングと設計能力は分子を臨床前研究から市場に出す成功可能性を増加させ、革新的な治療方法を可能にし、そして肝心な経路と標的をめぐって構築された豊富なパイプライン資産を支持する。著者らは高効率な目標識別と検証、複合材料設計とスクリーニング及び複合材料の最適化を誘導するシステムを構築した。発見段階では候補薬をテストして決定します
40
構造修飾を通じて化合物の吸収、分布、代謝、排泄と毒性性質を最適化し、最大の治療効果と最小の毒性を達成する。著者らの研究開発センターはターゲットの識別と検証、化合物の設計と最適化を効率的に完成できる的確な薬物発見とスクリーニングプラットフォームを支持している。
薬物発見段階において、著者らは新しい研究開発機会を探索し、実行可能性研究を行い、そして機会に対して評価を行った。著者らはまた新しい化合物を設計と製造し、新薬の製造技術と品質管理に対して系統的な研究を行い、そして技術プラットフォームを開発して関連技術を支持、管理と監督する。
化学·製造·制御
中央軍事委員会グループ
CMCグループは発見と臨床研究の間の重要な絆である。所望の物理的および化学的特性を有する医薬物質を製造し、最大バイオアベイラビリティおよび安定性を実現する製剤として製剤化するために、化学および製薬プロセスの開発を担当する。CMC段階では、各原料薬分子の合成に対して徹底的な研究を行い、薬物物質が所定の品質標準を達成でき、製造技術が安全、堅固、経済と環境保護を確保し、薬物製品は良好な安定性と適切な貯蔵条件と賞味期限を持つ。
臨床開発チーム
著者らの臨床開発チームは薬物開発の臨床試験を監督し、臨床事務のプログラム標準を確立し、そして臨床医学事務を処理した。著者らの臨床開発チームはまた臨床開発戦略、臨床試験方案の設計、臨床試験操作の協調、薬物警戒と臨床試験の品質管理に集中している。著者らの臨床開発チームのメンバーは臨床試験の設計、実施、薬物供給及び試験データの収集と分析を含む著者らの臨床試験のすべての段階を専門に管理している。著者らは多くの分野のトップレベルの臨床専門家と協力し、著者らの主要な研究者として、業界をリードする臨床研究機関の運営能力を利用し、中国と海外で有名な学術医療機関と臨床試験センターに依頼し、中国での臨床試験の高品質と効率的な実施を促進した。
臨床試験設計と実施
著者らの臨床開発チームは臨床試験のすべての段階を管理し、方案の設計、操作及び臨床データの収集と分析を含む。われわれの迅速な試験進展は,(I)優れた臨床前結果で臨床段階試験を開始する戦略決定,(Ii)厳しい試験設計,(Iii)異なる地域からの多くの病院や主要研究者との長期協力関係,および(Iv)高品質な実行のおかげである。著者らの臨床試験における豊富な知識と経験を利用して、著者らの臨床開発専門家は臨床試験で観察された異なる特性に基づいて、著者らの候補薬物のために独特な治療機会を識別し、そしてそれに応じて臨床計画を改善する。
競争
臓器繊維化市場は急速な変化の影響を受けている。私たちは強力な革新製品と候補薬物チャンネル、強力な販売とマーケティング能力、そして経験豊富な指導チームが私たちに競争優位を提供してくれると信じているが、私たちは多くの異なる源からの潜在的な競争に直面しており、これらの源は私たちの発売薬物や候補薬物に対する同じ適応に対する治療法の開発に努力している。その中には大手製薬会社,様々な規模の専門製薬やバイオテクノロジー会社,学術機関,政府機関,研究機関が含まれている。私たちが開発と商業化に成功した任意の候補薬物は、既存の薬物と将来出現する可能性のある任意の新薬と競争するだろう。
私たちの製品は主に治療効果、価格と医療専門家と病院が一般的に受け入れている市場認知度に基づいて、類似の条件で指示された製品と競争します。私たちの主な競争相手の識別は製品や候補薬によって異なり、場合によっては、私たちの競争相手は以下の点でより多くの財務資源と専門知識を持っている可能性があります
41
研究開発、製造、臨床前テスト、臨床試験を行い、監督管理の許可と販売許可を得た薬物の面で、私たちは私たちよりよくやった。
私たちの持続的な成功は主に私たちの革新製品と先進技術を開発する能力があり、技術をすべての生産ラインに応用し、広範な製品の組み合わせとパイプラインを絶えず開発し、私たちの既存と未来の製品を効果的に商業化とマーケティングし、私たちの流通ネットワークを拡大し、顧客関係を維持し、経験豊富で才能のある技術開発者を誘致し、維持し、高品質の標準を維持し、効率的な運営モードを維持し、監督部門の許可を得て維持すると信じている。
生産と品質管理
内部製造施設
我々の製造工場は中国河北省の北京と滄州に位置している.2022年12月31日と2023年12月31日までの年間で、私たちが販売している100%ピルフェニドンは北京と滄州の工場で生産されています。私たちの製造施設の設計と運行はcGMP規定に適合している。
品質管理
私たちは製品の品質が患者の安全を確保し、私たちの長期的な発展を実現する根本だと信じている。私たちの品質管理チームはNMPAの規定に基づいて私たちの運営の各段階を監視します。著者らは全体の生産過程において品質管理措置を実施し、サプライヤーの審査、原材料検査とテスト及び過程制御を含み、すべての製品は出荷前に徹底的な検査とテストを経た。
仕入品質管理
我々はすでに内部手続きを構築し、原材料サプライヤーの選択と品質管理を管理し、関連するcGMPと薬品登録法規の要求を満たす。私たちは各種の要素に基づいて私たちの原材料サプライヤーを選択して、彼らの経済状況、資本、名声、品質管理、生産規模と技術実力を含み、そして彼らの資質、私たちのアンケートに対するフィードバックと私たちの現場検査によって彼らを評価します。
物流と配送管理
私たちは第三者と物流サービス協定を締結した。この手配によると、物流サービス提供者は私たちの要求に応じて安全かつタイムリーに配達サービスを提供し、私たちは貨物の品質に責任があります。我々の物流サービスプロバイダは,輸送,荷役,輸送,交付を含む物流サービスを提供する際の不注意によるいかなる損失も,我々の物流サービスプロバイダが担当する.私たちの物流サービス提供者も保険についての連絡と処理を担当していますが、私たちは保険料と運賃の支払いを手配します。
在庫管理
私たちの在庫は主に原材料、製品、半製品(原料薬を代表する)と製造品を含みます。私たちは私たちの生産を絶え間なく維持するのに十分な在庫を合理的な水準に維持するために努力している。私たちは年間販売計画、生産計画、調達計画に基づいて供給協定を締結します。
販売、マーケティング、流通
私たちの内部販売とマーケティングチームは
42
2023年12月31日まで、私たちの内部販売とマーケティングチームは中国の30の省、自治区、直轄市の市場をカバーした。私たちの販売とマーケティングチームは主にカバーエリア内の店舗との関係の構築と維持を担当しています。
私たちの販売とマーケティングチームの相対的に高い医学知識と技能は、私たちの学術マーケティング方法を実施し、私たちのリードする製薬会社としての名声を維持するために非常に重要だと信じています。2023年12月31日まで、私たちの内部販売とマーケティングチームは391人の従業員を含み、平均9年以上の薬品販売経験を持っている。私たちのより経験のある職員たちもまた彼らの学術普及ネットワーク経験を定期的に共有するだろう。
従業員の経歴の詳細については、“を参照されたい従業員と人的資本“この節では。
学術昇進
私たちの販売とマーケティングでは、私たちは学術普及と患者サービスを強調する。我々は,医師や他の医療専門家を教育することで,ETUARY,我々の他の候補製品とそのそれぞれの適応を理解し,医学専門家における学術認知度とブランド意識を促進·強化することに取り組んでいる。私たちと医療専門家との協力関係は私たちの知名度を向上させ、医学界と患者のETUARYに対する認識を強化し、医療保健提供者の臨床能力を高め、そして私たちにETUARYを改善するための貴重な臨床データを提供し、これらはすべて私たちがETUARYをより効果的にマーケティングと販売することに役立つと信じている。
分布
ディーラーは私たちの直接顧客で、彼らは私たちの製品を病院、他の医療機関と薬局を含むサイトに転売します。流通業者は主に製品の引渡しと支払いを担当し、私たちの内部の販売とマーケティングチームは学術マーケティング活動やその他の販売促進活動を担当しています。
流通ルートの統合とディーラー業務管理の不安定さのため、私たちは時々一部のディーラーとの提携関係を更新しないことを終了または選択する。同時に、私たちが新しいディーラーを増加させるのは主に私たちの販売ネットワークが拡大して最適化されているからだ。全体的に、私たちは主要流通業者との関係が安定している。
製品定価
私たちは価格を決定する時に多くの要素を考慮して、その中に主に私たちの研究開発、生産とマーケティングコストと支出、製品の感知価値、私たちの市場シェアと競争構造を含む。また、私たちの価格設定戦略も医療保険の精算基準と医療と定価のやり方の監督管理を含む医薬業界に課せられた法規と政策の影響を受けている。著者らの商業化チームは中国医薬製品の定価に影響する新しい政策に密接に注目し、そして絶えず変化する監督管理環境に適応し、そして異なる省の現地政策と競争に対応して、私たちの製品の価格レベルを維持し、そして私たちの中国全体の売上を最大限に高めるために、絶えず私たちの定価戦略を更新している。詳細は“をご覧ください-我々の中国での業務:Gyre製薬−中国の規制要件−製薬業界に関する他の中国法規−価格制御”.
国家精算薬品目録
国家公共医療保険計画に参加する者はNRDLに含まれる薬品の調達価格を全額または部分精算する資格があり、その中には基本医療保険、労災保険、生育保険基金下の薬品支払い基準が規定されている。2017年から政府はNRDLの定期調整を開始し,ETUARYは同年NRDLへの進出に成功した。最新バージョンのNRDLは2023年3月1日から実施されている。詳細については“を参照されたい”我々の中国での業務:Gyre製薬−中国の規制要件−製薬業界に関する他の中国法規−国家医療保険計画のカバー範囲”.
43
2票制
2016年12月26日、国務委員、医療改革委員会、国家衛生計生委員会、国家発展改革委員会などの関係部門は共同で“公立医療機関で薬品購入2枚の領収書制度を推進する実施意見(試行)”を配布し、国家レベルで2枚の領収書制度を推進する実施細則を作成した。詳細は“をご覧ください-我々の中国での業務:Gyre製薬−中国の規制要求−その他製薬業界に関する中国法規−薬品流通と両発行制“と。関連規定を遵守するため、著者らは主に単層流通モデルを採用し、ディーラーが直接私たちの製品を病院と公立医療機関に転売した。ある流通業者は流通業者を招いて薬局に薬品を販売することができ、これらの薬局は2枚の領収書制度の制約を受けない。
集中入札プロセスと集中一括調達システム
中国では,公立病院や公立医療機関に売却される薬品の大部分の価格は省レベルや市レベルの競争的集中入札プログラムによって決定され,異なる条項やプログラムがある。集中入札過程では,薬品生産会社が落札価格で公立病院や他の公立医療機関にその製品を販売することを許可する。集中入札過程は代替製品或いは市場で代替製品と考えられる製品の中で定価圧力を引き起こし、そして中国の薬品定価と調達方式の重大な変化を招く可能性がある。
原材料と仕入先
私たちの“合格サプライヤーリスト”のサプライヤーを除いて、私たちのすべての重要な原材料サプライヤーは予備サプライヤーがあります。しかも、各材料には1つ以上のメーカーがあり、各メーカーには複数のディーラーがいる。このディーラーたちは合理的な在庫備蓄を持っている。もし私たちが新しいサプライヤーを探す必要があれば、比較研究を行い、確認した後、そのサプライヤーを私たちの合格サプライヤーカタログに追加して、製品供給を確保します。
中国の監督管理要求
中国政府当局は薬品と生物製品の研究、開発、テスト、製品の審査、製造、品質管理、製造変更、包装、貯蔵、記録保存、ラベル、普及、広告、販売、流通、マーケティング及び輸出入などの方面に対して広範な監督管理を行った。私たちの現在の候補製品は薬品として規制される予定だ。中国で規制承認を得る過程、および商業化前と商業化後に適用される法規と法規および他の監督管理機関を遵守することは、私たちの製品の生産とマーケティングおよび私たちの研究開発活動の重要な要素であり、大量の時間と財力を要する。
薬品監督制度
中国の薬品監督制度は全国人民代表大会常務委員会、国務院と国務院に属する多数の部·委員会と機構から構成され、その中に国家薬品監督管理局(その前身は中国食品薬品監督管理局)、国家衛生委員会(その前身は中華人民共和国国家衛生·計画出産委員会及び国家医療保障総局)を含む。
国家薬品監督管理局は国家市場監督管理総局(“SAMR”)の監督の下で、薬品、化粧品と医療設備の登録と監督を担当する監督機関である。
NHCは中国の首席医療監督機関であり、主に国家医療政策の起草を担当し、中国の公共衛生、医療サービスと健康応急制度を監督し、中国の医療改革を調整し、中国医療機関の運営と医療関係者の勤務を監督する。
44
衛生部は医療保険、生育保険と医療救助の政策、計画と標準の起草と実施を担当し、中華人民共和国の医療基金を管理し、統一的な薬品医療保険目録と支払い基準を制定し、医療用品と医療保健サービスを監督し、薬品と医療用品の入札募集政策を制定と管理する。
薬品に関する法律法規
医薬製品開発
中国では、国家薬品監督管理局は薬品及び医療機器と設備の管理を監視·監督している。中華人民共和国地方省級医療製品管理部門は本行政区域内の薬品監督管理を担当している。“中華人民共和国薬品管理法”(以下は“薬品管理法”と略称する)によると、薬品は人類疾病の予防、治療と診断、人体の生理機能を調節するための物品であり、中国の伝統薬品、化学薬物と生物製品を含む。“中華人民共和国薬品管理法”と“中華人民共和国薬品管理法実施条例”は薬品管理の法律枠組みを確立し、薬品研究、生産、経営、応用、監督管理に従事する単位と個人に適用する。
非臨床研究と動物試験
国家市場監督管理総局は臨床前データに輸入と国産薬品の登録申請を支持することを要求した。“非臨床実験室研究良好実験室操作規範認証管理方法に関する通知”によると,国家薬品監督管理局は中国非臨床研究機関の認証を担当し、地方省級医療製品管理部門は中国非臨床研究機関の日常監督管理を担当する。国家薬品監督管理局は、評価機関の組織管理、研究者、設備施設、非臨床薬物プロジェクトの運営管理を通じて、この機関が薬物非臨床研究を担う資格を持っているかどうかを決定する.もしすべての関連要求を満たしたら、国家食品薬品監督管理局はGLP証明書を発行し、この証明書も国家食品薬品監督管理局のウェブサイトで公表される。GLPの要求に適合する場合、国家薬品監督管理局は有効期間5年の薬品GLP証明書を承認し、発行する。
“実験動物管理条例”、“実験動物の良好な操作管理方法”、“実験動物証明書管理方法(試行)”は実験動物の使用と飼育を規範化し、動物に実験を行うには実験動物使用証を持つ必要がある。
新薬臨床試験の承認と改革
“薬品登録管理方法”、“中華人民共和国薬品管理法”と“中華人民共和国薬品管理法実施条例”によると、新薬申請は必ず臨床試験を行わなければならない。国家薬品監督管理局はすでに一連の措置を取って臨床試験申請の審査効率を高め、そして薬物試験の良好な臨床実践(“中華人民共和国GCP”)に対する監督と実行を著しく強化し、データの完全性を確保した。
“薬品登録管理方法”は一連の改革行動を確認したが、これらに限定されない:(一)上場授権者制度を全面的に実施し、臨床試験の開始を黙示する;(2)薬品、補助剤と包装材料に対して関連審査を実施する;(三)薬品迅速登録手続きを導入する。非臨床研究を完成した後、新薬登録申請は必ず臨床試験を行わなければならず、申請者は臨床試験を行う前に国家薬品監督管理局或いは疾病管理センターにINDの承認を申請しなければならない。
“国務院の薬品医療機器審査制度改革に関する意見”は薬品医療機器審査制度改革の枠組みを確立した。
45
中国薬品監督管理局は薬品登録審査のいくつかの政策に関する公告を発表し、更に薬品審査の流れを簡略化し、新薬INDが使い捨て傘式審査を実行することを規定し、再申告審査評価或いは分期審査の方式を採用しない。“国家薬品監督管理局の薬品臨床試験審査審査承認手続きの調整に関する公告”によると、INDが受理して費用を納付した後60日以内に、申請者がCDEの否定意見或いは質疑意見を受けていない場合は、提出された臨床試験方案に従って当該薬物の臨床試験を行うことができる。
“薬品発売許可優先審査と審査手続き(試行)”は更に明確にし、革新薬物は迅速なチャンネルIND或いは薬品登録経路を獲得する。
国際マルチセンター臨床試験について
国家薬監局が公布した“国際多センター臨床試験ガイドライン(試行)”によると、国際多センター臨床試験申請者は同じ臨床試験方案を用いて異なるセンターで同時に臨床試験を行うことができる。申請者が中国国内で国際多センター臨床試験を実施しようとしているのは、“薬品管理法”、“中華人民共和国薬品管理法実施条例”と“薬品登録管理方法”を守らなければならない。また,申請者はGCPを実行し,ICH−GCPなどの国際共通原則を参考にし,国際マルチセンター臨床試験に参加する国の法律法規を遵守しなければならない。申請者が国際多センター臨床試験データを使用して中国で薬品登録を許可しようとしているのは、申請は少なくとも中華人民共和国を含む2つの国に関連しなければならず、“国際多センター臨床試験ガイドライン(試行)の発表に関する通知”や“薬品登録管理方法”などの関連法律法規の臨床試験に対する要求に符合しなければならない。
薬物臨床試験登録
“薬品登録管理方法”によると、申請者は薬品監督管理局の許可を得た後、薬物臨床試験を行う前に、薬品臨床試験登録と情報公告プラットフォームに臨床試験方案情報を登録しなければならない。
“薬物臨床試験情報プラットフォーム公告”によると、国家薬品監督管理局の許可を得て中国で行われたすべての臨床試験は必ず臨床試験登録を完成し、そして薬物臨床試験情報プラットフォームを通じて試験情報を発表しなければならない。出願人は、第1の被験者が試験に参加することを登録する前に、試験の唯一の登録番号を取得し、いくつかの後続情報の登録を完了するために、INDの承認を得てから1ヶ月以内に試験事前登録を完了しなければならない。IND承認後1年以内に登録が完了していない場合は,出願人は説明を提出しなければならず,出願人の最初の提出が3年以内に完了していない場合,INDの承認は自動的に失効するであろう。
臨床試験段階およびCDEとのコミュニケーション
“薬品登録管理方法”によると、臨床試験は1期、2期、3期、4期と生物学的同等性試験に分けられる。研究内容は薬品の特徴と研究目的以外に、“薬品登録管理方法”が規定した臨床薬理研究、探索性臨床試験、検証性臨床試験と発売後の研究を含まなければならない。
“薬品研究,開発と技術審査コミュニケーション管理方法”によると,研究開発期間や革新新薬の登録申請(など)では,申請者はCDEとのコミュニケーション会議を提案することができる。コミュニケーション会議は三つのタイプに分けることができる。第一種類の会議を開催し、薬物臨床試験における肝心な安全問題と突破的治療薬物研究と開発中の肝心な技術問題を討論した。第二種会議は薬物の重要な研究と開発期間中に行われ、主にIND申請前の会議、第二段階試験終了時、第三段階試験開始前の会議を含む
46
新薬発売申請とリスク評価·制御会議を提出する前に。第3のクラス会議とは、第1のクラスまたは第2のクラスに属さない会議を意味する。
人類の遺伝資源の収集と保存
“中華人民共和国人類遺伝資源管理条例”は更に、薬品と医療器械の中国国内での上場許可について、人類遺伝資源材料を輸出していない臨床機関で中国人類遺伝資源を利用した国際臨床試験協力を展開し、承認する必要がないと規定している。しかし、双方は使用しようとする人類遺伝資源の種類、数量と用途を国務院科学技術行政部門に報告しなければならず、臨床試験を開始することができる。“人類遺伝資源サンプリング、採集、取引、出力或いは海外審査行政許可事項サービスガイドライン”(以下は“サービスガイドライン”と略称する)に基づいて、外商投資主催部門の人類遺伝資源のサンプリング、採集或いは研究活動は国際協力範疇に属し、中華人民共和国協力機構は中国人類遺伝資源管理弁公室の許可を得なければならない。
“中華人民共和国人類遺伝資源管理条例”によると、人類遺伝資源材料を輸出していない臨床機関で中国人類遺伝資源を利用した国際臨床試験協力を展開し、承認を得ることなく中国で関連薬品と医療機器の発売許可を得ることができる。しかし,双方は臨床試験開始前に,国務院科学技術行政部門に使用しようとするヒト遺伝資源の種類,数量,用途を申告しなければならない。
2023年6月1日、科学技術部は“人類遺伝資源管理条例実施細則”(“HGR実施細則”)を公表し、2023年7月1日から施行した。“人類遺伝資源管理実施細則”は“中華人民共和国人類遺伝資源管理条例”を細分化し、人類遺伝資源情報の定義を含むが、細分化し、外来実体の認定標準を明確にし、採集許可範囲を調整し、国際協力科学研究の審査手続きと行政監督管理規則を調整と完備した。
薬品の発売登録
“薬品登録管理方法”によると、申請者は薬学、薬理学、毒理学研究及び薬品臨床試験を完成しなければならない。申請者は品質標準を確定し、商業規模の製造技術を検証し、薬品登録審査を受ける準備をした後、提出要求に従って薬品上場許可申請と関連研究材料を提出しなければならない。申請が提出されると,CDEは薬剤師,医療専門家,他の技術専門家を集めてその薬剤の安全性,有効性,品質管理を分析する。総合審査を経た後、この薬品の発売を許可し、薬品登録証を発行した。
販売許可所有者制度
“薬品管理法”によると、国家は薬品管理に対して薬品上場授権者制度を実行する。薬品上場授権者は薬品登録証を取得し、薬品管理法の規定に従って薬品の非臨床研究、臨床試験、生産経営、発売後の研究、副作用のモニタリング、報告と加工を担当する企業或いは薬品開発機構である。
中国食品薬品監督管理局の“薬品上場許可所持者制度の試験的な関連事項の推進に関する通知”(“薬品上場許可所持者制度に関する通知”)によると、薬品発売許可所持者は必ず薬品品質保証システムを構築し、そして専門人を配備して独立して薬品品質管理を担当しなければならない。そのほか、薬品発売許可保持者は定期的に薬品メーカーと薬品流通業者の品質管理システムを審査し、その持続的な品質保証と制御能力を監督しなければならない。薬品発売許可所持者が自ら薬品を生産する場合は,“薬品経営許可通知”の規定に従って薬品生産許可証を取得しなければならない
47
保有者制度は、合格した薬品メーカーに依頼する。国務院薬品監督管理機構は薬品委託生産品質指針を制定し、薬品上場授権者を指導と監督し、生産企業に薬品品質保証義務を履行することを委託した。2023年10月17日から施行された“国家薬品監督管理局の薬品上場許可所持者の製造監督管理の強化に関する公告”は、薬品上場許可所持者の委託製造に対する監督管理の重要性を再確認し、薬品上場許可所持者が製造を委託する許可、品質、監督管理などの方面に対して更に厳格かつ細分化された要求を行った。
薬品登録分類
“薬品登録管理方法”によると、薬品は中華人民共和国薬品、化学薬品、生物製品とその他の薬品に分けられる。国家薬品監督管理局の“化学薬品登録分類改革方案の印刷発行に関する通知”に基づいて、化学薬品の登録分類を5種類に調整した。第一種類の薬物は世界のどこにも発売されていない革新的な化学薬物である。世界ではどこでも販売されていない改良された新しい化学薬は第二類に属する。国外ですでに発売されているが、中国国内で発売されていないオリジナル薬品と同等の品質と治療効果を有する模造薬は、第三種類の薬品とされている。元の薬品の品質と治療効果に相当し、すでに中国で発売された模造薬は第四種類の薬品に属する。第5種類の薬物はすでに国外で発売されているが、中国ではまだ承認されていない薬物である。1種類、2種類の薬品は必ず“薬品登録管理方法”の新薬登録申請プログラムに従って処理しなければならない;3種類、4種類の薬品は必ず模倣薬申請プログラムに従って処理しなければならない;5種類の薬品は必ず“薬品輸入管理方法”の申請と管理要求に従って処理しなければならない。
“化学薬物登録分類及び申請資料規定”によると,革新化学薬と改良新化学薬は5.1類に分類され,海外で発売されているはずであるが中国で承認されていない後発化学薬は5.2類に分類されている。
珍しい病気に対する薬品特別審査と迅速な審査
“珍しい病気名簿の制定手続きに関する通知”によると、珍しい病気の命名は同時に以下の4つの標準を満たさなければならない:(1)疾病は国内外で発病率或いは流行率が比較的に低い;(2)疾病は患者及びその家族に重大な影響がある;(3)明確な診断方法がある;(4)疾病は経済的に実行可能な方式で治療或いは干与を行うことができ、疾病に対して有効な治療或いは干与がなければ、すでにそれを国家科学研究プロジェクトに組み入れた。ある珍しい病気に対する薬物が国家珍しい病気のリストに登録されているため、1つの会社は国家薬品監督管理局のこれらの疾病の新薬に対する優先審査と許可を得る資格があるかもしれない。
“新薬登録特別審査管理規定”によると、以下の状況は新薬登録申請に適用される:(一)植物、動物、鉱物から抽出した薬物の有効成分及びその製剤は中国で発売されたことがなく、しかも新たに発見された原料薬及びその製剤を発見した;(二)薬品の化学原料及びその製剤と生物製品は許可されずに国内或いは国外で発売された;(三)エイズ、悪性腫瘍などの疾病に対して明らかな臨床治療優勢を有する新薬;または(4)現在有効な治療が不足している疾患に対する新薬。
“国務院の薬品医療機器審査制度改革に関する意見”に基づいて、革新薬品に対して特別審査制度を実行し、エイズ、癌、重大伝染病、珍しい病気などの疾病の予防と治療革新薬品審査審査の過程を加速する。
国家薬品監督管理局と国家薬品監督管理局の“薬品登録審査の最適化に関する公告”によると、CDEは優先審査審査許可範囲に組み入れられた登録申請の審査、審査と審査に資源を優先的に配置する。
良好な製造規範
48
薬品管理法により、薬品生産活動に従事するにはGMPを遵守し、健全なGMP管理制度を確立し、薬品生産全過程が法定要求と国務院薬品監督管理部門が薬品管理法に従って制定したGMP要求に符合することを確保すべきである。薬品生産企業の法定代表者と主要責任者は企業の薬品生産活動に対して全責任を負う。
“薬品良好生産規範”は製品の品質管理、組織と人員配置、生産場所と施設、設備、材料と製品、認可と検査、文書維持、生産管理、品質管理と品質保証、製品の契約製造と契約検査、製品交付とリコールに指導を提供した。
薬品生産許可証
国家薬品監督管理総局が公布した“薬品生産監督管理方法”によると、薬品生産活動に従事する人員は、薬品生産活動に従事する省、自治区、直轄市の薬品行政管理部門の許可を経て、国家薬品監督管理総局が公布した“薬品生産監督管理方法”に従って“薬品生産許可証”を取得し、薬品生産品質管理規範を厳格に遵守し、生産過程が常に法定要求に符合することを確保しなければならない。“薬品生産許可証”の有効期間は5年である。“薬品生産許可証”が満了しても薬品の生産を継続する必要がある場合は、“薬品生産許可証”の有効期限が6ヶ月に達する前に元の発行機関に“薬品生産許可証”の再発行を申請しなければならない。
“医薬品経営許可証”と“良好な供給規範”の要件
“薬品管理法”と“中華人民共和国薬品管理法実施条例”の規定によると、会社は薬品の卸売、小売業務に従事し、現地の薬品監督管理機構が発行した“薬品経営許可証”及び相応の“経営範囲”を取得し、そして国務院薬品監督管理局が公布した“薬品生産品質管理規範”を守らなければならない。2024年1月1日から施行される“薬品経営使用品質監督管理方法”によると、“薬品経営許可証”の有効期限は5年である。“薬品経営許可証”の保持者は、許可証の有効期限が満了する2ヶ月から6ヶ月以内に延期を申請しなければならない。そうでなければ、所有者は薬品監督管理機関が新しい“薬品経営許可証”を発行するまで、有効期限が満了した時に経営活動を停止しなければならない。
中華人民共和国の製薬業に関するその他の規定
薬品のリコール
“薬品リコール管理方法”によると、薬品リコールとは薬品上場許可所持者が規定のプログラムに従ってすでに発売されたが、品質問題或いはその他の安全上の危険が存在する薬品をリコールし、そして相応の措置を取って適時にリスクを管理管理し、危険を除去する活動である。品質問題或いはその他の安全上の危険は、薬品が研究開発、生産、貯蔵、ラベルなどの原因で、法定の要求に符合しない、或いは他の人体の健康と生命安全を脅かす可能性のある不合理なリスクが存在することである。
新薬の行政保護期と監視期
“中華人民共和国薬品管理法実施条例”によると、公衆の健康を保障する必要に基づいて、国家薬品監督管理局は薬品生産企業が生産した新薬に対して5年以下の観察期間を設定することができ、観察期間内に他の生産企業の生産或いは輸入を許可してはならない。
医薬製品の包装
49
薬品管理法の規定によると,薬品包装はラベルを印刷または貼付し,規定に基づいて文献を含まなければならない。“薬品包装管理方法”によると、薬品包装は必ず国家標準と業界標準に符合しなければならない。包装基準に適合する薬品は開発と承認されておらず、中国国内で販売または販売してはならない(軍用薬品を除く)。中華人民共和国薬品監督管理局の規定によると、研究製品の包装ラベルは臨床試験のみの情報、臨床試験情報と臨床試験に用いる薬物情報を明記しなければならないが、盲目的な試験状態を維持することができる。
挿入薬品説明書とラベル
“薬品説明書とラベル管理規定”の規定によると、薬品説明書とラベルは国家薬品監督管理局の審査を通過しなければならない。薬品説明書は薬品の安全性と有効性に関する重要な科学データ、結論と情報を含み、安全で合理的な薬品使用を指導すべきである。薬品の内ラベルは薬品の名称,適応あるいは機能,強度,用量と用途,生産日,ロット番号,有効期限,メーカーなどの情報を表記すべきであり,外ラベルは薬品の名称,成分,説明書,適応あるいは機能,強度,用量と用途,副作用,禁忌症,注意事項,貯蔵,生産日,ロット番号,有効期限,承認文番号とメーカーなどの情報を表記しなければならない。
医薬製品の広告宣伝
“薬品、医療機器、栄養補助食品、医療専用食品広告審査暫定管理方法”の規定によると、薬品広告の内容は国務院薬品監督管理部門が許可した薬品説明書に基づいていなければならない。薬品広告は薬品名称、適応或いは主要な作用、薬理作用に関連するものであり、説明書の範囲を超えてはならず、目立つ位置に禁忌症と副作用を表示しなければならない。処方薬広告は顕著な位置に“広告は医療や製薬専門家のみが読む”と表記しなければならず,非処方薬広告は顕著な位置に非処方薬ラベル(OTC)を表示し,顕著な位置には“薬品説明書や薬剤師の指導の下で薬品を購入·使用してください”と表示しなければならない。
薬物技術移転
薬品技術移転とは,所有者が薬品生産技術を譲渡先である薬品生産企業に譲渡し,譲渡先である薬品生産企業が“薬品技術譲渡登録管理規定”の規定に従って登録を申請することである.国家薬品監督管理局は“薬品技術移転登録管理規定”を公布し、薬品技術移転登録の申請、評価、審査、承認と監督を含む薬品技術移転登録プログラムを規範化した。薬品技術移転申請は必ず省薬品監督管理部門に提出しなければならず、国家薬品監督管理局は最終的に薬品審査評価センターの総合的な意見に基づいて承認決定を下す。条件を満たした申請は承認状と補充申請の薬品承認番号を受け取るだろう。
ネット薬品情報サービス
“インターネット薬品情報サービス管理方法”によると,経営的インターネット薬品情報サービスとは,インターネットを介して医療情報(医療機器を含む)などのサービスを提供する活動である.ウェブサイトがインターネット薬品情報サービスを提供するのは、必ず国家薬品監督管理局地方省級主管部門に申請し、国家薬品監督管理局の審査を経て、インターネット薬品情報サービス資格を取得することができる。“インターネット薬品情報サービス資格証明書”の有効期間は5年であり、関係部門の再審査により、有効期限が満了する前に少なくとも6ヶ月継続することができる。
薬品の集中調達と使用
50
“国務院弁公庁の国家薬品集中調達使用試験方案に関する通知”と“国家医療保障局の国家薬品集中調達使用試験医療保険セット方法に関する意見”に基づいて、11の試験都市を国家組織薬品集中調達使用試験都市として選択した。集中的に調達された薬品範囲は,後発薬に対応する後発薬名の中から品質と治療効果の一致性で評価された品種を選択した。試験地区の公立医療機関は調達を完了した後、中選薬品を優先薬品とし、試験購入期間内の中選薬品使用量は非中選薬品より低くない。
国家医療保障局、国家薬品監督管理局などの多部門が発表した“国家薬品集中調達使用試験地区の範囲拡大に関する実施意見”によると、国家組織薬品集中調達と使用数量集中調達モデルは全国で普及している。このモデルは国家組織薬品の集中調達と使用試験中の25種類の指定後発薬に適用される。
国家医療保険計画のカバー面
“国務院の都市従業員基本医療保険制度の構築に関する決定”によると、都市部のすべての使用者は従業員のために基本医療保険に加入し、使用者と従業員は共に保険料を納めなければならない。“国務院の都市住民基本医療保険試験に関する指導意見”によると、試験区の都市住民は自発的に都市住民の基本医療保険に参加することができ、都市従業員ではない。“国務院の都市·農村住民基本医療保険制度の統合に関する意見”は、都市住民基本医療保険と新型農村協力医療制度の統合を要求している。また、“国務院の試験都市住民基本医療保険に関する指導意見”は統一的な基本医療保険制度を構築し、農民工以外のすべての都市部住民と都市部従業員の基本医療保険に加入する柔軟な就業者をカバーした。計画参加者は,医療保険目録に含まれる薬品費用の全部または一部を精算する資格がある。
“都市従業員薬品基本医療保険カバー範囲管理暫定方法に関する通知”によると、医療保険目録に登録された薬品は必ず臨床必要、安全、有効、価格合理、使用便利、数量十分な薬品でなければならず、そして以下の要求の一つに符合しなければならない:(1)“中華人民共和国薬典”に登録し、(2)国家薬品監督管理局が公布した標準に符合し、(3)国家薬品監督管理局の許可を得て輸入薬品を輸入する。
中華人民共和国労働·社会保障部は他の政府部門と国家医療保険目録に組み入れられた薬品を確定する権利がある。国家医療保険目録に組み入れられた西洋薬と中成薬はA部分とB部分に分け、省級政府はすべて国家医療保険目録に登録されたA部分とB部分の薬品を本省医療保険目録に入れることを要求した。甲方薬品の使用による費用は,基本医療保険の規定で精算することができる。B部分を使用した費用は患者と基本医療保険が分担する。B一部の薬品の基本医療保険精算割合は地方当局によって規定されているため,中国では地域によって異なる可能性がある。
国家基本薬目録
“国務院弁公庁の国家基本薬物制度の完備に関する意見”“国家基本薬物目録管理方法”と“国家基本薬物目録(2018)”(“国家基本薬物目録”)に関する通知によると、政府が出資する基本医療機関(主に県級病院、県級中病院、農村診療所とコミュニティ診療所)は国家基本薬物目録に登録された薬品を貯蔵と使用しなければならない。“国家基本薬物目録”に登録された薬品は、集中入札調達を実行し、国家発改委価格管理を実行しなければならない。救済する
51
国家基本薬物目録中の薬品は国家基本医療保険目録に登録され、この薬品の購入価格は全額精算する権利がある。
医療保険清算基準
“国務院の都市従業員基本医療保険制度の構築に関する決定”、“新型農村協力医療制度の構築に関する意見”、“国務院の試験都市住民基本医療保険の構築に関する指導意見”、“国務院の都市·農村住民基本医療保険制度の統合に関する意見”に基づいて、都市と農村の従業員と住民の医療保険の全カバーを実現する。
“全国都市部従業員基本医療保険がカバーする医療サービス施設の診療管理、範囲と支払い基準に関する意見通知”によると、基本医療保険計画は一部の診療機器費用と診断検査費用をカバーする。精算範囲と精算料率は省級政策によって決定される。
国務院弁公庁が発表した“基本医療保険の支払い方式改革の更なる深化に関する指導意見”の主な目的は、診断関連グループ、一人当たりの上限と一人当たりのベッド日上限を含む多元化精算メカニズムを発展させることである。これらの新たな精算制度は、従来のサービス種別や製品価格に基づく精算方式に代わって全国的に実施されている。
価格規制
価格が市場によって決定される薬品に対して、“中華人民共和国薬品管理法”は、薬品価格は市場によって決定され、発売許可保持者、薬品生産経営者、薬品販売機関と医療機関は公平、合理、誠実信用、質価格が一致する原則に従って定価を行わなければならないと規定している。薬品と医療機関の上場許可所持者、生産経営者、ディーラーは国務院薬品価格主管部門が制定した薬品価格の薬品価格管理規定を遵守し、暴利、価格独占、価格詐欺などの行為を禁止しなければならない。
“中華人民共和国価格法”によると、薬品価格の確定は価値規則に従わなければならない。大多数の商品とサービスの価格は市場調節価格であり、ごく少数の商品とサービスの価格は政府指導価格或いは政府定価である。医薬製品の価格は主に市場競争によって決定される。政府は主に集中調達メカニズムを構築し、医療保険精算基準を改訂し、医療と価格行為に対する監督管理を強化することによって価格を制御し、直接の政府価格規制ではない。
国家医療保障局が公布した“現在有効に薬品価格管理を展開することに関する意見”は、薬品価格形成メカニズムを更に改善し、市場化された薬品定価メカニズムを強調することを求めている。麻酔薬品と第一種類の精神薬品は政府定価を実施しているが、他の薬品は薬品経営者が市場に基づいて定価している。同時に、国家と省級医療保障部門は第三者に薬品サプライヤーに対する価格コスト調査を実施或いは依頼することができ、調査結果は薬品が不公平な価格で販売されているかどうかを確定する根拠とすることができる。
薬品の流通と2発制
“公立医療機関の薬品購入2枚の領収書制度の推進に関する実施意見(試行)”(“2票制通知”)は薬品流通過程中に2枚の領収書しか発行しないことを要求する制度であり、即ち薬品生産企業は薬品販売店に1枚の領収書を発行し、薬品ディーラーは公立医療機関に別の領収書を発行する。2枚の領収書制度はチェーン流通業者による製品の販売を排除し、公共医療機関の最終薬品価格の上昇を招く。製薬会社は2枚の領収書制度を守らなければならず、公立病院との調達プロセスを行うことができる。
52
“国務院弁公庁の薬品生産流通使用政策の更なる改革完備に関する若干の意見”に基づいて、公立病院改革試験地区で2枚の発券制を普及させ、すでに2018年に全国で推進した。製薬会社は2枚の領収書制度を守らなければならず、公立病院との調達プロセスを行うことができる。
知的財産権に関する規定
特許
“中華人民共和国特許法”及び“中華人民共和国特許法実施細則”によると、発明創造とは、発明、実用新案又は意匠をいう。特許権を付与する発明及び実用新案及び発明創造は、新規性、創造性、及び実用性を有しなければならない。国家知的財産権局特許庁は特許出願の受理と承認を担当している。発明特許の保護期間は20年,実用新案特許の保護期間は10年,意匠特許の保護期間は15年であり,特許出願の日から計算される.特許権者又は利害関係者は,特許権者の許可を得ずに,特許の使用又は他の特許侵害を行う個人又は単位を人民法院に起訴することができ,侵害者に直ちに侵害行為を停止させるか,又は侵害者に罰金を科すように規制部門に請求することができる。犯罪を構成する者は,関連法律に従って特許侵害者の刑事責任を追及する.“中華人民共和国特許法”によると、公衆衛生の目的で、中華人民共和国国家知的財産権局は、特許薬品を生産し、中国が加入した関連国際条約がカバーする国又は地域に輸出することに対して強制許可を付与することができる。また,特許法によれば,いずれの組織や個人が外国で中国に設立された発明特許又は実用新案特許を出願しても,国家知的財産権局に秘密審査を報告しなければならない。
商標
“中華人民共和国商標法”と“中華人民共和国商標法実施条例”によると、登録商標の有効期間は10年であり、登録が承認された日から計算される。商標登録者が満了して登録商標を継続して使用する場合は,満了前12ヶ月以内に規定に従って更新手続きを行わなければならない。そうしなければ、商標登録者は6ヶ月の猶予を得ることができる。1回の継続期間は10年で、最後の有効期限が満了した日の翌日から計算されます。猶予期間内に更新手続きをしていない場合は,商標登録を取り消す。
著作権所有
中華人民共和国の著作権は“中華人民共和国著作権法”と“中華人民共和国著作権法実施条例”によって保護されている.これらの法律法規は作品の分類および著作権とその関連権利の取得と保護を規定している。
ドメイン名
ドメイン名は工業·情報化部が発表した“インターネットドメイン名管理方法”と中国相互接続ネットワーク情報センターが発表した“中国国家コードトップレベルドメイン登録実施細則”によって保護されている。工信部は中国インターネットドメインの管理を担当する監督管理機関である。中国相互接続ネットワーク情報センターは中国国家コードトップクラスドメイン登録管理を担当している。
商業秘密
“中華人民共和国反不正競争法”及び“最高人民法院の商業秘密民事事件適用法律の適用に関する最高人民法院の規定”によると、商業秘密とは、公衆に知られておらず、実用性があり、その合法的な所有者又は権利者のために商業利益又は利益を創出し、その合法的な所有者又は権利者が秘密を守る技術及び商業情報をいう。“中華人民共和国反不正競争法”によると,商人が他人の商業秘密を侵害することを禁止する
53
(一)窃盗、賄賂、詐欺、脅迫、電子侵入又はその他の不正手段で権利者から商業秘密を取得するもの、(二)前項に規定する方法で、権利者から取得した商業秘密を漏洩、使用又は使用を許可するもの、(三)他人が把握している商業秘密を漏洩、使用又は使用することを許可するもの、守秘義務又は権利者の商業秘密秘密に対する要求に違反するもの、(四)他人をそそのかし、誘惑し、他人の獲得、開示、使用に協力し、他人が権利者の商業秘密を使用することを許可することは、その守秘義務又は権利者が商業秘密を秘密にする要求に違反する。
“環境保全条例”
“中華人民共和国環境保護法”,“中華人民共和国建設プロジェクト環境保全管理条例”,“中華人民共和国環境影響評価法”,“中華人民共和国固体廃棄物汚染環境防止法”によると,企業が環境汚染をもたらして公衆に危害を及ぼす物質を並べて置く場合には,経営活動中に環境保全方法とプログラムを実施しなければならない。建設プロジェクト完成後に環境に影響を与える可能性がある場合,建築施工企業は環境保全に関する部門に環境影響報告書(表)あるいは環境影響登録表を提出しなければならない。法に基づいて環境影響報告書(表)の項目を作成する必要があり,その環境影響評価文書は環境保全部門の許可を得なければならず,そうでなければ建設を開始してはならない。“除染許可管理方法(試行)”と“除染許可証管理条例”によると、除染単位は法に基づいて除染許可証を持ち、排出許可証に従って汚染物質を排出しなければならない。どの単位でも汚染物質を排出するには,排出許可証を取得しなければならない。初めて発行された排出許可証の有効期限は3年、更新された許可証の有効期限は5年。
“国務院弁公庁の”汚染物質排出制御許可制度実施方案“と”固定汚染源排出許可分類管理目録(2019年版)“の発行に関する通知によると、国は排出企業と他の製造企業の排出量、排出量と環境破壊程度に基づいて、排出権に対して集中管理、簡略化管理と登録管理を実行する。薬剤と化学薬物を製造する製造用量(簡単に混合或いは分装した化学薬物投与量の製造を除く)は厳格な監督管理の業界に属し、規定された期限に従って排出許可を取得しなければならない。
危険化学品
危険化学品安全管理条例は危険化学品の安全生産、貯蔵、使用、操作及び輸送に対して管理要求を提出した。中華人民共和国政府は危険化学品の生産、貯蔵の統一計画と合理的な配置を厳格に制御し、危険化学品の生産、貯蔵建設プロジェクトの安全条件を厳格に審査する。危険化学品を生産·貯蔵する企業は,資質のある機関を指定し,3年ごとに安全生産条件を安全評価し,安全評価報告を作成しなければならない。
“危険化学品登録管理方法”によると、国は危険化学品の登録制度を実施している。危険化学品登録は企業申請、2級審査、統一発行、分級管理の原則を実行する。登録企業が危険化学品登録手続き又は危険化学品登録種類変更又はその生産、輸入された危険化学品に新たな危険特性を有しており、危険化学品登録内容変更手続きを行っていない場合は、登録企業は修正しなければならず、5万元以下の罰金を科すことができる。登録企業が拒否して修正しない場合は、5万元以上10万元以下の罰金を科す。筋が深刻な場合は,登録企業に生産停止と廃業整頓を命じた.
製品責任
54
“中華人民共和国製品品質法”によると、メーカーはその生産した製品の品質に対して責任を負うべきであり、製品の品質が法律の規定の要求に符合することを保証し、製品に不純物或いは模倣品を混入してはならず、偽物で劣り、次に補充してはならない。販売業者はその販売されている製品の品質を確保するための措置を取るように要求されている。製品に欠陥があり,製品自体以外の人身傷害や財産損害をもたらした場合,メーカーは賠償責任を負うべきであるが,メーカーは,(一)製品が流通していないこと,(二)製品流通時に傷害や損傷を与える欠陥が存在しないこと,(三)流通時の科学技術レベルが欠陥を発見するのに不十分であることを証明できる。
“中華人民共和国民法典”によると、製品欠陥が他人の人身、財産の安全に危害を及ぼす場合、生産者、販売者は法に基づいて民事責任を負わなければならない。
労働保護
“中華人民共和国労働法”によると、使用者は法に基づいて規則制度を制定·改善し、労働者が労働権利を享受することを保障し、労働義務を履行しなければならない。使用者は健全な労働安全衛生制度を構築し、国家労働安全規程とプログラムを厳格に執行しなければならない。使用者は労働安全事故を防止し、職業危害を減少しなければならず、労働安全衛生施設は必ず国家標準に符合しなければならない。使用者は労働者のために国家が規定する安全衛生条件に符合する必要な労働防護装備を配備し、職業病危害作業に従事する労働者に対して定期的に健康検査を行わなければならない。特殊作業に従事する労働者は専門訓練を経て、相応の従業資格を取得しなければならない。
“中華人民共和国労働契約法”と“中華人民共和国労働契約法実施条例”の規定によると、使用者と労働者は書面労働契約を締結しなければ、雇用関係を構築することができない。すでに労働関係が確立されているがまだ正式な契約が締結されていない場合は、従業員が仕事を開始した日から1ヶ月以内に書面労働契約を締結しなければならない。しかも、賃金は現地の最低賃金基準を下回ってはならない。
“労務派遣暫定規定”によると、使用者は臨時、補助又は代替職場の派遣労働者しか採用できず、派遣労働者の数を厳格にコントロールしなければならず、その労働者総数の10%を超えてはならない。
社会保険と住宅積立金条例
“中華人民共和国社会保険法”、“社会保険料徴収暫定条例”、“労災保険条例”、“失業保険条例”と“企業従業員生育保険試行方法”によると、使用者は基本養老保険、基本医療保険、生育保険、労災保険と失業保険などの社会保険計画に加入しなければならない。基本年金,医療,失業保険納付は使用者と従業員が共同で納付し,労災保険と生育保険納付は使用者のみが納付する。使用者が速やかに社会保険料を納付しなかった場合は,社会保険料徴収機関が期限の追納または追納を命じ,納付日から毎日0.05%で計算した滞納金に処せ,期限を過ぎて未納した場合は,関係管理部門が未納金額の1倍以上3倍以下の罰金を科す。
住宅積立金管理条例によると、使用者は必ず住宅積立金主管管理センターに登録し、主管管理センターの審査を経た後、従業員住宅積立金納付銀行の口座開設手続きをしなければならない。使用者はまた従業員を代表して速やかに住宅積立金を十分に納付しなければならない.“住宅積立金条例”に違反し,規定期限に沿って住宅積立金管理センターに到着して従業員のために住宅積立金口座を開設していない,あるいは口座を開設していない使用者
55
従業員のために住宅積立金の口座開設手続きを行う場合は、約1,412ドルから7,062ドル以下の罰金に処せられる。
外商投資
“中華人民共和国外商投資法”及び“外商投資法実施条例”は、外国の自然人、企業又はその他の組織が直接又は間接的に行う任意の投資活動に適用され、“外商投資法”の発効日前に設立された外商投資企業は、“中華人民共和国会社法”又は“中華人民共和国組合企業法”の規定に適合するようにその法律形態又は管理構造を調整し、2025年1月1日までに改正登録を完了しなければならない。“中華人民共和国外商投資法”によると、国家は外商投資に対して参入前内国民待遇とネガティブリスト管理制度を実行し、ネガティブリスト以外の外商投資に対して国民待遇を与える。また、外商投資法実施条例では、外商投資法の有効な実施を確保するための実施方法と細則が規定されている。
2019年12月30日、中国商務部と国家市場監督管理総局は共同で“外商投資情報申告方法”を発表し、2020年1月1日から施行された。この方法によると、国内の非外商投資企業の株式を購入することによる国内非外商投資企業の増資設立または引受とその後続変更を含む外商投資企業を設立するには、企業登録システムを通じて初期報告または変更報告を提出する必要がある。
“外商投資特別管理措置(2021年版)”(“ネガティブリスト”)は株式権や高級管理者要求などの外商投資参入特別管理措置を統一的に規定している。ネガティブリストに入っていない分野に対しては,内外資一視同仁の管理原則を実行する.国内企業はネガティブリストの投資禁止分野の業務に従事し、必ず国家の関係主管部門の審査許可を経た後、海外で株式を発行して上場取引することができる。海外投資家は企業の経営管理に参加してはならず、その持分比率は海外投資家の国内証券投資管理の関連規定を参照して実行される。
対外投資に関する規定
“対外投資管理方法”によると、商務部と省級商務主管部門は投資の実際の状況に基づいて、企業の海外投資に対して届出或いは確定権管理を実行する。敏感な国や地域、敏感な業界に関連する海外投資は確認管理を実施する。その他の場合の海外投資は、届出管理を実施します。
“企業対外投資管理方法”によると、中国国内企業(“中華人民共和国投資家”)は海外投資を行い、海外投資プロジェクト(“投資プロジェクト”)の確認、届出などの手続きを行い、関係状況を報告し、そして協力して監督検査を行うべきである。中国投資家が直接或いはそれによって制御する海外企業が行う敏感な投資プロジェクトは、承認すべきである;中国投資家が直接行う非敏感投資プロジェクト、即ち中国投資家の直接出資資産或いは権益或いは融資或いは担保を提供する非敏感投資プロジェクトに関連して、届出をしなければならない。前項敏感投資プロジェクトとは、敏感国、地域又は敏感業界に関連する投資プロジェクトをいう。発改委は“対外投資敏感業界目録(2018年版)”を公表し、敏感業界を詳しくリストアップした。
企業所得税
“中華人民共和国企業所得税法”(“企業所得税法”)及びその実施細則によると、中国住民企業は2008年1月1日から現行の統一税率で25%の企業所得税を徴収している
56
特定の資格基準があります。常駐企業とは、中国国内に設立された企業と、中国国外に設立され、中国国内に有効な管理場所を設けている企業のことである。
2018年、私たちは北京市政府が発行したハイテク企業資格証明書を取得し、2021年に証明書を更新した。この更新された証明書は、米国が2021年から2023年までの3年間に15%の所得税優遇税率を享受する権利があり、関連年がHNTEの地位を得るすべての基準を満たすことを前提としている。
非住民企業の税収手配
吾は中国の付属会社に直接オフショア親会社の配当金(あればある)を支払うには10%の税率で中国の源泉所得税を納めなければならないが、このような配当金はオフショア親会社の中国での任意の設立或いは場所と実際の関連がない。適用される税収条約又は税収手配の規定により、10%の源泉所得税税率を減免することができる。香港と中国大陸部は税務手配があり、いくつかの条件と要求(その中を含む)を満たす場合、香港住民企業が中国企業の少なくとも25%の持分を直接所有し、かつ配当の“実益所有者”である場合、配当金について5%の源泉徴収税を徴収しなければならないと規定されている。企業所得税法及びその実施規則によると、非住民企業が中国住民企業の株式を売却して得た収益は、10%の税率で中国の所得税を納付しなければならない。適用される税収条約または税収手配により、10%の源泉所得税税率を減免することができる。収益は販売収益と元の投資基盤との差額から計算される。もし中国住民企業の株式を直接譲渡する場合も、印紙税を払わなければならない。印紙税は譲渡価値の0.05%で計算され、譲渡者と譲受人がそれぞれ支払う。
“国家税務総局の非住民企業所得税源控除に関する問題に関する公告”(“第37号通知”)は、株式譲渡収入と課税根拠の定義、事前提出金額を計算する際に使用する外国為替レート及び事前提出義務の発生日などを含む上述の制度中のいくつかの問題を明確に実行することを目的としている。具体的には、第37号通知は、非中国住民企業の分割払いで取得した源から源泉徴収すべき移転所得は、先に以前の投資を回収したコストで処理することができ、全コストを回収した後、税金を控除することができると規定している。
付加価値税(“付加価値税”)
“中華人民共和国付加価値税暫定条例”及びその実施細則によると、関連法律法規が別に規定がある以外、中国国内で貨物販売、加工、修理と交換サービスの提供、販売サービス、無形資産と不動産及び輸入貨物に従事する企業と個人は、一般的に付加価値税を支払う必要がある。
“財政部、国家税務総局の付加価値税付加価値税税率の調整に関する通知”(“通知32”)によると、17%と11%の税率が適用されていた付加価値税販売行為または輸入貨物は、税率をそれぞれ16%と10%に調整した。税率17%と輸出税還付率17%を適用していた輸出貨物に対して、輸出還付率を16%に調整した。11%の税率、11%の輸出還付率が適用されていた輸出貨物と国境を越えた課税行為に対して、輸出還付率は10%に調整された。第32号通告は2018年5月1日に施行され、第32号通告と一致しない既存の規定の代わりになった。
“増値税改革政策の深化に関する公告”(“第39号通知”)によると、16%と10%の税率を適用していた付加価値税販売行為や輸入貨物に対して、税率はそれぞれ13%と9%に調整されている。16%税率と16%輸出還付率を適用していた輸出貨物に対して、輸出還付率を13%に調整した。10%税率と10%輸出還付率が適用されていた輸出貨物と国境を越えた課税行為に対して、輸出還付率は9%に調整された。第39号通告は2019年4月1日に施行され、第39号通告と一致しない既存の規定の代わりになった。“珍しい病気薬品の付加価値税政策に関する通知”によると、珍しい病気の薬品を生産、販売し、簡略化方法に従って3%の税率で付加価値税を納めることを計算する。
57
外国為替取引
“中華人民共和国外貨管理条例”によると、中国機関と個人の外貨収支と外貨業務経営、及び海外機関と個人が中国国内で行っている外貨収支と外国為替業務経営は、すべて外貨管理が適用される。人民元は、貿易·サービスに関連する外国為替取引や配当金支払いなどの経常項目の支払いに自由に両替することができるが、事前に外国為替局または現地対応機関の承認を得ない限り、中国国外での直接投資、ローンまたは証券投資などの資本支出項目を自由に両替することはできない。
“決済管理条例”によると、外商投資企業の経常項目での外貨収入は、外国為替局が規定する最大限に保留することができる。当該限度額を超えた部分は、外国為替指定銀行又は外国為替スワップセンターを通じて販売しなければならない。
“直接投資外貨管理政策の更なる簡略化と完備に関する通知”によると、銀行は直接審査して海外直接投資項目の下の外貨登録を行うべきである。国家外貨管理局及びその支店は銀行の直接投資の外貨登録に対して間接監督管理を実施する。
“国務院の一連の行政審査事項の廃止と調整に関する決定”に基づいて、外匯局及びその支店による海外上場募集資金の送金事項に対する行政審査を取り消した。“海外上場外国為替管理に関する問題に関する通知”によると、国内会社は海外上場発行が終了した日から15営業日以内に、設立地外匯局支店に海外上場登録をしなければならない。国内会社の海外上場募集資金は国内口座に振り込むことができ、海外口座に入金することもできるが、その用途は目論見書などの開示書類の内容と一致しなければならない。
“資本項目決済管理政策の改革と規範化に関する通知”によると、資本項目中の外貨収益は、外貨決済意欲を維持する関連政策に対して明確に実施されている(海外上場募集資金リコールを含む)ことは、国内機関の実際の業務に応じて銀行で決済業務を行うことができる。国内機関の資本項目の外貨収入の決済は暫定的に100%であり、外匯局は国際収支状況に応じて適時に調整することができる。
第37号通知は、中国住民が直接或いは間接的にオフショア実体を設立或いは間接的に制御して海外投資融資を行い、その合法的に所有している国内企業資産又は株式又はオフショア資産又は権益を外国為替局現地支店に登録しなければならないことを要求した。上記各項目の外管局登録要求を遵守できなかった場合、中国の法律に基づいて外国為替規制の法的責任から逃れることになる可能性がある。“直接投資外貨管理政策の一層の簡略化と完備に関する通知”は、銀行代表外為局が第37号通知に規定する外国為替初期登録と変更登録を直接審査·手続きすることを許可する。
株式インセンティブ計画に関する規定
外匯局は、海外上場企業の株式インセンティブ計画への国内個人の参加に関する通知“又は”株式オプション規則“を発表し、海外上場企業の株式インセンティブ計画に参加する中国公民又は中国国内に1年間連続して居住する非中国公民(外国の駐中国外交者及び国際組織の駐中国代表を除く)を規定し、当該会社の所属する国内会社を通じて国内機関(株式インセンティブ計画に参加する海外上場企業の中国関連機関であってもよい)を集団的に委託すべきである。あるいは当該会社は法律に基づいて資産信託業務資格を有する他の国内機関を指定して外貨登録を行い、海外機関にオプションの行使、売買などの事項を委託する
58
当該株式又は持分の譲渡及び当該資金の調達。また、株式インセンティブ計画が実質的に変化した場合、国内機関は株式インセンティブ計画の外国為替局登録を修正する必要がある。
配当分配に関する規定
中国における外商投資企業の配当分配を管理する主要な法律、法規と規則は“中華人民共和国会社法”と“外商投資法及びその実施条例”である。これらの要求に基づき、外商投資企業は中国の会計基準と法規に従って確定した累積利益の中から配当金しか支払うことができない。ある中国の会社は、数年前の累積赤字(あればある)を補った後、毎年少なくともその税引き後の利益の10%を支出し、これらの積立金の総額がその登録資本の50%に達するまで、ある資本積立金の資金としなければならない。
企業所得税法及びその実施細則は、2008年1月1日から、非中国住民企業に申告した配当金には、中国中央政府と非中国住民企業登録成立の他の国又は地域政府との間の条約又は手配に基づいて別途減免されない限り、10%の源泉所得所得税率が適用されることが規定されている。
香港と大陸部中国は税務手配があり、香港住民企業がいくつかの条件と規定を満たす時、配当金について5%の源泉徴収税を徴収しなければならないと規定されており、その中には当該香港住民企業が当該中国企業の最低25%の持分を直接所有し、かつ当該配当金の“実益所有者”であることが含まれている。
配当金·分配その他の譲渡
一般的に、現金は、当社を通して、以下のように当社に移転する:(I)香港の法律に基づいて設立された有限責任会社(“BJC株式会社”)、または他の国内株主から出資または株主ローンの形で、必要に応じてCPIから資金を移転する;および(Ii)吾らはBJC Limitedを介してCPIに配当金または他の分配を支払うか、または他の国内株主に配当金またはその他の分配を支払うことができる。
将来、海外融資活動から調達した現金収益は、出資や株主ローン(場合によっては)を通じて私たちに移ってくる可能性があります。
BCが成立してから本年報日まで、BJC Limited、BC、BC Biomedicalの間または投資家の間には譲渡、配当、または割り当てはない(上記開示者を除いて、株主出資は含まれていない)。私たちはすべての利用可能な資金と任意の将来の収益をその業務運営に保留するつもりで、予測可能な未来にその配当金について現金配当金を支払わないと予想される。それにもかかわらず、いかなる現金配当金の決定はGyre取締役会が適宜決定し、Gyreの経営業績、財務状況、未来の見通し、契約制限、適用法律の適用制限及びGyre取締役会が関連すると考えている他の要素を含む複数の要素に依存する。その他の情報については、ご参照くださいGyre治療会社が監査した財務諸表本年度報告書の他の部分に含まれています。
ケイマン諸島法律によると、CPIは資金額の制限を受けることなく、融資または出資によってその子会社に資金を提供することができ、このような資金がCPIの最適な利益に適合し、適切な目的に使用されることを前提としている。適用支払能力規定を遵守する場合、ケイマン諸島はBCが配当方式で割り当てた資金額をさらに制限することはないが、利益またはCPIの株式割増口座(いくつかの法定制限を受けて制限されている)を除いて、いかなる配当金も支払うことができない。ケイマン諸島は株主に配当金を支払うことに源泉徴収しない。
私たちの最大の株主はビージェイシー株式会社です。香港法律によれば、BJC Limitedが配当を発表することができる場合、この配当金は、香港の法律で許可された場合に、BJC Limitedがその分配可能利益(すなわち、以前に分配または資本化に使用されていなかった累積達成利益から、以前に資本減少または再編でログアウトしなかった累積達成損失を差し引く)からのみ支払うことができる。配当金は配当金から支払うことができない。香港の法律は改装に何の制限も制限もない
59
香港ドルを外貨に両替し、香港に通貨を送金します。香港税務署の現行のやり方によると。BJC Limitedが支払った配当金は香港で源泉徴収税を支払う必要がありません。
中国の法律と法規によると、私たちは親会社やアメリカの株主への移転を含む外国為替と国境を越えた現金の移転について制限されている。親会社や米国の株主に収益を分配する能力も限られている。中国の現行法規によると、吾等は中国の会計基準及び法規に基づいて定められた累積利益の中からBJC Limitedに配当金を支払うことしかできない。私たちは法定積立金の累計金額が私たちの登録資本の50%以上に達するまで、少なくとも10%の税引後利益を法定積立金として保留しなければならない。もしあれば、私たちの法定積立金に資金を提供しなければならない。これらの積立金は現金配当金として分配できない。また、私たちの収入と資産は一般的に人民元(“人民元”)で計算され、自由に他の通貨に両替することはできません。中国政府は人民元を外貨に両替することと、場合によっては通貨を中国に送金することを規制している。したがって、外貨不足は、これらのオフショア実体に配当金を支払うか、他のお金を支払うか、あるいは他の方法で外貨建ての債務を履行するために、私たちに関連するオフショア実体に十分な外貨を送金することを制限するかもしれない。我々のオフショア株主は,間接株主Gyreおよび香港法例に基づいて登録設立された有限会社ネパールホールディングス(“NEPENTH”)、英領バージン諸島に登録設立された有限責任会社Ratel Holdings Limited(“Ratel”)、香港法例登録により設立された有限責任会社Aling Limited(“Aering”)、英領バージン諸島に登録設立された会社Rossefinch Holdings Limited(“Rosefinch”)、CPIおよびそれぞれGyre全資が所有するもう1社の挑戦者である。
私たちは組織内のキャッシュフローのための厳格な統制と手続きを作った。実体間、国境を越え、米国株主への現金移転のたびに内部承認を受ける必要がある。現金振込を実現するためには、支払い領収書の発行、インターネットバンキングシステムへのログイン、その検証プロセスの完了、領収書のチェック、および支払いの実行を含むが、これらに限定されないいくつかのステップを実行する必要がある。一人の従業員は、現金転送の各段階を完了することを許可せず、プログラム全体の一部のみを完成させることを許可する。財政部門だけが現金送金をする権利がある。財務部門内では、支払い承認、支払い実行、記録保存、監査の役割は分離されており、リスクを最小限に抑える。
“中華人民共和国外商投資法”及びその実施細則に基づいて、外国投資会社の管理法律枠組みを共同で確立する“中華人民共和国外商投資法”の規定によると、外国投資家は他の適用法律に基づいて、その出資、利益、資本収益、資産処分所得、知的財産権、取得した特許権使用料、獲得した賠償或いは賠償及び清算によって得られたものを、人民元、人民元又は任意の外貨で中華人民共和国国内で自由に調達或いは呼び出しすることができ、いかなる単位又は個人は通貨、金額及び頻度においてこのような調達を不法に制限してはならない。中国会社法及びその他の中国の法律及び法規によると、吾等は彼などが中国の会計基準及び法規に基づいて定めたそれぞれの累積利益の中から配当金を派遣するしかない。また、私たちは毎年、その基金の総額がその登録資本の50%に達するまで、累積された税引後利益の少なくとも10%をある法定準備基金として計上しなければならない。法定積立金が前財政年度の損失を補うのに不十分であれば、法定積立金を引き出す前に、本財政年度の累積税引後利益を赤字補填に用いるべきである。吾らは適宜中国会計基準に基づいて当社の税引後プレミアムの一部を適宜準備基金に振り込むことができる。
人民元は自由に他の通貨に両替できません。中国政府は人民元を外貨に両替することと、場合によっては通貨を中国に送金することを規制している。利用可能な外貨の不足は、これらのオフショア実体に配当金を支払うか、他のお金を支払うか、あるいは他の方法で外貨建ての債務を履行するために、私たちに関連するオフショア実体に十分な外貨を送金することを制限するかもしれません。私たちのオフショア株主はGyreであり、これは間接株主であり、NEPENTH、Ratel、Aering、Rosefinch、CPI、およびさらなるChallengerであり、それぞれGyre全額によって所有されている。現在、人民元は“経常項目”の下で両替可能であり、“経常項目”には配当金、貿易、サービス関連の外貨取引が含まれているが、“資本項目”の下で両替することはできず、“資本項目”には外国直接投資と外貨債務が含まれており、岸子会社で獲得する可能性のある融資を含む。
60
現在、国家外国為替管理局や外国為替局の承認なしに、一定の手続き要求に応じて、外貨を購入して決済する“経常項目取引”を行うことができる。しかし、中国の関係政府当局は、将来的に外貨を購入して経常口座取引を行う能力を制限または撤廃する可能性がある。中国政府は引き続きその資本規制を強化する可能性があり、外管局は経常口座と資本口座に同時に属する国境を越えた取引に対して追加の制限と実質的な審査手続きを実施する可能性がある。いかなる既存と未来の通貨両替制限も、人民元で発生した収入を利用して、中国国外での業務活動に資金を提供したり、外貨で私たちの証券所有者に配当金を支払う能力を制限することができます。資本項目下の外国為替取引は依然として制限されており、外管局やその他の関連中国政府部門の承認や登録を得る必要がある。これは私たちが債務や株式融資を通じてBCとその子会社に外貨を獲得する能力に影響を及ぼす可能性がある。
従業員と人的資本
2023年12月31日まで、私たちは全部で593人、その中で北京は168人、滄州は40人、他の地区は385人で、主に全国各地での販売とマーケティング従業員です。私たちは多くの要素に基づいて従業員を募集し、仕事の経験、教育背景、関連ポストの空きの要求を含む。私たちは製品知識、プロジェクト開発、チーム建設など、私たちの管理者や他の従業員に様々な分野の内部と外部訓練を提供します。私たちは定期的に従業員にフィードバックを提供し、従業員の表現に基づいて従業員を評価し、彼らの給料、昇進と職業発展を確定する。
関連する中国労働法によると、私たちは私たちの従業員と個人雇用契約を締結し、内容は条項、給料、ボーナス、従業員福祉、職場安全、解雇理由などを含む。私たちの従業員の給与待遇には給料とボーナスが含まれており、一般的に彼らの経歴、業界経験、職位、表現によって決められています。私たちは、国内の競争相手と比較して、私たちの従業員の給与待遇に競争力があると思う。2022年12月31日と2023年12月31日までの年度内に,従業員に社会保険と住宅積立金を十分に納付した。
私たちはまた中国の安全法律と法規によって制限されている。私たちは、私たちの生産センターでの保護措置を含め、安全な作業環境を維持するために、様々な内部職業健康とセキュリティプログラムを実施し、私たちの設備や施設を定期的に検査して、安全上の危険を識別し、解決し、定期的に従業員の安全意識訓練を行っている。
2023年12月31日まで、私たちは私たちの従業員を代表するための労働組合を設立した。私たちは私たちが職員たちと良い仕事関係を維持していると信じている。2023年12月31日までの年度内に、当社は職業健康安全法律又は法規違反によりいかなる重大な請求、訴訟、処罰又は行政行動を受けておらず、当社の業務に重大な影響を与えるストライキ、労使紛争又は労働行動も経験していない。
中華人民共和国の税収
“中華人民共和国企業所得税法”第2条によると、住民企業とは、中国の法律に基づいて中国に設立された企業、又は外国(地域)法律に基づいて設立されたが、場所を有効に管理する中国にある企業をいう。中国税務について言えば、私たちは中国住民企業です。私たちは中国北京に登録した法人実体ですから。
吾らは中国住民企業であるため、企業所得税については、吾らが直接オフショア親会社に支払う配当金(あればある)は10%の税率で中国の予定所得税を納めなければならないが、このような配当金はオフショア親会社の中国での任意の設立或いは場所と実際の関連がない。また、非住民企業が中国住民企業の株式を売却して得た収益は、10%の税率で中国所得税を納付する。適用される税収条約又は税収手配の規定により、10%の源泉所得税税率を減免することができる。“”というタイトルの部分を参照リスク要因-私たちの中国での業務運営に関連するリスク-Gyre PharmPharmticalsは中国北京に登録された法人実体であるため、中国所得税については、同社はデフォルトで中国税務住民に分類され、この分類はGyre PharmPharmticalsとその非中国株主が不利な税収結果に直面している.”
61
株式を売却またはその他の方法で処分することにより実現された収益については、中国税務機関は、国家税務総局の“非住民企業間接財産譲渡徴収企業所得税の若干の問題に関する公告”又は“国家税務総局第7号通知”に規定されている間接譲渡規則に基づいて所得税を徴収することができるが、このような取引はその規定の避難港に属する可能性がある。“”というタイトルの部分を参照リスク要因-私たちの中国での業務運営に関するリスク-Gyre PharmPharmticalsとその株主は、中国住民企業の株式の間接譲渡や中国設立による非中国会社の他の資産、または中国で設立された非中国会社に帰属する他の資産に関する不確実性に直面している。中国税務機関が買収取引の審査を強化することは、Gyre PharmPharmticalsのオフショア再編取引やGyre PharmPharmticalsオフショア持ち株会社の株の売却や中国課税資産に関連する投資にマイナス影響を与える可能性がある.”
第1 A項。リスク要素です。
以下の部分には、私たちの業務や運営に悪影響を及ぼす可能性のある最も重要な要素が含まれています。私たちの普通株への投資を決定する前に、以下に説明するリスクと不確実性、および本10-K表年次報告に含まれるすべての情報をよく考慮しなければなりません。もし実際に以下のいかなるリスクが発生すれば、私たちの業務、財務状況、経営業績と成長見通しは重大な不利な影響を受ける可能性がある。この場合、私たちの普通株の取引価格は下がるかもしれません。あなたはあなたの全部あるいは一部の投資を失うかもしれません。
リスク要因の概要
私たちの証券に投資することは高度な危険と関連がある。以下は私たちが直面している主な危険の概要だ。以下ではこのような危険についてもっと全面的に議論するつもりだ。
62
私たちの財務状況と資本要求に関連するリスク
競争が激しい環境下で、私たちの業務は中国で販売されている製品ETUARYの販売に大きく依存しており、ETUARYの販売量、定価、収益力を維持または向上させることができないかもしれない。
私たちはバイオテクノロジー会社で、中国で許可されたETUARYとある模造薬の商業販売からのみ収入を得ている。私たちは一つの製品といくつかの模造薬しか商業販売できません。他の候補製品の開発はまだ初期段階にあります。私たちはETUARYの販売に大きく依存していますが、ETUARYの販売量、定価レベルあるいは利益率を維持できないかもしれません。2023年と2022年のETUARYの売上高はそれぞれ私たちの総収入の98.9%と96.9%を占めており、ETUARYの売上高は近い将来も総収入の大きな割合を占め続けると予想されています。したがって、ETUARY売上高や利益率のいかなる低下も、我々の業務や運営結果に実質的なマイナス影響を与えることになる。
そのほか、製薬業界の特徴は科学技術が日進月歩であり、工業技術が絶えず向上し、新製品が絶えず出現し、私たちの目標市場競争を激しくさせたことである。注目すべきは、中国IPF薬物市場の特徴は競争が日々激しくなっていることであり、私たちの製品ETUARY以外に、1種のピルフェニドン製品と1種の9チダニ製品が承認され、商業化されている。いくつかの候補薬剤はすでに第2段階またはより高度な臨床試験段階に入っている。IPF薬物浸透率の向上と全体市場の拡大に伴い,過去の新市場参加者を含めて,より多くの市場主体がIPF市場に参加することが予想されるため,我々の製品ETUARYの売上高は2021年の総市場シェアの55.3%から2021年の78.8%に低下し,可能である
63
減少を続ける。詳細は“をご覧ください-業務-私たちの製品と製品のパイプ—ETUARY:2011年に承認された国家1.1類IPF新薬—市場のチャンスと競争この年間報告書にあります。中国IPF市場の新規参入者は我々のETUARY平均販売価格に下振れ圧力を加える可能性があり、これはETUARYの販売および/または利益にマイナス影響を与える可能性がある。
私たちの多くの競争相手は、外国の製薬会社を含み、私たちよりも多くの臨床、研究、監督、製造、マーケティング、財務と人的資源を持っているかもしれない。私たちのいくつかの競争相手は、私たちの既存製品または既存製品のための候補製品または新しい適応を開発している分野で研究と開発に積極的に参加しているかもしれない。他の会社は私たちよりも早くまたはより成功的に製品を発見、開発、買収、または商業化するかもしれない。また、私たちの競争相手間の製薬業界も大きな統合が生じる可能性があり、あるいは競争相手間の合弁企業が彼らの市場シェアを増加させる可能性がある。さらに、私たちの競争相手は、私たちよりも早く中国、アメリカ、または他の国で、私たちの模倣薬、ETUARY、および候補製品と同じ期待用途を持つ製品の発売承認を申請し、獲得するかもしれない。関係当局、例えば国家薬品監督管理局、食品薬品監督管理局或いはその他の類似した外国監督管理機関は、同時に同じタイプの革新薬物の複数の上場申請を審査する能力が限られている可能性がある。したがって、競争相手の製品と同時に私たちの候補製品を審査する時、これらの当局は私たちの候補製品の審査が遅れる可能性があり、私たちの製品の登録過程が延長される可能性があります。
同一疾患に対する後発薬と他の製品や療法との市場競争に加えて,本稿で検討した多くの要因リスク要因一部の条項はETUARYの販売に不利な影響を与える可能性があり、政府政策と政府医療保険カバー範囲内或いはそれ以外による定価圧力、医療界の市場認知度、生産或いは流通中断、製品品質或いは副作用の問題、及び知的財産権紛争を含むが、これらに限定されない。また、私たちは努力したにもかかわらず、新しい製品を開発したり、獲得したりすることができず、私たちの業務を多様化し、ETUARYへの依存を減らすことができないかもしれません。
私たちの業務と財務結果の未来は私たちの候補製品の臨床と臨床前段階の進展と成功に大きく依存しており、例えばETUARYの中国における未来の適応、F 573は中国、F 351は中国と中国以外の他の市場にある。私たちはそれらの臨床開発を完成できず、必要な監督管理の許可を得たり、それらの上場を成功させるリスクに直面しており、そうでなければ、私たちはこれらの過程で重大な挫折を経験するかもしれない。
私たちは大部分の財務資源を研究開発に投入して、私たちの臨床前と臨床開発活動を含む。私たちが収入を創出し、利益を達成する能力は、私たちの候補製品の開発成功、必要な規制承認、および私たちの候補製品の製造と商業化にかかっています
64
私たちは上述した1つ以上の要素をタイムリーに達成できないかもしれないし、根本的に達成できないかもしれない。そのため、私たちは製品、ETUARY、候補製品の承認および/または成功した商業化の重大な遅延や承認を得ることができず、これは私たちが計画中のマイルストーンを実現できず、私たちの薬物開発の将来性に実質的な損害を与える可能性がある。医薬品開発に関連する多くのリスクおよび不確実性のため、費用を増加させる時間や金額、または私たちがいつ、または四半期または年間の収益性を維持または向上させることができるかどうかを正確に予測することはできない。収益性を維持または向上させることができないことは、私たちの普通株の価値を低下させ、資金調達、業務拡大、研究開発努力の維持、製品供給の多様化、さらには運営を継続する能力を弱める可能性がある。私たちの普通株価値の低下はまたあなたの投資損失の全部または一部を招く可能性があります。
私たちの研究開発活動や運営の成長を支援するためには、より多くの資金が必要であり、これらの資金は優遇された条件では得られないかもしれないし、全く得られない可能性もある。もし私たちが重要な時に必要な資金を得ることができなければ、私たちは私たちのいくつかの開発プロジェクト、市場導入計画、または他の操作側面を延期、削減または停止しなければならないかもしれない。
バイオ製薬製品と候補製品の開発、商業化、製造、マーケティング、販売と流通は資本集約型である。私たちはETUARYといくつかの模倣薬だけから中国の商業販売で収入を生成し、私たちが国家薬品監督管理局、FDAあるいは他の監督機関が私たちの他の候補製品の販売を許可する前に、私たちはETUARYといくつかの模倣薬を除いたいかなる製品収入も産生できないだろう。これまで、私たちはETUARYとある模造薬の商業販売からしか収入を得ておらず、他の候補製品が規制部門の承認を得るまで、これらの候補製品から何の収入も生じないと予想されていたため、私たちは、任意の新しい臨床試験、製品開発、第三者とのパートナー関係、戦略的協力を含む未来の研究開発を支援するために、大量の追加資本を調達する必要がある。われわれが臨床前·臨床開発活動を継続すれば,われわれの候補製品の臨床前·臨床開発に関する費用が生じ続ける。しかし、我々は、中国におけるETUARYの臨床開発を継続するために大量の追加資本を調達する必要があると予想され、F 573、F 528およびF 230は中国、およびF 351の中国および潜在的な中国以外の追加市場であり、資金の獲得性に応じて、私たちの候補製品の一部またはすべての開発を延期または停止する必要があるかもしれない。私たちがより多くの資本を集めても、私たちは私たちの努力を1つ以上の開発プロジェクトに集中させ、他の開発プロジェクトを延期または停止することを選択することができる。
私たちの策略と業務計画の実施は私たちが外部融資源に部分的に依存する必要があることを期待しているが、私たちは商業的に合理的な条件で追加資本を得ることができるかどうかは様々な要素の影響を受けることができ、その中の多くの要素は私たちがコントロールできることではなく、私たちの未来の財務状況、経営業績とキャッシュフロー、世界経済状況、業界と競争状況、金利、信用市場の現行状況、政府の融資政策を含む。私たちが追加的な資金が必要な時、私たちは受け入れ可能な条件でこのような資金を得ることができないかもしれない。十分な資金がなければ、私たちは私たちの研究または開発計画の一部または全部を延期、縮小、または廃止することを要求される可能性があり、現在想定されている戦略および業務計画を実行できない可能性があり、これは私たちの業務、財務状況、および運営結果に大きな悪影響を及ぼす可能性がある。
65
我々の候補製品の開発成功はまだ確定していないため,研究·開発を完了し,我々が開発している製品を商業化するために必要な実際の資金を見積もることはできない。私たちの将来の短期的かつ長期的な資金需要は多くの要素に依存するだろうが、これらに限定されない
さらに、私たちがより多くの資金を調達する能力は、世界の経済状況の潜在的な悪化の悪影響を受ける可能性があり、米国と世界各地の信用と金融市場は最近、公衆衛生危機、ロシアとウクライナの間の衝突、ハマス-イスラエル戦争、または紅海を横断する海上船の攻撃によって中断され、変動する可能性がある。中米関係リスクの詳細については、“を参照されたい私たちの中国での業務運営に関するリスク—中国政府の政治·経済政策や中米関係の変化は、我々の業務、財務状況、経営業績に影響を与える可能性がある。もし私たちが十分な追加資本を集めることができなければ、私たちは私たちが計画した業務と私たちの戦略の追求を縮小させることを余儀なくされるかもしれない。
もし私たちの取締役会が製品候補を探して開発することを選択した場合、本節で示した候補製品の発見、開発、商業化に関するリスクと、この部分で説明した他のリスクに直面しますリスク要因一節です。
証券の発行や許可手配による追加資金の調達は、株主に希釈を与え、私たちの業務を制限したり、所有権の放棄を要求したりする可能性があります。
私たちが株式または転換可能な債務証券を売却することによって追加資本を調達する場合、株主は希釈され、これらの新しい証券の条項は、清算または普通株主の権利に悪影響を及ぼす他の特典を含む可能性がある。Piper Sandler&と株式割当契約を締結しております
66
これにより、適用される米国証券取引委員会法規を遵守した場合、当時の市場価格に応じて、“市場別”取引方式で、最大5,000万ドルの普通株を発行することが可能となる。
債務融資に関連する可能性のあるプロトコルは、追加債務を招く、資本支出を行う、または配当を宣言するなど、私たちが具体的な行動をとる能力を制限または制限する契約を含む。もし私たちが第三者との協力、戦略連合、または許可手配を通じてより多くの資金を調達すれば、私たちは私たちの技術、候補製品、または未来の収入流の貴重な権利を放棄しなければならないかもしれないし、私たちに不利な条項に許可を与えなければならないかもしれない。私たちはまた条件が有利な時に公共資本や個人資本市場に入ることを求めるかもしれないが、その時でも私たちは追加的な資本を切実に必要としていない。私たちは必要があれば、私たちが追加的な支出を得ることができるということを保証できない。もし私たちが十分な資金をタイムリーに得ることができなければ、私たちは私たちの1つ以上または全部の開発計画を延期、削減、または廃止することを要求されるか、または私たちが自分で開発し、マーケティングすることをより望んでいた候補製品を開発し、マーケティングする権利を与えることができるかもしれない。
私たちは買収部分を通じて私たちの業務を拡大することができます。これは私たちの資本金要求を増加させ、私たちの株主を希釈し、私たちに債務や負債を発生させ、私たちの業務を管理する能力に大きな悪影響を与え、このような買収を成功させたり、将来の買収後の業績を向上させることができないかもしれません。
私たちの成長を促進し、私たちの製品開発、技術進歩、流通ネットワークを利益にするために、私たちは過去に業務、製品、技術またはノウハウを買収し続けたり、戦略的パートナー関係を構築したりすることが可能になった。達成された、進行中、または潜在的な買収または戦略的パートナーシップは、多くのリスクをもたらす可能性がある
私たちがこのような買収を通じて業務を発展させる計画は期待通りには実現しないかもしれません。
我々の2023年12月31日と2022年12月31日までの年間の高毛金利は持続不可能である可能性がある。
2023年12月31日と2022年12月31日までの年間で、高水準の毛金利を維持しました。2023年と2022年12月31日までの年間の利益率はそれぞれ95.9%と95.3%であり,これは我々の成熟した技術と規模効果がコストを大幅に低下させたためである。しかし私たちが維持することは保証できません
67
未来の毛利率も同じように高くなるだろう。様々な要素は私たちの毛金利に影響を及ぼすかもしれないが、その中の多くの要素は私たちがコントロールできない。例えば、関連市場競争構造の変化はETUARYの平均販売価格を低下させる可能性があり、これは私たちの毛金利にマイナス影響を与える可能性がある。しかも、私たちの毛金利は原材料コストのような私たちのコストの様々な構成要素の影響を受けるだろう。詳細は“をご覧ください”-製品の製造と供給に関するリスク-コスト増加を私たちの顧客に転嫁できなければ、私たちの原材料とエネルギー供給の価格変動、および私たちの生産過程に関連する他のコストは私たちに実質的な悪影響を及ぼす可能性がありますこのリスク要因の部分では
2023年12月31日と2022年12月31日までの数年間、私たち最大の5人の顧客が私たちの収入のかなりの部分を占め、これは私たちを集中度のリスクに直面させた。
私たちの5大顧客は、2023年12月31日と2022年12月31日までの数年間、かなりの収入を貢献してきた。したがって,我々は信用リスクに直面する可能性があり,顧客の信用状況の変化を適切に評価し,タイムリーに反応できる保証はない.私たちの信用リスクは2023年12月31日、2022年12月31日、2022年までに10%に集中しています。また、2023年12月31日現在、50.5%と85.5%の貿易売掛金および2022年12月31日現在の45.1%と78.3%の貿易売掛金は、それぞれ私たち最大の顧客と5つの最大顧客から来ています。もしこれらの顧客のキャッシュフロー、運営資金、財務状況あるいは経営業績が低下すれば、彼らは私たちに不足している貿易売掛金を迅速にまたは支払いたくないか、または支払いたくないかもしれない。いかなる重大な違約や遅延も、私たちのキャッシュフローに重大かつ不利な影響を与える可能性があり、もし私たちがこのような顧客の違約または遅延支払いによって私たちの顧客との関係を終了すれば、これは私たちのキャッシュフローと運営に不利で実質的な影響を与える可能性があります。
もし私たちの主要顧客が未来にETUARYの購入を停止したり、注文規模を大幅に減少させたりすれば、私たちはその顧客との契約関係の終了や修正によっても、私たちとは関係のない他のいかなる理由でも、ETUARYをタイムリーに識別して他の顧客に販売することができないかもしれません。したがって、私たちの業務と財政的業績は実質的な悪影響を受けるかもしれない。
私たちは在庫が古くなる危険に直面しているかもしれない。
私たちの在庫には原材料、製品、半製品、製品が含まれています。2023年12月31日と2022年12月31日まで、私たちの在庫価値はそれぞれ430万ドルと610万ドルです。2023年12月31日までおよび2022年12月31日までの年度中には、減値準備が必要な材料在庫項目は発見されておらず、適切な在庫レベルを維持することが市場ニーズをタイムリーに満たすのに役立つと信じています。私たちは通常、推定された需要と製造能力に基づいて物資を調達し、私たちの管理システムは倉庫プロセスの各段階をカバーしている。在庫とログの一貫性を維持するために、在庫の保管と分配を密接に監視します。しかし、私たちの業務の拡大に伴い、私たちの在庫レベルは増加する可能性があり、時代遅れのリスクはそれに応じて増加するかもしれません。さらに、供給中の予期しない材料の変動や顧客の選好の変化は、需要の減少と供給の滞貨を招き、時代遅れのリスクを増加させる可能性がある。
もし私たちの無形資産が減少すれば、私たちの経営業績と財務状況は不利な影響を受けるかもしれない。
我々は主に行われている製品開発、特許、技術ノウハウ、コンピュータソフトウェアからなる無形資産を持っており、2023年12月31日と2022年12月31日現在、これらの資産は私たちの総資産のかなりの部分を占めている。私たちの無形資産の価値は私たちの経営陣が作ったいくつかの仮定に基づいている。上記のいずれかの仮定が実現できなかった場合、または私たちの業務パフォーマンスがこれらの仮定と一致しない場合、私たちは私たちの無形資産の大部分をログアウトし、重大な減価損失を記録しなければならないかもしれない。また,無形資産が減値するかどうかの決定には,無形資産の帳簿価値と回収可能金額を見積もる必要がある。帳簿金額がその回収可能金額を超えると、私たちの無形資産が減値する可能性があり、これは私たちの業務、財務状況、経営業績に大きな悪影響を及ぼす可能性があります。
私たちは顧客から売掛金を受け取る時に信用リスクに直面する可能性があります。
2023年12月31日と2022年12月31日まで、私たちの売掛金はそれぞれ1,520万ドルと1,560万ドルで、その中で主に私たちの流通業者の売掛金残高です。私たちの流動性とキャッシュフローは直接影響を受けています
68
これは私たちの顧客が適時に私たちに支払う能力があるかどうかにかかっていますが、信用評価に努力しているにもかかわらず、私たちの顧客が将来約束を違反しない保証はありません。我々の売掛金回転日数は,2023年,2023年および2022年12月31日までの年間でそれぞれ50日および46日であった。
もし私たちのどの顧客の業務、キャッシュフロー、経営状況や業績が低下した場合、そのような顧客は私たちに借りた売掛金を迅速にまたは支払いたくないか、または支払いたくないかもしれません。私たちの主要顧客の倒産や信用状況の悪化はまた私たちの売掛金に実質的な悪影響を及ぼす可能性があります。私たちが直面している信用リスク集中に関するリスクの詳細については、“を参照されたい2023年12月31日と2022年12月31日までの数年間、私たち最大の5人の顧客が私たちの収入のかなりの部分を占め、これは私たちを集中度のリスクに直面させた。このリスク要因の部分ではもし私たちに巨額の借金を返済できなかったら、私たちは重大な衝撃が発生するかもしれません。私たちの流動資金とキャッシュフローは不利な影響を受けるかもしれません。
私たちは歴史的に政府の支出を受け、税金優遇を受けていたが、将来的には政府の財政的インセンティブを受け続けることはないかもしれない。
2023年12月31日と2022年12月31日までの年間で、私たちは従来、私たちのいくつかの研究開発と製造活動に関連した政府支出を受け入れ、他の収入と収益項目の政府支出がそれぞれ100万ドルと90万ドルであることを確認した。当社その他確認された収入の詳細については、ご参照くださいGyre治療会社が監査した財務諸表本年度報告で。ハイテク企業として、2023年12月31日と2022年12月31日までの年度ごとに15%の優遇企業所得税税率を享受している。また、私たちの製品ETUARYは中国で承認され、3%税率の付加価値税割引を受けます。しかし、このような優遇待遇を提供し続けることは保証されない。私たちが政府補助金と優遇税率を得る資格があるかどうかは様々な要素に依存しますが、既存技術の改善に対する評価、関連政府政策、異なる支出機関の資金獲得可能性を含むが、これらに限定されません。また、政府財政奨励の時間、金額及び基準は中国政府当局が適宜決定する。政府の財政的インセンティブは非日常的であり、私たちが政府の激励を受け続けるという保証はない。また、いくつかの政府の財政奨励は、適用される財政奨励協定の遵守とその中の特定のプロジェクトの完成を含むいくつかの条件を満たす必要があるかもしれないが、私たちはこれらの条件を満たすことができないかもしれない。私たちが現在受けている政府の財政的補償を減らしたり廃止したりすることは、私たちの財政状況に悪影響を及ぼす可能性がある。
私たちの業務運営と候補製品に関するリスク
中国で販売されている製品ETUARYおよび将来承認される可能性のある他の任意の製品は、医師、医療機関、薬局、患者、第三者支払者、およびより広範な医療コミュニティで十分な市場受容度を得ることができない可能性があり、これはそれらの商業生存に重要である。
私たちの既存および未来に承認された製品(あれば)の商業成功は、これらの製品が達成できる市場受容の程度に依存し、特に医師、病院、薬局、および他の医療機関において、これは多くの要素に依存する。現在または将来の承認製品に対する市場の受容度に影響を与えるこのような要因は、以下のことを含む可能性がある
69
もし我々の既存および将来承認された製品(ある場合)が広範な市場受容度を獲得または維持できなかった場合、または私たちの競争相手が発売した新製品が医療従事者および患者によってより有利で、より費用対効果的であると思われ、または私たちの製品を時代遅れにする場合、私たちの製品に対する需要が低下する可能性があり、私たちの業務および収益力は実質的かつ不利な影響を受ける可能性がある。
臨床薬物開発は長く高価な過程に関連し、結果は不確定であり、著者らは開発中の薬物の臨床試験を成功させることができず、中国の未来適応のETUARY、中国のF 573とF 351及び中国以外の他の市場を含む、あるいは著者らの候補製品の安全性と有効性を証明し、監督管理機関を満足させる。
規制機関の許可を得て私たちの候補製品を販売する前に、私たちはその安全性と有効性を証明するために広範な臨床試験を行わなければならないが、臨床薬物開発自体の予測不可能性のため、このような試験が適時あるいは費用効果のある方法で達成されることは保証されない。私たちが開発した製品ETUARYは、中国のIPFを治療するために使用され、規制部門の承認を得るだけで、私たちの既存の候補製品や将来開発を求める可能性のあるどの製品も決して規制部門の承認を得ないかもしれない。成功を妨げる可能性があり、または臨床開発をタイムリーに完了する可能性のあるイベントは、
70
中国におけるETUARYの将来の適応、中国におけるF 573、F 528およびF 230、および中国以外の潜在的な追加市場を含む追加の臨床前研究または臨床試験を行うことが要求された場合、候補製品の臨床試験、中国の将来適応を含むETUARY、中国のF 573、F 528およびF 230、F 351および中国以外の潜在的な追加市場または他のテストを成功させることができなければ、またはこれらの試験またはテストの結果が陽性でない場合、または軽微な陽性である場合、またはセキュリティ問題が存在する場合、私たちは以下のようになるかもしれない
そのため、臨床試験の完成のいかなる遅延も私たちのコストを増加させ、私たちの候補製品の開発と承認過程を延期し、私たちが承認した製品を商業化し、収入を創造する能力を脅かす可能性がある。
肝線維化は私たちが注目している分野であり、私たちのこの分野での重要な候補品はF 351である。私たちの未来のF 351の臨床試験は成功しないかもしれない。
私たちはF 351の研究開発に多くの精力と財力を投入する予定だ。F 351は現在中国で第3段階臨床試験が行われており,慢性B型肝炎に関連する肝線維化の治療に世界で初めて承認された薬剤となる可能性がある。F 351は2021年3月にNMPAのCDEに画期的な治療称号を付与され,われわれの3期臨床試験の患者登録は2022年1月に開始された。2023年10月までに、中国の248名の患者の3期臨床試験は全面的に組み入れられた。しかしながら、F 351のS突破的療法指定は、F 351が最終的に米国国家薬品監督管理局または他の同様の規制機関の承認を得る可能性を増加させない。最後の患者は2024年に退院し,2025年第1四半期にF 351のNMPA申請を中国に提出する予定である。
71
さらに私たちは IND申請を積極的に準備しており,2024年末に第二段階臨床試験を提出し,F 351治療にNASHに関連する進行肝線維化を評価する予定である。IND郭清後,2025年に米国で2 a期PoC臨床試験を開始し,非肝硬化性NASHに関連する進行肝線維化患者に対するF 351の安全性,耐性,PK,PDを評価する予定である。FDAはすでに計画中のF 351 2 a段階試験の設計についてIND前の提案を提供し、IND届出の内容について指導を提供した。F 351の2 a段階試験で積極的な傾向が観察されれば,米国でF 351の第2段階試験を開始する予定である。
私たちが製品販売から任意の収入を得る前に、F 351はより多くの臨床開発、臨床、臨床前と製造活動の評価、複数の司法管轄区でのマーケティング許可、大量投資と重大なマーケティング努力が必要となる。米国食品医薬品監督管理局、FDA、および同様の外国規制機関の上場承認を得るまで、F 351や他の候補製品のマーケティングや普及は許可されておらず、そのようなマーケティング承認は決して得られないかもしれません。
F 351の成功は様々な要因に依存する。私たちは臨床開発と監督提出過程のいくつかの側面、私たちの知的財産権に対する潜在的な脅威、および任意の未来の協力者の製造、マーケティング、流通と販売努力を含むその多くの要素を完全に制御することができない。したがって、承認されても、F 351を販売することで収入を得ることができることを保証することはできません。もし私たちがF 351の商業化に成功しなかったり、商業化の面で深刻に遅延したりすれば、私たちの業務は実質的な損害を受けるだろう。
もし私たちが臨床試験の開始或いは臨床試験の患者登録過程中に遅延或いは困難に遭遇すれば、私たちの監督許可は延期或いは阻止される可能性がある。
もし私たちがこれらの試験に参加するのに十分な数の条件を満たした患者を見つけ、登録し、維持することができなければ、私たちまたは私たちの協力者は、私たちの候補製品の臨床試験を開始または継続することができないかもしれない。これは、アメリカ国家薬品監督管理局、FDAまたは中国またはアメリカ以外の同様の規制機関の要求である。
また,NASH患者の募集には固有の困難があり,現在では肝臓生検で明確に診断するしかない。具体的には,既存の臨床指標が感受性と特異性に乏しい場合には,NASH生検登録基準を満たす可能性が最も高い患者を決定することが持続的な挑戦である。したがって,NASH試験は通常ベースライン肝生検の中央審査後に高レベルのスクリーニングに失敗し,登録者数が低い可能性がある。これらの困難と臨床試験におけるNASH患者の募集との激しい競争により,われわれや将来の協力者は必要な患者を募集して臨床試験を速やかに完成させることができないか,あるいはまったく募集できない可能性がある。また,我々の競争相手の中には,我々よりもはるかに多くの資源を持ち,同じ適応のための臨床試験を行っており,それらの研究への患者の参加を求めており,そうでなければわれわれの臨床研究や試験に参加する資格がある可能性がある。合格臨床研究者の数が限られているため,いくつかの競争相手が使用している同じ臨床試験地点でいくつかの臨床試験を行うことが予想され,これらの地点で臨床試験を行うことができる患者数をさらに減少させる可能性がある。他の承認された製品および臨床試験における他の製品の可用性は、我々の臨床試験に参加したい患者数を制限している可能性がある。
患者の入選は他の要素の影響を受けている
72
十分な数の患者を臨床試験に参加することはできません。これは深刻な遅延を招き、1つ以上の臨床試験を完全に放棄することを要求するかもしれません。我々が行った臨床試験の登録遅延は,候補製品の開発コストを増加させる可能性もあり,会社価値の低下を招き,追加融資を受ける能力を制限する。
私たちの研究開発への大量の投資は、私たちの製品、ETUARY、その他の候補製品を開発し、私たちの技術を強化し、最終的には実現できないかもしれない。
世界の薬品市場は絶えず発展しており、私たちは新しい技術と新しい方法の歩みについて、私たちの競争地位を維持しなければならない。私たちの将来の成功は、絶えず変化する市場需要を満たす新製品を発売する能力にある程度依存し、特に新しい疾病や疾病を効果的に治療できる新薬を発売することができる。しかし、私たちは私たちの製品の組み合わせを適時に改善することで、新興または発展の傾向に対応できるか、あるいは根本的にできないという保証はない。2023年12月31日と2022年12月31日までの年間で、我々の製品ETUARYや他の候補製品の研究開発に関する大量の支出が発生し、私たちは引き続き大量の人力と資本資源を投入して私たちの製品や候補製品を開発するとともに、私たちの技術を強化し、私たちのパイプライン製品を推進できるようにする予定です。また,薬物発見,開発,製造における技術力を強化していく予定であり,これは資金と時間集約型である。しかしながら、私たちが新しい技術および方法を開発、改善、または適応し、新しい技術機会の発見に成功するか、または新しいまたは強化された製品を開発し、市場に押し出すことができる保証はない。
我々が中国で承認された商業化製品ETUARYや任意の他の未来製品は,承認されれば,国,省,あるいは他の政府が援助する医療保険計画から除外される可能性がある。
中国の医療保険計画によると、患者はNRDL、関連省級精算薬品リスト或いは他の医療保険精算リストに記載されている薬品の全部或いは一部の費用の精算を受ける権利がある。しかし、このような組み入れは、臨床需要、使用頻度、治療効果、安全性および価格を含む様々な要素に基づいており、これらは私たちがコントロールできるものではないかもしれない。また、中国政府の関係部門は時々医療保険精算リストに記載されている製品の精算範囲を審査·改訂したり、精算範囲を変更したりすることができる。
我々の製品ETUARYは中国で承認され,2017年以降IPF適応でNRDLのB類薬剤とされているが,このように列挙し続けたり,精算範囲の変化の負の影響を受けない保証はない。もし私たちが将来承認した候補製品がどの医療保険精算リストにも含まれていない場合、あるいはそのような保険計画が変更またはキャンセルされた場合、そのような候補製品が関連医療保険精算リストから削除され、患者は選択することができ、病院、薬局、他の医療機関は代替治療方法を推薦することができ、これは私たちの製品、ETUARY、および未来の製品への需要を減少させる可能性があり、承認されれば、私たちの販売に悪影響を与える。
我々は,中国で承認された商業化製品ETUARYや任意の他の将来の製品(承認されれば)の価格を低下させる圧力に直面する可能性があり,医療保険の精算や市場競争による製品の取得資格がある。
我々は、これらの候補製品を医療保険精算リストに含めるために、中国で承認された商業化製品ETUARYの価格や、これらの候補製品を医療保険精算リストに含めるための他の将来の製品(承認された場合)の価格を低下させる圧力に直面する可能性があり、このような低価格および精算は必ずしも販売増加を招くとは限らない。価格低下や販売増加が収益性に及ぼす純影響を見積もることは困難であり、これ以上の販売増加なしに価格を大幅に下げると、将来的に製品の販売利益が低下する可能性がある。
また,代替製品の競争が激化し,病院や病院の入札過程により,薬品の価格が製品の全ライフサイクルにわたって低下するのが典型的である
73
政府主管部門、関連政府主管部門の定価政策または製薬会社は自発的に価格を調整する。市場競争により、私たちの既存または将来承認された製品のどの戦略的な価格引き下げも、私たちの業務や経営業績に大きな悪影響を及ぼす可能性があります。
また、我々が市場で販売しているETUARYが中国の集中一括調達計画に盛り込まれる可能性がある。詳細は“をご覧ください”-将来、中国政府が制定した集中一括調達政策は、私たちの商業化製品ETUARYおよび任意の他の未来製品をカバーする可能性があり、承認されれば、これらの製品の価格は低下する可能性があり、ひいては私たちの収入、財務状況、および経営業績に重大な悪影響を及ぼす可能性があるこのリスク要因の部分では
私たちは集中入札手続きを通じて私たちの商業化製品ETUARYや他の任意の未来製品(承認されれば)を中国の公立病院に売ることができないかもしれない。
私たちが流通業者に販売しているかなりの薬品は中国の公立病院や他の医療機関に販売されているので、私たちは集中入札過程で入札を提出して、これらの機関に私たちの商業化製品ETUARYと他の未来の製品を指定された価格で供給しなければなりません。中国の各公立医療機関は一般的に省級薬品集中調達プラットフォームを通じて薬品を調達し、そして集中入札プログラムを通じて実質的にそのすべての薬品調達を完成しなければならない。我々が集中入札過程で提出する入札は,通常,代替製品に対する価格やそのような代替製品の臨床的有効性,我々の製品やサービスの品質などの要因に基づいて考えられる。したがって、私たちの販売量とETUARYの収益性は私たちの製品を差別化することに成功できるかどうかにかかっていて、利益のあるレベルで集中入札過程で成功できるように入札価格を設定します。
しかし、著者らは各種の要素のために集中入札過程で落札できなかった可能性があり、これらの要素は関連製品に対する需要の減少、入札価格の競争性がない、ある品質要求を満たしていない、あるいは関連製品は臨床効果が競争製品より劣ると考えられる。もし私たちの商業化製品ETUARYや任意の他の未来の製品が承認されれば、1つ以上の地域の集中入札過程で選ばれなければ、私たちはこれらの地域の公立病院に関連製品を販売することが困難になる可能性があり、私たちの市場シェア、収入、収益力は悪影響を受ける可能性がある。
将来、中国政府が制定した集中一括調達政策は、私たちの商業化製品ETUARYと任意の他の未来製品(承認されれば)をカバーする可能性があり、そのような製品の価格は低下する可能性があり、更に私たちの収入、財務状況、経営業績に重大な悪影響を与える可能性がある。
中国政府当局はすでに、大量薬品調達制度を含む薬品の負担性をさらに高めるための政策を実施している。詳細については“を参照されたい業務-製品定価-集中入札プロセスと集中一括調達システムそして“商業.中華人民共和国の管理要求.製薬業に関連するその他の中華人民共和国条例.薬品調達と使用を集中するこの年間報告書にあります。
中国政府の将来の調達には、NRDLに列挙された巨大な市場需要と比較的に高い調達価格を持つ薬物が含まれる予定であり、このような未来の調達は中国の臨床使用に必要かつ品質が信頼できるすべてのタイプの国内販売薬物を段階的にカバーする予定である。したがって、すべての適切な薬品はこの条項に基づいて中国で調達することができる。適切な“孤児薬”と不足薬品の購入方法を積極的に探索し、安定供給を確保することができる。
私たちの発売製品ETUARYは現在集中的な一括購入プロセスの制約を受けていません。しかし、将来的に数量ベースの集中調達範囲が拡大するかどうかは不明であり、当社の製品ETUARY(中国で承認された)や他の候補製品が商業化されれば、小売価格の低下を招く可能性がある。さらに、私たちの製品、模倣薬、または候補製品(商業化されている場合)と比較可能または同様の製品が集中的な一括調達に組み込まれている場合、患者がそのような製品を使用する意思は実質的かつ不利な影響を受ける可能性があり、価格設定戦略を変更する必要があるかもしれない。上記のいずれかまたは全ての状況が発生した場合、私たちの販売収入は低下する可能性があり、これは逆に私たちの財務状況、収益性、および経営業績に重大な悪影響を及ぼすだろう。
74
私たちの製品と候補製品の真の市場潜在力は予想より小さいかもしれない。我々の拡張は,中国の現在および出現しているIPF患者数によって制限される可能性があり,中国および我々の他の候補製品におけるETUARYの将来の適応の拡張申請の承認と利益の発売を待つ。
私たちの現在と未来の研究開発計画および特定の適応候補製品への支出は、私たちの候補製品の市場機会が私たちが予想していたより小さいかもしれないので、商業的に実行可能な製品は何も生じないかもしれない。同様に、私たちが中国で承認された製品ETUARYの実際の市場規模は私たちが予想していたほど大きくないかもしれない。総潜在市場機会は医学界と患者のこの製品に対する受容度、製品定価と精算状況に依存する。さらに、潜在市場における患者の数は予想よりも低い可能性があり、患者は私たちの製品治療を受けることができないかもしれないし、新しい患者はますます識別または接触が困難になっている可能性がある。さらに、新しい研究は私たちの候補製品の目標疾患の推定発病率または流行率を変えるかもしれない。上記のいずれの不利な発展も、我々の業務、財務状況、および経営結果に重大な悪影響を及ぼす可能性がある。特に、中国の既存および新たに発見されたIPF患者症例が我々の予想よりも少ない場合、ETUARYおよび我々の他の候補製品(例えば、F 351)の拡大適応が承認され、利益を達成するまで、私たちの成長および財務状態は負の影響を受ける可能性がある。
Frost&Sullivanのデータによると、中国のIPF罹患率は2017年の83,002人から2022年には131,654人に増加し、2027年には214,664人、2031年には320,677人に増加すると予想されている。IPF罹患率は短期的に上昇するが、中国公共衛生システムの強化及び医療と技術の進歩に伴い、将来IPFの潜在リスクは低下或いは除去する可能性があり、更にIPFの中国における罹患率の相応の低下を招く可能性がある。そのため,IPFの中国での普及率は低下しており,ETUARYの市場規模に悪影響を与える可能性がある。
私たちは効果的な学術マーケティングを行うことができないかもしれない。
効果的なマーケティングと成功した販売は、中国におけるETUARYの市場浸透率を高め、私たちの病院、薬局、他の医療機関のカバー範囲を拡大し、新製品(あれば)を普及させるために重要である。特に,学術マーケティングを非常に重視し,学術マーケティングにより医療専門家,病院,薬局,その他の医療機関にETUARYを普及させている。私たちの販売とマーケティングチームは医療専門家、病院、薬局、その他の医療機関と積極的に協力して、ETUARYが競争相手の製品と比較した独特の特徴、優位性、安全性、有効性を紹介するように努力していますが、私たちは私たちの製品意識を高めることに成功できないかもしれません。
私たちは合格した販売とマーケティングチームを維持できないかもしれない。
私たちが中国で承認された商業化製品ETUARYおよび任意の他の未来製品(承認されれば)を成功的にマーケティングと販売するために、私たちの販売とマーケティングチームは比較的高い技術知識レベル、業界傾向の最新の理解、関連治療分野と製品の必要な専門知識、そして十分な宣伝とコミュニケーション能力を備えなければならない。しかし、十分な数の能力のある販売専門家が関連する稀な疾患知識および/または学術KOLまたは医師ネットワークを持って採用できることを保証することはできない。したがって、私たちの内部販売代表を効果的に訓練したり、彼らの学術マーケティング表現を監督して評価することができなければ、私たちの販売とマーケティングは予想された成功に及ばないかもしれません。
また、私たちは主に私たちの内部の販売チームに頼って、私たちの製品をマーケティングするために、十分な数の合格販売専門家の能力を引きつけ、激励し、維持することが特に重要です。経験のあるマーケティング、普及と販売人員に対する競争は非常に激しいため、私たちは十分な数のマーケティング、普及と販売専門家を誘致、激励、維持することができないかもしれない。もし私たちが合格した販売とマーケティングチームを維持できなければ、私たちの商業化製品ETUARYと任意の他の未来の製品の販売量が承認されれば、不利な影響を受ける可能性があり、私たちの病院、薬局、他の医療機関のカバー範囲を拡大したり、市場浸透率を増加させることができないかもしれません。
75
私たちは中国で承認された商業化製品ETUARYや任意の他の未来の製品(承認されれば)のために有効な流通ネットワークを維持または拡大したり、私たちの流通ルートをさらに拡大したりすることができないかもしれない。
私たちは主に私たちの流通業者ネットワークに依存して中国で商業化製品ETUARYを販売し、流通業者と協力して私たちの商業化製品ETUARYや任意の他の未来製品を販売し続けるつもりですので、承認されれば、予測可能な未来に、私たちの事業を維持し、発展させる能力は、十分な数の販売業者と広範な販売ネットワークを維持し管理する能力に依存して、私たちは様々な理由でこれを実現できないかもしれません。承認されれば、私たちの流通業者は彼らの販売ネットワークを維持したり拡大したりすることができない場合や、私たちの商業化製品ETUARYや任意の他の未来の製品を販売する際に困難に直面する可能性があります。私たちのディーラーは様々な理由で私たちと契約を更新しないことや、価格制御や他の要素など、私たちとの業務関係を終了することを選択するかもしれません。承認されれば、彼らは私たちの商業化製品ETUARYおよび任意の他の未来の製品を転売することで利益率を大幅に低下させることができます。また、承認されれば、私たちの商業化製品ETUARYや他の任意の未来製品のために適切なディーラーグループを見つけることができないかもしれません。あるいはそうするコストは尻込みになるかもしれません。私たちの流通ネットワークのいかなる中断も、私たちが関係を維持できなかったこと、新しい関係を形成したこと、または既存の流通協定を更新したことを含めて、商業化製品ETUARYおよび任意の他の未来の製品を販売する能力に負の影響を与える可能性があり、承認されれば、私たちの業務、運営結果、財務状況、および見通しに実質的な悪影響を及ぼす可能性がある。
私たちは製薬業界の臨床需要と市場変化に十分かつ適時に反応できないかもしれない。
医薬製品の臨床需要と市場条件は迅速かつ顕著に変化する可能性があり、著者らの成功はある程度私たちが製品の供給周期と需要を予測し、顧客の選好を識別し、私たちの製品をこれらの選好に適応させる能力に依存する。お客様のニーズ、販売傾向、その他の市場状況に応じて、私たちの研究開発計画、生産規模とスケジュール、製品の組み合わせと在庫レベルを調整する必要があるかもしれません。しかし、私たちが臨床需要と調達モードの変化に十分かつ迅速に反応しないことを保証することはできない。
公衆衛生危機、ロシアとウクライナの衝突、イスラエル-ハマス戦争のような地政学的事件と世界的な経済状況は、中国のETUARYの将来の適応、中国のF 573、F 528、F 230、中国以外の潜在的な追加市場を含む、我々の製品ETUARYおよび候補製品の第三者供給に影響を与える可能性があり、これは、私たちの商業化、マーケティング、開発努力を遅らせる、阻止、または損害するであろう十分な数のこのような製品、模倣薬または候補製品、または受け入れ可能なコストでこのような数量を得ることができないリスクを増加させる。
もし私たちの製品、ETUARYまたは候補製品(将来の適応のためのETUARY、中国のF 573、F 528とF 230、中国のF 351および中国以外の潜在的な追加市場を含む)の原材料供給が著しく遅れている場合、またはインフレと金利上昇、公衆衛生危機、ロシアとウクライナの間の衝突、ハマス-イスラエル戦争など、世界の経済状況の影響を含む任意の材料または任意の製品を生産する第三者が、インフレと金利上昇、公衆衛生危機、ロシアとウクライナの間の衝突、およびハマス-イスラエル戦争を含む任意の理由で供給を停止する場合、私たちが代替サプライヤーや製造業者を決定して同定すると、私たちはこれらのテストや試験を進める上で遅延に遭遇する可能性があり、私たちに有利な条項で代替供給を得ることができないかもしれない。さらに、私たちが十分な製品、模倣薬または候補製品、またはそれらを製造するための物質の供給を得ることができない場合、私たちの製品、模倣薬または候補製品(場合によっては)を商業化、マーケティング、または開発し、効果的に競争することはより難しいだろう。
私たちは現在、第三者サプライヤーへの依存が私たちの製品、模造薬、候補製品を開発する能力に悪影響を及ぼす可能性があり、私たちの臨床試験と開発計画およびマーケティングと商業化努力を延期する可能性があり、そうでなければ、私たちの運営と財務状況を損害し、私たちのコストと支出を増加させる可能性がある。参照してください“-第三者への依存に関連するリスク-私たちのいくつかの原材料は限られた数のサプライヤーに依存しているので、供給中断に遭遇する可能性があり、これは私たちの製品を製造する能力を損なう可能性があります.”
76
中米関係リスクの詳細については、“を参照されたい-中国での業務運営に関するリスク—中国政府の政治·経済政策や中米関係の変化は、我々の業務、財務状況、経営業績に影響を与える可能性がある。”
業務中断は私たちの将来の収入と財務状況を深刻に損害し、私たちのコストと支出を増加させるかもしれない。
当社の業務および当社の流通業者、サプライヤー、研究機関協力者および他の業務パートナーの業務は、自然災害や人為的災害、健康流行病、または業務中断の影響を受ける可能性があり、主にこれらの業務に自己保険を提供しています。火災、自然災害、衛生疫病、停電、通信障害、許可されていない進入、または他の事件は、私たちとパートナーの管理、開発、研究、製造、または記憶施設に損害または長時間の中断をもたらし、私たちと私たちのパートナーのコストと支出を増加させる可能性があり、私たちとパートナーのコストと支出を増加させる可能性があります。
私たちの保険範囲は限られていて、私たちの保険範囲を超えたすべてのクレームは巨額のコストと資源移転を招く可能性があります。
私たちは製薬産業を運営しており、これは多くの操作リスクと職業的危険と関連がある。私たちが維持している保険証は、適用された法律と法規と、私たちの運営需要と業界実践の評価に基づいて要求されます。しかし、既存の保険範囲が被害または発生した実際の損失を補償するのに十分であることは保証されない。さらに、戦争、テロ行為、健康または公共安全被害、地震、台風、洪水、および他の自然災害による損失のようないくつかの種類の損失があり、私たちは合理的な費用やこれらの損失のために保険を購入することができない。非保険損失又は保険限度額を超える損失が発生した場合、我々の業務、経営業績及び財務状況は実質的な悪影響を受ける可能性がある。製品責任クレームや環境責任が発生した場合の保険カバー範囲不足の具体的なリスクの詳細については、“を参照されたい”-製品責任クレームの影響を受ける可能性があります。これはコストと責任に直面する可能性があります“と”-環境、健康、安全の法律法規を遵守しない場合、罰金または処罰を受けることができ、または業務を損なう可能性のあるコストが発生する可能性があります“は,それぞれ本節で述べる.
私たちの候補製品の発見、開発、商業化に関するリスク
私たちは、より利益的または成功可能性の高い候補製品または適応を利用することなく、特定の候補製品または適応を追求するために限られたリソースを費やす可能性がある。
私たちの財務と管理資源が限られているので、私たちは私たちが決定した特定の適応の開発計画と候補製品に集中しなければならない。したがって、私たちは他の候補製品を探す機会を放棄または延期したり、これらの候補製品のために後により大きな商業潜在力が証明された他の兆候を探すことができるかもしれない。私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。現在と将来の開発計画および特定の適応の候補製品への支出は、いかなる商業的に実行可能な製品も生じない可能性がある。もし私たちが特定の候補製品の商業的潜在力や目標市場を正確に評価しなければ、私たちは協力、許可、または他の印税手配によって候補製品に価値のある権利を放棄するかもしれないが、この場合、候補製品の独占開発権と商業化権利を維持することは私たちに有利だ。
私たちは新しい候補製品を識別、発見、または開発できないかもしれないし、私たちの候補製品のための追加の治療機会を決定して、私たちの製品ラインを拡大または維持することができない。
私たちは私たちの現在のチャンネルを豊かにするために新しい候補製品を識別して開発し続けることができないかもしれない。我々の候補製品を開発してより多くの適応を獲得し、新しい候補製品と製品目標を決定するためには、研究計画には大量の技術、財政、人的資源が必要である。私たちがルートを構築することに成功しても、私たちが確定した潜在的な候補製品は臨床に適していないかもしれません
77
発展する。例えば、候補製品は有害な副作用や他の特徴を示す可能性があり、開発に成功する可能性が低いことを示しており、市場の承認を得て市場の承認を得ることはいうまでもない。もし私たちが私たちの方法に基づいて候補製品の開発と商業化に成功しなければ、私たちの将来の成長と見通しに実質的な悪影響を与え、これは私たちの財務状況に大きな損害を与え、私たちの株価に悪影響を及ぼすかもしれない。
前臨床試験あるいは早期臨床試験の結果は,われわれのETUARY,F 351およびF 573の前臨床試験や早期臨床試験の結果を含め,以降の試験では確認されない可能性があり,その後の臨床試験の成功も予測できない。
臨床前研究と早期臨床試験の結果は後期臨床試験の成功を予測できないかもしれない。我々の候補製品のより多くの患者における試験は類似した治療効果結果がない可能性があり、早期患者数の少ない試験では観察されない副作用を招く可能性がある。
私たちは厳格に制御された臨床試験を通じて、私たちの候補製品が安全で有効であることを証明して、それから私たちはその商業販売の市場承認を求めることができます。われわれのこれまでの臨床前研究において,有効性や許容可能な安全性特徴の提示は,将来の臨床試験で同様の結果が生じることを意味するものではない。例えば、ETUARYはIPFの治療のために中国で承認されているが、SSc−ILD、DM−ILD、塵肺またはDKD、または他の市場でのような他の適応の治療のために許可されていない可能性がある。また,F 351が中国で行われた前臨床研究や早期臨床試験のように将来の臨床試験に機能するかどうかは不明であり,F 351が米国のS一期試験で良好な耐性を示した証拠にもかかわらず,PKとわれわれの中国での第二期臨床試験は慢性B型肝炎関連線維化を逆転させた結果を示しているが,これまで有効な肝線維化臨床療法はなく,特定の治療製品が世界的に承認されていない。F 573が耐性やPKの第1段階臨床観察のようにALF/ACLFの第2段階臨床試験で現れるかどうかも不明である。候補製品はETUARY、F 351とF 573を含み、臨床前研究と早期臨床試験を通じて進展を得たが、後期臨床試験では十分な安全性と有効性を証明できない可能性があり、アメリカ国家薬品監督管理局、FDAとその他の類似した外国監督機関を満足させるのに十分である。規制当局はまた、早期試験で満足できる安全性または有効性の結果が証明されるまで、後期試験の範囲を制限する可能性がある。特に,ETUARYは中国でIPFの治療に許可されているにもかかわらず,SSc−ILDやDM−ILDを治療する第3段階臨床試験,塵肺疾患を治療する第3段階臨床試験あるいはDKD計画のETUARYの第1段階臨床試験に機能しない可能性がある。さらに私たちは IND申請を積極的に準備しており,2024年末に第二段階臨床試験を提出し,F 351治療に非肝硬化性NASHに関連する進行肝線維化を評価する予定である。IND郭清後,2025年に米国で2 a期PoC臨床試験を開始し,非肝硬化性NASHに関連する進行肝線維化患者に対するF 351の安全性,耐性,PK,PDを評価する予定である。FDAはすでに米国で計画されているF 351 2 a段階試験を審査し、設計、試験評価、IND文書の内容についてコメントを提供した。F 351の2 a段階試験で積極的な傾向が観察されれば,F 351でより大きな第2段階試験を開始することが予想される。われわれが中国で行った第二段階臨床試験における慢性B型肝炎患者に関する肝線維化データは,F 351が肝線維化を改善する潜在力を有していることを示しているにもかかわらず,NASHモデルの先行臨床前研究では,F 351の治療効果は将来の臨床試験で同様の結果が生じることを意味していない。
提供されたIND前指導に加えて、IND申請を審査する際に、米国国家薬品監督管理局、FDAまたは他の同様の外国規制機関は、ヒト被験者のリスクに関する十分かつ十分な情報を確保するために、IND文書を受ける前に追加の調査(非臨床)および分析(中国で行われる支持的臨床試験の分析を含む)を行うことを要求する可能性がある。このような追加的な要求は,IND申請と起動計画におけるNASH関連肝線維化2 a期試験のスケジュールを遅らせる可能性がある。また、米国国家薬品監督管理局またはFDAが、我々の臨床研究データと2 a期の臨床試験データを補充するために追加のデータが必要であると考えた場合、米国国家薬品監督管理局またはFDAは、より広い第2段階の臨床試験に拡張する前により多くの試験を行うことを要求するかもしれない。NMPA、FDA、および他の類似した外国規制機関が、米国で計画されている2 a期試験で期待されるデータを考慮することは、より大きな2期または検証性3期臨床試験においてF 351の開発を拡大させるのに十分であることを保証することはできない。私たちが期待していた時間内にこのような裁判を終わらせることができる保証はなく、このような裁判が肯定的な結果をもたらすという保証もない。私たちの臨床試験能力のいかなる制限も、規制部門の承認を延期または阻止することができ、あるいは承認されれば、私たちの候補製品を販売できる患者集団の規模を制限することができる。
78
製薬やバイオテクノロジー業界の多くの会社が早期開発で積極的な結果を得た後,後期ナッシュ臨床試験で大きな挫折を経験し,類似した挫折に直面する可能性がある。多くの会社は彼らの候補製品が臨床前研究と早期臨床試験で満足できると思っているが、まだ候補製品のマーケティング許可を得られなかった。私たちの候補製品の臨床試験結果は発売許可に値すると思っても、アメリカ国家薬品監督管理局、FDA或いは類似の外国の監督管理機関は同意しないかもしれないし、私たちの候補製品の発売を許可しないかもしれない。
いくつかの場合、多くの要素のため、同一候補製品の異なる臨床試験間の安全性或いは有効性結果は有意差が存在する可能性があり、方案中に規定された試験プログラムの変化、患者群の大きさとタイプの差異、投与方案と他の臨床試験方案の変化と遵守状況及び臨床試験参加者の退学率を含む。私たちが行う可能性のある任意の第2段階、第3段階、または他の臨床試験は、規制部門の許可を得て私たちの候補製品を市場に出すために必要な有効性と安全性を証明できないかもしれない。
著者らが時々発表或いは公表した臨床試験の初歩、“頂線”或いは中期データはより多くの患者データの獲得に従って変化し、監査と検証手続きの制約を受ける可能性がある。
私たちはすでに公開し、未来に私たちの臨床試験の初歩的または主要なデータを公開する可能性があり、これらのデータは当時利用可能なデータの初歩的な分析、結果、および関連する発見と結論はデータをより全面的に検討した後に変化する可能性がある。私たちはまた、これらのデータの分析の一部として、完全なデータを全面的かつ詳細に評価する機会がないという仮説、推定、計算、および結論を出した。したがって、私たちの報告書の初歩的または最も重要な結果は、同じ研究の将来の結果とは異なる可能性があり、または追加のデータを受信し、全面的な評価を行うか、またはその後に監査および確認手続きを行うと、異なる結論または考慮要素がこれらの結果を合格させる可能性がある。
最終データが利用可能になる前に、任意の予備データまたは主要データを慎重に確認しなければならない。私たちはまた時々臨床前研究と臨床試験の中期データを開示し、未来にも開示する可能性がある。中期データは、患者登録の継続とより多くの患者データの獲得に伴い、あるいは著者らの臨床試験の患者が他の治療を継続するにつれて、1つまたは複数の臨床結果が実質的に変化する可能性があるというリスクに直面している。さらに、他の人は、規制機関を含み、私たちの仮説、推定、計算、結論または分析を受け入れないか、またはデータの重要性を異なる方法で解釈またはトレードオフする可能性があり、これは、特定の計画の価値、特定の候補製品の承認または商業化の可能性、およびわが社の全体的な状況に影響を与える可能性がある。さらに、特定の臨床前研究または臨床試験に関する重要な情報または要約情報が時々開示される可能性がある。このような要約情報は、必然的により煩雑で広範な情報に基づいており、投資家または規制機関は、我々が重要であると考えているか、または他の方法で我々の開示に含まれるのに適した情報として決定することに同意しない可能性がある。もし私たちの報告書の初歩的、主要または中間データが実際の結果と異なる場合、または規制機関を含む他の人が結論に同意しない場合、私たちが承認を得て私たちの候補製品を商業化する能力が損なわれる可能性があり、これは私たちの業務、経営業績、将来性、または財務状況を損なう可能性がある。
私たちの候補製品を国際的にマーケティングすることに関連する様々なリスクは私たちの業務に実質的な悪影響を及ぼす可能性があります。
私たちはまた、中国のETUARY、中国のF 573とF 351、中国以外、中国とアメリカ以外の他の市場を含む規制機関の候補製品の承認を求める可能性がありますので、外国の異なる規制要件を含めて必要な承認を得られれば、外国での運営に関する追加リスクに直面することが予想されます。国際業務に関連するリスクは、私たちの業務、財務状況、運営結果に大きな悪影響を及ぼす可能性があります。
中国で承認された製品ETUARYと候補製品、中国の未来適応のETUARY、中国のF 573、F 528とF 230、中国のF 351および中国以外の潜在的な他の市場は、重大な有害事象、毒性または他の不良副作用を招く可能性があり、これらの副作用は安全状況の中断、遅延または臨床試験の停止を招き、監督部門の承認、上場承認または市場受け入れを延期または阻止し、それらの商業潜在力と承認された承認を制限する可能性がある
79
ラベルは、私たちの名声と経営結果に悪影響を与え、あるいは任意の規制承認後の重大な負の結果をもたらす。
ETUARY、F 351、F 573、F 528およびF 230を含む候補製品が、単独で使用または他の承認された製品またはINDと組み合わせて使用される場合、臨床前研究または臨床試験において予期しない特徴を有する場合、または他の承認された製品またはINDと一緒に使用される場合、副作用または他の特徴に関連して、または予期しない特徴を有する場合、それらの開発を中断、延期または放棄する必要があるかもしれない、またはその開発をより狭い用途またはサブ集団に制限する必要があり、リスク効果の観点から、これらの副作用または他の特徴はあまり一般的ではなく、あまり深刻ではない、またはより受け入れやすい。治療に関連する副作用は、患者が試験を完了する能力を募集したり、組み入れたり、あるいは潜在的な製品責任クレームを引き起こす可能性もある。多くの場合,研究候補製品が大規模な3期試験で試験を行った後,あるいは承認後に患者に商業規模の製品を提供した後にのみ,副作用を検出することができる場合がある。より多くの臨床経験が示す場合、私たちの現在の任意の候補製品および任意の未来の候補製品は、生命に深刻なまたは危険な副作用または他の副作用があり、潜在的な治療利益を超え、候補製品の開発が失敗または延期される可能性があり、または、候補製品が市場承認を得た場合、この承認は撤回される可能性があり、これは、私たちの業務、将来性、経営業績、および財務状況を損なうことになる。
私たちの候補製品は私たちの臨床試験で報告されたいかなる不良事件或いは深刻な不良事件は深刻な負の結果を招く可能性がある。このような結果には
不良あるいは予期せぬ副作用は、臨床テストで明らかにされていない潜在的な副作用、個別症例にはあまり見られないが深刻な副作用、私たちの品質管理システムでは検出されなかった欠陥製品、およびエンドユーザーの私たちの製品に対する誤用を含む多くのコントロールできない要素によって引き起こされる可能性がある。
80
さらに、他の製薬会社の製品、後発薬および将来の製品が、私たちの製品、後発薬および未来の製品と同じまたは類似した活性医薬成分、原材料または輸送技術を含む場合、承認された場合、深刻な副作用を引き起こす可能性があるか、または、規制機関または国際機関が、私たちの製品、後発薬および未来の製品と同じまたは類似した医薬成分を含む製品を決定した場合、承認された場合、深刻な副作用を引き起こす可能性があり、深刻な副作用を引き起こす可能性がある。我々の製品、後発薬、未来の製品は、承認されれば、深刻な副作用の原因がないか、決定的な確定が得られないため、深刻な副作用を招く可能性もあると考えられる。
全体的に言えば、F 351の期待臨床試験は末期肝繊維化患者を含み、彼らは更に肝硬変と悪化に発展するリスクがあるが、重篤な患者ではない。F 351で治療したB型肝炎肝繊維化患者の中で、一定の割合の患者は胃腸疾患、耳と迷路疾患、全身疾患、代謝と栄養疾患、皮膚と皮下組織疾患、心臓器官疾患と肝胆系疾患を含む不良事件を経験した。しかしながら、NASHにおけるF 351のリスク/利益は、B型肝炎肝線維症患者において示されるリスク/利益とは異なる可能性があり、有害事象の重症度および頻度が悪化する可能性のあるリスクが常に存在する可能性がある。“”というタイトルの部分を参照-ビジネス-F 351の概要.”
また,我々の様々な第3段階臨床試験では,我々の候補製品を用いて治療した患者群は重篤な疾患を有しており,重大な健康リスクの影響を受けやすい。したがって、これらの患者は深刻な不良事件を含む不良事件を経験する可能性がある。
薬物研究と開発を行う際、私たちは潜在的な責任に直面しており、特に私たちが重大な責任を招く可能性のある製品責任のクレームや訴訟を招く可能性がある。
もし私たちの候補製品が臨床試験中に損傷を与えたり、あるいは他の面で不適切であることが発見された場合、私たちは臨床試験による固有製品責任のリスクに直面する。是非曲直や最終結果にかかわらず、このような責任クレームは他のものを除いて、以下のような原因になる可能性がある
臨床試験によるこのような責任クレームを支払うために、私たちは私たちのすべての試験に臨床試験保険を提供し、これらの試験は私たちのパイプライン製品の商業化を承認するために必要である。しかし、私たちの責任は私たちの保険範囲を超えている可能性があり、あるいは私たちの保険は私たちに請求する可能性のあるすべての状況をカバーしないだろう。私たちはまた合理的なコストで保険範囲を維持できないかもしれないし、起こりうるいかなる責任を支払うのに十分な保険範囲を得ることができないかもしれない。
中国で承認された我々の商業化製品ETUARYと任意の他の未来製品の非ラベル使用による不良薬物反応と負の結果は、承認されれば、私たちの商業名声、製品ブランド名、財務状況に重大な損害を与え、私たちに責任を負わせる可能性がある。
医薬品市場で流通または販売されている製品は、非ラベル薬物使用に属する可能性があり、適応、用量、または規制部門によって承認された使用法およびラベルに適合しない剤形が処方される可能性がある。したがって、私たちの製品、ETUARY、および未来の製品は、承認されれば、非ラベル薬物によって使用される可能性があり、患者集団に処方されるか、または主管部門の承認されていない用量または剤形で、これは、私たちの製品、模倣薬、および未来の製品を、承認されれば、効果が悪いか、または完全に無効になり、薬物副作用を引き起こす可能性がある。このような事件のいずれも否定的な宣伝をもたらす可能性があります
81
商業名声、製品ブランド、商業運営と財務状況、私たちの株価を含む。これらの事件はまた私たちに責任を負わせ、あるいは私たちの臨床試験の進度の遅延を招き、最終的に私たちの候補製品が監督部門の承認を得られない可能性がある。
FDAが任意の候補製品に突破的な療法を指定することは、より速い開発や監督審査や承認過程を招くことがなく、候補製品が発売承認される可能性を増加させることもないかもしれない。
F 351は2021年3月に中国国家薬品監督管理局CDEに突破的治療称号を授与され、その3期臨床試験の患者登録は2022年1月に開始された。しかしながら、F 351のS突破的療法指定は、F 351が最終的に米国国家薬品監督管理局または他の同様の規制機関の承認を得る可能性を増加させない。
将来、私たちは臨床試験における臨床利益のロバスト性に基づいて、私たちの候補製品のためにアメリカまたは他の外国司法管轄区(例えば、ある)に突破療法の称号を申請するかもしれない。米国では、突破療法は、1つまたは複数の他の薬剤との単独または組み合わせによる重篤または生命に危険な疾患または状態を治療することを目的とした製品候補として定義され、予備臨床証拠は、製品候補が、1つまたは複数の臨床的重要終点において、臨床開発早期に観察される顕著な治療効果のような既存の治療法よりも有意な改善を示す可能性があることを示す。画期的な治療法として指定された候補製品に対して,FDAと試験スポンサーとの相互作用やコミュニケーションは,臨床開発の最も有効な方法を決定するのに役立つとともに,無効な制御案中の患者数を最小限に抑えることができる。FDAから画期的な治療法の候補品に指定され,NDA提出時に臨床データの支持を得ていれば,優先審査を受ける資格もある。
FDAはそれを画期的な治療法に指定する権利がある。したがって,我々の候補製品の1つが画期的療法として指定された基準に適合していると考えても,FDAは同意せず,このような指定を行わないことにした可能性がある。いずれの場合も、FDAの従来の手順に従って承認を考慮した候補製品と比較して、候補製品の突破療法指定を受けることは、より速い開発プロセス、審査または承認を招くことなく、FDAの最終承認を確保することもできない可能性がある。さらに、私たちの1つまたは複数の候補製品が画期的な療法の条件に適合していても、FDAは、候補製品がもはや資格条件に適合していないことを後で決定することができ、またはFDAの審査または承認の期間を短縮しないと決定する可能性がある。
私たちの製品や候補製品の製造と供給に関するリスク
大規模な商業生産医薬製品は非常に厳格で複雑で、私たちと私たちの第三者メーカーはこの過程で問題に直面するかもしれません。
医薬製品の製造は非常に複雑で、製造過程で様々な原因で問題が発生する可能性があるが、これらに限定されない
82
私たちは品質管理と保証システムと手続きがあるにもかかわらず、私たちはこのようなリスクを除去することができないかもしれません。これらのリスクは私たちの生産活動を遅延または一時的に一時的に停止する可能性があり、私たちは私たちが受け入れられる条項、品質とコストで、私たちの製品、模倣薬、または候補製品のために一時的な代替製造業者を見つけることができないかもしれません。もし私たちが上記の問題、私たちの臨床試験、および/または私たちが中国で承認された製品ETUARY、後発薬、および将来の製品(承認されれば)の商業販売が延期される可能性があり、これらの問題を修正し、私たちの製造施設で生産を維持するために、私たちの臨床試験および/または私たちが承認された製品を含むどんな生産問題に直面すれば、私たちは大量の時間とコストをかけるかもしれない。しかも、品質の問題のある製品は廃棄されなければならず、製品不足や追加費用を招く可能性がある。
また,生産過程や結果を最適化するために,生産方法や配合は候補製品の開発に伴い変化し,臨床試験から承認,商業化まで変化する場合がある。このような変更は、これらの予期される目標を達成できないリスクがあり、これらの変更のいずれの変更も、候補製品の表現を異なり、変更された材料を用いた計画臨床試験または他の将来の臨床試験の結果に影響を与える可能性がある。これは候補製品の商業化を遅らせる可能性があり、1つ以上の臨床試験を過渡的に研究または繰り返す必要があり、これは臨床試験コストの増加、製品の承認遅延を招き、そして私たちの製品販売の開始と収入を創出する能力を脅かす可能性がある。
われわれは薬明生物製F 351カプセルを使用する予定であり,将来的には海外のCROやCMOを使用し続ける可能性がある。薬明生物は、米国で計画されている2 a期臨床試験のF 351カプセルの生産を完了した。外国のCMOは、提案された生物安全法、制裁、貿易制限、および他の外国規制要件を含む米国の法律によって拘束される可能性があり、これらの要求は、コストを増加させるか、使用可能な材料の供給を減少させ、このような材料の調達または供給を延期するか、または私たちの潜在療法を購入する政府の重大な約束を得る能力に悪影響を及ぼす可能性がある。
例えば、中国のバイオ製薬業界は中国政府の厳しい規制を受けている。バイオ製薬会社に影響を与える中国法規や政府政策の変化は予測できず、私たちの中国におけるパートナーに重大な悪影響を与える可能性があり、それによって私たちの業務、財務状況、運営結果、将来性に悪影響を与える可能性がある。中国の公衆衛生、経済、政治、社会条件の変化、および中国と他の政府(例えばアメリカとイギリス)との関係をめぐる不確実性は、計画中の臨床試験のために私たちの候補製品を生産する能力にマイナス影響を与えるか、あるいは政府資金を得る能力に悪影響を与える可能性もあり、これは私たちの財務状況に悪影響を与え、臨床開発計画を延期する可能性がある。詳細については“を参照されたい”私たちの中国での業務運営に関するリスク—中国政府の政治·経済政策や中米関係の変化は、我々の業務、財務状況、経営業績に影響を与える可能性がある。”
また,第三者メーカーと我々の他の候補製品の薬物物質や薬物製品製造について様々な開発,製造,臨床供給サービス合意を達成する予定である。もし我々の第三者製造業者が十分な数または品質の候補製品をタイムリーに提供できない場合、または生産不足または他の供給遅延または公衆衛生危機または他の原因による中断であっても、我々の臨床前試験、臨床試験または規制承認(適用すれば)が延期される可能性がある。私たちの研究開発資源の大部分は製造業に集中している。もし私たちの任意の第三者製造業者が生産拡大に困難に直面した場合、あるいは汚染、設備故障、設備設置または操作の不適切、サプライヤーまたはオペレータのミス、または不適切な貯蔵条件によって製品損失が生じ、影響を受けた候補製品の潜在的な試験が延期され、さらには大幅に延期される可能性があり、これは私たちの業務に実質的かつ不利な影響を与える可能性がある。
私たちと私たちの契約メーカーは私たちの製品ETUARY(中国で承認された)と私たちの候補製品を製造する上で厳しい規制を受けるだろう。私たちと私たちの第三者製造施設の承認を遅延させて規制部門の承認を受けることは、私たちの開発計画や商業化努力を遅らせる可能性があります。
83
臨床研究または商業販売のための治療薬の準備に参加するすべての実体は、私たちと任意のETUARYと私たちの他の候補製品の契約メーカーを含めて、広範な監督管理を受けている。商業販売または末期臨床研究のために許可された完成治療製品の成分は、GMPに従って生産されなければならない。これらの条例は、調査製品および承認販売された製品の品質を制御し、確保するために、生産プロセスおよびプログラム(記録保存を含む)および品質システムの実施および動作を管理する。生産プロセスの不良な制御は、外来製剤または他の汚染物質の導入、または製品、模倣薬または候補製品の性能または安定性を無意識に変化させる可能性があり、これらの変化は最終製品試験では検出できない可能性がある。
我々の既存および計画中の製造施設、ならびに私たちの製造プロセス、ならびに私たちの第三者製造施設およびプロセスは、通常、上場承認を得るための前提条件であるGMPに適合することを確実にするために、米国食品薬品監督管理局、FDAまたは他の同様の規制機関の持続的な定期検査を受ける。また、私たちと私たちの第三者メーカーは、不動産開発の異なる段階で、関連行政部門から生産施設や他の場所の様々な許可証、証明書、その他の承認を得なければならない。計画許可証、建築許可証、土地使用権証明書、環境評価、消防評価、建築竣工検査、所有権証明書を含む。適用された法規を守らないことは、
その中のどれも私たちの業務に実質的で不利な被害をもたらす可能性がある。
私たちの生産拠点は深刻な妨害を受ける可能性があり、私たちの製品ETUARY(中国で承認された)や将来の製品(承認されれば)の生産に問題が生じる可能性があります。
私たちは中国北京と中国滄州での私たちの製造施設に依存している。多くの要因により、私たちの製造施設の持続的な運営と私たちの生産安全は大幅に中断される可能性があり、その多くの要素は私たちがコントロールできるものではありません。これらの要因は、火災、洪水、地震、停電、燃料不足、機械故障、テロおよび戦争または他の自然災害、ならびに免許、証明書および許可証の損失、これらの施設またはその近くの土地に対する政府の計画変化、および規制の変化を含む可能性がある。また,環境保全や公共活動を含めた政府政策や法規制により,我々の製造施設での生産活動は一時的に停止する可能性がある。もし私たちの任意の製造施設の運営が深刻な妨害を受けた場合、私たちはこれらの施設の設備や在庫を交換することができないかもしれないし、異なる場所または第三者請負業者を使用して、合法的、タイムリーかつ費用効果的な方法で、または私たちの生産を継続することができないかもしれない。吾等はいくつかの物件、機器及び設備及び吾等に対して所有、運営又は重要とされている他の資産維持財産保険であるが、中国の業界慣例によると、吾等は業務中断保険のような何らかの種別の保険をかけていない。私たちの保険金額と性質は、私たちのどの製造施設でも重大な中断が発生した場合、いかなる重大な損失を賠償するのに十分ではないかもしれません。
2021年9月以降,石炭供給不足に加え,メーカーの電力需要の高さに加えて,中国は広範囲の停電を経験している。中国政府はすでに、中国の複数の省の工場に対して電力規制を実施し、エネルギー需給不均衡の問題に対応する電力規制を実施している。2023年12月31日まで、私たちは政府関係部門から何の通知も受けていません。私たちは一時的に生産を停止したり、生産を制限したりすることを要求していません。私たちの北京と滄州生産センターもいかなる電力制限も受けません。中国政府が実施している電力規制は、2023年12月31日および2022年12月31日までの間に、我々の業務運営や財務表現に大きな悪影響はない。
84
私たちは中国で承認された商業化製品ETUARYと他の未来の製品の日々増加する需要を満たすことができないかもしれませんが、承認されれば、十分な製造能力を維持したり、私たちの予想成長を成功的に管理することができないかもしれません。
ETUARYと私たちのますます多くの候補製品を生産するためには、承認されれば、予想される市場需要の数量を満たす必要があると考え、新しい製造施設や生産ラインを建設することで、最初の生産レベルで生産能力を向上させる必要があるかもしれない。しかし、私たちが生産能力を高める拡張計画を実施することに成功した能力は、施工遅延や設備調達遅延のリスクを含むが、これらに限定されない多くのリスクと不確定要素の影響を受け、私たちは直ちに十分な合格従業員を募集して、私たちの生産能力の向上を支持する能力を提供する。もし私たちがこれができなくて、遅延して、経済的に実行不可能なコストに直面したり、第三者メーカーが見つからなければ、私たちはETUARYと私たちの未来に承認された候補製品を生産できないかもしれません。もしあれば、未来の需要を満たすのに十分です。また、私たちが生産能力を増加させる計画は大量の資本投資を必要とし、拡張計画の実際のコストは私たちの最初の見積もりを超える可能性があり、これは私たちの支出リターンに悪影響を及ぼすかもしれない。
また,我々の既存·計画中の製造施設の規模を考慮すると,運転開始後の合理的な時間内に十分に利用できない可能性がある。建設と生産開始期間中、製薬業界のマクロ経済は重大な変化が発生する可能性があり、その中には市場需要、製品と供給価格の傾向及び顧客の選好が含まれている。この分野のどんな不利な傾向も、私たちの施設の運営効率の低下と未使用の能力を招く可能性がある。
もし私たちがコスト増加を私たちの顧客に転嫁できなければ、私たちの原材料とエネルギー供給価格の変動、そして私たちの生産過程に関連する他のコストは、私たちに実質的な悪影響を及ぼすかもしれない。
私たちが中国で承認された商業化製品ETUARYおよび任意の他の未来製品(承認されれば)を生産するためには、十分な量の良質な原材料および安定したエネルギーと電力供給を商業的に許容可能な価格でタイムリーに獲得しなければならず、原材料価格の変動に関するリスクに直面しなければならない。このような材料の価格は多種の要素の影響を受ける可能性があり、市場需給、中国、アメリカ或いは国際環境と監督管理要求、自然災害及び全世界と現地の経済状況を含む。また、廃棄物処理コストなど、私たちの生産過程に関連する他のコスト変動の影響を受ける可能性があります。これらのコストは制御できません。私たちが適時に収入を増加させる能力は限られているかもしれませんが、このようなコストの大幅な増加は私たちの販売コストを増加させ、私たちの利益率にマイナスの影響を与えるかもしれません。
最適な在庫レベルを維持できなければ、私たちの運営コストが増加したり、顧客の注文が完了できなくなったりする可能性があります。
私たちは流通ネットワークからの需要を満たすために最適な在庫レベルを維持し、顧客の需要を満たすことに成功しなければならない。しかし、製品ライフサイクルの急速な変化、絶えず変化する臨床需要、製品開発と発売の不確定性、および中国の動揺した経済環境のため、私たちは中国で承認された商業化製品ETUARY、後発薬、その他の任意の未来製品の適切な在庫レベルを維持できないかもしれない。これらの傾向と事件を正確に予測できる保証はありません。私たちの商業化製品ETUARY、後発薬、その他の任意の未来の製品が承認された後の在庫不足を避けることができます。また、承認されれば、我々の商業化製品ETUARY、後発薬、および任意の他の未来製品への需要は、発注製品と交付準備との間で大きく変化する可能性がある。
在庫レベルが需要を超えると在庫減記、製品の期限切れや在庫保有コストの増加を招き、私たちの流動性に潜在的なマイナス影響を与える可能性がある。一方、需要を過小評価すると、在庫不足に遭遇する可能性があり、顧客の注文が履行されず、顧客関係に悪影響を与える可能性があります。
私たちの第三者への依存に関するリスクは
85
私たちは設立され、協力協定と戦略的パートナーシップを構築し続けることができる。しかし、私たちがこのような協力、連合、または許可協定から期待された利益を完全に達成する保証はなく、私たちの現在または未来のパートナーは衝突する可能性がある。
私たちは過去に、第三者との許可手配を含む戦略同盟、合弁企業、または他の協力を求め、構築することが可能であり、これは、私たちの既存の商業化製品ETUARY(中国で承認された)、模倣薬および候補製品(ETUARY、F 351、F 573、F 528、F 230を含む)、および私たちが開発する可能性のある任意の未来の製品の開発および商業化努力を補完または強化すると信じている。私たちのパートナーとの戦略的協力は多くの危険を含んでいる。私たちは取引から予想される収入とコストの協同効果を達成できないかもしれない。このような協同効果自体が不確定であり、重大な商業、経済と競争の不確定性と意外な事件の影響を受け、その多くは予測が困難であり、私たちのコントロールを超えているからである。また,我々のパートナーとの連携による相乗効果は,他の費用の増加,運営損失,あるいは我々の連携とは無関係な業務問題など,連携過程で生じる他のコストによって相殺される可能性がある.
しかも、私たちは現在または未来のパートナーとの間で紛争が発生するかもしれない。このような紛争または私たちのパートナーがその義務を十分に履行していないことは、我々の製品、模倣薬または候補製品の研究、開発または商業化の遅延または終了、またはコストの高い訴訟または仲裁を招き、管理職の注意および資源を分散させる可能性がある。特に、国際業務関係は、現地の司法管轄区域で契約条項を効率的に実行することの困難、および第三者パートナーが私たちの特許、商業秘密および他の知的財産権を正確に取得、維持、保護または実行できない可能性があり、私たちの製品、模倣薬または候補製品の規制の排他性、または私たちの知的財産権を使用して潜在的な訴訟または私たちの知的財産権を無効にする可能性のある知的財産権に関する訴訟を含む、私たちの利益業務の能力を達成または維持することに実質的な悪影響を及ぼす可能性がある追加のリスクに直面する可能性がある。
私たちは適切な協力者を探すことで激しい競争に直面している。我々が協力者と最終的な合意を達成できるかどうかは,他の事項に加えて,協力者の資源や専門知識の評価,提案協力の条項や条件,提案協力者のいくつかの要因の評価に依存する.これらの要因は、前臨床試験の設計または結果、米国国家食品薬品監督管理局、FDAまたは同様の外国規制機関が承認する可能性、および任意のこのような承認の規制方法、候補製品の潜在的な市場、製品の製造および患者への配送のコストおよび複雑性、および競争製品の潜在力を含む可能性がある。協力者はまた,代替製品,候補製品,または技術を考慮して,連携可能な類似適応を得ることができ,そのような連携が我々との連携よりも魅力的であるかどうかを考えることができる.私たちがどんな協力合意にも到達する保証もなく、どのような合意も有利な条件で行われる保証はない。
協力の交渉と記録は複雑で時間がかかる。また,最近では大手製薬会社間の業務合併数が大きく,将来の潜在的パートナー数の減少を招いている。私たちはタイムリーに基づいて受け入れ可能な条項で協力することができないかもしれないし、交渉することさえできないかもしれない。それができなければ、私たちが協力を求めている候補製品の開発を削減し、その開発計画や私たちの1つ以上の他の開発計画を減少または延期し、潜在的な商業化を延期したり、いかなる販売やマーケティング活動の範囲を縮小したり、私たちの支出を増加させ、自費で開発または商業化活動を行わなければならないかもしれない。もし私たちが私たちの支出を増やし、私たち自身の開発や商業化活動に資金を提供することを選択すれば、私たちは追加の資本を得る必要があるかもしれないし、これらの資本は受け入れ可能な条件で私たちに提供できないかもしれないし、全く得られないかもしれない。もし私たちが十分な資金がなければ、私たちは私たちの候補製品をさらに開発したり、それらを市場に出して製品収入を作ることができないかもしれない。
私たちはいくつかの候補製品を開発して商業化する権利は、他の会社が私たちに付与する許可証の条項と条件をある程度制限されている。
私たちのパートナーとの協力が成功するかどうかは、各当事者が関連する協力協定に基づいてそれぞれの義務を履行する状況にかかっている。このような合意は、米国に製品開発または商業化のための勤勉な義務、特定の開発または規制のマイルストーンおよび販売を実現する際の支払い義務、およびその他の義務を課す可能性がある。もし私たちが現在または未来の合意で規定されている義務を履行できなければ
86
私たちの取引相手はこれらの合意を終了する権利があるかもしれませんが、この場合、私たちは開発、製造、またはマーケティングプロトコルによってカバーされる候補製品を開発することができないかもしれません。これらの合意によって規定された許可または譲渡を終了するか、またはこれらの合意の下での私たちの権利を減少または廃止することは、私たちが条項の悪い新しい合意または修正された合意を交渉しなければならないことを招き、または重要な知的財産権または技術に対する私たちの権利を含むこれらの合意の下の権利を失うことになるかもしれない。
さらに、私たちは、私たちが許可を得たか、または第三者から譲渡された製品または候補製品をカバーする特許および特許出願の準備、提出、起訴、維持、強制執行、および弁護を制御する権利がないかもしれない。これらの特許および特許出願が我々の業務の最良の利益に適合する方法で準備、提出、起訴、維持、強制執行および弁護されなければ、関連する知的財産権に対する私たちの権利は減少またはキャンセルされる可能性があり、私たちの開発および商業化協定によってカバーされる製品、模倣薬、または候補製品の権利は悪影響を受ける可能性がある。
さらに、特定の特許権および他の知的財産権の許可に依存する第三者は、我々の製品、模倣薬または候補製品の開発、製造または商業化に重要または必要であり、これらの第三者自体は、他の第三者の上流ライセンスに依存する可能性がある。このようなサブライセンスは、私たちが開発または商業化したい候補製品のすべての関連使用分野またはすべての地域でカバーされた知的財産権を独占的に使用する権利を提供しない可能性があり、関連プロトコルの下での私たちの権利範囲のさらなる不確実性および複雑性を増加させる可能性がある。
さらに、私たちが未来に締結する許可または譲渡協定は複雑であり、そのようなプロトコルのいくつかの条項は様々な解釈の影響を受ける可能性がある。可能性のある契約解釈の相違の解決は、関連する知的財産権または技術に対する私たちの権利と考えられる範囲を縮小するか、または関連協定の下での私たちの財務または他の義務を増加させる可能性があり、いずれの場合も、私たちとそのパートナーとの協力関係を通じて私たちの発展に実質的な悪影響を及ぼす可能性がある。
著者らは第三者に依存して著者らの臨床前研究と任意の臨床試験のいくつかの方面を行い、これらの第三者の表現は満足できない可能性があり、締め切り前にこのような任務或いは試験を完成できなかったことを含む。
著者らはCRO、医療機関と臨床研究者が臨床前開発を行ういくつかの方面、例えばCRO、医療機関と臨床研究者が臨床前開発を行ういくつかの方面に依存し、分析開発とテストを含み、そして合格した患者を募集し、そして臨床試験を行い、監督と監督する。詳細については“を参照されたい-業務-私たちの研究と開発この年間報告書にあります。これらの第三者に対する臨床前と臨床開発活動への依存は,これらの活動の制御を減少させた。しかし、私たちのこれらの第三者への依存は、私たちの臨床研究が良好な臨床実践に適合することを確保し、関連する規制申請(例えばIND)に含まれる研究計画と方案を含む、私たちの監督責任を軽減しないだろう。また,我々と契約したCROは時間どおりに活動を達成できない可能性があり,あるいは法規の要求やわれわれの臨床研究設計に従ってわれわれの臨床前研究あるいは臨床研究を行っていない可能性がある。これらの第三者がその契約責任の履行に成功しなかった場合、または予想される締め切り前に完了しなかった場合、または我々の臨床レジメンまたは法規の要件を遵守できなかったためにCROまたは我々の研究者が取得した臨床データの品質または正確性に影響を与えるか、または生産された製品の品質がGMPに適合しない場合、製品、模倣薬および候補製品の開発および規制承認を達成し、それを商業化するための私たちの努力は延期または阻止される可能性がある。
また,われわれ,われわれの臨床プロジェクトCRO,われわれの研究者の臨床開発におけるすべての候補製品はGCPを遵守しなければならない。もし私たちまたは私たちの任意のCROまたは研究者が適用されたGCPに従わなかった場合、私たちの臨床試験で生成された臨床データは信頼できないと考えられるかもしれませんが、NMPA、FDA、または同様の規制機関は、私たちの上場申請を承認するかどうかを考慮する前に追加の臨床試験を行うことを要求するかもしれません。これは、規制部門の承認過程を遅らせることになります。
もし私たちの流通業者が関連協定に違反した場合、あるいは私たちが私たちと流通協定を締結していない流通業者が私たちの流通業者が遵守する政策と措置に同意しなければ、私たちの業務、将来性、名声は実質的で不利な影響を受ける可能性がある。
87
私たちは流通協定と私たちが制定した政策と措置によって私たちのディーラーを管理していますが、これらの合意、政策、措置が私たちのディーラーを効果的に管理することができるか、あるいは私たちのディーラーが私たちの合意と政策を遵守することを保証することはできません。もし私たちのディーラーが以下の1つまたは複数の行動をとる場合、私たちの業務、運営結果、将来性、および名声は不利な影響を受ける可能性があります
私たちの流通業者は、実際にまたは流通協定、私たちの政策、または任意の適用された法律と法規に違反または遵守しないと言われており、私たちの営業権が侵食され、私たちに責任を負わせ、私たちの流通ネットワークを混乱させ、私たちの製品、ETUARY、模造薬、および未来の製品の品質に対する大衆の不利な印象をもたらす可能性がある(承認されれば)。
また、私たちの一部の流通業者は私たちの製品を流通するために流通業者を招聘して、私たちはこれらの流通業者と直接接触しないし、彼らと契約関係を維持しない。代わりに、私たちは主に私たちの流通業者が法規の要求、私たちと流通業者との間の流通協定条項、そして私たちの流通業者に対する政策に基づいて、彼らのサブ流通業者を管理して制御することに依存している。私たちはこれらの流通業者に対する制御が限られているため、これらの流通業者が私たちの流通業者と合意した地域制限や私たちの流通協定や政策下の他の流通要求を遵守する保証はない。したがって、私たちは、私たちの業務を損なうかもしれないし、私たちの運営結果や名声に悪影響を与えないかもしれない、どんな流通業者のいかなる接近もタイムリーに発見または是正できる保証はありません。
私たちのいくつかの原材料は限られた数量のサプライヤーに依存しているため、私たちは供給が中断される状況に遭遇する可能性があり、これは私たちの製品を生産する能力を損なう可能性があります。
2023年12月31日と2022年12月31日までの数年間、私たちは少量のサプライヤーがいて、私たちは彼らが私たちと安定した関係があると信じています。しかし、私たちのサプライヤーの運営と業務戦略の安定性は私たちのコントロールを超えており、私たちの大型サプライヤーと安定した関係と質の高いアウトソーシング原材料やサービスを維持できる保証はありません。
従業員の事務、管理成長、私たちの業務運営に関するリスク
私たちの持続的な成功は私たちが肝心な幹部を維持し、採用し、熟練した専門家を維持し、激励する能力に依存する。
私たちが競争の激しい生物技術と製薬業界の中で競争できるかどうかは、私たちが高い素質の管理、科学と医療人員を引き付けることができるかどうかにかかっている。私たちは私たちの実行管理と科学者たちに非常に依存している。私たちはこのような個人や私たちの他のどんな従業員のためにも“キーパーソン”保険証書を維持しない。しかも、私たちは私たちの業務目標を達成するために人員を増やす必要がある。私たちの幹部、他の重要な従業員のサービスを失うこと、そして適切な代替者を見つけることができない、あるいは新しい臨床開発と製造者を募集することができず、製品開発の遅延を招き、私たちの業務を損なう可能性があります。
私たちはカリフォルニア州サンディエゴの工場でアメリカ業務を展開しています。この地域は多くの他の生物製薬会社と多くの学術·研究機関の本部である。私たちの市場は技術者に対する競争が非常に激しく、私たちが受け入れられる条件で高い素質の人員を採用し、維持する能力を制限する可能性があり、甚だしきに至っては根本的にはできない。中国では、他の製薬やバイオテクノロジー会社、大学、研究機関と合格した人材を争っている。適切な候補者は限られていて、現在の賃金レベルでは、十分な技術と経験豊富な科学者や他の技術者を採用して維持することができないかもしれません。提供する必要があるかもしれません
88
より高い報酬と他の福祉は、私たちの財務状況と運営結果に大きな悪影響を及ぼす可能性がある。
価値のある従業員を引き付けるために、賃金と現金インセンティブのほか、時間の経過とともに付与された株式オプションを提供しています。時間が経つにつれて、従業員に対する株式オプションの価値は、私たちの株価変動の大きな影響を受ける可能性があり、これらの変動は私たちの制御範囲を超えており、いつでも他社が提出したより利益のある見積もりを相殺するのに十分ではないかもしれない。私たちは価値のある従業員を引き留めるために努力しているにもかかわらず、管理チームと科学と発展チームのメンバーはすでに終了し、短時間で私たちとの雇用関係を終了する可能性がある。私たちの従業員は任意の雇用手配の下で仕事をしています。これは私たちのどの従業員も事前に通知するかどうかにかかわらずいつでも退職できることを意味します。人員を維持、置換、募集できなければ、私たちの業務を損なう可能性があります。
私たちの将来の業績は、新たに採用された幹部を私たちの管理チームに組み入れることに成功するかどうか、および上級管理職の間で効果的な仕事関係を築くことができるかどうかにある程度かかっています。私たちはこれらの個人を統合し、彼らと他の管理層のメンバーとの間に有効な仕事関係を構築することができず、私たちの製品、模倣薬と候補製品の開発と商業化効率の低下を招き、未来のマーケティング審査、私たちの製品、模倣薬と候補製品の販売、そして私たちの運営結果を損なう可能性がある。
私たちの従業員、主要な調査者、コンサルタント、ビジネスパートナーは、規制基準と要求を遵守しないこと、およびインサイダー取引を含む不正行為または他の不正活動に従事する可能性があります。
私たちは従業員、主要な調査者、コンサルタント、協力者の詐欺や他の不適切な行為のリスクに直面している。これらの当事者の不正行為は、意図的にアメリカ国家薬品監督管理局、アメリカ食品·薬物管理局、アメリカ証券取引委員会および非中国とアメリカ監督機関の規定を遵守できず、アメリカ国家薬品監督管理局、アメリカ食品と薬物管理局および非中国とアメリカの監督管理機関に正確な情報を提供することができず、中国、アメリカと海外の医療詐欺と乱用法律法規を遵守できず、財務情報やデータを正確に報告できなかった、あるいは許可されていない活動を私たちに開示することを含む可能性がある。特に、医療業界の販売、マーケティング、商業配置は、詐欺、不正行為、リベート、自己取引、その他の乱用行為を防止するための広範な法律法規によって制約されている。これらの法律法規は、広範な価格設定、割引、マーケティングおよび販売促進、販売手数料、顧客インセンティブ計画、および他のビジネス計画を制限または禁止する可能性があります。このような不正行為はまた不適切な使用が臨床研究期間中に得られた情報に関連する可能性があり、これは規制制裁を招き、著者らの名声に深刻な損害を与える可能性がある。従業員の不正行為を常に識別し、阻止することができるわけではなく、このような行為を発見し、防止するための予防措置は、未知または未管理のリスクや損失を効果的に制御することができないか、またはこれらの法律や法規を遵守できないことによる政府の調査または他の行動または訴訟から私たちを保護することができないかもしれない。もし私たちにこのような行動を取って、私たちが自分の権利を弁護したり、維持することに成功しなかったら、これらの行動は巨額の罰金や他の制裁を加えることを含めて、私たちの業務に大きな影響を与えるかもしれない。
上場企業の運営としては、引き続き大きなコストが発生し、我々の経営陣は上場企業の運営に関する法規を遵守するために多くの時間を投入する必要がある。
上場企業として、サバンズ-オキシリー法案とドッド·フランクウォール街改革と消費者保護法案、および米国証券取引委員会とナスダックが実施するルールを遵守するために、上場企業報告やコーポレートガバナンス要件に関連するコストを含む巨額の法律、会計、その他の費用を発生させ続けている。株主急進主義、政治環境、および現在の高度な政府介入と規制改革は、多くの新しい法規や開示義務を招く可能性があり、これは追加のコンプライアンスコストを招き、現在予想されていない方法で業務を運営する方法に影響を与える可能性がある。私たちの経営陣や他の人たちは上場企業の運営に関する法規を守るために多くの時間を投入する必要があります。また,これらの規制は,取締役や上級管理者責任保険を得ることが困難であり,コストも高く,現在のこのような保険レベルを維持するために大量のコストが必要となる可能性がある。上場企業として課せられた要求を守るために、毎年巨額の費用が発生すると予想しています。
私たちの管理チームはこれまでアメリカの上場企業を管理し、運営したことがありません。これらの幹部や他の人たちは、上場企業の報告要求と適用法律法規の遵守に関する専門知識を得るために多くの時間を投入して、私たちがこれらすべてを遵守することを保証する必要があります
89
要求します。これらの報告は要求、規則と規定に加え、上場会社が直面する可能性のある訴訟リスクの増加に加え、私たちが取締役会や取締役会委員会のメンバーになることを引き付け、維持することを困難にする可能性もあり、あるいは執行役員を務めたり、取締役と高級管理者保険を含むいくつかのタイプの保険を受け入れ可能な条件で獲得する可能性がある。
労働コストの増加は私たちの運営にマイナスの影響を与え、私たちの収益性に悪影響を与えた。
私たちの戦略と業務成長は私たちがより多くの従業員を雇用する必要があるかもしれないし、私たちは買収のためにより多くの従業員を募集するかもしれない。近年、インフレ、政府強制昇給、中国労働法の他の変化、製薬会社間の人材と適格社員の競争により、中国の平均労働力コストは着実に上昇している。したがって、労働コストの増加は私たちの成長にマイナス影響を与え、私たちの収益力を低下させる可能性がある。
私たちの知的財産権に関するリスクは
もし私たちまたは私たちの許可者が中国で承認された製品ETUARYおよび世界の任意の候補製品の十分な特許および他の知的財産権を確保、維持、擁護、または延長できなかった場合、またはこれらの知的財産権の広さが不足していれば、私たちの市場で効果的に競争する能力は影響を受ける可能性がある。
私たちは特許、商業秘密保護、秘密協定の組み合わせによって、私たちの製品、ETUARY、および候補製品に関連する知的財産権を保護します。ビジネスの重要性があると考えられる技術、製品、候補製品を保護するために、私たちはすでに中国、アメリカ、その他の国で特許出願を提出し続けている。しかしながら、特許保護の出願は高価で時間のかかるプロセスであり、私たちは必要または望ましいすべての特許出願を合理的なコストまたはタイムリーに成功裏に提出し、起訴することができないかもしれない。私たちは競争相手がこのようなすべての分野と地域で競争製品を開発して商業化することを阻止できないかもしれない。
バイオテクノロジーや製薬分野の特許実力は複雑な法律や科学問題に関連しており,不確定である可能性がある。例えば、科学または特許文献における発見と実際の発見および特許出願との間の遅延のため、私たちが私たちの特許または係属中の特許出願で主張された最初の発明であることは保証されない。中国と最近米国で採用されている“先出願”制度によると、合理的な調査を経ても、第三者が私たちの発明と同じまたは実質的に類似した発明について提出されたか、または私たちが提出した特許出願よりも早いか、または以前に提出された可能性があるので、私たちの製品、製品候補、プロセス、技術、改善および他の関連事項が特許不可になるかどうかを決定することができない可能性がある。第三者は私たちの特許の有効性、実行可能性、または範囲に疑問を提起する可能性があり、これはこれらの特許の範囲を縮小または失効させる可能性がある。私たちが持っている特許出願は、中国、アメリカ、または他の国/地域における私たちの製品および候補製品をカバーする権利を有する発行された特許を生成できないかもしれない。また、挑戦されていなくても、私たちの特許や特許出願は、私たちの知的財産権を十分に保護できず、私たちの製品や候補製品に排他性を提供したり、他の人が私たちの声明をめぐる設計を阻止したりすることができない可能性があります。私たちのいくつかの特許はまたプロセスを扱っており、実行することは難しいかもしれない。これらの結果のいずれも、第三者からの競争を防止する能力を弱める可能性があり、これらの競争は私たちの業務に悪影響を及ぼす可能性がある。
ETUARY、私たちのプログラムまたは候補製品の特許または特許出願が無効または発行されていない場合、またはそれらの保護の広さまたは強度が脅かされている場合、またはそれらが私たちの製品または候補製品に意味のある排他性を提供できない場合、将来の製品を商業化する能力を脅かす可能性がある。また、規制承認に遅延が生じた場合、特許保護の下で候補製品を販売する期間が短縮される可能性があります。しかも、特許の寿命は限られている。アメリカでは、特許の自然失効期間は一般的に申請後20年だ。中国では,“中華人民共和国特許法改正”(以下“改正された”中華人民共和国特許法“と呼ぶ)が特許期間延長と特許関連について規定している。改正された“中華人民共和国特許法”及び関連実施条例は訴訟理由を規定し、特許所持者が薬品監督審査過程中に明確な訴訟を提起することを許可し、その薬物が特許範囲に属するかどうかを確定し、これはアメリカの特許連動制度に匹敵する可能性がある。この制度は、NMPAが革新者が任意の訴訟を提起している間に潜在的な侵害の後続申請を引き続き審査することを要求する。しかし,国家特許商標局は後続の出願を承認せず,特許訴訟の解決を待つことができ,有利である
90
後続の出願または指定された時間帯は、より短い時間を基準とする。米国と同様に,改正された“中華人民共和国特許法”や関連実施条例も,特許権者の請求に応じて,新薬が規制審査過程で失った特許期間を延長することが規定されている。延長された特許期間は5年を超えず,新薬発売後の総特許期間は14年を超えない。しかし、私たちが中国で許可または所有している特許は、規制審査の過程で失われた任意の特許期間内に延長される資格がない可能性がある。様々な延期があることができる;しかし、特許の有効期間とその提供される保護は限られている。製品の特許有効期限が満了すると、私たちは模造薬の競争を受けるかもしれない。
特許提供の保護に加えて、私たちは、特許を出願できない、または特許を出願しない独自の技術を選択し、私たちの候補製品の発見および開発中に特許がカバーされていないノウハウ、情報、または技術に関する他の要素を保護するために、商業秘密保護および秘密協定に依存する。しかし、商業秘密は保護するのが難しいかもしれない。私たちは、従業員、コンサルタント、科学コンサルタント、請負業者と秘密保持協定を締結することで、私たちのノウハウやプロセスを保護することを求めています。我々はまた,我々のオフィスの物理的セキュリティおよび我々の情報技術システムの物理的および電子的セキュリティを維持することで,我々のデータとビジネス秘密の完全性とセキュリティを維持する.私たちはこのような個人、組織、そしてシステムに自信がありますが、合意や安全措置は違反される可能性があり、私たちはどんな違反にも対応する十分な救済措置がないかもしれません。しかも、私たちのビジネス秘密は競争相手に知られたり独立して発見されるかもしれない。
私たちは、すべての従業員およびコンサルタントが、その適用可能な発明を私たちに譲渡し、私たちのすべての従業員、コンサルタント、コンサルタント、および私たちのノウハウ、情報、または技術にアクセスできる任意の第三者が秘密協定を締結することを期待していますが、このようなすべての合意が正式に実行されているか、または私たちのビジネス秘密および他の機密固有情報が漏洩されないこと、または競争相手が他の方法で私たちのビジネス秘密または独立開発と実質的に同等の情報および技術を得ることができないことを保証することはできません。私たちのビジネス秘密を流用したり不正に開示したりすることは、私たちの競争地位を損なう可能性があり、私たちの業務に実質的な悪影響を及ぼす可能性があります。また,我々のビジネス秘密を守るための手順が不十分であると考えられれば,第三者による商業秘密の流用に対抗する十分な追跡権がない可能性がある.しかも、他の人たちは私たちの商業秘密と固有の情報を独立して発見するかもしれない。さらに、私たちの一部の従業員は、上級管理職を含めて、私たちの競争相手または潜在的な競争相手を含む他の製薬会社に雇われている可能性がある。これらの従業員たちは以前の仕事に関連した所有権、秘密、およびスポーツ禁止協定に署名したかもしれない。私たちは、これらの従業員がそのような従業員の任意の元雇用主の知的財産権(商業秘密または他の固有情報を含む)を使用または開示した疑いを受ける可能性がある。このようなクレームを訴訟する必要があれば、私たちは金銭的な損害を受け、貴重な知的財産権や人員を失う可能性がある。
また、世界のすべての国/地域で私たちの製品ETUARYと候補製品の出願、起訴、特許保護の費用は目を引くほど高くなり、私たちの中国とアメリカ以外のいくつかの国の知的財産権は中国とアメリカの知的財産権が広くない。また、一部の外国の法律は専有権の保護程度や方式も中国やアメリカの法律に及ばない。したがって、私たちは中国、アメリカ、海外の知的財産権の保護と保護に重大な問題に直面するかもしれない。もし私たちが私たちの技術に関連する非特許知的財産権の第三者への実質的な開示を阻止できず、私たちがこのような強制的に実行可能な商業秘密保護を持っている保証がなければ、私たちは私たちの市場で競争優位性を確立または維持することができないかもしれません。これは、私たちの業務、運営結果、および財務状況に大きな悪影響を及ぼす可能性があります。また、私たちは他の管轄区域の知的財産権侵害クレームと紛争に関連し、これらのクレームや紛争を弁護し、その是非にかかわらず、大量の訴訟費用を関連し、私たちの業務中の従業員資源を大量に移転する可能性がある。
私たちの特許保護の獲得と維持は、政府特許機関によって提出された様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
中国国家知的財産権局およびその他の政府特許機関は、特許出願中にいくつかのプログラム、文書、費用支払いおよびその他の同様の規定を遵守することを要求する
91
特許出願の流れ。例えば、特許有効期間内のいくつかの段階では、CNIPAおよび他の特許機関に定期保守費を支払わなければならない。多くの場合、適用された規則に基づいて、滞納金の支払いまたは他の方法で不注意を救済することができるが、規則を遵守しないことは、特許または特許出願の放棄または失効を招き、関連法ドメインの特許権の一部または全部を喪失させる可能性がある。このような規定を遵守しないイベントには、公式行動に迅速に対応していないこと、費用が支払われていないこと、および正式な文書が正しく提出されていないことが含まれる可能性がある。また、中国特許法によれば、いずれの出願人が外国で中国で完成した発明又は実用新案のために特許を出願するかは、国家知的財産権局に報告して秘密審査を行わなければならない。出願人が国家知的財産権局に秘密審査を行っていることを報告していない場合は,以後中国で出願した者は,特許権を付与してはならない。
私たちの特許保護範囲は不確定である可能性があり、第三者の知的財産権侵害または私たちの特許の発明権または所有権に挑戦するクレームは、私たちの開発および商業化努力を阻害または延期する可能性がある。
特許出願で要求されるカバー範囲は特許発行前に大幅に縮小することができ,特許発行後にその範囲を再解釈することができる.私たちが現在または将来許可または所有している特許出願が特許として発行されても、それらの発表形態は、競争相手や他の第三者が私たちと競争することを阻止し、または他の方法で任意の競争優位性を提供してくれることを提供してくれないかもしれない。また,医療機器会社の特許地位は一般に高い不確実性を有しており,複雑な法律や事実問題に関連しており,近年多くの訴訟の対象となってきた。したがって,我々の特許権の発行,範囲,有効性,実行可能性,商業的価値は高い不確実性を持っている.
私たちの商業的成功は第三者の特許と固有の権利の侵害を避けることにある程度依存する。中国と米国国内外では、特許侵害訴訟、妨害、CNIPA、USPTOまたは他の関連知的財産権局に以前の技術を提出した第三者、反対など、バイオテクノロジーや製薬業界に関連する特許やその他の知的財産権に関する訴訟が大量にある各方面間CNIPA、USPTO、および対応する外国特許庁の前の再審査手続きおよび認可後のプログラム、例えば、反対、派生、撤回、無効、再審または各方面間外国司法管轄区域において我々の発明の優先権又は我々の特許及び特許出願の他の特許的特徴に疑問を提起する審査、妨害手続又は同様の手続。我々が開発候補を求めている分野には,第三者が所有する米国や外国から発行された特許や係属中の特許出願が多く存在する.バイオテクノロジーや製薬業界の拡張やより多くの特許の発行に伴い,我々の製品,ETUARYおよび候補製品は第三者特許権侵害の告発を受けるリスクが増加する可能性がある。
第三者は、私たちの製品ETUARYと私たちの候補製品の製造、使用、または販売は、これらの第三者が持っている特許を侵害している、または私たちは彼らの独自技術を不正に使用したと主張するかもしれない。我々の製品、模倣薬および候補製品の使用または製造に関連する第三者特許または特許出願が存在する可能性があり、これらの特許または特許出願は、我々の製品、模倣薬および候補製品の成分、材料、配合、製造方法または治療方法に関連する。特許出願が発行されるまでに数年かかる可能性があるため、現在処理されている特許出願が存在する可能性があり、これらの出願は、我々の候補製品または現在の製品が発行された特許を侵害する可能性がある。
私たちにクレームを出した当事者は、私たちが侵害製品(これは不可能かもしれない)を再設計したり、適用特許によって許可されない限り、(これらの特許は商業的に合理的な条項では得られないかもしれない)、またはそのような特許が満了するまで、私たちの1つ以上の製品ETUARYおよび私たちの候補製品をさらに開発し、商業的に商業化することを効果的に阻止することができるかもしれない。
私たちは私たちの特許を保護したり強制したりする訴訟に巻き込まれるかもしれない。
競争相手は私たちの特許を侵害するかもしれない。権利侵害や不正使用に対抗するために、私たちまたは私たちの協力者は、費用がかかり、時間がかかるかもしれない侵害請求を要求されるかもしれない。さらに、侵害訴訟では、裁判所は、私たちの特許が無効であること、強制執行できないこと、および/または侵害されていないことを判断することができ、または、私たちの特許が関連技術をカバーしていないことを理由に、他方の関連技術の使用を阻止することを拒否することができる。第三者の提出、訴訟または訴訟の不利な裁決または結果は、特許権または排他性の喪失、または特許権利要件の縮小、無効または実行不能をもたらす可能性がある
92
これは、競争相手が同様の技術および製品を使用または商業化することを阻止する能力を制限するか、または私たちの技術、製品、ETUARY、および候補製品の特許保護期間を制限するかもしれない。
我々の特許または特許出願または我々の許可者の特許または特許出願に関連する発明の優先度を決定するために、第三者によって引き起こされるか、または我々によって提起された干渉手続が必要である可能性がある。不利な結果は、関連技術の使用を停止することを要求するか、または勝利者から権利許可を得ようとすることを要求するかもしれない。もし勝利者が商業的に合理的な条件で私たちにライセンスを提供しなければ、私たちの業務は損害を受ける可能性があります。私たちは単独でまたは私たちの許可者と共に私たちの知的財産権の盗用を防ぐことができないかもしれません。特に法律では中国やアメリカのようにこれらの権利を十分に保護していない国があるかもしれません。
さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。聴聞、動議、または他の一時的手続き、または事態発展の結果を公開することもできる。もし証券アナリストや投資家がこれらの結果がマイナスだと思っていれば、私たちの普通株の価格に実質的な悪影響を及ぼす可能性がある。
知的財産権訴訟は私たちに大量の資源を費やし、私たちの人員の正常な義務に対する注意を分散させるかもしれない。
解決策が私たちに有利であっても、知的財産権クレームに関する訴訟や他の法的手続きは、その是非にかかわらず、巨額の費用を発生させ、私たちの技術や管理者の正常な責任を分散させる可能性があります。私たちに対する侵害クレームが成功した場合、印税の支払い、侵害製品の再設計、または第三者から1つ以上の許可を得ることに加えて、3倍の損害賠償と故意に侵害された弁護士費を含む大量の損害賠償を支払わなければならないかもしれないし、多くの時間とお金の支出が必要かもしれない。また、公聴会、動議、または他の一時的な手続きまたは事態の発展の結果が公表される可能性があり、証券アナリストや投資家がこれらの結果がマイナスだと思う場合、私たちの普通株の価格に大きな悪影響を及ぼす可能性がある。このような訴訟または訴訟は、私たちの運営損失を大幅に増加させ、開発活動または任意の将来の販売、マーケティング、または流通活動に使用することができるリソースを減少させる可能性がある。私たちはそのような訴訟や訴訟手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らがより多くの財政資源を持っているので、私たちよりもこのような訴訟や訴訟の費用を効率的に負担するかもしれない。特許訴訟や他の訴訟の開始と継続による不確実性は、市場での私たちの競争能力を損なう可能性がある。
特許法の変更は特許の全体的な価値を低下させる可能性があり、これはETUARYと私たちの候補製品ラインを保護する能力を弱める可能性がある。
中国全国人民代表大会および国家知的財産権局の決定は、予測不可能な方法で特許を管理する法律および法規を変更する可能性があり、これは、私たちが新しい特許を獲得したり、私たちの既存特許および/または未来の特許を実行する能力に影響を与える可能性がある。米国は広範囲な特許改革立法を制定し、実施している。さらに、米国最高裁判所の最近の裁決は、場合によっては入手可能な特許保護範囲を縮小し、場合によっては特許所有者の権利を弱める。他の管轄区域法律の類似した変化は、私たちの特許権または他の知的財産権の価値に影響を及ぼす可能性がある。我々の将来の特許取得能力に関する不確実性の増加に加えて,いったん特許を取得する価値(あれば)に関する不確実性が存在する.中国と他の司法管轄区の特許に関する法律と法規の変化に伴い、これらの変化は私たちの知的財産権保護にマイナスの影響を与える可能性がある。
第三者から何らかの知的財産権の許可を得る必要があるかもしれませんが、このような許可は得られないかもしれませんし、商業的に合理的な条項では得られない可能性もあります。
第三者は特許権を含む私たちの候補製品開発に非常に重要または必要な知的財産権を持っている可能性がある。私たちは第三者の特許やノウハウを使用して私たちの候補製品を商業化する必要があるかもしれません。この場合、私たちは商業的に合理的な条項でこれらの第三者から許可を得ることを要求されます。そうでなければ、私たちの業務は損害を受ける可能性があり、実質的な可能性があります。
93
私たちは私たちの商標と商号を保護できないかもしれません。これは興味のある市場でブランド認知度を確立する能力にマイナスの影響を与えるかもしれません。
私たちは現在、発行された商標登録を有し、関心のある市場の潜在的なパートナーや顧客の間で知名度を確立するために、商標出願を処理している。しかし、このような商標登録と申請は私たちを商標の無効化、希薄化、侵害の危険に直面させる。私たちの商標登録と申請は、政府または第三者の反対を受ける可能性があり、疑問、侵害、回避、または汎用として発表される可能性がある。発表された商標登録または商標出願が成功して挑戦された場合、私たちは、そのような商標登録または出願を登録または維持することができない可能性がある。また、私たちの製品ETUARYが引き続き市場で販売されるにつれて、このような製品の私たちの商標への依存が増加し、競争相手と区別される可能性があります。私たちは、第三者が私たちの商標権の侵害、登録または使用の侵害、希釈、または他の方法で私たちの商標権を侵害する商標および商業的外観を採用することを阻止できないかもしれないし、不正な競争、誹謗、または私たちの商標権を侵害する他の行為に従事することができないかもしれない。さらに、我々の登録または未登録商標または商号変異体を含む他の登録商標または商標の所有者は、私たちに商号または商標侵害クレームを提出することができる。もし私たちが私たちの商標と商品名に基づいて名称を確立することができなければ、私たちは興味のある市場で効果的に競争できないかもしれません。私たちの業務は不利な影響を受けるかもしれません。
知的財産権は私たちの業務や競争優位が直面しているすべての潜在的な脅威を解決できないかもしれない。
私たちの知的財産権が提供する保護の程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できないか、あるいは競争優位性を維持することができるかもしれない。既存の知的財産権保護制度の限界には:
これらの事件が発生すると、それらは私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
我々の候補製品の規制承認とその他のコンプライアンス問題に関するリスク
我々の製品ETUARYの研究,開発,製造,商業化のすべての実質的な面で厳しい規制を受けており,この製品は中国で承認されている。
規制の承認を得て適用される法律や条例を遵守することは長く、高価で不確実な過程であり、多くの時間と財政資源がかかる。薬物開発過程または承認過程中または承認後のいつでも、適用の要求を守らなければ、行政または司法制裁を受ける可能性がある。これらの制裁は、規制機関が承認保留申請を拒否すること、承認撤回、免許取り消し、臨床封印、警告または無見出し手紙、自発的または強制的な製品リコール、製品差し押さえ、生産または流通の完全または部分的な一時停止、輸入警報、禁止、罰金、政府契約の拒否、原状回復、差し戻しまたは民事または刑事罰を含むことができるが、これらに限定されない。
アメリカ国家食品薬品監督管理局、FDAと類似の外国監督機関の承認手続きは広く、長引く、しかも本質的に不確定である。必要な承認を得られなかったり、承認過程で遅延に遭遇したりすると、中国におけるETUARYの将来の用途、F 573は中国で、F 351は中国と中国以外の他の市場で、これは私たちの収入創出に深刻な影響を与える可能性がある。
94
中国、アメリカと国外では、規制承認を得る過程は予測不可能で高価であり、承認されれば、通常臨床試験開始後数年が必要であり、関連する候補製品のタイプ、複雑性、新規性を含む様々な要素によって大きく変化する可能性がある。
いくつかの模倣薬を除いて、私たちは中国でIPFの治療に承認された商業化に成功した製品ETUARYしかない。ETUARYは現在中国でSSc-ILDとDM-ILDを治療する第三段階の臨床試験、塵肺疾患を治療する第三段階の臨床試験、及びDKD計画に対するETUARYの第一段階の臨床試験にある。ETUARYは中国で適応の治療に許可されているにもかかわらず,中国でETUARYを他の適応の治療に成功させることはできないかもしれない。
また,F 351は現在中国で慢性B型肝炎に関連する肝線維化の3期臨床試験を行っている。また、F 351は現在米国FDAで行われているIND申請があり、多種の慢性肝疾患に関連する肝繊維化の治療に用いられている。将来的には,NASHに関連する肝線維化に特化した追加のINDをF 351に提出することが予想され,将来の適応または将来の製品候補により多くのIND申請を提出する可能性がある。もしこのような未来のINDがFDAの適時な承認を得なければ、私たちの臨床開発スケジュールは負の影響を受ける可能性があり、任意の未来の臨床計画は延期または終了される可能性がある。したがって、私たちは規制部門の承認を受けたり、私たちの候補製品を成功的に商業化することができないかもしれない。
我々はまた、ALF/ACLF治療のためのF 573を含む早期臨床段階の製品ラインを有する。F 573はすでに中国で第二段階の臨床試験に入った。著者らは2022年7月に耐性とPKの第一段階臨床観察を完成し、2023年3月にF 573の第二段階臨床研究を開始した。著者らはまた分級の臨床前製品ラインを構築した。例えば,COPD治療のためのF 528を検討·開発している。また、私たちの候補製品F 230は現在臨床前段階にあり、動物試験でPAHを著しく緩和する潜在力を証明し、2024年3月13日、私たちは中国でF 230のIND申請を提出した。
もしあれば、いかなる臨床前研究と臨床試験が計画通りあるいは予定通りに完成することを保証することはできない。著者らの候補製品の臨床開発は薬物開発のどの段階においても失敗リスクの影響を受けやすく、臨床試験中或いは適切な患者集団で治療効果を証明できなかったこと、深刻な或いは医学的或いは商業的に受け入れられない不良事件の発生、規程或いは適用の法規要求を遵守できなかったこと、及びNMPA、FDA或いは任意の類似の外国監督管理機関が薬物製品の承認できないことを確定することを含む。私たちの1つまたは複数の候補製品が有益な効果を有していても、1つまたは複数の様々な要因(私たちの臨床試験の規模、持続時間、設計、測定、進行または分析を含む)により、臨床評価中にその効果が検出されない可能性がある。逆に,同様の要因により,われわれの臨床試験では,候補製品の有意な積極効果が実際の積極効果よりも大きいことが示唆されている可能性がある。
中国や米国で候補製品を商業化することはできません。米国国家食品薬品監督管理局またはアメリカ食品·薬物管理局の監督管理によって承認されない限り、同様に、外国規制機関のような規制承認を得ていない場合には、候補製品を中国や米国以外の場所で商業化することはできない。規制機関が私たちの候補製品の商業販売を承認する前に、私たちの候補製品は各目標適応に対して安全かつ有効であることを、長く、複雑で高価な臨床前研究と臨床試験によって証明しなければならない。監督管理の承認を得るためには,薬品製造過程に関する情報を関連監督機関に提出し,生産施設を検査する必要がある。また、我々の候補製品には、将来の適応のためのETUARY、中国のF 573、F 528およびF 230、および中国と中国以外の潜在的な追加市場のF 351が含まれており、効果がない可能性があり、適度に有効であるか、または不良または意外な副作用、毒性または他の特徴があることが証明される可能性があり、上場承認を阻害する可能性がある。
アメリカ国家薬品監督管理局、FDAと類似の外国の監督管理機関は審査過程中にかなりの自由裁量権を持っており、いかなる申請を受け入れることを拒否することができ、また私たちのデータが承認を得るのに十分ではなく、追加の臨床前、臨床或いはその他のデータが必要であることを決定することができる。私たちの候補製品は多くの理由で規制部門の承認を得ることができないかもしれません
95
候補製品の承認要求は管轄区域によって異なる可能性があるため、1つの管轄区で成功することは必ずしも他の管轄区で成功することを意味するわけではない。
以下に関連する遅延を含む計画中の臨床試験の完了を様々な理由で遅延させる可能性がある
もし医師が未解決の道徳的問題に遭遇した場合、患者を私たちの候補製品に組み入れる臨床試験を含むが、私たちの候補製品の核心適応の深刻な疾病の性質に基づいて安全性と有効性ファイルが確立された既存の治療方案を出すのではなく、私たちも承認を遅延させる可能性がある。さらに、臨床試験は、法規の要求またはその臨床規程に基づいて臨床試験を行うことができず、FDAまたは他の監督機関による臨床試験操作または試験地点の検査による臨床休止、予見できない安全問題または不良副作用、候補製品の使用を証明できない利点を含む、著者ら、試験を行っている機関のIRBs、試験のデータ監視委員会またはNMPA、FDAまたは他の監督機関が様々な理由で一時停止または終了することができる
96
政府法規や行政行為の変化或いは臨床試験を継続するのに十分な資金が不足している。さらに、FDAの審査·承認過程は、米国政府の将来のいかなる停止によっても延期される可能性があり、私たちの開発活動はそのために損害を受けたり延期されたりする可能性がある。もし私たちがどんな候補製品の臨床試験の終了や遅延の状況に遭遇すれば、候補製品を商業化する能力を損なうことになり、私たちが収入を作る能力は深刻な損害を受けるだろう。また、試験完了の遅延はコストを増加させ、私たちの製品開発と承認過程を延期し、製品販売を開始し、収入を創出する能力を弱める。臨床試験の開始または完了遅延をもたらす可能性がある多くの要因は、私たちの候補製品が規制の承認を拒否される可能性がある。
大量に開発されている薬の一部だけがNMPAを完成させました FDAや外国規制機関の承認手続きは、商業化されている。長い承認過程と将来の臨床試験結果の予測不可能性は、将来の適応のためのETUARY、中国のF 573、F 528とF 230、中国のF 351、中国以外の潜在的な追加市場を含む規制部門の承認を得られない可能性があり、これは私たちの業務、運営結果、および将来性を深刻に損なうだろう。
もし私たちが承認されれば、規制機関は、将来の適応のためのETUARY、中国のF 573、F 528およびF 230、および中国と中国以外の潜在的な追加市場のF 351、私たちが要求するよりも少ないまたは限られた適応を含む任意の候補製品を承認するかもしれません。最も商業的な見通しのある適応を承認できなかったことを含み、高価な発売後の臨床試験の表現に従って承認されるかもしれません、または候補製品の商業化に必要または必要なラベル声明の製品が含まれていない可能性があります。もし私たちが私たちの候補製品を得るために必要な規制承認を得ることができない場合、中国のETUARY、中国のF 573、F 528とF 230、中国のF 351、中国以外の潜在的な追加市場を含めることができなければ、私たちの候補製品を商業化することができない、あるいは商業化の過程で遅延し、私たちの収入を作る能力は大きな損害を受けるだろう。
NASHに関連する肝線維化の治療のためのF 351を開発している。国家食品薬品監督管理局はF 351の要求を承認した FDAと比較可能な外国規制機関は未知であり,予測が困難である可能性があり,時間の経過とともに変化する可能性があり,臨床開発の時間やコストや上場承認の可能性を予測することは困難である。
NASHに関連する肝線維化の治療のためのF 351を開発している。FDAはNASHを治療する薬物開発ガイドラインを発表しているにもかかわらず,米国ではF 351のような新候補製品の開発は他のより有名あるいは広く研究されている候補品よりも高価で時間がかかる可能性がある。他社はその潜在Nash療法の臨床試験の後期段階にあるため、これらの会社がその監督承認策略を完備し、監督機関と相互作用することに伴い、Nash療法の監督承認経路は短期的に発展し続ける可能性が予想される。この変化は今日予測できない方法で、試験規模と終点を含む未来の臨床試験設計に影響を与えるかもしれない。特に,肝生検データに対する規制当局の期待は変化する可能性があり,特に肝生検データの内在的変異性に関する情報発表に伴い。
FDAのコミュニケーションの後,われわれのいくつかの競争相手はNASH療法の規制に挫折した。これらの挫折がNASH療法やF 351の規制承認にどのような影響を与える可能性があるかは不明である(あれば)。
F 351または任意の将来の候補製品の開発が延期された場合、NMPA、FDAまたは他の同様の規制機関が、現在予想されている研究または試験以外に研究または試験を行うことを要求するか、または進行中または将来の臨床試験設計を変更することが要求されるので、我々の予想される開発コストが増加する可能性がある。また,われわれのNASH適応安全データベースを利用できなければ,追加的な試験が必要となる可能性があり,コスト増加を招き,われわれの臨床試験の時間や結果に影響する可能性がある。また,F 351は単一療法として開発されているのではなく,併用療法の一部として開発されている可能性があり,臨床開発の複雑さを増加させ,F 351のS開発をさらに遅延させ,我々のコストや経営陣の資源移転に影響を及ぼす可能性がある。
97
私たちは、私たちの業務に必要ないくつかの承認、免許、ライセンス、証明書を取得または更新することができず、私たちの業務、財務状況、および運営結果に実質的な悪影響を及ぼす可能性があります。
関連法律法規によると、私たちは関連部門から各種の承認、許可証、許可証と証明書を取得して維持して、私たちの業務を経営する必要があります。いくつかの承認、許可証、免許および証明書は、関係当局によって定期的に継続および/または再評価されなければならず、継続および/または再評価の基準は時々変更される可能性がある。当社の運営を取得または継続するために必要な任意の承認、免許、許可証および証明書を取得できない行為は、関連規制機関による当社の運営停止命令を含む法律執行行動を引き起こす可能性があり、資本支出または救済行動を要求する是正措置を含む可能性がある。既存の法律法規の解釈や実施が変化したり、新しい法規が発効したり、以前に必要とされていなかった追加の承認、許可、免許または証明書を取得することが要求された場合、そのような承認、許可、許可または証明書の取得に成功することは保証されない。
顧客と第三者支払者との関係は、刑事制裁、民事処罰、契約損害、名声損害、利益および将来の収益の減少に直面する可能性がある反リベート、詐欺および乱用、および他の医療法律法規の制約を受けることになります。
アメリカです
医療提供者、医師、第三者支払者は、私たちが市場で承認された任意の候補製品の推薦と処方において主な役割を果たすだろう。私たちの将来の第三者支払者と顧客との手配は、私たちの製品、ETUARYと未来の製品の業務または財務配置と関係を制限するかもしれない幅広い適用された詐欺や乱用、および他の医療法律や規制に直面するかもしれません。製薬会社としては,我々がいなくても医療サービスの転転を制御したり,Medicare,Medicaid,あるいは他の第三者支払者に直接請求書を発行したりすることはできないが,詐欺や乱用や患者の権利に関する連邦や州医療法律法規は現在も将来も我々の業務に適用されている。これらのルールには
98
私たちが第三者の業務配置と適用される医療法律や法規に適合するように努力することは、多くのコストに関連することを確実にします。政府当局は、私たちの業務実践が現在または未来に適用される詐欺および乱用または他の医療保健法律および法規に関連する現行または未来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。もし私たちの運営がこれらの法律または任意の他の適用可能な政府法規に違反していることが発見された場合、私たちは重大な民事、刑事、行政処罰、損害賠償、罰金、監禁、MedicareおよびMedicaidのような政府援助の医療計画から除外され、私たちの運営を削減または再構成されるかもしれない。もし私たちがそれと業務を展開する任意の医師や他の医療提供者や実体が適用法を遵守していないことが発見された場合、彼らは政府の援助された医療計画から除外されることを含む刑事、民事または行政制裁を受ける可能性がある。
中華人民共和国
我々の業務は、“中華人民共和国反不正競争法”、“中華人民共和国刑法”、“医師報酬陽光法律法規”を含む様々な詐欺と法律の乱用によって制限されているが、これらに限定されない。これらの要求を遵守する条件に曖昧な点があり、このような詐欺および法律違反行為は、罰、罰金および/または政府医療計画の排除または一時停止を含む刑事および/または民事制裁を受ける可能性があり、関連する司法管轄区域との契約を禁止する。法執行部門がこれらの法律をより重視するようになるにつれて、私たちが第三者の業務手配と適用される医療法律や法規に適合するようにする努力は巨額のコストに及ぶ可能性がある。
私たちは、米国の“海外腐敗防止法”や“海外腐敗防止法”や、中国と私たちが業務を経営している他の国の反腐敗·反賄賂法律、および米国と特定の外国の輸出規制、貿易制裁、輸入法律法規のような責任に直面する可能性がある。これらの法律の要求を遵守することは、特定の市場での私たちの競争能力を制限する可能性があり、私たちがこれらの法律に違反したと判断するいかなる行為も、私たちの業務または私たちの名声に実質的な悪影響を及ぼす可能性がある。
私たちの業務は“海外腐敗防止法”および中国と私たちが業務を展開している他の国の反賄賂や反腐敗法律、法規または規則のような制約を受けています。“海外腐敗防止法”およびその他の法律は、一般に、業務または他の利益を獲得または保留することを目的として、私たち、私たちの官僚、私たちの従業員、および仲介機関が非米国政府当局者に不正なお金を直接または間接的に提供、許可または支払いすることを禁止している。私たちは第三者を招いて中国とアメリカ以外で臨床試験を行い、私たちの商業化製品ETUARYおよび任意の他の未来製品(承認された場合)を海外に販売し、および/または必要な許可、許可証、特許登録、および他の規制承認を得ることができる。私たちは政府機関や政府付属病院、大学、その他の組織の役人や従業員と直接または間接的な相互作用を持っている。私たちの業務の拡大に伴い、“海外腐敗防止法”や他の反賄賂法の私たちの業務への適用性が増加します。もし私たちのプログラムと制御が
99
反賄賂コンプライアンスを監督することは、従業員や代理人の無謀なまたは犯罪行為から私たちを保護することができない場合、または私たちまたは私たちの従業員、代理、請負業者、または他の協力者が適用される反賄賂法律を遵守できない場合、私たちの名声は損なわれる可能性があり、私たちは刑事または民事処罰を受け、利益を渡し、他の制裁および/または重大な費用を発生させる可能性があり、これは、私たちの財務状況、運営結果、キャッシュフロー、および将来性を含む、私たちの業務に重大な悪影響を及ぼすかもしれません。
また、我々の商業化製品ETUARYや任意の他の未来製品は、承認されれば、米国や外国の輸出規制、貿易制裁、輸入法令の制約を受ける可能性がある。政府は、私たちの製品、ETUARY、未来の製品の輸出入を規制しており、承認された場合、あるいは必要なこのような製品の輸出入許可を得ることができなかった場合(適用すれば)、国際的または国内販売を損なう可能性があり、私たちの収入に悪影響を及ぼす可能性がある。我々の製品、ETUARY、および将来の製品輸出に関する適用法規の要求を遵守し、承認されれば、そのような製品の国際市場での発売を延期するか、または場合によっては、そのような製品の特定の国/地域への輸出を完全に阻止する可能性がある。また、米国の輸出規制法と経済制裁は、米国の制裁対象国、政府、個人に特定の製品やサービスを輸送することを禁止している。もし私たちが輸出入条例とこのような経済制裁を守らなければ、罰金および/または特定の輸出特権の剥奪を含む罰を受けるかもしれない。さらに、任意の新しい輸出または輸入制限、新しい立法、または既存の法規の実行または範囲内、またはこれらの法規が対象とする国、個人または製品のいずれの新しい方法も、私たちの製品、ETUARY、および将来の製品の使用量の減少(承認された場合)、または既存または潜在的な国際業務顧客にそのような製品を輸出する能力の低下をもたらす可能性がある。もし私たちの製品、ETUARY、未来の製品の使用を減らすことを承認したり、私たちがこのような製品を輸出したり販売する能力を制限したりすれば、私たちの業務に悪影響を及ぼす可能性があります。
私たちの運営結果は、現在と未来に取られる可能性のある医療立法と規制行動の悪影響を受けるかもしれない。
我々が研究、開発、製造、商業化活動を行ったすべての司法管轄区域はこれらの活動に対して深く、詳細な監督管理を行った。規制部門の承認を得ることは長くて高価で不確実な過程だ。私たちは私たちの業務を中国とアメリカの主要な市場に重点を置くつもりです。これらの地政学領域はすべて医療器械に対して厳格な監督管理があり、このようにする過程において、それらは大体似た監督管理策略を採用し、医療機器の製品開発、審査、製造、販売とマーケティング及び流通の監督管理を含む。しかし、地域によって規制制度が異なり、私たちのようにこれらの地域で事業を展開することを計画している会社の規制コンプライアンスはより複雑で、コストも高い。
アメリカです
政府の処方薬調達や精算計画に影響を与える立法や規制行動が頻繁に発生している。米国では,ACAは2010年に公布され,医療保険のカバー範囲を拡大している。その時以来、人々は腐敗防止法の全部または一部を廃止、改正、または行政制限するために多くの努力をしてきた。例えば、トランプ総裁が2017年に法律となった減税·雇用法案に署名して個人医療保険認可を廃止したことは、ACAの重要な構成要素と考えられている。2018年12月、テキサス州の連邦地域裁判所は、この判決が保留されて控訴を待っていたにもかかわらず、個人医療保険のライセンス違憲を理由にACAを覆した。ACAが直面している持続的な挑戦と新たな立法提案はACAの将来の生存能力と医療保険市場の不安定な不確実性を招いた。これによる私たちの業務への影響は不確実であり、実質的かもしれない。
処方薬の価格を統制する努力はまた私たちの業務に実質的な悪影響を及ぼす可能性がある。例えば、2018年、トランプ総裁と米国衛生·公衆サービス部長官は“米国患者優先青写真”を発表し、いくつかの部分の実施を開始した。このイニシアティブには,後発薬と生物類似薬の競争を増加させ,連邦医療保険計画が薬品価格をより直接交渉できるようにし,薬品価格の透明性を向上させ,我々消費者の自己負担コストを低減する方法がある。トランプ政権はまた、コスト決定の基準として“国際価格設定指数”を提案し、連邦医療保険B部での薬品の精算を制限する可能性がある。他の製薬業者に関する提案では、メーカーの壊滅的な段階での支払いを増やすために福祉構造を変更する法案が提案されている。前回の国会期間中,薬品定価に関する法案数が急激に増加したことから,我々の業務への影響は不確実であり,実質的である可能性がある。第118回国会がどの程度このようなやり方を続けるかはまだ定かではない。
100
IRAはCMSに処方薬と生物価格を直接交渉する能力を提供し、Medicare Part D参加者の自己支出を制限した。毎年、CMSは予め設定された数量の高価な薬物および生物製品を選択し、交渉するであろう。これらの薬物および生物製品は連邦医療保険B部分およびD部分に含まれ、模倣薬または生物類似の競争が不足している。D部分の価格交渉は2023年に始まる。アイルランド共和軍は2023年に発効し、連邦医療保険B部分またはD部分で規定されている薬物や生物の価格上昇幅がインフレ率よりも速い場合、薬品メーカーは連邦政府に新しい“インフレリベート”を支払わなければならないと規定している。アイルランド共和軍には薬品支出と連邦赤字を減らすための他の多くの条項が含まれており、アイルランド共和軍の競争と商業化への影響はまだ確定していないが、実質的な可能性がある。
さらに、多くの州は、バイオ製薬メーカーに独自の価格設定情報を開示すること、または国家機関が購入した薬品に対して最高価格上限を設定することを要求するなど、間接的または直接に薬品価格を規範化するための立法を提出または公布した。例えば、2017年、カリフォルニア州知事は、指定された敷居を超えるいくつかの薬品の値上げに対する処方薬メーカーの事前通知および解釈を要求する処方薬価格透明性州法案に署名した。国会や州立法機関は様々な法案を考慮しており,これらの法案は薬品調達と価格交渉を改革し,管理ツールを利用して連邦医療保険D部分のカバー範囲を制限し,米国以外からの低価格薬物の輸入を促進し,後発薬の使用を奨励することを許可している。もしアメリカで承認されれば、このような措置と立法は私たちの未来の製品に追加的な価格設定圧力を与えるかもしれない。
連邦や州レベルの医療補助計画の変化も我々の業務に実質的な悪影響を及ぼす可能性がある。米国で承認されれば、医療補助計画がカバーする薬剤を管理するための各州により大きな柔軟性を与えることや、米国で承認されれば、将来の製品の使用およびカバー範囲を制限する可能性があるため、将来の製品のカバー範囲や精算範囲の提案に影響を与える可能性がある。また,連邦基本医療補助税還付の増加により,州医療補助計画は我々の製品に対する追加的な補充還付を要求する可能性がある。ある程度、民間保険会社または管理式医療計画は医療補助カバー範囲と支払い状況を追跡し、米国で承認されれば、これらの増加したリベートの公布を利用して私たちの将来の製品に定価圧力をかけることができ、彼らがより低い支払いスケジュールを採用することは悪影響を拡大する可能性がある。
他の提案された影響メーカーの規制行動は私たちの業務に実質的な悪影響を及ぼすかもしれない。このような提案された立法や規制行動やそれによって生じる州行動が米国での将来の製品の使用や精算(承認されれば)に及ぼす影響を予測することは困難であるが,我々の運営結果は悪影響を受ける可能性がある。
中華人民共和国
国家薬品監督管理局の政策が変わる可能性があり、あるいは追加の政府法規が公布される可能性があり、これらの法規は私たちの候補製品に対する監督管理審査を阻止、制限または延期し、承認後の活動を制限または規範化し、私たちの利益能力に影響を与える可能性がある。規制環境が変化している中国の将来の立法や行政行動が生じる可能性のある政府政策や法規の可能性、性質、程度を予測することはできない。例えば、監督管理要求とガイドラインの変化が臨床試験方案を大幅に修正することを要求すれば、著者らはコストの増加或いは適時或いは根本的に臨床試験を完成できない状況に遭遇する可能性がある。薬品登録と承認に関連する政府法規の変化、例えば規制要求を緩和したり、簡略化された承認手続きを導入することは、潜在的な競争相手の参入ハードルを下げたり、規制要求を増加させたりする可能性があり、これらの要求を満たすことの難しさを増加させる可能性がある。
近年、中国は、より厳格なカバー基準を含む行政または立法措置の制定に努力し、私たちの製品、ETUARY、未来の製品の価格を下振れ圧力に直面させる可能性がある(承認されれば)。このような定価下圧力に関するリスクの詳細については、“を参照されたい私たちの業務運営と候補製品に関するリスク—我々は、中国で承認された商業化製品ETUARYおよび任意の他の未来製品の価格を低下させることを要求する圧力に直面する可能性があり、これらの製品は医療保険の精算や市場競争による資格を得ることができるこのリスク要因の部分では
101
また、中国の個人資料の収集と移転に関する法律と法規は、“中華人民共和国個人情報保護法”や“中華人民共和国人類遺伝資源管理条例”のいかなる変化も含めて、医療データを使用する能力に影響を与える可能性があり、このようなデータを以前に許可された目的に使用することで法的責任を負わせることができる。
中国政府または我々は、我々の商業化製品ETUARYおよび任意の他の未来製品を販売することを計画している国の他の政府当局は、中国で承認されれば、医薬品の販売に対して新しいまたは異なる法規を講じて、賄賂、腐敗または他の問題を解決することができる。新しいまたは異なる規制は、私たちの製品、ETUARY、および将来の製品(承認された場合)を販売する際に、私たち、私たちの従業員、または流通業者が発生するコストを増加させるか、または販売およびマーケティング活動に制限をかける可能性があり、これは逆に私たちのコストを増加させるかもしれない。
我々は,HIPAAとEU一般データ保護条例(EU)2016/679(“GDPR”)を含む変化するプライバシーとデータ保護法に支配されている.個人情報を保護できない場合、または既存または将来のデータ保護法規を遵守できない場合、私たちの業務、財務状況、運営結果、および将来性は大きな悪影響を受ける可能性があります。
多くの州および連邦の法律および法規は、個人情報の収集、伝播、使用、プライバシー、セキュリティ、セキュリティ、可用性、完全性、および他の処理を管理しています。HIPAAは、健康計画、医療情報交換センター、およびある医療保健提供者(保険エンティティと呼ばれる)と、このような保険エンティティとサービス契約を締結する商業パートナーとによって保護された健康情報(例えば、HIPAAの定義)“PHI”)を保護するための国家プライバシーとセキュリティ基準を確立した。HIPAAは、我々のような、行政、物理、および技術保障措置を講じてこのような情報を保護すること、およびデータ漏洩が発生したときのいくつかの通知要件を含む、電子PHIの保護、使用および開示に関するポリシーを策定し、維持することを含む、カバーされたエンティティおよびビジネスパートナーに要求される。
EU個人健康データとその他の個人データの収集と使用は,2018年5月に発効したGDPRの規定,EU個別加盟国に関するデータ保護法およびGDPRの欧州経済圏での実施によって管轄されている。GDPRは、個人データ(収集、分析、転送を含む)に適した処理(処理)に適した厳しい要件と制限を規定している即個人を識別するか、または個人を識別可能なデータ)、特に臨床試験および有害事象報告からの健康データに関する。GDPRは、処理の法的根拠(例えば、個人データに関連する個人の同意)、個人データを処理する前に個人に提供する情報、国家データ保護当局および/またはデータ当事者への通知義務(特にデータが漏洩した場合)、および個人データのセキュリティおよび秘密を含む要件を含む。EU加盟国はまた自国の立法を通じて健康、遺伝、そして生物統計データに追加的な要求を加えることができる。また、個人に様々な権利を提供します(例えば:個人データをアクセスまたは消去する権利)は、規定違反に対して、金額が大きい者を基準として、世界の年商の4%または2000万ユーロまでの罰金を科す可能性がある。GDPRに違反した場合、個人(例えば:経済的または非経済的損失の賠償を受ける権利もあります例えば:、遭難)。
GDPRを遵守しない場合やGDPRでの個人的権利を行使しない場合には,対象者で収集した臨床試験データを利用する能力を制限する可能性がある。また、EUは臨床試験データの公開を要求する傾向が高まっており、臨床試験健康データの処理に関する義務の複雑さを増加させている。このような公開開示義務は新しいEU臨床試験法規(EU CTR)、EMA開示イニシアティブと業界の自発的な承諾の中で規定されている。これらの義務を履行しないことは、政府の法執行行動や重大な処罰を招き、名声を損ない、業務や経営業績に悪影響を及ぼす可能性がある。CTRとGDPRなどの異なる規制フレーム間の相互作用の不確定性は更に複雑性を増加させた。
また、私たちはアメリカの複数の州の法律に支配されており、これらの法律は私たちのデータ処理実践と政策を修正し、これらの法律を遵守するために大量のコストと費用を発生させることを要求するかもしれない。
もし私たちが環境、健康、安全の法律法規を守らなければ、罰金や処罰を受けたり、私たちの業務を損なう可能性のあるコストが発生するかもしれません。
102
私たちの業務は危険な化学材料を使用し、危険な廃棄物を発生させる可能性があるため、私たちは空気排出、水排出、および危険材料と廃棄物の処理、使用、貯蔵、処理、処理に関する法律法規を含む多くの環境、健康と安全法律法規の制約を受けている。これらの材料や廃棄物を処分する危険廃棄物処理協定は第三者と締結されているが,これらの材料による汚染や傷害のリスクを解消することはできない。私たちの危険な材料を使用または処分して汚染または損傷をもたらす場合、私たちはそれによるいかなる損害に責任を負う可能性があり、いかなる責任も私たちの資源範囲を超える可能性がある。民事や刑事罰金やこのような法律や法規を遵守しない罰に関する巨額の費用を招く可能性もある。しかも、私たちは私たちが危険な材料や廃棄物を貯蔵、使用、または処分することによって、私たちが提起した環境責任や有毒侵害請求に保険を提供することはできません。
私たちは、危険材料の使用による従業員の負傷により生じる可能性のあるコストと費用を支払うために労働者賠償保険を維持しているが、この保険は十分な潜在的責任保険を提供できない可能性がある。しかし、私たちは私たちが提起した環境責任や有毒侵害請求に保険を提供することはできない。
私たちは私たちの商業化製品ETUARYに関する持続的な規制義務と持続的な規制審査を履行しなければならない。ETUARYは中国で承認され、承認されれば、私たちの将来の候補製品に関連するこのような義務と審査を受ける可能性があり、これは重大な追加費用を招く可能性があり、規制要求を遵守できなかったり、私たちの候補製品が予期しない問題に遭遇した場合、私たちは罰を受けるかもしれない。
我々が中国で承認された製品ETUARYおよび将来承認された任意の候補製品は、製造、ラベル、包装、貯蔵、流通、広告、販売促進、サンプリング、記録保存、上場後研究および安全、治療効果およびその他の発売後情報の提出を含む持続的な法規要求を遵守しなければならない。本年度報告“企業-政府監督管理”に記載されているように、中国の要求、米国の連邦と州要求、比較可能な外国監督管理機関の要求を含む。
さらに、規制承認は、特定の年齢層の使用制限、警告、予防措置または禁忌症に関連する重大な制限を含む可能性があり、重い承認後の研究またはリスク管理要件を含む可能性がある。例えば、FDAは、私たちの候補製品を承認するためにREMSを必要とすることができ、これは、制限された分配方法、患者登録、および他のリスク最小化ツールのような薬物ガイドライン、医師トレーニングおよびコミュニケーション計画、または安全な使用を保証する他の要素を必要とする可能性がある。また、私たちが中国で承認された商業化製品ETUARYと私たちの候補製品(承認されれば)およびその開発と商業化に関する活動は、その設計、テスト、製造、安全、効果、記録保存、ラベル、貯蔵、承認、広告、販売促進、販売、流通、輸出入は、それぞれ中国、アメリカ国家食品薬品監督管理局、FDAと他の規制機関、同様の外国の監督管理機関によって全面的に監督される。これらの要求には,安全性や他の上場後の情報や報告,登録の提出,および承認後に行った任意の臨床試験においてcGMPやGCPを継続的に遵守することが含まれている。薬品メーカー及びその施設はまた国家薬品監督管理局、食品と薬物管理局とその他の監督機関の持続的な審査と定期的な抜き打ち検査を受けて、cGMPに符合することを確保する。さらに、任意の候補製品の商業販売を承認した後、製造プロセスの変更および追加のラベル宣言のような製品のいくつかの変更は、NMPA、FDA、および/または同様の規制機関の追加的な審査および承認を受ける必要がある場合がある。
もし私たちが適用された規制要件を遵守できなかった場合、あるいは製品に安全や効果の問題がある場合、規制機関や法執行機関は他の措置をとる可能性がある
103
政府は違法の疑いのあるいかなる調査にも対応するために多くの時間と資源を必要とする可能性があり、否定的な宣伝が生じる可能性がある。現行の法規の要求を守らないいかなる行為も、私たちの製品の商業化と創造能力に重大な悪影響を及ぼす可能性がある。規制制裁を実施したり、規制承認を撤回したりすれば、わが社の価値と私たちの経営業績は悪影響を受けるだろう。
私たちの中国での業務運営に関するリスク
中国政府の法律、法規、規則の改正は、私たちの運営プロセスや業務方法を変化させる可能性がある。
中国政府は私たちの中国での業務行為に対して一定の監督と適宜決定権を持っており、政府が適切だと思って私たちの運営に介入したり、影響を与えたりして、規制、政治、社会目標をさらに実現する可能性がある。中国政府は最近、ある業界(例えば教育やインターネット業界)に重大な影響を与える新しい政策を発表し、将来的に私たちの業界に関する法規や政策を発表する可能性を排除することはできません。これらの法規や政策は、中国当局の許可を求めて中国で私たちの業務を経営し続けることを要求する可能性があり、これは私たちの業務、財務状況、経営業績に影響を与える可能性があります。また、中国政府の最近の声明には、“法に基づいて不法証券活動に厳しく打撃することに関する意見”と、2023年3月31日から発効する“国内企業の海外証券発行と上場試行管理方法”を含む中国政府が公表した意見募集の新規則が含まれており、届出に基づく新制度を構築し、国内会社の海外発行と上場を規範化している。もし私たちがいつでも法律の変化や他の予見できない原因で中国政府の直接介入や影響を受けるなら、私たちの中国での業務に大きな変化が必要かもしれません。
また、中国政府が中国での業務に随時関与または影響する可能性のあるリスクは、投資家に証券を提供または継続する能力を大幅に制限または完全に阻害する可能性があり、そのような証券の価値が大幅に縮小したり、一文の価値もなくなったりする可能性がある。
中国の製薬業界は厳しい規制を受けており、これらの法規は変化する可能性があり、これは私たちの製品、ETUARY、候補製品の承認と商業化に影響を与える可能性がある。
中国の製薬業は全面的な政府監督管理を受けており、新薬の審査、登録、製造、包装、許可とマーケティングを含む。中国における現在及び計画における業務の規制要件に適用される議論の詳細については、参照されたい-ビジネス-中国の規制要件この年間報告書にあります。私たちは私たちの戦略と方法が中国政府の政策と一致すると信じているが、私たちの戦略と方法が引き続き一致することを保証することはできない。近年,中国製薬業の規制枠組みに大きな変化が生じており,引き続き重大な変化が予想される。このような変更や修正は、以下のような結果になる可能性があります
104
中国当局は製薬業界の法律を実行する上でもますます警戒されており、適用される法律や法規を遵守できなければ、中国での業務活動の一時停止や終了を招く可能性がある。
中国政府の政治、経済、その他の政策の不利な変化は、中国全体の経済成長に重大な悪影響を及ぼす可能性があり、承認されれば、私たちの商業化製品ETUARYや任意の他の未来の製品への需要を減少させたり、他の方法で私たちの業務、運営、あるいは競争地位に重大な悪影響を与える可能性がある。
私たちの業務、経営業績、財務状況と見通しは中国の経済、政治、法律と社会条件の影響を大きく受ける可能性がある。中国経済は多くの方面で先進国の経済と異なり、政府が参加する数量、発展レベル、成長率、外貨規制、資源分配と絶えず発展する監督管理制度を含む。中国政府はすでに経済発展を奨励し、資源配置を誘導する様々な措置を実施しているが、その中のいくつかの措置は私たちにマイナスの影響を与える可能性がある。例えば、私たちの財務状況や経営結果は、政府の資本投資のコントロールや現在私たちに適用されている税収規制の変化の悪影響を受ける可能性があります。中国経済の成長は中国の異なる地域と異なる経済業界の間でずっと不均衡であり、将来の成長が類似した速度で持続または根本的に起こらないことは保証されない。国内や国際投資の観点から見れば、中国のビジネス環境や経済状況が悪化すれば、私たちの業務も悪影響を受ける可能性がある。
中国の法律、規則、法規の解釈と実行には不確実性がある。
中華人民共和国の法律体系は成文法典と成文法に基づく民法体系である。一般法制度と異なり、先の裁判所判決は説得力のある権威として援用される可能性があるが、その先例価値は限られている。20世紀70年代末以来、中華人民共和国政府は経済事務を全面的に管理する法律、法規と法規体系を公布した。しかしながら、これらの法律および条例は比較的新しいため、公表された決定の数は限られており、それらの解釈と実行は重大かつ確実性に関連しており、一致せず予測不可能である可能性がある。中国の行政と裁判所当局は法定·契約条項の解釈と実施に重大な情動権を持っているため、発達した法律制度よりも、行政や裁判所訴訟の結果や私たちが経験する可能性のある法的保護レベルを評価することは難しいかもしれない。これらの不確実性は、私たちが締結した契約を実行する能力を阻害し、私たちの業務、財務状況、および経営結果に実質的な悪影響を及ぼす可能性があります。
また、中国の法律法規は国有資産に対して重要な保護を提供している。国有資産の流失を招く可能性のある出資は主管部門の厳格な審査を受け、主管部門は関連法律法規の解釈と実施に大きな裁量権を持っている。もし私たちまたは私たちの関連会社が国有企業またはその関連会社と取引すれば、私たちは国有資産の潜在的損失に関連するリスクと不確定要素に直面し、これは私たちに債務を負担させ、私たちの業務、財務状況、経営業績に重大かつ不利な影響を与える可能性がある。
中国の法律制度はある程度政府政策と内部規則に基づいており、その中のいくつかは適時に公表されていないか、あるいは全く公表されておらず、追跡力を持っている可能性がある。したがって、私たちは違反が発生してから、私たちがこのような政策と規則に違反していることを認識しなければならないかもしれない。
中国で労働法律や法規を施行することは、私たちの業務や経営結果に悪影響を及ぼす可能性があり、中国の労働関連法律を完全に遵守できなければ、潜在的な責任と処罰に直面する可能性がある。
“中華人民共和国労働契約法”によると、使用者は労働契約の締結、最低賃金、報酬の支払い、従業員の試用期間の確定と一方的な労働契約の終了などの面でより厳しい要求がある。詳細な解釈規則と地方主管部門の広範な裁量権が不足しているため、労働契約法及びその実施細則がどのように中国の現行の就業政策とやり方に影響するかはまだ確定していない。私たちの雇用政策ややり方は労働契約法やその施行細則に違反する可能性があるため、関連する処罰、罰金、弁護士費を受ける可能性があります。
105
労働契約法とその実施細則を遵守することは、私たちの運営費用、特に私たちの人員費用を増加させる可能性があります。もし私たちの従業員の一部を解雇したり、他の方法で私たちの雇用や労働慣行を変更することを決定すれば、労働契約法およびその実施規則は、これらの変更を理想的または費用対効果的な方法で実施する能力を制限する可能性もあり、これは私たちの業務や運営結果に悪影響を及ぼす可能性がある。社会保険法によると、従業員は養老保険、労災保険、医療保険、失業保険、生育保険に加入しなければならず、使用者は従業員と一緒にあるいは単独で従業員のために社会保険料を納めなければならない。最近、中国政府は社会保険徴収に関する措置を強化しており、より厳しい法執行につながる可能性がある。
私たちはこのような法律法規の施行によって、私たちの労働コストが増加すると予想する。社会保険法とその施行細則を遵守することは、私たちの運営費用、特に私たちの人員費用を増加させる可能性があります。これらの法律·法規の解釈·実施はまだ発展中であるため、私たちの雇用実践政策がいつでも中国の労働に関する法律や法規に完全に適合しているとみなされることは保証されず、労使紛争や政府調査に直面する可能性がある。もし私たちが関連する労働法律法規に違反していると思われれば、私たちは私たちの従業員に追加の補償を要求されるかもしれません。私たちの業務、財務状況、運営結果は実質的な悪影響を受ける可能性があります。
為替レートの変動は外貨為替損失を招く可能性がある。
人民元の他の通貨に対する価値変動は変動する可能性があり、中国の政治·経済状況、中国の外貨政策および地元市場の需給変化の影響を受ける可能性がある。私たちは市場力や政府政策のリスクと、将来の人民元や他の通貨為替レートに対するそれらの影響に直面している。私たちのほとんどの収入とコストは人民元で計算され、私たちの大部分の金融資産も人民元で計算されます。人民元価値のいかなる重大な変動も、私たちの流動性とキャッシュフロー、そして私たちの金融資産価値に重大な悪影響を及ぼす可能性がある。
私たちの業務は中国の税務法律法規の変化の影響を受け、影響を受ける可能性がある。
中国政府は時々その税務法規を調整或いは改正し、未来の中国税務法規の調整或いは変更、及びそれによって生じるいかなる不確定性も、私たちの経営業績に不利な影響を与える可能性がある。私たちの製品ETUARYは中国で承認され、2019年3月から3%の税率の付加価値税優遇を受け、多種の珍しい病気薬に適用されます。しかし、我々が適用する付加価値税税率が一定または低下することは保証されず、将来の付加価値税政策の任意の変化は、ETUARYおよび将来承認された候補製品の販売価格に悪影響を及ぼす可能性がある。
また、改正された個人所得税法によると、中国に住所はありませんが、1つの納税年度内に中国に合計183日以上住んでいる外国人は、中国国内または海外で取得した所得は中国個人所得税を納めなければなりません。改正された個人所得税法は私たちの高技能外国科学者と研究技術者の中国での仕事の能力に重大な影響を与える可能性がある。吾らも中国税務機関が中国税務法律及び法規下の税務責任を履行するための定期的な審査を受けなければならず、しかも中国税務機関のいかなる当該等の審査が吾等の業務、財務状況及び経営業績及び私たちの名声に悪影響を与える可能性のある罰金、その他の処罰或いは行動につながらないことを保証することはできない。
私たちは私たちの科学データを海外に移したり、中国で収集した人類遺伝資源を使用することを制限されるかもしれない。
2018年3月17日、国務院弁公庁は“科学データ管理方法”(“科学データ方法”)を公布し、科学データを広義に定義し、科学データ管理の関連規則を規定した。“科学データ管理方法”によると、国家秘密に関連するいかなる科学データも、中国国内の企業は政府の許可を受けなければ、国外に移転したり、外国に移転したりすることができる。主管機関の許可を得た後、企業は規定に従って手続きを行い、科学データ使用者と秘密保護協定を締結しなければならない。また研究を行っている研究者は
106
少なくとも部分的に中国政府が援助した研究者はこの研究者が所属する実体管理のために関連科学データを提出しなければならず、その後、いかなる外国学術定期刊行物にもこのようなデータを発表することができる。“国家機密”という言葉は明確な定義がないことから、もし私たちの候補薬物の研究開発に関連して収集或いは生成したデータが関連政府当局が要求した科学データ措置及び任意の後続法律の制約を受けていれば、科学データ(例えば、私たちが中国国内で行った臨床前研究或いは臨床試験の結果)を国外或いは中国の外国パートナーに送信する時に常に関連する承認を得ることができることを保証することはできない。したがって、私たちはこのような政府当局から罰金と他の行政処罰を受けるかもしれない。
そのほか、“サービスガイドライン”に基づいて、臨床試験を通じて人類遺伝資源に対してサンプリング、採集或いは研究活動を行い、中国人類遺伝資源管理処にオンラインで報告する必要がある。そのほか、“中華人民共和国人類遺伝資源管理条例”(“人類遺伝資源条例”)は、中華人民共和国の重要な遺伝家族と特定地区の人類遺伝資源を採集し、あるいは国務院科学技術行政部門が規定した種類と数量に従って人類遺伝資源を採集し、中華人民共和国人類遺伝資源を保存し、科学研究に基礎プラットフォームを提供し、中華人民共和国人類遺伝資源を利用して国際科学研究協力を行い、中華人民共和国人類遺伝資源材料を国外に輸送することは、国務院科学技術行政部門の許可を得なければならないと規定している。
もし私たちが適時に必要な承認を得ることができなければ、規制要求を遵守することができず、甚だしきに至っては根本的にできなければ、候補薬物の開発が阻害される可能性がある。関連政府当局が我々の科学的データを送信したり、人間の遺伝資源を収集して使用したりすることが適用される中国の法律や法規の要求に違反していると判断すれば、これらの政府当局から罰金やその他の行政処罰を受ける可能性がある。また、法規の解釈と応用は著者らの臨床試験実践と一致しない可能性があり、人類の遺伝資源サンプルと関連データが没収され、行政罰金を科される可能性がある。
中国政府の政治·経済政策や中米関係の変化は、我々の業務、財務状況、経営業績に影響を与える可能性がある。
私たちの中国での業務により、私たちの業務、経営結果、財務状況は、中国の経済、政治、法律と社会条件、あるいは中国とアメリカあるいは他の政府との間の政府関係の変化のある程度の影響を受ける可能性がある。貿易政策、条約、政府法規、関税については、米国と中国との将来の関係には大きな不確実性がある。中国経済は多くの面で先進国の経済と異なり、政府が参加する数量、発展レベル、成長速度、外貨規制と資源配置の面を含む。中国経済は過去40年間で著しい成長を経験したが、異なる地域と異なる経済部門の成長はバランスがとれていない。中華人民共和国政府は経済発展を奨励し、資源配置を誘導する各種措置を実施した。その中のいくつかの措置は中国経済全体に有利かもしれないが、私たちにマイナスの影響を与えるかもしれない。また、過去、中国政府は利上げを含む何らかの措置を講じて経済成長速度をコントロールしていた。これらの措置は中国の経済活動を減少させる可能性があり、私たちの業務や経営業績に影響を与える可能性がある。
2023年と2022年12月31日までの年間で、特定の海外サプライヤーに直接·間接的に原材料を取得し、業務運営面で外国·地域実体との協力に直接·間接的に依存している。私たちはまた未来に外国と地域の実体とのパートナーシップを求めることができるかもしれない。そのため、私たちの業務は変化する国際経済、法規、社会と政治条件、外国と地域の現地条件の影響を受けている。そのため、中国とこれらの外国や地域との政治関係は、我々の製品、ETUARY、候補製品の開発と商業化に影響を与える可能性がある。
また、中国とこれらの外国や地域との政治関係も、現在と未来の第三者との関係に影響を与える可能性がある。私たちは私たちの既存または潜在的な協力者が政治関係の不利な変化によって彼らの見方や彼らの選好を変えないという保証はない
107
中国と国外の関連国或いは地区間の合意変更は、許可されれば、私たちの製品、ETUARY及び未来の製品に対する需要低下を招き、そして私たちの業務、財務状況、経営業績、現金フロー及び将来性に不利な影響を与える可能性がある。
2021年7月、中国政府は中国に本部を置く会社の中国海外融資に新たな指導を提供し、可変利益実体(“VIE”)と呼ばれる手配を含む。これらの発展を受けて、米国証券取引委員会は、米国証券取引委員会に証券登録を求める中国会社に対して、より厳しい情報開示要求を実施している。私たちはVIEアーキテクチャを持っていませんが、私たちの中国での業務のため、将来的には中国、アメリカあるいは他の規則や法規が中国で業務を持っている会社の融資や他の活動に制限を加え、私たちの業務や運営結果に影響を与える可能性があります。国内や国際投資の観点から見れば、中国のビジネス環境が悪化したり、中国と米国や他の政府との関係が悪化したりすれば、中国政府は中国と米国での私たちの運営や業務に介入する可能性がある。
アメリカと中国の法規の変化は私たちの業務、私たちの経営業績、私たちの融資能力に影響を与えるかもしれません。
米国証券取引委員会を含む米国政府が発表した声明と行動は、米国と国際関係の変化を招き、中国で製造されたいくつかの製品に数回の関税を課すこと、中国にある制裁と制限を実施すること、中国で何らかの業務を持っている会社の審査を強化することを示す声明を発表した。新しい立法、行政命令、関税、法律または法規、および新しい立法、行政命令、関税、法律または法規がどの程度通過するかは不明であり、このような行動が米国や中国、私たちの産業、または私たちと重大な関連がある会社にどのような影響を与えるかも分からない。私たちはアメリカと中国で研究活動と業務運営をしています。国境を越えた関係および/または国際貿易における政府のいかなる不利な政策も、中国でいくつかの業務を有する企業の審査、資本規制または関税の強化を含み、私たちの薬品、模倣薬および候補製品の競争地位、科学者および他の研究開発者の採用、私たちの薬品の需要、薬品開発に関連する原材料の輸出入、または私たちの融資能力に影響を与える可能性があり、または特定の国で私たちの薬品を販売することを阻止する。また、米国証券取引委員会は、主に中国で特定の業務を持っている会社を対象とした声明を発表している。例えば、2021年7月30日、米国証券取引委員会のジャンスラー議長は、中国の最近の事態の発展について投資家保護に関する声明を発表し、声明によると、ジェスラーは、米国証券取引委員会の従業員に、中国である業務を持っている会社の届出文書に対して的確な追加審査を要求したと述べた。声明はまた、VIE構造を持つ会社の固有のリスクについて言及した。私たちはVIE構造もなく、中国の外資所有権によって制限された業界にも属していない。しかし、米国証券取引委員会に提出された定期報告書や他の届出書類は、米国証券取引委員会の強化された審査を受ける可能性があり、このような追加的な審査は、米国での有効な融資能力に影響を与える可能性がある。
米国証券取引委員会の2021年7月30日の声明に応えるため、中国証券監督管理委員会(以下、証監会と略称する)は2021年8月1日に“と発表した[i]中米両国の監督管理機関は引き続き相互尊重、協力の原則に基づいて意思疎通を強化し、米上場企業の中国での監督管理問題を適切に解決し、安定した政策期待を形成し、市場のために良性のルール枠組みを創造すべきであると信じている。中国証監会は引き続き投資家、会社、関連部門を含む異なる利害関係者と密接に協力し、政策と実施措置の透明性と確実性をさらに促進するが、それはずっと開放されており、会社が関連法律法規に基づいて国際或いは国内市場で上場することを選択させることを強調している
もし新たな立法、行政命令、関税、法律および/または法規が実施されれば、既存の貿易協定を再交渉すれば、米国または中国政府が最近の米中緊張のために報復行動を取った場合、または中国政府が米国で行われた証券発行により多くの監督と制御を加える場合、これらの変化は私たちの業務、財務状況、運営結果、および私たちの融資能力に悪影響を及ぼす可能性がある。
108
中国の新しい“データセキュリティ法”、“ネットワークセキュリティ法”、“ネットワークセキュリティ審査方法”、“個人情報保護法”、ネットワークセキュリティ多層保護計画に関する法規と指針、その他の未来の法律法規を遵守することは重大な支出が生じる可能性があり、私たちの業務に影響を与える可能性がある。
中国はすでに規則を実施または実施し、データ保護に関する追加的な提案を考慮している。データセキュリティ法は、データを保護するためには、データ処理活動は“データ分類と階層保護システム”に基づいて行わなければならず、中国国内の実体が中国政府の事前承認を経ずに中国に格納されたデータを外国の法執行機関や司法機関に転送することを禁止すると規定されている。
また、中華人民共和国“ネットワークセキュリティ法”と“情報セキュリティ階層保護管理方法”は企業に一定の組織、技術と行政措置及びその他の必要な措置を取って、そのネットワークとそのネットワーク上に格納されたデータの安全を確保することを要求する。多段保護計画によれば、情報システムを運営するエンティティは、その情報およびネットワークシステムのリスクおよび状況を徹底的に評価して、エンティティの情報およびネットワークシステムのレベルを決定しなければならない。ネットワークセキュリティレベル保護等級と実施に関する一連の国家標準によると、これらのレベルは最低レベル1から最高レベル5まで様々である。評価結果は実体が守らなければならない安全保護義務を決定するだろう。二級以上の部門は等級を関係政府部門に報告して審査しなければならない.
中国ネットワークセキュリティ管理局はこのほど、複数の中国インターネット会社が米国証券取引所で初めて公募株(IPO)を提訴し、国家安全リスクが存在し、中国データ当事者の個人情報を収集して使用すべきではないと告発した。公式公告によると、この行動は“国家安全法”、“ネットワーク安全法”、“ネットワーク安全審査方法”に基づいて開始され、これらの法律法規は“国家データ安全リスクを防止し、国家安全を維持し、公共利益を守る”ことを目的としている
改訂された“CAC方法”によると、ネットワーク製品やサービスを調達するキー情報インフラ運営者、国家安全に影響を与える可能性のあるデータ処理活動を展開するネットワークプラットフォーム運営者(“改訂後の”CAC方法“草案中のデータ処理者に対して)は、”方法“の規定に従ってネットワークセキュリティ審査を行うべきである。また、100万人以上のユーザーの個人情報を持つオンラインプラットフォーム事業者は、外国株式市場への上場を求め、ネットワークセキュリティ審査を申請しなければならない。2021年11月14日、CACはさらに、その裁量の目的および方法に基づいてデータ処理活動を決定する個人および組織を指す“ネットワークデータセキュリティ管理条例(意見募集稿)”または“管理条例草案”を発表した。“管理条例”草案は、データ処理者が(I)100万人を超える個人情報を処理し、外国証券市場に上場することを計画していること、または(Ii)そのデータ処理活動が中国国家安全に影響を与える可能性があることを再確認し、ネットワークセキュリティ審査を受けるべきである。“管理条例”草案はまた、海外で上場するデータ処理業者は毎年自分で或いはデータ安全サービス機関を通じてそのデータ安全を評価し、評価報告を関連主管部門に提出しなければならないことを求めている。“管理条例”草案は公開意見のみを求めているため、その最終版や発効日は変更される可能性がある。
本年度の報告日まで、吾らは中国の監督管理機関から通知を受けておらず、吾らを“キー情報インフラ事業者”、“オンラインプラットフォーム事業者”あるいは“データ処理業者”と認定したり、改正されたCAC方法と管理条例草案に基づいてネットワークセキュリティ審査手続きを行うことを要求したりしている。改正されたCAC措置や管理条例草案の理解(例えば現在の提案通りに制定されている)によると、吾らは外国投資家への証券発行によってCACのネットワークセキュリティ審査を受けることはないと予想されている。なぜなら、(I)吾らが業務運営中に処理した臨床および臨床前データは、その性質や規模にかかわらず、通常中国国家安全に対する重大な懸念を引き起こすことはないこと、および(Ii)吾などはまだ処理されておらず、予測可能な将来に100万人を超えるユーザや個人の個人情報を処理することを期待していないからである。しかし、改正された食品典委措置と“管理条例”草案が現在の提案に従って公布されれば、どのように解釈または実行されるかには、まだ不確実性がある。また、中国の監督管理当局が新しい法律、法規、規則を通過できるかどうか、あるいは改正されたCAC措置と管理条例草案の詳細な実施と解釈については、まだ不確実性がある。私たちはこの分野で進化していく法律と法規に注目し、すべての合理的な措置を取るつもりですが
109
コンプライアンスリスクを低減するための措置は、改正されたCAC措置、管理条例草案、またはプライバシー、データ保護、情報セキュリティに関する他の法律法規の潜在的な影響を受けないことを保証することはできません。
また、“個人資料保護法”は個人資料を処理するために適用される資料のプライバシーと保護規定を一式提供し、資料保護コンプライアンス責任を中国国内の組織及び個人が個人資料を処理すること、及び中国海外人の個人資料を処理することを含むまで拡大した(例えばこのような処理の目的は中国国内の人々に製品とサービスを提供し、或いはその行為を分析及び評価することである)。個人情報保護法はまた、キー情報インフラ事業者と個人情報処理実体が処理する個人情報が中国サイバー空間規制機関が設定した数のハードルに達した場合には、中国で生成あるいは収集した個人情報を保存し、中国サイバー空間規制機関による当該などの個人情報の輸出に対する安全評価を行わなければならないと規定している。最後に、個人情報保護法は、深刻な違反に対して約720万ドルまたは前年の年収5%までの巨額の罰金を科すことを提案し、主管当局に関連活動の一時停止を命じられる可能性がある。私たちは保存せず、将来的には中国患者の個人的な身分健康情報を保存するつもりもない。
これらの法律、規則、条例の解釈、適用、執行は時々変化し、その範囲は新しい立法、既存の立法の修正または実行の変化によって変化していく可能性がある。中国の新しいネットワークセキュリティ法とデータセキュリティ法を遵守することは、私たちがサービスを提供するコストを大幅に増加させる可能性があり、業務を重大に変更し、さらには、私たちが現在運営しているか、将来運営可能な司法管轄区で何らかのサービスを提供することを阻止することが求められている。プライバシー、データ保護、情報セキュリティに関する適用法律、法規、その他の義務を遵守しようと努力しているにもかかわらず、私たちのやり方、製品、またはプラットフォームは、“ネットワークセキュリティ法”、“データセキュリティ法”、および/または関連実施条例が私たちに加えたすべての要求を満たすことができないかもしれません。吾等は、そのような法律又は法規又は私隠、資料保護又は情報セキュリティに関連する任意の義務、又は不正アクセス、個人識別資料又は他の資料の使用又は漏洩を招くいかなる安全損害、又は前述のタイプの失敗又は妥協が発生した認知又は指定を遵守することができず、吾等の名声を損なう可能性があり、新たな及び既存の取引相手が吾等と契約を結び、又は中国政府当局に調査、罰金、停職又はその他の処罰、並びに個人請求又は訴訟を引き起こす可能性があり、上記のいずれの事項も吾等の業務、財務状況及び経営業績に悪影響を及ぼす可能性がある。たとえ私たちのやり方が法的挑戦を受けなくても、プライバシー問題に対する見方は、有効かどうかにかかわらず、私たちの名声やブランドを損なう可能性があり、私たちの業務、財務状況、運営結果に悪影響を及ぼす可能性がある。また、“データ安全法”、改正された“反海外腐敗法”措置、最近の中国政府の行動による法的不確実性は、有利な条件で資本を調達する能力に悪影響を及ぼす可能性がある。
両替の制限には、現金を中国国外に移転するリスクが含まれており、Gyre PharmPharmticalsが外貨融資を有効に受け入れて使用する能力、あるいはGyre PharmPharmticalsや中国の他の潜在投資家から現金を転送する能力を制限する可能性がある。
GYRE PharmPharmticalsが通貨両替を獲得する能力は厳格な外貨規制を受けており、資本項目での取引であれば、中国政府当局(国家外国為替管理局を含む)の承認および/または登録を得る必要がある。特に、Gyre PharmPharmticalsがBJC Limitedや他の外国融資者から外債を借り入れることによって融資を行う場合、金額は法定限度額を超えてはならず、このような融資は外管局現地支店に登録しなければならない。Gyre製薬会社が追加出資方式で融資を行う場合は,国家市場監督管理総局又はその地方支局に登録し,商務部又はその地方支局に外商投資情報を報告するか,又は中国の他の政府主管部門に登録しなければならない。
中国法規が海外持株会社が中国実体の融資と直接投資に対して加えた各種の要求を受けて、Gyre PharmPharmticalsがGyre PharmPharmticalsの未来の融資或いは出資について必要な政府要求を適時に完成することができることを保証できない(もしあれば)。Gyre PharmPharmticalsがこのような要求を遵守できなかった場合、またはこのような承認を得ることができなかった場合、Gyre PharmPharmticalsがGyre PharmPharmticals中国業務(Gyre PharmPharmPharmticalsの技術開発を含む)を資本化したり、他の方法で資金を提供する能力が受けられる可能性がある
110
否定的な影響を受け、これはGyre PharmPharmticalsがGyre PharmPharmticalsに資金を提供し、業務を拡大する能力に重大な悪影響を及ぼす可能性がある。
Gyre TreeuticsはGyre PharmPharmticalsから資金を転出できない可能性があり、あるいはGyre Treeuticsが中国当局が加えた制限によって直ちに中国投資家に投資を募集することを決定すれば、Gyre Treeuticsは中国投資家から資金を移転する困難に直面する可能性がある。
中国住民がオフショア特殊目的会社を設立することに関する中国法規は、私たちが中国にいる中国住民の実益に責任を負わせたり、罰を受けたり、他の面で私たちに不利な影響を与える可能性がある。
“域内住民の海外投融資と特殊目的担体往復投資外国為替管理に関する問題に関する通知”又は“国家外匯局第37号通知”要求によると、中国住民はオフショア実体海外投融資を直接或いは間接的に制御し、その合法的に所有する国内企業資産又は株式又は海外資産又は権益は、国家外匯局第37号通書の中で“特殊目的担体”と呼ばれる。外管局第37号通達下の“制御権”という言葉は広義には中国住民が買収、信託、委託、投票権、買い戻し、交換可能債券或いはその他の手配などの方式で取得したオフショア特別目的ツール又は中国会社の経営権、受益権又は決定権と定義される。中国外管局第37号通達はさらに、特別目的担体の基本情報に任意の変化或いはそれに関連する任意の重大な変化が発生した場合、例えば中国住民の出資の増減、株式譲渡或いは交換、合併、分立或いはその他の重大な事件について、登録を改訂することを要求している。中国住民であるオフショア持ち株会社の株主が現地外匯局支店で登録を完了しなければ、中国子会社はオフショア親会社へのいかなる減資、株式譲渡或いは清算の利益と収益の分配を禁止される可能性があり、その後国境を越えた外貨活動を行い、オフショア親会社がその中国子会社に追加資本を注入する能力が制限される可能性がある。また、上記外管局の登録及び改正規定を遵守できなかった場合、中国の法律により、適用される外国為替制限から逃れることで責任を負う可能性がある。
いくつかの中国住民は当社が直接または間接的な権益を持っている可能性があり、吾らは当社が知っているように当社が直接または間接的な権益を持っている中国住民(あれば)に外管局第37号通達及びその他の関連規則の規定に従って必要な申請、届出及び改訂を提出することを要求する。しかしながら、吾等は、その等の登録を行うべき吾等の株主又は実益所有者の身分をいつでも完全に知ったり、通知したりすることはなく、また、吾等は、当該等の住民が吾等のいかなる適用登録の要求を遵守又は取得するか、又は外管局第37号通達又は他の関連規則の下での他の要求を遵守することを保証することはできない。もし私たちの中国住民の株主がこれらの規定に規定された登録手続きを遵守できなかった場合、私たちは罰金や法的制裁を受け、私たちの国境を越えた投資活動を制限するかもしれません。また、上記各種外貨登録要求を遵守しないことは、中国の法律に基づいて適用される外貨制限の責任を回避することを招く可能性がある。したがって、私たちの業務運営と私たちがあなたに製品を配布する能力は実質的な悪影響を受ける可能性があります。
私たち従業員の株式インセンティブ計画の登録要求に関する中国の法規を遵守できなかった場合、私たちは罰金や他の法律や行政処罰を受ける可能性があり、これは私たちの業務、財務状況、運営結果に悪影響を及ぼす可能性があります。
“国内個人が海外上場会社の株式激励計画に参加することに関する問題に関する通知”や“株式オプション規則”などの関連規則制度に基づいて、中国公民或いは中国国内に1年間連続して居住している非中国公民が海外上場会社のいかなる株式激励計画に参加するかは、少数の例外的な場合を除いて、当該海外上場会社の国内合格代理人(当該海外上場会社の中国子会社であってもよい)を介して外国為替局に登録し、特定の手続きを完了しなければならない。私たちの従業員は、中国公民や中国に1年以上連続して住んでいて、私たちの株式激励計画に参加して、この法規の制約を受けるだろう。私たちは私たちの従業員たちが彼らの株式奨励金を登録するのを助ける計画だ。しかし、私たちの株式インセンティブ計画によると、どの中国人個人実益所有者や株式奨励所有者も安全登録要求を遵守できず、罰金と法律制裁を受ける可能性がある。
111
Gyre PharmPharmticalsは中国北京に登録された法人実体であるため、中国所得税について言えば、同社はデフォルトで中国税務住民に分類され、このような分類はGyre PharmPharmticals及び非中国株主に不利な税務結果をもたらす。
“中華人民共和国企業所得税法”第二条によると、住民企業とは、中国国内に設立された企業又は中国国内に“事実上の管理機関”を設立する企業をいう。
中国税務について言えば、GYRE PharmPharmticalsは中国税務住民であることをデフォルトとしている。それは中国北京に登録されている法人実体であるからである。中国企業所得税について言えば、Gyre PharmPharmticalsは中国税務住民であるため、Gyre PharmPharmticalsの全世界収入は25%の税率で中国税を納めなければならず、これはGyre PharmPharmticalsの純収入を大幅に減少させる。また、Gyre PharmPharmticalsは中国税務住民所得税の申告義務を納めなければならない。
また、企業所得税については、Gyre PharmPharmticalsは中国の税務住民であるため、当該等の収益が中国からのものとみなされる場合、当該等の収益は中国税を納めなければならない可能性があり、非中国企業の税率は10%、非中国個人の税率は20%である(いずれの場合も税務条約の適用規定に制限されなければならない)。
Gyre PharmPharmticalsとその株主は、中国住民企業の株式を間接的に譲渡したり、中国で設立された非中国会社に帰属する他の資産、あるいは中国に帰属して設立された非中国会社の他の資産を間接的に譲渡する面で不確定性に直面している。中国税務機関が買収取引の審査を強化することは、Gyre PharmPharmticalsのオフショア再編取引やGyre PharmPharmticalsのオフショア持ち株会社の株式の売却や中国の課税資産に関連する投資にマイナス影響を与える可能性がある。
中国税務機関は“財政部、国家税務総局の企業改革経営活動に関する企業所得税処理に関する若干の問題に関する通知”(“第59号通知”)と“非住民企業株式譲渡企業所得税管理の強化に関する通知”(“第698号通知”)を発表することにより、非住民企業に対していくつかの課税資産(特に株式)を直接又は間接的に譲渡する審査を強化した。“企業所得税と非中国住民企業の間接譲渡資産問題に関する公告”(“公告7”)によると、非中国住民企業の“間接譲渡”資産(中国住民企業の株式を含む)は再同定可能であり、中国課税資産の直接譲渡とみなされ、この手配には合理的な商業目的がなく、中国企業所得税の納付を避けるために構築されていることが前提となっている。そのため、当該等の間接譲渡からの収益は中国企業所得税を払わなければならない可能性がある。
公告7によると、“中国課税資産”には、中国国内機関に帰属する資産、中国国内に位置する不動産及び中国住民企業の株式投資が含まれており、中国住民企業の直接所有者が当該等の資産を譲渡して得られた収益は中国企業所得税を納付しなければならない。取引手配に“合理的な商業目的”があるかどうかを確定する際に考慮すべき要素は、オフショア企業の株式に関する主要な価値が中国の課税資産に由来するかどうか、オフショア企業の資産が主に中国での直接或いは間接投資から構成されているかどうか、あるいはその収入が主に中国から来ているかどうか、オフショア企業及びその直接或いは間接的に中国課税資産を持っている子会社が真の商業性質を持っているかどうか、これはその実際の機能とリスク開放から証明できるかどうか;商業モデルと組織構造の存在期限;直接譲渡中国課税資産による取引の回復可能性;そしてこのような間接譲渡と適用される税金条約や似たような配置の税金状況。間接的にオフショア移転した中国機関の資産については、それによって生じた収益は移転された中国機関や営業地点の企業所得税申告に計上されるため、25%の税率で中国企業所得税を納めることになる。関連譲渡が中国国内に位置する不動産や中国住民企業の株式投資に関連し、当該不動産が非住民企業の中国での設立や営業場所と関係がない場合、税務条約や同様の手配を適用して得られる税収優遇の下で、10%の中国企業所得税が適用され、譲渡金を支払う義務がある側に源泉徴収義務がある。支払人が税金の納付を代行していない場合は,譲渡人は法定期限内に自ら税務機関に納付税を申告しなければならない.期限を過ぎて適用税金を納めると譲渡人に罰金を科される
112
違約利息。公告7これらの株式が公共証券取引所の取引により得られた場合は、投資者が公共証券取引所を介して株式を売却する取引には適用されない。
公告7は、Gyre PharmPharmticalsの一部のオフショア再構成取引に適用されるか、またはGyre PharmPharmticalsのオフショア持ち株会社の株式または中国課税資産に関連する投資として税務機関によって認定される可能性がある。譲渡者や譲受人は納税申告や源泉徴収代行税が必要である可能性があるが,Gyre製薬会社はこのような申告に協力することを求められている可能性がある。さらに、譲渡者または譲受人(源泉徴収代理人として)は、公告7または確定譲渡人に、Gyre製薬会社の以前および将来のGyre製薬会社のオフショア子会社の株式を再編または売却するために、貴重な資源を要することを要求される可能性がある。公告7によると、中国税務機関は譲渡された課税資産の公正価値と投資コストとの差額に基づいて課税資本利益を調整する権利がある。中国税務機関が公告7に基づいて取引の課税所得額を調整すれば、譲渡先が当該等の潜在的な買収や処分に関連する所得税コストが増加する。
Gyre PharmPharmticalsは非中国住民企業投資家がGyre PharmPharmticals株式を譲渡する未来の私募株式融資取引、株式交換或いはその他の取引の報告と結果の不確定性に直面している。中国税務機関はこの非住民企業の届出あるいは譲受人の源泉徴収義務を追及し、Gyre製薬に届出協力を要請することができる。したがって、このような取引中の非住民企業は、第59号公報又は第7号公報及び第37号公報に基づいて申告義務又は課税されるリスクに直面し、貴重な資源をかけて第59号公報、第7号公報及び第37号公報を遵守することが要求される可能性があり、又はその非住民企業はこれらの通告に基づいて課税すべきではないと判断される可能性がある。
SAT通告59、公告7及び公告37によると、中国税務機関は譲渡された課税資産の公正価値と投資コストとの差額に基づいて課税資本利益を調整する権利がある。Gyre PharmPharmticalsは現在中国や世界の他の地域でいかなる買収計画も行っていないが、Gyre PharmPharmticalsは将来複雑な会社構造に関連する可能性のある買収を行う可能性がある。Gyre PharmPharmticalsはデフォルトで中国税務住民であるため、中国税務機関がSAT通告59或いは公告7及び公告37によって取引の課税所得額を調整すれば、Gyre PharmPharmticalsはこのような潜在的買収に関連する所得税コストが増加し、これはGyre PharmPharmticalsの財務状況及び経営業績に不利な影響を与える可能性がある。
私たちの製品や候補製品の商業化に関連するリスク
私たちは医師、患者、第三者支払人、医学界の他の人の商業成功に必要な市場受容度を達成できないかもしれない。
もし私たちのすべての候補製品が市場の承認を得たら、私たちはまだ医者、患者、第三者支払人と医学界の他の人の十分な市場受容度を得ることができないかもしれない。例えば,これまで中国では塵肺疾患治療のための抗線維化製品は承認されておらず,ETUARYが市場承認を得ても,医師は塵肺疾患の治療薬としてETUARYを受け入れたり使用することはできない。同様に,これまでB型肝炎関連肝線維化を治療する特定の治療薬は世界的に承認されておらず,F 351が市販承認されても,医師は肝線維化治療薬としてF 351を受け入れたり使用したりしない可能性がある。もし私たちの候補製品が十分な受容度に達していなければ、私たちのどの候補製品への投資も著しい製品収入を生み出したり、何の見返りも得られないかもしれない。もし私たちの候補製品が商業販売のために承認されれば、市場の受け入れ度はいくつかの要素に依存する
113
我々の商業化製品ETUARYが中国で承認された市場受入度リスクの詳細については、“を参照されたい中国で販売されている製品ETUARYおよび将来承認される可能性のある他の任意の製品は、医師、医療機関、薬局、患者、第三者支払者、およびより広範な医療コミュニティで十分な市場受容度を得ることができない可能性があり、これはそれらの商業生存に重要である。”
私たちの多くの候補製品は規制部門の承認を得るためにあと数年かかる。
私たちの開発候補製品は数年以内にビジネスに投入されない予定ですが、もしあれば。さらに、候補製品の商業的成功は、他の会社が現在開発している競合製品を含む可能性がある当時利用可能な製品の治療および費用便益代替案として、医師、個人、第三者支払者、および他の重要な意思決定者がそれを受け入れるかどうかに依存する。“-”というタイトルのリスク要因を見てください私たちは激しい競争に直面しています。これは他の人が私たちよりも早く、あるいは成功的に製品を発見、開発、商業化することをもたらす可能性があります“もし私たちが開発に成功し、適時に規制承認を得ることができなければ(規制の変化や連邦政府の閉店など、私たちがコントロールできないため、FDAを含む)そして私たちの開発候補製品を商業化することができなければ、最終的に承認された任意の候補製品の製品販売から収入を得る能力は延期される可能性があり、私たちの業務、成長、財務募集説明書は実質的で不利な影響を受ける可能性がある。
例えば、中国では塵肺疾患の治療のための抗線維化製品は承認されていない。ETUARYは中国でIPFの治療に許可されているが、NMPA、FDA、または他の同様の規制機関が塵肺疾患のような他の適応の治療に使用することを許可することは決定できない。
また、中国、アメリカとEUの監督機関はNASHを治療するための製品をまだ許可していないが、アメリカ国家薬品監督管理局とFDAはNASHを治療する薬物開発ガイドラインを発表し、それぞれ薬物承認のためのNMPAとFDA代替終点表を発表したが、将来の承認の要求が変わるかどうか、あるいはNMPA或いはFDAが監督管理の前例に依存して未来の監督管理許可を行うかどうかはまだ不明である。このような変化はいずれも新たな試験を行う必要がある可能性があり,我々の時間枠を遅らせる可能性があり,F 351や任意の将来のNASH治療の候補品に関連する計画のコストを増加させる可能性がある。また,NASHの第3段階臨床試験やわれわれの候補製品の承認において,臨床あるいは規制機関がどのような治療効果の終点あるいはその提示形態を必要とする可能性があるかは決定できない。
国家薬監局、FDAまたは他の監督機関が私たちの候補製品を承認したとしても、承認はこのような製品の指定用途、使用条件、ラベル、広告、販売促進、マーケティングおよび/または生産に重大な制限を加える可能性があり、追加の研究開発と臨床試験を含む承認後の研究に持続的な承諾または要求を加える可能性がある。米国国家食品薬品監督管理局、FDA、その他の機関はまた、規制要求を遵守できなかった行為に対して、製品の承認を撤回することを含む様々な民事或いは刑事制裁を実施する可能性がある。
私たちの候補製品はこのような国で販売するために外国当局の規制承認を受ける必要がある。一つの規制機関の承認は他の管轄区域の規制機関も承認することを確実にすることはできない。例えば、ETUARYは中国で肺線維症の治療のために許可されているが、米国のような他の任意の司法管轄区では、このような適応のために許可されていない可能性がある。もし私たちが外国の管轄区域の承認を得ることができなければ、私たちの候補製品の地理的市場は制限されるだろう。
114
私たちは、私たちが中国で承認された商業化製品ETUARYを含む、他の人が私たちよりも早く、あるいは成功的に発見、開発、商業化、またはマーケティング製品を含む激しい競争に直面しているかもしれない。
生物技術と製薬業界の特徴は技術進歩が迅速で、競争が激しく、特許製品を高度に重視していることである。私たちは主要な製薬、専門製薬とバイオテクノロジー会社、学術機関と政府機関、そして公共と個人研究機関を含む多くの異なる源からの潜在的な競争に直面している。我々が開発と商業化に成功した任意の候補製品は,既存の療法や将来出現する可能性のある新しい療法と競争するであろう。
現在承認されていない肝線維化治療薬にもかかわらず,いくつかの会社が臨床研究における候補品を開発している。詳細は“をご覧ください”-ビジネス。-競争この年間報告書にあります。
著者らは現在の製品、模倣薬と候補製品の方面で競争に直面し、そして製薬、生物技術とその他の関連業界領域からの未来の候補製品の競争に直面し、これらの細分化業界は器官繊維化患者に対する標的治療、例えばIPF、NASH、SSC-ILD、DM-ILD、塵肺疾患、DKD、ALF/ACLF或いはCOPDを求めている。ETUARY、F 351、F 573、F 528、F 230、または将来の候補製品が競合製品に対する持続的な優位性を提供できない場合、私たちは現在および未来の競合他社との競争に成功できないかもしれない。
私たちの競争相手は、私たちよりも早く規制機関の製品の承認を得ることができるかもしれないし、特許保護や他の知的財産権を獲得する速度よりも速いかもしれません。これは、私たちの候補製品を開発または商業化する能力を制限します。私たちの競争相手はまた、私たちが中国で承認された製品ETUARYよりも有効で、より便利で、より広く、コストが低く、あるいはより安全性の高い薬物、および将来の製品(承認されれば)を開発する可能性があり、これらの競争相手は、その製品の製造とマーケティングにおいても私たちよりも成功するかもしれない。
もし競争相手が私たちの製品ETUARYや未来の製品よりも便利で、より効果的で、より安価で、より安全な製品または療法を開発して販売すれば、承認されれば、私たちの異なる適応でのビジネス機会は減少または消失するかもしれない。また、競争相手が我々よりも早くNMPA、FDA、または他の外国規制機関の承認を得ていれば、強力な市場存在や市場シェアを確立することができない可能性がある。私たちのすべての候補製品の成功を影響する重要な競争要素は、承認されれば、それらの治療効果、安全性、利便性、価格、模造薬の競争レベル、及び政府と他の第三者支払い者が精算できるかどうかである可能性が高い。我々の候補製品は,承認されたものがあれば,これらの既存の薬物や他の療法と競合する可能性があるが,価格的には競争力がない可能性がある。もし私たちの候補製品が承認されれば、それらの価格はブランド模造薬を含む競争相手の模造薬よりも著しく高いと予想されます。したがって、私たちが市場に進出することに成功したどの候補製品も市場の承認を得て、かなりの市場シェアを得ることが課題となる。
私たちと比較して、私たちが競争しているか、あるいは将来競争する可能性のある多くの会社は、研究開発、製造、臨床前テスト、臨床試験を行い、監督管理許可とマーケティング承認製品を獲得する上で、より多くの財務資源と専門知識を持っている。製薬、バイオテクノロジー、診断業界の合併と買収は、より多くの資源を私たちの数の少ない競争相手に集中させる可能性がある。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。これらの競争相手はまた、合格した科学と管理者を募集し、維持し、臨床試験場と臨床試験の個人登録を確立し、そして私たちの計画と相補的あるいは必要な技術を獲得する上で私たちと競争している。
私たちが中国で承認された商業化製品ETUARYおよび任意の他の未来製品は、承認されれば、不利な価格設定法規、第三者精算やり方、あるいは医療改革措置の制約を受け、私たちの業務を損なう可能性がある。
115
新薬製品の発売審査、定価、カバー範囲と精算を管理する規定は国によって異なる。現在と未来の立法は承認要求を大幅に変更する可能性があり、これは追加のコストに関連し、承認の遅延を招く可能性がある。一部の国は薬品の販売価格が承認されてから発売されることを要求している。多くの国で、定価審査期間はマーケティングまたは製品許可を承認した後から始まる。一部の外国市場では、処方薬の定価は初歩的な承認を得た後も、政府の持続的なコントロールを受けている。したがって、特定の国/地域での製品のマーケティング承認を得ることができるかもしれませんが、その後、価格法規の制約を受けて、これらの法規は私たちの製品の商業発表を延期し、長い時間遅延し、その国/地域で製品を販売することによって生じる収入に悪影響を与える可能性があります。不利な価格設定制限は、私たちの候補製品が市場承認を得ても、1つ以上の候補製品への投資を回収する能力を阻害する可能性がある。
アメリカです
私たちが任意の候補製品を商業化することに成功した能力はまた、政府衛生行政当局、個人健康保険会社、および他の組織がこれらの製品および関連治療に提供する保険範囲と十分な補償にある程度依存するだろう。政府当局と第三者支払者、例えば個人健康保険会社や健康維持組織は、どのような薬剤を支払うかを決定し、精算レベルを確立する。アメリカの医療業界と他の地域の主な傾向の一つはコストコントロールだ。政府当局と第三者支払者は,ある薬物のカバー範囲や精算金額を制限することでコストを抑制しようとしている。ますます多くの第三者支払人は製薬会社に価格に基づいて所定の割引を提供し、医療製品の定価に挑戦することを要求している。私たちまたは私たちの協力者が商業化しているどの製品に対しても、保険や精算を提供しないかもしれません。これらの製品が利用できても、精算レベルは満足できないかもしれません。精算は市場で承認された任意の候補製品の需要や価格に影響を及ぼす可能性がある。私たちの製品のために十分な精算を得て維持することは難しいかもしれない。私たちは保険と精算あるいは他の治療法に対する精算レベルが合理的であることを証明するために、高価な薬物経済学的研究を行う必要があるかもしれない。カバー範囲や十分な精算がない場合、あるいは精算が限られたレベルに限られている場合、マーケティングの承認を得た任意の候補製品を商業化することに成功しない可能性があります。
中華人民共和国
新たに承認された薬物の精算には大きな遅延がある可能性があり、カバー範囲は国家薬品監督管理局、FDA或いは中国とアメリカ以外の類似規制機関がこの薬物を承認する目的よりも限られている可能性がある。さらに、補償を受ける資格があることは、1つの薬剤がすべての場合に支払われることを意味するわけではなく、または支払いのレートは、研究、開発、製造、販売、および流通を含むそのコストをカバーするであろう。適用すれば,新薬の臨時精算レベルもその費用を支払うのに不十分である可能性があり,恒久的にはならない可能性がある。
販売率は薬物の使用や臨床環境によって異なる可能性があり,すでに低コスト薬物のために設定された精算レベルに基づいている可能性があり,他のサービスの既存支払いにも組み込まれている可能性がある。医薬品の正味価格は、政府の医療計画または個人支払者が要求する強制的な割引またはリベート、および将来の法律の任意の緩和によって低減することができ、これらの法律は、現在、中国または米国より低い価格で販売される可能性のある国からの医薬品の輸入を制限している。第三者支払者が自分の精算政策を設定する際には,通常連邦医療保険カバー政策と支払制限に依存する。私たちは政府の資金援助や個人支払者から開発された任意の承認された製品から保険と十分な販売率を迅速に得ることができず、これは私たちの経営業績、製品の商業化に必要な資金を調達する能力、および全体の財務状況に実質的な悪影響を及ぼす可能性がある。
また、我々が市場で販売しているETUARYが中国の集中一括調達計画に盛り込まれる可能性がある。詳細は“をご覧ください”-将来、中国政府が制定した集中一括調達政策は、私たちの商業化製品ETUARYおよび任意の他の未来製品をカバーする可能性があり、承認されれば、これらの製品の価格は低下する可能性があり、ひいては私たちの収入、財務状況、および経営業績に重大な悪影響を及ぼす可能性があるこのリスク要因の部分では
116
新たに承認された製品の保険カバー範囲と精算状況はまだ確定していない。新製品や既存製品のために十分な保険や精算を得ることができない場合、米国でこれらの製品をマーケティングする能力を制限し、収入を創出する能力を低下させる可能性がある。
政府と個人支払者の獲得可能性と精算範囲は,多くの患者が米国で高価な治療を負担できるために重要である。私たちの候補製品のアメリカと海外での販売は、私たちの候補製品の費用がどの程度健康維持、管理医療、薬局福祉、類似の医療管理組織によって支払われるか、あるいは政府の健康管理機関、個人健康保険会社、その他の第三者支払人によってどの程度精算されるかに大きく依存する。精算が得られない場合、あるいは限られたレベルに限られていれば、候補製品を商業化することに成功できないかもしれません。保険を提供しても、承認された精算金額が十分に高くない可能性があり、十分な投資リターンを実現するために十分な価格設定を確立したり維持したりするのに十分ではないかもしれません。
新たに承認された製品の保険カバーと精算に関する不確実性が大きい。また、米国や海外の政府や第三者支払者が医療コストの制限や低減に力を入れていることは、これらの組織が承認した新製品のカバー範囲や精算レベルを制限する可能性があるため、私たちの候補製品に十分な支払いを提供できない可能性がある。私たちは、管理型ヘルスケアの傾向、ヘルスケア組織のますますの影響力、および追加的な法的変化により、私たちの任意の候補製品の販売に関する価格設定圧力に直面すると予想される。全体的に,医療コストの下振れ圧力は非常に大きくなっている。そのため、新製品の参入にはますます高い壁が設けられている。
私たちの普通株に関するリスクは
私たちの普通株の市場価格は変動すると予想される。
私たちの普通株の市場価格は大きな変動の影響を受けるかもしれない。普通株市場の価格変動を引き起こす可能性があるいくつかの要素は
117
また、株式市場は全体的に大きな変動を経験しており、個別会社の経営業績とは無関係であることが多い。このような広範囲な市場変動はまた私たちの普通株の取引価格に悪影響を及ぼすかもしれない。さらに、マクロ経済状況、景気後退、不況、または他の持続的な不利な市場事件または他の要因は、私たちの業務および私たちの普通株の価値に実質的な悪影響を及ぼす可能性がある。従来、ある会社の証券市場価格が変動した後、株主はこのような会社に対して集団証券訴訟を起こすことが多かった。また,我々が維権者が我々の内在的価値を反映していないと考える市場推定値を経験すれば,市場変動は株主の維権活動を増加させる可能性がある.我々の戦略方向と競争したり衝突したり、取締役会構成の変更を求める過激な活動は、私たちの経営業績や財務状況に悪影響を及ぼす可能性があります。
経営業績の変動は私たちの普通株の価格に悪影響を及ぼす可能性があります。
私たちの経営業績は四半期ごとに大きく変動するかもしれません。このような変動は私たちの株価を下落させるかもしれない。経営結果の経時的変動を引き起こす可能性のあるいくつかの要因は、臨床前および臨床開発計画の範囲、進行、持続時間結果およびコスト、および候補製品および計画の非臨床研究および評価、再構成コスト、第三者との協力、許可、製造または他の重要な合意の実施または終了、任意のこのような合意の下での非日常的な収入または費用、規制コンプライアンス、承認または他の規制行動のコスト、時間および結果、規制承認の可能性、規制機関が規制承認および一般および業界固有の経済条件を与えていない場合、特に製薬業界に影響を与える場合、中国やアメリカのバイオ製薬やバイオテクノロジー産業です私たちの歴史と未来の財務業績の段階的比較には意味がないかもしれませんが、投資家はそれらを未来の業績の指標として依存すべきではありません。変動の損失は証券アナリストや投資家の予想に達しない可能性がある。もしこのような期待を達成できなければ、私たちの普通株の価格を下落させるかもしれない。
私たちの普通株の大量株を公開市場で売ったり、このような売却が発生する可能性があると考えたりすると、私たちの普通株の市場価格を下げる可能性があります。
私たちの現在の取引量は大きくなく、公開市場で私たちの普通株の相当数の株を売っているか、あるいはこれらの売却が発生する可能性があると考えて、市場価格の下落を招く可能性があります。これらの棚登録声明によると、私たちの普通株または他の証券の公開市場でのいかなる追加販売も、私たちの普通株の現行の市場価格に悪影響を及ぼす可能性がある。また、2023年12月31日現在、18,280,548株の普通株を購入する未償還オプションを有しており、加重平均行権価格は1.49ドルである。このような選択権を行使して株式を公開市場に売却すれば、当該等の売却も今後、適切と思われる価格で株式証券を売却することが困難になる可能性がある。これらの証券を私たちの普通株に転換したり行使したりする株式は、私たちの普通株の他の所有者に希釈され、このようなすべての株式が可能です
118
転換や行使後に公開市場で販売され、証券法の制限を受け、これにより我々普通株の市場価格が低下する可能性がある。
当社の登録証明書や定款、デラウェア州の法律の規定は、買収会社をさらに困難にする可能性があり、これは私たちの株主に有利になる可能性があり、私たちの株主が私たちの経営陣を交換または更迭しようとすることを阻止するかもしれません。
当社の会社登録証明書及び定款に含まれる条項は、株主が有利と思われる可能性のある会社の合併、買収又は他の統制権変更を阻止、延期、又は阻止する可能性があり、我々の普通株主が他の方法で割増取引を得る可能性があることを含む。これらの条項はまた、投資家が将来私たちの普通株に支払いたいかもしれない価格を制限し、それによって私たちの普通株の市場価格を下げる可能性がある。また、我々の取締役会は、我々の管理チームのメンバーに責任を持って命令するので、これらの規定は、株主が取締役会のメンバーを交換する難しさを増加させることによって、現在の経営陣の任意の試みを交換または罷免することを阻害または阻止する可能性がある。他の事項を除いて、これらの条文は以下のように含まれている
また、私たちはデラウェア州に登録して設立されているので、私たちはDGCL第203条の規定によって管轄されています。この条項は、合併または合併が規定された方法で承認されない限り、会社の15%以上の議決権のある株を発行した人が取引日後3年以内に会社と合併または合併しないことを一般的に禁止しています。
私たちの会社の登録証明書と定款は一般的に規定されており、デラウェア州衡平裁判所は私たちと株主の間のほとんどの紛争の独占法廷であり、これは私たちの株主が私たちまたは私たちの役員、高級管理者、または他の従業員との紛争において有利な司法フォーラムを得る能力を制限することができるかもしれない。
当社の登録証明書及び定款規定は、会社が書面で代替裁判所を選択することに同意しない限り、デラウェア州衡平裁判所(又は、衡平裁判所に管轄権がない場合、デラウェア州連邦地域裁判所又はデラウェア州他の州裁判所)は、以下のタイプの訴訟の唯一及び独占裁判所である:(1)私たちが提起した任意の派生訴訟又は法的手続を代表して、(2)私たちの任意の取締役、上級管理職、従業員又は株主が会社又は私たちの株主の受託責任を侵害すると主張するいかなる訴訟も、(3)“会社条例”のいずれかの条文に基づいて申索を提出する任意の訴訟、または“会社条例”に基づいてデラウェア州衡平裁判所に司法管轄権を付与する任意の訴訟、または(4)我々が再記載した会社登録証明書または当社の付例(それぞれの場合、時々改正することができる)のいずれかの条文または内務原則によって制限された任意の訴訟を提起する。このような裁判所条項の選択は、証券法、取引法、または連邦裁判所が排他的管轄権を有する任意の他のクレームを実行するために提出された義務または責任の訴訟には適用されない。
119
この排他的裁判所条項は、株主が別の司法管轄区を選択することを許可されているのと比較して、株主がクレームを出すコストをより高くする可能性があり、私たちまたは私たちの役員、幹部または他の従業員または株主との紛争に有利であると考えるクレームを司法裁判所で株主が提出する能力を制限する可能性があり、これは、私たちおよび私たちの取締役、役員、および他の従業員および株主に対するこのような訴訟を阻止する可能性がある。あるいは、裁判所が私たちの会社登録証明書および添付例に含まれる選択裁判所条項が訴訟で適用されないか、または実行できないことを発見した場合、私たちは、他の司法管轄区域でこのような訴訟を解決することに関連する追加費用を生じる可能性があり、これは、私たちの業務、財務状況、および運営結果に実質的かつ不利な影響を与える可能性がある。
私たちは小さな報告会社であり、より小さい報告会社に適用される開示要求の低減が、私たちの普通株の投資家に対する吸引力を低下させるかどうかを決定することはできません。
私たちは、1934年に改正された“証券取引法”(以下、“取引法”)で定義されてきた“小さな報告会社”であるため、提出された文書で簡略化された役員報酬開示を提供することが許可されている。私たちがアメリカ証券取引委員会に提出した文書では、私たちはまたいくつかの他の減少した開示義務を減らした。私たちは投資家が私たちがこれらの免除のいずれかに依存して私たちの普通株の吸引力が低下していると思うかどうか予測できない。したがって、一部の投資家が私たちの普通株の吸引力が低下していることを発見すれば、私たちの普通株はそれほど活発ではない取引市場がある可能性があり、私たちの株価はもっと変動するかもしれない。
私たちは予測可能な未来に、私たちはどんな現金配当金も支払わないと予想している。
現在の予想は、私たちは将来の収益を維持し、もしあれば、配当金を支払うのではなく、私たちの業務成長に資金を提供することだ。したがって、予測可能な未来に、私たちの普通株の資本増加(あれば)があなたの唯一の収益源になるだろう。
株式研究アナリストが我々、我々の業務又は我々の市場に関する研究又は報告を発表しない場合、又は不利な研究又は報告を発表しなければ、我々の株価及び取引量は低下する可能性がある。
私たちの普通株の取引市場は株式研究アナリストが発表した私たちと私たちの業務に関する研究と報告書の影響を受けるだろう。研究報告書の不足は私たちの普通株の市場価格に悪影響を及ぼすかもしれない。もし私たちが株式研究アナリストの報告書を持っていたら、私たちはアナリストや彼らの報告書に含まれている内容と意見を制御できないだろう。1人以上の株式研究アナリストが私たちの株式格付けを引き下げたり、他の不利なコメントや研究を発表したりすれば、私たちの普通株の価格は下落する可能性がある。1つ以上の株式研究アナリストが私たちの報告書を停止したり、私たちの報告書を定期的に発表できなかったりすれば、私たちの普通株に対する需要が減少する可能性があり、これは逆に私たちの株価や取引量を低下させる可能性がある。
私たちは統合に成功して寄付金の期待的な利益を達成できないかもしれない。
寄付金は両社の合併に関連しており、この2社は以前は独立会社として運営されていた。統合プロセスにかかる時間が予想より長い場合や費用が予想より高い場合、寄付金の一部またはすべての期待利益を達成できない可能性があります。
私たちが融合過程で遭遇する可能性のある困難は
統合プロセスはまた、私たちの経営陣の注意移転、私たちの業務の中断や中断、あるいは動力を失ったり、標準、制御、プログラム、および
120
政策は、いずれも、私たちが業務関係を維持する能力や、供給期待利益を達成する能力に悪影響を及ぼす可能性があり、または私たちの業務および財務業績に悪影響を及ぼす可能性がある。
一般リスク因子
純営業損失の繰越と税収控除を利用する能力が制限される可能性があります。
2023年12月31日現在、約1兆935億ドルの連邦と1050万ドルのカリフォルニア州の純営業損失繰越(“NOL”)が将来の課税収入を減らすために使用できる。“法典”第382節及び第383節及び州法律の規定によると、ある会社が所有権変更を経験した場合、変更前のNOL及び他の変更前の税収属性(例えば税収控除を検討する)を用いて変更後の収入を相殺する能力が制限される可能性がある。第382条“所有権変更”は、通常、特定株主によるその持分所有権の変化(価値別計算)が3年以内に50ポイントを超えると定義される。このような制限により,NOL繰り越しの一部が使用前に期限切れになる可能性がある。私たちは何度か所有権変更を経験した。約1兆565億ドルと7520万ドルのNOLがそれぞれ満期になり、連邦やカリフォルニアの目的には使われていない。これらの貢献は、私たちのその後の株式所有権の変化(その中のいくつかは私たちがコントロールできるものではない)のため、追加的な所有権の変化をもたらし、私たちは未来により多くの所有権の変化を経験するかもしれない。
さらに、私たちは潜在的な将来の課税収入と関連する所得税を相殺するために私たちのNOLを使用する能力は私たちの未来の課税収入の生成に依存し、私たちは私たちのNOLを使用するために私たちのNOLを使用するのに十分な課税収入がいつ生産されるかどうかを確定的に予測することができない。
私たちが利益を達成しても、私たちは私たちのNOLや他の税金属性の大部分を利用できない可能性があり、これはキャッシュフローや運営結果に実質的な悪影響を及ぼす可能性がある。州税法の似たような規定はまた私たちが累積的な州税収属性を使用することを制限するのに適用されるかもしれない。もう一つのリスクは、法規の変化、例えばNOLの使用停止、または他の予測不可能な理由により、私たちの既存のNOLが満期になるか、または将来の所得税債務を相殺するために使用できない可能性があるということだ。
税法またはその施行の変化は私たちの業務と財務状況に悪影響を及ぼすかもしれない。
立法手続きに参加した人員および米国国税局と米国財務省は、米国連邦、州、地方所得税に関する規則を審査してきた。税法の変化は私たちの業務や財務状況、または私たちの普通株の保有者に悪影響を及ぼすかもしれない。近年、このような変化は多く発生しており、未来も変化し続けるかもしれない。税務法律、法規および裁決がいつ、どのような形または発効日に公布されるか、または発表されるかどうかは予測できません。これは、私たちまたは私たちの株主の納税責任を増加させるか、または税法変化のいかなる悪影響を最大限に減少または軽減するために、運営方式を変更することを要求する可能性があります。潜在的投資家は税法の変化が私たちの業務および私たちの普通株の所有権と処置に対する潜在的な結果について彼らの税務顧問に相談しなければならない。
私たちはナスダック上場基準が指す“制御された会社”なので、資格があり、ある会社の管理要求を免除することに依存しています。私たちの株主はこのような要求を受けた会社の株主に同じ保護を提供していません。
GNI締約国は私たちが発行した普通株式の大部分の投票権を支配している。したがって、私たちはナスダック社の管理基準が指す“制御された会社”の資格に適合している。これらの規則によれば、個人、グループ、または他の会社が取締役選挙投票権の50%以上を有する上場企業が“制御された会社”であり、(I)私たちの取締役会の多くのメンバーが独立取締役で構成されていること、(Ii)取締役が独立取締役によって完全に選択または推薦されている取締役会、および(Iii)我々の報酬委員会が完全に独立取締役で構成されていることを含む、特定の会社管理要件を遵守しないことを選択することができる。
私たちはこの免除に依存している。したがって、私たちには独立取締役の多数はなく、取締役が指名した人は完全に独立取締役が選択または推薦して我々の取締役会に入るのではなく、私たちの報酬委員会も完全に独立取締役で構成されているわけではありません。同じ保護は得られないかもしれません
121
ナスダックのすべての会社のガバナンス要求に制約されている会社の株主に提供します。もし私たちがこれ以上“制御された会社”ではなく、私たちの株がナスダックに上場し続けるなら、私たちは適用される過渡期内にこのような規定を遵守することを要求されるだろう。
私たちの役員、役員、主要株主は、私たちの株主に提出されたすべての事項を制御または顕著に影響する能力があります。
2023年12月31日現在、私たちの役員、役員、主要株主の合計実益は、私たちが発行した普通株の大部分を持っています。したがって、これらの株主が共同行動を選択すれば、彼らは私たちの株主に提出されたすべての事項と、私たちの管理と事務を制御または顕著に影響することができるだろう。例えば、この人たちが共同行動を選択した場合、彼らは取締役の選挙と、私たちのすべてまたはほとんどの資産の任意の合併、合併、または売却の承認を制御または著しく影響するだろう。このような投票権の集中は他の株主が望む可能性のある条項で私たちを買収することを延期または阻止するかもしれない。
私たちは株主訴訟を含めてより多くの訴訟に直面する可能性があり、これは私たちの業務や運営に悪影響を及ぼす可能性がある。
私たちは時々、当社のサプライヤー、顧客、請負業者、ライセンシー、ビジネスパートナー、および私たちの業務運営のために採用された他の第三者と発生した様々な紛争またはクレームを含むが、これらに限定されない様々な訴訟、法律または契約紛争、調査または行政訴訟の影響を受けることができる。また、寄付の完了により、Catalystの業務とGyre製薬の業務の合併により、より多くの訴訟に直面する可能性があります。このような訴訟は、私たちの業務や運営結果に悪影響を及ぼす可能性があり、あるいは私たちの運営中断を招く可能性があります。また,過去にバイオテクノロジー会社株の市場価格が変動した後,株主はこれらの会社に対して集団訴訟を起こしていた。私たちにこのような訴訟を提起すれば、私たちに巨額の費用が発生し、経営陣の注意と資源を移転させる可能性があり、これは私たちの業務、財務状況、運営結果に実質的な悪影響を及ぼす可能性がある。
任意の裁決や裁決が私たちに不利である場合、または敵との和解に同意した場合、私たちは、重大な金銭的損害賠償の支払い、他の責任を負うこと、および/または関連する業務項目を一時停止または終了することを要求される可能性があります。訴訟、法的紛争、調査または行政訴訟による否定的な宣伝は、私たちの名声を損なう可能性があり、私たちのブランドや製品のイメージに悪影響を及ぼす可能性がある。
私たちは製品責任クレームの影響を受けるかもしれません。これは私たちをコストと責任に直面させるかもしれません。
著者らは中国、アメリカとその他の司法管轄区で薬品の開発、生産、マーケティング、普及と販売のために製品責任リスクに直面している。私たちの製品、ETUARY、および将来の製品(承認された場合)が、安全でない、無効であること、欠陥があること、または汚染されていることが証明された場合、または不十分または不適切な製品ラベルまたは不十分、不十分または誤解性を提供する副作用に関する警告または開示などの行為に従事していると告発された場合、そのようなクレームが発生する可能性がある。私たちが提起した製品責任クレームは、是非曲直や結果にかかわらず、名声損害や財務資源の緊張を招き、私たちの経営陣の時間と注意を消費する可能性があります。もし私たちがこのようなクレームを自己弁護することに成功できなければ、私たちは製品のリコール、私たちの製品、ETUARYと未来の製品(承認された場合)による人身傷害、死亡または他の損失の民事責任、刑事責任、そして私たちの営業許可証が取り消されるなどに直面するかもしれない。中国の法律と法規は現在私たちに要求されておらず、私たちは製品責任クレームをカバーするために責任保険の維持も要求しない。したがって、私たちは未来の製品責任クレームによる損失を取り戻すことができないかもしれない。
私たちの情報システムの抜け穴、故障、または中断は、私たちの業務に関連する敏感な情報を危険にさらし、私たちを責任や名声の損害に直面させる可能性があり、私たちが業務運営を効果的に管理する能力は不利な影響を受ける可能性があります。
私たちの情報システムは故障し、コンピュータウイルス、コンピュータハッカー、悪意のあるコード、従業員の誤りまたは汚職、窃盗または乱用、サービス拒否攻撃、複雑な民族国家および民族国家によって支持される行為者、許可されていないアクセス、自然災害、テロ、戦争、電気通信および電気故障、または他の危害のリスクに直面する可能性がある。データを中断する任意のシステムが破損したり、故障したりする
122
入力、検索、または転送またはサービス時間の増加は、完了したまたは将来の臨床試験の臨床試験データを失うことを含む、私たちの正常な運営を混乱させる可能性がある。臨床試験データの紛失は著者らの監督管理審査の遅延を招く可能性があり、そして著者らのデータの回復或いは複製のコストを著しく増加させる。私たちの情報システムの障害を効果的に処理できる保証はありません。あるいは、私たちの業務能力をタイムリーに回復することができるか、あるいは私たちの業務中断を根本的に避けることができません。もし任意の中断またはセキュリティホールが私たちのデータやアプリケーションを紛失または破損させ、または機密または固有の情報を適切に使用、開示、またはアクセスしない場合、私たちは責任を招く可能性があり、私たちの競争地位が損なわれる可能性があり、私たちの製品、ETUARY、および候補製品のさらなる開発および商業化は阻害または遅延される可能性がある。
私たちは正常な業務過程で敏感な個人データを収集して保存するかもしれない。詳細は“をご覧ください”-私たちの中国での業務運営に関するリスク-中国の新しいデータセキュリティ法、ネットワークセキュリティ法、ネットワークセキュリティ審査方法、個人情報保護法、ネットワークセキュリティ多段保護計画に関する法規およびガイドライン、および任意の他の将来の法律および法規を遵守することは、巨額の費用が発生し、私たちの業務に影響を与える可能性がありますこのリスク要因の部分ではもし個人データが私たちの情報が重大に漏洩した場合、市場の私たちの安全措置の有効性に対する見方が損なわれる可能性があり、私たちの名声と信頼性が損なわれる可能性がある。個人および団体が個人訴訟で提起した規制行動および/またはクレームの影響を受ける可能性があり、これらの訴訟は、データ収集および使用実践および他のデータプライバシー法律および法規に関連するプライバシー問題に関するものである。
あなたは私たちに不利な判決を得ることが難しいかもしれない。
私たちのほとんどの資産はアメリカ以外に位置している。私たちの業務の大部分と行政と会社の機能は中国で行われています。また、私たちの何人かの役員や役人はアメリカ以外の国の国民と住民です。この人たちの資産の大部分はアメリカ国外にある。したがって、アメリカとその中のいくつかの外国司法管轄区との間には互恵と条約が不足しているため、コストと時間制限を加えて、アメリカ内でこれらの人に法的手続き文書を送ることは難しいかもしれません。特に、我々の何人かの上級管理者や役員は通常中国に位置しており、これらの個人に対して責任や判決を執行することはさらに困難になる。
項目1 B。解決器VEDスタッフはコメントした。
ない。
プロジェクト1 C。ネットワークセキュリティです。
私たちは私たちの重要なデータ、ハードウェア、そして内部ネットワークをデジタル攻撃、窃盗、破壊から保護するために努力している。通常のビジネスプロセスでは、機密、敏感、独自、および個人情報をデジタル的に収集、使用、記憶、および送信します。これらの情報と我々の情報技術システムのセキュリティ維持は,我々の運営と業務戦略に非常に重要である.そのために、我々は、これらのシステムおよびその中に存在するデータの機密性、完全性、および利用可能性に悪影響を及ぼす可能性がある当社の情報技術システム上または当社の情報技術システムによって発生する潜在的な不正イベントのリスクを評価、識別、および管理することを実施した。Gyre Treeuticsでは、これらのプロセスは、第三者情報技術(“IT”)コンサルティング会社(“ホスト·サービスプロバイダ”)によって管理および監視され、当社の最高財務官によって監視される。Gyre製薬会社では,これらの流れは社長が率いる専門ITチームが管理·監視している。Gyre TreeuticsおよびGyre PharmPharmticalsのプロセスは、データ損失、盗難、誤用、またはデータに影響を与える他のセキュリティイベントまたは脆弱性を防止または低減することを目的とした機構、制御、技術、システム、および他のプロセスを含む。例えば,Gyre TreeuticsとGyre PharmPharmticalsはソフトウェアやハードウェア在庫を維持し,安全監視と警報を実行し,継続的なリスク評価を完了する.Gyre治療会社やGyre製薬会社では,ネットワークや情報セキュリティなどのテーマに関する従業員研修も定期的に行われている。また、両社は定期的に外部コンサルタントや専門家に相談し、将来の脅威や傾向の予測、会社のリスク環境への影響を含むネットワークセキュリティリスクの評価、識別、管理に協力している。
123
我々の最高財務官は、Gyre Treateuticsのネットワークセキュリティリスクの評価と管理を担当し、20年以上の情報技術とネットワークセキュリティ事務管理経験を有するITコンサルタントを招聘し、マイクロソフト認証システムエンジニア資格認証を取得したことを、ホスト·サービスプロバイダに直接報告する。我々のGyre PharmPharmticals IT部門上級主管はGyre PharmPharmticalsの社長に直接報告し,20年以上の情報技術とネットワークセキュリティ事務を管理した経験を持ち,Gyre PharmPharmticalsのネットワークセキュリティリスクの評価と管理を担当している.私たちGyre製薬会社の社長は取締役会に仕事を報告しました。私たちは企業リスク管理の枠組み全体でネットワークセキュリティと私たちが直面している他の重大なリスクを考慮している。前期には、これまで私たちに大きな影響を与えてきたサイバーセキュリティ事件を含め、既知のサイバーセキュリティ脅威からリスクは発見されていませんが、私たちはいくつかの持続的なサイバーセキュリティリスクに直面しており、これらのリスクを実現すれば、私たちに大きな影響を与える可能性が高いです。我々が直面しているネットワークセキュリティリスクに関するより多くの情報は,第1部1 A項“リスク要因”で議論されており,タイトルは“我々の情報システムにおけるあるいは我々の情報システムの破壊,障害,あるいは中断が我々の業務に関連する敏感な情報を危険にさらす可能性があり,責任や名声の被害に直面させ,我々が業務運営を効率的に管理する能力が悪影響を受ける可能性がある”としている
取締役会は全体と委員会レベルとして、私たちが直面している最も重大なリスクと、私たちがこれらのリスクを識別、優先的に処理、評価、管理、緩和する過程を監督している。監査委員会は完全に独立した役員で構成されており、私たちの取締役会がネットワークセキュリティリスクの監視を担当することを指定しています。監査委員会は定期的に我々の管理チームからネットワークセキュリティと情報技術事項および関連リスク暴露に関する最新の状況を受け取っている。監査委員会はまた、少なくとも毎年、ネットワークセキュリティリスクに関する管理職と監査委員会の最新の状況を受け取っている。
項目2.財産
GIRE治療会社の特性
私たちの会社の本社はカリフォルニア州サンディエゴにあります。そこで約一,六四三平方フィートのレンタル可能なオフィス空間を借りました。レンタルは2023年11月11日に開始され、2023年11月11日以降に開始された38番目の完全カレンダー月の最終日に満期になります。Gyreは商業的に合理的な条項でこの賃貸契約を更新したり、代替施設を獲得したりすることを望んでいる。
GYRE製薬会社の性質
自分に財産がある
GYRE PharmPharmticalsは中国北京市順義区及び中国河北省滄州市に位置する2枚の土地の土地使用権証明書を所有しており、総地盤面積は66,559平方メートル、及び6つの物件の建築物所有権証明書を有し、総床面積は12,206平方メートルである。ゲーリー製薬会社の2つの生産センターはそれぞれ中国北京と中国滄州に位置する。
賃貸物件
Gyre製薬会社は中国で21カ所の物件をレンタルした。Gyre PharmPharmticalsの21カ所の賃貸物件のうち,8カ所がオフィス,12カ所が従業員寮,1カ所が実験室として用いられている。
第3項.法的手続き
私たちは現在どんな実質的な法的手続きの当事者でもない。時々、私たちは正常な業務過程で発生した法的訴訟に巻き込まれるかもしれない。結果にかかわらず、訴訟は弁護と和解コスト、管理資源の移転、負の宣伝、名声損害などの要素によって私たちに不利な影響を与える可能性があり、有利な結果が得られる保証はない。
第四項鉱山安全情報開示
適用されません。
124
部分第2部:
項目5.登録者普通株式市場、関連株主事項、発行者権益株式証券の追跡。
普通株式市場情報
GYRE治療会社はナスダック資本市場に上場し、株式コードは“GIRE”である
普通株保有者
2024年3月19日現在、我々の普通株には約46名の登録株主がいる。我々の多くの普通株は仲介人や他の機関代表株主が保有しているため,これらの記録保有者が代表する株主総数を見積もることはできない.
配当政策
2022年9月20日、2022年9月6日終値時点で登録されている普通株主に約4500万ドル(または1株1.43ドル)の特別一次現金配当金を支払った。2023年1月12日、2023年1月5日の終値時に登録された普通株主に約760万ドル(または1株当たり0.24ドル)の特別使い捨て現金配当金を支払った。2023年6月、Vertex取引に関連してVertexから受信した費用を差し引いたプリフェッチ金額および潜在税務責任準備金を反映した2023年1月5日現在のCatalyst普通株式所有者(“CVR所持者”)に350万ドルを割り当てた。私たちは現在、私たちの業務のためにすべての未来の収益を維持し、予測可能な未来に私たちの普通株に現金配当金を支払わないつもりだ。将来的に現金配当を発表する任意の決定は、適用される法律に依存し、私たちの財務状況、経営結果、資本要求、一般業務状況、および私たちの取締役会が関連する他の要素に依存するかもしれない、私たちの取締役会が適宜決定する。
発行者および関連購入者が株式証券を購入する
ない。
第六項です[保留されている].
125
項目7.経営陣の以下の問題の議論と分析財務状況と経営結果。
以下の財務状況と経営結果の検討と分析、および当社の監査財務諸表と本年度報告書の他の場所に関する付記(Form 10-K)を読むべきです。本議論および本年度報告Form 10−Kの他の部分は、我々の計画、目標、期待、および意図の陳述のようなリスクおよび不確実性に関する前向きな陳述を含む。私たちの実際の結果はこのような前向きな陳述で議論された結果と大きく違うかもしれない。このような差異をもたらすか、または促進する可能性のある要因には、本報告の“リスク要因”と題する節で議論された要因が含まれるが、これらに限定されない。実際の結果が以下に説明する結果とは大きく異なる重要な要素をもたらす可能性があることを理解するために、本年度報告書Form 10−Kの“前向き陳述に関する警告説明”および“リスク要因”の部分をよく読まなければならない。
概要
私たちは財務的に持続可能な製薬会社であり、財務成功の記録があり、臓器疾患に対する小分子抗炎症と抗繊維化薬物の開発と商業化、特に臓器繊維化に集中している。線維性疾患とは,満たされていない医療ニーズを多く有する患者である。繊維化は複雑で多段階の過程であり、複数の経路がある。多くの潜在的な抗繊維化治療標的があるが、すでに確立されているものもあれば、出現しているものもあり、単一の分子経路を解決することは繊維化を予防、阻止或いは逆転するのに十分ではないかもしれない。
われわれの戦略は,ETUARY(ピルフェニドン)の開発成功と商業化に成功した経験を利用して,新たな適応に拡大し,類似した候補薬を開発することである。ピルフェニドンは日本、EU、アメリカと中国が特発性肺繊維化に使用することを許可した第一種の抗繊維化薬物であり、1種の小分子薬物であり、転化成長因子-1、腫瘍壊死因子-αと他の繊維化と炎症調節剤の合成を抑制できる。我々はすでに中国でIPF用ETUARY(ピルフェニドン)の承認を得た。
GYRE製薬会社は中国でピルフェニドンを研究開発から商業化に推進し、IPFの治療に成功した。ETUARYの年間売上高は毎年増加し続けており,2023年には1億121億ドルに達している。IPFを除くPirfenidoneはCTD−ILDの他の3つの3期研究を行っており,その適応と市場:SSC−ILD,DM−ILDとPDを拡大している。
F 351は,我々の主な開発候補であり,ETUARY(ピルフェニドン)の構造誘導体である。それは1種の新しい経口化学実体であり、抗繊維化、転化成長因子-1標的作用機序を有し、著者らは主要な市場で特許を持っている。研究により、F 351及びその主要な代謝物の薬物相互作用リスクは最も小さいことが分かった。IPFに潜在的な治療効果があるにもかかわらず、巨大な潜在市場と重大な満たされていない需要のため、私たちは肝線維化の治療にF 351を優先している。
GYRE製薬会社はすでにF 351が中国で慢性B型肝炎関連性肝繊維化を治療する第二段階試験を完成した。2期試験では,F 351の耐性は良好であり,明らかな毒性はなく,治療を受けた患者は統計的に有意な肝線維化改善を示し,270 mg/日の治療効果が最適であった。これらの結果から,中国では検証的な第3段階試験が行われている。検証性3期試験の248名の患者の募集は完了し,最後の患者は2024年に退院する予定であり,臨床結果は2025年初めに発表される予定である。
アメリカでは健康ボランティアにおけるF 351の第1段階臨床試験を完了しました私たちはIND申請を準備しており、2024年末に提出される予定だ。CHB関連肝線維化PRC第3段階試験の結果からINDの承認を待っており,2025年に2 a段階試験を開始し,F 351によるNASH関連肝線維化治療効果を評価する予定である。
F 351資産買収
Catalystは2022年12月26日にF 351プロトコルによりGNI日本およびGNI香港にF 351資産を購入したが,中国に位置する当該などの資産や知的財産権は除外した。
企業合併協定
126
2022年12月26日、CatalystはGNI USA、GNI Japan、GNI Hong Kong、SG(GNI USA、GNI JapanおよびGNI Hong Kongと合わせて“貢献者”と呼ばれ、それぞれ“貢献者”と呼ばれる)、いくつかの個人およびCPIが業務統合協定(2023年3月29日および2023年8月30日に改訂された)、いくつかの個人およびCPIを締結した. 2023年10月30日(“発効時間”)、出資(定義は以下参照)が発効し、Catalystは北京大陸医薬有限会社(経営名はGyre PharmPharmticals Co.)の間接持株権を取得した。寄付に関連して,Catalystは寄付が発効する前に登録証明書を修正し,その普通株の認可株式数を増加させ,1株当たり額面0.001ドル(“Catalyst普通株”)を100,000,000株から400,000,000株に増加させ,逆株式分割を実現し,その名称をGyre Treateutics,Inc.に変更した。
Catalyst普通株はこれまでナスダック資本市場に上場し、コードは“CBIO”だった。Catalystは合併後の会社としてナスダック(以下、ナスダック)に上場申請を提出した。2023年10月31日、Gyreの普通株はナスダック資本市場で取引を開始し、コードはGYREであり、株式の逆分割調整後の基礎に基づいている。
企業合併協定によると、出資発効時に、15株1株の逆株式分割を実施した後、
GyreはGNI USAの貢献により,CPIの株式100%を直接かつ間接的に保有している.GyreのCPIに対する所有権により,少数株主が出資する前に,GyreはGyre製薬会社56.0%の間接権益を持っている。少数株主の出資を完了した後、GyreはGyre製薬会社の追加間接権益を獲得し、合計65.2%の間接権益を持っている。
入金完了後,潜在希釈証券を含まず,GNI USAはGyre普通株約83.6%の流通株を持ち,Catalystの既存株主(GNI USAを除く)はGyre発行普通株約2.8%,少数株主はGyre普通株約13.7%の流通株を有している。転換可能優先株は供給が完了した後も返済されておらず、上記所有権のパーセンテージには含まれていない。
発効時、Gyre PharmPharmticalsは2021年株式激励計画(“2021年計画”)を中止し、Gyre PharmPharmticalsは2021年計画項の下でまだ行使されていない購入権(“Gyre PharmPharmticals Options”)を終了し、代わりにGyre 2023年総合激励計画(“2023年総合激励計画”)の項目の下で中国参加者のために付与した株式購入権は、このような購入株権はすべての重大な面でGyre PharmPharmticalsが先に2021年計画で行使しなかった株購入権とほぼ似ている。
発効時期に発行および発行されたCatalyst普通株1株およびCatalyst普通株購入のオプションは発行済みおよび発行済みの状態を維持しており,これらの株式およびオプションは出資の影響を受けない。
GYRE製薬会社は商業段階の生物製薬会社であり,2002年に中国に登録設立された。GYRE製薬会社は新薬の研究と開発,特発性肺線維化治療のためのETUARY(ピルフェニドンカプセル)や他の薬品の製造と商業化に取り組んでいる。Gyre製薬の登録事務所は中国北京市順義区順康路60号に位置する。
127
Gyre PharmPharmticalsの直接持株会社はBJContinent PharmPharmticals Limited(“BJC”)である。Gyre PharmPharmticalsの中間持株会社はCPIである。GNI USA寄付後、CPIの直接持ち株会社はGyreである。Gyreの大株主はGNI USA,GNI USAの大株主はGNI Japanである.
GNI USA供出は米国公認会計原則(“米国公認会計原則”)に基づいて資産買収とされ、CPIは会計買収側とみなされ、買収後報告目的の前身として列報されている。Catalystは合法的な購入者であるため,GNI USAの寄付は逆資産買収に計上される.この決定は、GNI USA出資に続いて、(I)GNI USA(GNI USA出資直前のCPIの親会社として)が合併後会社の圧倒的多数の投票権を有すること、(Ii)GNI USAが合併後会社の取締役会を制御する能力があること、および(Iii)Gyre製薬会社とGNI USAの上級管理者が合併後会社の高級管理において多数のキーポストを占めることを含む企業合併合意に基づく条項およびその他の要因である。GNI USA寄付が終了する前に,Catalystは企業の定義に適合しておらず,Catalystには組織的な労働力がないため,その創造産出能力に大きく貢献しており,その公正価値のほぼすべてが過程の研究と開発に集中している(“IPR&D”)。
GNI USA供出の決算日には,Catalystの純資産は購入日の相対公正価値に本年度報告表格10−K第II部第8項に記載されている総合財務諸表が計上されているが,GNI USA供出前に報告された経営業績はCPIの経営業績である。
少数株主の貢献は株式取引とみなされ、私たちはGyre PharmPharmticalsの追加的な間接権益を獲得し、Gyre PharmPharmticalsの制御を維持した。
あるいは価値のある権利協定
2022年12月26日に業務合併協定に署名すると同時に、Catalystは権利エージェント(CVR協定参照)と2023年3月29日に改訂されたまたは価値のある権利協定(“CVR協定”)に署名し、この合意によれば、売り手を除いて、各CVR所有者は、当社が所有するCatalyst普通株当たりに発行される契約または価値ある権利(“CVR”)を取得する。各CVRは、そのCVR所有者が将来いくつかの現金支払いを受ける権利を有するようにする。その他の資料については、別注13-を参照引受金とその他の事項本年度報告シート10-K第2部第8項に記載されている連結財務諸表。
株を逆分割する
私たちは寄付が発効する直前に15株1株の逆株分割を行った。逆株分割後,触媒普通株の額面は調整されず,1株当たり0.001ドルであった。Catalystのすべての発行済み株式および発行済み普通株式およびオプションは、すべての届出期間の逆株式分割を反映するように遡及調整されている。
他に説明がある以外に、すべての株式および1株当たりの資料はすべての提出期間の逆株式分割を有効にするために追跡的に調整されている。発行されたすべての購入株権及び株式承認証に行使或いは帰属する時、1株当たりの権利価格及び発行可能株式数は比例的に調整され、行使或いは当該等の購入持分及び株式承認証に帰属する時に保留して発行する普通株式数は割合で減少し、株式購入及び引受証については、当該等購入持分及び株式承認証の行権価格は比例的に減少する。
逆株式分割に関連する断片的な株式は発行されておらず、もともと株式の一部を取得する権利を持っていた株主は比例して支払われた現金を獲得した。
私募および証券購入協定
2023年10月27日、GNI USAと私募証券購入契約を締結しました。証券購入協定によると、GNI USAは(I)Gyre 811株の転換可能株の購入に同意した
128
優先株および(Ii)は最大811株の交換可能株の引受権証(“優先株権証”)を購入でき、総購入価格は500万ドルである。個人配給は寄付が終わったらすぐに終わります。
優先株式証は転換可能な優先株1株4915.00ドルの行使価格で行使でき、2033年10月30日に満期になる。1株当たり転換可能な優先株は約666.67株のGyre普通株に変換でき、逆株分割によって調整することができる。発行された優先株式証は独立した金融商品とみなされ、負債に分類される。
財務運営の概要
2023年12月31日までの年間で、純損失は8,550万ドル、普通株主は純損失9,290万ドルを占めるべきだ。2022年12月31日までの1年間の純収入は430万ドル、普通株主の純収入は230万ドルだった。2023年12月31日まで、私たちは累計8,550万ドル、現金および現金等価物3,350万ドルを赤字しました。2022年12月31日まで、私たちの利益剰余金は740万ドル、現金は2520万ドルです。
経営成果の構成部分
収入.収入
医薬製品の販売状況
私たちは主に中国でETUARYとある模造薬を販売することで収入を生み出している。ディーラーは私たちの直接顧客で、ディーラーに対する販売はETUARY収入の100%を占めている。これらの流通業者は、病院や他の医療機関、薬局を含むETUARYをあるサイトに販売している。
知的財産権許可
使用権を許可する制御権が顧客に譲渡されると,許可知的財産権の収入が確認される.
運営費
収入コスト
収入コストには主に販売コストが含まれており、製品が販売可能な状態に達しても生じる直接的·間接的なコストが含まれる。販売コストには、主に、(1)原材料コスト、(2)生産従業員の人的コスト、(3)生産に使用される財産や設備および無形資産に関する減価償却および償却、(4)税金および付加価値、(5)輸送コスト、(6)その他の雑コストがある。
販売とマーケティング費用
販売と市場普及支出は主に中国で私たちの製品ETUARYを販売と普及することに関連し、学術会議、シンポジウム及び座談会を主催して発生した支出を含む;病院でETUARYを使用することに関連する市場教育に関する普及支出;及び従業員コストは主に内部市場の普及と人員の給与と福祉を含む。
研究と開発費
研究·開発コストは発生時に費用を計上する。研究および開発のための商品またはサービスの払戻不可能な前払いは、最初に繰延され、前払い資産および他の流動資産の形態で資本化される。そして、資本化金額は、関連貨物がサービスを交付または提供する際に支出されるか、または貨物またはサービスが交付されることが予期されなくなるまで支出される。
129
研究開発コストは主に著者らの候補製品の臨床前と臨床開発に関連するコストを含み、その中には給料と他の人員に関連する費用、実験室用品と試薬、臨床前研究と臨床試験の契約研究開発サービス、材料とコンサルティングコスト、並びに施設分配、減価償却とその他の管理費用が含まれている。
一般と行政費用
一般及び行政支出は、(I)会計、情報科学技術、法律、行政及びその他の内部サービス者の支出、(Ii)職能従業員に付与された株式報酬、(Iii)専門サービス料、主に法律及び会計サービス、及び(Iv)その他の雑支出を含む。
買収した知的財産権の研究開発
すでに知的財産権の研究開発支出を買収するとは、知的財産権の研究開発を買収するが、他の未来の用途がないことによるコストであり、このなどのコストは買収時に直ちに列支する。
資産剥離損失
GNI USAが寄付する前に、私たちのGyre PharmPharmticalsの56.0%の間接所有権権益を除いて、私たちのほとんどの資産の剥離は赤字を記録した。この費用は私たちの総合業務表と全面(赤字)収益に反映されている。この損失は取引による直接コストを差し引いて列記される.
財産と設備処分損失
アメリカ国民総収入寄付の前に、私たちは財産と設備の処分について損失を記録した。この費用は私たちの総合業務表と全面(赤字)収益に反映されている。この損失は売却財産や設備に関する損失を差し引いて列記される。
その他の収入,純額
利子収入,純額
利息収入には主に私たちの長期預金で稼いだ利息が含まれています。利息収入は実際の利息法を用いて計上して確認し、金融商品の期待使用年数或いは短い期間(例えば適用)に適用して金融資産の帳簿純値を正確に割引して未来の現金収入の比率を推定する。
その他の収入
他の収入は主に政府支出で構成されている。贈与を受けることが合理的に保証され、すべての付加条件が遵守される場合、政府贈与はその公正価値で確認されます。贈与金が支出項目に関係している場合は,その補償しようとしている費用支出期間中に,システムによって収入と確認される。資産に贈与された場合は、公允価値を繰延収益帳に記入し、資産に関する期待使用年数内に等額年次分割払いで損益を計上するか、資産の帳簿金額から控除し、減価償却費用を減額するように損益を計上する。
権証責任の公正価値変動
私募については、対象証券が制御できない事件が発生した場合や償還が可能であるため、株式証負債を承認する独立金融商品として分類された優先株式証を発行した。優先株式証は発行時に公正価値で入金され、各報告期間が終了した時に再計量しなければならず、公正価値のいかなる変動も著者らの経営報告書の中で他の(収入)支出であることを確認しなければならない。
130
その他の費用
その他の費用には、権益法投資の損失のような非営業コストが含まれている。
所得税支給
所得税の支出は主に現在の所得税の支出(主に中国から利益を出すGyre製薬業務からの)と繰延所得税の支出から構成され、繰延所得税の支出は主にアメリカの税収目的の研究開発税収相殺と純営業損失の繰延の臨時差額の確認のための繰延税金、アメリカの税収目的のために制御された外国会社からの収入、及び推定免税額を差し引いた固定資産と無形資産を含むとみなされる。
経営成果
次の表は、2023年12月31日と2022年12月31日までの年間業務結果(変化率を除く千計)をまとめています
|
|
十二月三十一日までの年度 |
|
|
|
|
|
|
|
|||||||
|
|
2023 |
|
|
2022 |
|
|
(ドルを)変更する |
|
|
(%)変更 |
|
||||
収入.収入 |
|
$ |
113,450 |
|
|
$ |
102,290 |
|
|
$ |
11,160 |
|
|
|
10.9 |
% |
収入コスト |
|
|
4,636 |
|
|
|
4,793 |
|
|
|
(157 |
) |
|
|
(3.3 |
)% |
毛利 |
|
|
108,814 |
|
|
|
97,497 |
|
|
|
11,317 |
|
|
|
11.6 |
% |
収入コストは含まれていません |
|
|
|
|
|
|
|
|
|
|
|
|
||||
販売とマーケティング |
|
|
61,159 |
|
|
|
54,238 |
|
|
|
6,921 |
|
|
|
12.8 |
% |
研究開発 |
|
|
13,780 |
|
|
|
16,686 |
|
|
|
(2,906 |
) |
|
|
(17.4 |
)% |
一般と行政 |
|
|
14,662 |
|
|
|
17,370 |
|
|
|
(2,708 |
) |
|
|
(15.6 |
)% |
現在行われている研究と開発を買収する |
|
|
83,104 |
|
|
|
— |
|
|
|
83,104 |
|
|
** |
|
|
資産剥離損失 |
|
|
2,711 |
|
|
|
— |
|
|
|
2,711 |
|
|
** |
|
|
財産と設備処分損失 |
|
|
628 |
|
|
|
— |
|
|
|
628 |
|
|
** |
|
|
収入コストを含まない総運営費 |
|
|
176,044 |
|
|
|
88,294 |
|
|
|
87,750 |
|
|
|
99.4 |
% |
営業収入(赤字) |
|
|
(67,230 |
) |
|
|
9,203 |
|
|
|
(76,433 |
) |
|
** |
|
|
その他の収入(費用)、純額: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
利子収入,純額 |
|
|
1,044 |
|
|
|
726 |
|
|
|
318 |
|
|
|
43.8 |
% |
その他の収入 |
|
|
1,076 |
|
|
|
857 |
|
|
|
219 |
|
|
|
25.6 |
% |
株式証負債の公正価値変動を認める |
|
|
(9,261 |
) |
|
|
— |
|
|
|
(9,261 |
) |
|
** |
|
|
その他の費用 |
|
|
(2,594 |
) |
|
|
(1,374 |
) |
|
|
(1,220 |
) |
|
|
88.8 |
% |
所得税前収入 |
|
|
(76,965 |
) |
|
|
9,412 |
|
|
|
(86,377 |
) |
|
** |
|
|
所得税支給 |
|
|
(8,515 |
) |
|
|
(5,098 |
) |
|
|
(3,417 |
) |
|
|
67.0 |
% |
営業純収入 |
|
|
(85,480 |
) |
|
|
4,314 |
|
|
|
(89,794 |
) |
|
** |
|
|
非持株権の純収入に起因することができます |
|
|
7,453 |
|
|
|
2,012 |
|
|
|
5,441 |
|
|
|
270.4 |
% |
普通株主は純収入を占めなければならない |
|
$ |
(92,933 |
) |
|
$ |
2,302 |
|
|
$ |
(95,235 |
) |
|
** |
|
|
**意味がありません。
2023年12月31日までと2022年12月31日までの年度比較
収入.収入
2023年12月31日と2022年12月31日までの年間収入はそれぞれ1億135億ドルと1.023億ドルだった。上げ幅 主に中国地区の市場普及や販売活動の強化による医薬製品の売上高は1,260万ドル増加したが、この地域の2022年の売上高は低かったが、当社が2021年に締結したライセンス契約が2022年に確認した一次許可料に関する収入減少140万ドル分は上記の増加を相殺した。
131
収入コスト
2023年12月31日と2022年12月31日までの年間収入コストはそれぞれ460万ドルと480万ドル。T.T20万ドルを減らすのは主に有利な為替変動と20万ドル株ベースの報酬支出を減らす。売上高増加により収入コストが30万ドル増加し、これらの減少を部分的に相殺した。株式に基づく報酬の変化は2022年の完全付与オプションと2023年のオプション条項の修正と関係がある.
販売とマーケティング費用
2022年12月31日までの年度と比較して、2023年12月31日までの年間販売とマーケティング費用は690万ドル増加し、12.8%増となった。この増加は、主に従業員数の増加による販売とマーケティングの賃金コストが380万ドル増加し、販売促進費用が350万ドル増加し、販売人員出張費用が70万ドル増加したが、株式給与が130万ドル減少したことによって部分的に相殺された。株式に基づく報酬の変化は2022年の完全付与オプションと2023年のオプション条項の修正と関係がある.
研究と開発費
次の表は、2023年12月31日と2022年12月31日までの研究開発コスト(単位千、百分率変化を除く)を詳細に示している
|
十二月三十一日までの年度 |
|
|
|
|
|
||||||
|
2023 |
|
2022 |
|
(ドルを)変更する |
|
(%)変更 |
|
||||
直接計画支出: |
|
|
|
|
|
|
|
|
||||
臨床試験 |
$ |
4,228 |
|
$ |
4,132 |
|
$ |
96 |
|
|
2.3 |
% |
材料と公共事業 |
|
2,607 |
|
|
2,877 |
|
|
(270 |
) |
|
(9.4 |
)% |
臨床前研究 |
|
2,038 |
|
|
3,236 |
|
|
(1,198 |
) |
|
(37.0 |
)% |
間接費用: |
|
|
|
|
|
|
|
|
||||
株式ベースの報酬を含む人員関連のコスト |
|
4,092 |
|
|
5,716 |
|
|
(1,624 |
) |
|
(28.4 |
)% |
施設、減価償却その他 |
|
815 |
|
|
725 |
|
|
90 |
|
|
12.4 |
% |
研究開発費総額 |
$ |
13,780 |
|
$ |
16,686 |
|
$ |
(2,906 |
) |
|
(17.4 |
)% |
2023年12月31日までの1年間、研究·開発費は2022年12月31日までの年間290万ドル、または17.4%減少した。減少の主な原因は株式給与が180万ドル減少したことだ2022年の完全付与オプションと2023年のオプション条項の改正に関連している120万ドルの前臨床研究費が減少しました後者はいくつかの研究開発プロジェクトが2023年に臨床試験段階に入るか、あるいは応用段階に達した結果である。従業員数の増加により、研究·開発賃金コストは20万ドル増加し、この全体的な低下を部分的に相殺した。
一般と行政費用
2023年12月31日までの1年間は、2022年12月31日までの年度と比較して、一般·行政費が270万ドル、または15.6%減少した。減少の主な原因は株ベースの報酬が280万ドル減少したことだ。株式に基づく報酬の変化は2022年の完全オプション付与、2023年のオプション条項の改正、およびアメリカ国民総収入の入金後に新たなオプション贈与を提供することと関係があります.
買収した知的財産権の研究開発
2023年12月31日までの年間買収済み知的財産権研究開発費は8,310万ドルであり、知的財産権研究開発を買収するために発生したコストであり、このような支出はGNI USA寄付金が決済された時に直ちに支出される。
132
資産剥離損失
2023年12月31日までの年間で、Gyre PharmPharmticalsの56.0%の間接所有権権益を除いて、ほとんどの資産を剥離したことで270万ドルの損失を記録しました。この損失は取引による直接コストを差し引いて列記される.
財産と設備処分損失
2023年12月31日までの1年間に、60万ドルの財産と設備処分損失を記録した。この損失は売却財産や設備に関する損失を差し引いて列記される。
その他の収入,純額
2023年12月31日までの年間の利息収入は、2022年12月31日までの年間より30万ドル増加し、43.8%と増加しているが、これは主に長期預金への追加投資によるものである。
2022年12月31日までの年度と比較して,2023年12月31日までの年度別収入は20万ドル増加し,25.6%増加した。その他の収入増加は,主に2023年12月31日までに年度確認された中国政府補助金収入が1,000,000ドルであったのに対し,2022年12月31日までに年度確認された中国政府補助金収入は9,000,000ドルであったためである。
2023年12月31日までの1年間に、株式証負債の公正価値の変化は930万ドルであり、優先株式証負債の再計量と関係がある。2022年12月31日までの年度内に、株式を完済していない証はない。
2022年12月31日までの年度と比較して、2023年12月31日までの年度別支出は120万ドル増加し、88.8%増加した。この増加は主に権益法投資の損失が70万ドル増加したことと、現地慈善活動への賛助が50万ドル増加したことによるものだ。
所得税支給
所得税の課税額は、2023年12月31日と2022年12月31日までの年間それぞれ850万ドルと510万ドル。この増加は主にGyre製薬業務の利益増加と繰延税金資産の増加によるものである。
流動性と資本資源
流動資金源
2023年12月31日現在、私たちは3350万ドルの現金と現金等価物、2340万ドルの長期預金を持っており、運営に資金を提供することができ、累計赤字は8550万ドル。2023年12月31日までの年間純損失は8,550万ドルであるのに対し,運営活動が提供する現金は2,590万ドルである。私たちは、私たちの既存の現金と現金等価物、キャッシュフローを運営し、資本市場に入る機会は、本年度報告書の提出日から少なくとも12ヶ月以内に私たちの運営活動と債務に資金を提供するのに十分だと信じている。
将来の資金需要
私たちは運営のキャッシュフローを使用して、私たちの運営に資金を提供し、資本支出を含む、私たちの現在と未来の財務義務を履行する予定だ。私たちがこれらのお金を支払う能力は私たちの未来の表現にかかっていて、未来の表現は金融、商業、経済、規制、その他の要素の影響を受け、その多くの要素は私たちがコントロールできない。資金調達需要に影響を与える可能性のある要因には、これらに限定されない
133
将来の資本需要はまた、追加の補充業務、製品、技術にどの程度買収または投資するかにかかっている。
次の表は、列期間のキャッシュフロー(千単位)をまとめたものである
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
キャッシュフローデータ: |
|
|
|
|
|
|
||
経営活動が提供する現金純額 |
|
$ |
25,892 |
|
|
$ |
10,677 |
|
投資活動のための現金純額 |
|
|
(19,760 |
) |
|
|
(13,814 |
) |
融資活動が提供する現金純額 |
|
|
2,500 |
|
|
|
— |
|
現金および現金等価物に及ぼす為替レート変動の影響 |
|
|
(298 |
) |
|
|
(1,891 |
) |
現金と現金等価物の純変化 |
|
$ |
8,334 |
|
|
$ |
(5,028 |
) |
経営活動のキャッシュフロー
2023年12月31日までに、経営活動が提供する現金は2,590万ドルであり、我々の純損失は8,550万ドルであることを反映しているが、1.039億ドルの非現金プロジェクトに相殺され、非現金プロジェクトは主に買収されたGNI USA供出に関する知的財産権研究開発8,310万ドル、株式証明負債の公平値変動に関する930万ドル、株による報酬730万ドル、資産剥離損失270万ドル、および未合併連結会社の株式損失130万ドルが相殺された。また、業務活動が提供する現金は、750万ドルの純業務資産と負債の変化を反映している。
2022年12月31日までの年度,経営活動が提供する現金は1,070万ドルであり,我々の純収入を反映した430万ドル,非現金項目は1420万ドルであり,主に1340万ドルの株式ベースの報酬に関係しているが,純資産や負債の変化による780万ドルの現金純流出部分によって相殺されている。
投資活動によるキャッシュフロー
2023年12月31日までの年度の投資活動に用いられた現金は1,980万ドルであり,その中には購入預金1,570万ドル,購入物件および設備850万ドルおよび支払権益法が100万ドル投資され,GNI USA供出に関連して取得された560万ドルの現金が一部相殺されている。
134
2022年12月31日までの年度の投資活動用現金は1,380万ドルで、購入預金750万ドル、財産購入と設備500万ドル、権益法投資のための現金130万ドルが含まれている。
融資活動によるキャッシュフロー
2023年12月31日までの年間で、融資活動が提供した現金は250万ドルであったが、これはGNI USA寄付後、私募で転換可能な優先株と優先株証の収益分を発行したためである。
2022年12月31日までの年間で、融資活動のための現金は提供されていない。
契約義務その他の約束
賃貸借証書
(1)カリフォルニア州サンディエゴで本社のためのレンタル手配を締結しました。このレンタル契約は2023年11月11日以降に開始される38番目の完全カレンダー月の最終日に満了し、(2)中国のオフィスと実験室空間は2024年8月に満了します。2023年12月31日まで、私たちの固定賃貸支払い義務は40万ドルで、その中の20万ドルは12ヶ月以内に支払うべきです。
その他の契約義務と約束
2021年6月に、著者らは独立第三者南京保健薬業科学技術有限会社(“南京保健”)と譲渡協定を締結し、これにより、南京保健は慢性閉塞性肺疾患関連血小板減少症を治療するマレイン酸アバトーベ錠及びすべての関連技術を吾らに譲渡することに同意し、そして任意の研究或いは試験を完成し、そして著者らにCDE上場許可に必要なすべての材料を譲渡申請した。譲渡が完了した後、私たちは国家薬監局の許可を得て、マレイン酸アトモポ錠剤のマーケティング許可保持者になる予定だ。代わりにいくつかのマイルストーンに基づいて例えば:生物学的同等性の研究を完了したり,CDEに登録申請を行ったりする).我々は2022年8月1日に生物学的同等性研究を完成し、CDE検収を獲得し、2023年12月15日までに総額約180万ドルを支払った。
2022年9月、吾らは独立第三者新済源(北京)医薬科学技術有限会社(“新済源”)と譲渡協定を締結し、合意に基づき、新済源は中重度ざ瘡を治療する塩酸ミノサイクリン泡沫剤及びすべての関連技術を吾に譲渡することに同意し、製品開発を完成し、そして吾等への譲渡申請CDE上場許可に必要なすべての材料を申請した。譲渡完了後,国家薬監局の承認を得て,塩酸ミノサイクリン発泡スチロールの販売許可保持者となる予定である。交換として合計100万ドルを支払いますいくつかのマイルストーンに基づいて例えば:生物学的同等性の研究を完了したり,CDEに登録申請を行ったりする).工芸検証は既に完了した.2023年12月31日まで、私たちは全部で約70万ドルを支払った。
2022年12月、著者らは杭州百成薬業科学技術有限会社と独立第三者浙江CDMO薬業有限会社と譲渡協定を締結し、これにより百成は気道粘液の濃すぎるアセチルシステイン注射剤及びすべての関連技術を私たちに譲渡することに同意し、いかなる研究、試験などの必要なプログラムを完成することに協力し、そして私たちにCDE上場の承認を申請するために必要なすべての材料を譲渡することに同意した。この譲渡協定が完了した後、国家薬監局の承認を得て、アセチルシステイン注射剤のマーケティング許可保持者となることが予想される。私たちは2024年1月8日にCDEの検収を受けた。私たちは2023年12月31日までに、この合意に基づいて合計約50万ドルを支払った。CDEの最終承認を受けた後、私たちは40,000ドルを追加的に支払うつもりだ。
135
研究と開発計画
2023年12月31日までに、今後の様々なプロジェクトの研究開発活動に1270万ドルを支出することを約束しました。
財産と設備
2023年12月31日現在、私たちは購入と契約していますが、連結財務諸表に反映されていない財産や設備に関する引受金は280万ドルで、1年以内に発生する予定です。
重要な会計政策と試算
アメリカ公認会計原則に基づいて連結財務諸表と関連開示を作成し、私たちの財務状況と経営結果の討論と分析を行い、私たちの経営層に総合財務諸表と付記中の報告の金額に影響を与える判断、仮説と推定を行うことを要求する。我々が総合財務諸表を作成する際に採用した主な会計政策と方法は付記2-に記載されている重要会計政策の概要本年度報告シート10-K第2部第8項に記載されている連結財務諸表。経営陣は過去の経験や当時の状況で合理的と考えられた他の様々な仮定に基づいて推定し,これらの仮定の結果は資産や負債額面を判断する基礎となっている。実際の結果はこれらの推定とは異なる可能性があり、このような違いは実質的である可能性がある。
経営陣は、経営陣の判断と見積もりのより重要な分野に関連しているため、以下で議論する重要な会計政策と見積もりは、その歴史と将来の業績を知るために重要であると信じている。
収入確認
我々は、ASC主題606(“ASC 606”)“顧客との契約の収入”に従って収入を記録し、すなわち、顧客が約束された商品またはサービスの制御権を取得した場合、収入は、これらの商品またはサービスと交換するために予期される対価格を反映していることが確認される。1つの5ステップモデルは、(1)顧客契約の決定、(2)契約の履行義務の決定、(3)取引価格の決定、(4)取引価格を義務履行に割り当てる、(5)義務履行時に収入を確認する、というコア原則を実現するために使用される。私たちが顧客に転送する商品やサービスと交換するために、私たちが獲得する権利のある価格を受け取る可能性が高い時、私たちは5段階モデルを契約に適用します。
(A)薬剤製品の販売
医薬製品を販売して得られた収入は資産制御権が顧客に移行する際に確認され、一般的に顧客が薬品交付や品質検査を完了した時に確認される。医薬製品の販売については、私たちの顧客の多くは流通業者です。
製品販売収入は予想販売割引を差し引いて確認しました。これらの割引は、数ヶ月前の販売量と、各契約に規定されている割引率を適用して推定されます。
いくつかの医薬品販売契約には、製品の制御権および権利を顧客に移転した後に生成されるいくつかの販売リベートが含まれており、これらのリベートは、顧客への支払いの対価格を引き起こす。税金還付の具体的な額は収入確認時に最終的に確定していないため、歴史的経験に基づいて収入から差し引かれる。顧客への対価格を見積もる際には,どの方法が顧客が獲得する権利のある対価格額をより良く予測するかに依存する期待値方法や可能な金額方法を用いる必要がある.私たちはこれらの推定に関する情報を定期的に検討し、それに応じて金額を調整する。
136
将来のバックオフが予想される可変対価格を推定するために、可能な金額法は、単一の数の閾値を有する契約に適用され、期待値方法は、複数の数の閾値を有する契約に適用される。可変対価金額を最も予測できる選択された方法は、主に契約に含まれる数量閾値によって駆動される。可変対価格の拘束性推定の要求を適用し、顧客の売掛金から予想される将来のリベートを差し引く。
(B)知的財産権許可
使用権を許可する制御権がクライアントに譲渡された場合,許可プロトコルの収入が確認される.可変対価格に関する不確実性がその後解決された場合、マイルストーン支払いは可変対価格形式であり、取引価格に計上される範囲は確認された累計収入が大きく逆転しない可能性が高い程度である。マイルストーン支払いを含む各スケジュールの開始時に、マイルストーンが実現される可能性が高いと考えられるかどうかを評価し、可能な金額法を用いて取引価格に含まれる金額を推定する。承認を受ける前に、規制部門の承認のようなマイルストーン支払いは、実現不可能だと考えられている。
研究と開発費用と計上プロジェクト
研究·開発費用が我々の研究·開発サービスに関連しており、他の将来の用途がない場合、研究·開発費用は発生した費用に計上される。
臨床前と臨床試験コストは著者らの研究開発費用の重要な構成部分である。我々は第三者と契約を締結した歴史があり,これらの第三者は我々の候補製品の持続開発において我々を代表して様々な臨床前や臨床試験活動を行っている。臨床前および臨床試験に関する費用は,第三者がそれぞれの期間に提供する実際のサービスの見積もりに基づいて積算されている。
内部者や外部サービス提供者と議論し,外部サービス提供者から直接取得したサービスの進捗や完了段階に関する情報と,そのようなサービスに支払う取り決め費用により,完了した作業量を報告すると予想される.我々は,提供されているが領収書が発行されていない見積りサービスに基づいて研究開発活動のコストを記録し,これらのコストを総合貸借対照表中の計上費用と他の流動負債に計上し,これらのコストを総合経営報告書と全面収益(損失)における研究開発費に計上する.私たちは各報告期間の計算すべき残高を決定する時に重要な判断と推定をする。実際のコストの理解に伴い、私たちは計算計算量を調整した。
実際に発生した金額と実質的に差がないと予想されているにもかかわらず、提供されたサービスの実際の状態および時間に対する提供サービスの状態および時間の理解が異なる可能性があり、任意の特定の時期に私たちが報告する費用が高すぎるか、または低すぎる可能性がある。今まで、私たちは以前に推定された研究と開発費用について何の重大な調整もしていません。
所得税
負債法を用いて所得税を記録し、この方法では、繰延税項資産及び負債は、資産及び負債の財務報告と納税基礎との差に基づいて決定され、公布された税率と、差異予想が逆転した場合に発効する法律とを用いて計量される。一部または全部の税収割引が実現できない可能性があると判断された場合、推定値控除は、純営業損失と税収控除を含む繰延税金資産に計上される。
我々は、財務会計基準委員会(FASB)会計基準編纂(ASC)740の規定に基づいて、不確定な税収状況を会計処理する所得税それは.不確定な税収頭寸が存在する場合、税収頭寸の税収割引を確認し、税務機関が審査を行えば、その優遇が実現される可能性が高い。税収優遇がより実現可能かどうかの決定は,税収状況に基づく技術的利点および既存の事実や状況への考慮である。
137
未確認の税収優遇に関する利息及び罰金は所得税費用の構成要素として記録される(あれば)。
株に基づく報酬
私たちは、付与日奨励の公正価値に基づいて、従業員、非従業員、および取締役が持分ツールの奨励と交換するために受け取ったサービスのコストを計量し、従業員、非従業員または取締役が報酬の直線期間の関連費用と交換するためにサービスを提供する必要があることを確認した。業績条件を満たす可能性があると判断されると、業績条件を含む株式奨励の推定公正価値は奨励期間内に支出される。
付与日に株式奨励の公正価値を決定するには判断が必要である。私たちはブラック-スコアーズオプション価格モデルを使用して株式オプションの公正価値を決定する。オプション定価モデルを使用して、付与日オプションの公正価値を決定することは、私たちの普通株式の公正価値、オプション期待寿命内の期待普通株式価格変動性、株式オプションの期待期限、無リスク金利、および期待配当を含むいくつかの変数に対する私たちの仮定の影響を受ける。私たちは株ベースの報酬を没収された奨励金を差し引いた補償費用として記録する。私たちは没収が発生した時にそれを説明することを選択した。そこで、個別贈与の帰属条項に基づいて、その必要なサービス期間内に株式に基づく補償費用を確認する。このような仮定は固有の不確実性と重大な判断の適用に関するものだ。したがって、要因や予想結果が変化し、私たちが使用する仮説や推定が大きく異なる場合、私たちの株式ベースの報酬支出は大きく異なる可能性がある。私たちは、株式に基づく報酬費用計算のための予想に基づく仮定を評価するために判断を使用し続ける。付記12-を参照株に基づく報酬より多くの情報を理解する必要がある場合は、本年度報告第2部第8項のタブ10-Kの連結財務諸表を参照してください。
株式証法的責任
私募については、優先株式証を発行しました(付記3参照)公正価値計量と金融商品統合財務諸表は、本年度報告第II部第8項表格10-K)に記載されており、このような独立金融商品は、対象証券が吾等の制御可能なイベントが発生した場合や償還がある場合に償還されることができるため、株式証負債として分類される。優先株式証は発行時に公正価値で入金され、各報告期末に再計量しなければならない。私たちはブラック-スコアーズオプション定価モデルを使用して優先株式証の公正価値を決定し、これらの公正価値は私たちの多くの変数に対する仮定の影響を受け、これらの変数は私たちの転換可能な優先株の公正価値、オプション期待寿命内の期待株価変動、引受権証の期待期限、無リスク金利と期待配当金を含む。このような仮定は固有の不確実性と重大な判断の適用に関するものだ。したがって、要素や期待結果が変化し、私たちが使用する仮定や推定が大きく異なる場合、私たちの権証負債に対する推定公正価値は大きく異なるかもしれない。私たちは引き続き判断を使用して、私たちが将来に基づいて株式証明負債の公正価値を計算するために使用する仮説を評価します。
比較的小さな報告会社の状態
“取引法”の定義によると、私たちは“小さな報告会社”です。私たちは、規模の小さい報告会社が得ることができるいくつかの比例して開示された情報を利用して、私たちの非関連会社が保有する普通株が私たちの第2四半期の最後の営業日に2.5億ドルを下回るか、または最近終了した会計年度の年収が1.00億ドル未満であり、非関連会社が保有する私たちの普通株が私たちの第2四半期の最後の営業日に7.00億ドルを下回る限り、これらの比例開示された情報を利用することができるかもしれない。
最近の会計公告
注2-を参照重要会計政策の概要最近の会計声明、これらの声明を採用するタイミング、およびこれらの声明が我々の財務状況および経営成果に及ぼす潜在的な影響の評価(すでに評価していれば)を検討するために、本年度報告の第II部は、Form 10−K形式の第8項で連結財務諸表を評価した。
138
第七A項。定量と定性私は市場リスクを開示した。
市場リスクの定量的·定性的開示について
市場リスクとは、金融市場の価格や金利の不利な変化により、我々の財務状況に影響を及ぼす可能性のある損失リスクである。私たちの市場リスクの開放は主に為替レート、金利あるいはインフレの潜在的な変化による開放です。
外貨両替リスク
私たちの費用は通常私たちが業務を展開している司法管轄区の貨幣で計算されます。これらの管轄区は主にアメリカと中国にあります。そのため、私たちと将来の業務やキャッシュフローの結果は外貨為替レートの変化の影響を受けることになります。ドルの他の通貨に対する相対価値が10%上昇または低下すると仮定すると、我々の経営業績に実質的な影響を与えない。外貨為替レートは私たちの歴史経営業績に大きな影響がないため、デリバティブやヘッジ取引を行っていませんが、外貨への開放がもっと大きくなれば、未来にそうするかもしれません。
インフレリスク
全体的な経済への影響を除いて、インフレが私たちの業務、財務状況、あるいは経営結果に実質的な影響を与えているとは思いません。それにもかかわらず、もし私たちのコストがインフレ圧力の影響を受けたら、私たちは価格上昇によってこれらのより高いコストを完全に相殺することができないかもしれない。私たちはこれをできないかできないかは私たちの業務、財務状況、そして運営結果を損なうかもしれない。
139
プロジェクト8.財務統計員TSと補足データ
この表の10-K第F-1ページの連結財務諸表および付表インデックスを参照してください。
140
第9項.会計·財務開示面の変更と会計士との相違
ない。
第9条。制御とプログラムです
開示制御とプログラムの評価
我々の経営陣は、2023年12月31日現在、最高経営責任者と最高財務責任者の参加と監督の下で、我々の開示制御および手順を評価している(“取引法”規則13 a-15(E)および15 d-15(E)で定義されている)。取引法規則13 a-15(E)および15 d-15(E)に定義されている用語“開示制御および手順”は、取引法に従って提出または提出された報告において開示を要求する会社の情報が米国証券取引委員会規則および表によって指定された期間にわたって記録、処理、集約および報告されることを保証するための会社の制御および他のプログラムを意味する。開示制御および手続は、開示要求に関する決定をタイムリーに行うために、会社が取引所法案に基づいて提出または提出された報告書に開示を要求する情報が蓄積され、会社管理層(その主要幹部および主要財務官を含む)に伝達されることを保証するための制御および手順を含むが、これらに限定されない。経営陣は、どのような制御やプログラムも、どんなに設計や操作が良好であっても、その目標を実現するために合理的な保証を提供することしかできず、管理層は、可能な制御とプログラムの費用対効果関係を評価する際にその判断を運用しなければならないことを認識している。この評価に基づいて、我々の最高経営責任者およびCEOは、我々の開示制御および手続きが2023年12月31日から有効であり、合理的な保証を提供するために、取引所法案に提出または提出された報告書に基づいて、米国証券取引委員会規則および表が指定された期間内に記録、処理、まとめ、報告されることを要求し、これらの情報が蓄積され、我々の最高経営者および最高財務官を含めて、必要な開示に関する決定をタイムリーに行うために、我々の管理職に伝達されると結論した。
財務報告の内部統制の変化
2023年12月31日までの四半期内に、財務報告の内部統制(“外国為替法案”第13 a-15(F)および15 d-15(F)規則の定義に基づく)に大きな影響を与えなかったか、または合理的に財務報告の内部統制に大きな影響を与える可能性のある変化はなかった。
経営陣財務報告内部統制年次報告書
我々の経営陣は、財務報告書の十分な内部統制の確立と維持を担当している(取引法第13 a-15(F)条に記載されている)。財務報告内部統制は、米国公認の会計原則に基づいて財務報告の信頼性及び外部目的の財務諸表作成のための合理的な保証を提供するために、我々の経営陣(我々の最高経営者及び最高財務官を含む)の監督及び参加の下で設計されたプログラムである。
我々の経営陣は、2023年12月31日現在、テレデビル委員会後援組織委員会が内部統制-総合的な枠組みで提案した基準を使用して、財務報告書の内部統制に対する我々の有効性を評価している。この評価に基づき、我々の経営陣は、財務報告書の内部統制が2023年12月31日から有効であると結論した。
プロジェクト9 B。その他の入力隊形。
貿易手配
開ける
141
はい取引の手配に応じて全株式を売却する。2023年12月31日までの3ヶ月以内に、他の役員又は会社幹部はいない(取引法第16 a-1(F)条参照)
プロジェクト9 Cです。検査を阻害する外国司法管轄区域の開示に関する。
適用されません。
142
パー?パーT III
プロジェクト10.役員·役員休会氷と会社が管理します。
本第10項で要求される情報は、2024年株主総会のために提出された依頼書(“2024年依頼書”)の情報を参考にして格納されており、この依頼書は、“コーポレート·ガバナンス”というタイトルの下、S-K法規第405項に基づいて提出された“延滞第16(A)条報告”を含む2023年12月31日までの財政年度終了後120日以内に米国証券取引委員会に提出されると予想される。
我々は、我々のCEO、最高財務官、最高会計官または財務総監、または同様の機能を実行する者を含む、我々の役員、上級管理者、および従業員に適用される書面商業行為および道徳基準を採択した。このコードのコピーは、当社のウェブサイトwww.gyretx.comの“コーポレート·ガバナンス”で取得することができます。私たちは、改正または免除の日から4営業日以内に開示されなければならない商業行為および道徳基準の任意の改正または免除を私たちのウェブサイトで開示する予定です。
第11項.実行五、補償する。
本第11項に要求される情報は、タイトルの下に含まれる2024年の依頼書の情報を参照することによって本明細書に組み込まれる“役員報酬”そして“会社の管理”.
プロジェクト12.Certaの保証所有権すべての人たちと管理職と関連した株主について。
本第12条に要求される情報は、“いくつかの利益所有者および管理層の保証所有権”および“持分補償計画に従って発行された証券”というタイトルを含む、2024年の委託書の情報を参照して本明細書に組み込まれる
本第13項で要求される情報は、タイトルの下に含まれる2024年の依頼書の情報を参照することによって本明細書に組み込まれる“会社の管理”そして“特定の関係は関係者と取引する。”
第14項:元本口座NTANT料金とサービスです。
本プロジェクト14に要求された資料は、“独立監査員任命の承認”というタイトルの下に含まれている2024年の委託書に組み込まれている
143
部分IV.IV
プロジェクト15.展示と国際水泳連盟NCIAL文スケジュール。
連結財務諸表索引を参照して、本報告第2部第8項財務諸表と補足データを参照する。
すべての付表は,適用されない,あるいは必要な資料が第8項の下にあるため省略されている.本年度報告書“財務諸表及び補足資料”の表格10−K。
144
第十六項。
表10-Kの概要。
ない。
145
リスト.リスト展示品
|
|
|
|
引用で編入する |
|
||||||
展示品 違います。 |
|
展示品名 |
|
表 |
|
書類番号. |
|
展示品 違います。 |
|
提出日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
2.1(a)# |
|
資産購入協定は,2022年12月26日にCatalyst Biosciences,Inc.,GNI Group Ltd.,GNI Hong Kong Limitedによって署名された。 |
|
8-K |
|
000-51173 |
|
2.1 |
|
2022年12月27日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
2.1(b) |
|
Catalyst Biosciences,Inc.,GNI Group Ltd.とGNI Hong Kong Limited間の合意と資産購入プロトコル修正案は,2023年3月29日である. |
|
8-K |
|
000-51173 |
|
2.2 |
|
2023年3月30日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
2.2(a)# |
|
2022年12月26日までに、Catalyst Biosciences,Inc.,GNI USA,Inc.,GNI Group Ltd.,GNI Hong Kong Limited,上海ゲノム学有限会社,添付ファイルAに列挙された個人と大陸製薬会社が調印した業務合併協定。 |
|
8-K |
|
000-51173 |
|
2.2 |
|
2022年12月27日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
2.2(b) |
|
Catalyst生物科学会社、GNI米国社、GNIグループ有限会社、GNI香港有限会社、上海ゲノム会社、少数株主と大陸製薬会社との間の業務合併協定改正案は、2023年3月29日となっている。 |
|
8-K |
|
000-51173 |
|
2.1 |
|
2023年3月30日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
2.2(c) |
|
2023年8月30日現在、Catalyst Biosciences,Inc.,GNI USA,Inc.,GNI Group Ltd.,GNI Hong Kong Limited,Shanghai Genome,Inc.とContinent PharmPharmticals Inc.によって署名された業務統合協定第2修正案がある。 |
|
8-K |
|
000-51173 |
|
2.1 |
|
2023年8月31日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
3.1 |
|
第四に、発行された“会社登録証明書”を修正、再発行する。 |
|
S-8 |
|
333-133881 |
|
4.1 |
|
2006年5月8日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
3.2 |
|
第四条改正された会社登録証明書。 |
|
8-K |
|
000-51173 |
|
3.1 |
|
2015年8月20日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
3.3 |
|
4番目の改訂された会社登録証明書の2回目の改訂証明書。 |
|
8-K |
|
000-51173 |
|
3.1 |
|
2017年2月10日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
3.4 |
|
4番目の改訂された会社登録証明書の3回目の改訂証明書。 |
|
8-K |
|
000-51173 |
|
3.1 |
|
2023年10月30日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
3.5 |
|
定款を改訂して再制定する。 |
|
8-K |
|
000-51173 |
|
3.3 |
|
2023年10月30日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
3.6(a) |
|
Aシリーズ転換可能な優先株の指定優先株、権利、制限証明書は、2017年4月10日にデラウェア州国務長官に提出された。 |
|
8-K |
|
000-51173 |
|
3.1 |
|
2017年4月13日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
146
|
|
|
|
引用で編入する |
|
||||||
展示品 違います。 |
|
展示品名 |
|
表 |
|
書類番号. |
|
展示品 違います。 |
|
提出日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
3.6(b)* |
|
Aシリーズ優先株抹消証明書は、2024年3月25日にデラウェア州国務長官に提出された。 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3.7(a) |
|
Xシリーズ転換可能な優先株の指定優先株、権利、制限証明書は、2022年12月27日にデラウェア州国務長官に提出された。 |
|
8-K |
|
000-51173 |
|
3.1 |
|
2022年12月27日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
3.7(b) |
|
2023年10月30日にデラウェア州国務長官に提出されたXシリーズ転換可能優先株指定優先株、権利、制限証明書改正案。 |
|
8-K |
|
000-51173 |
|
3.2 |
|
2023年10月30日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
3.8(a) |
|
Y系列優先株の優先株、権利、制限指定証明書は、2023年6月20日にデラウェア州国務長官に提出され、Yシリーズ優先株に関連している。 |
|
8-K |
|
000-51173 |
|
3.1 |
|
2023年6月20日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
3.8(b) |
|
Yシリーズ優先株抹消証明書は、2023年8月31日にデラウェア州国務長官に提出された。 |
|
8-K |
|
000-51173 |
|
3.1 |
|
2023年8月31日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
4.1 |
|
証券説明。 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.2 |
|
2005年3月3日,シリコンバレー銀行にCatalyst Biosciences,Inc.の株式購入権証を発行した。 |
|
10-K |
|
000-51173 |
|
4.3 |
|
2016年3月9日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
4.3 |
|
Catalyst Biosciences,Inc.株を購入する引受権証は,転換可能な本チケットの購入者に発行される. |
|
10-K |
|
000-51173 |
|
4.5 |
|
2016年3月9日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
4.4 |
|
Xシリーズを購入すると優先株の引受権証形式に転換することができる。 |
|
8-K |
|
000-51173 |
|
4.1 |
|
2023年10月30日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.1(a)# |
|
または2022年12月26日にCatalyst Biosciences,Inc.および米国株譲渡によって信託会社LLCによって署名される価値のある権利協定。 |
|
S-3 |
|
333-273395 |
|
2.5 |
|
2023年7月24日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.2(b) |
|
Catalyst Biosciences,Inc.とAmerican Stock Transfer&Trust Company,LLCの間のまたは価値のある権利協定修正案は,2023年3月29日である。 |
|
8-K |
|
000-51173 |
|
2.3 |
|
2023年3月30日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.3# |
|
資産購入協定は、日付は2023年2月27日、Catalyst Biosciences,Inc.とGC Biopma Corpである。 |
|
8-K |
|
000-51173 |
|
10.1 |
|
2023年3月2日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.4 |
|
Catalyst Biosciences,Inc.とGNI USA,Inc.の間の証券購入プロトコルは,2023年10月27日である. |
|
8-K |
|
000-51173 |
|
10.1 |
|
2023年10月30日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.5+ |
|
改正および再署名された雇用協定は、2018年8月28日にCatalyst Biosciences,Inc.およびNassim Usman博士によって署名された. |
|
8-K |
|
000-51173 |
|
10.1 |
|
2018年8月31日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
147
|
|
|
|
引用で編入する |
|
||||||
展示品 違います。 |
|
展示品名 |
|
表 |
|
書類番号. |
|
展示品 違います。 |
|
提出日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.6+ |
|
Catalyst Biosciences,Inc.とNassim Usman博士の間の放棄合意は,2023年1月17日であった。 |
|
10-Q |
|
000-51173 |
|
10.2 |
|
2023年5月15日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.7+ |
|
Catalyst Biosciences,Inc.とDr.Grant Blouse間の放棄プロトコルは,2023年1月14日であった。 |
|
10-Q |
|
000-51173 |
|
10.3 |
|
2023年5月15日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.8+ |
|
Catalyst Biosciences,Inc.とSeline Millerさんの間の放棄合意は,2023年1月17日であった。 |
|
10-Q |
|
000-51173 |
|
10.4 |
|
2023年5月15日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.9+ |
|
Catalyst Biosciences,Inc.(前身はTargacept,Inc.)2015年株式インセンティブ計画(改正され再発効し、2016年6月9日から発効)。 |
|
定義14 A |
|
000-51173 |
|
付録A |
|
2016年4月25日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.10+ |
|
インセンティブ株式オプション奨励通知書フォーマット。 |
|
8-K |
|
000-51173 |
|
10.1 |
|
2017年7月14日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.11+ |
|
不合格株式オプション奨励通知書形式。 |
|
8-K |
|
000-51173 |
|
10.2 |
|
2017年7月14日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.12+ |
|
Catalyst Biosciences,Inc.2018年総合インセンティブ計画。 |
|
定義14 A |
|
000-51173 |
|
付録A |
|
2020年5月1日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.13+ |
|
Catalyst Biosciences,Inc.2018年従業員株式購入計画。 |
|
定義14 A |
|
000-51173 |
|
付録B |
|
2018年5月11日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.14+ |
|
株式オプションは合意形式を奨励する。 |
|
10-K |
|
000-51173 |
|
10.8 |
|
2022年3月31日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.15+ |
|
GYRE治療会社2023年総合インセンティブ計画 |
|
8-K |
|
000-51173 |
|
10.2 |
|
2023年10月30日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.16+ |
|
合意の形式を達成する |
|
8-K |
|
000-51173 |
|
10.3 |
|
2023年10月30日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.17+ |
|
雇用協定は,期日は2023年10月30日であり,会社とCharles Wuが署名した。 |
|
8-K |
|
000-51173 |
|
10.4 |
|
2023年10月30日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.18+ |
|
当社と陳若瑜との間の雇用協定は、2023年10月30日となっている。 |
|
8-K |
|
000-51173 |
|
10.5 |
|
2023年10月30日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.19+ |
|
別居協定は,期日は2023年10月30日であり,会社とナシム·ウスマン博士が署名した。 |
|
8-K |
|
000-51173 |
|
10.1 |
|
2023年11月2日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.20+ |
|
別居協定は、2023年10月30日に会社とセリーヌ·ミラーが署名した。 |
|
8-K |
|
000-51173 |
|
10.2 |
|
2023年11月2日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.21+ |
|
当社が韓英博士と締結した雇用契約は、2024年1月15日となっている。 |
|
8-K |
|
000-51173 |
|
10.1 |
|
2024年1月19日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
16.1 |
|
EisnerAmper LLCからアメリカ証券取引委員会への手紙は、期日は2023年11月2日である。 |
|
8-K |
|
000-51173 |
|
16.1 |
|
2023年11月2日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
21.1* |
|
当社の子会社リストです。 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
23.1* |
|
独立公認会計士事務所はすべて富会計士事務所が同意します。 |
|
|
|
|
|
|
|
|
|
24.1* |
|
授権書(本文書署名ページの一部とする). |
|
|
|
|
|
|
|
|
|
148
|
|
|
|
引用で編入する |
|
||||||
展示品 違います。 |
|
展示品名 |
|
表 |
|
書類番号. |
|
展示品 違います。 |
|
提出日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
31.1* |
|
2002年サバンズ-オキシリー法302節に基づいて可決された1934年“証券取引法”第13 a-14(A)及び15 d-14(A)条に基づいて発行された最高経営責任者証明書。 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
31.2* |
|
2002年サバンズ-オキシリー法302節に基づいて成立した1934年“証券取引法”第13 a-14(A)及び15 d-14(A)条に基づいて首席財務官の認証を行う。 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
32.1** |
|
取引法第13 a−14(B)条及び米国法第18編第1350条、及び2002年に“サバンズ·オックススリー法”第906条に基づいて可決された最高経営責任者の証明。 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
32.2** |
|
取引法第13 a-14条及び米国法第18編第1350条、及び2002年に“サバンズ·オックススリー法案”第906条に基づいて可決された首席財務官の証明。 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
97.1* |
|
会社の追跡政策 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.INS* |
|
XBRLインスタンスドキュメントを連結する |
|
|
|
|
|
|
|
|
|
101.Sch* |
|
イントラネットXBRL分類拡張アーキテクチャ文書 |
|
|
|
|
|
|
|
|
|
101.カール* |
|
インラインXBRL分類拡張計算リンクライブラリ文書 |
|
|
|
|
|
|
|
|
|
101.定義* |
|
インラインXBRL分類拡張Linkbase文書を定義する |
|
|
|
|
|
|
|
|
|
101.実験所* |
|
XBRL分類拡張ラベルLinkbase文書を連結する |
|
|
|
|
|
|
|
|
|
101.前期* |
|
インラインXBRL分類拡張プレゼンテーションLinkbaseドキュメント |
|
|
|
|
|
|
|
|
|
104 |
|
表紙相互データファイル(添付ファイル101に含まれるイントラネットXBRLのフォーマット) |
|
|
|
|
|
|
|
|
|
*アーカイブをお送りします。
**家具は用意されています。
第601(A)(5)項によれば、本展示品の添付ファイル、付表及びいくつかの展示品は省略されている。
#S-K法規第601(B)(10)項によれば、識別された秘密部分(I)は、当社が通常および実際に個人または機密と見なしているので、本展示品のいくつかの機密部分をスター番号で表記することによって省略され、(Ii)この情報は重要ではない。
+管理契約、補償計画、またはスケジュールを示します。
149
登録する解決策
1934年“証券取引法”第13又は15(D)節の要求に基づいて、登録者は、次の署名者が代表して本報告書に署名することを正式に許可した。
|
GYRE治療会社 |
||
|
|
||
日付:2024年3月27日 |
差出人: |
S/韓英、博士。 |
|
|
|
韓英、博士。 |
|
|
|
最高経営責任者 |
|
|
|
(首席行政主任) |
|
|
|
|
|
日付:2024年3月27日 |
差出人: |
投稿S/陳若瑜 |
|
|
|
|
陳若瑜 |
|
|
|
首席財務官 |
|
|
|
(首席財務会計官) |
権力があるのです弁護士
現在、韓英、博士及び陳若瑜をその真の合法的な事実受権者及び代理人に委任し、本人名義、本人名義及び以下に述べる各身分及び以下の各身分及び身分で署名し、本年度報告の任意及びすべての改訂を表格10-Kで提出し、本年度報告をすべての証拠物及びその他の関連文書とともに証券取引委員会に提出する。上記の事実代理人および代理人およびその各々に、物事の全ての権力および権力としてのすべての権利および権限を付与し、上記の事実代理人および代理人またはそれらのいずれかまたはそれらの代替者が、合法的にまたはそれに至るすべての行為および事柄を合法的に行うことができることを承認および確認する。
本報告は、1934年の証券取引法の要求に基づき、以下の者によって登録者として指定日に署名された。
サイン |
|
タイトル |
|
日取り |
|
|
|
|
|
S/韓英、博士。 |
|
取締役CEO兼最高経営責任者 |
|
2024年3月27日 |
韓英、博士。 |
|
|
|
|
|
|
|
|
|
投稿S/陳若瑜 |
|
首席財務官(首席財務·会計幹事) |
|
2024年3月27日 |
陳若瑜 |
|
|
|
|
|
|
|
|
|
/S/ローイン博士 |
|
取締役会議長 |
|
2024年3月27日 |
ロイン博士 |
|
|
|
|
|
|
|
|
|
/S/トーマス·イーストリング |
|
役員.取締役 |
|
2024年3月27日 |
トーマス·イーストリング |
|
|
|
|
|
|
|
|
|
/S/ゴードン·カーマイケル博士 |
|
役員.取締役 |
|
2024年3月27日 |
ゴードン·カーマイケル博士です |
|
|
|
|
|
|
|
|
|
投稿S/馬松江 |
|
総裁と役員 |
|
2024年3月27日 |
松江馬雲 |
|
|
|
|
|
|
|
|
|
/S/ロデニー·L·ヌスボム |
|
役員.取締役 |
|
2024年3月27日 |
ロデニー·L·ヌスボム |
|
|
|
|
|
|
|
|
|
/S/レナート·パリー博士 |
|
役員.取締役 |
|
2024年3月27日 |
レナート·パリー博士 |
|
|
|
|
|
|
|
|
|
150
/S/ナシム·ウズマン、博士 |
|
役員.取締役 |
|
2024年3月27日 |
ナシム·ウスマン博士です |
|
|
|
|
151
連結財務諸表索引
独立公認会計士事務所レポート(PCAOB ID# |
F-2 |
連結財務諸表 |
|
合併貸借対照表 |
F-4 |
連結経営報告書と全面的な収益 |
F-5 |
転換可能な優先株と株式合併報告書 |
F-6 |
統合現金フロー表 |
F-7 |
連結財務諸表付記 |
F-8 |
別表1-親会社の財務情報の概要 |
F-41 |
F-1
R独立公認会計士事務所報告
取締役会と株主
GYRE治療会社
財務諸表のいくつかの見方
Gyre Treateutics,Inc.(デラウェア州の1社)とその子会社(“当社”)2022年12月31日までの総合貸借対照表,2023年12月31日までの2年度の関連総合包括(赤字)収益表,転換可能な優先株と持分およびキャッシュフロー表,および15(A)項下の関連付記と財務諸表(総称して“財務諸表”と呼ぶ)を監査した。これらの財務諸表は,すべての重要な点で当社の2023年12月31日と2022年12月31日までの財務状況,および2023年12月31日までの2年度の経営結果とキャッシュフローを公平に反映しており,米国公認の会計原則に適合していると考えられる。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査することを求められておらず、私たちも招聘されて監査を行っていません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性について意見を述べるためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
以下に述べる重要な監査事項とは、財務諸表を当期監査する際に生じる、監査委員会に伝達または要求された事項であり、これらの事項は、(1)財務諸表に対して大きな意味を有する勘定または開示に関し、(2)私たちが特に挑戦的で主観的または複雑な判断に関するものである。重要監査事項の伝達は、財務諸表に対する私たちの全体的な意見をいかなる方法でも変えることはなく、次の重要な監査事項を伝達することによって、重要な監査事項又はそれに関連する勘定又は開示について個別の意見を提供することもない。
逆資産買収
財務諸表付記1と付記7にさらに記載されているように、会社は2023年10月30日にGNI USAの納付を完了した。会計目的については、GNI米国社の貢献は逆資産買収とされ、大陸製薬会社(“CPI”)は会計買収側とされている。私たちは逆資産買収が重要な監査事項だと思う。
(1)取引が資産買収であることの決定、(2)CPIが会計取得者であることの決定、および(3)を含む逆資産買収の主な考慮要因を決定する
F-2
関連する対価格の公正価値、特にブラック-スコアーズオプション定価モデルにおいて合併中に仮定された未償還株式オプションの公正価値を計算するために使用される予想変動率と期待期限仮定は、重要な監査事項であり、取引の複雑性と管理層の重大な判断であり、これは逆に監査師が実行手続きと監査証拠を評価する上で高度な判断と努力を招く。監査はまた専門的な技能と知識を持つ専門家の使用に関するものだ。
私たちの関連監査手続きには以下の内容が含まれている
優先株式証
財務諸表付記3及び11に記載されているように、当社は2023年10月に私募を完了し、転換可能優先株株式及び株式承認証を発行して転換可能優先株株式を購入する。株式承認証は負債ツールに分類され、各報告期間終了時に公正価値再計量が行われる。
権利証負債の公正価値は、Black-Scholesオプション定価モデルを使用して管理層によって推定される。同社の権証負債は、価格が観察されやすい活発な市場で取引されていないため、このモデルを使用して、権証負債の公正な価値を評価するために、多くの観察不可能な投入を含む。私たちは株式証明書負債が重要な監査事項だと確信する。
株式証負債を重要な監査事項とする主な考慮要素は、管理層が変動性と期待期限仮説を評価する際の重大な判断であり、これは逆に監査人がこれらの仮定に関連する監査証拠を実行し、評価する上で高度な判断と努力を招いた。監査はまた専門的な技能と知識を持つ専門家の使用に関するものだ。
私たちの関連監査手続きには以下の内容が含まれている
/S/ともに公認会計士事務所有限責任会社
2023年以来、私たちは会社の監査役を務めてきた。
2024年3月27日
F-3
GYRE治療会社
合併貸借対照表
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
|
2023年12月31日 |
|
|
2022年12月31日 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
売掛金と手形,純額 |
|
|
|
|
|
|
||
GNIのその他受取 |
|
|
|
|
|
— |
|
|
在庫、純額 |
|
|
|
|
|
|
||
前払い資産 |
|
|
|
|
|
|
||
その他流動資産 |
|
|
|
|
|
|
||
流動資産総額: |
|
|
|
|
|
|
||
財産と設備、純額 |
|
|
|
|
|
|
||
GCBPの長期売掛金 |
|
|
|
|
|
— |
|
|
無形資産、純額 |
|
|
|
|
|
|
||
使用権資産 |
|
|
|
|
|
|
||
土地使用権、純価値 |
|
|
|
|
|
|
||
繰延税金資産 |
|
|
|
|
|
|
||
長期預金証書 |
|
|
|
|
|
|
||
他の非流動資産 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
負債、転換可能な優先株、資本 |
|
|
|
|
|
|
||
流動負債: |
|
|
|
|
|
|
||
売掛金 |
|
$ |
|
|
$ |
|
||
収入を繰り越す |
|
|
|
|
|
|
||
関係者の都合で |
|
|
|
|
|
|
||
CVR期末超過現金対応 |
|
|
|
|
|
— |
|
|
費用とその他の流動負債を計算しなければならない |
|
|
|
|
|
|
||
所得税に対処する |
|
|
|
|
|
|
||
賃貸負債を経営し、流動 |
|
|
|
|
|
|
||
流動負債総額: |
|
|
|
|
|
|
||
非流動経営賃貸負債 |
|
|
|
|
|
|
||
繰延の政府支出 |
|
|
|
|
|
|
||
CVRは負債、非流動を誘導する |
|
|
|
|
|
— |
|
|
権証責任,非流動 |
|
|
|
|
|
— |
|
|
他の非流動負債 |
|
|
|
|
|
|
||
総負債 |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|||
転換可能優先株、$ |
|
|
|
|
|
— |
|
|
株本: |
|
|
|
|
|
|
||
普通株、$ |
|
|
|
|
|
|
||
追加実収資本 |
|
|
|
|
|
|
||
法定準備金 |
|
|
|
|
|
|
||
(累積損失)利益を残す |
|
|
( |
) |
|
|
|
|
その他の総合損失を累計する |
|
|
( |
) |
|
|
( |
) |
Gyre株主(損失)権益総額 |
|
|
( |
) |
|
|
|
|
非持株権益 |
|
|
|
|
|
|
||
総株 |
|
|
|
|
|
|
||
総負債、転換可能な優先株、資本 |
|
$ |
|
|
$ |
|
||
付記はこれらの連結財務諸表の構成要素である。
F-4
GYRE治療会社
合併状態経営プロジェクトと総合収入
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
収入.収入 |
|
$ |
|
|
$ |
|
||
運営費用: |
|
|
|
|
|
|
||
収入コスト |
|
|
|
|
|
|
||
販売とマーケティング |
|
|
|
|
|
|
||
研究開発 |
|
|
|
|
|
|
||
一般と行政 |
|
|
|
|
|
|
||
現在行われている研究と開発を買収する |
|
|
|
|
|
— |
|
|
資産剥離損失 |
|
|
|
|
|
— |
|
|
財産と設備処分損失 |
|
|
|
|
|
— |
|
|
総運営費 |
|
|
|
|
|
|
||
営業収入(赤字) |
|
|
( |
) |
|
|
|
|
その他の収入(費用)、純額: |
|
|
|
|
|
|
||
利子収入,純額 |
|
|
|
|
|
|
||
その他の収入 |
|
|
|
|
|
|
||
株式証負債の公正価値変動を認める |
|
|
( |
) |
|
|
— |
|
その他の費用 |
|
|
( |
) |
|
|
( |
) |
所得税前収入 |
|
|
( |
) |
|
|
|
|
所得税支給 |
|
|
( |
) |
|
|
( |
) |
営業純収入 |
|
|
( |
) |
|
|
|
|
非持株権の純収入に起因することができます |
|
|
|
|
|
|
||
普通株主は純収入を占めなければならない |
|
$ |
( |
) |
|
$ |
|
|
普通株主1株当たり純(損失)収益: |
|
|
|
|
|
|
||
基本的な情報 |
|
$ |
( |
) |
|
$ |
|
|
薄めにする |
|
$ |
( |
) |
|
$ |
|
|
普通株主の1株当たり純(損失)収益を算出するための加重平均株式数: |
|
|
|
|
|
|
||
基本的な情報 |
|
|
|
|
|
|
||
薄めにする |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
その他総合(赤字)収入: |
|
|
|
|
|
|
||
営業純収入 |
|
$ |
( |
) |
|
$ |
|
|
外貨換算調整 |
|
|
( |
) |
|
|
( |
) |
総合経営損失 |
|
|
( |
) |
|
|
( |
) |
非持株権の純収入に起因することができます |
|
|
|
|
|
|
||
非持ち株権の外貨換算調整に起因することができる |
|
|
( |
) |
|
|
( |
) |
非持株権益の総合収益(損失)に帰することができる |
|
|
|
|
|
( |
) |
|
普通株主は総合損失を占めなければならない |
|
$ |
( |
) |
|
$ |
( |
) |
付記はこれらの連結財務諸表の構成要素である。
F-5
GYRE治療会社
転換可能な優先株と株式合併報告書
(単位は千で、シェアは含まれていない)
|
|
転換可能優先株 |
|
|
普通株 |
|
|
その他の内容 |
|
|
法律を定める |
|
|
利益を残す |
|
|
積算 |
|
|
Gyre株主権益総額 |
|
|
非制御性 |
|
|
合計する |
|
|||||||||||||||||
|
|
株 |
|
|
金額 |
|
|
株 |
|
|
金額 |
|
|
資本 |
|
|
備蓄する |
|
|
(累積赤字) |
|
|
収入(損) |
|
|
(赤字) |
|
|
利子 |
|
|
|
|
|||||||||||
2021年12月31日現在の残高 |
|
|
— |
|
|
$ |
— |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|||||||||
法定備蓄金の支給 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
||||
外貨換算調整 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
純収入 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
||||
2022年12月31日現在の残高 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|
|
|
|
|
|
||||||||
GNI USA拠出金によるCatalyst前株主への普通株式発行と転換可能優先株と見なす |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||||||
少数株主の権益を買収する |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
|
|
( |
) |
|
|
— |
|
|||||
先送り時に優先株を発行する |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
||
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
||||
行使した株式オプション |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||||
外貨換算調整 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
純収益 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
|
|
|
|
|
( |
) |
|
2023年12月31日現在の残高 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
|
$ |
|
||||||||
付記はこれらの連結財務諸表の構成要素である。
F-6
GYRE治療会社
合併状態キャッシュフロープロジェクト
(単位:千)
|
十二月三十一日までの年度 |
|
|||||
|
2023 |
|
|
2022 |
|
||
経営活動 |
|
|
|
|
|
||
営業純収入 |
$ |
( |
) |
|
$ |
|
|
純(損失)収入を経営活動に提供する現金純額に調整する: |
|
|
|
|
|
||
現在行われている研究と開発を買収する |
|
|
|
|
— |
|
|
株に基づく報酬 |
|
|
|
|
|
||
減価償却および償却 |
|
|
|
|
|
||
非現金レンタル費用 |
|
|
|
|
|
||
土地使用権の償却 |
|
|
|
|
|
||
所得税を繰延し,純額 |
|
( |
) |
|
|
( |
) |
不良債権支出とその他の非現金項目 |
|
( |
) |
|
|
|
|
長期預金の受取利息 |
|
( |
) |
|
|
( |
) |
長期売掛金が価値変動を公正に許容する |
|
( |
) |
|
|
— |
|
派生負債の公正価値変動 |
|
|
|
|
— |
|
|
株式証負債の公正価値変動を認める |
|
|
|
|
— |
|
|
未合併関連会社の権益損失 |
|
|
|
|
|
||
資産剥離損失 |
|
|
|
|
— |
|
|
財産と設備処分損失 |
|
|
|
|
|
||
経営性資産と負債変動状況: |
|
|
|
|
|
||
売掛金と手形 |
|
|
|
|
( |
) |
|
GNIのその他受取 |
|
( |
) |
|
|
— |
|
棚卸しをする |
|
|
|
|
( |
) |
|
前払い資産とその他の資産 |
|
( |
) |
|
|
|
|
所得税に対処する |
|
|
|
|
( |
) |
|
売掛金 |
|
|
|
|
( |
) |
|
他の非流動負債 |
|
( |
) |
|
|
— |
|
関係者の都合で |
|
|
|
|
— |
|
|
費用とその他の負債を計算すべきである |
|
|
|
|
|
||
リース負債を経営する |
|
( |
) |
|
|
( |
) |
経営活動が提供する現金純額 |
|
|
|
|
|
||
投資活動 |
|
|
|
|
|
||
無形資産の買収 |
|
( |
) |
|
|
( |
) |
購入預金証書 |
|
( |
) |
|
|
( |
) |
財産と設備を購入する |
|
( |
) |
|
|
( |
) |
設備を売却して得た収益 |
|
|
|
|
— |
|
|
購買権益法投資 |
|
( |
) |
|
|
( |
) |
アメリカ国民総収入入金に関する現金と現金等価物 |
|
|
|
|
— |
|
|
仕入コストのための金 |
|
( |
) |
|
|
— |
|
処分済み子会社の現金残高 |
|
( |
) |
|
|
— |
|
投資活動のための現金純額 |
|
( |
) |
|
|
( |
) |
融資活動 |
|
|
|
|
|
||
私募株式交換可能株及び優先株証を発行して得られた金 |
|
|
|
|
— |
|
|
融資活動が提供する現金純額 |
|
|
|
|
— |
|
|
現金および現金等価物に及ぼす為替レート変動の影響 |
|
( |
) |
|
|
( |
) |
現金および現金等価物の純増加(減額) |
|
|
|
|
( |
) |
|
期初の現金と現金等価物 |
|
|
|
|
|
||
期末現金および現金等価物 |
$ |
|
|
$ |
|
||
|
|
|
|
|
|
||
非現金融資と投資活動の追加開示: |
|
|
|
|
|
||
GNI USA拠出金によるCatalyst前株主への普通株式発行と転換可能優先株と見なす |
$ |
|
|
$ |
— |
|
|
GNI USA寄付による転換可能な優先株と優先株証の前払 |
$ |
|
|
$ |
— |
|
|
GNIその他受取金の処分対価格 |
$ |
|
|
$ |
— |
|
|
関係者の買収コストを受け取るべきである |
$ |
|
|
$ |
— |
|
|
経営性リース負債と引き換えに使用権資産 |
$ |
|
|
$ |
|
||
他の受取金に含まれる株式オプションの収益を行使する |
$ |
|
|
$ |
— |
|
|
売掛金に入れた財産と設備を購入する |
$ |
— |
|
|
$ |
|
|
キャッシュフロー情報の追加開示: |
|
|
|
|
|
||
所得税の現金を納める |
$ |
|
|
$ |
|
||
付記はこれらの総合財務諸表の構成要素である
F-7
GYRE治療会社
総合新聞注意事項財務諸表
1. |
業務の組織と性質 |
業務説明
Gyre治療会社(“会社”,“Gyre”,または“合併会社”)は,前身はCatalyst Biosciences,Inc.(“Catalyst”)であり,生物製薬会社であり,1997年3月7日にデラウェア州に登録設立され,名称はTargacept,Inc.はプロテアーゼ工学に専門知識を持つ生物製薬会社である。2022年3月に研究と開発活動を停止する前に、Catalystはいくつかのプロテアーゼ資産を持ち、補体或いは凝固系の乱れの面で満足されていない医療需要を解決することを目的としている。
以下に述べる業務合併協定の下での取引を完了した後、Gyreは財務的に持続可能な製薬会社となり、財務的に成功した記録を持ち、臓器疾患に対する小分子抗炎症と抗繊維化薬を開発し、商業化し、特に臓器線維化に集中している。線維性疾患とは,満たされていない医療ニーズを多く有する患者である。
F 351資産買収
2022年12月26日、CatalystはGNI Group Ltd.(“GNI Japan”)及びGNI Hong Kong Limited(“GNI HK”)と2023年3月29日に改訂された資産購入協定(“F 351協定”)に署名及び完成し、主に非アルコール性脂肪性肝炎(“NASH”)(深刻な非アルコール性脂肪性肝疾患“(”NASH“)の臨床段階特許化合物に関連するすべての資産と知的財産権(総称して”F 351資産“)を購入するが、人民Republic of China(”中国“)に位置するこのなどの資産と知的財産権は含まれていない。
企業合併協定
2022年12月26日、CatalystとGNI USA、Inc.(“GNI USA”)、GNI Japan、GNI HK、Shanghai Genome,Inc.(“SG”およびGNI USA、GNI JapanおよびGNI HKの3つの“貢献者”およびそれぞれの“貢献者”)、いくつかの個人(“少数株主”および総称して“少数株主”と総称される)および大陸製薬有限会社(“CPI”)と業務統合協定(2023年3月29日および2023年8月30日改正)(“業務合併合意”)を締結した。2023年10月30日(“発効時間”)、出資(定義は以下参照)が発効し、Catalystは北京大陸医薬株式会社(“BC”)の間接持株権を取得した。寄付については,寄付が発効する前に,Catalystはその会社登録証明書を修正し,普通株の査定株式数,すなわち額面を増加させた$
Catalyst普通株はこれまでナスダック資本市場に上場し、コードは“CBIO”だった。Catalystは合併後の会社の上場申請をナスダック(以下、ナスダック)に提出した。2023年10月31日、Gyre普通株は逆株式分割後に調整した上で、ナスダック資本市場での取引を開始し、取引コードはGYREである。
“企業合併協定”によると、出資が発効した場合、15中1で発効した後
F-8
GyreはGNI USAの寄付により直接的かつ間接的に保有している
寄付が終わった後、潜在希釈証券を含まず、GNI USAはすぐに約
発効時には、BCは2021年株式インセンティブ計画(“2021年計画”)を終了し、2021年計画の下でまだ行使されていないオプション(“BCオプション”)も終了し、Gyre 2023年総合インセンティブ計画(“2023年総合インセンティブ計画”)の次の中国参加者のために設けられたサブ計画によって与えられたオプションは、すべての重大な点で、先に2021年計画で行使されていなかったBCオプションとほぼ類似している。
発効時期に発行および発行されたCatalyst普通株1株およびCatalyst普通株購入のオプションは発行済みおよび発行済みの状態を維持しており,これらの株式およびオプションは出資の影響を受けない。
BCは商業段階のバイオ製薬会社で、2002年に中国で登録設立された。BCは新薬の研究と開発,特発性肺線維化の治療のためのETUARY(ピルフェニドンカプセル)や他の薬品の製造と商業化に取り組んでいる。BCの登録事務所は中国北京市順義区順康路60号にある。
BCの直接持ち株会社はBJContinent PharmPharmticals Limited(“BJC”)である。ビーシーのミドルホールディングスはシーピーアイです。寄付の後、CPIの直接持ち株会社はGyreです。Gyreの直接ホールディングスはGNI USA,Gyreの最終ホールディングスはGNI Japanである。
国民総収入米国分担額は米国公認会計原則(“米国公認会計原則”)によって資産買収とされ、CPIは会計買収側とみなされ、買収後の米証券取引委員会報告目的の前身として列報された。Catalystは合法的な購入者であるため,GNI USAの寄付は逆資産買収に計上される.この決定は、GNI USA出資に続いて、(I)GNI USA(GNI USA出資直前のCPIの親会社として)が合併後会社の大部分の投票権を有すること、(Ii)CPIがGNI USAを介して合併後会社の取締役会を制御する能力があること、および(Iii)BCとGNI USAの上級管理職が合併後会社の上級管理職に多数のキーポストを有することを含む業務統合合意に基づく条項およびその他の要因である。Catalystは,寄付金が清算されるまで企業の定義に適合しておらず,Catalystにはその創造産出能力に大きく貢献する組織的な従業員チームがなく,その公正価値のほぼすべてが過程の研究と開発に集中している(“IPR&D”)からである。
GNI USA出資の締め切りには、Catalystの純資産はすでに買収日に当社に添付されている総合財務諸表に相対的な公正価値を記録しているが、GNI USA供出前に報告された経営業績はCPIの経営業績である。
少数株主の出資は株式取引とみなされ、会社は追加の間接権益を獲得し、ブリティッシュコロンビア州に対する支配権を維持した。
あるいは価値のある権利協定
F-9
Catalystは、2022年12月26日に業務統合協定に署名すると同時に、2023年3月29日に改正または価値ある権利協定(“CVR合意”)に署名し、この合意によれば、2023年1月5日までにCatalyst普通株を保有する各所有者(“CVR所有者”)(GNIを含まない)は、それが保有するCatalyst普通株1株について契約または価値ある権利(“CVR”)を得る。すべてのCVRの所有者は未来に一定の現金支払いを受ける権利がある。その他の資料については、別注13-を参照引受金とその他の事項.
株を逆分割する
同社は寄付金発効前に15株1株の逆株分割を実施した。逆株式分割後のCatalyst普通株の額面は調整されておらず、依然として$
他に説明がある以外に、すべての株式および1株当たりの資料はすべての提出期間の逆株式分割を有効にするために追跡的に調整されている。すべての発行済み株式オプション及び株式承認証を行使又は帰属する際の1株当たりの行使価格及び発行可能株式数を比例的に調整することにより、当該等の株式オプション及び承認証を行使又は帰属する際に予約して発行する当社の普通株式数は比例して減少し、株式オプション及び引受証については、当該等の株式オプション及び引受権証の行使価格が比例して減少する。
逆株式分割に関連する断片的な株式は発行されておらず、もともと株式の一部を取得する権利を持っていた株主は比例して支払われた現金を獲得した。
流動性
2023年12月31日現在,当社は純損失となっている$
2. |
重要会計政策の概要 |
陳述の基礎
添付されている総合財務諸表には、米国公認会計基準に基づいて作成された当社及びその制御された子会社の勘定が含まれている。適用されれば、会社間口座と取引は合併でキャンセルされた。
予算の使用
アメリカ公認会計原則に従って連結財務諸表を作成することは、合併財務諸表と付記中の報告の金額に影響を与えるために、経営陣に推定と仮定を要求する。管理層はその推定を継続的に評価し、収入確認、貸倒準備、長期売掛金、CVR由来負債、株式証負債、信用損失準備、超過或いは古い在庫準備、経営リース使用権資産及び負債、研究及び発展プロジェクトの進捗に基づいて研究開発費から適切な財務報告期までの推定、所得税、株式補償及び使用年数が確定した財産及び設備及び無形資産に関する推定を確認する。当社は当社が当時の状況で合理的と考えている様々な仮定に基づいて見積もりを出しています。実際の結果はこれらの推定とは異なる可能性がある。
非持株権益
F-10
当社は合併貸借対照表において子会社の非持株権益(“NCI”)を単独の権益構成要素として報告する。また、会社は合併経営報告書と全面収益(赤字)報告書の中で、それぞれ会社とNCIに帰属する純収益(赤字)と全面収益(赤字)部分を報告している。
細分化市場
運営分部は,独立した財務資料を持つ実体の構成要素と定義され,首席運営意思決定者(“CODM”)が個別支部にどのように資源を割り当てるかを決定し,業績を評価する際に定期的に査読する.会社のCODMはグループとして、Gyreの最高経営責任者(“CEO”)、BCの社長、Gyreの取締役会長(“Gyreの取締役会”)を含むそれは.その会社はその業務範囲を
リスクと不確実性
同社は類似の段階にある会社に関連する多くのリスクに直面しており、重要な個人への依存、比較的に大きいと成熟した会社からの競争、臨床結果の不確定性、成長を支持する十分な資金を得る能力、より多くの合格者を吸引し、維持して会社の予想成長を管理する能力、及び全体的な経済状況を含む。
濃度OF信用リスク
会社を集中的な信用リスクに直面させる可能性のある金融商品には、主に現金、売掛金、手形、長期預金が含まれる。
2023年12月31日と2022年12月31日まで、会社は現金を持っている$
米国連邦預金保険会社の保証金額を超える現金を持っている米国機関が違約すれば、当社は米国信用リスクに直面する。
2015年5月、中国政府は人民銀行が管理する新たな預金保険制度を実施した。中国大陸部でカードを持っている銀行の預金は預金保険計画によって保障され、最高限度額は人民元です
交流売掛金は通常無担保で、製品販売から来ます。当社は、未返済残高を継続的に監視し、支払履歴や信用に応じて発行される信用限度額を制限することで、売掛金に関する信用リスクを管理しています。歴史的に見ると、当社は信用期限内に顧客から売掛金を受け取り、重大な信用損失は発生していない。
顧客リスク集中
2023年12月31日と2022年12月31日までの年間で、顧客国薬控股株式有限公司が約半数を占めている
現金と現金等価物
F-11
同社は一部の超過現金を銀行預金に投資し、主に通貨市場共同基金で構成されている。購入日から、すべての原始満期日が3ヶ月以下の高流動性投資は現金等価物とみなされる。
公平である価値測定
当社は、連結財務諸表において公正価値で確認又は開示されたすべての経常的金融資産及び負債及び非金融資産及び負債に対して公正価値会計を採用する。
公正価値は、計量日に市場参加者間の秩序ある取引において資産または負債の元本または最も有利な市場の負債を移動させるために、受信される交換価格または支払いされる退出価格として定義される。
公正価値レベルは、公正価値を推定する際に観察可能な投入を最大限に利用することを1つのエンティティに要求する。公正価値レベルは、計量された資産または負債の公正価値の投入を推定するための市場観察可能性に基づく3つのクラスの分類を含む
レベル1-アクティブ市場における同じ資産または負債の見積もり。
レベル2同じ資産および負債のアクティブな市場オファー以外の観察可能な投入、非アクティブ市場の同じまたは同様の資産または負債のオファー、または実質的に全資産または負債期間の観測可能な、または観測可能な市場データによって確認可能な他の投入。
レベル3通常観察できない投入は、一般に、資産または負債のために価格を設定する際に市場参加者によって使用されるという管理層の仮定の推定を反映する。
財務諸表において公正価値で恒常的に確認された資産および負債について、当社は、各報告期間終了時に分類(公正価値全体の計量に重要な最低レベルの投入に基づく)を再評価することにより、階層構造における各レベル間に移行が生じているか否かを決定する。
デリバティブ金融商品
当社は、これらの契約が財務会計基準委員会(FASB)会計基準編纂(ASC)815のデリバティブ資格に適合しているか否かを判定するためにその契約を評価している派生ツールおよびヘッジそれは.負債入金としての派生金融商品については、派生ツールは、最初にその公正価値で入金され、各報告日に再評価される。公正価値の任意の変動は、各報告期間の非営業、非現金他の収入、または費用として記録される。派生ツール負債は、貸借対照表内で流動または非流動に分類され、これは、派生ツールが貸借対照表の日から今後12ヶ月以内に現金純額で決済可能であるかどうかに基づいている。
当社は,CVRプロトコル下のいくつかまたは有支払がASC 815に規定されているデリバティブ資格を満たしているため,負債として記録されていることを確認した。注3-を参照公正価値計量と金融商品そして付記13-引受金とその他の事項CVRデリバティブ責任に関する他の情報。
優先株式証
転換可能な優先株株を購入する引受権証(“優先株式証”)は、自社の総合貸借対照表上の引受権証負債に分類される独立した金融商品であり、対象証券は、自社が制御できない事件が発生した場合や償還が可能であるからである。優先株式証は発行時に公正価値で入金され、各報告期末に再計量しなければならない。優先株式証公正価値の任意の変動は他の収入(支出)、総合経営報告書と全面収益(損失)に計算して権利証負債の公正価値変動を計算する。当社は持分証の行使または満期になるまで優先持分証が価値変動を公正に許容する負債を調整する.
F-12
転換可能優先株
当社は発行当日の相対公正価値に応じて無投票権転換優先株の株式を記録しています。当社はASC 480-10-S 99-3 Aのガイドラインを実行し、米国証券取引委員会職員公告:償還可能証券分類計量転換可能優先株を計上する変換可能な優先株は、発行者の制御範囲内で不完全なイベントが発生したときに償還可能である可能性があるので、付随する総合貸借対照表では中間層権益に分類される。
長期預金証書
長期預金は2025年2月から2026年7月までの間に満期になる。預金証書は余剰コストによって入金され,公正な価値変動は調整されない.割増および割引は、実金利法を用いた収益の調整として、関連する満期日に固定された年間期間内に償却または累積する。“会社記録”
長期売掛金
Catalystは2023年2月に残されたまれな出血障害計画をGC Biophma Corp.(“GCBP”)に売却した。当社は、GCBP資産の売却による前払金が長期売掛金の条件を満たしていると認定している。売掛金は、会社が意図や能力を持って満期まで保有しているため、投資のための融資とされている。当社はすでに公正価値に応じて会計方法を選択して受取金に対応して会計処理を行い、公正価値のいかなる変動も利息とその他の収入を計上し、総合経営報告書と全面収益(損失)の中で純額を計上した。注3-を参照公正価値計量と金融商品長期売掛金に関するより多くの情報。
売掛金と手形,純額
売掛金が無条件支払権を有する場合、会社は売掛金を確認し、これは会社が取引中に受け取ると予想される金額を表す。当社の顧客との取引条件は主に売掛が主であり、信用期間は一般的に3ヶ月以内です。売掛金と手形は可換金算入で入金する。信用損失準備は、管理層が歴史データ、現在情報と未来の経済予測に対応する売掛金予想信用損失の最適な推定に基づいて決定した。入金は、延滞状態および顧客タイプに応じて資産プールにグループ化され、各プールに固定準備金百分率が設定される。準備金のパーセンテージは、顧客の歴史経験、監督管理と法律環境及び現在と未来の他の関連マクロ経済要素などの要素に基づいて決定される。
売掛金および手形純額は、示された日付(千計)に以下を含む
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
2022 |
|
|||
売掛金 |
|
$ |
|
|
$ |
|
||
受取手形 |
|
|
|
|
|
|
||
信用損失準備 |
|
|
( |
) |
|
|
( |
) |
売掛金と手形,純額 |
|
$ |
|
|
$ |
|
||
現在の信用損失準備の変化2023年12月31日および2022年12月31日には、以下の内容が含まれます(千単位)
|
|
2023 |
|
2022 |
|
|||
年初残高 |
|
$ |
( |
) |
|
$ |
( |
) |
信用損失を計上する |
|
|
— |
|
|
|
( |
) |
金の回収と核販売 |
|
|
|
|
|
|
||
外貨換算調整 |
|
|
|
|
|
|
||
年末残高 |
|
$ |
( |
) |
|
$ |
( |
) |
F-13
在庫、純額
在庫は原材料、製品と生産品からなり、コストまたは現金化可能な純価値の中の低い者に基づいて価格され、コストは先進先出法で確定される。当社は、初めて価値低下を確認している間、その販売コストにおける可変現純値を減記する。製品および完成品の平均コストは、主に材料、人工および製造間接費用(固定生産間接費用を含む)を含み、第三者サプライヤーのサービスおよび製品を含む。可変現価額は、推定販売価格から完成および処置によって生成された任意の推定コストを減算することによって計算される。同社はまた、歴史経験、歴史と予測販売傾向、特定在庫種別および既存在庫の満期日と年限の推定を利用して、既存と在途在庫数、および過剰と古い在庫の備蓄を定期的にモニタリングしている。連結貸借対照表に記載されている在庫は、超過と古い在庫準備金を差し引いた純額である。実際の状況や製品需要が想定より有利であれば、追加の在庫備蓄が必要になる可能性がある。
財産と設備、純額
建設中の工事を除いて、物件及び設備はコストから減価償却累計及びいかなる減価損失を引いて帳簿に記入する。財産および装置のコストは、その購入価格と、その予期される用途のためにその動作条件および位置に資産をもたらす任意の直接帰属可能コストとを含む。
財産や設備の投入使用後に発生する支出、例えばメンテナンスやメンテナンスは、通常、発生期間中に利益または損失を計上する。確認基準を満たした場合には、重大検査の支出を資産の帳簿金額に資本化し、代替とする。物件や設備の重要部分は一定期間ごとに交換する必要があり、当社はこのような部分を特定の耐用年数を有する個別資産として確認し、それに応じて減価償却を行う。
減価償却は、資産の推定耐用年数別に直線的に計算され、以下のようになる
建物.建物 |
|
賃借権改善 |
|
機械と電子機器 |
|
家具と固定装置 |
|
機動車 |
1つの財産および設備の部品が異なる使用寿命を有する場合、その部品のコストは、各部品間で合理的なベースで割り当てられ、それぞれ減価償却される。
最初に確認された任意の重要部分を含み、処置時またはその使用または処置が将来の経済的利益をもたらさない場合に確認を取り消されることを特徴とする財産および装置。販売収入純額と資産帳簿金額との差額は報告期間内に損益に計上され、関連資産は再確認されない。
建設中工事とは、建設中の建築物またはまだ稼動していない機械を指し、コストからいかなる減価損失(例えば適用)を減算し、減価償却を計算しない。コストには、建築や機械の直接コスト、および工事期間中に借り入れた関連資金の資本化借入コストが含まれる。建設工事が竣工して使用可能な場合には,新たに適切な種類の財産や設備に分類される.
資産グループの帳票金額が回収できない可能性があることを示すイベントや場合が発生した場合,財産や設備の減価を審査する.
無形資産、純額
単独で買収した無形資産は,初期確認時にコストで計測される.無形資産の使用寿命は限られたものまたは不確実なものとして評価される。限られた寿命を持つ無形資産は主に特許で構成されています
F-14
技術ノウハウとコンピュータソフトウェアはその後、利用可能な経済年限内に償却し、無形資産が減値可能であることを示す兆候がある場合に減値評価を行う。
特許と技術的ノウハウ
限られた耐用年数を持つ特許や技術ノウハウは,コストから任意の減値損失を差し引いて列報し,その経済効果を最も反映してどのように利用するかを反映した上で償却したり,その推定耐用年数内で実際の使用と実質的な差のない直線ベースで償却したりする
コンピュータソフト
購入したコンピュータソフトウェアはコストによっていかなる減価損失を引いて申告し、その推定使用年数内に直線的に償却します至れり尽くせり
賃貸借証書
当社は契約開始時に存在する独自の事実と状況に基づいて契約がリースであるか否かを評価します。契約が一定期間内に確定された資産の使用権を価格と交換するように制御した場合、契約はレンタルであるか、またはレンタルを含む。経営リース使用権(“ROU”)資産代表会社がリース期間内に対象資産を使用する権利は、経営リース負債代表会社がリースにより発生したリース金を支払う義務がある。経営リースは、付随する総合貸借対照表上の純資産、現在の経営リース負債、および長期経営リース負債に含まれる。運営リース負債は、発効日または賃貸改訂日(適用されるような)の予想レンタル期間内の将来の最低賃貸支払いの現在値で確認されます。レンタル契約に隠されている金利は一般的に確定しにくい。そこで,当社はその逓増借款金利,すなわち類似経済環境下で賃貸支払いに相当する金額を担保方式で類似期間内に借り入れることによる金利を利用している。支払いの初期直接コストや受信された報酬などの項目については、ROU資産を何らかの調整する必要がある場合がある。
当社は予想レンタル期間をテナントの取消不可期限と決定し、当社がそのオプションを行使することを合理的に決定した場合には、テナントの延長または終了の選択権を含むことができる。12ヶ月以下の賃貸契約は、総合貸借対照表では確認されません。当社はレンタル期間中に当該等の短期レンタルのレンタル料金を直線原則で確認しております。その会社の賃貸契約には残額保証は何も含まれていません。
同社は賃貸と非レンタル構成要素を単一の構成要素に統合することを選択した。最低レンタル支払いのレンタル料金は直線的に予想期間内に確認します。可変賃貸支払いは主に物件メンテナンス、税金、その他の使用状況に基づく支払いからなり、費用が発生した場合に確認します。
土地使用権網
大陸部中国のすべての土地は政府または集団の所有に属し、土地使用権は一定期間内に購入することができる。土地使用権購入価格はASCテーマ842項の経営リース前払いを代表する賃貸借証書また、総合貸借対照表に土地使用権、純資産と記載し、余剰賃貸期間内に償却する。
長期資産減価準備
長寿資産は、物件及び設備、有限年限無形資産、投資収益資産及び土地使用権を含み、いかなる事件や状況が当該などの資産の帳簿価値が回収できない可能性があることを示す場合、すべてそのような資産が減値可能かどうかを検討する。評価は資産グループレベルで行われ、キャッシュフローが他の資産および負債キャッシュフローとは大きく独立した最低レベルを識別することができる。これらの資産の回収可能性は,資産グループの帳票金額と資産予想を比較することで,使用および最終処分から生じる将来の未割引キャッシュフローによって測定される.もし審査が資産グループの帳簿金額が回収できないことを表明した場合、減価損失は帳簿価値と計量すべきです
F-15
資産グループの金額はその公正価値を超えているE.いずれの減価損失も当グループの長期資産に比例して割り当てられ、その等資産の相対額面を使用するが、個別資産の帳簿値はその公正価値を下回るまで減少することはできない。資産の公正な価値を計算することは重大な推定と仮定に関するものだ。これらの推定と仮定には,予想される将来のキャッシュフロー,リスク調整による割引率,将来の経済と市場状況,および適切な市場比較性の決定がある。これらの要因や用いた仮定の変化は,資産が減値とされている期間に確認された減値損失金額に大きな影響を与える可能性がある.“会社”ができた
所得税費用
所得税はバランスシート法により入金され、繰延税項資産及び負債は資産及び負債の財務報告及び課税基準間の差異に基づいて決定され、予想差が逆転したときに発効した制定された税率及び法律によって計量される。一部または全部の税収割引が実現できない可能性があると判断された場合、推定値控除は、純営業損失と税収控除を含む繰延税金資産に計上される。
当社は、ASC主題740の規定に基づいて、不確定な税収金を会計処理している所得税それは.不確定な税務倉位が存在する場合、当社は税務倉位の税務優遇を確認し、税務機関が審査すれば、当該税務優遇がより実現可能である。税収優遇がより実現可能かどうかの決定は,税収状況に基づく技術的利点および既存の事実や状況への考慮である。
未確認の税収割引に関する利息及び罰金(ある場合)は所得税費用の一構成要素として記録される。
収入確認
当社は、ASC主題606(“ASC 606”)に従って収入、すなわち顧客と契約を締結した収入、すなわち顧客が承諾した商品またはサービスに対する制御権を取得した場合、収入を確認した金額が、これらの商品またはサービスと交換するために予想される対価格を反映していることを確認する。1つの5ステップモデルは、(1)顧客契約の決定、(2)契約の履行義務の決定、(3)取引価格の決定、(4)取引価格を義務履行に割り当てる、(5)義務履行時に収入を確認する、というコア原則を実現するために使用される。エンティティが顧客に譲渡された商品またはサービスと交換するために獲得する権利のある対価格を受け取る可能性がある場合、会社は5ステップモードを契約に適用する。
(A)薬剤製品の販売
当社は主に中国の流通業者にその薬品を販売し、流通業者は最終的に製品を病院、他の医療機関、薬局に販売する。医薬製品を販売する収入は,製品制御権が顧客に移行する際に確認され,流通業者が薬品交付後に品質検査を完了して確認するのが一般的である。
同社は製品販売収入を記録し、推定された製品リターンを差し引く。お客様は製品を納品する際に発見された製品の破損や欠陥のみに限られた返品権利があります。当社は返品可能な製品販売金額を見積もり、この推定を関連製品収入確認期間の収入減少と負債の返金と記録しています。これまで、実際のリターンは会社の見積もりと実質的な差はなかった。
流通業者にリベートを提供することは製薬産業の接近と一致する。未払いまたは未開請求書のリベートの推定金額は、収入の減少(あれば)として記録される。推定返却点は契約料率と販売量に基づいて決定され、ディーラーの在庫にも基づいて決定される。当社はこれらの見積もりに関する情報を定期的に審査し、それに応じて金額を調整します。当社は期待値手法を用いてその獲得権のある対価格金額を推定します。
F-16
可変対価格推定の制限に関する要求を適用すること、すなわち、その後、可変対価格に関する不確実性を除去した場合、累積収入金額が確認された重大な収入逆転が発生せず、顧客の貿易売掛金から予想される将来のリベートを差し引く可能性が高い。
当社は、ASC 606による融資部分の評価について実際の便宜策を運用しており、貨物の納入から支払いまでの時間が一般的に1年以上であることから、重大な融資部分はないと結論した。
(B)知的財産権許可
ライセンスプロトコルの収入は,ライセンス使用権がクライアントに移行したときに確認する.他の承諾とバンドルされたライセンスについて、会社は、合併履行義務が時間の経過とともに履行されるか、ある時点で履行されるかを決定するために、合併履行義務の性質を評価するために判断を利用する。
取引価格に含まれる発展または規制マイルストーン支払いは、可変対価格に関する不確実性がその後解決された場合、確認された累計収入が大きく逆転しない可能性が高いことが条件だ。マイルストーン支払いを含む各手配の開始時に、当社はマイルストーンが実現可能であると考えられているかどうかを評価し、可能な金額法を用いて取引価格に含まれる金額を推定する。規制承認のような会社の統制範囲内ではない記念碑的支払いは、これらの承認を受けるまで実現可能ではないと考えられる。各報告期間の終了時に、会社は任意のマイルストーンを実現する可能性を再評価し、必要に応じて累積追跡原則に基づいて推定取引価格を調整する。
“会社”ができた
収入を繰り越す
繰延収入は、会社が関連商品やサービスを譲渡する前に顧客から支払いを受けたときに確認します。繰延収入は、会社が契約(関連商品またはサービスの制御権を顧客に移転する)を履行したときに収入と確認される。2023年12月31日現在、会社の繰延収入残高は$
収入コスト
収入コストには主に販売コストが含まれており、製品が販売可能な状態に達しても生じる直接的·間接的なコストが含まれる。販売コストには、主に、(1)原材料コスト、(2)生産従業員の人的コスト、(3)生産に使用される財産や設備および無形資産に関する減価償却および償却、(4)税金および付加価値、(5)輸送コスト、(6)その他の雑コストがある。
株に基づく報酬
同社は持分ツールを付与した日の公正価値に基づいて、持分ツールの奨励と交換するために獲得した従業員、非従業員と取締役サービスのコストを測定した。サービス条件のみを含む持分報酬については、会社は、従業員、非従業員、または取締役が報酬期間中の関連費用と交換するサービスを提供する必要があることを直線的に確認する。当社が業績条件を満たす可能性があると判断すると、業績条件を含む株式奨励の推定公正価値は奨励期間内に支出される。費用は相応の権益増加で確認される。
付与日に株式奨励の公正価値を決定するには判断が必要である。同社はブラック·スコアーズオプション定価モデルを使用して株式オプションの公正価値を決定した。オプション定価モデルを用いて、付与日オプションの公正価値がいくつかの変数に関する会社の仮定の影響を受けることを決定し、これらの変数は普通株式の公正価値、期待普通株価格の予想変動率を含む
F-17
オプションの期限、株式オプションの期待期限、無リスク金利、期待配当。会社は没収が発生した時にそれを説明することを選択した。
非市場表現やサービス条件が満たされていないため最終的に付与されていない報酬については,いかなる費用も確認しない.報酬が市場または非帰属条件を含む場合、取引は、すべての他の業績およびサービス条件が満たされる限り、市場または非帰属条件を満たすか否かにかかわらず、帰属とみなされる。
株式奨励の条項が修正され、奨励の元の条項が満たされた場合、条項が修正されていないかのように最低費用が確認される。さらに、株式ベースの支払いの総公正価値を増加させるか、または修正の日に従業員に有利な任意の修正は、費用を確認する。
配当決済報酬が廃止された場合、キャンセルの日に帰属するとみなされ、当該奨励金が確認されていない費用は直ちに確認される。これには、会社または従業員の制御範囲内の非帰属条件に適合しない任意の報酬が含まれる。ただし、取消された裁決が新たな裁決に置き換えられ、付与された日に代替裁決として指定された場合、前項で述べたように、取り消された裁決及び新たな裁決は、元の裁決の改正とみなされる。
買収した知的財産権の研究開発
当社は資産買収のコスト(取引コストを含む)に基づいて企業合併の資産買収とはみなされないことを計量し、確認した。営業権は資産買収で確認されていません。資産買収では、買収された知的財産権の研究開発に割り当てられ、将来的に他の用途のないコストは買収日に費用を計上する。注7-を参照“企業合併協定”下のGNI USA入金より詳細な説明を得るために。
研究と開発費
研究·開発コストは発生時に費用を計上する。研究および開発のための商品またはサービスの払戻不可能な前払いは、最初に繰延され、前払い資産および他の流動資産の形態で資本化される。そして、資本化金額は、関連貨物がサービスを交付または提供する際に支出されるか、または貨物またはサービスが交付されることが予期されなくなるまで支出される。研究及び開発費用には、賃金及びその他の関係者に関する費用、実験室用品及び試薬、契約研究及び開発サービス、材料及び相談費用、並びに施設分配及びその他の間接費用が含まれる。
販売とマーケティング費用
販売と市場普及支出は主にETUARYの販売と関係があり、学術会議、シンポジウム及び座談会を主催して発生した会議支出;病院で使用されているETUARY市場教育に関連する普及支出;及び主に内部市場の普及と普及人員の賃金と福祉からなる従業員コストを含む。
一般と行政費用
一般及び行政支出は、(I)会計、情報科学技術、法律、行政及びその他の内部サービス人員費用、(Ii)その機能従業員に付与された株式報酬、即ち引受権、(Iii)専門サービス料、主に法律及び会計サービス、及び(Iv)その他の雑支出を含む。
政府補助金
政府は贈与を受けることが合理的に保証され、すべての付加条件が遵守される場合、贈与はその公正価値で確認されます。贈与金が支出項目に関係している場合は,その補償しようとしている費用支出期間中に,システムによって収入と確認される。贈与が資産に関連している場合は、公許可価値を繰延収益口座に記入し、関連資産の予想耐用年数内に同額の年次分割払いまたは帳簿から差し引かれるように損益を計上する
F-18
金額減価償却費用を減らすことで利益や損失を計上する。贈与収入は総合業務表と総合(赤字)収入のうち他の収入に計上される。
利子収入
利息収入は実際の利息法を用いて計上して確認し、金融商品の期待使用年数或いは短い期間(例えば適用)に適用して金融資産の帳簿純値を正確に割引して未来の現金収入の比率を推定する。
総合収益(赤字)
純収益(損失)が付随しているものを含む全面収益(損失)のすべての構成要素を報告することを当社に要求する連結財務諸表を確認する際の財務諸表。全面収益(損失)は、一定期間内に非所有者源からの取引および他の事件および状況による権益変化と定義され、純収益、投資および外貨換算調整の未実現収益と損失を含む。
純収益(赤字)普通株主1株当たり収益(EPS)
当社はASCテーマ260に基づいて普通株主が基本的な1株当たり収益と希釈後の1株当たり収益を占めるべきであると計算した1株当たりの収益(“ASC 260”)、これは、各種類の株式(普通株および参加証券)の1株当たり収益が2種類の方法を使用して計算されることを要求する。二級法は、発表された又は累積した配当金及び未分配収益における株主の参加権に基づいて、各種類の普通株式及び参加証券の1株当たり収益を決定する。二段階法は、この期間のすべての収益が分配されたかのように、普通株式株主がその期間に獲得可能な収益を、普通株式と参加証券との間で、それぞれ配当を得る権利に基づいて分配することを要求する。ASC 260によれば、会社の転換可能な優先株は参加証券に分類される。転換優先株は契約に応じて当該等の株式の保有者を配当に参加させる権利があるが、契約に応じて当該等の株式の所有者に自社の損失を分担することを要求していない。したがって、本報告で述べた期間において、株主は純損失を占めるべきであり、これらの証券には割り当てられていない。
基本的な1株当たり収益の計算方法は、普通株主が純収益または損失を当期に発行された普通株の加重平均株式数で割るべきである。割り勘の1株当たりの収益は各提出期間内のすべての潜在的に薄くなった未返済証券によって計算される。会社の報告普通株株主が純損失を占めるべき期間中、普通株株主は希釈した1株当たり収益と普通株株主は基本的な1株当たり収益を占めるべきであり、潜在的な希薄化証券の影響は反希薄であるからである。付記16-を参照仕事がしやすいより多くの情報を得るために。
外貨換算と再計量
会社の本位貨幣はドルです。高度インフレの経済体を除いて、外国業務の現地通貨は通常、その機能通貨とされている。BCのビットコインは人民元です。各機能通貨の決定は、ASC主題830に記載された基準に基づく外貨事務それは.その会社はその報告通貨としてドルを使用している。
資産と負債は貸借対照表の日に為替レートで換算される。資本金額は歴史的為替レートに換算する。収入と支出は平均為替レートに換算する。これらの換算調整の影響は,合併貸借対照表と合併転換可能優先株と権益表における累計他の全面収益に列報し,換算収益(損失)は付随する総合経営表と包括(損失)収益のうち他の全面収益(損失)の単独構成要素として示した。2023年12月31日までの年間で、当社は$
関わった実体の本位貨幣以外の貨幣建ての取引による外貨損益を他の収入(費用)に計上し、純額を#年連結報告書に計上する
F-19
経営と総合収益。2023年12月31日まで及び2022年12月31日までの年度の外貨取引損益 どうでもいい。
外貨リスク
人民元は自由に両替できる通貨ではありません。国家外貨管理局は中国人民銀行の指導の下で、人民元と他の通貨の両替を管理する。人民元の貨幣価値は中央政府の政策変化及び中国外国為替取引システムの市場需給に影響する国際経済と政治動態の影響を受ける。2023年12月31日現在の会社現金と長期預金の99.5%$
最近採用された会計公告
2021年5月、FASBはASU 2021-04を発表した1株当たり収益(主題260)、債務修正および補償(主題470-50)、補償-株式補償(主題718)、派生ツールおよびヘッジ--エンティティ自身の資本契約(主題815-40):発行者による独立株式分類書面コールオプションのいくつかの修正または交換の会計処理。ASU 2021-04における改訂は、修正または交換後も持分分類を維持する発行者による独立持分分類書面コールオプション(例えば、株式承認証)の会計処理の多様性を明確にし、低減するための指針を提供する。本ASU 2021-04における修正案は、2021年12月15日以降のすべての年度およびこれらの年度内の移行期間に適用される。会社(The Company)
2020年8月にFASBはASU 2020-06を発表しました債務--転換可能な債務および他のオプション(主題470-20)と派生ツールおよびヘッジ--エンティティ自身の権益の契約(主題815-40)特定の金融商品の会計処理を単純化する。ASU 2020-06は、利益変換および現金変換機能を変換可能ツールから分離することを以前に要求していたモードを廃止し、エンティティ自己持分における契約の持分分類に関連する派生商品範囲例外指導を簡略化した。本ASUにおける改正案は,2021年12月15日以降の財政年度内に公共企業実体に対して有効である。会社(The Company)
FASBは2021年10月にASU 2021-08を発表した業務統合-契約資産と顧客契約との連絡負債の会計処理それは.この指導意見は,実践における多様性の問題を解決することにより,企業合併で顧客が獲得した収入契約の会計処理を改善することを目的としている。指導意見は、買収側が米国会計基準第606条に従って企業合併で買収した契約資産及び負債を確認及び計量し、買収日に公正な価値で計量するのではなく、開始契約とみなすことを要求する。会社(The Company)
2021年11月、FASBはASU 2021-10を発表した政府援助(テーマ832)−企業実体が政府援助状況を開示するそれは.本ASUにおける修正案は、(1)取引の種類、(2)取引の会計、および(3)取引が実体財務諸表に及ぼす影響を増加させるために、類比贈与または寄付会計モデルによって計算される政府との取引の開示を要求する。本ASUにおける改訂は、その範囲内のすべてのエンティティに適用され、2021年12月15日から発表される年次財務諸表に適用される。会社(The Company)
新しい会計公告-発表されたがまだ採用されていない
2023年11月、FASBはASU 2023-07を発表した報告可能部分に開示された改善(主題280)それは.本会計基準は、定期的にCODMに提供され、各報告の部門損益測定基準に含まれる重大な報告すべき部門支出の開示を要求することによって、報告すべき部門の開示要求を更新した。本ASUはまた、CODMとして決定された個人の肩書と職を開示し、CODMが報告された部門損益測定基準を使用して部門を評価する方法を説明することを要求している
F-20
性能と資源をどのように割り当てるかを決定する.ASUは2023年12月15日以降の年度期間と2024年12月15日以降の財政年度内の移行期間で有効である。ASUを採用することは、財務諸表に記載されている以前のすべての期間に適用されるべきである。早期養子縁組を許可する。当社は2024年1月1日にASU 2023−07の年度要求と関連更新を採用し,2025年1月1日にASU 2023−07の仮要求を採用する予定である。同社は現在、この基準を採用した連結財務諸表への影響を評価している。
FASBは2023年12月にASU 2023-09を発表しました所得税開示の改善(主題740)それは.ASUは,報告エンティティの有効税率入金に関する分類情報と,支払われた所得税に関する補足情報を提供することを要求している。ASUは2024年12月15日以降の年次期間に前向きである。早期養子縁組を許可する。採用されると、このASUは、必要な追加開示を会社の総合財務諸表に含めることになる。会社は2025年1月1日からASU 2023-09および関連更新を採用する予定だ。会社はこの基準を採用してその連結財務諸表に与える影響を評価するだろう。
3. |
公正価値計量と金融商品 |
公正価値階層と公正価値方法の説明については,付記2−を参照されたい重要会計政策の概要それは.2023年12月31日現在、会社の高流動性通貨市場基金は現金等価物に計上されている。
次の表は公正価値によって恒常的に計量された金融資産と負債の公正価値レベルであり、現在までである2023年12月31日(千)):
|
|
2023年12月31日 |
|
|||||||||||||
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
|
合計する |
|
||||
金融資産 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金(1) |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
GCBPの長期売掛金 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
||
金融資産総額 |
|
$ |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
金融負債 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
CVR派生負債 |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
||
権証責任,非流動 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
||
財務負債総額 |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
||
(1)
満期日が短いため、現金、売掛金及び手形、純売掛金、その他の売掛金、関連側帳簿金、CVR超過期末対応現金及び売掛金の帳簿金額はその公正価値に近い。
2023年12月31日までの年間で
いくつありますか
長期売掛金とCVR派生負債
長期売掛金及び対応するCVR派生負債は、非流動はGCBPとの資産購入プロトコルに関連する。この長期売掛金及び派生負債の公正価値は重大な観察できない投入に基づいており、これらの投入は公正価値体系中の第三級計量を代表する。非流動長期売掛金及びCVR由来負債の推定公正価値は、GCBP資産購入プロトコルによりGCBPから徴収される予想キャッシュフローの予想金額及び時間を割り出し、その現在値に割引し、割引率を推定するものである
F-21
対応するCVR派生負債は、非流動負債に利息とその他の収入を計上し、純額を総合経営表と全面収益(赤字)に計上する。
株式証法的責任
CatalystとGNI USAは2023年10月に私募について証券購入協定(“私募”)を締結した。私募は2023年10月30日の寄付終了直後に終了します。私募完了後,会社は発行した
優先持分証は独立した金融商品であり、会社総合貸借対照表上の権証負債に分類される。優先持分は各報告期間ごとに再評価され、公正価値変動は他の収入(支出)、総合経営報告書及び全面収益(損失)における権証負債の公正価値変動と表記される。
株式証負債の公正価値は根拠であるEブラック-スコアーズオプション定価モデルは、以下の加重平均仮定を採用する
株価.株価 |
|
$ |
|
|
行権価格 |
|
$ |
|
|
配当率 |
|
— |
|
|
無リスク利子 |
|
|
% |
|
期限(年) |
|
|
|
|
予想変動率 |
|
|
% |
|
以下の表に、会社3級金融資産と負債の推定公正価値変化(千単位)を示す
|
|
GCBPの長期売掛金 |
|
|
CVR派生負債 |
|
|
株式証法的責任 |
|
|||
2022年12月31日の残高 |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
この期間の新規プロジェクト |
|
|
|
|
|
|
|
|
|
|||
価値変動を公平に承諾する |
|
|
|
|
|
|
|
|
|
|||
2023年12月31日の残高 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
金融商品
現金等価物には、以下のものが含まれる(千計)
2023年12月31日 |
|
原価を償却する |
|
|
未実現収益総額 |
|
|
未実現損失総額 |
|
|
公正価値を見積もる |
|
||||
貨幣市場基金(現金等価物) |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
金融資産総額 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
分類は: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
現金と現金等価物 |
|
|
|
|
|
|
|
|
|
|
$ |
|
||||
金融資産総額 |
|
|
|
|
|
|
|
|
|
|
$ |
|
||||
4. |
貸借対照表の構成要素 |
F-22
在庫、純額
在庫、準備金を差し引いて純額#ドル
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
原料.原料 |
|
$ |
|
|
$ |
|
||
進行中の仕事 |
|
|
|
|
|
|
||
完成品 |
|
|
|
|
|
|
||
在庫、純額 |
|
$ |
|
|
$ |
|
||
2023年12月31日までと2022年12月31日までの年度の在庫と減記準備金はどうでもいい。
費用とその他の流動負債を計算しなければならない
計算すべき費用およびその他の負債には、以下の項目が含まれる(千で計算)
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
賃金総額と福祉を計算しなければならない |
|
$ |
|
|
$ |
|
||
仕入先が精算する |
|
|
|
|
|
|
||
費用を計算すべきである一般と行政費用 |
|
|
|
|
|
— |
|
|
販売割引を計算すべきである |
|
|
|
|
|
|
||
専門サービスに応じる |
|
|
|
|
|
— |
|
|
従業員が精算する |
|
|
|
|
|
|
||
計算すべき費用−研究と開発 |
|
|
|
|
|
|
||
課税費用--販売費用 |
|
|
|
|
|
|
||
繰延の政府支出 |
|
|
|
|
|
|
||
その他負債を計算すべき |
|
|
|
|
|
|
||
費用とその他の流動負債を計算しなければならない |
|
$ |
|
|
$ |
|
||
繰延の政府支出
繰延の政府支出とは、政府が研究開発、新施設の建設、あるいは既存の生産施設の改善に提供する資金のことである。2023年12月31日と2022年12月31日までの繰延政府贈与額は、政府贈与収入と確認された金額を差し引いた純額です。2023年12月31日及び2022年12月31日まで、当社は受け取りました$
以下は以下の日までの繰延政府支出である2023年12月31日および2022年12月31日(単位:千):
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
課税費用及びその他の流動負債に計上された政府財産及び設備補助金 |
|
$ |
|
|
$ |
|
||
当面延期された政府支出 |
|
|
|
|
|
|
||
政府の財産と設備に対する補助 |
|
|
|
|
|
|
||
非当期政府補助金を繰延する |
|
|
|
|
|
|
||
繰延政府支出総額 |
|
$ |
|
|
$ |
|
||
F-23
財産と設備、純額
財産と設備、純価値は以下の通り(千で計算)
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
建物.建物 |
|
$ |
|
|
$ |
|
||
建設中の工事 |
|
|
|
|
|
|
||
機械と電子機器 |
|
|
|
|
|
|
||
家具と固定装置 |
|
|
|
|
|
|
||
賃借権改善 |
|
|
|
|
|
|
||
機動車 |
|
|
|
|
|
|
||
財産と設備、毛額 |
|
|
|
|
|
|
||
減算:減価償却累計 |
|
|
( |
) |
|
|
( |
) |
財産と設備、純額 |
|
$ |
|
|
$ |
|
||
減価償却費用は$
5. |
無形資産 |
年限確定可能な会社無形資産の帳簿価値総額と累積償却2023年12月31日、2022年12月31日、2022年12月31日(単位:千):
|
2023年12月31日 |
|
|||||||||
|
総帳簿金額 |
|
|
累計償却する |
|
|
無形資産、純額 |
|
|||
寿命の限られた無形資産: |
|
|
|
|
|
|
|
|
|||
技術のこつ |
$ |
|
|
$ |
( |
) |
|
$ |
|
||
コンピュータソフト |
|
|
|
|
( |
) |
|
|
|
||
無形資産総額 |
$ |
|
|
$ |
( |
) |
|
$ |
|
||
|
2022年12月31日 |
|
|||||||||
|
総帳簿金額 |
|
|
累計償却する |
|
|
無形資産、純額 |
|
|||
寿命の限られた無形資産: |
|
|
|
|
|
|
|
|
|||
特許 |
$ |
|
|
$ |
( |
) |
|
$ |
|
||
技術のこつ |
|
|
|
|
( |
) |
|
|
|
||
コンピュータソフト |
|
|
|
|
( |
) |
|
|
|
||
無形資産総額 |
$ |
|
|
$ |
( |
) |
|
$ |
|
||
無形資産はコストから累積償却と減価(適用)を差し引いて計上され、償却費用は営業費用に計上される。2023年12月31日までの無形資産加重平均償却期間はい
償却費用は$
F-24
|
費用の償却を予定する |
|
|
2024 |
$ |
|
|
2025 |
|
|
|
2026 |
|
|
|
2027 |
|
|
|
2028 |
|
|
|
その後… |
|
|
|
合計する |
$ |
|
|
6. |
収入.収入 |
同社の製品収入は主にETUARYの販売から来ています。ETUARYの売上は
医薬製品の販売状況
同社の収入は主にETUARYとある模造薬の販売から来ている。ディーラーは会社の直接顧客で、ディーラーに対する販売は占めています
これまで製品の返品量は大きくありませんでしたが、当社は製品返品記録準備金を準備する必要はないと考えています。会社の製品収入は,基礎製品が顧客に渡されたときに確認された,すなわち顧客が製品制御権を獲得したときである.医薬製品の販売収入は$
知的財産権許可
使用権を許可する制御権が顧客に譲渡されると,許可知的財産権の収入が確認される.マイルストーン支払いは取引価格に含まれており、確認された累計収入が大きく逆転しない可能性が高い限り、可変対価格を表し、可変対価格に関する不確実性がその後解決された場合、この形態は収入として確認される。一里塚支払いを含む各項目の手配開始時に、当社はマイルストーンが実現可能であると考えられているかどうかを評価し、可能な金額法を用いて取引価格に含まれる金額を推定する。規制承認のような会社の統制範囲内ではない記念碑的支払いは、これらの承認を受ける前に、実現されない可能性が高いと考えられる。
当社の今年度までの製品別売上高2023年12月31日と2022年12月31日の状況は以下の通り(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
2022 |
|
||||
医薬製品の販売状況 |
|
$ |
|
|
$ |
|
||
知的財産権許可 |
|
|
— |
|
|
|
|
|
合計する |
|
$ |
|
|
$ |
|
||
7. |
“企業合併協定”下のGNI USA入金 |
企業合併協定(付記1参照)によると、GNI USAの出資は逆資産買収とされ、CPIは会計買収側、Catalystは合法買収側とされる。アメリカ公認会計原則によると、会社は統合された資産と活動が以下の条件を満たすかどうかを評価しなければならない
F-25
総資産の公正価値を評価することが主に単一資産または類似資産のセットに集中しているかどうかを評価する。この基準を満たしていれば、買収された業務は企業とみなされない。Catalystは業務定義に達していないが、その公正な価値は知的財産権研究開発資産に集中しているため、生産創造に大きな貢献をする組織的な労働力が不足しており、実質的な手続きも得られていない。そのため、アメリカ国民総収入寄付金は資産購入とみなされている。資産買収では、営業権が確認されず、買収された知的財産権研究·開発に割り当てられ、他の将来用途のコストが費用に計上されない。
以下にGNI USA払込みで支払われる購入価格(1株当たり金額を除く千単位)についてまとめた
|
発行済みCatalyst普通株 |
|
|
|
|
触媒株を乗じた1株当たりの公正価値(1) |
$ |
|
|
|
Catalyst株主が所有する普通株の公正価値 |
$ |
|
|
|
Catalyst株主が所有すべき優先株の公正価値(2) |
$ |
|
|
|
触媒の公正価値 |
$ |
|
|
|
CPIが想定するGNI前の米国寄与触媒株式オプション(3) |
$ |
|
|
|
発行された対価の公正価値 |
$ |
|
|
|
仕入コスト |
$ |
|
|
|
買い入れ価格 |
$ |
|
予想期限(年単位) |
|
|
|
波動率 |
|
% |
|
無リスク金利 |
|
% |
|
配当率 |
—% |
|
|
米国国民総収入入金で支払われた全購入価格は、有効時間の公正価値に基づいて得られた純資産と負担する負債に割り当てられている。以上のように,購入価格の割当ては以下のとおりである(千単位)
F-26
現金と現金等価物 |
$ |
|
|
GNIのその他受取 |
|
|
|
前払い資産 |
|
|
|
その他流動資産 |
|
|
|
GCBPの長期売掛金 |
|
|
|
他の非流動資産 |
|
|
|
知的財産権研究開発 |
|
|
|
売掛金 |
|
( |
) |
GNIから受け取った前金 |
|
( |
) |
費用とその他の流動負債を計算しなければならない |
|
( |
) |
CVR期末超過現金対応 |
|
( |
) |
CVRは負債、非流動を誘導する |
|
( |
) |
取得した純資産 |
$ |
|
少数株主の出資は株式取引とみなされ、会社は追加の間接権益を獲得し、ブリティッシュコロンビア州に対する支配権を維持した。
8. |
再編成する |
貢献する前にCPIはCPに入りました米国とGNI USAの株式購入協定ほとんどの資産を剥離しました
9. |
賃貸借証書 |
賃貸借契約を経営する
紀元前紀元前会社の本社、
同社はオフィスビルや従業員寮としての短期賃貸物件も複数持っている。同社は合わせて#ドルを記録した
2023年12月31日現在、同社が記録した総ROU資産は$
賃貸経営に関する賃貸料支出は$
経営リースに関する補足キャッシュフロー情報は以下の通り(単位:千):
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
賃貸負債に含まれる金額を計量するために支払う現金: |
|
|
|
|
|
|
||
**営業レンタルから営業キャッシュフローを取得 |
|
$ |
|
|
$ |
|
||
F-27
賃貸支払いの現在値を計算する際に使用する現在値を以下のように仮定する
|
十二月三十一日までの年度 |
|
||
|
2023 |
|
2022 |
|
加重平均残余レンタル期間 |
|
|
||
加重平均割引率 |
|
|
||
2023年12月31日現在、会社経営賃貸契約に割引されていない将来の最低支払いは以下の通り(千計)
|
|
金額 |
|
|
2024 |
|
$ |
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
未割引賃貸支払総額 |
|
|
|
|
差し引く:推定利息 |
|
|
( |
) |
リース総負債 |
|
|
|
|
差し引く:賃貸負債の当期分 |
|
|
( |
) |
賃貸負債、当期分を差し引く |
|
$ |
|
|
当社は以下の保証金を維持しなければならない$
土地使用権
2023年12月31日まで、会社は北京順義区2053年満期と河北滄州2067年から2071年までの2つの土地の土地使用権を持っている。これらのブロックの総面積は
10. |
株主権益 |
普通株
会社が改訂して再記載した会社登録証明書によると、会社はすでに
将来の発行のために予約された普通株式は以下のとおりである
|
十二月三十一日 |
|
|||||
|
2023 |
|
|
2022 |
|
||
発行済みと発行済みの転換可能優先株 |
|
|
|
|
— |
|
|
発行済みおよび未償還優先持分証 |
|
|
|
|
— |
|
|
既発行と未償還のオプション |
|
|
|
|
|
||
普通株式総数を保留する |
|
|
|
|
|
||
2021年ATM計画
Catalystは2021年10月15日、Piper Sandler&Co.(“Piper Sandler”)と販売代理として株式流通協定(“ATMプロトコル”)を締結し、この合意により、会社は時々提供·販売することができる
F-28
Piper Sandlerを通じて、会社の普通株式の額面は$
ATM機計画下の普通株の売却は、米国証券取引委員会が2021年5月3日に発効を発表したS-3表登録書(第333-253874号文書)および2021年10月15日に米国証券取引委員会に提出された関連目論見書補充書類に基づいて行うことができる。2023年12月31日まで及び2022年12月31日まで年度,
制限純資産
中国の法律法規によると、卑詩省は親会社や米国の株主への移転を含む外国為替や国境を越えた現金移転の面で制限されている。親会社や米国の株主に収益を分配する能力も限られている。中国の現行法規によると、BCは中国の会計基準と法規に基づいて定められた累計利益の中から北京競馬会に配当金を支払うことしかできない。制限された金額には、すでに入金された資本と卑詩省の法定準備金が含まれる。付属会社が分配できない純資産額を代表する制限資本及び法定備蓄金の総額は以下のとおりである$
上記の制限により、親会社の財務諸表のみが別表1に記載されている。
法定備蓄金
卑詩省は少なくとも残ることを要求された
紀元前のこのような備蓄に対する支出は$
11. |
転換可能優先株 |
当社は発行を許可されている
2022年12月Catalystが発表しました
2023年10月、寄付終了後、当社は直ちに発表しました
2023年11月、GNIは当社に通知を出し、それを要求した
転換可能優先株は2023年12月31日現在、会社の制御範囲内で完全にはない償還特徴が含まれているため、株主権益の外に分類されている。交換可能株の優先株は現在償還できないため、当社は交換可能株の優先株の帳簿価値をその償還価値に調整することはなく、この交換可能株の優先株も後日いかなる貸借対照表の期日に償還可能になることは不可能である。このような償還が発生する可能性がある場合にのみ、帳簿価値の後続調整が行われる。
F-29
転換可能な優先株の保有者は、普通株に変換した上で、会社の普通株が実際に支払った配当と同じ形で、実際に支払われた配当と同じ配当を得る権利がある。法律が別に規定されている以外に、転換可能な優先株は投票権を持たない。しかしながら、いかなる転換可能な優先株も発行されていない限り、当社は、(I)転換可能な優先株を付与する権限、優先株又は権利を不利に変更又は変更又は修正して転換可能な優先株の指定証明書を発行し、会社登録証明書又は会社定款の任意の規定を修正又は廃止し、又は任意の改正条項、指定証明書、優先株、制限及び任意の一連の優先株の相対的権利を提出してはならない。交換可能な優先株の権利、特権または権力、または交換可能な優先株の利益のために規定された制限;(Ii)交換可能な優先株をさらに発行するか、または交換可能な優先株の法定株式数を増加または減少させるか、または(Iii)上記の任意の事項について任意の合意を締結する。転換可能優先株は会社のいかなる清算、解散、または清算時に優先権を有していない。
2023年12月31日までに会社は
12. |
株に基づく報酬 |
BC 2021年株式インセンティブ計画
BC取締役会は2021年2月、BCのある従業員およびコンサルタントにオプションを付与する2021年計画を承認した。
2021年計画の契約期間は
2023年10月、BC取締役会は出資前に完全に帰属するBCオプションの満期日を延長する
2018総合インセンティブ計画
2018年6月、Catalyst株主は、2021年6月に改訂された2018年総合インセンティブ計画(“2018総合インセンティブ計画”)を承認した。発効直前の2018年総合インセンティブ計画に基づいて発行·発行されたCatalyst普通株購入ごとのオプションは、その条項に基づいて出資した後も返済されておらず、影響を受けていない。
2023年総合インセンティブ計画
2023年総合インセンティブ計画は、2023年8月にCatalystの株主によって承認され、2023年10月にGyre取締役会によって承認される。2023年総合インセンティブ計画は2023年10月30日に施行される。2023年総合インセンティブ計画では会社が最大の提供を可能にします
F-30
入金後、当社は当初付与されていたBCオプションの満期日を修正し、延長しました
2023年10月30日、寄付終了後、Gyre取締役会は寄付金を承認しました
次の表は株式オプション活動をまとめており、GNI USA入金完了後にBCオプションをGyreオプションに変換してGyre普通株の株を購入することを考えています
|
未償還オプション関連株式数 |
|
|
加重平均行権値 |
|
|
加重平均残契約期間(年) |
|
|
内在的価値を合計する |
|
||||
2022年12月31日に返済されていません |
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
没収される |
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
GNIによる米国献金はCatalystの前株主にオプションを発行するとされている |
|
|
|
$ |
|
|
|
|
|
|
|
||||
授与する |
|
|
|
$ |
|
|
|
|
|
|
|
||||
鍛えられた |
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
2023年12月31日現在の未返済債務 |
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2023年12月31日に行使できます |
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2023年12月31日まで年度内に付与された株式購入の加重平均授受日公正価値は$
2023年12月31日までの年間におけるオプション行使済みの内的価値の合計は$
推定値仮定
同社はブラック·スコアーズオプション定価式と単一オプション奨励方法を用いて付与された株式オプションの公正価値を推定した。関連履歴データが限られているため、当社はその変動率を見積もる際に複数の要因を考慮しており、比較可能な上場会社の変動率を使用することも含まれている。米国証券取引委員会従業員会計公告107によると、2023年総合激励計画に基づいて付与されたオプションの期待期限はすべて“普通”の条件を満たし、会社の限られた関連歴史のため、簡略化された方法に基づいて決定される。無リスク金利は、期限とオプションが一致する米国債収益率に基づいている。この公正な価値は、通常、帰属期間である報酬の必要なサービス期間内に比例的に償却されるであろう。
付与された従業員株式オプションの公正価値は、以下の加重平均仮定を用いて推定される
|
|
十二月三十一日までの年度 |
|
|
|
|
2023 |
|
|
期待変動率(%) |
|
|
||
無リスク金利(%) |
|
|
||
期待オプション寿命(年) |
|
|
||
期待配当率(%) |
|
|
— |
|
会社加重平均株価(ドル/株) |
|
$ |
|
|
F-31
確認された株式報酬総額は以下の通り(千で計算)
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
2022 |
|
||||
収入コスト |
|
$ |
|
|
$ |
|
||
販売とマーケティング |
|
|
|
|
|
|
||
研究開発 |
|
|
|
|
|
|
||
一般と行政 |
|
|
|
|
|
|
||
合計する |
|
$ |
|
|
$ |
|
||
2023年12月31日現在、会社が未確認の株式ベースの報酬支出は$
13. |
引受金とその他の事項 |
あるいは価値のある権利協定
CVRプロトコルの各CVR(注1参照-業務の組織と性質)所有者に権利を持たせる(1)Catalystレガシー資産の処分に関する純収益から何らかの現金支払いを得る,(2)
2023年8月、CatalystはVertex資産購入プロトコルに関するCVRを全額決済した。
2023年2月,Catalystはその残されたまれな出血障害計画をGCBPに売却した。そこで,当社はGCBP資産を売却して得られた現金純額を分配する$
2023年10月30日にCVR協定に基づき、当社は記録しました$
訴訟と法務
当社は正常な業務過程で発生したクレームや法的手続きの影響を受けます。当該等の事項自体に不確実性があり、いかなる当該等の事項の結果が当社に有利になるか、又は当該等の事項の解決が当社の総合財務諸表に重大な悪影響を与えない保証はない。
2023年4月、Catalystの異なる株主がデラウェア州衡平裁判所に提訴したブシャンスキーはCatalyst Biosciences,Inc.らを訴えたそしてScottはCatalyst Biosciences,Inc.,らを訴え,Catalystはデラウェア州の法律規定に違反した受託責任を告発し、提案業務合併協定に関する重要な情報を開示できなかった。202年2月にこの2つの訴訟はいずれも却下され,会社は訴訟に関する法律やその他の費用を株主に返済し,総金額は#ドルであった
F-32
♪the the the会社は訴訟と和解債務を記録し、金額は#ドルだった
購買承諾
財産と設備
当社が購入契約を締結しているが連結財務諸表に反映されていない財産·設備に関する引受金は以下のとおりである$
F351
2020年9月、卑詩省はGNI Japanとそのいくつかの完全子会社(“GNIグループ”)と知的財産権譲渡協定(“F 351譲渡協定”)を締結した。F 351譲渡協定に基づき、BCは中国大陸部での独占使用権及び全世界知的財産権の優先的要項権(“F 351知的財産権”)を取得した。
F 351譲渡プロトコルにより,F 351知的財産権の交換としてBCはGNI集団への支払いが義務付けられている$
研究と開発計画
F 351計画を除いて、2023年12月31日まで、会社は分配を約束した$
完済する
正常な業務過程において、当社は協定を締結し、取引或いはある事件と活動に関連するある責任を他人に賠償する可能性がある。補償された側が賠償条項に基づいて成功のクレームを出した場合、会社は損害賠償を要求される可能性がある。このような補償は一般的に様々な制限と制限を受けている。同社のこれらの合意下でのリスクは未知であり、将来同社にクレームを出す可能性があるが、まだクレームが出されていないことに関連しているからである。これまで、同社は何のクレームも支払っておらず、その賠償義務に関するいかなる訴訟も弁護されていない。
14. |
所得税 |
2023年10月、会社は卑詩省持株権の買収を完了した。出資の結果として、当社は支配されている複数の外国会社(“CFCs”)を直接または間接的に所有し、米国の税務目的に利用している。
当社の年度までの所得税準備金の構成要素2023年12月31日および2022年12月31日には、以下の内容が含まれます(千単位)
F-33
|
十二月三十一日までの年度 |
|
|||||
|
2023 |
|
|
2022 |
|
||
現行所得税規定: |
|
|
|
|
|
||
連邦制 |
$ |
— |
|
|
$ |
— |
|
状態.状態 |
|
|
|
|
|
||
外国-中国 |
|
|
|
|
|
||
当期所得税引当総額 |
$ |
|
|
$ |
|
||
|
|
|
|
|
|
||
繰延所得税準備: |
|
|
|
|
|
||
連邦制 |
$ |
— |
|
|
$ |
( |
) |
状態.状態 |
|
— |
|
|
|
— |
|
外国-中国 |
|
( |
) |
|
|
( |
) |
繰延所得税準備総額 |
$ |
( |
) |
|
$ |
( |
) |
所得税引当総額 |
$ |
|
|
$ |
|
||
連邦法定所得税率と会社の年度までの実税率との入金2023年12月31日と2022年12月31日の状況は以下の通りです
|
十二月三十一日までの年度 |
||
|
2023 |
|
2022 |
連邦法定税率で計算される税金 |
|
||
管轄区域の違いによる料率の違い |
- |
|
|
HNTE所得税割引税率 |
|
- |
|
差し引かれない費用--運営 |
- |
|
|
差し引かれない費用--入金に関する使い捨て費用 |
- |
|
|
R&D超過控除 |
|
- |
|
評価免税額変動 |
- |
|
|
従業員持株計画 |
- |
|
|
他にも |
- |
|
- |
実際の税率 |
- |
|
|
会社までの繰延税金資産の重要な構成要素2023年12月31日および2022年12月31日には、以下の内容が含まれます(千単位)
|
十二月三十一日までの年度 |
|
|||||
|
2023 |
|
|
2022 |
|
||
繰延税金資産: |
|
|
|
|
|
||
課税項目と準備金 |
$ |
|
|
$ |
|
||
収入を繰り越す |
|
|
|
|
— |
|
|
営業純損失繰り越し |
|
|
|
|
— |
|
|
その他株式投資 |
|
— |
|
|
|
|
|
税金の繰り越しを免除する |
|
|
|
|
— |
|
|
固定資産と無形資産 |
|
|
|
|
|
||
外国の会社からの影響 |
|
|
|
|
— |
|
|
資本化取引コスト |
|
|
|
|
|
||
賃貸負債 |
|
|
|
|
— |
|
|
未見積控除前の繰延所得税資産 |
|
|
|
|
|
||
繰延税金負債(ROU資産と略記) |
|
( |
) |
|
|
— |
|
繰延税金負債--固定資産 |
|
( |
) |
|
|
( |
) |
減算:推定免税額 |
|
( |
) |
|
|
— |
|
税金資産を繰延し,純額 |
$ |
|
|
$ |
|
||
推定免税額の変動状況は以下の通り(千で計算)
F-34
|
2023 |
|
|
2022 |
|
||
年初残高 |
$ |
— |
|
|
$ |
— |
|
評価免除額を変更する |
|
( |
) |
|
|
— |
|
年末の残額 |
$ |
( |
) |
|
$ |
— |
|
2023年12月31日の客観的証拠によると、当社は繰延税項目純資産が米国の税務目的で現金化される可能性が高いとは考えていない。そのため、当社は2023年12月31日の繰延税項目純資産に米国税務のための全額推定手当を提供している。
2023年12月31日現在、ある制限を考慮した後(後述)、会社は約$
2023年12月31日現在、同社は将来の約$を相殺するための税収免除繰越金を持っている
当社の3年間の所有権総変動が50ポイント(第382条所有権変動)を超えた場合、その変動前のNOL繰越の使用は米国国税法第382条の年次制限を受ける(カリフォルニアには類似した規定がある)。年間限度額の決定方法は,所有権変更前の会社株の価値に適用される長期免税税率を乗じることである。このような制限により,NOL繰り越しの一部が使用前に期限切れになる可能性がある。当社は、第382条に規定する所有権変更は、2007年12月31日、2015年8月20日、2017年4月13日、2018年2月15日、2020年2月18日、2022年12月26日に発生すると認定している。約$
すべての連邦研究開発信用は未使用の場合に満期になることができるが、カリフォルニアの研究開発信用は満期にならない。約$
2020年6月29日、カリフォルニア州知事はカリフォルニア州の法律となった議会85号法案(“A.B.85”)に署名した。A.B.85には、中型·大型企業の純営業損失の使用を3年間停止することを規定し、#ドル以下を相殺するために、商業インセンティブ税収控除に3年間の上限を設定する税収措置がいくつか含まれている
所得税における不確実性会計
当社は、関連税務機関がその技術的利点に基づいて監査を行った後に税額割引を維持する可能性がある場合にのみ、当該等の税額割引を確認します。不確実な税金状況が続く可能性が50%未満であれば、確認されないだろう。
その会社は約$
F-35
未確認税収割引の期初残高と期末残高の入金は以下の通り(千で計算)
2022年1月1日期初め残高 |
$ |
|
|
数年前に確認されなかった税収割引 |
|
|
|
今年度に関連する未確認税収割引の増加/(減少) |
|
|
|
2022年12月31日期末残高 |
|
|
|
2023年10月30日米国民総収入増加寄付金 |
|
|
|
数年前に確認されなかった税収割引 |
|
|
|
今年度に関連する未確認税収割引の増加/(減少) |
|
|
|
2023年12月31日期末残高 |
$ |
|
未確認の税収割引に関する利息と罰金は、法人税費用として会社の総合経営報告書に計上される。2023年12月31日と2022年12月31日まで会社が所有しています
同社は米国連邦、カリフォルニア州、フロリダ州に2023納税年度の所得税申告書を提出した。同社は2022年にフロリダ州に予備報告書を提出し、2021年にカンザス州、ミズーリ州、ニュージャージー州で最終報告書を提出した。その会社は現在連邦、州、あるいは他の管轄区域の所得税当局の審査を受けていない。2023年12月31日と2022年12月31日まで会社が所有しています
APB 23
一般に、ASC第740-30-25-17段落(APB意見第23号、所得税会計である特殊分野(“APB 23”)の無期限沖販売基準を満たしていれば、外国子会社に関する課税外部ベース差が確認されない可能性がある。1つのエンティティが無期限出荷基準を満たしていない場合、繰延税金負債が確認される。“米国会計基準”第740-30-25-17項には、子会社が無期限投資未分配収益、または収益が免税清算に送金されることを十分な証拠が示されている場合、すべての未分配収益が親会社に移転するという推定は克服でき、親会社は所得税を計算すべきではない。
会社は非アメリカ子会社の収益を会社に送金する計画はありません。しかし,当社がその中国業務に永久的に投資しない範囲では,DTLは約$である
15. |
関係者取引 |
F 351譲渡プロトコル
F 351譲渡プロトコル(付記13参照)によれば、代償の一部としてBCが支払う$
GNIを用いた研究と開発
2023年12月31日と2022年12月31日まで、会社は$
GNIのその他受取
F-36
当社は2023年12月31日までに記録しました$
16. |
仕事がしやすい |
発行済み株式オプションと引受権証の希薄化効果は在庫株方法を用いて計算した。株式オプションと引受権証は逆償却であり、行使価格が普通株の平均市場価格を超える場合は、普通株計算の希釈後の1株当たり純収入には含まれない。
以下の表は、基本的な1株当たり収益と希釈後の1株当たり収益(1株および1株当たりデータを除いて、千単位)を含む普通株主が1株当たり収益を占めるべき計算方法を示している
|
|
十二月三十一日までの年度 |
|
|
|||||
|
|
2023 |
|
|
2022 |
|
|
||
分子: |
|
|
|
|
|
|
|
||
営業純収入 |
|
$ |
( |
) |
|
$ |
|
|
|
より少ない収益:未分配収益を非持株権益に分配する |
|
|
|
|
|
|
|
||
普通株主は純収益を占めるべきである−基本収益と希薄収益− |
|
$ |
( |
) |
|
$ |
|
|
|
|
|
|
|
|
|
|
|
||
分母: |
|
|
|
|
|
|
|
||
発行された基本普通株: |
|
|
|
|
|
|
|
||
*加重平均普通株式が発行されました |
|
|
|
|
|
|
|
||
普通株主の1株当たり純(損失)収益を算出するための加重平均株式、基本 |
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
||
希釈性潜在普通株: |
|
|
|
|
|
|
|
||
*普通株式オプション加重平均値 |
|
|
— |
|
|
|
|
|
|
普通株主の1株当たり純(損失)収益の加重平均株式を計算し、希釈した後 |
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
||
普通株主1株当たり純(損失)収益: |
|
|
|
|
|
|
|
||
* |
|
$ |
( |
) |
|
$ |
|
|
|
*希釈後 |
|
$ |
( |
) |
|
$ |
|
|
|
各償却計算に含まれていない潜在的希薄化証券は以下の通りである
|
|
十二月三十一日 |
|
|
|
|
|
2023 |
|
|
|
普通株購入オプション |
|
|
|
|
|
転換可能優先株(換算後) |
|
|
|
|
|
優先株式証(転換済み) |
|
|
|
|
|
合計する |
|
|
|
|
|
17. |
従業員福祉計画 |
内地中国貢献計画
中国の関連法規によると、当社は中国省市政府組織の各種固定払込計画に貢献しなければならない。各従業員の支払いは、現地政府が要求した従業員の現在の報酬の一定の割合に基づいている。拠出金は中央年金法の規則に従って支払う際に利益または損失を記入する。総数
F-37
このような従業員の福祉の供給は$
固定払込貯蓄計画
米国では、同社は改正された1986年の国内収入法第401(K)条に基づいて固定払込貯蓄計画を維持している。この計画は最低年齢とサービス年限の要求に適合する従業員に適用される。2023年12月31日までの年間での貢献は微々たるものである。
18. |
市場情報を細分化する |
同社は合併された実体であり、2つの異なる経営部門から構成されている:BCとGyre。同社の報告可能部門は内部組織構造、管理業務の方式、CODMが部門業績を評価するための基準、単独財務情報の可用性及び全体的な重要性考慮要素に基づいている。Gyreのすべての業務は米国内にあり、BCのすべての業務は大陸中国にある。
紀元前紀元前
BCは1種の主要な商業薬物製品ETUARY、及びいくつかの臨床前と臨床開発の候補製品がある。BCの製品収入は主にETUARYといくつかの模造薬の販売から来ている。BCは主に中国の流通業者にその薬品を販売し、流通業者は最終的に製品を病院、他の医療機関、薬局に販売する。BCはまたライセンスプロトコルから収入を得る.しかし、ライセンス契約は2023年12月31日までの1年間何の収入も生じておらず、2022年12月31日までの年度でも実質的な収入は生じていない。
旋回する
GYREはバイオ製薬会社であり,米国でNASH関連肝線維化の治療に専念しているF 351の開発と商業化である。Gyreには、米国のF 351に関連する知的財産権を除いて、同社が出資終了前にすべての伝統的な知的財産権資産を売却したため、他に候補製品がない。寄付が終わった後、Gyreは何の収入も生じなかった。
2023年12月31日と2022年12月31日までの年度支部情報は以下の通り(単位:千)
F-38
|
|
2023年12月31日までの年度 |
|
|||||||||||||
|
|
紀元前紀元前 |
|
|
旋回する |
|
|
他にも |
|
|
統合された |
|
||||
収入.収入 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
収入コスト |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
毛利 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
収入コストは含まれていません |
|
|
|
|
|
|
|
|
|
|
|
|
||||
販売とマーケティング |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
研究開発 |
|
|
|
|
|
|
|
|
— |
|
|
|
|
|||
一般と行政 |
|
|
|
|
|
|
|
|
|
(1) |
|
|
||||
現在行われている研究と開発を買収する |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
資産剥離損失 |
|
|
— |
|
|
|
— |
|
|
|
|
(2) |
|
|
||
財産と設備処分損失 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
収入コストを含まない総運営費 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
営業収入(赤字) |
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
株式ベースの報酬を補充開示する |
|
|
|
|
|
|
|
|
|
|
|
|
||||
収入コスト |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
販売とマーケティング |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
研究開発 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
一般と行政 |
|
|
|
|
|
|
|
|
— |
|
|
|
|
|||
株に基づく報酬総額 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|||
(1) $
(2) $
|
|
2023年12月31日 |
|
|||||||||||||
|
|
紀元前紀元前 |
|
|
旋回する |
|
|
他にも |
|
|
統合された |
|
||||
総資産 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
|
|
2022年12月31日までの年度 |
|
|||||||||||||
|
|
紀元前紀元前 |
|
|
旋回する |
|
|
他にも |
|
|
統合された |
|
||||
財産と設備を購入する |
|
$ |
( |
) |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
( |
) |
|
|
2022年12月31日までの年度 |
|
|||||||||||||
|
|
紀元前紀元前 |
|
|
旋回する |
|
|
他にも |
|
|
統合された |
|
||||
収入.収入 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
収入コスト |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
毛利 |
|
|
|
|
|
— |
|
|
|
|
|
|
|
|||
収入コストは含まれていません |
|
|
|
|
|
|
|
|
|
|
|
|
||||
販売とマーケティング |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
研究開発 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
一般と行政 |
|
|
|
|
|
— |
|
|
|
|
(3) |
|
|
|||
収入コストを含まない総運営費 |
|
|
|
|
|
— |
|
|
|
|
|
|
|
|||
営業収入(赤字) |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
株式ベースの報酬を補充開示する |
|
|
|
|
|
|
|
|
|
|
|
|
||||
収入コスト |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
販売とマーケティング |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
研究開発 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
一般と行政 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
株に基づく報酬総額 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
(3) $
F-39
|
|
2022年12月31日 |
|
|||||||||||||
|
|
紀元前紀元前 |
|
|
旋回する |
|
|
他にも |
|
|
統合された |
|
||||
総資産 |
|
$ |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
|||
|
|
2022年12月31日までの年度 |
|
|||||||||||||
|
|
紀元前紀元前 |
|
|
旋回する |
|
|
他にも |
|
|
統合された |
|
||||
財産と設備を購入する |
|
$ |
( |
) |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
( |
) |
19. |
後続事件 |
転換優先株の転換
2023年11月22日、GNIは会社に通知を出し、
F-40
親会社その他の財務情報
財務諸表付表I
GYRE治療会社
簡明総合貸借対照表
(千単位で、株や1株当たりの金額は含まれていない)
|
|
2023年12月31日 |
|
|
2022年12月31日 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
関係者の売掛金 |
|
$ |
|
|
$ |
|
||
*流動資産全体 |
|
|
|
|
|
|
||
子会社への投資 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
負債、転換可能な優先株、株主権益 |
|
|
|
|
|
|
||
流動負債: |
|
|
|
|
|
|
||
関係者の都合で |
|
$ |
|
|
$ |
|
||
費用とその他の流動負債を計算しなければならない |
|
|
|
|
|
|
||
流動負債総額: |
|
|
|
|
|
|
||
転換可能優先株、$ |
|
|
|
|
|
|
||
株主(損失)権益: |
|
|
|
|
|
|
||
普通株、$ |
|
|
|
|
|
|
||
追加実収資本 |
|
|
|
|
|
|
||
法定準備金 |
|
|
|
|
|
|
||
(累積損失)利益を残す |
|
|
( |
) |
|
|
|
|
その他の総合損失を累計する |
|
|
( |
) |
|
|
( |
) |
株主権益総額 |
|
|
( |
) |
|
|
|
|
総負債、転換可能優先株、株主(損失)権益 |
|
$ |
|
|
$ |
|
||
付記はこのような簡明な総合財務諸表の構成要素である
F-41
GYRE治療会社
簡明総合業務報告書
(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
運営費用: |
|
|
|
|
|
|
||
一般と行政 |
|
$ |
|
|
$ |
|
||
資産剥離損失 |
|
|
|
|
|
— |
|
|
子会社未計権益の赤字収入 |
|
|
|
|
|
|
||
子会社権益収入 |
|
|
( |
) |
|
|
|
|
純収益 |
|
$ |
( |
) |
|
$ |
|
|
その他総合収入,税引き後純額: |
|
|
|
|
|
|
||
外貨換算調整 |
|
|
( |
) |
|
|
( |
) |
総合損失 |
|
$ |
( |
) |
|
$ |
( |
) |
付記はこのような簡明な総合財務諸表の構成要素である。
F-42
GYRE治療会社
キャッシュフロー表簡明連結報告書
(単位:千)
|
十二月三十一日までの年度 |
|
|||||
|
2023 |
|
|
2022 |
|
||
経営活動 |
|
|
|
|
|
||
純収益 |
$ |
( |
) |
|
$ |
|
|
純(損失)収入と経営活動のための現金純額を調整する: |
|
|
|
|
|
||
子会社権益損失 |
|
|
|
|
( |
) |
|
資産剥離損失 |
|
|
|
|
— |
|
|
経営性資産と負債変動状況: |
|
|
|
|
|
||
関係者の都合で |
|
|
|
|
|
||
経営活動が提供する現金純額 |
|
( |
) |
|
|
( |
) |
現金および現金等価物に及ぼす為替レート変動の影響 |
|
|
|
|
|
||
現金と現金等価物の純変化 |
|
— |
|
|
|
— |
|
期初の現金と現金等価物 |
|
— |
|
|
|
— |
|
期末現金および現金等価物 |
$ |
— |
|
|
$ |
— |
|
付記はこのような簡明な総合財務諸表の構成要素である
F-43
備考
F-44