アメリカ アメリカ
証券取引委員会
ワシントンD.C.,20549
表
締め切りの財政年度について
あるいは…。
への過渡期について
手数料ファイル番号:
(登録者の正確な名称は、その定款に規定されている名称と同じ)
| 番号:
| ||
(州や他の管轄区域 会社や組織) |
(I.R.S.雇用主 標識 番号) |
(主な実行機関アドレス )(郵便番号)
(登録者の電話番号、市外局番を含む)
同法第12条(B)に基づいて登録された証券:
| クラスごとのタイトル | 取引 記号 | 登録された各取引所の名称 | ||
同法第12(G)条により登録された証券:なし。
登録者が証券法規則405で定義されている有名な経験豊富な発行者であれば、再選択マークで を示してください。
はい
☐
登録者が当該法第13条又は第15条に基づいて報告書を提出する必要がない場合は,複選マークで示してください。
はい
☐
再選択マークは、登録者が、(1)過去12ヶ月以内(または登録者がそのような報告の提出を要求された短い期間内)に、1934年の証券取引法第13または15(D)節に提出されたすべての報告書を提出したかどうか、および(2) が過去90日以内にそのような提出要件に適合しているかどうかを示す。
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−Tルール(本章232.405節)405条に従って提出を要求した各対話データファイルを電子的に提出したか否かを示す。
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな申告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小型報告会社”、“新興成長型会社”の定義を参照してください
| 大型 加速ファイルサーバ | ☐ | 加速した ファイルマネージャ | ☐ | |
| ☒ | 小さな報告会社 |
新興成長型会社
もしbrが新興成長型会社である場合、登録者が延長された移行期間を使用しないことを選択したかどうかを再選択マークで示して、取引法第13(A)節に従って提供された任意の新しいまたは改正された財務会計基準を遵守してください
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オキシリー法案”(“米国法典”第15編7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価し、その監査報告を作成または発表した登録公共会計会社が評価する
証券が当該法第12(B)条に基づいて登録されている場合は、届出書類に含まれる登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かをチェックマークで示してください
これらのエラーのより真ん中に再記述があるかどうかをチェックマークで示すことは、登録者の任意の幹部が関連する回復中に受信したインセンティブベースの報酬を§240.10 D−1(B)に従って回復分析する必要があるかどうかを示す
登録者が空殻会社であるかどうかをチェックマークで表す(同法第12 b-2条で定義されている)。はい、違います
普通株の2023年6月30日の売却価格から計算すると,非関連会社が保有する投票権と無投票権普通株の総時価は約$である
2024年3月18日までに登録者の普通株は、額面0.0001ドル、発行されている。
参照により組み込まれた文書
MATINASバイオ製薬ホールディングス
表格10-K年次報告
2023年12月31日までの財政年度
カタログ表
| ページ | ||
| 第1部 | 1 | |
| 第 項1. | 業務.業務 | 2 |
| 1 a項目. | リスク要因 | 40 |
| 項目 1 B. | ネットワーク·セキュリティ | 68 |
| 第 項2. | 属性 | 69 |
| 第 項3. | 法律訴訟 | 69 |
| 第 項. | 炭鉱安全情報開示 | 69 |
| 第II部 | 69 | |
| 第 項5. | 登録者普通株市場、関連株主事項及び発行者による株式証券の購入 | 69 |
| 第 項6. | [保留されている] | 69 |
| 第 項7. | 経営陣の財務状況と経営成果の検討と分析 | 70 |
| 第 7 A項。 | 市場リスクの定量的·定性的開示について | 75 |
| 第 項8. | 財務諸表と補足データ | 76 |
| 第 項9. | 会計と財務情報開示の変更と相違 | 76 |
| 第 9 A項。 | 制御とプログラム | 76 |
| 第 9 B項。 | その他の情報 | 77 |
| 第三部 | 77 | |
| 第 項10. | 役員·幹部と会社の管理 | 77 |
| 第 項11. | 役員報酬 | 82 |
| 第 項12. | 特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 | 89 |
| 第 項13. | 特定の関係、関連取引、役員の独立性 | 90 |
| 第 項14. | 最高料金とサービス | 91 |
| 第4部 | 92 | |
| 第 項15. | 展示品と財務諸表の付表 | 92 |
| 第 項16. | 表格10-Kの概要 | 93 |
| 財務諸表 | F-1 | |
| i |
第 部分I
前向き陳述に関する警告的説明
本“Form 10-K”報告書には、“1995年個人証券訴訟改革法”(改正された“1933年証券法”第27 A節)及び“1934年証券取引法”(改正証券取引法“第21 E節)に基づく安全港条項による前向きな陳述が含まれている。展望性表現は、私たちの信念、計画、目標、目標、期待、予想、仮説、推定、意図と未来表現に関する表現を含み、既知と未知のリスク、不確定性およびその他の要素に関連し、これらのリスク、不確定性および他の要素は私たちがコントロールできない可能性があり、私たちの実際の結果、業績または成果はこのような前向き表現と明示的あるいは暗示的な未来の結果、業績または達成とは大きく異なる可能性がある。 歴史的事実以外のすべての陳述は前向き陳述である.これらの前向き陳述は、“可能”、“予想”、“仮定”、“すべき”、“表示”、“信じ”、“考慮”、“予想”、“求める”、“推定”、“継続”、“計画”、“指向”、“プロジェクト”、“予測”、“可能”などの言葉を使用することによって識別することができる。“ ”“意図”“目標”“潜在”や他の類似した言葉や未来を表す.
多くの重要な要素は、実際の結果が私たちが発表した任意の前向き声明で表現された結果と大きく異なる可能性がある。これらの要素にはこれらに限定されない
| ● | 私たちは追加資金を集めて、私たちの運営に資金を提供し、私たちの候補製品を開発することができます |
| ● | 私たちの臨床前開発、法規提出、臨床試験の開始と完成、および製品承認の予想時間 |
| ● | 私たちは設立以来毎年の営業赤字の歴史と、予測可能な未来に引き続き営業赤字を被ると予想されています |
| ● | 私たちはまだ初期開発段階にある候補製品に依存しています |
| ● | 私たちの独自の脂質ナノ結晶(LNC)プラットフォーム輸送技術への依存とロッグス大学(Rutgers University)が私たちに与えたいくつかの関連特許を独占的に許可しています |
| ● | 私たちの臨床前および臨床試験に必要な候補製品GMPロットを生産する能力、そしてその後、私たちの任意の製品が規制部門の許可を得たら、私たちは商業ロットを生産する能力 |
| ● | 私たちは私たちの主要な候補製品と他の候補製品に必要な臨床試験を完成し、異なる司法管轄区のFDAまたは他の監督機関の許可を得ることができる |
| ● | 私たちは中間体や最終製品レシピを生産する第三者や臨床試験を行う第三者契約研究機関を含む第三者に依存しています |
| ● | 私たちの特許と他の知的財産権の有効性を維持または保護する能力; |
| ● | 私たちはキーパーソンの能力を維持して募集します |
| ● | 私たちは内部で新しい発明と知的財産権の能力を開発しています |
| ● | 現在の法律の解釈と未来の法律の段落 |
| ● | もし私たちが単独でまたは潜在的な未来のパートナーを通じて規制部門の承認を得たら、私たちは販売とマーケティング組織と私たちが製品を商業化する能力が足りない |
| ● | 私たちは製品の商業化に成功する能力と、候補製品の将来の治療と商業潜在力への期待を持っている |
| 1 |
| ● | 私たちは費用、持続的な損失、未来の収入、資本需要、および追加融資の推定の正確さを得る必要があるか、または得ることができる |
| ● | 私たちの競争相手や私たちの産業に関連した発展と予測; |
| ● | 私たちの業務、業務、財務業績は武力衝突、流行病、地政学的不確実性による世界的不安定の悪影響を受ける可能性がある。 |
これらの展望的陳述は、我々の経営陣の未来の事件に対する信念と見方を反映し、本年度報告10-K表日までの推定と仮定に基づいて、リスクと不確実性の影響を受ける。私たちは“リスク要因”の節でその中の多くのリスクについてより詳細に議論した。また、我々の運営環境は競争が激しく、めまぐるしく変化しています。 新たなリスクがしばしば発生しています。私たちの経営陣はすべてのリスクを予測することができず、すべての要素が私たちの業務に与える影響を評価することもできません。あるいは任意の要素や要素の組み合わせは、実際の結果が私たちが行う可能性のある任意の前向き陳述に含まれる結果と大きく異なる程度をもたらす可能性があります。このような不確実性を考慮して、あなたはこのような展望的なbr陳述に過度に依存してはいけない。
本Form 10-K年次報告および私たちが引用して証拠品としてForm Form 10-K年次報告書に提出した文書を完全に読まなければなりません。私たちの将来の実際の結果は、私たちが予想していたものとは大きく異なる可能性があることを理解してください。我々は,これらの警告的声明により,本年度報告の10-K表中のすべての前向き陳述を限定した.法律が別に要求されない限り、私たち は、新しい情報、未来のイベント、または他の理由でも、いかなる前向きな陳述を公開更新する義務を負わない。
| 第 項1 | 業務.業務 |
会社 概要
私たちのbrは臨床段階の生物製薬会社であり、私たちの脂質ナノ結晶(LNC)プラットフォーム 輸送技術(LNCプラットフォーム)を用いて画期的な治療を提供することに集中している。我々は,LNCプラットフォームを利用した内部製品パイプラインの開発を求めており,小分子とオリゴヌクレオチドを成功させ,毒性を生じることなく所望の細胞や組織への標的と肝外輸送を促進する。また、LNCプラットフォームはまた、LNCプラットフォームの独特な特性を利用して、小分子、アンチセンスオリゴヌクレオチド(ASO)と沈黙或いは短干渉RNA(SiRNA)を含む複雑分子の伝達を促進、増強、最適化するために、新型調合の開発を求めるリード製薬会社のために外部製品導管を構築することを支持することができると信じている。
著者らの現在の主要な候補製品はMAT 2203(両性マイシンBの内服)であり、これは1種の高度に有効な抗真菌薬物であり、LNC送達を通じて、brはすでに経口、安全と耐性の良好な長期投与に製造され、生命を脅かす侵襲性真菌感染患者に応用されている。クリプトコッカス性髄膜炎治療のACT 2期試験に成功した後,MAT 2203は現在,新薬申請(NDA)を支援し,選択の限られた患者の侵襲性アスペルギルス症の治療に用いる単一の3期登録試験(ORALTO試験)を行う。
他の 内部発見計画が現在対象としているのは:
| 1. | 腫瘍学的応用に安全で有効な化学療法薬を提供する | |
| 2. | アンチセンスオリゴヌクレオチド(ASO)とサイレンシング或いは短干渉RNA(SiRNAs)などの小分子オリゴヌクレオチドを形成と伝達し、主な治療重点は炎症である。 |
我々 は,我々独自のLNCプラットフォームに関する価値を最大化することに取り組んでいる.この独自プラットフォーム技術は部分的にロッグス大学のある知的財産権に対する全世界の独占許可に依存し、ナノカプセル化化学と生物 ペイロードの方式で各種の分子の細胞内での安全、高効率と配向輸送を促進した。LNCは、小分子、ポリペプチド、タンパク質および小分子オリゴヌクレオチドを含む様々な治療用化合物のインビボ送達に成功している。重要なことは,LNCは他の脂質系ナノ粒子伝達技術とよく異なることである。
| 2 |
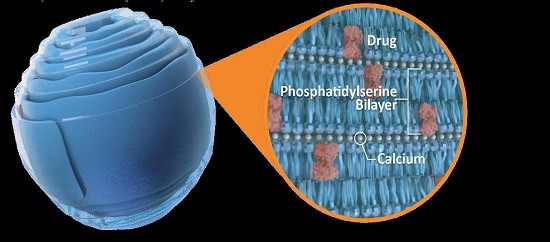
我々のLNCは主にリン脂質、例えばホスファチジルセリン(PS)、カルシウム(LNCを無傷に維持するために必要な)と指定された治療貨物から構成され、これらの貨物はLNC二重層内あるいは二層の間に閉じ込められ、高度に安定した結晶構造を形成し、貨物を保護し、胃腸条件下での経口生存を許可する。細胞外,例えば消化管液体や血液中では,正常な細胞外カルシウムレベルはこの結晶構造を維持しているが,組織ではLNCは専門的な貪食細胞や感染,炎症,悪性細胞に貪欲に吸収される。低カルシウムの細胞質環境では,LNCはその結晶構造を失い,細胞内で貨物を放出する。
LNC は新しい標的性を有し、PSを利用して膜融合とPS受容体を介したエンドサイトーシス作用を促進する。近接している治療標的によれば、貨物は特定の細胞内で直接影響を与える可能性があり、または、好中球などの血球は、感染または炎症の場合など、標的組織に担体として間接的に貨物を輸送することができる。LNCは、脂質ナノ粒子(LNP)送達とは異なり、中性免疫原性を有し、繰り返し投与することができ、脂質ナノ粒子(LNP)送達は、固有の細胞毒性により再投与できない可能性もある。ユニークなのは,LNCの高度に安定した構造が経口投与(LNP不可能)を可能にし,LNC構造内の貨物が保護されているため,胃腸の劣悪な環境の影響を受けないことである。
臨床前と臨床研究は一致して、LNCの経口投与は治療貨物の輸送に成功できることを表明した体内にある感染部位,炎症領域,腫瘍まで である。これらすべての標的組織は,その表面に曝露されたPS細胞(これは細胞膜との直接融合のための機会を創出した)と,組織が存在する専門的な貪食細胞上の特定のPS認識受容体を有する(これはエンドサイトーシスによる追加の細胞吸収に寄与する)。病変組織を標的とする能力と標的外効果を潜在的に回避する能力--融合とエンドサイトーシス機構を通じて細胞に入る能力、及びLNC被覆と広範な治療薬物を輸送する能力--広範な内部製品候補と外部パートナー関係を創立するために堅固な基礎を提供した。近年治療的核酸の輸送は大きく進歩しているが,これまで経口投与されてきたオリゴヌクレオチド療法は肝臓に対する結合オリゴヌクレオチド以外はほとんど進展していない。同様に,小分子(安全性と有効性を増強する潜在力を有する)や小オリゴヌクレオチドは腫瘍学分野においても大きな潜在的機会がある。
MAT 2203
著者らのLNCプラットフォームに基づく主要な候補薬物はMAT 2203であり、これは両性マイシンBの経口製剤であり、有名かつ高効率な抗真菌薬である。両性マイシンBは現在深刻な腎毒性に関連する静脈製剤でのみ使用可能であり、そしてすでにアメリカで の使用制限は2週間に達し、世界の大多数の地区では1週間しかなく、原因は毒性であり、その中で最も一般的なのは深刻な腎毒性である。これらの限界にもかかわらず、両性マイシンBはその効力のため、現在、様々な侵襲性および潜在的な致命的な真菌感染の治療に使用され、許可されている。著者らのLNCプラットフォームを用いて調製したMAT 2203はbrを保持し、両性マイシンBの治療効果を高める潜在力があり、同時に腎毒性リスクを除去し、更に便利かつ費用効果のある経口投与を提供した。MAT 2203のS製品プロファイルは,医師と患者がMAT 2203を使用する時間と範囲が,これまで外来環境で使用されていた両性マイシンBよりも長く,広いことを可能にしている。
MAT 2203はこれまで,米国国立衛生研究院(NIH)の国家アレルギー·感染症研究所(NIAID)の援助と財政支援の下で開発されてきた。MAT 2203は、侵襲性カンジダ症、アスペルギルス症、免疫抑制治療を受けている患者のIFIを予防するための迅速チャネル 状態を有する適格な感染症製品(QIDP)に指定されており、最近ではクリプトコッカス病治療孤児にも指定されている。著者らはより多くの孤児名brを求めてアスペルギルス症、侵襲性カンジダ症とある地方性真菌症の治療に応用する予定である。承認後、 MAT 2203は米国で最長12年間の規制やマーケティング排他性を得る資格がある。
MAT 2203の最初の適応は、侵襲性アスペルギルス症の治療に使用するために、治療選択が限られた患者において両性マイシンBを早期に中止することである。侵襲性アスペルギルス症は1種の深刻で、生命に危害を及ぼす侵襲性真菌感染であり、主に免疫機能が深刻に損傷した血液系悪性腫瘍患者と移植レシピエントに発生する。最初の降圧適応 は入門適応であり,MAT 2203を他の侵襲性真菌感染(IFI)の治療 に拡大することを計画しているため,免疫障害患者(例えば移植患者)のIFIの予防にも利用可能である。
| 3 |
制定(両性マイシン内服によるクリプトコッカス性髄膜炎治療の臨床試験)第2段階研究は、NIH NINDSによって援助され、100人のHIV陽性クリプトコッカス性髄膜炎患者におけるMAT 2203の安全性、耐性および有効性を評価する第2段階展望性、ランダム、開放ラベル、順序コホート研究である。ACT試験は合計4人の患者行列を含み,前の2つの待ち行列はMAT 2203試験を14日間誘導期に両性マイシンBを静脈内投与した後の早期降圧治療であり,後の2つの待ち行列試験MAT 2203は可能な全経口治療であった。各キュー内のすべての患者(活動または対照)の誘導期間は14日であり、その後、強化/維持中に周囲の治療(活動または対照) が追加的に行われる。キュー1と3はそれぞれキュー2と4の安全な誘導であり,キュー2と4は策定された重要な治療効果キューである.
ACTEの主要な終点は早期殺菌活性(EFA)であり、これは脳脊髄液真菌除去の測定基準である。EFAは抗真菌薬物の治療効果の有効な定量化指標であり、生存の重要な代替マーカーでもある。0.20対数未満のEFA10 毎日1ミリリットルあたりの脳脊髄液中のクリプトコッカスコロニー形成単位(CFU)は明らかに高い死亡率と比較的に悪い臨床結果と関係がある1それは.この閾値を超えて測定したEFAは臨床的意義があり、強力な真菌除去能力を代表している。ACTEの2番目のキューにおいて,MAT 2203を用いて治療した患者の平均EFAは0.38 logであった10CFU/ml/日、95%信頼区間 (0.30~0.46)は、予め指定された>0.20の主要終点閾値よりも有意に高い。MAT 2203を用いて誘導期を完了したすべての患者は、治療期間中(誘導期または早期強固段階にかかわらず)に無菌脳脊髄液培養を行った。MAT 2203治療期間から10週間の間、どの患者にも突破または再発を認めたクリプトコッカス感染の証拠は認められなかった。キュー2では,ランダムにMAT 2203治療を受けた40名の患者のうち,18週間後の総生存率は90%であった。
2022年10月,IDWeekはIDWeek上でMAT 2203(両性マイシンB内服)によるクリプトコッカス性髄膜炎(CM)治療の第2段階ACT研究第4キューの中期データを公表した。IDWeekの一部として,ACT要約は米国感染症学会から授与された優れた要約賞とIDSA賞を受賞した。ACT試験では,MAT 2203は早期殺菌活性(EFA)の主要終点閾値0.20 logを超えた10CFU/ml/日,平均EFAは0.30対数である10CFU/ml/日、95%信頼区間は0.22~0.38であった。
キュー4はまた、全体生存率および安全性を含む重要な二次終点を生成する。MAT 2203治療を受けた40名の患者のうち,総生存率は18週間で90%を維持していたが,2週目の生存率は95%であった(これは間もなく行われるクリプトコッカス性髄膜炎第三段階登録試験の主な終点である)。重要なことは,ACT試験全体において,従来のIV型両性マイシンB標準看護治療と比較して,MAT 2203治療は腎機能や貧血に関連する有害事象の発生率が有意に低下し,MAT 2203経口治療が6週間に及ぶ場合でも腎臓毒性の証拠がないことである。
2024年2月、我々はMAT 2203登録経路のすべての重要な要素 について合意(ORALTO試験)を含む、限られた侵襲性アスペルギルス症患者の治療選択においてMAT 2203の単一3期登録試験 を設計することを含む米国食品医薬品局(FDA)と合意したことを発表した。
ORALTO は3期ランダム、多中心、開放ラベル、審判者盲法研究であり、限られた侵襲性アスペルギルス症患者の治療選択の看護標準と比較し、AmBisome(リポソームIV-両性マイシンB)を用いた治療後の経口減少療法としてのMAT 2203の有効性と安全性を評価することを目的としている。主な治療効果の終点は研究42日目の全原因死亡率である。
主な の副次的な目標は:
1*臨床でDisに感染する2020;71(5):e45-49
| 4 |
| (a) | 治療関連毒性による治療変化(即ち、用量調整/中断或いは治療方案の変化)において、経口減量治療はアミノメチルジオキシピリミジンと比較した優位性を示した | |
| (b) | 研究84日目の全原因死亡率を用いてMAT 2203の長期生存効果を計算した | |
| (c) | MAT 2203が医療資源利用と生活の質に及ぼす影響を評価した。 |
登録brは、カビ活性アゾールの静脈内投与が受けられず、代替治療選択が限られているため、最近侵襲性アスペルギルス症と診断または確認された成人約216人を含むことが予想される。AmBisomeの最初の治療を受けた2日後には、条件を満たす研究参加者が研究に組み込まれ、mT 2203の経口投与またはAmBisome治療を2:1の割合でランダムに受け、標準看護を受けた。
すべての研究参加者はAmBisome治療を受けた初日から最長12週間の治療を受ける。すべての研究参加者が最初のAmBisome治療期間中に入院する予定である。MAT 2203内服後,研究参加者は退院する可能性があり,臨床状況に応じて外来治療を継続した。
独立したデータ審査委員会は、治療を無視し、臨床、放射線学、および真菌学的反応を含む主要かつ副次的な終点を裁くであろう。約75%の参加者が登録されると、独立したデータセキュリティ監視委員会は、サンプル量の仮定が合理的であることを保証し、研究が十分に支持されることを確実にするために、全体的に集約された全原因死亡率を盲目的に検討するであろう。集約されたイベントが期待レベルと大きく異なる場合には,実験に対してサンプル量調整を行うことができる.
ORALTO は米国,ヨーロッパ,南米,中東,アジア太平洋地域の約65地点で行われる。登録作業は2024年下半期に開始予定で、約24カ月かかる予定だ。
私たち は、MAT 2203の持続的な協力プロセスに積極的に参加しており、1つ以上の開発および/または商業化パートナーを探しています。 我々は、(I)協力取引の完了を要求するか、または(Ii) ORALTO試験を開始する前に追加資金を調達することを要求します。
ACT実験に加えて,MAT 2203慈悲/拡張使用アクセス計画を構築し,慈悲使用に基づいてMAT 2203を提供した.この計画に参加するためには、患者申請者はいくつかの資格基準を満たさなければならない
| ● | Br}患者には他の治療選択はなかった。 |
| ● | 侵襲性真菌感染は深刻であり、および/または生命に危害を及ぼす。 |
| ● | 患者はMAT 2203内服治療から利益を得、経口薬に耐えることができると予想される |
| ● | 患者の期待寿命は合理的であり,彼らの基本的な状況もコントロールされている。 |
これまでに19名の患者が複数の医療機関に登録されており,ミシガン大学,ジョン·ホプキンス大学,全国児童病院,希望の城,ファンデルビルト大学医療センター,国家衛生研究院,フィラデルフィア児童病院,スタローン·キャトリン癌センター,カリフォルニア大学サンディエゴ医学院を含む。登録されている患者の多くは移植後患者あるいは潜在的悪性腫瘍の治療を受けている。MAT 2203を使用して治療される感染には、様々な微生物(を含む)が含まれるコウジカビ, 毛かび属種, カンジダ症, 鎌菌 疑いを抱いています球胞子虫)脳、膀胱/結腸、骨、肺、副鼻腔および皮膚を含む複数の感染部位で発生する。多くの患者は登録前にAmBisome治療を受けているが,治療制限的な腎毒性が出現しており,多くの患者では薬剤耐性菌の治療が必要であったり,臨床的にアゾール系治療が行われておらず,他の治療選択はない。
この計画に参加した19名の患者のうち,15名の患者がフォローアップを受けており,4名の患者が最近開始またはMAT 2203による治療を開始している。
| 5 |
| ● | 15例の患者の中に12例の臨床症状は完全に緩和或いは感染指標が客観的に改善した(放射線学と真菌学を含む)。 | |
| ● | 完全な個人指定治療コース(2から1年,感染状況に依存)を達成した5名の患者のうち,すべての患者が完全な臨床緩解を得ており,再発や感染再発はなかった | |
| ● | 他の5名の患者の臨床指標は客観的に改善し,計画的にMAT 2203を継続して治療を行っている | |
| ● | 15名の患者のうち5名は、2名の患者の真菌感染が臨床的に有意に改善されたにもかかわらず、12個の全体的に成功した症例に含まれているにもかかわらず、それぞれの指定された治療コースを達成できなかった。 |
| – | 2名の患者は,悪性疾患が予期せぬ進展を示したため,MAT 2203治療開始直後に緩和ケアに移行した | |
| – | 1人の患者は、2日後に潜在的胃腸問題(すなわちクローン病)のために治療を中止した。 | |
| – | 1人の患者は、治療約8週間後に潜在的疾患の進行によって死亡した(しかし、以前の彼らの真菌感染は有意な臨床的改善があった) | |
| – | 1例の患者は治療10週間後に潜在的な胃腸問題(長期吐き気/嘔吐)のためMAT 2203治療を中止したが、彼らの真菌感染は改善した。 |
重要なことは,AmBisome治療後に腎毒性が出現したすべての患者が,MAT 2203治療に移行した後,腎機能はベースラインレベルに回復し,MAT 2203治療を延長する過程でさらなる腎臓副作用を受けなかったことである。
LNC プラットフォーム動作
MAT 2203をORALTO第三段階試験に進めるほか,LNCプラットフォームの使用を拡大し,小分子とオリゴヌクレオチドbrを感染症に応用したほか,炎症や腫瘍学についても検討した。
炎症が起こる
我々は,炎症性サイトカインIL−17 Aと腫瘍壊死因子α の2種類の小分子オリゴヌクレオチドに対する各種LNC製剤を検討し,一連の研究を行っている体外培養そして体内にあるミキノモート(Imiquimod)誘導マウス乾癬モデルにおいて、経口に関連する生物活性およびIL-17 Aノックアウトに関連する臨床的利益を評価する研究。
これらの初歩的な研究はサイトカイン抑制形式の生物活性を記録し、そしていくつかの証拠を提供し、このような定性の乾癬モデルにおいて、皮膚損傷外観(発赤、スケール)の改善は関連する有形臨床利益と関係があることを表明した。これらのデータはまだ評価中であり、分析の重点は、(A)サイトカイン阻害の強度および時間過程を明らかにすること、および(B)これらのモデルにおけるサイトカインmRNAレベルおよび特定の組織反応を評価して、これらのデータをよりよく理解し説明することである。
2023年12月に一連の発表を行いました体内にある研究は2種類のLNC配合の小一本鎖オリゴヌクレオチドの経口投与に成功し、この2種類の小一本鎖オリゴヌクレオチドは重要な炎症性サイトカイン腫瘍壊死因子αとIL-17 Aに特化し、ヒト疾患に出現する急性炎症反応の成熟と検証を模擬した動物モデルにおいて検証を行った。
| 6 |
急性大腸炎研究(“腫瘍壊死因子α”)
デキストラン硫酸ナトリウム誘導マウス大腸炎モデルを用いて、経口LNC送達の特異的に腫瘍壊死因子αm RNAを標的とした小分子オリゴヌクレオチドを評価した。リアルタイム定量ポリメラーゼ連鎖反応分析では,活性αを経口投与すると結腸組織における腫瘍壊死因子αの発現レベルが低下し,罹患しているが未治療動物と比較して血清中腫瘍壊死因子mRNA量が37%有意に低下した。重要なことは,研究における重要な時点の臨床疾患活動性スコアも活性LNC製剤を用いることにより有意に改善したことである。
急性乾癬研究(“IL-17 A”)
イミダキノモート(“ImQ”)を用いて誘導したマウス乾癬モデルは経口LNC送達の小分子オリゴヌクレオチドbrを評価するためにIL-17 A mRNAの合成を抑制することを目的とし、それは乾癬皮膚皮膚損傷の進展に重要な役割を果たしている。DSS大腸炎モデルと類似し、活性LNCを経口投与した後、ImQ乾癬モデル皮膚組織中のIL-17 A mRNAの発現レベルは単純ImQより低かった。このモデルでは,IL−17 A血清レベルは変わらないと予想されるが,臨床疾患の皮膚赤腫や鱗屑マーカーは改善され,さらにこれらの小分子オリゴヌクレオチドの生物活性が検証された。
腫瘍学
2023年11月に私たちは体内にあるドセタキセル経口LNC製剤の動物研究によると、ドセタキセルは1種の有名な化学療法薬物であり、多発性転移と切除できない腫瘍の治療に用いられる。14日目に、毎日LNC-ドセタキセルを内服する抗腫瘍効果はドセタキセル静脈注射に相当し、未治療対照群と比べ、腫瘍体積は明らかに減少し(LNC-63%高用量内服;低用量LNC-57%;ドセタキセル-68%静脈注射)であり、14日目に腫瘍重量は軽減した。全身毒性 は認められなかった。体重は治療期間中に安定し、血液学的パラメータは未治療対照群と類似している。
2024年3月,健常マウスで行ったもう一つのLNC−ドセタキセル体内研究の積極的な結果を発表した。この研究の目的は、LNC製剤ドセタキセルの経口投与がドセタキセルの通常の静脈内投与の全体的な安全性を改善できるかどうかを決定することである。br}この研究は24匹の健康なBALB/cマウスを含み、(1)生理食塩水対照群の経口投与、(2)ドセタキセルの静脈内投与(30 mg/kg、または0.6 mg/用量)、連続3週間、(3)LNC-ドセタキセル(37.5 mg/kg、または0.75 mg/用量)の経口投与、1日1回、3週間の経口投与を含む。この研究の主な終点は治療期間中の体重の変化であり,これはこのモデルにおける毒性の主な表現である。
主な のポイントは以下のとおりである
| ● | 22日目までに,LNC−ドセタキセル製剤を経口投与したドセタキセル総量は,ドセタキセル静脈内投与の8倍以上であった(最終静注用量は薬物動態の測定に用いられた)。 | |
| ● | ドセタキセル静脈注射治療を受けたすべてのマウスの体重(毒性)は明らかに低下し、平均ピーク減少量はその原始体重の20%であった。 | |
| ● | LNC−ドセタキセルを経口投与したマウスは体重を保持しており,生理食塩水を経口投与した対照群マウスの体重と統計学的に差はなかった。 | |
| ● | ドセタキセル静脈内投与群の1匹は研究終了前に死亡し,分析から削除され(最終測定では体重低下br}−32%),ドセタキセル静脈内投与群の曲線もドセタキセル静脈内投与後の予想7日間の回復を反映していた。 | |
| ● | 同遺伝子マウス黒色腫モデルにおいて、毎日LNC-ドセタキセルを経口投与する投与量はLNC-ドセタキセル投与量より50%高く、投与総量はLNC-ドセタキセル投与量の3.5倍であり、この研究はLNC-ドセタキセルの抗腫瘍活性がドセタキセル静脈注射と同等であることを証明した。 |
次のステップは、他の腫瘍モデルにおける現在のLNC-ドセタキセル製剤の治療効果を評価することを含む。また,小分子オリゴヌクレオチドLNC製剤の潜在的抗腫瘍活性を評価する予定である。
| 7 |
戦略.戦略
我々は、小分子、核酸、遺伝子療法、タンパク質/ペプチド、およびワクチンを安全かつ効率的に送達するための現在の課題を克服するために、我々のLNCプラットフォームおよびその応用によって核酸および小分子の細胞内送達 を再定義することに集中している。
私たちの戦略の重要な要素は:
| ● | Br}MAT 2203をORALTO試験に進め,開発および/または商業パートナーを獲得することにより,選択の限られた患者を治療する侵襲性アスペルギルス症を治療した。この初期適応は、必要な薬効学の架け橋を構築する門戸適応となることを目的とし、それによってMAT 2203の使用を他の適応に拡張し、505(B)(2)経路下の限られた追加の臨床仕事によって致命的な侵襲性真菌感染(例えば、毛カビ症、カンジダ症および地方性真菌症)を治療し、それによってMAT 2203を 製品のパイプラインにする |
| ● | Brは我々のLNCプラットフォームと他の小分子や小オリゴヌクレオチドの使用を炎症や腫瘍学分野に拡張し,差別化候補薬物の内部チューブを開発した。経口、肝外、非毒性の細胞内にこれらの分子を送達することは重大な進歩である |
| ● | 著者らのLNCプラットフォームを重点として、有力な製薬会社と外部協力チャネルを構築し、彼らの複雑な小分子と小さいオリゴヌクレオチドにASOとsiRNAを含む送達解決方案 を提供した |
脂質ナノ結晶(LNC)プラットフォームは
安全、高効率と的確な細胞内薬物輸送は依然として現在の製薬と生物技術業界が直面している最大の挑戦の一つである。細胞内の複雑な生物学に対する理解が深まるにつれ、細胞内代謝活動を駆動する遺伝機序に対する治療方法もますます複雑になってきている。しかし、挑戦は依然として存在し、特に重大な毒性を引き起こす小分子、および治療用オリゴヌクレオチドは以下の領域における挑戦である:安全かつ有効に貨物を細胞内部 に輸送し、肝臓以外の標的に輸送し、そして長期経口投与に利便性 を増加させ、潜在的な重大な薬物経済学的影響を産生する。
戦略的には、LNCプラットフォームの独特な機能とホスファチジルセリンの生物学的特性を利用して、効率的だが猛毒な小分子の細胞内送達および小オリゴヌクレオチドの経口投与標的送達のための潜在的な応用 を拡張することを求めている。
LNC -背景
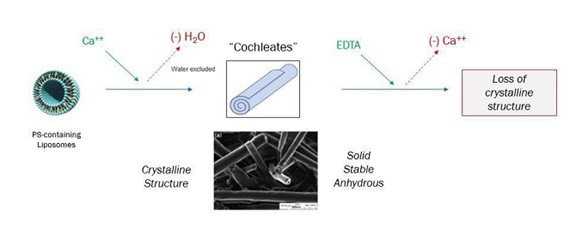
科学文献の最初の記述は,カルシウム添加により形成された複雑,円柱状,多層状構造である++水溶液中のホスファチジルセリン(PS)に関する超音波リポソーム製剤であって、そのシート層が水を含まずに螺旋結晶配置で折り畳まれている超音波リポソーム製剤。これらの構造 は十分な外部カルシウムが存在する場合には結晶状態を維持するが,これらの製剤にエチレンジアミン四酢酸(EDTA)(カルシウム除去) を添加すると安定した螺旋結晶構造が失われる(図1)。
| 8 |
図 1:ウォーム構造
我々はすでにウォームシェル組み立て時に貨物分子を埋め込む技術を開発している。これらの新しいキャリア構造(脂質ナノ結晶、LNCと呼ばれる)は、いくつかの異なる貨物分子を細胞に輸送することに成功している体外培養動物や人間にもそうです体内にあるそれは.その特殊な安定性(無水結晶構造)と独特の組成 (PSを含む二層)から,LNCはASOSやsiRNAのような様々な小分子 −タンパク質,ポリペプチド−および小オリゴヌクレオチドを細胞内で輸送するための有望な代替案であると考えられる。また,腸管中の正常な生理レベルのカルシウムはその結晶構造を維持できるため,包装されたbr貨物は劣悪な環境条件や酵素によって分解されないため,LNC製剤も経口投与される可能性がある(図2)。
図 2:LNCは無水環境で貨物をパッケージして保護する
ホスファチジルセリンの重要性
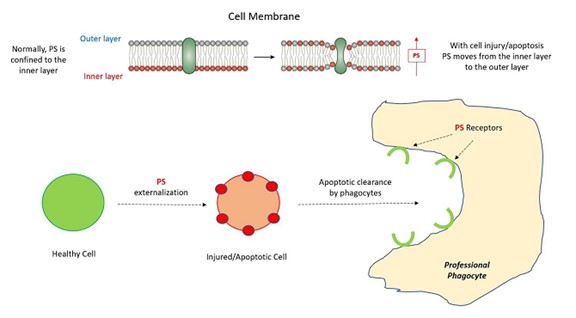
ホスファチジルセリン(PS)はほとんどの細胞に存在し,細胞膜の構成要素である。PSは通常活発な細胞過程で膜二重層の内部に位置づけられ,通常外部にさらされない(図3)。しかし、細胞が損傷および/またはアポトーシスを受けると、PSは内層から外層に移動し、専門的な貪食細胞を駆動して泡飲方式でアポトーシス細胞を除去する主要なシグナルである。
LNCを運搬する貨物に関する研究では,正常で健康な動物の組織では貨物の蓄積が不足しているが,感染した動物では影響を受けた組織に貨物が著しく輸送されていることが証明されている。LNCの潜在的安定性のもう1つの重要な側面は、血液中にその貨物を放出しないことであり(安定したカルシウムレベルが存在するため)、貨物の輸送は影響を受ける細胞および組織に限定される。
| 9 |
機序的には,LNCの細胞進入はPSによって駆動され,PSはアポトーシス細胞の細胞嘔吐除去と生理的細胞融合過程の重要な参加者である(図3)。指摘されているように,その表面にPSを発現するアポトーシス細胞は専門的な貪食細胞によって認識·除去され,正常な炎症反応を引き起こすことはなく,そうでなければ細胞死を招く可能性がある。プロ食細胞自体にはいくつかの非常に特異的なPS受容体があり,この除去に寄与している。ちなみに,エンベロープウイルスはこの機序を利用して免疫細胞のウイルス吸収を促進する“ウイルスアポトーシス擬態” −−エンベロープウイルスの“エンベロープ”は基本的にPSからなることに注意されたい。そのため、専門貪食細胞(いくつかの非専門貪食細胞)のLNCに対する貪食作用はLNC細胞内輸送の機序になっている。
図3:アポトーシス貪食におけるホスファチジルセリン(PS)の重要性
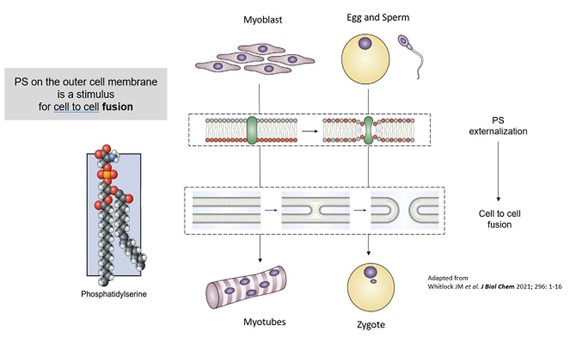
以上のように,貪食細胞としてアポトーシス細胞を吸収する“私を食べる”シグナルに加えて,PSは正常な生理的細胞融合過程において非常に重要な役割を果たしており,“融合私”シグナルとして(例えば,筋芽細胞が筋管を形成し,骨芽細胞が破骨細胞を形成し,精子受精が受精卵を形成して受精卵を形成する) さらには“私を修復する”シグナル(例えば,損傷や損傷細胞膜を修復した後の軸索融合)としている。したがって,LNC表面と標的細胞表面(損傷や炎症によりPSを発現する可能性がある)におけるPSの存在は,直接細胞融合による細胞内伝達のための追加の機会を創出する(図4)。
| 10 |
図4−ホスファチジルセリン(PS)の細胞融合における重要性
LNC薬物送達
その潜在的な安定性のため,LNCは血液中に貨物(安定したカルシウムレベルが存在するため)を放出することはなく,貨物の輸送は影響を受ける細胞や組織に限られる。放射性標識両性マイシンを用いたLNC製剤の研究では,brは正常な健康組織に蓄積されていないことが示されているが,多くの研究により,全身性真菌感染の場合,両性マイシンを携帯するLNCは臨床的意義のある組織レベルを有することが示唆されている。
経口投与後,LNCsは胃腸内の細胞を介して輸送される(細胞輸送)。その大きさによりLNCsは門脈循環に入らないため,肝臓の初過代謝は回避された。代わりに腸管リンパ管を介して胸腔カテーテルに輸送され,上大静脈を介して循環系に入る。それらが循環に入ると,LNCは専門的な貪食細胞(および他の非細胞機構を介して)に損傷や感染した場所に輸送され,そこでは感染/傷害した細胞に貪欲に吸収され,これらの細胞の細胞膜外層にPSがある。最後に,細胞内部の極低カルシウム環境に曝露した場合,LNC構造の安定維持を担当する力はそれほど強くなくなり,LNCはそれらの貨物を放出する。
LNP と区別する
血液中の通常のLNPsは,ApoEを認識する低密度リポ蛋白受容体によりApoEを表面に結合させ,メッシュ蛋白を介したエンドサイトーシス作用により細胞に入り,LNPsは早期の内包体内から内包膜を破壊し,細胞質に入る。LNPsの内体脱出は非常に非効率的な過程であり,通常内体脱出の割合は
| 11 |
治療用標的
感染
著者らの特許送達技術の治療応用は最初に、腎臓と聴力機能に対する不可逆的な毒性影響を含む治療制限の潜在毒性を有する高効率、有効な抗感染br薬物の提供に集中した。MAT 2203では,致命的な真菌感染の治療に従来の両性マイシンよりも長時間安全に使用できる毒性の低い経口生物利用型殺菌剤両性マイシンBを開発した。これは逆に従来の製剤では実現できない方法で両性マイシンBを使用する潜在的な機会を創出し、従来の両性マイシンが毒性が大きすぎることが証明された場合に両性マイシン治療の機会を創出し、より長期的な治療のための可能性を創出し、患者の全体的な結果を改善させ、brが顕著な薬物経済学的影響を有する。
炎症が起こる
LNCsが天然免疫細胞(単球,好中球,樹状細胞)に貪欲に摂取されることが観察されることにより,LNCsは炎症性疾患の治療に有用である可能性が信じられる。単一サイトカイン、特にIL-17 Aおよび腫瘍壊死因子を非常に選択的にノックダウンすることができるいくつかの小分子オリゴヌクレオチドが得られた。我々はこれらのオリゴヌクレオチドでプログラムを進めることに成功しており,まず成功した効率的なパッケージ化されたLNCレシピ,次いで である体外培養LNCレシピの有効性を検証した。最近では2つのLNC製剤の小さなオリゴマーを体内にある動物疾患モデル研究は、2つの異なるモデルの中で測定可能な生物活性と潜在的な治療効果を示すことに成功した。イミダモント誘導乾癬マウスモデルでは,組織IL−17 A mRNAの減少が認められ,皮膚の発赤とスケールの改善を伴っていた。同様に,炎症性腸疾患のDSSマウスモデルでは,LNC製剤を毎日経口投与した腫瘍壊死因子標的オリゴヌクレオチド は結腸腫瘍壊死因子VI遺伝子発現を減少させ,血清腫瘍壊死因子エンタルピーレベルは有意に低下し,疾患活動スコアは有意に改善した。
癌
MAT 2203は安全かつ有効な両性マイシン製剤の創造に成功した上で、著者らはLNC技術の応用を腫瘍学応用に拡張した。いくつかの腫瘍細胞系の表面PS発現レベルも相対的に高く、これはLNCを介した化学療法に対して陽性反応を示す腫瘍に潜在的な方向性を提供する。br}はMAT 2203に対する経験から、LNCは2つの特定の領域で主要な進展を提供できると考えられる
| 1) | 組織標的送達や低血中濃度により、毒性を低下させる可能性があります | |
| 2) | より効率的な配信により効率を向上させる潜在力 |
著者らはいくつかの化学療法薬物の初歩的な調合仕事を開始し、最終的にドセタキセルを選択した。私たちはすでに を示しました体外培養治療効果は従来のドセタキセルよりも優れており、迅速に移行していることが証明されています体内にあるBrを研究し、同遺伝子マウス黒色腫モデル(表面PS高発現が知られている)に入った。このモデルでは,われわれの経口LNC製剤であるドセタキセル は,従来のドセタキセル静注と同様の効果が証明されている。また,LNC製剤を毎日経口投与しているにもかかわらず,体重に悪影響はなく,血液毒性の兆候もなかった。このことから,毎日投与されているドセタキセルLNC経口製剤は,従来のドセタキセル静脈内投与に相当する治療効果を腫瘍に提供でき,毒性の兆候はないことが証明された。その他の内容体内にある健康マウスにおける研究により、より高用量のLNCドセタキセル(大50%)も同様に毒性がないが、ドセタキセル投与量の類似の増加は毒性指標を伴い、平均体重は約20%減少した。
| 12 |
LNC臨床分期資産:MAT 2203
著者らの主要な候補製品MAT 2203は両性マイシンBと呼ばれる広域抗真菌薬物のLNC経口製剤である。伝統的に、両性マイシンBは薬剤耐性カンジダ症、クリプトコッカス性脳膜脳炎とアスペルギルス症を含むトリアゾゾールとラチェット耐性に対する全身性真菌感染を治療するための静脈注射薬である。これまで臨床的に両性マイシンBの耐性報告はほとんど認められておらず,この化合物が将来的に真菌感染を治療する最後の手段となる可能性が最も高いことをさらに支持している。しかし,両性マイシンBの使用は相対的に限られており,現在静注製品としてしか提供されておらず,記録されている重篤な毒性(最も明らかなのは腎毒性)である。我々のLNCプラットフォームを用いて両性マイシンBをナノカプセル化することにより、この薬物の経口および標的送達を感染細胞および組織に機会を創出し、この薬物の副作用は現在利用可能な両性マイシンB静脈製剤よりも少なく、両性マイシン Bを安全に長時間投与できるため、潜在的により大きな治療効果を産生する可能性があると信じている。
著者らの両性マイシンBのLNC送達は生物分布を変化させ、感染部位の両性マイシンBレベルは比較的に高く、自由循環薬物のレベルは低い。循環薬の数を減らすことにより,我々のLNCプラットフォームは全体毒性を低下させる可能性がある。重要なのは,薬物を携帯した貪食細胞の炎症領域への移行特性により,薬物濃度は標的組織のみで高くなることである。brはこれまでに産生されたデータから,MAT 2203は安全性の向上と毒性低減の潜在力があると信じており,MAT 2203は異なる改善処方を提供し,両性マイシン Bを感染部位の標的細胞に直接輸送することができると信じている。
MAT 2203の動物毒性とヒト研究データは,観察された毒副作用において,MAT 2203が他の両性マイシンB製剤よりも有意に優れていることを示しており,2つの要因によって駆動されていると考えられる
| ● | LNCは固体結晶であり、循環過程中にその薬物内容物を著しく“漏洩”しない。結晶は標的細胞内でのみその薬物ペイロードを放出するため、MAT 2203の使用は、現在処方されている両性マイシンBを使用した場合に通常出現する腎臓器官外毒性を引き起こさないようである。 |
| ● | この標的送達により,両性マイシンB静注と比較してmg/kgをもとに治療窓 を増加させることができた。動物モデル研究では、比較的に低用量の同等の治療効果が観察され、最高耐容静脈用量の10倍までの経口投与量を使用することができた |
MAT 2203の特性は両性マイシンBの経口、標的、および非毒性投与に役立ち、IFIの治療モードを根本的に変える可能性があり、IFIは迅速に増加する全世界的な脅威である。
真菌病原菌が増加している問題
真菌 病原体と感染は日々深刻化する全世界の公衆衛生問題である。重篤な患者と免疫不全患者の医療管理面の進歩により、全世界の国際金融機関の数は増加している。薬剤耐性IFIの出現は入院時間を延長させ、高価でよく猛毒な二線抗真菌薬の使用を増加させた。
真菌による感染の種類は多いが、現在4種類の全身性抗真菌薬物(ポリエン、アゾール類、ラチェット類、ピリミジン)しかなく、経口製剤には2種類(アゾール類、ピリミジン類)しかない。これらの抗真菌薬物の使用は通常かなり専門的な専門知識が必要であり、これらの脆弱な患者の潜在毒性と複雑な薬物-薬物相互作用を管理するが、有効なポリエンとラチェットは長期入院して静脈投与する必要がある。そのため、より有効、耐性がより良く、より安全な経口抗真菌薬物に対する切実な需要はまだ満たされておらず、深刻で、生命に危害を及ぼす、通常薬剤耐性を有するIFI患者を治療する。
世界保健機関(WHO)は最近IFIが全世界の公衆衛生問題であることを認めた。2022年末、世界保健機関は真菌優先病原体リストを発表し、アスペルギルス、黄金色カンジダと白色カンジダを重要な優先グループ(即ち最高公共衛生脅威)とし、毛かび目、熱帯カンジダと近縁カンジダは高優先グループに属し、球状カンジダは中等優先グループに属する。アスペルギルス、カンジダ、球胞子菌とクリプトコッカス属も深刻かつ生命リスクを脅かす合格病原体であり、連邦法規第21-fdaタイトル317.2-CFR-Codeによって合格指定リストに登録されている。
| 13 |
我々はMAT 2203がIAとより広範な他のIFIを治療する一流療法になる可能性があると信じており、それは以下の重要な潜在的利点を提供する
| ● | 耐性病原体を治療する潜在力。MAT 2203は既存のアゾゾールとラチェット耐性の真菌感染を含む薬剤耐性真菌による真菌感染を予防と治療する潜在力があると信じており、これは両性マイシンBの殺菌性質と薬剤耐性菌株に対する効力、及び著者らのLNCプラットフォームが早期感染部位の治療においてより高い薬物暴露の潜在力を直接提供するためである。 |
| ● | 全口腔治療を実現する。侵襲性真菌感染の治療過程中に両性マイシンB製品を早期に中止する能力は、限られた経口治療選択しかない患者に顕著なメリットをもたらし、これらの患者の経口治療選択は顕著な安全性、耐性と高薬剤耐性傾向の問題があり、例えばアゾール系抗真菌薬物が存在する |
| ● | 入院期間はより短く,入院費用はさらに低く,外来費用も低かった。MAT 2203で医師や患者に経口投与可能な広域殺菌剤を提供することにより,病院コストを低減し,病院コストがIFI総治療コストの70%以上を占める潜在力があると信じられている。 |
MAT 2203開発履歴と計画
臨床前データ
MAT 2203はいくつかの動物モデルで経口投与した場合に抗真菌活性を示したクリプトコッカス, カンジダ?カンジダそして、そしてアスペルギルス 感染する[Zarifら2000; ペリン、2004; Luら、2019年]それは.これらの動物モデルにおける治療効果は,IV両性マイシンBに相当またはそれ以上の抗真菌活性を示すことが証明されているが,毒性は低い。
体内にあるMAT 2203は多くの感染マウスモデルにおいて治療効果を示した新生クリプトコッカスこれらの研究は国立衛生研究院のピーター·ウィリアムソン博士が行ったものです[Luら、2019年]それは.多くの研究により、MAT 2203は5 FCと併用する可能性があり、クリプトコッカス性髄膜炎の治療に有効な経口製剤を提供する。3日間遅延したマウスクリプトコッカス性脳膜脳炎モデルとA型強毒株の大量接種新生バチルスMAT 2203と5 FCの併用効果は両性マイシンBと5 FCの併用に相当し、フルコナゾール内服より優れ、しかもいかなる毒性反応も観察されなかった。治療を受けたマウスでは、脳における両性マイシン薬の有意なレベルに伴い、蛍光MAT 2203粒子も脳に転移し、免疫学的特徴は従来の両性マイシンB治療マウスと類似している。これらの研究は、知られている殺菌薬brが髄腔内クリプトコッカス病に対抗するために有効な経口製剤になる可能性があることを示している。したがって、MAT 2203はCMに有望な治療選択を提供する。
また,MAT 2203の臨床前研究を行い,他のIFIの治療法を検討した。経口MAT 2203は多くの非臨床研究において有効であることが証明されているそれは.離体する研究により、MAT 2203 LNC製剤は両性マイシンBの抗真菌活性に影響しないことが示され、MAT 2203経口MAT 2203はいくつかの系統性真菌感染のマウスモデルにおいて、カンジダ、アスペルギルスとアスペルギルスに有効であることが証明された。免疫機能低下マウスと免疫機能正常マウスのカンジダ感染モデルにおいて、MAT 2203の経口投与は感染動物の肺、肝臓、腎臓などの標的器官の組織負担を生存と軽減する上で両性マイシンBの腹腔注射の治療効果と相当する。アスペルギルス属マウス感染モデルにおいて、MAT 2203の経口投与は生存面で両性マイシンBの腹腔注射の治療効果と相当し、そして用量依存性に真菌組織負荷を減少させる。経口MAT 2203は も展示されています体内にある治療効果観察デルマルラクトバチルスあるいは…環状マイコバクテリアマウス肺感染におけるSの免疫抑制作用は,肺や脳中の真菌胞子の減少を招き,両性マイシンBリポソーム静注の結果に相当する。
| 14 |
第2段階試験を実施する
全面的な臨床前パケットに基づいて、アメリカ国立衛生研究院はミネソタ大学の支出申請を援助し、ウガンダでACTE 2 a/b期試験を行った。この研究は2019年10月にスタートし、クリプトコッカス性髄膜炎の誘導と維持治療にMAT 2203を使用することを探索し、クリプトコッカス性髄膜炎はHIV 患者の最もよく見られると日和見感染の一つである。HIV患者の中でCMに関連する高発病率と高死亡率を考慮すると、臨床で満足されていない需要は非常に高く、全世界の毎年の負担は100万例と推定されている。
法令 は2つの部分からなる.クリプトコッカス病歴のあるHIV陽性患者にACTE第1部を行い,MAT 2203の漸増経口投与量を評価し,試験第2部の安全最大耐容量を決定した。
ACTEの第2部分は展望性、ランダム、開放的な逐次行列臨床試験であり、SOCと比べ、100名のHIV陽性クリプトコッカス性髄膜炎患者におけるMAT 2203経口投与の安全性、耐性と有効性を研究することを目的とした。ACTE試験は全部で4群の患者を含み、前2群の試験MAT 2203は14日間の誘導期内にIV型両性マイシンBを用いて予備治療を行った後の早期降圧治療であり、後の2群の試験MAT 2203はすべて経口治療である可能性がある。各キュー内のすべての患者(活動または対照)の誘導期間は14日であり,その後,強固/維持期間中に周囲の治療(活動または対照) を行った。キュー1と3はそれぞれキュー2と4の安全な誘導であり、キュー2と4は策定された重要な治療効果キューである。
ACTEのコホート2は,MAT 2203経口投与によるCM感染治療の可能性を評価することを目的としており,両性マイシン静注治療2日後ただちに誘導治療を行った段階で,早期維持治療期間中にMAT 2203治療を6週間と多い。ACT第2部の主な治療効果の終点は早期殺菌活性(EFA)であり,脳脊髄液(CSF)中のクリプトコッカスの除去率(LOG)と定義した10コロニー形成単位[CFU]治療の最初の2週間に一連の定量的真菌培養によって測定した/ml/日。
Enactの キュー2では,以下の結果を報告している
有効な早期殺菌活性(EFA),脳脊髄液(CSF)滅菌は,MAT 2203使用治療期間中に画期的な感染の証拠はなかった
| ● | ACTEでは主な終点はEFAであり,これは脳脊髄液真菌除去量の測定である。EFAは十分に検証された抗真菌薬物の治療効果の定量評価基準であり、生存を評価する重要な代替指標でもある。1ミリリットル当たり脳脊髄液中のクリプトコッカスコロニー形成単位(CFU)が0.20 log 10未満のEFAは、有意に高い死亡率とより悪い臨床結果と相関している2 がこの閾値を超えて測定したEFAは臨床的意義があり、強い真菌除去能力を代表している。ACTEの第2のキューにおいて、MAT 2203を使用して治療された患者によって達成される平均EFAは、0.38 log 10 CFU/mL/日であり、95%の信頼区間(0.30~0.46) は、予め指定された主要終点閾値>0.20より有意に高い | |
| ● | MAT 2203治療を受けたすべての患者は,誘導段階終了後,治療期間(誘導段階でも早期強固段階でも)に無菌脳脊髄液培養を行った | |
| ● | MAT 2203~10週間の治療期間中には、クリプトコッカス感染を突破または再発した患者の証拠は認められなかった |
生きる
| ● | ランダムにMAT 2203治療を受けた40名の患者のうち,全体生存率は95%であった |
2*Disに臨床感染している。2020年;71(5):E 45-49
| 15 |
安全
| ● | MAT 2203はMAT 2203の腎臓毒性や電解質異常による証拠を示さず,重大な安全シグナルもなく,制限耐性の問題も使用されておらず,MAT 2203の長期治療が誘導段階から強固段階に延長してもbr}2週間から6週間に延長している |
Enactの キュー4において,以下の結果を報告した
効能 と全国民教育
| ● | 脳脊髄液酵母除去率はあらかじめ指定された>0.20の主要終点閾値目標を超え,平均EFAは0.30 logに達した10CFU/ml/日、95%信頼区間は0.22~0.38であった | |
| ● | 数名のベースライン真菌負荷の高いbr参加者はMAT 2203治療群において顕著な抗真菌活性を有し、その中に1名の 患者を含み、誘導期にスクリーニングを行う時、そのクリプトコッカス定量培養は915,000 cfu/mLに達し、これは真菌を有効に除去する重要な証拠であり、最も挑戦的な症例においても同様である |
生死存亡
| ● | MAT 2203治療を受けた40名の患者のうち,現在の一時生存率は90%であり,2週目の生存率は95%であった |
安全問題
| ● | SOC治療と比べ、MAT 2203患者の3級臨床有害事象(42%)は比較的に少ない(59%) | |
| ● | SOC治療と比較して、MAT 2203を服用した患者では、腎機能や貧血に関連する有害事象の発生率が著しく低下しており、MAT 2203を6週間服用した患者では腎臓毒性の証拠は認められなかった。 | |
| ● | キュー4に見られる良好な安全性および耐性データは、現在利用可能な両性マイシンB静注製剤に関連する毒性のため、より長期的な使用のための経口MAT 2203を支持する |
ACT試験結果は2023年8月の“臨床感染症雑誌”に発表され,編集選択の原稿とした。
拡張/思いやりのある アクセスプランの使用
ACTE試験完了後,2022年にわれわれの拡張/同情使用アクセス計画を開始し,MAT 2203が侵襲性真菌感染患者の治療に有意な影響を与えることを証明し,これらの患者の治療選択は限られているか,治療選択がないかは,第3段階臨床試験に参加すると考えられている患者と非常に類似している。
これまでに19名の患者が複数の医療機関に登録されており,ミシガン大学,ジョン·ホプキンス大学,全国児童病院,希望の城,ファンデルビルト大学医療センター,国家衛生研究院,フィラデルフィア児童病院,スタローン·キャトリン癌センター,カリフォルニア大学サンディエゴ医学院を含む。登録されている患者の多くは移植後患者あるいは潜在的悪性腫瘍の治療を受けている。MAT 2203を使用して治療される感染には、様々な微生物(を含む)が含まれるコウジカビ, 毛かび属種, カンジダ症, 鎌菌 疑いを抱いています球胞子虫)脳、膀胱/結腸、骨、肺、副鼻腔および皮膚を含む複数の感染部位で発生する。多くの患者は登録前にAmBisome治療を受けているが,治療制限的な腎毒性が出現しており,多くの患者では薬剤耐性菌の治療が必要であったり,臨床的にアゾール系治療が行われておらず,他の治療選択はない。
この計画に参加した19名の患者のうち,15名の患者がフォローアップを受けており,4名の患者が最近開始またはMAT 2203による治療を開始している。
| 16 |
| ● | 15例の患者の中に12例の臨床症状は完全に緩和或いは感染指標が客観的に改善した(放射線学と真菌学を含む)。 | |
| ● | 完全な個人指定治療コース(2から1年,感染状況に依存)を達成した5名の患者のうち,すべての患者が完全な臨床緩解を得ており,再発や感染再発はなかった | |
| ● | 他の5名の患者の臨床指標は客観的に改善し,計画的にMAT 2203を継続して治療を行っている | |
| ● | 15名の患者のうち5名は、2名の患者の真菌感染が臨床的に有意に改善されたにもかかわらず、12個の全体的に成功した症例に含まれているにもかかわらず、それぞれの指定された治療コースを達成できなかった。 |
| – | 2名の患者は,悪性疾患が予期せぬ進展を示したため,MAT 2203治療開始直後に緩和ケアに移行した | |
| – | 1人の患者は、2日後に潜在的胃腸問題(すなわちクローン病)のために治療を中止した。 | |
| – | 1人の患者は、治療約8週間後に潜在的疾患の進行によって死亡した(しかし、以前の彼らの真菌感染は有意な臨床的改善があった) | |
| – | 1例の患者は治療10週間後に潜在的な胃腸問題(長期吐き気/嘔吐)のためMAT 2203治療を中止したが、彼らの真菌感染は改善した。 |
重要なことは,AmBisome治療後に腎毒性が出現したすべての患者が,MAT 2203治療に移行した後,腎機能はベースラインレベルに回復し,MAT 2203治療を延長する過程でさらなる腎臓副作用を受けなかったことである。
我々は、これらの機会 が臨床試験環境の外でMAT 2203の安全性および有効性を示すことができると信じているので、MAT 2203の使用に同情する機会を提供することを評価し続ける予定であり、これはFDAおよび潜在的パートナーの検討のために重要な追加の患者データを表す。
ORALTO第3段階試験
2024年2月、同社はMAT 2203登録経路のすべてのキー要素について共通認識 を含むFDAとORALTO試験の設計について合意したことを発表した。
ORALTO は、制限された侵襲性アスペルギルス症患者の治療選択の標準的なケアと比較して、経口降圧性治療としてのMAT 2203の有効性と安全性を評価するための3期、ランダム、マルチセンター、開放ラベル、審判者盲法研究である。この研究はAmBisome(リポソームIV-両性マイシンB)治療後に行われる。主な治療効果の終点は研究42日目の全原因死亡率である。主な の副次的な目標は:
| ● | 治療関連毒性による治療変化(即ち、用量調整/中断或いは治療方案の変化)において、経口減量治療はアミノメチルジオキシピリミジンと比較した優位性を示した |
| ● | 研究84日目の全原因死亡率を用いてMAT 2203の長期生存効果を計算した |
| ● | MAT 2203が医療資源利用と生活の質に及ぼす影響を評価した。 |
登録brは、カビ活性アゾールの静脈内投与が受けられず、代替治療選択が限られているため、最近侵襲性アスペルギルス症と診断または確認された成人約216人を含むことが予想される。AmBisomeの最初の治療を受けた2日後には、条件を満たす研究参加者が研究に組み込まれ、mT 2203の経口投与またはAmBisome治療を2:1の割合でランダムに受け、標準看護を受けた。
| 17 |
すべての研究参加者はAmBisome治療を受けた初日から最長12週間の治療を受ける。すべての研究参加者が最初のAmBisome治療期間中に入院する予定である。MAT 2203内服後,研究参加者は退院する可能性があり,臨床状況に応じて外来治療を継続した。
独立したデータ審査委員会は、治療を無視し、臨床、放射線学、および真菌学的反応を含む主要かつ副次的な終点を裁くであろう。約75%の参加者が登録されると、独立したデータセキュリティ監視委員会は、サンプル量の仮定が合理的であることを保証し、研究が十分に支持されることを確実にするために、全体的に集約された全原因死亡率を盲目的に検討するであろう。集約されたイベントが期待レベルと大きく異なる場合には,実験に対してサンプル量調整を行うことができる.
ORALTO は米国,ヨーロッパ,南米,中東,アジア太平洋地域の約65地点で行われる。登録作業は2024年下半期に開始予定で、約24カ月かかる予定だ。
私たち は、MAT 2203の持続的な協力プロセスに積極的に参加しており、1つ以上の開発および/または商業化パートナーを探しています。 我々は、(I)協力取引の完了を要求するか、または(Ii) ORALTO試験を開始する前に追加資金を調達することを要求します。
ACTE第2と第4群で発生したデータ,われわれが行っている拡大参入計画のデータ,およびFDAとORALTO第3段階試験設計について合意した後,有限治療選択患者の侵襲性アスペルギルス症の予備適応としてMAT 2203のNDAを明確な経路で提出することができると信じている。われわれの全体的な発展戦略は,他のIFIの治療を含めてMAT 2203の適応を拡大することであり,さらに計画中のORALTO試験によりIV両性マイシンとの薬効学的架け橋を構築すると予防が可能である。
全体的な臨床パケット
MAT 2203を評価するための臨床研究は,健康ボランティアに対する2つの完成した1期研究(CAM−102とMB−70011の研究), と3つの類似2期の研究を含む:1つの完成した中重度外陰膣カンジダ症患者に対する研究,1つの標準非IV療法が無効または不耐性の皮膚粘膜カンジダ症患者に対する研究、および1つのクリプトコッカス性髄膜炎患者に対する2期研究である。
MAT 2203は、2024年3月までに、4つの臨床試験のうち合計202名の被験者に、我々の拡張/同情使用アクセス計画により、以下のように使用された
| ● | 52人の健康被験者(CAM-102を研究[N=36]およびMB-70011[N=16]) | |
| ● | HIV患者36例とCM患者101例(Enact Study MB-70007)、 | |
| ● | 91例の上室性頻脈(MB-70005) | |
| ● | 皮膚粘膜(食道と中咽頭)カンジダ症(MB−70004)4例 | |
| ● | 19人の患者が私たちの患者拡張/共感的使用アクセス計画に参加します |
これらの研究では、単回用量が2.0 gまでのMAT 2203および2.0 g/日までの繰り返し用量のMAT 2203および48カ月までの反復用量のMAT 2203が安全かつ耐性が良好であった。
治療選択の限られた患者における侵襲性アスペルギルス症について
侵襲性アスペルギルス症(IA)は1種の深刻で、生命に危害を及ぼす侵襲性真菌感染であり、主に免疫機能が深刻に損傷した悪性血液病患者と移植レシピエントに発生する。これらの患者の医療管理の進歩により,IAは世界的に増加しており,最近では世界的な公衆衛生問題として公認されている。2022年に世界保健機関は真菌優先病原体リストを発表しました煙曲カビ感染の最もよく見られる原因であり,重要な 優先群(すなわち感知された最高公衆衛生脅威)に属するコウジカビFDA合格指定 構成が深刻で生命リスクを脅かす病原体リストにも含まれている。
| 18 |
固形臓器移植を受けたIA患者の年間死亡率は41%であったが,幹細胞移植を受けた患者の年間死亡率は75%と高いことが報告されている。カビ活性アゾールの開発以来,結果は改善されているにもかかわらず,これらの薬物の使用は治療制限毒性,薬物と薬物の相互作用,最近出現した薬剤耐性により複雑になることが多い。
アメリカ伝染病学会(IDSA)の治療ガイドラインはIA患者が少なくともカビ活性のアゾール類薬物治療を6~12週間受けることを提案し、これは主に免疫抑制の程度と持続時間、発病部位と疾病証拠に依存する。カビ活性のアゾール類薬物は通常有効であるが、それらの使用は毒副作用と薬物と薬物の相互作用を管理するためにかなり高い専門知識を必要とし、これはよく治療持続時間を制限する。
IA はコウジカビアゾール系化合物の長期使用や農業でアゾール系殺菌剤が広く使用されているため,全世界でカビ活性アゾール系化合物に耐性を持つ種が増加している。この新たな耐性は特に懸念されていますIAはアゾールに対する耐性によるものですコウジカビ死亡率の高い命を脅かす病気です。
血液系悪性腫瘍患者と移植者は常にハイリスク時期にカビ活性アゾゾールの抗真菌予防を受け、感染を防止する。最近,抗真菌予防を受けた患者で画期的なIAが発生した症例が報告されている。これらの明らかな予防失敗の原因は,不コンプライアンス,吸収不良,薬物相互作用あるいは薬剤耐性に感染している可能性があるコウジカビ種です。
IDSA治療ガイドラインはAmBisomeのような両性マイシンBの静脈内投与を推奨し、カビ活性アゾール治療を受けられないIA患者の代替案とする。しかし、両性マイシンBの静脈注射は腎毒性と電解質異常を引き起こすことができ、通常入院して密接なモニタリングと電解質補充が必要である。両性マイシンBを静脈内投与する他の合併症は、呼吸困難、酸欠、胸背部痛、静脈内投与部位の静脈炎、貧血、肝毒性を含む急性輸液反応を含む。そのため、両性マイシンBの静脈内投与は数週間を超えると通常安全ではないか、または実行不可能である。利用可能な経口抗真菌薬は、これらの患者に推奨される6~12週間の治療を完了させることができる。そのため、これらのIA患者を治療するために有効かつ耐性の良好な経口抗真菌薬が切実に必要であり、この需要はまだ満たされていない。
MAT 2203規制指定
FDAは、免疫抑制治療を受けた患者のIFIを予防し、クリプトコッカス病を治療するためのMAT 2203合格感染症製品(QIDP)および侵襲性カンジダ症およびアスペルギルス症治療快速チャネルの称号を付与している。私たちは最近、クリプトコッカス病および関連CMを治療するMAT 2203に対する米国FDAおよびEMAの孤児薬の称号も取得した。FDAは、まれな疾患または疾患を治療するための候補製品を孤立薬として指定することができ、これは、一般に、米国での患者数が200,000人未満であるか、または米国での患者数が200,000人を超えると定義され、薬物開発のコストが米国の販売から回収されると合理的に予想できない。孤児の薬物名を有する製品がその後、孤児の薬物名を有する疾患の特定の活性成分に対するFDAの最初の承認を得た場合、孤児薬物名は、FDAの承認後に米国の孤児薬物独占経営権を得ることができる。合理的な仮定に基づいて希薬指定を得た製品brは、同じ適応のために許可された同じ薬物よりも臨床的に優れている製品に対して、承認された後に孤児薬物排他性を得るためには、同じ孤児適応のために許可された同じ薬物に対する臨床的優位性を証明しなければならない。br孤児薬物排他性は、NDAを含めて、同じ適応の同じ薬物を販売するために、FDAが7年以内に他の申請を承認しないことを意味する。限定された場合でなければ、例えばFDAが孤児薬物専有権の保有者を発見しない限り、それが疾患または指定された薬物に罹患している場合の患者の需要を満たすために十分な数の孤児薬を得ることができることを証明することができない。同様に、FDAが後者の薬剤が臨床的に良いと結論した場合、これは、後者の薬剤 がより安全で、より効果的であることを意味し、または患者ケアに大きな貢献がある場合、FDAは、その後、同じ活性部分を有する薬剤を同じ疾患のために排出期間内に承認することができる。孤児薬物指定はまた、一方が財政激励brを獲得する権利があり、例えば臨床試験費用の贈与資金の機会を獲得し、使用者費用の支払いを免除し、小児科患者に対する臨床研究を免除し、FDA法規が別の要求がなければ、及び臨床研究費用の税収控除を免除する。
| 19 |
“直ちに抗生物質を生成する奨励法案”または“利得法案”によって提供されるQIDP指定によれば、迅速なチャネル指定の資格を得ること、優先的に審査すること、およびFDAの承認を得た場合、追加の5年間の市場独占経営権を得る資格があることを含む、新しい抗菌または抗真菌薬の開発に何らかの報酬を提供する。高速チャネル指定は薬物開発と審査を加速するために、FDAとのより頻繁な相互作用 を実現することができる。高速チャネル指定は承認基準を変更することはなく、MAT 2203の高速チャネル指定を維持できる保証はありません。またはこのような指定は、より速い規制審査速度をもたらすことになります。孤児指定により提供された7年間の市場独占期間 が承認されれば、QIDP指定職MAT 2203が提供する追加5年間の市場独占経営権に加え、FDA承認時に米国で合計12年間の市場独占経営権を獲得する可能性がある。
抗真菌市場機会
2023年の全世界の抗真菌薬物市場の総価値は約158億ドルであり、2030年までに約205億ドルに達すると予想される。2021年、全世界の侵襲性真菌感染市場価値は72.1億ドルを超え、2030年には103.6億ドルに達すると予想される。これには,入院や外来環境における積極的な治療や予防(予防的)としての療法,入院患者の治療のための療法,退院患者の治療のための療法が含まれている。重要なのは,入院期間を延長したため,1回の受診個人保険コストは患者1人あたり約4万ドルから15万ドルまで様々である(2019年ベネディクト)。毎年150万例以上の異なる種のカンジダ、アスペルギルス菌そしてクリプトコッカスは世界で最もよく見られる3種類の侵襲性真菌病原体。米国では,これらの疾患の推定発症率は約46,000例の侵襲性カンジダ症,15,000例の侵襲性アスペルギルス症,4,900例のCMである。例えば,米国のみではアスペルギルス症に関連した入院治療コストは13億ドルを超え,間接コストは4.85億ドルと見積もられている。記録されているIFIに関連する疾患の迅速な発展と高い死亡率(20%-50%)は通常、疑い(未確定診断)症例に対する抗真菌治療、あるいは予防措置としてハイリスク患者に対して抗真菌治療を実施することを招く。そのほか、癌化学療法、臓器移植或いは自己免疫性疾患治療における免疫抑制薬物の応用は日々広くなり、IFIリスクに直面する患者の数は絶えず増加している。また,アゾール類,ラチェット類,ポリエン類からなる全身性抗真菌薬の種類が限られており,それらの広範な使用により,薬剤耐性菌株の感染者数が増加している。疾病コントロール·予防センター(CDC)はすでにフルコナゾールに対する耐性をリストに入れたカンジダ?カンジダ迅速かつ持続的な行動を必要とする深刻な脅威として,特に棘球虫耐性の上昇が認められた滑らかな偽糸酵母2022年、世界保健機関は新型クリプトコッカス、アスペルギルス菌と黄色ブドウ球菌と白色カンジダを含む真菌優先病原体リストを発表し、高度に満足されていない需要のため、それを抗真菌開発の重要な優先事項とした。新しい薬剤に対する切実なニーズを強調し,これらの薬剤は抗薬剤耐性菌株の活性を有し,br毒性を著しく低下させ,入院期間や関連コストを減少させるために早期退院する可能性があると考えられる。
LNC プラットフォーム拡張
MAT 2203をORALTO第三段階試験に進めるほか,LNCプラットフォームの使用を拡大し,小分子とオリゴヌクレオチドbrを感染症に応用したほか,炎症や腫瘍学についても検討した。
炎症が起こる
我々は,炎症性サイトカインIL−17 Aと腫瘍壊死因子α の2種類の小分子オリゴヌクレオチドに対する各種LNC製剤を検討し,一連の研究を行っている体外培養そして体内にあるミキノモート(Imiquimod)誘導マウス乾癬モデルにおいて、経口に関連する生物活性およびIL-17 Aノックアウトに関連する臨床的利益を評価する研究。
これらの初歩的な研究はサイトカイン抑制形式の生物活性を記録し、そしていくつかの証拠を提供し、このような定性の乾癬モデルにおいて、皮膚損傷外観(発赤、スケール)の改善は関連する有形臨床利益と関係があることを表明した。これらのデータはまだ評価中であり、分析の重点は、(A)サイトカイン阻害の強度および時間過程を明らかにすること、および(B)これらのモデルにおけるサイトカインmRNAレベルおよび特定の組織反応を評価して、これらのデータをよりよく理解し説明することである。
| 20 |
2023年12月に一連の発表を行いました体内にある研究は2種類のLNC配合の小一本鎖オリゴヌクレオチドの経口投与に成功し、この2種類の小一本鎖オリゴヌクレオチドは重要な炎症性サイトカイン腫瘍壊死因子αとIL-17 Aに特化し、ヒト疾患に出現する急性炎症反応の成熟と検証を模擬した動物モデルにおいて検証を行った。
急性大腸炎研究(“腫瘍壊死因子α”)
デキストラン硫酸ナトリウム誘導マウス大腸炎モデルを用いて、経口LNC送達の特異的に腫瘍壊死因子αm RNAを標的とした小分子オリゴヌクレオチドを評価した。リアルタイム定量ポリメラーゼ連鎖反応分析では,活性αを経口投与すると結腸組織における腫瘍壊死因子αの発現レベルが低下し,罹患しているが未治療動物と比較して血清中腫瘍壊死因子mRNA量が37%有意に低下した。重要なことは,研究における重要な時点の臨床疾患活動性スコアも活性LNC製剤を用いることにより有意に改善したことである。
急性乾癬研究(“IL-17 A”)
イミダキノモート(“ImQ”)を用いて誘導したマウス乾癬モデルは経口LNC送達の小分子オリゴヌクレオチドbrを評価するためにIL-17 A mRNAの合成を抑制することを目的とし、それは乾癬皮膚皮膚損傷の進展に重要な役割を果たしている。DSS大腸炎モデルと類似し、活性LNCを経口投与した後、ImQ乾癬モデル皮膚組織中のIL-17 A mRNAの発現レベルは単純ImQより低かった。このモデルでは,IL−17 A血清レベルは変わらないと予想されるが,臨床疾患の皮膚赤腫や鱗屑マーカーは改善され,さらにこれらの小分子オリゴヌクレオチドの生物活性が検証された。
腫瘍学
2023年11月に私たちは体内にあるドセタキセル経口LNC製剤の動物研究によると、ドセタキセルは1種の有名な化学療法薬物であり、多発性転移と切除できない腫瘍の治療に用いられる。現在、ドセタキセルは静脈投与されており、顕著な副作用と毒性に関係している可能性がある。この研究により、十分に検証されたマウス黒色腫モデルにおいて、腫瘍の大きさはドセタキセルの全身静脈内投与に相当することが示唆された。経口投与量の10日間,LNC製剤には何の毒性も認められなかった。治療コースは主に的確な治療効果であり、更なる研究と評価が必要であり、そして服薬時間を延長する必要がある。
毎日LNC-ドセタキセルを内服する抗腫瘍作用はドセタキセルの静脈注射と相当し、14日目は未治療対照群と比べ、腫瘍体積は明らかに縮小し(LNC-63%高用量内服;低用量LNC-57%;ドセタキセル-68%静脈注射)、14日目に腫瘍重量は軽減した。全身毒性は見られなかった。体重は治療期間中に安定し、血液学的パラメータは未治療対照群と類似している。
2024年3月,健常マウスで行ったもう一つのLNC−ドセタキセル体内研究の積極的な結果を発表した。この研究の目的は、LNC製剤ドセタキセルの経口投与がドセタキセルの通常の静脈内投与の全体的な安全性を改善できるかどうかを決定することである。br}この研究は24匹の健康なBALB/cマウスを含み、(1)生理食塩水対照群の経口投与、(2)ドセタキセルの静脈内投与(30 mg/kg、または0.6 mg/用量)、連続3週間、(3)LNC-ドセタキセル(37.5 mg/kg、または0.75 mg/用量)の経口投与、1日1回、3週間の経口投与を含む。この研究の主な終点は治療期間中の体重の変化であり,これはこのモデルにおける毒性の主な表現である。
主な のポイントは以下のとおりである
| ● | 22日目までに,LNC−ドセタキセル製剤を経口投与したドセタキセル総量は,ドセタキセル静脈内投与の8倍以上であった(最終静注用量は薬物動態の測定に用いられた)。 | |
| ● | ドセタキセル静脈注射治療を受けたすべてのマウスの体重(毒性)は明らかに低下し、平均ピーク減少量はその原始体重の20%であった。 | |
| ● | LNC−ドセタキセルを経口投与したマウスは体重を保持しており,生理食塩水を経口投与した対照群マウスの体重と統計学的に差はなかった。 |
| 21 |
| ● | ドセタキセル静脈内投与群の1匹は研究終了前に死亡し,分析から削除され(最終測定では体重低下br}−32%),ドセタキセル静脈内投与群の曲線もドセタキセル静脈内投与後の予想7日間の回復を反映していた。 |
同遺伝子マウス黒色腫モデルにおいて、毎日LNC-ドセタキセルを経口投与する用量はLNC-ドセタキセル投与量より50%高く、投与総量は3.5倍高く、この研究はLNC-ドセタキセルの抗腫瘍活性がIV-ドセタキセルに相当することを証明した。
次のステップは、他の腫瘍モデルにおける現在のLNC-ドセタキセル製剤の治療効果を評価することを含む。また,小分子オリゴヌクレオチドLNC製剤の潜在的抗腫瘍活性を評価する予定である。
LNCプラットフォームに係る戦略連携
我々のLNCプラットフォームは、様々な分子および薬物を再調製するために使用可能であり、これらの分子および薬物(I)は、体内の分子および薬物を有効に保護するために送達する必要があり、標的細胞の有効な送達および細胞取り込みから利益を得ることができると信じており、(Ii)は現在IV製剤でのみ提供されているか、または(Iii)顕著な毒性関連副作用を経験している。著者らはすでに概念検証動物研究において、著者らのLNCプラットフォームを用いて、オリゴヌクレオチド (mRNA、siRNA、DNAプラスミド)、ワクチン、抗炎症薬、非ステロイド性抗炎症薬とアトバノンを含む一連の再調製された薬物化合物をテストした。我々は,このユニークで破壊的なLNCプラットフォームの価値を最大化するために,単独であるいは他の製薬やバイオテクノロジー会社と協力して製品を開発する機会を求める予定である。
2019年12月、我々は羅氏社Genentechとの実行可能な協力を発表し、我々のLNCプラットフォームを用いていくつかのGenentech化合物の配合を評価した。最初の合意は、最大3種類の遺伝子テーク固有化合物を協力することを規定しています 体外培養テストします。そのうちの2つのプロジェクトが完了した。いずれもLNC製剤の小分子とオリゴヌクレオチドの細胞内送達に成功し,それに伴う毒性がないことが証明された。我々はこの協力を鈍化することを選択し,炎症におけるわれわれの内部オリゴヌクレオチド計画と腫瘍学的小分子計画に重点を置いた。
2020年12月,NIAIDと協力してギレドのレヒビルの経口製剤を開発することを発表し,現在は新冠肺炎に対する静脈療法のみとなっている。NIAIDは2021年と2022年の間にノースカロライナ大学教会山校流行病学科とともに2つの調査を行った体内にある我々のギレド科学社のRedesivir(LNC−RDV)のLNC配合を標準的なSARS−CoV−2トランスジェニックマウスモデルで試験した。これらの動物モデルでは,感染5日後にLNC−RDVを経口投与するとウイルス力価を低下させ,体重や充血スコアの臨床パラメータを改善し,その効果はレミキシビル皮下注射の効果と類似している。Gileadとデータbrを検討·検討した後,Gileadはその内部開発されたレバイビルプロドラッグに集中することが知られており,これらの薬剤の臨床応用が進んでいるためである。
2022年4月、我々は、BioNTechのmRNAフォーマットと独自のLNCプラットフォームとの組み合わせを評価するために、mRNA技術に焦点を当てたグローバル製薬会社BioNTech SEと共に独自の研究br協力を発表した。BioNTechとの合意条項によると,275万ドルの排他料を受け取り,BioNTechは協力に関するいくつかの研究費 を援助してくれた。私たちは密接に協力して開発し最適化し体外培養テストと単一の体内で この研究は2023年に行われた。♪the the the体内にある経口メッセンジャーリボ核酸の研究では経口投与の臨床前活性を示さなかった。このレシピ はmrnaの伝達に成功した体外培養複数の細胞系に入る前に体内にある健康なマウスで研究しています追加 内部体内にある類似した非脂質ナノ結晶(“LNC”)mRNA製剤の研究では,系統的(筋肉や腹腔内)投与時に確実に活性を示すことが示されている。また,これらの配合は4℃で少なくとも17週間の高度な安定性を示し,脂質ナノ粒子(LNPs)と比較して有利であり,Matinas とBioNTechとの研究協力は2023年5月に終了した体内にある結果を検討する。
2023年1月、我々はNational Resilience,Inc.(Resilience)と材料譲渡と評価プロトコルを締結し、識別された核酸の口頭送達の潜在力を探索することに重点を置いた。Resilienceに通知されており,小分子オリゴヌクレオチドにおける我々の内部作業を優先順位としているが,将来的にはこの協力を再考する可能性がある。
| 22 |
私たちは、他の関心のあるバイオテクノロジーおよび製薬パートナーとの他の潜在的な戦略的協力を評価し続ける。これらの早期概念検証評価は高効率、コストがもっと低い方法を提供し、革新医学領域で多くの戦略垂直市場を創立し、同時に成熟パートナーの開発専門知識と財務資源を利用することができる。これらの評価からのデータ は,多くの戦略パートナーが薬物開発のリスクとコストをよりよく吸収できるように,我々をLNCプラットフォームの許可者と位置づけることができるとともに,LNCプラットフォームの全体的な価値を最大限に実現しているため,我々の会社を前払い許可証,マイルストーン,特許権使用料支払いを可能にする特許使用料集積器とすることができる。
ロッグス大学と独占許可協定を締結する
Aquarius BioTechnologies Inc.の買収により、私たちはロッグス大学からLNCプラットフォームに関するいくつかの特許の許可を得た。 我々はその後、Aquarius BioTechnologies Inc.の名称をMatinas BioPharma NanoTechnologies,Inc.に変更し、2022年2月に 双方は第2回改訂と再署名した独占許可協定に同意した。このプロトコルでは,(1)このようなライセンス技術を用いた製品の純売上高の低桁と中央桁の間でステップ で印税を計算する,(2)ライセンス技術を用いた製品の売上が指定販売閾値に達した場合には,100,000ドルのマイルストーン費用を一度に支払うこと,および(3)ライセンス契約期間内に50,000ドルの年間ライセンス料を支払うことを規定している。協定により付与された専有特許権を第三者に再許可すると,ロッグス大学に支払われる対価格も減少する。ロッグス大学が改訂された許可協定で譲歩したことを考慮して、会社は2022年2月にロッグス大学に400,000株の普通株を発行した。私たちはまた、この技術に関連する特許訴訟と維持費を支払う責任を負担し続けることに同意した。
のいずれか一方が他の方法で終了しない限り、ライセンスの期限は、ライセンス技術を使用する国/地域で製品が初めて商業販売された日から、または合意によって許可された最終期特許権が満了した日から8年半以内に長い時間を基準としなければならない。もし私たちがbrの2回目の改正と再署名されたライセンス協定が発効した日から8年以内に少なくとも1つのライセンス技術を使用した製品の商業販売を開始しなければ、ロッグスは許可協定を終了する権利がある。
知的財産権
私たちの候補製品と私たちの発見計画、プロセス、技術ノウハウの独自の性質と保護は私たちの業務 に重要です。我々は、特許、商業秘密、ノウハウ、FDA排他性および契約開示制限の組み合わせによって、私たちの製品および関連技術の製造および開発を保護することを求めます。私たちの政策は特許権を追求し、維持し、守り、私たちの業務発展に重要な商業的意義を持つ技術、発明、改善を保護することです。私たちの成功は、私たちの業務に関連する重要な商業技術および発明およびノウハウの特許および他の固有保護 を取得し、維持する能力があるかどうかに大きく依存し、私たちの特許を保護し、私たちのビジネス秘密 の機密性を保護し、第三者が効果的かつ強制的に実行可能な特許および独自の権利を侵害することなく運営されるであろう。私たちはまた私たちの独自の地位を発展させて維持するためにノウハウと持続的な技術革新に深刻に依存している。
独自のLNCプラットフォームおよびMAT 2203に関する独自のライセンスとMatinasが所有する知的財産権
私たちは、ロッグス大学が独占的に許可した特許および特許出願から、私たちのいくつかのプロセスで使用される特許化学(Br)技術にいくつかの特許保護を提供し、私たちの脂質ナノ結晶および地塩カタツムリを製造し、この送達技術内で送達される活性薬物br成分を調製し、例えば、私たちの主導製品MAT 2203において、私たちの主導製品はLNCプラットフォームを構成した。我々のライセンス契約によれば、我々は、特許期間調整または延長を含まずに、係属中の米国非一時特許出願1つ、米国特許9件、および付与された49件の外国特許を含む2023年12月31日までのポートフォリオの権利を取得する。ライセンス特許は、ヨーロッパ、中国、インド、ブラジル、ロシア、カナダ、日本、韓国、オーストラリア、メキシコなどで許可されている。
マティナスが持っている特許の組み合わせは私たちのLNCプラットフォームとユーザーをカバーしている。この特許の組み合わせには、5つの被審査米国仮出願、2つの係属米国非仮出願、2つの係争PCT出願、13件の保留外国出願、20件の認可された外国特許が含まれる。外国の保留出願および認可された特許は、ヨーロッパ、中国、ブラジル、カナダ、日本、韓国、オーストラリア、メキシコなどの国に分布している
| 23 |
2023年12月31日現在、私たちは、オーストラリア、カナダ、ヨーロッパ、日本で発行された7つの外国特許と、LNCにおける活性物質の組織浸透を増強するための組成物および方法に関する韓国での未解決の外国特許出願とを有している。これらの特許は、2036年に満了する予定であり、係属中の出願または優先権を有する特許も、特許期間の調整または延長を含まずに2036年に満了する。
2023年12月31日現在、私たちはオーストラリア、ヨーロッパ、日本に9つの外国特許を持っており、マイコバクテリア感染を治療するLNC組成物および方法に関する。これらの特許は、特許期間の調整や延長を含まず、2036年に満了する予定だ。
2023年12月31日現在、我々は、米国が発行している特許、出願中の非一時的な米国特許、日本およびオーストラリアで取得された2つの外国特許、および中国、韓国、カナダ、欧州で出願中の4つの特許を有しており、クリプトコッカス感染を治療するLNC組成物および方法に関する。これらの特許は2037年に満了する予定であり,係属中の出願から発行または優先権を有すると主張されている特許も2037年に満了し,特許期限の調整や延長を含まない。
2023年12月31日現在、私たちは、クリプトコッカス感染を治療するLNC成分および方法に関する、係属中の米国の非臨時特許出願と、オーストラリア、ブラジル、カナダ、中国、ヨーロッパ、香港、日本およびメキシコの4つの係属中の出願を有している。係属中の出願から発行または要求された優先権を獲得した特許も2040年に満了する予定であるが,特許期限調整や延期は含まれていない。
2023年12月31日現在、LNC粒子サイズ制御方法に対するPCTアプリケーションを持っています。係属中の出願から発行または優先権を主張する特許も2043年に満了する予定であるが,特許期間の調整や延長は含まれていない。
2023年12月31日現在、LNC組成物および毛カビ症治療方法に対するPCT出願を有している。係属中の出願から発行されるか、または優先権を有すると主張される特許も、特許期限の調整または延長を含まずに2043年に満了する予定である。
2023年12月31日まで、著者らはLNCプラットフォームの改善に対する3つのアメリカ臨時申請を持っており、小分子オリゴヌクレオチドを含む治療用br薬物の送達のための。保留出願から発行されるか,または優先権を有すると主張される特許も2044年に満了する予定であり,特許期間の調整や延長は含まれていない。
2023年12月31日現在、私たちは核酸送達の脂質顆粒製剤に対する米国の一時的な申請を持っている。係属中の出願から発行または要求されて優先権を獲得した特許も2044年に満了する予定であるが,特許期限調整や延期は含まれていない。
2023年12月31日現在,我々はLNC組成物と癌治療法に対する米国の一時的な申請を持っている。係属中の出願から発行または要求されて優先権を獲得した特許も2044年に満了する予定であるが,特許期限調整や延期は含まれていない。
私たちは、私たちの任意の保留特許出願または将来所有または許可される可能性のある任意の特許出願が特許権を得ることを保証することができず、私たちの既存のいかなる特許または将来所有または許可される可能性のある任意の特許が私たちの技術を保護するのに役立つことを保証することもできない。このリスクおよび我々の知的財産権に関するより包括的なリスクについては,“リスク要因−我々の知的財産権や規制排他性に関連するリスク”を参照されたい
特許に加えて、私たちはビジネス秘密と技術ノウハウに依存して、私たちの競争地位を発展させ、維持しています。例えば、我々独自のLNCプラットフォームの重要な側面は、非特許に基づくビジネス秘密および技術的ノウハウである。ビジネス秘密とノウハウは を保護することは難しい.従業員、コンサルタント、科学コンサルタント、請負業者、ビジネスパートナーと守秘協定および発明譲渡協定を締結することによって、当社のノウハウおよびプロセスを保護することを求めています。これらのプロトコルは、我々の固有情報を保護し、発明譲渡プロトコルの場合、第三者との関係によって開発された技術の所有権を付与することを目的としている。私たちはまた、私たちの場所の物理的安全と私たちの情報技術システムの物理的および電子的安全を維持することによって、私たちのデータと取引機密の完全性と機密性を保護しようとしています。br}私たちはこれらの個人、組織、そしてシステムに自信がありますが、合意やセキュリティ措置は違反される可能性があり、私たちはどんな違反にも対応する十分な救済措置がないかもしれません。また,我々のビジネス秘密は 競争相手に知られたり独立して発見される可能性がある.私たちの請負業者が私たちのために働いているときに他人が所有している知的財産権を使用する場合、関連またはそれによって生じるノウハウおよび発明の権利によって紛争が生じる可能性がある。
| 24 |
我々はまた,適切な場合には,米国や米国以外の場所で商標保護を求める予定である。私たちはこれらの登録商標を私たちの薬品開発と私たちの候補製品に使うつもりです。
競争
生物技術と製薬業界の特徴は技術進歩が迅速で、競争が激しく、そして独自製品を高度に重視していることである。私たちは商業製薬とバイオテクノロジー企業、学術機関、政府機関、私営と公共研究機関を含む多くの異なる源からの競争に直面している。これらの会社の多くはより多くの人力と財力を持ち、より高度な開発段階にある候補製品がある可能性があり、多くの会社は私たちの候補製品の前に市場に投入するだろう。競争相手はまた、より効果的、より安全またはより安価な製品を開発するか、またはより良い耐性または利便性を有する可能性がある 。
私たちの独自のLNCプラットフォーム、経験、そして私たちの重点分野での知識は私たちに競争優位を提供してくれると考えていますが、潜在的な競争相手は私たちのビジネス機会を減らすかもしれません。私たちの多くの候補製品について、私たちは他の製品からの競争に直面することが予想されます。これらの製品は汎用ベースで提供され、価格は低いです。その中の多くの後発薬はすでに第三者によって長年販売されており、医師、患者と支払人に受け入れられている。
私たちbrは、MAT 2203と私たちが将来私たちの独自のLNCプラットフォームを使用して行う可能性のある任意の他の開発方案は、薬物送達分野における私たちの科学と開発専門知識に匹敵し、同業者に対する競争優位性を提供すると信じている。しかし、私たちはより規模が大きく、資金がより十分な製薬、専門製薬とバイオテクノロジー会社、および後発薬メーカー、学術機関、政府機関、および公共と個人研究機関からの競争を含む様々な源からの潜在的な競争に直面している。
MAT 2203は、主にポリエン、窒素およびラチェットを含む真菌およびカビ感染の治療のために許可された抗真菌薬と競合するであろう。これらの適応を承認するブランド療法としては,Cansidas(キャポフェン,メルク社が販売),Eraxis(ファイザー社製anidulafungin),Mycamine(ミカフィン浄,Astellas Pharma米国社製),地フッ素康(ファイザー社製フルコナゾール),ノサ非(ポサコナゾール,メルク社製),Vfend(ボリコナゾール,ファイザー社製),Sporanox (イトラコナゾール,Jansen製薬社製),simiilba(Astellas Pharma,Inc.,AmliB,両性)がある。Astellas Pharma US,Inc.マーケティング)Abelcet(脂質複合体両性マイシンBは,Sigma Tau製薬社製),Rezzayo(Rezafungin,Melinta Treeutics製),Brexafemme(グラクソ·スミスクライン製Ibrexafungerp),両性マイシンBデオキシコール酸塩(X−Gen製薬社製)である。MAT 2203市場の承認時に、現在、これらの製品のより多くの汎用バージョンが使用可能である可能性があり、これは追加の競争をもたらすであろう。承認された治療法に加えて,MAT 2203はolorofim(F 2 G,Ltd.開発),Fosmanogepix (Basileaによる開発),およびAM 2−19,両性マイシンBの誘導体がイリノイ治療社によって開発されるように,我々の知られている第三者臨床開発候補製品と競合する可能性が予想される。
製造業
我々の主要なLNCプラットフォーム候補製品MAT 2203の内部製造能力と、小分子と小さいオリゴヌクレオチド領域のLNCプラットフォームで計画された内部製造能力を発見し、運営している。我々が行っている臨床試験および可能な早期商業化に必要な製品を製造するのに十分な臨床用品 を製造するのに十分であるが、我々は、NDAの届出を支援し、その製品の商業生産を支援するために、尊敬される第三者契約製造業者との関係を検討している。私たちは将来また内部製造能力 を拡大する必要があるかもしれない。既存の製造施設を維持できなければ、MAT 2203と他の候補製品のためにより多くの内部製造能力brを開発して、これらの製品の商業化製品を生産することができなければ、私たちは第三者メーカーとの関係を発展させて候補製品を製造する必要があり、これは時間がかかり、コストがかかるかもしれません。
| 25 |
2022年第1四半期に、秘密協定が提出される可能性が予想されるため、MAT 2203の拡張と製造をサポートするためにThermo Fisher Scienceと合意し、合意しました。Thermo Fisher Scienceは全世界で65を超える地点で、すべての開発段階に集積したエンドツーエンド能力を提供し、原料薬、生物製剤、ウイルスベクター、cGMPプラスミド、配合、臨床試験解決方案、物流サービス及び商業製造と包装を含む。2022年の間、Thermo Fisherと協力して、MAT 2203配合と製造に使用されるいくつかのプロセスの技術移転の準備をしています。我々の決定 としてFDAの我々3期試験計画に対する監督フィードバックを優先することと,MAT 2203の開発を3期に進めるための薬品や政府パートナーを獲得したいという決定の一部として,2023年にThermo Fisherへの移行と計画に関する費用 を緩めた。私たちは、これらのソースのうちの1つ以上から追加資金を得ると、Thermo Fisherとの活動 を回復すると予想している。
両性マイシンBは著者らのリードする臨床段階の模倣活性薬物成分であり、いくつかの潜在的な第三者サプライヤー 製品の候補製品MAT 2203がある。これまで,両性マイシンBの十分な供給を確保し,我々のMAT 2203臨床計画を支援するための正式な供給プロトコルは締結されていないが,我々は,MAT 2203臨床計画を支援するために,1つ以上の第三者サプライヤーから両性マイシンBの供給を得ることができると信じている。我々の候補製品の開発に伴い,重要な活性薬物成分の長期供給スケジュールを達成したい。
販売 とマーケティング
私たち は現在販売やマーケティングインフラを持っていません。私たちは、特に集中的な専門販売チームを通じて市場に参入できる場合、私たちがマーケティング承認を得た候補製品のアメリカでのマーケティングと販売権または共同販売促進権を保持する予定です。市場に進出するために大量の販売チームが必要な場合には,米国以外の市場を考慮して,先行する製薬やバイオテクノロジー会社との協力手配により候補製品を商業化する予定である。
アメリカで薬品を審査·承認する
米国では,FDAは“連邦食品,薬物と化粧品法案”(FDCA)とその実施条例に基づいて薬品を規制している。監督管理の承認を得て、その後適切な連邦、州、地方と外国の法律法規を遵守する過程は大量の時間と財力を必要とする。製品開発プロセス、承認プロセス、または承認後の任意の時間に適用された米国の要求を遵守できなかったことは、FDAが係属中の申請の承認を拒否すること、承認の撤回、臨床棚上げの実施、警告状および他のタイプの手紙を発行すること、製品のリコール、製品の差し押さえ、生産または流通の完全または部分的な一時停止、禁止、罰金、政府契約の拒否、原状回復、利益の返還、利益の返還、利益の完全または部分的な一時停止、禁止、罰金を含む様々な行政または司法制裁を受ける可能性がある。あるいはFDAおよび司法省(DoJ)または他の政府エンティティによって提起された民事または刑事調査および処罰。
我々のbr候補製品はNDAあるいは生物製品許可証申請(BLA)を通じてFDAの承認を得なければならず,生物製品候補製品については,米国で合法的に発売されるためには,brの流れを経なければならない。米国での新薬製品の販売および流通の承認を求める出願人は、通常、以下の条件を満たさなければならない
| ● | FDAの良好な実験室操作規範(CGLP)に従って非臨床実験室テスト、動物研究と調合研究を完成した |
| ● | ヒト臨床試験開始前に発効しなければならない研究新薬出願(IND)をFDAに提出する |
| 26 |
| ● | 各臨床試験を開始する前に、各臨床サイトを代表する独立機関審査委員会(IRB)によって承認される |
| ● | 現在の良好な臨床実践(GCP)に基づいて十分かつ良好に制御された人体臨床試験を行い、各適応に対する提案薬物製品の安全性と有効性を確定する |
| ● | NDAまたはBLAをFDAに準備して提出します |
| ● | 適切な場合、または適切な場合には、FDA諮問委員会によって製品が検討される |
| ● | 現在の良好な製造仕様(CGMP)の要件に適合することを評価し、施設、方法、および製品の特性、強度、品質、および純度を維持するのに十分な施設、方法、および制御を保証するために、FDAによる生産製品またはその構成要素の1つまたは複数の製造施設の1つまたは複数の検査を達成することが好ましい |
| ● | 使用料を支払い、FDAがNDAまたはBLAを承認することを保証する |
| ● | 任意の承認後要求を遵守し、リスク評価と緩和策(REMS)、およびFDA要求の承認後の検討 を含む。 |
非臨床研究
非臨床br研究は、製造された医薬品または活性医薬成分および調製薬品または薬品の純度および安定性を実験室的に評価すること、および体外培養薬物の安全性及び活性を評価するための動物研究と、人体で予備試験を行い、治療使用の理論的基礎を確立するための動物研究とを備える。非臨床研究の進行はcGLP法規を含む連邦法規と要求の制約を受ける。非臨床試験の結果は,製造情報,分析データ,任意の利用可能な臨床データや文献,臨床試験計画などとともにINDの一部としてFDAに提出される。
会社のbr}は通常、生殖AEsや発ガン性の動物試験のような長期的な非臨床試験を完成させなければならず、また薬物化学と物理特性に関するより多くの情報を開発し、cGMP要求に基づいて商業大量生産薬物のプロセスを最終的に決定しなければならない。製造プロセスは品質が安定した候補薬物ロットを持続的に生産することができなければならず、その中で製造業者は最終薬物製品の特性、強度、品質、および純度をテストするための方法を開発しなければならない。また,適切な包装を選択·試験し,候補薬物が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
ヒトbrは規制承認の臨床試験を支持している
臨床試験は、GCP要求に従って合格した研究者の監督の下でヒト対象に研究製品を服用することを含み、すべての研究対象に任意の臨床試験に参加する前に書面でインフォームドコンセントを提供することを含む。臨床試験は書面による研究案に基づいて行い,その中で研究の目標,安全性モニタリングのためのパラメータ,評価すべき有効性基準を詳細に説明した。INDの一部として,各臨床試験の案および任意の後続案修正案をFDAに提出しなければならない。INDはFDAが受領してから30日後に自動的に発効し,それまでFDAが行う予定の臨床試験に懸念や問題を提起しない限り,試験は棚上げにした。この場合,INDスポンサーやFDAは臨床試験開始前に未解決の問題を解決しなければならない。したがって,INDの提出はFDAが臨床試験 の開始を許可する可能性もない可能性がある。
さらに、臨床試験に参加する各機関を代表するIRBは、その機関が任意の臨床試験を開始する前に計画を審査および承認しなければならず、IRBは少なくとも年に1回の継続的な審査および再承認を行わなければならない。 IRBは、研究対象に提供される研究案やインフォームドコンセント情報などを審査および承認しなければならない。 IRBはFDAの規定に従って動作しなければならない。いくつかの臨床試験に関する情報は,そのClinicalTrials.govサイト上で公開されるために,特定の時間範囲で米国国立衛生研究院に提出されなければならない。
| 27 |
米国国外で臨床試験を行うスポンサーはFDAの認可を得ることができるが,IND下で臨床試験を行うことが望ましい。ある国外の臨床研究がIND下で行われる時、放棄しない限り、FDAのすべてのIND要求を満たさなければならない。外国の臨床試験がINDに基づいて行われていなければ,臨床試験がGCPによって行われ,FDAが現場検査により臨床試験のデータを検証することができれば(FDAが必要と考える場合),スポンサーはNDAやINDを支援するために臨床試験のデータをFDAに提出することができる。
ヒトの臨床試験は通常3つの連続段階に分けて行われ、この3つの段階は重複または合併する可能性がある
ステップ 1:医薬は、最初に、少量の健康なヒト対象、または標的疾患(例えば、癌)または疾患を有する患者に導入され、その安全性、用量耐性、吸収、代謝、分布、排泄を試験し、可能であれば、その有効性の早期兆候が得られ、最適な用量が決定される。
ステップ 2:この薬剤は、可能な副作用および安全リスクを決定するために、より多くの試験参加者、数百人までに適用され、彼らは、一般に、可能な副作用および安全リスクを決定するために、実験薬物治療の疾患または状態を有し、特定の標的疾患に対する製品の治療効果を初歩的に評価し、用量耐性および最適用量を決定する。
ステップ 3:これらの臨床試験は一般に“キー”研究と呼ばれ、一般に、FDAまたは他の関連規制機関が薬物のデータを承認するかどうかを決定するために使用される研究を意味する。第三段階臨床試験では、この薬物は、制御された良好なbr臨床試験において、通常、地理的に分散した臨床試験地点でより多くの患者集団に適用され、承認のために製品の有効性および安全性を統計的に評価し、製品の全体的なリスク-収益プロファイルを確立し、製品のラベルに十分な情報を提供するのに十分なデータを生成する。
臨床試験結果の報告を詳細に説明する進展 は少なくとも年に1回FDAに提出しなければならず,重篤な副作用が発生するとより頻繁になる。 第1段階,第2段階,第3段階の臨床試験はどの指定された時間でも成功できないか,あるいは全く成功できない可能性がある。また、FDA あるいはスポンサーはいつでも様々な理由で臨床試験を一時停止或いは中止することができ、研究対象 が受け入れられない健康リスクに直面していることを発見することを含む。同様に、臨床試験がIRBの要求に従って行われない場合、または薬物が患者に意外な深刻な傷害を与えた場合、IRBは、その機関またはその代表機関の臨床試験の承認を一時停止または終了することができる。FDAは、通常、GCPおよび提出された臨床データの完全性を保証するために、1つまたは複数の臨床サイトを検査する。
FDAに秘密保持プロトコル を提出する
大多数の新薬或いは生物製品の監督管理許可は2つの十分かつ制御された良好な第三段階の臨床試験に基づいており、この2つの試験は新薬の安全性と有効性を提案する証拠を提供した。必要な臨床試験および他の要求を成功させると仮定すると, 非臨床および臨床試験の結果は,製品の化学,製造,br}制御および推奨されるラベルなどに関する詳細な情報とともにFDAに提出され,NDAの一部として,薬物製品 を1つまたは複数の適応に使用することの承認が求められる。連邦法によると、多くのNDAの提出には申請使用料がかかり、承認されたNDAのスポンサーは処方薬計画年会費と機関使用料を支払う必要がある。これらの費用は通常毎年増加します。
FDA は,NDAを受信してから60日以内にNDAの予備審査を行い,FDAが提出を受けて74日目にスポンサー申請が十分であるかどうかを通知し,実質的な審査を行うことができる。FDAは秘密協定の申請を受け入れるのではなく、より多くの情報を提供することを要求するかもしれない。この場合、申請は追加情報と共に再提出されなければならない。再提出された申請はFDAが届出を受ける前にも審査されなければならない。提出された申請が受け入れられると、FDAは深い実質的な審査を開始する。FDAはNDAの審査過程で具体的な業績目標を設定することに同意した。このような申請の多くは提出日から10ヶ月以内に審査され、多くの優先審査製品の申請は提出後6ヶ月以内に審査されます。FDAは、出願人によって提供された新しい情報を考慮して、最初の提出後にFDAによって発見された突出した欠陥を解決するために、出願人によって提供された新しい情報を考慮して、または明確にするために、様々な理由および異なる期間に審査手続きを延長することができる。
| 28 |
セキュリティ協定を承認する前に、FDAは、通常、製品を生産しているか、または生産する1つまたは複数の施設を検査する。これらの審査前検査はNDA提出に関連するすべての施設をカバーし、薬品成分製造(例えば活性製薬 成分)、完成品薬品製造と制御検査実験室を含む。FDAは、製造プロセスおよび施設がcGMP要件に適合していることを決定し、要求された仕様内で製品が持続的に生産されることを保証するのに十分でない限り、申請を承認しないであろう。さらに、セキュリティプロトコルを承認する前に、FDAは、通常、GCPに適合することを保証するために、1つまたは複数の臨床サイトを検査する。
FDAは新薬の申請を諮問委員会に提出したり,なぜこのような転任がないのかを説明することを求められている。通常,諮問委員会は臨床医や他の科学専門家を含む独立した専門家からなるグループであり,申請が承認されるべきかどうか,どのような条件で承認すべきかについて審査,評価,提言を担当する。FDAは諮問委員会の提案に制限されていないが、決定を下す時にこれらの提案を慎重に考慮する。
迅速な追跡、画期的な治療、優先的な指定の検討
FDA はある製品を指定して迅速な審査を行う権利があり、もしこれらの製品がbr}の深刻な或いは生命に危害を及ぼす疾病或いは状況を治療する時に満足されていない医療需要を満たすことを目的としている場合。これらの計画は,迅速チャネル指定,画期的治療指定,優先審査指定である。
特に、1つの製品が、深刻または生命に危険な疾患または状態を治療するために、単独でまたは1つまたは複数の他の薬剤と組み合わせて使用されることが意図されており、そのような疾患または状態の満たされていない医療要件を満たす可能性がある場合、FDAは、製品を迅速なチャネル審査に指定することができる。Fast-Track製品については,スポンサーがFDAとより多くのインタラクションを行う可能性があり,FDAは申請完了前にFast-Track製品のNDA部分を審査することができる.FDAがスポンサーから提出された臨床データを初歩的に評価した後にFast Track製品が有効である可能性があると判断すれば,スクロール審査が可能である。スポンサーはまた提供しなければならず、FDAは残りの情報の提出スケジュールを承認しなければならず、スポンサーは適用ユーザに 費用を支払わなければならない。しかし、FDAがFast Track申請の時間目標を審査するのは、NDAの最終部分が提出されるまで開始されます。 また、FDAがこの指定が臨床試験中に出現するデータの支持を得られなくなったと考える場合、FDAはFast Track指定を撤回する可能性があります。
次に,2012年,国会は“食品·薬物管理局安全·改善法案”を公布し,FDASIAと略称した。この法律は、“画期的な治療法”に指定された製品の審査を加速させるための新たな規制案を確立した。1つの製品が、深刻または生命に危険な疾患または状態を治療するために、単独または1つまたは複数の他の薬剤との併用が意図されており、初歩的な臨床証拠が、1つまたは複数の臨床的重要終点において、既存のbr療法よりも実質的に改善された効果を示す可能性があることを示す場合、例えば、臨床開発早期に観察された実質的な治療効果がある場合、製品はbr画期的療法として指定されることができる。Br FDAは、開発全体にわたってスポンサーとの会議を含むいくつかの行動をとる可能性があり、直ちに製品スポンサーに開発および承認に関する提案を提供する;より多くの上級者をbrプロセスの審査に参加させる;審査チームに1つの学際的なプロジェクト担当者を指定した;及び他のステップを取って効率的な方式で臨床試験を設計した。
第三に、薬物が重篤な疾患を治療し、承認された場合、安全性または有効性の面で有意に改善される場合、FDAは、製品を優先的に検討するように指定することができる。FDAは,他の利用可能な治療法と比較して,推奨されている薬物が有意に改善しているかどうかを詳細に決定している。顕著な改善は、ある疾患の治療の有効性の増加、治療を制限する薬物反応の除去或いは著しい減少、記録されている患者のコンプライアンスの向上、深刻な結果の改善、及び新亜群の安全性と有効性を招く可能性のある証拠として表現される可能性がある。優先度の指定は、このようなアプリケーションの評価に全体的な注意力とリソースを誘導し、FDAがマーケティングアプリケーションに行動する目標を10ヶ月から6ヶ月に短縮することを目的としている。
| 29 |
FDCA第524条によれば,FDAは法案の規定基準を満たすいくつかの熱帯病製品申請のスポンサーに優先審査証明書を付与する権利がある。優先審査証明書は、それを取得したスポンサーによって使用されてもよいし、他のスポンサーに転送されてもよく、後者は、異なる出願の優先審査を得るために使用されてもよい。優先査読クーポンは,候補製品の査読と承認時間を最大4カ月短縮することができる.熱帯疾患優先審査券を取得する資格を有するためには、 出願は、列挙された熱帯疾患;FDCA第505(B)(1)条または公衆衛生サービス法第351条に従って提出される;製品は、これらの法定条項に従って任意の他の出願で承認された有効成分を含まず、 であり、優先審査を受ける資格がなければならない。FDAは、ガイドラインにおいて、熱帯疾患の予防または治療のための製品適用を決定し、これらのアプリケーションは、優先審査証明書を取得する資格がある可能性がある。
Br承認経路を加速した
FDA は重症或いは生命を脅かす疾患のための薬物の承認を加速することができ、この薬物は患者に既存の治療よりも意義のある治療優勢を提供することができ、その根拠はこの薬物の代替終点に対する影響を決定することであり、この終点は合理的に臨床利益を予測する可能性が高い。この製品が中間臨床終点に影響を与え、不可逆的な発病率または死亡率またはIMMへの影響よりも早く測定することができ、そして病状の重症度、希少性または流行率、および代替治療の使用可能または不足を考慮すると、合理的に は不可逆的な発病率または死亡率または他の臨床的利益に対する影響を予測する可能性があり、FDAもこのような疾患の加速承認を許可することができる。加速的な承認を得た薬品は伝統的に承認された薬品と同じ安全と有効性法定基準に適合しなければならない。
承認の加速について言えば、代替終点は1つの標識であり、例えば実験室測定、放射画像、バイタルサイン或いは他の臨床利益を予測できると思われるが、それ自体は臨床利益の測定基準ではない。エージェント端末 は、通常、臨床端末よりも容易または迅速に測定を行う。中間臨床終点は治療効果の測定であり、それは合理的に薬物の臨床利益、例えばIMMへの影響を予測する可能性があると考えられている。中間臨床終点に基づく加速承認におけるFDAの経験は限られているが、治療効果が合理的に薬物の最終臨床効果を予測する可能性があると結論があれば、このような終点は通常加速承認 を支持することができ、終点で測定した治療効果自体が臨床利益と伝統的な承認の基礎ではないことを表明した。
Br}加速承認経路は、薬物の期待される臨床利益を測定するために時間を延長する必要がある環境において、代替或いは中間臨床終点への影響が早く発生しても、病気経過が比較的に長く、時間を延長する必要がある環境に最もよく用いられる。加速承認経路は、一般に、薬物の臨床的利益を検証および説明するために、スポンサーが勤勉な方法でbrの追加的な承認後の検証的研究を行うことに同意することに依存する。そのため、この基礎の上で承認された候補製品 は厳格な発売後のコンプライアンス要求を守らなければならず、4期或いは承認後の臨床試験を完成して臨床終点への影響を確認することを含む。必要な承認後研究を行わない場合や,発売後研究期間中に臨床 のメリットが確認できなければ,FDAがこの薬剤の市場からの撤回を加速させることが許される。加速法規により承認された候補製品のすべての販売促進材料 はFDAの事前審査を経なければならない。
FDA秘密保持プロトコルに関する 決定
NDAに対するFDAの評価および付帯情報によると、生産施設の検査結果を含み、FDAは承認書または完全な返信を発行することができる。承認書は、製品の商業マーケティングを許可し、特定の適応に対する具体的な処方情報を提供する。完全な返信手紙は、一般に、提出中の不足点 を概説し、FDAが出願を再検討するために、多くの追加のテストまたは情報を必要とする可能性がある。これらの不足点がNDAを再提出する際にFDAが満足した解決を得た後、FDAは承認書を発行するであろう。FDAは、含まれる情報タイプに応じて、そのような再提出された出願を2ヶ月または6ヶ月以内に検討することを約束している。この付加情報を提出しても, FDAは最終的にその申請が承認の規制基準を満たしていないと判定する可能性がある.
| 30 |
FDAがある製品を承認する場合、それは製品の承認適応を制限する可能性があり、製品ラベルに禁忌症、警告または予防措置を含むことを要求し、承認後の臨床試験を含む4期の臨床試験を要求し、承認後の薬物の安全性をさらに評価するために、製品の商業化後に製品を監視する試験および監視計画を要求するか、または製品の潜在的な市場および収益性に大きな影響を与える可能性のある他の条件を課す。また,承認の条件として,FDAは申請者にREMSの開発を要求する可能性がある。REMSは、製品の利益が潜在的リスクよりも大きいことを確実にするために、専門ラベル以外のリスク最小化戦略を使用する。REMSが必要かどうかを決定するために、FDAは、製品を使用する可能性のある集団の規模 ,疾患の重症度、製品の予期される利益、予期される治療持続時間、既知または潜在的な副作用の深刻さ、および製品が新しい分子実体であるかどうかを考慮するであろう。REMSは、薬物ガイドライン、医療専門家のための医師コミュニケーション計画、および安全な使用を確保する要素を含むことができ、処方または調剤のための特殊なトレーニングまたは認証、特定の場合のみの調剤、特殊な監視、および患者登録簿の使用を含むことができるが、これらに限定されない。FDAが製品の使用に関連する深刻なリスクを認識した場合、それは承認前または承認後のREMSを要求する可能性がある。REMSへの要求は製品の潜在的な市場と収益力に大きな影響を与える可能性がある。
FDA は発売後の研究或いはモニタリング計画の結果に基づいて製品のさらなるマーケティングを阻止或いは制限する可能性がある。承認された後、承認された製品の多くのタイプの変更、例えば、新しい適応の追加、製造変更、および追加のラベル宣言のように、 は、さらなるテスト要件およびFDAの審査および承認を受ける。
承認後に を要求する
FDAによって生産または流通を許可された医薬品brは、記録保存、定期報告、製品サンプリングおよび流通、広告および販売促進、および製品不良反応の報告に関連する要求を含むFDAによって普遍的かつ持続的に規制されている。承認された後、新しい適応または他のラベル宣言を追加するなど、承認された製品の大多数の変更は、FDAの審査および承認を事前に受ける必要がある。任意の上場製品やそのような製品を製造する機関には,継続的な年間使用料要件と,臨床データを有する補充応用に対する新規出願料がある。
また、医薬品メーカーと生産·流通承認薬品に参加する他のエンティティは、FDAと州政府機関にその機関を登録し、FDAとこれらの州政府機関の定期的な抜き打ち検査を受けて、cGMP要求に適合するかどうかを確認しなければならない。製造プロセスの変更は厳しく規制されており,通常FDAの事前承認が必要で が実施される。FDAの規定はまた、cGMPとのいかなる偏差も調査·是正し、スポンサーやスポンサーが使用を決定する可能性のある任意の第三者メーカーに報告や文書要求を行うことを要求している。そのため、メーカーはcGMPコンプライアンスを維持するために、生産と品質管理に時間、お金、労力をかけ続けなければならない。
承認後,規制要求や基準を遵守していない場合,あるいは製品発売後に問題が生じた場合,FDAは承認を撤回する可能性がある。予想されていない重症度または頻度の副作用(Br)、製造プロセス、または規制要件を遵守していないことを含む製品に以前に未知の問題が存在することが後に発見された場合、新しいセキュリティ情報を追加するために承認されたラベルの改訂をもたらす可能性がある;新しいbrの安全リスクを評価するために発売後研究または臨床試験を実施する;またはREMS計画に従って流通または他の制限を実施する。他の潜在的結果は その他の事項を含む:
| ● | 製品の販売または製造、製品の完全な市場撤退、または製品のリコールを制限する |
| ● | 承認後の臨床試験には、罰金、br警告、または無タイトル手紙または一時停止が科される |
| ● | FDAは、承認されるべきNDAまたは承認されたNDAの補充剤の承認を拒否するか、または製品承認を一時停止または撤回する |
| ● | 製品 は、製品の輸出入を許可することを拒否するか、差し押さえたり、差し押さえたりすることを拒否する |
| ● | 禁止されたり、民事または刑事処罰が適用される。 |
FDA 市場に投入された製品のマーケティング、ラベル、広告、販売促進を厳格に規範化します。承認された適応のみに基づいて,承認されたラベルの規定に基づいて薬物を普及させることができる。FDAや他の機関はラベル外使用の普及を禁止する法律や法規を積極的に実行しており,ラベル外使用を不当に普及させていることが発覚した会社は重大な責任を負う可能性がある。
| 31 |
そのほか、処方薬製品の流通は“処方薬販売法”(PDMA)の制約を受け、この法案 は連邦一級の薬品と薬品サンプルの流通を規定し、各州の薬品流通業者の登録と監督管理に最低基準を設定した。PDMA,州法ともに処方薬製品サンプルの分配を制限し し,分配責任確保の要求を規定している。
後発薬の新薬申請(ANDA)
1984年、FDAのHatch-Waxman修正案の採択に伴い、国会はFDAが法規のNDA条項に従って以前に承認した薬物と同じ模倣薬を承認することを許可した。模造薬の承認を得るためには,出願人は簡略化された新薬出願(ANDA)を当該機関に提出しなければならない。このような応用を支持するために、模倣薬メーカーは以前に機密協定によって許可された薬品による非臨床と臨床テストに依存することができ、参考発売薬物(RLD)と呼ばれる。
具体的には,ANDAを承認するためには,FDAは後発薬が有効成分,投与経路,剤形,薬物強度においてRLDと同様であることを発見しなければならない。同時に、FDAは後発薬と革新薬が生物学的同等性を有することを確定しなければならない。法規によると、もし模倣薬の吸収速度と程度が市販薬物の吸収速度と程度と明らかな差がなければ、この模倣薬は生物的にRLDに等しい。
ANDAが承認された後,FDAはその出版物“承認された治療同等性評価を有する薬物製品”(“オレンジブック”とも呼ばれる)で,このイミテーション製薬が治療においてRLDと同様であるかどうかを指摘している。医師と薬剤師は治療上同じ後発薬がRLDを完全に代替できると考えている。また,ある州の法律や多くの医療保険計画の実施により,FDAが指定した治療同等性は,薬剤師が処方医や患者が知らない場合やその同意を得ずに後発薬を自動的に代替することが多い。
Hatch-Waxman修正案によると、FDAは、RLDの任意の適用可能な非特許専門期間が満了するまでANDAを許可してはならない。FDCAは、新しい化学物質を含む新薬に5年間の非特許データ固有期間を提供する。排他性が付与された場合、ANDAは、5年の満了前にFDAにANDAを提出することができず、提出書類に第4段落の認証が添付されていない限り、出願人は、元の製品の承認後4年に出願を提出することができる。 NDAが1つ以上の新しい臨床研究(バイオアベイラビリティまたは生物学的同等性研究を除く)の報告を含む場合、FDCAはまた、3年間の排他性を規定する。これらの研究は、出願人または出願人のために行われるものであり、承認出願に重要である。この3年間の専門期間は、通常、新しい用量形態、投与経路、組み合わせ、または適応のような以前に承認された医薬製品の変化を保護する。
Hatch-Waxman 特許認証と30ヶ月の有効期限
NDAまたはその付録が承認された後、NDAスポンサーは、出願人の製品または承認された製品使用方法をカバーする各特許をFDAにリストしなければならない。NDAスポンサーにリストされたすべての特許はオレンジ色の本に発表されている。ANDA出願人がFDAにその出願を提出する場合、出願人は、“オレンジマニュアル”に記載されている参照製品に関連する任意の特許をFDAに証明しなければならないが、ANDA出願人が承認を求めていない使用方法に関する特許は除く。具体的には、出願人は、各特許証明について:
| ● | 必要なbrの特許情報が提出されていない; |
| ● | の発売特許は期限が切れている; |
| ● | 記載された特許はまだ満了していないが、特定の日に満了し、特許が満了した後に承認を求めることができる |
| 32 |
| ● | に記載されている特許は無効で、強制的に実行できないか、または新製品に侵害されない。 |
新製品が承認された製品の上場特許又はそのような特許を侵害しないか、又は強制的に実行できない認証を第4項認証と呼ぶ。出願人が列挙された特許に異議を唱えていない場合、または特許使用方法の承認を求めていないことを示す場合、ANDA出願は、参照される製品を要求するすべての特許が期限切れになるまで承認されないであろう。
ANDA出願人が第4項の認証をFDAに提供した場合、FDAがANDA届出を受けた後、出願人はまた、第4項の認証の通知をNDA及び特許所持者に送信しなければならない。次いで、NDAおよび特許所有者は、第4段落の認証の通知に対して特許侵害訴訟を提起することができる。第4項の認証を受けてから45日以内に特許侵害訴訟を提起すると、第4項の通知、特許満了または侵害事件においてANDA出願人に有利な裁決を受けてから30ヶ月後(早い者に準ずる)まで、FDAがANDAを承認することは自動的に阻止される。
小児科研究と排他性
“2003年小児科研究公平法”によれば、秘密協定またはその付録は、すべての関連する小児科亜群において医薬製品が主張する適応の安全性および有効性を評価し、製品に対して安全かつ有効な各小児科亜群の用量および投与をサポートするための十分なデータを含まなければならない。2012年の食品·薬物管理局安全·革新法案(FDASIA)の公布に伴い、スポンサーはデータを評価する前に小児科研究計画を提出しなければならない。これらの計画は、研究目標および設計、 の任意の延期または免除請求、および法規要件の他の情報を含む、出願人計画によって行われる1つまたは複数の小児科研究の概要を含む必要がある。そして、出願人、FDA、FDAの内部審査委員会は、提出された情報を審査し、相互に協議し、最終計画について合意しなければならない。FDAまたは出願人 は、いつでも計画の修正を要求することができる。
FDAは、申請者の要求に応じて、小児科データの一部または全部の提出を成人使用許可を得た後に延期するか、または小児科データ要件の一部または一部を免除することを許可することができる。延期要求と延期要求に関する付加要求 とプログラムはFDASIAにロードされる.
小児科専門権は、米国の別のタイプの非特許マーケティング専門権であり、付与された場合、任意の既存の法規専門権(非特許専門権を含む)の条項に追加6ヶ月の市場保護を追加することが可能である。もしNDAスポンサーが提出した小児科データがこのようなデータに対するFDAの書面請求に公平に応答した場合、この6ヶ月の専門権を付与することができる。これらのデータは,この製品が研究されている小児科群で有効であることを示す必要はなく,逆に臨床試験がFDAの要求に公平に応答していると考えられれば,追加的な保護が得られる。要求された小児科研究報告が法定期限内にFDAに提出され、FDAによって受け入れられる場合、製品の法定または規制専門期間または特許保護期間にかかわらず6ヶ月延長される。これは特許期間の延長ではないが、FDAが別の出願を承認できない規制期間を効果的に延長する。
孤児 指定と排他性
孤児医薬品法によれば、1つの医薬製品がまれな疾患または疾患の治療を意図している場合(通常、米国では200,000人未満に影響を与えることを意味するか、またはbrでは米国で疾患または疾患を治療するための医薬製品を開発および生産することができないコストが製品の販売から回収されることを意味する)、FDAはその医薬製品を“孤児薬物”として指定することができる。会社は機密協定を提出する前に孤児製品指定を申請しなければならない。 承認を要求した場合、FDAは治療薬の識別およびその潜在的用途を開示する。孤立製品命名 は監督管理審査と審査過程中にいかなる優勢を伝達することもなく、監督管理審査プロセスの持続時間を短縮することもない。
孤児状態を有する製品が、このような指定された疾患または状態を有するFDAによって初めて承認された場合、この製品は、孤児製品の専門権を得る権利がある。孤立製品排他性とは、いくつかの限られた場合を除いて、FDAが同じ製品の同じ適応を7年以内に承認しない可能性のある他の任意の申請 を意味する。競争相手はこの孤立製品が排他的な適応を持つことで異なる製品の承認 を得る可能性があり,同じ製品の承認を得る可能性があるが, は異なる適応の承認を得る可能性がある。孤立製品に指定された薬物や薬物製品が最終的に発売承認され,その適応範囲 が孤立製品申請で指定された範囲よりも広い場合には,独占経営権を得る権利がない可能性がある。
| 33 |
“21世紀の治癒法”
2016年12月13日,国会は第21回を採択したST世紀の治療法や治療法です“治療法案”は、医療保健を現代化と個性化し、革新と研究を刺激し、特定プロジェクトの連邦資金を増加させることによって、新しい治療法の発見と開発を簡略化することを目的としている。それは革新プロジェクトのためのFDAの資金を増加させることを許可する。新しい法律はまた“公衆衛生サービス法”を改正し、国家衛生研究院への援助を再認可と拡大する。この法案は、戦略計画の策定と実施、早期調査者、研究の費用を支払うためのNIH革新基金を設立しています。NIHはリーダーと協調拡大の小児科研究も担当しています。さらに、“治療法案”には、FDAに新しい臨床試験設計の使用、応用における真の証拠の使用、いくつかの適応の追加申請の要約レベル審査の利用可能性、および薬物開発ツールの資格に関する条項の評価および発行を要求する条項が含まれている。Cures Actが最近公布されたため,我々の業務への潜在的な影響は不明であるが,個人に対して拡張アクセス計画を提供する政策を公表することを要求する条項がある.このような条項はFDAがこのような政策を制定するのに数年かかることを可能にしているので、私たちに与える影響は延期されるかもしれない。
FDCAと公衆衛生サービス法(PHSA)の改正案により,“治療法”第3章では新薬や医療技術の発見·開発·交付の加速を求めている。そのため、他の条項を除いて、“治療法”は2026年前の稀な小児科疾患のある薬物を治療するための既存のbr}優先審査クーポン計画を再許可した;重大な国家安全脅威医学対策申請と決定された薬物申請のために新しい優先審査 金券計画を作成した;そして組合せ製品申請の審査を簡略化するために FDCAを改訂した。
“治癒法”第3042節は“限られた人口経路”を許可し、深刻な或いは生命に危害を及ぼす深刻な或いは生命に危害を及ぼす感染を治療することを目的とした抗菌製品の承認を加速し、これらの感染には満たされていない医療需要が存在する。この規定に基づいて承認された薬品は、顕著な“限られた人口”声明を含む特殊なラベル要求を遵守しなければならない。“治療法案”の実施状況を監視するが,その計画が我々の業務にどのように影響する可能性があるかは評価できない。
その他 医療法規
健康プライバシー法
私たち は、データ保護法令(すなわち、プライバシーおよびデータセキュリティに関連する法律法規)によって制約されています。米国では、州データ漏洩通知法、州健康情報プライバシー法、連邦と州消費者保護法(例えば、連邦貿易委員会法第5条)を含む多くの連邦と州法律法規が、健康に関する個人情報や他の個人情報の収集、使用、開示、保護を管理している。データ保護の法律および法規を遵守しないことは、政府の法執行行動を招き、私たちの責任(民事および/または刑事罰を含む可能性がある)、個人訴訟および/または不利なbr宣伝をもたらす可能性があり、これは私たちの経営業績および業務に負の影響を与える可能性がある。さらに、第三者 (例えば、私たちの臨床試験に参加する主要な研究者)から健康情報を取得することができ、これらの第三者は、“1996年健康保険携帯および責任法案”(HIPPA)(経済および臨床健康情報技術法案“(HITECH)改正)のプライバシーおよびセキュリティ要件に制約される。HIPAAは、一般に、保険エンティティ(ヘルスケア提供者、健康計画、および医療情報交換所) が、患者が保護された健康情報を開示する前に、患者の書面許可を取得することを要求する(br}許可要件の例外が適用されない限り)。許可が必要であり、患者が許可または許可を実行できなかった場合、必要なすべての条項が含まれていない場合、私たちは患者の情報にアクセスおよび使用できない可能性があり、私たちの研究の努力は損害または遅延を受ける可能性がある。さらに、有効な患者許可に基づいて提供される保護された健康情報の使用は、許可された規定によって制限される(例えば、製品の承認を得るために規制機関に研究および提出するために使用される)。他の事項に加えて、HITECHは、HIPAAのプライバシーおよびセキュリティ基準および遵守されていない様々な罰 を、カバーエンティティを表して、保護された健康情報の作成または使用、またはカバーエンティティにサービスを提供する特定の機能を含む独立請負業者またはエージェント を実行することに直接適用する。私たちは私たちがHIPAAの下の“ビジネスパートナー”だと信じていないが、規制機関は同意しないかもしれない。
| 34 |
2016年に採択された“一般データ保護条例”(GDPR)は、収集されたEU住民に関する個人データのセキュリティおよびそのような個人データのEU加盟国 国における国境を越えた流れを保護するための規制枠組みを構築し、個人データに関する個人同意の要求、個人への通知の性質および範囲、個人データのセキュリティおよびセキュリティ、データ漏洩通知 および個人データを処理する際に第三者プロセッサを使用することを含む。EU指令およびGDPRを遵守しない場合、私たちは規制制裁、臨床試験遅延、刑事起訴、および/または民事罰金または処罰を受ける可能性がある。また,GDPRは個々のデータエージェントが行動する直接的な原因を作成している.
詐欺と法の濫用
FDAの薬品マーケティングに対する制限以外に、近年、他のいくつかの州と連邦法律を応用して製薬業界のあるマーケティング行為を制限している。このような法律には反リベート規制と虚偽請求規制が含まれている。連邦医療計画反リベート法規は、購入、レンタル、注文または手配購入の見返りとして、Medicare、Medicaid、または他の連邦によって援助された医療計画によって精算可能な任意の医療項目またはサービスを誘導または注文することを意図的に提供、支払い、請求または受け取ることを禁止する。この法規は、医薬品メーカーと処方者、購入者、および処方マネージャーとの間の配置に適用されると解釈される。反リベート法規に違反した行為は、監禁、刑事起訴、民事罰金、連邦医療計画から除外されることができる。いくつかの法定免除と規制避風港はいくつかのよく見られる活動を起訴や他の規制制裁から保護しているが、これらの免除と安全避難港の範囲は限られており、処方、購入或いは推薦の報酬を誘導するためのやり方が免除或いは安全避難港の資格を満たしていなければ、審査を受ける可能性がある。
連邦虚偽請求法は、誰もが故意に連邦政府に提出するか、虚偽請求を提出させるか、または故意に虚偽陳述を行うか、または虚偽請求を支払いに導くことを禁止する。最近,いくつかの製薬会社や他のヘルスケア会社がこれらの法律により起訴されており,価格設定サービス機関に報告されている薬品の価格が上昇しており,これらの薬品は連邦医療保険や医療補助販売率を設定するために政府に使用されており,顧客がその製品の連邦計画に課金することを希望しているため,顧客に製品brを無料で提供することが告発されている。また、ラベル外販売促進を含むいくつかのマーケティング行為も虚偽申告法に違反する可能性がある。ほとんどの州にも連邦反リベート法規や虚偽申告法のような法規や法規があり、Medicaidや他の州計画に基づいて精算するプロジェクトやサービスに適用されたり、いくつかの州では支払者が誰であっても適用されます。
負担できる介護法案
2010年3月下旬、連邦政府は全面的な医療改革案、すなわち“平価医療法案”(ACA)を公布した。他の条項では、ACAは、個人および雇用主の健康保険要求を規定し、一定の保険補助金(例えば保険料や費用分担)を提供し、広範な保険市場改革を強制的に行い、新しい医療保険アクセスポイント(例えば、州と連邦の健康保険取引所に基づく)を作成し、医療補助計画を拡大し、異なる技術やプログラムの臨床的有効性の比較検討を促進し、医療保険計画が製品やサービスを清算する方法をいくつか変更する。ACA下の連邦虚偽請求法案改正案は,個人当事者が会社に対して訴訟を起こしやすくし,これらの訴訟により,告発者は政府に支払われた任意の金から一定の割合の報酬を得る権利がある可能性がある.
公布以来、司法と議会はACAのいくつかの側面に疑問と修正案を提出した。ACAの実施には、ACAのさらなる改正の可能性と、ACAの法的挑戦または 廃止への努力を含む 不確実性が依然として存在する。ACAが廃止またはさらに修正された場合、またはACAのいくつかの態様の実施が延期された場合、 のような廃止、修正、または遅延は、私たちの業務、戦略、見通し、経営業績、または財務状態に重大な悪影響を及ぼす可能性がある。現在、ACA実施中のいかなる廃止、修正、または遅延が私たちに全面的な影響を与えるかを予測することはできない。CMSや他の機関は、重大な規制改革を実施する必要があり、これらの改革を実施するために必要な多くのプロセスが必要となるため、どのような医療計画が連邦または州レベルで実施されるか、任意のこのような改革の時間 またはそのような改革、または任意の他の未来の立法または法規が私たちの業務に与える影響を予測することはできない。
| 35 |
適格感染症製品の指定と排他性
2012年、国会は“直ちに抗生物質を発生させるインセンティブ法案”または“利得法案”という立法を可決した。この立法は、深刻かつ生命に危害を及ぼす感染を引き起こす病原体の治療のための抗菌と抗真菌薬物製品の開発を奨励することを目的としている。そのため,法律ではFDAが適格感染症製品(QIDP)に指定された薬品がNDAの承認を得た後,5年間の市場排他性を追加的に与える。そのため、QIDPに対して、5年の新化学実体独占期、3年の新臨床研究独占期と7年の孤児薬物独占期はそれぞれ10年、8年、br}と12年になる。
Gain Actは、QIDPを“重大または生命を脅かす感染を治療するためのヒトによって使用される抗菌または抗真菌薬であって、-(1)新しいまたは新たに出現した感染性病原体を含む抗菌または抗真菌耐性病原体、または(2)いくつかの”適格病原体“によって引き起こされる感染を含む”と定義する。適格病原体“とは、公衆の健康に深刻な脅威となる可能性のある病原体(例えば、薬剤耐性グラム陽性病原体、多剤耐性グラム陰性細菌、多剤耐性結核、およびクロストリジウム·クロストリジウム)は、FDAが確立および維持するリストに含まれる。医薬品スポンサーは、セキュリティプロトコルを提出する前の任意の時間に、FDAにその製品を合格認証製品として指定することを要求することができる。FDAは指定申請後60日以内に合格認証決定を下さなければならない。QIDPに指定された製品はFDAの優先審査を取得し,“FAST Track”状態を取得する資格がある.
“ゲイン法案”によると、FDAによってQIDPに指定された薬品の追加5年間市場排他性は、2012年7月9日またはその後に初めて承認された薬物にのみ適用される。さらに、5年間の独占権延期は、FDCA第505(B)条に従って延期有効または期限切れの任意のQIDP申請のための補充申請 ;FDAによって承認された製品変更のために提出された後続の申請 には適用されず、この変更は、新しい適応、投与経路、投与スケジュール、用量 形態、投与システム、投与装置または強度、またはその承認に基づく使用が、第505(G)条のQIDPの定義された製品に適合しない。
特許期限の回復と延長
“ハッジ·ワックスマン法”によると、新薬製品を持つ特許は、製品開発およびFDA規制審査中に失われた特許期間が5年間に及ぶ特許を回復することを可能にする限られた特許期間延長を得る資格がある可能性があると主張している。承認された回復期は、通常、IND発効日とNDA提出日との間の時間の半分であり、NDA提出日と最終承認日との間の時間を加える。特許期限回復は,特許の残存期間の延長には利用できず,製品承認日から合計14年を超える。承認された薬品に適用される特許は1つのみ延期する資格があり,延期出願は関連特許が満了する前に提出しなければならない。承認を求める複数の薬物をカバーする特許は、そのうちの1つが承認された場合にのみ延期される。米国特許商標局は,FDAと協議した後,任意の特許期間の延長または回復の出願を審査·承認する。
EUの薬品審査と承認
アメリカ以外でいかなる製品を販売するためには、会社はまた他の国/地区と司法管轄区の品質、安全性と有効性に関する多くかつそれぞれ異なる法規要求を遵守し、そして臨床試験、マーケティング許可、薬品の商業販売と流通などに対して監督管理を行わなければならない。製品がFDAの承認を得ているか否かにかかわらず、同社は非米国規制機関のような必要な承認を得て、その後、これらの国または司法管轄区で臨床試験を開始したり、製品を販売したりすることができる。承認の流れは最終的に国と司法管轄区によって異なり、追加の製品テストと追加の行政審査期間に関連する可能性がある。他国および管轄地域で承認を得るのに要する時間は、FDA承認を得るのに要する時間とは異なり、FDA承認を得るのに要する時間よりも長くなる可能性がある。1つの国または司法管轄区で規制承認を得ることは、別の国または司法管轄区で規制承認を得ることを保証することはできないが、1つの国または司法管轄区で監督管理許可を得ることができなかったり、遅延したりすることは、他の国または司法管轄区の監督管理プロセスに悪影響を及ぼす可能性がある。
| 36 |
“欧州臨床試験指令”によると、EUはすでに加盟国の国家立法を通じて臨床試験審査制度を実施した。この制度によれば,申請者は臨床試験を行うEU加盟国の主管国当局の承認を得なければならない。また,出願人は主管倫理委員会が賛成の意見を発表した後にのみ臨床試験を開始することができる。臨床試験申請は,欧州臨床試験指令と加盟国に対応する国家法律で規定されている支持情報を持つ研究用薬品アーカイブを添付し,適用された指導文書の中でさらに詳細に説明しなければならない。
EUの監督管理制度に基づいて薬品の上場許可を得るためには、申請者は集中或いは分散プログラムに従って上場許可申請或いはMAAを提出しなければならない。
集中化手続きは、欧州委員会によってすべてのヨーロッパ EU加盟国に対して有効な単一マーケティング許可を付与することを規定している。特定の製品には、いくつかのバイオテクノロジープロセスによって生産された薬品、孤児薬品として指定された製品、高度な治療製品、およびある疾患を治療するための新しい活性物質を含む製品が含まれており、集中プロセスは強制的である。他の疾患を治療するための新しい活性物質を含む製品、および高度な革新性を有するか、または患者に有利な製品 については、集中化手順が任意である可能性がある。
集中手順により、ヨーロッパ薬品管理局(EMA)に設置された人用薬品委員会或いはCHMPが薬物の初歩的な評価を担当する。CHMPはまた、既存のマーケティング許可の修正または拡張を評価するなど、いくつかの許可後および保守活動を担当する。欧州連合の中央手続きによれば、出願人がCHMPの質問に答えるために追加的な情報または書面または口頭解釈を提供する必要がある場合、評価MAAの最長制限時間は、タイマポーズを含まない210日である。特殊な場合、公衆衛生の観点、特に治療革新の観点から見れば、1種の医薬製品は重大な価値があり、CHMPは加速評価を承認する可能性がある。この場合,EMAは150日以内にCHMPの意見を与えることを確保している.
複数のEU加盟国で製品を販売することを希望する申請者は、br分散プログラムを使用することができ、これらの国の製品 は、以前にどのEU加盟国でもマーケティング承認を得たことがない。分権手続きは、出願人によって指定された1つの加盟国(参照加盟国と呼ばれる)の出願に対する評価を、1つまたは複数の他の加盟国または関連加盟国によって承認されることを規定する。この手続きによれば、出願人は、製品特性概要草案、ラベル、および包装チラシ草案を含む、同じロールおよび関連材料に基づいて、参照加盟国および関連加盟国に出願を行う。会員国が有効な申請を受けてから210日以内に評価報告書の草稿と関連材料の草稿を作成することを参考にする。各関係加盟国は、加盟国の評価報告及び関連材料を参考にした日から90日以内に、評価報告及び関連材料を承認するか否かを決定しなければならない。
もし加盟国が公衆健康に対する潜在的に深刻な危害を理由に評価報告と関連材料を承認できない場合、 論争点は紛争解決メカニズムの制約を受け、最終的に欧州委員会に提出される可能性があり、その決定 はすべての加盟国に対して拘束力がある。
薬品のカバー範囲、定価と精算
FDAや他の政府機関が承認した製品の保証範囲や精算状態には大きな不確実性がある。製品の販売は、米国の政府医療計画、例えばMedicareおよびMedicaid、商業健康保険会社および管理医療組織などの第三者支払者が製品コストを支払う程度に部分的に依存するであろう。支払者が製品に保険を提供するかどうかを決定する流れbrは、価格または精算率を設定する流れとは独立している可能性があり、保険が承認されると、支払人は製品のために費用を支払う。第三者支払者は、保証範囲を承認済みリストまたは処方セット上の特定の製品に制限することができ、特定の適応のすべての承認された製品を含まない可能性がある。また、医療コストをコントロールすることは連邦と州政府の優先事項になっており、薬品価格はずっとこの仕事の重点である。アメリカ政府、州立法機関と外国政府は価格制御、精算制限と代替模造薬の要求を含むコスト制御計画の実施に大きな興味を示している。価格制御とコスト制御措置、および既存の制御と措置を持つ司法管轄区域でより厳しい政策をとることで、私たちの純収入と業績をさらに制限する可能性がある。
| 37 |
販売が承認される可能性のある任意の製品の保険·精算を確保するためには、会社は、製品の医療必要性および費用効果、およびFDAまたは他の同様の規制承認を得るのに必要なコストを証明するために、高価な薬物経済学的研究を行う必要があるかもしれない。支払者が製品に保険を提供することを決定することは、適切な販売率 を承認することを意味するわけではない。第三者決済は、製品開発投資の適切なリターンを実現するために、十分に高い価格レベルを維持するのに十分ではない可能性がある。
EUでは、定価と精算案は国/地域によって異なる。一部の国では、薬品は精算価格を合意した後にしか販売できないと規定されている。いくつかの国は追加的な研究を完成し、特定の候補製品のコスト効果を現在利用可能な治療法と比較することを要求するかもしれない。例えば、EUはその加盟国にオプションを提供し、その国の医療保険システムが精算を提供する薬品の範囲を制限し、人が使用する医療製品の価格を制御する。EU加盟国は薬品の具体的な価格を承認することができ、brは薬品を市場に投入する会社の収益力を直接または間接的に制御する制度をとることもできる。他の加盟国は会社が自分の薬品価格を決定することを許可するが、会社の利益を監視する。全体的には,医療コスト,特に処方薬の下り圧力が大きくなっている。そのため、新製品の参入にはますます高い壁が設けられている。また、一部の国/地域では、低価格市場からの国境を越えた輸入製品が競争圧力をもたらし、これは一国国内の定価水準を低下させる可能性がある。薬品に対して価格規制や精算制限を実施する国/地域のいかなる国/地域でも割引の精算と定価手配は許可されていない。
保健法律法規
医療保健の提供者、医師と第三者支払人は発売許可を得た薬品を推薦と処方する上で主要な役割を果たしている。第三者支払者や顧客との手配は、広く適用される詐欺や乱用、その他の医療保健法律法規の制約を受けている。 適用される連邦と州医療保険の法律と法規によると、このような制限は以下のように含まれる
| ● | 連邦医療保険逆控除法規は、他の事項に加えて、個人の転転または購入を誘導または奨励し、任意の商品またはサービスを注文または推薦するために、直接または間接的に現金または実物の形態で報酬を請求、提供、受信、または提供することを禁止し、個人の紹介または購入、注文または推薦を行うことができ、これらの商品またはサービスは、連邦医療保険および医療補助などの連邦医療計画に従って全部または部分的に支払うことができる |
| ● | 連邦虚偽請求法案は、連邦政府に故意に連邦政府に虚偽または詐欺的支払いクレームを提出したり、虚偽陳述によって連邦政府への支払い義務を回避、減少または隠蔽した個人または実体に対して民事処罰を実施し、通報者または準実体に対して民事訴訟を提起することを規定する |
| ● | HIPPAは、詐欺を実施する任意の医療福祉計画または医療事項に関する虚偽陳述を行う計画に刑事および民事責任を適用する |
| ● | HITECHとその実施条例改正されたHIPAAはまた、強制的な契約条項を含む個人が識別できる健康情報のプライバシー、安全、伝送を保護する義務を規定している |
| ● | 連邦虚偽陳述法規は、重大な事実を故意に偽造、隠蔽または隠蔽すること、または医療福祉、プロジェクトまたはサービスの交付または支払いに関連する任意の重大な虚偽陳述を行うことを禁止する |
| 38 |
| ● | ACA下の連邦透明性は、医薬品製造業者が、医師および教育病院への支払いおよび他の方法での価値の移転、ならびに医師の所有権および投資利益に関する情報を衛生公衆サービス部に報告することを要求し、報告された情報が検索可能なウェブサイト上で公表される;および |
| ● | 同様の州および外国の法律、例えば、州反リベートおよび虚偽クレーム法律は、販売またはマーケティング手配、ならびに非政府第三者支払者(個人保険会社を含む)によって精算される医療項目またはサービスに関するクレームに適用される可能性がある。 |
いくつかの州の法律は製薬会社に製薬業界の自発的コンプライアンスガイドラインと連邦政府が公布した関連コンプライアンスガイドラインを遵守することを要求し、それ以外に製薬業者に医者と他の医療保健提供者或いはマーケティング支出に支払うことに関する情報を報告することを要求する。場合によっては,州法や外国法は健康情報のプライバシーやセキュリティも管轄しており,その多くの法律は互いに大きく異なり,HIPAAに先を越されず,コンプライアンス作業を複雑化することが多い.
人的資源 資本資源
2024年3月18日現在、私たちは32人のフルタイム従業員がいる。私たちはどんな職員たちに対する集団交渉合意も持っていない。
私たちの成功は私たちが重要な人材を誘致、発展、維持する能力にかかっていると信じている。私たちは、肝心な従業員の技能、経験、業界知識は私たちの運営と業績に大いに役立つと信じています。
従業員の職場の健康と安全は私たちの核心価値観の一つであり、私たちは従業員に安全で信頼できる職場を提供します。
従業員 レベルを管理し、業務発展ペースと一致するように管理し、管理層は十分な人的資本を持って業務 の運営に成功すると信じている。
研究と開発
2023年と2022年12月31日までの年間の研究開発活動への支出はそれぞれ14,489ドルと16,678ドルであった。これらの費用には、私たちの抗感染候補製品MAT 2203およびMAT 2501、およびLNCプラットフォームをサポートし、強化することを含む、私たちの臨床および臨床前計画の開発に関連する現金および非現金費用が含まれています。我々の研究開発費 は,2023年12月31日と2022年12月31日までの年間で,CFF贈与協定に関するあるMAT 2501計画費用の精算金額がそれぞれ8.8万ドルと81.1万ドルであることを反映している。
会社 と既存の情報
我々 は2013年5月にMatinas BioPharma Holdings,Inc.の名称でデラウェア州に登録設立された。私たちには2つの運営子会社があります:Matinas BioPharma,Inc.これはデラウェア州の会社で、最初は2011年8月12日に設立され、名称はNereus BioPharma LLC;Matinas BioPharma NanoTechnologies,Inc., ,デラウェア州の会社で、最初に2015年1月29日に設立され、名称はAquarius BioTechnologies,Inc.である。
私たちの主な実行オフィスはニュージャージー州ベッドミンスター07921,302 Suit 3021545 Routo 206 Southにあります。私たちの電話番号は。私たちのサイトの住所はwww.matinnasbiopharma.comです。私たちのウェブサイトおよびウェブサイトに含まれている、または私たちのウェブサイトを介してアクセスできる情報は、参照によって本10-K表年次報告または米国証券取引委員会に提出または提供された任意の他の報告書に組み込まれているとみなされない。
我々は、これらの材料を米国証券取引委員会または米国証券取引委員会に電子的に提出または提供した後、合理的で実行可能な範囲内で、私たちの10-Kフォーム年次報告、10-Qフォーム四半期報告、8-Kフォーム現在の報告、および1934年の証券取引法第13(A)または15(D)節に提出または提供された報告書をできるだけ早く無料で提供する任意の修正案を提供する。私たちのアメリカ証券取引委員会の報告書は私たちのインターネットサイトの投資家の一部を通じてアクセスできる。さらに、本“Form 10−K”年間報告書のコピーは、ワシントンD.C.20549である米国証券取引委員会の公共資料室で見つけることができる。公共資料室の運営状況を知る必要があれば、アメリカ証券取引委員会:1-800-アメリカ証券取引委員会-0330に電話することができる。アメリカ証券取引委員会は、私たちが提出した書類に関する報告、依頼書、情報声明、その他の情報を含むサイトを維持しています。サイトはHttp://www.sec.gov。
| 39 |
| 1 a項目. | リスク要因 |
私たちの普通株への投資は投機的で、あなたの投資全体の損失リスクを含む高いリスクに関連しています。 私たちの普通株を購入する前に、以下に説明するリスクと今年度の報告書の他の情報をよく考慮しなければなりません。以下に説明するリスクと不確実性は私たちが直面している唯一の危険と不確実性ではない。他のリスクや不確実性も が私たちの業務運営に悪影響を及ぼす可能性があります。以下のリスク要因に記載されている任意のイベントが実際に発生した場合、私たちの業務、財務状況、または運営結果は深刻な影響を受ける可能性があります。この場合、私たちの普通株の価値は下がるかもしれません。あなたは私たちの普通株を購入する資金の全部または大部分を損失するかもしれません。
リスクファクターの概要
| ● | 私たち は設立以来大きな被害を受けた。私たちは今後数年で赤字になると予想していますが、決して を実現したり、利益を維持したりすることはできないかもしれません。 |
| ● | 私たちは多くの追加資金が必要になるだろう。もし私たちが必要な時に資金を集めることができなければ、私たちは私たちの製品開発計画や商業化努力を延期、減少、または廃止させることを余儀なくされるかもしれない。 |
| ● | 追加資本の調達は、株主に希釈、運営制限、または技術または候補製品の権利を放棄することを要求する可能性がある。 |
| ● | 私たちのbr株主は未償還オプションや引受権証の行使によって深刻な希釈を受ける可能性がある。 |
| ● | 私たちのこれまでの運営の歴史は、業務の成功度や将来の生存能力を評価することを困難にするかもしれません。 |
| ● | 我々の開発作業は初期段階であり,成功しない可能性がある. |
| ● | 私たちの候補製品が規制部門の承認を受けるかどうかを決定することはできません。そうでなければ、私たちは私たちの候補製品を販売することができません。承認過程のどんな遅延も私たちの業務を損なうだろう。 |
| ● | 私たちのbrの部分は、第三者が私たちに持っているか許可された技術に依存しており、これらの技術を失うことは、私たちの候補製品のさらなる開発を終了または遅延させ、私たちの名声を損なう、あるいはより高い印税を支払うように強要する。 |
| ● | 臨床薬物開発は長くて高価な過程と関連があり、結果は不確定である。 |
| ● | 私たちの臨床試験のどの方面の遅延も私たちのコストを増加させ、規制部門の私たちの候補製品に対する承認を得る能力を遅延または制限する可能性がある。 |
| ● | 私たちの供給と臨床研究義務を履行するために十分な数の製品がないか、得られないかもしれません。私たちの業務、財務状況、運営結果は不利な影響を受ける可能性があります。 |
| ● | もし私たちが私たちの製品を商業化することに成功できなければ、私たちが収入を作る能力は制限されるだろう。 |
| ● | もし私たちの臨床前と臨床研究に積極的な結果が生じなければ、もし私たちの臨床試験が延期された場合、あるいはこのような研究や試験期間中に深刻な副作用が発見された場合、私たちは遅延に遭遇し、追加のコストが発生し、最終的に私たちの候補製品を商業化できない。 |
| ● | 臨床試験を完了するのに十分な患者を募集できなければ,われわれの業務,財務状況,手術結果は悪影響を受ける可能性がある。 |
| ● | 我々の抗感染候補製品の孤立薬物指定や排他性を維持できない可能性がある。 |
| ● | FDAの任意の迅速なチャネル指定または優先審査地位の付与は、実際には、より速い開発または規制審査または承認プロセスをもたらすことはなく、FDAが私たちの候補製品を承認することを保証することもできません。また,我々の候補製品は が優先審査券資格を満たしていない適応を扱う可能性がある. |
| ● | FDAが私たちの候補製品に付与した任意のbr突破的療法指定は、より速い開発や規制の審査や承認過程を招くことはなく、私たちの候補製品が発売承認される可能性も増加しないかもしれない。 |
| ● | 我々の候補製品を合格した感染症製品として指定することは保証されず,いずれの場合も承認されても,必ずしもより速い開発や規制審査につながるとは限らず,FDAが我々の候補製品を承認する保証はない。 |
| 40 |
| ● | 私たちのbrは、私たちの任意の候補製品のために孤児薬指定、迅速チャネル指定、合格感染症指定、または突破的治療指定を獲得または維持できない可能性があり、承認されても、このような指定は本当に のより速い開発や規制審査を招くことはなく、FDAが私たちの任意の候補製品を承認することを保証することはできない。 |
| ● | もし私たちが他の候補製品の識別と開発に成功できなかったら、私たちの成長潜在力は影響を受けるかもしれない。 |
| ● | 私たち は現在販売とマーケティング組織がありません。もし私たちが満足できる販売とマーケティング能力を確立できなければ、 規制部門の承認を得ても、私たちの任意の候補製品を商業化することに成功できないかもしれない。 |
| ● | FDCA第505条(B)(2)条に従ってMAT 2203の承認を申請することができない場合、または505(B)(2)条による承認を得るためにセキュリティおよび有効性に関する追加データを生成する必要がある場合、予想される開発および商業化スケジュールを満たすことができない可能性がある。 |
| ● | 私たちは他のバイオテクノロジーと製薬会社からの競争に直面していますが、効果的に競争できなければ、私たちの経営業績は影響を受けます。 |
| ● | もし私たちが任意の候補製品のマーケティング承認を得たら、私たちは持続的な義務と持続的な規制審査の制約を受けるであろう これは多くの追加費用を招く可能性がある。また、私たちの候補製品はラベルや他の制限を受けて市場から撤退する可能性があります。もし私たちが規制要求を守らなかった場合、あるいは私たちの未来の製品が予期しない問題に遭遇したら、処罰されるかもしれません。 |
| ● | 将来の立法および/またはFDAが採用する法規および政策は、私たちが臨床試験を行って完成するのに要する時間とコストを増加させるかもしれない。 |
| ● | 医療保険法や施行条例の変化は我々に実質的な悪影響を及ぼす可能性がある。 |
| ● | 私たちの将来の成長は海外市場に進出する能力にある程度依存しており、そこでは追加の規制負担や他のリスクや不確実性の影響を受ける。 |
| ● | もし私たちが医療詐欺や法律乱用の方法で私たちの候補製品を販売したり、もし私たちが政府価格報告の法律に違反した場合、私たちは民事または刑事罰を受けるかもしれません。 |
| ● | 我々 は我々の協調プロトコルに大きく依存してMAT 2203を開発することが予想されており,我々 は第三者の性能に依存するリスクに直面している. |
| ● | 我々は第三者に依存して候補製品の臨床試験を行うことが予想され,第三者の性能に依存するリスクに直面している。 |
| ● | 我々の候補製品は現在も将来も完全に第三者に依存しており,これらの第三者がFDAや同様の外国規制機関の生産承認を得ることができず,十分な数の候補製品を提供できない場合,あるいは許容可能な品質レベルや価格で生産できない場合,我々の商業化努力は停止,延期,または利益低下する可能性がある. |
| ● | 不利な価格設定法規、第三者清算のやり方、あるいは医療改革の措置は私たちの業務を損なう可能性がある。 |
| ● | 伝染病の爆発は私たちの業務、財務状況、経営業績に実質的な悪影響を及ぼす可能性がある。 |
| ● | 経済的不確実性を含む不利な世界環境は、私たちの財務業績に悪影響を及ぼす可能性がある。 |
| ● | 私たち は私たちに許可されたいくつかの技術に依存する。私たちはこれらの技術を制御しないで、私たちはこれらの技術に対するいかなる権利の喪失も私たちの発見、開発、商業化候補製品を阻止することが可能です。 |
| ● | 私たちの知的財産権を保護することは難しくて高価で、私たちはこのような権利の保護を保障できない。 |
| ● | もし私たちが私たちの技術のために特許や商業秘密保護を取得したり、維持することができなければ、第三者は私たちの独自の情報を使用する可能性があり、これは私たちの市場での競争能力を弱化させ、私たちの収入を創出し、利益を達成する能力に悪影響を及ぼす可能性がある。 |
| ● | 私たちの候補製品は他人の知的財産権を侵害する可能性があり、これは私たちのコストを増加させ、私たちのbr開発と商業化努力を遅延したり阻害したりする可能性があります。 |
| ● | 私たちは私たちの組織の規模を拡大して私たちの業務を発展させる必要があり、私たちはこのような成長を管理する際に困難に直面する可能性がある。 |
| ● | もし私たちが高い素質の人材の誘致と維持に成功できなければ、私たちは私たちの業務 戦略を成功的に実施することができないかもしれない。しかも、特定の重要な従業員のサービスを失うことは私たちの業務の将来性に悪影響を及ぼすだろう。 |
| ● | 私たちに製品責任訴訟を提起すれば、私たちは大量の責任を負う可能性があり、候補製品の商業化を制限することが要求される可能性があります。 |
| 41 |
| ● | 我々の内部コンピュータシステム,あるいは我々のCROや他の請負業者やコンサルタントのシステムは,故障やセキュリティホールに遭遇する可能性があり,これは我々の製品開発計画を実質的に中断させる可能性がある. |
| ● | 私たちは将来的に事業や製品を買収したり、戦略同盟を結成したりするかもしれませんが、このような買収のメリットを意識していないかもしれません。 |
| ● | 私たちAシリーズの優先株条項によると、私たちは相当な印税を支払う義務があるかもしれない。 |
| ● | 普通株式保有者の権利は発行される可能性のある優先株によって損害を受ける可能性がある。 |
| ● | 私たちは予測可能な未来に私たちの普通株に配当金を支払うつもりはない。 |
| ● | 私たちの普通株の活発な公開取引市場は持続できないかもしれない。 |
| ● | 私たちの株価はずっと変動していて、引き続き変動するかもしれない。 |
| ● | もしbr証券や業界アナリストが私たちの業務に関する研究や報告を発表していない場合、あるいは彼らが私たちの株を不利に修正した場合、私たちの株価や取引量は低下する可能性がある。 |
| ● | 私たちはニューヨーク証券取引所アメリカ取引所から退市するかもしれません。これは私たちの株の流動性と私たちの資金調達能力を深刻に損なうかもしれません。 |
| ● | 当社が解散した後、あなたはすべてまたは一部の投資を回収することはできません。 |
| ● | 私たちの会社登録証明書は、株主のさらなる承認なしに新しい優先株シリーズを作成することを取締役会に許可しており、これは私たちの普通株式保有者の権利に悪影響を及ぼす可能性があります。 |
| ● | 我々の会社登録証明書、定款、デラウェア州法律の条項を逆買収することは、私たちを買収することをより困難にする可能性があり、これは私たちの株主に有利であり、私たちの株主が私たちの取締役会と経営陣の現メンバーを交換または罷免しようとすることを阻止するかもしれない。 |
| ● | 株主が私たちや私たちの役員、上級管理者や従業員と紛争したときに有利な司法フォーラムを得る能力が制限される可能性があります。 |
| ● | 純営業損失の繰越や他の税務属性を使用する能力が制限される可能性があります。 |
私たちの財務状況と追加資本需要に関するリスク
私たち は設立以来大きな被害を受けた。私たちは今後数年で赤字になると予想しており、永遠に実現できないかもしれないし、利益を維持しているかもしれない。
設立以来,我々 は毎年重大な運営損失が発生しており,予想される の将来純運営損失が予想される。2023年12月31日と2022年12月31日までの純損失はそれぞれ22,942,000ドルと20,997,000ドルです。2023年12月31日現在、私たちの累計赤字は175,573,000ドルです。私たちは私たちがいつ利益を出すのか分からない。これまで我々は製品販売から何の収入も得ておらず,私募や株式証券の公開により私たちの運営に資金を提供してきたが,嚢胞性線維化基金(CFF)や米国国立衛生研究院(NIH)の資金をわずかに利用してきた。私たちはほとんどの財力と精力を潜在的な候補製品の研究と開発に投入した。私たちのすべての候補製品は開発段階にあり、私たちはまだどの候補製品の開発も完了していません。私たちは今後数年も巨額の費用と運営損失が発生すると予想しています。私たちの純損失は四半期と年度によって大きく変動する可能性があります。純損失と負のキャッシュフローはすでに我々の株主赤字や運営資本に悪影響を与え続けている。私たちの費用は大幅に増加すると予想されています
| ● | 著者らの主要な候補LNC製品MAT 2203に対して更なる臨床と臨床前研究を行った |
| ● | このような研究の資金の一部が米国国立衛生研究院の非希釈性資金から来ていても、MAT 2203の更なる臨床研究を支持する |
| ● | より多くの候補製品を発見し開発するためにbrを求める; |
| ● | 臨床試験を成功させた任意の候補製品のために監督管理の承認を求める |
| ● | より多くの臨床開発と潜在的商業化候補製品の生産が要求される |
| ● | 私たちの知的財産権の組み合わせを維持し、拡大し、保護する |
| 42 |
| ● | 追加の臨床、品質管理、科学者を招聘し、 |
| ● | 私たちの製品開発と将来の商業化作業を支援し、上場企業の義務を履行するために必要な人員やインフラを支援する人を含む、br運営、財務、管理情報システムと人員を増加させます。 |
私たちが利益を達成して利益を維持する能力は私たちの収益力にかかっている。1つ以上の候補製品のマーケティング承認を得て商業化に成功するまで、相当な収入は生じないと予想される。これは、候補製品の臨床前テストと臨床試験の完成、他の候補製品の発見、これらの候補製品の監督管理許可、製造、マーケティングと販売を含む一連の挑戦的な活動で成功することを要求し、私たちが監督管理の許可を得る可能性のある任意の製品、任意の発売後の要求を満たし、個人保険或いは政府支払人から製品精算を得ることを含む。我々はこれらの活動の多くの初期段階 にとどまっており,他のこれらの活動は開始されていない.私たちは決してこのような活動で成功しないかもしれません。たとえ私たちが成功しても、利益を達成するのに十分な収入が生まれないかもしれません。
医薬品開発に関連する多くのリスクや不確実性のため,費用増加時間 や金額,いつあるいは利益が達成できるかどうかを正確に予測することはできない。米国食品医薬品局またはFDAまたは同様の非米国規制機関が現在予想外の研究を要求している場合、または臨床試験または任意の候補製品の開発を完了する上で何らかの遅延が生じた場合、私たちの費用は増加する可能性がある。
たとえ私たちが利益を達成したとしても、私たちは四半期や年間収益性を維持したり向上させることができないかもしれない。私たちが実現できず、利益を維持することは会社の価値を下げ、資金を調達し、業務を拡大し、私たちの研究開発努力を維持し、私たちの候補製品ルートを多様化し、甚だしきに至っては運営を継続する能力を弱める可能性がある。わが社の価値の下落はあなたの投資損失の全部または一部を招く可能性もあります。
私たちは多くの追加資金が必要になるだろう。もし私たちが必要な時に資金を集めることができなければ、私たちは私たちの製品開発計画や商業化努力を延期、減少、または廃止させることを余儀なくされるかもしれない。
私たちの費用は私たちの持続的な活動に従って増加することを予想して、特に私たちの候補製品に対してもっと多くの臨床研究を行う時、MAT 2203の潜在的な3期の臨床試験を含み、そしてもっと多くの臨床前と臨床試験 を行い、更に私たちのLNCプラットフォームを検証と拡張し、引き続き研究開発を行い、臨床試験を開始し、もし開発が成功すれば、 は監督管理機関が私たちの候補製品を承認することを求める。もし私たちが他の候補製品のために新しい研究と臨床前開発の努力を開始すれば、私たちの費用はさらに増加するかもしれない。また、いずれかの候補製品が規制部門の承認を得た場合、製品製造、マーケティング、販売、流通に関連した巨額の商業化費用が発生すると予想される。また、上場企業としての運営に顕著な追加コストが生じることが予想されます。したがって、私たちは私たちの持続的な運営に関連した多くの追加資金を得る必要があるだろう。もし私たちが必要な時や魅力的な条件下で資金を調達できなければ、私たちは私たちの研究開発計画や将来の商業化努力を延期、減少、または廃止させることを余儀なくされるかもしれない。
私たちのbrは、2023年12月31日まで、私たちの既存の現金、現金等価物、および取引可能な債務証券(限定現金を含まない)は13,756,000ドルであり、2024年第3四半期の運営費用と資本支出需要に資金を提供することができるが、超えてはいけないと信じている。私たちの推定は未来が間違っていることが証明される可能性があるという仮定に基づいており、私たちは現在予想されているよりも早く私たちの資本資源 を使用することができる。変化する環境は、私たちが現在の予想よりも資本を消費する速度を大幅に速くする可能性があり、 私たちがコントロールできない状況のため、私たちは現在の予想よりも多くの資金を必要とするかもしれない。私たちの将来の資本需要は、短期と長期を含め、多くの要素に依存するだろう
| ● | 私たちが行っている候補製品の臨床試験の進捗、時間、コスト、結果、 |
| ● | 他の候補製品(我々のLNCプラットフォームに基づく将来の候補製品MAT 2203を含む)の臨床試験範囲、進捗、時間、コストと結果、ならびに研究と臨床前開発作業、および私たちのLNCプラットフォームをさらに検証するために行われた任意の臨床前または臨床仕事を含む |
| 43 |
| ● | 私たちは、任意の協力、許可、または私たちが確立する可能性のある他の手配を達成する能力、および条項と時間を達成する |
| ● | 私たちが求めている他の候補製品の数量と開発要件は |
| ● | FDAと同様の非米国規制機関が私たちの候補製品を監督審査するコスト、時間、結果 |
| ● | 私たちが発売許可を得た任意の候補製品の将来の商業化活動のコストと時間は、製品製造、マーケティング、販売、流通を含む |
| ● | 私たちが発売許可を得た候補製品の商業販売収入(もしあれば) |
| ● | 私たちの研究開発とビジネスインフラの拡大に伴い、私たちの従業員数の増加と関連コスト |
| ● | 特許出願を準備し、提出し、起訴し、私たちの知的財産権を維持し、実行し、知的財産権に関連する任意のクレームを弁護するコストと時間 |
| ● | 他の製品や技術をどの程度手に入れているのか |
| ● | 上場企業としての運営コスト |
| ● | 技術と市場発展の相互競争の影響。 |
潜在的な候補製品を決定し、臨床前テストと臨床試験を行うことは時間がかかり、高価で不確定な過程であり、数年を要して完成する必要があり、しかも著者らは監督部門の許可を得て製品販売を実現するために必要なデータ或いは結果を永遠に生成できないかもしれない。しかも、私たちの候補製品が承認されれば、商業的成功を得られないかもしれない。私たちの商業収入は、もしあれば、私たちが何年も商業用途がないと予想している製品の販売から来ます。したがって、私たち は私たちの業務目標を達成するために追加的な融資に依存し続ける必要があるだろう。私たちは受け入れ可能な条項 で十分な追加融資を受けることができないかもしれないし、融資を受けることができないかもしれない。また、有利な市場条件や戦略的考慮のため、現在または将来の運営計画を実行するのに十分な資金があると考えても、追加の資本を求めることができる。
追加資本を調達することは、私たちの株主を希釈し、私たちの運営を制限したり、私たちの技術や候補製品に対する権利を放棄することを要求するかもしれません。
利益を達成するのに十分な製品収入を生成することができる前に、私たちは、公共または私募株式発行、債務融資、政府または他の第三者融資、協力、および許可手配の組み合わせによって、私たちの現金需要 を満たす予定です。国家衛生研究院からの限られた資金を除いて、私たちは外部資金源を何も約束していない。もし私たちが普通株、転換可能な証券、または他の株式証券を売却することによって追加資本を調達する場合、あなたの所有権資本は大幅に希釈される可能性があり、これらの証券の条項は清算または他の特典および逆希釈保護を含む可能性があり、これは普通株主としての権利に悪影響を及ぼす可能性がある。債務融資および優先持分融資(利用可能であれば)は、固定支払義務の増加をもたらし、我々が業務を展開する能力に悪影響を及ぼす可能性のあるbr債務、資本支出、または配当金などの特定の行動を宣言する能力を制限または制限する能力を含むいくつかの合意に関連する可能性がある。追加融資を獲得するには、私たちのbr経営陣が多くの時間と精力を投入する必要があり、彼らの注意を日常活動から移す可能性があり、これは私たちのbr経営陣が候補製品開発を監督する能力に悪影響を及ぼすかもしれない。
もし私たちが第三者との協力、戦略連合またはマーケティング、流通または許可手配によって追加の資金 を調達する場合、私たちは、私たちの技術、将来の収入フロー、研究計画、または候補製品に対する貴重な権利 を放棄しなければならないか、または私たちに不利になる可能性のある条項でライセンスを付与しなければならないかもしれない。もし私たちが必要な時に株式や債務融資を通じてより多くの資金を調達できない場合、私たちは私たちの製品開発や将来の商業化努力を延期、制限、減少、または中止する必要があるかもしれません。あるいは私たちは自分で開発し、マーケティングする候補製品をより望んでいた権利を与えています。
| 44 |
私たちのbr株主は未償還オプションの行使によって深刻な希釈を受ける可能性がある。
2023年12月31日まで、私たちは未返済オプションを持っていて、加重平均行権価格で1株0.83ドルで46,707,934株の普通株を購入することができます。このような未返済オプションを行使することは私たちの株の価値を希釈させるだろう。
私たちのこれまでの運営の歴史は、業務の成功度や将来の生存能力を評価することを困難にするかもしれません。
我々のbrは2013年から積極的に運営されており,我々の候補製品は臨床開発の初期段階にある。私たちは、私たちがどんな候補製品の規制承認を得ることに成功し、商業規模の製品を製造することに成功したか、あるいは第三者代表が私たちにそうするように手配したり、成功した製品の商業化に必要な販売とマーケティング活動を行うことができないことを証明していません。したがって、私たちの将来の成功または生存能力に対するあなたのいかなる予測も、私たちがより長い運営履歴 を持っている時のように正確ではないかもしれません。
さらに、私たちは予見できない費用、困難、合併症、遅延、および他の既知および未知の要素に直面する可能性がある。私たちが規制部門の承認を得ても、研究開発に集中している会社から、brのビジネス活動を支援できる会社に移行する必要があります。そのような移行で、私たちは成功しないかもしれない。
我々は,様々な要因により,我々の財務状況や経営業績が四半期間と年度間で大幅に変動し続けると予想しており,その多くの要因は制御できない.したがって、あなたは将来の運営業績の指標として、どんな四半期や年間業績にも依存してはいけません。
製品開発,規制承認,製造と商業化に関するリスク
我々の開発作業は初期段階であり,成功しない可能性がある.
私たちはまだ臨床開発の段階にあるので、現在と未来の候補製品の全体的な臨床開発経路、私たちが従う規制経路の時間とコストを決定している。私たちが製品収入を作る能力は、私たちの候補製品の成功開発と最終的な商業化に大きく依存します。製品収入は何年も出ないと予想しています。MAT 2203および私たちが開発可能な任意の他の候補製品の成功は、多くの要因に依存するであろう
| ● | 臨床前研究に成功しました |
| ● | Br登録に成功し、臨床試験を完了した: |
| ● | 安全性と有効性を示します |
| ● | 規制機関に適用された上場承認を受けた |
| ● | 臨床および商業製造能力を確立するか、または第三者製造業者との手配を行う |
| ● | 私たちの候補製品および技術のために特許および商業秘密保護および非特許専有権を獲得し、維持する |
| ● | 承認された場合、候補製品の商業販売を個別にまたは選択的に他者と協力して行うことができる |
| 45 |
| ● | 患者、医学界、および第三者支払者が製品候補を承認した場合、その製品を受け入れる |
| ● | 他の治療法と効果的に競争し |
| ● | 承認後の製品の持続的に許容可能なセキュリティプロファイル;および |
| ● | 知的財産権と権利主張を実行して擁護する。 |
もし私たちがこれらの目標のうちの1つまたは複数をタイムリーに達成できない場合、あるいは全く実現できない場合、私たちは重大な遅延に遭遇したり、私たちの候補製品を商業化することに成功できない可能性があり、これは私たちの業務を損なうだろう。
私たちの候補製品が規制部門の承認を受けるかどうかは確認できません。規制の承認がなければ、私たちは私たちの候補製品を販売することができません。承認過程のどんな遅延も私たちの業務を損なうだろう。
我々 は大部分の資本を我々のLNCプラットフォームの開発に投入する予定である.私たちは製品販売に関連した収入を創出する能力があるかどうか、私たちは少なくとも今後数年以内にこのような状況が起こらないと予想していますが、これは私たちの1つ以上の候補製品の開発成功と規制の承認にかかっています。私たちのすべての候補製品は商業化される前に規制審査と承認が必要です。私たちの候補製品の規制審査や承認のどの遅延も市場発表を延期し、私たちの現金需要を増加させ、追加の運営損失を招きます。規制部門の承認を得られなければ、私たちの候補製品は市場に投入できなくなり、私たちの業務に大きな悪影響を及ぼすだろう。
FDAおよび他の必要な規制承認(外国承認を含む)を得るプロセスは、一般に数年を要し、関連する製品のタイプ、複雑性、および新規性によって大きく異なる可能性がある。また,この承認プロセスはきわめて複雑で高価であり, と不確実性である.私たちは、私たちのいかなる製品にもアメリカで機密協定を提出することができないかもしれません。または外国司法管轄区でいかなるマーケティング承認申請を提出することもできません。私たちが任意の候補製品の上場承認を求めるために、任意の修正NDAまたは追加NDAを含むNDAをFDAに提出する場合、FDAは提出された届出を受け入れるか拒否するかを決定しなければならない。FDAがこれらの申請の中の任意の を受け入れて届出と審査を行うかどうかを確定することはできず、いかなる の他の監督管理機関に提出された上場審査申請がこれらの監督管理機関によって届出と審査を受けるかどうかを確定することもできない。私たちは、私たちが審査期間内に潜在的な規制行動をタイムリーに、または根本的に遅延させずに、いかなる規制要求に答えることができるかどうかを決定することはできません。私たちはまた、私たちの任意の候補製品がいかなるFDA諮問委員会や外国規制機関から有利な推薦を受けるか、またはFDAまたは外国監督機関の承認を得て上場するかどうかを決定することはできません。さらに、遅延承認またはマーケティング申請の拒否は、監督管理部門が追加的な分析、報告、データおよび研究を要求する規制問題、データおよび結果に関する規制問題、製品開発中の規制政策の変化 およびそのような候補製品に関する新しい情報の出現を含む多くの要素に基づく可能性がある。
前臨床研究と臨床試験から得られたデータbrは異なる解釈を受ける可能性があり、これは私たちの任意の候補製品に対するbrの監督審査或いは承認を延期、制限或いは阻止する可能性がある。さらに、安全性および有効性を証明するために必要なデータおよび結果に対する規制機関の態度は、時間の経過とともに変化する可能性があり、他の製品に関する情報、政策変化、機関資金、人員配置、リーダーなどの新たな情報の出現など、多くの要因の影響を受ける可能性がある。私たちは未来の規制環境の変化が私たちの業務の見通しに有利なのか不利なのか分からない。
また、時間が経つにつれて、私たちの規制を審査して文書を提出する環境も変わります。例えば、NDAに対するFDAの平均審査時間は近年変動しており、私たちが提出したどの文書の審査時間もどの規制機関 にも予測できません。審査時間は予算と資金レベル及び法定、規制とbr政策の変化を含む様々な要素の影響を受ける可能性がある。また、ある薬品の安全リスクに関する広く報道された事件を考慮して、監督管理機関、アメリカ政府担当局のメンバー、医療専門家と一般大衆は潜在的な薬品安全問題を懸念している。これらの事件は薬品の撤回を招き,薬品ラベルを改訂し,さらに薬品の使用を制限し,薬品流通を制限できるなどの再生可能エネルギー管理措置を策定した。薬物安全問題に対するより多くの関心はFDAが臨床試験においてより慎重な方法をとることを招く可能性がある。臨床試験のデータは、より厳しい安全性審査を受ける可能性があり、これは、FDAまたは他の規制機関が完了する前に臨床試験を終了する可能性が高く、またはより長い時間またはそれ以上の臨床試験を必要とする可能性があり、これは、多くの追加費用および遅延または失敗を承認brを得ることをもたらす可能性があり、または最初に求められたものよりも承認の適応が限られている可能性がある。
| 46 |
私たちのbrの部分は、第三者が私たちに持っているか許可された技術に依存しており、これらの技術を失うことは、私たちの候補製品のさらなる開発を終了または遅延させ、私たちの名声を損なう、あるいはより高い印税を支払うように強要する。
私たちはLNCプラットフォームと私たちがロゲス大学から独占的に許可されたいくつかの特許に深刻に依存している。これらのbr特許を取得できないことは、私たちの業務および将来の生存能力を深刻に損なう可能性があり、利用可能であれば、同等の技術が決定、許可および統合されるまで、私たちの候補製品および製剤の開発、導入、または維持を遅延させる可能性がある。さらに、私たちが許可している知的財産権のどの欠陥も、私たちの候補製品や配合の機能を実施したり、損害を与えたり、新製品や配合の発売を延期したり、私たちの名声を損なう可能性があります。もし私たちが第三者と交換技術の許可協定を締結することを要求されたら、私たちはもっと高い印税を支払う必要があるかもしれない。
私たちの供給と臨床研究義務を履行するのに十分な数の製品がないか、得られない可能性があり、私たちの業務、財務状況、運営結果は不利な影響を受ける可能性があります。
我々はこれまで,臨床開発に必要なLNCプラットフォームのために限られた内部製造能力のみを開発してきたが,我々の MAT 2203候補製品である。我々はThermoFisherの完全子会社Patheonと合意し、MAT 2203の商業化生産に備えている。私たちがLNCプラットフォーム候補製品のために持続的に開発された製品を生産するのに十分な長期製造能力を開発しておらず、規制部門の承認を得られれば、これらの製品の商業化は、少数の第三者メーカーに依存して候補製品を製造することになる。私たちは、これらの第三者のいずれかと長期合意を締結していないかもしれないが、もし彼らが何らかの理由で契約を履行できない場合、私たちは他の受け入れ可能な製造業者や処方業者を見つけることができないか、または彼らと有利な合意を達成することができないかもしれない。これらの第三者から十分な私たちの製品をタイムリーに得ることができなければ、臨床試験を延期し、経済的に効率的な方法で、あるいは適時に私たちの製品を開発することを阻止するかもしれない。また、私たちの候補製品のメーカーはcGMPと類似した海外 基準を守らなければならず、私たちのメーカーがこれらの規定を遵守することを制御できません。もし私たちの契約メーカーの一つがコンプライアンスを維持できなかったら、私たちの製品の生産が中断され、遅延と追加コストを招く可能性があります。また,これらのメーカーの施設が承認前または承認後の工場検査を通過していない場合,FDAは を承認せず,我々の製品のマーケティングや販売に制限を加える可能性がある.
私たち は私たちの臨床用品の需要を満たすために第三者メーカーとサプライヤーに依存するかもしれない。材料受信遅延、スケジュール、発表、顧客制御とコンプライアンスの問題は、著者らが提供する臨床試験を開始、維持或いは完成する能力に不利な影響を与える可能性がある。商業製造と供給協定はまだ確立されていない。規模拡大、環境コントロール、公衆衛生危機(例えば流行病と流行病)、設備要求或いはその他の要素による問題 は私たちの候補製品を生産する能力に悪影響を及ぼす可能性がある。
もし私たちが私たちの製品を商業化することに成功できなければ、私たちが収入を作る能力は制限されるだろう。
もし私たちの候補製品が規制部門の承認を得たら、私たちの長期生存と成長は製品の商業化の成功に依存しても、これは収入と利益をもたらすだろう。医薬製品開発は高価、高リスク、長い、複雑、資源集約型の過程である。成功するためには他のこと以外にも
| ● | 潜在的な候補医薬製品を決定する; |
| ● | 適切な実験室、臨床前、その他の研究を設計し、行います |
| ● | 臨床研究のために監督部門の承認を得ました |
| ● | 良好な実験室と良好な臨床実践に基づいて適切な臨床前と臨床研究を設計し、行う |
| 47 |
| ● | 臨床研究者を選抜し募集します |
| ● | を選択して研究対象を募集する; |
| ● | 私たちの研究のデータを収集し分析し正確に解読し |
| ● | マーケティングに対する監督部門の承認を得て提出し、 |
| ● | CGMPにより 候補薬品を生産した。 |
どんな特定の製品の開発計画も数年かかり、利益を創出する能力を遅延させるだろう。さらに、初期開発段階で有望に見える潜在的製品は、様々な理由で失敗する可能性があり、例えば、br製品は、大量の追加試験を必要とする可能性があるか、または最終的には、安全でないこと、無効であること、難しすぎること、または高価すぎて開発または製造できないこと、管理が難しすぎること、または不安定であることを証明することができる。もし私たちの製品が商業化に成功しなければ、私たちの業務、財務状況、運営結果に悪影響を及ぼすだろう。
もし私たちの臨床前と臨床研究が積極的な結果を得ていなければ、もし私たちの臨床試験が延期された場合、あるいはこのような研究や試験中に深刻な副作用が発見された場合、私たちは遅延に遭遇し、追加のコストが発生し、最終的に私たちの候補製品を商業化することができない。
監督部門の許可を得て私たちの候補製品を販売する前に、私たちは広範な臨床前のbrテストを行い、私たちの候補製品の動物上の安全性を証明し、臨床試験を行い、私たちの候補製品の人体における安全性と有効性を証明しなければならない。臨床前と臨床テスト費用が高く、設計と実施が困難であり、完成するのに数年かかるかもしれない。私たちの1つまたは複数の臨床前研究または臨床試験の失敗は、試験の任意の段階で起こる可能性がある。私たちは、前臨床試験および臨床試験中、または臨床試験中に多くの予見不可能な事件に遭遇する可能性があり、これらの事件は、規制部門の許可を得たり、候補製品を商業化したりすることを遅延または阻止する可能性がある
| ● | 私たちの前臨床試験または臨床試験は陰性または不確定な結果をもたらす可能性があり、私たちは決定または監督機関が追加の臨床前試験または臨床試験を要求するか、または私たちが期待する有望な項目を放棄することができるかもしれない |
| ● | 監督機関あるいは機関審査委員会は私たちが臨床試験を開始することを許可してはならない、あるいは予想試験の場所で臨床試験を行うことを許可してはならない |
| ● | FDAまたは任意の非アメリカ監督機関が私たちに適用した私たちの臨床試験範囲または設計に関する条件brは、規制環境の変化によって、私たちの臨床試験方案を機関審査委員会に再提出して再検査することを要求するかもしれない |
| ● | 私たちの臨床試験に必要な患者数は私たちが予想していたよりも多いかもしれないし、参加者は私たちの予想以上の比率で私たちの臨床試験を脱退するかもしれない |
| ● | 私たちの第三者請負業者または臨床研究者は、法規の要求を遵守できないか、または私たちに対する契約義務をタイムリーに履行できない可能性がある |
| ● | もし私たち、規制機関、または機関審査委員会が参加者が受け入れられない健康リスクに直面していると判断した場合、私たちは私たちの1つまたは複数の臨床試験を一時停止または終了しなければならないかもしれない |
| ● | 規制機関や機関審査委員会は様々な理由で、規制要件を満たしていないことを含む、臨床研究の一時停止、一時停止、または中止を要求する可能性がある |
| ● | 私たちの臨床試験のコストは予想以上に高いかもしれません |
| 48 |
| ● | 私たちの候補製品または臨床試験を行うために必要な他の材料の供給または品質が不足または不十分である可能性があり、または潜在的な臨床研究機関と受け入れられる条項と合意できない可能性がある; |
| ● | 私たちの候補製品の 効果は期待効果ではないかもしれないし、副作用が含まれている可能性があります。あるいは候補製品 は他の意外な特徴を持っている可能性があります。 |
さらに、もし私たちの候補製品に対して、現在予想されている以上の追加の臨床試験または他の試験を行うことを要求された場合、もし私たちが臨床試験または他の試験を成功させることができなければ、これらの試験または試験の結果が陽性でないか、または軽度陽性である場合、または安全問題がある場合、私たちは:
| ● | 1つまたは複数の候補製品のマーケティング承認を遅延させるか、または得ることができない場合がある |
| ● | 私たちが承認を申請した適応範囲とは異なる適応または完全に異なる適応の承認を得る; または |
| ● | 市場承認を得た後、この製品 を下積みします。 |
もし私たちがテストや承認に遅延があれば、私たちの製品開発コストも増加します。計画通りに任意の臨床前 テストあるいは臨床試験を開始するかどうか、再構成が必要かどうか、あるいは計画通りに完成するかどうかは分からない。重大な臨床前または臨床試験遅延も特許保護期間を短縮する可能性があり,その間に我々の候補製品を商業化する独占的な権利を持つ可能性がある。このような遅延は私たちの競争相手が私たちより先に製品を市場に出し、製品や候補製品を商業化する能力を弱めるかもしれない。
臨床試験を完了するのに十分な患者を募集できなければ,われわれの業務,財務状況,手術結果は悪影響を受ける可能性がある。
著者らの製品の臨床研究完成率は患者の入院率などの要素に依存する。患者登録brは、様々な要素の関数である
| ● | 調査員 識別と募集; |
| ● | 規制部門は研究場所の開始を許可した |
| ● | 患者数br |
| ● | 実験に用いるシナリオの性質; |
| ● | 患者brは臨床部位に近い; |
| ● | この研究の資格基準: |
| ● | 同じ患者集団に対する他社からの臨床研究の競争; |
| ● | 薬物/デバイスを比較する能力 を得る。 |
私たちは私たちの患者登録手続きが適切であると信じている;しかし、患者登録を延期することはコストを増加させ、私たちの製品の最終商業化と販売を延期する(あれば)。このような遅延は私たちの業務、財務状況、そして運営結果に大きな悪影響を及ぼすかもしれない。
| 49 |
我々の抗感染候補製品の孤立薬物指定や排他性を維持できない可能性がある。
私たち はすでにアメリカでMAT 2203の孤児薬物称号を獲得し、他の候補製品のために他の孤児薬物称号 を求めるかもしれない。米国やヨーロッパを含むいくつかの管轄区の規制機関は、患者数が比較的少ない薬物を孤児薬に指定する可能性がある。孤児医薬品法によれば、製品がまれな疾患または疾患を治療するための医薬である場合、FDAはそれを孤児薬として指定することができ、米国では、まれな疾患または疾患は、一般に患者数200,000人未満と定義される。一般に、孤児の薬物名を有する製品がその後、その名称を有するbr適応の最初の発売許可を得た場合、製品は、一定期間内に規制またはマーケティング排他期間を得る権利があり、その間、FDAまたはEMAは、薬物の同じ適応の別のマーケティング申請を承認することができない。合理的な仮定により希薬指定が得られた製品 では,その臨床効果は同じ適応のために承認された同じ薬物よりも優れており,承認された後にまれな薬排他性を得るためには,同一孤児の適応が承認されたbr}と同じ薬物に対する臨床的優位性を証明しなければならない。専門期間はアメリカでは7年、ヨーロッパでは10年。もし1つの薬物が孤児薬物指定の基準に適合しなくなった場合、あるいはその薬物の利益が十分に高く、市場の独占的地位を得る理由がなくなった場合、ヨーロッパの独占経営期間は6年に短縮されることができる。孤立薬物 FDAまたはEMAが指定要求に重大な欠陥があると判断した場合、または製造業者 が稀な疾患または疾患を有する患者の需要を満たすのに十分な数の薬剤を保証できない場合、排他性を失う可能性がある。
MAT 2203の孤児薬物指定申請または任意の他の候補製品に関連する任意の将来の出願を維持または承認することを保証することはできません。米国で孤児薬物指定を保持できなければ,孤児薬物指定による可能性のある市場独占期を得る資格もなく,孤児薬物指定に関する経済的奨励を得る資格もない。我々が製品の孤立薬物排他性を獲得しても,この排他性は競合から製品を効果的に保護できない可能性があり,異なる薬物が同じ条件で承認される可能性があるからである。孤児薬物が承認された後であっても、FDAが後の薬物の方が臨床的に良いと結論した場合、それがより安全で、より効果的であることが証明され、または患者ケアに大きな貢献があることが証明された場合、FDA はその後、同じ状況に対して同じ薬物を承認することができる。
FDAの任意の迅速なチャネル指定または優先審査地位の付与は、実際には、より速い開発または規制審査や承認プロセスをもたらすことはなく、FDAが私たちの候補製品を承認することを保証することもできません。また,我々の候補製品は優先審査券の資格に適合しない適応 を扱う可能性がある.
著者らは、侵襲性カンジダ症、アスペルギルス症、免疫抑制療法による侵襲性真菌感染の予防とクリプトコッカス病の治療のためのMAT 2203の迅速チャネル認証を取得し、いくつかの他の候補製品のために迅速チャネル認証 を求めるか、または特定の適応の製品候補承認申請を優先的に審査する可能性がある。もし薬物が深刻または生命に危険な疾患の治療に使用され、この薬物がこの疾患が満たされていない医療需要を満たす潜在力を示す場合、薬物スポンサーはFDA Fast Track認証を申請することができる。候補製品が治療において大きな進展を遂げた場合,FDAはその製品を指定して優先審査を受ける資格がある可能性がある。FDAはこれらの認証を付与するか否かを決定する幅広い裁量権を持っているため,特定の候補製品がこれらの認証を取得する資格があると考えても,FDAがこれらの認証を付与するかどうかを決定することは保証されない.私たちが高速チャネル指定または優先審査を受けたとしても、私たちは従来のFDAプログラムよりも速い開発過程、審査、または承認を経験しないかもしれない。FDAがわれわれの臨床開発計画のデータが指定 を支持しなくなったと考えると,Fast Track指定を撤回する可能性がある。
FDAが私たちの候補製品に付与したいかなる画期的な治療法の指定は、より速い開発や規制審査や承認過程を招くことはなく、私たちの候補製品が発売承認される可能性も増加しないかもしれない。
私たちのいくつかの候補製品に画期的な治療指定を求めることができるかもしれません。画期的な治療法は、単独または1つまたは複数の他の薬剤との併用治療が深刻または生命に危険な疾患または状態を治療することを意図した薬剤として定義され、予備臨床証拠は、1つまたは複数の臨床的意義を有するbrの終点で、臨床開発早期に観察されるような既存の治療法よりも有意な改善を示す可能性があることを示す。画期的な治療法として指定された薬物と生物製品に対して、FDAと試験スポンサー間の相互作用とコミュニケーションは最も有効な臨床開発経路の決定を助けることができ、同時に無効対照方案中の患者数を最低に下げることができる。関連基準を満たしていれば,FDAにより画期的な治療法に指定された薬物も加速承認を得る資格がある。
| 50 |
FDAは を画期的療法に指定する権利がある。したがって,我々の候補製品の1つが画期的療法として指定された基準に適合していると考えても,FDAは同意せず,このような指定を行わないことにした可能性がある。いずれの場合も、 は、FDAの従来の手順に従って承認を考慮した薬剤と比較して、候補製品に対する画期的な治療指定を受けることは、より速い開発プロセス、審査または承認をもたらすことができない可能性があり、FDAの最終承認を保証することもできない。さらに, 我々の1つまたは複数の候補製品が画期的な療法の条件を満たしていても,FDAは今後これらの製品が資格条件を満たしていないことを決定したり,FDAの審査や承認の期間を短縮しないことを決定する可能性がある。
我々の候補製品を合格した感染症製品として指定することは保証されず,いずれの場合も承認されても,必ずしもより速い開発や規制審査につながるとは限らず,FDAが我々の候補製品を承認する保証はない。
我々 は,ある適応に対する適格感染症製品やQIDP認証を受けており, は将来の候補製品をQIDPとして指定する資格がある可能性がある.QIDPは、新たに出現した感染性病原体またはいくつかの“合格した病原体”を含む抗菌または抗真菌耐性病原体によって引き起こされる感染を含む、深刻または生命を脅かす感染を治療するための抗菌または抗真菌薬である。QIDPとして指定された製品は、FDAの優先審査を取得し、“高速チャネル”状態を取得する資格がある可能性がある。FDAがQIDPとして指定された薬品のセキュリティプロトコルが承認された後、この製品は資格が得られる任意の他の規制独占期間に加えて、5年間の規制独占期間 を得る。FDAは広範な裁量権を持ち,これらの称号を付与するかどうかを持つため,ある特定の候補製品がこのような称号や地位を獲得する資格があると考えても,FDAはその称号を付与しないことを決定することができる。また、このような認証を確実に取得しても、従来のFDAプログラムと比較して、より速い開発過程、審査または承認を経験しない可能性があり、 は、我々の候補製品がQIDPとして決定されてもFDAの承認を得ることが保証されない。
もし私たちが他の候補製品の識別と開発に成功できなかったら、私たちの成長潜在力は影響を受けるかもしれない。
規制部門のMAT 2203や私たちが開発する可能性のある他の候補製品の承認を得ても、私たちはこれらの製品を商業化することに成功できないかもしれません。私たちがその販売から得られる収入は限られている可能性があります。
発売が承認された場合、MAT 2203または私たちが開発する可能性のある任意の他の候補製品の商業的成功は、医師、患者、および医療支払者を含む医学界によって受け入れられるかどうかに依存する。MAT 2203またはそのような他の候補製品の市場受容度は、いくつかの要因に依存する
| ● | この候補製品の臨床的安全性と有効性を示す |
| ● | 管理に対する利便性と使いやすさ; |
| ● | どのような悪影響の流行率や深刻さ |
| ● | 医師がこの候補製品を処方する意思と目標患者群が新しい療法を試みる意欲は |
| ● | 価格設定と費用効果; |
| ● | 適用される治療ガイドラインには、このような候補製品が含まれるか、または省略される |
| ● | 私たちまたは未来のパートナーの販売およびマーケティング戦略の有効性 |
| ● | FDA承認タグに含まれる制限または警告; |
| ● | 私たちは、政府医療計画(MedicareおよびMedicaid、個人健康保険会社、および他の第三者支払者を含む)から十分な第三者保険または精算を取得し、維持することができる |
| ● | 患者は第三者保険や精算なしに費用を自己負担する意思がある。 |
| 51 |
もし私たちが開発可能なMAT 2203または任意の他の候補製品が承認されたが、医師、医療支払者、および患者の十分な受け入れ度を得られなければ、十分な収入を生み出すことができず、利益を達成したり維持したりすることができないかもしれない。我々の教育医療界や第三者支払者がこのような候補製品のメリットを知る作業には大量の資源が必要である可能性があり,決して成功しない可能性がある。
また、規制部門の承認を得ても、どの承認の時間や範囲も、このような候補製品を商業化に成功させる能力を禁止または低下させる可能性がある。例えば、承認過程が長すぎると、予想される市場機会を逃し、他の 会社に競争製品を開発したり、市場主導的な地位を確立したりする能力がある可能性がある。私たちが最終的に得たどんな規制承認も限られているかもしれないし、制限されたり、承認された後に約束された制限を受けて、そのような候補製品が商業的に実行できないようにすることができる。例えば、規制当局が承認する可能性のある候補製品は、私たちが要求するものよりも少ないか、または制限されている可能性があり、候補製品に対して請求しようとしているbr価格を承認しないかもしれないし、高価な発売後の臨床試験の表現によって承認される可能性があり、または、そのラベルがこの適応の商業化に必要または必要なラベル宣言を含まないことを承認する可能性がある。さらに、FDAは、薬物の安全な使用を確保するために、潜在的な要求またはリスク管理計画、およびREMSへの要求を含む承認時に条件を追加する可能性がある。FDAがREMSが必要であると判断した場合,NDAのスポンサーは提案したREMSを提出しなければならず,必要であればFDAは承認されていないREMSでNDAを承認しないであろう。REMSは、限定された配布方法、患者登録表、および他のリスク最小化ツールのような、薬物使用ガイド、医師のコミュニケーション計画、または安全な使用を保証する要素を含むことができる。これらの承認またはマーケティング制限のいずれも、そのような候補製品の商業普及、流通、処方、または配布を制限することができる。また、製品が規制基準を満たしていない場合や、製品が最初に発売された後に問題が発生した場合、製品承認が撤回される可能性がある。上記のいずれの場合も、このような候補製品の商業成功に重大な損害を与える可能性がある。
私たち は現在販売とマーケティング組織がありません。もし私たちが満足できる販売とマーケティング能力を確立できなければ、規制部門の承認を得ても、私たちの任意の候補製品を商業化することに成功できないかもしれません。
現在、私たちは販売やマーケティングをしていません。商業販売のための製品の商業化を承認するためには,販売やマーケティングインフラを開発したり,そのような商業インフラを持つ第三者と協力したりしなければならない.もし私たちが自分の販売とマーケティング組織を設立することを選択したら、私たちは最初にFDAに機密協定を提出する時に販売とマーケティング担当者の募集を開始するつもりで、そしてFDAがMAT 2203または私たちの他の候補製品を承認するまで、アメリカで自分の販売組織を設立するつもりはありません。
私たちは経済的に効率的な方法で直売チームを作ることができないかもしれないし、この投資の正のリターンを実現することもできないかもしれない。また、私たちは老舗で資金の豊富な製薬と生物技術会社と競争しなければならず、採用、育成、育成と販売とマーケティング人員を維持しなければならない。戦略的パートナーや許可者がいない場合、米国でMAT 2203または任意の他の候補製品を商業化することを阻害する可能性がある要因は、
| ● | 私たちは十分な数の効果的な販売とマーケティング担当者を募集して維持することができない |
| ● | 販売員は十分な数の医者が私たちの未来の製品を出すように接触したり説得することができません |
| ● | 販売員はセット製品が不足しており、これは製品ラインの広い会社に対して競争劣勢になる可能性がある |
| ● | 独立した販売およびマーケティング組織の作成に関連する予測不可能なコストおよび費用。 |
| 52 |
もし私たちが販売およびマーケティング担当者の募集に成功しなかった場合、または販売およびマーケティングインフラを構築することができなかった場合、または適切な連携計画を達成できなかった場合、MAT 2203または私たちが開発する可能性のある他の候補製品の商業化に成功することは困難であり、これは私たちの業務、運営結果、および財務状況に悪影響を及ぼすだろう。米国以外では、製薬パートナーと協力協定を締結することで、私たちの候補製品を商業化することができる。私たち は私たちが受け入れられる条項や根本的にこのような合意に到達できないかもしれない。また,我々がこのような関係を築いても, 我々のこれらの第三者の販売,マーケティング,流通活動の制御は限られており,制御さえしていない可能性がある.私たちの未来の収入はこのような第三者の努力の成功に大きく依存するかもしれない。
FDCA第505条(B)(2)条に従ってMAT 2203の承認を申請することができない場合、または505(B)(2)条に従って承認されるためにセキュリティおよび有効性に関する追加データを生成する必要がある場合、予想される開発および商業化スケジュールを満たすことができない可能性がある。
我々がMAT 2203にNDAを提出する現在の計画は、候補製品のマーケティング承認を得るために生成されるデータを最大限に削減し、開発時間を短縮することを含む。両性マイシンBの治療効果の歴史に依存する予定であり,2019年,2021年,2022年にFDAと面会して我々のMAT 2203の開発計画を検討したが,FDAが505(B)(2)でMAT 2203を承認するという要求を満たす保証はない。MAT 2203セキュリティプロトコルの提出と審査のスケジュールは,FDCA第505条(B)(2)条に基づいてセキュリティプロトコルを提出する計画に基づいており,公共分野や他の場所のbrデータに部分的に依存することができる.私たちはまだ505(B)(2)条に基づいて任意の候補製品に秘密協定を提出していません。FDAによって承認されたデータが必要とされる可能性があり、その中のいくつかのデータは、FDAによって承認された製品に関連する可能性がある。依存データがFDAによって承認され、第三者特許によってカバーされた製品に関連している場合、私たちは、私たちが列挙された特許を侵害していないか、またはそのような特許が無効であるか、または強制的に実行できないことを証明するように要求されるであろう。認証の結果,第三者 は我々の認証通知を受けてから45日以内に我々に行動することができる.
このような認証に対して訴訟が提起されれば,我々がこのような訴訟に対して抗弁している間,我々の秘密保持協定の承認は30カ月以上の制限を受ける可能性がある.したがって、505(B)(2)条による私たちの候補製品の承認は、特許 の独占満了まで延期されるか、またはこれらの特許の私たちの候補製品への適用性に挑戦することに成功する可能性があります。あるいは、私たち は、候補製品の承認をトリガする可能性のあるデータ に依存しないように、十分な他の臨床データを生成することを選択することができる。FDAは505(B)(2)条に従って我々のアプリケーションに適用されなくても、幅広い裁量権を有しており、我々の候補製品の安全性および有効性に関する追加データを生成して、私たちが依存することが許容される可能性のある第三者データを補完することを要求しています。いずれの場合も、私たちは、どの候補製品の市場承認を得る前に、私たちが現在製品候補の承認を得るために計画している活動ではなく、大量の新しい研究開発活動を行うことが要求される可能性があります。 このような追加的な新しい研究と開発活動は高価で時間がかかるだろう。
我々 は我々の任意の候補製品の開発時間を短縮できない可能性があり,FDAは提出されたデータの審査に基づいて我々のセキュリティプロトコル を承認しない可能性がある.我々が必要とする参照上場医薬製品が何らかの安全上の理由でFDAによって市場から撤回された場合、私たちはこのような製品を参照して候補製品の505(B)(2)セキュリティ協定をサポートすることができない可能性があり、505(B)(1)節のより広い要求を満たす必要があるかもしれません。承認を支援するための追加データを生成する必要がある場合、予想される開発および商業化スケジュールを満たすことができない可能性があり、合理的なコストで追加データを生成できない場合や、追加データを生成することができず、主要候補製品の市場承認を得ることができない可能性があります。
私たちは他のバイオテクノロジーと製薬会社からの競争に直面しています。もし私たちが効果的に競争できなければ、私たちの経営業績は影響を受けます。
バイオテクノロジーと製薬業界は競争が激しく、迅速で重大な技術変革の影響を受けている。私たちは複数の管轄区に競争相手がいて、その中の多くの司法管轄区の知名度、商業インフラ、財務、技術と人的資源は私たちよりずっと大きいです。私たちは商業薬品と生物技術企業、学術機関、政府機関及び私営と公共研究機関を含む多くの異なる源からの競争に直面している。古い競争相手brは巨大な資金を投入して新しい化合物を迅速に発見し、開発する可能性があり、これらの化合物はMAT 2203または私たちが開発する可能性のある任意の他の候補製品を時代遅れまたは経済的にしないかもしれない。承認された製品と競争するすべての新製品は、商業的に成功するために、効果、コスト、利便性、耐性と安全性の面で納得できる優位性を示す必要があるかもしれない。模倣薬競争を含む他の競争要因は、価格を低下させるか、または販売減少をもたらす可能性があり、特に第三者によって長年販売され、医師、患者、および支払人によって良好に受け入れられている製品を引き起こす可能性がある。また、他社が開発した新製品はMAT 2203や私たちの任意の他の候補製品の競争相手になる可能性があります。もし私たちが現在と未来の競争相手と効果的に競争できなければ、私たちの業務は成長できず、私たちの財務状況と運営は影響を受けるだろう。
また、 独自のLNCプラットフォーム、経験、および私たちの重点分野での知識が競争優位を提供してくれると考えていますが、MAT 2203の潜在的なライバルは私たちのビジネス機会を減らす可能性があります。
| 53 |
もし私たちが任意の候補製品のマーケティング承認を得たら、私たちは持続的な義務と持続的な規制審査の制約を受けるであろう これは多くの追加費用を招く可能性がある。また、私たちの候補製品はラベルや他の制限を受けて市場から撤退する可能性があります。もし私たちが規制要求を守らなかった場合、あるいは私たちの未来の製品が予期しない問題に遭遇したら、処罰されるかもしれません。
米国の監督管理機関がMAT 2203または私たちが開発する可能性のある任意の他の候補製品の承認を得ても、FDAはその指示の用途やマーケティングまたは承認条件に重大な制限を加えたり、高価で時間がかかる可能性のある承認後の研究と発売後の監視に持続的な要求を提出して、安全性と有効性を監視する可能性がある。私たちの未来の製品はまた持続的な法規要求を遵守し、企業とその他の上場後の情報の製造、ラベル、包装、貯蔵、流通、安全監視、広告、宣伝、記録保存と報告を規範化する。これらの要件は、FDAで として登録されることと、承認後に行われる任意の臨床試験について、現在の良好な臨床実践法規またはcGCPを継続的に遵守することとを含む。また、医薬品製造業者およびその施設は、現在の良好な生産実践、cGMP、品質管理に関する要求、br}品質保証、およびそれに対応する記録およびファイル維持を保証するために、FDA および他の規制機関の持続的な審査および定期検査を受けることを含む。
FDA は、REMSをNDAの一部として要求する権利があるか、または承認された後に、承認された薬剤の配布または使用にさらなる要求または制限を適用することができ、例えば、専門的に訓練された特定の医師または医療センターに処方を制限し、治療をいくつかの安全な使用基準を満たす患者に制限するか、または患者にテスト、監視、および/または登録に を登録することを要求することができる。
私たちまたは任意の未来のパートナーの販売およびマーケティング活動については、広告および販売促進材料は、米国の他の適用可能な連邦、州および現地法律、および他の国/地域の類似した法律要求に従うほか、FDA規則brを遵守しなければならない。米国では、医師に製品サンプルを配布することは、米国の処方薬営業法の要求に適合しなければならない。変更された性質によると、申請保持者はFDAの承認を得なければ製品や製造を変更することができません。私たちはまた、米国の反リベート法規、米国の虚偽請求法案、および同様の州法律を含む様々な詐欺や乱用の法律の制約を直接または間接的に受けることができます。これらの法律は他に加えて、私たちが提案している販売、マーケティング、科学/教育支援計画にも影響を与えます。もし私たちがアメリカ医療補助薬品リベート計画、アメリカ退役軍人事務部の連邦供給スケジュールあるいは他の政府薬品計画に参加すれば、私たちは報告と支払い義務に関する複雑な法律とbr法規の制約を受ける。これらのすべての活動はまた、米国連邦と州消費者保護および不正競争法によって制約される可能性がある。他の国の多くのこのような分野にも似たような要求がある。
また、私たちの製品ラベル、広告、販売促進は、規制要件と持続的な規制審査の制約を受けます。FDAは処方薬に関する販売促進声明を厳格に規制します。製品は、製品の承認されたラベルに反映されるFDA承認されていない使用に使用されてはならない。もし私たちの候補製品が市場の承認を得たら、br先生はまだ合法的に承認されたラベルと一致しない方法で彼らの患者に私たちの製品を出すことができます。もし私たちがこのようなラベル外の用途を普及させることが発見されたら、私たちは重大な責任と政府の罰金に直面するかもしれません。FDAや他の機関は、非ラベル用途の普及を禁止する法律法規を積極的に実行しており、不正な方法で非ラベル用途を普及させた企業は、その上場承認の撤回を含む重大な制裁を受ける可能性があることが発見された。連邦政府は不正販売促進の疑いのある会社に巨額の民事と刑事罰金を科し、いくつかの会社がラベル外販売促進に従事することを禁止している。FDAはまた、企業に永久禁止の同意法令を締結することを要求しており、これらの法令により、具体的な販売促進行為が変更または減少される。
| 54 |
もし私たちまたは監督機関が製品に以前に知られていない問題、例えば予想されていない深刻さや頻度の不良事件、その製品を生産する施設に問題があることを発見した場合、あるいは私たちまたは私たちの製造業者は適用された規制要求を遵守できなかった場合、私たちは以下の行政または司法制裁を受ける可能性がある
| ● | 製品の販売または製造を制限し、市場から製品をリコールするか、または自発的または強制的に製品をリコールすること |
| ● | 警告状や無見出しの手紙を出す |
| ● | 臨床的に堅持する; |
| ● | 民事または刑事罰または罰金を禁令または適用する |
| ● | 規制承認の一時停止または撤回; |
| ● | 進行中の臨床試験を一時停止します |
| ● | 私たちが提出した保留申請または承認された申請を承認することを拒否するか、または製品許可証の承認を一時停止または取り消し; |
| ● | 費用の高い新しい製造要件を含む、一時停止または運営に制限を加えること;または |
| ● | 製品 は製品の輸入または輸出の許可を差し押さえ、差し押さえ、または拒否する。 |
上述した任意のイベントまたは処罰が発生すると、MAT 2203または私たちの任意の他の候補製品を商業化し、収入を創出する能力を抑制することができる。不利な監督管理行動は、承認前も承認後も、製品責任クレーム を招き、私たちの製品責任開放を増加させる可能性がある。
将来の立法および/またはFDAが採用する法規および政策は、私たちが臨床試験を行って完成するのに要する時間とコストを増加させるかもしれない。
FDA は薬品開発と審査の流れを管理する法規を確立し、外国の監督管理機関も同様である。FDAと他の規制機関の政策は変わる可能性があり、追加の法律や政府法規を公布して、防止、制限、遅延を防止するかもしれないが、私たちの候補製品に対する監督審査も加速するかもしれない。例えば、2016年12月、“治療法案”が法律として署名された。その他の事項以外に、“治療法案”は薬品監督管理の現代化を実現し、革新を刺激することを目的としている。Br治療法やFDAの既存または将来の任意の指導が我々の候補製品の開発にどのような影響を与えるかを予測することはできない。
医療保険法や施行条例の変化は我々に実質的な悪影響を及ぼす可能性がある。
アメリカと一部の外国司法管轄区では、すでに医療保健システムに関する多数の立法と法規変更及び 提案された変更が継続され、これらの変更は候補製品の上場承認を阻止或いは延期し、承認後の活動を制限或いは監督し、そして利益方式でマーケティング審査を獲得した候補製品を販売する能力に影響を与える可能性がある。
米国や他の地方のbr政策立案者や支払者の中で,医療システムの変革を推進することに大きな興味があり,その既定目標は医療コストの抑制,質の向上,および/または参入拡大である。アメリカでは、製薬業界はこれらの努力の重点であり、重大な立法計画の重大な影響を受けてきた。例えば,米国では,2010年の患者保護·平価医療法案(“ACA”)は,医療が政府や民間保険会社が資金を提供する方式を大きく変え,製薬業に著しく影響している。ACAの多くの条項は生物製薬業界に影響を与え、生物製薬製品がMedicare Part BとMedicaid計画に従って連邦精算brを獲得するため、あるいはアメリカ政府機関に直接販売するために、メーカーは割引を公衆衛生サービス法(PHS)によって薬品定価計画に参加する資格のある実体に拡大しなければならない。
| 55 |
また、2022年に発効する“2022年インフレ率低減法案”には、薬品価格に直接影響を与え、連邦政府の薬品支出を減少させるための政策も含まれている。この立法には実質的な薬品定価改革が含まれており、アメリカ衛生と公衆サービス部内に薬品価格交渉計画を構築し、メーカーに連邦医療保険が保証する特定の薬品に対して協議の“最高公平価格”を徴収することを要求し、あるいは規定を守らないために消費税を支払うために、連邦医療保険BとD部分に支払ういくつかの薬品のメーカーに税金還付支払い要求を確立し、インフレを超える価格brを処罰し、メーカーにD部分薬品に割引を提供することを要求することを含む。
薬品コストを制御する立法、行政と個人支払人の努力は一連の提案をカバーし、薬品価格交渉、Medicare D部分の再設計、薬品価格インフレリベート、国際メカニズム、模倣薬普及と反競争行為、メーカー報告、及び加速承認経路の使用に影響を与える可能性のある療法の改革を含む。ACA、インフレ低減法案あるいは他の連邦と州医療政策改革努力の最終内容、時間或いは効果 を予測できず、薬品定価に対する改革努力を含む。連邦や州医療改革が私たちの将来の業務や財務結果に悪影響を与えないことは保証されず、将来の連邦や州が医療政策に関連する立法、司法、あるいは行政改革が私たちの業務にどのように影響するかを予測することもできない。
私たちのbrは、より多くの立法改正が公布されるかどうか、あるいは政府法規、指導または解釈が変更されるかどうか、あるいはこのような変更が私たちの候補薬物や製品(ある場合)の上場承認、販売、定価または精算にどのような影響を与えるかを決定することができません。私たちは、これらの措置と将来取られる可能性のある他の医療改革措置は、より厳しい保険基準を招き、私たちが受け取った任意の承認薬の価格に追加の下振れ圧力をもたらす可能性があると予想している。br連邦医療保険または他の政府が計画している清算のいかなる減少も、個人支払者支払いのような減少を招く可能性がある。コスト抑制措置や他の医療改革を実施することは、私たちが収入を創出し、利益を達成し、あるいは私たちの薬品を商業化することを阻止するかもしれない。
さらに、FDAは、我々の業務および製品に大きな影響を与える可能性がある方法でFDA法規およびガイドラインを修正または再解釈する可能性がある。任意の新しい法規またはガイドライン、または既存の法規またはガイドラインの改訂または再解釈は、我々の候補製品の追加コストを増加させるか、またはFDAの審査時間を延長する可能性がある。私たちは、規制、法規、政策、br、または説明がいつ、発表、実施、または採用された変化が私たちの将来の業務にどのように影響する可能性があるかを決定することができません。他の事項に加えて、このような変更は、以下のような必要がある場合があります
| ● | 承認を得る前に追加の臨床試験を行う; |
| ● | 製造方法 ; |
| ● | 1つ以上の製品のリコール、交換、または生産停止; |
| ● | その他 記録保存. |
このような 変更には時間がかかり、大きなコストをもたらす可能性があり、あるいは私たちの候補製品の潜在的なビジネス価値 を低下させる可能性があります。さらに、他の製品の規制許可や承認を遅延させたり、取得できなかったりすることは、当社の業務、財務状況、および運営結果を損なうことになります。
精算費用brレートは、製品の使用および使用の臨床環境によって異なる可能性があり、精算されたより低いコスト製品によって許容される支払いに基づいて、他の製品またはサービスの既存の支払いに統合される可能性があり、これらのレートを計算するための連邦医療保険または医療補助データにおける予算制限および/または欠陥を反映する可能性がある。製品純価格は政府医療計画が要求する強制的な割引やリベートによって低下する可能性がある。このような立法または同様の規制の変化、または他の国/地域からの製品の輸入制限を緩和する法律は、私たちが将来販売する任意の製品の純価格を低下させる可能性がある。したがって、私たちの未来の製品は最終的に費用効果があると思われないかもしれない。私たちは私たちのどの候補製品にも精算を提供するかどうかを確信できない。しかも、私たちは清算政策が未来の製品の需要や支払いの価格を下げないということを確認できない。精算が得られない場合や限られた基礎の上で精算を提供できなければ、私たちが開発した任意の候補製品の商業化に成功できないかもしれません。
| 56 |
私たちの将来の成長は海外市場に進出する能力にある程度依存しており、そこでは追加の規制負担や他のリスクや不確実性の影響を受ける。
私たちの将来の収益性は候補品を海外市場で商業化する能力にある程度依存し、第三者との協力に依存するつもりです。MAT 2203または海外市場で開発される可能性のある他の候補製品を商業化すれば、追加のリスクと不確実性に直面するだろう
| ● | 私たちのbr顧客は海外市場で私たちの候補製品を精算することができます |
| ● | 私たちは第三者に依存しているので、ビジネス活動を直接統制することはできません |
| ● | 複雑で変化の多い外国の監督管理、税務、会計、法律要求の負担を遵守する |
| ● | 外国の異なる医療実践と風俗習慣は市場受容度に影響を与える |
| ● | 輸入または輸出許可要件; |
| ● | 売掛金入金時間が長い ; |
| ● | 出荷の納期がもっと長い; |
| ● | 言語障害 技術訓練; |
| ● | 海外の一部の国の知的財産権の保護を減少させる |
| ● | 外国通貨の為替レートの変動 |
| ● | 契約紛争が発生した場合、外国の法律によって管轄される契約条項の解釈。 |
私たちの候補製品の海外販売は、政府規制、政治的、経済的不安定、貿易制限、関税変化の悪影響を受ける可能性もあり、いずれも私たちの運営結果に悪影響を及ぼす可能性があります。
もし私たちが医療詐欺や法律乱用の方法で私たちの候補製品を販売したり、もし私たちが政府価格報告の法律に違反した場合、私たちは民事または刑事罰を受けるかもしれません。
FDA は法律法規を施行し,薬品販売促進は承認された処方情報と一致しなければならないことが求められている。医師は承認された製品をいわゆる“非ラベル”用途に処方する可能性があるが,製薬会社はその承認されたラベルと一致しない方法で製品が違法であることを宣伝しており,このような行為に従事している会社はいずれも同社に重大な責任を負わせる可能性がある。同様に、EUや他の外国司法管轄区の業界法規は、会社がラベル外販売促進活動に従事することを禁止し、各国の監督管理機関は法規違反行為に対して民事処罰を行うことを禁止している。br}我々の販売促進材料が私たちのラベルと一致することを保証しようとしているが、規制機関は私たちの評価に同意しない可能性があり、無見出し書簡、警告状、または他の民事または刑事法執行手続きを提起する可能性がある。FDA の薬品マーケティングの制限以外に、近年、他のいくつかのタイプの州と連邦医療詐欺と乱用法律を実施し、製薬業界のあるマーケティング行為を制限している。これらの法律には米国反リベート法規、米国虚偽申告法、類似の州法律が含まれている。これらの法律の広さと安全港の狭さのため、私たちのいくつかの商業活動はそのうちの1つ以上の法律の挑戦を受けるかもしれない。
| 57 |
米国の“反リベート条例”は、Medicare、Medicaid、または他の連邦政府によって援助された医療計画に従って精算可能な任意の医療項目またはサービスを誘導または見返りとして購入、レンタル、購入、レンタルまたは注文を誘導または見返りとして提供、支払い、請求、または徴収することを意図的に禁止している。この法規は、医薬品メーカーと処方者、購入者、処方管理者の間の手配に適用されると広く解釈されている。いくつかの法定免除と規制安全港保護のいくつかの一般的な活動は起訴されていないが、免除と安全港の範囲は狭く、処方、購入または推薦の報酬を誘導するためのやり方が免除または安全港の資格を満たしていなければ、審査される可能性がある。すべての場合、私たちの接近は安全港を反リベート責任から保護するすべての基準を満たしていないかもしれない。しかも、最近の医療改革法案はこのような法律を強化した。例えば、他の事項に加えて、ACAは、米国の反リベート法規および刑事医療詐欺法規の意図要件を修正し、個人またはエンティティは、この法規またはこの法規に違反する特定の意図を実際に理解する必要がなくなった。また、ACAは、米国の“虚偽請求法”について、政府は、“米国反リベート条例”違反による物品やサービスを含むクレームが虚偽または詐欺的クレームを構成していると断言できると規定している。連邦虚偽請求法律は、誰もが故意に連邦政府に提出するか、虚偽請求を提出することを引き起こすか、または虚偽請求支払いを得るために虚偽陳述を行うか、または虚偽請求支払いを引き起こすことを禁止する。
過去数年間、いくつかの製薬と他の医療保健会社は、例えば、処方者に無料旅行、無料商品、虚偽相談費と贈与、および他のbrの金銭的利益を提供した疑いがある;定価サービスに虚高な平均卸売価格を報告し、その後、連邦計画によって販売率を設定するために使用された;ラベル外販売促進に従事して、MedicareまたはMedicaidにクレームを提出して非引受保険、br}ラベル外用途に使用した。Medicaid返却計画に誇張された最適な価格情報を提出して、Medicaid返却点の責任を減少させます。 の多くの州にも米国反リベート法規や米国虚偽申告法のような法規や法規があり、Medicaidや他の州の計画によって精算されるプロジェクトやサービスに適用され、あるいはいくつかの州では、支払者にかかわらず適用されます。これらの連邦や州法律によると、制裁には巨額の民事罰金、政府計画に基づいてメーカーの製品を清算範囲から除外し、巨額の刑事罰金、監禁を含む可能性がある。
我々 は我々の協調プロトコルに大きく依存してMAT 2203を開発することが予想されており,我々 は第三者の性能に依存するリスクに直面している.
MAT 2203の研究·開発活動を展開する際には、現在、大学、政府機関、非営利組織との協力協定に依存して、戦略と財政資源を獲得していく予定である。これらの合意の鍵は,我々が米国国立衛生研究院とMAT 2203の開発について合意したことである。もし私たちまたは私たちのbrパートナーがどんな適用可能な合意や手配に従って職責を履行できなかったか、または私たちの候補製品のためにより多くの合意を得ることができなかった場合、 は、私たちが行っていることと予想されている臨床試験を含む、私たちの研究開発活動を大幅に中断または遅延させるであろう。このような損失は、私たちの費用を増加させ、私たちの業務、財務状況、および運営結果に実質的な損害を与える可能性がある。
我々は第三者に依存して候補製品の臨床試験を行うことが予想され,第三者の性能に依存するリスクに直面する。
私たちは、NIHのような第三者CROまたは政府エンティティと合意して、私たちの臨床プロジェクトを実施し、管理することを望んでいます。私たちは、MAT 2203および私たちの他の候補製品の臨床研究を実行するためにこれらの機関に深刻に依存しており、彼らの活動のいくつかおよび非常に限られた側面しかコントロールできません。しかし、私たちは私たちのすべての研究が適用された合意、法律、法規、科学的基準に従って行われていることを確実にする責任があり、私たちのNIHやCROへの依存は私たちの規制責任を軽減しない。我々、NIH、および我々のCROは、FDA、欧州経済圏加盟国の主管当局および同様の外国規制機関によって臨床開発中の任意の製品に対して実行される法規およびガイドラインであるcGCPの遵守を要求されるであろう。FDAは,試験スポンサー,主要調査者,試験地点を定期的に検査することでこれらのCGCP規定を実行している。もし私たちまたはNIHまたは私たちのCROが適用されたCCCPを遵守できなかった場合、私たちの臨床試験で生成された臨床データは信頼できないと考えられる可能性があり、FDAまたは同様の外国の規制機関は、私たちの上場申請を承認する前に追加の臨床試験を行うことを要求するかもしれない。FDA は検査後にいずれの臨床試験がCCCPに適合しているかどうかを確認することは保証できません。また,われわれの臨床試験はcGMP法規により生産された製品を用いて行わなければならず,多くの試験対象が必要となる。私たちがNIHあるいは私たちのCROがこれらの規定を遵守できなかったかどうかは、私たちが臨床試験を繰り返す必要があるかもしれません。これは規制の承認過程を延期し、民事と刑事罰を含む法執行行動に直面させる可能性があります。
| 58 |
したがって、私たちの薬物開発プロジェクトの多くの重要な側面は私たちの直接制御の範囲内ではない。また,NIHやCROは我々との手配や法規の要求に応じてそのすべての義務を履行しない可能性がある.NIHまたはCROが満足できる方法で臨床試験を行うことができず、私たちに対する義務に違反したり、法規要件を遵守できなかった場合、 MAT 2203または私たちが開発する可能性のある任意の他の候補製品の開発および商業化が遅れる可能性があり、または私たちの開発 計画が実質的かつ不可逆的な損害を受ける可能性がある。私たちはこれらのCROが私たちのbr計画または候補製品に使用される資源の数量と時間を制御することができない。われわれCROが収集した臨床データに依存できなければ,繰り返し,臨床試験の持続時間を延長したり,臨床試験の規模を増加させたりすることが要求される可能性があり,商業化が著しく遅れ,有意に大きな支出が必要となる可能性がある。これらの第三者CROとの任意の関係が終了すれば、代替CROとスケジュール を達成できない可能性がある。上述したように、私たちの財務業績とMAT 2203と私たちの他の候補製品の商業見通しは損なわれ、私たちのコストは増加するかもしれません。私たちの収益能力は遅れるかもしれません。
我々の候補製品は現在も将来も完全に第三者に依存しており、これらの第三者がFDAや同様の外国規制機関の生産承認を得ることができず、十分な数の候補製品を提供できない場合、または許容可能な品質 レベルまたは価格で商業化できない場合、私たちの商業化作業は一時停止、延期、または利益低下する可能性がある。
私たちは今のところありませんし、MAT 2203または私たちの任意の候補製品において活性医薬成分brまたは原料薬を生産する能力またはインフラを得ることも、私たちの臨床試験または商業化製品に使用することも意図していません。したがって、 我々は、開発過程全体にわたって契約メーカーに依存し、MAT 2203または任意の候補製品が商業化承認されれば を得る。私たちはまだいかなる契約製造業者とも商業供給契約を締結しておらず、私たちに有利な条項、またはすべての場合、契約製造業者とMAT 2203または私たちの任意の候補製品について商業的に供給することができないかもしれません。
私たちの契約メーカーが私たちの任意の候補製品を生産するための施設はFDAの承認を得なければなりません。検査は私たちがFDAに機密協定を提出した後に行われます。私たちは、契約製造パートナーの生産過程を制御せず、契約製造パートナーが活性医薬物質および完成薬を生産する際にcGMPを遵守することに完全に依存している。これらのcGMP規定は、私たちの候補製品に関連する製造、テスト、品質管理、記録保存のあらゆる面をカバーしている。もし私たちの契約メーカーが、私たちの規格やFDAや他の機関の厳格な規制要件に適合した材料を生産することに成功しなければ、彼らは、その製造施設の規制承認を確保および/または維持することができないであろう。FDAまたは同様の外国の規制機関が、これらの施設が候補製品を生産するために承認しない場合、または将来的にそのような承認を撤回する場合、代替製造施設を探す必要があるかもしれないが、これは、私たちが規制承認を獲得し、許可を得たり、そのような候補製品を開発、販売する能力に著しく影響するだろう。
我々の契約メーカーは、cGMPや類似の法規要件に適合しているかどうかを知るために、FDAおよび対応する州と外国機関の持続的な定期抜き打ち検査を受ける。私たちは私たちの契約製造業者がこのような規制と基準を遵守しているかどうかを統制できない。もし私たちの任意の契約製造業者が適用された法規を遵守できなかった場合、罰金、禁止、民事処罰、私たちの任意の製品の発売を許可できなかったbr候補製品、遅延、一時停止または承認撤回、運営制限、および刑事起訴を含む制裁を私たちに加える可能性があり、いずれも深刻なbrをもたらし、私たちの業務に悪影響を及ぼす可能性がある。しかも、私たちの契約製造業者が十分な品質管理、品質保証、合格者の能力を維持することを制御することはできません。もし私たちの契約製造業者がこれらのbr規格のいずれかを遵守または維持できない場合、私たちの開発、規制部門の承認、または私たちの任意の候補製品を開発、またはマーケティングする能力に悪影響を及ぼす可能性がある。
| 59 |
もし、 がいかなる理由でも、これらの第三者が契約を履行できないか、または履行したくない場合、私たちは彼らとの合意を終了することができない可能性があり、私たちは代替製造業者や処方業者を見つけることができないか、または彼らと有利な合意を達成することができない可能性があり、 のような第三者が将来の要求を満たすことができるかどうかを決定することができない。これらの製造業者または任意の代替薬品完成品製造業者が、それぞれの原料薬または完成品の製造プロセスにおいて重大な困難に遭遇した場合、または私たちとの業務往来を停止すべきである場合、私たちは、製品供給が深刻に中断されている場合に遭遇する可能性があり、または候補製品の供給を全く創出できない可能性がある。もし私たちが製造問題に直面したら、私たちが十分な製品供給を生産する能力は否定的な影響を受けるかもしれない。私たちは私たちの第三者製造パートナーの努力を調整できない、あるいは私たちの第三者製造パートナーは利用可能な生産能力が不足しており、これは私たちが必要なbrレベルで任意の候補製品を供給する能力を弱めるかもしれない。新たな大口または完成品メーカーの資格を得るためには、重要な規制要件を満たす必要があるため、現在の製造パートナーとこれらまたは他の困難に直面していれば、これらの困難に対応するために生産を1つまたは複数の代替メーカーに移行することを決定すれば、製品供給が深刻に中断される可能性がある。
どんな製造問題や契約メーカーの損失も、私たちの運営を中断し、販売損失を招く可能性があります。また、私たちは第三者に依存して私たちの潜在製品を生産するために必要な原材料を提供します。サプライヤーへのいかなる依存も のいくつかのリスクに関連する可能性があり、重要な材料が得られない可能性があり、生産コスト、納品計画、信頼性と品質の制御低下を含む。サプライヤー問題は未来の契約メーカーによるいかなる意外な中断も私たちの候補製品の出荷を遅延させ、私たちの商品を販売するコストを増加させ、販売損失を招く可能性があります。
私たちの製造と供給パートナーが時間の経過とともに任意の候補製品の商業規模製造コストを下げることができる保証はありません。ビジネス規模の製造コストが予想以上であれば,これらのコストは我々のbr}運営業績に著しく影響する可能性がある。コストを下げるために、私たちはプロセスの改善を開発して実施する必要があるかもしれない。しかし、そのためには、規制機関に時々通知したり、規制機関に意見を提出したりする必要があり、改善には規制機関の承認が必要かもしれない。私たちは私たちがこれらの必要な承認を受けるかどうか、あるいはこれらの承認が適時に承認されるかどうかを確認することはできません。私たちはまた、私たちの商業製造過程で生産量を強化し、最適化することができるという保証はありません。私たちがbrを上げて生産量を最適化できなければ、時間が経つにつれてコストを下げることができないかもしれない。
伝染病の爆発は私たちの業務、財務状況、経営業績に実質的な悪影響を及ぼす可能性がある。
私たちは衛生流行病や伝染病の爆発と関連した危険に直面している。私たちのいくつかのビジネスパートナーは、私たちの有効な薬物成分の生産運営、アジアまたは他の地方の感染症の発生、またはそのような疫病が発生する可能性があると考えられ、影響を受ける国/地域政府が講じた措置は、私たちの業務、財務状況、または運営結果に悪影響を及ぼす可能性があるため、米国、中国、および他のアジア諸国/地域にいない。例えば、疫病は私たちが中国内外を旅行したり、材料を輸送する能力を制限し、私たちが依存する施設を一時的に閉鎖させ、私たちの業務を著しく混乱させる可能性がある。
経済的不確実性を含む不利な世界環境は、私たちの財務業績に悪影響を及ぼす可能性がある
世界的な状況、金融市場の混乱、あるいはインフレは私たちの業務に悪影響を及ぼすかもしれない。また、グローバルマクロ経済環境は、グローバル経済市場の不安定さ、米国と他の国との貿易関税や貿易紛争の増加、グローバル信用市場の不安定さ、サプライチェーンの弱さ、地政学的環境と政治的緊張情勢の不安定、および外国政府債務懸念の負の影響を受け続ける可能性がある。これらの課題は、地域経済や世界金融市場で不確実かつ不安定になり続けている可能性があり、これは私たちの業務に悪影響を及ぼす可能性があります。
我々の知的財産権と規制排他性に関するリスク
私たち は私たちに許可されたいくつかの技術に依存する。私たちはこれらの技術を制御しないで、私たちはこれらの技術に対するいかなる権利の喪失も私たちの発見、開発、商業化候補製品を阻止することが可能です。
私たちのLNCプラットフォームとロッグス大学が私たちに独占的に許可してくれたいくつかの特許に依存している。私たちはLNCプラットフォームをサポートする部分特許を独占的に持っていない。私たちが独占的に許可した特許を使用する権利は、ロゲス大学との交渉、継続、そして私たちの許可協定の条項に依存する。私たちとロッグス大学の許可協定条項によると、私たちは私たちが許可している特許の起訴、維持または届出、および第三者に対してこれらの特許を強制的に執行することを制御します。しかしながら、私たちのいくつかの特許および特許出願は、他の会社からこれらの特許および特許出願を取得するか、第三者から許可を得るか、別の会社から取得されるかである。したがって、このような特許と特許出願は私たちまたは私たちの弁護士が書いたものではなく、私たちはこの特許のいくつかのbrの起草と起訴を制御することができない。もし私たちが特許と出願の所有者であり、起草と起訴を制御すれば、前の特許所有者と私たちの許可者は、私たちのようにこのような特許と出願の起草と起訴に同じ関心を持たないかもしれない。私たちは、許可特許および特許出願の起草および/または起訴が適用された法律法規を遵守するか、または効果的かつ強制的に実行可能な特許および他の知的財産権を生成することを許可者が決定することはできない。
| 60 |
我々 が我々が許可した技術を使用する権利は,所有者の知的財産権の有効性に依存する.私たちのライセンス特許または抗弁を実行するか、またはこれらの特許が無効であると主張する任意の声明は、一般に、私たちのライセンシーの制御または協力によって制限される。私たちが許可している知的財産権の所有者に法的訴訟を提起することができ、このような法的行動における不利な結果は、このような会社または機関が私たちの業務を継続して許可するために必要かもしれない知的財産権 を許可することを阻止する可能性があるので、私たちの業務を損なう可能性がある。また、このようなライセンス者は、彼らに利益を与える方法でこのような訴訟を解決する可能性がありますが、私たちの製品にライセンス技術を使用する能力に悪影響を与えます。
私たちとロッグス大学との合意に含まれるいくつかのbr許可条項は、許可者が以下の場合に許可を終了することを可能にする:(I)私たちの は、合意中の任意の支払い義務または他の重要な条項に違反し、書面終了通知後の一定時間内に違反行為を是正することができなかった;(Ii)私たちまたは私たちの任意の付属会社、被許可者または二次許可者は、任意のライセンス特許の有効性、実行可能または延期に直接または間接的に疑問を提起するか、または(Iii)私たちは破産または解散を宣言する。私たちのライセンス下での権利は、ライセンスの下で満期になった印税を支払うことを含む、ライセンスの条項を守り続けることに依存します。これらのライセンスの終了は、我々がリードしている抗感染製品候補製品MAT 2203を含むLNCプラットフォームに基づく候補製品の発見、開発、商業化を阻止する可能性がある。許可の範囲や関連する印税義務を決定することは困難である可能性があり,我々と許可側とのトラブルを招く可能性がある.このような紛争の不利な解決は,ライセンスに応じて支払われる印税の増加を招く可能性がある.許可側が許可に応じて支払うべき印税を支払っていない,あるいは許可条項を遵守していないと判断した場合,許可側は許可の撤回を試みる可能性がある.このような試みが成功すれば、我々の先行する抗感染候補製品 を含むLNCプラットフォームに基づく候補製品の発見、開発、商業化が禁止される可能性がある。
私たちの知的財産権を保護することは難しくて高価で、私たちはこのような権利の保護を保障できない。
私たちのビジネス成功は、私たちの技術、製品、およびプロセスのための特許保護にある程度依存し、 はこれらの特許を第三者の挑戦から保護することに成功し、これらの特許を第三者の競争相手に対抗することに成功する。br}製薬会社の特許地位は高度に不確定である可能性があり、複雑な法律、科学と事実の問題に関連しているため、重要な法律原則はまだ解決されていない。特許法または特許法解釈の変化は我々の知的財産権の価値を低下させる可能性がある。したがって、私たちは、私たちの特許(私たちが所有し許可されている特許を含む)において許容または強制的に実行される可能性のある特許の広さを予測することができない。私たちは現在、私たちのLNCプラットフォームに関連する30個の発行された特許の権利、 および私たちのLNCプラットフォームの未解決特許出願を持っているか、または所有しています。これらの特許は、米国または外国特許庁の承認を得ることができないかもしれません。 また、私たちのどの技術の既存の特許出願も、最終的に発行される可能性のある任意の特許は、第三者によって挑戦され、無効または回避される可能性があり、同様の製品や技術を有する競争相手から私たちを保護できないかもしれません。
将来の私たちの所有権に対する保護の程度は不確定であり、法的手段は限られた保護しか提供できないため、私たちの権利を十分に保護できないかもしれないので、私たちが私たちの競争優位性を獲得したり維持したり、あるいは私たちにいかなる競争優位性を提供することもできないかもしれない。私たちは、第三者が所有するいかなる特許出願が、私たちが提出した特許出願よりも優先されないか、または米国または外国特許庁の妨害、反対または無効訴訟手続きに巻き込まれないと判断することはできません。
| 61 |
私たちはまた、技術を保護するために商業秘密に依存しています。特に特許保護が適切でないと考えられたり、入手できない場合には。 しかし、商業秘密は保護が困難です。我々は、従業員、学術協力者、コンサルタント、および他の請負業者 にセキュリティ協定を締結することを要求しているが、私たちは、私たちのビジネス秘密または他の独自または許可情報 を十分に保護することができない可能性がある。通常,研究協力者や科学コンサルタントは,我々がアクセスする権利がある可能性のあるデータや情報を発表する権利がある.もし私たちが私たちのノウハウや他の機密情報を秘密にすることができなければ、私たちが特許保護を受ける能力と私たちが持っている価値のある情報を保護する能力が脅かされるかもしれない。第三者br}エンティティに我々の任意の商業機密を不正に取得して使用することを強制することは高価で時間がかかり、結果は予測できない。しかも、裁判所は特許よりも商業秘密を保護することを望んでいないことがある。また,我々の競争相手は同等の知識,方法,ノウハウを自主的に開発することができる.
もし私たちが私たちの技術のために特許や商業秘密保護を取得したり、維持することができなければ、第三者は私たちの独自の情報を使用する可能性があり、これは私たちの市場での競争能力を弱化させ、私たちの収入を創出し、利益を達成する能力に悪影響を及ぼす可能性がある。
私たちの製品と競争相手の製品を区別するために商標を開発することも可能です。私たちは私たちまたは私たちの業務パートナーが提出したどの商標申請も承認されることを保証できない。第三者はまた、このような商標出願に反対するか、または他の方法で商標の使用に挑戦することができる。もし私たちが使用した商標が成功的に挑戦されれば、私たちは私たちの製品を再命名することを余儀なくされるかもしれません。これはブランド認知度の低下を招き、広告やマーケティングの新しいブランドに資源を投入する必要があるかもしれません。また、競争相手が私たちが使用している商標を侵害しない保証はありませんし、これらの商標を実行するのに十分な資源がある保証はありません。
私たちの候補製品は他人の知的財産権を侵害する可能性があり、これは私たちのコストを増加させ、私たちの開発と商業化努力を遅延したり阻害したりする可能性があります。
私たちの成功は他人を侵害することを避けるためのノウハウにある程度かかっている。製薬業の特徴は特許と他の知的財産権に関する訴訟が頻繁であることだ。特許検索は、特許間の用語の違い、データベースの不完全さ、および特許請求の範囲の意味の評価が困難であるため、我々のノウハウに関連する可能性のある第三者特許権を識別することは困難である。さらに、特許出願 は、出願が発行されるまで秘密にされているため、MAT 2203または任意の将来の候補製品が商業化されて侵害される可能性のある第三者特許を知らない可能性がある。MAT 2203または任意の将来の候補製品を研究、開発または商業化するために、発行された特許および特許出願が許可を要求する可能性があるかもしれないが、これらの特許および特許出願が商業的に合理的な条項で許可を得ることができるかどうか、または全く知られていない。第三者による特許侵害請求は非常に時間がかかり、可能性があります
| ● | その結果、訴訟費用が高かった |
| ● | 私たちの技術者と管理者の時間と注意力を移します |
| ● | 特許が満期になるまで製品を商業化することを阻止し、または法廷で最終的に無効または侵害されないと判断されるまで、 |
| ● | この技術の使用を停止または修正すること、および/または非侵害技術を開発することを要求する;または |
| ● | 私たちは印税や許可協定を締結することを要求します。 |
第三者が私たちに権利侵害請求をしていないにもかかわらず、他の人はMAT 2203 の上場を阻止する可能性のある独自の権利を持っている可能性がある。損害賠償を要求し、MAT 2203または私たちのプロセスに関連する商業活動を禁止しようとする特許関連法律訴訟は、潜在的な損害賠償責任を負わせる可能性があり、現在の候補製品または任意の未来の候補製品を製造または販売し続けるために許可証を得ることを要求する可能性がある。私たちがこのような行動のいずれかで勝利するかどうか、またはこれらの特許に必要な任意の許可が商業的に受け入れられる条項で提供されるかどうかを予測することはできない。さらに、必要であれば、MAT 2203または任意の将来の候補製品またはプロセスを再設計して権利侵害を回避することができるかどうかを決定することはできません。したがって、司法や行政訴訟で不利な裁決を下したり、必要な許可を得られなかったりすると、MAT 2203または将来の候補製品の開発および商業化を阻止する可能性があり、これは私たちの業務、財務状況、br、および経営業績を損なう可能性があります。
| 62 |
私たちの競争相手は、新しい技術のための特許保護や特許 技術を規制部門に提出する努力に時々反対するかもしれないと予想しています。競争相手は、承認プロセスを遅延させたり、私たちが付与した特許に挑戦したりするために、例えば、米国特許商標局またはUSPTOに私たちの特許の再審査を要求することによって、または反対または挑戦がほとんど価値がなくても、承認プロセスを遅延させたり、反対意見を提出することによって、私たちの特許出願に反対しようと試みるかもしれない。このような訴訟は、通常、技術含有量が高く、費用が高く、時間がかかり、このような挑戦が、私たちが挑戦を受けているいかなる特許の範囲を縮小または完全に撤回しないかを保証することはできない。
一般 会社関連リスク
私たちは私たちの組織の規模を拡大して私たちの業務を発展させる必要があり、私たちはこのような成長を管理する際に困難に直面する可能性がある。
2024年3月18日まで、私たちは32人の従業員を持っている。私たちの発展と商業化計画と戦略の発展に伴い、管理、開発、運営、販売、マーケティング、財務、その他の資源の従業員規模を拡大する必要があるだろう。将来の成長は、確定、採用、メンテナンス、インセンティブ、br、およびより多くの従業員を統合する必要があることを含む、管理層メンバーに大きな追加責任をもたらすだろう。また、私たちの経営陣は、私たちの日常活動から不比例な注意を移し、これらの成長活動を管理するために多くの時間を投入しなければならないかもしれない。私たちの将来の財務業績 と候補製品を商業化する能力と効果的な競争の能力は、私たちの将来の任意の成長を効果的に管理する能力にある程度依存するだろう。
もし私たちが高い素質の人材の誘致と維持に成功できなければ、私たちは私たちの業務 戦略を成功的に実施することができないかもしれない。しかも、特定の重要な従業員のサービスを失うことは私たちの業務の将来性に悪影響を及ぼすだろう。
もし私たちが高い素質の人材の誘致と維持に成功できなければ、私たちは私たちの業務 戦略を成功的に実施することができないかもしれない。また、最高経営責任者Jerome D.Jabbourとbr}総裁、最高開発者Theresa Matkovits、最高技術官劉輝博士を含むいくつかの重要な従業員のサービスを失うことは、私たちのビジネスの将来性に不利な影響を与えるだろう。
競争の激しい製薬業界における競争能力は、高い素質の管理、科学、医療者を誘致する能力に大きく依存している。価値のある従業員を引き付けるために、私たちは従業員に時間とともに付与された株 オプションを提供するつもりです。時間の経過とともに、従業員に付与された株式オプション価値は、私たちの株価変動の大きな影響を受け、私たちはこれらの変動を制御することができず、他社が提供するより利益のあるオファーを相殺するのにいつでも十分ではない可能性があります。
私たちと適格人材を競争する他のbr製薬会社は、より多くの財務と他の資源、異なるリスク状況、 および私たちよりも長い業界歴史を持っている。彼らはまた、より多様な機会とより良い職業昇進の機会を提供する可能性がある。これらの特徴のいくつかは、私たちが提供するより高い素質の候補者を引き付ける可能性がある。もし私たちが引き続き高い素質の人材を吸引し、維持することができなければ、私たちが候補製品を開発と商業化する速度と成功率は制限される。
私たちに製品責任訴訟を提起すれば、私たちは大量の責任を負う可能性があり、候補製品の商業化を制限することが要求される可能性があります。
MAT 2203または任意の将来の候補製品の臨床試験のため、私たちは潜在的な製品責任リスクに直面しており、MAT 2203または任意の他の未来の製品を商業化すれば、より大きなリスクに直面する。例えば、私たちが開発した任意の製品または私たちが製品で使用している任意の材料がダメージを与えると言われている場合、または製品テスト、製造、マーケティング、または販売中に不適切が発見された場合、起訴されるかもしれません。このような製品責任クレームには、製造欠陥、設計欠陥、製品固有の危険、不注意、厳格な責任、保証違反の告発が含まれている可能性があります。国家消費者保護法に基づいてクレームを出すこともできる。もし私たちが製品責任クレームで自分を弁護することに成功できなければ、私たちは巨額の責任を負うか、MAT 2203の商業化を制限することを要求されるかもしれません。成功的な防御であっても、多くの財政的で管理された資源が必要だ。事件がどうであっても最終的な結果がどうであろうと、賠償責任は次のようになるかもしれない
| ● | MAT 2203または私たちが開発する可能性のある未来の製品の需要を減少させる |
| 63 |
| ● | 私たちの名声を損なう |
| ● | 臨床試験参加者は脱退しました |
| ● | 関連訴訟を弁護する費用 ; |
| ● | 管理職を移転する時間と私たちの資源 |
| ● | 試験参加者または患者に巨額の金銭的報酬を支給する |
| ● | 製品 リコール、撤回またはラベル付け、マーケティングまたは販売促進制限; |
| ● | 収入損失 ; |
| ● | 候補品を商業化することはできません |
| ● | 私たちの株価は下落しています。 |
私たち は、潜在的な製品責任クレームを防止するために、許容可能なコストで十分な製品責任保険を得ることができません。これは、私たちが開発した製品の商業化を阻止または阻害する可能性があります。私たちはすでに私たちのbr臨床試験のために製品責任保険を購入して、総金額は500万ドル以上です。私たちはこのような保険を維持しますが、私たちに対するいかなるクレームも、裁判所の判決や和解の金額が私たちの保険範囲内にない、あるいは私たちの保険範囲を超えてしまう可能性があります。私たちの保険リストにも様々な例外があります。私たちは製品責任クレームの影響を受けるかもしれませんが、私たちは保険範囲を持っていません。私たちは、私たちの保険範囲の制限を超えているか、または私たちの保険カバー範囲内にない任意の裁判所によって裁決された金額、または和解合意で交渉された任意の金額を支払う必要があるかもしれません。私たちは、これらの金額を支払うために十分な資本を得ることができないか、または十分な資本を得ることができません。
我々の内部コンピュータシステム、または我々のCROまたは他の請負業者またはコンサルタントのシステムは、故障したり、セキュリティホールに遭遇したりする可能性があり、これは、我々の製品開発計画を実質的に中断させる可能性がある。
セキュリティ対策が実施されているにもかかわらず、我々の内部コンピュータシステムおよび私たちのCROおよび他の請負者およびコンサルタントのコンピュータシステム は、コンピュータウイルス、ソフトウェアエラー、許可されていないアクセス、自然災害、テロ、戦争、ならびにbr電気通信、デバイスおよび電気故障の破壊または破壊を受けやすい。我々の知る限り,これまで重大なシステム障害,br事故やセキュリティホールを経験したことはないが,このようなイベントが発生して我々の運営が中断されると,我々の計画が深刻に中断される可能性がある.例えば、私たちの任意の候補製品の完了または行われている臨床試験における臨床試験データの損失は、我々の規制承認作業を遅延させる可能性があり、データを回復または複製するコスト を著しく増加させる。また,我々の情報セキュリティシステムや我々CROの情報セキュリティシステムも法律法規に制約されており,我々の業務で収集·使用されているいくつかの情報のプライバシーやセキュリティを保護するための措置が求められている.例えば、HIPAA及びその実施条例は他の要求のほかに、個人健康情報のプライバシーと安全に対していくつかの規制と契約要求を提出した。EUでは、一般データ保護条例(GDPR)は、符号化システムによって隠蔽された情報を含むすべての個人情報に対して、より厳しい制限を有している。HIPAAとGDPRを除いて、多くの他の連邦と州法律は、州安全違反通知法、州健康情報プライバシー法、および連邦と州消費者保護法を含むが、個人情報の収集、使用、開示および保存を管理する。任意の中断またはセキュリティホールが、私たちのデータやアプリケーションの紛失または破損、または機密または独自の情報の不適切な開示または盗難に至った場合、私たちは責任を負う可能性があり、私たちの候補製品のさらなる開発が遅延する可能性があり、 私たちの競争地位が損なわれる可能性があり、または私たちの商業的名声が損なわれる可能性があります。
| 64 |
私たちは将来的に事業や製品を買収したり、戦略同盟を結成したりするかもしれませんが、このような買収のメリットを意識していないかもしれません。
私たちは、より多くの業務や製品を買収し、戦略連合を結成したり、第三者と合弁企業を設立したりする可能性があり、これらの業務は私たちの既存の業務を補完または拡大すると考えられます。市場や技術の見通しの良い企業を買収すれば、既存の業務や会社文化との統合に成功できなければ、これらの企業を買収するメリットを実現できないかもしれません。私たちは、戦略連合や買収によって発生した任意の新製品を開発、製造、マーケティングする際に、多くの困難に直面する可能性があり、それによって、予想される利益を達成したり、私たちの業務を強化したりすることを遅延または阻止することができます。このような買収の後、取引が合理的であることを証明するために、予想される相乗効果を達成することを保証することはできません。
私たちの証券に関するリスク
私たちAシリーズの優先株条項によると、私たちは相当な印税を支払う義務があるかもしれない。
私たちAシリーズの優先株の割引、権利、指定証明書(“指定証明書”) を制限する条項によると、毎年3500万ドルまでの特許使用料を支払う必要があります。FDAまたはEMAのMAT 2203および/またはMAT 2501の承認を得た場合(2027年までは発生しないと予想される)、および/またはこのような製品の販売が発生した場合、またはMAT 2203またはMAT 2501の許可または他の処置から任意の収益を得る場合、私たちAシリーズの優先株のいくつかの前所有者brに合計(I)の純売上高の4.5%(指定証明書に定義されているような)に相当する特許使用料を支払わなければならず、brはすべての場合において例年2500万ドルの上限を超えてはならない。および(Ii)許可報酬の7.5%( 指定証明書で定義されるように),いずれの場合も上限は例年1,000万ドルである.印税支払権は適用製品の特許 が満期になり、現在2033年と予想されています。
普通株式保有者の権利は発行される可能性のある優先株によって損害を受ける可能性がある。
私たちの会社規約は、私たちの取締役会が1つ以上のシリーズの優先株を指定して発行することを可能にします。したがって、取締役会は、株主の承認なしに、投票権、配当、転換、清算、または他の普通株式保有者の相対投票権および持分に悪影響を及ぼす可能性のある権利を含む新たな系列優先株を発行する可能性がある。追加発行された優先株は、1株当たり1票以上の権利を有する可能性があり、制御権変更を阻止、延期、または防止する効果がある可能性がある。買収試みへの影響は我々普通株の価格 に悪影響を及ぼす可能性がある。私たちは現在、どんな新しい優先株シリーズを指定したり、どんな優先株を発行したりするつもりはありませんが、私たちは未来にそうするかもしれません。
私たちは予測可能な未来に私たちの普通株に配当金を支払うつもりはない。
取締役会は私たちの財務状況、経営業績と資本要求及びその他の要素を考慮した後、自ら私たちの配当政策を決定します。私たちは予測可能な未来に私たちの普通株に現金配当金を支払わないと予想して、あなたは配当収入を期待して私たちに投資してはいけません。
私たちの普通株の活発な公開取引市場は持続できないかもしれない。
私たちの普通株はニューヨーク証券取引所アメリカ取引所に上場していますが、私たちの株式市場は異なる程度の取引活動を示しており、活発な取引市場が続くことを保証することはできません。活発な市場の不足はあなたが売りたい時やあなたが合理的だと思う価格で私たちの普通株を売る能力を弱めるかもしれません。活発な市場の不足はまた私たちの普通株の価格を下げるかもしれない。不活発な市場は、株を売却することで本来資金を調達する能力を弱める可能性もあり、普通株を対価格で他社や技術を買収する能力を弱める可能性もある。
| 65 |
私たちの株価はずっと変動していて、引き続き変動するかもしれない。
私たちの普通株の市場価格は歴史的に経験しており、大幅な変動を経験し続ける可能性がある。私たちの候補製品の開発における進展、政府法規が私たちの製品と業界に与える影響、株主は私たちの普通株を大量に売る可能性があり、私たちの四半期の経営業績、経済あるいは金融市場の全体的な状況の変化、そして私たちあるいは私たちの競争相手に影響を与える他の事態の発展は私たちの普通株の市場価格を大幅に変動させ、同時に重大な市場損失をもたらす可能性があります。もし私たちの株主が大量の普通株を売却し、特にこれらの売却が短時間で行われた場合、これらの売却は私たちの普通株の市場価格に悪影響を与え、私たちの資金調達能力を弱める可能性がある。また、近年、株式市場は明らかな価格と出来高変動を経験している。この変動 は,会社の経営業績に関係なく多くの会社が発行する証券の市場価格に影響を与え,我々の普通株の価格に悪影響を及ぼす可能性がある.また、株価変動によって証券集団訴訟を受ける可能性があり、これは大量のコストおよび経営陣の注意と資源の移転を招き、株価、業務、将来性、運営結果、財務状況を損なう可能性があります。
もしbr証券や業界アナリストが私たちの業務に関する研究や報告を発表していない場合、あるいは彼らが私たちの株を不利に修正した場合、私たちの株価や取引量は低下する可能性がある。
私たちの普通株の取引市場は、業界や証券アナリストが発表した私たちまたは私たちの業務に関する研究と報告の影響を受けるだろう。私たちの産業と金融アナリストの研究カバー範囲は現在限られている。たとえ私たちのアナリストカバー率が増加しても、私たちを追跡する1つ以上のアナリストが私たちの株式格付けを引き下げたら、私たちの株価は下落するかもしれない。もし1人以上のアナリストが私たちの会社の報道を停止したり、私たちの報告書を定期的に発表できなかった場合、私たちは金融市場での可視度を失い、ひいては私たちの株価や取引量を低下させる可能性がある。
私たちはニューヨーク証券取引所アメリカ取引所から退市するかもしれません。これは私たちの株の流動性と私たちの資金調達能力を深刻に損なうかもしれません。
私たちは過去に私たちの普通株がニューヨーク証券取引所アメリカに上場する要求を満たすことは困難でしたが、将来も困難になるかもしれません。もし私たちがこのようなコンプライアンスを回復または維持できなければ、私たちはもうナスダックで取引する資格がないかもしれない。この場合:
| ● | 我々 は,場外掲示板や“粉ミルク”のような認知度や認知度の低い市場で取引しなければならない可能性がある |
| ● | 我々普通株の株式 の流動性や販売可能性が低下する可能性があり、株主が歴史的な速度と価格で私たちの株を売買する能力を低下させる可能性がある。もし私たちの株が“格安株”として取引されれば、私たちの株の取引はもっと難しく煩雑になるだろう。 |
| ● | 私たちのbrは、優遇条項で資本を得ることができないか、あるいは資本を得ることができないかもしれません。別の市場で取引されている会社は、魅力が低く、関連リスクの高い投資とみなされる可能性があるので、既存または潜在的な機関投資家は、私たちの普通株への投資にあまり興味がないか、または投資が禁止されているかもしれません。これはまた私たちの普通株の市場価格の下落を招くかもしれない。 |
私たちの普通株の逆株式分割は予想された結果を生じないかもしれない。
2023年11月1日、我々のbr株主は、わが社の登録証明書の改正案を承認し、2株1株から1株50株の割合で逆株式分割を行うことを取締役会が適宜決定し、普通株の認可株式総数をbr 5億株から2.5億株に減少させ、それぞれ逆株式分割と認可株式削減と呼ぶ。私たちは、株式の逆分割は私たちの普通株の市場価格を上昇させ、私たちの普通株の金融界に対する吸引力を強化し、私たちの普通株の取引流動性を改善し、私たちの普通株をより高いレベルの証券取引所に入れることができ、特定の生物技術と医薬取引指数と取引所取引基金に入れる資格を持つことができると信じている。
しかし、逆株式分割を実施すれば、私たちの普通株の市場価格が比例して増加して、私たちが発行した普通株の数を減らすことを含めて、私たちの普通株の市場価格が比例して増加することを保証することはできません。また,逆株分割 は我々全体の時価を低下させる可能性がある.逆株分割が機関投資家や投資基金の1株当たりの株価を引き付けることを保証することはできず、その株価が機関投資家や投資基金の投資ガイドのbrを満たしたり、私たちの普通株の取引流動性を改善したりする保証はない。
また、逆株式分割を実施すると、逆株式分割が許可株式数に対する普通株の流通株数を減少させるため、普通株発行可能株式数が増加する。このような利用可能な株は、将来の買収、投資機会、協力または他の戦略的合意の確立、株式または変換可能な債務証券に関する融資取引、将来の普通株式市場での将来の発行、br、または現在または将来の従業員持分計画に従って発行されること、およびそのような取引に関連する株式証券の発行 が、現在の株主の私たちの所有権利益に対する潜在的な重大な希釈をもたらす可能性がある将来の会社の目的に使用されることができる。
当社が解散した後、あなたはすべてまたは一部の投資を回収できないかもしれません。
当社に清算、解散、または清算が発生した場合、任意であっても非任意であっても、当社の収益および/または資産は、取引発効後に残り、私たちのすべての債務および負債の支払いは、まず私たちの優先株保有者に割り当てられ、その後、普通株主(私たちの優先株保有者を含む)に比例して割り当てられます。当社の清算、解散、または清算時に、利用可能な資産を普通株式保有者または任意の金額に支払うことは保証できません。この場合、あなたの は投資の一部または全部を損失する可能性があります。
| 66 |
私たちの会社登録証明書は、株主のさらなる承認なしに新しい優先株シリーズを作成することを取締役会に許可しており、これは私たちの普通株式保有者の権利に悪影響を及ぼす可能性があります。
私たちの取締役会は優先株の相対的な権利と優先株を決定して決定する権利がある。私たちの取締役会 は株主がこれ以上承認することなく、最大10,000,000株の私たちの優先株を増発する権利があります。したがって、私たちの取締役会は、清算時に私たちの資産に対する優先権、配当金が普通株式所有者に割り当てられる前に配当支払いを得る権利、および私たちの普通株を償還する前に株を償還する権利およびプレミアムを付与する一連の優先株の発行を許可することができる。また、我々の取締役会 は、私たちの普通株よりも大きな投票権を有する一連の優先株の発行を許可することができ、または私たちの普通株に変換することができ、これは、私たちの普通株の相対投票権を低下させたり、既存の株主の株式を希釈したりする可能性がある。
私たちの会社の登録証明書、定款、デラウェア州の法律の条項は私たちを買収することをより困難にするかもしれません。これは私たちの株主に有利であり、私たちの株主が私たちの取締役会と経営陣の現メンバーを交換または罷免しようとすることを阻止するかもしれません。
私たちが改訂して再記述した会社登録証明書および定款のいくつかのbr条項は、あなたの株式が割増取引を得る可能性があることを含む、合併、買収、または株主が有利と考える可能性のある他の支配権変更を阻止、延期または阻止する可能性があります。さらに、これらの規定は、私たちの株主が私たちの取締役会メンバーを交代または罷免しようとしていることを阻止または挫折させるかもしれない。これらの条項はまた、投資家が将来私たちの普通株に支払いたいかもしれない価格を制限し、それによって私たちの普通株の市場価格を下げる可能性がある。このような取引に参加することを望む株主はそうする機会がないかもしれない。他にも、これらの規定には、
| ● | “方法”は、株主特別会議は取締役会総裁または弊社取締役会長のみが開催することができ、または少なくとも50%の発行済みおよび発行済み議決権普通株を有する登録株主の書面要求を経てのみ開催されることが規定されている |
| ● | それらの は役員選挙で累積投票を行う規定を含まない。累積投票により、十分な数の株式を持つ少数株主が1人以上の取締役を選出することを確保できる可能性がある。累積投票権の不足は、少数の株主が取締役会を改革する能力を制限する可能性がある |
| ● | それらの は、株主の承認なしに最大10,000,000株の優先株を発行することを可能にします(これらのすべての優先株はまだ発行可能です) これは私たちの普通株式保有者の権利と権力に悪影響を及ぼす可能性があります。 |
また、特定のbr基準に適合しない限り、大株主、特に私たちの普通株の15%以上の投票権を有する株主が、所定の時間内に私たちと合併または合併することを禁止する可能性がある“デラウェア州会社法”(DGCL)第203節の規定によって管轄されている。
株主が我々または我々の役員、上級管理者または従業員とトラブルが発生した場合に有利な司法フォーラムを得る能力が制限される可能性がある。
私たちの会社登録証明書は、デラウェア州衡平裁判所は、(I)私たちが提起した任意の派生 訴訟または訴訟を代表する独占フォーラムであり、(Ii)私たちの任意の取締役、br}上級管理者または他の従業員が私たちまたは私たちの株主に対して受託責任を有するクレームを主張する任意の訴訟、(Iii)デラウェア州一般会社法、当社の会社登録証明書または私たちの定款の任意の規定に基づいてクレームを提起する任意の訴訟、または(Iv)内部事務原則によって管轄されるクレームを主張する任意の訴訟であることが規定されている。私たちの会社登録証明書はまた、アメリカ合衆国連邦地域裁判所 は、証券法に基づいて提出された任意の訴因を解決する独占裁判所であり、デラウェア州がこのような独占裁判所に対して規定する実行可能な最終裁決に依存すると規定している。これらの排他的なbrフォーラム条項は、司法フォーラムにおいて、私たちまたは私たちの役員、役員、または他の従業員とのトラブルのクレームに有利であると考える能力を株主が提出することを制限する可能性があり、これは、私たちと私たちの役員、役員、および他の従業員に対するこのような訴訟を阻止する可能性がある。例えば、衡平裁判所にクレームを出した株主は、特にデラウェア州またはその近くに住んでいなければ、そのようなクレームを提起する際に追加の訴訟費用に直面する可能性がある。衡平裁判所と連邦地域裁判所はまた、訴訟の株主がいる可能性があるか、訴訟を提起する裁判所を選択することを含む他の裁判所とは異なる判決または結果を得る可能性があり、このような判決または結果は、私たちの株主よりも有利かもしれない。連邦地域裁判所フォーラムのような選択条項を採用する会社は現在、デラウェア州裁判官br裁判所の訴訟を受けており、株主はこの条項が実行できないと主張している。もし裁判所が私たちの会社の登録証明書に含まれるいずれかの裁判所条項が訴訟で適用されないか、または実行できないことを発見した場合、私たちは他の管轄区域でこのような訴訟を解決することに関連する追加費用 を生じる可能性があり、これは私たちの業務および財務状況に悪影響を及ぼす可能性がある。例えば、デラウェア州衡平裁判所は最近、アメリカ合衆国連邦地域裁判所が“証券法”に基づいて提出された任意の訴因を解決する独占法廷規定は強制的に執行できないと判断した。この決定のため,我々は現在,わが社の証明書中の連邦フォーラム選択条項を実行するつもりはなく,控訴時にその決定を覆さない限りである.しかし、この決定が控訴時に再審され、最終的にデラウェア州最高裁判所によって覆された場合、私たちは連邦地域裁判所排他的フォーラム条項を実行する。
| 67 |
純営業損失の繰越や他の税務属性を使用する能力が制限される可能性があります。
Aquarius BioTechnologies,Inc.との合併により,米国連邦純営業損失,繰越,米国連邦税収控除を利用する能力は,改正された1986年国税法第382節に制限される可能性がある。382節と383節で定義された“所有権変更”が発生すれば,制約 を適用する.通常、適用されるテスト期間内(通常は 3年)、1つまたは複数の直接的または間接的に“5%株主”が所有する株式価値のbr}パーセントがその最低所有権パーセントよりも ポイント以上増加した場合、所有権変更が発生する。さらに、私たちの株式所有権の将来の変化は、私たちの制御範囲内ではなく、“所有権の変化”を引き起こす可能性があり、第382条および第383条の制限を招く可能性がある。したがって,課税収入の純額を稼ぐと,変動前の純営業損失の繰越や他の税収属性を用いて米国連邦課税収入を相殺する能力が制限される可能性があり,将来の納税義務が増加する可能性がある。また、税法は他にも、利息控除に重大な追加制限を加え、純営業損失(NOL)控除を2017年12月31日以降に開始する課税年度に生じる損失の80%に制限しています。
| 項目 1 B. | ネットワーク·セキュリティ |
ネットワークセキュリティリスク管理と戦略
私たちは業界内の他の会社と同様に、私たちの業務に関連するいくつかのネットワークセキュリティリスクに直面している。これまで,我々の業務戦略,運営結果,財務状況はネットワークセキュリティ脅威リスクの影響を受けていない.報告期間中、私たちはどんな重大なネットワーク事件も経験しなかったし、開示されなければならない一連の非実質的な事件も経験しなかった。
私たちの通常のビジネスプロセスでは、従業員、試験参加者、パートナー、顧客、br、およびサプライヤーのデータを含むデータを使用、保存、および処理します。我々は,このデータと我々のシステムに対するネットワークセキュリティ脅威によるリスクを識別,評価,緩和するためのネットワークセキュリティリスク管理計画を実施した.我々のネットワークセキュリティリスク管理計画は、br情報セキュリティ計画の評価、自動化ツールを用いたネットワークリスクと脅威の継続的な監視、書面イベント応答と災害復旧政策とプログラム、および従業員訓練を含む複数の構成要素を含む。私たちの最高財務官の指導の下で、私たちのネットワークリスク管理計画は、30年以上の技術工学経験と複数の高度な学位を持つ第三者ITコンサルタントが指導します。また、他の評判の良いネットワークセキュリティソフトウェアと共に端末検出ソフトウェアおよびデバイス管理を展開しています。また、すべてのシステムにわたってマルチファクタ認証を行う必要があります。
我々 は定期的に第三者を招いてリスク評価や他の脆弱性分析を行う。最後に、私たちの計画はすべての職員たちのためのサイバーセキュリティ訓練を含む。半年ごとの訓練の重点はネット釣りを含むネット脅威意識だ。
ネットワークセキュリティリスクに関するガバナンス
最高経営責任者(“CEO”)の最終指示によると、取締役会は、必要に応じて、任意の重要なネットワークセキュリティリスク、持続的なネットワークセキュリティ計画および戦略、適用される規制要件および業界基準を含む、必要に応じて当社のネットワークセキュリティリスク管理計画を更新する。最高経営責任者はまた、取締役会に任意のネットワークセキュリティイベント(疑似または実際)を通報し、必要に応じてイベントやネットワークセキュリティリスク緩和活動に関する最新の状況を提供する。
| 68 |
| 第 項2. | 属性 |
施設
私たちの行政事務室はニュージャージー州ベッドミンスターの約8,900平方フィートのオフィス空間で構成されており、レンタル契約は2029年6月に満期になります。ニュージャージー州ブリッジウォルトで約14,000平方フィートの実験室空間を借りました2027年7月に満期になります
| 第 項3. | 法的訴訟 |
私たちは現在いかなる法的手続きの当事者でもなく、私たちは私たちに対するクレームや訴訟が未解決であるか、あるいは脅かされていることを知らない。今後、私たちは時々私たちの正常な業務過程で発生したクレームに関する訴訟に巻き込まれるかもしれない。
| 第 項. | 鉱山安全開示 |
は適用されない
第 第2部分
| 第 項5. | 登録者普通株,関連株主事項及び発行者が株式証券を購入する市場 |
私たちの普通株はニューヨーク証券取引所アメリカ取引所で看板取引され、コードは“MTNB”です。
“ニューヨーク証券取引所MKT”によると、2024年3月18日、私たちの普通株の終値は1株0.27ドルで、私たちの普通株は約102人の記録的な保有者がいる。記録保持者の数は我々の譲渡エージェントの記録から決定され,普通株の受益所有者は含まれておらず,その株は各種証券仲介人,取引業者,登録決済機関の名義で保持されている.Vock Transfer,LLCは我々の普通株式の譲渡エージェントと登録業者である.
配当をする
私たちは私たちの普通株についていかなる現金配当金を支払うか発表したことがありません。私たちは予測可能な未来に私たちの普通株に対していかなる現金配当金も支払わないと予想しています。私たちはすべての利用可能な資金と任意の未来の収益を維持して、私たちの業務の発展と拡張に資金を提供するつもりです。 将来配当金を派遣するかどうかは私たちの取締役会が適宜決定し、多くの要素に依存して、私たちの運営結果、財務状況、将来の見通し、契約制限、法律適用制限 及び私たちの取締役会が関連していると思う他の要素を含む。
最近販売されている未登録証券
ない。
発行者と関連購入者株証券の買い戻し
本年報で述べた期間、私たちは何の登録証券も購入していません。
| 第 項6 | [保留されている] |
| 69 |
| 第 項7. | 経営陣の財務状況と経営結果の検討と分析 |
以下、我々の財務状況と経営結果の検討と分析は、我々の財務諸表および本年度報告書10-K表の他の部分に関する説明と一緒に読まなければならない。本議論および分析に含まれるいくつかの情報または本10-Kフォーム年次報告書の他の部分に列挙された情報には、我々の業務計画および戦略および融資ニーズに関する情報が含まれる。リスクおよび不確定要因を含む前向き陳述は、本年度報告の10-K表の“リスク 要因”の部分と共に読まれ、議論は、以下の議論および分析に含まれる前向き陳述に記載または示唆された結果とは大きく異なる重要な要素をもたらす可能性がある。 は、様々な要因のため、本年度報告および米国証券取引委員会に提出された他の報告で議論された内容を含む、これらの前向き陳述で予想される結果とは大きく異なる可能性がある。 特に“リスク要因”の下のものである.1株当たりのデータを除いて、表および段落形式のすべてのドル金額は千元で表されるか、または他の方法で説明される。
概要
私たちのbrは臨床段階の生物製薬会社であり、私たちの脂質ナノ結晶(LNC)プラットフォーム 輸送技術(LNCプラットフォーム)を用いて画期的な治療を提供することに集中している。LNCプラットフォームを利用した内部製品パイプラインの開発を求めており、小分子とオリゴヌクレオチドを成功的に包み、br毒性なしに所望の細胞組織への標的および肝外送達を促進している。
私たちの戦略の重要な要素は:
| ● | Br}MAT 2203をORALTO試験に進め,開発および/または商業パートナーを獲得することにより,選択の限られた患者を治療する侵襲性アスペルギルス症を治療した。この初期適応は、必要な薬効学の架け橋を構築する門戸適応となることを目的とし、それによってMAT 2203の使用を他の適応に拡張し、505(B)(2)経路下の限られた追加の臨床仕事によって致命的な侵襲性真菌感染(例えば、毛カビ症、カンジダ症および地方性真菌症)を治療し、それによってMAT 2203を 製品のパイプラインにする | |
| ● | Brは我々のLNCプラットフォームと他の小分子や小オリゴヌクレオチドの使用を炎症や腫瘍学分野に拡張し,差別化候補薬物の内部チューブを開発した。経口、肝外、非毒性の細胞内にこれらの分子を送達することは重大な進歩である | |
| ● | 著者らのLNCプラットフォームを重点として、有力な製薬会社と外部協力チャネルを構築し、彼らの複雑な小分子と小さいオリゴヌクレオチドにASOとsiRNAを含む送達解決方案 を提供した |
2023年12月31日と2022年12月31日までの年間の純損失はそれぞれ22,942ドルと20,997ドルであった。成立以来,我々は個々の 期間に損失が発生し,予見可能な未来にさらに多くの損失が予想される.手元の現金,現金等価物,販売可能債務証券は,本年度報告提出日から今後12カ月の計画運営に資金を提供するには不十分であると考えられる。私たちは公開または私募株式発行、債務融資、政府または他の第三者資金、協力、許可手配を通じて私たちの運営に資金を提供することを求めます。受け入れ可能な条項では、私たちはこのような資金調達代替案を得ることができないかもしれないし、根本的にできないかもしれない。そのため、私たちが経営を続けている企業として継続する能力が大きく疑われています。
財務 運営概要
収入.収入
2023年12月31日までの1年間に、BioNTech SEとGenentech Inc.との研究協力により1,096ドルの契約研究収入が生まれました。2022年12月31日までの1年間に、BioNTech SEとの研究協力により3,188ドルの契約収入が生まれました。私たちが製品収入を作る能力は、私たちの初期候補製品の成功と最終的な商業化に大きく依存します。これは何年も起こらないと予想されます。
| 70 |
研究と開発費
研究と開発費用には、候補製品MAT 2203の開発と我々のLNCプラットフォームのアップグレードによるコストが含まれており、 は:
| ● | 臨床前と人体臨床試験材料を取得し、開発し、製造するコスト |
| ● | 化学および製造制御(CMC)、臨床前および臨床活動、および規制操作に関するコンサルタントおよび請負業者の費用 ; |
| ● | われわれの臨床前または臨床試験を行う契約研究組織またはCRO(NIHを含む)との合意による費用 ; |
| ● | 研究開発過程に参加した従業員の賃金および株式報酬支出を含む従業員関連の費用 |
| ● | CFF報酬プロトコルに関する費用の 精算. |
下表は,我々が2023年12月31日と2022年12月31日までの年度内に候補製品と開発プラットフォームに支払う直接研究開発費をまとめたものである。私たちの直接研究開発費用は、主に外部コスト、例えば、私たちの開発作業に関連して請負業者、コンサルタント、分析実験室およびCROおよび/またはNIHに支払われる費用を含む。我々は通常,我々の従業員とインフラ資源を用いて臨床試験材料を作成し,製品分析,研究案開発 および外部サプライヤーの監督を行う。以下の“内部人員編成、管理費用およびその他”には、実験室空間、用品、研究開発(R&D)従業員費用(株式オプション費用を含む)、出張、医学教育費用が含まれる。
| 十二月三十一日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| 直接研究開発費: | ||||||||
| 製造プロセス開発 | $ | 1,182 | $ | 2,523 | ||||
| 臨床前試験 | 852 | 720 | ||||||
| 臨床発展 | 1,696 | 2,175 | ||||||
| 監督管理 | 581 | 737 | ||||||
| 内部人員の配置、管理費用、その他 | 10,178 | 10,523 | ||||||
| 総研究開発 | $ | 14,489 | $ | 16,678 | ||||
研究と開発活動は私たちの業務モデルの核心だ。我々の研究開発費は時間とともに増加すると予想されるが,臨床開発後期段階の候補製品は通常臨床早期開発段階の候補製品よりも開発コストが高いため,これは主に後期人体試験の規模と持続時間が増加しているためである。しかし、2024年の研究·開発費は、MAT 2203の第3段階登録試験の開始とLNCプラットフォーム交付技術の向上を支援するために、2023年に発生する費用よりも低くなると予想されています。
一般料金 と管理費用
一般費用と行政費用には主に行政·財務機能者の賃金と関連費用が含まれる。その他の一般および行政費用には、施設コスト、保険、投資家関係費用、法律専門費用、特許審査、コンサルティング、会計/監査サービスが含まれています。2023年に発生した費用と比較して,2024年の一般·行政費用はやや低下すると予想される。
| 71 |
販売純営業損失(NOL)と税収控除
2023年12月31日と2022年12月31日までの年間で、ニュージャージー州技術業務税務証明書に基づいて未使用の純営業損失(NOL)を販売する計画と研究開発税収控除による収入は、それぞれ484ドルと3491ドルだった。2023年に記録された収入には2022年の納税年度に関する売上高が含まれ、2022年に記録された収入には2021年と2020年の納税年度に関する売上高が含まれる。
その他 純収入
その他の収入は,純額は主に利息収入(費用)と配当金からなる。
キー会計政策と会計推定の応用
重要な会計政策は、我々の財務状況や経営結果を記述するために重要であり、管理層が最も困難で、最も主観的または最も複雑な判断を行う必要があるのは、通常、本質的に不確実な事項の影響を評価する必要があるからである。
我々の重要な会計政策の説明については、“注3”を参照されたい重要会計政策の概要“これらの政策では、以下は、(I)研究開発コスト、および(Ii)営業権および他の無形資産を必要とするので、我々の総合財務諸表を理解するために重要であると考えられる。
現在の 運営傾向
著者らの現在の研究開発は臨床開発を通じて著者らのリードLNC候補製品MAT 2203を推進し、内部努力と第三者との協力を通じて著者らのLNCプラットフォームの応用を拡大することに集中している。我々の研究開発費には,br製造作業やこのような作業で使用される活性薬物成分や補剤のコスト,コンサルタントに支払われる臨床試験設計や規制活動に関する費用,提供者に支払われる様々な臨床研究やそのような研究結果の分析費用,および我々の薬物の潜在的有効性と安全性に関連する他の医学研究の費用が含まれている。製品開発における重大な投資は競争の必要であると考え,これらの投資を継続して,候補製品やノウハウの潜在的な潜在力を実現する予定である。
私たちのbrは近い将来、私たちの研究開発費の大部分が私たちの現在と未来の臨床前と臨床開発計画を支援するために使用されると予想されている。このような支出は完了時間と費用に関連する多くの不確実な要素の影響を受ける。著者らは大量の臨床前研究において化合物の安全性、毒理学と有効性をテストした。適切な時期に,規制部門の承認を得られれば,早期臨床試験を行う予定である。私たちはこのような実験を自分で支援し、連邦支出、契約、または他の合意の助けを借りてbrを支援する可能性があると予想される。私たちが試験から結果を得た時、私たちはより将来性のある製品に資源を集中させるために、brを停止したり、いくつかの製品の臨床試験を延期することを選択するかもしれない。臨床試験の完成には数年の時間が必要である可能性があり、時間の長さは候補製品のタイプ、複雑性、意外性と期待用途によって大きく異なる。
著者らの製品の臨床試験の開始と完成は多くの要素によって延期される可能性があり、臨床試験期間中の治療効果の不足、予見できない安全問題、参加者の募集速度が予想より遅い、資金不足或いは政府遅延を含む。また、多種の要素のため、著者らの候補製品の予想安全性或いは有効性を支持しない結果、臨床試験設計中の既知の欠陥及び製品開発中の監督管理政策の変化を含む規制遅延或いは拒否に遭遇する可能性がある。これらのリスクと不確実性のため,我々の臨床開発計画の具体的な時間やコスト,あるいは候補製品の現金流入時間(あれば)を正確に見積もることはできない。私たちの業務、財務状況、および運営結果は、私たちの臨床試験の任意の遅延または終了、またはFDAが規制部門の承認を証明するのに不十分な状況によって重大な悪影響を受ける可能性があり、関連薬物または計画の現金流入が延期または発生しないことを前提としている。
| 72 |
運営結果
2023年12月31日と2022年12月31日までの年度
次の表は、2023年12月31日と2022年12月31日までの年間運営費をまとめています
| 十二月三十一日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| 収入.収入 | $ | 1,096 | $ | 3,188 | ||||
| 費用: | ||||||||
| 研究開発 | $ | 14,489 | $ | 16,678 | ||||
| 一般と行政 | 10,373 | 11,100 | ||||||
| 運営費 | $ | 24,862 | $ | 27,778 | ||||
| 販売純営業損失(NOL) | $ | 484 | $ | 3,491 | ||||
収入。 2023年12月31日と2022年12月31日までの会計年度は、それぞれ1,096ドルと3,188ドルの収入を創出した。2023年の収入には,BioNTech SEとの研究協力とGenentechとのフィージビリティスタディ協定による契約研究収入があり,2022年の収入は完全にBioNTech SEとの研究協力によるものである。
研究と開発費用。2023年12月31日と2022年12月31日までの年間の研究開発費はそれぞれ14,489ドルと16,678ドルであった。brが2,189ドル減少したのは,臨床試験費用が1,302ドル減少し,専門·相談費が541ドル減少したためであり,主にMAT 2203開発計画を一時停止したことと,2022年に記録されたライセンス契約改訂費用が291ドル減少したためである。
一般 と管理費用です。2023年12月31日と2022年12月31日までの年度のG&A費用はそれぞれ10,373ドルと11,100ドルである。brが前年より727ドル減少した主な原因は,ある保険料の減少と,前年からのある項目に関する相談費やbr施設費の低下である。
販売純営業損失(NOL)と税収控除。同社は2023年12月31日と2022年12月31日までの年間で、それぞれ484ドルと3491ドルを確認し、ニュージャージー州技術営業税証明書に基づいて第三者への州純営業損失の売却と州研究開発控除に関連していることを確認した。2023年に記録された収入には2022年の納税年度に関する売上高が含まれ、2022年に記録された収入には2021年と2020年の納税年度に関する売上高が含まれる。
流動性 と資本資源
流動資金源
私たちbrは設立以来、主に私たちの優先株、普通株、普通株式承認証を私募することで、私たちの運営に資金を提供してきました。2023年12月31日現在、2013年の設立以来、株式証券の売却により合計156,851ドルの総収益と144,141ドルの純収益を集めてきた。
2023年12月31日まで、私たちは現金、現金等価物、売却可能な債務証券を持っています。限定現金を含まず、総額は13,756ドルです。
2020年市場販売協定
2020年7月2日,吾らはBTIG,LLC(“BTIG”)と市販プロトコル(“販売プロトコル”)を締結し,この合意により,吾らはBTIG(販売エージェントおよび/または依頼者として)を通して合計発行価格5,000万ドルに達する普通株 株式を随時発売·販売することができ,ただ販売契約に掲載されている吾などの提供および販売可能な普通株数に関する若干の制限に制限されなければならない。BTIGは毎回販売された毛収入から3%の手数料を得るだろう。私たちはいつでも販売プロトコルを終了することができます;BTIGはいくつかの限られた場合に販売プロトコルを終了することができます。2023年または2022年の間、私たちは販売契約に基づいていかなる普通株式も売却していません。2023年12月31日現在、販売契約の利用可能容量は44,247ドルである。
| 73 |
キャッシュフロー
以下の各時期の主要な現金源と用途を以下の表に示す
| 12月31日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| 経営活動用の現金 | $ | (15,278 | ) | $ | (19,156 | ) | ||
| 投資活動が提供する現金 | 13,242 | 4,877 | ||||||
| 融資活動のための現金 | (7 | ) | 79 | |||||
| 現金と現金等価物および制限的現金純減少 | $ | (2,043 | ) | $ | (14,200 | ) | ||
操作 活動
2023年12月31日までの1年間、経営活動で使用された現金純額は15,278ドルだったが、前年は19,156ドルだった。期間中の純損失が3,878ドル減少したのは,主に日常業務過程における受取時間により運営資本調整が6,456ドル減少したが,今期の純損失が1,945ドル増加し,株による報酬支出が280ドル減少したこと,および2022年とのライセンス契約費用が291ドル減少したことで部分的に相殺された。2024年の運営で使用される現金数はやや高くなると予想されるが,我々の候補製品やLNCプラットフォームの開発サイクルを進めていくからである。
投資 活動
2023年12月31日までの年度,投資活動が提供する現金純額は13,242ドルであるのに対し,2022年12月31日までの年度,投資活動が提供する現金純額は4,877ドルである。投資活動が8,365ドル増加したのは,主に有価証券の購入と満期日が前年比7,691ドル減少し,設備·リース改善の購入が674ドル減少したためである。
活動に資金を提供する
2023年12月31日と2022年12月31日までの年間、融資活動および融資活動による現金純額はそれぞれ(7ドル)、79ドル。融資活動が提供する現金が86ドル減少したのは、主に2022年に株式オプションを行使して受け取った99ドルの収益が減少したためだ。
資金需要やその他の流動資金の問題
我々 は予測可能な未来に巨額の費用と増加していく運営損失が続くと予想している。私たちは次のような状況に伴い、私たちの費用が大幅に増加すると予想している
| ● | 著者らの主要な候補製品MAT 2203の更なる臨床研究を行い、甚だしきに至ってはこのような研究は主にNIHの非希釈性資金によって援助されている |
| ● | より多くの候補製品を発見し開発するためにbrを求める; |
| ● | 臨床試験を成功させた任意の候補製品のために監督管理の承認を求める |
| ● | より多くの臨床開発と潜在的商業化候補製品の生産が要求される |
| ● | 私たちの知的財産権の組み合わせを維持し、拡大し、保護する |
| ● | 追加の臨床、品質管理、科学者を招聘し、 |
| 74 |
| ● | 私たちの製品開発と将来の商業化作業を支援し、上場企業の義務を履行するために必要な人員やインフラを支援する人を含む、br運営、財務、管理情報システムと人員を増加させます。 |
私たちの既存の現金、現金等価物、および売却可能な債務証券は、本年度報告書の提出日から今後12ヶ月以内の運営費用および資本支出需要を支払うのに十分であるとは思いません。そのため、同社の継続経営企業としての継続経営能力が大きく疑われている。
私たちが利益を達成するのに十分な製品収入を生み出すことができる前に、私たちは私募と公募株式発行、債務融資、政府または他の第三者融資、協力、許可手配を通じて、私たちの現金需要に融資する予定です。 私たちが普通株、転換可能証券、または他の株式証券を売却することによって追加資本を調達する場合、私たちの株主の所有権権益は大幅に希釈される可能性があり、これらの証券の条項には、清算またはあなたの普通株主権利に悪影響を及ぼす他の特典が含まれている可能性がある。債務融資および優先持分融資が利用可能であれば、固定支払義務の増加を招き、特定の行動をとる能力を制限または制限する契約を含む契約に関連する可能性があり、例えば、追加債務を招く、資本支出を行う、または配当を宣言することができ、これらは、私たちの業務を展開する能力に悪影響を及ぼす可能性がある。追加融資を得るには、私たちの経営陣が多くの時間と精力を投入する必要があるかもしれませんが、彼らの注意を日常活動から比例しない位置に移す可能性があり、私たちの経営陣が候補製品開発を監督する能力に悪影響を及ぼす可能性があります。
もし私たちが第三者との協力、戦略連合またはマーケティング、流通または許可手配によって追加の資金 を調達する場合、私たちは、私たちの技術、将来の収入フロー、研究計画、または候補製品に対する貴重な権利 を放棄しなければならないか、または私たちに不利になる可能性のある条項でライセンスを付与しなければならないかもしれない。もし私たちが必要な時に株式や債務融資を通じてより多くの資金を調達できない場合、私たちは私たちの製品開発や将来の商業化努力を延期、制限、減少、または中止する必要があるかもしれません。あるいは私たちは自分で開発し、マーケティングする候補製品をより望んでいた権利を与えています。
私たちの財務状況と運営結果はまた、グローバル·サプライチェーンの中断、世界的な貿易紛争、および/または政治的不安定など、他の私たちがコントロールできないかもしれない要素の影響を受ける可能性がある。金利上昇、特に政府支出の減少や金融市場の変動に加えて、経済の不確実性をさらに増加させ、これらのリスクを悪化させる可能性がある。また、上昇するインフレ率は、従業員関連コストや臨床試験費用など、運営費用を増加させることで、私たちの運営結果に悪影響を及ぼす可能性があります。
契約義務と約束
付記10-を参照“約束”連結財務諸表の付記では、会社の契約義務及び承諾を検討する。
表外手配 表内手配
提出期間中、我々brは、現在、未合併エンティティまたは金融パートナーシップ企業との関係など、米国証券取引委員会規則で定義された表外手配もなく、これらのエンティティは、一般に、我々の貸借対照表に反映される必要のない融資取引を促進するために、構造的融資または特殊な目的エンティティと呼ばれる。
最近の会計声明
注釈3-“を参照してください重要会計政策の概要、“連結財務諸表付記では、最近の会計声明を検討します。
| 第 7 A項。 | 市場リスクの定量的·定性的開示について |
は適用されない.
| 75 |
| 第 項8 | 財務諸表と補足データ |
私たちの財務諸表は、独立公認会計士事務所の財務諸表と共に、EisnerAmper LLP(PCAOB ID:EisnerAmper LLP:PCAOB ID:EisnerAmper LLP:EisnerAmper LLPを含む)の第15項“証拠物、財務諸表明細書”から引用されています
| 第 項9. | 会計·財務開示面の変更と会計士との相違 |
は適用されない.
| 第 9 A項。 | 制御 とプログラム |
開示制御とプログラムの評価
制御とプログラムの開示:
2023年12月31日現在、我々の最高経営責任者及び最高財務責任者の監督·参加の下で、我々の開示制御及び手順(例えば、1934年の証券取引法改正規則13 a−15(E) 及び15 d−15(E)で定義されている)の設計及び運用の有効性を評価している。この評価に基づき,我々の最高経営責任者および最高財務責任者は,2023年12月31日までに,我々の開示制御および手順が合理的なbr保証レベルで有効であると結論した。
我々のbr}開示制御およびプログラムは、取引法に基づいて提出または提出された報告書 で開示を要求した情報が、米国証券取引委員会規則および表が指定された時間帯に記録、処理、まとめ、報告されることを保証するために合理的な保証を提供することを目的としている。開示制御および手続きは、取引所法案に従って提出された報告書に開示すべき情報が蓄積され、我々の管理層(最高経営責任者および最高財務責任者を含む)に伝達されることを確実にするための制御および手順を含むが、開示要求に関する決定をタイムリーに行うために、限定されない。
財務報告の内部統制に関する経営陣の報告書:
私たちの経営陣は財務報告書の十分な内部統制を確立して維持する責任がある。財務報告の内部制御は著者らの主要な行政人員と主要な財務官が設計或いはその監督下で設計したプログラムであり、目的は公認された会計原則に基づいて、財務報告の信頼性と外部目的の総合財務諸表の作成に合理的な保証を提供することである。私たちの財務報告に対する制御は、(1)合理的で詳細かつ正確かつ公平に反映された私たちの資産の取引および処分の記録を維持することに関連している、(2)公認された会計原則に基づいて財務諸表を作成するために必要な取引が記録されている合理的な保証を提供し、私たちの収入と支出は私たちの経営陣と取締役の許可によってのみ行われる、という政策と手続きを含む。(3)連結財務諸表に重大な影響を与える可能性のある不正な取得、使用、または処理を防止またはタイムリーに発見する合理的な保証を提供する。
固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来の期間に行われる任意の有効性評価の任意の予測は,条件の変化により制御措置が不足したり,政策やプログラムの遵守度が悪化したりする可能性がある.
経営陣 は、テレデビル委員会(COSO)後援組織委員会が発表した“内部統制-総合枠組み(2013)”で確立された基準に基づいて、財務報告書の内部統制に対する有効性を評価した。この評価に基づき、経営陣は、財務報告の内部統制において重大な弱点として決定された欠陥が認められなかったため、2023年12月31日現在、財務報告の内部統制に有効であると結論した。
重大な欠陥は財務報告内部制御の欠陥或いは欠陥の組み合わせであるため、合理的な可能性があり、年度或いは中期合併財務諸表の重大な誤報は予防或いは適時に発見されない。
| 76 |
財務報告内部統制の変化 :
本報告がカバーする2023年12月31日までの四半期において、1934年の“証券取引法”(改正“証券取引法”)規則13 a-15(E)および15 d-15(E)(D)段落に基づいて要求される評価は、財務報告の内部統制にbr}が変化しておらず、これが我々の財務報告の内部統制に大きな影響を与えたり、大きな影響を与えたりする可能性が高い。
| 第 9 B項。 | その他 情報 |
ない。
| 第 9 C項. | 検査阻止に関する外国司法管轄区域を開示する |
ありません
第 第3部分
| 第 項10. | 役員·役員·会社管理 |
すべての役員の任期は1年で、彼らの後継者が当選して資格を得るまで。上級管理者は我々の取締役会 によって任命され,適用される雇用協定に基づいて取締役会が適宜決定する.次の表に私たちの上級管理職と取締役会のメンバーに関する情報を示します。
| 名前.名前 | 年ごろ | ポスト | ||
| エリック·エンダー | 55 | 取締役取締役会長 | ||
| ジェローム·D·ジャボル | 49 | 取締役CEOと社長 | ||
| ジェームズ·J·ファーガソン | 70 | 首席医療官 | ||
| トーマス·J·フーバー | 54 | 首席ビジネス官 | ||
| キース·クチンスキー | 54 | 最高財務官 | ||
| 劉慧 劉 | 56 | 首席技術官 | ||
| テレサ·マッテコヴィッツ | 56 | 首席開発官 | ||
| ハーバート·コンラッド | 91 | 役員.取締役 | ||
| キャサリン·コルゾ | 63 | 役員.取締役 | ||
| ナターシャ·ゾダンノ | 63 | 役員.取締役 | ||
| ジェームズ·S·シベルタ | 59 | 役員.取締役 | ||
| マシュービクター | 74 | 役員.取締役 |
管理する
ジェローム·ジャボルJD2018年3月にCEOに任命された。彼は2016年3月から私たちの総裁を務めている。これまでは、2013年10月から執行副総裁、首席商務官、総法律顧問兼秘書を務め、2012年4月から2013年11月までの間に取締役の一人を務めていた。ジャブルはMatinas BioPharmaの共同創始者でもある。私たちの管理チームに参加する前に、2012年から2013年10月までMediMedia USA執行副総裁兼総法律顧問を務め、同社は個人持株の多元化医療サービス会社であった。MediMediaに入社する前は、グローバル製薬·バイオテクノロジー会社ウォークハート(Wockhardt)有限会社のグローバル法務担当上級副社長(2008年から2012年)と、Reliant社の高級弁護士兼アシスタント秘書(2004年から2008年)を務めていた。brは、キャリアの初期にAlPharma,Inc.でビジネス法律顧問(2003年から2004年)、Lowenstein(Br)Sandler LLPで企業アシスタント(1999年から2003年)を務めていた。ジャブルさんはニュージャージー州のセトンホール大学法学部で法学博士号を取得し、ボルチモアのロヨラ大学で心理学の学士号を取得した。
| 77 |
ジェームズ·J·ファーガソン医学博士2019年2月に首席医療官に任命された。当社に入社する前に、2016年から2019年まで多国籍バイオ製薬会社安進(ナスダック:AMGN)で米国の医療事務心血管と骨治療地域担当を務めていた。安進に加入する前に、ファーガソン博士は多国籍製薬と生物製薬会社のアスリーカンで多くの高級職を務めており、アメリカ心血管医療と科学対外関係部副総裁、心血管グローバル医療事務部治療領域副総裁、ブリンタ米国発展ブランド責任者を含む®アスリーカンに入社する前は、製薬会社の外科·集中治療部の総裁副主任だった。そのほか、ファーガソン博士は20年以上の学術経験を持ち、テキサス心臓研究所臨床心臓病学研究役員副研究員、ヒューストンサンルーク聖公会病院心臓病学奨学金訓練プロジェクトの連合取締役br、ベラー医学院医学助教授とヒューストンテキサス大学健康科学センター臨床アシスタント教授を務めたことがある。ファーガソン博士はハーバード大学で生物学学士号(優秀な成績で卒業)を取得し、ペンシルバニア大学医学院で医学博士号を取得し、ミシガン大学医学センター、ミシガン州アナベルク、マサチューセッツ州ボストンベースイスラエル病院で大学院生訓練を完了した。
トーマス·J·フーバーMBA2021年12月から首席商務官を務めてきた。当社に入社する前に、フーバーさんは2016年から2021年にかけてミレンドラ治療会社(現在は嵐治療会社(ナスダック·コード:TPST))のチーフ商務官を務めていました。br}はミレンドーに加入する前に、フーバーさんは世界的にバイオ製薬会社Sunovion製薬会社の新製品計画と企業開発許可部の社長副社長でした。胡さんは2001年にグラクソ·スミスクラインで彼の製薬業のキャリアを開始し、世界的なビジネス戦略グループで働いた。彼のキャリアの初期、胡仏はボストンコンサルティンググループで働いていた。フーバーさんは、ノースカロライナ大学の商工管理修士号、ハーバード大学の学士号を持っている。
ケス·クチンスキーMBA公認会計士2019年1月に首席財務官に任命された。彼は最近2018年に民間医療相談機関RemedyOneの首席財務官 を務めた。これまで,2009年から2015年までPAR製薬会社で副総裁兼財務担当を務めており,PAR製薬会社は有力な後発薬と特殊ブランド製薬会社Endo International plcの運営会社であった。また、クシンスキーさんは、バル製薬会社において、取締役財務·企業開発上級主管および取締役財務アシスタント兼上級財務担当者を含む複数の職務を歴任しています。クシンスキーさんは公認会計士です。彼は聖母大学から工商管理会計学士号を取得し、ニューヨーク大学レナード·N·ストーン商学院から金融·管理工商管理修士号を取得した。
劉慧、博士、工商管理修士2020年12月以来首席技術官を務めてきた。当社に入社する前に、劉博士は2017年から2020年まで個人持株のグローバルインフルエンザと大流行対応分野のリーダーSeqirus USA Inc.が取締役br製剤と交付担当者を務めた。br}はSeqirusに加入する前に、2017年にプライベート持株の発展段階でバイオ製薬会社Cellics Treeutics,Inc.でCMCの取締役を務め、2015年から2017年までグローバル眼科ケアリーダーのエルコン社(Six/NYSE:ALC)で高級技術担当を務めた。彼のキャリアの初期、劉博士は個人持株のバイオテクノロジー会社Cellics Treeutics、Inc.とAllerganで職務を担当したことがある。劉博士はミシガン大学のポリマー化学博士号、マサチューセッツ大学アーマースター校の工商管理修士号、中国科学技術大学の理科学士号を持っている。
Theresa Matkovits博士2018年10月から首席開発官を務めてきた。彼女は当社に入社する前、2015年から2018年まで臨床段階バイオ製薬会社の康輝製薬(ナスダック:CTRV)(現和鵬製薬)の首席運営官を務めた。2013年から2015年まで、Matkovits博士はNPS PharmPharmticalsのグローバルプロジェクト担当者を務め、NPS PharmPharmticalsは専門製薬会社であり、2015年にShireに買収された。NPSに加入する前、マット·コヴィッツ博士は薬品会社革新主管総裁副主任だった。彼女のキャリアの初期に、Matkovits博士はノ華全世界開発部とアメリカ商業組織で複数のグローバル指導職を務めたことがあり、アメリカ医療と薬品監督事務部の戦略計画と運営主管を担当したことがある。Matkovits博士は羅氏分子生物学と器官研究所で彼女のキャリアを開始し、そこで彼女は女性の健康と不妊分野の研究で臨床開発職を務めた。マテコヴィッツ博士はAppili Treeutics(トロント証券取引所コード:APLI;OTCQX:APLIF)の取締役会メンバーであり、非上場製薬会社GoodCap PharmPharmticalsの会長でもある。Matkovits博士はニュージャージー州医学と歯科大学で生化学と分子生物学博士号を取得した。
| 78 |
役員.取締役
エリック·エンダー2017年4月から私たちの取締役会に勤めており、2022年10月にわが社の取締役会長に選ばれました。エンダー博士はエンダー生物医学コンサルティンググループの総裁であり、個人持株のコンサルティング会社であり、生命科学会社の資金調達を支援し、許可パートナーを探し、会社の構造を最適化し、生命科学業界の顧客のためにプライベートと公共投資機会を分析することに集中しており、彼は2009年以来この職を務めている。また、エンダー博士は、森林実験室、遺伝子工学会社、生物遺伝研究会社、アミリン社の取締役会メンバーを任命する過程でイカン企業会社に相談した。エンダー博士は2010年から遺伝子工学会社(ナスダックコード:GENZ)の取締役会や監査·リスク管理委員会のメンバーを務め、2011年までセノフィ社に買収された。別の次元権活動を通じて、エンダー博士は2019年から腫瘍会社Progenics PharmPharmticals,Inc.の取締役会メンバーを務め、2020年までLantheus Holdings,Inc.によって買収され、報酬委員会の議長と監査委員会と科学委員会のメンバーを務めた。エンダー博士はバイオ製薬会社Avadel製薬会社の取締役会のメンバーも務め、指名と会社管理委員会の議長、監査と報酬委員会のメンバーを務めている。エンダー博士は現在、西奈山革新パートナー会社の技術移転委員会に勤め、Egenix,Inc.破産を監督する無担保債権者委員会の議長を務めている。2002年から2008年まで、エンダー博士は美林の高級バイオテクノロジーアナリストである。2000年から2002年まで,エンダー博士は米国銀行証券の高級バイオテクノロジーアナリストであり,1997年から2000年までリーマン兄弟のバイオテクノロジーアナリストであった。Ende博士は1997年にニューヨーク大学ステインビジネススクール金融と会計工商管理修士号を取得し、1994年にシナイ山医学院医学博士号を取得し、1990年にエマレー大学生物学と心理学学士号を取得した。私たちは恩徳博士が私たちの取締役会に勤めている資格があると信じています。彼はbr業界の経験があり、恩徳生物医学コンサルティンググループの総裁と生物技術アナリストを務め、彼が以前公開上場した会社の取締役会経験を含むからです。
ジェローム·D·ジャブルです“管理”の下の説明を参照されたい
ハーバート·コンラッド2013年7月から2022年10月まで取締役会長を務め、2012年10月から2022年10月までMatinas BioPharma,Inc.の取締役会長を務めた。彼は今日私たちの取締役会のメンバーを続けている。生物製薬会社Celldex Treateutics,Inc.(ナスダックコード:CLDX)の取締役も務め,免疫療法や他の標的生物製品の開発と商業化に専念し,西奈山病院シーバー自閉症センターの顧問を務めている。コンラッド·さんは、1982年から1993年まで、米国の製薬事業部の社長を務めていた。これまで,米国の羅氏製薬会社で複数の役割が増加してきた職を務めてきた。Conradさんは、Arbutus Biophma Corporation(ナスダック·コード:ABUS)、PharmAsset,Inc.(代表取締役社長)、Savient PharmPharmticals,Inc.(ナスダック: SVNT)、デュラ製薬会社、UroCor,Inc.,GenVec,Inc.(代表取締役社長)、SICOR,Inc.,bone Care International,Inc.(代表取締役社長)、サファイア治療会社(代表取締役社長)、Henry Schein Inc.(ナスダック:HSIC)の医療コンサルタント委員会に在籍しており、彼は取締役の一員であり、Relant製薬会社の共同創業者でもある。2011年、PharmAssetはGilead Sciences、Inc.は110億ドルで買収され、Repantは2007年にグラクソ·スミスクラインに16.5億ドルで買収された。彼はブルックリン薬学部から学士と修士号を取得し、長島大学から人道主義文学の栄誉博士号を取得した。Conradさんは、生命科学分野で豊富な専門知識と経験を有し、取締役会経験が豊富であるため、当社の取締役会に勤務する資格があると信じています。
キャサリン·コルゾ2021年9月から取締役会に勤務しており、現在はbit.bioの首席運営官である。Bit.bioに加入する前,Corzoさんは武田製薬有限会社の企業投資部門武田ベンチャー会社のパートナーであり,これまではグローバルバイオ製薬会社武田製薬有限会社(東京証券取引所コード:4502/ニューヨーク証券取引所コード:TAK)腫瘍学細胞療法開発担当者であり,2020年からこの職を務めてきた。武田に加入する前に、Corzoさんは2010年から2019年までの間にセノフィ(Sanofi)のグローバル特殊介護業務部門Genzymeでますます多くの責任職を務めてきた。セノフィに加入する前に、Corzoさんは1989-2010年の間にホフマン-羅氏、羅氏分子システム、礼来会社とSyndaxで仕事をし、その間、彼女は多種の治療性製品と適応の運営、全世界の臨床開発、医療事務、業務開発、市場参入とブランド管理における経歴は絶えず向上した。私たちはCorzoさんが生命科学分野で豊富な経験を持っているので、彼女は私たちの取締役会のメンバーを務める資格があると信じている。
ナターシャ·ゾダンノそれは.ゾダンノさんは2020年9月から私たちの取締役会のメンバーを務めてきた。佐丹奴さんは2016年1月から取締役(ナスダックコード:PLXP)の最高経営責任者兼CEOを務めている。これまで、ゾダンノさんは2015年5月から2015年11月までの間に個人持株の学習·訓練プラットフォーム会社ClearPoint Learning,Inc.の最高経営責任者を務めていた。彼女は2009年12月から2015年11月までClearPoint取締役会のメンバーを務めている。これまで、佐丹奴さんは2014年1月から2014年8月までリードするヘルスケアプロバイダーの米国ヘルスケア会社(NYSE:HCA)の最高経営責任者を務めていた。2009年6月から2012年8月まで、佐丹奴さんはXanodyne PharmPharmticals,Inc.の最高経営責任者、CEO、取締役会のメンバーを相次いで務めてきた。Xanodyne PharmPharmticals,Inc.は個人持株のブランド専門製薬会社であり、痛み管理と女性の健康管理に専念する開発と商業能力を持っている。これまで、彼女は2007年から2008年まで全世界技術サービス会社Cegedim Dendite(前身はDendite International Inc.)でアメリカ区総裁を務め、2004年から2007年までCegedim Denditeグローバル顧客業務部高級副総裁 を務めた。ゾダンノさんはワグナー学院看護学学士号を持っている。私たちはゾダンノさんが取締役を務める資格があると信じています。商業化の経験が豊富で、一般的な管理や製薬や医療業界への理解があるからです。
| 79 |
ジェームズ·S·シベルタ2013年11月から私たちの取締役会のメンバーを務めてきた。シベッタは現在Rock Springs Capitalの上級コンサルタントであり,長期的に偏った医療に集中した公共株基金を管理している投資会社である。ヒベルタさんは、2021年から2023年までの間、免疫ID企業のプライベート·ホールディングスのCEOですこのbrは既存の抗体反応を用いて免疫系の複雑さを迅速に明らかにし,brの正確で変革的な治療への経路を明らかにする。免疫IDの前に2017年から2021年にかけてMaverick Treeuticsの最高経営責任者であり、発展段階の免疫腫瘍学会社であり、後に武田薬業有限会社に買収された。Maverickに加入する前に、専門製薬会社Pacira PharmPharmticals,Inc.(ナスダックコード:PCRX)の首席財務官兼首席財務官であり、2015年10月以来この職を務めてきた。これまで、西ベータさんは2008年以降Paciraの首席財務官を務めてきた。2008年8月にPaciraに加入する前に、2007年にBioenvision Inc.(ナスダックコード:BIVN)をGenzymeに売却した後、Genzyme Corporationの顧問を務めた。2006年から2007年まで、ゼータさんはBioenVisionの首席財務官を務めた。2001年から2006年まで、彼はナスダック:MACK薬業会社の執行副総裁兼首席財務官を務めた。ヒベルタさんは、以下の生命科学会社の取締役会に勤務していた:Nephros Inc.(ナスダック·コード:NEPE)、Merrimack PharmPharmticals、Labopharm Inc.。管理経験を得る前に、ヒベルタさんは、投資銀行で10年以上働いており、広範な公共および民間の医療保健·生命科学会社の調達および取引を担当していた。セベータ·さんは、フィンケ森林大学の物理学士号、ミシガン大学の工商管理修士号を取得しています。私たちは、製薬業界で豊富な経営経験、彼の投資銀行の経験、およびいくつかの上場企業で首席財務官や監査委員会のメンバーとして働いている経験があるので、ゼータさんが私たちの取締役会に勤めている資格があると信じています。
マシューA.ビクター2018年1月から私たちの取締役会のメンバーを務めてきました。Wikler博士は現在私営コンサルティング会社の伝染病技術発展コンサルティング会社(IDTD Consulting)の責任者を務め、伝染病の治療と予防の新技術を開発する会社に臨床、医療と監督管理戦略見解を提供し、2015年以来ずっとこの職を担当している。これに先立ち、2012年から2015年にかけて、Wikler博士は生物製薬会社The Medicines Company(ナスダックコード:MDCO)に在任し、新業務リスク投資副総裁と医療取締役感染症看護副総裁を務めた。彼のキャリアの中で、Wikler博士は複数の製薬会社で、RIB-X製薬会社の首席開発官、br}Inc.、個人持株の生物製薬会社、新しい抗生物質を開発し、生命に危害を及ぼす感染の治療により大きなカバー範囲を提供し、安全で便利なbr},社長とIASO Pharma Inc.の最高経営責任者、IASO Pharma Inc.,抗菌と抗真菌療法の開発に専念する個人持株の臨床段階バイオテクノロジー会社、One World Health研究所、501(C)(3)非営利薬物開発組織、Mpex製薬会社、Mpex製薬会社の高級指導者を務めた。それらのパートナーには、グラム陰性生物に対する抗生物質耐性療法の研究開発と生産に取り組む民間会社半島製薬会社、生命にかかわる感染を治療する抗生物質の研究開発および商業化に専念する私営生物製薬会社半島製薬会社(買収側はジョンソン(ニューヨーク証券取引所コード:JNJ))、ナスダック(ニューヨーク証券取引所コード:VPHM)、百時美施貴宝社(ニューヨーク証券取引所コード:BMY)、正泰製薬会社(ジョンソン傘下子会社)がある。Wikler博士はSmith Kline&French/Smith Kline Beechamで彼のキャリアを始め、そこで彼は10年間ますます責任が大きくなってきた職を務めた。ビクター博士はFDAで様々な職務を担当しており、取締役抗感染薬物製品部の副総監を含む。Wikler博士はフランクリンとマーシャルカレッジで化学学士号を取得し、タンプル大学医学院で医学博士号を取得し、ペンシルバニア大学ウォートン商学院でMBA学位を取得した。彼はペンシルバニア大学病院で伝染病奨学金を修了し、アメリカ伝染病学会の会員である。Wikler博士は製薬業界で豊富な管理経験と、彼の臨床、薬物開発と監督経験を持っているため、私たちの取締役会に在任する資格があると信じている。
私たちの取締役や役員の間には家族関係はありません。
| 80 |
科学顧問委員会
私たちbrは伝染病領域の科学と臨床指導者を求め、誘致することが私たちの発展に提案と支持を提供すると信じている。私たちはMAT 2203のために彼らが選択した分野の専門家と多くの学術栄誉と賞の受賞者からなる科学顧問委員会を設立した。
取締役会 委員会
私たちの取締役会には三つの常設委員会-監査委員会、報酬委員会、指名、そして会社管理委員会があります。
監査委員会 それは.監査委員会は、我々の財務報告の流れ及び内部制御システムを監督·監視し、我々の公認会計士による監査を審査·評価し、監査過程で発見された任意の実質的な問題を取締役会に報告する。監査委員会は、私たちが登録した独立した公共会計士の任命、報酬、仕事の監督を直接担当します。監査委員会は関連者とのすべての取引を検討して承認する。ジェームズ·シベッタ、ハーバート·コンラッド、ナターシャ·ゾダンノは現在監査委員会のメンバーを務め、シベータが議長を務めている。監査委員会のすべてのメンバーは、財務に精通していると決定され、ニューヨーク証券取引所の上場基準および適用される米国証券取引委員会規則および法規によって定義された独立取締役とみなされている。ゼータさんは、“米国証券取引委員会”がこの言葉の定義を定めているため、監査委員会の“財務の専門家”になる資格を持っている。監査委員会は2023年の間に4回の会議を開催した。私たちの取締役会は監査委員会の規定を通過しました。以下のサイトで見ることができますWww.matinnasbibiharma.com.
報酬委員会 それは.報酬委員会は、従業員の報酬、福祉計画、役員報酬などの分野について取締役会に提案と提案を提供する。報酬委員会はまた、私たちの最高経営責任者の報酬を含めて私たちの役員を審査し、取締役会全体に提案します。キャサリン·コルゾ、ジェームズ·シベタ、マシュー·ビクターが現在報酬委員会のメンバーを務め、シベタが議長を務めている。報酬委員会のすべてのメンバーは、ニューヨーク証券取引所MKT上場基準によって定義された独立取締役とみなされている。給与委員会は2023年の間に6回の会議を開催した。私たちの取締役会は報酬委員会の規約を採択しましたWww.matinnasbibiharma.com.
Brとコーポレートガバナンス委員会を指名する.私たちの株主によって選出された個人を指名して会社管理委員会に指名して取締役会に入ります。指名·コーポレートガバナンス委員会は、我々の定款に規定された手順に基づいて、株主の提案 を考慮し、考慮されたすべての人に同じ基準 を適用する。ハーバート·コンラッド、キャサリン·コルゾー、エリック·エンデ、ナターシャ?ゾダンノは現在、指名と会社統治委員会のメンバーで、ゾダンノさんが議長を務めている。ニューヨーク証券取引所MKTの上場基準によると、指名と会社管理委員会のすべてのメンバーは独立取締役とされている。指名と会社統治委員会は2023年の間に4回の会議を開催した。当社の取締役会はすでに指名及び企業管理約章を通過しており,この約章はWww.matinnasbibiharma.com.
ビジネス行為と道徳的基準
私たちは、私たちのCEO、最高財務責任者、会計官、または同様の機能を実行する人を含む、私たちの役員、高級管理者、および従業員に適用される書面商業行為および道徳基準を通過しました。コードコピーは弊社サイトのコーポレート·ガバナンス部分に掲示されており、サイトはwww.matinasbibiharma.comです。もし私たちが任意の上級管理者または役員の商業行為および道徳規範に対して任意の実質的な改正または免除を行う場合、私たちのウェブサイトでこのような改正または免除の性質を開示します。
| 81 |
インサイダー取引政策
我々は,連邦証券の法律,規則と法規およびニューヨーク証券取引所の規則と法規の遵守を促進するためのインサイダー取引政策(“取引政策”)を採用した。取引政策は,機密情報を持っている場合の取引と,我々の証券や他の上場会社の証券の取引を招くMatinasの基準を規定している.それは、場合によっては取引を禁止し、私たちのすべての役員、高級管理者、および従業員、ならびにMaatinasの重要な非公開情報にアクセスできる独立請負業者またはコンサルタントに適用される。また、私たちの取引政策 は私たちのすべての役員と役員に特別な追加取引制限を加えました。貿易政策 は添付ファイルとして本年度報告に添付されている。
| 第 項11. | 役員報酬 |
概要 給与表-2023
次の表には、2023年12月31日現在、最高経営責任者と最高報酬の2人の役員が、様々な身分で提供されているサービスによって獲得、獲得、支払いされた総報酬情報を示しています。この人たちは私たちが2023年に任命された執行官たちだ。
| 名称と主要ポスト | 年.年 | 賃金.賃金 ($) | ボーナス.ボーナス ($) | オプション大賞 ($) (1) | 他のすべての補償(ドル) | 合計する ($) | ||||||||||||||||||
| ジェローム·D·ジャブル | 2023 | 598,000 | 244,375 | 757,176 | - | 1,599,551 | ||||||||||||||||||
| 最高経営責任者 | 2022 | 575,000 | 306,500 | 792,707 | - | 1,674,207 | ||||||||||||||||||
| ジェームズ·J·ファーガソン | 2023 | 468,000 | 153,000 | 259,603 | - | 880,603 | ||||||||||||||||||
| 首席医療官 | 2022 | 450,000 | 169,920 | 249,179 | - | 869,099 | ||||||||||||||||||
| テレサ·マッテコヴィッツ | 2023 | 430,000 | 140,080 | 281,237 | - | 851,317 | ||||||||||||||||||
| 首席発展官 | 2022 | 412,000 | 189,000 | 249,179 | - | 850,179 | ||||||||||||||||||
| (1) | 金額 は、2023年と2022年に会計基準コードテーマ718によって付与されたオプション報酬の付与日公正価値を反映している。これらの額は、指定実行幹事が確認する実際の価値と一致しない。 |
Narrative 報酬集計表開示
私たちが指定した役員と雇用協定を締結します
ジャボル
2018年3月22日、我々はJabbourさんと雇用契約を締結しましたが、この協定は2023年3月3日に改訂されました。Jabbourさんの雇用契約条項によると、Jabbourさんは84,000ドルの契約ボーナスと年間350,000ドルの基本給を取得しました。 Jabbourさんの現在の給与は598,000ドル。さらに、Jabbourさんは、年間ボーナスを取得する資格があります。目標は、基本給の50% ですが、私たちの報酬委員会は、彼の個人的なパフォーマンスと、私たちの全体的なパフォーマンス とに基づいて調整することができます。Jabbourさんは、私たちの報酬委員会が適宜決定するオプションを獲得する資格があります。Jabbourさんは、私たちの報酬委員会によって適宜決定されるオプションの贈与と持分の贈与を受ける資格があります。もしわれわれが理由なくJabbourさんの雇用を打ち切るか、あるいはJabbourさんが理由なく辞任する(支配権の変更はない)場合、我々は彼にその基本給のために最大12ヶ月の解散費と、12ヶ月のコブラの福利厚生と、彼が終了日までに比例して計算した年間目標年次ボーナスを支払う必要がある。 また、12月31日までに支給される未償還オプションの50%が付与される、終了後、2021年には完全に加速されます ,Jabbourさんは、その既得株式オプション行の権利分離日の後2年に延長されます。もし我々が支配権変更後24か月以内に理由なくJabbourさんの雇用を中止したり,あるいはJabbourさんがコントロール権変更後24か月以内に十分な理由で辞任した場合は,我々は彼に18か月の基本給の解散費と1.5倍の目標年度ボーナスと18か月のコブラ福祉を支払うよう要求されるであろう。さらに、Jabbourさんの未償還オプションがすべて付与されることになり、Jabbourさんは、その既得株式オプション行の権利期間の分離日 を取得した後、2年延期することになります。Jabbourさんはまた、通例の守秘協定を遵守しなければならない。この合意に基づき、Jabbourさんは、その在任中と雇用終了後の18ヶ月以内に、eスポーツ禁止条項を遵守することに同意した。
| 82 |
ファーガソン
2019年2月22日、我々はFergusonさんと2019年2月25日に施行される雇用契約を締結し、その後、2023年3月3日に改訂されました。ファーガソンの雇用契約条項によると、ファーガソンの基本給は年間375,000ドル、現在は468,000ドルである。さらに、ファーガソンさんは、基本賃金の35%を目標とする年間ボーナスを得る資格がありますが、当社の報酬委員会は、彼の個人的なパフォーマンスと私たちの全体的なパフォーマンスに応じて調整する可能性があります。ファーガソンさんは、私たちの報酬委員会が適宜付与するオプションを得る資格があります。もし私たちが理由なくファーガソンさんの雇用を中止したり、ファーガソンさんが正当な理由で辞任したりすれば、我々は彼に最大12ヶ月の基本給の解散費と福祉を支払う必要があります。また,2021年12月31日までに発行された50%未償還オプションの帰属は終了後に全面的に加速される.もし我々がコントロール権の変更直後の12か月以内に理由なくFergusonさんへの雇用を打ち切ったり,あるいはJabbourさんがコントロール権の変更に続いて12か月以内に辞任する十分な理由があれば,我々は彼に12か月の基本給と目標年次ボーナスの解散費と12か月のコブラの福利を支払わなければならない。また, 彼の未返済オプションはすべて付与される.ファーガソンさんはまた、通例の守秘契約に拘束されており、この合意によると、ファーガソンさんは、雇用期間中および終了後18ヶ月以内に、eスポーツ禁止条項を遵守することに同意している。
マット·コヴィッツ
2018年9月25日、2018年10月15日に施行され、2023年3月3日に改訂されたMatkovitsさんと雇用協定を締結しました。Matkovitsさんの雇用協定条項によると、Matkovitsさんの基本給は年間35万ドルで、現在43万ドルだ。しかも、Matkovitsさんは年間ボーナスを得る資格があり、ボーナスの目標は彼女の基本給の35%だが、私たちの給与委員会は彼女の個人的な表現と私たちの全体的な表現に基づいて調整されるかもしれない。 Matkovitsさんは私たちの給与委員会が適宜決定したオプション付与を得る資格がある。もし私たちがMatkovitsさんの雇用を理由なく終わらせたり、Matkovitsさんが辞任する十分な理由があれば、私たちは彼女の最大12ヶ月の基本給と福祉の解散費を支払うことを要求されるだろう。また、2021年12月31日までに発行された50%未償還オプションの帰属は終了後にすべて加速される。もし私たちが直ちにコントロール権変更後12ヶ月の間にMatkovitsさんの雇用を理由なく中止したり、Matkovitsがコントロール権変更に続く12ヶ月の間に十分な理由で辞任した場合、私たちは彼女の12ヶ月の基本給の解散費と彼女の目標年間ボーナスに12ヶ月のコブラ福祉を支払うことを要求されます。しかも、彼女の未返済オプションはすべて付与されるだろう。Matkovitsさんはまた慣例的な秘密保持協定を守らなければならない。この協定によると、Matkovitsさんはその在任中と雇用終了後の18ヶ月以内に競業禁止条項を遵守することに同意した。
未償還のbr 2023年度年末表における持分奨励
次表は,任命された幹部1人あたり2023年12月31日までに保有する普通株関連流通株 オプションの数をまとめたものである。
| オプション大賞 | ||||||||||||||||
| 名前.名前 | 量 証券 潜在的な 体を鍛えていない オプション(#) 練習可能である | 量 証券 潜在的な 体を鍛えていない オプション(#) 行使できない | 選択権 トレーニングをする 値段(ドル) | 選択権 満期になる 日取り | ||||||||||||
| ジェローム·D·ジャブル | - | 3,500,000 | $ | 0.25 | 2033年12月14日 | |||||||||||
| 538,432 | 1,449,868 | $ | 0.53 | 2032年12月19日 | ||||||||||||
| 863,558 | 794,542 | $ | 0.92 | 2031年12月13日 | ||||||||||||
| 1,166,667 | 433,333 | $ | 1.36 | 2030年12月31日 | ||||||||||||
| 979,167 | 20,833 | $ | 2.27 | 2029年12月31日 | ||||||||||||
| 750,000 | - | $ | 1.08 | 2029年2月10日 | ||||||||||||
| 1,000,000 | - | $ | 0.98 | 2028年3月21日 | ||||||||||||
| 400,000 | - | $ | 3.32 | 2027年2月20日 | ||||||||||||
| 350,000 | - | $ | 0.43 | 2026年2月4日 | ||||||||||||
| 175,000 | - | $ | 0.41 | 2025年1月27日 | ||||||||||||
| 350,000 | - | $ | 1.28 | 2024年7月20日 | ||||||||||||
| ジェームズ·J·ファーガソン | - | 1,200,000 | $ | 0.25 | 2033年12月14日 | |||||||||||
| 169,250 | 455,750 | $ | 0.53 | 2032年12月19日 | ||||||||||||
| 306,029 | 281,571 | $ | 0.92 | 2031年12月13日 | ||||||||||||
| 419,271 | 155,729 | $ | 1.36 | 2030年12月31日 | ||||||||||||
| 489,584 | 10,416 | $ | 2.27 | 2029年12月31日 | ||||||||||||
| 350,000 | - | $ | 1.09 | 2029年2月24日 | ||||||||||||
| テレサ·マッテコヴィッツ | - | 1,300,000 | $ | 0.25 | 2033年12月14日 | |||||||||||
| 169,250 | 455,750 | $ | 0.53 | 2032年12月19日 | ||||||||||||
| 260,406 | 239,594 | $ | 0.92 | 2031年12月13日 | ||||||||||||
| 309,896 | 115,104 | $ | 1.36 | 2030年12月31日 | ||||||||||||
| 342,709 | 7,291 | $ | 2.27 | 2029年12月31日 | ||||||||||||
| 350,000 | - | $ | 1.08 | 2029年2月10日 | ||||||||||||
| 350,000 | - | $ | 0.79 | 2028年10月14日 | ||||||||||||
| 83 |
2013年株式報酬計画
一般情報
2013年8月2日、当社取締役会は、本稿で述べた条項に基づいて2013年株式報酬計画を採択した。2013年の株式報酬は2013年8月7日に株主の承認を得る計画だ。2014年5月8日から、取締役会と株主の許可を経て、著者らは2013年の株式補償計画に対して改訂と再記述を行い、主に“常青樹”条項に加入し、この条項の規定は2015年から、この計画によって発行可能な普通株式数は毎年1月1日に自動的に増加し、前の例年12月31日に発行された普通株式数の4%または取締役会が決定したより少ない数の普通株に相当する;“公允 時価”の定義を修正する。この計画の奨励制限を向上させます改訂·再記述された2013年株式補償計画 は、本稿では“2013計画”と呼ばれる
2013年計画の全体的な目的は、私たちの業務の将来の成長を共有できるように、私たちの従業員、役員、コンサルタント、コンサルタントを激励することです。私たちの取締役会は、株式オプション、制限株式奨励、br}非制限株式奨励、および同様の株式ベースの報酬が管理層の連続性を促進し、インセンティブとbrを増加させることが、主に私たちの長期計画の策定と実行、私たちの成長と財務成功を確保する人員の会社福祉に対する個人的利益を保証する責任があると信じている。
私たちの取締役会は、2013年計画は、(A)私たちの成功に大きな貢献をすることができる従業員、コンサルタント、取締役、コンサルタントを誘致し、維持すること、(B)これらの貢献をした従業員、コンサルタント、取締役、コンサルタントを奨励すること、および(C)私たちの株式を保有することによって、従業員、コンサルタント、取締役、コンサルタントが私たちの長期的な利益を考慮することを奨励することを、私たちの能力を強化することで促進すると信じている。
2013年度株式報酬計画の説明
以下の“2013年計画”の主な条項の説明は要約であり、全文は“2013年計画”の全文に制限されており、全文は本文書の添付ファイル10.1参照。
| 84 |
行政管理2013年計画は、取締役会全体が報酬委員会の代わりに行動できることを前提としていますが、2013年計画で提出されたいくつかの要求を遵守しなければなりません。給与委員会は、私たちの普通株、株式付加価値権、株式単位、私たちのbr}普通株を購入する制限的な株、業績株、業績単位、インセンティブボーナス、その他の現金奨励、その他の株式奨励のオプションを付与することができます。報酬委員会はまた、各オプションまたは他のタイプの報酬の条項および条件を決定し、2013年計画の管理規則および条例を改正、廃止するための幅広い権力を持っている。適用される法律に適合する場合、報酬委員会は、2013年計画で定義されたように、1人以上の通報者または他の役人に報酬を付与することができる(通報者または報酬委員会に特別に報酬を付与する他の官僚を除く)。当社の取締役会が“2013計画”を採択した10周年の当日または後には、“2013計画”に基づいていかなる奨励も付与されてはなりませんが、この10周年まで授与された奨励は、その日の後まで継続することができます。
資格2013年度計画によると、報酬は、当社または任意の子会社である従業員、上級管理者、取締役、コンサルタント、コンサルタントまたは他の個人サービスプロバイダの任意の個人、または報酬委員会が、当社または任意の子会社の将来の従業員、高度管理者、取締役、コンサルタント、コンサルタントまたは他の個人サービスプロバイダとして認定された任意の個人に付与することができる。
2013年計画に制約された株 それは.2024年3月18日現在、2013年計画により付与された奨励により、発行可能な普通株式総数は62,984,400株であり、株式分割、株式配当又はbrのような取引の慣例調整(“初期限度額”)を受けている。2013年計画によると、これらすべての 株に対して奨励的株式オプションを付与することができる。2013年の計画によると、発行可能な普通株式数は毎年1月1日に自動的に増加し、10年間、2015年1月1日から前年の12月31日に発行された普通株式総数の4%(4%)に相当する(“年度増加”)。上記の規定にもかかわらず、取締役会は、その例年の株式積立金が増加してはならないこと、または当該例年の普通株式積立金の年間増額が、前の文に基づいて発生すべき普通株式数よりも少なくなければならないことを規定している。株式オプション発行を奨励する普通株式数は現在の限度額に等しく、毎年1月1日にこのカレンダー年度の に従って増加することができる。
2013年計画に基づいて普通株式で支払われた任意の報酬が没収され、キャンセルされ、帰属要求を満たしていない、または他の没収事件が発生したために当社を返却する場合、またはそれ以外の場合に終了し、その支払いに基づいていない場合、カバーされた普通株式は、2013年計画下の将来の付与に使用することができる。普通株 は、株式オプションを行使する際に発行されるべきであるか、または任意の他の形態の奨励金を支払う際に発行されるべきであり、 は、その株式オプションの行使またはそのような支払いを行うために源泉徴収された税金が必要な場合に提出された普通株式を支払いまたは部分的に支払う場合には、2013年計画下の将来の付与にも使用することができる。
オプション条項と条件 それは.2013年計画に基づいて付与されたオプションは、改正された1986年の“国税法”(以下、“規則”)422節の要求を満たすための“奨励的株式オプション”であってもよいし、この守則422節の要求に適合しない“不適格株式オプション”であってもよい。給与委員会は2013年計画に基づいて付与されたオプションの行使価格を決定するだろう。株式オプションの発行価格は、付与された日に私たちがオプションを行使して発行可能な普通株式1株当たりの公正時価を下回ってはならない(10%株主に付与された奨励的オプションであれば、公正時価の110%を下回らない)。
普通株が付与された日に証券取引所または国家市場システムに上場する場合、市場価値を開示することは、通常、その日までの終値であるか、またはその日に取引記録がない場合は、その取引記録の前の日である。付与された日に普通株が場外取引市場で取引される場合、公平市場 は、通常、その日までの普通株株の終値と要件の平均値、または、その日に普通株の終値および要件がなければ、その終値および要件を得る日までの直近の日の普通株の入札および要件の平均値である。普通株が国家証券取引所または国家市場システムで上場していない場合、または場外取引市場で取引されていない場合、公平な市場価値は、改正された1986年の国内税法第409 a条の方法で報酬委員会によって決定されなければならない。前述の規定にもかかわらず、普通株が付与された日に証券取引所に上場するか、または全国市場システムでオファーされる場合、または場外取引市場で取引される場合、そして、賠償委員会は、株式オプション付与の行使価格又は株式付加権付与の基準価格のみを決定するために、付与前又は付与後の最後の売却、付与前の取引日又は付与後の取引日の終値、前の取引日又は付与された取引日の高値及び低価格の算術平均値に応じて、又は普通株取引所が取引所又は市場で報告された普通株の実際の取引状況の任意の他の合理的な方法を使用することができる。また、大蔵省条例第1.409 A-1(B)(5)(Iv)節で許可された他の任意の方法を用いて公平な市場価値を決定することもできる。
| 85 |
付与された日から10年以内には何のオプションも行使できない(10%株主の奨励的株式オプションを付与する場合は5年)。2013年計画に基づいて付与されたオプションは、付与時報酬委員会が規定した時間または複数の時間に行使される。どの従業員も毎年、初めて行使可能な奨励的株式オプションを得ることができず、金額は100,000ドルを超えている。報酬委員会は、非適格株式オプション所有者が、そのオプションが他の方法で行使可能になる前にオプションを行使することを適宜許可することができ、この場合、受給者に発行された普通株式は、行使前にそのオプションの帰属要求に適用される制約を受け続ける。
一般に、オプション価格は、現金、銀行小切手、または報酬委員会が受け入れる可能性のある他の方法で支払うことができる。奨励協議に記載されたまたは補償委員会が他の方法で決定したように、付与時または付与後に、付与時または後に普通株式の形態でオプションの一部を全額支払いまたは支払いすることができる部分行使価格:(A)参加者が保有する普通株式の形態 で、補償委員会が会計または他の目的に適していると考えられている間は、その株の公正な市場価値推定値を行使し、(Ii)オプションを行使する際に受信すべき普通株を会社に提出する。(Iii)2013年計画に対して報酬委員会によって実施されたキャッシュレス行使計画;および/または(Iv) は、報酬委員会によって承認され、認可プロトコルに規定された他の方法。
遺言または相続·分配法を除いて、いかなる選択権も譲渡してはならず、受給者が生きている間に、選択権は受給者または受給者の保護者または法定代表者がしか行使できない。しかしながら、補償委員会は、制限されていない株式オプション、株式で決済された株式付加価値権、制限された株式報酬、業績株または株式で決済された他の株式ベースの奨励を許可することができ、(A)2013年計画で定義されたように、(A)文書を介して参加者の直系親族に譲渡することができ、(B)投資者または遺言信託(または他のエンティティ)に文書を介して譲渡することができ、報酬は参加者の指定された受益者に伝達されるか、または(C)慈善団体に贈与されることができる。報酬委員会は、株式オプション所有者がサービス終了後にどの程度オプションを行使できるかを決定するだろう。
株増価権利それは.報酬委員会は、オプションまたはオプションとは独立した株式付加価値権を付与することができる。報酬委員会は、株式付加価値権に適用される条項を決定する。株式付加価値権の基本価格は補償委員会によって決定されるが、その株式付加権を付与した日の普通株の公正時価の100%を下回らない。2013年計画により付与された任意の特別行政区の最長期限は、授与日から10年 である。一般的に、香港特別行政区の株式付加価値権は、参加者が行使時に以下の金額に相当する金額を獲得する権利を持つことになる
| ● | 株式付加価値権行使日普通株の公允時価が当該株式付加価値基準価格を超える に乗じる |
| ● | 株式付加価値権を行使する株式数。 |
支払いは、私たちの普通株の株であってもよいし、現金であってもよいし、一部は普通株であってもよく、部分は現金であり、これらはすべて報酬委員会によって決定される。
制限された株と株式単位. 報酬委員会は、2013年計画に従って制限された普通株式および/または株式単位を付与することができる。限定的な株式奨励には参加者に譲渡された株が含まれており、特定の条件を満たさなければ、これらの株は制限され、 没収につながる可能性がある。株式単位は、将来報酬委員会が指定した特定の条件に達したときまたはその後の将来の日に普通株、現金または株と現金の組み合わせを受け入れる権利を与えてくれます。 報酬委員会は、業績に基づく条件が含まれる場合がある制限株式またはbr株単位を付与するために適用される制限および条件を決定するであろう。制限株式に関連する配当金は、株主に配当金を支払う際に株式所有者に支払うことができ、制限株式報酬の帰属または他の支払い時に株式所有者に支払うこともできる。br}株式単位報酬は、配当等価権と共に付与することができ、これらの配当等価権は蓄積することができ、追加の 株式単位に再投資するとみなされ、報酬委員会によって適宜決定されることができる。株単位報酬が制限されながら任意の配当等価物を支払う場合、配当協定が別途規定されていない限り、配当等価物は対象株式 単位と同じ譲渡制限を遵守しなければならない。給与委員会が別の決定がない限り、制限された株の保有者は株に投票する権利があるだろう。
| 86 |
パフォーマンスシェアとパフォーマンス単位. 報酬委員会は、Br 2013計画に従って成績表および/または業績単位を付与することができます。パフォーマンスシェアおよびパフォーマンス単位は、特定のパフォーマンス期間中に報酬委員会によって決定されたパフォーマンス基準に達した場合に得られる報酬である。報酬委員会は、各業績株および業績単位報酬に適用される制限および条件を決定します。
奨励賞ボーナス奨励それは.給与委員会は2013年計画に基づいて奨励金を付与することができる。奨励ボーナスは、会社または子会社の特定の業績レベルに基づいて、予め確立された目標業績基準に従って 報酬委員会を適宜決定することができる。奨励ボーナスは、奨励協定の規定に従って、現金または普通株の形で支払われる。
他のbr株と現金奨励. 報酬委員会は、2013年計画に従って、私たちの普通株の非限定的な株を付与または要約することを含み、普通株式価値に基づく金額 を現金または他の方法で支払うことを含む他のタイプの株式または現金報酬を付与することができる。
ある会社の取引の影響 それは.賠償委員会は、(1)行使の加速または延長、帰属または任意の裁決の実現の期限、(2)裁決の業績または他の条件の取り消しまたは修正、(3)賠償委員会によって決定された同値現金価値に基づいて、現金で裁決を決済することを含む、裁決を付与する際に、(2013年計画で定義されたように)任意の裁決への制御権変更の影響を規定することができる。または(Iv)補償委員会は、制御権変更時または後に参加者の権利および利益を維持して保護するために、適切な他の修正または裁決調整を考える。補償委員会は、制御権変更の発生に応じて、適宜、(A)任意またはすべての未行使のオプションおよび株式付加価値権を直ちに全部または部分的に行使可能にすることと、(B)任意の他の ボーナスの全部または一部を没収できないようにすることと、(C)代替オプションと交換するために、任意のオプションまたは株式付加価値権をキャンセルすることと、のうちの1つまたは複数をとることができる。(D)制限株、株式単位、業績株または業績単位への報酬を取り消して、任意の相続人会社の株式の類似報酬brと交換する;(E)任意の制限株、株式単位、業績株または業績単位 を償還し、現金および/または他の代替対価で、その価値は、制御権変更の日の私たちの普通株の非制限株の公平な市場価値に相当する。(F)任意のオプションまたは株式付加価値を廃止して、現金および/または他の代替対価格 と交換して、制御権に応じて日付に応じて私たちの普通株式価値を変更する,(G) 制御権変更の影響を受けた参加者が保有する任意の株式単位または業績単位を取り消して、現金および/または他の代替価格と交換し、その価値は、制御権変更当日の普通株の1株当たり公平な市価に等しいか、または(H)報酬委員会が必要または適切であると考えている他の未払い報酬に対して 修正、調整または修正する。
修正、 終了それは.補償委員会は、2013年計画に抵触しないいかなる方法でも奨励条項を修正することができるが、未参加者の同意がなく、いかなる修正も未解決の報酬に関する参加者の権利に悪影響を与えてはならないことが条件である。また、当社取締役会は、2013年計画を随時修正、一時停止または終了することができるが、条件は、(I)いかなる参加者の同意もなく、このような修正、一時停止または終了は、任意の係属中の裁決項目における任意の参加者の権利に重大な悪影響を与えてはならないこと、および(Ii)任意の適用法律、法規または証券取引所規則を遵守するために必要かつ適切な範囲内で、2013計画は株主の同意を得ることを要求することである。任意の計画改訂は、2013年計画の下で発行可能な普通株式数を増加させたり、資格 を獲得して奨励を受ける資格のある人やカテゴリを変更したりする場合は、株主の承認を得なければならない。
| 87 |
税金を前納する
Br社は、法律または法規の要求を満たす連邦、州と地方税収(国内または海外)を支払うために、参加者に最低法定金額 を控除または控除または要求する権利がある。
役員報酬
私たちのbrは政策を維持し、この政策によると、私たちの非従業員役員は年化された報酬を得るだろう。この政策は、以下のような補償金額を現金で支払うこと、または非従業員取締役が選択した後に制限されない普通株株で支払うことを規定している:(I)各非従業員取締役は50,000ドルの年会費を得る権利があり、(Ii)会長は追加の年費25,000ドルを得る権利があり、(Iii)副会長は追加の年会費20,000ドルを得る権利がある。(Iv)私たちの監査委員会議長は15,000ドルの年会費を得る権利があり、私たちの監査委員会の他のメンバーは$7,500ドルの年会費を得る権利があり、(V)私たちの給与委員会議長は10,000ドルの年会費を得る権利があり、私たちのbr給与委員会の他のメンバーは6,000ドルの年会費を得る権利があり、(Vi)私たちの指名とコーポレートガバナンス委員会の議長は$8,000ドルの年会費を得る権利があり、他のメンバーは4,000ドルの年会費を得る権利がある。
各株主周年総会の日から、各非従業員取締役はオプション付与を獲得し、付与された日に私たちの既存の持分インセンティブ計画または私たちが将来取る可能性のある任意の他の持分インセンティブ計画の下でブラック·スコアーズ方法で決定された80,000ドルの普通株式を購入し、この計画は12ヶ月に分けて分割払いになる。
取締役補償政策下のすべてのbr費用は季節ごとに延滞し、毎回の会議費用は支払わない。すべての費用は役員選挙時に無制限普通株で支払うことができます。取締役会や委員会会議に参加することによる非従業員取締役の合理的な費用も精算します。
役員報酬表-2023年
次の表は私たちの非従業員役員の2023年の年間給与をまとめています。
| 名前.名前 | 現金補償 ($) | 選択権 賞.賞 ($) (1) | 合計する ($) | |||||||||
| ハーバート·コンラッド | 61,500 | 80,000 | 141,500 | |||||||||
| キャサリン·コルゾ | 60,000 | 80,000 | 140,000 | |||||||||
| エリック·エンダー | 79,000 | 80,000 | 159,000 | |||||||||
| ナターシャ·ゾダンノ | 65,500 | 80,000 | 145,500 | |||||||||
| ジェームズ·S·シベルタ | 75,000 | 80,000 | 155,000 | |||||||||
| マシュー·ビクター | 56,000 | 80,000 | 136,000 | |||||||||
| (1) | 金額 は、会計基準編集テーマ718によって2023年に付与された株式奨励およびオプション奨励の付与日公正価値を反映している。これらの金額は役員が確認する実際の価値と一致しません。 |
報酬委員会と内部関係者の参加
取締役会報酬委員会は現在、以下の3人の非従業員取締役から構成されている:キャサリン·コルゾー会長、ジェームズ·シベッタ会長、マシュー·ビクター氏。給与委員会のメンバーは前財政年度内にbr社の管理者や従業員ではなかった。また、当社の取締役のいずれかが役員を務める会社は、その報酬委員会や取締役会は、当社の執行者が担当することはできません。第13項を参照。
| 88 |
| 第 項12. | 安全 いくつかの実益所有者と管理層の所有権、および関連する株主事項。 |
次の表は、2024年3月18日までに以下の会社実益が所有する普通株式数を示しています
| ● | 私たちが知っている実益は私たちの普通株の5%以上の株主を持っています |
| ● | 私たちのすべての執行官は |
| ● | 私たちすべての役員は |
| ● | チームとして、私たちのすべての役員と現職の幹部。 |
利益を得るbr所有権は米国証券取引委員会の規則によって決定される。個人 が株式を投票および/または処分する権利がある場合、その人は株式の実益所有権を所有する。このような権力は単独でまたは共有されてもよく、直接的または間接的であってもよい。次の表に適用される所有権パーセンテージ は、2024年3月18日現在の217,482,830株の発行済み株に基づいています。ある人が実益所有している株式の数とその人の所有権パーセントを計算する際には、その人が保有するオプションまたは株式承認証に拘束され、2024年3月18日またはその後60日以内に行使可能な普通株式が発行された普通株式として計算される。しかしながら、他の人(S)の持株率を計算する場合、これらの株式は、発行済み株式には計上されない。本表の脚注 には別の説明があるほか,適用されるコミュニティ財産法により,表中で指名された各人は,その名前に対する普通株式に対して独占投票権と処分権を持つ.以下に説明しない限り、以下に列挙される各個人のアドレスは、c/o Matinas BioPharma Holdings,Inc.,1545 Road 206 South,Suite 302,Bedminster,NJ 07921である。
| 実益所有者の氏名または名称 | 実益所有株式数 | 実益所有株式の割合 | |||||||
| 役員および行政員 | |||||||||
| ジェローム·D·ジャブル(1) | 6,912,028 | 3.1 | % | ||||||
| ハーバート·コンラッド(2) | 6,057,976 | 2.8 | % | ||||||
| キャサリン·コルゾ(3) | 549,186 | * | % | ||||||
| エリック·エンダー(4) | 1,519,169 | * | % | ||||||
| ナターシャ·ゾダンノ(5) | 776,515 | * | % | ||||||
| ジェームズ·シベルタ(6) | 1,937,535 | * | % | ||||||
| マシュー·ビクター(7) | 1,515,549 | * | % | ||||||
| ジェームズ·J·ファーガソン(8歳) | 1,905,501 | * | % | ||||||
| トーマス·J·フーバー(9) | 681,758 | * | % | ||||||
| キース·A·クチンスキー(10歳) | 1,909,578 | * | % | ||||||
| 劉慧(11) | 894,770 | * | % | ||||||
| テレサ·マテコヴィッツ(12歳) | 1,927,578 | * | % | ||||||
| 役員及び行政幹事(12名)(13名) | 26,587,143 | 11.2 | % | ||||||
* 1%未満
(1) は、6,451,704株がオプション行使時に発行可能な普通株式を含む。5,707,196株普通株 オプションは含まれていない。
(2) オプション行使後に発行可能な1,363,410株の普通株式を含む.182,185株の普通株式 オプションは含まれていない。
(3) オプション行使後に発行可能な549,186株の普通株式を含む.212,478株普通株式 オプションは含まれていない。
(4) オプション行使後に発行可能な1,375,077株普通株を含む.182,185株の普通株式 オプションは含まれていない。
(5) は,行使時に発行可能な776,515株の普通株式を含む.182,185株の普通株式のオプションは含まれていない。
(6) オプション行使時に発行可能な1,287,577株普通株式を含む.182,185株の普通株式 オプションは含まれていない。
(7) オプション行使時に発行可能な1,225,077株普通株式を含む.182,185株の普通株式 オプションは含まれていない。
(8) オプション行使後に発行可能な1,905,501株の普通株式を含む。1,932,099株普通株 オプションは含まれていない。
(9) オプション行使後に発行可能な691,758株普通株式を含む.1,643,242株の普通株式 オプションは含まれていない。
(10) オプション行使時に発行可能な1,815,078株普通株式を含む.1,684,922株普通株 オプションは含まれていない。
(11) オプション行使後に発行可能な894,770株の普通株式を含む.1,605,230株式普通株式 オプションは含まれていない。
(12) オプション行使時に発行可能な1,927,578株普通株式を含む.1,972,422株の普通株式 オプションは含まれていない。
(13) 付記(1)~(12)を参照。
| 89 |
株式補償計画に基づいて発行された証券
次の表は、2023年12月31日現在の私たちの株式給与計画の情報をまとめています。
| 計画種別 | 株式数 発行された普通株 未償還オプションを行使する (a) | 加重平均 行使価格: 未平倉オプション (b) | オプション数 持分補償計画によると残りは将来発行することができる((A)欄に反映された証券を除く) (c)(2) | |||||||||
| 株主が承認した株式報酬計画(1) | 46,707,934 | $ | 0.83 | 2,905,170 | ||||||||
| 株主の承認を得ない持分補償計画 | — | — | — | |||||||||
| 合計する | 46,707,934 | $ | 0.83 | 2,905,170 | ||||||||
| (1) | 今回表示されたbr金額には、Matinas BioPharma Holdings、Inc.改訂と再予約された2013年株式インセンティブ計画(“2013計画”)下の証券が含まれている。 |
| (2) | 我々の2013年計画中の“常青樹”条項によると、2024年の第1取引日には、2023年12月31日の発行済み株式数の4%に相当する追加8,690,581株が自動発行される。これらのbr}株は今回の計算には含まれていない。 |
| 第 項13. | いくつかの関係、関連取引、取締役独立性 |
ある 関係と関連先取引
私たちが指定した役員や役員の報酬スケジュールを除いて、2023年1月1日以来、取引や一連の類似した取引に参加したり、参加したりすることはありません
| ● | 関連する金額は、(I)12万元および(Ii)の過去2つの完全財政年度年末総資産平均値の1%を超えるか、または超えることになり、両者は少ない者を基準とする |
| ● | 当社の任意の役員、役員、または5%以上の株式を保有する者、または上記の人の任意の直系親族は、かつてまたは直接的または間接的な重大な利益を持つことになる。 |
賠償協定
私たちは私たちの役員と役員と賠償協定を締結しました。賠償協定は、被保険者が脅威、未決または完了した訴訟、訴訟または他の訴訟によって実際かつ合理的に発生した費用、判決、罰金、罰金を賠償することを規定しているが、いくつかの制限を受けている。賠償協定はまた、最終的な、控訴できない判決または他の裁決の前に、訴訟に関連する費用を前借りすることができるが、条件は、被保険者が最終的に被保険者が私たちの賠償を受ける権利がないことが発見された場合、前払いした任意の金を返済することである。賠償協定は,賠償請求や前借り費用の提出と応答の手続きと,被賠償者との間で賠償合意により生じる任意の紛争に適用される紛争解決手続きを規定している.
関係者取引の政策と手順
私たちは、私たちの役員、取締役、取締役として指名された候補者、任意の種類の普通株の実益所有者が5%を超える、上記の人のいずれかの直系親族、およびその中で雇用され、パートナーまたは依頼者として、または同様の職にあるか、または5%以上の実益所有権を有する会社、会社、または他のエンティティを総称して関連者と呼ぶ政策をとっている。当社の取締役会が審査委員会を介して行動したり、場合によっては審査委員会議長の事前同意を得ていない場合は、当社との取引を行うことはできません。私たちが関係者との取引を要求する任意の請求は、100,000ドルを超える金額に関連しており、その関連側には直接的または間接的な利益があり、まず私たちの監査委員会に提出されるか、または場合によっては私たちの監査委員会の議長に提出されて審査、考慮、承認されなければならない。そのような提案を承認または拒否する場合、我々の監査委員会または私たちの監査委員会議長は、取引条項が、同じまたは同様の場合に非関連第三者が通常得ることができる条項 ,私たちへの利益の程度、他の類似した製品またはサービスのソースがあるかどうか、および関連側の取引における利益の程度を下回っていないかどうかを含むが、これらに限定されない取引の重要な事実を考慮するであろう。
取締役 独立
我々の取締役会は、各取締役の要求と提供に応じて、Herbert Conradさん、Eric Endeさん、James Scibettaさん、Matthew Wiklerさん、およびMsesさんを決定しました。Kathryn CorzoおよびNatasha Giordanoは、ニューヨーク証券取引所MKTのコーポレートガバナンス要求規則および1934年に発行された証券取引法(改正)規則10 A-3に定義されている“独立取締役”である。
| 90 |
| 第 項14. | 依頼者 は課金とサービス |
下表は、当社の独立公認会計士事務所EisnerAmper LLPが2023年12月31日と2022年12月31日までの財政年度に当社に徴収した費用総額を示しています。
| 十二月三十一日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| 料金を審査する | $ | 270 | $ | 265 | ||||
| 監査関連費用 | - | - | ||||||
| 税金.税金 | - | - | ||||||
| 総費用 | $ | 270 | $ | 265 | ||||
監査費用 当社の年次財務諸表、監査財務報告書の内部統制、中期総合財務諸表、慰め、同意書の審査のための専門サービス費用が含まれています。
監査に関する費用 関連サービスを保証するために提供される専門サービスにかかる費用が含まれており、これらのサービスは私たちの金融サービスの監査または審査の業績 と合理的に関連している。
税 手数料主に税務相談と税務計画に関連した税務サービスだ。
監査委員会は、取引法第10 A(H)条に基づいて独立公認会計士事務所が提供を許可されているすべての監査サービス及び任意の非監査サービスを予め承認している。監査委員会は、事前承認をそのメンバーに依頼することができるが、このような許可が行われた場合、監査委員会全員が次回の定期会議でそのメンバーが行った任意の事前承認決定を提出しなければならないことが条件である。
| 91 |
第4部
| 第 項15. | 表と財務諸表明細書 |
| 添付ファイル 番号: | 説明する | |
| 2.1 | 当社、Matinas Merger Sub,Inc.とMatinas BioPharma,Inc.の間で2013年7月11日に署名された合併協定(合併内容は、2014年2月7日に米国証券取引委員会に提出された会社S-1表登録説明書修正案1添付ファイル2.1を参照)。 | |
| 2.2 | デラウェア州のAquarius BioTechnologies,Inc.(以下“Aquarius”と略す),デラウェア州の完全子会社Saffron Merge Sub,Inc.(以下,“合併子会社”と略す)および株主代表J.Carl Craftが署名した合併協定と合併計画(“合併協定”)(“合併協定”)は,2015年1月30日に米国証券取引委員会に提出された8−K表の現在報告されている添付ファイル2.1を参考にした。 | |
| 3.1 | 会社登録証明書(会社が2014年2月7日に米国証券取引委員会に提出したS-1表登録説明書第1号修正案添付ファイル3.1参照)。 | |
| 3.2 | 会社定款(会社が2014年2月7日に米国証券取引委員会に提出したS-1表登録説明書第1号修正案添付ファイル3.2参照)。 | |
| 3.3 | 2015年10月29日の会社登録証明書の改訂証明書。(米国証券取引委員会に2015年11月5日に会社が提出した現在のForm 8-K報告書を参照することによって本明細書に組み込まれる)。 | |
| 4.1 | 普通株式サンプル(2017年3月31日に米国証券取引委員会に提出された会社の2016年12月31日までの年次報告Form 10−Kの添付ファイル4.1を参照して合併)。 | |
| 4.6 | 証券説明* | |
| 10.1 | Matinas BioPharma Holdings,Inc.は2013年の持分補償計画を改訂·再策定した(同社が2015年3月31日に提出したForm 10−K年度報告書の添付ファイル10.6を引用した) | |
| 10.2 | 株式オプションインセンティブ協議表(2014年2月7日に米国証券取引委員会に提出された会社登録説明書S−1号修正案添付ファイル10.7を参照して編入)。ガンギエイ |
| 92 |
| 10.3 | 無留保株式オプション協定表(2014年2月7日に米国証券取引委員会に提出されたS-1表会社登録説明書修正案第1号添付ファイル10.8を参照) | |
| 10.4 | 賠償協議表(会社が2014年2月7日に米国証券取引委員会に提出したS−1レジストリ第1号修正案添付ファイル10.14を参照して編入)。ガンギエイ | |
| 10.5 | リースは2013年11月4日に施行され、会社とA-K Bedminster Associates,L.P.(会社が2014年2月7日に米国証券取引委員会に提出したS-1表登録説明書修正案第1号添付ファイル10.17を参照して設立された)。 | |
| 10.6 | ロッグス大学、ニュージャージー州立大学、Aquarius BioTechnologies,Inc.の間で2015年1月29日に署名された独占許可協定(2015年3月31日に会社が提出したForm 10-K年報添付ファイル10.18)+を改訂·再署名した | |
| 10.8 | リース契約は、2016年12月15日にCIPII/AR Bridgewater Holdings LLCとMatinas BioPharma Holdings,Inc.が締結された(これに合併し、2017年4月28日に米国証券取引委員会に提出された会社の現在8-Kレポートの添付ファイル10.1を参照)。 | |
| 10.9 | 会社とジェローム·D·ジャブルとの雇用契約は、2018年3月22日となっている(2018年3月27日に米国証券取引委員会に提出された会社8-Kフォームの添付ファイル10.1合併を参照することにより) | |
| 10.10 | Matinas Biophma Holdings,Inc.およびTheresa Matkovitsが2018年10月15日に署名した雇用協定(2018年10月15日に米国証券取引委員会に提出された会社の現在の8-K表報告書の添付ファイル10.1として)。ガンギエイ | |
| 10.11 | Matinas Biophma Holdings,Inc.およびKeith Kucinskiが2019年1月3日に署名した雇用協定(2019年1月3日に米国証券取引委員会に提出された会社の現在の8-K表報告書の添付ファイル10.1として提出) | |
| 10.12 | Matinas Biophma Holdings,Inc.およびJames J.Ferguson IIIが2019年2月25日に署名した雇用協定(2019年2月25日に米国証券取引委員会に提出された会社の現在の8-K表報告書の10.1添付ファイルとして)。ガンギエイ | |
| 10.13 | Matinas BioPharma Holdings,Inc.とBTIG,LLCが2020年7月2日に署名した市場販売協定(会社が2020年7月2日に米国証券取引委員会に提出した8−K表の添付ファイル1.01を引用した)。 | |
| 10.14 | 治療開発賞協定は,2020年11月19日にMatinas BioPharma Holdings,Inc.と嚢胞性線維化財団が締結された(引用会社が2021年3月29日に提出したForm 10−K年度報告書の添付ファイル10.1により組み込まれている)。 | |
| 10.15 | Matinas Biophma Holdings,Inc.と劉輝の間で2020年12月1日に締結された雇用協定。(添付ファイル10.16を参照して会社に編入して2022年3月8日に提出されたForm 10-K年間報告書) | |
| 10.16 | Matinas Biophma Holdings,Inc.とThomas Hooverの間で2021年12月6日に締結された雇用協定。(添付ファイル10.16を参照して会社に編入して2022年3月8日に提出されたForm 10-K年間報告書) | |
| 会社がラファエル·J·マンニーノと締結したコンサルティング契約は、2022年8月8日となっている(会社が2022年11月2日に提出したForm 10-Q四半期報告の添付ファイル10.1を参考に合併した)。 | ||
| 10.17 | Matinas Biophma Holdings,Inc.とJerome Jabbourの雇用協定改正案は,2023年3月3日(引用会社により2023年3月15日に提出されたForm 10−K四半期報告書の添付ファイル10.17に編入された) | |
| 10.18 | Matinas Biophma Holdings,Inc.とTheresa Matkovitsが2023年3月3日に署名した雇用協定改正案(会社が2023年3月15日に提出したForm 10−K四半期報告書の添付ファイル10.18を引用して合併した) | |
| 10.19 | Matinas Biophma Holdings,Inc.とKeith Kucinskiが2023年3月3日に締結した雇用協定修正案(合併内容参考会社が2023年3月15日に提出したForm 10−K四半期報告添付ファイル10.19) | |
| 10.20 | Matinas Biophma Holdings,Inc.とJames Fergusonとの間の雇用協定改正案は,2023年3月3日(会社が2023年3月15日に提出したForm 10−K四半期報告書の添付ファイル10.20を引用して編入されている) | |
| 19.1 | Matinas BioPharma Holdings Corp.インサイダー取引政策* | |
| 21.1 | 子会社指数* | |
| 23.1 | EisnerAmper LLP同意* | |
| 31.1 | 2002年のサバンズ·オキシリー法第302条に基づいて総裁と最高経営責任者証明書が発行された* | |
| 31.2 | 2002年のサバンズ·オクスリ法第302条による代理財務官の証明* | |
| 32.1 | 第1350節認証** | |
| 97.1 | Matinas BioPharma Holdings,Inc.報酬回収政策* | |
| 101 | 以下は、XBRL (拡張可能ビジネス報告言語)形式で作成された2023年12月31日現在の年次報告における以下の財務情報である:(I)2023年12月31日現在と2022年12月31日現在の総合貸借対照表、(Ii)2023年12月31日および2022年12月31日までの総合経営報告書と全面赤字報告書、(Iii)2023年12月31日および2022年12月31日までの総合株主権益(赤字)変動表。(4)2023年12月31日現在、2023年12月31日現在、2022年12月31日現在の総合キャッシュフロー表;および(V)総合財務諸表を付記する | |
| 104 | 本年度報告の表紙はForm 10−K,フォーマットはイントラネットXBRLである。 |
| + | 改正された1934年の証券取引法で公布された規則24 b-2に基づき、本展示品のいくつかの条項の秘密保護処理が求められている。 |
| † | 管理契約または報酬計画、契約または手配を表します。 |
| * | 同封してアーカイブする。 |
| ** | 同封で提供します。 |
| 第 項16. | 表 10-K要約 |
ない。
| 93 |
サイン
証券法第13節または15(D)節の要件によれば、登録者は、2024年3月27日にニュージャージー州ベイドミンスター市で、以下の署名者代表登録者が本報告書に署名することを正式に許可している。
| MATINASバイオ製薬ホールディングス | ||
| 差出人: | /S/ジェローム·D·ジャブル | |
| 名前: | ジェローム·D·ジャボル | |
| タイトル: | CEO | |
| 差出人: | /S/ キース·A·クチンスキー | |
| 名前: | キース·クチンスキー | |
| タイトル: | 最高財務官 | |
1934年の証券取引法の要求によると、本報告は、以下の者によって登録者として指定日に署名された。
| 人は… | 容量 | 日取り | ||
| /S/ジェローム·D·ジャブル | 最高経営責任者兼取締役 | 2024年3月27日 | ||
| ジェローム·D·ジャボル | (CEO ) | |||
| /S/ キース·A·クチンスキー | 最高財務官 | 2024年3月27日 | ||
| キース·クチンスキー | (最高財務会計官 ) | |||
| /S/ エリック·エンデ | 取締役会議長 | 2024年3月27日 | ||
| エリック·エンダー | ||||
| /S/ ハーバート·コンラッド | 役員.取締役 | 2024年3月27日 | ||
| ハーバート·コンラッド | ||||
| /S/ キャサリン·コルゾ | 役員.取締役 | 2024年3月27日 | ||
| キャサリン·コルゾ | ||||
| /S/ ナターシャ·ゾダンノ | 役員.取締役 | 2024年3月27日 | ||
| ナターシャ·ゾダンノ | ||||
| /S/ ジェームズ·S·シベルタ | 役員.取締役 | 2024年3月27日 | ||
| ジェームズ·S·シベルタ | ||||
| S/ マシュー·A·ビクター | 役員.取締役 | 2024年3月27日 | ||
| マシューA.ビクター |
| 94 |
独立公認会計士事務所報告{br
その会社の取締役会と株主
Matinas BioPharmaホールディングス
財務諸表に対する意見
Matinas BioPharma Holdings,Inc.およびその子会社の2023年12月31日と2022年12月31日までの総合貸借対照表,およびこの日までの各年度までの関連総合経営報告書と全面赤字,株主権益とキャッシュフロー,および関連付記(総称して“財務諸表”と呼ぶ)を監査した。上記財務諸表は,米国公認の会計原則に従って,当社の二零一年十二月三十一日,二零二三年及び二零二二年の財務状況,及び当該日までの各年度の経営実績及びキャッシュフローを各重大な面で公平に列報していると考えられる。
注目を行っている
添付財務諸表の作成は、当社が継続的に経営を継続する企業であると仮定しています。財務諸表付記 2で述べたように,当社には経常的純損失と純現金流量が運営に用いられており,継続経営企業としての能力が大きく疑われている。付記2は、これらの事項における経営陣の計画も説明しています。財務諸表には、このような不確実性の結果によって生じる可能性のあるいかなる調整も含まれていません。
意見を求める根拠
これらの財務諸表は会社経営陣が担当しています。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会(SEC)とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たち はPCAOBの基準に従って監査を行います。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。会社 はその財務報告の内部統制を監査する必要はなく、私たちを招いて監査を行うこともありません。私たちの監査の一部として、財務報告の内部統制を知る必要がありますが、社内財務報告の内部統制の有効性について意見を述べるためではありません。したがって、私たちはそのような意見を表現しない。
我々のbr監査には、財務諸表の重大な誤報のリスクを評価するための実行プログラムが含まれており、エラーによるものであっても詐欺であっても、これらのリスクに対応するプログラムを実行する。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。私たちの監査には、経営陣が使用する会計原則の評価と重大な推定、財務諸表の全体的なレポートの評価も含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重大監査事項
以下に述べる重要な監査事項とは、当期に財務諸表を監査する際に生じる事項であり、この事項は、監査委員会に伝達または要求されたものである:(1)財務諸表に対して重大な意義を有する勘定又は開示に関するものであり、(2)特に挑戦性、主観性又は複雑性に係る判断である。重要な監査事項のコミュニケーションは、財務諸表全体に対する私たちの意見をいかなる方法でも変えることはなく、以下の重要な監査事項 を伝達することによって、重要な監査事項またはそれに関連する勘定または開示について個別の意見を提供することもない。
| F-1 |
研究開発費は計算項目 と前払い残高
総合経営報告書に開示されているように、2023年12月31日現在、当社では大量の研究·開発(“研究開発”)支出が発生しており、総額は約1,450万ドルである。当社は2023年12月31日現在、総合貸借対照表に約90万ドルの研究開発費を計上している。同社はまた、総合貸借対照表に前払い研究開発費約70万ドルを記録している。会社の研究開発費の大部分は、契約研究組織(CRO)や契約製造組織(CMO)に支払われるサービス料である。 とこれらのCROやCMOの研究開発活動は契約契約に記録されており、通常は長い時間で実行される。このような研究開発契約の総合貸借対照表に記録されている金額は、会社が当時知っていた事実と状況に基づいて未支払いと前払い研究開発費の推定であり、CROとCMOの適時かつ正確な報告に依存する。これらの合意に基づき、会社が財務報告期間内に完了した作業進捗の推定に基づいて、確認すべき研究開発費金額は、判断及び推定に係る。
我々のbrは、研究開発費の計算と前払い残高に対する管理層の推定を重要な監査事項として決定し、これらの費用は財務諸表に対して重要な意義があるため、管理層は臨床研究或いは提供するサービスの進捗或いは完成状態を決定する際に重大な判断と推定を行う必要がある。したがって、経営陣が作成した推定に関する監査証拠を評価するためには、監査人の高度な判断と追加的なテストが必要である。
この問題を解決することは、連結財務諸表に対する私たちの全体的な意見を形成するための実行手順および評価監査証拠に関するものである。研究開発費の計上や前払い残高に関する監査プログラムには、(I)経営陣の流れを理解し、臨床研究の状態からこれまでに発生した費用を推定する過程を含む、その計算すべき計算および前払い研究費推定の制御措置の設計·実施を評価したものが含まれている。(Ii)CROおよびCMOとの精選プロトコルおよび契約修正案を読み、上記の重大な仮定と、研究開発試算および貸借対照表の日未払いおよび前払い金額を作成および再計算する際に使用する方法を評価し、(Iii)いくつかのCROおよびCMOと契約承諾および完了、支払いおよび未払い金額を確認し、(Iv)プロジェクト状態について会社研究者に直接問い合わせた。年末後に受け取った領収書と他の文書、および管理職の使用時にCROとCMOとの通信を検査し、その研究開発支出推定を作成する。
/S/ EisnerAmper LLP
私たちは2011年から当社の監査役を務めています
2024年3月27日
| F-2 |
Matinas BioPharmaホールディングス
合併貸借対照表
(単位:千、共有データを除く)
| 十二月三十一日 | ||||||||
| 2023 | 2022 | |||||||
| 資産: | ||||||||
| 流動資産: | ||||||||
| 現金と現金等価物 | $ | $ | ||||||
| 売却可能債務証券 | ||||||||
| 制限された現金--保証金 | ||||||||
| 前払い費用と他の流動資産 | ||||||||
| 流動資産総額 | ||||||||
| 非流動資産: | ||||||||
| レンタル改善と設備--純額 | ||||||||
| 賃貸使用権資産を経営する--純額 | ||||||||
| 融資リース使用権資産-純額 | ||||||||
| 現在行われている研究と開発 | ||||||||
| 商誉 | ||||||||
| 制限された現金--保証金 | ||||||||
| 非流動資産総額 | ||||||||
| 総資産 | $ | $ | ||||||
| 負債と株主資本: | ||||||||
| 流動負債: | ||||||||
| 売掛金 | $ | $ | ||||||
| 費用とその他の負債を計算すべきである | ||||||||
| レンタル負債を経営しています--流動負債 | ||||||||
| 融資リース負債-流動 | ||||||||
| 流動負債総額 | ||||||||
| 非流動負債: | ||||||||
| 繰延税金負債 | ||||||||
| 賃貸負債を扱う--当期分を差し引く | ||||||||
| 融資リース負債--当期分を差し引く | ||||||||
| 非流動負債総額 | ||||||||
| 総負債 | ||||||||
| 株主権益: | ||||||||
| 普通株式額面$一株一株2023年12月31日と2022年12月31日にそれぞれ認可された株2023年12月31日までと2022年12月31日までそれぞれ発行と未返済 | ||||||||
| 追加実収資本 | ||||||||
| 赤字を累計する | ( | ) | ( | ) | ||||
| その他の総合損失を累計する | ( | ) | ( | ) | ||||
| 株主権益総額 | ||||||||
| 総負債と株主権益 | $ | $ | ||||||
付記はこれらの連結財務諸表の構成要素である。
| F-3 |
Matinas BioPharmaホールディングス
合併経営報告書と全面赤字
(単位は 千で、1株当たりおよび1株当たりのデータは含まれていない)
| 12月31日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| 収入: | ||||||||
| 契約収入 | $ | $ | ||||||
| コストと支出: | ||||||||
| 研究開発 | ||||||||
| 一般と行政 | ||||||||
| 総コストと費用 | ||||||||
| 運営損失 | ( | ) | ( | ) | ||||
| ニュージャージー州の純営業損失と税金控除 | ||||||||
| その他の収入、純額 | ||||||||
| 純損失 | $ | ( | ) | $ | ( | ) | ||
| 1株当たり純損失--基本損失と赤字 | $ | ) | $ | ) | ||||
| 加重平均発行された普通株式: | ||||||||
| 基本的希釈の | ||||||||
| その他総合損益、税引き後純額 | ||||||||
| 証券売却可能な未実現損益 | ( | ) | ||||||
| その他総合損益、税引き後純額 | ( | ) | ||||||
| 総合損失 | $ | ( | ) | $ | ( | ) | ||
付記はこれらの連結財務諸表の構成要素である。
| F-4 |
Matinas BioPharmaホールディングス
合併 株主権益変動表
(単位:千、共有データを除く)
| 普通株 | 余分な実収 | 積算 | 他の総合を累計する | 株主合計 | ||||||||||||||||||||
| 株 | 金額 | 資本 | 赤字.赤字 | (赤字)/収入 | 権益 | |||||||||||||||||||
| バランス、2021年12月31日 | $ | $ | $ | ( | ) | $ | ( | ) | $ | |||||||||||||||
| 株に基づく報酬 | — | |||||||||||||||||||||||
| オプション行使時に普通株式を発行する | ||||||||||||||||||||||||
| 株式承認証と引き換えに普通株式を発行する | ||||||||||||||||||||||||
| 許可協定改正案に基づいて普通株式を発行する | ||||||||||||||||||||||||
| その他総合損失 | — | ( | ) | ( | ) | |||||||||||||||||||
| 純損失 | — | ( | ) | ( | ) | |||||||||||||||||||
| バランス、2022年12月31日 | $ | $ | $ | ( | ) | $ | ( | ) | $ | |||||||||||||||
| 株に基づく報酬 | — | |||||||||||||||||||||||
| その他総合収益 | — | |||||||||||||||||||||||
| 純損失 | — | ( | ) | ( | ) | |||||||||||||||||||
| バランス、2023年12月31日 | $ | $ | $ | ( | ) | $ | ( | ) | $ | |||||||||||||||
付記はこれらの連結財務諸表の構成要素である。
| F-5 |
Matinas BioPharmaホールディングス
統合されたキャッシュフロー表
(単位:千)
| 12月31日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| 経営活動のキャッシュフロー: | ||||||||
| 純損失 | $ | ( | ) | $ | ( | ) | ||
| 純損失と経営活動で使用される現金純額の調整: | ||||||||
| 減価償却および償却 | ||||||||
| 設備処分損失 | ||||||||
| 株に基づく報酬費用 | ||||||||
| 経営的リース使用権資産の償却 | ||||||||
| 融資リース使用権資産の償却 | ||||||||
| 債券割引償却 | ||||||||
| 許可協定改正案に基づいて発行された株 | ||||||||
| 経営性資産と負債変動状況: | ||||||||
| 前払い費用と他の流動資産 | ( | ) | ||||||
| 売掛金 | ( | ) | ( | ) | ||||
| 費用とその他の負債を計算すべきである | ( | ) | ||||||
| リース負債を経営する | ( | ) | ( | ) | ||||
| 経営活動のための現金純額 | ( | ) | ( | ) | ||||
| 投資活動によるキャッシュフロー: | ||||||||
| 売却可能な債務証券を購入する | ( | ) | ||||||
| 有価証券を売却して得られる収益 | ||||||||
| レンタル改善と設備の購入 | ( | ) | ( | ) | ||||
| 投資活動が提供する現金純額 | ||||||||
| 資金調達活動のキャッシュフロー: | ||||||||
| オプション行使で得られた収益 | ||||||||
| 資本賃貸負債の支払い--元金 | ( | ) | ( | ) | ||||
| 融資活動が提供する現金純額 | ( | ) | ||||||
| 現金、現金等価物、および限定的な現金純減少 | ( | ) | ( | ) | ||||
| 期初現金、現金等価物、および限定現金 | ||||||||
| 期末現金、現金等価物、および制限現金 | $ | $ | ||||||
| 非現金融資と投資活動を補完します | ||||||||
| 取引可能債務証券の未実現収益/(損失) | $ | $ | ( | ) | ||||
| 現金行使前払い費用とその他の流動資産における権利証 | $ | $ | ( | ) | ||||
| 経営性リース変更による負債と引き換えに使用権資産を使用する | $ | $ | ( | ) | ||||
| 資産を使用して融資リース負債と交換する | $ | $ | ||||||
付記はこれらの連結財務諸表の構成要素である。
| F-6 |
注 1-業務説明
Matinas BioPharma Holdings Inc.(“Holdings”)はデラウェア州の会社で、2013年に設立された。ホールディングスはMatinas BioPharma,Inc.(“BioPharma”)とMatinas BioPharma NanoTechnologies,Inc.(“NanoTechnologies”,前身はAquarius BioTechnologies,Inc.)とその運営子会社(“NanoTechnologies”,“Holdings”と“BioPharma”,“当社”)の親会社である。同社は臨床段階のバイオ製薬会社で、新たな薬物製品の識別と開発に注力している。
注: 2-流動資金、運営計画、持続経営
Br社は設立以来,時期ごとの運営に純損失と負キャッシュフローが出現している。2023年12月31日現在、同社の累積損失は$
2011年以来、 社は脂質ナノ結晶(“LNC”)プラットフォーム輸送技術と関連する 候補製品(MAT 2203とMAT 2501を含む)のパイプラインの開発に取り組んできた。これまで、同社は規制機関のいかなる候補製品の承認も得ておらず、製品販売から何の収入も得られておらず、その候補製品の開発を完了するために巨額の費用が発生すると予想されている。当社は米国や国際的にいかなる兆候でも規制部門の任意の候補製品に対するマーケティング承認を得ることができないかもしれませんし、当社が収入を生み出したり、利益を達成したりする保証もありません。
同社の1つ以上の候補製品が米国食品医薬品局(“FDA”)の承認を得た場合、同社は、同社が商業投入を実現すると、その費用が増加し続けると予想している。同社はまた,現在の候補製品のより多くの臨床研究とより多くの候補製品の開発に伴い,その研究·開発費用が増加し続けると予想している。そのため、会社は予見可能な未来に引き続き巨額の損失を被り、これらの損失は引き続き増加すると予想される。
2023年12月31日現在、会社は現金と現金等価物$を持っています
当社の持続経営企業としての持続経営能力は,その運営費用の制御と,BTIG,LLCと締結された市販協定(“販売協定”)による将来の普通株売却の期待収益に依存する。追加的な融資を受けています当社はこの戦略の実行可能性を信じ、当社が現在とっている行動がその継続的な経営に機会を提供していると信じているが、当社が成功する保証はない。特に、市場状況により、販売プロトコルの使用が不可能である可能性があり、新しい融資は受け入れ可能な条項で獲得できないかもしれない、あるいは全く存在しないかもしれない。当該等の総合財務諸表には、回収可能及び資産分類や負債分類に関する調整は一切含まれておらず、当該等の資産金額や負債の金額及び分類は、当社が経営を継続できない場合に必要である可能性がある。
注: 3-重要会計政策の概要
列報根拠と合併原則
添付されている連結財務諸表は、Holdingsおよびその完全子会社BioPharmaおよびNanoTechnologiesの合併勘定を含む。添付されている連結財務諸表は、米国公認会計原則(“米国公認会計原則”)に基づいて作成され、当社とその完全子会社の経営状況を反映しています。br}すべての会社間取引は合併で抹消されています。
| F-7 |
財務的にコントロールできないイベントの影響
私たちの財務状況と運営結果は、流行病、グローバルサプライチェーンの中断、世界的な貿易紛争、および/または政治的不安定など、私たちがコントロールできないかもしれない要素の影響を受ける可能性がある。金利上昇、特に政府支出の減少や金融市場の変動に加えて、経済の不確実性をさらに増加させ、これらのリスクを悪化させる可能性がある。また、上昇するインフレ率は、従業員関連コストや臨床試験費用など、運営費用を増加させることで、私たちの運営結果に悪影響を及ぼす可能性があります。
社の2023年12月31日までの年間財務業績は、我々がコントロールできない要因の大きな影響を受けておらず、 は上述したように。しかしながら、会社員の持続的な健康状態、サービスプロバイダおよびサプライヤーが運営および交付を継続する能力、会社が運営を維持する能力、およびこれらの要因に対して取られた任意の政府および/または公共行動を含むが、これらの要因に限定されないため、会社は、上述した任意の要因が将来の業績または会社の資金調達能力に及ぼす影響を予測することができない。
見積もりを使った
アメリカ公認会計原則に基づいて連結財務諸表を作成することは管理層に推定と仮定を要求し、即ち: は財務諸表の日付の資産と負債額及び報告期間内の収入と費用に影響する。実際の結果はこれらの推定とは異なる可能性がある。
このような推定や仮定の影響を受ける重大な項目には,当社の研究開発費 や営業権および無形資産減価評価が含まれているが限定されない。
細分化市場 と地理情報
運営部門は企業の構成要素として定義され、その独立した離散情報は、首席運営意思決定者または意思決定グループが資源をどのように割り当てるかを決定し、業績を評価する際に評価することができる。会社はbrの運営を見て管理しています
現金、現金等価物、および制限された現金
当社はすべての元の満期日が3ヶ月以下の高流動性金融商品を現金と現金等価物と見なし、購入日から満期日が3ヶ月を超えるすべての投資は販売可能な債務証券に分類される。現金および現金等価物には、銀行小切手および預金口座内の現金、通貨市場基金、および決済日の3ヶ月以内に満期になる短期米国債が含まれる。会社はキャッシュフロー表に限定的な現金および現金と現金等価物 を列挙した。制限された現金とは、会社が予約しなければならない資金であり、建物の経営リースや他の用途を支払うためのものである。会社の現金、現金等価物、および限定的な現金の完全な開示については、4-現金、現金等価物、制限された現金、および取引可能な債務証券を付記して参照されたい。
販売可能な債務証券
販売可能な債務証券は、アメリカ国債、アメリカ政府手形と会社債務証券を含み、すべて公正価値に基づいて計算され、損益報告が累積の他の総合損失として実現されていないが、非一時的減値と確定された損失は除外される。購入時に生成される任意の割増または割引は、手形の有効期間内に償却され、および/または利息収入および/または費用が増加される。当社は定性と定量的な要素を用いて、公正な価値がコストを下回る低下が信用に関連する損失または他の要素に起因するかどうかを決定するために、売却可能な債務証券の組み合わせを審査する。損益と非一過性と判定された価値低下を純損失に計上する決定を実現し,他の収入,純額を計上した。公正な価値は報告日の市場見積もりに基づいている。証券売却可能な利息とbr配当金は他の収入純額に計上される。会社の取引可能債務証券の完全な開示については、付記4-現金、現金等価物、制限された現金、および取引可能債務証券を参照されたい。
| F-8 |
信用リスク集中度
会社が集中的な信用リスクに直面している金融商品は主に現金、現金等価物、制限された現金と取引可能な債務証券を含む。当社の投資政策は、高信用品質基準に適合する機関のみに投資し、任意の単一の取引相手との投資金額や満期時間に制限を設定しています。残高
は米国金融機関に保存されており,連邦預金保険会社(“FDIC”)
$の保険限度額を時々超える可能性がある
改善と設備のレンタル
レンタル改善と設備はコストから減価償却と償却を減価償却します。設備減価償却は資産の推定耐用年数内に直線法で計算され、推定耐用年数の範囲は至れり尽くせり
廃棄または販売の場合、資産を処分するコストおよび関連する減価償却と償却は口座から が差し引かれ、それによって生じる任意の収益や損失は運営損失に計上される。修理とメンテナンス費用は発生した費用に計上される。当社のリース改善および設備の完全な開示については、付記6-リース改善および設備を参照してください。
営業権とその他の無形資産
商誉 は、被買収エンティティのために支払われる対価格が買収された純資産の公正価値を超える場合に計上される。営業権は、brを償却するのではなく、報告単位で少なくとも年に1回の減値を評価するか、またはイベントおよび状況が営業権が減値可能であることを示すときに、減値をより頻繁に評価する。アメリカ公認会計原則は、会社は定性評価を行うことを選択して、報告単位の公正価値がその帳簿価値より低い可能性があるかどうかを確定することができる。当社の確定状況が確かであれば、当社はさらに定量化分析を行い、確認すべき営業権減価損失金額(あれば)を決定し、計量することができる。
報告単位は、運営部門であるか、または1つの運営部門より1つ低いレベルである。歴史的に見ると、当社は単一の運営部門と報告機関で業務を展開してきた。
2023年12月31日までの年度では,主に当社の継続的な経営能力に大きな疑問があるため,当社はその報告単位を定量的に分析して営業権減値を評価している。数量化審査の一部として、当社はその公正価値(市場参加者が当社を潜在的に買収する際に支払いたい価格で決定する)がその帳簿価値(営業権を含む)を超えるかどうかを考慮する。当社の評価結果によると、当社はその営業権に欠陥がないと判断した。
当社は、2022年12月31日までの年度について、その報告ユニットを定性分析することにより、営業権減価を評価しています。定性的審査の一部として、当社は関連事件や状況を考慮し、当社がその時価によって決定した公正価値がその帳簿価値よりもはるかに高いことを強調しています。当社の評価結果によると、当社はその営業権が損害を受けていない可能性が高いことを確認しました。
不確定なbr}普通無形資産は、現在行われている研究開発(“IPR&D”)からなり、買収時に技術的実行可能性が達成されていない、あるいは監督管理の承認が不足している業務グループで買収されたプロジェクトを代表する。 これらのIPR&D資産は毎年減値審査を行い、事件や状況変化が資産の帳簿価値が回収できない可能性があることを示す場合、技術実行可能性や監督管理許可を確定した後により早い審査を行う。減価損失(あれば)は,資産の公正価値とその帳簿価値を比較することで計算される.資産の帳票価値がその公正価値を超えると,差額に減値損失を計上し,その帳簿価値はそれに応じて減少する.営業権減価テストと同様に、当社は、確認されるべき減価損失金額を決定して測定するために、数量化分析が必要であるかどうかを決定するために、無期限減値無形資産を定性的に審査することができる。
| F-9 |
当社は2023年12月31日までにキャッシュフロー割引手法を用いて定量化分析を行い,公平なbr価値を決定し,無期限減価資産は減値していないと判断した。当社は2022年12月31日までに定性 方法を採用し,無期限普通資産が減値していない可能性が高いと判断した。
賃貸借証書
財務会計基準委員会(“FASB”)会計基準編纂(“ASC”)テーマ842、“レンタル”、“br}は、テナントに12ヶ月を超えるすべてのレンタルROU資産およびレンタル負債を保証することを要求する使用権(”ROU“)モデルを構築した。リースは財務リースや経営的リースに分類され,分類は損益表における費用確認のモデルや分類に影響を与える。新しい基準の下でのレンタル人会計は基本的に変わらない。追加的な定性的で定量的な開示も必要だ。当社の賃貸借契約の完全開示については、付記8-借約を参照されたい。
所得税 税
繰延税項は負債法によると、繰延税項資産は差し引くことができる一時的な差異であることが確認され、営業損失及び税項相殺繰越及び繰延税項負債は課税の一時的な差異であることが確認された。一過性差異 は,報告されている資産と負債額とその税基との差である。経営陣が繰延税金資産の一部または全部が現金にならない可能性が高いと考えている場合、繰延税金資産はbrの推定値を引いて準備される。繰延税項資産及び負債は、公布日を含む期間内に、税法及び税率変動の影響に応じて調整される。
同社は、ASC 740−10の規定を採択し、申告書の提出が義務付けられている可能性のある司法管轄区における2023年および2022年の申告書を分析した。当社は、その所得税申告状況と控除額は監査後も継続的に存在すると信じており、その財務状況に重大な変化を招く調整は何もないと予想されている。したがって,不確定な
所得税頭寸記録準備金はない。当社の政策は、所得税支出において所得税に関する利息及び/又は罰金
を確認することです。その会社は所有している
会社設立以来の納税年度ごとに純営業損失が発生しているため、2014年から2022年までの所得税申告書は、納税属性を使用した年から少なくとも3年以内に国税局の審査と調整を受ける。
公正価値計測
ASC 820“公正価値計量”で定義されているように、公正価値計量は、公正価値レベルの3つのレベルでそれぞれ開示されなければならない。公正価値で記録された資産と負債については,会社の政策は既定の公正価値レベルに基づいて 公正価値計量を策定する際に,観察可能な 投入(活発な市場のオファー)を最大限に使用し,観察できない投入(会社の仮定)を最大限に減らすことである。会社の公正価値計量の完全な開示については、付記5-公正価値計量を参照してください。
株式ベースの従業員報酬には、株式オプション付与と制限株が含まれる。当社はASC 718-10の規定により株式報酬を計算します報酬--株式報酬従業員、非従業員 及び取締役に支払われるすべての株式ベースの支払いは、株式オプション及び制限株式の付与を含み、総合経営報告書で を確認し、付与された日に必要なサービス期間(通常は奨励に関する帰属期間)の公正価値で全面赤字を確認しなければならないことが規定されている。没収は発生状況に応じて計算されます。一般に,当社が発行する株式オプション奨励は 個のサービス類付与条件のみであり,これらの報酬の費用を直線的な方法で記録する.会社が株式に基づく報酬費用を分類する方式は、受賞者の給料やサービスプロバイダのコストを分類する方式と同じだ。
| F-10 |
会社はASCテーマ505小テーマ50の規定により、非従業員に発行された株式ツールを会計処理する非従業員に持分ベースの支払いを支払う対象ツールの公正価値に基づいている.株式ツールはコンサルタントに付与された株式オプションからなり,ブラック·スコアーズ推定モデルを用いて評価を行った。会社はブラック-スコアーズ定価モデルを用いて付与オプションの公正価値を計算し、推定された公正価値或いは普通株から制限株の公正価値を推定した。
従業員と非従業員報酬による補償費用は、通常、奨励に必要な サービス期間内に直線的に確認される。
1株当たり純損失情報は2種類の方法で決定され、期間内に発行された普通株と他の証券の加重平均株式数を含む。
2種類の法の下で、普通株株主が1株当たりの基本純損失を占めるべき計算方法は、普通株株主は純収益を期間中に発行された普通株の加重平均株式数で割るべきである。普通株株主は当株当たり純損失 を占めるべきであり、(1)2種類の法或いは(2)IF割引法の中で希釈程度が高い方法で計算すべきである。
2023年および2022年12月31日までの年度中に、1株当たりの普通株収益が普通株1株当たりの基本収益と同じになるのは、当社が各届出期間中に純損失を出したため、発行されたすべての引受権および引受権証の行使による潜在的な希薄化証券が逆償却作用が生じると仮定したためである。2023年12月31日、2023年、2022年までの希釈株式残高は以下の通り
| 12月31日まで | ||||||||
| 2023 | 2022 | |||||||
| 株式オプション | ||||||||
| 株式承認証 | ||||||||
| 合計する | ||||||||
収入 確認
主題606によれば、当社が確認した収入は、これらの商品またはサービスと交換するために、エンティティが獲得する権利が予想される対価格を反映する顧客への譲渡承諾された商品またはサービスの金額を記述する。このコア原則を達成するために、主題606は、i)顧客との契約を決定するステップ、ii)契約における履行義務を決定すること、iii)取引価格を決定すること、iv)契約中の個々の履行義務に取引価格を割り当てること、およびv)義務を履行する際に義務履行に関連する収入を確認することを含む、顧客契約からの収入を確認する5つのステッププロセスについて概説する。
Br契約の開始時に、会社は、各契約で約束された貨物またはサービスを評価し、各約束された貨物またはサービスが異なるかどうかを評価し、どれが義務を履行するかを決定する。そして、会社は、履行義務を履行する際にそれぞれの履行義務に割り当てられた取引価格の金額が収入であることを確認する。
会社の収入には,2023年と2022年12月31日までの年間で,会社独自のLNCプラットフォームを利用したメッセンジャーリボ核酸フォーマットの組み合わせを評価することを目的としたBioNTechプロトコルからの契約研究収入が主に含まれている。会社収入確認の完全開示 については、付記9-収入確認、協力協定、その他の研究·開発プロトコルを参照してください。
2019年12月12日、会社は遺伝子テーク社(“遺伝子テーク”)とフィージビリティスタディ協定(“プロトコル”)を締結した。このフィージビリティスタディは、会社のLNCプラットフォームを用いて経口製剤を開発することで、様々な納入困難な分子
を開発することができる。合意条項によると、遺伝子テイクは同社に合計#ドルを支払った
| F-11 |
連携 プロトコル
同社は、協力プロトコルがASCトピック808に支配されているかどうかを評価している協力手配(主題808)それらが共同経営活動に関連しているかどうか、および双方が手配に積極的に参加し、重大なリスクおよびリターンに直面しているかどうかに基づく。このスケジュールが主題808の範囲に属する場合、会社は、主題606の会計指導単位をクラスプッシュ的に適用して、異なる履行義務を決定し、その後、各異なる履行義務に顧客関係 が存在するかどうかを決定する。会社が合意における履行義務が顧客に関連していると判断した場合、会社は主題606における指導を適用する。プロトコル内の異なるバンドルされた商品またはサービスの一部がクライアントに属さない場合、課金単位は主題606の範囲内になく、課金単位の確認および計量は、権威ある会計文書に対する類比に基づくべきであるか、または適切な類比がない場合、合理的、理性的、および一貫して適用される会計政策選択に基づくべきである。
このような手配の条項は、一般に、前払い費用、開発および規制支払い、製品供給サービス、研究開発コスト精算、利益共有スケジュール、およびいくつかの製品が商業化に成功した後の印税のうちの1つまたは複数を会社に支払うことを含む。これらの手配の会計計算の一部として,会社は契約で決定された契約義務ごとの独立販売価格を決定する必要があると判断する仮定 を策定している.これらの重要なbr}仮説は、予測された収入、臨床開発スケジュールおよびコスト、人員コストの販売率、割引率、および技術および法規が成功する確率を含む可能性がある。
前払いライセンス料 :当社の知的財産権許可が合意で決定された他の履行義務 とは異なると判定された場合、当社は、許可が被許可者に譲渡され、被許可者が許可を使用して利益を得たときに許可に割り当てられた払戻不可能な前払い費用の収入 を確認することを確認し、これは、通常、契約開始時または契約開始近くに発生する。他の約束とバンドルされたライセンスについて、会社は、合併履行義務が一定期間内に履行されているか、またはある時点で履行されているかを決定するために、判断を利用して、合併履行義務の性質を評価し、時間が経過するにつれて、払い戻し不可能な前払い費用収入を確認するために、進行を測定する適切な方法を決定する。当社は各報告期間終了時に進捗測定基準を評価し、必要に応じて業績測定基準と関連収入確認を調整する。
研究 と開発マイルストーン支払い:開発マイルストーンの支払いを含む各手配が開始された場合,会社 はマイルストーンが達成可能であると考えられるかどうかを評価し,可能な金額法を用いて取引価格に含まれる金額を推定する.大きな収入逆転が起こらない可能性が高い場合,関連するマイルストーン 値は取引価格に含まれる.承認に関する不確実性が解決されるまで,会社や被許可者の制御範囲内にない記念碑的支払い(例えば規制承認)は実現可能とはみなされない.そして,取引価格は相対的に独立した販売価格で契約履行義務ごとに割り当てられ,会社は契約項下の履行義務を履行する際に収入を確認する.その後の各報告期間が終了した時点で、当社はこのような開発と規制マイルストーンを実現する可能性と、任意の関連変数 の価格制限を再評価し、必要に応じて当社の全体取引価格の見積もりを調整します。いずれの調整 も累積追跡をもとに記録されている.
研究と開発費の精算:同社の協力計画には、将来の臨床開発と薬物安全サービスの承諾と、いくつかの合同委員会への参加が含まれる可能性がある。このようなサービスが顧客またはパートナーに提供され、これらのサービスが会社の連携パートナーに提供されるライセンスとは異なる場合、これらの約束は、個別の履行義務として入金され、会社は、発生した内部開発コストと、合意期限 による予測を用いてこれらの義務を推定する。当社はこれらのサービスの収入を記録しており,進捗に応じてこれらのサービスの履行義務が時間の経過とともに履行されているためである.しかし,会社がその連携パートナーがこれらの連携研究や開発活動の顧客ではないと結論すれば,このような支払いを研究開発費削減として提出する.
| F-12 |
研究と開発計画:我々とCFFプロトコルの研究開発プロトコルの条項によると、当社はこのスケジュールを主題606に基づいて説明していません。しかし、同社は製品が成功し、商業化された後に得られるリスクとリターンを分担するため、協力協定のパートナー
であることを確認している。そこで,本プロトコル条項により受け取ったbr資金を研究·開発コストの精算として入金し,会社の経営報告書と全面赤字報告書の中で研究·開発費用を削減する。累積コスト額が大きく逆転する可能性が高い場合、会社は合意に関連するある材料や他の研究開発コストの精算を記録する。2023年12月31日及び2022年12月31日まで、当社は確認しました
研究開発費
研究と開発費用には、主に、我々の候補製品の組み合わせの臨床前および臨床開発に関するコストが含まれている
| ● | 契約研究機関(“CRO”)や他のサプライヤーおよび契約製造組織(“CMO”)が薬物や薬品の生産について手配した外部研究開発費; および |
| ● | 従業員に関連する費用には、賃金、福祉、株式給与費用が含まれる。 |
研究および開発費用には、製品ライセンスおよび関連技術権利(将来他の用途がない場合)を取得するコスト、研究開発のためのプロトタイプコスト、顧問料、および私たちのいくつかのパートナーに支払われる金額も含まれる。
FASB ASCテーマ730によると、研究開発で発生したすべての研究開発費は運営に計上されています。当社は将来の研究開発活動で使用している貨物とサービスの前払いを返金しません。 サービスが完了した場合、または荷物を受け取った場合、会社はこれらの前払いを費用として記入するのではありません。
研究開発費を計上する
会社の財務諸表作成プロセスの一部として、会社はその計算費用を見積もる必要があります。 この流れは、見積書と契約を審査し、会社を代表して提供されたサービスを決定し、会社が領収書を発行していない場合や他の方法で実際のコストを通知している場合に、提供されるサービスのレベルとサービスによるコストを見積もることに関連しています。会社のあるサービス提供者は毎月会社に借金の領収書を発行して、提供されたサービスまたは契約マイルストーンに達したときの借金を支払う。当社は、当社が当時知っている事実と状況に基づいて、その財務諸表の中で貸借対照表毎の日付までの課税費用を推定します。会社は定期的にサービスプロバイダとその推定の正確性を確認し、必要に応じて調整する。当社が計算すべき研究開発費の重大な見積もりは、CRO、CMO、その他のサプライヤーが研究開発と製造活動に関連する費用と関係がある。
CROやCMOに関する会社の費用は,見積とそれを代表する研究開発·製造活動を行うサプライヤーとの契約に基づいて,受信したサービスとかかる作業量を推定している。これらの合意の財務条項 は協議が必要であり、契約によって異なり、支払いの流れが不均一になる可能性があります。場合によっては、会社のサプライヤーに支払われるお金は、提供されたサービスレベルを超え、適用される研究開発または製造費用の前払いをもたらす可能性がある。サービス料を計算する際には,会社はサービスを提供する時間帯 と時間ごとにかかる作業量を見積もる.実際にサービスを実行する時間または努力の程度がその推定値と異なる場合、会社はそれに応じて計算または前払い費用を調整する。実際に発生した金額と実質的に差がないと予想されているにもかかわらず、提供されたサービスの実態や時間に対する会社の提供状態や時間の理解が異なる可能性があり、任意の特定の時期に当社が報告した金額が高すぎたり、低すぎたりする可能性がある。本報告に掲げる期間の見積り数は実質的に変化しなかった。
| F-13 |
特許費用
特許取得·保護による法的費用やその他の直接コストも発生時に一般費用と 総合経営報告書における管理費用を計上する。
その他 全面赤字
その他の全面赤字には、売却可能な販売可能債務証券の純収益/(赤字)と未実現損失が含まれており、合併経営報告書と全面赤字に列報されている。
最近の会計声明
FASBは2023年11月、会計基準更新(ASU)第2023-07号を発表した支部報告(テーマ280):改善可報告分部開示 (“ASU 2023-07”)。本ASUは,定期的に首席運営決定者(“CODM”)に提供される重大な支部費用,説明 を構成する他の支部項目を持つ金額,およびCODMの名称や役職の開示を要求する。ASU 2023-17は、2023年12月31日以降の年度期間と2024年12月15日以降の財政年度内の移行期間に適用される。早期採用を許可し、このASUを財務諸表に示された各時期に遡及適用すべきである。当社は1つの運営および報告支部でその運営およびそのbr業務を検査·管理しているため、当社はこの追加開示はその総合財務諸表や関連開示に影響を与えないと信じている。したがって、私たちは2024年第4四半期にこのASUの規定を採用するつもりだ。
FASBは2023年12月にASU 2023-09を発表しました所得税(特集740):所得税法の改善(“ASU 2023−09”). 本ASUは,公共業務エンティティが毎年表レート台帳に特定のカテゴリを開示し,5%数の閾値に達した入金項目に の他の情報を提供することを要求する.また、ASUは、すべてのエンティティ に、連邦、州および外国税収に分類された納付済み所得税金額、および納付済み所得税総額の5%以上の個別司法管轄区域を開示することを要求する。ASU 2023-09は、2024年12月31日以降の年間期間に適用されます。早期採用を許可し、予想に基づいてこのASUを適用し、財務諸表に遡及応用を許可すべきである。会社は現在、新しい基準が私たちの連結財務諸表と関連開示に及ぼす影響を評価しています。したがって、私たちは2025年第4四半期にこのASUの規定を採択するつもりだ。
注: 4-現金、現金等価物、制限された現金、および取引可能な債務証券
当社はすべての元の満期日が3ヶ月以下の高流動性金融商品を現金と現金等価物と見なし、購入日から満期日が3ヶ月を超えるすべての投資は販売可能な債務証券に分類される。現金および現金等価物には、銀行小切手および預金口座内の現金、通貨市場基金、および決済日の3ヶ月以内に満期になる短期米国債が含まれる。
現金、現金等価物、制限された現金
Br社は、キャッシュフロー表に限定的な現金および現金および現金等価物を列挙します。2023年12月31日と2022年12月31日の制限現金はそれぞれ$
| F-14 |
次の表では、連結貸借対照表で報告されている現金、現金等価物および制限現金を、2023年12月31日、2022年12月31日および2021年12月31日までの連結現金フロー表の総額と照合する
十二月三十一日 2023 | 十二月三十一日 2022 | 十二月三十一日 2021 | ||||||||||
| 現金と現金等価物 | $ | $ | $ | |||||||||
| 流動/非流動資産に含まれる制限された現金 | ||||||||||||
| 現金フロー表の現金、現金等価物、および制限現金 | $ | $ | $ | |||||||||
販売可能な債務証券
同社は、売却可能な債務証券への投資を売却可能および流動資産に分類している。当社は取引可能な債務証券への投資を公正価値で計上し、収益と損失を株主権益の単独構成要素として計上していない。未実現損失と収益は他の総合(損失)/収入に分類され,コストは特定の確認に基づいて決定される.取引可能債務証券の実現済み収益と損失は他の収益純額に記入する。当社は2023年12月31日および2022年12月31日までの年間損益は何も実現していません。当社は2023年12月31日および2022年12月31日までに未実現収益/(損失)$を記録した
次の表は、2023年12月31日までの会社の有価証券をまとめたものです
| 償却する | 実現していない | 実現していない | ||||||||||||||
| コスト | 利得 | (損をする) | 公正価値 | |||||||||||||
| アメリカ国債 | $ | $ | $ | ( | ) | $ | ||||||||||
| アメリカ政府債券 | ( | ) | ||||||||||||||
| 取引可能債務証券総額 | $ | $ | $ | ( | ) | $ | ||||||||||
売却可能に分類されたすべての債務証券は2023年12月31日の1年以内に満期になる。
| 原価を償却する | 実現していない 利得 | 未実現(赤字) | 公正価値 | |||||||||||||
| アメリカ国債 | $ | $ | $ | ( | ) | $ | ||||||||||
| アメリカ政府債券 | ( | ) | ||||||||||||||
| 会社債務証券 | ( | ) | ||||||||||||||
| 取引可能債務証券総額 | $ | $ | $ | ( | ) | $ | ||||||||||
売却可能な債務証券に分類される満期日 は以下のとおりである
| 公正価値 | ||||
| 1年以内に満期になる | $ | |||
| 1年から5年後に期限が切れなければならない | ||||
| $ | ||||
社は2023年12月31日と2022年12月31日までの未実現(赤字)と収益が一時的であることを決定した。未実現(損失)および収益 は、通常、発行者や基礎資産の信用品質が根本的に弱いことによるキャッシュフローの不利な変化の結果ではなく、市場参加者が要求するリスクプレミアム増加の結果である。
| F-15 |
注: 5-公正価値計量
会社は公正な価値レベルを使用してその金融商品の価値を測定する。公正価値階層は、観察可能または観察不可能な公正価値を計量するための推定技術の投入に基づいている。観察可能な入力は、市場参加者が独立したソースから得られた市場データが資産または負債の価格設定に基づいて使用されるという仮定を反映し、観察できない入力は、報告エンティティがそれ自身の市場仮定に基づいて行う定価を反映する。この階層構造内の各 レベルの公正価値計測の基礎は以下のとおりである
| ● | レベル 1-アクティブ市場における同じ資産または負債の見積もり。 |
| ● | レベル 2-非アクティブな市場における同じまたは同様の資産および負債の見積もり;またはその投入が直接または間接的に見られる他のモデル由来の推定値、またはその重要な価値駆動要因が見られる。 |
| ● | 第 レベル3-推定モデルの1つまたは複数の重要入力では が観察されず、管理層推定に基づいて仮説を使用する推定技術からの推定値。 |
当社は推定技術を採用し、観察可能な投入を最大限に利用し、観察できない投入を最大限に減少させ、公正な価値を評価する際に取引相手の信用リスクを考慮する。
これらのツールの短期的な性質のために、現金等価物、制限された現金の現在の部分、前払い費用および他の流動資産、支払すべき負債、賃貸負債の現在の部分、および計算されるべき費用の帳簿金額は、公正な価値に近い。
上記の階層構造に従って公正価値に基づいて入金された資産と負債の概要は以下のとおりである
| 公正価値階層構造 | ||||||||||||||||
| 2023年12月31日 | 合計する | (レベル1) | (レベル2) | (レベル3) | ||||||||||||
| 資産 | ||||||||||||||||
| 取引可能債務証券: | ||||||||||||||||
| アメリカ国債 | $ | $ | $ | $ | ||||||||||||
| アメリカ政府債券 | ||||||||||||||||
| 合計する | $ | $ | $ | $ | ||||||||||||
| 公正価値階層構造 | ||||||||||||||||
| 2022年12月31日 | 合計する | (レベル1) | (レベル2) | (レベル3) | ||||||||||||
| 資産 | ||||||||||||||||
| 取引可能債務証券: | ||||||||||||||||
| アメリカ国債 | $ | $ | $ | $ | ||||||||||||
| アメリカ政府債券 | ||||||||||||||||
| 会社債務証券 | ||||||||||||||||
| 合計する | $ | $ | $ | $ | ||||||||||||
米国債は、アクティブ市場で同じ 資産の見積市場価格を用いて推定されるため、公正価値レベルの第1級に分類される。米国政府手形と会社債務証券からなる取引可能債務証券は2級に分類され、非アクティブ市場の見積市場価格を用いて推定される。
| F-16 |
注: 6-レンタル改善と設備
レンタルの主なカテゴリー別にまとめられた内装と設備には、2023年12月31日と2022年12月31日までの年度が含まれています
十二月三十一日 2023 | 十二月三十一日 2022 | |||||||
| 装備 | $ | $ | ||||||
| 賃借権改善 | ||||||||
| 合計する | ||||||||
| 減算:減価償却累計と償却 | ||||||||
| レンタル改善と設備、正味価値 | $ | $ | ||||||
2023年12月31日までと2022年12月31日までの年度の減価償却と償却費用は$
注: 7-費用とその他の負債を計算すべきである
計上すべき費用とその他の負債は、2023年12月31日と2022年12月31日終了年度の以下の項目を含む主要カテゴリ別にまとめられている
| 12月31日まで | ||||||||
| 2023 | 2022 | |||||||
| 賃金総額とインセンティブ | $ | $ | ||||||
| 一般と行政費用 | ||||||||
| 研究開発費 | ||||||||
| 繰延収入* | ||||||||
| その他繰延負債** | ||||||||
| 合計する | $ | $ | ||||||
| * | |
| ** |
注: 8-賃貸借証書
Br社は、オフィススペース、実験室、製造施設、各種設備を含む様々なレンタル契約を締結しています。
経営リースおよび融資リースは、会社の総合貸借対照表にリース使用権資産、現在の賃貸負債、長期賃貸負債として示されている。私たちがレンタルした資産と負債は、レンタル開始日 レンタル期間内の残り賃貸支払いの現在値確認に基づいて、当社の逓増借款金利や暗黙的なbr金利(確定しやすい場合)を使用します。初期期間が12ヶ月以下の短期賃貸は貸借対照表 に記録されない。当社の経営リースは暗黙的な金利を提供していないため、当社はその増額借入金金利(信用状況が似ている会社の長期借入コストに応じて決定される)を利用してレンタル義務を記録している。同社の融資リースは決定しやすい隠れた金利を提供する。経営性賃貸については、当社は賃貸手配の固定構成要素に基づいて、最低賃貸料支出 を直線原則で確認した。会社はレンタル開始日からのレンタル期間内にこの費用を償却します。
| F-17 |
運営 レンタル義務
2013年11月1日、当社は
2016年12月15日、当社は締結しましたレンタルニュージャージー州ブリッジウォルトの実験室と製造スペース。レンタルは2017年8月から。月極からの価格は約$です
Br社はその運営リースによりレンタル費用が発生した$
融資リース
会社の融資リースによる利息支出は#ドルです
次の表には、2023年12月31日までの会社の経営リースと融資リースによる負債額と時間を示しています
| 賃貸負債満期日 | リース負債を経営する | 融資リース負債 | ||||||
| 2024 | $ | $ | ||||||
| 2025 | ||||||||
| 2026 | ||||||||
| 2027 | ||||||||
| 2028 | ||||||||
| その後… | ||||||||
| 未割引経営賃貸支払総額 | $ | $ | ||||||
| 差し引く:推定利息 | ||||||||
| リース負債現在価値を経営する | $ | $ | ||||||
| 加重平均残存賃貸年限(年) | ||||||||
| 加重平均割引率 | % | % | ||||||
次の表には、2022年12月31日までの会社の経営リースと融資リースによる負債額と時間を示す
| 賃貸負債満期日 | リース負債を経営する | 融資リース負債 | ||||||
| 2023 | $ | $ | ||||||
| 2024 | ||||||||
| 2025 | ||||||||
| 2026 | ||||||||
| 2027 | ||||||||
| その後… | ||||||||
| 未割引経営賃貸支払総額 | $ | $ | ||||||
| 差し引く:推定利息 | ||||||||
| リース負債現在価値を経営する | $ | $ | ||||||
| 加重平均残存賃貸年限(年) | ||||||||
| 加重平均割引率 | % | % | ||||||
| F-18 |
注: 9-収入確認、協力協定、その他の研究開発協定
BioNtech研究協力
2022年4月8日、会社は、会社独自のLNCプラットフォームを利用したメッセンジャーリボ核酸フォーマットの組み合わせを評価するためにBioNTechプロトコルを締結した。BioNTech協定の条項によると、会社は#ドルの排他的費用を受け取った
同社はASC 808に基づきBioNTechプロトコルを評価した協力手配ASC 606と顧客と契約を結んだ収入 (“ASC 606”)、手配構造に基づいて結論を出すと、契約相手側BioNTech SEは、手配構造に基づく顧客である。(1)独占研究許可証および(2)臨床研究サービスを付与する2つの重大な約束を契約に基づいて決定した。しかし、承諾の性質を考慮して、ライセンスと研究サービスは契約の範囲内で互いに異なるとはみなされていない。したがって、会社は許可証と研究サービスの両方に合併の履行義務があると結論した。
$
2023年12月31日までの年度は$
嚢胞性線維症財団治療発展賞
2020年11月19日,同社は嚢胞性線維化財団(CFF)と奨励協定(“CFF協定”)を締結し,同協定により$までの治療発展賞を受賞した
2023年12月31日までに会社は$を受け取りました
| F-19 |
遺伝子テーク実行可能性研究プロトコル
2019年12月12日、会社は、会社のLNCプラットフォームを用いた経口製剤の開発に関する遺伝子テーク協定を締結した。遺伝子テイク協定の条項によると、遺伝子テイクは同社に合計#ドルを支払った
ライセンス プロトコル
Aquariusを買収することにより,会社はロゲス大学,ニュージャージー州立大学(ニュージャージー医学と歯科大学のbr権益相続人)のLNCプラットフォームに関するいくつかの特許の許可(“許可br}協定”)を取得した
注: 10-支払いを引き受ける
印税支払権
当社は、A系列優先株の割引、権利、指定証明書(“指定証明書”)
を制限する条項により、最高$を支払う必要がある場合があります
雇用契約
Br社はまた、ある従業員と雇用契約を締結しており、ある事件が発生した場合、支配権変更、無断解雇、退職などの場合、特定の金額の資金を提供する必要がある。
その他 正常業務運営プロトコル
また,正常業務運営過程において,会社は契約サービスプロバイダと協定を締結し,brの研究開発や製造活動の展開に協力している。これらの第三者への支出は臨床開発の大きなコストを表しており、前金と長期現金約束が必要かもしれない。拘束力のある調達注文に要求される通知期間及び義務に適合する場合、会社は、これらの合意項目の下での作業を停止することを随時選択することができる。
法的手続き
会社は現在いかなる法的手続きの一方でもなく,会社はその業務に対するクレームや訴訟が未解決または脅かされていることを知らない.将来、会社は時々私たちの正常な業務過程におけるクレームに関する訴訟に巻き込まれるかもしれない。
| F-20 |
注: 11-所得税
会社はバランスシート法を採用して繰延所得税を計算する。この方法によれば、繰延税金負債と資産 が帳簿金額と資産および負債の課税ベースとの間の一時的な差によって生じる予想される将来の税項結果が確認される 。既存の証拠の量に基づいて、繰延税金資産の一部または全部が現金化できない可能性が高い場合、繰延税金資産の推定値で準備する。当社の政策は税収が確定していない利息と罰金を所得税費用に計上することです。当社は2023年12月31日と2022年12月31日まで、重大な 不確定な税務状況が存在するとは考えていません。したがって、税金状況が不確定であるため、利息と罰金は計上されていない。
所得税に規定されているbrの構成要素は以下のとおりである
| 十二月三十一日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| 当期費用(福祉): | ||||||||
| 連邦制 | $ | $ | ||||||
| 状態.状態 | ||||||||
| 外国.外国 | ||||||||
| 総当期費用(収益): | $ | $ | ||||||
| 繰延費用(福祉): | ||||||||
| 連邦制 | $ | $ | ||||||
| 状態.状態 | ||||||||
| 外国.外国 | ||||||||
| 繰延費用(収益)合計: | $ | $ | ||||||
| 所得税総支出(福祉): | $ | $ | ||||||
繰延所得税は、財務報告目的のための資産および負債の帳簿価値と所得税目的のための金額との間の一時的な差異の純影響を反映する。米国の法定連邦税率と会社の有効税率 の入金は以下の通りである
| 十二月三十一日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| アメリカの法定税率で計算される収入 | % | % | ||||||
| 連邦福祉を差し引いた州税 | % | % | ||||||
| 恒久的差異 | ( | )% | ( | )% | ||||
| 税金控除 | % | % | ||||||
| 評価税免除額 | ( | )% | ( | )% | ||||
| % | % | |||||||
一般および行政費用に含まれるある国の最低税額を除いて、会社は現在所得税に対応していない。
当社の2023年と2022年の繰延税金資産(負債)の重要な構成要素は、以下の通りです
| 十二月三十一日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| 株式ベースの報酬 | $ | $ | ||||||
| 減価償却および償却 | ( | ) | ( | ) | ||||
| 負債を計算すべきである | ||||||||
| 営業純損失繰り越し | ||||||||
| 研究開発信用繰り越し | ||||||||
| 研究開発費第174条 | ||||||||
| 他にも | ( | ) | ||||||
| 知的財産権研究開発 | ( | ) | ( | ) | ||||
| ROU資産 | ( | ) | ( | ) | ||||
| ROU責任 | ||||||||
| 繰延税金資産総額 | $ | $ | ||||||
| 推定免税額 | ( | ) | ( | ) | ||||
| 繰延税項目純資産(負債) | $ | ( | ) | $ | ( | ) | ||
| F-21 |
2020年3月27日、“コロナウイルス援助、救済、経済安全(CARE)法案”が法律となり、国内税法が何度も改正された。これらの変化には,控除可能な利息支出額を増加させる制限,br社のある純営業損失の繰越を許可すること,および課税所得額を相殺するために使用可能な純営業損失繰越額を増加させることが含まれるが,これらに限定されない。この法案における税法の変化は会社の所得税規定に実質的な影響を与えていない。
繰延税金資産の現金化能力を評価する際には、当社は繰延税金資産の一部または全部が現金化できない可能性が高いかどうかを考慮する。繰延税金資産の最終現金化は、将来の純控除可能金額を代表する臨時差額が減税期間に生じる課税所得額に依存し、当社が損失を当社の課税所得額に繰り越す能力の影響を受ける。当社は赤字の歴史と他の肯定的な証拠が不足して課税収入を支持しているため、当社は現金化しないことが期待されている繰延税金資産計について評価を準備しています。推定免税額は$である
2023年12月31日現在、会社の連邦純営業損失は$に転換している
以前に発生あるいは将来発生する可能性のある会社の所有権変更により、繰り越しの営業純損失と一般営業税の繰越免除の使用は1986年の“国税法”第 382と383節の重大な制限を受ける可能性がある。これらの所有権変更は、毎年それぞれ将来の課税収入と税収を相殺する純営業損失と一般営業税の繰越免除金額 に使用できる可能性があります。一般に、第(Br)382節の定義によれば、所有権変更とは、3年以内に、ある株主や公共団体の会社株における所有権が50ポイント以上増加した取引を意味する。当社は設立以来所有権変更 を経験しているかどうかを確認するために研究を完了していない。
“2017年減税·雇用法案”によると、研究開発コストは完全に控除されることはなく、2022年1月1日から米国の税収目的に資本化·償却しなければならない。義務資本化要求は私たちの繰延税金資産を増加させる。
販売純営業損失(NOL)と税収控除
会社確認$
注: 12-株主権益
市場の株式発行
当社は2020年7月2日にBTIG,LLC(“BTIG”)と市販プロトコル(“販売プロトコル”)を締結し,この合意により,当社はBTIGを介して販売代理および/または依頼者として普通株式br}株式を随時発売および販売することができ,総発行価格は最高$に達する
| F-22 |
普通株 株
2022年2月8日会社発表第二次改正及び再署名された独占許可協定に基づき、その普通株式の未登録株式を一部の代償としてニュージャージー州立大学ロッグス大学(“ロッグス大学”)に売却する
優先株
“会社登録証明書”によると,会社は発行する権利がある額面$の優先株.
株式承認証
当社は2023年12月31日現在、株式承認証を発行して普通株を購入したものは何もない。2023年12月31日と2022年12月31日までの未結権証要約 は以下のとおりである
| 株 | ||||
| 2021年12月31日現在の未返済債務 | ||||
| 発表されました | ||||
| 鍛えられた | ( | ) | ||
| 期限が切れる | ( | ) | ||
| 2022年12月31日に返済されていません | ||||
| 発表されました | ||||
| 鍛えられた | ||||
| 期限が切れる | ( | ) | ||
| 2023年12月31日現在の未返済債務 | ||||
注: 13-累計その他総合収益/(損失)
次の表は、2023年12月31日、2023年12月31日、2022年12月31日までの年度内の構成要素別累計その他全面収益/(赤字)の変化をまとめています
| 証券売却可能な未実現純収益/(損失) | 累計その他総合収益/(損失) | |||||||
| バランス、2021年12月31日 | $ | ( | ) | $ | ( | ) | ||
| 売却可能証券は純損失を実現していない | ( | ) | ( | ) | ||||
| バランス、2022年12月31日 | $ | ( | ) | $ | ( | ) | ||
| 証券売却可能な未実現純収益 | ||||||||
| バランス、2023年12月31日 | $ | ( | ) | $ | ( | ) | ||
その他の総合収益/(損失)を累積したすべての 組成物は税引き後純額である。
| F-23 |
会社が改正し再確認した2013年株式報酬計画(“計画”)は、条件を満たす従業員、高級管理者、非従業員取締役、その他の個人サービス提供者に奨励的株式 オプション、非制限株式オプション、制限株式単位、業績単位、株式購入権を付与することを規定している。この計画下のオプションには取締役会報酬委員会が決定した授与日の株式数。報酬委員会は、計画で定義されたいくつかの制限に基づいてオプションが行使可能な期限を決定し、 現在返済されていないオプションは通常付与されるあるいは…何年もですそれは.2023年12月31日までに会社は本計画に基づいて発行された普通株式を許可します。
取締役会と多数の株主の承認を経て、2014年5月8日から、この計画の改訂と再記述が行われた。修正案は,毎年1月(取締役会の承認を経て)から,その計画に基づいて発行可能な普通株式数 を自動的に増加させることを規定している最大4%に達します%)前年12月31日に発行された普通株式総数
| 十二月三十一日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| 研究と開発 | $ | $ | ||||||
| 一般と行政 | ||||||||
| 合計する | $ | $ | ||||||
| 贈られた賞を保留する | 発行·行使された裁決 | 授与可能な賞 | ||||||||||
| 2013株式報酬計画(単位:千) | * | ** | ||||||||||
| * | |
| ** |
株 オプション
| オプション数 | 加重平均行権値 | 加重平均契約期間(年) | ||||||||||
| 2022年1月1日に返済されていません | $ | |||||||||||
| 授与する | ||||||||||||
| 鍛えられた | ( | ) | ||||||||||
| 没収される | ( | ) | ||||||||||
| 期限が切れる | ( | ) | ||||||||||
| 2022年12月31日に返済されていません | $ | |||||||||||
| 授与する | ||||||||||||
| 鍛えられた | ||||||||||||
| 没収される | ( | ) | ||||||||||
| 期限が切れる | ( | ) | ||||||||||
| 2023年12月31日現在の未返済債務 | $ | |||||||||||
| F-24 |
| 行権価格区間 | 突出した数字 | 1株あたりの加重平均行権値 | ||||||
| $ - $ | $ | |||||||
| $ - $ | $ | |||||||
| $ - $ | $ | |||||||
| $ - $ | $ | |||||||
| $ | ||||||||
2023年12月31日現在,発行済みオプションの既得株数は加重平均で行重み $それは.2023年12月31日現在、未返済現金オプションの総内的価値はそれは.合計内的価値は当社株終値$となる2023年12月31日、オプション価格にオプション数を乗じた。2023年12月31日までに未確認の株式の報酬総額に基づく。 このようなコストは加重平均期間で確認される予定であり、加重平均期間は約何年もです。
発送日から計算する.2018年までに従業員に付与されたオプションは3年以内に月額分割払い です。2018年から、従業員に付与されたオプションは4年以内に授与される付与された最初の年度(Br)周年に帰属する株式の%は、残りの株式はその後3年以内に月分36回の均等額で帰属する。
株式オプションによる補償費用は,一般に報酬の必要なサービス期間内に直線的に確認される.以下の加重平均は、比較期間中の株式単位で計算された報酬を計算するものとする
| 12月31日までの年度 | ||||||||
| 2023 | 2022 | |||||||
| 波動性 | % - | % | % - | % | ||||
| 無リスク金利 | % - | % | % - | % | ||||
| 配当率 | % | % | ||||||
| 期待寿命 | 数年前 | 数年前 | ||||||
会社には十分な歴史情報がなく、将来のトレーニングパターンや退職後の雇用終了行為を合理的な期待にすることはできません。 そこで、当社は“従業員会計公告”(SAB) 107に記載の“簡略化方法”を用いて、引受権付与の期待期間を推定する。
期待株価変動率会社の歴史的株価変動率に基づくと仮定する.
注: 15-後続事件
ない。
| F-25 |