CCCC-2023123100016625792023会計年度誤りHttp://Fasb.org/us-GAAP/2023#CollaborativeArrangementMemberHttp://Fasb.org/us-GAAP/2023#CollaborativeArrangementMemberP 5 YP 3 M00016625792023-01-012023-12-3100016625792023-06-30ISO 4217:ドル00016625792024-02-14Xbrli:共有00016625792023-12-3100016625792022-12-31ISO 4217:ドルXbrli:共有00016625792022-01-012022-12-310001662579アメリカ-アメリカ公認会計基準:普通株式メンバー2021-12-310001662579US-GAAP:AdditionalPaidInCapitalMembers2021-12-310001662579アメリカ公認会計原則:他の総合収入メンバーを累計2021-12-310001662579アメリカ-公認会計基準:前払いメンバーを保留2021-12-3100016625792021-12-310001662579アメリカ-アメリカ公認会計基準:普通株式メンバー2022-01-012022-12-310001662579US-GAAP:AdditionalPaidInCapitalMembers2022-01-012022-12-310001662579アメリカ公認会計原則:他の総合収入メンバーを累計2022-01-012022-12-310001662579アメリカ-公認会計基準:前払いメンバーを保留2022-01-012022-12-310001662579アメリカ-アメリカ公認会計基準:普通株式メンバー2022-12-310001662579US-GAAP:AdditionalPaidInCapitalMembers2022-12-310001662579アメリカ公認会計原則:他の総合収入メンバーを累計2022-12-310001662579アメリカ-公認会計基準:前払いメンバーを保留2022-12-310001662579アメリカ-アメリカ公認会計基準:普通株式メンバー2023-01-012023-12-310001662579US-GAAP:AdditionalPaidInCapitalMembers2023-01-012023-12-310001662579アメリカ公認会計原則:他の総合収入メンバーを累計2023-01-012023-12-310001662579アメリカ-公認会計基準:前払いメンバーを保留2023-01-012023-12-310001662579アメリカ-アメリカ公認会計基準:普通株式メンバー2023-12-310001662579US-GAAP:AdditionalPaidInCapitalMembers2023-12-310001662579アメリカ公認会計原則:他の総合収入メンバーを累計2023-12-310001662579アメリカ-公認会計基準:前払いメンバーを保留2023-12-31CCCC:細分化市場0001662579CCCC:実験室装置のメンバー2023-12-310001662579US-GAAP:ComputerEquipmentMembers2023-12-310001662579CCCC:OfficeEquipmentFurnitureand FixturesMembers2023-12-310001662579アメリカ公認会計基準:MoneyMarketFundsMembers2023-12-310001662579アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ公認会計基準:MoneyMarketFundsMembers2023-12-310001662579アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-公認会計基準:公正価値入力レベル2メンバー2023-12-310001662579アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ公認会計基準:MoneyMarketFundsMembers2023-12-310001662579アメリカ-公認会計基準:アメリカ証券メンバー2023-12-310001662579アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-公認会計基準:公正価値入力レベル1メンバー2023-12-310001662579アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-公認会計基準:公正価値入力レベル2メンバー2023-12-310001662579アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-公認会計基準:公正価値投入レベル3メンバー2023-12-310001662579アメリカ-公認会計基準:会社債務証券メンバー2023-12-310001662579アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-公認会計基準:会社債務証券メンバー2023-12-310001662579アメリカ-公認会計基準:会社債務証券メンバーアメリカ-公認会計基準:公正価値入力レベル2メンバー2023-12-310001662579アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-公認会計基準:会社債務証券メンバー2023-12-310001662579アメリカ-公認会計基準:会社債務証券メンバー2023-12-310001662579アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-公認会計基準:会社債務証券メンバー2023-12-310001662579アメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-公認会計基準:会社債務証券メンバー2023-12-310001662579アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-公認会計基準:会社債務証券メンバー2023-12-310001662579アメリカ-公認会計基準:アメリカ政府債務証券メンバー2023-12-310001662579アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-公認会計基準:アメリカ政府債務証券メンバー2023-12-310001662579アメリカ-公認会計基準:アメリカ政府債務証券メンバーアメリカ-公認会計基準:公正価値入力レベル2メンバー2023-12-310001662579アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-公認会計基準:アメリカ政府債務証券メンバー2023-12-310001662579アメリカ-公認会計基準:アメリカ証券メンバー2023-12-310001662579アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-公認会計基準:アメリカ証券メンバー2023-12-310001662579アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-公認会計基準:公正価値入力レベル2メンバー2023-12-310001662579アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-公認会計基準:アメリカ証券メンバー2023-12-310001662579アメリカ-公認会計基準:公正価値入力レベル1メンバー2023-12-310001662579アメリカ-公認会計基準:公正価値入力レベル2メンバー2023-12-310001662579アメリカ-公認会計基準:公正価値投入レベル3メンバー2023-12-310001662579アメリカ公認会計基準:MoneyMarketFundsMembers2022-12-310001662579アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ公認会計基準:MoneyMarketFundsMembers2022-12-310001662579アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-公認会計基準:公正価値入力レベル2メンバー2022-12-310001662579アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ公認会計基準:MoneyMarketFundsMembers2022-12-310001662579アメリカ-公認会計基準:アメリカ証券メンバー2022-12-310001662579アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-公認会計基準:公正価値入力レベル1メンバー2022-12-310001662579アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-公認会計基準:公正価値入力レベル2メンバー2022-12-310001662579アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-公認会計基準:公正価値投入レベル3メンバー2022-12-310001662579アメリカ-公認会計基準:会社債務証券メンバー2022-12-310001662579アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-公認会計基準:会社債務証券メンバー2022-12-310001662579アメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-公認会計基準:会社債務証券メンバー2022-12-310001662579アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-公認会計基準:会社債務証券メンバー2022-12-310001662579アメリカ-公認会計基準:アメリカ政府債務証券メンバー2022-12-310001662579アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-公認会計基準:アメリカ政府債務証券メンバー2022-12-310001662579アメリカ-公認会計基準:アメリカ政府債務証券メンバーアメリカ-公認会計基準:公正価値入力レベル2メンバー2022-12-310001662579アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-公認会計基準:アメリカ政府債務証券メンバー2022-12-310001662579アメリカ-公認会計基準:アメリカ証券メンバー2022-12-310001662579アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-公認会計基準:アメリカ証券メンバー2022-12-310001662579アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-公認会計基準:公正価値入力レベル2メンバー2022-12-310001662579アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-公認会計基準:アメリカ証券メンバー2022-12-310001662579アメリカ-公認会計基準:公正価値入力レベル1メンバー2022-12-310001662579アメリカ-公認会計基準:公正価値入力レベル2メンバー2022-12-310001662579アメリカ-公認会計基準:公正価値投入レベル3メンバー2022-12-310001662579アメリカ-公認会計基準:会社債務証券メンバー2023-12-31CCCC:安全0001662579アメリカ-公認会計基準:アメリカ政府債務証券メンバー2023-12-310001662579アメリカ-公認会計基準:会社債務証券メンバー2022-12-310001662579アメリカ-公認会計基準:アメリカ政府債務証券メンバー2022-12-310001662579アメリカ-公認会計基準:アメリカ証券メンバー2022-12-310001662579CCCC:実験室装置のメンバー2022-12-310001662579アメリカ公認会計基準:レンタルとレンタル改善メンバー2023-12-310001662579アメリカ公認会計基準:レンタルとレンタル改善メンバー2022-12-310001662579アメリカ-GAAP:家具と固定機器のメンバー2023-12-310001662579アメリカ-GAAP:家具と固定機器のメンバー2022-12-310001662579アメリカ-GAAP:OfficeEquipmentMembers2023-12-310001662579アメリカ-GAAP:OfficeEquipmentMembers2022-12-310001662579US-GAAP:ComputerEquipmentMembers2022-12-310001662579アメリカ-アメリカ公認会計基準:建設中のメンバー2023-12-310001662579アメリカ-アメリカ公認会計基準:建設中のメンバー2022-12-310001662579CCCC:水城レンタル会員CCCC:オフィスと実験室空間のメンバー2017-07-310001662579CCCC:水城レンタル会員CCCC:オフィスと実験室空間のメンバー2017-07-012017-07-31CCCC:期間0001662579CCCC:水城レンタル会員CCCC:オフィスと実験室空間のメンバー2018-04-300001662579CCCC:水城レンタル会員CCCC:新しいレンタルスペースのメンバー2022-01-012022-01-310001662579CCCC:水城レンタル会員CCCC:新しいレンタルスペースのメンバー2022-01-310001662579CCCC:水城レンタル会員CCCC:新しいレンタルスペースのメンバー2022-12-310001662579CCCC:水城レンタル会員CCCC:新しいレンタルスペースのメンバー2023-12-31CCCC:契約0001662579CCCC:水城レンタル会員2022-01-31Xbrli:純0001662579CCCC:メルクプロトコルのメンバー2023-12-112023-12-110001662579CCCC:メルクプロトコルのメンバーCCCC:研究·開発マイルストーンのメンバー2023-12-110001662579CCCC:メルクプロトコルのメンバー2023-12-11CCCC:ターゲット0001662579CCCC:メルクプロトコルのメンバー2023-12-310001662579CCCC:BettaAgreementメンバー2023-05-292023-05-290001662579CCCC:研究·開発マイルストーンのメンバーCCCC:BettaAgreementメンバー2023-05-290001662579CCCC:DrugApplicationApprovalMemberCCCC:BettaAgreementメンバー2023-05-290001662579CCCC:BettaAgreementメンバー2023-05-290001662579SRT:最小メンバ数2023-05-292023-05-290001662579CCCC:BettaAgreementメンバー2023-12-310001662579CCCC:BettaAgreementメンバー2023-01-012023-12-310001662579アメリカ-公認会計基準:外国人メンバー2023-01-012023-12-310001662579CCCC:RestatedRocheAgreementメンバー2018-12-012018-12-310001662579CCCC:RestatedRocheAgreementメンバー2020-11-300001662579CCCC:RestatedRocheAgreementメンバー2023-12-310001662579CCCC:RestatedRocheAgreementメンバーCCCC:リード·シリーズの認識成果メンバー2020-11-012020-11-300001662579CCCC:RestatedRocheAgreementメンバー2020-11-012020-11-300001662579CCCC:RestatedRocheAgreementメンバーCCCC:研究開発とビジネスマイルストーン支払いのメンバーSRT:最大メンバ数2020-11-300001662579CCCC:RestatedRocheAgreementメンバーSRT:最大メンバ数CCCC:OneTimeSalesBasedPaymentsメンバー2020-11-300001662579CCCC:RestatedRocheAgreementメンバーSRT:最小メンバ数2023-01-012023-12-3100016625792018-12-31CCCC:パフォーマンス無効化0001662579CCCC:RestatedRocheAgreementメンバー2018-12-310001662579CCCC:研究·開発目標メンバー2023-12-310001662579CCCC:OptionRightsMember2023-12-310001662579CCCC:生物生成研究とライセンスプロトコルメンバーSRT:最大メンバ数2020-02-012020-02-29CCCC:タンパク質0001662579CCCC:生物生成研究とライセンスプロトコルメンバー2020-02-012020-02-290001662579CCCC:生物生成研究とライセンスプロトコルメンバーCCCC:研究·開発マイルストーンのメンバーSRT:最大メンバ数2020-02-012020-02-290001662579CCCC:生物生成研究とライセンスプロトコルメンバーSRT:最大メンバ数CCCC:OneTimeSalesBasedPaymentsメンバー2020-02-012020-02-290001662579SRT:最小メンバ数CCCC:生物生産許可プロトコルのメンバー2023-01-012023-12-310001662579CCCC:生物生産許可プロトコルのメンバー2023-12-31CCCC:パフォーマンス_義務0001662579CCCC:研究開発サービスのメンバーCCCC:生物生産許可プロトコルのメンバー2023-01-012023-12-310001662579CCCC:研究開発サービスのメンバーCCCC:生物生産許可プロトコルのメンバー2023-12-310001662579CCCC:Calicoライセンス契約メンバー2017-03-012017-03-310001662579CCCC:Calicoライセンス契約メンバー2021-08-012021-08-310001662579CCCC:Calicoライセンス契約メンバー2021-09-012021-09-300001662579CCCC:Calicoライセンス契約メンバー2017-03-012020-06-300001662579CCCC:Calicoライセンス契約メンバーCCCC:潜在力研究開発とビジネスマイルストーン支払いメンバーSRT:最大メンバ数2017-03-012017-03-310001662579CCCC:Calicoライセンス契約メンバーSRT:最大メンバ数CCCC:OneTimeSalesBasedPaymentsメンバー2017-03-012017-03-310001662579CCCC:研究開発サービスのメンバーCCCC:Calicoライセンス契約メンバー2023-01-012023-12-310001662579CCCC:RocheAgreementメンバー2023-01-012023-12-310001662579CCCC:RocheAgreementメンバー2022-01-012022-12-310001662579CCCC:生物遺伝子プロトコルのメンバー2023-01-012023-12-310001662579CCCC:生物遺伝子プロトコルのメンバー2022-01-012022-12-310001662579CCCC:CalicoAgreementメンバー2023-01-012023-12-310001662579CCCC:CalicoAgreementメンバー2022-01-012022-12-310001662579CCCC:BettaAgreementメンバー2022-01-012022-12-310001662579CCCC:RocheAgreementメンバー2023-12-310001662579CCCC:生物遺伝子プロトコルのメンバー2023-12-310001662579CCCC:RocheAgreementメンバー2022-12-310001662579CCCC:生物遺伝子プロトコルのメンバー2022-12-310001662579CCCC:CalicoAgreementメンバー2022-12-310001662579Cccc:RestatedRocheaccement生物生成許可プロトコルとCalicoライセンスプロトコルメンバー2023-12-310001662579CCCC:CreditAgreement WithPerceptiveLifeScience MasterFundLTDMメンバCCCC:TermLoanMember2020-06-050001662579CCCC:CreditAgreement WithPerceptiveLifeScience MasterFundLTDMメンバCCCC:TermLoanMemberCCCC:TrancheOneMembers2020-06-050001662579CCCC:CreditAgreement WithPerceptiveLifeScience MasterFundLTDMメンバCCCC:TermLoanMemberCCCC:TranscheTwoMembers2020-06-050001662579CCCC:CreditAgreement WithPerceptiveLifeScience MasterFundLTDMメンバCCCC:TermLoanMemberCCCC:TrancheOneMembers2020-06-3000016625792020-10-31CCCC:投票0001662579CCCC:AtmOfferingsMembersSRT:最大メンバ数CCCC:EquityDistributionAgreement WithCowenAndCompanyLLCMメンバー2021-11-012021-11-300001662579CCCC:AtmOfferingsMembersCCCC:EquityDistributionAgreement WithCowenAndCompanyLLCMメンバー2023-01-012023-12-310001662579CCCC:AtmOfferingsMembersCCCC:EquityDistributionAgreement WithCowenAndCompanyLLCMメンバーアメリカ公認会計基準:副次的事件メンバー2024-01-012024-01-310001662579CCCC:AtmOfferingsMembersCCCC:EquityDistributionAgreement WithCowenAndCompanyLLCMメンバー2021-11-012023-12-310001662579CCCC:AtmOfferingsMembersCCCC:EquityDistributionAgreement WithCowenAndCompanyLLCMメンバー2023-12-310001662579CCCC:AtmOfferingsMembersCCCC:EquityDistributionAgreement WithCowenAndCompanyLLCMメンバー2022-01-012022-12-310001662579CCCC:2,000,15株オプションと付与計画メンバー2015-12-280001662579SRT:最小メンバ数CCCC:2,000,15株オプションと付与計画メンバー2015-12-282015-12-280001662579CCCC:2,000,15株オプションと付与計画メンバーSRT:最大メンバ数2015-12-282015-12-280001662579CCCC:2,000,15株オプションと付与計画メンバー2015-12-282015-12-280001662579CCCC:2千と22の株式オプションと計画メンバー2020-09-300001662579CCCC:2千と22の株式オプションと計画メンバー2023-12-310001662579CCCC:2,000,15株オプションと付与計画メンバー2023-12-310001662579CCCC:2千と22の株式オプションと計画メンバー2021-01-012021-01-010001662579CCCC:2千と22の株式オプションと計画メンバーアメリカ公認会計基準:副次的事件メンバー2024-01-010001662579米国-公認会計基準:研究·開発費メンバー2023-01-012023-12-310001662579米国-公認会計基準:研究·開発費メンバー2022-01-012022-12-310001662579アメリカ-公認会計基準:一般と行政費用メンバー2023-01-012023-12-310001662579アメリカ-公認会計基準:一般と行政費用メンバー2022-01-012022-12-310001662579CCCC:2千と22の株式オプションと計画メンバー2022-12-310001662579CCCC:2千と22の株式オプションと計画メンバー2022-01-012022-12-310001662579CCCC:2千と22の株式オプションと計画メンバー2023-01-012023-12-310001662579SRT:最小メンバ数CCCC:2千と22の株式オプションと計画メンバー2023-01-012023-12-310001662579CCCC:2千と22の株式オプションと計画メンバーSRT:最大メンバ数2023-01-012023-12-310001662579SRT:最小メンバ数CCCC:2千と22の株式オプションと計画メンバー2022-01-012022-12-310001662579CCCC:2千と22の株式オプションと計画メンバーSRT:最大メンバ数2022-01-012022-12-310001662579米国-GAAP:制限株式単位RSUメンバー2022-01-310001662579米国-GAAP:制限株式単位RSUメンバー2022-02-280001662579米国-GAAP:制限株式単位RSUメンバー2023-12-310001662579米国-GAAP:制限株式単位RSUメンバー2022-12-310001662579米国-GAAP:制限株式単位RSUメンバー2023-01-012023-12-310001662579CCCC:TimeBasedRestratedStockUnitsMember2023-02-280001662579CCCC:TimeBasedRestratedStockUnitsMember2023-01-310001662579CCCC:TimeBasedRestratedStockUnitsMember2023-12-310001662579CCCC:TimeBasedRestratedStockUnitsMember2022-12-310001662579CCCC:TimeBasedRestratedStockUnitsMember2023-01-012023-12-310001662579米国-公認会計基準:従業員株式オプションメンバーアメリカ-GAAP:ShareBasedPaymentArrangementEmployeeMembers2023-07-310001662579米国-公認会計基準:従業員株式オプションメンバーアメリカ-GAAP:ShareBasedPaymentArrangementEmployeeMembers2023-07-012023-07-310001662579米国-公認会計基準:従業員株式オプションメンバーアメリカ-GAAP:ShareBasedPaymentArrangementEmployeeMembers米国-GAAP:共有による補償補償TracheOneMember2023-07-012023-07-310001662579米国-公認会計基準:従業員株式オプションメンバーアメリカ-GAAP:ShareBasedPaymentArrangementEmployeeMembers2023-12-310001662579CCCC:従業員2千20万人株式調達計画メンバー2020-09-012020-09-300001662579CCCC:従業員2千20万人株式調達計画メンバー2023-01-012023-12-310001662579CCCC:従業員2千20万人株式調達計画メンバー2023-12-310001662579CCCC:従業員2千20万人株式調達計画メンバー2021-01-012021-01-010001662579CCCC:従業員2千20万人株式調達計画メンバー2021-01-010001662579CCCC:従業員2千20万人株式調達計画メンバー2022-01-010001662579米国-公認会計基準:従業員株式オプションメンバーアメリカ-GAAP:ShareBasedPaymentArrangementEmployeeMembersUS-GAAP:共有ベースの補償報酬送信2人のメンバ2023-07-012023-07-310001662579米国-GAAP:国内/地域メンバーアメリカ-公認会計基準:研究メンバー2023-12-310001662579米国-GAAP:国内/地域メンバーアメリカ-公認会計基準:研究メンバー2022-12-310001662579アメリカ-公認会計基準:研究メンバーアメリカ-公認会計基準:州と地方法律法規のメンバー2023-12-310001662579アメリカ-公認会計基準:研究メンバーアメリカ-公認会計基準:州と地方法律法規のメンバー2022-12-310001662579アメリカ公認会計基準:国際収入サービスIRSMメンバー2023-12-310001662579CCCC:公共株を購入するオプションメンバー2023-01-012023-12-310001662579CCCC:公共株を購入するオプションメンバー2022-01-012022-12-310001662579SRT:最大メンバ数2023-01-012023-12-310001662579アメリカ公認会計基準:副次的事件メンバー2024-01-092024-01-090001662579アメリカ公認会計基準:副次的事件メンバー2024-01-09 アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
_____________________________________
表10-K
_____________________________________
(マーク1)
| | | | | |
| x | 1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
本財政年度末まで十二月三十一日, 2023
あるいは…。
| | | | | |
| o | 1934年証券取引法第13条又は15条に基づいて提出された移行報告 |
日本から日本への過渡期については、日本は中国から日本に移行し、中国は日本から日本に移行する
手数料書類番号001-39567
_____________________________________
C 4治療会社
(登録者の正確な氏名はその定款に記載)
_____________________________________
| | | | | |
| デラウェア州 | 47-5617627 |
(明またはその他の司法管轄権
会社や組織) | (税務署の雇用主 識別番号) |
アーセナル通り490番地, スイートルーム120 水城, 体積量 | 02472 |
| (主にオフィスアドレスを実行) | (郵便番号) |
登録者の電話番号、市外局番を含む:(617) 231-0700
_____________________________________
同法第12条(B)に基づいて登録された証券:
| | | | | | | | | | | | | | |
| クラスごとのタイトル | | 取引
記号 | | 登録された各取引所の名称 |
| 普通株、1株当たり0.0001ドル | | CCCC | | ナスダック世界ベスト市場 |
同法第12条(G)に基づいて登録された証券:ありません
_____________________________________
登録者が証券法第405条に規定する有名な経験豊富な発行者である場合は、再選択マークで表示してください。はいo 違います。x
登録者がこの法第13又は15(D)条に従って報告書を提出する必要がないか否かを、再選択マークで示す。はい、そうですo 違います。x
登録者が(1)過去12ヶ月以内(または登録者がそのような報告を提出する必要があるより短い期間)に1934年の証券取引法第13条または15(D)条に提出されたすべての報告書を再選択マークで示すかどうか、および(2)このような提出要求を過去90日以内に遵守してきたはい、そうですx違いますo
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示すはい、そうですx違いますo
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
| | | | | | | | | | | | | | |
| 大型加速ファイルサーバ | o | | ファイルマネージャを加速する | o |
| 非加速ファイルサーバ | x | | 規模の小さい報告会社 | x |
| 新興成長型会社 | o | | | |
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守するo
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われるo
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用するo
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に第240.10 D−1(B)条に従って受信されたインセンティブベースの補償に従って回復分析を行う必要があるかどうかを再選択マークで示すo
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用するo
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に第240.10 D−1(B)条に従って受信されたインセンティブベースの補償に従って回復分析を行う必要があるかどうかを再選択マークで示すo
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)。はい、そうですo違いますx
登録者の非関連会社が保有する投票権と無投票権を有する普通株の総時価は、普通株のナスダック株式市場における2023年6月30日の終値に基づいて、$となる129,573,953.
2024年2月14日現在、登録者が発行した普通株の数は68,600,669.
引用で編入された書類
登録者は、第14 A条に基づいて、2023年12月31日までの財政年度終了後120日以内に提出された2023年株主総会の最終委託書の一部の内容に基づいて、本年度報告の第3部分Form 10−Kを参照して組み込む。その範囲は、本明細書に記載された範囲内である。
| | | | | | | | |
監査役事務所ID:185 | 監査役の名前:ピマウェイ会計士事務所 | 監査役位置:アメリカマサチューセッツ州ボストン |
カタログ表
| | | | | | | | |
| | ページ |
第1部 | | |
第1項。 | 業務.業務 | 1 |
第1 A項。 | リスク要因 | 36 |
項目1 B。 | 未解決従業員意見 | 78 |
第二項です。 | 属性 | 79 |
第三項です。 | 法律訴訟 | 79 |
第四項です。 | 炭鉱安全情報開示 | 79 |
| | |
第II部 | | |
五番目です。 | 登録者普通株市場、関連株主事項及び発行者による株式証券の購入 | 80 |
第六項です。 | [保留されている] | 80 |
第七項。 | 経営陣の財務状況と経営成果の検討と分析 | 80 |
第七A項。 | 市場リスクの定量的·定性的開示について | 89 |
第八項です。 | 財務諸表と補足データ | 89 |
第九項です。 | 会計と財務情報開示の変更と相違 | 89 |
第9条。 | 制御とプログラム | 89 |
プロジェクト9 B。 | その他の情報 | 90 |
プロジェクト9 Cです。 | 検査妨害に関する外国司法管区の開示 | 90 |
| | |
第三部 | | |
第10項。 | 役員·幹部と会社の管理 | 91 |
第十一項。 | 役員報酬 | 91 |
第十二項。 | 特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 | 91 |
十三項。 | 特定の関係や関連取引、取締役の独立性 | 91 |
14項です。 | 最高料金とサービス | 91 |
| | |
第4部 | | |
第十五項。 | 展示·財務諸表明細書 | 92 |
プロジェクト16 | 表格10-Kの概要 | 94 |
前向き陳述に関する特別説明
本年度報告には,Form 10−KまたはForm 10−Kの形で,“経営陣の財務状況や経営結果の検討と分析”と題する章が含まれており,経営陣の信念や仮定,管理職が現在把握している情報の明示的または示唆に基づく前向きな陳述が含まれている。私たちはこれらの展望性陳述に反映される予想は合理的であると考えているが、これらの陳述は未来の事件或いは私たちの未来の運営或いは財務表現に関連し、既知と未知のリスク、不確定性とその他の要素に関連し、私たちの実際の結果、業績或いは業績はこれらの前向き陳述と明示的或いは暗示する任意の未来の結果、業績或いは成果とは大きく異なる可能性がある。本10−Kテーブルの前向きな陳述は、以下の態様に関する説明を含むことができるが、これらに限定されない
•私たちの研究と開発計画、ならびに私たちの現在と未来の臨床前研究と臨床試験の開始、時間、進捗、結果、安全性と有効性およびコストは、研究または試験の開始と完了時間、試験結果がどのくらいの時間で利用できるか、および私たちの研究と開発計画に関する声明を含む
•私たちは候補製品のさらなる開発、製造、商業化を達成するために、私たちの運営のために必要な資金を得ることができる
•私たちは現在または未来の任意の候補製品のために規制部門の承認を得て維持することができる
•私たちの既存の現金と現金等価物および有価証券は、私たちの運営費用および資本支出要件を支払うのに十分な時間帯になると予想される
•他の病気の適応を治療するための候補製品を識別し開発する能力は
•私たちの候補製品の潜在的な属性と利点は
•私たちが開発する可能性のある候補製品の市場受容度と臨床的実用性の速度と程度
•私たちの候補製品の価格設定と精算は、承認されたら、私たちの製品が2022年の“インフレ低減法案”や他の適用法に基づいて医療保険や医療補助サービスセンターと強制的な価格交渉をすれば、製品の価格を下げる可能性があります
•競争が私たちの現在または未来の任意の候補製品に与える影響と、私たちの業界の現在と未来の競争相手の革新
•私たちの業務戦略計画、私たちが開発する可能性のある製品候補品、そして私たちの魚雷を実行します® (T目標.目標あるいは…。すでに貸したP腐っているEはいD漸進器OPtimizer)プラットフォーム;
•私たちの第三者戦略パートナーは、F.Hoffmann-La Roche LtdとHoffmann-La Roche Inc.,Roche,Biogen MA,Inc.,Biogen,Betta PharmPharmticals,Co.,Betta Pharma,Merck Sharp&Dohme LLCまたはMerckの既存の協力プロトコル推進計画の能力、または他の新しい協力協定を含む、私たちの候補製品に関する研究、開発、製造活動の能力と意思を継続している
•私たちの候補製品のための知的財産権保護範囲を確立することができます
•私たちの将来の支出、収入、資本需要、および追加融資需要の推定
•承認されれば、将来的に第三者と私たちの候補製品の製造と商業化について合意します
•私たちの候補製品の市場規模と成長潜在力と私たちがこれらの市場にサービスする能力は
•私たちの財務業績は
•アメリカや他の国の規制動向は
•私たちは第三者サプライヤーと製造業者と契約を締結する能力と彼らが契約を十分に履行する能力
•競争療法の成功や可能性があります
•私たちは重要な科学や管理者の能力を引き付けて維持しています
•私たちの競争相手や業界の発展と
•第1部1 A項である本テーブル10−Kにおけるリスク要因で議論されているリスクおよび不確実性を含む他のリスクおよび不確実性
場合によっては、前向き表現は、すべての前向き表現がこれらの語を含むわけではないが、“可能”、“予想”、“意図”、“計画”、“目標”、“予想”、“信じ”、“推定”、“予測”、“潜在”、“継続”またはこれらの用語の否定または他の同様の用語によって識別することができる。これらの声明はただ予測に過ぎない。あなたは展望的な陳述に過度に依存してはいけません。それらは既知と未知のリスク、不確実性、および他の要素に関連しているので、これらの要素は場合によっては私たちがコントロールできず、結果に大きな影響を与えるかもしれません。実際の結果が現在の予想と大きく異なる可能性がある要素は、他の事項を除いて、“リスク要因”と題する節と本10-K表の他の部分に列挙された要素を含む。これらのリスクまたは不確実性のうちの1つまたは複数が発生する場合、または私たちの基本的な仮定が正しくないことが証明された場合、実際のイベントまたは結果は、前向き陳述における明示的または暗示的なものとは大きく異なる可能性がある。どんな展望的な陳述も未来の業績に対する約束や保証ではない。
本10-K表における前向き陳述は,我々の本10-K表発行日までの観点を表す.私たちはその後に発生した事件と事態の発展が私たちの観点を変化させると予想している。しかし、私たちは未来のある時点でこのような前向きな陳述を更新することを選択するかもしれないが、私たちは現在、法律の要求が適用されない限りそうするつもりはない。したがって、あなたはこの10-K表の日付までの私たちの任意の日付の観点を表すために、これらの前向きな陳述に依存してはいけません。
本10-K表は、業界出版物および研究、調査、および第三者による研究から得られた統計データおよび他の業界および市場データを含むことができる。業界出版物および第三者研究、調査および研究は一般に、彼らの情報はこのような情報の正確性または完全性を保証しないにもかかわらず、信頼できると考えられるソースから得られたものであることを示している。私たちはこのようなソースに含まれている情報を独立的に確認していない。
リスク要因の概要
私たちが業務戦略を実施する能力は多くのリスクの影響を受けており、投資決定を下す前に、これらのリスクを意識すべきです。これらのリスクは、第1部1 A項である本表格10-Kにおけるリスク要因により全面的に記載されている。これらのリスクには
•私たちは臨床段階の生物製薬会社で、運営の歴史は限られており、設立以来ずっと重大な損失を受けている。今まで、私たちは製品販売から何の収入も得ていません。少なくとも今後数年以内に、巨額の費用と増加していく運営損失が生じ続け、永遠に実現または利益を維持しない可能性があると予想している。2023年、2023年、2022年12月31日までの年度の純損失はそれぞれ1.325億ドルと1.282億ドルだった。
•私たちは私たちの業務目標を達成し、私たちの運営を継続するために多くの追加資金を必要とするだろう。もし私たちが必要な時に資金を集めることができなければ、私たちは私たちの研究や製品開発計画や将来の商業化努力を延期、制限、減少、または中止することを要求されるかもしれない。
•我々は魚雷プラットフォームに基づいて候補製品を発見·開発する方法が検証されておらず、いかなる製品の開発に成功した時間、コスト、可能性を予測することは困難である。
•著者らは臨床段階の会社であり、いくつかの候補製品の臨床試験を開始したが、著者らはいずれの候補製品の臨床試験を完成したことがない。もし私たちが規制機関の候補製品の承認を得られず、および/またはそれを商業化することができなければ、あるいは私たちがこれらのことをする上で大きな遅延に遭遇した場合、私たちの業務は損なわれる可能性がある。
•著者らは著者らの臨床前試験と臨床試験の適時な完成或いは結果を確定することができない。また,前臨床研究の結果は臨床試験の結果を予測できない可能性があり,われわれが開始したいずれの早期臨床試験の結果も後期臨床試験の結果を予測できない可能性がある。
•私たちの臨床前研究と臨床試験は、私たちの任意の候補製品の安全性と有効性を十分に証明できないかもしれません。これは、私たちの現在と未来の候補製品を開発、規制し、商業化するために、追加の研究または分析を阻止、延期、または必要とするでしょう。
•私たちは羅氏、Betta Pharma、メルク社と持続的な協力協定があり、BiogenとCalicoとも協力協定があり、この2社の研究期間はそれぞれ2023年6月30日と2023年3月13日に満了した。私たちはまた、将来的に第三者とより多くの協力を求めて、私たちのいくつかの候補製品を開発および/または商業化することができます。しかし、私たちは、このような既存または潜在的な協調配置の下でのすべての潜在的な利点を決して達成できないかもしれない。
•私たちは、私たちよりも他の人が同じ適応および/または患者集団のための製品を発見、開発、または商業化することに成功する可能性がある激しい競争に直面している。
•私たちは依存し、臨床前および臨床試験のための当社の候補製品の生産、および商業生産のための第三者の生産に依存し続けることが予想されます(任意の候補製品が市場の承認を得たら)。このような第三者への依存は、十分な数の候補製品をタイムリーに、または許容可能なコストまたは品質で供給できないリスクを増加させる可能性がある。
•もし私たちの候補製品のために必要なマーケティング承認、商業化、製造、特許保護を獲得し、維持したり、市場の承認を得ることができなければ、もし私たちがそうする上で重大な遅延があったら、私たちの業務は深刻な損害を受け、私たちが製品販売から収入を得る能力は深刻な損害を受けるだろう。
•もし私たちの技術や製品のために特許保護を獲得して維持することができない場合、または取得された特許保護範囲が十分に広くない場合、または実行可能でなければ、私たちの競争相手は、私たちと似ているか、または同じ技術、候補製品、および製品を開発し、商業化する可能性があり、私たちの技術、候補製品、および製品を商業化することに成功した能力は損なわれる可能性がある。
会社推薦人についての説明
文意が別に指摘されている以外に、本10-K表の用語“C 4治療”、“会社”、“私たち”、“私たち”と“私たち”はC 4治療会社とその合併の子会社を指す。
商標に関する説明
当社の会社名、C 4治療、当社のロゴ、当社の魚雷技術プラットフォームの名前、および当社のBIDACおよびMONODACタンパク質分解器候補製品の名前を含む、当社のビジネス運営に関連する様々な商標、サービスマーカーおよび商品名を所有または使用する権利があります。本テーブル10-Kはまた、第三者の商標、サービスマーク、および商標名を含むことができ、これらは、それぞれの所有者の財産である。私たちは、本募集説明書において、第三者の商標、サービスマーク、商号、または製品を使用または展示しており、私たちと私たちとの関係、または私たちの裏書きまたは賛助を暗示しないためでもありません。便宜上、本明細書で言及されている商標、サービスマーク、および商号は使用しなくてもよいTMあるいは…SMしかし、このような参照を省略することは、適用法律に従って私たちの権利またはこれらの商標、サービスマーク、および商号の適用所有者の権利を最大限に主張しないことを意味するわけではない。
第1部
プロジェクト1.ビジネス
概要
著者らは臨床段階の生物製薬会社であり、標的タンパク質分解(TPD)科学の約束を実現し、患者の生活を変える新世代の小分子薬物を創造することに取り組んでいる。我々独自の魚雷プラットフォームを利用することにより,小分子タンパク質分解器を効率的に設計·最適化することができ,人体破壊に不要なタンパク質の自然過程を利用することで,その期待目標を高度に活性化することができる。私たちが信じているのは新型口腔製品候補製品阻害剤によく出現する薬剤耐性を克服し,現在の“投与できない”標的を狙い,患者の結果を改善する可能性がある。これまで,我々はいくつかのタンパク質分解剤の設計に成功し,それを臨床に応用し,一連の目標種別をカバーしており,われわれの臨床試験のデータによると,われわれの候補製品はすでに強力な目標分解性を示している。
我々の魚雷プラットフォームは2種類のタンパク質分解器を設計する能力があり、この2種類の分解器方法はすべて著者らの臨床導管に反映されている。著者らは第一種類の分解剤をMonoDAC分解剤と呼び、業界の一部の人はそれを“分子ガム”と呼んでいる。MonoDAC分解物はE 3リガーゼに結合し、E 3リガーゼ上に新しい表面を作製することによって、E 3リガーゼと標的タンパク質の結合を増強し、作用を発揮する。著者らは第二タイプの分解剤をBiDAC分解剤と呼び、業界の一部の人もそれを異二機能分解剤と呼んでいる。BiDAC分解物の設計は,分子の一端が原因標的蛋白に結合し,他端がE 3リガーゼに結合することである。これらの分解方法のいずれも,E 3リガーゼを原因タンパク質に十分近くすることで,E 3リガーゼが破壊されたラベルを貼り付けることができるという同じ終点を産生することを目的としている
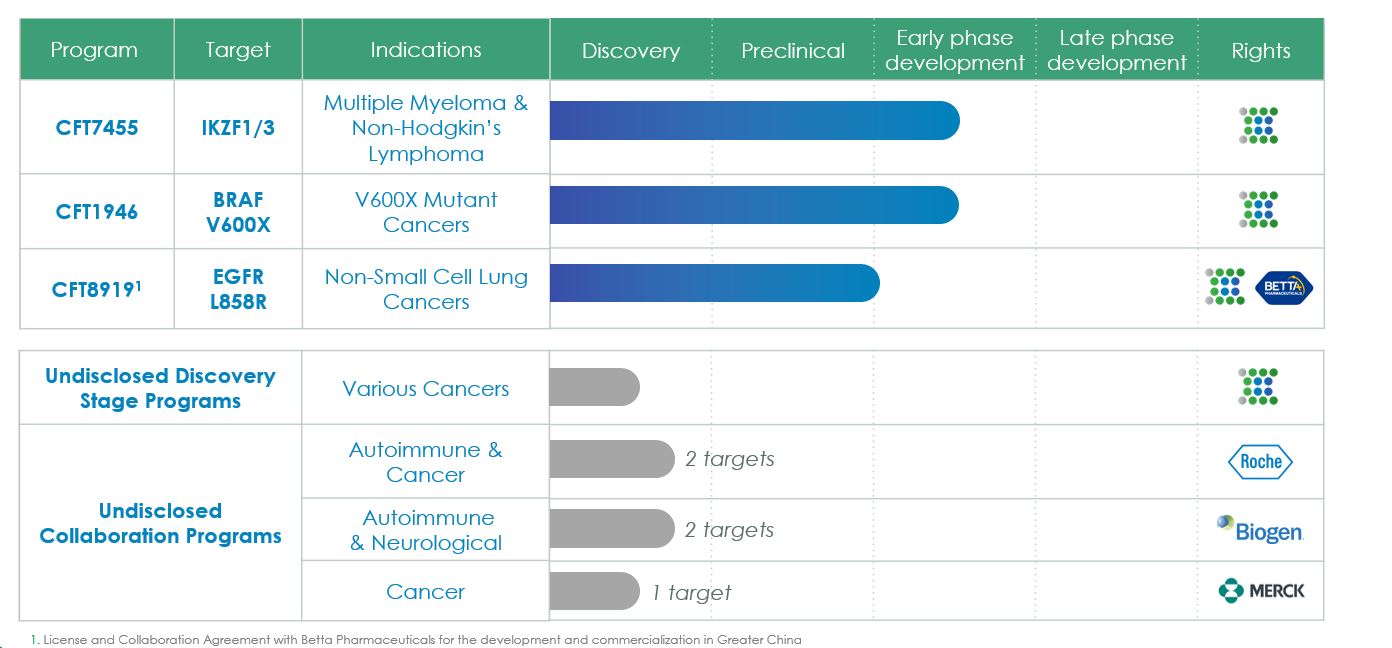
著者らの臨床前と臨床パイプラインにはMonoDACとBiDAC分解物が含まれており、これらの分解物は各種の癌と他の適応の原因蛋白に対して、患者の需要を満たしていない領域に使用される可能性がある。我々の全資本が持つパイプラインは腫瘍学に集中しており,我々の協力戦略は他の適応を探索できるようにしている。私たちのチャネル全体は、次の図に示すように、パートナーシップを含む。
著者らの最先端の候補製品CFT 7455は1種の経口生物使用可能なMonoDAC分解剤であり、IKZF 1とIKZF 3と呼ばれ、現在多発性骨髄腫(MM)と非ホジキンリンパ腫(NHL)の臨床開発中である。IKZF 1とIKZF 3を標的とするのは非常に強い機序の基礎と明確な生物学的定義があり、1種の新型分解剤を標的として重要な満足されていない需要を解決する可能性がある。われわれの臨床前研究では,この候補製品は良好な薬理学的特性を有する有効かつ選択的なタンパク質分解を示している。2021年6月、著者らはこの候補製品のために初のヒト1/2期臨床試験を開始した。2021年8月、米国食品医薬品局(FDA)はCFT 7455をMM治療のために承認した。2023年12月、CFT 7455 1/2期試験用量増加部分の陽性臨床データを提供し、単一療法としてデキサメタゾンと併用してMMを治療した。MMとNHLが行っている1/2期臨床試験を引き続き進めている
著者らの次の最も先進的な候補製品CFT 1946は、黒色腫、非小細胞肺癌、結腸直腸癌、およびこのような変異を含む他の悪性腫瘍の治療に有効かつ選択的に抗することを目的とする経口生物学的に利用可能なBiDAC分解剤である。臨床前研究では,CFT 1946はCRCやNSCLC BRAF V 600 X異種移植モデルおよび黒色腫患者由来の異種移植またはPDX,BRAF阻害剤耐性モデルで標準看護療法よりも有効であった。また、CFT 1946は臨床前転移性黒色腫中枢神経系(CNS)モデルにおいて活発である。2023年1月、我々はCFT 1946の最初のヒト1/2期臨床試験を開始し、非小細胞肺癌、結腸直腸癌、および黒色腫を含むBRAF V 600 X変異固形腫瘍の治療に使用した。2024年1月、実施中の1/2段階試験の最初の2つの用量アップグレードキューのPKおよびPDデータを共有し、BRAFの深分解に関連する用量比曝露および経口バイオアベイラビリティを示した。黒色腫、非小細胞肺癌、結腸直腸癌を含むBRAF V 600 X癌の1/2期臨床試験を継続した
また,経口生物利用,アロステリック,変異選択性の上皮増殖因子受容体またはEGFRのBiDAC分解剤であり,非小細胞肺癌にL 858 R変異を有するCFT 8919を開発している。臨床前研究において、BA/F 3細胞モデルにおいて、CFT 8919はEGFR抑制に耐性のEGFR変異(L 858 R-C 797 S、L 858 R-T 790 MとL 858 R-T 790 M-C 797 Sを含む)はL 858 R単一変異と同様の抗増殖活性を示した体外培養それは.2023年5月、私たちはBetta Pharmaと許可と協力協定を締結し、CFT 8919の大陸部中国、香港特別行政区、マカオ特別行政区、台湾の開発と商業化について協力し、私たちは世界の他の地域でCFT 8919を開発および商業化する権利を保留した。また、2023年6月、FDAはCFT 8919の研究新薬或いはIND申請を許可し、2023年12月、倍他医薬は中国指導の国家医療製品管理局のCFT 8919に対する臨床試験申請或いはCTA許可を得た
これらの最初の候補品に加えて、私たち自身の特許パイプラインと、私たちがメルク、生物遺伝、羅氏と協力して開発したパイプラインに対して、臨床検証と現在使用できない目標に対する新しい分解剤を開発し、更に私たちのパイプラインを多様化した。我々が設計した分解物は臨床前研究において血液脳関門の透過に成功しており,腫瘍学的脳転移や神経変性疾患などの治療領域を治療する可能性のある薬物開発の重要な一歩である。多くの治療分野や適応において,我々の魚雷プラットフォームを用いて新しい分解剤を開発することが有利である可能性が信じられている
私たちの戦略
我々は,原因タンパク質を破壊する新しい療法を発見,開発,商業化することにより,癌や他の疾患の治療法を変更することに取り組んでいる
私たちの戦略の重要な要素は
•我々の臨床口腔腫瘍降下剤計画を推進した我々独自の魚雷プラットフォームを用いて,癌治療のための新製品候補製品が生成されており,これらの製品が患者の予後を改善する可能性があると信じている。われわれの臨床試験結果に基づき,FDAと協力し,我々に適した候補製品の潜在的加速開発と承認経路の加速を検討する
•われわれの内部チャネルをTPD法から利益を得ると考えられる腫瘍学的目標に集中し続けた我々の標的識別過程を通じて、著者らはTPDがどこで患者に大きな影響を与えることができるか、それを通じて既存の治療方式の潜在力を改善するか、あるいは現在抑制剤に“薬品使用不可能”とされている標的を選択することによって、これらの標的を分解することは疾病に影響を与える可能性があることを生物学的に表明した。研究活動のためにどのような目標を選択するかを考える際には,生物学が重要な役割を果たしている。私たちはある病気と明確な遺伝的関連がある目標を選択する。降下剤が阻害剤や他の治療選択よりも良い利点をどのように提供するかも考えられる。
また,臨床開発を目標選択過程の一部とした。潜在的な目標を検討し,患者を前向きに選択して臨床試験を行い,登録された経路を決定できるようにした。利用可能なバイオマーカー分析を評価し,どの患者が研究治療から利益を得る可能性があるかを決定することを支援した。われわれは我々のパートナーと現在の独自計画を選定した腫瘍学的適応や他の適応に集中している
•パートナーと協力して、魚雷プラットフォームのすべての価値を実現する我々は羅氏、生物遺伝、メルク社と既存の戦略協力を行い、これらの協力の下で、我々は分解物-抗体結合体やDACなどの新しい方法を含む複数の治療領域と目標上で新しい分解物を識別し、開発するために努力している。これらのパートナーシップは私たちの研究を進めて加速させることができ、より多くの能力を得ることができ、私たちの魚雷の用途を拡大することができます ホームです
タンパク質分解研究概要
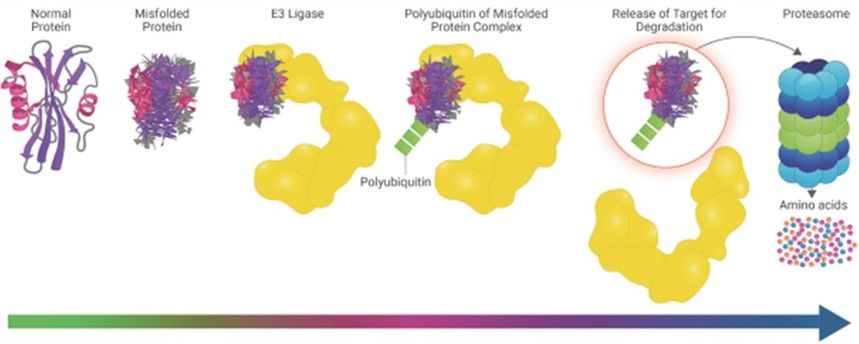
蛋白質分解
タンパク質は大きな複雑な分子であり、人体内で多くの重要な役割を果たしている。それらの生物機能における中心的な作用により、タンパク質相互作用は健康と疾病状態を引き起こす機序を制御している。疾患は通常突然変異によるものであり,これらの変異はタンパク質の正常な機能を変化させ,さらにタンパク質の機能障害を招き,さらに疾患を引き起こす。最近の科学的進歩は,特定のタンパク質の多様な疾患状態における役割を示唆し続けている。
細胞内のタンパク質レベルはその合成速度と分解速度の間の平衡によって制御される。タンパク質分解は、タンパク質レベルの安定したバランスを維持し、老化または欠陥タンパク質を除去するか、または特定のシグナルに対する特定の調節タンパク質の反応を迅速に除去するための自然な機序を提供する。人体には高度に保守的な分解機序があり、ユビキチンプロテアソーム系と呼ばれ、UPSと略称され、タンパク質を認識し、それをその組成のアミノ酸に分解することができる。この過程はある程度E 3リガーゼと呼ばれるタンパク質ファミリーによって媒介される。E 3リガーゼの主な役割は品質管理検査員として機能し、古い、損傷、誤った折り畳み、あるいは分解を準備すると考えられる他のタンパク質を識別することである。E 3リガーゼが分解する目的タンパク質を決定すると、ユビキチン化と呼ばれる過程にユビキチンと呼ばれる分子タグが付加される。このユビキチン化過程は通常,目的タンパク質が複数のユビキチンが標識されるまで続く,いわゆるポリユビキチン化である。標的蛋白がポリユビキチン化されるとE 3リガーゼによって放出され,すぐにプロテアソームに認識され,プロテアソームは細胞の回収工場である。プロテアソームはポリユビキチン化したタンパク質をその組成のアミノ酸に分解し,これらのアミノ酸は循環的に新たなタンパク質を形成したり,細胞に排泄されたりする。この過程を図に示す.
ヒト遺伝子の約5%はユビキチン-プロテアソーム系の構成要素を特異的にコードしている。そのほか、多くの治療意義のある蛋白質は通常E 3リガーゼによって制御される。結論的に、これらの要素はE 3リガーゼが正常細胞機能において果たす重要な役割を強調し、それらがどのように治療用タンパク質標的に対して使用されるかを強調した。
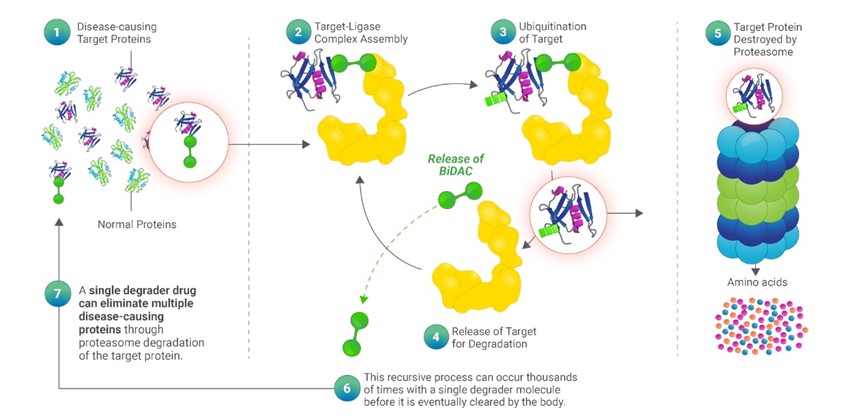
標的蛋白分解物は新しい方法を代表し、このような自然分解機序を利用して病原性標的蛋白を破壊しようとし、そうでなければE 3リガーゼはそれを破壊の目標としない。我々の分解物を介した標的タンパク質分解過程を以下の図に示す。
我々のMonoDAC分解物とBiDAC分解物は同じ触媒過程に従い、最初のステップは天然E 3リガーゼ、分解物と目的蛋白の間に複合体を形成することであり、私たちは三元複合体と呼ぶ。BiDAC降圧器の場合、これは上の図のステップ2に示される。ステップ3において、ユビキチン化が可能な適切な三元複合体の形成は、標的タンパク質のマルチユビキチン化をもたらす。標的タンパク質の1分子のマルチユビキチン化プロセスが完了すると、分解物が放出され、ステップ4に示すように、ステップ5でプロテアソームによって標的タンパク質が分解される。分解物の放出は不変であるため、この再帰的プロセスは、標的タンパク質に結合し、E 3リガーゼと三元複合体を形成し、マルチユビキチン化および放出する-分解を行うために、単一の分解物分子と共に数千回発生し、最終的に身体によって除去される可能性がある。重要なことは,天然のタンパク質分解過程と我々の分解物を介した標的タンパク質の分解が速く起こり,最初の標的−リガーゼ相から多ユビキチン化に遭遇し,プロテアソームへの分解が約数ミリ秒であることである。これにより,タンパク質分解物は自然過程の触媒として機能し,この過程を触媒循環と呼び,これは分解物と従来のタンパク質阻害剤の重要な違いであり,目的タンパク質と結合しなければ有効ではない。
従来のタンパク質阻害剤に対する標的タンパク質分解の利点
現在の多くの標的治療は,標的タンパク質の生物機能を抑制する小分子に基づいている。阻害剤に基づく治療の主な限界の1つは,タンパク質の生物機能を抑制し,治療効果を達成するために持続的な目標占有率レベルが必要であることである。薬物の薬理作用は薬物暴露状況によって決定されるため、薬物作用の全体時間と持続時間は薬物の吸収、分布と除去に依存する。これらの曝露は実現が困難である可能性があり,目標から大きく外れた副作用の可能性を増加させる可能性がある。この方法のもう1つの制限は,タンパク質上の特定の活性部位に結合する化合物を見つける必要があり,目的タンパク質には小分子が結合できる部位が多いが,全体の機能に影響を与えないため機能抑制を引き起こすことである。
標的タンパク質分解は新しい方法であり、従来の小分子阻害剤方法と比較して、改善と持続的な効力、迅速かつ再帰的な触媒効果、高選択性、広い標的化の将来性を含む顕著な潜在的利点を提供することができると考えられる
効果を高め続けています
分解物は,単一の分解物分子が大量の目的タンパク質に再帰的に作用するため,倍拡大の効果を提供することができる。分解物分子がその触媒循環を複数回繰り返すことができる能力を触媒増幅と呼ぶ。これに対し,従来のタンパク質阻害剤は阻害剤分子と目的タンパク質との1対1結合に依存し,阻害剤が結合した場合にのみタンパク質が不活化する。これは,タンパク質分解剤と同様の治療効果を達成するためには,より高濃度のタンパク質阻害剤薬が必要であることを意味している
タンパク質阻害剤よりもはるかに少ない薬物が必要であるほか,目的タンパク質機能に対する分解剤の影響は従来の阻害剤よりも持続する。これは,阻害物が標的蛋白に結合しなくなると標的蛋白の活性が回復し,分解物がその標的蛋白を完全に除去し,原因活性が阻止され,細胞が代替蛋白を合成できるまでに数時間あるいは数日かかる可能性があるためである。だから、
典型的な可逆阻害剤とは異なり,分解剤の効果は体内から除去して良好に保持することができる。分解剤がその標的を完全に除去する能力は、伝統的な阻害剤に対して効力を高めるもう一つの機序である。これは,1つのタンパク質が通常様々な機能を有しており,それぞれの機能が異なるドメインによって媒介されているためである.阻害物はその結合領域に直接影響を受けるタンパク質機能しか妨害できない。原因タンパク質では,多くの機能が原因活性に貢献し(例えば,酵素活性異常や多タンパク質複合体を形成する能力),分解物はすべての機能を除去するため,1つの機能のみを阻止する阻害剤よりも分解物の方が深い影響を与える。これらのすべての要素は、分解剤がより持続的な生物効果とより良い臨床結果を実現するのに役立つ可能性があることを意味する
高選択性
タンパク質抑制の主な課題の1つは、正常細胞或いは他のタンパク質に有害な影響を与えることなく、癌細胞又は病原性タンパク質のみを対象とした分子の識別と開発を試みること、すなわち通常言われている脱標的効果である
タンパク質分解触媒サイクルにおける各ステップは、循環全体で行われる目的タンパク質とE 3リガーゼの特定の局在を必要とし、これらの局在要求は、分解物分子の選択性を増加させるためのフィルタとして必要であり、それにより、分解物が複数のタンパク質に結合しても、最終的には目的タンパク質のみが分解される。例えば,分解者は目的蛋白と相互作用するだけでなく,E 3リガーゼを標的とした方式で行わなければならず,生産性目的蛋白ユビキチン化に適合した三元複雑コンホメーションを仮定している。したがって,分解物が非標的蛋白に結合しても,得られた三元複合体はユビキチン化とその後の分解を促進するコンホメーションに適していない可能性がある。ユビキチン−プロテアソーム蛋白分解経路のこれらの固有特性を利用して分解物を設計し,原因蛋白に対して高い選択性を持たせることができる
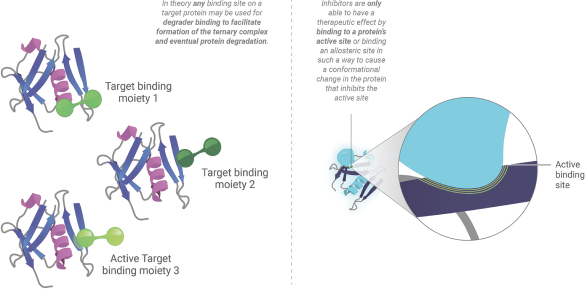
広大な目標景観
従来の阻害剤は,その機能を妨害する原因タンパク質上のある部位に密接に結合した場合にのみ治療効果を得ることができる。これには,阻害剤がタンパク質の活性部位に直接結合したり,コンホメーション変化を引き起こすようにアロステリック部位に結合したりして,タンパク質の活性を低下させる必要がある。これは、多くの潜在的な結合部位が機能を妨害するタンパク質領域ではないので、従来の阻害剤でアドレス指定可能な薬物標的の数を内在的に制限する。逆に,分解者はタンパク質上の任意の結合部位を用いてE 3リガーゼとの三元複合体の形成を促進し,その破壊を招くことができる。また、阻害物はその結合部位との高い親和性で占有と遮断機能を維持する必要があるにもかかわらず、分解物は相対的に弱い親和性と短い方法で相互作用することができ、依然としてタンパク質をユビキチン化と破壊させることができる。的確な蛋白質分解は潜在的な標的池を大きく拡大し、現在薬品使用不可能とされている大部分を含む。
私たちの方法は
著者らは1種の全面的な方法を用いて、候補製品を発見、選択と開発し、著者らの蛋白質分解剤の潜在的な治療効果を最大限に発揮する。著者らは高い価値を持つ蛋白質標的の適応を探し、これらの標的は分解者から最大の利益を得る可能性があり、触媒分解回転率は蛋白質分解を評価する重要な指標である。そのため、著者らは実験ツール、計算と予測モデル及びチームの専門知識に大量の資金を投入し、著者らの魚雷プラットフォームを通じて著者らの分解器の触媒能力を分析と最適化した。さらに,我々のプラットフォームを用いて,分解者がユビキチン−プロテアソームタンパク質分解周期を開始し,その機能を予測する能力を最適化する体内にあるそれは.魚雷は迅速な最適化を可能にしているので プラットフォームと私たちの能力は
降格効果を予測するプラットフォーム体内にある我々は,目標決定段階から候補開発段階までプロジェクトを迅速かつ効率的に進めることができる
私たちの魚雷 ホーム.ホーム
我々独自の魚雷プラットフォーム1組の実験方法とツールの集合であり、私たちは分解剤の性能を設計、分析、予測することができ、これは私たちが分子レベルで分解剤を設計して効力と選択性を増強することができるこのプラットフォームの重要な要素は、
•触媒効率に注目してください我々の分析技術と予測モデルを分解剤特性と最終タンパク質分解との関係に集中させることで触媒分解回転率を最適化することを求めた。著者らの分解器はE 3リガーゼを活性化し、標的蛋白結合とユビキチン化を促進し、全体の目標の迅速な分解を招くことを目的とした。我々の分解者はこの過程を再帰的に繰り返すことができ,同じ分解者分子は目的タンパク質の多くのコピーと相互作用し,我々の候補製品を最適化し,触媒分解回転を実現し,より大きな治療効果を有する候補製品を創出することができる。我々のMonoDAC分解剤とBiDAC分解剤は短い三元錯体形成を開始するのに十分な結合親和性を得る必要があるが,従来の阻害剤とは異なり,長期安定な結合を実現して期待される生理効果を達成する必要はない。われわれのいくつかの前臨床研究では,結合剤が弱くても非常に有効な分解剤を産生することが観察されており,より高い触媒分解回転率を許容する可能性があるため,より高い活性を実現することを優先している
•Cereblonへの投資:我々の鉛分解者はE 3リガーゼとしてCereblonを利用した。ヒトプロテオームには600種類以上のE 3リガーゼがあり,その中で生物学的特性が最も良いのは50種類以下である。Cereblon、VHL、MDM 2、IAPsおよびオスミウム-TRCPを含む少数のE 3リガーゼは、標的タンパク質分解に適していることが報告されている。Cereblonを我々のタンパク質分解方法とするE 3リガーゼ標的を選択した理由はいくつかある
◦すでに許可された薬物サリドマイド、レナドアミンとポマ度アミンの広範な臨床経験により、Cereblonの使用は人類疾病の標的分解に影響することを表明した。これらの分子の作用機序は,Cereblonと錯体を形成させることにより疾患標的,特にIKZF 1とIKZF 3を分解することである。ドナドアミンとポマドアミンはすべて承認された薬物であり、2006年と2013年にそれぞれ承認されて以来、MM治療の看護標準の一部である。つまり,この経験はCereblonがE 3リガーゼであり,有効な薬剤の生産に有用であることが臨床的に確認されている
◦Cereblonは組織に広く発現し、細胞質と核を含むすべての細胞室に存在し、Cereblonを介した標的蛋白の広範な臨床環境と潜在標的での分解を可能にする可能性がある
◦我々は様々な独自のCereblon接着剤を開発しており,経口バイオアベイラビリティ,溶解性,透過性,安定性を向上させるような薬物特性を改善するために設計されており,我々のすべての候補製品や計画は,我々の独自Cereblon接着剤のこれらの特性から利益を得ている。我々のCereblonルーズリーフライブラリは,分解器発見に独自かつ機能的なキットを提供している.このCereblon接着剤キットは、これらの接着剤カテゴリのそれぞれが異なる類似薬物の属性をコードしているため、よりモジュール化された方法で分解剤を識別および最適化することができ、重要なことは、タンパク質分解後にCereblon表面の独特の“脱退軌跡”から、より良い目標分解回転を促進することができる。
触媒効率とCereblonの利用に専念することにより,TPD分野独自の能力を確立した。これらの機能によって私たちは
•デシリアライザの性能を設計、分析、予測する:私たちは計算方法とツールに大量の投資を行い、私たちが合理的に分解器を設計する能力を強化したシリカゲルの中でモデルは私たちのパイプの中でより強い効力と選択性を持つ分解器を設計することを可能にする。また,我々の細胞分解分析は高品質なデータを提供し,基本酵素学的原理に基づく独自かつ独自の枠組みを用いて解析を行い,任意の用量での目標分解の深さと持続時間を予測することができた体内にある.
◦我々は,定量データを生成し,分解剤濃度と目的タンパク質分解との関係を示すための高スループットの細胞分解分析を開発した。この方法は、従来のウエスタンブロット法と比較して、より高い精度とより高いスループットでタンパク質分解を定量することができる。私たちの独自モデルへの実験データの応用
そして、蛋白質分解動力学を予測でき、そして迅速に候補分解剤を反復と改善でき、触媒分解回転の性能を最適化するために設計を行った。
◦著者らはまた、分解物が蛋白質分解を誘導する重要な動力学パラメータを決定するために、分解物濃度、時間と目的蛋白質分解との関係を評価と平衡化する酵素学的枠組みを構築した。私たちはこのフレームワークを独自のPK/PDモデルに拡張しましたこれらのモデルはこれらの動力学的パラメータを新陳代謝とPK曝露を組み合わせて予測しています体内にある性能が低下する。劣化性能の予測は通常以下のように検証される体内にあるPD実験は、標準的なウエスタンブロット分析法を用いて腫瘍試料中の標的分解を測定した。これらのモデルは細胞分析と予測を結びつけています体内にある性能はわれわれの発見過程を著しく加速し,臨床前モデルから臨床への移行に成功する可能性を増加させると信じている
•MonoDACとBiDAC分解器の開発:私たちのプラットフォームは、異なる種類の病原性タンパク質を量的にカスタマイズすることで解決する柔軟性を提供する。リガンド結合部位が存在する標的タンパク質については,我々のCereblonツールキットを用いてBiDAC分解剤を開発することができる。リガンド結合部位が存在しないか、または転写因子のような十分な特異性を欠く標的タンパク質については、7,000種類以上の化合物を含む我々固有のMonoDACライブラリーを用いて、標的に対する一致をスクリーニングすることができる。私たちが目標に命中したことを確認すると、私たちは私たちの魚雷プラットフォームの他の要素を利用してMonoDACやBiDACデコーダーを最適化することができる。
これらの機能は,我々のプラットフォームが最低生物や毒性リスクを示すと考えられる候補製品の創出に重点を置くとともに,未満足の医療ニーズを解決している。
私たちの候補品である効率的かつ選択的な標的タンパク質分解剤
我々は現在CFT 7455とCFT 1946の最初のヒト1/2期臨床試験を行っている。これらの案は,既存療法がまだ十分に治療されていない目標を対象としている
CFT 7455:多発性骨髄腫および非ホジキンリンパ腫治療のIKZF 1/3分解剤
我々は、MMおよびNHLの治療のためのCFT 7455、IKZF 1/3に対する経口生物分解剤を開発している。私たちは強力なメカニズム基礎と明確な生物学的定義を持っているので、私たちの初期分解目標としてIKZF 1とIZKF 3を選択した。臨床前研究において、CFT 7455は多発性骨髄腫(MM)、末梢T細胞リンパ腫(PTCL)とマントル細胞リンパ腫(MCL)皮下移植マウスモデルにおいて強力な活性を示し、臨床前概念検証を提供した。CFT 7455の差別化薬理作用は,その高い効力を含めて,我々が開発を行っている適応ごとに臨床結果の改善に移行する可能性が信じられている
2023年12月、我々は進行中のCFT 7455段階1/2臨床試験の臨床データを公表し、単一療法としてデキサメタゾン併用によるMM治療を行った。データによると、14日間の計画/14日の非計画が最適であり、データ締め切りまでCFT 7455の耐性が良好であり、好中球が減少し、予想される標的毒性であり、最も一般的な3段階以上の有害事象が観察された。さらに、CFT 7455はB細胞成熟抗原(BCMA)療法を含む、以前に多発性骨髄腫治療を受けた患者に対する抗骨髄腫活性および国際骨髄腫ワーキンググループ(IMWG)の反応を示した
IKZF 1およびIZKF 3はよく知られているいくつかの血液癌の生物標的である
IKZF 1とIKZF 3はリンパ髄系多能性前駆細胞が成熟免疫細胞によって分化するコア転写因子であり、T細胞と形質細胞、例えばB細胞を含む。特に、B細胞の成熟を阻止することにより、B細胞駆動血液癌、例えば多発性骨髄腫、B細胞リンパ腫と骨髄異形成症候群に対して抗増殖作用がある。これらの細胞のIKFZ 1/3によるB細胞の成熟促進に対する内在依存性以外に、第三者研究により、IKZF 1とIKZF 3の分解はT細胞IL-2発現を増強させ、これはIKZF 1/3の分解もT細胞活性を誘導でき、そして抗癌作用を発揮する可能性があることを意味する。また,レナドアミンとポアマイドは主にIKZF 1/3をその作用機序としているため,IKZF 1/3はすでに臨床現場で標的として検証されている
私たちの最初の人間段階の1/2実験は
2021年6月、我々はCFT 7455の最初のヒト1/2期臨床試験を開始した。この試験は主に安全性、耐性と抗腫瘍活性を研究することを目的としている。副次的および探索的目標は,CFT 7455のPKとPD特徴を記述することである.この実験では,NHL患者の単一療法としてCFT 7455を評価し,デキサメタゾンと併用して再発や難治性MMを治療しており,第1段階用量漸増試験に参加する2つの分枝を募集している。
多発性骨髄腫
米国では,多発性骨髄腫は全新癌症例の1.8%近くを占めている。国立癌研究所では,2023年に米国で35730例の多発性骨髄腫新症例があり,12590人がこの疾患で死亡したと推定されている。アメリカ国家衛生研究院(NIH)のデータによると、MM患者の全体予後は過去数十年間に大きく改善したにもかかわらず、MM患者の予後は非常に悪く、予測された中位5年相対生存率は57.9%であった。したがって、まだ満たされていない多くの需要がある
多くの多発性骨髄腫患者は治療に初歩的な反応がある。骨髄に対する蛍光in situハイブリダイゼーション或いはFISH研究により、患者はハイリスク或いは標準リスク分類に分類された。造血細胞移植条件に適合するハイリスク患者は、一般に、幹細胞採取前に腫瘍細胞数を減少させるために、IKZF 1/3標的薬、例えばレナドアミンを含む併用レジメンの誘導治療を受ける。代替的に、造血細胞移植条件に適合しない患者は、通常、IKZF 1/3標的薬およびステロイド、通常はデキサメタゾンを含む3~4種類の薬剤を含む組み合わせレジメンを直ちに受け入れ、病状が進行するまで、または受け入れられない毒性が出現する
しかしながら、異なる治療選択があるにもかかわらず、大多数の患者は最終的に進行および/または連続再発を経験する。我々のCFT 7455臨床計画では、私たちは最初に、レナドアミン、ポアマイド、プロテアソーム阻害剤、および抗CD 38モノクロナル抗体を含む少なくとも3つの特定の治療を受けた再発または難治性MM患者に注目した。進行中のCFT 7455 1/2期試験の用量増加データは,CFT 7455が患者に有意なメリットをもたらすことを証明している可能性が考えられる。
末梢T細胞リンパ腫
PTCLは異質性、典型的な侵襲性NHLである。アメリカ国立衛生研究院のモニタリング、疫学と最終結果計画(SEER Program)は2023年にアメリカで新たにNHL病例80,540例、死亡20,180例と推定された。アメリカとヨーロッパでは、PTCLはすべてのNHL診断の約5%から15%を占めている。PTCL患者の5年総生存率は30%−50%であった
PTCLは多くのサブタイプを有する異質性悪性腫瘍であり,これらのサブタイプの結果はそれぞれ異なるが,多くのPTCL患者の予後は悪い。例えば,亜型が定義されていないPTCL患者では,通常PTCL(別途指定されていないPTCL)やPTCL−NOSと呼ばれ,5年総生存率は約20%から32%である。そのほか、血管免疫芽細胞腫、ナチュラルキラー細胞/T細胞リンパ腫、成人T細胞白血病/リンパ腫、肝脾、腸病型或いはALK末梢T細胞リンパ腫患者の中位5年の総生存率はすべて50%未満であった。化学療法の初期総有効率は約40%から75%であったが,多くの患者は最終的に再発する。化学療法後の中位無進展生存期間(PFS)は12~14ケ月であり、中位5年生存率は約20%から30%であった。ドナドアミンはPTCLの2期臨床試験で評価されており,その総有効率は22%から26%であることが示された。しかし,レナドアミンなどの脳アルブミン調節剤は広く使用されておらず,PTCLの治療にも承認されていない。われわれの臨床前データによると,CFT 7455はこれらの患者のために有意義な利点を創出し,承認されればこれらの患者の既定の看護基準となる可能性があると信じている
マントル細胞リンパ腫
アメリカ癌協会のデータによると、MCLは成熟したB細胞NHLであり、すべてのNHL診断の約5%を占める。米国国立衛生研究院のデータによると、毎年20万人に1例が発生している。標準治療を受けた患者の中位総生存率は4年から5年であった。臨床試験でテストした薬物以外に、第一線の治療選択は通常通常の化学免疫治療、リツキシマブと放射線治療のある種の組み合わせを含む。多くのMCL患者は連続再発の経験があり、IKFZ 1/3標的薬、BTK阻害剤またはbcl 2阻害剤ventoclaxを含む様々な薬物の治療を受けている。レボドアミンは、以前の2つの療法(1つはポートゾミを含む)後に再発または進行するMCL患者のために承認され、部分的に観察された総応答率に基づいて約26%であった。しかし,ドナドアミンはMCLの治療に広く用いられていない。したがって,承認されればCFT 7455は結果を著しく改善し,これらの患者の既定の看護基準となる可能性があると信じている
CVT 1946:BRAF V 600 X分解による黒色腫、結腸直腸癌、および非小細胞肺癌の治療
CFT 1946,BRAF V 600 Xの経口生物分解剤を開発している。我々が目標としてBRAF V 600 Xを選択したのは,その強力な機械原理,良好な生物学的定義,満たされていない需要のためである。米国ではBRAF変異が約5%の癌で発生しており,毎年約10万人の患者がBRAF変異癌と診断されていることを意味する。すべてのBRAF変異において,約70%~90%が別のアミノ酸をV 600で置換している。我々は、まず3つのタイプの癌においてCFT 1946の開発を行う予定である:黒色腫であって、BRAF V 600 X変異が約35%の末期患者で発生し、結腸直腸癌であって、BRAF V 600 X変異が約5%~10%の患者で発生し、非小細胞肺癌であり、BRAF V 600 X変異が約1%~2%の患者で発生する、黒色腫。患者には
これは、承認されたBRAF阻害剤の後に再発したり、それに反応しない人にとっては、依然として高度に満たされていない需要である。著者らは様々なBRAF V 600 X固形腫瘍におけるCFT 1946の応用を評価しており、黒色腫、結腸直腸癌および非小細胞肺癌を含み、これらの特定の適応の現在の看護標準BRAF阻害剤は第一線に設置されたDradfenibとトリメチニブの併用による黒色腫であり、無進展生存期間(PFS)は11.4ケ月であり、Enorafenibとセツキシマブの併用によるセツキシマブ治療二線レンタルCRC、PFSは4.2ケ月、及びDradfenibと曲美チニブの併用によるNSCLCの治療、PFSは15.2ケ月である。変異特異的BRAF V 600 X分解剤は,現在利用可能なBRAF阻害剤よりも有意な機械的利益を提供し,臨床結果を有意に改善する可能性が考えられる
私たちの最初のヒト1/2期臨床試験は
2023年1月,CFT 1946の最初のヒト1/2期臨床試験を開始し,進行中の臨床試験に患者を募集し続けた。1/2期臨床試験は主に安全性、耐性および抗腫瘍活性を調査し、二次および探索的目標はCFT 1946のPKとPD特徴を特徴付けることである。この研究の第1段階の初期段階は、前にBRAF阻害剤治療を受けた後、結腸直腸癌、黒色腫、および非小細胞肺癌患者を含む単一薬物治療BRAF V 600 X固形腫瘍患者としてCFT 1946を評価するであろう。第一段階試験の進展に伴い、私たちはCFT 1946とトリメチニブの併用によるBRAF V 600 X黒色腫と以前のBRAF阻害剤治療後の非小細胞肺癌患者を評価するために追加のARMを増加させる可能性があり、私たちはまた前のBRAF阻害剤治療後のCFT 1946とセツキシマブの併用治療BRAF V 600 X結腸直腸癌の以前のBRAF阻害剤治療後の治療効果を評価するために別のARMを増加する可能性がある
BRAF V 600 Xはよく知られている発癌変異である
BRAFはダウンへのシグナル伝達に関与して細胞増殖を開始するいくつかのプロテインキナーゼの一つであり,このシグナル経路はマイトジェン活性化プロテインキナーゼ,あるいはMAPKと呼ばれる。MAPK経路は細胞外の増殖シグナルを核に伝達し,細胞増殖のシグナルを発する。多くの癌の特徴はこのMAPK経路の各構成部分の活性化突然変異であり、BRAF V 600 X突然変異を含み、それはMAPK経路の構造的活性化を与え、発癌転化を促進し、そして腫瘍の成長を招くことができる。
BRAF遺伝子の600番目のコドン上のアミノ酸Valineの単塩基置換はV 600 Xと呼ばれ,I類変異と呼ばれ,これらのV 600 X変異がグルタミン酸置換Valine(最もよく見られるこのような変異)を引き起こす場合,V 600 E変異と呼ばれる。
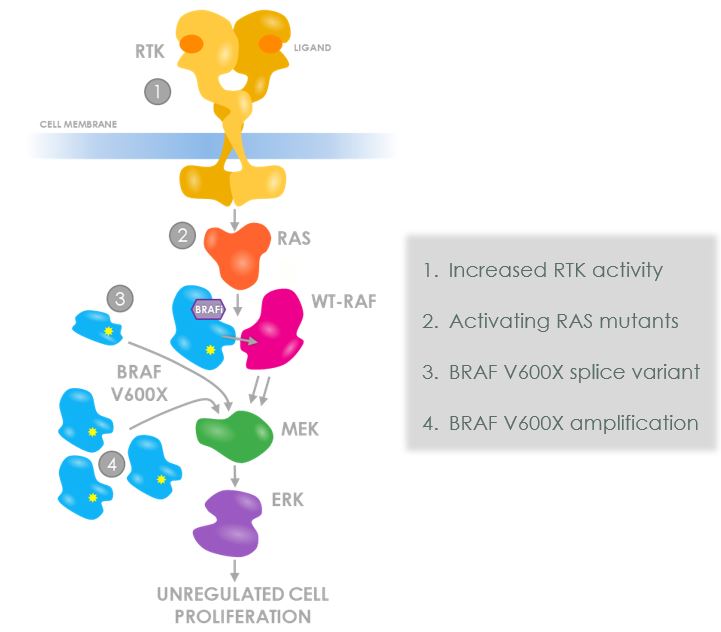
BRAF V 600 X変異体はMAPK経路を構造的に活性化し,これは細胞の増殖がこの経路を正常に活性化するために必要な細胞外増殖シグナルを受けずに活性化されることを意味する。構造的活性化が起こるのは,BRAF V 600 X変異体が単一タンパク質やモノマーとしてシグナルを発することができるのに対し,BRAFとCRAFを含む野生型RAFタンパク質は,下流シグナル伝達を行うためには,2種類のタンパク質の複合体や二量体を形成しなければならないからである。このような構造的活性化はMAPK細胞増殖経路の過剰活性化を招き、腫瘍細胞の増殖と腫瘍の成長を招く。許可されたBRAF V 600 E小分子阻害剤であるビモラファニ、ダプラファニとエンコラファニー-阻害変異BRAFモノマーのMAPK経路に対する構造的活性化。しかし,これらの分子のBRAF抑制は様々な抵抗性機構の出現を招くことは避けられない。他のMAPK経路標的とは異なり,この耐性はATP結合ポケットの変異によるものではない。対照的に、第1世代阻害剤は、BRAF V 600 Xと他のRAFパートナーとの間の二量体形成を促進する抵抗性機構の影響を受けやすく、これらのパートナーは、BRAF V 600 X自体、野生型BRAF、または野生型CRAFを含むことができる。これは,これらの阻害剤が矛盾活性化と呼ばれる現象の影響を受け,RAFダイマーを背景に2種類のRAF蛋白を同時に抑制できないためである。したがって,BRAF V 600 Xの阻害形態は,RAFダイマー形成を促進する細胞環境において非薬物結合RAFパートナーの活性化に寄与することができる。以下は,腫瘍細胞が第1世代BRAF阻害剤の存在下でこの脆弱性を利用して耐性を産生する4つの方式である。
•RTK活動の増加:増強されたRTKシグナルはRASを活性化することができ、それによって薬物結合BRAF V 600 Xと野生型RAFタンパク質との間の二量体形成を促進する。BRAF V 600 Xのみが阻害剤で占められているため,野生型RAFシャペロンは同時に二量体によって活性化されMAPK経路を上昇させる
•RAS変異体を活性化する:上流RTKシグナルが乏しい場合、RASの変異は、薬物結合を促進するBRAF V 600 Xと野生型RAFタンパク質との間に二量体を形成する能力を活性化することができる。以上のように,非阻害物質が結合した野生型RAFパートナーがMAPK活性を上昇させる。
•BRAF V 600 Xコネクタ変種:RAF V 600 Xサブタイプを欠くRAF V 600 Xサブタイプ、またはエクソン3-5によってコードされるRBDを選択的にスプライシングすることによって産生される。RBDがない場合、これらのBRAFサブタイプは、低レベルのRAS活性の存在下でも二量体化され、BRAF V 600 Xダイマーが形成されると、阻害剤は、一方のBRAF V 600 Xパートナーを阻止し、占有されていない別のBRAF V 600 Xパートナーを活性化し、MAPK活性を自由に上昇させることしかできない
•BRAF V 600 X拡張版:遺伝子改変はBRAF V 600 X遺伝子の多くの追加コピーをもたらす--この現象は遺伝子増幅と呼ばれる。これは逆にBRAF V 600 X蛋白濃度の上昇を招く
高濃度では、BRAF V 600 X蛋白は上流RTKやRAS活性化を必要とせずに二量体を形成する。もう一度、第一世代阻害剤は、2つのBRAF V 600 X対を同時に結合し、抑制することができず、占有されていないBRAF V 600 X二量体対が活性化され、MAPK活性を自由に上昇させることができる
BRAF V 600 X変異の標的タンパク質分解は,分解剤が一般阻害剤よりも優れているため,分解変異体BRAFが様々なBRAFダイマーに基づく抵抗性機構に組み込まれている可能性を除去し,これらの機序がMAPK経路の矛盾した活性化をもたらすため,既存のBRAF阻害剤を根本的に改善する可能性を提供していると考えられる。したがって,CFT 1946は適応によって異なる薬剤耐性機序を克服する可能性がある。黒色腫および非小細胞肺癌では,獲得性薬剤耐性の発生は,BRAF V 600 Xモノマーが抑制されているにもかかわらず,上述したMAPK経路によって活性化されている場所である。結腸直腸癌において、もう一つの追加の複雑な要素、即ちMAPKシグナル伝達の抑制は内在的な薬剤耐性機序であるEGFRの活性化を招き、これは標準的な看護治療方法がBRAF阻害剤と抗EGFR抗体の組み合わせ、例えばセツキシマブを含む理由である。
黒色腫
国立がん研究所のデータによると、2023年に米国では約97,610人の患者が黒色腫と診断され、そのうちの約5%、すなわち毎年約4,800人の患者が転移性疾患を患っている。さらに、末期黒色腫患者の約35%がBRAF V 600 X変異を有し、その約90%がBRAF V 600 E変異である
BRAF V 600 X変異の切除不能または転移性黒色腫患者に対して、推奨される一線治療方案は、pembrolizumabまたはnivolumabなどの抗PD-1単薬、またはダプラファニブ、ビモラフェニまたはEnorafenibなどのBRAF阻害剤、およびトリメチニブ、cobimetinibまたはbinimetinibなどのMEK阻害剤との併用治療である。しかし,このような併用治療を受けた患者の相当数は,この治療に対する耐性が出現したため,十分な反応や持続的な反応がなかった
現在、BRAF V 600 E/K黒色腫患者の看護治療標準はBRAFとMEK阻害剤の組み合わせであり、ダプラファニブとトリメチニブであり、この方案の中位PFSは約11.4ケ月である
結腸直腸癌
国立がん研究所のデータによると,2023年,米国では約153,020名の患者が結腸直腸癌と診断された。これらの患者のうち,約20%(約3万名)が転移性疾患,約25%(約3.7万名)が局所疾患と診断され,これらの疾患は転移に伴い再発する。約5%~10%のCRC患者がBRAF V 600 Eを有するか、または年間約11,000例のCRC BRAF V 600 E症例が発生する
BRAF V 600 E変異の患者は,従来の治療で進展すれば,Enorafenibとセツキシマブの併用治療が可能であり,両薬剤とも阻害剤治療である。しかし、この方案の治療効果は限られており、中位PFSは約4.2ケ月である
非小細胞肺癌
国立癌研究所のデータによると,2023年に米国では約238,340名の患者が非小細胞肺癌と診断され,そのうち約53%,すなわち126,320名の患者が転移性疾患と診断され,現地で診断された腫瘍に比べて再発率が高かった。約1~2%の非小細胞肺癌患者はBRAF V 600 X変異を有する
BRAF V 600 E突然変異を有する新しい診断NSCLC患者は第一線でダプラファニブとトリメチニブ治療を受けた時、典型的な中位PFSは15.2ケ月であった。ダプラファニブやトリメテニブ治療を受ける前に,化学療法や他の標的薬治療を受けた患者の中位PFSはわずか9カ月程度であった。
臨床前発展
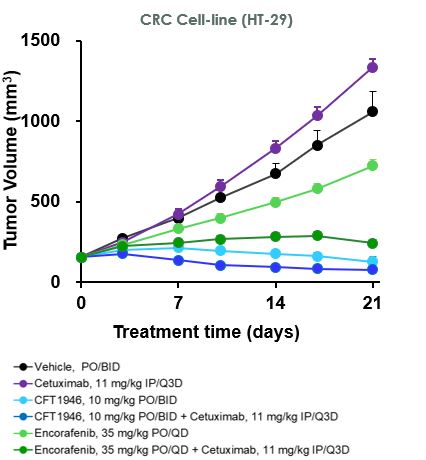
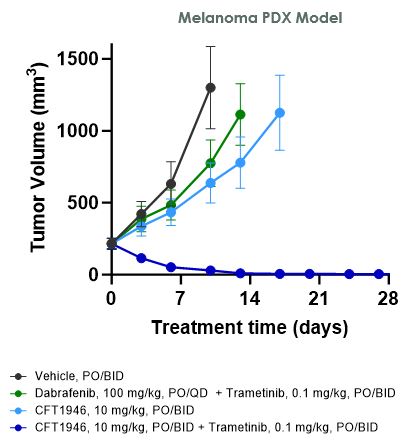
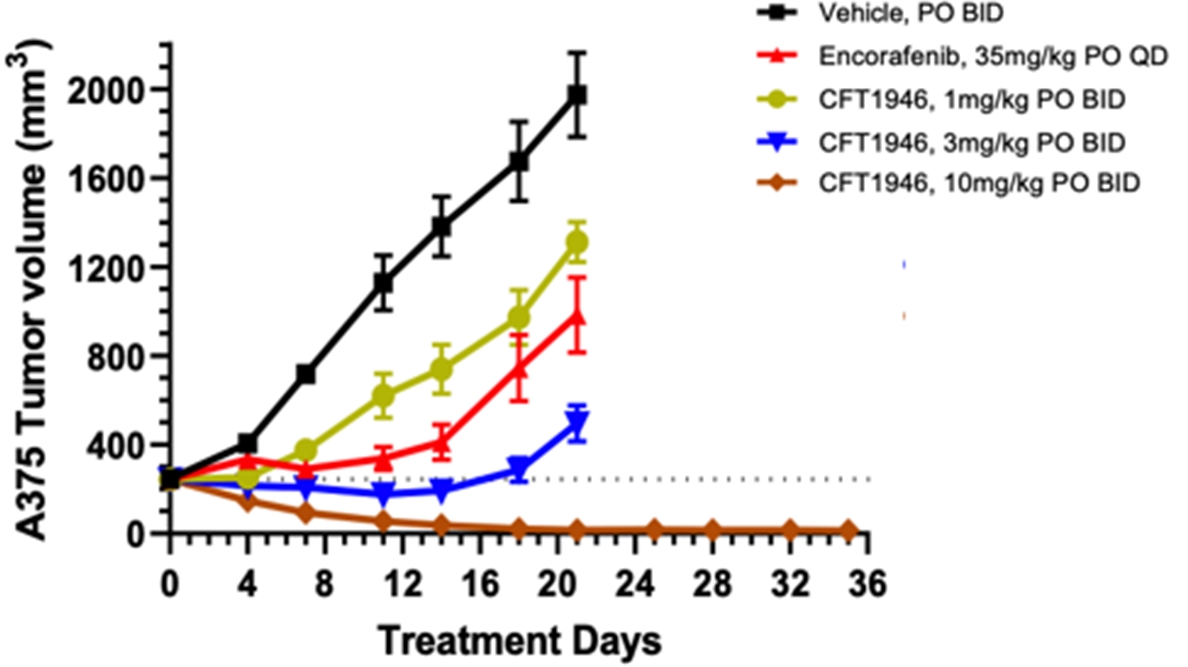
前臨床研究において、CFT 1946はBRAF V 600 X結腸直腸癌、非小細胞肺癌および黒色腫の治療に対して標準治療よりも有効であることが証明された。以下の図に示すように,HT−29大腸癌細胞株異種移植モデルでは,CFT 1946を単一療法およびセツキシマブと併用した場合,セツキシマブ単剤,エンコラフェニ単薬,エンコラフェニとセツキシマブの併用治療よりも奏効率が強いことが示された。非小細胞肺癌PDX異種移植モデルでは,CFT 1946は単一療法としてダプラファニブとトリメチニブの併用治療に対して増強した反応を示した。また、黒色腫PDX BRAF阻害剤耐性モデルでは、CFT 1946とトリメチニブの併用は、ダプラファニブ連合トリメチニブと比較して腫瘍深度の消退を示した
A 375 BRAF V 600 E黒色腫モデルでは、CFT 1946の腫瘍成長に対する用量依存性の抑制作用が認められ、10 mg/kgを1日2回経口投与した最低有効用量で腫瘍の消退を示した。また,CFT 1946は2 mg/kgから(経口および1日2回)ともFDA承認のBRAF V 600 E阻害剤Enorafenibよりも優れた活性を示した。また,以下に示す異種移植研究では副作用は認められなかった(体重の有意な減少による評価)
CFT 1946は臨床前転移性黒色腫中枢神経系(CNS)モデルにおいて活発であることが証明された。また,CFT 1946はエンケラフェニと比較して生存期間を延長し,中枢神経系の負担を軽減した。
CFT 8919:高効率、経口、アロステリック、突然変異選択性EGFR L 858 R分解剤
我々は、非小細胞肺癌の治療のためのCFT 8919の経口生物的に利用可能なEGFR L 858 Rアロステリック分解剤を開発している。著者らがEGFRを標的とすることを選択したのは、その明確な生物学的特性、及びEGFRキナーゼ阻害剤が直面している局限性であり、著者らの分解剤方法はこれらの局限性を克服できる可能性があると信じている。私たちの最初のターゲットはEGFR L 858 R駆動非小細胞肺癌患者であり、彼らはオシメチニブを含む承認されたEGFR阻害剤治療を受けた後に進行した。著者らは、蛋白質分解の独特な優勢により、CFT 8919は標準看護EGFR阻害剤に対する耐性を克服し、それによってより深いレベルとより持続的な反応を産生する可能性があると信じている。2023年5月、我々はBetta PharmaとCFT 8919の大陸部中国、香港特別行政区、マカオ特別行政区、台湾地区を含む中国地区における開発と商業化について独占許可および協力協定を締結した。このパートナー関係は、大中国地区のEGFR L 858 R駆動の非小細胞肺癌の高発によるEGFR駆動薬剤耐性突然変異の患者の中でCFT 8919の全体開発を加速させ、CFT 8919から利益を得る可能性のある患者数を最大限に増加させることができると信じている。そのため、一期臨床研究は闘魚製薬によって大中国地区で行われる。Betta Pharmaが第1段階試験から結果を出すまで,大中国以外で臨床研究を開始するつもりはなかった。私たちは大中国の外でCFT 8919を開発して商業化するすべての権利を保持している
EGFRは既知の薬剤耐性機序を有する腫瘍標的蛋白である
EGFRは受容体チロシンキナーゼであり、細胞分裂と生存の制御に参与する細胞シグナル経路である。EGFR遺伝子の突然変異はあるタイプの癌細胞中のEGFR蛋白シグナル異常を招き、1組の非小細胞肺癌患者を含む。既知のEGFRチロシンキナーゼドメイン変異では、エクソン19の欠失またはエクソン21の点変異が約90%発生し、後者は858コドン(L 858 R)上のアルギニン置換ロイシンをもたらす。エクソン21のL 858 R活性化突然変異はEGFR突然変異NSCLCの約25%~45%を占める。EGFRチロシンキナーゼ阻害剤或いはTKIはすでに開発され、顕著な臨床利益を提供した。しかし、患者は最終的に薬剤耐性を産生し、通常EGFRの第二次薬剤耐性突然変異を獲得する。T 790 Mは第一世代と第二世代EGFR TKIに続く最もよく見られる薬剤耐性突然変異であり、ゲフィチニブ、エルロチニブ、アファチニブとダコミチニブを含む。第三世代共有結合EGFR阻害剤osimertinibはこのような薬剤耐性機序を克服することができ、現在すでに第一線の治療への応用が許可されているが、獲得性薬剤耐性は依然として1つの問題である。オシメチニブ治療後に進展した患者は有効な治療選択が不足しているが、EGFR C 797 S突然変異は最もよく見られる標的薬剤耐性機序である
CFT 8919はEGFR L 858 Rに対する突然変異選択的降下物であり、薬剤耐性二次突然変異(T 790 M及び/又はC 797 S)の設定において依然として有効である。CFT 8919は標準蛋白阻害剤に対する分解剤の優勢のため、より深いレベルとより持続的な反応を実現することを期待していると信じている。また、30%-40%の突然変異のEGFR非小細胞肺癌患者は脳転移が発生するため、中枢神経系への浸透はこの区域の治療効果を推進するのに十分であり、著者らはCFT 8919を開発候補として選択する重要な要素である。
非小細胞肺癌
アメリカ国立癌研究所のデータによると、2023年にアメリカでは約238,340人の患者が非小細胞肺癌と診断され、その中の10%~15%の患者は突然変異EGFRまたはmEGFRを患っている。EGFR突然変異はアジア血統を有する非小細胞肺癌患者において特によく見られる。中国では約693,000人の患者が診断されました
毎年のNSCLCでは、約50%の診断はEGFR突然変異によって駆動される。そのほか、30%から40%のEGFR突然変異患者は脳転移が発生する
EGFR L 858 R突然変異は第二種の最もよく見られるEGFR活性化突然変異であり、アメリカと中国のEGFR診断において約40%の突然変異を発見した。標準的なEGFR阻害剤治療を受ける時、EGFR L 858 R突然変異を持った患者はエクソン19欠損を示す患者と比べ、治療結果はもっと悪い。EGFR L 858 R患者の一線治療にオシメチニブを使用した場合,中位PFSは14.4カ月であったが,エクソン19欠損患者は21.4カ月であった。この患者群の中で、EGFRを介したオシメチニブに対する薬剤耐性が最も多い機序はC 797 S突然変異である。この比較的に短いPFS中央値と薬剤耐性はL 858 R患者群の医療需要がまだ満たされていないことを表明した。
臨床前発展
著者らはすでに臨床前実験を行い、著者らのEGFR分解剤の活性特徴を表現し、そしてCFT 8919は1種の有効、高選択性EGFR L 858 R経口生物分解剤であり、広範な標的薬剤耐性突然変異と頭蓋内活性を有することを証明した。
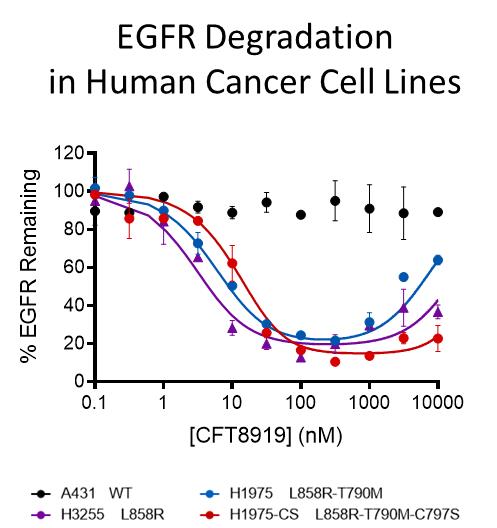
ヒト癌細胞系では体外培養CFT 8919は、低ナノモル濃度でEGFR L 858 Rの有効な分解を誘導し、10μM以下では野生型分解を誘導しないことが観察された。重要なことは、CFT 8919は、T 790 MおよびT 790 M−C 797 Sなどの二次抵抗性変異の存在下でも活性を維持することである。これは次の図に反映される.
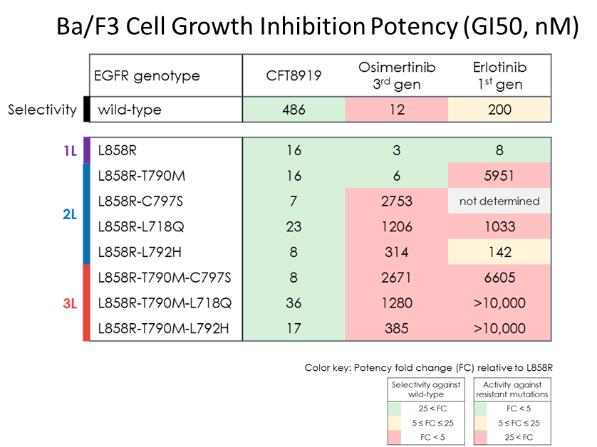
また、我々はBA/F 3細胞モデルにおいて、広範なEGFR耐性突然変異のグループに対してCFT 8919を評価した体外培養それは.異なる濃度のEGFR変異形質転換したBA/F 3細胞の72時間以内の増殖を測定することにより、細胞増殖抑制力価GI 50を決定した。以下の表に示すように、L 858 R単一突然変異と比べ、CFT 8919は大グループのEGFR二次突然変異に対して同等の抗増殖活性を示し、これらの突然変異はすでに許可されたEGFR阻害剤(例えばosimertinibとerlotinib)に対する獲得性耐性の産生を招く
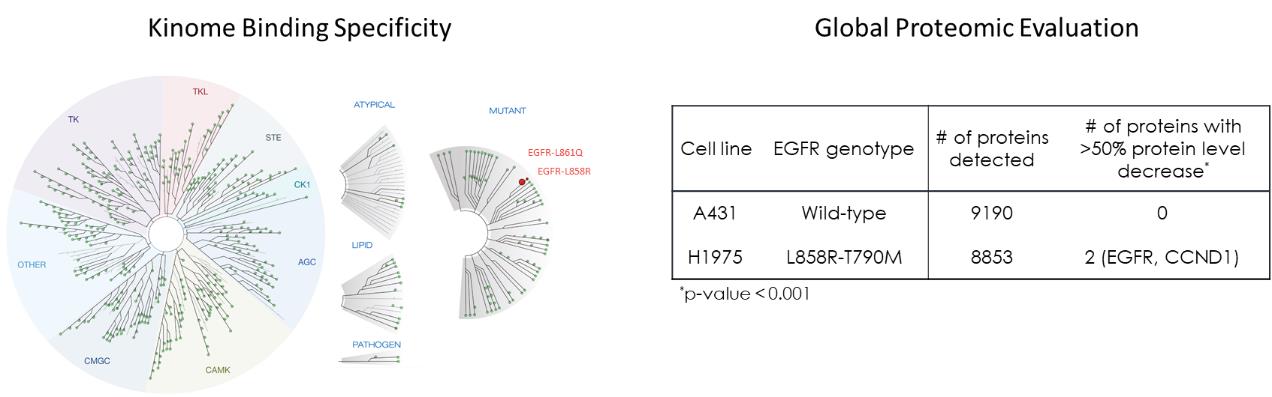
Kinomeプロファイルと全世界プロテオミクスによりCFT 8919の結合と分解選択性を評価したが,CFT 8919は有意な標的外活性は認められなかった。Kinome選択性は
DiscoveryX KINOMEスキャンで468個のキナーゼを検出し,左図に示す。CFT 8919を100 nmの単一濃度で試験したところ,50%のカットオフ点で野生型キナーゼが結合を示さなかった。CFT 8919は顕著な結合を示す唯一のキナーゼは標的上のエクソン21 EGFR活性化変異体L 858 RとL 861 Qである。また,A 431 EGFR−WTとH 1975 EGFR−L 858 R−T 790 M細胞系を300 nM CFT 8919で6時間処理した後のプロテオームの分解選択性を評価した体外培養それは.右表に示すように,8000個を超えるタンパク質の測定では,EGFR−L 858 R−T 790 MとCCND 1のタンパク質レベルのみがCFT 8919処理後に50%以上低下し,高い選択的分解特徴を示した。CCND 1蛋白の喪失はEGFR抑制の生物学的効果によるものであり,直接分解ではなく,この変化がosimertinib EGFR阻害剤治療においても観察されたためと考えられる
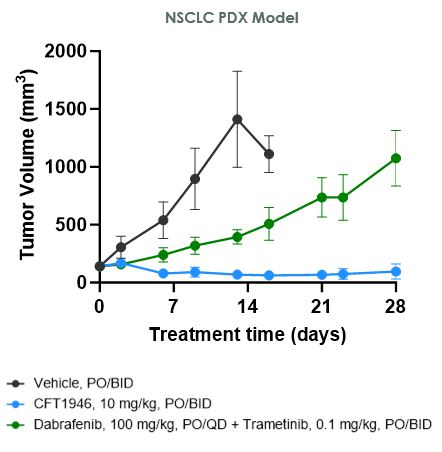
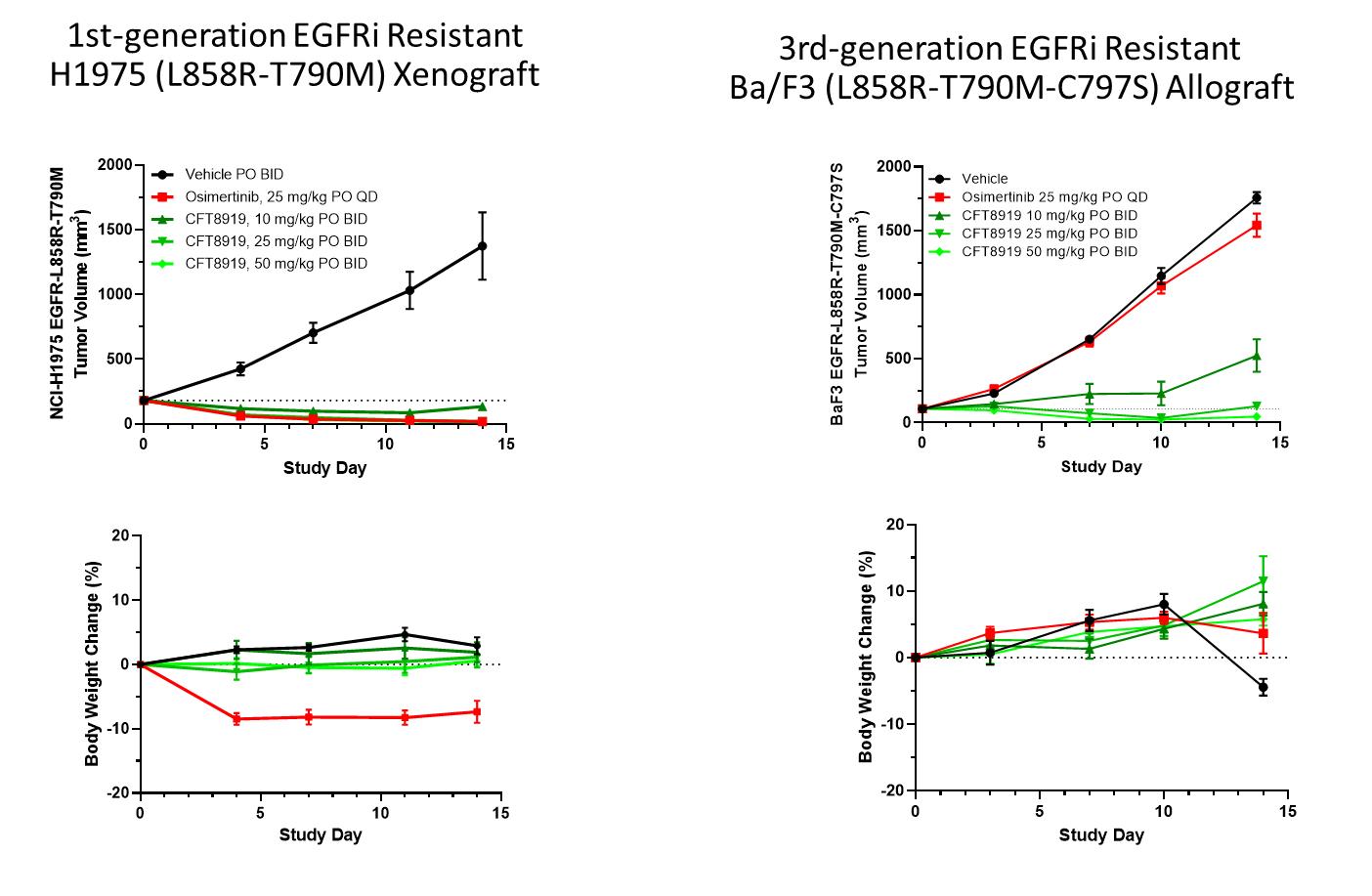
生体内H 1975 EGFR-L 858 R-T 790 M異種移植腫瘍におけるCFT 8919の活性測定(1STTKI耐性)とBaF 3 EGFR−L 858 R−T 790 M−C 797 S(耐オルシメチニブ)同種移植モデル。H 1975異種移植モデルでは,CFT 8919(BID)を1日2回経口投与して用量依存性の活性を示し,10 mg/kgと低い用量で腫瘍の消退を認めた。投与量>25 mg/kgのCFT 8919はBID>25 mg/kgのCFT 8919をBA/F 3、EGFR-L 858 R-T 790 M-C 797 S同種移植腫瘍に対して消退させることができ、オシメチニブは予期したように不活性化した。いずれの用量も耐性は良好であり,体重の有意な低下はなかった。これは次の図に反映される.
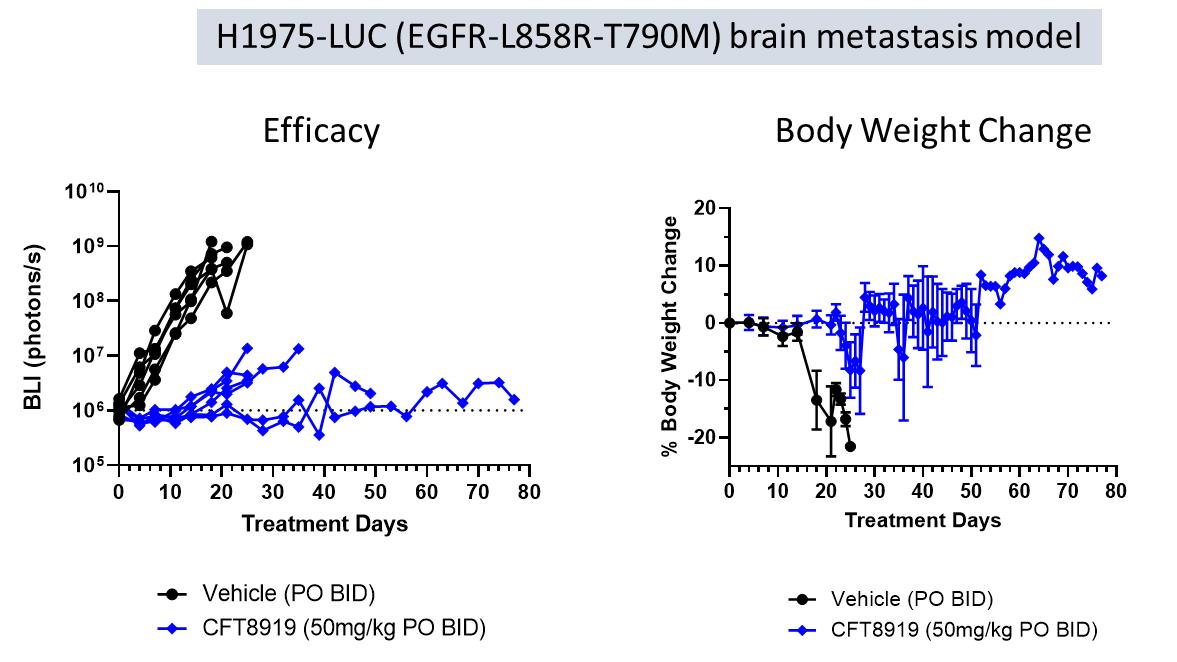
H 1975−Luc脳転移モデルではCFT 8919の脳内活性を評価した。雌性BALB/cヌードマウス頸動脈にH 1975(EGFR L 858 R-T 790 M)ルシフェラーゼ発現細胞を注射した。生物発光イメージング技術(BLI)を用いて腫瘍の成長状況を追跡した。以下の図に示す
CFT 8919は経口投与後,腫瘍負担が急速に有意に減少し,体重減少が最小であることを示し,中枢神経系に活性を有することが示唆された。
私たちの他の探索計画は
上で議論した項目のほかに,他の様々な発見段階のパイプラインプロジェクトを進めている.我々の戦略によれば,我々が開発可能な分解剤が同一の疾患を治療する基準や他の方法ではなく,納得できる差別化方法を提供するかどうかを1つの目標ごとに評価し,まれな疾患を含む生物学的および毒性リスクを最小限に抑えるポイントと一致し,まれな疾患を含む高度に満たされていない医療ニーズに重点を置く。これらの早期発見計画には,すでに臨床前モデルにおいて適切な場合に血液脳関門を横断できることを示す化合物が含まれている。私たちの探索計画は、私たちが完全にコントロールして持っている内部計画と、私たちのパートナーと協力する計画の組み合わせです。
協力と許可協定
メルク許可と協力協定
2023年12月、私たちはメルク社と許可および協力協定、またはメルク許可プロトコルと呼ばれ、分解物-抗体結合体(DAC)を協力して開発し、それを商業化し、DACは癌細胞中の病原性タンパク質を選択的に標的とすることを目的とした新興の方法である。ライセンス契約の条項によると、私たちは私たちのいくつかの知的財産権に基づいて、最初に開示されなかった腫瘍学目標に対するDACを開発、製造、商業化するために、メルク社に全世界独占許可を付与した。メルク社はすべての開発、監督管理、承認、製造、商業化コストを担当している。メルク許可協定の条項によると、メルクは1000万ドルの現金を前払いした。初期目標に対するDACについては,合計約6億ドルの記念碑的支払いを獲得し,純売上高の分級印税を得る資格がある。また、協力の一部として、他の3つの目標に対するDACを開発、製造、商業化するために、いくつかの知的財産権の下でグローバル独占許可を得る選択権をメルク社に付与し、各目標は価格を支払う必要があります。メルク社がこれらのオプションを行使する場合、これらの追加の計画はまた、メルク許可協定で詳細に説明された特許権使用料の期間内に追加の潜在的マイルストーンおよび特許権使用料を提供するだろう。メルク社が協力延長のすべての選択肢を行使すれば、協力全体で約25億ドルにのぼる潜在的な支払いを得る資格があるだろう。
Betta Pharma許可と協力協定
2023年5月、吾らはBetta Pharma或いはBetta Pharma許可協定と許可及び協力協定を締結し、CFT 8919の開発と商業化について協力し、CFT 8919は1種の経口生物使用可能なBiDAC分解剤であり、大陸部の中国、香港特別行政区、マカオ特別行政区及び台湾からなる大中国地区に有効及び選択的に対抗することを目的とし、この地区は大陸部中国、香港特別行政区、マカオ特別行政区及び台湾からなり、我々は全世界の他の地域で開発及び商業化する権利を保持する。
Betta Pharma許可協定の条項によると、私たちは、大中国で人間のためのCFT 8919を開発、製造、商業化するために、私たちのいくつかの知的財産権に基づいてBetta Pharmaに独占許可を与える。ベータ製薬は大中国地域でのすべての開発、規制承認、製造、商業化コストを担当しているが、ベータ製薬は大中国地域における私たちの代理店として参加している
私たち。協力の一部として、Betta Pharmaは1,000万ドルの現金を前払いし、3.57億ドルにのぼるマイルストーン支払いを得る資格があり、CFT 8919の大中華区における中国の純売上高の等級別特許権使用料を得る資格がある。Betta Pharmaが私たちに支払った特許使用料は低いものから中2桁まで様々だが、Betta Pharmaライセンス契約に記載されている特定の場合には減少する可能性がある。また,協力の一部として,米国食品医薬品局から承認されたCFT 8919新薬申請を受けた後,Betta Pharmaに4,000万ドルまでの記念碑的支払いを支払うことに同意し,この記念碑的金額は,Betta Pharmaが組み入れた予想臨床試験における患者率と承認された治療経路から得られた。また、当地域(中国以外の世界の他の地域)CFT 8919の純売上高についてBetta Pharmaに低い1桁パーセント範囲の階層的特許権使用料を支払うことに同意したが、Betta Pharma許可プロトコルで説明されているようにいくつかの削減を行う場合もある。Betta Pharmaライセンス契約下のすべての予想される特許使用料の使用期限は、(I)当該製品が国/地域で初めて商業販売されて12(12)周年、(Ii)当該製品の国/地域における任意の規制排除期間が満了することをカバーし、(Iii)当該製品をカバーする最後のライセンス特許が当該国/地域で満了した日から、個々の製品および国/地域に基づいて終了しなければならない。また、2024年1月、Betta Pharmaの付属会社は以下のように2500万ドルで私たちの普通株を購入した。
生物遺伝研究協力と許可協定
2018年12月、私たちはBiogenと共同研究と許可協定、すなわちBiogenプロトコルに署名し、協定によると、私たちはBiogenと協力し、私たちの独自のタンパク質分解器プラットフォームを使用して小分子タンパク質分解器を研究、開発、識別することに同意した。2020年2月、我々は生物遺伝協定の修正案に署名し、生物遺伝の標的結合部分に対する所有権をさらに明らかにし、これらの結合部分は分子の一部であり、協力目標に対する任意の関連知的財産権を特定した。この改正はさらに規定されており、Biogenは、Biogenプロトコルの下で予想される研究および開発活動を行うために、これらのBiogen標的拘束性部分および任意の関連知的財産権を使用する権利を私たちに許可する。
“生物遺伝協定”によると,我々は知的財産権の下で生物遺伝の独占的許可を付与し,複数のレベルで再許可を行う権利があり,(A)各方面が合意した研究開発計画に基づいて候補開発活動を行うことを目的としており,(B)すべての分解物や製品の世界での任意の用途を利用することを目的としている
“生物遺伝的プロトコル”の条項に基づき,プロトコルに規定されている目標選択や置換プログラムに基づいて,生物遺伝的選択のいくつかの目標の研究·開発活動を担当する.我々は,候補開発活動を実行するために必要なすべての資源を提供し,これらの活動を適用法律と“生物遺伝研究プロトコル”に基づいて合理的な慎重さと技能で実行し,適用開発計画に規定された活動を達成し,一連のあらかじめ定義された基準を満たすように生物遺伝研究会社に一定数の目標に対する分解剤を渡す必要がある。我々とBiogenはまたBiogenの目標選択過程に情報を提供する研究活動を担当しており,Biogenはそのために自分のコストを支払い,我々のコストを精算し,最高で一定の金額に達する.生物遺伝研究プロトコルの研究期間は2023年6月に終了した。
生物遺伝会社が生物遺伝研究会社が選択した各目標に対して分解剤のIND-Enabling研究を開始する際、生物遺伝研究会社は商業的に合理的な努力を担当し、使用することに同意し、ある地域ではこのような目標ごとに少なくとも1つの製品のさらなる開発、規制事務、製造、商業化を行う
Biogen協定に署名した時、Biogenは候補者開発活動の前払いとして4500万ドルの前金を私たちに支払った。生物遺伝会社が目標ごとの分解剤を受け取り,あらかじめ定義された基準を満たすと,目標ごとに200万~500万ドルの支払いを受ける資格がある。生物遺伝会社が目標ごとの開発候補に対して第1回イネーブルINDの研究を開始した後,生物遺伝会社は800万ドルを支払う必要がある。個々の目標に対して,生物遺伝会社は(A)合計3500万ドルの開発と商業化マイルストーン支払い,および(B)合計2600万ドルの販売マイルストーン支払いを我々に支払う必要があり,これらの目標に対するすべての製品の一定額の純売上高を達成し,1件あたり一定の減少幅がある。生物遺伝会社が協力期間を延長し、より多くの目標を選択すれば、総開発、商業化、販売マイルストーン支払いが増加する。また生物遺伝研究会社は製品の印税を支払うように要求されています-製品ごとの純売上高を基準に1桁の中央値パーセンテージで計算しているが、一定幅の減額が必要である
より早く終了しない限り、“生物遺伝的合意”は、適用国/地域適用製品の承認されたラベルに使用方法からなる特許権を含む最後の有効な権利主張が満了した日から、製品および国/地域の最後の日に失効する。我々と生物遺伝はそれぞれ,(A)1つまたは複数の開発候補,製品または協力目標,あるいは生物遺伝の場合のみプロトコル全体を終了することができ,他方の未治癒材料がその義務に違反していること,および(B)そのすべてが原因である
他の当事者が破産して、資金が借金をしない、あるいは似たような手続きの時。便宜上、Biogenはまた、1つまたは複数の開発候補、製品、または協力目標のためにBiogenプロトコルを完全に終了するか、または終了することができる。
羅氏は許可協定を改訂と再署名した
2016年3月に羅氏とライセンス契約を締結し、その後、2016年6月、2017年3月、2018年12月を含めて何度も修正した。私たちはこの修正されて再説明された合意をロ氏協定と呼ぶ。羅氏協定によると,我々は羅氏と協力し,我々の独自魚雷プラットフォームを用いて癌や他の適応を治療し,標的拘束性分解剤薬を研究,開発,製造し,商業化することに同意した。羅氏合意の条項に基づき,我々は合意に規定された目標選択と置換手順に基づいて,羅氏が選択したいくつかの目標に対して臨床前研究と開発活動を行うことを担当している。特定の目標の製品に対する第一段階臨床試験を行い,研究計画の適用に関する製造活動を担当しているが,羅氏は予定時間に製造責任を負う権利がある。私たちと羅氏はそれぞれこれらの研究活動の費用を分担する
羅氏協定によると、我々は羅氏に独占的なグローバル許可の独占的選択権を付与し、複数のレベルで再許可を行い、協力目標ごとの製品を開発して商業化する権利がある。特定の目標に対する選択権を行使する際、羅氏はその目標に対する製品の製造、開発、商業化を担当し、費用は自負している。しかし,特定の目標に対する製品の共同開発を選択することができ,その場合には,このような共同開発製品に関する部分開発コストを担当し,そのような共同開発製品の販売からより多くの印税を得る資格がある.我々が共同開発選択権を行使した製品を連携して詳細に説明することも選択できる.もし私たちが共通細部選択権を行使すれば、私たちは共通細部費用の一部に責任を負うだろう。私たちは一般的にこのような共同開発と共同詳細化活動に参加しないことを選択する権利がある。
2020年11月には、羅氏協定の更なる修正案に署名し、羅氏と共通目標終了協定を締結することにより、目標ごとに羅氏合意の終了に相互に同意することができる仕組みを提供する。このような性質の終了後、修正されたロー氏合意は、その行動パターンとして抑制された製品(ロー氏領域と呼ばれる)をサポートするすべてのノウハウおよび知的財産権がロー氏に回復され、その行動モードに格下げされた製品(C 4 T領域と呼ばれる)をサポートするすべてのノウハウおよび知的財産権のすべての権利が私たちの手に回復されることを規定する。また、この修正案は、共通目標終了協定を締結した後、羅氏は、羅氏分野の協力によって生じる任意のノウハウおよび知的財産権に対して権利および責任を有し、C 4 T分野に適合する協力によって生じる任意のノウハウおよび知的財産権に対して権利と責任を有することを規定している。このような権利分配を支援するために、修正案によれば、羅氏は、永久的、撤回不可能、全額納付十分な、独占的(授権者に対しても同様)、相互目標終了プロトコルに従って一方に割り当てられた特許の再許可(多層を含む)許可、および相互終了合意に従って一方に割り当てられることができる、永久的、撤回不可能、非排他的、再許可可能な(多層を含む)独自技術許可を提供する
2020年11月、この修正案に参加することにより、我々は羅氏と共同で目標EGFRに関する羅氏合意を終了することに同意した。また、2021年11月、2022年7月、2023年12月に、我々は羅氏と共同で、BRAFに関するロ氏合意および他の2つの不開示目標を終了することに同意した。したがって,羅氏は現在,羅氏分野でこれらの目標を自由に追求することができ,我々はC 4 T分野でこれらの目標を自由に追求することができ,羅氏分野のこれらの目標に関連するすべてのノウハウや知的財産権の権利と責任は羅氏当事者に返還され,C 4 T分野のこれらの目標に関連するすべてのノウハウや知的財産権の権利と責任は我々に返還され,羅氏はC 4 T分野の特許を譲渡してくれた。2023年9月、羅氏は羅氏合意に基づいて新たな未開示目標を指定し、協力に追加した。このような努力のせいで、現在もこの協力の一部である二つの目標がある。2023年12月、私たちは羅氏と共同で、羅氏が線量範囲のデータパケットを受信した後に開始するために、羅氏の残りの2つの目標に対する選択権の時間を調整するために、羅氏合意をさらに修正することに同意した
羅氏協定に署名した後、私たちは羅氏から4000万ドルの前金を受け取った。また、我々は羅氏から各積極的な研究計画の年間研究資金を獲得し、いくつかの目標の予定研究と開発成功基準を達成したときに追加支払いを受ける資格がある。改正された羅氏合意によると、羅氏が残りの2つの目標のいずれかに対して選択権を行使すれば、羅氏は800万ドルの権利料を支払う義務がある。羅氏が行使した各目標オプションについて、私たちは対応する製品に関連するいくつかの研究、開発、商業マイルストーンの実現時に2.73億ドルに達するマイルストーン支払いを得る資格があるが、知的財産権カバー範囲に基づくいくつかの減免と排除によって制限されている。羅氏はまた、各目標の1.5億ドルに達する一度の販売ベースのマイルストーン支払いを支払う必要があります
ある特定の目標を達成した製品の純売上高。最後に,羅氏が行使した選択権に応じて販売される製品純売上高の1桁中央値から10代程度のパーセンテージまでの階層特許権使用料を得る資格があるが,一定の減免が必要である.我々が共同開発権を行使する製品の販売については,適用される特許権使用料は低い1桁の割合で増加する
以前に終了しない限り、ロー氏合意は、ロー氏合意の下の特許使用料または他の支払い義務が満了していないか、または満了する日に終了する。我々と羅氏はそれぞれ完全にまたは1つずつ目標ごとにまたは製品ごとに羅氏協定を終了することができ、我々の場合、他方が羅氏協定に規定された義務に違反したこと、または他方が破産し、債務を返済しないか、または同様の手続きに違反したため、羅氏合意を1つずつ中止することができる。便宜上、ロー氏は、ターゲットごと、製品毎、または国ごとにロー氏プロトコルを終了することができる。羅氏の競争相手に買収されれば、羅氏は羅氏合意に基づいて私たちの研究、開発、共同で活動を詳しく説明することを要求する権利があり、その後、このような活動終了の支払いを受ける資格がありません。
競争
生物技術と製薬業界の特徴は技術進歩が迅速で、競争が激しく、知的財産権と独自製品を非常に重視していることである。私たちは私たちの技術、専門知識、科学知識と知的財産権が私たちに競争優位を提供すると信じているが、私たちは主要な製薬、専門製薬、生物技術会社、学術機関、政府機関及び研究を行い、特許保護を求め、研究、開発、製造と商業化協力手配の公共と個人研究機関を含む多くの異なる源からの潜在的な競争に直面している。私たちはタンパク質分解に集中している他の会社と競争しなければならないだけでなく、開発と商業化に成功した任意の候補製品は、既存の療法や将来出現する可能性のある新しい療法と競争する。しかも、私たちの産業の特徴は特許数が多く、特許侵害疑惑が頻繁だということだ
私たちの重点は私たちの魚雷プラットフォームを使ってタンパク質分解療法を発見し開発することだ。タンパク質分解のためのキメラ小分子を開発する他の会社としては、Arvinas,Inc.,BioTheryX,Inc.,Captor Treateutics,Inc.,Cullgen Inc.,Foghorn Treateutics,Inc.,Frontier Medicines Corporation,GluBioTreateutics,Inc.,Haisco製薬集団,Kymera Treateutics,Inc.,Monte Rosa Treateutics,Inc.,Nurix Treateutics,Inc.,Orum Treateutics,Inc.,PhoreMtreLtets,Plexc.,Inlarium,Phartics,Inc.(SKバイオ製薬株式会社の子会社)及びVividiion治療会社(バイエル株式会社の子会社)。また、いくつかの大手製薬会社は、安進会社、アステラス製薬会社、アスリコン社、百時美施貴宝社(およびその子会社Celgene社)、グラクソ·スミスクライン社、遺伝子テーク社、ノワール国際会社を含むこの分野への臨床前投資を開示している。他のタンパク質分解療法からの競争に加えて、我々が開発したどの製品も、小分子、抗体、T細胞または遺伝子療法のような他のタイプの療法からの競争に直面する可能性がある
私たちの主な候補製品は腫瘍適応に適している。最もよく見られる腫瘍適応患者を治療する方法は手術、放射線と薬物治療であり、化学療法、ホルモン治療、細胞治療と標的薬物治療を含む。市販されている癌薬物療法は多種多様である。多くの場合、これらの薬物は治療効果を向上させるために併用される。現在承認されているいくつかの薬物療法はブランド薬物であり、特許保護されており、他の薬物は模倣薬に基づいて提供されている。これらの承認された薬物の多くは治療薬であり,医師,患者,第三者支払者に広く受け入れられている。全体的に言えば、過去数十年来癌の治療は著しい進歩を得たにもかかわらず、現在市場での治療方法も多くの患者にメリットを提供したが、これらの治療方法はある程度その治療効果と不良事件の発生率の制限を受け、しかもすべての患者の治療に成功する方法はない。そのため、癌の発病率と死亡率は依然として高い
現在発売されている薬物のほかに,いくつかの候補製品が臨床前開発されており,腫瘍学的適応の治療に用いられている。これらの開発中の製品は,現在市販されている治療法では提供できない治療効果,安全性,利便性,その他の利点を提供する可能性がある。したがって、それらは私たちが市場承認を受けた任意の候補製品に大きな競争になるかもしれない
もし私たちの候補製品が私たちが期待する臨床試験の適応に使用されることが承認されれば、それらは上記の治療法や現在発売されている薬物、および開発されている可能性のある任意の薬物と競争するだろう。他の生物や薬物からの競争や,他のタイプの療法からの競争に直面する可能性もある
私たちの多くの既存または潜在的な競争相手は、単独またはパートナーと協力しても、研究開発、製造、臨床前テスト、臨床試験を行い、監督管理許可とマーケティング承認を得た製品の面で、私たちよりも多くの財務資源と専門知識を持っている。これらの競争相手も私たちと競争しています
合格した科学と管理人員を募集と維持し、臨床試験場を創立し、臨床試験のために患者を登録し、及び著者らの計画と相互補完或いは必要な技術を獲得する。製薬とバイオテクノロジー産業の合併と買収は、私たちの数の少ない競争相手により多くの資源を集中させる可能性がある。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。これらの競争相手はまた合格した科学と管理人員を募集と維持し、臨床試験場を創立し、臨床試験の患者登録を確立し、著者らの計画と相互補完或いは必要な技術を獲得する方面で私たちと競争している。もし私たちの競争相手が私たちが開発する可能性のあるどの製品よりも安全で、より効果的で、副作用が少なく、より便利で、より安い製品を開発し、商業化すれば、私たちのビジネス機会は減少または消失するかもしれない。私たちの競争相手も私たちよりも早くFDAや他の規制機関のその製品の承認を得ることができ、これは私たちの競争相手が私たちが市場に入る前に強力な市場地位を確立することをもたらすかもしれない。また、多くの場合、私たちの競争能力は、後発薬の使用を奨励することを求める保険会社または他の第三者支払者の影響を受ける可能性がある。現在,我々が求めているいくつかの適応に対する模倣薬が市販されており,今後数年でより多くの模倣薬が発売されることが予想される。もし私たちの候補製品が承認されたら、私たちはそれらの価格が競争相手の模造薬より著しく高いと予想する
私たちのすべての計画の成功を影響する重要な競争要素は、承認されれば、それらの有効性、安全性、利便性、価格、模造薬の競争レベルと精算可能である可能性が高い。
製造業
私たちは所有したり運営したりしないし、現在は製造施設を設立する計画もない。我々は依存し,引き続き第三者契約製造組織であるCMOに依存し,薬物物質や完成品に利用することが予想される
私たちは現在注文を調達する方法でこれらのメーカーから私たちの供給を得ています。私たちの候補製品と他の材料に対して長期的な約束供給手配はありません。もしこれらのメーカーのいずれかがどんな理由でも私たちに提供できなければ、私たちはこのような代替製品を決定して同定する時にいくつかの遅延があるかもしれないが、多くの潜在的な代替製品があると信じている。もっと知りたいのは“リスク要因”というタイトルを参照してください第三者依存に関するリスクである製薬製品の生産は非常に複雑であり,様々な原因で製品の遅延や損失を招く可能性がある。我々は第三者と契約を結び,臨床前試験や臨床試験のための候補製品を生産し,商業化のために継続していく予定である。このような第三者への依存は、私たちが十分な数の候補製品や製品を持っていないリスクを増加させるか、または許容可能なコストまたは品質で、私たちが望むまたは要求される数のリスクを適切な時間に得ることができないであろう。これは、私たちの開発や商業化努力を延期、阻止、または損害する可能性がある。”
我々のすべての候補薬物は低分子有機化合物であり,生物製薬界では通常小分子と呼ばれているが,我々のBiDAC分解物は従来の小分子療法よりも大きいことが多い。これらの化合物を選択することは,それらの潜在的な臨床活性と耐性だけでなく,それらの物理的性質にも基づいている。私たちの候補製品は信頼できる、再使用可能な合成技術を採用し、既製の原料を原料とし、拡大しやすい化学を基礎としている。契約製造施設で費用対効果の高い方法で生産できる候補薬剤の開発を継続したい。
商業化計画
私たちの候補製品はまだ臨床開発段階にあるので、私たちはまだ自分の商業組織や流通能力を確立していません。私たちの協力協定によって制約されているプロジェクトを除いて、私たちは開発中のすべてのプロジェクトの完全な商業化権利を保持しています。私たちの任意の候補製品が発売承認される前に、適切な時に、アメリカや他の重要な市場で商業化する計画を立てる必要がある。私たちは現在、マーケティング承認を得た候補製品の米国での商業化を支援するために、独自の集中、専門的な販売、マーケティング組織を構築し、これらの製品をそのような能力で商業化することができると予想している。我々は、1つまたは複数の第三者との様々なタイプの協力、共同販売促進、流通、および他のマーケティングスケジュールを利用して、私たちの候補製品を米国以外の市場またはより大規模な販売およびマーケティング組織が必要とする場合に商業化したい
候補製品が私たちのチャンネルで進展するにつれて、私たちの商業計画は変わるかもしれない。私たちのいくつかの研究計画は潜在的なより大きな適応を対象としている。データ,開発計画の規模,目標市場の規模,商業インフラの規模および製造需要は,米国,ヨーロッパ,世界の他の地域における我々の戦略に影響を与える可能性がある。
知的財産権
私たちのビジネス成功は、私たちの魚雷プラットフォーム、私たちが独占的に所有している候補製品、私たちが羅氏と共同で所有している候補製品、私たちの業務に関連する技術ノウハウを含む、私たちのタンパク質分解技術の特許および他の独自保護の能力を確保し、維持する能力にある程度依存する。私たちのコア技術と製品を保護するためには、必要に応じて私たちの知的財産権、特に私たちの特許権を起訴、弁護することに成功し、私たちのビジネス秘密を保護し、他人の効果的かつ実行可能な知的財産権を侵害することなく運営する必要があります。我々の候補製品については,物質成分,薬物成分,使用方法(併用療法を含む),製造プロセス,プロセス中間体をカバーする特許保護が一般的に求められている。私たちは新技術と候補製品を開発すると同時に、私たちの知的財産権戦略を評価し、改善していく。我々は現在、我々の知的財産権戦略に基づいて、適切な場合には、競争に適応したり、私たちのビジネス機会の改善を求めたりする分野を含めて、より多くの特許出願を提出することを計画している
私たちのような生物製薬会社の特許地位は通常不確定であり、複雑な法律、科学、事実の問題に関連している可能性がある。また,新たな司法裁決の発表や新たな法律,規則や条例により,知的財産権保護を管理する法律は時間の経過とともに変化する可能性がある。さらに、特許出願において要求されるカバー範囲は、特許発行前に著しく減少することができ、その範囲は、発行後であっても再解釈および質疑することができる。したがって、私たちは私たちのすべての候補製品が効果的で強制的に実行可能な特許によって保護されるか、または引き続き保護されることを保証できない。私たちが現在求めている特許出願が任意の特定の司法管轄区域で特許として発行されるかどうか、または任意の発行された特許の権利主張が競争相手から攻撃されるのに十分な特許保護を提供するかどうかも予測できない。私たちが持っているどんな特許も第三者によって挑戦され、回避され、無効に発表される可能性がある
私たちの特許の排他的条項はそれらを獲得する国/地域の法律にかかっている。我々が現在出願を提出している国では,特許期間は非臨時特許出願が最初に提出された日から20年である。米国特許の期間は、規制機関が薬物の販売を許可するのに要した時間(特許期間延長と呼ばれる)または米国特許商標局が特許訴訟中に遭遇した遅延(特許期間調整と呼ばれる)を補償するために延長することができる。例えば、“ハッジ·ワックスマン法”は、FDAによって承認された新しい化学物質薬物の特許期間を、承認薬物またはその用途をカバーする特許の通常満期日よりも最大5年間延長することを可能にする。特許期間の延長の長さは,薬物が審査過程で規制審査と職務調査にある時間の長さと関係がある。米国では、特許期間の延長は、製品承認日から合計14年間を超えることができず、承認された薬物に関する特許のみ、またはその使用方法を延長することができる。補完保護証明書と呼ばれる類似の特許延期は、ヨーロッパにもある。いくつかの他の法域では、特許の期間を延長するために法的枠組みを使用することもできる。私たちは現在、私たちが合格特許を持っていて、延長期間を得ることができる任意の管轄区域で、私たちの任意の発行された特許出願に特許期間を延長するつもりです;しかし、米国のFDAを含む適用可能な規制機関は、このような性質の延長を承認すべきかどうか、これらの延長の長さを承認すべきかどうかの評価に同意します。さらに、我々の任意の特許が延長または調整されても、これらの特許は、それらの特許の延長または調整部分を含み、米国または外国の最終管轄権裁判所によって無効または強制的に実行できないと判断される可能性がある。
特許と特許出願
2023年12月31日現在,発行された米国特許17件,米国特許40件以上(仮および米国実用プログラム出願を含む),特許協力条約(PCT)に基づいて出願された7件の特許出願,250件以上の外国で未解決の特許出願を有している
我々の特許の組み合わせは、一般に、我々の独自の魚雷プラットフォームに関連する発明、およびタンパク質標的固有分解器の特許出願をカバーすることを目的としたプラットフォーム特許出願の2つに分類され、以下では、各カテゴリについてより詳細に説明する。
魚雷プラットフォーム組合せ
私たちは私たちの専用魚雷プラットフォームを使用して設計された私たちのプラットフォーム特許権を独占的に持っている。2023年12月31日現在、私たちのプラットフォーム特許の組み合わせは、発行された13件の米国特許、18件の係属中の米国特許出願、1つのPCT特許出願、および75件を超える未決外国特許出願を含む。この特許の組み合わせは,Cereblon E 3ユビキチンリガーゼやCRBNに結合するか,MonoDAC分子の一部として単独でするか,BiDAC分子の一部として,疾患修飾タンパク質標的とのタンパク質リガンドを含むBiDAC分子の様々なリガンドをカバーしている
具体的には、プラットフォーム組み合わせは、異なるクラスのCRBNリガンドおよびそれによって誘導される分解剤のための物質請求項を含む魚雷プラットフォームをカバーする19個の特許ファミリーからなる
関連する使用方法、医薬組成物及び製造方法。これらの家族における特許は,公開·維持されていれば,潜在的な特許期間の延長や調整を考慮することなく,2037年から2044年の間に満了する
製品と目標の組み合わせ
我々の候補製品を含む特定の標的分解物に対する特許出願は、物質の組成、薬物組成、使用方法、および製造過程に集中し、原因タンパク質を分解するための新規化合物に関する。2023年12月31日現在、我々は、発行された4つの米国特許、24件の係属中の米国特許出願、6つのPCT特許出願、および我々の分解剤および候補製品をカバーする175以上の外国特許出願を有している。
具体的には、2023年12月31日現在、4つの特許シリーズ(米国特許2件、米国特許3件、PCT特許出願1件、外国特許出願54件)を有し、IKZF 1/3タンパク質標的分解を引き起こす化合物に物質組成および薬物組成要件、および関連する癌治療および製造過程の使用方法を提示する。これらの特許ファミリーの3つの特許シリーズは、物質組成物に対する請求項、特にCFT 7455、我々の主要候補製品のうちの1つおよび関連する使用方法を含み、すべての必要な費用を支払うことによって発行および維持される場合、可能な特許期間の延長または調整を考慮することなく、2040年、2041年および2043年にそれぞれ満了する。我々のIKZF 1/3分解器をカバーする第4の特許シリーズは、最初の3つのシリーズでカバーされたものとは異なる特許シリーズを対象としており、可能な特許期間の延長または調整を考慮することなく、すべての必要な費用を支払ってライセンスおよびメンテナンスを行う場合、2039年に満了する
2023年12月31日現在、我々は、我々のCFT 8634候補製品、ならびに関連する医薬組成物、使用方法、および製造方法を含む3つの特許シリーズ(1つの米国特許、4つの米国特許出願、PCT特許出願、および24個の外国特許出願)を有している。これらの特許出願よりも優先的であると主張する米国及び外国特許は、すべての必要な費用を支払うことにより付与及び維持される場合、可能な特許期間の延長又は調整を考慮することなく、それぞれ2041年、2042年及び2044年に満了する
2023年12月31日現在、我々は、4つの特許シリーズ(5つの米国特許出願、1つのPCT特許出願、28件の外国特許出願)を有しており、その請求項は、我々のBRAF分解器およびそれらの関連使用方法、医薬組成物および製造方法に関連する物質組成物を有する。2つの特許系列は、CFT 1946、我々の主要候補BRAF製品、および関連する使用方法をカバーする物質組成に対する一般的および具体的な特許要件を含み、これらの特許出願よりも優先的であると主張する米国および外国特許は、可能な特許期間の延長または調整を考慮することなく、すべての必要な費用の付与および維持を支払うことによって、それぞれ2042年および2043年に満了する。我々のBRAF分解器をカバーする他の2つの特許シリーズは、これらの特許出願の米国および外国特許よりも優先的であると主張し、最初の2つのシリーズがカバーする特許系列から分離することを意図しており、可能な特許期間の延長または調整を考慮することなく、すべての必要な費用を支払うことによって承認および維持される場合、それぞれ2041年および2044年に満了することになる。
2023年12月31日現在、我々は、我々のCFT 8919候補製品、ならびに関連する使用方法、医薬組成物、および製造プロセスを含む、我々のEGFR分解器をカバーする物質組成物に関する2つの特許シリーズ(1つの米国特許、2つの米国特許出願、および50個の外国特許出願)を有している。これらの特許出願よりも優先的であると主張する米国及び外国特許は,すべての必要な費用を支払うことにより付与及び維持される場合,可能な特許期間の延長又は調整を考慮することなく,それぞれ2040年及び2042年に満了する
2023年12月31日現在、我々は、我々のRET分解剤およびそれらの関連する使用方法、医薬組成物および製造プロセスに関連する物質組成物を必要とする2つの特許シリーズ(2つの米国特許出願および9つの外国特許出願)を有している。これらの特許出願よりも優先的であると主張する米国及び外国特許は,すべての必要な費用を支払うことにより付与及び維持される場合,可能な特許期間の延長又は調整を考慮することなく,それぞれ2041年及び2044年に満了する。
2023年12月31日現在、我々は、8つの米国特許出願、3つのPCT特許出願、および12つの外国特許出願を有しており、これらの特許出願の請求項は、分解剤の組成物、不開示の目標および関連する使用方法、医薬組成物および製造方法を含む。これらの特許出願よりも優先的であると主張する米国及び外国特許は,すべての必要な費用を支払って付与·維持する場合には,可能な特許期間の延長や調整を考慮することなく,2040年から2044年の間に満了する。
羅氏と共同所有している協力特許出願
羅氏は、2023年12月31日まで(上述したように、羅氏は、2022年7月および2023年12月に、開示されていない目標に関連する特許の各権利を譲渡している)、羅氏と共同でいかなる特許出願または特許も有していない。もし今後行われている協力活動に関連する新しい特許出願が提出された場合、このような将来の特許出願の任意の権利は、上述したロー氏協定によって管轄されるであろう。
目標プラットフォーム協力
私たちは戦略的パートナーと協力して、羅氏、Calico、Biogen、そしてメルクを含む私たちのプラットフォームの潜在力を拡大する。これらのパートナーとの合意によれば、我々は、これらの合意に基づいて開発されている製品の経済的利益と引き換えに、我々単独または共同発明の開発候補特許権を適用パートナーに譲渡するのが一般的である
商業秘密
私たちはまた、ビジネス秘密、技術ノウハウ、持続的な革新に依存して、私たちの競争優位性を発展させ、維持している。私たちが彼らと締結した合意によると、私たちの従業員やコンサルタントは、どの会社が所有する特許出願でも識別され、彼らがどのような特許出願でも所有する可能性のある任意の権利を私たちに譲渡するだろう。私たちはまた、従業員、コンサルタント、他のコンサルタントと締結された秘密または他の合意に依存して、私たちの固有の情報を保護します。私たちの政策は、私たちの機密および商業秘密情報の適切な保護を含む、重要な機密情報を受け取った第三者が、私たちと秘密または他の合意を締結することを要求することです
商標
私たちはアメリカと海外で様々な登録されていない商標とサービス商標を持っています。C 4 Treeutics、私たちの家の標識、私たちの魚雷プラットフォームの名前、私たちのBIDAC分解器とMONNODAC分解器の名前を含みます
政府の監督管理
FDAと州と地方司法管轄区及び外国の類似監督管理機関は薬品の臨床開発、製造とマーケティングに対して実質的な要求を提出した。これらの機関と他の連邦、州、地方実体管理研究と開発活動、ならびに私たちの製品のテスト、製造、品質管理、安全、有効性、ラベル、貯蔵、包装、記録、追跡、承認、輸入、輸出、流通、広告、販売促進
アメリカ政府の薬品の監督管理
米国では,FDAは連邦食品,薬物と化粧品法案(FDCA)とその実施条例に基づいて薬品を規制している。規制の承認を得て、その後、適用される連邦、州、地方、外国の法規と条例を遵守する過程には、多くの時間と財力が必要だ。製品開発プロセス、承認プロセス、または承認後の任意の時間において、出願人が適用される米国の要求を遵守できない場合、FDAが係属中のNDAの承認を拒否する、承認の撤回、臨床棚上げの実施、警告状の発行、製品のリコール、製品の差し押さえ、生産または流通の完全または部分的な一時停止、禁止、罰金、政府契約の拒否、原状回復、返還、または民事または刑事罰のような様々な行政または司法制裁を受ける可能性がある
FDAが候補製品が米国で発売される前に必要とされるプログラムは、一般に以下のような態様を含む
•GLPに従って行われなければならない非臨床実験室や動物試験
•臨床試験開始前に発効しなければならない研究用新薬またはIND出願をFDAに提出する
•各臨床サイトまたは各試験が開始される前に、独立した機関審査委員会またはIRBによって集中的に承認される;
•提案された候補製品の安全性と有効性を決定し、その期待用途のために、良好な臨床実践またはGCPに従って行うために、十分かつ良好に制御されたヒト臨床試験を提供する
•FDAにセキュリティプロトコルを提出し、使用料を支払う
•適用されれば、FDA諮問委員会の審査が満足的に完了する
•現在の良好な生産実践またはcGMPとGCPに適合しているかどうかを理解するために、生産施設および選択された臨床研究者に対して承認前検査を行う
•GCPおよび臨床データの完全性を保証するために、臨床試験場所に対するFDAの監査を満足に完了させること;および
•FDAは、特定の適応のビジネスマーケティングを可能にするためにNDAを審査し、承認する
テストと承認過程には多くの時間、エネルギー、そして財力が必要だ
臨床前研究
臨床前研究は薬物化学、薬理学、毒性と薬物製剤の実験室評価、及び潜在的安全性と有効性を評価する動物研究を含む。候補製品の第1回臨床試験を開始する前に,スポンサーは臨床前試験と臨床前文献の結果,製造情報,分析データおよび任意の利用可能な臨床データや文献,その他の必要な情報をINDの一部としてFDAに提出しなければならない。IND提出後も,いくつかの臨床前研究は継続する可能性がある。INDはFDAが30日以内に臨床試験の実施に対して安全懸念または問題を提起し、臨床一時停止を強制しない限り、FDA受信後30日以内に自動的に発効する。この場合,INDスポンサーやFDAは臨床試験開始前に未解決の問題を解決しなければならない。したがって,INDの提出はFDA認可による臨床試験の開始につながらない可能性がある
臨床試験
臨床試験はGCP要求に符合する合格研究者の監督の下で、人類被験者に研究用新薬を服用することに関連する。製品開発期間中に行われた後続臨床試験ごと,従来提出されていた臨床試験の改正は,既存のINDに単独で提出しなければならない。また,臨床試験に参加する各機関の独立IRBは,臨床試験がその場所で開始される前に,任意の臨床試験の計画,そのインフォームドコンセント,その他の研究対象との通信を審査·承認しなければならない。臨床試験が行われている間、IRBは研究計画のいかなる変化も含めてそれを監視し続けなければならない
規制当局、IRBまたはスポンサーは、対象が受け入れられない健康リスクに直面していることを発見すること、臨床試験がFDAまたはIRBの要求に従って行われていないこと、または薬物が対象が予期せぬ深刻な損傷を受ける場合を含む、様々な理由で臨床試験を随時一時停止または中止することができる。いくつかの研究は、臨床試験中に非盲目的データへの特別なアクセスを受けるデータ安全監視委員会をさらに含み、それが被験者に受け入れられない安全リスクまたは他の理由があると判断した場合、治療効果証明がなければ、スポンサーに臨床試験の停止を提案する可能性がある
人体臨床試験は通常3つの連続段階に分けて行われ、これらの段階は重なる可能性があり、合併する可能性もある
•第1段階−最初に研究を行ったのは、健康ボランティアまたは目標疾患または条件を有する対象における製品の安全性、用量耐性、構造−活性関係、作用機序、吸収、代謝、分布および排泄を試験するためである。可能であれば,一期臨床試験は製品の有効性を得るための初歩的な適応にも利用可能である
•段階2-対照研究は、初歩的な治療効果、最適な用量および投与計画、および安全な拡張証拠を評価するのに十分なデータを提供するために、特定の疾患または状態を有する被験者のセットに対して行われる。より大規模かつより広範な3期臨床試験を開始する前に、複数の2期臨床試験を行い、情報を得ることができる
•段階3-これらの臨床試験は、一般に、統計学的に有意な有効性証拠を提供し、複数の臨床試験地点の拡大対象群において安全性をさらに試験するために、より大きな対象集団で行われる。これらの臨床試験は製品の全体的なリスク/利益概況を確定し、製品ラベルに十分な基礎を提供することを目的としている。これらの臨床試験は米国以外の試験地点で行うことができ,グローバル地点でも米国人口を代表し,グローバル地点で行われている研究はGCP遵守などFDAの規定や指導に適合している。
通常,これらの開発段階では,臨床試験はFDAや外国規制機関と協議して設計されている。開発されている適応は,臨床試験中に採用されている研究設計,例えば第一線の癌治療適応に影響を及ぼす可能性があり,臨床的優位性や既存の治療法よりも劣勢であることを証明する対面データが必要となる可能性がある。規制当局は、利用可能かつより早い治療法があるにもかかわらず、癌の進行を加速させることを望んでいる人が、利用可能かつより早い治療方法があるにもかかわらず、癌適応開発計画のスケジュールが三線以上に求められている適応よりも長い可能性がある。そのため、多くの新しい腫瘍学製品は最初に三線治療の適応を求め、これは任意の腫瘍学的適応の中で比較的に小さい応用可能な治療群、そして任意の後に求めた承認である
より大きな治療群に対する早期治療シリーズ製品は追加の臨床試験を行う必要があるかもしれない
FDAはすでに腫瘍学候補製品開発に影響を及ぼす可能性のある取り組みを実施している。例えば、2021年にスタートした最適化プロジェクトは計画であり、腫瘍学薬物開発中の投与量選択と用量最適化モデルを改革し、薬物治療効果を最大限に高めるだけでなく、その安全性と耐性を最大限に高める1つ以上の投与量を選択することを強調する。Optimusプロジェクトは,腫瘍学的薬物の開発者に進行中の計画中に戦略を実施し,用量選択に非臨床と臨床データを利用し,試験中に一連の用量をランダムに評価する潜在的な需要を含め,これらの研究を開発計画中に早期に行うことを求めている
そのほか、FDAは2022年3月に“拡張行列:最初のヒト臨床試験のために腫瘍薬物と生物製品の開発を加速するために使用する”と題する最終ガイドラインを発表し、その中で薬物開発者がどのように腫瘍薬物開発の初期段階(即ち最初の人体臨床試験)で通常シームレス試験設計と呼ばれる適応性試験設計を利用して、伝統的な3段階の試験を拡張行列試験と呼ばれる連続試験に圧縮するかについて概説した。個人拡張キュー設計をサポートする情報は、IND出願に含まれ、FDAによって評価される。コホート試験を拡大することは薬物開発の効率を向上させ,開発コストや時間を削減する可能性がある。
FDAは要求するかもしれないし、会社は製品が承認された後に追加の臨床試験を行うかもしれない。これらのいわゆる4期実験は承認後に満たすべき条件となる可能性がある.4期試験の結果,候補製品の有効性が確認でき,重要なセキュリティ情報を提供することができた
臨床試験はGCP要求に適合する合格した研究者の監督の下で行わなければならず、その中には、すべての研究対象に書面で任意の臨床試験に参加することを要求するインフォームドコンセントを提供し、IRBによってこの研究を審査し、承認することが含まれている。調査者はまた,スポンサーがFDAに具体的な財務情報を開示することを可能にするために臨床試験スポンサーに情報を提供しなければならない。臨床試験は,試験目標,試験手順,安全性モニタリングのためのパラメータ,評価すべき治療効果基準,統計分析計画などを詳細に説明する案に基づいて行った。試験および試験結果の記述を含むいくつかの臨床試験の情報は、そのウェブサイト上で公開されるために、特定の時間枠内でNIHに提出されなければならない。臨床試験結果を詳細に説明する進捗報告は少なくとも毎年FDAに提出しなければならず,深刻な有害事象が発生すればより頻繁に提出される
ヒト臨床試験のための研究薬物の製造はcGMPに要求される制約を受けている。米国に輸入された研究薬や活性薬物成分もFDAのラベルや流通に関する規制を受けている。また,米国以外の研究用薬物製品の輸出は受け入れ国の規制要求や米国“食品薬品規制法”下の輸出要求の制約を受けなければならない。臨床試験結果を詳細に説明する進捗報告は少なくとも毎年FDAやIRBに提出されなければならず,重篤な副作用が発生すればより頻繁に提出される
臨床試験と同時に,会社は通常追加の動物研究を完了し,候補製品の化学的および物理的特性に関する追加情報を開発し,cGMP要求に基づいて商業量産製品を最終的に決定するプロセスを開発しなければならない。製造過程は一貫して高品質の候補製品ロットを生産できる必要があり、特に最終製品の特性、強度、品質、純度を試験するための方法を開発しなければならない。また,適切な包装を選択·テストし,候補製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない
FDAは審査と承認計画を加速させる
FDAには、迅速チャネル指定、突破的治療指定、加速承認および優先審査を含む様々な計画があり、これらの計画は、深刻または生命に危険な疾患または状態を治療するための薬物の開発およびFDA審査プロセスを加速または簡略化し、満たされていない医療需要を解決する潜在力を示すことを目的としている。これらの計画の目的は,FDA標準審査プログラムよりも早く患者に重要な新薬を提供することである
迅速チャネル計画によれば,新薬候補のスポンサーは,薬剤候補INDの提出と同時にまたはその後,特定の適応の候補薬を迅速チャネル薬として指定することをFDAに要求することができる。迅速なチャンネル認証を取得する資格があるため、FDAはスポンサーの要求に基づいて、深刻な或いは生命に危害を及ぼす疾病或いは状況を治療することを目的とした製品を確定し、満足されていない医療需要を満たす潜在力を示しなければならない。FDAは、製品が存在しない療法を提供するか、または治療効果または安全要因に基づく潜在的に既存の療法よりも優れた治療法を提供する場合、満たされていない医療需要を満たすことを決定するであろう。高速チャネル指定は、FDAレビューチームとの相互作用により多くの機会を提供し、NDAのスクロール審査を可能にする可能性があります
完全な出願を提出する前に、スポンサーが機密協定部分を提出するスケジュールを提供した場合、FDAは、機密協定の一部を受け入れることに同意し、スケジュールが許容可能であると判断し、スポンサーは、秘密協定の第1の部分を提出する際に必要な使用料を支払う。しかしながら、FDA審査申請の期間目標は、NDAの最後の部分が提出されてから開始される。FDAが資格基準がこれ以上適用されないと判断した場合、それは高速道路指定を撤回することを決定するかもしれない
さらに、1つの薬剤が、深刻または生命に危険な疾患または状態を治療するために、単独でまたは1つまたは複数の他の薬剤と併用することが意図されている場合、初期臨床証拠は、1つまたは複数の臨床的に重要な終点において、臨床開発早期に観察された実質的な治療効果のような既存の療法よりも実質的な改善を示す可能性があり、スポンサーが画期的な治療指定を申請することができることを示す。画期的療法に指定された薬物は,FDAの効率的な薬物開発計画に関する密な指導を得る資格があり,製品開発と審査に対する組織承諾は,高度管理者の参加や,Fast Track製品と同様にNDAの転動審査を得る資格もある。関連基準に適合する場合、迅速チャネルおよび突破的治療製品は、加速承認および/または優先審査を受ける資格がある
FDAの加速的承認経路によれば、FDAは、患者に既存の治療よりも意義のある治療利益を提供する深刻なまたは生命に危険な疾患に対する薬剤を承認する可能性があり、その基礎は、臨床的利益を合理的に予測する可能性のある代替終点、または不可逆的な発症率または死亡率よりも早く測定することができる臨床終点であり、不可逆的な発症率または死亡率または他の臨床的利益への影響を合理的に予測することができ、同時に、病状の重症度、希少性または流行率、および代替療法の利用可能性または不足を考慮することである。この基礎の上で承認した候補薬物は必ず厳格な発売後のコンプライアンス要求を遵守しなければならず、4期或いは承認後の臨床試験を完成し、臨床終点への影響を確認することを含む。2022年食品·薬物総合改革法案(FDORA)によると、FDAは現在、これらの試験を承認前または承認を加速させた製品の承認日後の特定の期間で行うことを適宜要求されている。FDORAによれば、FDAは、例えば、検証試験が製品の予期される臨床的利益を検証することができない場合、承認の加速下で承認された薬物または適応の承認を撤回することができるように、手続きを加速させる権限を増加させる。加速承認条例により承認された候補薬物の宣伝材料は,FDAが別途スポンサーに通知しない限り,FDAの事前審査を経なければならない
重篤な疾患を治療するための製品のNDAが提出されると、FDAが製品が承認された場合、安全性または有効性の面で著しい改善を提供すると判断した場合、FDAは優先審査指定を指定する可能性がある。優先審査は、FDA審査申請の目標が6ヶ月であり、“処方薬使用料法案”(PDUFA)ガイドラインに基づく標準審査10ヶ月ではないことを意味する。現在のPDUFA業績目標によると、この6ヶ月と10ヶ月の審査期間は、受信日ではなく、新しい分子実体の60日の提出日から計算され、これは、通常、提出日から審査のタイムラインを約2ヶ月増加させる
1つの製品がこれらの計画のうちの1つまたは複数に適合していても、FDAは、製品がもはや資格条件に適合していないことを後で決定することができ、またはFDAの審査または承認を決定する期間が短縮されないことができる。さらに、深刻または生命に危険な疾患を治療する研究薬の製造業者は、例えば、そのウェブサイトにアクセス拡大要求に応答するためのその政策を掲示することによって提供を要求される。また,迅速チャネル指定,画期的治療指定,加速承認,優先審査は承認の基準を変更することはなく,最終的に開発や承認過程を加速させない可能性もある
食品医薬品局が提出した秘密保持協定と審査
必要な臨床と前臨床試験,その他の項目,製品開発の結果,化学,製造と制御,非臨床研究と臨床試験を含め,提案されたラベルとともにFDAに提出し,NDAの一部とすることに成功したと仮定する。秘密協定を提出するには相当な使用料がFDAに支払われなければならない。これらの使用料は、申請がスクロールして提出されていても、初めて申請を提出する時に提出されなければならない。場合によっては、費用を免除または減少させることができる。出願料免除の1つの根拠は、出願人が雇用した従業員が500人未満である場合、付属会社の従業員を含む場合、出願人は、州間商業に導入または交付された製品マーケティング出願を承認しておらず、出願人は、その付属会社を含めて、その第1のマーケティング出願を提出している。孤児指定薬品も申請料を無料にします。
“小児科研究公平法”によれば、新しい有効成分、適応、剤形、投与レジメンまたは投与経路のNDAまたは補充NDAは、すべての関連する小児科亜集団において主張される適応の安全性および有効性を評価し、安全かつ有効な各小児科亜集団に対するこの製品の用量および投与をサポートするための十分なデータを含まなければならない。法規が別途要求されない限り、PREAは、PREAが分子上の孤児によって指定された場合、PREAが元のNDAに新しい有効成分を申請しない限り、孤児薬剤として指定された適応薬には適用されない
成人癌を治療するための標的癌製品であって、FDAに対して小児癌の成長または進行に密接に関連する分子標的を決定する。FDAは、成人のために使用されるか、または小児科データの要求を完全にまたは部分的に免除するために製品が使用されることが承認されるまで、申請者の要求に応じて、または小児科データの一部または全部の提出を延期することができる
FDAは、これまでFDAの承認を得ていなかった活性成分(活性成分のエステルまたは塩を含む)を含む薬物申請を諮問委員会に提出する可能性がある。FDAは安全性,純度あるいは有効性の問題を提起した薬物を諮問委員会に提出することも可能である。諮問委員会は通常、臨床医と他の専門家からなるグループであり、申請を審査、評価し、申請を承認すべきかどうか、どのような条件下で提案すべきかについて提案する。FDAは諮問委員会の提案に制限されていないが、それは決定を下す時にこれらの提案を慎重に考慮するだろう
FDAは、製品がその予期される用途に対して安全かつ有効であるかどうかを決定し、製造制御が製品の特性、強度、品質、および純度を確保および維持するのに十分であるかどうかを決定するために出願を審査する。秘密協定を承認する前に、FDAは製品を生産する1つまたは複数の施設を検査するだろう。FDAは、cGMP要件に適合し、要求された仕様で製品が一貫して生産されることを確実にするのに十分な製造プロセスおよび施設を決定しない限り、契約製造業者および下請け契約を含む申請を承認しないであろう。さらに、NDAを承認する前に、FDAは、GCPに適合することを確実にするために、通常、1つまたは複数の臨床試験地点を検査する
FDAが申請を受けると、申請を受ける前に、NDAを審査して、実質的な審査が可能になったかどうかを決定するために60日間の時間がある。提出された申請が受け入れられると、FDAはNDAの深い審査を開始する。FDAがPDUFAで達成した目標と政策によると,FDAの新分子実体またはNMEに対する標準NDAの審査目標は60日の出願日から10カ月であった。優先審査申請の場合、NME NDAに対するFDAの審査目標は、60日の申請日の6ヶ月以内である。このようなデッドラインをPDUFA日付と呼ぶ.PDUFA日付は1つの目標にすぎず,FDAはつねにそのPDUFA日付を達成するわけではない.FDA要件またはNDAスポンサーが審査中に元の出願を修正するために補足情報を提供するか、または明確にする場合、審査プロセスおよびPDUFA日も延長することができる
FDAによる申請の審査が完了すると、FDAは、完全な返信またはCRLまたは承認手紙を発行する。CRLは,申請の審査周期が完了していることを示しており,申請は承認の準備ができていない.CRLは、一般に、NDAの最終承認を保証するために満たされなければならない特定の条件を含む声明を含み、FDAが再提出されたNDAにおいて出願を再検討するために、追加の臨床または臨床前テストまたは他の情報または分析が必要とされる可能性がある。追加の情報が提出されても、FDAは最終的にその申請が承認された規制基準を満たしていないと決定する可能性がある。もしこのような条件がFDAの満足を得たら、FDAは承認書を発行するかもしれない。この薬物の商業マーケティングを許可し、特定の適応の具体的な処方情報を提供する
適用される規制基準を満たしていない場合、FDAは、製品の安全性または有効性を監視するために、追加の試験または情報を要求する追加の試験または情報を遅延または拒否することができ、および/または流通制限または他のリスク管理メカニズムを含む他の条件を適用することができる。例えば、FDAは、任意の決定または疑われる深刻なリスクを軽減し、薬物の安全な使用を保証するために、承認または承認後の条件として、リスク評価および緩和戦略、またはREMSを要求することができる。FDAは上場後の研究またはモニタリング計画の結果に基づいて、製品のさらなる販売を阻止または制限し、あるいは追加の上場後の要求を適用することができる。承認後、新たな適応の追加、製造変更、および追加のラベル宣言など、承認された製品のいくつかのタイプの変更は、さらなるテスト要件、FDA通知、およびFDA事前審査および承認の制約を受けることになる。さらに、新しいセキュリティ情報が発生した場合、追加のテスト、製品タグ変更、またはFDA通知が必要となる可能性がある
製品が規制部門の承認を得た場合、このような承認は、製品が発売される可能性のある指定された用途を制限することができ、または禁忌症、パッケージ警告、または製品ラベル内の他の警告または予防措置を含む可能性があり、それによってパッケージ警告をもたらす可能性がある。ブロック警告は、薬物が深刻な危険に関連していることを示す合理的な証拠がある場合、FDAが医薬品ラベルにおいて提示する最も厳しい警告である。FDAはまた、申請者が求めるすべてのラベルクレームをその中に含めることを許可しないかもしれない。承認されると、発売前と発売後の規制基準の遵守が維持されていない場合、あるいは製品が市場に進出した後に問題が発生した場合、FDAは製品承認を撤回する可能性がある。また、FDAは、承認製品の有効性或いは安全性を監視するために、第4段階上場後研究を行うことを要求する可能性があり、これらの発売後の研究結果に基づいて、当該製品のさらなる販売を制限する可能性がある
アメリカの承認後に要求する
我々は、FDAによって製造または流通を許可された任意の製品は、定期報告、製品サンプルおよび流通、広告、販売促進、薬品不足報告、4期臨床試験またはREMSのような承認条件として適用される任意の承認後要求を遵守し、不良経験を含む記録および報告要件を含むFDAの持続的な規制を受けるであろう
承認後、新たな適応または他のラベル宣言を追加するなど、承認された製品の多くの変更は、FDAの審査および承認を事前に受けなければならない。承認された製品に対しては,継続的な年間計画費用要件と,臨床データを持つ補充アプリケーションの新規申請料がある。医薬品メーカーおよびその下請け業者、および製品、成分および成分を提供する会社は、FDAといくつかの州機関に登録し、その薬品をリストし、FDAとこれらの州機関のcGMPおよび他の要求に対する定期的な発表と抜き打ち検査を受けなければならず、これらの要求は手続きと文書要求を加えている
製造プロセスの変更は厳しく規制されており,通常FDAの事前承認や通知が必要である。FDAの規定はまた、cGMPおよび規範との任意の偏差を調査および是正し、スポンサーおよびスポンサーが使用を決定する可能性のある任意の第三者メーカーに報告および文書要求を提出することを要求する。そのため、メーカーはcGMPコンプライアンスを維持するために、生産と品質管理に時間、お金、労力をかけ続けなければならない。
製品はその後、予期しない深刻度や頻度の不良事件、または製造技術、あるいは規制要求を遵守できなかったことを含む以前の未知の問題を発見し、新しいセキュリティ情報または他の制限を追加するために上場承認の撤回、強制改訂のラベルを招く可能性があり、新しい安全リスクを評価するために発売後研究或いは臨床試験を実施するか、あるいはREMS計画に従って流通或いはその他の制限を実施するなどの結果を招く可能性がある
FDAは薬品のマーケティングと普及を厳格に規制している。1社はFDAが承認したラベルと一致した安全性と有効性に関する声明しか提出できない。医師はその独立した専門医学に基づいて判断することができ、製品ラベルに説明されておらず、著者らがテストし、FDAによって許可された用途とは異なる合法的に利用可能な製品のために処方を発行することができる。しかし、製造業者と彼らを代表して行動する第三者は、承認されたラベルと一致しない方法で薬品を販売または普及させることを禁止された。FDAや他の機関はラベル外用途の普及を禁止する法律や法規を施行しており,ラベル外用途の普及が不適切であることが発見された会社は重大な責任を負う可能性がある
FDAのいかなる要求も遵守しないことは重大な不利な法執行行動を招く可能性がある。これらの制裁には、承認保留申請の拒否、免許の取り消しまたは免許取り消し、承認の撤回、臨床試験の一時停止または終了、警告状、見出しなし手紙、宣伝材料またはラベルの修正、製品の押収または拘束、輸入または輸出の許可の拒否、生産または流通の完全または部分的な一時停止、禁止、禁止、罰金、同意法令、会社の誠実な合意、政府契約および既存の契約下の新しい命令の拒否、罰金および監禁を含む連邦および州医療保健計画への参加の禁止、賠償、返還、返還または民事または刑事罰など、様々な行政または司法制裁が含まれることができる。処方薬の普及に関するFDAの要求を守らなければ,連邦や州医療詐欺や乱用などの法律や州消費者保護法に違反していると告発される可能性もある。このような制裁のいずれかは否定的な宣伝と他の不利な結果をもたらす可能性がある
アメリカマーケティング排他性
FDAは非特許規制専門期間を規定しており、これは承認されたNDAの保有者に限られた保護を提供し、FDAがNDAを承認してから3年または5年以内にその承認された薬物に代表される革新の市場での新しい競争から守ることができる。新しい化学実体やNCEは5年間の排他性を得ることができる。NCEは,FDAが任意の他のNDAで承認した活性部分を含まない薬物である。活性部分は、薬物をエステル、塩、水素または配位結合を有する塩、または原子間で電子対を共有する非共有結合を含まない他の分子、誘導体、例えば分子の錯体(すなわち、2つの化合物の化学作用から形成される)、キレート(すなわち、化合物)またはケージ化合物(すなわち、捕捉分子のポリマー骨格)を含まない医薬物質の治療活性を担当する分子またはイオンを意味する。排他的期間内に、FDAは、別の会社によって提出された簡略化された新薬出願またはANDAまたは505(B)(2)NDAを受け入れないか、または承認しない可能性がある
それは以前に承認された能動的な部分を含む。しかしながら、第4項の認証が提出された場合、ANDAまたは505(B)(2)の出願は、NCE排他的満了の1年前に提出することができる
孤児医薬品法によれば、1つの医薬製品がまれな疾患または疾患の治療を目的としている場合、FDAは、医薬製品を“孤児薬”として指定することができる(一般に、米国では、疾患または疾患を治療するための医薬製品を米国で開発および製造するコストが製品の販売から回収されることが合理的に予想できない場合、米国で影響を受ける人は20万人以下であることを意味する)。会社は秘密保持協定を提出する前に孤児薬物指定を申請しなければならない。この要求が承認された場合、FDAは、治療薬の識別情報およびその潜在的用途を開示するであろう。孤立製品の指定は、規制審査·承認過程においていかなる利点も伝達されず、規制審査·承認過程の継続時間を短縮することもない
孤児として指定された薬物が、そのような指定された疾患または条件を有するFDAの最初の承認を得た場合、またはその治療として指定されたまれな疾患または条件下での選択された適応または使用の場合、製品は、通常、孤児薬物排他性を得るであろう。孤立薬物排他性は、いくつかの限られた場合を除いて、FDAが7年以内に同一の製品の同一の適応を承認することができない他の任意の出願を意味する。孤児が指定した薬物が市販承認された場合,その適応の範囲はその孤児製品申請で指定された適応よりも広く,排他性を得る資格がない可能性がある。場合によっては、孤児特許も、より良い治療効果または安全性を有するため、または患者ケアに大きな貢献をしているので、同じ適応を有する同じ有効成分を有する後続製品が、より臨床的に承認された製品よりも良いことが証明された場合、または孤児薬物特許を有する会社が市場ニーズを満たすことができない場合を含む別の製品の承認を阻害しない場合がある。また、FDAは、これらの製品が異なる有効成分を含む限り、1つ以上の製品を同一の孤児適応または疾患に使用することを許可することができる。また,競争相手は同一製品を承認する可能性があるが,孤立製品に対して排他的な異なる適応が承認される可能性がある
また,FDCAはスポンサーが小児科群で薬物研究を行うことを奨励している。スポンサーが自発的に小児科研究を完了し、FDAが発表した書面請求に公平に応答した場合、薬物は小児科独占特許を取得する資格がある可能性がある。承認された場合、小児科専有権は、小児科専門権が付与された活性部分を含む承認された医薬製品を含むすべての既存の規制専門期間および特許条項のために6ヶ月増加する。
アメリカ以外の規制
私たちの候補製品のアメリカ以外での開発については、EUと中国を含む類似の外国法律法規の制約を受ける
EU薬物開発
連合では、私たちの候補製品もまた広範囲な規制要求を受けるだろう。アメリカと同様に、医薬製品は主管監督機構のマーケティング許可を得た後にのみ発売でき、しかもEUの臨床前と臨床研究の各段階は厳格な監督管理によって制御されている。
2014年4月、EUはEU第536/2014号臨床試験条例を採択し、この条例は2022年1月31日に臨床試験指令2001/20/ECに代わった。臨床試験条例の一過性条項では,2025年1月31日までに行われているすべての臨床試験はこの条例に移行しなければならないと規定されている。新しい臨床試験条例の主要な特徴は:臨床試験情報システムを通じて単一入口点を通じて申請プログラムを簡略化すること;申請のために単一文書を準備と提出すること、及び臨床試験スポンサーの報告プログラムを簡略化すること;臨床試験申請評価の統一プログラムは2つの部分に分けられる(第1部分は科学と医薬製品文書を含み、第2部分は国家と患者レベルの文書を含む)。第1部は、加盟国が作成した報告書を参照して臨床試験許可申請(関係加盟国)が提出されたすべてのEU加盟国主管当局の協調審査によって評価される。二番目の部分は関連する各会員国によって個別的に評価される。臨床試験申請の評価には厳しい期限が設定されている。関連倫理委員会の評価手続きにおける役割は依然として関連EU加盟国の国家法律によって管轄されているが、全体的な関連スケジュールは“臨床試験条例”によって決定されている。“臨床試験条例”は施行日から3年間の過渡期がある。
EUでは、小児科委員会またはEMAのPDCOは、(1)特定の製品免除、(2)カテゴリ免除、または(3)PIPに含まれる1つまたは複数の措置を遅延させない限り、マーケティング許可申請またはMAAを提出する前に、小児科調査計画またはPIPを承認しなければならない。PIPは製薬会社が小児科群の中で新医薬製品を調査する戦略について概説した。MAAを提出したり、既存のマーケティング許可を修正したりする前に、EMA評価会社は、各関連PIPに記載されている合意された研究および措置を遵守しているかどうかを評価する。出願人がすべてのEU加盟国で上場許可を取得した場合、または欧州委員会が集中手続きで付与された上場許可を取得し、小児科群に対する研究結果が製品情報に含まれている場合、否定的であっても、追加的に6-を取得する資格がある
この延期出願は、製品のSPC出願を提出すると同時に、又はSPCの満了前のいつでも提出される限り、任意の補充保護証明書又はSPCの期限を延長して合格特許保護の1ヶ月の期限を延長することができる。孤児薬品の場合、孤児市場の独占権を2年間延長することができる。この小児科奨励は特定の条件の制約を受け,PIPに適合したデータを開発·提出する際に自動的に獲得されない。
欧州連合は審査と発展を加速させる
PRIMEまたは優先医学は、満たされていない医療需要に対する薬物開発の支援を強化し、重大な革新を代表する製品に加速評価を提供することを目的としたEMAによって提供される計画であり、MAAは集中手順によって行われる。Primeの資格を得るためには,候補製品は早期臨床証拠が必要であり,この療法が既存療法よりも大きな治療優位を提供し,あるいは治療選択のない患者に利益を与える可能性があることを証明した。中小企業の製品は大企業よりも早くPrime計画に参加する資格があるかもしれません。Primeの利点は、EMAの科学委員会から調査委員を任命し、MAAの前に継続的な支援と知識の蓄積を支援し、重要な発展マイルストーンで早期対話と科学的提案を行うことと、申請過程がより早い時期に製品の資格鑑定を加速して審査する可能性があることを含む。Prime指定を受けると承認基準は変更されませんが、開発や承認過程が加速する可能性があります。製品が開発過程で資格基準を満たしていなければ、Prime計画下の支援を撤回する可能性がある。
EUの薬品審査と承認
EUでは、医薬製品はマーケティング許可やMAを得た後にのみ商業化されることができる。二つのタイプのMAがあります。ヨーロッパ共同体が欧州薬品管理局人用薬品委員会(CHMP)の意見に基づき,集中プログラムにより発行された集中式MAは,EU全体およびEEAの他の加盟国(アイスランド,リヒテンシュタイン,ノルウェー)で有効である。特定のバイオテクノロジーによって生産された医薬品、孤児薬として指定された製品、高度な治療薬(すなわち、遺伝子療法、体細胞療法または組織工学薬)、およびHIV、エイズ、癌、神経変性疾患、糖尿病、自己免疫および他の免疫機能障害およびウイルス疾患を治療するための新しい活性物質を含む薬剤のようないくつかのタイプの製品については、集中プログラムが実行されなければならない。EUで許可されていない新しい活性物質を含む製品、または重大な治療、科学的または技術革新、またはEUの公衆衛生上の利益に適合する製品については、集中手順がオプションである。
国家MAはEU加盟国の主管当局によって発行され、それぞれの領土のみをカバーし、集中プログラムの強制範囲に属さない製品に適用される。1つの製品がEU加盟国で販売されることが許可されている場合、その国MAは、相互認識手順によって別の加盟国で承認を得ることができる。この製品が申請時にどの加盟国でも国MAを取得していない場合は、分散手続きによって各加盟国で同時に承認を得ることができる。分権手続きに従って、MAを求める各加盟国の主管当局に同じ書類を提出し、申請者はそのうちの1つを参考加盟国、またはRMSとして選択する。RMS主管当局は、評価報告草案、製品特性概要草案またはSmPC、およびラベルおよび包装チラシ草案を作成し、他の加盟国(関係加盟国を指す)に承認を送付する。関連加盟国が公衆健康に対する潜在的に深刻な危害に基づいて、RMSによって提案された評価、SmPC、ラベル、またはパッケージに異議を唱えなかった場合、製品はその後、すべての加盟国(すなわち、RMSおよび関連加盟国)で国MAを取得する。
上記の手順により、MAを付与する前に、EMA又はEU加盟国主管当局は、製品の品質、安全性及び有効性に関する科学的基準に基づいて、製品のリスク効果バランスを評価する。
小児科用マーケティング許可、またはPUMAは、すでに許可された薬剤のために使用することができ、SPCまたはSPC資格に適合する特許によってカバーされず、小児開発にのみ使用されるであろう。PUMAは小児科群に特化して開発された医薬製品の適応と配合をカバーする専門MAであり,この開発が承認されたPIPに基づいて行われる場合である。申請PUMAには,製品が本来そのプログラムの強制範囲に属していなくても,集中プロセスに入ることを含む様々なインセンティブがある.
EU孤児の称号
欧州連合では、欧州委員会は、EMA孤児薬物製品委員会の意見を受けた後、そのスポンサーが、(1)生命または慢性衰弱にかかわる疾患の診断、予防または治療を目的としていることを証明できる場合、(2)または(I)この場合、欧州委員会は、その製品について孤児の称号を与えることを証明する
申請が提出されると、EUにおける製品の影響は10,000人中5人以下であるか、または(Ii)孤児のアイデンティティによる利益がなければ、その開発に必要な投資が合理的であることを証明するためにEUで十分な見返りを与えることは不可能であり、(3)このような疾患を満足できる診断、予防または治療方法がEUで販売されていないこと、または、そのような方法が存在する場合、その疾患の影響を受けている人に大きな利益をもたらすであろう
EUでは,孤児指定は一方が費用を削減したり費用を免除したりし,MA付与後10年間の市場排他性を付与するなど,経済的インセンティブを得る権利がある。この市場排他期間内に、欧州医薬品管理局、欧州委員会またはEU加盟国のどの主管機関も申請を受け入れることができず、“医薬製品”のような市場参入を承認することもできない。類似医薬製品“の定義は、承認された孤児医薬製品に含まれる1つ以上の類似活性物質を含む医薬製品であり、同じ治療適応のためのものである。5年目の終了時に、製品の利益が市場排他性の維持が合理的であることを証明するのに十分でないことを含む孤児指定基準に適合しないと判定された場合、この期間は6年に短縮されることができる。非常に特別な場合、許可された孤児製品と同様の医薬製品に許可書を付与することができ、例えば、(I)同様の医薬製品が許可された製品よりも安全で、より効果的であるか、または臨床的に良好であることを決定することができ、(Ii)孤児製品を許可するMA保持者は、同様の医薬製品の許可に同意するか、または(Iii)孤児製品を許可するMA保持者は、十分な孤児医薬製品を供給することができない。MA申請を提出する前に,孤児としての指定を要求しなければならない。孤児指定は、規制審査および承認過程においていかなる利点も伝達されず、規制審査および承認過程の継続時間を短縮することもない。
中国の薬品開発
中国で行われている上場承認を求めるすべての臨床試験は,中国国家医療製品管理局の承認を得,GCP要求に適合した病院で行わなければならない。開発を支持する独立中国試験以外に、輸入薬品申請者は中国臨床サイトを国際多中心試験(IMCT)に入れることができる。国内で生産された薬物は外国の承認の要求を受けず,従来のやり方とは異なり,国家薬品監督管理局はこれらの薬剤もIMCTによる開発を許可することを決定した。
2019年に改正された“中華人民共和国薬品管理法”は、現在すでに新薬の臨床試験に対して黙示審査制度を採用している。60営業日後,出願人が薬物評価センター(CDE)から何の反対意見も受けていなければ,従来のように臨床試験の事前承認過程が長く,出願人は肯定的な承認を待たなければならない。GCP認証制度の廃止と裁判地点により簡略化された通知手順に従うことを要求することにより,裁判地点は裁判地点の数も拡大した。
NMPAは2021年11月に臨床価値をガイドとする腫瘍学薬物研究開発ガイドラインを決定し、その政策の一部として、重大な臨床価値を有する革新腫瘍学薬物の研究と開発を奨励し、そして重複研究と患者に対する臨床価値が最も低い或いは臨床価値のない薬物の開発を阻止することを目的とした。
中国で行われている臨床試験は薬物臨床試験情報プラットフォーム(www.chinadrugtrials.org.cn)を通じて登録し、発表しなければならない。出願人は、臨床試験の承認を得てから1ヶ月以内に試験情報を事前登録して、試験の唯一の登録番号を取得し、最初の被験者が試験に参加することを登録する前に、いくつかの後続情報の登録を完了する必要がある。臨床試験の承認を得てから1年以内に上記の事前登録と登録を取得していない場合は,出願人は説明を提出しなければならず,3年以内に手続きが完了していない場合は,臨床試験の承認は自動的に失効する。
他の医療保険法
医療提供者、医師、第三者支払者は、私たちが市場で承認された任意の製品の推薦と処方において主な役割を果たすだろう。私たちの業務運営および現在または将来の第三者支払者、医療提供者、医師との任意の手配は、私たちが上場許可を得た任意の薬物の業務または財務的配置および関係を制限する可能性があり、広範に適用される詐欺や乱用、および他の医療法律および法規に直面する可能性があります。米国では、これらの法律は、州および連邦反リベート、虚偽声明、医師透明性、および患者データプライバシーおよびセキュリティ法律法規を含むが、これらに限定されないが、これらに限定されない
•他の事項に加えて、個人および実体が直接または間接的に、現金または実物で直接または間接的に現金または実物で直接または間接的に請求、提供、支払い、受け入れまたは提供されること(任意のリベート、花嫁またはいくつかのリベートを含む)を現金または実物で直接または間接的に要求、提供、支払い、または提供することを禁止し、個人の紹介または購入または購入または推薦に誘導または補償するために、任意の商品またはサービスを提供することができ、連邦医療計画に基づいて、それを全部または部分的に支払うことができる
医療保険と医療補助です。個人や実体は,連邦の“反リベート法令”やその法令違反の具体的な意図を実際に知る必要がなく,違法行為を実施することができる.いくつかの法的例外と規制避難所がいくつかの一般的な活動を保護することは起訴されない。違反行為は民事と刑事罰金に処せられ、違反ごとに罰金が科され、最高3倍の報酬、監禁、および政府医療計画から除外される
•個人または実体が虚偽、架空または詐欺的な支払いまたは承認請求を意図的に提出または提出することを禁止する民事虚偽請求法案またはFCAを含む連邦民事および刑事虚偽請求法、虚偽陳述または記録材料の故意、使用、または使用を禁止し、虚偽または詐欺的なクレームまたは連邦政府への金銭または財産の支払いまたは移転の義務を禁止するか、または連邦政府に金銭または財産を支払う義務を故意に隠蔽または故意に回避または減少させる。FCAによると、メーカーが政府支払人に直接クレームを提出していなくても、虚偽や詐欺的なクレームを“原因”とされていれば、責任を問われる可能性がある。また、政府は、民事虚偽請求法については、連邦反リベート法規違反による物品やサービスを含むクレームが虚偽または詐欺的クレームを構成していると断言することができる。FCAはまた、FCA違反行為を告発し、いかなる金銭的回収も共有する“密告者”としての個人代表が連邦政府を代表して訴訟を起こすことを許可した
•連邦民事罰金法は、他の事項を除いて、連邦医療保険又は州医療保健計画受益者に報酬を提供又は移転又は支払いする行為に対して民事罰金を科し、もしこの人が知っている又は知るべきであれば、例外が適用されない限り、連邦医療保険又は州医療保健計画精算サービスの特定の提供者、従事者又はサプライヤーに対する受益者の選択に影響を与える可能性がある。また、連邦民事罰金法によると、連邦反リベート法規違反による物品やサービスのクレームが虚偽または詐欺的クレームを構成していると断言することができる
•1996年の“健康保険携行性と責任法案”、またはHIPAAは、計画を知りながら故意に実行したり、計画を実行しようとしたり、個人支払者を含む任意の医療福祉計画を詐欺したり、医療福祉計画を故意に流用または盗み取ったり、医療保健違法行為に対する刑事調査を故意に阻害したり、重大な事実を偽造、隠蔽または隠蔽したり、あるいは医療福祉、プロジェクトまたはサービスの提供または支払いに関連する任意の重大な虚偽陳述を行い、刑事および民事責任を規定する。連邦の“反リベート法令”と同様に、個人または実体は、その法令またはその法令に違反する具体的な意図を実際に知る必要がなく、違法行為を実施することができる
•HIPAAは2009年の“衛生情報技術促進経済と臨床健康法”及びそのそれぞれの実施条例改正を経て、その他のほか、保険実体及びその業務パートナーに対して単独で識別可能な健康情報のプライバシーと安全に関する具体的な要求を提出し、強制的な契約条項とこのような情報に対する技術保障の必要な実施を含む。HITECHはまた新しい民事罰金等級を作成し、HIPAAを修正し、民事と刑事処罰がある場合に商業パートナーに直接適用され、州総検察長に新しい権力を与え、連邦裁判所に民事訴訟を提起し、損害賠償または禁止令を要求して連邦HIPAA法律を実行し、連邦民事訴訟に関連する弁護士費と費用を求めることができる
•“医師支払い陽光法案”は、2010年に“医療·教育和解法案”によって改正され、または総称して“医療保険法案”と呼ばれる“患者保護·平価医療法案”の一部であり、これらのメーカーに医師(医師、歯科医師、検眼師、足科医および脊椎マッサージ師を含む定義)、他の免許を有する医療従事者および教育病院、およびこれらの医師およびその直系親族が持つ所有権および投資権益を要求し、いくつかの支払いおよび医師(医師、歯科、検眼師、足科医、脊椎マッサージ師を含む)に費用を支払うこと、およびこれらの医師および直系親族が所有する所有権および投資権益を要求する“医師支払い陽光法案”の一部である
•同様の州および外国の法律および法規、例えば、州反リベートおよび虚偽請求法は、販売またはマーケティング計画および非政府第三者支払者(個人保険会社を含む)によって償還される医療項目またはサービスに関するクレームに適用可能であり、範囲は連邦同等の法律よりも広い可能性があり、州および外国の法律は、製薬会社が製薬業の自発的コンプライアンスガイドラインおよび連邦政府によって発行された関連コンプライアンスガイドラインを遵守することを要求するか、または他の方法で医療保健提供者への支払いを制限することを要求する。州と外国の法律は、薬品製造業者に薬品の定価と支払いに関する情報を報告し、医者と他の保健提供者に他の価値移転を行い、マーケティングのやり方を制限し、あるいはマーケティング支出と定価情報の開示を要求する;州と地方の法律は登録を要求する
医薬品販売代表;場合によっては健康情報のプライバシーと安全を管理する州と外国の法律。これらのデータプライバシーやセキュリティ法律は大きく異なる可能性があり,HIPAAに先制されないことが多く,コンプライアンス作業を複雑化させる可能性がある
また,製薬業者は連邦や州消費者保護や不正競争法律や法規の制約を受ける可能性があり,これらの法律や法規は市場活動を広く規制し,消費者を損なう可能性がある
薬品と生物製品の流通は広範な記録保存、許可、貯蔵と安全要求を含む追加の要求と条例を遵守し、許可されていない薬品の販売を防止しなければならない
これらの法律のすべての範囲や執行は不確実であり,現在の医療改革環境の急速な変化の影響を受けている。連邦と州法執行機関は引き続き医療会社と医療保健提供者の間の相互作用の審査を強化し、これは医療保健業界の一連の調査、起訴、有罪判決と和解を招いた。政府当局は、私たちの業務やり方が現在または未来に適合していないと結論するかもしれないが、詐欺および乱用または他の医療保健法律および法規の適用に関する現行または未来の法規、法規または判例法に関連している。もし私たちの運営がこれらの法律または任意の他の私たちに適用される可能性のある政府法規に違反していることが発見された場合、私たちは重大な民事、刑事と行政処罰、損害賠償、罰金、監禁、返還、政府援助の医療計画から除外された(例えばMedicareおよびMedicaid)、名声損害、追加の監督、報告義務に直面する可能性があり、もし私たちが会社の誠実な合意または同様の和解の制約を受けて、これらの法律違反に関する疑惑を解決し、私たちの業務を削減または再編する。もし私たちがそれと業務往来があることを期待している任意の医師や他の医療提供者や実体が適用法を遵守していないことが発見された場合、彼らは類似した行動、処罰、そして制裁を受けるかもしれない。ビジネス手配が適用される医療保険法に適合していることを確保し,政府当局が行う可能性のある調査に対応することは,時間や資源がかかる可能性があり,会社の業務への関心を分散させる可能性がある
保証と精算を請け負う
米国や他国の市場では,自分の病状に応じて処方治療を受けている患者や処方サービスを提供する提供者は,通常第三者支払者に依存してすべてまたは一部の関連医療費を精算している。したがって、候補製品が承認されても、製品の販売は、MedicareおよびMedicaid、商業健康保険会社および管理型医療機関などの米国の政府医療計画を含む第三者支払者にある程度依存し、その製品に保険を提供し、十分な精算レベルの程度を確立する。米国では、第三者支払者の間に統一された薬品保険や精算政策はない。そのため、薬品の保証範囲と精算範囲は支払人によって異なる。第三者支払者が製品に保険を提供するか否かを判定する過程は、価格又は販売率を設定する過程と分離することができ、保険が承認されると、支払者は製品に料金を支払うことができる。第三者支払者は、徴収された価格に挑戦し、医療の必要性を審査し、医療製品およびサービスの費用対効果を審査し、コストを管理するために制御を実施することが増えている。第三者支払者は、保証範囲を承認リスト上の特定の製品に制限することができ、特定の適応のすべての承認製品を含まない可能性がある配合表とも呼ばれる
販売が承認される可能性のある任意の製品の保険·精算を確保するためには、企業は、製品の医療必要性および費用効果、およびFDAまたは他の同様の規制承認を得るために必要なコストを証明するために、高価な薬物経済学的研究を行う必要があるかもしれない。また、会社は購入者、個人健康計画、または政府医療計画に割引を提供する必要があるかもしれない。それにもかかわらず、候補製品は医学的に必要または費用効果があると考えられないかもしれない。製品が承認されると、第三者支払者が製品を保証しない決定は医師の使用率を減少させ、販売、私たちの運営、財務状況に実質的な悪影響を及ぼす可能性がある。また、第三者支払者が製品に保険を提供することを決定することは、十分な販売率を承認することを意味するわけではない。また、1人の支払人が1つの製品に保険を提供することを決定することは、他の支払者もその製品に保険や精算を提供することを保証することはできず、支払者によって保険や精算レベルが大きく異なる可能性がある
医療コストの抑制は連邦、州と外国政府の優先順位となっており、製品価格はこの努力の重点である。各国政府はコスト制御計画の実施に大きな興味を示し、価格制御、精算制限と代替後発薬の要求を含む。価格制御及びコスト制御措置を講じ、既存の統制及び措置を講じている司法管区においてより厳しい政策をとることにより、任意の承認された製品の販売から得られる収入をさらに制限することができる。保証政策と第三者支払人の販売率はいつでも変化する可能性があります。1社またはその協力者が規制承認を受けた1つまたは複数の製品が有利な引受·精算状態を獲得しても、将来的にはあまり有利ではない保証政策や精算料率が実施される可能性がある
医療改革
米国や一部の外国司法管轄地域では、医療システムに関する複数の立法·規制改革および提案された改革が継続して存在する可能性があり、医療の獲得性を拡大し、医療の質を向上させ、医療のコストをコントロールまたは低減することを目的としている。例えば、2010年、米国議会は、政府医療計画下の製品のカバー範囲の変更と支払い方法を含むACAを公布した。ACAには私たちの潜在的な製品候補に非常に重要な条項が含まれています
•指定されたブランドの処方薬および生物製品を生産または輸入する任意のエンティティに控除できない年間費用を徴収し、特定の政府医療計画におけるこれらのエンティティの市場シェアに基づいてこれらのエンティティ間で分担する
•医療補助計画の資格基準を拡大し、メーカーの医療補助リベート責任を潜在的に増加させた
•医療補助薬品還付計画下のメーカーの税金還付責任を拡大した
•340 B薬品割引計画に適合する実体タイプを拡大した
•次の生物製品の許可フレームワーク
•CMSに医療保険と医療補助革新センターを設立し、革新的な支払いとサービス交付モードをテストして、医療保険と医療補助支出を低減し、処方薬支出を含む可能性がある
•連邦医療保険D部分引受切欠き割引計画を構築し、メーカーに保証間隔期間内に条件を満たす受益者に適用ブランド薬品交渉価格70%の販売時点割引を提供することを要求し、メーカーの外来薬物として連邦医療保険D部分で保険を受ける条件を提供した
•患者を中心とした結果研究所を作成し、優先事項を監督、確定し、臨床有効性比較研究を行い、このような研究に資金を提供した
ACAが公布されて以来、アメリカはまた他の立法改正を提案し、採択した。2011年8月、2011年予算制御法には、他にも、医療保険提供者に支払う医療保険総金額を前期ごとに2%削減することが含まれている。これらの削減は2013年4月1日に施行され、その後法規の立法改正が行われたため、これらの削減は2031年まで有効となる。2013年1月、“2012年米国納税者救済法”が法律に署名され、病院、画像形成センター、癌治療センターを含むいくつかの医療サービス提供者に支払う医療保険をさらに減少させ、政府が医療サービス提供者に多額のお金を支払う訴訟時効を3年から5年に延長した。2021年3月11日、総裁·バイ登は“2021年米国救援計画法案”に署名し、2024年1月1日から、単一源と革新多源薬物に対する法定医療補助薬品還付上限を廃止し、現在この上限は薬品メーカー平均価格の100%である。2010年の法定現金給付法案、2021年の米国救援計画法案、および後続立法による予算赤字推定が増加したため、これ以上の立法がない場合、2025年から提供者に支払われる医療保険金額はさらに減少する。これらの法律および法規は、医療保険や他の医療資金のさらなる減少を招き、規制承認を得る可能性のある任意の候補製品の価格に影響を与えるか、またはそのような候補製品を開発または使用する頻度に影響を与える可能性がある。
また、支払い方法は医療立法と規制措置の影響を受ける可能性がある。例えば、CMSは、バンドル支払いモードのような新しい支払いおよび配信モードを開発することができる。また、最近政府はメーカーがその商業製品の価格設定の方式に対してより厳格な審査を行い、国会が数回の調査を行い、州と連邦立法を提出し、製品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、政府の薬品に対する計画補償方法を改革することを提出し、公布した。例えば、連邦レベルでは、バイデン総裁は処方薬のコストを低減するための複数の行政命令を発表した。2023年2月、厚労省はまた、バイデン総裁が2022年10月に発表した行政命令に応答する提案を発表した。提案された処方薬の価格設定モデルを含み、目標とした医療保険支払い調整が、承認経路を加速することで承認された薬品の実証試験を完成させるのに十分かどうかをテストする。いくつかの措置および他の提案された措置は発効するために追加の立法によって許可される必要があるかもしれないが、バイデン政府はこれらの措置を撤回または他の方法で変更する可能性があるが、バイデン政府と国会は薬品コストを制御するための新しい立法措置を求め続けると表明している。
2022年8月、アイルランド共和軍は法律に署名した。IRAは、2025年からMedicare Part D受益者の自己負担上限を2,000ドルに引き下げ、Medicare Part D中のいくつかの薬物に新しいメーカーの財務責任を適用し、アメリカ政府が模造薬や生物類似競争のないいくつかの高コスト薬物と生物製品のMedicare Part BとPart D価格上限について交渉することを許可し、会社にある価格がインフレよりも速い薬品のためにMedicareにリベートを支払うことを要求し、薬局福祉マネージャーが受け取ることができる費用のリベート規則を延期することを含む、私たちの業務に異なる程度の影響を与える可能性のあるいくつかの条項を含む。また,IRAによると,孤児薬は連邦医療保険薬品価格交渉計画の制限を受けないが,孤児疾患名があることを前提としており,唯一承認された適応はこの疾患や条件に対するものである。1つの製品が複数のまれな疾患の指定または複数の承認された適応を得た場合、孤児薬免除を受ける資格がない。アイルランド共和軍の実施は現在行われている訴訟を受け,アイルランド共和軍の医療保険薬品価格交渉計画の合憲性を疑問視している。Ireland共和軍が私たちの業務と医療産業全体に及ぼす影響はまだ明確ではない。
米国の個別州も、価格または患者の精算制限、割引、特定の製品への参入およびマーケティングコスト開示の制限および透明性措置を含む、立法および実施によって薬品の価格を制御するための法規をますます多く実施しており、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。例えば,承認されれば,米国で薬品の価格規制を行う外国療法からの候補薬物の開発や研究薬物の競争に直面する可能性がある。米国では、FDAは、最初は外国で販売しようとしていたが、外国での販売を許可しているが、FDAによって承認された薬物のために追加の国家医薬品コードを得る方法について概説する最終指導文書を発表した。現在、最終的な指針の市場影響はまだ分からない。麻薬再輸入の支持者たちは立法を通じて、場合によっては再輸入を直接許可しようと努力するかもしれない。立法や規制が薬物の再輸入を許可すれば、私たちが開発する可能性のある任意の製品の価格を低下させ、将来の収入や利益の見通しに悪影響を及ぼす可能性がある。また、法律で規定されている第三者支払者の支払金額の価格制御又はその他の制限は、我々の業務の見通し、財務状況、運営結果に悪影響を及ぼす可能性がある。また,地域医療当局や個別病院では,どの薬品やサプライヤーが処方薬や他の医療計画に含まれるかを決定するために入札プログラムが使用されるようになってきている。これは私たちの候補製品に対する最終的な需要を減らすか、あるいは私たちの製品の価格設定に圧力を与えるかもしれない。
2018年5月30日、“裁判権法案”が法律に署名された。他の事項以外に、この法律はある患者に連邦フレームワークを提供し、彼らが第一段階の臨床試験を完成し、FDAの許可を得た研究用新薬製品を獲得するために調査を行っていることを許可した。場合によっては、条件を満たす患者は、臨床試験に参加することなく、FDA拡大参入計画に従ってFDA許可を得ることなく治療を求めることができる。“試用権法案”によると、薬品メーカーはその薬品を条件に合った患者に提供する義務がない
米国以外では、製品のカバー範囲の確保と十分な支払いも課題に直面している。多くの国で、処方薬の価格設定は政府によって規制されている。例えば、カナダでは、特許薬品の価格規制立法が現在大きな変化を経験しており、カナダで製品を販売している会社の収益性に大きな影響を与える可能性がある。政府当局との価格交渉は製品が監督管理の許可を得る範囲をはるかに超えている可能性があり、臨床試験を行い、製品のコスト効果を他の利用可能な治療法と比較する必要があるかもしれない。このような臨床試験を行うことはコストが高く、商業化の遅延を招く可能性がある
人的資本資源
2023年12月31日現在、私たちは145人の常勤従業員を持っており、そのうち61人は医学博士および/または博士号を持っている。これらの常勤従業員のうち,105名が研究·開発活動に従事し,44名が一般·行政活動に従事している。2024年1月9日、運営コストを低減し、従業員チームを業務需要と一致させるための計画を実施したため、従業員数は約30%減少した。私たちは現在9人の上級指導者がいて、その中の7人が執行幹事を担当している。私たちの職員たちは労働組合代表もなく、集団交渉協定のカバー範囲もない。私たちは私たちが従業員と仲がいいと思う。
私たちは私たちの未来の成功は私たちが高い技能職員たちを引きつけて維持し続ける能力にかかっていると信じている。私たちは従業員に競争力のある給料とボーナス、株式機会、持続的な学習と成長を支援する発展計画、医療保健、退職計画、有給休暇を含む彼らの生活のあらゆる福祉を促進する穏健な雇用プランを提供する。私たちは人材を向上させ、維持するための努力の一部として、持続的な発展にも投資している。
私たちは、包容的で公平な環境を持つ多様な労働力が企業成功の重要な構成要素であると信じているので、職場の多様性、公平性、包摂性に取り組んでいる。私たちの目標は、すべての従業員が繁栄し、貢献し、最善を尽くす文化を育成することです。この重点の下で、私たちは目標を立て、私たちの労働力チーム全体で多様性、公平、包摂的なイニシアティブを推進し、マネージャーや従業員と協力して戦略を制定し、彼ら自身の偏見を理解し、包摂的なリーダーシップを学ぶことから、異なる背景の指導者の進歩を促進し、私たちの地域コミュニティを支援することで、サービス不足コミュニティの機会格差を埋めるために努力している。2022年に設立された多様性、公平、そして包括的な理事会の助けを借りて、私たちは私たちの多様性、公平、そして包括的な計画を私たちの日常的な仕事に集中し続けている。
企業情報
私たちは2015年10月にデラウェア州の法律に基づいて登録された。私たちの主な実行事務室はマサチューセッツ州ウォータータウン、02472、アーセナル路490号、Suite 120にあります。私たちの電話番号は(617231-0700)。完全子会社、C 4 T証券会社、マサチューセッツ州の会社があります。
利用可能な情報
私たちのサイトの住所はwww.c 4 Treatutics.comです。私たちのサイト上の情報は参考にここに含まれていません。私たちは、材料を電子的にアーカイブまたは米国証券取引委員会に提供した後、合理的で実行可能な範囲内で、私たちの10-Kフォーム年次報告、10-Qフォーム四半期報告、8-Kフォームの現在の報告、証拠品を含む、および取引法第13(A)または15(D)節に提出または提供された報告書の任意の修正をできるだけ早く無料で提供します。米国証券取引委員会は、米国証券取引委員会に電子的に提出された報告書、依頼書、情報声明、その他の発行者に関する情報を含むインターネットサイトを有する。
当社のコーポレートガバナンス基準、ビジネス行為および道徳基準、および(I)監査委員会、(Ii)組織、リーダーシップおよび報酬委員会、および(Iii)指名およびコーポレートガバナンス委員会規約のコピーは、当社のウェブサイトwww.c 4 Treateutics.comの“コーポレート·ガバナンス”のタイトルの下に掲示されており、コピーを必要とする人は電話(617231-0700)に電話で連絡したり、C 4治療会社、C 4治療会社、住所:490 Arsal Way、Suite 120、Wattown、Massachusetts 02472、宛先:会社秘書に連絡することができます。
重大な非公開情報を開示する手段として、プレスリリース、当社のサイト(私たちの投資家関係サイトを含む)、および当社のLinkedInおよびTwitterアカウント(以下に示す)を使用し、FD法規下での開示義務を遵守する予定です。
 Wwwww.linkedin.com/Company/c 4-Treatutics-Inc
Wwwww.linkedin.com/Company/c 4-Treatutics-Inc. Http://twitter.com/C 4 Treateutics
Http://twitter.com/C 4 Treateutics
第1 A項。リスク要因です
私たちの普通株に投資することは高い危険と関連がある。当社を評価する際には、“経営陣の財務状況および経営結果の議論および分析”と題する部分、および本Form 10-K年度報告書の末尾の財務諸表および関連注釈を含む、以下のリスクおよび本Form 10-K年次報告書の他のすべての情報をよく考慮しなければなりません。以下、米国証券取引委員会に提出された他の文書に記載されているリスクおよび不確実性は、私たちが直面している唯一のリスクおよび不確実性ではないかもしれない。次のいずれかの事件又は事態の発生は、本当に発生すれば、我々の業務、財務状況、経営結果及び成長の見通しを損なう可能性がある。したがって、私たちの普通株の市場価格は下がるかもしれません。あなたは私たちの普通株へのすべてまたは一部の投資を失うかもしれません
私たちの財務状況と追加資本需要に関連するリスク
私たちは臨床段階の生物製薬会社で、設立以来すでに重大な損失が発生している。私たちは少なくとも今後数年以内に損失が出て、永遠に利益を達成したり維持したりしないかもしれないと予想している
私たちは臨床段階の生物製薬会社で、運営の歴史は限られている。2023年、2023年、2022年12月31日までの年度の純損失はそれぞれ1.325億ドルと1.282億ドルだった。2023年12月31日までの累計赤字は5.284億ドルだった。今まで、私たちは製品販売から何の収入も得ていません。主に私たちの株式を売却することで、私たちの普通株の公開発行、私たちの協力収益、債務融資を含む私たちの運営に資金を提供します。私たちはまだ候補製品開発の初期段階にいる。したがって、私たちはまだ数年かかると予想され、もしあれば、規制部門の承認と商業化のための候補製品を持つことができる。私たちはこのような活動で決して成功しないかもしれないし、たとえ私たちが成功しても、利益を達成するのに十分な収入が生まれないかもしれない。利益を実現し、維持するためには、マーケティング承認を成功させ、大量の収入を生み出した製品を商業化しなければならない。これは、我々が候補製品の臨床前研究と臨床試験を成功させ、より多くの候補製品を発見し、第三者と臨床試験を行う手配を確立し、臨床および商業規模の製造を調達し、候補製品のマーケティング承認を得ること、マーケティング承認を得る可能性のある任意の製品を製造、マーケティング、販売することを含む一連の挑戦的な活動において成功することを要求し、パートナーが決定した候補製品または既存の候補製品の他の用途を開発するためにパートナーを決定し、パートナー候補製品の開発を成功させることができる
少なくとも今後数年以内に、巨額の費用と増加する運営損失を招き続けることを予想している。私たちの費用は大幅に増加すると予想されています
•私たちの候補製品の最初の人類臨床試験と後期臨床試験を開始し、行い、成功し、そして著者らの独自の研究と開発組み合わせの範囲を拡大した
•我々の魚雷プラットフォームを用いてより多くの候補製品を識別し、それを臨床前と臨床開発に投入する
•私たちの魚雷プラットフォームの能力を拡張し
•臨床試験に成功した候補製品のために市場承認を求める
•最終的に販売、マーケティング、流通インフラを構築し、外部製造能力を拡大し、市場の承認を得ることが予想される任意の製品を商業化する
•私たちの知的財産権の組み合わせを推進し、拡大し、維持し、保護する
•我々がより多くの候補製品を発売し、および/または既存の候補製品を引き続き開発することに伴い、管理者は絶えず変化する業務需要を満たすように需要を構成する。
また、重大な法律、会計、保険、投資家関係、その他の費用を含む上場企業の運営に関する追加コストが引き続き発生すると予想される
FDA、欧州医薬品局、または他の規制機関が現在予想されている基礎の上で試験を行うことを要求する場合、あるいは私たちの任意の候補製品のための適切な製造手配を確立したり、臨床試験または臨床開発を完成させる上でいかなる遅延に遭遇した場合、私たちの費用は私たちの予想を超えるかもしれない
医薬品開発に関連する多くのリスクや不確実性のため、私たちが発生する費用の増加時間や金額、あるいはいつ利益を達成できるかを正確に予測することはできません。私たちが利益を達成しても四半期を維持したり向上させることができないかもしれません
年ごとに計算する。もし私たちが実現して利益を維持できなければ、会社の価値を下げ、資金を調達し、研究開発努力を維持し、業務を拡大し、あるいは運営を継続する能力を弱める可能性がある。わが社の価値や私たちの普通株の価値が下がっても、あなたの全部または一部の投資損失を招く可能性があります
もし私たちが開発した1つ以上の候補製品が商業販売のために承認されれば、これらの承認された候補製品の商業化に関連する巨額のコストが発生すると予想される。たとえ承認された製品の販売から収入を得ることができても、利益を得ることができない可能性があり、運営を継続するために追加の資金が必要かもしれない
私たちには大量の 私たちの業務目標を達成し、私たちの運営を継続するために追加資金を提供する。もし私たちが必要な時に資金を集めることができなければ、私たちは私たちの研究や製品開発計画や将来の商業化努力を延期、制限、減少、または中止することを要求されるかもしれない
私たちが行っている活動に関連する費用は大幅に増加することが予想され、特に私たちが行っているおよび計画中の候補製品の人類第1段階1/2臨床試験の準備と起動、進行と完了、私たちの魚雷プラットフォームの推進と研究開発活動の継続、独自の研究開発の組み合わせの拡大、および現在と未来の臨床前計画の臨床試験を開始し、継続し、市場承認を求めることが可能である。また、いずれかの候補製品が市場承認されれば、製品製造、マーケティング、販売、流通に関連した巨額の商業化費用が発生すると予想される。また、上場企業の運営に関連した追加コストが引き続き発生すると予想される。したがって、私たちは私たちの持続的な業務と関連した多くの追加資金を得る必要があるだろう。もし私たちが必要な時や魅力的な条件下で資金を調達できない場合、私たちは私たちの研究、製品開発計画、あるいは未来の商業化努力を延期、制限、減少または終了することを要求されるかもしれません。あるいは私たちが自分で開発し、マーケティングすることをより望んでいた候補製品の権利を与えることができます
2023年12月31日現在、私たちは約2兆817億ドルの現金、現金等価物、有価証券を持っている。私たちは、これらの資金は、私たちが2024年1月初めに受け取った資金とともに(I)私たちの“市場別”に基づいて発売された株式を決済し、(Ii)先に開示された株式購入契約に従ってBetta Pharmaの関連会社への株式売却を完了し、(Iii)私たちの2023年12月の許可と協力協定に基づいてメルクから得られた前払い、および再編から節約されると予想されるコストは、2027年までに計画された運営費用を支払うのに十分だと信じている。私たちは間違っていることが証明される可能性があるという仮定に基づいてこの推定をして、私たちは現在予想されているよりも早く既存の資本資源を枯渇させるかもしれない。私たちの将来の資本需要は多くの要素に依存します
•私たちが行っているおよび計画中の候補製品のヒト第1段階1/2臨床試験の時間、進行、コスト、および結果、およびこれらの候補製品の任意の未来の臨床開発
•臨床開発段階計画と私たちの他の候補製品と開発計画の範囲、進捗、コストと結果
•私たちが求めている他の候補製品の数量と開発要件は
•私たちの持続的な協力の成功は
•私たちの候補製品に対する規制審査のコスト、時間、結果
•将来の商業化活動のコストと時間、製品製造、マーケティング、販売、流通、および上場承認を得たり予想したりする任意の製品の将来の商業化活動の時間とコストを含む
•私たちが上場許可を得た候補製品の商業販売から得られた収入(ある場合)およびそのような収入を受信した時間;
•私たちが臨床前研究、臨床試験および/またはサプライチェーンで遭遇した任意の遅延または中断は、任意の全世界の衛生流行病による遅延を含む
•特許出願を準備し、提出し、起訴し、私たちの知的財産権を維持し、実行し、知的財産権に関する任意のクレームを弁護するコストと時間;
•私たちは、有利な条件下で他のバイオテクノロジーや製薬会社と協力して、私たちの候補製品を開発したり、それを商業化したり、私たちの魚雷プラットフォームに入ることができます
私たちの現在の現金、現金等価物、および有価証券は、規制機関の承認によって私たちの任意の候補製品に資金を提供するのに十分ではないだろう。したがって、私たちは大量の追加資本を集めて完成させる必要があるだろう
私たちの候補製品の開発と商業化。潜在的な候補製品の確定及び臨床前研究と臨床試験を行うことは時間がかかり、高価と不確定な過程であり、完成するのに数年かかる。私たちは市場の承認を得て製品販売を実現するために必要なデータや結果を決して生成しないかもしれない。しかも、私たちの候補製品が承認されれば、商業的成功を得られないかもしれない。私たちの商業収入は、もしあれば、私たちが数年以内に商業使用できないと予想している製品を販売します。もしなければ。受け入れ可能な条件で、私たちは十分な追加資金を得ることができないかもしれないし、全くないかもしれない。また、有利な市場条件や戦略的考慮により、現在または将来の運営計画を実行するのに十分な資金があると考えても、追加の資本を求めることができる
もし私たちが開発した1つ以上の候補製品が商業販売のために承認されれば、これらの承認された候補製品の商業化に関連する巨額のコストが発生すると予想される。たとえ承認された製品の販売から収入を得ることができても、利益を得ることができない可能性があり、運営を継続するために追加の資金が必要かもしれない
私たちはまだ開発ライフサイクルの初期段階にありますが、これは私たちの業務のこれまでの成功度と私たちの未来の生存能力を評価することが難しいかもしれません
私たちは2015年末に運営を開始し、これまで、私たちの活動は組織と配備会社の人員、業務計画、資金調達、発見と研究活動、特許申請の提出、潜在的な候補製品の決定、私たちの魚雷プラットフォームの開発と推進、臨床前研究を行い、第三者と私たちの候補製品を製造する最初の数量について手配し、早期臨床試験を準備し、行うことに限られている。いくつかの臨床試験が行われているが,他のすべての候補製品はまだ発見段階にある。私たちはまだ私たちがどんな臨床試験を成功させ、市場の承認を得て、直接あるいは第三者を通じて商業規模の製品を製造したり、成功した製品の商業化に必要な販売、マーケティング、流通活動を行うことができるかを証明していません。したがって、もし私たちがより長い運営歴史を持っているなら、あるいは私たちが過去にいくつかまたはすべてのタイプの活動を成功的に達成した場合、あなたは私たちの未来の成功または生存能力に対するいかなる予測もそんなに正確ではないかもしれない
また,バイオ製薬会社としては,予見できない費用,困難,合併症,遅延などの既知と未知の挑戦に遭遇する可能性がある。私たちはある時点で研究開発に集中している会社からビジネス活動を支援できる会社に転換する必要がありますが、私たちはこの転換に成功できないかもしれません
様々な要因により、私たちの財務状況と経営業績は四半期ごとと毎年大幅に変動し続け、その多くの要素はコントロールできないと予想しています。したがって、今後の経営業績の指標として、いかなる四半期や年度の業績にも依存してはいけません
追加資本の調達は私たちの株主に希釈し、私たちの運営を制限したり、私たちの技術や候補製品に対する権利を放棄することを要求するかもしれません
私たちが製品販売から相当な収入を得ることができる前に、私たちは株式発行、債務融資、協力、戦略連合、マーケティング、流通、または許可手配を通じて私たちの現金需要に資金を提供する予定です。私たちは私たちの協力に基づいて潜在的な未来の支払いを受けるかもしれないが、私たちは現在何の約束も外部資金源を持っていない。もし私たちが株式または転換可能な債務証券を売却することで追加資本を調達する場合、あなたの所有権権益は希釈され、私たちが将来発行する可能性のある任意の証券の条項は清算または他の特典を含む可能性があり、普通株主としての権利に悪影響を及ぼす可能性がある。債務融資および優先株融資に関与する可能性のある合意には、追加債務を招く、買収、資本支出、または配当の発表など、私たちが具体的な行動をとる能力を制限または制限する契約が含まれる。
もし私たちが第三者との協力、戦略連合またはマーケティング、流通または許可手配を通じてより多くの資金を調達するならば、私たちは私たちの技術、将来の収入源、研究計画、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になる可能性のある条項でライセンスを付与しなければならないかもしれない
私たちの候補製品の発見と開発に関するリスク
我々は魚雷プラットフォームに基づいて候補製品を発見·開発する方法が検証されておらず,どの製品の開発にも時間,コスト,開発成功の可能性を予測することは困難である
標的タンパク質分解を利用して疾病を治療することは新しい治療モードである。私たちの未来の成功はこの新しい治療法の成功にかかっている。我々の魚雷プラットフォームによって開発された製品のような配向タンパク質分解を用いた小分子候補製品は、人体で試験されることは少なく、我々の魚雷プラットフォームによって開発された製品候補製品は、米国、ヨーロッパ、または任意の他の司法管轄区域で承認されていない。これらのタイプの治療製品の開発の可能性の背後にあるデータは初歩的です
そして限られています目的タンパク質分解物の他の開発者が何か不利な学習をすれば,我々の候補製品の開発は実質的な影響を受ける可能性がある。ユビキチン-プロテアソーム経路を利用してタンパク質標的を分解する小分子の発見と発展は以下の要素によって大きく阻害される:ユビキチン-プロテアソーム系特定成分の機能、生化学と構造生物学の複雑かつ限られた理解、標的蛋白ユビキチン化に参与するE 3リガーゼ及びそれに必要な補助蛋白、及び蛋白質-蛋白質相互作用を促進する工学化合物の挑戦を含む
我々が魚雷プラットフォームの下で分解器候補製品を開発する基礎を構成する科学研究が行われており、魚雷プラットフォーム派生療法の開発可能性を支持する科学的証拠は初歩的であり、限られている。また、一部の癌患者は承認された原因蛋白を抑制する薬物に対して固有の一次耐性を示し、他の患者はこれらの阻害剤に対して二次性耐性を産生している。著者らの候補製品は現在市場で販売されている病原性酵素阻害剤に対して薬剤耐性の特定の突然変異を分解する能力があると信じているが、患者は著者らの候補製品に対する任意の固有の一次或いは獲得性二次耐性はそれらの臨床利益を阻止或いは弱めることができ、もし著者らの努力の基礎を構成する科学研究が矛盾であることが証明されれば、状況はこのようになる
いくつかの臨床試験が行われているが,現在のところ候補製品の臨床試験は完了していない。そのため、私たちはまだ患者に対する主要な候補製品の安全性を評価し始めたばかりで、私たちはまだ私たちの他の初期候補製品の人体上の安全性を評価していません。我々のいくつかの早期候補製品は動物研究において明らかな結果を得たにもかかわらず、それらの動物への影響の安全性データセットは限られている。さらに、これらの候補製品は、ヒトにおいて同じ化学的および薬理特性を示さない可能性があり、予測不可能、無効または有害な方法でヒト生物系と相互作用する可能性がある。したがって、私たちの現在または未来の任意の候補製品の治療は、私たちが現在予測できない悪影響を及ぼすかもしれない
さらに、我々のような新製品候補製品の規制承認プロセスは、他のより有名で広く研究されている候補製品よりも高く、時間がかかる可能性がある。他社でも標的タンパク質分解に基づく療法が開発されているにもかかわらず,規制機関はこのような性質の療法を承認していない。したがって,我々の候補製品を開発する時間やコストを予測することは困難であり,我々の魚雷プラットフォームや類似あるいは競合相手のタンパク質分解プラットフォームの応用が上場により承認された候補製品の開発につながるかどうかも予測できない.私たちが未来に遭遇する私たちの魚雷プラットフォームや私たちの任意の研究計画に関連する開発問題は、重大な遅延や意外なコストを招く可能性があり、あるいは商業的に実行可能な製品の開発を阻害する可能性がある。これらの要因のいずれも、臨床前研究または開始可能な任意の臨床試験を完了することを阻止することができ、タイムリーまたは利益に基づいて開発される可能性のある任意の候補製品の商業化を阻止することができる
私たちは臨床段階のバイオテクノロジー会社で、私たちはすでに私たちのいくつかの候補製品の臨床試験を始めましたが、私たちの多くの他の候補製品はまだ発見段階にあります。私たちが臨床開発、開発、規制部門の候補製品の承認を得て商業化することができなければ、あるいはこの点で重大な遅延があれば、私たちの業務は実質的な損害を受ける可能性がある
私たちは臨床段階のバイオテクノロジー会社で、いくつかの臨床試験が行われていますが、私たちの他の候補製品の多くは現在発見段階にあります。したがって、彼らは失敗する危険が高い。私たちはほとんどの精力と財力を投じて私たちの魚雷プラットフォームを構築し、私たちのリードプロジェクトを含む現在の候補製品の臨床前開発を確定し、展開しています。私たちが製品販売から収入を得る能力は、私たちの1つ以上の候補製品の成功開発と最終商業化に大きく依存します。この状況は数年以内には起こらないと予想されます。私たちの候補製品の成功は以下のいくつかの要素に依存するだろう
•私たちの財政と他の資源が十分であるかどうか
•臨床試験の開始に成功しました
•成功した患者を登録し、臨床試験を行い、完成させる
•適用された規制機関から上場許可を得た領収書と関連条項
•私たちの候補製品のために特許または商業秘密保護と規制の排他性を獲得し、維持する
•第三者製造業者と私たちの候補製品の臨床と商業供給について適切に手配します
•期待され、その予想される適応に適した治療特性を実現するための候補製品を開発する
•販売、マーケティング、流通能力を確立し、承認を得た後、単独でまたは他人と協力して私たちの製品の商業販売を開始します
•もし患者、医学界、第三者支払者が私たちの製品を承認したら、私たちの製品を受け入れます
•第三者保険と適切な補償を受けて維持します
•私たちの製品の持続可能な安全状況を確立し、承認後にこの状況を維持する
•他の治療法と効果的に競争し
•我々の第三者協力パートナーは,我々の候補製品(S)をタイムリーに開発する市場で上記のいずれかのスキルと成功を実現している
もし私たちがこれらの要素のうちの1つまたは複数をタイムリーにまたは根本的に達成できなければ、私たちは重大な遅延に遭遇したり、私たちの候補製品を商業化することに成功しなかったりする可能性があり、これは私たちの業務に実質的な損害を与えるかもしれない。しかも、もし私たちが規制部門の承認を得なければ、私たちは運営を続けることができないかもしれない
ある会社として、著者らは臨床研究を完成する前にINDの保存を実現し、INDの提出或いは起動、登録と臨床試験を行う経験は相対的に限られている
会社として,INDを有効にする前臨床研究を完成させた経験は,4種類の候補製品の臨床開発を開始したことから来ている。この仕事はかなりの進歩を表しているが,これまで会社としての開始,登録,臨床試験の経験は比較的限られていた。一部の理由は,われわれがこの分野で進展を続けているにもかかわらず,われわれが計画した臨床試験が時間どおりに開始,登録あるいは完了するかどうかを確認できないからである。また,適用された規制機関がINDを初めて提出する際に我々INDに規定されている臨床試験の設計と実施に同意しても,これらの規制機関が将来彼らの要求を変えないことは保証されない。これらの考慮要因は,上記のIND,我々が将来提出する可能性のある追加IND,および既存または新しいIND修正案として提出される可能性のある新しい臨床試験に適用される
そのほか、大規模な臨床試験は大量の追加の財務と管理資源を必要とし、第三者臨床研究者、契約研究組織或いはCROと顧問に依存する。第三者臨床研究者、CROと顧問に依存することは著者らがコントロールできない遅延に遭遇する可能性があり、現在臨床開発中にあるすべての候補製品に対して、著者らはすべて1名のCROを招聘して著者らの初の人類1/2段階臨床試験を指導した。私たちの臨床前研究或いは臨床試験において第三者に依存することは、彼らが研究或いは試験方案を十分に遵守できないか、あるいは私たちの計画を監督機関に提出する任意の研究或いは試験に必要な良好な実験室実践或いは良好な臨床実践或いはGCPを十分に遵守できない可能性があるというリスクに直面する可能性がある。臨床試験開始後に確定し、過去にCROを決定する必要があるかどうかを決定することもできないかもしれません。既存または潜在的な将来CROなしに、他の候補製品が必要な場合には、タイムリーまたは完全に受け入れ可能な条項で交渉し、適切な契約手配を達成することができる保証はありません。
私たちの臨床前研究と臨床試験は私たちの任意の候補製品の安全性と有効性を十分に証明できない可能性があり、これは開発、監督管理許可と商業化を阻害或いは延期する。また,臨床前研究の結果は今後の研究や試験の将来の結果を予測できない可能性があり,臨床試験の初歩的な成功はこれらの試験完了時あるいは後期臨床試験で得られた結果を示すことができない可能性がある。
規制機関が私たちの任意の候補製品の商業販売を承認する前に、私たちの候補製品は各目標適応において安全かつ有効であることを、長く、複雑で高価な臨床前研究と臨床試験によって証明しなければならない。このテストは費用が高く、完成するのに数年かかるかもしれない。しかも、このような活動の結果は本質的に不確実だ。失敗は臨床開発過程中のいつでも発生する可能性があり、著者らの多くの候補製品は早期開発段階にあり、人体でテストを行ったことがないため、失敗のリスクが高い。さらに、標的タンパク質分解器は比較的新しい候補製品カテゴリであるため、このカテゴリ中の他の開発者が臨床前或いは臨床テストで見たいかなる失敗或いは不良結果は著者らの計画の成功に実質的な影響を与える可能性がある。私たちは絶対に適切な製品を開発することに成功しないかもしれない
著者らの候補製品の臨床前研究と早期臨床試験の結果も後期臨床試験の結果を予測できない可能性がある。臨床前と臨床データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品は
それにもかかわらず、臨床前研究と臨床試験はまだその製品の発売許可を得られなかった。候補製品は臨床前研究および早期臨床試験において有望な結果を示す可能性があるが、その後の臨床試験では有効または安全ではないことが証明される可能性がある。我々が行っているおよび計画中の候補製品のヒト第1段階1/2臨床試験の用量増加部分の結果は、これらの候補製品または任意の他の候補製品のさらなる臨床試験の結果を予測できない可能性があり、1/2段階臨床試験の第2段階まで行うのに十分ではないかもしれない。動物で実験を行う条件は人間で実験を行う条件とは異なるため,動物研究の結果は人間の体験を正確に予測できない可能性がある
臨床前研究と臨床試験による候補製品の失敗は通常極めて高い流出率を招く。臨床前研究と/或いは初歩的な臨床試験を通じて成功したにもかかわらず、臨床試験後期段階の候補製品は必要な安全性と有効性を示すことができないかもしれない。同様に、早期の比較的小規模な臨床試験は大規模な肝心な臨床試験の最終的な安全性或いは有効性を予測できない可能性がある。特に,われわれが計画しているこれらの試験設計の早期臨床試験では,患者数が少なく,これらの試験の結果がその後の臨床試験の結果に対してそれほど予測性を持たない可能性がある。生物製薬業界の多くの会社は高級臨床試験において重大な挫折を受け、原因は治療効果の不足、治療効果の持続性不足或いは受け入れられない安全性問題であり、早期の試験で良好な結果を得たにもかかわらず。臨床開始前研究と臨床試験の候補製品の多くは適切な製品として承認されたことがない。私たちの臨床開発において、このような性質の挫折はすべて私たちの業務、財務状況、運営結果と将来性に実質的な損害を与える可能性がある
また,我々の候補製品の第1回臨床試験はオープンラベル研究であり,患者や研究者は患者が研究製品候補を受け入れているかどうか,あるいは既存の承認薬やプラセボを受け入れているかどうかを知っていると予想される。これは我々が行っている第1回ヒト臨床試験であり,現在臨床開発が予定されている他の候補製品の第1回ヒト臨床試験でもある。開放ラベル臨床試験は通常研究製品のみをテストし、時々異なる用量レベルでテストを行う。開放ラベル臨床試験は様々な制限を受けており,これらの制限は任意の治療効果を誇張する可能性があり,開放ラベル臨床試験中の患者が治療を受ける際に知られているからである。また,オープンラベル臨床試験は,“調査者偏見”の影響を受ける可能性があり,すなわち臨床試験の生理結果を評価·審査する人は,どの患者が治療を受けているかを知り,その知識を知っている場合に治療群の情報をより有利に解釈することが可能である
私たちが可能またはすでに行った任意の臨床前研究または臨床試験は、規制部門の許可を得て、私たちの候補製品を市場に発売するために必要な安全性と有効性を証明しないかもしれない。もし私たちが行っているあるいは未来の臨床前研究或いは臨床試験の結果が私たちの候補製品の安全性と有効性に対して定説がなければ、もし目標降格の証拠が臨床治療効果と関係がなければ、もし私たちが統計と臨床意義を持つ臨床終点に達していなければ、あるいは私たちの候補製品に安全問題がある場合、私たちはこれらの候補製品の発売承認を阻止或いは延期する可能性がある。ある場合、多くの要素のため、同一候補製品の異なる臨床前研究と臨床試験の安全性或いは有効性結果は著しい差が存在する可能性があり、方案中に規定された試験プログラムの変化、患者群の大きさとタイプの差異、臨床試験方案の変化と遵守及び臨床試験参加者の退学率を含む
我々はすでにいくつかの候補製品の臨床試験を開始しており、その中のいくつかはまだ行われているが、私たちはまだ他の候補製品の臨床試験を始めていない。すべての薬物の場合と同様に、私たちの候補製品を使用することは、化学ベースの毒性を含む標的毒性、標的外毒性、または他の薬物毒性機序に関連する副作用を生じる可能性がある。われわれの臨床試験結果は,このような性質の副作用の重症度と普遍性を示す可能性があり,受け入れられない。許容できない毒性レベルが観察された場合、または私たちの候補製品が他の予期しない特徴を有する場合、それらの開発を放棄し、用量レベルおよび/または用量スケジュールまたは他の方法で、または開発をより狭い用途またはサブ集団に制限する必要があるかもしれないが、不良副作用または他の特徴は、リスク-収益の観点からそれほど一般的ではなく、それほど深刻ではなく、またはより容易に受け入れられる。例えば,観察された安全信号により,我々は以前CFT 7455が行っている1/2期臨床試験で用量計画を修正し,この臨床試験を進めてきたからである。さらに、許容できない副作用レベルが観察された場合、または他の類似のタンパク質分解剤を標的とする開発者が、その候補薬剤の副作用が深刻または一般的に受け入れられないことを発見した場合、私たちの試験は一時停止または終了する可能性があり、FDAまたは同様の外国の規制機関は、私たちの任意またはすべての標的適応の候補製品の開発を停止または拒否するように命令することができる。薬物に関連する副作用は、患者が進行中の試験を完了する能力を募集したり、取り入れたり、潜在的な製品責任クレームを引き起こす可能性もある。多くの化合物は最初は癌治療の早期テストで希望を示したが,その後副作用が生じることが発見され,この化合物のさらなる発展を阻止した。このようなどんな状況でも、私たちの業務、財政状況、そして見通しを深刻に損なうかもしれない
著者らの臨床試験で公表或いは公表された仮頂線と初歩データによる結論と分析は更に多くの患者データの獲得に従って変化する可能性がある。また、私たちが提供するすべての一時的なデータは、最終的なデータに大きな変化をもたらす可能性がある監査とチェック手続きを遵守しなければなりません。
時々、私たちは臨床試験の中期または最高の予備データを発表するかもしれない。我々が行う可能性のある臨床試験の中期データは、患者登録の継続とより多くの患者データの獲得に伴い、1つまたは複数の臨床結果が実質的に変化する可能性があるというリスクに直面している。さらに、初期または最も重要なデータは、監査および確認手続きを受ける必要があり、これは、最終データと、私たちが以前に発表または公表した予備データとに大きな差がある可能性がある。したがって、最終データを得る前に、中期と予備データを慎重に見なければならない。初期または中期データと最終データとの間の不利な差は、私たちの名声、業務、財務状況、運営結果、および将来性を深刻に損なう可能性があります。
薬物開発は長くて高価な過程であり、結果は不確実だ。私たちは私たちの候補製品の開発と商業化を完成したり、最終的に完成できなかったりする過程で意外なコストや遅延が生じるかもしれません
いくつかの候補製品の臨床試験を開始していますが、そのうちの1つは閉鎖を選択し、パートナーのBetta Pharmaを通じてCFT 8919の臨床試験が開始されると予想されていますが、私たちの他のすべての候補製品は現在も発見段階にあり、すべての候補製品が失敗するリスクは依然として高いです。私たちの候補製品がいつ、人体で有効または安全を証明するか、あるいは発売許可を得るかどうかを予測することはできません。規制部門の許可を得て任意の候補製品を販売する前に、私たちの候補製品の人体における安全性と有効性を証明するために、広範な臨床試験を行わなければならない
臨床試験費用が高く、設計と実施が困難であり、登録と完成まで数年かかる可能性があり、時間と結果を確定しない。1つまたは複数の臨床試験の失敗は、そのプロセスの任意の段階で発生する可能性がある。臨床試験中または臨床試験の結果として、私たちは多くの予見できない事件に遭遇する可能性があり、これは、上場許可を得たり、私たちの候補製品を商業化する能力を延期または阻止する可能性があります
•遅延或いは監督機関と臨床試験設計について合意できなかったか、或いは受け入れ可能な臨床前結果を産生して人体臨床試験に入ることができなかった
•候補製品または臨床試験を行うために必要な他の材料の供給または品質が不足しているか、または不十分である可能性があり、候補製品を試験、検証、製造、および臨床現場への臨床現場への送達のために、私たちと契約していくつかの機能を実行する第三者の遅延を含む
•遅延または予期される試験場所またはCROと許容可能な臨床試験契約または臨床試験方案と合意することができなかった
•規制機関または機関審査委員会またはIRBsは、私たちまたは私たちの研究者が予想される試験場所で臨床試験を開始したり、臨床試験を行ったりすることを許可できなかった
•臨床試験の設計とまだよく研究されていない疾病のための終点を選択することの困難、及び疾病の自然歴史と病気経過についてあまり知られていない
•いくつかの臨床終点を選択するには、これらの終点は比較的に長い臨床観察または結果データ分析を必要とする可能性がある
•私たちの候補製品の臨床試験に必要な患者の数は私たちが予想していたより多いかもしれません。これらの臨床試験の登録人数は私たちの予想より遅いかもしれません。参加者がこれらの臨床試験から退出する割合は私たちの予想よりも高いかもしれません。あるいは治療後のフォローができないか、あるいは適切な患者を募集できないかもしれません
•私たちの候補製品は副作用や他の予期しない特徴がある可能性があり、私たちまたは私たちの研究者、監督機関、またはIRBsが私たちの臨床試験を一時停止または終了させる可能性があります
•様々な理由で、参加者が受け入れられない健康リスクにさらされていることを発見することを含む、候補製品の臨床試験を一時停止または終了しなければならないかもしれない
•私たちと契約を締結した第三者は、規制要件を適時に遵守したり、私たちに対する契約義務を履行したり、根本的に遵守しなかったりする可能性がある
•規制機関またはIRBsは、規制要件に適合していない、または受け入れられない安全リスクを含む、様々な理由で臨床試験を一時停止または終了することを私たちまたは私たちの研究者に要求する
•私たちの候補製品の臨床試験は否定的または不確定な結果をもたらす可能性があり、私たちは決定したり、監督機関が追加の臨床試験を要求するかもしれません投与量レベルおよび/または用量スケジュールや他の態様のような開発計画を修正し製品開発計画を放棄したり
•私たちの候補製品の臨床試験コストは予想以上に高いかもしれません
•人員不足、著者らが臨床試験を行う機関が訓練されているか、経験豊富な臨床研究アシスタント或いは医療従事者が不足しているか、あるいは現場請負と活性化に参加するこれらの機関は十分な支持者が不足していることを含むが、遅延を招く可能性があり、あるいは私たちが適時に効果的に臨床試験を行うことに他の挑戦を引き起こす可能性がある
•深刻な不良事件、ある種類の候補製品に対する懸念或いは著者らの臨床試験操作、試験場所或いは製造施設の検査のため、監督管理機関は強制的に臨床一時停止を実施した
•候補製品に関連する深刻な有害事象の発生は、その潜在的な利点を超えていると考えられている
•全世界の衛生流行病による中断、例えば最近の新冠肺炎の大流行は、私たちがこのような困難に遭遇する可能性を増加させ、あるいは私たちが起動、登録、進行或いは完成計画中の臨床試験に他の遅延を招く可能性がある
アメリカあるいは他の管轄区域の絶えず変化する監督管理政策のため、私たちは臨床開発プロジェクトにおいても挑戦に直面する可能性がある。例えば,2021年にFDAの腫瘍学卓越センターが最適化プロジェクトを開始し,腫瘍学薬物開発における用量選択を改革するイニシアティブであり,このイニシアティブはまだ実施中である。FDAが私たちの候補製品のために選択された1つまたは複数の用量が治療効果および安全性および耐性を最大限に向上させるかどうかを十分に決定していない場合、FDAは、追加の臨床試験を行うことを要求するか、または追加の用量関連情報を生成することを要求する可能性があり、これは、私たちの臨床開発計画の費用を著しく延期および/または増加させる可能性がある。
もし私たちが現在考慮している候補製品に対して追加の臨床試験または他の試験を行うことを要求された場合、もし私たちが候補製品の臨床試験または他の試験を成功的に登録または完了できなければ、これらの試験または試験の結果が陽性でないか、または軽微な陽性である場合、または候補製品に関連する安全問題が存在する場合、私たちは可能である
•私たちの候補製品の市場承認を遅延させる(もしあれば)
•承認された適応や患者集団は期待や期待ほど広くない
•重大な使用または流通制限または安全警告を含むラベルの承認を得ること;
•上場承認を支援するための追加的な臨床試験が求められている
•規制部門に承認を撤回または一時停止させるか、またはリスク評価および緩和戦略(REMS)の形態で候補製品の流通に制限を加える
•追加の上場後のテスト要求または製品管理方法の変更を受ける;または
•市場の承認を得た後、私たちの製品を市場から撤去します
もし私たちが臨床前研究或いは臨床試験或いは発売承認を得る上で遅延に遭遇すれば、私たちの製品開発コストも増加する。いくつかの候補製品の臨床試験を開始し,パートナーのBetta Pharmaを介してCFT 8919の臨床試験を開始する予定であるが,われわれの他の任意の臨床試験が計画通りに開始されるかどうか,再構成が必要かどうか,計画通りに完了するかどうか,あるいは全く知られていない。重大な臨床試験遅延は、候補製品を商業化する独占的な権利を持つ私たちの任意の期限を短縮するか、または私たちの競争相手が私たちの前に製品を市場に出すことを可能にし、候補製品を商業化することに成功する能力を弱める可能性があり、これは私たちの業務、運営結果、財務状況、将来性を損なう可能性がある
さらに、癌治療は、1線、2線、または3線として記述されることがある。FDAは通常,最初に三線またはそれ以降の新たな腫瘍学的療法のみを承認するが,これは2つ以上の他の治療に失敗した後に使用されることを意味する。癌が十分に早く発見されたとき、第一線の治療は、通常、全身抗癌治療(例えば、化学療法)、手術、放射線治療、またはこれらの療法の組み合わせであり、治癒ではなく、癌を治癒または延長するのに十分である場合がある。以前の治療が無効であることが証明された場合,患者に対して二線と三線治療を行う。私たちが行っている早期臨床試験は、1つ以上の以前の治療を受けた患者に対して、最初に規制機関に私たちの主要な候補製品を二線または二線として承認することを求める予定です
三線治療です。その後、十分有益であることが証明された製品については、もしあれば、一次治療としての承認を求めることが予想されるが、私たちが開発した任意の候補製品は、二線治療または三線治療のために承認されても、一次治療のために承認されない可能性があり、および/または一次治療のいずれかの承認を求める前に、追加の臨床試験を行わなければならないかもしれない
ターゲットを絞ったタンパク質分解は新しい方法であり、既存と新興のバイオテクノロジーと製薬会社の大きな興味を引き続けている。したがって、私たちは、私たちよりも他の人が同じ適応および/または患者集団に対する製品を発見、開発、または商業化することに成功する可能性がある激しい競争に直面している
生物技術と製薬業界の特徴は技術進歩が迅速で、競争が激しく、そして独自製品を高度に重視していることである。我々は、タンパク質分解、抗体治療、阻害性核酸、免疫治療、遺伝子編集または遺伝子治療開発プラットフォーム、および小分子阻害剤のようなより伝統的な治療方式に専念する会社を使用して、第三者からの競争に直面し、引き続き直面するであろう。私たちが直面し、直面する競争は多種の源から来る可能性があり、主要な製薬、専門製薬と生物技術会社、学術機関、政府機関及び公共と個人研究機関を含む
標的タンパク質分解は新興の治療方式であり、患者の結果を改善する治療方法を提供することが可能である。そのため,一部のバイオテクノロジーや製薬会社は分解に基づく療法の開発に努めており,この分野に参入する企業数は増加し続けている。Arvinas,Inc.,Astellas Pharma Inc.,BioTheryX,Inc.,Captor Treateutics,Inc.,Cullgen Inc.,Foghorn Treateutics,Inc.,Frontier Medicines Corporation,Glubio Treateutics,Inc.,Kymera Treateutics,Inc.,Monte Rosa Treateutics,Inc.,Nurix Treeutics,Inc.,Orum Treateutics,Inora Tretuticc.,Inora Treateutics,Inc.,Nurix Treeutics,InTreTreutics,InorePhars,InLivals,InLifaris社がいくつか開発されていることが知られている。会社(バイエル株式会社の子会社)。また、いくつかの大手製薬会社と学術機関は、安進、アスリーカン、百時美施貴宝会社(及びその子会社Celgene Corporation)、グラクソ·スミスクライン、遺伝子テーク社、ノワール国際会社を含むこの分野での投資と研究を開示している。他のタンパク質分解療法からの競争に加えて、我々が開発したどの製品も、小分子、抗体、T細胞または遺伝子療法のような他のタイプの療法からの競争に直面する可能性がある。
私たちの多くの既存または潜在的な競争相手は、単独またはパートナーと協力しても、研究開発、製造、臨床前テスト、臨床試験を行い、監督管理許可とマーケティング承認を得た製品の面で、私たちよりも多くの財務資源と専門知識を持っている。これらの競争相手はまた合格した科学と管理人員を募集と維持し、臨床試験場と臨床試験患者登録を確立し、著者らの候補製品と相補或いは必要な技術を獲得する方面で著者らと競争を展開している。製薬とバイオテクノロジー業界の合併と買収は、より多くの資源が私たちの少ない競争相手に集中する可能性があり、これらの競争相手の規模は競争しにくいかもしれない。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。もし私たちの競争相手が私たちが開発する可能性のある任意の候補製品よりも安全で、より効果的で、副作用が少なく、より便利で、より安い製品を開発し、商業化すれば、私たちのビジネス機会は減少または消失するかもしれない。私たちの競争相手も私たちよりも早くFDAや他の規制機関のその候補製品の承認を得ることができ、これは私たちの競争相手が私たちが市場に入る前に強力な市場地位を確立することをもたらすかもしれない。また、多くの場合、私たちの競争能力は、後発薬の使用を奨励することを求める保険会社または他の第三者支払者の影響を受ける可能性がある。現在,我々が求めているいくつかの適応に対する模倣薬が市販されており,今後数年でより多くの模倣薬が発売されることが予想される。もし私たちの候補製品が承認されたら、私たちはそれらの価格が競争相手の模造薬より著しく高いと予想する
私たちは純営業損失の繰越と研究開発税収の繰越を免除する能力が限られている可能性があります
2023年12月31日現在、2.149億ドルの連邦純運営損失が繰り越し、3.153億ドルの米国州純運営損失が繰り越しており、その一部は2043年までの異なる期日で満期となっている。現行法によると、2017年以降に開始された納税年度に生じる連邦純営業損失(あれば)が満期にならず、無期限繰越になる可能性がありますが、2020年12月31日以降に開始される納税年度にこのような連邦純営業損失を差し引く能力は、純営業損失繰越や会社調整後の課税所得額の80%のうち小さいものに限られます(1986年改正“国税法”第382節)。各州がどの程度連邦税法を遵守するかはまだ確定されていない。また、州レベルでは、一定期間使用を一時停止したり、他の方法で純営業損失を制限したりする可能性があり、これは州の課税税を加速または永久的に増加させる可能性がある
2023年12月31日まで、私たちはアメリカ連邦と州の研究開発税収控除はそれぞれ1260万ドルと520万ドルであり、これらの相殺は2043年まで続く。これらの税金の繰越免除は満期になる可能性があり、未使用で、私たちの未来の所得税債務を相殺することはできません
また、改正後の1986年の“国内税法”第382条及び383条、又は同法規及び州法の対応規定によると、1社が3年以内に持分所有権価値が50%を超える“所有権変更”を経験した場合、当該会社は変更前純営業損失繰越及び他の変更前税収属性を用いて変更後の収入又は税収を相殺する能力が制限される可能性がある。2021年,成立から2020年12月31日までの所有権変更研究を完了し,規則382節で定義した所有権変更を経験したと結論した.しかし、有限繰越や満期未使用の繰越には純営業損失はない。私たちはまだこの研究を更新していません。2022年から2023年の間に所有権変更が発生したかどうかを評価します。私たちは他のまだ確定されていない所有権の変化を経験しているかもしれません。これは私たちの純営業損失と税収控除の満期を招いて、それから使用することができて、私たちはその後の株式所有権の変化を経験するかもしれません。その中のいくつかは私たちがコントロールできません。したがって、もし私たちが純課税所得額を稼いで、所有権が変化したと決定すれば、私たちは歴史的な純営業損失と税収の繰越免除能力を利用して実質的に制限され、これは私たちの未来の納税義務を効果的に増加させることで、私たちの未来の経営業績を損なうことになる
もし私たちが開発する可能性のある任意の候補製品の開発中に深刻な有害事象、副作用または意外な特徴または結果が発見された場合、私たちはこれらの候補製品のさらなる臨床開発を修正、放棄、または制限する必要があるかもしれない
いくつかの候補製品の臨床試験を開始し、パートナーのBetta Pharmaを通じてCFT 8919の臨床試験を開始することが予想されていますが、私たちの他のすべての候補製品は現在まだ発見段階にあり、これはまだ私たちの他の候補製品に対して人体臨床試験を行っていないことを意味します。私たちは私たちが開発する可能性のある任意の候補製品がいつ、あるいは人間にとって安全であることが証明されるか予測できない。我々の魚雷プラットフォームを介して開発された任意の候補製品が副作用を引き起こさないことは保証されず、これらの副作用は臨床前または臨床開発過程のいつでも出現する可能性がある
我々の魚雷プラットフォームによって開発された候補製品または任意のタンパク質分解製品候補製品の潜在的リスクは、健康なタンパク質または分解を標的としないタンパク質が分解されるか、または標的タンパク質自体の分解が有害事象、副作用または意外な特徴または結果をもたらす可能性があることである。我々の魚雷プラットフォームにより開発された候補品を用いて治療を行った後,不良事象を遅延させる潜在的なリスクもある。
私たちが開発した任意の候補製品が深刻な有害事象または副作用に関連している場合、または他の意外な特徴または結果を有する場合、私たちは、用量レベルおよび/または用量スケジュールまたは他の方法で、または開発を有害事象、副作用または他の特徴があまり一般的ではない、あまり深刻ではない、またはリスク効果の観点からより受け入れやすい特定の用途またはサブ集団に選択または放棄する必要があるかもしれない。このような事件の発生は、私たちの業務、財務状況、運営結果、および将来性に悪影響を及ぼすだろう。最初に癌や他の疾患を治療する早期テストで希望を示した候補製品の多くは、後に副作用が生じることが発見され、候補製品のさらなる臨床開発を阻害したり、市場での競争力を制限したりする。例えば,単一のBRAF阻害剤は角化皮膚腫と呼ばれる二次性悪性腫瘍を引き起こすことができ,BRAFが阻害剤と結合した際に矛盾して活性化することによる皮膚癌である
もし私たちが臨床試験で患者を募集する時に遅延や困難に遭遇した場合、私たちは申請を提出し、必要な上場承認を得る時間が遅れる可能性があり、あるいは上場承認を得ることを完全に阻止される可能性がある
FDAや米国以外の類似規制機関の要求に応じて、十分な数の合格患者を見つけて募集することができなければ、私たちの候補製品のために臨床試験を開始することができないかもしれない。著者らはすでに3種類の候補製品CFT 7455、CFT 8634とCFT 1946をそれぞれ2021年6月、2022年5月と2022年12月に初の人体臨床試験を行い、CFT 7455とCFT 1946の臨床試験は現在進行中であり、CFT 8919で行う予定である。進行·計画中の各臨床試験のために十分な数の患者を募集できると信じているが,患者を募集して試験に参加することがどの程度困難であるかを予測することはできず,その中にはまれな適応である試験もある。該当する患者を識別して募集し、候補製品の臨床試験に参加する能力は限られていることが証明されるかもしれません。あるいはこれらの試験を募集する速度は私たちが予想していたよりも遅いかもしれません。また,我々の競争相手の中には候補製品の臨床試験を行っており,これらの候補製品は我々の候補製品と同様の適応を持っているため,われわれの臨床試験に参加する資格のある患者が参加を選択する可能性がある
私たちの競争相手の候補製品に対して臨床試験を行います。患者登録が臨床試験に参加することも他の要素の影響を受けている
•調査中の病気の重症度は
•試験に関する資格基準
•臨床試験で提供される候補製品の既知のリスクおよび利益;
•臨床試験に参加するための努力を促進し
•医者の患者の回診のやり方
•臨床試験場所に適切で十分なスタッフがいるかどうか
•臨床試験レジメンに必要なプログラムの範囲および侵襲性が患者にもたらす負担のため、いくつかのプログラムは不便および/または不快である可能性がある
•治療中および治療後に患者の能力を十分に監視する;
•潜在的患者のための臨床試験場所の距離および利用可能性を提供する
•全世界の衛生流行病の影響、例えば最近の新冠肺炎の大流行は、潜在登録の緩和或いは臨床試験の合格患者数の減少、或いは患者が臨床試験現場に戻って必要なモニタリング、プログラム或いは後続の仕事を行う能力を妨害することを含む臨床試験の進行に影響を与える可能性がある
私たちは十分な数の患者を臨床試験に参加することができない、あるいは私たちは適時にそうすることができなくて、重大な遅延を招き、1つ以上の臨床試験を完全に放棄することを要求するかもしれない。私たちの臨床試験の登録遅延は、私たちの候補製品の開発コストを増加させる可能性もあり、これはわが社の価値を低下させ、追加融資を受ける能力を制限する
私たちは、より利益的または成功可能性の高い候補製品または適応を利用することなく、特定の候補製品または適応を追求するために限られたリソースを費やす可能性がある
私たちの財務と管理資源が限られているため、私たちは私たちが確定した特定の適応の研究プロジェクトと製品に重点を置いています。したがって、私たちは他の候補製品を探す機会を放棄または延期するか、または後により大きな商業潜在力を有することが証明された他の兆候を探す機会を放棄または延期するかもしれない。私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。私たちの現在と未来の研究開発計画および特定の適応候補製品への支出はいかなる商業的に実行可能な製品も発生しないかもしれない。もし私たちが特定の候補製品の商業的潜在力や目標市場を正確に評価しなければ、私たちは協力、許可、または他の印税手配によって候補製品に価値のある権利を放棄するかもしれないが、この場合、候補製品の独占開発権と商業化権利を維持することは私たちに有利だ
私たちまたは私たちのパートナーは私たちの候補製品を他の薬物と組み合わせて開発するかもしれない。FDAや米国以外の同様の規制機関がこれらの他の薬剤を承認しない場合、またはこれらの他の薬剤の承認を撤回するか、または私たちの候補製品と組み合わせて評価する薬剤を選択した場合、安全性、有効性、製造、または供給の問題が発生した場合、私たちは候補製品の承認を得ることができないかもしれないし、市場に押し上げることができないかもしれない
著者らの多くの候補製品の研究設計によると、著者らの最初のヒト1/2段階臨床試験の用量増加部分から推薦投与量を決定すると、著者らは通常この臨床試験の一部を1種以上の他の薬物と組み合わせて行う予定である。私たちは現在承認されている薬物の開発や市場承認を得ていませんし、私たちの候補製品と組み合わせて研究する可能性のある薬剤も製造または販売していません。FDAまたは米国以外の同様の規制機関が、私たちの候補製品と組み合わせて提供しようとしている1つ以上の薬物の承認を撤回した場合、私たちの候補製品を撤回された薬剤と組み合わせて販売することはできないだろう
これらの薬剤のいずれかに安全性や有効性の問題が生じた場合、重大な規制遅延に遭遇する可能性があり、FDAまたは米国以外の同様の規制機関は、いくつかの臨床試験の再設計または終了を要求する可能性がある。我々が使用している薬物が候補製品選択の適応のための看護基準に置き換えられれば,FDAや米国以外の類似規制機関は追加の臨床試験を要求する可能性がある。また、製造やその他の問題が私たちの候補製品と組み合わせて使用することを決定した薬物供給が不足すれば、現在のスケジュールで私たちの候補製品の臨床開発を完成させることができず、甚だしきに至っては完成できない可能性がある
私たちの候補製品が他の既存の薬剤と併用するために発売承認または商業化されていても、FDAまたは米国以外の同様の規制機関が、私たちの候補製品と併用した薬物の承認を取り消す可能性があるか、またはこれらの既存の薬剤が安全性、有効性、製造、または供給問題に直面する可能性があるリスクに直面する
併用療法は通常癌の治療に用いられており,我々が我々の任意の他の候補製品を開発し,他の薬物や癌以外の適応と併用することを選択すれば,類似したリスクに直面する。これは私たち自身の製品が市場から撤退されたり、商業的にあまり成功しないことをもたらすかもしれない
私たちは他の潜在的な候補製品を発見したり発見したりすることに成功しないかもしれない
著者らの現在の臨床段階計画は腫瘍学目標に集中しているが、著者らの戦略の重要な要素は著者らの魚雷プラットフォームを用いて広範な目標と新しい治療領域に対する候補製品を開発することであり、例えば神経退行性変化、老年病と伝染病である。我々が行っている治療発見活動は,癌や他の疾患の治療に有用な候補製品を決定することに成功しない可能性がある。我々の研究計画は当初、潜在的な候補製品の決定に希望を示す可能性があるが、様々な原因で臨床開発のための候補製品を生成できなかった
•さらなる研究の後、潜在的な候補製品は、有害な副作用または他の特徴を有することが証明される可能性があり、上場承認または市場許容を得る薬剤である可能性が低いことを示している
•潜在的な候補製品は、その標的疾患の治療において無効である可能性がある;または
•潜在候補製品の目標適応の市場規模が時間の経過とともに縮小する可能性があるのは,看護基準の向上により,これ以上発展する必要がないためである
新製品候補製品を確定する研究プロジェクトには大量の技術、財力と人的資源が必要である。私たちは私たちの努力と資源を最終的に成功しないことが証明された潜在的な製品候補に集中することを選択するかもしれない。臨床前と臨床開発に適した候補製品を決定できなければ、将来的に製品販売から収入を得ることができなくなり、私たちの財務状況に重大な損害を与え、株価に悪影響を与える可能性がある
もし私たちが発表し、予想された時間枠内で予想された開発目標を達成できなければ、私たちの製品の商業化は延期される可能性があり、したがって、私たちの株価は下落する可能性がある
時々,様々な科学,臨床,法規,その他の製品開発目標の期待達成時間を見積もることができ,マイルストーンと呼ぶことがある。これらのマイルストーンは臨床前研究と臨床試験の開始或いは完成、及び監督文書の提出を含む可能性がある。私たちは時々いくつかのマイルストーンの予想時間を公開するかもしれない。このようなマイルストーンのすべての現在と未来は無数の仮定に基づいている。私たちの推定と比較して、このようなマイルストーンの実際の時間は大きく異なる可能性があり、場合によっては、原因は私たちの統制を超えている。もし私たちが公開発表されたこれらのマイルストーンに達していない、あるいは全く達成されていなければ、私たちの収入は予想を下回るかもしれないし、私たちの製品の商業化は延期されるかもしれないし、永遠に実現されないかもしれないので、私たちの株価は下落するかもしれない
第三者に依存するリスク
第三者による将来の臨床試験を行う予定であり,締め切りまでにこのような試験を完了できないことを含めて,これらの第三者の表現は満足できない可能性がある
われわれは現在CROによる臨床試験を行っているが,現在われわれのどの候補製品に対しても独立した臨床試験を行う予定はないからである。また,第三者研究サイトと契約を結び,我々の臨床試験を行わなければならない。私たちがベータ医薬に依存して大中国でCFT 8919を効率的に開発しているように、私たちは将来的にも他の第三者パートナーに依存して特定の期間内に異なる地域で私たちの1つまたは複数の製品を開発することにも依存するかもしれない。これらのCRO、サイト、他の第三者とのプロトコルは、第三者が履行されていないことを含む様々な理由で終了する可能性があります。代替計画を達成する必要があれば,あるいは行っている臨床試験のためにCROを交換する必要があれば,われわれが過去に行ったように,臨床開発活動に遅延が生じる可能性がある
我々の研究や開発活動へのCROへの依存は,これらの活動に対する我々の制御を減少させるが,これらの活動をどのように実行するかの責任は免除されない.例えば,われわれの個々の臨床試験が適用されるINDにおける一般的な研究計画や案に沿って行われることを確保していきたい。さらに、FDAは、データおよび報告の結果が信頼性および正確であることを保証し、試験参加者の権利、安全、および機密性を保護するために、臨床試験結果を行い、記録し、報告する基準を遵守することを要求し、一般にGCPと呼ばれる。GCP適合性は以下の項目の発起人だけでなく
臨床研究に限らず,臨床研究に関与するCROやサイトを含む第三者も含まれている。同様に、世界各地の他の監督機関も類似の基準を遵守することを要求し、これらの基準はCROと臨床試験場所などの臨床試験スポンサーと他の第三者にも適用される。
さらに、これらのCROまたはサイトは、他のエンティティと関係がある可能性があり、そのうちのいくつかは、私たちの同業者または競合相手である可能性がある。私たちと協力しているCROやサイトが規制要件や規定された規程に従ってその契約責任を成功裏に履行できない場合、予想される期限内に私たちの臨床試験を完了または行うことができない場合、私たちは候補製品のマーケティング承認を得ることができないか、遅延する可能性があり、候補製品の商業化に成功する努力を得ることができないか、遅延する可能性があります
薬品生産は複雑な仕事であり、各種の原因で製品の遅延或いは損失が発生しやすい。我々は第三者と契約を結び,臨床前試験や臨床試験のための候補製品を生産し,商業化のために継続していく予定である。このような第三者への依存は、私たちが十分な数の候補製品や製品を持っていないリスクを増加させるか、または許容可能なコストまたは品質で、私たちが望むまたは要求される数のリスクを適切な時間に得ることができないであろう。これは、私たちの開発や商業化努力を延期、阻止、または損害する可能性がある
私たちは所有したり経営したりしないし、現在は製造施設を設立する計画もない。私たちはCMOに依存して薬物物質と完成品を提供し続けると予想される。このような第三者への依存は、私たちが十分な数の候補製品や製品を持っていないリスクを増加させるか、または許容可能なコストまたは品質で所望または要求される数量のリスクを得ることができない可能性があり、これは、承認前検査または製造場所の検査を必要とすることを含む、私たちの開発または商業化努力を延期、阻止または損害する可能性があり、FDAは、任意の理由で審査期間内にこれらの要求された検査を完了することができない。
私たちはCMOと合意できないかもしれないし、受け入れられる条件でそうすることもできないかもしれない。CMOと合意できても、第三者メーカーに依存することは追加的なリスクをもたらす
•規制、コンプライアンス、品質保証、製造成功の面で第三者に依存する
•第三者CMOは製造協定に違反する可能性がある
•CMOは、規制問題、財務破綻、適用法律またはその他の理由により、私たちに必要なサービスの提供を一時的または永久的に停止するか、または業務を完全に閉鎖するリスクがある
•私たちのビジネス秘密およびノウハウを含む私たちの固有情報を盗用する可能性があります
•サードパーティは、コストが高いとき、または私たちに不便なときにプロトコルを終了または更新しないか、またはCMOが必要なときに製造期間を提供することができません
私たちの候補製品については、私たちは限られた技術移転協定しかありません。これらの既存の計画は商業供給には適用されません。私たちは注文を購入した上で多くの重要な材料を得た。したがって、私たちは私たちの候補製品と他の材料に対する長期的な約束計画を持っていない。もし私たちが任意の候補製品の上場承認を受けたり、獲得することが予想される場合、私たちは1つ以上の第三者と商業生産協定を確立または確立した必要があるだろう
第三者製造業者は、現在の良好な製造実践またはcGMP、法規、または米国以外の同様の規制要件を遵守できない可能性がある。我々のいくつかの分子は非常に有効であり,追加の安全データなしに高い職業曝露帯域,すなわちOEBを得るであろう。これらの指定されたOeBは,我々の候補製品製造の一部として講じなければならない抑制や他の予防措置を規定しており,高いOEBラベルを有する分子については,我々の分子を製造する資格のあるCMOの数を制限している。私たちまたは私たちのCMOは、私たちのCMOが私たちの効率的な材料および関連する安全協定を使用する能力を含む適用された法規を遵守することができず、臨床封印、罰金、禁止、民事処罰、遅延、一時停止または許可の撤回、許可証の取り消し、候補製品または製品の差し押さえまたはリコール、運営制限および刑事起訴を含む制裁を実施する可能性があり、いずれも私たちの製品供給に重大な悪影響を及ぼす可能性がある。
私たちの候補製品と私たちが開発する可能性のあるどの製品も他の候補製品や製品と製造施設を競争する可能性があります。したがって、私たちはこのような施設を優先的に使用することができないかもしれないし、全く使用できないかもしれない。CGMP法規下で運営されるメーカー数は限られており,我々のために生産できるメーカー数は限られており,特に場合によっては我々の化合物の効力やOEBを考慮している
私たちの既存または未来のメーカーのどんな業績失敗や業績遅延も、臨床開発またはマーケティング許可を延期する可能性があります。例えば、過去に、我々と協力したCMOの一人は、我々のCFT 7455候補製品の生産運転中の製造ステップにおいて機械的問題が発生した。この問題は
もし私たちが最終的にCFT 7455 INDの提出時間を延期しなければ、将来私たちは製造問題に直面するかもしれません。これは私たちの候補製品の開発に実質的な影響を与え、このような事件の発生は私たちがコントロールできることではありません。私たちは現在薬物や薬物製品の余分な供給や第二の供給源のための手配をしていない。もし私たちの既存のCMOが約束通りに義務を履行できなければ、私たちは彼らを交換しなければならないかもしれない。候補製品の一部または全部を生産できる潜在的な代替サプライヤーをいくつか決定しましたが、代替メーカーを選択して同定する際に、サプライヤーの交換は著しい追加コストと私たちの運営遅延を招く可能性があり、私たちは私たちが選択できるサプライヤーの制限、特により高いOEB標識を有する化合物、または代替メーカーが受け入れられる条項と合意できないかもしれません
私たちの現在と未来の他人が私たちの候補製品や製品を生産することへの依存は、私たちの将来の利益率と、マーケティングの承認をタイムリーかつ競争力的に獲得する製品の商業化能力に悪影響を及ぼす可能性があると予想されています
また、著者らは現在単一源供給業者が著者らの臨床前と臨床試験に供給するサプライチェーンの一部に依存している。もし私たちの現在或いは未来の供給者が、原材料、薬物物質、薬品にかかわらず、著者らの臨床前研究と臨床試験に十分な材料を提供することができなければ、私たちは新しいサプライヤー或いはメーカーを探し、鑑定する時に開発仕事の遅延に遭遇する可能性がある
私たちが依存する第三者製造業者は、彼ら自身の独自のプロセスを私たちの候補製品製造プロセスに統合するかもしれない。第三者の独自製造過程に対する私たちの統制と監視は限られている。第三者製造業者がそのプロセスを修正する場合、これらの修正は、追加生産または製造業者の製品損失または故障を必要とすることを含む、私たちの製造に悪影響を及ぼす可能性があり、両方の場合は、私たちの候補製品のコストを著しく増加させ、生産を著しく遅らせる可能性があります
著者らの候補製品が臨床前研究と臨床試験を通じて次第に承認と商業化されることに伴い、著者らは製品開発と製造過程の各方面が絶えず発展し、過程と結果の最適化に努力することを予想している。一部の製品および製造プロセスの変更は、第三者のノウハウの使用に関連する可能性があり、第三者からの許可を得る必要がある可能性があります。さらに、これらのタイプの変更は、私たちの規制申請を修正する必要があるかもしれません。これは、私たちの任意の候補製品に対する修正された製造プロセスの時間枠の使用をさらに遅らせる可能性があります
また、私たちの候補製品を後期臨床試験に推進し、候補製品の潜在的な商業化を計画するにつれて、より多くの薬品および/または薬物供給者を必要または適切に導入する必要があることが決定される可能性があり、これは、私たちの候補製品の製造プロセスを変化させる可能性があり、規制機関により多くの情報を提供することを要求するかもしれない。もし私たちの候補製品に追加のCMOを導入する場合、私たちはまた、分析の比較可能性を証明し、および/または追加の移行研究または試験を行うことを要求される可能性があり、これらはすべて追加の時間と費用を必要とする
私たちは第三者と既存の協力を持っていて、これらの協力の下で、いくつかの候補製品の研究、開発、商業化に従事しています。もしこのような協力のいずれも成功しなければ、私たちはこれらの候補製品の市場潜在力を利用できないかもしれない。しかも、このような協力は私たちの知的財産権に影響を及ぼすかもしれない。
2023年12月31日までに、私たちの研究計画に関する4つの持続的な協力があります
•我々は2015年12月に羅氏と合意した協力協定を締結し、2018年12月にこの合意を改訂し、その後定期的にさらに改訂し、現在2つの目標について協力活動を展開している
•2017年3月にCalicoと合意し、2021年9月に1つのプロジェクトについて延長し、その研究期間は2023年3月に満了した
•我々が2018年12月にBiogenと締結した協力協定は、2020年2月に改訂され、“生物遺伝協定”の想定では、2023年6月の研究期限終了後、指名目標に関するいくつかの研究活動が継続される
•我々は2023年12月にメルク社と協力協定を締結し、1つの初期目標に対して分解物-抗体結合体またはDACを開発し、それを商業化し、メルク社は所定の期間内に最大3つの目標を増加させることを選択することができる。
これらの協力協定によると,我々は通常,パートナーが選択した目標に応じて,我々の魚雷プラットフォームを用いて候補薬物の開発を担当している。また、これらの合意は、私たちのパートナーが
その選定と保留のためのターゲット開発降下器の独占的権利を持つ.したがって、私たちは単独でまたは他のパートナーと関心がある可能性のある目標を追求することを許可されず、その目標はこれらの制限された制約を受ける。
さらに、もし私たちの協力が製品の開発と商業化に成功しなかった場合、または私たちの協力者が私たちとの合意を終了したり、協力を継続しない計画を選択した場合、私たちはその協力の下で、またはその終了した計画について未来の研究資金、マイルストーン、または印税支払いを得ることができないかもしれません。このような状況が発生した場合、私たちはその計画を放棄するか、または自ら計画を推進することを決定するかもしれないが、これは私たちが未来にその計画に追加的な資源を投入する必要があるだろう。さらに、もし私たちのある協力者が便宜上、90日から270日以内に私たちとの合意を終了したり、特定の目標または指定された時間内に解決されていない重大な違約行為に関する合意について、私たちは新しい協力者を引き付けることが難しくなり、私たちの開発計画が延期される可能性があり、あるいは商業および金融界での私たちの見方が悪影響を受ける可能性があるかもしれない。製品開発やマーケティングに関するすべてのリスク この報告書に記載されている承認と商業化は私たちの協力者の活動に適用される。
私たちの協力者はまた、私たちの許可手続きによって生成された知的財産権または固有の権利を正確に取得、維持、強制、または保護することができない場合があり、または私たちの固有の情報を危険にさらす可能性があるか、または私たちの固有の情報を無効にするか、または私たちが潜在的な訴訟に直面するように私たちの固有の情報を使用する可能性がある。例えば、適用される協力協定によると、羅氏、生物遺伝、Calico、およびメルクは、特定の許可プログラムのために特定の知的財産権を実行し、保護する優先的な権利を持っており、私たちの協力者がそうしなければ、私たちはこれらの知的財産権の実行と保護を負担する権利があるかもしれないが、私たちがそうする能力は彼らの行動によって損なわれる可能性がある。さらに、任意の許可されたプログラムが後に回復された場合、私たちがプログラムに関連する任意の知的財産権または他の固有の権利を保護する能力は、プログラムが回復する前に私たちの協力者が提出した知的財産権出願または取られた他のステップの影響を受けるであろう。また、私たちの協力者は、彼らとの協力によって生成された製品の知的財産権を持っているか、または共同で所有している可能性があり、適用された場合、私たちは協力知的財産権を商業化する独占的な権利を持っていません
私たちは将来的に協力や戦略同盟を形成したり、追加の許可手配を追加したりするかもしれないが、私たちはこれらの協力、連合、または許可手配の利点を達成できないかもしれない
2023年5月にBetta PharmaとBetta Pharmaライセンス契約を締結しましたこの合意に基づいて協力していますCFT 8919の大中華区での開発と商業化については,CFT 8919を世界の他の地域で開発·商業化する権利を保持している。同様に、未来には、wEは、戦略的同盟を結成したり、合弁企業または他の協力を作成したり、または第三者と追加の許可合意を達成したりする可能性があり、これは、候補製品および将来開発可能な任意の候補製品の開発および商業化に関する私たちの努力を補完または強化すると信じています。私たちが達成する可能性のある他の協力計画の中で、私たちの可能な協力者は大中型製薬会社とバイオテクノロジー会社を含む。しかし、私たちはこのような性質の協力合意に到達できないかもしれないし、任意の潜在的な新しい協力計画の条項は有利ではないかもしれない
例えば、MMまたは他の適応分野におけるCFT 7455候補製品を推進するために、または特定の地理的領域で候補製品を開発し、商業化することができるように、または我々がCFT 8919およびBetta Pharmaとの協力でやったように、協力計画を達成または達成することを求めることができるかもしれない。また,我々がメルク社との協力協定で行っているように,他社が我々の魚雷プラットフォームにアクセスして利用することで,我々のパートナーが選択した目標に対する薬剤を開発できるように協力合意を求めることができるかもしれない。これらの関係のいずれも、非日常的な費用と他の費用を発生させ、私たちの短期的かつ長期的な支出を増加させ、私たちの既存の株主を希釈したり、私たちの管理と業務を混乱させたりする証券を発行することを要求することができるかもしれない。
また、適切な戦略的パートナーを探す上で激しい競争に直面しており、このような取引の交渉過程は時間がかかり複雑で、コストが高い。また、私たちの候補製品のための戦略的パートナーシップや他の代替手配を構築する努力は成功しないかもしれません。なぜなら、協力努力の開発段階が早すぎると考えられる可能性があるので、第三者は、私たちの候補製品が安全性と有効性を示し、市場の承認を得るために必要な潜在力を持っているとは思わないかもしれません。さらに、我々の既存のパートナーは、私たちの業務の見通し、財務状況、および運営結果に悪影響を及ぼす可能性がある他の開発目標タンパク質分解剤または私たちの候補製品に対する目標または適応に対する会社との買収または協力を決定する可能性がある
したがって、もし私たちが追加的な協力協定と戦略的パートナー関係を締結すれば、私たちの候補製品ができるかもしれません。もし私たちがこれらの取引を私たちの既存の運営や会社文化と組み合わせることに成功できなければ、私たちはこれらの取引の利点を実現できないかもしれません
私たちの候補製品の商業化に関するリスク
たとえ私たちのすべての候補製品が市場の承認を得ても、医師、患者、第三者支払人、医学界の他の人が商業成功に必要な市場受容度を得ることができない可能性がある
もし私たちのすべての候補製品が市場の承認を得たら、それはまだ医者、患者、第三者支払人と医学界の他の人の十分な市場受容度を得ることができないかもしれない。例えば,現在の癌治療法,例えば化学療法や放射線治療は,医学界では成熟しており,医師はこれらの治療法に依存し続けている可能性がある。もし私たちの候補製品が十分な受容度に達していなければ、私たちは製品販売から相当な収入を得ることができないかもしれませんし、利益を上げることができないかもしれません。もし私たちの候補製品が商業販売に使用されることが許可されれば、市場の受け入れ度は多くの要素に依存するだろう
•代替療法と比較した治療効果と潜在的優位性
•特に代替療法と比較して副作用の発生率や重症度は
•私たちは競争力のある価格で私たちの製品を販売する能力と、政府当局が私たちの製品の定価とこれらの強制的な交渉の時間を交渉する能力を要求してくれました
•代替療法と比較して投与の利便性と簡易性
•対象患者群が新たな治療法を試みる意思と,これらの患者を治療する医師がこれらの治療法を処方する意欲
•マーケティング、販売、流通支援の実力
•第三者保険のカバー範囲と適切な精算状況
•他の製品の承認に関連する任意の上場承認の時間;
•患者の権利擁護団体の支援
•私たちの製品が他の薬と一緒に使用されるすべての制限。
会社として、私たちは現在マーケティングや販売組織もなく、製品をマーケティングした経験もありません。もし私たちがマーケティングと販売能力を確立できない場合、あるいは第三者と合意して私たちの候補製品をマーケティングして販売することができなければ、承認されれば、製品収入を生み出すことができないかもしれない
会社として、私たちは現在、販売、マーケティング、あるいは流通能力がなく、製品をマーケティングした経験もありません。私たちは内部マーケティング組織と販売チームを発展させるつもりで、これは大量の資本支出、管理資源と時間を必要とするだろう。私たちは他の製薬とバイオテクノロジー会社と競争して、採用、採用、研修、マーケティングと販売員を維持しなければならないだろう
もし私たちが内部販売、マーケティング、流通能力を確立しないことを決定できない場合、承認されれば、第三者販売、マーケティング、流通パートナーと私たちの製品の販売とマーケティングの合意を求めることになります。しかし、私たちが有利な条件でこれらのタイプの手配を確立または維持できることを保証することができないか、または私たちがそうすることができれば、これらの第三者手配が効果的な販売力やマーケティングおよび流通能力を提供することを保証することはできない。私たちが得たどんな収入もこのような第三者の努力にかかっていて、これは成功しないかもしれない。私たちはこれらの第三者のマーケティングや販売活動に対してほとんどコントロール権がないかもしれません。私たちが製品販売から得た収入は、私たち自身が候補製品を商業化した収入よりも低いかもしれません。私たちは私たちの候補製品の販売とマーケティングを助けるために第三者を探している時も競争に直面しています
米国または海外で任意の製品を商業化するために、内部販売および流通能力を発展させたり、第三者パートナーと関係を構築したり、維持したりすることは保証されない
私たちの候補製品の市場機会は、最初に他の承認を受ける資格のない治療または以前の治療に失敗した患者のために承認されるだけであることが予想されるので、比較的小さい可能性がある。しかも、私たちの目標患者群の流行率の推定は正確ではないかもしれない
我々は癌に対する候補品を開発しているが,癌療法は1線,2線,3線または後続線として記述されることがあるが,FDAは通常最初に特定の用途に対する新しい療法のみを承認する。癌が十分に早く発見された時、第一線の治療は治愈を必要とすることなく、癌を治癒したり、生命を延長するのに十分であることがある。第一線の治療--通常化学療法、抗体薬物、腫瘍標的小分子、免疫治療、ホルモン治療、放射線治療、手術、他の標的治療、またはこれらの治療の組み合わせであることが証明された場合、二次治療が行われる可能性がある。二線療法には通常より多くの
化学療法、放射線治療、抗体薬物、腫瘍標的小分子またはそれらの組み合わせ。三線治療は化学療法、抗体薬物と小分子腫瘍標的治療、より侵襲性のある手術形式と新しい技術を含むことができる。我々は最初に我々の候補製品の承認を求め,多くの場合二線あるいは三線療法として再発や難治性癌患者に利用される予定である。その後、十分な安全かつ有益であることが証明された候補製品(もしあれば)については、二線療法および潜在的な一次療法としての承認を求めることが予想されるが、第二、第三、またはその後の治療経路として承認されても、より早い経路治療のために承認されることは保証されない。また,われわれの製品が早期シリーズ治療で任意の承認を得る前に,追加の臨床試験を行わなければならないかもしれない
我々の目標癌を有する人数、彼らの腫瘍を遺伝子配列決定する可能性のある人数、および特定の治療経路を受けることができ、私たちの候補製品治療から利益を得る可能性があるこれらの癌患者のサブセットの予測は、私たちの合理的な信念と推定に基づいている。これらの推定は様々な源から来ており、科学文献、診療所調査、患者基金会或いは市場研究を含み、不正確或いは時代遅れであることが証明される可能性がある。また,新たな治療法は,我々が狙っている癌の推定発症率や流行率を変える可能性がある。したがって、私たちの候補製品が二線または三線治療のために承認されても、私たちの候補製品を使用して治療を行う資格がある患者の数は予想よりもはるかに少ないかもしれない。また,各腫瘍タイプに対して異なる承認療法があれば,治療医が複数の腫瘍タイプのために承認された製品をどのように開発することが期待されるかを決定するための市場研究は行われていない。
私たちまたはCFT 8919の場合でも、Betta Pharmaは私たちの任意の候補製品のマーケティング承認を得て、私たちの製品は不利な価格設定法規、第三者精算慣行、または医療改革措置の影響を受ける可能性があり、そのいずれも私たちの業務に影響を与えるだろう
新薬の発売審査、定価、カバー範囲と精算を管理する規定は国によって異なる。現在と未来の立法は承認要求を大幅に変更する可能性があり、これは追加のコストに関連し、承認の遅延を招く可能性がある。一部の国は薬品の販売価格が承認されてから発売されることを要求している。多くの国で、定価審査期間はマーケティングまたは製品許可を承認した後から始まる。一部の国で精算或いは定価の承認を得るためには、私たちの候補製品のコスト効果を他の利用可能な治療法と比較する臨床試験を行う必要があるかもしれない。一部の外国市場では、処方薬の定価は初歩的な承認を得た後も、政府の持続的なコントロールを受けている。したがって、私たちまたはCFT 8919の場合、Betta Pharmaはある候補製品の特定の国/地域でのマーケティング承認を得る可能性があるが、その後、価格法規の制約を受け、これらの法規は製品の商業発表を延期し、長い時間があるかもしれないが、これは私たちがその国/地域で製品を販売することによって生じる収入(あれば)に負の影響を与えるだろう。したがって、不利な価格設定制限は、私たちの候補製品が市場承認を得ても、1つ以上の候補製品への投資を回収する能力を阻害する可能性がある
我々の場合、CFT 8919については、Betta Pharmaが任意の候補製品の商業化に成功するかどうかは、政府医療計画、個人健康保険会社、および他の組織がこれらの製品および関連治療に提供する保険範囲および十分な補償にある程度依存するであろう。政府当局と第三者支払者、例えば個人健康保険会社や健康維持組織は、どのような薬剤を支払うかを決定し、精算レベルを確立する
アメリカの医療業界と他の地域の主な傾向の一つはコストコントロールだ。連邦医療保険薬品価格交渉計画は、2022年インフレ低減法案(通常IRAと呼ばれる)の一部としてCMSによって管理されており、もし私たちの製品が選択されて交渉すれば、この計画は私たちの製品に適用される可能性があり、これらの製品が承認されれば、私たちの製品から得られる収入を大幅に減少させることができるかもしれない。政府当局と第三者支払者は,特定の薬物のカバー範囲や精算金額を制限することでコストを抑制しようとしている。政府当局や第三者支払人は、価格に基づいて所定の割引を提供し、医療製品の定価に挑戦することを製薬会社に要求するようになってきている。私たちが商業化しているどの製品も保険や精算を受けることができないかもしれません。あっても、精算レベルは満足できないかもしれません。精算は私たちが市場で承認された任意の候補製品の需要や価格に影響を及ぼすかもしれない。私たちの製品のために保証範囲と適切な精算を獲得して維持することは難しいかもしれません。私たちは、カバー範囲と精算または他の治療法に対する精算レベルを証明するために、高価な薬物経済学的研究を要求されるかもしれない。また、IRAの要求に応じて、承認されれば、連邦医療保険と私たちの候補製品の定価について交渉する必要があるかもしれません。これらの交渉価格は、製品が承認されてから9年後に発効します。カバー範囲や十分な精算がない場合、あるいは精算が限られたレベルに限られている場合、マーケティングの承認を得た任意の候補製品を商業化することに成功しない可能性があります
新たに承認された薬物の保険と補償に大きな遅延がある可能性がある。さらに、カバー範囲は、FDAや米国以外の同様の規制機関がこの薬剤を承認する目的よりも限られている可能性がある。さらに、保険や精算を受ける資格があるということは、すべての場合に薬物の費用を支払うことを意味するのではなく、研究、開発、知的財産権、製造、販売、流通費用を含む私たちのコストを支払うことを意味するわけではない。もし適用されれば、新薬の一時精算レベルも私たちのコストを支払うのに十分ではないかもしれないし、永久的にならないかもしれない。販売率は薬物の使用や臨床環境によって異なる可能性があり,すでに低コスト薬物のために設定された精算レベルに基づいている可能性があり,他のサービスの既存支払いにも組み込まれている可能性がある。医薬品の正味価格は、政府医療計画または個人支払者が要求する強制的な割引またはリベート、および将来、米国価格よりも低い価格で販売される可能性のある国から医薬品を輸入することを制限する法律の緩和によって低下する可能性がある
米国では、第三者支払者は製品の保証と精算に対して統一的な政策を持っていない。そのため、私たちの製品の保証範囲と精算範囲は支払人によって違います。支払者が製品に保険を提供するかどうかを決定するプロセスは、支払者が製品のために支払う支払率を設定するプロセスと分離することができる。1人の支払人が1つの製品に保険を提供することを決定し、他の支払者もその製品に保険と補償を提供することを保証することはできない。第三者支払者は、承認されたリストまたは処方表上の特定の製品に保証範囲を制限することもでき、FDAによって承認された特定の適応のすべての製品を含まない可能性がある。第三者支払者が自分の精算政策を設定する際には,通常連邦医療保険カバー政策と支払制限に依存する。私たちは政府援助や個人支払人から開発された承認された製品のために保険と十分な販売率を迅速に得ることができません。これは私たちの経営業績、製品の商業化に必要な資金を調達する能力、そして私たちの全体的な財務状況に悪影響を及ぼすかもしれません
私たちに対する製品責任訴訟は、私たちが重大な責任を負い、私たちが開発する可能性のある任意の製品の商業化を制限する可能性があります
私たちは人体臨床試験で私たちの候補製品をテストすることに関連する固有の製品責任リスクに直面しています。もし私たちが開発する可能性のある任意の製品を商業的に販売すれば、私たちはより大きなリスクに直面します。もし私たちが私たちの候補製品や製品に被害を与えるクレームを自己弁護することに成功できなければ、私たちは大きな責任を招くだろう。是非曲直や最終結果にかかわらず、賠償責任は
•すべての候補製品や私たちが開発する可能性のある製品の需要が減少した
•臨床試験を中止し
•承認された薬品の発売許可、製品のリコール、制限承認または“ブラックボックス”警告または禁忌症を撤回する
•臨床試験参加者の脱退
•関連訴訟の重大な弁護費用および/または増加した製品責任保険費用
•実験参加者や患者に多額の報酬を与え
•収入損失
•私たちの名声とメディアの深刻な否定的な関心を損なう
•経営陣の資源を減らして業務戦略を推進する
•私たちが開発する可能性のあるどんな製品も商業化できない
私たちは現在、私たちの臨床開発活動を支援するために製品責任保険を維持している。私たちは私たちの臨床試験を拡大したので、追加の製品責任保険を購入する必要があるかもしれません。もし私たちが候補製品を商業化し始めたら。保険範囲はますます高くなっています。私たちは可能などんな責任にも対応するために、合理的な費用や十分な金額で保険範囲を維持することができないかもしれない
私たちの知的財産権に関するリスクは
もし私たちの技術、候補製品、および製品のために特許保護を獲得し、維持することができない場合、または取得された特許保護範囲が十分に広くない場合、または実行可能でなければ、私たちの競争相手は、私たちと同様または同じ技術、候補製品および製品を開発し、商業化する可能性があり、私たちの技術、候補製品、および製品を商業化することに成功した能力が損なわれる可能性があり、あるいは市場で効果的に競争できない可能性がある
私たちは特許、商業秘密保護、および秘密保護協定の組み合わせによって私たちの知的財産権を保護し、他の人が私たちのプラットフォーム技術、私たちが開発している候補薬物製品、私たちが開発する可能性のある任意の未来の候補薬物製品、およびその使用または製造を防止する
私たちのビジネス成功は、私たちの独自技術、候補製品、および製品に関連する米国および他の国で特許および他の特許保護を取得し、維持する能力にある程度依存する。私たちは、私たちの新技術や候補品に関する特許出願を米国と海外に提出することで、私たちの独自の地位を保護することを求めています。私たちの機密固有情報を第三者に開示したり盗用したりすることは、競争相手が私たちの技術的成果を迅速にコピーしたり、超えたりして、市場での私たちの競争地位を侵食する可能性がある。さらに、私たちが所有している、共同所有、または許可されている特許出願は、米国または他の国で特許を発行できない可能性がある
特許訴訟過程は高価で時間がかかり、私たちはすべての必要または望ましい特許出願を合理的なコストまたは適時に提出、起訴、および維持することができないかもしれない。我々は,特許保護を得るのが遅くなるまで,我々の研究開発成果で特許を申請できることを確認できない可能性もある.さらに、場合によっては、私たちは、第三者から許可された技術または私たちが私たちの協力者に許可してくれた技術を含む特許出願の準備、提出および起訴を制御する権利がありません。したがって、このような特許および出願は、私たちの業務の最適な利益に合った方法で起訴され、強制されてはならない
生物製薬業界の特許地位は通常高度に不確定であり、複雑な法律と事実問題に関連し、多くの訴訟のテーマとなってきた。しかも、外国の法律はアメリカの法律のように私たちの権利を保護しないかもしれない。科学文献で発表された発見は往々にして実際の発見に遅れており、米国と他の司法管轄区の特許出願は通常、申請18ヶ月後に発表され、時には全く発表されないこともある。したがって、私たちは、私たちが所有している、共同所有または許可されている特許または係属中の特許出願において最初に要求された発明であるかどうか、またはこれらの発明のために特許保護された最初の発明者であるかどうかを正確に知ることはできない。したがって,我々または我々の協力者の特許権の発行,範囲,有効性,実行可能性,商業的価値は高度に不確定である.私たちの未解決および未来の特許出願は、私たちの技術、候補製品または製品の全部または一部を保護するために、または他社が競争技術、候補製品、および製品を商業化することを効果的に阻止するために、特許の発行を招くことができないかもしれない。米国および他の国の特許法または特許解釈または他の法律の変化は、私たちの特許および潜在的応用の価値を低下させ、私たちの特許保護範囲を縮小するか、または第三者に使用料を支払うことを要求される可能性がある。さらに、私たちの特許および特許出願によって提供される保護の広さまたは強度が脅かされている場合、会社が私たちと協力し、現在または将来の候補製品を許可、開発、または商業化することを阻止するかもしれない
私たちが所有し、共同所有し、許可している特許産業は主に特許出願からなり、その多くは起訴の初期段階にある。私たちが所有し、共同所有し、許可した特許出願が特許の形で発表されても、それらの発表形態は私たちに意味のある保護を提供してくれ、競争相手が私たちと競争することを阻止したり、他の方法で私たちにどんな競争優位性を提供してくれたりしないだろう。私たちの競争相手は、類似または代替技術、候補製品、または製品を非侵害的に開発することによって、私たちが所有している、共同所有、または許可された特許を回避することができるかもしれない
特許の発行は、その発明性、範囲、有効性、または実行可能性に対して決定的ではなく、私たちが所有している、共通して所有し、許可されている特許または私たちの協力者が獲得した特許は、米国および海外の裁判所または特許庁で挑戦される可能性がある。これらの課題は、独占的または経営の自由を失うこと、または特許主張の全部または部分的な縮小、無効、または実行できないことをもたらす可能性があり、これは、他人が同様の技術、候補製品および製品を使用または商業化する能力、または私たちの技術、候補製品、および製品の特許保護期間を制限することを制限する可能性がある。新製品候補製品の開発、テスト、規制審査に要する時間を考慮すると、候補薬物製品を保護する特許は商業化前または直後に満期になる可能性がある。したがって、私たちが所有し、共同所有し、許可している特許の組み合わせ、または私たちの協力者の特許の組み合わせは、他の会社が私たちと似ているか同じ製品を商業化することを排除するために十分な権利を提供してくれないかもしれない
特許法や特許法の変更は、私たちの特許の全体的な価値を低下させたり、私たちの特許に対する第三者の挑戦を増加させ、私たちの候補製品を保護する能力を弱めるかもしれません
特許改革立法は、私たちの特許出願をめぐる起訴と、私たちが発表した特許の実行または保護の不確実性とコストを増加させるかもしれない。2011年9月16日、“ライシー·スミス米国発明法”または“ライシー·スミス法案”が法律となり、米国特許法が多くの重大な改正を行った。このような変化は特許出願起訴方式に影響を与える条項を含み、特許訴訟に影響を与える可能性もある。米国特許商標局(USPTO)は、Leahy-Smith法案の管理を管理するための新たな法規や手続きを制定し、Leahy-Smith法案に関連する多くの特許法が実質的に改正され、初めて発明者を申請する条項を含め、2013年3月16日に発効した。Leahy-Smith法案とその実施は、私たちの特許出願をめぐる起訴と、私たちが発行した特許の実行または保護の不確実性とコストを増加させる可能性があり、これらはすべて私たちの業務と財務状況に悪影響を及ぼす可能性がある。最初に書類を提出した人
ライシー·スミス法案の規定は、常に第三者が特許出願を提出する可能性があるリスクがあり、これが私たちの特許出願を阻害する可能性があるので、発明から特許出願提出までの間に迅速に行動することを要求する。しかし、迅速に行動しようとしていても、状況は、私たちの発明に関する特許出願を迅速に提出または起訴することを阻止する可能性がある。ライシー·スミス法案はまた、従来技術に適合した開示範囲を拡大し、発明特許保護を得る能力に影響を与える可能性がある
ライシー·スミス法案は初めて新しいプログラムを作成し、このプログラムによると、第三者はライセンス後審査を含む米国で発行された特許に挑戦することができる各方面間審査と派生プログラムは,これらすべてが米国特許商標局で行われている対抗性プログラムである.“ライシー·スミス法案”が発効して以来、一部の第三者は、その競争相手が発行した特許の一部または全部の権利要件のキャンセルを求め、達成するために、これらのタイプの訴訟を利用してきた。Leahy-Smith法案によると、優先日が2013年3月16日以降の特許(私たちのすべての特許出願はそうである)については、第三者は、特許発行時から9ヶ月の窓口内の任意の時間に許可後審査請願書を提出することができる。また,優先日が2013年3月16日以降の特許については,第三者は出願を提出することができる各方面間支出を提出した後に請願書を審査する九ヶ月の期限が満了した後に審査を行います。贈与後の審査手続きはどんな疑問の理由でも提起することができます各方面間公開された既存技術に基づいて疑問を提起してこそ、検討手続きを提起することができる。適用法によると,USPTOによるこのような対抗訴訟の審査基準は,米国特許の有効性を推定せずに行われ,第三者が米国連邦裁判所に訴訟を提起することで特許を無効にしようとする場合には,この基準が適用される。USPTOは2018年11月11日に最終規則を発表し、現在米国連邦裁判所で現在使用されている同じ請求項解釈を使用することを発表した-これは使用されている言葉の簡単で一般的な意味である-これらのUSPTO訴訟における特許請求の範囲を解釈する。このような規制構造のため、もし私たちの任意の特許がこのような性質のUSPTO訴訟で第三者の挑戦を受けた場合、私たちが疑問視された特許の一部または全部を守ることに成功する保証はありません
この立法により、私たちまたは私たちの協力者の特許権の発行、範囲、有効性、実行可能性、および商業的価値は非常に不確定であり、これは私たちの業務、財務状況、運営結果、および将来性に悪影響を及ぼす可能性がある
私たちは私たちの特許、ライセンシーの特許、または他の知的財産権を保護または強制する訴訟に巻き込まれるかもしれません。これは高価で、時間がかかり、成功しないかもしれません
競争相手は私たちが発行した特許、私たちの許可者または協力者の特許、または私たちの他の知的財産権を侵害するかもしれない。権利侵害や不正使用に対抗するために、私たちは費用がかかり、時間がかかり、予測できないかもしれない侵害請求を要求されるかもしれない。私たちは侵害者と思われるいかなるクレームも、私たちが彼らの特許を侵害していると主張するように、これらの当事者たちに反クレームを促す可能性があります。さらに、特許侵害訴訟では、裁判所は、私たちまたは私たちのライセンシーまたは協力者の特許の全部または一部が無効または実行不可能であると判断し、その特許の権利要件を狭く解釈するか、または私たちの特許が関連する技術を含まないことを理由に、他方の関連技術の使用を阻止することを拒否することができる。任意の訴訟手続きにおける不利な結果は、私たちの1つまたは複数の特許を、または実際に無効にされるか、強制的に実行できないか、または狭く解釈される危険性に直面させる可能性がある。私たちが私たちの特許を主張することに成功しても、裁判所は私たちの損失を補償するのに十分な救済措置を与えないかもしれない。さらに、私たちは侵害者とみなされることに対抗するために、私たちの特許を十分に実行するために十分な財政的または他の資源を求めていないかもしれませんが、これは私たちの製品の収益性に実質的な悪影響を及ぼすかもしれません。
私たちは第三者から知的財産権の許可を得る必要があるかもしれないし、このような性質の許可は得られないかもしれないし、商業的に合理的な条項では得られない可能性がある
第三者は、特許権を含む、当社の製品または私たちの協力者の製品の開発または製造に非常に重要または必要な知的財産権を保有することができる。したがって、私たちは、第三者の特許または独自技術を使用して、私たち自身または私たちの協力者の技術または製品を商業化する必要があるかもしれません。この場合、私たちまたは私たちの協力者は、その第三者から許可を得ることを要求されます。この知的財産権の許可は得られないかもしれないし、商業的に合理的な条項では得られない可能性があり、これは私たちの業務や財務状況に悪影響を及ぼす可能性がある
第三者知的財産権の許可と取得は競争的な接近だ。私たちよりも成熟したり、より多くの資源を持っている可能性のある企業も、私たちの候補製品を商業化するために、必要または魅力的だと思う第三者知的財産権を許可したり、獲得したりする戦略をとっているかもしれません。より成熟した会社は、彼らのより大きな規模と現金資源、あるいはより強い臨床開発と商業化能力のため、私たちより競争優位にあるかもしれない。私たちはこのような交渉を成功させ、最終的に私たちが買収を求める可能性のある他の候補製品をめぐる知的財産権を得ることができないかもしれない
第三者は、私たちが彼らの知的財産権を侵害したことを告発する法的訴訟を提起する可能性があり、その結果は不確実であり、私たちの業務成功に悪影響を及ぼす可能性がある
私たちのビジネス成功は、私たちと私たちの協力者が私たちの候補製品を開発、製造、マーケティング、販売し、第三者の独自の権利を侵害することなく、当社の独自技術を使用する能力に依存します。生物製薬業界にはかなりの知的財産権訴訟と、再審、認可後審査、特許に挑戦する行政訴訟がある各方面間米国特許商標局の審査、派生手続又は介入手続、並びに外国司法管轄区域の異議及びその他の同様の手続
私たちは、派生、再審、許可後の審査、国際的に 部品.部品米国特許商標局の訴訟手続きを審査または介入する。第三者は既存の特許または将来付与される可能性のある特許に基づいて私たちに侵害請求をするかもしれない。バイオ製薬業界の拡張や特許の発行に伴い,我々の候補製品は他人の特許権侵害のクレームを引き起こすリスクが増加する可能性がある。私たちが現在知らない第三者特許、すなわち、私たちの候補薬剤の使用または製造に関連する材料、製剤、製造方法、または治療方法の請求項が存在する可能性がある。特許出願が発行されるまでに数年かかる可能性があるため、現在処理されている特許出願が存在する可能性があり、これらの出願は、我々の候補製品が発行された特許を侵害する可能性がある。さらに、第三者は将来的に特許を取得し、私たちの候補製品や私たちの技術を使用してこれらの特許を侵害したと主張するかもしれない
もし私たちが管轄権のある裁判所に第三者の知的財産権を侵害していると認定された場合、私たちは適用された第三者知的財産権所有者からライセンスを取得して、私たちの候補製品、製品、技術の開発とマーケティングを継続することを要求されるかもしれない。しかし、私たちは商業的に合理的な条項や必要な許可証を得ることができないかもしれない。我々が許可を得ることができても,非排他的である可能性があり,我々の競争相手が我々に許可された同じ技術にアクセスできるようにする.私たちは裁判所の命令を含めて権利侵害技術や製品の商業化を停止させることを余儀なくされるかもしれない。さらに、もし私たちが故意に特許を侵害したことが発見された場合、私たちは3倍の損害賠償と弁護士費を含む金銭損害賠償責任を負われる可能性がある。権利侵害の発見は、候補製品を商業化することを阻止したり、いくつかの業務運営を停止させたりする可能性があり、これは私たちの業務に実質的な損害を与える可能性がある。我々が第三者の機密情報や商業秘密を盗用したと主張することは,我々の業務に類似した負の影響を与える可能性がある
多くの他の会社や大学や他の組織は、私たちの製品と同じ分野で特許を出願し、特許を取得しています。これらの分野は、標的タンパク質分解剤または私たちのプラットフォーム技術であり、これらの特許出願は、将来的に私たちまたは私たちの協力者に告発される可能性があり、これは、私たちの業務成功に悪影響を及ぼす可能性があり、成功すれば、高価な訴訟を引き起こす可能性があり、私たちの製品の収益性に影響を与え、および/または私たちの製品の販売または使用を禁止する可能性があります
我々のMonoDACとBiDAC候補製品は小分子薬物であり,特定のタンパク質を分解することができる。多くの会社や機関はこの一般分野で特許を出願しており,例えばAccutar Biotech,Inc.,安進,Amphista Treateutics,Ltd.,Arax Pharma,LLC,Arvinas,Inc.,Astellas Pharma Inc.,アスリコン,Aurigen Discovery Technologies,Ltd.,バイエル株式会社(およびその子会社VividTreion ateutics,Inc.,百済神州株式会社,BioTheryX,Inc.,Briehringer Inglhenational International GmbH,BMySers Squers社,InoTheryX,Inc.,Briehringer InglhebH,BMySers Squers,InCaputicany,Capgycer,Inc.会社、フォグホーン治療会社、Frontier Medicines社、グラクソ·スミスクライン、遺伝子テーク社、Glubio治療会社、Hinova製薬会社、Janssen Biotech社、Kymera治療会社、モンテローサ治療会社、ノワール国際会社、Nurix治療会社、オレム治療会社、大塚製薬会社、Phoremore社、Plexium社、Prelude治療会社、羅氏社、Salarius製薬会社、Salarius製薬会社、種子治療会社、四川Haisco製薬会社、SK生命科学実験室、Haisco製薬会社、SK Science Labs、会社(SKバイオ製薬株式会社の子会社)、ミシガン大学医学部、Vertex製薬会社など。もしこれらの会社または機関のいずれかまたは他の本リストに登録されていない会社または機関が、そのある特許が私たちが開発または使用または製造する可能性のある任意の候補製品または製品によって侵害されたと主張した場合、私たちまたは私たちの協力者はコストの高い訴訟に巻き込まれる可能性があり、これは私たちの業務の将来性、財務状況、運営結果に悪影響を及ぼす可能性があり、私たちの管理チームのメンバーと多くの従業員が多くの時間を費やし、彼らを心配させる必要がある。また、このような性質の訴訟が成功すれば、我々製品の収益性に実質的な悪影響を与えたり、それらの販売を禁止したりする可能性がある。私たちは現在または将来私たちの業務や製品の特許請求に影響を及ぼす可能性があることを知らないかもしれない。特許出願は、通常、提出後6~18ヶ月以内に発行され、未解決の出願において新たな特許請求が提出された場合には、一定期間内に公衆(我々を含む)には見えない場合がある。さらに、特許出願が公開された後であっても、その特許出願をまだ見ていない可能性があるので、提出および発行された特許出願の特許請求の範囲または範囲を知らない可能性がある。だから私たちは何も提供できません
我々の一般的な技術分野で業務に従事する第三者が、我々の1つまたは複数の候補製品または製品またはその使用または製造方法をカバーする特許主張を提起しないか、または提示しないことを保証する。もしこのような状況が発生した場合、私たちまたは私たちの協力者は、適用された特許または出願を無効にしようとするステップを取らなければならないかもしれないし、このような性質の場合、私たちまたは私たちの協力者はそうしないことを選択するかもしれない、または私たちの試みは成功しないかもしれない。例えば、2023年5月1日、私たちはUSPTO特許裁判および控訴委員会(PTAB)に、米国特許番号11,414,416(‘416特許と呼ばれる)のすべての権利要件のライセンス後審査を要求する請願書を提出し、2023年10月23日、米国特許番号11,560,381(“381特許と呼ばれる)のすべての権利要件のライセンス後審査を要求する請願書をPTABに提出した。416特許および381特許の両方は、BRD 9関連疾患の治療のための化合物に関する。特許所有者は、2023年8月21日に“416特許のすべての権利要件を放棄し、”416特許のすべての合法的権利を放棄し、416特許挑戦を成功的に解決するための法定免責声明を提出した。特許所有者は、2024年1月17日に“381特許出願のすべての法定免責声明を提出し、”381特許のすべての権利および“381特許下のすべての権利を放棄し、”381特許挑戦を解決することに成功した。もし、第三者の特許または特許出願のライセンスが必要であると判断した場合、合理的な条項でライセンスを取得できないか、またはライセンスが全くない可能性があり、これは、私たちまたは私たちの協力者が製品を販売したり、私たちの独自技術を使用することを阻止するかもしれないことを発見するかもしれない。
もし私たちの候補製品が承認されれば、米国の1984年の“薬品価格競争と特許期限回復法”の制約を受けることになり、この法案は“ハッチ·ワックスマン法案”とも呼ばれ、これは私たちの製品を販売しようとしている模倣薬会社と訴訟を起こすリスクを増加させ、特許保護を失う可能性がある
我々の臨床候補薬物はFDA薬物評価·研究センターによって審査される薬物分子であるため,商業化後,米国ではハーチ−ワックスマン法(Hatch−Waxman Act)特許訴訟手続きの制約を受け,模倣製薬会社がFDAに簡略化された新薬申請(ANDA)を提出することを可能にし,生物学的同等性データのみを用いて我々の薬物を販売する後発薬バージョンの承認を得る。“ハッジ·ワックスマン法案”によると、我々の薬物製品またはそのそれぞれの使用方法をカバーする特許をFDAの“承認された薬物製品と治療同等性評価”コーパスに記載し、オレンジブックと呼ばれることもある。
特許に関する詳細な規則と要求があり、これらの特許はオレンジ本に列挙するためにFDAに提出される可能性がある。私たちはオレンジブックの発売要件を満たす1つ以上の特許を含む私たちの候補製品をカバーする特許を得ることができないかもしれない。たとえ私たちがOrange Bookに特許を提出しても、FDAはその特許のリストを拒否するかもしれないし、模倣薬メーカー、アメリカ連邦貿易委員会、または他の実体は上場に疑問を提起するかもしれない。私たちの候補製品のうちの1つが承認され、候補製品をカバーする特許がオレンジブックに記載されていない場合、模倣薬製造業者は、リストされていない特許について、FDAに提出された任意のANDA出願を事前に通知する必要がなく、候補製品の模造バージョンを販売する許可を得ることができる。
現在、米国では、FDAは新しい化学実体またはNCEに5年間のデータ排他性を付与する可能性があり、NCEは任意の他の新薬申請またはNDAにFDA承認の有効部分が含まれていない薬物である。私たちはすべての製品がNCEの資格に適合すると予想しているが、FDAはこの薬物のマーケティング申請を審査する前に、NCE状態を評価しない。イミテーション製薬会社は私たちがNCEに指定した任意の医薬製品が承認されて4年後にFDAにANDAを提出することができる。模造製薬会社がANDAを提出することは特許侵害の技術行為とされている。模造製薬会社は、それは私たちの上場特許が自然期限が切れる日まで待って、私たちの製品の模造薬バージョンを販売することができますか、あるいは私たちの1つ以上の上場特許が無効で、強制執行できない、あるいは侵害されていないことを証明することができます。もし後発薬メーカーが後者を選択すれば、私たちは45日間模造製薬会社に対して特許侵害訴訟を提起するだろう。もし私たちがそうすれば、模倣薬製造業者が私たちがリストした特許が無効で、強制執行できない、または侵害されていないと思うので、私たちOrange Bookに列挙された1つ以上の特許に挑戦する可能性が高い。訴訟が提起された場合、FDAは、私たちが模倣薬製造業者の認証通知を受けてから30ヶ月以内に、または主審裁判所が当事者のいくつかの行為または裁判所の最終判断に基づいて、私たちが主張する特許主張が無効、実行不可能、または侵害されていないより短いまたはそれ以上の時間前に、FDAが後発薬に対するANDAの最終承認を発行することを禁止してはならない。もし私たちがOrange Bookに私たちの関連特許を正確に列挙していなければ、あるいはANDAによって模倣薬会社の認証を直ちに提訴しなければ、あるいは私たちがそこで発生した特許訴訟で勝利しなければ、私たちは特許保護に基づく特許市場から利益を得る能力を失う可能性があり、私たちは医者が私たちの薬物製品の模造薬の開発と分配に転換することを発見するかもしれない。しかも、私たちがオレンジブックで私たちの関連特許を正確に列挙し、直ちに訴訟を提起し、訴訟で勝利しても、模倣薬訴訟は私たちに多大な費用をもたらすかもしれない。弁護士費と従業員時間の面でも、長い間そうだ。また,1つ以上の模倣製薬会社が創新者の薬物を同時に販売しようとするのは一般的であるため,後発薬メーカーからの複数の訴訟のコストと気晴らしに同時に直面する可能性がある。疑似製薬会社が私たちに入ることを可能にする必要があると確信しているかもしれません
私たちの特許が満期になる前に、または他の方法で私たちの特許の強度、有効性、または実行可能性に悪影響を及ぼす市場。
多くの製薬会社は米国連邦貿易委員会あるいは他の国の相応機関の厳格な審査の対象であり、審査の根拠はそれらが製薬製品に関連する特許訴訟を行う方法である。実は、いくつかの検討は反独占違反の疑いを招き、時々罰金や権利喪失を招く。私たちは私たちがこのような性質の審査を受けないか、あるいはこのような性質の審査の結果が私たちに有利であるか、あるいはどんなこのような性質の審査が罰金や処罰につながらないのかを確認することができない。
過去数年,米国連邦貿易委員会(Federal Trade Commission,略称FTC)は連邦裁判所に複数の訴訟を起こし,反競争を理由に革新者会社と後発薬会社との間で成立したANDA訴訟和解合意に挑戦した。例えば、連邦貿易委員会は、支払いの有無にかかわらず、どんな価値のあるものも支払いであるという急進的な立場を取っている。彼らのやり方によれば、革新者が特許和解の一部として、最初の模造製薬会社が革新者薬をカバーするOrange Book上場特許に挑戦することに同意した180日以内に、ライセンス模倣薬の発売を発売または延期しないか、または支払いなしに交渉が延期されることに同意すれば、連邦貿易委員会はこれを受け入れられない逆支払いだと考えるかもしれない。製薬業界の会社は,これらのタイプの合意は薬物革新者がリスクに対応するための理性的な商業決定であるため,和解条項が特許の排他的潜在力の範囲内であれば,これらの和解協定は反独占攻撃から守るべきであると弁明している。2013年、米国最高裁判所は賛成5票、反対3票で裁決されたFTCはActavis,Inc.を訴える.いわゆる逆支払いに関する製薬業と連邦貿易委員会の論点を却下した。逆に、最高裁は、遅延進入と引き換えに価格に関連する“逆支払い”和解が反競争分析を受けるべきかどうかは、(A)競争に真の悪影響を与える可能性、(B)支払いの理由、(C)特許権者が反競争損害をもたらす能力、(D)支払い金額が特許弱点の実行可能な代替物であるかどうか、の5つの側面の考慮に依存すると考えている。(E)多額の不合理な支払いに対する反独占責任は、例えば、模倣薬がブランド薬物の特許が満了する前に市場に参入することを許可し、特許権者が後発薬製造業者に費用を支払わないように、訴訟当事者がその訴訟を解決することを妨げるものではない。さらに、逆支払いが合理的かどうかは、その大きさ、特許権者が予想する将来の訴訟費用の規模、およびそれが支払いを表す可能性のある他のサービスの独立性に依存する(例えば、アトビス)と、他の納得できる理由が不足しています。逆に、最高裁は、逆支払い和解は独占禁止法に違反する可能性があり、標準的な反独占理由ルール分析の制約を受け、協定が連邦貿易委員会で不正であることを証明する責任があるとしている。この判決を下す際、最高裁はこの理由解析の仕組みを下級裁判所に残した。
もし私たちが後発薬会社とのHatch-Waxman訴訟を含む薬品特許訴訟に直面すれば、私たちはFTCのこのような性質の挑戦に直面するかもしれません。このような挑戦は私たちがどのように事件を解決するか、あるいは解決するかに影響を与えるかもしれません。たとえ私たちがFTCの立場に強く反対しても、私たちは巨額の費用や罰金に直面する可能性があります。私たちは“ハッジ·ワックスマン法案”に基づいて後発医薬品会社と合意したいかなる訴訟和解も、保険会社、直接購入者、または自分が和解に悪影響を受けていると思う他の人のような第三者支払者の挑戦を受ける可能性がある。このような後続訴訟は集団訴訟かもしれないし、費用が高いかもしれないし、長年続くかもしれない。もし私たちがこのような性質の訴訟に直面すれば、私たちはこれらのクレームを破ることに成功できないかもしれないので、私たちは巨額の支払い義務の制約を受ける可能性があり、私たちはこれらの義務を全部または部分的に満たすことができないかもしれない
私たちは米国の1984年の“薬品価格競争と特許期間回復法”によって特許期間を延長することができない可能性があるため、私たちの候補製品が承認されれば、十分な時間で特許保護が得られない可能性がある。
米国では,1984年の“薬品価格競争と特許期限回復法”は,各製品の特許を正常満期後に最大5年間延長することを許可しており,治療法特許に関連していれば,承認された適応に限られている。特許期間延長の長さは、通常、臨床試験期間の半分にFDA審査NDA期間の全時間を加えて計算され、これらの期間内の任意の遅延時間を減算する。特許期間の延長にも制限があり,薬品承認日から14年を超えない。したがって、もし私たちが最近提出·発表された特許の特許期間延長を選択して付与された場合、私たちは可能な特許期間延長からすべてのメリットを得ることができないかもしれない。例えば、適用期間内に出願できなかったこと、関連特許が満了する前に出願できなかったこと、または他の多くの適用要件のいずれかを満たすことができなかったため、特許期間の延長が全く付与されない可能性もある。また、FDAによって承認された製品の有効成分がFDAによって承認された以前の製品において規制審査および承認された場合、FDA承認された製品の規制審査期間は、特許期間を延長するための基礎となってはならない。また、適用当局は、米国のFDAおよびUSPTOおよび他の国/地域の任意の同等の規制機関を含み、このような性質の延期があるかどうかの評価に同意しない可能性があり、私たちの特許延期の承認を拒否するか、または私たちが要求したよりも限られた延期を承認する可能性がある。このような状況が発生すれば、私たちの競争相手は私たちの特許が満期になった後、私たちの臨床と臨床前データを参考にすることで競争製品の承認を得て、他の場合よりも早く彼らの製品を発売するかもしれない。このような状況が発生すれば、私たちが製品収入を作る能力に悪影響を及ぼすかもしれない。
ヨーロッパでは、補充保護証明書は、規制審査中に失われた特許期間を補償するために、特許期間を最大5年に延長することができ、合意された小児科調査計画に従って臨床試験データを取得すれば、この期間を5年半に延長することができる。ヨーロッパのすべての国は補充保護証明書を提供しなければならないが,ヨーロッパ諸国間には統一された立法がないため,薬物開発者は国ごとに補充保護証明書を申請しなければならない。したがって、1つの会社は、すべての関連国でこれらの証明書を申請して受け取るために大量の資源を必要とする可能性があり、いくつかの国ではないが(あれば)それらを取得することができる。
特許法の弱化や米国と外国裁判所の法執行は私たちが市場を保護する能力に影響を及ぼすかもしれない。
過去数年間、米国最高裁判所は特許事件においていくつかの意見を発表し、多くの人はこれらの意見が米国の特許保護を弱める可能性があると考えているか、ある場合に利用可能な特許保護範囲を縮小し、いくつかのタイプの革新は特許を申請できないと考えているか、または通常は他の方法で裁判所が特許を無効にしやすいようにしている。また,最近では米国や他の国の特許法をより多く改正することが提案されており,採択されれば,我々のノウハウのために特許保護を受ける能力,あるいは我々のノウハウを実行する能力に影響を与える可能性がある。米国議会、米国裁判所、米国特許商標局、および他の国の関連立法機関の将来の行動によれば、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、それによって、私たちが新しい特許を獲得したり、私たちの既存の特許と将来獲得可能な特許を実行する能力を弱める可能性がある。
一部の外国司法管轄区の法律は知的財産権の保護程度はアメリカに及ばず、多くの会社は外国司法管轄区の保護とこのような権利を守る面で重大な困難に直面している。もし私たちが知的財産権を保護する上でこのような困難に遭遇したり、他の理由で外国の管轄区域で私たちの知的財産権を効果的に保護できない場合、私たちのビジネスの見通しは深刻な損害を受ける可能性があります。例えば、私たちは、欧州特許庁、または外国裁判所での特許訴訟や他の手続のような外国の手続に反対する側になることができる。もしそうであれば,このような訴訟手続きの開始と継続による不確実な要因は,我々が市場で競争する能力に悪影響を与える可能性がある.外国対抗訴訟の費用も高い可能性があり、多くの外国司法管轄区では、敗訴側は勝訴側の弁護士費を支払わなければならない。
私たちは、私たち、私たちの従業員、コンサルタント、または請負業者が適用される第三者の知的財産権を流用したと主張したり、私たち自身の知的財産権の所有権を要求したりする第三者のクレームを受けるかもしれません。
私たちが雇った個人は以前、私たちの競争相手や潜在的な競争相手を含む大学や他のバイオテクノロジーや製薬会社に雇われていた。私たちは、協力者、潜在的な許可者、および契約守秘および非使用義務に制限される可能性のある他の第三者から機密および独自の情報を受け取りました。私たちは、私たちの従業員が私たちのために働いているときに他人の固有情報やノウハウを使用しないことを保証するために努力しているが、私たちは、これらの従業員または私たちが商業秘密または他の固有情報を含む任意のそのような従業員の前の雇用主の知的財産権を使用または開示しているという疑惑を受ける可能性がある。私たちはまた、前雇用主または他の第三者が私たちの特許に対して所有権を持っているというクレームに直面する可能性がある。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。私たちはこれらのクレームを弁護することに成功できないかもしれません。もし私たちがこのようなクレームを弁護できなければ、金銭損害賠償の支払いに加えて、価値のある知的財産権の独占所有権や使用権のような貴重な知的財産権を失う可能性があります。私たちが成功しても、訴訟は巨額のコストと名声損失を招き、私たちの経営陣や他の従業員の注意を分散させる可能性がある
また、私たちの政策は、知的財産権開発に参加する可能性のある従業員、コンサルタント、請負業者が合意に署名し、それによって生じる知的財産権を私たちに譲渡することを要求していますが、実際に私たちが自分と見なしている知的財産権を開発しているそれぞれと同様の合意を実行することができない可能性があります。このような性質の譲渡協定は自動的に実行されないかもしれないし、違反される可能性があり、私たちが私たちの知的財産権の所有権とみなされていることを決定するために、第三者にクレームを請求したり、私たちに提起される可能性のあるクレームを正当化させられたりする可能性があります。さらに、従業員または請負業者は発明を作成することができるが、私たちに通知することなく、この場合、私たちは発明のメリットを失う可能性があり、従業員または請負業者は他の場所に行って発明を開発するために離れてしまう可能性がある。
知的財産権訴訟は私たちに大量の資源を費やし、私たちの人員の正常な義務に対する注意を分散させるかもしれない。
解決策が私たちに有利であっても、知的財産権クレームに関連する訴訟や他の法的手続きは、巨額の費用を発生させ、私たちの技術や管理者の正常な責任を分散させる可能性があります。また,公聴会,動議や他の一時的な手続きや事態の発展の結果が公表される可能性があり,証券アナリストや投資家がこれらの結果を負のものと考えると,大きな影響を与える可能性がある
私たちの普通株価格に悪影響を及ぼす。このような性質の訴訟または訴訟は、私たちの運営損失を大幅に増加させ、開発活動または任意の将来の販売、マーケティング、または流通活動に使用することができるリソースを減少させる可能性がある。私たちはそのような訴訟や訴訟手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らがより多くの財政資源を持っているので、私たちよりも効率的に訴訟やこのような訴訟の費用を負担するかもしれない。特許訴訟や他の訴訟の開始と継続による不確実性は、市場での私たちの競争能力を損なう可能性がある。
特許保護の獲得と維持は,政府特許庁が提出した様々なプログラム,文書,費用支払い,その他の要求を遵守することに依存し,これらの要求を遵守しなければ,我々の特許の保護は減少またはキャンセルされる可能性がある。
発行された特許の定期維持費は,特許出願及びそれによって生成された任意の特許の有効期間内にいくつかの段階で米国特許商標局及び外国の特許庁に支払われなければならない。米国特許商標局や外国特許庁は,特許出願過程において多くのプログラム,書類,費用支払いなどの要求を遵守することを要求している。多くの場合、適用規則に従って滞納金を支払うことによって、または他の方法でミスを訂正することができるが、場合によっては、規定を遵守しないことは、特許または特許出願が放棄または失効される可能性があり、それにより、関連法ドメインの特許または特許権の一部または全部の喪失をもたらす可能性がある。特許または特許出願の放棄または失効をもたらす可能性のある不正事件には、規定された期限内に公式行動に応答できなかったこと、費用が支払われていなかったこと、および適切に合法化され、正式な文書が提出されなかったことが含まれるが、これらに限定されない。この場合、私たちの競争相手が市場に参入する可能性があり、これは私たちの業務に悪影響を及ぼすだろう
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの商業と競争の地位は損なわれるだろう
私たちのいくつかの候補技術と製品のための特許を申請するほか、私たちは、私たちの競争地位を維持するために、特許を取得していないノウハウ、技術、および他の独自の情報を含む商業秘密に依存している。私たちは、これらの商業秘密を保護することを求めています。一部は、これらの商業秘密にアクセスする権利のある当事者と秘密保護協定を締結することによって、例えば、私たちの従業員、会社協力者、外部科学協力者、契約メーカー、コンサルタント、コンサルタント、および他の第三者です。私たちはまた私たちの従業員とコンサルタントと秘密と発明または特許譲渡協定を締結します。これらの合意は、機密情報の開示を効果的に阻止することができない可能性があり、知的財産権を効率的に譲渡することができず、機密情報の不正開示や他の合意違反の場合には、十分な救済措置を提供できない可能性がある。しかも、他の人たちは私たちの商業秘密と固有の情報を独立して発見するかもしれない。この場合、私たちはその第三者に対するいかなる商業秘密権利も主張できない。このような努力にもかかわらず、どちらも合意に違反し、私たちのビジネス秘密を含む私たちの固有の情報を漏洩する可能性があり、私たちはこのような違反について十分な救済措置を得ることができないかもしれない。一方の当事者が商業秘密を不正に開示したり流用したりする主張を実行することは困難であり,高価で時間がかかり,このような性質の紛争の結果は本質的に予測できない.私たちは私たちの固有の権利の範囲を強制的に執行し、決定するために高価で時間のかかる訴訟が必要かもしれないが、私たちが商業秘密保護を獲得したり維持できなかったりすることは、私たちの競争ビジネスの地位に悪影響を及ぼすかもしれない。しかも、アメリカ以外のいくつかの裁判所は商業秘密をあまり望んでいないし、保護したくない。2016年の“商業秘密擁護法”は、商業秘密の所有者が商業秘密が流用された際に連邦裁判所に訴訟を起こすことを許可する米国連邦法である。国会はこの法律を可決し、商業秘密所有者の権利を強化しようとしており、これらの人々の貴重な資産は無許可で持ち去られた。もし私たちの任意の商業秘密が競争相手によって合法的に取得または独立して開発された場合、私たちは彼らまたは彼らが情報を伝達する人がその技術や情報を使用して私たちと競争することを阻止する権利がないだろう。もし私たちのビジネス秘密が競争相手に漏れたり、競争相手によって独立して開発されたりすれば、私たちの競争地位は損なわれるだろう。
私たちのいくつかの特許については、私たちは限られた地理的保護だけで、私たちは世界各地で私たちの知的財産権を保護できないかもしれない。
世界のすべての国で私たちの候補品をカバーしている特許は、申請、起訴、維持、保護されるほど高価になるだろう。したがって、私たちのアメリカ以外のいくつかの国での知的財産権は、私たちがアメリカで得た保護ほど広くないかもしれない。もし私たちが本当にこのような許可内の機会があれば、私たちが世界のすべての国の候補製品をカバーする許可内特許は同じように尻込みするほど高いかもしれない。さらに、私たちが私たちの候補製品を開発または商業化する司法管轄区域内でも、特許を許可または届出、起訴、維持、擁護することは、尻込み的または非現実的に高価になる可能性がある。競争相手は私たちが特許保護を受けていないかもしれない司法管轄区域で私たちと私たちのライセンシーの技術を使用して彼ら自身の製品を開発することができ、さらに私たちと私たちのライセンシーが特許保護を持っている地域に他の侵害製品を輸出することができますが、法執行力はアメリカに及ばないか
EUです。これらの製品は私たちの候補製品と競争する可能性があり、私たちまたは私たちの許可者の特許または他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。
しかも、私たちはまだ未解決のままであるので、国と地域の特許出願を放棄することを決定するかもしれない。各国又は地域特許の付与手続は独立した手続であり,関連特許庁が出願を拒否し,実質的に類似した出願が他の機関によって承認されている場合を招く可能性がある。例えば、中国は他国よりも特許性の詳細な記述が要求されている。さらに、模倣薬製造業者または他の競争相手は、私たちまたは私たちのライセンシーの特許の範囲、有効性、または実行可能性に疑問を提起し、私たちまたは私たちのライセンシーに複雑で長く、高価な訴訟、または他の訴訟を要求するかもしれない。後発薬メーカーは私たちの製品の模造薬バージョンを開発し、承認を求めて発売することができる。同様に一般的な場合は、国によって同じ候補製品または技術の特許保護範囲が異なる可能性があることである
一部の司法管轄区の法律は知的財産権の保護程度はアメリカとEUの法律或いは法規に及ばず、多くの会社はこれらの司法管轄区の保護と専有権の保護に重大な困難に直面している。さらに、特定の国の法律制度、特に特定の発展途上国の法律制度は、特許、商業秘密、または他の形態の知的財産権の実行を支持しておらず、特定の管轄区域の競争相手が私たちの専有権を侵害する方法で競合製品を販売することを阻止することを困難にする可能性がある。
外国司法管轄区域で我々の特許権を強制的に執行する訴訟は,成功するか否かにかかわらず,巨額のコストを招き,我々の努力と注意を我々の業務の他の側面から移行させる可能性があり,また,我々または我々のライセンシーの特許が無効または偏狭に解釈されるリスクに直面させる可能性があり,我々または我々の許可者の特許出願が発行されないリスクを増加させるか,あるいは第三者からのクレームを引き起こす可能性がある.私たちは私たちが起こしたいかなる訴訟にも勝てないかもしれないし、敵に損害賠償や他の救済措置を支払うかもしれないが、これは大きなビジネス的意義を持つかもしれない。もし私たちが勝訴すれば、私たちに与えられた損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれません。したがって、私たちが世界各地で私たちの知的財産権を実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネスメリットを得るのに十分ではないかもしれません。また、私たちが予想している重要な市場で私たちの知的財産権を保護するつもりですが、候補製品をマーケティングしたいかもしれないすべての司法管轄区域で同様の努力を開始したり維持したりできることを保証することはできません。したがって、これらの国で私たちの知的財産権を保護するための努力は十分ではないかもしれませんが、これは私たちが予想されるすべての重要な海外市場で私たちの候補製品を商業化することに成功した能力に悪影響を及ぼすかもしれません。もし私たちまたは私たちのライセンシーがこれらの管轄区域で保護困難に遭遇したり、私たちの業務に重要な知的財産権を効果的に保護できなければ、これらの権利の価値は低下する可能性があり、私たちはこれらの管轄区域で追加の競争に直面するかもしれない。
いくつかの司法管轄区域では、強制許可法は特許権者に第三者に許可を付与させる。さらに、いくつかの国は、政府機関または政府請負業者に対する特許の実行可能性を制限している。これらの国では,特許権者の救済措置は限られている可能性があり,このような特許の価値を大幅に低下させる可能性がある。もし私たちまたは私たちの任意の許可者が私たちの業務に関連する特許の下で第三者に許可を付与することを余儀なくされた場合、または私たちまたは私たちの許可者が第三者に対して特許権を執行することを阻止された場合、私たちの競争地位はこれらの管轄区域で深刻な損害を受ける可能性があります。
規制事項に関するリスク
FDAと外国規制機関から規制承認を得ることは長く、時間がかかり、本質的に予測不可能であり、もし私たちが最終的に私たちの候補製品のためにマーケティング承認を得ることができなければ、私たちの業務は深刻な損害を受けるだろう。
FDAと外国の監督管理機関の承認を得るのに要する時間は予測できないが、通常は臨床試験開始後数年後に必要であり、監督管理機関のかなり大きな適宜決定権を含む多くの要素に依存する。また、承認基準、法規、あるいは承認を得るために必要な臨床データのタイプと数量は候補製品の臨床開発過程で変化する可能性があり、司法管轄区域によって異なる可能性がある。私たちはまだ候補製品のマーケティング承認を得ていません。私たちの既存の候補製品または私たちが将来開発を求める可能性のある任意の候補製品(独立または私たちのパートナーのうちの1つ)は、決してマーケティング承認を得ないかもしれません。
私たちの候補製品は、以下の理由を含む、多くの理由でマーケティング承認を得ることができないかもしれません
•FDAや外国の規制機関は、ここではそれぞれ衛生当局と呼ばれ、私たちの臨床試験の設計や実施に同意しないかもしれない
•私たちは、その提案の適応について、候補製品が安全で有効であるか、または衛生当局の基準に基づいて、十分な強度、識別または品質を有することを衛生当局に証明することができないかもしれない
•臨床試験結果は衛生部門が許可した統計的有意水準に達しない可能性がある
•候補製品の臨床的および他の利益がその安全リスクよりも大きいことは証明できないかもしれない
•衛生当局は前臨床研究や臨床試験データの説明に同意しないかもしれません
•私たちの候補製品の臨床試験から収集されたデータは、FDAへの機密協定の提出をサポートするのに十分ではないかもしれないし、外国の監督管理機関に他の申請を提出するか、または米国または他の任意の国または司法管轄区で上場承認を得るのに十分ではないかもしれない
•衛生当局は、私たちと臨床および商業用品契約を締結した第三者メーカーの製造プロセスまたは施設に欠陥があるか、または承認されないことを発見する可能性がある
•衛生当局の承認基準、政策或いは法規は重大な変化が発生する可能性があり、著者らの臨床データは承認に不十分である。
この長い薬物開発過程、および将来の臨床試験結果の予測不可能性は、規制部門の承認を得ることができず、私たちのすべての候補製品を販売できなくなる可能性があり、これは私たちの業務、運営結果、将来性を深刻に損なうだろう。FDAおよび他の衛生当局は、承認過程においてかなりの裁量権を有し、承認を加速させた場合を含めて、いつまたは任意の候補製品の規制承認を得るかどうかを決定する。我々の候補製品の臨床試験から収集されたデータが有望であると信じていても,これらのデータはFDAや任意の他の衛生当局の承認を支持するには不十分である可能性がある
さらに、私たちが承認されても、規制機関は、私たちの要求された範囲以下またはそれ以上の適応を承認することができ、私たちが製品のために徴収しようとしている価格を承認しないかもしれないし、承認は高価な発売後の臨床試験の表現に依存するかもしれないし、候補製品を承認する可能性のあるラベルは、候補製品の商業化に成功するために必要または望ましいラベル宣言を含まないかもしれない。上記のいずれの場合も、私たちの候補製品のビジネス見通しに実質的な損害を与える可能性があります
たとえ私たちの候補製品が米国でFDAの承認を得ても、私たちはいかなる他の管轄区域でも承認されたり、商業化されたりすることはありません。これは私たちがそのすべての市場潜在力を達成する能力を制限するだろう。
任意の特定の管轄区域で任意の製品を販売するためには、安全性と有効性に関する多くのかつ異なる規制要件を各国に基づいて確立し、遵守しなければならない。
米国FDAの承認は、他の国や管轄区域の規制機関も承認することを確保していない。しかし、一つの管轄区域で承認を得ることができなかったことは、私たちが他の場所で承認を得る能力に悪影響を及ぼすかもしれない。また、一国で行われた臨床試験は他の国の規制機関に受け入れられない可能性があり、一国の規制承認は他の国の規制承認を保証することはできない
承認の流れは国/地域によって異なり、追加の製品テストと検証、および追加の行政審査期限が含まれる可能性があります。外国の監督管理機関の承認を求めることは私たちに困難とコストをもたらす可能性があり、追加の臨床前研究或いは臨床試験が必要であり、これは高価で時間がかかるかもしれない。各国の規制要求には大きな違いがある可能性があり、私たちの製品がこれらの国で発売されることを延期または阻止する可能性がある。私たちは国際市場を含めてどの司法管轄区でも候補製品の販売を承認しておらず、私たちは会社としても国際市場で規制承認を受けた経験がありません。もし私たちが国際市場の規制要求を遵守できなかったり、必要な承認を得て維持できなかったり、あるいは国際市場の規制承認が延期されれば、私たちの目標市場は減少し、私たちが開発したどの製品も市場の潜在力を十分に発揮する能力は達成できないだろう。
私たちがどんな候補製品の規制承認を得ても、私たちは持続的な規制義務と持続的な規制審査の制約を受けることになり、これは多くの追加費用を招く可能性があり、もし私たちが規制要求を遵守できなかったり、私たちの候補製品が予期しない問題に遭遇したら、私たちは処罰されるかもしれない
もし私たちの任意の候補製品が承認された場合、それらは製造、ラベル、包装、貯蔵、広告、販売促進、サンプリング、記録保存、上場後研究、および安全性、有効性およびその他の上場後情報を提出することを含む持続的な法規要求を遵守し、アメリカ連邦と州の要求、比較可能な外国監督管理機関の要求を含む。さらに私たちは進行を要求されるかもしれません
進行や完了が困難な特殊な人の中で承認を行った後に検討する.われわれが承認後に行ったいずれの臨床試験についても,cGMPやGCP要求を遵守していきたい。
メーカーとメーカーの工場は、品質管理と製造プロセスがcGMP法規と適用される製品追跡と追跡要求に適合することを確保することを含む、FDAと同様の外国規制機関の広範な要求に適合しなければならない。したがって、私たちと私たちのCMOは、cGMPの遵守状況、および任意のNDA、他のマーケティング申請、および以前の検査観察に対する応答に対する承諾の遵守状況を評価するために、持続的な審査および検査を受ける。したがって、私たちと私たちと協力している他の人たちは、製造、生産、品質管理を含む規制コンプライアンスのすべての分野に時間、お金、エネルギーをかけ続けなければならない。
私たちの候補製品のために得られた任意の規制承認は、その製品が発売される可能性のある承認指示用途の制限または承認条件によって制限される可能性があり、または候補製品の安全性および有効性を監視する第4段階の臨床試験および監視を含む、コストの高い上場後試験の要求を含む可能性がある。FDAはまた、REMS計画を私たちの候補製品を承認する条件として要求することができ、これは、患者の長期フォローアップ、薬物使用ガイドライン、医師コミュニケーション計画、または配布方法の制限、患者登録、および他のリスク最小化ツールのような安全な使用を保証する他の要素の要求を必要とする可能性がある。似たような外国の規制機関もまたREMSと似たような計画を持っているかもしれない。また、FDAや同様の外国規制機関が私たちの候補製品を承認した場合、安全および他の上場後の情報の提出、報告および登録を含む要求を遵守しなければなりません。
規制要件や基準が守られていない場合、あるいは製品発売後に問題が発生した場合、FDAは同意法令を強制的に実施したり、承認を撤回したりする可能性がある。その後、私たちの候補製品には、予期されない重症度または頻度の有害事象、または私たちの第三者CMOまたは製造プロセス、または規制要件を遵守できなかったことが、新たなセキュリティ情報を追加するために承認されたラベルの改訂につながる可能性があることが発見された;発売後研究または臨床試験を実施して新しいセキュリティリスクを評価するため、またはREMS計画に従って流通制限または他の制限を実施する。他の他の潜在的な結果には
•私たちの製品の販売や製造を制限し、製品を市場からリコールするか、または自発的または強制的に製品をリコールする
•罰金や警告状や臨床試験を一時停止した者は
•FDAは、処理すべき出願の承認を拒否するか、または私たちが提出した承認された出願の追加を拒否するか、またはライセンス承認を一時停止または撤回する
•製品を差し押さえたり抑留したり、私たちの候補製品の輸入または輸出を許可することを拒否したり;
•民事または刑事処罰を禁令または適用する。
FDAは市場に投入された製品のマーケティング、ラベル、広告、販売促進を厳格に規制している。製品は承認された適応と承認されたラベルの規定でしか販売促進できません。FDAや他の機関はラベル外用途の普及を禁止する法律や法規を積極的に実行しており,ラベル外用途の普及が不適切であることが発見された会社は重大な責任を負う可能性がある。しかし、医師はその独立した医学判断に基づいて、ラベル外で使用される合法的に使用可能な製品のために処方することができる。FDAは医師が治療を選択する行為を規範化していないが,FDAは製品ラベル外使用問題におけるメーカーのコミュニケーションを制限している。FDAと同様の外国規制機関の政策は変わる可能性があり、私たちの候補製品に対する規制承認を阻止、制限、または延期するための追加の政府法規が公布されるかもしれない。米国や海外の将来の立法や行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することはできない。もし私たちが既存の要求の変化に緩やかに適応できない場合、あるいは新しい要求や政策を採用することができない場合、または規制適合性を維持できない場合、私たちは私たちが得る可能性のあるマーケティング承認を失う可能性があり、利益を達成したり維持することができないかもしれない。
FDAが指定した画期的な治療法は、私たちのどの候補製品にも与えられても、より速い開発や規制審査や承認過程を招くことはなく、私たちの候補製品が発売承認される可能性を増加させることはありません。
私たちは、CFT 7455およびCFT 1946を含む、現在および未来のいくつかまたはすべての候補製品のための画期的な治療法の称号を求めるかもしれない。突破療法は、単独または1つまたは複数の他の薬剤との併用治療が深刻または生命に危険な疾患または状態を治療することを意図した薬剤として定義され、初歩的な臨床証拠は、1つまたは複数の臨床的に重要な終点において、例えば、臨床開発早期に観察される実質的な治療効果のような既存の療法よりも有意な改善を示す可能性があることを示す。入手したものに適用します
画期的な治療法として、FDAと試験スポンサー間の相互作用とコミュニケーションは臨床開発の最も有効な方法を確定することができ、同時に無効なコントロール方案中の患者数を最低に下げることができる。FDAで画期的な治療法に指定されている薬物も,承認の加速を含めて他の迅速な承認計画を得る資格がある可能性がある。
FDAはそれを画期的な治療法に指定する権利がある。したがって,我々の候補製品の1つが画期的療法として指定された基準に適合していると考えても,FDAは同意せず,このような指定をしないことにする可能性がある。いずれにしても、画期的な治療法指定は、このような指定された薬物の開発および審査を加速させることを目的としているにもかかわらず、非迅速FDA審査手順に従って承認を考慮した候補製品と比較して、候補製品の画期的な治療指定を受けることは、より速い開発過程、審査または承認を招くことなく、FDAが最終的に候補製品を承認することを確保することができない可能性がある。さらに、私たちの1つまたは複数の候補製品が突破療法の条件を満たしていても、FDAは後でその製品が資格条件を満たしていないと決定する可能性がある。したがって,われわれの主要候補品や各種癌治療の一部またはすべての将来の候補品のためにブレークスルー療法の称号を求めるつもりであっても,ブレークスルー療法の称号を得ることは保証されない。
FDAの迅速なチャネル指定は、私たちの1つまたはすべての主要候補製品、または私たちの現在または未来の任意の他の候補製品に付与されても、より速い開発や規制審査または承認過程を招くことはなく、私たちの候補製品が上場承認される可能性を増加させることはない
様々な場合、私たちは私たちの1つまたは複数の候補製品のための高速チャネル認証を求めることができる。重症または生命に危険な疾患の治療に使用される医薬が使用され、そのような疾患が満たされていない医療需要を解決する可能性を示す場合、薬物スポンサーは、特定の適応のためにFDA迅速チャネル指定を申請することができる。私たちは私たちの1つまたはすべての主要な候補製品および/またはいくつかの未来の候補製品のために高速チャネル認証を求めることができるかもしれないが、FDAがこの地位を私たちが提案した任意の候補製品に与える保証はなく、私たちは何度も申請した後にのみ、FDAのある候補製品に対する迅速なチャネル認証に成功することができるかもしれない。FDAが提供する政策やプログラムによると、Fast Track開発製品のスポンサーが提出したマーケティング申請には資格優先審査がある可能性があるが、Fast Track指定を受けてこのような資格やFDAが最終的に上場を承認することは保証されていない。FDAは広範な裁量権を持ち,高速チャネル認証が付与されているかどうかを持つため,特定の候補製品がその認証を取得する資格があると考えても,FDAが付与を決定する保証はない。Fast Track認証を確実に取得しても,Fast Track認証がこのような認証を得るための薬物の開発や審査を加速させることを目的としていても,従来のFDAプログラムと比較して,より速い開発過程,審査や承認を経験することはなく,Fast Track認証を得ることでFDAの最終承認が保証されない可能性がある。また,FDAが我々の臨床開発計画のデータが高速チャネル指定をサポートしなくなったと考えた場合,その指定を撤回する可能性がある。また,FDAはいつでもFast Trackの指定を取り消すことができる.
私たちはCFT 7455の孤児薬物指定を取得しており、もし私たちが他の現在または未来の候補製品のために孤児薬物指定を求めることを決定すれば、私たちは市場排他性の潜在力を補充することを含む、孤児薬物指定に関連する利点を維持できないかもしれない
2021年8月、FDAはCFT 7455によってMMを治療する孤児薬物称号を付与した。私たちは、現在または将来の1つまたは複数の候補製品のために孤児薬物称号を求めることができるかもしれない。米国やEUを含むいくつかの管轄区の規制機関は、患者数が比較的少ない薬物を孤児薬に指定する可能性がある。孤児医薬品法によれば、FDAは、米国では患者数が20万人未満であるか、または米国では患者数が20万人を超えるが、合理的な期待がなく、米国で薬物を開発および提供するコストであると定義されている稀な疾患または状態を治療するための薬剤の称号を孤児に与えることができる。この薬のアメリカでの販売から回復します。米国では、孤児薬物の称号を獲得することは、臨床試験費用、税収優遇、ユーザー費用の減免に贈与資金を提供する機会など、当事者に財政奨励を得る権利がある。FDAが孤児薬物指定を承認した後、FDAは、この薬剤の模倣薬識別情報およびその潜在的孤児用途を開示する。孤児薬物指定は、まれな疾患や疾患に対する薬物開発を促進することを目的としているが、孤児薬物指定は、規制審査および承認過程においていかなる利点も伝達されず、規制審査および承認過程の持続時間を短縮することもない。また,孤児薬の指定を受けることは,FDAが小児科群での研究を放棄する義務を招く可能性があるが,この免除は腫瘍薬には適用できない可能性がある
孤児薬物指定を有する製品が、その後、このような指定された疾患を有する特定の活性成分に対するFDAの最初の承認を得た場合、この製品は、孤児製品の排他性を得る権利があり、これは、FDAがNDAを含む他の出願を承認しない可能性があり、以下の特徴を有する製品に対する臨床的利点を示すような限られた場合でなければ、7年以内に同じ適応の同じ薬剤を販売することを意味する
孤児薬物排他性、またはFDAは、指定された薬物が対象とする疾患または状態の患者の需要を満たすために十分な数の孤児薬物を得ることができることを証明していないことを発見した。したがって,我々の候補製品が孤児薬の排他性を獲得しても,FDAは同じ適応や疾患の治療に異なる有効成分を含む他の薬剤を承認することができる。また、私たちが十分な製品供給を生産できなければ、FDAは孤児薬の独占経営権を放棄することができる。
私たちはまた、私たちの他の主要な候補製品および/または私たちの現在または未来の一部またはすべての他の候補製品のための孤児薬物指定を求めることができ、これらの候補製品を使用するための医学的に信頼できる基礎がある他の孤立適応のために使用することができる。孤児薬物指定を受けた場合であっても、孤児指定適応よりも広い適応の承認を求めると、米国での独占営業権が制限される可能性があり、FDAが後に指定要求に重大な欠陥があると判断した場合、またはまれな疾患や疾患患者の需要を満たすのに十分な数の製品をメーカーによって保証できない場合には、独占営業権を失う可能性がある。また、私たちが他の候補製品のために孤児薬の指定を求めても、私たちは決してこれらの指定を得られないかもしれない。例えば、FDAは、組織不知性療法に適した孤児薬物指定の規制考慮を懸念しており、FDAは、孤児薬物指定または孤児薬物独占特許を得る能力を制限または阻止するために、FDCAおよびその孤児薬物法規を解釈する可能性があり、私たちの候補製品が承認されれば、私たちの標的適応のために使用される。
FDAの承認加速経路に基づいて、適用された場合に候補製品の承認を求めることができます。このアプローチは、より速い開発や規制審査や承認過程をもたらすことはないかもしれませんし、私たちの候補製品が上場承認される可能性も増加しません。
我々は,我々の主要候補製品の承認速度を加速させ,適用可能な場合には,FDAの加速承認プロセスを用いて将来の候補製品の承認を求める予定である.製品が重篤または生命に危険な疾患を治療し、一般に既存の療法よりも有意な利点を提供する場合、加速承認を得る資格がある可能性がある。さらに、それは、臨床的利益を合理的に予測する可能性のある代替終点への影響、または不可逆的な発病率または死亡率またはIMMよりも早く測定することができる臨床終点への影響を証明しなければならず、IMMまたは他の臨床的利益を合理的に予測することができる。食品·薬物総合改革法案によれば、一般にFDORAと呼ばれ、FDAは、承認前または承認が加速された製品の承認日後の特定の期間内に、1つまたは複数の承認後の検証的研究を行うことを適宜要求することが可能である。FDORAは,登録目標の実現を含めて180日ごとにFDAにこれらの研究の最新状態を送信することも求められており,FDAはこれらの情報を迅速に公開しなければならない。FDAはまた,スポンサーがこのような活動をタイムリーに展開できなかった場合には,必要な最新情報をFDAに送信したり,このような承認後に薬物の期待された臨床的利益を検証できなかった場合には,FDAは迅速に承認を撤回し,職務調査ができなかった会社に対して罰金などの行動をとったり,速やかにFDAに進捗状況報告を提出したりする権限をFDAに与えている。さらに、FDAは、通常、加速された承認を得ることを要求する製品の販売促進材料が事前に承認されなければならず、これは、製品の商業発売時間に悪影響を及ぼす可能性がある。したがって,どの候補製品にも加速承認経路を利用することを求めても,加速承認を得ることができない可能性があり,加速承認を得ても,その製品はより速い開発や規制審査や承認過程を経験できない可能性がある.また,加速承認を得ることは,製品の加速承認が最終的に従来の承認に変換される保証はない.
FDAは書面で小児科情報が承認された候補製品に有利であることを決定し,小児科研究を依頼することができる。これらの研究を行わないことを選択することができますあるいは、これらの研究を行うことを選択すれば、それらを完成させることができないかもしれません。あるいは、これらの研究によって生成されたデータはFDAに受け入れられないかもしれません。
“食品、薬物及び化粧品法”又は“食品、薬物及び化粧品法”の第505条(A)条は、児童に対して薬物研究を行う医薬品メーカーにインセンティブを提供する。この法律は“小児科排他性条項”と呼ばれ,製薬業者に追加的な6カ月の非特許排他性を提供しており,これらの製薬業者は新薬や現在発売されている薬物に対して許容可能な小児科研究を行い,FDAの書面請求により小児科データが有利となる。したがって,FDAの小児科研究書面の請求を受け,小児科臨床研究を行い,法定期限内にFDAが受け入れた報告書を提出すれば,他のすべてのタイプの特許と非特許排他性以外に6カ月の規制排他性を追加することができれば,我々のすべての承認された医薬製品に対して,小児科排他性特許を付与する活性部分が含まれていれば,追加6カ月の規制排他性を得ることができる。しかし、FDAから1つ以上の医薬製品の小児科研究に対する書面の要請を受けても、FDC法案505(A)節に適合する小児科研究を行わないか、または検討することができないかもしれません
この目的のためにFDAによって受け入れられない。もしこのような状況が発生したら、私たちは追加的な6ヶ月の規制排他的延期を得ないだろう
顧客、医療提供者、第三者支払者との関係は、外国、連邦、州医療詐欺および乱用法律、虚偽クレーム法律、医療情報プライバシーおよび安全法律、および他の医療法律法規の制約を直接または間接的に受けている。もし私たちがこのような法律を遵守できないか、または完全に遵守できなければ、私たちは巨額の処罰に直面するかもしれない。
米国や他の地域の医療提供者および第三者支払者は、マーケティングの承認を得た任意の候補製品を推薦し、処方する上で主な役割を果たす。私たちは現在と未来の医療専門家、首席調査者、コンサルタント、顧客、第三者支払人との手配により、私たちは様々な連邦と州詐欺と乱用法律、その他の医療保健法律の制約を受けて、もし私たちがマーケティングの許可を得たら、これらの法律は私たちの研究、販売、マーケティング、流通、私たちの候補製品の業務あるいは財務手配と関係を制限するかもしれません。特に、我々の候補製品の研究、および医療プロジェクトおよびサービスの普及、販売およびマーケティング、ならびに医療業界のいくつかの商業的配置は、(I)詐欺、リベート、自己取引および他の乱用行為を防止するための広範な法律的制約、(Ii)健康情報の安全およびプライバシーを保証すること、および(Iii)医師、教育病院および医薬品、医療機器および生物学的製剤製造業者間の財務関係の透明性を増加させることを目的としている。これらの法律法規は、幅広い価格設定、割引、マーケティングおよび販売促進、構造および手数料(S)、特定の顧客インセンティブ計画、および他の商業または財務スケジュールを制限または禁止する可能性があります。タイトルを見て“ビジネス−その他医療保健法“と”ビジネス-医療改革私たちの2022年年次報告書にあります。
第三者との業務配置ややり方が適用される医療法律や法規に適合していることを確保し、コストが高くなる可能性がある。政府当局は、私たちの業務実践が現在または未来に適用される詐欺および乱用または他の医療保健法律および法規に関連する現行または未来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。もし私たちの運営がこれらの法律または任意の他の私たちに適用される可能性のある政府法規に違反していることが発見された場合、私たちは重大な民事、刑事と行政処罰、損害賠償、罰金、返還、監禁、政府援助に参加する医療計画(例えばMedicareおよびMedicaid)から除外され、追加の報告要件および監督に直面する可能性があり、もし私たちが会社の誠実な合意または同様の合意の制約を受けて、これらの法律違反に関する告発、契約損害、名声損害、および私たちの業務の削減または再編を解決することができます。持続的に変化するコンプライアンス環境、および異なるコンプライアンスまたは報告要件を有する複数の司法管轄区域の必要性に適合するために、穏健かつ拡張可能なシステムを確立し、維持することは、医療会社が1つまたは複数の要件と衝突する可能性を増加させる。
もし私たちがそれと業務を展開することを期待している医師または他の提供者または実体が適用法律を遵守していないことが発見された場合、彼らは政府の援助された医療計画から除外されることを含む重大な刑事、民事または行政制裁を受ける可能性がある。解決策が私たちに有利であっても、医療法令に関連する訴訟や他の法的手続きは、巨額の費用を発生させる可能性があり、私たちの技術や管理者の正常な責任を分散させる可能性があります。さらに、公聴会、動議、または他の一時的な手続き、または事態の発展の結果が発表される可能性がある。もし証券アナリストや投資家がこれらの結果がマイナスだと思っていれば、私たちの普通株の価格に大きな悪影響を及ぼす可能性がある。このような性質の訴訟または訴訟は、私たちの運営損失を大幅に増加させ、開発、製造、販売、マーケティング、または流通活動に使用できる資源を減少させる可能性がある。適用される医療法律法規に関連する訴訟または他の訴訟の開始および継続による不確実性は、市場での競争能力に悪影響を及ぼす可能性がある。
私たちの候補製品のアメリカと海外での商業化は、政府当局と個人健康保険会社を含め、保険と十分な補償レベルをどの程度提供するか、私たちの候補製品に有利な価格設定政策を実施する第三者支払者に部分的に依存する。もし私たちの候補製品がカバー範囲を獲得または維持し、十分な精算を得ることができなければ、承認されれば、これらの製品をマーケティングする能力を制限し、収入を創出する能力を低下させる可能性がある
私たちが規制承認を受ける可能性のあるどの製品のカバー範囲や精算状態にも、大きな不確実性がある。米国や他の国では,治療を受けた患者は通常,第三者支払者によって彼らの治療に関連する費用の全部または一部を精算する。第三者支払者(政府医療計画(例えば、米国のMedicare、MedicaidまたはTRICARE)、管理型ヘルスケア提供者、個人健康保険会社、健康維持組織、および他の組織を含む)は、私たちの製品の保証範囲および精算の十分性を含み、多くの患者が医療サービスおよび医薬品(例えば、私たちの候補製品)を負担することができるために重要である。第三者支払者はどんな薬を使うかを決定します
支払いと清算水準が確立されるだろう。“”というタイトルの部分を参照保証と精算を請け負う私たちの2022年年次報告書にあります。
私たちが私たちの候補製品を商業化することに成功するかどうかは、第三者支払人が私たちの製品と関連治療に提供するカバー範囲と十分な補償にある程度依存するだろう。また、支払者が製品に保険を提供することを決定することは、十分な返済率を承認することを意味するものではない。保険や十分な精算が得られない場合、あるいは限られたレベルに限られていれば、候補製品を商業化することに成功できないかもしれません。保険を提供しても、承認された精算金額が十分に高くない可能性があり、十分な投資リターンを実現するために十分な価格設定を確立したり維持したりするのに十分ではないかもしれません。第三者支払人が私たちの製品を使用している医療製品や治療を保証しないか、単独で精算しないと決定すれば、承認されると、医師の私たちの製品への使用が減少する可能性があります。
私たちは、米国および他の国/地域の保険および精算が、私たちの現在または未来の候補製品に適用されるか、または私たちの現在または未来の候補製品を使用する任意の手続きに適用され、得られる可能性のある任意の精算が不十分であるか、または将来的に減少またはキャンセルされる可能性があることを保証することはできない。
米国では、第三者支払者は製品の保証と精算に対して統一的な政策を持っていない。そのため、私たちの製品の保証範囲と精算範囲は支払人によって違います。支払者が製品に保険を提供するかどうかを決定するプロセスは、支払者が製品のために支払う支払率を設定するプロセスと分離することができる。1人の支払人が1つの製品に保険を提供することを決定し、他の支払者もその製品に保険と補償を提供することを保証することはできない。第三者支払者は、承認されたリストまたは処方表上の特定の製品に保証範囲を制限することもでき、FDAによって承認された特定の適応のすべての製品を含まない可能性がある。アメリカでは、新薬精算に関する主な決定は通常連邦医療保険と医療補助サービスセンター(CMS)によって行われ、CMSはアメリカ衛生と公衆サービス部(HHS)の一つの機関である。CMSは私たちの製品がどの程度連邦医療保険の下でカバーと精算されるかを決定し、個人支払者はしばしばCMSに大きく従う。支払人が精算を決定する際に考慮する要素は、製品があるかどうかに基づいている
•健康計画の下で保障された福祉
•安全で効果的で医学的に必要です
•特定の患者に適しています
•費用対効果があります
•実験的でも調査的でもない。
私たちは私たちの候補製品の保証範囲と精算範囲が利用できることを保証することができません。あるいは私たちの候補製品の潜在的な収入を正確に推定することができません。また、私たちが開発する可能性のあるどの製品も保証と精算を得ることができません。
また、米国や海外の第三者支払者が医療コストの制限や低減に力を入れていることは、支払人組織が新承認製品の保証範囲や精算レベルを制限する可能性があるため、候補製品の支払いや十分な支払いを提供できない可能性がある。販売が承認される可能性のある製品の保証範囲や精算を確保するためには、我々の製品の医療必要性と費用効果を証明し、FDAや同様の規制承認を得るのに必要なコストを証明するために、高価な薬物経済学的研究を行う必要があるかもしれない。さらに、購入者、個人健康計画、または政府医療計画に割引を提供する必要があるかもしれない。それにもかかわらず、私たちの候補製品は医学的に必要で費用効果的だと思われないかもしれない。第三者支払者が、製品が他の利用可能な療法と比較して費用対効果があると思わない場合、彼らは承認後にその製品をその計画の下の福祉としてカバーしないかもしれない、または、支払いレベルが会社にその製品を販売させて利益を得るのに十分ではない可能性があると思う場合がある。私たちは、私たちの任意の候補製品を潜在的に販売する時、第三者支払人からの定価圧力に遭遇すると予想しています。
最後に、いくつかの外国の国では、薬品の提案価格は必ず承認されなければならず、合法的に発売されることができる。各国の薬品定価に対する要求は大きく異なる。例えば、EU加盟国は、その国の健康保険制度が精算を提供する医療製品の範囲を制限することができ、人が使用する医療製品の価格を制御することができる。精算または定価の承認を得るために、その中のいくつかの国は臨床試験の完成を要求する可能性があり、特定の候補製品のコスト効果を現在利用可能な治療法と比較する。EU加盟国は医薬製品の具体的な価格を承認することができ、医薬製品を市場に投入する会社の収益力に対して直接或いは間接的に制御する制度を採用することもできる。連合加盟国間の接近法には食い違いが発生している。例えば,フランスでは,有効な市場参入は病院との合意によって支援され,製品は社会保障基金が精算する可能性がある。薬品価格は保健品経済委員会と協議した。薬品に対して価格規制や精算制限を実施することが保証されない国は有利なことを許可するだろう
私たちのすべての候補製品の精算と定価の手配。歴史的に見ると、EUで発売された製品は米国の価格構造に従わず、通常EUの価格は米国の価格より明らかに低いことが多い。
今後の医療立法は、臨床計画の推進や候補製品のマーケティング承認および商業化の難しさやコストを増加させ、設定可能な価格に影響を与える可能性があります。
米国や他の管轄地域では,医療システムの複数の立法や規制面の改革や提案中の改革が継続されることが予想され,将来の運営結果に影響を与える可能性がある。特に,米国連邦や州レベルでは,医療コストの低減と医療の質の向上を図る取り組みが継続されている。米国の個別州も、価格または患者の精算制限、割引、いくつかの製品参入およびマーケティングコスト開示の制限、および透明性措置を含む、価格または患者の精算制限、割引、特定の製品参入およびマーケティングコスト開示の制限および透明性措置を含む法律を立法し、実施することが増えており、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。“”というタイトルの部分を参照企業−政府規制−医療改革私たちの202年度報告書にあります。
将来的にはより多くの州と連邦医療改革措置がとられ,いずれも州と連邦政府が特定の医療製品やサービスをカバーする程度を制限し,連邦と州政府が医療製品やサービスに支払う金額を制限する可能性があると予想される。これは、私たちが開発した任意の候補製品に対する需要の減少、または追加の価格設定圧力を招く可能性がある。
米国以外の市場では,精算や医療保険支払い制度は国によって異なり,多くの国で特定製品や療法に価格上限が設定されている。米国以外の価格規制法規は特定市場の収益力に大きな影響を与える可能性があり、これらの法律が変化すれば、さらなる不確実性をもたらす。
私たちは米国や任意の他の司法管轄区域の将来の立法や行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することができない。政府、保険会社、医療組織、その他の医療サービスを管理する支払人は、医療コストの抑制または低減、および/または価格規制の実施に努力し続けており、悪影響を及ぼす可能性がある
•私たちの候補製品の需要は、もし私たちが規制部門の承認を得たら
•私たちは承認された製品のために公平だと思う価格を設定することができます
•私たちは収入を作ったり利益を達成したり維持したりします
•私たちが払わなければならない税金レベル
•資金の入手可能性。
もし私たちまたは私たちが接触する可能性のある任意の第三者が既存の要求の変化にゆっくりまたは適応できない場合、または新しい要求や政策を採用することができない場合、または私たちまたはこれらの第三者が規制適合性を維持できない場合、私たちの候補製品は得られた可能性のあるいかなる規制承認も失う可能性があり、私たちは利益を達成したり維持することができないかもしれない。
もし私たちが賛助した臨床試験から識別可能な患者の健康情報を得て、適用されたプライバシー法によって、私たちはアメリカと他の司法管轄区で潜在的な責任に直面する可能性がある。
ほとんどの医療提供者は、私たちがそれから患者の健康情報を得ることができるいくつかの研究機関を含み、1966年に公布された“健康保険携帯性と責任法案”(HIPAA)によって公布されたプライバシーおよび安全法規に基づいて制限され、この法案は“経済および臨床健康情報技術法案”によって改正されている。事実および状況によると、HIPAAによって許可されていない方法でHIPAAによってカバーされているエンティティによって維持されている個別に識別可能な健康情報を取得、使用、または開示する場合、私たちは民事、刑事、および行政処罰を受ける可能性がある。また,州法の制約を受け,個人情報が漏洩した場合に影響を受けた個人や州規制機関に通知することが要求される可能性があり,HIPAAが保護している健康情報よりも広い情報種別である。
世界のデータ保護構造は急速に発展しており、私たちは多くの連邦、州と外国の法律法規、および規制ガイドラインの制約または影響を受ける可能性があり、これらの法規と法規は個人データの収集、使用、開示、転送、安全と処理を管理しており、例えば、私たちが収集した臨床試験に関連する参加者および医療保健提供者の情報を管理している。予測可能な未来には、基準および法執行慣行を実施することはまだ不確定である可能性があり、これは、私たちの業務に不確実性をもたらし、私たちまたは私たちのサービスプロバイダがいくつかの司法管轄区域で個人データを収集、保存、移転、使用、および共有する能力に影響を与え、私たちが責任を負うか、または追加のコンプライアンスまたは他のコストを適用することをもたらすかもしれない。私たちは連邦、州、外国を守ることができなかったと思われています
法律または自律基準は、否定的な宣伝、管理時間と精力の移転、そして政府の実体または他の人たちが私たちに訴訟を提起することをもたらす可能性がある。
米国では、2018年にカリフォルニア消費者プライバシー法、またはCCPAが、2020年1月に施行される。CCPAは、消費者に新しいデータプライバシー権を提供し、消費者または家庭の特定の個人データを処理するエンティティにより多くのプライバシーおよびセキュリティ義務を課すことを含む、会社に新しい運営要件を提供する。このような要求は私たちのコンプライアンス費用と潜在的な責任を増加させるかもしれない。CCPAはカリフォルニア住民により大きな権利を与え,彼らの個人情報にアクセスして削除し,ある個人情報を共有しないことを選択し,彼らの個人情報がどのように使用されるかに関する詳細な情報を受信することができる.CCPAは違反行為に対する民事処罰と,データ漏洩に対する個人訴権を規定しており,データ漏洩訴訟が増加すると予想される。現在HIPAAと臨床試験法規に制約されている保護された健康情報は例外的な状況があるが、CCPAは著者らのいくつかの業務活動に影響を与える可能性がある
また、カリフォルニアプライバシー権法案(CPRA)は2023年1月1日に施行されたCCPAを改正した。CPRAが提出した修正案は、カバーする企業に追加義務を課し、消費者権利を拡大し、特定の敏感な個人情報に新たな義務を課すことを含むCCPAを大幅に修正した。CPRAはまた、CCPAを実施し実行する権利を有する新しい国家機関を作成した。改正された“海外腐敗防止法”の影響は重大である可能性があり、私たちのデータ収集や処理のやり方や政策を修正し、規制法および/または訴訟における私たちの潜在的なリスクを遵守し、増加させるために、大量のコストと支出を発生させる必要があるかもしれない。
また,多くの州では広範な消費者プライバシー法が成立しており,これらの法律は多くの点でCCPAと類似しており,多くの他州が類似した法律を提出するにつれて,他の州が追随する可能性が高く,包括的なプライバシーを重視した立法も通過する可能性が高い。このような立法が通過する場合、このような立法は、複雑性、要求のさらなる変化、制限、および潜在的な法的リスクを増加させる可能性があり、コンプライアンス計画に追加のリソースを投入する必要があり、データ収集戦略および以前の有用なデータの利用可能性に影響を与え、コンプライアンスコストの増加および/またはビジネス実践および政策の変化をもたらす可能性がある。アメリカの異なる州に全面的なプライバシー法が存在することは、私たちのコンプライアンス義務をより複雑で費用を高くし、私たちが法執行行動を受けたり、他の方法で規則に合わないことで責任を負う可能性を増加させるかもしれない。さらに、州レベルの全面的な法律に加えて、いくつかの州はプライバシー特定のための法律を提出または通過してきた。例えば,ワシントン州ではHIPAAがカバーしていない医療や健康関連情報のプライバシーを保護する範囲の広い法律が可決され,少数の州では生体識別情報に特化した法律が成立している。
プライバシーおよびデータ保護法の数および複雑さの増加、ならびに世界の法律または法規の他の変化、特に私たちの臨床試験からの医療データまたは他の個人情報のようないくつかのタイプの敏感なデータの保護の強化に関連する変化は、政府が私たちに対して法執行行動および重大な処罰をとることを招き、私たちの業務、財務状況、または運営結果に大きな悪影響を及ぼす可能性がある。
アメリカ以外にも、私たちは厳格なプライバシーとデータ保護法の挑戦に直面している。例えば、欧州経済地域(EEA)の立法者は、EUまたはEU一般データ保護条例(EU GDPR)とEU GDPRを通過し、これを連合王国、イギリスGDPRに変換し、総称してGDPRと呼ぶ。GDPRは,ヨーロッパ経済地域とイギリスに位置する被験者の個人データのコントローラやプロセッサに,健康,生体認識,遺伝情報を含む“特殊クラスデータ”の特殊保護を含むより厳しいデータ保護コンプライアンス要求を加え,不適合に対する重大な処罰を規定している。さらに、GDPRは、健康、遺伝子、および生体認証データ処理に関連する法律など、EEA加盟国に広範な権利を提供し、このようなデータの使用および共有能力をさらに制限するか、またはコスト増加をもたらし、私たちの業務および財務状態を損なう可能性がある。GDPRには、我々の業務に適用される可能性のあるコンプライアンス義務が含まれており、これは、ビジネス実践を変更し、違反行為に対する経済的処罰を増加させる可能性があります(最悪の違反に対して最高2000万ユーロ(イギリスGDPRは1750万ポンドと規定されている)の罰金を科す可能性があり、前会計年度の世界の年商の4%と、GDPR第82条のいずれかの個人が要求する経済的または非経済的損害賠償の権利を含む)。このような罰金に加えて、私たちは訴訟および/または否定的な宣伝に直面する可能性があり、これは私たちの名声や業務に実質的な悪影響を及ぼすかもしれない。
GDPRはまた、データ主体と消費者協会に個人訴訟権利を与え、監督当局に苦情を提出し、司法救済を求め、GDPR違反による損害について賠償を受けることができる。さらに、GDPRは、国境を越えたデータ転送の制限を含む。GDPRは,GDPRに拘束された個人データの処理における我々の責任と責任を増加させる可能性がある.はい
さらに、個別国によって実施されるGDPR要件を含むGDPRの遵守を保証するための追加のメカニズムを確立する必要があるかもしれない。
GDPRは,個人資料をどのように処理するかと,彼らの権利をどのように行使するかを資料当事者に通知し,個人資料を処理するための有効な法的基盤を確保し(これが同意すれば,同意を得る要求にはより高いハードルがある),敏感な個人データ(すなわち健康データ)が大規模に処理された場合にデータ保護官を任命することを要求する.また、GDPRは、欧州経済圏およびイギリス全体に強制的なデータ漏洩通知要求を導入し、我々の処理活動の記録を保存し、高リスク処理が存在する場合にデータ保護影響評価を記録することを要求し、サービスプロバイダと契約を締結する際に追加的な義務を課し、個人データを保護するための適切な技術的および組織的措置をとることを要求し、政策、プログラム、訓練、データ監査を含む適切なプライバシー管理を行うことを要求する。私たちは適切で適用された場合にGDPRを遵守する措置を取っているが、これは持続的なコンプライアンス過程だ。GDPRを遵守することは、厳格で時間のかかるプロセスとなり、私たちのビジネスコストを増加させたり、私たちのビジネス慣行を変更することを要求したりする可能性があります。GDPRや他の適用可能なヨーロッパ経済圏やイギリスの法律法規を遵守する努力が成功しなかったり、成功しないと考えられたりすれば、欧州経済地域および/またはイギリスにおける私たちの業務に悪影響を及ぼす可能性がある。
GDPRは、個人データが欧州経済地域およびイギリスから欧州経済区/イギリス以外の他の地域または第三国に移行するために厳しいルールを実施しており、場合によっては、主管データ保護当局(米国を含む)は、これらの地域または第3国が十分な国際移転保障措置(例えば、EU委員会が承認した標準契約条項、またはEU SCC、およびイギリス国際データ転送プロトコル/付録、またはイギリスIDTA)が存在しない限り、“十分な”プライバシー保護を提供していないと考えていることに留意されたい。EUのSCCまたはイギリスIDTAに依存してデータ転送を行う場合には、EU SCCおよびイギリスIDTAによる転送影響評価をケースベースで要求されることもあり、受信国の法律が送信された個人データを保護するために“実質的に同じ”保護を提供することを保証し、欧州経済圏やイギリスの規定と同様であり、この基準に適合しない場合には、補完的な措置が必要となる可能性があるEEAとイギリスのデータ保護制度下の国際移転義務は、多くの努力とコストを必要とし、EEAとイギリスの個人データの位置と、どのサービスプロバイダを利用してEEAとイギリスの個人データを処理することができるかを戦略的に考慮する必要があるかもしれませんデータ保護法に従って個人データをヨーロッパ経済圏から米国に移すことができない行為は、私たちの実験能力を阻害し、私たちの業務や財務状況に悪影響を及ぼす可能性があります。
英国はEU GDPR下の第3の国の一つとされているが、欧州委員会は追加的な保障措置を必要とすることなく、欧州経済圏加盟国から英国へのデータの移転を可能にするイギリスに有利な十分な決定を採択した。イギリス政府は、イギリスからヨーロッパ経済圏への個人データ転送が依然として自由に流動していることを確認した。イギリスとEUのデータ保護法のある方面の関係はまだ不明であり、イギリスのデータ保護法律と法規が中長期的にどのように発展するか、イギリスに出入りするデータ転送がどのように長期的に規制されるかも不明である。また、イギリス政府はイギリス立法手続きにデータ保護とデジタル情報法案、あるいはイギリス法案を導入している。イギリス法案の目的はイギリスの離脱後のデータ保護制度を改革することだ。可決されれば、英国法案の最終版は英国と欧州経済圏データ保護制度との類似性をさらに変更し、欧州委員会の英国充足率の決定を脅かす可能性がある。また、欧州経済圏加盟国は、GDPRから部分的にずれている可能性のあるGDPRを実施するための国家法律を採択した。また,欧州経済圏加盟国の主管当局のGDPR義務の解釈は国によってやや異なる可能性があるため,欧州経済圏は統一された法的環境では動作しないと予想される。EU GDPRと英国GDPRのそれぞれの条項と法執行が将来さらに分化する可能性は、追加の規制課題と不確実性をもたらしてくれた。このような将来のイギリスの法律法規とEUの法律法規との相互作用に対する明瞭性の欠如は、私たちの個人データおよび私たちのプライバシーとデータ安全コンプライアンス計画を処理する法的リスク、複雑さ、コストを増加させ、イギリスとヨーロッパの経済地域に対して異なるコンプライアンス措置を実施することを要求するかもしれない。
米国やヨーロッパ以外では、CROや他の方法で業務を展開している多くの司法管轄区域も考慮および/または全面的なデータ保護立法が制定されている。しかし、私たちはGDPRと適用される欧州経済圏加盟国とイギリスプライバシー法に基づいて、私たちがそれらを遵守するための任意の措置を取って責任、費用、コスト、および他の運営損失を招くかもしれない
私たちのヨーロッパ経済区とイギリスで個人データを処理する司法管轄区域内では、私たちの業務活動は、ヨーロッパ経済区またはイギリスの個人行動の監視(例えば、臨床試験を行う際)を含む現地データ保護当局によって監視される可能性がある。私たちは多くの第三者に依存して私たちのサービスを提供し、その中のいくつかの第三者代表は私たちのEEAおよび/またはイギリス個人の個人データを処理します。私たちはすべてのこのようなプロバイダと契約を締結したり、契約を締結しようとしています。これらの手配によると、彼らは契約上、私たちの指示だけに基づいて個人資料を処理する義務があり、十分な技術と組織の安全措置が適切であることを保証するために最善を尽くしています。
さらに、いくつかの健康プライバシー法、データ漏洩通知法、消費者保護法、および遺伝子試験法は、私たちおよび/または私たちの協力者の運営に直接適用される可能性があり、個人の健康情報を収集、使用、および伝播することに制限を加えることができる。私たちまたは私たちの協力者が健康情報を取得する可能性のある患者、およびこの情報を私たちと共有する可能性のある提供者は、私たちの情報の使用および開示の能力を制限する法定または契約権利を持っている可能性がある。私たちは適用されるプライバシーとデータセキュリティ法律を持続的に遵守することを保証するために、多くの資本と他の資源を費やす必要があるかもしれない。私たちがプライバシー権を侵害したり、私たちの契約義務に違反していると主張して、私たちが責任がないことが発見されても、弁護は高価で時間がかかる可能性があり、否定的な宣伝を招く可能性があり、私たちの業務を損なう可能性があります。
もし、私たちまたは第三者CMO、CROまたは他の請負業者またはコンサルタントが適用される連邦、州/省または地方規制要件を遵守できない場合、私たちは一連の規制行動の影響を受ける可能性があり、これらの規制行動は、私たちまたは私たちの請負業者が私たちの候補療法を開発および商業化する能力に影響を与える可能性があり、商業化できる任意の影響を受ける療法の販売を損害または阻止する可能性があり、または私たちの療法の開発、商業化、およびマーケティングのコストおよび支出を大幅に増加させる可能性がある。いかなる脅威や実際の政府の法執行行動も否定的な宣伝を生む可能性があり、そうでなければ、これらの資源は私たちの業務の他の側面に使用できるように大量の資源を投入することが要求される。ソーシャルメディアをますます使用することは、責任、データセキュリティが破壊され、または名声を損なう可能性がある。
また,上記の各医療法に支配されている州は海外と同等の法律であり,その中にはいくつかの法律の範囲が広く,支払者が誰であろうと適用される可能性がある。
もし私たちまたは私たちの第三者製造業者とサプライヤーが環境、健康、安全の法律法規を遵守できなかった場合、私たちは罰金や処罰を受けたり、私たちの業務の成功に悪影響を及ぼす可能性のあるコストが発生する可能性があります。
私たちは多くの環境、健康と安全法律法規の制約を受けて、それらの研究室の手続きと危険材料と廃棄物の処理、使用、貯蔵、処理と処理を管理する法律と法規を含む。我々の研究·開発活動は,生物や危険材料を使用し,危険廃棄物製品を生成することに関するものである。私たちは一般的に第三者と契約を結び、このような材料と廃棄物を処理する。私たちはこれらの材料の汚染や傷害リスクを除去することができません。これは私たちの商業化努力、研究開発努力、業務運営の中断を招き、環境破壊は高価な整理を招き、適用された法律と法規に従ってこれらの材料と指定廃棄物の使用、貯蔵、処理、処分に責任を負うことができます。我々の第三者CMOがこれらの材料を処理·処分する際に使用するセキュリティプログラムは,これらの法律法規が規定する基準にほぼ適合していると信じているが,状況が確かにそうである保証はなく,これらの材料の意外な汚染や傷害のリスクを解消することもできない。このような事件が発生した場合、私たちはそれによって引き起こされた任意の損害に責任を負う可能性があり、この責任は私たちの資源範囲を超える可能性があり、州、連邦、または他の適用機関は、いくつかの材料の使用を制限し、および/または私たちの業務運営を中断するかもしれない。また,環境法律法規は複雑で変化が頻繁であり,より厳しくなる傾向にある。私たちはこのような性質のどんな変化の影響も予測できないし、私たちの未来のコンプライアンスを決定することもできない。また、現在または未来の環境、健康、安全法律法規を遵守するためには、巨額のコストが生じる可能性がある。このような現行または未来の法律法規は私たちの研究、開発、または生産努力を損なうかもしれない。このような法律法規を遵守しないことはまた巨額の罰金、処罰、または他の制裁につながる可能性がある。
私たちは私たちのコストと支出を支払うために労働者補償保険を維持していますが、危険材料の使用や他の仕事に関する傷害により従業員が負傷する可能性がありますが、潜在的な責任に十分な保険を提供できない可能性があります。私たちは生物学的または危険な廃棄物の暴露または汚染による損害と罰金を含む特定の生物廃棄物または危険廃棄物保険、労働者補償または財産と死傷者および一般責任保険を保証しない
私たちはアメリカとある外国の輸出入規制、制裁、禁輸、反腐敗法律、反マネーロンダリング法律と法規の制約を受けている。このような法律基準を遵守することは国内と国際市場での私たちの競争能力を弱めるかもしれない。私たちは違反によって刑事責任と他の深刻な結果に直面するかもしれないし、これは私たちの業務を損なうかもしれない。
我々は、米国輸出管理条例、米国税関条例、米国財務省外国資産規制弁公室によって実施された様々な経済·貿易制裁条例、1977年に改正された米国反海外腐敗法、米国連邦法典第18編201節に含まれる米国国内賄賂法規、米国旅行法、2001年米国愛国者法、および私たちが活動している国の他の州と国の反賄賂および反マネーロンダリング法を含む輸出規制と輸入法律の制約を受けている。腐敗防止法は、会社およびその従業員、代理人、請負業者、および他の協力者が、公的または民間部門の受給者に不正な支払いまたは任意の他の価値のあるものを直接または間接的に許可、約束、提供、または提供することを禁止すると広く解釈されている。将来的にはアメリカ以外の場所で第三者を招いて臨床試験を行うかもしれません
私たちが商業化段階に入って、および/または必要な許可、許可証、特許登録、その他の規制承認を得ると、私たちは私たちの製品を海外に販売する権利があります。また、政府機関や政府付属病院、大学、その他の組織の役人や従業員と直接または間接的なインタラクションを行うことも可能である。私たちは、私たちが明確に権限を持っていなくても、または実際にこれらの活動を理解していなくても、従業員、代理、請負業者、および他の協力者の腐敗や他の不法活動に責任を負わなければならないかもしれない。上記の法律および法規に違反するいかなる行為も、重大な民事および刑事罰金および処罰、監禁、輸出入特権の喪失、資格取り消し、税収の再評価、契約違反および詐欺訴訟、名誉損害、およびその他の結果を招く可能性がある。
従業員事務、管理成長、運営事務に関するリスク
私たちは私たちの重要な人たちに強く依存して、新しいキーパーソンを募集する予定だ。もし私たちが高い素質の人材を誘致し、維持することに成功できなければ、私たちは私たちのビジネス戦略を成功的に実施することができないかもしれない
競争の激しいバイオテクノロジーと製薬業界での競争能力は私たちが高い素質の管理、科学、医療、販売とマーケティング人員及び他の人員を誘致し、維持する能力にかかっている。私たちは私たちの総裁と最高経営責任者、最高科学官、首席医療官、最高財務官、最高法務官、最高人事官、最高業務官を含む私たちの経営陣、科学、医療従事者に高度に依存しています。どんな幹部、他の重要な従業員、他の科学と医療コンサルタントのサービスを失うこと、そして適切な代替者を見つけることができないことは、製品開発の遅延を招き、私たちの業務を損なう可能性がある。新たに任命された幹事や管理者を統合すれば、秩序ある移行プロセスに参加することが予想されるが、管理者の注意を業務関心から移行させ、他のキーパーソンを引き留めることができなかったり、機関知識を失ったりするなど、管理移行に関する様々なリスクや不確定要因に直面している。
私たちはマサチューセッツ州ウォータータウンの施設で事業を展開しています。マサチューセッツ州地区は多くの他の生物製薬会社及び多くの学術と研究機関の本部である。私たちの市場は技術者に対する競争が非常に激しく、私たちが受け入れられる条件で高い素質の人員を採用し、維持する能力を制限する可能性があり、甚だしきに至っては根本的にはできない。米国の移民と仕事認可法律法規の変化は、科学と専門人材の流動を制限する法律法規を含み、政治力と経済活動レベルの重大な影響を受ける可能性がある。移民やビザの法律法規の立法または行政変更が私たちの採用プロセスや目標、または非米国市民に関連するプロジェクトを損害した場合、私たちの業務は実質的な悪影響を受ける可能性がある。
価値のある従業員がわが社に残ることを奨励するために、賃金や現金奨励のほか、時間の経過やマイルストーンの実現に基づいて付与できる株式オプションや他の持分奨励を提供します。時間が経つにつれて、私たち従業員に付与された株式奨励の価値は、私たちの株価変動の大きな影響を受ける可能性があり、これらの変動は私たちの制御範囲を超えており、いつでも他社が提供するより利益のある見積もりを相殺するのに十分ではないかもしれない。マイルストーンの業績に基づいて付与された株式奨励についても、状況はそうかもしれない。私たちは価値のある従業員を引き留めるために努力しているにもかかわらず、私たちの管理、科学、開発チームのメンバーは短時間で私たちとの雇用関係を終了するかもしれない。私たちは私たちの役員職員と雇用協定を持っていますが、これらの雇用協定は自由に雇用できることを規定しています。これは、私たちのどの幹部従業員も通知の有無にかかわらず、いつでも私たちの職場を離れることができることを意味します。私たちの成功はまた、私たちが引き続き高技能の初級、中級と高級管理者、及び初級、中級と高級科学、医療、一般と管理者を吸引、維持、激励する能力があることにかかっている。
また、私たちは科学と臨床コンサルタントを持っていて、私たちの発展と臨床戦略の制定を助けてくれます。これらのコンサルタントは私たちの従業員ではなく、他の実体と約束、相談、または相談契約を持っているかもしれません。これは彼らの私たちに対する利用可能性を制限するかもしれません。また、私たちのコンサルタントは他社と合意し、これらの会社が私たちと競争する可能性のある製品や技術を開発するのを助けるかもしれません。
私たちの内部コンピュータシステムまたは私たちの任意の協力者、サプライヤー、請負業者、またはコンサルタントのコンピュータシステムは、故障したり、セキュリティホールに遭遇したりする可能性があり、これは、私たちの製品開発計画が実質的に破壊され、私たちの名声を損なうか、または私たちに責任を負わせ、私たちの業務および財務業績に悪影響を及ぼす可能性があります
我々の内部コンピュータシステムおよび任意の協力者、サプライヤー、請負業者またはコンサルタントのコンピュータシステムは、コンピュータウイルス、許可されていないアクセス、自然災害、テロ、戦争、および電気通信および電気故障の破壊を受けやすい。これまで、重大なシステム障害、事故、セキュリティホールは経験していませんが、このような事件が発生して私たちの運営が中断された場合、私たちのビジネス機密や他の固有情報の損失によるものであっても、他の同様の中断によるものであっても、私たちの開発計画や業務運営の大きな中断を招く可能性があります。例えば、完成したまたは未来の臨床試験における臨床試験データの損失は、私たちの市場承認作業の遅延を招き、データを回復または複製するコストを著しく増加させる可能性がある。どんな中断やセキュリティホールも、私たちのデータやアプリケーションが失われたり、破損したりします
機密や独自の情報を適切に開示しなければ、私たちは責任を招く可能性があり、私たちの競争地位が損なわれる可能性があり、私たちの候補製品のさらなる開発と商業化が延期される可能性がある。しかも、私たちは私たちが保存している第三者情報にデータセキュリティ義務があるかもしれない。このような第三者データまたは情報の不正アクセスまたは使用は、罰金または他の処罰をもたらす可能性があり、これらの第三者との関係および私たちの運営に影響を与える可能性があります
私たちのプラットフォーム、システム、およびネットワークの任意の実際または感知されたセキュリティホールは、私たちの名声とブランドを損なう可能性があり、私たちを訴訟と可能な責任リスクに直面させ、セキュリティホールによる問題に対応し、緩和するために大量の資本と他の資源を必要とする。私たちが十分なサイバー犯罪と責任保険を維持する能力は減少するかもしれない。いくつかの管轄区域では、あるタイプの個人データのデータセキュリティホールに関連する場合に個人に通知することを要求する法律が制定されており、特定のパートナーと合意した合意は、セキュリティ事件が発生した場合に通知することを要求している。このようなタイプの強制開示はコストが高く、否定的な宣伝を招く可能性があり、私たちのパートナーが私たちのデータセキュリティ対策の有効性に自信を失う可能性がある。これらの事件のいずれも、私たちの名声を損なうか、または私たちに責任を負わせ、私たちの業務と財務業績に実質的で不利な影響を与える可能性がある。私たちはネットワーク責任保険を維持しているにもかかわらず、私たちはその保険範囲が実際に発生した責任を補うのに十分かどうか、あるいは経済的に合理的な条項で保険を受け続けるかどうか、あるいは根本的にはできない。
私たちの従業員、独立請負業者、サプライヤー、主要な調査者、CROとコンサルタントは規制基準と要求、およびインサイダー取引法律を遵守しないことを含む不当な行為または他の不適切な活動に従事する可能性があります。
私たちが直面しているリスクは、私たちの従業員、独立請負業者、サプライヤー、主要な調査者、CRO、CMO、およびコンサルタントが詐欺または他の不法活動に従事する可能性があるということだ。他にも、これらの当事者の不適切な行為には、以下のようなものがある
•意図的、無謀または不注意な行為、または研究および試験プログラムまたはFDAまたは外国の規制機関に規定されたような不正活動の開示;
•アメリカと海外の医療詐欺と法律法規の乱用
•私たちの普通株式取引に関連するアメリカ連邦証券法に違反し
•財務情報やデータを正確に報告できなかった
特に、医療業界の販売、マーケティング、商業配置は、詐欺、不正行為、リベート、自己取引、その他の乱用行為を防止するための広範な法律法規によって制約されている。これらの法律法規は、広範な定価、割引、マーケティングと販売促進、販売手数料、顧客インセンティブ計画、その他の業務手配を規範化している。その他の形式の不当行為は臨床試験過程で得られた情報を不当に使用すること、或いは著者らの臨床前研究或いは臨床試験中に虚偽データを作成することに関連する可能性があり、これは規制制裁を招き、著者らの名声に深刻な損害を与える可能性がある。私たちは私たちのすべての従業員に適用される商業行為と道徳基準、他の会社の管理とコンプライアンス文書、政策、定款を採択した。しかし、従業員と他の第三者の不適切な行為を常に識別して阻止できるわけではない。さらに、このような活動を発見し防止するための予防措置は、未知または未管理のリスクまたは損失を効果的に制御することができないか、またはこれらの法律や法規を遵守できないことによる政府の調査または他の行動または訴訟から私たちを保護することができない可能性がある。さらに、私たちは誰かが詐欺や他の不適切な行為を告発する可能性があるという危険に直面している。もし私たちにこのような訴訟が提起された場合、私たちは私たちの権利を弁護または維持することに成功できませんでした。これらの訴訟は、民事、刑事および行政処罰、損害賠償、罰金、連邦医療保険、医療補助および他の連邦医療保健計画から除外される可能性があり、契約損害、名声損害、利益および将来の収益の減少、および/または私たちの業務を削減することを含むかもしれません。そのいずれもが、私たちの業務の見通し、財務状況、および運営結果に悪影響を及ぼす可能性があります
私たちの普通株に関するリスクは
もし私たちが未来にもっと多くの資本を集めることに決めたら、あなたの投資は希釈されるだろう。
私たちは将来、市場状況、戦略的考慮、運営要求に応じて、株式または他の株に変換可能な証券を売却することで追加資本を調達することを選択するかもしれない。ある程度、私たちはこのような方法で追加資本を調達し、私たちの株主は希釈されるだろう。将来的には、我々普通株や他の株式証券の発行、あるいはこのような性質の売却が発生する可能性があると考え、我々普通株の取引価格に悪影響を及ぼす可能性があり、将来的に株式や株式証券を発行することで資金を調達する能力を弱める可能性がある。将来の普通株の売却や将来売却可能な普通株が我々の普通株の取引価格に影響を与えるかどうかは予測できない
現在、私たちの普通株はナスダック世界の精選市場で発売されている。しかし、このような市場にはあなたが持っている私たちの普通株を売ることができるように十分な流動性がないかもしれません。
現在、私たちの普通株はナスダック世界の精選市場で発売されている。もし私たちの株の活発な取引市場が持続できなければ、あなたはあなたの株を迅速にあるいは市価で売ることができないかもしれません。私たちは投資家の私たちに対する興味がどの程度活発で流動性の強い取引市場を維持することにつながるのか予測できない。また、活発でない市場は、普通株を売却することで資金を調達する能力を弱める可能性があり、戦略的パートナーシップを達成したり、私たちの普通株を対価格で会社や製品を買収する能力を弱める可能性があります。
証券や業界アナリストが私たちの業務に関する研究や報告を発表または停止しない場合、または彼らが私たちの株式に不利または誤った意見を発表した場合、私たちの株価や取引量は低下する可能性がある
私たちの普通株の取引市場は、業界や証券アナリストが発表した、私たちの業務または標的タンパク質分解空間に関する研究と報告の影響を受け続けている。私たちはこのようなアナリストを統制できない。既存のアナリストが研究記事を提供し続けることは保証されないし、新しいアナリストが研究報告を提供し始める。私たちはすでにアナリストの報告を得ているにもかかわらず、もし私たちのどのアナリストが私たち、私たちのビジネスモデル、私たちの知的財産権、あるいは私たちの株式表現に否定的または誤った意見を発表した場合、あるいは私たちの臨床前研究と未来の臨床試験と運営結果がこれらのアナリストの予想を満たしていなければ、私たちの株価は下落する可能性がある。もし一人以上のアナリストが私たちの報告書を停止したり、私たちに関する報告書を定期的に発表できなかった場合、私たちは金融市場での可視性を失う可能性があり、これは逆に私たちの株価や取引量を低下させる可能性がある。
私たちの普通株の価格は大きく変動する可能性があり、これは私たちの普通株の購入者に大きな損失をもたらすかもしれない。
私たちの普通株の株式取引価格はずっと不安定で、様々な要素の広範な変動を受け続ける可能性があり、その中のいくつかの要素は私たちがコントロールできない。株式市場,特に小さいバイオ製薬会社の市場は,極端な変動を経験しており,この変動は特定の会社の経営業績とは無関係であることが多い。この変動のため、普通株を獲得したか、あるいはそれ以上の価格で普通株を売ることができないかもしれません。私たちの普通株の市場価格は多くの要素の影響を受けるかもしれません
•競争力のある製品または技術の成功度または看護基準の変化;
•私たちの候補製品や競争相手の臨床前研究と臨床試験結果
•私たちの臨床開発活動の時間と進展と臨床試験データを発表する時間
•アメリカや他の国の法規や法律の発展
•特許出願、発行された特許または他の固有の権利に関連する開発または紛争;
•キーパーソンの採用や退職
•私たちの任意の候補製品または臨床開発計画に関連する費用レベルと、私たちが持っている現金、現金等価物、および有価証券の価値
•私たちは他の候補技術や製品の結果を発見し開発し、得るために努力している
•財務結果、発展スケジュール、または証券アナリストの提案に関する推定の実際または予想変化;
•私たちの財務業績や私たちと似ていると思われる会社の財務業績の違い
•医療支払い制度の構造を変え
•製薬とバイオテクノロジー部門の市場状況
•最近の新冠肺炎の大流行のような公衆衛生危機、流行病と流行病の影響
•一般経済、産業、市場状況;
•“リスク要因”の節で述べた他の要因
上記のいずれかの要素が私たちの業務、見通し、または運営にマイナス影響を与える可能性があると考えられる場合、または私たちの経営業績が投資家や証券アナリストの予想より低い場合、私たちの普通株の価格は大幅に下落する可能性がある。かつてある会社の証券市場価格の変動に伴い、証券は
その会社に対する集団訴訟はよく提起される。私たちにこのような性質の訴訟を提起すれば、これらのクレームを弁護するための巨額の費用を発生させ、経営陣の注意と資源を移転させる可能性があり、これは私たちの業務、財務状況、運営結果、将来性を深刻に損なう可能性がある。また、私たちの役員や上級職員責任保険コストはこのような訴訟によって増加する可能性があり、私たちの保険会社が任意の保険を提供することを要求される前に、私たちの保険免除額は大きいかもしれません
私たちは私たちが集めた資本を使用する上で広範な自由裁量権を持っており、私たちの資本を有効に使用できないかもしれない。
私たちの経営陣は、私たちが以前に融資した純収益を運用する上で、私たちの初公募株と後続の公開発行を含む幅広い裁量権を持っており、得られた資金を私たちの運営業績を改善したり、私たちの普通株の価値を高めたりしない方法に使うことができます。私たちの経営陣がこれらの資金を有効に使用できなかった場合、財務損失を招く可能性があり、これは私たちの業務に悪影響を与え、私たちの普通株価格の下落を招き、候補製品の開発を延期する可能性があります。使用する前に、私たちは私たちの融資活動の純収益を収入や切り下げが生じないように投資するかもしれない。
我々の役員、取締役、主要株主は、株主に承認された事項を制御または著しく影響する能力があるだろう
私たちの役員と取締役、そして私たちの株主を加えて、彼らはアメリカ証券取引委員会に提出した書類を通じて、彼らは全部で私たちが発行した普通株の5%以上を持って、私たちのかなりの割合の株式を保有していると報告しました。したがって、私たちの役員と取締役は、私たちの5%以上の株主に加えて、この所有権地位を通じて私たちを統制することができます。これらの株主が共同で行動すれば、私たちの株主に提出された承認事項、そして私たちの管理と事務を引き続き制御します。例えば、この人たちが一緒に行動することを選択した場合、彼らは取締役の選挙と、私たちのすべてまたはほとんどの資産の任意の合併、合併、または売却の承認を制御するだろう。このような所有権制御の集中可能性:
•制御権の変更を延期、延期、または阻止する
•私たちの経営陣や取締役会を強化する
•私たちの他の株主が望む可能性のある合併、合併、接収、または他の業務合併を妨げる。
私たちの定款文書とデラウェア州法律における反買収条項は、制御権の変更を延期または阻止する可能性があり、これは私たちの普通株の市場価格を制限し、私たちの株主の現在の経営陣の変更を阻止または罷免する試みを阻止または挫折させる可能性がある
当社の会社登録証明書の改訂及び再記載の定款に含まれる条項は、わが社の支配権変更を延期または阻止することができ、または当社の株主が有利と考える取締役会の変動を遅延または阻止する可能性があります。いくつかの規定には
•取締役会は3つのレベルに分かれ、3年間の任期を交錯させ、その結果、すべての取締役会メンバーが1回の選挙で選出されるのではないか
•株主が書面での同意で行動することを禁止した結果、すべての株主の行動は私たちの株主会議で行われなければならない
•株主特別会議は、当時取締役を務めていた多数の賛成票で可決された決議に基づいて取締役会のみが開催されることを要求した
•株主指名と指名が取締役会に入る事前通知要求;
•私たちの株主は、法律の要求の他の投票でなければ、取締役会のメンバーを罷免しないことを要求し、当時取締役選挙で投票する権利があった私たちの議決権のある株の3分の2以上の流通株の承認を得なければならない
•株主行動によって任意の附例または会社登録証明書の特定の条項を修正するために、議決権を有する株の3分の2以上の全流通株の承認を要求する
•取締役会が株主の承認を受けていない場合には、取締役会が決定した条項に従って優先株の権力を発行し、優先株は普通株式保有者の権利よりも高い権利を含むことができる。
また、私たちはデラウェア州で登録されているので、私たちはデラウェア州会社法第203条の規定によって管轄されています。この条項は、株主が議決権を持って発行された株の15%以上のいくつかの業務統合を禁止する可能性があります。これらの反買収条項と私たちが改訂して再記載した証明書の中の他の条項
会社の設立および定款の改訂と再記述は、株主や潜在的な買収者が私たちの取締役会に対する支配権を得ることを困難にしたり、当時の取締役会が反対する行動を起こしたりして、わが社の合併、買収要約、代理権に関する争いを延期または阻害する可能性もある。これらの規定はまた、代理権競争を阻止し、あなたと他の株主があなたが選択した取締役を選ぶことを難しくしたり、あなたが希望する他の会社の行動を取らせたりすることができます。制御権の変更、取引、取締役会の変動を遅延または阻止することは、私たちの普通株の市場価格の下落を招く可能性がある
上場企業としては、引き続き追加コストを発生させ、我々の経営陣はコンプライアンスイニシアチブやコーポレートガバナンス実践に多くの時間を投入する必要があるだろう
上場企業としては、大量の法律、会計、その他の費用を負担し続け、民間会社としては、そうする必要はありません。2002年のサバンズ-オキシリー法案、またはサバンズ-オクスリ法案、ドッド·フランクウォール街改革および消費者保護法、ナスダック株式市場有限責任会社の上場要件および他の適用される証券規則および法規は、効率的な開示、財務制御、およびコーポレートガバナンス慣行の確立と維持を含む上場企業に対して様々な要求を提出した。私たちの経営陣と他の人たちはこのようなコンプライアンス計画に多くの時間を投じた。しかも、このような規則と法規は私たちの法律と財務コンプライアンスと保険コストを増加させ、いくつかの活動をより時間と費用を増加させる。例えば、これらの規制は、私たちが取締役や上級管理職責任保険を獲得することをより困難かつ高価にし、逆に合格した取締役会メンバーを引き付け、維持することを困難にする可能性がある。
私たちは、私たちが生成する可能性のある追加のコスト金額やこれらのコストの時間を常に予測したり、推定したりすることができないこのような規制を評価し続けている。これらの規則や条例もしばしば異なる解釈を受け,特殊性に欠ける場合が多いため,規制機関や理事機関が新たな指導意見を提供するにつれて,実践における適用は時間とともに変化する可能性がある。これは遵守事項に関する持続的な不確実性と、開示と統治慣行を絶えず修正するために必要なより高いコストをもたらす可能性がある。
サバンズ·オキシリー法404条または404条によると、私たちは、財務報告書の内部統制に関する経営陣の報告書を提出しなければならない。しかし、“より小さな報告会社”として、我々の独立公認会計士事務所が発行する財務報告内部統制の認証報告を含めて、私たちがもはや小さな報告会社ではなくなるまで要求されないであろう。2023年12月31日までの財政年度終了時には、“1934年証券取引法”(改正証券取引法)または“取引法”で定義されている“非加速申告会社”と“小さな報告会社”の資格を満たしている。私たちは第404条の規定に従うと、大量の会計費用を発生させ、多くの管理努力をかけなければならない。
内部資源を提供し続ける必要があり、外部コンサルタントを招聘することが可能であり、財務報告内部統制の十分性を評価·記録するための詳細な作業計画により、適宜ステップ改善制御プログラムを採用し、制御措置がファイルのように機能しているかどうかをテストにより検証し、財務報告内部統制の継続報告及び改善手順を実施する。私たちが努力したにもかかわらず、私たちは規定された時間枠内で、または根本的に結論を出すことができないかもしれない、すなわち、私たちは財務報告書の内部統制に有効であり、第404条の要求に適合する。さらに、私たちが過去または未来に取った措置が、将来の財務報告の内部統制に重大な欠陥や重大な欠陥が生じることを防止することを保証することはできません。将来的に1つ以上の重大な弱点を発見すれば、金融市場の不良反応を招き、将来的に資本市場に参入する機会を制限する可能性があり、統合財務諸表の簡素化の信頼性に自信を失っているからである。
私たちの改正と再記述の定款は、私たちの株主が起こしうるいくつかの訴訟の独占的なフォーラムとして特定の裁判所を指定し、これは、私たちの株主が有利な司法フォーラムを得ることが私たちとの紛争を処理する能力を制限することができる
当社の改正及び再記載された付例によれば、吾等が書面で別の裁判所を選択することに同意しない限り、デラウェア州衡平裁判所は、以下の件について州法律クレームを提出する唯一及び独占裁判所である:(I)吾等を代表して提起された任意の派生訴訟又は法律手続き;(Ii)吾等の任意の取締役、高級職員又は他の従業員が吾等又は当社株主に信頼された責任を有する訴訟を主張する任意の訴訟;(Iii)“デラウェア州会社法”又は当社が改正及び再記載された会社登録証明書又は改正及び再記載された付例のいずれかの規定に基づいて任意の訴訟を提起する。(Iv)私たちが改正および再記載した会社登録証明書または改正および再記載された定款の有効性を決定する任意の訴訟、または(V)内務原則によって管轄されるクレームを主張する任意の訴訟。私たちは私たちが改正して再説明した定款でこの条項をデラウェアフォーラム条項と呼ぶ。デラウェアフォーラム条項は、1933年の“証券法”(改正された)、“証券法”または“1934年取引法”(改正された)または“取引法”によって生じたいかなる訴訟原因にも適用されない
私たちが書面で代替裁判所を選択することに同意しなければ、米国マサチューセッツ州地域裁判所は任意の苦情を解決する唯一のかつ独占的な裁判所となることが、私たちの改正と再記述の付例によってさらに規定されている
証券法によると、私たちの本部はマサチューセッツ州ウォトタウンにあるので、訴訟理由が提起されます。私たちは改正されて再記述された規定でこの条項を連邦フォーラム条項と呼ぶ。また、私たちが改正して再記載した定款規定は、私たちの株式株式の任意の権益を購入または他の方法で獲得した個人または実体は、デラウェアフォーラム条項および連邦フォーラム条項を通知し、同意したとみなされる。
私たちが改訂して再記述した定款におけるデラウェアフォーラム条項と連邦フォーラム条項は、株主に任意のこのようなクレームを求める際に追加の訴訟費用をもたらす可能性がある。さらに、これらのフォーラム選択条項は、私たちの株主が司法フォーラムで、私たちまたは私たちの役員、役員、または従業員との紛争のクレームに有利だと思う能力を提出することを制限し、私たちと私たちの役員、役員、および従業員に対する訴訟を阻止する可能性があり、訴訟が成功すれば、私たちの株主に利益をもたらすかもしれません。また、デラウェア州最高裁判所は2020年3月に、デラウェア州法律によると、証券法に基づいて請求を要求する連邦裁判所選択条項は表面的に有効であると判断したが、他の裁判所が我々の連邦フォーラム条項を実行するかどうかには不確実性がある。もし連邦フォーラムの条項が実行できないことが発見されたら、私たちはこのような問題の解決に関連した追加費用を発生させるかもしれない。連邦フォーラムの条項はまた、その条項が実行不可能または無効だと主張する株主に追加的な訴訟費用を適用するかもしれない。デラウェア州衡平裁判所および米国マサチューセッツ州地域裁判所もまた、訴訟の株主が存在するか、または訴訟を提起する裁判所を選択する可能性があり、このような判決は、私たちの株主に多かれ少なかれ有利である可能性があることを含む、他の裁判所とは異なる判決または結果を下す可能性がある
私たちは予測可能な未来に私たちの株に現金配当金を支払わないと予想されているので、資本増加(あれば)があなたの唯一の収益源になるだろう。
私たちは私たちの株の現金配当金を発表したり支払ったりしたことがない。私たちは現在、私たちの未来のすべての収益を維持するつもりで、もしあれば、私たちの業務の成長と発展に資金を提供します。したがって、私たちの普通株の資本付加価値は(もしあれば)あなたが予測可能な未来に唯一の収益源になるだろう。
不安定な市場状況や経済·市場状況の低迷は、我々の業務、財務状況、株価に深刻な悪影響を及ぼす可能性がある
私たちの経営業績は世界経済と世界金融市場の全体的な状況の悪影響を受ける可能性があります。例えば、世界金融危機は資本と信用市場の極端な変動と混乱をもたらした。同様に、新冠肺炎の疫病による激しい変動と最近の地政学的緊張情勢は、ロシアとウクライナ及びイスラエルとハマスの間の衝突を含め、すべて資本と信用市場で深刻な不安定と混乱をもたらした。世界経済状況は引き続き不安定であり、米国や海外の状況も不確定である。我々の業務は、より高いインフレ率、上昇する金利、サプライチェーン中断、景気後退、貿易制限、関税増加または潜在的な新しい関税、および米国が実施する経済禁輸を含む、市場経済と政治的変化の悪影響を受ける可能性がある。深刻または長期的な経済低迷は、私たちの候補製品に対する需要が弱まることを含め、私たちの業務に様々なリスクをもたらす可能性があり、必要に応じて許容可能な条件で追加資本を調達する能力(あれば)にも影響を与える可能性がある。我々の一般的なビジネス戦略は、このような性質の景気後退、不安定なビジネス環境、または持続的な予測不可能かつ不安定な市場状況のいずれかの悪影響を受ける可能性がある。現在の株式や信用市場が悪化している場合、あるいは改善されていない場合、必要な債務や株式融資をより困難にし、コストが高く、希釈度が高く、あるいは全く得られない可能性がある
適時に有利な条件で任意の必要な融資を得ることができなければ、私たちの成長戦略、財務業績と株価に悪影響を与える可能性があり、臨床開発計画の延期、修正、或いは放棄を要求する可能性がある。さらに、私たちの現在の1つまたは複数のサービスプロバイダ、製造業者、および他のパートナーは、この困難な経済時期を乗り切ることができないかもしれません。これは、私たちが時間通りに予算で運営目標を達成する能力に直接影響を与える可能性があります。
歴史的に見ると、証券集団訴訟は通常、ある会社の証券市場価格が下落した後に提起される。このリスクは,バイオテクノロジーや製薬会社が近年大幅な株価変動を経験しているため,我々と特に関連している。私たちが起訴されれば、巨額のコストや経営陣の注意力や資源の移転を招く可能性があり、これは私たちの業務の見通し、財務状況、運営結果に悪影響を及ぼす可能性がある。
金融サービス業の不利な事態の発展に影響を与え、例えば流動性、違約又は金融機関又は取引相手側が義務を履行しない実際の事件又は懸念は、会社の現在及び予想される業務運営及びその財務状況及び運営結果に悪影響を及ぼす可能性がある。
流動性が限られている、契約違反、業績が悪い、または金融サービス業または金融サービス業の他の不利な発展に影響を与える実際の事件、または任意のこのような事件または他の類似のリスクに対する懸念または噂は、過去および未来に市場全体の流動性問題を引き起こす可能性がある。例えば2023年3月10日シリコンバレー銀行(SVB)
カリフォルニア金融保護·革新部によって閉鎖され、同部は連邦預金保険会社またはFDICを担当者に指定している。同様に,2023年3月12日,Signature BankとSilvergate Capital Corp.はそれぞれ破産管理プログラムに巻き込まれた.財務省、FRB、連邦預金保険会社の声明によると、SVBのすべての預金者は、閉鎖されたわずか1つの仕事後に、無保険預金口座内の資金、信用協定下の借り手、信用協定下の借り手、信用協定下の信用状およびいくつかの他の金融商品、署名銀行またはFDICが接収した任意の他の金融機関を含むすべての資金を引き出すことができるが、その中で抽出されていない金額を抽出できない可能性がある。これらの銀行の倒産は当社に大きなリスクを与えていませんが、もし私たちの融資者やどのようなツールの取引相手も接収された場合、私たちはこれらの機関が持っている資金を得ることができないかもしれません。さらに、私たちの任意のパートナー、サプライヤー、または私たちと業務往来のある他の当事者が、そのようなツールやそのような金融機関との融資スケジュールに基づいて資金を得ることができない場合、これらの当事者が私たちに債務を支払い、または追加金を私たちに支払うことを要求する新しいビジネス計画を達成する能力は悪影響を受ける可能性がある。このような点で、より広範な金融サービス業の流動性に対する懸念には依然として不確実性がある。同様の影響は、例えば2008-2010年の金融危機の間に過去にも発生したことがある。
インフレと金利の急速な上昇は、以前に発行された金利が現在の市場金利よりも低い国債の取引価値を低下させる。財務省、連邦預金保険会社、連邦準備委員会は、このようなツールの売却による潜在的損失のリスクを低減するために、金融機関が保有するいくつかのこのような政府証券を担保とする金融機関に250億ドルまでの融資を提供する計画を発表しているが、広範に存在する顧客引き出し需要や金融機関の即時流動性の他の需要は、このような計画の能力を超える可能性がある。また、米国財務省、連邦預金保険会社、連邦準備委員会が将来他の銀行や金融機関が閉鎖された場合に未保険資金のルートを提供すること、あるいはタイムリーにそうすることは保証されない。
私たちは、私たちがこれらの関係の中で適切な多様性を確保することを含む、私たちが必要だと思う銀行と他の関係を定期的に評価する。それにもかかわらず、我々が獲得した資金源および他の信用手配は、現在および予想される将来の業務運営に資金または資本化を提供するのに十分であり、当社、当社と直接信用協定や手配を締結した金融機関、または金融サービス業全体または経済全体に影響を与える要因の深刻な影響を受ける可能性がある。他にも、これらの要因には、流動性の緊張または失敗、様々な金融、信用または流動資金協定または手配された義務を履行する能力、金融サービス業または金融市場の中断または不安定、または金融サービス業会社の将来性に対する懸念または否定的な予想が含まれる可能性がある。これらの要因は、当社と財務や業務関係にある金融機関や金融サービス業界会社に関連する可能性があるが、金融市場や一般金融サービス業界に関連する要因も含まれている可能性がある
項目1 B。未解決の従業員のコメント
適用されません。
プロジェクト1 C。ネットワークセキュリティです。
ネットワークリスク管理と戦略
取締役会監査委員会の監督の下で、私たちはネットワークリスクを識別、評価、緩和することに重点を置いて、持続的なネットワークセキュリティリスク管理計画を実施し、維持した。我々は、持続的なセキュリティコンサルティングサービスからセキュリティ監視および応答管理までの様々なサービスを提供する複数の第三者プロバイダと交渉する。また、サプライヤーアンケートおよび契約セキュリティ要件を適宜使用することを含む第三者プロバイダおよびサービスプロバイダのネットワークセキュリティアプローチを評価して検討するプログラムもある。これらの努力に加えて,ネットワークセキュリティリスクの識別,評価,対応のためのプロセスを含む継続的な企業リスク管理計画を実施した。我々のネットワークセキュリティは、ネットワークセキュリティ技術によってサポートされる定期的、的確なリスク評価を含む業界基準に準拠しており、これらの技術には、ネットワークセキュリティリスクを監視、識別、対応するための第三者セキュリティ解決策および監視ツールが含まれている
また、上場企業として、私たちの情報技術システムの制御及び財務諸表やシステムへの影響を含む、私たちの内部統制をめぐる様々な規制要求の制約を受けています。我々は,ネットワークセキュリティに関する制御措置や,関連するリスクを低減する戦略など,第三者サプライヤーを招いて,これらの要求を遵守するためのアドバイスを提供している.ネットワークセキュリティリスクを構成する制御欠陥を見つけるには、首席財務官や監査委員会に報告し、是正行動計画を適宜提出する。
ネットワークセキュリティリスクに関するガバナンス
我々のネットワークリスク管理計画および関連運営とプロセスは我々の取締役情報技術部が管理し,法律や人的資源チームと協議している。現在,取締役の情報技術職は17年以上のネットワークセキュリティ,情報技術,システム工学経験を持つ個人が担当している.情報技術部役員は首席人事官に報告します。
情報技術部役員は定期的に首席人事官と面会し,我々のネットワークセキュリティリスク管理プロセスの成果をモニタリング·検討し,ネットワークセキュリティリスク管理戦略に関する事項を検討·処理している。情報技術部は、首席法務官や首席人事官とともに、監査委員会に定期報告を提出し、監査委員会は、ネットワークセキュリティリスクを含む会社のリスク管理プロセスの審査·監督を担当する。首席財務官、首席人事官および首席法務官および/または法律チームの他の上級メンバーが監査委員会会議に参加し、監査委員会会議は、通常、首席財務官によって指導され、取締役会全体会議に参加する
私たちの企業リスク管理プロセスは首席法務官と首席財務官が監督します。企業リスク情報を収集する際には、ネットワークセキュリティが具体的にリスク種別とされており、我々の企業リスク評価過程の結果、ネットワークセキュリティに関するリスクも含めて、監査委員会や上級管理職と定期的に検討されている。
項目2.財産
私たちは現在マサチューセッツ州ウォトタウンで約111,611平方フィートのオフィスと実験室空間をレンタルしています。レンタル契約は2032年に満期になります。2025年に満期になる転貸手配によると、私たちはマサチューセッツ州ウォトタウンで約31,039平方フィートのオフィスと実験室空間を転用した。私たちは予測可能な未来に、私たちの施設は私たちの現在の需要を満たすのに十分であり、必要な時に適切な追加空間を提供すると信じている
項目3.法的訴訟
私たちは現在どんな実質的な法的手続きの当事者でもない。私たちは時々様々な法的手続きやクレームの影響を受けるかもしれません。これらの訴訟とクレームは私たちの正常な業務活動の過程で発生します。結果にかかわらず、弁護と和解コスト、管理資源の移転などの要因により、訴訟は私たちに実質的な悪影響を及ぼす可能性がある
第4項鉱山安全情報開示
適用されません。
第II部
第5項:登録者普通株市場、関連株主事項、発行者が株式証券を購入する
市場情報
我々の普通株は2020年10月2日からナスダック全世界精選市場で公開取引され、取引コードはCCCCである。その前に、私たちの普通株は市場を公開しなかった。
所持者
2023年2月15日現在、私たちの普通株は約60人の登録保有者がいます。この数字には,その株式が街中の被指名者が所有する実益所有者は含まれていない.
配当をする
設立以来、私たちはどんな現金配当金も発表したり支払ったりしたことがない。私たちは将来の収益(あれば)を残し、私たちの業務の運営と拡張に資金を提供し、予測可能な未来に普通株式保有者に現金配当金を支払うことはないつもりです。
株式補償計画に基づいて発行された証券
私たちの持分補償計画に関する情報は、第12項を参照して本明細書に組み込まれる特定の実益所有者の担保所有権及び経営陣及び関連株主の事項本年度報告の表格10−K。
最近売られている未登録証券
ない。
発行者または関連購入者が株式証券を購入する
2023年12月31日までの年間において、吾らまたは任意の関連バイヤーまたは吾を代表する任意の代表または関連バイヤーを代表して行動する者は、当社の普通株のいかなる株式も購入していない
第六項です[保留されている]
適用されません。
第7項:経営陣の財務状況と経営成果の検討と分析
以下、我々の財務状況と経営結果の検討と分析は、我々の連結財務諸表および本年度報告書の他の部分がForm 10-K形式で含まれるこれらの報告書に関する付記とともに読まなければならない。歴史財務情報に加えて、以下の議論と分析には、リスク、不確実性、および仮説に関連する前向きな陳述が含まれている。新聞の掲載を容易にするために、本明細書のいくつかの数字は四捨五入された。我々の実際の結果は,これらの前向き陳述で予想された結果と大きく異なる可能性があり,これは,本年度報告10−K表の第1 A項(リスク要因)で議論された要因を含む多くの要因の結果である。
概要
著者らは臨床段階の生物製薬会社であり、TPD科学の約束を実行し、患者の生活を変える新世代の小分子薬物を創造することに力を入れている。我々独自の魚雷プラットフォームを利用することにより,小分子タンパク質分解器を効率的に設計·最適化することができ,人体破壊に不要なタンパク質の自然過程を利用することで,その期待目標を高度に活性化することができる。我々の新型経口製品候補製品は,阻害剤がよく出現する薬剤耐性を克服し,現在の“投与できない”標的を目指し,患者の結果を改善する潜在力があると信じている。
著者らの臨床前と臨床パイプラインにはMonoDACとBiDAC分解物が含まれており、これらの分解物は各種の癌と他の適応の原因蛋白に対して、患者の需要を満たしていない領域に使用される可能性がある。我々の全資本が持つパイプラインは腫瘍学に集中しており,我々の協力戦略は他の適応を探索できるようにしている。
著者らの最先端の候補製品CFT 7455は1種の経口生物使用可能なMonoDAC分解剤であり、IKZF 1とIKZF 3と呼ばれ、現在多発性骨髄腫(MM)と非ホジキンリンパ腫(NHL)の臨床開発中である。IKZF 1とIKZF 3を標的とするのは非常に強い機序の基礎と明確な生物学的定義があり、1種の新型分解剤を標的として重要な満足されていない需要を解決する可能性がある。われわれの臨床前研究では,この候補製品は良好な薬理学的特性を有する有効かつ選択的なタンパク質分解を示している。2021年6月、著者らはこの候補製品のために初のヒト1/2期臨床試験を開始した。2021年8月、米国食品医薬品局(FDA)は、MM治療の孤児薬としてCFT 7455を承認した。2023年12月、私たちは
CFT 7455段階1/2試験の用量増加部分の陽性臨床データを提供し,単一療法としてデキサメタゾン併用によるMM治療を行い,進行中のMMとNHLの1/2段階臨床試験を進めている
著者らの次の最も先進的な候補製品CFT 1946は、黒色腫、非小細胞肺癌、結腸直腸癌、およびこのような変異を含む他の悪性腫瘍の治療に有効かつ選択的に抗することを目的とする経口生物学的に利用可能なBiDAC分解剤である。臨床前研究では,CFT 1946はCRCやNSCLC BRAF V 600 X異種移植モデルおよび黒色腫患者由来の異種移植またはPDX,BRAF阻害剤耐性モデルで標準看護療法よりも有効であった。また、CFT 1946は臨床前転移性黒色腫中枢神経系(CNS)モデルにおいて活発である。2023年1月、我々はCFT 1946の最初のヒト1/2期臨床試験を開始し、非小細胞肺癌、結腸直腸癌、および黒色腫を含むBRAF V 600 X変異固形腫瘍の治療に使用した。2024年1月、実施中の1/2段階試験の最初の2つの用量アップグレードキューのPKおよびPDデータを共有し、BRAFの深分解に関連する用量比曝露および経口バイオアベイラビリティを示した。黒色腫、非小細胞肺癌、結腸直腸癌を含むBRAF V 600 X癌の1/2期臨床試験を継続した
また,経口生物利用,アロステリック,変異選択性の上皮増殖因子受容体またはEGFRのBiDAC分解剤であり,非小細胞肺癌にL 858 R変異を有するCFT 8919を開発している。体外の臨床前研究において、CFT 8919はEGFR抑制に対する突然変異(L 858 R-C 797 S、L 858 R-T 790 MとL 858 R-T 790 M-C 797 Sを含む)はL 858 R単一突然変異と同じ抗増殖活性を示した。2023年5月、私たちはBetta Pharmaと許可と協力協定を締結し、CFT 8919の大陸部中国、香港特別行政区、マカオ特別行政区、台湾の開発と商業化について協力し、私たちは世界の他の地域でCFT 8919を開発および商業化する権利を保留した。また、2023年6月、FDAはCFT 8919の研究新薬或いはIND申請を許可し、2023年12月、倍他医薬は中国指導の国家医療製品管理局のCFT 8919に対する臨床試験申請或いはCTA許可を得た
これらの最初の候補品に加えて、私たち自身の特許パイプラインと、私たちがメルク、生物遺伝、羅氏と協力して開発したパイプラインに対して、臨床検証と現在使用できない目標に対する新しい分解剤を開発し、更に私たちのパイプラインを多様化した。我々が設計した分解物は臨床前研究において血液脳関門の透過に成功しており,腫瘍学的脳転移や神経変性疾患などの治療領域を治療する可能性のある薬物開発の重要な一歩である。多くの治療分野や適応において,我々の魚雷プラットフォームを用いて新しい分解剤を開発することが有利である可能性が信じられている。
最新の発展動向
2024年1月9日、私たちは2024年に私たちの戦略計画に基づいて実行する業務優先事項を発表しました。これらの改正された優先順位のため、私たちは私たちの業務を再構成し、約30%の従業員を削減した。また,会社の市場(またはATM)メカニズムにより,13,686,743株の普通株を5.42ドルの平均購入価格で売却することを発表し,純収益は7180万ドルであった。
2024年1月4日、我々は、先に発表されたBetta Pharmaの完全子会社との株式購入合意に基づいて想定される取引を完了することを発表した。終値時、Betta Pharmaの付属会社の5,567,928株の普通株を1株4.49ドルの買い取り価格で売却し、総投資額は2500万ドルだった。買い取り価格は、2023年5月29日までの株式購入契約締結前の2営業日の60取引日の出来高加重平均値より25%割増した。
2023年12月12日、単一療法としてCFT 7455を発表し、デキサメタゾン併用によるMMの1/2期試験の用量増加部分の陽性臨床データを発表した。MMとNHLが行っている1/2期臨床試験を進めていく
2023年12月12日、私たちは、マク社と独占的な許可および協力合意を達成し、分解物-抗体結合体を開発し、DACと略称し、癌細胞中の病原性タンパク質を選択的および中和することを目的とした新興の方法であることを発表した
2023年11月20日、私たちはオーウェン·ヒューズを取締役会メンバーに任命することを発表した。
2023年11月1日、CFT 8634を臨床試験の第1段階用量アップグレード部分に進めないポートフォリオ優先度決定を発表した。
経営成果の構成部分
収入.収入
これまで、私たちは製品販売から何の収入も得ておらず、予測可能な未来にも製品販売から何の収入も得られないだろう。今まで、私たちの収入は研究協力と許可協定から来た。私たちは各合意に基づいて予想業績期間の収入を確認します。私たちが望むのは
今後数年間の収入は主に私たちの現在の協力協定と私たちが未来に達成する可能性のある任意の追加的な協力から来るだろう。今まで、私たちは既存の協力協定の下でどんな印税も受け取っていない
メルク、Betta Pharma、羅氏、Biogen、Calico Life Science LLCやCalicoとの協力協定の説明については、注8を参照されたい協力と許可協定本年度報告10-K表の連結財務諸表に記載されている。
研究開発費
研究と開発費用は主に私たちの研究活動によって発生するコストを含み、私たちの発見努力と私たちの候補製品開発を含む
•研究開発に従事する者の賃金、福祉、その他の関連費用は、株式ベースの報酬費用を含む
•契約研究組織および私たちを代表して研究、臨床前および臨床活動を行う他の第三者、および臨床前および臨床試験のための候補製品を製造する第三者を含む、第三者との合意に基づいて発生する費用
•外部コンサルタントの費用には彼らの費用と関連する旅費が含まれている
•臨床前研究と臨床試験の実験室用品と材料購入費用;
•CFT 8634の段階的オフ活動に関連するコスト;
•施設に関する費用は、設備の直接減価償却費用、施設賃貸料及び整備費及びその他の業務費用の分配費用を含む
•第三者許可料
私たちは発生した費用に応じて研究と開発費用を支払う。外部開発活動のコストは,具体的なタスク達成の進捗の評価に基づいて,我々のサプライヤーが提供してくれた情報を用いて確認した.これらの活動の支払いは、発生したコストモデルとは異なる可能性がある個別合意に基づく条項であり、私たちの連結財務諸表に前払いまたは計算された研究および開発費用として反映される可能性がある。将来的に研究および開発活動のために受信される貨物またはサービスの払戻不可能な前払いは、前払い費用として記録され、関連貨物が提供またはサービスを提供する際に支出される。
われわれが計画している臨床前と臨床開発活動により,われわれの研究開発費は引き続き大幅に増加することが予想される。
一般と行政費用
一般的および行政費用は、主に株式ベースの給与を含む行政、財務、法律、業務発展と行政機能者の賃金およびその他の関連コストを含む。一般および行政費用には、会社の事務に関連する法律費用、会計、監査、税務およびコンサルティングサービスの専門費用、保険料、出張費用、直接減価償却コスト、施設賃貸料および維持分配費用、および他の運営コストが含まれる施設関連費用も含まれる
私たちは今後、より多くの研究開発活動を支援するために、私たちの一般的かつ行政的費用が増加し続けることを予想している。これらの増加には、より多くの人員の雇用に関連するより高いコスト、外部コンサルタント、弁護士、および会計士に支払う費用、および投資家および広報費用が含まれるかもしれない。
その他の収入(費用)
その他の収入(費用)、純額は主に:
•長期債務の利息支出と償却
•債務返済損失
•私たちの現金、現金等価物と有価証券から稼いだ利息と他の収入、及び有価証券の割引が増加します。
行動の結果
2023年12月31日までと2022年12月31日までの年次比較
収入.収入
私たちの協力とライセンス契約収入には以下のものが含まれています(千計)
| | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | |
| 協力協定からの収入: | | | | | |
| 生物遺伝協定 | $ | 10,623 | | | $ | 19,214 | | | |
| 羅氏協定 | 9,063 | | | 4,939 | | | |
| | | | | |
| 印刷布協定 | 1,070 | | | 6,943 | | | |
| 協力協定からの総収入 | $ | 20,756 | | | $ | 31,096 | | | |
2022年12月31日までの年度と比較して、2023年12月31日までの年間収入は1030万ドル減少した
•“生物遺伝プロトコル”により確認された収入が860万ドル減少した要因は,“生物遺伝プロトコル”の研究期限が2023年6月に終了し,研究期限終了後の一定期間で指名された目標を追加的に研究活動することであり,これらはすべて“生物遺伝プロトコル”が想定していることである
•印紙協議により確認された収入が590万ドル減少したのは,2023年3月31日現在,履行義務と取引価格が完全に満たされているためである。
羅氏合意により確認された収入が410万ドル増加したことがこれらの増加を相殺したのは,2023年に指定された目標の研究活動を完了したためである。
研究と開発費
次の表に私たちの研究開発費(単位:千)をまとめます
| | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | |
| 研究開発費: | | | | | |
| 人員費 | $ | 42,706 | | | $ | 41,068 | | | |
| 臨床前と開発費用 | 28,496 | | | 42,033 | | | |
| 臨床費用 | 19,238 | | | 10,643 | | | |
| 施設と用品 | 13,594 | | | 13,978 | | | |
| 専門費 | 8,605 | | | 7,047 | | | |
| 知的財産権やその他の費用 | 5,067 | | | 3,072 | | | |
| | | | | |
| 研究開発費総額 | $ | 117,706 | | | $ | 117,841 | | | |
2023年12月31日までの年間で,研究·開発費が2022年12月31日までの年間より10万ドル減少したのは,主に臨床段階に入る追加計画に関する臨床前·開発コストが1350万ドル減少したためである。
これらの費用は以下の部分によって相殺される
•進行中のCFT 7455とCFT 1946の1/2期臨床試験およびCFT 8634の1期臨床試験により,臨床費用は860万ドル増加した
•知的財産権や他の会社の支出は200万ドル増加しました
•人員支出が170万ドル増加し、賃金と福祉コストを代表しているのは、従業員数が増加して私たちの臨床計画を支援しているからである
•専門費用は160万ドル増加した。
一般と行政費用
次の表は、私たちの一般と行政費用(千単位)をまとめています
| | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | |
| 一般的かつ行政的費用: | | | | | |
| 人員費 | $ | 30,244 | | | $ | 30,546 | | | |
| 専門費 | 7,365 | | | 8,576 | | | |
| 施設と用品 | 2,076 | | | 2,371 | | | |
| その他の費用 | 2,396 | | | 1,296 | | | |
| 一般と行政費用総額 | $ | 42,081 | | | $ | 42,789 | | | |
2022年12月31日までの年度と比較して,2023年12月31日までの年度一般·行政費は70万ドル減少し,主に専門費用が120万ドル減少し,人員·施設コストが60万ドル減少したが,その他の費用は110万ドル増加し,その低下を部分的に相殺した。
その他の収入(費用)
次の表は、私たちの他の収入(支出)(単位:千):
| | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | |
| その他の収入,純額 | | | | | |
| 利息とその他の収入,純額 | $ | 9,812 | | | $ | 3,575 | | | |
| 長期債務関連側の利子支出と償却 | (1,373) | | | (2,216) | | | |
長期債務返済損失 | (621) | | | — | | | |
| その他の収入を合計して純額 | $ | 7,818 | | | $ | 1,359 | | | |
2022年12月31日までの年度と比較して、2023年12月31日までの年間その他の収入(支出)は650万ドル増加し、2023年の私たちの投資利息の増加により利息やその他の収入が620万ドル増加したためだ。
所得税費用
所得税支出は2023年12月31日までの1年間で130万ドルだったが、2022年12月31日までの1年間は所得税支出はなかった。これは,主に中国の税務機関に支払われた1,000,000ドルの源泉徴収税が,Betta許可協定に従って受け取った前金と,1月に受信したBettaマイルストーン支払いに関する源泉徴収税2万ドルに関連しているためである(付記8および付記12参照)。
流動資金と資本資源
流動資金源
設立以来、私たちは重大な運営損失が発生した。私たちは、予測可能な未来に、私たちのプロジェクトの継続に伴い、巨額の費用と運営損失が生じると予想している。私たちは現在どんな承認された製品もなく、製品販売から何の収入も得たことがない。これまで、私たちは主に優先株の売却、私たちの普通株の公開、パートナーの支払いを通じて、私たちの運営に資金を提供してきました。自分から2023年12月31日私たちは約2億817億ドルの現金、現金等価物、有価証券を持っている。
2021年11月、自動発効したS-3表登録説明書を米国証券取引委員会に提出し、数量不明な普通株、優先株、債務証券、権証及び/又はそれらの任意の組み合わせの単位の発行、発行及び販売を登録した。我々は同時に販売代理であるCowen and Company,LLCと株式分配協定を締結し,登録声明と登録声明やATM計画とともに提出された関連募集説明書に基づいて,2億ドルまでの普通株を随時発行·販売する.同社は合計13,686,743株の普通株を売却し、平均価格は1株5.42ドル、手数料と費用を差し引いた収益は7180万ドルだった。2023年12月31日までの1年間に、ATM計画により決済された普通株は11,186,142株、純収益は5,770万ドル。2024年1月には2500,601株が決済され、純収益は1410万ドルだった。(付記10参照)。
Betta Pharmaライセンス契約の署名について、当社のBetta PharmaとBetta Pharmaの関連会社(Betta Investment(Hong Kong)Limited、またはBetta Investment)は#年の株式購入契約を締結した2023年5月29日又は闘魚株式購入協定によれば、闘魚投資は、当社5,567,928株普通株又は株式を購入することに同意し、総購入価格は約2,500万ドル、又は1株当たり4.49ドルであり、闘魚株式購入協定発効日前の2取引日前の60取引日の出来高加重平均終値より25%割増する。“闘魚株式購入協定”は、このような協定にいくつかの慣用的な制限があるBetta株購入契約による取引は2024年1月に発生した(付記8参照).
キャッシュフロー
次の表は、本報告に記載されている間の私たちの現金源および用途(千計)をまとめています
| | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | |
| 現金、現金等価物、および限定的な現金の純変化: | | | | | |
| 業務活動におけるNETのアプリケーション | $ | (106,838) | | | $ | (105,939) | | | |
| 投資活動提供の現金純額 | 158,349 | | | 58,422 | | | |
| 融資活動が提供する現金純額 | 45,489 | | | 1,147 | | | |
| 現金、現金等価物、および限定的な現金純変化 | $ | 97,000 | | | $ | (46,370) | | | |
経営活動
2023年12月31日までの年度、経営活動のための現金純額は主に以下のような要因によって駆動されている
•純損失は1兆325億ドル
•売掛金は1,030万ドル増加し
•経営リース負債は470万ドル減少した。
これらの金額は以下の項目の一部によって相殺される
•株式補償費用、減価償却、償却に関する非現金費用3530万ドル、および私たちの資産を使用する帳簿金額の減少
•前払い費用と流動と長期資産は270万ドル減少した
•私たちの新しい協力協定のおかげで繰延収入は380万ドル増加した
•費用と他の流動負債は130万ドル増加しなければならない
2022年12月31日までの年間経営活動で使用される現金純額は、主に以下の要因によって駆動される
•純損失は1億282億ドル
•繰延収入が2,270万ドル減少した理由は、私たちの協力協定に基づいて収入を確認することである
•売掛金は330万ドル減少しなければならない。
これらの金額は以下の項目の一部によって相殺される
•3,760万ドルの非現金支出は、株式補償費用、減価償却、および使用権資産の帳簿金額の減少に関連している
•費用とその他の負債は510万ドル増加しなければならない
•売掛金は420万ドル減少した。
投資活動
2023年12月31日までの年間で、投資活動が提供した1億583億ドルの現金純額は、売却可能証券の満期収益1.601億ドル(購入を差し引く)によるものであり、財産や設備を購入した170万ドルで相殺される。
2022年12月31日までの年度、投資活動が提供した5840万ドルの現金純額は、6,390万ドルの有価証券収益(購入を差し引く)に起因し、財産と設備を購入した550万ドルで相殺される
融資活動
2023年12月31日までの年度,融資活動が提供する4550万ドルの現金純額は主に5770万ドルの市場発行収益によって推進されているが,前払い感知信用ホールディングスIII,LP定期融資の残存元金残高1250万ドルはこの影響を相殺している。
2022年12月31日までの1年間、融資活動が提供した110万ドルの現金純額は、主に株式オプションを行使する80万ドルの収益によって推進された。
資金需要
私たちの設立以来、私たちはすでに重大な運営損失が発生しており、予測可能な未来に、候補製品の臨床前と臨床開発を進めるにつれ、重大な支出と増加していく運営損失を招き続けると予想される。また、上場企業として、運営に関連したコストが引き続き発生すると予想される
具体的には、私たちは未来の支出が増加すると予想している
•私たちが行っている人間の第1段階の1/2実験を続けて
•より多くの候補製品が臨床前と臨床開発段階に入るように推進する
•独自の魚雷プラットフォームに投資し続けています
•私たちの知的財産権の組み合わせを推進し、拡大し、維持し、保護する
•私たちがより多くの候補製品を発売したり、既存の候補製品を開発し続けたりすることに伴い、管理者の需要は絶えず変化する業務需要を満たす。
•臨床試験に成功した候補製品のために市場承認を求める
•最終的に販売、マーケティング、流通インフラを構築し、外部製造能力を拡大し、マーケティングの承認を得ることが可能な任意の製品を商業化する
私たちの候補製品の開発と商業化に関連する多くのリスクと不確実性のため、私たちは現在と期待されている臨床前と臨床開発に関連する増加した資本と運営コストを見積もることができない。私たちの将来の資本需要は多くの要素に依存します
•私たちの主要な候補製品は、人間の第一段階の1/2試験の進行、コスト、結果、およびこれらの先行候補製品の任意の未来の臨床開発を行っており、計画されている
•私たちの他の候補製品と開発計画の臨床前と臨床開発の範囲、進捗、コストと結果
•私たちが求めている他の候補製品の数量と開発要件は
•私たちがマイルストーンを達成した後にパートナーから追加の研究支援やマイルストーン支払いを得るかどうかを含む、私たちの既存と未来の第三者パートナーとの協力の進展と成功
•私たちの候補製品に対する規制審査のコスト、時間、結果
•特許出願を準備し、提出し、起訴し、私たちの知的財産権を維持し、実行し、知的財産権に関する任意のクレームを弁護するコストと時間;
•私たちは、現在またはより多くの将来の候補製品を開発または商業化するために、有利な条件で他のバイオテクノロジーまたは製薬会社とより多くの協力計画を確立することを望んでおり、能力がある
•私たちが市場承認を得た任意の候補製品について、将来の商業化活動のコストと時間は、製品製造、マーケティング、販売、および流通を含む
•私たちの候補製品の商業販売から得られた収入(あれば)は、マーケティング承認を得ました
上記の予想支出のため、私たちは私たちの持続的な運営を支援し、長期業務計画を実行するために、多くの追加資金を得る必要があるだろう。私たちは製品販売から相当な収入を得ることができる前に、株式発行、債券発行、協力、戦略連合、マーケティング、流通、許可手配を通じて私たちの現金需要に資金を提供する予定です。私たちは羅氏、Biogen、Calico、Betta Pharma、およびメルクとの協力を通じて潜在的な未来のマイルストーンと特許使用料支払いを得るかもしれないが、2023年12月31日まで、私たちは約束された外部資金源を持っていない。受け入れ可能な条件で、私たちは十分な追加資金を得ることができないかもしれないし、全くないかもしれない。もし私たちが必要な時や魅力的な条件下で資金を調達できない場合、私たちは私たちの研究、製品開発、または将来の商業化努力を延期、制限、減少または終了することを要求されるかもしれません。あるいは私たちが自分で開発し、マーケティングすることをより望んでいた候補製品の権利を与えることができます。
私たちが株式証券を売却することで追加資本を調達する場合、各投資家の所有権権益は希釈され、これらの証券の条項は清算または他の特典を含む可能性があり、普通株主としての権利に悪影響を及ぼす。優先株融資が可能であれば、関連する可能性のある協定は、買収または資本支出を行うこと、または配当を発表することなど、特定の行動をとる能力を制限または制限する契約を含む。
もし私たちが第三者との協力、戦略連合またはマーケティング、流通または許可手配を通じてより多くの資金を調達するならば、私たちは私たちの技術、将来の収入源、研究計画、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になる可能性のある条項でライセンスを付与しなければならないかもしれない。
契約義務
以下は、2023年12月31日現在の重要な契約義務の概要(単位:千)です
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 合計する | | 少ないです
1年 | | 1~3
年.年 | | 4~5個
年.年 | | 超過
5年間 |
| | | | | | | | | |
賃貸承諾額を経営する (注6参照) | $ | 89,046 | | | $ | 8,828 | | | $ | 18,459 | | | $ | 21,008 | | | $ | 40,751 | |
| | | | | | | | | |
| | | | | | | | | |
我々は正常業務過程において第三者CROと臨床試験、臨床前研究、製造とその他のサービス及び運営目的のための製品の契約を締結した。これらの契約は一般に通知後しばらく後に終了することが規定されているので、私たちはこれらの合意の下での撤回不可能な義務は実質的ではなく、それらは上の表に含まれていないと考えられる。マイルストーンまたは特許使用料支払いまたは他の契約支払い義務の時間および金額が未知または不確定である場合、私たちはそれを上の表に含まなかった。
肝心な会計見積もり
経営陣の財務状況や経営結果の検討·分析は、我々の総合財務諸表に基づいており、これらの報告書は米国公認の会計原則に基づいて作成されている。我々の連結財務諸表および関連開示を作成する際には、我々の総合財務諸表における資産および負債、収入、コストおよび費用の報告金額、または資産および負債の開示に影響を与える推定および仮定を作成する必要がある。我々は,歴史的経験,既知の傾向や事件,および当時の状況で合理的と考えられる様々な他の要因に基づいて推定し,これらの要因の結果は資産や負債の帳簿価値の判断の基礎を構成しているが,これらの資産や負債の帳簿価値は他の源から容易に見られるものではない.私たちは持続的な基礎の上で私たちの推定と仮定を評価する。異なる仮定や条件の下で、私たちの実際の結果はこれらの推定とは異なる可能性がある。
私たちの重要な会計政策は付記2にもっと詳しく説明されていますが重要会計政策の概要本年度報告書10−K表の総合財務諸表については、以下の会計政策が総合財務諸表の作成に用いる判断と見積もりが最も重要であると考えられる。
契約収入
会計基準に基づいてASCや606を編纂しました取引先と契約した収入またはASC 606。ASC 606によれば、そのクライアントがコミットメント商品またはサービスの制御権を取得すると、エンティティは、エンティティがこれらの商品またはサービス交換から得られると予想される対価格を反映する収入を確認する。エンティティがASC 606の範囲内に属するスケジュールの収入確認を決定するために、エンティティは、プロトコル開始時またはプロトコルの実質的な修正時に、(1)クライアントとの契約を決定するステップ(S)、(2)契約中の履行義務を決定するステップ、(3)決定するステップの5つのステップを実行する
取引価格は,可変対価格(ある場合),(4)契約中の履行義務に取引価格を割り当てる,(5)エンティティが履行義務を履行した場合(または)収入を確認する
上記(5)ステップにおける履行債務の履行状況は重要な会計推定であると考えられる。より具体的には,サービス履行義務の実現度を検討·開発し,その満足度モデルは,これまでに発生した費用を将来発生·発生予定の総費用と比較して測定しており,これは鍵となる会計推定によって推進されている。
将来予想される費用を見積もる際には、管理層は、その最新の予算と長期計画を使用し、任意の関連情報に基づいて調整する。これは報告期間までの最適な見積もりであるが,研究開発活動の範囲や時間が時間とともに大きく変化する可能性があるため,将来発生すると予想されるコストは経営陣の判断が必要である。候補製品の進歩を支援するために必要な追加の作業や試験における患者数の変化など、いくつかの要因に応じて研究開発活動の範囲を調整することができる。また,研究·開発サービスは我々のいくつかのプロジェクトのように協調プロトコルの範囲に属さなくなる可能性がある.研究開発コストが発生することが予想される時間は、製造またはサプライチェーンによる遅延、または患者募集の困難、または計画の優先順位のような内部要因によって変更される可能性がある。我々の研究開発サービスの範囲と時間の実際の範囲と時間に対する見積りは収入確認に大きな影響を与える可能性がある.
前払いと応算の研究·開発費用
連結財務諸表の作成の一部として、我々が計上すべき研究と開発費用を見積もる必要がある。このプロセスは、見積書および契約を審査し、提供されているサービスを決定し、領収書を受け取っていない場合や他の方法で実際のコストを通知している場合に、提供されるサービスレベルおよびサービスに生じる関連コストを推定することを含む。私たちは、私たちが当時知っていた事実と状況に基づいて、私たちの連結財務諸表において、各貸借対照表の日付までの課税費用を推定します。さらに、場合によっては、供給者に支払われるお金は、提供されたサービスレベルを超え、料金の前払いをもたらす可能性があり、この場合、そのような金額は、前払い料金および他の流動資産として反映される。サービス料を計算する際に、サービスを提供する時間帯と各時間の努力度を見積もります。サービスの実行時間や努力の程度が私たちの見積もりと異なる場合、それに応じて計算すべき費用や前払い費用の金額を調整します実際に発生した金額と実質的に異なることはないと予想されるが,提供されたサービスの実態や時間に対する提供サービスの状態や時間の理解が異なり,報告された金額に大きな影響を与える可能性がある.
将来的に研究·開発活動のための貨物またはサービスを受け取る払戻不可能な前払いが延期され、前払い費用および他の流動資産の形態で資本化される。資本化金額は関連貨物の納入またはサービス提供時に費用を計上する。
経営リース(借入金金利を逓増)
ASCテーマ842に基づいてレンタルを計算しました賃貸借証書またはASC 842。ASC 842によれば、レンタルスケジュールの開始または修正時に、私たちのレンタル負債および対応する使用権資産の再計量を要求される可能性があります。賃貸負債は、逓増借款金利を用いて賃貸手配下の賃貸支払いの現在値を計算することで計測される。逓増借款金利とは、私たちが支払わなければならない金利であり、担保に基づいて、借入金の金額は類似期限内の賃貸支払いに相当し、経済環境下での賃貸期限に相当する。
私たちのレンタルに隠されている増加借金金利は確定しにくいため、開始日に得られる情報に基づいた推定逓増借入金金利を用いて、レンタル支払いの現在値を計算するための割引率を決定します。私たちは最近の対外借款を持っていないので、増額金利は金融機関が提供する価値と借入期限に基づいて決定され、会社と市場特定要因に基づいて調整されて提供される借入金金利に関する情報に基づいて決定される。この決定には,同業者群の決定,我々自身のリスクプロファイル,会社や市場特定要因による調整を含む経営陣の判断が必要である。
増額借入金利の見積もりは合理的な敏感性の範囲内で大きな差は生じないことが予想されるが、適切な金利を選択する際には判断が必要であり、私たちの賃貸選択の金利は総合貸借対照表における賃貸負債とそれに応じた使用権資産の価値に影響を与える。
株式オプション
私たちは公正な価値で従業員と非従業員に付与されたすべての株式奨励を株式報酬支出として会計処理します。私たちの株式ベースの支払いには、帰属が必要な普通株を含む株式オプション、制限株式単位、普通株の付与が含まれている。報酬の計量日は付与日とし,株式の補償コストに基づいて必要なサービス期間(通常は授権期間)の費用を直線的に確認する.株式による補償費用は,関連サービスを提供する機能ごとに合併経営報告書と総合損失表に分類される。私たちは一部の奨励金が付与された株式ベースの報酬支出を確認した。没収は発生時に記録された。各株式オプション付与の公正価値は、付与された日にBlack-Scholesオプション定価モデルを用いて推定される。ブラック·スコアーズオプション定価モデルは、報酬の予想期間、奨励予想期間内の予想変動率、および予想無リスク金利、期待配当金支払い、および株式報酬に基づく普通株の公正価値を含む様々な仮定を含む。
私たちは予想変動率が重要な会計推定だと思う。私たちは十分な取引履歴がないので、私たちはBlack-Scholesオプション定価モデルのために、代表的な上場生物製薬会社(私たち自身を含む)の平均履歴変動率を使用して予想変動率を計算した。この仮定は私たちの最適な推定を反映しているが、それは市場状況に基づく内在的な不確実性に関連しており、これらの市場状況は通常私たちのコントロールを受けていない。したがって、異なる変動率を用いれば、株による報酬コストが実質的な影響を受ける可能性がある。
新会計公告
新会計基準に関する情報は、付記2を参照されたい重要会計政策の概要本年度報告Form 10−Kには我々の総合財務諸表を加えた
第七A項。市場リスクに関する定量的で定性的な開示
私たちは正常な業務過程で市場リスクに直面している。このような危険は主に金利敏感性を含む。我々の生息資産には現金,現金等価物,有価証券がある。私たちの利息収入は一般金利水準の変化に敏感で、主にアメリカ金利です。2023年12月31日現在、私たちは1.551億ドルの有価証券を持っている。私たちの有価証券は短期証券で、加重平均満期日は0.6年です。したがって、これらの利息を稼ぐツールはある程度の金利リスクを持っているが、利息収入の歴史的変動は私たちにとって顕著ではない。
項目8.財務諸表と補足データ
本項目8の要求に基づいて提出された財務諸表は、本年度報告のテーブル10−Kの後に添付される。これらの財務諸表のインデックス項目15を参照してください展示品と財務諸表の付表本年度報告の表格10−K。
第九項会計及び財務開示に関する変更と相違。
ない。
第9条。制御とプログラムです
情報開示制御とプログラムの評価
我々の経営陣は、我々の最高経営責任者及びCEOの参加の下、本年度報告書10−K表に含まれる期間が終了するまでの間の開示制御及び手続の有効性を評価し、これらの制御及び手続は、取引法第13 a−15(E)及び15 d−15(E)条の規定に適合する。取引法規則13 a-15(E)および15 d-15(E)に定義されている用語“開示制御および手順”は、取引法に従って提出または提出された報告において開示を要求する会社の情報が米国証券取引委員会規則および表によって指定された期間にわたって記録、処理、集約および報告されることを保証するための会社の制御および他のプログラムを意味する。開示制御及び手続は、会社が“取引法”に基づいて提出又は提出された報告書において開示を要求する情報が蓄積され、その主要幹部及び主要財務官を含む会社経営者に伝達されることを保証することに限定されるものではないが、必要な開示に関する決定をタイムリーに行うために、同様の機能を果たす者の制御及び手続を適宜行う。経営陣は、どのような制御やプログラムが、どんなに設計や操作が良くても、その目標を実現するために合理的な保証を提供するしかないことを認識しており、管理部門は、可能な制御とプログラムのコスト-利益関係を評価する際にその判断を運用しなければならない。
2023年12月31日までの開示統制及び手続の評価によると、我々の最高経営責任者及び最高財務官は、その日までに、我々の開示制御及び手続が合理的な保証レベルで有効であると結論した。
経営陣財務報告内部統制年次報告書
私たちの経営陣は私たちの財務報告書に対する十分な内部統制を確立して維持する責任がある。“取引法”に基づいて公布された規則13 a-15(F)および15 d-15(F)における財務報告の内部統制は、財務報告の信頼性と米国公認会計原則に基づいて外部目的の財務諸表を作成するための合理的な保証を提供するために、我々の主要幹部および主要財務官が設計またはその監督の下で、財務報告に関する信頼性を提供するために、我々の取締役会、管理層および他の人員によって実施されるプログラムと定義される。私たちの財務報告書に対する内部統制は以下の政策と手続きを含む
•私たちの資産に対する私たちの取引および処置を合理的、詳細、正確かつ公平に反映した記録を保存することと関連がある
•アメリカ公認会計原則に基づいて財務諸表を作成するために、取引が必要と記録されていることを保証する合理的な保証を提供し、私たちの収入と支出は、私たちの経営陣と取締役の許可のみに基づいて行われます
•当社の財務諸表に重大な影響を及ぼす可能性のある不正な取得、使用、または処理を防止またはタイムリーに発見することができる合理的な保証を提供します。
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。将来的にどのような有効性評価を行うかの予測には,条件の変化により制御が不十分になる可能性がある,あるいは政策やプログラムを遵守する程度が悪化する可能性があるというリスクがある.
我々の経営陣は、2023年12月31日までの財務報告内部統制の有効性を評価した。この評価を行う際には,経営陣はトレデビル委員会後援組織委員会(COSO)が2013年の内部統制−総合枠組みで提案した基準を用いた。我々の評価によると、我々の経営陣は、2023年12月31日現在、財務報告に対する内部統制がこれらの基準に基づいて有効であると結論している。
財務報告の内部統制の変化
私たちは内部統制の効率性と効力を向上させることを絶えず求めている。これは会社全体の流れを改善することにつながる。2023年12月31日までの年間で、財務報告の内部統制(“外国為替法案”第13 a-15(F)および15 d-15(F)条の定義による)に大きな影響を与えなかったか、または合理的に財務報告の内部統制に大きな影響を与える可能性のある変化はなかった。
制御措置の有効性の固有の制限
いかなる財務報告内部制御制度の有効性は、私たちの内部制御制度を含めて、内在的に制限されており、設計、実施、運営と制御とプログラムを評価する時に判断力を行使し、不正行為を完全に除去できないことを含む。したがって、どのような財務報告書の内部統制制度も、私たちの制度を含めて、設計と運営がどんなに良くても、絶対的な保証ではなく、合理的な保証しか提供できない。また,将来的に任意の有効性評価を行う予測は,条件の変化により制御不足のリスクが生じたり,政策やプログラムの遵守度が悪化したりする可能性がある.私たちは、私たちの業務の必要性や適切な内部統制の監視とアップグレードを継続するつもりですが、これらの改善が財務報告に対する効果的な内部統制を提供するのに十分である保証はありません。“リスク要因”を見てください上場企業として、追加コストを発生させ続け、私たちの経営陣は、新たなコンプライアンスやコーポレートガバナンスを実施するために多くの時間を投入する必要があります。”
プロジェクト9 B。他の情報。
ない。
プロジェクト9 Cです。検査を妨害する外国司法管轄区域を開示する。
ない。
第三部
プロジェクト10.取締役、行政、企業管理
第10項に要求される情報は、本年度報告書10-K表に含まれる年度終了後120日以内に提出され、参照によって本明細書に組み込まれる、米国証券取引委員会または米国証券取引委員会に提出された2024年年次総会に関する最終委託書に含まれる。
第11項.行政職報酬
第11項に要求される情報は、本年度報告書に含まれる2024年年次総会に関する10-K表年次報告書に含まれる年度終了後120日以内に提出され、参照によって本明細書に組み込まれる米国証券取引委員会に提出される最終委託書に含まれる。
第12項:特定の実益所有者の保証所有権及び管理職及び関連株主事項。
第12項に要求される資料は、本年度10-K表に含まれる2024年株主総会に関する年次報告書に含まれる年次報告後120日以内に米国証券取引委員会に提出された最終委託書に含まれ、引用されて本明細書に組み込まれる。
第十三条特定関係及び関連取引、並びに取締役の独立性。
第13項に要求される情報は、本年度報告書に含まれる2024年年次総会に関する10-K表年次報告書に含まれる年度終了後120日以内に提出され、参照によって本明細書に組み込まれる米国証券取引委員会に提出される最終委託書に含まれる。
第14項目主要会計費用とサービス
第14項に要求される情報は、本年度報告書に含まれる2024年年次総会に関する10-K表年次報告書に含まれる年度終了後120日以内に提出され、参照によって本明細書に組み込まれる米国証券取引委員会に提出される最終委託書に含まれる。
第4部
項目15.物証、財務諸表付表
1.財務ファクトシート
ここに掲げる財務諸表のリストについては、参照されたい連結財務諸表索引本年度報告表格10−Kに記載されているF−1ページは,参照により本プロジェクトに組み込まれる。
2.財務諸表の添付表
財務諸表の添付表は、必須でないか、適用されないか、またはこれらの情報が連結財務諸表またはその付記に含まれているので省略されている。
3.展示品
| | | | | | | | | | | | | | | | | | | | |
展示品
番号をつける | 展示品説明 | 表 | ファイル.ファイル
番号をつける | 日取り
保存する | 展示品
番号をつける | 保存済み
ここから声明する |
| 3.1 | 5番目の現行有効な登録者登録証明書 | 8-K | 001–39567 | 10/06/2020 | 3.3 | |
| | | | | | |
| 3.2 | 登録者の2回目の改訂及び別例の書式の再改訂 | S-1 | 333–248719 | 09/10/2020 | 3.5 | |
| | | | | | |
| 4.1 | 登録者、その権利証保持者といくつかの株主との間の“投資家権利協定”が改正され、再署名され、日付は2020年6月5日である | S-1 | 333–248719 | 09/10/2020 | 4.1 | |
| | | | | | |
| 4.2 | 普通株式証明書サンプル形式 | S-1/A | 333–248719 | 09/28/2020 | 4.3 | |
| | | | | | |
| 4.3 | 1934年改正証券取引法第12条に基づいて登録された証券説明 | 10-K | 001-39567 | 03/11/2021 | 4.4 | |
| | | | | | |
| 10.1# | 改訂された2015年株式オプション及び付与計画及びその付与協定のフォーマット | S-1 | 333–248719 | 09/10/2020 | 10.1 | |
| | | | | | |
| 10.2# | 2020年株式オプションとインセンティブ計画とその奨励協定のフォーマット | 10-K | 001-39567 | 02/23/2023 | 10.2 | |
| | | | | | |
| 10.3# | 2020年従業員株購入計画 | S-1/A | 333–248719 | 09/28/2020 | 10.3 | |
| | | | | | |
| 10.4# | 上級管理職現金奨励金計画 | S-1 | 333–248719 | 09/10/2020 | 10.4 | |
| | | | | | |
| 10.5# | “役員賠償協議”形式 | S-1 | 333–248719 | 09/10/2020 | 10.5 | |
| | | | | | |
| 10.6# | 上級乗組員の合意のフォーマット | S-1 | 333–248719 | 09/10/2020 | 10.6 | |
| | | | | | |
| 10.7# | 行政人員採用協議形式 | S-1 | 333–248719 | 09/10/2020 | 10.7 | |
| | | | | | |
| 10.8# | 登録者とアンドリュー·ヘシーとの雇用協定は2020年9月6日 | S-1 | 333–248719 | 09/10/2020 | 10.8 | |
| | | | | | |
| 10.9† | 登録者とBiogen MA,Inc.との間の共同研究と許可協定は,2018年12月28日である | S-1 | 333–248719 | 09/10/2020 | 10.10 | |
| | | | | | |
| 10.10† | 登録者とBiogen MA,Inc.との間の共同研究及び許可協定の第1号改正案は,期日は2020年2月25日である | 10-K | 001-39567 | 03/11/2021 | 10.11 | |
| | | | | | |
| 10.11† | 登録者,F.Hoffmann-La Roche LtdとHoffmann-La Roche Inc.の間の許可協定の改訂と再署名は2018年12月20日である | S-1 | 333–248719 | 09/10/2020 | 10.11 | |
| | | | | | |
| | | | | | | | | | | | | | | | | | | | |
展示品
番号をつける | 展示品説明 | 表 | ファイル.ファイル
番号をつける | 日取り
保存する | 展示品
番号をつける | 保存済み
ここから声明する |
| 10.12† | 登録者,F.Hoffmann-La Roche LtdとHoffmann-La Roche Inc.の間で改訂·再署名された許可協定の第1改正案は,2020年11月12日である | 10-K | 001-39567 | 03/11/2021 | 10.13 | |
| | | | | | |
| 10.13† | 登録者とCalico生命科学有限責任会社との協力と許可協定は,2017年3月13日となっている | S-1 | 333–248719 | 09/10/2020 | 10.12 | |
| | | | | | |
| 10.14 | 480アーセナグループ有限責任会社が登録者に付与した賃貸契約は、日付が2017年7月5日で、改訂されました | S-1 | 333–248719 | 09/10/2020 | 10.14 | |
| | | | | | |
| 10.15 | 第三次改正賃貸借契約、期日は2021年11月24日、C 4治療会社とコロンビアマサチューセッツ州アーセナルオフィス物件会社との間の賃貸契約 | 8-K | 001-39567 | 11/30/2021 | 10.1 | |
| | | | | | |
| 10.16† | ライセンスと協力協定は,2023年5月29日にC 4治療会社とBetta製薬株式会社が署名した。 | 8-K | 001-39567 | 05/30/2023 | 10.1 | |
| | | | | | |
| 10.17† | C 4治療会社、Betta製薬有限公司とBetta投資(香港)有限公司との間の株式購入契約は、2023年5月29日となっている。 | 8-K | 001-39567 | 05/30/2023 | 10.2 | |
| | | | | | |
| 10.18† | 許可と協力協定は、期日は2023年12月11日で、C 4治療会社とメルク·シャープ·ダム有限責任会社が署名した | | | | | X |
| | | | | | |
| 10.19# | 非限定株式オプション協定インセンティブの形式 | | | | | X |
| | | | | | |
| 21.1 | 登録者の子会社 | | | | | X |
| | | | | | |
| 23.1 | 独立公認会計士事務所ピマウェイ会計士事務所同意 | | | | | X |
| | | | | | |
| 31.1 | 2002年サバンズ-オキシリー法第302節で可決された1934年“証券取引法”第13 a-14(A)及び15 d-14(A)条に従って発行された最高経営責任者証明書 | | | | | X |
| | | | | | |
| 31.2 | 2002年サバンズ-オキシリー法第302節で可決された1934年“証券取引法”第13 a-14(A)及び15 d-14(A)条による首席財務官の証明 | | | | | X |
| | | | | | |
| 32.1* | 2002年にサバンズ·オクスリ法案第906条で可決された“米国法典”第18編1350条による主要行政官の証明 | | | | | X |
| | | | | | |
| 32.2* | 2002年サバンズ·オキシリー法第906条で可決された“米国法典”第18編1350条による首席財務官の証明 | | | | | X |
| | | | | | |
| 97 | 役員報酬回収政策 | | | | | X |
| | | | | | |
| 101.INS | XBRLインスタンスドキュメントを連結する | | | | | X |
| | | | | | |
| 101.書院 | イントラネットXBRL分類拡張アーキテクチャ文書 | | | | | X |
| | | | | | | | | | | | | | | | | | | | |
展示品
番号をつける | 展示品説明 | 表 | ファイル.ファイル
番号をつける | 日取り
保存する | 展示品
番号をつける | 保存済み
ここから声明する |
| | | | | | |
| 101.カール | インラインXBRL分類拡張計算リンクライブラリ文書 | | | | | X |
| | | | | | |
| 101.def | インラインXBRL分類拡張Linkbase文書を定義する | | | | | X |
| | | | | | |
| 101.介護会 | XBRL分類拡張ラベルLinkbase文書を連結する | | | | | X |
| | | | | | |
| 101.Pre | インラインXBRL分類拡張プレゼンテーションLinkbaseドキュメント | | | | | X |
| | | | | | |
| 104 | 表紙相互データファイル(添付ファイル101に含まれるイントラネットXBRL文書のフォーマット) | | | | | |
_____________________________________
| | | | | |
| # | 契約または任意の補償計画、契約または手配を管理することを指す。 |
| † | 米国証券取引委員会の規定により、本展示品の一部の内容(星番号で示す)は省略される。 |
| * | 証拠品32.1および32.2は提供されており、取引法第18条の目的として提出されてはならない、または他の方法でこの条項の責任を負うものとみなされてはならないし、引用によって証券法または取引法に基づいて提出された任意の登録声明または他の文書に引用によって記載されていない限り、言及されてはならない。 |
項目16.表格10-Kの概要
適用されません。
サイン
改正された1934年証券取引法第13又は15(D)節の要求に基づいて、登録者は、正式に許可された次の署名者がその代表を代表して本報告書に署名することを正式に手配した.
| | | | | | | | |
| C 4治療会社 |
| | |
日付:2024年2月22日 | 差出人: | /S/アンドリュー·J·ヘシー |
| | アンドリュー·J·ヘシー |
| | 社長と最高経営責任者 |
本報告書は、改正された1934年の証券取引法の要求に基づき、以下の登録者によって登録者として指定日に署名された。
| | | | | | | | | | | | | | |
| 名前.名前 | | タイトル | | 日取り |
| | | | |
| /S/アンドリュー·J·ヘシー | | 最高経営責任者総裁と役員 | | 2024年2月22日 |
| アンドリュー·J·ヘシー | | (首席行政主任) | | |
/S/ケンドラ·R·アダムス | | 首席財務官兼財務主管 | | 2024年2月22日 |
ケンドラ·R·アダムス | | (首席財務官) | | |
寄稿S/マーク·モスラー | | 首席会計官 | | 2024年2月22日 |
マーク·モスラー | | (首席会計主任) | | |
| /S/ブルース·ドニー | | 会長兼取締役 | | 2024年2月22日 |
| ブルース·ドニー | | | | |
| /S/ケネス·C·アンダーソン医学博士 | | 役員.取締役 | | 2024年2月22日 |
| ケネス·C·アンダーソン医学博士 | | | | |
| /S/ローラ·ベソン医学博士 | | 役員.取締役 | | 2024年2月22日 |
| ローラ·ベソン医学博士 | | | | |
| 寄稿S/グレン·デュビン | | 役員.取締役 | | 2024年2月22日 |
| グレン·デュビン | | | | |
| /S/ドナ·グロガン医学博士 | | 役員.取締役 | | 2024年2月22日 |
| ドナ·グローガン医学博士 | | | | |
寄稿S/オーウェン·ヒューズ | | 役員.取締役 | | 2024年2月22日 |
オーウェン·ヒューズ | | | | |
| /S/Utpal Koppikar | | 役員.取締役 | | 2024年2月22日 |
| Utpal Koppikar | | | | |
| 寄稿S/マルコム·ソルト | | 役員.取締役 | | 2024年2月22日 |
| マルコム·ソルト | | | | |
連結財務諸表索引
| | | | | |
独立公認会計士事務所報告 | F-2 |
| |
合併貸借対照表 | F-3 |
| |
合併経営報告書と全面赤字 | F-4 |
| |
株主権益合併報告書 | F-5 |
| |
統合現金フロー表 | F-6 |
| |
連結財務諸表付記 | F-8 |
独立公認会計士事務所報告
株主や取締役会に
C 4治療会社:
連結財務諸表に対するいくつかの見方
C 4治療会社とその子会社(当社)の2023年12月31日現在と2022年12月31日までの連結貸借対照表、同年度までの関連総合経営報告書と全面赤字、株主権益と現金流量、および関連付記(総称して総合財務諸表と呼ぶ)を監査した。総合財務諸表は、すべての重要な面で、会社の2023年12月31日と2022年12月31日までの財務状況、および2023年12月31日と2022年12月31日までの年度の経営結果とキャッシュフローを公平に反映しており、米国公認会計原則に適合していると考えられる。
意見の基礎
これらの連結財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいてこのような連結財務諸表に意見を発表することだ。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、連結財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査することを求められておらず、私たちも招聘されて監査を行っていません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性について意見を述べるためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、連結財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであっても詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれる。これらの手続きは、連結財務諸表中の金額および開示に関する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、合併財務諸表の全体列報の評価も含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
重要な監査事項とは、連結財務諸表を当期監査する際に生じた、伝達された、または監査委員会に伝達された事項である:(1)総合財務諸表に対して重大な意義を有する勘定または開示に関連する;(2)私たちが特に挑戦的で、主観的または複雑な判断に関連する。私たちは重要な監査事項が存在しないと確信する。
/s/ピマウェイ法律事務所
2016年以来、当社の監査役を務めてきました。
ボストン、マサチューセッツ州
2024年2月22日
C 4治療会社
合併貸借対照表
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
| | | | | | | | | | | |
| 十二月三十一日 |
| 2023 | | 2022 |
| 資産 | | | |
| 流動資産: | | | |
| 現金と現金等価物 | $ | 126,590 | | | $ | 29,754 | |
| 流通有価証券 | 127,091 | | | 246,399 | |
| 売掛金 | 11,799 | | | 1,473 | |
| 前払い費用と他の流動資産 | 5,709 | | | 9,931 | |
| 流動資産総額 | 271,189 | | | 287,557 | |
| 非流通有価証券 | 28,008 | | | 60,962 | |
| 財産と設備、純額 | 7,132 | | | 7,400 | |
| 使用権資産 | 63,956 | | | 70,116 | |
| 制限現金 | 3,443 | | | 3,279 | |
| その他の資産 | 2,723 | | | 1,526 | |
| 総資産 | $ | 376,451 | | | $ | 430,840 | |
| | | |
| 負債と株主権益 | | | |
| 流動負債: | | | |
| 売掛金 | $ | 1,446 | | | $ | 1,172 | |
| 費用とその他の流動負債を計算しなければならない | 20,630 | | | 19,769 | |
| 収入を繰延し,当期 | 15,471 | | | 16,618 | |
| 賃貸負債を経営し、流動 | 5,219 | | | 4,700 | |
| 長期債務、債務割引関連先を差し引くと、当期 | — | | | 2,287 | |
| 流動負債総額 | 42,766 | | | 44,546 | |
| 繰延収入は当期を差し引く | 21,814 | | | 16,895 | |
| 賃貸負債を経営し,当期純額 | 65,757 | | | 70,970 | |
| 長期債務、債務割引関連者を差し引いた純額、当期純額 | — | | | 9,195 | |
| 総負債 | 130,337 | | | 141,606 | |
| 引受金及び又は事項(付記6及び付記9参照) | | | |
| 株主権益: | | | |
優先株、額面は$0.0001一株一株10,000,000ライセンス株、そして違います。2023年と2022年12月31日現在発行または発行済み株 | — | | | — | |
普通株、額面$0.0001一株一株150,000,000ライセンス株、そして60,467,188そして48,966,2162023年12月31日と2022年12月31日までの発行済株式 | 6 | | | 5 | |
| 追加実収資本 | 774,618 | | | 689,256 | |
| その他の総合損失を累計する | (127) | | | (4,137) | |
| 赤字を累計する | (528,383) | | | (395,890) | |
| 株主権益総額 | 246,114 | | | 289,234 | |
| 総負債と株主権益 | $ | 376,451 | | | $ | 430,840 | |
付記はこれらの連結財務諸表の構成要素である。
C 4治療会社
合併経営報告書と全面赤字
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
| | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | |
| R協調プロトコルから収益を得る | $ | 20,756 | | | $ | 31,096 | | | |
| 運営費用: | | | | | |
| 研究開発 | 117,706 | | | 117,841 | | | |
| 一般と行政 | 42,081 | | | 42,789 | | | |
| 総運営費 | 159,787 | | | 160,630 | | | |
| 運営損失 | (139,031) | | | (129,534) | | | |
| その他の収入,純額 | | | | | |
| 長期債務関連側の利子支出と償却 | (1,373) | | | (2,216) | | | |
| 債務損失を繰り上げ返済する | (621) | | | — | | | |
| 利息とその他の収入,純額 | 9,812 | | | 3,575 | | | |
| | | | | |
| その他の収入を合計して純額 | 7,818 | | | 1,359 | | | |
| 所得税前損失 | (131,213) | | | (128,175) | | | |
| 所得税費用 | (1,280) | | | — | | | |
| 純損失 | $ | (132,493) | | | $ | (128,175) | | | |
| 1株当たり純損失--基本損失と赤字 | $ | (2.67) | | | $ | (2.62) | | | |
| 加重-平均株式数-基本株式と希釈株式 | 49,640,505 | | | 48,861,665 | | | |
| | | | | |
その他総合収益(損失): | | | | | |
| 有価証券の未実現収益 | 4,010 | | | (3,362) | | | |
| 総合損失 | $ | (128,483) | | | $ | (131,537) | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
付記はこれらの連結財務諸表の構成要素である。
C 4治療会社
株主権益合併報告書
(単位は千で、シェアは含まれていない)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 普通株 | | その他の内容
支払い済み
資本 | | 積算
他にも
全面的に
収入(損) | | 積算
赤字.赤字 | | 合計する
株主の
権益 |
| 株 | | 金額 | | | | |
| 2021年12月31日現在の残高 | 48,688,875 | | | $ | 5 | | | $ | 658,091 | | | $ | (775) | | | $ | (267,715) | | | $ | 389,606 | |
| 株式オプションの行使と株式単位の解放 | 235,450 | | | — | | | 777 | | | — | | | — | | | 777 | |
| 2020年ESPPによる普通株式発行 | 31,402 | | | — | | | 372 | | | — | | | — | | | 372 | |
| 株に基づく報酬 | — | | | — | | | 29,858 | | | — | | | — | | | 29,858 | |
| 有価証券は赤字を実現していない | — | | | — | | | — | | | (3,362) | | | — | | | (3,362) | |
| 純損失 | — | | | — | | | — | | | — | | | (128,175) | | | (128,175) | |
| 他にも | 10,489 | | | — | | | 158 | | | — | | | — | | | 158 | |
| 2022年12月31日現在の残高 | 48,966,216 | | | $ | 5 | | | $ | 689,256 | | | $ | (4,137) | | | $ | (395,890) | | | $ | 289,234 | |
| 市場配当案に基づいて普通株を発行し,純額 | 11,186,142 | | | 1 | | | 57,672 | | | — | | | — | | | 57,673 | |
| 株式オプションの行使と株式単位の解放 | 13,096 | | | | | 58 | | | — | | | — | | | 58 | |
| 制限株単位に帰属するときに普通株を発行し,源泉徴収税を差し引いて買い戻した株式 | 117,745 | | | — | | | (110) | | | — | | | — | | | (110) | |
| 2020年ESPPによる普通株式発行 | 133,784 | | | — | | | 368 | | | — | | | — | | | 368 | |
| 株に基づく報酬 | — | | | — | | | 27,235 | | | — | | | — | | | 27,235 | |
| 有価証券の未実現収益 | — | | | — | | | — | | | 4,010 | | | — | | | 4,010 | |
| 純損失 | — | | | — | | | — | | | — | | | (132,493) | | | (132,493) | |
| 他にも | 50,205 | | | — | | | 139 | | | — | | | — | | | 139 | |
| 2023年12月31日現在の残高 | 60,467,188 | | | $ | 6 | | | $ | 774,618 | | | $ | (127) | | | $ | (528,383) | | | $ | 246,114 | |
付記はこれらの連結財務諸表の構成要素である。
カタログ表
C 4治療会社
統合現金フロー表
(単位:千)
| | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | |
| 経営活動で使われているキャッシュフロー: | | | | | |
| 純損失 | $ | (132,493) | | | $ | (128,175) | | | |
| 純損失と業務活動で使用されている現金の照合の調整: | | | | | |
| 株に基づく報酬費用 | 27,235 | | | 30,016 | | | |
| 減価償却および償却 | 1,880 | | | 1,676 | | | |
| 資産を使用する帳簿金額を減らす | 6,159 | | | 5,896 | | | |
| 有価証券の割増割引 | (3,784) | | | 714 | | | |
| 債務割引関連の償却 | 405 | | | 713 | | | |
| 債務損失を繰り上げ返済する | 621 | | | — | | | |
| 他にも | 77 | | | — | | | |
| 経営性資産と負債変動状況: | | | | | |
| 売掛金 | (10,327) | | | 4,244 | | | |
| 前払い費用その他の流動と長期資産 | 2,723 | | | 388 | | | |
| 売掛金 | 273 | | | (3,333) | | | |
| 費用とその他の流動負債を計算しなければならない | 1,316 | | | 5,083 | | | |
| リース負債を経営する | (4,695) | | | (507) | | | |
| 収入を繰り越す | 3,772 | | | (22,654) | | | |
| 経営活動のための現金純額 | (106,838) | | | (105,939) | | | |
| 投資活動が提供するキャッシュフロー: | | | | | |
| 有価証券満期日収益 | 289,955 | | | 283,445 | | | |
| 有価証券を購入する | (129,898) | | | (219,527) | | | |
| 財産と設備を購入する | (1,708) | | | (5,496) | | | |
| | | | | |
| 投資活動が提供する現金純額 | 158,349 | | | 58,422 | | | |
| 資金調達活動が提供するキャッシュフロー: | | | | | |
| 市場配当案に基づいて普通株を発行して得られた収益,純額 | 57,673 | | | — | | | |
| 支払長期債務関連者 | (12,500) | | | — | | | |
| 株式オプションを行使して得られる収益 | 58 | | | 777 | | | |
| 源泉徴収のための普通株買い戻し支払い | (110) | | | — | | | |
| 他にも | 368 | | | 370 | | | |
| 融資活動が提供する現金純額 | 45,489 | | | 1,147 | | | |
| | | | | |
| 現金、現金等価物、および限定的な現金純変化 | 97,000 | | | (46,370) | | | |
| 期初現金、現金等価物、および限定現金 | 33,033 | | | 79,403 | | | |
| 期末現金、現金等価物、および制限現金 | $ | 130,033 | | | $ | 33,033 | | | |
| | | | | |
| 現金、現金等価物、および制限現金の入金: | | | | | |
| 年末現金、現金等価物、制限現金 | $ | 130,033 | | | $ | 33,033 | | | |
| 差し引く:制限された現金 | (3,443) | | | (3,279) | | | |
| 年末現金および現金等価物 | $ | 126,590 | | | $ | 29,754 | | | |
付記はこれらの連結財務諸表の構成要素である。
カタログ表
C 4治療会社
統合現金フロー表--継続
(単位:千)
| | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | |
| キャッシュフロー情報の追加開示: | | | | | |
| 売掛金繰延収入 | $ | 10,000 | | | $ | — | | | |
| 利息関連先に支払われた現金 | $ | 990 | | | $ | 1,624 | | | |
| 所得税の現金を納める | $ | 1,003 | | | $ | — | | | |
| | | | | |
| 非現金投資と融資活動の追加開示: | | | | | |
| 売掛金と売掛金における資本支出 | $ | 18 | | | $ | 473 | | | |
| 使用権資産の取得による経営リース負債 | $ | — | | | $ | 44,067 | | | |
付記はこれらの連結財務諸表の構成要素である。
注1業務的性質
C 4治療会社またはその子会社である当社とともに、臨床段階の生物製薬会社であり、指向性タンパク質分解(TPD)科学の約束を実現し、患者の生活を変える新世代小分子薬物を創造することに取り組んでいる。その独自の魚雷プラットフォームを利用することにより,同社は小分子タンパク質分解剤を効率的に設計·最適化し,人体破壊に不要なタンパク質の自然過程を利用することで,目標の高度な活性化を期待している。その会社はそれを信じている新型口腔製品候補製品阻害剤によく出現する薬剤耐性を克服し,現在の“投与できない”標的を狙い,患者の結果を改善する可能性がある。同社は2015年10月7日にデラウェア州に登録設立され、主な事務所はマサチューセッツ州ウォータータウンにある
流動資金と資本資源
設立以来、同社の主な活動は、研究·開発活動、会社の知的財産権の構築、人員募集、資金調達に集中しており、これらの活動を支援してきた。これまで、同社の運営資金は主に償還可能な転換可能な優先株の売却、協力協定による公開会社の普通株の公開、債務融資によるものだった。
同社は設立以来、純損失#ドルを含む経常赤字が発生している132.5百万ドルとドル128.22023年12月31日までと2022年12月31日までの年度はそれぞれ100万ドル。また、2023年12月31日現在、会社の累計損失は1ドルとなっている528.4百万ドルです。同社はこれまで、候補商品が商業化されていないため、製品販売から何の収入も得ていない。同社は予見可能な未来に引き続き営業赤字が生じると予想している
同社は現金、現金等価物、有価証券を$と予想している281.72023年12月31日までの100万ドルは、これらの連結財務諸表の発行日から少なくとも今後12ヶ月の運営に資金を提供するのに十分である。だから、 総合財務諸表の作成は、会社が継続的に経営する企業であると仮定し、正常な業務過程で資産および負債の返済と約束を実現することを考慮している。
リスクと不確実性
同社は初期開発段階で他の生命科学会社と同様のリスクに直面しており、追加資金を調達する能力の不確実性、製品開発と商業化、ライバル開発の新技術革新の不確実性、キーパーソンへの依存、製品の市場受容度、マーケティング·販売の歴史の欠如、製品責任、ノウハウや知的財産権の保護、食品·薬物管理局(FDA)や他の政府法規の遵守を含む。もし同社がその計画を人体臨床試験に進めることに成功せず、人体臨床試験を通じてその任意の候補製品の商業化に成功した場合、あるいは他社との協力により、製品収入を生成したり、利益を達成することができない可能性がある。会社の研究開発作業が成功することは保証されず、会社の知的財産権が十分に保護されることは保証されず、開発されたどの製品も必要な政府規制の承認を得る保証はなく、いかなる承認された製品も商業的に実行可能であることは保証されない。同社の製品開発事業が成功しても、その会社がいつ(あれば)製品販売から相当な収入を得るかは定かではない。同社は技術の急速な変化と製薬やバイオテクノロジー会社からの激しい競争の環境で運営されている。
注2重要会計政策の概要
陳述の基礎
当社はすでに米国公認会計原則または米国公認会計原則に従って付随する総合財務諸表を作成した。本説明における適用ガイドラインへの任意の言及は、財務会計基準委員会(FASB)の会計基準編纂(ASC)と会計基準更新(ASU)における権威ある米国公認会計原則を指す。
合併原則
同社の総合財務諸表には、C 4治療会社とその完全子会社、マサチューセッツ州証券会社C 4 T証券会社の勘定が含まれている。すべての重大な会社間残高と取引は合併で販売された。
予算の使用
米国公認会計原則に基づいて連結財務諸表を作成することは、連結財務諸表日までの資産及び負債額、又は有資産及び負債の開示、並びに報告期間内の収入及び支出の報告金額に影響を与えるために、管理層に推定及び仮定を要求する。当社は過去の経験とその当時の状況で合理的と考えられる様々な要素に基づいて見積もりと仮説を立てています。この過程は,実際の結果と総合財務諸表を作成する際に用いる見積り金額が大きく異なる可能性があり,これらの結果が歴史的経験と異なる場合,あるいは他の仮説が実質的に正確ではないことが証明されていれば,たとえそのような仮説が作成時に合理的であってもよい.経営陣が主観的判断を使用する分野には、当社の研究開発協力計画に基づいて確認された収入の金額および時間、前払いおよび計算される研究開発費、長期資産使用寿命の推定、長期資産減価の評価、使用権資産および賃貸負債の計量、賃貸負債を計量するための逓増借款率、株式に基づく報酬支出の推定および確認が含まれるが、これらに限定されない。当社は継続的な基準で推定を評価し、過去の経験、既知の傾向、合理性を信じている他の様々な仮定に基づいて推定し、その結果、資産および負債額面を判断する基礎を構成する。実際の結果はこのような推定とは大きく異なるかもしれない。
細分化市場
運営部門は企業の構成要素として定義され、その独立かつ離散的な情報は、首席運営決定者が資源をどのように割り当てるかを決定し、業績を評価する際に評価することができる。その会社は所有している1つは運営部門です。会社の運営意思決定者兼最高経営責任者が総合管理会社の運営を担当し、資源を分配することを目的としている。同社のすべての長期資産はアメリカで保有しています。
現金等価物
当社はすべての購入時満期日が三ヶ月以下の高流動性投資を現金等価物と見なしています。当社の現金等価物は公正価値で恒常的に計量されています。
制限現金
制限現金には、付記6にさらに記載されているように、マサチューセッツ州ウォータータウン施設賃貸契約条項の要求に基づいて、個別制限銀行口座に入金される現金が含まれている賃貸借証書.
有価証券
会社は購入日の残り期限が3カ月を超える有価証券を売却可能証券に分類している残り満期日が1年以上の有価証券は非流動資産に分類される売却可能な証券は公正価値に従って勘定し、損益計上は累計他の総合(損失)収入を計上しておらず、株主権益の構成部分として、実現までとしている。購入時に生成される任意の割増または割引は、手形の有効期間内に償却され、および/または利息収入および/または費用が増加される。実現した損益は特定の確認方法で決定し,他の収入(費用)に計上する。
信用リスクが集中する
会社を深刻な集中信用リスクに直面させる可能性のある金融商品は主に現金、現金等価物、有価証券、制限された現金を含む。当社の金融機関での預金は政府の保険限度額を超えることができます。当社は重大な信用リスクはないと信じており、その預金は管理層が高い信用要素を持つと考えられている金融機関に保管されているが、当社はこのような預金によって何の損失も受けていない。また、同社は信用格付けと満期日に関するガイドラインを策定し、元金残高の保障と流動性の維持を目指している。同社はその投資政策に基づいてその基金を維持し、この政策は許可された投資を定義し、信用品質基準を規定し、任意の単一発行者に対する信用リスクを制限することを目的としている。
金融商品の公正価値
ASCテーマ820、公正価値計量またはASC 820は、公正価値計量のためのツールのための公正価値階層構造を構築し、市場データに基づく仮説(観察可能な投入)と会社自身の仮説(観察不可能な投入)とを区別する。観察可能な投入は、市場参加者が資産価格を設定する際に使用する投入または
当社から独立したソースから得られた市場データの責任に基づく。観察できない投入は、市場参加者が資産や負債の価格設定のために使用されるという会社の仮定を反映し、その時点で入手可能な最適な情報に基づいて制定される。ASC 820は、公正価値を交換価格または退出価格として決定し、市場参加者間の秩序ある取引において資産を売却することによって受信された金額または移転負債によって支払われた金額を表す。公正価値計量において市場参加者の仮説を考慮する基礎として、ASC 820は以下の項目を区別した三級価値階層構造を構築した
•第1レベル--アクティブ市場における同じ資産または負債の活発な市場オファーを反映した観察可能な投入
•第2レベル−市場で直接または間接的に観察される第1レベル投入以外の投入
•第3レベル-観察不可能な投入は、市場活動の支援が少ないか、またはサポートされておらず、市場参加者が使用可能な仮説を反映した会社によって作成された仮説の推定を使用することが可能である
ある程度、推定値は市場で観察または観察できないモデルや投入に基づいており、公正価値の決定にはより多くの判断が必要である。そのため、当社が価値を公平に判断する際に下した判断は、第3級に分類されたツールに対する判断度が最も高く、公正価値レベル内の金融商品レベルは、公正価値計量に大きな意味を持つ任意の投入の中で最低レベルに基づいている。当社は各報告期間の終了時に異なるレベル間の移行を評価します。
財産と設備
財産と設備はコストから減価償却累計を引いて入金されます。修理と保守支出は発生時に費用を計上する。資産が廃棄又は処分された場合には、資産及び関連する減価償却が勘定からキャンセルされ、それによって生じる収益又は損失のいずれも純損失の確定に計上される。
設備減価償却は、資産の推定耐用年数内に直線法を用いて以下のように計算される
| | | | | | | | |
| 資産種別 | | 使用寿命を見込む |
| 実験室装置 | | 5年.年 |
| コンピュータ装置 | | 3年.年 |
| 事務設備、家具、固定装置 | | 5年.年 |
| 賃借権改善 | | 耐用年数または残存賃貸期間が短いもの |
賃貸借証書
当社はASCテーマ842に基づいてレンタルを会計処理している賃貸借証書またはASC 842。手配開始時に、当社は、当時の特定の事実及び状況、確認された資産(S)の存在(あれば)及び当社の確認済み資産(S)の使用制御権(適用)に基づいて、その手配が賃貸に属するか否か又は賃貸を含むか否かを決定する。ASC 842の規定によると、会社は賃貸と非レンタル構成要素をすべてのレンタルの単一構成要素に統合することを選択する。経営リース負債及びそれに応じた使用権資産は、予想リース期間内の将来の賃貸支払いの現在値に基づいて入金される。受け取った報酬などの項目については、使用権資産を何らかの調整する必要がある可能性がある。同社は、レンタルスケジュールを評価する際には、通常、初期レンタル期間のみを含み、同社が継続すると合理的に判断されない限り、レンタルを更新するオプションは評価に含まれていない。レンタル契約に隠されている金利は一般的に確定しにくい。そこで、賃貸負債を算出する際には、当社は、賃貸金に相当する金額を抵当に入れるような経済環境下で発生した金利を逓増借款金利を採用している。レンタル料金は予想レンタル期間内に直線的に確認します。
長期資産減価準備
事件や環境変化がある資産の帳簿価値が回収できない可能性があることを示すたびに、当社はその長期資産の減値を評価する。保有·使用する資産の回収可能性は,資産の帳簿価値と資産予想による将来の未割引現金流量を比較することで測定した。当該等の資産は減値とみなされ、確認すべき減値は、当該資産の帳簿価値が当該資産の公正価値を超える金額で計量される。処分すべき資産を帳簿または公正価値から売却コストの中の低いものを引いて申告する。
引受金とその他の事項
クレーム、評価、訴訟、罰金および罰金およびその他の出所によって発生したまたは損失のある負債は、負債が発生し、金額が合理的に推定できる可能性が高い場合に記録される。損失の有無に関する法的費用は発生時に費用を計上する。
収入確認
会社はASCテーマ606に基づいて収入を確認した取引先と契約した収入またはASC 606。本基準は、顧客と締結されたすべての契約に適用され、他の基準範囲に属する契約は除外される。ASC 606によれば、エンティティは、そのクライアントが約束された商品またはサービスの制御権を取得したときに収入を確認し、その金額は、エンティティがこれらの商品またはサービスと交換するために予期される対価格を反映する
同社は、戦略パートナーとASC 606の範囲内の協力および許可協定を締結し、この合意によれば、その候補製品の研究、開発、製造、および商業化の権利を第三者に独占的に許可することができる。これらの取り決めの条項は、一般に、(1)払い戻し不可能な前払い許可料、(2)特定の費用の精算、(3)追加商品またはサービスの顧客選択費、(4)発展マイルストーン支払い、(5)規制および商業マイルストーン支払い、および(6)特許製品の純売上の特許権使用料のうちの1つまたは複数の費用を会社に支払うことを含む
各合意に規定された義務を履行する際に確認すべき適切な収入額を決定する場合、会社は、(1)契約で約束された貨物またはサービスを決定するステップと、(2)契約で異なるか否かを含む承諾された貨物またはサービスが契約義務であるかどうかを決定するステップと、(3)可変対価格の制限を含む取引価格の計量と、(4)取引価格を履行義務に割り当てるステップと、(5)会社が各履行義務を履行する場合(または履行義務とする場合)収入を確認するステップとを実行する。この等手配の会計手配の一部として、当社は、(A)上記(Ii)ステップの決定による履行責任数、(B)上記(Iii)ステップ目の取引価格、(C)上記(Iv)ステップ目の取引価格割当契約によって決定された各履行責任の独立販売価格、及び(D)上記(V)ステップ目の履行責任の契約条項及び履行モードに基づいて判断しなければならない。当社は、以下に述べるように、特許使用料を除くマイルストーンまたは他の可変対価格が取引価格に含まれるべきかどうかを決定するために判断を使用します。取引価格は相対独立販売価格で契約履行義務ごとに割り当てられ、会社は契約項目の履行義務を履行する際に収入を確認する
収入確認基準を満たすために当社に不足している金又は提携合意条項に基づいて支払う契約金は、当社の総合貸借対照表に売掛金として入金されています。収入確認基準を満たす前に受け取った金額は繰延収入として会社の総合貸借対照表に記録されている。貸借対照表の後日12カ月以内に収入と確認された金額は当期繰延収入に分類されると予想される。貸借対照表後12カ月以内に収入が確認されなかった金額は繰延収入に分類され、当期分を差し引く
許可証料を前払いする
会社の知的財産権の許可が合意で決定された他の約束または義務とは異なると判断された場合、許可が顧客に譲渡され、顧客が許可を使用して利益を得ることができる場合、会社は、許可に割り当てられた払戻不可能な前払い費用の収入を確認する。承諾または義務履行が他の約束と異なるかどうかを評価する際に、会社は、顧客の研究、製造、および商業化能力、会社が任意の重要な権利を保持すること、および一般市場における関連専門知識の利用可能性を考慮する。また、当社は、顧客が残りの約束を受けずに、その期待目的について約束から利益を得ることができるかどうか、約束の価値が未履行の承諾に依存するかどうか、他のサプライヤーが残りの約束を提供できるかどうか、およびその約束が残りの約束とは別に識別できるかどうかを考慮する。他の承諾と組み合わせたライセンスについて、会社は、合併履行義務の性質を評価して、合併履行義務が時間の経過とともに履行されるか、ある時点で履行されるかを決定する判断を行使し、時間が経過すれば、収入を確認するための進展を測定するための適切な方法を決定する。当社は報告期間ごとに進捗指標を評価し、必要に応じて業績指標と関連収入確認を調整します
顧客オプション
1つのスケジュールが、顧客が追加の商品またはサービスを取得することを可能にする顧客選択権を含むと判定された場合、顧客選択権の基礎となる商品およびサービスは、手配開始時に契約義務とみなされない。当社は、無料または割引で追加商品またはサービスを得る実質的な権利または選択権に対する顧客の選択を評価します。顧客選択権が実質的な権利を表すと決定された場合、実質的な権利は、手配の開始時に別個の履行義務として確認される。当社は、決定された割引と顧客がオプションを行使する可能性に基づいて決定された相対独立販売価格に基づいて材料権利に取引価格を割り当てる。重大な権利に割り当てられた金額は,最も早く選択権を行使するまで収入として確認されない.オプションが行使されていない場合、目標が終了され、会社は実質的な権利履行義務に関連するすべての残りの収入を加速して確認するだろう
研究と開発サービス
会社の協力協定の下での約束には、会社が顧客または顧客を代表するために提供する研究·開発サービスが含まれる可能性がある。会社の研究開発作業による支払いや精算は、会社がこのような仕事の依頼者であるため、毛数でサービスを提供していることが確認された。顧客からの精算や顧客への支払いは、顧客との連携関係の結果であり、共同開発活動などの顧客関係ではなく、研究·開発費用の削減記録として用いられる
一里塚払い
開発マイルストーンの支払いを含む各手配の開始時に、当社はマイルストーンが実現可能であると考えられているかどうかを評価し、可能な金額法を用いて取引価格に含まれる金額を推定する。大きな収入逆転が起こらない可能性が高い場合、関連するマイルストーン価値は取引価格に含まれるだろう。規制承認のような会社または被許可者の統制範囲内にない記念碑的支払いは、これらの承認を受ける前に実現可能であるとは考えられない。同社はこの評価における特定のマイルストーンを実現するために、科学、臨床、監督管理、商業、その他克服しなければならないリスクなどの要素を評価した。重大な収入逆転が発生しない可能性があるかどうかを決定する際には,かなりの判断が必要である。その後の各報告期間が終了した時点で、当社はすべての制限されたマイルストーンの実現確率を再評価し、必要に応じて全体の取引価格の推定を調整する。いずれの調整も累積追跡をもとに記録されており,調整期間中の収入や収益に影響を与える.
連携収入会計処理の更なる検討については、参照のこと注8.連携および許可プロトコル。
研究開発費
研究開発コストは発生した費用に基づいて計上され、臨床前研究と臨床試験を行うことに関連するコストを含み、主に給料、株式ベースの給与と他の従業員福祉支出、実験室関連用品と会社の研究開発活動に関連する他の運営コストを含み、分配された施設関連の支出と招聘による研究開発活動を行う外部サプライヤーの外部コストを含む。
臨床試験費用は同社の研究と開発費用の重要な構成部分である。会社の候補製品の持続的な開発過程で,会社は従来,会社を代表して様々な臨床試験活動を行ってきた第三者と契約を結んできた。連結財務諸表作成過程の一部として、当社は研究と開発費用を見積もりなければならない。会社は、その時点で知られている事実および状況に基づいて、連結財務諸表において、各貸借対照表の日付までの計算費用を推定し、未平倉契約および調達注文を審査することを含み、その人員とコミュニケーションして、会社を代表して提供されたサービスを決定し、企業が領収書を受信していない場合、または実際のコストを通知していない場合に、提供されるサービスレベルおよびサービスに生じる関連コストを推定する。第三者と締結された契約金額が修正された場合(例えば、臨床試験レジメンまたは実行されるべき作業範囲の変化のため)、会社は、予想に基づいて、関連する計算すべき項目を修正するであろう。契約範囲の改正は、修正を引き起こした事実が合理的に確定している間に費用を計上する。
また、会社のサプライヤーに支払われる金額は、提供されたサービスレベルを超え、前払い費用をもたらす可能性があり、この場合、このような金額は、前払い費用および他の費用として反映される
流動資産。サービス料を計算する際には、当社はサービスを提供する時間帯と時間ごとにかかる努力度を見積もる。実際にサービスを実行する時間や努力の程度が見積もりと異なる場合、会社はそれに応じて費用または前払い費用の金額を調整する。将来的に受信された研究および開発活動のための貨物またはサービスの払戻不可能な前払いは延期され、前払い費用および他の流動資産の形態で資本化される。資本化金額は関連貨物の納入またはサービス提供時に費用を計上する。
株に基づく報酬
当社はASCテーマ718を適用している報酬--株式報酬またはASC 718は、株式ベースの支払いを示す。ASC 718によれば、会社は報酬が分類され、責任報酬または株式奨励に計上されるべきかどうかを決定する。会社が従業員に付与するすべての株式奨励は株式奨励に分類され、会社の財務諸表では、付与日の公正価値に基づいて確認され、公正価値はブラック·スコルスオプション定価モデルを用いて計量される。ブラック·スコアーズオプション定価モデルは、株式に基づく報酬の期待期限、予想変動率、無リスク金利、配当率、および普通株の公正価値を使用して、株式報酬の公正価値を推定する
•当社は、“簡略化”方法を用いて、期待期限、すなわち期待期間がオプションの帰属期限と契約期間の算術平均値に等しいと推定する
•十分な会社の特定の歴史と隠れた変動率データが不足しているため、当社の予想変動率計算は代表的な上場企業の歴史変動率に基づいており、これらの会社は製品開発段階と生命科学業界の重点を含む当社と類似した特徴を持っている
•オプション期待期間内の無リスク金利は、オプション付与期間中に有効な米国債収益率曲線から算出される
•期待配当収益率はゼロ当社は配当金を派遣したことがないため、現在もその普通株についていかなる配当金も派遣する計画はない。
•株式に基づく奨励の普通株の公正価値は、会社普通株が付与された日の市場オファーである
•会社は没収行為が発生した場合に確認します。
報酬の公正価値は、報酬の必要なサービス期間(通常は帰属期間)内で直線原則で株式ベースの補償支出として確認される。業績とサービスの帰属条件に基づく制約を同時に受けた報酬については、管理層の最適な推定によれば、業績条件が実現可能であると考えられた場合には、残りのサービス期間内の株式ベースの報酬支出を確認する。株式に基づく補償費用の総合業務と総合損失報告書における分類方式は,受賞者の賃金コスト分類や受賞者サービス支払分類の方式と同様である。
株式報酬のいかなる条項や条件のいかなる変化も報酬の修正とみなされる。逓増補償コストとは、修正された裁決の公正価値が、その条項が修正される直前の元の裁決の公正価値を超える部分(ある場合)であり、修正日の裁決の公正価値及び他の関連要素に基づいて計算される。既得奨励については、会社は修正発生期間中の増分補償コストを確認した。帰属されていない補償については、当社は、残りの必要なサービス期間内の費用、増分補償コスト、および修正日の元の補償の残りの未確認補償コストの和を確認する。修正された裁決の公正価値が改正前の元の裁決の公正価値よりも低い場合、会社が確認した最低補償コストは、元の裁決のコストである。
所得税
当社は、連結財務諸表または当社納税申告書で確認された事件の将来の税務結果を予想する繰延税金資産と負債の確認を要求する貸借対照法を用いて所得税を計算する。この方法によると、繰延税項資産及び負債は、総合財務諸表と資産及び負債の税ベースとの差額に基づいて決定され、予想差額に応じて繰り返される年度の現行税率が決定される。繰延税金資産と負債の変動を所得税に計上する準備。当社は、その繰延税金資産が将来の課税収入から回収される可能性を評価し、既存の証拠の重みに基づいて繰延税金資産の全部または一部が現金化できない可能性があると考えた場合に、推定支出を確立する
当社は、ASCトピック740の規定を使用して、その不確定な税務状況を評価する所得税またはASC 740は、財務諸表確認および計量納税申告書において採用されるまたは予想される税金状態の“より可能な”基準を使用する税務状況が総合財務諸表において確認される前に満たされる必要がある確認閾値を規定する。所得税の準備には、それによって生じる税収準備金または未確認の税収割引の影響が適切であると考えられ、関連する純利息および罰金が含まれる。
総合損失
総合損失は、企業が一定期間内に非所有者由来の取引や他のイベントや状況によって発生する権益変化と定義される。総合損失には純損失及び株主権益の他の変化が含まれ、その中には純損失以外に含まれないいくつかの権益変化が含まれている。同社の他の全面収益の唯一の構成要素は有価証券の未実現収益と損失である。
1株当たり純損失
1株基本と償却純損失は当期発行普通株の加重平均を用いて計算した。当社が1株当たりの償却(赤字)収益を計算する際には、期間内に発行された株式オプションや無帰属制限株の希薄化効果を考慮し、当該等の証券が逆償却作用を持たない限り、赤字を抑制する
普通株等値株に逆償却作用があれば、計算希釈後の1株当たり純損失には含まれない。当社が純損失期間を報告している間は,希釈後の1株当たり純損失は通常1株当たりの純損失とほぼ同じであり,希釈性普通株の効果が逆薄であれば発行されているとは仮定しないからである。
経営を続ける企業
各報告期間内に、当社が条件や事件があるかどうかを評価し、当社が総合財務諸表が発行された日から1年以内に経営を継続する能力に大きな疑いを抱かせます。当社が重大な疑いがあると判断した場合、当社の計画はその疑いを軽減していない場合や、その計画が鈍化して当社の持続的な経営能力に対する重大な疑いがある場合には、当社はいくつかの追加開示をしなければならない。同社の評価は、予想される経営予算と会社の現金需要の予想予測を分析し、これらの需要を現在の現金、現金等価物、有価証券残高と比較する必要がある。
最近発表された会計基準
FASBは、2023-09、所得税(主題740):改良所得税開示を発表した。またはASU 2023-09。ASU 2023-09は、会社の年次財務諸表に、料金対帳に一致するカテゴリとより多くの情報分類を含め、管轄区域別に所得税を支払うように要求している。ASU 2023−09は2025年12月15日以降に開始された会社年度報告期間内に有効である。採択された方法は展望的か、完全に追跡された移行方法だ。早期養子縁組を許可する。会社は現在、ASU 2023-09を採用して連結財務諸表に及ぼす影響を評価している。
注3公正価値計量
以下の表は、当社が公正価値で常時計測している金融資産の情報を示し、2023年12月31日現在の公正価値を決定するための公正価値レベル(千単位)を示している
| | | | | | | | | | | | | | | | | | | | | | | |
| 公正価値 | | レベル1 | | レベル2 | | レベル3 |
| 現金等価物: | | | | | | | |
| 貨幣市場基金 | $ | 103,564 | | | $ | 103,564 | | | $ | — | | | $ | — | |
| アメリカ国債 | 14,972 | | | — | | | 14,972 | | | — | |
| 会社債務証券 | 7,588 | | | — | | | 7,588 | | | — | |
| 有価証券: | | | | | | | |
| 会社債務証券 | 128,705 | | | — | | | 128,705 | | | — | |
| アメリカ政府債務証券 | 20,428 | | | — | | | 20,428 | | | — | |
| アメリカ国債 | 5,966 | | | — | | | 5,966 | | | — | |
| 現金等価物と有価証券総額 | $ | 281,223 | | | $ | 103,564 | | | $ | 177,659 | | | $ | — | |
次の表は、2022年12月31日に公正価値別に会社が区分した金融資産の公正価値(単位:千)を示している
| | | | | | | | | | | | | | | | | | | | | | | |
| 公正価値 | | レベル1 | | レベル2 | | レベル3 |
| 現金等価物: | | | | | | | |
| 貨幣市場基金 | $ | 28,705 | | | $ | 28,705 | | | $ | — | | | $ | — | |
| | | | | | | |
| アメリカ国債 | 799 | | | — | | | 799 | | | — | |
| 有価証券: | | | | | | | |
| 会社債務証券 | 234,327 | | | — | | | 234,327 | | | — | |
| アメリカ政府債務証券 | 47,641 | | | — | | | 47,641 | | | — | |
| アメリカ国債 | 25,393 | | | — | | | 25,393 | | | — | |
| 現金等価物と有価証券総額 | $ | 336,865 | | | $ | 28,705 | | | $ | 308,160 | | | $ | — | |
当社はその通貨市場基金を公正価値レベル内の1級資産に分類しており、これらの基金は活発な市場の見積もりに基づいて推定されており、推定調整は行われていない
有価証券には、米国債、米国政府債務証券、会社債務証券が含まれ、ASC 320によると、これらの証券はすべて売却可能に分類されている投資--債務と持分証券それは.有価証券が公正価値レベルの第2レベルに分類されるのは、定価投入がアクティブ市場のオファーとは異なり、報告日に直接または間接的に観察されることができ、公正価値はモデルまたは他の推定方法を使用して定期的に決定されるからである。
注4有価証券
2023年12月31日現在の有価証券には、以下のものが含まれています(千単位)
| | | | | | | | | | | | | | | | | | | | | | | |
| 償却する コスト | | 毛収入 実現していない 収益.収益 | | 毛収入 実現していない 損 | | 公平である 価値がある |
| 有価証券、現在: | | | | | | | |
| 会社債務証券 | $ | 100,903 | | | $ | 16 | | | $ | (221) | | | $ | 100,698 | |
| アメリカ政府債務証券 | 20,457 | | | 14 | | | (43) | | | 20,428 | |
| アメリカ国債 | 5,965 | | | 1 | | | — | | | 5,966 | |
| 非流通有価証券 | | | | | | | |
| 会社債務証券 | 27,901 | | | 120 | | | (14) | | | 28,007 | |
| | | | | | | |
| | | | | | | |
| 流通と非流通有価証券総額 | $ | 155,226 | | | $ | 151 | | | $ | (278) | | | $ | 155,099 | |
2022年12月31日現在の有価証券には、以下のものが含まれています(千単位)
| | | | | | | | | | | | | | | | | | | | | | | |
| 償却する コスト | | 毛収入 実現していない 収益.収益 | | 毛収入 実現していない 損 | | 公平である 価値がある |
| 有価証券、現在: | | | | | | | |
| 会社債務証券 | $ | 183,270 | | | $ | 2 | | | $ | (2,068) | | | $ | 181,204 | |
| アメリカ政府債務証券 | 40,986 | | | — | | | (1,184) | | | 39,802 | |
| アメリカ国債 | 25,650 | | | — | | | (257) | | | 25,393 | |
| 非流通有価証券 | | | | | | | |
| 会社債務証券 | 53,592 | | | 2 | | | (471) | | | 53,123 | |
| アメリカ政府債務証券 | 8,000 | | | — | | | (161) | | | 7,839 | |
| | | | | | | |
| 流通と非流通有価証券総額 | $ | 311,498 | | | $ | 4 | | | $ | (4,141) | | | $ | 307,361 | |
現物に分類された有価証券の満期日は1年未満である。非流動証券に分類される有価証券とは、(I)満期日が1年を超えることと、(Ii)これらの資金が使用可能であるにもかかわらず、今後12ヶ月以内に清算されることを意図していない証券とを意味する。利用可能なものはありません
2023年12月31日現在保有する販売待ち債務証券または2021年12月31日の残り期限が5年を超える。
いままで赤字を達成していない有価証券2023年12月31日以下の部分からなる(単位:千、証券数を除く):
| | | | | | | | | | | | | | | | | |
| 量
証券 | | 公平である
価値がある | | 毛収入
実現していない
損 |
| 12ヶ月連続で赤字を達成していない有価証券: | | | | | |
| 会社債務証券 | 64 | | $ | 68,563 | | | $ | (146) | |
| アメリカ政府債務証券 | 9 | | 11,914 | | | (22) | |
| | | | | |
| 12ヶ月以上連続して赤字を達成していない有価証券: | | | | | |
| 会社債務証券 | 18 | | 22,138 | | | (89) | |
| アメリカ政府債務証券 | 3 | | 5,979 | | | (21) | |
| | | | | |
| 未実現損失の有価証券総額 | 94 | | $ | 108,594 | | | $ | (278) | |
いままで赤字を達成していない有価証券2022年12月31日以下の部分からなる(単位:千、証券数を除く):
| | | | | | | | | | | | | | | | | |
| 量
証券 | | 公平である
価値がある | | 毛収入
実現していない
損 |
| 12ヶ月連続で赤字を達成していない有価証券: | | | | | |
| 会社債務証券 | 63 | | $ | 134,027 | | | $ | (1,262) | |
| アメリカ政府債務証券 | 6 | | 15,748 | | | (245) | |
| アメリカ国債 | 4 | | 5,575 | | | (11) | |
| 12ヶ月以上連続して赤字を達成していない有価証券: | | | | | |
| 会社債務証券 | 36 | | 82,375 | | | (1,277) | |
| アメリカ政府債務証券 | 7 | | 31,892 | | | (1,100) | |
| アメリカ国債 | 4 | | 19,817 | | | (246) | |
| 未実現損失の有価証券総額 | 120 | | $ | 289,434 | | | $ | (4,141) | |
歴史的経験、市場データ、発行者特定要素、現在の経済状況などの要素に基づいて、彼の会社はできた違います。It‘2023年12月31日に信用損失準備金を記録しないでください2022年12月31日にはこれらの証券に関連している。
注5財産と設備
財産と設備は以下の部分から構成される(千計)
| | | | | | | | | | | |
| 12月31日まで |
| 2023 | | 2022 |
| 財産と設備 | | | |
| 実験室装置 | $ | 8,042 | | | $ | 8,757 | |
| 賃借権改善 | 4,712 | | | 4,682 | |
| 家具と固定装置 | 1,422 | | | 1,181 | |
| 事務設備 | 621 | | | 529 | |
| コンピュータ装置 | 98 | | | 191 | |
| 建設中の工事 | — | | | 183 | |
| 総資産と設備 | 14,895 | | | 15,523 | |
| 減算:減価償却累計 | (7,763) | | | (8,123) | |
| 財産と設備の合計 | $ | 7,132 | | | $ | 7,400 | |
財産や設備に関する減価償却費用は以下の通り(千で計算)
| | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | |
| 減価償却費用 | $ | 1,578 | | | $ | 1,676 | | | |
注6賃貸借証書
同社は2017年7月、マサチューセッツ州ウォータータウン本社オフィスとラボスペースの賃貸契約、またはウォータータウン賃貸を締結した。ウォータータウンのレンタルにはキャンセルできない期限があります10年拡張可能なオプションを持つ1つはその他の内容5年制レンタル期間全体で、レンタル料は上昇するだろう。また、ウォータータウンリース会社は同社に担保を提供することを要求しており、これらの担保は総合貸借対照表に限定的な現金と記載されている。水城レンタルは2018年4月に開始された。水城賃貸は経営賃貸に分類され、2018年4月開始時に当社は#ドルの賃貸負債を記録した15.1100万ドルの使用権資産16.7$を含む百万ドル1.5百万ドルの建設コストは会社が出資します。最初にリース負債と使用権資産を計算する際には、会社は追加のものを含まない5年制経営陣は当社がこのオプションを行使すると信じていないため、そのオプションを行使することを合理的に把握している。レンタル料のほか、当社はレンタルスペースに関する公共エリアメンテナンス、不動産税、財産保険による比例配分の費用を比例的に支払うことを担当しており、これらの費用は可変賃貸コストに計上されている。
2021年11月に、当社は水城賃貸借契約または改訂された賃貸契約を改訂した。レンタルの改訂は、当社の既存のレンタルスペースまたは既存のレンタルスペースのレンタル期間を延長し、追加のオフィススペースおよびラボスペースまたは新しいレンタルスペースを提供することを目的としています。改訂賃貸は二零二年一月から発効しましたが、当社は新賃貸空間で賃貸料を支払う責任は二零二年三月から発効し、新賃貸料責任の金額は既存の賃貸空間に対する当社の賃貸料支払いの継続責任に加入しています。改訂された賃貸契約は2032年3月に終了する10新しいレンタルスペースから借りた日から年を数えます。改訂されたレンタルは固定レンタル料で追加しなければなりません。最高で$に達します2.6改訂されたリース期間を延長するための選択を当社に提供します1つはその他の内容5年制ピリオド。改訂賃貸借契約を締結した後,当社は担保を$に増加させる3.3この現金は、2023年12月31日、2023年12月31日、2022年12月31日まで、添付されている連結資産負債表に限定的な現金とされている。賃貸料のほか、改訂されたレンタル条項に基づいて、当社もレンタルスペースに関する公共エリアのメンテナンス、不動産税及び財産保険による比例分担コストを比例的に支払うことを担当しており、既存のレンタルスペース及び2022年1月に新たにレンタルするスペースを含み、このような金額は変動レンタルコストに計上されている。
改訂賃貸借契約の会計処理
改訂借款により既存のレンタルスペースの年間期間を延長し、新しいレンタルスペースを使用する通路を提供するため、ASC 842の規定により、当社は改訂賃貸契約を入金します二つ単独の契約:1)既存賃貸空間のレンタル期間を延長するために既存賃貸契約を修正すること、および2)新規賃貸空間の使用権を延長する新しい賃貸契約。
当社は改訂賃貸契約を締結して既存の賃貸空間の制御を維持しているため、当社は使用権資産および賃貸負債を#ドル増加させた20.1改訂賃貸署名後、現有のレンタル空間は延長期間内の新しい賃貸料支払い及び現在の増加借入金利に関連する百万ユーロである。既存の賃貸空間のレンタル負債と使用権資産の計算には追加のものは含まれていません5年制当社は合理的な把握がこの購入株権を行使すると信じていないため、当社はこのオプションを行使する合理的な把握はしていない。
以上のように、新たなレンタルスペースのレンタルは2022年1月に開始される。そのため、会社は#ドルの使用権資産を記録した44.4百万ドル、それに応じたレンタル負債は#ドルです44.1新しいレンタルスペースのために100万ドルです。新規賃貸住宅の賃貸負債と使用権資産の計算には追加のものは含まれていません5年制改訂賃貸借契約によって提供される期間選択権によると、当社はこの選択権が行使される合理的な確実性を信じていないためである。以上のように、改正された賃貸借規定は最高$に達する2.62022年12月31日までの年間に全額使用されるテナント改善手当百万円。
2022年5月、当社は第三者と転貸契約を締結し、マサチューセッツ州ウォータータウンアーセナル路490号にあるSuite 200のオフィスと実験室スペースの一部を転用した。転貸期間は2025年6月に終了する。
レンタル料金の構成は以下の通り(千で計算)
| | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | |
| レンタル料: | | | | | |
| リースコストを経営する | $ | 10,030 | | | $ | 10,031 | | | |
| 可変リースコスト | 1,979 | | | 2,026 | | | |
| 転貸収入 | (2,577) | | | (1,481) | | | |
| 総賃貸コスト | $ | 9,432 | | | $ | 10,576 | | | |
次の表は、レンタル負債を計算するためのレンタル期間と逓増借入金金利をまとめています
| | | | | | | | | | | |
| 12月31日まで |
| 2023 | | 2022 |
| 残借期 | 8.1年.年 | | 9.1年.年 |
| 増額借款金利 | 5.3 | % | | 5.3 | % |
2023年12月31日までの取消不可レンタルでは、2023年12月31日までの年間の将来賃貸支払いは以下の通り(千単位)
| | | | | |
| 未割引のレンタル支払い: | |
| 2024 | $ | 8,828 | |
| 2025 | 9,093 | |
| 2026 | 9,366 | |
| 2027 | 9,646 | |
| 2028 | 11,362 | |
| その後… | 40,751 | |
| 未割引賃貸支払総額 | 89,046 | |
| 差し引く:推定利息 | (18,070) | |
| 2023年12月31日までの総経営賃貸負債 | $ | 70,976 | |
注7費用とその他の流動負債を計算しなければならない
計算すべき費用および他の流動負債には、以下の項目が含まれる(千で計算)
| | | | | | | | | | | |
| 12月31日まで |
| 2023 | | 2022 |
| 費用とその他の流動負債を計算しなければならない | | | |
| 計画に応じて研究·開発する | $ | 11,243 | | | $ | 9,824 | |
| 報酬と福祉に計上すべきである | 7,344 | | | 6,831 | |
| | | |
| 他にも | 2,043 | | | 3,114 | |
| 費用とその他の流動負債総額を計算しなければならない | $ | 20,630 | | | $ | 19,769 | |
2023年12月31日までに3.1CFT 8634の逐次終了活動に関するコストのうち1.6億ドルは計算すべき研究開発残高に含まれている.
注8協力と許可協定
メルク許可と協力協定
2023年12月11日、同社はメルク社またはメルク協定と独占的許可および協力協定を締結し、癌細胞中の病原性タンパク質を選択的に標的とすることを目的とした分解物-抗体結合体(DAC)を開発した。
メルク合意の条項によると、会社は1部を受け取りました$10.0百万2024年1月に前払いします。同社とメルク社は,最初に開示されなかった腫瘍学的目標に対するDACを協力開発することは,協力独自のものである。この初期目標に対するDACでは,会社は合計約$を取得する資格がある600600万ドルと将来販売される等級版税です。この協定はまた、メルク社に協力範囲を拡大するオプションを提供した三つ協力独自の他の目標であり、これはオプション行権支払いおよび潜在的なマイルストーンおよび特許使用料を生じる可能性がある。もしメルク社がすべての延長協力の選択権を行使すれば、同社は最高約$を得る資格があるだろう2.5協力過程全体での潜在的な支払い金額は2000億ドルに達する。
協力は、メルク社と同社の代表者で構成された共同研究委員会またはJRCによって管理されている場合によっては、各当事者はまた、セキュリティ中断、特許挑戦、または他方の重大な違約を規制することを含むが、いくつかの条件によって制限される必要があるが、様々な停止権を有する合意を終わらせるには司法員叙用委員会の決定に依存しなければならない。
メルク合意会計
同社は、メルクプロトコル開始時に、(1)独占ライセンスと、(2)最初に開示されなかった腫瘍学目標と共同研究計画の研究活動とを含む履行義務を決定した。当社は、ライセンスと研究活動はお互いに区別がないと認定していますが、当社が行っている研究活動がなければ、ライセンスの価値は限られています。メルク共同研究センターに参加してメルク合意に関する研究活動や技術移転を監督するために数量的にも品質的にも無関係であることが決定されたため,義務履行から除外された。当社はこの履行義務に割り当てられた取引価格を確認し,入力法で提供される研究·開発サービスを用いるため,研究開発目標ごとにこれまでに発生したコストは,基本義務履行のための発生と将来発生予定の総コストに比例する。統制権の移譲はこの時期に発生し、経営陣から見れば、履行義務の履行に進展した最良の尺度である。
2023年12月31日までの取引総価格は$10.0百万研究開発サービス実績義務と$10.0百万割り当てられた出来高の一部はまだ満たされていない。
当社がまだ受け取っていない金を売掛金とし、受け取った収入が確認されていない金を当社の総合貸借対照表で繰延収入とする。
Betta Pharma許可と協力協定
2023年5月29日、会社はBetta Pharmaとライセンスおよび協力契約、またはBetta Pharmaライセンス契約を締結して協力するCFT 8919の大中国地域での開発と商業化については、
当社は大陸部中国、香港特別行政区、マカオ特別行政区及び台湾からなり、大中国又はC 4 T地域以外の世界他地域のCFT 8919の権利を保持している。
Betta Pharmaライセンス契約の条項によると、当社は当社のいくつかの知的財産権に基づいてBetta Pharmaに独占許可を与え、中国大区人類のすべての用途のためのCFT 8919を開発、製造、商業化する。ベータ製薬は大中国地域でのすべての開発、規制承認、製造、商業化コストを担当しているが、会社が協賛するグローバル試験で大中国地域での会社の代理を担当している場合は除く。Betta Pharmaは協力の一部として1ドルの現金を前払いしました10.0100万ドルを会社に渡し最高で357.0合計マイルストーンで100万ポンドを支払うほか、中国地区の純売上高CFT 8919の分級特許権使用料を増大させる。これらの金は人民Republic of China国家税務機関が代行して支払う.Betta Pharmaが会社に支払う特許使用料は低いものから中2桁のパーセンテージまで様々であり、Betta Pharmaライセンス契約に記載されている場合には一定の減免がある可能性がある。また、提携の一部として、会社はBetta Pharmaに記念碑的なお金を支払うことに同意しており、金額は最高$に達する40.0同社がFDA承認のCFT 8919新薬申請を受けた後,この記念碑的な金額は,Betta Pharma社が登録した予想臨床試験における患者のパーセンテージと承認された治療経路に基づいている。さらに、同社は、C 4 T地域CFT 8919の純売上高について、Betta Pharmaに階層的特許使用料を低い1桁パーセントの範囲で支払うことに同意したが、Betta Pharmaライセンス契約に記載されている場合もあれば減少する可能性がある
Betta Pharmaライセンス契約の署名について、当社のBetta PharmaとBetta Pharmaの関連会社(Betta Investment(Hong Kong)Limited、またはBetta Investment)は#年の株式購入契約を締結した2023年5月29日または、Betta株式購入プロトコル、およびBetta Pharma許可プロトコルまたはBettaプロトコルは、このプロトコルに従って、Betta Investmentが購入に同意する5,567,928会社の普通株、または株式、総購入価格は約$です25.0百万ドル、あるいはドル4.49これは1株当たり25闘魚購入契約発効日前の2取引日の60取引日の出来高加重平均終値より割増%であった。“闘魚株式購入協定”は、このような協定にいくつかの慣用的な制限がある闘魚株購入協議での取引は2024年1月4日まで完了したため、2023年12月31日現在、闘魚株購入協議での取引は記録されていない。
協力は私のものですBetta Pharmaと同社の代表で構成された共同指導委員会が管理を担当している。CFT 8919第1段階試験の用量増加段階が完了した後、便宜上、Betta Pharmaは、少なくとも以下の場合、Betta Pharma許可プロトコルを終了することができる90数日前に書面でお知らせします。場合によっては、各当事者はまた、セキュリティ中断、特許挑戦、または相手の実質的な違約を規制することを含むが、いくつかの条件によって制限される必要があるが、様々な停止権を有する
ベータ合意会計
同社はBettaプロトコルに基づいて、ライセンス契約である収入のタイプを確認する予定だ。BETTAプロトコルには,(1)知的財産権許可,(2)臨床製造供給プロトコル,(3)製造技術移転,(4)商業製造供給プロトコルが含まれる。自分から2023年12月31日現在の取引総額は$です12.0何百万ドルもあります10.0百万ドルの前払い現金対価格と2.0Betta Pharmaライセンス協定によると、2023年12月に100万個のマイルストーンが実現された。Bettaプロトコルに関する収入確認は,臨床製造供給プロトコルに基づいて臨床供給が交付された時点で開始される予定であり,この合意は現在までである2023年12月31日それは.同社はすでに純額#ドルを受け取っている9.0Betta Pharmaの前払金から100万ドルを獲得し、関連する源泉徴収税$を支払った1.0中国税務機関に税金を納める(付注12参照)。先日まで何の収入も確認されていない2023年12月31日Bettaプロトコルに割り当てられた取引価格について
当社がまだ受け取っていない金を売掛金とし、受け取った収入が確認されていない金を当社の総合貸借対照表で繰延収入とする。
ロ氏の協力と許可協定
2016年3月、当社は羅氏と2016年6月に改訂され、2017年3月に再改訂されたライセンス契約を締結した。当社と羅氏は2018年12月にこの合意を改訂し、再記述した(このように改訂された)。この修正されて再記述された協定はロ氏協定と呼ばれる。羅氏協定によると、同社と羅氏社は協力し、会社独自の魚雷プラットフォームを用いて癌やその他の適応を治療し、標的拘束力のある分解薬を研究、開発、製造、商業化することに同意した。羅氏合意によると、会社はいくつかの共同開発権に加入することを選択することができ、この場合、会社は
この目標に対する製品からより高い将来の製品販売の印税を得る。また、会社がいくつかの共通詳細権を選択すれば、いくつかの商業化コストの補償を受ける権利もある。ロ氏協定を締結した後、会社は追加の前払い費用#ドルを受け取りました40.0百万ドルです
2020年11月、当社は各方面が発展することを規定する更なる改正案に署名した5人潜在的な目標として、羅氏はこれらの目標に対する製品許可と商業化の選択権を保持している。2020年11月の改正では、当社と羅氏が共通目標終了協定を締結することで、目標ごとに羅氏合意の終了に相互に同意することができる仕組みも提供されている。共通目標終了協定が締結されると、ロー氏協定は、その行動パターンとして抑制された製品のノウハウおよび他の知的財産権をサポートするすべての権利および責任をロ氏の所有とし、その行動モードに格下げされた製品のノウハウおよび他の知的財産権をサポートするすべての権利および責任をロー氏が所有することを規定する。この権利割り当てを支援するために、ロー氏は、相互の目標に基づいて当事者に割り当てられた特許およびノウハウを終了するために、永久的に撤回不可能で、全額支払いが十分である、独占的(許可者であっても)、再許可(複数のレベルを含む)可能な特許およびノウハウを当社に提供する。羅氏との研究活動は時間の経過とともに進歩と発展し,現在では二つ各方面が引き続き協力する目標は、羅氏はその許可と商業化を維持し、これらの目標に対する製品の選択権を維持する二つ目標です。2023年12月、当社はロ氏協定の第二次改正案に署名し、その効果は協定の条項を更新することである二つ各方面が引き続き協力する目標。“ロ氏合意”の第2項改正案によると、羅氏はこれらの目標に対する製品の許可と商業化の選択権を保持しているが、その選択権の時間は羅氏が線量範囲でパケットを発見した後に開始するように調整されている。羅氏協定の第2次改正は2023年の会計に実質的な影響を与えなかった
改訂された羅氏合意によると、会社は年間研究計画の支払いを受けた#ドル1.0すべての肯定的な研究計画は100万ドルを持っている。上には二つ各方面がまだ協力している目標に対して、羅氏は会社に#ドルの費用を支払う必要がある2.0目標がリーダーシリーズ決定達成段階に進んだ時、100万ドルの資金があるだろう。羅氏がこれらの目標の一つに対してオプションを行使すれば、羅氏は会社に#ドルのオプション行権料を支払わなければならない8.0百万ドルです
改正された羅氏合意によると、羅氏が行使した目標オプションごとに、会社は#ドルまでのマイルストーン支払いを受ける資格がある273.0相応の製品に関連するいくつかの開発マイルストーンが実現された後、知的財産権のカバー範囲に基づいていくつかの減少と排除を行うことができる。羅氏はまた、#ドルまでの費用を会社に支払うことを要求された150.0一次販売に基づくマイルストーン支払いでは、目標ごとにその目標に対する製品が指定された純売上高レベルに達したときに百万ドルを支払う。最後に、羅氏は羅氏が行使した選択権によって販売された製品の純売上高の1桁の中桁から10代中期までの階層特許権使用料を会社に支払う必要があるが、一定の減幅が必要である。会社が共同開発権を行使する製品の販売については、適用される特許権使用料税率は低い1桁の割合で増加する。
その協力は共同研究委員会(JRC)によって管理されている。ロ氏が特定の目標に対してそのオプションを行使する前に、会社はJRCに対して支配権を持ち、その後羅氏はJRCを接収し、羅氏はいくつかの場合、一つの目標または製品ごとに羅氏協定を終了することができる90数日前に書面でお知らせします
羅氏合意会計
開始時に同社は確定した12個“ロ氏合意”における履行義務は,6人潜在的な研究開発目標は当時協力に含まれていました羅氏が各目標に対して持っていた選択権です6人目標です。同社の知的財産権を用いた研究開発活動やJRC参加の非独占免印税許可が承諾されたサービスとして決定された。しかし、当社は、研究開発許可証と研究開発サービスは互いに区別されておらず、JRCへの参加は数量的にも品質的にも関係ないと考えている。
ロ氏合意の取引総価格は履行義務の相対独立販売価格に応じて履行義務に割り当てられる。割り当てられた取引価格は,以下の2つの方式の1つで連携プロトコルからの収入であることが確認された
•研究·開発目標:当社は個々の研究·開発実績義務に割り当てられた取引価格部分を確認し,1つの投入方法で研究·開発サービスを提供する場合でも,研究開発目標ごとにこれまでに発生したコストと基本義務履行のために発生·将来発生予定の総コストとの割合
研究開発目標に関連しています統制権の移譲はこの時期に発生し、経営陣から見れば、履行義務の履行に進展した最良の尺度である。
•オプション:オプションに割り当てられた取引価格は実質的な権利と考えられ、ロー氏がオプションを行使または行使しないことが潜在的な研究および開発目標を許可し、商業化されている間に確認される。
この手配で決定された履行義務に対する総取引価格の分配状況と、2023年12月31日現在満たされていない取引価格金額(千単位)をまとめた
| | | | | | | | | | | |
| 取引記録 値段 分配された | | 取引記録 値段 満足していない |
| 契約履行義務: | | | |
| 研究開発目標 | $ | 61,149 | | | $ | 17,394 | |
| オプション権 | 6,757 | | | 1,670 | |
| 合計する | $ | 67,906 | | | $ | 19,064 | |
当社がまだ受け取っていない金を売掛金とし、受け取った収入が確認されていない金を当社の総合貸借対照表で繰延収入とする。
生物遺伝研究協力と許可協定
2018年12月、同社は生物遺伝会社と共同研究·許可協定、または生物遺伝協定を締結した。2020年2月、当社とBiogenは、Biogenの目標結合部分(分子の一部)に対する所有権、および協力目標または協力目標にバインドされた任意の関連知的財産権をさらに明確にするためにBiogenプロトコルを改訂した。この改正はさらに,Biogenは当社がこのようなBiogen目標拘束性部分および任意の関連知的財産権(必要に応じて)を使用して,Biogenプロトコル項で期待される研究および開発活動を行うことを許可している。生物遺伝子プロトコルの条項によると,同社と生物遺伝子会社は協力して研究活動を行うことに同意し,標的タンパク質分解(TPD)をその作用モデルとする薬物に依存することにより,アルツハイマー病やパーキンソン病などの神経疾患を治療する新しい療法を開発し,これらの薬剤はすべて会社の分解器技術を用いて作成した。生物遺伝協定の条項に基づき,同社は分解剤技術を利用したTPD療法の開発に取り組んでいる5人標的タンパク質は一定期間内に54数ヶ月、2023年6月に終わります。個々の目標に基づいて,所定の目標評価期間を成功させた後,生物遺伝会社は個々の目標を発展させ続けるすべての権利と責任を負う
Biogenの非独占的研究許可証と$を交換するために45.0会社はすでに許可を得て、各目標に関連する製品を開発、商業化し、生産することができ(これは合意を取り消さないことに依存する)、薬物発見に初歩的な研究サービスを提供し、その知的財産権に非独占研究と商業許可を提供し、共同指導委員会或いは生物遺伝研究連合委員会に参加する。同社には他の潜在目標の早期研究活動や砂箱活動に参加する義務があり、生物遺伝研究会社が選択し、最高金額は超えてはならない。これらのサービスのためのいかなる仕事も生物遺伝研究会社が精算し、生物遺伝研究会社は会社にあるFTE費用を精算する。2021年8月31日現在、砂箱活動下での会社の義務が完了した。任意の目標について、開発候補基準を達成した後、INDの研究をサポートする任意の前に、生物遺伝会社は、すべての費用および支出を負担し、生物遺伝研究プロトコルの下で開発されている任意の分解物およびその分解物を含むすべての製品に関連するさらなる活動の唯一の裁量および決定権を有するであろう。生物遺伝会社は高い$を会社に支払うことも要求されています35.0発展のマイルストーンの目標ごとの百万ドルとドルは26.0目標ごとに一度の売上に基づく支払いを第一製品とし、一定レベルの純売上を実現する。また,生物遺伝会社はグローバル製品の純売上高に応じて,製品ごとにライセンス使用料を会社に支払う必要がある。すべての記念碑的支払いおよび販売ベースの支払いは、企業がその目標を達成するための共同研究計画に規定された基準を達成した後に支払われ、生物遺伝会社は商業化目標に関連する製品を制御し、これらの支払いの受信は、会社が追加的な研究および開発努力を行うことなく、生物遺伝会社の商業化目標に対する製品のさらなる開発に依存する。
協力はBiogen JSCによって管理され、BiogenはJSCに対して制御権を有し、Biogenはいくつかの場合、ターゲット毎または製品毎にBiogenプロトコルを終了することができ、少なくともここでは90数日前に書面でお知らせします。
生物遺伝協定会計
生物遺伝研究と開発サービスプロトコルによると、同社が確認した収入は以下の通り
•研究開発サービス:同社は1つは“生物遺伝的プロトコル”開始時の履行義務は,(1)ライセンス,(2)すべての人の目標評価段階に向けた研究活動を含む総合履行義務である5人目標と(3)目標ごとの共同研究計画段階.当社は、ライセンスと研究活動はお互いに区別がないと認定していますが、当社が行っている研究活動がなければ、ライセンスの価値は限られています。生物遺伝研究センターの監督研究活動への参加や生物遺伝ライセンスプロトコルに関する技術移転は数量的にも品質的にも重要ではないことが決定され,義務履行から除外された。当社は,この履行義務に割り当てられた取引価格を確認し,入力法を用いて提供する研究·開発サービスは,研究開発目標ごとにこれまでに発生したコストと,前記研究·開発目標に関する基本的な義務を履行するために発生·将来発生予定の総コストに比例する。統制権の移譲はこの時期に発生し、経営陣から見れば、履行義務の履行に進展した最良の尺度である。
2023年12月31日までの取引総価格はドルです55.0研究開発サービスの業績義務と#ドルのために百万ドル0.8分配された出来高のうち100万はまだ満たされていない。
当社がまだ受け取っていない金を売掛金とし、受け取った収入が確認されていない金を当社総合貸借対照表の繰延収入に記入する。
Calico協力と許可協定
2017年3月、当社はCalicoと協力および許可協定、またはCalico協定を締結し、この合意に基づき、当社はCalicoと協力して小分子タンパク質分解剤の開発と商業化に同意し、癌を含む老年性疾患の治療に利用した5年制締め切りは2022年3月。2021年8月、同社はCalicoにあるプロジェクトの研究期間を最大1年延長する選択権を提供した1年制締め切りは2023年3月です。2021年9月にCalicoは選択権を行使して1ドルを生成しました1.0会社に百万の支払いを延期します。また、Calicoは研究段階によって、特定の市場料率でFTEを会社に返済し続ける。2023年3月13日現在,印紙協議の研究期限が満了しており,同社の同協定に関する研究活動はほぼ完了している。
Calico協定によると、Calicoは#ドルを前払いした5.0百万元と数年間の支払いは合計$である5.02020年6月30日までに、研究段階によって、指定された市場料率で目標起動費用を支払い、いくつかのFTEを会社に精算する。すべての目標に対して、会社は最高$を得る資格があります132.0潜在開発とビジネスマイルストーン支払いは百万ドルで、協力努力によるすべての製品の販売から来ています。Calicoは会社に最高#ドルの支払いを要求されました65.0一度に百万元の販売をベースにした最初の製品は一定レベルの純売上を実現します。また、Calicoはライセンス製品ごとに、グローバル製品の純売上高を1桁の中央値の割合で会社に印税を支払う必要がある。すべての記念碑的な支払いと販売ベースの支払いは、会社がその目標の共同研究計画に規定された基準を達成した後に行われ、Calicoは商業化目標に関連する製品を制御し、会社がこれらの支払いを受け取るかどうかは、会社が追加的な研究と開発努力を必要とすることなく、Calicoの商業化製品目標のさらなる発展に依存する。
2023年12月31日までの取引総価格はドルです13.0研究開発サービス履行義務には100万ドルが割り当てられ,取引価格はすべて分配され満足されている。
当社がまだ受け取っていない金額に対応するために当社の簡明総合貸借対照表に売掛金と記入します。
連携プロトコルから確認した収入まとめ
連携プロトコルからの収入は以下のとおりである(千単位)
| | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | |
| 協力協定からの収入: | | | | | |
| 羅氏協定 | $ | 9,063 | | | $ | 4,939 | | | |
| 生物遺伝協定 | 10,623 | | | 19,214 | | | |
| 印刷布協定 | 1,070 | | | 6,943 | | | |
| “ベータ協定” | — | | | — | | | |
| 協力協定からの総収入 | $ | 20,756 | | | $ | 31,096 | | | |
連携·許可協定に関する補足情報には、2023年12月31日現在の総合貸借対照表の以下が含まれる(千計)
| | | | | | | | | | | | | | | | | | | | | | | |
| 勘定.勘定 売掛金 | | 延期する 収入は 現在のところ | | 延期する 収入は 海流純額 | | 延期する 収入は 合計する |
| 補足情報: | | | | | | | |
| 羅氏協定 | $ | — | | | $ | 2,667 | | | $ | 11,814 | | | $ | 14,481 | |
| “ベータ協定” | 1,799 | | | 4,000 | | | 8,000 | | | 12,000 | |
| メルク協定 | 10,000 | | | 8,000 | | | 2,000 | | | 10,000 | |
| 生物遺伝協定 | — | | | 804 | | | — | | | 804 | |
| | | | | | | |
| 合計する | $ | 11,799 | | | $ | 15,471 | | | $ | 21,814 | | | $ | 37,285 | |
連携·許可協定に関する補足情報には、2022年12月31日現在の総合貸借対照表の以下が含まれる(千計)
| | | | | | | | | | | | | | | | | | | | | | | |
| 勘定.勘定 売掛金 | | 延期する 収入は 現在のところ | | 延期する 収入は 海流純額 | | 延期する 収入は 合計する |
| 補足情報: | | | | | | | |
| 羅氏協定 | $ | 417 | | | $ | 4,649 | | | $ | 16,895 | | | $ | 21,544 | |
| 生物遺伝協定 | — | | | 11,427 | | | — | | | 11,427 | |
| 印刷布協定 | 1,056 | | | 542 | | | — | | | 542 | |
| 合計する | $ | 1,473 | | | $ | 16,618 | | | $ | 16,895 | | | $ | 33,513 | |
連携およびライセンス契約に関する補足財務情報には、(千計):
| | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | |
| 期初に契約負債を計上した確認済収入 | $ | 20,185 | | | $ | 25,412 | | | |
| 以前各期の全部または部分的に履行義務を履行して確認された収入 | 43 | | | 518 | | | |
2023年12月31日現在,ロ氏プロトコル,ベータプロトコル,メルクプロトコル,生物遺伝プロトコルにより履行義務に割り当てられた部分が満たされていない取引価格総額はドルである41.9百万ドルです。
注9長期債務と権利証の関係者
長期債務に関する側は以下の部分からなる(千計)
| | | | | | |
| | 十二月三十一日
2022 |
定期融資関係者は、未償却債務発行コストと債務割引#ドルを差し引いた純額1,0182022年12月31日 | | $ | 11,482 | |
| マイナス:現在の部分 | | 2,287 | |
| 長期債務関連先の総額、当期と割引を差し引く | | 9,195 | |
2020年6月5日、Bラウンド融資を完了すると同時に、当社はPerceptive Advisors LLCまたはPerceptiveの関連会社Perceptive Credit Holdings III、LPと信用協定を締結し、元金借款総額が最高$に達することを規定した20.0100万ドルを2回に分けて提供します12.5百万ドルとドル7.5百万ドルです。クレジットプロトコル開始時の当社普通株に対する所有権を感知することに基づいて、感知は当社の関連先とみなされる
2020年6月、当社は最初の#ドルを抽出しました12.5100万ドルや定期的なローンです同社は2021年6月30日に満期となった第2弾の資金を抽出しないことを決定した
2023年5月17日、当社は信用協定修正案を締結し、これにより、当社とその融資者はLIBOR基準の代わりにSOFRを採用することに同意し、SOFRはニューヨーク連邦準備銀行の監督を受けた。金融市場行為監督局は2023年6月30日にLIBORを段階的に淘汰する計画であるため、同社はその信用協定を修正した。SOFR為替レートの使用は2023年7月1日から施行される
2023年7月、信用協定の条項により、当社の前払い費用の支払い義務が取り消されました。2023年7月26日、会社は定期ローンを返済した残りの未返済元金残高を選択した。定期融資の未返済残高については、会社のほとんどの資産に対する留置権を解除した。会社に$が発生しました0.6この債務の返済損失は2023年12月31日までの年度で100万ポンドである。
2023年12月31日と2022年12月31日までの年度の利息支出は$1.0百万ドルとドル1.5百万ドルです。
注10株主権益
普通株
2020年10月、当社が発行を許可した優先株は10,000,000発行可能な法定普通株を150,000,000株、両方とも$を持っている0.0001一株当たりの額面。
普通株保有者には権利がある1つは全株主会議において株式毎に投票し、規定された会議の代わりに書面で行動する。優先株保有者が獲得権のあるすべての優先配当金を全部払った場合、普通株式保有者は合法的に利用可能な資金から配当を得る権利がある。もし会社に清算、解散または清算が発生した場合、支払会社のすべての債務と負債および優先株保有者が資産分配を清算して獲得する権利があるすべての優先金額を支払いまたは準備した後、普通株式保有者は会社の残りの分配可能な資産を比例的に共有する権利がある。普通株保有者には優先引受権や他の引受権がなく、普通株の償還や債務返済基金に関する規定もない。
市場の株式計画
2021年11月、当社は、自動発効したS-3表登録説明書を米国証券取引委員会に提出し、発行、発行及び販売額が不明な普通株、優先株、債務証券、権証及び/又はそれらの任意の組み合わせの単位を登録する。また,会社は販売代理であるCowen and Company,LLCと最高$の発行と売却を規定する持分配分協定を締結した200.0登録声明と登録声明やATM計画とともに提出された関連目論見書によると、時々“市場で”発行される普通株は数百万株に達する。2023年12月31日までの年間11,186,142純収益$の株式57.7100万ドルです。現金自動支払機で決済する計画です。2024年1月2,500,601株式決済の純収益は#ドルです14.11000万ドルです。上記2023年12月と2024年1月の活動に基づき、当社は共に販売しました13,686,743普通株の平均価格は$です5.421株当たりの収益は、手数料と手数料を差し引いて$となります71.81000万ドルです違います。販売はATM計画に基づいて2022年12月31日までの年度内に完了した。
注11株に基づく報酬
2015年奨励株式オプションと奨励計画
2015年12月28日、会社取締役会は2015年度奨励株式オプションと付与計画、または2015年度計画を採択し、保留した2,525,327本計画に基づいて発行された普通株
2015年計画認可取締役会または取締役会委員会は、条件を満たす会社員、外部取締役、およびコンサルタントに奨励株式オプション、非制限株式オプション、および制限株式奨励を付与する予定です。2015年計画によると付与されたオプションは5人あるいは…8年崖があります1年その後の四半期帰属および失効または没収されたオプションは再び付与されることができる。2015年計画に基づいて付与されたすべての代替案の契約期限は10年授与の日から効力を発揮する
2020年株式オプションとインセンティブ計画
2020年9月8日、取締役会はC 4治療会社の2020年株式オプションとインセンティブ計画、または2020年計画を採択し、2020年9月30日に発効した。養子縁組された後6,567,1442020年計画に基づいて保留された普通株を発行する。当社の取締役会、取締役会報酬委員会、および場合によっては、当社の最高経営責任者は、2020年計画に基づいて、株式オプション、株式付加価値権またはSARS、制限株式報酬またはRSA、制限株式単位またはRSU、業績奨励および株式配当金を会社の上級管理者、従業員、取締役、コンサルタントを含む他のキーパーソンに奨励する権利がある
2020年計画が発効した後、当社は2015年計画下の贈与を停止します。しかし、2015年には、その計画に基づいて付与された係属中の裁決の条項や条件を管理し続ける予定だ。2015年計画に基づいて奨励された普通株株は、2015計画終了後に没収やその他の方法でこのような奨励を受けなければ、2020年計画に基づいて発行することができる。2023年12月31日までに3,358,3362020年計画によると、将来付与される株式を提供することができる。
2020年計画では毎年増加することが規定されており,2021年1月1日から財政年度ごとの初日に増加し,2020年まで計画が満了するまで続いている(I)5(二)2020年度計画管理人(会社取締役会又は取締役会報酬委員会)が決定したより少ない数の普通株式。2024年1月1日、2020年計画の年間成長は追加的な3,023,359発行された株式は2020年計画に追加された。
株式ベースの報酬支出は以下の通り(単位:千):
| | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | |
| 株式ベースの報酬費用: | | | | | |
| 研究開発 | $ | 11,826 | | | $ | 13,108 | | | |
| 一般と行政 | 15,409 | | | 16,908 | | | |
| 株式に基づく報酬総支出 | $ | 27,235 | | | $ | 30,016 | | | |
次の表は、2023年12月31日までの年間会社持分奨励計画における株式オプション活動をまとめたものである
| | | | | | | | | | | | | | | | | | | | | | | |
| 量
オプション | | 加重平均
行権価格 | | 加重平均
残り
契約書
期限(年) | | 骨材
固有の
価値がある
(単位:千) |
| 2022年12月31日現在の未返済債務 | 6,993,052 | | | $ | 21.57 | | | 7.78 | | $ | 1,031 | |
| 授与する | 4,180,270 | | | 3.87 | | | | | |
| 鍛えられた | (13,096) | | | 4.46 | | | | | |
| 没収/期限切れ | (1,685,217) | | | 18.48 | | | | | |
| 2023年12月31日現在の未返済債務 | 9,475,009 | | | $ | 14.34 | | | 7.61 | | $ | 8,261 | |
| 2023年12月31日から行使可能 | 3,838,707 | | | $ | 20.54 | | | 6.68 | | $ | 482 | |
| 帰属しており、2023年12月31日に帰属する予定です | 9,475,009 | | | $ | 14.34 | | | 7.61 | | $ | 8,261 | |
同社のオプション活動に関するその他の情報は以下のとおりである
| | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | |
| 付与されたオプションの加重平均公平価値 | $ | 2.84 | | | $ | 13.44 | | | |
| オプションの内在的価値(千単位) | $ | 22 | | | $ | 1,796 | | | |
2023年12月31日現在,未返済オプションに関する未確認補償コストは1ドルである51.6100万ドルで加重平均期間中に確認される予定です2.1何年もです
ブラック·スコアーズオプション定価モデルにおける従業員に付与された株式オプション公正価値を決定するための仮定を以下の表にまとめた
| | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | |
| 期待オプション寿命(年) | 5.50 - 6.11 | | 5.50 - 6.11 | | |
| 無リスク金利 | 3.45% - 4.73% | | 1.47% - 4.22% | | |
| 予想変動率 | 81.99% - 86.15% | | 81.36% - 88.75% | | |
| 期待配当収益率 | 0.00% | | 0.00% | | |
業績に基づく限定株式単位
2022年1月と2月に会社取締役会が発行を許可しました563,500業績に基づく限定的な株式単位、またはPSUは、2020年計画下の会社指導チームメンバーを含む特定の従業員に付与される。PSUの帰属はある発見と臨床マイルストーンの成果の決定、あるいは特定の市場条件の満足に依存する。付与された場合、各PSUは自動的に1つは会社の普通株のシェア。
次の表は、2023年12月31日までの年度、会社の株式計画におけるPSUに関する活動をまとめています
| | | | | | | | | | | |
| | 株 | | 加重平均
授与日
公正価値 |
| 2022年12月31日現在の未返済債務 | 465,291 | | | $ | 25.09 | |
| 授与する | — | | | — | |
既得(1) | (135,301) | | | 26.52 | |
| 没収される | (175,990) | | | 28.91 | |
| 2023年12月31日現在の未返済債務 | 154,000 | | | $ | 19.47 | |
(1) 付与されたPSUは17,556会社が保有している株式は、法定最低源泉徴収税を支払うために使用される。
2023年12月31日現在,未返済PSUに関する未確認賠償コストは#ドルである3.1100万ドル加重平均期間中に確認される予定です1.2何年もです。
時間に基づく制限株式単位
2023年1月と2月に会社取締役会が発行を許可824,6002020年計画に基づき、従業員に時間に基づく制限株式単位(RSU)を提供する。RSUの帰属は、時間に基づく帰属条件に依存する。この等RSUは授出日に日関連株式の時価値を授与する。付与された場合、各RSUは自動的に1つは会社の普通株のシェア。
以下の表は、会社の株式計画における2023年12月31日までの年間RSUに関する活動をまとめたものである
| | | | | | | | | | | |
| | 株 | | 加重平均
授与日
公正価値 |
| 2022年12月31日現在の未返済債務 | — | | | $ | — | |
| 授与する | 824,600 | | | 5.37 | |
| 既得 | — | | | — | |
| 没収される | (74,390) | | | 5.67 | |
| 2023年12月31日現在の未返済債務 | 750,210 | | | $ | 5.35 | |
2023年12月31日現在,RSU未返済に関する未確認賠償コストは#ドルである3.5100万ドル加重平均期間中に確認される予定です3.2何年もです。
奨励補助金
2023年7月の2つの異なる日に、会社取締役会は、購入のための非制限株式オプションの付与を許可した351,000会社の普通株を新入社員2人に分ける。これらの奨励によって付与されたオプションは以下の期限内に付与される4年崖があります1年その後四半期ごとに帰属を行う.これらの株式オプションは、2023年12月31日現在、帰属または没収されていない351,000このような奨励賞の下で、オプションはまだ明確にされていない。
2020年従業員株購入計画
2020年9月、会社取締役会はC 4治療会社の2020年従業員株購入計画、あるいは2020年ESPPを採択した。資格に適合した従業員は給料を差し引くことができて、最高で15要件期間内にその合格報酬の%を取得します。会社が持つことができる1つは毎年またはそれ以上の募集期間では、従業員は2020年のESPPに基づいて株を購入することができるだろう。その会社は発行した133,7842023年12月31日までの年度内の株式
2023年12月31日までに会社は1,673,1292020年ESPPによると、将来発行可能な株。2020年ESPPでは,2021年1月1日から2030年1月1日まで,各財政年度の初日に年度昇給が増加し,(I)に相当することが規定されている1前年12月31日普通株式流通株の割合、(2)656,714株式、または(Ii)2020年ESPP管理人によって決定されるより少ない株式数。2024年1月1日、2020年ESPPの年間成長は追加的な604,671発行を許可された株は2020年にESPPに追加されるだろう。
注12所得税
所得税(福祉)費用は以下の項目からなる(千計)
| | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | |
| 現行の税収規定: | | | | | |
| 現行連邦条項 | $ | — | | | $ | — | | | |
| 現在の国の規定 | 76 | | | — | | | |
| 現行の外国条項 | 1,204 | | | — | | | |
| 総当期に準備する | 1,280 | | | — | | | |
| 繰延税の準備: | | | | | |
| 連邦準備を延期する | — | | | — | | | |
| 繰延状態準備金 | — | | | — | | | |
| 繰延外貨準備金 | — | | | — | | | |
| 総税額を支出する | $ | 1,280 | | | $ | — | | | |
税率入金
総合財務諸表に反映される法定連邦税率で計算される予想所得税(福祉)費用と所得税の入金は以下のとおりである
| | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | |
| 連邦法定税率で計算される所得税割引 | 21.0 | % | | 21.0 | % | | |
| 州税-連邦税網 | 4.2 | % | | 8.0 | % | | |
| 連邦信用 | 3.5 | % | | 1.4 | % | | |
| 国家信用 | 1.4 | % | | 0.1 | % | | |
| 他の恒久的差異 | (0.2) | % | | (0.8) | % | | |
| 税金を前納する | (0.9) | % | | — | % | | |
| 株に基づく報酬 | (2.8) | % | | (3.0) | % | | |
| | | | | |
| | | | | |
| 推定免税額 | (27.2) | % | | (26.7) | % | | |
| 合計する | (1.0) | % | | — | % | | |
当社の2023年12月31日までの有効税率は、Betta許可協定に関する前払金やマイルストーン支払いに関係する中国の源泉徴収義務の影響を受けています(付記8参照)。これは#ドルの税金支出を生む1.2百万ドル、有効税率は(1.02023年12月31日までの割合
繰延税金の重要な構成要素
繰延所得税は、(A)財務報告のための資産および負債の帳簿金額と所得税用金額との間の一時的な差、および(B)営業損失および税収の純税収影響を反映する
信用繰越。会社の繰延税金資産と繰延税金負債の重要な構成要素は以下の通り(千計)
| | | | | | | | | | | |
| 12月31日まで |
| 2023 | | 2022 |
| 繰延税金資産: | | | |
| 純営業損失 | $ | 61,036 | | | $ | 54,300 | |
| 資本化研究と実験支出 | 51,401 | | | 27,568 | |
| リース負債を経営する | 20,611 | | | 21,443 | |
| 研究開発と投資税控除 | 18,049 | | | 11,519 | |
| 株に基づく報酬 | 9,061 | | | 6,296 | |
| 収入を繰り越す | 3,881 | | | 8,457 | |
| 外国の税収控除 | 1,204 | | | — | |
| 資本化起動コスト | 555 | | | 699 | |
| 未実現損益 | — | | | 1,090 | |
| 他にも | 27 | | | 354 | |
| 繰延税項目の総資産総額 | 165,825 | | | 131,726 | |
| 繰延税金負債: | | | |
| 使用権資産 | (18,573) | | | (19,868) | |
| 固定資産 | (1,663) | | | (1,738) | |
| | | |
| 減算:推定免税額 | (145,589) | | | (110,120) | |
| 繰延税金純額 | $ | — | | | $ | — | |
当社は繰延税金資産の現金化に影響するプラスとマイナスの証拠を評価しました。2023年12月31日現在、2023年12月31日現在、2022年12月31日現在、当社の歴史的営業損失により、当社はその繰延税金資産の利益が実現できない可能性が高いと結論した。そのため、当社は2023年12月31日、2023年12月31日および2022年12月31日までの繰延税金資産に全額評価を提供する準備をしています。2023年12月31日と2022年12月31日までの繰延税金資産推定免税額は#ドル145.6百万ドルとドル110.1それぞれ100万ドルです推定手当純額は#ドル増加した35.5百万ドルとドル34.72023年12月31日と2022年12月31日までの年間でそれぞれ100万ドル。2023年12月31日までの年度変動は、主に純営業損失及び税額控除の繰越増加、研究及び実験支出の資本化、及び年内に確認された繰延収入の減少によるものである
同社は2023年12月31日と2022年12月31日まで214.9百万ドルとドル171.6米国連邦純営業損失総額、あるいはNOLは、それぞれ繰り越し、将来の課税収入を相殺するために用いることができる。2017年12月に公布された減税·雇用法案(TCJA)は、通常、2017年以降に発生する連邦損失の無期限繰越を許可しているが、NOL繰越額は小さい者や会社の課税所得額の80%に制限されるのが一般的である(1986年に改正された米国国税法(IRC)第382条の制約)。また、2017年以降に発生した損失は繰り越すことはないだろう。2018年までに発生した損失は、一般的に会社のNOL繰越や会社の課税所得額の中で小さい範囲で控除され、赤字が発生した日から20年以内に控除されます。米国連邦所得税の目的で、同社の2017年以降に発生した連邦NOLは$214.9100万ドルで、この債券は期限が切れないだろう。当社には2019年までに発生した利用可能なNOLは何もありません。これらのNOLは2019年にすべて使用されているからです。コロナウイルス援助、救済、経済安全法案、またはCARE法案は、2018、2019年、2020年に発生した純運営損失を上位5納税年度に繰り越すことを一時的に許可している。また、ここ数年で発生した純営業損失は、必要なく、前年の課税収入を完全に相殺することができます80TCJAが規定する課税所得額の%です
同社の米国州純営業損失総額は、2023年12月31日、2023年12月31日、2022年12月31日までの$となっている315.3百万ドルとドル273.6それぞれ100万ドルで、2043年に異なる日に満期になった将来の課税収入を相殺するために使用できる
2023年12月31日、2023年12月31日、2022年12月31日に、会社は米国連邦研究信用繰越$を持っている12.6百万ドルとドル9.1それぞれ100万ドルで、将来の連邦所得税債務を相殺することができ、これらの債務は2043年までの異なる日に満期になる。2023年12月31日、2023年12月31日、2022年12月31日に、会社はアメリカ州研究信用繰越$を持っています5.2
百万ドルとドル2.9それぞれ100万ドルで、2038年までに異なる日に満期になる将来の税収負担を減らすことができる
純営業損失と税収控除繰越は年次制限を受ける可能性があり,重要株主の所有権権益が3年間の何らかの累積変化が50%を超える場合は,それぞれIRC第382条と383条および同様の国の規定により定義される。これは、将来の課税収入または納税義務を相殺するために毎年使用できる税金属性の数を制限することができるかもしれない。年間限度額の金額は、所有権変更直前の会社価値に基づいて決定される。その後の所有権変更は今後数年間の制限にさらに影響を及ぼすかもしれない。2022年、会社は設立から2020年12月31日までの所有権変更研究を完了し、設立以来所有権変更が発生したかどうか、あるいは複数回の所有権変更が発生したかどうかを評価する。この研究の結果,会社は国税法第382条で定義された所有権変更を経験したが,あることが分かった違います。純営業損失の繰越は限定的であり、このような所有権変更により満期になって未使用となる。赤字が続いている状況を受けて、当社は現在、IRC第382条に基づいて2020年12月31日以降のいかなる活動も分析していない。すでに当社の営業純損失及び税額控除の繰延に関する繰延税項資産について全数推定値を準備しており、もし調整が必要であれば、この調整は推定支出の調整によって相殺される。
当社は所得税支出において未記録の税収割引に関する利息及び/又は罰金を確認する。2023年、2023年、2022年12月31日まで、会社は違います。記録されていない税金優遇に関連するものは利息または罰金を計算しなければならない
同社はアメリカ、カリフォルニア州、フロリダ州、マサチューセッツ州で所得税申告書を提出した。連邦と州所得税申告書は通常、2020年12月31日までの納税年度を税務審査しなければならない。当社が税務属性の繰越を持っている場合には、当該等の属性を発生させた納税年度は、国税局又は国家税務機関の審査を経て将来の期間中に使用できる程度に調整することができる。その会社は現在どんな税務機関の審査も受けていない。
注131株当たり純損失
注2で述べたように、重要会計政策の概要しかし、会社が純損失を報告している期間については、その影響が逆薄になるため、潜在的な希薄化証券は1株当たりの純損失の計算から除外されている。したがって,基本1株当たり純損失と希釈後の1株当たり純損失を計算するための発行済み普通株加重平均は同じである期間の希釈後の1株当たり純損失を計算する場合、同社は期末既発行金額に基づいて以下の潜在普通株を計上しない。これらの株式を計上すると逆償却効果が生じるからである
| | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | |
| 普通株購入オプション | 9,475,009 | | | 6,993,052 | | | |
| | | | | |
1株当たり基本損失と希釈損失の計算方法は、純損失を発行された加重平均普通株(単位は千株、株と1株当たりのデータを含まない)で割ることである
| | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | |
| 分子: | | | | | |
| | | | | |
| | | | | |
| 純損失−基本損失と希釈損失 | $ | (132,493) | | | $ | (128,175) | | | |
| 分母: | | | | | |
| 加重平均発行済み普通株式-基本と希釈後の普通株式 | 49,640,505 | | | 48,861,665 | | | |
| 1株当たり純損失--基本損失と赤字 | $ | (2.67) | | | $ | (2.62) | | | |
注14固定払込計画
会社には401(K)退職計画、すなわち401(K)計画があり、この計画によると、すべての従業員は最大で支払うことができる90彼らの合格補償の%は、最高でアメリカ国税局が規定した最高許容金額に達する。会社がそれにマッチする100401(K)計画に対する払込率3%と、もう一方と50限度額を超えない出資比率5%、合計
潜在的マッチング4従業員一人当たりの割合です会社は2023年12月31日,2023年12月31日,2022年12月31日までの各年度に約$を貢献した1.1百万ドルとドル1.0それぞれ401(K)計画に100万ドルを提供した.
注15引受金とその他の事項
法律手続き
当社は現在いかなる重大な法的手続きにも関与していません。報告日ごとに、当社は、潜在損失金額又は潜在損失範囲が権威的な案内処理又は事項の規定によって可能かつ合理的に評価されているか否かを評価する。当社はこのような法律訴訟に関連する費用が発生した場合に支出します。
注16後続事件
2024年1月9日、同社は運営コストを低減し、従業員チームをその業務ニーズにより良く適応させる計画を実施した。コスト削減計画により,同社は約をリストラした30%です。同社は約#ドルの使い捨て再構成費用を発生させると推定しています2.7従業員解散費、福祉、関連解雇費用を含む百万ドルで、会社は2024年第1四半期にその大部分の費用を支払う予定だ。