アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
本財政年度末まで
あるいは…。
1934年証券取引法第13条又は15(D)条に基づいて提出された |
手数料書類番号
(登録者の正確な氏名はその定款に記載)
(明またはその他の司法管轄権 会社や組織) |
(税務署の雇用主 識別番号) |
(主にオフィスアドレスを実行) |
(郵便番号) |
登録者の電話番号、市外局番を含む:(
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
|
取引 記号 |
|
登録された各取引所の名称 |
|
|
|||
|
|
|
|
|
同法第12条(G)に基づいて登録された証券:ありません
登録者が証券法規則405で定義されている経験豊富な発行者である場合は、再選択マークで示してください。はい。☐
登録者が当該法第13条又は第15条(D)に従って報告書を提出する必要がないか否かを、再選択マークで示す。はい。☐
登録者が(1)過去12ヶ月以内(または登録者がそのような報告の提出を要求されたより短い期間)に、1934年の証券取引法第13または15(D)節に提出されたすべての報告を提出したかどうか、および(2)このような提出要求を過去90日以内に遵守してきたかどうかを、再選択マークで示す
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
大型加速ファイルサーバ |
|
☐ |
|
ファイルマネージャを加速する |
|
☐ |
|
|
|
|
|||
|
☒ |
|
規模の小さい報告会社 |
|
||
|
|
|
|
|
|
|
新興成長型会社 |
|
|
|
|
|
|
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引法第13(A)節に提供された任意の新しいまたは改正された財務会計基準を遵守する.
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に第240.10 D−1(B)条に従って受信されたインセンティブベースの補償に従って回復分析を行う必要があるかどうかを再選択マークで示す☐
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)。はい違います
2023年6月30日現在、ナスダック資本市場における普通株の終値は1株20.76ドルで計算され、登録者非関連会社が保有する普通株総時価は約$
登録者が発行する普通株式数はoである2024年2月29日は
カタログ表
|
|
ページ |
第1部 |
|
|
第1項。 |
業務.業務 |
1 |
第1 A項。 |
リスク要因 |
42 |
項目1 B。 |
未解決従業員意見 |
93 |
プロジェクト1 C。 |
ネットワーク·セキュリティ |
93 |
第二項です。 |
属性 |
94 |
第三項です。 |
法律訴訟 |
94 |
第四項です。 |
炭鉱安全情報開示 |
94 |
|
|
|
第II部 |
|
|
五番目です。 |
登録者普通株市場、関連株主事項及び発行者による株式証券の購入 |
95 |
第六項です。 |
[保留されている] |
95 |
第七項。 |
経営陣の財務状況と経営成果の検討と分析 |
96 |
第七A項。 |
市場リスクの定量的·定性的開示について |
107 |
第八項です。 |
連結財務諸表と補足データ |
108 |
第九項です。 |
会計と財務情報開示の変更と相違 |
108 |
第9条。 |
制御とプログラム |
108 |
プロジェクト9 B。 |
その他の情報 |
108 |
プロジェクト9 Cです。 |
検査妨害に関する外国司法管区の開示 |
108 |
|
|
|
第三部 |
|
|
第10項。 |
役員·幹部と会社の管理 |
109 |
第十一項。 |
役員報酬 |
115 |
第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
122 |
十三項。 |
特定の関係や関連取引、取締役の独立性 |
125 |
14項です。 |
最高料金とサービス |
126 |
|
|
|
第4部 |
|
|
第十五項。 |
展示·財務諸表明細書 |
127 |
第十六項。 |
表格10-Kの概要 |
127 |
i
前向き陳述と市場データに関する警告説明
この10-K表年次報告(“年次報告”)には、1934年証券取引法(“取引法”)第21 E節及び1933年証券法(“証券法”)第27 A条(“証券法”)の意味に適合する前向きな陳述が含まれている。本年度報告に含まれる歴史的事実の陳述を除いて、本年度報告に含まれるすべての陳述は、私たちの将来の運営結果または財務状況、研究開発計画、私たちが進行している計画中の臨床前研究および計画における臨床試験の予想時間、コスト、設計と進行に関する声明、候補製品の規制申告と承認の時間と可能性、候補製品を商業化する能力(承認されれば)、将来の候補製品開発の潜在力、戦略協力の潜在的メリット、成功のタイミングと可能性、将来の運営の管理計画と目標、および予想製品開発事業の将来の結果などに関する声明を含み、前向きな声明である。場合によっては、“可能”、“そうなる”、“すべき”、“可能”、“予想”、“意図”、“目標”、“計画”、“予想”、“信じ”、“推定”、“予測”、“潜在”、“継続”などの用語、またはこれらの用語の否定または他の同様の表現によって識別することができる。このような展望的な陳述はただ予測に過ぎない。これらの展望的な陳述は主に私たちの現在の未来の事件と財務傾向の予想と予測に基づいており、私たちはこれらの事件と財務傾向が私たちの業務、財務状況と経営結果に影響を与える可能性があると考えている。これらの前向き陳述は、本年度報告が発表された日までの状況のみを代表し、第1部1 A項“リスク要因”に記載されているリスク、不確定要因、仮説を含むいくつかのリスク、不確定要素、仮説の影響を受ける可能性がある。著者らの展望性陳述に反映された事件と状況は実現できない或いは発生できない可能性があり、実際の結果は展望性陳述中の予測結果と大きく異なる可能性がある。法的要件が適用されない限り、私たちは、任意の新しい情報、未来のイベント、状況の変化、または他の理由による、本明細書に含まれる任意の前向きな陳述を公開または修正するつもりはありません。すべての展望的陳述は1995年の個人証券訴訟改革法の安全港条項に基づいて作られたこの警告的声明である。
本年度報告書には、他の組織の財産に属する商標、商号、サービスマークが含まれている。便宜上、本年度報告で言及された商標および商標名は、和?記号を持たないが、これらの参照は、適用法に基づいて、これらの商標および商標名に対する私たちの権利を最大限に主張しないこと、または適用されるすべての人がその権利を主張しないことを意味するわけではない。
本年度報告はまた、私たち自身の内部推定および研究、ならびに独立した市場研究、業界および一般出版物および調査、政府機関および公開から得られる情報からの業界、市場、および競争地位データを含む。いくつかの場合、私たちはこのようなデータの出所を明確に言及しなかった。この点で、私たちがどの段落でもこのようなデータの1つまたは複数のソースを言及した場合、他の平文規定または文脈に別の要求がない限り、同じ段落に出現するこのような他のデータは、同じソースからのものであると仮定すべきである。また,本報告に含まれる業界,市場,競争状況データは信頼でき,合理的な仮定に基づいていると考えられるが,このようなデータにはリスクや不確定要因が含まれており,“リスク要因”と題する章で議論されている要因も含めて様々な要因によって変化する可能性がある。これらの要素および他の要素は、結果が独立した当事者または私たちが推定した結果とは大きく異なる可能性がある。
II
第1部
情報技術EM 1.ビジネス。
概要
我々は臨床段階の生物製薬会社であり,選択的翻訳調節阻害剤(STRI)と呼ばれる新しい腫瘍学薬の開発に専念している。翻訳は細胞中のタンパク質の産生が遺伝子配列に含まれる情報によって指導される過程である。我々は独自の選択的翻訳制御技術プラットフォームを利用して、内部で一連の小分子STRI候補製品を発見した。我々の候補製品はeIF 4 F複合体及びその活性化キナーゼ、マイトジェン活性化プロテインキナーゼ相互作用キナーゼ(“MNK”)に対して。EIF 4 F複合体は癌の中で最も突然変異が最も頻繁な2つのシグナル経路であるPI 3 K-AKTとRas-MEK経路の中心ノードであり、それらは集まって、選択メッセンジャーRNA(“mRNA”)を蛋白質への翻訳を活性化し、蛋白質は肝心な疾病駆動過程中によく発生する張本人である。これらの標的のいずれかを抑制することは、複数の疾患キネシンの発現を同時に低下させ、それらを産生する。我々の各候補は、癌タンパク質(その異常機能が癌を引き起こす可能性のあるタンパク質)、T細胞における免疫抑制タンパク質、および腫瘍の成長、生存および免疫逃避を共同制御する複数の機能関連タンパク質の発現を駆動することができる単一のタンパク質に作用することを意図している。
著者らの主要な候補製品TomivosertibはMNKの阻害剤であり、現在KEYTRUDA(Pembrolizumabとも呼ばれる)と連合して評価を行っており、KEYTRUDAはFDAが許可したプログラム性細胞死蛋白1(PD-1)阻害剤であり、転移性非小細胞肺癌患者の2 b期無作為臨床試験に用いられている。著者らの第二の候補製品であるゾチフェンはeIF 4 Aの阻害剤、eIF 4 F複合体の一成分であり、現在ある固形腫瘍患者の1/2期の臨床試験で評価されている。著者らはすでにこの試験の初期用量増加部分といくつかの適応の初期第2段階拡張部分を完成し、ゾタフェンとフルビストランとアベシル(“ZfA三連”)の併用によるER+乳癌患者の治療効果を評価することを含む。これまでにER+乳癌患者を含む5つのキューのデータが報告されており,これらのデータはゾタフェンの通常耐性が良好であることを示し,大量に前処理されたER+乳癌患者の部分反応を含む活性シグナルを示している。著者らはすでにファイザー会社と著者らの最初の段階計画eIF 4 E阻害剤について全世界の協力と許可協定を締結し、ファイザー社は現在この計画のために研究性新薬応用(IND)研究を行っている。私たちのすべての候補製品は患者の予後を改善し、チェックポイント阻害剤や標的治療などの癌治療の効用を拡大する潜在力があると信じている。
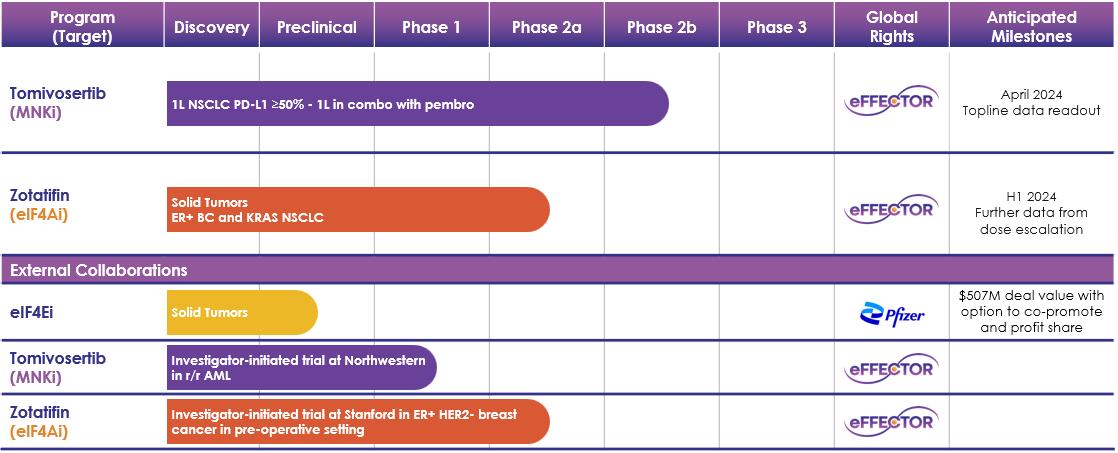
次の表は私たちの現在の計画をまとめています
図1:私たちのチャネル
1
標的eIF 4 F複合体とMNK
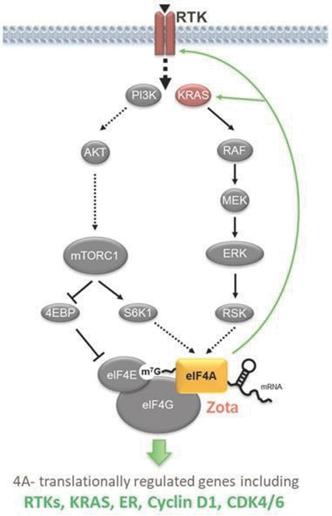
EIF 4 F複合体はある細胞成長と分裂を促進する蛋白質の産生に重要な役割を果たしている。正常な細胞機能において、細胞外因子、例えば増殖因子或いは抗原は細胞表面受容体例えば受容体チロシンキナーゼ(RTK)とT細胞受容体(TCR)に結合し、後者はPI 3 K-AKTとRAS-MEK経路を通じてシグナルを開始し、成長を刺激する。正常ではRTKとTCRは細胞成長や増殖が必要な場合にのみ一時的な刺激を受ける。しかし、eIF 4 Fが過剰かつ持続的に活性化する時、PI 3 K-AKTとRAS-MEK経路を活性化する癌遺伝子突然変異によるのも、腫瘍中のTCR抗原の持続的な伝達によりも、これは蛋白質合成の持続的な上昇を招く。この持続的な上昇は腫瘍細胞の制御されない成長とT細胞の枯渇を招き、T細胞がそれほど有効でない癌戦士になる。EIF 4 F複合体中の阻害標的は、疾患キネシンを合成する前にそれらの産生を低下させる。EIF 4 F複合体によって制御される腫瘍細胞中のタンパク質は、(1)現在標的治療によって対象とされている複数の癌タンパク質、例えばRTKおよびKRAS、(2)MYCおよびCyclin D 1などの現在標的治療に利用可能な癌タンパク質が存在しないこと、および(3)Cyclin D 1、CDK 4/6、RTKsおよびKRASのような薬剤耐性機序として標的治療を標的とする際にしばしば上昇するいくつかのタンパク質を含む。T細胞において、MNKはPD-1、PD-L 1、TIM 3、LAG 3とIL-10を含む多種の免疫抑制因子の産生を制御し、これらの因子はT細胞を枯渇させ、免疫反応を弱めるために使用される。
MNKとeIF 4 F複合体にはいくつかの潜在的な優位性があると考えられる。まず,腫瘍細胞が成長,増殖,生存のためにハイジャックした多くの重要な疾患キネシンの産生抑制に同時に取り組むことができる。我々の各候補は、単一の標的癌タンパク質を阻害するのではなく、癌細胞において一般的に共通して産生される複数の標的癌タンパク質、または活性化T細胞において産生される複数の免疫抑制因子を下方制御することを意図している。そのほか、著者らの候補製品は多くのフィードバック経路によって過剰に産生される蛋白質を下方制御することを目的としており、これらの蛋白質は腫瘍の標的治療に対する反応が比較的に悪い薬剤耐性機序である。また、著者らの候補製品はMYCとCyclin D 1のようないくつかの疾患キネシンを下方制御することを目的としており、これらの標的の細胞位置と複雑な形状のため、現在いかなる既存の市場代理によって解決されていない。最後に、著者らの候補製品は正常な細胞機能を保護すると同時に、腫瘍細胞の殺傷力を増強することを目的としているが、これらの過剰生産された原因蛋白はMNKとeIF 4 F複合体の産生に依存し、正常細胞と比較して、それらは腫瘍細胞の成長と生存に更に重要である。
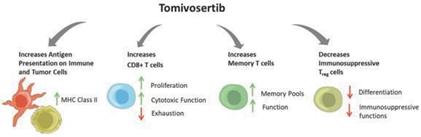
高効率で高選択性のMNK阻害剤トミフセチブ
著者らの主要な候補製品Tomivosertibは経口MNK小分子阻害剤であり、著者らはPD-1とプログラム化細胞死リガンド1(PD-L 1)の阻害剤と結合して開発されており、総称して抗PD-(L)1療法と呼ばれ、固形腫瘍患者の治療に応用されている。MNKはリン酸化学基の酵素添加によってリン酸化または修飾されたキナーゼであり、リン酸化学基はeIF 4 F複合体中の重要なタンパク質である。MNK、Tomivosertibを抑制することによって、多種の免疫抑制蛋白の産生を低下させ、そしてT細胞を再プログラミングして不全と機能障害を遅延させ、それらの腫瘍細胞に対する能力を増強することを目的としている。臨床前研究において、TomivosertibはすでにPD-1、PD-L 1、TTM 3、LAG 3とIL-10を含む多種の免疫抑制蛋白の産生を下げることが証明された。MNKは多くの腫瘍の発展において重要な役割を果たしており、協調的な方法で多種の因子の発現を制御し、それによって免疫反応を弱めることを含む。免疫減弱は正常な生物過程であり、免疫系への過度な刺激を防止することができる。しかし、腫瘍はよく減衰過程を利用して免疫制御から逃れる。臨床前研究において、トミフゼチブはMNKを抑制し、免疫系が腫瘍を攻撃する能力を増強した。免疫検査点、例えばPD-1、PD-L 1、TIM 3とLAG 3は、免疫と腫瘍細胞に発現するシグナル分子であり、多種の機序を活性化して抗腫瘍免疫反応を弱めることができる。過去数年間、チェックポイント阻害剤と呼ばれる薬物は、主に抗PD-(L)1療法であり、すでに癌を治療する重要な新しい治療法となり、これらの免疫チェックポイント経路を遮断することができる。2022年、抗PD-(L)1療法の全世界市場価値は350億ドルを超え、その半分近くが転移性非小細胞肺癌患者の治療に応用されている。検査点阻害剤治療は様々な癌の患者に非常に有効であるが、これらの薬物は通常治愈できず、多くの患者は最終的にチェックポイント阻害剤治療の進展を得て、新しい第一線の連合治療が必要であることを強調し、これは長期的な持続的な利益を経験した患者の割合を増加させ、他の癌の進展を遅らせる可能性がある
2
病人です。バイオマーカーとしてPD−L 1発現レベル50%以上(“PD−L 150%”)を用い,進行中のトミフザチブのKickStart 2 b期臨床試験への参加を募集した。米国では毎年27,000名の新たな転移性非小細胞肺癌患者のPD−L 1が50%と推定されており,米国では40億ドルの市場機会を代表していると推定されている。TomivosertibはPD−L 1発現レベル1−49%の患者の治療にも利用できると信じており,米国では別の50億ドルの市場機会を代表していると予想される。
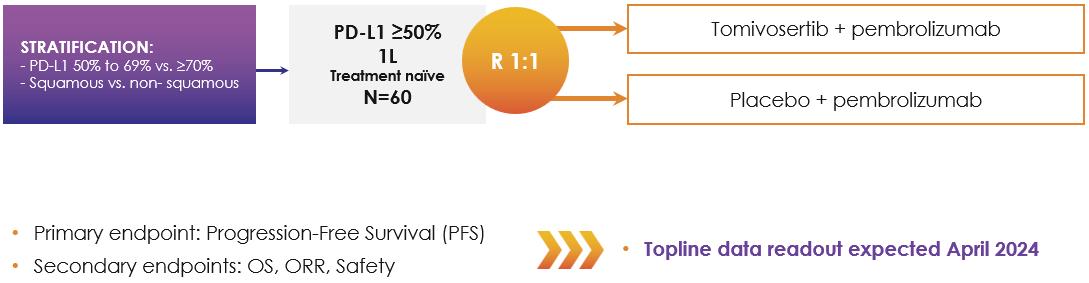
著者らの2 a期臨床試験の鼓舞的な結果に基づいて、著者らはKickStartをスタートし、これは二重盲検、無作為、プラセボ対照の2 b期試験であり、トミソチブとペブロズモノクロナル抗体の併用によるPD-L 1 50%の転移性非小細胞肺癌患者を治療した。Pembrolizumabはメルク社が所有し、マーケティングし、第一線の非小細胞肺癌と他のいくつかの適応に応用されている。我々は2021年第2四半期にこの試験を開始し,2024年4月初めにKickStart試験の背線データを報告する予定である。
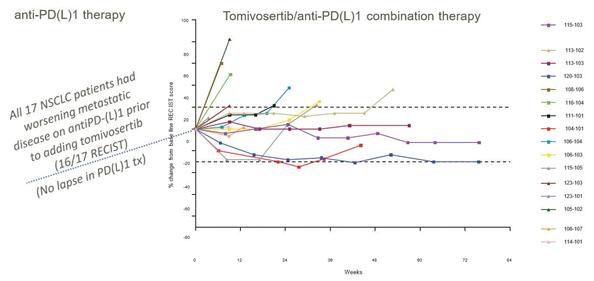
著者らが完成した2 a期CPI-A臨床試験において、17名の転移性肺癌患者に対してTomivosertib連合抗PD-(L)1治療の評価を行い、TomivosertibはMPFSを明らかに延長し、即ち以前抗PD-(L)1治療が無効な患者の中で、患者は生存を維持し、疾病進展がない時間であり、その定義は彼らの腫瘍評価が20%以上増加した或いは新しい病変が出現したことである。そのほか、2020年9月までに研究が完成した時、この17名の患者の中の2名(12%)はすでに部分的な緩和を確認し、或いは腫瘍評価はベースラインレベルより30%以上低下し(“PR”)、その中の1人の患者は引き続き確認された完全な緩和を実現し、或いは検出されなかった腫瘍病変(“CR”)、3人目の患者の腫瘍は28%消退した。この臨床試験では,トミフザチブの耐性は一般に良好であった。この17例の患者のうち,抗PD−(L)1治療にトミフサーチブを加えて何の変化や中断もなく,20週間のMPFSがあった。この試験では,PD−L 1発現陽性患者のMPFSは53週であったのに対し,PD−L 1陰性患者のMPFSは9週であった。PD−L 1発現はT細胞が腫瘍に浸透するバイオマーカーであり,治療期間中に行った診断分析の既存データを特別に分析して決定した。KickStart試験に先立ち,Tomivosertibは200名以上の患者の完成研究でテストを行い,Checkpoint阻害剤と併用した患者約80名を含め,単一薬としてCheckpoint阻害剤と併用したところ,Tomivosertibは良好な有害事象特徴を示した。
非小細胞肺癌患者をPD−L 1 50%のKickStart試験に組み込んだ。第一線の環境下で、PD-L 1が50%の患者は臭化リズマブ治療に最も敏感である。この場合,pembrolizumab単一療法は標準看護であり,pembrolizumabは最も広く用いられているチェックポイント治療である。この初期起動患者集団に加えて、PD-L 1発現が1~49%であるTomivosertib非小細胞肺癌患者に対して追加の臨床試験を行い、他のPD-(L)1治療を標準治療とする他の適応に適用する予定であり、腎臓癌、膀胱癌、三陰性乳癌、または高マイクロサテライト不安定性を有する腫瘍(“マイクロサテライト-H”)などの他のPD-(L)1治療許可を得た癌を含む。
著者らの臨床前研究により、トミフセテと抗PD-(L)1の併用治療は検査点阻害剤に対する耐性機序を克服でき、それによって増強と持続性の敏感性があることを表明した。また,われわれの臨床前データでは,トミフザ置換は単一薬物としても抗PD−1治療と併用しても,薬物治療中止後に持続的に存在する抗腫瘍免疫を促進することが示唆された。われわれの方法の重要な利点の1つは,MNKを抑制することにより,Tomivosertibが複数の免疫チェックポイントを協調的に下方制御し,サイトカイン蛋白の産生を抑制し,腫瘍に対する免疫反応を活性化することであると考えられる。著者らの臨床前データにより、Tomivosertibの活性は以下の方式により検査点阻害剤の薬剤耐性の重要な機序を解決した
結論的に、これらの作用は腫瘍の識別を増加させ、免疫反応を回復し、反応の持続性を高め、抗腫瘍免疫を保護することによって、検査点阻害剤を補充することができる。
3
ゾチフェンは高効率で選択的なeIF 4 Aヘリカーゼ阻害剤です
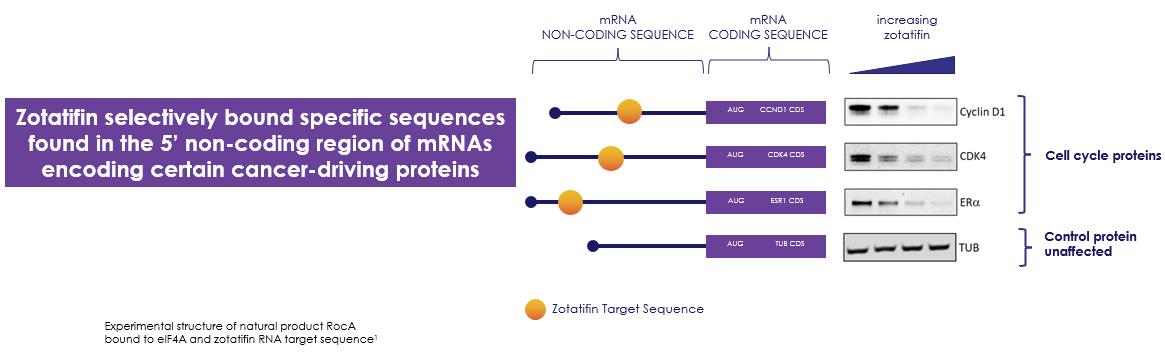
著者らの第二の候補製品であるゾタフェンはeIF 4 Aを抑制することを目的とした小分子であり、現在固形腫瘍患者の1/2期臨床試験で評価されている。EIF 4 Aは複雑な二次構造を解くことを担当し、あるmRNAの5‘非翻訳領域(“UTR”)に発見されたヘリカーゼである。この解離は1つの調節制御ステップであり、重要な蛋白質の有効な刺激産生を招き、正常細胞を成長シグナルに反応させ、そして腫瘍細胞の中で上昇させる。腫瘍細胞中のeIF 4 Aによって制御されているタンパク質は、現在標的治療によって解決されている多種の癌蛋白を含み、現在標的治療に利用可能な癌蛋白、および薬剤耐性機序としての標的治療がよく上昇するいくつかの蛋白がない。これらの癌蛋白のいくつかは垂直シグナル経路で協同作用し、腫瘍の成長、増殖と癌の生存を推進することができ、ある乳癌と非小細胞肺癌を含む。著者らの臨床前研究において、著者らは多くのゾチフェンによって阻害された蛋白質がその識別されたmRNA 5‘端非コード領域に共通の異なる翻訳開始制御エレメントを有することを発見した。著者らの臨床前データは、体外生理濃度のゾタフェン抑制は細胞中の約5%のmRNAの翻訳にしか影響しないことを示した。また,これらの翻訳開始制御エレメントは,タンパク質合成に含まれるアミノ酸のコード配列を決定する前のmRNAに位置し,コード配列とは独立しているため,それらの阻害はタンパク質変異変異体とは独立している。著者らはすでにER+乳癌、FGFR+乳癌とKRAS突然変異NSCLCの中で多数の2 a期拡張行列を起動し、ゾタフェンは単一薬物であり、連合標的薬物でもあり、現在ER+乳癌の連合治療に集中している。つまり,米国では,我々2 a期拡張キュー登録基準に適合する乳癌と非小細胞肺癌患者の推定目標人口総数は約82,000人であると信じている。
EIF 4 E−ファイザーとのグローバル協力
我々の第3のプロジェクトはeIF 4 Eの阻害剤の開発に集中しており,現在ファイザーによるファイザープロトコルによる開発が行われている。ファイザーは現在主要候補製品のIND起用研究を行っている。これまで、ファイザー協定によると、4200万ドルを受け取り、将来的には4.65億ドルの記念碑的支払いと潜在的な販売特許使用料を受け取ることができるかもしれない。
私たち独自のSTRI-私たちの翻訳仕様技術プラットフォームを使って
私たちは私たちの独自の選択的翻訳規制技術プラットフォームを使用して私たちの候補製品を発見した。また、eIF 4 Fの複雑で成長に依存した翻訳制御の専門家である創始者と協力者チームを結成した。翻訳制御の疾患における重要性は製薬業界でますます認識されており,STRIに重点を置いた癌治療法の開発の最前線にあると信じている。著者らの独自の選択的翻訳制御技術プラットフォームを利用して、著者らは多種の腫瘍タイプと他の疾病における翻訳上昇の遺伝子の理解を開発した。これにより,意義のある臨床効果を有する可能性のある治療介入点を決定し,これらの関与点で作用する可能性のある候補製品に最も反応する可能性のある患者集団を決定することができる。著者らは、翻訳制御生物学に対する深い理解に加え、著者らの複雑かつ集中的な構造に基づく設計と薬物化学の計算化学方法に加えて、著者らは翻訳制御療法という新興領域の先鋒領域で重要な優勢を獲得し、そして巨大な進入障壁を創造したと信じている。私たちは現在、私たちの既存の候補製品の臨床開発に資源を集中させる予定です。
私たちは強力な物質構成と他の知的財産権を持っていて、私たちの候補製品とその用途をカバーして、世界の主要市場の知的財産権を通じて私たちの候補製品と私たちの技術プラットフォームを保護するように努力しています。
4
戦略.戦略
STRIの開発を継続し,最終的に商業化し,多様なタイプの癌の治療に利用することを目標としている。私たちの目標を達成するために、私たちは次のような戦略を取るつもりだ
5
わが社の起源とチーム
著者らは2012年にDavide Ruggero博士とKevin Shokat博士実験室の先駆的な研究に基づいて著者らの会社を設立し、その後カリフォルニア大学サンフランシスコ校の翻訳解析技術の独自応用許可を得た。著者らの科学創始者と管理チームは業界のベテランから構成され、彼らは腫瘍とその他の疾病領域のすでに発売された小分子薬物、単抗療法と細胞療法の発見と開発に重要な役割を果たし、Adcetris、アバスティン、Cabometyx、Cellcept、Cotellic、Inlyta、Tecentriq、ToradolとViraceptを含む。
翻訳制御とeIF 4 F複合体の腫瘍における作用
図2:遺伝子発現の過程.
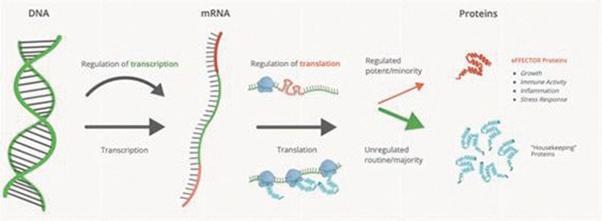
ヒトゲノムに含まれる情報は,遺伝子発現と呼ばれるプロセスによって細胞行動を指導し,この過程でRNAにコードされる命令がタンパク質の合成を指示するために用いられる。遺伝子発現の2つの重要なステップは転写と翻訳である(上図2参照)。転写はDNA配列をメッセンジャーリボ核酸に複製するが,翻訳はその後メッセンジャーリボ核酸配列を用いてタンパク質合成を指導する。60年以上前、DNAからRNA、そしてタンパク質への情報の一方向流動がフランシス·クリークによって分子生物学の中心教条と命名されて以来、生物学者は転写を遺伝子発現制御の主なポイントとしてきた。最近、私たちと私たちの科学創始者は、翻訳が細胞中の一部のタンパク質の過剰生産の重要な制御ステップであり、薬物開発に適していることを証明した。翻訳制御は通常機能関連蛋白の発現を制御し、これらの蛋白は細胞生理に深い影響を与えることができ、細胞成長、分裂と免疫細胞機能を駆動する細胞外と細胞内シグナルに対する反応を含む。これらの効果蛋白発現を破壊する翻訳制御は多くの疾病の発生と発展を招くことができる。
癌では,eIF 4 F複合体の異常活性化により,ある厳格に制御されたmRNAの翻訳がしばしば失調し,発癌蛋白の産生を招き,制御されない成長,免疫逃避と転移を特徴とする悪性腫瘍を引き起こす。われわれの治療法は,腫瘍が自身の利益のためにハイジャックする過程の翻訳制御を回復し,正常な細胞機能を維持することができると信じている。翻訳制御の過程は腫瘍細胞とT細胞に発生し、身体の免疫系からの逃避を含む腫瘍の生存と成長に重要である。腫瘍細胞および/またはT細胞に作用することによって、我々の候補製品は、癌の進行を推進する多くのタンパク質の産生を調節することができ、単一の治療剤に複数の標的治療および/または免疫治療の利点を結合することが可能である。
著者らはすでに、免疫反応を弱める複数の過程を担当し、検査点蛋白の上昇と抗原提示蛋白の下方制御を含み、翻訳調節によって制御されていることを発見した。健康な組織では,免疫活性化後にバランス,非自己破壊の基調を保つためには,免疫反応の能力を弱めることが重要であるが,腫瘍を免疫検出や破壊から脱出させることも可能である。T細胞を再プログラミングし、腫瘍を免疫媒介破壊から脱出させる因子の翻訳を阻止することにより、腫瘍をより効率的に攻撃するために、患者の免疫系を放出することができると信じている。
6
EIF 4 F複合体は癌中の2つの最も頻繁に突然変異するシグナル経路であるPI 3 K-AKTとRas-MEK経路--の中心接続点であり、1つの中央ノードを代表し、SELECT mRNAを蛋白質に翻訳することを担当し、蛋白質は重要な疾病駆動過程中によく発生する原因である(以下図3参照)。また,T細胞におけるMNKとeIF 4 F複合体の持続的な活性化は不全や機能障害を引き起こす。著者らの独自の選択的翻訳制御技術プラットフォームはすでに、ある疾患状態、例えば癌は、MNKとeIF 4 F複合体の大幅な上昇を招き、それらは共に多種の腫瘍成長と増殖を推進する癌蛋白の産生を活性化し、及びT細胞不全と機能障害を招く免疫抑制蛋白の産生を引き起こすことを証明した。これらの病原性蛋白は重要な翻訳制御因子によって制御され、著者らの3つの標的:MNK、eIF 4 AとeIF 4 Eを含む。MNKはシグナルと生存において重要な役割を果たすキナーゼであり、腫瘍免疫反応を減少或いは下方制御することが知られている遺伝子を調節する。MNKはリン酸化eIF 4 Eの末端キナーゼであり、eIF 4 Eは翻訳開始を担う複合体の重要な成分であり、eIF 4 Aは翻訳前にmRNA構造を解くことを担当する。著者らは、MNK、eIF 4 A、およびeIF 4 Eの各々が、腫瘍生物学および疾患サブセットの異なる態様に影響を与える機会を提供するために、非常にユニークなmRNAサブセットの翻訳を選択的に調節することを発見した。
図3:エフェクターの標的は、発癌シグナル経路とそれらが産生するタンパク質との間の重要なノードに位置する。
細胞外信号
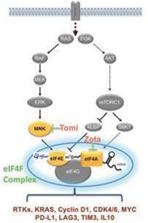
EIF 4 F複合体中のキー翻訳調節因子を標的とした癌治療により治療結果を改善する高い潜在力が見られた。これらの翻訳調節因子を標的として癌を治療することは,単一翻訳蛋白活性を抑制するための計画に対して,我々の候補製品が腫瘍により広範な治療影響を与えると信じている。腫瘍成長と身体の腫瘍に対する反応を駆動する機能関連蛋白質の発現を調節することによって、著者らの候補製品は腫瘍と免疫細胞に影響を与えることを目的とし、著者らはこれらの影響は現在の標的或いは免疫療法の多くの局限性を解決できると信じている。
我々の発展計画は
我々はeIF 4 F複合体に対する一連の選択的小分子STRIを開発しており,これらの複合体は現在の治療が限られているか,あるいは利用できないいくつかの重要な癌タイプの現在の標的または免疫療法のいくつかの限界を克服する可能性があると信じている。
7
主要な候補製品:高効率で高選択性のMNK阻害剤Tomivosertib
Tomivosertibの概要
Tomivosertibは1種の経口小分子MNK阻害剤であり、開発中であり、抗PD-(L)1の併用による固形腫瘍患者の治療に用いられている。MNKはeIF 4 F複合体を活性化し、T細胞の多種の免疫抑制因子の産生を制御し、PD-1、PD-L 1、TTM 3、LAG 3とIL-10を含む。MNKを阻害することによって、Tomivosertibは、不全および機能障害を遅延させるためにT細胞を再プログラミングするように設計され、腫瘍細胞に対するより強い能力を提供する。われわれが完成した2 a段階CPI−A臨床試験では,17名の非小細胞肺癌患者に対してトミフザ置換併用抗PD−(L)1治療の評価を行った結果,トミソチブは以前の抗PD−(L)1治療が無効な患者のMPFSを有意に延長できることが示唆された。また,2020年9月までに研究が完了し,この17名の患者のうち2名(12%)にPRが確認され,そのうちの1名はその後CRが確認され,3名目の患者は腫瘍が28%消退した。この臨床試験では,トミフザチブの耐性は一般に良好であった。これらの結果に基づいて、2021年第2四半期に、著者らはKickStartの患者登録を開始し、これはトミフサーチブとペブロリズマブの併用による転移性非小細胞肺癌患者の二重盲検、無作為、プラセボ対照の2 b期試験である。2024年4月初めにKickStart試験の背線データを報告する予定である。
市場のチャンス
肺癌は米国で2番目によく見られる癌(皮膚癌を含まない)であり、癌死亡の主な原因でもある。国立癌研究所では,2022年の新規肺癌症例は23.7万例を超えると推定されている。非小細胞肺癌は最もよく見られる肺癌亜型であり、すべての肺癌診断の82%を占める。現在、約75%の非小細胞肺癌患者の腫瘍は1種の特定の操作可能な突然変異が不足しており、この突然変異はすでに許可された突然変異特異的標的治療に役立つかもしれないが、これらの患者は抗PD-(L)1を転移性非小細胞肺癌の第一線の治療の一部として受ける資格があるかもしれない。2022年、抗PD-(L)1療法の全世界市場価値は350億ドルを超え、その半分近くが転移性非小細胞肺癌患者の治療に応用されている。検査点阻害剤治療は各種の癌患者に対して非常に有効であるが、これらの薬物は通常治愈できず、大多数の患者は最終的に検査点抑制剤治療の方面で進展を得て、新しい第一線の連合治療が必要であることを強調し、これは長期的な持続的な利益を得る患者の割合を増加させ、そして他の患者の進展を遅らせる可能性がある。米国では約27,000名の転移性非小細胞肺癌患者のPD−L 1発現が50%と推定されており,米国では40億ドルの市場機会を代表していると推定されている。TomivosertibはPD−L 1発現レベル1−49%の患者の治療にも利用できると信じており,米国では別の50億ドルの市場機会を代表していると予想される。
PD-L 150%の設定は米国と世界で巨大な市場機会を提供し、膀胱癌、腎細胞癌またはMSI-H癌などの他のチェックポイント敏感癌を含むTomivosertibの開発を拡大する機会が多いと信じている。
高選択性MNK阻害剤Tomivosertibの発明の概要
著者らは広範な薬物化学研究を行い、構造に基づく薬物設計を結合し、トミフザチブが著者らの主要な候補製品であることを確定した。Tomivosertibは酵素分析においてMNK亜型MNK 1とMNK 2に対して効率的かつ選択的なMNK阻害作用を示し,その半数抑制濃度(IC 50)は1−2ナノモル(1リットル当たり10億分の1モル)であり,可逆的なATP競合作用機構によりこのキナーゼを抑制した。TOMIVOSERTIBで腫瘍細胞株を処理したところ,eIF 4 Eセリン209位のリン酸化レベルは用量依存的に低下し(IC 50=1.4−21.5 NM),これは先の研究結果と一致する,すなわちこの部位のリン酸化はMNKに完全に依存する。また、インビトロ試験において、トミフザチブは、MNKに対して高い選択性を有する阻害剤であることが証明され、MNKに対する効力は、2種類のプロテインキナーゼCLK 4およびDRAK 1に対する効力よりも約100倍高く、残りの412個の試験されたキナーゼ標的に対する効力よりも1000倍以上高い。
Tomivosertibの設計目的はMNKを抑制し、それによってT細胞中のeIF 4 Eのリン酸化とMAPKシグナル下流eIF 4 F複合体の活性化を阻止し、そして選択的に特定のmRNA sの蛋白翻訳を調節することである。トミフザチブ抑制作用の影響を全面的かつ定量的に測定した
8
多種の腫瘍細胞系と免疫細胞タイプに発現するmRNAの翻訳。この研究において、MNKは既知の重要な免疫チェックポイント蛋白とサイトカインの発現を制御することによって、抗腫瘍免疫反応の調節に重要な役割を果たし、これらの蛋白とサイトカインは免疫抑制の腫瘍微小環境を創造し、共に免疫細胞の機能を制限した。
また、免疫能を有するマウスで行われた同遺伝子マウスモデルおよび遺伝子工学マウス癌モデル、およびヒト細胞の増殖を可能にするために免疫系が損傷したマウスに移植された複数の異種移植モデルを含む、複数の体内腫瘍モデルにおいてTomivosertibを試験した。この群の体内前臨床試験により、著者らはトミフゼチブの単一薬物治療として免疫活性マウスモデルにおいて広範な抗腫瘍免疫反応を誘発し、トミソチブ投与量停止後の持続的な抗腫瘍免疫を誘導することを証明した。
Tomivosertibの作用機序:免疫系を刺激して腫瘍殺傷力を増強する
これまで、著者らが収集した臨床前と臨床データにより、トミフセテと抗PD-(L)1阻害剤の併用は検査点阻害剤に対する耐性機序を克服でき、それによって検査点阻害剤に対する敏感性を増強することができることを表明した。著者らの臨床前研究により、MNKの体内と体外での抑制はT細胞エフェクター反応を広く増強できることを表明した。重要な細胞内シグナル経路を遮断することによりT細胞を再プログラミングする機序により,Tomivosertibが証明されている
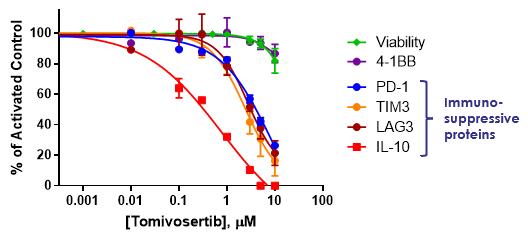
臨床前研究において、臨床関連濃度のトミフザ置換はPD-1、PD-L 1、TTM 3、LAG 3とIL-10を含む多種の免疫抑制蛋白を同時に低下させることが証明され、以下の図4に示す。特に,活性化された初代ヒトT細胞インキュベーション濃度と増加したトミフサーチペアはT細胞枯渇に関連する複数の検査点に対して用量依存性の抑制を示し,統計学的意義(P)を達成した
図4:Tomivosertibは複数のチェックポイント蛋白と免疫抑制IL-10を下方制御した。
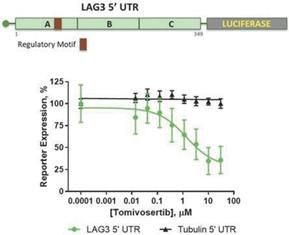
下方制御の分子機構を探索するために,LAG 35‘UTR配列をルシフェラーゼレポーター遺伝子解析に置き,Tomivosertibが増加した場合にT細胞におけるルシフェラーゼの発現を評価した結果,ルシフェラーゼ蛋白レベルはTomivosertib濃度の増加とともに低下し,統計学的意義(P)に達することが分かった
9
図5:Tomivosertib下方制御タンパク質収量は、チューブリン5‘非翻訳領域と比較してLAG 3 5’非翻訳領域に対して選択的であった。
TomivosertibのT細胞機能への影響を知るために,マウスまたはヒトT細胞集団を用いて,SIINFEKL,オボアルブミンから抽出したポリペプチド,あるいはヒト蛋白CD 19をそれぞれ認識し,以下の図6に示す。マウスでは,CD 44とCD 62 Lを高発現する細胞をマーカーとして,トミフザチブ濃度を増加させた場合に工程T細胞からなる脾細胞を刺激し,T細胞の中央記憶を増加させ,オボアルブミンポリペプチドを担持する標的細胞へのキラーを増加させることができる。同様に,CD 19を標的としたヒト工学T細胞がトミフザチブ濃度を増加させると刺激され,幹細胞記憶T細胞プールが増加し,これはヒト表面マーカーCD 45 RA+CD 27+で定義され,CD 19を発現する標的細胞へのキラーが増加する。
図6:Tomivosertibは中央記憶と幹細胞記憶T細胞バンクを増加させ、標的細胞殺傷を増強する。
マウス.マウス
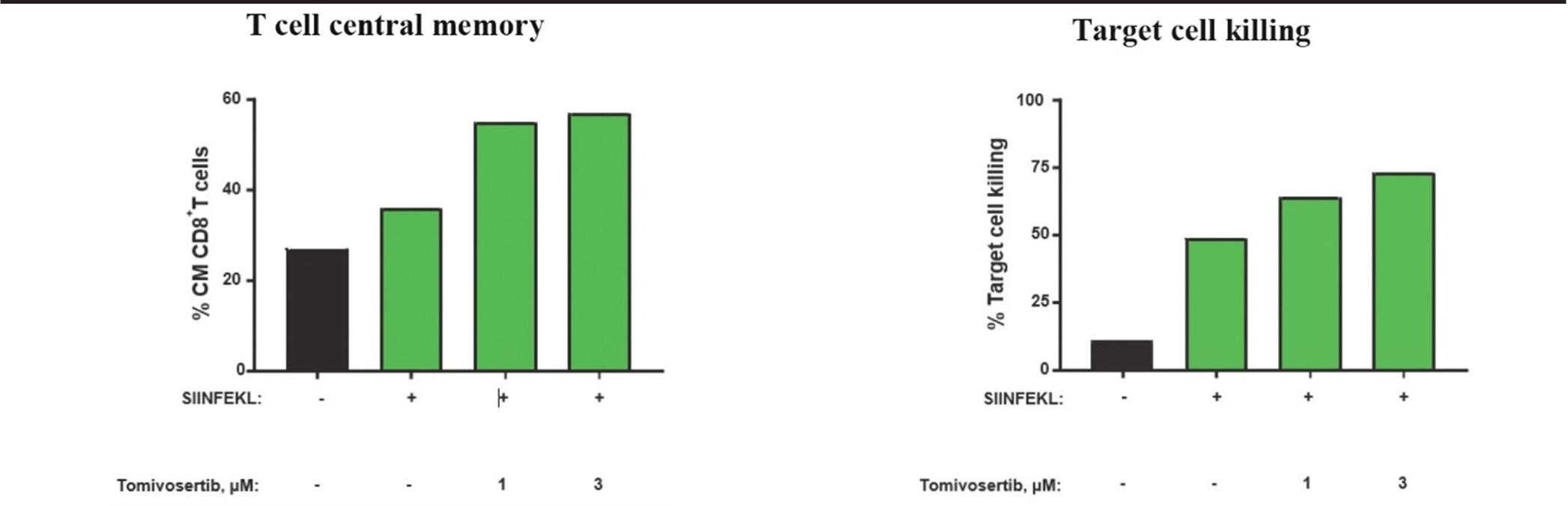
10
人間
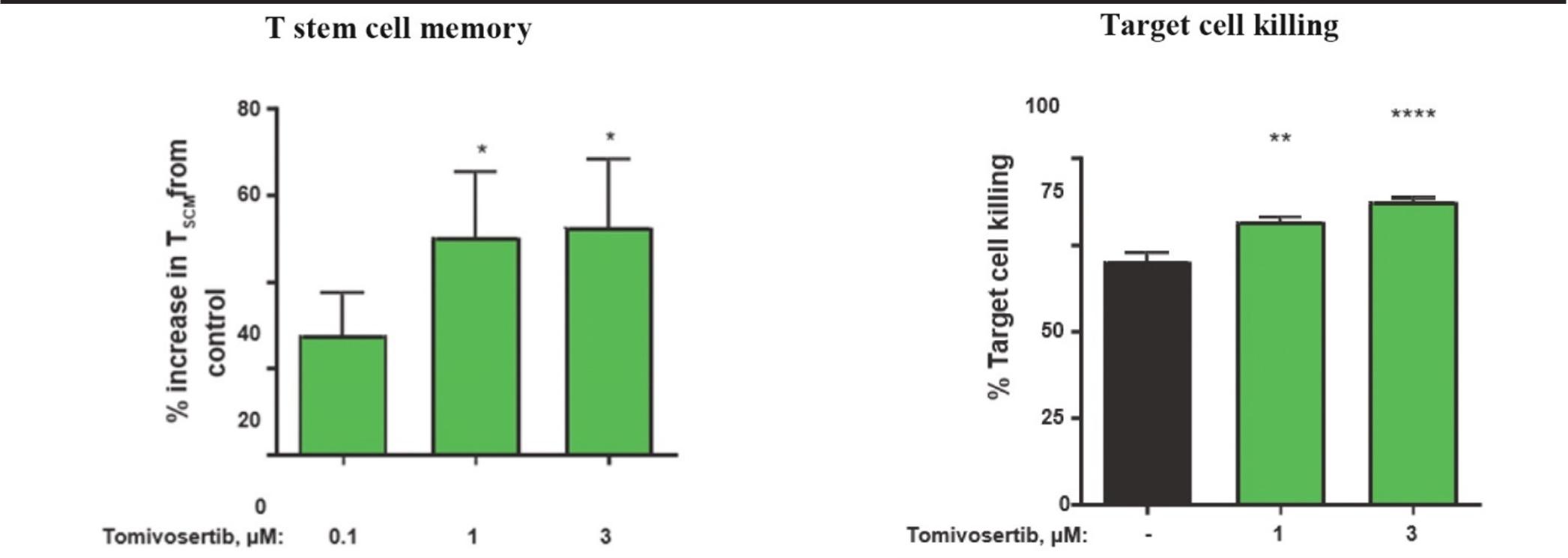
* p
したがって,トミフザチブの治療は,T細胞不全や機能障害を引き起こす多様なタンパク質や因子の産生を抑制し,記憶T細胞プールを増加させ,T細胞の標的細胞への殺傷を増加させることが証明されている。
臨床前モデルにおいて、Tomivosertibは単一薬物として免疫記憶を誘発し、抗PD-1活性を増強することが証明された
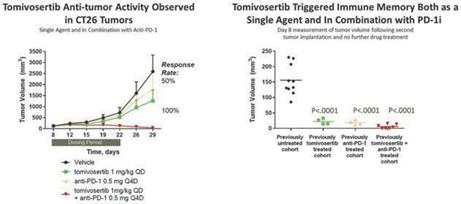
Tomivosertibの免疫増強特性が抗腫瘍活性を招くことができることを証明するため、著者らは免疫系の完全なマウスに臨床前研究を行い、そして同遺伝子腫瘍CT 26をそれらの体内に移植した。これらの腫瘍マウスは、その後、トミフセテ、マウスバージョンの抗PD-1抗体、または2つの薬剤の組み合わせを投与される(以下図7参照)。投与期間はいずれも2週間であった。これらの実験は,対照未治療マウスと比較して,トミフザチブ単独または抗PD−1で治療したマウス腫瘍が約50%減少し,両者の結合によりすべての治療を受けたマウスの腫瘍が完全に消退したことを示している。免疫記憶の作用を証明するために、さらなる薬物治療なしに、腫瘍消退を示す各キュー内のマウスが再挑戦され、追加のCT-26腫瘍細胞が対側翼に注入された。その結果、これらのマウスは、単一トミフザチbで前処理したマウスと、トミフザチbと抗PD-L 1を併用して治療したマウスを含み、その後の腫瘍攻撃に抵抗することができ、免疫記憶の増強が新しい移植腫瘍の成長を阻止できることを示した。CT 26モデル中の更なる薬効学的バイオマーカーは、トミフザ置換治療はまた腫瘍内効果CD 8+細胞毒性T細胞と免疫抑制制御性T細胞FOXP 3+の比率を増加させ、そして腫瘍内免疫抑制M 2マクロファージの減少を招くことを示した。つまり,これらのデータはトミフザチブが免疫系を増強し,腫瘍成長の持続的な抑制を招いていることを示している。マギル大学の独立した研究者が最近発表した論文でも、T細胞および他の免疫細胞をさらに活性化することによって、MNKを遮断することは、いくつかの黒色腫マウスモデルにおいて強力な免疫活性化および腫瘍消退をもたらすことができることを示している(JCI,2021)。
11
図7:臨床前の研究により、トミフザチブガ抗PD-1阻害剤はすべての動物の退化を招くことができ、そして腫瘍の再攻撃時に免疫記憶を維持することができ、単一薬物としても同様である。
Tomivosertibは様々な種類の細胞に作用し、免疫反応を推進する
我々のデータおよびマギル大学からの最新のデータは、複数の免疫細胞タイプを遮断するMNKが免疫系による癌細胞の死滅に広く関与していることを示している(以下図8参照)。これらの免疫活性を促進する機序は、(1)T細胞上の複数のチェックポイント蛋白と免疫抑制サイトカインを低下させること;(2)樹状細胞の抗原提示を増加させること;(3)CD 8+T細胞の細胞毒機能を増強し、T細胞の枯渇/機能障害を阻止すること;(4)T細胞記憶池を拡大することを含む。つまり、これらの作用は、腫瘍の識別を増加させ、免疫反応を回復させ、反応の持続性を向上させ、免疫持続性を維持することによって、検査点阻害剤を補充することができる。
図8:Tomivosertibは多種の細胞タイプに作用し、免疫反応を推進することを目的としている。
これらの知見から,トミフセ代替は,患者のチェックポイント阻害剤使用のメリットを拡大し,チェックポイント阻害剤の反応を停止した患者へのメリットを回復させることで,現在の癌免疫治療を改善する可能性が考えられる。
癌患者の第1期用量逓増試験と健康ボランティアの食物効果研究
著者らはそれぞれ固形腫瘍とリンパ腫の中で2つの独立した第1段階用量逓増臨床試験を行い、トミフザチブの安全性、薬物動態学、薬効学と腫瘍制御を評価した。各試験の主な終点は、MTDを確立し、推奨される2期用量(“RP 2 D”)を決定することである。
著者らの固形腫瘍の第一段階の用量増加試験において、著者らは標準看護治療の方面で進展を得た転移性固形腫瘍患者を募集した。この試験からRP 2 Dを200 mg,1日2回(BID),絶食時に服用したカプセル製剤を決定し,その後,この用量レジメンを用いて2 a期CPI−A検討を行った。このような患者では,トミフ司チブ単一療法の耐性が一般的に良好であることが分かった。最もよく見られる急診副作用は吐き気、嘔吐、無力、便秘、消化不良と振戦である。投与量がRP 2 Dを超えると、これらのAEsの発生率と重症度はもっと高い。全体的な薬物動態学的暴露は用量の増加に従って増加し、半減期は約12時間であり、BID方案を支持する。
12
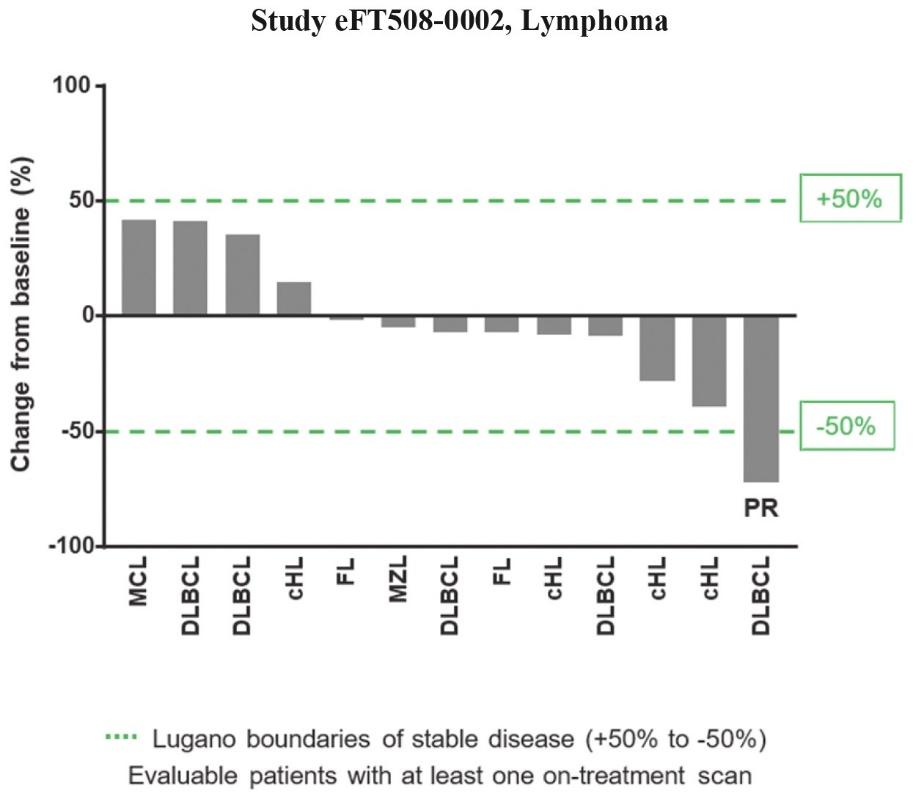
著者らのリンパ腫の第一段階用量増加試験において、著者らはB細胞悪性腫瘍患者を募集し、主にリンパ腫患者であり、彼らは標準看護治療方面で進展を得た。この試験は最初に2つの用量レベルの安全性をテストし、1日300 mgまたは450 mg(QD)と200ミリグラムBIDまたは300 mg BID、その後RP 2 D、200 mg BIDカプセルで限られた拡張を行った。本研究では,MTD 200 mg Bidカプセル絶食を確立した。RP 2 D拡張キューにおいて、患者が経験する最もよく見られる副作用は、吐き気、嘔吐、高カルシウム血症、および疲労である。200ミリグラムBIDカプセル製剤を受けた患者は、リンパ腫のルガノール基準に従って、治療後のスキャンと比較して、腫瘍サイズが少なくとも50%減少したことを確認したPRを得た(以下図9参照)。この患者は以前R-CHOP(メロワ、シクロホスファミド、アドリアマイシン、ビンクリスチンとプレドニゾロンの化学療法連合方案)と自己幹細胞移植の放射線学的進展を経験した。8人の患者の病状が安定していることは、彼らの腫瘍評価がリンパ腫のベースラインレベル+/-50%より既定の限界内に維持され、新しい病変が出現せず、その中の何人かの患者が最初に腫瘍体積の縮小を示したことを意味する。入選した19名の患者のうち,13名の患者が少なくとも1回の治療走査を行い,放射線学的評価が可能であった。彼らの全体的な最適応答を以下の図9に示す.
図9:第1段階用量増加試験は複数の腫瘍消退患者を含む。
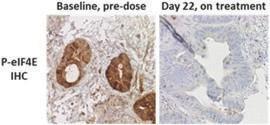
また,末梢血単球および治療前および治療中の腫瘍生検組織における薬効学的結果は,MNK阻害マーカーリン酸化eIF 4 EがRP 2 Dで有効に抑制されていることを示している。RP 2 Dでは,治療前と治療中の生組織標本(以下図10参照)のリン酸化eIF 4 E免疫組織化学(“IHC”)のMNK標的に対する抑制程度は90%から100%であることが観察された。
13
図10:患者の生検サンプルから、MNK活性化マーカーが腫瘍において下方制御されていることを示す
また,トミフサーチブの絶食および摂食条件下での薬物動態分布を評価するために,健康ボランティアの2つの健康ボランティアの食物効果研究を完成させた。これらは、同じ患者の2つの異なる用量のトミフザチブ(100 mgまたは200 mg)における薬物曝露状況を評価する単回用量交差研究であり、各用量は食品と一緒に服用するか、または食物と一緒に服用しない。これらの研究の結果,食物はトミフゼチブの血液曝露濃度を約2倍に増加させることが確認され,食物とともに100 mgを服用したトミフザチブ曝露は,食物を服用しない200 mgに相当することが示唆された。患者を容易にするために,われわれの2 b期KickStart試験では,トミフザチブのRP 2 D投与量は100 mgとなり,BIDは食物とともに服用した。
Tomivosertib併用チェックポイント阻害剤の2 a期試験
抗PD−(L)1単一療法を開始したが反応しなかった被験者におけるトミフザチブの作用,すなわちRECISTの治療基準,すなわち進行性疾患(PD)への進展,あるいは標的腫瘍サイズの20%以上の増加,あるいは12週間の抗PD−(L)1治療を受けたが,PRやCRの証拠はない2 aCPI−A期試験を行った。この試験では,異なる腫瘍タイプを有し,これまでにFDAが承認した抗PD−(L)1療法の適応がある39名の患者を募集した。これらの患者のうち,17名の抗PD−(L)1治療を受けた患者のうち17名の原発癌型の非小細胞肺癌と進行転移があり,そのうち16名はトミフザ置換に加入する前にRECISTのPD基準に適合していた。我々のレジメンによれば、被験体は、彼らのバッグ挿入に従って抗PD-(L)1治療を継続し、治療レジメンを中断せず、次いで、彼らの次の所定の抗PD-(L)1治療7日前の絶食7日前にトミフセテ200 mg、2回/dの投与を開始した。この試験の主な目標は安全性と抗腫瘍活性を評価し,PFSとORRで測定することである。全体的に言えば、トミフセテと抗PD-(L)1の併用治療の全体的な耐性は良好であった。発生した副作用はトミフセチブと抗PD-(L)1単剤治療のAE値とほぼ一致した。最もよく見られる副作用は吐き気、無力、振戦、嘔吐、及びアスパラギン酸トランスアミナーゼとアラニンアミノトランスフェラーゼの上昇であり、この2種類の代謝酵素の血液中のレベルは肝機能の測定基準として追跡されている。これらのAEsの重症度は一般に1級または2級である。
2 a段階研究では、87%(39人中34人)の被験者がトミフザ置換に関連する可能性のある有害事象を経験した。最もよく見られる有害事象は>20%の対象で発生し、トミソスタットに関連する有害事象は、吐き気、16(41.0%)対象、15(38.5%)対象振戦、11(28.2%)対象疲労、9(23.1%)対象嘔吐を含む。2 a段階研究では,28%の対象者がトミフサーチリングに関与する可能性のある3段階有害事象を経験した。トミソス置換に関する3級以上の特定不良は発生しなかった。2名の患者は、アラニントランスアミナーゼの上昇、クレアチンホスホキナーゼ(1種の代謝酵素、その血液レベルが筋肉組織に対する薬物作用の潜在的な指標と評価されている)の増加、および皮疹の3級有害事象を経験した。
合計登録された39名の患者のうち,3名(7.7%)がRECIST 1.1基準によりPRを確認したり,腫瘍評価が30%以上減少したりした。確定診断されたPR患者のうち、2人の患者は非小細胞肺癌を有し、1人の患者は腎臓癌を患っている。また,胃癌患者として登録されている患者は1名のみであり,トミフザチブを加えたところ,標的病変は66%減少した。腎癌として登録された5名中1名(20%)がPRと診断された。17名の非小細胞肺癌患者のうち,トミフザ置換は,以前抗PD−(L)1治療を行っていた患者のMPFSを大幅に延長した。また,2020年9月までに研究が完了し,17名の非小細胞肺癌患者のうち2名(12%)にPRが確認され,そのうちの1名は研究完了後も確認されたCRを獲得し続け,3名目の患者は腫瘍消退28%であった。NSCLC患者は研究開始前に多くの治療を受けており,中央値はこれまでの2つの治療,16
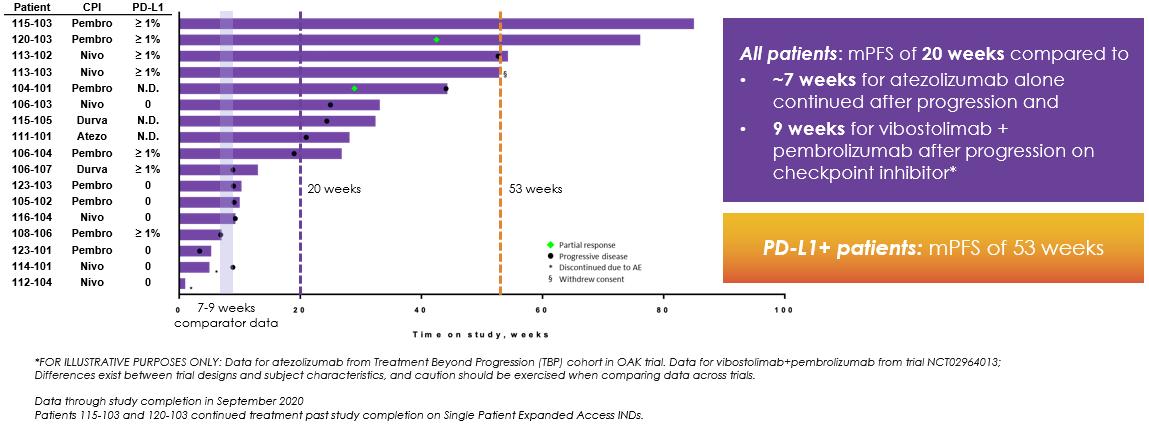
14
非小細胞肺癌患者17名(94%)では,トミフザ置換を加える前にPD−(L)1がRECIST進展し,もう1名の腫瘍サイズが13%増加し,RECIST進展に分類されなかった。17例の非小細胞肺癌患者のMPFSは20週間であった(以下図11参照)。また,PD−L 1発現レベルが1%(PD−L 1>1%)より大きいことが知られている患者のMPFSは53週であり,免疫反応性腫瘍であることが示唆された。われわれのデータベースから,患者の特徴はPd−L 1=0,Pd−L 1>1%あるいはPd−L 1未知である。対照的に,FDAによるPD−L 1承認をきたした阻害剤であるアゾールズマブを二線プラスとして非小細胞肺癌患者を治療した3期OAK試験では,初期RECISTを超えて進展しアトゾールモノクロナル抗体治療を継続した患者(n=168)の平均利益期間は約7週間であった。そのため、非小細胞肺癌患者にトミフザチブを添加した後に観察された利点は歴史対照患者のほぼ3倍であった。また,もう一つの免疫抑制チェックポイント蛋白TIGITに対するVibostolimabをPembrolizumabを服用した患者のPembrolizumabに加えたところ,9週間のMPFSが認められた。しかし,トミフザチブはこれらの薬剤との対面臨床試験では検討されていないため,研究案,条件,患者群の違いにより,これらのデータを直接比較することはできない可能性がある。
図11.水泳者のグラフは,非小細胞肺癌患者のトミソスタット抗PD−(L)1併用治療の時間長を示している。
TomivosertibのP 2 A試験における活性の重要な一点は,Tomivosertibを添加する前に抗PD−(L)1治療を行った患者の腫瘍成長軌跡を変化させる能力があることである。以下の図12に示すように,多くの患者がトミフザチブ添加後に標的腫瘍病変が安定しているか消退しており,17名中9名(53%)が少なくとも6カ月のPFS延長を経験しており,一般に臨床的意義があると考えられている。
15
図12:クモ図はトミフザチブに抗PD(L)1単剤治療を加えた標的腫瘍病変の軌跡を示している。
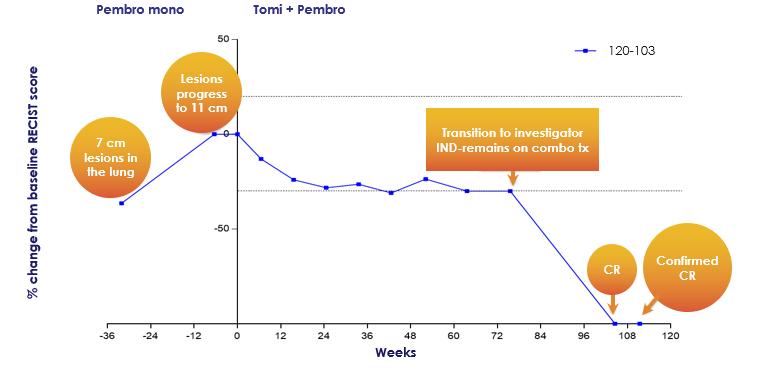
以下の図13に示すように,われわれのNSCLC患者の1人は,われわれのP 2 A試験で約80週間の確認PRを経験し,試験終了前であった。われわれの試験終了後,この患者はトミフザチブとペブロリズマブの使用を継続し,研究者が後援した同情的使用レジメンの下で併用治療を行い,その後確認されたCRを経験したことは,合計約24カ月の併用治療後,2回のスキャンで完全な緩解が確認されたことを意味している。本患者のPD-L 1>50%であった。
[図13]併用治療2年後、トミフザチブおよびペブロリズマブの併用治療が完全に緩和された患者の腫瘍軌跡。
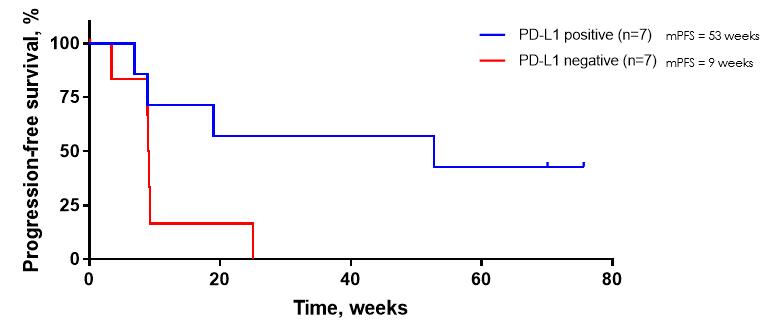
われわれの2 a期試験では,腫瘍がバイオマーカーPD−L 1を発現することが知られている患者は,腫瘍がPD−L 1発現を有さないことが知られている患者に対して優先的な治療反応を示した。非小細胞肺癌患者の抗PD-(L)1治療感受性に対するPD-L 1状態の影響を評価するため、著者らはPD-L 1陽性患者とPD-L 1陰性患者に対してカプラン-マイヤー(KM)分析を行った。17人の患者のうち14人の患者のPD-L 1状態が利用可能であった。KM分析では,PD−L 1陰性患者のMPFS 9週と比較して,PD−L 1陽性患者のMPFS 53週のトミフザチブのメリットが最も高く,この値は他と最も高かった
16
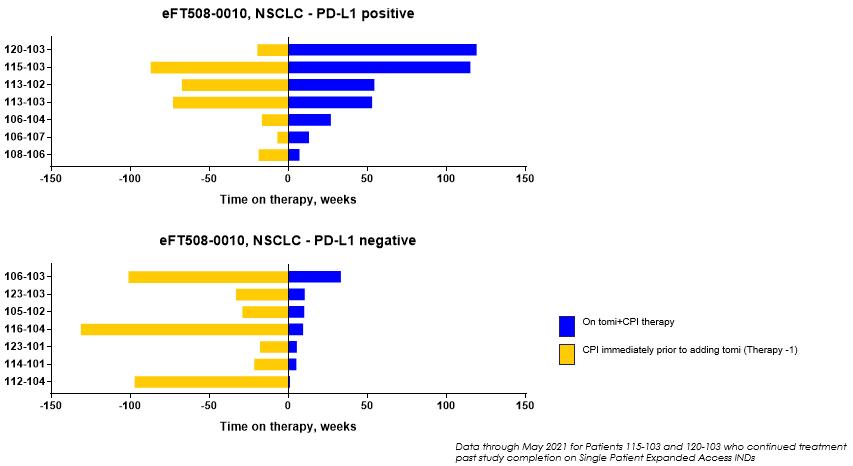
進行後に抗PD−(L)1治療のみを受けた患者の試験(以下図14 a参照)。この相関はトミオスタットのT細胞枯渇逆転と免疫系の再活性化の機序と一致していると考えられ,PD−L 1陰性患者はわれわれの2 b期KickStart試験から除外した。Tomivosertib治療に対するPD−L 1状態の影響とは逆に,Atezolizumab単剤治療のOAK研究の超越進展行列では,PD−L 1状態は治療効果とは無関係であり,われわれの2 b期KickStart試験では,PD−L 1陰性患者の排除はプラセボ+臭化リズマブと比較してTomivosertib+Pembrolizumab群の治療効果を有意に増強する可能性が示唆された。PD−L 1陽性患者のトミフザチブ利得濃度はさらに竜巻図に示され,トミフザチブ添加後の治療時間は,それに続く抗PD−(L)1治療時間と比較して示されている。PD−L 1陽性患者ではトミフザ置換添加後の治療時間は以前の治療時間と同等かそれ以上であったが,PD−L 1陰性の患者ではトミフザ置換添加後の治療時間は前の治療時間よりも短かった(以下図14 b参照)。
図14 a:われわれのP 2 A試験において,PD−L 1陽性とPD−L 1陰性患者との差を示すKaplan Meier曲線。
17
図14 b:われわれのP 2 A試験において,竜巻グラフはPD−L 1陽性とPD−L 1陰性患者の差を示している。
KickStart−PD−L 1評価のランダム2 b期実験≥50%
著者らは現在転移性NSCLC患者のKickStartを評価しており、これはランダム、二重盲検、プラセボ対照の2 b期臨床試験であり、以下の図15に示す。著者らはこの試験を行い、第一線の臭化リズマブによるPD-L 1 50%の非小細胞肺癌患者の治療にトミフセチブを加えた治療効果と安全性を評価している。KickStart試験では,これまで抗PD−(L)1治療を受けていなかった患者を含め,転移性非小細胞肺癌治療を受けたことのない患者を含め,無作為に1:1に分けてトミフザ置換と臭化リズマブまたはプラセボ+臭化リズマブの治療を受けた。
KickStart実験では54名の患者を募集し,最初の目標は60人,主な終点はPFSであった。また、OS、セキュリティ、およびORRはサブエンドポイントとして評価される。私たちは2024年4月初めにバックラインデータを報告する予定だ。この分析は37個のPFSイベントの後に行われ,これらのイベントは79%の電力を提供して危険比0.65のpを観察する
図15:非小細胞肺癌一次治療適応で行われているKickStart P 2 b試験模式図。
18
SU 2 C乳癌試験
マギル大学Nahum Sonenberg博士が率いる研究では、Tomivosertibは転移性乳癌合併化学療法の2 a期単腕臨床試験でもテストを行った。私たちはこの実験にトミソサチブカプセルを提供しました。他のすべての費用はカナダ抗癌協会(SU 2 C)の寄付によって全額援助されました。この組織は最初に40人もの転移性乳癌患者を募集する予定であり、これらの患者は承認され、利用可能な治療方法は癌をコントロールする上で無効である。TomivosertibはパクリタキセルやNaB−パクリタキセルと併用した。この試験の主な目的は,単一療法とパクリタキセルとの併用としてのトミフザキの安全性と耐性を評価し,トミソ治療の生物活性指標としての薬効学的効果を評価することである。このグループは19名の患者のみを募集することを選択し,調査者は積極的な薬効学的データを報告するのに十分であると考えている。
西北大学の研究者による急性骨髄性白血病患者の試験
2023年、著者らは、再発/難治性急性骨髄性白血病(AML)患者におけるトミソチブの使用を評価するために、研究者によって開始された第1段階用量漸増試験を開始することを発表した。この試験は西北大学ロバート·H·ルリ総合癌センターで行われ,医学助教授,医学博士ヒラ·ダナーが司会を務める。レオニダス·プラタニヤス医学博士、取締役会社、ロバート·H·ルーリー総合癌センター医学(血液学と腫瘍学)及び生化学と分子遺伝学教授及び医学(血液学と腫瘍学)医学教授ジェシカ·ウルトラマンが試験の共同議長を務める。この試験は,先に発表された結果を利用することを目的としており,これらの結果はトミフザ置換が急性骨髄性白血病モデルにおいて臨床前活性を有することを示している。トミフサーブのAMLへの適切な用量が決定されると、研究者はトミソと万カーネーションとアゾシチジンの併用を測定するために、試験範囲を拡大することを望んでいる。
固形腫瘍におけるTomivosertibの追加探索実験
我々のCPI−AとKickStart実験の前に,Tomivosertibはいくつかの追加的な2 a期試験で評価を行った。2019年、著者らはファイザーとメルクKGaAとの臨床試験と供給プロトコルを通じて、マイクロサテライト安定型結腸直腸癌(MSS CRC)患者におけるPD-L 1阻害剤Avelumabの連合試験を完成した。多発性硬化症結腸直腸癌は免疫学薬物に対して一般的に反応しない。この実験では,最初の10名の患者を含む55名の患者を募集し,トミフザチ連合Avelumabの標準看護用量,最初にTomivosertibを単一治療として受けた15名の患者,TomivosertibとAvelumabの組み合わせの交差使用,TomivosertibとAvelumabの組み合わせを受けた30名の患者を認めた。トミフザチブとアビルモノクロナルの併用耐性は良好であり、投与量は200 mg、1日2回、絶食し、RP 2 Dとした。確定診断されたPRを1例観察し,この典型的な免疫難治性患者群では,25%の患者が12週間を超えて研究を継続した。しかし,Tomivosertibの作用機序に基づいて,Tomivosertibの将来開発をより免疫反応性のある癌に重点を置くことを選択した。この試験に先立ち,16名の患者を抗去勢前立腺癌(CRPC)の単一治療試験に参加させた。このCRPC試験ではPRやCRSは認められず,16名中7名(44%)にSDが認められた。活性が限られているため,この試験を中止し,検査点阻害剤のさらなる開発に重点を置いた。
ゾチフェンは高効率で選択的なeIF 4 Aヘリカーゼ阻害剤です
ゾタファンの概要
我々が臨床開発した第2の候補製品であるゾチフェンはeIF 4 Aの小分子阻害剤であり、eIF 4 F複合体のサブユニットであり、細胞増殖蛋白の翻訳を調節し、これは癌の発癌駆動因子である可能性がある。EIF 4 Aは1種のヘリカーゼであり、SELECT mRNA 5‘端非コード領域中の複雑なmRNA二次構造を解明し、有効なリボソーム結合を許可し、その後mRNAを重要なタンパク質に翻訳する。ゾチフェンは多種の癌蛋白を下方制御することを目的とし、その中のいくつかの癌蛋白の上昇は特徴の良好なフィードバック経路の一部であり、特定の標的治療に対する薬剤耐性を招く。著者らの臨床前実験により、ゾタフェンは単一薬物として作用を発揮することもできるし、いくつかの標的治療と結合して薬剤耐性を防止することもでき、いくつかの乳癌腫瘍タイプとKRAS突然変異のNSCLCなどの重要な適応を含む。私たちはこの試験の初期用量増加部分を完成し、現在ゾタフェンが評価されています
19
ER+乳癌患者のためのフォービスターおよびアベシル(“ZfA三聯体”)の併用は、2 a期開放ラベル拡張キューで使用された。1/2期臨床試験で観察された良好な安全性結果とこれまでに産生された標的接触データを考慮して、著者らはゾチフェンとフルビセトロンの併用によるER+乳癌患者への投与量の増加を回復し、将来の臨床研究においてより高用量のゾチフェンを使用できるかどうかを決定した。2024年第1四半期、ZF二聯体の用量アップグレードが終了し、ZF二結合体のRP 2 Dとして0.2 mg/kgゾタフェンQ 2 Wを確定した。
これまでにER+乳癌患者を含む5つのキューのデータが報告されており,これらのデータはゾタフェンの通常耐性が良好であることを示し,大量に前処理されたER+乳癌患者の部分反応を含む活性シグナルを示している。われわれは6月の米国臨床腫瘍学会(ASCO)2023年年次総会でER+乳癌にZfA三聯体行列を完全に組み入れたTOPLINE結果を報告し,2023年5月3日までにZfA三連体治療を受けた評価可能患者19名中5名(26%)に部分緩解を認め,確定診断された部分緩解4名と未確認部分緩解1例を含めた。また,初めて回復したZF二重用量漸増行列では,3名中1名(33%)に部分反応が認められた。ZfAとZFの組み合わせは全体的に耐性が良好で、多くの有害事象は1級または2級である。12月に開催された2023年サンアントニオ乳癌シンポジウム(SABCS)でZfA三胞胎行列の成熟データを報告し、2023年11月17日までの締め切り、中位PFSは7.4月である。
市場のチャンス
国立癌研究所では,2022年に米国で28.7万例を超える浸潤性乳癌と23.7万例を超える肺癌新例が発生したと推定されている。ER+乳癌はすべての乳癌の約60%或いはそれ以上を占め、非小細胞肺癌は最もよく見られる肺癌亜型であり、すべての肺癌診断の82%を占める。KRAS突然変異肺癌は非小細胞肺癌の約25%を占めると推定されている。転移性ER+乳癌では,現在患者はエストロゲン受容体(ER)やCDK 4/6の阻害剤を用いて治療を行うのが一般的であるが,多くの患者は最終的に進歩する。したがって,治療法を改善する必要がある。KRAS突然変異肺癌において、KRAS G 12 C突然変異亜型に対する選択性阻害剤は、すでに2種類の薬物がFDAの許可を得たが、薬剤耐性が出現し、この薬物は他のKRAS突然変異亜型、例えばG 12 A、G 12 D或いはG 12 Vに対して無効である。
著者らは現在ER+転移性乳癌におけるゾタフェンの研究進展に注目している。われわれの臨床前研究によると,ゾチフェンとER阻害剤(例えばfulvestrant)の組み合わせでER+乳癌患者を治療できる可能性が考えられ,米国では年間約4.2万人の患者がいる。fulvestrantは生体模倣薬であり,アスリコンを含むいくつかの会社で販売されており,後者はFaslodexのブランドで販売されており,乳癌治療に用いられている。また,ゾチフェン,フルビストロング,CDK 4/6阻害剤の三重組み合わせがより良い活性を提供する可能性が考えられる。現在市販されているCDK 4/6阻害剤には,アベシル(Verenzio),Palbociclib(Ibrance),リボリボソーム(KISSALI)がある。また,ゾタフェンは乳癌の他の亜群でもFGFR+とHER 2+亜群を含めて活発であると考えられた。FGFR 2とHER 2はRTKであり,癌を推進する重要なタンパク質である。現在市場にはHER 2、EGFRとFGFRの阻害剤などの多種のRTKの阻害剤がある。しかしながら、RTKおよび/または下流エフェクタータンパク質の産生を変異または上方制御することによって、単一のRTK阻害剤に対する緊急耐性を産生することは依然として挑戦である。臨床前データに基づいて、ゾタチフェンはあるER+乳癌に対して単一の薬物活性を有し、これらの乳癌もHER 2或いはFGFR突然変異が存在し、著者らは全部で約17,000名の患者がいると推定した。
NSCLCでは、著者らはKRAS G 12 Cを抑制する薬物のような単一薬物としてZotatifin KRAS変異NSCLCを開発することを計画している。NSCLC患者の約25%はKRAS活性化変異を発生し、これらの患者を治療するために使用できる薬物は限られている。KRASは多種の活性化突然変異亜型があり、G 12 A、G 12 C、G 12 DとG 12 Vを含み、著者らはアメリカに2.8万名のKRAS突然変異のNSCLC患者がいると推定した。
TNBCにおいて、ベラー医学院が行った臨床前研究により、ゾタフェンはSox 4とFGFR 1の産生を抑制し、インターフェロン関連経路の誘導と腫瘍免疫微小環境の再構築を招き、それによって細胞増殖を抑制することが示唆された。また、ゾチフェンはカルボプラチン(TNBC治療のための化学療法薬)と協同作用し、TNBCの腫瘍進展を抑制することができる。この研究はTNBCにおけるゾチフェンの応用を評価するために重要な理論的根拠を提供し、これは高度に満足されていない需要適応である。
20
ゾチフェンの発明の概要−天然産物を設計とした永続化学的取り組み−
元素.元素
その結果、ゾタフェンが臨床候補薬物として決定された発見過程は、シビスターとログラムA(“ROC A”)で発見された1つの核心薬効団から始まり、この2種類の天然製品は面白い生物活性を示したが、ある類似薬物の特性が不足していた。我々は既存の情報に対して、eIF 4 AとRNAに結合したROC Aの結晶構造を含む複雑かつ全面的な計算分析を行った。さらに、変異解析を使用して、eIF 4 Aにおける結合に必要な重要なアミノ酸、および我々の初期プログラム化合物間の構造-活性関係を決定して、コア薬効団におけるいくつかの置換基の優先配向を決定する。これらの知見は,合成および試験をeIF 4 Aと強い親和性を保持する高い機会の化合物に制限するために有効な過程を行うことができるようにした。これにより,成熟したプログラム化合物類似薬物の特性を与える持続的な発見計画に資源を集中させることができる。
ゾチフェンの作用機序:一丸中で多種の原因蛋白を下げる
EIF 4 AはeIF 4 F複合体の触媒サブユニットであり、それは細胞増殖蛋白の翻訳を調節し、細胞増殖蛋白は癌の発癌駆動要素である可能性がある。EIF 4 Aは1種のヘリカーゼであり、SELECT遺伝子5‘非コード領域中の複雑な二次構造を解くことを担当する。この解離は1つの調節制御ステップであり、重要な蛋白質の有効な産生を招き、正常細胞が成長シグナルに反応し、腫瘍細胞の中で上昇することができる。
以下の図16に示すように、eIF 4 Aは2つの重要な細胞成長と増殖経路PI 3 K-AKTとRAS-MEK経路の交差点の中心ノードに位置し、選択メッセンジャーmRNAの蛋白質への翻訳を活性化し、蛋白質は重要な疾病駆動過程中によく発生する張本人である。EIF 4 Aは多種の成長関連蛋白の産生を調節し、細胞の成長、増殖と生存に参与する。これらの蛋白の多くは発癌の駆動因子であり,癌ではよく上昇する。腫瘍細胞におけるeIF 4 Aによって制御されるタンパク質は、
(1)FDAはER、HER 2、FGFRとKRAS G 12 Cなどの標的治療の多種の癌蛋白を許可した
(2)MYCとCyclin D 1などの標的治療方法の癌タンパク質はまだない;
(3)薬剤耐性機序としての標的治療は、Cyclin D 1、CDK 4/6、RTKとKRASなどのいくつかのタンパク質をよく上方制御する。
これらの癌蛋白のいくつかは垂直シグナル経路で協同作用し、腫瘍の成長、増殖と癌の生存を推進することができ、ある乳癌と非小細胞肺癌を含む。1つまたは複数の垂直シグナル経路および/または1組の相乗経路内の複数の癌タンパク質を同時に阻害することは、普及可能な癌治療方法として公認されている。ゾチフェンは同一の垂直シグナル経路中の複数の癌蛋白、および一連の協同作用の経路を同時に調節するため、これは単一の薬物活性を招く可能性がある。また,同一経路に作用する別の標的治療と組み合わせることにより,ゾチフェンは複数の発癌駆動因子の抑制を増強し,ゾチフェンや補充薬物に対する反応を深めたり拡大したりする可能性がある。これらの標的治療に対する耐性は、標的蛋白及び他にもeIF 4 Aによって制御される経路蛋白を上昇させることによって発生することができる。
21
図16:eIF 4 Aは1つの重要なノードであり、複数のRTKとKRASによって活性化され、多くの癌駆動蛋白の産生を制御している。
以下の図17に示すように,ゾチフェンによって阻害されたタンパク質の多くは,mRNAの5‘UTRにおいて共通の異なる翻訳開始制御エレメントを有しており,これらの要素はゾチフェンが認識していることが分かった。これらの共通の調節エレメントは多くの癌を引き起こす重要な癌蛋白のメッセンジャーリボ核酸に存在する。また,これらの蛋白の多くは標的治療に過剰発現し,薬剤耐性を招いている。重要なことは,われわれの臨床前データは,体外生理濃度のゾタフェン抑制が細胞中の約5%のmRNAの翻訳にしか影響しないことを示しており,これらの濃度では全世界のタンパク質合成が影響を受けていないことを示している。また,これらの翻訳開始制御エレメントは,タンパク質合成に含まれるアミノ酸のコード配列を決定する前のmRNAに位置し,コード配列とは独立しているため,それらの阻害はタンパク質変異変異体とは独立している。例えば、臨床前研究において、ゾタチフェンはその共通の翻訳起動調節要素のため、多種の活性化突然変異亜型、例えばG 12 C、G 12 VとG 12 DにおけるKRASの産生を抑制する。
22
図17:ゾチフェンは腫瘍成長および耐性を駆動するタンパク質に対して選択的である。
ゾタフェンの臨床前研究
臨床前の実験により、2つ以上の発ガン駆動要素を選択するモデルの中で、ゾチフェンが最も活発であり、これらの発癌駆動要素は直接ゾチフェンによって下方制御された。また、臨床前モデルも、ゾタフェンと同じ垂直経路における特定のタンパク質に対する薬剤(HER 2、FGFR、AKTまたはPI 3 Kの阻害剤)や、これらの経路によって活性化される標的(例えばERおよびCDK 4/6)に対する薬剤と併用した場合の腫瘍増殖阻害活性を示している。現在のところ、Cyclin D 1やMYCに直接標的となる薬剤はなく、これらのタンパク質によって駆動される癌の魅力的な候補薬である可能性が信じられる。
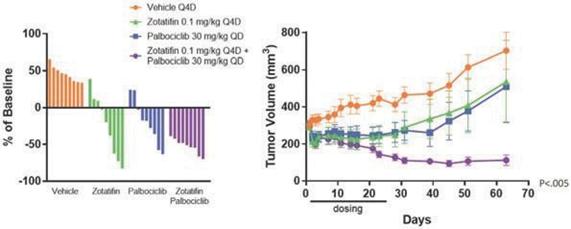
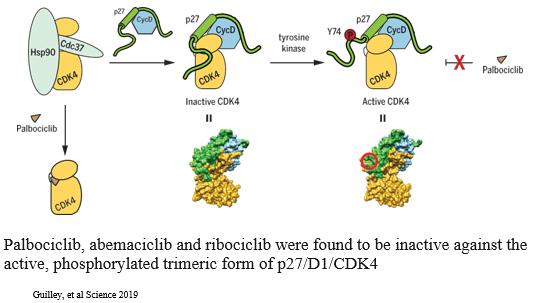
著者らは、ゾチフェンはER、Cyclin D 1、CDK 4/6 FGFRとHER 2を含む特定の乳癌腫瘍タイプに関連する多種の疾病駆動蛋白を下方制御する可能性があり、これはこれらの腫瘍タイプの患者に重要な治療選択を提供する可能性があると考えている。著者らの臨床前研究において、ゾタチフェンはいくつかの乳癌マウスモデルにおいて治療効果を示した。例えば,乳癌モデルのMDA−MB−361,ER+HER 2+PIK 3 CA変異体では,ゾチフェンやCDK 4/6の阻害剤Palbociclibで治療した結果,類似した効果であった。興味深いことに,ゾタフェンとパポシリ併用治療は強い連合作用を示し,併用投与中止後に腫瘍消退は40日以上持続した(以下図18 a参照)。多種の臨床前モデルにおいて、ゾタチフェンは細胞周期蛋白D 1の発現を低下させる。CDK 4、Cyclin D 1とp 27は細胞分裂を促進する活性三量体複合体を形成でき、CDK 4/6阻害剤に対して無効であり、遊離Cyclin D 1を上昇させることはCDK 4/6抑制に対する抵抗力を増強できる。そのため、著者らはゾタチフェンとCDK 4/6阻害剤の併用はER+転移性乳癌患者の有望な治療選択になる可能性があると信じている(以下図18 b参照)。
図18 a:乳癌の臨床前モデルにおいて、ゾタチフェンはパポシリに相当する単剤活性を示し、パポシリーと併用し、納得できる腫瘍消退を示した。
23
図18 b:Zotatifin下方制御Cyclin D 1はp 27/D 1/CDK 4三量体の形成に拮抗することが期待される。
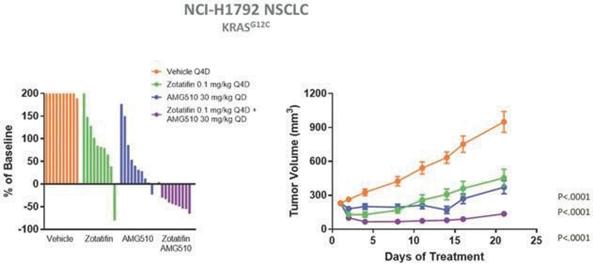
評価された約100個の細胞株において、ゾタチフェン治療は、KRAS変異を活性化する多くの細胞株のアポトーシスをもたらす。また,細胞増殖とアポトーシス解析では,ゾタテフェンと安進社製KRAS G 12 C阻害剤AMG 510やsotorasibの併用が強い活性を示し,最近規制部門の承認を得た。著者らは、ゾタチフェンはCyclin D 1とあるRTKを下方制御し、一からのKRAS蛋白の産生を抑制することによって、KRAS G 12 C阻害剤の薬剤耐性機序を克服する可能性があると考えられる。著者らの臨床前研究により、NCI-H 1792 KRAS G 12 C変異型NSCLCモデルにおいて、ゾタフェンはAMG 510と類似した腫瘍成長抑制活性を有することが示唆された。この臨床前データはまた、ゾタフェンとAMG 510の組み合わせにより、ほとんどの被験動物の腫瘍が消退することを示している(以下図19参照)。KRAS突然変異腫瘍の臨床前モデルにおいて、ゾタチフェンはKRAS、Cyclin D 1といくつかのRTKの発現を低下させる。これらの蛋白の上昇はすでにKRAS抑制に対する抵抗力を高めることが証明されたため、著者らはゾタフェンとKRAS阻害剤の結合はKRASG 12 C非小細胞肺癌患者の有望な治療選択になる可能性があると信じている。
図19:KRASG 12 C非小細胞肺癌の臨床前モデルにおいて、ゾタテフェンはKRAS G 12 C阻害剤AMG 510に相当する単剤活性を示し、AMG 510と併用し、腫瘍消失率を有意に低下させた。
24
1/2期臨床試験観察と計画
著者らはゾチフェンに対して1/2期臨床試験を行い、ある固形腫瘍患者に応用している。著者らは、この試験中に最初に計画された用量増加部分と、ゾタフェンとフルビストランとアベシル(“ZfA三連”)の併用によるER+乳癌患者の治療効果の評価を含むいくつかの適応の初期第2段階拡張部分を完成した。試験第1段階の主な目標は、安全性の評価と静脈投与のRP 2 Dゾタフェン(“IV”)の選択を含む。試験の第一段階の用量増加部分で、著者らは37名の患者を募集し、投与量は毎週0.005 mg/kgから毎週0.1 mg/kgの静脈注射まで、治療2週間後に1週間中止する改善方案を含む。投与量増加の過程で、2級血小板減少のDLTを含む3つのDLTが観察され、毎週0.035 mg/kg静注キューで観察されたDLTウィンドウ全体の治療の完了を阻止し、2つのDLTは毎週0.1 mg/kg静注後2週間と1週間後に観察された。1人の患者は3級貧血のDLTを経験し,もう1人の患者は2級血小板減少下で3級胃腸出血を経験した。このデータから,0.1 mg/kg用量がMTDを超えていると結論した。すべての用量レベルの全体的な副作用は主にレベル1とレベル2の吐き気、嘔吐と貧血を含む。ゾタフェンは投与量の割合の薬物動態学的暴露を示し、半減期は比較的に長く、約4日であった。投与量が0.035 mg/kg以上の時、ゾタフェンのヒト血液中の暴露用量はマウス研究における臨床前活動のレベルと同等である。
2021年6月,われわれの1/2期臨床試験の1期用量増加分のデータ評価に基づき,RP 2 Dとして21日周期の1日目と8日目に0.07 mg/kgを投与することを選択した。
われわれは,2 a期適応特定拡張キューにおいてゾタフェンの臨床評価を開始した。2 a期キューの主な目標は、セキュリティをさらに決定し、バイオマーカー固有の患者集団において最初の治療効果シグナルを決定することである。4つの拡張キューを立ち上げました3つはMBC、1つはNSCLCです具体的には,ER+/FGFR+MBCとしてのゾタフェンを評価する単一療法,FDA承認されたER阻害剤fulvestrantとのER+MBCにおける併用療法,FDA承認CDK 4/6阻害剤abemaciclibとのER+/HER 2−MBCにおける併用治療,およびFDAが承認したKRAS阻害剤sotorasibとのKRAS G 12 C NSCLCにおける併用治療の列を開始した。我々の2 a期拡張キューは,サイモンの2段階設計に従って構造設計されており,そのうち7名の患者が第1段階試験に登録され,第2段階試験に入る前に活動評価が行われる。
2022年6月、進行中の2 a段階拡張キューの4つの中間データを報告しました。2022年3月4日までの締め切りで、中期結果は、ゾタフェンは0.07 kg/ミリグラム用量で全体的な耐性が良好であり、ゾタフェンに関連する緊急副作用(“TEAE”)の多くは軽微で、コントロールしやすく、可逆的であり、疲労、貧血、下痢、嘔吐と吐き気を含むことを示した。推奨された第2段階用量を受けた25名の患者では,ゾタテフェンに関連するレベル3,レベル4またはレベル5 TEAEは出現しなかった。ER+乳癌コホート評価ゾタフェンとフルビスター(“ZF Doublet”)では,部分緩解7例中1例と安定した7例中3例を報告し,患者総数を7人から18人に拡大した。ER+/FGFR増幅乳癌コホートでは,ゾタフェンを単一療法として評価し,7名に1名の患者が安定しており,コホートの規模を拡大するには不十分であった。KRAS G 12 C非小細胞肺癌行列中でゾタフェンとソトラシブを評価し、ある患者は一時データ遮断日に登録し、そして病状が安定していることを報告した。ZfA三連結体を評価するER+乳癌キューでは、2人の患者が中間データ遮断日に登録され、そのうちの1人は部分的に緩和したと報告されている。
2023年1月、私たちはZF双子とZfA三胞胎キューの最新の中期データを報告した。2022年12月15日までの締め切りまで、ゾタチフェンは引き続き全体的な耐性が良好であり、ZF二連群(n=18)中に4つの3級+TEAEがあり、ZfA三連群(n=7)は4つの3+TEAEがあった。ZF二結合体キューでは部分緩解1例と長期安定患者1例,ZfA三聯体キューでは部分応答2例と長期安定患者1例を報告した。
2023年6月,完全に組み込まれた拡大コホート患者(n=20)に関する新たな中期データが公表され,これらの患者は21日周期の1日目と8日目にZfA三連体治療を受け,用量は0.07 mg/kgであった。厳重な予備治療を受け,4種類の転移性疾患治療の中央値を受けた。2023年5月3日までの締め切りで,RECIST評価を受けた19名中5名(26%)が部分寛解(PR)であり,確定診断患者4名と未確認患者1名を含む。PRを受けた5人のすべての患者は以前にCDK 4/6およびFulvestrant治療を受けており、5人の患者全員が1つ以上の以前の化学療法を受けたことがある。CDK 4/6、内分泌および/または化学療法後、これらの大量の前治療を受けた患者の治療効果の結果は、fulvestrantとabemaciclib(“FA Doublet”)に対する期待を超えていた。ZfA三胞胎の全体的な耐性は良好であり、3名の患者は任意の原因の不良事件(AEs)のため使用を中止し、大多数のAEsは1級或いは2級である。
25
2023年12月、私たちはサンアントニオ乳癌シンポジウムでER転移性乳癌患者におけるゾタフェンの用量増加および2期拡張キューの新しい積極的な中期データを発表した(図20参照)。ZfA三連群では,転移性疾患の治療中央値が4本の線の患者は21日周期の1日目と8日目に0.07 mg/kgのゾタフェン治療を受け,フルビストロングとアベルミリブと併用した。このキューでは,2023年11月17日までの締め切りで,中位無進展生存期間(MPFS)は7.4カ月(95%信頼区間は2.8から計り知れない)であった。
図20:水泳者のグラフは,ゾタファンガフォヴィストとアベシル(ZfA三聯体)の服薬時間を示している。
1/2期臨床試験で観察された良好な安全性結果とこれまでに産生された標的参加データを考慮して、著者らは1週間ごとに投与するゾタフェン投与量の増加を回復し、最初はエストロゲン受容体陽性乳癌患者のZF二重結合において、未来の臨床研究においてより高用量のゾチフェンを使用できるかどうかを確定した。その後、ZfA三胞胎で投与量の増加を開始した。2024年第1四半期、ZF二聯体の用量アップグレードが終了し、0.2 mg/kgゾタフェンQ 2 Wを二結合体のRP 2 Dとして確定した。私たちは2024年上半期に用量上昇に関するより多くのデータを報告する予定だ。
ゾチフェンが十分な安全性と十分な活性シグナルを示し続けていれば,関連対照対照群との無作為試験の組み合わせとなる可能性があるゾチフェンの臨床開発を継続する予定である。我々は現在,2024年上半期に投与量とスケジュールを決定した後,無作為試験でZfA三連体の計画をテストしている。2023年に受信した高速車線称号を用いてFDAと発展戦略について相互作用する予定である。
スタンフォード大学の研究者によるER陽性乳癌患者の無作為二期臨床試験
スタンフォード大学はER+乳癌患者に対する二期臨床試験を賛助している。この試験はスタンフォード医学院医学アシスタント教授Jennifer Caswell-jin医学博士が指導し、スタンフォード医学院医学、遺伝学と生物医学データ科学教授Christian Curtis博士とスタンフォード医学院人工知能と癌ゲノム学取締役が行った仕事の基礎の上で、乳癌総合亜群の科学を臨床に導入する。ゾタフェンは、標準的なリスク患者と、再発を予測する特定のマーカーを有するハイリスク患者とを含む、特定のゲノム定義サブセットにおいて試験される。 これらの特にリスクの高い患者に臨床的利益を提供する可能性のある治療方法を決定するために、カストウェル-金博士は傘形、ランダムな術前試験を指導し、ER+/HER 2-乳癌の総合サブタイプ標的治療をテストする。ゾタフェンは、1組の細胞周期タンパク質D 1と線維芽細胞増殖因子3の過剰発現を含み、これは癌の成長と生存を促進する1組のタンパク質であり、1組の単独のものである、高再発リスクのある患者の中で研究する
26
線維芽細胞増殖因子受容体1の過剰発現や再発標準リスクのある患者列においても同様である。この2つのキューでは、患者は手術の14日前にランダムに単回用量のゾチファンガフビストを受けるか、またはファーヴィストのみを受けるであろう。この研究の主な目標は腫瘍増殖状態の変化を評価することであり,Ki 67染色測定によりベースラインから術前治療後14日まで,いずれのレジメンを採用してもよい。
EIF 4 E-ファイザーとのグローバルパートナー関係
2019年12月、私たちはファイザー協定に署名し、協定に基づいて、私たちは最初の段階の計画であるeIF 4 E阻害剤である世界的な許可をファイザーに授与した。EIF 4 Eは1種の癌遺伝子であり、その発現は多種のヒト癌において発現が増加或いは上昇し、不良予後とある治療薬剤耐性と関係がある。EIF 4 EはBRAF、MYC、mTOR、PI 3 K、AKTとPTENを含む多種の重要な癌遺伝子と腫瘍抑制蛋白のシグナルを統合し、そしてMNKとeIF 4 Aとは異なる標的mRNAの翻訳を選択的に調節する。これは,選択的翻訳調節療法から利益を得る可能性のある潜在的な患者集団を拡大する可能性がある。ファイザー社と協力して、INDを支援する研究に入るために、私たちの主な開発候補を選びました。ファイザー社はこの計画の更なる開発を担当し、INDの提出と一期用量逓増臨床試験の開始を含む。ファイザープロトコルの説明については、以下の“-ファイザー研究協力とeIF 4 E阻害剤許可プロトコル”を参照されたい。
Davide Ruggero博士によって行われ、2015年7月に“細胞”誌に発表された研究により、一部のmRNAはeIF 4 Eレベルの低下に敏感であることが明らかになった。EIF 4 Eの敏感なmRNAは腫瘍転化、腫瘍成長刺激とアポトーシス経路の抑制に関連する蛋白質をコードし、eIF 4 Eは魅力的な癌治療標的であることが示唆された。また,eIF 4 Eを過剰発現する腫瘍は,頭頸部扁平上皮癌,リンパ腫,乳癌を含め,重要な臨床機会を代表していると考えられる。
EIF 4 Eの天然リガンドは高度に帯電した実体であり,5‘キャップと呼ばれているため,細胞内でこのタンパク質を抑制する候補製品を決定することは歴史的に困難であった。我々の独自の構造および断片に基づく薬物設計の専門知識を利用して、5‘キャップと同じ位置に結合し、それと競合するいくつかのeIF 4 Eの小分子阻害剤を発明した。ファイザーと連合して、著者らは1種の主要な候補製品を選択し、臨床前モデルにおいてこの候補製品は有効で選択的なeIF 4 E阻害剤であることを示した。この候補薬剤は腫瘍細胞検出で活性を示し,体内で大量の抗腫瘍活性を示した。
EIF 4 Eの主要候補製品阻害剤の開発進捗を引き続き評価し、必要に応じて販売·マーケティングインフラの構築を考慮して、データの発展に伴い米国での共同普及と利益共有の選択権を行使することを支援する。
私たち独自の翻訳規範技術プラットフォーム
私たちは私たちの独自の選択的翻訳規制技術プラットフォームを使用して私たちの候補製品を発見した。私たちのプラットフォームには私たちのリボソームマップ技術と最先端の化学設計戦略が含まれています。著者らのリボソームスペクトル技術は細胞中に発現するmRNA上のリボソーム密度を全面的かつ定量的に測定することができ、それによって翻訳速度を予測し、それによって腫瘍中で上昇した標的を識別することができ、これらの標的の産生は著者らの候補製品の選択性抑制に敏感である。どのメッセンジャーリボ核酸の翻訳が私たちの候補製品によって抑制できるかに関する情報は、著者らが臨床研究の腫瘍タイプと患者集団を選択する過程における重要な部分である。リボソームは巨大分子機器であり,メッセンジャーリボ核酸中の指令に基づいてタンパク質の合成を担当する。このスペクトルは細胞或いは組織におけるmRNAの翻訳効率を評価し、遺伝子発現の転写と翻訳制御を区別し、治療標的、バイオマーカーと薬物感受性或いは薬剤耐性の背景を確定することができる。各種の正常と疾病状態での翻訳効率を測定することにより、どのタンパク質が翻訳制御されているかを決定することができ、より重要なのは、どのタンパク質が様々な疾病状態で上昇しているかである。我々はカリフォルニア大学サンフランシスコ校から許可を得た原始技術を統合し,強化し,その工業化を我々の内部薬物発見と開発に応用した。この技術の応用は、多種の腫瘍タイプにおける翻訳失調の遺伝子の固有理解を産生し、治療干与の特定点を決定することができるようにした。
27
我々の候補製品を開発するために,薬物化学に対して集中的な方法を行い,断片と構造に基づく設計技術を組み合わせた。私たちはまた原子相互作用に関する専門知識を応用し、これは強力で特異的な薬物標的相互作用に潜在力を提供した。著者らは薬物標的の選択性と有効な阻害剤を識別する方法であり、部分的には物理化学的性質のバランスと高い結合親和性に基づいている。これらの薬物設計能力を外部合成化学努力と組み合わせて,効率的かつ効率的な方法で有効かつ選択的な鉛候補製品を識別する能力を向上させた。
製造業
私たちは所有も運営もしていないし、今のところ製造施設を設立する計画もない。私たちは現在依存しており、今後の私たちの候補製品とそのコンポーネント原材料、私たちの臨床前開発と臨床試験、および将来の候補製品の任意の商業化のために、第三者メーカーに十分な数の私たちの候補製品とそのコンポーネント原材料を生産することに依存し続けることが予想される。私たちの第三者メーカーは私たちの候補製品を製造するために必要な原材料を得る責任があり、私たちはこれらの原材料が1つ以上のソースから得られると信じている。より多くの第三者製造業者が、充填、ラベル、包装、および流通研究のために使用されるだろう。この方法は,より効率的なインフラを維持することができるとともに,我々の製品開発に専門知識を集中させることができる.私たちは私たちの候補製品と関連した原材料を生産するための複数の潜在的な供給源を持っていると信じているが、私たちは現在、TomivosertibとZotatifinの異なる側面を得るために、Curia Global(旧AMRI)、Catalent、Cordenを含む単一製造業者に依存している。
商業化計画
私たちは現在、販売、マーケティング、商業製品の流通能力もなく、製品を商業化した経験もない。時間が経つにつれて、私たちは自分の商業化組織と能力を構築し、北米で任意の承認された製品を販売するつもりだ。この商業組織の規模は適度であることができ、目標は私たちの目標市場に特化した腫瘍学者が相対的に少ないと信じている。北米以外では、製薬会社と協力して、それらの商業化能力を利用して、私たちの候補製品の潜在力を最大限に発揮することができるかもしれません。
私たちの候補製品が開発段階で進展するにつれて、私たちのビジネス計画は変わるかもしれません。臨床データ,開発計画の規模,我々の目標市場の規模,商業インフラの規模および製造需要は,我々の米国,ヨーロッパ,世界の他地域の商業化戦略に影響を与える可能性がある。
私たちの協力と許可協定は
ファイザー研究協力とeIF 4 E阻害剤許可プロトコル
2019年12月,我々はeIF 4 Eを標的とした小分子を研究·開発するファイザー合意に達した。ファイザー協定によれば、私たちはファイザーに世界的な独占許可を与え、私たちのいくつかの特許、技術および材料に基づいて任意およびすべての適応に対する使用、開発、製造、商業化、または他の方法でeIF 4 Eをターゲットとした化合物または製品を開発する権利がある。ファイザー協定によると、ファイザーは私たちに選択権を与え、アメリカで損益配当の手配で共同出資と共同で単一のこのような許可製品を普及させる。このオプションは、最初の患者が臨床試験に参加すると予想される前の指定された時間前に行使することができ、この臨床試験は、上場承認を得るためにNDAをサポートすることを目的としている。
ファイザー協定によると、Efftorはファイザーとの協力の初歩的な研究を担当し、ファイザーはINDの提出とすべての臨床開発と商業化活動を含むこの資産のすべての更なる開発を担当する。ファイザーには、商業的に合理的な努力でライセンス製品を開発し、規制機関の承認を求め、ファイザーが規制の承認を得た場合に、ライセンス製品を米国や他の特定の国·地域で商業化する義務がある。共同援助と共同普及オプションを行使すれば,共同指導委員会は共同開発製品の開発計画と予算を監督し,ヘルスケア提供者への製品マーケティングプレゼンテーションの一部を担当する。
28
ファイザー協定によると、私たちはファイザーから1500万ドルの使い捨て、払い戻し不可能、貸切不可の前金を受け取った。ファイザーには、研究所で発生した費用を精算する義務があり、最高限度額は2桁の百万ドル。特定の早期開発と規制マイルストーンに達すると、ファイザーは合計8000万ドルまでの費用を支払う義務がある。他の非早期開発マイルストーンについては,ファイザーの我々に対する支払い義務は,共同出資と共同推進選択権を行使しているかどうかにかかっている:1)選択権を行使しなければ,非早期開発支払い総額は1.65億ドルに達する可能性がある,2)選択権を行使すれば,非早期開発支払い総額は7000万ドルに達する可能性がある。指定された販売マイルストーンに達した後、ファイザーには合計2.35億ドルにのぼる階段式マイルストーン支払いが義務付けられている。個々の製品に加えて、ファイザーは各ライセンス製品の年間純売上高のために数桁までの印税を支払うことも要求される。もし私たちが共同販売促進と共同援助オプションを行使すれば、印税支払いはアメリカでの販売は含まれていません。アメリカで販売されているライセンス製品の利益をファイザーと共有します。
ファイザーのすべての支払い義務が満了するまで、ファイザー協定は早期に終了しない限り有効である。米国を除いて、私たちが共同援助と共同販売促進選択権を行使すれば、与えられた国/地域の任意の許可製品に印税を支払う義務が満了し、すべての満期金額を支払った後、ファイザーのその国/地域での許可は全額、永久、取消不能、印税免除となる。便宜上、ファイザーは書面通知後にファイザーとの合意を終了することができる。他方が係争のない実質的な違約が所定の期限内に是正されていない場合、又は他方が破産に関する事件により所定の期限内に解除通知されていない場合には、いずれもファイザープロトコルを終了することができる。
カリフォルニア大学サンフランシスコ校と独占ライセンス契約を締結しました
2013年5月、カリフォルニア大学サンフランシスコ校と協定を締結し、カリフォルニア大学サンフランシスコ校が最初にカリフォルニア大学サンフランシスコ校で開発した翻訳模倣実験室技術に関連するいくつかの発明の独占特許権(“カリフォルニア大学サンフランシスコ校翻訳特徴特許権”)を、カリフォルニア大学サンフランシスコ校と共同で所有しているいくつかの特許権を含む協定を提供した。協定によると、私たちは、カリフォルニア大学サンフランシスコ校の翻訳プロファイル特許権を利用して発見および開発された製品を研究、開発、製造、販売することを許可され、私たちは許可製品と呼ばれ、カリフォルニア大学サンフランシスコ校の翻訳プロファイル特許権を利用するいくつかの許可プロセスを使用し、このような許可製品およびプロセスを再許可する。私たちの排他性は、カリフォルニア大学サンフランシスコ校が保持しているいくつかの研究権利によって制限され、“米国法典”第35編第200-212節で述べたように、米国政府の権利(あれば)によって拘束されている。この法律によれば、米国政府は、カリフォルニア大学サンフランシスコ校の翻訳プロファイルに記載された発明を、米国政府または米国政府を代表してカリフォルニア大学サンフランシスコ校の翻訳プロファイルに記載された発明を世界各地で実践することができる非排他的で譲渡不可能な支払済みライセンスを取得している可能性がある。いくつかの条件の下で、私たちは許可を得たカリフォルニア大学サンフランシスコ校の翻訳プロファイル特許権について潜在的な商業的意義を持つ特許侵害請求を提起する権利がある。
協定によると、著者らは指定された時間内に特許製品と関連するいくつかの指定発展、監督管理及び商業マイルストーンを達成するために、商業上の合理的な努力をしなければならない。協定によって私たちに与えられた権利を考慮して、私たちはカリフォルニア大学サンフランシスコ校に50,000ドルの使い捨てライセンス発行費現金を支払った。2021年7月には,ライセンス契約への合併の影響を確認するためにライセンス契約を改訂し,この合意に基づき,合併完了後,UCSFに約100万ドルの現金を一度に支払った。私たちはまた、許可製品のいくつかの臨床と規制マイルストーンが完了した後、UCSFに現金マイルストーン支払いを要求された。
私たちは今までにカリフォルニア大学サンフランシスコ校に合計40,000ドルの現金マイルストーン支払いを支払いました。残りの潜在的マイルストーン支払い総額は約375,000ドルだ。また、私たちまたは私たちの付属会社が販売している最初の2つのライセンス製品の純売上である1%未満の特許使用料をUCSFに支払うことに同意し、毎年最低15,000ドルの特許権使用料(最低支払当年に支払うべき特許権使用料を免除することができます)を支払い、場合によっては他の調整を行う。我々の各ライセンス製品又はサービスに対する印税義務は,適用許可製品又はサービスをカバーする最後のライセンス特許が満了する,すなわち2034年2月まで継続され,特許期間の調整や延長はない。
私たちが合意のいかなる実質的な条項を履行または違反することができず、カリフォルニア大学サンフランシスコ校の通知を受けた60日以内に、または私たちが債務を返済しない場合、そのような不履行または違反を是正できない場合、UCSFは合意を終了することができる。私たちは現在合意されたすべての実質的な条項を遵守している。
29
カリフォルニア大学サンフランシスコ校に60日間の書面通知を出した後に合意を終了することができ、カリフォルニア大学サンフランシスコ校に書面通知を出すことにより、特許請求、特許毎、およびカリフォルニア大学サンフランシスコ校の翻訳概況特許権を国ごとに終了することができる。事前に終了しない場合、この合意は、カリフォルニア大学サンフランシスコ校翻訳概況特許権に含まれる最長寿命特許権の満期日まで継続される。2016年5月、カリフォルニア大学サンフランシスコ校ライセンス契約の条項に基づいて、PRPS-2の癌治療を抑制する方法によって、将来の製品に対する任意の権利を放棄すると主張する特許出願に関連する特許訴訟費用の支払い義務を終了することを選択したことを示す通知を提供した。私たちがこの選択をした時、私たちはまだEfftorやUCSFにこのような製品がないことを知らなかった。
知的財産権
私たちは、内部開発でも第三者から許可を得ても、特許権を求め、維持し、擁護することを含む、当社の業務に重要なビジネス的意義を有するノウハウ、発明、改善を保護し、強化するために努力しています。私たちは私たちの主要候補製品Tomivosertibに関連した発行された特許と特許出願を持っている。私たちの政策は、米国および米国以外の司法管轄区域に当社の独自技術、発明、改善、および候補製品のための特許出願を提出することを含む、私たちの独自の地位の保護を求めることであり、これらの特許は私たちの業務の発展と実施に非常に重要です。著者らはまた著者らの独自技術と製品候補に関連する商業秘密と技術ノウハウ、持続的な革新及び許可内の機会に依存して、免疫腫瘍学とeIF 4 A阻害剤標的治療領域における著者らの独自の地位を強化し、維持した。データ独占性,市場独占性,特許期間の延長(利用可能であれば)に依存する予定である。私たちのビジネス成功は、(1)私たちの技術、発明、および改善のために特許および他の固有保護を取得し、維持すること、(2)私たちの商業秘密を秘密にすること、(3)第三者が所有する知的財産権を使用するライセンスを取得し、維持すること、(4)将来所有可能な任意の特許を含む私たちの独占権を擁護し、実行すること、および(5)効果的かつ強制的に実行可能な特許および第三者の他の独自の権利を侵害することなく運営される、私たちの商業的成功は、私たちの能力にある程度依存するだろう。
2024年2月1日現在、私たちは、MNK阻害剤(Tomivosertibを含む)、eIF 4 A mRNAヘリカーゼ阻害剤(ゾチフェンを含む)、および私たち独自の選択的翻訳調節プラットフォームの様々な用途、ならびに私たちのいくつかの独自技術、発明、改善、または他の候補製品のための許可、所有、および共同所有の特許の組み合わせを取得した。我々はまた、関連する製造プロセスおよび技術を含む、私たちの候補製品の開発および商業化に関連する多くの技術的ノウハウおよび商業秘密を有し、および/または許可している。
具体的には、私たちの特許の組み合わせは以下のシリーズを含む
30
通常の業務過程で開発および商業化しようとしている候補製品やプロセスについては、成分、使用方法、用量、処方をカバーする可能性のある場合に特許保護を求める予定です。私たちはまた製造と薬物開発過程と技術の面で特許保護を求めることができる。
発行された特許は、特許出願の提出日、特許発行日、および特許を取得した国における特許の法的期限に依存する異なる時期の保護を提供することができる。一般に、米国で出願された出願に発行された特許は、最初の有効出願日から20年の排他的権利を提供することができる。さらに、場合によっては、FDA承認された製品をカバーまたは主張する発行された米国特許の期限は、FDA規制審査期間によって実際に失われた部分期間を再取得するために延長することができ、これは、特許期限延長と呼ばれる。回復期は5年を超えてはならず,回復期を含む総特許期はFDA承認後14年を超えてはならない。米国以外の特許の期限は外国司法管轄区の法律によって異なるが,通常は最初に有効出願された日から20年である。しかしながら、特許によって提供される実際の保護は、製品によって異なり、国によって異なり、特許のタイプ、そのカバー範囲、規制に関連する延長の利用可能性、特定の国における法的救済の利用可能性、および特許の有効性および実行可能性を含む多くの要因に依存する。
私たちのような会社の特許地位は通常不確実であり、複雑な法律と事実の問題に関連している。免疫腫瘍学分野特許で許可されている特許請求の範囲については,米国では一致した政策は認められていない。関連特許法及び米国以外での解釈も確定していない。米国および他の国の特許法またはその解釈の変化は、私たちの技術または製品候補製品および私たちが許可した特許権を保護する能力を弱める可能性があり、このような知的財産権の価値に影響を与える可能性がある。特に、我々は、我々の知的財産権を侵害する製品を製造、使用、販売、提供、または輸入することを阻止する能力は、私たちの技術、発明、および改善をカバーする特許主張を成功的に獲得し、実行できるかどうかにある程度依存する。ライセンスおよび会社のすべての知的財産権について、私たちは、私たちの任意の未解決特許出願または将来提出される可能性のある任意の特許出願が特許を取得することを保証することはできません。私たちはまた、将来私たちに付与される可能性のある任意の特許が、私たちの製品、使用方法、または製品製造を保護する上で商業的に有用であることを保証することはできません。
31
また、私たちが許可した特許が発行されても、製品の商業化において私たちの技術を実践する権利は保証されません。製薬とバイオテクノロジー分野の特許と他の知的財産権は多くのリスクと不確定要素に関連して進化している。例えば、第三者は、候補製品を商業化し、私たちのノウハウを実践することを阻止するために使用されることができる阻止特許を有することができる。私たちが許可した特許と将来発表される可能性のある特許は挑戦、失効、または回避される可能性があり、これは競争相手の関連製品のマーケティングを阻止する能力を制限するか、あるいは私たちの候補製品の特許保護期間を制限するかもしれない。さらに、発行された任意の特許によって付与された権利の範囲は、同様の技術を有する競合他社のための保護または競争優位性を提供してくれない可能性がある。さらに、我々の競争相手は、同様の技術を独立して開発することができ、これらの技術は、私たちが所有しているか、または許可された範囲内の任意の発行された特許の下で付与された権利の範囲内ではない。このような理由で、私たちは私たちの候補製品の面で競争に直面するかもしれない。さらに、潜在的製品の開発、テスト、および規制審査に要する時間が長いため、任意の特定の候補製品が商業化できる前に、その製品に対する任意の特許保護は、商業化後非常に短い期間で満了または有効に維持される可能性があり、それにより、特許提供の商業的利点が減少する。
競争
生物技術と製薬業界の特徴は技術進歩が迅速で、競争が激しく、そして独自製品を高度に重視していることである。私たちは私たちの技術、知識、経験と科学資源が私たちに競争優位を提供すると信じているが、私たちは主要な製薬、専門製薬とバイオテクノロジー会社、学術機関と政府機関、そして公共と個人研究機関を含む多くの異なる源からの潜在的な競争に直面している。我々が開発と商業化に成功した任意の候補製品は,既存の療法や将来出現する可能性のある新しい療法と競争するであろう。私たちの多くの競争相手は、単独でも彼らとの協力者でも、私たちよりも多くの財務、技術、製造、マーケティング、販売、供給資源または経験を持っている。そのため、著者らの競争相手は研究開発、製造、臨床前テスト、臨床試験を行い、治療許可を獲得し、広範な市場受け入れを獲得する方面で著者らより成功する可能性があり、それによって著者らの治療方法を時代遅れ或いは競争力がない。バイオテクノロジーとバイオ製薬業界のM&A活動は、私たちの数の少ない競争相手により多くの資源を集中させる可能性がある。これらの会社はまた合格した科学と管理人員を募集と維持し、臨床試験のために臨床試験場と患者登録を確立し、著者らの計画と相互補完或いは必要な技術を獲得する方面で私たちと競争している。私たちはまた、未来の潜在的なパートナーを探して、私たちの候補製品に協力するために、これらの会社からの競争に直面しています。もし私たちの競争相手が私たちの同類製品よりも効果的で、より安全で、毒性が低く、より便利で、あるいはより安い製品を開発し、商業化すれば、私たちのビジネス機会は大きく制限されるかもしれない。私たちのすべての候補製品の成功に影響を与える重要な競争要素は、承認されれば、それらの効果、安全性、利便性、価格、模造薬とその他の競争のレベル、及び政府と他の第三者支払人が精算できるかどうかである可能性が高い。
もし我々の任意の候補製品が非小細胞肺癌または乳癌などの腫瘍学的適応で承認された場合、それらは小分子療法、生物学的製剤、細胞療法および伝統的な化学療法または任意のそのような方法の組み合わせと競合するであろう。トミフセテについては,AUM生物科学社が固形腫瘍用MNK阻害剤AAM 001を開発していることが知られている。また,FDAが承認したPD−1やPD−L 1阻害剤のいくつかの新しい組み合わせがNSCLCで開発されている。これらの薬物には,百時美施貴宝社,ジリッド科学社,グラクソ·スミスクライン社,メルク社,ノワール製薬会社,羅氏社製抗TIGITと抗LAG 3薬が含まれている。ゾチフェンについては,PIC治療会社が真核細胞翻訳開始因子(EIF)に対する臨床前小分子を開発していることが知られている。また,現在いくつかのER+乳癌治療の発売薬や候補製品はゾチフェンと競合する可能性があり,アスリカンが販売しているfulvestrant,Menariniが販売するelacestrant,アスリコンが販売するaketertib,Arvinas社が開発した経口SERDS,アスリコン,礼来,衛材株式会社,羅氏社,Zentaris製薬会社などがある。
32
政府の監督管理
アメリカ連邦、州と地方各級及びその他の国の政府当局は薬品の研究、開発、テスト、製造、品質管理、承認、ラベル、包装、貯蔵、記録保存、販売促進、広告、流通、マーケティングと輸出入などの方面に対して広範な監督管理を行った。一般的に、新薬が発売される前に、その品質、安全性、有効性を証明するデータを大量に取得し、各規制機関の特定のフォーマットに組織し、審査を提出し、監督機関の承認を得なければならない。新薬はNDAプログラムを通じてFDAの承認を得なければ,米国で合法的に発売されることができない。私たちは、任意の第三者請負業者と共に、私たちの製品および候補製品の研究または承認を求める国/地域規制機関の様々な臨床前、臨床、および商業承認要件を満たすことを要求されるだろう。規制の承認を得て、その後、適用される連邦、州、地方、外国の法規と条例を遵守する過程には、多くの時間と財政資源が必要だ。
アメリカの薬物開発プロセスは
米国では,FDAは連邦食品,薬物,化粧品法案(“FDCA”)とその実施条例に基づいて薬物を規制している。規制の承認を得て、その後、適切な連邦、州、地方、外国の法規と条例を遵守する過程には、多くの時間と財政資源が必要だ。FDAが米国で発売される前に必要とされるプログラムは、一般に以下のような態様を含む
臨床前開発段階は、通常、薬物化学、配合および安定性の実験室評価、および動物におけるこの分子の毒性を評価する研究を含み、これはその後の臨床試験を支持する。臨床前研究の進行はある毒理学研究に対するGLP法規を含む連邦法規と要求の制約を受けている。スポンサーは,臨床前研究の結果を,生産情報,分析データ,任意の利用可能な臨床データや文献,提案された臨床案とともにINDの一部としてFDAに提出しなければならない。INDはFDAがヒトに対する研究薬物製品の使用を許可する要求である。IND提出の中心焦点は臨床研究の全体的な研究計画と方案(S)である。INDはまた、製品の毒性学、薬物動態学、薬理学および薬効学的特徴を評価する動物およびインビトロ研究結果、化学、製造および制御情報、および研究製品の使用を支援するための任意の利用可能なヒトデータまたは文献を含む。長期的な前臨床試験、例えば生殖や
33
発ガン性は,IND提出後も存在し続ける可能性がある。INDはヒト臨床試験が始まる前に発効しなければならない。INDはFDAが30日以内に提案された臨床試験に対して安全懸念または問題を提起しない限り、FDA受信後30日以内に自動的に発効する。この場合,INDは臨床的に放置される可能性があり,INDスポンサーやFDAは臨床試験が開始される前に未解決の問題や問題を解決しなければならない。FDAは、安全考慮または規定を満たしていないために、研究前または研究期間中の任意の時点で臨床一時停止を強制的に実施することもできる。したがって,INDの提出はFDAが臨床試験の開始を許可する可能性もない可能性がある。
臨床試験は、合格した調査者の監督の下で、GCPに従ってヒト被験者に研究製品を提供することに関連し、これらの調査者は、通常、試験スポンサーによって雇用されているか、または試験スポンサーによって制御されていない医師であり、他を除いて、すべての研究対象に任意の臨床研究への参加についてインフォームドコンセントを提供することを要求することが含まれる。臨床試験は、研究目標、用量プログラム、被験者選択および排除基準、被験者の安全性を監視するためのパラメータ、および評価すべき有効性基準を詳細に説明するレジメンに基づいて行われる。製品開発中に行われる各後続の臨床試験および後続の任意のレジメン修正は、既存のINDに個別に提出されなければならない。INDは活発であるが、前回の進展報告以来行われた臨床試験と非臨床研究結果の進展報告をまとめ、他の情報以外に、少なくとも毎年1回の書面IND安全報告をFDAに提出しなければならず、その中でFDAと調査者に書面IND安全報告を提出しなければならず、深刻かつ意外な疑わしい不良事件を理解し、他の研究結果は同じ或いは類似の薬物に暴露することは人類に対して重大なリスクがあることを表明し、動物或いは体外試験結果は人類に対して重大なリスクがあることを表明し、及び方案或いは研究者のマニュアルに列挙された深刻な疑わしい不良反応の発生率と比較して、任意の臨床重要な疑わしい副作用の発生率は増加する。
また,臨床試験を推奨する各地点の独立IRBは,その地点で臨床試験を開始する前に任意の臨床試験の計画とそのインフォームドコンセントを審査·承認しなければならず,完成まで研究を監視しなければならない。いくつかの研究はまた、臨床研究スポンサーによって組織された独立した合格専門家グループの監視を含み、このグループは、研究のいくつかのデータへのアクセスに基づいて、研究が指定されたチェックポイントで行うことができるかどうかを許可するデータ安全監視委員会と呼ばれ、被験者に受け入れられない安全リスクまたは他の理由があると判定された場合、治療効果を示さない場合、臨床試験を停止する可能性がある。その規約によれば、当該グループは、試験のあるデータへのアクセスに基づいて、試験が指定されたチェックポイントで行うことができるか否かを決定することができる。FDAあるいはスポンサーは研究対象や患者が受け入れられない健康リスクに直面していることを発見することを含む、様々な理由で臨床試験を一時停止することができる。同様に、1つの臨床試験が委員会の要求に従って行われない場合、または薬剤が患者に予期せぬ深刻な傷害を与えた場合、IRBは、その所在機関の臨床試験の承認を一時停止または終了することができる。現在行われている臨床研究や臨床研究結果を公的登録機関に報告することに関する要求もある。
人体臨床試験は通常3つの連続段階に分けて行われ、これらの段階は重複或いは合併する可能性がある
34
場合によっては、FDAは、承認適応内の製品に関するより多くの情報を得るために、製品が承認された後に追加の臨床試験を行うことを要求またはスポンサーに要求する可能性がある。これらのいわゆる4期研究は,最初の市場承認後に行われる可能性があり,治療適応が予想される患者の治療からより多くの経験を得るために利用される可能性がある。場合によっては,FDAはNDAを承認する条件として4期臨床試験を強制的に実行することができる。
また,新薬開発期間中,スポンサーはいつかFDAと会う機会がある。これらの要件は、INDを提出する前、第2段階の終了時、および秘密協定の提出前にある可能性がある。他の時間に会議を開催することを要求することができます。これらの会議は,スポンサーにこれまで収集してきたデータに関する情報を共有する機会を提供し,FDAにアドバイスを提供し,スポンサーとFDAに次の段階の開発について合意することができる。
臨床試験と同時に、会社は通常追加の動物研究を完成し、薬物化学と物理特性に関する追加情報を開発し、cGMPの要求に基づいて最終的に商業量産製品のプロセスを決定しなければならない。製造過程は一貫して高品質の候補製品ロットを生産できる必要があり、また、メーカーは最終薬物の身分、強度、品質、純度をテストする方法を開発しなければならない。また,適切な包装を選択·テストし,候補製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
アメリカの審査と承認の流れ
すべての適用された法規要件に基づいて必要なすべてのテストが成功したと仮定すると,製品開発の結果は,臨床前および他の非臨床研究や臨床試験の結果,製造過程の記述,薬物化学の分析テスト,提案されたラベル,その他の関連情報を含み,その製品の発売承認を要求するセキュリティプロトコルの一部としてFDAに提出される。データは、製品使用の安全性および有効性を試験するための会社によって支援された臨床研究から来ることができ、独立した調査者によって開始された研究を含む多くの代替源から来てもよい。秘密協定の提出には相当な使用料が必要であり、いくつかの限られた場合には、そのような費用を免除することができる。また,孤児薬に指定されている非処方薬については,使用料は何も評価せず,申請しない限り非孤児適応が求められている。
そのほか、小児科研究公平法(PREA)はスポンサーに大多数の薬物、新しい有効成分、新しい適応、新しい剤形、新しい投与方案或いは新しい投与経路に対して小児科臨床試験を行うことを要求している。PREAによれば、元のNDAおよびいくつかのサプリメントは、スポンサーが延期または免除を受けていない限り、小児科評価を含まなければならない。要求された評価は、すべての関連する小児科亜群において適応の安全性および有効性を主張する製品を評価し、その安全に有効であると考えられる各小児科亜群の用量および投与を支持しなければならない。スポンサーまたはFDAは、小児科亜群の一部または全部の小児科臨床試験の延期を要求することができる。延期は、小児科臨床試験が完了する前に、成人で使用を許可する準備ができていることを発見するか、または小児科臨床試験が開始される前に追加の安全性または有効性データを収集する必要があることを含むいくつかの理由があるかもしれない。FDAは、必要な評価を提出できなかった、延期された最新の状況を維持し、または小児科処方承認要求を提出できなかった任意のスポンサーに、規定に適合しない手紙を送信しなければならない。
FDAは、届出を受ける前に、提出後の最初の60日以内にすべてのNDAを予備審査して、それらが十分に完全であるかどうかを決定し、FDAの実質的な審査を可能にすることは、NDA届出を受け入れるのではなく、より多くの情報を提供することを要求することができる。この場合、秘密協定と追加情報を再提出しなければならない。再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。提出されると、FDAは、製品がその予期される用途に対して安全かつ有効であるかどうかを決定し、その製造がcGMPに適合するかどうかを決定して、製品の特性、強度、品質、および純度を保証および維持するためにNDAを検討する。現在施行されている“処方薬使用料法案”ガイドラインによると,FDAは申請日から10カ月以内に新しい分子実体薬のNDAの標準審査を完了することを目標としている。この審査には通常12カ月の時間が必要であり,NDAがFDAに提出された日から計算すると,FDAは申請提出後約2カ月で“届出”決定を下すためである。
35
FDAは新薬の申請を諮問委員会に提出するかもしれない。諮問委員会は,臨床医や他の科学専門家を含む独立した専門家からなるグループであり,申請を審査,評価し,申請を承認すべきかどうか,どのような条件でアドバイスを提供すべきかを担当する。FDAは諮問委員会の提案に制限されていないが、それは決定を下す時にこれらの提案を慎重に考慮するだろう。
秘密協定を承認する前に、FDAは通常、製品を生産する1つまたは複数の施設を検査する。FDAは、製造プロセスおよび施設がcGMPに適合していることを決定し、要求された仕様で製品が一貫して生産されることを保証するのに十分でない限り、申請を承認しないであろう。さらに、NDAを承認する前に、FDAは、GCPに適合することを確実にするために、1つまたは複数の臨床場所を検査することができる。
FDAがNDAを評価し、研究製品および/またはその薬物を生産する製造施設を検査した後、FDAは承認書または完全な返信(CRL)を発行することができる。承認書は、製品の商業マーケティングを許可し、特定の適応に関する具体的な処方情報を提供する。CRLは、一般に、FDAによって決定されたNDAにおける特定の欠陥を記述し、追加の臨床試験、または臨床試験、非臨床研究、または生産に関連する他の重要で時間のかかる要件を含む追加の臨床データを必要とする可能性がある。CRLを発行すると,スポンサーはNDAを再提出し,手紙で確定したすべての不足点を解決したり,申請を撤回したりしなければならない.このようなデータや情報を提出しても,FDAはNDAが承認基準を満たしていないと認定する可能性がある。
1つの製品が規制部門の承認を受けた場合、このような承認は特定の適応が付与され、製品が発売される可能性のある指定用途の制限をもたらす可能性がある。例えば、FDAは、製品の利点がそのリスクよりも大きいことを確実にするために、リスク評価および緩和戦略(“REMS”)を有するNDAを承認する可能性がある。REMSは、既知または潜在的な薬物に関連する深刻なリスクを管理し、薬物の安全な使用を管理することによって、患者がそのような薬物を継続的に得ることができるようにするための安全戦略であり、制限された分配方法、患者登録、および他のリスク最小化ツールのような薬物ガイドライン、医師のコミュニケーション計画、または安全使用を保証する要素を含むことができる。FDAはまた,提案されたラベルを変更したり,適切な制御や仕様を作成したりすることを条件に承認することも可能である.FDAはまた、製品の商業化後の安全性と有効性をさらに評価し、監視するために、1つまたは複数の第4段階上場後の研究とモニタリングを要求する可能性があり、これらの発売後の研究の結果に基づいて製品のさらなる販売を制限する可能性がある。
開発と審査計画を加速する
FDAは合格した候補製品に一連の迅速な開発と審査計画を提供した。例えば、迅速チャネル計画は、深刻または生命に危険な疾患または状態を治療することを目的とした新製品の審査プロセスを加速または促進し、疾患または状態を解決する満たされていない医療需要の潜在力を示すことを意図している。高速チャネルは,候補製品と研究中の特定の適応に適した組合せを指定する.Fast Track候補製品のスポンサーは,製品開発期間中に適用されるFDA審査チームとより頻繁なインタラクションを行う機会があり,機密協定が提出されると,申請は優先審査を受ける資格がある可能性がある.高速チャネル候補製品のセキュリティプロトコルは、スクロール審査を行う資格がある可能性もあり、この場合、FDAは、完全な出願を提出する前に、セキュリティプロトコルを考慮する審査部分をスクロールさせることができ、スポンサーがセキュリティプロトコル部分を提出するスケジュールを提供した場合、FDAは、セキュリティプロトコルの部分を受け入れることに同意し、スケジュールが許容可能であると判断し、スポンサーは、セキュリティプロトコルの第1の部分を提出する際に任意の必要な使用料を支払うことができる。
重篤または生命に危険な疾患や状況を治療しようとする候補品も,その開発や審査を加速するための画期的な療法指定を受ける資格がある可能性がある。予備臨床証拠が、候補製品が単独または1つまたは複数の他の薬物または生物製品と組み合わせて使用される可能性があることを示す場合、候補製品は、1つまたは複数の臨床重要終点において、既存の治療法よりも実質的な改善を示す可能性があり、例えば、臨床開発早期に観察された実質的な治療効果を示す場合、候補製品は突破的治療称号を得ることができる。この指定には、Fast Track計画のすべての機能と、第1段階で開始されたより密集したFDA相互作用および指導と、高度な管理者の参加を含む候補製品開発および審査を加速する組織約束とが含まれる。
36
迅速なチャネル指定および/または画期的な治療指定を有する候補製品を含むFDA承認を提出する任意の薬物マーケティング申請は、FDAの他のタイプの計画の資格に適合する可能性があり、これらの計画は、優先審査のようなFDAの審査および承認プロセスを加速させることを目的としている。候補製品が、深刻なまたは生命を脅かす疾患または状態の治療を意図しており、承認された場合、そのような疾患または状態の既存の代替品と比較して、安全性または有効性の点で有意な改善を提供する場合、NDAは、優先審査を受ける資格がある。新しい分子実体NDAについて、優先審査指定は、FDAの目標が、60日の出願日の6ヶ月以内にマーケティング申請に行動することであることを意味する。
さらに、臨床研究に適用される設計によれば、深刻または生命に危害を及ぼす疾患または状況の治療において安全性と有効性研究を行う候補製品は、迅速な承認を得る可能性があり、条件は、この製品が臨床利益を合理的に予測する可能性のある代替終点に有効であるか、または不可逆的な発病率または死亡率よりも早く測定することができる中間臨床終点に有効であり、この中間臨床終点は、不可逆的な発病率または死亡率または他の臨床利益への影響を合理的に予測することが可能であり、同時に病状の深刻性、希少性または流行度、および代替治療の有用性または不足を考慮することである。承認を加速する条件として、FDAは、通常、不可逆的な発症率または死亡率または他の臨床的利益に対する予想される影響を検証および説明するために、スポンサーに十分かつ制御された検証的臨床研究を要求し、承認加速の承認前にそのような検証的試験を行うことを要求する可能性がある。スポンサーが必要な検証的研究をタイムリーに行うことができなかった場合、またはそのような研究が予測の臨床的利益を検証できなかった場合、加速的な承認を得た製品は、迅速な脱退手順によって制限される可能性がある。また、FDAは現在、承認を加速させる条件として宣伝材料を事前承認することを求めており、製品商業発売の時期に悪影響を及ぼす可能性がある。
高速チャネル指定、画期的な治療指定、優先審査、承認の加速は承認基準を変更することはありませんが、開発や承認過程を加速する可能性があります。製品候補が1つまたは複数の計画の条件に適合していても、FDAは、その製品がもはや資格条件を満たしていないと後で決定するか、またはFDAの審査または承認を決定する期間が短縮されない可能性がある。
孤児薬の指定と排他性
孤児医薬品法によれば、FDAは、まれな疾患または状態を治療するための薬剤を孤児として指定することができ、このような疾患または状態は、一般に、(I)米国における患者数が20万人未満であるか、または(Ii)米国における患者数が20万人を超え、米国での薬剤開発および提供コストが米国での販売から回収されることが合理的に予想できない場合のうちの1つの疾患または状態として定義される。秘密保持協定を提出する前に、指定孤児薬を申請しなければならない。FDAが孤児薬物指定を承認した後、FDAは、治療薬の模倣薬識別情報およびその潜在的孤児の使用を開示する。
孤児薬物指定を有する製品が、その後、このような指定された疾患または疾患の特定の有効成分を有するFDAの最初の承認を得た場合、この製品は、孤児製品の排他性を得る権利があり、これは、FDAが、承認日から7年以内に同じ疾患または疾患の同じ薬剤を販売するために、限定された場合を除いて、完全なNDAを含む他の出願を許可しないことを意味する。例えば、孤児薬物排他性を有する製品に対する臨床的優位性を示すか、またはFDAは、指定された薬物の疾患または状態を有する患者の需要を満たすために十分な数の孤児薬を得ることができることを証明していないことを発見する。孤児薬物排他性は、FDAが同じ疾患または状態に対する異なる薬物、または異なる疾患または状態に対する同じ薬物を承認することを阻止しない。孤児薬を指定する他の利点は、いくつかの研究の税金控除とNDA申請使用料の免除を含む。指定された孤児薬物が孤児として指定された疾患または状況よりも広い使用のために許可された場合、孤児薬物排他性を得ることはできない。さらに、FDAが後に指定要求に重大な欠陥があると判断した場合、または上述したように、第2の出願人が、その製品が孤児排他性を有する承認製品よりも臨床的に優れていることを証明する場合、または製品を承認する製造業者が、まれな疾患または疾患患者の需要を満たすのに十分な数の製品を保証できない場合、米国における孤児薬の独占営業権を失う可能性がある。
37
承認後に要求する
FDAによって生産または流通を許可された薬品は、記録保存、副作用報告、定期報告、製品サンプリングと流通、および製品の広告と販売促進に関連する要求を含むFDAの普遍的かつ持続的な規制を受けなければならない。承認後、承認された製品の大多数の変更は、新たな適応または他のラベル宣言を追加するなど、FDAの事前審査および承認を経なければならない。どんな発売された製品についても、継続的な年間計画費用があります。医薬品メーカーおよびその下請け業者は、FDAとある州機関に彼らの機関を登録し、FDAとある州機関の定期的な抜き打ち検査を受けて、cGMPのコンプライアンスを理解しなければならない。これは、私たちと私たちの第三者メーカーにいくつかの手続きと文書要求を加えている。製造プロセスの変更は厳しく規制されており,変更の重要性により,FDAが事前に承認して実施する必要がある可能性がある。FDAの規定では,cGMPから外れた状況を調査·是正し,報告要求を行うことも求められている。そのため、メーカーは生産と品質管理の分野で時間、お金、精力をかけ続け、cGMPやその他の法規遵守性を維持しなければならない。
規制要件や基準が守られていない場合、あるいは製品発売後に問題が発生した場合、FDAは承認を撤回する可能性がある。その後、製品には、予期されない深刻度または頻度の不良事象、または生産プロセス、または規制要件を遵守できなかったことを含む、以前に未知の問題が存在することが発見され、新しいセキュリティ情報を追加するために承認されたラベルの改訂を招く可能性がある;新しい安全リスクを評価するために発売後研究または臨床研究を実施すること、またはREMS計画に従って流通制限または他の制限を実施することが可能である。他の他の潜在的な結果には
FDAは薬品のマーケティング、ラベル、広告、販売促進を密接に規制している。1社はFDAが承認したラベルの規定に基づいて、安全性と有効性、純度、効力に関する声明しか提出できない。FDAと他の機関は非ラベル用途の普及を禁止する法律法規を積極的に施行している。これらの要求を守らないことは、否定的な宣伝、警告状、改正広告、および潜在的な民事と刑事罰を招く可能性がある。医師は彼らの独立した専門医学判断に基づいて、製品ラベルに記載されていない用途と、私たちのテストとFDAが許可した用途とは異なる合法的に利用可能な製品のために処方することができる。医師は,異なる場合,このような非ラベル使用が多くの患者の最適な治療法であると考えるかもしれない。FDAは医者が治療を選択する時の行動を規範化しない。しかし、FDAは製品ラベルの外使用問題に対する製造業者のコミュニケーションを制限した。しかしながら、会社は、FDAによって承認された製品ラベルと一致する真で誤解されない情報を共有するかもしれない。
38
そのほか、処方薬製品の流通は“処方薬販売法”(“PDMA”)の制約を受け、この法は連邦一級の薬品と薬品サンプルの流通を規定し、各州の薬品流通業者に対する登録と監督管理に最低基準を設定した。PDMA,州法ともに処方薬製品サンプルの配布を制限し,配布中の責任の確保を求めている。
マーケティング排他性
FDCAによって許可された市場排他性条項は、いくつかのマーケティング申請の提出または承認を延期する可能性がある。FDCAは新しい化学実体秘密協定の承認を得た最初の申請者に5年間のアメリカ国内の非特許データ排他期を提供した。FDAが以前に同じ活性部分を含む他の新薬を承認していなければ,薬物は新しい化学実体であり,活性部分は薬物物質の作用を担う分子やイオンである。排他的期間内に、FDAは、505(B)(2)条(“505(B)(2)NDA”)に従って提出された簡略化新薬出願(“ANDA”)またはNDA(“505(B)(2)NDA”)を承認しないか、または受け入れない可能性があり、その薬剤が元の革新薬と同じ適応であるか、または他の適応のために使用されるかにかかわらず、出願人が合法的な参照権を有していない場合、または承認に必要なすべてのデータを参照する。しかしながら、出願がイノベーターNDA所有者がFDAに記載された特許のうちの1つを含む特許が無効または未侵害証明である場合、4年後に出願することができる。
FDCAはまた、NDAに3年間の非特許排他性を提供するか、または出願人が行ったり賛助したりする新しい臨床研究(バイオアベイラビリティ研究を除く)がFDAによって承認出願に重要であると考えられる場合に、既存の薬物の新しい適応、用量、または強度のような既存のNDAの補充を提供する。この3年間の排他性は、この薬剤が新しい臨床研究に基づいて承認された修正のみを含み、FDAがANDAまたは505(B)(2)NDAを許可することは禁止されておらず、元の適応または使用条件の有効成分を含む薬剤のために使用される。5年と3年の排他性は完全な秘密協定の提出や承認を延期したり承認したりしないだろう。しかしながら、完全なセキュリティプロトコルを提出する出願人は、安全かつ有効であることを証明するために、任意の臨床前研究および十分かつ良好に制御された臨床試験を参照するために必要な権利を行うか、または得ることを要求されるであろう。
小児科専門権は米国で利用可能な別のタイプのマーケティング専門権である。小児科排他性規定は,スポンサーがFDAの書面請求に応じて児童に臨床試験を行う場合,別の既存の規制排他期間または特許期間に追加6カ月の排他性を付加することを規定している。書面出願の発表はスポンサーに述べた臨床試験を要求しない。また、上述したように、孤児薬の独占性は7年間の市場独占期間を提供することができるが、場合によっては除外される。
他の医療法律法規
私たちのような製薬会社は連邦政府およびそれらが業務を展開している州と外国司法管轄区域当局の追加医療監督と法執行を受けている。このような規制は、私たちがマーケティングの承認を得た任意の製品の財務配置と関係を制限するかもしれません。このような法律には、連邦反バックル法規および連邦民事虚偽請求法案のような連邦および州の反リベート、詐欺および乱用、および連邦反バックル法規および連邦民事虚偽請求法案のような虚偽クレーム法律、ならびに医薬品定価および医薬品製造業者の医師および他の医療保健提供者への支払いおよび他の価値移転を処理する透明性法律および法規、例えば連邦医師が日光法案を支払うことが含まれるが、これらに限定されない。このような任意の法律または任意の他の適用可能な政府法規に違反することは、行政、民事および刑事罰、損害賠償、罰金、返却、削減または再構成業務、誠実な監督および報告義務を含むが、違反容疑を解決し、連邦および州医療保健計画(例えば、MedicareおよびMedicaid)から除外され、監禁されることを含む重大な処罰を招く可能性がある。
39
保証範囲·定価·精算
任意の承認された薬品の販売は、連邦、州と外国政府医療保健計画、商業保険とホスト医療組織、および第三者支払人のこのような製品に対する第三者支払人の清算レベルにある程度依存する。提供されるべき補償範囲と金額に関する決定は個々の計画に基づいて行われる。第三者決済者は医療製品、薬品、関連製薬会社サービスの保証と精算をますます減少させている。また、米国政府、州立法機関、外国政府は価格制御、カバー範囲と補償の制限及び模造薬代替の要求を含むコスト制御計画を継続して実施している。価格制御とコスト制御措置、および既存の制御·措置を有する管轄区域でより厳しい政策をとることで、1つ以上の管轄区域で上場許可を得る可能性のある任意の製品の販売をさらに制限することができる。いかなる製品の第三者精算または第三者支払者が製品を保証しないことを決定することは、医師の使用量や患者の製品に対する需要を減少させ、販売に実質的な悪影響を及ぼす可能性がある。
また、あるアメリカ連邦医療保健計画に参加し、製品をその中に含める条件として、例えば連邦医療保険と連邦医療補助は、製薬業者に連邦医療補助平均メーカー価格(AMP)と最適価格、連邦医療保険平均販売価格、340 B上限価格、退役軍人事務部に報告した非連邦AMP、および連邦医療補助受益者使用メーカーの製品について法定リベートを支払うように製薬業者に計算し、政府に報告することを要求する連邦法律と法規の制約を受ける可能性がある。
このような法律と法規を遵守するためには大量の資源が必要であり、私たちの収入に実質的な悪影響を及ぼすかもしれない。
医療改革
また,前述したように,米国のヘルスケア業界や他の地域の主な傾向はコストコントロールである。政府当局や他の第三者支払者は、特定の医療製品やサービスのカバー範囲や精算金額を制限し、医療保険や他の医療資金を削減し、新たな支払い方法を適用することでコストを抑制しようとしている。例えば,米国では,ACAが2010年3月に公布され,政府や民間保険会社が医療保健に資金を提供する方式を大きく変更し,製薬業に大きな影響を与えている。ACAには、連邦医療計画の登録、補償調整、および詐欺および乱用の法律の改正を含むいくつかの条項が含まれている。別の例として、2020年12月27日に法律となる2021年総合支出法案に署名し、2022年1月1日からすべての連邦医療保険B部分に含まれる医薬品メーカーに製品の平均販売価格を連邦政府に報告し、民事罰金により強制執行することを含む幅広い医療条項と既存法の改正を盛り込んだ。ACAのいくつかの側面は公布以来、行政、司法、そして国会の挑戦を受けてきた。2021年6月17日、米国最高裁はいくつかの州がACAに対して提出した最新の司法挑戦を却下したが、ACAの合憲性を具体的に裁くことはなかった。
ACAが公布されて以来、2021年の米国救援計画法案を含む他の立法改正も提出され、2024年1月1日に発効し、薬品メーカーが医療補助薬品還付計画(MDRP)に基づいて不足している税金還付金額の法定上限を廃止した。これまで,保険外来薬のリベート上限はAMPの100%であった。
また、政府はメーカーがその販売する製品に価格を設定する方式に対してより厳格な審査を行い、国会で数回の調査を行い、連邦と州立法を提出し、製品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、政府の薬品の精算方法を改革することを提出し、公布した。最近は2022年8月16日に“2022年インフレ削減法案”(IRA)が署名されて法律となっている。他の事項を除いて、アイルランド共和軍はある薬品のメーカーに連邦医療保険との価格交渉(2026年から)を要求し、価格は交渉できるが上限があり、連邦医療保険B部分と連邦医療保険D部分に基づいてリベートを実施し、インフレを超える価格上昇を処罰し(2023年に初めて満了)、D部分のカバーギャップ割引計画の代わりに新しい割引計画を用いる(2025年から)。
40
アイルランド共和軍は,衛生·公衆サービス部(HHS)秘書が最初の数年間,規制ではなく指導によってその多くの規定を実施することを許可した。これらの計画の実施に伴い,HHSは指導意見を発表·更新し続けている。2023年8月29日、HHSは、連邦医療保険薬品価格交渉計画が現在法的挑戦を受けているにもかかわらず、価格交渉を受ける上位10種類の薬物のリストを発表した。このような理由と他の理由で、アイルランド共和軍がどのように実施されるのかは不明だ。
州レベルでは、立法機関は、価格または患者の精算制限、割引、特定の製品参入の制限、マーケティングコスト開示および透明性措置を含む、医薬品および生物製品の価格設定を制御するための法規を立法および実施することが増えており、場合によっては、他の国からの輸入および大量購入を奨励することを目的としている。2020年12月、米国最高裁判所は、連邦法律は各州の薬品福祉マネージャー(PBM)と医療保健と薬品サプライチェーンの他のメンバーの能力を妨害しないと一致し、この重要な決定は各州がこの分野で更なるかつ積極的な努力をすることを招く見通しである。
政府も新冠肺炎の流行に対応するために行動する可能性がある。将来的には,より多くの州と連邦医療改革措置がとられ,いずれも連邦や州政府および他の第三者支払者が医療製品やサービスに支払う金額に影響を与える可能性が予想される。
データのプライバシーとセキュリティ
多くの州、連邦、そして外国の法律は個人情報の収集、使用、開示と保護を規範化している。アメリカでは、データ漏洩通知法、健康情報プライバシーと安全法、および消費者保護法律と法規を含む多くの連邦と州の法律法規が、健康に関連する個人情報と他の個人情報の収集、使用、開示と保護を規範化している。また、いくつかの外国法は、健康に関するデータを含む個人データのプライバシーおよびセキュリティを管理する。プライバシーとセキュリティ法律、法規、その他の義務は絶えず変化し、互いに衝突し、コンプライアンス作業を複雑化させ、調査、訴訟あるいは行動を招き、重大な民事および/または刑事罰およびデータ処理の制限を招く可能性がある。
人力資本
2024年2月29日現在、私たちは14人のフルタイム従業員がいて、アルバイト従業員はいません。これらの従業員のうち,4人は博士または医学博士号を持ち,8人は研究開発に従事している。私たちの職員たちは労働組合代表もなく、集団交渉協定のカバー範囲もない。私たちは私たちが従業員と仲がいいと思う。
私たちの人的資本目標は識別、採用、維持と激励、私たちの管理チームと私たちの臨床、科学と他の従業員と顧問を含む。私たちの株式と現金インセンティブ計画の主な目的は、私たちの利益と株主の利益が私たちの従業員とコンサルタントの利益と一致するように、株と現金に基づく報酬奨励を付与することによって、従業員を引き付け、維持し、激励することである。
企業情報
我々は2020年10月2日にデラウェア州法により設立され,名称はLocust Walk Acquisition Corp(“LWAC”)である。エフェクター治療会社は2012年5月1日にデラウェア州の法律登録に基づいて設立された。2021年8月25日、私たちは合併を完了し、この合併によって、Locust Walk Acquisition Corp.の完全子会社はEfftor Treateutics Operations,Inc.(前身はEfftor Treateutics,Inc.)と合併した。(“旧効力者”)、エフェクター治療運営会社は当社の完全子会社となった(“業務合併”)。合併が完了した後、私たちはEfftor Treateuticsと改名し、Inc.当社の本社は現在カリフォルニア州ソラーナビーチBスイート142 North Cedross 142 North Cedros、郵便番号:92075、電話番号は(858)925-8215です。
文意が別に指摘されているほか、本節で言及した“私たち”、“私たち”、“私たち”または“効果者”は、効果者治療会社の業務合併完了前の業務を指し、これは業務合併完了後の業務である。
41
利用可能な情報
私たちのインターネットアドレスはwww.ffector.comです。私たちの投資家関係サイトはhttps://investors.ffector.com/にあります。我々は、米国証券取引委員会(“米国証券取引委員会”)にこのような資料を提出または提出した後、我々の投資家関係サイト上の“米国証券取引委員会届出”の項の下で、我々の年間Form 10-K報告、Form 10-Q四半期報告、Form 8-K現在の報告、取締役および上級管理者第16条の報告、およびこれらの報告の任意の修正を無料で提供する。これらはアメリカ証券取引委員会のサイトwww.sec.govで無料で入手することもできます。
我々は,重要な非公開情報を開示する手段として我々の投資家関係サイトを用い,FD法規下での開示義務を遵守している.投資家は,我々のニュース原稿,米国証券取引委員会の届出文書,公開電話会議やインターネット放送に注目するほか,このようなサイトにも注目すべきである.コーポレートガバナンスに関する情報は私たちの投資家関係サイトにも含まれています。米国証券取引委員会及び当サイトにおける情報又は当サイトを通じて取得された情報は、本出願に組み込まれることもなく、本出願の一部ともみなされない。
第1 A項。リスク要因です
私たちの普通株式または株式承認証に投資する前に、以下のリスク要因と、当社の財務諸表および関連説明、ならびに“経営陣の財務状況および運営結果の検討および分析”を含む本年度報告に含まれる他の情報をよく考慮しなければなりません。私たちはあなたに次の危険要素で議論されたどんな事件も起こらないということを保証できません。これらのリスクは、私たちの業務、運営結果、財務状況、および成長見通しに実質的な悪影響を及ぼす可能性がある。このような状況が発生した場合、私たちの普通株式または株式証明書の取引価格は低下する可能性がある。私たちは今知らないか、あるいは私たちが現在どうでもいいと思っている他のリスクと不確実性はまた私たちの業務運営や財務状況を損なう可能性がある。 本節では,まず我々が直面している主要なリスクと不確実性について概説し,次に全リスク要因を提供し,より詳細な議論を行う.
リスク要因の概要
42
私たちの限られた経営歴史、財務状況、資本要求に関するリスク
私たちの経営の歴史は限られており、設立以来ずっと重大な運営損失を受けており、予測可能な未来に重大な損失が予想されている。私たちは製品販売からどんな収入も得たり、利益を達成したりしないかもしれません。あるいは、もし私たちが利益を達成すれば、私たちは持続できないかもしれません。
私たちは臨床段階の生物製薬会社で、運営の歴史が限られていますので、それに基づいて私たちの業務と将来性を評価することができます。私たちは2012年に運営を開始した。これまで、著者らは主に資金調達に集中し、潜在的な候補製品を確定し、私たちの知的財産権の組み合わせを構築し、臨床前研究と臨床試験を行い、第三者と私たちの候補製品と関連原材料の生産について手配を確立し、そしてこれらの業務に一般と行政支持を提供する。我々の技術プラットフォームに基づいて候補製品を発見·開発する方法は検証されておらず,規制部門の商業的価値のある製品の開発や承認を得ることができるかどうかは分からない.また,われわれの臨床開発では2種類の候補製品,トミフサーチブとゾタフェンのみであった。私たちはまだ重要な臨床試験を成功させ、監督管理の許可を得て、商業規模の製品を製造したり、第三者代表が私たちにそうするように手配したり、成功した製品の商業化に必要な販売とマーケティング活動を行うことができないことを証明していません。したがって、もし私たちが薬品の開発と商業化に成功した歴史があれば、私たちの未来の成功や生存能力に対するいかなる予測もそれらができるほど正確ではないかもしれない。
ファイザー社との研究協力やライセンス契約(“ファイザー合意”)による収入を除いて、設立以来大きな運営損失を受けており、将来的には大きな損失が予想される。私たちはどんな製品も販売を許可されておらず、設立以来何の製品収入も発生していない。もし私たちが候補製品の開発に成功し、必要な承認を得ることができなければ、私たちは製品販売から何の収入も得られないかもしれない。2022年12月31日までの年度の純損失は2270万ドルで、2023年12月31日までの年度の純損失は3580万ドルです。2023年12月31日までの累計赤字は1兆794億ドルだった。我々のほとんどの運営損失は,我々の候補製品や開発計画の研究や開発に関する費用,および我々の運営に関する一般的かつ行政コストから来ている.私たちのすべての候補製品は大量の追加の開発時間と資源が必要で、それから私たちは規制部門の承認を申請したり、製品販売から収入を得ることができます。私たちは予測可能な未来に損失が出ることを予想し、私たちが開発を継続し、監督部門に許可された候補製品の承認を求め、商業化する可能性があることに伴い、これらの損失は大幅に増加する。
利益を実現し、維持するためには、大量の収入を生む製品を開発し、最終的に商業化しなければならない。これは、私たちの候補製品の臨床試験と臨床前研究を完成させ、これらの候補製品の規制承認を得ること、および私たちが規制承認を得る可能性のある任意の製品を含む、一連の挑戦的な活動で成功することを要求するだろう。私たちはただこのような活動の大多数の初期段階にいるだけだ。私たちはこのような活動で決して成功しないかもしれないし、たとえ私たちが成功しても、利益を達成するのに十分な収入が生まれないかもしれない。また,我々は,企業が新たかつ急速に発展する分野でしばしば遭遇する多くのリスクや不確実な要因を克服することに成功する能力,特に生物製薬業界ではまだ示されていない。薬品開発に関連する多くのリスクや不確実性により、費用が増加する時間や金額、あるいは利益を達成できるかどうかを正確に予測することはできない。たとえ私たちが確実に利益を達成したとしても、私たちは四半期や年度の収益性を維持したり向上させることができないかもしれない。
43
私たちが達成できず利益を維持していることは、わが社の価値に悪影響を及ぼす可能性があり、資金調達、業務拡大、研究開発努力の維持、候補製品の多様化、さらには運営を継続する能力を弱める可能性があります。わが社の価値の低下はあなたの投資損失の全部または一部を招く可能性もあります。
私たちは、私たちの運営に資金を提供するために大量の追加資本が必要であり、必要な場合に受け入れ可能な条件で必要な資本を得ることができない場合、あるいは必要な資本を全く得ることができない場合、私たちの開発計画、商業化努力、または他の運営を延期、制限、減少、または終了させることができるかもしれない。
候補薬品の開発は資本集約型である。設立以来、私たちの業務は大量の現金を消費した。私たちが行っている活動に関連する費用が増加することが予想され,特に私たちが行っている臨床試験と計画されている臨床試験を行い,規制部門のトミフセテやゾタフェンの承認を求めた場合である。また、ファイザーは現在私たちのeIF 4 E計画の開発を担当しているにもかかわらず、ファイザー協定の条項に基づいて共同援助を行使し、この計画の選択権を共同推進すれば、追加費用が発生する。また、いずれかの候補製品が規制部門の承認を得た場合、製品製造、マーケティング、販売、流通に関連した巨額の商業化費用が発生すると予想される。いかなる臨床試験或いは臨床前研究の結果はすべて高度に不確定であるため、著者らは著者らの候補製品の開発と商業化に成功するために必要な実際の数量を合理的に見積もることができない。また、私たちは上場企業の運営に関連した追加コストが発生すると予想される。したがって、私たちは私たちの持続的な業務と関連した多くの追加資金を得る必要があるだろう。もし私たちが必要な時や魅力的な条件下で資金を集めることができなければ、私たちは私たちの研究開発計画や将来の商業化努力を延期、減少、または廃止することを余儀なくされるかもしれない。
私たちの現在の運営計画によると、私たちは既存の現金、現金等価物、短期投資が2025年第1四半期の運営に資金を提供できると信じている。私たちは間違っていることが証明される可能性があるという仮定に基づいてこの推定をして、私たちは現在予想されているよりも早く私たちの資本資源を使用することができる。私たちの現在知られていない多くの要素のため、私たちの運営計画と私たちの現金資源に対する他の需要は変化する可能性があり、私たちは計画よりも早く追加資金を求める必要があるかもしれません。潜在的な協力、ライセンス、および他の同様の手配を含む、公的またはプライベートエクイティまたは債務融資または他の資本源を通じて。また、有利な市場条件や戦略的考慮により、現在または将来の運営計画のために十分な資金があると考えても、追加の資本を求めることができる。より多くの資金を得ようとすることは、私たちの経営陣の日常活動への注意をそらす可能性があり、これは候補製品を開発する能力に悪影響を及ぼす可能性がある。
2022年1月、私たちはリンカーンパーク資本基金有限責任会社(“リンカーンパーク”)と株式購入契約(“購入契約”)を締結し、購入契約の36ヶ月以内にリンカーン公園に最大5,000万ドルの普通株を販売することを規定したが、いくつかの条件の制限を受け、2023年12月31日までに310万ドルを売却した。
2022年9月に,吾らはCantor Fitzgerald&Co(“Cantor”)と制御持分発売契約(“販売合意”)を締結し,この合意に基づき,吾らは我々のS-3表登録声明(“ATM発売計画”)に基づいて総生産5,000,000ドルの普通株を随時販売することができる.2023年12月31日までの1年間に、合計537,200株の普通株を売却し、総収益は720万ドルだった。販売エージェントが当時の市場条件や適切と考えられる数量や価格に応じて将来の販売に成功する保証はない.また、米国証券取引委員会の現行規定によると、本年度報告書10-K表を提出した時点で、私たちの公衆持株量は7,500万ドル未満であり、米国証券取引委員会の規定によると、私たちの公衆持株量が依然として7,500万ドル未満である限り、私たちは任意の12ヶ月間に保留登録声明を使用して、初公開証券の公開によって調達した資金は、私たちの公衆保有量の3分の1を超えてはならないということは、乳児棚上げ規則と呼ばれている。2024年3月15日現在、我々の公開流通株は約6,560万ドルであり、非関連会社が保有する3914,309株に基づいて普通株を発行しており、価格は1株当たり16.95ドルであり、2024年3月4日に我々の普通株が最後にナスダック資本市場で発表された販売価格である。私たちの公開流通株が7500万ドルを下回るので、私たちの公開流通株が7500万ドルを超えるまで、私たちは乳児棚規則によって制限され、これは私たちが任意の12ヶ月の間に棚登録声明に従って最大3分の1の公開流通株しか販売できないことを意味する。2024年3月15日までの12ヶ月間、棚登録声明に基づいて合計1620万ドルの製品を販売しており、現在の棚登録声明に基づいて株式を売却する能力を制限することになります。
44
私たちの未来の資本需要は多くの要素に依存するだろうが、これらに限定されない
臨床試験と臨床前研究を行うことは時間がかかり、高価と不確定な過程であり、数年の時間をかけて完成する必要があり、しかも著者らは永遠に監督部門の許可を得て、著者らの候補製品を商業化するために必要なデータ或いは結果を生成できないかもしれない。しかも、私たちの候補製品が承認されれば、商業的成功を得られないかもしれない。私たちの商業収入は、もしあれば、数年以内に商業用途がないと予想される製品の販売から来ます。したがって、私たちは追加的な資金調達に依存して私たちの業務目標を達成し続ける必要があるだろう。私たちは受け入れ可能な条項で十分な追加融資を受けることができないかもしれないし、根本的にできないかもしれない。
追加資本の調達は私たちの株主に希釈し、私たちの運営を制限したり、私たちの技術や候補製品に対する権利を放棄することを要求するかもしれません。
これまで、相当な製品収入を生成することができれば、潜在的な追加協力、ライセンス、および他の同様の手配を含む、株式発行、債務融資、または他の資本源によって私たちの現金需要を満たすことが予想されています。私たちは私たちのATM製品計画やリンカーン公園購入契約に基づいて潜在的な販売を除いて、外部資金源を提供していません。私たちが株式または転換可能な債務証券を売却することによって追加資本を調達する場合、あなたの所有権資本は希釈され、これらの証券の条項は清算または他の特典を含む可能性があり、普通株主としての権利に悪影響を及ぼす可能性がある。私らとOxford Financial LLC(以下、“Oxford LSA”と呼ぶ)との融資および保証プロトコルには、追加債務を招く、資本支出を行う、または配当を宣言するなど、特定の行動をとる能力を制限または制限するチノを含む任意の将来の債務融資および優先株融資(例えば、可能性がある)に関連するプロトコルが含まれる。これらの制限は、私たちが業務を展開し、業務計画を実行する能力に悪影響を及ぼす可能性がある。
45
もし私たちが追加的な協力、許可、および他の同様の計画を通じて資金を調達するならば、私たちは私たちの技術、将来の収入源、研究計画、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利であり、および/または私たちの普通株の価値を低下させる可能性がある条項で許可を付与しなければならないかもしれない。もし私たちが必要な時や私たちが受け入れた条件下で株式や債務融資または他の手配を通じて追加資金を調達できない場合、私たちは私たちの製品開発または将来の商業化努力を延期、制限、減少または終了すること、または私たちが開発とマーケティングをより望んでいた候補製品を開発し、マーケティングする権利を付与することを要求されるだろう。
リンカーン公園購入協定の条項はリンカーン公園に発行できる普通株の数を制限します。これは私たちがこの計画を利用して現金資源を強化する能力を制限するかもしれません。
購入契約には、(I)売却が325,357株の普通株(購入契約が実行される直前に発行された普通株の約19.99%に相当し、2024年1月12日に完了した25株の逆株式分割調整後に相当する)(“取引所上限”)、または(Ii)が売却されると、リンカーン公園およびその関連会社の実益が9.99%を超える発行および発行された普通株を有することを含む、リンカーンパークへの普通株の売却能力の制限が含まれる。2023年12月31日までに、我々は購入契約に基づいて合計29,221株の普通株を発行し、取引所の上限を296,136株に引き下げた。したがって、今回の発行で全5,000万ドルの普通株を売却できる保証はありません。もしこれらの制限によって、リンカーン公園が購入を約束したすべての株を売ることができなければ、私たちはもっとコストが高く、より時間のかかる方法で資本市場に入る必要があるかもしれません。これは私たちの流動性と現金状況に実質的な悪影響を及ぼすかもしれません。
オックスフォードLSAの条項は私たちの運営と財政的柔軟性に制限を加えた。もし私たちが債務融資を通じて追加資本を調達すれば、どんな新しい債務の条項も私たちが業務を運営する能力をさらに制限するかもしれない。
私たちは2023年12月31日まで、オックスフォードLSAで元金2000万ドルの未返済定期ローンを持っている。オックスフォード大学LSAは、2023年6月30日までにいくつかの臨床開発マイルストーンを達成したときに1,000万ドルを追加的に抽出することを可能にした(“B期ローン”)。我々が先に2023年1月に発表したKickStart試験の1つのキューが終了したため,我々は2023年6月30日までに臨床開発マイルストーンの1つを実現しなかったため,B期ローンでの追加1,000万ドルを得ることはできなくなった。定期ローンは留置権を担保とし、私たちのほとんどの個人財産、権利と資産をカバーしており、知的財産権を含まず、知的財産権は負の質権の制約を受けている。オックスフォードLSAは私たちに適用される慣用的な肯定と否定の契約と違約事件を含む。これらの肯定的な条約には、政府の承認を維持し、いくつかの財務報告書を提出し、保険カバー範囲を維持し、重大な知的財産権を保護することを要求する条約が含まれている。負の条約は、担保の譲渡、追加債務の招く、合併または買収、現金配当金の支払い、または他の分配、投資、留置権の設立、資産の売却、および二次債務の支払いの任意の支払いの制限を含み、いずれの場合もいくつかの例外的な状況によって制限される。オックスフォード法律サービス協定の制限条項は、私たちが私たちまたは私たちの株主が有益だと思うビジネス機会を追求できないかもしれない。さらに、オックスフォード大学は、それがオックスフォードLSAで定義された重大な不利な変化と考えられるいかなる事件が発生したときに違約を宣言することができる。私たちは持続的な経営企業としての持続的な経営能力に大きな疑問があると考えているため、オックスフォード法律サービス協定の下で重大な不利な変化が生じる可能性があるかどうかの評価は私たちの制御範囲内ではなく、融資が違約とみなされる可能性のあるリスクを増加させることが確認された。もし私たちがオックスフォードLSAの下で約束を破ったら、オックスフォードは私たちのすべての返済義務を加速させ、私たちの資産を統制するかもしれないが、これは私たちが合意の中で私たちにあまり有利ではない条項を再交渉するか、または直ちに運営を停止する必要があるかもしれない。さらに、もし私たちが清算された場合、オックスフォード大学が返済を得る権利は、私たちの普通株式所有者が清算から任意の収益を得る権利よりも優先されるだろう。オックスフォードの違約事件に対するいかなる声明も私たちの業務と見通しを深刻に損なう可能性があり、私たちの普通株の価格下落を招く可能性がある。もし私たちが追加的な債務融資を調達すれば、このような追加債務の条項は私たちの運営と財政的柔軟性をさらに制限するかもしれない。
2023年12月31日現在、我々の経営陣と我々の独立公認会計士事務所は、2023年12月31日現在の財政年度と2023年12月31日現在の財政年度の財務諸表から、経営を継続する企業として経営を継続する能力に大きな疑問があると結論している。
我々が2023年12月31日までの財政年度監査を受けた財務諸表は、継続的に経営する企業として存続すると仮定して作成したものである。述べられた持続的な経営基盤は、私たちが予見可能な未来に経営を継続し、私たちの資産を正常に現金化し、私たちの負債を返済することができると仮定する
46
このような調整には、我々が経営を継続できないことによる資産または金額の回収可能性および分類および負債分類がもたらす可能性のある将来の影響を反映するための調整は含まれていない。2023年12月31日現在、我々の経営陣は、予想される運営損失と負のキャッシュフローに基づいて、財務諸表発表後12ヶ月以内に経営を継続する能力があるかどうかに大きな疑問があると結論した。私たちが経営を続ける能力は、購入契約に基づいて将来リンカーン公園に普通株を売却する可能性と、ATM発売計画に基づいて普通株を売却することを含む、株式発行や債務融資によって追加資本を調達する能力に依存する。さらに、ファイザー協定によると、私たちは追加的なマイルストーン支払いを受けるかもしれない。しかし、私たちはタイムリーまたは優遇的な条件で追加的な融資を得ることができないかもしれないし、もしあれば、何の記念碑的な支払いも得られないかもしれない。もし私たちが経営を続けることができなければ、私たちは私たちの資産を清算しなければならなくなり、これらの資産の私たちの財務諸表に対する価値を得ることができ、私たちの株主は彼らの私たちへの投資の一部または全部を失うかもしれない。もし私たちが将来のビジネス活動を支援するための追加資金を求めている場合、私たちが継続的に経営している企業としての能力には依然として大きな疑問があり、投資家や他の融資源は商業的に合理的な条項や追加の資金を提供したくないかもしれない。
我々の候補製品の発見,開発,規制承認に関するリスク
われわれはトミフセテとゾチフェンの成功に大きく依存しており,この2種類の薬剤は第二段階の臨床開発にある。もし私たちまたは私たちの協力者が開発に成功し、規制機関の私たちの候補製品の承認を得て商業化することができなかったら、あるいはこの点で重大な遅延があったら、私たちの業務は深刻な損害を受けるだろう。
我々の開発はまだ初期段階であり,臨床開発には2種類の候補製品であるトミフザチブとゾタフェンしかない。私たちとファイザーの協力の下で、私たちのもう一つのeIF 4 E阻害剤に焦点を当てた開発プロジェクトはまだ臨床前段階にある。私たちが製品収入を作る能力は私たちの候補製品の成功と最終商業化に大きく依存します。私たちは製品収入が何年も起こらないと予想しています。私たちの候補製品の成功は以下のいくつかの要素に依存するだろう
47
上に挙げられた多くの要素は私たちがコントロールできないことであり、私たちが重大な遅延に遭遇したり、規制部門の許可を得たり、私たちの候補製品を商業化することを阻止したりする可能性がある。もし私たちまたは私たちの協力者が私たちの候補製品に対する規制部門の承認を得ることができなければ、あるいは承認されれば、私たちは私たちの業務を継続するのに十分な収入を生むことができないかもしれない。
我々の技術プラットフォームに基づいて候補製品を発見·開発する方法は検証されておらず,ビジネス価値のある製品を開発できるかどうか,あるいは競争方法が候補製品の商業価値を制限するかどうかは分からない.
私たちの業務の成功は、主に私たちの独自の選択的翻訳法規技術プラットフォームに基づいて、私たちの候補製品を識別、開発、商業化する能力にかかっています。また,我々の候補製品はいくつかの疾患キネシンを下方制御することを目的としており,いずれの承認された治療法もこれらの問題を十分に解決していないことは,これらの標的の位置や複雑さによるものと考えられる。我々は、我々の技術プラットフォームに基づく候補製品に関連する良好な臨床前研究および早期臨床試験結果が観察されたと信じているが、私たちはまだ、臨床試験において任意の候補製品の有効性および安全性を証明することに成功していないかもしれないし、FDAまたは他の規制機関のマーケティング承認を得たり、その候補製品を商業化することに成功していないかもしれない。我々の独自の選択的翻訳技術プラットフォームに基づく任意の候補製品は、有害な副作用を有することが証明される可能性があり、または他の特徴を有する可能性があり、追加の臨床試験が必要であるか、または候補製品が販売できないか、または上場承認を得ることができない可能性がある。特に、著者らはeIF 4 F複合体及びその活性化酵素、マイトジェン活性化プロテインキナーゼ(“MAPK”)相互作用蛋白(“MNK”)の構成要素を標的とし、同時に複数の疾病キネシンを下方制御する新しい方法は予期しない結果を産生する可能性があり、著者らの候補製品の開発と承認に成功できない不良事件を含む。さらに、私たちの現在のすべての候補製品および開発計画はeIF 4 F ComplexおよびMNKに集中しているため、私たちの候補製品または開発計画の不利な発展は、私たちの他の候補製品または開発計画の実際または予想成功可能性および価値に重大な悪影響を及ぼす可能性がある。
しかも、バイオテクノロジーと生物製薬産業の特徴は技術が急速に進歩していることだ。私たちの未来の成功は私たちが私たちの科学的方法で競争地位を維持できるかどうかにある程度かかっているだろう。私たちが技術変革の最前線に立って、私たちの方法を利用してSTRI候補製品を創造し開発することができなければ、効率的に競争できないかもしれません。私たちの競争相手は私たちの方法を時代遅れにするかもしれないし、既存の技術的方法の進歩や新しい方法や異なる方法の開発によって私たちの候補製品の商業的価値を制限し、それによって、私たちの方法から得られたと考えられる薬物発見過程における利点を潜在的に除去することができる。対照的に、類似した方法を使用しようとする他の会社の不利な発展は、私たちの候補製品の実際または知覚的価値と潜在力に悪影響を及ぼすかもしれない。上記のいずれかの事件が発生した場合、1つまたは複数の計画に対する開発作業の延期、修正、または放棄を余儀なくされる可能性があり、これは、私たちの業務に大きな悪影響を与え、運営を停止させる可能性があります。
臨床と臨床前開発は1つの長く高価な過程に関連し、結果は不確定であり、臨床前研究と早期臨床試験の結果は必ずしも未来の結果を予測できるとは限らない。我々のどの候補製品も今後の臨床試験で有利な結果がない可能性があり,あれば,あるいは速やかに監督部門の承認を得ることができる(あれば)。
臨床や臨床前の開発費用は高価であり,完成までに数年かかる可能性があり,その結果自体も定かではない。著者らはいかなる臨床試験或いは臨床前研究が計画通りに或いは予定通りに完成することを保証できず、しかも臨床前研究或いは臨床試験過程中のいつでも失敗する可能性があり、著者らがコントロールできない要素を含む。また,臨床試験データ読み出しの期待時間枠を満たすことができない可能性がある。臨床前或いは臨床結果の将来性は有望であるが、任意の候補製品は臨床前或いは臨床開発の任意の段階で意外に失敗する可能性がある。歴史的障害率は
48
私たちの業界の製品候補は高いです。同じ種類の候補製品或いは競争相手候補製品の臨床前研究或いは臨床試験の結果は著者らの候補製品の後続の臨床試験の結果を予測できない可能性があり、臨床試験の中期、背線或いは初歩的な結果は必ずしも最終結果を代表するとは限らない。臨床前研究と初歩的な臨床試験で進展を得たが、臨床試験後期段階の候補製品は期待した安全性と有効性特徴を示すことができないかもしれない。特に,トミフゼチブのいくつかの前臨床研究や早期臨床試験が行われているが,トミソッサーがこれらの先行研究のように進行中や将来の臨床試験に機能するかどうかは不明である。臨床試験では前臨床研究と早期臨床試験に基づく予期せぬ結果が観察されることは珍しくなく,多くの候補製品は臨床試験で失敗しており,早期結果は非常に有望であるにもかかわらず。製薬と生物技術業界のいくつかの会社は臨床開発において重大な挫折を経験し、早期の研究においても奮い立つ結果を得た。
これらの理由から,われわれが進行·計画中の臨床試験や臨床前研究が成功するかどうかは確認できない。私たちの目標適応のいずれの臨床試験で観察される任意の安全問題も、これらの適応および他の適応において規制部門の承認を得る見通しを制限する可能性があり、これは、私たちの業務、財務状況、および運営結果に大きな悪影響を及ぼす可能性がある。
現在または計画中の臨床試験の開始または完了過程における任意の困難または遅延、または任意の終了または一時停止は、私たちのコスト増加を招き、私たちの収入を創出する能力を延期または制限し、私たちのビジネスの将来性に悪影響を及ぼす可能性がある。
FDAの許可を得て新薬を発売するために、著者らは著者らの候補製品の人体上の安全性と有効性を証明し、FDAを満足させなければならない。これらの要求を満たすためには,十分かつ良好な制御の臨床試験が必要である。臨床テストは高価で時間がかかり、不確実性も存在する。
私たちまたは私たちの協力者が候補製品の臨床試験を開始することができる前に、私たちまたは彼らは、INDまたは同様の規制提出の一部として、候補製品の化学、製造および制御、および提案された臨床試験案に関する情報を含む臨床前研究結果および他の情報をFDAまたは同様の外国規制機関に提出しなければならない。FDAまたは同様の外国の規制機関は、任意の候補製品に対する追加の臨床前研究を私たちまたは私たちの協力者に要求し、その後、任意のINDまたは同様の規制提出書類に従って臨床試験を開始することを可能にするかもしれません。これは、私たちの臨床前開発計画の遅延とコストを増加させる可能性があります。また,これらの試験が開始されても,規制当局のこのような臨床試験の一時停止や終了を招く可能性がある。我々が行っていることや計画中の臨床試験の開始や完成のいかなる遅延も,我々の製品開発スケジュールや製品開発コストに大きな影響を与える可能性がある。
私たちは私たちの計画中の実験が時間通りに始まるかどうか、あるいは予定通りに完了するかどうか分からない。臨床試験の開始、データ読み出し、完了は様々な原因で遅延する可能性があり、以下に関連する遅延を含む
49
臨床試験はFDAや他の適用規制機関の法的要求,法規やガイドラインに基づいて行われ,これらの政府機関や臨床試験を行う医療機関の道徳委員会やIRBsの監督を受けなければならない。臨床試験が我々,このような試験を行っている機関のIRBs,そのような試験のデータ安全監視委員会やFDAなどの外国規制機関によって一時停止または終了されれば,我々も遅延に遭遇する可能性がある。このような主管部門は一連の要素のために臨床試験を一時停止または終了する可能性があり、これらの要素は、監督管理要求または著者らの臨床規程に従って臨床試験、FDAまたは同様の外国の監督機関の臨床試験操作または試験場所の検査を行うことができなかったことによる臨床一時停止、予見できない安全問題または副作用、ある種の薬物の使用の利益を証明できなかった、政府法規または行政措置の変化、または十分な資金が不足して臨床試験を継続することを含む。また、規制要求と政策は変化する可能性があり、私たちはこれらの変化に適応するために臨床試験方案を修正する必要があるかもしれない。修正案は,われわれの臨床試験案をIRBsに再提出して再検査することが要求される可能性があり,臨床試験のコスト,時間,あるいは成功に影響する可能性がある。
また,われわれの海外での臨床試験は追加的なリスクをもたらし,われわれの臨床試験の完成を遅らせる可能性がある。これらのリスクには、外国に登録された患者が医療サービスや文化的慣習の違いにより臨床案を遵守できなかったこと、外国規制計画に関連する追加行政負担の管理、戦争を含むこれらの外国に関連する政治的·経済的リスクが含まれる。
さらに、臨床試験の終了または一時停止、または臨床試験の開始または完了遅延をもたらす多くの要因も、最終的に候補製品の規制承認を拒否する可能性がある。私たちは候補製品の処方または生産変更を行うかもしれませんが、この場合、これらの新しい処方から得られた結果が以前の結果と一致することを証明するために、追加の臨床前研究および/または臨床試験を行う必要があるかもしれません。したがって、私たちの臨床試験に生じるどんな遅延も、候補製品を商業化する独占的な権利を持つ可能性のある任意の期限を短縮することができ、私たちの競争相手は私たちの前に製品を市場に出すかもしれません。私たちの候補製品の商業的可能性は著しく低下するかもしれません。このようなどんな状況でも、私たちの業務、財政状況、そして見通しに大きな被害を及ぼす可能性がある。
50
私たちの臨床試験に参加する患者を募集することは難しいかもしれない。われわれが臨床試験で被験者を募集する困難に遭遇した場合,われわれの臨床開発活動は延期されたり,他の悪影響を受けたりする可能性がある。
FDAや米国以外の同様の規制機関の要求に応じて、これらの試験に参加するのに十分な数の合格者を決定し、募集することができなければ、候補製品の臨床試験を開始または継続できない可能性がある。被験者登録は臨床試験スケジュール中の重要な要素であり、それは多くの要素の影響を受け、それは患者群の規模と特徴、患者と臨床場所の接近程度、試験の資格と排除標準、臨床試験の設計、登録した患者が臨床試験を完成できないリスク、著者らは適切な能力と経験を持つ臨床試験研究者を募集する能力、著者らは患者の同意を得て維持する能力、医師の患者の転換やり方、治療期間と治療後に患者の能力を十分に監視する能力を含む。競合する臨床試験および他の既存療法に対する研究中の候補製品の潜在的優位性およびリスクに対する臨床医および患者の見方は、我々が研究している適応のために承認される可能性のある任意の新製品および開発されている任意の候補製品を含む。
私たちは私たちのすべての臨床試験のために十分な数の被験者を決定し、募集することを要求される。任意の計画された臨床試験の潜在的被験体は、私たちの標的疾患として十分に診断されていないか、またはそのような試験の進入基準に適合していない可能性がある。われわれが進行·計画している臨床試験に適した疾患段階の患者を決定·募集することや,治療期間や治療後にこれらの患者を十分にモニタリングすることも困難である可能性がある。また,NSCLCや乳癌患者および我々が評価しようとしている他の癌患者の大量の臨床試験を募集することが求められており,十分な数の患者,特に我々の特定の登録基準に適合する患者の募集が遅れたり困難になったりする可能性があり,計画通りに試験を完了する(あれば)。FDAや同様の外国の規制機関が要求する臨床試験に参加するのに十分な数の合格者を見つけることができなければ、臨床試験を開始または継続できない可能性がある。また,患者を発見し診断する過程は,現在も高価であり続ける可能性がある。著者らの臨床試験の時間スケジュールは著者らが患者を募集して試験に参加する速度、及び必要な後続時期の完成状況にある程度依存する。私たちの臨床試験の資格基準はさらに利用可能な試験参加者を制限した。患者が何らかの理由で私たちの試験に参加したくない場合、類似した患者集団に対する並行臨床試験が存在し、承認された治療方法が存在するか、または十分な数の患者を募集することが困難である場合、被験者を募集し、研究を行い、規制部門の私たちの候補製品に対する承認を得るスケジュールが遅れる可能性がある。例えば,2023年1月には,われわれのトミフザ置換併用転移性非小細胞肺癌患者のKickStart試験のPD−L 1>50%キューとPD−L 1>1%キューを維持する人員問題と臨床サイトを介した他の試験の登録挑戦を発表した。そこで,PD-L 1>1%のキューへの登録を停止し,PD-L 1>50%のキューに重点を置いた.2024年4月初めにPD−L 1>50%キューのバックラインデータを報告する予定である。また,われわれの臨床試験は進行/転移性癌患者を募集する可能性があるため,これらの患者は通常疾患の末期にあり,我々の候補製品とは独立した臨床疾患の進展を経験する可能性があり,臨床試験では評価できず,追加の患者登録が必要となる。私たちは将来のどの臨床試験のために十分な数の被験者を募集することができず、重大な遅延を招くか、あるいは1つ以上の臨床試験を完全に放棄する必要があるかもしれない。また,将来の臨床試験が適切かつタイムリーに行われることをCROと臨床試験地点で確保する予定であり,彼らのサービスを管理する協定を締結しているが,彼らの実際の表現への影響は限られている。予想される臨床試験スケジュールを決定するための仮定が正しいか、あるいは登録遅延に遭遇しないことは、このような試験の完了が予想されるスケジュールの後まで遅延することを保証することはできません。
私たちの候補製品の使用は、副作用、有害事象、または他の性質または安全リスクに関連する可能性があり、これは承認を延期または阻止する可能性があり、私たちが臨床試験を一時停止または停止させ、候補製品を放棄し、承認されたラベルの商業イメージを制限すること、または他の深刻な負の結果を招き、私たちの業務、将来性、経営業績、財務状況を深刻に損なう可能性がある。
腫瘍薬の一般的な場合と同様に,我々の候補品の使用に関連する副作用や有害事象がある可能性がある。われわれの臨床試験結果は副作用や予期せぬ特徴の重症度と流行度を示す可能性がある。私たちの候補製品を単独で使用したり、他の承認された薬物や研究試薬と組み合わせて使用した場合に生じる副作用は、私たちまたは規制機関の臨床試験の中断、延期、または停止を招き、より厳しいラベルをもたらす可能性があり、またはFDAまたは同様の外国の規制機関が規制承認を延期または拒否する可能性がある。麻薬関係の
51
副作用は患者の募集や患者の試験完了能力に影響し、あるいは潜在的な製品責任クレームを招く可能性がある。このようなどんな状況でも、私たちの業務、財政状況、そして見通しに大きな被害を及ぼす可能性がある。
さらに、私たちの候補製品が臨床試験において副作用に関連している場合、または予期しない特徴を有する場合、私たちは、それらの開発を放棄することを選択するか、またはそれらの開発をより狭い用途または集団に制限することが可能であり、リスク-利益の観点から、副作用または他の特徴は、それほど一般的ではなく、それほど深刻ではなく、またはより容易に受け入れられ、これは、候補製品の商業的期待を制限する可能性がある。われわれが行っている臨床試験の結果に基づいてわれわれの研究計画を修正することも求められるかもしれない。
著者らの固形腫瘍患者のトミフセテ1期用量増加試験において、カプセル製剤を使用し、最もよく見られる治療に関連する有害事象(TRAE)は吐き気、嘔吐、疲労、便秘、消化不良と振戦である。推奨されている第二段階用量(“RP 2 D”)を超えると,TRAEの発生率と重症度がより高いことが観察された。われわれのリンパ腫患者トミソチブ1期用量増加試験では,RP 2 D拡張キューにおいて,患者が経験した最もよく見られるTRAEは嘔気,嘔吐,高カルシウム血症,疲労であった。トミソチブと抗PD-(L)1薬物を併用した2 a期試験において、最もよく見られるTRAEは吐き気、無力、振戦、嘔吐、アスパラギン酸トランスアミナーゼの上昇とアラニントランスアミナーゼの上昇である。これらのTRAEの重症度は一般的に1級あるいは2級であるが、2例の患者のアラニンアミノトランスフェラーゼの上昇、血クレアチンホスホキナーゼの上昇と皮疹はすべて3級であった。
著者らの1/2期臨床試験において、ゾタチフェンはある突然変異を有する固形腫瘍患者に対して完成した第一段階用量増加部分を行い、著者らは3種類の用量制限毒性(DLTS)を観察した。週0.035 mg/kg静注キューで観察された最初のDLTは2級血小板減少症であり,DLTウィンドウ全体の治療継続を阻害した。2週間後と1週間後に0.1 mg/kgを静脈内投与し,2回目と3回目のDLTを観察した。したがって,0.1 mg/kgの投与量は最大耐容量(“MTD”)を超えた。1人の患者は3級貧血のDLTを経験し,もう1人の患者は2級血小板減少下にDLTの3級胃腸出血を経験した。すべての用量レベルの全体的な副作用(“副作用”)は主にレベル1と2級の吐き気、嘔吐と貧血を含む。
われわれが行っている臨床試験の結果に基づいて,われわれの開発と臨床試験計画を修正する必要があるかもしれない。多くの化合物は当初癌治療の早期テストで希望を示したが,その後副作用が認められ,この化合物のさらなる進展を阻止したり,より大きな患者群では統計的に有意な効果を示すことはできなかった。さらに、規制部門は、不利な安全発見をさらに探索するために、異なる結論を出したり、追加的なテストを要求したりする可能性がある。
私たちがより大きく、より長く、より広範な臨床試験で私たちの候補製品を試験する場合、異なる用量レジメンの使用を含む場合、またはこれらの候補製品の使用がより広くなる(規制部門の承認を得た場合)被験者は、早期試験で観察された疾患、傷害、不快感、および他の有害事象、および以前の試験で発生しなかったか、または検出されなかったことを報告する可能性がある。これらの副作用が開発後期に、または承認された後に知られていれば、これらの発見は、私たちの業務、財務状況、および将来性に大きな損害を与える可能性がある。また,われわれが行っている計画中のトミフザ置換連合プログラム細胞死蛋白1(PD−1)とプログラム細胞死リガンド1(PD−L 1)の阻害剤(総称して抗PD−(L)1治療)の臨床試験は,併用治療による有害事象を引き起こす可能性があり,このような臨床試験で報告されている有害事象に悪影響を及ぼす可能性がある。抗PD-(L)1療法はすでに肝臓と他の器官系の免疫関連不良事象を含む不良事象の存在が証明されており、これは著者らの臨床試験の最大用量を制限するか、あるいは他の方法で著者らの連合臨床試験に負の影響を与える可能性がある。私たちの候補製品の治療を受けた患者はまた手術、放射線と化学療法を受けている可能性があり、これは私たちの候補製品とは関係のない副作用或いは不良事件を招く可能性があるが、依然として著者らの臨床試験の成功に影響を与える可能性がある。重篤な患者を著者らの臨床試験に組み入れることは死亡或いはその他の不良医療事件を招く可能性があり、原因はこれらの患者は他の治療方法或いは薬物を使用している可能性があり、或いはこれらの患者の病状が深刻であるためである。例えば、私たちの臨床試験に登録されたいくつかの患者は、私たちの臨床試験中に、あるいはそのような試験に参加した後に死亡したり、重大な臨床イベントを経験することが予想される。
さらに、もし私たちの1つまたは複数の候補製品が発売承認され、私たちまたは他の人が後にこのような製品による不良副作用を発見した場合、多くの潜在的な重大な負の結果を招く可能性がある
52
これらの事件のいずれも、特定の候補製品に対する市場の受け入れ度を達成または維持することを阻止することができ、承認されれば、私たちの業務、運営結果、および将来性を深刻に損なう可能性がある。
組織として、私たちは重要な臨床試験を完成したことがなく、私たちのすべての候補製品の臨床試験を完成できないかもしれない。
FDAや同様の外国の規制機関の承認を得て、候補製品を市場に出すために、私たちが計画した臨床試験および後期と重要な臨床試験を成功させる必要があります。後期臨床試験と提出に成功したNDAは複雑な過程である。組織として、私たちは以前に何の重要な臨床試験も行っておらず、マーケティング申請の準備と提出の面で経験が限られており、候補製品の機密協定や他の類似した外国監督申請も提出されていない。また,FDAとのインタラクションは限られており,我々の候補製品に対してどれだけの追加の臨床試験が必要か,あるいはこれらの試験をどのように設計すべきかは決定できない。したがって、私たちは必要な臨床試験を成功的かつ効率的に実行し、完成することができず、規制部門が私たちの候補製品を提出し、承認することにつながるかもしれない。私たちは私たちの競争相手よりも多くの時間とより多くのコストを必要とするかもしれないし、私たちが開発した候補製品の規制承認を得ることに成功しないかもしれない。私たちの計画した臨床試験を開始または完了または遅延させることができず、候補製品のNDAを提出し、商業化することを阻止または遅延する可能性がある。
私たちは他の療法と組み合わせて使用するために私たちの候補製品を開発しており、これは私たちをより多くのリスクに直面させるだろう。
我々は、fulvestrant、HER 2阻害剤、Herceptin、KRAS G 12 C阻害剤およびCDK 4/6阻害剤abemaciclibなどのER阻害剤との併用のために、現在承認されている1つまたは複数の抗PD-(L)1療法およびゾチフェンとの併用を開発している。Fulvestrantは後発薬であり,アスリコンを含むいくつかの会社で販売され,アスリコンはFaslodexのブランドで販売され,乳癌治療に用いられている。ヘキセチンは遺伝子テークが所有し販売されており,乳癌や他の癌の治療に用いられている。2種類のKRAS G 12 C阻害剤は現在非小細胞肺癌の治療に許可されている。AbemaciclibはER+/Her 2−乳癌の治療にVerzenioという名称で礼来社によって販売されている。したがって、TomivosertibまたはZotatifinが共同使用のために発売承認または商業化されていても、FDAまたは同様の外国規制機関がPD-(L)1療法またはER、HER 2またはKRAS G 12 C阻害剤とそれぞれTomivosertibまたはZotatifinとの併用承認を撤回する可能性があり、またはこれらの併用治療に安全性、有効性、製造または供給の問題が生じる可能性があるというリスクを負い続ける。併用療法は通常癌の治療に用いられており,他の候補品を開発して他のカテゴリーの腫瘍療法と併用すれば,類似したリスクに直面する。承認された抗PD−(L)1療法やER,HER 2およびKRAS G 12 C阻害剤を用いて併用療法を開発し,トミフザチブとゾタフェンをそれぞれ計画しているように,われわれが開発可能な任意の併用療法における各有効成分の安全性と有効性を証明するためにデータを収集する必要があるなど,臨床や開発に関連するリスクをより多く直面させている。さらに、トミフザチブ、ゾタフェンまたは他の候補製品と、FDAまたは同様の外国規制機関の承認を得ていない1つまたは複数の他の癌療法との組み合わせを評価することも可能である。候補製品をマーケティングして販売することはできないかもしれません
53
このような未承認癌療法と組み合わせて使用するために使用されていますが、これらの療法は最終的に自分のマーケティング承認を得ません。
FDAまたは同様の外国の規制機関がこれらの他の併用薬を承認したり、その承認を撤回したりしない場合、または私たちの候補製品と共同評価する薬剤に安全性、有効性、製造または供給の問題が生じることを選択した場合、私たちはトミソ社、ゾチフェン、または他の候補製品の承認を得ることができないかもしれない。
さらに,われわれの臨床試験で1つ以上の組み合わせ薬を使用することは,このような臨床試験のコストを増加させる。さらに、私たちの候補製品と組み合わせて使用されている療法または開発中の療法の第三者プロバイダが、臨床試験または候補製品の商業化のために十分な数を生産できない場合、または併用療法のコストが高すぎる場合、私たちの開発および商業化努力が損なわれることは、私たちの業務、財務状況、運営結果、および成長の見通しに悪影響を及ぼすであろう。
私たちは、より利益があるか、またはより成功する可能性のある候補製品または適応を利用することなく、特定の候補製品を追求するために限られた資源を使うかもしれない。
私たちの財務と管理資源が限られているので、私たちは特定の候補製品と特定の適応に集中している。したがって、私たちは他の候補製品を探す機会を放棄したり延期したりするかもしれないが、これらの機会はより大きなビジネス潜在力を持つことができる。具体的には,eIF 4 F複合体とその活性化キナーゼMNKに特化した候補製品を開発しており,これらの目標に対する抑制に敏感な適応の候補製品を優先的に開発している。例えば,マイクロサテライト安定型結腸直腸癌患者におけるTomivosertibとPD−L 1阻害剤Avelumabの併用試験が完了した後,Tomivosertibの将来の開発はより免疫反応性の強い癌に重点を置くことを選択した。同様に,著者らは去勢耐性前立腺癌患者におけるトミフザ置換の臨床試験を中止し,トミソと抗PD−(L)1併用治療の開発に重点を置いた。私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。現在および将来の研究開発計画および特定の適応の候補製品への支出は、いかなる商業的に実行可能な候補製品も生じない可能性がある。もし私たちが特定の候補製品の商業的潜在力や目標市場を正確に評価していなければ、私たちは協力、許可、その他の同様の手配を通じて候補製品に価値のある権利を放棄する可能性があり、この場合、候補製品の独占開発権と商業化権利を維持することは私たちに有利である。
著者らは時々発表或いは公表した臨床試験と臨床前研究の中期、主要と初歩データは更に多くの患者データの獲得に従って変化する可能性があり、そして監査と検証プログラムの影響を受け、これらのプログラムは最終データの重大な変化を招く可能性がある。
著者らは時々著者らの臨床試験と臨床前研究の中期、背線或いは初歩データを公開開示する可能性があり、これらのデータは当時使用可能なデータの初歩的な分析に基づいて、特定の試験に関連するすべてのデータを全面的に分析した後、結果及び関連する発見と結論は変化する可能性がある。私たちはまた、私たちのデータ分析の一部として、すべてのデータを全面的かつ詳細に評価する機会がないか、または受け取る機会がないかもしれないという仮説、推定、計算、および結論を出した。したがって、追加のデータが受信され、十分に評価されると、私たちの報告書の中期、バックライン、または予備結果は、同じ試験の将来の結果とは異なる可能性があり、または異なる結論または考慮要因が、これらの結果を合格させる可能性がある。バックラインと初歩データは依然として監査とチェック手続きを受けなければならず、これは最終データが以前公表された予備データと大きく異なる可能性がある。したがって、最終データが利用可能になる前に、バックラインおよび予備データは慎重に表示されなければならない。
私たちはまた私たちの臨床試験の中間データを開示するかもしれない。著者らが完成する可能性のある臨床試験の中期データは、患者登録の継続とより多くの患者データの獲得に伴い、1つ以上の臨床結果が実質的に変化する可能性があるというリスクに直面している。中間データ、バックラインデータ、または予備データと最終データとの間の不利な差は、私たちのビジネスの将来性を深刻に損なう可能性があります。
さらに、他の人は、規制機関を含み、私たちの仮定、推定、計算、結論または分析を受け入れないか、またはデータの重要性を異なる方法で解釈またはトレードオフする可能性があり、これは、特定の計画の価値、特定の製品の承認または商業化に影響を与える可能性がある
54
候補者や製品と私たちの全体的な業務。さらに、私たちが開示する特定の研究または臨床試験に関する開示を選択する情報は、一般に広範な情報に基づいており、あなたまたは他の人は、私たちが開示すべき重大な情報または他の適切な情報を含むことを決定することに同意しない可能性があり、私たちが開示しないことを決定した任意の情報は、最終的には、特定の薬剤、候補製品または私たちの業務に関する将来の決定、結論、観点、活動、または他の側面に対して大きな意味を有すると考えられるかもしれない。もし私たちが報告した中期、バックライン、または予備データが実際の結果と異なる場合、または規制機関を含む他の人が結論に同意しない場合、私たちの候補製品が承認され、それを商業化する能力が損なわれる可能性があり、私たちの業務、経営業績、見通し、または財務状況が損なわれる可能性がある。
候補製品の製造または処方を変更する方法は、追加のコストや遅延を招く可能性がある。
候補製品が臨床試験から発売承認と商業化まで発展することに伴い、開発計画の各方面、例えば製造方法と調合は、この過程で常に変化し、安全性、有効性、生産量と生産ロットを最適化することに努力し、最大限にコストを下げ、一致した品質と結果を実現する。私たちは未来に候補製品に対するいかなる変更もこれらの候補製品の表現の違いを招く可能性があり、そして未来の臨床試験の結果に影響を与えるかもしれない。このような変化或いは関連する不利な臨床試験結果は追加の臨床試験の開始或いは完成を遅らせる可能性があり、接続研究或いは臨床試験或いは1つ以上の研究或いは臨床試験を繰り返し、開発コストを増加させ、潜在的な市場承認を延期或いは阻止し、私たちの候補製品の商業化(承認された場合)と収入を創造する能力を脅かす必要がある。
私たちは、承認を加速するルートを使用することで、FDAや同様の外国規制機関の承認を得ることを試みるかもしれない。もし私たちがそのような承認を得ることができなければ、私たちは予想以上の追加臨床試験を要求される可能性があり、これは必要な上場承認を得る費用を増加させ、必要な上場承認を遅らせる可能性がある。FDAの加速承認を得ても,我々の検証的試験が臨床的利益を証明していない場合,あるいは厳格な上場後の要求を守らなければ,FDAは加速承認の撤回を求める可能性がある。
私たちは未来に私たちの1つ以上の候補製品のための加速的な承認を求めるかもしれない。加速承認計画によれば、FDAは、代替終点または中間臨床終点に対して臨床的利益を合理的に予測することができる候補製品を決定する場合に、既存の治療法よりも意義のある治療利益を提供する、深刻または生命に危険な疾患の治療のための候補製品の承認を加速することができる。FDAは臨床利益は特定の疾病の背景下で臨床意義のある積極的な治療効果であり、例えば不可逆的な発病率或いは死亡率であると考えている。承認を加速するために、代替終点は1つの標識であり、例えば実験室測定、放射画像、バイタルサイン或いは他の臨床利益を予測できると考えられる指標であるが、それ自体は臨床利益の測定基準ではない。中間臨床終点は不可逆的発病率或いは死亡率への影響の前に測定できる臨床終点であり、それは不可逆的な発病率或いは死亡率或いは他の臨床利益に対する影響を合理的に予測する可能性がある。加速承認経路は既存療法に対する新薬の優位性は直接の治療優位ではないかもしれないが、患者と公衆衛生の観点から見ると臨床上重要な改善状況である。承認された場合、承認を加速することは、一般に、薬剤の臨床的利益を確認および説明するために、勤勉な方法で追加の検証的研究を行うことにスポンサーが同意することに依存する。もしこのような承認後の研究がこの薬物の臨床治療効果を確認できなかった場合、或いは適時に完成しなかった場合、FDAは迅速にこの薬物の承認を撤回することができる。また、2022年12月、総裁·バイデンは2023年度まで米国政府に資金を提供する総合支出法案に署名した。総合法案には、患者が以前加速された承認を受けた無効薬物の潜在的リスクを軽減するために、他に加えて、FDAに新たな法定権力を付与する2022年の食品·薬物総合改革法案が含まれている。これらの規定により、FDAは、承認を加速させる製品を求めるスポンサーに、承認前に検証的試験を行うことを要求することができる。
私たちの任意の候補製品の加速承認を求める前に、FDAのフィードバックを求めるつもりです。そうでなければ、私たちが承認を求めて加速させる能力を評価します。フィードバックや他の要因を評価した後、加速的な承認または任意の他の形態の加速開発、審査、または承認を得るために機密協定を求めたり提出したりすることは保証されません。さらに、私たちが候補製品の加速承認または受信申請を提出することを決定した場合、そのような申請が受け入れられるか、または加速された開発、審査、または承認がタイムリーに承認されるか、または全く保証されない保証はない。FDAや他の似たような外国の規制機関も私たちにさらなることを要求することができます
55
私たちの申請を考慮したり、任意の種類の申請を承認する前に検討します。もし私たちの候補製品が加速承認または任意の他の形態の加速開発、審査または承認を得られなかった場合、候補製品の商業化の時間がより長くなり(あれば)、候補製品の開発コストを増加させ、市場での競争地位を損なう可能性がある。
資金不足や世界的な健康問題によるFDAや他の政府機関の中断は、重要な指導部や他の人員の採用、保留、配置の能力を阻害する可能性があり、あるいは新製品や修正された製品のタイムリーまたは開発、承認、商業化を他の方法で阻止することは、私たちの業務にマイナスの影響を与える可能性がある。
FDAが新製品を審査および承認する能力は、政府予算および資金レベル、法規、法規および政策の変化、FDAのキーパーソンの雇用および保留、ユーザ費用の支払いを受ける能力、およびFDAが通常の機能を履行する能力に影響を与える可能性のある他の事件を含む様々な要素の影響を受ける可能性がある。FDAの平均審査時間は近年変動している。また,他の研究開発活動を援助する政府機関への政府の援助は政治過程の影響を受けており,この過程は本質的に不安定で予測不可能である。FDAおよび他の機関の中断は、新薬または承認された薬物の修正が必要な政府機関による審査および/または承認に要する時間を遅らせる可能性もあり、これは私たちの業務に悪影響を及ぼすだろう。例えば、ここ数年間、米国政府は何度か閉鎖されており、FDAのようないくつかの規制機関は、FDAのキー従業員を休暇にし、キー活動を停止しなければならない。
また、新冠肺炎の流行に対応するため、アメリカ食品薬品監督管理局は国内外の異なる場所の製造施設の大部分の検査を延期した。FDAは標準的な検査操作を回復したにもかかわらず、ウイルスのいかなる巻き返しや新しい変種の出現も行政や検査遅延を招く可能性がある。政府が長期的に停止している場合、または世界的な健康問題がFDAまたは他の規制機関の定期検査、審査または他の規制活動を阻害した場合、FDAまたは他の規制機関が私たちが提出した規制文書を適時に審査して処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に重大な悪影響を及ぼす可能性がある。
私たちは将来のいくつかの候補製品のための孤児薬物指定を求めるかもしれないが、私たちはそのような指定を得ることができないか、または市場排他性を含む孤児薬物指定に関連する利点を得ることができないかもしれないが、これは私たちの製品収入を減少させる可能性がある。
私たちは私たちのいくつかの候補製品のために孤立した製品指定を求めるかもしれないが、私たちは決してそのような指定を受けないかもしれない。孤児医薬品法によると、医薬品がまれな疾患や疾患を治療しようとしている場合、FDAは孤児薬に指定することができる。まれな疾患または疾患の定義は、米国では患者数が20万人未満、または米国では患者数が20万人を超えているが、米国では薬物開発のコストは米国での販売によって回収できないことである。秘密保持協定を提出する前に、指定孤児薬を申請しなければならない。米国では,孤児薬を指定することで一方が臨床試験費用,税収割引,申請費の減免のために贈与資金を提供する機会などの財政的奨励を受ける権利がある。FDAが孤児薬物指定を承認した後、FDAは、この薬剤の模倣薬識別情報およびその潜在的孤児用途を開示する。
さらに、1つの製品が孤児の資格を有する疾患または条件に対するFDAの最初の承認を得た場合、この製品は、孤児の独占製品に対する臨床的優位性を示す権利がある場合、または製造業者が孤児患者集団に十分な製品数を提供することが保証されない限り、限定された場合、例えば孤児独占製品に対する臨床的利点を示すか、または製造業者が孤児患者集団に十分な製品数を提供することを保証できないことを意味する。私たちまたは私たちの協力者が孤児によって指定された疾患または条件よりも承認範囲の広い疾患または条件を求める場合、米国で独占営業権を得ることができない可能性があり、FDAが後に指定要求に重大な欠陥があると判断した場合、独占営業権を失う可能性がある。
孤児薬の称号を得ても,医薬品開発に関する不確実性により,特定の孤児の疾患や状況に対する上場承認を得た最初の会社ではない可能性がある。また,候補製品の孤立薬物排他性を獲得しても,この排他性は異なる薬物が同じ疾患や疾患のために承認される可能性があるため,競合から製品を効率的に保護することができない可能性がある。孤児薬が承認された後であっても、FDAが後者の薬物が臨床的により安全で、より効果的であると結論した場合、
56
患者の看護への主な貢献。孤児薬物を指定することは薬物の開発時間や監督審査時間を短縮することもなく、監督審査或いは承認過程において薬物にいかなる利点をもたらすこともない。
FDAの迅速なチャネル指定は、私たちに任意の候補製品を付与しても、より速い開発や規制審査や承認過程を招くことはなく、私たちの製品候補が規制承認を得る可能性を増加させることはありません。
2023年11月、FDAは、エストロゲン受容体陽性(ER+)/ヒト上皮増殖因子陰性(HER 2-)末期または転移性乳癌患者の治療のための二線治療およびCDK 4/6阻害剤治療を受けた後の病状の進行のためのフルビスターおよびアベミリブ(ZfA三連体)との併用を許可し、これらの患者は、内分泌治療およびCDK 4/6阻害剤治療を受けた後に病状が進行し、我々の一部またはすべての他の候補製品のためにそのような指定を求めることができる。Fast Track計画は、特定の基準に適合する候補製品の審査プロセスを加速または促進することを目的としている。具体的には、薬物および生物学的製品が、1つまたは複数の医薬または生物学的製品と共に、深刻または生命に危険な疾患または状態を単独で治療することが意図されており、疾患または状態が満たされていない医療需要を満たす可能性が示されている場合、迅速なチャネル指定を受ける資格がある。高速チャネルは,候補製品と研究中の特定の適応に適した組合せを指定する.Fast Track候補製品のスポンサーは,製品開発期間中に適用されるFDA審査チームとより頻繁なインタラクションを行う機会があり,機密協定が提出されると,申請は優先審査を受ける資格がある可能性がある.高速チャネル候補製品のために提出されたNDAもスクロール審査を行う資格がある可能性があり、この場合、FDAは、完全な出願を提出する前に、NDAを考慮する審査部分をスクロールさせることができ、スポンサーがNDA部分を提出するスケジュールを提供した場合、FDAは、NDAの部分を受け入れることに同意し、スケジュールが許容可能であると判断し、スポンサーは、申請の第1の部分を提出する際に任意の必要な使用料を支払うことができる。
FDAは広い自由裁量権を持っていて、この称号が与えられているかどうか。ある特定の候補製品がこの認証を受ける資格があると信じていても、FDAがこの認証を承認することを保証することはできません。たとえ私たちが任意の候補製品の迅速なチャネル認証を得たとしても、これらの候補製品は、従来のFDAプログラムよりも速い開発プロセス、審査、または承認を経験しない可能性がある。FDAが我々の臨床開発プロジェクトのデータが高速チャネルの指定をサポートしなくなったと考える場合,その指定を取り消す可能性もある。また、このような指定は、Fast Trackによって指定された可能性のある他の他の候補製品が米国で規制承認される可能性を増加させることはありません。Fast Trackによって指定された候補製品の多くは最終的に承認されません。
もしFDAが私たちの任意の候補製品を承認する際にセットの診断テストの承認を得ることを要求し、私たちがFDAの診断テストの承認を得ていないか、または遅延していなければ、私たちはこの候補製品を商業化することができず、私たちの収益能力は実質的に損なわれるだろう。
もし私たちの候補製品の安全と有効な使用が体外培養もし私たちの診断製品に他の商業的用途がない場合、FDAは通常、FDAが私たちの候補製品を承認すると同時に、その診断、いわゆるセット診断を承認または承認することを要求する。FDAの指導によれば、FDAが随伴診断テストを決定することが新しい治療製品または適応を安全かつ有効に使用するために不可欠である場合、随伴診断もこの適応への使用が承認または承認されていない場合、FDAは通常、その治療製品または新しい治療製品の適応を承認しない。商業的に満足できる診断がない場合、私たちは規制部門の承認を受けて要求された診断の開発または獲得を要求される可能性がある。このような診断プログラムを取得または作成するプロセスは時間も費用もかかる。
随伴診断は関連製品の臨床計画とともに開発され,医学テストとしてFDAや類似規制機関の規制を受けており,これまでFDAは癌治療に伴う診断が発売前に承認されることが求められてきた。診断に伴う診断を治療製品ラベルの一部として承認し,治療製品の使用を診断に伴う検出のための特定の遺伝子変化を発現する患者に制限する。FDAまたは同様の規制機関が私たちの任意の候補製品の承認を要求する場合、上場承認を得る前であっても後であっても、私たちおよび/または将来の協力者は、その候補製品の承認を開発および獲得する際に困難に直面する可能性がある。我々または第三者協力者が規制機関の承認を得たキット診断プログラムの任意の遅延または失敗は、そのような候補製品の承認または営業継続を延期または阻止する可能性がある。また、セット診断のために持続可能で反復可能で拡張可能な製造プロセスを開発する際に遅延に遭遇したり、プロセスを移行したりすることも可能である
57
商業パートナーまたは保険補償計画を交渉し、これらのすべては、承認され、適時または利益があれば、私たちの臨床試験の完成を阻止するか、または候補製品を商業化することができる。
私たちの第三者への依存に関するリスクは
私たちは第三者に依存して臨床試験と臨床前研究を行う。これらの第三者がその契約義務の履行に成功しなかった場合、適用された法規要件を遵守し、または予想される期限内に完了した場合、私たちの開発計画および候補製品のために規制部門の承認を求めたり、商業化したりする能力が延期される可能性がある。
私たちは第三者に依存して臨床試験と臨床前研究を行う。具体的には,我々は使用し依存しており,医療機関,臨床研究者,CROとコンサルタントを継続して使用·依存し,われわれの臨床規程や法規の要求に基づいてわれわれの臨床前研究と臨床試験を行う予定である。これらのCRO、調査者、その他の第三者はこれらの実験の進行とスケジュール、その後のデータ収集と分析に重要な役割を果たしている。私たちはすでに合意して、私たちの第三者請負業者の活動を規範化しますが、彼らの実際の表現に対する影響力は限られています。しかし、私たちは私たちのすべての臨床試験が適用された方案と法律、法規、科学的基準に基づいて行われていることを確実にする責任があり、私たちのCROと他の第三者への依存は私たちの規制責任を免除しない。私たちと私たちのCROはGCP要求を遵守しなければならない。これらの要求は、FDAと同様の外国規制機関が私たちの臨床開発におけるすべての候補製品に対して実行する法規とガイドラインである。規制当局は,試験スポンサー,主要調査者,試験地点を定期的に検査することでこれらのGCPを実行している。もし私たちまたは私たちの任意のCROまたは試験サイトが適用されたGCPに従わなかった場合、私たちの臨床試験で生成された臨床データは信頼できないと考えられるかもしれませんが、FDAまたは同様の外国の規制機関は、私たちのマーケティング申請を承認する前に追加の臨床試験を行うことを要求するかもしれません。また,われわれの臨床試験はcGMP法規により生産された製品を用いて行わなければならない。私たちがこれらの規定を遵守しないことは、私たちが臨床試験を繰り返す必要があるかもしれないが、これは規制部門の承認過程を遅らせるだろう。
我々のCRO、調査者、または他の第三者が、そのような試験または研究に十分な時間および資源を投入するか、または契約要件に従って行われることは保証されない。もしこれらの第三者のいずれかが予想される最終期限内に達成できなかった場合、私たちの臨床方案を遵守したり、規制要求を満たしたり、あるいは他の方法で不合格を示した場合、私たちの臨床試験は延長、延期、または終了される可能性がある。さらに、私たちと契約した多くの第三者も、私たちの競争相手を含む他のビジネスエンティティと関係があるかもしれません。彼らは、これらのエンティティのための臨床試験や、私たちの競争地位を損なう可能性のある他の開発活動を行う可能性もあります。また,われわれの臨床試験の首席研究員は時々私たちの科学コンサルタントやコンサルタントを務め,このようなサービスに関する報酬を得ることが可能である。これらの関係と任意の関連する賠償が知覚的または実際の利益の衝突をもたらす場合、またはFDAは、財務関係が研究の解釈に影響を及ぼす可能性があると結論し、適用される臨床試験場所で生成されるデータの完全性が問われる可能性があり、臨床試験自体の効用が脅かされる可能性があり、これは、FDAが私たちが提出した任意のNDAを延期または拒否する可能性がある。そのような遅延や拒否は私たちが候補製品を商業化することを防ぐことができる。
もし治癒されていない重大な違約が発生したら、私たちのCROは私たちとの合意を終わらせる権利がある。また、私たちの臨床試験に参加した被験者の安全が合意を終了する必要があることを合理的に証明できれば、債権者の利益のために一般的に譲渡するか、または清算されれば、私たちのCROのいくつかは私たちとの合意を終了することができる。もし私たちがこれらの第三者との任意の関係が終わったら、私たちは商業的に合理的な条項や代替の第三者と合意できないかもしれない。追加のCRO、調査者、および他の第三者が追加のコストを伴う追加のCROを交換または増加させるには、私たちの管理職の時間と労力が必要です。しかも、新しいCROが仕事を始める時、自然な過渡期がある。したがって,遅延が生じ,期待される臨床開発スケジュールを満たす能力に実質的な影響を与える可能性がある。CRO、調査者、その他の第三者との関係を慎重に処理しているにもかかわらず、将来的に挑戦や遅延に遭遇しない保証はなく、これらの遅延や挑戦が私たちの業務、財務状況、および見通しに実質的な悪影響を与えないことを保証することはできない。
私たちは第三者に依存して私たちの臨床と臨床前に候補製品を開発し、予測可能な未来にこのようにしていきたい。このような第三者への依存は、許容可能なコストで十分な数の候補製品または製品またはそのような数を得ることができないリスクを増加させ、これは、私たちの開発または商業化努力を延期、阻止、または損害する可能性がある。
58
我々は製造施設を所有したり運営したりすることもなく,自分の臨床や商業規模の製造能力を開発する計画もない。私たちは依存し、引き続き第三者が私たちの候補製品と臨床および臨床前開発のための関連原材料を生産し、商業生産に依存することが予想される(任意の候補製品が市場の承認を得たら)。第三者メーカーが私たちの候補製品を生産するための施設は、FDAおよび任意の類似した外国規制機関の承認を得なければならず、検査は、私たちがFDAに機密協定を提出するか、または外国規制機関に任意の同様の提出を提出した後に行われる。我々は第三者メーカーの製造過程を制御せず、第三者メーカーに完全に依存してcGMP要求製品の生産を遵守している。これらの第三者製造業者が、我々の規格およびFDAまたは外国規制機関のような厳格な規制要件に適合する材料の生産に成功しない場合、彼らは、その製造施設の規制承認を確保および/または維持することができないであろう。また、第三者製造業者が十分な品質管理、品質保証、合格者の能力を維持することを制御することはできない。FDAまたは任意の同様の外国規制機関が、これらの施設が私たちの候補製品を生産するために承認されていない場合、または将来的にそのような承認を撤回すれば、代替製造施設を探す必要があるかもしれません。これは、私たちが規制機関の承認を得たり、私たちの候補製品を販売したりする能力に深刻な影響を与えます。私たちまたは私たちの第三者製造業者が適用された法規を遵守できなかったことは、臨床封印、罰金、禁止、民事処罰、遅延、承認の一時停止または撤回、候補製品または製品の差し押さえまたはリコール、運営制限、および刑事起訴を含む制裁を実施する可能性があり、いずれも私たちの製品供給に重大な悪影響を及ぼす可能性があります。
私たちまたは第三者が商業的に合理的な条項で私たちの製造要件を実行できず、cGMPまたは他の法規要件を遵守しない場合、様々な態様で私たちのビジネスに悪影響を及ぼす可能性があります
しかも、私たちは第三者製造業者と長期的な約束や供給協定を持っていない。私たちは、私たちの第三者製造業者といかなる供給合意も確立できないかもしれないし、許容可能な条項でそうすることができないかもしれません。これは、十分な数の候補製品をタイムリーに獲得するか、または許容可能なコストでそのような数を得るリスクを増加させます。たとえ第三者製造業者と合意できても、第三者メーカーに依存することは、追加的なリスクをもたらす
私たちの候補製品と私たちが開発する可能性のあるどの製品も他の候補製品や製品と製造施設を競争する可能性があります。CGMP法規下で運営されているメーカー数は限られており,我々のために生産できるメーカー数は限られており,特にゾタチフェンの高い効力によるものである。私たちの既存または未来のメーカーのどんな性能故障も臨床開発や市場承認を延期する可能性があり、どんな関連する救済措置もコストが高いか、時間がかかる可能性がある
59
実施する。私たちはまだ私たちの候補製品を生産するために必要なすべての原材料に余分な供給源や二番目の供給源を提供するように手配していません。もし私たちの既存または未来の第三者製造業者が合意通りに実行できなければ、私たちはこれらのメーカーの交換を要求される可能性があり、私たちはそれらをタイムリーにまたは根本的に交換できないかもしれない。特に、私たちのメーカーのどの交換にも大量の努力と専門知識が必要であり、合格した交換数は限られている可能性があるからです。場合によっては、我々の候補製品を製造するために必要な技術的スキルまたは技術は、元の製造業者固有または独自である可能性があり、そのようなスキルまたは技術を他の第三者に譲渡することは困難である可能性があり、実行可能な代替案が存在しない可能性がある。また、私たちのいくつかの候補製品と私たち自身の独自の方法は会社以外で生産または実施されたことがありませんので、これらの候補製品や方法のための新しい第三者製造計画を構築しようと試みると、私たちの開発計画が遅延する可能性があります。
私たちの現在と未来の他人が私たちの候補製品や製品を生産することへの依存は、私たちの将来の利益率と、マーケティングの承認をタイムリーかつ競争力的に獲得する製品の商業化能力に悪影響を及ぼす可能性があると予想されています。
私たちの第三者への依存は、私たちのビジネス秘密を共有することを要求し、これは、競争相手がこれらの秘密や私たちのビジネス秘密が流用または開示されていることを発見する可能性を増加させる。
私たちは現在、第三者に依存して候補製品を生産し、品質テストを行っており、様々な組織や学術機関と協力して私たちのいくつかの開発プロジェクトを進めているため、ビジネス秘密を含む独自技術や機密情報を彼らと共有しなければならない場合があります。独自の情報の研究または開示を開始する前に、当社の協力者、コンサルタント、従業員およびコンサルタントと秘密協定および材料譲渡協定、共同研究協定、コンサルティング協定、または他の同様の合意を締結することによって、当社のノウハウを保護することを求めています。このような協定は一般に第三者が私たちの機密情報を使用または開示する権利を制限する。第三者と協力する際に契約条項が採用されているにもかかわらず、商業秘密および他の機密情報を共有する必要は、そのような商業秘密が私たちの競争相手に知られ、意図的にまたは意図的に他の人に組み込まれているか、またはこれらの合意に違反する方法で開示または使用されるリスクを増加させる。私たちの独自の地位が私たちのノウハウおよびビジネス秘密にある程度基づいていることを考慮すると、私たちはビジネス秘密を保護しようと努力しているにもかかわらず、競争相手は私たちのノウハウや機密情報、または他の許可されていない使用または開示が私たちの競争地位を損なうことを発見し、私たちの業務、財務状況、運営結果、および将来性に大きな悪影響を及ぼす可能性がある。
我々はeIF 4 Eの小分子阻害剤を発見、開発、商業化するためにファイザープロトコルに依存している。ファイザーは便宜のために一方的に合意を終了する可能性があり,これは我々の業務に実質的な悪影響を与える可能性がある.
2019年12月,我々の最初の段階計画であるeIF 4 E阻害剤−についてファイザー合意が成立し,ファイザーは現在この計画のINDイネーブル研究を行っている。ファイザー協定によると、著者らはファイザーとの協力の初歩的な研究を担当し、ファイザーはINDの提出とすべての臨床開発と商業化活動を含むeIF 4 E開発計画のすべての更なる開発を担当する。ファイザー協定の条項によると、ファイザーは主に開発活動を制御し、もし私たちがこのような活動を制御しなければ、私たちのeIF 4 E計画の開発と商業化に遅延或いはその他の困難を招く可能性がある。ファイザーとのいかなるトラブルも、この計画の開発や商業化の遅延または終了を招く可能性があり、コストの高い訴訟を招き、私たちの管理層の日常活動に対する注意と資源を移転し、私たちの業務、財務状況、運営結果、将来性に悪影響を及ぼす可能性がある。
さらに、ファイザーは(便宜上を含む)ファイザープロトコルを終了することができ、ファイザーがファイザープロトコルを終了する場合、私たちは、プロトコルに従っていかなる開発資金、マイルストーン支払い、特許権使用料、および他の福祉を得る資格がもはやないであろう。また、ファイザーとファイザー協定の終了のいかなる決定も、国民の私たちの候補製品に対する見方にマイナス影響を与える可能性があり、これは私たちの普通株の市場価格に不利な影響を与える可能性がある。私たちはファイザーとの協力の成功について何の保証も提供できない。上記のいずれの事件も、当社の業務、財務状況、運営結果、および見通しに重大な悪影響を及ぼす可能性があります。
60
私たちは他の協力、許可、他の類似した手配を求めることができるかもしれないが、成功しないかもしれないし、私たちが成功しても、私たちは貴重な権利を放棄する可能性があり、このような関係の利点を実現できないかもしれない。
候補製品の開発や商業化に必要な資本コストや製造制限により、我々の候補製品を開発または商業化するために、他の協力、合弁企業、許可、および他の同様の手配を達成することが求められる可能性がある。このような連携発見努力は,我々のパイプラインに追加的な開発や製品候補を生成しない可能性がある.私たちの候補製品のためのこのような協力を確立または維持する努力は成功しないかもしれません。なぜなら、私たちの研究開発ルートが不十分である可能性があり、私たちの候補製品は協力努力の開発段階が早すぎると思われるかもしれません。あるいは第三者は私たちの候補製品が安全性と有効性や重大なビジネス機会を示す必要な潜在力を持っていないと思うかもしれません。また、適切な戦略的パートナーを探す上で激しい競争に直面しており、交渉過程は時間がかかり複雑かもしれない。そのような任意の手配の一部として、私たちは、私たちの将来の収入源、研究計画、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になる可能性のある条項で許可を与えなければならないかもしれません。このような計画は、他の潜在的なパートナーとの追加的な合意を制限するかもしれません。私たちは協力、許可、あるいは戦略的取引の後、私たちが経済的利益を得て、このような取引が合理的であることを証明することを確信できない。
私たちがこのような協力を成功させたとしても、私たちが合意した条項は私たちに不利になるかもしれないが、例えば、候補製品の開発や承認が延期され、候補製品の安全性が疑問視されたり、承認された候補製品の販売が満足できない場合、私たちはこのような協力を維持できないかもしれない。
しかも、どんな潜在的な未来の協力も私たちの戦略的パートナーによって中止されるかもしれないし、私たちはこのような合意の下で私たちの権利を十分に保護できないかもしれない。また、戦略的パートナーは、(承認されれば)候補製品の開発および商業化に関する決定を制御するために、いくつかの権利について交渉することができ、これらの活動を私たちと同じ方法で行わない可能性がある。もし私たちが将来協力を終了したり、私たちの候補製品に関する協力を遅延させたりすれば、候補製品の開発と商業化を延期し、それらの競争力を低下させる可能性があり、それらが市場に進出すれば、私たちの業務、財務状況、運営結果に大きな悪影響を及ぼす可能性がある。
私たちの候補製品の商業化に関するリスクは
私たちがどんな候補製品の規制承認を得ても、私たちは持続的な規制義務と持続的な規制審査の制約を受けることになり、これは多くの追加費用を招く可能性がある。
私たちまたは私たちの既存または未来のパートナーが入手する可能性のある任意の候補製品に対する規制承認は、製品の安全性および有効性を監視するために、規制機関および監督に報告書を提出することを要求し、特定の年齢層の使用制限、警告、予防措置または禁忌症に関連する重大な制限を含む可能性があり、重い承認後の研究またはリスク管理要件を含む可能性がある。例えば、FDAは、REMSを私たちの候補製品を承認する条件として要求することができ、薬学的ガイドライン、医師コミュニケーション計画、または安全な使用を保証する他の要素の要件、例えば、制限された分配方法、患者登録、および他のリスク最小化ツールを含むことができる。さらに、FDAまたは同様の外国規制機関が私たちの候補製品を承認すれば、私たちの製品の製造プロセス、ラベル、包装、流通、有害事象報告、貯蔵、広告、販売促進、輸入、輸出、記録保存は広範で持続的な規制要求を受けるだろう。これらの要求には,安全性や他の上場後の情報や報告,登録の提出,および我々が承認後に行った任意の臨床試験がcGMPやGCP要求を遵守し続けているかどうかが含まれている。承認された製品およびその施設の製造業者は、cGMP法規および基準に適合することを確実にするために、FDAおよび他の規制機関の持続的な審査および定期的な抜き打ち検査を受ける。その後、予想されていない深刻度や頻度の有害事象、または当社の第三者製造業者または製造プロセス、または規制要件を遵守できなかったことが、以下のような状況を引き起こす可能性がある以前に未知の問題が存在することが分かった
61
上記のいずれの事件や処罰が発生しても、製品を商業化し、収入を創出する能力を抑制することができ、対応するために多くの時間と資源を必要とする可能性があり、負の宣伝が生じる可能性がある。
FDAや他の規制機関の政策は変わる可能性があり、私たちの候補製品に対する規制承認を阻止、制限、または延期するための追加の政府法規が公布されるかもしれない。米国や海外の将来の立法や行政行動によって生じる可能性のある政府規制の可能性、性質、程度も予測できない。もし私たちが既存の要求の変化に緩やかに適応できない場合、あるいは新しい要求や政策を採用することができない場合、あるいは規制コンプライアンスを維持できない場合、私たちは法執行行動の影響を受ける可能性があり、利益を達成したり維持したりすることができないかもしれない。
FDAと他の規制機関は非ラベル使用の普及を禁止する法律法規を積極的に実行している。
もし私たちの候補製品が承認され、私たちがこれらの製品のラベル外用途を不正に普及させたことが発見されたら、私たちは重大な責任を負うかもしれない。FDAや他の規制機関は、承認されれば、処方製品(例えば、私たちの候補製品)に対する販売促進主張を厳格に規制することができる。特に、製品は、製品が承認されたラベルに反映されるように、FDAまたは他の規制機関によって承認されていない使用に使用されてはならない。もし私たちが候補製品のマーケティング承認を得たら、医者は承認されたラベルと一致しない方法で患者に処方するかもしれない。もし私たちがこのようなラベル外の使用を普及させることを発見されたら、私たちは重大な責任を負うかもしれない。米連邦政府は、ラベル外使用の不当な普及の疑いがある会社に巨額の民事と刑事罰金を科し、いくつかの会社がラベル外販売促進に従事することを禁止している。政府はまた、企業に同意法令に署名したり、永久禁止を実施したりすることを要求し、これらの禁止令に基づいて、特定の販売促進行為が変更または制限される。もし私たちが私たちの候補製品の普及を成功的に管理できなければ、承認されれば、私たちは重大な責任を負う可能性があり、これは私たちの業務や財務状況に実質的な悪影響を及ぼすだろう。
私たちの候補製品の商業成功は、承認されれば、医師、患者、医療保険支払人と医学界の他の人のこれらの候補製品に対する市場受け入れの程度に依存する。
私たちの候補製品は、承認されれば、商業的に成功しないかもしれない。私たちのすべての候補製品が規制部門の承認を得ても、それらは医師、患者、医療支払者、あるいは医療界の市場で受け入れられない可能性がある。私たちの現在または未来の任意の候補製品の商業成功は、承認された適応を得るために、医師および患者の最終製品の広範な採用および使用に大きく依存するであろう。私たちの製品に対する市場の受け入れ度は多くの要素に依存するだろう
62
任意の候補製品が承認されたが、医師、病院、医療支払者、または患者の十分な受容度に達していない場合、私たちはその製品から十分な収入を得ることができず、利益を達成または維持できない可能性がある。私たちの教育医療界と第三者支払者が私たちの製品のメリットを知る努力には大量の資源が必要かもしれませんし、決して成功しないかもしれません。
私たちの候補製品の商業化に成功し、承認されれば、政府当局と健康保険会社が構築した保険範囲、十分な補償レベル、優遇された価格設定政策にある程度依存する。もし私たちの製品が保証範囲を獲得したり維持したりして十分な精算を得ることができなければ、これらの製品をマーケティングする能力を制限し、収入を創出する能力を低下させるかもしれません。
承認されれば、政府医療計画(例えばMedicareおよびMedicaid)、個人健康保険会社および他の第三者支払者が提供する保険範囲および精算の十分性は、多くの患者が私たちの候補製品などの処方薬を負担することができるために重要である。第三者支払者の私たちの製品に対するカバー範囲と受け入れ可能な精算レベルを実現することができるかどうかは、これらの製品を商業化することに成功する能力に影響を与えます。したがって、私たちは承認されたすべての候補製品のカバーと精算戦略を成功的に実施する必要がある。第三者支払者が特定の製品の保険を獲得したとしても,それによる精算支払率が十分に高くない可能性があり,あるいは患者が受け入れられないほど高い共同支払いが必要である可能性がある。医師の監督下で管理されている製品については、このような薬物が高い価格に関連することが多いため、保険および適切な補償を得ることは特に困難である可能性がある。また,製品自体やその製品を使用した治療やプログラムは単独では精算できない可能性があり,医師の使用に影響を与える可能性がある。私たちが開発する可能性のあるどんな製品もアメリカ、EU、あるいは他の場所で保険と精算を受けることができることを保証することはできません。将来的にはどんな可能な精算も減らしたりキャンセルしたりする可能性があります。
第三者支払者は、生物製薬製品およびサービスの価格に挑戦することが増えており、同等の模倣薬またはより安い治療法が利用可能な場合、多くの第三者支払者は、特定の薬物への保険および精算を拒否する可能性がある。第三者支払者は,我々の製品が代替可能であると考え,価格の低い製品のみを患者に精算することを提案するかもしれない。たとえ私たちの製品が治療効果を高めたり、投与の利便性を改善したことを証明したとしても、既存の薬物の定価は製品に対する費用を制限するかもしれません。これらの支払者は、特定の製品の精算状態を拒否またはキャンセルしたり、新製品や既存市場製品の価格を低すぎるレベルに設定したりして、製品開発投資から適切なリターンを実現することができない可能性がある。精算が得られない場合や限られたレベルでしか精算が得られない場合、私たちの製品を商業化することに成功できない可能性があり、私たちが開発する可能性のある製品から満足な財務リターンを得ることができないかもしれません。
第三者支払者カバー範囲と新たに承認された製品および既存療法に組み合わせて添加された製品の精算には大きな不確実性がある。米国では,連邦医療保険や医療補助計画のような第三者支払者,個人や政府支払者を含め,新薬のカバー範囲を決定する上で重要な役割を果たしている。一部の第三者支払者は、新しいまたは革新的な設備または薬物療法の保証範囲を事前に承認し、その後、このような治療法を使用する医療提供者に精算する必要があるかもしれない。第三者決済者が私たちの製品の保証範囲と精算についてどのように決定するか予測するのは難しいです。
63
獲得とメンテナンスの精算状態は時間がかかり、コストも高く、確定していない。連邦医療保険や医療補助計画は,個人支払者や他の政府支払者がどのように薬品保険や精算政策を策定するかのモデルとして利用されるようになってきている。しかし、米国の第三者支払者では、製品のカバーや精算に統一された政策はない。そのため、製品の保証範囲と精算範囲は支払人によって異なる。したがって、保証範囲の決定プロセスは、通常、時間がかかり、高価なプロセスであり、保証範囲および十分な補償が一致するか、または最初に得られることを保証することなく、各支払人にそれぞれ私たちの製品を使用する科学的および臨床的支援を提供する必要がある。また、精算に関する規則や条例は常に変化しており、場合によっては短時間で通知されており、これらの規則や条例が変わる可能性があると考えられる。
アメリカ以外では、国際業務は一般的に広範な政府価格制御と他の市場監督管理を受けており、ヨーロッパと他の国のコスト制御措置の日々の重視はすでに私たちの製品の定価と使用に圧力を与え続けると信じている。多くの国では、国家衛生システムの一部として、医療製品の価格は異なる価格制御メカニズムの制約を受けている。他の国は会社が自ら医療製品を価格設定することを許可しているが、会社の利益を監督してコントロールしている。追加の外国価格規制や定価規制の他の変化は私たちの製品に対する料金を制限するかもしれません。そのため、米国以外の市場では、米国に比べてわが製品の精算が減少する可能性があり、商業的に合理的な収入や利益を生み出すには不十分である可能性がある。
また、米国や海外の政府や第三者支払者が医療コストの制限や低減に力を入れていることは、これらの組織が新承認製品のカバー範囲や精算レベルを制限する可能性があるため、私たちの製品に十分な保険を提供したり、十分なお金を支払うことができない可能性がある。管理型ヘルスケアの傾向、健康維持組織の日々の影響力、および追加の立法変化により、どの製品の販売も価格設定圧力に直面することが予想される。全体的に,医療コストの下振れ圧力は非常に大きくなり,特に処方薬,外科手術,その他の治療が行われている。そのため、新製品の参入にはますます高い壁が設けられている。
新たな治療法や技術プラットフォームの開発を含む癌候補製品を開発または開発する可能性のある実体からの激しい競争に直面している。もし私たちの競争相手が技術や候補製品を開発する速度が私たちより速い、あるいは彼らの技術がもっと効果的であれば、私たちの業務および私たちが製品を開発して商業化する能力は不利な影響を受ける可能性がある。
生物技術と生物製薬業界の特徴は技術が迅速に進歩し、競争が激しく、特許と新製品及び候補製品を高度に重視していることである。我々の競合他社は、PD-(L)1阻害剤との併用も推奨される可能性のある製品を含む、我々の候補製品と競合する製品、候補製品およびプロセスを開発しているか、または開発している可能性がある。我々が開発と商業化に成功した任意の候補製品は,既存の療法や将来出現する可能性のある新しい療法と競争するであろう。かなりの数の製品が現在開発中であり,将来ビジネスに投入され,開発を試みる可能性のある候補製品の適応を治療するために使用される可能性があると信じている。特に,腫瘍学分野での競争は非常に激しい。私たちの競争相手はより規模が大きく、資金が豊富な製薬、生物製薬、バイオテクノロジー、治療会社を含む。また,大学や他の研究機関と競合する可能性もあり,これらの大学や研究機関は腫瘍学研究分野で活躍し,我々と直接競争する可能性がある。また、これらの組織と競合して管理者、科学者、臨床開発者を募集しており、これは私たちの専門レベルや業務計画を実行する能力に悪影響を及ぼす可能性があります。私たちはまた臨床試験場を設立し、臨床試験のための被験者を募集し、新製品の候補を確定と許可する上で競争に直面する。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。
もし我々の任意の候補製品が非小細胞肺癌あるいは乳癌などの腫瘍学的適応で承認されれば、それらは小分子療法、生物製剤、細胞療法および伝統的な化学療法と競争する。類似適応に対する他の治療法と競合するほか,我々の候補製品に類似した標的や/あるいは同じ適応を治療する異なる科学的方法に集中している他の会社や学術機関も多い。これらの会社はAUM生物科学会社、ブリンガーインゲルハイム社、礼来社、Exelixis、ノワ製薬、セルヴィタ社を含み、それらの計画はMNK社を対象としている。FDAが承認したPD-1またはPD-L 1阻害剤を有する会社は、アスリコン社、百時美施貴宝社、メルク社、ファイザー/メルク社、Regeneron製薬会社、羅氏集団/遺伝子技術会社である。また、抗体療法、小分子阻害剤、腫瘍溶解ウイルス、癌ワクチン、細胞ベース療法など、検査点阻害剤と新規免疫調節剤との結合を積極的に試験している会社もある。
64
私たちの多くの競争相手は私たちよりも多くの財務、技術、製造、マーケティング、販売、資源または経験を持っています。もし私たちが任意の候補製品の承認を得ることに成功したら、私たちは私たちの製品の安全性と有効性、私たちの製品の管理の容易さ、患者が比較的新しい投与経路を受け入れる程度、これらの製品が規制の承認を受ける時間と範囲、製造、マーケティングと販売能力の可用性とコスト、価格、精算範囲、特許地位を含む多くの異なる要素に基づく競争に直面するだろう。私たちと競争する製品は、私たちが開発する可能性のある任意の製品よりも効率的で、安全で、より便利で、より安価で、またはより効率的なマーケティングおよび販売を含む、より良い治療代替案を提供することができます。私たちが候補製品の開発と商業化の費用を回収する前に、競争製品方法は私たちが開発したいかなる製品も時代遅れまたは競争力を持たないかもしれない。もし私たちが効果的に競争できなければ、私たちが開発する可能性のある製品を販売することから収入を得る機会は不利な影響を受けるかもしれない。
私たちの候補製品の市場機会は、以前の治療を受ける資格がない患者または以前の治療を通過できなかった患者に限定される可能性があり、規模が小さいか、または私たちの推定とは異なる可能性がある。
癌治療は,治療経路および治療−幼稚あるいは以前の治療状態の患者によって定義される。通常、新しい療法の最初の承認は遅い生産ラインであり、その後のより早い生産ラインでの承認は不可能である可能性がある。癌が十分に早く発見された時、第一線の治療は治愈を必要とすることなく、癌を治癒したり、生命を延長するのに十分であることがある。標的治療、免疫治療、化学療法、ホルモン治療、手術またはこれらの療法の組み合わせが成功しないことが証明された場合、二次治療を行うことができる。二次治療は、一般に、追加の化学療法、放射線、抗体薬物、腫瘍標的小分子、またはこれらの薬剤の組み合わせを含む。三線治療は骨髄移植、抗体と小分子標的治療、より侵襲性のある手術形式と新しい技術を含むことができる。承認された療法の市場では,我々の候補品が承認されても,二線や一線療法への使用が承認される保証はない。これは私たちの潜在的な市場機会を制限するかもしれない。また、二線または一線治療の承認を得る前に、追加の臨床試験を行わなければならないかもしれない。
私たちの目標癌患者数の予測と、末期治療を受け、私たちの候補製品治療から利益を得る可能性があるこれらの癌患者のサブセットは、私たちの信念と推定に基づいている。これらの推定は、科学文献、公開利用可能な臨床分子報告、患者基礎或いは市場研究を含む様々な源から来ており、不正確であることが証明されている可能性がある。また,新たな試験や情報はこれらの癌の推定発症率や流行率を変化させる可能性がある。米国、他の主要市場、および他の地域の患者数は予想を下回る可能性があり、患者は私たちの製品治療を受けることができないかもしれない、あるいは新しい患者はますます識別しにくくなったり、接触したりする可能性があり、これらすべては私たちの運営結果および業務に悪影響を及ぼすであろう。また、私たちの候補製品のためにかなりの市場シェアを得ても、私たちのいくつかの潜在的なターゲット層が非常に少ないので、私たちは将来的に利益を達成できないかもしれない。
私たちは現在マーケティングや販売組織がなく、会社としても製品を商業化した経験がなく、これらの能力を開発するために大量の資源を投入しなければならないかもしれません。マーケティングや販売能力を確立できない場合や、第三者と合意して私たちの製品をマーケティングして販売することができなければ、製品収入を生み出すことができないかもしれません。
私たちは内部販売、マーケティング、流通能力もなく、製品を商業化していない。もし私たちの任意の候補製品が最終的に規制部門の承認を得たら、主要市場でこのような各製品を商業化するために、技術的な専門性と流通能力を支援するマーケティング·販売組織を構築しなければならない。これは高価で時間がかかる、あるいは直接販売チームや流通システムを構築する第三者と協力して、私たち自身の販売チームや流通システムを強化し、あるいは私たち自身の販売チームや流通システムの代わりになるだろう。ある会社として、私たちは以前生物製薬製品のマーケティング、販売と流通の面で経験がなく、販売組織の構築と管理は重大なリスクに関連しており、私たちは合格者を雇用、維持と激励し、十分な販売手がかりを生成し、販売とマーケティング人員に十分な訓練を提供し、異なる地理的な位置に分散した販売とマーケティングチームを有効に管理する能力を含む。私たちの内部販売、マーケティング、流通能力の発展のいかなる失敗や遅延も、これらの製品の商業化に悪影響を及ぼすだろう。私たちは、受け入れ可能な財務条項で協力することができないか、またはコンサルタントまたは外部サービスプロバイダを招いて、私たちの販売、マーケティング、および流通機能を支援することができないかもしれません。また、私たちが第三者に依存してこれらの機能を実現すれば、私たちの製品の収入と収益力(あれば)は、私たちが開発したどの製品のマーケティング、販売、流通よりも低くなる可能性があります。私たちはこのような第三者に対して支配権がほとんどないかもしれません。彼らのいずれも必要な資源と注意力を投入して、私たちの製品を効果的に販売し、マーケティングすることができないかもしれません。もし私たちが私たちの製品を商業化することに成功できなければ、私たち自身でも、1つ以上の第三者との手配によっても、私たちは未来にどんな製品収入も発生できないかもしれないので、私たちは重大な追加損失を招くだろう。
65
私たちの将来の成長は海外市場における私たちの運営能力にある程度依存するかもしれませんが、そこでは追加の規制負担や他のリスクや不確実性の影響を受けることになります。
私たちの将来の成長は私たちの候補製品を海外市場で開発し商業化する能力にある程度依存するかもしれない。海外市場適用規制機関の規制承認を得るまで、私たちは私たちの候補製品のマーケティングや普及は許可されておらず、私たちはいかなる候補製品の規制承認も得られないかもしれない。多くの他の国で単独の監督管理許可を得るためには、安全性と有効性、及び私たちの候補製品の臨床試験、商業販売、定価と流通などの方面の監督管理を含む多くの異なる監督管理要求を守らなければならない。もし私たちが規制機関の候補製品の承認を得て、最終的に私たちの製品を海外市場で商業化すれば、私たちは追加のリスクと不確実性に直面します
私たちの業務運営や業界に関連するリスク
私たちの経営業績は大幅に変動する可能性があり、これは私たちの将来の経営業績を予測しにくくし、私たちの経営業績が予想を下回ったり、私たちが提供する可能性のあるいかなる指導を招く可能性があります。
私たちの四半期と年度の経営業績は大きく変動する可能性があり、将来の経営業績を予測することは困難です。これらの変動は様々な要因によって引き起こされる可能性があり、その多くの要因は、これらに限定されないが、我々が制御できるものではない
66
これらの要因の累積影響は我々の四半期や年度経営業績に大きな変動と予測不可能を招く可能性がある。したがって、異なる時期に私たちの経営業績を比較することは意味がないかもしれない。投資家たちは私たちの過去の業績を私たちの未来表現の指標として依存してはいけない。
このような変化性および予測不可能性はまた、業界や金融アナリスト、または投資家の任意の時期に対する期待を満たすことができない可能性がある。もし私たちの収入や経営業績がアナリストや投資家の予想よりも低い場合、または私たちが市場に提供するいかなる予測よりも低い場合、または私たちが市場に提供する予測がアナリストや投資家の予想よりも低い場合、私たちの普通株の価格は大幅に下落する可能性がある。私たちが提供する可能性のある以前に公開された収入や収益案内に到達したとしても、このような株価下落は起こる可能性がある。
私たちの成功は私たちが高い素質の管理者と他の臨床と科学者を引き付ける能力にかかっている。
著者らの成功はある程度私たちが引き続き吸引、維持、管理と激励する高素質の管理、臨床と科学者の能力に依存し、著者らは経験豊富な人員に対する激しい競争に直面している。私たちは私たちの上級管理職と私たちのベテラン科学者と管理チームの他のメンバーに非常に依存している。これらの人の誰もがサービスを失っても、私たちの製品パイプラインの成功開発を遅延または阻止し、私たちの臨床試験と臨床前研究、あるいは私たちの候補製品の商業化を開始または完成させる可能性がある。我々はすでに我々の高度管理チームの各メンバーと雇用協定や招聘書に署名しているにもかかわらず、これらの合意は通知または通知なしに任意に終了することができるため、予想通りに彼らのサービスを保留することができない可能性がある。私たちは現在、私たちの役員や従業員の生命のために“キーパーソン”生命保険を保有していません。保険不足は私たちがこのような個人のサービス損失を補うために十分な賠償を受けられない可能性があるということを意味する。
私たちの臨床開発と商業化努力に成功するために、私たちの管理、運営、財務、その他の資源を拡大し、効果的に管理する必要があるだろう。生物製薬、生物技術とその他の業界の合格人材に対する激しい競争のため、特にサンディエゴ地区で、私たちは未来に私たちの独特な会社文化を維持することに成功できないかもしれず、引き続き合格の管理、臨床と科学者を誘致或いは維持する。近年、私たちの業界の管理職の流出率は高い。もし私たちが必要な人員を誘致、統合、維持、激励することができなければ、私たちはいくつかの制限に直面するかもしれません。これらの制限は、私たちの発展目標の実現、追加資本を調達する能力、そして私たちが業務戦略を実施する能力を深刻に阻害します。
私たちは私たちの成長を管理し、私たちの業務の成功を拡大する困難に直面するかもしれない。
2023年12月31日まで、私たちは15人のフルタイム従業員がいます。私たちが引き続き私たちの候補製品の潜在的な商業化を開発し、追求することに伴い、上場企業として、私たちの財務、開発、監督、製造、マーケティング、販売能力を拡大し、あるいは第三者と契約を締結し、これらの能力を提供する必要があるだろう。私たちの業務の拡大に伴い、様々な戦略パートナー、サプライヤー、他の第三者とのより多くの関係を管理する必要があると予想されます。私たちの将来の財務業績と、私たちの候補製品を開発し、商業化し、効果的に競争する能力は、将来の任意の成長を効果的に管理する能力にある程度依存するだろう。
67
私たちと処方者、購入者、第三者支払者と患者との関係は、適用される反リベート、詐欺と乱用、および他の医療法律と法規の制約を受けることになり、これは私たちを刑事制裁、民事処罰、契約損害、名声損害、利益と未来の収入の減少に直面させるかもしれない。
私たちの業務運営と調査者、医療専門家、コンサルタント、第三者支払人、患者組織と顧客との現在と未来の手配は、私たちが広く適用される外国、連邦、州詐欺と乱用、その他の医療とプライバシーに関する法律法規に直面させます。これらの法律は、私たちがマーケティング、販売、流通をどのように研究、マーケティング、販売、流通するかを含む、私たちが業務を展開する業務または財務配置と関係を制約するかもしれません。これらの法律には
68
我々の現在と将来の第三者の業務配置が適用される医療·プライバシー法令に適合することを確保する努力は、持続的な巨額のコストに関連する。政府当局は、株または株式オプションの形で補償された医師とのいくつかの科学顧問委員会の手配を含む、私たちのビジネス実践は、現在または将来的に詐欺および乱用または他の医療保健法律および法規の適用に関する現行または将来の法規、法規または判例法に適合しない可能性があると結論するかもしれない。もし私たちの運営がこれらの法律または任意の他の私たちに適用される可能性のあるすべての政府法規に違反していることが発見された場合、私たちは民事、刑事および行政処罰、損害賠償、罰金、返還、監禁、政府援助から除外された医療計画(MedicareおよびMedicaidを含む)、誠実な監督と報告義務、契約損害、名声損害、利益減少および将来の収益減少、ならびに私たちの業務の削減または再編を含む重大な処罰を受ける可能性がある。このような行動を防御するには費用がかかり、時間がかかる可能性があり、大量の財政と人的資源が必要かもしれない。したがって、私たちが私たちに提起される可能性のあるいかなる訴訟も防ぐことに成功しても、私たちの業務は損害を受ける可能性がある。さらに、私たちがそれと業務を展開することを期待している任意の医師または他のヘルスケア提供者またはエンティティが、適用された法律に適合していないことが発見された場合、彼らは、政府が援助する医療計画から除外されることを含む重大な刑事、民事または行政制裁を受ける可能性がある。
最近公布された立法、将来の立法、医療改革措置は、候補製品の市場承認を獲得し、それを商業化する難しさとコストを増加させ、設定可能な価格に影響を与える可能性がある。
米国や一部の外国司法管轄地域では、医療システムは、コスト制御措置を含む複数の立法·規制改革を継続しており、これらの措置は、新薬の保証範囲や精算範囲を減少または制限し、上場承認された任意の候補製品を収益性のある方法で販売する能力に影響を与える可能性がある。特に,米国連邦や州レベルでは,医療コストの低減と医療の質の向上を図る取り組みが継続されている。
例えば、2010年3月、米国では“医療·教育和解法案”(“平価医療法案”と総称する)で改正された“患者保護·平価医療法案”が公布された。ACAの中で私たちの潜在的な製品候補に重要な意義を持つ条項の中で、ACAは任意の指定ブランドの処方薬と生物製剤の実体に対して控除できない年間費用を設立した;メーカーの医療補助リベート責任を連邦医療補助管理に参加する医療機関に割り当てられた個人の保険薬品に拡大した;医療補助計画の資格基準を拡大した;公共衛生サービス薬品定価計画の下で割引を受ける資格がある実体を拡大した;医療補助薬品リベート計画によってメーカーが支払わなければならない法定最低リベートを高めた;新しい連邦医療保険D部分カバー割引計画を作成した;新しい患者を中心とした結果研究所を設立し、監督、優先事項を確定し、臨床治療効果の比較研究を行い、そしてこのような研究に資金を提供した;そしてCMSで連邦医療保険と医療補助革新センターを創立し、革新的な支払いとサービス交付モードをテストし、連邦医療保険と医療補助支出を下げる。
ACAのいくつかの側面は公布以来、行政、司法、そして国会の挑戦を受けてきた。2021年6月17日、米国最高裁はいくつかの州がACAに対して提出した最新の司法挑戦を却下したが、ACAの合憲性を具体的に裁くことはなかった。
さらに、ACAが公布されて以来、他の立法改正も提案され、採択された。2011年8月2日、2011年予算制御法が署名され、提供者への医療保険の支払いを減らすことを含む法律となり、2013年4月1日に施行され、この法規の後続立法改正により、国会が追加的な行動を取らない限り、2020年5月1日から2022年3月31日まで一時的に一時停止される2032年まで有効となる。また、2013年1月2日、2012年の米国納税者救済法が法律に署名し、病院を含むいくつかの医療サービス提供者に支払う医療保険をさらに減らし、政府が提供者に多額の支払いを取り戻す訴訟時効を3年から5年に延長した。
また、処方薬のコストが上昇していることを受けて、米国政府は薬品定価のやり方の審査を強化した。このような審査は最近の国会調査を招き、製品価格の透明性の向上、価格設定とメーカー患者計画との関係を検討するための連邦と州立法を提出し、公布した
69
政府製品計画の精算方法を改革する.例えば,2021年3月11日,2021年米国救援計画法案が署名されて法律となり,2024年1月1日から単一源と革新者多由来薬物の法定医療補助薬品還付上限が廃止された。これまで,還付の上限は薬品メーカーの平均価格の100%であった。最近は2022年8月16日に“2022年インフレ削減法案”(IRA)が署名されて法律となっている。他の事項を除いて、アイルランド共和軍はある薬品のメーカーに連邦医療保険との価格交渉(2026年から)を要求し、価格は交渉できるが上限があり、連邦医療保険B部分と連邦医療保険D部分に基づいてリベートを実施し、インフレを超える価格上昇を処罰し(2023年に初めて満了)、D部分のカバーギャップ割引計画の代わりに新しい割引計画を用いる(2025年から)。アイルランド共和軍は,衛生·公衆サービス部(HHS)秘書が最初の数年間,規制ではなく指導によってその多くの規定を実施することを許可した。これらの計画の実施に伴い,HHSは指導意見を発表·更新し続けている。2023年8月29日、HHSは、連邦医療保険薬品価格交渉計画が現在法的挑戦を受けているにもかかわらず、価格交渉を受ける上位10種類の薬物のリストを発表した。このような理由と他の理由で、アイルランド共和軍がどのように実施されるのかは不明だ。
州レベルでは、立法機関は、価格または患者の精算制限、割引、特定の製品参入の制限、マーケティングコスト開示および透明性措置を含む、医薬品および生物製品の価格設定を制御するための法規を立法および実施することが増えており、場合によっては、他の国からの輸入および大量購入を奨励することを目的としている。法律で規定されている第三者支払者の支払金額の価格制御またはその他の制限は、私たちの業務、運営結果、財務状況、見通しを損なう可能性があります。2020年12月、アメリカ最高裁判所は、連邦法律は各州が医療保健と薬品サプライチェーン中の薬品福祉マネージャー(PBM)と他のメンバーの能力を監督することを妨げないと一致して判断し、この重要な決定は各州がこの領域で更なるかつ積極的な努力をすることを招く見通しである。また,地域医療当局や個別病院では,どの薬品やサプライヤーが処方薬や他の医療計画に含まれるかを決定するために入札プログラムが使用されるようになってきている。承認されれば、これは私たちの候補製品に対する最終的な需要を減少させるか、あるいは私たちの製品価格に圧力を与える可能性があり、これは私たちの業務、運営結果、財務状況、見通しにマイナスの影響を与える可能性がある。
ACA、これらの新しい法律、および将来取られる可能性のある他の医療改革措置は、連邦医療保険および他の医療保険資金のさらなる減少、より厳しいカバー基準、新しい支払い方法、および私たちが受けた任意の承認製品の価格のさらなる低下の圧力をもたらす可能性があると予想される。連邦医療保険や他の政府が計画している精算のいかなる減少も、個人支払者の支払いの同様の減少を招く可能性がある。コスト抑制措置や他の医療改革を実施することは、承認されれば、収入を創出し、利益を達成することができ、あるいは候補製品を商業化することができるかもしれない。
実際または予想されるように、適用されるデータ保護、プライバシーおよびセキュリティ法律、法規、基準、およびその他の要求を遵守できないことは、私たちの業務、運営結果、および財務状況に悪影響を及ぼす可能性があります。
世界のデータ保護構造は急速に変化しており、私たちは多くの州、連邦と外国の法律、要求、法規の制約を受けているかもしれません。これらの法律、要求、法規は個人情報の収集、使用、開示、保留、安全を管理しています。予測可能な未来には、実施基準および法執行実践は依然として不確定である可能性があり、私たちはまだ未来の法律、法規、基準、またはその要求に対する見方が私たちの業務に与える影響を決定することができない。このような変化は私たちの業務に不確実性をもたらす可能性があり、私たちがある司法管轄区域で業務を展開し、あるいは個人情報を収集、保存、移転、使用、共有する能力に影響を与え、私たちの契約でより重い義務を受ける必要があり、私たちが責任を負うか、または追加コストをかける必要がある。これらの法律のすべては裁判所と政府機関の異なる解釈を受け、複雑なコンプライアンス問題をもたらしている。もし私たちが適用された法律と法規を遵守しなければ、私たちは政府の調査および/または法執行行動、罰金、民事または刑事罰、個人訴訟、または否定的な宣伝に直面する可能性があり、これらは私たちの業務、財務状況、および運営結果に悪影響を及ぼすかもしれない。例えば、私たちが故意に保証エンティティから個人識別可能な健康情報を取得または開示し、この方法が2009年の“健康情報技術促進経済および臨床健康法案”によって改正された“健康保険携帯および責任法案”およびその下で実施される法規または適用された州法の許可または許可されていない場合、私たちは刑事罰を受ける可能性がある。
70
私たちに製品責任訴訟を提起すれば、私たちは重大な責任を負い、私たちの製品の商業化を制限することを要求されるかもしれません。
私たちの候補製品の臨床試験のため、私たちは固有の製品責任リスクに直面して、私たちの候補製品を商業化すれば、私たちはもっと大きなリスクに直面します。例えば、私たちの候補製品が製品テスト、製造、マーケティング、または販売中にダメージを与えたり、不適切なことが発見されたと言われた場合、私たちは起訴されるかもしれない。また,既存または将来の協力者の候補製品開発における行為によって責任を負う可能性がある.このような製品責任クレームは、製造欠陥、設計欠陥、候補製品固有の危険について警告、不注意、厳格な責任、保証違反の告発を含む可能性がある。臨床試験参加者、患者、または他の使用、管理、または将来承認される可能性のある製品を販売する人は、私たちにクレームをつけるかもしれない。州消費者保護法によると、クレームも主張することができる。
もし私たちが製品責任クレームで自分を弁護することに成功できなければ、私たちは重大な責任を招いたり、私たちの製品の商業化を制限または停止することを要求されるかもしれません。成功的な防御であっても、多くの財政的で管理的な資源が必要だ。事件がどうであっても最終的な結果がどうであろうと、賠償責任は次のようになるかもしれない
私たちは現在全部で約500万ドルの製品責任保険を持っていて、毎回発生する制限はありません。私たちの臨床試験の拡大に伴い、あるいは候補製品を商業化し始めたら、私たちの保険カバー範囲を増やす必要があるかもしれません。保険範囲はますます高くなっています。潜在的な製品責任クレームを防止するために、許容可能なコストで十分な製品責任保険を得ることができず、これは私たちの候補製品の商業化を阻止または阻害する可能性がある。私たちはこのような保険を維持しますが、私たちに対するいかなるクレームも、裁判所の判決や和解の金額が私たちの保険範囲内にない、あるいは私たちの保険範囲を超えてしまう可能性があります。私たちの保険証書も様々な排除があります。私たちは製品責任クレームの影響を受けるかもしれませんが、私たちは保険範囲を持っていません。私たちは私たちの保険範囲の制限を超えたり、私たちの保険カバー範囲内でない裁判所の裁決または和解合意で達成された任意の金額を支払う必要があるかもしれません。私たちはこれらの金額を支払うために十分な資本を持っていないか、または得ることができません。
私たちの保険証書は高くて、いくつかの商業リスクからだけ保護されています。これらのリスクは私たちに多くの未保険の責任を負わせます。
私たちは私たちの業務が直面する可能性のあるすべての種類のリスクに保険をかけない。私たちが現在維持しているいくつかの保険書は財産、一般責任、雇用福祉責任、ビジネス自動車、労働者賠償、製品責任、悪意が私たちの電子システムに侵入し、臨床試験、役員と高級管理者の雇用やり方と受託責任保険を含む。しかし、私たちは私たちが十分な保険カバー水準を維持できるかどうか分からない。保険引受人がクレーム発生後に保険のキャンセルや拒否を求めない保証はありません。いかなる重大な未加入責任も私たちが多額の金額を支払う必要があるかもしれないが、これは私たちの財務状況と運営結果に悪影響を及ぼすだろう。
71
私たちと私たちの現在または未来の任意のパートナーは、私たちが承認したいかなる製品が不良医療事件を引き起こしたり、何もしなければ制裁を招き、それによって私たちの業務に実質的な損害を与えることを規制機関に報告することを要求されるだろう。
もし私たちまたは私たちの現在または未来の任意の潜在的な協力者が私たちの製品を商業化することに成功すれば、FDAおよび外国規制機関は、これらの製品がこれらの不良事件を引き起こしたり、促進したりする可能性があれば、これらの協力者に不良医療事件に関するいくつかの情報を報告することを要求するだろう。私たちの報告義務の時間は私たちが不良事件とイベントの性質を認識した日によって触発されるだろう。私たちと私たちの潜在的な未来のパートナーやCROは規定された時間範囲で不良事件を報告できないかもしれない。もし私たちまたは任意の既存または潜在的なパートナーまたはCROがそのような報告義務を遵守できなかった場合、FDAまたは外国規制機関は、刑事起訴、民事罰金の適用、私たちの製品の差し押さえ、または将来の製品の承認を延期または承認することを含む行動をとることができる。
情報技術システム障害,ネットワーク攻撃やネットワークセキュリティ欠陥が発生すると,我々の業務や運営が影響を受ける可能性がある.
通常の業務プロセスでは、知的財産権、独自の業務情報、臨床試験データおよび個人情報を含むが、これらに限定されないが、総称して秘密情報と呼ばれる大量のデータを収集、記憶、送信、および他の方法で処理する。セキュリティ対策が実装されているにもかかわらず、我々の情報技術システム(インフラを含む)および私たちの現在および未来の任意のCROおよび他の請負業者、コンサルタント、第三者サービスプロバイダおよび協力者のシステムは、コンピュータウイルス、ネットワークセキュリティの脅威(例えば、サービス拒否攻撃、恐喝ソフトウェア、サプライチェーン攻撃、ネットワーク攻撃またはインターネット上のネットワーク侵入、誤った構成、“エラー”または他の脆弱性、ハッカー攻撃、ネットワーク釣りおよび他の社会工学攻撃)、許可されていないアクセスまたは使用、自然災害、テロ、戦争、電気通信および電気故障の攻撃、破壊、および中断を受けやすい。私たちのシステムはまた、窃盗、誤用、許可されていないアクセス、または従業員、サプライヤー、および他の合法的に私たちのシステムにアクセスする第三者の他の不適切または意外な行為のような内部脅威にさらされるだろう。第三者はまた、ユーザ名、パスワード、または他の情報など、私たちの従業員および請負業者に敏感な情報を詐欺的に漏洩させることを試みるか、または他の方法で、私たちのデータにアクセスするために、私たちの電子システム、ネットワーク、および/または物理施設の安全に害を及ぼす可能性がある。また、持続的な混合作業環境のため、私たちはインターネット技術への依存と私たちの遠隔作業の従業員の数によって、サイバー犯罪者の抜け穴を利用するためのより多くの機会を作るかもしれないので、より多くのネットワークセキュリティリスクに直面する可能性がある。
情報技術中断の時間、性質、および範囲の予測不可能性を考慮すると、我々または我々の第三者パートナーおよびサービスプロバイダによって実施される任意のセキュリティプログラムおよび制御措置が、ネットワーク攻撃の発生を防止するのに十分であることは保証されない。妥協の遅延は一般的に数ヶ月で測定されるが、数年かもしれないし、私たちは妥協をタイムリーに検出できないかもしれない。目標に対して新しい技術を開始する前に、私たちはこれらの技術を識別できない可能性があり、これらの技術またはイベントを予測し、その深刻性または影響を評価し、タイムリーに反応または適切な反応を行うか、または十分な予防措置を実施することができず、潜在的なデータ損失をもたらすか、または我々の情報技術システムに他の損害を与える可能性がある。発見されても、攻撃者が法医学証拠の検出を回避し、検出を回避し、除去し、または混同するためのツールおよび技術を使用するために、事件または違反行為を十分に調査または修復することができない可能性がある。
私たちはこれまでどんな重大なシステム障害、事故、またはセキュリティホールを経験してきたとは思いませんが、セキュリティホールが発生して私たちの運営を中断させたり、個人の身分情報や個人の健康情報の不正流出やアクセスを招いたり(何らかのプライバシー法に違反する可能性があります)場合、私たちのビジネス機密の紛失や他の同様の中断にかかわらず、私たちの開発計画や業務運営が実質的に妨害される可能性があります。連邦、州、および外国政府のいくつかの要求は、会社が特定の個人識別情報に関連する個人にセキュリティホールを通知する義務があることを含み、これらの脆弱性は、私たちまたは私たちのサプライヤー、請負業者、または私たちと戦略的関係にある組織が経験した脆弱性によるものかもしれない。いかなるセキュリティホールや他の事件も、実際に発生しても感知されても、私たちの名声に影響を与える可能性があり、法律費用を含む巨額のコストが発生し、顧客の信頼を損ない、新しい市場への私たちの拡張を損害し、修復費用を発生させたり、既存の顧客を失ったりする可能性がある。例えば、臨床試験における臨床試験データの紛失は、私たちの監督管理の承認作業を遅延させ、私たちの回復を著しく増加させる可能性があります
72
データをコピーする。私たちはまた、私たちの候補製品を生産するために第三者に依存しており、彼らのコンピュータシステムに関連する類似のイベントも、私たちの業務に実質的な悪影響を及ぼす可能性がある。実際または予想される中断またはセキュリティホールが私たちのシステム(または私たちの第三者協力者、サービスプロバイダ、請負業者またはコンサルタントのシステム)に影響を与える場合、または機密情報の紛失や意外、不正または許可されていないアクセス、使用、配布、または他の処理をもたらしたり、私たちのデータやアプリケーションを破損したり、機密情報を不適切に開示したりする場合、私たちは責任を招く可能性があり、私たちの候補製品のさらなる開発および商業化は延期される可能性があり、私たちは特定のプライバシーおよびセキュリティ法律に違反するいかなる行為によっても巨額の罰金、処罰または責任を受ける可能性がある。私たちは ネットワーク責任保険を維持する;しかし、この保険は、私たちのシステムの中断または破壊によって生じる可能性のある財務、法律、業務、または名声損失をカバーするのに十分ではない可能性がある。
私たちの業務は訴訟、政府調査、そして法執行行動の影響を受けるかもしれない。
私たちは現在、規制の高い業界の複数の管轄区域で業務を展開しており、私たちは米国で様々な問題について訴訟、政府調査、法執行行動を受ける可能性がある。または、知的財産権、監督管理、製品責任、環境、告発者、虚偽声明、プライバシー、リベート、反賄賂、証券、商業、雇用、および他のクレームおよび法的手続きを含むが、これらに限定されない。
法的手続き、政府調査、そして法執行行動は費用が高くて時間がかかるかもしれない。このような訴訟、調査、または法執行行動によって生じる不利な結果は、重大な損害賠償、罰金、処罰、連邦医療計画から除外されたこと、医療禁止、禁止救済、製品リコール、名声損害、および私たちの業務慣行の修正をもたらす可能性があり、これは私たちの業務および運営結果に実質的な悪影響を及ぼす可能性がある。
私たちの従業員と独立請負業者は、首席調査者、CRO、コンサルタント、サプライヤーを含み、規制基準と要求を遵守しないことを含む、不当な行為またはその他の不当な活動に従事する可能性がある。
私たちは、首席調査者、CRO、コンサルタント、サプライヤーが不正行為または他の不正活動に従事する可能性があるリスクを含む、私たちの従業員と独立請負業者に直面しています。これらの当事者の不正行為は、(I)FDAの法律および法規および他の同様の規制要件に違反する、意図的、無謀、および/または不注意な行為、または許可されていない活動を私たちに開示すること、(Ii)cGMP要件を含む製造基準、(Iii)米国および海外の連邦および州データプライバシー、セキュリティ、詐欺および乱用、および他の医療保健法律法規、または(Iv)財務情報またはデータを真に、完全かつ正確に報告することを要求する法律を含む可能性がある。これらの法律に拘束された活動はまた、不適切な使用或いは歪曲臨床試験過程で得られた情報を含み、著者らの臨床前研究或いは臨床試験中に虚偽のデータを作成し、或いは薬物製品を不法に流用し、これは規制制裁を招き、著者らの名声に深刻な損害を与える可能性がある。従業員や他の第三者の不正行為を常に識別し、阻止することができるわけではなく、このような活動を発見し、防止するための予防措置は、未知または管理不可能なリスクや損失を効果的に制御することができないか、またはそのような法律や法規を遵守できないことによる政府調査または他の行動または訴訟から私たちを保護することができない可能性がある。さらに、私たちは、一人や一人の政府が、起こらなくても、このような詐欺や他の不適切な行為を告発する可能性があるというリスクに直面している。もし私たちにこのような訴訟を提起した場合、私たちは私たちの権利を弁護したり、維持することに成功しませんでした。これらの行動は、重大な民事、刑事および行政処罰、損害賠償、罰金、引き渡し、連邦医療保険、医療補助および他の連邦医療保健計画から除外される可能性があり、監禁、契約損害、名声損害、利益および将来の収益の減少、追加の報告要件および監督を含むかもしれません(当社の誠実な合意または同様の合意の制約を受けて、これらの法律に違反した疑いを解決し、私たちの業務を削減すれば)。いずれも私たちの業務運営能力と運営結果に悪影響を及ぼす可能性があります。
73
私たちは戦略的取引に従事するかもしれません。これは私たちの流動性に影響を与え、私たちの費用を増加させ、私たちの経営陣に大きな妨害を与えるかもしれません。
我々は、企業の買収、資産の購入、および知的財産権、製品または技術の対外許可または内部許可のような戦略的取引を時々考慮するかもしれない。私たちが将来考慮する可能性のある潜在的な取引は、剥離、戦略パートナーシップ、合弁企業、再編、資産剥離、業務合併と投資を含む様々な商業手配を含む。将来のいかなる取引も、私たちの短期的および長期的な支出を増加させ、私たちの株式証券(私たちの普通株を含む)の潜在的な希釈発行、または債務、または負債、償却費用、または買収が行われている研究開発費を発生させる可能性があり、これらのいずれも、私たちの財務状況、流動性、および運営結果に影響を与える可能性がある。将来の買収はまた私たちが追加的な融資を受ける必要があるかもしれないし、これらの融資は優遇条項で提供されないかもしれないし、全くないかもしれない。このような取引は決して成功しないかもしれないし、私たちの経営陣が多くの時間とエネルギーを投入する必要があるかもしれない。また、私たちが将来買収する可能性のある任意の業務の統合は、私たちの既存の業務を混乱させる可能性があり、複雑で、リスクとコストの高い努力である可能性があり、買収のすべてのメリットを実現することができないかもしれません。したがって、私たちが上記の性質の任意の追加取引を行うか、成功するかは保証できませんが、私たちが実際に達成した任意の追加取引は、私たちの業務、運営結果、財務状況、および見通しに重大な悪影響を及ぼす可能性があります。
私たちは純営業損失の繰越と他の税務属性を使用する能力が限られている可能性がある。
私たちの歴史の中で、私たちは大きな損失を受けて、近い将来に利益を達成することを期待していないし、永遠に利益を達成しないかもしれない。課税損失が生じ続ける限り、未使用損失は繰り越し、制限された場合には将来の課税収入を相殺し、あれば、このような未使用損失が満期になるまで(2017年12月31日以降の課税年度に発生した連邦純営業損失(NOL)繰越は満期の影響を受けないにもかかわらず)。2023年12月31日まで、私たちの連邦とカリフォルニアNOLの繰り越し金額はそれぞれ約2.09億ドルと9380万ドルで、連邦とカリフォルニアの研究開発信用の繰越金額は約1140万ドルで、その中に連邦孤児薬品税収の控除免除金額と480万ドルが含まれている。
規則第382節によると、わが社の所有権に何らかの累積的な変化が生じた場合、私たちの連邦NOL繰り越しは年間制限を受ける可能性があります。“規則”第382条の規定によると、1つ以上の会社の株式の少なくとも5%を保有する株主又は株主団体が3年間のスクロール期間内の持株比率がその最低持株割合より50ポイント以上増加した場合、“所有権変更”が発生する。似たような規則は州税法に適用されるかもしれない。私たちはまだ私たちの所有権の累積変化金額を正式に決定していない、あるいは私たちはNOL繰越と他の税金属性の能力を利用して、何の制限も受けていない。しかし,所有権の変更により,将来の課税収入や税金負債を相殺するNOL繰越や他の税収属性を利用する能力が制限される可能性があると考えられる。また、私たちのNOL繰り越しはアメリカ国税局と州税務機関の審査と可能な調整を受けることになります。また、2017年12月31日以降の課税年度に発生する連邦NOL繰り越しは、通常、2020年12月31日以降の課税年度課税所得額の80%を相殺するためにしか利用できない。もし私たちが課税収入を稼げば、これらの制限は私たちの将来の所得税負担を増加させる可能性があり、私たちの将来のキャッシュフローは悪影響を受けるかもしれない。我々のNOL繰延資産や他の繰延税金資産が最終的に将来の収益を実現する不確実性により、このような資産に関する全額推定準備が記録されている。
私たちの知的財産権に関するリスクは
私たちの成功は私たちの知的財産権とノウハウを保護する能力にかかっている。
私たちのビジネス成功は、私たちの候補製品、ノウハウ、およびその用途のために特許保護および商業秘密保護を取得および維持する能力、および他人の固有の権利を侵害することなく運営する私たちの能力にある程度依存する。私たちは一般に、私たちの候補製品、ノウハウ、およびその用途に関連する特許出願を米国および海外に提出することによって、私たちの固有の地位を保護しています。これらの特許は、私たちの業務に非常に重要です。我々の特許出願は、そのような出願が特許を取得するまで、およびそのような出願が特許を取得するまで、発行された特許請求の範囲に限定されない限り、そのような出願において主張される技術を実施する第三者に対して実行することはできない。私たちの特許は保証できません
74
私たちまたは私たちの許可者の出願は、追加の特許が発行されることをもたらしますか、または発行された特許は、同様の技術を有する競争相手に対して十分な保護を提供することになり、発行された特許が第三者によって侵害され、設計され、失効しないことを保証することもできません。発行された特許が後に無効であると認定されたとしても、強制的に執行されないか、または第3の方向の各特許庁または裁判所が提起した訴訟において修正または撤回される可能性がある。未来の私たちの所有権に対する保護の程度は不確実だ。限られた保護だけを提供するかもしれないし、私たちの権利を十分に保護できないかもしれないし、私たちがどんな競争優位性を獲得したり維持したりすることを可能にするかもしれない。もし私たちが獲得した任意の特許保護の範囲が十分に広くない場合、あるいは私たちまたは私たちの許可されたいかなる特許保護も失った場合、私たちは競争相手の能力が悪影響を受けることを阻止するだろう。私たちの候補製品に関連する知的財産権を適切に保護できなかったことは、私たちの財務状況や運営結果に大きな悪影響を及ぼす可能性があります。私たちは米国でTomivosertibおよびZotatifinに対する許可された特許を持っているが、私たちの他の米国で決定されている特許出願、対応する国際特許出願、特定の外国地域のTomivosertibおよびZotatifinに対する特許出願中の権利要件、または私たちの他の候補製品に対する任意の特許出願は、米国特許商標局(USPTO)米国裁判所または外国特許庁および裁判所によって特許出願可能とみなされ、挑戦されれば、私たちが発行した特許中の権利要件が無効または実行不可能であると認定されないことも決定できない。
特許出願プロセスは、多くのリスクおよび不確実性の影響を受け、私たちまたは私たちの現在または未来のどのパートナーも、特許を取得して保護することによって、私たちの候補製品を保護することに成功することを保証することはできない。これらのリスクと不確実性には
特許訴訟プロセスも高価で時間がかかり、私たちは合理的なコストで、またはタイムリーに、または商業的利点を持つ可能性のあるすべての司法管轄区域を保護して、すべての必要または望ましい特許出願を提出し、起訴することができないかもしれない。我々は,特許保護を得るのが遅くなるまで,我々の研究開発成果で特許を申請できることを確認できない可能性もある.さらに、場合によっては、カリフォルニア大学サンフランシスコ校とのライセンス契約に基づいて、特許出願の準備、提出、起訴を制御する権利があり、または第三者から許可された技術に対する特許を維持する権利がありません。私たちはまた、許可された特許権を実行するために許可側の協力が必要である可能性があり、このような協力は提供されない可能性がある。したがって、このような特許および出願は、私たちの業務の最適な利益に合った方法で起訴され、強制されてはならない。私たちは、私たちの許可者の特許起訴および維持活動が適用された法律および法規を遵守しているか、またはそのような特許またはそのような出願が発行される可能性のある任意の特許の有効性および実行可能性に影響を及ぼす可能性があることを確認することができない。もし彼らがそうしなければ、これは私たちが許可している任意の知的財産権を適用する権利を失う可能性がありますので、私たちが製品や候補製品を開発して商業化する能力は不利な影響を受ける可能性があり、競争相手が競合製品を製造、使用、販売することを阻止できないかもしれません。
75
さらに、私たちが所有しているおよび許可中の特許権は、1つまたは複数の第三者の権利によって保持される可能性がある。例えば,カリフォルニア大学サンフランシスコ校とのライセンス契約によると,特許権を発生させる研究部分は米国政府が援助している.したがって,政府はこれらの特許権や技術に対して一定の権利,あるいは進行権を持つ可能性がある.政府資金で新しい技術を開発する場合、政府は、政府が非商業目的のために発明を使用することを許可する非独占的許可を含む、生成された任意の特許のいくつかの権利を得ることができる。これらの権利は、政府が第三者に私たちの機密情報を開示し、第三者が私たちが許可した技術を使用または許可する先行権を行使することを可能にするかもしれない。さらに、このような発明に対する私たちの権利は、そのような発明を含む製品を米国で製造するいくつかの要件によって制約される可能性がある。政府がこのような権利を行使することは、私たちの競争地位、業務、財務状況、経営結果、そして将来性を損なう可能性がある。
さらに、私たちは、私たちの従業員、会社協力者、外部科学協力者、CRO、第三者メーカー、コンサルタント、コンサルタント、および他の第三者などと、私たちの研究開発成果を得る権利のある特許に関する当事者と秘密保持および秘密協定を締結しているにもかかわらず、いずれもこのような合意に違反し、特許出願を提出する前にそのような成果を開示し、特許保護を求める能力を危険にさらしている可能性がある。
また、生物製薬会社の特許地位は通常高度に不確定であり、複雑な法律や事実問題に関連しており、近年多くの訴訟のテーマとなってきている。特許の発行は、その発明性、範囲、有効性または実行可能性に関する確実な結論ではなく、もし私たちの特許が発行され、あるいは私たちが他人から許可した特許権が、米国や海外の裁判所または特許庁で挑戦される可能性がある。このような挑戦は、排他的な喪失または特許主張の縮小、無効、または実行不能をもたらす可能性があり、これは、他人が類似または同じ製品を使用することを阻止するか、またはそれを商業化する能力を制限するか、または私たちの製品および候補製品の特許保護期間を制限する可能性がある。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちの知的財産権は他の人たちが私たちと似ているか同じ製品を商業化することを排除するために十分な権利を提供してくれないかもしれない。
私たちのビジネスの成功は、第三者の特許や他の固有の権利を侵害することなく運営する私たちの能力に大きく依存する。第三者は私たちが彼らの専有権を侵害し、損害賠償責任を招いたり、私たちの開発と商業化努力を阻害したり延期したりする可能性があると主張している。
私たちの商業的成功は第三者の特許と固有の権利の侵害を避けることにある程度依存する。他のエンティティは、特許または独自の権利を有しているか、または取得することができ、これは、私たちの候補製品および将来承認される可能性のある製品を製造、使用、販売、提供、または輸入する能力を制限し、または私たちの競争的地位を損なう可能性がある。米国国内外では、特許侵害訴訟、干渉、異議、再審、当事者間の審査手続き、および米国特許商標局および/または対応する外国特許庁に提起された認可後審査(“PGR”)プログラムを含むバイオテクノロジーおよび製薬業界特許および他の知的財産権に関する訴訟が大量にある。我々が候補製品を開発している分野には,第三者米国や外国から発行された特許や係属中の特許出願が多く存在する.我々の候補製品の使用または製造に関連する第三者特許または材料、配合、製造方法または治療方法の請求項に記載の特許出願が存在する可能性がある。
バイオテクノロジー産業の拡張とより多くの特許の発行に伴い、我々の候補製品は第三者特許権侵害の告発を受けるリスクが増加する可能性がある。特許出願は一定期間秘密であるため、関連出願が公表される前に、任意の候補製品の商業化が第三者特許を侵害する可能性があることを知らない可能性があり、製品候補又は技術に関連する特許出願を初めて提出した会社であることも確認できない。さらに、特許出願が発表されるまでに数年かかる可能性があるため、現在処理されている特許出願がある可能性があり、後で私たちの候補製品が発行された特許を侵害する可能性がある。さらに、特許検索は、特許間の用語の違い、データベースの不完全さ、および特許請求の意味を評価することが困難であるため、我々の技術に関連する可能性のある第三者特許権を識別することは困難である。私たちが知っていない保証もありませんが、私たちの業務とは関係のない既存技術だと思いますが、それにもかかわらず、これらの技術は最終的には、私たちが将来承認される可能性のある製品を製造、使用、販売、提供、または輸入する能力を制限したり、私たちの競争的地位を損なう可能性があります。第三者が主張するいかなる特許侵害請求も非常に時間がかかり、可能性がある
76
本年度報告の日現在、第三者は私たちの特許を侵害していると主張していませんが、他の人は私たちの候補製品の発売を阻止する可能性のある独自の権利を持っている可能性があります。私たちの特許関連法律訴訟に対して損害賠償を要求し、私たちの候補製品やプロセスに関連する商業活動を禁止しようと努力しても、故意に侵害されたと判断された場合、3倍賠償し、私たちの候補製品を製造またはマーケティングする許可証を取得することを含む潜在的な損害賠償責任を負わせる可能性があります。これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、当社の業務における従業員資源を大量に移転することになる。私たちがこのような訴訟に勝つかどうかは予測できませんし、これらの特許に必要ないかなる許可が商業的に受け入れられる条項で提供されるかどうかも予測できません。また,我々や将来の戦略的パートナーが許可を得ることができても,これらの権利は非排他的である可能性があり,これは我々の競争相手に同じ知的財産権を獲得させる可能性がある.さらに、必要であれば、権利侵害を避けるために、私たちの候補製品やプロセスを再設計できるかどうかを決定することはできません。したがって、司法や行政訴訟で不利な裁決を下したり、必要なライセンスを取得できなかったりすることで、私たちの候補製品の開発と商業化を阻止することができ、これは私たちの業務、財務状況、経営業績を損なう可能性があります。また、知的財産権訴訟は、その結果にかかわらず、負の宣伝をもたらす可能性があり、私たちの候補製品や技術をマーケティングまたは他の方法で商業化することを禁止する可能性があります。
私たちは私たちの特許を保護または強制的に執行することができるかもしれない特許の訴訟に巻き込まれるかもしれないが、これは高価で時間がかかり、成功しないかもしれない。さらに、法廷で疑問が提起された場合、私たちが発行した特許は無効または実行不可能と認定される可能性がある。
競争相手は私たちの知的財産権や私たちの許可側の知的財産権を侵害するかもしれない。権利侵害や不正使用を防ぐために、私たちは権利侵害請求を要求されるかもしれません。これは高価で時間がかかるかもしれません。さらに、特許侵害訴訟では、裁判所は、私たちが所有しているまたは許可中の特許が無効であること、強制執行できないこと、および/または侵害されていないと判断することができる。もし私たちまたは私たちの現在または未来の任意の協力者が第三者に対して法的訴訟を提起して、私たちの候補製品のうちの1つに対する特許を強制的に執行する場合、被告は私たちの特許の全部または一部が無効であり、および/または強制的に執行できないと反訴することができる。米国の特許訴訟では,被告が無効および/または実行不可能と主張する反訴が一般的である。有効性を疑問視する理由には、新規性の欠如、明らかな、または実施できないことを含む、いくつかの法定要件のいずれかを満たすことができなかったと言われている。主張を実行できない理由には,特許訴訟に関連する者が米国特許商標局に関連情報を隠蔽したり,起訴中に誤った陳述をしたりした疑惑が含まれている可能性がある.第三者も米国特許商標局に類似したクレームを出すことができ、訴訟範囲外であっても同様である。
法的宣言が無効であり、強制執行できない結果は予測不可能であり、従来技術は、我々の特許または我々の許可者の特許を無効にするか、または私たちの1つまたは複数の係属中の特許出願が特許を発行することを阻止する可能性がある。私たちの特許および特許出願に関連するすべての潜在的に関連する以前の技術が発見されたことは保証されない。また、私たちの特許が挑戦されていなくても、それらは私たちの知的財産権を十分に保護し、私たちの候補製品に排他性を提供し、他の人が私たちの主張をめぐる設計を阻止したり、競争優位を提供したりすることができない可能性がある。被告が無効および/または強制執行できない法的主張に勝った場合、私たちは候補製品の少なくとも一部またはすべての特許保護を失うだろう。さらに、私たちの特許および特許出願によって提供される保護の広さまたは強度が脅かされている場合、会社が私たちと協力し、現在または将来の候補製品を許可、開発、または商業化することを阻止するかもしれない。このような特許保護の喪失は私たちの業務に実質的な悪影響を及ぼすだろう。
77
被告が無効および/または強制執行できない法的主張に勝った場合、私たちは候補製品の少なくとも一部またはすべての特許保護を失うだろう。このような特許保護の喪失は私たちの業務に実質的な悪影響を及ぼすだろう。
我々の特許または特許出願または我々の許可者の発明に関連する発明の優先権を決定するために、第三者によって引き起こされるか、または米国特許商標局によって発表された干渉または派生プログラムが必要である可能性がある。不利な結果は、私たちが関連技術の使用を停止することを要求するか、または勝利者から許可を得ようとすることを要求するかもしれない。もし勝利者が商業的に合理的な条件で私たちにライセンスを提供しなければ、私たちの業務は損害を受ける可能性があります。私たちの訴訟、妨害、または派生訴訟の弁護は失敗する可能性があり、成功しても巨額のコストを招き、私たちの経営陣や他の従業員の注意を分散させる可能性がある。また、訴訟に関連する不確実性は、私たちの臨床試験を継続するために資金を調達し、私たちの研究計画を継続し、第三者から必要な技術的許可を得ること、または候補製品を市場に出すための開発またはパートナー関係の製造に役立つ能力の確立に大きな悪影響を及ぼす可能性がある。
解決策が私たちに有利であっても、私たちの知的財産権に関する訴訟や他の法的手続きは、私たちに巨額の費用を発生させ、私たちの技術や管理者の正常な責任を分散させる可能性があります。さらに、公聴会、動議、または他の一時的な手続きまたは事態の発展の結果を公開発表する可能性があり、証券アナリストまたは投資家がこれらの結果がマイナスであると考える場合、私たちの普通株式価格に大きな悪影響を及ぼす可能性がある。このような訴訟または訴訟は、私たちの運営損失を大幅に増加させ、開発活動または任意の将来の販売、マーケティング、または流通活動に使用することができるリソースを減少させる可能性がある。私たちはそのような訴訟や訴訟手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らがより多くの財政資源を持っているので、私たちよりもこのような訴訟や訴訟の費用を効率的に負担するかもしれない。特許訴訟や他の訴訟の開始と継続による不確実性は、市場での私たちの競争能力を損なう可能性がある。
さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。聴聞、動議、または他の一時的手続き、または事態発展の結果を公開することもできる。もし証券アナリストや投資家がこれらの結果がマイナスだと思っていれば、私たちの普通株の価格に実質的な悪影響を及ぼす可能性がある。
しかも、特許の発行は私たちに特許発明を実践する権利を与えない。第三者は私たち自身の特許製品の販売を阻止し、私たち自身の特許技術を実践する特許を持っているかもしれない。私たちが特許権を行使する能力はまた私たちが侵害行為を検出する能力にかかっている。その製品やサービスに関するコンポーネントや方法を宣伝しない侵害者を検出することは困難である可能性がある.さらに、競争相手または潜在的な競争相手の製品またはサービス侵害の証拠を得ることは困難または不可能である可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、もし私たちが勝てば、得られた損害賠償や他の救済措置は商業的な意味がないかもしれない。
特許改革立法は、私たちの特許出願をめぐる起訴と、私たちが発表した特許の実行または保護の不確実性とコストを増加させるかもしれない。
2011年9月16日、“ライシー·スミス米国発明法”(略称“ライシー·スミス法案”)が法律に署名された。“ライシー·スミス法案”は米国特許法を多くの重大な改正を行った。これらの条項には,2013年3月以降に提出された特許出願が起訴される方法に影響を与える条項が含まれており,特許訴訟に影響を与える可能性がある。特に、“ライシー·スミス法案”によれば、米国は2013年3月に“最初に特許出願を提出した発明者”制度に移行し、すなわち最初に特許出願を提出した発明者が特許を取得する権利を有することになる。第三者は、米国特許商標局が特許を発行する前に既存技術を提出することを許可され、許可された後の審査、派生、再審査、当事者間の審査または妨害プログラムを含み、私たちの特許権または他の人の特許権に挑戦することが可能である。このような提出、訴訟、または訴訟における不利な裁決は、私たちの特許権の範囲を縮小したり、実行可能にしたり、無効にしたりして、私たちの競争的地位に悪影響を及ぼす可能性があります。
78
USPTO手続きの証拠基準は、米国連邦裁判所が特許請求の無効を宣言するために必要な証拠基準よりも低いため、第三者は、USPTO手続において、USPTOが権利請求を無効にするのに十分な証拠を提供する可能性があり、同じ証拠が最初に地域裁判所訴訟で提出された場合には、権利請求を無効にするのに十分ではない。したがって,第三者は米国特許商標局の手続きを用いて我々の特許主張の無効を宣言しようとする可能性があり,第三者が地域裁判所訴訟で最初に被告として疑問を提起すれば,我々の特許主張は無効ではない.したがって、“ライシー·スミス法案”およびその実施は、私たちの将来の特許出願または私たちの現在および未来のライセンシーの特許出願をめぐる起訴、および私たちが将来発行する特許または私たちの現在および未来のライセンシーの特許の実行または保護の不確実性およびコストを増加させる可能性があり、これらは、すべて私たちの業務、財務状況、運営結果、および見通しに大きな悪影響を及ぼす可能性がある。
また、2023年6月には、欧州特許に関する訴訟に単一の汎欧州単一特許と新たな欧州統一特許裁判所を提供することを目指して、欧州特許パッケージ又はEU特許パッケージ条例が実施された。UPCは新しい裁判所制度であるため、裁判所には前例がなく、UPC以前のいかなる訴訟の不確実性も増加した。現在提案されているEU特許セットによると、私たちは裁判所が存在する7年前に私たちの特許をUPCから離脱することを選択する権利があるが、そうすることは私たちが新しい統一裁判所の利点を達成することを阻止するかもしれない。
もし私たちが既存のライセンス契約や任意の未来のライセンスプロトコルの下での任意の義務を履行できなかったり、これらの合意に関連した紛争が発生した場合、私たちの業務と私たちの知的財産権に悪影響を及ぼす可能性があります。
私たちはカリフォルニア大学サンフランシスコ校と締結されたライセンス契約の締約国であり、第三者と追加のライセンス契約を締結する可能性があり、私たちに適用、職務調査、開発、商業化スケジュール、マイルストーン支払い、特許使用料、保険、その他の義務が適用されるかもしれません。私たちが許可された知的財産権を使用する権利はこのような合意条項の継続と遵守にかかっている。第三者から許可された知的財産権の権利については、これらに限定されないが、議論が生じる可能性がある
我々が許可したり、第三者から取得した知的財産権および他の権利をめぐる紛争が、許容可能な条項で現在の許可スケジュールを維持する能力を阻害または弱化した場合、影響を受けた候補製品の開発に成功し、商業化することができない可能性がある。現在または将来のライセンス契約の義務を履行できない場合、これらのプロトコルは終了される可能性があり、またはこれらのプロトコルの下での私たちの権利範囲が縮小される可能性があり、これらのプロトコルによって許可された任意の製品を開発、製造、または販売することができない可能性がある。
79
私たちは競争相手から従業員を誤って雇用したか、または私たちまたは私たちの従業員が彼らの前の雇用主の機密情報や商業秘密を間違って使用または開示したという非難を受けるかもしれない。
製薬業界では、私たちの従業員のほかに、コンサルタントを招いて候補製品の開発を手伝ってくれています。これは製薬業界でよく見られます。これらのコンサルタントの多く、および私たちの多くの従業員は、以前に他の製薬会社に雇用されていたか、または以前に他の製薬会社に提供されていた可能性があり、または現在、私たちの競争相手または潜在的な競争相手を含む他の製薬会社にコンサルティングサービスを提供している可能性がある。私たちは、私たち、私たちの従業員またはコンサルタントが無意識に、またはその前の雇用主またはその前の顧客または現在の顧客の商業秘密または他の固有の情報を使用または漏洩したという疑惑の影響を受けるかもしれない。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭損害賠償の支払いに加えて、貴重な知的財産権を失う可能性があり、これは私たちの業務に悪影響を及ぼす可能性がある。私たちがこれらのクレームを弁護することに成功しても、訴訟は巨額のコストを招き、私たちの管理チームの注意を分散させる可能性がある。
また、私たちの政策は、知的財産権の概念や開発に参加する可能性のある私たちの従業員と請負業者が合意に署名し、このような知的財産権を私たちに譲渡することを要求しているにもかかわらず、実際に私たちが自分の知的財産権と見なしているすべての当事者とこのような合意を実行することができないかもしれない。知的財産権の譲渡は自動的に実行されない可能性があり、あるいは譲渡協定が違反される可能性があり、私たちは第三者にクレームをつけさせられたり、私たちが私たちの知的財産権の所有権だと思うことを決定するために、私たちが提起する可能性のあるクレームを弁護したりすることができます。このようなクレームは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。
私たちは私たちの特許と他の知的財産権の発明権または所有権のクレームに疑問を受けるかもしれない。
私たちはまた、元従業員、協力者、または他の第三者が私たちの特許または他の知的財産権に対して所有権権益を持っているというクレームを受ける可能性がある。訴訟は、これらと他の挑戦在庫または所有権のクレームに対抗するために必要かもしれない。もし私たちがこのようなクレームを弁護することができなければ、お金の損害賠償を支払う以外に、私たちは貴重な知的財産権を失う可能性がある。このような結果は私たちの業務に実質的な悪影響を及ぼすかもしれない。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。上記のいずれも、当社の業務、財務状況、経営結果、見通しに重大な悪影響を及ぼす可能性があります。
特許条項は候補製品に対する私たちの競争地位を十分に長く保護するのに十分ではないかもしれない。
特許の寿命は限られている。米国では、すべての維持費が適時に支払われる場合、特許の自然失効時間は、通常、米国で最初の非臨時出願日から20年である。様々な延期があるかもしれないが、特許の有効期限とその提供される保護は限られている。私たちの候補製品が特許を取得しても、特許有効期限が切れると、生物模倣薬を含む競争製品からの競争に直面するかもしれません。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちが持っている特許と許可された特許の組み合わせは、他社が私たちと似ているか同じ製品を商業化することを排除するために十分な権利を提供してくれないかもしれない。
関連する第三者特許を識別できないか、または第三者特許の関連性、範囲、または満了時間を誤って解釈することができない可能性があり、これは、候補製品を開発およびマーケティングする能力に悪影響を及ぼす可能性がある。
私たちは、関連特許の識別、特許請求の範囲、または関連特許の満了を含めて、私たちのいかなる特許検索または分析も保証することはできず、私たちは、米国および海外が現在および未来の製品および候補製品の商業化に関連しているか、または必要なすべての第三者特許および未解決特許出願を識別していることを保証することもできない。特許請求の範囲は、法律の解釈、特許における書面開示、および特許の起訴履歴に依存する。問題の関連性や範囲の説明は
80
特許または出願中の特許は正しくない可能性があり、これは私たちの製品を販売する能力に悪影響を及ぼす可能性がある。私たちは、私たちの製品または候補製品が第三者特許のカバー範囲内にないかどうかを誤って決定するか、またはサードパーティ係属中の特許出願が関連範囲の特許請求を提出するかどうかを誤って予測する可能性がある。私たちが関連する特許の満期日の決定は正しくないかもしれませんが、関連特許を識別して正確に解釈できなければ、私たちが製品を開発し、マーケティングする能力に悪影響を及ぼすかもしれません。上記のいずれも、当社の業務、財務状況、経営結果、見通しに重大な悪影響を及ぼす可能性があります。
もし私たちが私たちの候補製品のために特許期間を延長しなければ、私たちの業務は実質的な損害を受けるかもしれない。
FDAが我々の候補製品の上場承認の時間、期限、および詳細に基づいて、私たちの1つ以上の米国特許は、1984年の“薬品価格競争および特許期限回復法”(“Hatch-Waxman修正案”)に従って限られた特許期限回復を得る資格がある可能性がある。ハッジ·ワックスマン改正案は、製品開発およびFDA規制審査中に失われた特許期間の補償として、特許回復期間を最長5年とすることを許可している。1つの特許期間の延長は、製品が承認された日から14年を超えてはならず、1つの特許を延長することしかできず、承認された薬物、その使用方法又はその製造方法に関する請求項を延長することしかできない。一部の外国司法管轄区域にも、規制審査による商業化遅延を補償するための同様の特許期間回復条項があり、例えば欧州では、補充保護証明書(SPC)に基づいている。しかし,適用の最終期限内に出願を提出できなかったこと,関連特許の満了前に出願を提出できなかったことや適用の要求を満たしていなかったことなどにより延期が得られなかった可能性がある.しかも、特許保護の適用期間や範囲は私たちが要求しているものよりも短いかもしれない。もし私たちが特許期間の延長や回復を得ることができない場合、あるいはどのような延長の期限が私たちが要求したよりも短い場合、私たちの競争相手は私たちの特許が満期になった後に競争製品の承認を得ることができ、私たちの収入は減少する可能性があり、実質的である可能性がある。また、このような状況が発生すれば、私たちの競争相手は私たちの開発と試験への投資を利用して、私たちの臨床と臨床前データを参考にして、他の場合よりも早く彼らの製品を発売するかもしれない。
もし私たちの商標と商号が十分に保護されていなければ、私たちは興味のある市場で知名度を作ることができないかもしれません。私たちの業務は悪影響を受けるかもしれません。
私たちの登録または未登録商標または商号は、疑問、侵害、回避、または汎用商標として発表されるか、または他の商標を侵害していると認定される可能性がある。商標登録過程で、私たちは米国特許商標局または他の外国司法管轄区から私たちの出願の拒絶を受けることができる。私たちはこの拒否に答える機会があるにもかかわらず、私たちはそれらを克服できないかもしれない。また,米国特許商標局や多くの外国司法管轄区の類似機関では,第三者は係属中の商標出願に反対し,登録商標の取り消しを求める機会がある。私たちの商標に反対したり取り消したりするかもしれませんが、これらの商標は存在し続けることができないかもしれません。また、私たちは、私たちが登録したかどうかにかかわらず、私たちの候補製品と共に使用されるどんな名前もFDAの承認を受けなければならないと提案しています。ヨーロッパにも似たような要求がある。FDAは、通常、他の製品名と混同される可能性がある可能性を含む、提案された製品名を検討する。FDAまたは外国司法管轄区域の同等の行政機関が私たちが提案した任意の独自製品名に反対する場合、私たちは、適用商標法の資格に適合し、第三者の既存の権利を侵害せず、FDAのために受け入れられる適切な代替名を決定するために、多くの追加資源を必要とするかもしれません。さらに、多くの国では、商標登録を所有し維持することは、高級商標所有者がその後に提起した侵害クレームに対して十分な弁護を提供できない可能性がある。
私たちはこれらの商標や商品名に対する私たちの権利を保護できないかもしれません。私たちは私たちが関心のある市場の潜在的なパートナーや顧客の中で知名度を確立するために必要です。時々、競争相手は私たちと似たような商品名や商標を採用して、ブランド表示を確立する能力を阻害し、市場の混乱を招く可能性があります。さらに、他の登録商標または商標の所有者は、我々の登録または未登録商標または商号の変異体を含む商号または商標侵害クレームを提出することができる。長期的には、私たちの商標や商号に基づいて名称を確立することができなければ、効果的に競争できない可能性があり、私たちの業務は悪影響を受ける可能性があります。商標、商業秘密、ドメイン名、著作権または他の知的財産権に関連する専有権を実行または保護する努力は無効である可能性があり、大量のコストおよび資源移転を招き、私たちの財務状況または運営結果に悪影響を及ぼす可能性があります。
81
米国特許法や他の国の法律の変化は、特許の全体的な価値を低下させ、候補製品を保護する能力を弱める可能性がある。
他の生物製薬会社と同様に、私たちの成功は知的財産権、特に特許に大きく依存している。生物製薬産業で特許を取得して実施することは高度な技術と法律の複雑さに関連している。したがって、生物製薬特許を取得して実行することは高価で、時間と固有の不確実性だ。米国や他国の特許法または特許法解釈の変化は,我々の知的財産権の価値を低下させる可能性がある。私たちは、私たちの特許または第三者特許で許容または実行される可能性のある特許請求の範囲の広さを予測することができない。しかも、国会や他の外国立法機関は私たちに不利な特許改革立法を通過するかもしれない。例えば、米国最高裁判所は近年、いくつかの特許事件に対して裁決を下しているが、場合によっては利用可能な特許保護範囲を縮小するか、場合によっては特許所有者の権利を弱めるかである。我々の将来の特許取得能力に関する不確実性の増加に加えて,このようなイベントの結合は,いったん特許を取得する価値に関する不確実性をもたらしている.米国議会、米国連邦裁判所、米国特許商標局または外国司法管轄区の類似機関の決定によると、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、それによって、私たちが新しい特許を獲得したり、私たちの既存特許と将来獲得可能な特許を実施する能力を弱める可能性がある。
私たちは世界各地で私たちの知的財産権を保護できないかもしれない。
私たちは米国でTomivosertibおよびZotatifinに対する発行された特許を持っているにもかかわらず、米国および他の国ではTomivosertib、Zotatifin、および他の候補製品に対する未解決の特許出願を持っているが、Tomivosertib、Zotatifin、および他の候補製品の特許は世界のすべての国で提出され、起訴され、擁護されているが、私たちは米国以外のいくつかの国での知的財産権は米国ほど広くないかもしれない。また、いくつかの外国の法律は知的財産権の保護の程度はアメリカの連邦や州法律に及ばない。したがって、私たちは、米国以外のすべての国/地域で第三者が私たちの発明を実施したり、米国または他の管轄区域で私たちの発明を使用して製造された製品を販売または輸入することを阻止することができないかもしれない。さらに、欧州、日本、および中国などのいくつかの司法管轄区域の特許可能性基準は、例えば、元の特許出願において請求項が字面的なサポートを有することを要求することと、元の特許出願に存在しないサポートデータの使用を制限することとを含む、米国よりも高い可能性がある。これらのより高い特許性要件によると、米国や他の管轄地域で同じまたは同様の特許保護を受けることができても、特定の管轄区で十分な特許保護を受けることができない可能性がある。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの技術を使用して自分の製品を開発することができ、また、他の侵害製品を私たちが特許保護を持っているが法執行力がアメリカに及ばない地域に輸出することもできる。これらの製品は私たちの候補製品と競争するかもしれませんが、私たちの特許や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。
さらに、米国および外国の地政学的行動は、私たちの特許出願または任意の既存または将来のライセンシーの特許出願をめぐる起訴または維持、ならびに私たちが発行した特許または任意の既存または未来のライセンシーの特許の維持、実行または保護の不確実性およびコストを増加させる可能性がある。例えば、ウクライナでのロシアの衝突に関連する米国と外国政府の行動は、ロシアでの特許出願の提出、起訴、維持を制限または阻止する可能性がある。政府の行動はまたロシアで発行された特許を維持することを阻止するかもしれない。これらの行動は、ロシアにおける特許権の一部または全部の喪失を招く可能性がある私たちの特許または特許出願を放棄または失効させる可能性がある。このような事件が発生すれば、私たちの業務に実質的な悪影響を及ぼす可能性がある。また、ロシア政府は2022年3月に、ロシアの会社や個人が同意または補償なしに米国から特許権者が所有する発明を実施することを許可する法令を採択した。したがって、私たちは、第三者がロシアで私たちの発明を実践したり、私たちの発明を使用して製造された製品をロシア国内で販売したり輸入したりすることを防ぐことができないだろう。したがって、私たちの競争地位は損なわれる可能性があり、私たちの業務、財務状況、経営業績、見通しは不利な影響を受ける可能性があります。
82
多くの会社は外国の管轄区域の知的財産権の保護と保護に重大な問題に直面している。多くの国の法制度は、特許や他の知的財産権保護の強制執行を支持しておらず、これは、私たちの特許の侵害を阻止したり、私たちの独自の権利を侵害する方法で競合製品をマーケティングすることを困難にする可能性がある。外国の管轄区域で私たちの特許権を強制的に執行する訴訟手続きは、巨額のコストを招き、私たちの努力と注意を私たちの業務の他の側面から移転させる可能性があり、私たちの特許が無効または狭義に解釈されるリスクに直面する可能性があり、私たちの特許出願を発表できないリスクに直面させ、第三者からのクレームを引き起こす可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。したがって、私たちが世界各地で私たちの知的財産権を実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネスメリットを得るのに十分ではないかもしれません。多くの国にも強制許可法があり,これらの法律により特許権者は第三者に強制的に許可を付与される可能性がある。また、多くの国は政府機関や政府請負業者に対する特許の実行可能性を制限している。これらの国では,特許権者の救済措置は限られている可能性があり,このような特許の価値を大幅に低下させる可能性がある。もし私たちが第三者に私たちの業務に関連する任意の特許の許可を与えることを余儀なくされた場合、私たちの競争地位は損なわれる可能性があり、私たちの業務、財務状況、運営結果、および将来性は不利な影響を受ける可能性がある。
知的財産権訴訟は不利な宣伝を招き、私たちの名声を損ない、私たちの普通株の市場価格を下落させる可能性がある。
任意の知的財産権訴訟において、提訴の公告及び聴聞結果、動議及び裁決及び訴訟における他の臨時手続がある可能性がある。もし証券アナリストや投資家がこれらの声明が否定的だと思うなら、私たちの既存製品、計画、または知的財産権の知覚価値は低下する可能性がある。したがって、私たちの普通株の市場価格は下がるかもしれない。このような声明はまた私たちの名声や私たちの未来の製品の市場を損なう可能性があり、これは私たちの業務に実質的な悪影響を及ぼすかもしれない。
私たちの特許保護の獲得と維持は、法規および政府特許機関によって提出された様々な手続き、文書、費用支払い、および他の要求に依存しており、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
定期維持費、継続費、年会費、ならびに特許および/または出願に関連する様々な他の政府費用は、我々の特許および/または出願の有効期間内の異なる時間点で米国特許商標局および各外国特許庁に支払われる。私たちはシステムが私たちにこれらの費用を支払うことを注意して、私たちは私たちの外部特許年金サービスが満期になった時にこれらの費用を支払うことに依存します。また、米国特許商標局および各外国特許庁は、特許出願過程において、いくつかのプログラム、文書、費用支払い、および他の同様の条項を遵守することを要求する。私たちは名声の良い法律事務所や他の専門家を招いて私たちの遵守を助け、多くの場合、不注意は滞納金を支払うことによって、あるいは特定の管轄区域に適用される規則に従って他の方法で是正することができる。しかしながら、場合によっては、規定を遵守しないことは、特許または特許出願の放棄または失効をもたらす可能性があり、それにより、関連する管轄区域の特許権の一部または全部が失われる可能性がある。このような事件が発生すれば、私たちの業務に実質的な悪影響を及ぼす可能性がある。
知的財産権は必ずしも私たちの競争優位に対するすべての潜在的な脅威を解決できるとは限らない。
私たちの知的財産権が提供する未来の保護の程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できないか、あるいは競争優位性を維持することができるかもしれない。例えば:
83
このような事件が発生した場合、私たちの業務、運営結果、そして将来性を深刻に損なう可能性がある。
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの商業と競争の地位は損なわれるだろう。
さらに、私たちは、非特許技術的ノウハウ、技術、および他の独自の情報を含む、私たちのビジネス秘密を保護することによって、私たちの競争地位を維持します。第三者との秘密協定の締結、従業員、コンサルタント、コンサルタントとの秘密情報および発明協定の締結を含む、当社の商業秘密および非特許ノウハウを保護するための措置を講じています。私たちがこのような努力をしたにもかかわらず、私たちはこのようなすべての合意が正式に実行されていることを保証することはできず、どちらも合意に違反し、私たちのビジネス秘密を含む私たちの固有の情報を開示する可能性があり、私たちはこのような違反について十分な救済措置を得ることができないかもしれない。強制執行側が商業秘密を不正に開示したり流用したりする主張は困難であり、高価で時間がかかり、結果は予測できない。しかも、米国国内外のいくつかの裁判所は商業秘密をあまり望んでいないか、または保護したくない。さらに、第三者は、この情報を取得する可能性があり、またはこの情報または同様の情報を独立して取得する可能性があり、私たちは、彼らが技術または情報を使用して私たちと競合することを阻止する権利がない。私たちは、私たちの現在および未来のビジネスパートナー、協力者、請負業者、および商業秘密が盗まれるリスクの高い他の国に位置する他の他の人と、個人または外国の行為者を介した直接侵入、および国家行為者に関連しているか、または国家行為者によって支配されている人を含む商業秘密を含む私たちの固有の情報を共有する必要があるかもしれない。もしこのような事件が発生した場合、あるいは私たちが私たちのビジネス秘密の保護を失った場合、これらの情報の価値は大幅に低下し、私たちの競争的地位は損なわれるかもしれない。もし私たちが特許発表前に特許保護を申請しない場合、または私たちの独自技術および他の機密情報を他の方法で秘密にすることができない場合、私たちが特許保護または私たちの商業秘密情報を保護する能力が脅かされる可能性がある。
私たちの許可者は彼らが私たちと締結した知的財産権許可協定の条項と条件に違反する可能性があり、私たちは彼らがこれらの協定を遵守することを強制することに成功できないかもしれない。
私たちは、プリペイド、マイルストーン、印税、および他の考慮事項と交換するために、特定の候補製品の特許、ノウハウ、およびノウハウの権利を第三者に付与します。私たちの被許可者は、彼らが製品の権利を持っている国で許可製品の承認を得ることができないかもしれません。許可製品が承認されれば、彼らはその許可製品を商業化するために努力しないかもしれません。この場合、私たちは印税や印税を受けないだろう。私たちの許可者は、許可製品がその許可地域内に安全問題が発生する可能性があり、許可地域以外の国/地域で許可製品にマイナスの影響を与える可能性があり、名声被害、許可製品の名声の損傷、または私たちの名前を損なうか、許可地域以外の当局による直接規制行動に影響を与える可能性がある。我々の許可者は、安全、患者およびデータプライバシー、反独占、賄賂および腐敗に関連する法律および法規を含む許可地域のいくつかの法律および法規に違反する可能性があり、それによって巨額の罰金、刑事調査および責任を招き、または規制当局が許可製品を市場から除去する可能性がある。もしこのような事件のいずれかが発生したら、私たちは私たちの譲受人または許可者から私たちの予想された財政的代価を受け取ることができず、私たちは彼らのいかなる違反や私たちのことに指名されて関与するかもしれない
84
譲受人または許可された人は私たちの関連損害と責任を賠償してはいけません。もし私たちの譲受人やライセンシーが合意に違反した場合、私たちは法廷または合意された論争解決メカニズムを通じてそのような合意の条項と条件を成功的に実行することができないかもしれません。たとえ私たちがこのような紛争に勝っても、救済措置は私たちの損失を補償するのに十分ではないかもしれません。
未来の任意の候補製品の特許保護と特許起訴は第三者にかかっているかもしれない。
私たちは第三者に依存して特許出願を提出して起訴し、いくつかの現在および未来の許可協定に従って特許または他の方法で許可された知的財産権を保護するかもしれない。このような配置の下で、私たちは、いくつかの特許特許または特許出願および他の知的財産権のこれらの活動に対して主要な制御権を持たないかもしれない。私たちは、第三者のこのような活動が適用された法律および法規に準拠しているか、または効果的かつ強制的に実行可能な特許または他の知的財産権をもたらすだろうと判断できない。さらに、現在および将来の許可者は、いかなる特許権の起訴、保守、または実行においても、私たちと完全に協力または同意しない可能性があり、このような特許権を損なう可能性がある。私たちは将来ライセンス契約を締結するかもしれません。ライセンス者は、私たちのライセンス特許の実施を制御したり、これらの特許が無効であると主張する任意のクレームを抗弁する権利があるかもしれません。私たちがこのような強制執行や抗弁を許可されていても、私たちはライセンシーの協力が必要になります。私たちは、私たちの許可者が十分な資源を割り当てているかどうか、またはそのような特許の実行またはこのような主張の弁護を優先して、許可特許における私たちの利益を保護するかどうかを決定することはできません。たとえ私たちがこれらの法的行動の一方でなくても、不利な結果は私たちの業務を運営するために必要かもしれない知的財産権を許可し続けることを阻止するかもしれないので、私たちの業務を損なう可能性がある。もし私たちの任意のライセンシーまたは任意の未来のライセンシーまたは現在または未来の協力者が任意の未来の候補製品をカバーする特許保護を適切に起訴し、維持することができない場合、私たちがこれらの候補製品を開発および商業化する能力は悪影響を受ける可能性があり、私たちは競争相手が競合製品を製造、使用、販売することを阻止できないかもしれない。
さらに、私たちが特許出願の起訴または第三者から取得または許可された特許の実行を制御する権利があっても、私たちの前任者は、私たちがこのような活動を制御する前の人およびその弁護士の行動または非作為の悪影響または損害を受ける可能性がある。第三者は、非商業的学術および研究用途に基礎技術を使用する権利、その技術に関連する研究の一般的な科学的発見を発表する権利、および技術に関連する情報を従来の科学および学術的に開示する権利を含む、彼らに許可された技術のいくつかの権利を保持することができる。私たちの先輩を監視することは難しいかもしれませんが、彼らの技術の使用をこれらの用途に制限しているかどうか、悪用すると、許可技術の権利を強制的に執行するために多くの費用が発生する可能性があります。
取得または許可された技術を利用する能力が制限されている場合、または重要な許可内技術の権利を失った場合、私たちは私たちの製品を開発、許可、マーケティング、販売することに成功できないかもしれません。これは新製品の発売を阻止または延期する可能性があります。私たちの業務戦略はライセンスと買収の技術を商業製品に開発することに成功しています。したがって、これらの技術を利用する能力に対するいかなる制限も、私たちの候補製品を開発、許可、またはマーケティング、販売する能力を損なう可能性があります。
私たちは買収と許可内で任意の未来の候補製品の必要な権利を獲得または維持することに成功できないかもしれない。
私たちの開発計画は将来的に他の第三者が持っている独占権を使用する必要があるかもしれないので、私たちの業務の成長は、これらの第三者の独占権を取得、許可、または使用する能力にある程度依存するかもしれません。私たちは、将来の任意の候補製品と考えられる任意の成分、使用方法、プロセス、または他の第三者知的財産権を第三者から得ることができないかもしれません。第三者知的財産権の許可·買収は競争分野であり、一部のより成熟した企業も魅力的と考えられる第三者知的財産権許可または買収戦略を求めている。これらの老舗会社は私たちより競争優位を持っているかもしれません。それらの規模、現金資源及びより強い臨床開発と商業化能力のためです。しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。私たちはまた私たちの投資が適切な見返りを得るための条項の許可や第三者知的財産権を得ることができないかもしれない。私たちが必要な第三者知的財産権を成功させたり、既存の知的財産権を維持したりすることができなければ、私たちはこのプロジェクトの発展を放棄しなければならないかもしれません。私たちの業務や財務状況は影響を受ける可能性があります。
85
私たちの普通株式と引受権証に関連するリスク
私たちの普通株と株式承認証の市場価格はいつもそうで、高度に変動し続ける可能性があります。あなたは投資の一部または全部を損失する可能性があります。
私たちの普通株式と株式認定証の市場価格はすでに高度に変動し続ける可能性があり、以下の要素を含む様々な要素の広範な変動を受ける可能性がある
また、株式市場は極端な価格と出来高変動を経験し、多くの会社の株式証券の市場価格に影響を与え続けている。これらの変動は往々にしてこれらの会社の経営業績に関係なく、または比例しない。広範な市場と業界要素、及び一般的な経済、政治、監督管理と市場条件は、私たちの実際の経営業績にかかわらず、私たちの普通株と株式承認証の市場価格にマイナスの影響を与える可能性がある。
私たちの株価の変動は私たちを証券集団訴訟に直面させるかもしれない。
従来、証券集団訴訟は、ある会社の証券市場価格が下落した後に提起されることが多かった。このリスクは,バイオテクノロジーやバイオ製薬会社が近年大幅な株価変動を経験しているため,特に我々と関連している。私たちがこのような訴訟に直面すれば、巨額のコストを招き、経営陣の注意と資源を移転させる可能性があり、これは私たちの業務を損なう可能性があります。
86
もし私たちがナスダック資本市場の持続的な上場要求を満たすことができなければ、私たちの普通株と権利証は取られるかもしれない。
もし私たちがナスダック資本市場の持続的な上場要求、例えば最低終値、株主権益或いは一括所有者要求或いは会社管理要求を満たすことができなければ、ナスダックは措置を取って、私たちの普通株式及び/又は株式承認証を退市するかもしれない。
私たちは現在ナスダック資本市場の持続的な上場要求に符合しているにもかかわらず、私たちは過去にナスダックから通知を受けて、私たちはいくつかの上場要求を遵守していないと言って、私たちは未来にこのような要求を守り続けることができることを保証することができません。私たちの普通株がナスダック資本市場から撤退し、他の市場や取引所でオファーまたは上場する資格がない場合、私たちの普通株は場外取引市場または未上場証券のために設立された電子掲示板(例えば、粉票または場外取引掲示板)でしか取引できません。この場合、私たちの普通株を売却したり、正確な普通株のオファーを得ることがより困難になる可能性があり、証券アナリストやニュースメディアによる私たちの報道も減少する可能性があり、これは私たちの普通株価格をさらに下落させる可能性がある。しかも、私たちが大型取引所に上場しなければ、私たちは追加的な資本を調達することが難しいかもしれない。
証券や業界アナリストが私たちに関する研究や報告を発表しない場合、あるいは否定的な報告を発表しなければ、私たちの株価や取引量は低下する可能性がある。
私たちの普通株式と権利証の取引市場は、証券や業界アナリストが発表した私たちに関する研究と報告にある程度依存している。私たちはこのアナリストたちに何の統制権も持っていない。もし私たちの財務業績がアナリストの期待に達していない場合、あるいは私たちのアナリストを追跡して1つ以上のアナリストが私たちの普通株式や権利証の格付けを引き下げたり、彼らの観点を変えたりすれば、私たちの普通株式と権利証の取引価格は低下する可能性がある。もしこれらのアナリストのうちの1人以上が私たちの報告書を停止したり、私たちに関する報告書を定期的に発表できなかった場合、それは金融市場で可視性を失う可能性があり、これは私たちの普通株式と引受権証の取引価格や取引量を低下させる可能性がある。
私たちは予測可能な未来に現金配当金を支払わないと予想されるので、資本付加価値(あれば)があなたの唯一の収益源になるだろう。
私たちは現在、事業の発展、運営、拡張のために未来の収益を保留し、予測可能な未来に現金配当金を発表または支払うことはないと予想している。したがって、予測可能な未来に、私たちの普通株の資本増加は、あなたがこのような株に投資する唯一の収益源になるだろう。
わが社の登録証明書やデラウェア州法律の条項は、株主が有利と思う買収を阻止し、経営陣の強固化を招く可能性がある。
当社の登録証明書や定款に含まれる条項は、私たちの株式の価値を大幅に低下させ、潜在的な買収対象にしたり、私たちの取締役会の同意を得ていない統制権の変更や経営陣の変更を遅延または阻止したりする可能性があります。私たちの憲章の文書には以下の内容が含まれている
87
私たちはまた“デラウェア州会社法”第203条に記載されている反買収条項を守らなければならない。第二百三十条によれば、一般的に、会社は、その株式の15%以上を保有する株主と業務合併を行ってはならない。当該株式を保有する株主が当該株式を三年間保有している場合を除き、又はその他の例外を除いて、取締役会は、当該取引を承認した。
我々の会社登録証明書は、デラウェア州衡平裁判所は、私たちと私たちの株主との基本的なすべての紛争の独占フォーラムとなり、連邦地域裁判所は、証券法に基づいて提出された訴訟原因の任意のクレームを解決する独占フォーラムであり、これは、私たちの株主が有利な司法フォーラムを獲得することを制限し、私たちまたは私たちの役員、上級管理者または従業員または引受業者との紛争、またはそのようなクレームを引き起こす任意の要約を処理する可能性がある。
私たちの会社登録証明書は、デラウェア州衡平裁判所は、私たちを代表して提起された任意の派生訴訟または訴訟、受託責任違反を主張する任意の訴訟、デラウェア州会社法、当社の会社登録証明書または私たちの定款に基づいて私たちにクレームを提起する任意の訴訟、または私たちの内部事務原則によって管轄されているクレームを提起する任意の訴訟の独占法廷であることが規定されています。さらに、私たちの会社登録証明書は、私たちが書面で代替裁判所を選択することに同意しない限り、アメリカ連邦地域裁判所は、その訴状に指名された任意の被告に対して主張されたすべての訴因を含む、証券法に基づいて提起された任意の訴因を解決するための独占的なフォーラムとなることを規定している。上記の規定にもかかわらず、本裁判所選択条項は、“取引法”に規定されているいかなる責任又は義務を強制的に執行するための訴訟にも適用されず、米国連邦地域裁判所が排他的管轄権を有する他のいかなるクレームにも適用されない。疑問を生じないために、本条文は、吾等、吾等の上級職員及び取締役、任意の株式引受業者、及び任意の他の専門実体(その専門が当該者又は実体になされた声明を許可し、目論見の基礎となる文書の任意の部分を準備又は証明した)に恩恵を受け、本条文を強制的に執行することができるようにすることを目的としている。これらの裁判所条項の選択は、私たちまたは私たちの役員、役員、または他の従業員との紛争に有利であると考えられるクレームを司法裁判所で株主が提出する能力を制限する可能性があり、これは、私たちおよび私たちの役員、役員、および他の従業員に対するこのような訴訟を阻止し、投資家のクレームのコストを増加させる可能性がある。しかし、この条項に同意することで、株主は連邦証券法とその下の規則と条例の遵守を放棄したとみなされないだろう。また,他社の会社登録証明書の中で類似した場所条項を選択する実行可能性が法的手続きで問われており,裁判所はこれらのタイプの条項が適用されないか実行不可能であると考える可能性がある.当社の登録証明書の中から選択された裁判所条項が訴訟で適用されないか、実行できないことが裁判所によって発見された場合、他の管轄区域でこのような訴訟の解決に関連する追加費用が発生する可能性があり、これは私たちの業務や財務状況に悪影響を及ぼす可能性があります。
88
私たちは新興成長型会社と小さな報告会社であり、新興成長型会社やより小さい報告会社に適した報告要求を下げることが投資家に対する私たちの株の魅力を低下させるかどうかを決定することはできません。
私たちはJumpStart Our Business Startups Act(“JOBS法案”)で定義されているように、新興の成長型会社ですこれは…。そして、(2)前3年間に10億ドルを超える転換不能債券を発行した日。
また、雇用法案によれば、新興成長型企業は、これらの基準が民間企業に適用されるまで、新たなまたは改正された会計基準の採用を延期することができる。私たちは、この延長された過渡期間を利用して新しい会計基準または改正会計基準を遵守することを選択したので、他の非新興成長型企業の公衆会社のように同じ新しい会計基準または改正会計基準を遵守することはない。
取引法の定義によると、私たちも規模の小さい報告会社です。新興成長型企業の資格に適合しなくなった後も、規模の小さい報告会社になる資格があり、404条に準拠した監査人認証要件の免除、本年度報告書と定期報告書および委託書における役員報酬に関する開示義務の削減など、多くの同じ開示要件を利用することができるようになる。私たちの非関連会社が保有する投票権と無投票権普通株が第2四半期の最終営業日に2.5億ドルを下回るか、または最近終了した会計年度の年収が1.00億ドル未満であれば、非関連側が保有する投票権と無投票権普通株が第2四半期の最終営業日に7.00億ドルを下回る限り、これらを利用して大規模に開示することができる。
私たちは投資家がこれらの免除に依存する可能性があるため、その普通株の吸引力が低下していることを発見する可能性があるかどうかを予測できない。したがって、一部の投資家が私たちの普通株の吸引力が低下していることを発見すれば、普通株の取引市場はそれほど活発にならない可能性があり、私たちの市場価格はもっと変動する可能性がある。
もしあなたが“現金なし”で公共株式証明書を行使すれば、このような行使から得られる普通株は、あなたがこの株式承認証を現金と交換した時に得られる普通株よりも少ないだろう.
場合によっては、現金なしで公共株式承認証を行使することを要求または許可することができる。われわれの普通株が国家証券取引所に上場されていない引受権証をいつでも行使する場合は、証券法第18(B)(1)条の“担保証券”の定義に適合するように、証券法第3(A)(9)条の規定により、その株式証を行使する公共株式証保有者にキャッシュレスに基づいてそうすることを要求することができ、もしこのように選択すれば、有効な登録声明の提出や維持を要求されないであろう。免除されない場合には、適用される青空法律に基づいて株式の登録や資格認証を行うために最善を尽くします。第三に、私たちが公衆に引受権証の償還を要求すれば、私たちの経営陣は、引受証を行使したいすべての所有者に無現金でそうすることを要求する権利があるだろう。無現金に基づいて行使する場合、保有者が株式承認証の行権価格を支払う方法は、この数量の普通株式承認株式証を提出することであり、この数は株式承認証関連普通株式数に承認株式証の行権価格と“公平市価”(定義は次の文を参照)の間の差額に(Y)公平市価を乗じて得られた商数に等しい。“公平市価”とは,持分証代理人が行使通知または償還通知を受けて承認持分証所持者に送付される日前の第3取引日までの10取引日内の普通株の平均最終販売価格を意味する。したがって、このような株式から得られる普通株式は、株式承認証を行使して現金と交換するよりも少なくなる。
89
当時少なくとも65%の未発行の公共株式証所有者の承認を得た後、私たちは所有者に不利な方法で公開株式証の条項を修正する可能性がある。
私どもの引受権証は株式承認証代理である大陸株式譲渡信託会社と私どもとの間の権証合意に基づいて登録形式で発行されています。株式承認協定は、株式証を承認する条項は、いかなる所有者の同意なしに改訂して、いかなる曖昧なところを是正し、あるいはいかなる欠陥のある条文を訂正することができるが、当時少なくとも65%が発行されていない公共株式証所有者の承認を得なければならず、公共株式証登録所有者の利益に不利な影響を与える任意の変更を行うことができる。したがって、当時少なくとも65%が発行されていなかった公共株式証を持っていた所有者が修正に同意した場合、私たちは所有者に不利な方法で公共株式証の条項を修正することができる。我々は、当時少なくとも65%の発行済み公共株式証明書の同意を得た場合、公開株式証の条項を改訂する能力は無限であるが、このような改正例には、株式承認証の行使価格の向上、株式承認証を現金または株に変換すること、行使期間を短縮すること、または引受証を行使する際に購入可能な普通株数を減少させることが含まれる可能性がある。
私たちはあなたに不利な時間にあなたの未満期の公共株式証明書を償還して、公共株式証明書を一文の価値もなくするかもしれません。
私たちは、発行された公共株式証明書が行使可能になった後および満期前のいつでも、普通株式の最終報告販売価格(私たちの普通株が任意の特定の取引日に取引されていない場合、私たちの普通株式の終値)が、適切な償還通知が発行される日前の第3の取引日までの30取引日以内の20取引日以内に、1株当たり450.00ドル(株式分割、株式配当、再編などの調整後)に等しいか、またはそれを超えることを前提とする能力がある。私たちが償還通知を出した日以降、私たちが公共株式証明書を償還するまでの期間内に、私たちは証券法に基づいて、公共株式証明書を行使する際に発行可能な普通株式と、それに関連する最新の株式募集説明書を含む有効な登録声明を持っています。株式証を公開して私たちが償還することができれば、私たちはすべての適用された州証券法に基づいて対象証券を登録したり、売却資格に適合させることができなくても、償還権を行使することができます。発行された公共株式証明書を償還することは、(I)公共株式証を行使し、あなたに不利になる可能性がある時間にそのために行使価格を支払うことを強制する可能性があります。(Ii)公共株式証を保有したい場合は、その時の市場価格で公共株式証明書を売却します。または(Iii)名義償還価格を受けて、公共株式証の償還を要求した場合、名義償還価格はあなたの公共株式証の時価を大きく下回る可能性があります。
一般リスク因子
上場企業として、私たちの運営コストは高く、私たちの経営陣は新しいコンプライアンスを実施するために多くの時間を投入する必要があります。
上場企業として、私たちは多くの法律、会計、その他の費用を負担していますが、これは私たちが民間会社として起きていないことです。私たちは、私たちが米国証券取引委員会に私たちの業務と財務状況に関する年間、四半期、現在の報告書を提出しなければならないことを含む取引法の報告要件を遵守しなければならない。また、サバンズ-オクスリ法案と、米国証券取引委員会とナスダックが後にサバンズ-オクスリ法案条項を実施するために採択された規則は、有効な情報開示と財務制御の確立と維持を要求し、コーポレートガバナンスのやり方を変更することを含む上場企業に重大な要求を提出した。また、2010年のドッド·フランクウォール街改革と消費者保護法によると、米国証券取引委員会は、これらの分野で追加の規則や規定を採択し、例えば、私たちがもはや新興成長型企業ではない場合には、私たちの強制的な“報酬発言権”投票要求に適用される。株主急進主義、現在の政治環境、および現在の高度な政府介入と規制改革は、大量の新しい法規と開示義務を招く可能性があり、これは追加のコンプライアンスコストを招き、現在予見できない方法で業務を運営する方法に影響を与える可能性がある。
上場企業に適用される規則や法規は、私たちの法律と財務コンプライアンスコストを大幅に増加させ、いくつかの活動をより時間的で高価にすることを予想しています。これらの要求が私たちの経営陣と従業員の注意を他の業務から移すと、それらは私たちの業務、財務状況、運営結果に実質的な悪影響を及ぼす可能性がある。増加したコストは私たちの純額を減少させます
90
これは私たちの収入を増加させ、あるいは私たちの純損失を増加させ、他の業務分野のコストを下げたり、製品やサービスの価格を向上させることを要求するかもしれません。例えば、これらの規則や条例は、取締役や上級管理者責任保険をより難しく、より高価にすることが予想され、同じまたは同様の保険範囲を維持するために多くのコストを発生させることが要求される可能性がある。私たちは、これらの要求に応答して生成される可能性のある追加コストの金額や時間を予測または推定することができない。これらの要求の影響は、私たちの取締役会、私たちの取締役会、または役員に参加することをより難しくし、合格した人を引き付け、維持することを難しくするかもしれない。
もし私たちが財務報告に対して適切かつ効果的な内部統制を維持できなければ、私たちが正確かつタイムリーな財務報告を作成する能力が損なわれる可能性があり、投資家は私たちの財務報告に自信を失う可能性があり、私たちの普通株の取引価格は低下する可能性がある。
サバンズ·オキシリー法404条によると、私たちの経営陣は、財務報告書の内部統制に対する私たちの有効性を報告しなければならない。“新興成長型企業”の地位を失い、申告加速のハードルに達した場合、独立公認会計士事務所は、財務報告に対する内部統制の有効性を証明することを求めることになる。私たちの経営陣が財務報告の内部統制を評価するために達成しなければならない基準のルールは複雑で、大量の文書、テスト、可能な救済措置が必要です。取引法における報告会社としての要求に適合するためには、私たちの情報技術システムをアップグレードする必要があるかもしれません。追加の財務および管理制御、報告システム、およびプログラムを実施し、追加の会計および財務者を招聘する必要があります。もし私たちまたは私たちの監査人が財務報告の内部統制に有効であると判断できなければ、投資家は私たちの財務報告に自信を失う可能性があり、私たちの普通株の取引価格は低下する可能性がある。
私たちはアメリカとある外国の輸出入規制、制裁、禁輸、反腐敗法律、反マネーロンダリング法律法規の制約を受けている。私たちは違反によって刑事責任と他の深刻な結果に直面するかもしれないし、これは私たちの業務を損なうかもしれない。
我々は、米国の輸出管理条例、米国税関条例、米国財務省外国資産規制事務所が管理する様々な経済·貿易制裁条例、改正された米国の1977年の“海外腐敗防止法”、米国“米国法典”第18編201節に掲載された米国国内賄賂法規、米国旅行法、アメリカ愛国者法案と他の州と国の反賄賂と反マネーロンダリング法は私たちが活動している国にあります。腐敗防止法は、会社およびその従業員、代理人、CRO、請負業者および他の協力者およびパートナーが直接または間接的に許可、承諾、提供、提供、誘致、または公共または民間部門の受取人の不当な支払い、または任意の他の価値のあるものを禁止することを広く解釈されている。私たちは第三者を招いてアメリカ以外で臨床試験を行い、商業化段階に入った後に私たちの製品を海外に販売し、および/または必要な許可、許可証、特許登録、その他の規制承認を得ることができる。私たちは政府機関や政府付属病院、大学、その他の組織の役人や従業員と直接または間接的な相互作用を持っている。私たちが私たちの従業員、代理、CRO、請負業者、および他の協力者およびパートナーの腐敗や他の不法活動を明確に許可したり、実際に理解していなくても、私たちは責任を追及される可能性がある。私たちはまた、米国の他の輸出規制に関する法律と法規、特定の国と個人に対する経済制裁と禁輸を受けている。
上記の法律および条例に違反するいかなる行為も、重大な民事と刑事罰金と処罰、監禁、輸出入特権の喪失、資格取り消し、税額の再評価、契約違反および詐欺訴訟、名誉損害、およびその他の結果を招く可能性がある。
また、米国の輸出規制法と経済制裁は、米国の制裁対象国、政府、個人に特定の製品やサービスを提供することを禁止している。他の国の軍事衝突により、米国がすでにまたは実施可能な制裁は、このような制裁がカバーされる地域内で将来の臨床試験地点で活動し続ける能力に影響する可能性がある。もし私たちが輸出入条例とこのような経済制裁を守らなければ、罰金および/または特定の輸出特権の剥奪を含む罰を受けるかもしれない。このような輸出入規制と経済制裁はまた私たちのサプライチェーンに悪影響を及ぼすかもしれない。
91
私たちおよび私たちの任意の第三者製造業者またはサプライヤーは、強力な化学剤および危険材料を使用する可能性があり、これらの材料の不適切な処理、貯蔵または処置に関連するいかなるクレームも、非常に時間的または費用がかかる可能性がある。
私たちと私たちの任意の第三者製造業者またはサプライヤーと、現在または潜在的な将来のパートナーは、生物学的材料、強力な化学剤を使用し、化学品、生物学的製剤および化合物を含む危険な材料を使用する可能性があり、これらは、人間の健康および環境安全に危険をもたらす可能性がある。私たちの業務と私たちの第三者製造業者とサプライヤーの業務も危険な廃棄物製品を発生させます。連邦、州、地方の法律法規はこれらの材料と廃棄物の使用、発生、製造、貯蔵、処理、処理を管理する。適用される環境法律や法規を遵守することは費用がかかる可能性があり、現在または未来の環境法律と法規は私たちの製品開発努力を損なう可能性がある。しかも、私たちはこのような材料や廃棄物が意外なダメージや汚染をもたらす危険を除去することができない。私たちは特定の生物或いは危険廃棄物保険を受けていません。私たちの財産、意外と一般責任保険は生物或いは危険廃棄物の暴露或いは汚染による損害と罰金は明確に含まれていません。汚染や傷害が発生した場合、損害賠償責任を請求されたり、私たちの資源を超えた罰金が科されたりする可能性があり、私たちの臨床試験や監督管理の承認は一時停止される可能性があります。私たちは従業員のために危険材料や他の労災による傷害によって招く可能性のあるコストと支出に労働者補償保険を維持しているにもかかわらず、この保険は潜在的な責任に十分な保険を提供できないかもしれない。私たちは私たちが生物、危険、あるいは放射性物質を貯蔵したり処分したりすることによって、私たちに提起された有毒侵害に対する保険を維持することはできません。
また、現在または将来の環境、健康、安全法律および法規を遵守するためには、時間の経過とともにより厳しくなることが多い巨額のコストが生じる可能性があります。このような現行または未来の法律法規は私たちの研究、開発、または生産努力を損なうかもしれない。これらの法律および法規を遵守しないことは、巨額の罰金、処罰、または他の制裁または責任をもたらす可能性もあり、これは、私たちの業務、財務状況、運営結果、および将来性に重大な悪影響を及ぼす可能性がある。
業務中断は私たちの将来の収入と財務状況を深刻に損害し、私たちのコストと支出を増加させるかもしれない。
私たちの業務は地震、電力不足、電気通信故障、水不足、洪水、ハリケーン、台風、火災、極端な天気条件、戦争、テロ、医療流行病、その他の自然または人為的災害または業務中断の影響を受ける可能性があり、私たちは主に自己保険です。私たちは私たちの候補製品を生産するために第三者メーカーに依存している。これらのサプライヤーの運営が人為的または自然災害または他の業務中断の影響を受ける場合、候補製品の臨床供給を得る能力が妨害される可能性がある。また、当社の本社はカリフォルニア州ソラナビーチにあり、主な地震断層と火域に近く、主要地震断層と火域の近くに位置し、ある地理的地域で固められたことが私たちに与える最終的な影響は不明です。このような業務中断の発生は、私たちの運営と財務状況を深刻に損害し、私たちのコストと支出を増加させる可能性がある。
不安定な市場や経済状況は、私たちの業務、財務状況、株価に深刻な悪影響を及ぼす可能性がある。
世界の信用と金融市場は時々、流動性と信用供給の深刻な減少、金利とインフレ率の上昇、消費者自信の低下、経済成長の低下、失業率の上昇、経済安定の不確定を含む極端な変動と破壊を経験する。将来の信用と金融市場の悪化と経済状況に対する自信が起こらない保証はない。私たちの全体的な業務戦略は、このような経済低迷、不安定なビジネス環境、または持続不可能で不安定な市場状況のいずれかの悪影響を受ける可能性がある。金融市場と世界経済はまた、ロシアとウクライナの間で持続的な衝突、テロ、または他の地政学的事件を含む軍事衝突の現在または予想される悪影響を受ける可能性がある。米国や他の国がウクライナ紛争を含むこのような紛争に対応するために実施している制裁は、金融市場や世界経済に悪影響を及ぼす可能性もあり、影響を受けた国や他の国のいかなる経済対策も市場や経済の不安定を悪化させる可能性がある。現在の株式と信用市場が悪化すれば、任意の必要な債務や株式融資をより困難にし、コストがより高く、希釈程度をより高くする可能性がある。有利な条件で必要な融資を速やかに得ることができなければ、
92
これは私たちの成長戦略、財務業績、株価に重大な悪影響を及ぼす可能性があり、臨床開発計画の延期または放棄を要求する可能性がある。さらに、我々の現在の1つまたは複数のサービスプロバイダ、製造業者、および他のパートナーは、経済低迷の中で生き残ることができない可能性があり、これは、私たちが時間通りに、予算通りに運営目標を達成する能力に直接影響する可能性がある。
アメリカ税法の変化は私たちの財務状況、経営業績、キャッシュフローに重大な悪影響を及ぼす可能性があります。
新しい収入、販売、使用、または他の税金法律、法規、規則、法規または条例は、いつでも公布されるか、または私たちに不利な解釈、変更、修正、または適用される可能性があり、これらはいずれも、私たちの業務運営および財務業績に悪影響を及ぼす可能性があります。特に、米国政府は、企業所得税税率の引き上げ、特定のタイプの収入に対して最低税率または付加税を徴収することを含む商業実体の税収を重大に改革する可能性がある。このような変化が公布されたり施行されたりする可能性はまだ不明だ。私たちは現在このような変化が起こるかどうか予測できない。もしこのような変化が公布されたり施行されたりすれば、私たちは現在私たちの業務に最終的な影響を予測できない。私たちは投資家に税法のいかなる変化と私たちの普通株に投資する潜在的な税金結果について彼らの法律と税務顧問と協議することを促す。
情報技術EM 1 B.未解決の従業員のコメント。
ない。
プロジェクト1 C。ネットワークセキュリティです。
ネットワークセキュリティリスク管理と戦略
我々は,我々のキーシステムと情報の機密性,完全性,可用性を保護するためのネットワークセキュリティリスク管理計画を策定し実施した.私たちのサイバーセキュリティリスク管理計画にはサイバーセキュリティ事件対応計画が含まれている。
私たちは国際標準化機関27002標準に基づいて私たちの計画を設計して評価します。これは、私たちが任意の特定の技術基準、規範、または要求を満たすことを意味するわけではありませんが、私たちは、私たちの業務に関連するネットワークセキュリティリスクを識別、評価、管理するのを助けるために、国際標準化組織27002をガイドラインとして使用しています。
我々は外部資源を利用して,我々の既存のネットワークセキュリティ実践と組織リスク評価システムに貢献し,独立した評価を提供する.第三者が会社の材料情報を処理、所有、処理、記憶する際には、第三者サービスプロバイダのリスクを識別、評価、管理するための既定のプロセスを使用する。
我々のネットワークセキュリティリスク管理計画は、(I)我々のキーシステム、情報、製品、サービス、およびより広範な企業情報技術環境における重大なネットワークセキュリティリスクを識別することを支援するための政策と、(Ii)外部サービスプロバイダを使用して、セキュリティ制御を管理、評価、テスト、および他の方法で支援するための様々な態様と、(Iii)ネットワークセキュリティイベントに対応するプログラムを含むネットワークセキュリティイベント応答計画とを含む。
私たちは、私たちの運営、業務戦略、運営結果、または財務状況を含む、既知のネットワークセキュリティ脅威(これまでの任意のネットワークセキュリティイベントを含む)から、私たちに重大な影響を与えるか、または合理的に私たちに重大な影響を与える可能性があるリスクを決定していません。しかし、私たちのネットワークセキュリティ予防と緩和努力が常に成功することは保証されず、サイバーセキュリティ脅威は将来、私たちの業務、運営、または財務状況に実質的な悪影響を及ぼす可能性がある。参照してください“リスク要因-当社の業務運営や業界に関連するリスク-当社の業務および運営は、情報技術システムの故障、ネットワーク攻撃、またはネットワークセキュリティ欠陥の場合に影響を受ける可能性があります。“本テーブルの10-Kの”リスク要因“というタイトルの下で。
サイバーセキュリティ·ガバナンス
当社取締役会は、ネットワークセキュリティリスクをそのリスク監督機能の一部とし、監査委員会(“委員会”)にネットワークセキュリティや他の情報技術リスクの監督を依頼している。同委員会は、経営陣と外部情報技術サービスプロバイダを監督し、我々のネットワークセキュリティリスク管理計画を実施している。
93
委員会は私たちのサイバーセキュリティリスクに関する管理職の最新の状況を定期的に受け取っている。また,管理層は,必要に応じて任意の重大なネットワークセキュリティイベントおよび任意の影響の小さいイベントの最新状況を委員会に通報する.
委員会はまた私たちのネットワークリスク管理計画に関する経営陣のブリーフィングを聞いた。委員会メンバーは、上場企業に影響を与えるテーマに関する取締役会の継続教育の一部であるネットワークセキュリティテーマに関する我々の財務部門の紹介を受け、首席財務官や外部専門家を含む。
私たちの管理チームは、私たちの最高財務官と、私たちの外部情報技術サービスプロバイダを含めて、私たちのネットワークセキュリティ脅威からの重大なリスクの評価と管理を担当しています。我々は,我々の全体ネットワークセキュリティリスク管理計画の実施と日常機能を我々の第三者情報技術サービスプロバイダにアウトソーシングした.私たちの管理チームは私たちの内部人員と私たちが招いた外部ネットワークセキュリティ顧問を監視する。
我々の管理チームは、財務部門または外部情報技術者のプレゼンテーション、政府、公共またはプライベートソース(私たちが招聘した外部コンサルタントを含む)から得られた脅威情報および他の情報、ならびに情報技術環境に配置されたセキュリティツールによって生成された警報および報告を含む様々な手段によって、ネットワークセキュリティリスクおよびイベントを予防、発見、緩和および修復する作業を監視する。
情報技術EM 2.財産.
私たちの会社の本社はカリフォルニア州ソラーナビーチにあります。敷地は一,八百平方フィートです。2024年1月、本社の賃貸契約を3年間延長し、新たな満期日は2027年10月31日とした。私たちは私たちの既存の施設が私たちの短期的な需要を満たすのに十分だと信じている。今後必要があれば、商業上の合理的な条件に従って、適切な追加的あるいは別の用地を提供することができると信じています。
イットM 3.法的訴訟
私たちは現在どんな実質的な法的手続きの当事者でもない。しかし、私たちは時々法的手続きに巻き込まれたり、私たちの正常な業務過程でクレームを受けるかもしれない。結果にかかわらず、このような訴訟またはクレームは、弁護および和解費用、資源移転、および他の要因によって私たちに悪影響を及ぼす可能性があり、有利な結果が得られる保証はない。
情報技術EM 4.鉱場安全を開示する
適用されません。
94
第II部
情報技術EM 5.登録者普通株、関連株主事項、発行者が株式証券を購入する市場
市場情報
2021年8月25日に私たちはバッタの行買収会社と業務合併を完了して以来、私たちの普通株と権利証はすでにナスダック資本市場で公開取引され、コードはそれぞれ“EFTR”と“EFTRW”である。
普通株保有者
2024年2月29日現在、私たちの普通株は約53人の登録保有者がいます。この数字は私たちの株主記録から来ていて、私たちの普通株の利益所有者は含まれていません。彼らの株は様々な取引業者、決済機関、銀行、ブローカー、および他の受託者の名義で保有されています。
配当政策
私たちは私たちの株のどんな現金配当金も発表したり支払ったりしたことがない。私たちは将来の収益(あれば)を残して、私たちの業務運営に資金を提供し、予見可能な未来には何の現金配当金も支払わないつもりです。配当政策に関する任意の未来決定は、当社取締役会が当社の財務状況、経営業績、資本要求、業務見通し及び取締役会が関連する他の要素を考慮した後に適宜行い、任意の未来融資ツールに記載されている制限によって制限される。さらに、私たちはオックスフォード金融有限責任会社との融資と保証協定は私たちの債務を制限し、私たちが現金配当金を申告して支払う能力を制限した。
株式補償計画に基づいて発行された証券
当社の株式報酬計画に関する情報は、本年度報告第3部第12項を参照して、参照により本明細書に組み込まれています。
[パフォーマンスチャート]
適用されません。
最近の未登録証券の売却と収益の使用
ない。
発行者は株式証券を買い戻す
ない。
情報技術イーエム六です[保留されている]
95
イット経営陣の財務状況と経営成果の議論と分析。
以下の議論及び分析は、本年度報告書の他の部分に含まれる2023年12月31日現在、2022年12月31日までの監査総合財務諸表と共に読まなければならない。本討論と分析は、私たちの現在の信念、計画と期待に基づく展望的陳述を含み、リスク、不確定性と仮説に関連する。様々な要因により、“リスク要因”や本年度報告の他の部分に記載されている要因が含まれているため、我々の実際の結果は、これらの前向き陳述で予想される結果とは大きく異なる可能性がある。
概要
我々は臨床段階の生物製薬会社であり,STRIと呼ばれる新しい腫瘍学薬の開発に専念している。翻訳は細胞中のタンパク質の産生が遺伝子配列に含まれる情報によって指導される過程である。我々は独自の選択的翻訳制御技術プラットフォームを利用して、内部で一連の小分子STRI候補製品を発見した。我々の候補製品はeIF 4 F複合体及びその活性化キナーゼ、即ちマイトジェン活性化タンパク質相互作用キナーゼ(“MNK”)に対して。EIF 4 F複合体は癌中の2つの最も頻繁に突然変異するシグナル経路であるPI 3 K-AKTとRas-MEK経路の中心ノードであり、それらは集まって、SELECT mRNAから蛋白質への翻訳を活性化し、蛋白質は重要な疾病駆動過程中によく発生する張本人である。これらの標的のいずれかを抑制することは、複数の疾患キネシンの発現を同時に低下させ、それらを産生する。我々の各候補は、癌タンパク質(その異常機能が癌を引き起こす可能性のあるタンパク質)、T細胞における免疫抑制タンパク質、および腫瘍の成長、生存および免疫逃避を共同制御する複数の機能関連タンパク質ネットワークの発現を駆動する単一のタンパク質に作用することを意図している。
著者らの主要な候補製品Tomivosertibは経口MNK小分子阻害剤であり、著者らは抗PD-(L)1療法の阻害剤と併用して固形腫瘍患者の治療に応用している。2021年第2四半期に、著者らはKickStart投与量を開始し、これは著者らの2 b期無作為臨床試験であり、トミフザチ連合ペブロリズマブによるPD-L 1発現レベルが50%(PD-L 150%)以上の転移性非小細胞肺癌患者を評価した。Pembrolizumabはメルク社が所有し、マーケティングし、第一線の非小細胞肺癌と他のいくつかの適応に応用されている。2024年4月初めにKickStart試験の背線データを報告する予定である。この2 b期臨床試験から積極的な結果が得られれば,この試験後に後続の3期登録試験を行う予定である。
著者らの第二の候補製品であるゾチフェンはeIF 4 Aの阻害剤、eIF 4 F複合体の一成分であり、現在ある固形腫瘍患者の1/2期の臨床試験で評価されている。著者らはすでにこの試験の初期用量増加部分といくつかの適応の初期第2段階拡張部分を完成し、ゾタフェンとフルビストランとアベシル(“ZfA三連”)の併用によるER+乳癌患者の治療効果を評価することを含む。1/2期臨床試験で観察された良好な安全性結果とこれまでに産生された標的参加データを考慮して、著者らは1週間ごとに投与するゾタフェン投与量の増加を回復し、最初はエストロゲン受容体陽性乳癌患者のZF二重結合において、未来の臨床研究においてより高用量のゾチフェンを使用できるかどうかを確定した。その後、ZfA三胞胎で投与量の増加を開始した。2024年第1四半期、ZF二聯体の用量アップグレードが終了し、0.2 mg/kgゾタフェンQ 2 Wを二結合体のRP 2 Dとして確定した。私たちは2024年上半期に用量上昇に関するより多くのデータを報告する予定だ。
これまでにER+乳癌患者を含む5つのキューの中期データが報告されており,これらのデータはゾタフェンが全体的に耐性が良好であることを示し,大量に前処理されたER+乳癌患者の部分反応を含む活性シグナルを示している。われわれは6月の米国臨床腫瘍学会(ASCO)2023年年次総会でER+乳癌にZfA三胞胎行列を完全に組み込んだTOPLINE結果を報告し,ZfA三胞胎治療を受けた評価可能患者19名中5名(26%)に部分反応を認めた。また,初めて回復したZF二重用量漸増行列では,3名中1名(33%)に部分反応が認められた。ZfAとZFの組み合わせは一般的に耐性が良好であり、副作用の多くは1級または2級である。12月に開催された2023年サンアントニオ乳癌シンポジウム(SABCS)でZfA三胞胎行列の成熟データを報告し、中位PFSは7.4月であった。
ゾチフェンが十分な安全性と十分な活性シグナルを示し続けていれば,関連対照対照群との無作為試験の組み合わせとなる可能性があるゾチフェンの臨床開発を継続する予定である。我々は現在,2024年上半期に投与量とスケジュールを決定した後,無作為試験でZfA三連体の計画をテストしている。2023年に受信した高速車線称号を用いてFDAと発展戦略について相互作用する予定である。
96
著者らはまた1 b期の臨床試験を完成し、ゾタフェンを抗SARS-CoV-2の抗ウイルス薬として評価した。この研究は二重盲検、無作為、プラセボ対照試験であり、単用量ゾチフェンの安全性と抗ウイルス活性を評価した。この実験では,ゾタフェンは安全性と耐性が良好であることが発見され,プラセボと比較したいくつかのウイルス駆除評価で有利な傾向を示した。著者らはすでにファイザー会社と著者らの最初の段階計画eIF 4 E阻害剤について全世界の協力と許可協定を締結し、ファイザー社は現在この計画のために研究性新薬応用(IND)研究を行っている。
2012年の設立以来、著者らはほとんどの資源を投入して資金を調達し、潜在的な候補製品を確定し、私たちの知的財産権の組み合わせを構築し、臨床前研究と臨床試験を行い、第三者と私たちの候補製品と関連原材料の生産について手配を確立し、そしてこれらの業務に一般と行政支持を提供する。私たちはどんな製品も販売を許可されていないし、製品販売から何の収入も得ていない。2023年12月31日現在、転換可能優先株の売却·発行の総収益1.5億ドル、2021年8月の業務合併に関連する普通株発行の総収益6700万ドルを含む計3億258億ドルを調達し、ファイザーとの研究協力·許可協定(“ファイザー協定”)での協力収入4200万ドル、クレジット融資項目で3500万ドルを支援している。2023年5月登録直接発売(定義は後述)と2023年6月登録直接発売(定義は後述)普通株を売却して得られた毛収入は1,620万ドルであり、Cantor Fitzgerald&Co(“Cantor”)との制御持分発行販売協定(“販売契約”)に基づいて普通株を売却して得られた毛収入750万ドル、カリフォルニア大学取締役会がサンフランシスコキャンパス(“カリフォルニア大学サンフランシスコ校”)と締結した研究分賞協定による500万ドルの贈与収入に基づいて、リンカーンパーク資本基金有限責任会社(“リンカーンパーク”)との株式購入協定(“購入契約”)に基づいて普通株を売却して得られた毛収入310万ドル。2020年にファイザー協定による収入による純収入と、2021年に利益負債の推定値の変化による2021年の純収入を除いて、我々は設立以来大きな運営損失を発生している。当社の各期間の純損失は、2023年12月31日および2022年12月31日までの純損失はそれぞれ3,580万ドルおよび2,270万ドルです。2023年12月31日と2022年12月31日までの累計赤字はそれぞれ1兆794億ドルと1兆436億ドルだった。私たちのほとんどの運営損失は,我々の候補製品の研究や開発に関する費用と我々の運営に関する一般的かつ行政コストによるものである。
私たちは少なくとも今後数年以内に、私たちは巨額の費用と損失を招き続けると予想している。私たちが引き続き開発し、規制部門が承認された候補製品の承認を求め、それを商業化し、より多くの人を雇用し、私たちの知的財産権を保護し、上場企業に関連する追加コストを発生させることに伴い、私たちの費用は大幅に増加すると予想される。われわれの臨床試験と臨床前研究の時間および他の研究や開発活動への我々の支出によると,われわれの純損失は四半期と四半期と年度との間に大きく変動する可能性がある。2023年12月31日現在、私たちは1840万ドルの現金、現金等価物、短期投資を持っている。さらなる運営に資金を提供するために、私たちは追加的な資本を調達する必要があるだろう。私たちの現在の資本資源は、私たちの任意の候補製品の臨床開発を完成させるのに十分ではないか、またはFDAや同様の外国規制機関の承認を得る可能性のある候補製品を商業化する準備ができているだろう。したがって、私たちは、潜在的な追加協力、ライセンス、および他の同様の手配を含む、株式発行、債務融資、または他の資本源の組み合わせによって、私たちの現金需要を満たす予定だ。受け入れ可能な条件で、私たちはもしあれば十分な資金を得ることができないかもしれない。私たちは必要な時に資金を調達したり、このような他の手配を達成することができず、私たちの財務状況に負の影響を与え、私たちの研究開発計画や他の業務を延期、制限、減少または終了させるか、あるいは私たちが自分で開発とマーケティングをより望んでいた候補製品を開発し、マーケティングする権利を与える可能性があります。
株を逆分割する
2024年1月9日、私たちはデラウェア州州務卿に私たちが改訂と再登録した会社証明書の修正証明書を提出し、2023年6月22日に私たちの2023年年次総会の株主の許可に従って、25株1株の割合で私たちの普通株を逆株式分割した。私たちは2024年1月12日に逆株式分割を実施した。私たちが発行を許可された普通株数は1,000,000,000株から40,000,000株に比例して減少し、その普通株額面は1株当たり0.0001ドルと変わらない。
本年度報告における株式と1株当たりの金額は,逆株分割が発効した後に列報される。1株当たりの行使価格と発行可能普通株数の割合調整を行った
97
未償還株式オプションと引受権証に基づいて。また、株式インセンティブ報酬計画のために予約された普通株式数の割合調整を行った。
財務運営の概要
収入.収入
私たちは現在販売を許可されていない製品で、2022年に確認された収入は贈与収入からです。将来、私たちは、私たちの候補製品とすでに締結されているか、または可能性のある協力、許可または許可契約、および任意の承認された製品の製品販売から追加収入を得ることができるだろう。私たちが製品収入を作る能力は私たちの候補製品の成功的な開発と最終商業化にかかっているだろう。もし私たちの候補製品の開発を適時に完成させたり、規制機関の私たちの候補製品の承認を得ることができなければ、私たちの将来の収入を創出する能力と私たちの運営業績と財務状況は重大な悪影響を受けるだろう。
ファイザー協定
2019年12月,我々はeIF 4 Eを標的とした小分子を研究·開発するファイザー合意に達した。ファイザー協定によれば、私たちはファイザーに世界的な独占許可を与え、私たちのいくつかの特許、技術および材料に基づいて任意およびすべての適応に対する使用、開発、製造、商業化、または他の方法でeIF 4 Eをターゲットとした化合物または製品を開発する権利がある。合意により,我々はファイザーとの連携の予備研究を担当し,ファイザーはIND提出とすべての臨床開発と商業化活動を含むこの開発計画のすべてのさらなる開発を担当した。
ファイザー協定によると、私たちはファイザーから1500万ドルの使い捨て、払い戻し不可能、貸切不可の前金を受け取った。ファイザーには、研究所で発生した費用を精算する義務があり、最高限度額は2桁の百万ドル。特定の開発、規制、販売マイルストーンを実現した後、ファイザーは合計4億8千万ドルの費用と、許可製品ごとの年間純売上高の1桁パーセントまでの印税を支払う義務がある。本プロトコルに関するより多くの情報は、本プロトコルに従って我々に支払い可能なお金を含み、本年度報告に含まれるForm 10-Kに含まれる“業務-当社の連携および許可プロトコル”を参照されたい。
DARPA下請けプロトコル
2021年、私たちはカリフォルニア大学サンフランシスコ校と研究賞協定(“分賞協定”)を締結し、この協定によると、ゾタフェンに関連する臨床と生産活動は500万ドルに達する新冠肺炎治療費用を精算することができる。下請け契約の条項によると、私たちは定期的にカリフォルニア大学サンフランシスコ校に財務と技術報告を提供する義務がある。私たちは2022年に下請け合意で規定された500万ドルのすべての許容費用を使い果たした。2023年2月、著者らはゾタフェンによる新冠肺炎治療の1 b期臨床試験のプラスの結果を報告し、この試験は分賞協定によって一部の資金を提供した。今回の実験で発生したデータは新冠肺炎プロジェクトを後期開発段階に入れることができると考えられるが,我々は新冠肺炎が非希釈資金源を得ない限り,このプロジェクトをさらに開発するつもりはない腫瘍学的資産の開発に注力している。
運営費
研究と開発費
研究と開発費用には,主に我々の候補製品の臨床前と臨床開発に関するコストが含まれている。私たちの研究開発費は
98
私たちは発生した費用に応じて研究と開発費用を支払う。サービスが完了または貨物を受け取った場合、将来の研究開発活動で費用として使用される貨物およびサービスの前払いは払い戻しできません。私たちは開発計画と他の計画の特定に基づいて外部費用を追跡する。しかし,これらのコストは主に人員や施設に関係しているため,具体的な計画の内部コストは追跡されておらず,これらの人員や施設は複数の開発中の計画に配置されている.
次の表に,指定された時期における我々の研究と開発費用(千単位)をまとめる.
|
十二月三十一日までの年度 |
|
|||||
|
2023 |
|
|
2022 |
|
||
外部開発計画費用: |
|
|
|
|
|
||
トミフ·サーチット(EFT 508) |
$ |
9,460 |
|
|
$ |
8,918 |
|
ゾチフェン(EF 226) |
|
6,422 |
|
|
|
6,695 |
|
EIF 4 E |
|
— |
|
|
|
8 |
|
未分配の内部研究開発費: |
|
|
|
|
|
||
関係者 |
|
5,211 |
|
|
|
5,390 |
|
他にも |
|
1,826 |
|
|
|
2,302 |
|
研究開発費総額 |
$ |
22,919 |
|
|
$ |
23,313 |
|
予測可能な未来には,我々の候補製品の開発が継続されるにつれて,特に臨床開発の後期段階に入ると,我々の研究·開発費用は大幅に増加し,臨床開発のコストは通常より高くなると予想される。臨床試験と臨床前研究は監督管理部門の許可を得るために必要であり、この過程は高価で時間がかかる。私たちは私たちのどんな候補製品も市場承認を得ることに絶対に成功しないかもしれない。現在,我々の任意の候補製品の余剰開発を完了するための努力の性質,時間やコストを合理的に見積もることはできず,これらの候補製品の現金純流入が開始可能な時期(あれば)を見積もることもできない。私たちは、臨床および臨床前の結果、法規の発展、各製品の候補および計画の商業潜在力の持続的な評価、および私たちが協力に参加する能力に基づいて、どの候補製品と計画を追求するか、および各候補製品および計画にどれだけの資金を提供し続けるかを継続的に決定し、協力者の資源または専門知識が所与の候補製品または計画に有利になることを決定することを予想する。
以下の要因により,我々の開発コストは大きく異なる可能性がある
99
一般と行政費用
一般および行政費用には、株式ベースの報酬および福祉、財務、会計、および他の行政機能に基づく相談費が含まれる賃金および他の関連費用が主に含まれる。その他のコストには、特許や会社の事務に関する法的費用、保険、施設コストが含まれており、そうでなければ研究開発費には含まれていない。
予測可能な未来には,臨床開発による候補製品の推進に伴い,我々の一般的かつ管理費が大幅に増加することが予想される。我々も上場会社の運営によって追加支出を発生し、アメリカ証券取引委員会規則及びナスダック上場規則の遵守に関連する支出、追加保険料、投資家関係活動及びその他の行政及び専門サービスを含む。また、いずれかの候補製品が規制部門の承認を得た場合、これらの候補製品を自ら商業化することを選択すれば、販売·マーケティングチームの設立に関連した費用が発生することが予想される。
その他の収入(費用)
利子収入
利息収入には現金等価物と短期投資から稼いだ利息が含まれています。
利子支出
2023年12月31日までおよび2022年12月31日までに年間収録された利息支出には、オックスフォード金融有限公司(“オックスフォード”)での未返済定期融資額が含まれています。
その他の収入(費用)
業務合併取引に関する私募株式証は,報告日ごとに負債として入金し,公正価値に応じて再計量する必要があると仮定し,公正価値の変動は他の収入(支出)の一部報告とした。
2022年1月に、吾らはリンカーン公園と購入契約を締結し、リンカーン公園に発行された普通株承諾株に関する他の支出を取引中に記録した。
収益負債の公正価値変動
吾らは,既存の旧発効株主に収益株式を発行するか債務が会計基準に基づいて編集(“ASC”)815−40が我々の株式にリンクしていないため,負債として入金し,報告期間ごとに公正価値を再計量し,公正価値変動を他の収入(支出)の構成要素として報告する必要があると判断した。トリガイベントは、業務統合終了日から2年以内に発生していないため、私たちの利益達成期間は2023年8月25日に満了します。そのため、2023年8月25日から、旧効果株主とオプション保有者は株式を稼ぐ資格がなくなった。
100
経営成果
2023年12月31日までと2022年12月31日までの年度比較
次の表に2023年12月31日までと2022年12月31日までの年度の経営成果(単位:千)を示す
|
十二月三十一日までの年度 |
|
|
|
|||||
|
2023 |
|
2022 |
|
変わる |
|
|||
奨学金収入 |
|
— |
|
|
3,553 |
|
|
(3,553 |
) |
運営費用: |
|
|
|
|
|
|
|||
研究開発 |
|
22,919 |
|
|
23,313 |
|
|
(394 |
) |
一般と行政 |
|
10,925 |
|
|
12,643 |
|
|
(1,718 |
) |
総運営費 |
|
33,844 |
|
|
35,956 |
|
|
(2,112 |
) |
運営損失 |
|
(33,844 |
) |
|
(32,403 |
) |
|
(1,441 |
) |
その他の収入,純額 |
|
(1,967 |
) |
|
9,738 |
|
|
(11,705 |
) |
純損失 |
$ |
(35,811 |
) |
$ |
(22,665 |
) |
$ |
(13,146 |
) |
贈与収入
2023年12月31日と2022年12月31日までの年間で、贈与収入はそれぞれゼロと360万ドル。贈与収入が減少したのは,2022年12月31日までに終了した贈与完了に関連した時間によるものである。
研究と開発費
2023年12月31日と2022年12月31日までの年度の研究開発費はそれぞれ2290万ドルと2330万ドル。研究開発費は40万ドル減りました これは,2022年12月31日までの1年間と比較して,ゾタフェン新冠肺炎計画が200万ドル減少し,コンサルタント費用が50万ドル減少し,関係者関連コストが20万ドル減少したためである。これらの費用はeFT 226−002(ゾチフェン腫瘍学)試験関連費用の170万ドル増加とTomivosertib計画60万ドル増加によって部分的に相殺され,これは主にeFT 508−011(KickStart)試験関連費用の増加によるものである。
一般と行政費用
2023年12月31日と2022年12月31日までの年度の一般·行政費はそれぞれ1090万ドルと1260万ドル。この間,一般·行政費用が170万ドル減少したのは,主に保険料に関する他の一般·行政費用が100万ドル減少し,相談者や監査に関する費用が60万ドル減少したためである。また、2022年12月31日までの年度と比較して、人事に関する費用は20万ドル減少した。特許関連費用は2022年12月31日までの年度と比較して10万ドル増加し,これらのコストを部分的に相殺した。
その他の収入(費用)
2023年12月31日までの年度,その他の支出は200万ドル,2022年12月31日までの年度,その他の収入は970万ドルであった。 1,170万ドル減少した要因は,2022年12月31日までの1年間に,収益負債と株式証明負債の公正価値変動収益が,2022年に記録されたリンカーン公園購入協定に関する120万ドルの他の費用によって部分的に相殺されたためである。
流動性と資本資源
流動資金源
会社設立から2023年12月31日まで、転換可能な優先株の売却と発行の毛収入1.5億ドル、業務関連の普通株発行の毛収入6700万ドルを含む3億258億ドルの運営資金を調達した
101
合併2021年8月には、ファイザーとの研究協力·許可協定での協力収入4,200万ドル、クレジット手配下の融資3,500万ドル、2023年5月登録直接発売と2023年6月登録直接発売で普通株を販売する毛収入1,620万ドル、ATM発売計画下で普通株を売却する毛収入750万ドル、カリフォルニア大学サンフランシスコ賞との合意での贈与収入500万ドル、購入契約によりリンカーン公園に普通株を販売する毛収入310万ドル(12月31日現在、購入契約下でも販売可能な4,690万ドル)2023年)。
2023年12月31日現在、私たちの現金と現金等価物および短期投資総額は1840万ドルです。私たちの業務に使用する必要がある前に、私たちは通常、私たちの現金を高流動性投資に投資し、購入日にいつでも元の期限が1年以下の現金に変換することができます。我々は,認可された金融機関の口座にのみ残高を保持することで,我々の現金や現金等価物や投資に関するリスクをできるだけ少なくしようとしているため,一般商業銀行関係に関連する正常信用リスク以外の異常信用リスクの影響を受けているとは考えられない.
オックスフォードローンの手配
2021年3月に、吾らはオックスフォードと融資及び担保協定(“オックスフォードLSA”)を締結し、この合意によると、吾らは最大3,000万ドルを借りることができ、2回に分けて発行することができ、それぞれ2,000万ドル(“A期ローン”)及び1,000万ドル(“B期ローン”)であり、総称してオックスフォードローンと呼ぶことができる。2021年3月、私たちは2000万ドルのA期ローンを借りた。A期ローンには利息のみの期限があり、借金の日から毎月の初日に満期になって利息を支払います。
2022年2月22日、オックスフォードLSAに対して修正案を行い、この改正案によると、A期ローンの利子期間は2023年5月1日ではなく、2024年3月1日に終了する。改正案に関連して、期限Aローンの満期日は2026年3月18日から2027年2月1日に延長される。2022年12月31日と2023年12月31日まで、オックスフォード融資の下で満期になった元本と関連する最終支払いは、オックスフォード融資項目の下での重大な不利な変化条項が私たちの制御範囲内でないと評価するため、流動負債に分類されている。この報告日まで、貸手はまだ私たちに違約事件を知らせてくれなかった。
B期ローンはある臨床開発マイルストーンに達した時に使用可能であり、(I)2023年6月30日、(Ii)この臨床開発マイルストーンの発生後45日と(Iii)違約事件が発生する前に有効である。我々は2023年6月30日まで臨床開発マイルストーンを実現していないため,B期ローンでの追加1,000万ドルを得ることはできなくなった。
私たちは満期時に各資金部分の5.5%に相当する最終支払いを要求され、このお金は債務割引として記録され、債務手配の期限内に償却された。
リンカーン公園との購入契約
2022年1月24日、私たちはリンカーン公園と購入契約を締結し、購入契約の36ヶ月の期間内に、ある条件に基づいて、リンカーン公園に最大5,000万ドルの普通株を販売することを規定した。購入契約については,リンカーン公園は22,304株の普通株に相当する300万ドルの普通株を初歩的に購入し,購入契約締結に関する承諾費としてリンカーン公園に5,717株の普通株を発行した。2023年12月31日現在、前払い額を除いて1200株の普通株が売却されており、これらの株は2022年6月30日までの3カ月間で売却されている。リンカーン公園に株を売ることができる最低価格は一株一万00ドルです。私たちは私たちが購入合意に従って任意の追加の普通株を売却することを保証することができません。あるいは、もし私たちがそうすれば、私たちが売却した普通株の価格または金額、あるいはそのような売却の日について保証することはできません。
102
コントと共同で発売した市場販売計画
2022年9月に,吾らはCantorと販売協定を締結し,この合意により,吾らは総発行価格5,000,000ドルまでの普通株式を随時販売することができる(“ATM発売計画”)。普通株株の売却は売却時の現行市場価格で行うか、カント社と別途合意する。私たちはCantorに販売協定に従って売却された任意の普通株総収益の3.0%の手数料を支払うつもりだ。2023年12月31日までの1年間に、ATM機発売計画に基づき、1株13.48ドルの加重平均価格で合計537,200株の普通株を売却し、総収益は約720万ドルだった。2023年12月31日までの1年間に、ATM発売計画に関する発売コストが発生し、手数料を含めて約40万ドルとなった。
登録された直売製品
当社は2023年5月に単一機関投資家と証券購入協定を締結し、登録直接発売方式で188,000株の普通株を発行した(“2023年5月登録直接発売”)。2023年5月の登録直接発行については,予融資権証の形で270,015株の普通株を発行し,直ちに行使した。同時に行われた私募では、株式引受証を発行し、1株13.25ドルの使用価格で最大458,015株の普通株を購入した(“2023年5月普通株式承認証”)。2023年5月の普通株式承認株式証は発行時に行使でき、2028年11月30日に満期になり、即ち発行日から5年半になる。また,H.C.ウェインwright&Co.,LLC(“配給代理”)承認株式証を発行し,1株20.47ドルの使用価格で最大32,060株の普通株を購入した(“2023年5月配給代理承認株式証”)。2023年5月の配給代理承認株式証は、2028年5月26日に満期、つまり2023年5月に登録して直接発売して販売を開始してから5年になる。私たちが2023年5月に登録直接発売から得た総収益は750万ドルで、80万ドルの手数料と他の取引コストを差し引くと、純収益は約670万ドルです。
2023年6月、同一機関投資家と追加の証券購入契約を締結し、そのうち238,000株の普通株が登録直接発売方式で発行された(“2023年6月登録直接発売”)。2023年6月の登録直接発行については,予融資権証の形で72,578株の普通株を発行し,直ちに行使した。同時に行われた私募では、株式引受証を発行し、1株25.00ドルの使用価格で最大310,577株の普通株を購入した(“2023年6月普通株式承認証”)。2023年6月の普通株式承認株式証は発行時に行使でき、2028年12月8日に満期になり、即ち発行日から5年半になる。また,配給代理権証を発行し,1株35.1575ドルの使用価格で最大21,739株の普通株を購入した(“2023年6月配給代理権証”)。2023年6月の配給代理権証は2028年6月6日に満期、つまり2023年6月に登録直接発売が発売されてから5年になる。私たちが2023年6月に登録直接発売から得た総収益は870万ドルで、90万ドルの手数料や他の取引コストを差し引くと、純収益は約780万ドル。
2024年1月、338,000株の普通株を直接発売する新規単一機関投資家と追加証券購入契約を締結した(“2024年1月登録直接発売”)。2024年1月の登録直接発売については,予融資権証として1,150,834株の普通株を発行し,そのうち497,834株がその後行使された。我々は同時に株式引受証を発行し、1株9.95ドルの使用価格で最大1,488,834株の普通株を購入した(“2024年1月普通株式承認証”)。2024年1月の普通株式承認株式証は発行時に行使でき、2027年7月29日に満期になり、即ち自発的な行日から3年半になる。また、配給代理権証を発行し、1株あたり12.5938ドルの使用価格で最大104,218株の普通株(“2024年1月配給代理権証”)を購入した。2024年1月の配給代理権証は2027年7月29日に満期、すなわち2024年1月に登録直接発売されてから3年半。2024年1月に直接発売されて得られた毛収入は1,500万ドルであり、140万ドルの手数料や他の取引コストを差し引いた純収益は約1,360万ドルである。
103
2023年7月31日現在、我々の公募株は7,500万ドルを超え、非関連会社が保有している2,427,820株に基づいて普通株を発行し、1株当たり34.75ドルであり、これは私たちの普通株の2023年6月6日の終値であり、これは私たちの普通株が2023年7月31日以降60日間にナスダック資本市場で報告された最高販売価格である。私たちの公開流通株が7500万ドルを超えているので、私たちは一般的な指示I.B.6に制限されなくなった。S-3表の規定によると,我々が任意の12カ月間に初公開株式公開により募集した資金は,公募資金総額の3分の1を超えてはならない,いわゆる乳児保有ルールである.しかし、2024年3月15日現在、我々の公開流通株は約6,560万ドルであり、非関連会社が保有する3914,309株に基づいて普通株を発行しており、価格は1株16.95ドルであり、2024年3月4日に我々の普通株が最後にナスダック資本市場で発表された販売価格である。私たちの公開流通株が7500万ドルを下回るので、私たちの公開流通株が7500万ドルを超えるまで、私たちは乳児棚規則によって制限され、これは私たちが任意の12ヶ月の間に棚登録声明に従って最大3分の1の公開流通株しか販売できないことを意味する。2023年5月の登録直接発売と2023年6月の登録直接発売の証券販売は、ベビーラック規則に基づいて行われ、このような発行12ヶ月周年に達するまで、現在のラック登録声明に基づいて株式を売却する能力が制限されます。2024年1月に登録直接発売された証券販売は、乳児棚ルールの制限を受けず、現在の棚登録声明に基づいて容量を制限することもありません。私たちは、私たちの公衆所有量が7,500万ドルを超えるまで、S-3表棚登録声明における乳児棚規則の制約を受け続け、その際、S-3表登録声明の下で販売できる証券の数は、乳児棚規則によって制限されなくなる。
資金需要
2023年12月31日現在、私たちは1840万ドルの現金と現金等価物と短期投資を持っており、2024年1月に登録直接発売された1360万ドルの純収益に加え、現在の運営計画によると、2025年第1四半期の運営に資金を提供するのに十分だと推定されています。しかし、将来予想される現金流入を考慮せずに、2023年12月31日までの年次財務諸表発表後12カ月以内に継続して経営を継続する企業としての能力があるかどうかに大きな疑問があることを示すキャッシュフロー予測を用意している。私たちは間違っていることが証明される可能性があるという仮定に基づいてこの推定をして、私たちは予想よりも早く私たちの資本資源を使用することができる。しかも、私たちの運営計画は変わるかもしれないし、私たちは計画よりも早い追加資金が必要かもしれない。また,臨床試験では候補製品を試験する過程コストが高く,これらの試験の進展時間も不確定である。私たちの将来の資本需要は予測が難しく、多くの要素に依存するだろうが、これらに限定されない
104
リンカーンパークとの購入契約とコントーとのATM発売計画下の潜在的な未来販売以外に、私たちには他に約束された資金源がありません。私たちが十分な製品収入を生成して私たちの現金需要を満たすことができる前に、もしあれば、私たちは主に株式発行、債務融資、または他の資本源を通じて、潜在的な追加協力、ライセンス、その他の同様の手配を含む将来の現金需要を満たすことが予想される。しかし、私たちは必要に応じて優遇条件で、または追加資金を調達できないか、またはそのような他の計画を達成することができないかもしれない。私たちが株式または転換可能な債務証券を売却することによって追加資本を調達する場合、私たちの株主の所有権権益は希釈されるか、または希釈される可能性があり、これらの証券の条項は、清算または他の私たちの普通株主の権利に悪影響を及ぼす特典を含む可能性がある。債務融資が可能であれば、追加債務を招く、資本支出を行う、または配当を宣言するなど、私たちが具体的な行動をとる能力を制限または制限する契約を含むいくつかの合意が含まれる可能性がある。もし私たちが第三者との他の協力や許可手配を通じてより多くの資金を調達すれば、私たちは私たちの技術、将来の収入源、研究計画、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になる可能性のある条項で許可を付与しなければならないかもしれない。もし私たちが必要な時に株式や債務融資を通じてより多くの資金を調達できない場合、私たちは私たちの研究開発計画や他の業務を延期、制限、減少または終了することを要求されるかもしれません。あるいは、私たちが自分で開発し、マーケティングしたいと思っていた第三者が候補製品を開発し、マーケティングする権利を与えることができます。
キャッシュフロー
次の表は,期間ごとのキャッシュフロー純額活動(千計)をまとめたものである
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
提供された現金純額(使用): |
|
|
|
|
|
|
||
経営活動 |
|
$ |
(29,550 |
) |
|
$ |
(25,899 |
) |
投資活動 |
|
|
14,484 |
|
|
|
(17,860 |
) |
融資活動 |
|
|
21,233 |
|
|
|
2,765 |
|
現金純増(マイナス) |
|
$ |
6,167 |
|
|
$ |
(40,994 |
) |
経営活動
2023年12月31日までの年度における経営活動に用いられた現金純額は2960万ドルであり、運営資産と負債の変化および非現金費用調整後の純損失3580万ドルの結果となった。非現金費用およびその他の調整には、460万ドルの株式ベースの給与、50万ドルの投資割引、償却純額、30万ドルの非現金利息支出、10万ドルの減価償却支出が含まれている。 経営資産と負債の変化には、上場会社の保険の償却に関する前払い費用やその他の資産が80万ドル減少することと、領収書の支払いスケジュールによる売掛金が90万ドル増加することがある。
2022年12月31日までの年間で、経営活動に用いられる現金純額は2590万ドルであり、運営資産と負債の変化および非現金費用調整後の純損失2270万ドルの結果となった。非現金費用およびその他の調整は、利益負債公正価値変化から記録された収益1210万ドル、株式ベースの補償530万ドル、リンカーン公園の購入協定に関連する他の支出120万ドル、負債分類から株式証明公正価値変化に記録された収益60万ドル、および非現金利息支出30万ドルを含む。 経営資産と負債の変化には、前払い上場会社の保険償却に関する前払い費用やその他の資産が180万ドル減少し、前払い研究·開発残高が減少し、売掛金が100万ドル増加する。
105
投資活動
2023年12月31日までの年度内に、短期投資満期により、投資活動が提供する現金純額は1,450万ドルであるが、期間の購入部分によって相殺される。
2022年12月31日までの年間で、投資活動のための現金純額は1790万ドルで、短期投資を購入した結果となったが、期間内の満期資金部分で相殺された。
融資活動
2023年12月31日までの年間で、融資活動が提供する現金純額が2,120万ドルであるのは、主に2023年5月と2023年6月に登録直接発売中に発行された普通株の純収益1,450万ドルと、同期にATM発売計画に基づいて普通株を発行する純収益660万ドルのためである。
2022年12月31日までの1年間に,融資活動が提供する現金純額は280万ドルであり,購入合意によりリンカーンパークに普通株を発行する純収益と,同期にATM発売計画に基づいて普通株を発行する純収益の結果である。
重要な会計政策と試算
米国証券取引委員会は、キー会計政策を、経営陣が我々の財務状況や経営結果を記述するために重要であると考えている政策と定義し、経営陣に判断を求めている。経営陣の財務状況と経営結果の議論と分析は、米国公認の会計原則(“GAAP”)に基づいて作成された我々の総合財務諸表に基づいている。これらの連結財務諸表を作成するには、連結財務諸表における資産、負債及び費用の報告金額、又は有資産及び負債の開示に影響を及ぼす推定及び判断が必要である。継続的な基礎の上で、計算すべき費用および株式ベースの報酬に関する推定および判断を含む、我々の推定および判断を評価する。我々は過去の経験,既知の傾向や事件,および様々な当時の状況に属すると考えられる合理的な要素から推定しているが,これらの要因の結果は資産や負債の帳簿価値を判断する基礎を構成しており,当該などの資産や負債の帳簿価値は他の出所から容易に見られるものではない。異なる仮定または条件では、実際の結果は、これらの推定値とは異なる可能性がある。
我々の重要な会計政策は、本年報の他の部分の総合財務諸表付記2により全面的に記述されているが、以下の会計政策は、我々の財務状況や経営結果を全面的に理解し評価するために最も重要であると考えられる。
臨床試験は費用と前臨床研究を計算しなければならない
われわれは,サプライヤーやコンサルタント,CROと臨床サイトとの契約に基づき,臨床試験や臨床前研究に関する義務に伴う費用を記録した。これらの契約の財務条件は、契約によって異なり、支払流量がこれらの契約に基づいて材料またはサービスを提供する期限と一致しない可能性がある交渉を行う必要がある。我々は,臨床試験と臨床前研究費用をサービスや努力支出の時間と整合させることにより,これらの費用を我々の合併財務諸表に反映させた。著者らは臨床試験或いは臨床前研究の進展によってこれらの費用を計算し、これらの進展は臨床試験、臨床前研究或いは関連活動の各方面の時間によって測定する。我々は,関連契約の審査と財務モデルの準備を行い,臨床や他のキーパーソンおよび第三者サービスプロバイダとの臨床試験,臨床前研究あるいは進行中の他のサービスの進展状況との通信を考慮して,算定すべき推定を決定した。臨床試験や前臨床研究過程において,実際の結果がわれわれの見積もりと異なれば,費用認知率を調整する。今まで、私たちの推定は発生した金額と実質的な差はなかった。しかし、見積もりの性質のため、私たちの臨床試験、臨床前研究、あるいは他の研究活動の状態や進行に関するより多くの情報を知った場合、将来的に私たちの推定を変更しないことを保証することはできません。
106
新興成長型企業と小さな報告会社の地位
業務合併後、“雇用法案”によると、新興成長型会社になる資格がある。したがって、私たちは延長された過渡期を利用して新しい会計基準や改正された会計基準を遵守することができる。これは、これらの基準が民間企業に適用されるまで、新興成長型企業が特定の会計基準の採用を延期することを可能にする。この免除を利用して新たな会計基準または改正会計基準を遵守することを選択しているため、我々の総合財務諸表は、上場企業の発効日までに新しい会計公告または改正会計公告を遵守している会社と比較できない可能性がある。我々はまた、“雇用法案”によって提供される他の免除に依存する予定であり、これらに限定されず、サバンズ·オキシリー法案第404(B)節の監査人認証要件の遵守が要求されない。
(I)2026年12月31日まで、(I)2026年12月31日まで、(Ii)財政年度の最終日まで、私たちの年間総収入は少なくとも12.35億ドルに達する。(Iii)財政年度の最終日、取引法により、ルール12 b-2で定義されている“大型加速申告会社”とみなされ、この年度第2四半期の最終営業日までに、非関連会社が保有する私たちの普通株の時価が7000万ドルを超えると、このような状況が発生する。あるいは(Iv)私たちは前3年の間に10億ドルを超える転換不可能債務証券を発行した日。
取引法の定義によると、私たちも規模の小さい報告会社です。私たちがもう新興成長型会社ではなくても、私たちは規模の小さい報告会社であり続けるかもしれない。私たちの非関連会社が保有している投票権と非投票権普通株が第2四半期の最後の営業日に2.5億ドルを下回る限り、私たちの非関連会社が保有している投票権と非投票権普通株が第2四半期の最終営業日に7.00億ドルを下回る限り、これらの比例して開示された情報を利用することができる。
最近の会計公告
最近の会計声明に関する情報は、本年度報告の他の部分に掲載されている総合財務諸表付記2を参照されたい。
第七A項オファー市場リスクに関する実質的で定性的な開示。
金利リスク
我々の金融商品や財政状況に固有の市場リスクは、金利の不利な変動による潜在的損失を代表している。私たちは2023年12月31日現在、無利子運営口座、利息通貨市場基金、米国国債を含む1,490万ドルの現金と現金等価物を持っている。2023年12月31日現在、米国債を含む350万ドルの短期投資がある。私たちの市場リスクに対する主な開口は金利感受性であり、金利敏感性はアメリカの金利全体の水準変化の影響を受けている。我々の通貨市場基金と短期投資はいずれも短期的、低リスクであるため、金利を直ちに100ベーシスポイント調整することは、私たちの現金等価物の公平な市場価値に実質的な影響を与えない。
私たちのオックスフォード条項Aローンの変動金利は(I)7.7%と(Ii)最優遇金利プラス4.45%の両方の大きな者に等しい。市場金利変動100ベーシスポイントの影響は、我々の財務状況及び/又は経営業績に実質的な影響を与えることはない。
外貨両替リスク
私たちの報告書と機能通貨はドルです。私たちは現在外貨に対する開放が大きくありません。外貨契約、オプション契約あるいは他の外貨ヘッジ手配を持っていないからです。私たちの業務は将来外貨為替レートの変動の影響を受けるかもしれません。
インフレの影響
インフレは一般的に労働力と研究開発費用を増加させることで私たちに影響を及ぼす。私たちは、この報告書で述べた間、インフレは私たちの経営業績に実質的な影響を与えないと信じている。
107
イットM 8.連結財務諸表および補足データ。
本プロジェクトの要求に基づいて作成された連結財務諸表は本年度報告項目15に掲載され,F−1ページから列報される。
情報技術EM 9.会計及び財務開示面の変更と会計士との相違。
ない。
ITEM 9 Aです。制御とプログラムです
開示制御とプログラムの有効性に関する結論
我々は、取引所法案に基づいて提出または提出された報告書に開示すべき情報が、米国証券取引委員会規則および表で指定された期間内に記録、処理、集約および報告され、これらの情報が、必要な開示に関する決定をタイムリーに行うために、我々の経営陣に蓄積され、伝達されることを保証するために、開示制御および手続きを維持する。開示制御およびプログラムを設計·評価する際に、管理層は、任意の制御およびプログラムは、設計および動作がどんなに良好であっても、予想される制御目標を達成するために合理的な保証を提供することしかできず、管理層は、その判断を用いて、可能な制御およびプログラムのコスト−収益関係を評価しなければならないことを認識する。本報告で述べた期間終了までの開示制御および手順(取引所法案規則13 a−15(E)および15 d−15(E)で定義されている)の有効性を、CEOおよび最高財務官を含む経営陣の監督·参加の下で評価した。この評価に基づき、我々の最高経営責任者および最高財務責任者は、2023年12月31日までに、我々の開示統制および手続きが合理的な保証レベルで有効であると結論した。
経営陣財務報告内部統制年次報告書
我々の経営陣は、取引所法案第13 a-15(F)及び15 d-15(F)条の規定に基づいて、財務報告の十分な内部統制を確立·維持する責任がある。我々の財務報告に対する内部統制は、財務報告の信頼性と公認会計基準に基づいて外部目的の財務諸表の作成に合理的な保証を提供することを目的とした過程である。その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来的にどのような有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムの遵守度が悪化する可能性がある.
我々の経営陣は、2013年の内部統制-総合的な枠組みでテレデビル委員会後援組織委員会が提案した基準に基づいて、2023年12月31日までの財務報告書の内部統制に対する有効性を評価した。この評価に基づき、我々の経営陣は、財務報告書の内部統制が2023年12月31日から有効であると結論した。
公認会計士事務所認証報告
雇用法案による“新興成長型会社”の免除により、本年度報告にはわが公認会計士事務所の認証報告は含まれていません
財務報告の内部統制の変化
2023年12月31日までの四半期内に、財務報告の内部統制に大きな影響を与えたり、合理的に財務報告の内部統制に大きな影響を与える可能性のある変化はありません。
プロジェクト9 B他の情報。
ない。
プロジェクト9 Cです検査妨害に関する外国司法管区の開示
適用されません。
108
第三部
情報技術EM 10.取締役、行政者、会社管理。
経営陣と取締役会
以下に、2024年2月15日現在のいくつかの情報を示し、これらの情報は、当社の役員および取締役会のメンバーを担当する者に関するものである。
名前.名前 |
|
年ごろ |
|
|
ポスト |
|
行政員 |
|
|
|
|
|
|
スティーヴン·T·ウォラン博士 |
|
|
66 |
|
|
取締役最高経営責任者総裁 |
マイケル·バーンズ |
|
|
47 |
|
|
首席財務官 |
ダグラス·ワーナー医学博士 |
|
|
52 |
|
|
首席医療官 |
非従業員取締役 |
|
|
|
|
|
|
小ブライアン·M·ガラゲル博士(1)(3) |
|
|
54 |
|
|
椅子 |
エリザベス·P·バート(3) |
|
|
56 |
|
|
役員.取締役 |
クリス·エルリッヒ(1)(2) |
|
|
54 |
|
|
役員.取締役 |
クリスチャン·ハリントン·スミス(2) |
|
|
51 |
|
|
役員.取締役 |
バーバラ·クロンク医学博士(2) |
|
|
66 |
|
|
役員.取締役 |
キャロライン·ロイド(1) |
|
|
58 |
|
|
役員.取締役 |
行政員
スティーヴン·T·ウォラン博士2012年5月の設立以来、私たちの総裁兼最高経営責任者と旧効果器取締役会のメンバーを務め、2021年8月以来取締役会のメンバーを務めてきた。これまで、ウォランド博士は2007年8月からAnadys PharmPharmticals,Inc.の最高経営責任者と取締役会メンバーを務め、2011年に羅氏ホールディングスに買収された。アナディス製薬会社の最高経営責任者に任命される前に、ウォランド博士は同社の首席科学官、総裁博士は製薬会社の首席科学官を務めた。アナディス製薬会社に入社する前、ウォランド博士はファイザー社で副総裁兼抗ウイルス研究主管を務め、ワーナー-ランバート社で総裁副主任を務め、全世界の抗感染薬物戦略を担当した。ウォランド博士は2015年2月からTracon PharmPharmticals,Inc.の取締役を務め、2017年4月以来黒石医薬会社の取締役を務めている。ウォランドはハーバード大学でアメリカ国立衛生研究院分子生物学博士を務め、カリフォルニア大学バークレー校で化学博士号を取得した。彼は最高の栄誉でミシガン大学の生化学学士号を取得した。ウォランダー博士の私たちの業務に対する広範な理解と、バイオテクノロジーと製薬業界での彼の豊富な経験は、私たちの取締役会の結論、すなわち彼がわが社の取締役を務めるべきだと結論しました。
マイケル·バーンズ2020年12月以来、私たちの最高財務責任者を務めてきた。これまで、バーンズは2020年1月から2020年9月までセノフィに買収されるまで、Prciia Biophma,Inc.の財務担当上級副総裁を務めてきた。これまで、Byrnesさんは、Mallinckrodt PharmPharmticalsによって、2018年5月から2020年1月までの間、Alkahest,Inc.のチーフ財務官を務め、2014年12月から2017年12月まで、Ocera Treateutics,Inc.のチーフ財務官を担当していました。Byrnesさんは、2010年3月から2014年12月までの間に、NeurogesX,Inc.、Lipid Sciences,Inc.およびフィリップス医療システム会社の傘下のADAC実験室、Inc.が、NeurogesX,Inc.,Lipid Sciences,Inc.およびフィリップス医療システム会社の傘下のADAC実験室,Inc.において、MaxyrnesさんをMaxygen,Inc.の財務ディレクターに務めた。バーンズさんは、2024年2月以来、CERO療法会社(取締役コード:CERO)に勤務している。バーンズさんは、セントクララ大学でファイナンスの学士号、カリフォルニア州立大学ヘイワード校のビジネスマネジメント修士号を取得しています。
109
ダグラス·ワーナー医学博士です2022年8月以来私たちの首席医療官を務めてきました。これまでワーナー博士は安進会社に18年間勤務しており,そこで腫瘍学と一般医学の複数の適応の広範な臨床開発プロジェクトを監督してきた。最近のポストでは、ワーナー博士は取締役グループ製品区域主管を務め、第一段階から発売製品までの広範な固形腫瘍免疫腫瘍学と経路阻害剤開発計画に開発指導と監督を提供した。これまでワーナー博士はVectibix,XGEVA,Proliaを含む複数の製品のグローバル開発担当であった。このポストで、ワーナー博士は証拠生成を指導し、そして研究開発の各段階の設計、実行と分析を監督し、大型全世界3期試験を含み、そして全世界の主要な監督申告文書の臨床開発担当者である。ワーナー博士は複数の同業者評議文章の共著者であり、“柳葉刀”、“柳葉刀腫瘍学”と“臨床腫瘍学雑誌”に発表された文章を含む。彼はペンシルバニア大学で学士号を取得し、デューク大学医学部で医学博士号を取得し、カリフォルニア大学ロサンゼルス校アンダーソン管理学院で工商管理修士号を取得した。
非従業員取締役
ブライアン·M·ガラゲル博士2020年以来Old Efftorの取締役会メンバーを務め、2020年以来取締役会長を務め、2021年8月以来取締役会長を務めている。現在,ガラゲル博士はTrekk Venture Partnersの管理パートナーと共同創業者であり,早期バイオテクノロジーに焦点を当てたベンチャー企業である。ガラゲル博士は2018年から2022年までAbingworth LLPのパートナーを務めていた。これまで、2010年から2018年まで、ガラゲル博士はSR Oneのパートナーだった。現在Slate Bioの取締役メンバー、2020年から2022年まで取締役Q 32 Bioの取締役、2018年から2022年までAbingworth Management Inc.の取締役会メンバー、2011年から2018年までNimbus治療会社の取締役メンバー、2012~2017年にRiver Vision(Horizon Pharmaに買収された)の取締役メンバー、2011年から2019年まで翻訳生物会社の取締役メンバー、2010年から2018年まで副翼治療会社の取締役、2014年から2018年までNavitor Pharmticalsの取締役、2013~2018年にCalciMedicaの取締役、2010年から2018年まで星座製薬会社の取締役観察員、2010年から2014年までDicerna製薬会社の取締役会観察員、その他の上場企業の取締役を務めた。SR One以前、ガラゲル博士はSirトリス製薬会社に勤め、会社がGSKに買収された会社の発展、運営、合併後の統合を担当していた。彼のキャリアの初期には、ガラゲル博士はAlantos製薬会社(安進社に買収された)や衛材会社の運営と研究開発で重要な役割を務めていた。ガラゲル博士はミシガン大学有機化学博士号とマサチューセッツ大学化学学士号を持ち、マサチューセッツ大学でシャピロ学者を務めていた。彼は25件以上の特許と応用の発明家であり、有名な定期刊行物に複数の論文を発表したベテラン著者である。彼は現在、ミシガン大学生物医学リスク基金投資顧問委員会と、ミシガン薬物発見と元ニューヨーク大学医学院治療連盟の顧問委員会に勤めている。ガラゲル博士のベンチャー投資家としての経験と,他の生物製薬会社で取締役を務めるサービスは,我々の取締役会がわが社の会長を務めるべきであるとの結論につながった。
エリザベス·P·バート2021年1月からLWACの独立取締役を務め、業務合併後も取締役会メンバーを継続している。パットさんは2022年6月以来、Septerna,Inc.の首席運営官を務めてきた。2019年9月から2022年5月まで、バートさんは上場臨床段階のバイオ製薬会社応用分子輸送会社(ナスダック:AMTI)の首席業務と戦略官を務めている。これまで、パットさんはバイオ製薬会社Achaogen,Inc.に勤務し、2018年7月から2019年6月まで首席運営官、2017年9月から2019年6月まで首席商務官を務めていた。2019年4月、Achaogenは破産申請を連邦裁判所に提出し、破産法第11章に基づいて保護を求めた。Achaogenに加入する前に、バートさんは2006年7月から2017年9月まで研究を基礎とした上場生物製薬会社ジリッド科学会社(ナスダックコード:GILD)で様々な職務を担当し、2016年1月から2017年9月まで企業発展部副総裁を務め、2011年5月から2015年12月まで取締役企業発展部高級主管を務めた。バートさんはポモナ学院の化学学士号、カリフォルニア大学サンディエゴ校の生物医学修士号、西北大学ケロッグ管理学院のMBA学位を持っている。バートさんの深い科学的背景、生物科学技術業界内で各種の技術職務を担当した経験、及び彼女が生物科学技術会社を評価、投資と監督した経験は、私たちの取締役会の結論を促進し、つまり彼女はわが社の取締役を務めるべきである。
クリス·エルリッヒ2020年10月から2021年8月までの間にLWAC最高経営責任者と取締役を務め、業務合併後も取締役会メンバーを継続している。エルリヒは2021年10月からフェニックスバイオテクノロジー買収会社のCEOを務めてきた。エルリヒは2013年からバッタ行動パートナー会社で様々な職務を担当し、まず取締役高級取締役社長とバッタ行動パートナーグローバル生物医学チーム責任者を2021年まで務め、2021年からバッタ行動買収会社の最高経営責任者を務めている。
110
エルリッヒさんは、2013年にLocust Walk Partnersに参加する前に、2000年から2013年にかけて、ヘルスケアや情報技術に特化したベンチャーキャピタルInterWest Partnersで取締役社長を務めました。InterWestでは,個人持株製薬会社凱製薬(2012年に安進に買収),バイオテクノロジー会社Biomimtic Treateutics,Inc.(2013年にライト医療技術社に買収),Inc.(2018年にStrykerに買収された医療技術会社),バイオ製薬会社Xenon製薬会社(ナスダック:XENE)の取締役を務めている。インターWestに入社する前に、エルリッヒさんは、プライベート製薬会社普渡製薬のライセンスおよび業務開発担当役員であり、重要な知的財産のライセンスを含む生物腫瘍学のフランチャイズ権の開発を担当し、バイオテクノロジー会社との連携を確立し、管理し、バイオテクノロジー会社の普渡バイオ製薬のビジネス運営をリードする。バイオテクノロジー社に入社する前に,EhrlichさんGenentech社でビジネス開発に従事し,米国ロシア投資ファンドでベンチャーキャピタルに従事し,L.E.K.コンサルティング会社でバイオテクノロジー戦略開発に従事していました。エルリッヒは2014年以降、診断会社の前立腺管理診断会社の取締役会メンバー、ピーター·マイケル財団顧問委員会のメンバー、西北大学ケロッグ諮問委員会のメンバーを務めてきた。ピーター·マイケル財団は前立腺癌に専念する慈善団体で、2012年から同財団の上級顧問を務めている。彼はダートマス学院で学士号を取得し、西北大学ケロッグ管理学院で工商管理修士号を取得した。彼はFINRAの登録代表でもあり、彼のシリーズ79、63、24ライセンスを持っている。エルリヒさんは、バイオテクノロジー産業の幅広い経験、ならびにベンチャーキャピタルおよびビジネス開発における豊富な経験を有しており、取締役会の結論に寄与しており、すなわち当社の取締役を務めることになりました。
クリスチャン·ハリントン·スミス2022年2月以来取締役会に勤めている。2022年11月以来、ハリントン-スミスさんは上場生物製薬会社のアディーク治療会社の高級副総裁兼首席商務官を務めてきた。ADC治療会社に入社する前、ハリントン-スミスさんは免疫遺伝子会社の首席商務官を務めていたが、そこで彼女はこの組織が彼らの初めてのビジネス発売の準備を手伝ってくれた。免疫遺伝子会社に入社する前に,ハリントン−スミスさんは2000年7月からノワ製薬会社でますます多くの役割を務めており,最近の職務は2020年から2021年まで米国血液学ビジネス担当,2016年から2020年まで副総裁と米国CAR−T担当者を務めている。彼女はウィリアムズ大学の学士号とノースカロライナ大学教会山校ケナン-フラグラードビジネススクールのMBAの学位を持っています。ハリントン-スミスさんの製薬業界での豊富なビジネス経験は、彼女がわが社の取締役会社の役員を務めるべきだという私たちの取締役会の結論につながった。
バーバラ·クロンク医学博士2021年11月以来取締役会に勤めています。Klencke博士は現在もTscan治療会社(ナスダックコード:TCRX)、Xencor(ナスダックコード:XNCR)と免疫ONCの取締役会メンバーを務め、2017年から2020年まで同社がFoundation Medicineに買収される前にLexent Bioの取締役会メンバーを務めたことがある。彼女は以前Sierra Oncology Inc.の首席医療·首席開発官を務めており,同社は上場した臨床段階バイオ製薬会社であり,2015年から2022年までGSKに買収された。2011年から2015年にかけて、Klencke博士は2013年に安進社に買収されたOnyx製薬会社でグローバル発展上級副総裁を務めた。2003年から2011年まで、彼女は遺伝子技術会社で働いている間、各種の早期と末期腫瘍学プロジェクトを指導した。彼女はカリフォルニア大学サンフランシスコ校で内科と血液学/腫瘍学研修を修了し,そこで腫瘍学アシスタント教授を務め,1995年から2002年まで臨床研究に専念した。クロンク博士はインディアナ大学の学士号とカリフォルニア大学デイビス校の医学博士号を持っています。Klencke博士のバイオテクノロジーにおける重要な科学専門長は、彼女がわが社の取締役を務めるべきであるという私たちの取締役会の結論に寄与した。
キャロライン·ロイド2023年9月以来取締役会メンバーを務めてきた。Loewyさんは上場会社の取締役会に勤め、生命科学会社に戦略コンサルティングサービスを提供し、生物製薬業界で25年以上の経験を持っている。2015年から2017年にかけて、彼女は人と共同で専門製薬会社Aucture Life Science,Inc.の首席財務官と首席商務官を務めた。生命科学の学位を取得する前に、2012年から2014年までのTobira治療会社、2008年から2011年までのCorcept治療会社、2006年から2008年までのPoniard製薬会社を含む複数の生命科学会社の首席財務官を務めた。これまで、Loewyさんは2000年から2004年までモルガン·スタンレー社で高級バイオテクノロジー株研究アナリストを務め、1996年から1999年まで保誠証券会社で高級バイオテクノロジー株研究アナリストを務めた。彼女のキャリアはアメリカの銀行会社の金融アナリストから始まった。ロイドさんは全世界の遺伝子プロジェクトとKCNQ 2治癒連盟基金会の創立取締役会のメンバーである。Loewyさんは2016年12月からCymaBay治療会社(ナスダックコード:CBAY)の取締役会メンバーを務め、2018年7月から2023年9月までPhaseBio製薬会社の取締役メンバー、2021年10月から2024年2月までフェニックスバイオテクノロジー買収会社の取締役、2020年9月から2022年3月までZgenix,Inc.の取締役会メンバー、2018年4月から2022年6月までAptose Biosciences社の取締役会メンバー、2021年1月から2021年8月までLocust Walk Acquisition Corp.の取締役会メンバーを務めた。ロイドさんはカリフォルニア大学バークレー校の学士号とカーネギーメロン大学の工商管理修士/理学修士号を持っている。
111
家族関係
当社の取締役会と当社のどの行政者との間には家族関係はありません。
取締役会と役員選挙
役員は自主独立している
ナスダック上場規則の要求は、ナスダック上場会社の取締役会の過半数は独立取締役から構成されなければならず、独立取締役は一般に会社或いはその子会社の幹部、従業員或いは任意の他の関係のある個人以外の人と定義され、会社取締役会は独立取締役会が取締役が取締役の職責を履行する際に独立判断を行使することを妨害すると考えている。当社の取締役会は、上場規則および取引所法案第10 A-3条に基づき、エリヒ·さん、バート夫人、ガラゲル博士、ハリントン·スミスさん、クロンケル博士およびロイドさんを独立ナスダック取締役とすることを決定しました。これらの決定を下す際に、我々の取締役会は、各非従業員取締役の現在と以前と私たちとの関係、および取締役会が独立性の決定に関連すると考えているすべての他の事実および状況を考慮しており、各非従業員取締役の私たちの普通株に対する実益所有権、および“いくつかの関係および関連取引”節に記載されている彼らに関連する取引を含む
分類取締役会
わが社の登録証明書の条項によると、私たちの取締役会は3種類に分類され、交錯する3年間の任期です。毎回株主総会では、任期満了の取締役が再任され、再任後の第3回年次会議まで再任する資格がある。私たちの取締役は以下の3つに分類されます
わが社の登録証明書は、取締役会が決議を採択して初めて、許可された取締役数を変更することができると規定しています。取締役数の増加により増加した任意の取締役職は、各レベルが可能な限り3分の1の取締役で構成されるように、この3つのレベルに割り当てられる。私たちの取締役会を3つのレベルに分け、3年間の任期を交錯させることで、私たちの取締役会の交代や会社の支配権の変更を延期または阻止する可能性があります。私たちの役員は当時取締役選挙で投票する権利のある少なくとも三分の二の議決権のある株を発行した保有者が賛成票を投じた場合にのみ免職されます。
取締役会の指導構造
私たちの取締役会は現在ガラゲル博士が議長を務めている。我々の取締役会は、会社の持続的な発展に伴い、経営陣の独立した監督を確保するために、最適な取締役会指導構造を決定することが非常に重要であることを認識している。私たちはこの2つの役割の違いを認識するために、CEOと取締役会長の役割を分けた。CEOは会社の戦略方向や会社の日常的な指導や業績の策定を担当し、取締役会議長は最高経営責任者に指導を提供し、全体取締役会会議を主宰する。私たちは、このような職責分離が管理取締役会と監督会社にバランスのとれた方法を提供すると信じている。
私たちの取締役会の結論は、私たちの現在のリーダーシップがこの時点で適切だということだ。しかし、私たちの取締役会は、私たちのリーダーシップを定期的に検討し続け、将来的に適切だと思う変化をするかもしれません。
112
リスク監視過程における取締役会の役割
私たちの取締役会は、私たちのリスク管理手続きを監督し、定期的に経営陣と私たちの主要なリスクの開放、それらが私たちの業務に与える潜在的な影響、そして私たちが取った管理ステップを討論する責任があります。リスク監督手続きには、取締役会委員会と上級管理職メンバーの定期的な報告を受け、私たちの取締役会が潜在的な重大なリスク分野に関するリスク識別、リスク管理とリスク緩和戦略を理解できるようにし、運営、財務、法律、監督、戦略と名声リスクを含む。
監査委員会は流動性と運営に関する情報を審査し、金融リスクの管理を監督する。監査委員会はリスク評価、リスク管理、損失防止、コンプライアンスに関する私たちの政策を定期的に検討している。監査委員会の監督には、私たちの外部監査師と直接コミュニケーションをとり、経営陣と重大なリスクの開放及び管理層がこのような開口を制限、監視或いは制御するための行動を討論することが含まれている。報酬と人材委員会は、私たちの任意の報酬政策や計画が過度な冒険を奨励する可能性があるかどうかを評価する責任がある。指名と会社管理委員会は、取締役会の独立性、会社開示やり方、潜在的利益衝突に関するリスクを管理する。各委員会は何らかのリスクを評価し、これらのリスクの管理を監督する責任があるが、取締役会全体が定期的に委員会の報告書を通じてこれらのリスクを理解している。重大な戦略的リスク事項は当社取締役会全体で考えています。
取締役会各委員会
私たちの取締役会の常務委員会は、監査委員会、報酬委員会、指名、会社管理委員会を含み、各委員会は私たちの取締役会が承認した定款に基づいて運営されています。
監査委員会
監査委員会の主な機能は、私たちの会計と財務報告手続きを監督し、私たちの総合財務諸表の監査を監督することです。他の事項を除いて、この委員会の義務は以下のことを含む
113
私たちの監査委員会のメンバーは、エルリッヒさん、ガラゲル博士、およびロイドさんです。ロイドさんはその委員会の議長です。私たちの監査委員会のすべてのメンバーは、アメリカ証券取引委員会とナスダックが適用する規制の金融知識に対する要求に適合しています。当社の取締役会はすでに、Loewyさんはアメリカ証券取引委員会の上場規則を適用して定義された“監査委員会財務専門家”であり、ナスダック上場基準を適用して定義された必要な財務経験を備えていると認定した。当社取締役会は、米国証券取引委員会とナスダックの適用規則に基づき、エルリッヒさん、ガラゲル博士、ルイ女史がそれぞれ独立していることを決定しました。
報酬委員会
私たちの給与委員会は私たちの官僚たちと職員たちの給与と福祉に関する政策を承認した。報酬委員会は、我々のCEOや他の役員の報酬に関連する企業目標および目標を承認し、これらの目標および目標に基づいてこれらの役員のパフォーマンスを評価し、これらの評価に基づいてこれらの役員の報酬を承認する。報酬委員会はまた私たちの株式計画に基づいて株式オプションと他の奨励金の発行を承認する。報酬委員会は、報酬委員会がその定款を遵守している場合を含む、報酬委員会及びそのメンバーの業績を少なくとも毎年検討して評価する。
私たちの報酬委員会のメンバーは、エルリッヒさん、ハリントン·スミスさん、クロンケル博士です。エルリッヒさんは同委員会の議長だ。当社の取締役会は、適用されるナスダック上場基準に基づき、エリヒ·さん、ハリントン·スミス、クレンク博士がいずれも独立した者であり、取引所法案の第16 B-3条の規則によって定義された“非従業員取締役”に属することを決定しました。
指名と会社管理委員会
指名及び会社管理委員会は、合資格者の取締役会メンバーへの就任を物色すること、当社年度株主総会(又は取締役を選出する株主特別会議)で取締役候補を選出すること、候補者を選択して当社取締役会及びその任意の委員会の空きを埋めることを含む、当社取締役会の取締役会の職責履行に協力する責任を負う。また、指名及び会社管理委員会はわが社の管理政策を監督し、管理事項について取締役会に報告及び提案を提出し、取締役会の評価を監督する。私たちが指名して会社統治委員会のメンバーはバートさんとガラゲル博士だ。バートさんはその委員会の議長を務めている。当社の取締役会は、適用されるナスダック上場基準に基づいて、バートさんとガラゲル博士が独立していることを認定した。
報酬委員会は内部の人と連動して参加する
私たちの給与委員会のメンバーの中の一人も私たちの職員や従業員ではない。当社には、当社の取締役会又は報酬委員会のメンバーの実体である取締役会又は報酬委員会のメンバーとして、現在、又は1人以上の行政者を務めている行政者はいない。
取締役会の多様性
著者らの指名及び企業管理委員会は毎年著者らの取締役会と一緒に取締役会全体及び個別メンバーに必要な適切な特質、技能及び経験を検討することを担当している。個別候補(新候補や現メンバーを含む)が当選または委任に適しているかどうかを評価する際には、指名および会社管理委員会や取締役会は、複数の要因を考慮する
114
我々の取締役会は、取締役会全体を背景に各個人を評価し、業務成功を最大限に向上させ、これらの異なる分野での異なる経験を利用して合理的な判断を行うことで株主の利益を代表するグループを構築することを目的としている。
“行動規範”と“道徳的規範”
我々は、我々のCEO、最高財務官、最高会計官または財務総監、または同様の機能を実行する者を含む、我々の役員、上級管理者、および従業員に適用される書面商業行為および道徳基準を採択した。私たちのビジネス行為と道徳基準は私たちのサイトの会社管理部分で得ることができます。サイトはwww.ffector.comです。さらに、私たちは、本規則の任意の条項の任意の改正または免除に関連するすべての開示を、法律またはナスダック上場基準によって要求されるすべての開示を私たちのウェブサイトで公表する予定です。我々のサイトアドレスへの引用は,我々のサイトに含まれる情報や我々のサイトを参照することで得られる情報の統合を構成していない.私たちはただ私たちのウェブサイトのアドレスを非活動的なテキストとして参照する。
情報技術EM 11.役員報酬。
本節では、以下の“報酬集計表”で指名された役員報酬計画の主要な構成要素について議論する。2023年、私たちの“指名された幹部”とその地位は以下の通りです
本議論は、私たちの現在の計画、考慮事項、予想、および将来の報酬計画の決定に基づく前向きな陳述を含むことができる。私たちが将来採用する実際の報酬計画は、本議論でまとめた現在の計画計画とは大きく異なる可能性がある。
2024年1月12日、同社はその普通株に対して25株1株の逆株式分割(“逆株分割”)を行った。別の説明がない限り、本プロジェクト第11項に規定される1株当たり金額および行権価格は、逆株式分割に対してトレーサビリティを有する。
115
報酬総額表
次の表は,2023年12月31日終了年度と2022年12月31日終了年度の間に,指名された実行幹事がサービス提供により獲得,稼いだり支払ったりした報酬総額をまとめたものである.
名称と主要ポスト |
|
年.年 |
|
|
給料(元) |
|
|
ボーナス(ドル)(1) |
|
|
オプション賞(ドル)(2) |
|
|
非持分インセンティブ計画報酬(ドル)(3) |
|
|
その他すべての補償(元)(4) |
|
|
合計する |
|
|||||||
スティーヴン·T·ウォラン博士 |
|
|
2023 |
|
|
|
594,700 |
|
|
|
— |
|
|
|
343,544 |
|
|
|
237,880 |
|
|
|
41,381 |
|
|
|
1,217,505 |
|
社長と最高経営責任者 |
|
|
2022 |
|
|
|
566,500 |
|
|
|
— |
|
|
|
2,987,797 |
|
|
|
254,295 |
|
|
|
34,460 |
|
|
|
3,843,052 |
|
マイケル·バーンズ |
|
|
2023 |
|
|
|
450,000 |
|
|
|
— |
|
|
|
171,776 |
|
|
|
171,000 |
|
|
|
33,817 |
|
|
|
826,593 |
|
首席財務官 |
|
|
2022 |
|
|
|
435,735 |
|
|
|
— |
|
|
|
1,351,611 |
|
|
|
156,865 |
|
|
|
31,090 |
|
|
|
1,975,301 |
|
ダグラス·ワーナー医学博士 |
|
|
2023 |
|
|
|
474,308 |
|
|
|
— |
|
|
|
85,880 |
|
|
|
151,779 |
|
|
|
34,073 |
|
|
|
746,040 |
|
首席医療官 |
|
|
2022 |
|
|
|
179,135 |
|
|
|
50,000 |
|
|
|
407,562 |
|
|
|
64,356 |
|
|
|
10,909 |
|
|
|
711,962 |
|
補償表に対する叙述的開示
基本給
指名された行政人員の給与は一般的に毎年年初に決定と承認され、あるいは比較した場合、行政人員が雇用を開始する時に当社の取締役会或いは報酬委員会が決定し、承認する。以下は、任命された執行幹事が2023年に発効した基礎給である。
名前.名前 |
|
2023年の年間基本給(ドル) |
|
|
スティーヴン·T·ウォラン博士 |
|
|
594,700 |
|
マイケル·バーンズ |
|
|
450,000 |
|
ダグラス·ワーナー医学博士 |
|
|
474,308 |
|
116
ボーナス補償
年間現金激励ボーナスは私たちの総給与計画の重要な構成部分であり、幹部を維持するために必要な激励を提供すると考えられる。指名された行政人員1人当たり、指定された目標年間ボーナス金額に基づいて、業績別に計算された年間現金ボーナスを獲得する資格があり、その金額は指名された行政者の基本給の割合で表される。以下は、任命された実行幹事の2023年の基本給の目標率である
名前.名前 |
|
2023年の目標率 |
スティーヴン·T·ウォラン博士 |
|
50% |
マイケル·バーンズ |
|
40% |
ダグラス·ワーナー医学博士 |
|
40% |
毎年のボーナスは通常次の年の第1四半期に確定されて支給される。2023年に、我々の取締役会は、当社の目標を検討し、検討後、2023年におけるこれらの目標に対する我々の進展の評価に基づいて、任命された幹部に目標の80%に年間ボーナスを支払うことを決定した。これらの目標は、臨床、財務、監督、企業発展目標に基づいている。バーンズ2023年の年間ボーナスは、個人の2023年のパフォーマンスに応じてさらに目標の95%に引き上げられた。指定された役員に支払われた2023年の年間ボーナスは、上記の給与要約表に反映されます。
持分激励賞
私たちの株式インセンティブ奨励は、私たちの利益と株主の利益を私たちの従業員やコンサルタント(任命された役員を含む)の利益と一致させることを目的としています。取締役会は株式贈与を承認する責任がある。
Old Efftorは、業務合併が完了する前に、2013年の株式インセンティブ計画(“2013計画”)を維持している。2013年計画によると、Old Efftorは、私たちが指定した役員を含む適格サービスプロバイダに株式オプション報酬を提供して、その普通株を購入します。業務合併完了とエフェクター治療会社による2021年インセンティブ奨励計画(“2021年計画”)については、2013年計画では他の奨励は付与されなくなる。
すべてのオプションの発行価格は、付与株当日の私たちの普通株の公平な市価を下回らない。私たちの株式オプション奨励は、一般に4年以内に付与され、以下の“任命された幹部との雇用配置”で説明されるように、いくつかの終了および統制権変更事件の下で加速的に付与および行使される可能性がある
2023年1月ウォランダー博士、バーンズさん、ワーナー博士私たちの2021計画によると、一人当たり毎年それぞれ40,000,20,000件と10,000件の株式オプションを獲得します。株式オプションの発行価格は1株11.75ドルで、これは私たち普通株の付与日の終値です。当該等株式オプションは、授出日から4年以内に帰属し、2023年1月17日の毎月記念日に当該等オプションに拘束されている株式総数の1/48に帰属するが、適用される帰属日内に吾等にサービスを提供し続ける必要がある。
2024年2月ウォランダー博士、バーンズさん、ワーナー博士私たちの2021計画によると、一人当たり毎年それぞれ80,000,40,000件と25,000件の株式オプションを獲得しています。株式オプションの発行価格は1株11.32ドルで、これは私たち普通株の付与日の終値です。この等株式オプションは、授出日から4年以内に帰属し、2024年2月19日の毎月1周年日に当該オプションに拘束されている株式総数の1/48に帰属するが、適用される帰属日内にサービスを提供し続けなければならない。
任命された行政員との採用手配
私たちは私たちが指定したすべての幹部と雇用協定を締結する側だ。この等の手配は一般的にいかなる特定の条項がない場合に任意に採用することができ、任命された行政人員の初期基本賃金、ボーナス潜在力、資格に符合する従業員福祉及び資格に符合して雇用を終了する時の解散費福祉を明らかにすることができるが、この従業員は吾などと離職協定に署名しなければならない。
117
Worland博士との雇用契約は
私たちはウォラン博士、私たちの総裁、最高経営責任者と雇用協定を締結した。彼の合意によると、Worland博士は年間基本給と目標年間奨励金を得る権利があり、それぞれの場合は私たちの取締役会の報酬委員会が時々制定している。
彼の雇用合意によると、Worland博士が“支配権変更”の前または“支配権変更”後12ヶ月(それぞれの雇用合意における定義)を超えて無断で雇用を終了した場合、(1)12ヶ月の基本給の支払いを継続する権利と、(2)解雇された後にCOBRA下の持続健康福祉の保険料を支払う権利があり、最長12ヶ月となる。
彼の雇用合意によると、“支配権変更”後12ヶ月以内に“原因”がない場合や“十分な理由”があればWorland博士の雇用を終了する権利があり、(1)18ヶ月の基本給の支払いを継続する権利があり、(2)解雇された後、COBRAにより最大18ヶ月の持続健康福祉保険料を支払うこと、(3)今年度の目標ボーナスに相当する150%の金額を支払うこと、(4)未返済の持分奨励金の帰属を全面的に加速させることができる。
Worland博士の福祉の条件の1つは,1年間の非募集義務を含む合意規定の終了後義務を遵守し,我々に有利な包括的クレーム声明に速やかに署名することである。
バーンズさんとの雇用契約
我々はすでに我々の最高財務官バーンズさんと雇用契約を締結しました。彼の合意によると、Byrnesさんは、当社の取締役会の報酬委員会によって時々策定される年間基本給および目標年次報酬ボーナスを得る権利があります。
彼の雇用契約によれば、Byrnesさんが“支配権変更”の前または“支配権変更”後12ヶ月(それぞれその雇用契約で定義される)を超えて理由なく雇用を終了した場合、彼は、(1)9ヶ月間の基本給の支払いを継続し、(2)彼が解雇された後、COBRAの下で最大9ヶ月の持続的健康保険料を彼に支払う権利がある。
彼の雇用契約によれば、バーンズさんが“支配権変更”の日またはその後12ヶ月以内に理由なく採用を終了したり、バーンズさんによって“十分な理由”で採用を終了したりすれば、(1)12ヶ月間の基本給の支払い継続、(2)解雇後COBRAによる最長12ヶ月間の持続的健康福祉保険料の支払い、(3)現在の年間目標ボーナスに相当する金額の支払い、および(4)すべての未償還配当金の帰属を全面的に加速させる権利がある。
このほかの事項のほか、バーンズさんは、契約終了後の彼の契約の規定を遵守する義務、非募集義務を含む契約終了後の義務、および当社にとって有利な包括的なクレーム声明書に速やかに署名することを条件としています。
ワーナー博士との雇用協定
私たちは私たちの首席医療官ワーナー博士と雇用協定を締結した。ワーナー博士の合意によると、ワーナー博士は年間基本給と目標年度奨励金を得る権利があり、両者とも私たちの取締役会の報酬委員会が時々制定している。彼の雇用協定によると、ワーナー博士は5万ドルの一次契約ボーナスを獲得した。
彼の雇用合意によると、ワーナー博士が“支配権変更”の前に無断で雇用を中止されたり、“支配権変更”の後12ヶ月を超えたり(いずれも彼の雇用協定で定義されている)ならば、(1)12ヶ月の基本給の支払いを継続する権利と、(2)解雇された後、COBRAにより最大12ヶ月の持続健康福祉の保険料を支払う権利がある。
118
彼の雇用契約によると、ワーナー博士が“支配権変更”の日またはその後12ヶ月以内に無断で採用を中止されたり、ワーナー博士に“十分な理由”で採用を中止されたりすれば、(1)12ヶ月の基本給の支払いを継続する権利があり、(2)終了後12ヶ月以内にCOBRAによる持続健康福祉保険料の支払い、(3)今年度の目標ボーナスに相当する金額の支払い、および(4)すべての未償還持分奨励の帰属を全面的に加速させる権利がある。
ワーナー博士の福祉の条件の1つは,1年間の非募集義務と,我々に有利な包括的クレーム声明にタイムリーに署名することを含む合意規定を遵守した契約終了後の義務である。
財政年度終了時の優秀株奨励
次の表は、任命された幹部に付与された2023年12月31日現在も返済されていない株式報酬に関するいくつかの情報を示しており、これらの情報は、逆株式分割に関連するこのような報酬の変換を反映するように調整されている。
|
|
オプション賞(1)(2) |
||||||||||||||
名称と主要ポスト |
|
授与日 |
|
行使可能な未行使オプション対象証券数 |
|
|
行使できない未行使オプションの証券数(#) |
|
|
オプション取引権価格 |
|
|
オプション期限 |
|||
スティーヴン·T·ウォラン博士 |
|
12/4/2014 |
|
|
8,691 |
|
|
|
— |
|
|
|
18.25 |
|
|
12/4/2024 |
|
|
1/8/2016 |
|
|
24,760 |
|
|
|
— |
|
|
|
28.50 |
|
|
1/8/2026 |
|
|
2/17/2016 |
|
|
5,794 |
|
|
|
— |
|
|
|
28.50 |
|
|
2/17/2026 |
|
|
8/21/2017 |
|
|
11,588 |
|
|
|
— |
|
|
|
41.50 |
|
|
8/21/2027 |
|
|
1/20/2022 |
|
|
9,979 |
|
|
|
10,847 |
|
|
|
166.25 |
|
|
1/20/2032 |
|
|
6/20/2022 |
|
|
15,620 |
|
|
|
5,207 |
|
|
|
37.25 |
|
|
6/20/2032 |
|
|
1/17/2023 |
|
|
9,166 |
|
|
|
30,833 |
|
|
|
11.75 |
|
|
6/20/2032 |
マイケル·バーンズ |
|
12/10/2020 |
|
|
10,139 |
|
|
|
3,380 |
|
|
|
59.75 |
|
|
12/10/2030 |
|
|
1/20/2022 |
|
|
4,514 |
|
|
|
4,907 |
|
|
|
166.25 |
|
|
1/20/2032 |
|
|
6/20/2022 |
|
|
7,066 |
|
|
|
2,355 |
|
|
|
37.25 |
|
|
6/20/2032 |
|
|
1/17/2023 |
|
|
4,583 |
|
|
|
15,417 |
|
|
|
11.75 |
|
|
6/20/2032 |
ダグラス·ワーナー医学博士 |
|
8/8/2022 |
|
|
8,242 |
|
|
|
16,484 |
|
|
|
22.44 |
|
|
8/8/2032 |
|
|
1/17/2023 |
|
|
2,291 |
|
|
|
7,708 |
|
|
|
11.75 |
|
|
6/20/2032 |
補償の他の要素
追加手当、健康、福祉
任命された役員は,私たちの医療,歯科,視力,団体人寿,障害や事故死および肢解保険計画を含む私たちの従業員福祉計画に参加する資格があり,いずれの場合も,これらの計画の基礎は我々他のすべての従業員と同じである。
119
私たちは一般的に任命された幹部に追加手当や個人福祉を提供しない。しかし、私たちは指定された幹部を含めて、私たちのすべての従業員に定期生命保険と障害保険の保険料を支払っています。もし私たちの取締役会がそうすることが私たちの最適な利益に合っていると思うなら、私たちの取締役会は将来的に保留または保留のある福祉計画を採用することを選択することができます。
401(K)計画
私たちは401(K)計画を提供し、条件に合ったアメリカ人従業員に税金優遇に基づいて退職貯蓄の機会を提供する。条件に適合する従業員は、条件を満たす報酬を毎年更新される特定のコード制限に延期することができる。401(K)計画の目的は、規則第401(A)節の規定に適合することであり、関連信託計画は、規則501(A)節の規定により免税である。納税条件に適合した退職計画として、401(K)計画への供出金は、作成時に差し引くことができ、401(K)計画から抽出または分配する前に、これらの金額の供出および収入は、通常、従業員に納税すべきではない。私たちは、401(K)計画によって退職退職貯蓄を繰延するツールを提供することによって、私たちの役員報酬スキームの全体的な収益性を増加させ、私たちの給与政策に基づいて、私たちが指定した役員を含む私たちの従業員をさらに激励すると信じている。
非限定延期補償
私たちは不合格の固定支払い計画や他の不合格の繰延給与計画を維持しない。もし私たちがそうすることが私たちの最適な利益に合致すると思うなら、私たちは未来に私たちの職員たちおよび他の従業員に非限定固定貢献または他の非限定繰延補償福祉を提供することを選択することができる。
無税毛収入
私たちは、私たちが支払ったり提供したりする報酬や福祉に関連する指定された役員の個人所得税を支払わない。
払戻政策
私たちは、テレス·フランク法に基づいて実施された新アメリカ証券取引委員会規則とニューヨーク証券取引所上場基準に基づいて要求された、2023年10月2日以降に受けたいくつかの誤った支払いのインセンティブ報酬を取り戻すための補償回収政策をとっている。
役員報酬
下表は、非従業員取締役が2023年12月31日までの年間で受け取った報酬をまとめたものである。私たちのCEO兼最高経営責任者総裁博士も私たちの取締役会のメンバーですが、従業員として得られた報酬を除いて、取締役としてのサービスは何の追加報酬も得られません。ウォランダー博士の報酬は上にさらに説明されている。
名前.名前 |
|
稼いだ費用や |
|
|
オプション大賞 |
|
|
株式大賞 |
|
|
他のすべての補償(ドル) |
|
|
合計する |
|
|||||
ブライアン·ガラゲル |
|
|
81,500 |
|
|
|
28,320 |
|
|
|
— |
|
|
|
— |
|
|
|
109,820 |
|
ジョン·スミス(3) |
|
|
40,653 |
|
|
|
23,730 |
|
|
|
— |
|
|
|
— |
|
|
|
64,383 |
|
キャロライン·ロイド(4) |
|
|
16,914 |
|
|
|
18,786 |
|
|
|
— |
|
|
|
— |
|
|
|
35,700 |
|
バーバラ·クロンク |
|
|
45,000 |
|
|
|
20,871 |
|
|
|
— |
|
|
|
— |
|
|
|
65,871 |
|
クリス·エルリッヒ |
|
|
57,500 |
|
|
|
23,420 |
|
|
|
— |
|
|
|
— |
|
|
|
80,920 |
|
エリザベス·バート |
|
|
52,000 |
|
|
|
21,484 |
|
|
|
— |
|
|
|
— |
|
|
|
73,484 |
|
クリスチャン·ハリントン·スミス |
|
|
45,000 |
|
|
|
20,871 |
|
|
|
— |
|
|
|
— |
|
|
|
65,871 |
|
120
名前.名前 |
|
2023年12月31日現在返済されていない証券標的オプション数 |
|
|
ブライアン·ガラゲル |
|
|
5,237 |
|
ジョン·スミサー |
|
|
3,692 |
|
キャロライン·ロイド |
|
|
1,600 |
|
バーバラ·クロンク |
|
|
4,325 |
|
クリス·エルリッヒ |
|
|
4,637 |
|
エリザベス·バート |
|
|
4,400 |
|
クリスチャン·ハリントン·スミス |
|
|
4,058 |
|
役員報酬計画
我々の取締役会は非従業員役員報酬計画(“役員報酬計画”)を承認した。役員報酬計画は非従業員取締役に年間事前招聘費と長期持分奨励を提供する。2024年1月、私たちの取締役会は、以下に述べるように、取締役会議長/取締役独立首席メンバーおよび報酬委員会メンバーの年間事前招聘費の増加を許可しました。
役員報酬計画は以下の部分からなる
現金補償
年間現金費用は四半期分期に滞納し、サービスの任意の部分カレンダー四半期に比例して計算される。
121
持分補償
2024年2月19日、取締役会は非従業員メンバーごとに2,000株を購入する追加株式オプション奨励を獲得した。これらの奨励は授与日から12ヶ月以内に月等額で分割払いになるが、非従業員取締役が適用されるまでサービスを継続する必要がある。
取締役会が適宜決定しない限り、株主総会又は株主周年総会の前6ヶ月以内に初当選した非従業員取締役は、当該年次総会に関する年間奨励を受けることなく、予備奨励のみを受けることになる。また、各初期奨励及び年間奨励、並びに2023年1月に非従業員取締役に付与された奨励は、“2021年計画”で定義された制御権変更が発生する直前に全数が当時完成していない程度に帰属しなければならない。
以上のように、我々役員報酬計画下の報酬は、2021年計画で規定されている非従業員役員報酬の年間制限を受ける。
情報技術EM 12.特定の実益所有者の保証所有権および管理層および関連する株主事項。
証券の実益所有権
次の表は、2024年2月29日までの普通株式の利益所有権を示している
普通株の実益所有権は、2024年2月29日現在の3,687,309株に基づいて発行された普通株。
実益所有権は、現在2024年2月29日から60日以内に行使可能なオプションおよび引受権を含む、1人が1つの証券に対して単独または共有された投票権または投資権を有する場合、その証券の実益所有権を有することが一般的に規定されている米国証券取引委員会の規則に基づいて決定される。
122
他に説明がない限り、私らは次の表に列挙されたすべての人々がその実益を持つ投票権のある証券が唯一の投票権と投資権を持っていると信じている。
実益所有者の氏名又は名称及び住所(1) |
|
株式数 |
|
|
所有権の割合 |
|
5%保有者 |
|
|
|
|
|
|
アビンワース生物ベンチャー会社VI,L.P.(2) |
|
|
193,195 |
|
|
5.2% |
役員および行政員 |
|
|
|
|
|
|
スティーヴン·ウォランド博士(3) |
|
|
130,652 |
|
|
3.5% |
マイケル·バーンズ(4) |
|
|
34,848 |
|
|
* |
ダグラス·ワーナー医学博士(5) |
|
|
14,469 |
|
|
* |
エリザベス·バート(6) |
|
|
4,116 |
|
|
* |
クリス·エルリッヒ(7) |
|
|
11,128 |
|
|
* |
ブライアン·ガラゲル(8歳) |
|
|
5,792 |
|
|
* |
クリスチャン·ハリントン·スミス(9) |
|
|
3,146 |
|
|
* |
バーバラ·クロンク(10歳) |
|
|
3,591 |
|
|
* |
キャロライン·ロイド(11歳) |
|
|
1,005 |
|
|
* |
全役員及び執行幹事(9名)(12名) |
|
|
208,747 |
|
|
5.4% |
*1%未満
123
株式補償計画に基づいて発行された証券(2023年12月31日現在)
次の表は、2023年12月31日現在、私たちの株式証券の発行を許可した補償計画に関する情報を提供します
計画種別 |
|
未償還オプション,株式承認証及び権利を行使する際に発行される証券数 |
|
|
未満期オプションの加重平均行権価格 |
|
|
株式補償計画の下で将来発行可能な証券の数 |
|
|
|||
持分補償計画 |
|
|
540,019 |
|
(2) |
$ |
51.67 |
|
(3) |
|
155,593 |
|
(4) |
株式報酬計画はありません |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
合計する |
|
|
540,019 |
|
|
$ |
51.67 |
|
|
|
155,593 |
|
|
2021年計画によると、発行可能な普通株式数は、2031年までの例年の1月1日に年ごとに増加し、額は(I)前年の最終日に発行された株式の5%または(Ii)が自社取締役会で決定した少ない株式に相当する。2024年1月1日から、2021年計画により発行可能な株式数は149,733株増加し、上の表に反映されていない。
また、ESPPによると発行可能な普通株式数は、2031年までの例年の1月1日に毎年増加し、額は(A)前の日付の最終日に発行された普通株式数の1%または(B)当社取締役会が決定した少ない数に相当する。2024年1月1日から、ESPPにより発行可能な株式数は29,946株増加し、上の表に反映されていない。
124
情報技術EM 13.何らかの関係や関連取引,および取締役の独立性.
以下は、2022年1月1日以降に我々が参加している取引の概要であり、金額が120,000ドルを超えるか、またはそれを超える(2023年12月31日を下回る場合、私たちの総資産平均金額の1%を超える)、これらの取引のうち、任意の取締役、役員、または私たちの知る限り、5%を超える株式の実益所有者または前述の者のいずれかの直系親族または直接または間接的な重大な利益(持分およびその他の報酬を除く)、終了、支配権変更、およびその他の手配について説明する。また、以下では、取締役、役員、株主との他の取引についても説明します。
登録権協定
業務合併完了後,吾等の5%を超える普通株を持ついくつかの役員,上級職員及び実体は改訂及び再予約された登録権協定を締結し,この合意により,彼等は吾等が保有する任意の証券の転売について登録する権利を有する。また、リンカーン公園との購入協定について、リンカーン公園は登録権協定を締結し、この協定によると、彼らは登録権を獲得する権利があり、彼らが持っている任意の証券の転売を登録することを要求している。登録権協定に関するその他の情報は、添付の“登録証券説明”添付ファイルを参照してください。
会社登録証明書と改訂された付例による賠償
我々の規約では,DGCLが許容する最大限に我々の役員や上級管理者に賠償を行うが,定款に含まれるいくつかの例外は除外する。また、会社登録証明書は、私たちの取締役は受託責任に違反した金銭損害賠償責任を負わないと規定しています。
私たちはまた私たちのすべての幹部と役員と賠償協定を締結した。賠償協定は、賠償、立て替え費用および精算を得るために被賠償者に契約権利を提供するが、DGCLが許容する最大範囲内であるが、これらの合意に含まれるいくつかの例外は除外される。より多くの情報については、“我々の証券説明−上級管理者及び取締役の責任制限及び賠償”を参照されたい
関連者取引政策
我々の取締役会は、書面による関係者取引政策を採択し、関連者取引の審査及び承認又は承認のための政策及び手続を規定している。本政策は、任意の取引、手配または関係、または吾などがかつてまたは参加しようとしていた任意の類似の取引、手配または関係をカバーするが、証券法の下でS-K条例第404項に記載されている例外を除いて、関連する金額は120,000ドルを超えるか、または過去の2つの完全会計年度の私たちの総資産平均値の1%を超え、関係者は、関連者または実体が商品またはサービスを購入することを含むが、その関係者または実体がこれらの取引、手配または関係において重大な利益、債務、またはサービスを有することがあるが、これらに限定されない。私たちは関係者たちの債務と雇用に対する保証を提供する。このような取引を承認する際に、我々の監査委員会の任務は、取引の条項が公平な取引の条項に相当するか否か、および取引における関係者の権益の程度を含むすべての関連する事実および状況を考慮することである。
役員は自主独立している
取締役独立性に関する情報は、本年度報告第3部第10項を参照して、引用により本明細書に組み込む。
125
イットM 14.主な会計費用およびサービス。
独立公認会計士事務所は有料です
安永会計士事務所(“安永会計士事務所”)は2013年からEnfftorの独立公認会計士事務所を務めている。
次の表には、安永が2023年度と2022年度に徴収した費用と支出総額を示しています
|
|
2023 |
|
|
2022 |
|
||
課金(1) |
|
|
549,523 |
|
|
|
763,800 |
|
税金(2) |
|
|
31,209 |
|
|
|
33,990 |
|
他のすべての費用 |
|
|
— |
|
|
|
— |
|
合計する |
|
|
580,732 |
|
|
|
797,790 |
|
承認前の政策と手順
我々の監査委員会は、我々の独立公認会計士事務所が提供するすべての監査及び許可された非監査サービスが事前に監査委員会の承認を受ける政策を策定しており、このようなサービスは、2021年12月31日までの会計年度にこの政策に基づいて予め承認されている。これらのサービスは、監査サービス、監査関連サービス、税務サービス、および他のサービスを含むことができる。監査委員会はすべての非監査サービスを提供することが私たちの監査人の独立性を維持することに適合しているかどうかを考慮している。事前承認は特定のサービスやサービス種別に関する詳細な説明であり,通常特定予算の制約を受ける.我々の独立公認会計士事務所及び経営陣は、独立公認会計士事務所が本予め承認したサービスの程度、及びこれまで提供されてきたサービスの費用を監査委員会に定期的に報告しなければならない。
126
第4部
情報技術EM 15.証拠物、財務諸表明細書
本年度報告はF−1ページから,Efftor Treateutics,Inc.の合併財務諸表および独立公認公共会計士事務所安永会計士事務所の報告を含む。
その中に記載されている資料が連結財務諸表または付記に適用または表示されないことが要求されるので、すべての付表は省略される。
本年度報告署名ページの前の“展示品索引”には、展示品リストが記載されており、参照されて本明細書に組み込まれている。
情報技術EM 16.表格10-Kの概要
ない。
127
エフェクター治療会社
財務諸表索引
|
ページ |
|
|
独立公認会計士事務所レポート(PCAOB ID: |
F-2 |
合併貸借対照表 |
F-3 |
合併経営報告書と全面赤字 |
F-4 |
合併株主権益報告書(損失) |
F-5 |
統合現金フロー表 |
F-6 |
連結財務諸表付記 |
F-7 |
F-1
独立公認会計士事務所報告
エフェクターは会社の取締役会と株主を治療する。
財務諸表のいくつかの見方
効果治療会社(当社)の2023年12月31日と2022年12月31日までの連結貸借対照表、2023年12月31日までの2年間の毎年の関連総合経営と全面赤字報告書、株主権益(損失)と現金流量および関連付記(総称して“総合財務諸表”と呼ぶ)を監査した。総合財務諸表は、すべての重要な点で、2023年12月31日、2023年12月31日、2022年12月31日までの会社の財務状況、および2023年12月31日までの2年度の経営結果とキャッシュフローを公平に反映しており、米国公認会計原則に適合していると考えられる。
会社が経営を続ける企業として経営を続ける能力
添付されている総合財務諸表の作成仮説会社は引き続き経営を継続する企業となる。財務諸表付記1で述べたように,2023年の会社純損失は,設立以来の経営活動によるキャッシュフローは負であり,会社の持続経営企業としての継続経営能力には大きな疑問があることを示している。付記1はまた、これらの事項に関する経営陣のイベントや条件の評価、経営陣の計画について説明しています。合併財務諸表には、このような不確実性の結果生じる可能性のある調整は含まれていません。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査することを求められておらず、私たちも招聘されて監査を行っていません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性に対する意見を表明するためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
/s/
2013年以来、当社の監査役を務めてきました。
2024年3月26日
F-2
第I部--FIN金融情報
イットM 1.財務諸表
エフェクター治療会社
合併貸借対照表
(千単位、株式及び額面データを除く)
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
短期投資 |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
流動資産総額 |
|
|
|
|
|
|
||
財産と設備、純額 |
|
|
|
|
|
|
||
経営的リース使用権資産 |
|
|
|
|
|
|
||
その他の資産 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
負債と株主権益(赤字) |
|
|
|
|
|
|
||
流動負債: |
|
|
|
|
|
|
||
売掛金 |
|
$ |
|
|
$ |
|
||
費用を計算する |
|
|
|
|
|
|
||
当期ローン,純額 |
|
|
|
|
|
|
||
定期ローン末期の支払いを計算しなければならない |
|
|
|
|
|
|
||
賃貸負債、流動部分 |
|
|
|
|
|
|
||
流動負債総額 |
|
|
|
|
|
|
||
その他の非流動計算負債 |
|
|
|
|
|
|
||
負債をかせぐ |
|
|
|
|
|
|
||
非流動株式証負債 |
|
|
|
|
|
|
||
非流動賃貸負債 |
|
|
|
|
|
|
||
総負債 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
株主権益(赤字): |
|
|
|
|
|
|
||
優先株、$ |
|
|
|
|
|
|
||
普通株、$ |
|
|
|
|
|
|
||
追加実収資本(1) |
|
|
|
|
|
|
||
その他の総合損失を累計する |
|
|
|
|
|
( |
) |
|
赤字を累計する |
|
|
( |
) |
|
|
( |
) |
株主権益合計 |
|
|
( |
) |
|
|
|
|
総負債と株主権益(赤字) |
|
$ |
|
|
$ |
|
||
(1)
付記はこれらの連結財務諸表の構成要素である。
F-3
エフェクター治療会社
合併経営報告書と全面赤字
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
奨学金収入 |
|
$ |
|
|
$ |
|
||
運営費用: |
|
|
|
|
|
|
||
研究開発 |
|
|
|
|
|
|
||
一般と行政 |
|
|
|
|
|
|
||
総運営費 |
|
|
|
|
|
|
||
営業損失 |
|
|
( |
) |
|
|
( |
) |
その他の収入(費用) |
|
|
|
|
|
|
||
利子収入 |
|
|
|
|
|
|
||
利子支出 |
|
|
( |
) |
|
|
( |
) |
その他の収入,純額 |
|
|
( |
) |
|
|
( |
) |
収益負債公正価値変動 |
|
|
|
|
|
|
||
その他収入合計 |
|
|
( |
) |
|
|
|
|
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
総合的な損失: |
|
|
|
|
|
|
||
純損失 |
|
|
( |
) |
|
|
( |
) |
その他全面収益(赤字) |
|
|
|
|
|
( |
) |
|
総合的な損失: |
|
$ |
( |
) |
|
$ |
( |
) |
1株当たり基本と希釈して純損失(1) |
|
$ |
( |
) |
|
$ |
( |
) |
加重平均発行済み普通株式、基本普通株式、希釈後普通株(1) |
|
|
|
|
|
|
||
(1)金額は、2024年1月12日に発効した25株1株逆分割(付記1.連結財務諸表付記の組織および列報基礎参照)を反映するためにさかのぼって記載されている。
付記はこれらの連結財務諸表の構成要素である。
F-4
エフェクター治療会社
合併株主権益報告書(損失)
(単位:千、共有データを除く)
|
|
|
|
|
|
|
|
積算 |
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
その他の内容 |
|
|
他にも |
|
|
|
|
|
合計する |
|
||||||
|
|
普通株(1) |
|
|
支払い済み |
|
|
全面的に |
|
|
積算 |
|
|
株主の |
|
|||||||||
|
|
株 |
|
|
金額 |
|
|
資本(1) |
|
|
収入(損) |
|
|
赤字.赤字 |
|
|
権益(赤字) |
|
||||||
2021年12月31日の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||||
株式オプション権 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
普通株発行は発行コストを差し引く |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
短期投資の未実現損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2022年12月31日の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
株式オプション権 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
普通株発行、発行コストを差し引く--登録直接発行 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
前払い資金承認証を行使する--登録直接発売 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
普通株発行は発行コストを差し引く |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
短期投資の未実現収益 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2023年12月31日の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
||||
(1)
付記はこれらの連結財務諸表の構成要素である。
F-5
エフェクター治療会社
統合現金フロー表
(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
経営活動: |
|
|
|
|
|
|
||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
純損失と業務活動で使用されている現金の照合の調整: |
|
|
|
|
|
|
||
減価償却および償却費用 |
|
|
|
|
|
|
||
投資割引と償却保険料を増やし,純額 |
|
|
( |
) |
|
|
( |
) |
株に基づく報酬 |
|
|
|
|
|
|
||
資産の損失(収益)を処分する |
|
|
( |
) |
|
|
|
|
株式証負債の公正価値変動収益 |
|
|
|
|
|
( |
) |
|
収益負債公正価値変動収益 |
|
|
( |
) |
|
|
( |
) |
持分購入契約に関するその他の費用 |
|
|
|
|
|
|
||
非現金利子支出 |
|
|
|
|
|
|
||
経営性資産と負債変動状況: |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
他の非流動資産 |
|
|
|
|
|
|
||
売掛金 |
|
|
|
|
|
|
||
費用を計算する |
|
|
|
|
|
( |
) |
|
経営的リース使用権資産と負債純額 |
|
|
( |
) |
|
|
|
|
経営活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
投資活動: |
|
|
|
|
|
|
||
固定資産購入状況 |
|
|
( |
) |
|
|
( |
) |
短期投資満期日 |
|
|
|
|
|
|
||
短期投資を購入する |
|
|
( |
) |
|
|
( |
) |
投資活動提供の現金純額 |
|
|
|
|
|
( |
) |
|
融資活動: |
|
|
|
|
|
|
||
債務発行コストを支払う |
|
|
|
|
|
( |
) |
|
普通株式オプションと引受権証を行使して得られた金,純額 |
|
|
|
|
|
|
||
普通株式に登録された直接発行の収益,純額 |
|
|
|
|
|
|
||
ESPPを含む普通株の収益を発行し、発行コストを差し引く |
|
|
|
|
|
|
||
融資活動が提供する現金純額 |
|
|
|
|
|
|
||
現金および現金等価物の純増加(減額) |
|
|
|
|
|
( |
) |
|
期初現金及び現金等価物 |
|
|
|
|
|
|
||
期末現金および現金等価物 |
|
$ |
|
|
$ |
|
||
キャッシュフロー情報の追加開示: |
|
|
|
|
|
|
||
支払の利子 |
|
$ |
|
|
$ |
|
||
非現金投資と融資活動を追加開示します |
|
|
|
|
|
|
||
承諾株を発行する |
|
|
|
|
|
|
||
売掛金と売掛金に掲げる固定資産の購入 |
|
|
|
|
|
|
||
付記はこれらの連結財務諸表の構成要素である。
F-6
エフェクター治療会社
違います。TESから連結財務諸表まで
業務説明
バッタ買収会社(LWAC)は当初設立されました
2021年5月26日、LWACは、LWACの完全子会社、デラウェア州社Locust Walk Merge Sub,Inc.およびデラウェア州のEfftor Treateutics,Inc.(“Old Efftor”)と合併協定および合併計画(“合併協定”)を締結した。
合併合意の条項によると、LWACとOld Efftorの間の業務統合はSubとOld Efftorの合併によって実現され、Old Efftorは既存の会社とLWACの完全子会社となり、名称はEfftor Treeutics Operations,Inc.である。2021年8月25日、LWACは業務合併の終了に伴い、Efftor Treeutics,Inc.(“Efftor”または“会社”)と改称された。すべての旧エフェクターの発行済み優先株を1:1の割合で旧エフェクターの普通株に変換し、その後、これらの優先株を旧エフェクターのすべての発行済み普通株と共に適用することにより約
同社は臨床段階の生物製薬会社であり,新規腫瘍薬の開発に注力しており,選択的翻訳調節抑制薬(“STRI”)と呼ばれている。同社の主な業務は米国で、カリフォルニア州のソラーナビーチに本部を置いている。同社はすでにそのほとんどの資源を資金調達に応用し、潜在的な候補製品を確定し、その知的財産権の組み合わせを確立し、臨床前研究と臨床試験を行い、第三者とその候補製品と関連原材料の生産について手配を確立し、そしてこれらの業務に一般と行政支持を提供する。2023年12月31日現在、同社は許可と許可収入を除いて、その主要業務から収入を生み出していない。
株を逆分割する
2024年1月9日、会社は会社の普通株の逆分割を実現し、額面を$とするために、会社の改訂と再登録証明書の改訂証明書をデラウェア州州務卿に提出した
同社は2023年12月31日と2022年12月31日までの連結財務諸表に1株と1株当たりの金額をさかのぼって再報告した。すべての発行済み株式オプションと引受権証によって発行可能な普通株の1株当たりの発行権価格と株式数の割合調整を行った。また、会社の株式インセンティブ給与計画のために予約された普通株式数の割合調整を行った。総合株主権益(損失)表と貸借対照表は逆株式分割の影響を反映しており、“普通株”を“追加実収資本”に再分類し、金額は逆株式分割による株式減少の額面に等しい。
陳述の基礎
会社の総合財務諸表は米国公認会計原則(“GAAP”)に基づいて作成されている。
流動性
同社の経営歴史は限られており、会社の業務や市場の販売や収入の潜在力はまだ確認されていない。添付の総合財務諸表の作成仮説会社は、このような不確実性がもたらす可能性のある資産または金額の回収可能性および分類、ならびに負債分類の将来的な影響を反映するために、通常の業務プロセスにおいて資産現金化および負債補償を考慮するための継続的な経営企業として継続すると仮定する。
F-7
経営陣は持続経営企業としての持続経営能力を2段階分析する必要がある。経営陣は、会社の持続経営企業としての継続経営能力に重大な疑いを与える条件や事件が存在するかどうかをまず評価しなければならない(ステップ1)。経営陣が実質的な疑いを出したと結論すれば、経営陣はその計画がこの疑いを解消したかどうかも考慮する必要がある(ステップ2)。
同社の累積赤字は#ドルです
同社の持続的な経営企業としての持続的な経営能力は、追加資本を得る能力にかかっている。経営陣は、潜在的な追加協力、ライセンス、その他の同様の手配を含む、株式発行または他の資本源を通じてより多くの資本を調達しようとしている。さらに、会社は、ファイザー社との研究協力および許可協定(付記11参照)、リンカーンパーク資本基金有限責任会社(“リンカーンパーク”)との株式購入プロトコル(“購入契約”)から普通株を発行するか、またはCantor Fitzgerald&Co(“Cantor”)と市場発売計画(付記8に記載)に従って普通株を発行することによって、追加のマイルストーン支払いを得ることができる。しかし、同社はタイムリーまたは割引の条件で追加融資を受けることができない可能性があり、あれば何の記念碑的な支払いも得られない可能性がある。追加資本がない場合、会社は、いくつかの研究開発活動または他の業務の延期、削減、またはキャンセルを余儀なくされる可能性があり、運営を継続するために十分な資金を提供するために製品開発を延期することができ、または合併または買収戦略をとることが要求される可能性があり、これらは、その株主の持株または権利に悪影響を及ぼす可能性がある。
予算の使用
会社の総合財務諸表は、米国公認の会計原則(“公認会計原則”)に基づいて作成されている。当社の総合財務諸表を作成する際には、当社の総合財務諸表及び付記に報告されている資産、負債及び費用金額及び負債の開示に影響を与えるための推定及び仮定を行う必要がある。同社の連結財務諸表の中で最も重要な見積もりはその臨床試験費用を計算すべきであることと関係がある。経営陣はその見積もり数を評価し続けている。これらの見積りは,会社の歴史的経験,現在の事件の理解,将来とりうる行動に基づいているが,実際の結果は最終的にはこれらの見積りや仮定とは大きく異なる可能性がある.
現金·現金等価物証券取引と短期投資
現金と現金等価物
当社はすべての金利リスクが大きくなく原始満期日が三ヶ月以下の高流動性投資を現金等価物と見なしています。現金と現金等価物には、通貨市場基金と購入日の元の満期日が3カ月未満の米国債が含まれる。
短期投資
短期投資には米国債が含まれ、売却可能な証券に分類され、満期日は3カ月を超えるが1年に満たない。同社は、売却可能なすべての証券を貸借対照表上の流動資産に分類しており、これらの証券は高流動性証券と考えられているため、現在の業務に利用可能である。当社は公正価値に応じてこれらの証券を保有し、未実現収益と損失を累計他の全面収益(損失)の単独構成要素として報告している。割増または割引の割引を購入し、総合経営報告書および全面的な損失に計上された利息収入を増加させる。
F-8
金融商品の公正価値
このような項目は短期的な性質に属するため、すべての現金等価物、前払い支出及びその他の資産、支払すべき帳簿及び計算すべき負債の額面はすべてその公正価値に対する合理的な推定である。当社の現行の類似条項ローンの借入金利によると、当社は定期ローンの公正価値がその帳簿価値に近いと信じている(付記6参照)。
信用リスクその他のリスクと不確定要因が集中している
金融商品は主に現金と現金等価物および短期投資からなり、会社をかなり集中的な信用リスクに直面させる可能性がある。同社の連邦保険の主要金融機関での預金は連邦保険の限度額を超えている。当社は当該等の口座で何の損失も被っておらず、経営陣は、当該等の預金を保有する預金機関の財務状況により、当社は重大な信用リスクに直面しないと信じている。
同社は、他の生物製薬会社と同様のリスクに直面しているが、これらに限定されないが、十分な追加資金を得る必要があり、現在または将来の臨床前研究または臨床試験は失敗する可能性があり、第三者またはパートナーに依存して臨床試験を行う必要があり、その候補薬物のための規制およびマーケティング承認またはパートナー依存を得る必要があり、ライバルは新しい技術革新を開発しており、その候補薬物の商業化および市場受け入れに成功する必要があり、会社に付与されたライセンスの条項および条件に基づいて候補薬を開発および商業化する権利、独自技術の保護、マイルストーンの能力を得る必要がある。任意の許可または協調協定に基づいて支払われるべき特許権使用料または他の支払い、ならびに第三者との適切な製造スケジュールの確保および維持の必要性。もしその会社がその候補薬物の商業化または協力に成功しなかった場合、製品収入を生成したり、利益を達成することができないだろう。
財産と設備
財産及び設備をコスト別に記録し、資産の推定耐用年数内に直線法で減価償却又は償却する(一般至れり尽くせりあるいは、あるいは一般に、実験室装置、コンピュータおよびオフィス機器、家具および固定装置、ならびにレンタル改善が含まれる。修理とメンテナンス費用は発生時に費用を計上します。
長寿資産
当社は、そのすべての長期資産(物件及び設備を含む)の帳簿価値及び推定耐用年数を定期的に検討し、帳簿価値又は推定耐用年数を調整する必要がある減値指標が存在するか否かを決定する。減値が存在すれば,減値損失は資産公正価値の帳簿価値から帳簿価値を超えて計測される.その会社は所有している
賃貸借証書
リース開始日には,当社はリース負債を確認し,リース金の支払い責任,および使用権資産(“ROU資産”)を代表し,レンタル期間内に関連資産を使用する権利を代表する。レンタル負債はレンタル期間内のレンタル支払いの現在価値に応じて計測されます。当社の賃貸借は通常暗黙金利を提供していないため、当社はレンタル開始日に得られる情報に応じて漸増借入金金利を使用しています。ROU資産は、リース負債および会社によって発生する初期直接コストの初歩的な計量を含むが、リースインセンティブは含まれていない。純収益資産は経営リース純資産に計上され、リース負債は貸借対照表の流れと非流動経営賃貸負債に計上される。
贈与収入
同社の2022年の贈与収入は、米国防高等研究計画局(DARPA)がカリフォルニア大学サンフランシスコ校(UCSF)を通じて提供した贈与から来ている。同社はDARPAの贈与収入が予算期間内に発生した精算可能な贈与コストであり、あらかじめ承認された奨励限度額に達していることを確認した。これらの償還に関する費用は,添付された2022年総合業務報告書と全面赤字に研究·開発費用の一部として反映されている。収入を超えた請求書は,売掛金として総合貸借対照表における前払い費用や他の流動資産に計上される。
研究開発コスト
研究と開発費用には,主に同社の候補製品の臨床前と臨床開発に関するコストが含まれている。研究·開発コストは発生時に費用を計上する。
F-9
臨床試験は費用と前臨床研究を計算しなければならない
同社は,サプライヤーやコンサルタント,CROと臨床サイトとの契約による臨床試験や臨床前研究に関する費用を記録している。これらの契約の財務条件は、契約によって異なり、支払流量がこれらの契約に基づいて材料またはサービスを提供する期限と一致しない可能性がある交渉を行う必要がある。同社は,臨床試験や臨床前研究費用をサービスや努力支出の時間に合わせることで,財務諸表にこれらの費用を反映している。当社は,臨床試験や臨床前研究の進展状況に基づき,臨床試験,臨床前研究あるいは関連活動の各方面のスケジュールにより,これらの費用を会計処理している。同社は、基本契約、臨床および他のキーパーソンおよび第三者サービスプロバイダとの通信、行われている臨床試験、臨床前研究または他のサービスの進展状況、およびこれまでに領収書または支払いを発行した金額に基づいて、推定すべきカウントを決定する。臨床試験や前臨床研究過程において,実際の結果が見積り結果と異なれば,会社は費用認知率を調整する。
特許費用
出願及び特許出願に係る費用は、一般及び行政費用として記録され、このような費用の回収が不確定であるため、発生した費用として計上される。
収入確認
会社は、連携手配における課金単位が仕入先と顧客関係の特徴を表現しているか否かを決定するために、連携手配を評価する。顧客関係にある手配や会計単位に対して、会社は収入確認指導を適用する。会社は約束した貨物やサービスを顧客に譲渡する際の収入を確認し、その額は、会社がこれらの貨物やサービスの対価格と交換する権利があることを反映している。顧客との契約の収入確認を決定するために,会社は,(1)顧客との契約を決定する(S),(2)契約中の履行義務を決定する,(3)取引価格を決定する,(4)取引価格を契約に割り当てる履行義務,および(5)会社が履行義務を履行した場合に収入を確認する,の5つのステップを実行する.契約開始時に、会社は、各契約で約束された貨物またはサービスを評価し、各約束された貨物またはサービスが独特であるかどうかを評価し、どれが履行義務であるかを決定する。そして、会社は、履行義務を履行する際にそれぞれの履行義務に割り当てられた取引価格の金額が収入であることを確認する。
会社の知的財産権許可が合意で決定された他の履行義務とは異なると判定された場合、許可が被許可者に譲渡され、被許可者が許可を使用して利益を得ることができる場合には、会社は、許可に割り当てられた払戻不可能な前払い費用の収入を確認する。他の履行義務とバンドルされたライセンスについて、当社は、合併履行義務が一定期間またはある時点で履行されているか否かを決定するために、判断を用いて合併履行義務の性質を評価し、時間が経過すれば、収入を確認するための進展を測定する適切な方法を決定する。当社は報告期間ごとに進捗指標を評価し、必要があれば、予想に基づいて業績指標および関連収入確認を調整します。
提携プロトコルにより実行される研究開発サービスについては,その履行義務が時間の経過とともに履行され,会社は入力法を用いて活動の進捗を測定している.使用する入力方法は,関連履行義務を履行するための努力や発生コストに基づいている.当社の見積もりにかかる作業量には、当社が活動完了に要する時間を見積もることや、特定の期間内に発生するコストを含めて、履行義務を履行するために推定される総作業量やコストに対してコストが含まれています。これは、各期間に確認された収入額を決定するために、研究および開発サービスに割り当てられた対価格を百分率に乗じることになる。この方法は重大な判断を推定して使用する必要がある。提携中に変化すると推定または判断される場合、それらは、当期および将来の間に確認された収入の時間および金額に影響を与える可能性がある。
一里塚
マイルストーン支払い(可変対価格)を含む各手配の開始時に、会社はマイルストーンが達成可能であると考えられているかどうかを評価し、最も可能な金額法を用いて取引価格に含まれる金額を推定する。大きな収入逆転が起こらない可能性が高い場合、関連するマイルストーン価値は取引価格に含まれるだろう。規制承認のような当社または当社の協力パートナーの制御範囲内での一里塚的な支払いは、通常、これらの承認を受けるまでは実現可能ではないと考えられる。そして、取引価格は相対的に独立した販売価格で契約履行義務毎に割り当てられ、会社は契約項下の履行義務を履行する際に収入を確認する。その後の各報告期間終了時に、当社はこのようなマイルストーンを実現する可能性や任意の制限を再評価し、必要に応じて当社の全体取引価格の見積もりを調整します。いずれの調整も,累積追跡に基づいて履行された履行と部分的に履行された履行義務に割り当てられ,履行期間中に継続履行債務に割り当てられた対価格を確認する.
F-10
所得税
当社は、連結財務諸表に含まれる事件の将来の税務結果を予想した繰延税金資産と負債を確認することを要求する貸借対照法に基づいて所得税を計算する。この方法によると、繰延税項資産及び負債は、総合財務諸表と資産及び負債の税ベースとの差額に基づいて、予想差額を用いて返送される年度の現行税率を決定する。税率変動が繰延税金資産や負債に及ぼす影響は、公布日を含む期間の収入で確認されている。
当社が繰延税金資産の範囲を確認したのは、当社はこれらの資産がより現金化する可能性があると考えています。このような決定を下す際には、管理層は、既存の課税の一時的な差の将来逆転、将来の課税収入の予想、税務計画戦略、最近の経営の結果を含む、利用可能なすべてのプラスおよび負の証拠を考慮する。経営陣が、当社が将来その記録金額を超える繰延税金資産を実現できると判断した場合、管理層は繰延税金資産の推定値を調整し、所得税の支出を減らすことになる。
2023年12月31日現在、2023年12月31日現在、2022年12月31日現在、会社は“より可能性が高い”実現のハードルに達していないと結論したため、会社は繰延税金資産の評価準備を維持している。推定免税額が所得税引当金で確認された場合、推定免税額の変化は、推定された年間実質税率の変化を招く可能性がある。
当社は、(1)経営陣が税務倉庫位の技術的優位性に基づいて当該等の税務倉位を維持する可能性が高いか否かを決定する不確定な税務倉位、及び(2)より確認のハードルに達する可能性のある税務倉位について、管理層が最終的に関連税務機関と和解した後に実現可能な50%を超える最大税務割引額を確認する。同社は所得税支出で未確認の税収割引に関する利息と罰金を確認した。どんな課税利息と罰金も関連された納税義務に含まれている。
了解衝撃損失
総合損失には純損失と売却投資可能な未実現収益または損失が含まれる。当社は全面赤字とその構成要素を総合経営報告書と全面赤字の一部として列報しています。
細分化市場報告
経営分部は企業の構成要素として確認され、その独立した離散財務情報は、首席運営決定者が資源配分に関する意思決定と業績評価を行う際に評価することができる。会社及びその主な経営意思決定者は、会社の運営状況を確認し、会社の業務を管理する
株に基づく報酬費用
株式ベースの報酬支出とは、付与日のコストであり、従業員株式オプション付与の公正価値は、奨励に必要なサービス期間(通常は授権期間)内で直線的に確認される。同社はブラック·スコアーズオプション定価モデルを用いて株式オプション付与の公正価値を推定している。同社は公正価値方法を用いて非従業員に付与された株式オプションを会計処理した。
ブラック·スコアーズオプション定価モデルは、無リスク金利、予想株価変動率、株式オプション予想期限、および期待配当収益率を含む主観的仮定の使用を要求する。当社の歴史的株式オプション活動は限られているため、付与された株式オプションの期待期限は、簡略化方法を用いて推定され、株式オプションの契約期間とその加重平均帰属期間の平均値を表す。ブラック·スコアーズオプション定価モデルで使用される基本普通株の公正価値は、付与日の普通株の終値に基づく。
公共·個人配給持分証
業務合併完了後、当社はLWACが2021年1月に初公開発売時に発行した公開及びプライベート配給承認株式証を負担し、これにより、公開及びプライベート配給承認株式証の所有者が自社の普通株を買収する権利を有する。同社の結論は、株式証明書の公開は株式分類である。私募株式証の決済価値部分は,受取時に誰が引受権証を持っているかに依存するため,会社株にリンクしているとはみなされないため,負債として記録されている。負債に分類された権証は,発行当日にその推定公正価値で入金され,その後資産負債表ごとに日ごとに再推定され,公正価値が他の収入(支出),添付総合経営報告書純額および全面赤字確認に変動することが明らかになった。同社はBlack-Scholesオプション定価モデルを用いてこれらの株式承認証の公正価値を推定した。
F-11
普通株および配給代理承認持分証
2023年5月と2023年6月、当社は2回の独立した登録直接発行を完了し、普通株式証と配給代理権証を発行し、当社の普通株を買収する権利がある。株式承認証の適切な会計処理を決定するために、会社は会計基準編纂(ASC)815-40項での指数付け指導を考慮した。当社の結論は、2023年5月及び2023年6月に発行された普通株式承認株式証及び配給代理権証はいずれも株式種別であり、この等承認持分証は自社株とリンクされているとみなされているため、株式分類を妨げる決済条項は何も含まれていない。
株を稼ぐ
統合協定によると
当社は,既存の旧発効株主に利益株式を発行するか,ASC 815−40に基づいて自社株にリンクする義務がないことを決定したため,持分処理を排除した。増発株式発行を決定するトリガイベントには,我々の普通株に完全にリンクしていない条項が含まれているため,負債分類を行う必要がある.負債に分類された権益リンクツールは、発行当日にその推定公正価値で入金され、その後資産負債表ごとに日ごとに再評価され、公正価値が他の収入(支出)、添付総合経営報告書の純額および全面赤字に変動することが確認された。
当社は、既存の旧発効者オプション所有者に発行または株式ベースのASC 718の補償の範囲に属することを決定しており、オプション所有者がイベントが発生するまでサービスの継続を要求されているためである。オプション保有者が株式を稼いだ公正価値は,モンテカルロシミュレーション推定モデルの派生サービス期間内に株式による補償と記載されており,研究·開発では,一般と行政費用が総合経営報告書と全面損失で確認されていることが確認された。
トリガイベントは、業務統合終了日から2年間発生しておらず、利益の実現期限は2023年8月25日に満了します。そのため、2023年8月25日から、旧発効株主とオプション保有者は株式を取得する資格がなくなった。
最近の会計公告
新しい会計声明は、財務会計基準委員会(“FASB”)または他の基準策定機関によって時々発表され、指定された発効日から当社が採択される。別の議論がない限り、当社は、その予備評価に基づいて、最近発表されたまだ発効していない基準の影響が、その財務状況や採用後の経営結果に実質的な影響を与えないと考えている。
2023年11月、財務会計基準委員会(“FASB”)は、年度·中期支部開示に関する最新の会計基準を発表した。他の事項を除いて、最新の会計基準はいくつかの重大な部分費用の開示を要求する。2024年12月31日までの年次報告Form 10−Kに最新の会計基準を採用する。私たちは新しい会計基準を採用する影響が私たちの支部開示に実質的な影響を与えないと予想しています。
FASBは、2023-09、所得税(主題740):改良所得税開示を発表した。この更新は公共企業実体が毎年使用パーセンテージと貨幣金額の表フォーマットの為替レート台帳を開示し、それを特定のカテゴリに細分化することを要求し、もしある帳簿項目が規定の敷居を超えた場合、性質と管轄権によって更に細分化する。また、すべての実体は、納付された所得税、受信した返金後の純額、および司法管轄区に区分された返金を受けた純額を開示しなければならず、金額が所得税の支払総額の少なくとも5%を占める場合は、受け取った返金を差し引かなければならない。ASUの通過は,改正案の前向きまたはトレーサビリティ適用を許可し,2024年12月15日以降の年度期間に有効であり,早期採用を許可している。会社の合併財務諸表に対するASU 2023-09の影響の評価はまだ完了していない。私たちは、最近発表されたがまだ発効していない他の会計声明は、採択されれば、私たちの総合財務諸表や開示に大きな影響を与えるとは思わない。
F-12
純損失1株当たり
1株あたりの基本純損失の計算方法は,純損失を当期発行普通株の加重平均で割ったものであり,潜在的な希釈証券は考慮しない。1株当たり純損失の算出方法は,純損失を普通株の加重平均に期内に発行された潜在希薄化証券の潜在薄化影響を加えた和で割ったものである。このような組み入れの影響が逆希釈であれば、潜在的な希釈証券は希釈収益または1株当たり損失から除外される。当社の潜在的希薄化証券は、1株当たり純損失に対して逆償却作用があるため、1株当たりの純損失の計算から除外されている。列報のすべての期間において、会社の純損失状況により、基本と希釈後の流通株を計算するための株式数に差はなかった。
潜在希釈証券、現在まで2023年12月31日と2022年12月31日は以下の通り(普通株等値株は、1株当たり逆株式分割を反映するように調整されています)
|
|
12月31日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
|
|
|
|
|
|
|
||
普通株式引受証 |
|
|
|
|
|
|
||
配給代理授権証 |
|
|
|
|
|
|
||
株式証を公開する |
|
|
|
|
|
|
||
私募株式証明書 |
|
|
|
|
|
|
||
株を稼ぐ |
|
|
|
|
|
|
||
保険者株に帰属していない |
|
|
|
|
|
|
||
未償還株式オプション |
|
|
|
|
|
|
||
合計する |
|
|
|
|
|
|
||
会計指針は公正価値を定義し、公正価値を計量するために一致した枠組みを構築し、公正価値の経常性或いは非日常性に基づいて計量する各主要資産と負債カテゴリの開示範囲を拡大した。公正価値は、市場参加者間の秩序ある取引において資産を売却して受信した金額または移転負債によって支払われた金額を表す脱退価格として定義される。したがって、公正価値は市場に基づく計量であり、資産または負債の定価のために市場参加者が使用する仮定に基づいて決定されるべきである。このような仮定を考慮する基礎として、会計基準は三級公正価値レベルを確立し、公正価値を計量する際に使用する投入の優先順位は以下の通りである
第1レベル:アクティブ市場の未調整見積は、計量日に同じ、制限されていない資産又は負債の見積を得ることができる。
第2レベル:アクティブ市場における同様の資産および負債のオファー、非アクティブ市場におけるオファー、または資産または負債の全期間内に直接または間接的に観察可能な投入。
第3級:価格や推定技術は公正な価値計量に重大な意義があるが観察できない投入が必要である(すなわち、市場活動の支持が少ないか、全くない)。
会社の現金等価物は、見積された市場価格、仲介人または取引業者のオファー、または合理的な価格透明性を有する代替価格源を使用して推定されるので、公正価値レベルの第1レベルの投入を使用して分類される。当社のいかなる非金融資産または負債も公平な価値に基づいて非日常的に記録されていないお姉ちゃん
必要があれば、当社は、以下の情報に基づいて、各報告日にBlack-Scholesオプション定価モデルを使用して、発行およびその後の再計量時に、その株式証負債の公正価値を推定する:無リスク金利、期待配当率、株式証の残り契約期間、対象株式の公正価値、および対象株式価格の予想変動を推定する。これらの見積りはある程度主観的な仮定に基づいており,将来は大きく異なる可能性がある.これらの仮定の変化および報告日における会社株の公正価値は株式証負債の公正価値に重大な影響を与える可能性がある。
F-13
下表は,会社が経常的公正価値に応じて計量する必要がある資産と負債と,公正価値レベルに基づくそれぞれの投入レベルをまとめたものである2023年12月31日および2022年12月31日(単位:千):
|
|
|
|
|
公正価値計量使用 |
|
||||||||||
|
|
十二月三十一日 |
|
|
オファー |
|
|
意味が重大である |
|
|
意味が重大である |
|
||||
|
|
2023 |
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
||||
資産 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
現金等価物: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
||
アメリカ国債 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
短期投資: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
総資産 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
負債.負債 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
私募株式証責任 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
総負債 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
|
|
|
|
|
公正価値計量使用 |
|
||||||||||
|
|
十二月三十一日 |
|
|
オファー |
|
|
意味が重大である |
|
|
意味が重大である |
|
||||
|
|
2022 |
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
||||
資産 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
現金等価物: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
||
短期投資: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
|
|
|
|
— |
|
|
$ |
|
|
|
— |
|
||
総資産 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
負債.負債 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
私募株式証責任 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
負債をかせぐ |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
総負債 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
現金等価物と短期投資
公正な価値に応じて恒常的に計量される金融資産には、会社の現金等価物および短期投資が含まれる。同社は、その投資マネージャーから定価情報を取得し、一般に、報告された取引、仲介人/取引業者のオファー、および入札および/または要約を含む、標準的に観察可能な投入を使用して投資証券の公正価値を決定する。
投資のオファーが同じ証券の活発な市場で利用可能である場合、投資は公正価値レベルで第1レベルに分類される。通貨市場基金への投資は#ドル
有価証券への投資は第二レベル投入を用いて評価する。二級証券は最初に取引価格で評価され、その後、直接或いは間接的に観察可能な見積もり以外の投入を利用して評価と報告を行い、例えば第三者定価サプライヤーからのオファーを行う。二次投入によって確定された公正価値は、見積もり、金利、収益率曲線などの観察可能なデータ点を利用して、判断と使用推定を行う必要があり、見積もりが変化すれば、会社の財務状況と運営結果に重大な影響を与える可能性がある。短期投資に関する課税利息は#ドルである
F-14
次の表は、会社が2023年12月31日まで、2022年12月31日までの売却証券として入金可能な短期投資(単位:千)をまとめています
|
|
|
|
|
|
|
2023年12月31日 |
|
||||||||||
|
|
成熟性 |
|
償却する |
|
|
実現していない |
|
|
実現していない |
|
|
推定数 |
|
||||
|
|
(単位:年) |
|
コスト |
|
|
収益.収益 |
|
|
損 |
|
|
公正価値 |
|
||||
アメリカ国債 |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|||||
|
|
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
|
|
|
|
|
|
|
2022年12月31日 |
|
||||||||||
|
|
成熟性 |
|
償却する |
|
|
実現していない |
|
|
実現していない |
|
|
推定数 |
|
||||
|
|
(単位:年) |
|
コスト |
|
|
収益.収益 |
|
|
損 |
|
|
公正価値 |
|
||||
アメリカ国債 |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
私募株式証責任
業務合併については、当社は付記2に記載の公開及び個人配給承認持分証を負担する資本処理を計上せず、会社の普通株にリンクしているとみなされないため、負債として記録する。私募株式証負債は公正価値によって計量し、観察可能と観察できない情報を総合的に使用する。私募株式証負債の公正価値変動は、経営報告書及び全面赤字の他の収入(支出)に記録されている
|
十二月三十一日 |
|
十二月三十一日 |
|
||
普通株価格 |
$ |
|
$ |
|
||
予想変動率 |
|
% |
|
% |
||
無リスク金利 |
|
% |
|
% |
||
予想期限(年単位) |
|
|
|
|
||
期待配当収益率 |
|
|
|
|
||
以下の表には、2023年12月31日までの1年間における私募株式証負債の活動を示し、重大な観察不可能な3級投入を採用し、公正価値で計量する(千計)
|
|
私募株式証責任 |
|
|
2022年12月31日の残高 |
|
|
|
|
価値変動を公平に承諾する |
|
|
|
|
2023年12月31日の残高 |
|
$ |
|
|
財産と設備、純額は以下の部分からなる(千計)
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
|
|
|
|
|
|
|
||
実験室装置 |
|
$ |
|
|
$ |
|
||
コンピュータとオフィス機器 |
|
|
|
|
|
|
||
家具と固定装置 |
|
|
|
|
|
|
||
賃借権改善 |
|
|
|
|
|
|
||
建設中の工事 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
減価償却累計と償却を差し引く |
|
|
( |
) |
|
|
( |
) |
|
|
$ |
|
|
$ |
|
||
会社が記録した減価償却と償却費用は#ドルです
F-15
計算すべき費用には、以下の項目が含まれる
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
従業員報酬 |
|
$ |
|
|
$ |
|
||
研究開発 |
|
|
|
|
|
|
||
専門と外部サービス |
|
|
|
|
|
|
||
利子 |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
|
|
$ |
|
|
$ |
|
||
オックスフォード定期ローン
Old Efftorは2021年3月にOxford Finance LLC(“Oxford”)と融資および保証協定(“Oxford LSA”)を締結し、この合意により、当社は最大$を借りることができます
2022年2月22日、当社はオックスフォードLSA改正案を締結し、この改正案によると、A期ローンの利子期間はわずかに終了した
オックスフォードローンの変動金利は(I)の大きな者に等しい
その所有する知的財産権を除いて、オックスフォード法律サービス協定の下での当社の義務は、その現在と将来のほとんどの資産の優先保証権益を保証する。同社には、同意なしに知的財産権資産を差し押さえる能力の制限を含む他の様々な習慣条約の遵守が義務付けられている。
同社は#ドルの債務割引を記録した
F-16
会社A期ローンの未返済元本金額に基づいて、次の表は年ごとに会社の現在までを示しています2023年12月31日(千):
2023年12月31日まで |
|
|
|
|
2024 |
|
$ |
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 |
|
|
|
|
必要な将来元金支払い |
|
$ |
|
|
未償却債務割引 |
|
|
( |
) |
今期のローンは、2023年12月31日までの純額 |
|
$ |
|
|
株式承認証と私募株式承認証
業務合併が完了した後、公開株式証及び私募株式証所持者はすべて当社の普通株を買収する権利がある。株式承認証は2022年1月12日から行使可能であり、LWAC初公募株終了まで12カ月である。1部の株式承認証は登録所有者に行使価格$で普通株を購入する権利を持たせる
株式公開承認証及び個人配給株式承認証が行使されると、当社は未発行の引受権証の一部ではなくすべての権利があり、価格は$となる
私募持分証は公開株式証と同じであり、ただ保証人或いはその譲渡許可者だけが保有しなければならない:(I)当該等株式承認証は当社が償還しない;(Ii)この等株式証は所有者がキャッシュレス基礎で行使することができる;及び(Iii)当該等株式承認証は登録権規程を受けなければならない。
私募株式証は責任によって分類され(付記3参照)、公開株式証は権益によって分類される
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
|
行権価格 |
|
|
期日まで |
|||
株式証を公開する |
|
|
|
|
|
|
|
$ |
|
|
||||
私募株式証明書 |
|
|
|
|
|
|
|
$ |
|
|
||||
株式承認証-登録直接発売
2023年5月に、当社は発行を含む登録直接発売(“2023年5月登録直接発売”)を完了しました
当社は2023年6月に登録直接発売(“2023年6月登録直接発売”)を完了し、発行を含めて
F-17
一般権利証と配給代理権証は株式によって分類される
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
|
行権価格 |
|
|
期日まで |
|||
2023年5月普通株式承認証 |
|
|
|
|
|
|
|
$ |
|
|
||||
2023年5月配給代理承認株式証 |
|
|
|
|
|
|
|
$ |
|
|
||||
2023年6月普通株式承認証 |
|
|
|
|
|
|
|
$ |
|
|
||||
2023年6月配給代理承認株式証 |
|
|
|
|
|
|
|
$ |
|
|
||||
調達協定
当社は2022年1月24日にリンカーン公園と購入契約および登録権協定を締結し、リンカーン公園に売却される金額は最高で$に達することを規定している
“購入契約”によると、ある条件を満たす場合、会社は会社が選定した任意の営業日にリンカーン公園に購入する権利があります
定期購入を除いて、ある条件および制限の下で、会社は、(I)そのように定期的に購入された株式の数の3(3)倍または(Ii)まで、リンカーン公園に各購入日に次の営業日に購入することを要求することを自ら決定することができる
購入契約によると、会社がリンカーン公園に売却できる株式の総数は超えてはいけません
購入協定には慣例陳述、保証、チェーノ、成約条件、賠償、終了条項が含まれている。会社はいつでも自分で購入契約を終了することを決定することができて、何の費用や罰金を支払う必要がなく、リンカーン公園に一営業日の通知を出すだけでいいです。さらに、リンカーン公園はまた、直接または間接的に空売りしたり、普通株を売却しないことを約束した。購入契約は、収益、財務又は業務契約の使用に制限はなく、将来の融資に制限はない(ただし、当社が期限内に類似したタイプの合意又は株式信用限度額を達成する能力の制限を除き、登録ブローカーとの市場取引を除く)、優先購入権、参加権、罰金又は違約金。
F-18
市場に計画を提供する
当社は2022年9月にCantorと制御持分発売契約(“販売契約”)を締結し、この合意により、当社は合計発行価格50ドルまでの自社普通株株式を随時販売することができる
当社は2023年12月31日までに販売しております
当社は2022年12月31日までに販売します
登録された直売製品
2023年5月の登録直接発行には発行と販売の合計が含まれています
2023年5月の登録直接発売については,同社は独占配給代理であるH.C.Wainwright&Co.,LLCに相当する支払いを行った
2023年6月の登録直接発行には発行と販売の合計が含まれています
2023年6月の登録直接発売については,同社は独占配給代理であるH.C.Wainwright&Co.,LLCに相当する支払いを行った
優先株
企業合併取引が完了したときは、改訂された会社登録証明書の条項に基づいて、
F-19
保証人株
企業合併の終了について、LWACスポンサーが受け取りました
従業員株購入計画
ESPPは6ヶ月の入札期限を規定しており、各入札期限が終了した時点で、従業員は
2013持分インセンティブ計画
Old Efftorは、業務統合の前に、2013年の株式インセンティブ計画(“2013計画”)を維持し、この計画によると、Old Efftorは従業員、取締役、非従業員コンサルタントに奨励的株式オプション、制限株式奨励、その他の株式ベースの奨励を付与する。業務合併が完了した後、当社は2013年計画下の奨励の付与を停止し、以下に述べるように、2013年計画下のすべての奨励は、同じ条項と条件で2021年計画下の奨励に転換する。2021年8月25日現在、企業合併取引の前に、
2021年株式インセンティブ計画
2021年8月25日の業務合併完了に合わせて、取締役会は“2021年株式激励計画”(“2021年計画”と略称する)を採択することを許可した。デシemまで2023年9月31日
2021年計画に基づいて付与された購入権は、付与時に決定された異なる期日に行使することができ、授与日から10年を超えないか、またはいくつかの非法定オプションについては、付与日から10年以内に満期とすることができる。各オプションの実行権価格は取締役会がオプションを付与した日の会社株の公正市場価値に基づいて決定され、公正市場価値は会社普通株の終値と定義される。奨励的株式オプションを実施する者は,執行権価格を下回ってはならない
F-20
これらの計画によると、会社の株式オプション活動の概要は以下のとおりである(千では、株式および1株当たりの金額および年は含まれていない)
|
|
株 |
|
|
重み付けの- |
|
|
重み付けの- |
|
|
骨材 |
|
||||
2022年12月31日に返済されていません |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
授与する |
|
|
|
|
|
|
|
|
|
|
|
|
||||
鍛えられた |
|
|
( |
) |
|
|
|
|
|
— |
|
|
|
|
||
キャンセルまたは没収 |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
2023年12月31日現在の未返済債務 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2023年12月31日に帰属して行使可能です |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2023年12月31日までの既得オプションの公正価値総額は$
株に基づく報酬費用
当社は株式に基づく報酬支出が具体的に関連していることを確認した$の株式オプション
|
|
十二月三十一日までの年度 |
||
|
|
2023 |
|
2022 |
無リスク金利 |
|
|
||
予想変動率 |
|
|
||
予想期限(年単位) |
|
|
||
期待配当収益率 |
|
|
||
無リスク金利です無リスク金利は米国債に基づいていると仮定しており、その条項は会社株オプションの期待期限と一致している。
予想される波動性当社の経営歴史は限られており、会社に特定された歴史や隠れた変動率が不足しているため、予想変動率は株価が公開されている業界同業者の歴史的波動性を研究することによって決定されると仮定している。
期限を見込む株式オプションの予想期間は、株式オプション期待未償還の加重平均期間を表す。当社はアメリカ証券取引委員会が提供する簡略化方法を用いて期待期間を推定しています。簡略化された方法は、期待期限をオプションの帰属時間および契約期間の加重平均として計算する。
配当収益率を期待する期待配当仮定は、会社の歴史と配当金の予想に基づいている。当社はまだ支払っていませんし、配当金を支払うつもりもありません。
没収するそれは.当社は発生期間中に実際に没収された株式ベースの補償費用を削減します。
2023年12月31日現在,OUTに関する未確認補償コスト常備従業員オプションは$
F-21
未来発行の普通株を確保する
将来の発行のために予約された普通株式には以下が含まれる2023年12月31日:
|
|
十二月三十一日 |
|
|
発行済みおよび未償還株式オプション |
|
|
|
|
発行および未償還の公共持分証 |
|
|
|
|
発行済みおよび未発行の私募株式承認証 |
|
|
|
|
発行済み及び発行済み普通株式承認証 |
|
|
|
|
発行済みおよび未発行の配給代理承認株式証 |
|
|
|
|
保険者株に帰属していない |
|
|
|
|
将来の株式奨励またはオプション付与のための許可 |
|
|
|
|
ESPPの許可により将来発行される |
|
|
|
|
合計する |
|
|
|
|
統合協定によると
2022年12月31日現在,株主とオプション保有者は約を取得する資格がある
|
十二月三十一日 |
|
|
株価.株価 |
$ |
|
|
予想変動率 |
|
% |
|
無リスク金利 |
|
% |
|
予測期間(年) |
|
|
|
権益コスト |
|
% |
|
老効果株主
当社は,既存の旧発効株主に利益株式を発行するか,ASC 815−40に基づいて自社株にリンクする義務がないことを決定したため,持分処理を排除した。増発株式発行を決定するトリガーイベントには、当社の普通株に完全にリンクしているわけではない条項が含まれているため、負債分類が必要となる。当社は株主利益株式の発行日の公正価値を推定し、各報告期間内に負債を再評価し、価値変動を公平に総合経営報告書及び全面損失に計上する。合併協定によると、トリガ事件が発生する前にサービス提供を停止した旧発効人の株式購入所有者は、稼いだ株式を占めて残りの合資格株主及び株式購入所有者に再分配しなければならない。企業合併終了日後の2年期は2023年8月25日に満期となったため、利益負債は約#ドル
利益が満了する前に既存の旧効果者オプション所有者に利益株を発行するか、またはオプション所有者がトリガイベントが発生するまでサービスを継続することが要求されるので、ASC 718の株式ベースの補償の範囲に属することが義務付けられている。業務合併完了日に、株式所有者が株式を購入して獲得した公正価値は約$である
F-22
二零一三年五月、当社はカリフォルニア大学董会(“カリフォルニア大学董会”)と合意を結び、カリフォルニア大学董会が最初に開発した翻訳略字実験室技術に関するいくつかの発明(“カリフォルニア大学董会翻訳略字特許権”)について当社に独占的に特許権を取得した。この合意によれば、同社は、特許権発見および開発された製品をカリフォルニア大学サンフランシスコ校を用いて翻訳解析し、ライセンス製品と呼び、カリフォルニア大学サンフランシスコ校を用いて特許権を翻訳解析するいくつかの許可プロセスを使用し、これらの許可製品およびプロセスの再許可を可能にするために、研究、開発、製造および販売を許可される。
2021年7月、会社は、業務合併が許可協定に与える影響を確認するための許可協定改正案を署名し、業務合併の終了に関連して、会社はUCSFに約#ドルの現金を一度に支払うことを含む
同社が毎年支払う最低使用料は#ドルです
2019年12月、同社はeIF 4 Eに対する小分子を研究·開発するために、ファイザー社と研究協力·許可協定(“ファイザー協定”)を締結した。
ファイザー協定によると、同社はファイザーとの協力の初歩的な研究を担当し、ファイザーは試験的新薬申請の提出とすべての臨床開発と商業化活動を含むこの計画のすべての更なる開発を担当する。ファイザーには、商業的に合理的な努力でライセンス製品を開発し、規制機関の承認を求め、ファイザーが規制の承認を得た場合に、ライセンス製品を米国や他の特定の国·地域で商業化する義務がある。同社が共同出資と共同推進オプションを行使すれば、共同指導委員会が共同開発製品の開発計画と予算を監督し、医療提供者への製品マーケティングプレゼンテーションの一部を担当する。
ファイザー協定によると、同社は一度に、返却できない、貸記できない金#ドルを受け取った
初期成約価格は$
2021年4月、会社はカリフォルニア大学サンフランシスコ校と研究分賞協定(“分賞協定”)を締結し、この協定によると、最高で$に達する
F-23
賃貸借証書
2021年9月、当社の参入は撤回できません
当社は2023年12月31日および2022年12月31日まで毎年$を支給しています
次の表は,以下の日までのリースに関する補足貸借対照表情報をまとめたものである2023年12月31日および2022年12月31日(単位:千):
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
資産: |
|
|
|
|
|
|
||
経営的リース使用権資産 |
|
$ |
|
|
$ |
|
||
使用権資産総額 |
|
|
|
|
|
|
||
負債.負債 |
|
|
|
|
|
|
||
賃貸負債を経営し、流動 |
|
|
|
|
|
|
||
非流動経営賃貸負債 |
|
|
|
|
|
|
||
リース負債総額を経営する |
|
$ |
|
|
$ |
|
||
自分から2023年12月31日、既存経営賃貸に基づき、将来最低年度賃貸支払いは以下の通り(単位:千、加重平均残存期間と加重平均割引率を除く):
2024 |
|
|
|
|
余剰賃貸支払総額 |
|
|
|
|
差し引く:推定利息 |
|
|
( |
) |
リース負債総額を経営する |
|
|
|
|
マイナス:現在の部分 |
|
|
( |
) |
長期経営賃貸負債 |
|
$ |
|
|
加重平均-残りのレンタル期間(年単位で) |
|
|
|
|
加重平均割引率 |
|
|
% |
同社には、条件に合った従業員が使用できる固定拠出金401(K)計画がある。この計画の条項によると、従業員は自発的に報酬のパーセンテージを支払うことができるが、連邦税収法規が許可する最高額を超えてはならない。会社はその歴史において分泌は,401(K)計画に貢献する可能性がある。2023年12月31日までに同社は
F-24
当社は2023年12月31日および2022年12月31日までに所得税の支出を記録していない。
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
繰延税金資産: |
|
|
|
|
|
|
||
純営業損失が繰り越す |
|
$ |
|
|
$ |
|
||
無形資産 |
|
|
|
|
|
|
||
研究と開発資本化 |
|
|
|
|
|
|
||
補償すべきである |
|
|
|
|
|
|
||
単位 |
|
|
|
|
|
|
||
固定資産 |
|
|
|
|
|
|
||
その他、純額 |
|
|
|
|
|
|
||
使用権責任 |
|
|
|
|
|
|
||
繰延税金資産 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
繰延税金負債: |
|
|
|
|
|
|
||
使用権資産 |
|
|
( |
) |
|
|
( |
) |
繰延税金負債 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
||
繰延税項目純資産 |
|
|
|
|
|
|
||
推定免税額 |
|
|
( |
) |
|
|
( |
) |
繰延税項目純資産 |
|
$ |
|
|
$ |
|
||
連邦法定税率で計算される所得税と2023年12月31日現在と2022年12月31日までの所得税費用(福祉)との入金は以下の通り(単位:千):
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
法定税率で課税する |
|
$ |
( |
) |
|
$ |
( |
) |
連邦福祉を差し引いた州所得税 |
|
|
|
|
|
( |
) |
|
評価免除額を変更する |
|
|
|
|
|
|
||
不確定税収状況 |
|
|
|
|
|
|
||
収益負債公正価値変動収益 |
|
|
( |
) |
|
|
( |
) |
永久的な相違やその他の |
|
|
|
|
|
|
||
取引コスト |
|
|
|
|
|
|
||
資本化R&D |
|
|
|
|
|
|
||
LWAC純運営損失 |
|
|
( |
) |
|
|
( |
) |
単位 |
|
|
( |
) |
|
|
( |
) |
所得税費用 |
|
$ |
|
|
$ |
|
||
経営陣は、既存の繰延税金資産を使用するために十分な将来の課税収入が生じるかどうかを推定するために、すべての利用可能な証拠を評価する。会社は設立以来純損失(2021年12月31日までと2020年12月31日までの年度を除く)を経て、当時の純収入はそれぞれファイザー社の研究協力と許可協定に関する研究協力と許可協定に関する公正価値確認収益と非経常収入の収益によって実現された)、会社業務と市場の収入と収入潜在力は実証されていない。会社の持続的な研究と開発活動により、会社は予見可能な未来に純損失が続くと予想される。したがって、当社は繰延税金資産が現金化される可能性が高いとは断定できない。推定免税額は#ドルです
F-25
純営業損失(“NOL”)と研究開発(“R&D”)信用繰越の使用は重大な年度制限を受ける可能性があり、原因は所有権変更制限がすでに発生または将来発生する可能性があるためであり、1986年の国内税法(“規則”)第382と383節及び国と外国の類似規定が要求したように。これらの所有権の変化は、毎年それぞれ将来の課税収入と税収を相殺するために使用できるNOLとR&D信用繰越金額を制限する可能性がある。一般的に、“規則”第382条に規定されている“所有権変更”とは、3年以内に行われる1回または一連の取引により、ある株主による会社の発行済み株式の所有権変更が50ポイントを超えることを意味する。当社は設立以来、株式を発行することで何度も資金を調達してきたが、株式を発行すること自体や、当該株式を購入した株主がその後に当該等の株式を売却することと結合して、上記の所有権変更を招いたり、将来の所有権変更を招いたりする可能性がある。
同社は創立から2020年12月31日までの予備コード第382と383節の研究を完了し、結論を出した
同社の連邦とカリフォルニアNOLの繰り越し額は約$
締め切り:2023年1月31日、同社は連邦とカリフォルニアの研究開発(“R&D”税)を持って繰越契約$を免除した
2022年8月16日に企業代替最低税(CAMT)を含む2022年インフレ低減法案が署名された。これらの変化は2022年12月31日以降に開始される納税年度に影響を及ぼす。新税は会社に連邦所得税の目的のために2回の単独計算を計算し、新しい最低税額またはその通常の納税義務のうちの大きい1つを支払うことを要求する。同社は、2022年12月31日以降の数年間に会社に影響が出るかどうかを確認するために、同法案の影響を監視する。その法案は会社に実質的な影響を与えないと予想される。
当社は不確定税務状況からの税務利益を確認し、技術的な是非曲直を前提としており、この状況はいかなる関連控訴や訴訟手続きの解決も含めて審査後に維持される可能性が高い。所得税の額は発効日に確認に適合しなければならない可能性が確認を満たしていない可能性よりも大きい。
2023年と2022年の税収割引が確認されていない期間初めと期末金額(利息および罰金を含まない)は以下のように入金されます(千で計算)
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
年初残高 |
|
$ |
|
|
$ |
|
||
税収の増加/(減少)-前年 |
|
|
|
|
|
|
||
今年度のポストに関する増加 |
|
|
|
|
|
|
||
年末の残額 |
|
$ |
|
|
$ |
|
||
2023年12月31日現在、当社は利息及び罰金を約$と確認しています
同社は現在カリフォルニア州とアメリカ国税局に所得税申告書を提出している。当社には現在管轄区域で審査されている税期はありません。経営損失の繰越純額が存在するため、当社の設立以来のすべての税期はすべての司法管区の税務機関の審査に開放されています。
2023年12月31日現在の未確認税収割引残高には$が含まれている
F-26
2024年1月29日、当社は発行と販売の合計を含む2024年1月の登録直接発行を完了しました
また、当社は配給代理の指定者に最大購入のための引受権証を発行しています
2024年1月の登録直接発売については,同社は独占配給代理であるH.C.Wainwright&Co.,LLCに相当する支払いを行った
142
EXhibitインデックス
|
|
|
引用で編入する |
||||
展示品 番号をつける |
|
説明する |
表 |
|
展示品 |
|
提出日 |
|
|
|
|
|
|
|
|
2.1 |
|
2021年5月26日現在、Locust Walk買収会社、Locust Walk合併子会社会社、Efftor Treateutics社が署名した合併協定と計画。 |
8-K |
|
2.1 |
|
5/27/2021 |
|
|
|
|
|
|
|
|
3.1 |
|
エフェクター治療会社の登録証明書を改訂し、再署名した。 |
8-K |
|
3.1 |
|
8/31/2021 |
|
|
|
|
|
|
|
|
3.2 |
|
修正されたエフェクター治療会社の登録証明書を改訂し、再修正します。 |
8-K |
|
3.1 |
|
6/23/2023 |
|
|
|
|
|
|
|
|
3.3 |
|
修正されたエフェクター治療会社の登録証明書を改訂し、再修正します。 |
8-K |
|
3.1 |
|
1/10/2024 |
|
|
|
|
|
|
|
|
3.4 |
|
エフェクター治療会社の付例を改訂·再作成した。 |
8-K |
|
3.2 |
|
8/31/2021 |
|
|
|
|
|
|
|
|
4.1 |
|
普通株式証明書サンプル。 |
S-4/A |
|
4.1 |
|
8/5/2021 |
|
|
|
|
|
|
|
|
4.2 |
|
株式権証契約を承認し、期日は2021年1月7日であり、大陸株譲渡信託会社とLocust Walk Acquisition Corpである。 |
8-K |
|
4.1 |
|
1/13/2021 |
|
|
|
|
|
|
|
|
4.3 |
|
証券説明書を登録する。 |
10-K |
|
4.3 |
|
3/16/2022 |
|
|
|
|
|
|
|
|
4.4 |
|
あらかじめ出資して株式証の書式を承認する |
8-K |
|
4.1 |
|
5/30/2023 |
|
|
|
|
|
|
|
|
4.5 |
|
共同授権書の書式 |
8-K |
|
4.2 |
|
5/30/2023 |
|
|
|
|
|
|
|
|
4.6 |
|
ウィンライトは株式証のフォーマットを承認する |
8-K |
|
4.3 |
|
5/30/2023 |
|
|
|
|
|
|
|
|
4.7 |
|
あらかじめ出資して株式証の書式を承認する |
8-K |
|
4.1 |
|
6/8/2023 |
|
|
|
|
|
|
|
|
4.8 |
|
共同授権書の書式 |
8-K |
|
4.2 |
|
6/8/2023 |
|
|
|
|
|
|
|
|
4.9 |
|
ウィンライトは株式証のフォーマットを承認する |
8-K |
|
4.3 |
|
6/8/2023 |
|
|
|
|
|
|
|
|
4.10 |
|
あらかじめ出資して株式証の書式を承認する |
8-K |
|
4.1 |
|
1/26/2024 |
|
|
|
|
|
|
|
|
4.11 |
|
共同授権書の書式 |
8-K |
|
4.2 |
|
1/26/2024 |
|
|
|
|
|
|
|
|
4.12 |
|
ウィンライトは株式証のフォーマットを承認する |
8-K |
|
4.3 |
|
1/26/2024 |
|
|
|
|
|
|
|
|
10.1 |
|
2021年1月7日の合意書は、バッタ行買収会社とバッタ行買収会社のある証券所持者、高級管理者、取締役との間で署名された。 |
8-K |
|
10.1 |
|
1/13/2021 |
|
|
|
|
|
|
|
|
10.2 |
|
投資管理信託協定は、期日は2021年1月7日であり、大陸株式譲渡信託会社とLocust Walk Acquisition Corpである。 |
8-K |
|
10.2 |
|
1/13/2021 |
|
|
|
|
|
|
|
|
10.3 |
|
改正および再署名された登録権協定は、2021年8月25日に、Efftor Treeutics,Inc.,Efftor Treeutics Operations,Inc.,Locust Walkスポンサー,LLCとある株主との間で達成される。 |
8-K |
|
10.3 |
|
8/31/2021 |
|
|
|
|
|
|
|
|
10.4 |
|
Locust Walk Acquisition Corp.とLocust WalkスポンサーLLCの間で署名された単位引受プロトコルは,2021年1月7日である. |
8-K |
|
10.4 |
|
1/13/2021 |
|
|
|
|
|
|
|
|
10.5 |
|
行政サービス協定は,期日は2021年1月7日であり,バッタ走行買収会社とバッタ歩行スポンサー有限責任会社が署名した。 |
8-K |
|
10.5 |
|
1/13/2021 |
|
|
|
|
|
|
|
|
10.6 |
|
バッタ買収会社賠償協議フォーマット |
S-1 |
|
10.5 |
|
12/18/2020 |
|
|
|
|
|
|
|
|
10.7 |
|
スポンサー支援協定は、2021年5月26日にバッタ走行買収会社とバッタ歩くスポンサー有限責任会社が署名した。 |
8-K |
|
10.1 |
|
5/27/2021 |
|
|
|
|
|
|
|
|
10.8 |
|
スポンサーロック協定は、2021年5月26日にバッタ走行スポンサー有限責任会社が署名した。イナゴ歩行者天国で会社を買収しました |
8-K |
|
10.2 |
|
5/27/2021 |
|
|
|
|
|
|
|
|
10.9 |
|
承認プロトコルフォーマットは,日付は2021年5月26日であり,Locust Walk Acquisition Corp.とある加入者が署名した. |
8-K |
|
10.3 |
|
5/27/2021 |
|
|
|
|
|
|
|
|
143
10.10+ |
|
エフェクター治療会社の2021年インセンティブ奨励計画とその下の株式オプションプロトコルフォーマット。 |
8-K |
|
10.10 |
|
8/31/2021 |
|
|
|
|
|
|
|
|
10.11+ |
|
効果器治療会社。2021年従業員株購入計画。 |
8-K |
|
10.11 |
|
8/31/2021 |
|
|
|
|
|
|
|
|
10.12+ |
|
賠償協議形式。 |
S-4/A |
|
10.12 |
|
8/5/2021 |
|
|
|
|
|
|
|
|
10.13# |
|
独占許可協定は、2013年5月9日にEfftorとカリフォルニア大学の取締役会が署名した。 |
S-4 |
|
10.13 |
|
6/14/2021 |
|
|
|
|
|
|
|
|
10.14# |
|
研究協力と許可協定は、2019年12月20日にEfftorとファイザーが署名した。 |
S-4 |
|
10.14 |
|
6/14/2021 |
|
|
|
|
|
|
|
|
10.15 |
|
融資·担保協定は、日付が2021年3月19日であり、有効者とオックスフォード金融有限責任会社とその他の貸主との間で締結される。 |
S-4 |
|
10.15 |
|
6/14/2021 |
|
|
|
|
|
|
|
|
10.16 |
|
転貸協定は,日付は2020年8月24日であり,Efftorとカーディフ腫瘍会社が締結し,両者の間で締結されている。 |
S-4 |
|
10.16 |
|
6/14/2021 |
|
|
|
|
|
|
|
|
10.17+ |
|
効果器治療会社の2013年株式激励計画は、改訂され、及びオプション合意の形式を経た。 |
8-K |
|
10.17 |
|
8/31/2021 |
|
|
|
|
|
|
|
|
10.18+ |
|
効果器治療会社は非従業員役員報酬計画を改訂し、再起動した。 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
10.19+ |
|
スティーヴン·ウォランド博士と執行者の間の二番目の改正と再署名された雇用協定。 |
S-4 |
|
10.19 |
|
6/14/2021 |
|
|
|
|
|
|
|
|
10.20+ |
|
マイク·バーンズと執行者の間で改正され再署名された雇用協定。 |
S-4 |
|
10.21 |
|
6/14/2021 |
|
|
|
|
|
|
|
|
10.21# |
|
“独占許可協定改正案”は、2021年7月12日にEfftorとカリフォルニア大学取締役会の間で署名された。 |
S-4/A |
|
10.23 |
|
7/19/2021 |
|
|
|
|
|
|
|
|
10.22 |
|
Efftorとリンカーンパーク資本基金有限責任会社との間の購入協定は、2022年1月24日。 |
8-K |
|
10.1 |
|
1/24/2022 |
|
|
|
|
|
|
|
|
10.23 |
|
登録権利協定は、2022年1月24日に、Efftorとリンカーンパーク資本基金有限責任会社によって達成される。 |
8-K |
|
10.2 |
|
1/24/2022 |
|
|
|
|
|
|
|
|
10.24 |
|
合同·第1修正案融資·担保協定は、2021年9月7日と、有効者とオックスフォード金融有限責任会社との間で行われる。 |
8-K |
|
10.1 |
|
2/24/2022 |
|
|
|
|
|
|
|
|
10.25 |
|
“融資·担保協定第2修正案”は、2022年2月22日とし、有効者とオックスフォード金融有限責任会社が共同で完成させる。 |
8-K |
|
10.2 |
|
2/24/2022 |
|
|
|
|
|
|
|
|
10.26 |
|
“融資·担保協定第3修正案”は、2022年4月28日とし、有効者とオックスフォード金融有限責任会社が共同で完成させる。 |
10-Q |
|
10.1 |
|
5/10/2022 |
|
|
|
|
|
|
|
|
10.27 |
|
被支配持分発行SMEfftorとCantor Fitzgerald&Co.が締結した販売協議販売協定は,2022年9月1日である。 |
S-3 |
|
1.2 |
|
9/1/2022 |
|
|
|
|
|
|
|
|
10.28+ |
|
雇用協定は,2022年8月8日に発効し,EfftorとDouglas Warnerによって署名され,両者の間で発効した。 |
10-K |
|
10.33 |
|
3/8/2023 |
|
|
|
|
|
|
|
|
10.29+ |
|
雇用協定は,EfftorとMayank Gandhiによって署名され,2022年9月1日から発効する。 |
10-K |
|
10.34 |
|
3/8/2023 |
|
|
|
|
|
|
|
|
10.30+ |
|
クレームは2024年2月13日に全面的に公表され,EfftorとMayank Gandhiが提出し,両者の間で行われた。 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
10.31 |
|
証券購入契約フォーマットは,期日は2023年5月26日であり,発効者とその内で指名された購入者との間で署名される。 |
8-K |
|
10.1 |
|
5/30/2023 |
|
|
|
|
|
|
|
|
10.32 |
|
証券購入契約フォーマットは,期日は2023年6月6日であり,発効者とその内で指名された購入者との間で署名される。 |
8-K |
|
10.1 |
|
6/8/2023 |
|
|
|
|
|
|
|
|
10.33 |
|
証券購入契約のフォーマットは、日付が2024年1月24日であり、有効者とその中に列挙された購入者との間で署名される。 |
8-K |
|
10.1 |
|
1/26/2024 |
|
|
|
|
|
|
|
|
23.1 |
|
独立公認会計士事務所の同意 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
144
31.1** |
|
2002年のサバンズ·オキシリー法第302節で可決された1934年の証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて発行された首席執行幹事証明書。 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
31.2** |
|
2002年サバンズ−オキシリー法第302節で可決された1934年証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて首席財務幹事が認証された。 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
32.1** |
|
2002年にサバンズ·オクスリ法案第906条で可決された“米国法典”第18編1350条に規定されている最高経営責任者証明書。 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
32.2** |
|
2002年の“サバンズ-オックススリー法案”906節で採択された“米国法典”第18編1350条による首席財務官の認証。 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
97 |
|
誤って判決された賠償を追討する政策 |
|
|
|
|
* |
|
|
|
|
|
|
|
|
101.INS |
|
連結されたXBRLインスタンス文書−インスタンス文書は、XBRLタグがイントラネットXBRL文書に埋め込まれているので、相互作用データファイルには表示されない。 |
|
|
|
|
* |
101.書院 |
|
Linkbase文書を組み込んだインラインXBRL分類拡張アーキテクチャ |
|
|
|
|
* |
104 |
|
表紙インタラクションデータファイル(イントラネットXBRL文書に埋め込む) |
|
|
|
|
* |
S-K規則第601(A)(5)項の規定により、本展示品のある展示品及びスケジュールは省略されている。会社はアメリカ証券取引委員会のすべての漏れた展示品とスケジュールのコピーの提供を要求しなければならない。
+管理契約または補償計画を示します。
#法規S-K 601(B)(10)(Iv)項によれば、本添付ファイルの一部は省略されています。米国証券取引委員会の要求に応じて、登録者は、編集されていない本展示品のコピーを提供することに同意する。
*アーカイブをお送りします。
**この証明書は、証券取引法第18条の規定に基づいて提出されていないものとみなされず、証券法または取引法に引用された出願書類とみなされてはならない。
145
サイン
改正された1934年証券取引法第13又は15(D)節の要求に基づいて、登録者は、正式に許可された次の署名者がその代表を代表して本報告書に署名することを正式に手配した.
|
|
エフェクター治療会社 |
|
|
|
|
|
日付:2024年3月26日 |
|
差出人: |
寄稿S/スティーブン·ウォランド |
|
|
|
スティーヴン·ウォランド博士 |
|
|
|
社長と最高経営責任者 |
本報告書は、改正された1934年の証券取引法の要求に基づき、以下の登録者によって登録者として指定日に署名された。
名前.名前 |
|
タイトル |
|
日取り |
|
|
|
|
|
/S/スティーヴン·ウォランド、博士 |
|
*社長およびCEO 行政主任(首席行政主任) |
|
2024年3月26日 |
スティーヴン·ウォランド博士 |
|
|
|
|
|
|
|
|
|
寄稿S/マイケル·バーンズ |
|
*最高財務責任者 首席財務会計官(首席財務会計官) |
|
2024年3月26日 |
マイケル·バーンズ |
|
|
|
|
|
|
|
|
|
S/ブライアン·M·ガラゲルJr.博士 |
|
*取締役および取締役会長 |
|
2024年3月26日 |
ブライアン·M·ガラゲル博士 |
|
|
|
|
|
|
|
|
|
/S/エリザベス·P·バート |
|
役員.取締役 |
|
2024年3月26日 |
エリザベス·P·バート |
|
|
|
|
|
|
|
|
|
寄稿S/クリス·エルリッヒ |
|
役員.取締役 |
|
2024年3月26日 |
クリス·エルリッヒ |
|
|
|
|
|
|
|
|
|
/S/クリスチャン·ハリントン-スミス |
|
役員.取締役 |
|
2024年3月26日 |
クリスチャン·ハリントン·スミス |
|
|
|
|
|
|
|
|
|
/S/バーバラ·クロンク |
|
役員.取締役 |
|
2024年3月26日 |
バーバラ·クロンク |
|
|
|
|
|
|
|
|
|
/S/キャロライン·ロイド |
|
役員.取締役 |
|
2024年3月26日 |
キャロライン·ロイド |
|
|
|
|
|
|
|
|
|
146