
アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
本財政年度末まで
あるいは…。
1934年証券取引法第13条又は15(D)条に基づいて提出された |
手数料書類番号
(登録者の正確な氏名はその定款に記載)
(明またはその他の司法管轄権 会社や組織) |
(税務署の雇用主 識別番号) |
(主にオフィスアドレスを実行) |
(郵便番号) |
登録者の電話番号、市外局番を含む:(
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
|
取引 記号 |
|
登録された各取引所の名称 |
|
|
♪the the the |
同法第12条(G)に基づいて登録された証券:ありません
登録者が証券法第405条に規定する有名な経験豊富な発行者である場合は、再選択マークで表示してください。はい☐
登録者がこの法第13又は15(D)条に従って報告書を提出する必要がないか否かを、再選択マークで示す。はい☐
登録者が(1)過去12ヶ月以内(または登録者がそのような報告を提出する必要があるより短い期間)に1934年の証券取引法第13条または15(D)条に提出されたすべての報告書を再選択マークで示すかどうか、および(2)このような提出要求を過去90日以内に遵守してきた
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
大型加速ファイルサーバ |
|
☐ |
|
ファイルマネージャを加速する |
|
☐ |
|
|
|
|
|||
|
☒ |
|
規模の小さい報告会社 |
|
||
|
|
|
|
|
|
|
新興成長型会社 |
|
|
|
|
|
|
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に第240.10 D−1(B)条に従って受信されたインセンティブベースの補償に従って回復分析を行う必要があるかどうかを再選択マークで示す☐
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)。はい☐違います
登録者非関連会社が保有する議決権と議決権なし普通株の総時価は,ナスダック普通株の2023年6月30日の終値に基づいている
2024年3月21日までに
引用で編入された書類
登録者が2024年株主総会の最終委託書(“委託書”)について本年度報告書に含まれる10−K表に含まれる財政年度終了後120日以内に証券取引委員会に提出する場合、委託書の一部の内容は、本年度報告の第3部分表10−Kに引用的に組み込まれる。依頼書が120日以内に提出されていない場合、登録者は、本年度報告書の改訂を120日以内に提出するであろう。この改訂は、本年度報告第III部分に格納または組み込む必要がある情報を含む。
監査役事務所ID: |
監査役の名前: |
監査役位置: |
カタログ表
|
|
ページ |
第1部 |
|
|
第1項。 |
業務.業務 |
4 |
第1 A項。 |
リスク要因 |
36 |
項目1 B。 |
未解決従業員意見 |
92 |
プロジェクト1 C。 |
ネットワーク·セキュリティ |
92 |
第二項です。 |
属性 |
93 |
第三項です。 |
法律訴訟 |
93 |
第四項です。 |
炭鉱安全情報開示 |
93 |
|
|
|
第II部 |
|
|
五番目です。 |
登録者普通株市場、関連株主事項及び発行者による株式証券の購入 |
93 |
第六項です。 |
保留されている |
94 |
第七項。 |
経営陣の財務状況と経営成果の検討と分析 |
94 |
第七A項。 |
市場リスクの定量的·定性的開示について |
100 |
第八項です。 |
財務諸表と補足データ |
100 |
第九項です。 |
会計と財務情報開示の変更と相違 |
100 |
第9条。 |
制御とプログラム |
100 |
プロジェクト9 B。 |
その他の情報 |
101 |
プロジェクト9 Cです。 |
検査妨害に関する外国司法管区の開示 |
101 |
|
|
|
第三部 |
|
|
第10項。 |
役員·幹部と会社の管理 |
102 |
第十一項。 |
役員報酬 |
102 |
第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
102 |
十三項。 |
特定の関係や関連取引、取締役の独立性 |
102 |
14項です。 |
最高料金とサービス |
102 |
|
|
|
第4部 |
|
|
第十五項。 |
展示品と財務諸表の付表 |
103 |
プロジェクト16 |
表格10-Kの概要 |
105 |
i
このForm 10-K年間報告書では、用語“私たち”、“会社”および“ProKidney”はProKidney Corp.(前社会資本Suvretta Holdings Corp.III)とその子会社を意味する。二零二二年七月十一日(“締め切り”)には、ケイマン諸島法律登録の免除会社社会資本Suvretta Holdings Corp.III(“SCS”及び本稿で述べた業務合併後の“当社”)は、二年一月十八日の業務合併協定(“業務合併協定”)の条項に基づき、SCSと有限責任組合企業ProKidney LP(“PKLP”)が業務合併(“業務合併合意”)を完了する。業務合併及び業務合併プロトコルが期待する他の取引(総称して“取引”及びこのような取引の“完成”と呼ぶ)によって、PKLPはSCSの付属会社となり、SCSはProKidney Corpと改名した。
前向き陳述に関する警告説明
以下、我々の財務状況と経営結果の検討と分析は、当社の財務諸表と本報告の他の部分に関する付記と併せて読まなければなりません。歴史財務情報以外に、以下の討論には、私たちの計画、推定、仮説、信念を反映した前向きな陳述が含まれている。私たちの実際の結果は展望的陳述で議論された結果と大きく違うかもしれない。これらの差異をもたらすか、または促進する可能性のある要因は、以下および本報告の“第1部分である第1 A項目リスク要因”の下で議論される要因を含む。展望的陳述は、私たちの可能性または仮定された将来の経営結果、業務戦略および経営、融資計画、潜在的成長機会、潜在的市場機会、私たちの薬物開発努力または試験の潜在的結果、および競争影響に関する情報を含む。前向き表現は、すべての非歴史的事実の表現を含み、“予想”、“信じ”、“可能”、“求める”、“推定”、“予想”、“意図”、“可能”、“計画”、“潜在”、“予測”、“プロジェクト”、“すべき”、“将”、“将”または同様の表現、およびこれらの用語の否定によって識別することができる。このような不確実性を考慮して、あなたはこのような前向きな陳述に過度に依存してはいけない。また、前向きな陳述は、私たちの経営陣の本報告日までの計画、推定、仮説、信念だけを代表しています。法的要件がない限り、私たちは、未来に新しい情報があっても、これらの前向き陳述を公開的に更新する義務がない、または実際の結果を更新することは、未来に新しい情報があっても、これらの前向き陳述で予想される結果と大きく異なる原因である可能性がある。
1
主なリスク要因の概要
私たちの業務は、私たちの業務目標の達成を阻害したり、私たちの業務、財務状況、運営結果、キャッシュフロー、将来性に悪影響を及ぼす可能性があるリスクを含む多くのリスクに直面しています。以下“第1部”
私たちの財務状況と追加資本の需要
腎臓自家細胞療法またはRipparencelの発展と私たちの未来の候補製品
Reactionの製造と未来の製品候補は
Reactionの商業化と未来の製品候補は
私たちの第三者への依存は
2
法律と規制コンプライアンス事項
我々の知的財産権は
私たちの業務と運営を管理します
私たちの組織構造は
3
パー?パーT I
プロジェクト1それは.公事です。
概要
2022年7月11日まで、私たちはケイマン諸島の法律に基づいて登録された空白小切手会社であり、1つ以上の企業との合併、株式交換、資産買収、株式購入、再編、または同様の業務合併を目的としています。2022年7月11日、PKLPと締結した2022年1月18日の企業合併協定に基づいて企業合併を完了しました。事業合併が完了した後、私たちは“ProKidney Corp.”と改名した。PKLPの業務は私たちの業務となります
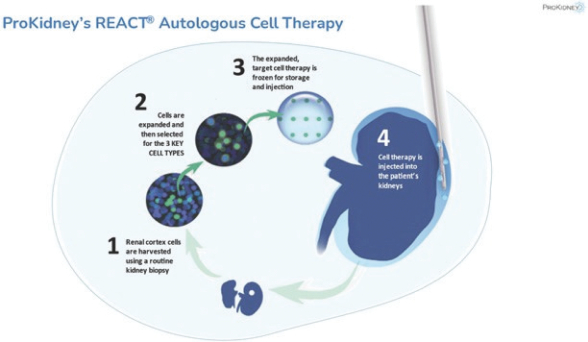
我々は臨床段階のバイオテクノロジー会社であり,変革性の特許細胞治療プラットフォームを有しており,患者自身が治療しようとしている患者から分離した細胞を用いて多様な慢性腎臓疾患を治療することが可能である。われわれの方法は,慢性腎臓疾患(CKD)の治療を再定義し,腎不全の管理から保存(改善しなければ)腎機能に重点を移すことを試みている。著者らの主要な候補製品Rilparencel、著者らは時々Reactionと呼ばれ、慢性腎臓病患者の病変腎臓の腎機能を保護することを目的としている。Reactionは、患者自身の(自家)腎臓細胞から調製された自己選択腎細胞(SRC)を含む製品である。SRCは、患者の腎臓に再注入するための製品として調製され、必要に応じて反復可能な低侵襲外来プログラムを使用する。Reactionは個人化治療であるため、患者自身の腎臓から作製した細胞から構成され、患者が別の(異遺伝子)ドナーの腎臓移植を受ける時、患者の一生の間に免疫抑制治療を行う必要はない。
著者らは現在、重度糖尿病腎症(“DKD”)被験者の反応に使用するための全世界第三段階開発計画と複数の第二段階臨床試験を行っている。先天性腎臓や尿路異常(“CAKUT”)によるCKD患者(“CAKUT”)に対する第1段階臨床試験も完了し,前回の被験者訪問は2023年1月,臨床研究報告は2023年12月にFDAに提出された。これまで,1期と2期の臨床試験では,中等度から重度のCKDを有する被験者はREPACTに対する耐性が良好であった。
我々の特許技術には,細胞治療製品の製造や医療交付における多くのブレークスルーが含まれている。長い間、人体は再生能力を有する細胞を含むと考えられてきたが、我々の技術は患者自身の腎臓細胞を拡大することによって、重要な前駆細胞あるいはSRCを準備し、同じ患者に再注射し、慢性病によって失われた腎臓機能を保存または改善しようとしている。患者が罹患した腎臓の小片や生検が私たちの製造工場に送られると、私たちの過程が始まった。生検から抽出した細胞を処理し,再生能を有する特定の細胞を選択することができる。これらのSRCは、損傷した腎臓(S)を再注入するための個人化された製品として調製される。これまで、臨床研究により、一定期間内に、Reactionは腎機能に積極的な影響を与えることができ、このことは安定に推定した糸球体濾過率(“EGFR”)或いは2型糖尿病によるCKD患者のEGFR低下の速度を遅らせることによって反映されている。
われわれは当初,米国では2型糖尿病による中から重度のCKD患者へのREACTIONの開発を求めていた。著者らは現在約3600万から3700万人の成人が慢性腎臓病を患っており、アメリカの成年人口の約14%を占めると推定した。慢性腎臓病は5つのCKD段階に分けられ、軽度(CKD段階1)から重症(CKD段階5或いは腎不全)まで。主に糖尿病による3 b期と4期CKD患者では,米国では約120万から180万人がREPACT治療を受ける資格があると推定されている。第三段階発展計画には、北米、ヨーロッパ、ラテンアメリカ、南米、アジア太平洋地域の他の国が含まれる。
我々は現在,FDAと欧州薬品管理局(EMA)の品質基準に適合し,生組織検査の材料から反応治療を生産する製造工場を運営している。この施設は米国ノースカロライナ州ウィンストン-セレムに位置し,我々の世界第3段階計画および潜在商業発射の第1段階を供給するのに十分な潜在能力を有しており,規制部門の承認を得るべきである。
4
私たちのパイプは
我々は,糖尿病およびCAKUTによるCKD腎不全を阻止または遅延させるために,我々の細胞治療技術を用いて候補製品を開発している。以下の表に我々が現在行っている臨床研究をまとめた
Reactionは研究療法であり,参加者自身の腎組織から作製した腎臓細胞の混合物である。最初の腎組織は専門研修を受けた医師による標準腎生検であった。この組織から,腎臓の治癒に重要であると考えられる腎臓細胞は,高度に特化した実験室施設で培養·増殖している。数週間後、十分な細胞が利用可能になると、細胞の反応は臨床試験場所に送り返される準備ができ、そこで参加者自身の腎臓に直接注射することができる。時間の経過とともに,これらのSRCは罹患した腎臓の安定あるいは機能改善を助ける可能性があり,腎機能を温存することにより透析や移植の需要を潜在的に遅らせることができると信じられている。
臨床研究により、2型糖尿病による慢性腎臓病患者において、ReactionはEGFRの安定或いはEGFRの低下率を緩和することによって、腎機能に積極的な影響を与えることができる。我々は、我々のReaction第3段階試験(REGEN−006およびREGEN−016と呼ばれる)および2型糖尿病によるCKD患者の治療のための製品の長期保存を可能にする低温保存バージョンのReactionバージョンを開発した。凍結保存したReaction製剤に加えて,われわれの第2段階試験(RMCL−002とREGEN−003と呼ぶ)と先天性腎臓と尿路異常の第1段階試験(“CAKUT”)(REGEN−004と呼ぶ)にゼラチン系ハイドロゲル配合を用いた。反応の低温保存バージョンは、一般に、単一の生検試料から最大5~10剤を調製することを可能にする。われわれは2つの臨床前計画(Reaction/GenとReaction/Universalと呼ぶ)があり,遺伝子組換えの生物活性腎臓細胞群を用いて罹患した腎臓に再生効果を提供する予定である。臨床前計画では“汎用ドナー”細胞集団の創出が求められており,遺伝子編集は免疫反応を生じない新たな腎臓細胞集団を産生するために用いられており,“既製”製品として使用可能である。
私たちのチームと会社の歴史は
著者らは経験豊富な内部研究開発チームを持ち、著者らの腎臓疾患経路の深い理解を利用して、様々な経路に対するマルチモード機序を有する細胞に基づく新しい治療法の発見と開発に集中している。私たちの設立以来、私たちは私たちのチームを拡大し、必要に応じてより多くの専門知識を取り入れて、私たちが完全に統合されたバイオ製薬会社になる目標を達成しました。著者らは腎臓疾患、細胞治療、開発、監督管理事務、運営、品質、製造と商業の方面で専門知識を持つ肝心な管理チームのメンバーを集めた。
ProKidney LLCの前身はPKLP(“ProKidneyバミューダ”)の完全子会社で、2018年12月に設立され、バミューダ有限責任会社として製薬業界の投資家たちによって設立された。
ProKidney(前身はRegenMed(Cayman)Ltd.(d/b/a inRegen)(“ProKidney-KY”)は2015年12月21日にケイマン諸島会社法(改正)(“ケイマン諸島会社法”)によって正式に免除会社に登録された。2020年、ProKidney-KYはRegenMed(ケイマン)有限公司からProKidneyと改名した。プロキッドニ有限責任会社
5
(前身はTwin City Bio LLC)(“ProKidney-US”)は、Twin City Bio LLCからProKidney,LLCと改名した。ProKidney-USはデラウェア州の有限責任会社で、2015年12月18日に設立された。2019年1月、ProKidneyバミューダはProKidney-KYとProKidney-USの全株式を買収した。
ProKidneyバミューダはProKidney−KYの株を買収し,その腎臓自己細胞療法を開発した。ProKidneyバミューダはProKidney-USを買収し、ProKidney-KYに契約開発と製造サービスを提供した。
PKLPは2021年8月5日、アイルランド1907年の“有限パートナーシップ法”(“アイルランドパートナーシップ法”)と適用された1890年にアイルランドパートナーシップ法に基づいて有限パートナーシップとして設立され、ProKidneyバミューダはPKLPの完全子会社となった。2021年9月、ProKidneyバミューダはProKidney-KYとProKidney-USでの持分を割り当てた
PKLPを買収し,ProKidney−KYとProKidney−USをPKLPの直接完全子会社とした。原腎
百慕は2022年10月に解体された。
“ProKidney”や“会社”は,一般に再編後のPKLPと閉鎖後のProKidney Corp.を指す.
私たちの戦略
我々の目標は,全面的に統合された生物製薬会社となり,CKD治療の先駆者となることである。私たちのビジネス戦略の主な構成要素は
|
|
|
監督管理部門の許可を得て、REPACTの商業化に成功し、最初は2型糖尿病によるCKD患者の治療に応用されたEMAとFDAが共同審査した世界第3段階臨床開発計画により,REACTの臨床開発を進めていく予定である。私たちは2021年第4四半期に私たちの第1の3期臨床試験のために第1のサイトREGEN-006(私たちは“PROACT 1”とも呼ばれる)を活性化し、最初のインフォームドコンセントに署名し、2022年第1四半期にランダムに第1の被験者を試験に入れた。同社は2024年初めに規制機関にこの研究の案修正案を提出し、2024年上半期に科目募集を再開する予定だ。我々の第2段階3試験REGEN-016(“PROACT 2”とも呼ぶ)は,2024年通年でサイトを活性化する予定である.REGEN-016の募集は2024年下半期に始まる予定だ。2023年第4四半期に、私たちの試験の一部としてREACTを受けた被験者に対して、REGEN-008長期追跡試験を開始した。 |
|
|
|
我々の細胞治療法を用いて,腎臓疾患治療のための他の候補製品を発見·開発した私たちのチームは腎臓疾患について豊富な発見研究経験と深い専門知識を持っている。その開発と商業化REPACTの主要な任務を遂行するとともに,我々のチームは腎臓疾患に関連する他の疾患経路を調査し,関与の重要な目標を決定し,これらの目標に対して候補製品を生成する。第三者から許可を得たり,第三者と協力したり,第三者を買収したりして候補製品を開発することも可能であり,腎臓疾患や治療経路の理解に基づいて,これらの製品は有望な治療法であると考えられる。 |
|
|
|
私たちの製品製造に関する複雑な内部専門知識を維持し、改善していきますそれは.現在の良好な製造実践(“cGMP”)に沿って運営するための製造工場を開発·建設しており,その中で臨床試験のための反応を製造し,必要な規制承認を得ることを前提として最終商業製造過程のための目的を開発していく予定である。私たちの現在の製造施設は私たちの第三段階の臨床試験のために製品を製造し、私たちの商業発射施設とすることができます。私たちは商業化反応の需要を満たすために私たちの生産拠点を拡大する予定だ。 |
腎疾患の概要
慢性腎臓病はアメリカとEUで非常に流行している。2018年の全国健康·栄養検査調査の既存米国データによると、米国とEUのCKD総人口は2020年の約7000万人から2030年の約8000万人、2040年の約9300万人に増加すると予想される。成人CKDの最もよく見られる原因は糖尿病、高血圧と糸球体疾患であり、児童群の中で、CAKUTは最もよく見られる原因である。米国では,年間約320万人の患者が3 b期または4期CKDを有しているとされており,そのうち120万人から180万人の患者がREPACT治療を受ける資格がある可能性がある。
私たちの方法:体細胞療法で腎臓機能の保護に努めています
自家細胞療法とは,ヒト自身の細胞を用いてヒト疾患を予防または治療することであり,これらの細胞は体外で選択,増殖,調製され,細胞由来の患者に注入される。我々の技術は,患者自身のSRCを用いることで腎機能を保護する可能性があると信じている。
Reactionは自己相同細胞混合物であり、これは、各個体の対象自身の腎臓生検組織から得られた拡大SRCから作られることを意味する。
6
病変腎臓の生検がわれわれの実験室に送られ,そこで腎臓細胞増幅とSRC選択が行われると,準備反応が開始された。我々は患者自身の健康前駆細胞を識別し,それを個人化製品に調製することができる。SRCは約100×10の濃度で低温保存製品に調製した6細胞/mlは、凍結して臨床現場に搬送され、そこで患者の損傷した腎臓の皮質に注射することができる。早期のREPACT注射中に重篤な出血が発生したため,非切断針設計のREPACTプログラムに変更した。臨床前研究によると、生産された反応製品が罹患腎臓の皮質に注射されると、製品の前駆細胞は腎臓全体に迅速に分布し、損傷した腎単位と間質に統合される。これまで、臨床研究により、2型糖尿病によるCKD患者に対するREPACT治療はEGFRの安定或いはEGFR低下率の緩和を通じて腎機能に積極的な影響を与える可能性が示唆された。臨床前および臨床反応治療で観察された他の改善には,代謝やろ過上の利点,UACRの安定および/または減少,腎臓皮質厚の増加,ヘモグロビンレベルの改善,リン酸カルシウムやビタミンD 3の改善がある。
反応の作用機序
SRCの移植は内因性腎臓修復機序を活性化するために分子と機序の基礎を提供すると考えられているが、内因性腎臓修復機序は慢性疾患腎臓において依然として活発である。多種のCKD動物モデル上の非臨床研究により、SRCは直接腎皮質に注射され、直接移植或いは組織置換、及び可能な傍分泌機序を通じて、腎単位の複数の位置で再生反応を産生することができ、分泌因子の影響に関与する。CKDの動物モデルにおいて、SRC治療は腎機能を保留し、蛋白尿を減少させ、著しい生存利益を提供した。また,臨床前モデルからの証拠は,SRC投与が骨やミネラル代謝や造血に関連する他の重要な腎機能を増強する可能性が示唆された。CKDの動物モデルでは,SRCはCKD二次性副甲状腺機能亢進による破骨細胞性骨吸収の進展を最大限に減少させ,赤血球産生に関連する骨髄細胞密度を改善することが証明されている。
私たちの候補製品
REPACTは現在第3段階開発にあり,2型糖尿病による中から重度CKDの治療に用いられている第2段階臨床試験が行われている。これらの試験は米国,ヨーロッパ,アジア,ラテンアメリカの約150の臨床地点で行われる予定である。これまで,2型糖尿病による慢性腎症患者の臨床研究により,REACTION治療はEGFRを安定させることで腎臓代替療法(“RRT”)を延期あるいは阻止する可能性が示唆されてきた。早期研究で観察されたREACTの他の潜在的改善には,UACRの安定,腎臓皮質厚の増加,血清炭酸水素塩の改善およびヘモグロビンレベルの安定がある。REACTIONの潜在的利益は、2つの十分な対照臨床試験を含む第3段階計画において評価されるであろう。
現在行われている臨床開発は,経皮的腎臓注射法を用いて,外来当日のプログラムで意識的鎮静を用いて行う予定である。この手術は一般的に耐性が良好であり,最もよく観察される手術に関連するイベントは嘔気,嘔吐,発熱と腎生検血尿である。RMCL-002では
7
この試験では,我々の現在の第3段階試験で使用されている異なる配合の反応製品と異なるプログラムを用いて,一人の参加者が瘢痕形成や線維化および腎機能低下を含む深刻な有害事象を経験した。第2の被験者は、比較増強されたコンピュータ断層撮影(CT)画像において腎臓血流量の減少および腎機能の低下を認めた。また、注射関連の痛み、腎臓に関連する事件、例えば血腫、腎血管事件、EGFR低下と急性腎臓損傷を含む深刻な不良事件を報告した。その他の深刻な不良事件は、急性心筋梗塞、急性呼吸不全、末期腎症と冠状動脈疾患もすでに報告されており、通常は2型糖尿病患者の併存と関係がある。
背景と未満足の需要
慢性腎病
慢性腎臓病の特徴は進行性疾患であり,治療介入がなければ,患者が死亡あるいは終末期腎臓疾患(ESRD)に達するまで病態が悪化する。CKD患者の腎臓は進行性損害と機能喪失があり、実験室検査により、EGFR減少及び/或いは尿アルブミン排泄増加は明らかであることを発見した。CKDの全世界の成人罹患率は10%と推定され、異なる人群の範囲は8-16%である。慢性腎臓病は通常糖尿病などのかなり大きな合併症と関連し、そして常に潜在疾患状態と/或いはリスク要素(例えば心血管疾患、高血圧と糖尿病)の不良結果を伴い、死亡リスクの増加を招く。97%の中から重度の慢性腎臓病患者の無症状の疾患であり,この段階の慢性腎臓病と 心血管疾患のリスクは2倍から4倍に増加し、同時に各種の原因の死亡率は明らかに増加した。一部のCKD患者だけが末期腎症(即ち5期疾病)に進展したが、人類の期待寿命の延長はますます多くの慢性病患者と末期臓器不全患者を招いた。高価な治療を受けても,末期腎症患者の発症率と死亡率は高い。生存のためには,終末期腎症患者は腹膜透析,血液透析あるいは腎移植による腎臓代替治療(RRT)が必要である。CKDの進展を予防或いは遅延し、内科合併症の発生と治療合併症を遅延させることはCKD治療の重要な策略である。許可された慢性腎臓病患者を治療する方法はすでにある患者の慢性腎臓病の進展を遅らせることができることが証明された。しかし、多くの患者は引き続き腎機能を失い、末期腎症に進展した。
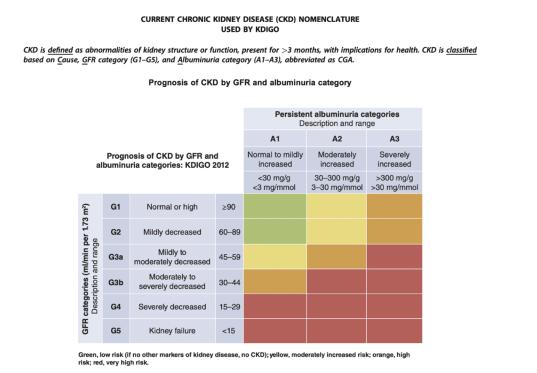
成人慢性腎臓病の主要な病因は糖尿病と高血圧である。CKD症例のほぼ半分は糖尿病によるものであり,高血圧の有無にかかわらずである。慢性腎臓病の発病率は引き続き増加し、これは主に全世界の2型糖尿病と代謝症候群の発病率の増加によるものである。腎機能の分期と分級はEGFRと尿アルブミン排泄(“アルブミン尿”)の測定に依存する。KDIGO 2012慢性腎臓疾患評価と管理臨床実践ガイドラインは全科医師と腎臓科医師が成人と児童慢性腎臓病に対する評価、分類と管理を助けるためのガイドラインを提供した。以下に示すように、図1は2種類のEGFR測定結果に基づいてESRDのリスクを“低”から“極めて高い”に分類し、範囲は>90 ml/分/1.73メートルからなる2至れり尽くせり2蛋白尿の分類は300 mg/gから様々ですEGFRが到達すると23ヶ月以上持続し、通常透析または腎移植治療が行われる。
8
CKD分類推定要約
CKD患者の治療の重点は合併症(例えば糖尿病と高血圧)と内科合併症を治療し、心血管リスクを下げ、腎機能低下を緩和し、そして腎機能不全或いは腎臓代替治療の準備である。多くの患者にとって、慢性腎臓病は心血管疾患と2型糖尿病などの複雑な共病群の一部である。
医薬治療は、アンギオテンシン変換酵素阻害剤および/またはアンギオテンシン受容体遮断薬、ナトリウム-グルコース共輸送体-2(SGLT-2)阻害剤、非ステロイド性鉱質コルチコイド受容体アンタゴニスト、血糖コントロールおよび脂質低下治療のためのインスリンおよび/または抗糖尿病薬のいずれかまたは全部を含むことができる。
患者が終末期腎症に達した場合,通常腎透析や移植の形でRRTを行い,延命する必要がある。アメリカとその他の先進国の大多数の透析患者は主に血液透析、腹膜透析或いは家庭血液透析の形式で透析を受けている。透析は正常機能のない腎臓に蓄積した毒素を除去することができ,腎不全時には透析も液体と電解質のバランス回復に寄与する。血液透析は多種の深刻な合併症及び生活の質に対する妨害と関係がある。腎臓移植は現在依然として末期腎症を治療する最も有効なRRT形式であるが、器官は長期的に不足している。患者が移植した腎臓の安全を確保できれば,拒絶反応を防ぐために長期的な免疫抑制治療を行う必要がある。免疫抑制剤の使用はより高い感染率をもたらし,長期的には何らかのタイプの癌を引き起こすこともある。異種移植は未来にヒト器官、組織と細胞需給不足を埋める有望な代替方法である可能性があるが、現在免疫障害は臨床異種移植の制限要素である。
患者は既存の治療時に腎機能を喪失し続けているが,CKD治療のコストは高く,CKDとESRDの発生率および関連支出は引き続き上昇すると予想される。アメリカの医療保健支出の主要な源として、CKD受益者の毎年の医療保険支出は約860億ドルである。ESRD受益者の連邦医療保険支出は約520億ドル,連邦医療保険優勢は約180億ドルであり,1患者あたり毎年透析に用いる総コストは9.5万ドルを超えている。
臨床発展
著者らが完成した臨床試験と現在行われているREACTION臨床試験を以下にまとめ,試験中に試験された候補製品の名称はNeo−Kidney Augment(“NKA”)のいくつかの試験が完了した後にREACTIONに変更された。
以下の要約には一時的な結果も含まれており,その根拠は当時利用可能なデータの初歩的な分析であり,その結果,関連結論,結論は具体的な実験に関するデータをより網羅的に審査した後に変更される可能性がある.特に私たちは発表しました仮われわれが行っているRMCL−002第二段階臨床試験の中期結果は2023年11月に発表される。完成したREGEN−003第2段階臨床試験とREGEN−004第1段階臨床試験の最終結果を提出した
9
臨床試験は2023年末にFDAに提出される。われわれはすでにREGEN−006第3段階臨床試験の募集を開始しており,REGEN−016第3段階臨床試験は2024年に開始予定である。我々が完成する可能性のある臨床試験の中期結果は必ずしも未来のデータからの結果を示すとは限らず、またより多くの患者データの獲得に伴い、1つ以上の臨床結果が実質的に変化するリスクが発生する可能性がある。初歩的な結果あるいはベースラインの結果は依然として監査とチェック手続きを守らなければならず、これは最終データが私たちが以前公表する可能性のあるいかなる初歩的なデータと大きく異なることを招く可能性がある。したがって、最終データを得る前に、中期と予備データを慎重に見なければならない。
糖尿病による慢性腎臓病
第1段階臨床開発(TNG−CL 010とTNG−CL 011)
TNG−CL 010はREACT(従来NKAと呼ばれていた)に対するオープンラベル安全と送達最適化研究であり,スウェーデンでCKD被験者に行った。TNG-CL 010は2013年4月に着工し、2014年12月に完成した。TNG-CL 011も米国で行われた2型糖尿病およびCKDによって引き起こされるCKD対象に対する開放ラベルの安全性および送達最適化の研究である。TNG−CL 011は2014年2月に最初の科目の登録を開始し,2014年12月に完成した。
これらの試験の主な目的は、1つの腎臓に注入した場合の反応の安全性と伝達を評価することである。6人のスウェーデン患者(TNG-CL 010)および1人のアメリカ患者が選ばれ、年齢53-70歳、EGFRレベル19-34(平均25+/-2、Cystatin C)、ヨードハイアルコール除去量15-39(平均26+/-3)。TNG−CL 011には2型糖尿病による慢性腎症患者が登録されている。
第1段階試験の結果,腎臓に投与した場合,REPACTの耐性は良好であり,REPACTからの有害事象はなかった。投与前と投与後の腎機能低下を比較したところ,この段階1試験でREACTIONを受けた被験者の透析遅延は約1.5年であり,EGFRが投与前よりベースライン低下速度が減少したことに基づいていた。投与した腎臓皮質厚は注射時の平均14 mmから1年後の約16 mmに増加した。反応注射後,被験者のACRによりヨードハイアルコール除去により腎機能を温存した。ベースライン貧血を有する被験者(7人中3人)はReaction投与後にヘモグロビンレベルが改善し,残りの被験者は研究期間中に正常レベルを維持した。REACT投与後の前6カ月では,6名中3名の患者の降圧薬が減少した。
TNG−CL 011からのデータは、第2段階に進行した臨床データパケットの一部としてFDAによって受け入れられる。
第二段階臨床開発(RMCL−001、RMCL−002、REGEN−003およびREGEN−007)
RMCL-001:
RMCL-001は2型糖尿病による慢性腎症患者に対する2期、開放ラベルの安全性と有効性の研究である。この研究は2016年5月に始まり、2017年5月に終了した。
この研究の主な目的は,第二次反応注射の安全性と有効性を評価することであり,覚醒鎮静下で行われる低侵襲経皮操作を,同日の外来操作として用いた。個々の被験者のEGFRは14 ml/分/1.73 mであった2上記第1段階研究(TNG−CL 011)から登録されている。第2剤反応は,第1段階腎生検から得られた凍結保存腎細胞から作製された。被験者は3 x 10の投与量を投与された6Cells/g-KWESTそれは.被験者のEGFRは約20 ml/分/1.73メートルに増加した28カ月後,被験者の腎機能は急激に低下し,血液透析を開始した。臨床試験の発起人は透析後にこの研究を終了し,研究RMCL−002に資源を移した。
RMCL-002:
RMCL-002は2型糖尿病と慢性腎臓病患者に対して行った2期展望性、ランダム、両腕、治療延期、開放ラベル、重複投与、安全性と有効性の研究である。最初の被験者は2017年2月に本研究に入り、すべての被験者はすでにフォローアップを完成した。
この研究の主な目的は、2回の反応性注射の安全性と有効性を評価することであり、これらの注射間隔は6ケ月(目標日後4週間)で、eGFRsの20から50 ml/min/1.73 mの間の2型糖尿病によるCKDを治療することである2外来低侵襲経皮経路を用いて、覚醒鎮静下で90分未満で2種類の用量の腎臓生検を完成した。患者は2用量の3 x 10の反応を受けた6Cells/g-KWESTみんなです。
患者は腎臓生検後にランダム(1:1)に積極治療群と遅延治療群(即ち対照群)に分けられた。能動治療群の被験者は、反応製品を製造し、臨床現場に搬送した後、直ちに第1回の反応注射を受けた。6カ月後(目標日後4週間以内)、適宜2回目の注射を行った。対照的に,治療延期群の被験者は腎生検後12カ月の観察期間を受けた。延期治療群は積極治療群と比較して,非曝露群の腎機能変化率と合併症の評価を許可した。その間彼らは同期の標準看護を受けました
10
CKDを治療すると同時に3ケ月ごとにフォローアップ評価を行い、積極的な治療群の被験者に類似した。12ヶ月後、治療を延期した群の被験者は、6ヶ月間隔(目標日の+/-4週間)間隔で最大2回の反応注射を受け、状況に応じた。そのため、研究設計は前12ケ月に標準看護治療を受けたランダム対照群と同一時間帯に最大2回の反応注射と後続評価を受けるランダム積極治療群を含む。
第二段階臨床試験に参加した被験者の総数は83人であった。2名の被験者を中止および/または交換した後、2020年12月までに81名の被験者が入選し、そのうち41名の被験者が積極治療群に入り、42名の被験者が延期治療群に入った。
私たちは安全性と有効性の最終結果が2024年上半期に発表されると予想している。能動治療群の腎機能進展率は延期治療群の腎機能進展率と比較し,最終反応注射後24カ月以内にEGFRを予備ランダム系列測定することにより評価した。また,履歴と臨床データから得られた被験者ごとのEGFR低下率ベースライン比率を,個別被験者のREPACT最終投与後24カ月間のEGFR低下率と比較した。一次反応注射を受けた被験者(あれば)の腎機能進展率は,2回の反応注射を受けた被験者と比較することができた。試験第1部では,最終反応注射後24カ月の追跡調査を行った。本研究のオープンラベル拡張部分(第2部分)は、すべての被験者をさらに3年間追跡するために2021年2月に増加した。訪問は3カ月ごとに行い,最後のReaction注射後に合計5年間のフォローアップを行った(第1部+第2部)。
この研究の最新中期データは2023年11月に発表され,その中には研究に取り入れられた83名の患者のデータが含まれている。一時データ集が示した安全性は以前に報告された早期1期と2期試験データと一致し、耐性は常規の腎臓生検に類似している。全体的に、最新の試験データにより、2型糖尿病による末期CKD患者の中で、REACTIONは腎機能を保護する潜在力があり、最も顕著な潜在的利益は腎機能不全のリスクが最も高い患者(CKD段階4、中から重度の蛋白尿がある)であることを示した。
2024年上半期にRMCL−002第2段階研究の安全性と有効性の最終結果を提供する予定である。一時的なデータの結果は未来のデータの結果を表さないかもしれない。
Regen-003:
Regen-003はすでに完成した第2段階展望性開放ラベル、単腕、安全性と耐性研究であり、2型糖尿病による慢性腎臓病患者、特にそれらの高リスクの末期4 DKD患者の研究に用いられる。この研究は2018年3月に1人目の患者を募集した。早期研究結果は2023年1月にネット上で発表された血液浄化雑誌“2型糖尿病合併末期糖尿病関連慢性腎臓疾患の腎臓自己細胞療法(REACT):試験設計と早期分析”(DOI:DOI:DOI.org/10.1159/10.1159)と題する原稿にある。臨床研究結果は2023年9月8日にFDAに提出された。
この研究の主な目的は2回の反応注射の安全性と有効性を評価することであり、これらの注射間隔は6ケ月(目標日後4週間)で2型糖尿病による慢性腎不全患者を治療し、そのeGFRsは14から20 ml/min/1.73 mの間である2そして低侵襲経皮経路を用いて生検した腎臓に輸送し、この経路は覚醒鎮静下で90分未満で輸送することができる。被験者のEGFRは14~20 ml/分/1.73 mであった2それは.被験者は最大2剤3 x 10の反応を受けた6Cells/g-KWESTみんなです。
この研究では,大人10名(男性5名と女性5名)を募集した。経皮的腎生検とSRC体外拡張形成反応後,CT画像ガイド下に反応産物を生検腎の皮質に注入した。9人の参加者は6ヶ月ごとに2回のReaction製品の注射を受け、1人の参加者は1回のみ注射を受けた。試験前に6ケ月の観察期間を要し、患者自身のベースラインとCKD進展率を確定した。細胞製品に関連した深刻な有害事象は報告されていない。3名の参加者は,急性腎損傷,慢性腎臓病進展,腎動静脈瘻,血腫を含むこの反応手順に関連する腎臓関連の重篤な有害事象を報告しており,いずれも輸血や血管造影介入なしに観察する必要がある。結局,平均透析期間は19.4カ月(四分位数範囲13.3−27.9)であった。2名の患者(20%)は研究を完了したが(2回目の注射後24カ月),RRTには入らなかった。1人の患者はCOVIDに関連する合併症で死亡し、もう1人の被験者は登録約18カ月後に心筋梗塞で死亡した。この研究の結果,ハイリスクの4期と5期のDKD患者では,Reactionが透析を遅らせる可能性が示唆された。
Regen-007:
Regen-007は進行中の第二段階展望性、ランダム、開放ラベル、反復用量、2つの列、1型或いは2型糖尿病と慢性腎臓病患者に対する安全性と有効性研究である。
コホート1の主な目標は、eGFRが20~50の間の1型糖尿病および2型糖尿病によって引き起こされる慢性腎症患者において、3ヶ月間隔(目標日後60日)に最大2回の反応注射を行う安全性および有効性を評価することである
11
Ml/分/1.73 m2低侵襲経皮経路を用いて生組織と非生組織の対側腎臓に輸送した。また、Cohort 2の主な目標は単回反応注射を実施し、その後、2回目の反応注射をモニタリングし、実施する可能性があり、生検されていない対側腎臓に注射し、EGFRの20%低下および/または尿アルブミン/クレアチニン比(UACR)の30%増加を誘発し、事件発生後約60日以内に増加させることである。これまでの第2段階研究では,同一腎臓にREPACTを2回投与した。従来研究で観察された全体的に良好な安全性から,REGEN−007では現在両腎内投与Reactionを行っている。REGEN-007によって生成されたデータは、2つの腎臓注射による腎機能への影響をよりよく知ることができると予想され、これは著者らの第3段階で研究した用量レジメンである。
2つの腎臓を注射することにより、患者は最大限に反応細胞に暴露し、より大きな割合の腎臓腫瘍に影響を与える可能性がある。さらに、2つの腎臓を注射すると、再生治療を受ける糸球体(腎臓の濾過ユニット)の数が有効に2倍に増加した。
被験者は3 x 10のReaction注射を最大2回受けます6Cells/g-KWEST1剤当たり。この研究では、30歳から80歳までの年齢、EGFR>20、50 ml/分/1.73メートルの被験者を募集します2それは.被験者は腎生検前にランダム(1:1)に2群に分けられた。第1群は3カ月間隔で2回Reaction注射を受けた。キュー2は1回目の反応注射を受け、以下に述べるように、1回目の注射から3ヶ月以上後に第2回反応注射を行う資格があるため、トリガー条件を満たさなければならない。
キュー1の各被験者は、REPACT製品が生産され、臨床現場に搬送された直後に最初のREPACT注射を受ける。3ヶ月後、被験者は適宜2回目の注射を受ける。コホート2の被験者もREPACT製品を生産して臨床現場に搬送した直後にREPACT注射を受ける。キュー2では、被験者が1つ以上の臨床代替マーカー基準に適合している場合にのみ、第2回反応注射が行われる。第2の反応注射は、第1の注射後3ヶ月以上、再用量トリガーに達してから30日以内(目標日後最大4週間)にキュー2内の対象に注射される。再投与のトリガー因子は、(1)EGFRが30日以内に持続的に低下し、ベースラインより少なくとも20%低下する;(2)ベースラインUACRが少なくとも30%増加し、30 mg/gを超え、ベースライン測定後30日以内に測定することを含む。2回目の注射を受けたすべての被験者について,2回目の注射は生検されていない対側腎臓で行われる。
その間、プラトゥーン2の被験者は、1回目の注射後1、14(目標日後7日)、28日(目標日後7日)に同期の標準ケア治療を受け、その後、1回目の注射後3ヶ月(目標日後10日)に追跡評価を行い、キュー1の被験者と類似している。すべての被験者は、最終注射後3ヶ月毎に18ヶ月間の長期フォローアップを行う。また,各被験者のベースライン腎臓衰退率は,初回投与REPACTの24カ月前に得られた十分な歴史と臨床データに基づいて,一定期間の腎機能不全進展率をモニタリングする比較指標とした。REGEN-007の主要な治療効果の終点は腎機能衰退率の減弱であり、これは18ケ月以来のEGFR総(急性+慢性)傾斜と注射前基準値の変化によって示された。主な安全終点は最後のREPACT注射後18カ月以内に発生した治療緊急不良事件である。Regen-007は非盲検法研究であり、その中でキュー1患者は著者らの第3段階計画中の患者と同じ治療方案を受け、これらの患者はランダムに有効なARMに割り当てられる。
REGEN−007は,我々の第3段階計画で観察された臨床的利益にいくつかの知見を提供する可能性があると信じている。
私たちはすでにREGEN-007の募集を完了し、合計53科目です。著者らは2024年にこの研究の中期データを報告し、限られた数量のCohort 1患者の12ケ月のフォローアップを反映し、2025年上半期にCohort 1の完全な12ケ月のデータを報告する予定である。
第三段階臨床開発:Proact 1とProACT 2(REGEN−006とREGEN−016):
我々は2021年10月28日にREPACTのRMAT称号を申請し,FDAの承認を得た。RMAT指定の想定とFDAの指示により,REPACTの臨床前と臨床開発と製造状況を審査し,候補製品承認を支援するための計画臨床計画を検討するために,FDAと包括的·多学科B型会議を開催することが求められている。FDAは我々が会議要求で提起した質問に対して詳細な書面回答を提供し,クラスB会議は2022年3月に開催された。その会議の結果として、第3段階試験設計と端末を修正した
|
|
|
REGEN−006およびREGEN−016の計画サンプル量を500人から600人に増加させる |
|
|
|
REGEN−006およびREGEN−016の主要複合終点から、90日間持続したランダム尿微量アルブミン/尿クレアチニン比を用いて、UACRの少なくとも30%および少なくとも30 mg/gの増加を除去した |
|
|
|
REGEN−016の設計では、偽制御アームおよび単一ブラインドアセンブリが追加され、以下からなる複合イベント間隔時間が追加される |
|
|
|
2009年CKD-EPI血中クレアチニン方程式を使用して、30日間持続し、EGFRは少なくとも40%減少した |
12
|
|
|
EGFR.EGFR |
|
|
|
腎臓や心血管疾患で死亡します |
我々は引き続き米国の臨床開発計画を推進し、FDAの登録計画に対する期待と要求の解像度を高め、承認に必要な試験の設計、生産分析、比較可能性研究を含む、以下に述べる。
REGEN-016/PROACT 2:
Regen-016(PROACT 2)は計画した3期ランダム、二重盲検、偽対照、両側腎投与量、対照治療効果の研究であり、2型糖尿病(特にEGFRは20 ml~44 ml min/1.73 mの間)による3 bと4 CKD期の被験者の反応に用いられる2中から重度の蛋白尿(UACRは300から5,000 mg/gの間)がある。この研究はヨーロッパ、ラテンアメリカ、アジア太平洋地域、アメリカのいくつかの臨床センターで行われる。
この研究の主な目的は最大2回から3ケ月離れた反応注射を評価し、低侵襲経皮経路を用いてそれを生組織検査と非生組織検査の対側腎臓に輸送することである。登録された対象は、疾患段階および看護基準に従って階層化され(例えば、SGLT 2および/またはMRAを使用して)、次いで、腎臓生検の前にランダム(1:1)に治療群または“覆面”偽対照群に割り当てられる。計画募集総人数は600門である.
治療群の被験者は3 x 10のReaction注射を2回受けます6Cells/g-KWESTそれは.この研究は30歳から80歳の間の研究対象を募集してEGFRを行います>20とMl/分/1.73 m2.
治療群の各被験者は、腎生検後約14週間以内に第1回反応注射を受ける。3カ月後,対側腎臓に2回目の注射を行う予定である。対照群の被験者は2回の偽注射を受け,1回目は偽生検後12週,2回目は1回目の偽注射後3カ月で行った。その間、対照群の被験者は治療群の対象と同じ時間間隔のフォローアップ評価を受けながら、同期の標準看護治療を受ける。すべての被験者は、世界的な実験終了日が発表され、研究訪問が終了するまで、この研究を継続する。
主な複合ゴールは最初の注射から最初の時間までです
|
|
|
2009年CKD-EPI血中クレアチニン方程式を使用して、30日間持続し、EGFRは少なくとも40%減少した |
|
|
|
EGFR.EGFR |
|
|
|
腎臓や心血管疾患で死亡します |
同社の契約制合格者(QP)は最近、EUでREACTを発行·配布する準備状況を評価する監査を行い、EUの臨床場所に製品を配布·配布する前に、解決すべき品質管理システムファイルにいくつかの欠陥があることを発見した。したがって、EUと世界標準の第3段階計画を満たすために私たちの能力を最適化し、商業製造に移行しようとすると、製造は一時停止される。一時停止しなければならない安全事件は何もない。PROACT 2は2024年下半期に募集を開始することが予想される.
REGEN-006/PROACT 1テスト:
REGEN-006(PROACT 1)は進行中の3期ランダム、盲法、両側腎投与量、偽対照、対照治療効果の研究であり、2型糖尿病による慢性腎症患者の反応を研究するために用いられる。この研究方案は修正されており、4期CKD患者のサブセット(EGFRは20 ml~30 ml min/1.73 mの間にある)に重点を置いている2)および末期3 b CKD(EGFRは30 ml~35 ml min/1.73 mの間である2)に蛋白尿を伴う。この研究は米国,カナダ,オーストラリア,メキシコ,台湾,イギリスの臨床センターで行われる。
EUおよび世界標準の第3段階計画を満たすために生産を一時停止し、その能力を最適化するために、企業は、RMCL−002の中間データおよび支払者および健康技術評価(HTA)の専門家からのフィードバックに基づいて、REGEN-006を積極的に修正して、成功の可能性を増加させるためにREGEN-006を積極的に修正している。同社はEGFR登録範囲を現在の20~50 ml/分/1.73平方メートルから新たな20~35 ml/分/1.73 mの範囲に修正する2それは.この修正は腎臓疾患の進展リスクが最も高い患者への関心を反映している。医療看護に関する大きなコスト負担により,これらの患者も特に支払者の興味を引いている。臨床サイトのフィードバックによると、同社はより多くの行政改革を取り入れ、運営効率と臨床サイトの参加度を高める予定である。このような合意にどんな変更も必要なセキュリティ問題はない。同社は2024年初めに規制機関にこの研究案修正案を提出し、2024年上半期に被験者の登録を再開する予定だ。
13
この研究の主な目的は最大2回から3ケ月離れた反応注射を評価し、低侵襲経皮経路を用いてそれを生組織検査と非生組織検査の対側腎臓に輸送することである。この研究で最初に計画された600名の被験者は,すでに登録されている約85名の被験者を含め,総目標登録者数は約685名の患者に増加する。治療群の被験者は3 x 10のReaction注射を2回受けます6Cells/g-KWEST.
主な複合ゴールは最初の注射から最初の時間までです
|
|
|
2009年CKD-EPI血中クレアチニン方程式を使用して、30日間持続し、EGFRは少なくとも40%減少した |
|
|
|
EGFR.EGFR |
|
|
|
腎臓や心血管疾患で死亡します |
被験者はランダム(1:1)を治療群と偽手術対照群に分け,腎生検前に行った。
治療群の各被験者は、腎臓生検後約14週間以内に第1回反応注射を受ける。3カ月後,第2剤を対側腎臓に注射する予定である。対照群の被験者は2回の偽注射を受け,1回目は偽生検後12週,2回目は1回目の偽注射後3カ月で行った。その間、対照群(Sham)の被験者は、同期の標準看護治療を受けながら、1回目の注射後1、14および28日(+7日)、その後、1回目の注射後3カ月(+10日)で後続評価を行い、治療群の被験者と類似した。すべての被験者は、世界的な実験終了日が発表され、研究訪問が終了するまで、この研究を継続する。
他の臨床発展研究
REGEN-008
REGEN−008は進行中の第3段階,展望性,開放ラベル,観察性主方案研究であり,糖尿病と慢性腎症患者を対象としており,これらの患者は以前に登録されREPACT治療を受けている。REGEN-008メインプロトコルの下で2つのサブ研究がある:
Regen-008亜研究1は多中心、展望性、非介入性、長期観察性延長研究であり、被験者は以前の介入性臨床研究にFresh Reaction製剤を登録し、服用した。参加者たちは5年間にわたる監視を受けるだろう。
Regen−008サブ研究2は,研究参加者が以前に受けた凍結保存製剤の反応を追跡することを目的としている。この研究では,参加者は5年(60カ月)に追跡され,前回の研究アクセスプロトコルが終了したことから計算される.
REGEN-015
多用量研究REGEN−015は,1型または2型糖尿病による慢性腎症患者の反応に関する計画の第1段階開放ラベル研究である。この研究の期待目的は,以前にREACT治療を受けた参加者にREPACT注射を補充する安全性を評価することである。同社はREGEN−015が臨床開発計画全体の価値を増加させたとは考えず,この研究を行わないことにした。患者は反応を受けず,この研究は2024年に本格的に終了する。
REGEN-004
Regen-004は完全な第1段階展望性、開放ラベル、単腕、安全性、耐性と早期治療効果研究であり、CAKUTの慢性腎臓病被験者の反応に用いられる。
この研究の主な目的は,最大2回6カ月(目標日後4週間)離れた反応注射の安全性と有効性を評価し,低侵襲経皮経路を用いて,覚醒鎮静下でCAKUTによるeGFRsが14~50 ml/min/1.73 m 2のCKD患者に90分未満で投与することである。5人の被験者がこの実験に参加した。被験者は2剤3 x 10の反応を受けた6Cells/g-KWEST.
全5名の被験者が1回目の注射を受け,4名の被験者が2回の注射を受けた。試験は2023年1月に終了し、最後の患者は最後に診察を受けた。研究期間のほとんどの時点で、EGFRの平均レベルはベースラインよりやや低下した。
14
その結果、EGFR低下率(標準偏差)は-0.4(2.28)mL/min/1.73 mと推定された2注射前の間、毎年-2.1(1.35)ml/分/1.73 m2および-1.4(0.93)ml/分/1.73 ml/分2注射後毎年。
この研究の最終結果は2023年12月にFDAに報告された。その会社は現在これ以上この兆候を求めないだろう。
競争
生物技術と製薬業界の特徴は技術進歩が迅速で、競争が激しく、知的財産権を強調することである。我々は、尿細管および糸球体細胞薬物調節剤(例えば、SGLT 2阻害剤)、抗線維化薬(例えば、鉱物皮質ホルモン受容体アンタゴニスト(MRAs))、グルコース依存性インスリン放出刺激剤(例えばGLP-1アゴニスト)、誘導多能性細胞、他の自家間葉系幹細胞および機械的腎補助装置(例えば、埋め込み型およびウェアラブル腎透析機)の開発者、ならびに腹膜透析および家庭透析の進展を含む、主要かつ専門的な製薬およびバイオテクノロジー会社を含む異なる源からの潜在的競争に直面している。他社は世界的に細胞による臨床試験を行っており,臍帯,脂肪,骨髄由来の間葉系幹細胞を用いて慢性腎症を治療している。日本ではヒト誘導多能性幹細胞による腎臓疾患治療の早期段階が行われている。
私たちが開発と商業化に成功したどの候補製品も、現在の治療法と将来出現する可能性のある新しい療法と競争するだろう。私たちのすべての候補製品の成功に影響を与える重要な競争要素は、治療効果、安全性、用量、コスト、販売促進支援の有効性と知的財産権保護を含むと信じている。具体的な反応については,承認されれば,その成功に影響を与える重要な競争要因には,予想される患者集団,用量および投与の相対的利便性および有効性が含まれると予想される。
CKDを制御するための薬物を開発した他の他の単独またはパートナーと協力して、研究および開発の面で明らかに多くの財務資源および専門知識を持っている 研究開発、臨床前テスト、臨床試験、製造とマーケティングはすべて私たちよりよくできている。我々の主な競争相手はSGLT 2阻害剤の開発者であり,Canagliflzin(楊森製薬会社などで販売されている名称はInvokana),ダパリフジン(アスリコン社や百時美施貴宝社などで販売されているブランドFarxigaやForxiga)やempagliflzin(社が販売しているJardiance),MRAsは最近慢性腎臓病の進行リスクを低下させるための小分子療法,特にフェナノン(バイエル株式会社を含む販売会社名)であると考えられる。最近,2型糖尿病や肥満症の治療のために承認されたグルカゴン様ペプチド−1アゴニストは,業界協会の強力な支持のもと,2型糖尿病,末期慢性腎臓病,終末期腎症患者の全因死亡率を低下させるために使用されており,Semagluide(ノワノード社によるOzempic,Rybelsus,Wegoyのブランド販売)やtirzepatide(礼来社が販売しているブランド)を含む。将来の協力、合併、買収は、より少ない数の競争相手に資源をさらに集中させる可能性がある。
もし私たちの競争相手が私たちが開発する可能性のある製品よりも安全で、より効果的で、副作用が少なく、より便利で、より安い製品を開発し、商業化すれば、私たちのビジネス潜在力は減少または解消されるかもしれない。私たちの競争相手も私たちよりも早くFDAや他の規制機関からその製品の承認を得ることができ、これは私たちの競争相手が私たちが市場に入る前に強力な市場地位を確立したり、私たちの開発をもっと複雑にしたりする可能性がある。これらの競争相手はまた臨床試験のために類似の合格科学と管理人材バンク、場所と患者集団、及び著者らの計画のために補充或いは必要な技術を奪い合う可能性がある。
供給と製造
私たちは過去20年間、化学、製造、制御(CMC)の製造技術を開発したと信じている。このようにする過程で、私たちはかなりの知的財産権を構築し、広範な製造技術を結合して、一致した品質で製品を製造することができるようにした。
高いレベルの製造と監督管理の専門知識の支持の下で、私たちの内部製造能力は臨床試験で迅速に進歩できるようにした。私たちの現在の製造能力は、私たちの臨床試験を供給するのに十分な数の臨床試験材料を提供することができると信じている。私たちが私たちの候補製品を開発し続けるにつれて、私たちは私たちの製造能力を拡大する必要があるかもしれない。ノースカロライナ州ウィンストン-セレムに位置する製造施設と品質システムは全世界の品質基準を満たすことを目的としている。臨床反応製品の生産には通常約12週間を要する。これまでに,我々の製造チーム,施設,生物処理能力は200種類以上の細胞療法を生産してきた。
同社の契約制合格者(QP)は最近、EUでReactionを発行·配布する準備状況を評価する監査を行い、品質管理システムファイルにいくつかの欠陥があることを発見した
15
EUの臨床サイトに製品を発表して配布します。したがって、EUと世界標準の第3段階計画を満たすために私たちの能力を最適化し、商業製造に移行しようとすると、製造は一時停止される。一時停止しなければならない安全事件は何もない。
我々の生物プロセスはFDAとEMAの審査を通過しており,検証活動が行われており,Reactionの潜在的な発売に向けたビジネス準備が予定されている。私たちは必要に応じて日々増加する需要を満たすために私たちの製造能力を拡大する計画だ。
私たちのビジネス戦略の重点は、必要な規制承認を得られれば、予想される市場反応を満たすために規模を拡大することです。
必要な規制承認を得たと仮定して,商業反応製品の製造コストをさらに低減するために生物プロセス開発を改善する予定である。培地は反応処理の中で最も高いコストであり,生物プロセスや自動化による培地使用量の削減を模索している。さらに私たちの最終的なビジネス反応製品は 計画は冷凍保存のレシピであり,我々の新しい反応レシピと比較して製造コストを低減することが予想される。大口調達の積極的な交渉材料の定価を利用して、コスト低減をさらに推進する見通しだ。しかし,REPACTをビジネス規模で生産した場合,我々の第3段階試験の製造コストは,我々が最近完成した第2段階RMCL−002研究よりも実際に低い保証はない。多くの要因は、我々が期待している自動化効率、サプライチェーンにおけるコスト超過または効率低下を達成できなかったこと、およびReactionの製剤または生物処理をコストを低減する方法で改善できなかったことを含む、これらのコスト低減を達成できない可能性がある。
知的財産権
私たちの成功は、私たちの候補製品、技術、およびノウハウのために独自の保護を獲得し、維持し、他人の固有の権利を侵害することなく運営し、他の人が私たちの固有の権利を侵害することを防止する能力があるかどうかにある程度依存する。私たちの政策は、アメリカやアメリカ以外の司法管轄地域で当社のノウハウ、発明、改善、候補製品に関する特許保護を求め、獲得することを求めることであり、これらは私たちの業務の発展と実施に非常に重要です。例えば、私たちは、各候補製品のために物質組成をカバーする特許を出願しているか、または、各臨床試験使用方法をカバーする特許保護を求めている。私たちはまた、ビジネス秘密、技術ノウハウ、持続的な技術革新、潜在的な許可内の機会に依存して、私たちの独自の地位を発展させ、維持することができます。また、適切な場合には、規制当局が私たちの製品を承認するのに要する時間を補うために、規制機関が私たちの製品を承認するのに要する時間を補うために、米国、ヨーロッパ、他の国の法的枠組みから利益を得ることが予想される。
私たちは新技術と候補製品を開発すると同時に、私たちの知的財産権戦略を評価し、改善していく。私たちは、適切な時期に、私たちが競争に適応したり、ビジネス機会を改善したりすることを求めているところを含めて、私たちの知的財産権戦略に基づいてより多くの特許出願を提出する予定です。また、私たちが開発した新しい技術を保護するために、私たちが適切だと思う場合に特許出願を提出する予定です。
使用方法のような我々の独自技術、独自の細胞ベース反応製品、および関連方法をカバーするために、19個の特許シリーズを代表する特許出願を提出した。2023年12月31日現在、我々の完全所有特許産業には、発行または未解決の特許出願317件、うち9件が発行されている米国特許、係属中の米国非仮特許出願10件、係属中の米国仮特許出願5件、係属中の特許協力条約(PCT)2件、発行された外国特許172件、および異なる外国司法管轄区における未解決外国特許出願119件が含まれている。
特に、我々の特許シリーズは、発行された34の特許および8つの係属中の特許出願を含む、Reaction、移植可能な構造、および腎機能を改善するために使用されるまたは腎臓疾患を治療するための細胞のための請求項に記載されている。この一連の特許は、米国(3つのライセンス特許)、欧州(2つのライセンス特許、各特許はそれぞれ7カ国/地域で検証されている)、中国、日本、韓国を含む9つの司法管轄区で許可されている。特許期間の調整または延長がない場合、発行された特許と、その家族の8つの保留出願から発行される可能性のある任意のさらなる特許とは、2029年に満了する予定である。
私たちはまた、私たちの反応製剤と製剤の製造方法に対する2つの特許シリーズを持っている。これらの家族のうち、米国、中国、日本、韓国、カナダを含む複数の管轄区で16件の特許を取得した。私たちはまた5つの特許出願がヨーロッパ、カナダ、メキシコ、オーストラリアを含む複数の管轄区域で決定されている。この2つの特許ファミリーの特許は、未決定出願で発行される可能性のある任意の特許を含み、2031年から2038年の間に満了すると予想され、具体的にはそれぞれの出願日に依存し、特許期限の調整または延長はない。
さらに、我々は、我々の独自の方法で調製されたReaction中の腎細胞が治療作用を示す表現型および機能的特徴を有することを保証するために、品質管理方法を採用することを要求する2つの特許ファミリーを有する
16
活動します。第1の特許家族では、米国(2つのライセンス特許)、欧州(2つのライセンス特許、うち1つは21カ国で検証され、もう1つは20カ国で検証されている)、中国(2つのライセンス特許)、日本(2つのライセンス特許)、韓国(2つのライセンス特許)、香港(2つのライセンス特許)、オーストラリア(2つのライセンス特許)、ニュージーランド(2つのライセンス特許)を含む61件の特許ライセンスを有している。米国、ヨーロッパ、オーストラリア、中国、韓国を含む10件の特許出願が複数の司法管轄区域で決定されている。発行された特許、および一連の10個の保留出願から発行される可能性のある任意のさらなる特許は、いかなる特許期間の調整または延長も行うことなく、2033年に満了すると予想される。私たちの第二の特許シリーズでは、私たちは16件の特許出願がアメリカ、ヨーロッパ、日本、中国、カナダを含む複数の司法管轄区域で審査されています。これらの出願から発行されるいずれの特許も、特許期間の調整や延長を行うことなく、2041年に満了することが予想される。
我々はまた、DKDまたは先天性異常に起因する腎臓疾患のような腎機能の改善および/または腎臓疾患の治療方法に関する3つの特許ファミリーを有する。これらの家族のうち、私たちは、米国、ヨーロッパ、香港、中国、韓国、日本、オーストラリア、ブラジル、メキシコ、イスラエル、カナダ、メキシコを含む米国特許と、15の管轄区で提出された29件の未解決特許を含む3つの発行された特許を持っている。私たちが発行する米国特許は、特許期限の調整や延長を行うことなく、2037年に満期になる見通しだ。これら3つの特許シリーズの特許が発行されれば、それぞれの出願日に依存し、特許期限の調整や延長はなく、2036年から2042年までの間に満了すると予想される。
さらに、私たちは私たちの革新的な側面についてもっと多くの申請をする予定で、これらの方面の特許期限はこれらの日を超えるかもしれない。
個別特許の期限は、このような特許を取得した国の法律にかかっている。我々が現在出願を提出している国では,特許期間は優先出願である非臨時特許出願の最初の提出日から20年である。しかしながら、米国特許の期間は、規制部門の承認を得て薬物の販売を許可するのに要する時間(特許期間の延長)または米国特許商標局(USPTO)(略称USPTO)が特許訴訟中に遭遇した遅延による遅延を補償するために延長することができる 特許期間調整)。例えば、ハッジ·ワックスマン法は、FDAが承認した薬物が特許満了後に最大5年間の特許期間を延長することを許可する。特許期間の延長の長さは,薬品が審査過程で監督審査と職務調査にある時間の長さと関係がある。特許期間の延長は、製品が承認された日から14年を超えることができず、承認された薬物またはその使用方法に関する特許を延長することしかできない。補完保護証明書と呼ばれる類似の特許延期は、ヨーロッパにもある。特定の他の管轄区域でも、特許の有効期間を延長するための法的枠組みがある。私たちは現在、私たちが合格特許を持っていて、延長期間を得ることができる任意の司法管轄区域内で、私たちが発行した任意の特許のために特許期間の延長を求めるつもりですが、米国のFDAを含めて適用可能な規制機関は保証できません。このような延長を承認すべきかどうか、延長期間が承認されても評価することに同意します。さらに、私たちの特許が延長されても、その特許は、特許の延長部分を含めて、米国または外国の最終管轄権裁判所によって無効または強制執行不可能と判断される可能性がある。
他のバイオテクノロジーや製薬会社と同様に、私たちが候補製品や技術に対する特許地位を獲得し、維持できるかどうかは、これらの未解決特許の有効な特許主張を成功させ、承認された場合にこれらの特許主張を実行するかどうかにかかっている。しかし、私たちが係属中の特許出願、および将来第三者から提出または許可される可能性のある任意の特許出願は、特許の発行につながらないかもしれません。私たちはまた私たちの特許で許可または実行される可能性のある特許請求の範囲の広さを予測することができない。また、我々の競争相手は、類似の作用機序を有する製品を独立して開発し、商業化し、私たちの特許を侵害することなく、私たちの治療方法や戦略を複製することができるかもしれない。私たちが開発する可能性のある治療製品の臨床開発と規制審査には多くの時間がかかるため、私たちのどの製品が商業化できる前に、どの関連特許も商業化後の短い期間で期限が切れたり、有効に維持されたりして、どのような特許のいかなる利点も弱める可能性がある。また、私たちが発行した特許であっても、臨床候補者の商業化において私たちの技術を実践する権利がある保証はありません。製薬分野の特許と他の知的財産権は絶えず発展する分野であり、多くのリスクと不確定性を有し、第三者は臨床候補薬物の商業化を阻止する特許を持っている可能性がある。
バイオテクノロジーと製薬産業の特徴は特許と他の知的財産権に関する広範な訴訟だ。私たちが私たちの候補製品と技術のために私たちの特許地位を獲得し、維持することができるかどうかは、承認または承認可能なクレームを成功的に実行できるかどうかにかかっている。しかし、私たちの承認された治療薬市場を保護するために私たちが依存する可能性のある特許を含む私たちの任意の特許は、最終管轄権の裁判所によって無効または実行不可能と判断されるかもしれない。あるいは、私たちは私たちの特許の期限に影響を与えるか、実行可能な方法で訴訟を解決することが私たちの利益に合致すると決定することができる。米国や他の国の特許法または特許法解釈の変化は、私たちの発明を保護し、私たちの知的財産権を実行する能力を弱める可能性がある。したがって、私たちは、私たちの特許または第三者特許に付与された特許請求の範囲の広さまたは実行可能性を予測することができない。
17
商業秘密
特許に加えて、私たちは非特許の商業秘密とノウハウ、および持続的な技術革新に依存して、私たちの競争地位を発展させ、維持することができる。私たちは、私たちのビジネスパートナー、協力者、従業員、コンサルタントとの秘密協定、および私たち従業員との発明譲渡協定の一部を使用して、私たちの独自の情報を保護することを求めています。これらのプロトコルは、我々の独自の情報を保護し、発明譲渡プロトコルの場合、第三者との関係によって開発された技術の所有権を付与することを目的としている。このような合意は違反されるかもしれないし、私たちはどんな違反にも対応する十分な救済策がないかもしれない。しかも、私たちのビジネス秘密は競争相手に知られたり独立して発見されるかもしれない。私たちのビジネスパートナー、協力者、従業員、およびコンサルタントが、私たちのために働いているときに他人が所有している知的財産権を使用する場合、関連するまたはそれによって生じるノウハウおよび発明の権利に関する紛争が生じる可能性がある。
政府の監督管理
アメリカでは、生物製品は、細胞ベースの再生療法製品を含み、連邦食品、薬物と化粧品法(“FD&C法”)、公衆衛生サービス法(“PHS法”)と他の連邦、州、地方と外国法規の規制を受けている。その他の事項以外に、“食品と薬物規制法”と“小霊通法案”及びその相応の法規はすべて生物製品に関連する研究、開発、臨床試験、テスト、製造、安全、効果、ラベル、包装、貯蔵、記録保存、流通、報告、広告とその他の宣伝方法を管理する。細胞に基づく治療製品のすべての臨床試験レジメンはFDAによって審査されなければならない。どんな生物製品も販売する前に、FDAの承認を受けなければならない。規制承認を得る過程と、その後適切な連邦、州、地方、外国法規を遵守する過程には多大な時間と財力がかかり、私たちは必要な規制承認を得ることができないかもしれない。
遺伝子または細胞療法、遺伝子テストおよび遺伝子研究の倫理、社会および法律の懸念は、追加の法的規制、または使用可能なプロセスを制限または禁止する可能性がある。連邦と州立法機関、機関、国会委員会、そして外国政府はバイオテクノロジーをさらに規制することに興味を示している。より厳しい法律法規や既存の法律や法規の解釈、あるいは私たちの候補製品が安全でないと主張したり、危険を構成したりすることは、任意の製品を商業化することを阻止するかもしれません。新しい政府要求を制定し、規制部門が私たちが開発している候補製品を承認することを延期または阻止するかもしれない。立法が変わるかどうか,条例,政策やガイドラインが変わるかどうか,機関や裁判所の解釈が変わるかどうか,あるいはこれらの変化の影響(あれば)が何になるかは予測できない。
アメリカの生物製品開発プロセス
米国食品医薬品局が生物製品が米国市場に参入する前に必要なプログラムには、通常、以下のような態様が含まれる
|
|
|
良好な実験室操作規範(“GLP”)および適用された実験動物の人道使用要件または他の適用法規に基づいて非臨床実験室テストおよび動物研究を完成させる |
|
|
|
ヒト臨床試験が開始される前に発効しなければならない新薬(“IND”)の申請をFDAに提出する |
|
|
|
各臨床試験が開始される前に、各臨床試験地点の研究審査委員会(“IRB”)または倫理委員会の承認案および関連文書 |
|
|
|
適用されたIND法規、良好な臨床実践或いは良好な臨床実践(“GCP”)と他の臨床試験に関連する法規に基づいて十分かつ制御された人体臨床試験を行い、各提案適応に対する研究生物製品の安全性と有効性を評価する |
|
|
|
市販承認のためにFDAに生物製品ライセンス申請(“BLA”)を提出し、非臨床試験および臨床試験による結果を含む、提案された生物製品の各提案された適応に対する安全性、純度および効力を証明する十分な証拠を含む |
|
|
|
ヒト細胞および組織製品の使用に関するFDAの現行の良好な組織規範(CGTP)を遵守するために、施設、方法、および生物製品の特性、強度、品質および純度を維持するのに十分な制御を確保するために、cGMPへの適合性を評価するために、バイオ製品を製造するための1つまたは複数の製造施設の検査を良好に完了させる |
|
|
|
FDAはGLPとGCPに適合することを保証し、BLAを支持するために提出された臨床データの完全性を確保するために、非臨床研究と臨床試験場所を検査する可能性がある |
|
|
|
候補製品は、適切な場合、または適切な場合には、FDA諮問委員会によって検討される |
|
|
|
FDAがBLAの使用料を審査する(適用費用免除が適用されない限り) |
|
|
|
FDAによるBLAの審査および承認、またはライセンス。 |
18
臨床前試験
細胞ベースの再生治療製品を含む任意の生物学的候補製品を人体で試験する前に、候補製品が臨床前試験段階に入る。臨床前試験は、非臨床研究とも呼ばれ、候補製品の生物学的特性、化学、毒性、安定性と調合の実験室評価、及び候補製品の潜在的安全性と活性を評価する動物試験を含む。スポンサーは,臨床前研究の結果を,生産情報,分析データ,任意の利用可能な臨床データや文献,提案された臨床案とともにINDの一部としてFDAに提出しなければならない。INDはFDAがヒトに研究製品の使用を許可する要求であり,ヒト臨床試験が開始される前に発効しなければならない。2022年12月29日に法律となった2023年総合支出法案(P.L.117-328)に署名し、薬物や生物製品の非臨床試験を規定する連邦食品、薬物、化粧品法案(FDCA)と公衆衛生サービス法案を改正したが、必要ではない体内にある動物実験です。修正された言語によると、スポンサーは様々なことを行うことができます体外培養分析(例えば、細胞ベースの分析、器官チップ、または微生理システム)シリコン片研究(すなわち、コンピュータモデリング)、人間または非ヒト生物学に基づく他のテスト(例えば、生物印刷)、または体内にある動物実験です。
臨床前試験の進行はGLPを含む連邦法規と要求に適合しなければならない。いくつかの長期的な臨床前テスト、例えば生殖不良事件と発ガン性の動物テストは、候補薬物のINDを研究してFDAに提出し、人体臨床試験を開始した後に引き続き行う可能性がある。
血中乳酸を支持するヒト臨床試験
臨床試験は合格研究者の監督の下で、GCP要求と規程に基づいて投与することに関連し、これらの要求と規程は研究の目標、評価する安全性と有効性標準をモニタリングするためのパラメータを詳しく説明した。すべてのプログラムはINDの一部としてFDAに提出されなければならない。対象者は臨床試験に参加する前にインフォームドコンセントに署名しなければならない。行っている臨床試験や臨床試験結果を公的登録機関に報告することも求められている。
INDは、認可されていない候補製品が州間商業で臨床研究のために輸送されることを可能にするFD&C法案の制約を受けず、FDAに対してこのような研究製品をヒトに使用することを許可する要求でもある免除を提供する。このような許可は、州間で許可されていないBLAテーマのバイオ製品候補製品を輸送し、管理する前に取得されなければならない。IND出願を支援するためには,出願人は各臨床試験の案を提出しなければならず,任意の後続の案修正はINDの一部としてFDAに提出されなければならない。また,前臨床試験の結果は,生産情報,分析データ,任意の利用可能な臨床データや文献,臨床試験計画などとともに,INDの一部としてFDAに提出されなければならない。FDAは各IND提出後30日間の待機期間を求め,臨床試験を開始することが求められている。この待機期間は、ヒト研究対象が不合理な健康リスクに直面するかどうかを決定するために、FDAがINDを審査することを可能にすることを目的としている。INDはFDAが受信した30日後に自動的に発効し、それ以前にFDAが提案試験の継続を明確に許可する通知を発表しない限り、または1つまたは複数の提案された臨床試験に懸念または問題を提起し、臨床試験を保留する。FDAが懸念を提起したり、試験を保留したりすれば、INDスポンサーと機関は提案された試験が開始される前に任意の懸案問題を解決しなければならない。
したがって,INDの提出はFDAが臨床試験の開始を許可しない可能性がある。
臨床試験開始後、FDAもこの試験を臨床保留或いは一部の臨床保留を行うことができる。臨床保留はFDAがスポンサーに発表した命令であり,提案された臨床研究の延期や進行中の研究の一時停止が要求されている。一部の臨床保留はIND要求の一部の臨床仕事を遅延或いは一時停止することである。臨床保留或いは一部の臨床保留を実施した後30日を超えない後、FDAはスポンサーに棚上げ根拠に関する書面解釈を提供する。臨床棚上げや一部の臨床放置を発表した後,FDAがスポンサー調査が継続可能であることを通知した後にのみ,調査を回復することができる。
スポンサーは選択できるが,必要ではなく,IND下で外国臨床試験を行う。IND下で外国臨床試験を行う場合,放棄しない限り,FDAのすべてのIND要求を満たさなければならない。海外の臨床試験がIND下で行われていない場合、スポンサーはこの研究があるものに適合することを確保しなければならない FDAの規制要件は、この研究をINDまたは上場承認が申請可能かもしれないサポートとして使用するために使用される。特に,このような研究はGCPに従って行われなければならず,IRBや独立倫理委員会(IEC)の審査·承認,被験者のインフォームドコンセントを含め,十分な患者集団規模や統計能力など,他の臨床試験要求を満たさなければならない。FDAが必要と判断すれば,FDAは現場検査によりデータを検証できる必要がある。
臨床試験に参加する各機関を代表するIECまたはIECは、臨床試験が開始される前に、任意の臨床試験の計画を審査および承認しなければならず、IRBまたはIECは、少なくとも毎年継続的な審査および再承認を行わなければならない。IRBやIECは,対象者に提供する研究案やインフォームドコンセント情報などを審査·承認しなければならない。IRBやIECの運営はFDAの規定に適合しなければならない。IRBまたはIECは一時停止または
19
臨床試験がIRBの要求に沿って行われていない場合、あるいは候補製品が患者に予期せぬ深刻な被害を受けた場合、その機関またはその代表機関の臨床試験の承認を終了する。
いくつかの試験は、試験スポンサーによって組織された合格専門家からなる独立したグループによって監督され、このグループはデータ安全監視委員会(“DSMB”)と呼ばれる。このグループは,そのグループのみが保守している研究から利用可能なデータへのアクセスに基づいて,許可試験が指定されたチェックポイントで行えるかどうかを判定する.
いくつかの臨床試験に関する情報は、プログラムの詳細および最終的な研究結果を含み、ClinicalTrials.govデータレジストリ上で公開されるために、特定の時間枠内で国家衛生研究院に提出されなければならない。臨床試験登録の一部として,臨床試験の製品,患者群,調査段階,研究場所や研究者,その他に関する情報が公開されている。スポンサーも完成後にその臨床試験の結果を開示する義務がある。場合によっては、これらの裁判結果の開示は、裁判が完了した日から2年に延期されることができる。対象の臨床研究や法律規定の研究結果を適時に登録できなかったことは民事罰金を招く可能性があり、また違反側が連邦政府の将来の支出を獲得することを阻止する。米国衛生·公衆サービス部(DHHS)の最終規則と米国国立衛生研究院(NIH)のClinicalTrials.gov登録と報告要求に関する対応政策は2017年に発効し、NIHとFDAはいずれも規則外の臨床試験スポンサーに対して法執行行動を提起した。
臨床試験は通常3つの連続段階に分けて行われ、これらの段階は重複または合併する可能性がある
|
|
|
ステップ1それは.この候補製品は最初に健康な人体に導入され、安全性、用量耐性、吸収、代謝、分布と排泄に対してテストを行った。いくつかの深刻または生命を脅かす疾患の候補製品の場合、特に候補製品がその固有の毒性のために健康ボランティアに道徳的に服用できない可能性がある場合、最初の人体テストは通常患者に行われる。 |
|
|
|
第二段階それは.限られた患者集団の中で候補製品を評価して、可能な副作用と安全リスクを確定し、候補製品の特定の目標疾患に対する治療効果を初歩的に評価し、そして用量耐性、最適用量と用量計画を確定する。 |
|
|
|
第3段階それは.臨床試験は地理的に分散した臨床試験地点で患者群を拡大し、投与量、臨床治療効果、効力と安全性を更に評価するためである。これらの臨床試験は、候補製品の全体的なリスク/利益比率を決定し、適切な場合に承認と製品ラベルに十分な基礎を提供することを目的としている。これらの試験は、プラセボおよび/または他の対照治療との比較を含むことができる。治療の持続時間が常に延長され、製品のマーケティング期間中の実際の使用をシミュレートする。 |
承認後の臨床試験は,4期臨床試験と呼ばれることがあり,最初の上場承認後に行われる可能性がある。これらの臨床試験は,治療適応が予想される患者の治療から追加的な経験を得るためのものであり,特に長期安全なフォローアップのためである。場合によっては,FDAはBLAを承認する条件として4期臨床試験を強制的に実行することができる。
2023年総合支出法案では,国会でFDCAが改正され,3期臨床試験や上場許可された新薬や生物を支援する他の“キー研究”のスポンサーにこのような臨床試験の多様性行動計画の提出が求められている。行動計画には,スポンサーの多様な学生募集目標と,目標の理由やスポンサーがこれらの目標をどのように達成するかの記述が含まれなければならない。スポンサーはスポンサーが試験案をFDA審査に提出する前にFDAに多様な行動計画を提出しなければならない。FDAは多様な行動計画の一部またはすべての要求を免除することができる。多様性行動計画が第3段階試験計画やスケジュールにどのように影響するかは不明であり,FDAがこのような計画の中でどのような具体的な情報を期待するかも不明であるが,FDAがスポンサーの多様性行動計画に反対し,スポンサーに計画の修正や他の行動を要求すれば,試験開始を遅らせる可能性がある。
臨床開発のすべての段階において、監督管理機関はすべての臨床活動、臨床データと臨床試験調査人員に対して広範なモニタリングと監査を行うことを要求している。臨床試験結果を詳細に説明する年次進展報告はFDAに提出しなければならない。書面のIND安全報告書は、深刻かつ予期せぬ有害事象、他の研究の任意の発見、実験室動物、または研究者を理解するために、FDA、担当IRBs、および調査者に迅速に提出しなければならない体外培養実験により、人類被験者に対して重大なリスクがあり、或いは方案或いは研究者マニュアルに記載された試験と比べ、深刻な不良反応の発生率は臨床上重要な意義がある。スポンサーは15日以内にINDセキュリティ報告書を提出し,スポンサーがその情報有資格報告を確定した後でなければならない。スポンサーはまた、スポンサーがこのような情報を初めて受け取ってから7日以内に、任意の意外、致命的、あるいは生命に危害を及ぼす疑いのある副作用をFDAに通知しなければならない。第1段階、第2段階、および第3段階の臨床試験は、もしあれば、任意の指定された時間で成功しない可能性がある。
人体細胞を基礎とした製品が直接腎臓組織に入ることは新しい治療方法である。これは比較的に新しく、絶えず拡大する新型治療干与措置領域であるため、研究期間の長さ、FDAがヒト細胞に基づく治療製品の安全性、純度と有効性を確定するために研究に組み入れることを要求する患者の数を保証することができず、これらの研究で産生されたデータがFDAによって受け入れられ、発売承認を支持することも保証されない。
20
臨床試験と同時に、会社は通常追加の動物研究を完成し、生物製品の物理的特徴に関する追加情報を開発し、cGMP要求に基づいて商業大量生産製品のプロセスを最終的に決定しなければならない。PHS法案では,生物製品を用いた外来製剤導入のリスク低減を支援するために,属性が正確に定義できない製品の製造制御の重要性を強調している。製造過程は一貫して高品質の候補製品ロットを生産することができなければならず、他の以外に、スポンサーは最終生物製品の特性、強度、品質、効力と純度をテストする方法を開発しなければならない。また,適切な包装を選択·試験し,候補生物製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
アメリカの承認プロセス
候補生物製品の臨床試験が完了した後,生物製品の商業マーケティングの前に,FDAのBLAの承認を得なければならない。BLAは製品開発、実験室と動物研究、人体研究、製品製造と成分の情報、提案のラベル、その他の関連情報を含まなければならない。上場承認を支持するために、提出されたデータは、研究製品の安全性、純度、効力および有効性を決定して、FDAのその提案に対する1つまたは複数の適応の要求を満たすために、品質と数量で十分でなければならない。テストや承認過程には多大な時間と労力が必要であり,FDAがBLAの届出を受ける保証はなく,届出してもどの承認もタイムリーに承認される保証はない.
改正された“処方薬使用料法案”(“PDUFA”)によると、各BLAは相当な使用料を伴わなければならない。FDAは毎年PDUFAユーザ料金を調整する。PDUFAは登録された生物製品メーカーにも年会費を徴収している。場合によっては、小企業が初めて出願した出願料を免除することを含む、費用を免除または減免することができる。
出願が提出されてから60日以内に、FDAは、FDAが提出を受け入れる前に実質的に完了したかどうかを決定するために、提出されたBLAを審査する。FDAは、それが不完全であるか、または提出時に適切に審査できないと考えられる任意のBLAの提出を拒否することができ、より多くの情報の提供を要求することができる。この場合,BLAおよび付加情報を再提出しなければならない.再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。提出された申請が受け入れられると、FDAは開始します “法案”に対する深い実質的な検討が行われた。FDAは、提案された製品が安全、純粋、有効、およびその提案に適合する1つまたは複数の適応であるかどうか、および製品の持続的な安全性、純度、および効力を保証するために、cGMPに従って生産されているかどうかを決定するためにBLAを審査する。
FDAがPDUFAで実施した業績目標と政策によると、元のBLASに対して、FDAの目標は、提出日から10ヶ月以内に標準出願の予備審査を完了し、出願人に応答し、提出日から6ヶ月間優先審査を行う出願である。FDAは常にPDUFAの目標日を達成するわけではなく、FDAがより多くの情報を提供したり、明確にすることを要求しているため、審査過程はしばしば著しく延長される。FDAは、新しい情報を考慮するために、審査プロセスおよびPDUFA目標日をさらに3ヶ月延長することができ、または出願人が明確な説明を提供する場合には、FDAが最初の提出後に発見された係属中の欠陥を解決することができる。
BLAを承認する前に、FDAは通常、この製品を生産する施設を検査する。FDAは、製造プロセスおよび施設がcGMP要件に適合していることを決定し、要求された仕様の下で製品が一貫して生産されることを保証するのに十分でない限り、この製品を承認しないであろう。細胞による治療製品については,メーカーがcGTPSに適合していなければ,FDAもこの製品を承認しない。これらは、ヒト細胞、組織、および細胞および組織ベースの製品(“HCT/Ps”)を製造するための方法および使用のための施設および制御措置を管理するFDAの法規であり、これらの製品は、ヒトレシピエント内に移植、移植、注入または転移するためのヒト細胞または組織である。CGTP要求の主な目的は,細胞や組織に基づく製品の製造方式の確保であり,感染症の導入,伝播,伝播を防止することを目的としている。FDAの規定はまた、組織機関がFDAに彼らのHCT/Pを登録して列挙することを要求し、適用された場合、適切なスクリーニングおよびテストによってドナーを評価する。さらに、BLAを承認する前に、FDAは通常、IND研究要求およびGCP要求に従って臨床試験が行われることを確実にするために、1つまたは複数の臨床場所を検査する。CGMP,CGTP,GCPに適合することを確保するためには,申請者は訓練,記録保存,生産,品質管理に多大な時間,お金,労力をかけなければならない。
さらに、FDAは、安全性または有効性の問題を提起する新しい生物候補の申請を含む任意のBLAを諮問委員会に提出することができる。通常,諮問委員会は臨床医や他の科学専門家を含む独立した専門家からなるグループであり,申請を審査,評価し,申請を承認すべきかどうか,どのような条件でアドバイスを提供すべきかを担当する。FDAは諮問委員会の提案に制限されていないが、最終承認決定を下す際にこれらの提案を考慮する。FDAは臨床試験データを再分析する可能性があり,FDAや出願人の審査過程で広く議論される可能性がある。FDAはまた、REMSがそうであると判断した場合、リスク評価および緩和策(“REMS”)の提出を要求する可能性がある
21
薬物のメリットがそのリスクよりも大きいことを確保し,薬物やバイオ製品の安全な使用を確保する必要がある。REMSは、制限された分配方法、患者登録、または他のリスク最小化ツールのような薬物ガイドライン、医師コミュニケーション計画、評価計画、および/または安全使用を保証する要素を含むことができる。FDAは具体的な状況に応じてREMSに対する要求および具体的なREMS条項を決定する。FDAがREMSが必要であると結論した場合,BLAのスポンサーは提案したREMSを提出しなければならない。必要であれば、FDAはREMSのないBLAを承認しないだろう。
小児科研究公平法(“PREA”)により,新製品のBLAやBLA(例えば,新たな活性成分,新たな適応など)を補充する。すべての関連小児科亜群において適応が主張される候補生物製品の安全性および有効性を評価し、安全かつ有効な各小児科亜群に対する投与および投与を支持するためのデータを含まなければならない。FDAはデータの提出を延期することを許可するか、またはすべてまたは部分的な免除を与えることができる。PREAは,新活性成分,新適応,新剤形,新投与案または新投与経路を含む製品のマーケティング申請を含むスポンサーに初期小児科研究計画を提出することを要求している2期会議終了後60日以内に、またはそのような会議がない場合には、3期または2/3期臨床試験を開始する前に、確実な範囲で早期に提出する。最初のPSPは、試験目標および設計、年齢群、関連する終点および統計方法、またはそのような詳細な情報を含まない理由、ならびに小児科研究データおよび支援情報の提供を延期または完全または部分的に免除することを要求する任意の要件を含む、スポンサー計画によって実施される1つまたは複数の小児科研究の概要を含まなければならない。FDAとスポンサーはPSPについて合意しなければならない。臨床前研究,早期臨床試験あるいは他の臨床開発計画から収集したデータから小児科計画の変化を考慮する必要があれば,スポンサーは合意した初期PSPに対する修正案を随時提出することができる。
FDAは、製品が安全で、純粋かつ有効であるかどうか、およびその製造、加工、包装、または保持されている施設が、製品の持続的な安全、純度および効力を保証するための基準に適合しているかどうかを決定するためにBLAを審査する。承認過程が長く、しばしば困難であり、適用された規制基準が満たされていない場合、または追加の臨床または他のデータおよび情報が必要とされる可能性がある場合、FDAはBLAの承認を拒否する可能性がある。FDAは、製造施設の検査結果を含むBLAの評価および付随する情報に基づいて、FDAは、承認書または完全な返信(CRL)を発行することができる。承認書は、製品の商業マーケティングを許可し、特定の適応に関する具体的な処方情報を提供する。CRLは,申請の審査周期が完了したことを示しており,申請は現在の形で承認されない.CRLは、通常、提出中の不足点を列挙し、FDAが申請を再検討するために、大量の追加のテストまたは情報を必要とする可能性がある。CRLは、追加の臨床または他のデータ、追加の重要な第3段階臨床試験(S)、および/または臨床試験、非臨床研究、または生産に関連する他の重要で時間のかかる要件を必要とする可能性がある。CRLが発行された場合,申請者は,手紙で決定されたすべての不足点を解決するBLAを再提出するか,または申請を撤回することを選択することができる.これらの不足点がBLAの再提出時にFDAによって満足的に解決された場合、FDAは承認書を発行する。FDAは、含まれる情報のタイプに応じて、発行されたCRLに応答するために、そのような再提出を2ヶ月または6ヶ月以内に検討することを約束している。しかしながら、この補足情報を提出しても、FDAは最終的に、その申請が承認された規制基準を満たしていないと決定する可能性がある。
1つの製品がFDAの規制によって承認された場合、承認は、申請に記載された使用条件(例えば、患者数、適応)に限定される。さらに、必要に応じて解決される特定のリスク(S)に応じて、FDAは、承認後に製品の安全性をさらに評価するための承認後試験(4期臨床試験を含む)を要求するために、製品ラベルに禁忌症、警告または予防措置を含むことを要求することができ、試験および監視計画は、製品の商業化後に製品を監視することを要求するか、または販売および使用制限またはREMS下の他のリスク管理機構を含む他の条件を適用することができ、これらの条件は、製品の潜在的な市場および利益に大きな影響を与える可能性がある。FDAは発売後の試験或いはモニタリング計画の結果に基づいて、製品の更なるマーケティングを阻止或いは制限することができる。承認後、新たな適応の追加、製造変更、および追加のラベル宣言など、承認された製品のいくつかのタイプの変更は、さらなるテスト要件およびFDAの審査および承認を受けることになる。
高速チャネル、RMAT、および優先レビュー指定
FDAには、深刻または生命に危険な疾患または状態を治療するための薬物および生物製品の開発またはFDA審査プロセスを加速または簡略化することを目的とした迅速チャネル指定、RMAT指定、および優先審査を含む様々な計画がある。これらの計画は承認基準を変更することはありませんが、開発や承認過程の加速に役立つ可能性があります。
迅速なチャネル指定を得る資格があるために、FDAはスポンサーの要求に基づいて、新薬或いは生物製品を決定し、深刻な或いは生命に危害を及ぼす疾患を治療することを目的とし、そして存在しない治療法或いは1種の治療効果或いは安全要素に基づく治療法を提供することによって、既存の治療法よりも優れている可能性があることを証明し、満たされていない医療需要を満たす可能性がある。高速チャネル指定は、FDA審査チームとより頻繁な相互作用を行うための機会を提供し、製品の開発と審査を加速する。FDAはまた、NDAまたはBLAの高速チャネル製品部分を審査するために、完全な出願を提出する前にスクロールすることができ、スポンサーおよびFDAが出願部分のスケジュールについて合意し、スポンサーがNDAまたはBLAの第1の部分を提出する際に任意の必要な使用料を支払うことを前提とすることもできる。はい
22
また,迅速チャネルの指定が臨床試験中に出現したデータの支持を得なくなった場合,スポンサーがその指定を撤回したり,FDAが指定を撤回したりする可能性がある。
2016年12月に公布された“21世紀治療法案”(以下、“治療法案”)の一部として、国会は、再生医学高度療法(RMATs)の効率的な開発計画を促進し、細胞および遺伝子療法、治療用組織工学製品、ヒト細胞および組織製品、およびこのような任意の療法または製品を使用した組み合わせ製品を含む審査速度を加速させる“FD&C法案”を改正した。ReactionはFDAのRMAT称号を受けた。RMAT指定には、“小霊通法案”第361条及び“連邦法規”第1271部にのみ規定されているHct/Pは含まれていない。この計画は、深刻なまたは生命に危害を及ぼす疾患または状況を治療、修正、逆転または治癒することを目的とした再生医学療法の有効な開発と審査の加速を促進することを目的としている。スポンサーは、INDの提出と同時にまたはその後の任意の時間に、FDAに薬剤をRMATとして指定するように要求することができる。FDAは60個のカレンダー日があり、この薬物が標準に符合するかどうかを決定し、初歩的な臨床証拠があるかどうかを含み、この薬物は深刻な或いは生命に危害を及ぼす疾病或いは状況の未満足の医療需要を満たす可能性があることを表明した。RMAT指定された再生医学療法を取得したBLAは、長期臨床的利益を合理的に予測する可能性のある代替物または中間終点を使用することによって、または大量のサイトから取得されたデータに依存して、優先審査または加速承認を得る資格がある可能性がある。RMATを指定する利点はまた、承認の加速をサポートするための任意の潜在的代替品または中間終点を検討するために、FDAとの早期相互作用を含む。加速され、承認され、承認された後に要求されるRMAT指定された再生医学療法は、電子健康記録のような臨床試験、患者登録または他の真の証拠源からの臨床証拠の提出、より大きな検証データセットの収集、または承認前にそのような治療を受けたすべての患者を承認後に監視することによって、これらの要求を満たすことができる。
最後に、FDAは、重篤な疾患を治療する薬剤または生物学的製品である場合、承認されれば、安全性または有効性の面で顕著な改善を提供する優先審査製品を指定することができる。FDAがマーケティング申請を提出する際には,具体的な状況から,他の既存療法と比較して,推奨されている薬物が治療,予防または疾患診断において有意に改善されているかどうかを決定した。著明な改善は,ある疾患の治療の有効性の向上,治療反応の制限の解消や大幅な減少,記録されている患者のコンプライアンスの向上,重篤な結果の改善,あるいは新亜群の安全性と有効性の証拠につながる可能性があることから説明できる。優先審査指定の目的は、全体的な関心およびリソースをこのような申請の評価に誘導し、FDAがマーケティング申請に行動する目標を10ヶ月から6ヶ月に短縮し、申請を提出した日から元のBLAまたは新分子実体NDAに行動することである。
1つの製品がこれらの計画のうちの1つまたは複数に適合していても、FDAは、製品がもはや資格条件に適合していないことを後で決定することができ、またはFDAの審査または承認を決定する期間が短縮されないことができる。また,迅速チャネル指定,RMAT治療指定,優先審査は承認の基準を変更することはなく,最終的に開発や承認過程を加速させない可能性もある。
承認のルートを速める
さらに、研究された製品は、深刻または生命を脅かす疾患の治療における安全性および有効性、ならびに既存の治療方法よりも意義のある治療利益を提供する製品は、FDAの加速承認を得る可能性があり、十分かつ制御された臨床試験に基づいて承認される可能性があり、これらの試験は、製品が臨床的利益を合理的に予測する可能性のある代替終点に影響を及ぼすことを証明している。中間臨床終点に対する製品の影響が不可逆的発病率または死亡率(IMM)への影響よりも早くすることができ、病状の重症度、希少性または流行率、および代替治療が利用可能または不足していることを考慮すると、IMMまたは他の臨床的利益への影響を合理的に予測する可能性があり、FDAはこのような薬物または生物学的製剤の許可を加速することも許可することができる。承認の1つの条件として、FDAは、IMMまたは他の臨床終点に対する予期される効果を検証および記述するために、承認を加速させた薬物のスポンサーに発売後の臨床試験を要求することができ、迅速な退出手順を必要とする可能性がある。加速的に承認された薬品および生物製品は、従来承認された薬物および生物製品と同じ安全と有効性法定基準に適合しなければならない。
承認を加速するために、代替終点は、例えば実験室測定、放射画像、バイタルサイン、または他の臨床的利益を予測することができると考えられるが、それ自体は臨床的利益の測定基準ではない標識である。代替終点は通常、臨床終点よりも容易または迅速に測定される。中間臨床終点は治療効果の測定であり、1種の薬物の臨床利益、例えばIMMに対する効果を合理的に予測することが可能であると考えられる。FDAは中間臨床終点に基づく加速審査の経験は限られているが、終点で測定した治療効果自体が臨床利益と伝統的な承認の基礎ではなく、もし基礎が治療効果を得て1種の薬物の最終長期臨床利益を合理的に予測する可能性があれば、このような終点は通常加速承認を支持できることを表明した。
加速承認経路は、代替物または中間体に対する薬物の影響であっても、薬物の予期される臨床的利益を測定するために、疾患の経過が長く、時間を延長する必要がある環境において最もよく使用される
23
臨床の終点が早く現れた。例えば、加速承認は、様々な癌を治療するための薬剤の開発および承認に広く使用されており、治療の目標は、通常、生存率を向上させること、または発症率を低下させることであり、典型的な病気経過の持続時間は、臨床または生存上の利点を証明するために長い、場合によっては大型の臨床試験を必要とする。
承認を加速する方法は、一般に、薬物の臨床的利益を検証および説明するために、勤勉な方法で追加的な承認後の検証的研究を行うことにスポンサーが同意することに依存する。そのため、この基礎の上で承認された候補製品は必ず厳格な発売後のコンプライアンス要求を守らなければならず、4期或いは承認後の臨床試験を完成し、臨床終点への影響を確認することを含む。必要な承認後研究を行わない場合、あるいは発売後の研究期間中にこの製品の期待される臨床的利益が確認できなければ、FDAがこの薬剤の承認を撤回することを許可する。2023年の総合支出法案の一部として、国会はマーケティングを継続する前に加速的に承認された無効薬の患者への潜在的リスクを軽減するために、FDAに追加の法定権力を提供した。この法案のfdcaに対する修正案によると,fdaは加速承認を得た製品のスポンサーに承認前に検証的実験を行うことを求めることができる.スポンサーはまた、試験が完了するまで、検証性試験の進捗報告を6ヶ月ごとに提出しなければならず、これらの報告はFDAのウェブサイトに発表されている。修正案はまた、FDAがスポンサーの検証性試験が製品主張の臨床的利益を検証できなかった場合に、迅速なプログラムを使用して製品承認を撤回することを選択することを可能にする。
加速承認手続きにより検討·承認されているすべての候補製品の宣伝材料はFDAの事前審査を経なければならない。
承認後に要求する
適用される連邦、州、地方法規を基本的に守るには、多くの時間と財力が必要だ。新製品が承認された後、製造業者および承認された製品は、監視および記録活動、製品の不良体験の報告、製品のサンプリングおよび流通制限、販売促進および広告要件の遵守を含むFDAの一般的かつ持続的な規制を受ける。
FDAの規定は,製品は特定の承認された施設で生産され,cGMPに適合しなければならないことを要求している。CGMP条例には、人員、建物および施設、設備、部品および製品容器および閉鎖的な制御、生産およびプロセス制御、包装およびラベル制御、保有および分配、実験室制御、記録および報告、ならびに返品または回収された製品に関する要件が含まれる。我々は,品質管理と品質保証および記録とファイルの保守を含むcGMPやCGTP法規における適用要求を遵守しなければならない.生産·流通承認に関与する生物製品やヘキサクロロエチレン/ポリ塩化ビフェニルの実体は,FDAとある国機関および適用された外国同業者にその機関を登録し,これらの政府当局の定期的な抜き打ち検査を受け,cGMP,CGTP,その他の法律を遵守することを確保しなければならない。したがって,我々はcGMPのコンプライアンスを維持するために,生産や品質管理に時間,お金,労力をかけ続けなければならない。政府当局の将来の検査では、我々の工場に存在するコンプライアンス問題が発見される可能性があり、これらの問題は生産や流通を混乱させたり、是正するために大量の資源が必要になる可能性がある。さらに、cGMPまたはCGTPに適合しないことを含むこれらの規則違反の条件が発見され、法執行行動を引き起こす可能性があり、承認後に製品問題が発見されると、以下に説明する自発的なリコールおよび規制制裁を含む製品、製造業者または承認されたBLA所有者への制限を引き起こす可能性がある。
他の生物製品の承認後要求に適しており、報告は、配布製品の識別、効力、純度および全体的な安全性に影響を与える可能性のあるcGMP偏差、記録保存要求、不良反応の報告、最新の安全および効果情報の報告、および電子記録および署名要件の遵守を含む。BLAが承認された後、この製品はまた正式なロット発表が必要になる可能性がある。製造プロセスの一部として、製造業者は、製品の各ロットに対していくつかのテストを行うことを要求され、その後、流通のために発表することができる。製品がFDAによって正式に発表されなければならない場合、製造業者は、各製品のサンプルをFDAに提出し、バッチの生産履歴および製造業者がバッチについて行ったすべての試験結果の要約を示す発行プロトコルを提示する。FDAはまた、ウイルスワクチンのような多くの製品に対していくつかの検証性テストを行い、その後、メーカーがロットを発表して流通する可能性がある。また、FDAは生物製品の安全性、純度、効力と有効性規制基準に関する実験室研究を行っている。
また、FDAの広告および販売促進要件、例えば、消費者向け広告、業界スポンサーの科学的および教育活動、ならびにインターネットに関連する販売促進活動に関連する要求、および製品承認のラベルに記載されていない使用のための宣伝製品の使用、または患者集団における使用(“非ラベル使用”と呼ばれる)を禁止しなければならない。医師はラベル外の用途のために合法的な製品を処方する可能性があるが、メーカーはこのような用途を販売したり普及させたりしてはならない。FDAや他の機関はラベル外用途の普及を禁止する法律法規を積極的に実行しており,ラベル外用途の普及が不適切であることが発見された会社は重大な責任を負う可能性がある。
24
製品に適応、ラベルまたは製造プロセスまたは施設の変化を含む修正がある場合、出願人は、新しいBLAまたはBLAサプリメントの承認を得るために提出および提出を要求される可能性があり、これは、出願人に追加のデータの開発を要求するか、または追加の臨床前研究および臨床試験を行うことを要求する可能性がある。FDAはまた、製品の安全な使用を保証するためにREMSを要求することを含む、承認時に他の条件を追加することができる。REMSは、制限された分配方法、患者登録、および他のリスク最小化ツールのような薬物ガイドライン、医師のコミュニケーション計画、または安全な使用を確保する要素を含むことができる。承認またはマーケティングに関するこれらの制限は、製品の商業普及、流通、処方、または配布を制限する可能性がある。製品承認は、規制基準を満たしていないことや、初期マーケティング後に問題が発生したことで撤回される可能性があります。
薬剤が承認または承認されると、規制要件および基準の遵守が維持されていない場合、または製品発売後に問題が発生した場合、FDAは承認を撤回する可能性がある。その後、製品に以前に未知の問題が存在することが発見された場合、意外な深刻性または頻度の不良事象、または生産プロセス、または法規要件を遵守できなかった場合、新たなセキュリティ情報を追加するために承認されたラベルを強制的に改訂し、新しい安全リスクを評価するために発売後または臨床試験を実施するか、またはREMS計画に従って流通または他の制限を実施することにつながる可能性がある。他の他の潜在的な結果には
|
|
|
製品の販売や製造を制限し、市場から製品を完全に撤回したり、製品をリコールしたりする |
|
|
|
承認された臨床試験には、罰金、警告状、または実行に関連する他の手紙または臨床保留が科される |
|
|
|
FDAは、承認されるべきNDA/BLAまたは承認されたNDA/BLAの補充の承認を拒否するか、または製品の承認を一時停止または撤回する |
|
|
|
製品の差し押さえ、差し押さえ、あるいは製品の輸出入を許可しないことを拒否した |
|
|
|
民事又は刑事処罰の禁止又は適用;及び |
|
|
|
法令、会社の誠実な合意に同意し、連邦医療計画の禁止を取り消したり、除外したり、宣伝材料やラベルを強制的に修正したり、訂正情報を発表したりする。 |
また、処方薬製品の流通は“処方薬販売法”の制約を受け、この法は各州の薬品流通業者に対する登録と監督管理の最低基準を規定している。また、アメリカで流通されているいくつかの処方薬を識別し、追跡するための電子システムを構築することを目的とした“医薬品サプライチェーン安全法”(DSCSA)が公布された。DSCSAは薬品メーカー,卸売業者,流通業者に10年以内に段階的と資源集約型の義務を負うことを求め,最終的に2023年11月に終了した。最近、FDAは1年の安定期から2024年11月まで、DSCSAに制約されたエンティティにより多くの時間を与えて相互操作可能な追跡システムを決定し、サプライチェーンの連続性を確保することを発表した。時々、新しい立法と法規が施行される可能性があり、これらの法規はFDA規制製品の承認、製造、マーケティングの法定条項を著しく変える可能性がある。さらなる立法または規制の変化が公布されるかどうか、あるいはFDAの法規、ガイドライン、または解釈が変わるかどうか、またはこれらの変化の影響が(あれば)何になるかは予測できない。
アメリカの特許期限回復と市場排他性
FDAが私たちの候補製品の使用を許可した時間、期限、具体的な状況によると、私たちのいくつかのアメリカ特許は、ハッジ·ワックスマン修正案に基づいて限られた特許期間の延長を受ける資格がある可能性がある。ハッジ·ワックスマン改正案は、製品開発およびFDA規制審査中に失われた特許期間の補償として、特許回復期間を最長5年とすることを許可している。しかし,特許期限の回復は特許の残存期間を延長することはできず,製品承認日から合計14年を超えることはできない。特許期間回復期間は、一般に、INDの発効日とBLA提出日との間の時間の半分に、BLA提出日とその出願が承認された日との間の時間を加える。承認された生物製品に適用される特許は1つのみ延期する資格があり,延期出願は特許が満了する前に提出されなければならない。しかも、一つの特許は一度しか延期できず、単一製品だけを対象としている。米国特許商標局はFDAと協議し,任意の特許期間の延長または回復の出願を審査·承認する。将来、私たちは、臨床試験の期待長と関連BLAの提出に関連する他の要因に依存して、現在の満期日以降の特許寿命を延長するために、私たちの特許出願のための特許期間を回復することを意図しているかもしれない。
生物製品は米国で小児科市場の排他性を獲得する可能性もある。小児科専有権は、米国で入手可能な非特許マーケティング専有権であり、承認された場合、任意の既存の規制専有権または上場特許の期間に追加的な6ヶ月の市場保護を追加することを規定する。NDAスポンサーから提出された小児科データがこのようなデータに対するFDAの書面要求に公平に応答すれば,この6カ月の排他性を与えることができる。これらのデータは,この製品が研究されている小児科群で有効であることを証明する必要はなく,逆に臨床試験がFDAの要求に公平に応答していると考えられれば,追加的な保護が得られる。要求された小児科研究報告が法定期限内にFDAに提出され、FDAによって受け入れられた場合、製品の法定または規制排他性または特許保護期間にかかわらず6ヶ月間延長される。これは特許期間の延長ではないが、規制期間を効果的に延長した
25
その間、FDAは別の申請を承認することができない。書面申請を出すことは、スポンサーにその研究を要求しない。
生物製品の参考製品排他性
2010年3月、米国は“生物製品価格競争と革新法案”(以下、“生物製品価格競争と革新法案”)を含む“患者保護と平価医療法案”(以下、“患者保護と平価医療法案”)を公布した。BPCIAは公衆衛生サービス法(PHSA)を改正し、FDAが許可した参考生物製品の生物と類似或いは交換可能な生物製品のために短い承認経路を作った。これまで、FDAはいくつかの生物模倣薬を承認し、多くの生物模倣薬はすでにヨーロッパで承認された。FDAはまたいくつかの指導文書を発表し,その生物模倣薬と交換可能な生体模倣薬を審査·承認する方法について概説した。
生物類似製品は、臨床不活性成分に微小な差があるにもかかわらず、参考製品と高度に類似しており、しかも製品の安全性、純度と効力について、生物製品と参考製品の間に臨床上意義のない差異と定義されている。交換可能製品は、任意の所与の患者において参照製品と同じ臨床結果を生成することが予想され、複数回投与された製品の場合、製品および参照製品は、安全リスクを増加させることなく、またはそのような代替または切り替えを行うことなく、安全リスクを増加させることなく、または参照生物製品を独占的に使用することに対して治療効果のリスクを低減することなく、以前の投与後に交互にまたは交換することができる生物学的類似製品である。FDAの承認後、基準製品の衛生保健提供者の介入を開くことなく、交換可能な生物類似体で参照製品を代替することができる。FDAは2021年に交換可能なモノクロナル抗体生物類似体を含む最初の交換可能な生物模倣薬を承認した。
生物類似申請者は、以下のデータに基づいて、製品の生物類似を証明しなければならない:(1)生物類似製品が参考製品と高度に類似していることを示す分析研究、(2)動物研究(毒性を含む)、および(3)1つまたは複数の適切な参考製品の承認使用条件下での安全性、純度、および効力を証明するための1つまたは複数の臨床研究。さらに、出願人は、ラベル、投与経路、用量および強度における生物学的類似製品および参照製品の使用条件が同じ作用機序を有し、生産施設が製品の安全、純度、および効力を確保するための基準に適合しなければならないことを証明しなければならない。
製品が初めて許可を得た日から、参考生物製品は12年のデータ独占期が授与され、最初に承認された交換可能な生物製品は1年に及ぶ独占期が授与され、この独占期間は最長で初の商業発売後の1年となる。2023年の総合支出法案の一部として、国会は同じ日に承認された複数の交換可能な製品が獲得され、この1年の排出期から利益を得ることを可能にするためにPHSAを改正した。FDAが書面で小児科研究を行い、受け入れなければならない場合、12年の専門期間はさらに6ケ月延長される。さらに、FDAは、参照製品が最初に許可された日から4年後まで、参照生物製品に基づく生物学的類似または交換可能な製品の申請を受け入れないであろう。“初許可”とは、一般に、米国で特定の製品が許可された初期日を意味する。最初の許可日は、基準製品の同じ発起人または製造業者(おそらく人、利害関係者、または他の関連エンティティ)がその後に提案した変更(生物物品の構造の変更を含まない)は、新しい適応、投与経路、投与スケジュール、剤形、投与システム、投与装置または強度の変化をもたらすか、または生物学的製品の構造を変更して安全性、純度または効力の変化を引き起こさない後に出願される参照製品の補充物の許可日をもたらす(および新しい特定期間は適用されない)。したがって、新製品が以前の許可製品の構造の修正を含むかどうかを決定し、それにより、安全性、純度、または効力の変化をもたらし、新製品の許可がそれ自身の排他期間をトリガする最初の許可であるかどうかを評価する必要がある。その後の出願は、承認された場合には、生物製品としての“第一次許可”の排他性が保証されるか否かは、具体的な状況やスポンサーが提出したデータに依存する。
BPCIAは複雑であり、FDAはまだ説明して実施している。また、政府の最近の提案は12年間の参考製品専門期間を短縮しようとしている。BPCIAの他の面では,そのいくつかはBPCIAの排他的規定に影響を与える可能性があり,訴訟のテーマでもある.したがって、協約の最終的な影響、実行、そして意味には大きな不確実性がある。
法規を付加する
これらの規定に加えて,環境保全や有害物質に関する州や連邦法は,“職業安全と健康法”,“資源節約と回収法”,“有毒物質制御法”を含めて,我々の業務に影響を与える。これらの法律と他の法律は私たちの様々な生物、化学、放射性物質の使用、処理と処理、これらの物質は私たちの行動、そして私たちの行動によって生成された廃棄物を規範化している。もし私たちの運営が環境汚染を招いたり、個人を危険物質に曝露させたりすれば、損害賠償と政府罰金の責任を負う可能性があります。私たちは、私たちが適用される環境法律を実質的に遵守し、これらの法律を遵守し続けることは、私たちの業務に実質的な悪影響を与えないと信じている。しかし、私たちはこのような法律の変化が私たちの未来の運営にどのように影響するか予測できない。
26
また、いくつかの国はすでにあるいはヒト遺伝物質、細胞或いは組織の輸出入に対して法的制限を実行することを考慮している。例えば、1998年6月、科学技術部と元衛生部は中国で共同で“中国人類遺伝資源管理暫定方法”を制定した。2015年7月、衛生部は“人類遺伝資源のサンプリング、採集、取引、出力サービスガイドライン”を発表し、その中で、外国実体は臨床試験中に患者の人類遺伝資源を収集と使用し、そのオンラインシステムを通じて人類遺伝資源管理局(HGRAO)に報告すべきであると規定した。
2017年10月、科学技術部は“人類遺伝資源の行政審査の最適化に関する通知”を発行し、中国薬品の上場許可を求めることを目的として人類遺伝資源の収集と使用の審査プロセスを簡略化した。
2019年5月、中国の国務院は“人類遺伝資源管理条例”(以下は“条例”と略称する)を発表し、国内外の部門が研究協力を展開する審査条件を規定した。この新規定によると,臨床研究地点で中国患者の生物試料を用いて国際臨床試験を行う際には,このような生物試料を中国以外の地域に輸出することには触れない新たな届出制度(従来の事前承認方法とは逆)が実施される。このような臨床試験を行う前に,バイオ殺虫剤のタイプ,数量,用途が規定された通知書類をHGRAOに提出する必要がある。輸出に関する基礎科学研究の国際協力では,中国患者の生物試料を収集,使用,外転移するにはHGRAOの事前承認が必要である。
2020年10月、全人代常務委員会は“中国生物安全法”を公布し、2021年4月15日から施行された。中国生物安全法は“中国生物安全条例”が規定した監督管理要求を再確認するとともに、中国人類の遺伝資源の収集、保存或いは輸出の疑いのある外国実体に対する行政罰金を大幅に高める可能性がある。
アメリカの“海外腐敗防止法”
我々が受けている米国の“反海外腐敗法”(以下、“反海外腐敗法”と略称する)は、会社や個人が何らかの活動に従事して業務を取得または保留したり、公的な身分で仕事をしている人に影響を与えることを禁止している。いかなる外国の政府職員、政府職員、政党または政治候補者に支払いを提案し、任意の価値のあるものを支払うことを提案して、業務を獲得または保留しようとしているか、または他の方法で公的身分で働く人に影響を与えることは、不法である。
アメリカ以外の政府規制
アメリカの法規のほかに、研究と開発、臨床試験、テスト、製造、安全性、有効性、 ラベル、包装、保存、記録保存、配布、報告、広告、および生物製品に関する他の販売促進活動、ならびに私たちの製品の許可と承認。生物由来の原材料は独特の汚染リスクに直面しているため、それらの使用はいくつかの国で制限される可能性がある。
臨床試験、製品許可、定価と精算を指導する要求と手続きは国によって異なる。いずれの場合も,臨床試験はGCPおよび“ヘルシンキ宣言”からの適用法規要求と倫理原則に基づいて行われなければならない。もし私たちが適用される外国の規制要求を遵守できない場合、私たちは罰金、臨床試験の一時停止、監督管理の許可の一時停止または撤回、製品のリコール、製品の差し押さえ、運営制限、刑事起訴などに直面する可能性がある。
EU臨床試験条例
私たちの製品がFDAの承認を得ているかどうかにかかわらず、私たちは臨床試験を開始する前に、またはこれらの国でその製品を販売する前に、外国の規制機関の必要な承認を得なければならない。米国以外のある国にも類似したプログラムがあり,ヒト臨床試験開始前に臨床試験申請を提出することが求められており,INDに似ている。例えば、EUでは、各臨床試験は、FDAおよびIRBと非常に類似している各国の国家主管当局(“NCA”)および少なくとも1つのIECに臨床試験申請(“CTA”)を提出しなければならない。CTAが一国の要求に応じて承認されると,それに応じた臨床試験を継続することができる。現在の制度(EU臨床試験指令2001/20/ECと対応する国家法律)によると、臨床試験期間中に調査された薬物に対して発生するすべての疑わしい意外な深刻な副作用は、これらの反応が発生した加盟国のNCAとECSに報告しなければならない。
2022年1月31日に施行された(EU)第536/2014号臨床試験条例によると、EU加盟国の主管当局が先頭に立って申請の第1部を審査する集中的な申請手続きがある
27
それは科学と医薬製品文書を含み、他の国家当局は限られた参加しかない。二番目の部分は国と患者レベルの文書を含み、各EU加盟国によって個別に評価される。CTAに提出された試験案または他の情報のいずれかの重大な修正は、関係主管当局および道徳委員会に通知するか、またはその承認を得なければならない。臨床試験で使用される薬品は良好な生産実践に従って生産されなければならない。他の国と連合の範囲の規制要件も適用される可能性がある。
現在、臨床試験がどの程度臨床試験条例の監督を受けるかは、臨床試験がいつスタートするか或いは行っている試験の持続時間に依存する。2023年1月から、すべての新しい臨床試験は臨床試験条例に符合しなければならない。また,2023年1月1日にすでに行われている臨床試験と,“臨床試験規則”の適用日(すなわち2025年1月31日)から3年間を超える臨床試験が継続され,その際に“臨床試験規例”がこの臨床試験に適用されるようになる。
欧州医薬品局による薬品の審査と承認
ヨーロッパ経済区(“EEA”)では(EU加盟国にノルウェー、アイスランドとリヒテンシュタインを加えて構成されている)、医療製品、高級治療薬(“ATMP”)を含み、すべてヨーロッパ経済区と国家レベルの監督管理機関の広範な発売前と発売後の監督管理を受けている。(EC)第1394/2007号法規(“ATMP法規”)に基づいて規制され、ATMPは遺伝子治療製品、体細胞治療製品と組織工学製品を含む。猫(以下の定義)は組織工学製品に指定されている.遺伝子治療製品は遺伝子を体内に送り込み、それによって治療、予防或いは診断効果を産生する。私たちはこの反応が欧州経済地域でATMPとして規制されると予想している。
欧州経済区規制制度の下でATMPの規制承認を得るためには、欧州経済区管理局が管理する中央手続きに基づいて上場許可申請(MAA)を提出しなければならない。米国がBLAを提出するために使用する出願は、ATMP法規に記載されているいくつかの特定の要件、例えば、MAAに含まれなければならないいくつかの追加の製品特性情報である点で、EUが要求する出願手順と同様である。集中化手続きは、欧州委員会によってすべてのヨーロッパ経済地域に有効な単一マーケティング許可を付与することを規定している。ATMP条例の規定によると,ATMPのMAASの科学的評価は主に高度治療委員会(“CAT”)と呼ばれる専門的な科学委員会によって行われている。拷問禁止委員会はATMPの品質、安全性、有効性に関する意見草案を用意しており、これがテーマです MAAの一部は,人用薬品委員会(“CHMP”)に送付され最終承認された。そして、CHMP提案はすべての欧州経済圏加盟国に対して拘束力がある決定を採択した欧州委員会に送信された。ATMPのMAA評価の最長期限は、有効なMAAを受信した日から210日であり、拷問禁止条約および/またはCHMPの質問に回答した場合の出願人の補足情報または書面または口頭解釈は含まれていない。タイミングを停止することは、MAAを評価する時間フレームを210日を超えるように大幅に延長する可能性がある。CHMPが肯定的な意見を与えた場合、EMAは、EMAの提案を受けて67日以内にマーケティング許可を付与する最終決定を下し、発表するサポートファイルと共にEU委員会に提供する。特殊な場合、公衆衛生の観点、特に治療革新の観点から見ると、1種の医薬製品が重大な利益を持っている場合、CHMPは加速評価を承認することができる。CHMPがそのような要求を受け入れる場合、210日間の評価期限は150日(クロック停止を含まない)に減少するが、CHMPが申請が加速評価にもはや適していないと判断した場合、中央プログラムの標準時限に回復する可能性がある。
2023年4月、欧州委員会は既存のヒト薬物立法枠組みを改正し、代替する提案を発表した。現在の提案に従って採択され、実施されれば、これらの改正はEUの医薬製品の開発と承認のいくつかの側面を著しく変えるだろう。
また、連合王国(大ブリテンおよび北アイルランド連合王国を含む)がEUから離脱したため、イギリスは集中マーケティング許可のカバーを受けなくなる(北アイルランド議定書によると、北アイルランドでは集中マーケティング許可を認め続ける)。現在の集中マーケティング許可を有するすべての医薬製品は、2021年1月1日にイギリスのマーケティング許可に自動的に変換される。しかし,2021年1月1日以降にイギリスで新医薬製品を商業化するためには,英国薬品·保健製品規制機関(MHRA)に単独で申請する必要がある
データとマーケティングの排他性
ヨーロッパ経済地域はまた市場排他性の機会を提供する。ヨーロッパ薬品管理局のマーケティング許可を得た後、革新的な医薬製品は通常8年間のデータ独占権と他の2年間の市場独占権を獲得する。もし承認を得た場合、データ排他性は模倣薬或いは生物類似申請者がヨーロッパ薬品管理局が初めて参考製品を許可した日から8年以内に模倣薬或いは生物類似製品の発売許可を申請する時、参考製品ファイルに含まれる革新者の臨床前と臨床試験データを参照する。追加の2年間の市場独占期間内に、模倣薬または生物学的に似たマーケティング許可を提出することができ、イノベーターのデータは
28
しかし、市場の排他期が終わる前に、いかなる模倣薬や生物類似製品も発売できない。この10年の最初の8年間に、マーケティング許可所有者が1つまたは複数の新しい治療適応の許可を得た場合、10年全体の期間は最大11年に延長され、許可前の科学的評価では、これらの適応は既存の療法と比較して有意な臨床的利益をもたらすことができると考えられる。1つの革新的な医薬製品が所定のデータ独占期間を取得しても、別の会社がMAAベースのマーケティング許可、および薬物試験、臨床前試験および臨床試験の完全な独立したデータパケットを取得した場合、同社はその製品の別のバージョンを販売することができる。しかし、1つの製品がEU監督機関によって革新的な医薬製品とみなされることは保証されないため、製品はデータ排他性を得る資格がない可能性がある。
承認後にコントロールする
承認を得た後、上場許可の保有者は医薬製品の製造、マーケティング、普及と販売に適用される一連の要求を守らなければならない。これらの措置には
|
|
|
上場許可所持者は薬物警戒システムを構築·維持し,資格のある個人を指定して薬物警戒を担当し,このシステムの監督を担当しなければならない。主な義務は深刻な副作用の疑いの報告の加速と定期的な安全更新報告(“PSURs”)の提出を含む。 |
|
|
|
すべての新しいMAAは、企業が実施するリスク管理システムを記述し、製品に関連するリスクを最小限に防止または低減するための措置を記録するリスク管理計画(RMP)を含まなければならない。規制部門はまた、上場許可の条件として特定の義務を規定することができる。このようなリスク最小化措置または認可後の義務は、追加の安全監視、PSURsのより頻繁な提出、または追加の臨床試験または認可後の安全性研究を含む可能性がある。RMPおよびPSURsは通常,アクセスを要求する第三者に利用可能であるが,限られた編集を行う必要がある. |
|
|
|
許可された医薬製品の製造はまた、第2001/83/EC号指令、第2003/94/EC号指令、(EC)第726/2004号条例、および欧州委員会の良好な製造規範ガイドラインを含む、適用されるEUの法律、法規とガイドラインを厳格に遵守しなければならない。これらの要件には,医薬製品や活性医薬成分を生産する際にEU cGMP基準を遵守することが含まれており,EU以外で活性医薬成分を製造して活性医薬成分をEUに輸入しようとすることも含まれている。 |
|
|
|
製品のすべての広告および販売促進活動は、承認された製品特性要約(SmPC)と一致しなければならないため、すべてのラベル外の販売促進活動は禁止される。連合はまた消費者に直接向けた処方薬の広告を禁止する。薬品広告や販売促進の一般的な要求はEU指令に基づいて制定されているが、詳細はEU加盟国ごとの法規によって管轄されており、各国が異なる可能性がある。 |
EUおよびEU加盟国が臨床試験、生産承認、医薬製品の上場許可およびこのような製品のマーケティング(上場許可を承認する前と後)、薬品製造、法定医療保険、賄賂および反腐敗または他の適用法規の要求に適用される法律を遵守しない場合、行政、民事または刑事罰を受ける可能性がある。このような処罰には、臨床試験の遅延または拒否、または上場許可の付与、製品の撤回およびリコール、製品の差し押さえ、一時停止、マーケティング許可の撤回または変更、生産の完全または部分的な一時停止、流通、製造または臨床試験、経営制限、禁止、免許一時停止、罰金、および刑事罰が含まれる可能性がある。
イギリスの離脱とイギリスの規制枠組み
2021年1月1日から、EU法律はイギリスに直接適用されなくなった。連合王国は既存のEU薬品条例を採択し、独立した連合王国立法として、上場許可及び他の規制規定に関連する手続き及びその他の要求を反映するためのいくつかの改正を行った。
MHRAはイギリスの医薬製品市場(大ブリテンおよび北アイルランド連合王国)の管理を担当している。イギリスで薬品を販売するために、製造業者はイギリスの許可を持っていなければならない。2021年1月1日、すべてのEUマーケティングライセンスはイギリスマーケティングライセンスに変換されましたが、メーカーは撤退を選択しなければなりません。連合王国はすでに規制提出と医薬製品MAのために単独のイギリスに対するプログラムを導入しており、MHRAガイドラインは、連合王国はEU分散と相互承認プログラムによるマーケティング許可を考慮する権利があると規定している。2024年1月1日、MHRAは、EMAまたはFDAのような信頼されたパートナー機関から積極的なマーケティング許可決定を得た製品に迅速な許可プログラムを提供する国際認可プログラム(IRP)を開始した。国際専門家諮問計画の下で、2つの評価と認められる方法がある
|
|
|
承認経路A−提出書類発効後60日 |
29
|
|
|
|
申請は2年前の基準規制(RR)、マーケティング許可に基づいていなければならない |
|
|
|
|
|
RRが承認した品質プロファイルと大きく異なる場合には,承認方法Bによる評価が必要である |
|
|
|
|
|
提出時には生産場所がGMP要求に適合する証拠を提供しなければならない |
|
|
|
|
|
経路Bを識別するいかなる条件も満たさない |
|
|
|
|
承認経路B-提出発効から110日目には,申請者が審査中に発見された問題に対応することを可能にするために,70日目に計時(最大60日)を停止する予定である |
||
|
|
|
|
申請は過去10年間のRRマーケティング許可に基づいていなければなりません |
|
|
|
|
|
他の事項以外に、承認を得る必要がある基準Bは、以下のことを含む |
|
|
|
|
|
|
RRは、条件付きまたは特別な場合のマーケティング許可を付与する |
|
|
|
|
|
出願に含まれる他の製造地点はRRによって評価されていないか、または製造場所はGMP認証を通過していない |
|
|
|
|
|
RR承認のプロセスに比べて製造プロセスが大きく変化している |
|
|
|
|
|
いくつかの製品タイプ(例えば、高度治療薬、孤児薬、非処方薬) |
|
|
|
|
|
リスク管理計画はRRで評価されていない |
|
|
|
|
|
RRは製品の1つ以上のライセンス後の安全研究を要求している |
|
|
|
|
|
製品を正しく使用するにはセットの診断プログラムが必要です |
“2021年薬品と医療機器法”によると、イギリスの薬品立法は将来的に規制が変化する可能性がある。その法案は薬品条例を採択するための新しい枠組みを作った。
北アイルランド議定書施行後、北アイルランドでは異なる規則が適用され、この議定書によると、EU中央マーケティング申請は北アイルランドに適用され続けている。しかし、2023年3月、イギリス政府と欧州委員会は北アイルランド議定書、すなわちウィンザー枠組みの代わりに規制枠組みについて合意した。ウィンザー枠組みは2025年1月1日から適用される予定であり、英国の薬品規制を含む北アイルランド議定書下の既存制度を変更する。具体的には、MHRAはイギリス(すなわちイギリスと北アイルランド)で販売しようとしているすべての薬物を承認することを担当し、EMAは北アイルランドで販売しようとしている薬物の承認に参加しなくなる。
“貿易·協力協定”は2021年1月1日に発効し、EUと連合王国間のパートナーシップのための枠組みを定めた。欧州連合と連合王国間の“貿易と協力協定”には、医薬製品の薬品供給を促進し、公衆の健康を促進し、消費者を保護することを目的とした医薬製品に関する添付ファイルが掲載されている。添付ファイルには良好な製造規範(GMP)検査と証明書の相互承認が規定されており,これは製造施設が両市場の重複検査を必要としないことを意味する。添付ファイルは、“貿易·協力協定”の項目の下での事項を処理し、協力を促進し、技術的議論を行うための医薬製品ワーキンググループを設置する。“貿易·協力協定”のテーマに属さない規制分野について、薬物警戒を含めたさらなる二国間議論が続く見通しだ。貿易·協力協定には、ロット検出認証を認める互恵的な取り決めも含まれていない。しかし、イギリスは欧州経済地域を含む承認された国をリストしており、これにより、イギリスの輸入業者と卸売業者が特定の認証と規制基準を認めることができるようになる。欧州委員会はまだそのような承認手続きを採択していない。
連合王国に過渡的な承認手続きがあるにもかかわらず、単独の連合王国認可システムが構築されることが予想され、追加の規制費用につながる。また、ロット検査と関連規制措置の相互承認が不足しているため、追加の規制コストが生じる可能性がある。
これに関連して、イギリスのEU離脱に伴い、“一般データ保護ルール”(以下、“一般データ保護ルール”)がイギリス(すなわちイギリス“一般データ保護ルール”)で実施されている。英国GDPRは、改正された2018年の英国データ保護法と並んで、EU GDPRのいくつかの減税を英国法に盛り込んでいる。イギリスGDPRによると、イギリスに設立されていないが、イギリスで個人への商品やサービスの提供に関する個人データを処理したり、その行動を監視したりする会社は、イギリスのGDPRの制約を受ける--その要求はEU GDPRの要求とほぼ一致するため、類似のコンプライアンスおよび運営コストを招き、潜在的な罰金は1,750万ポンドまたは世界売上高の4%に達する可能性がある。2021年6月28日、欧州委員会は、連合王国がEU GDPRによってEUからイギリスに移行した個人データが十分に保護されることを確保する決定を発表した。2021年6月、欧州委員会は、2025年6月27日に落山し、これ以上の行動を取らない決定を発表し、EU GDPRによるEUから英国への個人データの十分な保護を確保することを英国に求めた。また、イギリス議会は現在、2018年データ保護法、イギリスGDPR、プライバシー、電子通信法規を1つの立法枠組みで統一するためのデータ保護とデジタル情報法案を審議している。
30
他の保健法やコンプライアンス要件は
もし私たちの候補製品がアメリカで承認されたら、私たちは反リベート法と虚偽声明法律、規則、法規を含む、医療詐欺や乱用に関連する様々なアメリカ連邦と州の法律、規則、法規を遵守しなければならない。詐欺や法の濫用は以下のように処罰される 刑事·民事制裁は、連邦医療保険や医療補助を含む連邦·州医療保健計画から除外される場合も含まれる。これらの法律には
|
|
|
他の事項に加えて、直接的または間接的、公開的または隠蔽的に現金または実物で直接または間接的に、公開的または間接的に請求、受信、提供または提供または任意の報酬(任意のリベート、賄賂またはリベートを含む)を誘導または交換として、個人を紹介するか、または購入、レンタル、注文、手配、または連邦医療保険および医療補助計画のような連邦医療保険および医療補助計画に従って支払い可能な任意の商品、施設、物品またはサービスを推薦することを禁止する連邦反バックル法規(AKS)。個人またはエンティティは、AKSまたはAKSの具体的な意図に違反することを実際に知る必要がなく、違反を実施することができる。さらに、政府は、連邦虚偽請求法(FCA)または連邦民事罰金法規に基づいて、AKS違反による物品またはサービスのクレームが虚偽または詐欺的クレームを構成することを含むと断言することができる |
|
|
|
FCAを含む連邦民事および刑事虚偽申告法および民事罰金法であって、個人または実体が虚偽または詐欺的な支払い申請の提出または提出を意図的に禁止するか、またはMedicare、Medicaidまたは他の連邦医療保健計画によって承認され、虚偽記録または報告書の作成、使用または作成または使用をもたらすことは、虚偽または詐欺的クレームまたは連邦政府への資金の支払いまたは移転の義務に重要な意味を持ち、または故意に隠蔽または承知して、連邦政府に金銭を支払う義務を不当に回避または減少または隠蔽することを含む、連邦民事および刑事虚偽申告法および民事罰金法。メーカーが直接政府にクレームを提出していなくても、虚偽や詐欺的なクレームを“原因”とされていれば、FCAによってメーカーは責任を問われる可能性がある。FCAはまた、FCA違反行為を告発し、いかなる金銭的回収も共有する“密告者”としての個人代表が連邦政府を代表して訴訟を起こすことを許可した |
|
|
|
民事経済罰金法(受益者誘導法)は、他に加えて、医療保険または医療補助受益者に無料または公平な市場価値よりも低い任意の方法で物品またはサービス(限られた例外)を譲渡することを含むが、これらの物品またはサービスが、連邦または州政府案によって精算可能な物品またはサービスを選択する特定のサプライヤーに影響を与える可能性があることを知っているか、または知るべきである |
|
|
|
1996年の“健康保険携帯性および責任法案”(HIPAA)は、支払人(例えば、公共または個人)にかかわらず、支払人(例えば、公共または個人)にかかわらず、詐欺または詐欺的な言い訳、陳述または約束によって、支払人(例えば、公共または個人)にかかわらず、任意の医療福祉計画が所有または保管または制御された任意の金銭または財産を故意にまたは意図的に実行または実行しようとすることを禁止する新しい連邦刑法を制定し、詐欺または装置の重要な事実を故意に偽造、隠蔽または隠蔽または隠蔽し、または任意の重大な虚偽、架空を可能にする。または保健事務に関連する保健福祉、プロジェクトまたはサービスの提供または支払いに関連する詐欺的陳述または陳述;AKSと同様に、個人またはエンティティは、法規または法規違反の具体的な意図を実際に知る必要がなく、違反を実施することができる |
|
|
|
ACA下の連邦透明性要件は、連邦医療保険、医療補助または児童健康保険計画に従って支払いを受けることができる適用可能な薬品、器具、生物製品および医療用品の製造業者が、米国医療保険および医療補助サービスセンター(CMS)に毎年米国医療保険および医療補助サービスセンター(CMS)に報告され、医師(医師、歯科医、検眼師、足科医および脊医を含むと定義される)、医師アシスタント、看護師勤務者、臨床看護師、登録看護師麻酔科医、麻酔科医アシスタント、登録助産師および教育病院への支払いまたは他の価値移転に関する情報を含む、一般的に医師支払い法と呼ばれる条項およびその実施条例を含む。医師とその直系親族が所有している所有権と投資権そして |
|
|
|
“海外腐敗防止法”および他の反腐敗法律法規は、私たちの財務関係および外国政府関係者との相互作用に関連しており、米国企業およびその従業員、役人および代表が任意の外国政府職員(私たちが製品を運営または販売している国の医療専門家を含むことができる)、政府職員、政党または政治候補者への支払い、支払い、承諾または許可を提出して、業務を獲得または維持し、または他の方法で優遇待遇を求めることを禁止する。 |
また、他の以外にも、上述の各医療法律と法規を遵守する州は国外と同等の法律と法規を遵守しなければならず、その中のいくつかの法律と法規の範囲はもっと広く、支払者にかかわらず適用可能である。米国の多くの州はAKSやFCAと類似した法律を採用しており、研究、流通、販売またはマーケティング手配、および非政府支払人(個人保険会社を含む)によって精算される医療項目やサービスに関するクレームを含むが、私たちのビジネス実践に適用可能である。また、一部の州では、製薬会社に2003年4月の総監察長室の製薬メーカーおよび/または製薬研究に関するコンプライアンス計画ガイドラインの遵守を求める法律が可決された
31
アメリカの保健専門家と相互作用しているメーカーですいくつかの州はまた、他のマーケティング制限を実施したり、製薬会社にその州へのマーケティングまたは価格開示を要求したりしている。このような州の要求を守るために何が必要なのかは曖昧で、もし私たちが適用された州の法律要求を守らなければ、私たちは処罰されるかもしれない。
これらの法律の範囲が広く、法定例外状況と選択可能な避難港が限られていることから、私たちのいくつかの商業活動は1つ以上のこのような法律の挑戦を受けるかもしれない。
詐欺および法律違反行為は、懲罰、罰金、監禁および/または連邦および州医療保健計画(例えば、MedicareおよびMedicaid)の排除または一時停止、および米国政府との契約を禁止することを含む刑事および/または民事制裁を受ける可能性がある。また、個人的には米国政府を代表してFCAやいくつかの州の虚偽請求法に基づいて訴訟を起こす能力がある。
法執行部門は医療詐欺や法律の乱用をますます重視しており、私たちのいくつかのやり方はこれらの法律の挑戦を受けるかもしれない。私たちの現在と将来の第三者との業務配置と、私たちの業務が全体的に適用される医療法律や法規に適合していることを確保するために努力しており、多くのコストがかかります。もし私たちの業務が、私たちと医師および他のヘルスケア提供者との手配を含めて、私たちに適用される任意のこのような法律または任意の他の政府法規に違反していることが発見された場合、私たちは、行政、民事および刑事罰、損害、罰金、返還、契約損害、名声損害、利益減少および将来の収益、私たちの業務の縮小または再編、連邦および州医療保健計画(例えばMedicareおよびMedicaid)から除外され、監禁を含むかもしれませんが、これらはいずれも、私たちの業務運営能力や財務業績に悪影響を及ぼす可能性があります。我々の米国以外の任意の候補製品の承認と商業化も,上記の医療保健法や他の外国法の外国等価物の制約を受けるであろう。
もし私たちがそれと業務を展開することを期待している任意の医師や他の医療提供者や実体が法律に適合していないことが発見された場合、彼らは政府の援助された医療計画から除外されることを含む刑事、民事または行政制裁を受ける可能性があり、これはまた私たちの業務に悪影響を及ぼす可能性がある。
これらの法律の多くは規制部門や裁判所によって十分に解釈されていないため、これらの法律に違反するリスクが増加していることが発見され、それらの条項には様々な解釈があるかもしれない。このような法律に違反して私たちに取った行動は、たとえ私たちがそれを防御することに成功しても、可能です 私たちは巨額の法的費用を招き、私たちの経営陣の業務運営への関心を移すことになった。常に変化するコンプライアンス環境、および異なるコンプライアンスおよび報告要件を有する複数の司法管轄区域の需要に適合するために強力なシステムを確立し、維持することは、医療保険会社が1つまたは複数の要件に違反する可能性を増加させる。私たちが第三者の業務配置と適用される医療法律や法規に適合するように努力することは、多くのコストに関連することを確実にします。
データのプライバシーとセキュリティ
健康情報や個人情報のプライバシーや安全を管理する連邦,州,外国の法律があり,その多くの法律は互いに大きく異なり,同時に適用され,コンプライアンス作業を複雑にしている。
衛生情報技術による経済と臨床健康(HITECH)及びその実施条例の改正を促進したHIPAAは個人が健康情報を識別できるプライバシー、安全と伝送に関連する要求を強化し、拡大した;そしてある個人と監督管理機関が個人が健康情報の安全を識別できることに違反した場合に影響を受けた個人と監督管理機関に通知することを要求した。
HITECHはHIPAAを強化·拡大し,違反に対する罰を増加させた.HITECHによると、監督されたエンティティは連邦政府と州総検察長の強制執行を受け、彼らはHITECHによるHIPAAの実行を許可された。一部の州の法律では、HIPAAよりも厳しいプライバシー保護と、医療情報以外の情報に適用されるデータセキュリティ要件が実施されている(例えば、2018年カリフォルニア消費者プライバシー法案(CCPA))。これらの州の法律は法執行レベルを増加させ、違反事件が発生した時に追加的な報告書を提出することを要求するかもしれない。私たちと協力して私たちの製品を開発してテストするほとんどのアメリカのヘルスケア提供者はHIPAAと適用される州法律を守らなければなりません。私たちはこれらの法律に直接支配されていないかもしれないが、私たちはこれらの法律に従って私たちの活動を組織して、衛生情報を取得して使用して、私たちの研究、開発、その他の活動を支援できることを確実にしなければならない。私たちがこれらのプライバシーおよびセキュリティ法律を遵守していないか、または健康情報または個人データに違反することは、当社のヘルスケアプロバイダーパートナーのための法執行を促進し、当社の第三者責任および/または当社に重大な財務的または名声的損害をもたらす可能性があります。
私たちはまた追加的なプライバシー制限を受ける可能性がある。欧州経済地域の個人に関する個人データを収集、使用、記憶、開示、移転、または他の方法で処理し、個人健康データを含み、2018年5月25日に施行されたGDPRによって管轄される。“個人資料開示条例”は範囲が広く、個人資料を処理する会社に複数の規定を加え、健康及びその他の敏感な資料の処理、個人資料に関連する個人の明確な同意の取得、個人に資料処理活動に関する資料を提供し、個人資料の安全と秘密を保障するための保障措置を実施し、資料違反事項について通知を行い、及び
32
第三者加工業者と交渉する時にいくつかの措置を取る。GDPRはまた、EUや欧州経済圏以外の国(米国を含む)への個人データの移転に厳しいルールを実施し、データ保護当局が2000万ユーロまたは世界の年収の4%に達する可能性のある罰金を含むGDPR違反行為に巨額の罰金を科すことを許可している。GDPRはまた、データ主体と消費者協会に個人訴訟権利を与え、監督当局に苦情を提出し、司法救済を求め、GDPR違反による損害について賠償を受けることができる。さらに、GDPRは、国境を越えたデータ転送の制限を含む。GDPRを遵守することは厳格で時間のかかるプロセスであり、私たちの業務コストを増加させたり、私たちに業務やり方の変更を要求したりする可能性があり、私たちはこれらの努力をしたにもかかわらず、私たちは私たちのヨーロッパ活動に関連した罰金と処罰、訴訟、名声損害のリスクに直面するかもしれない。
2023年7月、欧州委員会は、EUから米国にデータを転送する新しいメカニズムであるEU-米国データプライバシーフレームワークに関する十分な決定を採択し、EU内の個人に、そのデータにアクセスする権利を取得する権利、または誤ったまたは不正に処理されたデータの訂正または削除の権利を取得することを含むいくつかの新しい権利を提供する。十分な決定を下す前に、EUは、Schrems IIという事件の判決で提起されたいくつかの点を解決するために、新たな拘束力のある保障措置を導入する行政命令に署名した。この事件は、以前のEU-米国のプライバシー盾を無効にした。新たな義務は、米国の情報機関が必要かつ見合った程度しかデータにアクセスできないことを確保し、国家安全目的のためのデータ収集に関する欧州人の苦情を処理するための独立かつ公正な救済メカニズムを構築することを目的としていることに留意されたい。欧州委員会は十分な決定を下しながら、米国の事態を検討していくだろう。事態が適用法域の保護レベルに影響すれば,十分な決定を調整あるいは撤回することができる.連合データ保護当局の未来の行動は予測が難しい。一部の顧客または他のサービスプロバイダは、これらの変化する法律および法規に応答して、私たちができない、またはしたくないいくつかのプライバシーまたはデータに関連する契約約束をすることを要求するかもしれません。これは既存または潜在的な顧客または他のビジネス関係を失うことになる可能性がある。
医療改革
米国や一部の外国司法管轄地域では、医療システムに関するいくつかの立法や規制改革や提案された改革が継続されており、これらの改革は、製品や治療候補薬の発売承認を阻止または延期し、承認後の活動を制限または規範化し、上場承認された製品や治療候補薬の利益のある販売能力に影響を与える可能性がある。FDAおよび他の規制機関の政策は変わる可能性があり、規制部門の私たちの製品および候補治療薬の承認を阻止、制限、または延期するための追加の政府法規が公布されるかもしれないもし私たちが既存の要求の変化に適応できない場合、あるいは新しい要求や政策を採用することができない場合、あるいは規制コンプライアンスを維持できない場合、私たちが本来得る可能性のあるマーケティング承認を失う可能性があり、私たちは利益を達成したり維持することができない可能性があり、これは私たちの業務、将来性、財務状況、運営結果に悪影響を及ぼすだろう。
また,医療費の抑制が連邦や州政府の優先順位となっており,治療薬の価格がこの努力の重点となってきた。アメリカ政府、州立法機関と外国政府は政府が支払う医療コストの増加を制限するためにコスト制御計画を実施することに強い興味を示し、これらの計画はそれぞれ価格制御、精算制限及びブランド処方薬の代わりに模造薬と生物類似製品の使用を要求することを含む。近年,米国議会では医師が管理する薬品や生物製品の医療保険精算レベルを下げることが考えられている。CMSは連邦医療保険や医療補助計画を管理する機関であり,精算料率を改正し,多くの薬物や生物製品にカバー制限を実施する権利もある。立法や法規によって実施されるコスト削減措置やカバー範囲の変化は、将来販売される可能性のある任意の承認された製品の使用率や精算を減少させる可能性があります。連邦医療保険法規は連邦医療保険受益者の薬品福祉にのみ適用されるが,個人支払者は自分の精算料率を設定する際には通常連邦医療保険カバー政策と支払制限に従う。したがって、連邦立法や法規によるいかなる精算減少も、個人支払者の支払いの同様の減少を招く可能性がある。
“衛生保健と教育負担能力協調法”の改正されたACAは2010年に公布され、アメリカ政府と私営保険会社が衛生保健に資金を提供する方式を大きく変え、製薬業に重大な影響を与えた。ACAは医療保険の許容性を拡大し、医療支出の増加を減少或いは制限し、医療詐欺と濫用に対する救済措置を強化し、医療保健と医療保険業界の新しい透明性要求を増加させ、製薬業者に対して新しい税費を徴収し、追加の医療政策改革を実施することを目的としている。生物製薬製品の面で、他の事項以外に、ACAは1種の新しい方法を提案し、即ち吸入、輸液、点滴、移植或いは注射の治療薬物に対して、メーカーがMedicaid薬品リベート計画の下で獲得すべきリベートを計算し、Medicaid薬品リベート計画の下でメーカーが不足している最低Medicaidリベートを増加し、そしてリベート計画をMedicaid管理の看護組織に登録した個人に拡大し、あるブランドの処方薬メーカーに対する年会費を創立し、新しいMedicare Part Dカバーギャップ割引計画を作成し、そして340 B薬品割引計画を拡大した。別の例として2021年には
33
2020年12月27日に署名された法律となる総合支出法案は、連邦医療保険B部分でカバーされているすべての薬品·生物製品メーカーに2022年1月1日から製品の平均販売価格を国土安全保障省に報告し、民事罰金により強制執行することを含む幅広い医療条項と既存の法律の改正を盛り込んでいる。
LACA下の規制および規制の変化は依然として可能であるが、任意のこのような変化または任意の法律がどのような形態をとるか、およびそれがバイオ製薬業界全体または私たちの将来の業務にどのように影響を与える可能性があるかどうかは不明である。ACA,MedicareおよびMedicaid計画の変化や増加,および他の医療改革措置による変化,特に個別州の医療参入,融資または他の立法面での変化は,米国の医療産業に実質的な悪影響を及ぼす可能性が予想される。
また,政府はバイオ製薬メーカーがその上場製品に価格を設定する方式についてもより厳しい審査を行っている。このような審査はアメリカ議会が最近数回の調査を行い、連邦と州立法を提出し、公布し、薬品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、連邦医療保険下の薬品のコストを下げ、政府の薬品に対する計画精算方法を改革することを目的とした。注目すべきは、2019年12月20日、“2020年更なる総合支出法”が法律(第116-94号法律)となり、“2019年に同等のサンプルを取得する法案の作成と回復”(“Creates Act”)という両党立法が含まれていることである。Creates ActはFDAと他の業界人が表明した懸念、すなわちいくつかのブランドメーカーがその製品の流通を適切に制限しないことを解決し、ある製品のREMSの存在を引用して、模倣薬と生物類似製品の開発業者がブランド製品のサンプルを獲得することを阻止することを含む。模倣薬と生物類似製品開発業者はFDA要求のいくつかの比較テストを行うためにサンプルを必要とするため、一部の人は直ちにサンプルを得ることができないが、模倣薬と生物類似製品の市場進出遅延によるものである。この懸念を解消するために、Creates Actは、模倣薬または生物類似製品開発業者がブランドメーカーを起訴することを許可し、“商業的に合理的で市場に基づく条項”で必要なサンプルを提供することを強要する個人訴訟理由を確立した。模倣薬と生物類似製品の開発及びこの新しい道をどのように使用するか、及びCreates Act条項に対する任意の法律挑戦が生じる可能性のある結果は、依然として高度に不確定であり、それが私たちの未来の商業製品に対する潜在的な影響も未知である。
最近、2022年8月に総裁·バイデンは2022年の“インフレ低減法案”(略称“アイルランド共和軍”)に署名し、法律にした。他の点では、アイルランド共和軍には複数の条項があり、連邦医療保険計画や米国全体に販売されている薬品の価格に影響を与える可能性がある。2023年から、薬品の価格上昇速度がインフレ率よりも速い場合、連邦医療保険BまたはD部分がカバーする薬物または生物製品のメーカーは連邦政府にリベートを支払わなければならない。この計算は薬品製品をもとに行われており、連邦政府に不足している税金還付額は、連邦医療保険B部分またはD部分が支払う薬物製品の数に直接依存する。また、CMSは2026年から毎年、選定された数量の単一源D部分薬物について薬品価格交渉を行い、13年に発売された、模倣薬や生物類似競争のない生物製品を含む。CMSはまた,2028年の支払年度から発売13年の生物製品を含む選定された数のB部分薬物について薬品価格を交渉する。CMSが1つの薬物を選択して交渉すれば,このような薬物による収入は減少すると予想される。CMSはこれらの新しい許可の実施を開始し、2023年10月に製薬業者と第1セットの合意を締結し、価格交渉を行った。しかし,アイルランド共和軍の米国製薬業への影響は不明であり,一部の原因は複数の大手製薬会社や他の利害関係者(例えば米国商会)がCMSに対して様々な理由で違憲,その他の苦情を訴えていることである。このような訴訟は現在も進行中だ。
米国の州レベルでも、立法機関は、価格や患者の精算制限、割引、ある製品への参入の制限、マーケティングコスト開示および透明性措置を含む薬品の価格設定を制御するための法規を立法し、実施することが増えており、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。例えば,近年,いくつかの州で処方薬負担能力委員会(“PDAB”)が設立されている。アイルランド共和軍の薬品価格交渉計画のように,これらのPDABは公共·商業健康計画においてそれぞれの州で販売されている薬品に対して支払上限(UPL)を実施しようとしている。2023年8月、コロラド州のPDABは、負担能力の審査を受ける5種類の処方薬を含むリストを発表した。これらの努力の効果はまだ確定しておらず、いくつかの連邦訴訟の結果が待たれ、これらの訴訟は州政府が処方薬の支払い限度額を規制する権力に挑戦している。また、2020年12月、米国最高裁判所は、連邦法律は各州の薬品福祉マネージャーと医療保健と薬品サプライチェーンの他のメンバーの能力を阻害しないと一致して判断し、この重要な決定は各州がこの分野で更なるかつ積極的な努力をすることを招く可能性がある。2022年中に、連邦貿易委員会はPBM業界のやり方に対して全面的な調査を展開し、これはこのような実体の運営、薬局ネットワーク或いは財務手配に対するより多くの連邦と州立法或いは規制提案を招く可能性がある。アメリカに現在存在しているPBM業界を変える重大な努力は薬品サプライチェーンと他の利益関係者の業務に影響を与える可能性があり、著者らのような生物製薬開発業者を含む。また,地域医療当局や個別病院では,どの薬品やサプライヤーが処方薬や他の医療計画に含まれるかを決定するために入札プログラムが使用されるようになってきている。これらの措置が承認されると、私たちの製品に対する最終的な需要を下げたり、私たちの製品の価格設定に圧力をかけるかもしれません。
34
米国や海外の将来の立法や行政または行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することはできない。私たちは将来、より多くの連邦、州、外国の医療改革措置を取ることが予想され、そのいずれも連邦と州政府が医療製品とサービスのために支払う金額を制限する可能性があり、これは限られたカバー範囲と精算、そして私たちの製品に対する需要の減少、承認されると、あるいは追加の価格設定圧力を招く可能性がある。
保証と精算を請け負う
我々が規制承認を得る可能性のある任意の細胞による再生療法のカバー範囲や精算状態には,大きな不確実性がある。米国や他の国の市場では、規制部門の承認を得て商業販売を行う任意の細胞ベース療法の販売は、保証範囲の可用性および支払者の精算にある程度依存する。支払者には、政府当局、医療サービス提供者、個人健康保険会社、その他の組織が含まれている。自分の病状に応じて処方治療を受けた患者やそのような治療処方を処方した提供者は,通常,これらの第三者支払者に依存して,治療や他の関連医療費の全部または一部を精算する。支払者が医薬品、装置、または生物製品に保険を提供するかどうかを決定するためのプロセスは、支払人が製品のために支払うべき販売率を設定するためのプロセスから分離することができる。支払者は、承認されたリストまたは処方内の特定の製品に保証範囲を制限することができ、FDAによって承認された特定の適応のすべての製品を含まない可能性がある。もし支払人が私たちの細胞ベースの治療法を保証しないことを決定すれば、私たちの製品が承認されれば、医師のこれらの製品の使用を減少させ、私たちの販売、運営結果、財務状況に実質的な悪影響を与える可能性がある。しかも、支払人が商品に保険を提供することを決定することは十分な意味ではありません 販売率は承認されるだろう。十分な第三者精算が得られない可能性があり、製品開発や製造コストへの適切な投資リターンを実現するために十分な価格レベルを維持することができる。
また、製品の保証範囲と精算範囲は支払人によって異なる。1つの支払人は、ある特定の医療製品又はサービスを保証することを決定し、他の支払者も当該医療製品又はサービスに保険を提供することを確保することができず、又は適切な販売率で保険を提供することができる。米国では,新薬精算に関する主な決定は通常CMSによって行われる。CMSは新薬がどの程度連邦医療保険の下でカバーと精算されるかを決定し、個人支払者はよくCMSに大きく従う。
また、保証範囲の決定過程は、各支払人にそれぞれ私たちの製品を使用した科学的および臨床的支援を提供することが要求され、これは時間のかかるプロセスになるだろう。
支払者は、価格、医療製品およびサービスの医療必要性および費用効果、ならびにそれらの安全性および有効性を検討することにますます挑戦している。任意の製品の保険や精算を獲得し維持するためには,このような製品の医療必要性とコスト効果,規制承認に要するコストを証明するために高価な証拠生成研究を行う必要があるかもしれない。支払者が、1つの製品が現在の介護基準と比較して費用効果があると思わない場合、彼らは、彼らの計画に基づいて製品を福祉としてカバーしないかもしれない、または、彼らがそうした場合、支払いレベルは、会社にそのコストまたは利益を補うのに十分ではないかもしれない。
連邦医療保険B部分とD部分(上記参照)がカバーする薬品と生物製品に下振れ圧力を加えることを目的としたアイルランド共和軍のほか、バイデン総裁は2022年10月に14087号行政命令を発表し、CMSに潜在的な支払いと交付モードに関する報告書を準備し、提出することを要求し、これらのモードはIRAを補充し、薬品コストを下げ、革新薬物の獲得を促進する。2023年2月、CMSはその報告を発表し、その中に3種類の潜在的なモードを記述し、重点的に負担可能性、獲得性と実施の実行可能性であり、CMS革新センターの更なるテストに供する。2024年2月まで、CMS革新センターは提案したモデルを引き続きテストし、各州とメーカーがある製品タイプ(例えば細胞と遺伝子療法)にアクセスするモデルテストの計画を開始した。将来的にはより多くの州および連邦医療改革措置が取られることが予想され、いずれも連邦および州政府が医療製品およびサービスのために支払う金額を制限する可能性があり、これは特定の生物製薬製品に対する需要の減少または追加の価格設定圧力をもたらす可能性がある。
アメリカ以外の多くの国では、薬品の定価は政府によってコントロールされている。例えば、連合では、様々な国の価格設定と補償プログラムの差が大きい。一部の国では、政府当局と補償価格を合意した後にのみ、製品を販売することができると規定されている。さらに、いくつかの国は、精算または価格設定の承認を得るために、いわゆる衛生技術評価の一部として、特定の治療法の有効性および/またはコスト効果を現在の看護基準と比較することを追加的な研究の完了を要求する可能性がある。また,診断関連グループシステム下の病院環境で管理されている製品に関するプログラムの精算活動を確保する必要がある可能性があり,記帳コードが存在しない可能性や,現在プログラムの費用を支払うのに不十分である可能性がある。その他の場合,国は製品数をモニタリング·制御し,医師に指導意見を発表し,治療政策の形で処方を制限することができる。各国が衛生保健支出を管理しようとしていることに伴い,薬品や医療機器の価格や使用を抑える努力が続く可能性がある。
35
人的資本資源
ProKidneyは2023年12月31日まで163人のフルタイム従業員を持っている。その中に研究開発部門は53個、製造、業務、品質管理と品質保証部門は83個、一般と行政機能部門は27個であった。私たちは従業員たちと集団交渉合意に到達しなかったし、何の中断も経験しなかった。私たちは私たちが従業員と仲がいいと思う。
私たちの執行担当者と役員に関する情報
名前.名前 |
|
ポスト |
行政員 |
|
|
ブルース·カールトン医学博士 |
|
取締役CEO兼最高経営責任者 |
ジェイムズ·クールストン公認会計士 |
|
*最高財務責任者 |
ダリン·J·ウェーバー博士 |
|
*最高経営責任者、上級副社長、規制事務、品質、バイオメトリクス、および市場参入 |
トッド·C·ジロラモ |
|
*最高法務官 |
非従業員取締役 |
|
|
パブロ·レゴレタ |
|
--取締役会議長 |
ウィリアム·F·ドイル |
|
役員.取締役 |
ジェニファー·フォックス |
|
役員.取締役 |
ホセ·イグナチオ·ヒメネス·サントス |
|
役員.取締役 |
アラン·M·ロトヴィン医学博士 |
|
役員.取締役 |
ジョン·M·マラガノ博士です |
|
役員.取締役 |
ブライアン·J·G·ペレイラ医学博士 |
|
役員.取締役 |
ウマ·シンハ博士です |
|
役員.取締役 |
インターネット上で提供される情報は
私たちのインターネットアドレスはhttp://www.proystney.comで、私たちはよく私たちのニュース原稿のコピーと私たちに関するより多くの情報を公開します。本年度報告Form 10-Kと我々のForm 10-Q四半期報告、Form 8-K現在の報告、およびこのような報告のすべての改訂は、米国証券取引委員会に電子的に提出するか、または米国証券取引委員会に電子的に提出した後、合理的で実行可能な範囲内でできるだけ早く当サイトの投資家部分を介して無料で提供することができます。米国証券取引委員会は、米国証券取引委員会に電子的に提出された報告書、依頼書、情報声明、その他の発行者に関する情報を含むインターネットサイトを有する。私たちは本年度報告書に私たちのサイトアドレスを含めて、非アクティブなテキストとしてのみ参考にします。我々のサイトに含まれる情報は、本報告書または米国証券取引委員会に提出された他の文書の一部を構成していません。
第1 A項それは.リスク要因です
私たちの財務状況と追加資本需要に関連するリスク
成立以来、重大な純損失が発生しており、予測可能な未来に重大な純損失が続くことが予想される。
私たちが設立されて以来、私たちは重大な純損失を発生させた。私たちは私たちの持続的な運営に関連した巨額の研究開発と他の費用を発生させ続けている。2023年,2022年,2021年12月31日までの年度において,我々が報告した非持株権益前純損失はそれぞれ1.354億ドル,1.481億ドル,5510万ドルであった。2023年12月31日と2022年12月31日までの累計赤字はそれぞれ11.397億ドルと11.041億ドルだった。私たちはほとんどの資源と努力を研究と開発に投入しており、製品販売から収入を得るのにあと数年かかると予想される。主要候補製品REPACTのマーケティング承認を得て商業化しても、より多くの潜在的候補製品を開発·マーケティングするために、多くの研究開発やその他の費用が発生し続けることが予想される。
私たちの候補製品Reactionはまだ臨床試験段階にある。私たちは予測可能な未来に大きな損失を被ると予想していますもし私たちの費用が大幅に増加すると予想しています
|
|
|
臨床開発によりREACTや他の任意の将来候補製品の開発を進め,成功すれば後期臨床試験も行う |
|
|
|
将来の任意の臨床前研究、私たちの現在の臨床試験、私たちが依存している第三者サービスプロバイダまたは私たちのサプライチェーンからサービスを受ける遅延または中断は、健康危機またはイベントまたは私たちがコントロールしていない状況による遅延を含む |
36
|
|
|
将来臨床試験に成功する可能性のある候補製品のために監督管理の承認を求める |
|
|
|
商業化反応と任意の未来の候補製品が承認されれば |
|
|
|
候補製品と生産ラインの延長を発見し開発するために、研究と開発活動の数量を増加させる |
|
|
|
臨床試験に必要な材料を生産するか、または必要な規制承認を受けた後、私たちの製造施設で商業販売を行う |
|
|
|
商業規模のcGMP製造施設を構築し、検証し、契約開発·製造組織(CDMO)と協力する |
|
|
|
商業化インフラを構築し、内部と外部の製造·流通能力を拡大し、規制の承認を得る可能性のある任意の候補製品を商業化する |
|
|
|
臨床開発、監督管理、製造、品質管理、品質保証、科学、公共/投資家関係の一般及び行政と管理人員の面でより多くの行政人員を募集する |
|
|
|
私たちの臨床開発と製造努力、一般と行政機能、上場企業としての私たちの運営を支援する人員を含む、私たちの運営、財務、管理システムを拡大し、人員を増加させる |
|
|
|
国内と世界的な販売、マーケティング、医療事務、流通インフラを構築し、マーケティングの承認を得て、単独または第三者と共同で商業化しようとしている任意の製品を商業化する |
|
|
|
私たちの知的財産権の組み合わせを維持し、拡大し、保護する |
|
|
|
他の候補技術や製品に投資したり許可したりする。 |
利益を実現し、維持するためには、巨大な市場潜在力を持つ製品を開発し、最終的に商業化しなければならない。これは私たちが一連の挑戦的な活動の中で成功を得ることを要求し、非臨床研究と臨床試験を完成し、Reactionと任意の未来の候補製品のためにマーケティング許可を獲得し、製造、マーケティングと販売はマーケティング許可を得る可能性のある製品を獲得し、そして任意の発売後の要求を満たすことを含む。バイオ製薬製品開発に関連する多くのリスクや不確実性のため、費用を増加させる時間や金額、あるいはいつ、または利益を達成できるかどうかを正確に予測することはできない。私たちは決してこのような活動やすべての活動で成功しないかもしれないし、たとえ私たちが成功しても、私たちは利益を達成するのに十分な収入を得られないかもしれない。もし私たちが確実に利益を達成したら、私たちは四半期や年度の収益性を維持したり向上させることができないかもしれない。もし私たちが実現して利益を維持できなければ、会社の価値を下げ、資金を調達し、研究開発努力を維持し、業務を拡大し、あるいは運営を継続する能力を弱める可能性がある。
私たちは引き続き私たちの運営に資金を提供するために多くの追加資本を必要とするだろう。必要に応じて、または許容可能な条件下でそのような資金を調達できない場合、私たちは、私たちの1つまたは複数の研究および製品開発計画、将来の商業化努力、または他の運営の延期、減少、および/またはキャンセルを余儀なくされる可能性がある。
臨床試験を含む生物製薬製品を開発することは、非常に時間がかかり、高価で不確定な過程であり、完成するのに数年かかる。設立以来、私たちの業務は大量の現金を消費した。私たちは、私たちが行っている活動に関連する費用が増加することを予想しています。特に、Reactionと私たちが開発する可能性のある任意の未来の候補製品に対して行われている計画された臨床試験、Reactionと私たちの未来の候補製品のための規制承認を求め、私たちが規制承認を受けた任意の製品を製造、発売、商業化する場合。私たちはまた上場企業の運営に関する追加コストが発生すると予想している。したがって、私たちは私たちの持続的な業務を維持するために多くの追加資金を得る必要があるだろう。もし私たちが必要な時や受け入れ可能な条件で資金を調達できなければ、私たちは私たちの1つまたは複数の研究および製品開発計画または将来の商業化努力を延期、減少または廃止することを余儀なくされるかもしれない。
2023年12月31日現在、私たちは約3.63億ドルの現金、現金等価物、短期投資を持っている。私たちの現在の運営計画によると、私たちの現金、現金等価物、短期投資は、2025年第4四半期までの運営費用と資本支出需要を支払うのに十分だと信じています。しかし、これは市場状況のため、私たちが既存の現金、現金等価物、および投資の一部を得ることができない可能性があることを反映していない。例えば、2023年3月10日、連邦預金保険会社(FDIC)はシリコンバレー銀行を引き継ぎ、シリコンバレー銀行の係に任命された。他の銀行や金融機関が将来、銀行システムや金融市場の金融状況に影響を与えて破産手続きや破産に入った場合、既存の現金、現金等価物、投資を取得する能力が脅かされ、私たちの業務や財務状況に大きな悪影響を及ぼす可能性がある。また、私たちの将来の資本需要と既存資源が私たちの運営を支援する時間は私たちの予想と大きく異なるかもしれませんが、いずれにしても、私たちの現在の任意のプロジェクトの臨床開発を完成させるために追加の資本が必要になります。
私たちの毎月の支出水準は新しいものと進行中の開発や会社活動によって異なります。Reactionや任意の将来の候補製品の開発に関する時間や活動の長さは高度に不確実であるため,我々はできない
37
私たちは開発、マーケティング、商業化活動に必要な実際の資金を調達すると予想される。私たちの将来の短期的かつ長期的な資金需要は多くの要素に依存するだろうが、これらに限定されない
|
|
|
Reactionおよび任意の未来の候補製品の臨床試験の開始、進行、時間、コスト、および結果 |
|
|
|
これらの候補製品のための臨床開発計画は |
|
|
|
私たちが開発した候補製品の数量と特徴は |
|
|
|
FDA、EMA、および他の類似外国規制機関が制定した規制要求の結果、時間、コストを満たす |
|
|
|
このような合意の下での支払条項および時間を含む協力協定を締結して維持することができるかどうか |
|
|
|
私たちの特許主張と他の知的財産権の提出、起訴、弁護、そして実行のコスト |
|
|
|
知的財産権紛争の弁護コストは、第三者が私たちに提起した特許侵害訴訟を含めて、私たちの未来の任意の製品候補に反応します |
|
|
|
競争の臨床、技術、市場発展の影響 |
|
|
|
私たち自身のビジネス規模のcGMP製造施設を維持するコストは |
|
|
|
私たちが私たちの製品を商業化することを選択した地域では、規制の承認を得ることができる任意の候補製品の販売、マーケティング、流通能力を確立するコスト |
|
|
|
Reactionおよび私たちが上場承認を得た任意の将来の候補製品の商業販売から得られる収入(ある場合)、および |
|
|
|
上場企業の運営コストとして。 |
私たちは現在、私たちの開発努力を支援するための約束された外部資金源や他の支援を持っておらず、私たちは受け入れ可能な条件で追加資金を提供するかどうか、または根本的にはできないと確信できない。私たちが私たちの現金需要を満たすのに十分な収入を生み出すことができる前に、私たちは公開または私募株式発行、債務融資、協力、戦略連合、許可手配、および他のマーケティングまたは流通手配を通じて、将来の現金需要に資金を提供する予定です。公開または私募株式発行によってより多くの資金を調達する場合、これらの証券の条項には、清算または他の株主権利に悪影響を及ぼす特典が含まれている可能性があります。さらに、私たちが普通株式または転換可能な証券または普通株に交換可能な証券を販売することによって追加資本を調達する場合、あなたの所有権資本は希釈されます。さらに、いかなる債務融資も、追加債務を招く、資本支出を行う、または配当を発表するなど、私たちが具体的な行動を取る能力を制限または制限する固定支払義務と契約を負担させる可能性がある。もし私たちがマーケティングおよび流通手配、または第三者との他の協力、戦略連合、または許可手配によって追加の資本を調達する場合、私たちは、いくつかの価値のある知的財産権または他の反応権利、ならびに任意の未来の製品候補、技術、将来の収入フローまたは研究計画、または私たちに不利になる可能性のある条項でライセンスを付与しなければならないかもしれない。私たちはまた、Reactionまたは私たちの任意の未来の候補製品のために協力者を探すか、または私たちの反応権を放棄し、私たちが開発または開発を求めていた任意の未来の製品候補または技術を放棄することをより早い段階で要求されるかもしれない私たち自身を商業化しています市場変動はまた私たちが必要な時に資本を得る能力に悪影響を及ぼすかもしれない。もし私たちが受け入れられる条件で十分な追加資本を調達できない場合、私たちはReactionまたは私たちの未来の任意の候補製品または1つまたは複数の他の研究開発計画の開発または商業化を大幅に延期、削減または停止しなければならないかもしれない。上記のどの事件も、私たちの業務、将来性、財務状況と経営結果を深刻に損害し、私たちの普通株価格の下落を招く可能性があります。
Reactionと私たちの将来の候補製品開発に関するリスク
私たちの運営の歴史は限られていて、今まで何の収入も生まれておらず、永遠に利益を上げないかもしれない。
私たちは臨床段階の生物製薬会社で、運営の歴史は限られている。私たちは2018年に設立され、商業販売が許可されていないし、何の収入も生まれていない。これまで,我々の業務は組織と我々を備えた会社,業務計画,資金調達,非臨床研究,臨床試験,キーオピニオンリーダーネットワークの発展およびReactionの研究·開発に限られてきた。候補製品を発見し開発する方法は実証されておらず、ビジネス価値のある製品を開発できるかどうかもわかりません。規制の承認を申請または取得し、製品販売から収入を得ることができる前に、私たちが開発した他の任意の候補製品は、多くの追加の開発と臨床研究時間と資源を必要とするだろう。私たちはまだ後期臨床試験を通じて任意の候補製品を進展させ、それによって成功したマーケティング許可を得ることができることを証明していない。規制部門の承認を得ることができず、商業規模の製品を作ることができず、第三者代表を配置することができず、保険会社や医療保健提供者の市場参入や認可を得ることができず、成功した製品の商業化に必要な販売やマーケティング活動を行うこともできないかもしれない。
生物製薬製品開発への投資は非常に高い投機性があり、それは大量の前期資本支出を必要とし、そして重大なリスクが存在するため、即ちいかなる潜在的な候補製品は十分な治療効果或いは受け入れ可能な安全性を証明できず、監督管理部門の許可を得ることができず、商業上実行可能ではない。もし私たちの製品候補の一つが
38
規制部門の承認が得られれば、研究開発に専念する会社から、ビジネス活動を支援できる会社に転換する必要があるだろう。そのような移行で、私たちは成功しないかもしれない。また、経営歴史が限られている企業として、私たちは迅速な変化と複雑領域の早期生物製薬会社がよく遭遇する予見不可能な費用、困難、合併症、遅延などの既知と未知の要素とリスクに直面する可能性がある。したがって,我々の業務を評価する意味のある運営履歴はなく,より長い運営履歴や医療製品の開発·商業化の歴史があれば,私たちの将来の成功や生存能力の予測はそれほど正確ではないかもしれない。
これらの活動に関連する不確実性とリスクのため、私たちは収入の時間と金額、さらなる損失の程度、または私たちがいつ利益を達成することが可能かどうかを正確かつ正確に予測することができない。私たちはこのような活動で決して成功しないかもしれません。たとえ私たちが私たちの1つ以上の候補製品を商業化することに成功しても、私たちは利益を達成するために十分な収入を生成しないかもしれません。もし私たちが確実に利益を達成すれば、四半期や年度の収益性を維持または向上させることができないかもしれませんが、より多くの候補製品を開発し、マーケティングするために、大量の研究開発や他の支出を生み出し続けます。もし私たちが達成して利益を維持できなければ、私たちの株の価値を下げ、資金を調達し、研究開発努力を維持し、業務を拡大し、あるいは運営を継続する能力を弱めるかもしれない。
生物製品と腎臓疾患治療の市場競争が激しい。もし私たちが効果的に競争できなければ、私たちの業務、財務状況、経営業績は影響を受けるだろう。
私たちは生物製品市場と腎臓疾患治療分野で激しい競争に直面している。私たちは専門と専門を含む細胞療法の開発と製造会社からの競争に直面しています製薬と生物技術会社、尿細管と糸球体細胞薬物調節剤、抗繊維化薬物、誘導多能性細胞、その他の自己間葉系幹細胞と機械的腎補助設備(例えば移植と着用可能な腎透析機)の開発者、及び腹膜透析と家庭透析の進展。他社は世界的に細胞による臨床試験を行っており,臍帯,脂肪,骨髄由来の間葉系幹細胞を用いて慢性腎症を治療している。日本ではヒト誘導多能性幹細胞による腎臓疾患治療の早期段階が行われている。我々の主な競争相手にはSGLT 2阻害剤とMRAsの開発者があり,最近CKD進展リスクを低下させるための小分子療法が承認されていると考えられる。
私たちの現在の多くの競争相手は私たちより競争優位を持っているかもしれません。研究開発、臨床前テスト、臨床試験、製造とマーケティングの面で私たちよりもっと多くの財務資源と専門知識を持っています。
私たちは目標市場の主な競争要因は
|
|
|
敏感性と特異性、および結果の再現性を含む正確性 |
|
|
|
顧客の中での名声 |
|
|
|
製品や製品の革新は承認されれば |
|
|
|
治療効果と安全性の概要; |
|
|
|
コスト; |
|
|
|
支援の有効性を宣伝する |
|
|
|
知的財産権保護 |
|
|
|
患者数を予測しています |
|
|
|
薬をあげるのと投与するのが比較的便利です。 |
承認されても、既存の競争相手や目標市場に進出した新会社から発売された新製品や技術のますます激しい競争に直面しても、我々の製品は有利な競争を得ることができない可能性があり、成功しない可能性がある。われわれはGLP−1アゴニストからの追加的な競争に直面する可能性があり、GLP−1アゴニストは2型糖尿病および肥満の治療のために許可されており、末期CKDおよびESRD患者の死亡率を低下させ、CKDの進行を緩和し、長期ダイエットを引き起こす可能性が証明されていることに留意されたい。GLP-1アゴニストまたは他の新しいまたは革新的な技術、薬物または他の治療方法の持続的な使用および増加は、私たちの予想される患者集団の増加速度に影響を与えるか、または潜在市場の規模を縮小する可能性がある。新しい技術または革新的な技術、薬物、治療、または他の態様の発展によっても、私たちの予想される患者の人口増加率または私たちの製品の需要のいかなる持続的または著しい低下も、私たちの業務に悪影響を及ぼす可能性がある。さらに、私たちの競争相手が現在または将来所有または開発する可能性のある製品または技術は、彼らが私たちよりも能力的または低コストの競争力のある製品を生産することを可能にするだろう。効果的な競争ができなかった場合は、私たちの業務、財務状況、経営業績に実質的な悪影響を及ぼす可能性があります。
私たちの業務は、私たちの主要候補製品REPACTの成功と、臨床開発に進む可能性のある任意の他の未来候補製品に強く依存しています。Reactionと私たちの未来の候補品は大量に必要になるでしょう
39
規制部門の製品の承認を求め、それを商業応用に投入することができるまで、追加の臨床開発と資金が必要である。
私たちは現在商業販売が許可されていない製品で、決して適切な製品を開発できないかもしれません。私たちの主要候補製品Reactionは臨床開発の第三段階にある。私たちはいかなる保証も肯定的な予測を提供することはできず、いかなる場合でも、予想される時間内に、このような第3段階の臨床開発は成功し、このような第3段階の臨床開発仕事から積極的な臨床データを獲得するか、あるいは監督機関はREPACTの発売を許可する。また,REPACTの規制承認を得ても,REPACTがCKDに利用可能な患者タイプを制限したり,特定の警告やタグ言語を必要としたりする可能性があり,いずれもREPACTのビジネス潜在力を低下させる可能性がある。承認されても、私たちはReactionを商業化することに成功しないかもしれない。もし私たちがREPACTの規制承認を得られなかった場合、あるいはこのような規制承認後にREPACTの商業化に成功できなかった場合、私たちの業務や財務状況は実質的な損害を受ける可能性があり、私たちは私たちの他の治療計画の成功にもっと依存するかもしれない。
組織として,我々は以前何の後期あるいは肝心な臨床試験を行ったことがなく,準備,提出,監督申告を求める上で経験が限られており,候補製品のBLAも提出されていない。私たちは1つ以上の司法管轄区域で臨床開発、規制審査、承認を完了しなければならず、その後、私たちの先行候補製品、REPACT、または私たちの将来の任意の候補製品の販売から任意の収入を得ることができます。私たちはまた私たちの持続的な運営を支援し、私たちの成長戦略を実施するために多くの追加資金を得る必要がある。また、REACTや将来の任意の候補製品が承認された場合には、十分なビジネス製造能力を確保し、任意のビジネス発表において大きなマーケティング努力を行わなければなりません。このような努力は大量の投資を必要とし、私たちはReactionや私たちの未来の任意の製品候補製品を開発し続ける財力がないかもしれない。
私たちは挫折し、規制部門が任意の未来の候補製品を商業化、反応または商業化する能力を承認または阻止することを遅延または阻止する可能性がある
|
|
|
私たちの臨床試験の陰性または非決定性結果、または私たちに似た候補製品の他の臨床試験の陽性結果は、追加の臨床前試験または臨床試験を決定または要求するか、または計画を放棄することをもたらす |
|
|
|
患者または対象が、私たちの臨床試験において、または個人的に、私たち、FDA、他の規制機関、または他の人がReactionまたは将来の任意の候補製品の開発に関連する薬物または療法を使用すると考えているときに、製品に関連する副作用または有害事象を経験する; |
|
|
|
INDまたは同様の外国出願の提出を遅延させるか、または臨床試験を開始するために規制機関の必要な承認を得ることができなかったか、または臨床試験が開始されると一時停止または終了した |
|
|
|
FDAまたは同様の外国当局は、私たちの臨床終点、および任意の追加的な検証試験の要件を含む、私たちの臨床試験の範囲または設計について適用された条件である |
|
|
|
FDAによるGCP、国際調整会議のガイドライン(“ICHガイドライン”)、GLPおよびcGTPSを含む被験者の臨床試験への参加と臨床試験の完了を遅延させる |
|
|
|
CGMPを含み、他のプロセスを効率的に遵守するための法規要件を維持することができない |
|
|
|
臨床試験の被験者の中退率は高い |
|
|
|
Reactionまたは将来の候補製品または臨床試験を行うために必要な他の材料の供給または品質が不足している |
|
|
|
臨床試験費用は予想以上だった |
|
|
|
他の治療法とは競争できません |
|
|
|
臨床試験期間中、Reaction或いは未来の候補製品の治療効果は良くない |
|
|
|
実験結果の時間は予想よりも長かった |
|
|
|
実験は詐欺、データ取得失敗、または他の技術故障の影響を受け、私たちの実験結果は無効になった |
|
|
|
私たちはEU、アジア太平洋地域、ラテンアメリカで条件付き承認申請の裁判結果を支持しない |
|
|
|
FDAや他の監督機関の臨床試験場の検査と審査に不利である |
|
|
|
私たちの第三者請負業者または調査者は、監督要求をタイムリーにまたは根本的に遵守しなかったか、またはその契約義務を履行しなかった |
|
|
|
臨床開発または特に私たちの技術に追加的な監督管理を適用することを含む、規制要件、政策、およびガイドラインの遅延および変化 |
|
|
|
FDAと同様の外国の規制機関のデータの異なる解釈 |
|
|
|
政府当局と共同で衛生技術評価(“HTA”)プログラムを完成させる |
|
|
|
REPACTに対するCMSの任意の政策レベルの検討; |
|
|
|
私たちが行っていることや未来の他のプロジェクトの資金 |
40
|
|
|
細胞療法と生物学的処理の科学的発見と技術;または |
|
|
|
製造自動化は淘汰され、これは、交換部品の品質を確保するために部品または設備を再設計する必要がある可能性があり、交換部品の遅延は、製造の重大な遅延および販売損失をもたらす可能性がある。 |
私たちは、臨床開発および規制提出プロセスのいくつかの態様、私たちの知的財産権および私たちの製造、マーケティング、流通および販売努力、または任意の未来のパートナーへの潜在的な脅威を含む多くの要素を完全に制御することができません。
REACTは新しい技術に基づいており、製品開発およびその後の規制承認の時間およびコストを予測することは困難である。
FDA、EMAとその他の監督機関の臨床試験要求及びこれらの監督管理機関は候補製品の安全性と有効性を確定するための標準は、潜在製品のタイプ、複雑性、意外性及び期待用途と市場によって大きく異なる。我々のような候補製品の規制承認プロセスは、他のより有名またはより広く研究されている医薬品または他の製品の候補製品よりも高価であり、時間がかかる可能性がある。
米国および他の国の細胞治療製品に対する規制要求は変化しており、FDAまたは他の規制機関は要求を変更したり、REPACTまたは私たちの未来の任意の候補製品を承認するために異なる規制経路を決定したりする可能性がある。例えば,FDAは生物製品評価·研究センター(“CBER”)内に組織·高度療法オフィス(“CBER”)を設置し,細胞療法や関連製品の審査を統合し,細胞,組織,遺伝子療法諮問委員会を設置し,必要に応じてCBERの審査にアドバイスを提供している。時間の経過とともに、新しいまたは異なる部門が確立されるか、または我々の再生細胞ベース製品を含む細胞治療製品を管理する責任が付与される可能性がある。さらに、心臓-腎臓諮問委員会のより多くの規制参加を含むFDA諮問機関は、さらなる調査が必要な追加的な提案を検討したり、提出したりする可能性がある。したがって、私たちは、私たちの規制戦略を変更したり、私たちの規制承認申請を修正することを要求されるかもしれません。これは、非臨床的および臨床開発と製造を完了し、規制承認、反応、または任意の未来の候補製品を得る能力を遅延させ、弱める可能性があります。規制機関および顧問グループの変化、または彼らが発行した任意の新しい要求またはガイドラインは、規制審査プロセスを延長し、追加の研究を要求し、私たちの開発と製造コストを増加させ、規制経路、立場および解釈の変化を招き、Reactionまたは任意の未来の候補製品の承認および商業化を延期または阻止し、あるいは重大な承認後の制限または制限を招く可能性がある。
我々の研究と開発努力は,再生腎細胞による治療法の利用に集中している。FDAはこれまで,比較的少ない細胞ベース療法を商業化することを承認しており,再生腎による細胞療法の商業用途への認可はなされていない。FDAまたは他の適用可能な規制機関によって課せられたプロセスおよび要件は、Reactionまたは任意の将来の候補製品の上場許可承認を得る上での遅延および追加コストをもたらす可能性がある。著者らのプラットフォームは新しいため、細胞に基づく療法は比較的に新しいため、監督管理機関はReactionなどの候補製品を評価する経験が不足している可能性がある。この新規性は、FDAがIND申請を提出する際に我々のIND申請を審査するのに要する時間を含み、当社の開発コストを増加させ、REPACTの商業化を延期または阻止することを含む規制審査プロセスを延長する可能性がある。また、発展しつつある新しい慢性腎臓病療法は、私たちに大きな挑戦をもたらしている
|
|
|
教育医療従事者はREPACTの潜在的副作用の概況を理解し,臨床開発計画の進展に伴い,REPACTの観察された副作用を知る |
|
|
|
REPACTを正確に使用して伝達する医療従事者を訓練する |
|
|
|
臨床試験に十分な数の被験者を募集し |
|
|
|
REPACTの臨床開発を支援するために製造プロセスの開発を継続した。 |
私たちはこのような挑戦を克服して、私たちが開発、商業化、そして生産的に対応できるようにしなければならない。
我々の反応推進に伴い,FDAや他の規制機関との協議が求められ,反応はFDA諮問委員会が審査する可能性がある。私たちはまた適用された要求を守らなければならない。もし私たちがそうしなければ、私たちはRESPECTの開発を延期または停止することを要求されるかもしれない。潜在的な製品を市場に投入するために必要な規制承認の遅延や意外なコストを獲得したり、監督管理の承認を得られなかったりすることは、業務を維持するために十分な製品収入を生成する能力を弱める可能性がある。
さらに、細胞治療製品の分野で他の人が行った臨床前研究または臨床試験の不利な発展は、FDA、EMA、および他の規制機関が、私たちが開発する可能性のある任意の候補製品の承認要求を修正し、他の方法でREPACTまたは将来の候補製品を開発および商業化する能力に悪影響を及ぼす可能性がある。同様に、欧州委員会は、細胞療法の開発およびマーケティング許可に関する新しいガイドラインを発表し、将来開発される可能性のある任意の候補製品を支援するために、または私たちの候補製品が承認されない上場承認を成功させるために、追加の研究または臨床試験を必要とする可能性があるこれらの新しいガイドラインを遵守することを要求するかもしれない。
41
上述した規制審査委員会および諮問グループおよびその発表された新しいガイドラインは、追加的な研究または臨床試験を行うことを要求し、私たちの開発コストを増加させ、規制の立場や解釈の変化を招き、これらの候補治療薬の承認および商業化を延期または阻止し、または承認後の重大な制限または制限を招く可能性がある。我々の研究計画や将来の候補製品の開発を進める際には、これらの規制·諮問チームと協議し、適用されるガイドラインを遵守することが求められる。もし私たちがこれをできなかったら、私たちは私たちが決定して開発した任意の候補製品の開発を延期または停止することを要求されるかもしれない。
臨床開発は長い、複雑かつ高価な過程に関連し、不確定な結果、非臨床研究の結果、以前の臨床試験或いは行われている臨床試験の中期結果、及び私たちの未来の任意の候補製品は未来の結果を予測できないかもしれない。また、私たちはReactionと私たちの未来の任意の候補製品の開発を完了する上で大きな遅延に直面するかもしれません。
我々の候補製品Reactionは臨床開発段階であり,失敗のリスクが高い。承認されれば、Reactionまたは私たちの将来の任意の候補製品の臨床試験、製造、およびマーケティングは、米国および他の国/地域の多くの政府機関の広範かつ厳格な審査および規制を受け続けており、これらの国および地域では、Reactionおよび私たちの将来の任意の候補製品をテストし、マーケティングする予定である。Reactionまたは私たちの将来の任意の候補製品の商業販売の規制承認を得る前に、私たちの候補製品が各目標適応において安全かつ有効であることを、長い、複雑で高価な試験および臨床試験によって証明しなければならない。特に,Reactionは生物製品として規制されているため,安全,純粋かつ有効であり,その目標適応に利用可能であり,潜在的な不良細胞の影響はないことを証明する必要がある。Reactionおよび我々が開発可能な任意の他の候補製品は、その目標患者集団および目標用途において十分なリスクおよび収益プロファイルを証明しなければならない。投与の過程には,腎臓組織の小範囲の生検を行うことがある。生検に関連するリスクとしては,出血,疼痛,血腫や鬱傷,瘢痕や梗塞,あるいは機能喪失をきたす血液供給喪失がある。
臨床試験費用は高価であり,完成まで数年かかる可能性があり,その結果自体も確定していない。特に,FDAが新たな治療法を承認する一般的な方法は,関連患者集団において関連製品を2回十分かつ良好に制御する臨床試験のデータを含むことができる。我々の第3段階開発計画は1~2千人の患者に関連する可能性があり,コストが高く,完成には数年かかる。候補製品は試験のどの段階でも失敗する可能性があり,早期の非臨床研究や臨床試験で有望な活性シグナルが観察された後でも同様である。Reactionと我々の将来の候補製品の非臨床研究と早期臨床試験の結果は第三段階登録開発計画の成功を予測できない可能性があり、臨床試験の中期結果も最終結果を予測できるとは限らない。そのほか、臨床試験の初歩的な成功はこれらの試験が完成した後に得られた結果を代表しないかもしれない。通常,候補製品が臨床試験で失敗することによる自然流出率は極めて高い。非臨床研究と初歩的な臨床試験で進展を得たが、臨床試験後期段階の候補製品は必要な安全性と有効性を示すことができないかもしれない。生物製薬業界のいくつかの会社は高級臨床試験で重大な挫折を受け、早期の試験で良好な結果を得たが、治療効果の不足或いは受け入れられない安全性の問題が存在する。大多数の臨床試験を開始する候補製品は治療用製品として承認されたことがなく、また私たちの未来のいかなる臨床試験が最終的にReaction或いは未来の任意の候補製品の更なる臨床開発に成功或いは支持することを保証することができない。細胞療法や組織工学製品の候補製品や納入方法は開発の初期段階で有望に見えるが,いくつかの理由で市場に参入できない可能性がある
|
|
|
非臨床または前臨床研究または臨床試験は、候補製品の有効性が予想よりも低いことを示す可能性がある(例えば、臨床試験はその主要な終点(S))に到達できない可能性がある)、または製品または投与方法に関連する許容できない副作用または毒性を有する; |
|
|
|
適用監督機関が臨床上意義と関連性があると考えられる臨床終点を確立できなかった |
|
|
|
必要な規制の承認を得ていない |
|
|
|
製造コスト、配合問題、定価または補償問題、行動メカニズム、後方勤務制限、または候補製品を経済的にしない他の要因; |
|
|
|
他の会社とその競争製品と技術の独占権は、これらの製品と技術は私たちの候補製品の商業化を阻止するかもしれない。 |
また,早期臨床試験と後期臨床試験の試験設計の違いは,早期臨床試験の結果を後期臨床試験に外挿することが困難である。そのほか、臨床データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品が臨床試験で満足できると考えているが、しかし依然としてその製品の発売許可を得られなかった。また,われわれの早期試験はオープンラベル研究であり,被験者も調査者も被験者が反応や標準看護治療を受けているかどうかを知っている。最も典型的なのは,オープンラベル臨床試験は候補の研究製品のみをテストし,異なる用量レベルでテストを行う場合がある。オープンラベル臨床試験は様々な制限や偏見を受けており,いずれの治療効果も誇張される可能性があり,開放ラベル臨床試験の被験者は治療を受けていることを知っているからである。またオープンラベル臨床試験は遵守する必要があるかもしれません
42
研究者偏向“とは、臨床試験の生理的結果を評価およびレビューする人が、どの被験者が治療を受けたかを知っており、その知識を知っている場合に、治療群の情報をより有利に説明することができることを意味する。我々の早期試験には開放ラベル用量設計が含まれていることから,われわれの試験は客観的評価方法を用いてわれわれの終点を測定していると考えられるため,被験者や研究者の偏見のいかなる影響を受ける可能性は低いが,開放ラベル設計がその製品や他の候補製品の将来の臨床試験結果を予測できないかどうかは不明であり,制御された環境で研究あるいは客観的終点のみを使用する場合,われわれはこれらの候補製品に対して開放ラベル臨床試験を行う。
また、FDAや同様の外国規制機関が規制反応に使用する基準は判断が必要であり、時間の経過とともに変化する可能性があり、これらの基準がどのように適用されるかを確定的に予測することは困難である。著者らは非臨床と臨床活動からのデータに対して行ったいかなる分析も監督機関の検証と解釈を経なければならず、これは監督部門の承認を延期、制限、あるいは阻止する可能性がある。新しい政府規定のせいで、私たちはまた予期しない遅延や費用増加に遭遇する可能性がある。このような規制の例は、将来の立法または行政行動、または製品開発およびFDA規制審査中のFDA政策の変化を含む。具体的には,中国のようないくつかの国では,ヒト遺伝物質,細胞,組織への輸出入制限の制定が検討されている。このような法律法規は,我々がヒト細胞や細胞に基づく療法を輸出入する能力を弱める可能性があり,我々の業務に実質的な悪影響を及ぼす可能性がある。私たちは立法変化が公布されるかどうか、FDAや外国の法規、指導や解釈が変わるかどうか、あるいはこれらの変化の影響が(あれば)何になるかもしれないと予測できない。FDAはまた、承認を支援する安全性および有効性データの十分性を審議する専門家グループ、すなわち諮問委員会を要求することができる。諮問委員会の意見には拘束力はないが、我々が開発した任意の候補製品が承認される能力に大きな影響を与える可能性がある。
私たちはこれまでREPACTを承認するための重要な実験を完了していない。いずれの臨床試験を行う上で遅延が生じる可能性があり,再設計が必要であり,時間どおりに被験者を募集·募集したり,時間どおりに完成したり,まったく必要ない。臨床試験は様々な原因で遅延、一時停止或いは終了することができ、以下の方面に関連する原因を含む
|
|
|
高度臨床試験に適用される製造技術と品質標準の十分な開発、確定、標準化或いは制御に遅延が発生した |
|
|
|
ある候補製品の患者試験資格をスクリーニングするために適切な分析方法を開発することに遅延があった |
|
|
|
FDA、EMA、または他の規制機関との臨床試験の設計または実施について合意した |
|
|
|
将来の臨床試験を行うための追加的な規制許可を得る |
|
|
|
より多くの/未来の臨床試験場所または未来のCROと受け入れ可能な条項と合意し、これらの条項は広範な交渉を行うことができ、異なる臨床試験場所は大きく異なる可能性がある |
|
|
|
IRBまたは道徳委員会の承認は、各追加/未来の試験場所で得られる |
|
|
|
適切な患者を臨床試験に参加させる |
|
|
|
被験者に臨床試験を完成させ、或いは治療後のフォローアップを行った |
|
|
|
適用された監督機関が臨床試験場所或いは操作を検査し、或いは臨床一時停止を実施する |
|
|
|
臨床サイト、CRO、または他の試験レジメンから外れているか、または試験を終了する第三者; |
|
|
|
FDAのGCP要件、または他の国/地域の適用法規要件を含む適用される法規要件に適合しない |
|
|
|
候補製品または送達プロセスに関連する有害事象の発生を含み、これらの有害事象がその潜在的な利点を超えると考えられる試験中に出現する患者の安全問題を解決する |
|
|
|
十分な数の臨床試験点を増やし |
|
|
|
臨床試験のための十分な数の候補製品を生産します |
|
|
|
私たちのサプライチェーンの中断、これは、私たちの製品コンポーネントの適切な貯蔵、輸送、または開発条件をもたらす可能性があり、これらのコンポーネントの処理は、時間および温度に敏感であり、患者によって異なる;または |
|
|
|
私たちの製造過程が中断され、これは私たちが適切に治療できないかもしれない。 |
臨床試験中または臨床試験の結果では、私たちは、上場許可を得ることを遅延または阻止するか、Reactionまたは将来の任意の候補製品を商業化するか、またはそのような試験のコストを著しく増加させる可能性がある多くの予測不可能なイベントに遭遇する可能性がある
|
|
|
監督要求やガイドラインの変化、あるいは監督機関からのフィードバックを受けて、臨床試験の設計を修正することを要求した |
|
|
|
Reactionまたは将来の候補製品の臨床試験は否定的または不確実な結果をもたらす可能性があり、私たちは決定または監督機関が追加の臨床試験を要求するか、または開発計画を放棄することを要求するかもしれない |
|
|
|
Reactionや将来の候補製品の臨床試験に必要な被験者の数は、私たちが予想していたよりも多かったかもしれません。これらの臨床試験の登録速度は、私たちが予想していたよりも遅いかもしれません |
43
|
|
|
私たちの第三者請負業者は、法規の要求を適時に遵守したり、私たちに対する契約義務を履行できなかったり、全く守らなかったりする可能性があります |
|
|
|
私たちまたは私たちの研究者は、規制要件に適合していないこと、Reactionまたは私たちの将来の候補製品が副作用または他の意外な特徴を持っていることを発見すること、または対象が受け入れられない健康リスクに曝露されていることを発見することを含む、様々な理由でReactionまたは将来の候補製品の臨床試験を一時停止または終了しなければならないかもしれない |
|
|
|
Reactionや将来の候補製品の臨床試験コストは私たちが予想していたよりも高いかもしれませんこれらのコストを支払う資金がないかもしれません |
|
|
|
Reactionまたは私たちの将来の候補製品または私たちの候補製品の臨床試験に必要な他の材料の供給または品質が不足または不十分である可能性がある |
|
|
|
規制当局は、REPACTまたは私たちの未来の製品候補製品を承認する要求を修正するかもしれないし、これらの要求は私たちの予想とは異なるかもしれない |
|
|
|
将来臨床試験を行ういずれの協力者も上記のいずれかの問題に直面する可能性があり、彼らが自分に有利だと思っているが私たちに不利な方法で臨床試験を行う可能性がある。 |
もし私たちがReactionまたは私たちの任意の未来の候補製品に対して、現在予想されている以上の追加の臨床試験または他の試験を行うことを要求された場合、もし私たちがReactionまたは私たちの未来の製品候補または他の試験の臨床試験を成功させることができなければ、これらの試験または試験の結果が陽性でないか、またはわずかな陽性である場合、または安全問題がある場合、私たちは:
|
|
|
計画外費用を招く |
|
|
|
Reactionまたは将来の任意の候補製品の上場承認を遅延させるか、または上場承認を全く得ていない |
|
|
|
一部の国では発売が許可されていますが他の国ではありません |
|
|
|
市場で承認された適応や患者集団は期待や期待ほど広くない |
|
|
|
マーケティング承認は、ボックス警告またはREMSを含む重大な使用または流通制限またはセキュリティ警告を含むタグによって取得される |
|
|
|
追加の上場後のテスト要求を受けます |
|
|
|
製品の使用方法の変化に支配されています |
|
|
|
上場許可を得た後、規制部門はその製品の承認を撤回、一時停止したり、その販売を制限したりする。 |
臨床試験がわれわれ、このような試験を行う機関のIRBs、そのような試験のためのDSMBまたはFDAまたは他の規制機関によって一時停止または終了されれば、遅延に遭遇する可能性がある。このような主管部門は、様々な要素のために臨床試験を一時停止または終了する可能性があり、これらの要素は、規制要求または私たちの臨床規程に従って臨床試験を行っていない;FDAまたは他の監督機関の臨床試験操作または試験場所の検査による臨床一時停止の強制実施;予見できない安全問題または副作用;REACTまたは私たちの未来の候補製品の使用によるメリットを証明できなかった;政府法規または行政措置の変化、または臨床試験を継続するのに十分な資金が不足していることを含む。
著者らの主要な候補製品ReactionはBLA或いはMAAを監督部門の承認のために提出する前に、広範な臨床テストを行う必要がある。REPACTの臨床開発が完了する可能性があるかどうかを確実に予測し、REPACTの規制承認のためにBLAまたはMAAを提出するか、またはそのようなBLAまたはMAAが承認されるかどうかを確実に予測することはできない。私たちはまた、FDA、EMA、または他の規制機関が私たちの臨床開発計画に対するフィードバックを求めることができ、FDA、EMA、またはそのような監督機関はそのようなフィードバックをタイムリーに提供できない可能性があり、またはそのようなフィードバックは有利ではないかもしれず、これは私たちの開発計画をさらに延期するかもしれない。
私たちは現在海外とアメリカで臨床試験を行っている。もし私たちが引き続き外国で臨床試験を行うか、あるいは外国の司法管轄区で上場許可を求めるならば、私たちは臨床試験の進行、製造とマーケティング許可、定価と第三者精算を含む多くの外国の監督管理要求を守らなければならない。外国監督審査手続きは国/地域によって異なり、上述したFDA承認に関連するすべてのリスクと、外国司法管轄区の現地法規を満たすことによるリスクが含まれる可能性がある。また、外国規制機関から承認を得るのに要する時間は、FDA承認を得るのに要する時間とは異なる可能性がある。FDAの承認は米国以外の規制機関の承認を確保しておらず、その逆も同様である。各候補製品について、臨床試験の成功は、FDAにマーケティング申請を提出し、同様のマーケティング申請を比較可能な外国規制機関に提出するための前提条件であり、したがって、任意の候補製品を最終的に承認し、商業マーケティングを行うための前提条件でもある。私たちは否定的または不確実な結果に直面する可能性があり、これは、私たちが規制当局によってより多くの臨床研究や試験を要求されたり、私たちの製品開発計画の一部または全部を放棄したりすることを招き、私たちの業務に実質的な悪影響を及ぼす可能性がある。
44
我々の非臨床研究および臨床試験の任意のバックラインデータまたは中期分析は、より多くのデータの出現に伴い時々公表または公表される可能性があり、引き続き監査と検証手続きの制限を受け、これらのプログラムは最終データの重大な変化を招く可能性がある。
我々は、いくつかの実施されている臨床試験の中期分析を開示し、将来的に、その非臨床研究および臨床試験の中期または主要なデータを開示し続ける可能性がある。これらの仮更新は,当時利用可能なデータの初歩的な分析をもとに,特定の研究や実験に関するデータをより網羅的に審査した後,結果や関連する調査結果や結論が変化する可能性がある.私たちは私たちのデータ分析の一部として、私たちがすべてのデータを全面的かつ詳細に評価する機会がないか、または受け取る機会がないかもしれない、仮説、推定、計算、および結論を要求されるだろう。したがって、私たちが報告する可能性のある中期またはバックライン結果は、同じ研究または試験の将来の結果とは異なるかもしれないし、またはより多くのデータを受信して十分に評価されると、異なる結論または考慮要素がこれらの結果を合格させる可能性がある。一時的なデータとバックラインのデータは引き続き監査と確認手続きを受けることになり、これは最終データが私たちが以前に公表したデータと実質的に異なる可能性がある。したがって、最終データが利用可能になる前に、任意の中期またはバックラインデータを慎重に表示すべきである。私たちが完成する可能性のある臨床試験の中期データは、患者登録の継続とより多くの患者データの獲得に伴い、1つまたは複数の臨床結果が実質的に変化する可能性があるというリスクに直面する。
さらに、規制機関を含む他の人は、私たちの仮説、推定、計算、結論または分析を受け入れないか、またはデータの重要性を異なる方法で解釈またはトレードオフする可能性があり、これは、特定の計画の価値、特定の候補製品、および私たちの全体的な商業化の程度に影響を与える可能性がある。さらに、私たちが開示する特定の研究または臨床試験に関する情報を選択することは、一般に、広範な情報に基づいており、投資家または他の人は、私たちが決定した重要な情報または他の適切な情報が私たちの開示に含まれることに同意しない可能性があり、私たちが開示しないことを決定した任意の情報は、最終的には、特定の候補製品または私たちの業務に関する将来の決定、結論、観点、活動、または他の側面に対して大きな意味を有すると考えられるかもしれない。もし私たちが報告した中期またはバックラインデータが実際の結果と異なる場合、あるいは規制機関を含む他の人が結論に同意しない場合、私たちは承認を得て私たちの候補製品を商業化する能力が損なわれる可能性があり、これは私たちの業務、経営業績、見通し、または財務状況を損なう可能性がある。
FDA、EMAと外国機関のような規制承認過程は長く、時間がかかり、本質的に予測できない。もし私たちがReaction、私たちの主要候補製品、または私たちの未来の任意の候補製品に必要な規制承認を得ることができなければ、私たちの業務は実質的で不利な影響を受けるかもしれない。
FDA、EMA、および同様の外国当局の承認または他の上場許可を得るのに要する時間は予測不可能であり、通常、臨床試験開始後数年後に必要であり、監督当局のかなり大きな裁量権を含む多くの要素に依存する。また、承認政策、法規および承認を得るために必要な臨床データのタイプと数量は候補製品の臨床開発過程で変化する可能性があり、司法管轄区域によって異なる可能性がある。私たちはまだ候補製品の規制承認を得ていませんし、私たちは将来開発を求める可能性のあるどの製品も規制部門の承認を得られないかもしれません。私たちがFDAの規制承認を得るまで、私たちおよび現在または未来のパートナーは、米国でいかなる候補生物製品の販売も許可しておらず、私たちがFDAの承認やEMAのMAAの承認または他の必要な規制承認を得るまで、私たちはEUでBLAを販売することができない国です。これまで、私たちはアメリカ、EU、アルゼンチン、イスラエル、カナダ、ブラジルの監督管理機関とのみ臨床開発計画あるいはこれらの司法管轄区域内の任意の候補製品の規制承認について限られた討論を行った。
承認された任意の候補生物製品を米国または海外で商業化する前に、厳格に制御された臨床試験によって大量の証拠を提供し、FDA、EMAまたは他の外国の規制機関に、これらの候補製品がその予期される用途に対して安全かつ有効であることを満足させなければならない。非臨床或いは臨床前研究と臨床試験の結果は異なる監督管理機関によって異なる解釈がある可能性がある。REPACTの非臨床的または臨床的データが有望であると信じていても,これらのデータはFDAや他の規制機関の承認を支持するのに不十分である可能性がある。FDAはまた,承認前または後に追加の非臨床研究や臨床試験を行うことや,われわれの臨床開発計画の内容に反対することを要求する可能性がある。
Reactionは、以下のような理由を含む、多くの理由で規制部門の承認を得ることができないかもしれない
|
|
|
FDAまたは同様の外国の規制機関は、私たちの臨床試験の設計または実施に同意しないかもしれない |
|
|
|
私たちは、候補製品がその提案に対する適応が安全で有効であることをFDAまたは同様の外国の規制機関に証明することができないかもしれない |
|
|
|
私たちの臨床試験結果はFDAなどの外国の規制機関が許可した統計的意義レベルに適合していないかもしれない |
|
|
|
候補製品の臨床的および他の利益がその安全リスクよりも大きいことは証明できないかもしれない |
|
|
|
FDAまたは同様の外国の規制機関は、臨床および商業供給契約を締結する第三者サプライヤーの製造プロセスまたは施設を承認できない可能性がある |
45
|
|
|
FDAや同様の外国機関の承認政策や法規は大きく変化する可能性があり、私たちの臨床データは承認を得るのに十分ではない。 |
バイオ製品メーカーが開発した大量の候補製品のうち,一部のみがFDAや外国規制機関の承認手続きに成功し,商業化されている。長い承認とマーケティング許可プロセス、および将来の臨床試験結果の予測不可能性は、市場反応または将来の任意の候補製品に対応するために、規制部門の承認およびマーケティング許可を得ることができない可能性があり、これは、私たちの業務、財務状況、運営結果、および将来性を深刻に損なうことになります。
私たちはReactionの開発に多くの時間と財力を投入した。我々の業務は,非臨床·臨床開発を成功させ,規制部門の承認を得,承認された場合には,REPACTや任意の将来の候補製品の商業化に成功しているかどうかに依存する。
我々が最終的に臨床試験を完了し、Reactionまたは任意の将来の候補製品に対するBLAまたは海外マーケティング申請の承認を得たとしても、FDA、EMAまたは適用される外国の監督管理機関は、高価な追加の臨床試験(上場後の臨床試験を含む)の表現に基づいて、他のマーケティング許可を承認または承認する可能性がある。FDA、EMA、または適用される外国規制機関もまた、私たちが最初に要求したよりも限られた適応または患者集団の候補製品の販売を承認または許可することができ、FDA、EMAまたは適用される外国規制機関は、候補製品の商業化に成功するために必要または望ましいラベルであると考えている可能性がある。適用可能な規制承認や他のマーケティング許可を得るか得られないかのいずれの遅延も、候補製品の商業化を延期または阻止し、私たちの業務および将来性に重大な悪影響を及ぼすだろう。
さらに、FDA、EMA、および他の規制機関は、彼らの政策を変更し、追加の法規を発表したり、既存の法規を修正したり、他の行動を取ったりする可能性があり、これは、私たちの将来の候補製品が適時に承認されることを阻止または延期するかもしれない。このような政策や規制の変化は、私たちに追加的な要求を加えるかもしれません。承認された能力を延期したり、コンプライアンスコストを増加させたり、私たちが獲得した可能性のある任意のマーケティング許可を維持する能力を制限したりするかもしれません。
我々の非臨床研究および臨床試験は、Reactionまたは将来の候補製品の安全性および有効性を証明する実質的な証拠、またはReactionまたは私たちの任意の未来の候補製品の開発中に深刻な不良または許容できない副作用を発見することができない可能性があり、これは、Reactionまたは私たちの任意の未来の候補製品の規制承認範囲を阻止、延期または制限し、それらの商業化を制限し、我々のコストを増加させるか、またはReactionまたは将来の候補製品の開発を放棄または制限する必要があるかもしれない。
Reactionと私たちの未来の任意の候補製品の商業販売に必要な規制承認を得るためには、私たちの候補製品が安全で純粋かつ有効であり、各目標適応に使用できることを、長く、複雑で高価な非臨床試験と臨床試験を通じて証明しなければならない。非臨床試験と臨床試験は高価で時間がかかり、その結果自体は確定しない。開発中、故障はいつでも発生する可能性がある。非臨床研究や早期臨床試験の結果はその後の臨床試験の結果を予測できない可能性があり,臨床試験の中期結果も最終結果を予測できるとは限らない。そのほか、非臨床と臨床データはよく異なる解釈と分析の影響を受けやすい。多くの会社は彼らの候補製品が臨床前研究と臨床試験で満足できると思っているが、まだその候補製品のマーケティング許可を得られなかった。
十分かつ良好に制御された試験から大量の証拠を得ることができず、FDAまたは同様の外国の規制機関に、その期待される用途に対して、反応が安全かつ有効であることを満足させることができるかもしれない。
自己細胞治療製品を用いた治療に出現する可能性のある副作用としては,血小板減少,身震い,貧血,好中球減少症,下痢,好中球減少症,嘔吐,低血圧,呼吸困難,サイトカイン放出症候群,神経毒性がある。副作用は医療従事者によって識別や処理が不適切ではない可能性があり,反応細胞による治療に慣れていないため無関係と考えられる。Reaction治療は腎臓生検を行って組織を得て生物活性成分を製造し、その後注射してReaction産物を腎臓に堆積させる必要がある。すべての干与はよく知られている有害事象のリスク、例えば腎臓出血、皮質瘢痕、腎機能低下或いは他の入院、輸血或いは血管造影干与を必要とする可能性のある不良事件をもたらす。
RMCL−002試験では,我々が現在3期試験で使用している異なる配合の反応製品と異なるプログラムを用い,1人の参加者は瘢痕形成や線維化および腎機能低下を含む深刻な有害事象を経験した。第2の被験者は,コンピュータ断層撮影(CT)画像で腎臓血流量の減少と腎機能の低下を認めた。もし他の有害事象や他の予期しない深刻な不良事件が発生した場合、著者らの臨床試験は一時停止または終了する可能性がある。現在および計画されている臨床試験に登録された被験者が経験した有害事象がREACT候補製品またはプログラムによって引き起こされないことを証明できない場合、FDA、EMAまたは他の外国規制機関は、任意またはすべての適応を標的とする候補製品のさらなる開発または承認を拒否するように命令することができる。現在および計画されている臨床試験に登録された被験者が経験した深刻な有害事象が製品とは無関係であることを証明することができても、このような事件は患者募集や
46
被験者を募集して試験を完了させる。さらに、もし私たちが私たちの任意の候補製品の任意の将来の臨床試験を開始、延期、一時停止、または終了しないことを選択または要求された場合、その候補製品の商業的将来性が損なわれる可能性があり、これらの候補製品から製品収入を創出する能力は延期またはキャンセルされる可能性がある。これらの状況のいずれも、他の候補製品を開発する能力を損なう可能性があり、私たちの業務、財務状況、および将来性を深刻に損なう可能性があります。
さらに、Reactionまたは私たちの未来の任意の候補製品が非臨床研究または臨床試験において悪影響に関連している場合、または予期しない特徴がある場合、決定または要求される可能性がある追加の非臨床研究を実行するか、または私たちの候補製品のさらなる臨床開発を停止または延期するか、またはその開発をより狭い用途または集団に制限するか、リスク-利益の観点から、不良副作用または他の特徴はあまり一般的ではなく、それほど深刻ではない、またはより容易に受け入れられ、これは、候補製品の商業的期待を制限する可能性がある(承認された場合)。FDA、EMA、および他の衛生機関、IRBまたはIECはまた、私たちの候補製品の安全性と有効性を理解するために、安全情報に基づいて、私たちの臨床試験を一時停止、停止または制限すること、または私たちの候補製品の安全性と有効性を理解するために追加的な動物または人体試験を行うことを要求するかもしれない。これらの発見は、規制当局が私たちの候補製品にマーケティング許可を提供できないか、または承認された場合、承認の適応の範囲を制限することをさらに招く可能性がある。早期テストで最初に希望を示した候補製品の多くは後に副作用が生じることが発見され,その候補製品のさらなる開発を阻害している。
さらに、REACTまたは将来の任意の候補製品が発売承認された場合、私たちまたは他の人は、そのような製品による許容できない副作用が、多くの潜在的な重大な負の結果をもたらす可能性があることを発見した
|
|
|
規制部門は、このような製品の承認を一時停止、撤回、または制限することができ、またはその製造または流通を禁止する禁止を求めることができる |
|
|
|
規制部門はラベルに警告を追加することを要求するかもしれない |
|
|
|
私たちは、患者に配布するために、またはREMSの他の要件に適合するために、そのような副作用のリスクを要約するための薬物ガイドラインの作成を要求されるかもしれない |
|
|
|
私たちは製品の投与方法を変えたり、追加の非臨床研究や臨床試験を行うことを要求されるかもしれません |
|
|
|
私たちは起訴され、患者への傷害に責任を負うかもしれない |
|
|
|
私たちはこの製品を市場から撤退させることに決めたかもしれません |
|
|
|
第三者支払者保険と十分な精算を実現したり維持したりすることはできないかもしれません |
|
|
|
私たちは罰金、禁止、あるいは民事または刑事処罰を受けるかもしれない;そして |
|
|
|
私たちの名声と医者や患者の私たちの製品に対する受容度は影響を受けるかもしれない。 |
FDAまたは外国規制機関が任意の製品に関連する有害事象に関連する任意の問題をタイムリーまたは完全に解決することは保証されない。さらに、これらの事件のいずれも、特定の候補製品に対する市場受容度を達成または維持することを阻止または維持することができ、承認されれば、私たちの業務、運営結果、および将来性を深刻に損なう可能性がある。
負の世論やREPACTを用いた自己細胞療法の規制審査の強化は,現在および将来の候補製品の開発や商業成功に悪影響を及ぼす可能性がある。
Reactionの臨床と商業成功は公衆の自己細胞療法による腎臓疾患治療に対する受容程度にある程度依存する。REACTIONの使用に対する公衆のいかなる不利な態度も,われわれの臨床試験登録能力に悪影響を及ぼす可能性がある。さらに、私たちの成功は、医師が処方した処方および彼らの患者が受けることを望む治療に依存し、これらの治療は、彼らがよく知っている既存の治療方法を代替または補充するために、私たちが開発可能な候補製品を使用することに関連し、より多くの臨床データを得ることができる。
より厳格な政府法規または負の世論は、私たちの業務または財務状況に負の影響を与え、Reactionまたは私たちの将来の任意の候補製品の開発および商業化を遅延または損害する可能性があり、または承認されると、任意の製品の需要を満たす可能性がある。私たちや他の人の不良事件は臨床試験は、最終的に私たちの候補製品によるものでなくても、それによって生じる宣伝は、政府の規制の増加、不利な公衆の見方、Reactionまたは私たちの将来の候補製品をテストまたは承認する際に生じる可能性のある規制遅延、承認された候補製品に対するより厳しいラベル要求、およびそのような候補製品の需要が減少する可能性があり、これらはすべて私たちの業務および運営に負の影響を与える。
我々は第1段階3期臨床試験を行っており,これらの試験や将来の臨床試験を成功させることはできないかもしれない。
3期臨床試験の進行は複雑な過程である。我々の管理チームメンバーは過去に他社勤務時に3期臨床試験を行っているにもかかわらず,会社としては現在我々の最初の3期開発計画を行っているため,予想以上の時間とより大きなコストが必要かもしれない。正しい治療法、完了または遅延を含むことができなかった第3段階の臨床試験は、開始を阻止または延期するかもしれません
47
未来のREPACTの臨床試験、監督部門の許可を得て商業化することは、著者らの財務業績に不利な影響を与える。また,我々の競争相手の中には現在,同じ適応をREPACTと見なした候補製品の臨床試験を行っており,本来われわれの臨床試験に参加する資格のある患者は,競争相手の候補製品の臨床試験に参加することができる。
われわれは患者を臨床試験に参加させる上で困難に直面し続けている可能性があり,われわれの臨床開発活動は延期されているか,あるいは他の方法で悪影響を受け続けている可能性がある。
臨床試験の成功とタイムリーな完成には十分な数の患者を募集する必要がある。患者登録は臨床試験時間の重要な要素であり、患者集団の大きさと性質、および競争相手と私たちの臨床試験に参加する資格のある患者に対する競争を含む多くの要素の影響を受け、競争相手は開発中の候補製品に対して臨床試験を行って、私たちの1つ以上の候補製品と同じ適応、あるいは私たちが候補製品を開発する条件に対して承認された製品を治療している可能性がある。
臨床試験は、患者登録にかかる時間が予想されるよりも長いか、または予想される対象よりも多く撤退するために遅延される可能性がある。FDAや外国規制機関の要求に応じて十分な数の合格患者を見つけて募集することができなければ,REACTや将来の候補製品の臨床試験を開始または継続することができない可能性がある。将来の臨床試験で被験者を募集する成功度を予測することはできません患者の登録は多くの要素に依存している
|
|
|
協定に規定されている患者資格と排除基準 |
|
|
|
臨床試験の主要な終点を分析し、患者の過程を決定するために必要な患者群の大きさと人口統計を分析した |
|
|
|
被験者と臨床試験地点との距離 |
|
|
|
実験の設計 |
|
|
|
私たちは適切な能力と経験を持つ臨床試験研究者を募集することができます |
|
|
|
研究中の候補製品の他の利用可能な治療法に対する潜在的な利点およびリスクに対する臨床医および患者の見方は、私たちが調査中の適応のために承認される可能性のある任意の新製品を含む |
|
|
|
市場で入手可能な競争療法および他の競争候補製品の臨床試験の利用可能性; |
|
|
|
臨床試験の被験者のインフォームドコンセントを得て維持する能力は |
|
|
|
臨床試験に参加した被験者は,試験完了前に試験を終了するリスクである。 |
例えば,我々は最初に糖尿病や先天性腎臓や尿路異常によるCKDを治療するためのREPACTを開発している。アメリカでは、慢性腎臓病の影響は3800万人を超える成人を超えると推定されている。われわれがReactionの臨床試験で被験者を募集することは困難であり,困難に直面し続ける可能性があり,一部の原因は,被験者の厳密な基準,治療法の新規性,物理侵襲的プログラムに関与していることである。また,われわれの臨床試験は,REPACTと同じ治療分野の候補製品を他の臨床試験と競合しており,この競争は,われわれの試験に参加することを選択する可能性のある患者の中には,我々の競争相手の1つによる試験への参加を選択する可能性があるため,使用可能な患者数やタイプを減少させ続けている可能性がある。合格臨床研究者の数は限られているため、著者らはいくつかの競争相手が使用している同じ臨床試験地点で著者らのいくつかの臨床試験を行うことが予想され、これはこのような臨床試験地点で臨床試験を行うことができる患者数を減少させる可能性がある。
十分な数の患者を募集して臨床試験を行うことはできません。これは深刻な遅延を招き、1つ以上の臨床試験を完全に放棄することを要求するかもしれません。患者募集の遅延は、コスト増加を招き続ける可能性があり、将来の臨床試験の時間または結果に影響を与え続ける可能性があり、これは、これらの試験の完了を阻止し、Reactionまたは任意の将来の候補製品の開発能力の推進に悪影響を及ぼす可能性がある。また,CROと臨床試験地点によりわれわれの臨床試験の適切かつタイムリーな進行を確保することが予想され,それらの発現への影響は限られる。
さらに、追跡期間および最長研究持続期間が5年(60カ月)であること、および現場訪問の要求により、被験者は、私たちが予想していたよりも高い比率で我々の臨床試験を脱退するか、またはREPACTおよび任意の未来の候補製品と同じ適応を有する競合他社によってスポンサーされる代替臨床試験に参加することを選択する可能性がある。私たちの臨床試験のために十分な数の被験者を募集することができても、私たちの臨床試験でこれらの被験者の登録を維持することは困難かもしれない。
また,国会は最近FDCAを改正し,3期臨床試験のスポンサー,あるいはマーケティング認可を支援する新薬や生物の他の“重要な研究”を要求し,このような臨床試験のための多様な行動計画の設計と提出を求めている。行動計画は,学生募集の適切な多様性目標と,これらの目標の理由やスポンサーがこれらの目標をどのように達成するかの記述を記述しなければならない。我々の第3段階REGEN−006およびREGEN−016は、この要求が発効する前に開始されるが、我々が計画している今後の第3段階試験については、免除を受けることができない限り、INDの一部として、このような第3段階または重要な研究案の計画を提出する際に、INDの一部として検討しなければならない
48
多様な行動計画の一部またはすべての要求。多様性行動計画が私たちの候補製品の未来の第三段階試験の計画と時間スケジュールにどのように影響するかはまだ不明であり、FDAがこのような計画の中でどのような具体的な情報を期待するかも不明である。しかしながら、FDAが私たちの候補製品のために提案した任意の第3段階試験の多様な行動計画に反対する場合、このような試験の開始は延期される可能性があり、多様な被験者を募集する際に困難に遭遇し、承認された任意の多様な行動計画の要求を満たすことを試みる可能性がある。
我々が行っていることや将来の臨床試験の設計や実行は上場承認をサポートしていない可能性がある。
臨床試験の設計或いは実行はその結果が上場承認を支持するかどうかを確定することができ、臨床試験の設計或いは実行中の欠陥は臨床試験の進展が良好になるまで明らかにならない可能性がある。さらに、場合によっては、多くの要因により、同じ候補製品の異なる試験間の安全性または有効性結果に有意差が存在する可能性があり、試験レジメン、患者集団の大きさおよびタイプの差、研究者の可変コンプライアンス、および臨床試験被験者間のレジメン要件および中退率を含む。私たちが行った任意の臨床試験が、市場反応または将来の任意の候補製品の発売承認を得るために、一致または十分な有効性と安全性を証明するかどうか分からない。
さらに、FDAおよび同様の外国規制機関は、承認中およびReactionをいつまたは獲得するかどうか、または将来の任意の候補製品の上場承認を決定する上で、かなりの裁量権を持っている。われわれの第三段階臨床試験や登録試験で主要な終点に達しても,Reactionは承認されない可能性がある。FDA或いは類似の外国の監督機関は著者らの試験設計と著者らの非臨床研究或いは臨床試験データの解釈に同意しないかもしれない。さらに、これらの規制機関のいずれも、重要な段階3期または登録臨床試験の案を審査し、コメントまたは提案を提供した後であっても、候補製品の承認要求を変更することができる。さらに、これらの規制機関のいずれかは、私たちが要求するよりも少ないまたは限られた適応の候補製品を承認することもでき、または高価な発売後の臨床試験の表現に基づいて承認される可能性がある。FDAまたは同様の外国規制機関は、承認されれば、Reactionまたは私たちの将来の任意の候補製品の商業化に必要または望ましいと考えられるラベル宣言を承認しないかもしれない。
私たちはFDAからREPACTのRMAT指定を取得しましたが、これはより速い開発や規制審査過程を招くことはなく、このような指定は私たちの候補製品の米国での上場承認の可能性を増加させず、FDAはそのような指定を撤回する可能性があります。
我々は、REACTが追加的な迅速開発経路を得る資格があるかどうか、または規制機関が関連名を承認または保持することを許可するかどうかを決定することができないにもかかわらず、迅速な開発経路を用いてREACTを行うことができるように、規制ポリシーを評価する予定である。
RMAT指定は、深刻なまたは生命を脅かす疾患および満たされていない需要を治療するための設計の候補製品の開発および検討を加速することを目的としており、“予備臨床証拠は、候補製品が1つまたは複数の臨床的重要終点において、例えば臨床開発早期に観察される実質的な治療効果のような既存の治療法よりも実質的な改善を示す可能性があることを示すとき”である。迅速に指定された反応を指定することは、FDAとのより頻繁な会議の開催、候補製品の開発計画の検討、支援承認に必要な適切なデータの収集を確保すること、FDAと臨床試験の設計やバイオマーカーの使用などの問題についてより頻繁に書面通信を行うこと、最初の段階から有効な細胞治療計画について密な指導を提供すること、高度な管理者が参加する組織承諾、BLA提出時に臨床データ支援があれば、スクロール審査および優先審査を行う資格があることを含む、潜在的な利点を提供する。
細胞と遺伝子療法、治療用組織工学製品、ヒト細胞と組織製品、および任意のこのような治療法或いは製品の組み合わせ製品を使用して、深刻な或いは生命に危害を及ぼす疾患或いは状況を治療、修正、逆転或いは治愈することを目的とし、すべてFDAによってRMATsに指定される資格がある。RMATの指定は、承認の加速を支援するための任意の潜在的代替品または中間終点を検討するために、優先的な審査または承認を加速する資格を提供すること、およびFDAとの早期相互作用を提供することによって、再生医学療法の効率的な開発および検討を促進することを意図している。
我々は2021年10月28日にREPACTのRMAT称号を申請し,FDAの承認を得た。RMAT指定の想定とFDAの指示により,REPACTの臨床前と臨床開発と製造状況を審査し,候補製品承認を支援するための計画臨床計画を検討するために,FDAと包括的·多学科B型会議を開催することが求められている。FDAは我々が会議要求で提起した質問に対して詳細な書面回答を提供し,クラスB会議は2022年3月に開催された。その会議の結果として,米国におけるREACTの臨床開発計画を進めるとともに,承認に必要な試験の設計,生産分析,比較可能性研究を含むFDAの登録計画への期待と要求の明瞭性を向上させる。FDAの意見は“第三段階臨床開発(REGEN−006とREGEN−016)”と題する部分でより全面的に述べられている第1部--第1項、業務。”
49
2021年10月にRMAT認証を取得したにもかかわらず,このような認証は製品承認の基準を変えることはなく,この認証が審査や承認を加速する保証もなく,承認の適応がRMAT認証がカバーする適応よりも狭くならない保証もない。したがって,RMATがREACT用に承認されても,従来のFDAプログラムと比較して,より速い開発過程,審査,上場承認を経験しない可能性がある.FDAがこの製品がもはや合格基準を満たしていないと思った場合、RMATの指定を取り消す可能性がある。FDAはわれわれの試験設計にさらなる投入を提供する可能性もあり,この場合,われわれのREPACT臨床開発完了のスケジュールが遅れる可能性がある。もし私たちがこのような他の開発と規制を加速させる方法を利用できなければ、私たちの業務は損害を受けるかもしれない。
私たちは、より利益的または成功可能性の高い候補製品または適応を利用することなく、特定の候補製品または適応を追求するために限られたリソースを費やす可能性がある。
私たちの財務と管理資源が限られているので、私たちは私たちが決定した特定の適応の開発計画と候補製品に集中しなければならない。そこで,CKDやCAKUT治療のためのReactionの開発のみに注目した。したがって、私たちは他の候補製品を探す機会を放棄または延期したり、これらの候補製品のために後により大きな商業潜在力が証明された他の兆候を探すことができるかもしれない。私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。現在と将来の開発計画および特定の適応の候補製品への支出は、いかなる商業的に実行可能な製品も生じない可能性がある。もし私たちが特定の候補製品の商業的潜在力や目標市場を正確に評価しなければ、私たちは協力、許可、または他の印税手配によって候補製品に価値のある権利を放棄するかもしれないが、この場合、候補製品の独占開発権と商業化権利を維持することは私たちに有利だ。
我々は将来的に米国国外でより多くの臨床試験を継続する可能性があり,FDAや同様の外国規制機関はその管轄外の地点で行われているこのような試験のデータを受け入れない可能性がある。
私たちはすでにアジア太平洋地域、EU、ラテンアメリカでREACTIONの他の臨床試験を行い、将来は南米、オーストラリア、ニュージーランドあるいは他の外国の管轄区域を含む米国以外の地域で臨床試験を継続する可能性がある。FDAが米国国外で行った臨床試験を受けたデータは何らかの条件によって制限される可能性があり,拒否される可能性もある。米国国外で行われた臨床試験のデータが米国での上場承認の唯一の根拠となることを意図している場合、FDAは通常、(I)このデータが米国の人口および米国の医療実践に適用されない限り、外国のデータのみに基づいて申請を承認しない。(Ii)この試験は、公認能力を有する臨床研究者によってGCPに従って行われる。および(Iii)データは、FDAによる現場検査を必要とすることなく、有効であると考えることができ、または、FDAがこのような検査を行う必要があると考える場合、FDAは、現場検査または他の適切な手段でデータを検証することができる。そのほか、十分な患者群と統計能力を含むFDAの臨床試験要求を満たさなければならない。多くの外国の規制機関もまた似たような承認要求を持っている。また、このような外国臨床試験は、試験を行う外国司法管轄区に適用される現地法律に制限される。FDAや類似した外国の規制機関が米国や適用司法管轄区域以外で行われた臨床試験のデータを受け入れることは保証されない。FDAまたは同様の外国の規制機関がこのようなデータを受け入れない場合、追加の臨床試験が必要となり、これは高価で時間がかかり、私たちの業務計画の様々な側面を遅延させ、適用司法管轄地域でREPACTが商業化承認または許可を得ることができない可能性がある。
私たちが未来にもっと多くの候補製品を発見したり発見したりする努力は成功しないかもしれない。
我々の研究計画は当初、潜在的な候補製品の決定に希望を示す可能性があるが、様々な原因で臨床開発のための候補製品を生成できなかった
|
|
|
所望の薬理特性や魅力的な薬物動態を持つ候補製品を設計することはできません |
|
|
|
適切な製造プロセスを設計し開発することはできません |
|
|
|
さらなる研究では、潜在的な候補製品は、有害な副作用または他の特徴を有することが証明される可能性があり、上場承認を得て市場から受け入れられる薬剤である可能性が低いことを示している。 |
新製品候補製品を確定する研究プロジェクトには大量の技術、財力と人的資源が必要である。臨床開発に適した化合物を決定できなければ、将来的に製品収入を得ることができず、私たちの財務状況に大きな損害を与える可能性がある。
50
私たちの資源と資金を得るルートが限られているので、私たちはいくつかの計画と候補製品に資源を割り当てることを決定しなければならない;これらの決定は間違っていることが証明され、私たちの業務に悪影響を及ぼす可能性がある。
我々の財力と人的資源は限られており,最初は研究プロジェクトや製品候補に重点を置き,限られた適応を得るつもりであった。したがって、私たちは、他の候補製品または後により大きなビジネス潜在力またはより大きな成功可能性を有することが証明された他の兆候を求める機会を放棄または延期するかもしれない。また、早期研究が完了する前にいくつかのReactionまたは未来の候補製品の臨床試験を開始することを含む、我々の開発スケジュールを加速させることが求められるかもしれない。この方法は、その後、早期臨床試験に合格できない1つまたは複数の候補製品の後期試験を準備および実施するために大量の資源を投入することをもたらす可能性がある。したがって、私たちの資源配分決定は、実行可能な商業製品や利益のある市場機会を利用できない、あるいは実行不可能な候補製品に資源を費やすことにつながる可能性がある。
Reactionや将来の候補製品のためにより多くの治療機会を決定することができるか、または内部研究計画によって適切な潜在的候補製品を開発することができることは保証されず、これは、私たちの将来の成長および将来の見通しに実質的な悪影響を及ぼす可能性がある。私たちは私たちの努力と資源を潜在的な候補製品または最終的に成功しないことが証明された他の潜在的な計画に集中するかもしれない。
もし私たちが発表し、予想された時間枠内で私たちの予想された開発目標を達成しなければ、私たちの製品の商業化は延期されるか、永遠に達成されないかもしれない。
時々,様々な科学,臨床,法規,製造,その他の製品開発目標の完成時間を見積もることができ,マイルストーンと呼ぶことがある。これらのマイルストーンは、IND提出を含む臨床試験の開始または完了および規制文書の提出を含む可能性がある。私たちは時々いくつかのマイルストーンの予想時間を公開するかもしれない。このようなすべてのマイルストーンは様々な仮定に基づいており、未来にもそうだ。私たちの推定と比較して、このようなマイルストーンの実際の時間は大きく異なる可能性があり、場合によっては、原因は私たちの統制を超えている。私たちが行った任意の未来の臨床試験の間、または私たちが行った任意の臨床試験のために、私たちは多くの予見できない事件に遭遇する可能性があり、これらの試験は、上場承認または商業化反応または将来の任意の候補製品の能力を遅延または阻止する可能性がある。
REACT製造に関するリスクと将来の候補製品
細胞療法は複雑で製造が困難であり,我々は製造問題を経験し続けている可能性があり,これらの問題は我々の主要候補製品Reactionの開発や商業化遅延,あるいは他の方法で我々の業務を損なう可能性がある。
細胞治療製品の製造技術は複雑で、大量の専門知識と資本投資が必要である。予見できない事件による生産困難は著者らの臨床試験に必要な材料の供給を遅らせる可能性がある。また,Reactionの予想規模の市場で生産されている他の細胞療法があることは知られていない。Reactionが商業販売のために許可された場合、その製造過程は複雑なプロセスに関連するので、市場のこの製品に対する需要を直ちに満たすことができないかもしれない。
また,臨床試験や商業販売のための治療薬の準備に参加するすべての実体は,広範なcGMP,州,連邦法規,適用時の外国要求を遵守しなければならない。 2023年10月、同社の契約合格者(QP)は、EUにおけるREACTの発行および配布の準備状況を評価するための監査を行い、EUの臨床場所で製品を配布する前に解決すべき品質管理システム文書にいくつかの欠陥があることを発見した。それに応じて、私たちは、私たちの第3段階計画のEUと世界的な基準を満たすために、私たちの能力を最適化するために生産を一時停止し、商業製造への移行に備えている。製造活動は2024年6月末までに再開される予定だ。
商業販売または末期臨床試験のための完成治療製品の使用が許可された成分は、cGMP規定に従って生産されなければならない。これらの条例は、調査製品および承認販売された製品の品質を制御し、確保するために、生産プロセスおよびプログラム(記録保存を含む)および品質システムの実施および動作を管理する。生産プロセスの不良制御は、外来試薬または他の汚染物質の導入をもたらす可能性があり、または最終製品試験では検出できない可能性のある反応特性または安定性の意図しない変化をもたらす可能性がある。私たちはBLAやMAAをサポートするために必要なすべてのファイルをタイムリーに提供しなければならない。我々の施設および品質システムは、私たちと契約して最終製品の任意の重要な部品を製造する任意の第三者の施設および品質システムを含み、規制部門としてREACTまたは私たちの任意の他の潜在的製品の条件を承認するために、適用される法規に適合するために、承認前の検査を通過しなければならない。さらに、FDAおよび他の規制機関は、REACTまたは私たちの他の潜在的製品または関連品質システムの製造に関連する製造施設が、当施設で行われる活動に適した法規に適合しているかどうかを随時審査または検査することができ、私たちの施設または私たちが契約した第三者の施設がこのような監査または検査を通過していない場合、彼らは、私たちの1つまたは複数の臨床試験を一時停止することができる。もしこれらの施設が承認前の工場検査を通過しなければ、FDAはこの製品を承認しないだろう。守ることができなかった人や
51
このような規制要求に適合する文書は、FDA、EMAまたは外国の監督管理機関の臨床試験または法執行行動に対する反応遅延または中断を招く可能性がある。もし私たちまたは私たちのサプライヤーがFDA、EMA、または他の規制機関の要求を遵守できなかった場合、FDA Form 483の検査観察通知の発行、警告状または無見出し手紙、臨床封印、罰金、禁止、民事処罰、遅延、一時停止または許可の撤回、許可証の取り消し、候補製品または製品の差し押さえまたはリコール、運営制限および刑事起訴を含む規制行動または制裁を受ける可能性があり、いずれも反応の供給に重大な悪影響を及ぼす可能性がある。私たちの将来の他者のReaction生産への潜在的な依存はまた、私たちの将来の利益率と、Reactionまたはタイムリーで競争力に基づいて監督管理によって承認された将来の候補製品の商業化能力に悪影響を及ぼす可能性がある。
生物製品はもともと製造が難しい。Reactionは技術的に複雑な技術を用いて製造され、専門的な設備と施設、高度に特定の原材料、患者から収集した自己細胞と試薬が必要であり、この技術は各種の生産制限に関連している。私たちの目標は可能な限り予備の原材料と試薬供給があることであるにもかかわらず、私たちの主要な供給源が利用できなければ、私たちはこれらの供給が十分かどうかを確認することができない。肝心な原料或いは試薬の不足、或いは生産過程における技術問題は、臨床開発或いは商業化計画の遅延を招く可能性がある。私たちが使用している原材料コンポーネントの製造過程のどんな変化も、私たちの製造過程に予期せぬ影響や不利な影響をもたらし、遅延を招く可能性があります。
細胞療法製造の遅延や失敗により,患者が細胞治療を受けられないか,あるいは再製造が必要となる可能性があり,それ自体が他の患者の製造遅延を招く。いかなる遅延、失敗或いは適時な生産ができないことは患者の予後に不利な影響を与える可能性があり、そして著者らの臨床試験のスケジュールを延期する。このような遅延、故障、または製造できないことは、以下の理由による可能性がある:
|
|
|
製造プロセス自体の故障、例えば、製造装置のエラーまたは試薬故障、製造プロセスの任意のステップの故障、cGMP環境を維持できなかった、または製造に適した品質システムの故障、無菌故障またはプロセス中の汚染による; |
|
|
|
患者の自己細胞または他のサンプルを収集すること、分析実験室への材料の搬送、および最終細胞療法をコールドチェーンを使用して配送された場所に戻すことに関連する物流問題、患者の出発材料との差異、細胞成長の不整合性および製品特性の変異性に関連する製造問題による製品損失または故障; |
|
|
|
製造過程自体は信頼性や再現性が不足し、細胞療法の最終的な製造の多変性を招き、これは監督当局が臨床試験を一時停止することを招き、あるいはこの過程に関するさらなる情報を提供することを要求する可能性があり、これは逆に臨床試験の遅延を招く可能性がある |
|
|
|
患者の体細胞または特徴との差、加工中断、汚染、所定の時間範囲内で患者分離材料を提供できなかった(例えば、輸入または輸出によって阻害された)またはサプライヤーエラーに関連する問題を含む、物流問題による製品損失または故障 |
|
|
|
患者が生産を必要とする場合、患者のための細胞療法を生産するのに十分な生産槽を有することができない |
|
|
|
原料や製造原料を調達できません |
|
|
|
もし私たちのサプライチェーンが中断された場合、これは、REACTを生産するために必要な1つまたは複数のコンポーネントのための代替製造業者またはサプライヤーを見つける必要があるかもしれません。これは、逆に、メーカーまたはサプライヤーがBLAおよび/またはMAAを介して資格を補充することを要求し、これは、新しいメーカーに依存して商業生産を行う場合、メーカー交換に関連する巨額のコストおよび遅延に関連する可能性がある規制機関に追加的な研究を要求する可能性がある |
|
|
|
私たちの細胞療法を生産するための任意の製造施設を失ったり閉鎖したり、代替製造能力をタイムリーに見つけることができません |
|
|
|
患者の出発材料の損失または汚染は、患者から出発材料を再取得するか、または製造プロセスを再開する必要がある |
|
|
|
任意の製造プロセスの修正または変更が要求され、これは、必要な修正または任意の必要な比較可能なテストを実行する能力を遅延させるために、比較可能なテストを行う必要がある可能性があり、さらなる規制承認が必要であるか、または生産を継続するためにCDMOに技術を成功裏に譲渡する必要がある。 |
これらの要素は臨床試験、監督提出、必要な承認或いはReaction商業化の遅延を招く可能性があり、私たちはより高いコストを発生させ、Reactionを商業化に成功させることを阻止する(承認されれば)。
必要な規制承認を得たと仮定して,商業反応製品の製造コストを低減するために生物プロセス開発を改善する予定である。培地は反応処理の中で最も高いコストであり,生物プロセスや自動化による培地使用量の削減を模索している。また,我々の最終商業反応製品計画は冷凍配合であり,我々の新鮮な反応製剤と比較して製造コストを低減することが予想される。大口調達の積極的な交渉材料の定価を利用して、コスト低減をさらに推進する見通しだ。しかし,我々の第3段階試験がREPACTを商業規模で生産した場合の製造コストが実際に我々の第2段階RMCL−002研究よりも低いことは保証されない。多くの要素が無能力を招く可能性があります
52
これらのコスト低減を実現するためには、我々が期待している自動化効率を実現できなかったこと、サプライチェーンにおけるコスト超過や効率低下、およびReactionの製剤または生物処理をコスト低減で改善できなかったことが含まれる。
私たちは自分の製造能力を持っていて、これは私たちが発生する費用を増加させるかもしれない。
我々のReaction製造工場はノースカロライナ州のウィンストン−セレム工場内にあり,現在われわれの臨床試験のためのSRCを生産している。規制機関、特にFDAは、ウィンストン-セレム工場でSRCまたは他の細胞療法を生産する能力を継続的に承認しないかもしれない。
私たちが合理的な時間内と現在予想されているコスト内でウィンストン-セレム工場が私たち自身の細胞療法を成功的に生産できるかどうかは、多くの要素に依存している
|
|
|
私たちは適切なレベルと経験を持ち、所定の時間範囲で必要な従業員を募集し、これらの必要な従業員の採用を維持することができる |
|
|
|
私たちは規制機関の施設と施設での細胞療法の承認を得て、規制当局の要求を満たし続けることができる |
|
|
|
私たちは細胞療法を確実かつ反復的に製造し、必要な患者を十分な時間で投与することができる |
|
|
|
我々の細胞療法を生産する能力は、FDAおよび州規制機関によって実行されるcGMPを含む米国およびEUに適用される要求を含む適用された法規要件に適合している |
|
|
|
私たちは内部品質管理とプロセスを開発し、私たちのウィンストン-セレム工場が細胞療法を生産し供給できるようにすることができます |
|
|
|
私たちは現在使用されている製造プロセスと比較可能性を確立し、このような比較可能なデータを適切な規制機関に受け入れることができる |
|
|
|
私たちは私たちの施設で細胞療法に必要な設備の製造に成功することを含め、進行中の開発に資金を提供することができる。 |
2023年10月、同社の契約合格者(QP)は、EUにおけるREACTの発行および配布の準備状況を評価するための監査を行い、EUの臨床場所で製品を配布する前に解決すべき品質管理システム文書にいくつかの欠陥があることを発見した。それに応じて、私たちは、私たちの第3段階計画のEUと世界的な基準を満たすために、私たちの能力を最適化するために生産を一時停止し、商業製造への移行に備えている。
私たちの工場で生産されたいかなる更なる遅延或いは失敗は著者らの臨床試験の細胞療法の供給遅延を招く可能性がある。臨床試験のための細胞療法を生産できない場合や,必要なレベルの細胞療法を生産できなければ,代替製造能力を確保する前に,このような臨床試験を支援することはできない。
契約開発·製造組織のユニット製造能力には限界があり、我々の業務の長期的な成長見通しを抑制する可能性がある。
私たちは現在ノースカロライナ州ウィンストン-セレムの施設で臨床試験のために材料を生産しています私たちの製品に対する需要は既存の製造能力を超える可能性があります。我々自身の細胞治療開発計画の進展や細胞治療サービスに対する業界の需要の拡大に伴い,将来的には我々の細胞治療サービスや製品の製造サプライヤーを拡大する必要がある可能性が予想され,大量の資本を投入して規制部門の承認を得る必要があるかもしれない。メーカーが日々増加している製品やサービス需要をタイムリーに満たすことができない場合や,cGMP/CGTPコンプライアンス基準を維持できなければ,我々自身のプロジェクトの進捗が影響を受ける可能性が高く,我々の開発プロジェクト全体の成功に実質的な悪影響を与える可能性がある.
商業販売や晩期臨床試験のための治療製品の成分はcGMPで生産されなければならず,細胞に基づく候補製品のメーカーはcGTPSに適合しなければならない。さらに、治療製品の製造業者は、規制要件に応答するために、その製造プロセスを時々修正することを要求される可能性がある。生きたハニカム製品の製造は複雑であり,大きな規制負担がかかり,この負担は時間の経過とともに変化する可能性がある。私たちの限られた製造経験のため、私たちは私たちの候補製品を生産する時に困難に直面するかもしれない。
私たちの自己細胞治療製品は患者によって異なり、正しい製品が正しい患者に適用されることを確保する必要があります。
自家細胞療法の管理は患者に対するものと個人化された薬剤である。このプロセスは、追跡プロセスに誤りがないことを保証し、患者が患者に固有の製品および障害安全追跡を慎重に処理する必要がある
53
患者から採取し、製造と再投与まで同一患者に投与し、サンプルを追跡する。このようなメカニズムはすでに整っているが、追跡プロセスが失敗した場合、私たち自身の施設、第三者施設、製造プロセスおよびサプライチェーンのいずれにおいても、患者は別の患者のSRCを受け取り、深刻な毒性および潜在的な患者の死亡をもたらす可能性がある。我々は、障害の安全な追跡をさらに確保するために、バーコードおよび電子識別チェーンおよび監視チェーンシステムのような強化されたシステムに投資する必要がある。どんなそのようなシステムでも、いつも失敗の危険がある。許容可能な障害安全追跡方法および処理制度を開発または採用できないことは、私たちが規制部門の承認を得ることを遅延または阻止する可能性があり、および/または患者が別の患者のSRCを受け入れた場合、重大な毒性および潜在的な患者の死亡を招く可能性がある。自己細胞療法を制御または賛助しない臨床試験に使用すると、このようなリスクが増加する可能性があり、このような臨床試験においてわれわれの自己細胞療法の管理に誤りが生じた場合、これは、故障安全追跡を保証するために、我々自身の臨床試験および製造過程に必要なステップに影響を与える可能性がある。患者の安全管理をさらに確保するために必要な追跡システムはまた、ヨーロッパのデータ保護要件のような他の規制要件を満たすために、プログラムおよび管理を強化する必要があるかもしれない。追跡システムを確保する必要がありますこれらの追加的な法規要件を遵守することは、臨床試験の開始遅延をもたらす可能性があり、またはそのような試験を開始する前に追加の法規制許可または同意を得る必要がある。
生産反応の製造過程や施設の規制承認や製造過程の中断を遅延させることは、我々の商業化努力を遅らせるか、撹乱する可能性がある。米国では,cGMP細胞治療メーカーがFDAの承認を得て承認された細胞治療製品を生産することは少ない。
REPACTや将来の任意の候補製品の商業化生産を開始できる前に、当社の製造プロセスや製造を行う施設に対するFDAの規制承認を得なければなりません。EUが商業化される前に、適切なEU規制機関から製造許可を得なければならない。米国では,ごく少数のcGMP細胞治療メーカーのみがFDAの承認を得ており,承認された細胞治療製品を生産することができるため,我々の候補製品の規制承認に要する時間枠組みは不確定である。また、REACTの任意のコンポーネントを生産する施設を含むFDAおよび他の関連規制機関による製造施設の承認前検査を経て、REPACTまたは将来の任意の細胞を介して製品候補製品の発売承認を治療しなければならない。政府が長期的に停止している場合、または世界的な健康問題がFDAまたは他の規制機関の定期的な検査、審査、または他の規制活動を阻害し続けている場合、FDAが私たちが提出した規制文書を適時に審査し、処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。また、今回の発行完了後、上場企業としての運営において、将来の政府閉鎖は、適切に資本化し、運営を継続するために、公開市場に参入し、必要な資本を得る能力に影響を与える可能性があります。
承認を得るためには、私たちのすべてのプロセス、品質システム、方法、設備、政策、およびプログラムがcGMPおよび他の適用法規に適合することを保証し、サプライヤー、契約実験室、サプライヤーを広範に監査する必要があります。もし私たちの任意のサプライヤー、契約実験室、またはサプライヤーがcGMPまたは他の反応に関連する適用法規に適合していないことが発見された場合、私たちがこれらの第三者と協力して違反を是正したり、適切な代替サプライヤーを探そうと努力した場合、私たちは製造過程で遅延や中断に遭遇する可能性があります。CGMP要求は,他の事項に加えて,製造過程の品質管理およびファイルポリシーやプログラムを規定している。CGMP規定を遵守するとともに,製品が適用される規格やその他の要求に適合するように,生産,記録保存,品質管理に時間,金銭,労力をかけることが義務付けられている。もし私たちがこれらの要求を遵守できなかったら、私たちは規制された法執行行動や他の法的制裁を受け、私たちが開発する可能性のある任意の製品の販売を禁止されるかもしれない。
私たちの製造プロセスや施設におけるどんな問題も、潜在的なパートナー(大きな製薬会社や学術研究機関を含む)の魅力の低いパートナーになる可能性があり、より多くの魅力的な開発プロジェクトを獲得することを制限する可能性があります。
複雑なサプライチェーンを管理したり、製造業に関する規制要求を満たしたりする会社として、私たちは経験がありません。
FDA、EMA、および他の外国規制機関は、任意のロットの承認製品のサンプルをいつでも提出し、適用ロットのリリース試験結果を示すプロトコルを要求することができます。場合によっては、FDA、EMA、または他の外国規制機関は、関連機関が発行を許可する前に製品ロットを配布しないように要求する可能性があります。製造過程中の微小な偏差は、それらの品質属性と安定性に影響する偏差を含み、細胞治療製品の受け入れられない変化を招く可能性があり、それによって大量故障或いは製品リコールを招く可能性がある。大量失敗や製品リコールは製品の発表や臨床試験を延期する可能性があり、これは高い代価を払わせるかもしれません。そうでなければ、私たちの業務、財務状況、運営結果、将来性を損なうことになります。私たちの製造過程の問題は私たちの製品に対する市場の需要を満たす能力を制限するかもしれない。
54
自己体外細胞治療サプライチェーンを管理することは非常に複雑である。患者の細胞由来材料は収集され、準備され、製造施設に輸送されなければならず、凍結保存された治療製品は、気相液体窒素輸送容器を使用して患者に服用するために、治療センターに戻さなければならない。また、私たちは高度に専門化されたサプライヤーに依存して、私たちの製造過程に原材料と部品を提供します。
患者から収集されると、細胞由来材料は規定された手順に従って準備されて保存されなければならない。治療センターのプロセスを標準化する予定であるが,プロセスにばらつきが生じると,患者の細胞由来材料が悪影響を受け,製造失敗につながる可能性がある。患者の細胞材料は、材料を十分に低い温度に維持し、一般に収集後4日以内に送達および処理しなければならない輸送コンテナを使用して製造施設に輸送されなければならない。信頼性の良い配達員や代理を使用してこのような材料を輸送する予定であるが、輸送コンテナが開いたり破損したりして、適切な貯蔵/輸送温度を維持できない場合、細胞由来の材料は不利な影響を受ける可能性があり、患者のための細胞治療製品の生産は不可能である可能性がある。同様に、悪天候、誤った経路、税関で遅延された、または他のイベントによって輸送が遅延された場合、細胞由来材料は、細胞治療製品の製造に成功することを可能にする時間ウィンドウ内で送達できない可能性がある。
同様に,患者の自家細胞治療製品は臨床現場に戻らなければならず,専用の輸送容器を用いて材料を非常に低い温度に保って患者に投与しなければならない。信頼性の良い配達員や代理店を用いて製品を輸送する予定であるが,輸送コンテナが開いたり破損したりして極めて低い温度を保つことができない場合には,細胞療法製品が悪影響を受ける可能性があり,患者への使用が不可能である可能性があり,あるいは使用すると患者に被害を与える可能性がある。同様に,悪天候,経路ミス,税関での遅延や他の事件により貨物が遅延し,極めて低温度を維持した時間内に臨床現場に搬送されなかった場合,細胞療法製品は悪影響を受け,投与できない可能性があり,あるいは投与すると患者に傷害を与える可能性がある。
私たちのビジネススタート戦略の一部として、私たちは私たちの目標地域の治療センターを遅延または確定できず、参加し、成功したり、資格鑑定を行ったりすることができます。これは患者を延期または阻止する可能性があります細胞治療を受け、承認されれば。もし私たちの治療センターが満足できなければ、私たちは名声、運営、業務の損害を受ける可能性があります。
上記のいずれの事件が発生すると、私たちの発展スケジュールおよび私たちの業務、財務状況、運営結果、および見通しに悪影響を及ぼす可能性があります。
我々は、臨床試験を行うために必要な材料を第三者サプライヤーに提供することに依存しており、これらの第三者サプライヤーは私たちの主要な候補製品であり、これらの第三者サプライヤーを失ったり、十分な数の材料を提供することができなかったり、受け入れ可能な品質レベルでこれらの材料をタイムリーに提供することができず、私たちの業務を損なう可能性がある。
Reactionの製造は私たちの主要な候補製品であり、多くの試薬が必要であり、これらの試薬は私たちの製造過程で化学反応や生物反応を引き起こすための物質、および他の特殊な材料と設備であり、その中のいくつかは資源と経験の限られた小さな会社によって製造または供給され、商業生物製品の生産を支持する。私たちは現在、限られた数のサプライヤーに依存してREACTを製造するために使用されるいくつかの材料とデバイスを提供しています。その中のいくつかのサプライヤーは生物製薬会社がcGMP下で生産した臨床試験と商業製品を支持する能力がないかもしれない、あるいは装備が不足している可能性があり、私たちの需要を支持できないかもしれない。私たちもこれらのサプライヤーの多くと供給契約を締結していません。受け入れ可能な条件や供給契約をまったく得られないかもしれません。したがって、臨床的または商業的製造を支援する重要な材料および設備を受け取ることにおいて、遅延または中断に遭遇する可能性がある。コンポーネントまたはコンポーネント供給の任意の重大な遅延または中断、または代替ソースから代替コンポーネント、コンポーネントまたは材料を許容可能な価格でタイムリーに得ることができない場合、お客様のニーズを満たす能力を弱める可能性があり、ビジネスを損なう可能性があります。
これらの試薬、装置、および材料のいくつかについては、私たちは依存しており、将来的には独占的なソースの供給者または限られた数の供給者に依存する可能性がある。REPACTの製造に必要な試薬や他の特殊材料や設備の供給は随時減少または中断する可能性がある。この場合、代替サプライヤーを探して採用することは遅延を招く可能性があり、私たちは受け入れ可能な条件で他の許容可能なサプライヤーを見つけることができないか、あるいは全く見つからないかもしれない。サプライヤーの交換は大量のコストを伴う可能性があり、私たちが期待する臨床およびビジネススケジュールの遅延を招く可能性がある。もし私たちがサプライヤーを交換して商業生産を行う場合、適用される規制機関は私たちに追加的な研究や試験を要求するかもしれない。もし重要な供給者を失った場合、あるいは材料供給が減少したり、供給を停止したりすれば、私たちはタイムリーかつ競争力的に開発、製造、市場反応を行うことができず、甚だしきに至っては全くできないかもしれない。これらのサプライヤーのうちのいずれかから製品を調達し続けることができないのは、サプライヤーの規制行動または要求、サプライヤーが経験した不利な財務または他の戦略的発展、労使紛争または不足、意外な需要または品質の問題を含む多くの問題が、Reaction需要を満たす能力に悪影響を及ぼす可能性があり、これは、私たちの製品販売および運営結果、または私たちの臨床試験を行う能力に悪影響および実質的な影響を与える可能性があり、いずれも私たちの業務を深刻に損なう可能性があるからである。
55
私たちが私たちの製造過程を発展させ、拡大するにつれて、私たちはこの過程の一部として、いくつかの材料と設備の権利と供給を得る必要があると予想される。私たちは商業的に合理的な条項でそのような材料や設備の権利を得ることができないかもしれないし、これらの権利を全く得ることができないかもしれません。もし私たちがそのような材料や設備を使用したり、適切な代替品を見つけることを避けるために商業的に可能な方法で私たちのプロセスを変えることができなければ、これは私たちの業務に大きな悪影響を与えます。他の材料や設備を使用するために私たちのプロセスを変えることができても、このような変化は、私たちの臨床開発および/または商業化計画の遅延をもたらす可能性がある。すでに臨床開発段階にある候補製品にこのような変更が発生した場合、変更にはこの2つの操作を同時に実行する必要があるかもしれません離体するより高度な臨床試験を行う前に比較可能な研究を行い,被験者から追加的なデータを収集した。これらの要因は,非臨床研究や臨床試験,規制提出,必要な承認またはREPACTの商業化の遅延を招く可能性があるあるいは私たちが開発した未来の候補製品は、より高いコストを招き、私たちの候補製品の商業化に成功することを阻止します。
私たちの細胞ベースの製品製造過程における任意の微生物汚染、原材料不足、あるいは私たちの任意の重要なサプライヤーが必要なコンポーネントを渡すことができなかったことは、私たちの臨床開発やマーケティング計画の遅延を招く可能性がある。
細胞製品製造の性質に鑑み,微生物汚染のリスクがある。どの微生物汚染も、私たちの時間通りに生産、放出、あるいは私たちの細胞療法を管理する能力に悪影響を及ぼす可能性があるため、私たちの運営結果を損害し、名声を損なう可能性がある。
私たちの製造過程で必要ないくつかの原材料は生物源から来ている。このような原材料は入手が難しく、汚染されたりリコールされる可能性がある。原料不足、汚染、リコール或いは使用制限生物由来物質の製造Reactionは商業生産或いは臨床材料生産に不利な影響或いは中断を与える可能性があり、これは著者らの開発スケジュールと著者らの業務、財務状況、運営結果と将来性に不利な影響を与える可能性がある。
REACTは臨床部位で特定の貯蔵,処理,管理による超低温保存が必要である。
REACTは低温保存が必要であり,ただちに使用されるまで非常に低い温度で専用の液体窒素貯蔵タンクや専用輸送コンテナに貯蔵しなければならない。投与では,冷凍保存した製品は慎重に倉庫から取り出し,制御された温度条件下で患者のベッドサイド付近の領域で急速に解凍し,ただちに患者に注射しなければならない。凍結細胞治療製品の処理、解凍および投与は、特定の説明に従って行われなければならず、一般に特定の使い捨て用品が使用され、いくつかのステップは、特定の期間内に行われなければならない。製品を正しく処理していない、解凍および投与説明に従っていない、または規定された解凍後の期限内に投与しないことは、製品の効果および/または安全性に負の影響を与える可能性がある。
候補製品の製造または処方を変更する方法は、追加のコストや遅延を招く可能性がある。
候補製品の臨床試験から発売承認と商業化に伴い、開発計画の各方面、例えば製造方法と製品調合は、この過程で変化する可能性があり、生産量の最適化、大量生産、最大限にコストを下げ、一致した品質と結果を実現する。このような変化は予想された目標を達成できない危険をもたらす。これらのいずれの変化も、REPACTまたは私たちの将来の任意の候補製品の表現が異なり、計画中の臨床試験または変更された材料を用いた他の将来の臨床試験の結果に影響を与える可能性がある。これは私たちが行っているか計画中の臨床試験の完成を遅らせる可能性があり、過渡的な臨床試験を実行するか、または1つ以上の臨床試験を繰り返し、臨床試験コストを増加させ、Reactionまたは未来の任意の候補製品に対する任意の潜在的な承認を延期し、Reactionまたは任意の未来の候補製品を商業化し、収入を創造する能力を脅かすことが要求される。
そのほか、臨床試験或いは商業流通の技術開発と大規模生産に関連するリスクがあり、その中にはコスト超過、技術拡大の潜在問題、技術再現性、安定性問題、cGMP、ロット一致性と原材料の適時可獲得性が含まれている。我々の任意の候補製品が市場承認を得ても、FDAや他の同様の外国規制機関が受け入れ可能な規格に従って承認された製品を生産して、その製品の潜在的な商業投入または将来の潜在的需要を満たすのに十分な数の製品を生産することができる保証はない。また,生物製品の製造プロセスは小分子よりも複雑で高価であるこれらの開発計画のために臨床試験用品を生産するためには、より多くの製造サプライヤーが必要かもしれない。もし私たちが臨床試験や商業化に十分な数を生産できなければ、私たちの開発と商業化努力は損なわれ、これは私たちの業務、財務状況、運営結果、成長の見通しに悪影響を及ぼすだろう。
私たちの現在の業務はノースカロライナ州の製造工場を含む複数の場所に集中している。私たちまたは私たちが依存している第三者は、地震、野火、または他の自然災害のような悪影響を受ける可能性がある
56
そして流行病、流行病、その他の事件、私たちの業務の連続性と災害復旧計画は私たちを深刻な災害から十分に保護できないかもしれません。
洪水、火災、爆発、地震、極端な天気状況、医療流行病または流行病、電力不足、通信故障またはその他の自然または人為的事故または事件などの計画外事件は、私たちの施設を十分に利用できず、私たちの業務運営能力に重大かつ不利な影響を与える可能性があり、特に日常的な基礎の上で、私たちの財務と運営状況に重大なマイナス影響を与える可能性がある。これらの施設へのアクセスを失うことは、コスト増加、Reaction、または将来の任意の候補製品の開発遅延、または当社の業務運営中断をもたらす可能性があります。地震、野火、またはその他の自然災害は、私たちの運営をさらに混乱させ、私たちの業務、財務状況、運営結果、見通しに実質的で不利な影響を与える可能性がある。もし自然災害、停電、または他の事件が私たちの製造施設の全部または大部分を使用できないようにしたり、他の方法で運営を中断したりする場合、私たちは難しいかもしれないし、場合によってはかなり長い間私たちの業務を継続することはできないかもしれない。契約製造業者およびそれに関連する技術移転および品質問題の内在的リスクを使用することを含む、我々の既存の災害復旧および業務連続性計画は、深刻な災害または同様の事件が発生した場合には不十分であることが証明される可能性がある。私たちの災害復旧と業務連続計画の限られた性質のため、私たちは大量の費用を発生する可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性があります。私たちのリスク管理政策の一部として、私たちは私たちの業務に適していると思うレベルに保険範囲を維持します。しかし、これらの施設で事故や事件が発生した場合、保険金額がどんな損害や損失を補うのに十分な保証はできません。もし私たちの施設が事故や事件、あるいはどんな他の理由でも作動できなければ、短い時間であっても、私たちのいかなる研究開発プロジェクトも損害を受ける可能性があります。どの業務中断も、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。
Reactionの商業化に関連するリスクと将来の候補製品
我々が開発したReactionや将来の候補製品が市場承認を得ても,医師,患者,第三者支払人,医学界の他の人がビジネス成功に必要な市場受容度を達成できない可能性がある。
Reactionまたは私たちが開発した任意の他の候補製品が市場の承認を得ても、それは医師、患者、第三者支払人(例えばMedicareとMedicaid計画と管理看護組織)および医学界の他の人の十分な市場受容度を得ることができない可能性がある。また,第三者支払者が提供する保険は,既存および将来の医療コスト低減のための医療改革措置の影響を受ける可能性がある。もし私たちが開発した候補製品が十分な受容度に達していなければ、著しい製品収入が生じないかもしれませんし、利益を上げることができないかもしれません。
すべての候補製品が商業販売のために承認された場合、市場の受け入れ度は多くの要素に依存する
|
|
|
代替療法と比較して、私たちの現在または未来の候補製品の治療効果と潜在的な利点 |
|
|
|
FDA、EMA、または他の外国規制機関の製品ラベルまたは製品挿入要件は、任意のブラックボックス警告またはREMSを含む製品承認ラベルに含まれる任意の制限または警告を含む |
|
|
|
現在または未来の候補製品が承認された臨床適応 |
|
|
|
将来承認されたり商業的に導入された代替療法の供給状況 |
|
|
|
私たちの製品を提供することができ、承認されたら競争力のある価格で販売することができる |
|
|
|
代替療法よりも便利で投与しやすい |
|
|
|
対象患者群が新たな療法を試みる意思と,われわれの治療法が承認されれば,有害事象リスクの増加に関与していると考えられる場合を含め,医師がこれらの治療法を処方する意欲 |
|
|
|
私たちと私たちの候補製品に適した様々な科学組織が発表したガイドラインの私たちの候補製品に関する提案 |
|
|
|
有力なマーケティングと流通支援 |
|
|
|
私たちの製品を他の薬と一緒に使用する制限はありません |
|
|
|
私たちはアメリカで販売チームの能力を雇用し維持しています |
|
|
|
必要な精算コードを含む十分な第三者保険と十分な精算を得ることができます |
|
|
|
副作用の流行率や重症度は |
|
|
|
適切なサービスプロバイダ補償のために、現在のプログラム用語(“CPT”)コードおよびリソースベースの相対的価値基準を得ることができる |
|
|
|
世界保健機関から疾患名の指定された国際疾患分類(“ICD-10”)コードを取得する能力; |
|
|
|
提供者プログラム主義者は、プログラム関連または細胞ベースの有害事象に医療責任を増加させる可能性がある侵襲的腎臓プログラムを実行することを望んでいる |
57
|
|
|
候補製品の配送のための高度な手続き訓練を提供することができる。 |
細胞ベースの製品の販売はまた、医師が治療処方を出す意志に依存し、これは、これらの医師が製品が安全で、治療が有効であり、費用効果があると判断したことに基づく可能性がある。また,異なる医師団体による治療ガイドラインや影響力のある医師の観点から製品を取り入れたり排除したりすることは,他の医師が治療処方を処方する意思に影響する可能性がある。医師、医師組織、病院、他の医療提供者、政府機関、あるいは民間保険会社が、私たちの製品が競争相手の治療法と比較して安全で、治療が有効で、費用効果があるかどうかを予測することはできません。任意の候補製品が承認されたが、このような各当事者の十分な受容度に達していない場合、候補製品から十分な収入を生成または得ることができず、利益を達成または維持することができない可能性がある。政府や他の第三者支払者が商業化されたどの製品にも保険と十分な補償レベルを提供しなければ、市場受容度や商業成功は低下するだろう。Reactionは腎臓に経皮的に注射され,追加のプログラム的技術訓練が必要であり,認証を継続的に維持する可能性がある。REACTを交付する施設には、州、連邦、または実験室認証機関が追加のバッテリベースの許可を提供する必要があり、適切な技術および在庫を有するデバイスが必要となる場合があります。
私たちは現在マーケティングや販売組織がなく、会社としても製品を商業化した経験がなく、これらの能力を開発するために大量の資源を投入しなければならないかもしれません。マーケティングや販売能力を確立できない場合や、規制の承認を得た製品をマーケティング·販売するために第三者と合意できない場合、製品収入を生み出すことができない可能性があります。
私たちは内部販売、マーケティング、流通能力もなく、製品を商業化していない。REACTまたは将来の任意の候補製品が最終的に規制部門の承認を得た場合、主要市場でこのような各製品を商業化するために、技術的な専門性と製造·流通能力を支援するマーケティング·販売組織を構築することが予想される。会社として、私たちは以前、生物製薬製品のマーケティング、販売、流通の面で経験がなく、販売組織の構築と管理は重大なリスクに関連していた。例えば、販売チームの採用と訓練は高価で時間がかかり、どんな製品の発表も延期される可能性がある。販売チームを募集し、マーケティング能力を確立する候補製品の商業発表が何らかの理由で延期または発生していない場合、これらの商業化費用を早期または不必要に発生させる。これは費用が高いかもしれません。もし私たちが私たちの販売とマーケティング担当者を維持したり再配置できなければ、私たちの投資は損失します。
私たちが製品を販売するのを阻害するかもしれない要素は
|
|
|
十分な数の効果的な販売とマーケティング担当者を募集し、訓練し、維持することはできません |
|
|
|
承認されると、販売員は、製品管理および製品交付を含む私たちの候補製品を医師に紹介するために、医師に接触することができない |
|
|
|
販売者が提供するセット製品の不足は、より広い製品ラインを持つ会社と比較して競争劣勢になる可能性がある |
|
|
|
独立した販売およびマーケティング組織の作成に関連する予測不可能なコストおよび費用。 |
私たちの内部販売、マーケティング、流通能力の発展のいかなる失敗や遅延も、これらの製品の商業化に悪影響を及ぼすだろう。直接販売チームや流通システムを構築する第三者と協力して、自分たちの販売チームや流通システムを強化したり、自分たちの販売チームや流通システムの代わりにしたりすることも選択できます。私たちは、受け入れ可能な財務条項で協力することができないか、またはコンサルタントまたは外部サービスプロバイダを招いて、私たちの販売、マーケティング、および流通機能を支援することができないかもしれません。また、私たちが第三者に依存してこれらの機能を達成すれば、私たちの製品収入と収益力(あれば)は、私たち自身のマーケティング、販売、流通、規制部門の承認を得た任意の製品を開発して得ることができるかもしれません。私たちはこのような第三者に対して支配権がほとんどないかもしれません。彼らのいずれも必要な資源と注意力を投入して、私たちの製品を効果的に販売し、マーケティングすることができないかもしれません。もし私たちが私たちの製品を商業化することに成功できなければ、私たち自身でも、1つ以上の第三者との手配によっても、私たちは未来にどんな製品収入も発生できないかもしれないので、私たちは重大な追加損失を招くだろう。
Reactionまたは私たちの未来の候補製品の影響を受けている人たちは、私たちまたは第三者が現在予測しているものよりも少ないかもしれませんが、これはReactionまたは私たちの未来の候補製品の潜在的な市場に影響を与える可能性があります。
私たちが治療を求めている疾患を有する患者の数の予測と、REPACTまたは私たちの将来の任意の候補製品から利益を得る可能性があるこれらの疾患患者のサブセットは、これらの疾患に対する私たちの理解と理解の推定に基づいている。これらの推定は不正確であることが証明される可能性があり、新しい研究、薬物或いは医療実践はこのような疾病の推定発病率或いは流行率をさらに低下させる可能性がある。米国、EU、および他の地域の患者数は予想を下回る可能性があり、REPACT治療を受けられない可能性があり、または将来の任意の候補製品または患者はますます識別および接触が困難になる可能性があり、これらはすべて私たちの業務、財務状況、運営結果、および見通しに悪影響を及ぼすだろう。また私たちが得ても
58
REACTまたは将来の任意の候補製品の承認のために、FDAまたは他の規制機関は、REACTまたは任意の将来の候補製品開発のための私たちのより狭い用途またはサブグループに、その承認の適応を制限する可能性がある。
Reactionまたは私たちの未来の任意の候補製品の総アドレス可能な市場機会は、最終的には、最終ラベルに含まれる診断および治療基準を含む多くの要因に依存するであろう特定の適応下での販売、医学界の受け入れ、患者参入及び製品の定価と精算を許可した。発病率と流行率の推定はよく不正確と可能性の不適切な情報と仮説に基づいており、方法は展望性と投機性がある。われわれが目標適応のために発症率や流行範囲を推定する際に用いる過程は,限られたデータを複数のソースから整理することに関連している。したがって,本年度報告に含まれる発症率と流行率推定数を慎重に見るべきである。また、本年度報告で使用されるデータおよび統計情報は、これによって得られた推定を含み、我々の競合他社が作成した情報および推定とは異なるか、または独立したソースで行われている現在または将来の研究とは異なる可能性がある。
Reactionまたは将来の任意の候補製品の規制承認を1つの管轄区域で取得し、維持することは、他の管轄区域でReactionまたは将来の候補製品の規制承認を得ることに成功することを意味するわけではない。
1つの管轄区域でReactionまたは将来の任意の候補製品の監督管理承認を獲得し、維持することは、任意の他の管轄区域で規制承認を得ることができるか、または維持することができる保証はなく、1つの管轄区で監督管理承認を得ることができないか、または遅延することは、他の管轄区域の規制承認過程に悪影響を及ぼす可能性がある。例えば、FDAが候補製品の発売を承認しても、同様の外国規制機関は、この候補製品のこれらの国での製造、マーケティング、普及を承認しなければならない。承認および許可手続きは司法管轄区域によって異なり、1つの管轄区域で行われる臨床試験は他の管轄区の監督機関によって受け入れられない可能性があるので、米国とは異なるか、または米国よりも大きい要求および行政審査期間に関連する可能性がある。米国以外の多くの管轄区では、候補製品は先に精算許可を得てから、その管轄区で販売を許可することができる。場合によっては、私たちが私たちの製品のために受け取る価格もまた承認されなければならない。
私たちはまた他の国でマーケティング申請を提出することができる。米国以外の管轄区域の監督管理機関は候補製品の承認に要求があり、私たちはこれらの管轄区が発売される前にこれらの要求を守らなければならない。同様の外国規制の承認を得て、類似した外国規制要求を遵守することは、私たちに重大な遅延、困難、コストをもたらす可能性があり、私たちの製品が特定の国/地域で発売されることを延期または阻止する可能性がある。私たちは国際市場を含めてどの司法管轄区でも候補製品の販売を承認しておらず、国際市場で規制承認を受けた経験もありません。もし私たちが国際市場の規制要求を遵守できず、および/または適用されたマーケティング承認を得ることができなければ、私たちの目標市場は減少し、REPACTまたは将来の任意の候補製品の市場潜在力を十分に発揮する能力は損なわれるだろう。
ラベルの外で使用したり、私たちの製品を誤用したりすることは、私たちの市場での名声を損なう可能性があり、コストの高い製品責任訴訟のダメージを招く可能性があり、および/または私たちが法規の要求を遵守できなかったり、任意の製品の意外な問題に遭遇した場合、私たちは処罰を受けるだろう。
もし私たちが承認された候補製品があれば、私たちの製品ラベル、広告、販売促進は規制要求と持続的な規制審査を受けるだろう。米国では、FDA及び連邦貿易委員会(以下、“連邦貿易委員会”と呼ぶ)は、このような製品に関するいかなる主張も規制機関の承認と一致し、誤解性或いは虚偽ではないことを保証し、十分な臨床データ支持を有することを保証するために、薬剤製品に関する販売促進主張を厳格に規制する。虚偽、誤解、未確認または未承認(またはラベル外)用途の方法で薬物または生物製品を宣伝することは、FDA、FTCおよび他の規制機関が実行書を発行し、問い合わせと調査を行い、民事と刑事制裁を実施する可能性がある。特に、製品は、当該製品が承認されたラベルに反映されるように、FDA承認されていない用途に使用されてはならない。もし私たちが候補製品のマーケティング承認を得たら、医者は承認されたラベルと一致しない方法で患者に処方するかもしれない。もし私たちがこのようなラベル外の使用を普及させることを発見されたら、私たちは重大な責任を負うかもしれない。FDAなどは非ラベル用途の普及を禁止する法律法規を積極的に施行し、ある会社に不正があることを発見した非ラベル使用を普及させることは、重大な制裁を受ける可能性があり、連邦および州法規に基づいて提起された虚偽クレーム訴訟を引き起こす可能性があり、これは、法令、民事罰金、原状回復、刑事罰金および監禁、および連邦医療保険、医療補助、および他の連邦および州医療保健計画から除外される可能性がある。連邦政府は不正販売促進の疑いのある会社に巨額の民事と刑事罰金を科し、いくつかの会社がラベル外販売促進に従事することを禁止している。政府はまた、企業に同意法令および/または永久禁止令を締結し、これらの禁止に基づいて特定の販売促進行為を変更または制限することを要求する。Reactionまたは私たちの未来の製品候補製品のラベル外使用は、医師と患者における私たちの市場名声を損なう可能性がある。もし医師が私たちの製品をこれらの未承認の用途に使用しようとすれば、患者の負傷のリスクを増加させる可能性もあり、これは製品責任訴訟を招き、大量の財務と管理資源を必要とし、私たちの名声を損なう可能性がある。
59
また、米国国外で承認された候補製品の広告や販売促進活動は、比べものにならない外国の規制機関や利害関係者の厳しい審査を受ける。
バイオ製品として、承認を求める反応と将来の候補製品は予想よりも早く競争に直面する可能性があります。もし私たちが効果的に競争できなければ、私たちの経営業績は影響を受けるでしょう。
競争相手の前に候補製品を商業化する規制承認を得ることに成功したとしても、REPACTまたは将来の任意の候補製品は、生物学的類似製品からの競争に直面する可能性がある。米国ではREACTは生物製品としてFDAによって規制されることが予想され,BLA経路に応じてREACTの承認を求める予定である。2009年の生物製品価格競争および革新法(“BPCIA”)は、以前に許可された参考製品に基づいてFDAが生物類似および交換可能な生物製品を承認するための短い道を開いた。BPCIAによると,生物類似生物製品の出願は,最初の参考生物製品がBLAによる承認を得てから12年後にのみFDAの承認を得ることができる。この法律は複雑であり、FDAはまだ説明して施行している。したがって、その最終的な影響、実施、そして意味には不確実性がある。FDAがBPCIAを実施するためのこのようなプロセスをいつ完全に採用するかは不明であるが、どのようなプロセスもREACTの将来のビジネス見通しに重大な悪影響を及ぼす可能性がある。
BLAにより生物製品として承認された我々の現在または将来のいずれの候補製品も,参考生物製品の使用に利用可能な12年の専門期間を得る資格があると考えられる。しかしながら、国会の行動や他の理由により、このような排他性が短縮される可能性があり、またはFDAは、競合製品に関するBPCIAの排他的条項の解釈に基づいて、我々の製品候補を参考生物製品と見なすことはなく、予想よりも早く後発薬後続生物類似競争のための機会を創出する可能性がある。さらに、承認されると、生物類似製品が私たちのいずれかの参照製品をどの程度置換するか、その方法は非生物製品の伝統的な模倣薬代替と類似しており、これは、将来の競争相手が私たちの製品のうちの1つの生物類似製品の互換性指定を求めるかどうかを含む、まだ発展中のいくつかの市場および規制要素に依存するかどうかは不明である。BPCIAと国家薬剤法によると、交換可能な生物類似製品のみが参考生物製品の代替と考えられ、原始生物製品を処方する衛生保健提供者の介入を必要としない。しかし,患者−提供者関係や患者特定の医療ニーズを背景にしたすべての処方決定と同様に,衛生保健提供者はラベル外で生物類似製品を処方することは制限されていない。また、競争相手は、生物類似製品の簡略化された承認手続きを放棄することを決定し、自分の非臨床研究と臨床試験を完了した後に完全なBLAを提出して製品許可を得ることができる。この場合,BPCIAにより我々が獲得する資格がある可能性のあるいかなる排他性も,競争相手がその生物製品が承認された直後にその生物製品を販売することを阻止しない。
また、Creates Actは2019年末に公布され、FDAと他の業界人が表明した懸念、すなわちいくつかのブランドメーカーがその製品の流通を不適切に制限し、いくつかの製品のREMSの存在を引用して、後続の製品開発者がブランド薬物または生物製品のサンプルを得ることを阻止することを含む。後続製品開発者はFDA要求のいくつかの比較テストを行うためにサンプルを必要とするため、一部の人は後続製品の進入遅延によるサンプルを適時に得ることができない。このような懸念を解消するために、“創造法”は、後続製品開発業者がブランドメーカーを起訴することを許可し、“商業的に合理的で市場に基づく条項”で必要なサンプルを提供することを強制する私的訴因を確立した。したがって、後続の開発者は、後続の生物類似バージョンをサポートするために比較テストを行うために市場の承認を得た場合、私たちのReaction候補製品サンプルを要求することができ、もし私たちがそのような要求を拒否する場合、私たちはCreates Actに従って訴訟を受ける可能性がある。“創建法”が公布されて以来、“創建法”に基づいて訴訟が提起されているにもかかわらず、これらの訴訟は個人的に解決されているが、そのため、これまで連邦裁判所の審査や論評という法律言語はなく、法律の範囲と適用にはまだ不確実性がある。
ヨーロッパでは、欧州委員会は、過去数年間に発表された生物類似承認に関する一般的かつ特定の製品カテゴリに関するガイドラインに基づいて、いくつかの生物類似製品のマーケティング許可を承認した。また、会社は他の国で生物類似製品を開発する可能性があり、承認されれば、これらの製品は私たちの製品と競争する可能性がある。もし競争相手が私たちの候補製品を参照する生体模倣薬のマーケティング承認を得ることができれば、もし承認されれば、私たちの将来の製品は、それらが交換可能に指定されているかどうかにかかわらず、競争圧力と潜在的な不利な結果に伴うこのような生物模倣薬の競争を受ける可能性がある。これらの競争力のある製品は、私たちの候補製品が承認される可能性のあるすべての指標で直ちに私たちと競争するかもしれない。
競争相手の会社または病院は、EUのルールを利用して、許可されていない薬品を個人患者に販売することを許可し、マーケティング許可なしに競合製品を販売することができる。
EU薬品規則は個別加盟国がマーケティング許可なしに特殊な需要を満たすために医薬製品を供給することを許可し、もし製品が真の能動的な注文に基づいて供給された場合、この注文は衛生保健専門家の規格に基づいて制定され、個人患者がその直接個人の指導の下で使用するために提供する
60
責任です。いくつかの国では、これは、特定の患者またはグループの患者を治療するために輸入されたEU以外の国で生産された製品にも適用されるかもしれない。さらに、指定されたATMPが非通常ベースで調製され、個々の患者の医療処方に従って同じEU加盟国の病院で使用される場合、マーケティング許可は必要とされない。
これらの免除は、特にこれらの競合他社が関連EU加盟国に細胞処理施設を有する場合、マーケティング許可および臨床試験費用を得ることなく、EUでの販売を可能にすることができる。同じように、いくつかの病院はこのような規則に基づいて私たちと競争するかもしれない。どのような販売もマーケティング許可なしに行われるので、競争相手会社または病院は、私たちのマーケティング許可ファイルの臨床データを参照する必要がありませんので、私たちの製品のために得られたいかなるデータも、このような競合販売を阻止することはできません。
承認されれば、Reactionまたは将来の候補製品は、いくつかの細分化された市場でのカバー範囲および精算範囲が限られているか、または利用できない可能性があり、これは、任意の候補製品を利益的に販売することを困難にするかもしれない。
米国や他の国では,病状に応じて処方治療を受けた患者は,通常第三者支払者に依存して治療に関連する費用の全部または一部を精算している。私たちが規制承認を受ける可能性のあるどの製品のカバー範囲や精算状態にも、大きな不確実性がある。米国では、規制マーケティングの承認を得ることができる任意の製品の販売は、保険の可用性や第三者支払者の精算にある程度依存する。第三者支払者は、Medicare、Medicaid、TRICARE、退役軍人管理局、管理式看護提供者、個人健康保険会社、その他の組織などの政府機関を含む。政府医療保健計画(例えば連邦医療保険や医療補助)や商業支払者のカバー範囲と十分な補償は新製品の受容度に重要である。私たちのどの製品も規制部門の承認を得ても、患者は保険を提供しない限り、これらの製品を使用することはあまり不可能であり、かなりの費用を支払うのに十分な費用を精算する。もし私たちのどの製品が承認されれば、私たちのどの製品も保険と精算を受けることができることを保証することができません。あるいは私たちのどの製品の潜在的な収入を正確に推定することもできませんし、私たちが開発する可能性のあるどの製品も保険と精算を受けることができる保証はありません。新たな細胞療法やCKDの新たな適応により,REPACTは資源の相対価値単位割当てとICD−10名に基づくCPTコードを作成する必要がある可能性がある。いずれも異なるプログラムで得られており,未知時間長の償還遅延を招く可能性がある.
政府当局と他の第三者支払人は彼らがどのような治療と精算金額をカバーするかを決定する。第三者支払者の製品使用状況の決定を含む第三者支払人の保証範囲と精算は多くの要素に依存する可能性がある
|
|
|
健康計画の下で保障された福祉 |
|
|
|
安全で効果的で医学的に必要なものです |
|
|
|
特定の患者に適しています |
|
|
|
同業評議の医学定期刊行物の支持を得た |
|
|
|
臨床実践ガイドラインに含まれています |
|
|
|
費用対効果があります |
|
|
|
実験的でも調査的でもない。 |
私たちが規制部門の承認を得た任意の製品を商業化することに成功できるかどうかは、政府医療計画や個人健康保険会社を含む、これらの製品と関連治療がどの程度第三者支払者から保険と十分な補償を受けることができるかにある程度依存する。また,支払者が生物製薬製品に保険を提供することを決定することは,適切な償還率を承認することを意味するものではない。保険や十分な精算が得られない場合、あるいは限られたレベルに限られている場合、Reactionや将来の任意の候補製品を商業化することに成功することができないかもしれません。保険を提供しても、承認された精算金額が十分に高くない可能性があり、十分な投資リターンを実現するために十分な価格設定を確立したり維持したりするのに十分ではないかもしれません。
米国では、第三者支払者の間に統一された製品保証や精算政策はない。そのため、私たちの製品の保証範囲と精算範囲は支払人によって違います。したがって、政府や他の第三者支払人から製品の保証と精算承認を得ることは時間がかかり、費用がかかるプロセスであり、これは、各支払人に科学的、臨床的、費用効果的なデータを提供して、個々の支払人に基づいて私たちの製品を使用することを支援する必要があるかもしれないが、保証され、十分な精算を得ることは保証されないかもしれない。特定の製品の保険を受けても,それによる精算支払率は利益を実現したり維持したりするのに不十分である可能性があり,あるいは患者が受け入れられないと考える高い共済額が必要となる可能性がある。さらに、承認されると、第三者支払者は、候補製品を使用した後に必要な長期的な後続評価をカバーすることができず、十分な補償を提供しない可能性がある。承認されれば、第三者決済者はReactionまたは私たちの未来の任意の候補製品の保証範囲と精算についてどのような決定を下すのか予測することは難しい。
現行法および将来取られる可能性のある州や連邦医療改革措置の変更は、医療保険や他の医療資金のさらなる減少を招き、他の方法で入手可能な任意の製品の価格に影響を与える可能性がある
61
私たちは規制によって承認された候補製品またはそのような任意の候補製品の処方または使用頻度を得ることができる。
私たちに製品責任訴訟を提起すれば、私たちは大量の財務または他の責任を負う可能性があり、Reactionや私たちの将来の候補製品の商業化を制限することが要求されるかもしれない。
私たちは臨床試験におけるテスト、反応、あるいは私たちの未来の任意の候補製品によって固有の製品責任リスクに直面しており、もし私たちが任意の製品を商業化すれば、私たちはより大きなリスクに直面する。例えば、臨床試験、製造、マーケティング、または販売中に、Reactionまたは私たちの将来の任意の候補製品が傷害をもたらすか、または不適切であることが発見された場合、起訴される可能性がある。このような製品責任クレームは、製造欠陥、設計欠陥、製品固有の危険について警告、不注意、厳格な責任、または保証違反の告発を含む可能性がある。州消費者保護法によると、クレームも主張することができる。もし私たちが製品責任クレームで自分自身を弁護することに成功できなければ、私たちは重大な責任を招くかもしれないし、Reactionや私たちの未来の任意の候補製品の商業化を制限することが要求されるかもしれない。このようなクレームを正当化することに成功しても、多くの財政と管理資源が必要だ。事件がどうであっても最終的な結果がどうであろうと、賠償責任は次のようになるかもしれない
|
|
|
候補品を市場に出すことはできません |
|
|
|
私たちの製品への需要が減少しました |
|
|
|
私たちの名声を損なう |
|
|
|
臨床試験対象が退出し、臨床試験を継続することができない |
|
|
|
規制当局が調査を開始しました |
|
|
|
関連訴訟の巨額の抗弁費用 |
|
|
|
私たちの経営陣の資源を減らして、私たちの業務戦略を推進します |
|
|
|
対象者に多額の金銭的報酬を提供する |
|
|
|
製品のリコール、撤回またはラベル付け、マーケティング、または販売促進制限; |
|
|
|
収入損失 |
|
|
|
利用可能なすべての保険と私たちの資本資源を枯渇させる |
|
|
|
私たちが開発するかもしれない製品を商業化することはできません |
|
|
|
私たちの株価は下落した。 |
潜在的な製品責任クレームを防止するために、許容可能なコストで十分な製品責任保険を得ることができず、私たちが開発した製品の商業化を阻止または阻害する可能性がある。REACTの継続臨床開発とより多くの候補製品の臨床進出に伴い、著者らは臨床試験のために追加の保険を得る必要がある。しかし、私たちは臨床試験保険を受けることができないかもしれないし、不利な条項で私たちの任意の臨床試験の任意の責任をカバーするのに十分な金額の臨床試験保険を得ることができないかもしれない。私たちの保険証書にも様々な例外があるかもしれません。私たちは製品責任クレームの影響を受けるかもしれません。私たちは保険範囲を持っていません。私たちは私たちの保険範囲の制限を超えたり、私たちの保険カバー範囲内でない裁判所の裁決または和解合意で達成された任意の金額を支払う必要があるかもしれません。私たちはこれらの金額を支払うために十分な資本を持っていないか、または得ることができません。私たちが未来の会社のパートナーと合意しても、私たちは損害賠償を受ける権利があります。何かクレームがあれば、この賠償は利用できないか十分かもしれません。
もし私たちが現在または将来雇用している任意の契約製造業者やサプライヤーが環境、健康、安全の法律法規を遵守できなければ、罰金や罰金を受けたり、私たちの業務に重大な損害を与える可能性のあるコストが発生する可能性があります。
私たちが現在または未来に接触している任意のCDMOと供給者は、多くの連邦、州と地方の環境、健康と安全の法律、法規、許可要件、それらの管理実験室手続き、危険と規制された材料と廃棄物の発生、処理、使用、貯蔵、処理および処理、地面、空気および水中への危険物質の排出と排出、ならびに従業員の健康と安全を守らなければならない。私たちの行動は化学物質と生物学的材料を含む危険な材料の使用に関するものだ。私たちの業務はまた危険な廃棄物を発生させるだろう。私たちは一般的に第三者と契約を結び、このような材料と廃棄物を処理する。私たちはこのような材料が汚染や傷害をもたらす危険を除去することができない。もし私たちが危険な材料を使用して汚染や損傷をもたらしたら、私たちはそれによるいかなる損害に責任を負い、いかなる責任も私たちの資源の範囲を超える可能性がある。いくつかの環境法により、私たちは現在または過去の施設および第三者施設の任意の汚染に関する費用に責任を負わなければならないかもしれない。私たちはまた民事や刑事罰金と処罰に関連した巨額の費用を発生させるかもしれない。
適用される環境法律と法規を遵守することはコストが高い可能性があり、現在または未来の環境法律と法規は私たちの研究、製品開発、製造努力を損なう可能性がある。しかも、私たちはこのような材料や廃棄物が意外なダメージや汚染をもたらすリスクを完全に除去することはできない。危険材料の使用による従業員の負傷により生じる可能性のあるコストや支出を支払うために労働者補償保険を維持しているが、潜在的な責任を支払うのに十分ではない可能性がある。私たちは特定の生物あるいは危険廃棄物保険を受けません。私たちの財産、死傷者と一般責任保険は明確に排除します
62
生物または危険廃棄物に接触または汚染することによる損害と罰金を保証する。そこで私たちは汚染や傷害が発生すると、損害賠償責任を請求されたり、私たちの資源を超えた罰金が科されたりする可能性があり、私たちの臨床試験や監督管理の承認が一時停止される可能性があり、これは私たちの業務に実質的な損害を与える可能性があります。
私たちの第三者への依存に関するリスクは
私たちは第三者に依存して、私たちの研究、非臨床試験、および臨床試験の特定の部分を監視し、これらの第三者が契約義務の履行、法規の要求を遵守し、または他の方法で満足できるように実行できない場合、私たちは規制の承認を得ることができない、あるいはReactionを商業化することができないかもしれない、あるいはこのような承認または商業化が遅れる可能性があり、私たちの業務は実質的な損害を受ける可能性がある。
大学、医療機関、CRO、戦略パートナー、および他の機関との合意に基づいて、私たちは第三者に依存して、あるいは将来、第三者に依存して、私たちの非臨床研究および臨床試験のいくつかの側面を行い、データを監視し、管理する可能性がある。これらの第三者と予算や契約を交渉しなければならないことが予想され、これにより、私たちの開発スケジュールが延期され、コストが増加する可能性があります。これらの臨床試験は,臨床CRO,医療機関,臨床研究者を含めて第三者に依存し続ける予定である。もし私たちがこれらの第三者CROまたは他の人との任意の関係が終了すれば、私たちはCROまたは他の第三者と合意することができないか、または商業的に合理的な条項でそうすることができないかもしれない。CROを交換または追加することは、追加のコストをもたらし、管理時間と労力を必要とします。しかも、新しいCROが仕事を始める時、自然な過渡期がある。そのため,遅延が生じる可能性があり,期待される開発スケジュールを満たす能力に大きな影響を与える可能性がある.私たちはCROとの関係を慎重に管理しているにもかかわらず、私たちが未来に似たような挑戦や遅延に遭遇しないこと、またはこれらの遅延や挑戦が私たちの業務、財務状況、および見通しに実質的な悪影響を与えないという保証はない。
私たちの非臨床研究や臨床試験を実行する任意の第三者は私たちの従業員ではなく、私たちとこのような第三者との合意によって私たちに提供される可能性のある救済措置を除いて、彼らが私たちの非臨床研究と臨床試験に十分な時間と資源を投入するかどうかを制御することができない。これらの第三者はまた、私たちの競争相手を含む他の商業実体と関係があるかもしれません。彼らはまた、これらの実体のための臨床試験や他の製品開発活動を行っているかもしれません。これは、彼らが私たちを代表する表現に影響を与えるかもしれません。これらの第三者がその契約の義務または義務を成功裏に履行できなかった場合、または予想された期限内に完了できなかった場合、または交換する必要がある場合、または彼らが取得した非臨床または臨床データの品質または正確性が、私たちのプログラムまたは法規の要求または他の理由に従わなかった場合、臨床開発スケジュールを含む我々の製品開発スケジュールは、延長、延期または終了される可能性があり、REPACTの開発を完了し、規制部門の承認を得ることができないか、または商業化に成功する可能性がある。したがって、私たちの財務業績とReactionのビジネス見通しは損なわれ、私たちのコストは増加する可能性があり、私たちの収入を創出する能力は延期されるかもしれない。
われわれの臨床試験過程では,特に第三者に依存し,臨床研究者への制御が限られており,彼らの日常活動への可視性も限られており,承認された臨床試験案を遵守している場合も含まれている。しかし、私たちは私たちのすべての臨床試験が適用された方案、法律と法規の要求、そして科学的な基準に従って行われ、私たちの第三者への依存が私たちの規制責任を免除しないことを確実にする責任がある。我々およびこれらの第三者は、FDAおよび同様の外国規制機関が臨床開発において候補製品に対して実行する法規およびガイドラインであるGCP要求を遵守することを要求されている。監督管理機関は定期的に試験スポンサー、臨床研究者、臨床試験地点を検査することによって、これらのGCP要求を実行する。もし私たちまたはこれらの第三者のいずれかが適用されたGCP要件を遵守できなかった場合、私たちの臨床試験で生成された臨床データは信頼できないと考えられる可能性があり、FDAまたは同様の外国の規制機関は、これらの試験の一時停止または終了を要求するか、または私たちのマーケティング申請を承認する前に追加の非臨床研究または臨床試験を行うことができる。検査後、これらの規制機関が私たちの臨床試験がGCP要求に適合しているかどうかを確認することはできません。また,われわれの臨床試験はcGMPと可能なCGTP法規により生産された生物製品を用いて行わなければならず,多くの試験対象が必要となる。CROを含む私たちまたは契約の第三者は、これらの規定を遵守できなかったか、または十分な数の被験者を募集できなかった場合、規制承認プロセスを遅延させる臨床試験を繰り返す必要があるかもしれない。さらに、これらの第三者が連邦または州詐欺および乱用または虚偽声明法律法規または医療プライバシーおよび安全法律に違反した場合、私たちの業務は巻き込まれる可能性がある。
また,指定された時間範囲で行われているいくつかの臨床試験を登録し,政府が支援しているデータベースClinicalTrials.gov上でいくつかの完了した臨床試験の結果を公表することも求められている。そうしないと罰金、否定的な宣伝、そして民事と刑事制裁につながるかもしれない。
また,われわれの臨床試験の首席研究員は時々私たちの科学コンサルタントやコンサルタントを務め,このようなサービスに関する報酬を得る可能性がある。場合によっては、私たちはその中のいくつかの関係をFDAに報告することを要求されるかもしれない。FDAは結論を出すかもしれないが,我々と主要研究者との経済関係は利益衝突をもたらしたり,他の方法で実験の解釈に影響を与えたりしている。したがって,FDAは適用された臨床試験地点で生じるデータの完全性を疑問視する可能性があり,臨床試験自体の効用が脅かされる可能性がある。これはおそらく
63
FDAは私たちの上場申請の承認を遅延または拒否し、最終的にReactionまたは私たちの将来の任意の候補製品の上場承認を拒否する可能性があります。
私たちはまた、他の第三者に依存して、私たちの臨床試験の製品供給を保存し、配布したい。私たちのディーラーのどんな業績ミスもReactionや未来の候補製品の臨床開発やマーケティング承認、あるいはReactionあるいは私たちの未来の候補製品の商業化を延期する可能性があり、追加の損失をもたらし、私たちの潜在的な収入を奪うことができます。
私たちは組織サンプルを含む第三者に私たちの研究開発活動に必要な材料を提供することに依存して、これらの第三者と合意できなければ、私たちの研究開発活動は延期されます。
私たちは第三者に依存して、主に病院、衛生診療所、学術機関であり、私たちの研究と開発活動に必要な組織サンプルと他の材料を提供します。これらの材料を得るには、様々な承認を得る必要があり、病院または他の材料サプライヤーと受け入れ可能な条件でビジネス合意を達成する必要がある。私たちは現在、組織サンプルを受け取る機関と合意していますが、私たちはこれらのソースと排他的な取り決めがなく、ビジネス的に合理的な条項でこのような合意を維持または継続できる保証はありません。もし私たちがこのような合意を維持したり更新できなければ、私たちは新しい病院、診療所、あるいは衛生機関と新しい手配を求めさせられるだろう。もしそうなら、私たちは他のパートナーと合意したり、私たちが受け入れられる条件でそうすることができないかもしれない。もし私たちがこのような合意に到達できなければ、私たちの研究開発活動は延期され、私たちの発展戦略の重要な部分を実施する能力は影響を受けるだろう。
私たちは将来、Reactionおよび/または未来の候補製品を開発し、商業化するために第三者との協力を求めるかもしれませんが、私たちの将来の協力は私たちの業務に非常に重要になるでしょう。もし私たちが協力できない場合、あるいはこれらの協力が成功しなければ、私たちの業務は不利な影響を受ける可能性がある。
我々の戦略の一部は、パートナーが重大なビジネスおよび/または開発能力を増加させることができると考えられる標識および地理的位置におけるパートナーシップを考慮することである。しかも、私たちの製品の能力は限られています開発は、まだ商業化能力を備えていない。そのため、私たちは将来的に他社と協力して、私たちの計画や技術に重要な技術と資金を提供することが可能になりました。
私たちが未来に行うどんな協力も、以下のリスクを含む多くのリスクをもたらすかもしれない
|
|
|
協力者は彼らが適用する努力と資源を決定する上で大きな自由裁量を持っている |
|
|
|
協力者は期待通りに義務を履行していないかもしれない |
|
|
|
協力者は、規制部門の承認を得た任意の候補製品の開発および商業化を行ってはならず、臨床試験結果、協力者の戦略的重点または利用可能な資金の変化または外部要因(例えば、資源の移転や競争優先度を作成する可能性のある戦略取引)に基づいて、開発または商業化計画または許可スケジュールを継続または更新しないことを選択してはならない |
|
|
|
協力者は臨床試験を延期し、臨床試験計画に資金不足を提供し、臨床試験を停止或いは候補製品を放棄し、新しい臨床試験を繰り返し或いは行うことができ、或いは臨床試験候補製品の新しい調合を要求することができる |
|
|
|
協力者は、競争力のある製品がより成功する可能性があると考えているか、または私たちよりも経済的に魅力的な条項で商業化できることを前提として、第三者開発と直接または間接的に我々の製品および候補製品と競合する製品を独立して開発または間接的に開発することができる |
|
|
|
私たちと協力して発見された候補製品は、私たちの協力者によって彼ら自身の候補製品または製品と競争するとみなされるかもしれません。これは、協力者がReactionまたは未来の候補製品の商業化に資源を投入することを停止させる可能性があります |
|
|
|
協力者は、候補製品または製品の開発、製造、流通、またはマーケティングに関する適用法規要件を遵守できない可能性がある |
|
|
|
マーケティングおよび流通の権利を有するパートナーまたは規制の承認を得た1つまたは複数の将来の候補製品は、これらの製品または製品をマーケティングおよび流通するのに十分なリソースが投入されていない可能性がある |
|
|
|
協力者は、将来の許可協定に基づいて、開発進捗や活動に関するタイムリーかつ正確な情報を提供することができない可能性があり、これは、投資家に進捗を報告し、Reactionまたは将来の候補製品の開発能力を計画することに悪影響を及ぼす可能性がある |
|
|
|
特許権、契約解釈、または第一選択開発プロセスにおける相違を含む協力者との相違は、候補製品の研究、開発または商業化の遅延または終了をもたらす可能性があり、候補製品に対して追加的な責任を負うこと、または訴訟または仲裁を引き起こす可能性があり、いずれも時間的で高価である可能性がある |
64
|
|
|
協力者は、私たちの知的財産権を正確に維持したり、守ったりすることができないかもしれないし、何らかの方法で私たちの固有の情報を使用して、それによって訴訟を引き起こし、それによって、私たちの知的財産権または固有の情報を無効にしたり、私たちを潜在的な訴訟に直面させたりすることができる |
|
|
|
協力者は第三者の知的財産権を侵害する可能性があり、これは私たちを訴訟と潜在的な責任に直面させるかもしれない |
|
|
|
もし私たちの協力者が業務統合に参加した場合、その協力者は、私たちの許可された任意の候補製品の開発または商業化を弱化または終了する可能性がある |
|
|
|
協力者は協力を終了することができ、終了すれば、適用される候補製品をさらに開発または商業化するために、より多くの資金を集める必要があるかもしれない。 |
もし私たちが未来に行ったどんな協力も候補製品の発見、開発、商業化に成功しなかった場合、または私たちの協力者が私たちとの合意を終了した場合、私たちは未来の研究資金やマイルストーンや特許使用料を受け取ることができないかもしれない。本年度報告書に記載されている製品開発、規制承認、商業化に関するすべてのリスクは、我々の協力者の活動にも適用される。
さらに、もし私たちの協力者が私たちとの合意を終了したら、私たちは新しい協力者を引き付けることがもっと難しいことを発見するかもしれませんし、ビジネスや金融界での私たちの見方は不利な影響を受けるかもしれません。
我々はReactionと未来の候補製品のために適切なパートナーを探す上で激しい競争に直面しており、交渉過程は時間がかかり複雑である。Reactionまたは私たちの未来の任意の候補製品のための協力関係を成功させるためには、潜在的な協力者は、私たちが求めている条項と他の会社が許可することができる製品に基づいて、これらの候補製品は彼らが魅力的だと思う市場で経済的価値があると考えなければならない。協力の交渉と記録は複雑で時間がかかる。また,最近では大手バイオ製薬会社間の大量の業務合併により将来の潜在的パートナー数が減少している。我々が協力について最終的な合意を達成できるかどうかは,他の事項に加えて,協力者の資源や専門知識の評価,協調の条項や条件,提案された協力者のいくつかの要因の評価に依存する.もし私たちが適時に、受け入れ可能な条項によって、または適切なパートナーと合意できない場合、私たちは候補製品の開発を削減し、その開発計画または私たちの1つまたは複数の他の開発計画を減らしたり、その潜在的な商業化を延期したり、いかなる販売またはマーケティング活動の範囲を縮小したり、または私たちの支出を増加させ、自費で開発または商業化活動を行わなければならないかもしれない。もし私たちが私たちの支出を増やし、私たち自身の開発や商業化活動に資金を提供することを選択すれば、私たちはより多くの専門知識と追加の資本を得る必要があるかもしれないが、これらは私たちが受け入れられない条件であるか、あるいは全く得られないかもしれない。もし私たちが将来の協力を達成できなかったり、必要な開発や商業化活動を展開するのに十分な資金や専門知識がなければ、REPACTや未来の候補製品をさらに開発し、それらを市場に出し、そのような製品の販売から収入を得ることができないか、あるいは私たちの技術を開発し続けることができなければ、私たちの業務は実質的に悪影響を受ける可能性がある。私たちが新たな戦略協力を成功させたとしても、私たちが合意した条項は私たちに不利になる可能性があり、例えば、候補製品の開発や承認が延期または承認された製品販売に失望した場合、私たちはこのような戦略的協力を維持できないかもしれない。Reactionや私たちの将来の候補製品に関連する新しい戦略協力協定の任意の遅延は、それらの開発および商業化を延期し、市場に進出しても、それらの競争力を低下させる可能性がある。最後に、第三者との任意の協力を求めるには、会社の投入時間と資源が必要となり、経営陣の注意を分散させ、会社が新たな戦略協力合意を達成できない場合や締結できない場合に業務を分散させる可能性がある。
法律と規制コンプライアンスに関するリスク
顧客、ヘルスケア提供者、医師、処方者、購入者、第三者支払者、慈善組織および患者との関係は、適用されるリベート、詐欺および乱用、および他のヘルスケア法律および法規の制約を受けることになり、これは、私たちを刑事制裁、民事処罰、契約損害、名声損害、および利益および将来の収入の減少に直面させる可能性があります。
私たちは現在何の製品も発売されていませんが、Reactionや私たちの将来の任意の候補製品が商業化されると、承認されれば、アメリカ連邦と州政府、そして私たちが業務を展開している司法管轄区の外国政府の追加医療法律法規の要求と監督を受けます。米国や他の地域の医療提供者は,医師を含め,バイオ製薬製品の推奨や処方の中で主な役割を果たしている。第三者支払者および顧客との合意は、AKSおよびFCAを含むが、AKSおよびFCAを含むが、これらの法律および法規は、これらの会社のバイオ製薬製品の販売、マーケティング、および流通の業務または財務配置および関係を制限する可能性がある、広範に適用される詐欺および乱用、ならびに他の医療法律および法規に直面する可能性がある。はい特に、Reactionまたは私たちの将来の任意の候補製品の研究、ならびに医療プロジェクトおよびサービスの普及、販売およびマーケティング、ならびに医療業界のいくつかの商業的配置は、詐欺、リベート、自己取引、および他の乱用行為を防止するための広範な法律によって制限されている。これらの法律法規は、広範な価格設定、割引、マーケティング、および
65
普及、組織、委託(S)、特定の顧客インセンティブ計画、その他の一般的な業務手配。これらの法的拘束を受けた活動は,患者を募集して臨床試験を行う過程で得られた情報の不正使用に関するものである。
私たちに影響を与える可能性のある医療法は、AKSを含む連邦詐欺および乱用法、FCAおよび民事通貨懲罰法を含む虚偽請求および民事罰金法、HITECHによって改正されたHIPAAを含む連邦データプライバシーおよびセキュリティ法、前年に医師(医師、歯科医、視光師、足科医および脊医を含む)、いくつかの非医師の健康従事者および教育病院による支払いおよび/または他の価値移転、および医師が持っているいくつかの所有権および投資権益を報告することを要求する日光法を含む。また,多くの州には類似した法律法規があり,他の州や連邦法律とは大きく異なり,コンプライアンス作業を複雑化させている可能性がある。また、いくつかの州は、生物製薬会社に生物製薬業界の自発的コンプライアンスガイドラインと連邦政府が公布した関連コンプライアンスガイドラインを遵守することを要求し、医師および他の医療保健提供者やマーケティング支出への支払いおよび他の価値移転に関する情報をメーカーに報告することを要求する可能性がある。また、いくつかの州と地方の法律はこの司法管轄区に生物薬品販売代表を登録することを要求している。
これらの法律の範囲も執行も不確定であり、現在の衛生保健改革環境において、特に適用の前例や条例が乏しい場合には、迅速な変化が生じる可能性がある。商業手配が適用される医療保険法に適合することを確保することや,政府当局が行う可能性のある調査に対応することは,時間や資源がかかる可能性があり,会社の業務その他への注意をそらす可能性がある。
政府と法執行当局は、私たちの商業行為は、解釈が適用される詐欺や乱用、または他の医療保健法律および法規の現在または未来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。もし私たちにこのような訴訟が提起された場合、私たちは私たちの権利を弁護したり、維持することに成功しませんでした。これらの行動は、重大な民事、刑事および行政処罰、損害賠償、罰金、返還、監禁、名誉被害、連邦および州政府が援助する医療計画から除外される可能性があり、契約損害、および私たちの業務の削減または制限、およびもし私たちが会社の誠実な合意または他の合意の制約を受けて、これらの法律違反に関する告発、および追加の報告義務と監督を解決することを含む、私たちの業務に重大な影響を与えるかもしれません。さらに、それと業務を展開する任意の医師または他の医療提供者または実体が、適用法に適合していないことが発見された場合、彼らは、政府が援助する医療計画から除外されることを含む重大な刑事、民事または行政制裁を受ける可能性がある。これらの法律に違反するいかなる行為も、正当化に成功しても、バイオ製薬メーカーが巨額の法的費用を招き、経営陣の業務運営への注意をそらす可能性がある。したがって、私たちが私たちに提起される可能性のあるいかなる訴訟も防ぐことに成功しても、私たちの業務は損害を受ける可能性がある。今後発売される製品の販売を禁止または制限または撤回することは、不利な方法で業務に大きな影響を与える可能性がある。
私たちがどんな候補製品の規制承認を得ても、私たちは持続的な監督監督と持続的な規制審査を受けることになり、これは多くの追加費用を招く可能性があり、もし私たちが規制要求を遵守できなかったり、Reactionや任意の未来の製品候補製品の意外な問題に遭遇した場合、私たちは処罰を受けるかもしれない。
REACTまたは将来の任意の候補製品が承認された場合、製品の製造、ラベル、包装、貯蔵、広告、販売促進、サンプリング、記録保存などの活動が行われます広範囲で持続的な規制要件に支配されている。これらの要求には、セキュリティおよび他の上場後の情報および報告の提出、登録、およびcGMPおよびCGTP法規の継続が含まれています。バイオ製薬メーカーおよびいかなる製品製造プロセスを担当するCDMOも、品質管理と製造プロセスがcGMPとCGTP法規および任意の適用される外国同等法規に適合することを確保することを含むFDAと類似の外国規制機関の広範な要求を遵守しなければならない。したがって、私たちと私たちが将来採用する可能性のある任意のCDMOは、cGMPおよびCGTPの遵守状況、および任意のBLA、他のマーケティング申請、および以前の検査観察に対する応答に対するコミットメントの遵守状況を評価するために、持続的な審査および検査を受けるであろう。したがって、私たちと私たちと協力している他の人たちは、製造、生産、品質管理を含む規制コンプライアンスのすべての分野に時間、お金、エネルギーをかけ続けなければならない。
FDA或いは類似の外国の監督管理機関もまた、製品の安全性或いは有効性を監視するために、高価な発売後の非臨床研究或いは臨床試験(通常は4期試験と呼ばれる)と発売後のモニタリングを要求する可能性がある。もし私たちまたは監督機関が製品に以前に未知の問題、例えば意外な深刻性または頻度の不良事件、生産問題または製品生産または加工施設の問題、例えば製品汚染または適用されるcGMP法規に深刻に違反していることを発見した場合、規制機関はその製品、製造施設、または私たちに制限を加えるかもしれない。もし私たちまたは第三者サプライヤーが適用された法規を完全に遵守できなかったら、私たちは私たちの製品をリコールしたり、撤回したりすることを要求されるかもしれません。
66
Reactionまたは私たちの未来の任意の候補製品には、予期せぬ重症度または頻度の不良事象、または私たちの製造プロセス、または法規要件を遵守できなかった以前の未知の問題が存在することが後に発見され、他に加えて、以下の結果をもたらす可能性がある
|
|
|
製品の製造、承認された製造業者または製造プロセスの制限; |
|
|
|
製品のラベルやマーケティングの制限 |
|
|
|
製品の流通や使用の制限; |
|
|
|
発売後の研究や臨床試験が求められている |
|
|
|
製品が市場から撤退した |
|
|
|
製品をリコールする |
|
|
|
FDAからの警告状または無タイトル手紙または外国規制機関からの同様の違反通知; |
|
|
|
FDAまたは他の適用可能な規制機関が処理すべき出願または承認された出願の追加を承認することを拒否する; |
|
|
|
罰金、利益または収入の返還、 |
|
|
|
上場承認の一時停止または撤回 |
|
|
|
私たちが行っているどんな臨床試験も一時停止します |
|
|
|
輸入または輸出の許可を差し押さえ、差し押さえたり、または拒否したり; |
|
|
|
法令、禁止、または民事または刑事処罰に同意する。 |
さらに、FDAやEMAのような規制機関の政策が変化する可能性があり、Reactionまたは私たちの将来の任意の候補製品の規制承認を阻止、制限、または延期するための追加の政府法規が公布される可能性がある。もし私たちが既存の要求の変化に適応できない場合、あるいは新しい要求や政策を採用することができない場合、または他の方法で法規遵守を維持できない場合、私たちは私たちが得る可能性のあるマーケティング承認を失う可能性があり、これは私たちの業務、将来性、利益を達成または維持する能力に悪影響を及ぼすだろう。
安全監視や薬物警戒に関するEUの要求を守らないことも重大な経済的処罰を招く可能性がある。同様に、個人情報保護に関するEUの要求を守らないことはまた重大な処罰と制裁につながる可能性がある。
FDAの政策は変わる可能性があり、Reactionまたは私たちの将来の任意の候補製品の上場承認を阻止、制限または延期するための追加の政府法規が公布されるかもしれない。もし私たちが既存の要求の変化に適応できない場合、あるいは新しい要求や政策を採用することができない場合、または私たちがコンプライアンスを維持できない場合、私たちは私たちが得る可能性のあるマーケティング承認を失う可能性があり、これは私たちの業務、将来性、利益を達成または維持する能力に悪影響を及ぼすだろう。
医療コストを低減するための立法措置を含む医療政策、法律、法規の変化は、承認されれば、Reactionまたは将来の任意の候補製品を商業化する能力に影響を与える可能性がある。
私たちの業務のすべての方面は、研究開発、製造、マーケティング、定価、販売、訴訟と知的財産権を含み、広範な法律と規制を受けている。適用されるアメリカ連邦と州の法律と機関法規および外国の法律法規の変化は私たちの業務に実質的なマイナス影響を与える可能性がある。米国および他のいくつかの司法管轄地域では、医療システムに関する立法および法規の変更および提案された変更は、私たちの候補製品または将来の任意の潜在的候補製品の上場承認を阻止または延期し、承認後の活動を制限または規範化し、またはマーケティング承認を得た任意の候補製品を収益的に販売する能力に影響を与える可能性がある。アメリカ議会のFDA承認過程に対するより厳格な審査は上場承認を著しく延期或いは阻止する可能性があり、そして私たちはより厳しい製品ラベルと上場後のテストとその他の要求の制約を受けることができる。議会はまた、FDAのユーザ費用計画を5年ごとに再認可し、これらの計画を常に修正しなければならず、また、定期的な再認可プロセスの一部として、FDAと業界利害関係者は、政策または手続き上の変更を交渉する可能性がある。国会が最近ユーザー料金計画を再認可したのは2022年9月であり、実質的な政策変化はなかった。
米国や他の地域の政策立案者や支払者の中で,医療システムの改革を推進することに大きな興味があり,医療コストを抑え,質の向上および/またはカバー範囲の拡大を目指している。米国では、製薬業はこれらの努力の重点であり、重大な立法計画の大きな影響を受けてきた。2010年3月,議会はACAを採択し,政府や民間保険会社が医療に資金を提供する方式を大きく変え,米国製薬業に大きな影響を与えた。
67
ACA,MedicareおよびMedicaid計画の変化や増加,および他の医療改革措置による変化,特に個別州の医療参入,融資または他の立法面での変化は,米国の医療産業に実質的な悪影響を及ぼす可能性が予想される。
DSCSAは2024年11月に全面的に発効し、適用され、薬品メーカーに製品追跡と追跡に関する義務が課せられている。さらに、2022年2月、FDAは、各州の薬品卸売業者に対する許可国家基準を改訂するための提案された法規を発表し、州許可第三者物流プロバイダのための新しい最低基準を確立し、州計画なしに使用するための連邦システムを作成し、各計画はDSCSAによって強制的に実行された。承認後の要求を拡大し、薬品の販売や販売促進活動を制限するための他の立法や規制提案も提出された。私たちは、より多くの立法変化が制定されるかどうか、あるいは既存の法規、指導、解釈が変更されるかどうか、またはこれらの変化が私たちの業務に何の影響を与えるかどうかを決定しない。
また,処方薬や生物製品コストの上昇を考慮して,米国政府はバイオ医薬品定価の見直しを強化している。このような審査は,最近の数回の国会調査を招き,製品定価の透明性の向上,価格設定とメーカー患者計画との関係の審査,政府計画の製品精算方法の改革を目的とした連邦や州立法を提案·採択した。例えば、総裁·バイデン2022年10月に発表された14087号行政命令は、アイルランド共和軍を補完し、薬品コストを低減し、革新的な薬物の獲得を促進するための報告書をCMSに準備し、ホワイトハウスに提出することを要求した。2023年2月、CMSはその報告を発表し、その中に3種類の潜在的なモードを記述し、重点的に負担可能性、獲得性と実施の実行可能性であり、CMS革新センターの更なるテストに供する。2024年2月まで、CMS革新センターは提案したモデルを引き続きテストし、各州とメーカーがある製品タイプ(例えば細胞と遺伝子療法)にアクセスするモデルテストの計画を開始した。
州レベルでは、立法機関は、価格または患者の精算制限、割引、いくつかの製品参入およびマーケティングコスト開示の制限、および透明性措置を含む薬品の価格設定を制御するための法規を立法および実施することによって、場合によっては、他の国からの輸入および大量調達を奨励することを目的としている。2020年12月、アメリカ最高裁判所は、連邦法律は各州の薬品福祉マネージャー(PBM)と医療保健と薬品サプライチェーンの他のメンバーの能力を妨害しないと一致し、この重要な決定は各州がこの分野で更なるかつ積極的な努力をすることを招く可能性がある。そして、2022年中に、連邦貿易委員会はPBM業界のやり方に対して全面的な調査を展開し、これはこのような実体の運営、薬局ネットワーク或いは財務手配に対するより多くの連邦と州立法或いは規制提案を招く可能性がある。また,過去数年間にいくつかの州でPDABが設立され,それぞれの管轄範囲で販売されている薬品に対してUPLを実施する権利がある。しかし,いくつかの懸案の連邦訴訟は各州がUPLを強制実施する権威に挑戦している。
最近は2022年8月に総裁·バイデンが2022年インフレ削減法案、あるいはアイルランド共和軍に署名した。他の点では、アイルランド共和軍には複数の条項があり、連邦医療保険計画や米国全体に販売されている薬品の価格に影響を与える可能性がある。2023年から,連邦医療保険BやD部分がカバーする薬物や生物製品の価格上昇速度がインフレ率よりも速い場合,メーカーは連邦政府にリベートを支払わなければならない。この計算は薬品製品をもとに行われており、連邦政府に不足している税金還付額は、連邦医療保険B部分またはD部分が支払う薬物製品の数に直接依存する。また、CMSは2026年から毎年、模造薬や生物類似競争を含むことなく、選定された数量の単一源D部分薬物について薬品価格交渉を行う。CMSはまた,選定数のB部分薬の薬品価格を2028年から交渉する。CMSが1つの薬物を選択して交渉すれば,このような薬物による収入は減少すると予想される。
任意の追加の連邦または州医療改革措置は、第三者支払いが将来の医療製品およびサービス支払いの金額を制限する可能性があり、逆にいくつかの開発プロジェクトの期待価値を著しく低下させ、私たちの収益力を低下させる可能性がある。
米国食品医薬品局、米国証券取引委員会、その他の政府機関の資金不足は、重要な指導部や他の人員の採用と維持の能力を阻害し、新製品やサービスの適時な開発や商業化を阻止するか、あるいはこれらの機関が私たちの業務運営に依存する可能性のある正常な業務機能を履行することを阻止する可能性があり、これは私たちの業務に負の影響を与える可能性がある。
FDAが新製品を審査·承認する能力は、政府予算と資金レベル、肝心な人員の雇用と維持及びユーザー費用の支払いを受ける能力、及び法律、法規と政策の変化を含む様々な要素の影響を受ける可能性がある。したがって、その機関の平均検討時間は近年変動している。また、政府が米国証券取引委員会や我々の業務に依存する可能性のある他の政府機関に提供する資金は、研究開発活動に資金を提供する機関を含め、政治プロセスの影響を受けており、政治プロセス自体が不安定で予測不可能である。
FDAや他の機関の中断も、新製品が必要な政府機関によって審査および/または承認されるのに要する時間を遅らせる可能性があり、これは私たちの業務に悪影響を及ぼすだろう。例えばここ数年で
68
米国政府は何度か閉鎖されており、食品·医薬品局や米国証券取引委員会などのいくつかの規制機関は、肝心な従業員を休暇にし、重要な活動を停止させなければならない。
政府が長期的に停止している場合、または世界的な健康問題がFDAや他の規制機関の定期検査、審査、または他の規制活動を阻害した場合、FDAが提出した規制文書を適時に審査して処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に大きな悪影響を及ぼす可能性がある。また、将来的に政府の閉店は、適切な資本化と事業継続のために、私たちが公開市場に進出し、必要な資本を得る能力に影響を与える可能性がある。
EUの薬品マーケティングと清算法規は私たちのヨーロッパ加盟国の製品マーケティングと保証能力に重大な影響を与えるかもしれない。
私たちはアメリカと選定された外国司法管轄区で市場反応の承認を求めるつもりだ。もし私たちが1つ以上の外国の管轄区域で承認されたら、私たちはこのような管轄区域の規制によって制限されるだろう。一部の外国国、特にEU諸国では、薬品や細胞療法の定価は政府規制や他の市場によって規制されており、これはREPACTの価格設定や使用に圧力をかける可能性がある。これらの国では、候補製品の市場承認を得た後、政府当局との定価交渉にかなりの時間がかかる可能性がある。また,REACTに対する市場の受け入れや販売は,REACTが十分な保証範囲と第三者支払者の補償を持っているかどうかに大きく依存し,既存や将来の医療改革措置の影響を受ける可能性がある。また,精算に関する国際規制構造は不確定であり,REPACTを商業化できるまで発展していく可能性がある。
米国の連邦AKS禁止令のように、医師に福祉や利点を提供し、処方、推薦、裏書き、購入、供給、注文または医療製品の使用を誘導または奨励することは、EUでも禁止されている。医師への福祉や利益の提供は,EU加盟国の国家収賄法およびイギリス(EU加盟国ではなく)の“2010年英国反収賄法”によって管轄されている。このような法律に違反することは巨額の罰金と監禁につながるかもしれない。
いくつかの連合会員国で医者に支払われた費用は公開されなければならない。また、医師との合意は、通常、医師の雇用主、医師の主管専門組織、および/またはEU加盟国の監督当局に事前に通知し、承認しなければならない。これらの要件は、EU加盟国に適用される国家法律、業界規則、または専門行為規則に規定されている。このような要求を守らないことは、名声のリスク、公開非難、行政処罰、罰金、または監禁につながる可能性がある。
また、ヨーロッパ薬品管理局を含む大多数の外国国では、薬品の提案定価は承認されなければ、合法的に発売されない。各国の薬品定価と精算に対する要求は大きく異なる。例えば、欧州連合は、その国の健康保険制度が補償を提供する医療製品の範囲を制限し、人が使用する医療製品の価格を制御するために、その加盟国に様々な選択を提供している。EUの各加盟国が採用している参考定価と平行分配、あるいは低価格と高価な加盟国の間で裁定を行うことで、価格をさらに下げることができる。加盟国は医薬製品の具体的な価格を承認することができ、医薬製品を市場に投入する会社の収益力に対して直接或いは間接的に制御制度をとることもできる。ある国では、私たちは臨床研究や他の研究を行う必要があるかもしれません。私たちの任意の候補製品の費用効果を他の利用可能な治療法と比較して、精算または価格設定の承認を得たり維持したりする必要があります。しかも、このような規制は変化しており、私たちが商業的に反応できる前に変化するかもしれない。生物製薬製品に対して価格制御や精算制限を実施することを保証できない国は、私たちのいかなる製品に対しても有利な精算と定価手配を許可する。歴史的に見ると、EUで発売された製品はアメリカの価格構造に従わず、通常価格ははるかに低くなることが多い。第三者支払者や主管当局が割引を公表することは、公布国や他の国の価格や補償レベルにさらなる圧力を与える可能性がある。もし価格設定が満足できないレベルに設定されている場合、あるいは私たちの製品が精算できない場合、範囲や金額が限られていれば、私たちの販売収入およびこれらの国/地域における私たちの候補製品の潜在的な収益力はマイナスの影響を受けるだろう。
私たちは、変化するグローバルデータ保護法律や法規を遵守しようと努力する過程で巨額のコストが生じる可能性があり、私たちがこのような法律や法規を遵守できないことは、私たちの業務と運営を損なう可能性があると考えられています。
全世界のデータ保護構造は急速に変化しており、私たちは多くの連邦、州と外国の法律法規、および規制ガイドラインの制約または影響を受ける可能性があり、これらの法律法規と法規は個人データの収集、使用、開示、転送、安全と処理を管理しており、例えば、私たちが収集した臨床試験に関連する被験者および医療保健提供者の情報を管理している。予測可能な未来には、基準および法執行慣行を実施することはまだ不確定である可能性があり、これは、私たちの業務に不確実性をもたらし、私たちまたは私たちのサービスプロバイダがいくつかの司法管轄区域で個人データを収集、保存、移転、使用、および共有する能力に影響を与え、私たちが責任を負うか、または追加のコンプライアンスまたは他のコストを適用することをもたらすかもしれない。どんなものでも
69
私たちは連邦、州、または外国の法律または自律基準を遵守できなかったか、または遵守できなかったと考えられ、否定的な宣伝、管理時間と精力の移転、および政府の実体または他の人が私たちに訴訟を提起する可能性がある。
アメリカでは連邦健康情報プライバシー法を含む多くの連邦や州の法律があります例えば:HIPAA、HITECHで改正された州データ漏洩通知法、州健康情報プライバシー法、連邦と州消費者保護法(例えば:連邦貿易委員会法第5条)によれば、健康に関する個人情報及びその他の個人情報の収集、使用、開示及び保護は、我々の業務又は我々の協力者の業務に適用される可能性がある。さらに、HIPAA(HITECH改訂)や他のプライバシーおよびデータセキュリティ法のプライバシーおよびセキュリティ要件によって制限される第三者(臨床試験データを取得する研究機関を含む)から健康情報を取得することができる。事実および状況によると、もし私たちがHIPAAによってカバーされているエンティティによって維持されている保護された健康情報を故意に取得、または許可されていない方法で取得、使用、または開示する場合、私たちは刑事罰を受ける可能性がある。しかし,保護された健康情報が適用されたプライバシー基準や我々の契約義務に従って処理されているかどうかを決定することは複雑であり,変化する解釈の影響を受ける可能性がある.
もし私たちが保護された健康情報や私たちが持っている他の個人、敏感または機密情報のプライバシーおよび安全を適切に保護できなければ、私たちは私たちの契約に違反していることが発見されるかもしれない。また、適用されるHIPAAプライバシーおよびセキュリティ基準を含む適用されるプライバシー法を遵守しなければ、重大な行政、民事、刑事罰に直面する可能性があります。法執行活動はまた財務的責任と名声の損害を招く可能性があり、このような法執行活動に対する反応は大量の内部資源と外部資源を消費する可能性がある。また、州総検察長は民事訴訟を起こし、禁止令や損害賠償を求め、州住民のプライバシーを脅かす侵害行為に応える権利がある。法執行活動や潜在的な契約責任に関連するリスクに加えて、私たちは連邦や州レベルで変化する法律と法規を遵守するために努力している可能性が高く、私たちの政策、手続き、システムを絶えず修正する必要があります。
多くの州の法律は,特定の場合に個人情報やデータのプライバシーやセキュリティを管理しており,その多くの法律は互いに大きく異なり,HIPAAに先制されておらず,HIPAAよりも禁止的な効果があり,コンプライアンス作業を複雑化している可能性がある.例えば、2020年1月に発効したCCPAは、消費者に新たなデータプライバシー権を提供し、会社に新たな運営要件を提供し、コンプライアンスコストや潜在的な責任を増加させる可能性がある。CCPAはカリフォルニア住民により大きな権利を与え,彼らの個人情報にアクセスして削除し,特定の個人情報を共有しないことを選択し,彼らの個人情報がどのように使用されるかに関する詳細な情報を受信することができる.CCPAは違反行為に対する民事処罰と,データ漏洩に対する個人訴権を規定しており,データ漏洩訴訟が増加すると予想される。現在は健康を守る例外がありますがHIPAAと臨床試験法規の制約を受けた情報は、現在の書面規定により、CCPAは著者らのいくつかの業務活動に影響を与える可能性がある。また、最近、消費者権益保護法の内容を強化するために“カリフォルニア消費者権利法案”(以下、“CPRA”)が公布され、2023年1月1日に施行された。他のいくつかの州も同様の包括的プライバシー法を公布したり、類似したプライバシー提案を考慮したりしている。コロラド州プライバシー法案、コネチカット州個人データプライバシー、オンライン監視法案、ユタ州消費者プライバシー法案、バージニア州消費者データ保護法が2023年に施行されました。モンタナ州、オレゴン州、テキサス州のプライバシー法は2024年に施行される。また,米国の他州の法律は2024年後に施行され,米国の他州の提案も考慮されている。これらのプライバシー法は,我々のビジネス活動に影響を与える可能性があり,我々の業務が変化する個人データに関する規制環境における脆弱性を示している.
私たちのアメリカでの業務は、医療や他の健康情報や他の個人情報のプライバシーや安全に関する法律の制約を受ける可能性があるほか、私たちは行っています。そして、私たちは将来EEAで臨床試験を行い、追加のヨーロッパデータプライバシー法律、法規、ガイドラインの制約を受ける可能性があります。一般データ保護条例は、(EU)2016/679(“GDPR”)が2018年5月25日に施行され、個人健康データを含む欧州経済地域個人に関する個人データの収集、使用、記憶、開示、移転、または他の処理に関する。GDPRは,GDPRに拘束されている会社に対して,個人を識別可能な個人情報を処理し,このような情報を欧州経済地域以外に移行させる米国を含め,これらの個人にその個人の健康や他の敏感なデータを処理する詳細を提供し,個人データに関する個人の同意を得,個人情報の安全を確保し,個人情報を処理する第三者とデータ処理プロトコルを締結し,個人がその個人情報に対して権利を行使する要求に応じ,担当国データ保護当局や影響を受けた個人報告に個人データに関するセキュリティ違反を報告し,データ保護官を任命することを含む広範な厳しい要求を行っている。データ保護影響評価を行い,記録を保存する.GDPRは、いくつかの比較的軽い犯罪に対して、最高1000万ユーロまたは世界の年商の2%の罰金を科すことができ、深刻な犯罪に対しては、最高2000万ユーロまたは私たちの世界の年商の4%に達することを含む、規定を守らずに受ける可能性のある罰を大幅に向上させます。金額が大きい者を基準にしてください。GDPRはまた、データ主体と消費者協会に個人訴訟権利を与え、監督当局に苦情を提出し、司法救済を求め、GDPR違反による損害について賠償を受けることができる。さらに、GDPRは、国境を越えたデータ転送の制限を含む。
また,欧州連合加盟国の国家法律はGDPRの要求に応じて調整されており,GDPRから部分的に逸脱する可能性のある国家法が施行され,国によって異なる義務が課せられているため,欧州経済圏は統一された法的環境では動作しないと予想される。また、処理と転送に関連しているため
70
遺伝子データについては,GDPRは国家法律がより多く,より具体的な要求や制限を加えることを明確に許可しているが,ヨーロッパの法律はこの分野で従来大きく異なり,追加的な不確実性を招いている。
GDPRはGDPRが管轄する個人データをいわゆる第三国に移行する問題も規定しており,これらの国は欧州委員会に適切なデータ保護を提供していると認定されていない。ヨーロッパの法律開発はこのような譲渡の複雑さと不確実性をもたらした。たとえば,2020年7月16日,EU裁判所(略称CJEU)は,いわゆるSchrems II判決により,EU-米国プライバシー盾フレームやプライバシー盾が無効であることを宣言し,この判決により,個人データは欧州経済区からプライバシー盾計画に基づいて自己認証を行う米国エンティティに移行することができる.しかし、2023年7月10日、欧州委員会は、そのデータにアクセスし、不正確または不正に処理されたデータを修正または削除する権利を含むいくつかの新しい権利をEU個人に提供し、EU個人データを自由に受信するために、EU個人データを自由に受信するために、EUから米国にデータを転送する新しいメカニズムであるEU-米国データプライバシーの枠組みに関する十分な決定を採択した。この十分な決定は、Schrems II判決で提起された問題を解決するために、新たな拘束力のある保障措置を導入した行政命令に署名した後に行われる。欧州委員会は十分な決定を下しながら、米国の事態を検討していくだろう。
英国のEU離脱(すなわち英国の離脱)と、2020年12月31日に終了した英国の離脱移行期間の終了に伴い、EU GDPRはすでに英国(すなわち英国GDPR)で実施されている。英国GDPRは2018年の英国データ保護法と並んで、EU GDPRのいくつかの減税を英国法に盛り込んでいる。イギリスGDPRによると、イギリスに設立されていないが、イギリス個人への商品やサービスの提供に関する個人データを処理したり、その行動を監視したりする会社は、イギリスGDPRの制約を受ける-その要求(現在)はEU GDPRでの要求とほぼ一致するため、類似のコンプライアンスおよび運営コストを引き起こす可能性があり、潜在的な罰金は1,750万ポンドまたは世界売上高の4%に達する。2021年6月、欧州委員会は、EU GDPRによるEUから英国への個人データの十分な保護を確保するために、2025年6月27日に日没する決定を発表した。事態が適用法域の保護レベルに影響すれば,十分な決定を調整あるいは撤回することができる.また、イギリス議会は現在、2018年データ保護法、イギリスGDPR、プライバシー、電子通信法規を1つの立法枠組みで統一するためのデータ保護とデジタル情報法案を審議している。
我々はEEAで臨床試験を行っており,GDPRはGDPRに拘束された個人データの処理における責任と責任を増加させ,個別の国で実施されているメカニズムや保障措置を含めてGDPRの遵守を確保するための追加的なメカニズムや保障措置を確立する必要がある。GDPRを遵守することは厳格で時間のかかるプロセスであり、これは私たちの業務コストを増加させ、あるいは私たちに業務やり方を変更することを要求し、私たちはこれらの努力をしたにもかかわらず、私たちは私たちのヨーロッパ活動に関連した罰金と処罰、訴訟、名声損害のリスクに直面する可能性がある。私たちは、私たちが欧州プライバシー法で規定されているいかなる義務を遵守する努力が十分かどうかという不確実性に直面し続けると予想する。もし私たちがヨーロッパデータ保護機関の調査を受けたら、私たちは罰金と他の処罰に直面するかもしれない。欧州データ保護当局のこのような調査または告発は、私たちの既存の業務および新しい顧客または生物製薬パートナーを引き付け、維持する能力にマイナスの影響を与える可能性がある。いくつかのデータ保護機関が現行法(GDPRを含む)を解釈する際に適用される現在(特に未来)のデータ保護義務がリスクをもたらす可能性があるので、私たちはまた、ヨーロッパまたは多国籍サプライヤーまたはバイオ製薬パートナーが私たちの製品を使用し続ける際の躊躇、不本意または拒否に遭遇する可能性がある。このようなサプライヤーやバイオ製薬パートナーも他のコンプライアンス方法が高すぎると思うかもしれません負担が重く、法的に不確実や反感があるので、私たちと商売をしないことにしました。上記のいずれも、我々の業務、見通し、財務状況、および経営結果に実質的な損害を与える可能性がある。
私たちの国際業務に関連する法律、政治、経済的不確実性は、私たちの業務に負の影響や制限を与える可能性があります。
2016年の国民投票結果に続き、英国の離脱は2020年1月31日に発効した。英国とEUが合意した正式な離脱手配によると、2021年1月1日から、英国はEU規則の継続適用の過渡期(過渡期)に支配されなくなった。過渡期終了後、連合王国と欧州連合は連合王国と欧州連合との税関や貿易関係について交渉を継続する予定だ。
イギリスの規制枠組みの大部分は私たちの業務に適用されているが、私たちの主要な候補製品REACTはEUの指令と法規から来ているため、過渡期が過ぎた後、イギリスの離脱はイギリスまたはEUのREACTの開発、製造、輸入、承認、商業化に関する規制制度に大きな影響を与える可能性がある。例えば、イギリスの離脱をめぐる不確実性のため、EMAはロンドンからアムステルダムに移った。移行期間後、イギリスはEMAからEU範囲内のマーケティング許可を取得する集中プログラムによってカバーされなくなり、具体的な合意が達成されない限り、イギリスは別個のハニカム製品許可プログラムを必要とし、その潜在的な手続きは現在のところ不明である。イギリスの離脱や
71
そうでなければ、私たちがイギリスやEUで反応することを阻止し、私たちが収入を創出し、利益を達成し、維持することを制限するだろう。さらに、私たちは税金や関税を支払うことを要求されたり、EUの輸入反応に関連する他の障害を受けたり、EUに製造工場を設立することで費用が発生して、これらの障害物を迂回することができるかもしれない。上記のいずれかの結果が生じた場合、私たちは、イギリスまたはEUで規制承認を求めて反応する努力を制限または延期することを余儀なくされるか、または私たちの業務を運営するための重大な追加費用を生成することができ、これは、私たちの収入を生成したり、事業の利益を達成する能力に重大で実質的な損害または遅延をもたらす可能性がある。イギリスの離脱やその他の理由で、国際貿易、関税、輸出入法規の任意のさらなる変化は、予期しない関税コストまたは他の非関税障壁をもたらすかもしれない。これらの事態が発展したり、そのいずれかが起こりうるとの見方は、世界貿易、特に影響を受けている国と連合王国との貿易を大幅に減少させる可能性がある。イギリスの離脱は、特にEUから来た従業員たちのために、私たちが従業員を引き付け、維持する能力にマイナスの影響を与える可能性もある。
さらに、私たちは私たちがその中で運営することを計画している各司法管轄区域の多くの法律と法規を遵守するためにもっと多くの資源を投入しなければならない。“海外腐敗防止法”は、いかなる米国人または商業実体も、任意の外国人官僚、政党または候補者に直接的または間接的に支払い、提供、許可支払い、または任意の価値のあるものを提供することを禁止し、その個人または企業が業務を獲得または保持することを支援するために、外国の実体の任意の行為または決定に影響を与えることを目的としている。
“反海外腐敗法”を遵守することは高価で困難であり、特に腐敗は公認問題である国である。また、“海外腐敗防止法”は、多くの国で病院が政府によって運営されているため、医師や他の病院従業員が外国人官僚とされているため、バイオ製薬業界に特別な挑戦をもたらしている。臨床試験やその他の仕事に関連した病院や衛生保健提供者への何らかの支払いは,政府関係者への不正金と考えられ,“海外腐敗防止法”の法執行行動につながっている。
様々な法律、法規、および行政命令はまた、国家安全目的のために秘密にされている情報製品およびこれらの製品に関連するいくつかの製品、技術および技術データの米国国外での使用および伝播、またはいくつかの非米国国民との共有を制限する。私たちが拡大するにつれて世界的に業務を展開するために、これらの法律を遵守するために追加の資源を投入することが要求されます。これらの法律は、米国以外の地域で特定の製品や候補製品の開発、製造、または販売を阻止する可能性があり、これは私たちの成長潜在力を制限し、私たちの開発コストを増加させるかもしれません。
国際ビジネス慣行に関する法律を遵守しなければ、重大な民事·刑事罰を受け、政府契約の資格を一時停止または廃止する可能性がある。米国証券取引委員会は、発行者が海外腐敗防止法の会計規定に違反したため、発行者の米国取引所での証券取引を一時停止または禁止する可能性もある。
私たちはアメリカと外国のいくつかの反腐敗、反マネーロンダリング、輸出規制、制裁、その他の貿易法律と法規の制約を受けている。私たちは違反によって深刻な結果に直面するかもしれない。
その他の事項を除いて、米国および外国の反腐敗、反マネーロンダリング、輸出規制、制裁およびその他の貿易法律および法規(総称して“貿易法”と総称される)は、会社およびその従業員、代理人、臨床研究組織、法律顧問、会計士、コンサルタント、請負業者および他のパートナーの許可、約束、提供、提供、誘致、直接または間接的に公共または民間部門の受取人の腐敗または不当な支払い、または任意の他の価値のあるものを受け入れることを禁止する。貿易法違反は、大量の刑事罰金と民事処罰、監禁、貿易特権の喪失、資格取り消し、税収の再評価、契約違反と詐欺訴訟、名声損害、その他の結果を招く可能性がある。私たちは政府機関や政府付属病院、大学、その他の組織の役人や従業員と直接または間接的な相互作用を持っている。我々は、第三者を招いて臨床試験および/または必要な許可、許可、特許登録、および他の規制承認を得ることを計画しており、私たちは、このような活動を明確に許可していなくても、事前にそのような活動を知っていても、私たちの人員、代理またはパートナーの腐敗または他の不正活動に責任を負わなければならないかもしれない。
私たちの役員、役員、証券所有者およびその関連会社には、私たちの利益と衝突する競争的な金銭的利益が存在する可能性があります。
吾等は政策を制定しておらず、吾等の役員、行政者、証券所持者又は共同経営会社が吾等が買収又は処分する任意の投資において、又は吾等が参加又は権益を有するいかなる取引においても、直接又は間接的な金銭又は財務利益を有することを明文禁止している。私たちはまたこのような人々が私たちが行っている商業活動に自ら従事することを禁止する政策明文を持っていない。したがって、このような個人や実体は彼らの利益と私たちの利益の間で衝突するかもしれない。
改正及び重述された第二の組織定款の大綱及び細則(“定款”)は、吾等は、法律で許容される最大範囲内で、当社従業員でない任意の取締役に提供される任意の会社の機会における権益を放棄し、又はいずれかの取締役が関係会社の機会を知ることができ、その機会が明確に当該人のみが当社の取締役メンバーとして当該人に提供されなければならず、かつその機会は法律及び契約が私たちに享受することを許可するものである
72
約束しなければ、私たちには合理的だ。また、私たちの憲章には、法律で許容される最大範囲で、その人たちがいかなるビジネス機会やそのようなビジネス機会を提供できないことを認識しているため、当社に生じる可能性のある任意の責任、義務または責任について、そのような人々を免除し、保障する条項が掲載されている。
ケイマン諸島法律によると、私たちの役員と上級管理者の個人と財務的利益は利益衝突を招く可能性があり、彼らが私たちに対する受託責任に違反する可能性があり、私たちまたは私たちの株主は私たちの株主の権利を侵害してそのような個人にクレームを出す可能性があります。しかし、私たちは最終的にそのような理由で彼らに何のクレームもしないかもしれない。
私たちはケイマン諸島の法律登録によって成立したので、あなたはあなたの利益を保護する上で困難に直面するかもしれません。あなたがアメリカ連邦裁判所を通じてあなたの権利を保護する能力は制限されるかもしれません。
私たちはケイマン諸島の法律登録に基づいて設立された免除会社だ。したがって,投資家は米国内で我々の役員や幹部に法的手続き文書を送達したり,米国裁判所で得られた我々の役員や幹部に対する判決を実行することは困難である可能性がある.
私たちの会社の事務は私たちの憲章、ケイマン諸島会社法、そしてケイマン諸島一般法によって管轄されている。私たちはまたアメリカ連邦証券法の制約を受けている。ケイマン諸島法律によると、株主が役員を提訴する権利、少数株主の訴訟、および取締役の私たちに対する受託責任はケイマン諸島普通法によって大きく管轄されている。ケイマン諸島の一般法の一部は,ケイマン諸島の比較的限られた司法判例およびイギリス一般法に由来しており,その裁判所の裁決には説得力があるが,ケイマン諸島の裁判所には拘束力がない。私たちの株主の権利とケイマン諸島の法律の下での私たちの役員の受託責任はアメリカのある司法管轄区域の法規や司法前例によって規定されているのとは違います。特に、ケイマン諸島は米国とは異なる会社法や証券法を有しており、ある州、例えばデラウェア州は、より完全かつ司法解釈の会社法機関を持っている可能性がある。また、ケイマン諸島会社は米連邦裁判所で株主派生訴訟を起こす資格がないかもしれない。
私たちのケイマン諸島法律顧問は、ケイマン諸島の裁判所が(I)米国または任意の州連邦証券法の民事責任条項に基づく米国の裁判所の判決を認めたり実行したりすることは不可能であり、(Ii)ケイマン諸島で提起された原告訴訟では、米国または任意の州連邦証券法の民事責任条項に基づいて、そのような条項が適用される責任が刑事責任である限り、私たちに法的責任を課すことを教えてくれる。この場合、ケイマン諸島は米国で取得した判決を法的に強制執行していないにもかかわらず、ケイマン諸島裁判所は管轄権を有する外国裁判所の外国資金判決を認め、執行する。その根拠は、外国主管裁判所の判決規定は、ある条件を満たせば、債務者が判決を下した金を支払う義務があると判定することである。ケイマン諸島で外国判決を執行するためには、このような判決は最終的かつ決定的でなければならず、かつ弁済された金額でなければならず、税収、罰金または処罰に触れてはならず、ケイマン諸島の同一事項に関する判決と一致してはならず、詐欺を理由に弾劾されてはならず、何らかの方法で得られてはならないし、自然正義またはケイマン諸島公共政策に違反する強制執行タイプに属してはならない(懲罰的または多重損害賠償の裁決は公共政策違反と判断される可能性が高い)。もし同時に他の場所で訴訟が提起されれば、ケイマン諸島裁判所は実行手続きを保留することができる。
このような理由から、株主は、経営陣、取締役会メンバー、あるいは持株株主の行動に直面した場合、米国会社の株主としてよりも自分の利益を保護することが困難になる可能性がある。
私たちの知的財産権に関するリスクは
私たちの成功は私たちの知的財産権を保護する能力にある程度かかっている。私たちの固有の権利と技術を保護することは難しくて高価で、私たちはそれらの保護を保障できないかもしれない。
私たちの業務は、私たちのノウハウに対する特許、商標および商業秘密保護、ならびに私たちの主要な候補製品、それらのそれぞれの構成要素、合成中間体、製剤、共同療法、それらを製造するための方法および治療方法の反応を取得し、維持し、これらの特許を第三者の挑戦から保護することに成功することに大きく依存するであろう。許可されていない第三者の製造、使用、販売、販売または輸入反応を阻止する能力は、これらの活動をカバーする有効かつ実行可能な特許の下で当社が所有する権利の程度に依存します裁判所は強制救済措置を発表するだろう。もし私たちが開発したいかなる製品や技術のために特許保護を獲得して維持することができない場合、あるいは獲得した特許保護範囲が十分でなければ、私たちの競争相手は私たちと類似または同じ製品や技術を開発し、商業化する可能性があり、私たちが開発する可能性のある任意の候補製品の商業化の能力は不利な影響を受ける可能性がある。
73
特許出願プロセスは高価で時間がかかり、私たちはすべての必要または望ましい特許出願を合理的なコストまたはタイムリーに提出して起訴することができないかもしれない。特許を申請する過程には多くのリスクがあり、私たちが私たちが申請した特許を成功的に獲得することは保証されない。しかも、私たちはすべての関連市場で特許保護を追求、獲得、または維持しないかもしれない。我々は,特許保護を得るのが遅くなるまで,我々の研究開発成果で特許を申請できることを確認できない可能性もある.さらに、場合によっては、私たちは、第三者から許可を得て、第三者に与えられ、私たちのライセンシーまたはライセンシーの技術に依存することができるかもしれないことを含む、特許出願の準備、提出および起訴を制御する権利がないかもしれない。
バイオテクノロジーやバイオ製薬分野の特許の強度は複雑な法律や科学的問題に関連しており,不確実である可能性がある。私たちの所有または許可中の特許出願は、米国または他の国/地域におけるそれの反応または使用をカバーする発行された特許を生成することができない可能性がある。特許が確実に発行されたとしても、第三者はその有効性、実行可能性、または範囲に疑問を提起する可能性があり、これは、このような特許が縮小され、無効にされ、または強制的に実行されない可能性がある。さらに、それらが挑戦されていなくても、私たちの特許および特許出願は、私たちの特許における特許要件をめぐる他の人の設計を反応または阻止することを含む、私たちの技術を十分に保護することができないかもしれない。もし私たちが持っている特許出願が提供する保護の広さや強度が脅かされていれば、会社が私たちと協力して開発することを阻止し、私たちの商業化と反応の能力を脅かすかもしれない。また,臨床試験で遅延に遭遇すれば,特許保護下で市場反応が可能な時間帯が短縮される。
私たちがReactionを含む私たちの技術に関連する特許出願を初めて提出した会社であることは確認できません。もし私たちがそうでなければ、私たちはReactionを含む私たちの技術のための特許保護から除外される可能性があります。
私たちは私たちが最初の発明未解決特許出願でカバーされた発明者であることを確認することができません。もし私たちがそうでなければ、私たちは優先権紛争の影響を受けるかもしれません。さらに、すべての特許請求の範囲が2013年3月16日までに優先日を得る権利がある米国出願については、第三者が介入手続きを開始するか、または米国特許商標局によって訴訟を提起して、誰が我々が出願した特許請求の範囲に含まれる任意の主題を最初に発明したかを決定することができる。同様に、特許請求の範囲の少なくとも1つが2013年3月16日前に優先日を有する権利を有していない米国出願については、特許請求の範囲の主題が以前の発明者の開示から由来しているかどうかを決定するために、派生プログラムを提起することができる。
私たちは特定の特許の一部または全部の期間を放棄することを要求されるかもしれない。私たちが知らない従来技術は、特許または特許請求の範囲の有効性または実行可能性に影響を及ぼす可能性があるかもしれない。我々が知っているかもしれないが,クレームの有効性や実行可能性に影響を与えるとは考えられない既存技術があるが,最終的にはクレームの有効性や実行可能性に影響を与えることが発見される可能性がある。もし挑戦を受けた場合、私たちの特許は裁判所によって有効または強制執行可能であると宣言されるか、または有効かつ強制的に実行可能であることが発見されても、私たちの特許は私たちの技術または製品を十分に保護するか、または競争相手の技術または製品の侵害を受けると裁判所によって判断されることを保証することはできない。私たちの活動に関連していると考えられる競争相手の特許や特許出願を分析し、REPACTに関する操作を自由に行うことができると考えられるかもしれませんが、私たちの競争相手は、私たちが無関係だと思う特許を含む、発表されたクレームを実現する可能性があり、これは、私たちの努力を阻害したり、REPACTや私たちの活動がこのようなクレームを侵害したりする可能性があります。他社は、当社の特許または他の知的財産権を侵害しない独立した上で、当社の製品と同じ効果を有する製品を開発するか、または当社の製品をカバーする可能性のある特許の特許要件を中心に設計される可能性があります。
最近または将来の特許改革立法は、私たちの特許出願をめぐる起訴と、私たちが発表した特許の実行または保護の不確実性とコストを増加させるかもしれない。2013年3月以降に制定された“ライシー·スミス米国発明法”(略称“米国発明法”)によると、米国は先発明から先出願制度に転換した。“先提出書類”制度の下で、他の特許性要件が満たされていると仮定すると、最初に特許出願を提出した発明者は、通常、他の発明者がいるか否かにかかわらず、その発明の特許を取得する権利がある。米国発明法には、特許出願起訴方式に影響を与える条項を含む米国特許法の他のいくつかの重大な変化が含まれており、既存技術を再定義し、新たなライセンス後審査制度を確立する。また,裁判所はその多くの条項や“米国発明法”や本明細書で議論されている特定特許に関する新条例の適用性を扱っておらず,これらの問題は未確定であり,審査が必要である.しかしながら、米国発明法およびその実施は、私たちの特許出願をめぐる起訴および私たちが発行した特許の実行または保護の不確実性およびコストを増加させる可能性があり、これらすべては、私たちの業務および財務状況に実質的な悪影響を及ぼす可能性がある。
法的手段は限られた保護しか提供できず、私たちの権利を十分に保護できないか、あるいは私たちの競争優位性を獲得または維持することができるかもしれないので、未来の私たちの所有権の保護の程度は不確定だ。例えば:
|
|
|
他の会社はReaction成分と類似した化合物を製造または使用することができるかもしれないが、私たちの特許はこれらの化合物をカバーしていないかもしれない |
|
|
|
米国政府の資金を使用して発明または開発された任意の認可内の特許および特許適用については、私たちまたは私たちの許可者は、米国政府に対する義務を履行できず、特許権の損失を招く可能性がある |
|
|
|
私たちまたは私たちのライセンス者は、これらの発明のために特許出願を提出した最初の人ではないかもしれない |
|
|
|
他の会社は類似または代替技術を独立して開発したり、私たちの任意の技術をコピーしたりすることができる |
74
|
|
|
私たちが処理している特許出願は発行された特許を生成しない可能性がある |
|
|
|
私たちまたは私たちのライセンシーの特許(場合によっては)または私たちまたは彼らの一部の特許は、事前の開示によって無効になる可能性があります |
|
|
|
他の人たちは私たちが持っている特許や許可された特許を迂回するかもしれない |
|
|
|
未公表の出願または特許出願が秘密にされている可能性があり、今後、我々の製品または技術と同様の権利要件が提示される可能性がある |
|
|
|
外国の法律はアメリカの法律のように私たちまたは私たちのライセンシーの固有の権利を保護しないかもしれない |
|
|
|
私たちが所有しているまたは許可されていない特許または特許出願の請求項は、発行された場合、反応を含まない可能性がある |
|
|
|
私たちが所有しているまたは許可されていない特許は、競争優位性を提供してくれないかもしれないし、範囲を縮小したり、第三者の法的挑戦によって無効または実行不可能と認定されたりする可能性がある |
|
|
|
私たちが所有または許可している特許または特許出願の発明者は、競争相手と交渉し、私たちの特許を中心に設計された製品またはプロセスを開発するか、または私たちまたは彼らが発明者として指定された特許または特許出願に敵意を持っている可能性がある |
|
|
|
私たちの所有または許可中の特許または特許出願は、発明者として登録されるべき個人または発明者として登録されてはならない個人を見落としている可能性があり、これにより、これらの特許またはこれらの特許出願から発行された特許が無効または強制的に実行できないと認定される可能性がある |
|
|
|
私たちは過去に科学協力に参加して、未来もそうし続けるだろう。このような協力者は私たちと隣接したり競合したりする製品を開発するかもしれませんこれらの製品は私たちの特許の範囲内ではありません |
|
|
|
特許保護を受けることができる他のノウハウを開発することはできない |
|
|
|
私たちが開発した候補製品または診断テストは、第三者特許または他の独占的権利によって保護される可能性がある |
|
|
|
もし私たちが所有しているまたは許可中の任意の特許または出願がアメリカ政府の資金で行われている場合、アメリカ政府は、私たちまたは私たちの許可者に第三者に許可を与え、要求された発明を実践することを可能にするために、いくつかの入場権利を主張するかもしれない |
|
|
|
他の人たちの特許は私たちの業務に悪影響を及ぼすかもしれない。 |
私たちは未来に許可や他の協力協定を締結するかもしれないし、これらの協定は私たちにいくつかの義務を加えるかもしれない。第三者と締結されたこのような未来の合意に規定された義務を履行できなければ、将来の業務に非常に重要である可能性のある許可権を失う可能性があります。
私たちの候補製品チャネルを拡大するために努力するために、私たちは将来、他の候補製品に権利を付与するために、いくつかの許可または他の協力協定を締結するかもしれない。このような合意は、様々な職務調査、マイルストーン支払い、特許権使用料、保険、または他の義務を私たちに強要するかもしれない。もし私たちがこれらの義務を履行できなかった場合、私たちの許可者またはパートナーは、関連合意を終了する権利がある可能性があり、この場合、私たちは、そのような許可またはプロトコルがカバーする製品を開発またはマーケティングすることができません。
さらに、ライセンス契約によると、知的財産権に関する紛争が生じる可能性があります
|
|
|
ライセンス契約に従って付与された権利範囲および解釈に関連する他の問題; |
|
|
|
反応、私たちの主要候補製品または任意の他の候補製品の技術およびプロセスが、許可プロトコルに拘束されていない許可者の知的財産権をどの程度侵害しているか |
|
|
|
私たちの協力開発関係に基づいて、特許と他の権利を再許可する |
|
|
|
私たちのライセンス契約の下での義務と、どのような活動がこれらの職務義務を満たしていますか |
|
|
|
私たちのライセンス者、私たちおよびパートナーが知的財産権を共同で創造または使用することによって生成された発明およびノウハウの発明および所有権; |
|
|
|
特許技術発明の優先権。 |
さらに、どのプロトコルによって第三者の知的財産権または技術を許可するかは複雑であり、そのようなプロトコルのいくつかの条項は様々な解釈の影響を受ける可能性がある。可能性のある契約解釈の相違の解決は、関連する知的財産権または技術に対する私たちの権利と考えられる範囲を縮小するか、または関連協定の下での私たちの財務または他の義務を増加させる可能性があり、いずれも、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。さらに、我々が許可している知的財産権をめぐる紛争が、商業的に許容可能な条項で現在の許可手配の能力を維持していることを阻害または損害すれば、影響を受けた候補製品の開発および商業化に成功することができない可能性があり、これは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
さらに、私たちは、これらの許可内の特許および特許出願の維持および起訴、または私たちの許可内の知的財産権に関連する任意の他の知的財産権の維持および起訴について限られた支配権を有する可能性がある財産です。例えば、私たちはどんな未来の許可者のこのような活動がすでに適用されたか、または適用される法律を遵守するかどうかを決定することはできない
75
規制または効果的で強制的に施行可能な特許と他の知的財産権をもたらすだろう。私たちは、ライセンシーが知的財産権の第三者侵害者に対して侵害訴訟を提起したり、私たちに許可されたいくつかの知的財産権を弁護する方法に限られた支配権を持っています。ライセンシーの権利侵害訴訟や弁護活動は私たち自身が行うほど激しくないかもしれない。
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの技術的価値は否定的な影響を受ける可能性があり、私たちの商業と競争地位は損なわれるだろう。
特許保護に加えて、私たちは、特に特許保護が不適切であると考えられている場合に、当社の機密および独自の情報を保護するために、当社の従業員、コンサルタント、および第三者と締結された秘密協定および発明譲渡協定に深刻に依存している。契約措置に加えて、私たちは物理的および技術的セキュリティ措置を使用して、私たちの固有の情報の機密性を保護しようと努力している。例えば、従業員またはアクセス許可権限を有する第三者が商業秘密を盗用する場合、このような措置は、私たちの独自の情報に十分な保護を提供しない可能性がある。私たちの安全措置は、従業員やコンサルタントが私たちのビジネス秘密を盗用して競争相手に提供することを阻止できないかもしれませんが、このような不正行為に対する追跡権は、私たちの利益を十分に保護するための十分な救済措置を提供できないかもしれません。強制執行側が商業秘密を不正に開示したり流用したりする主張は困難であり、高価で時間がかかる可能性があり、結果は予測できない。また,ビジネス秘密は,我々の法的追跡を阻止するように他の人によって独立して開発されている可能性がある.1つまたは複数の第三者がこれらの技術を取得または他の方法で複製することができる場合、我々の臨床開発戦略の重要な機能および差別化は、潜在的な競争相手に利用可能である。もし私たちのビジネス秘密のような私たちの任意の機密または独自の情報が漏洩または流用された場合、またはそのような情報が競争相手によって独立して開発された場合、私たちの競争地位は損なわれる可能性がある。
しかも、アメリカ以外の裁判所は商業機密を保護することをあまり望まないこともある。もし私たちが第三者が私たちのどんな商業秘密を使用するのを阻止するために法廷に訴えることを選択すれば、私たちは巨額の費用を生むかもしれない。私たちが勝訴しても、このような訴訟は私たちの時間と他の資源を消費するかもしれない。私たちは、当社の従業員やコンサルタントと契約を締結することを含む、当社の独自情報およびビジネス秘密を保護する措置をとっていますが、第三者は、実質的に同じ独自の情報および技術を独立して開発したり、他の方法で私たちのビジネス秘密を取得したり、当社の技術を開示したりすることができます。
したがって、私たちは私たちの商業秘密を意味的に保護することができないかもしれない。私たちの政策は、私たちの従業員、コンサルタント、外部科学協力者、協賛研究者、および他のコンサルタントに、私たちとの雇用や相談関係を開始する際に秘密協定を実行することを要求します。これらの合意は、当方との関係過程において、関係個人又は実体に開示されたすべての当方の業務又は財務に関連する機密情報を秘密にしなければならず、特定の場合を除き、第三者に開示してはならないと規定している。従業員の場合、合意は、個人的に構想された、私たちの現在または計画中の業務または研究開発に関連するもの、または通常の勤務時間内、私たちのオフィス内で行われる、または私たちの設備または独自の情報を使用するすべての発明が、私たちの固有財産であることを規定している。また、第三者が私たちのノウハウを盗用することを防止するために、物理的および技術的セキュリティ対策のような他の適切な予防措置を講じている。また,政策を講じて訓練を行い,ビジネス秘密保護への期待を指導し,ベストプラクティスについて提案する予定である。
第三者の知的財産権侵害に対するクレームはコストが高く、時間がかかる可能性があり、私たちの製品の発見、開発と商業化努力を阻害または延期する可能性がある。
私たちのビジネス成功は、私たちの主要候補製品Reactionを開発、製造、マーケティング、販売する能力にある程度依存し、Thirdの独自の権利を侵害することなく、私たちの独自技術を使用しますパーティーです。バイオテクノロジーやバイオ製薬業界では、特許や他の知的財産権に関する訴訟や、干渉、派生、派生を含む特許に挑戦する行政訴訟が数多くある各方面間米国特許商標局の再審、付与後再審及び再審手続、又は外国司法管轄区の異議及びその他の類似手続。私たちは、反応および/またはノウハウが彼らの知的財産権を侵害していると主張する特許または他の知的財産権を持つ第三者の将来の訴訟の脅威に直面するか、または受ける可能性がある。我々がREPACTを開発している分野には,第三者が所有する米国や外国から発行された特許や係属中の特許出願が多く存在する.バイオテクノロジーやバイオ製薬業界の拡張や特許の発行に伴い,このような反応のリスクが増加し,他人の特許権侵害のクレームを引き起こす可能性がある。
2020年12月に公布された“紫書継続性法案”第2章325条は、承認された生物製品データベースであるいくつかの特許情報を“紫書”に公開するようFDAに初めて指示した。具体的には、参照製品スポンサー(“RPS”)は、公衆衛生サービス法第351(L)(3)(A)または(L)(7)条に従ってRPSが351(K)出願人に初期リストを提供してから30日以内に、特許リストおよび対応する満了日(ここでは“初期リスト”と呼ぶ)をFDAに提供しなければならない。したがって,RPSは,後続の開発者や生物類似体と特許舞踊を行った後にのみ,紫書に登録するためにFDAにその特許情報を提供することができる.常にはっきりしているわけではありません
76
業界参加者は、我々を含み、特許は、様々なタイプの医薬品、製品またはその使用または製造方法、特に製品発見および開発の初期段階をカバーする。したがって,我々の分野では大量の特許が発行され,特許出願が提出されているため,第三者が我々の候補製品,技術または方法を含む特許権を持っていると主張するリスクがある可能性がある.
もし第三者が私たちが知的財産権を侵害していると主張すれば、私たちはいくつかの問題に直面するかもしれませんが、これらに限定されません
|
|
|
権利侵害と他の知的財産権のクレームは、事件の状況にかかわらず、訴訟を提起するのは高価で時間がかかる可能性があり、そして私たちの管理層の核心業務に対する注意力を移す可能性がある |
|
|
|
権利侵害の実質的な損害賠償は、もし裁判所が紛争製品または技術侵害または第三者の権利に違反すると判断した場合、私たちは支払わなければならないかもしれない。もし裁判所が侵害が故意であることを発見した場合、私たちは3倍の損害賠償金と特許権者の弁護士費の支払いを命じられる可能性がある |
|
|
|
裁判所は、第三者がその製品の権利を私たちに許可しない限り、私たちの開発、製造、マーケティング、またはREPACTの販売を禁止し、そうする必要はありません |
|
|
|
第三者から許可を得る場合、私たちは大量の使用料、前払い費用、および/または私たちの製品の知的財産権に交差許可を付与する必要があり、任意の利用可能な許可は非排他的である可能性があり、これは私たちの競争相手が同じ知的財産権を獲得する可能性があります |
|
|
|
それらが侵害されないように反応またはプロセスを再設計することは不可能かもしれないし、大量のお金の支出と時間を必要とする可能性がある。 |
また、公聴会、動議または他の一時的な手続きまたは事態の発展の結果が公表される可能性があり、証券アナリストや投資家がこれらの結果がマイナスであると考える場合、私たちの証券価格に大きな悪影響を及ぼす可能性がある。このような種類の訴訟や訴訟は、私たちの運営損失を大幅に増加させ、開発活動に利用できる資源を減少させる可能性がある。私たちはそのような訴訟や法的手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らがより多くの資源を持っているので、私たちよりも複雑な特許訴訟の費用を効果的に受けるかもしれない。さらに、いかなる訴訟の開始と継続に生じるいかなる不確実性も、運営を継続するために必要な資金を調達する能力に重大な悪影響を及ぼす可能性があり、あるいは私たちの業務、運営結果、財務状況と見通し。さらに、知的財産権訴訟や行政訴訟に関連する大量の開示要求により、私たちのいくつかの機密情報が開示によって漏洩される可能性がある。
第三者は私たちが許可されていない状況で彼らのノウハウを使用していると主張するかもしれない。
現在知られていない第三者特許があるかもしれないが、Reactionの組成、使用、または製造を含む保護物質の成分、材料、配合、製造方法または治療法が要求される。現在、私たちが現在知らない係属中の特許出願がある可能性があり、これらの出願は、発行された特許に反応するか、またはそれらの使用または製造が侵害される可能性がある。さらに、第三者は将来的に特許を取得し、私たちの技術を使ってこれらの特許を侵害したと主張するかもしれない。管轄権のある裁判所が、REACT、REACTまたは私たちの材料を製造するための中間体、私たちの処方または使用方法の態様をカバーするために、任意の第三者特許を所有している場合、そのような特許の所有者は、許可を得ない限り、またはその特許が満了するまで、または最終的に無効または実行不可能であると判定されるまで、候補製品を開発し、それを商業化する能力を阻止することができるかもしれない。いずれの場合も、そのような許可は商業的に合理的な条項や全く存在しないかもしれない。もし私たちが商業的に合理的な条項で第三者特許の必要な許可を得ることができない場合、あるいは根本的にできなければ、私たちの商業化反応能力は損害や遅延を受ける可能性があり、これは逆に私たちの業務を深刻に損なう可能性がある。我々が許可を得ても,我々の競争相手が我々に許可された同じ技術にアクセスできるように非排他的である可能性がある.さらに、私たちの特許および特許出願によって提供される保護の広さまたは強度が脅かされている場合、会社が私たちと協力し、現在または将来の候補製品を許可、開発、または商業化することを阻止するかもしれない。
私たちにクレームを出した当事者は、禁止または他の公平な救済を求めて得ることができ、これは、私たちのさらなる開発と商業化反応の能力を効果的に阻止することができる。これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、当社の業務における従業員資源を大量に移転することになる。もし私たちに対する侵害クレームが成功した場合、私たちは故意に侵害した3倍の損害賠償金と弁護士費、第三者から1つ以上の許可証を取得し、印税を支払うか、または私たちの侵害製品を再設計することを含む大量の損害賠償を支払わなければならないかもしれないし、大量の時間とお金の支出が必要かもしれない。私たちはそのようなライセンスがあるかどうか、あるいは商業的に合理的な条項で提供されるかどうかを予測できない。また,訴訟がない場合でも,第三者から許可を得て我々の研究やREPACTの商業化を進める必要がある可能性がある.私たちはもしあれば、合理的な費用または合理的な条項でこのような許可証のいずれかを得ることができないかもしれない。このような状況では、私たちはこれ以上開発と商業化反応ができなくなり、これは私たちの業務を深刻に損なう可能性がある。
77
第三者は、我々の従業員やコンサルタントが機密情報を誤って使用または漏洩したり、商業秘密を流用したりしたと主張する可能性がある。
バイオテクノロジーやバイオ製薬業界でよく見られるように、私たちが雇った個人は以前、私たちの競争相手や潜在的な競争相手を含む大学や他のバイオテクノロジーやバイオ製薬会社に雇われていた。現在、私たちに対するクレームが解決されていないにもかかわらず、私たちの従業員およびコンサルタントが、私たちのために働いているときに他人の固有情報またはノウハウを使用しないことを確実にするために努力しているにもかかわらず、私たちまたは私たちの従業員、コンサルタントまたは独立請負業者は、商業秘密または他の独自情報を含む、以前の雇用主または他の第三者の知的財産権を不注意または他の方法で使用または開示する可能性がある。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭的損害賠償を支払う以外に、私たちは貴重な知的財産権や人員を失う可能性がある。このようなクレームに対抗することに成功しても、知的財産権クレームに関連する訴訟や他の法的手続きは、巨額の費用を発生させる可能性があり、私たちの技術や管理者の正常な責任を分散させる可能性があります。また、公聴会、動議または他の一時的な手続きまたは事態の発展の結果が公表される可能性があり、証券アナリストや投資家がこれらの結果がマイナスであると考える場合、私たちの証券価格に大きな悪影響を及ぼす可能性がある。このような種類の訴訟や訴訟は、私たちの運営損失を大幅に増加させ、開発活動に利用できる資源を減少させる可能性がある。私たちはそのような訴訟や法的手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らの財政資源がはるかに大きいので、私たちよりもこのような訴訟や訴訟の費用を効果的に負担するかもしれない。特許訴訟または他の知的財産権関連訴訟の開始および継続によって生じる不確実性は、市場での競争能力に悪影響を及ぼす可能性がある。
私たちは、関連する第三者特許を識別しないか、または第三者特許の関連性、範囲、または失効を誤って解釈する可能性があり、これは、私たちが製品を開発およびマーケティングする能力に悪影響を及ぼす可能性がある。
私たちは、関連特許の識別、特許請求の範囲、または関連特許の満了を含めて、私たちのいかなる特許検索または分析も保証することはできず、私たちはまた、どの司法管轄区域でReaction商業化に関連する可能性があるか、または必要な米国および海外のすべての第三者特許および係属中の出願を識別したことを確実にすることはできない。
特許請求の範囲は、法律の解釈、特許における書面開示、および特許の起訴履歴に依存する。特許または保留出願の関連性または範囲の解釈は正しくない可能性があり、これは私たちの製品を販売する能力に悪影響を及ぼす可能性がある。私たちの製品が第三者特許のカバー範囲内にないか、または第三者の保留出願が関連範囲のクレームを出すかどうかを誤って予測する可能性がある。米国や海外のいずれかに関連する特許の満期日の決定は正しくない可能性があり、これは私たちの開発能力や市場反応に悪影響を及ぼす可能性がある。私たちは関連特許を識別して正確に解釈することができず、私たちが製品を開発し、マーケティングする能力に悪影響を及ぼすかもしれない。
私たちは、許容可能な条件で任意の未来の候補製品を開発するために、必要な知的財産権を獲得または維持することに成功しないかもしれない。
Reaction、私たちの現在の候補製品は、効率的かつ効率的に動作するために特定の処方が必要となる可能性があり、これらの権利は他の人によって保持されている可能性がある。私たちは私たちの化合物と予め存在する生物製薬化合物を含む製品を開発するかもしれない。私たちは、当社の業務運営に必要または重要であると考えられる任意の成分、使用方法、プロセス、または他の第三者知的財産権を第三者から得ることができないかもしれません。私たちは合理的な費用または合理的な条項でこれらのライセンスのいずれかを得ることができないかもしれないが、もしあれば、これは私たちの業務を損なうだろう。私たちは、このような第三者知的財産権がカバーする成分や方法の使用を停止する必要があるかもしれませんし、このような知的財産権を侵害しない代替方法の開発を求める必要があるかもしれません。たとえこのような不可能な代替方法を開発することができても、追加のコストと開発遅延を招く可能性があります。我々が許可を得ることができても,非排他的である可能性があり,我々の競争相手が我々に許可された同じ技術にアクセスできるようにする.この場合、私たちは代替技術を開発または許可するために多くの時間と資源を必要とするかもしれない。
第三者知的財産権の許可·買収は競争分野であり、私たちよりも成熟したり、より多くの資源を持っている企業も、商業化反応のために必要または魅力的だと思う第三者知的財産権許可または買収戦略をとる可能性がある。より成熟した会社は私たちより競争優位にあるかもしれません。それらの規模、現金資源、そしてより強い臨床開発と商業化能力のためです。このような交渉を成功させ、最終的に買収を求める可能性のある他の候補製品をめぐる知的財産権を得ることができる保証はない。
78
私たちは、私たちの特許が人かもしれない特許を保護または強制する訴訟に巻き込まれたり、他人の特許権に挑戦したりすることができます。これは高価で、時間がかかり、成功しないかもしれません。
競争相手は私たちの特許や私たちの現在または未来の許可者の特許を侵害するかもしれない。権利侵害や不正使用に対抗するために、私たちは侵害請求を要求されるかもしれません。これは高価であるかもしれません時間がかかります。さらに、侵害訴訟では、裁判所は、私たちの1つまたは複数の特許が無効または強制的に実行できないと判断するか、または私たちの特許が関連技術または他の理由をカバーしないことを理由に、他方の関連技術の使用を阻止することを拒否することができる。任意の訴訟または弁護手続きにおける不利な結果は、私たちの1つまたは複数の特許が無効であることが宣言されるか、強制的に実行できないか、または狭義の解釈に直面するリスクに直面する可能性があり、私たちの特許出願を発行できないリスクに直面させる可能性がある。これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、当社の業務における従業員資源を大量に移転することになる。
米国特許商標局に一方的な再審で特許請求書を審査することを請求することにより、第三者が米国特許請求書の特許性に挑戦することを選択することができる各方面間審査または支出後に手続きを検討する。このような手続きは高価で、私たちの時間や他の資源を消費するかもしれない。欧州特許庁(“EPO”)または他の外国特許庁の特許反対手続において第三者の特許に挑戦することを選択することができる。このような訴訟に反対する費用は巨大かもしれないし、私たちの時間や他の資源を消費するかもしれない。もし私たちが米国特許商標局、ヨーロッパ特許庁、または他の特許庁で有利な結果を得ることができなかった場合、私たちはReactionまたは私たちのノウハウが私たちの特許を侵害している可能性があると非難する第三者の訴訟に直面する可能性がある。
さらに、米国のいくつかの特許出願は特許発行前に秘密にされている可能性があるため、米国および多くの外国司法管轄区域の特許出願は通常、提出されてから18ヶ月後に公表され、科学文献の出版物は実際の発見よりも遅れており、他の人が私たちが所有して許可している発行特許または私たちが審理中の出願に含まれている技術について特許出願を提出していないか、または私たちまたは適用すれば許可者が最初にその技術を発明した人であるとは判断できない。私たちの競争相手はすでに提出されている可能性があり、将来的に特許出願を提出することができ、私たちと類似した製品または技術をカバーすることができる。このような任意の特許出願は、私たちが所有している特許出願または特許よりも優先される可能性があり、これは、そのような技術をカバーする発行された特許を取得する権利を要求することができる。他方が私たちによって所有されているか、または許可されている発明に米国特許出願を提出したような場合、米国における発明の優先度を決定するために、USPTOが発表した干渉または派生プログラムに参加しなければならないかもしれない。もし私たちまたは私たちの許可者のうちの1つが妨害または派生プログラムの一方であれば、私たちが所有している発明または私たちに許可された発明に対する米国特許出願に関連して、私たちが成功しても、私たちは巨額のコストを招き、管理職を移転する時間と他の資源を費やす可能性がある。
我々の特許または特許出願または我々の許可者の発明に関連する発明の優先権を決定するために、第三者によって引き起こされるか、または米国特許商標局によって発表された干渉または派生プログラムが必要である可能性がある。不利な結果は、現在の特許権の喪失を招く可能性があり、関連技術の使用を停止したり、勝利者から許可権を得ようとしたりすることが要求される可能性があります。もし勝利者が商業的に合理的な条項で私たちに許可を提供しない場合、または許可を全く提供しない場合、または非排他的な許可が提供され、私たちの競争相手が同じ技術を獲得した場合、私たちの業務は損なわれる可能性がある。訴訟や介入手続きは、私たちの利益に不利な決定を招く可能性があり、私たちが成功しても、巨額のコストを招き、私たちの経営陣や他の従業員の注意を分散させる可能性があります。私たちは私たちの商業秘密や機密情報の盗用を単独でまたは私たちの許可者と一緒に防ぐことができないかもしれません。特に法律ではアメリカのようにこれらの権利を十分に保護していないかもしれません。
さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。さらに、公聴会、動議、または他の一時的な手続き、または事態の発展の結果が発表される可能性がある。もし証券アナリストや投資家がこれらの結果がマイナスだと思っていれば、私たちの証券価格に重大な悪影響を及ぼす可能性がある。
私たちの特許保護の獲得と維持は、政府特許機関によって提出された様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
私たちが所有しライセンスしている特許および特許出願の定期維持費、継続費、年会費、および様々な他の政府費用は、特許有効期間内に米国特許商標局および外国特許代理機関に段階的に支払われる。米国特許商標局および各種外国政府特許機関は、特許出願中および特許発行後に、いくつかのプログラム、文書、費用支払い、およびその他の規定を遵守することを要求する。多くの場合、適用規則に従って滞納金を支払うか、または他の方法で不注意を是正することによって失効を是正することができるが、場合によっては、規定を遵守しないことは、特許または特許出願が放棄または失効され、関連法ドメインの特許権の一部または全部の喪失をもたらす可能性がある。特許または特許出願の放棄または失効をもたらす可能性のある不正事件には、規定された期限内に公式行動に応答できなかったこと、費用が支払われていなかったこと、および適切に合法化され、正式な文書が提出されなかったことが含まれるが、これらに限定されない。場合によっては
79
不用意な不正事件であっても永久的かつ取り返しのつかないように特許権を危険にさらす可能性がある。この場合、私たちの競争相手が市場に参入する可能性があり、これは私たちの業務に実質的な悪影響を及ぼすだろう。
法廷または米国特許商標局で疑問が提起された場合、REACTIONをカバーするいくつかの特許は、無効または実行不可能であることが発見される可能性がある。
私たちまたは私たちの許可者のうちの1つが特許カバー反応を強制的に実行するために第三者に対する法的手続きを開始した場合、被告は、適用される場合、特許カバー反応が無効および/または実行不可能であることを反訴することができる。米国の特許訴訟では、被告が特許の無効および/または強制執行不可能と主張する反訴はありふれており、第三者は多くの理由から特許が無効または強制的に実行できないと断言することができる。第三者も米国や海外の行政機関に類似したクレームを出すことができ、訴訟範囲外でも同様である。この仕組みには再検討が含まれています各方面間審査、承認後の審査と外国司法管轄区における同等の手続き(例えば、反対手続)。このような訴訟は、RESPECTをこれ以上カバーしないように、私たちの特許が撤回されたり修正されたりするかもしれない。法的に無効と実行不可能と断言された後の結果は予測できない。例えば、有効性の問題について、私たちは無効な以前の技術がないことを確認することはできないが、私たち、私たちの特許弁護士、特許審査員は起訴中にこれを知らない。もし被告が無効および/または強制執行できない法律で勝訴した場合、または私たちが他の方法で私たちの権利を十分に保護できない場合、私たちはREPACTに関する特許保護の少なくとも一部、さらには全てを失うだろう。このような特許保護の喪失は、私たちの業務および私たちの技術および候補製品を商業化または許可を得る能力に実質的な悪影響を及ぼす可能性がある。
私たちの最初の特許は私たちの最初の製品がアメリカや外国の管轄区域で発売承認される前か近いうちに満期になるかもしれません。私たちの既存の特許が満期になると、私たちは他の人たちを排除してこのような発明を実施する権利を失うかもしれない。これらの特許の満期は、私たちの業務、運営結果、財務状況、および将来性に類似した大きな悪影響を及ぼす可能性があります。
米国特許法の変化、政府の法律解釈の変化、あるいは他の管轄区域法律の変化は、特許の全体的な価値を低下させ、製品を保護する能力を弱める可能性がある。
米国又は他の管轄区域における特許法又は特許法解釈の変化は、特許出願をめぐる起訴及び発行された特許の実行又は弁護の不確実性及びコストを増加させる可能性がある。他の特許性要件を満たしていると仮定すると,2013年3月16日までは,米国では,最初に発明して保護された発明を要求した者が特許を有しているが,米国以外では,最初に特許出願を提出した者が特許を有している。2013年3月16日、2011年9月に公布された“米国発明法”によると、米国は初めての発明家に変わった特許可能性の他の要件が満たされていると仮定して、第1の特許出願を提出した発明者が、第三者が第1の発明が保護された発明を発明したか否かにかかわらず、発明の特許を取得する権利がある文書制度。したがって、2013年3月16日以降に米国特許商標局に特許出願を提出することができるが、我々の前の第三者は、当該第三者が発明を行う前に本発明を作成したとしても、我々の発明をカバーする特許を付与することができる。これは私たちが発明から特許出願までの提出時間を認識することを要求するだろう。米国およびほとんどの他の国/地域の特許出願は、提出後または発行前の一定期間秘密であるため、私たちまたは私たちのライセンス者が、私たちの候補製品に関連する任意の特許出願または(Ii)発明または私たちのライセンシーの特許または特許出願に要求される任意の発明を最初に提出することを決定することはできない。
米国の発明法にはいくつかの重大な変化も含まれており、これらの変化は特許出願の起訴方法に影響を与え、特許訴訟に影響を与える可能性もある。これらの措置には、第三者が特許訴訟中に米国特許商標局に以前の技術を提出することを可能にすることと、特許の有効性を攻撃するために、ライセンス後審査を含む米国特許商標局によって管理される認可後手続きとが含まれる各方面間審査と派生手続き。USPTO訴訟における証拠基準は、米国連邦裁判所が特許請求の無効を宣言するために必要な証拠基準よりも低いため、第三者は、USPTO手続きにおいて、USPTOが権利要求を無効とするのに十分な証拠を提供する可能性があり、同じ証拠が最初に地域裁判所訴訟で提出された場合には、権利請求を無効にするのに十分ではない。したがって,第三者は米国特許商標局の手続きを用いて我々の特許主張の無効を宣言しようとする可能性があり,第三者が地域裁判所訴訟で最初に被告として疑問を提起すれば,我々の特許主張は無効ではない.したがって、米国発明法およびその実施は、私たちの所有または許可をめぐる特許出願の起訴および私たちが所有または許可した発行された特許の実行または弁護の不確実性およびコストを増加させる可能性があり、これらのすべては、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
また、生物薬品開発と商業化における企業の特許地位は特に不確定である。米国最高裁判所の最近の裁決は、場合によっては入手可能な特許保護範囲を縮小し、場合によっては特許所有者の権利を弱める。この一連の事件は特許取得後の有効性と実行可能性の面で不確実性をもたらしている。米国議会、連邦裁判所、および米国特許商標局の将来の行動によると、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、私たちの既存の特許の組み合わせおよび私たちの将来の知的財産権の保護と実行能力に重大な悪影響を及ぼす可能性がある。
80
私たちは限られた外国の知的財産権を持っていて、世界各地で私たちの知的財産権を保護して実行できないかもしれない。
私たちはアメリカ以外の国で複数の特許を持っているにもかかわらず、CKDが盛んなアメリカ以外のすべての潜在市場で知的財産権を持っているわけではありません。世界各国で候補製品特許を出願、起訴、擁護する費用は目を引くほど高く、米国以外のいくつかの国での知的財産権は米国ほど広くないかもしれない。また、いくつかの外国の法律は知的財産権の保護の程度はアメリカの連邦や州法律に及ばない。したがって、私たちは、米国以外のすべての国/地域で第三者が私たちの発明を実施したり、米国または他の管轄区域で私たちの発明を使用して製造された製品を販売または輸入することを阻止することができないかもしれない。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの技術を使用して自分の製品を開発することができ、また、他の侵害製品を私たちが特許保護を持っているが法執行力がアメリカに及ばない地域に輸出することもできる。これらの製品は、私たちが発行した特許を持っていない司法管轄区域で私たちの製品と競争するかもしれませんが、私たちの特許主張や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。
多くの会社は外国の管轄区域の知的財産権の保護と保護に重大な問題に直面している。特定の国の法制度、特に発展途上国の法律制度特許、商業秘密、および他の知的財産権保護の強制執行には賛成できず、特に生物製薬製品に関連する特許、商業秘密、および他の知的財産権保護は、私たちの特許の侵害や第三者の競争製品の販売に対する行為を阻止することを困難にする可能性があり、これらの行為は私たちの独占権を普遍的に侵害する。第三者が外国の管轄区域で訴訟を起こし、私たちの特許権の範囲または有効性に挑戦し、巨額のコストを招く可能性があり、私たちの努力と注意を私たちの業務の他の側面から移すことができます。外国の管轄区域で私たちの特許権を強制的に執行する訴訟手続きは、巨額のコストを招き、私たちの努力と注意を私たちの業務の他の側面から移転させる可能性があり、私たちの特許が無効または狭義に解釈されるリスクに直面する可能性があり、私たちの特許出願を発表できないリスクに直面させ、第三者からのクレームを引き起こす可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。したがって、私たちが世界各地で私たちの知的財産権を実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネスメリットを得るのに十分ではないかもしれません。
特許条項は、Reactionまたは将来の候補製品における私たちの競争地位を十分に長く保護するのに十分ではないかもしれません。もし私たちがハッジ-ワックスマン修正案および同様の非米国立法によって保護されていなければ、Reactionまたは私たちの将来の候補製品をカバーする特許期間を延長するために、私たちの業務は実質的に損害を受ける可能性があります。
特許の寿命は限られている。米国では、すべての維持費が適時に支払われれば、特許の自然失効時間は、通常、最初に主張された米国の非臨時出願日から20年である。特許期限調整および/または延長などの様々な延長は利用可能である可能性があるが、特許の有効期間およびその提供される保護は限られている。Reactionまたは将来の任意の候補製品をカバーする特許を取得しても、特許有効期限が満了すると、競争製品からの競争を受け入れることができるかもしれません。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。
FDAが私たちが開発する可能性のある任意の候補製品の上場承認の時間、期限、および詳細によれば、私たちの1つ以上の米国特許は、1984年の“薬品価格競争および特許期限回復行動”(“ハッジ-ワックスマン修正案”とも呼ばれる)に従って限られた特許期間を延長する資格がある可能性がある。ハッジ·ワックスマン修正案は、FDA規制審査中に失われた特許期間の補償として、満期日以降に最大5年間の特許延長期間を延長することを許可している。1つの特許期間の延長は,製品承認日から計14年の期限を超えてはならず,1つの特許しか延長できない.しかし、私たちは試験段階や規制審査中に職務調査が行われなかったために全面的な延期を受けることはできないかもしれない。また,適用の最終期限内に出願を提出できなかったこと,関連特許の満了前に出願を提出できなかったこと,適用の要求を満たしていなかったことなどにより,何の延期も得られなかった可能性がある.しかも、特許保護の適用期間や範囲は私たちが要求しているものよりも短いかもしれない。もし私たちが特許期間の延長を得ることができない場合、あるいはこのような延長の期限が私たちが要求したものよりも短い場合、私たちはその製品に対して特許権を行使することができる期限が短縮され、競争相手はより早く承認され、競争製品市場に入るかもしれません。したがって、私たちは適用製品からの収入が減少する可能性があり、私たちの業務に実質的な悪影響を及ぼす可能性がある。
私たちが取得したまたは取得可能ないかなる商標も侵害される可能性があり、または他の方法で違反または成功して挑戦し、それによって私たちの業務に損害を与える可能性がある。
商標による手段として,上場が許可された場合にはREPACTを我々の競争相手の製品と区別したい.私たちが新しい商標を選択して登録を申請すると、私たちの商標申請は承認されないかもしれない。私たちはこのような拒否に答える機会があるだろうが、私たちはこの拒否を克服できないかもしれない。第三者は、私たちの商標出願または商標をキャンセルしようとしたり、他の方法で商標の使用に挑戦しようとしたりすることができる。はい
81
もし私たちの商標が成功的に挑戦されれば、私たちは私たちの製品ブランドを再形成することを余儀なくされるかもしれません。これはブランド認知度の喪失を招き、広告と新しいブランドをマーケティングするために資源を投入する必要があるかもしれません。私たちの競争相手私たちの商標を侵害または他の方法で侵害する可能性があり、私たちは私たちの商標を実行するのに十分な資源がないかもしれない。このようなどんな事件も私たちの業務に重大な悪影響を及ぼす可能性がある。また,米国でREACTとともに使用される予定の任意の名称は,登録されているか否かや商標として登録されているか否かにかかわらず,FDAの承認を得なければならない。FDAは、通常、他の製品名と混同される可能性がある可能性を含む、提案された製品名を検討する。FDAが私たちが提案した任意の独自製品名に反対する場合、私たちは、適用商標法の資格に適合し、第三者の既存の権利を侵害せず、FDAのために受け入れられる適切な代替名を決定するために、多くの追加資源を必要とするかもしれません。
管理業務と運営に関するリスク
私たちは、私たちの臨床開発、研究、規制能力を拡大し、私たちの製造と管理能力を拡大し、販売、マーケティング、流通能力を実施することが可能であるため、私たちは私たちの成長を管理する上で困難に直面する可能性があり、これは私たちの運営に悪影響を及ぼすかもしれない。
2023年12月31日までに、約163人のフルタイム従業員がいます。私たちの臨床開発と商業化計画と戦略の発展、そして私たちの上場会社運営への移行に伴い、私たちは私たちの管理、臨床、監督、製造、販売、マーケティング、財務、開発と法的能力を拡大し、あるいは第三者と契約を締結し、これらの能力を提供する必要がある。私たちの業務の拡大に伴い、様々な戦略パートナー、サプライヤー、他の第三者とのより多くの関係を管理する必要があると予想されます。私たちの将来の成長は、経営陣のメンバーにより多くの重大な責任を負わせるだろう
|
|
|
より多くの従業員を識別し、採用し、統合し、維持し、激励する |
|
|
|
Reactionおよび任意の他の候補製品に対する臨床およびFDA審査プロセスを含む、我々の開発および商業化作業を効率的に管理し、同時に、請負業者および他の第三者に対する契約義務を遵守し、 |
|
|
|
私たちの業務、財務、管理制御、報告システム、そして手続きを改善する。 |
私たちが開発と商業化反応を続ける能力は、未来のどんな成長の能力を効果的に管理するかにある程度依存するだろう。私たちの経営陣はまた、これらの成長活動の管理に多くの時間を使用するために、不比例な注意を日常活動から移しなければならないかもしれない。
現在、予測可能な未来に、私たちはいくつかの独立した組織、コンサルタント、コンサルタントに大きく依存していくつかのサービスを提供するだろう。必要な場合には、独立した組織、コンサルタント、コンサルタントのサービスがタイムリーに提供される保証はなく、合格した代替者を見つけることも保証されない。さらに、私たちのアウトソーシング活動を効率的に管理できない場合、または提供されるサービスの品質、正確性、または数が何らかの理由で影響を受ける場合、私たちの臨床試験は延長、延期、または終了される可能性があり、私たちは規制部門の承認を得る過程でReactionまたは私たちの将来の任意の候補製品を獲得するか、または他の方法で私たちの業務を促進することができないかもしれない。私たちは私たちが既存のコンサルタントを経済的に合理的な条件で管理したり、他の適任な外部請負業者やコンサルタントを見つけることができるか、あるいは全く保証できないという保証はない。
新入社員を雇用したり、コンサルタントや請負業者チームを拡大したりすることで、私たちの組織を効果的に拡大することができなければ、Reactionや他の候補製品のさらなる開発や商業化に必要な任務を実行することに成功しない可能性があり、我々の研究、開発、商業化目標を実現できない可能性がある。
もし私たちが重要な管理職を失った場合、あるいは私たちがより多くの高技能者を募集できない場合、私たちは新製品候補製品を開発したり識別したりする能力が損なわれ続け、これは市場シェアや市場シェアを失い、私たちの競争力を低下させる可能性があります。
私たちが競争の激しい生物技術と生物製薬業界の中で競争できるかどうかは、私たちが高い素質の管理、科学と医療人員を引き付けることができるかどうかにかかっている。私たちは私たちの経営陣、科学、医療者に高度に依存して、どんな幹部、他の重要な従業員、他の科学と医療コンサルタントのサービスを失い、適切な代替者が見つからなければ、製品開発の遅延を招き、私たちの業務を損なう可能性があります。
私たちはアメリカとケイマン諸島を含む世界的に多くの場所で事業を展開している。私たちの業界は技術人材に対する競争が非常に激しく、私たちが受け入れられる条件で高い素質の人員を採用と維持する能力を制限する可能性があり、甚だしきに至っては根本的にできない。
価値のある従業員をわが社に引き付けるために、給料と現金インセンティブのほか、時間とともに付与された株式奨励を提供する予定で、その中のいくつかは未登録株の形態である可能性があり、投票権を希釈する可能性があります
82
私たちの株主の経済的権利。時間が経つにつれて、このような株式奨励の従業員に対する価値は、私たちの株価変動の大きな影響を受ける可能性があり、これらの変動は私たちの制御範囲を超えており、いつでも他社が提出したより利益のある見積もりを相殺するのに十分ではないかもしれない。私たちは価値のある従業員を引き留めるために努力しているにもかかわらず、私たちの管理、科学、開発チームのメンバーは短時間で私たちとの雇用関係を終了するかもしれない。私たちの重要な従業員は好きなような従業員であり、これは私たちのどの従業員も通知または通知せずにいつでも離職できることを意味します。私たちの成功はまた私たちが引き続き高技能の初級、中級と高級科学と医療人員を吸引、維持、激励できるかどうかにかかっている。
当社の従業員、独立請負業者、コンサルタント、協力者、首席調査員、CRO、サプライヤー、およびサプライヤーは、規制基準および要件を遵守しないことを含む、不適切な行為または他の不適切な活動に従事する可能性があります。
私たちは、従業員、独立請負業者、コンサルタント、協力者、主要な調査者、CRO、サプライヤー、およびサプライヤーが詐欺または他の不正活動に従事する可能性があるリスクに直面しています。これらの当事者の不適切な行為は、真、完全かつ正確な情報をFDAに報告することを要求する法律、製造基準、連邦および州医療保健法律法規、および真の、完全かつ正確な財務情報またはデータの報告を要求する法律を含むFDAの規定に違反する故意、無謀、および/または不注意を含む可能性がある。特に、衛生保健業界の販売、マーケティング、商業手配は、詐欺、リベート、自己取引、その他の乱用行為を防止するための広範な法律と法規の制約を受けている。これらの法律法規は、広範な価格設定、割引、マーケティングおよび販売促進、販売手数料、顧客インセンティブ計画、および他のビジネス計画を制限または禁止する可能性があります。これらの各方面の不当な行為はまた個人識別情報の不適切な使用に関連する可能性があり、臨床試験過程で得られた情報を含むが、これに限定されず、これは規制制裁と著者らの名声に深刻な損害を与える可能性がある。不正行為を識別し、阻止することは常に可能ではなく、私たちがそのような活動を発見し、防止するための予防措置は、未知または未管理のリスクまたは損失を効果的に制御することができないか、またはそのような法律または法規を遵守できないことによる政府の調査または他の行動または訴訟から私たちを保護することができないかもしれない。もし私たちにこのような訴訟を起こした場合、私たちは私たちの権利を弁護したり、維持することに成功しませんでした。これらの行動は、損害賠償、罰金、返還、監禁、連邦医療保険や医療補助などの政府医療保健計画から除外されたが、当社の誠実な合意や同様の合意の制約を受けて、これらの法律違反に関する告発を解決し、私たちの業務を削減または再編することを含む、私たちの業務に重大な影響を与える可能性があります。
私たちの内部コンピュータシステム、または私たちの協力者や他の請負業者やコンサルタントのシステムは、故障したり、セキュリティホールに遭遇したりする可能性があり、これは、私たちの製品開発計画が実質的に破壊される可能性があります。
私たちが運営する業務運営、製造、臨床研究の名声と能力は、私たちのコンピュータシステムと、私たちが運営に使用している第三者システムの性能と安全性に依存します。これらのシステムおよび任意の将来の協力者および他の請負者またはコンサルタントのシステムは、コンピュータウイルス、許可されていないアクセス、自然災害、テロ、戦争、ならびに電気通信および電気故障の破壊を受けやすい。このようなイベントが発生し、当社の運営中断を招く場合、我々のビジネス機密や他の独自情報の損失によるものであっても、他の同様の中断によるものであっても、我々の開発計画や業務運営中断を招く可能性がある。例えば、未来の臨床試験における臨床試験データの紛失は著者らの監督管理の承認作業の遅延を招く可能性があり、そして著者らのデータの回復或いは複製のコストを著しく増加させる。もしどんな中断やセキュリティホールが私たちのデータやアプリケーションを紛失したり破損したり、機密または独自の情報を適切に開示しない場合、私たちは責任を招く可能性があり、私たちの競争地位が損なわれる可能性があり、Reactionまたは私たちの未来の任意の候補製品のさらなる開発と商業化が延期される可能性がある。
会社やサプライヤーの情報システムやネットワークで維持されている情報には,我々従業員や研究対象の個人情報,および会社やサプライヤーの機密データが含まれており,流用,誤用,漏洩,改ざんや故意や意外な情報漏洩や紛失によるリスクを受ける可能性がある.さらに、外部の当事者は、私たちまたは私たちのサプライヤーのシステムを浸透させようとしたり、詐欺的な手段で私たちの人員または私たちのサプライヤーの人員に敏感な情報を開示させて、私たちのデータおよび/またはシステムにアクセスしようとするかもしれません。私たちのデータとシステムは悪意のあるコードとウイルス、ネット釣り、および他のサイバー攻撃を含む脅威にさらされる可能性がある。時間が経つにつれて、このような脅威の数と複雑さは増加している。もし私たちの情報技術システムやサプライヤーの情報技術システムに重大な破壊や意外や故意にデータが失われた場合、市場の私たちのセキュリティ対策の有効性に対する市場の見方が損なわれる可能性があり、私たちの名声と信頼性が損なわれる可能性がある。私たちは情報システムやネットワークを修復したり交換したりするために多くの資金と他の資源が必要かもしれない。さらに、個人訴訟における規制行動および/または個人および団体からのクレームを受ける可能性があり、これらの訴訟は、データの乱用または不正開示に対するクレーム、および不公平または詐欺的なやり方を含むデータ収集および使用慣行および他のデータプライバシー法律および法規に関連するプライバシー問題に関するものである。我々はこれらの事件の発生を防止するためのシステムと制御を開発·維持し、脅威を識別·緩和するプロセスもあるが、これらのシステム、制御、プロセスの開発と保守はコストが高く、技術の変化とセキュリティ対策を克服する努力がますます複雑になり、継続的に監視·更新する必要がある。しかも、私たちが努力したにもかかわらず、このような事件が発生する可能性を完全になくすことはできない。より多くの情報システムをサプライヤーにアウトソーシングし、支払人や患者とより多くの電子取引を行うことに伴い、
83
より多くのクラウドベースの情報システムは、関連するセキュリティリスクが増加し、私たちの技術と情報システムを保護するために追加の資源が必要になるだろう。さらに、システム障害時の障害、サービス中断、データの悪化または損失から私たちを保護するのに十分なセキュリティおよび制御措置を実施するために、私たちの内部情報技術システムまたは私たちの第三者請負業者のシステム、または私たちのコンサルタントが十分なセキュリティおよび制御措置を実施するための努力を保証することは、ネットワーク攻撃、セキュリティホール、産業スパイ攻撃、または金融、法律、商業または名声被害をもたらす可能性のある内部脅威攻撃時にデータが盗まれたり破損したりすることを防止するのに十分である。
私たちはクラウドサービスに依存して私たちの業務のいくつかの側面を運営して、私たちのクラウドサービスの使用に対する中断や干渉は私たちの運営に影響を与え、私たちの業務は不利な影響を受けるだろう。
私たちが使用しているクラウドサービスは、私たちの業務運営に分散コンピューティングインフラプラットフォームとアプリケーションホスティングを提供します。クラウドベースのサービスが提供するアプリケーションホスト、記憶機能、通信、および他のサービスを利用するために、我々のソフトウェアおよびコンピュータシステムを設計した。このため、私たちのこのようなサービスの使用に対するいかなる干渉や干渉も私たちの運営に影響を与え、私たちの業務も不利な影響を受けるだろう。
健康およびデータ保護法律法規を遵守しないことは、政府の法執行行動(民事または刑事罰を含む可能性がある)、個人訴訟および/または負の宣伝をもたらす可能性があり、私たちの経営業績および業務に負の影響を与える可能性がある。
私たちと任意の潜在的な協力者は、連邦、州、および外国のデータ保護法律および法規(すなわち、プライバシーおよびデータセキュリティに関する法律および法規)によって制約される可能性がある。アメリカでは、連邦健康情報プライバシー法、州データ漏洩通知法、州プライバシー及び健康情報プライバシー法、連邦及び州消費者保護法(例えば、連邦貿易委員会法第5条)、管理収集、使用、開示及び健康に関する個人情報や他の個人情報を保護することは、私たちの業務または私たちの協力者の業務に適用することができます。また,HIPAAのプライバシーやセキュリティ要求に拘束された第三者(臨床試験データを取得した研究機関を含む)から健康情報を取得する可能性がある。事実および状況によると、HIPAAによって許可されていないまたは許可されていない方法でHIPAAによってカバーされているエンティティによって維持されている個別に識別可能な健康情報を取得、使用または開示する場合、民事または刑事罰を受ける可能性がある。
GDPRを含む国際データ保護法は,米国国外で収集された健康に関する情報や他の個人情報にも適用可能である。GDPRは個人データを処理する上での私たちの責任と責任を増加させ、新しいEU(EEAを含む)データ保護ルールに遵守することを保証するために追加的なメカニズムを構築する必要があるかもしれません。しかも、イギリスの離脱はイギリスのデータ保護規制により多くの不確実性をもたらした。連合王国は2018年のデータ保護法改正案と並んで英国法にGDPRを保持している。欧州連合は欧州連合から連合王国にデータを移すための十分な決定を採択した。しかも、連合王国から欧州連合への移転には新しい要求がない。しかしながら、将来を展望すると、欧州連合と連合王国のデータ保護ルールは異なる可能性があり、データ転送は不可能である可能性があり、および/または新たな配置を作成する必要があるかもしれない。特に、連合王国の制度がどの程度GDPRに逆行し始めているのかは不明であり、連合王国に出入りするデータ転送をどのように管理するかも不明である。
さらに、カリフォルニア州は最近CCPAを公布し、カリフォルニアの消費者のために新しいプライバシー権(法律で定義されているような)を創造し、消費者または家庭の個人データを処理するエンティティに対してより多くのプライバシーとセキュリティ義務を規定した。CCPAは2020年1月1日に施行され,CPRAの大幅な改正により2023年1月1日に施行された。他にも、CPRAはCPRAに基づいて新しい法規を制定し、法執行権を拡大することを任務とする新しい規制機関であるカリフォルニアプライバシー保護局を設立した。2023年にはバージニア州、コロラド州、コネチカット州、ユタ州の全面的なプライバシー法が施行され、モンタナ州、オレゴン州、テキサス州の法律が2024年に施行される。また,米国の他州の法律は2024年後に施行され,米国の他州の提案も考慮されている。これらのプライバシー法は,我々のビジネス活動に影響を与える可能性があり,我々の業務が変化する個人データに関する規制環境における脆弱性を示している.
米国と国際データ保護法律法規を遵守することは、私たちがデータを収集、使用、開示する能力を制限し、または場合によってはいくつかの司法管轄区域で運営する能力に影響を与える契約でより重い義務を負うことを要求するかもしれない。米国および国際データ保護の法律および法規を遵守しないことは、政府の法執行行動(民事または刑事罰を含む可能性がある)、個人訴訟、および負の宣伝を招き、私たちの経営業績および業務に負の影響を与える可能性がある。また,我々または我々の潜在的協力者が情報を取得する臨床試験対象,およびこれらの情報を共有する提供者は,我々の情報の使用や開示能力を契約的に制限する可能性がある。私たちがプライバシー権を侵害していると主張し、データ保護法を遵守できなかったり、私たちの契約義務に違反したりして、私たちが責任を負わなくても、弁護の費用が高く、時間がかかり、否定的な宣伝を招き、私たちの業務を損なう可能性があります。
84
税法や政策の変更は、私たちの実際の税率と税負担、または普通株式保有者が支払うべき税金を増加させる可能性があり、どれも私たちの業務、財務状況、経営業績に重大な悪影響を及ぼす可能性があります。
適用される税金法律、法規、またはその行政解釈の変化は私たちに悪影響を及ぼすかもしれない。例えば、米国が2017年12月に公布した連邦税法は、一般に減税と雇用法案(以下、税法)と呼ばれ、改正された1986年の米国国税法(以下、“税法”と略称する)に根本的な変化が生じ、その中には、連邦企業所得税税率の引き下げ、企業の利息支出の一部制限の控除、ある役員と役員の給与支出の制限の控除、純営業赤字の繰越と繰り越しの制限、米国の国際業務収益への課税の範囲と時間に関する変化が含まれる。その後の立法“コロナウイルス援助、救済、経済安全法”(2020年3月27日に公布された“注法”は、純営業損失の使用と繰り越しの制限、商業利息支出控除の制限を含む税法がある課税年度に加えるいくつかの制限を緩和した。税法やCARE法案が今後数年間の確実な影響を定量化することは難しいが、これらの変化は私たちの投資家、私たちの顧客が投資している会社、あるいは私たちに実質的な影響を与える可能性がある。米国の立法提案が可決されれば、企業所得税税率と資本利益税税率を引き上げる。また、後日他の改訂を制定し、利息減額をさらに制限し、付随権益に対してより重い税項目を徴収したり、その他の実施が私たちの業務、経営業績及び財務状況に重大な悪影響を与える可能性のある変化を実施する可能性がある。
私たちの有効税率は変動するかもしれません。私たちが税務管区で発生した債務は計算金額を超えるかもしれません。
私たちはいくつかの税務管轄区で税金を支払うことができます。したがって、私たちの有効税率は私たちが経営している地域別の適用税率を組み合わせたものです。私たちの財務諸表を作成する際には、このような場所ごとに税金を支払う必要があると予想されます。しかし、多くの要素のため、私たちの実際の税率は過去と異なる可能性があります。新しく公布された税法の通過、私たちの収益力の組み合わせの司法管轄区から司法管轄区への変化、私たちの税務申告の審査と監査結果、私たちは税務当局と受け入れ可能な合意を達成したり維持できないこと、所得税会計計算の変化、税法の変化を含むかもしれません。これらの要因のいずれも、私たちの実際の税率が前の時期または現在の予想と大きく異なり、納税義務が私たちの財務諸表の課税金額を超えることをもたらす可能性があります。
私たちは受動的な外国投資会社や“PFIC”かもしれませんが、これはアメリカ投資家に不利なアメリカ連邦所得税の結果をもたらすかもしれません。
ProKidneyは,米国連邦所得税の目的でPFICに分類される可能性が高いとしている。もし私たちがAクラスの普通株式米国保有者の保有期間内の任意の課税年度(またはその一部)を含むPFICである場合、米国所有者は不利な米国連邦所得税結果の影響を受け、追加の報告要件の制約を受ける可能性がある。本課税年度以降のいずれの課税年度においても、PFICの地位は保証されません。また,いずれの課税年度における実PFICの地位もこの課税年度終了後に決定される。我々がいかなる課税年度のPFICであるか(保証できない)と判断すれば,米国の保有者が“適格選挙基金”選挙を行うことができるように,米国国税局が要求すべきPFIC年度情報報告書を含む米国国税局に要求可能な情報を提供するように努力する。しかし、ProKidneyがこのような情報をタイムリーに提供することは保証されない。私たちはアメリカの投資家に適用可能なPFIC規則について彼ら自身の税務顧問に相談することを促す。
実際の税率の意外な変化や私たちの収入や他の納税申告書の審査による不利な結果は、私たちの財務状況や経営業績に悪影響を及ぼす可能性があります。
私たちは異なる管轄区域で直接または間接的に所得税を納付し、私たちの納税義務は異なる管轄区域の費用分配の影響を受けるだろう。私たちの将来の実際の税率は変動したり、複数の要因の悪影響を受ける可能性があります
|
|
|
繰延税金資産と負債の推定値の変化 |
|
|
|
任意の税金推定免税額の時間と金額が発行される予定です |
|
|
|
株式報酬の税収効果 |
|
|
|
会社間の再編に関連する費用 |
|
|
|
税金の法律、法規とその解釈の変更 |
|
|
|
法定税率の低い司法管轄区における将来の収益は予想を下回り、法定税率の高い司法管轄区での将来の収益は予想よりも高い。 |
85
しかも、私たちは税務機関の私たちの所得税、販売税、そして他の取引税に対する監査を受けるかもしれない。このような監査の結果は私たちの財務状況と運営結果に悪影響を及ぼすかもしれない。
私たちの主要株主は、株主の承認を必要とする決定への影響を含め、私たちに大きな影響を与え、彼らの利益はProKidney Corp.A類普通株保有者の利益と衝突する可能性がある。
資本制御会社(“資本制御”)が2022年2月14日に締結した契約(“投票合意”)によると、当社の任意の取締役株式を選挙、委任または罷免する場合、資本制御会社は、取引終了3周年まで、資本制御会社が保有していない他のProKidney B類普通株の投票方式で投票することが規定されている。そのため、選挙、任命、または任意の取締役の罷免において、トーランティア有限責任会社は実際にProKidney Corp.の多数の投票権を支配している。また、パブロ·レゴレッタは取締役会長としてトレランティアに所属し、同社の多数の株式を所有·制御している。そのため、トレランシア及びその共同経営会社は当社の管理及び事務に重大な影響力を持ち、共同行動を通じて、任意の取締役の選挙、任命又は罷免を効果的に制御し、任意の合併、合併又は売却を含む重大な会社取引の承認を間接的に制御し、場合によっては取締役会の承認が必要である限り、株式を発行又は償還する場合がある。
トラティアとCECの利益は常に私たちの利益と重なるわけではなく、ProKidney Aクラスの普通株の所有者を含む私たちの利益と私たちの他の株主の利益と衝突する可能性がある。投資家は利益衝突が存在または発生する可能性があると考えているため、このような所有権の集中は私たちProKidney A類普通株の現行の市場価格にも影響を与える可能性がある。したがって、このような所有権集中はあなたの最善の利益に合わないかもしれない。
また,これらの株主はProKidney Corp.を介して我々の業務における経済的利益を持つのではなくPKLPを介しているため,彼らの利益はProKidney A類普通株保有者の利益とさらに衝突する可能性がある.ProKidney Corp.におけるこれらの所有者の大量の所有権とそれによって生じる効果的な制御能力は、誰かがProKidney Corp.に重大な株式投資を行うことを阻止するかもしれないし、ProKidney A類普通株を含む所有者がその株式がプレミアムを獲得して当時の市場価格よりも高い取引をする可能性があることを阻止するかもしれない。
私たちはナスダック規則の意味での“制御された会社”であるため、私たちの株主は非制御会社の株主が享受できるいくつかの会社管理保護を有していないかもしれない。
取締役選挙の投票権の50%以上を個人、グループ、あるいは他社が保有している限り、私たちはナスダック社のガバナンス基準の意味での“制御された会社”になる資格があります。投票合意の条項によると、どの取締役を選挙、委任または罷免するかについては、トレランティアは実際に私たちが発行した普通株の多数の投票権を支配している。したがって、私たちはナスダック社のガバナンス基準が指す“制御された会社”であり、(I)私たちの取締役会の多数のメンバーは独立取締役で構成されており、(Ii)ナスダック上場規則第5605条(B)(2)条の例外的な状況に基づいて、私たち取締役会には、少なくとも2人のメンバーからなる報酬委員会があり、各メンバーは独立した取締役であり、その委員会の目的と責任を説明する書面規約があり、(Iii)取締役が取締役選考のために著名人を選出または推薦しなければならない。取締役会の独立取締役の多数を占める独立取締役が投票を行い,独立取締役のみが投票に参加するか,あるいは完全に独立取締役からなる指名と会社管理委員会が投票を行い,この委員会は書面で定款を持ち,当該委員会の趣旨と責任を述べている。業務合併協定の規定によると、“ナスダック上場規則”については、取締役会の大部分の取締役は“独立”取締役であるが、業務合併後少なくとも一定期間内に、吾等は上記他の免除を使用することができる。
上記の2つの場合、将来の株式発行又はそれ自身が自社株式を売却する行動により、トーランティアの当社における権益が希釈される可能性があり、これにより、ナスダック上場規則により得られた“制御された会社”免除が失われる可能性がある。そして、私たちはナスダック市場で要求された条項を遵守することを要求されるだろう。
憲章に含まれる反買収条項や、ケイマン諸島の法律の規定は、買収企図を損なう可能性がある。
私たちの憲章にはいくつかの条項が含まれており、株主がその最適な利益に合致すると考えることができる自発的な買収提案を阻止するかもしれない。これらの規定は、経営陣の解除作業をより困難にする可能性があり、我々の証券の現行市場価格よりも高い割増を支払うことに関連する可能性のある取引を阻害する可能性がある。これらの規定には以下のようなものが含まれる
|
|
|
役員選挙では投票権が蓄積されず、小株主が取締役候補を選挙する能力を制限している |
86
|
|
|
3年間の任期を交錯させた分類取締役会は、株主が取締役会の多数のメンバーの能力を変更することを延期する可能性がある |
|
|
|
特別決議を採択してこそ取締役会から取締役を解任することができる |
|
|
|
取締役会が取締役会を選挙する権利は、取締役会の拡大または取締役が場合によっては辞任、死亡または罷免によって生じた取締役会の空きを埋めることで、株主が取締役会の穴を埋めることを阻止する |
|
|
|
株主が特別株主総会を開催することを禁止し、株主総会が取締役会メンバーのみで開催されることを規定することは、我々の株主が提案や行動(取締役罷免を含む)を強制的に考慮する能力を遅延させる可能性がある |
|
|
|
取締役会には投票権及びその他の優先株権を発行·設定する権利があり、これは普通株式保有者の投票権及びその他の権利に悪影響を及ぼす可能性がある。 |
JOBS法案は,我々のような“新興成長型会社”が,他の非新興成長型会社の上場企業に適用される各種報告要求の何らかの免除を利用することを許可している。
我々は現在、“雇用法案”改正後の“新興成長型会社”である“証券法”第2(A)(19)節の定義に適合している。したがって、私たちが新興成長型企業であり続ける限り、(I)財務報告書の内部統制に関するSOX第404条による監査人認証要求の免除、(Ii)報酬発言権、頻度発言権、およびゴールドパラシュート投票要求の免除、および(Iii)当社の定期報告および依頼書における役員報酬に関する開示義務の減少を含む、他の非新興成長型企業に適用される上場企業の様々な報告要件のいくつかの免除を利用する。したがって、私たちの株主は彼らが重要だと思ういくつかの情報にアクセスできないかもしれない。(A)2026年7月2日以降、私たちの初公募株(Social Capital Suvretta Holdings Corp.III)5周年、(B)私たちの年間総収入は少なくとも12.35億ドル。又は(C)大規模加速申請者とみなされ、これは、前第2四半期の最終営業日における非関連会社が保有するA類普通株の時価が7億ドル以上であり、(Ii)前3年間に10億ドルを超える転換不能債券を発行した日を意味する。
また、雇用法第102条(B)(1)条免除新興成長型企業は、民間企業(すなわち、施行が宣言されていない証券法登録声明又は“取引法”に基づいて登録されていない証券種別)が、新たな又は改正された財務会計基準の遵守を要求されるまで、新たな又は改正された財務会計基準を遵守する。JOBS法案では,会社は延長からの移行期間を選択し,非新興成長型会社に適用される要求を遵守することができると規定しているが,いずれの選択脱退も撤回できない。私たちは、移行期間を延長することを選択しないことを選択した。これは、基準が発表または改正された場合、その基準が上場企業または民間企業に異なる適用日があれば、私たちは新興成長型企業として、民間会社が新たな基準または改正基準を採用する際に新たなまたは改正された基準を採用することができることを意味する。これにより、私たちの財務諸表を、使用する会計基準の潜在的な違いのために、延長された移行期間を使用しないことを選択するために、新興成長型会社でも新興成長型会社でもない別の上場企業と比較することができるかもしれない。
また,S−K条例第10(F)(1)項で定義された“小さな報告会社”である。規模の小さい報告会社は、2年間の監査済み財務諸表のみを提供することを含む、いくつかの減少した開示義務を利用する可能性がある。私たちは、本会計年度の最終日、すなわち(I)前の会計四半期の最終営業日まで、非関連会社が保有する私たちの普通株式の時価が2.5億ドルを超えるまで、または(Ii)完成した会計年度において、私たちの年収が1億ドル以上であり、前の第2四半期の最終営業日までに、非関連会社が保有する当社の普通株の時価が7億ドル以上になるまで、規模の小さい報告会社となる。
私たちは投資家がこれらの免除に依存して私たちのA種類の普通株の吸引力が低下していることを発見するかどうか予測できない。もし一部の投資家が私たちのA類普通株の吸引力が低下していることを発見すれば、私たちのA類普通株はそれほど活発でない取引市場が出現する可能性があり、私たちの株価はもっと変動する可能性がある。
財務報告の内部統制は有効ではないかもしれませんが、独立公認会計士事務所はその有効性を証明できない可能性があり、これは私たちの業務や名声に大きな悪影響を及ぼす可能性があります。
上場企業として、我々は、改正された2022年サバンズ·オキシリー法案第302及び404条を実施するために、米国証券取引委員会規則を遵守しなければならず、これらの条項は、経営陣に、我々の四半期及び年間報告書において財務その他の情報を認証し、財務報告の内部統制の有効性に関する年間管理報告を提供することを要求する。上場企業の要求に応えるためには私たちは、より多くの内部統制と手続きを実施し、より多くの会計や内部監査者を雇用するなど、様々な行動をとる必要があるかもしれない。新興成長型企業として私たちの独立は
87
私たちが新興成長型会社でなくなる前に、公認会計士事務所は、第404条による財務報告書に対する内部統制の有効性を正式に証明する必要はない。この時、私たちの独立公認会計士事務所が私たちの制御措置の記録、設計、あるいは操作レベルに満足していなければ、不利な報告書を発行するかもしれません。
これらの統制をテストして維持することは、私たちの経営陣の関心を私たちの業務運営に重要な他の問題から移すことができる。我々の財務報告内部統制に重大な弱点があることが発見された場合、または第404条の要求を遵守できない場合、または財務報告内部統制が有効であると断言したり、独立公認会計士事務所が新興成長型企業の資格を有していなくなった場合には、財務報告内部統制の有効性について意見を述べることができず、投資家は我々の財務報告の正確性および完全性に自信を失う可能性があり、我々の普通株の市場価格は負の影響を受ける可能性があり、米国証券取引委員会や他の規制機関の調査を受ける可能性があり、追加の財務·管理資源が必要となる可能性がある。
価格上昇とインフレは私たちの利益率の表現と財務業績に否定的な影響を与えるかもしれない。
インフレが激化し、原材料、部品、送料、包装、労働力、エネルギー価格の上昇、私たちの製品の製造と流通のコストが増加し、これらのコストを私たちの顧客に転嫁できないかもしれません。また、包装、運賃、労働力、エネルギー価格などの他のコスト変動の影響も受けている。もしこれらのコストのインフレが運営効率などの措置を実施することで制御する能力を超えていれば、価格を高めることができず、顧客の需要にマイナスの影響を与えることなく、様々なコスト増加の影響を十分に相殺し、利益率の表現と運営結果にマイナスの影響を与える可能性がある。
地政学的リスクは市場の変動性と不確実性の増加を招く可能性があり、これは私たちの業務、財務状況、運営結果にマイナスの影響を与える可能性がある。
地政学的衝突による敵対行動の不確定な性質、範囲、規模、持続時間は、このような敵対行動の潜在的な影響と、このような敵対行動が世界経済と市場に対して取る制裁、禁輸、資産凍結、サイバー攻撃、その他の行動を含み、世界市場を混乱させ、市場の変動性と不確実性を悪化させ、これは私たちの業務とサプライチェーンに影響を与えるマクロ経済とその他の要素に悪影響を及ぼす可能性がある。サプライチェーンのどんな中断も私たちの収入を減少させ、私たちの財務業績に悪影響を及ぼす可能性がある。この干渉は、ウクライナとイスラエルの持続的な衝突、および公共事業および他のサービスの中断を含む、軍事衝突、地政学的事態の発展、戦争またはテロを含むが、これらに限定されない多くの事件によって引き起こされる可能性がある。十分な納品が得られない場合や、代替供給源を探す必要がある場合や、内部でそのようなコンポーネントを製造、組み立て、テストする必要がある場合は、製品を出荷する能力を著しく遅らせる可能性があり、これは、既存および潜在的な顧客との関係を損なう可能性があり、私たちの名声やブランドを損なう可能性があり、私たちの業務、財務状況、および運営結果に悪影響を及ぼす可能性があります。
持続的な衝突や衝突による経済制裁がエスカレートし、より広範な経済·安全懸念を招くかどうかも知ることはできず、私たちのサプライチェーン、サプライヤー、顧客、および潜在的な顧客に悪影響を及ぼす可能性がある。これらの紛争のより広範な結果を予測することはできず、その中には、さらなる制裁、禁輸、地域不安定、地政学的変化、およびマクロ経済条件、材料、供給、労働力、通貨為替レートおよび金融市場の獲得可能性およびコストへの悪影響が含まれる可能性があり、これらすべては、私たちの業務、財務状況、および運営結果に実質的な悪影響を及ぼす可能性がある。
私たちの組織構造に関連するリスクは
我々はPKLPの有限パートナーであるが,場合によっては有限責任の利益を失う可能性がある.
我々はPKLPの有限パートナーであり,PKLPはアイルランド法に基づいて登録された有限パートナーである。
アイルランド有限責任組合法によると、アイルランド有限責任組合の有限パートナーは、その出資額を超える債務や義務を負わない。しかし、アイルランドの有限責任会社法案もまた、(I)有限パートナー(例えばProKidney Corp.)であれば、このような有限責任は失われる可能性があると規定している。PKLPの業務管理に参加し、(Ii)PKLPが有限責任組合として登録できなかったか、またはPKLPの登録詳細に変更があり、PKLPの名称、PKLPの一般的な業務性質、PKLPの主な営業場所、PKLPのパートナーまたは任意のパートナーの名前、PKLPの性質、PKLPの性質、任意の有限パートナーの出資金額、または任意のパートナーが通常のパートナーではなく、有限パートナーまたは有限パートナーから有限パートナーになるための法的責任を含む。(Iii)有限責任パートナーは、その部分または一部の資本を抽出し、この場合、彼または彼女またはそれは、商号の債務および義務に対して法的責任を負うことになり、このように抽出された額に達することができる。
88
私たちは持株会社で、私たちの唯一の重要な資産はPKLPにおける私たちの権益であるため、私たちは子会社の分配に依存して税金を納め、課税協定に基づいてお金を支払い、配当金を支払います。
私たちはPKLPでの私たちの所有権権益以外に実質的な資産がない持株会社です。したがって、私たちは収入やキャッシュフローを作る独立した手段を持っていない。業務合併(“課税契約”)が終了する前に,社会資本Suvretta Holdings Corp.III,TRA当事者代表(“受取税金協定”の定義参照)とPKLP所持者との間で税金を支払い,期日2022年7月11日の課税税金合意支払い,および配当金(あれば)を支払う能力は,PKLPとその子会社の財務業績とキャッシュフローおよびPKLPから得られた割り当てに依存する.どのような理由でも、PKLPおよびその子会社の財務状況、収益、またはキャッシュフローの悪化は、このような分配を支払う能力を制限または弱める可能性がある。さらに、資金が必要であり、PKLPおよび/またはその任意の子会社が、適用される法律または法規または任意の融資スケジュールの条項に従ってこのような割り当てを制限されている場合、またはPKLPがそのような資金を提供できない場合、私たちの流動性および財務状態に大きな悪影響を及ぼす可能性がある。
PKLPは米国連邦所得税の目的で引き続き共同企業とみなされるため,通常はいかなる実体レベルの米国連邦所得税も納付する必要はない。逆に、PKLPの課税収入は、ProKidney Corp.を含むPKLP共通単位(“ProKidney Common Units”)の所有者に分配される。そのため、PKLPの任意の課税所得額における課税所得額に所得税を支払う必要がある可能性がある(例えば、このような課税所得額の純額が米国での貿易や事業の展開に有効に関連している場合、米国連邦所得税および支店利得税)。PKLPの第2次改正および再改訂された有限組合協定(“第2改正および再改正ProKidney有限組合協定”)の条項によると、PKLPはProKidney Common Units(ProKidney Corp.を含む)の所有者に税金配分を行う義務がある。いくつかの仮定税率で計算されました。税務支出に加えて、課税契約の下での支払い義務(およびそのような支払い義務を管理するコスト)を含む、当社の業務に関連する費用が発生します。これは、PKLPによって償還される可能性があります(課税協定の下での支払い義務は含まれていません)。私たちはPKLPに比例してProKidney Common Unitsの所有者に割り当てを促すつもりで、適用される所得税(想定税率で計算)、関連運営費用、課税協定に基づいて支払わなければならないお金、そして私たちが発表した配当金(あれば)をカバーするのに十分な金額です。しかしながら、以下に説明するように、PKLPがこのような割り当てを行う能力は、債務プロトコルまたは任意の適用可能な法律を含むPKLPの当時の当事者である任意の契約またはプロトコルに違反するか、またはPKLPを破綻させる効果があるかもしれないが、割り当ての制限を含むが、これらに限定されない様々な制限および制限を受ける可能性がある。もし吾等の現金資源が課税項目合意下の義務の履行や吾等の債務に資金を提供するのに不十分であれば、吾等は当該等の金を支払うために必要な流動資金を提供するための追加の債務を招く可能性があり、これは吾等の流動資金や財務状況に重大な悪影響を与え、吾等に当該等の貸金人に加えられる様々な制限を受ける可能性がある。もし私らが何らかの理由で課税項目合意に基づいてお金を支払うことができなかった場合、その等は支払いを遅延させ、支払い前に利息を計算することになるが、指定された期間に支払わない場合は、課税項目合意の下での重大な責任に対応する重大な違反となる可能性があり、課税項目合意下での対応を加速させる可能性があり、これらの金額はかなり膨大である可能性がある。
さらに、PKLPは一般に実体レベルの米国連邦所得税を支払う必要がないにもかかわらず、逆の選挙がなければ、連邦税法に従ってその納税申告書を調整する責任がある可能性がある。PKLPの課税収入計算が正しくなければ、そのメンバーはProKidney Corp.を含み、今後数年間にこの連邦立法とその関連指導によって重大な責任を負う可能性がある。
ある時期には,PKLPから得られる割当てが,課税プロトコルによって支払われる実際の納税義務や義務を超える可能性が予想される.取締役会は、ProKidney Aクラスの普通株式の配当金を支払うことを含む、このように累積された超過現金をどのように使用するかを適宜決定することができる。私たちはこれらの現金(または発表された配当金以外の他の利用可能な現金)を私たちの株主に割り当てる義務はありません。
ProKidney Aクラスの普通株の配当金は取締役会によって適宜決定され、取締役会は、私たちの業務、経営業績、財務状況、現在および予想される現金需要、拡張計画、および私たちがこのような配当能力を支払う任意の法律または契約制限を考慮するだろう。融資手配には、株主に配当金を支払う能力や他の分配を行う能力を制限する制限的な契約が含まれる可能性がある。Ireland有限責任パートナー法によると、PKLPの有限パートナーがPKLPの一部または一部に彼または彼女またはその貢献を抽出した場合、その有限責任者は有限責任を失う可能性があり、この場合、彼または彼女または彼女はProKidneyの債務および義務に責任を負い、最高抽出可能な金額に達する。
ProKidneyの子会社がProKidneyに流通する能力は一般的に似たような法的制限を受けている。もしProKidneyが分配するのに十分な資金がなければ、私たちは現金配当金を発表して支払う能力も制限されたり損害されたりする可能性がある。
89
場合によっては,PKLPは我々やProKidney Common Unitsの他の所持者に配布することが要求され,PKLPが要求される配布は大量である可能性がある.
通常,PKLPは一定の想定税率に比例してProKidney Common Unitsの他の所持者に比例して現金を割り当てることが要求され,割り当てられた金額は,PKLPのそれぞれの課税所得額における我々とProKidney Common Unitsの他の所持者の課税所得分を支払うのに十分である.(I)ProKidney Common Unitsの他の所有者に割り当てられる課税所得額の潜在的な違いから、(Ii)会社に適用される税率が個人未満であること、(Iii)私たちの非米国個人アイデンティティ、および(Iv)PKLPの分配義務を計算する際に仮定税率(ニューヨーク個人または会社住民のために規定されている米国連邦、州、地方最高有効限界税率)を使用することにより、私たちが受け取る可能性のある税収分配は、課税協定下での納税義務および支払い義務を大幅に超える。私たちは、配当金、課税項目協定の下の責任の支払い、および他の支出の支払いを含む、このように累積された超過現金の適切な用途を適宜決定する。私たちはProKidney Aクラスの普通株式の所有者にこのような余分な現金(または発表された配当金以外の他の利用可能な現金)を割り当てる義務はないだろう。ProKidney普通株によるProKidney Aクラスの普通株の償還または交換比率は、以下のような理由で調整されない:(I)私たちが送ったいかなる現金配当金も、(Ii)私たちが保持し、株主に割り当てられていないいかなる現金もない。これらの余分な現金をProKidney A類普通株の配当金として分配するのではなく、このような現金残高を保有したり、PKLPに貸したりすれば、ProKidney普通株の所有者は、ProKidney普通株を償還または交換した後、ProKidney A類普通株を所有することによって生じる、このような現金残高に起因するいかなる価値からも利益を得ることができる。
政府当局は、私たちの有効税率を高めたり、他の方法で私たちの業務を損なうために、私たちの会社間移転価格政策を疑問視したり、彼らの法律を変更したりするかもしれません。
国際構造を有する会社として、資金の流れと子会社との間の利益分配に関する法律を含む米国と外国の税収·譲渡価格法の制約を受ける。税務機関が私たちの会社間移転定価に疑問を提起すれば、私たちの経営はマイナス影響を受ける可能性があり、私たちの有効税率は上がるかもしれません。税率は国によって異なりますが、規制当局が1つの管轄区域での利益が増加すべきだと認定すれば、他の管轄区域の税収支出のいかなる関連増加も完全に相殺できない可能性があり、これは私たちの実際の税率を増加させます。また、経済協力開発機構(“OECD”)/20カ国グループのBEPS(“税ベース侵食と利益移転”)に関する包摂的な枠組みでは、140以上の司法管轄区域が最低税率の実施に同意している。私たちの有効税率は最低税率の実施によって変化するかもしれません。これは私たちの構造と未来のグローバル業務の足跡にかかっています。最後に、私たちはこれらの法律を理解し、遵守するために努力しているにもかかわらず、すべての適用された税関、外国為替規制、付加価値税、譲渡定価法律を常に遵守しているわけではないかもしれない。この場合、私たちは私たちの操作手順を調整する必要があるかもしれないし、私たちの業務は悪影響を受けるかもしれない。
課税協定によると、私たちはProKidney Corp.ProKidneyの普通株式とProKidney Aクラスの普通株といくつかの他の税務優遇との交換によって増加したProKidney資産税基準によって節約されたいくつかの税金の85%を支払わなければならないが、このような支払いは相当なものかもしれない。
取引終了前に、PKLP所有者(“決済ProKidney単位所有者”)は、ProKidney A類普通株と交換するために、吾ら、PKLPおよび決済ProKidney単位所有者間で2022年7月11日に締結された交換協定(“交換協定”)に基づいて追加のProKidney普通株を交換したり、いくつかの制限を受けた場合、後日ProKidney A類普通株または現金(“交換協定”)を現金で交換することが可能であるが、合意および改訂および改訂された2つ目のProKidney有限契約に記載されているいくつかの条件および譲渡制限によって制限されなければならない。これまで発生した交換は、ProKidney Corpの登録地が非課税司法管轄区域であるため、PKLPの有形無形資産の納税に基づいて分配可能なシェアを増加させなかった。しかしながら、場合によっては、将来の交換は、PKLP有形無形資産の納税に基づく割り当て可能なシェアを増加させる可能性がある。これらの税ベースの増加は(税収目的で)減価償却や償却減額を増加させる可能性があるため、このような交換が発生しなければ、将来的に支払う必要のある所得税やフランチャイズ税の金額を減少させる。
業務合併については、吾らは一般に、吾等が課税項目合意を締結することにより増加した課税基準及びPKLPのある他の税務属性及び税務優遇によるいくつかの節税金額(あればある)の85%を支払うことを規定している。このお金はPKLPの義務ではなくProKidney Corp.だ。ProKidneyは、その資産中の税金ベースで私たちが分配できるシェアの実際の増加と、課税契約によって支払われる任意の金額の金額と時間は、交換の時間、交換時のA類普通株の市場価格、このような交換の課税度、私たちの収入を確認する金額と時間など、多くの要素によって異なります。課税契約によると、私たちはどれだけのお金を支払うか、多くの要素は私たちのコントロール範囲内ではありません。これらのお金(あれば)は額が大きく、私たちの財務状況に大きな悪影響を及ぼす可能性があります。仮定しても他の点では
90
関連税法に実質的な変化はなく,PKLPの企業価値は等しい事業統合は、すべてのProKidney汎用単位を交換する際や、ProKidney汎用機関が将来的に大量の償還や交換がある場合、課税契約に基づいて支払われるお金は大きな影響を与えないと予想される。PKLPは現在、(I)事業を米国に移す計画がないため、または(Ii)ProKidney汎用機関の償還や交換によって米国国外で得られる税収割引が期待され、これは、PKLPの米国国外での予想される業務に基づいて課税協定に基づいて負担される義務をトリガする。また,PKLPは現在米国では業務運営がないため,近い将来大量の営業収入が生じないことが予想され,もしあれば,近い将来課税協定によって支払われる金は実質的ではないと予想される.現在予想されている業務運営や戦略とは逆に,業務運営が米国に移行し,米国以外の業務運営が変化したり,関連税法が大きく変化したりすると,課税協定による支払いは重大である可能性がある。私たちが受け取る税金協定によって支払われるいかなる金額も通常私たちが獲得する可能性のある全体的なキャッシュフロー金額を減少させます。もし私らがどんな理由でも課税項目合意に基づいて適時に支払うことができなかった場合、未納金は繰延され、支払い前に利息を計算します。また、将来的には課税契約による支払い義務が買収目標への吸引力を低下させる可能性があり、特に買収側が課税契約により実現されたとみなされる可能性のある税収割引の一部または全部を使用できない場合には、特に買収側が課税契約による支払い義務を使用できない場合には、買収目標に対する吸引力を低下させる可能性がある。
場合によっては、課税契約に基づいて支払われるお金は、私たちが達成した、または加速する可能性のある実際の税金割引を超える可能性があります。
課税されるべき契約の下での支払いは、私たちが確定した納税申告の立場に基づいており、アメリカ国税局または任意の他の税務機関は、私たちのすべてまたは任意の部分の税収ベースの増加と、私たちが取った他の税金の立場に疑問を提起するかもしれません。裁判所はそのような挑戦に耐えるかもしれません。もし私たちが最初に申請した任意の税金優遇が拒否された場合、期末ProKidney単位所有者は、例えば、税務機関の審査によって生じる調整のような、課税協定に従って支払う前に支払われた超過金を返済する必要がないだろう。逆に、当該等所持者に支払われる超過金は、その等超過金を決定した後、当社が支払わなければならない任意の将来の現金金(ある場合)から差し引かれる。しかし、吾等の最初に請求されたいかなる税務優遇に対する疑問は、最初に当該等を支払ってから数年以内には生じない可能性があり、あるいは早期に異議を唱えても、当該等の超過現金支払いは、課税契約条項吾等によって支払われる可能性のある将来の現金支払金額を超える可能性があるため、将来の現金支払可能額はない可能性がある。したがって、場合によっては、課税契約に基づいて、私たちの実際の収入やフランチャイズ税を超えて節約されたお金を支払うことができ、これは私たちの財務状況を深刻に損なう可能性があります。
また、課税契約は、(I)吾等が課税契約に従って早期終了権利を行使した場合、(Ii)課税契約が破産事件において法律施行により拒否された場合、(Iii)ProKidney Corp.のいくつかの支配権が変化した場合(課税協定に記載されているような)又は(Iv)課税契約に基づいて満了した金が3ヶ月を超えることを延滞している(私等がこのような金を支払うのに十分な資金がないと誠実に判断しない限り)、又は他の方法で課税協定の下でのいかなる重大な義務にも深刻に違反し、私たちは期末ProKidney単位所有者に、課税契約に従って支払われる将来の支払いを予測するすべての現在値に相当する使い捨て現金支払いを直ちに支払うことを要求されます。この一括払いは、私たちの将来の課税所得額に関する仮定を含むいくつかの仮定に基づいています。期末ProKidney単位所有者に一度に支払う金額は大きいかもしれませんが、このお金を支払った後に実現された実際の税金割引を超える可能性があります。この金額を計算する際には、今後数年で想定される潜在的な税金優遇を使用できると仮定し、私たちに適用される税率はその年を終了するのと同じになるからです。
課税契約に基づいて支払われた金が我々が実現した実際の収入や特許経営税節約を超えていれば、我々の流動性に実質的な負の影響を与える可能性がある。また,課税契約による支払い義務には,何らかの合併,資産売却,その他の形態の業務合併や他の制御権変更の遅延,遅延または阻止の効果が生じる可能性がある.もし私たちの現金資源が時間差やその他の理由で課税契約の下での義務を履行するのに十分でない場合、私たちは未収税金協定での支払いを支払うために追加の債務を発生する必要があるかもしれません。このような債務は私たちの財政状況に実質的な悪影響を及ぼすかもしれない。
最後に,我々は持ち株会社であるため,自分の業務がないため,課税契約に基づいて金を支払う能力はPKLPが我々に割り当てる能力に依存する.もし吾らが何らかの理由で課税項目合意に基づいて支払うことができなかった場合、その等の支払いは遅延し、支払い前に利息を計上することになり、当社の経営業績に悪影響を与え、当社の当該等の支払い期間中の流動資金に影響を与える可能性がある。
91
私たちはケイマン諸島の免税会社です。私たちの株主の権利はアメリカ司法管轄区域の法律によって管轄されている株主の権利とは異なるかもしれない。
私たちはケイマン諸島の免税会社です。私たちの会社の問題は私たちの憲章とケイマン諸島の法律によって引き続き管轄されるだろう。株主の権利および取締役会メンバーの責任は、米国司法管轄区域の法律によって管轄されている会社の株主権利および取締役の責任とは異なる可能性がある。支払能力のあるケイマン諸島免除会社の取締役会は、その職責を履行する際に、その会社の最大の利益を考慮しなければならず、これはその1つまたは複数の個人株主の利益とは異なる可能性がある。
プロジェクト1 Bそれは.未解決の従業員のコメント。
ない。
プロジェクト1 C。ネットワークセキュリティです。
不正アクセス、ネットワークセキュリティ攻撃、およびハッカーによって実装されたセキュリティイベント、およびハードウェアおよびソフトウェアシステムへの意図しない破損または中断、データ損失、および機密情報が流用されるなど、ネットワークセキュリティに関連するリスクに直面しています。ネットワークセキュリティ脅威からの重大なリスクを識別と評価するために、著者らは、合理的に予測可能なネットワークセキュリティリスクと脅威を識別、保護、検出と応答し、管理するための企業範囲の情報セキュリティ計画を維持した。我々の情報システムをネットワークセキュリティの脅威から保護するために、私たちは様々なセキュリティツールを使用して、迅速な予防、識別、アップグレード、調査、識別された脆弱性とセキュリティ事件を解決し、そこから回復するのを助ける。
当社の業務、製造運営、臨床試験、プライバシーおよびコンプライアンスの問題に関連するネットワークセキュリティリスクは、第三者評価、内部IT監査、ITセキュリティ、リスクおよびコンプライアンス審査によって識別および解決されます。ネットワークセキュリティイベントを防御、検出、対応するために、我々はシステムとアプリケーションに対して能動的なネットワークセキュリティ審査を行い、外部第三者ツールと技術を用いて浸透テストを実行してセキュリティ制御をテストし、従業員訓練を行い、データ保護と情報セキュリティに関連する新しい法律法規を監視し、適切な変更を実施する。
私たちのプロセスはまた、第三者サービスプロバイダの使用に関連するネットワークセキュリティ脅威リスクを解決し、第三者サービスプロバイダは、私たちのプロバイダおよび製造業者を含むか、または患者および従業員データまたは私たちのシステムにアクセスすることができる人を含む。また、ネットワークセキュリティ面の考慮は、第三者サービスプロバイダの選択と監視にも影響を与える。我々は,我々のシステム,データ,またはそのようなシステムやデータを格納する施設にアクセスできる第三者を調査し,そのような調査によって発見されたネットワークセキュリティ脅威リスクを監視し続ける.
国家標準·技術研究所(NIST)イベント応答の枠組みに基づくイベント応答とデフォルト管理プロセスを実施した。これには,1)明確な役割と事故応答起動プロセス,2)事故検出と分析,3)抑制,根絶と回復,および4)事故後分析がある。このようなイベント応答と関連するネットワークセキュリティ事務は、私たちの情報技術、製造、臨床運営、監督と法律チームの指導者によって監督される。
私たちは、定期的なネットワークと端末監視、監査、脆弱性評価、浸透テストを含む一連のツールとサービスを使用して、私たちのリスク識別と評価に情報を提供します。また、私たちは外部監査人とコンサルタントを招いて、私たちの内部ネットワークセキュリティ計画と適用実践と基準の遵守状況を評価します。
セキュリティイベントとデータイベントを評価し、重大度順にソートし、応答と修復の優先度を決定する。重要性および運営および業務影響を決定し、プライバシーへの影響を検討するためにイベントを評価する。
上記の流れの一部として,外部監査人とコンサルタントを招いて,我々の内部ネットワークセキュリティ計画および適用実践と基準の遵守状況を評価する.
我々は、“我々の内部コンピュータシステム、または私たちの協力者または他の請負者またはコンサルタントのコンピュータシステムに障害が発生したり、セキュリティホールに遭遇したりする可能性があり、これは、我々の製品開発計画の重大な中断をもたらす可能性がある”というタイトルの下で、識別されたネットワークセキュリティ脅威(これまでの任意のネットワークセキュリティイベントを含む)からのリスク、および私たちの業務戦略、運営結果、または財務状況を含む私たちにどのように大きな影響を与えるかを記述する。
私たちはどんな重大なサイバーセキュリティ事件も経験したことがなく、私たちがサイバーセキュリティ事件によって発生した費用も取るに足らない。
サイバーセキュリティ·ガバナンス
ネットワークセキュリティは私たち全体のリスク管理プロセスの重要な構成部分であり、私たちの取締役会と経営陣が注目している分野でもある。私たちの取締役会の監査委員会は、私たちのネットワークセキュリティリスクを監督し、四半期ごとに上級副総裁情報技術部門の報告を受けます。これには、既存および新しいネットワークセキュリティリスク、管理層がどのように解決するか、および/または
92
これらのリスク、ネットワークセキュリティ、データプライバシーイベント(あれば)、重要な情報セキュリティの状況、業界傾向、その他の重要な分野を低減します。
私たちはまた、私たちの上級副総裁、情報技術、首席財務官、首席法務官が率いる情報技術管理委員会を設立した。この委員会のメンバーは、上述のネットワークセキュリティリスク管理と戦略プロセスを管理と参加することによって、私たちのイベント対応計画の運営を含めて、ネットワークセキュリティイベントの予防、緩和、検出と修復を理解し、監視する。
プロジェクト2それは.財産です。
2026年9月から2029年8月までの期限が切れた賃貸契約によると、ノースカロライナ州ウィンストン-セレムで合計約110,700平方フィートのオフィス、製造、研究空間を借りた。これは私たちの主な実行オフィスとしてウィンストン-セレムのオフィス空間を含む。また,2027年7月と2028年3月にそれぞれ満期になった賃貸契約に基づき,ノースカロライナ州の研究三角公園地域で合計約12,400平方フィートのオフィスと実験室空間をレンタルした。最後に、私たちはマサチューセッツ州ボストンで約7400平方フィートのオフィス空間を借りました。レンタル契約は2029年1月に満期になります。
2023年の間、私たちはノースカロライナ州グリーンスバーレで210,000平方フィートの施設と約22エーカーの土地を購入して、Reactionが規制部門の承認を得ないように、私たちの商業製造需要の準備をしています。
第3項それは.法律訴訟。
時々、私たちは訴訟や他の法的手続きに巻き込まれるかもしれない。私たちは現在、私たちの経営陣が私たちの業務に重大な悪影響を及ぼす可能性があると考えている訴訟や法的手続きに参加していません。結果にかかわらず,弁護や和解コスト,管理資源分流などにより,訴訟は我々に悪影響を与える可能性がある。
プロジェクト4それは.炭鉱の安全情報開示。
適用されません。
第II部
第5項それは.登録者普通株、関連株主事項及び発行者が株式証券を購入する市場。
市場情報
私たちのA類普通株はナスダック資本市場に上場して、コードはPROKです。
配当政策
今まで、会社は普通株式権益について現金配当金を発表したり、支払ったりしたことがない。
所持者
2024年3月21日現在,約61,621,330株のA類普通株が39名の記録保持者から発行·保有されており,約167,722,201株B類普通株は3名の記録保持者が発行·保有している。我々のA類普通株の大部分はブローカー,被著名人,他機関代表株主が保有しているため,これらの記録保持者に代表される株主総数を見積もることはできない.私たちのB種類普通株は公開市場を持っていない。
未登録証券販売
適用されません。
発行人が株式証券を購入する
次の表は、2023年12月31日までの四半期における会社A類普通株買い戻しに関する情報を報告した
93
期間 |
|
購入株式総数(1) |
|
|
1株平均支払価格 |
|
|
公開発表された計画または計画の一部として購入した株式総数 |
|
|
計画や計画に基づいて購入可能な最大株式数 |
|
||||
2023年10月1日-31日 |
|
|
– |
|
|
$ |
– |
|
|
|
– |
|
|
|
– |
|
2023年11月1日-30日 |
|
|
7,256,367 |
|
|
$ |
1.309 |
|
|
|
– |
|
|
|
– |
|
2023年12月1日-31日 |
|
|
– |
|
|
$ |
– |
|
|
|
– |
|
|
|
– |
|
*合計 |
|
|
7,256,367 |
|
|
|
|
|
|
– |
|
|
|
– |
|
|
(1)2023年11月19日、ProKidney Corp.(“貴社”)とSC PIPE Holdings LLC及びSC Master Holdings,LLC(“売却株主”)と株式買い戻し協議(“株式買い戻し協議”)を締結することにより、当社は売却株主にA類普通株7,256,367株を購入することに同意し、1株当たり額面0.0001ドル、1株購入価格は1.309ドル(“株式買い戻し”)である。同社が株式買い戻しで支払った総価格は約950万ドル。株式買い戻しは2023年11月21日に終了した。株式買い戻しはProKidney取締役会の満場一致で承認され、既存の株買い戻し計画の一部ではない。
プロジェクト6それは.保留します。
プロジェクト7それは.経営陣の財務状況と経営結果の検討と分析。
本10-K表年次報告で使用される“会社”、“登録者”、“私たち”または“私たち”は、ProKidney社およびその子会社を意味する。以下、我々の財務状況と経営結果の検討と分析は、当社の財務諸表と本報告の他の部分に関する付記と併せて読まなければなりません。歴史財務情報以外に、以下の討論には、私たちの計画、推定、仮説、信念を反映した前向きな陳述が含まれている。私たちの実際の結果は展望的陳述で議論された結果と大きく違うかもしれない。これらの差異をもたらすか、または促進する可能性のある要因は、本報告の他の部分“第1の部分である第1のリスク要因”の下で議論される要因を含む。展望的陳述は、私たちの可能性または仮定された将来の経営結果、業務戦略および経営、融資計画、潜在的成長機会、潜在的市場機会、私たちの薬物開発努力または試験の潜在的結果、および競争影響に関する情報を含む。前向き表現は、すべての非歴史的事実の表現を含み、“予想”、“信じ”、“可能”、“求める”、“推定”、“予想”、“意図”、“可能”、“計画”、“潜在”、“予測”、“プロジェクト”、“すべき”、“将”、“将”または同様の表現、およびこれらの用語の否定によって識別することができる。このような不確実性を考慮して、あなたはこのような前向きな陳述に過度に依存してはいけない。また、前向きな陳述は、私たちの経営陣の本報告日までの計画、推定、仮説、信念だけを代表しています。法的要件がない限り、私たちは、未来に新しい情報があっても、これらの前向き陳述を公開的に更新する義務がない、または実際の結果を更新することは、未来に新しい情報があっても、これらの前向き陳述で予想される結果と大きく異なる原因である可能性がある。
概要
我々は臨床段階のバイオテクノロジー会社であり,変革性の特許細胞治療プラットフォームを有し,患者自身が治療しようとしている患者から分離した細胞を用いて多様な慢性腎臓疾患を治療することができる。著者らの方法は慢性腎臓疾患(CKD)の治療を再定義し、腎不全の治療から腎機能の回復、保存或いは改善に重点を移し、CKDの進展を阻止或いは遅延させることを試みた。著者らの主要な候補製品はREACT(Rilparencel)と呼ばれ、CKD患者の罹患腎臓の腎機能を保護することを目的としている。Reactionは、患者自身の自己腎細胞から調製された精選腎細胞(“SRC”)を含む製品である。SRCは、必要に応じて繰り返すことができる低侵襲外来プログラムを用いて患者の腎臓に再注入することができる製品として調製される。Reactionは患者自身の腎臓から作製した細胞からなる個人化治療であるため,免疫抑制療法を用いる必要はなく,患者が別の異遺伝子ドナーの腎臓移植を受ける場合,患者は一生の間免疫抑制療法を使用する必要があるからである。
われわれは現在,重度糖尿病腎症(“DKD”)までの被験者の反応に用いられる3期開発計画と複数の2期臨床試験を行っている。REPACTはすでにアメリカ食品と薬物管理局(FDA)の再生医学高度療法(RMAT)の称号を獲得した。これまで,1期と2期の臨床試験では,中から重度のDKDを有する被験者はREPACTに対する耐性が通常良好であった。著者らは最近、先天性腎臓と尿路異常患者(“CAKUT”)の治療に用いられる臨床試験を完成した。
設立以来、私たちはほとんどの資源を資金調達、組織、会社員、商業と科学計画の配備、発見と研究活動の展開、候補製品の買収や発見、
94
著者らの知的財産権の組合せを確立と保護し、開発と進展反応を行い、臨床試験の準備を行い、第三者と部品材料の製造について手配を確立し、そしてこれらの業務に一般と行政支持を提供する。私たちは候補製品の販売が許可されていないし、製品販売から何の収入も得られていない。
他の傾向や不確実性は
2022年、世界各国の中央銀行(米国連邦準備委員会を含む)が次々と利上げを行った。これらの金利引き上げはこれまで会社に大きな悪影響を与えていないが、このような金利引き上げが金融市場や経済全体に与える影響は、合理的な条項や必要に応じて資金を得ることをより困難かつ高価にすることを含む将来的に会社に悪影響を及ぼす可能性がある。また、世界経済は高インフレとグローバルサプライチェーンの中断を経験し続けていくだろう。私たちはこれらのサプライチェーン、インフレ、金利要因、そして全体の経済環境による不確実性を監視し続けている。
また、私たちはロシア、ベラルーシ、ウクライナ、イスラエル、パレスチナで業務や直接業務をしていないにもかかわらず、ロシア-ウクライナの軍事衝突が世界経済に悪影響を与えているため、グローバルサプライチェーンが中断され、供給面で限られた制限があり、いくつかの材料や用品を得るのに必要なコストも増加している。今まで、私たちの業務は紛争の実質的な影響を受けていませんでした;しかし、紛争の継続や悪化に伴い、それは私たちの業務、財務状況、または運営結果に悪影響を及ぼす可能性があります。
財務運営の概要
収入.収入
私たちは設立以来何の収入も生まれていませんし、近い将来に製品販売から何の収入も生まれたくありません。もしあれば。Reactionや任意の他の候補製品の開発に成功して市場承認を得た場合、または第三者と協力または許可協定を締結した場合、将来的に製品販売またはそのような合意の支払いから収入を得ることができるかもしれません。
費用.費用
研究と開発費
研究と開発費用には,主に我々の研究や開発活動に関するコストが含まれており,Reactionの開発を含む.
研究開発コストには
私たちは発生した費用に応じて研究と開発費用を支払う。我々は,サプライヤが提供してくれた情報に基づいて特定のタスク達成の進捗を評価し,外部開発コストを確認した.これらの活動の支払いは、発生したコストパターンとは異なる個別合意の条項に基づく可能性があり、私たちの総合貸借対照表には、前払い臨床費用として、または計算されるべき総費用および他の費用の一部として反映される。将来的に研究·開発活動のための貨物やサービスの前金は返金されず,研究·開発が将来的に他の用途がなくても延期して資本化しなければならない。資本化金額は前払い臨床費用として記録され,関連貨物交付またはサービス実行時に費用を計上する。
研究開発活動は私たちのビジネスモデルの核心だ。予測可能な将来,REPACTが臨床開発の後期に入るにつれて,我々の研究や開発費用は大幅に増加することが予想される。
95
Reactionの成功開発と我々が将来開発可能などの候補製品も大きな不確実性を持っている.したがって、私たちは、私たちの任意の候補製品の開発および商業化に必要な努力の性質、時間、および見積もりコストを合理的に推定または知ることができない。いつ(あれば)Reactionや潜在的な未来候補製品の販売から大量の現金が純流入するかも予測できないが,承認されれば。これは、開発候補製品に関連する多くのリスクおよび不確実性が原因であり、その中の多くのリスクおよび不確実性は、以下の不確実性を含む我々の制御範囲内にないからである
これらの変数の結果の任意の変化は、我々の候補製品開発に関連するコストおよび時間の大きな変化を意味する可能性がある。例えば、FDAまたは他の規制機関が、私たちが予想以上に候補製品の臨床開発を完了するために必要な臨床試験を要求した場合、あるいは患者登録やその他の理由で私たちの臨床試験に重大な遅延が生じた場合、私たちは大量の追加の財政資源と時間をかけて臨床開発を完成させることを要求される。私たちのどんな候補製品も規制部門の承認を受けることができないかもしれない。
一般と行政費用
一般及び行政支出は主に人事に関連するコストを含み、執行、財務、会社及び行政機能に関連する個人の賃金、ボーナス、福祉及び持分補償支出、及び外部専門サービスの支出は、法律、監査、会計及び税務関連サービス及びその他の顧問費を含む;施設に関連する支出は、減価償却コスト及びその他分配された施設賃貸料及び修理費用、保険料、募集費用、出張支出及びその他の一般行政支出を含む。
私たちは、予見可能な未来に、私たちの業務の拡大とより多くの人員を募集して私たちの運営を支援することに伴い、私たちの一般的かつ行政的費用が大幅に増加すると予想しています。また、法律、監査、会計、投資家·広報、税務関連サービス、役員·高級社員保険の費用、米国証券取引委員会(“米国証券取引委員会”)の規則や規定の遵守、全国的な証券取引所に上場する企業に適用される上場基準に関する規制費用など、上場企業に関連する費用が増加することも予想される。
96
その他の収入(費用)
他の収入は主に現金、現金等価物、および有価証券の利息収入を含む。
所得税給付
所得税支出は我々の子会社が米国所得税を目的としてC社のために獲得した収入を組織した連邦税と州税を反映している。
経営成果
本節では,2023年12月31日までの年度と2022年12月31日までの年度の経営結果について検討する。2022年12月31日までの年度と2021年12月31日までの年度比較については、2023年3月28日に米国証券取引委員会に提出されたForm 10−K年度報告書の“経営陣の財務状況と経営結果の検討と分析”の部分を参照されたい。
2023年12月31日までと2022年12月31日までの年次比較
次の表は、2023年12月31日と2022年12月31日までの年間運営結果(単位:千)をまとめたものです
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
変わる |
|
|||
運営費用: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
$ |
106,707 |
|
|
$ |
82,070 |
|
|
$ |
24,637 |
|
一般と行政 |
|
|
44,815 |
|
|
|
70,937 |
|
|
|
(26,122 |
) |
総運営費 |
|
|
151,522 |
|
|
|
153,007 |
|
|
|
(1,485 |
) |
運営損失 |
|
|
(151,522 |
) |
|
|
(153,007 |
) |
|
|
1,485 |
|
利子収入 |
|
|
22,083 |
|
|
|
5,983 |
|
|
|
16,100 |
|
利子支出 |
|
|
(12 |
) |
|
|
(215 |
) |
|
|
203 |
|
税前純損失 |
|
|
(129,451 |
) |
|
|
(147,239 |
) |
|
|
17,788 |
|
所得税費用 |
|
|
5,996 |
|
|
|
896 |
|
|
|
5,100 |
|
非持株権益前純損失 |
|
|
(135,447 |
) |
|
|
(148,135 |
) |
|
|
12,688 |
|
非持株権益は純損失を占めなければならない |
|
|
(99,979 |
) |
|
|
(40,103 |
) |
|
|
(59,876 |
) |
A類普通株株主が獲得できる純損失 |
|
$ |
(35,468 |
) |
|
$ |
(108,032 |
) |
|
$ |
72,564 |
|
研究開発費
2022年12月31日までの年度と比較して、2023年12月31日までの年間の研究·開発費は約2460万ドル増加しており、主な原因は以下の通りである
一般と行政費用
2022年12月31日までの年度と比較して、2023年12月31日までの年度の一般·行政費は約2610万ドル減少しており、主な原因は以下の通りである
97
利子収入
2022年12月31日までの年度と比較して,2023年12月31日までの年度の利息収入が約1600万ドル増加したのは,平均現金と有価証券投資の利息増加および金利上昇によるものである。
所得税費用
2022年12月31日までの年度と比較して,2023年12月31日までの年間所得税支出が約510万ドル増加したのは,主に推定免税額の増加によるものであり,合格研究や開発コスト控除期間の影響である。
流動性と資本資源
流動資金源
私たちが設立して以来、私たちは何の収入も確認せず、運営損失と運営キャッシュフローが負になる状況が発生した。私たちはまだ何の製品も商業化していません。もしあれば、数年以内にどの製品の販売からも収入が生じないと予想しています。私たちが設立してから2023年12月31日まで、主にPKLP所有者の出資と業務合併と関連私募融資によって得られた収益を通じて、私たちの運営に資金を提供しています。
2024年1月に当社は公開市場販売協定を締結したSM(“販売契約”)Jefferies LLCを販売代理とすることにより、当社はA類普通株をJefferiesを通して随時発売することができ、1株当たり額面0.0001ドル、証券法第415(A)(4)条で定義されている“市場別発売”(“ATM発売”)のいずれの方式でも発売でき、総発行価格は最高100,000,000ドルに達する。当該等株式は、当社S-3用紙の棚上げ登録声明に基づいて発売及び販売されています。
2023年12月31日現在保有している既存の現金、現金等価物、有価証券は、2025年第4四半期の運営費と資本支出需要に資金を提供できるようになると予想されています。私たちの推定は間違っていることが証明される可能性のあるいくつかの仮定に基づいており、私たちは予想よりも早く私たちの資本資源を枯渇させるかもしれない。
私たちは私たちの費用が大幅に増加すると予想しています
また、業務合併が終了して以来、重大な法律、監査、会計、投資家と公共関係、規制、税務関連、役員および役員保険料、その他の費用を含む上場企業の運営に関連する追加コストが発生し始めている。臨床試験を行うことを含む薬物製品の開発は、時間がかかり、高価で不確定なプロセスであり、完成するのに数年かかり、私たちはいかなる候補製品の発売許可を得るために必要なデータや結果を得ることができないかもしれない、あるいは私たちが発売承認を得る可能性のある候補製品の販売から収入を得ることができる。しかも、私たちの候補製品が承認されれば、商業的成功を得られないかもしれない。私たちの商業収入は、もしあれば、少なくとも数年以内に商業使用に投入できないと予想される製品の販売から来ます。
98
したがって、私たちは私たちの持続的な運営を支援し、私たちの成長戦略を実施するために多くの追加資金を必要とするだろう。製品販売から相当な収入を得ることができる前に、株式、政府または個人団体の贈与、債務融資、または他の資本源(他社との潜在的な協力や他の戦略取引を含む)を公開または私的に売却することで、私たちの運営に資金を提供する予定です。私たちが株式または転換可能な債務証券を売却することによって追加資本を調達する場合、私たちの株主の所有権権益は希釈されるか、または希釈される可能性があり、これらの証券の条項は、清算または他の私たちの株主の権利に悪影響を及ぼす特典を含む可能性がある。債務融資および持分融資に関連する可能性のあるプロトコルは、追加債務を招く、資本支出を行う、または配当を宣言するなど、私たちが具体的な行動をとる能力を制限または制限する契約を含む。もし私たちが追加資金を得ることができない場合、私たちは研究開発計画の一部または全部の延期、減少、または廃止、製品の組み合わせ拡張、または任意の商業化努力を余儀なくされる可能性があり、これは私たちの業務の将来性に悪影響を及ぼすかもしれないし、運営を継続できないかもしれない。もし私たちが第三者との戦略的協力や他の同様の計画を通じて資金を調達する場合、私たちは私たちの技術、将来の収入源、研究計画または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になる可能性があり、および/または私たちの株式価値を低下させる可能性がある条項でライセンスを付与しなければならないかもしれない。私たちがより多くの資金を調達する能力は、潜在的な世界経済状況の悪化や米国や世界各地の信用·金融市場の中断や変動の悪影響を受ける可能性がある。製品開発に関連する多くのリスクや不確実性のため、費用増加の時間や金額を予測することはできず、永遠に利益を上げたり、経営活動から正のキャッシュフローが生じる保証もありません。
キャッシュフロー
2023年12月31日まで及び2022年12月31日までの年間現金流量
次の表は、2023年12月31日と2022年12月31日までの年間のキャッシュフロー情報(単位:千)を提供しています
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
経営活動のためのキャッシュフロー純額 |
|
$ |
(90,069 |
) |
|
$ |
(77,089 |
) |
投資活動のためのキャッシュフロー純額 |
|
|
(329,983 |
) |
|
|
(1,738 |
) |
融資活動が提供するキャッシュフロー純額( |
|
|
(9,551 |
) |
|
|
548,521 |
|
現金と現金等価物の純変化 |
|
$ |
(429,603 |
) |
|
$ |
469,694 |
|
経営活動
2023年12月31日までの1年間で、経営活動に用いられた現金純額は約9010万ドルで、非持株権益を差し引く前の純損失は約1兆354億ドルだった。運営資金変動約1,670万ドルおよび非現金費用と投資収益2,870万ドルで上記用途を相殺した。非現金費用には、主に3080万ドルの株式ベースの給与支出と390万ドルの減価償却と償却支出が含まれる。運営資金の変化は,主に我々のサプライヤーへのサービス提供時間の支払いに関するものである.
2022年12月31日までの1年間で,経営活動に用いられた現金純額は約7710万ドル,非持株権益を差し引く前の純損失は1.481億ドルであり,主に運営資本の変化により約650万ドルであった。このような用途は7750万ドルの非現金費用によって部分的に相殺される。非現金費用には、主に7450万ドルの持分ベースの給与支出と、300万ドルの減価償却と償却支出が含まれる。運営資金の変化は,主に我々のサプライヤーへのサービス提供時間の支払いに関するものである.
2022年12月31日までの年度と比較して,2023年12月31日までの年間で,経営活動のための現金が約1300万ドル増加したのは,主に非現金費用を差し引いて非制御権益を差し引く前の純損失の増加と,約3610万ドルの投資収益がサプライヤーへの支払い時間に推進された運営資本変化の影響によって相殺されたためである。
投資活動
2023年12月31日と2022年12月31日までの年度の投資活動用現金純額はそれぞれ約3億3千万ドルと170万ドルだった。2023年12月31日までの1年間、投資活動のための現金は、主に業務合併で調達された収益の一部を有価証券に投資し、ノースカロライナ州グリーンズベリーで土地と建物を2550万ドルで購入するために使われている。
融資活動
2023年12月31日と2022年12月31日までの年間、融資活動と融資活動による現金純額はそれぞれ960万ドルと5億485億ドルだった。2023年12月31日までの年度の融資活動の主な推進力は買い戻しである
99
2022年12月31日までの年度の融資活動は、業務合併で得られた金を反映している。
重要な会計政策と重大な判断と見積もり
私たちの経営陣の財務状況と経営結果の議論と分析は、私たちの合併財務諸表に基づいています。私たちの連結財務諸表は公認会計基準に基づいて作成された。我々の連結財務諸表および関連開示を作成する際には、総合財務諸表における資産、負債、コストおよび費用に影響を及ぼす報告金額、または資産および負債の開示に影響を及ぼす推定および判断を行う必要がある。我々は,歴史的経験,既知の傾向や事件,および当時の状況で合理的と考えられる様々な他の要因に基づいて推定し,これらの要因の結果は資産や負債の帳簿価値の判断の基礎を構成しているが,これらの資産や負債の帳簿価値は他の源から容易に見られるものではない.私たちは持続的な基礎の上で私たちの推定と仮定を評価する。異なる仮定や条件の下で、私たちの実際の結果はこれらの推定とは異なる可能性がある。
我々の重要な会計政策は本年報の他の部分の審査総合財務諸表付記2に掲載されている。
最近の会計公告
本年度報告書に含まれる総合財務諸表付記2は、最近発表された当社の財務状況及び経営結果に影響を及ぼす可能性のある会計声明の説明を開示している。
雇用法案会計選挙
私たちは改正された2012年にJumpStart Our Business Startups Act(“JOBS Act”)で定義されたように、新興の成長型会社です。雇用法案では、新興成長型企業は、上場企業に適用される新しいまたは改正された会計基準を遵守するために長い過渡期を利用することができ、これらの基準が民間企業に適用されるまでこれらの基準の採用を延期することを可能にする。雇用法案に基づいて、私たちはこの延長された過渡期を使用することを選択した。したがって、我々の総合財務諸表は、上場企業に適用される新しい会計基準や改正会計基準の発効日を遵守しなければならない会社の財務諸表と比較できない可能性があり、投資家に対する私たちの普通株の吸引力を低下させる可能性がある。
第七A項それは.市場リスクに関する定量的で定性的な開示。
取引法第12 b-2条の定義によると、我々は小さな報告会社であり、本プロジェクトに要求される他の情報を提供する必要はない。
プロジェクト8それは.財務諸表と補足データ。
本プロジェクトに必要な資料は,本年度報告第4部第15項に掲げる財務諸表及び補足資料に掲載した。
プロジェクト9それは.会計や財務開示における会計士との変更と食い違い。
適用されません。
第9 A項それは.制御とプログラムです
情報開示制御とプログラムの評価
我々の最高経営責任者と最高財務責任者の監督·参加の下で、経営陣は、2023年12月31日までの開示制御および手順(1934年証券取引法規則13 a-15(E)または15 d-15(E)で定義されているような)の設計および運営の有効性を評価した。この評価に基づき、我々の最高経営責任者および最高財務官は、2023年12月31日まで、我々の開示制御およびプログラムが、我々(我々の連結子会社を含む)に関する重要な情報を管理層にタイムリーに記録、処理、まとめ、報告させることを効果的に促し、米国証券取引委員会開示義務に基づいて、公開開示の品質と即時性を確保すると結論した。
私たちの経営陣は、私たちのCEOやCEOを含めて、私たちの開示制御や手続きがすべてのミスやすべての詐欺を防ぐことができることを期待していません。発想や動作がどんなに良くても、絶対的な保証ではなく、合理的な保証しか提供できず、制御システムの目標が実現されることを確保する制御システム。また,制御システムの設計は,資源制約が存在し,そのコストに対する制御の利点を考慮しなければならないという事実を反映しなければならない.
100
すべての制御システムの固有の限界により,どの制御評価も会社のすべての制御問題や不正事件(あれば)が発見されていることを絶対に保証することはできない.これらの固有の限界には,意思決定における判断が誤りである可能性があり,簡単な誤りや誤りによって故障が発生する可能性があるという現実がある.また、ある人の個人的な行為、2人以上の談合や管理層のコントロールを凌駕することは、コントロールを回避することができる。
任意の制御システムの設計も、将来のイベント可能性のいくつかの仮定に部分的に基づいており、どの設計もすべての潜在的な未来の条件でその目標を成功的に達成することが保証されていない。時間の経過とともに,制御は条件の変化や政策やプログラムへの遵守度の悪化により不十分になる可能性がある.コスト効果のある制御システムの固有の制限により,誤りや詐欺による誤った陳述が発生する可能性があり,発見されない可能性がある.
経営陣財務報告内部統制年次報告書
経営陣は、取引法第13 a-15(F)条の規定に従って、財務報告の十分な内部統制を確立·維持する責任がある。財務報告の内部統制は、財務報告の信頼性を合理的に保証することを目的としたプログラムであり、公認された会計原則に基づいて外部報告目的の財務諸表を作成する。財務報告書に対する私たちの内部統制は以下の書面政策と手続きを含む
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来的にどのような有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムの遵守度が悪化する可能性がある.
経営陣は、財務報告書の内部統制に対する2023年12月31日までの我々の有効性を評価した。経営陣は、テレデビル委員会後援組織委員会(COSO)が発表した“内部統制--総合枠組み(2013)”に記載されている基準に基づいてこの評価を行った。この評価に基づき、経営陣は、2023年12月31日現在、財務報告に対して有効な内部統制を維持していると判断した。
財務報告の内部統制の変化
最近の財政四半期では、財務報告の内部統制に大きな影響を与えていないか、または合理的に財務報告の内部統制に大きな影響を与える可能性のある変化が発生しています。
報告書と他社の管理情報のサイト可用性
会社は全面的な会社管理計画を持っており、取締役会の会社管理基準、取締役独立性を評価する取締役会準則及び監査委員会、指名と会社管理委員会及び人材と報酬委員会の定款を含む。同社には、会社のウェブサイトwww.prokinney.comが設置されており、株主および他の興味のある人は、同社の管理材料および特定の米国証券取引委員会の届出書類を無料で閲覧することができ、これらの材料は、通常、米国証券取引委員会に届出書類を提出するのと同じ営業日に米国証券取引委員会のウェブサイトで閲覧することができる。私たちのウェブサイトの内容は今年度の報告書の一部ではない。
プロジェクト9 Bそれは.他の情報。
適用されません。
プロジェクト9 Cです。検査を妨害する外国司法管轄区域を開示する。
適用されません。
101
パ.パRT III
第10項それは.役員、幹部、会社が管理する。
本プロジェクトに必要な情報は、2023年12月31日までの会計年度120日以内に米国証券取引委員会に提出される2024年株主年次総会依頼書に引用することにより、“管理と会社管理”、“延滞16(A)条報告”、“道徳及び商業行動準則”と題する議論に組み込まれている。依頼書が2023年12月31日までの財政年度終了後120日以内に提出されなければ,本プロジェクトに要求される情報は10-K/Aテーブルに含まれる.
プロジェクト11それは.役員報酬。
本プロジェクトに要求される情報は,2023年12月31日までの財政年度の120日以内に米国証券取引委員会に提出される2024年株主総会の依頼書に引用により組み込まれ,これに対する議論を参考にして,“役員と米国証券取引委員会報酬”と題する。依頼書が2023年12月31日までの財政年度終了後120日以内に提出されなければ,本プロジェクトに要求される情報は10-K/Aテーブルに含まれる.
プロジェクト12それは.いくつかの実益所有者の保証所有権及び管理層及び関連株主事項。
本プロジェクトに要求される情報は、2023年12月31日までの財政年度120日以内に米国証券取引委員会に提出される2024年株主総会の依頼書に引用により組み込まれ、2023年12月31日までの財政年度内に米国証券取引委員会に提出される2024年株主総会の委託書に引用され、タイトルは“ある利益所有者と経営層の担保所有権”と“持分補償計画情報”である。依頼書が2023年12月31日までの財政年度終了後120日以内に提出されなければ,本プロジェクトに要求される情報は10-K/Aテーブルに含まれる.
第13項それは.特定の関係と関連取引、そして役員の独立性。
本プロジェクトに必要な情報は,2023年12月31日までの財政年度120日以内に米国証券取引委員会に提出される2024年株主総会の依頼書に引用により組み込まれ,これに関する議論を参考にして“何らかの関係と関連者取引”と“管理とコーポレート管理”と題する。依頼書が2023年12月31日までの財政年度終了後120日以内に提出されなければ,本プロジェクトに要求される情報は10-K/Aテーブルに含まれる.
プロジェクト14それは.首席の料金とサービス料です。
本プロジェクトに要求される情報は,2023年12月31日までの財政年度から120日以内に米国証券取引委員会に提出された2024年株主周年総会依頼書の中で独立公認会計士事務所の選択を承認することに関する検討を参考にした。依頼書が2023年12月31日までの財政年度終了後120日以内に提出されなければ,本プロジェクトに要求される情報は10-K/Aテーブルに含まれる.
102
第4部
プロジェクト15それは.展示品と財務諸表明細書。
(A)(1)財務諸表
以下のファイルは、本ファイルに添付されているF−1~F−26ページに含まれ、本年度レポートテーブル10−Kの一部として提出される
独立公認会計士事務所報告 |
F-2 |
|
|
2023年12月31日と2022年12月31日までの連結貸借対照表 |
F-3 |
|
|
2023年12月31日まで、2022年と2021年12月31日までの年度の総合業務報告書 |
F-4 |
|
|
2023年12月31日現在、2022年12月31日と2021年12月31日までの合併全面損失表 |
F-5 |
|
|
2023年12月31日まで、2022年12月31日と2021年12月31日までの年度償還可能非持株権益と株主赤字/メンバー権益変動表 |
F-6 |
|
|
2023年12月31日現在、2022年12月31日現在、2021年12月31日までの統合現金フロー表 |
F-10 |
|
|
連結財務諸表付記 |
F-11 |
(A)(2)財務諸表付表
適用されません。
(A)(3)展示品リスト
展示品 番号をつける |
|
説明する |
|
|
|
2.1 |
|
業務合併協定は,2022年1月18日にProKidney Corp.(前身はSocial Capital Suvretta Holdings Corp.III)とProKidney LPが署名した。(引用合併によって2022年1月21日に米国証券取引委員会に提出された表8-K)(文書番号001-40560)。 |
|
|
|
2.2 |
|
新しいGP加盟日は、2022年6月7日(添付ファイルを参照して2.2~2022年7月15日に米国証券取引委員会に提出された8-K表)(文書番号001-40560)である。 |
|
|
|
3.1 |
|
2回目の改訂及び再改訂されたProKidney Corp.の組織覚書及び規約(2022年7月15日から米国証券取引委員会に提出された表3.1~Form 8−K)を引用して提出する(文書番号001−40560)。 |
|
|
|
4.1* |
|
証券登録の説明 |
|
|
|
10.1 |
|
課税課税協定は、期日は2022年7月11日であり、ProKidney Corp.(前身は社会資本Suvretta Holdings Corp.III)、TRA党代表(その中で定義)とTRA当事者(引用合併により、2022年7月15日に米国証券取引委員会に提出された8-K表の第001-40560号文書)によって締結される。 |
|
|
|
10.2 |
|
交換協定は、2022年7月11日、ProKidney Corp.(前身は社会資本Suvretta Holdings Corp.III)、ProKidney LP(その一般パートナーProKidney Corp.GP Limitedにより行動)、その中で指定された特定の所持者(引用により表10.2から引用により、2022年7月15日に米国証券取引委員会に提出された8-K表)が締結される(文書番号001-40560)。 |
|
|
|
10.3 |
|
ロックアップ協定は,期日は2022年7月11日であり,ProKidney Corp.(前身はSocial Capital Suvretta Holdings Corp.III),SCSスポンサーIII LLC,保証人鍵保持者(ここで定義するように)とProKidney所有者(引用合併により2022年7月15日に米国証券取引委員会に提出されたForm 8−K表)によって締結される(文書番号001−40560)。 |
|
|
|
10.4 |
|
改訂·再署名された登録権協定は、2022年7月11日、ProKidney Corp.(旧社会資本Suvretta Holdings Corp.III)、SCSスポンサーIII LLC、ProKidney所有者(定義中)、Marc Semgran、Uma Sinha、Sukumar Nagendran、David明鏡、および投資家株主(定義はその中を参照)(添付ファイル10.4を参照して統合され、2022年7月15日に米国証券取引委員会に提出されたForm 8-K)(文書番号001-40560)である。 |
|
|
|
10.5* |
|
2回目の改訂と再署名されたProKidney LPという有限責任組合契約は、2022年7月11日、改訂日は2023年11月14日、Tholantia,LLC、資本制御会社S.A.de C.V.,ProKidney Management Equity LLC,ProKidney Corp.(前身はSocial Capital Suvretta Holdings Corp.III),ProKidney Corp.GP LimitedとProKidney GP Limitedとの間の合意改訂である。 |
|
|
|
103
10.6 |
|
機関投資家引受契約表は,期日は2022年1月18日であり,ProKidney Corp.(前身は社会資本Suvretta Holdings Corp.III)とその引受先(添付ファイル10.1から引用合併により,2022年1月21日に米国証券取引委員会に提出された8−K/A表)(書類第001−40560号)が署名された。 |
|
|
|
10.7 |
|
個人投資家引受契約表は,期日は2022年1月18日であり,ProKidney Corp.(前身は社会資本Suvretta Holdings Corp.III)と引受側(引用合併により組み入れられ,2022年1月21日に米国証券取引委員会に提出された8−K/A表)(文書番号001−40560)が署名された。 |
|
|
|
10.8 |
|
保証人支援協定は、2022年1月18日、ProKidney Corp.(前身は社会資本Suvretta Holdings Corp.III)、SCSスポンサーIII LLC、ProKidney LP、およびその中で言及された役員および上級管理職によって署名された(添付ファイル10.3から引用により、2022年1月21日に米国証券取引委員会の8-K/A表に提出された)(文書番号001-40560)。 |
|
|
|
10.9 |
|
会社単位所有者支援協定は,2022年1月18日にProKidney Corp.(前身は社会資本Suvretta Holdings Corp.III),ProKidney LP,その中で言及された者(引用合併により表10.10から2022年7月15日に米国証券取引委員会に提出された8-K表)によって署名された(文書番号001-40560). |
|
|
|
10.10 |
|
ProKidney Corp.2022年インセンティブ株式計画(添付ファイル10.11を参照して、2022年7月15日に米国証券取引委員会に提出された8-K表を形成する)(文書番号001-40560)。 |
|
|
|
10.11 |
|
ProKidney Corp.従業員株式購入計画(添付ファイル10.12~2022年7月15日に米国証券取引委員会に提出された8-K表を参照することにより)。 |
|
|
|
10.12 |
|
ProKidney Corp.とその役員と幹部の間で2022年7月11日に提出された賠償協定表(添付ファイル10.13から引用により、2022年7月15日に米国証券取引委員会に提出されたForm 8−K)(文書番号001−40560)。 |
|
|
|
10.13 |
|
雇用協定は,James CoulstonとProKidney,LLCによって署名され,2022年12月3日(添付ファイル10.14から引用合併により,2023年3月28日に米国証券取引委員会に提出されたForm 10−K)(文書番号001−40560)である。 |
|
|
|
10.14 |
|
取締役株式オプション奨励表(添付ファイル10.17~2023年3月28日に米国証券取引委員会に添付された10-K表を参照して組み込むことにより)。 |
|
|
|
10.15 |
|
従業員インセンティブ株式オプション奨励表(添付ファイル10.18~2023年3月28日に米国証券取引委員会に提出された10-K表を参照することによって提出される)(番号001-40560)。 |
|
|
|
10.16 |
|
ProKidney Corp.2022年取締役賠償政策(添付ファイル10.20を参照して、2023年3月28日に米国証券取引委員会に提出された10-K表を形成する)(文書番号001-40560)。 |
|
|
|
10.17 |
|
ProKidney短期インセンティブ業績計画(添付ファイル10.1~2023年1月19日に米国証券取引委員会に提出された8−Kフォームを参照することにより)(ファイル番号001−40560)。 |
|
|
|
10.18 |
|
ProKidney,LLCとBruce Culletonの間の雇用協定は,2023年12月3日である。(参照合併によって2023年12月5日から米国証券取引委員会に提出された表8-K/A)(文書番号001-40560)。 |
|
|
|
10.19 |
|
George Clinic Pty LimitedとProKidney(前身はRegenMed(Cayman)Ltd.)が2021年2月15日に締結した主サービス協定。(表10.13より引用して、S-1を形成し、2022年8月9日に米国証券取引委員会に届出する)(アーカイブ番号333-266683)。 |
|
|
|
10.20 |
|
研究、開発、工事サービスと許可証の覚書と協定、日付は2022年1月16日、ProKidney(前身はRegenMed(Cayman)Ltd.)デカ製品有限責任組合企業(添付ファイル10.14から引用合併し、2022年8月9日に米国証券取引委員会に届出したS-1を構成する)(文書番号333-266683). |
|
|
|
10.21 |
|
ProKidney(前身はRegenMed(Cayman)Ltd.)が2020年4月2日に署名した臨床試験サービス主契約Frenova,LLC(引用合併により,2022年8月9日に米国証券取引委員会に提出されたS-1に形成された)(写本第333-266683号). |
|
|
|
10.22 |
|
主サービス協定は、日付は2019年5月1日であり、PPD Development、LPとProKidney(前身はRegenMed(Cayman)Ltd.)によって署名された。(添付ファイル10.17を参照して合併し、S-1を形成し、2022年8月9日に米国証券取引委員会に届出する)(アーカイブ番号333-266683). |
|
|
|
10.23 |
|
2015年8月14日にCTI臨床試験サービス会社とRegenMedTX,LLC(表10.18から参照により組み込まれ、2022年8月9日に米国証券取引委員会に提出されたS-1)の間で締結された主サービス協定(文書番号333-266683)が形成された。 |
|
|
|
10.24 |
|
実験室サービス協定は、日付は2016年8月16日であり、Covance中央実験室サービス有限会社、Covance中央実験室サービスSAVI RLとProKidney(前RegenMed(ケイマン)有限会社)が署名した。(引用編入によりS-1が形成され、2022年8月9日に米国証券取引委員会に届出される)(アーカイブ番号333-266683)。 |
|
|
|
104
10.25 |
|
実験室サービス協定は、日付は2017年8月1日であり、Covance中央実験室サービス有限会社、Covance中央実験室サービスSA≡RLとProKidney(前身はRegenMed(Cayman)Ltd.)によって署名された。(表10.20より引用して、S-1を形成し、2022年8月9日に米国証券取引委員会に届出する)(アーカイブ番号333-266683)。 |
|
|
|
10.26 |
|
Covance中央実験室サービス有限会社、Covance中央実験室サービスSA≡RLとProKidney(前身はRegenMed(Cayman)Ltd.)が署名した実験室サービス協定は2019年6月21日である。(表10.21より引用し、S-1を形成し、2022年8月26日に米国証券取引委員会に届出する)(アーカイブ番号333-266683)。 |
|
|
|
10.27 |
|
実験室サービス協定は、期日は2021年9月16日であり、Labcorp中央実験室サービス有限会社、Labcorp中央実験室サービスSA≡RLとProKidney(前身はRegenMed(Cayman)Ltd.)によって署名された。(表10.22より引用して、S-1を形成し、2022年8月26日に米国証券取引委員会に届出する)(アーカイブ番号333-266683)。 |
|
|
|
10.28 |
|
コンサルティングサービス協定は、2020年1月1日まで、ProKidney(前身はRegenMed(Cayman)Ltd.(d/b/a in Regen))とNefro Health(添付ファイル10.30を参照することにより、2023年3月28日に米国証券取引委員会に提出されたForm 10-K)(文書番号001-40560)を提供する |
|
|
|
10.29 |
|
コンサルティングサービス協定は、2020年1月1日現在、ProKidney,LLC(Twin City Bio LLC)とNefro Health(添付ファイル10.31を参照することにより、2023年3月28日に米国証券取引委員会のForm 10−Kに提出される)(ファイル番号001−40560)。 |
|
|
|
10.30 |
|
改訂された購入契約は、2023年3月29日にProKidney Corp.と73 BCI 2 LLCによって署名された。(添付ファイル10.1に参照することによって、2023年8月10日に米国証券取引委員会に提出された10-Qテーブルを形成する)(文書番号001-40560)。 |
|
|
|
10.31 |
|
2023年7月17日にProKidney Corp.とProKidney Acquisition Company,LLCが署名した譲渡と売買契約を担当します。(添付ファイル10.2に参照することによって、2023年8月10日に米国証券取引委員会に提出された10-Qテーブルを形成する)(文書番号001-40560)。 |
|
|
|
10.32 |
|
公開市場販売プロトコルSMは、2024年1月19日、ProKidney Corp.およびJefferies LLC(参照により表1.1から統合され、2024年1月19日に米国証券取引委員会に提出されたForm 8−K)(文書番号001−40560)である。 |
|
|
|
21.1* |
|
登録者の子会社 |
|
|
|
23.1* |
|
安永法律事務所が同意した |
|
|
|
31.1* |
|
2002年のサバンズ·オキシリー法第302節で可決された1934年の証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて発行された首席執行幹事証明書。 |
|
|
|
31.2* |
|
2002年サバンズ−オキシリー法第302節で可決された1934年証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて首席財務幹事が認証された。 |
|
|
|
32.1* |
|
2002年にサバンズ·オクスリ法案第906条で可決された“米国法典”第18編1350条に規定されている最高経営責任者証明書。 |
|
|
|
32.2* |
|
2002年の“サバンズ-オックススリー法案”906節で採択された“米国法典”第18編1350条による首席財務官の認証。 |
|
|
|
97* |
|
ProKidney Corp政策を取り戻す |
|
|
|
101* |
|
以下の材料は、会社の2023年12月31日までの10-K表年次報告から抜粋し、フォーマットはiXBRL(内連拡張可能商業報告言語):(I)合併貸借対照表、(Ii)連結経営報告書、(Iii)非持株権益と株主損失を償還可能な合併変動表、(Iv)合併現金フロー表と(V)連結財務諸表に付記し、テキストブロックでラベルを表示し、詳細ラベルを含む。 |
|
|
|
104* |
|
本年度報告の表紙はForm 10−K,フォーマットはイントラネットXBRL,フォーマットは2023年12月31日までの年度である。 |
|
|
|
契約または補償計画または手配を管理する
*アーカイブをお送りします。
プロジェクト16それは.表格10-Kの概要
ない。
105
サイン
改正された1934年証券取引法第13又は15(D)節の要求に基づいて、登録者は、正式に許可された次の署名者がその代表を代表して本報告書に署名することを正式に手配した.
|
|
PROKIDNEY社です。 |
|
|
|
|
|
日付:2024年3月21日 |
|
差出人: |
/S/ブルース·カールトン |
|
|
|
ブルース·クルトン |
|
|
|
最高経営責任者 |
本報告書は、改正された1934年の証券取引法の要求に基づき、以下の登録者によって登録者として指定日に署名された。
名前.名前 |
|
タイトル |
|
日取り |
|
|
|
|
|
/S/ブルース·カールトン |
|
取締役CEO兼最高経営責任者 |
|
2024年3月21日 |
ブルース·クルトン |
|
(首席行政主任) |
|
|
|
|
|
|
|
/寄稿S/ジェームズ·コルストン |
|
*最高財務責任者 |
|
2024年3月21日 |
ジェームズ·クルストン |
|
(首席財務会計官) |
|
|
|
|
|
|
|
/S/パブロ·レゴレッタ |
|
彼は社長だ |
|
2024年3月21日 |
パブロ·レゴレタ |
|
|
|
|
|
|
|
|
|
/S/ウィリアム·F·ドイル |
|
役員.取締役 |
|
2024年3月21日 |
ウィリアム·F·ドイル |
|
|
|
|
|
|
|
|
|
/S/ジェニファー·フォックス |
|
役員.取締役 |
|
2024年3月21日 |
ジェニファー·フォックス |
|
|
|
|
|
|
|
|
|
/S/ホセ·イグナシオ·ヒメネス·サントス |
|
役員.取締役 |
|
2024年3月21日 |
ホセ·イグナチオ·ヒメネス·サントス |
|
|
|
|
|
|
|
|
|
書名/作者Alan M.Lotvin,M.D。 |
|
役員.取締役 |
|
2024年3月21日 |
アラン·M·ロトヴィン医学博士 |
|
|
|
|
|
|
|
|
|
/S/ジョン·M·マラガノール博士 |
|
役員.取締役 |
|
2024年3月21日 |
ジョン·M·マラガノ博士です |
|
|
|
|
|
|
|
|
|
書名/著者Brand J.G.Pereira,M.D. |
|
役員.取締役 |
|
2024年3月21日 |
ブライアン·J·G·ペレイラ医学博士 |
|
|
|
|
|
|
|
|
|
/S/ウーマ·シンハ、博士 |
|
役員.取締役 |
|
2024年3月21日 |
ウマ·シンハ博士です |
|
|
|
|
106
連結財務諸表索引
独立公認会計士事務所レポート(PCAOB ID:42) |
F-2 |
|
|
2023年12月31日と2022年12月31日までの連結貸借対照表 |
F-3 |
|
|
2023年12月31日まで、2022年と2021年12月31日までの年度の総合業務報告書 |
F-4 |
|
|
2023年12月31日現在、2022年12月31日と2021年12月31日までの合併全面損失表 |
F-5 |
|
|
2023年12月31日まで、2022年12月31日と2021年12月31日までの年度償還可能非持株権益と株主赤字/メンバー権益変動表 |
F-6 |
|
|
2023年12月31日現在、2022年12月31日現在、2021年12月31日までの統合現金フロー表 |
F-10 |
|
|
連結財務諸表付記 |
F-11 |
F-1
代表者独立公認会計士事務所のORT
ProKidney Corpの株主と取締役会へ
財務諸表のいくつかの見方
添付されているProKidney Corp.(当社)2023年12月31日現在、2023年12月31日と2022年12月31日までの総合貸借対照表、2023年12月31日までの3年度の関連総合経営表、全面赤字、償還可能非持株権益変化と株主赤字/メンバー持分と現金流量、および関連付記(総称して“総合財務諸表”と呼ぶ)を監査した。総合財務諸表は、すべての重要な点で、2023年12月31日、2023年12月31日、2022年12月31日までの会社の財務状況、および2023年12月31日までの3年度の経営結果とキャッシュフローを公平に反映しており、米国公認会計原則に適合していると考えられる。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査することを求められておらず、私たちも招聘されて監査を行っていません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性に対する意見を表明するためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
/S/安永法律事務所
2019年以来、当社の監査役を務めてきました。
ノースカロライナ州ローリー市
2024年3月21日
F-2
ProKidney Corp
統合された貸借対照表
(単位:千、共有データを除く)
|
2023年12月31日 |
|
|
2022年12月31日 |
|
||
資産 |
|
|
|
|
|
||
現金と現金等価物 |
$ |
|
|
$ |
|
||
有価証券 |
|
|
|
|
|
||
受取利息 |
|
|
|
|
|
||
前払い資産 |
|
|
|
|
|
||
前払い臨床 |
|
|
|
|
|
||
その他流動資産 |
|
|
|
|
|
||
流動資産総額 |
|
|
|
|
|
||
|
|
|
|
|
|
||
固定資産、純額 |
|
|
|
|
|
||
使用権資産、純額 |
|
|
|
|
|
||
無形資産、純額 |
|
|
|
|
|
||
総資産 |
$ |
|
|
$ |
|
||
|
|
|
|
|
|
||
負債と株主赤字/メンバー権益 |
|
|
|
|
|
||
売掛金 |
$ |
|
|
$ |
|
||
賃貸負債 |
|
|
|
|
|
||
費用その他を計算する |
|
|
|
|
|
||
所得税に対処する |
|
|
|
|
|
||
流動負債総額 |
|
|
|
|
|
||
|
|
|
|
|
|
||
所得税を納め,当期分を差し引いた純額 |
|
|
|
|
|
||
賃貸負債、当期分を差し引く |
|
|
|
|
|
||
総負債 |
|
|
|
|
|
||
|
|
|
|
|
|||
償還可能な非持株権益 |
|
|
|
|
|
||
|
|
|
|
|
|
||
株主赤字/会員権益: |
|
|
|
|
|
||
A類普通株、$ |
|
|
|
|
|
||
B類普通株、$ |
|
|
|
|
|
||
追加実収資本 |
|
|
|
|
|
||
その他の総合損失を累計する |
|
|
|
|
|
||
赤字を累計する |
|
( |
) |
|
|
( |
) |
株主赤字総額/メンバー権益 |
|
( |
) |
|
|
( |
) |
総負債と株主赤字/メンバー権益 |
$ |
|
|
$ |
|
||
付記はこれらの連結財務諸表の構成要素である。
F-3
ProKidney Corp
連結業務報告書
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
運営費 |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
一般と行政 |
|
|
|
|
|
|
|
|
|
|||
総運営費 |
|
|
|
|
|
|
|
|
|
|||
営業損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|||
その他の収入(支出): |
|
|
|
|
|
|
|
|
|
|||
利子収入 |
|
|
|
|
|
|
|
|
|
|||
利子支出 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
所得税前純損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
所得税費用 |
|
|
|
|
|
|
|
|
|
|||
非持株権益前純損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
非持株権益は純損失を占めなければならない |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
A類普通株株主が獲得できる純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
|
|
|
|
|
|
|
|
|
|||
発行されたAクラス普通株式加重平均:(1) |
|
|
|
|
|
|
|
|
|
|||
基本的希釈の |
|
|
|
|
|
|
|
|
|
|||
A類普通株1株当たり純損失:(1) |
|
|
|
|
|
|
|
|
|
|||
基本的希釈の |
|
$ |
( |
) |
|
$ |
( |
) |
|
|
|
|
(1)
付記はこれらの連結財務諸表の構成要素である。
F-4
ProKidney Corp
合併全面損失表
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
非持株権益を含めた純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
他の全面的な収入: |
|
|
|
|
|
|
|
|
|
|||
有価証券の未実現収益 |
|
|
|
|
|
|
|
|
|
|||
その他総合収益 |
|
|
|
|
|
|
|
|
|
|||
非持株権益を含めた全面赤字総額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
差し引く:非持株権益による全面的な損失総額 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
A類普通株株主は総合損失総額を占めなければならない |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
付記はこれらの連結財務諸表の構成要素である。
F-5
ProKidney Corp
非持株権益と株主損失·メンバー権益総合変動表の償還が可能
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
F-6
|
|
2023年12月31日までの年度 |
|
||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
甲類単位 |
|
|
B類単位 |
|
|
A類普通株 |
|
|
B類普通株 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||
|
|
償還可能な非持株権益 |
|
|
|
職場.職場 |
|
|
金額 |
|
|
利益と利息 |
|
|
株 |
|
|
金額 |
|
|
株 |
|
|
金額 |
|
|
追加実収資本 |
|
|
その他の総合損失を累計する |
|
|
赤字を累計する |
|
|
株主赤字総額/会員権益 |
|
||||||||||||
2020年12月31日の残高 |
|
$ |
– |
|
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
– |
|
|
$ |
– |
|
|
$ |
– |
|
|
$ |
– |
|
|
$ |
– |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||||
出資する |
|
|
– |
|
|
|
|
|
|
|
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
|
|||
株式ベースの報酬 |
|
|
– |
|
|
|
|
– |
|
|
|
– |
|
|
|
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
|
||
純損失 |
|
|
– |
|
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
( |
) |
|
|
( |
) |
2021年12月31日現在の残高 |
|
|
– |
|
|
|
|
|
|
|
|
|
|
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
|
|
|
( |
) |
|
|
|
|||||
出資する |
|
|
– |
|
|
|
|
– |
|
|
|
– |
|
|
|
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
|
||
企業合併前の持分報酬/支払い |
|
|
– |
|
|
|
|
– |
|
|
|
– |
|
|
|
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
|
||
企業合併前純損失 |
|
|
– |
|
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
|
|
|
( |
) |
|
|
( |
) |
|
企業合併の影響、株式売却の純収益を含む |
|
|
|
|
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
– |
|
|
|
|
|
|
( |
) |
|
|
( |
) |
||||||
企業合併後の持分報酬 |
|
|
|
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
|
|
|
– |
|
|
|
– |
|
|
|
|
|||
F-7
クラスB限定株式権利の帰属 |
|
|
– |
|
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
償還可能な非制御的権益に及ぼす持分取引の影響 |
|
|
( |
) |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
|
|
|
– |
|
|
|
– |
|
|
|
|
||
企業合併後の純損失 |
|
|
( |
) |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
|
|
|
( |
) |
|
|
( |
) |
|
2022年12月31日現在の残高 |
|
|
|
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
– |
|
|
|
( |
) |
|
|
( |
) |
||||||
株式ベースの報酬 |
|
|
|
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
|
|
|
– |
|
|
|
– |
|
|
|
|
|||
A類普通株を発行する |
|
|
– |
|
|
|
|
|
|
|
– |
|
|
|
– |
|
|
|
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
||
クラスB限定株式権利の帰属 |
|
|
– |
|
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
B類普通株A類普通株と交換する |
|
|
( |
) |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
|
|
|
|
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
– |
|
|
|
– |
|
|
|
|
||||
A類普通株買い戻し |
|
|
– |
|
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
( |
) |
|
|
( |
) |
|
|
– |
|
|
|
– |
|
|
|
( |
) |
|
|
– |
|
|
|
– |
|
|
|
( |
) |
償還可能な非制御的権益に及ぼす持分取引の影響 |
|
|
( |
) |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
|
|
|
– |
|
|
|
– |
|
|
|
|
||
有価証券は赤字を実現していない |
|
|
|
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
|
|
|
– |
|
|
|
|
|||
純損失 |
|
|
( |
) |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
( |
) |
|
|
( |
) |
F-8
非持株権益償還価値変動 |
|
|
|
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
– |
|
|
|
( |
) |
|
|
( |
) |
|
2023年12月31日現在の残高 |
|
$ |
|
|
|
|
– |
|
|
$ |
– |
|
|
$ |
– |
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
付記はこれらの連結財務諸表の構成要素である。
F-9
ProKidney Corp
合併報告書キャッシュフロー
(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
経営活動のキャッシュフロー |
|
|
|
|
|
|
|
|
|
|||
非持株権益前純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
非制御性利息前純損失と使用済みキャッシュフロー純額の調整 |
|
|
|
|
|
|
|
|
|
|||
減価償却および償却 |
|
|
|
|
|
|
|
|
|
|||
株式ベースの報酬 |
|
|
|
|
|
|
|
|
|
|||
有価証券収益純額 |
|
|
( |
) |
|
|
|
|
|
– |
|
|
設備処分損失 |
|
|
|
|
|
|
|
|
|
|||
経営性資産と負債の変動 |
|
|
|
|
|
|
|
|
|
|||
受取利息 |
|
|
( |
) |
|
|
|
|
|
– |
|
|
前払い資産とその他の資産 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
売掛金と売掛金 |
|
|
|
|
|
|
|
|
|
|||
所得税に対処する |
|
|
|
|
|
|
|
|
|
|||
経営活動のためのキャッシュフロー純額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|||
投資活動のためのキャッシュフロー |
|
|
|
|
|
|
|
|
|
|||
設備を売却して得た収益 |
|
|
|
|
|
|
|
|
|
|||
SCSからの純現金 |
|
|
|
|
|
|
|
|
|
|||
有価証券を購入する |
|
|
( |
) |
|
|
|
|
|
|
||
有価証券の販売 |
|
|
|
|
|
|
|
|
|
|||
設備の購入と施設の拡張 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
投資活動のためのキャッシュフロー純額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|||
融資活動によるキャッシュフロー |
|
|
|
|
|
|
|
|
|
|||
融資リースの支払い |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
企業合併による収益、パイプライン融資、関連コストの控除 |
|
|
|
|
|
|
|
|
|
|||
関係者支払手形項下の借入金 |
|
|
|
|
|
|
|
|
|
|||
A類普通株買い戻し |
|
|
( |
) |
|
|
|
|
|
|
||
関係者支払手形を償還する |
|
|
|
|
|
( |
) |
|
|
|
||
現金純寄付 |
|
|
|
|
|
|
|
|
|
|||
融資活動が提供するキャッシュフロー純額( |
|
|
( |
) |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|||
現金と現金等価物の純変化 |
|
|
( |
) |
|
|
|
|
|
|
||
期初の現金 |
|
|
|
|
|
|
|
|
|
|||
期末現金 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|||
キャッシュフロー情報の追加: |
|
|
|
|
|
|
|
|
|
|||
期間中にお支払いいただいた所得税の現金は、返金後の純額を差し引いております |
|
$ |
|
|
$ |
|
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|||
非現金投資と融資活動を追加開示します |
|
|
|
|
|
|
|
|
|
|||
賃貸義務と引き換えの使用権資産 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
B類普通株の交換 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
償還可能な非持株権益に及ぼす持分取引と補償の影響 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
非持株権益償還価値変動 |
|
$ |
|
|
$ |
|
|
|
|
|||
設備と施設の拡張には支払すべき帳簿と |
|
$ |
|
|
$ |
|
|
$ |
|
|||
付記はこれらの連結財務諸表の構成要素である。
F-10
ProKidney Corp
メモ:連結財務諸表
注1:業務説明と届出根拠
業務説明
ProKidney Corp.(“会社”または“ProKidney”)は当初設立された社会資本Suvretta Holdings Corp.III(“SCS”)。SCSは空白小切手会社で、#年にケイマン諸島免除会社として登録されています
SCSは2022年1月18日、アイルランドの法律下の有限責任組合企業ProKidney LP(“PKLP”)と最終的な業務合併協定(“業務合併協定”)に署名した。業務合併協定の条項によると、PKLPはSCSの付属会社となり、傘式組合会社(“UP-C”)構造で組織され、持分所有者が最終的にその過去の権益をA類普通株に交換する時、PKLPはSCSに潜在的な未来の税収割引を提供する。取引は2022年7月11日(“成約日”)に完了した(“成約”)。取引が完了した後、SCSはProKidney Corp.と改名した。
SCSとPKLP間の業務統合(“業務統合”)は約#ドルの毛収入を生み出している
業務統合は,共同制御下のエンティティ間の逆資本再構成取引とみなされ,この取引により,PKLPは会計購入者と前身実体とみなされる.業務統合はPKLPがSCSに相当する純資産発行株に反映され,同時に資本再編が行われ,商誉や無形資産は確認されない.
ProKidney Corp.は、その運営子会社ProKidneyとProKidney LLCを通じて、腎臓自己細胞療法(Rilparencel)の開発に専念している。ProKidneyはケイマン諸島会社法(改正)に基づいて登録されて設立された免除会社(“ProKidney-KY”)、ProKidney LLCはデラウェア州法に基づいて設立された有限責任会社(“ProKidney-US”)であり、慢性腎臓疾患患者の腎機能を保護し、透析や臓器移植の需要を延期または除去する可能性がある。
合併原則
ProKidney Corp.は持株会社であり、その主要資産はPKLP及びその完全運営子会社ProKidney-KYとProKidney-USの持株権である。当社はPKLPが会計上の可変利息実体であることを決定しており、ProKidneyはPKLPの主要な受益者であり(PKLPにおけるその管理員の権益や、ProKidneyの上級管理職もPKLPの上級管理職でもある)、PKLPの経済表現に最も重要な活動を含むPKLPのすべての活動を指導する権利があるからである。したがって、同社は、会計基準に従って主題810“統合”を符号化し、その連結財務諸表にPKLPの結果を統合した。2023年12月31日まで異なる所有者はPKLPにおいて無投票権を持つ権利を持ち,代表
すべての会社間取引と残高は無効になりました。
F-11
再分類する
各期間の情報を比較しやすいように,前期額を何らかの再分類し,今期に該当する列報方式とした.
注2:重要会計政策
予算の使用
公認会計原則に基づいて、連結財務諸表を作成することは、合併財務諸表の日付の資産および負債の報告金額、または負債の開示および報告期間の費用金額に影響を与えるいくつかの推定および仮定を管理層に要求する。これらの総合財務諸表のいくつかの推定は、研究および開発費用、権益に基づく報酬費用、および所得税の支出または利益の計算に関連する。当社は、場合によっては経営陣がその場合に合理的と考えている将来予測を含む過去の経験や様々な他の仮定に基づいて推定しています。実際の結果はこれらの推定とは異なる可能性がある。推定数の変化は既知期間の報告結果に反映されている。
現金等価物と有価証券
当社は購入日原始満期日が90日以下のすべての高流動性投資を現金等価物と見なしている。これらの項目の短期的な性質により,現金と現金等価物の帳簿価値は公正価値に近い.
その会社の販売可能な債務証券への投資は分類され、販売可能と記されている。同社は現在の業務に利用可能であるため、販売可能な債務証券を短期債券に分類している。証券売却のコストは特定の識別方法を用いて決定される。
当社はすべての入手可能な証拠を考慮して信用損失があるかどうかを評価し、存在すれば信用損失準備金を確認する。
信用リスクの集中度
現金及び現金等価物は当社が保有する主要な金融商品であり、信用リスクの集中の影響を受ける可能性がある。同社の現金や等価物は大手金融機関の口座に保管されており、これらの金額は連邦保険の限度額を超える可能性がある。
費用を計算する
総合貸借対照表に記載された応算費用2023年12月31日および2022年12月31日には、以下の内容が含まれます(千単位)
|
2023年12月31日 |
|
|
2022年12月31日 |
|
||
補償する |
$ |
|
|
$ |
|
||
解散費 |
|
|
|
$ |
|
||
臨床研究関連費用 |
|
|
|
|
|
||
施設関連コスト |
|
|
|
|
|
||
法律費用を計算する |
|
|
|
|
|
||
製造改善コスト |
|
|
|
|
|
||
相談料と専門費を計算します |
|
|
|
|
|
||
その他の課税費用 |
|
|
|
|
|
||
費用とその他の費用総額を計算しなければならない |
$ |
|
|
$ |
|
||
研究開発コスト
研究·開発コストは発生時に費用を計上する。研究開発費には,研究開発活動を行うことによるコスト,賃金,福祉,第三者許可費,製造と臨床前開発活動および臨床試験を招聘された外部サプライヤーの外部コストがある。
同社は,多くの契約研究機関と締結した契約に基づいて,受け取ったサービス,かかる努力や借金の見積もりに基づいて,計上項目を記録している。正常な業務過程において、会社は第三者と契約を結び、潜在製品を持続的に開発する過程で様々な臨床研究活動を行っている。このような合意の財務条件は契約によって異なる可能性があり、不均衡な支払いの流れを招く可能性がある。契約下の支払い
F-12
いくつかの事件の完成と一部の臨床研究或いは類似状況の完成のようないくつかの要素に依存する。同社の権責発生制政策の目標は,その財務諸表中の費用記録を実際に受け入れたサービスや費用の努力にマッチさせることである。したがって,臨床研究に関連する計算すべき費用は,特定の臨床研究で指定された1つまたは複数の事象の完成度の見積もりに基づいて会社が確認した。
同社は,将来の研究·開発活動に支払われた払戻不可能な前払い記録を前払い費用としている。会社が関連貨物やサービスを受け取った場合、前払い費用は合併経営報告書と全面赤字で費用として確認されます
許可を得た技術が技術的可能性に達しておらず,将来的に他の用途がない場合には,技術許可を得ることによるコストは,購入が行われている研究·開発費用に計上される。
固定資産
固定資産はコストから減価償却累計を引いて列記する。通常、維持·修理費用は費用に記入し、重大な改善または交換は資本化に記入する。当社は直線法を用いて資産推定耐用年数内の減価償却と償却を計算します。レンタル改善は、レンタル年数またはレンタル改善の推定耐用年数の中で短い時間で償却する
建物.建物 |
|
コンピュータ装置及びソフトウェア |
|
家具と設備 |
|
賃借権改善 |
固定資産には以下の内容が含まれる(千計)
|
2023年12月31日 |
|
|
2022年12月31日 |
|
||
土地 |
$ |
|
|
$ |
|
||
建物.建物 |
|
|
|
|
|
||
賃借権改善 |
|
|
|
|
|
||
家具と設備 |
|
|
|
|
|
||
コンピュータ装置及びソフトウェア |
|
|
|
|
|
||
建設中の工事 |
|
|
|
|
|
||
減算:減価償却累計 |
|
( |
) |
|
|
( |
) |
固定資産総額,純額 |
$ |
|
|
$ |
|
||
2023年12月31日現在、2022年と2021年12月31日までの年度減価償却費用は
無形資産
無形資産は得られた集合労働力からなり、これらの労働力はASC 350-無形資産-営業権その他に従って会計処理を行う。得られた集合労働力は5年間の耐用年数で直線的に償却される.
|
|
2023年12月31日 |
|
|
2022年12月31日 |
|
||
総帳簿金額 |
|
$ |
|
|
$ |
|
||
累計償却する |
|
|
|
|
|
|
||
帳簿純額 |
|
$ |
|
|
$ |
|
||
集合労働力無形資産に関する償却費用は$
長期資産減価準備
事件や環境変化がある資産の帳簿金額が回収できない可能性があることを示す場合には,償却すべき固定資産や無形資産などの長期資産が減値審査を行う。保有·使用すべき資産の回収可能性は,資産の帳簿価値を予想される未割引将来の現金フローと比較することで測定される
F-13
資産から生成される。1つの資産の帳票金額がその推定された将来のキャッシュフローを超えた場合、その資産の帳票金額が当該資産の公正価値を超えた金額について減価費用を確認する。2023年12月31日現在,2022年および2021年12月31日まで年度減価費用は記録されていない.
所得税
当社は、“米国会計基準”第740テーマである所得税の要求に応じて、負債法を用いて所得税を会計処理し、この方法によれば、繰延税金資産および負債は、既存の資産と負債の帳簿金額の財務諸表とそれぞれの税基準との差による将来の税収結果として記録される。税金資産及び負債を繰延して税率計量を策定し、その等の一時的な差額を回収又は決済する予定の年度の課税収入に適用されることが予想される。税率変動が繰延税金資産や負債に及ぼす影響は、公布日を含む期間の経営実績で確認されている。繰延税金資産が現金化される可能性が高い場合を除き、繰延税金資産の帳簿金額を減少させるための推定値を提案する。繰延税金資産の最終的な現金化は、これらの一時的差額控除可能期間中に生成された将来の課税所得額に依存する。経営陣はこの評価を行う際に、繰延税金負債の予定沖販売、繰り越し期間の利用可能な税項目、将来の課税収入および税務計画策を考慮する。そのため、当社は2023年12月31日および2022年12月31日の繰延税項目純資産を相殺するための全額推定手当を提供している。
所得税に関する利息及び罰金は、会社総合経営報告書の所得税収益(費用)に含まれる。本報告で述べたいずれの期間においても、当社は所得税に関連する重大な利息または罰金を生じていない。
公正価値計量
公正価値は、計量日の市場参加者間の秩序ある取引において、資産または負債が元金または最も有利な市場で負債を譲渡するために課金または支払いされる価格として定義される。公正価値を計量するための投入を優先順位付けする三級公正価値レベルは以下のとおりである.公正価値を計量するための3種類の投入レベルは以下のとおりである
公正価値に記録された資産と負債について、当社の政策は、公正価値レベルに基づいて公正価値計量を策定する際に、観察可能な投入を最大限に使用し、観察できない投入を最大限に減少させることである。有限または観察可能な市場データが存在する場合、資産および負債の公正な価値計量は、主に推定に基づいており、一般に、経済および競争環境、資産または負債の特徴および他の要因に基づいて計算される。そのため、公正価値計量は正確に決定できず、実際に資産や負債を実際に販売または直ちに決済する際に実現できない可能性がある。また,いずれの計算方法にも固有の弱点が存在する可能性があり,使用する基本的な仮定の変化は,割引率や将来のキャッシュフローの推定を含めて,計算の現在または将来の公正価値に大きな影響を与える可能性がある.当社は公正価値計量を採用して、ある資産と負債に対する公正価値調整を記録し、公正価値開示を確定した。
これらのツールの短期的な性質により,現金等価物,売掛金,売掛金の帳簿価値は公正価値に近い.
賃貸借証書
会社は最初から賃貸契約かどうかを確定しています。当社の運営および融資リースに関する残高は、総合貸借対照表に計上されている使用権資産、純額および賃貸負債に計上されていることが確認された。使用権資産およびリース負債は、発効日レンタル期間内の将来最低賃貸支払いの現在値で確認します。資産使用に直接関連する可変支払いおよび指数による支払いの将来調整が発生または変化している間に確認し、初期計量日の賃貸支払いに計上しない。レンタル条項には、会社が選択権を行使することを決定する理由があることを前提として、レンタルの延長または終了の選択権を含むことができる。当社の大部分の借款は暗黙的な金利を提供していないため、当社は発効日に得られた資料に基づいて、逓増借款金利を採用して将来の支払いの現在値を決定します。使用権資産には、レンタル報酬および生成された初期直接コストは含まれていない支払いされた任意のレンタル金額も含まれる。当社は実際の便宜的な方法を選択し、そのレンタルと非レンタル構成部分を分離するのではなく、それらを単独のレンタル構成要素として計算した。12ヶ月以下の賃貸は貸借対照表に計上されません。
F-14
最低レンタル料金のレンタル料金はレンタル期間内に直線的に確認します。短期賃貸のリース支払いは直線法で営業費用に計上され、可変賃貸支払いはこれらの支払いが発生した債務が発生した期間に計上される。
あるいは負債がある
1つの資産が財務諸表日に減値されたか、または負債が発生した可能性があり、損失金額が合理的に推定される場合、当社は負債準備金を計上するか、または負債準備金を計上する。
株式ベースの報酬
発行された株式補償奨励の補償費は、付与された日の公正価値に基づいている。当社は株式ベースの報酬報酬が没収されたことを記録しています。会社株補償計画が付与する奨励は、1)業務合併終了時にペア権益に変換されるProKidney LPの利益権益、(I)B類普通株またはB類制限株式権利と(Ii)ProKidney LPの普通株または制限普通株(総称して“レガシー利益権益”と呼ぶ)、2)SCS発行の制限株式単位(“Legacy SCS Awards”);3)ProKidney Corp.発行の既得株式オプション、および4)ProKidney Corp.発行の市場既得株式オプションを含む。
時間付与株式オプション奨励の付与日公允価値は、ブラック·スコアーズオプション定価式を用いて推定される。付与日レガシー利益権益およびレガシーSCS奨励の公正価値は、付与日発行者の株式またはメンバー単位の時価に基づく(場合によって)。時間付与株式オプション、レガシー利益権益、レガシーSCS奨励に関する報酬支出は、適用されるサービス期間内に直線的に確認される。
幾何ブラウン運動/モンテカルロモデルを用いて市場既得株式オプション付与日の公正価値を推定した。当該等報酬に関する株式補償支出は、派生サービス期間内に各帰属部分を比例して確認し、報酬が市況に達したか否かにかかわらず、サービス条件を満たしていない範囲で調整するだけである。
自社株に関する十分な歴史的取引情報が不足しているため、当社は、自身と類似した市場や経済的特徴を持つと考えられる選定会社の株式組合の履歴変動率に基づいて期待変動率を推定する。無リスク金利は、付与時に発効した米国債収益率曲線に基づいている。過去の行権データが不足しているため、市場条件のない奨励について、当社はスタッフ会計公告テーマ14.D.2に規定されている簡略化方法を用いて、未償還株式オプションの期待寿命を推定している。
定義するNED支払い計画
細分化市場
その会社は
注3:投資
現金等価物と有価証券は公正価値によって計量し、そして公正価値構造の第2級以内にあり、著者らはできるだけ見積もり市場価格或いはその他の定価源とモデルを使用して、市場で観察できる資料を利用して公正価値を決定するためである。
次の表は我々の現金等価物と公正価値で恒常的に計量された有価証券をまとめたものである2023年12月31日(千):
F-15
|
公正価値階層構造 |
|
原価を償却する |
|
|
未実現収益総額 |
|
|
未実現損失総額 |
|
|
公正価値 |
|
|
現金等価物 |
|
|
有価証券 |
|
||||||
貨幣市場基金 |
レベル2 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||||
定期預金 |
レベル2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
商業手形 |
レベル2 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||||
政府債券 |
レベル2 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||||
会社債務証券 |
レベル2 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||||
合計する |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
|
|
$ |
|
|||||
次の表は契約満期日に会社の現金等価物と有価証券の公正価値を示し,現在までである2023年12月31日(千):
|
2023年12月31日 |
|
|
1年以下の期間で満期になる |
$ |
|
|
1年から5年以内に満期になります |
|
|
|
合計する |
$ |
|
|
下表には,他の総合損失を累計した公正価値と未実現損失総額を計上し,種別と個別証券までの持続損失時間の合計を示した2023年12月31日(千):
|
12ヶ月以下です |
|
|
12ヶ月以上 |
|
|
合計する |
|
|||||||||||||||
|
公正価値 |
|
|
未実現損失 |
|
|
公正価値 |
|
|
未実現損失 |
|
|
公正価値 |
|
|
未実現損失 |
|
||||||
定期預金 |
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||||
商業手形 |
|
|
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|
( |
) |
||||
政府債券 |
|
|
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|
( |
) |
||||
会社債務証券 |
|
|
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|
( |
) |
||||
合計する |
$ |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
||||
当社は信用品質の高い会社の債務証券を保有しており,その債務証券の信用リスクは2023年12月31日までの年度中に大きな変化はないことが確認されたそれは.そのため同社は
ProKidneyは免除されたケイマン諸島会社とされており,現在ケイマン諸島や米国の所得税や所得税申告要求に制約されていない。
当社の子会社PKLPは有限組合の形で組織されており,米国所得税では組合企業に分類されているため,その組織がC社であるか米国連邦所得税で会社とみなされる子会社を選択するかについては,連邦と州所得税のみを記録して支出している。
同社の子会社ProKidney−USとProKidney Acquisition CompanyはC級会社とされているため,連邦と州の税収準備が記録されている。
当社の付属会社ProKidney-KYはすでにケイマン諸島政府と行政会議と1つの承諾書に基づいて税務優遇を与え、同社がその運営或いは相続税或いは相続税の性質について徴収するいかなる利益、収入、収益或いは付加価値税を免除し、20年間、2016年1月20日から発効する。米国の税金の目的で,ProKidney−KYは所有者とは無関係な実体と見なすことを選択したため,所得税の支出を記録していない。
所得税支出準備金は以下の部分からなる2023年12月31日まで、2022年12月31日、2021年12月31日までの年度(単位:千):
F-16
|
2023年12月31日 |
|
|
2022年12月31日 |
|
|
2021年12月31日 |
|
|||
現在: |
|
|
|
|
|
|
|
|
|||
連邦制 |
$ |
|
|
$ |
|
|
$ |
|
|||
状態.状態 |
|
|
|
|
|
|
|
( |
) |
||
当期所得税支出総額 |
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|||
延期: |
|
|
|
|
|
|
|
|
|||
連邦制 |
|
|
|
|
|
|
|
|
|||
状態.状態 |
|
|
|
|
|
|
|
|
|||
繰延所得税支出(福祉)合計 |
|
|
|
|
|
|
|
|
|||
所得税費用 |
$ |
|
|
$ |
|
|
$ |
|
|||
アメリカの法定金利の法定金利と
|
2023年12月31日 |
|
|
2022年12月31日 |
|
|
2021年12月31日 |
|
|||
現在: |
|
|
|
|
|
|
|
|
|||
法定税率で所得税を徴収する |
|
% |
|
|
% |
|
|
% |
|||
連邦福祉を差し引いた州税 |
|
|
|
|
|
||||||
未納税所得額 |
|
( |
) |
|
|
( |
) |
|
|
( |
) |
連邦信用 |
|
|
|
|
|
|
|
|
|||
株式ベースの報酬 |
|
( |
) |
|
|
( |
) |
|
|
|
|
評価免除額を変更する |
|
( |
) |
|
|
( |
) |
|
|
( |
) |
他にも |
|
( |
) |
|
|
( |
) |
|
|
( |
) |
有効所得税率 |
|
( |
)% |
|
|
( |
)% |
|
|
% |
|
統合貸借対照表に含まれる会社繰延税金資産および負債の構成要素は以下のとおりである(千で計算)
|
2023年12月31日 |
|
|
2022年12月31日 |
|
||
繰延税金資産: |
|
|
|
|
|
||
補償すべきである |
$ |
|
|
$ |
|
||
連邦信用繰り越し |
|
|
|
|
|
||
賃貸借証書 |
|
|
|
|
|
||
株式ベースの報酬 |
|
|
|
|
|
||
研究と実験コスト資本化 |
|
|
|
|
|
||
純営業損失が繰り越す |
|
|
|
|
|
||
起動コスト |
|
|
|
|
|
||
減価準備前の繰延税金資産 |
|
|
|
|
|
||
推定免税額 |
|
( |
) |
|
|
( |
) |
繰延税金資産総額 |
$ |
|
|
$ |
|
||
|
|
|
|
|
|
||
繰延税金負債: |
|
|
|
|
|
||
無形資産 |
$ |
|
|
$ |
|
||
固定資産 |
|
|
|
|
|
||
前払い費用 |
|
|
|
|
|
||
繰延所得税負債総額 |
|
|
|
|
|
||
繰延税項目純資産 |
$ |
|
|
$ |
|
||
付記6で述べたように、当社は関連側と課税項目合意を締結し、この協定は、当社が完了(“ProKidney単位所有者”)を終了する前に、当社がある取引によって実際に現金化された(または場合によっては、当社は現金化とみなされる)米国連邦、州および地方所得税または特許経営税節約金額(ある場合)の85%をPKLP所持者に支払うことを規定している。本プロトコルの下の責任をトリガする可能性のある取引は何も発生していませんので、2023年12月31日現在、当社は本プロトコルに関する責任は何も確認していません。
繰延税金資産の現金化能力を評価する際には、管理層は、一部の繰延税金資産をより現金化する可能性があるかどうかを考慮する。繰延税金資産の最終的な現金化は、これらの一時的差額控除可能期間中に生成された将来の課税所得額に依存する。経営陣が計画を考えているのは
F-17
繰延税金負債の償却、繰越期間中の利用可能な税額、予測された将来の課税所得額、およびこの評価を行う際の税務計画戦略したがって、経営陣は、繰延税金資産の可能性が高くないことを確認し、会社は#ドルの推定手当を提供していると結論した
同社は純営業損失が#ドルに繰り越した
終了年度税引き総額が確認されていない期初と期末金額を照合する2023年12月31日および2022年12月31日には、以下の内容が含まれます(千単位)
|
2023年12月31日 |
|
|
2022年12月31日 |
|
||
未確認税収割引(毛額): |
|
|
|
|
|
||
年初の福祉 |
$ |
|
|
$ |
|
||
前年の納税状況に関する増加 |
|
|
|
|
|
||
前年度の納税状況に関する減少額 |
|
|
|
|
|
||
今年度の納税状況に関する増加 |
|
|
|
|
|
||
年末福祉 |
$ |
|
|
$ |
|
||
いくつありますか
2020年から2023年までの納税年度は依然として連邦と州当局の審査を受ける必要がある。
注5:賃貸借契約
FASBは2016年2月、ASU 2016-02:レンタル(テーマ842)を発表した。このASUは,テナントがその貸借対照表上で多くの経営的リースの使用権資産とリース負債を確認することを要求している。ASU 2016−02は、2018年12月15日以降に開始された年度と中期に対して有効であり、これらの会計年度内の中期を含む。2018年7月、FASBはASU 2018-11、レンタル(テーマ842):的確な改善を発表し、会社に追加のオプション移行方法を提供し、累積効果調整により採用日に発効したレンタルに新基準を適用しました。当社は2021年1月1日から新たな賃貸基準を採用し、改正された遡及移行法を採用している。
当社は、既存の賃貸契約の初期直接コストを再評価することなく、ASU 2016-02で参照されている一括実際の便宜的な計を選択し、既存の賃貸契約の識別と分類を保留することを可能にした。同社はまた実際の便宜策を選択し,初期レンタル期間を免除した
同社には不動産(主にオフィススペース)とある設備の経営賃貸契約があり、レンタル期間はそれぞれ異なる。同社はまた、材料とみなされないいくつかの設備に融資リースを提供している。当社の運営リースコストは、2023年、2022年および2021年12月31日までの年度まで
F-18
下表は,以下の日までの会社総合貸借対照表における経営と融資リース資産および債務の分類をまとめたものである2023年12月31日および2022年12月31日(単位:千):
|
|
2023年12月31日 |
|
|
2022年12月31日 |
|
||
経営リース: |
|
|
|
|
|
|
||
資産使用率 |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
*賃貸負債、流動 |
|
$ |
|
|
$ |
|
||
非流動賃貸負債 |
|
|
|
|
|
|
||
リース負債総額を経営する |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
融資リース: |
|
|
|
|
|
|
||
資産使用率 |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
*賃貸負債、流動 |
|
$ |
|
|
$ |
|
||
非流動賃貸負債 |
|
|
|
|
|
|
||
融資リース負債総額 |
|
$ |
|
|
$ |
|
||
当社の運営および融資リースの賃貸負債満期日は以下の通りである2023年12月31日(千):
|
|
賃貸借契約を経営する |
|
|
融資リース |
|
|
合計する |
|
|||
2024 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
2025 |
|
|
|
|
|
|
|
|
|
|||
2026 |
|
|
|
|
|
|
|
|
|
|||
2027 |
|
|
|
|
|
|
|
|
|
|||
2028 |
|
|
|
|
|
|
|
|
|
|||
その後… |
|
|
|
|
|
|
|
|
|
|||
賃貸支払総額 |
|
|
|
|
|
|
|
|
|
|||
差し引く:推定利息 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
賃貸負債現在価値 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
経営リースの加重平均残存レンタル期間は
備考6:関連先取引
交換協定
当社は締め切り時に,PKLPおよびいくつかの決済ProKidney単位所有者(“交換プロトコル”)と交換プロトコル(“交換プロトコル”)を締結し,このプロトコルに基づいて,任意の契約販売禁止期間(ロックプロトコル(以下参照)を含む)の免除または満了後、交換プロトコルによって定義された合併後の元腎普通株保有者(またはいくつかの譲渡許可者)は、取引終了後180日後に、その合併後の元腎普通株および同等数の会社B類普通株を時々1対1で交換して、会社A類普通株(“取引所”)と交換する権利がある。ただし、いくつかの例外を除いて、いくつかの2回目の発売を除いて、当社はA類普通株出来高に応じて平均価格(“VWAP”)を現金で取引所の全または一部の株式を決済することができ、ただいくつかの制限によって制限されなければならない。交換協定は、一般事項として、もし当社が合併後の元腎臓共同単位所有者が法律或いは法規によって禁止されることを確定した場合、或いは当社及びその付属会社との他の合意に違反し、第二次改正と再起動した元腎臓有限組合契約と交換協定を含む場合、合併後の元腎臓共同単位所有者は合併後の元腎臓共同単位を交換する権利がない。
販売禁止協定
開ける締め切りには,当社,SCSキーパーIII LLCおよびいくつかの決済ProKidney単位保持者がロックプロトコル(“ロックプロトコル”)を締結した。販売禁止協定にはSCSに関するいくつかの譲渡制限が含まれている
F-19
スポンサー?スポンサーIII LLCとその閉幕したProKidney Unitholders党。これらの制限は、終値から始まり、(I)終値後180日の日付と、(Ii)(A)33%の禁売株(引受株式および私募株式を含まない)の日、すなわちあるカテゴリの最終報告販売価格の日(早い者)を終了する
2023年1月には販売禁止期間
課税課税協定
当社は締め切り日に、決済ProKidney単位所有者と課税契約(“課税契約”)を締結した。課税契約によると、その他の事項を除いて、当社は期末ProKidney単位の所持者に支払わなければなりません
割増権益
終値には,ある株主が合計を獲得した
関連側債務
2022年1月18日、当社は“企業合併協定”を履行するために本票を締結した。この約束票を通じて、チケットを持っている人は#ドルまでの資金を提供することができる
約束手形の引き出しは#ドルの倍数とすることができます
2022年12月31日までの年間で、当社は$を借入します
ProKidney-KYとNefro Healthの間のコンサルティングサービスプロトコル
2020年1月1日、ProKidney-KY(前身はinRegen)は、Nefro Health(“Nefro”)とコンサルティングサービス協定を締結し、Nefro Health(“Nefro”)は、会社の傘下のPablo Legorretaさんによって支配され、多数の持分を保有するアイルランドの共同企業であり、この合意によると、Nefroは、北米およびEUでの臨床試験、設計、およびProKidney候補製品の製造および商業化前の活動を含むコンサルティングサービスを提供する。合意によると、Nefroは$を得るだろう
F-20
協定は2020年12月31日まで続き、いずれか一方が予定終了日の少なくとも90(90)日前に他方に書面通知を出して終了しない限り、毎回1年間の自動更新を許可する条項に基づいて更新される。他方が合意規定の義務を履行する際に実質的な違約が発生した場合、又は非違約者の書面通知を受けてから30(30)日以内にいかなる条項、陳述、保証又は契約面の違約を是正していない場合、いずれか一方は本合意を終了することができる。さらに、いずれか一方は、30(30)日前に他方に問い合わせサービスプロトコルの終了を通知した後、任意の理由でプロトコルを終了することができる。このような終了の場合,ProKidney−KYは終了日から稼いでいるが支払われていない相談料をNefroに支払う義務がある。
ProKidney-USとNefro Healthの間のコンサルティングサービスプロトコル
2020年1月1日、ProKidney-US(前身はTwin City Bio,LLC)はNefroとコンサルティングサービス協定を締結し、この協定によると、Nefroは北米とEUでの臨床試験、設計と製造会社の候補製品および商業化前活動を含む会社候補製品の研究と開発にコンサルティングサービスを提供する。合意によると、Nefroは$を得るだろう
備考7:非持ち株権を償還できる
当社は未償還株式を代表する合併後の原腎共通単位に関する交換協定を遵守しなければならない
償還可能な非持株権益は、(1)その初期公正価値に非持株権益に関連する累積収益/損失又は(2)資産負債表までの償還価値の中の高い者を加えて確認する。2023年12月31日償還可能な非持株権益はその初期公正価値に非持株権益に関連する累積損失を加えて入金することができ、この金額は貸借対照表の日の償還価値より高く、約$である
当社がPKLPの持株権を保持している場合、当社のPKLPにおける所有権権益の変動は持分取引に計上され、当社は当該等の変動について非持株権及び権益を調整しなければならない
F-21
|
この年度までに |
|
|
2022年7月11日から2022年12月31日まで |
|
||
A類普通株株主が獲得できる純損失 |
$ |
( |
) |
|
$ |
( |
) |
(増加)/ProKidney Corp.の累積赤字減少の影響 |
|
|
|
|
|
||
(増加)/ProKidney Corp.追加実収資本の削減 |
|
( |
) |
|
|
|
|
(増加)/ProKidney Corp.ホームのための追加実収資本の減少 |
|
( |
) |
|
|
( |
) |
A類普通株株主の純損失の変動及び変動 |
$ |
( |
) |
|
$ |
( |
) |
注8:株主権益
ビジネス統合と組み合わせて
事業合併後、会社の法定配当金は
A類普通株の権利
A類普通株の保有者一人一人に権利がある
任意の発行済み優先株の優先株割引に適用される規定の下で、A類普通株式保有者は、取締役会が時々発表する配当金(ある場合)を比例して受け取る権利があり、これらの配当金は合法的に分配可能な資金から振り出すことができる。ケイマン諸島の法律によると、すべての配当金はいくつかの制限によって制限されており、すなわち、吾などは利益または株式割増口座から配当金を支払うことしかできず、いかなる場合でも、配当金が正常な業務過程で満期になった債務を返済できない場合は、配当金を支払うことができない。
任意の任意の自発的または非自発的な清算、解散または清算が発生した場合、会社Aクラス普通株の所有者は、私たちの債務および他の負債を償還した後に残ったすべての資産を比例的に共有する権利があるが、優先株または会社A類普通株に優先する任意のカテゴリまたは系列株(ある場合)の優先分配権は制限される。
B類普通株の権利
会社B類普通株の所持者一人一人に権利があります
任意の自発的又は非自発的な清算、解散又は清盤が発生した場合、当社B類普通株の保有者は、当該等のB類普通株が吾等の債務及びその他の負債を償還した後の残存資産に相当する実納資本の課税額を得る権利があるが、優先株又は当社が当時発行したB類普通株優先の任意の種類又は系列株式の優先分配権に制限されなければならない。会社のB類普通株は私たちの利益や資産を共有する他の権利を持っていません。
割増権益
業務合併が終了した時点で,いくつかの株主が配給合弁を獲得した
F-22
a 終値直後の5年間で,1株あたりの価格はこれらと同じVWAPの敷居を超えていた.取得されると、プレミアム権利は、統合されたProKidney普通株式およびクラスB普通株式に自動的に変換される。
株式取引入金として割増権を発行する。オーバーフロー権は、終了ProKidney単位所有者(すなわち、企業合併における会計買収側)に発行されるため、オーバーフロー権手配の会計は、会計基準編纂(“ASC”)主題805(企業合併)または主題718(株式補償)には属さない。
また、この手配が負債に分類されるべきかどうかを決定するために、米国会計基準第480号特別テーマ“負債と権益とを区別する”の下でオーバーフロー権の会計が評価された。この分析により,オーバーフロー権は負債入金としての基準を満たしていないことが決定された.また,ASCトピック815では,派生ツールの下でオーバーフロー権を評価した。この分析の一部として、オーバーフロー権が派生ツールの定義に適合することを決定した;しかし、それらは実体自身の株式と明らかに密接に関連し、株式処理基準に符合するため、範囲例外基準を満たしている。
注9:1株当たり純損失
1株当たりの基本純損失の計算方法は、A類普通株株主が純損失をA類発行済み普通株の加重平均株式で割るべきであり、潜在希薄化証券の対価格を含まない。1株当たり純損失を希釈するとは、すべての潜在的希薄化株式の影響を計上するように調整された1株当たり基本純損失である。1株当たりの純損失はすべての期間の1株当たりの赤字とほぼ同じであり、潜在的な発行可能株式に組み入れることは逆償却作用があるからである。
当社は2022年7月11日の業務合併前の1株当たり純損失の計算を分析し、その発生価値が連結財務諸表の使用者に意味がないことを確定し、業務合併により資本構造が完全に変化した。そのため、1株当たりの純損失情報は業務合併までの一定期間は報告されていない。A類普通株株主は、2022年12月31日までに、1株当たり基本および償却純損失は業務連結後のみを代表して2022年12月31日までとする。
次の表は、2023年12月31日までの年度および2022年7月11日から2022年12月31日までの1株当たり基本と希釈後の純損失の計算(千単位で、1株および1株当たりの金額を含まない)を示している
|
十二月三十一日までの年度 |
|
|
2022年7月11日から12月31日まで |
|
||
|
2023 |
|
|
2022 |
|
||
分子.分子 |
|
|
|
|
|
||
純損失 |
$ |
( |
) |
|
$ |
( |
) |
差し引く:非持株権益は純損失を占めるべき |
|
( |
) |
|
|
( |
) |
ProKidney Corp.Aのような一般株主が得る純損失は |
$ |
( |
) |
|
$ |
( |
) |
|
|
|
|
|
|
||
分母.分母 |
|
|
|
|
|
||
加重平均Aクラスの普通株式やProKidney Corp.が発行されました |
|
|
|
|
|
||
|
|
|
|
|
|
||
A類単位の純損失 |
|
|
|
|
|
||
ProKidney Corp.A類普通株1株当たり基本と希釈後の純損失 |
$ |
( |
) |
|
$ |
( |
) |
|
12月31日まで |
|
|
12月31日まで |
|
||
|
2023 |
|
|
2022 |
|
||
逆希釈証券 |
|
|
|
|
|
||
ProKidney Corp.B類普通株 |
|
|
|
|
|
||
無帰属限定株式権利 |
|
|
|
|
|
||
割増権益 |
|
|
|
|
|
||
従来のSCS制限株式単位 |
|
|
|
|
|
||
2022年株式インセンティブ計画に基づいて付与された株式オプション |
|
|
|
|
|
||
注10:権益ベースの報酬
F-23
2022年インセンティブ株式計画
2022年7月11日、会社株主は、会社員、非従業員取締役、個人コンサルタント、コンサルタント、および他のサービスプロバイダに株式ベースの奨励を行うことを規定するProKidney Corp.2022年インセンティブ株式計画(以下、2022年計画と略す)を承認した。2023年12月31日までに
会社は2022年計画に基づいて、会社のある従業員と非従業員取締役に奨励性と非制限株式オプション奨励を支給した。当社はその繰延税金資産について全額評価準備を設けていることから、当社はこのような奨励に関する税務優遇は何も確認していません。
時間授与賞
会社はブラック·スコアーズオプション定価モデルを用いて付与時間の株式オプションの公正価値を計算した。これらの奨励は一般に3年または4年以内に比例して授与され、オプション奨励は付与された日から10年後に満期になる
|
|
十二月三十一日までの年度 |
||
|
|
2023 |
|
2022 |
予想変動率 |
|
|
||
オプションの期待寿命は年単位である |
|
|
||
無リスク金利 |
|
|
||
期待配当収益率 |
|
|
||
下表は、当社が2022年計画に基づいて付与したタイミング株式オプション奨励の活動をまとめたものです2023年12月31日までの年度:
|
|
株式数 |
|
|
加重平均行権値 |
|
||
2023年1月1日現在の未返済期間帰属オプション |
|
|
|
|
$ |
|
||
授与する |
|
|
|
|
|
|
||
没収される |
|
|
( |
) |
|
|
|
|
2023年12月31日までの未返済期間帰属オプション |
|
|
|
|
$ |
|
||
2023年12月31日行使可能期間既得オプション |
|
|
|
|
$ |
|
||
加重平均残存契約寿命 |
|
|
|
|
|
|||
帰属し、2023年12月31日に帰属する予定の時間帰属オプション |
|
|
|
|
$ |
|
||
加重平均残存契約寿命 |
|
|
|
|
|
|||
2023年12月31日までの年度、会社は$を確認しました
2023年12月31日現在完成していない現金時間報酬の総内在価値はい$です
市場はすでに入賞した
当社は2022年12月31日までの前任行政総裁にも授与されています
F-24
のです$
予想変動率 |
|
|
次善運動倍数 |
|
|
無リスク金利 |
|
|
期待配当収益率 |
|
前首席執行幹事が2023年に雇用を終了した場合、以前に確認されたこれらの賠償に関する補償費用が戻される。これにより約#ドルが売れました
残された利益と利益
ProKidney LPは2021年8月5日に締結した有限組合契約(“有限組合契約”)が改訂及び再予約されたProKidney LLC有限責任会社協定の代わりに当社の親実体の管理文書となり、当社の従業員、取締役、その他のサービス提供者及びPKLPの1つ或いは複数の乙類単位(定義は有限組合契約参照)の形式で値を計算した他の人に利益権益を支給することを許可する(定義は有限組合契約参照)。
有限組合協定により,PKLP(“Legacy GP”)の前普通パートナーProKidney GP Limitedはすでに発行されたプレミアム権益の条項と条件を決定する.各贈与に割り当てられたしきい値は,付与された日PKLPの公平な市場価値を下回ってはならない
2022年1月17日、PKLPは、一部の保留された分配イベントにより、利益権益保持者がPKLPから不比例な分配を獲得し、当該等所有者ごとの推定値の敷居がゼロになるまで、当該等所有者の累積分配総額に比例するシェアを“追いつく”ことで、前述の規定に従って利益保持者に十分な分配を行うと、PKLPの関連B類単位が自動的にPKLPのA類単位に変換されることを規定している有限パートナーシップ協定(“改訂及び再予約された有限組合協定”)を改訂し、再記載した。
付記1で述べた業務統合が完了すると,PKLPの既存のBクラスとB-1クラスユニットは“追いつかれ”され,PKLPのAクラスユニットに変換される.これによって生成された帰属および帰属されていないPKLP Aクラス単位は、その後、それぞれ資本が合併後の原腎一般単位または会社の制限された一般単位に再構成される。この資本構造調整は各参加者が持っている賞の数を減少させる。したがって、これらの財務諸表内の利益、利息、および関連単位価値の数は、この資本再構成を反映するように調整されている。資本再編後、制限された共通単位は、元の利益利息奨励に関する帰属スケジュールを維持する。
下表は当社の今年度までの利益利息奨励に関する活動をまとめたものです2023年12月31日:
|
|
株式数 |
|
|
加重平均付与日公正価値 |
|
||
2023年1月1日までに無許可の賠償 |
|
|
|
|
$ |
|
||
既得 |
|
|
( |
) |
|
|
|
|
没収される |
|
|
( |
) |
|
|
|
|
2023年12月31日現在の未帰属裁決 |
|
|
|
|
$ |
|
||
2023年12月31日までこれらの賠償に関する未確認賠償支出は#ドルである
F-25
B-1 単位は、資本再編の影響に応じて調整された。いくつありますか
2023年12月31日現在利益権益に帰属していない総内在価値はい$です
利子賞を改訂する
2022年1月17日、“有限責任組合協定”は、ある制限された分配イベントが、利益権益保持者がPKLPから比例しない分配を得ることを規定し、このような各所有者のドア値がゼロになるまで、その保持者が総累積分配において比例的に割り当てられたシェアに“追いつく”ように改正され、前述の規定に従って利益権益保持者に十分な分配が行われると、関連するBクラス単位は自動的にAクラス単位に変換される。
この修正は,修正日までに完了していないPKLPにおけるB-1クラスユニットの修正を構成し,ASCトピック718による規定である.まだ支払われていない株式ベースの報酬を修正することについて、会社は追加の報酬支出総額が#ドルであることを確認します
当社は、2023年12月31日までの年度内に、ある人が退職したときに彼らに発行した未返済時間型株式オプションの帰属条項も改正した。この修正は、ASCトピック718に規定されている賠償金の改正を構成し、#ドルを追加承認することになる
サービス提供者に利益権益を支給する
当社は2022年12月31日までに年度中に発送します
PKLPにおけるB-1クラス単位の購入
会社の従業員や取締役会のメンバーやサービスプロバイダが購入しました
利益と利子の公正価値推定
業務統合前にPKLPは個人所有であり,その持分ツールは活発な公開市場を持っていなかった.そのため、財務報告の目的のため、管理層は同期推定値を用いてPKLP持分(利益利息を含む)の推定1株当たり公正価値を決定した。これらの同期の推定値は、米国公認会計士協会の勤務援助と一致した方法で行われている補償として発行された個人持株会社株式証券の推定値また,学習支援とも呼ばれる.
2022年12月31日までに年度内に付与されたオーバーフロー利息奨励、推定方法は混合方法を採用し、オプション定価方法(“OPM”)及び確率加重予想リターン方法(“PWERM”)を含む。将来発生する可能性のある流動性イベント(業務統合を含む)の予想可能性に応じて、OPMおよびPWERM方法に重みを割り当てる。
OPM方法によれば、各態様において、PKLPの総株式の公正価値は、まず、逆解アルゴリズムを使用して推定され、配当価値は、PKLP自身の証券の最近の取引から導出され、次いで、利益利息を含む各株式カテゴリの特定の権利に基づいて、OPMまたは棚布法を使用して資本構造の各構成要素に総株式価値が割り当てられる。OPMは1つの情景下でPKLPの総株式の公正価値を起点とし、期待リターンと変動性に対する仮定を取り入れ、これらの仮定は市場参加者の予想と一致し、知る市場参加者が予想する可能性のあるイベント範囲をカバーする持分価値分布を生成した。このプロセスは、シーン間およびシーン内で一連の権益価値を作成する。公正価値
F-26
測定は観測不可能入力の変化に敏感である.このような投入の変化は公正な価値計量の増加または減少を招く可能性がある。
PWERM方法は情景に基づく分析であり、普通株の予想される未来の権益価値の確率に基づいて現在値を加重し、様々な可能な未来の流動性イベント情景(提案された業務合併を含む)の下で、各カテゴリと一連の株式の権利と選好(市場流通性の不足によって割引された利益権益を含む)に基づいて、普通株の各株の価値を推定する。
これらの評価を行う際に、管理層は、PKLPの各評価日における業務状況、見通し、経営業績の最適な推定を含む、関連すると考えられるすべての客観的かつ主観的要因を考慮した。評価には,一連の要因,仮説,方法を用いた.重要な要素は業界内の傾向、PKLPがそのA級単位を販売する価格、測定日ごとにB級単位に対するA級単位の権利と選好、運営結果、財務状況、研究と開発仕事の状況、発展段階と業務戦略、単位が活発な公開市場が不足していること、及び当時の市場状況に応じて撤退を実現する可能性を含む。
以下に,各推定シナリオで用いられる重要な仮定を反映する
|
|
OPM |
|
|
PWERM |
|
||
総株価値(千) |
|
$ |
|
|
$ |
|
||
総株式予想変動率 |
|
|
% |
|
|
|||
市場不足で割引する |
|
|
% |
|
|
|||
事件終了予定の時間 |
|
|
|
|
||||
伝統SCS賞
2021年、スラグ銀行は許可することに同意した
会社が付与したRSUはASC 718の範囲に属する。ASC 718によれば、株式分類奨励に関連する株式ベースの報酬は、付与日に公正価値で計算される。付与されたRSUは業績条件(すなわち業務統合の発生)によって制限される.この場合にのみ、適用される会計ファイルの下で履行条件が発生する可能性がある場合にのみ、RSUに関する補償費用が確認される。取引完了時には、これらの奨励の業績条件が満たされ、業務合併が発生したため、株主は合格した株式計画を承認した。したがって、このような奨励に関連するすべての株式ベースの報酬支出は$となる
補償費用
株式奨励に関する報酬支出は、研究開発費と一般管理費に含まれており、具体的には以下のようになる(単位:千)
|
十二月三十一日までの年度 |
|
|||||||||
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
研究開発 |
$ |
|
|
$ |
|
|
$ |
|
|||
一般と行政 |
|
|
|
|
|
|
|
|
|||
株式に基づく報酬支出総額 |
$ |
|
|
$ |
|
|
$ |
|
|||
注11:後続活動
2024年1月に当社は公開市場販売協定を締結したSM(“販売契約”)は,Jefferies LLCを販売エージェントとすることにより,当社はJefferiesを通してそのA類普通株株式を随時提供および販売することができ,額面は$とすることができる
F-27
2024年2月、当社は発表
F-28