カタログ表
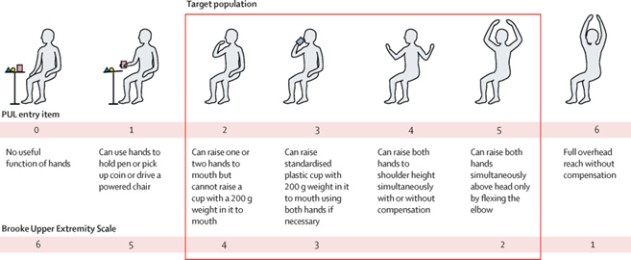
上肢動作(PUL入門項目) (1)
(CAP-1002現在のDMDターゲット群)
| (1) | HOPE-2からの画像柳の葉刀出版(2022年3月) |
B群の登録は進行中であり,この案は約44名の参加者を募集することを目的とし,ランダムにCAP−1002群あるいはプラセボ群に分け,割合は1:1であった。CAP-1002或いはプラセボを4回服用した後、12ケ月目に各個体列に対して初歩的な治療効果と安全性分析を行う。私たちは2024年第2四半期にB組の学生募集を完成させる予定です。キューBは私どもサンディエゴ工場製の製品を使用しております。
HOPE−3研究の主な結果指標は上肢(“Mul”)v 2.0の表現であり,これは検証されたツールであり,高(肩),中(肘)と遠位(手首と手)機能を評価するために設計され,その概念フレームは上肢機能の虚弱進展を反映している。HOPE−3はまた,心機能評価を含めた様々な副次的終点を測定する
我々のRMAT指定の下,2023年第3四半期にBクラス会議でFDAと面会し,我々の製造計画を検討し,BLA申請を提出する可能性があると予想した。今回の会議では,我々の第3段階であるHOPE−3計画との整合性を確認した。また,我々の力価分析や他の商業化を支援する製品発表基準を含めた商業製造活動の計画についても検討した。我々は2024年第1四半期にFDAと面会し,BLAを獲得する方法を検討していく予定である。これから来るBクラス会議では,我々のさらなるCMCビジネス発射計画を検討する予定であり,承認されれば,BLA提出の経路を加速させることを目的としている.我々の最終目標は,我々のサンディエゴ工場製CAP−1002商用製品の使用を許可するBLAを提出することである。
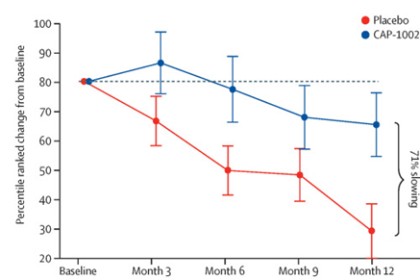
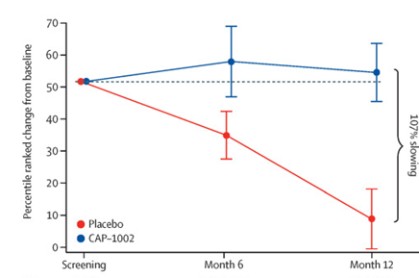
第二段階HOPE-2臨床試験: HOPE-2はアメリカの多くの地点で行われたランダム、二重盲検、プラセボ対照臨床試験であり、2021年に完成した。この臨床試験はCAP-1002の反復静脈注射による骨格筋損傷の証拠がある男の子と若い男性に対する安全性と有効性を評価することを目的としており、彼らの活動状態にかかわらず。この研究では,約90%の患者が非臥床患者であり,すべての患者が安定したステロイド治療を受けている。2つの治療群の間の人口統計学とベースライン特徴は類似している。HOPE-2の最終年結果が発表されました“柳葉刀”2022年3月、この試験はPUL v 1.2の中級次元の主要な治療効果終点(p=0.01)および完全PUL v 2.0の追加陽性終点(p=0.04)を達成したことを示した。中層のPUL v 1.2は試験のための主要な終点であるが,我々もPUL v 2.0を用いて分析を行ったが,FDAはBLAを支持する主要な奏効率の終点として更新されたPUL v 2.0を提案しているためである。プラセボを服用した患者群では、左室駆出率は時間の経過とともに低下したが、CAP 1002を服用した患者は改善し、心臓病の進行速度が107%(p=0.002)低下したことを示した(p=0.002)。左室駆出率は心臓ポンプ機能の世界的な測定基準である。また,これらのデータは,指数容量(LVESV,LVEDV)で測定した心機能が全体的に改善されていることを示している。これらはいずれも心機能の代替測定であり,長期的な結果に関与していると考えられる。また,バイオマーカーCK−MBは減少し,この酵素は心筋細胞損傷がある場合にのみ放出されることが示されている。正常人では,通常血液中にCK−MBは検出されない。DMD時に持続する筋細胞損傷は心筋細胞喪失に関連する病的高酵素レベルを引き起こすことが知られている。これは,DMDの中で初めて心臓機能安定と細胞障害を減少させるバイオマーカーとを関連づけた臨床研究であることが知られている。ステロイド以外にDMDの機能保持は一般的ではない。プラセボ患者の結果は自然病歴と一致したが,治療群では,1年間の治療期間中,多くの患者がこれらの終点で安定または改善した。全体の研究過程において、CAP-1002は全体的に安全であり、耐性は良好である。アレルギーを除いて
7