アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
本財政年度末まで
あるいは…。
1934年証券取引法第13条又は15(D)条に基づいて提出された |
手数料書類番号
(登録者の正確な氏名はその定款に記載)
(国や他の司法管轄権 会社や組織を設立する) |
(税務署雇用主 識別番号.) |
|
|
(主要執行機関の住所) |
(郵便番号) |
登録者の電話番号は市外局番を含んでいます
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
取引 記号 |
各取引所名 それに登録されている |
同法第12条(G)により登録された証券:なし
登録者が証券法第405条に規定する有名な経験豊富な発行者である場合は、再選択マークで表示してください
登録者がこの法第13又は15(D)条に従って報告書を提出する必要がないか否かを、再選択マークで示す。はい☐
登録者が(1)過去12ヶ月以内(または登録者がそのような報告を提出する必要があるより短い期間)に1934年の証券取引法第13条または15(D)条に提出されたすべての報告書を再選択マークで示すかどうか、および(2)このような提出要求を過去90日以内に遵守してきた
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T法規第405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
☒ |
ファイルマネージャを加速する |
☐ |
|
|
|
|
|
非加速ファイルサーバ |
☐ |
規模の小さい報告会社 |
|
|
|
|
|
|
|
新興成長型会社 |
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する☐
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する。
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に§240.10 D−1(B)に従って受信されたインセンティブベースの報酬に従って回復分析を行う必要があるかどうかを、再選択マークで示す。 ☐
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)。はい☐違います
登録者の非関連会社が保有する投票権及び無投票権普通株の総時価は$
登録者の普通株出資額2024年2月20日まで、丁俊暉
引用で編入された書類
登録者は,第14 A条に基づいて米国証券取引委員会に提出された2024年年次総会最終委託書のForm 10−K部分を,本年度報告の第III部(第10,11,12,13及び14項)に組み込む。
カタログ表
|
|
ページ |
前向き陳述に関する警告説明 |
1 |
|
商標 |
2 |
|
市場と業界データ |
2 |
|
リスク要因の概要 |
2 |
|
|
|
|
第1部 |
|
|
第1項。 |
業務.業務 |
5 |
第1 A項。 |
リスク要因 |
46 |
項目1 B。 |
未解決従業員意見 |
110 |
プロジェクト1 C。 |
ネットワーク·セキュリティ |
110 |
第二項です。 |
属性 |
111 |
第三項です。 |
法律訴訟 |
111 |
第四項です。 |
炭鉱安全情報開示 |
111 |
|
|
|
第II部 |
|
|
五番目です。 |
登録者普通株市場、関連株主事項及び発行者による株式証券の購入 |
112 |
第六項です。 |
[保留されている] |
114 |
第七項。 |
経営陣の財務状況と経営成果の検討と分析 |
115 |
第七A項。 |
市場リスクの定量的·定性的開示について |
126 |
第八項です。 |
財務諸表と補足データ |
126 |
第九項です。 |
会計と財務情報開示の変更と相違 |
126 |
第9条。 |
制御とプログラム |
126 |
プロジェクト9 B。 |
その他の情報 |
129 |
プロジェクト9 C |
検査妨害に関する外国司法管区の開示 |
129 |
|
|
|
第三部 |
|
|
第10項。 |
役員·幹部と会社の管理 |
130 |
第十一項。 |
役員報酬 |
130 |
第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
130 |
十三項。 |
特定の関係や関連取引、取締役の独立性 |
130 |
14項です。 |
最高料金とサービス |
130 |
|
|
|
第4部 |
|
|
第十五項。 |
展示·財務諸表明細書 |
131 |
第十六項。 |
表格10-Kの概要 |
134 |
前向きな警告について説明するINGレポート
このForm 10-K年度報告書には、1933年の証券法(改正)第27 A条または“証券法”および改正された“1934年証券取引法”第21 E条または“取引法”の意味に適合する前向きな陳述が含まれている。これらの展望性陳述は私たちの現在の予想と予想の経営結果を反映し、これらはすべて既知と未知のリスク、不確定性とその他の要素の影響を受けることができ、これらのリスク、不確定性とその他の要素は私たちの実際の結果、業績或いは業績、市場傾向或いは業界結果はこのような展望性陳述の明示或いは暗示とは大きく異なる可能性がある。したがって、本明細書に含まれる任意の非歴史的事実の陳述は前向きな陳述である可能性があり、評価されるべきである。前述の場合に限定されることなく、“予想”、“予想”、“提案”、“計画”、“信じる”、“予定”、“プロジェクト”、“予測”、“推定”、“目標”、“予測”、“すべき”、“可能”、“会する”及びその否定および類似の言葉および表現は、前向き陳述を識別することを目的としている。これらの展望性陳述は、本報告の第1部分1 A項“リスク要素要約”および“リスク要因”に記載されたリスク、不確定要素および仮説を含むいくつかの重要なリスク、不確定性および仮説の影響を受ける。法的要求がない限り、私たちは、そのような前向き情報に影響を与える実際の結果または要因の変化を反映するために、そのような前向き情報を更新する義務がない。このような前向きな陳述は他の事項を反映している
1
本Form 10−K年次報告において、用語“BEAM”、“会社”、“私たち”、“私たち”または“私たち”を使用する場合、文脈が別に説明されていない限り、総合ベースのBEAM治療会社およびその子会社を指す。
商標
私たちは、米国および/または他の国/地域における商標としてBEAMおよび他のマークを使用する。このForm 10-K年間報告書は、私たちの商標およびサービスマーク、および他のエンティティに属する商標およびサービスマークへの参照を含む。便宜上、本報告で言及された商標および商品名は、ロゴ、イラスト、および他の視覚的表示を含み、商標記号またはTM記号がない可能性があるが、このような参照は、適用法に従って、これらの商標および商品名に対する私たちの権利または適用許可者の権利を最大限に主張しないことをいかなる方法でも示すものではない。私たちは、任意の他のエンティティとの関係を示唆するために、または任意の他のエンティティによって裏書されたり、後援されたりするために、他のエンティティの商号、商標またはサービスマークを使用または提示するつもりはない。
市場と業界データ
別の説明がない限り、本10-K表年次報告に含まれる私たちの業界と私たちが経営している市場に関する情報は、私たちの一般的な期待、市場地位、市場機会を含み、すべて私たちの経営陣の推定と研究、ならびに業界と一般出版物、ならびに第三者による研究、調査、研究に基づいている。本年報に掲載されている第三者刊行物,研究,調査および研究所が提供する資料は信頼できると信じている。経営陣の見積りは,公開的に得られる情報,彼らの我々の業界に対する理解,およびそれらの情報や知識に基づく仮定に基づいており,これらの情報や知識は合理的であると考えられる.これらのデータは、本報告の第1の部分1 A項“リスク要因要約”および“リスク要因”に記載された要因を含む様々な要因に関連しており、これらの仮定および制限は、必然的に高い不確実性およびリスクを有する。このような要素と他の要素は私たちの未来の業績が私たちの仮定と見積もりと大きく違うことをもたらすかもしれない。
リスク要因の総和メアリー
私たちの普通株に投資するのは危険がある。私たちの普通株に投資する前に、以下のリスクをよく考慮しなければなりません。これらのリスクは、“1.A.リスク要因”でより包括的な議論と、本10-K表年次報告に含まれる他のすべての情報を考慮しなければなりません。これらのリスクは以下のリスクを含むが、これらに限定されない
2
3
4
部分 I
項目1.B役に立つ。
概要
私たちはバイオテクノロジー会社で、リードし、全面的に統合された精密遺伝子薬物プラットフォームの構築に力を入れている。私たちのビジョンは深刻な病気を患っている患者に生涯治療を提供することだ。このビジョンを実現するために、遺伝子編集と交付技術のセットと内部製造能力を含むプラットフォームを組み立てた。
我々の遺伝子編集技術キットは我々の独自の塩基編集技術を基礎としており、これはDNA二本鎖切断を招くことなく、ゲノム中の単一塩基に対する差別化精密遺伝薬を可能にする可能性がある。この方法は、標的配列上で正確で予測可能かつ効率的な遺伝結果を生成することを目的とする化学反応を使用する。著者らの独自の塩基編集プログラムは2つの主要な構成部分がある:(I)RNA結合を誘導するクラスター形成の規則間隔短回文反復配列或いはCRISPR蛋白質であり、それはCRISPRが確立したDNA標的能力を利用するが、改造された二本鎖切断を招くことはない;及び(Ii)塩基編集酵素、例えばデアミナーゼであり、それは必要な標的DNA塩基の化学修飾を実行する。この設計は、従来の遺伝子編集方法と比較して、より正確かつ効率的な編集を実現するのに役立ち、遺伝子編集の影響を著しく増加させる可能性があると信じている。私たちはこの2つの配送モデルを探しています離体するそして体内にある術式は,組織型に依存する。塩基編集方法の優雅さは組織特定の送達方式と結合し、方向性、高効率、正確かつ高度に汎用的な遺伝子編集システムに基礎を提供し、このシステムはいくつかの遺伝子を同時に遺伝子修正、遺伝子サイレンシング、遺伝子活性化、遺伝子修正および/または多重編集を行うことができるように設計されている。
我々の目標は,異なる遺伝子検証された編集目標に対して,広範で多様な基礎エディタの組合せ,および革新的なプラットフォームビジネスモデルを推進し,我々のプログラムのカバー範囲をより多くの患者に拡大することである.全体的に言えば、著者らはリードする正確な遺伝医学総合プラットフォームを構築することを求めており、これは広範な治療適用性と正確な遺伝医学領域を変える潜在力を持っているかもしれない。
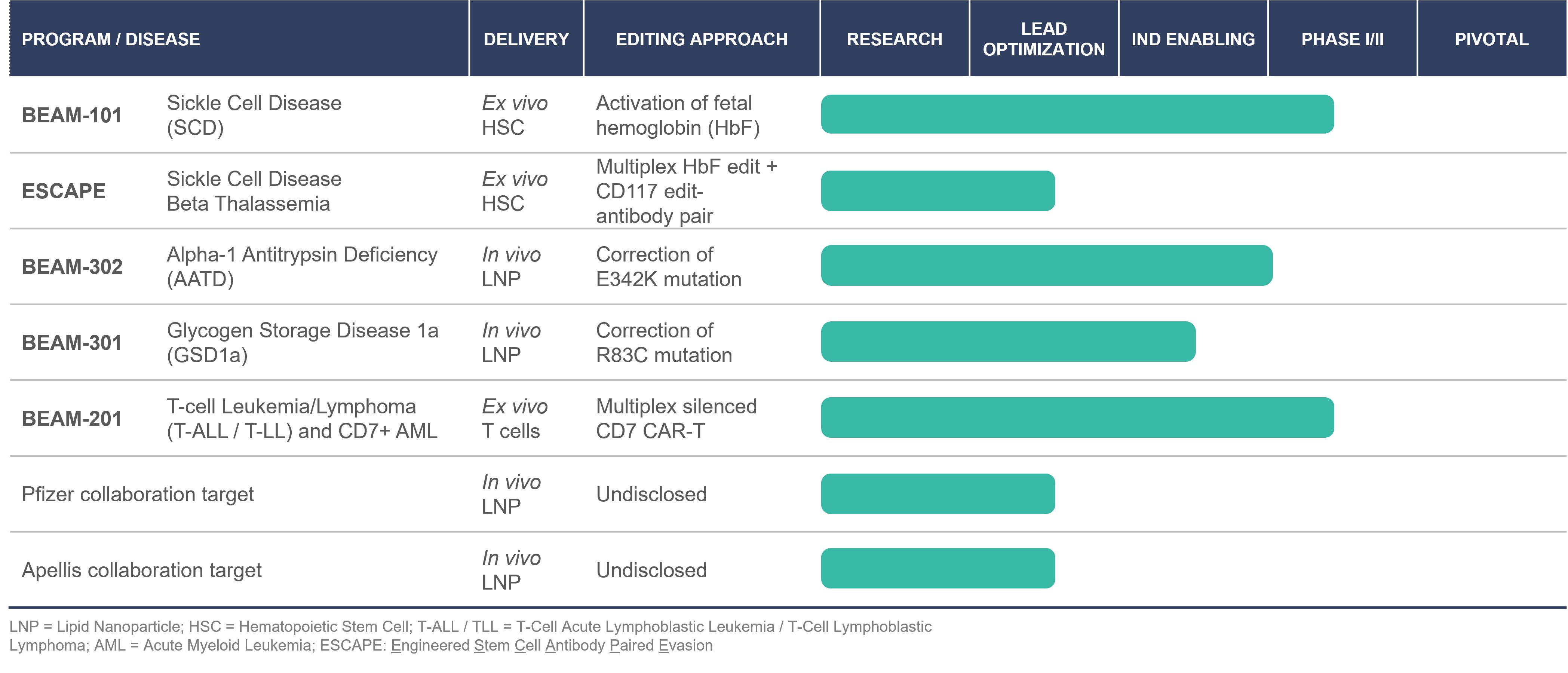
著者らは現在血液学と遺伝病投資組合の中で主要な項目を優先しており、各項目は高度に満足されていない医療需要を有する重要な患者群に差別化治療を提供する可能性がある:
私たちは他の血液学遺伝病免疫/腫瘍学プロジェクトを進めています
5
ヌクレアーゼ編集の限界
遺伝子編集の動作原理はゲノムの自然環境において遺伝子を妨害、挿入、あるいは修正することである。確立された遺伝子編集方法の多くはヌクレアーゼという酵素に依存し、DNAを標的位置で二本鎖切断させる。ヌクレアーゼ編集にはいくつかの明らかな制限がある。
まず,ヌクレアーゼで遺伝子配列を変化させた場合,遺伝結果は予測可能性に乏しい。所望の結果が遺伝子全体をノックアウトまたは閉鎖する場合には、ヌクレアーゼ編集は有効である可能性があるが、標的位置を正確に制御することは許されない特定の遺伝結果であり、その影響は個人によって異なる可能性がある。
第二に、細胞死反応の活性化および/またはゲノム不安定のような二本鎖切断に関連する潜在的毒性が存在する。また,二本鎖切断が誤った位置で発生すると,切断が不必要な遺伝子破壊を招く可能性もある。二本鎖切断を用いた多重編集はこの問題を複雑化させ,大規模なゲノムシフトや再配置を招く可能性があり,ヌクレアーゼによる手法の多重編集への適用性を潜在的に制限している。
第三に,ヌクレアーゼで遺伝子を破壊することは有効であるが,特定の配列を変化させることで遺伝子を訂正または修正することは依然として無効である。それはまた、必要な正しい遺伝子配列を含む別のDNAテンプレートを同時に送達する必要があり、このテンプレートは、二本鎖切断が生じる正確な位置に位置する必要がある。追加のDNAテンプレートの要求は配送の複雑さを大幅に増加させる。
最後に,成人体内の多くの細胞が未分裂であることから,ヌクレアーゼ編集は未分裂細胞中の遺伝子を是正することを許さず,さらにそれらの応用を制限している。
塩基編集:潜在的な差別化遺伝子編集
著者らの塩基編集技術は差別化された遺伝子編集治療方法であり、遺伝情報の基礎レベルである単一塩基--DNA二本鎖切断を招くことなく、ヒトゲノムを変化させる可能性がある。この方法の優雅さと簡単さは“鉛筆”と考えることができ、誤りを消して正しい文字を書くことができる。
私たちの基礎編集プラットフォームは、既存の遺伝子編集方法と比較して、意味のある利点を提供すると信じています
私たちの基礎編集技術は
我々の独自のDNA塩基エディタには2つの主成分があり,それらは融合したり相互に結合して単一タンパク質を形成することができる.第一成分はCRISPR関連蛋白である。これらのタンパク質はヒト細胞中の特定のゲノム位置を対象に改変され改造されている。CRISPR蛋白の標的能は保持されているが,切断能力は修正され,CRISPR蛋白がDNA中で二本鎖切断を起こさないようにした。私たちの基礎編集の第二の構成部分はヒトデアミナーゼであり、これは自然に産生される酵素である。我々のシトシン塩基エディタ(CBE)とアデニン塩基エディタ(ABES)はそれぞれ異なる遺伝子工学デアミナーゼを用いており,我々が設計したこのデアミナーゼは一本鎖DNAにのみ作用する.アデニン(A)やシトシン(C)塩基上のアミン基をデアミナーゼが予測可能な化学修飾を行い,脱アンモニア作用と呼ぶ。
6
CBE中のデアミナーゼはCのアミン基に変換し,ウラシル(U)を形成し,DNAポリメラーゼはウラシルをチミン(T)塩基と読む。この鎖が編集されると,中間DNAは編集後の鎖と未編集の鎖からなり,編集後の鎖は目的部位にUを含み,未編集の鎖はグアニン(G)塩基を持つ.U:Gは不整合であり、編集を保留するために、未編集DNA一本鎖を切断するためにCRISPRを修正し、ノッチと呼ばれる。ノッチは、細胞が未編集の鎖ではなく、新たに編集された鎖を修復テンプレートとして使用することを誘導することによって、編集効率を向上させ、それによって最小転座のU:A対を産生することを目的としている。DNA修復または複製時に、UはTと読み、T:A対を生成し、C:G塩基対からT:A塩基対への永久変換を完了する。同様に、CBEではなくABEが使用される場合、Aのアミン変換はイノシンを産生し、DNAポリメラーゼはイノシンをGと読み、その後、A~Gの変化をもたらす。その結果,A:T対はG:C対に変換される.DNAは二本鎖であるため,非コード鎖を標的とすることにより,コード鎖中のT:A対をC:G,G:C対をA:T対に変換することも可能である.
我々の基礎エディタのモジュールや単独コンポーネントは,特定の疾患に対してカスタマイズすることが可能であり,顕著な開発効率を持つ新しいプログラムを作成することが可能である.例えば,誘導RNAおよび/またはCRISPRタンパク質を変化させることにより,塩基編集された遺伝子配列に応じて異なるゲノム位置にリダイレクトすることができる。デアミナーゼを変化させることで,どの塩基が編集されているか(例えば,CやA)を再配置することができる。したがって,我々の塩基編集技術は汎用性,高効率性,スケーラビリティが高く,将来的に新たな候補薬物の発見に利用できると信じている。
私たちの基地編集プラットフォームは
塩基編集の独特な利点である単塩基編集精度,予測可能な編集結果,高い編集効率と二本鎖切断を回避することは注目される方法となり,広範な治療応用に適していると信じている。これには,遺伝子補正,遺伝子修飾,遺伝子サイレンシングと遺伝子活性化,およびいくつかの遺伝子を同時に多重編集することが含まれる。
我々の次世代遺伝子編集技術を補充するために、著者らはまた一連の交付技術に重大な投資を行い、遺伝子編集或いは他の核酸ペイロードを適切な細胞に伝達し、潜在的な根治療法を実現することを目的とした。これらの技術には離体する電気穿孔や体内にある形態,たとえばLNPsである.私たちの計画では、最初はこれらの技術の応用に重点を置いていましたが、これらの技術の伝達能力は第三者の臨床検証を受けています離体する血液幹細胞を編集し,LNPを肝臓に輸送する。長期的には、OrbitalTreateutics、Inc.または次世代メッセンジャーリボ核酸および非ウイルス送達技術に集中するOrbaryとの協力を含む、より革新的な送達選択にも投資している。また,自己および同種細胞治療のためのメッセンジャーリボ核酸製造および細胞処理などの重要なイネーブル能力を開発した。
高品質製造及び生産タイミングと技術ノウハウの制御が重要であるため、著者らはノースカロライナ州研究三角園区に100,000平方フィートの製造工場を設立し、広範な臨床プロジェクトを支持することを目的とした。この施設は2023年末にcGMP運営を開始し、私たちの製造を支援することを目指しています離体する 血液学と細胞治療計画は体内にある 肝臓と肝臓介在性疾患に対する非ウイルス伝達計画は、潜在的な商業供給を支持するために規模を拡大する能力がある。われわれの予備臨床試験では,主に我々の内部製造能力と,遺伝子薬物に関して製造経験のあるCMOに依存することが予想される。この投資は私たちのポートフォリオと能力の価値、私たちが技術的に成功する可能性、そして私たちが患者に潜在的な生涯治療の速度を提供することを最大限に高めると信じています。
要するに、私たちの遺伝子編集能力と先進的な交付と製造能力を組み合わせた統合プラットフォームを構築することは、新しいプロジェクトを迅速に開発し、私たちのコアプロジェクトのライフサイクルを改善するために、持続可能なポートフォリオを柔軟に開発できると信じています。
私たちの内部パイプを除いて、私たちは技術プラットフォームの広さと深さを統合して、私たちが他の会社と協力するハブを作る機会を与えることができて、これは私たちのビジネスモデルの重要な構成要素です。このモデルは、私たちが重点的に注目している核心領域以外の多くの応用を含む、より広範な可能な応用領域で精密遺伝医学のすべての潜在力を放出するのに役立つと信じている。
私たちのこれらのプラットフォーム活動の全体的な目標は、私たちが遺伝子薬物技術とチームを獲得する機会を拡大し続けることであり、これらの技術とチームは私たちの長期的な価値創造と患者への影響を最大化する。
私たちの基礎編集ブリーフケースは
我々は,臨床開発により複数のプロジェクトを並行して推進する予定であり,各項目が遺伝子定義された患者集団の第1段階臨床試験で概念検証を提供し,加速経路上で承認される可能性がある。われわれの主な項目は鎌状細胞疾患やAATDに集中しており,他の遺伝病や免疫学/腫瘍学的プロジェクトも進められている。
次の表は私たちの主な計画の状態をまとめたものである
7
血液学
著者らは血液学的基礎編集計画を進めており、この計画では、患者から造血幹細胞を収集し、電気穿孔を用いて編集を行い、電気穿孔は治療性構造を採取した細胞に送達し、その後、清髄性調節方案後に患者内、例えば白花丹治療に輸血するための臨床検証技術であり、これは現在造血幹細胞移植の標準看護である。再注入すると,造血幹細胞はインプラントと呼ばれる過程で骨髄の一部を再充填し始める。移植、編集されたHSCsは、正しい遺伝子配列を有する前駆細胞タイプを産生する。これを展開しています離体する私たちのBEAM-101とEASH基地編集プログラムに近い。
鎌状細胞病は1種の深刻な遺伝性血液疾患であり、βグロブリン遺伝子の一点突然変異E 6 Vによって引き起こされる。この変異によりHBSの変異形式は長い剛性分子に集積し,低酸素条件下で赤血球を鎌状に屈曲させた。鎌状細胞は血管を閉塞し、早期に死亡し、最終的に貧血、深刻な痛み(危険像)、感染、脳卒中、臓器不全と早期死亡を招く。鎌状細胞病は米国で最もよく見られる遺伝性血液疾患であり,100,000人が影響を受けると推定されており,その大部分はアフリカ系アメリカ人(1:365生まれ)である。β−地中海貧血はもう一つの遺伝性血液疾患であり,β−グロブリン発現不足による機能性ヘモグロビン産生減少による重篤な貧血を特徴とする。輸血依存型β−地中海貧血(TDBT)はこの疾患の最も深刻な形式であり,通常毎年何度も輸血が必要である。TDBTを有する患者はすくすくと成長できず、持続的な感染と生命に危害を及ぼす貧血を有する。全世界で症状のあるβ−地中海貧血の発症率は1:100,000と推定され,その中でヨーロッパは1:10,000である。米国では,新生児10万人あたり0.7人が影響を受ける出生発生率と,生存率が向上していることから,この疾患の影響を受ける個人数は1400人を超え,上昇していると予想される。
著者らは著者らの血液病治療の基礎編集方法のために長期、段階的な発展戦略を追求しており、その中には著者らのリードを推進することを含む離体するBEAM-101計画は、第1波において、第2波において患者条件調整スキームを改善し、体内にある第3波では,LNPにより基礎編集を患者のHSCに直接伝達している.この技術である基礎編集,改善の条件反射と体内にある著者らの鎌状細胞疾患計画の患者への潜在的適用性を最大限に向上させ、他の多くの深刻な遺伝性血液疾患の治療のためのプラットフォームを作成することができる造血幹細胞の編集を提供する。
第1波:BEAM-101を用いた自己移植を行うEX Vivo塩基編集
我々は,鎌状細胞疾患やβ−地中海貧血の治療のために塩基編集を用いてBEAM−101を開発している。BEAM-101は患者に対する自己HSC研究療法であり、潜在的な同種の最適なプロファイルを提供することを目的とし、遺伝性胎児ヘモグロビン持続性(HPF)個体に見られる一塩基多型をシミュレートすることを目的とした塩基編集を結合する。BEAM−101は,胎児ヘモグロビンを増加させることにより鎌状細胞病やβ地中海貧血の影響を緩和することを目的としており,機能的ヘモグロビン産生を増加させ,鎌状細胞病を有する場合にはヘモグロビンSの重合を抑制することが予想される。
8
2024年1月、私たちは、鎌状細胞症に対するBEAM-101治療の安全性および有効性を評価することを目的とした1/2期臨床試験の第1の患者が投与され、移植に成功したことを発表した。Beacon試験は,最初の3人の患者を含め,1回の治療で1人の患者を治療し,移植成功を確認し,45人までの患者に投与量治療を行う予定である。この臨床試験は、18歳から35歳までの深刻な鎌状細胞疾患を有する患者を最初に組み込むことを目的としており、彼らは以前、少なくとも1つの疾患改良剤の治療を受けていたが、反応が良くなかったか、または耐えられなかった。BEAM-101の動員、調節、および治療後、患者は、好中球および血小板移植を含む安全性および耐性評価を受けるであろう。また患者の治療効果を評価し、治療効果の終点は深刻な血管閉塞事件とベースラインの変化、輸血要求、ヘモグロビンFレベルと生活の質評価を含む。2024年上半期に歩哨キュー中の残りの2名の患者投与量を予定し,Beacon試験の拡張コホート患者で投与を開始し,2024年下半期にBeacon試験における複数人の患者の予備データを報告する予定である。
胎児形式のヘモグロビンあるいはHBFは,成人ヘモグロビン変異を補償する有益な効果が初めてHPF患者で確認された。変異を持っている人は通常,β−地中海貧血や鎌状細胞疾患患者となるが,HPFも罹患しており,症状がないか,あるいははるかに軽微な疾患形態を経験している。HPFはHBG 1とHBG 2遺伝子調節領域の単塩基変化によるものであり,1つ以上のリプレッサー蛋白の結合を阻止し,HBF四量体を構成するガンマグロブリンの発現を増加させる。
塩基編集を用いて,これらの特定の自然発生するガンマグロブリン遺伝子制御エレメントの塩基変化を再現し,リプレッサー蛋白の結合を防止し,ガンマグロブリン発現の再活性化を図り,ガンマグロブリンレベルを向上させようとしている。私たちの臨床前研究は体外培養そして体内にあるBEAM-101の特徴離体するDeliveryはヒトCD 34+HSPCに対する精確かつ高効率な編集を実現し、長期移植とマウス体内の標的遺伝子発現の治療関連増加を招いた。また,この番組については,ガイドに依存することやガイドに依存しないターゲットからの逸脱イベントは観察されなかった.
第2波:非遺伝毒性条件反射
第1弾の開発とともに,造血幹細胞移植を受けた患者の移植コンディショニング案の改善,造血幹細胞移植標準看護に関する毒性挑戦の減少に取り組んでいる。条件反射は患者の体の準備をして受け入れることです離体する編集後の細胞は患者の骨髄を移植しなければ有効ではない。しかし、今日のコンディショニング方案は非特異的化学療法或いは放射線治療に依存し、これは顕著な毒性と関係がある。HSCTにおける遺伝子毒性プレコンディショニングの潜在的代替案として,我々のEASH計画を進めている。Escapeは抗体に基づく調節を多遺伝子編集HSCsと結合することによって、自己造血幹細胞移植前に現在利用可能な鎌状細胞疾患とβ-地中海貧血患者に対する調節方案に関連する毒性挑戦を避けることを目的としている。血液や免疫系の他の疾患では造血幹細胞移植が潜在的なメリットをもたらす可能性があるが,現在の標準的な看護条件療法の毒性はESPECTの応用を制限している。
2022年12月,われわれは2022年米国血液学会年次総会と博覧会でわれわれのEASH計画に関する臨床前データを提出した。鎌状細胞病からの脱出計画は、鎌状細胞病のHGB 1/2遺伝子に対する治療的編集およびCD 117の追加編集を含む多重塩基編集造血幹細胞を含む。ASHに提出された研究結果は最初の体内にある臨床前 脱出計画のデータは2022年以前に共有されたデータに基づいており、このデータは、脱出抗体が野生型CD 117に結合し、そのリガンドのマウス体内での結合を阻止することを示している。また,脱出抗体を服用したマウスの未編集細胞の枯渇と編集細胞の豊富さを観察した。
私たちはすでに私たちの脱出プラットフォームに大量の投資を行い、2024年に第一段階-逃亡鎌状細胞病計画を支持する臨床前研究を開始する予定である。
第3波:生活の中で HSCに対するLNPによる基礎編集
私たちはまだ潜在的な体内にある造血幹細胞に対するLNPsを注入することにより塩基編集を患者に伝達することにより,移植の必要性を完全に解消した鎌状細胞疾患に対する塩基エディタ。この方法は患者により容易な選択を提供することができます特に離体する治療は挑戦的です。前臨床研究では体内にある最も効果的なHSCガイドのLNPを検証して示しました
9
遺伝病
LNPsは臨床的に検証された核酸ペイロードを肝臓に送達する技術である。LNPは多成分の粒子であり、塩基編集分子mRNAと1つ以上のガイドを封入し、外部環境で分解されないように保護し、塩基編集分子の瞬時伝達を実現している体内にあるそれは.1つの治療コースで1剤の基礎編集療法しか必要としない可能性があるため,LNPsは適切な送達様式であり,オリゴヌクレオチドの送達やリボ核酸の遺伝子治療を行う際に観察される合併症など,LNPsの長期使用時に出現する合併症に直面する可能性は低いと考えられる。LNPのすべてのコンポーネントおよび塩基エディタをコードするmRNAの定義が明確であり,合成製造が可能であり,拡張可能な製造に機会を提供している。我々は現在、LNPsを使用してBEAM−302およびBEAM−301を進めている。
BEAM-302:生体内 AATDのLNP肝標的治療
BEAM-302は肝臓に対するLNP塩基編集試薬製剤であり、E 342 K点変異(PIZ型)を是正するための一次治療を提供することを目的としており、これは深刻なAATDを引き起こす主要な原因である。AATDは遺伝性疾患であり、早発性肺気腫と肝臓疾患を引き起こすことができる。最も深刻なAATDは,患者がアミノ酸342位のSERPINA 1遺伝子の2つのコピーにやや変異している(E 342 K,PIZ変異や“Z”対立遺伝子とも呼ばれる)。この点突然変異はα-1アンチトリプシン或いはAATの誤ったフォールディングを引き起こし、分泌ではなく肝細胞内に蓄積し、循環中のAATレベルが非常に低い(10%-15%)ことを招く。PIZ AAT蛋白変異体は,低いレベルをもたらすことに加え,野生型AAT蛋白に比べて酵素活性も低かった。その結果,肺は好中球エラスターゼによって保護されず,肺気腫などの肺内進行性の破壊的変化を招き,肺移植が必要となる可能性がある。変異したAAT蛋白も肝臓に蓄積し,肝臓の炎症や硬化を招き,最終的に肝不全や癌をきたす可能性があり,肝移植を受ける必要がある。米国では約10万人が2つのZ対立遺伝子のコピーを持っていると推定されている。AATD患者に対する根治療法は現在のところない。
我々の塩基編集の高効率と精度により,我々のABESを用いてE 342 K点変異を精度良く是正し,機能性AAT蛋白の生産を回復することを目標としている。2020年には,塩基編集によるAATDの変異を直接訂正する能力を示し,提供した体外培養そして体内にあるこの疾患を是正するための基礎編集の臨床前概念検証に用いられる。2024年1月にBEAM−302にCTAを提出したことを発表し,CTAが受け入れられれば,BEAM−302の安全性と有効性を評価するために,2024年上半期に米国以外の試験地点で1/2期の臨床試験を開始する予定である。
BEAM−301:体内肝臓標的GSD 1 aのLNP研究
BEAM-301は肝臓に対するLNP塩基編集試薬の調合であり、R 83 C突然変異を是正することを目的とし、これはGSD 1 aの最もよく見られる発病突然変異であり、最も深刻な形式を招くGSD 1 aの突然変異でもある。GSD 1 aは常染色体劣性遺伝病であり、G 6 PC遺伝子突然変異によって引き起こされ、この突然変異は重要な酵素G 6 Paseを破壊し、G 6 Paseはグルコース安定性の維持に重要である。G 6 Pase活性の抑制は空腹時血糖値の低下を招き、てんかん発作と致命的である。この突然変異の患者は通常コーンスターチを継続的に服用する必要があり,コーンスターチがなければ1~3時間以内に低血糖ショックに入る可能性がある。
著者らはGSD 1 a患者を治療する方法はLNP伝達に塩基編集を応用することによって、このような疾病を引き起こす2つの最もよく見られる突然変異、R 83 CとQ 347 Xを修復することである。米国では,この2つの点変異はそれぞれ300名と500名の患者を占めると推定されている。
2023年10月、私たちは新しい臨床前データを公表し、BEAM-301がR 83 C変異を直接是正する能力を証明した。これらのデータは単一用量のBEAM-301が回復したことを示しています体内にあるげっ歯類疾患モデルは少なくとも1年間持続する。2024年上半期には、BEAM-301によるGSD 1 a治療の効果を米国で選択されたいくつかの試験地点で評価するIND申請を提出する予定です。
免疫学·腫瘍学
塩基編集は強力なツールであり、核酸酵素と同時に編集した意外な標的効果を生じることなく、多くの遺伝子を同時に多重編集することができ、二本鎖切断を生じるためであると信じている。T細胞において大量の多重編集を作成する能力はCAR-T細胞と他の細胞療法の一連の機能の組み合わせを与えることができ、それによって血液病或いは固形腫瘍の治療におけるそれらの治療潜在力を極めて大きく増強する可能性がある。
10
BEAM-201:汎用CD 7標的CAR-T細胞
BEAM-201は健康ドナーからのT細胞からなり、これらのT細胞はTRAC、CD 7、CD 52とPDCD 1で同時に編集し、その後、抗CD 7 CARをコードするレンチウイルスと形質導入され、このCARはCAR-T細胞に対する同種異体CD 7を産生することを目的とし、殺菌剤にも免疫抑制にも抵抗できる。再発/難治性T-ALL/T-LL患者におけるBEAM-201の安全性および有効性を評価することを目的とした、ヒト第1段階1/2臨床試験において、第1の患者にBEAM-201を投与した。試験の肝心な安全終点は緊急治療と治療に関連する不良事件を含み、肝心な治療効果の終点は完全或いは部分反応の患者の割合、造血幹細胞移植条件に符合する割合及び最小残留疾患陰性状態に達する割合を含む。私たちは引き続き1/2期臨床試験に参加しており、2024年下半期にBEAM-201の予備臨床データセットを報告し、この潜在的パートナーおよび他の潜在的パートナーのために潜在的パートナーを求める予定である離体する我々が行っている多重塩基編集を用いた次世代同種細胞療法の研究も含めたCAR−T計画である。
私たちの精密遺伝子編集技術の組み合わせは
私たちはいくつかの分野のために他の3つの相補的な遺伝子編集技術の組み合わせ-Prime編集、Cas 12 bヌクレアーゼ編集、およびRNA塩基編集を許可した。塩基編集と結合して、著者らはすでに広範かつ多機能な次世代遺伝子編集技術の組み合わせを組み立て、多くの深刻な疾病の潜在的治療に応用した。
我々はPrime Medicine,Inc.のPrime編集ライセンスを持っている.Prime編集は目標位置でDNA短配列の書き換えを実現できる可能性がある.Prime編集はCRISPR蛋白を用いてDNA中の突然変異位置を標的とし、標的DNAの一本鎖を切断する。誘導RNAは、CRISPRタンパク質が誘導RNAに相補的なDNA配列を認識することを可能にし、逆転写のためのプライマーおよび置換鋳型も担持する。逆転写酵素はノッチ部位のテンプレート配列を複製し,編集を実装する.塩基編集と同様に,モチーフ編集は標的DNAの二本鎖切断を招くことはなく,挿入と欠損率は二本鎖切断に依存する遺伝子編集技術よりも低い。
私たちは、任意の単一塩基変換変異の作成または修正、および鎌状細胞疾患の治療のための任意の編集のための主要編集技術を開発するための独占的な権利を有する。変換変異(すなわちAからG、GからA、CからTまたはT~C)は、現在の塩基エディタのすべての目標を含む疾患に関連する最大の遺伝子変異である。
私たちはまた、ブロイド研究所またはブロス研究所と許可プロトコルを締結し、Cas 12 bヌクレアーゼファミリーへのアクセスを可能にし、これは、二本鎖切断を必要とするいくつかのアプリケーションに適用可能な“切断”編集を可能にし、またはCas 12 bの汎用遺伝子標的能力を他の遺伝子編集アプリケーションに使用することを可能にする。
我々の遠大な研究所許可証はまたRNA塩基編集技術を使用することができ、これは2つの部分からなるモジュール化システムであり、RNA指導のCRISPRタンパク質を用いてRNA鎖を標的とし、デアミナーゼを用いて編集を行う。このCas 13と呼ばれるCRISPRタンパク質は修飾されており,RNA鎖を切断することができず,RNA鎖中の特定の目的位置で単一塩基編集が可能なデアミナーゼと融合している。
協力する時間です。国家統計局
私たちの塩基編集、遺伝子編集と交付技術集合は広範な遺伝病領域で巨大な潜在力を持っていると信じている。この潜在力を十分に実現するために、先駆的な会社や有力な学術·研究機関との革新的な協力、許可、戦略連盟を構築し、計画している。また,臨床前研究と開発の関係を加速させる可能性があることを継続して発展させる予定である。これらの関係は,基地編集の潜在力を最大限に発揮し,重篤な疾患を有する患者に生涯治療を提供するというビジョンを積極的に追求することができると信じている。
ファイザー社
2021年12月にファイザー社と4年間の研究協力協定を締結しました体内にある肝臓、筋肉と中枢神経系の3種類の稀な遺伝病に対する基礎編集プログラム。合意条項によると、我々は、既存の計画に含まれていない3つの予め指定された未開示の目標のために開発候補者を選択することで、すべての研究活動を行う。ファイザーは、各候補開発プロジェクトに独占的なグローバルライセンスを付与することを選択することができ、その後、このような候補開発プロジェクト毎のすべての開発活動、および潜在的な規制承認および商業化を担当する。私たちは、第1/2期臨床試験終了時に、選択権行使費用を支払った後、協力によって許可された薬物に関する世界共同開発と共同商業化プロトコルを加える権利があり、この合意によると、ファイザーとファイザーは純利益および開発と商業化コスト(ビリオン/ファイザー)を35%/65%の割合で共有する。
11
アペリス製薬会社は
2021年6月、私たちはApellis PharmPharmticals、Inc.またはApellisと研究協力プロトコル、すなわちApellisプロトコルを締結し、私たちの塩基編集技術を用いて補体系によって駆動される疾患の新しい療法を発見することに重点を置いた。アペリスプロトコルの条項によると,異なる器官における補体系における特定の遺伝子に対して目,肝臓,脳を含む6つの塩基エディタの臨床前研究を行う。Apellisはこの6つのプログラムのいずれか1つまたは全部を許可する独占的選択権を持ち、後続開発の責任を負うだろう。私たちはApellisと協力許可の下の1つのプロジェクトについてそれぞれ50%を占める米国の共同開発と共同商業化協定を締結することを選択するかもしれない。
Verve治療会社
2019年4月、心血管疾患治療遺伝子編集に専念したVerve Treateutics,Inc.またはVerveと協力および許可プロトコルであるVerveプロトコルを締結し、2022年7月にVerveとVerveプロトコルを改訂した。改訂されたVerveプロトコル条項によると、我々は、これらの標的に対して我々の基礎編集技術を使用すること、およびその中の2つの標的に対して我々のいくつかの遺伝子編集技術を使用することを含む、合計3つの肝臓媒介心血管疾患標的に対するヒト治療応用のためのVerveグローバル独占許可を我々のいくつかの編集技術に付与する。交換として、私たちはVerve普通株の株式を取得した。
2023年10月、私たちは、PCSK 9、Angptl 3、および開示されていない肝臓媒介心血管標的のためのプログラムを含むVerveプロトコル下のいくつかの資産および他の権利を取得し、Verveの各心血管疾患基礎編集プログラムを共同開発および共同商業化する権利を含む礼来会社または礼来会社と譲渡および委託契約を締結した。また、礼来会社はVerveプロトコルに従って任意の未来のマイルストーンまたは特許権使用料支払いを私たちに支払う権利を獲得した。礼来会社協定の条項によると、私たちは2億ドルの支払いを受け、いくつかの臨床、監督、連盟活動が完了した後、3億5千万ドルまでの潜在的な未来開発段階で支払いを受ける資格がある。2023年10月には、礼来社が1株24.94ドルで礼来社に2,004,811株の普通株または普通株を売却·発行することを規定しており、購入契約日前の30取引日の私たちの普通株の出来高加重平均株価割増15%に相当し、総購入価格は約5,000万ドルである。購入協定はすべての当事者の慣例的な陳述、保証、そしてチェーノを含む。
サナバイオテクノロジー
2021年10月、我々はSana Biotech,Inc.またはSanaとオプションおよびライセンス契約、またはSana協定を締結し、この協定に基づいて、私たちのCRISPR Cas 12 b技術にSanaの非独占的研究開発および商業権を付与し、ヌクレアーゼ編集をある程度実行する離体する遺伝子工学細胞治療計画ですサナ協定の条項によれば、許可製品は、特定の遺伝的標的のための特定の同種T細胞および幹細胞由来製品を含むが、サナは、そのような製品および標的を添加および置換するいくつかの限られた権利を含む。サナ協定は、任意の梁制御を付与するベース編集を実行する権利を排除する。2023年、SANAはSC 291の一期臨床試験を開始し、SC 291はCD 19に対する同種異体CAR-T細胞療法であり、各種のB細胞悪性腫瘍患者に応用された。2023年11月、SANAはFDAがそのIND申請を許可し、各種B細胞を介した自己免疫疾患患者におけるSC 291の初のヒト試験を開始したことを発表した。2024年1月、SANAはFDAがそのIND申請を許可し、SC 262の初の人体試験を開始し、SC 262はそのCD 22が指導した同種異体CAR-T細胞療法であり、再発或いは難治性B細胞悪性腫瘍患者に応用した。上記のすべての事件について、サナはサナ協定に基づいて私たちに非実質的なマイルストーン支払いを支払った。
眼窩治療学
2022年9月、我々は軌道会社と許可および研究協力協定または軌道協定を締結し、この協定によれば、軌道協定締結後3年以内に制御されたいくつかの技術にそれぞれ他の許可を付与し、これらの技術は、非ウイルス送達またはヒト疾患の予防、治療または診断のためのRNAの設計または製造に必要または合理的に有用である。我々の軌道許可は我々の排他的領域以外のすべての領域に適用され,我々の既存計画の目標と基本的にすべての適応も排除されている.私たちの専属領域は、細胞移植のための遺伝子編集または条件作用中に作用するすべての製品および生物製品、または任意のそのような製品または生物製品と組み合わせた製品および生物製品を含む。軌道会社の私たちの許可は軌道会社の専属分野を除くすべての分野に適用されます。Orbitの専門分野は、ワクチンとして使用される製品および生物学的製品および治療用タンパク質を含むが、治療用タンパク質は、(I)遺伝子編集を使用し、(Ii)調節のための、(Iii)再生医学のための、(Iv)CAR-T、CAR-NKおよびCAR-マクロファージ組成物を含む自動車免疫療法として使用され、(V)t細胞受容体療法として使用されるか、または(Vi)特定の免疫反応を調節するための自動車免疫療法として使用される。ライセンスは3年以内にそれぞれの排他的領域で独占的であり、その後これらの分野では非独占的である。我々と軌道会社は、“軌道協定”を締結してから3年以内に、限られた例外を除いて、相手の排他的な分野で製品または生物製品を研究、開発、商業化したり、研究、開発、商業化のライセンスを付与したりしないことに同意している。
12
競争
製薬と生物技術業界は、遺伝子薬物領域を含み、技術の迅速な進歩、競争が激しく、知的財産権を高度に重視する特徴がある。私たちは差別化された技術、科学的専門家、知的財産権の地位が競争優位を提供してくれると信じていますが、私たちはこれらの分野で様々な会社からの潜在的な競争に直面しています。これらの業界では、既存の大手製薬会社、専門製薬会社、バイオテクノロジー会社と競争する。
他にもCRISPR/Cas 9ヌクレアーゼ技術が使用されており,Cariou Biosciences,Editas Medicine,CRISPR Treeutics,Intellia Treeutics,Arbor BioTechnologies,Metagenomiがある。他のいくつかの会社では,他のCASヌクレアーゼ,亜鉛指,ArcusおよびTALヌクレアーゼ,Sangamo Biosciences,Precision BioSciences,Bluebird Bio,Alallgene Treeutics,Mammoth Bioscience,Cellectisを含む他のヌクレアーゼ系遺伝子編集技術を用いている。また,Prime Medicine,Tessera Treeutics,Scribe Treeutics,Tome Biosciences,Life EDIT(ElevateBio社),PerkinElmer(前Horizon Discovery社),Intellia Treeuticsなど,他の遺伝子編集方式も出現している。PerkinElmer,Metagenomi,Revrate,Intellia Treeuticsは基本編集技術を開発しているが,Tessera Treeuticsは移動可能な遺伝子を用いて遺伝子編集を行っている。また,様々な遺伝子薬,エピジェネティック調節,オリゴヌクレオチド,CAR−T治療法などを利用した会社からの競争に直面している。
私たちが注目している疾患分野では,治療法が承認された競合会社,治療法を開発している会社,将来出現する可能性のある他の会社も意識している。ヘモグロビン疾患については,これらの会社は全世界血液治療会社(ファイザー社の一部),CRISPR治療会社,Vertex製薬会社,ブルーバード生物会社,ノワ製薬会社,Editas医薬会社,Kamau治療会社,支点治療会社,Agios製薬会社を含む。T細胞悪性腫瘍では,これらにはicell遺伝子治療会社,PersonGen,Wu genがある。より広く言えば、免疫腫瘍学細胞治療領域では、これらの会社は異遺伝子治療会社、Cellectis社、CRISPR治療会社、Adicet Bio社、百時美施貴宝会社、Fate治療会社、Gilead Sciences社、ノワ製薬会社、ポセイダ社、伝奇生物会社とAutolus治療会社を含む。われわれの肝臓標的治療には,Intellia Treateutics,Editas Medicines,CRISPR Treateutics,浪潮生命科学社,Moderna,Korro Bio,矢印製薬会社,Dicerna製薬会社,Ultragenyx,矢印製薬会社,LogicBio治療社,Generation Bio,Vertexがある。
私たちが開発と商業化に成功した任意の候補製品は、既存の治療法や将来発売される可能性のある新しい療法と競争し、これらの治療法および新しい療法は、承認される可能性のある製品候補疾患の治療に承認される。これには、小分子療法、抗体療法および/またはタンパク質療法などの他のタイプの療法が含まれる可能性がある。
また、私たちの多くの既存または潜在的な競争相手は、単独であっても、パートナーと協力しても、研究開発、製造、臨床前研究と臨床試験を行い、製品の承認を求める上で、今日よりも多くの財務資源と専門知識を持っている。製薬、バイオテクノロジー、遺伝子治療業界の合併と買収は、私たちの少数の競争相手により多くの資源を集中させる可能性がある。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。著者らはまた合格した科学と管理人材を採用、採用と維持し、臨床試験のために臨床試験場と患者登録を確立し、CMOで製造槽を獲得し、著者らのプロジェクトと相補或いは必要な技術を獲得する面でこれらの会社と競争している。もし私たちの競争相手が私たちが開発する可能性のある任意の製品よりも安全で効率的な製品を開発し、商業化すれば、特にそれらが治療法を代表し、副作用がより少なく、より深刻ではなく、より便利で、より安価であれば、私たちのビジネス機会は減少または消失するかもしれない。私たちの競争相手も私たちよりも早くFDAや他の規制機関のその製品の承認を得ることができ、これは私たちの競争相手が私たちが市場に入る前に強力な市場地位を確立することをもたらすかもしれない。私たちのすべての計画の成功に影響を与える重要な競争要素は、それらの有効性、安全性、利便性、精算の利用可能性である可能性がある。
知的財産権
私たちの成功は、私たちの業務に関連するプラットフォーム技術、私たちのプログラム、およびノウハウのために独自の保護を獲得し、維持し、私たちの知的財産権、特に私たちの特許権を擁護し、実行する能力があるかどうかにある程度依存し、私たちのビジネス秘密を秘密にし、他人のいかなる効果的かつ実行可能な知的財産権を侵害、流用、または他の方法で侵害することなく運営される。私たちは、私たちのプラットフォーム技術、既存および計画中の計画、および私たちの業務発展に非常に重要な改善に関連する米国およびいくつかの外国特許出願を独占的に許可し、提出することによって、私たちの独自の地位を保護することを求めており、これらの分野では、特許保護を得ることができる。私たちはこれらの努力をしたにもかかわらず、私たちが許可または提出した任意の特許出願または将来可能な許可または提出された任意の特許が特許を付与されることを保証することはできず、私たちが許可した任意の特許または将来許可または許可される可能性のある特許が挑戦されないか、無効または回避されないか、またはこれらの特許が商業的に私たちの技術を保護するために使用されることを保証することはできない。我々の知的財産権に関連するリスクに関するより多くの情報は、第1 A項を参照されたいリスク要因-私たちの知的財産権に関するリスクこのForm 10-K年間報告書にあります
13
私たちの完全な所有と許可された特許と特許出願は、私たちの基本編集プラットフォームと私たちのプログラムの様々な側面をカバーしています
CRISPR/CAS 9システムに関連する特許および特許出願を許可することも可能である。私たちは、物質組成、使用方法、およびプロセスクレームを含む、可能な場合に、私たちのプラットフォーム技術の各コンポーネントと私たちの製品の組み合わせにおける計画のための追加の特許保護を求め続けるつもりです。2023年12月31日現在、私たちの完全特許組み合わせには、発行された米国特許5件と、米国以外の管轄区で発行された6つの特許が含まれている。PCT出願、仮特許出願、および上述した米国および外国特許に対応する出願を含む約387件の処理される特許出願がある。また、ピム社は、ボイド研究所会社、ロンドン大学学院商業有限公司とアペリス製薬会社の間の米国特許と26件の未解決特許出願を共同で所有している。米国以外の特許および特許出願は、オーストラリア、ブラジル、カナダ、中国、欧州、香港、インド、日本、韓国、シンガポール、南アフリカを含む多くの司法管轄区で提出されている。我々が持つ多くの特許および特許出願は、増強活性または新たな特性を有すると主張する塩基編集変異体を含む、我々のDNA塩基編集技術に関連しており、この塩基編集方法を用いて、この塩基編集を適応治療に用いる方法、免疫細胞における多重塩基編集離体する塩基編集を標的としたRNAを治療に関連するDNA配列に導き、塩基編集特異性を評価する方法。私たちが持っているいくつかの特許と特許出願はウイルスと非ウイルス伝達技術と関連がある。米国特許が発行された場合、適切な維持費が支払われた場合、米国特許は、追加の特許期限調整または特許期限延長を含まず、2039年から2044年の間に満了すると予想される。
2023年12月31日現在、私たちのライセンス特許の組み合わせは、約45件の許可された米国特許と、米国以外の司法管轄区域で許可された約145件の特許とを含む。PCT出願、仮特許出願、および上述した米国および外国特許に対応する出願を含む約274件の処理される特許出願がある。米国以外の特許および特許出願は、オーストラリア、カナダ、中国、ヨーロッパ、香港、インド、イスラエル、日本、韓国、ニュージーランド、ロシア、シンガポールを含む多くの司法管轄区域で提出されている。我々のDNA塩基編集許可製品の組み合わせの特許および使用は、新規塩基編集のための請求項、塩基編集のための工学的デアミナーゼ(例えば、進化TADA)の請求項、塩基編集または構成要素としての工学的デアミナーゼを含む組成物、このような塩基編集を使用する方法、このような塩基編集を適応の治療に使用する方法、および塩基編集を標的とするRNAを誘導して関連DNA配列を治療することを含む、我々のDNA塩基編集許可製品の組み合わせの特許および使用を含む。ライセンス内の特許および出願はまた、使用を含むプラットフォーム技術に関連する様々な態様をカバーしている化膿性連鎖球菌カス9、黄色ブドウ球菌Cas 9、Cas 9 PAM変形、Cas 9の非アクティブ形態および/またはCas 9ニックネーム、および基本エディタを提供するためのシステム。我々のRNA塩基編集許可製品の組み合わせの特許および使用は、塩基編集を構成要素とする組成物、塩基編集を治療関連RNA配列の誘導RNAとして使用すること、およびこのような塩基編集を使用して適応を治療するための方法を含む新規塩基編集の請求項を含む、新規塩基編集の請求項を含む。我々のCas 12 bが製品の組み合わせを許可する特許および出願を編集することは、Cas 12 bを使用してDNA(例えば、DNAのヌクレアーゼ切断)を修飾する方法、およびCas 12 bを構成要素として含む工学的および/または非自然に存在する組成物を含む。我々が許可した送達技術の組み合わせの特許および使用は、新規脂質系送達システムおよび組成物、ウイルス系送達システムおよび組成物の請求項、およびそのようなシステムおよび組成物を使用して塩基編集を送達する方法を含む。我々のプラットフォーム平衡認可製品の組み合わせの特許および使用は、荷電塩基編集タンパク質を細胞に送達するための組成物および方法の請求項、塩基編集システムの修正および改善、塩基編集複合体のヌクレオチド結合タンパク質成分、RNA成分および塩基編集酵素成分の誘導の改善、遺伝子標的および塩基編集効率を評価する方法、ならびに主要編集組成物および方法を含む。私たちの現在の特許と特許出願は
14
適切な維持費を支払うことは、追加の特許期間調整または特許期間延長(または対応する外国等価物)を含まず、2034年から2040年の間に満了すると予想される。私たちが許可した知的財産権に関する情報は、“-知的財産権ライセンス”という次の項目を参照してください
我々は研究活動を行う非独占的許可を有しており,何らかの特許やEditasのCas 9やCas 12 aに対する特許出願を独占的に許可する権利があり,Editasは様々な学術機関からこのような特許の許可を得ている.Cas 9のケースでは、多くの米国特許は特許商標局によって発表された妨害を受け、いくつかの欧州特許は1つまたは複数の反対の対象である。我々の知的財産権に関するリスクに関するより多くの情報は、第1項を参照されたいビジネス−知的財産権−知的財産権ライセンスプロジェクト1 Aリスク要因-私たちの知的財産権に関するリスクこのForm 10-K年間報告書にあります
個別特許の期限は,特許を付与した国の特許法的期限に依存する。米国を含む多くの国では,特許期間は適用国の非臨時特許出願の最初の要求提出日から20年である。しかしながら、特許によって提供される実際の保護は、国によって異なり、特許のタイプ、そのカバー範囲、規制に関連する延長の利用可能性、特定の国の法的救済の利用可能性、および特許の有効性および実行可能性を含む多くの要因に依存する。米国では、場合によっては、特許期間は、特許期限調整またはPTAによって延長することができ、このような調整は、米国特許商標局による特許の審査および付与に関する特許権者の行政遅延を補償することができ、または短縮することができる(例えば、).もし、1つの特許がより早い満期日を有する共通所有特許によって最終的に放棄される場合)。場合によっては、そのような特許協定は、米国特許期間を、米国特許に関連する非臨時特許出願を提出する最初の日から20年以上延長させる可能性がある。1984年の“医薬品価格競争および特許期限回復法”(通称“ハッジ·ワックスマン法”)によれば、特許期間延長またはPTEは、FDA規制審査中に失われた特許期間の補償として、FDAによって承認された薬物をカバーする特許にも適用可能である。ハッジ·ワックスマン法は特許満了後最大5年間のPTEを許可する。PTEの長さは薬物規制審査を受ける時間の長さと関係がある。1つの特許の残り期間は、製品承認日から合計14年間延長することができず、承認された薬物、その使用方法または製造方法に適した特許を延長することしかできない。ヨーロッパおよび他のいくつかの管轄区域にも同様の規定があり、承認された薬物をカバーする特許の期限を延長する。将来、私たちの製品が規制部門の承認を得たら、私たちはそのような製品の特許出願PTEをカバーする資格があるかもしれませんが、米国FDAを含む適用当局が、このようなPTEが付与されるべきかどうか、そのようなPTEが付与された場合の期限の評価に同意する保証はありません。我々の知的財産権に関連するリスクに関するより多くの情報は、第1 A項を参照されたいリスク要因-私たちの知的財産権に関するリスクこのForm 10-K年間報告書にあります
私たちはまた、ビジネス秘密、技術ノウハウ、持続的な技術革新、および機密情報に依存して、当社の独自の地位を発展させ、維持し、特許保護から当社の業務を保護しているか、または特許保護に適していないと考えている側面を保護しています。従業員、コンサルタント、科学コンサルタント、請負業者と秘密保護協定を締結することで、私たちのノウハウやプロセスを保護することを求めています。また、私たちは私たちのビルの実体安全と、私たちの情報科学技術システムの実体と電子安全を維持して、私たちの資料と商業秘密の完全性とセキュリティを維持することに力を入れています。私たちは私たちのビジネス秘密を保護して保存する措置を実施しているが、これらの措置は違反される可能性があり、私たちはこのような違反に対応する十分な救済措置がないかもしれない。しかも、私たちのビジネス秘密は競争相手に知られたり独立して発見されるかもしれない。我々の知的財産権に関連するリスクに関するより多くの情報は、第1 A項を参照されたいリスク要因-私たちの知的財産権に関するリスクこのForm 10-K年間報告書にあります
私たちの会社名と関連設計はまた商標保護に依存している。私たちの登録商標の組み合わせは、2023年12月31日現在、米国および特定の海外司法管轄区域における登録·許可商標および処理すべき商標の約35個の出願を含む。
知的財産権許可証
私たちは多くのライセンス契約の当事者であり、これらの合意に基づいて、私たちは第三者から特許、特許出願、その他の知的財産権を許可します。許可された知的財産権部分は,CRISPRに関連する物質成分とその基礎編集のための状況をカバーしている。このような許可証は私たちに様々な勤勉さと財政的支払い義務を課している。私たちは未来にこのような種類の許可協定を締結し続けると予想する。私たちは次の許可協定が私たちの業務に必須的だと思う。
総裁やハーバード大学のアカデミー会員とのライセンス契約
2017年6月、私たちはハーバード大学と改訂された許可協定またはハーバード許可協定を締結し、ハーバード大学が所有または制御している特定の特許権に基づいて、ヒト生殖系の修正および非ヒト動植物用途のための製品を含まず、独占的、世界的に、印税を徴収する再許可可能な許可を取得し、任意およびすべてのヒト疾患および疾患の予防または治療の分野で製造、製造、販売、販売、および輸入許可製品を提供することができる。我々はまた,非独占的,世界的に印税を負担する再許可可能な研究,研究,開発,ハーバード特許権に関する非許可製品の“イネーブル”製品を取得した。
15
他に加えて、付与された特許は、特定の疾患および条件の治療、およびより一般的な基礎編集のためのC−to−T、A−to−G、およびC−to−G基礎編集のためのものである。
ハーバード許可協定によると、私たちが準備してハーバード大学に提出した開発計画に従って、ライセンス特許がカバーする基本編集技術を含む製品を商業的に合理的に開発しなければなりません。開発計画には、私たちが達成しなければならないいくつかの発展マイルストーンと、これらのマイルストーンを完成させるスケジュールが含まれており、私たちは私たちの善意の判断に基づいて、時々発展計画を更新して、これらのマイルストーンを実現することができます。もし私たちがどの国でも規制部門の承認を得ることに成功し、許可製品をその国の商業市場に導入することができれば、私たちはまた、その許可製品を商業化し、その許可製品を合理的に大衆に提供するための商業的合理的な努力をしなければならない。もし私たちが開発マイルストーンの任意の期限内に完了できなかった場合、ハーバードは故障の性質と影響を受けたマイルストーンに基づいて“ハーバード許可協定”を終了したり、適用された許可製品(S)の許可を終了したりする可能性がありますが、いくつかの例外的な状況と私たちがこのような失敗を是正する機会の制限を受けています。また,いくつかのサブカテゴリライセンス特許がカバーするライセンス製品の開発のマイルストーンを達成しなければならない.これらのサブカテゴリの面でマイルストーンを実現することができず、ハーバードはこれらの失敗したサブカテゴリの下で第三者に非独占的な許可を与える権利がある。
ハーバード許可協定に基づいて私たちに付与された許可は、ハーバードおよびいくつかの第三者が所有するいくつかの以前に存在する権利から明確に制限されている。例えば、いくつかの許可特許は、Howard Hughes医学研究所の従業員によって開発され、その後、ハーバード大学に割り当てられたが、ハーバード大学とHoward Hughesとの間の非独占的許可の制約を受けており、この許可に基づいて、Howard Hughesはハーバード大学から研究目的のためのいくつかの許可特許の許可を取得し、非営利組織および政府エンティティに再許可する権利がある。さらに、いくつかのライセンス特許要件または米国政府が後援する研究によって生成された発明をカバーし、適用される米国法律に基づいて、米国政府は、そのようなライセンス特許に関連するいくつかの権利を保持する。また、ハーバード大学は、自分と他の非営利研究組織が研究、教育、学術目的で特許を許可する限られた権利を実践していることを保持している。さらに、ハーバードは、米国またはいくつかのヨーロッパ諸国または地域の規制機関が任意の特許製品を承認してからしばらくの間、第三者に非独占的権利を付与して、開発、製造、製造、輸入、販売、またはそのような特許製品またはその等価物を他の方法で流通または流通させ、私たちが規制承認を求めることを計画していない特定の発展途上国で現地で負担される基礎販売のためにのみ提供する。
ハーバード許可協定に基づいて付与された許可は独占的であるにもかかわらず、ハーバードは、許可された特許に基づいて、限られた場合に、この分野の1つまたは複数の特定の目標のための製品または提案された製品を研究、開発および商業化するために、第三者に許可を付与することができる。私たちが競争相手を指定した第3の方向でなければ、ハーバードがこのような許可を問い合わせ、ハーバードが私たちに推薦して再許可協定を締結しようとしたが、しばらくしてできなかった場合、ハーバードに提案された開発と商業化を記述するいくつかの情報を含む提案を提出した場合、ハーバードはその提案を私たちに通知するかもしれない。もし私たちが提案された製品を研究、開発、または商業化していない場合、私たちはハーバード大学に、その提案された製品を開発することに興味があるかどうか、第三者と再許可協定を締結してその提案された製品を開発すること、または他の第三者と再承認して同じ提案を開発できる製品を開発することができるかどうかを通知することができる。もし私たちがハーバードにこの提案の製品を開発することに興味があることを通知すれば、私たちはハーバード許可協定の下での開発計画と類似した開発計画を用意して、その提案した製品を開発する。もし私たちがハーバードに通知した場合、私たちは、第三者が許可特許下の従属許可を取得して、提案された製品を開発するために従属許可協定を達成することに興味がある場合、指定された時間に当該従属許可協定を締結し、合理的な証拠を提供する。このような提案された製品を研究、開発または商業化していない場合、開発計画を提供することができない場合、または指定された期間内にそのような提案された製品について再許可協定を達成することができない場合、ハーバードは、許可特許の下で適用される第三者が提案した製品を研究、開発、商業化することができる。
私たちは、第3者と達成されたこのような再許可協定が“ハーバード許可協定”の条項を引き続き遵守しなければならないことを前提として、“ハーバード許可協定”の下で私たちの権利を第三者に再許可することをさらに許可され、“ハーバード許可協定”に基づいて私たちに付与されたいくつかの権利は、再許可しか与えられない善意の私たちと協力して1つ以上の許可製品を開発する協力パートナー。さらに、このような再許可協定には、私たちが“ハーバード許可協定”を遵守できることを確実にするためのいくつかの条項が含まれなければならない。私たちはまた、適用される従属許可者が再許可協定に違反するいかなる行為にも責任を負い、もしこのような違反が“ハーバード許可協定”に実質的に違反した場合、違反を是正し、または私たちが従属許可を終了する権利を実行しようと努力すれば、たとえ“ハーバード協定”に対する重大な違反を招いても、従属許可者の違約によってハーバードで終了しないことを前提としている。
16
ハーバード許可協定に基づいて私たちに与えられた許可の交換として、私たちは最初にハーバードに101,363株の普通株を発行し、その後、ハーバード許可協定の逆希釈権利に基づいて765,549株の私たちの普通株を発行した。特定のカレンダー年度に応じて、ハーバード大学に毎年低から中五桁から下位六桁までのライセンス維持費を支払うことも求められている。ハーバード大学はまた中高八桁の範囲内の潜在的な臨床と監督管理のマイルストーンを獲得し、私たちの普通株の公平な市場価値の増加によって成功報酬を得る権利がある。もし私たちがハーバード許可協定の間に統制権変更が発生したら、いくつかの記念碑的な支払いが増加するだろう。AラウンドとBラウンドの融資を終えた後、私たちは合計900万ドルをハーバード大学に支払った。
2021年5月、第1回成功支払測定を行い、ハーバード大学に不足している金額は1500万ドルと算出された。私たちは普通株で支払うことを選択し、2021年6月10日に174,825株の私たちの普通株を発行してこの債務を終わらせた。私たちはまたハーバード大学に9000万ドルの費用を追加的に借りているかもしれない。
我々,我々の付属会社または我々の再許可者が許可製品を販売する場合,ハーバードは,(I)適用許可製品をカバーするライセンス特許の最後の有効権利主張が満了する最後の満了まで,(Ii)当該ライセンス製品の当該国/地域における排他性に関連する時期,または(Iii)当該ライセンス製品が当該国/地域で初めて商業販売された後の一定年数までライセンス製品の純売上高から低い1桁の印税を得る権利がある。特定の国/地域のライセンス製品について、私たちは、これらの使用料のいくつかの減免および補償を得る権利があり、もし私たち、私たちの関連先、または再ライセンス者が任意のライセンス特許に関連する特許挑戦を提起した場合、私たちは、いくつかの増加を得る権利がある(そのような増加を遅延および/または回避するために、適用可能な行動が取られた従属許可を終了および/または終了するために努力する能力があるように制限される)。もし私たちが“ハーバード許可協定”に基づいて製品を開発または商業化する権利を第三者に再許可し、非特許権使用料の再許可収入を受け取った場合、ハーバードはこのような対価格の一定の割合を得る権利があり、高いビット数から上位10位までの金額は様々であり、具体的にはその再許可契約を締結した日と当時の私たちの許可製品の開発段階に依存する。
ハーバードは、私たちがこのような起訴と維持活動に対して慣用的な相談、論評、審査権を持っていることを前提として、すべての許可された特許の起訴と維持を担当している。私たちはハーバードがこのような起訴や保守に関する検証可能な自己負担費用を担当しているが、ハーバードが第三者と許可協定を締結し、この協定に基づいて当分野以外の許可された特許の下での第三者の許可を付与する場合、ハーバードはこの協定に条項を加え、このような許可特許の起訴と維持費用の分担を規定するために合理的な努力をしなければならない。特定のライセンス特許の訴訟やメンテナンス費用を支払わないことを選択した場合、このような支払い義務は免除されますが、そのライセンス特許に関するライセンスも終了します。
事前に終了しない限り、“ハーバードライセンス協定”は、ライセンス特許の最後の有効権利要件の満了または最後のライセンス使用料期限が終了するまで有効である。ハーバードに書面で通知した後、私たちは便利な時にハーバード許可協定を終わらせることができる。いずれも他方の重大な違約行為により“ハーバード許可協定”を終了することができるが、通知と救済期限を守らなければならない。もし私たちが破産したり、借金をしなければ、あるいは私たちが保険を購入して維持できなかったら、ハーバードもハーバード許可協定を中止することができます。“ハーバード許可協定”の満了又は終了時には、付与された許可は終了し、付与された特許権の下のすべての権利はハーバード所有となる。
Editas Medicine,Inc.と締結されたライセンスプロトコル.
2018年5月に、吾らはEditasとライセンス契約またはEditasライセンス契約を締結し、Editasが所有または制御するいくつかの特許権利に基づいて、Editasに対しても独占的(Editasに対しても)、印税負担、再許可可能なグローバルライセンスを取得し、これらの特許権は、ヒト疾患または状態を治療するためのいくつかの基本編集技術およびCRISPR技術に関連し、開発、商業化、製造、製造、使用、販売、販売、および輸入される。私たちが受け取った許可証は特定の指定された目標に対して非排他的だ。私たちの許可分野は、Editasの他のパートナーに許可されたいくつかの使用分野でいくつかの遺伝子編集技術を使用することは含まれておらず、Editasとこれらのパートナーとの合意の終了、満了、または修正によって他のEditasパートナーに許可された領域が減少または修正されれば、私たちの許可領域が拡大する可能性があることを前提としている。また,Editasが所有または制御する1組の単独の特許権により,印税免除,再許可不可,非独占許可を取得し,我々が許可した分野で研究活動を行うことができ,Editasから独占許可を得ることを選択することができる。
Editasライセンス契約に基づいて私たちに付与されたいくつかの特許は、ブロド研究所とハーバード大学から得られたEditasであり、私たちが許可を得る権利があるいくつかの特許は、マサチューセッツ州総合病院(MGH)からEditasに許可される。したがって、Editasライセンスプロトコルによって付与されたライセンスは、遠大研究院、ハーバードとEditasとの間のライセンス特許または遠大/ハーバードHead許可の各ライセンスプロトコルに規定された条項および条件、ならびにMGHとEditasとの間で許可を得る権利がある特許またはMGH Headによって許可される各ライセンスプロトコルに規定される条項および条件に関するものである。
17
上述したように、Editasは、個々の特許家族に基づいて、いくつかの特許下の独占的許可を得ることができる独占的選択権を付与する。もし我々がそのようなオプション特許の特許族について選択権を行使すれば,Editas許可協定に従って我々に独占的に許可された他の特許の範囲と同じ範囲でその特許族の独占許可を得ることになる。これらのオプション特許の特許族に対してオプションを行使するためには,特定のオプションを行使する日に応じて8桁のオプション使用料を支払う.
Editasライセンス契約によると、私たちはアメリカ、日本、イギリス、イギリス、ドイツ、フランス、イタリア、スペインの許可分野で商業的に合理的な努力を使ってライセンス製品を開発しなければなりません。もし私たちがどの国でも許可製品の規制承認を得ることに成功すれば、その許可製品をその国/地域で商業化するビジネス上の合理的な努力もしなければならない。私たちはまた特許製品開発に関連するすべての規制活動に対して独占的な統制権と責任を持っている。
私たちは、第三者にEditasライセンス協定の下でのいくつかの権利をさらに再許可することを許可され、第三者と達成された任意のこのような再ライセンス合意は、Editasライセンス合意および遠大/ハーバードHead許可およびMGH Head許可(適用される場合)の条項に引き続き適合しなければならないことを前提としている。また、適用される従属許可受信者が従属許可協定に違反するいかなる行為にも責任を負い、そのような従属許可を操作することによって支払われるべきEditasライセンスプロトコルに従って支払われるべきすべてのお金を担当する。Editasライセンス協定に署名した後、私たちは遠大研究所とハーバード大学から得たライセンス特許を第三者に再許可する権利をさらに獲得し、私たちがEditasであるように、いくつかの他の慣例条件を守らなければならないということを前提として、私たちは遠大/ハーバードHeadライセンス協定の下でいくつかの再許可要求を守らなければならない。MGHからそのような権利はまだ取得されておらず、ライセンス特許の下の権利をさらにMGHから第三者に再許可することを可能にし、そのような権利を第三者にさらに再許可したい場合には、書面による同意が必要である。
Editasライセンス契約に署名した後、私たちはEditasに18万ドルの前払い費用を支払った。Editasに1,833,333株A-1シリーズ優先株と1,222,222株A-2シリーズ優先株も発行した。さらに、許可製品に関連する任意の商業、規制、開発、または販売活動が、Editasが遠大/ハーバードHead許可またはMGH Head許可の下でEditasのマイルストーン支払いまたは再許可収入を借りていることをトリガした場合、私たちはEditasにマイルストーン支払いまたは再許可収入の全額を支払わなければならない。前提は、Editasに任意のオプション行使費用を支払うことによって生じる再許可収入をEditasに支払わないことを前提とする。Editasライセンスプロトコルによれば、いくつかの基礎編集技術およびCRISPR技術に関連する権利を使用して開発および商業化された各製品のマイルストーンの総金額は6880万ドルに達する可能性があり、Editasライセンスプロトコルに従って許可または選択された追加特許ファミリーの権利要件によってカバーされる製品を開発および商業化する場合、マイルストーン支払い総額は各製品7400万ドルに達する可能性がある。Editas許可協定によると、私たちは不足している再許可収入の割合をゼロから10%から20%の間にする。また、Editasが遠大/ハーバードHead許可およびMGH Head許可および特許許可に基づいて発生または不足する年間許可維持費、および起訴および保守費用の一部を支払うことに同意する。Editasライセンス契約によると、Editasに支払われる前払い費用、持分発行、オプション行使支払いには、Editasライセンス契約に基づいて私たちに付与されたライセンスの対価格と、ライセンス特許の起訴とメンテナンス費用の精算も含まれています。
私たち、私たちの付属会社、または私たちの再許可者が許可製品を販売するためには、適用されるIn-許可の下で遠大研究所、ハーバード大学またはMGHの印税を借り、ライセンス製品の純売上高の追加的な1桁から1桁の特許使用料に相当する金額をEditasに支払う必要があり、このライセンス製品がEditasが所有する特許によってカバーされ、所与のカレンダー年間ライセンス製品のグローバル純売上高合計に基づいているかどうかに依存する。特定の国/地域のライセンス製品について、私たちは、これらの印税のいくつかの減免および補償を得る権利があり、Editasが関連する遠大/ハーバードヘッドライセンスまたはMGH Headライセンスからその印税支払い義務に関連する任意の減免または補償を受ける権利がある場合、Editasは、このような減免を利用するために合理的に努力し、これは、Editasライセンス契約下での印税支払い義務を減少させるであろう。ライセンス使用料の期間は、以下の時間において、ライセンス製品および国/地域で終了する:(I)その国/地域では、任意の適用可能な遠大/ハーバードHead許可またはMGH Headライセンスに従って最終的に満了されるライセンス使用料期限、および(Ii)当該製品がもはや当該国/地域のライセンスEditasのすべての特許の有効な権利要件によってカバーされない日。
双方の間で,Editasはすべての許可された特許を起訴·維持する責任があり,特定の許可された特許に対して一定の情報,評論,審査権を持つことを前提としている.
18
Editasライセンス契約は、事前に終了しない限り、各国/地域のライセンス製品および国/地域のライセンス製品の適用印税期間が満了した後に満了します。Editasに書面で通知した後にEditasライセンス契約を終了することができますが、指定された通知期間を守らなければなりません。どちらも他方の重大な違約行為によりEditas許可プロトコルを終了することができるが,通知と救済期限を守る必要がある.もし私たちがいかなる許可特許の有効性に疑問を提起すれば、EditasはEditas許可協定を終了することもできるが、慣例的な彫刻によって制限されている。Editasライセンスプロトコルがすべて満了または終了した場合、私たちに付与されたライセンスは、直ちに終了するか、または満了または終了する特許シリーズは終了する(場合によって決定される)、しかし、Editasライセンスプロトコルに実質的に違反するため、Editasライセンスプロトコルを終了する権利がある場合、Editasライセンスプロトコルでの印税支払い義務を特定の割合で減少させることを選択することができる。
ブロド研究所,Inc.と締結したライセンス契約.
2018年5月、当社の付属会社Blink Treateutics Inc.またはBlinkは、遠大研究所と改訂されたライセンス契約または遠大ライセンス契約を締結しました。2021年9月にBlinkはビムと合併し、Blinkの独立会社の存在を停止させたが、ビムは存続している会社およびBlinkに関する遠大な許可プロトコルの合併後継者とし続けた。広範な許可プロトコルに基づいて、以下でさらに詳細に説明すると、RNA編集プラットフォームの救助を含むRNA塩基編集技術のいくつかの権利を獲得した修復していますCas 13を用いてデアミナーゼに連結し,それぞれRNA転写産物の単塩基A−to−IまたはC−U編集,およびCas 12 bヌクレアーゼファミリーの遺伝子編集酵素を提供する。
より具体的には、遠大ライセンスプロトコルによれば、遠大研究所は、遠大研究所が所有または制御する範囲内で(マサチューセッツ工科大学またはマサチューセッツ工科大学およびハーバード大学との機関間プロトコルを含む)特定の特許権に基づいて、特定の特許権に基づいて、(I)核酸編集を標的とする新しいCRISPPR酵素およびシステム(DNA切断に関連するシステムを含む)またはシステム、方法および組成物のいくつかの特許権を要求または開示することを含む遠大研究所の所有または制御の範囲内であり、それぞれの場合、これらの特許がカバーする製品を開発するための(Ii)核酸編集、標的核酸編集、核酸標的編集のための方法および組成物の特定の製品の特許権を要求または開示することを含む。いずれの場合も、当該等の特許がカバーする塩基編集製品及び(Iii)一般に遺伝子標的に関連するいくつかの特許権を利用して、当該等の特許がカバーする塩基編集製品を利用して、複数の許可された製品を製造、製造、カプセル販売、販売、販売及び輸入する。
遠大ライセンス協定によれば、我々はまた、(I)遠大ライセンス協定に基づいて、2021年5月までに開始された研究または発見計画において、これらの特許を使用したり、遠大研究所によって譲渡された材料を使用して、実用価値を有するものを製造、発見、開発または決定するために、2021年5月までに開始された研究または発見計画において使用された特許または遠大研究所によって譲渡された材料を使用することによって、実用価値を有するものを製造、発見、開発、または決定することができるが、これらの特許は、ライセンス特許の範囲内ではなく、(Ii)私たちが独占的に許可されているすべての特許項目の下での非独占的、世界的範囲内、特許使用権および再許可可能な内部ライセンスを提供する。ブロド研究所が付与したすべてのライセンスは、ヒト生殖系の修正、特定の遺伝子または植物または動物個体群内の特徴を刺激する偏見遺伝的刺激、およびタバコ植物のいくつかの修正を含み、ブロイド研究所、ハーバードおよびマサチューセッツ工科大学、ならびに米国連邦政府のいくつかの保持権利によって制限されている。ボイド研究所はまた、自分、ハーバード大学とマサチューセッツ工科大学及びその他の非営利研究組織が研究、教育及び学術目的で特許を許可する限られた権利を実践している。
遠大許可協定によると、著者らは遠大な準備と遠大研究院に提出した発展計画に基づいて、商業上の合理的な努力を用いて許可製品を開発しなければならない。開発計画には,我々が達成しなければならないいくつかの発展マイルストーンと,これらのマイルストーンを達成するためのスケジュールが含まれており,誠実に判断すれば,これらの発展マイルストーンを実現する能力を向上させるために開発計画を更新する必要があると考え,開発計画を随時更新することができる.遠大研究院に合理的な解釈と計画を提供しない場合、著者らはこのような開発マイルストーンスケジュールを延期することができず、さらに、いかなるマイルストーンスケジュールの延長が指定年数を超えても、遠大研究院はその合理的な適宜決定権でこの解釈と計画を承認する必要があると規定した。もし私たちがどの国でも規制部門の承認を得ることに成功し、許可製品をその国の商業市場に導入することができれば、私たちはまた、その許可製品を商業化し、その許可製品を合理的に大衆に提供するための商業的合理的な努力をしなければならない。
また、私たちは、ある許可特許サブカテゴリに含まれている、要求または開示された技術の実行可能性を商業的に合理的に追求しなければならず、有効なクレームがカバーされた許可製品を開発するための発見計画を開始しなければならない、または“遠大許可協定”に署名した後の特定の期間内に、その許可特許サブカテゴリを使用することによって一般的に実現され、その期間内にその許可特許サブカテゴリのために遠大研究院が合理的に受け入れた最新の開発計画と開発マイルストーンを提出しなければならない。このような技術の実行可能性を追求するために特定の期間にわたって商業的に合理的な努力を使用することができなかった場合、または発見計画を開始したり、更新された開発計画を提出したりすることができなかった場合、許可特許サブカテゴリ下の許可は終了し、許可特許サブカテゴリが塩基編集特許権を含む場合、遺伝子標的ライセンス特許に関する権利は、そのような権利が終了した塩基編集許可特許のために許可されることができるように非排他性に変換されなければならない。
19
ブロイド研究所、マサチューセッツ工科大学、およびハーバード大学はまた、特定の場合に製品の研究、開発および商業化を希望する第三者(指定されたエンティティを除く)にさらなる許可を付与する権利を保持しており、そうでなければ、ブロダー研究所、ハーバード、およびマサチューセッツ工科大学の包括的革新モデルによれば、この製品は、ブロイド研究所およびハーバード大学によって付与された独占的な許可範囲に属する。特定の時間の後、このような第三方向遠大研究所はこのような許可を問い合わせ、提案製品の提案開発と商業化を記述する情報を含む提案書を遠大研究所に提出した場合、遠大研究院は提案製品が対象とする適用遺伝子を含む提案製品の性質を含むことを私たちに通知することができる。博大研究院は申請者が私たちに提供してくれた包括的な革新モデルに関する他の情報を共有する必要はない。もし私たちがこの提案製品の研究、開発、あるいは商業化を行っていなければ、私たちは遠大研究院に通知することができて、私たちは誠実にこの提案製品を開発することに興味があるかどうか、要求した第三者と再許可協定を締結してこの提案製品を開発することができますか、他の第三者と締結してこの提案製品を開発することができるかどうか、あるいは上述したいかなる事項にも興味がありません。もし我々が遠大研究院にこの提案製品の開発に興味があることを通知すれば、その範囲は遠大許可プロトコルでの開発計画と類似しており、この提案製品を開発し、指定された期限内にその提案製品の開発計画を起動しなければならない開発計画を用意する。もし私たちが遠大研究院に通知して、私たちは再許可協定を達成することに興味があり、この合意に基づいて、問い合わせの第三者または他の第三者が私たちから許可特許下の再許可を得て、この提案された製品を開発することに興味があれば、この再許可協定を締結し、その間に合理的な証拠を提供することができる。もし我々が前述の活動を行うことを拒否したり、指定された期限内にこのような活動を完了しなかった場合、遠大研究院は許可特許項の下の適用第三者に許可を付与し、その提案した製品を研究、開発して商業化することができ、遠大研究院の選択の下で、このような適用特許権に対する許可は、提案された第三者製品のテーマとなる遺伝子に対して終了する。
我々は,許可された特許を関連会社や第三者に再許可することが許可されており,いずれもこのような再許可協定は遠大な許可協定の条項を遵守して適合しなければならないことを前提としている.さらに、このような再許可協定は、私たちが遠大な許可協定を遵守することができることを確実にするために、いくつかの習慣条項を含まなければならない。また、適用される従属許可者が従属許可協定に違反するいかなる行為にも責任を負い、そのような従属許可を操作することによって、遠大な許可協定の下でのすべての対応金を支払う責任がある。
遠大許可協定によって付与された権利の部分対価格として、遠大研究院は1,940,000株のBlinkの普通株を獲得した。2018年9月25日、私たちのBlinkの買収について、遠大研究院に発行された株は865,240株の私たちの普通株に両替された。
遠大許可協定によると、低から中五桁から低から六桁までの年間許可維持費も遠大研究所に支払う必要があり、具体的には特定のカレンダー年度に依存する。ブロド研究所はまた、合計中高八桁の範囲内の臨床と規制マイルストーンを獲得し、私たちの普通株の公平な市場価値の増加によって支払いに成功する権利がある。
2021年5月,第1回成功支払測定を行い,遠大研究所を借りた金額は1,500万ドルと算出した。私たちは普通株で支払うことを選択し、2021年6月10日に174,825株の私たちの普通株を発行してこの債務を終わらせた。私たちはまた遠大な学院に9,000万ドルの支払いに成功しているかもしれない。
我々はまた,ライセンス特許がカバーする製品に低い1桁の印税を支払うことを要求されており,ライセンス特許または譲渡材料によって実現された製品については,印税は一定の割合を低下させるが,ライセンス特許のカバー範囲には含まれていない.私たちが支払うべき特許使用料は、特定の国/地域の製品に関連するこれらの特許権使用料の常習減免および補償に依存する。国/地域における製品の印税期間は、(I)適用製品をカバーする最後のライセンス特許が満了したときに終了し、(Ii)その国/地域に関連する製品の排他的期間、または(Iii)当該製品が国/地域で初めて商業販売された後の期間内に終了する。もし著者らが遠大許可プロトコルに基づいて製品を開発或いは商業化許可製品の権利を第三者に再許可し、非特許権使用料の再許可収入を獲得すれば、遠大研究院はこのような対価格の一定の割合を獲得する権利があり、比較的に高い1桁から低い上位10桁まで様々であり、具体的には遠大許可協定下で製品の再許可実行時の開発段階に依存する。
遠大研究院はすべての許可特許の起訴と保守を担当し、このような独占許可特許権の起訴と維持活動に対して一定のコンサルティング、評論、審査権を持つことを前提としている。
事前に終了しない限り、遠大ライセンス契約は、我々のライセンス製品をカバーするライセンス特許の最後の満了有効主張または最後の満期ライセンス使用料期限が終了するまで有効である。便宜のため、著者らは遠大学院に書面で通知した後、遠大許可協定を終了することができるが、指定された通知期間を守らなければならない。いずれも他方の重大な違約行為により“遠大許可協定”を終了することができるが、通知と救済期限が必要である。私たちが破産したり、債務を返済しない場合、もし私たちが保険を購入して維持できなかった場合、あるいは私たち、私たちの関連会社または再許可者が任意の許可された特許に特許挑戦(適用行動を取った従属許可者の治療期間を終了することによって制限されている)であれば、遠大研究所も遠大な許可協定を終了することができる。
20
Bio Palette Co.,Ltd.とライセンス契約を締結する.
2019年3月27日、Bio Palette Co.,Ltd.またはBio Paletteとライセンス契約、またはBio Paletteライセンス契約を締結し、Bio Paletteが所有または制御する基礎編集に関連するいくつかの特許権に基づいて、独占的(Bio Paletteおよびその付属会社に対しても)、再許可可能な許可を取得し、その研究、製造、製造、輸入、輸出、流通、使用、使用、販売、販売またはカプセル販売、他の方法で世界各地でヒト疾患治療のための製品を開発したが、アジア微生物グループ分野の製品は含まれていない。また,我々が所有または制御している塩基編集や遺伝子編集に関するいくつかの特許権付与Bio Palette独自(我々およびその付属会社に対しても)の許可に基づき,研究,製造,製造,輸入,輸出,流通,使用,使用,販売,販売または要約販売,その他の方法でアジア微生物グループ分野の製品を開発し,吾らの一任を受けてBio Paletteの許可(および適用される印税義務)を地域全体に拡大する権利を適宜決定した.“バイオパレットプロトコル”のいずれも、全世界の微生物グループ分野で製品を開発·製造する非排他的権利を保持しており、その唯一の目的は、その領土でこれらの製品を開発することである。いずれか一方が微生物群領域での権利を利用しないと判断した場合,いずれも微生物群領域の何らかの協調義務を負うことに同意する。
Bio Paletteが任意のヒト疾患または疾患の治療、診断または予防に有用な任意の他の特許権の制御下に入り、特定の限定された領域および特定の限定された地域で特許権付与許可に基づいて、これらの分野および地域において特許権に基づいて独占的な許可を得るための独占的交渉権を有することが意図されている。特定の限定分野において有用な他の任意の特許権を制御し,ある限定分野においてその特許権に基づいて許可を付与しようとする場合,Bio Paletteはこれらの分野や地域でその特許権に基づいて独占的な許可を得る独占交渉権を持つ.
Bio Paletteライセンスプロトコルの一部として,科学諮問委員会を設立すれば,Bio PaletteはBio Paletteライセンス契約発効日から5年後に終了するまで,2人の代表をこの委員会に任命する権利がある.また,Bio Paletteとは潜在的な基地編集協力について日本でのコミュニケーションを行うことに同意した.
私たちは商業的に合理的な努力でアメリカ、日本、イギリス、フランス、ドイツ、イタリア、スペインでライセンス製品を開発することを要求された。私たちの許可分野や地域で規制の承認を受けたいかなる許可製品についても、関連国/地域で商業化するために商業的に合理的な努力を使用しなければならない。Bio Paletteは商業的に合理的な努力を用いて日本製製品を開発する必要がある。規制許可を得た任意の許可製品については、Bio Paletteは、このような許可製品を関連国/地域で商業化するために商業的に合理的な努力を使用しなければならない。
Bio Paletteライセンスプロトコルによって我々に付与されたいくつかの特許は,我々がKobe Headライセンスと呼ばれるライセンスプロトコルに基づいて神戸大学からBio Paletteにライセンスされている.したがって,Bio Paletteライセンス協定により我々に付与された許可は,いくつかの権利を規定する政府当局を含む第三者が保持する条項を含む神戸ヘッドライセンスに規定されている条項や条件によって拘束される.
Bio Palette許可プロトコルとKobe Head許可の適用条項がこれらの付属会社と第三者に適用される限り,我々もBio Paletteも許可された特許を関連会社と第三者に再許可することが許可される.また、再許可者は、適用される再許可者がそのような条項に違反する任意の行為に責任を負い、そのような再許可を操作するために“バイオパレット許可協定”の項目の下で支払われるべきすべてのお金を責任を負う。
Bio Paletteライセンス契約に署名した後、私たちはBio Paletteに50万ドルの費用を前払いした。 Bio Palette許可協定に署名した時、私たちはBio Paletteに私たちの普通株16,725株を発行し、引用されたBio Palette特許がアメリカで発行されれば、私たちはより低い6桁で私たちの普通株式を追加発行するという合意に達した。2020年6月に米国であるBio Palette特許が発行された後,Bio Paletteに200万ドルの記念碑的支払いを支払い,2020年7月にBio Paletteに175,000株の普通株を発行し,30万ドルの価値がある。私たちはまた、Bio Paletteが私たちに許可してくれた特許がカバーする製品の純売上高に零点数ポイントの印税を支払うことに同意し、Bio Paletteは私たちがBio Paletteに許可した特許がカバーする製品の純売上高に零点数ポイントの印税を支払うことに同意した。1つの国/地域における製品のライセンス使用料期間は、(I)当該国/地域における特許に基づくライセンス製品の排他性または(Ii)当該ライセンス製品の規制排他性が満了したときに終了する。
“バイオパレットライセンス契約”に基づいて行われる活動によって生成される任意の知的財産権は、このような知的財産権を発明する側に所有される。Bio Paletteは、Bio Paletteによって許可されたすべての特許の起訴と維持を担当しており、特定のPCT出願の国家エントリについてのみ、このような起訴および保守活動に対して慣用的な相談、コメント、および審査権を有することを前提としている。私たちは私たちがBio Paletteに権限を与えた特許を起訴して維持する権利がある。
21
Bio Paletteライセンス契約は、事前に終了しない限り、このようなライセンス製品および国/地域の適用印税の期限が満了したときに、ライセンス製品および国/地域によって期限切れになります。各当事者は、所定の通知期間内に当事者に付与された許可を終了するために、生物パレット許可プロトコルを終了する権利がある。どちらも他方の許可の付与に関するBio Palette許可プロトコルを終了することができ,その許可は他方の重大な違約行為によって終了するが,指定された通知と救済期限を守る必要がある.さらに、他方が破産または資金が相殺されない場合、または他方、その関連会社または分被許可者が任意の許可された特許に特許挑戦を行った場合、いずれか一方も“バイオパレット許可協定”を終了することができる(ただし、分割被許可者がそのような特許挑戦を提起した場合、その当事者は、適用行動をとった被許可者との合意の治療期間を終了する)。
政府の監督管理
アメリカ連邦、州と地方の各レベル及びEU或いはEUを含む他の国と司法管轄区の政府当局は、その他のほか、生物製品を含む薬品の研究、開発、テスト、製造、包装、ラベル、貯蔵、記録保存、精算、広告、販売促進、流通、審査後の監視と報告及び輸出入、定価と精算などの方面に対して広範な監督管理を行っている。製品開発過程や承認後のいつでも,適用される法規の要求を守らなければ,上場承認スポンサーは開発や承認に遅延が生じ,行政や司法制裁を受ける可能性がある.
米国や他の国や管轄地域でマーケティング承認を得、承認前と承認後に適用される法規や法規の要求を遵守し、精算地位を得る過程には、引き続き多くの時間と財力が必要となる。生物製品の開発、承認とマーケティングに適用する監督管理要求は変化する可能性があり、法規と行政指導はよく機関によって私たちの業務に重大な影響を与える可能性がある方法で修正または再解釈される。遺伝子治療、遺伝子テスト、および遺伝子研究の倫理、社会および法律の懸念は、追加の規制または使用可能なプロセスを禁止する可能性がある。
アメリカの生物製品の許可と規制
アメリカでは、私たちの候補製品は生物製品あるいは生物製品として“公衆衛生サービス法”(PHSA)、“連邦食品、薬物と化粧品法”(FDCA)、FDA施行条例、その他の連邦、州と地方法規の規制を受けている。
FDAはアメリカで発売されるために、治療適応の候補製品を承認しなければならない。このような製品の臨床開発計画の発起·管理を担当する会社,機関,あるいは組織をスポンサーと呼ぶ。米国での新生物の販売と流通の承認を求めるスポンサーは、通常、以下の各ステップを満足させなければならない
22
臨床前研究と研究性新薬の応用
人体で任意の研究用生物製品をテストし、候補遺伝子編集製品を含む前に、候補製品は臨床前テストを経なければならない。臨床前試験は製品の化学、調合と安定性の実験室評価、及び動物実験において治療効果と毒性潜在力を評価する研究を含む。これらの研究は一般にINDを支援する研究と呼ばれる.臨床前試験の進行および試験に用いる化合物の配合は、GLP規制および基準、米国農務省の動物福祉法(適用される場合)を含む連邦法規および要求に適合しなければならない。臨床前試験の結果および生産情報と分析データはIND申請の一部としてFDAに提出された。
INDはFDCAの免除であり、未承認の薬物或いは生物製品の州間商業での輸送を許可し、臨床研究に使用する。このような許可は、承認されていない任意の新薬出願またはNDAの候補製品を州間輸送および管理する前に取得されなければならない。IND申請を支援するためには,申請者は各臨床試験に1つの案を提出しなければならず,どの後続の案修正もINDの一部としてFDAに提出されなければならない。INDは、ヒト研究対象が不合理な健康リスクに直面することを懸念すること、または提案された製品の化学、製造および制御またはCMCに関連する任意の問題を含む、FDAの受信後30日後に自動的に有効であり、それ以前にFDAが提案された臨床試験の製品または懸念または問題を提起しなければならない。この場合,INDスポンサーやFDAは臨床試験開始前にFDAの未解決の問題を解決しなければならない。IND提出後も,臨床前または非臨床試験は通常継続している。
IND下での臨床試験開始後,FDAもこの試験を臨床放置あるいは一部の臨床放置を実施することができる。臨床保留はFDAがスポンサーに発表した命令であり,提案された臨床研究の延期や進行中の研究の一時停止が要求されている。一部の臨床保留はIND要求の一部の臨床仕事を遅延或いは一時停止することである。例えば、一部の臨床的保留は、特定のプロトコルまたはプロトコルの一部が継続できないことを宣言する可能性があり、プロトコルの他の部分または他のプロトコルはそうすることができる。臨床保留或いは一部の臨床保留を実施した後30日を超えない後、FDAはスポンサーに棚上げ根拠に関する書面解釈を提供する。臨床保留あるいは一部の臨床保留を発表した後、FDAがスポンサー調査が継続可能であることを通知した後にのみ、臨床調査を回復することができる。FDAは、スポンサーによって提供された情報に基づいて、調査が継続または再開可能であることを決定し、これらの情報は、上述した欠陥を修正するか、または他の方法でFDAを満足させるであろう。
治療のための薬を研究する機会を広げる
使用を拡大することは、“同情的使用”と呼ばれることがあり、臨床試験以外に研究製品を使用し、比較可能または満足できる代替治療案がない場合には、重篤または直ちに生命を脅かす疾患または条件を有する患者を治療する。FDAの法規は、個別患者(緊急時および非緊急時に治療された単一患者IND申請)、中規模の患者集団、および治療レジメンまたは治療INDに従って研究製品の使用を申請したより大きな集団のために、会社または治療医がINDの研究製品を治療目的で使用することを可能にする。
製造業者に研究製品を得るためのもっと多くの機会を提供することは要求されていない。しかしながら、製造業者がその研究製品を参入拡大に使用することを決定した場合、FDAは、参入拡大の要求を審査し、治療が可能かどうかを決定する。以下のすべての基準が適用される場合、アクセスを拡大することは適切である可能性がある:患者(S)は、深刻または直ちに生命を脅かす疾患または状態を有しており、疾患または状態を診断、監視または治療するための比較可能または満足できる代替療法がない;潜在的な患者利益は、治療の潜在的リスクが合理的であることを証明し、潜在的リスクは、治療される場合には不合理ではなく、要求された治療のために研究薬物を拡大することは、製品の発売承認を支持するか、または他の方法で製品の潜在的開発に影響を与える可能性のある臨床研究の開始、進行、または完了を妨げることはない。
FDCAによれば、重篤な疾患(S)または病状(S)を治療するための1つまたは複数の研究製品のスポンサーは、個々の患者参入を拡大する要求を評価および応答するために、それらの政策を公開しなければならない。スポンサーは、2期または3期の研究開始が早いとき、または研究薬または生物が突破的療法、迅速チャネル製品または再生医学高度療法として指定された後15日以内にそのような政策を開示しなければならない。
23
また、2018年5月30日には、“裁判権法案”が法律に署名された。他の事項に加えて、この法律は、生命を脅かす疾患を有する患者に追加の機序を提供しており、これらの患者は、承認された治療法を使い切っており、いくつかの研究製品を得るために臨床試験に参加することができず、これらの製品はI期臨床試験を完了しており、活発なINDの対象であり、調査を受けており、FDAの承認を待っている。上述した拡張されたアクセスフレームワークとは異なり、Pathを試用する権利は、FDAが研究製品の使用を審査または承認する要求を要求しない。“試用権法案”によると,メーカーは条件を満たす患者にその研究製品を提供する義務はない。
血中乳酸を支持するヒト臨床試験
臨床試験は、GCP要求に応じて、合格した主要な研究者(通常は試験スポンサーに雇用されていないか、または試験スポンサーによって制御されている医師)の監督の下で、研究製品を健康ボランティアまたは治療を受ける疾患患者に候補することを含み、すべての研究対象に参加に対するインフォームドコンセントを提供することを含む。臨床試験は研究案に基づいて行われ,その中で研究の目標,組み入れと排除基準,安全性モニタリングのためのパラメータおよび評価すべき有効性基準が詳細に説明されている。INDの一部として,各臨床試験の案と任意の後続の案修正案をFDAに提出しなければならない。
米国で行われている臨床試験にはINDが必要であり,いずれの臨床試験も臨床試験を行う機関のIRBが集中的あるいは単独で審査·承認されなければならない。委員会が考慮する事項は,臨床試験設計,患者インフォームドコンセント,倫理的要因,被験者の安全,および機関が担う可能性のある責任である。IRBの運営はFDAの規定に適合しなければならない。臨床試験はまた広範なGCP規則と被験者のインフォームドコンセントを得る要求に符合しなければならない。FDA、IRBまたは臨床試験スポンサーは、臨床試験がFDAの要求(GCPを含む)に従って行われていないこと、または対象または患者が受け入れられない健康リスクに直面していることを発見することを含む、様々な理由で臨床試験を随時一時停止または中止することができる。
さらに、いくつかの臨床試験は、データ安全監視委員会または委員会と呼ばれる臨床試験スポンサーによって組織された独立した合格専門家グループによって監督される。このグループは,計画的に研究を継続し,研究進行を変更したり,研究のあるデータへのアクセスに応じて,指定されたチェックポイントで研究を停止することを提案することができる.最後に,組換えや核酸分子の合成に関するNIHの研究ガイドラインによると,感染剤,危険化学品,組換えDNAおよび遺伝子組換え生物や製剤に関する研究活動は,機関生物安全委員会(IBC)の審査と承認を受ける必要がある可能性がある。
臨床試験は通常3つの連続段階に分けて行われるが、これらの段階は重複或いは合併する可能性がある。承認された後に追加的な研究が必要かもしれない。
24
場合によっては、FDAは候補製品のBLAを承認する可能性があるが、承認後の候補製品の安全性または有効性をさらに評価するために、スポンサーに追加の臨床試験を行うことが要求される。このような試験は通常承認後あるいは発売後の臨床試験と呼ばれる。これらの研究は、予期される治療適応患者の治療から追加の経験を得るために使用され、加速承認条例によって承認された生物学的製品の場合に臨床的利益を証明するために使用される。承認後や発売後の臨床試験で職務調査を行うことができなかったことは,製品の承認を撤回する可能性がある。FDAは通常、スポンサーが長期追跡研究中に潜在遺伝子治療に関連する遅延不良事件の被験者を観察することを提案し、統合ベクターは15年に達し、潜伏期を確立できるヘルペスウイルスベクターは15年に達し、既知の持続感染を確定する微生物ベクターは15年に達し、遺伝子編集製品は15年に達し、AAVベクターは5年に達する。FDAは,これらの長期追跡研究に少なくとも5年間の年次健康診断を含め,残りの観察期間を年次調査し,対面でも電話でも書面でも行うことを提案している。
2022年12月、食品·薬物総合改革法案(FDORA)の成立に伴い、国会は、各新薬または生物製品の第3段階臨床試験または任意の他の“重要な研究”のための多様な行動計画の制定と提出をスポンサーに要求した。これらの計画はより多くの異なる患者群がFDA監督製品の後期臨床試験に参加することを奨励することを目的としている。具体的には,行動計画には,スポンサーの登録目標,これらの目標の基本原理,スポンサーがこれらの目標をどのように実現しようとしているかの解釈が含まれなければならない。このような要求に加えて、立法はFDAに多様な行動計画に関する新しいガイドラインを発表するように指示した。FDAは2024年1月に指導意見草案を発表し、臨床試験において人種と民族データを収集する政策を示した。
2023年6月、FDAはガイドライン草案を発表し、GCPに対する提案を更新し、臨床試験の設計と現代化を目指した。これらの更新は,より効率的な臨床試験の道を開き,医療製品の開発を促進することを目的としている。このガイドライン草案は国際調整理事会(ICH)が最近更新したE 6(R 3)ガイドライン草案から採択されたものであり、このガイドライン草案の開発は迅速に発展した技術と方法の革新を臨床試験企業に組み込むためである。また,FDAはガイドライン草案を発表し,分散臨床試験の実施について概説した。
臨床試験結果を詳細に説明する進捗報告は少なくとも毎年FDAに提出しなければならず,深刻な有害事象が発生すればより頻繁に提出される。さらに、以下のうちの1つのINDセキュリティ報告書は、深刻かつ意外な疑わしい副作用、他の研究または動物からの報告書に提出されなければならない体外培養この製品に接触する人体は重大なリスクのテストが存在することを表明した;及び方案或いは研究者マニュアルに列挙された情況と比べ、臨床上深刻な不良反応の発生が疑われるいかなる重要な増加である。第1段階、第2段階、および第3段階の臨床試験は、任意の指定された時間内に成功しないか、または全く成功しない可能性がある。FDAは、通常、GCPおよび提出された臨床データの完全性を保証するために、1つまたは複数の臨床場所を検査する。
処方薬を含むいくつかのFDA規制製品の臨床試験のスポンサーは、NIHによって維持されている共通レジストリに特定の臨床試験情報を登録して開示しなければならない。特に,臨床試験登録の一部として,臨床試験の製品,患者群,調査段階,研究場所,調査者,その他に関する情報が公開されている。スポンサーも試験完了後に臨床試験結果を開示する義務があるが,場合によっては結果の開示が試験完了日後2年に延期される可能性がある。NIHのClinicalTrials.gov登録と報告要件に関する最終規則が2017年に施行されました。FDAは過去2年間、自発的な是正行動の事前通知と規定を遵守しないいくつかの通知を発表した。これらの違反通知は民事罰金を招くことはないが,FDCAの規定により,Clinicaltrials.govに臨床試験情報を要求通りに提出できなかったことは禁止されており,違反行為は毎日10,000ドルまでの民事罰金を受け続ける可能性がある。
臨床開発計画中のFDAとの相互作用
INDが承認され臨床試験を開始した後,スポンサーは臨床開発計画の特定の時点でFDAと会う機会があった。スポンサーとFDAの間には5種類の会議がある。Aクラス会議は,もともと停滞していた製品開発計画を継続したり,重要なセキュリティ問題を解決したりするために必要な会議である.クラスB会議には,IND前会議とNDA前会議,第2段階終了会議のような段階終了会議がある.クラスC会議とは,AクラスまたはBクラス会議以外のいずれかの製品開発やレビューに関する会議である.D類会議は一連の狭い問題に集中し、FDAの3つ以上の学科或いは部門に意見を提供することを要求すべきではない。最後に,インタラクティブ会議は,研究製品の早期開発において独自の挑戦を提示する新製品と開発計画を目指している.FDAは、議事録および諮問書簡で伝達された応答は、スポンサーに対する提案および/または提案のみを構成するため、スポンサーは、そのような提案および/または提案の制約を受けないと述べている。しかし,実践的には,スポンサーがFDAの提案に沿って臨床計画を設計していないことは,その計画を大きな失敗リスクに直面させる可能性がある。
25
FDAが承認したアメリカの海外臨床研究をサポートします
われわれの臨床開発計画では,米国以外の地点で試験を行う予定である。IND下で海外臨床試験を行う場合,放棄しない限り,すべてのIND要求を満たさなければならない。外国の臨床試験がINDの下で行われない場合、スポンサーは試験をIND或いは上場承認申請の支持として使用するために、試験がFDAのある規制要求に適合することを保証しなければならない。具体的には,独立倫理委員会やIECの審査·承認を受けること,被験者のインフォームドコンセントを求めて受けることなど,GCPに従って行わなければならない。GCPは臨床試験の倫理とデータ完全性基準を含むことが要求される。FDAの規定は非IND外国臨床試験に参加した人体被験者の保護、及び結果データの品質と完全性の確保を助けることを目的としている。これらは,非IND外国試験の進行方式がIND試験に必要な方式に相当することを確保するのにも役立つ.
FDAは米国国外で行われた臨床試験の試験データを受けて米国の承認が何らかの条件で制限される可能性があり,まったく受け入れられない可能性もある。外国の臨床試験からのデータが米国での上場承認の唯一の根拠となることを意図している場合、FDAは通常、(I)データがアメリカの人口とアメリカの医療実践に適用されない限り、外国のデータのみに基づいて申請を承認しない。(Ii)試験は公認能力を有する臨床研究者によって行われ、GCP規定に適合する。および(Iii)データは、FDAによる現場検査を必要とすることなく、有効であると考えることができ、またはFDAがこのような検査を行う必要があると考えた場合、FDAは、現場検査または他の適切な手段によってデータを検証することができる。
また,外国試験データが承認の唯一の根拠としようとしなくても,FDAはこれらのデータを上場承認申請としての支援を受けず,試験設計が良好でない限り,GCP要求に応じて良好な操作を行うことができ,FDAは必要と考えた場合に現場検査により試験データを検証することができる。多くの外国の規制機関もまた似たような承認要求を持っている。また、このような外国裁判は、裁判を行う外国司法管轄区に適用される現地法によって管轄されている。
小児科研究
2003年の“小児科研究公平法”またはPREAによれば、BLAまたはそのサプリメントは、すべての関連する小児科亜集団において主張される適応の安全性および有効性を評価するのに十分なデータを含み、安全で有効な各小児科亜群に対する製品の用量および投与をサポートしなければならない。スポンサーは、第2段階会議終了後60日以内に、またはスポンサーがFDAと合意した場合に、小児科研究計画またはPSPを提出しなければならない。PSPは提案された小児科研究または彼らの計画による研究を概説し、研究目標と設計、任意の延期または免除要求、および法規要求の他の情報を含む。FDAはその後、提出された情報を審査し、スポンサーに相談し、最終計画について合意しなければならない。FDAやスポンサーはいつでも計画の修正を要求することができる。2023年5月、FDAは新しいガイドライン草案を発表し、さらにPREA下の小児科研究要求を述べた。
深刻な或いは生命に危害を及ぼす疾病或いは状況を治療することを目的とした研究製品に対して、FDAはスポンサーの要求に応じて会議を開催し、初期小児科研究計画の準備或いは討論を討論し、小児科評価を延期或いは放棄しなければならない。また,FDAは開発過程の早期に会議を開催し,小児科研究計画をスポンサーと検討し,FDAは重篤あるいは生命に危険な疾患よりも遅くない第1段階会議が終了する前と,FDAが研究計画を受け取ってから90日後にスポンサーと面会しなければならない。FDAは、成人のために製品が承認されるまで、または特定の場合には小児科データ要件を完全にまたは部分的に免除するまで、スポンサーの要求に応じて、承認を先送りし、小児科データの一部または全部の提出を延期することができる。法規が別途要求されない限り、小児科データ要求は孤児の称号を有する製品には適用されない。
FDAは、成人のために製品が使用されるか、または小児科データ要件を完全にまたは部分的に免除するまで、スポンサーの要求に能動的にまたは対応することができ、承認は、小児科データの一部または全部の提出を延期することができる。延期は、小児科試験が完了する前に、製品または候補治療薬が成人で使用を許可する準備ができていることを発見するか、または小児科試験が開始される前に追加の安全性または有効性データを収集する必要があることを含むいくつかの理由があるかもしれない。現在、法律は、FDAが、PREA要求を提出できなかった小児科評価、延期または延期を求めることができなかったか、または必要な小児科処方の承認を要求できなかったスポンサーにPREA不適合書簡を送信することを要求する。それはさらにFDAにPREAコンプライアンスとスポンサーの反応を公開することを要求する。規制が別途要求されない限り、小児科データ要件は、孤児として指定された製品には適用されないが、FDAは最近、PREAにおいてこの法定免除を乱用すると考えられる行為を制限する措置を講じているにもかかわらず、まれな小児科亜群に追加的な孤児薬物指定を付与することを意図していないことを宣言し、一般的な疾患である。FDAはまた、児童人口中の疾病罹患率が比較的に低いため、PREA要求を免除する疾病リストを保留した。
26
遺伝子治療製品に関する特別規定とガイドライン
遺伝子治療製品および細胞治療製品に応用されるプログラムおよび標準は、著者らが開発可能な任意のCRISPR/Cas 9候補製品に適用可能であるが、現在のところ確定されていない。FDAは、遺伝子治療製品を、転写および/または翻訳によって転移された遺伝物質および/または宿主ゲノムに統合し、核酸、ウイルスまたは遺伝子工学微生物として使用する製品として定義する。この製品は細胞を修飾するために使うことができる体内にある独房に移されたり離体する受取人を管理する前に。FDAの生物製品評価と研究センター(CBER)は遺伝子治療製品の監督管理を担当している。CBER内部では,遺伝子治療と関連製品の審査が治療製品オフィス(OTP)に統一されており,FDAはすでに細胞,組織,遺伝子治療諮問委員会を設立し,その審査についてCBERにアドバイスを提供している。
FDAは遺伝子療法に関する多くの指導文書を発表している。FDAの指導文書には法的拘束力がないにもかかわらず、いくつかの態様の規定を遵守することは、私たちが開発した任意の候補製品が承認されるために必要である可能性が高い。これらのガイドラインは、FDAが遺伝子治療開発の各段階で考慮する因子について提案およびより多くの明瞭性を提供し、これらの要因は、遺伝子治療の適切な臨床前評価、IND出願に含まれるべきCMC情報、INDまたはBLA出願をサポートする製品の有効性を測定するために正確に設計テスト、研究遺伝子治療に曝露された対象の遅延副作用を観察するための措置、およびまれな疾患を治療するための遺伝子治療製品を提供する。
遺伝子治療試験がNIHの助成を受けた組換えまたは合成核酸分子研究に関連する機関で行われるか,あるいはその助成が行われる場合,この試験はNIHの組換えDNA分子に関する研究ガイドラインに基づいて行われなければならない。このような機関で行われている組換えまたは合成核酸分子または組換えまたは合成核酸分子からのDNAまたはRNAをヒト被験者に転移させることに関する研究は、開始前に国際生物倫理委員会の審査および承認を経なければならない。多くの会社や他のNIHガイドラインに拘束されていない機関は自発的にこれらのガイドラインに従っている。
CGMPとCGTPの要求に合致する
FDAの規定では,薬品は特定の承認施設で生産され,cGMPに適合しなければならないことが求められている。CGMP条例には、人員、建物および施設、設備、アセンブリおよび薬品容器および閉鎖的な制御、生産およびプロセス制御、包装およびラベル制御、保有および分配、実験室制御、記録および報告、ならびに返品または回収された製品に関する要件が含まれる。医薬品の生産·流通承認に関与するメーカーおよび他のエンティティは、FDAおよびいくつかの州機関にその工場を登録し、cGMPおよび他の要求を遵守することを確実にするために、FDAの定期的な抜き打ち検査を受けなければならない。検査は“リスクに基づくスケジュール”に従わなければならないが、これはいくつかの機関がより頻繁に検査されることを招く可能性がある。
メーカーはまた、その工場に関する電子または実物記録の提供を要求しなければならないかもしれない。FDAの検査の延期、拒否、制限、または拒否は、製品が偽とみなされる可能性がある。承認された製品の製造プロセス,仕様や容器閉鎖システムの変更は厳しく規制されており,通常はFDAの承認を得て実施する必要がある。FDAの法規はまた、cGMPとの任意の偏差を調査および是正し、NDAスポンサーおよび承認製品の生産に参加する任意の第三者製造業者に報告および文書要求を提出することを要求する。
BLAを承認する前に、FDAは通常、製品を生産する1つまたは複数の施設を検査する。FDAは、製造プロセスおよび施設がcGMP要件に完全に適合していることを決定し、要求された仕様の下で製品が一貫して生産されることを保証するのに十分であると判断しない限り、申請を承認しないであろう。PHSAは,属性が正確に定義できない生物製品などの製品の製造制御の重要性を強調している。承認後、製造設備、位置、またはプロセスにおける材料変更は、追加の規制審査および承認をもたらす可能性があります。2022年12月に公布された“大流行病予防法”は、1種の薬物または生物が米国に輸入または提供される前に、米国国外の別の機関でさらなる製造、製造、繁殖、複合または加工を行っても、外国の薬品製造機関は登録と発売要求を守らなければならないことを明らかにした。
遺伝子治療製品については,メーカーがCGTPに適合していなければ,FDAもこの製品を承認しない。これらの基準は、ヒト細胞、組織、および細胞および組織に基づく製品またはHCT/Pを製造するための方法および施設および制御を管理するFDAの法規および指導文書において見つけることができ、これらのHCT/Pは、ヒトレシピエントに移植、移植、注入または転移するためのヒト細胞または組織である。GTP要求の主な目的は,細胞や組織に基づく製品の製造方式の確保であり,感染症の導入,伝播,伝播を防止することを目的としている。FDAの規定はまた、組織機関がFDAに彼らのHCT/Pを登録し、適用した場合にスクリーニングとテストを通じてドナーを評価することを要求する。
27
製造業者や他の製品の製造と流通に参加する人たちもまたFDAと特定の州機関に彼らの工場を登録しなければならない。国内でも海外の製造企業でも,最初に生産過程に参加する際には,FDAに登録して追加的な情報を提供しなければならない。登録されていない工場で製造または輸入されたどの製品も、外国でも国内でも、FDCAの下に誤ったブランドが貼られているとみなされている。これらの生産施設は,cGMPや他の法律に適合することを確保するために,政府当局の定期的な抜き打ち検査を受ける可能性がある。製造施設が製品が承認されたときに適用される法規および要件に実質的に適合していない場合、その施設からの製品の出荷を禁止し、および/または以前に出荷された製品のリコールを禁止する警告状または禁止令を発行することが含まれる規制法執行行動をとることができる。
“大同法”を提出する
必要な臨床試験、臨床前研究および臨床試験の結果、ならびに製品CMCおよび提案ラベルに関する情報が、製品候補製品を1つまたは複数の適応に押し出すことの承認を要求するために、申請の一部としてFDAに提出されると仮定する。“処方薬ユーザー費用法案”(PDUFA)によるBLA提出に要する費用は高く(例えば、2024年度、申請費用は約405万ドル)、承認されたBLAのスポンサーは年間計画費を支払う必要があり、2024年度の合格処方薬製品あたりの年会費は416,734ドルとされている。これらの費用は、一般に年に1回調整され、場合によっては、公衆の健康を保護するために免除が必要であるなど、免除および免除がある可能性があり、費用は革新に大きな障害となるか、またはスポンサーは、審査のためにその最初の人間治療申請を提出する小企業である。
FDAは、BLAを受信してから60日以内にBLAを予備審査し、その時点で実質的な審査を可能にするためにスポンサー申請が十分であるかどうかを通知しなければならない。関連部分において、FDAの出願に関する規定は、FDAがすべての関連情報及びデータを受信する前に、出願は提出されたとみなされてはならない。FDAが申請がこの基準を満たしていないと判断した場合、それはスポンサーに提出拒否またはRTF決定を発行する。一般に、技術移転フレームワークの根拠は、明らかに情報または必要な情報の部分を見落としているような行政上の不完全さであり、安全性、純度および効力の評価を見落としたり、説明を適切に使用するために必要なキーデータ、情報または分析を提供するなど、科学的な不完全さ、または情報の内容、紹介または組織が不十分であり、実質的かつ意義のある審査を行うことができない。FDAはBLAの届出を受け入れるのではなく、より多くの情報を提供することを要求するかもしれない。この場合、申請は追加情報と共に再提出されなければならない。再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。
出願が受け入れられた後、FDAは出願の深い実質的な審査を開始した。FDAは、BLAを受け入れたときに、または74にこれは…BLAを受け取った日。その後、FDAは、機関がBLAを審査する過程でスポンサーに“情報要求”を提出することができる。FDAは、提案された製品がその予期される用途に対して安全に有効であるかどうか、許容可能な純度プロファイルを有するかどうか、および製品がcGMPに従って製造されているかどうかを決定するためにBLAを審査する。FDAがPDUFAで達成した目標と政策によると、FDAは10ヶ月間、新分子実体である研究製品の標準出願の予備審査を完了し、“優先審査”を有する出願については、FDAが6ヶ月間で出願を完了した。FDAは、新しい情報を考慮するために、またはスポンサーが明確な提供を提供する場合に、FDAが最初の提出後に発見した突出した欠陥を解決するために、審査プロセスをさらに3ヶ月延長することができる。これらの審査目標にもかかわらず,FDAによるBLAの審査がPDUFA目標日を超えることは珍しくない。
BLAを承認する前に、FDAは、INDおよびGCP要件に適合することを保証し、FDAに提出された臨床データの完全性を保証するために、スポンサーおよび1つまたは複数の臨床試験場所を検査することができる。FDORAの通過に伴い,国会はFDAに提出された臨床や非臨床研究の準備,進行や分析に係る施設,研究記録を持ったり研究過程に参加したりする他の人の検査を明確に許可し,FDAが検査を行う権限を明らかにした。
さらに、FDAは、安全性または有効性の問題を提起する新製品候補申請を含むBLAを、審査、評価、提案を諮問委員会に提出して、申請を承認すべきかどうか、およびどのような条件で承認すべきかを決定する可能性がある。通常,諮問委員会は臨床医や他の科学専門家を含む独立した専門家からなるグループであり,申請を審査,評価し,申請を承認すべきかどうか,どのような条件でアドバイスを提供すべきかを担当する。FDAは諮問委員会の提案に制限されていないが、最終承認決定を下す際にこれらの提案を考慮する。臨床試験のデータは常に決定的ではなく,FDAやその諮問委員会は異なる方法でデータを解釈する可能性があり,NDAスポンサーは同じデータを解釈する可能性がある。FDAは臨床試験データを再分析する可能性もあり,FDAやスポンサーの審査過程で広く議論される可能性がある。
28
BLAに関するアメリカ食品医薬品局の決定は
FDAはBLAを審査し、製品が安全で、純粋で効果的かどうかを決定する。この決定を達成するために、FDAは、提案製品の期待利益が患者に対する潜在的リスクよりも大きいことを決定しなければならない。このような“利益−リスク”評価は、製品の安全性、純度、および効力に関するBLAにおける大量の証拠によって提供される。この評価はまた他の要素の影響を受け、潜在疾病の深刻性及び現有の治療法がどの程度患者の医療需要を満たしているか;発売前の臨床試験証拠はどのようにこの製品の発売後の環境における実際の使用状況の不確定性を推定するか;及びリスク管理ツールが特定のリスクを管理する必要があるかどうかを含む。
FDAは一般的に強力なセキュリティデータベースと大量の製品有効性の証拠を要求する。FDAの“実質的証拠”という言葉の解釈は,製品の有効性を確認するためには,少なくとも2回の十分かつ良好な制御の臨床調査が必要である。しかしながら、場合によっては、FDAは、いくつかの特徴および追加情報を有する単一の実験がこの基準を満たす可能性があることを示している。このやり方は1998年に国会で認められました立法は関連部分で規定されています[アメリカ食品医薬品局は]関連する科学的決定に基づいて、良好な臨床調査からのデータおよび確認性証拠(調査の前または後に得られる)が有効性を決定するのに十分である場合、FDAは、そのようなデータおよび証拠を実質的な証拠と見なすことができる。2019年12月、FDAはガイドライン草案を発表し、有効性の実質的な証拠を確立するために必要な研究をさらに説明した。このガイドラインはまだ最終的に決定されていないが、FDAは確かに2023年9月にガイドライン草案を発表し、その中で二回目の臨床試験ではなく検証性証拠に依存して治療効果を証明する考慮要素を概説した。
さらに、FDAは、申請を承認する前に、製品の製造、加工、包装、または保有施設が製品の持続的な安全を確保するための基準に適合しているかどうかを決定する。承認過程が長く、しばしば困難であり、適用された規制基準が満たされていない場合、または追加の臨床または他のデータおよび情報が必要とされる可能性がある場合、FDAはBLAの承認を拒否する可能性がある。申請およびすべての関連情報を評価した後、諮問委員会の提案(ある場合)および製造施設および臨床試験場所の検査報告書を含む、FDAは、承認書または完全な返信、またはCRLを発行する可能性がある。
CRLは,申請の審査周期が完了したことを示しており,申請は現在の形で承認されない.CRLは、通常、提出中の不足点を列挙し、FDAが申請を再検討するために、大量の追加のテストまたは情報を必要とする可能性がある。CRLは、追加の臨床または他のデータ、追加の重要な第3段階臨床試験(S)、および/または臨床試験、臨床前研究または生産に関連する他の重要で時間のかかる要件を必要とする可能性がある。CRLを発行すれば,スポンサーはFDAが決定した欠陥に1年間応答することができ,FDAは申請が撤回されたと考えたり,スポンサーの追加延長を適宜承認したりすることができる.FDAのCRL決定に挑戦しようとする人に対して、FDAは、スポンサーがCRLについての正式な公聴会を要求することができるか、または協議要求または紛争解決の正式な要求を行うことができることを示している。製品承認過程において、FDAはまた、製品の安全な使用を確保するためにREMSが必要かどうかを判断する。FDAがREMSが必要であると結論した場合,BLAのスポンサーは提案したREMSを提出しなければならず,必要であればFDAはREMSのないBLAを承認しないであろう。
一方,その製品の商業マーケティングを承認し,特定の適応に関する具体的な処方情報を提供する。すなわち、承認は、FDA承認のラベルに記載された使用条件(例えば、患者数または適応)に限定される。さらに、解決すべき具体的なリスク(S)に応じて、FDAは、承認後に製品の安全性をさらに評価するための承認後試験を含む承認後試験の実施を要求する製品ラベルに禁忌症、警告または予防措置を含むことを要求することができ、試験および監視計画は、製品の商業化後に製品を監視することを要求するか、または販売および使用制限またはREMS下の他のリスク管理メカニズムを含む他の条件を適用することができ、これらは製品の潜在的な市場および利益に大きな影響を与える可能性がある。FDAは発売後の試験或いはモニタリング計画の結果に基づいて、製品の更なるマーケティングを阻止或いは制限することができる。承認後、新たな適応の追加、製造変更、および追加のラベル宣言など、承認された製品のいくつかのタイプの変更は、さらなるテスト要件およびFDAの審査および承認を受けることになる。
迅速チャネル、画期的な治療、優先審査、再生性高度治療指定
FDAには薬物と生物製品の開発と承認を加速するためのいくつかの計画があり、これらの薬物と生物製品は深刻な或いは生命に危害を及ぼす疾病或いは状況の治療を目的としている。これらの計画は、迅速チャネル指定、突破的治療指定、優先審査指定、再生医学高度治療(RMAT)指定を含む。これらの指定は相互排他的ではなく、候補製品はそのうちの1つまたは複数の計画の資格に適合する可能性がある。これらの計画は製品開発と承認を加速させることを目的としているが、FDA承認の基準を変えることはない。
製品が深刻または生命に危険な疾患または状態の治療のために使用されることが意図されており、非臨床的または臨床的データが、そのような疾患または状態の満たされていない医療需要を満たす可能性があることを示す場合、FDAは製品迅速チャネル称号を付与する可能性がある。Fast Track製品については、スポンサーがFDAとより多くの相互作用を有する可能性があり、場合によっては、FDAは、申請が完了する前に、Fast Track製品マーケティング申請の部分の審査を開始する可能性がある。FDAがこの製品が資格基準を満たしていないと判断した場合、高速チャネル資格は撤回される可能性がある。
29
1つの製品が、深刻なまたは生命に危険な疾患または状態を治療することを意図しており、初歩的な臨床証拠が、製品が1つまたは複数の臨床的重要終点において既存の療法よりも実質的に改善されている可能性があることを示す場合、製品は突破的療法として指定することができる。画期的な治療法の場合、FDAは、開発過程全体にわたってスポンサーとの会議を行うこと、製品スポンサーに開発と承認に関する提案をタイムリーに提供すること、より多くの上級者を審査過程に参加させること、審査チームに学際的なプロジェクト担当者を割り当てること、およびスポンサーが効果的に臨床試験を設計することを助ける他のステップを含むいくつかの行動をとる可能性がある。製品がこれ以上資格基準を満たしていなければ、画期的な指定が撤回される可能性がある。
2016年12月の“21世紀治療法案”の成立に伴い、国会は再生医学高度療法の追加迅速計画を承認した。製品が、深刻なまたは生命を脅かす疾患または状態を治療、修正、逆転または治癒することを目的とした再生医学療法であり、初歩的な臨床証拠が、製品がそのような疾患または状態の満たされていない医療需要を満たす可能性があることを示す場合、製品はRMAT称号を得る資格がある。RMATを指定する利点は、代替または中間終点に基づく優先審査および承認を加速する潜在的資格を含む画期的な治療の利点を含む。もし製品がこれ以上資格基準を満たしていなければ、RMAT資格は撤回されるかもしれない。
製品が重篤な疾患を治療し、承認された場合、このような疾患の治療、予防または診断の安全性または有効性の面で有意な改善を提供する場合、FDAは、製品を優先的に検討するように指定することができる。優先指定の目的は、このようなアプリケーションの評価に全体的な注意とリソースを誘導し、FDAがマーケティング申請に行動する目標を10ヶ月から6ヶ月に短縮することである。
承認のルートを速める
FDAは、患者に既存の治療よりも意義のある治療利点を提供する深刻または生命に危険な疾患の承認を加速する可能性があり、これは、製品が臨床的利益を合理的に予測する可能性のある代替終点に影響を及ぼすことを決定することに基づく。中間臨床終点に対する製品の影響が不可逆的な発病率または死亡率またはIMMへの影響よりも早いことができ、このような状況の重症度、希少性または流行率、および代替治療が利用可能または不足している場合を考慮すると、IMMまたは他の臨床的利益への影響を合理的に予測することが可能である場合、FDAはこのような状況の加速承認を許可することもできる。
加速承認経路は病気経過が比較的に長く、製品の期待される臨床利益を測定するために時間を延長する必要がある環境に最もよく用いられ、代用或いは中間臨床終点への影響は非常に速い。したがって、加速承認は様々な癌を治療するための製品の開発と承認に広く使用されており、その中で治療の目標は通常、生存率を向上させること、または発病率を低下させることであり、典型的な病気経過の持続時間は長く、時には大型の試験を必要とし、臨床または生存利益を証明することである。
加速された承認を得るために、FDAは、一般に、製品の臨床的利益を検証および説明するために、追加の承認後の検証的研究を勤勉な方法で行うことをスポンサーに要求する。職務調査を行うために必要な承認後研究は行われておらず,承認後研究期間中に臨床的利益が確認されていない,あるいは虚偽や誤解性を伝播する宣伝材料は,FDAが製品承認を迅速に撤回することを許可する。FDAがスポンサーに別途通知しない限り、加速承認の下で承認された候補製品の販売促進材料はFDAの事前審査を経なければならない。
2022年12月のFDORAの成立に伴い、国会は薬品と生物製品の承認を加速するためのいくつかの条項を改正した。具体的には、新しい立法許可FDAは、(I)スポンサーが加速承認を得る前に検証的臨床試験を行うことを要求し、(Ii)加速承認された製品のスポンサーは、6ヶ月毎に承認後の研究の進捗報告(研究完了まで)をFDAに提出し、(Iii)実証試験が製品の臨床的利益を検証できなかった後、迅速なプログラムを使用してNDAまたはBLAの加速承認を撤回することを要求する。また,FDORAは,承認加速後にこのような研究を要求しないことを決定した場合には,そのサイト上で“なぜ承認後研究に適していないのか,あるいは不要な理由”を公表することを求めている。2023年3月、FDAはガイドライン草案を発表し、現在加速承認設計、試験データの構想と分析を行い、腫瘍治療薬物の加速承認を支持することを目的とした。
承認後法規
製品発売の規制承認または既存製品の新しい適応が得られた場合、スポンサーは、すべての通常の承認後の規制要件、およびFDAが承認プロセスの一部として適用される任意の承認後要求を遵守することを要求されるであろう。スポンサーは、いくつかの不良反応や生産問題をFDAに報告し、最新の安全性および有効性情報を提供し、広告や販売促進ラベル要求に関する要求を遵守することを要求される。製造業者およびそのいくつかの下請け業者は、FDAおよびいくつかの州機関に彼らの工場を登録し、製造業者にいくつかのプログラムおよび文書要件を適用するcGMP法規を含む現行の法規要件に適合しているかどうかを知るために、FDAおよびいくつかの州機関の定期的な抜き打ち検査を受けなければならない。したがって、スポンサーおよびその第三者メーカーは、cGMP法規や他の法規要件の遵守を維持するために、生産および品質管理の分野で時間、お金、エネルギーをかけ続けなければならない。
30
製品はまた正式なロット発表が必要である可能性があり、これはメーカーが製品が発表される前に、製品の各ロットに対していくつかのテストを行わなければならないことを意味する。製品が正式なバッチ発行を必要とする場合、製造業者は、各バッチのサンプルをFDAに提出し、バッチの生産履歴要約および製造業者がバッチに対して行ったすべての試験結果を示す放出スキームを提示しなければならない。さらに、FDAは、いくつかの製品のバッチに対していくつかの検証的試験を行い、その後、これらのロットを流通させるかもしれない。最後に、FDAは薬品の安全性、純度、効力と有効性に関する実験室研究を行う。
承認された場合、規制要求や基準の遵守が維持されていない場合、または製品発売後に問題が発生した場合、FDAは承認を撤回する可能性がある。その後、製品には、予期されない深刻度または頻度の不良事象、または生産プロセス、または規制要件を遵守できなかったことを含む、以前に未知の問題が存在することが発見され、新しいセキュリティ情報を追加するために承認されたラベルの改訂を招く可能性がある;新しい安全リスクを評価するために発売後研究または臨床試験を実施すること、またはREMS計画に従って流通または他の制限を実施することが可能である。他の他の潜在的な結果には
FDAは生物製品を含む処方薬製品の広告やラベルを厳格に規制している。他にも、この規定には、消費者向け直接広告、未承認用途に関する通信、業界スポンサーに関する科学的および教育活動、ならびにインターネットおよびソーシャルメディアに関連する販促活動の基準および規定が含まれる。薬物が承認されるまで,薬物の安全性や有効性の宣伝は禁止されている。さらに、米国承認薬物のスポンサーは、医師が医学的慣行に従ってラベル外使用の薬剤を処方することができるにもかかわらず、未承認またはラベル外の使用のための薬剤を宣伝してはならない。非常に具体的で狭い条件下では、製造業者が、科学または医学定期刊行物情報の配信のようなラベル外情報に関する非販売促進、非誤解的伝播に従事することを可能にすることができる。また,2022年12月に“承認前情報交換法”(PIE Act)が成立するにつれ,承認されていない製品のスポンサーは,製品承認後に患者の接触を加速させるために,開発中の製品に関する何らかの情報を支払者に能動的に伝達することができる。これまで,このような通信はFDAの指導の下で許可されていたが,新たな立法はスポンサーの保護を明確にしており,これらのスポンサーは未承認の製品用途を含む開発中の製品に関するいくつかの情報を支払者に伝えてきた。また,FDAは2023年10月にガイドラインを発表し,未承認用途に関する科学的情報を医療保健提供者に配布する拘束力のない政策を管理して概説した。本ガイドライン草案は、このような通信が真実で、誤解性がなく、事実と偏らないことを要求し、医療保健提供者が許可されていない使用に関する情報の利点および欠点、ならびに有効性および実用性に必要なすべての情報を説明することを含む。
もしある会社がラベル外の使用を促進したことが発見された場合、それはFDA、米国司法省、HHS監察長事務室、および州当局の行政と司法執行を受ける可能性がある。これは、民事および刑事罰金、および会社の薬品の宣伝または流通を実質的に制限する方法の合意を含む一連の重大な商業的影響を及ぼす可能性のある一連の処罰を企業に受ける可能性がある。連邦政府は、不当な販売促進の疑いのある会社に対して巨額の民事と刑事罰金を科し、企業に同意法令または永久禁止令を締結し、これらの法令または永久禁止に基づいて、特定の販売促進行為を変更または制限することを要求する。
承認後、承認された製品のいくつかのタイプの変更、例えば、新しい適応または投与レジメンの追加、変更または追加のラベル宣言の製造は、FDAのさらなる審査および承認を受ける。また、FDAは、商業化された承認製品の効果を監視するためにテストおよび監視計画を要求する可能性があり、FDAは、これらの上場後計画の結果に基づいて、製品のさらなる販売を阻止または制限する権利がある。
31
規制要求や基準が守られていない場合、あるいは製品発売後に問題が発生した場合、FDAは製品承認を撤回する可能性がある。その後、製品には、予期しない深刻度または頻度の不良イベントまたは製造プロセスの問題を含む以前に未知の問題が存在することが発見され、新しいセキュリティ情報を追加するために承認されたラベルの改訂を招く可能性がある;発売後の研究または臨床試験を実施して新しい安全信号を評価する;またはREMS計画に従って流通または他の制限を実施する。他の他の潜在的な結果には
最後に、製品に適応、ラベル、または製造プロセスまたは施設の変化を含む修正がある場合、スポンサーは、新しいBLAまたはBLAサプリメントの承認を得るために提出および提出を要求される可能性があり、これは、スポンサーに追加のデータの開発を要求するか、または追加の臨床前研究および臨床試験を行うことを要求する可能性がある。FDAが新しい適応を承認することを確保する過程は、元の適応を承認する過程と類似しており、新適応における製品の安全性および有効性を証明するために、十分かつ制御された臨床試験データを提出する必要がある。このような実験を行っても,FDAはタイムリーに使用するためにラベル適応のいかなる拡張も承認しないか,あるいは全く承認しない可能性がある。継続的な年間使用料要求もあり,現在は何らかの承認された薬物の計画費用として評価されている。
孤児薬の指定と排他性
米国の孤児薬物指定は,スポンサーにまれな疾患や疾患の治療のための製品の開発を奨励することを目的としている。米国では、法律は、まれな疾患または疾患を、米国で20万人未満に影響するか、または米国で20万人を超える影響を与えると定義しており、疾患または疾患に対する製品の開発および提供のコストが米国での販売から回収されることを合理的に予想することができない。
孤児薬物指定は会社が特定の税金免除を受ける資格があるようにする。さらに、孤児薬物指定を有する候補薬剤が、その後、このような指定された疾患を有する薬物に対するFDAの最初の承認を得た場合、この製品は、孤児薬物排他性を得る権利があり、これは、FDAが、その後の候補製品が臨床的利点を有することが証明されない限り、製品承認後7年以内に同じ適応を有する同じ薬剤を販売するために、他の出願を許可することができないことを意味する。臨床的優位性がない場合、FDAは、スポンサーの同意やスポンサーが十分な数を提供できない限り、他のメーカーによって生産された同じ製品を同じ適応のために市場独占期間内に承認することができない。
スポンサーは、以前承認されていなかった製品を孤児薬として指定したり、すでに発売されている製品のために新たな孤児適応を申請することを要求することができる。さらに、1つの製品が他の態様で承認された孤児薬物と同じ製品である場合、製品が信頼できる仮定を提示することができる場合、すなわち、その製品が第1の薬剤よりも臨床的に優れている可能性がある場合、製品の発起人は、同じ稀な疾患または疾患の後続製品に対する孤児薬物名を求めることができ、取得することができる。複数のスポンサーは、同じ製品のために同じまれな疾患または疾患の孤児薬物指定を得ることができるが、孤児薬物指定を求める各スポンサーは、完全な指定申請を提出しなければならない。しかし,孤児の排他的治療を受ける資格があるためには,これまでに承認された製品,すなわち同じ疾患の同じ薬物よりも臨床的に優れていなければならない。
遺伝子治療製品は、いつ2つの製品が孤児の排他的目的であるかを評価するための新しい問題を提起した。FDAは2021年9月、孤児の排他的目的における現在の遺伝子治療製品と別の製品の“同じ”という考え方を記述した最終的な指導文書を発表した。この指導の下で、2つの遺伝子治療製品間の遺伝子組換えまたはベクターが異なり、“微小な”差を反映しない場合、この2つの製品は孤児薬物の排他的目的のための異なる薬物とみなされる。FDAはケースに基づいて、同じウイルスクラスからの2つのベクターが同じかどうかを決定し、評価と同時に他の重要な特徴を考慮する可能性がある。このガイドラインは、FDAが“同一性”を評価する方法に対していくつかの追加的な解明を提供するが、FDAが同じカテゴリのウイルスベクター、ベクター、またはトランスジェニックのどのような差が微小であると考えられ、どのような追加の特徴が考慮される可能性があるかについては、依然として重大な曖昧性および不確実性が存在する。
32
専門期間はFDAが上場申請を承認した日から,この製品が指定した適応にのみ適用される。FDAは、同じ製品の第2の出願を異なる使用のために承認することができ、または同じ使用のために製品の臨床的により優れたバージョンを申請することができる。場合によっては、孤児薬物排他性は、孤児薬物排他性を有する会社が市場需要を満たすことができない場合、または同じ場合に同じ薬物を使用する後続製品が、より良い治療効果または安全性に基づいて臨床的に承認された製品よりも優れていることが証明された場合、または患者ケアに大きく貢献することを含む、別の製品の承認を阻止しない。孤児薬物法案は孤児薬物の排他性を認めることをFDAに明確に要求していたが,その臨床的優位性にかかわらず,早い時期の裁判所の意見であった。2020年12月に署名された総合立法によると、製品の臨床的優位性を示す要求は、2017年のFDA再認可法案(FDARA)公布前に孤児薬物指定を受けたが、FDAの承認または許可を得ていない薬物および生物製品に適用される。
2021年9月,第11巡回控訴裁判所は,排他的範囲を決定するために,法規中の“同一疾患または状況”という言葉は指定された“まれな疾患または状況”を指し,その機関はそれを“適応または使用”と解釈することができないと判断した。したがって,裁判所は孤児薬物は“適応や使用”ではなく,指定された疾患や状況全体に排他的に適用されると結論した。FDAがこの裁判所の判決をどのように実行するのかまだ分からない。この決定を覆すための立法提案があったが、それらはまだ法律になっていない。FDAは2023年1月23日、裁判所の命令範囲を超える事項において、FDAは、孤児薬の独占性と孤児薬の使用または適応のために許可された使用または適応とを束ねて、その既存の法規を適用し続けると発表した。
小児科排他性
小児科排他性はアメリカのもう一つのタイプの非特許規制排他性である。具体的には,“小児最適薬品法”はさらに6カ月の排他性を付加することを規定しており,小児科排他性が付与された場合には,任意の残りの規制排他性の期限に付加される。BLAスポンサーから提出された小児科データがこのようなデータに対するFDAの書面要求に公平に応答した場合、これらのデータが研究された小児科群において有効であることを示さなくても、この6カ月の排他性が付与される可能性がある。
生物模倣薬と排他性
2010年3月に法律となった2010年の患者保護·平価医療法案(PPACA)には、2009年のバイオ医薬品価格競争および革新法案(BPCIA)という副題が含まれている。BPCIAは、FDAが生体模倣薬と交換可能な生物模倣薬を許可することを許可する規制方案を確立した。
BPCIAによれば、製造業者は、以前に承認された生物製品または“参照製品”“生物学的に類似している”または“交換可能”と一致する生物製品のライセンス申請を提出することができる。FDAに生物類似製品を承認させるためには、参考製品と提案された生物類似製品が安全性、純度と効力の面で臨床的に意義のある差がないことを発見しなければならない。FDAが生物類似製品を参照製品と交換することができるようにするために、この機関は、生物学的類似製品が参照製品と同じ臨床結果を生成することが期待でき、(複数回投与された製品のための)生物学的製剤および参照生物製剤は、安全リスクを増加させることなく、または参照生物製剤の独占的使用と比較して治療効果のリスクを低下させることなく、以前の投与後に交換可能であることを発見しなければならない。
BPCIAによると,生物類似製品の申請は参考製品が承認された日から4年後にFDAに提出される。FDAは参考製品が初めて許可された日から12年まで生物類似製品を承認しない可能性がある。この12年間の独占期間は、生物類似製品の承認を禁止する参考製品固有期間と呼ばれるが、特に完全なBLAによる競合製品の承認は阻止されない(すなわち、スポンサー自身の臨床前データおよび十分かつ良好に制御された臨床試験からのデータを含み、この製品の安全性、純度および有効性を証明する)。BPCIAはまた、交換可能な製品として承認された生物模倣薬のためのいくつかの排他的期限を設けている。2022年12月、FDAは、これらの製品がそのような製品が基準製品と交換可能であると承認された初日に承認される限り、複数の第1の交換可能な生物類似生物製品を承認することができるFDORAによって明らかにされた。この法律はまた、革新者生物と生物類似物製造業者に生物類似物を承認する前に特許侵害、有効性、および実行可能性について訴訟を提起させる広範な手続きを含む。
BPCIAが成立して以来、多くの州では、生物模倣薬の使用を規範化するために州政府によって規制された薬局のやり方を管理する法律を含む法律が改正された。
33
特許期限の回復と延長
ハッジ·ワックスマン法案によると、新生物製品を持つ特許は、製品開発およびFDA規制審査中に失われた特許期間の補償として、製品の単一特許を承認して最大5年間の回復を可能にする限られた特許期間延長を受ける資格がある可能性があると主張している。製品をカバーする特許の回復期は、通常、人間に関する臨床研究が発効した日から市場申請提出日までの半分の時間から、発起人が職務調査を行っていない1銭を減算し、出願提出日から最終承認日までの間の時間から、発起人が職務調査を行っていない金を差し引くものである。特許期間回復は特許の残存期間の延長には利用できず,製品承認日から合計14年を超える。承認された製品に適用される特許は、延期する資格があり、承認された薬剤、その使用方法、または製造方法に関する権利要件のみを延期することができ、延期出願は、関連特許が満了する前に提出されなければならない。複数の製品をカバーする特許は、そのうちの1つの承認に関連して延期することしかできない。米国特許商標局は,FDAと協議した後,任意の特許期間の延長または回復の出願を審査·承認する。
FDAは診断を承認しました
FDAは2014年8月に最終指導意見を発表し、承認治療製品への適用と体外培養診断を伴う。ガイドラインによれば、新薬のための、キット診断装置およびその対応する治療装置は、製品ラベルにおいて示される治療のためのFDAの承認または承認を同時に得るべきである。キット診断装置の承認または許可は、装置が十分に評価され、ターゲット集団において十分な性能特徴を有することを保証するであろう。FDAは2016年7月、薬物治療および治療のスポンサーを支援するためのガイドライン草案を発表した体外培養製品の共同開発に関する問題に関するキット診断装置。
また,FDAは2020年4月に追加のガイドラインを発表し,適切な多様な薬物や生物腫瘍学製品の指定用途を支援するためのマーカーセット診断装置の開発とマーカーの考慮事項を述べた。本ガイドラインは,診断に伴うラベルに関する既存の政策に基づいている。FDAはその2014年のガイドラインで、診断に伴う診断が特定のカテゴリーの治療製品と共に使用するのに適していると結論するのに十分な証拠があれば、診断に伴う期待用途/使用適応は、特定の製品ではなく、特定の治療製品群と命名すべきであることを示している。2020年ガイドラインは、2014年ガイドライン中の政策声明を拡張し、セット診断開発者は、彼らのテストを開発することができるかどうかを決定する際に、一連の要因を考慮することを提案するか、または、単一の治療製品(S)をリストするのではなく、特定のグループのための腫瘍学治療製品のようなより広いラベル宣言をサポートするために、承認されたセット診断のラベルを追加修正することができる。
FDCAのもとでは体外培養診断は,随伴診断を含め,医療機器として規制されている。アメリカでは、医療機器の設計と開発、臨床前と臨床試験、発売前の承認或いは承認、登録と発売、製造、ラベル、貯蔵、広告と販売促進、販売と流通、輸出と輸入及び発売後の監督などの事項は、すべてアメリカ食品薬品監督管理局及びその実施条例及びその他の連邦と州法規と条例によって管理されている。適用免除が適用されない限り、診断テストは商業流通の前に市場許可またはFDAの承認を得る必要がある。
FDAは以前体外培養随伴診断は、候補製品に反応して発売前承認またはPMAを得る患者を選択することを目的とし、同時に治療製品候補を承認することを目的としている。臨床および臨床前データの収集、およびFDAおよびFDAへの審査の提出を含むPMAプロセスは、数年またはそれ以上を要するかもしれない。これには厳格な上場前審査が含まれており、その間、スポンサーは設備の安全性と有効性の合理的な保証、設備設計、製造、ラベルなどに関する設備とその部品の情報を準備してFDAに提供しなければならない。PMA申請は申請費を支払う必要があり、2024年の連邦財政年度の申請料は483,560ドル、小企業の申請料は120,890ドルである。
PMA申請は通常、臨床試験を必要とし、一部の症例では、FDAは510(K)の提出をサポートするための臨床研究を必要とする可能性がある。この装置に関する臨床研究を行いたいメーカーはFDAのIDEで規定されている。IDE規制は重大と非重大リスク装置研究を区別していますが、承認されて研究を開始するプログラムもそれに応じて違います。さらに、いくつかのタイプの研究はIDE規制によって制限されません。重大なリスク装置は被験者の健康、安全あるいは福祉に深刻なリスクをもたらす可能性がある。重大な危険設備とは疾病の診断、治癒、軽減或いは治療或いは人類の健康被害を防止する上で非常に重要な設備である。臨床研究を開始する前に,重大なリスクを有する設備の研究にはFDAとIRBの承認が必要である。多くの随伴診断は重大なリスクツールと考えられており,疾患や状況の診断に機能しているためである。非重大危険装置とは,人体に重大な危険を構成しない装置である。非重大危険装置研究は臨床研究開始前にIRBの承認を得るだけでよい。
34
設備が市場に投入された後、それは依然として厳格な規制要求を受けている。医療機器の販売は,その許可や承認の用途や適応にしか利用できない。デバイス製造業者はまたFDAに登録とデバイスリストを確立しなければならない。医療機器メーカーおよびその供給者の製造過程は、医療機器の設計、テスト、生産、プロセス、制御、品質保証、ラベル、包装および輸送の方法および文書を含む品質体系法規の適用部分を遵守しなければならない。米国食品医薬品局は定期的に国内工場の記録や製造プロセスを不定期に検査する。FDAはまた、米国に製品を輸出する外国施設を検査する可能性がある。
連邦と州のデータプライバシーとセキュリティ法
複数のプライバシーやデータセキュリティ法律は、米国や他の私たちが実験を行ったり、将来業務を展開する可能性のある国のビジネス活動に影響を与える可能性があります。このような法律は変化しており、私たちの義務と未来の規制リスクを増加させるかもしれない。一般的に、医療業界では、1996年の連邦健康保険携帯性および責任法に基づいて、HHSは、いくつかの医療提供者、健康計画、および医療情報交換所を含む保護された健康情報(PHI)のプライバシーおよびセキュリティを保護するための法規を発表している。HIPAAはまた、保護された健康情報を取得した保証エンティティの商業パートナーに対して、保証エンティティまたは代表的な保証エンティティにサービスを提供する際に何らかの義務を課す。HIPAAは場合によっては私たちに適用される可能性があり、私たちのビジネスパートナーにも適用される可能性があり、その方法は私たちと彼らの関係に影響を与える可能性があります。我々が行ったどの臨床試験も45 CFR 46 A支部によって規制され,この支部は共通ルールとも呼ばれ,プライバシーに関する具体的な条項も含まれている。連邦プライバシー法規のほかに、多くの州の法律が健康情報のセキュリティと安全性を管理しており、これらの法律は私たちの業務に適用される可能性がある。州総検察長は、HIPAA違反行為に対して連邦民事·刑事処罰を行う可能性があるほか、連邦裁判所に民事訴訟を提起し、損害賠償や禁令を求めてHIPAAを執行し、連邦民事訴訟に関連する弁護士費と費用を求める権利がある。また、州総検察長(個人原告とともに)は、HIPAAのプライバシーや安全ルール違反の疑いで禁止令や損害賠償を求める民事訴訟を起こしている。州総検事長はまた州のプライバシーと安全法を執行する権利がある。さらに、将来的にはプライバシーと安全に関する新しい法律法規が採択される可能性がある。
州レベルでは、2018年、カリフォルニア州では、2020年1月1日に施行され、カリフォルニア州住民の個人情報を処理する企業に、収集された彼らに関する情報およびそのような情報の使用および共有方式に関する通知をデータ主体に提供し、そのような個人情報へのアクセスを要求する権利をデータ主体に提供し、場合によってはそのような個人情報の削除を要求する“カリフォルニア消費者プライバシー法”が可決された。CCPAはまた,カリフォルニア住民にその個人情報を販売しないことを選択する権利を与えている。CCPAにはその要求に違反した会社に対する重大な処罰が含まれている。また、カリフォルニア州住民に個人情報に関する漏洩事件において法定損害賠償を求める能力を含む個人訴訟権利を提供している。CCPAを遵守することは厳格で時間のかかる過程であり、ビジネスコストを増加させたり、会社にそのビジネスやり方を変更して、完全な遵守を確保したりする可能性がある。2020年11月3日、カリフォルニア州有権者は、CCPAを拡大し、カリフォルニア住民の個人情報の使用、保持、共有を要求することを含む追加の条項を含むカリフォルニアプライバシー権法案を可決し、収集または処理の目的に応じて、敏感な個人情報の追加的な保護を提供し、住民の保留情報の通知に関する情報のより多くの開示を要求する。CPRAはまた,第三者と個人情報を共有して広告に利用しないことを選択する権利の作成,企業が持つ個人情報知る権利の回顧期間の延長,第三者が持つ情報の消去権の拡大など,カリフォルニア住民の個人情報権を拡大する.CPRA条項の多くは、2022年1月1日以降に収集された任意の個人情報に適用されるが、2023年1月1日に施行される。このような規定は私たちのいくつかの商業活動に適用されるかもしれない。しかも、他の12州は州プライバシー法を採択した。例えば、ワシントン州は2023年に“私の健康私のデータ法案”を可決し、この法案はHIPAAルールによって規制されていない健康情報を専門的に規制している。他の州は似たような法律を採択して、他の州は未来にそうするかもしれない。しかも、議会は連邦プライバシー法を採択することについて討論した。これらの法律は、私たちの研究対象の決定、業務パートナーとの関係、最終的に私たちまたは私たちの協力者が規制およびマーケティングの許可を得た任意の製品のマーケティングおよび流通を含む、私たちの業務活動に影響を与える可能性があります。
連合とイギリスの薬品承認に関する規制と手続き。
アメリカ国外で任意の製品をマーケティングするために、会社はまた他の国と司法管轄区域の品質、安全性と有効性及び製品に対する臨床試験、マーケティング許可、商業販売と流通などの方面の多くの異なる監督管理要求を守らなければならない。製品がFDAの承認を得ているか否かにかかわらず、スポンサーは外国の監督管理機関のような必要な承認を得なければならず、その後、これらの国あるいは司法管轄区でその製品の臨床試験やマーケティングを開始することができる。具体的には,EUの医薬製品承認の流れは米国とほぼ同じである。製品の安全性と有効性の各提案の適応を決定するために、臨床前研究と十分かつ良好に制御された臨床試験を満足できるように完成させる必要がある。また、関連主管当局にマーケティング許可申請、またはMAAを提出し、これらの主管部門からマーケティング許可を付与し、その後、製品をEUで販売·販売することができるように要求されている。
35
非臨床研究
非臨床研究を行うのは,新たな化学や生物物質の健康や環境安全性を証明するためである。非臨床(薬物-毒性)研究は、EU指令2004/10/ECに規定されている良好な実験室慣行またはGLPの原則を遵守しなければならない(特定の医薬製品、例えば放射性ラベル目的のための放射性薬物前駆体については、別の正当な理由がない限り)。特に非臨床研究は両者が体外培養そして体内にあるGLP原則に従って計画、実行、監視、記録、報告とアーカイブを行わなければならず、GLP原則は組織過程の品質体系と非臨床研究の条件として一連の規則と標準を規定した。このようなプロス基準は経済協力と開発組織の要求を反映する。
臨床試験許可
2022年1月31日,新しい臨床試験条例(EU)第536/2014号,あるいはCTRは,従来の臨床試験指令2001/20/ECに代わってEUで発効した。新しい規定はEUの臨床試験の許可、進行と透明性を簡略化し、簡素化することを目的としている。新しい臨床試験承認調整手続きによると、1つ以上のEU加盟国またはEU加盟国で行われる臨床試験の発起人は、承認申請を提出するだけでよい。提出された材料は臨床試験情報システムを介して提出され、これはEMAが監督する新しい臨床試験ポータルサイトであり、臨床試験スポンサー、EU加盟国の主管当局と公衆に使用することができる。
手続きの簡略化に加えて,CTRには,申請のための書類のセットと,簡略化された臨床試験スポンサー報告プログラムと,臨床試験申請評価の統一プログラムが含まれており,このプログラムは2つに分けられている。第1部は、臨床試験許可申請が提出されたすべてのEU加盟国または関連加盟国の主管当局によって評価される。二番目の部分は関連する各会員国によって個別的に評価される。臨床試験申請の評価には厳しい期限が設定されている。評価手続きにおける道徳管理委員会の役割は、関連加盟国の国家法律によって引き続き管轄されるだろう。しかし、全体的に関連するスケジュールはCTRによって定義されるだろう。
CTRは、スポンサーが事前に臨床試験を行うEU加盟国主管国当局の承認を得なければならないという、以前に存在した要求を変えていない。もし臨床試験が異なるEU加盟国で行われた場合、これらのEU加盟国の主管当局は臨床試験の承認を提供しなければならない。また,スポンサーは適用された倫理委員会が賛成の意見を発表した後にのみ,特定の臨床地点で臨床試験を開始することができる。
CTRは3年間の過渡期が予想される。進行中の臨床試験と新たな臨床試験がCTRによってどの程度制御されるかはそれぞれ異なる。2022年1月31日までに“臨床試験指令”に基づいて申請を提出した臨床試験,または(Ii)は2022年1月31日から2023年1月31日までの間であり,スポンサーが“臨床試験指令”を適用する臨床試験を選択しており,2025年1月31日まではこの指令の管轄を受けている。この日以降,すべての臨床試験(行われている臨床試験を含む)はCTR条項に拘束される。
米国と同様に,ある臨床試験を行う締約国はEU臨床試験登録簿にEUの臨床試験情報を公表しなければならない。
マーケティング許可
EUの監督管理制度の下で遺伝子治療製品のマーケティング許可或いはMAを獲得するためには、スポンサーはヨーロッパ薬品管理局(EMA)が管理する中央プログラムを通じて申請を提出しなければならない。具体的には、EUでは、遺伝子療法医薬製品のような生存可能なヒト組織または細胞を含む製品のためのMAが発行され、高級治療医薬製品に関する第1394/2007/EC号条例によって管轄されており、この条例は、欧州議会および欧州理事会の第2001/83/EC号指令と組み合わされており、この指令は一般に欧州共同体医薬製品規則と呼ばれている。第1394/2007/EC条例は遺伝子治療薬物製品、体細胞治療薬物製品と組織工学製品の許可、監督と薬物警戒について具体的な規則を規定した。高級治療薬のメーカーはEMAの高級治療委員会にその製品の品質、安全性と有効性を証明しなければならず、この委員会は上場許可申請に関する意見草案を提供し、EMAの人用薬品委員会の最終的な承認が待たれる。欧州委員会はこの最終承認に基づいてマーケティング許可を承認または拒否する。
EUの中央手続きによると、MA申請の評価の最長期限は210日であり、スポンサーが人用医薬品委員会(CHMP)の質問に答える際に追加情報または書面または口頭で説明する時間は含まれていない。特殊な場合、公衆衛生の観点、特に治療革新の観点から見ると、1種の医薬製品が重大な意義を持っている場合、CHMPは加速評価を承認することができる。CHMPがそのような要求を受けた場合、210日間の制限時間は150日に減少するが、CHMPが加速評価にもはや適していないと判断した場合、集中手順の標準時限に回復する可能性がある。
36
条件付き承認
特定の場合、EU立法(第14条-a条例(EC)第726/2004号((EU)2019/5号条例及び(EC)507/2006号条例による人用薬品に関する条件付きMA改正)は、スポンサーが全面的な上場許可を申請するために必要な全面的な臨床データを得る前に条件付きMAを取得することを可能にする。以下の場合、候補製品(孤児薬として指定された薬剤を含む)に条件付き承認を与えることができる:(1)候補製品は、深刻な衰弱または生命に危険な疾患を治療、予防または医学的に診断するために使用される、(2)候補製品は、患者が満たされていない医療需要を満たすことを目的としている、(3)全面的な臨床データを提出する前に、即時発売に関するメリットが依然として補充データが必要であるという事実に固有のリスクよりも大きいことを前提として、上場許可を承認することができる、(4)候補製品のリスク-利益バランスが正である。また,(5)スポンサーは必要な包括的な臨床試験データを提供できる可能性が高い。条件付き販売許可には,現在行われているあるいは新たな研究の完了や薬物警戒データの収集義務など,販売許可保持者が履行しなければならない具体的な義務が含まれていることができる。条件付きマーケティング許可の有効期間は1年であり、リスク−収益バランスが正のままであり、条件または特定の義務を付加または修正する必要性が評価された後、毎年更新することができる。上記の集中プログラムのスケジュールは、条件付きマーケティング許可申請の審査にもCHMPに適用される。
特殊な事情
“特別な場合”では、出願人が、製品が許可され、特定の手順に従った後であっても、正常な使用条件下での有効性及び安全性に関する包括的なデータを提供することができず、MAを付与することができる。特に期待される適応は非常にまれであり,現在の科学的知識状態では網羅的な情報を提供することが不可能である場合や,データを生成することが一般的に受け入れられている倫理原則に違反する可能性がある場合がある.このMAは、重篤な疾患または満たされていない医療需要のために承認されるべき医薬製品を保持し、出願人は、MAに付与するために必要な合法的な完全データセットを有さないので、条件付きMAに近い。しかしながら、条件付きM&Aとは異なり、出願人は必要とせず、将来的には欠落したデータを提供することもない。“特殊な場合”のMAは最終的に承認されているが,毎年薬品のリスク−収益バランスが審査されており,リスク−収益比が有利でなければMAは撤回される。これらのプログラムにより,MAを付与する前に,EMAまたは加盟国主管当局は,製品の品質,安全性,有効性に関する科学的基準に基づいて,製品のリスク−利益バランスを評価する。MAの初期期間は条件付きMAを除いて5年である.この5年後、リスク-収益バランスを再評価した上で許可を更新することができる。
EUの規制データ排他性
EUでは,改正(EC)第726/2004号条例と改正された2001/83/EC指令に基づき,完全な独立パケットによって承認された新しい化学実体に基づいて,8年間のデータ独占と追加2年間の市場独占を得る資格がある。データ排他性防止EUの規制当局は、8年以内にイノベーターのデータを参考にして汎用(略語)アプリケーションを評価する。これは生体模倣薬にも適用される。追加の2年間の市場専門期間内に、後発薬の発売許可申請を提出することができ、革新者のデータを参考にすることができるが、市場専門権が満期になるまで、いかなる後発薬も発売できない。この10年の最初の8年間に、マーケティング許可所有者が1つまたは複数の新しい治療適応の許可を得た場合、10年全体の期間は最大11年に延長され、許可前の科学的評価では、これらの適応は既存の療法と比較して有意な臨床的利益をもたらすことができると考えられる。また,小児科調査計画が受け入れられた場合,より1年間の市場排他性(あるいは代替的に,さらに6カ月延長された特許延期(SPC))を得ることが可能である。孤児医薬製品では期限が異なり,合計10年間のデータ排他性があるため,PIPがあればその10年間の期間をさらに2年間延長することができる。化合物が新しい化学または生物学的実体であると考えられていても、イノベーターが所定のデータ独占期間を取得し、別の会社がMAAベースのマーケティング許可を取得した場合、同社は、薬物試験、臨床前試験、および臨床試験を含む完全な独立したデータパケットを有している場合、同社は、その製品の別のバージョンを販売することができる。
授権期間と継続期間
原則として、上場許可の有効期間は5年であり、5年後にEMAまたはライセンス加盟国の主管当局によるリスク-収益バランスの再評価に基づいて更新することができる。そのため、販売許可保持者は、販売許可が失効する少なくとも6ヶ月前に、販売許可が付与されてから導入されたすべての変化を含む、品質、安全、および有効性に関する文書の統合バージョンをEMAまたは主管当局に提供しなければならない。一旦更新されると、上場許可の有効期限は無期限であり、欧州委員会または主管当局が薬物警戒に関する正当な理由に基づいて、追加の5年間の継続を決定しない限り。認可が失効した後3年以内にEU市場に薬品を投入しない(集中手続きの場合)、または認可加盟国の市場に投入されたいかなる許可もない。
37
上場認可後の規制要件
承認を得た後、上場許可の保有者は医薬製品の製造、マーケティング、普及と販売に適用される一連の要求を守らなければならない。これらの措置には,EUの厳格な薬物警戒や安全報告規則の遵守が含まれており,これらの規則により,認可後の研究や追加的なモニタリング義務を実施することができる。また、許可製品の製造も欧州薬品管理局のGMP要求とEUの他の規制機関の類似した要求を厳格に遵守しなければならず、これらの要求は薬品の製造、加工、包装に使用される方法、施設、制御措置を規定し、その安全性と身分を確保する。改訂された2001/83 EC指令によると、EUは許可製品のマーケティングと普及、業界賛助の継続医学教育と薬品処方者及び/或いは一般公衆に対する広告を含み、厳格な監督管理を行った。
EUのトップクラスの称号
EUには優先薬品計画、即ちPrimeがあり、満たされていない医療需要領域の薬物開発を奨励し、集中プログラムの下で審査する重大な革新を代表する製品に対して加速評価を提供することを目的としている。中小企業の製品は大企業よりも早くPrime計画に参加する資格があるかもしれません。Primeの称号を持つ候補製品のスポンサーは多くのメリットを得ることができ、しかし限らず、早期にEMAと積極的な監督対話を行い、臨床試験設計とその他の開発計画要素を頻繁に討論し、及びファイルを提出した後にマーケティング許可申請の評価を加速する。
小児科研究
EUのマーケティング許可を得る前に、スポンサーは、EMAが特定の製品の免除、カテゴリ免除、またはPIPに含まれる1つまたは複数の措置を承認しない限り、EMA承認に適合するすべての小児科集団をカバーするPIPに含まれるすべての措置を証明しなければならない。(EC)第1901/2006号条例、いわゆる“小児科条例”は、すべての販売許可手続のそれぞれの要求を規定する。会社が認可された薬物のために新たな適応,薬物形式あるいは投与経路を増加させようとした場合にも,この要求は適用される。EMAの小児科委員会、またはPDCOは、ある薬物の開発を延期することを承認する可能性があり、会社が成人に対する有効性および安全性を証明するのに十分な情報があるまで、小児薬の開発を延期することを可能にする。小児薬の開発を必要としない場合、または適切でない場合、PDCOは、高齢者人口のみに影響を与える疾患のような免除を与えることもできる。MAAを提出するか、または既存のマーケティング許可を修正する前に、EMAは、会社が各関連PIPに列挙された合意された研究および措置を実際に遵守していると判断する。
孤児薬の指定と排他性
条例(EC)第141/2000号及び条例(EC)第847/2000号によると、製品は、そのスポンサーが診断、予防又は治療を目的としていることを前提として、欧州委員会によって孤児薬として指定されることができ、(1)申請時にEUの影響が万分の5を超えない生命又は長期虚弱疾患に影響を及ぼすか、又は(2)EUで生命を脅かし、深刻な虚弱又は深刻な慢性疾患を脅かし、インセンティブ措置がなければ、EUでの販売に十分な見返りを与えることができず、必要な投資が合理的であることを証明することができる。上記のいずれの場合においても、スポンサーは、EUによって許可されていない関連疾患の診断、予防または治療の好ましい方法を証明しなければならない、または、そのような方法が存在する場合、薬剤は、その疾患の影響を受ける人に大きな利益をもたらすであろう。
小児科排他性
スポンサーがすべてのEU加盟国でマーケティング許可を取得した場合、または欧州委員会が集中手続きで付与されたマーケティング許可を取得し、小児科集団に対する研究結果が製品情報に含まれている場合、否定的であっても、補充保護証明書(SPC)の期限を延長することによって追加6ヶ月の合格特許保護期間を得る資格がある。
EUと他の管轄区域の特許期間の延長
EUはまたSPCを通じて特許期間を延長することを規定している。SPC獲得のルールと要求は米国と類似している。最高特許委員会は、特許の有効期限を予定期限の後5年まで延長することができ、1種の薬物に最大15年の市場排他性を提供することができる。場合によっては、小児科専門権が取得された場合、これらの期間は、さらに6ヶ月延長することができ、これは以下で詳細に説明される。SPCはEU全体で使用可能であるにもかかわらず、スポンサーは各国に基づいて申請しなければならない。EU以外のいくつかの他の外国司法管轄区域にも同様の特許期間延長権が存在する。
38
製品の定価決定を承認する
連合では、様々な国の価格設定と補償プログラムの差が大きい。一部の国では、補償価格を合意した後にのみ、製品を販売することができると規定されている。いくつかの国は、特定の候補製品のコスト効果を現在利用可能な治療法またはいわゆる衛生技術評価と比較して、精算または価格設定の承認を得るために、追加の研究を達成することを要求するかもしれない。例えば、EUは、その国の健康保険制度が精算を提供する製品範囲を制限し、人が使用する医療製品の価格を制御するための様々な選択をEU加盟国に提供している。EU加盟国は製品の具体的な価格を承認することができ、製品を市場に投入する会社の収益力を直接または間接的に制御する制度をとることもできる。他のEU加盟国は会社が自分の製品価格を固定することを許可したが、処方量を監視と制御し、医師に指導を発表して処方を制限した。最近、EUの多くの国が薬品割引要求を高め、各国が医療支出を管理しようとしていることに伴い、これらの努力は継続される可能性があり、特にEUの多くの国が深刻な財政危機と債務危機を経験した場合である。全体的には,医療コスト,特に処方薬の下り圧力が大きくなっている。そのため、新製品の参入にはますます高い壁が設けられている。政治、経済、規制面の事態は定価交渉をさらに複雑化させる可能性があり、補償を受けた後、定価交渉が継続される可能性がある。EUの各加盟国が使用する参考価格と平行貿易(すなわち、低価格と高価な加盟国間の裁定)は、価格をさらに下げることができる。薬品に対して価格規制や精算制限を実施することが保証されていない国は、これらの国で承認されれば、任意の製品に対して有利な精算と定価手配を許可する。
一般データ保護法規
米国の法律と同様に、ヨーロッパや他の国にも重要なプライバシーやデータセキュリティ法が適用されている。欧州経済地域または欧州経済圏に位置する個人の個人データ(個人健康データを含む)の収集、使用、開示、移転または他の処理、および欧州経済地域で行われる個人データの処理については、EUの一般データ保護法規(GDPR)の制約を受け、2018年5月25日に施行される。GDPRの範囲は広く,個人データを処理する会社に多くの要求があり,健康や他の敏感なデータを扱う会社に対してより高い要求が出されており,たとえば多くの場合,企業にはこのようなデータを処理する前に,敏感な個人データに関する個人の同意を得なければならないことが要求される.GDPRがGDPRの範囲内に属する個人データを処理する会社に課す義務は,個人へのデータ処理活動に関する情報の提供,個人データのセキュリティおよび秘密保護の保障措置の実施,データ保護官の任命,データ違反通知の提供,および第三者プロセッサの採用時に何らかの措置をとることである.GDPRは、米国を含む欧州経済圏以外の国への個人データの移転にも厳格な規定を実施し、データ保護当局がGDPR違反行為に巨額の罰金を科すことを許可しており、金額が大きい者を基準として、2,000万ユーロまたは世界の年収の4%に達する可能性のある罰金を含む。GDPRはまた、データ主体と消費者協会に個人訴訟権利を与え、監督当局に苦情を提出し、司法救済を求め、GDPR違反による損害について賠償を受けることができる。GDPRを守ることは厳格で時間のかかる過程であり,ビジネスコストを増加させたり,会社にそのビジネスやり方を変更して完全な遵守を確保したりする可能性がある.
人々は企業が個人データをEUから他の国に移す能力を心配してきた。2020年7月、EU裁判所はEU-米国プライバシー保護枠組み(以下、プライバシー保護枠組み)の無効を宣言し、プライバシー保護枠組みは、個人データを欧州経済区から米国に移転するための合法化メカニズムの一つである。CJEUの決定も、欧州経済区から米国への個人データの転送方式である標準契約条項の長期的な可能性に疑問を提起させた。CJEUのこの決定は、EUから米国へのデータ転送の一般的なより厳しい審査を招き、データプライバシー立法を遵守するコストと、サプライヤーや業務パートナーと適切なプライバシーとセキュリティ協定を交渉するコストを増加させた。
CJEUが決定した後,2022年10月,総裁·バイデンはEU−米国プライバシーの盾に代わるEU−米国データプライバシー枠組みを実施する行政命令に署名した。欧州委員会は2022年12月にEU-米国データプライバシーの枠組みのための十分な決定を開始し、現在ではEUから米国へのデータの送信を可能にする十分な決定を採択している。この開発はこの枠組みの下でこの点でデータ転送を可能にし、より広く言えば、国際データ転送をより直接的にするが、これらの規定は法廷で挑戦されている。この問題をめぐる持続的な不確実性はEUでの私たちの業務運営にさらに影響を及ぼすかもしれない。
2016年6月23日、イギリスの有権者はEU離脱、いわゆるイギリスの退欧に賛成票を投じた。他のイギリスの離脱に関する問題のように、個人データがイギリスでどのように保護されるか、個人情報がEUからイギリスに移行できるかどうかについては、懸案の問題がある。イギリスのEU離脱後、2018年のイギリスデータ保護法は、イギリスで行われている個人データ処理に適用され、GDPRが規定している義務と平行した義務が含まれている。英国の2018年のデータ保護法が2018年5月23日に王立で承認され、英国で施行されたが、“GDPR”によると、欧州経済圏から英国にデータを移すことが合法であるかどうかは不明である。この法案は“GDPR”の実施と補充である。英国政府は、英国からEU/欧州経済圏へのデータが影響を受けないことを保証するために、EU 27カ国および欧州経済圏加盟国のすべてがデータ保護において十分であると判断している。また、欧州委員会の最近の決定は、この決定が将来再評価される可能性があるにもかかわらず、イギリスがEUからイギリスにデータを移す上で“基本的に十分だ”と考えているようだ。イギリスとアメリカは枠組みについても合意しました
39
イギリスとアメリカの間で個人データを転送することは、英米データブリッジと呼ばれている。英米データ橋は未来に挑戦されるかもしれない。これらのデータ転送の持続的な不確実性は、将来の変化の可能性を含めて、我々の業務運営に影響を与える可能性がある。
GDPRに加えて,世界ではプライバシーやデータセキュリティ法が制定されている国が増えている.多くの法律はGDPRに倣って手本としているが、他の法律は異なるまたは互いに衝突する条項を含む。これらの法律は、私たちの臨床試験と任意の最終的な商業製品の販売と流通能力を含む、私たちの業務活動を展開する能力に影響を与えるだろう。
イギリスの離脱とイギリスの規制枠組み。
EUと英国は、2021年1月1日に臨時発効し、2021年5月1日に発効する貿易·協力協定における新たなパートナーシップについて合意した。貿易·協力協定は主に自由貿易に注目し、医療製品を含む商品貿易に関税や割当量を徴収しないことを確保する。その後、EUとイギリスは2つの異なる規制と法制度によって管理される2つの独立した市場を形成する。そのため、“貿易と協力協定”は貨物貿易障壁を最小限にすることを求めるとともに、イギリスが単一市場の一部ではないため、国境検査は避けられないことを認めている。2021年1月1日より,薬品·保健品規制機関(MHRA)はイギリスの薬品や医療機器の規制を開始し,国内法によるとイギリスはイングランド,スコットランド,ウェールズを含み,北アイルランドは北アイルランド議定書に基づいてEUが制定した規則に拘束され続けている。
2023年2月27日、イギリス政府と欧州委員会は、北アイルランド議定書、すなわち“ウィンザー枠組み”と呼ばれる新たな手配の代わりに、原則的な政治的合意を発表した。この新しい枠組みは、イギリスの医薬製品の規制を含む北アイルランド議定書の下の既存の制度を根本的に変えた。特に、MHRAはイギリス市場に輸送されたすべての医薬製品を承認する責任があり、EMAは北アイルランドへの医薬製品の承認に何の役割も果たすことはないだろう。MHRAは、イギリスで販売されているすべての医薬製品に単一の全英国MAを付与し、単一のパッケージでイギリス全土で販売できるようにする。ウィンザー枠組みは2023年3月24日にEU-イギリス連合委員会の承認を得たため、イギリス政府とEUは立法措置を制定し、法律にする。2023年6月9日,MHRAはウィンザーフレームワークの薬品について2025年1月1日から適用すると発表した。“2012年人類薬品条例”(SI 2012/1916)(改正された)はイギリスの薬品監督管理の主要な法律文書である。“人類薬品条例”はすでにイギリスSのEU離脱前から存在する医薬製品を管理するEU法律文書を国内法律に組み入れた。
EU法は二次立法により英国法に転換され、“保留されたEU法”として適用され続けている。しかし、CTRのような新しい立法はイギリスには適用されないだろう。イギリスの薬品規制枠組みの大部分は薬品の品質、安全性と有効性、臨床試験、MA、商業販売、薬品流通に関連しているため、EUからの指示と法規によって、イギリスの離脱はイギリスにおける私たちの候補製品の開発、製造、輸入、承認、商業化の規制制度に実質的な影響を与える可能性がある。例えば、イギリスはEMAからEU範囲内のMAを獲得する集中的な手続きのカバーを受けない。新たな国際認可枠組みは2024年1月1日から発効し,この枠組みにより,MHRAは新たなイギリスMAの申請を決定する際に,EMAと何らかの他の規制機関によるMAの承認決定を考慮する。
カバー範囲、定価、精算
FDAや他の政府機関の規制承認が求められる可能性のある任意の候補製品のカバー範囲や精算状態には、重大な不確実性がある。米国や他国の市場では,自分の病状に応じて処方治療を受けている患者や処方サービスを提供する提供者は,通常第三者支払者に依存してすべてまたは一部の関連医療費を精算している。患者は、保険を提供し、そのような候補製品を支払うのに十分なコストの大部分を清算しない限り、私たちが開発する可能性のある任意の候補製品を使用することはあまりできない。私どもの製品の販売は保証範囲の可用性と第三者支払者の精算が十分かどうかに大きく依存します。
米国では、第三者支払者は、MedicareおよびMedicaidのような政府当局または政府医療計画、管理型医療組織、個人健康保険会社および他の組織のようなプライベートエンティティを含む。第三者支払者が製品に保険を提供するかどうかを決定するプロセスは、支払者が薬品のために支払うべき販売率を設定するプロセスと分離することができる。第三者支払者は、承認されたリストまたは処方表上の特定の製品に保証範囲を制限することができ、FDAによって承認された特定の適応のすべての製品を含まない可能性がある。一部の第三者支払者は、特定の処方の保証範囲の事前承認(“事前許可”と呼ばれる)を要求することによって、特定の製品の使用を管理することができる(支払人が医療の必要性を評価することを可能にする)。また、第三者支払者が薬品に保険を提供することを決定することは、適切な販売率を承認することを意味するものではない。製品開発への投資の適切な見返りを実現するために、十分な第三者精算が得られない可能性があり、十分な純価格レベルを維持することができる。また、薬品の保証範囲と精算範囲は支払人によって異なる。1つの第三者支払人は、ある特定の薬品又はサービスを保証することを決定し、他の支払人も当該薬品に保険を提供することを保証することができないか、又は適切な販売率で保険を提供することを保証することができない。
40
安全性と有効性に加えて、第三者支払者は、価格に挑戦し、新製品やサービスの費用対効果を検討することが増えている。現在または将来の製品の保険や精算を獲得または維持するためには、私たちの製品の医療の必要性と費用効果を証明するために、高価な薬物経済学研究を行う必要があるかもしれない。このような研究は規制部門の承認を得るために必要な研究の補完になるだろう。第三者支払者が、製品が他の利用可能な療法と比較して費用対効果があると思わない場合、彼らは承認後にその製品をその計画の下の福祉としてカバーしないかもしれない、または、支払いレベルが会社にその製品を販売させて利益を得るのに十分ではない可能性があると思う場合がある。したがって、清算状態を獲得して維持するのは時間もかかるし、費用もかかる。
上述したように、政府や他の第三者支払者が保険および十分な補償を提供できない場合、規制部門の承認を得て商業販売を行う任意の候補製品の適正性が影響を受ける可能性がある。米国はコスト制御措置を強調し、私たちは薬品価格の圧力が増加すると予想している。保証政策と第三者精算料率は随時変化する可能性がある。1人以上の第三者支払者から規制承認を得た1つまたは複数の候補製品が有利な保証範囲と精算状態を獲得しても、将来的にはあまり有利ではない保証政策や精算率が実施される可能性がある。
もし私たちが将来適切な承認を得てアメリカで私たちの現在の任意の候補製品を販売すれば、私たちは連邦医療計画(例えばMedicaid)の保険を得るために、政府医療計画に基づいて、または特定の政府および個人購入者に割引またはリベートを提供する必要があるかもしれない。このような計画に参加することは私たちが特定の薬品価格を追跡して報告する必要があるかもしれない。もし私たちがそのような価格を正確に報告しなければ、私たちは罰金と他の処罰を受けるかもしれない。
アメリカ以外では、私たちが開発可能な任意の候補製品に十分なカバー範囲と支払いを提供することを確保することは挑戦に直面するだろう。多くの国で、処方薬の価格設定は政府によって規制されている。政府当局との価格交渉は、製品の監督マーケティング承認を受ける範囲をはるかに超えている可能性があり、私たちが開発する可能性のある任意の候補製品の費用対効果を他の利用可能な治療法と比較するために臨床試験を要求することができるかもしれない。このような臨床試験を行うことはコストが高く、私たちの商業化努力の遅延を招く可能性がある。
EUでは、これはまだ統一されたEU法律のテーマではないため、各国の定価と補償案の差が大きい。多くの国は、補償価格を合意した後にのみ、製品を販売することができると規定している。一部の国は追加の研究の完成を要求する可能性があり、特定の候補製品のコスト効果を現在利用可能な治療法と比較して(いわゆる衛生技術評価)、精算または価格設定の承認を得て、他の国はその価格を他のバスケットの国と連結することを要求するかもしれない。EU加盟国は製品の具体的な価格を承認することができ、会社が製品を市場に投入する収益力を直接または間接的に制御する制度を採用することもできる。いくつかの加盟国は価格をコントロールするほか、処方量をモニタリングし、制御し、医師に指導意見を発表し、処方を制限する。最近、EUの多くの国が薬品割引要求を高め、各国が医療支出を管理しようとしていることに伴い、これらの努力は継続される可能性があり、特にEUの多くの国が深刻な財政危機と債務危機を経験した場合である。全体的には,医療コスト,特に処方薬の下り圧力が大きくなっている。そのため、新製品の参入にはますます高い壁が設けられている。政治、経済、規制の発展は定価交渉をさらに複雑化させる可能性があり、補償を受けた後、定価交渉は継続される可能性がある。EU加盟国が使用する参考価格と平行貿易(低価格と高価な加盟国間の裁定)はさらに価格を下げることができる。薬品に対して価格制御や精算制限を実施することが保証されていない国は、これらの国で承認されれば、私たちのいかなる製品に対しても有利な精算と定価手配を許可する。
41
医療保健法律法規
医療保健提供者と第三者支払人は推薦と処方が発売承認された薬品の面で主要な役割を果たしている。提供者、コンサルタント、第三者支払者および顧客との間の配置は、広く適用される可能性のある詐欺および乱用、リベート、虚偽請求法律、医療提供者への支払いの報告、および患者のプライバシー法律法規および他の、我々の業務および/または財務的手配を制限する可能性のある医療法令の制約を受ける可能性がある。私たちが発売されている製品がある場合にのみ適用されるいくつかの法律と法規を含む、連邦および州医療保健法律および法規の制限を適用する
医療やその他の改革は
アメリカの医療業界と他の地域の主な傾向の一つはコストコントロールだ。過去数年間、連邦と州政府は薬品と生物製薬製品の定価制限、薬品と他の医療製品のカバー範囲と精算、政府制御及びアメリカ医療保健システムの他の改革に関する多くの提案を提出した。
2010年3月、米国議会は、2010年に“医療·教育調整法案”によって改正され、または総称して“PPACA”と呼ばれる“患者保護·平価医療法案”を公布し、これに加えて、政府医療計画下の薬品のカバー範囲や支払い方法の改正を含む。PACAが公布されて以来、他の立法改正も提案され、採択された。2011年8月、“2011年予算制御法案”(Budget Control Act Of 2011)などの法案は国会のための支出削減措置を制定した。赤字削減合同特別委員会の任務は、2013年から2021年までの間に少なくとも1.2兆ドルの赤字削減を提案することであるが、同委員会は必要な目標を達成できず、立法をいくつかの政府プロジェクトに自動的に削減することを触発した。これらの変化には、各年度に提供者に支払われる連邦医療保険総額が2%に達し、2013年4月に発効し、2031年まで続くことが含まれている。
現在の立法によると、医療保険支出の実質減幅は2022年の1%から本自動減額の最終年度の3%まで様々になる。総合支出法案は2022年12月に総裁·バイデンが署名して法律となり、医療保険計画の自動減額にいくつかの改正がなされた。総合支出法案第1001条は、2010年の4%の法定現金支払法またはPAYGO自動減額を2024年末まで2年延期する。“2021年米国救援計画法案”の公布により,医療保険計画を4%削減する計画が2023年1月に発効する。総合支出法案の医療補償タイトルには、2011年連邦医療保険自動減額の2%予算制御法案を6ヶ月~2032年度に延長し、2030年度と2031年度の支払減少率を低減する4163節が含まれる。
42
2012年の“米国納税者救済法”は,いくつかの医療サービス提供者に支払う連邦医療保険を減少させ,政府が提供者に多額の金を取り戻す訴訟時効を3年から5年に延長した。これらの法律は、連邦医療保険および他の医療資金のさらなる減少をもたらし、他の方法で、規制によって承認される可能性のある任意の候補製品の価格、またはそのような任意の候補製品の処方または使用頻度に影響を与える可能性がある。
PACAが公布されて以来、この法律の条項を廃止し、代替するために、多くの法的挑戦や国会行動が続くだろう。例えば、トランプ総裁が2017年12月22日に署名した“2017年減税·雇用法案”の公布に伴い、国会は“個人強制令”を廃止した。この条項は、ほとんどのアメリカ人が最低レベルの医療保険を購入することを要求する条項が2019年に施行される。2021年6月17日、米国最高裁はいくつかの州がPPACAに提出した最新の司法挑戦を却下したが、PPACAの合憲性を具体的に裁くことはなかった。PACAをめぐる訴訟と立法は継続される可能性があり、結果は予測不可能で不確実だ。
2021年1月、新たな行政命令は、連邦機関に、米国人の医療保険取得を制限するルールや他の政策を再検討し、この機会を保護し強化するための行動をとることを考慮するよう指示した。この命令によると、連邦機関は、新冠肺炎に関連する合併症を含む以前の疾患を有する人の保護の政策を弱めること、連邦医療補助および“全米政治行動計画”によるデモおよび免除は、仕事要件を含むカバー範囲または破壊計画を減少させる可能性のある政策、健康保険市場または他の医療保険市場を破壊する政策、連邦医療補助および“全米政治行動計画”への参加の難しさを増加させる政策、および扶養者への負担能力を含む保険または経済援助の負担能力を低減する政策を含む再検討を指示されるであろう。
薬品価格
アメリカでも処方薬の価格はずっと話題になっています。アメリカ議会は最近数回の調査を行い、州と連邦立法を提案し、公布し、薬品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、連邦医療保険と医療補助下の薬品コストを下げることを目的とした。2020年には、総裁·トランプ氏が処方薬のコスト削減を目的としたいくつかの行政命令を発表し、これらの命令のいくつかが条例に盛り込まれている。これらの規定には、価格最恵国モデルが実施され、連邦医療保険B部分のある医師が管理する薬品に対する支払いを他の経済先進国が支払う最低価格とリンクさせ、2021年1月1日から発効する臨時最終規則が含まれている。しかし、この規定は全国的な予備禁止令によって制限され、2021年12月29日、医療保険·医療補助サービスセンター(CMS)はそれを廃止するための最終規則を発表した。CMSは、この規則の発表に伴い、それは価値をMedicare B部分の薬品支払いのすべての選択に組み入れ、そして受益者が根拠に基づく看護を獲得する機会を改善することを探索する。
また、2020年10月、HHSとFDAは、各州と他のエンティティが804条の輸入計画、すなわちSIPを制定し、いくつかの処方薬をカナダから米国に輸入することを許可する最終規則を発表した。この規定は米国薬物研究やメーカー協会(PhRMA)の訴訟で挑戦されたが,裁判所でPhRMAがHHSを起訴する資格がないことが発見された後,2023年2月に連邦地域裁判所に却下された。9つの州(コロラド州、フロリダ州、メイン州、ニューハンプシャー州、ニューメキシコ州、ノースダコタ州、テキサス州、バーモント州、ウィスコンシン州)はカナダからの麻薬の輸入を許可する法律が採択された。いくつかの州は第804条輸入計画提案を提出しており、FDAの承認を待っている。FDAは2023年1月5日にフロリダ州のカナダ薬物輸入計画を承認した。
また、2020年11月20日、HHSは薬品メーカーのD部分下でスポンサーの値下げを計画する安全港保護を廃止し、法律が値下げを要求しない限り、直接或いは薬局福祉マネージャーを通過する法規を決定した。この規定はまた、販売時点での値下げを反映するための新しい避難港を作成し、薬局福祉マネージャーと製造業者との間のいくつかの固定料金手配のための避難港を作成する。裁判所の命令により,上記安全港の除去と増加は延期され,最近の立法は同規則の実施を2026年1月1日に一時停止した。2022年のインフレ削減法案、あるいはアイルランド共和軍と呼ばれ、さらにこの規定の実施を2032年1月1日に延期する。
2021年9月,総裁·バイ登が署名した行政命令により,衛生·公衆サービス部は薬品価格を下げる計画を発表した。この計画の主な特徴は,(A)メーカーとの薬品価格交渉を支援することにより,薬品価格がすべての消費者や医療システム全体により負担と公平になること,(B)サプライチェーンの強化を支援し,生体模倣薬や後発薬を促進し,透明性を増加させる市場改革により,処方薬業界全体の競争を改善·促進すること,(C)公共·民間研究を支援し,市場インセンティブを確保することで価値と入手可能な新しい療法の発見を促進し,科学的革新を促進し,より良い医療保健と健康改善を促進することである。
43
2022年8月16日、“アイルランド共和軍”は総裁·バイデンによって法律に署名された。新しい立法は連邦医療保険D部分に影響を与え、D部分は計画であり、連邦医療保険A部分または連邦医療保険B部分に加入する個人は毎月外来処方薬保険料を支払うことを選択することができる。他の事項を除いて、アイルランド共和軍はある薬品のメーカーに連邦医療保険との価格交渉(2026年から)を要求し、価格は交渉できるが上限があり、連邦医療保険B部分と連邦医療保険D部分に基づいてリベートを実施し、インフレを超える価格上昇を処罰し(2023年に初めて満了)、D部分のカバーギャップ割引計画の代わりに新しい割引計画を用いる(2025年から)。アイルランド共和軍は衛生·公衆サービス部長官が最初の数年に規制ではなく指導によってその多くの規定を実施することを許可した。
具体的には、価格交渉において、国会はMedicareが、競合する模倣薬または生体模倣薬を有さず、Medicare B部分およびD部分によって精算されるいくつかの高価な単一由来薬剤および生物製品のための低い価格を交渉することができる。CMSは、2026年からMedicare D部分によって支払われる10種類の高コスト薬剤の価格を交渉することができ、その後、2027年の15種類のD部分薬剤、2028年の15種類のB部分またはD部分薬剤、および2029年以降の20種類のB部分またはD部分薬剤の価格を交渉することができる。この規定は、少なくとも9年間承認された医薬品および許可13年の生物製品に適用されるが、単一のまれな疾患または疾患のための許可された医薬および生物製品には適用されない。また、この立法は、製薬業者が提供する価格が法律で規定された交渉で達成された“最高公平価格”以下でない場合、または値上げ幅がインフレを超える場合、製薬業者は民事罰金と潜在的な消費税を受けることになると規定している。この立法はまた、メーカーに連邦医療保険D部分の価格上昇幅がインフレを超えた薬品にリベートを支払うことを要求している。新法律では,2024年の連邦医療保険の自己払い薬品コスト上限は年間4000ドルと規定されており,その後2025年から年間2000ドルが上限となっている。
2023年6月6日,メルク社はHHSとCMSを提訴し,アイルランド共和軍の連邦医療保険に対する薬品価格交渉計画が憲法第5改正案に違反し,無償獲得となったと主張した。その後、他の一部の当事者は、アメリカ商会、百時美施貴宝会社、アメリカ薬物研究とメーカー、ノとノド会社、楊森製薬会社、ノワ製薬、アスリコンとブリンガー-インゲルハイム国際有限会社を含み、異なる裁判所にも訴訟を提起し、HHSとCMSに対して類似した憲法クレームを提出した。私たちはIreland共和軍のこのような条項と他の条項に関連した訴訟が継続され、結果的に予測可能で不確実だと予想する。
州レベルでは、各州はますます積極的に、価格または患者の精算制限、割引、ある製品への参入の制限、マーケティングコストの開示および透明性措置を含む薬品および生物製品の価格設定を制御するための法規を立法と実施し、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。さらに,地域医療機関や個別病院では,どの薬品やサプライヤーが処方薬や他の医療計画に含まれるかを決定するために入札プログラムが使用されるようになってきている。これらの措置が承認されると、私たちの製品に対する最終的な需要を下げたり、私たちの製品の価格設定に圧力をかけるかもしれません。私たちは将来、より多くの州と連邦医療改革措置を取ることが予想され、そのいずれも連邦と州政府が医療製品やサービスのために支払う金額を制限する可能性があり、これは私たちの候補製品に対する需要の減少または追加の価格設定圧力を招く可能性がある。
連合では、様々な国の価格設定と補償プログラムの差が大きい。一部の国では、補償価格を合意した後にのみ、製品を販売することができると規定されている。いくつかの国は、特定の候補薬物の費用効果を現在利用可能な治療法またはいわゆる衛生技術評価と比較して、精算または価格設定の承認を得るために、追加の研究を達成することを要求するかもしれない。例えば、EUは、その国の健康保険制度が補償を提供する製品範囲を制限し、人が使用する医療製品の価格を制御するためのオプションをその加盟国に提供している。EU加盟国は製品の具体的な価格を承認することができ、その製品を市場に投入する会社の収益力を直接または間接的に制御する制度をとることもできる。他のEU加盟国は会社が自分の製品価格を固定することを許可したが、処方量を監視と制御し、医師に指導を発表して処方を制限した。最近、EUの多くの国が薬品割引要求を高め、各国が医療支出を管理しようとしていることに伴い、これらの努力は継続される可能性があり、特にEUの多くの国が深刻な財政危機と債務危機を経験した場合である。全体的には,医療費,特に処方薬の下り圧力が大きくなっている。そのため、新製品の参入にはますます高い壁が設けられている。政治、経済、規制面の事態は定価交渉をさらに複雑化させる可能性があり、補償を受けた後、定価交渉が継続される可能性がある。EUの各加盟国が使用する参考価格と平行貿易、すなわち低価格と高価な加盟国の間の裁定は、価格をさらに下げることができる。薬品に対して価格規制や精算制限を実施することが保証されていない国は、これらの国で承認されれば、任意の製品に対して有利な精算と定価手配を許可する。
44
人的資本資源
チームのメンバー
2023年12月31日現在、私たちには436人の常勤チームのメンバーがおり、そのうち116人が医学博士号を持っている。これらのグループメンバーのうち,193人が研究と開発活動に従事し,40人が臨床と監督活動に従事し,65人が技術操作に従事し,48人が品質職,90人が一般·行政職を担当している。私たちのグループのメンバーたちは労働組合代表もなく、集団交渉協定のカバー範囲もない。
人的資本戦略
私たちの人的資本戦略は私たちの価値観から始まります
これらの価値観は,重篤な疾患を有する患者に生涯治療を提供するビジョンの実現に専念するチームを構築するのに役立つ。
私たちは次の優先順位がこのビジョンを達成するための鍵だと思う
婚約する
私たちは彼らの声が聞こえることを確実にするために定期的にフィードバックを収集する高度に参加したグループを持っている。私たちは参加調査、毎週のチーム会議、一対一の相互作用、そして公開フォーラムを通じてこれをします。2023年に内部敬業度調査を行い,91%のチームメンバーが調査に参加した。
総報酬(報酬と福祉)
私たちは人材を誘致して維持し続けるために私たちのチームメンバーを奨励するために努力している。私たちは私たちの報酬計画が競争力を持っていることを確実にするために市場評価を定期的に行うことでこれをする。私たちはまた定期的にチームメンバーと接触して、彼らが重視している利点を理解する。このようなフィードバックは私たちのグループの要求に能動的に対応するために、私たちの総報酬を発展させることができる。
健康度
私たちの人的資本戦略を実行するために、私たちのチームメンバーの福祉が第一だ。そのため,身体,精神,経済面の健康に重点を置いたいくつかの福祉を提供した。例えば、新冠肺炎の流行中、私たちは私たちのチームのメンバーとその家族を保護するために業務を改革した。このような変化は柔軟な労働時間と私たちが提供し続ける技術的支援を含む。私たちは引き続き私たちの健康製品を検討し、それを修正して、私たちのチームメンバーと私たちの業務の需要と一致することを保証します。
包括性多様性帰属感
私たちは引き続き包容と多様な文化を建設し、独特の視点を許容し、すべての人のために成長と発展の機会を創造し、関連患者コミュニティの需要を反映する。私たちの包括性、多様性、帰属感チームは、私たちのチームメンバーのために毎月のプロジェクトを制定して、彼らが参加するために、外部スピーカーとグループ討論を主宰し、異なる現地企業をサポートし、私たちの年間記念活動のためにコミュニケーションを作ります。私たちの毎月の番組を除いて、当社の聞く旅の直接投入によって構築され、私たちの価値観につながっている目標を達成するために行動しています
45
利用可能な情報
私たちのサイトはwww.beamTx.comで、私たちの投資家関係サイトはInvestors.beamTx.comです。私たちのサイト上の情報は参考にここに含まれていません。私たちは、材料を電子的にアーカイブまたは米国証券取引委員会に提供した後、合理的で実行可能な範囲内で、私たちの10-Kフォーム年次報告、10-Qフォーム四半期報告、8-Kフォームの現在の報告、および取引法第13(A)または15(D)節に提出または提供された報告書の任意の修正をできるだけ早く無料で提供します。米国証券取引委員会は、米国証券取引委員会に電子的に提出された報告書、依頼書、情報声明、その他の発行者に関する情報を含むインターネットサイトを有する。
投資家および他の人は、私たちのウェブサイトの“投資家およびメディア”の部分を含むが、これらに限定されない、米国証券取引委員会の記録文書、プレスリリース、および当社のウェブサイトのうちの1つまたは複数の方法で投資家に重要な情報を発表することに注意しなければならない。我々は,これらのチャネルや,XやLinkedInなどのソーシャルメディアチャネルを用いて,公衆に広く,非排他的に情報を配信することを実現し,FD法規による開示義務を遵守している.私たちが会社のウェブサイトや他のソーシャルメディアで発表した情報は重要な情報とみなされるかもしれない。そこで、私たちは、投資家、メディア、およびわが社に興味を持っている他の人が、会社のサイトの“投資家センター”の部分と私たちのソーシャルメディアチャネルで発表されている情報を見ることを奨励します。しかし、わが社のサイトやソーシャルメディアチャネルの内容は、本年度報告Form 10-Kの一部ではありません。
第1 A項。リスク要因です
わが社を評価する際には、当社の連結財務諸表と本10-K年度報告書の末尾の関連付記を含む、以下に説明するリスクおよび不確実性、および当社の10-K年次報告書に含まれる他のすべての情報をよく考慮しなければなりません。以下に説明するいかなる事件や発展が発生した場合、私たちの業務、見通し、経営業績、財務状況は重大な影響を受ける可能性があり、私たちの普通株の取引価格は低下する可能性があります。以下に説明するリスクと不確実性は私たちが直面している唯一の危険と不確実性ではない。私たちは今知らないか、あるいは私たちが現在どうでもいいと思っている他のリスクと不確実性はまた私たちの業務に悪影響を及ぼすかもしれない。
私たちの財務状況と追加資本需要に関連するリスク
設立以来、私たちは大きな損失を受けた。私たちは予測可能な未来に損失が出て、永遠に達成されたり利益を維持したりしないかもしれないと予想している。
設立以来、私たちは重大な運営損失が発生した。2023年、2022年、2021年12月31日までの年度の純損失はそれぞれ1.325億ドル、2.891億ドル、3兆706億ドルだった。2023年12月31日現在、私たちの累計赤字は12億ドルです。私たちは主に私たちの優先株を私募し、私たちの普通株を売却する収益と協力収入を通じて私たちの運営に資金を提供します。私たちは基本的にすべての努力を研究と開発に投入した。予測可能な未来には、巨額の費用と増加する運営損失が引き続き発生する見通しだ。私たちの純損失は四半期ごとに大きく変動するかもしれません。私たちの費用は大幅に増加すると予想されています
候補製品の臨床試験はまだ完了しておらず,まだ何年もかかると予想され,あれば候補製品の商業化を許可することができる。利益を維持するためには発展し直接または通過しなければなりません
46
協力者は、最終的に1つ以上の巨大な市場潜在力を持つ薬物を商業化する。これは私たちが一連の挑戦的な活動の中で成功することを要求し、候補製品を決定し、候補製品の臨床前研究と臨床試験を完成し、これらの候補製品のマーケティング許可を獲得し、製造、マーケティングと販売がマーケティング許可を得る可能性のある薬物を獲得し、任意の発売後の要求を満たすことを含む。私たちはこのような活動で決して成功しないかもしれないし、たとえ私たちが成功しても、利益を達成するのに十分な収入や十分な収入が生まれないかもしれない。基礎編集製品の開発に関連する多くのリスクや不確実性のため,将来の損失の程度やいつ利益が実現するかは予測できない(あれば).もし私たちが確実に利益を達成したら、私たちは四半期や年度の収益性を維持したり向上させることができないかもしれない。もし私たちが実現して利益を維持できなければ、会社の価値を下げ、資金を調達し、研究開発努力を維持し、業務を拡大し、あるいは運営を継続する能力を弱める可能性がある。
47
私たちは多くの追加資金が必要になるだろう。もし私たちが必要な時に資金を集めることができなければ、私たちは私たちの研究と製品開発計画や将来の商業化努力を延期、減少または廃止することを余儀なくされるだろう。
私たちは最近、私たちの運営費用、特に研究開発活動を減らす措置を取っていますが、これらの費用は過去に大幅に増加しており、候補製品の臨床試験を拡大し、マーケティング承認を求めるにつれて、これらの費用は再び増加する可能性があります。また、私たちが開発可能な任意の候補製品のためにマーケティング承認を得た場合、このような販売、マーケティング、製造、流通は協力者の責任ではないので、製品販売、マーケティング、製造、流通に関連する巨額の商業化費用が発生すると予想される。したがって、私たちは私たちの持続的な業務と関連した多くの追加資金を得る必要があるだろう。私たちは最近、潜在的な短期価値駆動要素と長期成長を支援するために、いくつかの研究と製品開発計画を延期、減少、廃止した。もし将来私たちが必要な時や魅力的な条件下で資金を集めることができなければ、私たちは再びプロジェクトの延期、減少、キャンセル、あるいは商業化努力の削減を迫られるかもしれない。
2023年12月31日現在、私たちの現金、現金等価物、有価証券は12億ドルです。私たちは、私たちの既存の現金、現金等価物、および有価証券が、少なくとも今後12ヶ月の運営費用と資本支出需要に資金を提供できるようにすると信じている。しかし、現在不明な要素のため、私たちの運営計画は変化する可能性があり、私たちは計画よりも早く追加資金を求める必要があるかもしれない。私たちの将来の資本需要は多くの要素に依存します
潜在的な候補製品を確定し、臨床前研究と臨床試験を行うことは時間がかかり、高価かつ不確定な過程であり、数年を要して完成する必要があり、しかも著者らは永遠に市場の許可を得て製品販売を実現するために必要なデータ或いは結果を生成できないかもしれない。また、候補製品の決定と開発に成功し、これらの候補製品が承認されても、ビジネス成功は得られない可能性がある。私たちの商業収入は、もしあれば、薬品の販売から来ます。これらの薬品は何年も商業化されないと予想されます。したがって、私たちは追加的な資金調達に依存して私たちの業務目標を達成し続ける必要があるだろう。私たちは受け入れ可能な条項で十分な追加融資を受けることができないかもしれないし、根本的にできないかもしれない。
どんな追加的な資金調達努力も、私たちの経営陣の日常活動に対する関心を移す可能性があり、これは私たちの候補製品を開発し、商業化する能力に悪影響を及ぼすかもしれない。私たちは受け入れ可能な条件で私たちに追加資金を提供するかどうか、あるいは根本的にできないかどうかを確認することができない。私たちは約束された追加資本源を持っていません。もし私たちが十分な量の追加資本をタイムリーに調達できない場合、あるいは私たちが受け入れられる条項で追加資本を調達することができなければ、私たちは開発を大幅に延期、削減、または停止しなければならないかもしれません。あるいは承認されれば、私たちの候補製品や他の研究開発計画は商業化されなければなりません。例えば、2023年10月には、いくつかのパイプライン計画の一時停止またはキャンセルをもたらすコスト削減措置を含む、ポートフォリオの再順序付けおよび戦略的再構成を発表した。
48
もし私たちが合意項目の下での支払いや他の義務を履行できなければ、私たちは現在、任意の未来の許可協定と協力協定も終了する可能性がある。私たちは、他の場合ではなく、私たちが開発する可能性のある候補製品のためのパートナーを探すことを要求されるかもしれないし、他の方法よりも不利な条項でパートナーを探したり、不利な条項で私たちが開発する可能性のある候補製品の権利を放棄したり、許可したりすることができます。そうでなければ、私たちは自分で開発または商業化された市場を求めることができるかもしれません。
もし私たちがタイムリーに資金を得ることができなければ、私たちは予想通りに私たちの業務を拡大したり、他の方法で私たちのビジネスチャンスを利用することができないかもしれません。これは私たちの業務、財務状況、運営結果に大きな影響を与えるかもしれません。上記のどの事件も、私たちの業務、将来性、財務状況と経営結果を深刻に損害し、私たちの普通株価格の下落を招く可能性があります。
追加資本の調達は私たちの株主に希釈し、私たちの運営を制限したり、私たちが開発する可能性のある技術や製品の権利を放棄することを要求するかもしれません。
これまで、相当な製品収入を生み出すことができれば、株式発行、債務融資、協力、戦略連合、許可手配の組み合わせで現金需要を満たす予定です。私たちは約束された外部資金源を持っていない。ある程度、私たちは株式または転換可能な債券を売却することで追加資本を調達し、あなたの所有権権益は希釈されるだろう。これらの証券の条項は、清算または他の特典を含むことができ、普通株主としてのあなたの権利に悪影響を及ぼすことができます。債務融資が可能であれば、追加債務を招く、資本支出を行うこと、配当金を発表すること、および可能な他の制限を含むような、特定の行動をとる私たちの能力を制限または制限する契約を含むことができる。
もし私たちが追加的な協力、戦略連合、または第三者との許可手配を通じて資金を調達すれば、私たちは私たちの技術、将来の収入源、研究計画、または私たちが開発する可能性のある候補製品の貴重な権利を放棄しなければならないかもしれないし、私たちは私たちに不利な条項で許可を与えなければならないかもしれない。もし私たちが必要な時に株式や債務融資を通じてより多くの資金を調達できなければ、私たちは私たちの製品開発や将来の商業化努力を延期、制限、減少または終了することを要求されるかもしれません。あるいは私たちが自分で開発し、マーケティングすることをより望んでいた候補製品の権利を与えることができます。また、将来的に協力·買収合意に達する可能性があり、この合意によれば、将来のマイルストーン支払い義務に関連した追加普通株を発行しなければならない。これらと未来に私たちのパートナーや協力者に発行される他の株は私たちの株主に深刻な希釈をもたらすかもしれない。
私たちの短い経営の歴史は、私たちの業務のこれまでの成功度を評価することが難しく、私たちの未来の生存能力を評価することも難しいかもしれません。
私たちは初期段階にある会社です。私たちは2017年1月に設立され、2017年7月に運営を開始した。今まで、私たちの業務は組織と会社のために人員、業務計画、資金を調達し、買収と開発、潜在的な候補製品を確定し、臨床前研究と臨床試験を開始することに限られている。我々の多くの製品開発プロジェクトはまだ臨床前や研究開発段階にあり、失敗のリスクが高い。私たちはまだ大規模、肝心な臨床試験、上場許可を得て、商業規模の薬物を生産したり、第三者代表を手配してこのようにしたり、商業化に成功するために必要な販売とマーケティング活動を行うことを含む任意の臨床試験を成功させる能力があることを証明していない。通常,新薬の開発は発見から患者の治療に利用可能になるまで約10年から15年を要する。したがって、私たちの未来の成功や生存能力に対するあなたのどんな予測も、私たちがもっと長い運営歴史を持っている時のように正確ではないかもしれません。
私たちの限られた運営の歴史、特に急速に発展する塩基編集や遺伝子編集の分野を考慮すると、私たちの技術や業界を評価し、私たちの将来の表現を予測することが困難になるかもしれません。私たちの運営会社としての短い歴史は、私たちの未来の成功や生存能力のどの評価も重大な不確実性の影響を受けるようにした。私たちは急速に発展する分野でスタートアップ企業がよく直面するリスクと困難に直面するだろう。もし私たちがこのような危険にうまく対応できなければ、私たちの業務は影響を受けるだろう。
また、新しい業務として、私たちは他の予見できない費用、困難、合併症、遅延などの既知と未知の要素に遭遇する可能性がある。私たちは最終的に研究に集中した会社からビジネス活動を支援できる会社に転換する必要がある。そのような移行で、私たちは成功しないかもしれない。
私たちは製品販売から収入を得たことがなく、永遠に利益を上げないかもしれない。
私たちが製品販売から収入を得て利益を達成する能力は、私たちが単独でまたはパートナーと開発に成功し、開発のために可能な候補製品の商業化に必要な規制承認を得る能力に依存します。私たちは今後数年は製品販売から収入を得ないと予想しています。もしあれば。私たちが製品販売から将来の収入を得る能力は、私たちまたは私たちの協力者が以下の目標を達成することに成功した能力に大きく依存する
49
私たちまたは私たちの協力者が開発する可能性のある1つ以上の候補製品が商業販売のために承認されても、任意の承認された候補製品の商業化に関連する巨額のコストが生じることが予想される。FDA、EMA、あるいは他の規制機関が現在予想されている基礎の上で臨床と他の研究を行うことを要求すれば、私たちの費用は予想を超える可能性がある。たとえ承認された製品の販売から収入を得ることができても、利益を得ることができない可能性があり、運営を継続するために追加の資金が必要かもしれない。
たとえ私たちが確実に利益を達成したとしても、私たちは四半期や年度の収益性を維持したり向上させることができないかもしれない。もし私たちが実現して利益を維持できなければ、会社の価値を下げ、資金を調達し、研究開発努力を維持し、業務を拡大し、あるいは運営を継続する能力を弱める可能性がある。
50
私たちが将来純営業損失の繰越といくつかの他の税務属性を利用する能力は限られているかもしれない。
私たちの歴史の中で、私たちは大きな損失を受けて、私たちは決して利益を達成しないかもしれない。引き続き課税損失が発生した場合、2017年以降に発生したいかなる未使用損失も無期限に繰り越して将来の課税収入を相殺し、2018年までに発生したいかなる課税損失も生成年度から20納税年度に繰り越す。また,研究開発税収控除を含めて営業税控除を継続しており,通常は生成年度から20納税年度を繰り越して,将来の部分納税義務(あれば)を相殺することができる。また、改正された1986年の国税法第382条及び383条は、企業が“所有権変更”を経験した範囲で税収属性を利用する能力を制限している。“所有権変更”は、通常、3年間のスクロール試験期間内に、5%以上の株主間の所有権変化が50%を超える(価値で測定される)と定義される。もし会社が所有権変更を経験した場合、所有権変更前の税収属性(例えば純営業損失と一般営業税控除)を利用して所有権変更後の課税所得額或いは税金が年間制限されていることを相殺し、通常は会社の所有権変更前の権益価値と計算し、ある規定の調整を経て、アメリカ国税局が毎月公表した長期免税率を乗じる。2023年12月31日現在、第382節の研究を完了し、2017年6月、2018年12月、2021年12月に歴史的な所有権変更を経験したことを確認しました。さらに、私たちは未来に株式所有権の変化のために所有権の変化を経験するかもしれないが、その中のいくつかの変化は私たちの制御範囲内ではない。したがって、もし私たちが課税純収入を稼ぐ場合、私たちは所有権変更前の純営業損失や他の税収属性を利用してアメリカ連邦の課税収入や税金を相殺する能力が制限される可能性があり、将来の納税義務が増加する可能性があります。私たちの会社の構造のため、私たちのいくつかの子会社或いは制御された実体による純営業損失或いはその他の税項属性は他の付属会社或いは制御された実体の課税収入或いは税金を相殺できない可能性がありますので、私たちは私たちの純営業損失及びその他の税務属性を利用して将来の課税収入或いは税務項目を相殺する能力は追加制限を受ける可能性があります。
もう一つのリスクは、法規の変化や他の予見できない原因により、私たちの既存の純営業損失または営業税控除が満期になる可能性があり、あるいは他の方法で将来の所得税負債を相殺できないということだ。以下の“全面税改正立法は、私たちの業務および財務状況に悪影響を及ぼす可能性がある”で述べられているように、2017年の減税および雇用法案、または米国連邦税率および純営業損失管理規則の変更を含むコロナウイルス援助、救済、経済保障法案またはCARE法案によって改正された税法は、純営業損失を利用して将来の課税収入を相殺する能力に著しく影響する可能性がある。州レベルでは、純営業損失や営業税控除の一時停止や他の方法での使用を制限する時期もある可能性があり、州の課税税を加速または永久的に増加させる可能性がある。また、一方の州で発生した純営業損失は、別の州で発生した収入を相殺するために使用することはできない。これらの理由で、利益を達成しても、私たちの純営業損失や税金控除を使用することで税金優遇を実現できないかもしれません。
ポートフォリオの優先順位付けや戦略的再編が期待されるコスト節約と収益を実現できなければ、私たちの業務の見通しや財務状況は悪影響を受ける可能性がある。さらに、優先順位と再構成は私たちの業務中断を招くかもしれない。
2023年10月、潜在的な短期的価値駆動要因および長期成長を支援するためのポートフォリオ優先順位および戦略再編を発表した。軽重緩急および再構成の実際の節約または収益は、予想を下回るか、または予想を大幅に下回る可能性がある。再構成活動は連続性の喪失,知識の蓄積,効率の低下を招く可能性もある.また、内部の軽重緩急や再編には、経営陣や他の従業員が多くの時間と労力を要する可能性があり、運営への関心を分散させる可能性がある。さらに、軽重緩急および再構成は、予期しない支出または負債および/またはログアウトをもたらす可能性がある。優先順位および再構成が予想されるコスト節約および利益の一部または全部を達成できない場合、私たちの現金資源は予想されたほど長く続かない可能性があり、私たちの業務、運営結果、および財務状況は実質的で不利な影響を受ける可能性がある。
51
発見·開発·商業化に関するリスク
塩基編集はまだ臨床でヒト治療に応用されていない新しい技術である。我々が講じている新しい療法の発見と開発方法は検証されておらず,適切な製品が生じない可能性がある。
我々は塩基編集技術を用いた潜在的な治療効果のある薬物の開発に焦点を当てている。近年遺伝子治療や遺伝子編集の分野で大きな進展が得られているが,塩基編集技術は新たであり,ほとんど検証されていない。私たちが許可を得て開発している技術はまだ何の臨床試験も完了していない。これらの技術開発候補製品の実行可能性に基づく科学的証拠を支持することは初歩的であり、限られており、その基礎編集と交付方式は斬新である。治療薬を安全に人体内に輸送する標的細胞を含む多くの問題を解決する必要があります離体するこれらの候補製品の効率および特異性を設定し、最適化し、これらの候補製品の治療選択性を確保する。基本編集薬を治療活性を有する薬物に変換するためには,いくつかの生物学的ステップが必要である。これらの処理手順は,個体によって異なり,ターゲット組織に基づいて異なる可能性がある.これらの差異は治療蛋白の異なるレベル、異なる活性、免疫原性或いは異なる組織分布を招き、更に基礎編集薬物の開発に固有のリスクを増加させる可能性がある。私たちがこれらのすべての問題を成功的に解決することを保証することはできず、予想されたスケジュールに従って私たちの臨床前研究あるいは臨床試験を進めることができる保証はない。
著者らは最近治療学を臨床に導入したが、著者らの未来の成功は塩基編集技術、細胞送達方法とこの技術の治療応用の成功開発に高度に依存している。他の遺伝子編集技術は臨床試験により進展しているが,それらは依然として様々な制限を受けており,これらの制限は我々の将来の成功に影響を与える可能性がある。我々は我々の最初の計画を変更または放棄することを決定するかもしれないが,新たなデータが利用可能であるため,基礎編集療法の開発で経験を得た。例えば、2022年11月に、私たちは、私たちの直接矯正“Makassar”方法、および私たちの鎌状細胞疾患計画のための私たちのHPFH方法の第2の波および第3の波を最適化することを決定したと発表した。私たちの技術が満足できる製品を生成することは確認できません。私たちの初期適応または私たちが追求している任意の他の適応において、これらの製品は安全で効果的、拡張可能、または利益があります。
基地編集分野の開発活動は現在,ある知的財産権の所有権や使用に関するリスクに直面しており,これらのリスクは米国で特許干渉訴訟を受け,ヨーロッパでは反対訴訟を受けている。私たちおよび私たちのライセンス者に適用可能な知的財産権のリスクに関する他の情報は、“-私たちの知的財産権に関するリスク”というタイトルの節を参照してください。
私たちは潜在的な候補製品を探して開発する努力で成功しないかもしれない。もしこのような努力が成功しなければ、私たちは決して商業舞台会社にならないかもしれないし、何の収入も生まれないかもしれない。
私たちの業務の成功は主に私たちの遺伝子編集プラットフォームに基づいて候補製品を識別、開発、商業化する能力にかかっている。私たちのいくつかの製品開発プロジェクトはまだ研究あるいは臨床前開発段階にあります。様々な理由から,われわれの研究計画は臨床開発の潜在的候補製品を決定できない可能性がある。私たちの研究方法は潜在的な候補製品の決定に成功できないかもしれません。私たちの潜在的な候補製品は臨床前に有害な副作用があることが証明される可能性があります体外培養実験または動物モデル研究では、それらはこのような実験または研究において有望な治療効果信号を示さない可能性があり、あるいはそれらは他の特性を有する可能性があり、候補製品が生産に適用されない、販売できない、または発売承認を得ることができない可能性がある。
また,基礎編集は我々の候補製品の組合せを迅速に拡張し,現在開発可能な候補製品を超えるようにすると信じているが,我々は候補製品の開発に成功しておらず,製品の組合せを拡張する能力は決して実現されないかもしれない.
いずれかの状況が発生した場合、私たちは、研究または開発事業を放棄し、1つまたは複数のプロジェクトに参加することを余儀なくされる可能性があり、これは、私たちの業務、財務状況、運営結果、および将来性に大きな悪影響を及ぼすだろう。新製品候補製品を決定する研究計画には大量の技術、財政、人的資源が必要である。私たちは最終的に成功しないことが証明された潜在的な計画や製品候補に私たちの努力と資源を集中させるかもしれないが、これは高価で時間がかかるだろう。
52
遺伝子編集分野は比較的新しい分野であり、急速に発展している。我々の研究開発は主に塩基編集技術を用いた遺伝子編集に集中しているが,他の遺伝子編集技術が塩基編集よりも著しく優れており,我々の業務に実質的な被害を与える可能性がある.
我々の研究開発は主に塩基編集を用いた遺伝子編集技術に集中している.他社も遺伝子編集技術の研究·開発に従事しており,亜鉛指ヌクレアーゼ,エンジニアリングヌクレアーゼ,転写活性化物質様エフェクターヌクレアーゼ,Cas 9ヌクレアーゼ,トランスポゾン編集,質点編集,“遺伝子作成”,特定部位標的素子によるプログラム可能添加などを用いている。塩基編集技術が遺伝子薬物の開発を招くことは肯定できない、あるいは他の遺伝子編集技術は薬物開発により良いあるいは魅力的とは考えられない。また,塩基編集に関する技術ではなく,主に遺伝子編集技術に注目することにした場合,これらの技術の権利を得ることができるかどうかは決定できない.私たちは現在、基礎編集技術の分野でコンサルティングやコンサルティングサービスを提供してくれたすべての創始者が、彼らが提供してくれたサービスについて発明義務を譲渡しているにもかかわらず、これらの発明義務の譲渡は制限されており、他の分野での彼らの仕事にも適用されず、それぞれの学術·研究機関によって作られた知的財産権にも適用されていない。これらの創始者がこれらの機関に割り当てられた知的財産権を得るためには、商業的に合理的な条項で得られないか、あるいは全く得られない可能性があるライセンス契約をこれらの機関と締結する必要がある。また,我々の創業者3人にはそれぞれのコンサルティングプロトコルにeスポーツ禁止条項があるが,eスポーツ禁止義務は人間の治療学の基礎編集分野に限られており,我々の創始者はすでに開発しており,将来的にはそのeスポーツ禁止義務を超えて我々の業務に競争力を持つ可能性のある新しい技術が開発される可能性がある.例えば,マサチューセッツ工科大学とブロード研究所のDavid·劉,馮章と彼らのそれぞれのチームは,トランスポゾン編集,塩基編集,良質編集技術を含む新たな遺伝子編集技術を開発しており,これらの技術は彼らのeスポーツ禁止義務分野を超えており,我々の業務と競合する製品を開発するために使用される可能性がある.これらの要素のいずれも、私たちのビジネス機会を減少または除去することができ、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
私たちの開発はまだ初期段階にあります。私たちの候補製品はまだ臨床前開発或いは早期臨床開発段階にあり、私たち或いは私たちの協力者は候補製品を商業化するのに数年の時間を要する。もし私たちの候補製品を臨床開発に進め、臨床開発を完成することができなければ、監督部門の承認を得られず、最終的に私たちの候補製品を商業化することができなければ、あるいはその過程で重大な遅延に遭遇すれば、私たちの業務は実質的に損害を受けるだろう。
私たちの開発はまだ初期段階にあり、私たちの将来の成功は私たちの基礎編集製品候補製品の成功開発と私たちの臨床試験結果に大きく依存しており、これらの結果はまだ完成していない。私たちが製品収入を作る能力は、私たちの候補製品の成功と最終商業化に大きくかかっていますが、これは決して起こらないかもしれません。製品収入は何年も起こらないと予想しています。私たちは現在どんな製品の販売からも収入を得ていません。私たちは永遠に適切な製品を開発したり商業化することができないかもしれません。
アメリカで臨床試験を開始することはFDAが著者らの試験新薬申請或いはINDを受けることに依存し、FDAと他の監督機関との討論に基づいて最終的に試験設計を確定する。FDAは過去と将来に再び私たちに追加の臨床前研究を完成することを要求し、そして著者らの臨床試験に対するFDAの他の要求を満たし、このような試験の開始或いは進展が延期されるかもしれない。例えば、FDAは2022年7月にBEAM-201 INDが臨床的に放置されていることを通知した。著者らはその後、FDAの正式な臨床座礁信頼を受け取り、FDAは臨床前研究といくつかの非標的編集実験の更なる分析に追加の対照データを提供することを要求した。我々は2022年11月にFDAに応答を提出し,2022年12月にFDAが臨床棚上げを中止したと発表した。
同様に、EUやEUでは、研究を行っている各加盟国の国家主管当局からCTAを獲得し、独立道徳委員会の積極的な意見を得なければならない。CTAが承認され,倫理委員会の積極的な意見が得られると,臨床試験はその特定の加盟国で行うことができる。欧州国家主管機関または他の規制機関が、臨床試験を開始する前に追加の臨床前研究を完了するように要求されている場合、または欧州国家主管機関または他の規制機関の他の要求を満たすことが要求された場合、そのような臨床試験の開始は延期される可能性があり、欧州国家主管機関または他の規制機関は、候補製品の臨床開発を許可しない可能性がある。
私たちがこれらの規制機関の指導意見を受け取り、組み入れても、FDA、ヨーロッパ国家主管機関、または他の規制機関は、彼らが臨床試験を開始する要求を満たしていることに同意しないかもしれないし、私たちのデータ、試験設計、または選択された臨床終点に対する受容可能な立場を変更することは、より多くの臨床前研究または臨床試験を完成させる必要があるかもしれないし、承認に対して私たちが現在予想しているよりも厳しい要求を提出する必要があるかもしれない。
53
さらに、開発中の市場承認政策の変化、追加法規、法規またはガイドラインの制定または公布の変化、または各提出された製品申請に対する規制審査の変化は、申請の承認または拒否の遅延を招く可能性がある。例えば、2022年12月、FDORAの採択に伴い、国会は、各新薬または生物製品の3期臨床試験または任意の他の“重要な研究”のための多様な行動計画を策定し、提出することをスポンサーに要求する。これらの計画はより多くの異なる患者群がFDA監督製品の後期臨床試験に参加することを奨励することを目的としている。また,2022年1月には,従来の臨床試験指令2001/20/ECの代わりに,新たな臨床試験条例(EU)第536/2014号がEUで施行された。この規定はEUの臨床試験の許可、進行と透明性を単純化して簡素化することを目的としている。臨床試験承認の調整手続きによると、1つ以上のEU加盟国で行われる臨床試験のスポンサーは、承認申請を提出するだけでよい。提出された材料は臨床試験情報システムを通じて提出され、これはヨーロッパ薬品管理局(EMA)が監督する臨床試験ポータルサイトであり、臨床試験スポンサー、EU加盟国の主管部門と公衆に使用することができる。
私たちが開発する可能性のある候補製品の商業化には追加の臨床前と臨床開発が必要になる;私たちの候補製品は、FDAが米国市場と欧州委員会に対してEMAに対してヨーロッパ経済地域で積極的な利益/リスク評価を提供すること、製造供給、能力、専門知識を得るか、または創造すること、商業組織を構築すること、および重大なマーケティング努力を含む、私たちの候補製品を販売する任意の司法管轄区域で規制およびマーケティング承認を得ることになる。私たちが決定し、開発した候補製品の成功は、以下の要素を含む多くの要素に依存する
もし私たちがこれらの活動のうちの1つまたは複数をタイムリーにまたは完全に成功させることができなければ、私たちは私たちが開発する可能性のある任意の候補製品を商業化することに成功したり、私たちの業務に実質的な損害を与えるかもしれない重大な遅延に遭遇するかもしれない。もし私たちの候補製品が規制部門の承認を得られなければ、私たちは運営を続けることができないかもしれない。
54
もし私たちが開発する可能性のある任意の候補製品または私たちが依存して管理するこれらの候補製品の配送方法が深刻な不良イベント、不良副作用または意外な特性をもたらす場合、このようなイベント、副作用または特性は、規制部門の候補製品の承認を遅延または阻止し、商業潜在力を制限し、または任意の潜在的なマーケティング承認後に重大な負の結果をもたらす可能性がある。
私たちはまだ私たちの候補製品の人体臨床試験を終えていません。また,限られた数の臨床試験のみが我々の技術と類似した塩基編集技術の使用に関与している。私たちが開発したすべての候補製品がいつ、あるいは人体で安全であることが証明されるかどうかを予測することはできない。遺伝医学領域では、過去にいくつかの遺伝子治療の重大な不良事件が発生し、報告された白血病、深刻な血液疾患と死亡病例を含む。患者のDNAおよび他の影響の不適切な編集は、リンパ腫、白血病または他の癌、他の重篤な疾患または症候群または他の機能異常の細胞を引き起こす可能性があるため、基本編集技術または私たちの候補製品のコンポーネントまたは送達方法が副作用を引き起こさないことを保証することはできない。
任意の基礎編集製品候補製品の1つの重大なリスクは、深刻な有害事象、不良副作用、または予期しない特徴を引き起こす可能性がある“非目標”編集が発生する可能性があることである。例えば,二次元左らである。マウス胚でテストを行うと、シトシン塩基編集は大量の非標的編集、すなわちDNA上の予期しない位置で編集を行うことが報告されている。この無意識の編集は“偽の脱アミノ”と呼ばれる。著者らは著者らの計画或いは未来のいかなる臨床研究においても非標的編集が発生しないことを確定することはできず、臨床前研究で観察されなかった副作用はこのような副作用が人類の臨床研究で発生しないことを保証することはできない。我々は,このような編集の発生頻度が非常に低くても,非ターゲット編集を検出できる解析手法を開発した.これらの分析を用いて,ベース編集製品候補における非目標編集が観察された.これらの分析の感度が増加するにつれて,このような非目標編集をより多く検出し続ける可能性がある.我々がこれまで観察してきた非目標編集が候補製品の安全性または利益に実質的な悪影響を与えているとは考えられないが,候補製品の非目標編集が安全性または有効性に悪影響を及ぼすことが将来的に発見されれば,候補製品を治療用製品として開発する能力は悪影響を受ける可能性がある。
DNAの恒久的編集や遺伝物質の候補品を携帯するための他の成分は,塩基編集療法に曝露した後に有害事象を遅延させる潜在的なリスクがある。また,塩基編集は恒久的に変化するため,副作用を認めた後でも治療を取り消すことはできない。またRees et al.はGrunewaldらと。我々は現在,C塩基エディタやA塩基エディタにおいてDNA塩基編集に用いられているデアミナーゼもRNA意外変異を引き起こすことが報告されており,このエディタが細胞中に存在すれば。
実験室環境で基礎編集を設計してその編集の特異性を向上させる能力を他の人たちと示しているにもかかわらず、私たちのプロジェクト努力が私たちが開発する可能性のある任意の候補製品で効果的であることを保証することはできません。たとえば,必要な変更を行うためのエディタを設計できない場合や,傍観者の編集が我々の編集の有効性を低下させる可能性がある.
私たちのいくつかのプログラムでは、LNPを使用して私たちの基本エディタを提供する予定です。LNPsは一定量で肝臓で酸化ストレスを誘導し,全身炎症反応を引き起こすことが証明されており,場合によっては致命的である可能性がある。我々のLNPsの最適化を継続することを目標としているが,我々のLNPsが悪影響を与えない保証はない.我々のLNPは、免疫反応、注入反応、補体反応、オプソニン反応、IgA、IgM、IgEまたはそれらの何らかの組み合わせを含む抗体反応、またはLNPに関連するいくつかの脂質またはポリエチレングリコールのポリエチレングリコールに対する反応のうちの1つまたは複数を促進することができる。著者らは薬物のいくつかの方面はmRNA或いは脂質からの免疫反応、及び肝臓経路内の副作用或いはmRNA或いはLNPの分解を引き起こす可能性があり、その中のいずれも著者らの現在或いは未来の1つ以上の臨床試験中に重大な有害事象を発生させる可能性がある。このようなタイプの副作用の多くは残されたLNPsに見られている。このような有害事象の根本的な原因には不確実性が存在する可能性があり,将来の臨床試験で副作用を正確に予測することは困難であり,われわれの計画の著しい遅延を招くであろう。
AAVまたはレンチウイルスを含むいくつかの基本エディタで使用される可能性があるいくつかのウイルスベクターは、疾患治療のための比較的新しい方法であり、既知の副作用もあり、将来的に追加のリスクが生じる可能性がある。過去に他の人による非AAVウイルスベクターによる臨床試験では,遺伝子治療が報告されている白血病や死亡例を含めていくつかの著明な副作用をきたしている。例えば,2021年2月,ブルーバード生物社はその第1/2期臨床試験で急性骨髄性白血病に意外な重篤な副作用が疑われることと,骨髄異形成症候群のSUSAR,LentiGlobinがレンチウイルスベクターを用いて鎌状細胞疾患を治療する遺伝子療法であることを報告し,fdaはこの試験を一時的に保留し,ZYNTEGLO(Beti−cel)の条件付きマーケティングを一時停止し,12歳以上の輸血依存型β地中海貧血を有する患者にも用いられ,これらの患者はβ0/β0遺伝子型を持たず,造血幹細胞移植に適している。しかし、現在まだヒト白血球抗原に関連するHSCドナーはいない。ウイルスベクターの他の潜在的な副作用は、免疫反応と挿入発癌、すなわち細胞成長或いは分裂に重要な遺伝子の近傍に機能遺伝子を挿入して細胞分裂を暴走させる過程を含む可能性があり、これは悪性転化のリスクを増加させる可能性がある。私たちが使用したベクターが同様の副作用または他の有害事象を示す場合、私たちは、このような技術を使用する任意の潜在的候補製品のさらなる臨床開発を停止または延期することを要求されるかもしれない。また,FDAは,レンチウイルスベクターは不良事象を遅延させる可能性の高いリスクがあるという特徴を有することを示している。このような遅延された有害事象は、AAVベクターを含む他のウイルスベクターにより低い比率で現れる可能性がある。
55
私たちの候補製品によって引き起こされる副作用および有害事象に加えて、BEAM-101で使用されるコンディショニング投与プロセスまたは関連するプログラム、および可能な他の離体する候補製品はまた不良副作用と有害事象を引き起こす可能性がある。また,代替調整案を開発しており,これらの調整案が我々の候補製品と互換性があるかどうかは予測できない.将来、これらの有害事象が使用されたコンディショニングレジメン、投与プロセス、または関連プロセスによって引き起こされないことが証明できない場合、FDA、欧州委員会、EMA、または他の規制機関は、さらなる開発を停止するように命令することができ、または、私たちの候補製品がそのようなスキーム、プログラム、またはプログラムを使用して任意またはすべての目標適応の承認を行うことを拒否または制限することができる。たとえ不良事象が候補製品または候補製品の投与と関係がないことを証明できても、このような事件は患者の募集、入選患者の臨床試験を完成する能力、あるいは監督管理の許可を得た任意の候補製品の商業実行可能性に影響を与える可能性がある。
もし私たちが開発した任意の候補製品が深刻な有害事象、副作用、または予期しない特徴に関連している場合、私たちはその開発を放棄する必要があるかもしれないし、その開発を深刻な有害事象、副作用、または他の特徴があまり一般的ではない、あまり深刻ではない、またはリスク収益の観点からより受け入れやすい特定の用途またはサブグループに制限する必要があり、いずれも私たちの業務、財務状態、運営結果、および見通しに実質的な悪影響を及ぼす。多くの最初に癌や他の疾患を治療する早期テストで希望を示した候補製品の多くはその後副作用が生じることが発見され,これらの候補製品のさらなる臨床開発を阻害した。
将来私たちが上記のいかなる有害事象が私たちの候補製品以外の要素によって引き起こされているかを証明できなければ、FDA、EMA、または他の規制機関は、私たちが任意のまたはすべての目標適応に対して開発できる任意の候補製品の開発を停止または拒否するように命令することができる。将来のすべての深刻な有害事象が製品に関連していないことを証明できても,このような事件は患者募集や入選患者の試験完了能力に影響を与える可能性がある。さらに、私たちが私たちが開発する可能性のある任意の候補製品の臨床試験を延期、一時停止、または終了することを選択または要求された場合、その候補製品の商業的将来性が損なわれる可能性があり、これらの候補製品から製品収入を生成する能力は延期またはキャンセルされる可能性がある。これらの状況のいずれも、候補製品を識別し開発する能力を損なう可能性があり、私たちの業務、財務状況、運営結果、および将来性を深刻に損なう可能性があります。
もし私たちが候補製品の開発に成功し、上場承認を得た場合、国際統一基準に基づいて定期的な利益リスク評価報告(PBRER)で利益-リスクプロファイルを評価することができるように、承認製品のリスクおよび承認適応における利益に関する新しいまたは出現している情報の全面的、簡潔かつ重要な分析を提出する必要がある。さらに、FDAまたは同様の規制機関(例えば、EMA)は、そのような候補製品を使用した治療の利点が各潜在的患者に対するリスクよりも大きいことを保証するために、リスク評価および緩和戦略(REMS)、またはリスク管理計画(RMP)のような同様の要件を採用することを要求することができ、製品が患者に配布されるリスクを概説する薬物ガイドライン、医療従事者向けのコミュニケーション計画、広範な患者監視、または高度に制御され、制限性が強く、典型的な業界よりもコストが高い流通システムおよびプロセスを含むことができる。FDA、EMA、または他の同様の規制機関は、候補製品の臨床治療効果および/または安全性をさらに特徴付けるために、特定の承認後試験を要求する可能性がある。さらに、もし私たちまたは他の人が後に私たちが開発した任意の候補製品による不良副作用が発見されれば、いくつかの潜在的な重大な負の結果を引き起こす可能性がある
これらの事件のいずれも、私たちが決定し開発する可能性のある任意の候補製品に対する市場の受容度を達成または維持することを阻止することができ、私たちの業務、財務状況、および運営結果に重大な悪影響を及ぼす可能性がある。
56
著者らはまだ臨床試験で著者らが提案した多くの送達方式と候補製品をテストしておらず、いかなる有利な臨床前結果も臨床試験で観察される可能性のある結果を予測することができない。
私たちはまだ臨床試験で私たちが提案した多くの送達方法をテストしていない。例えば,我々のいくつかの基本エディタでは,LNPを使用していくつかの基本エディタを提供する予定である.LNPsはいくつかの承認された治療法に使用されているが、塩基編集などの承認された遺伝子編集療法には使用されていない。そのほか、多くのウイルスを介した遺伝子治療方法と同様に、ある臨床試験患者の免疫システムは分娩成功を阻止し、潜在的にこれらの患者の治療結果を制限する可能性がある。たとえ私たちのすべての候補製品の初歩的な臨床試験が成功したとしても、これらの候補製品は臨床開発の後期段階で期待される安全性と有効性を示すことができないかもしれないが、これらの製品は臨床前研究と予備臨床試験に成功したにもかかわらず。
臨床試験中の薬物や生物製品の失敗率が高かった。製薬と生物技術業界のいくつかの会社は後期臨床試験で重大な挫折を経験し、早期臨床試験においても奮い立つ結果を得た。臨床前と臨床活動から得られたデータは異なる解釈を受ける可能性があり、FDA、EMA或いは他の監督管理機関は私たちの解釈に同意しない可能性があり、これは規制承認を延期、制限、或いは阻止する可能性がある。また、製品開発中の規制政策の変化を含む多くの要因により、規制の遅延や拒否に遭遇する可能性がある。
どのような有害事象も、進行中または計画中の臨床試験を延期、制限、または終了させる可能性があり、そのいずれも、私たちの業務、財務状況、運営結果、および将来性に重大な悪影響を及ぼす。
また,臨床前研究あるいは臨床試験の結果は後の臨床前研究あるいは臨床試験の結果を予測できない可能性がある。このような結果は,今後の臨床前研究や臨床試験でも類似した結果がある保証はない。同様に、たとえ私たちが現在と未来の候補製品のいずれかの予備臨床試験に成功しても、それらは臨床開発の後期段階で必要な安全性、純度と有効性データを生成できない可能性があり、それらは臨床前研究と予備臨床試験に成功したにもかかわらず。そのほか、臨床前と臨床データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品が臨床前研究と臨床試験で満足できると考えているが、しかし依然としてその候補製品のマーケティング許可を得られなかった。
もし私たちが臨床試験で患者の登録や治療に遅延や困難に遭遇した場合、必要な規制承認を受けることは延期または阻止される可能性がある。
FDA、EMA、または米国以外の他の同様の規制機関の要求に応じて、または必要に応じて所与の試験に適切な統計データを提供することができない場合、私たちまたは私たちの協力者は、私たちが決定または開発した任意の候補製品のために臨床試験を開始または継続することができず、これらの試験において十分な数の合格患者を見つけ、登録し、治療することができないかもしれない。著者らが最先端の計画の中でいくつかの稀な遺伝子定義疾患、及び著者らの小児科人群に対するいくつかの候補製品に対して、多くの要素、患者数が少ないこと及び患者資格のスクリーニングとテスト要求を制限することを含むため、登録は特に挑戦性があるかもしれない。さらに、患者がバイオテクノロジー、遺伝子治療または遺伝子編集領域に関連する有害事象の負の宣伝、患者集団のような競争的臨床試験、競合製品の臨床試験または他の原因で私たちの基礎編集試験に参加したくない場合、私たちが開発した任意の候補製品の募集患者、研究を行い、監督管理の承認を得る時間が遅れる可能性がある。また,我々の競争相手のいくつかは現在と将来候補製品の臨床試験を行っている可能性があり,これらの候補製品は我々が開発中と将来開発可能な候補製品と同じ適応を有しており,本来我々の臨床試験に参加する資格のある患者は競争相手の候補製品の臨床試験に参加することができる。登録された患者の治療も、著者らの実験の複雑さを含む多くの要素によって遅延または阻止される可能性がある。例えば,我々のBeacon臨床試験では,幹細胞を取得して編集や移植を行うための動員プログラムが求められている。患者は過去と未来に複数回の動員が必要かもしれないが、これは治療を遅延させるだろう。さらに、私たちのいくつかの試験にホイッスル行列および交差治療レジメン、例えば私たちのBeacon試験を含むことが要求されるので、1つの患者の治療における任意の遅延は、他の患者の治療遅延をもたらす可能性がある。
臨床試験患者の入選は他の要素の影響を受けている
57
私たちが外国で臨床試験を開始、登録、完成することに成功する能力は、外国で業務を展開するために独自の多くのリスクの影響を受ける
私たちの臨床試験における登録や治療遅延は、私たちが開発する可能性のある任意の候補製品の開発コストを増加させる可能性があり、これはわが社の価値を低下させ、追加融資を受ける能力を制限する。私たちまたは私たちの協力者が計画通りに臨床試験を行うのに十分な数の患者を募集または治療することが困難な場合、進行中または計画中の臨床試験を延期、制限または終了する必要があるかもしれませんが、いずれも私たちの業務、財務状況、運営結果、および将来性に悪影響を与えます。
私たちが決定し、開発した任意の候補製品の臨床試験が、規制機関が満足できる安全性および有効性を証明できなかった場合、または他の方法で積極的な結果が生じなければ、これらの候補製品の開発および商業化を完了する過程で追加のコストが生じたり、遅延が生じたり、最終的には達成できない可能性がある。
規制部門が決定し、開発した任意の候補製品の販売承認を得る前に、臨床前開発を完成させ、その後、広範な臨床試験を行い、人体における安全性と有効性を証明しなければならない。臨床試験は費用が高く,設計と実施が困難であり,完成まで数年かかる可能性があり,結果はまだ確定していない。1つまたは複数の臨床試験の失敗は、試験の任意の段階で発生する可能性がある。臨床前研究と早期臨床試験の結果は後の臨床試験の成功を予測できない可能性があり、臨床試験の中期結果も必ずしも最終結果を予測できるとは限らない。
そのほか、臨床前と臨床データはよく異なる解釈と分析の影響を受けやすい。多くの会社は彼らの候補製品が臨床前研究と臨床試験で満足できると思っているが、まだその候補製品のマーケティング許可を得られなかった。
私たちと私たちの協力者は、臨床試験中または臨床試験の結果において、多くの予見不可能な事件に遭遇する可能性があり、これらの事件は、私たちの上場承認を延期または阻止するか、または私たちが決定し、開発した任意の候補製品を商業化することを含むかもしれない
58
もし私たちまたは私たちの協力者が、私たちが開発した任意の候補製品に対して追加の臨床試験または他のテストを行うことを要求された場合、もし私たちまたは私たちの協力者が、私たちが開発した任意の候補製品の臨床試験または他のテストを成功させることができない場合、またはこれらの試験またはテストの結果が陽性でない場合、または適度な陽性である場合、または安全問題がある場合、私たちまたは私たちの協力者は、
もし私たちまたは私たちの協力者が臨床試験や他のテスト、あるいは市場の承認を得る上で遅延に遭遇すれば、製品開発コストも増加するだろう。いずれの臨床試験が計画通りに開始されるかどうか,再構成が必要かどうか,あるいは予定通りに完成するかどうか,あるいは全く知られていない。重大な臨床試験遅延は、私たちが開発する可能性のある任意の候補製品を商業化するための私たちの独占的な期限を短縮する可能性があり、私たちの競争相手が私たちの前に製品を市場に出すことを可能にし、私たちが開発する可能性のある任意の候補製品を商業化することに成功する能力を弱めるかもしれません。いずれも私たちの業務、財務状況、運営結果、将来性を損なう可能性があります。
59
私たちは、より利益的または成功可能性の高い候補製品または適応を利用することなく、特定の候補製品または適応を追求するために限られたリソースを費やす可能性がある。
私たちの財務、科学と管理資源が限られているため、私たちは研究プロジェクトと製品候補に重点を置いて、これらのプロジェクトと製品は私たちが多くの潜在的な選択の中で確定した特定の適応である。したがって、私たちは他の候補製品を探す機会を放棄または延期するか、または後により大きな商業潜在力を有することが証明された他の兆候を探す機会を放棄または延期するかもしれない。私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。私たちの現在と未来の研究開発計画と特定の適応候補製品への支出はいかなる商業的に実行可能な薬物も発生しないかもしれない。特定の候補製品の商業的潜在力や目標市場を正確に評価していなければ、協力、許可、または他の印税手配によって候補製品に価値のある権利を放棄する可能性があり、この場合、候補製品の独占開発権と商業化権利を保持することが私たちに有利になるかもしれない。例えば2023年10月に戦略的再編を実施しました離体するそして体内にあるBEAM-101や私たちの脱出調節策や体内にある造血幹細胞への輸送計画や体内にある塩基編集BEAM−302はα−1アンチトリプシン欠損症の治療に開発されている。私たちはまた、BEAM-201および他の潜在的なプロジェクトを含むベストプロジェクトを開発するための協力機会を探索する計画を発表した離体するCAR−T療法。私たちはその中のいくつかの計画を進めるための新しい協力パートナーを見つけるかもしれないが、私たちはタイムリーで受け入れ可能な条項、またはこれを根本的に成功させることができないかもしれない。そうでなければ、私たちはこれらの計画を推進するために十分な追加資金を集めることができないかもしれないし、内部資源分配や他の理由で、私たちはこれらの計画を開発することを放棄することにしたかもしれない。したがって、私たちはこのような計画の潜在力を利用する貴重な機会を逃すかもしれない。ある治療分野の候補製品に内部資源を割り当てることも可能であり,その分野では,実行可能なビジネス機会がないことが協力や事実上有利であることが証明されている。私たちの財政と人的資源を有効に活用できなかったいかなるものも、私たちの業務と運営を損なう可能性がある。
私たちはアメリカ以外の場所で臨床試験を行う予定です。FDAはこれらの地点で行われた試験のデータを受け入れない可能性があり,米国以外で試験を行うことで余分な遅延や費用に直面する可能性がある。
私たちはアメリカ以外の1つまたは複数の試験地点で1つまたは複数の臨床試験を行う予定だ。FDAや他の規制機関がその管轄外で行われた臨床試験を受けた試験データは,何らかの条件によって制限される可能性があり,まったく受け入れられない可能性もある。外国の臨床試験からのデータが米国での上場承認の唯一の根拠となることを意図している場合、FDAは通常、(I)データがアメリカの人口とアメリカの医療実践に適用されない限り、外国のデータのみに基づいて申請を承認しない。(Ii)試験は公認能力を有する臨床研究者によって行われ、GCP規定に適合する。および(Iii)データは、FDAによる現場検査を必要とすることなく、有効であると考えることができ、またはFDAがこのような検査を行う必要があると考えた場合、FDAは、現場検査または他の適切な手段によってデータを検証することができる。
また,外国試験データが承認の唯一の根拠としようとしなくても,FDAはこれらのデータを上場承認申請としての支援を受けず,試験設計が良好でない限り,GCP要求に応じて良好な操作を行うことができ,FDAは必要と考えた場合に現場検査により試験データを検証することができる。多くの外国の規制機関もまた似たような承認要求を持っている。また、このような外国裁判は、裁判を行う外国司法管轄区域に適用される現地法によって管轄される。FDAや同様の外国規制機関が米国や適用司法管轄区域以外で行われた試験データを受け入れることは保証されない。FDAまたは同様の外国の規制機関がこれらのデータを受け入れない場合、追加の試験が必要になり、これは高価で時間がかかる可能性があり、私たちが開発する可能性のある現在または未来の候補製品が適用される司法管轄区域で商業的な承認を得ることができない可能性がある。
アメリカ以外で臨床試験を行うことで、以下のようなリスクを含む多くのリスクに直面することになります
60
私たちが必要な臨床試験を終えても、いつ、あるいは規制部門の承認を得るかどうかは予測できません。アメリカや他の任意の管轄区域で開発された候補製品を商業化していますが、どのような承認も私たちが求めている適応範囲よりも狭いかもしれません。
適切な規制機関が候補製品を審査して承認するまで、私たちは候補製品を商業化することができない。私たちが開発可能な任意の候補製品が臨床試験でその安全性と有効性の終点に達しても、監督部門はその審査過程を適時に完成できない可能性があり、あるいは監督部門の承認を得ることができない可能性がある。FDA諮問委員会、EMA人が医薬品委員会またはCHMPまたは他の規制機関を使用して承認または制限を提案しない場合、追加の遅延を招く可能性がある。さらに、将来の立法や行政行動における追加的な政府規制、または製品開発、臨床試験、審査過程における規制機関の政策の変化による遅延や拒否に遭遇する可能性がある。
規制当局はまた、要求よりも限られた適応の候補製品を承認することができ、または狭い適応、警告またはREMSまたはRMPの形態で重大な制限を加える可能性がある。これらの規制機関は、使用条件に関連する予防または禁忌症を含むラベルを要求することができ、あるいは高価な発売後の臨床試験の表現に基づいて承認される可能性がある。また、規制部門は、私たちが開発する可能性のある任意の候補製品の商業化に必要なまたは望ましいラベル宣言を承認しないかもしれない。上記のいずれの状況も、私たちが開発する可能性のある任意の候補製品のビジネス見通しに重大な損害を与え、当社の業務、財務状況、運営結果、および将来性に重大な悪影響を及ぼす可能性があります。
EEAにおける米国FDAまたはEMAの上場承認が得られた場合、他の国または管轄区域規制機関の承認を得ることは確実ではない。また、一国で行われた臨床試験は他の国の規制機関に受け入れられない可能性があり、一国の規制承認は他の国の規制承認を保証することはできない。承認の流れは国/地域によって異なり、追加の候補製品テストと検証、および追加の行政審査期限が含まれる可能性があります。外国の監督管理機関の承認を求めることは私たちに困難とコストをもたらす可能性があり、追加の臨床前研究或いは臨床試験が必要であり、これは高価で時間がかかるかもしれない。各国の規制要求には大きな違いがある可能性があり、これらの国で開発された候補製品の発売が延期または阻止される可能性がある。外国規制機関の承認過程はFDA承認に関連するすべてのリスクに関連する。私たちは国際市場を含めてどの司法管轄区でも候補製品の販売を承認しておらず、国際市場で規制承認を受けた経験もありません。もし私たちが国際市場の規制要求を遵守できなかったり、必要な承認を得て維持できなかったり、あるいは国際市場の規制承認が延期されれば、私たちの目標市場は減少し、私たちの候補製品のすべての市場潜在力を達成する能力は達成できないだろう。
私たちが開発する可能性のあるすべての候補製品が市場の承認を得ても、それらは医師、患者、医療保険支払者、医学界の他の人が商業成功に必要な市場受容度を達成できないかもしれない。
私たちが開発する可能性のある任意の候補製品のビジネス成功は、それが医師、患者、第三者支払人、および医学界の他の人に受け入れられる程度に依存するだろう。遺伝子薬物の倫理、社会、法律への懸念、特に塩基編集技術は、追加の法規制を招き、我々が開発する可能性のある候補製品のマーケティングを制限または禁止する可能性がある。たとえ私たちが開発可能なすべての候補製品が市場の承認を得ても、それらは医師、患者、医療保険支払人、医学界の他の人の十分な市場受容度を得ることができないかもしれない。もし私たちが開発したすべての候補製品が商業販売に使用されることが許可された場合、市場のその製品に対する受け入れ度は複数の要素に依存する
61
私たちが開発する可能性のあるすべての候補製品が承認されても、これらの製品は十分な受容度を得ることができないかもしれません。私たちは著しい製品収入が生じないかもしれません。私たちは利益を上げることができないかもしれません。
将来的には、我々が開発する可能性のある任意の候補製品を販売およびマーケティングするために、販売およびマーケティング能力を確立することができない場合、これらの候補製品が承認された場合、商業化に成功できないかもしれない。
私たちは医薬製品を販売したり、マーケティングしたり、流通した経験もありません。私たちが販売とマーケティング責任を保留している承認された薬品を商業的に成功させるためには、販売とマーケティング組織を構築し、あるいはこれらの機能を第三者にアウトソーシングしなければならない。将来、私たちが開発したいくつかの候補製品が承認されれば、集中的な販売、マーケティング、および商業支援インフラを構築して、これらの製品を販売したり、私たちの協力者と一緒に販売活動に参加したりすることを選択するかもしれません。
私たち自身のビジネス能力の確立と第三者とのこれらのサービス提供の合意はリスクに関連している。例えば、販売員の募集と訓練や専門員の精算は高価で時間がかかり、どんな製品の発表も延期される可能性がある。販売チームを募集し、マーケティングや他の商業化能力を確立する候補製品の商業発表が何らかの理由で延期または発生していない場合には、これらの商業化費用を早期または不必要に発生させる。これは費用がかかるかもしれないし、もし私たちが私たちの商業化者を維持したり再配置できなければ、私たちの投資は損失するだろう。
私たちが開発した候補製品を商業化しようと努力するのを阻害するかもしれません
もし私たちが第三者と合意して、販売、マーケティング、商業支援、流通サービスを行えば、私たちの製品収入またはこれらの製品収入が私たちにもたらす収益性は、私たち自身が開発した任意の薬をマーケティングし、販売することよりも低いかもしれません。また、私たちは第三者との合意に成功し、私たちの候補製品を商業化することができないかもしれません。私たちは私たちに有利な条項で開発したり、そうできないかもしれません。私たちはこれらの第三者に対してほとんどコントロール権がないかもしれません。彼らのいずれも必要な資源と注意力を投入して、私たちの薬品を効果的に販売し、マーケティングすることができないかもしれません。もし私たちが商業化能力を構築することに成功できなければ、私たち自身も第三者と協力しても、私たちが開発可能な候補製品を商業化することに成功しないだろう。
62
技術が急速に変化する環境では、私たちは激しい競争に直面しており、私たちの競争相手は、私たちの前に規制部門の承認を得たり、私たちよりも安全で、先進的で、より効果的な治療法を開発する可能性があり、これは私たちの財務状況を損なう可能性があり、私たちが開発する可能性のある任意の候補製品の能力を成功させたり、商業化したりすることができる。
新薬製品の開発と商業化競争は激しい。そのほか、基礎編集と交付技術領域の特徴は技術が日進月歩で、競争が激しく、知的財産権に対する高度な重視である。私たちが将来開発や商業化を求める可能性のある任意の候補製品は、世界各地からの主要な製薬会社、専門製薬会社、バイオテクノロジー会社の競争に直面するだろう。潜在的な競争相手はまた学術機構、政府機関とその他の公共と個人研究組織を含み、これらの組織は研究を展開し、特許保護を求め、研究、開発、製造と商業化のための協力手配を確立する。
現在,多くの大手製薬やバイオテクノロジー会社が製品をマーケティング·販売しているか,あるいは研究計画のある疾患適応を治療するための製品が開発されている。競争力のある製品および療法のいくつかは、私たちの方法と同じまたは類似した科学的方法に基づいており、他のいくつかは完全に異なる方法に基づいている。
他にもCRISPR/Cas 9ヌクレアーゼ技術が使用されており,Cariou Biosciences,Editas Medicine,CRISPR Treeutics,Intellia Treeutics,Arbor BioTechnologies,Metagenomiがある。他のいくつかの会社では,亜鉛指,ArcusおよびTALヌクレアーゼ,Sangamo Biosciences,Precision BioSciences,Bluebird Bioscience,allgene Treeutics,Mammoth Bioscience,Cellectisを含む他のヌクレアーゼ系遺伝子編集技術も用いられている。また,Prime Medicine,Tessera Treeutics,Scribe Treeutics,Tome Biosciences,Life EDIT(ElevateBio社),PerkinElmer(従来のHorizon Discovery),Intellia Treeuticsなど,新たな遺伝子編集モデルが出現している。PerkinElmer,Metagenomi,Revrate,Intellia Treeuticsは基本編集技術を開発しているが,Tessera Treeuticsは移動可能な遺伝子を用いて遺伝子編集を行っている。また,様々な遺伝子療法,エピジェネティック調節,オリゴヌクレオチド,CAR−T治療法を利用した会社からの競争に直面している。
私たちが開発と商業化に成功したどの候補製品も、既存の治療法や将来発売される可能性のある新しい療法と競争し、これらの療法と新しい療法は、私たちが開発する可能性のある候補製品によって治療される同じ疾患の治療に承認される。これには、小分子療法、抗体療法および/またはタンパク質療法などの他のタイプの療法が含まれる可能性がある。
私たちの多くの既存または潜在的な競争相手は、単独またはパートナーと協力しても、研究開発、製造、臨床前テスト、臨床試験、監督管理許可およびマーケティング承認を得た製品の面で、私たちよりも多くの財務資源と専門知識を持っているかもしれない。製薬、生物技術と遺伝子治療業界の合併と買収はより多くの資源が私たちの少数の競争相手に集中する可能性がある。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。これらの競争相手はまた、合格した科学と管理者を募集と維持し、臨床試験場と臨床試験の患者登録を確立し、そして私たちの計画と相補的或いは必要な技術を獲得する上で私たちと競争している。もし私たちの競争相手が開発し商業化する候補製品が私たちが開発する可能性のある任意の候補製品よりも安全で、より効果的で、副作用がより少なく、より深刻ではなく、より便利で、あるいは私たちが開発する可能性のある任意の候補製品を時代遅れにしたり、競争力を持たなかったりすれば、私たちのビジネス機会は減少または消失するかもしれない。私たちの競争相手はまた、私たちよりも早く彼らの候補製品のためにFDA、EMA、または他の規制機関の承認を得るかもしれません。これは、私たちの競争相手が市場に入る前に強力な市場地位を確立することをもたらすかもしれません。また、私たちの競争相手が開発した技術は、私たちの潜在的な候補製品を経済的あるいは時代遅れにする可能性があり、私たちは私たちが開発する可能性のあるいかなる競争相手の候補製品もうまくマーケティングできないかもしれません。
さらに、我々の特許権の満了または成功の挑戦により、競争相手製品に関連する特許の有効性および/または範囲のより多くの訴訟に直面する可能性がある。私たちの競争相手の製品供給は、私たちが開発と商業化する可能性のある任意の候補製品の需要と私たちが受け取ることができる価格を制限するかもしれません。
63
公衆の遺伝子薬物に対する否定的な見方、特に遺伝子編集と塩基編集は、監督管理部門が著者らの潜在製品の承認及び/或いは需要に負の影響を与える可能性がある。
私たちの潜在的な治療製品にはヒトゲノムの編集が含まれている。我々の潜在製品の臨床と商業成功は遺伝子編集療法を用いたヒト疾患の予防或いは治療に対する公衆の理解と受け入れにある程度依存するであろう。公衆の態度は遺伝子編集の不安全、不道徳あるいは不道徳な説の影響を受ける可能性があるため、私たちの候補製品は公衆或いは医学界の受け入れを得ることができない可能性がある。例えば,1999年に1人の患者が遺伝子治療臨床試験中に死亡した後,公衆は遺伝子治療に強く反対した。臨床試験被験者の死亡はAAVメディアの投与に関連する合併症によるものであった。また,2020年,Audentes Treeuticsの臨床試験では,AT 132(AAV投与による遺伝子治療候補製品)によるX連鎖筋管性ミオパチー(XLMTM)の治療を検討した3名の患者が死亡した。両例の直接死因は敗血症,3例目は胃腸出血であり,いずれもAT 132服用後4−6週間以内に進行性肝機能障害が発生し,標準治療には反応しなかった。公衆の悪い態度は私たちが臨床試験を募集する能力に悪影響を及ぼすかもしれない。さらに、私たちの成功は、医師が処方した処方および彼らの患者が受けることを望む治療に依存し、これらの治療は、彼らがよく知っている既存の治療方法を代替または補充するために、私たちが開発可能な候補製品を使用することに関連し、より多くの臨床データを得ることができる。
そのほか、遺伝子編集技術を人類胚胎或いは人類生殖系に応用することに関する倫理問題のため、遺伝子編集技術は公衆討論ともっと厳格な監督管理審査を受けた。例えば,米国を含むいくつかの国の学術科学者は,基礎研究の一部としてヒト胚遺伝子を編集しようとする試みを報告している。また、報告によると、2018年11月、深セン南方科技大学生物系助教授、中国生物物理学研究員の何建奎博士は、最初のヒト遺伝子編集の赤ちゃん、双子の女の子を作ったと主張している。この説と、もう一つの説、すなわち何博士は第二の遺伝子編集者の妊娠を助け、その後中国当局の実証を得て、公衆、特に科学界の人々の負の反応を得た。この声明が提出された後、世界保健機関は、ヒト遺伝子編集のグローバル·ガバナンス·監督基準を策定するための新しい諮問委員会を設立し、ヒト遺伝子編集の研究を追跡するための新しいグローバル登録機関の計画を発表した。再生医学連盟はまた、遺伝子編集技術を使用する会社が認可した治療応用に遺伝子編集を使用する原則を発表した。
遺伝子編集技術に対する監督管理は司法管轄区によって異なる。米国では,2015年12月にFDAがこのような活動を禁止して以来,臨床応用のための生殖系編集が明確に禁止されてきた。イギリス、ヨーロッパの大部分の地域、中国、世界の多くの他の国も禁止令を持っている。米国では、米国国家衛生研究院は、この機関は、ヒト胚におけるいかなる遺伝子編集技術の使用にも援助しないと発表し、既存の複数の立法および規制機関が、研究目的またはヒト胚を破壊するための研究にヒト胚を創出するための資金を使用することを禁止するディキ-ウィック修正案を含むこのような仕事を禁止することを指摘した。イギリスの法律では遺伝子組換え胚を女性に移植することは禁止されているが,2016年以来,イギリスではミトコンドリア代替療法が許可されている。また,イギリスでは,ヒト受精や胎生学管理局の許可を得た研究実験室で胚を変化させることができる。他のいくつかのヨーロッパ諸国では、胚の研究はもっと厳格に統制されている。
また、2016年2月に米議会に提出された年間世界的脅威評価報告では、米国国家情報院取締役は、欧米諸国とは異なる規制基準で行われた遺伝子編集研究は、大量破壊兵器を含む潜在的有害生物剤や製品を産生するリスクを増加させる可能性があると述べている。遺伝子編集技術の分布が広く、コストが低く、発展速度が加速していることから、その故意或いは無意識の濫用は経済と国家安全に深い影響を与える可能性があると指摘した。
私たちは私たちの技術を使用してヒト胚胎またはヒト生殖系を編集しないが、このようなヒト胚胎における遺伝子編集技術の使用に関する公開討論およびより厳格な規制審査は私たちの候補製品の開発を阻止または延期する可能性がある。より厳格な政府法規または否定的な世論は、私たちの業務や財務状況に負の影響を与え、候補製品の開発および商業化、または私たちが開発する可能性のある任意の候補製品の需要を遅延または損害する可能性がある。我々の前臨床研究または臨床試験または我々の競争相手または遺伝子編集技術を使用する学術研究者の有害事象は、最終的には、我々が識別し開発する可能性のある候補製品に起因することができなくても、遺伝子宣伝は政府規制の増加を招く可能性があり、公衆は、私たちが識別し開発する可能性のある潜在的候補製品の試験または承認に対して規制遅延が生じる可能性があり、承認された候補製品に対してより厳しいラベル要求を提出し、そのような任意の候補製品に対する需要を減少させる可能性がある。第三者や政府が遺伝子編集技術を利用して米国の国家安全を脅かす生物製剤や製品を開発することも,我々に同様の負の影響を与える可能性がある。
64
いずれの候補製品を商業化することができても,これらの製品は不利な定価法規,第三者精算やり方,医療改革の影響を受ける可能性があり,業務を損なうことになる。
新薬の発売承認、定価と精算を管理する規定は国によって異なる。一部の国は薬品の販売価格が発売前に承認されることを要求している。多くの国で、定価審査期間はマーケティングまたは製品許可を承認した後から始まる。一部の外国市場では、処方薬の定価は初歩的な承認を得た後も、政府の持続的なコントロールを受けている。そのため、ある種の薬物の特定の国/地域での発売許可を得ることができるかもしれないが、その後、価格法規の制約を受ける可能性があり、これらの法規は私たちの薬品の商業発売を延期し、長期にわたって持続する可能性があり、その国/地域でこの薬品を販売することによって生じる収入に負の影響を与えるかもしれない。不利な価格設定制限は、私たちが開発する可能性のある任意の候補製品が市場承認を得ても、私たちが開発する可能性のある1つ以上の候補製品への投資を回収する能力を阻害するかもしれない。
私たちがいかなる薬物を商業化することに成功した能力はまた、政府当局或いは医療保健計画、個人健康計画、その他の組織によるこれらの薬物と関連治療の清算程度にある程度依存するであろう。政府当局と第三者支払者、例えば個人健康計画は、彼らがどのような薬物を支払い、精算レベルを確立するかを決定する。アメリカの医療業界と他の地域の主な傾向の一つはコストコントロールだ。政府当局と第三者支払者は,特定の薬物のカバー範囲や精算金額を制限することでコストを抑制しようとしている。例えば、2022年の“インフレ低減法案”(IRA)には、Medicare Part D中のすべての薬品に新しいメーカーの財務責任を適用する条項が含まれており、米国政府がある高コストの薬物と生物製品の定価について交渉することを可能にする条項、模倣薬や生物学的に類似した競争の条項がなく、会社がインフレよりも速い薬品のためにMedicareにリベートを支払うことを要求する条項と、福祉薬局マネージャーを受益者にリベートすることを要求するリベート規則とが含まれている。私たちはまたアイルランド共和軍が私たちの業務と医療産業全体に及ぼす影響を予測できない。
ますます多くの第三者支払者も医療製品の価格に挑戦しており、製薬会社に価格に基づいて予め定められた割引を提供することを求めている。新たな医療製品がカバーされれば,強化された使用管理制御を受ける可能性があり,医療が必要な場合にのみ製品が使用されることを確保することを目的としている。このような利用管理制御は,処方に関する管理負担や処方者や患者にカバー範囲の不確実性を増加させ,処方や医療製品の使用を阻害する可能性がある。私たちは私たちが商業化されたどんな薬でも精算できることを確実にすることができない。もし精算ができれば、精算レベルが十分かどうか。精算は私たちが市場で承認された任意の候補製品の需要や価格に影響を及ぼすかもしれない。清算が得られない場合や限られたレベルに限られていれば、マーケティングの承認を得た任意の候補製品を商業化することに成功できないかもしれません。
新たに承認された薬物の精算には大きな遅延がある可能性があり,カバー範囲はFDA,EMAあるいは米国以外の他の規制機関がこの薬物を承認する目的よりも限られている可能性がある。さらに、精算を受ける資格があることは、すべての場合に任意の薬物が支払われることを意味するわけではなく、または支払いの費用は、研究、開発、製造、販売、および流通を含む私たちのコストをカバーする。適用すれば、新薬の一時精算レベルも私たちの費用を支払うのに十分ではないかもしれませんし、恒久的にならないかもしれません。販売率は薬物の使用や臨床環境によって異なる可能性があり,すでに低コスト薬品のために設定された精算レベルに基づいている可能性があり,他のサービスの既存支払いにも組み込まれている可能性がある。政府医療計画又は個人支払者が要求する強制割引又はリベート、並びに将来的に米国価格以下で販売される可能性のある国から医薬品を輸入することを制限する法律の緩和により、薬品の純価格が低下する可能性がある。私たちは政府援助と個人支払者から、私たちが開発する可能性のある任意の承認された薬物の保証範囲と利益の支払率を迅速に得ることができません。これは、私たちの経営業績、薬品の商業化に必要な資金を調達する能力、および私たちの全体的な財務状況に実質的な悪影響を及ぼすかもしれません。
私たちの技術の新規性、および私たちが開発する可能性のある任意の候補製品が単一投与または限られた数の投与において治療利益を提供する潜在力のため、私たちはこれらの候補製品の価格設定と精算に関連する不確実性に直面している。
我々の最初の目標患者数は比較的少ないため,我々が開発する可能性のある任意の候補製品の定価や精算は,承認されれば必要な商業インフラを支援するのに十分でなければならない。もし私たちが十分な精算レベルを得ることができなければ、私たちがこのような候補製品をマーケティングして販売する能力は不利な影響を受けるだろう。我々が開発可能な任意の候補製品に関するサービス(例えば,患者に私たちの候補製品を管理する)の精算方法やレベルも重要である。このようなサービスの精算不足は、医師や支払人のボイコットを招き、私たちが開発する可能性のある候補製品をマーケティングしたり販売したりする能力に悪影響を及ぼす可能性がある。しかも、私たちは十分な価値を達成するために新しい精算モデルを開発する必要があるかもしれない。支払者はこのような新しいモデルを採用できないか望まない可能性があり,患者はこのモデルが負担する費用の一部を負担することができないかもしれない。もし私たちがこのような新しいモデルが必要だと思うなら、私たちは開発に成功していない、あるいはそのようなモデルが支払人に採用されていなければ、私たちの業務、財務状況、経営結果、見通しは不利な影響を受けるかもしれない。
65
遺伝子薬が規制部門の承認を得た場合,例えば開発を求めている薬剤は,1回の投与コストが膨大になると予想される。政府や個人支払者のカバーと補償が,多くの患者がこれらの治療を負担できる鍵となると予想される。したがって、このような候補製品の販売は、私たちが開発可能な任意の候補製品のコストが、国内でも海外でも、政府当局、個人健康計画、および他の第三者支払者によってどの程度支払われるかに大きく依存するであろう。支払者たちは単一の行政管理のために高い価格を支払いたくないかもしれない。第三者支払者の製品使用状況の決定を含む第三者支払人の保証範囲と精算はいくつかの要素に依存する可能性がある
第三者支払人から製品の保険と精算を得ることは時間がかかり高価な過程であり、支払人に科学的、臨床的、費用効果を支援するデータを提供する必要があるかもしれない。新たに承認された製品の第三者カバーや精算に関する不確実性が大きい。私たちは保証と補償の承認を得るのに十分なデータを提供できないかもしれない。保険や精算が得られない場合、あるいは限られたレベルに限られていれば、私たちが開発する可能性のある任意の候補製品を商業化することに成功できないかもしれません。保険を提供しても、承認された精算金額は、私たちの投資に十分な見返りを達成するのに十分ではないかもしれません。
また,医療コストは全体的に下り圧力が大きくなり,特に処方薬,外科手術,その他の治療が行われている。そのため、私たちのような新製品候補製品の参入にはますます高い壁が設けられている。もし私たちが十分な精算レベルを得ることができなければ、私たちが開発する可能性のある候補製品のマーケティングと販売に成功する能力は損なわれるだろう。
もし私たちが開発する可能性のある候補製品の市場機会が私たちが思っているより小さいなら、私たちの潜在収入は不利な影響を受けるかもしれません。私たちの業務は影響を受けるかもしれません。私たちが開発する可能性のある多くの候補製品のターゲット患者群が少ないため、利益と成長を維持するために、患者を成功的に識別し、顕著な市場シェアを得ることができなければならない。
我々は,まれな遺伝子定義疾患の治療に大部分の研究と製品開発を集中させている。我々が開発可能な多くの候補製品は単一変異に対するものであることが予想され,したがって,関連する患者集団は小さい可能性がある。これらの疾患を有する人数および我々が開発する可能性のある候補製品治療から利益を得る可能性のあるこれらの疾患患者のサブセットの予測は、推定に基づいている。これらの推定は正しくないことが証明される可能性があり,新しい研究はこれらの疾患の推定発症率や流行率を変化させる可能性がある。米国、ヨーロッパ、および他の地域の患者数は予想を下回る可能性があり、患者は私たちが開発する可能性のある候補製品の治療を受け入れられないかもしれない、あるいはますます識別や獲得が困難になる可能性があり、これらはすべて私たちの業務、財務状況、運営結果、および将来性に悪影響を及ぼすだろう。また,我々が開発したどの候補製品も標的疾患を治癒する可能性があるため,患者から経常的収入を得ることができず,根治療法により患者群の罹患率を枯渇させる可能性がある。
どの患者が私たちが開発した任意の候補製品の治療から利益を得ることができるか、あるいはその過程で重大な遅延に遭遇する可能性があるかどうかを決定することができなければ、私たちは私たちが開発する可能性のある任意の薬物のすべての商業的潜在力を達成できないかもしれない。
私たちの成功は、私たちが開発した任意の薬物治療から利益を得る可能性のある患者を識別する能力にある程度依存する可能性があり、これは、特定の配列が存在するかどうかを決定するために、これらの潜在的患者のDNA分析を行う必要がある。私たちまたは私たちに協力を依頼した第三者がそのような患者の識別に成功しなかった場合、またはこの操作を実行する際に遅延があった場合、:
66
我々が開発したどの候補製品も、治療から利益を得る可能性のある患者を識別するために、セット診断を使用する必要があるかもしれない。もし私たちが開発可能な任意の候補製品の安全かつ有効な使用がセット診断製品に依存する場合、私たちが開発、識別、識別、または私たちの候補製品と一緒に使用されるセット診断製品の規制承認または許可を得ることができない場合、上場承認を得ることができないか、またはマーケティング承認が延期される可能性がある。診断プログラムをセットにしたメーカーを決定し、メーカーと合意しても候補製品の開発が遅れる可能性があります。
これらの要素のため、私たちは私たちが確定し、開発する可能性のある任意の候補製品の商業潜在力を成功的に開発し、実現できない可能性があり、私たちの業務、財務状況、経営業績、将来性は重大な悪影響を受けるだろう。
私たちに対する製品責任訴訟は、私たちが重大な責任を負うことを招き、私たちが開発する可能性のある任意の薬物の商業化を制限することができるかもしれない。
我々が開発する可能性のある任意の候補製品が人体臨床試験で試験されることに関連する固有製品責任曝露リスクに直面しており,開発可能な任意の薬物を商業化販売すれば,より大きなリスクに直面するであろう。もし私たちが自分自身を弁護することに成功できなければ、私たちの候補製品や薬物による傷害のクレームに反対すれば、私たちは重大な責任を招くかもしれない。是非曲直や最終結果にかかわらず、賠償責任は
私たちは製品責任保険を維持しているが、それは私たちが発生する可能性のあるすべての責任をカバーするのに十分ではないかもしれない。臨床試験を開始し,任意の薬物の商業化に成功した場合,保険カバー範囲を増やす必要があると予想される。保険範囲はますます高くなっています。私たちは可能などんな責任にも対応するために、合理的な費用や十分な金額で保険範囲を維持することができないかもしれない。
もし私たちまたは私たちが雇用した任意のCMOおよびサプライヤーが環境、健康、安全法律法規を遵守できなかった場合、私たちは罰金や処罰を受けたり、コストを発生させたりする可能性があり、これは私たちの業務成功に実質的な悪影響を及ぼす可能性がある。
私たちと私たちが雇用しているどんなCMOと供給者も、多くの連邦、州と地方環境、健康と安全の法律、法規、許可要件、それらの管理実験室手続き;危険で規制された材料と廃棄物の発生、処理、使用、貯蔵、処理と処理、地面、空気と水中への危険物質の排出と排出、そして従業員の健康と安全を守らなければならない。私たちの行動は化学物質、生物学的、そして放射性物質を含む危険で燃えやすい材料の使用に関するものだ。私たちの業務はまた危険な廃棄物を発生させるだろう。私たちは一般的に第三者と契約を結び、このような材料と廃棄物を処理する。私たちはこのような材料が汚染や傷害をもたらす危険を除去することができない。もし私たちが危険な材料を使用して汚染や損傷をもたらしたら、私たちはそれによるいかなる損害に責任を負い、いかなる責任も私たちの資源の範囲を超える可能性がある。いくつかの環境法により、私たちは現在または過去の施設および第三者施設の任意の汚染に関する費用に責任を負わなければならないかもしれない。私たちはまた民事や刑事罰金と処罰に関連した巨額の費用を発生させるかもしれない。
適用される環境法律や法規を遵守することは費用がかかる可能性があり、現在または未来の環境法律と法規は私たちの研究と製品開発努力を損なう可能性がある。しかも、私たちはこのような材料や廃棄物が意外なダメージや汚染をもたらすリスクを完全に除去することはできない。私たちは私たちのコストと支出を支払うために労働者補償保険を維持していますが、危険材料の使用で従業員が怪我をする可能性がありますが、潜在的な責任を支払うのに十分ではないかもしれません。また、私たちは生物または危険廃棄物保険を保証していますが、このような保険範囲は損失を補うのに十分ではないかもしれません。私たちの財産、傷害、一般責任保険は生物または危険廃棄物の暴露や汚染による損害と罰金は明確に含まれていません。したがって、汚染や傷害が発生した場合、私たちは損害賠償責任を負うことを要求されるか、または私たちの資源を超えた罰金に処せられる可能性があり、私たちの臨床試験または監督管理の承認が一時停止される可能性があり、これは私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
67
また、現在または未来の環境、健康および安全法律、法規、許可要件を遵守するためには、巨額のコストが生じる可能性がある。これらの現行または未来の法律、法規、許可要件は、私たちの研究、開発、または生産努力を損なう可能性がある。これらの法律、法規、および許可要件を遵守しないことは、巨額の罰金、処罰、または他の制裁または業務中断を招く可能性もあり、これは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
私たちが雇用している任意の第三者契約製造業者とサプライヤーもまたこれらと他の環境、健康、安全法律法規の制約を受けるだろう。彼らがこれらの法律と法規に基づいて発生した債務は、重大なコストや運営中断を招く可能性があり、これは私たちの業務、財務状況、運営結果、将来性に重大な悪影響を及ぼす可能性がある。
遺伝子薬は新しいもので、私たちが開発したどの候補製品も複雑で、製造が難しいかもしれません。私たちは、規制機関の要求を満たす遅延や生産問題に遭遇し、私たちの開発や商業化計画の遅延、私たちが開発する可能性のある候補製品の供給を制限したり、他の方法で私たちの業務を損害したりする可能性があります。
私たちが開発可能などの候補製品も、ほとんどの化学薬に必要な加工工程よりも複雑な加工工程を必要とするかもしれない。また,化学薬とは異なり,生物の物理的·化学的性質は,我々が開発しようとしている候補製品のように,通常完全には表現できない。したがって、候補完成品の分析は、候補製品が予期される方法で動作することを確実にするのに十分ではない可能性がある。製造過程の問題は、正常過程との微小な偏差であっても、すべて製品欠陥或いは製造失敗を招く可能性があり、それによって大量故障、製品リコール、製品責任クレーム、在庫不足、或いは著者らの潜在的なIND届出の進展を遅延させる可能性がある。もし私たちが候補製品の開発に成功したら、私たちは問題に直面する可能性があり、十分な数量と品質の臨床レベルの材料を得ることができません。これらの材料はFDA、EMAあるいは他の同様の適用外国標準や規範に適合し、一致して受け入れ可能な生産生産量とコストを持っています。さらに、我々が開発する可能性のある候補製品は、電気穿孔、LNPsまたはウイルスベクターのような複雑な輸送方法を必要とし、それぞれが製造中に追加の複雑さを導入する。
さらに、FDA、EMA、および他の規制機関は、任意のロットの承認製品のサンプルをいつでも提出し、適用試験結果を示すプロトコルを要求することができるかもしれない。場合によっては、FDA、EMA、または他の規制機関は、機関が発行を許可する前に大量の製品を配布しないように要求するかもしれません。製造過程中の微小な偏差は、品質属性や安定性に影響を与える偏差を含み、製品に許容できない変化を招き、大量故障や製品リコールを招く可能性がある。大量失敗や製品リコールは臨床試験や製品発表を延期する可能性があり、これは私たちに高い代価を払わせるかもしれません。そうでなければ、私たちの業務、財務状況、運営結果と将来性を損なうことになります。
また,新たなウイルス技術を用いて候補製品の塩基エディタやRNA構造を提供する予定であるが,これらの技術に基づく候補製品開発の可能性を支援する科学的証拠は初歩的であり,限られている.
私たちはまた、私たちの製造プロセスを管理するために必要な経験豊富な科学、品質管理、製造者の雇用と維持の問題に直面する可能性があり、これは私たちの生産が遅れたり、適用された法規要件を守ることが困難になる可能性があります。
生物製品生産の性質を考慮して、生産過程において汚染リスクが存在する。どの汚染も私たちが計画通りに候補製品を生産する能力を深刻に損なう可能性があり、私たちの運営結果を損害し、名声の損害をもたらす可能性がある。私たちは製造過程で必要ないくつかの原材料が生物源から来ると予想している。このような原材料は入手が難しく、汚染されたりリコールされる可能性がある。材料不足、汚染、リコール、または私たちが開発する可能性のある任意の候補製品の生産に生物由来物質を使用することは、商業製造または臨床材料の生産に悪影響または中断をもたらす可能性があり、これは、私たちの開発スケジュールおよび私たちの業務、財務状況、運営結果、および将来性に実質的な損害を与える可能性がある。
私たちの製造プロセスや私たちと契約した施設のいずれの問題も、潜在的なパートナー(大きな製薬会社や学術研究機関を含む)の魅力の低いパートナーになる可能性があり、これは、より多くの魅力的な開発プロジェクトを獲得することを制限する可能性があります。第三者製造プロセスまたは施設における問題は、私たちが行っているまたは計画されている任意の臨床試験に十分な臨床材料を提供する能力を確保し、私たちが開発および商業化する任意の候補製品の市場需要を満たすことを制限する可能性もある。
68
私たちの第三者との関係に関するリスクは
我々は、我々が開発、臨床試験、および研究および臨床前試験のいくつかの態様の候補製品を開発し、行う可能性のあるコンポーネントを製造するために第三者に依存し続けることが予想されるが、これらの第三者の表現は、締め切り前にこのような試験、研究または試験を完了できないことを含む満足できない可能性がある。
著者らは引き続きCMO、CRO、臨床データ管理組織、医療機関と臨床研究者などの第三者に依存し、著者らの研究と臨床前テストのいくつかの方面を行い、著者らの候補製品のコンポーネントを製造し、そして著者らの臨床試験を行う。いくつかの基準によると、このような第三者のいずれかは、いつでも私たちとの契約を終了することができる。もし私たちが代替計画を達成する必要があれば、私たちの製品開発活動を延期するかもしれない。
これらの第三者研究開発活動への依存は,これらの活動に対する我々の制御を減少させたが,我々の責任を軽減していない.例えば、私たちは私たちのすべての臨床試験が試験の全体的な研究計画と方案に従って行われることを確実にする責任がある。また、FDA、EMAと他の監督機関は、データと報告の結果が信頼性と正確であることを保証し、試験参加者の権利、完全性と機密性を保護するために、臨床試験結果を行い、記録し、報告する基準を遵守することを要求している。米国では,行われている臨床試験を一定期間登録し,完成した臨床試験結果を政府助成データベースClinicalTrials.govに発表することも求められている。そうしないと罰金、否定的な宣伝、そして民事と刑事制裁につながるかもしれない。
著者らは著者らの候補製品のために臨床試験を設計したが、著者らはCROによる一部或いはすべての臨床試験に依存し、引き続き予定している。したがって、私たちの開発計画の多くの重要な側面は、それらの実施とタイミングを含めて、私たちの直接制御範囲内ではないだろう。著者らの第三者による臨床前研究と臨床試験への依存も、臨床前研究と臨床試験によって開発されたデータ管理をそれほど直接制御しないことを招き、私たち自身の従業員に完全に依存するのではない。外部の当事者とのコミュニケーションも挑戦的である可能性があり、ミスや協調活動の困難を招く可能性がある。外部の当事者は
これらの要素は第三者が著者らの臨床前研究と臨床試験を行う意志或いは能力に重大な不利な影響を与える可能性があり、そして著者らのコントロール範囲を超える意外なコスト増加に直面させる可能性がある。CROや他の第三者が満足できる方法で臨床前研究と将来の臨床試験を行わなかった場合、彼らの私たちの義務に違反したり、規制要求を遵守できなかったりすれば、私たちの候補製品の開発、規制承認、商業化が遅れる可能性があり、私たちは規制承認を得られず、私たちの候補製品を商業化することができないかもしれません。あるいは私たちの開発計画は実質的で不可逆的な損害を受ける可能性があります。もし私たちのCROおよび他の第三者が収集した臨床前および臨床データに依存できなければ、私たちが行った任意の臨床前研究または臨床試験の規模を繰り返し、延長または増加させる必要があるかもしれないが、これは商業化を著しく延期し、より大きな支出を必要とするかもしれない。
私たちは第三者と契約を結び、私たちの研究計画、臨床前研究、臨床試験の製造と材料を提供し、私たちの将来の研究計画、臨床前研究と臨床試験の少なくとも一部、そして私たちが開発する可能性のある任意の候補製品の商業化において、私たちはこのように続けていくと予想される。このような第三者への依存は、私たちがそのような材料、候補製品、または私たちが開発および商業化する可能性のある任意の薬物を十分な数のリスクを増加させるか、または許容可能なコストでそのような供給を得ることができないリスクを増加させ、これは、私たちの開発または商業化努力を延期、阻止または損害する可能性がある。
私たちは現在、第三者が私たちの一部の臨床前研究と臨床試験のために材料を製造し、供給することに依存しており、将来の研究計画、臨床前研究、臨床試験、および私たちが開発する可能性のある任意の候補製品の商業供給のために引き続きそうする可能性があり、私たちまたは私たちの協力者は市場の承認を得ている。私たちはいかなる第三者サプライヤーとも長期供給協定を締結していません。私たちは注文に必要な供給を購入します。
私たちは私たちを支援するための製造施設を設立しました離体する血液学と腫瘍学の細胞治療計画と体内にあるノースカロライナ州研究三角園区の肝臓疾患非ウイルス送達計画では、cGMPコンプライアンスを維持し、私たちの内部製造能力を拡大し、あるいは私たちの計画の製造需要を満たすことができるかどうかを確定することができない。
私たちは第三者サプライヤーと長期供給協定を確立できないかもしれないし、受け入れ可能な条件でそうすることができないかもしれない。たとえ第三者と長期供給協定を確立することができても、第三者に依存することは追加的なリスクをもたらす
69
第三者サプライヤーはcGMP法規や米国以外の他の法規要件を遵守できない可能性がある。私たちまたは第三者サプライヤーが適用法規を遵守できないことは、罰金、禁止、民事処罰、遅延、承認の一時停止または撤回、許可証の取り消し、候補製品や薬品の差し押さえまたはリコール、運営制限、刑事起訴を含む制裁を実施する可能性があり、いずれも私たちの薬品供給に重大な悪影響を与え、私たちの業務、財務状況、運営結果、将来性を損なう可能性があります。
例えば,様々なCROにより非ヒト霊長類,あるいはHEPを獲得し,臨床前開発に用いている。2023年2月,Charles River実験室,あるいはCharles Riverと呼ばれ,我々のNHPの主要サプライヤーの1つは,カンボジアからNHPを輸入することに関する米国司法省の召喚状を受け取ったことを発表した。Charles Riverはさらに,現在自発的にカンボジアからのNHPの出荷を一時停止していると発表した。我々は現在,我々の最近の需要を満たすのに十分なNHP供給を得ることができると信じているが,この供給はサプライチェーン制限の悪影響を受ける可能性がある。Charles Riverや他のCROから十分なNHP供給を得ることができない場合,あるいはNHP不足によりNHP価格が大幅に上昇すると,われわれのいくつかの臨床前開発が延期され,発見プロジェクトや臨床前開発活動を行うコストが大幅に増加する可能性がある。このような遅延やコスト増加は,われわれの発見や臨床前開発活動およびわれわれの業務に実質的な悪影響を及ぼす可能性がある。
私たちが開発したどんな薬も他の候補製品や製品と生産施設や供給を競争する可能性がある。CGMP法規下で運営されているメーカー数は限られており,遺伝子編集に必要な薬物成分や薬物製品を生産する能力がある可能性がある。私たちの現有或いは未来のサプライヤーのいかなる業績ミスも臨床前或いは臨床開発或いはマーケティング承認を延期する可能性がある。私たちは今私たちの遺伝子編集製品候補に必要なすべての薬物成分と薬物製品を余分に供給するように手配していません。私たちの現在の契約製造業者またはサプライヤーのいずれかが合意を履行できない場合、私たちはその製造業者またはサプライヤーの交換を要求される可能性があります。いくつかの潜在的な代替サプライヤーが私たちが開発する可能性のある任意の候補製品を支持することができると信じていますが、そのような代替製品を決定して同定する際に、追加のコストと遅延が生じる可能性があります。
私たちは現在、将来的に私たちが開発する可能性のある任意の候補製品を他人に依存して生産し、私たちの将来の利益率と、私たちが適時かつ競争力のある上場承認された薬物商業化の能力に悪影響を及ぼすかもしれない。
私たちの薬物開発パイプラインの増加と成熟に伴い、私たちの施設や第三者の臨床用品に対する需要の増加は私たちの運営能力に影響を与える可能性がある。私たちはサプライチェーン全体の生産能力を増加させる必要があるだろう。さらに、私たちは、製造または試験サービスを提供するサービスプロバイダを含む多くのサービスプロバイダに依存しており、これらすべてのサービスプロバイダの運営には、私たちの運営に悪影響を及ぼす可能性のある内在的なリスクが存在する。
私たちの臨床試験を完成させ、私たちの候補製品を商業化するためには、私たちの候補製品を十分な生産量で製造するために、施設を獲得したり開発したりする必要があり、承認されれば、商業規模の生産を行う。われわれは臨床試験を支援するために必要ないずれの候補製品の生産にも経験が限られており,商業販売を支援するために必要な量の生産には経験がない。これらの能力を確立する努力は、進度、拡大規模、再現性、生産量、純度、コスト、効力或いは品質など、最初の予想に達しない可能性がある。また、他社は、その多くが豊富な資源を持っており、私たちと競争して私たちの候補製品を生産するために必要な材料を獲得しています。
我々は現在,第三者を利用して原材料,部品,部品,消耗品を製造し,品質テストを継続する予定である。塩基編集や他の遺伝子薬物分野が拡大し続けると,これらの材料やサービスに対する日々の激しい競争に遭遇する可能性がある。第三者製造や試験施設への需要増加速度は既存生産能力よりも速い可能性があり、これは、私たちの候補製品を製造するために必要な原材料、部品、消耗品を生産できる第三者メーカーの能力を探し、保持することを妨げる可能性がある。サービス提供者とサプライヤーを使用することは、私たちをリスクに直面させるかもしれないが、これらに限定されない
70
第三者製造業者への私たちの依存は、私たちの運営に悪影響を与えたり、予見できない遅延や他の私たちがコントロールできない問題を招く可能性があります。契約の制限と専門知識を持つ第三者メーカーの数が限られているため、臨床規模と商業規模で私たちの候補製品を生産するためには、監督管理の承認と施設が必要であり、メーカーの交換は高価で時間がかかる可能性があり、そして私たちの候補製品の生産中断を招く可能性がある。第三者製造業者たちはまた生産で困難に直面する可能性がある。これらの問題には
したがって、どんな遅延や中断も、私たちの業務、財務状況、または運営結果に実質的な悪影響を及ぼす可能性がある。
私たちは将来的に第三者と協力して、私たちが開発したいくつかの候補製品の研究、開発、商業化を行うことが可能になった。もしそのような協力が成功しなければ、私たちはこの候補製品の市場潜在力を利用できないかもしれない。
私たちは、将来的に開発されたいくつかの候補製品の研究、開発、商業化のために第三者パートナーを探すことが可能になった。私たちが達成した合意と、私たちが将来任意の第三者と達成する可能性のある任意の合意に基づいて、私たちは、私たちの協力者が開発または商業化に取り組んでおり、彼らと開発した任意の候補製品の資源の数と時間が限られるかもしれないことを求めています。私たちがこのような計画から収入を創出する能力は、私たちの協力者がこれらの手配の中で彼らに割り当てられた機能を成功的に履行する能力に依存するだろう。私たちは私たちが参加したどんな協力も成功するかどうかを予測できない。
私たちの研究計画や私たちが開発する可能性のある任意の候補製品の協力は、私たちに多くのリスクをもたらしてくれます
71
もし私たちの協力が候補製品の開発と商業化に成功しなかった場合、または私たちの協力者が私たちとの合意を終了した場合、私たちは協力によって未来の研究資金、マイルストーン、印税支払いを得ることができないかもしれない。また、私たちがこのような支払いを受けても、私たちの許可者との許可協定によると、それらは支払い義務を招く可能性があり、これはかなり大きい可能性がある。もし私たちがこれらの協力協定に従って所望の資金を受け取っていない場合、あるいはこれらの資金が許可側への支払い義務によって大きく相殺された場合、私たちの候補製品の開発が延期される可能性があり、候補製品を開発するために追加の資源が必要かもしれません。さらに、もし私たちの協力者が私たちとの合意を終了したら、私たちは適切な代替協力者を見つけたり、新しい協力者を引き付けることがもっと難しいことを発見するかもしれません。私たちの開発計画は延期されるかもしれません。あるいは商業と金融界での私たちのイメージは不利な影響を受けるかもしれません。本Form 10−K年次報告で述べた製品開発,規制承認,商業化に関するすべてのリスクは,我々の協力者の活動に適用される。
これらの関係、または同様の関係は、非日常的な費用や他の費用の発生、短期および長期支出の増加、既存の株主を希釈した証券の発行、または私たちの管理や業務を混乱させることを要求することができる。また、適切な協力者を探す上で、私たちは激しい競争に直面する可能性があり、交渉過程は時間がかかり複雑である。我々が最終的な協調合意を達成する能力は,協力者の資源や専門知識の評価,協調の条項や条件の提案,提案した協力者のいくつかの要因の評価に依存する.もし私たちがどんな候補製品にも許可すれば、私たちは私たちまたは私たちの協力者が開発する可能性のある製品を開発するかもしれません。もし私たちがこれらの取引を私たちの既存の運営と会社文化と組み合わせることに成功できなければ、私たちはこのような取引のメリットを達成できないかもしれません。
もし私たちが私たちの協力者や戦略的パートナーと衝突すれば、これらの当事者たちは私たちに不利な方法で行動し、私たちが戦略を実施する能力を制限するかもしれない。
もし私たちの企業や学術パートナーや戦略パートナーが私たちと衝突した場合、相手は私たちに不利な方法で行動し、私たちの戦略を実施する能力を制限するかもしれません。私たちのいくつかの協力者と戦略的パートナーは私たちが協力する各分野と複数の製品開発を行っています。しかし、私たちの協力者または戦略パートナーは、単独で、または他の人と一緒に関連分野の製品を開発することができ、これらの製品は、私たちが開発する可能性のある候補製品と競争力を持ち、これらの候補製品は私たちと協力するテーマである。協力者または戦略パートナーによって開発された競合製品であっても、協力者または戦略パートナーが権利を有する競合製品であっても、パートナーが我々が開発する可能性のある候補製品の支援を撤回する可能性がある。
私たちのいくつかの協力者や戦略的パートナーたちはまた私たちの未来の競争相手になるかもしれない。私たちの協力者や戦略パートナーは競争製品を開発し、私たちがその競争相手との協力を阻止し、適時に規制の承認を得ることができず、私たちとの合意を早期に終了したり、製品の開発と商業化に十分な資源を投入することができなかった。このような進展のいずれも私たちの製品開発努力を損なう可能性がある。
もし私たちが商業的に合理的な条件下で協力を作ることができなければ、私たちは私たちの開発と商業化計画を変えなければならないかもしれない。
私たちの製品開発と研究計画、そして私たちが開発する可能性のある任意の候補製品の潜在的な商業化には、費用を支払うために多くの追加の現金が必要になります。私たちが開発する可能性のあるいくつかの候補製品については、他の製薬やバイオテクノロジー会社と協力してこれらの候補製品を開発し、商業化することを決定するかもしれない。
私たちは適切な協力者を探すことで激しい競争に直面している。我々が協力について最終的な合意を達成するかどうかは,協力者の資源や専門知識の評価,協調の条項や条件の提案,提案された協力者のいくつかの要因の評価に依存する.これらの要因は、臨床試験の設計または結果、FDA、EMAまたは米国国外の類似規制機関の承認の可能性、候補研究製品の潜在的市場、そのような候補製品の製造と患者への配送のコストと複雑性、競争製品の潜在性、技術所有権の不確実性、挑戦の是非曲直を考慮せずにこのような所有権に挑戦し、存在する可能性のある不確実性、および一般的な業界および市場状況を含むことができる。協力者はまた、同様の協力可能な兆候を得るために候補製品または技術を代替することと、そのような連携が私たちとの連携よりも魅力的であるかどうかを考慮することができる。
72
既存の協力協定によれば、潜在的なパートナーと特定の条項について未来の合意を締結することができないように制限される可能性もあります。協力の交渉と記録は複雑で時間がかかる。また,最近では大手製薬会社間の業務合併数が大きく,将来の潜在的パートナー数の減少を招いている。
私たちはタイムリーに基づいて受け入れ可能な条項で協力することができないかもしれないし、交渉することさえできないかもしれない。それができなければ、私たちが協力を求めている候補製品の開発を削減し、その開発計画や私たちの1つ以上の他の開発計画を減らしたり延期したり(承認されれば)、潜在的な商業化を延期したり、販売やマーケティング活動の範囲を縮小したり、私たちの支出を増やしたり、自費で開発や商業化活動を行ったりしなければならないかもしれない。もし私たちが私たち自身の開発や商業化活動を支援するために私たちの支出を増加させることを選択すれば(承認されれば)、私たちは追加の資本を得る必要があるかもしれないし、これらの資本は受け入れ可能な条件で私たちに提供できないかもしれないし、全く得られないかもしれない。もし私たちが十分な資金がなければ、私たちは候補製品を開発したり、それらを市場に出して製品収入を作ることができないかもしれない。
私たちは新しい会社である軌道治療会社の設立を支援し、私たちが期待していた優位性を実現していないかもしれないことを含め、新会社の設立に関連するリスクに直面している。
2022年9月、著者らはARCH Venture Partnersと協力し、軌道治療会社の発売を支援し、リボ核酸(RNA)薬物の発展を推進することを目標とした。軌道発射に関しては、我々は、軌道会社と許可および協力協定、または軌道協定を締結しており、この協定によれば、我々および軌道会社は、それぞれ、その制御のいくつかの技術に他の許可を付与し、これらの許可は、非ウイルス送達またはヒト疾患の予防、治療または診断のためのRNAの設計または製造に必要または合理的に有用である。また、私たちが軌道協定に署名すると同時に、軌道会社は7500万株の普通株を発行してくれた。Arch Venture PartnersはOrbaryの株主でもあり、私たちはArch Venture Partnersと関連する2人の取締役Kristina BurowとJohn Maraganoreと私たちのCEO John EvansはOrbaryの取締役メンバーであり、私たちの総裁博士Giuseppe Ciaramella博士はOrbaryの臨時CEOと取締役会メンバーである。
私たちはOrbaryの少数の株式を持っているので、私たちはその業務運営のコントロールの程度が低く、追加の財務、法律、運営、そしてコンプライアンスのリスクに直面する可能性があります。さらに、軌道会社の持株株主または経営陣は、私たちと一致しないビジネス利益、戦略、または目標を持っている可能性がある。これらのリスクは:Orbaryまたは他の株主の経済的または商業的利益または目標が私たちの経済的または商業的利益または目標と一致しないか、または一致しない;私たちが私たちの指示、要求、政策または目標に反する行動をとる可能性がある;私たちに予期しない責任やリスクを負わせる;私たちの投資リターンを下げる行動をとる;私たちの重要な特許権利、または私たちの所有または許可された重要な知的財産権または他の権利を損なう方法を取ること、または私たちの名声を損なう、または私たちの経営業務能力を制限する行動をとることを含む。また、私たちは軌道会社の所有権を持っているので、私たちは将来、私たちの総合財務業績に軌道会社の財務情報を含むことを要求されるかもしれません。私たちは以前、私たちの財務諸表に少数の株式子会社を含まなかったので、軌道財務諸表を正確に述べ、それを私たち自身の財務諸表に含めることは、米国証券取引委員会への書類提出の遅延、重大または重大な弱点の発見などを招く可能性がある。これは、私たちの連結財務諸表の信頼性に自信を失ったことによる金融市場の不良反応など、私たちの業務に有害な結果をもたらす可能性があります。
私たちの知的財産権に関するリスクは
もし私たちが開発した任意の候補製品や私たちのプラットフォーム技術のために特許や他の知的財産権保護を獲得して維持することができない場合、あるいは取得した特許や他の知的財産権保護の範囲が十分でなければ、私たちの競争相手は私たちと似ているか同じ製品や技術を開発して商業化する可能性があり、私たちが開発する可能性のある任意の候補製品を商業化する能力に成功すれば、私たちのプラットフォーム技術は不利な影響を受ける可能性がある。
私たちのビジネス成功は、それらを製造するための交付プラットフォーム技術方法および処理方法、ならびに私たちの特許および他の知的財産権を第三者挑戦から保護することができるかどうか、私たちの基礎編集プラットフォーム技術、候補製品および他の技術の特許、商標、商業秘密および他の知的財産権保護を獲得し、維持する能力があるかどうかに大きく依存する。私たちの基礎編集プラットフォーム技術と候補者を保護することは難しくて高価で、私たちは彼らの保護を確保できないかもしれない。私たちが許可されていない第三者が、私たちが開発する可能性のある候補製品を製造、使用、販売、提供、輸入、または他の方法で商業化することを阻止する能力は、これらの活動をカバーする有効かつ実行可能な特許または商業秘密の下で当社が所有する権利の程度に依存する。
私たちは、私たちのプラットフォーム技術に関連する知的財産権を付与し、私たちの基礎編集プラットフォーム技術、プラットフォーム技術の配信、および私たちの業務に重要な候補製品に関する特許出願を米国および海外に提出することで、私たちの独自の地位を保護することを求めています。私たちまたは私たちのライセンシーが、私たちの基礎編集プラットフォーム技術、配信プラットフォーム技術、および私たちが開発する可能性のある候補製品について特許保護を獲得または維持することができない場合、または取得した特許保護範囲が十分でない場合、私たちの競争相手は、私たちと類似または同じ製品および技術を開発し、商業化する可能性があり、私たちが開発する可能性のある任意の候補製品の商業化能力は悪影響を受ける可能性がある。
73
特許訴訟過程は高価で、時間がかかり、複雑であり、私たちは合理的なコストまたはタイムリーな提出、起訴、維持、強制執行、またはすべての必要または望ましい特許出願を許可することができないかもしれない。しかも、私たちはすべての関連市場で特許保護を求めたり、獲得しないかもしれない。我々の研究開発成果における出願可能な特許の側面をタイムリーに決定できず,特許保護を得ることができない可能性もある.私たちは、私たちの研究開発成果にアクセスする権利のある秘密または特許可能な当事者と、当社の従業員、会社の協力者、外部科学協力者、CRO、契約製造業者、コンサルタント、コンサルタント、および他の第三者のような秘密および秘密協定を締結していますが、これらの当事者のいずれかは、合意に違反し、特許出願を提出する前にそのような成果を開示し、特許保護を求める能力を危険にさらしている可能性があります。さらに、有効かつ強制的に実行可能な特許を取得して保持する能力は、我々の発明と従来技術との差が、我々の発明が従来技術よりも特許を出願することを可能にするか否かに依存する。また、科学文献で発表された発見は実際の発見よりも遅れがちであり、米国および他の司法管轄区の特許出願は通常、出願18ヶ月後に公表され、時には全く公表されない場合もある。したがって、私たちまたは私たちのライセンス者が、私たちが所有しているまたは任意のライセンス特許または係属中の特許出願で主張されている発明を最初に提出した人であるか、または私たちまたは私たちのライセンス者が、そのような発明のために特許保護を申請した最初の人であることを確認することはできません。
バイオテクノロジーと製薬会社の特許地位は通常高度に不確定であり、複雑な法律と事実問題に関連しており、近年多くの訴訟のテーマとなってきた。遺伝子編集分野,特に塩基編集技術分野は,広範な特許活動や訴訟のテーマであった。したがって,我々の特許権の発行,範囲,有効性,実行可能性,商業的価値には高い不確実性があり,複雑で高価な訴訟に巻き込まれる可能性がある.私たちの未解決および将来の特許出願は、私たちの基礎編集プラットフォーム技術、配信プラットフォーム技術、および私たちが開発する可能性のある候補製品を保護する特許を発行すること、または他社が競争技術および候補製品を商業化することを効果的に阻止することにつながる可能性があります。
遺伝子編集分野で許容されるクレーム範囲については,塩基編集技術を含め,米国ではまだ一致した政策は見られていない。アメリカ以外の地域の特許保護範囲も確定されていない。米国および他の国の特許法またはその解釈の変化は、私たちの発明、獲得、維持、実行、そして私たちの知的財産権を保護する能力を弱める可能性があり、より広く言えば、私たちの知的財産権の価値に影響を与えたり、私たちが所有し許可している特許権の範囲を縮小したりする可能性がある。ライセンス内および所有する知的財産権については、私たちおよび私たちのライセンシーが現在求めている特許出願が任意の特定の司法管轄区域で特許として発行されるかどうか、または任意の発行された特許の権利主張が有効かつ実行可能であり、競争相手の影響を受けずに十分な保護を提供するかどうかを予測することはできない。
また,特許出願に要求されるカバー範囲は特許発行前に大幅に縮小することができ,その範囲は特許発行後に再解釈することができる.私たちが現在または将来許可または所有している特許出願が特許の形態で発表されても、それらは、私たちに任意の意味のある保護を提供し、競争相手または他の第三者が私たちと競争することを阻止し、または他の方法で私たちに任意の競争優位性を提供する形で発表されることはない。私たちが持っているまたは許可されているどの特許も、第三者によって挑戦、縮小、回避、または無効にされる可能性がある。したがって、私たちが開発する可能性のあるいかなるプラットフォームの進歩や候補製品が効果的で強制的に実行可能な特許によって保護されているか、または引き続き保護されているかどうかは分からない。私たちの競争相手または他の第三者は、非侵害的に類似または代替技術または製品を開発することによって、私たちの特許を回避することができるかもしれない。
さらに、新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちの知的財産権は他の人たちが私たちと似ているか同じ製品を商業化することを排除するために十分な権利を提供してくれないかもしれない。さらに、私たちが所有し、許可されたいくつかの特許と特許出願は、私たちが第三者と共同で所有しており、未来に共通して所有されているかもしれない。例えば,我々の潜在的なHBG 1およびHBG 2製品候補に対する特許出願は,我々,ハーバード大学の総裁,フィロスおよびブロス研究所が共同で所有している。現在、私たちはハーバード大学やブロイド大学の所有権権の許可を得ていない。もし私たちがこのような特許または特許出願におけるこれらの第三者共同所有者の権利の独占的な許可を得ることができない場合、これらの共通所有者は、私たちの競争相手を含む他の第三者に権利を許可することができ、私たちの競争相手は競争製品および技術を販売することができる。さらに、私たちは、第三者に対してそのような特許を強制的に実行するために、私たちの特許のどのような共通所有者の協力も必要とするかもしれませんが、そのような協力は私たちに提供されないかもしれません。上記のいずれも、我々の競争地位、業務、財務状況、経営結果、および見通しに重大な悪影響を及ぼす可能性がある。
私たちの基礎編集プラットフォーム技術と候補製品の権利は、他人が私たちに付与したライセンスの条項と条件にある程度制限されています。
私たちは第三者から許可された知的財産権に依存しており、私たちの許可者たちはいつも私たちの最善の利益に従っているわけではないかもしれない。もし私たちが知的財産権の許可下での私たちの義務を履行できなかった場合、許可が終了された場合、またはこれらの許可に関する紛争が発生した場合、私たちは私たちの業務に重要な重要な権利を失うかもしれない。
74
私たちは、当社の基礎編集技術および候補製品の開発に非常に重要または必要な第三者からの特定の特許権およびノウハウに許可し、依存しています。たとえば,遠大研究院,Editas,ハーバード大学,Bio Paletteなどとライセンス契約を締結し,これらの合意に基づき,我々の基盤編集プラットフォーム技術と候補製品にキー特許と特許応用許可(それぞれ遠大ライセンスプロトコル,Editas許可プロトコル,ハーバード許可プロトコル,Bio Paletteライセンスプロトコル)を付与した.このような許可協定は私たちに様々な勤勉さ、マイルストーン支払い、特許権使用料、保険、そして他の義務を課している。もし私たちがこれらの義務を履行しなければ、私たちの許可者は私たちの許可を終了する権利があるかもしれません。この場合、私たちは、私たちの基本編集プラットフォームやこれらの合意によって許可された知的財産権がカバーする任意の他の候補技術や製品を開発またはマーケティングすることができません。例えば、ハーバード許可協定によると、いくつかのサブライセンス特許カテゴリによってカバーされるライセンス製品を開発するためには、いくつかの開発マイルストーンを達成しなければならない。もし私たちがこのようなマイルストーンを達成できなければ、私たちのサブカテゴリに対する特許の権利は終わるだろう。
これらの許可および他の許可は、すべての関連する使用分野および将来、私たちの基礎編集プラットフォーム技術および候補製品のすべての地域でこのような知的財産権および技術を使用することを望む可能性がある独占的な権利を提供しないかもしれません。私たちに与えられたいくつかの許可は、許可者またはいくつかの第三者が所有するいくつかの以前に存在する権利に明示的に制限されている。したがって、競争相手がある地域や分野で競合製品を開発し、商業化することを阻止することはできないかもしれない。例えば、Howard Hughes Medical InstituteまたはHHMIの従業員によって開発されたいくつかの許可特許は、その後、ハーバード大学に割り当てられ、ハーバード許可協定に従って私たちに許可され、これらの特許は、依然としてハーバード大学とHHMIとの間の非独占的許可によって制限される。Editasライセンスプロトコルは,我々の使用領域は,ある工学的T細胞によるある遺伝子編集技術をヒト癌の診断,治療,予防に用いる使用を除外し,これらのT細胞はJuno治療会社(百時美施貴宝社の子会社)の許可を得ていると規定している。もし私たちの任意の候補製品を商業化し、または私たちの競争優位性を維持するために必要なこのような排除分野の権利を決定すれば、私たちの候補製品の開発、製造、またはマーケティングを継続するために、第三者から許可を得る必要があるかもしれない。私たちは独占的に基づいて、商業的に合理的な条項やそのような許可を得ることができないかもしれません。これは、候補製品を商業化することを阻止したり、私たちの競争相手や他の人が私たちの業務に重要な技術を得る機会を得ることができるかもしれません。
広範なライセンスプロトコルによれば、私たちに付与される権利は、Cas 12 bまたはCas 13のためのいくつかの米国に限定される特許出願を含む。これらの特許出願の共同所有者は、ブロイド研究所、ハーバード大学、マサチューセッツ工科大学、ニュージャージー州立大学またはロゲス大学、スコルコボ科学技術研究所またはSkoltech、および米国国立衛生研究院を含む。現在、私たちはロッグス大学、Skoltech、またはNIH所有権のライセンスを持っていない。もし私たちがこのような特許出願におけるロッグス大学、Skoltech、NIHの独占許可を得ることができなければ、ロッグス大学、Skoltech、およびNIHはその権利を私たちの競争相手を含む他の第三者に許可することができ、これらの第三者は競争製品や技術を販売することができるかもしれない。また,これらの特許出願によって発行された特許を第三者に強制的に実行するためには,ロッグス大学,Skoltech,あるいは米国国立衛生研究院の協力が必要となる可能性があり,この協力は提供されない可能性がある。上記のいずれも、我々の競争地位、業務、財務状況、経営結果、および見通しに重大な悪影響を及ぼす可能性がある。
さらに、私たちとブロドカレッジとのライセンス契約およびハーバード大学とのライセンス契約によれば、特定の場合(それぞれの場合)、ブロド大学またはハーバード大学(場合によっては)は、そのライセンスプロトコルの対象に属する特許を、その特許が私たちの分野と同じ分野に許可されている第三者に付与することができる。この第三者は、遠大な許可プロトコルまたはハーバード許可プロトコル(状況に応じて)のすべての権利を持っている可能性があり、これは、私たちの競争地位に影響を与え、第三者が私たちの将来の潜在的な候補製品および技術と類似した製品を商業化することを可能にするかもしれない。この場合、第三者に付与されるいかなる権利も、ブロード研究所および/またはハーバード大学から取得した特許および特許出願の専有権範囲を縮小するであろう。
私たちは、特許および特許出願の準備、提出、起訴、維持、実行、および弁護に関して完全な制御権を有しておらず、これらの特許および特許出願は、第三者から許可を得る技術をカバーしている。例えば、私たちが遠大研究所、ハーバード大学、Editas、Bio Paletteと締結したすべての知的財産権ライセンスによると、私たちのライセンシーは、準備、提出、起訴、および維持に対する制御権を保持し、場合によっては、その特許および特許出願の実行および弁護の制御権も保持する。私たちのライセンシーが侵害者に対して特許を実行したり、有効性の挑戦または実行可能な主張を弁護する力は、私たち自身が実行するよりも強くないかもしれないし、私たちの最適な利益に合わないかもしれない。私たちはこのような特許と特許出願の準備、提出、起訴、維持、強制執行、そして弁護が私たちの業務の最高の利益に合致しているかどうかを決定することができない。もし私たちの許可者がこれらの特許を起訴、維持、強制執行および保護できなかった場合、またはこれらの特許または特許出願の権利を失った場合、私たちが許可した権利は減少またはキャンセルされる可能性があり、私たちが開発する可能性のある任意の製品候補製品の開発および商業化の権利は悪影響を受ける可能性があり、私たちは競争相手が競合製品を製造、使用、販売することを阻止できないかもしれない。
75
私たちのライセンス者は第三者コンサルタントや協力者または第三者からの資金に依存する可能性がありますので、私たちのライセンス者は私たちが許可を得た特許の唯一および独占所有者ではありません。他の第三者が私たちの許可内の特許を所有している場合、このような許可を付与するために共通の所有者の同意が必要な司法管轄区域内では、私たちの許可は無効になる可能性があり、これらの共通の所有者は、このような特許許可を私たちの競争相手に与える可能性があり、私たちの競争相手は競争製品および技術を販売することができる。さらに、ライセンス内特許および特許出願に対する私たちの権利は、そのような認可内特許と特許出願の共同所有者との間の機関間または他の運営プロトコルにある程度依存する。1つまたは複数の共通の所有者がそのような機関間または運営プロトコルに違反している場合、私たちは、このようなライセンス内の特許および特許出願の権利に悪影響を受ける可能性がある。これらの事件のいずれも、私たちの競争地位、業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
さらに、私たちのいくつかのライセンス特許と特許出願に含まれる発明は、アメリカ政府の資金を用いて行われている。私たちは、私たちが許可した特許および特許出願に関連する義務である、このような資金によって生成された適用義務、例えばタイムリーな報告を遵守することを保証するために、我々の許可者に依存する。もし私たちの許可者がその義務を履行できなかった場合、関連特許の権利喪失または強制執行を招く可能性がある。例えば、米国政府は、このような許可内の特許に対して、米国政府が発明を使用することを許可するか、または米国に代わって米国に発明を使用させる非排他的許可を含むいくつかの権利を有することができる。もしアメリカ政府がこのような権利を行使することを決定すれば、私たちをその請負業者として雇う必要はない。米国政府の権利はまた、援助された発明および技術を第三者に開示し、米国政府の資金開発を使用する許可技術を第三者が使用または使用することを許可する先行権を行使することを可能にするかもしれない。米国政府が、我々または我々の許可者が米国政府が支援する技術の実用化を実現できなかったため、健康や安全需要を緩和し、連邦法規の要求を満たすために行動する必要があると判断した場合、または米国工業を優先することができれば、米国政府もそのデモ権利を行使することができる。さらに、このような許可された米国政府によって援助された発明の権利は、米国でそのような発明を含む候補製品を製造するために、いくつかの要件によって制限される可能性がある。上記のいずれも、私たちの業務、財務状況、経営結果、見通しに重大な損害を与える可能性があります。
もし私たちの第三者許可者が認定した場合、私たちは努力したにもかかわらず、許可協定または許可協定の下のいくつかの義務に深刻に違反し、適用可能な許可プロトコルを終了するか、または場合によっては適用許可プロトコルの下の1つまたは複数の許可を終了することを選択することができ、この終了は、おそらくカバー可能な候補製品および技術を開発および商業化することができなくなるであろう。第三者入網許可が終了した場合、あるいは第三者入網許可下の基礎特許が予想される排他性を提供できなかった場合、競争相手は規制部門の承認を求め、私たちと同じ製品を市場に出す権利がある。これらの事件のいずれも、私たちの競争地位、業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
私たちが持って許可された特許と特許出願は、私たちのプラットフォーム技術、私たちの候補製品、そして私たちの未来の候補製品に十分な保護を提供することができないかもしれません。
我々は、塩基編集および遺伝子標的技術、および私たちの交付プラットフォーム技術を含む多くの発行された米国特許および特許出願を許可した。我々は、我々の基礎編集プラットフォーム技術および特定の疾患および疾患の治療に関連する用途を専門的にカバーすることを目的とした臨時特許出願または特許協力条約(PCT)出願を出願しており、現在発行されている3つの米国特許を有している。私たちは、私たちの交付プラットフォーム技術を専門的にカバーすることを目的とした臨時特許出願またはPCT出願を出願しているが、現在は発行された米国特許は何も持っていない。いずれの米国仮特許出願も、適用された仮特許出願が提出された日から12ヶ月以内に非仮特許出願を提出しない限り、発行された特許となる資格がない。この時間内に非一時的特許出願を提出できなかったいかなる行為も、関連する一時特許出願に開示された意図のために特許保護を得る能力を失う可能性がある。もし彼らがそうすれば、これらの特許は、私たちの基本編集プラットフォーム技術、交付プラットフォーム技術、または私たちの候補製品をカバーまたは十分に保護するかどうか、またはこれらの特許が挑戦、縮小、回避、無効、または実行できないかどうか、これらの特許出願のいずれかが特許として発行されるかどうかを決定することはできない。私たちの基本編集プラットフォーム技術、交付プラットフォーム技術と候補製品に関連する特許保護を獲得または維持できなかったことは、私たちの業務、財務状況、運営結果と成長の将来性に重大な不利な影響を与える可能性がある。
私たちが持っている特許および特許出願および私たちが許可した特許および特許出願は、当基地が候補製品を編集するための物質からなる声明と、そのような候補製品を用いた遺伝子治療のための方法とを含む。使用方法特許は、競争者または他の第三者が特許方法の範囲を超える指示のために同じ製品を開発または販売することを阻止しない。さらに、使用方法特許に関しては、競合他社または他の第三者が、私たちが特許を取得する可能性のあるターゲット化された適応または用途に対して彼らの製品を積極的に宣伝していなくても、サプライヤーは、ラベルの外でこれらの製品を使用することを患者に提案するか、または患者自身がそうすることを提案する可能性がある。
76
バイオテクノロジーや製薬分野の特許実力は複雑な法律や科学問題に関連しており,不確定である可能性がある。私たちが所有しているまたは許可されている特許出願は、発行された特許を生成することができない可能性があり、その声明は、私たちの候補製品または米国または他の国/地域におけるその使用をカバーする。例えば、私たちの特許出願保留中に、私たちは、第三者によって米国特許商標局またはUSPTOに既存技術を予め提出しておくか、妨害または派生手続きに巻き込まれたり、外国の管轄地域で同等の手続きを行ったりする可能性がある。特許が確実に発行されても,第三者は異議申立,撤回,再審査,付与後と各方面間訴訟手続きを審査する。このような提出、訴訟、または訴訟における不利な裁決は、私たちが所有しているまたは許可内の特許権の範囲を縮小したり、無効にしたり、実行することができず、第三者が私たちの技術または候補製品を商業化し、私たちに支払うことなく、直接競争することを可能にしたり、第三者特許権を侵害することなく製品を製造または商業化することができなくなる可能性があります。さらに、私たちまたは私たちのライセンシーのうちの1つは、発明の優先権を決定するために、米国特許商標局が発表した干渉手続に参加しなければならないか、または付与された挑戦手続において、例えば、外国特許庁の反対意見において、私たちが所有または許可している特許および特許出願について、私たちまたは私たちのライセンス者の発明優先権または他の特許可能な特徴に挑戦しなければならない可能性がある。このような挑戦は、特許権の喪失、排他性の喪失、または特許主張の縮小、無効または実行不能をもたらす可能性があり、これは、他人が類似または同じ技術および製品を使用することを阻止するか、またはそれを商業化する能力を制限するか、または我々の技術および候補製品の特許保護期間を制限する可能性がある。また,それらが挑戦されていなくても,我々の特許や特許出願は,我々の知的財産権を十分に保護したり,我々の主張をめぐる他の人の設計を阻止したりすることができない可能性がある.もし私たちが持っている特許と特許出願が提供する保護の広さや強度が脅かされている場合、あるいは私たちが許可している私たちの基礎編集プラットフォーム技術、配信プラットフォーム技術および候補製品に関連する特許および特許出願が脅かされていれば、会社が私たちと協力して私たちの候補製品を開発することを阻止し、私たちがそれを商業化する能力を脅かすかもしれない。また、新製品候補の開発、テスト、規制審査に遅延があれば、特許保護の下で候補製品を販売できる時間が短縮されるだろう。
米国および他の国の特許出願が提出されてから一定期間秘密であることを考慮すると、いつでも、私たちまたは私たちの許可者が過去または将来、私たちの基礎編集技術、交付プラットフォーム技術または候補製品に関連する任意の特許出願を最初に提出した会社であることを決定することはできない。さらに、米国のいくつかの特許出願は特許が発行されるまで秘密にされているかもしれない。したがって、私たちまたは私たちの許可者が知らない既存技術が存在する可能性があり、これらの技術は特許主張の有効性または実行可能性に影響を与える可能性があり、私たちまたは私たちの許可者は優先権紛争の影響を受ける可能性がある。私たちのライセンス内特許の組み合わせについては、私たちのライセンシーに依存して在庫を決定し、優先出願をPCT出願に変換する前に優先出願の発明者譲渡を取得して保存する。これがタイムリーにできなければ,このようなPCT申請から国有化された外国出願の優先権に挑戦する可能性がある。例えば、欧州特許庁、又は欧州特許庁、反対部、又は欧州特許庁反対部は、我々が選択したブロッド研究所特許欧州特許番号を撤回した。EP 2771468は、第三者がその優先度に挑戦した後である。この特許は優先権の喪失によって撤回された。われわれ又はわれわれの許可者は、欧州又は他の外国司法管区の訴訟手続又は優先権紛争の影響を受け、将来的に訴訟手続又は優先権紛争の当事者となる可能性がある。このようなヨーロッパ特許の優先権を失ったり、これらの特許を失ったりすることは、私たちの業務行動に重大な悪影響を及ぼす可能性がある。
私たちは特定の特許または特許出願の一部または全部の期間を放棄することを要求されるかもしれない。私たちが知らないいくつかの既存技術は、特許請求の範囲の有効性または実行可能性に影響を及ぼす可能性があるかもしれない。私たちまたは私たちのライセンシーが知っているかもしれませんが、私たちまたは私たちのライセンス者は、クレームの有効性や実行可能性に影響を与えるとは考えていませんが、最終的には、クレームの有効性または実行可能性に影響を与えることが発見される可能性があります。もし私たちの特許が挑戦された場合、私たちの特許は裁判所、特許庁または他の政府当局によって有効または強制執行可能であると宣言されるか、または有効かつ強制的に実行可能であることが発見されても、競争相手の技術または製品は裁判所によって私たちの特許を侵害したと認定されることは保証されない。私たちは、私たちの活動に関連すると考えられる競争相手の特許または特許出願を分析し、私たちの候補製品に対して自由に運営できると考えているかもしれませんが、私たちの競争相手は、私たちが関係ないと思う特許を含むクレームを出すかもしれません。これらのクレームは、私たちの努力を阻害したり、私たちの製品候補や私たちの活動がこのようなクレームを侵害したりする可能性があります。私たちの競争相手は特許出願を提出した可能性があり、将来的には私たちと類似した製品または技術をカバーする特許出願を提出する可能性がある。これらの特許出願は、我々が所有している特許出願および認可内の特許出願または特許よりも優先する可能性があり、これは、そのような技術をカバーする発行された特許を取得する権利を要求することができる。他の会社が、私たちの特許または他の知的財産権を侵害することなく、私たちの候補製品と同じ効果を有する製品を開発するか、または私たちの特許出願または私たちの許可内の特許または私たちの候補製品をカバーする特許出願の権利要件をめぐり設計される可能性もある。
77
同様に、我々が現在所有している特許および特許出願(特許として発行されている場合)および許可中の特許および特許出願(特許として発行されている場合)は、可能な特許期限の調整または延長を考慮することなく、我々の独自基礎編集技術および我々の候補製品について、可能な特許期限の調整または延長を考慮することなく、2034年から2044年までの期限が予想される。私たちが持っているまたは許可中の特許は、私たちの最初の候補品がアメリカまたは外国司法管轄区で発売承認される前または直後に満期になるかもしれません。さらに、米国特許商標局または関連外国特許庁が、現在または将来所有しているまたは許可されている任意の未解決特許出願を承認することは保証されない。私たちの既存の特許が満期になると、私たちは他の人たちを排除してこのような発明を実施する権利を失うかもしれない。これらの特許の満期はまた、私たちの業務、財務状況、運営結果、および将来性に類似した大きな悪影響を及ぼす可能性があります。
我々が所有する特許及び特許出願及びライセンス内の特許及び特許出願及びその他の知的財産権は、優先権紛争又は在庫紛争及び同様の訴訟の影響を受ける可能性がある。もし私たちまたは私たちのライセンシーがこれらの訴訟で失敗した場合、私たちは第三者から許可を得ることを要求されるかもしれません。これらの許可は商業的に合理的な条項では得られないかもしれません。あるいは私たちが開発する可能性のある1つ以上の候補製品の開発、製造、商業化を停止することは、私たちの業務に大きな悪影響を及ぼす可能性があります。
特定の特許とCas 9およびCas 12 aに対するEditasからの特許出願を独占的にライセンスすることができ、Editasはブロス研究所を含む様々な学術機関からこのような特許の許可を得ることができるが、私たちは現在そのような特許および特許出願のライセンスを持っていない。私たちが選択権を持っているいくつかのアメリカ特許と1つのアメリカ特許出願はブロッド研究所とマサチューセッツ工科大学が共同で所有しており、場合によってはブロダー研究所、マサチューセッツ工科大学、ハーバードが共同所有しており、私たちは総称してボストン許可側と呼ばれ、カリフォルニア大学、ウィーン大学、エマニュエル·チャペンティエが共同所有する米国特許出願に対する米国の干渉に参加しており、私たちは総称してカリフォルニア大学と呼ばれている。2018年9月10日、連邦巡回控訴裁判所(CAFC)は、事実介入がないと判断した米国特許商標局(PTAB)の特許裁判と控訴委員会を確認した。干渉は、異なる当事者によって提出された特許請求の範囲の標的の発明優先権を決定することを目的とする、米国特許商標局内のプログラムである。
2019年6月24日、PTABは、カリフォルニア大学が共同所有する10件の米国特許出願(米国シリアル番号15/947,680;15/947,700;15/947,718;15/981,807;15/981,808;15/981,809;16/136,159;16/136,165;16/136,168および16/136,175)と、13件の米国特許および1つの米国特許出願(米国特許8,697,359;8,771,945;8,795,965;8,865,406;8,871,445;8,889,356;895,8308;8,916,916;916,916,932;916,916,932;916,916,916,916,932;916,916,916,916,918)を発表した。8,945,839;8,993,233;8,999,641;および9,840,713,および米国シーケンス番号14/704,551))は,ボストンライセンス側が共同で所有しており,Editasライセンスプロトコルに従って選択する権利がある.発表された介入では、カリフォルニア大学は一次政党、ボストン許可側は高級政党に指定された。
干渉を宣言した結果,PTAB前に米国特許商標局で対抗プログラムが起動され,このプログラムは優先度,具体的には,どちらがまず保護が要求されるテーマを最初に発明したかを最終的に決定するためであることを宣言した.干渉は通常2段階に分けられる.第1段階は運動段階または予備運動段階と呼ばれ、第2段階は優先段階と呼ばれる。第1段階では、各当事者は、一方の当事者が既存技術、書面記述、および許可された権利要件に基づく特許性に関連する問題を含むが、これらに限定されない問題を提起することができる。一方の当事者は,より早い優先権利益を求めたり,まず干渉が適切であるかどうかを宣言したりすることも可能である.干渉の第2段階で優先権を決定するか、または誰が一般的に保護を必要とする発明を最初に発明したかを決定する。カリフォルニア大学の10特許出願およびボストンライセンス者が共同所有する13件の米国特許および1つの米国特許出願は、一般に、CRISPR/Cas 9システムまたは融合または共有結合RNAを有するCRISPR/Cas 9システムを含む真核細胞および真核細胞におけるそれらの使用に関する。2022年2月28日、PTABは、カリフォルニア大学に対して真核細胞で機能する単一RNA CRISPR-Cas 9システムの発明優先権を有するボストン許可者の裁決を発表した。この決定は控訴されている。アメリカの介入が控訴時にボストン許可側に有利な解決を得る保証はありません。米国の介入結果がカリフォルニア大学に有利である場合、またはボストン許可側の特許および特許出願が縮小され、無効または強制的に実行できない場合、選択可能な特許および特許出願を許可する能力を失う可能性があり、候補製品をカバーする関連第三者特許の許可を得ることができなければ、候補製品を商業化する能力は悪影響を受ける可能性がある。私たちは商業的に合理的な条項や必要な許可証を得ることができないかもしれない。たとえ私たちが許可を得ることができても、それは非排他的である可能性があり、それによって、私たちの競争相手と他の第三者が私たちに許可された同じ技術にアクセスできるようにすることができ、これは私たちが大量の許可と印税を支払う必要があるかもしれない。もし私たちが商業的に合理的な条項で第三者特許の必要な許可を得ることができなければ、私たちの基礎編集プラットフォーム技術や候補製品を商業化できないかもしれません。あるいはこのような商業化努力は著しく遅れる可能性があり、逆に私たちの業務を深刻に損なう可能性があります。
78
私たちまたは私たちの許可者たちは未来に似たような妨害を受けるかもしれないが、その危険は上述したように。例えば、2020年12月14日、PTABは14件の米国特許および2つの米国特許出願(米国特許番号8,697,359;8,771,945;8,795,965;8,865,406;8,871,445;8,889,356;8,889,418;8,895,308;8,906,616;8,932,814;8,945,839;8,993,233;8,999,641;および9,840,713およびボストンライセンスによって共同所有される米国特許出願番号14/704,551および15/330,876)と、ToolGen,Inc.またはToolGenによって所有される米国特許出願(米国特許番号14/685,510)。宣言された介入では,ボストン許可側は初級側,ToolGenは高級側に指定された。2021年3月、PTABは、ボストン許可者およびToolGenによって提案されたいくつかの動議を部分的に承認および部分的に拒否する予備動議に関する命令を発行した。2022年9月12日、米国第106,126号妨害案優先段階の口頭公聴会が行われた。2022年9月28日、PTABは、ボストン許可者およびToolGenによって提案されたいくつかの動議を拒否または却下する予備動議に関する決定を発表し、干渉優先段階の手続きを一時停止する命令を発行した。私たちはいつ決定が下されるのか確実に予測できない。米国干渉106,126号に関連するボストンライセンスが共同所有する14の米国特許および2つの米国特許出願は、一般に、CRISPR/Cas 9システムまたは融合または共有結合を有するRNAを含むCRISPR/Cas 9システムを含む真核細胞および真核細胞におけるそれらの使用に関する。
2021年6月21日、PTABは、同じ14の米国特許および2つの米国特許出願(米国特許番号8,697,359;8,771,945;8,795,965;8,865,406;8,871,445;8,889,356;8,889,418;8,895,308;8,906,616;8,932,814;8,945,839;8,993,233;8,999,641;そして、Sigma-Aldrich Co.,LLCまたはSigma-Aldrichによって所有される米国特許出願(米国特許番号15/456,204)。発表された介入では,ボストン許可側が初級党,Sigma−Aldrichが高級党に指定された。2021年9月、PTABは、ボストン許可者およびSigma-Aldrichによって提案されたいくつかの動議を承認、延期、却下、または拒否する予備動議に関する命令を発行した。2022年11月21日,米国第106,133号介入優先段階の口頭公聴会が開催された。2022年12月14日、PTABは、ボストン許可者およびSigma-Aldrichによって提出されたいくつかの動議の予備動議を拒否または却下することに関する決定を発表し、干渉優先段階の手続きを一時停止する命令を発行した。私たちはいつ決定が下されるのか確実に予測できない。
私たちまたは私たちの許可者はまた、元従業員、協力者または他の第三者が、発明者または共同発明者として、私たちが所有している特許または特許出願、許可内の特許または特許出願または他の知的財産権において権利を有するクレームの制約を受ける可能性がある。もし私たちがこのような特許または特許出願における第三者共同所有者の権利の独占的許可を得ることができない場合、これらの共通所有者は、私たちの競争相手を含む他の第三者に権利を許可することができるかもしれない。さらに、私たちは、そのような特許出願から発行された任意の特許を第三者に強制的に実行するために、このような共同所有者の協力が必要である可能性があり、そのような協力は私たちに提供されないかもしれない。
私たちまたは私たちのライセンシーが、私たちまたは彼らが直面している任意の妨害訴訟または他の優先権、有効性(任意の特許異議を含む)または発明権紛争に失敗した場合、私たちは、私たちが所有している、許可されている、またはオプションの1つまたは複数の特許を失うことによって、貴重な知的財産権を失うことができ、またはそのような特許主張が縮小され、無効にされたり、強制的に実行されないか、または私たちが所有しているまたは許可内の特許の独占所有権または独占使用権を失うことによって失われる可能性がある。このような紛争が特許権の喪失を招いた場合、そのような妨害手続または他の優先権または在庫紛争に参加する当事者を含む第三者から許可を取得し、維持することを要求される可能性がある。このような許可は商業的に合理的な条項では得られないかもしれないし,まったく存在しない,あるいは非排他的である可能性がある.もし私たちがそのようなライセンスを取得して維持できなければ、私たちが開発する可能性のある1つまたは複数の候補製品の開発、製造、商業化を停止する必要があるかもしれない。排他性の喪失や私たちの特許主張の縮小は、他の人が類似または同じ候補技術および製品を使用または商業化することを阻止する能力を制限するかもしれない。我々または我々のライセンシーが介入手順や他の同様の優先権や発明権紛争で勝訴しても、巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。上記のいずれも、当社の業務、財務状況、経営結果、または将来性に重大な悪影響を及ぼす可能性があります。
私たちは限られた外国の知的財産権を持っており、世界各地で私たちの知的財産権と独自の権利を保護できないかもしれない。
私たちのアメリカ以外の知的財産権は限られている。世界のすべての国で候補製品の申請、起訴、特許保護の費用は目を引くほど高く、私たちのアメリカ以外のいくつかの国での知的財産権はアメリカほど広くないかもしれない。また、外国の法律は知的財産権の保護の程度は米国の連邦や州法に及ばない。しかも、私たちの知的財産権許可協定は常に世界的な権利を含むわけではないかもしれない。したがって、私たちは、米国以外のすべての国/地域で第三者が私たちの発明を実施したり、米国または他の管轄区域で私たちの発明を使用して製造された製品を販売または輸入することを阻止することができないかもしれない。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの技術を使用して自分の製品を開発することができ、また、他の侵害製品を私たちが特許保護を持っているが法執行力がアメリカに及ばない地域に輸出することもできる。これらの製品は私たちの候補製品と競争するかもしれませんが、私たちの特許や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。
79
多くの会社は外国の管轄区域の知的財産権の保護と保護に重大な問題に直面している。特定の国の法律制度、特に特定の発展途上国の法律制度は、特許、商業秘密、および他の知的財産権保護、特にバイオテクノロジーおよび医薬製品に関する保護の強制執行を支持しておらず、これは、私たちの特許の侵害や第三者の競争製品の販売に対する行為を阻止することを困難にする可能性があり、これらの行為は、全体的に私たちの知的財産権と専門権を侵害する。外国の管轄区域で私たちの特許と知的財産権を強制的に執行する訴訟手続きは巨額のコストを招く可能性があり、私たちの努力と注意を私たちの業務の他の方面から移すことは、私たちの特許を無効または狭義に解釈されるリスクに直面させる可能性があり、私たちの特許出願を発行できないリスクに直面させ、第三者が私たちにクレームを提起する可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。また、第三者は、外国管轄区域における特許権の範囲または有効性が巨額のコストを招く可能性があることに挑戦し、私たちの努力と注意を私たちの業務の他の側面から移すための訴訟を開始した。したがって、私たちの知的財産権および独自の権利を世界各地で強制的に実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネス的優位性を得るのに十分ではないかもしれません。
多くの国に強制許可法があり,これらの法律により特許権者は第三者に強制的に許可を付与される可能性がある。また、多くの国は政府機関や政府請負業者に対する特許の実行可能性を制限している。これらの国では,特許権者の救済措置は限られている可能性があり,このような特許の価値を大幅に低下させる可能性がある。もし私たちまたは私たちの任意の許可者が第三者に私たちの業務に関連する任意の特許の許可を付与することを余儀なくされた場合、私たちの競争的地位が損なわれる可能性があり、私たちの業務、財務状況、運営結果、および見通しは不利な影響を受ける可能性があります。
もし私たちが第三者に知的財産権を許可する合意の義務を履行できなかった場合、あるいは私たちとライセンス者との業務関係が妨害された場合、私たちは私たちの業務に非常に重要な許可権を失う可能性があります。
私たちは第三者とライセンス契約を締結しており、私たちの研究を進めたり、私たちが開発可能な候補製品の商業化を可能にするために、既存のライセンシーや他のライセンシーから追加の許可を得る必要があるかもしれません。私たちは合理的な費用や合理的な条項で任意の追加のライセンスを得ることができないかもしれない。いずれの場合も、私たちは、私たちの技術、候補製品、またはそれらを製造する方法を再設計するために、多くの時間および資源を必要とすることができ、または代替技術を開発または許可することは、技術的にも商業的にも不可能である可能性がある。もし私たちがそれができなければ、影響を受けた候補製品を開発したり商業化することができないかもしれません。これは、私たちの業務、財務状況、運営結果、将来性を深刻に損なう可能性があります。基礎編集技術、交付プラットフォーム技術、製造方法、候補製品、または私たちの製造または将来の販売を禁止することをもたらす未来の方法または製品、または私たちの将来の販売について、第三者に印税および/または他の形態の賠償を支払う義務があることは、私たちの現在の技術に対して強制的に実行される可能性のある第三者特許が存在しない保証はありません。
私たちのすべてのライセンス協定で、私たちは一般的に私たちが許可した特許を侵害する任意の第三者に対する訴訟を担当します。私たちのいくつかのライセンス協定はまた、製品の開発と商業化のためのスケジュールを設定することを含む、開発のハードルを達成して許可を維持することを要求しています。私たちの努力にもかかわらず、私たちの許可側は、このようなライセンス協定の下での義務に深刻に違反していると結論する可能性がありますので、ライセンス契約を終了し、これらのライセンスプロトコルに含まれる製品や技術を開発し、商業化する能力を廃止または制限する可能性があります。これらの許可が終了した場合、または基礎特許が予期される排他性を提供できなかった場合、競争相手または他の第三者は、規制部門の承認を求め、私たちと同じ製品を市場に出す権利がある場合、開発および商業化または基礎編集プラットフォーム技術、配信プラットフォーム技術、または候補製品の開発および商業化を要求される可能性がある。上記のいずれも、我々の競争地位、業務、財務状況、経営結果、成長見通しに重大な悪影響を及ぼす可能性がある。ライセンス契約によると、知的財産権に関する論争が発生する可能性があります
80
また,我々が現在第三者から知的財産権や技術許可を得ているプロトコルは複雑であり,このようなプロトコルのいくつかは様々な解釈の影響を受ける可能性がある.起こりうる任意の契約解釈の相違の解決は、関連する知的財産権または技術に対する私たちの権利と考えられる範囲を縮小したり、ライセンシーが私たちの知的財産権および技術に対する権利であると考えられる範囲を拡大したり、関連協定の下での私たちの財務または他の義務を増加させる可能性があり、いずれも私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。また、我々が許可している知的財産権をめぐる紛争が、商業的に許容可能な条項で現在の許可手配の能力を維持していることを阻害したり、損害を与えたりすれば、影響を受けた候補製品の開発に成功し、商業化することができない可能性がある。したがって、私たちの知的財産権許可の任意の終了または論争は、私たちの基礎編集プラットフォーム、配信プラットフォーム、または他の候補製品を開発および商業化する能力を失う可能性があり、または私たちは他の重要な権利を失う可能性があり、いずれも、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。場合によっては、第三者はまた、いくつかの同じ技術の限られた許可を得る可能性がある。
私たちはキーテクノロジーや私たちが開発する可能性のある任意の候補製品の必要な権利を取得したり許可したりすることに成功できないかもしれない。
我々は現在知的財産権の権利を有しており,第三者の許可により候補製品を決定·開発し,我々の候補製品パイプラインの拡大,内部許可のキー技術を部分的に通過する権利を求める予定である.私たちのビジネスの将来の成長は、私たちが許可する能力があるかどうか、または他の方法でより多くの候補製品および技術を得る権利があるかどうかにある程度依存するだろう。私たちは過去に第三者許可者(ハーバード大学、遠大研究院、Editas、Bio Paletteを含む)から技術許可を得ることに成功しましたが、許容可能な条項または全く受け入れられない条項で第三者から任意の候補製品または技術の許可または権利を得ることができることを保証することはできません。
例えば、私たちがある第三者許可者と締結したプロトコルは、私たちの使用領域は特定の領域を含まず、例えば、特定の遺伝子編集技術を使用して、特定の工学的T細胞を用いてヒト癌の診断、治療、および予防を行い、これらのT細胞は、第三者に独占的または非独占的に許可されることを規定している。もし私たちがこれらの分野の権利が私たちの候補薬物の商業化または私たちの競争優位性を維持するために必要であると判断した場合、私たちは、私たちの候補薬剤を開発、製造、またはマーケティングを継続するために、第三者からライセンスを取得する必要があるかもしれない。私たちは独占的に、商業的に合理的な条項、またはそのような許可を得ることができないかもしれません。これは、候補薬物を商業化することを阻止したり、私たちの競争相手や他の人が私たちの業務に重要な技術を得る機会を得ることを可能にするかもしれません。
また、遺伝子編集·交付技術分野には広範な特許出願活動が存在し、製薬会社、バイオテクノロジー会社、学術機関は遺伝子編集·交付技術分野で我々と競合しているか、または遺伝子編集·交付技術分野で我々と競合することが予想され、当社の業務に関連する可能性のある特許出願を提出し、いくつかの第三者特許を知っており、発行された場合には第三者が特許権を回避することを可能にする可能性がある特許出願を提供する。例えば、発行されれば、我々の基礎編集技術、配信技術、および候補製品をカバーすると解釈される可能性がある第三者特許および特許出願がいくつかあることが知られている。私たちの候補製品をマーケティングするためには、このような第三者知的財産権所有者から許可を得ることが必要または慎重であることが分かるかもしれません。しかし、私たちはこのような許可を得ることができないかもしれないし、他の方法で第三者から取得することができないかもしれませんが、私たちが開発する可能性のある候補製品に必要な任意の成分、使用方法、プロセス、または他の知的財産権は、編集および技術に基づいていると考えられます。また、我々が開発および将来開発可能な候補製品のための他の交付方法を含めて、第三者から他の非基礎編集技術のライセンスを取得する必要があるかもしれない。さらに、私たちが持っているいくつかの特許および特許出願および授権中の特許および特許出願は、第三者と共同で所有されている。第三者と共同所有するいかなる特許についても、私たちはこのような共通の所有者のこのような特許の利益に許可を提供する必要があるかもしれない。もし私たちがこのような特許または特許出願における第三者共同所有者の権利の独占的な許可を得ることができない場合、これらの共通所有者は、私たちの競争相手を含む他の第三者に権利を許可することができ、私たちの競争相手は競争製品および技術を販売することができる。さらに、私たちは、第三者に対してそのような特許を強制的に実行するために、私たちの特許のどのような共通所有者の協力も必要とするかもしれませんが、そのような協力は私たちに提供されないかもしれません。
また,学術機関と協力し,これらの機関と合意した書面により,われわれの臨床前研究や開発を加速する可能性がある。場合によっては、これらの機関は、協力によって生成された機関の技術的な任意の権利について交渉することができるオプションを提供してくれる。たとえ私たちがこのような選択肢を持っていても、私たちは交渉できないかもしれない
81
指定された時間範囲内で、または私たちが受け入れることができる条項の下で機関から許可を得る。もし私たちがそれができなければ、その機関は他の人たちに知的財産権を提供するかもしれないし、私たちの計画を実行し続けることを阻止するかもしれない。
また、第三者知的財産権のライセンスまたは買収は競争の激しい分野であり、一部の成熟した企業も魅力的または必要と考えられる第三者知的財産権ライセンスまたは買収戦略をとっている。これらの老舗会社はその規模、資本資源及び更に強い臨床開発と商業化能力のため、私たちより競争優位を持っているかもしれない。しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。私たちはまた私たちが投資から適切な見返りを得ることを可能にする条項に従って許可したり、第三者知的財産権を取得することができないかもしれない。もし私たちが必要な第三者知的財産権を得ることに成功したり、私たちの既存の知的財産権を維持することができなければ、関連計画や候補製品の開発を放棄しなければならないかもしれません。これは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。
遺伝子編集技術(塩基編集および交付技術を含む)をめぐる知的財産権構造は高度に動的であり、第三者は法的訴訟を提起する可能性があり、私たちが彼らの知的財産権を侵害、流用、または他の方法で侵害することを告発する可能性があり、その結果は不確定であり、私たちの製品発見と開発を阻止、延期、または妨害する可能性がある。
遺伝子編集分野、特に基地編集技術分野はまだ初期段階であり、基地編集製品候補はまだ発売されていない。私たちと私たちの競争相手を含むいくつかの会社がこの分野や技術分野で緊張した研究開発を行っているため、知的財産権の構造が変化し、変化しており、今後数年間も不確定である可能性がある。将来的には知的財産権に関する重大な訴訟や、私たちが所有しているものや許可されていない他の第三者、知的財産権、独自の権利に関する訴訟があるかもしれない。
私たちのビジネス成功は、私たちの能力と、私たちの協力者およびライセンシーが、第三者の知的財産権および独自の権利を侵害、流用、または他の方法で侵害することなく、任意の候補製品を開発、販売する能力に依存しています。生物技術と製薬業界の特徴は、特許とその他の知的財産権に関する広範な訴訟と特許に挑戦する行政訴訟であり、干渉、派生、各方面間米国特許商標局の再審、認可後の再審及び再審手続又は外国司法管轄区の異議及びその他の同様の手続。私たちの基礎編集プラットフォーム技術、配信プラットフォーム技術、私たちが開発する可能性のある任意の候補製品に関する知的財産権について、私たちは、妨害プログラム、許可後の審査、各方面間米国特許商標局の審査及び派生手続、並びに欧州特許庁の反対意見のような外国司法管轄区域の類似手続。我々が我々の候補製品を開発している分野には,第三者が所有する米国や外国から発行された特許や係属中の特許出願が多く存在し,それらは既存特許または将来付与される可能性のある特許に基づいて,その是非曲直にかかわらず侵害請求を行う可能性がある.
生物技術と製薬業界の拡張とより多くの特許の発行に伴い、私たちの基地編集プラットフォーム技術、交付プラットフォーム技術と製品候補は他人の特許権侵害のクレームを引き起こすリスクが増加する可能性がある。さらに、私たちを含む業界参加者は、どの特許が様々なタイプの療法、製品、またはそれらの使用または製造方法をカバーするかを常に明確にしているわけではない。私たちはいくつかの第三者特許と特許出願を知っており、発表されれば、私たちの基本編集技術、交付技術、および候補製品をカバーすると解釈されるかもしれない。我々が現在知らない第三者特許、すなわち、私たちの候補製品の使用または製造に関連する技術、製造方法、または治療方法の請求項もあるかもしれない。特許出願が発行されるまでに数年かかる可能性があるため、現在処理されている特許出願が存在する可能性があり、これらの出願は、我々の候補製品が発行された特許を侵害する可能性がある。さらに、第三者は将来的に特許を取得し、私たちの技術を使ってこれらの特許を侵害したと主張するかもしれない。
82
我々が候補製品を開発している分野には,第三者米国や外国から発行された特許や係属中の特許出願が多く存在する.私たちの候補製品はCRISPRに基づく技術を使用しており、これは特許出願が非常に活発な分野である。CRISPRおよびCAに関連する大量の特許出願は、我々の基礎編集プラットフォーム技術および候補製品およびそれらの使用または製造をカバーすることができる関連特許および係属中の出願の範囲を全面的に評価することを困難にする。我々の基礎編集プラットフォーム技術および候補製品の使用または製造に関連する第三者特許または特許出願、これらの特許または特許出願は、材料、処方、製造方法または治療方法を使用または製造することができる。例えば、カリフォルニア大学、ウィーン大学、およびEmmanuelle Charpentierによって共同所有される特許の組み合わせ、または複数の特許および遺伝子編集のための係属中の出願を含むカリフォルニア大学の組み合わせが知られている。The University of California portfolio includes,for example,U.S.Patent Nos.10,266,850;10,227,611;10,000,772;10,113,167;10,301,651;10,308,961;10,337,029;10,351,878;10,407,697;10,358,659;10,358,658;10,385,360;10,400,253;10,421,980;10,415,061;10,428,352;10,443,076;10,487,341;10,513,712;10,519,467;10,526,619;10,533,190;10,550,407;10,563,227;10,570,419;10,577,631;10,597,680;10,612,045;10,626,419;10,640,791;10,669,560;10,676,759;10,752,920;10,774,344;10,793,878;10,900,054;10,982,230;10,982,231;10,988,780;10,988,782;11,001,863;11,008,589;11,008,590;11,028,412;11,186,849;11,242,543;11,274,318;11,293,034;11,332,761;11,401,532;11,473,108;11,479,794;11,549,127;11,634,730;11,674,159;11,814,645項目は、2033年3月頃に満了すると予想され、追加の特許期限調整(PTA)または特許期限延長(PTE)および放棄された任意の端末免責条項を含まない。カリフォルニア大学のポートフォリオには他にも多くの未解決の特許出願が含まれている。これらの特許出願が特許として発行された場合、それらは、PTA、PTE、および放棄された端末免責条項を含まず、2033年3月頃に満了すると予想される。上述したように、カリフォルニア大学ポートフォリオのいくつかの出願は、現在、ボストンライセンス者によって共同所有されているいくつかの米国特許および米国特許出願に関する米国106,115号によって妨害されており、私たちはEditasライセンスプロトコルに従ってこれらの特許を選択する権利がある。特定の特許とCas 9およびCas 12 aに対するEditasからの特許出願を独占的にライセンスすることができ、Editasはブロス研究所を含む様々な学術機関からこのような特許の許可を得ることができるが、私たちは現在そのような特許および特許出願のライセンスを持っていない。カリフォルニア大学ポートフォリオのいくつかのメンバーはヨーロッパで複数の政党に反対されている。例えば、欧州特許庁反対部は、2033年3月に満了することが予想される欧州特許番号EP 2,800,811 B 1およびEP 3,241,902 B 1およびEP 3,401,400 B 1に対して反対手続を開始している(いかなる特許期限の調整または延長も含まれていない)。
欧州特許庁の反対手続は,欧州特許付与日から9ヶ月以内に付与された欧州特許の有効性に疑問を提起することを1つ以上の第三者に許可する。異議訴訟に係る可能性のある問題には,優先権,関連特許請求の特許性,および特許出願の提出に関する手続が含まれるが,これらに限定されない。反対手続の結果として,反対部は特許を取り消し,付与された特許を維持したり,修正された形で特許を維持したりすることができる。欧州特許EP 2,800,811 B 1の大部分の権利要件は野党部門の修正を受けていないが,この決定は控訴している。2021年4月、欧州特許EP 3,241,902 B 1の権利要件はすべて取り消され、この決定は上訴されなかった。2022年2月、欧州特許EP 3,401,400 B 1の権利は、修正された形で野党に維持されることが要求され、この決定は控訴している。これらの特許が反対部によって維持されている場合、その権利主張は現在反対されている特許と類似しており、これらの特許の許可を得ていなければ、候補製品を商業化する能力は悪影響を受ける可能性がある。私たちは商業的に合理的な条項や必要な許可証を得ることができないかもしれない。たとえ私たちが許可を得ることができても、それは非排他的である可能性があり、それによって、私たちの競争相手と他の第三者が私たちに許可された同じ技術にアクセスできるようにすることができ、これは私たちが大量の許可と印税を支払う必要があるかもしれない。もし私たちが商業的に合理的な条項で第三者特許の必要な許可を得ることができなければ、私たちの基礎編集プラットフォーム技術や候補製品を商業化できないかもしれません。あるいはこのような商業化努力は著しく遅れる可能性があり、逆に私たちの業務を深刻に損なう可能性があります。
他の第三者は、免疫治療またはキメラ抗原受容体に関する遺伝子編集、誘導核酸、PAM配列変異体、分裂イントロン、Cas 12 bまたは遺伝子編集に関する多くの他の特許および特許出願を提出している。
私たちの分野では大量の特許が発行され、特許出願が提出されているので、第三者は彼らが私たちの候補製品、技術、または方法を含む特許権を持っていると主張するかもしれない。第三者は、私たちが彼らのノウハウを不正に使用していると主張し、特許侵害請求または訴訟を提起する可能性があります。もし私たちがこのような第三者の特許侵害が発見された場合、私たちは損害賠償金の支払い、権利侵害技術の商業化の停止、またはこのような第三者から許可を得ることを要求されるかもしれません。これらの許可は商業的に合理的な条項や全く得られないかもしれません。
83
もし私たちが商業的に合理的な条項で私たちの候補製品、配信プラットフォーム技術、または基礎編集プラットフォーム技術をカバーする関連第三者特許の許可を得ることができなければ、私たちが米国と海外で候補製品を商業化する能力は不利な影響を受ける可能性がある。第三者の知的財産権主張に法的根拠がないと考えても,裁判所が侵害,有効性,実行可能性あるいは優先権問題において我々に有利な裁決を下す保証はない.管轄権のある裁判所は、これらの第三者特許が有効で、強制的に実行可能であり、侵害されていると判断する可能性があり、これは、開発可能な任意の候補製品および主張される第三者特許がカバーする任意の他の候補製品または技術を商業化する能力に実質的な悪影響を及ぼす可能性がある。連邦裁判所でこのような米国特許の有効性に挑戦することに成功するためには,有効性推定を克服する必要がある。この負担が重いため,このような米国特許主張の無効について明確で納得できる証拠を提出することが求められているため,管轄権のある裁判所がこのような米国特許の主張の無効を宣言する保証はない.もし私たちが第三者の知的財産権を侵害していることが発見され、そのような特許が無効または強制的に実行できないことを成功的に証明できなかった場合、私たちは、開発、製造、およびマーケティングの可能性のある任意の候補製品および私たちの技術を継続するために、第三者からライセンスを取得することを要求されるかもしれない。しかし、私たちは商業的に合理的な条項や必要な許可証を得ることができないかもしれない。私たちが許可を得ることができても、それは非排他的である可能性があり、私たちの競争相手や他の第三者が私たちに許可された同じ技術にアクセスできるようにするためには、大量の許可と印税を支払う必要があるかもしれない。もし私たちが商業的に合理的な条項で第三者特許の必要な許可を得ることができなければ、私たちの基礎編集プラットフォーム技術、配信プラットフォーム技術、あるいは候補製品を商業化することができないかもしれません。あるいはこのような商業化努力は著しく遅れる可能性があり、逆に私たちの業務を深刻に損なう可能性があります。私たちはまた裁判所の命令を含む権利侵害技術または候補製品の開発、製造、商業化を停止させることを余儀なくされる可能性がある。さらに、私たちが特許や他の知的財産権を故意に侵害していることが発見された場合、私たちは3倍の損害賠償と弁護士費を含む重大な金銭損害に責任があると判断される可能性がある。第三者の機密情報や商業秘密を盗用したと主張することは、当社の業務、財務状況、運営結果、見通しに類似した重大な悪影響を及ぼす可能性があります。
公金の流用や知的財産権侵害の第三者クレームの弁護には大量の訴訟費用がかかり、管理職と従業員の時間と資源に大量に分流される。いくつかの第三者は、彼らがより多くの資源を持っているので、私たちよりも複雑な特許訴訟の費用を効果的に維持することができる。さらに、いかなる訴訟の開始および継続に生じるいかなる不確実性も、運営を継続するために必要な資金を調達する能力に重大な悪影響を及ぼす可能性があり、または私たちの業務、財務状況、運営結果、および見通しに重大な悪影響を及ぼす可能性がある。公聴会、動議、または他の一時的な手続きや事態の発展の結果も公表される可能性があり、証券アナリストや投資家がこれらの結果がマイナスだと思う場合、私たちの普通株の価格に大きな悪影響を及ぼす可能性がある。上記のいずれの事件も、私たちの業務、財務状況、経営結果、見通しに重大な悪影響を及ぼす可能性があります。
私たちは、私たちの特許、将来の特許、または私たちの許可者の特許を保護または強制する訴訟に巻き込まれる可能性があり、これは高価で、時間がかかり、成功しない可能性があり、そのような特許が実行不可能または無効であることを発見する可能性がある。
競争相手は私たちの特許、未来の特許、またはパートナーの特許を許可するかもしれないし、あるいは私たちは侵害クレームに対する抗弁を要求されるかもしれない。さらに、私たちの特許、将来の特許、または私たちの許可パートナーの特許も関連し、将来的には発明権、優先権、有効性、または実行可能な紛争に関連する可能性がある。このような疑いに反撃したり弁護したりすることは費用が高くて時間がかかるかもしれない。侵害訴訟では、裁判所は、私たちが所有または許可した特許が無効または強制執行できないと判断することができ、または、私たちが所有している特許が関連技術をカバーしていないことを理由に、他方の関連技術の使用を阻止することを拒否することができる。任意の訴訟手続きにおける不利な結果は、私たちが所有しているまたは許可された1つまたは複数の特許を無効または狭義に解釈されるリスクに直面させる可能性がある。
米国の特許訴訟では、被告が特許の無効および/または強制執行不可能と主張する反訴はありふれており、第三者は多くの理由から特許が無効または強制的に実行できないと断言することができる。第三者も米国や海外の行政機関に類似したクレームを出すことができ、訴訟範囲外でも同様である。これらのタイプの仕組みには再審査、贈与後の審査、各方面間審査手続き、介入手続き、派生手続き、外国司法管轄区の同等の手続き(例えば、反対手続き)。このような種類の訴訟は私たちの特許が撤回されたり修正されたりして、私たちの候補製品をカバーしないかもしれない。法的に無効と実行不可能と断言された後、どの特定の特許の結果も予測できない。有効性の問題については、例えば、無効な以前の技術がないことを確認することはできませんが、私たちのライセンシーは、私たちの特許弁護士及び特許審査員が起訴中に知られていません。もし被告が無効および/または強制執行できない法的主張で勝訴した場合、または他の方法で私たちの権利を十分に保護できない場合、私たちは私たちの技術および/または候補製品の少なくとも一部またはすべての特許保護を失うだろう。これらのタイプのクレームを弁護し、その是非にかかわらず、巨額の訴訟費用がかかり、私たちの業務の従業員資源を大量に移転する。
代わりに、私たちはアメリカ特許商標局に再審査、許可を要求することで審査することができます各方面間審査手続き、介入手続き、派生手続き、外国司法管轄区の同等の手続き(例えば、反対手続き)。我々は現在,欧州特許庁や他の外国特許庁の特許反対手続において第三者特許に挑戦しており,将来的にも挑戦を選択する可能性がある.たとえそうであっても
84
成功すれば、このような反対訴訟の費用は巨大で、私たちの時間や他の資源を消費するかもしれない。もし私たちが米国特許商標局、欧州特許庁、または他の特許庁で有利な結果を得ることができなかった場合、私たちは候補製品、基礎編集プラットフォーム技術、交付プラットフォーム技術、または他のまたは独自技術が私たちの特許を侵害している可能性があるという第三者の訴訟に直面する可能性がある。
例えば、上述したように、カリフォルニア大学特許組合の要素はヨーロッパで様々に反対され、私たちは反対手続きに参加している。欧州特許庁反対部は、2033年3月に満了すると予想される欧州特許(特許期間調整または延長を含まない)に反対訴訟を起こしており、これらの特許はカリフォルニア大学が共同で所有している。欧州特許庁の反対手続は,欧州特許付与日から9ヶ月以内に付与された欧州特許の有効性に疑問を提起することを1つ以上の第三者に許可する。異議訴訟に係る可能性のある問題には,優先権,関連特許請求の特許性,および特許出願の提出に関する手続が含まれるが,これらに限定されない。反対手続の結果として,反対部は特許を取り消し,付与された特許を維持したり,修正された形で特許を維持したりすることができる。反対部がいつ、あるいはどのような方法でこれらの欧州特許の反対手続きに行動するかはまだ確定していない。これらの特許が反対部によって維持されている場合、その権利主張は現在反対されている特許と類似しており、これらの特許の許可を得ていなければ、候補製品を商業化する能力は悪影響を受ける可能性がある。私たちは商業的に合理的な条項や必要な許可証を得ることができないかもしれない。たとえ私たちが許可を得ることができても、それは非排他的である可能性があり、それによって、私たちの競争相手と他の第三者が私たちに許可された同じ技術にアクセスできるようにすることができ、これは私たちが大量の許可と印税を支払う必要があるかもしれない。もし私たちが商業的に合理的な条項で第三者特許の必要な許可を得ることができなければ、私たちの基礎編集プラットフォーム技術、配信プラットフォーム技術、あるいは候補製品を商業化することができないかもしれません。あるいはこのような商業化努力は著しく遅れる可能性があり、逆に私たちの業務を深刻に損なう可能性があります。
解決策が私たちに有利であっても、知的財産権クレームに関連する訴訟や他の法的手続きは、巨額の費用を発生させ、私たちの人員の正常な責任を分散させる可能性があります。さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。また、公聴会、動議、または他の一時的な手続きまたは事態の発展の結果が公表される可能性があり、証券アナリストや投資家がこれらの結果がマイナスだと思う場合、私たちの普通株の価格に大きな悪影響を及ぼす可能性がある。このような訴訟または訴訟は、私たちの運営損失を大幅に増加させ、開発活動または任意の将来の販売、マーケティング、または流通活動に使用することができるリソースを減少させる可能性がある。私たちはそのような訴訟や訴訟手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らがより多くの財政資源とより成熟して発展した知的財産権の組み合わせを持っているので、このような訴訟や法的手続きの費用を私たちよりも効率的に負担するかもしれない。特許訴訟または他の訴訟の開始と継続によって生じる不確実性は、市場での競争能力に重大な悪影響を及ぼす可能性がある。
私たちの特許保護の獲得と維持は、政府特許機関によって提出された様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
私たちが所有または許可している特許および出願の有効期間内には、米国特許商標局および米国以外の外国特許代理機関に定期維持費、継続費、年会費、および様々な他の特許および出願政府費用を支払わなければならない。場合によっては、私たちは私たちの許可パートナーに依存して、これらの費用をアメリカと非アメリカの特許機関に支払う。米国特許商標局および外国特許代理機関は、特許出願中にいくつかのプログラム、文書、費用支払い、および他の同様の条項を遵守することを要求する。私たちはまた、私たちが許可した知的財産権に関するこれらの要求を遵守するために、私たちの許可側が必要な行動を取ることに依存している。適用規則によれば、滞納金の支払いや他の方法で過失を是正することができるが、規定を遵守しないことにより、関連法域の特許権の一部または全部が失われる可能性がある。不適切な事件が発生した場合、私たちの競争相手は類似または同じ製品または技術で市場に参入する可能性があり、これは私たちの業務、財務状況、運営結果、および将来性に重大な悪影響を及ぼす可能性がある。
米国と非米国管轄地域の特許法の変化は、特許の全体的な価値を低下させ、私たちのプラットフォーム技術と候補製品を保護する能力を弱める可能性がある。
他のバイオテクノロジーや製薬会社と同様に、私たちの成功は知的財産権、特に特許に大きく依存している。生物製薬業界で特許を獲得と実施することは技術上の複雑性と法律上の複雑性にも関連するため、コストが高く、時間がかかり、しかも内在的な不確定性を持っている。
85
特許法または特許法解釈の変化は、特許出願をめぐる起訴および我々が発行した特許の実行または保護の不確実性およびコストを増加させる可能性がある。例えば、2013年3月、“ライシー-スミス米国発明法”(Leahy-Smith America Invents Act)、または“米国発明法”(America Invents Act)に基づいて、米国は“先発明”から“先出願”特許制度に移行した。“先行出願”制度の下で、他の特許性要件が満たされていると仮定すると、最初に特許出願を提出した発明者は、通常、他の発明者がいるか否かにかかわらず、1つの発明の特許を取得する権利がある。2013年3月以降に米国特許商標局に特許出願の第三者を提出したが、我々の前に、当該第三者が発明を行う前に本発明を作成したとしても、我々の発明をカバーする特許を付与することができる。これは私たちが発明から特許出願までの提出時間を認識することを要求するだろう。米国およびほとんどの他の国/地域の特許出願は、提出後または発行前の一定期間秘密であるため、私たちまたは私たちのライセンス者が、私たちの技術または製品候補に関連する特許出願または発明を最初に提出した私たちまたは私たちのライセンシーの特許または特許出願において主張されている任意の発明の会社であることを確認することはできない。米国発明法には、特許出願起訴方式に影響を与える条項を含む米国特許法の他のいくつかの重大な改正も含まれており、第三者が既存技術の提出を許可し、ライセンス後審査を含む新たな認可後審査制度を確立している各方面間審査と派生手続き。USPTO手続きの証拠基準は、米国連邦裁判所が特許請求の無効を宣言するために必要な証拠基準よりも低いため、第三者は、USPTO手続において、USPTOが権利請求を無効にするのに十分な証拠を提供する可能性があり、同じ証拠が最初に地域裁判所訴訟で提出された場合には、権利請求を無効にするのに十分ではない。したがって,第三者は米国特許商標局の手続きを用いて我々の特許主張の無効を宣言しようとする可能性があり,第三者が地域裁判所訴訟で最初に被告として疑問を提起すれば,我々の特許主張は無効ではない.米国特許商標局が“米国発明法”に関連する新しい法規やプログラムを引き続き公布することに伴い、その中のいくつかの変化の影響は現在のところ不明である。また,裁判所はその多くの条項を扱っておらず,この法案や本出願で議論されている特定特許に関する新法規の適用性は未定であり,審査が必要である。しかしながら、“米国発明法”およびその実施は、我々の特許出願をめぐる起訴および我々が発行した特許の実行または保護の不確実性およびコストを増加させる可能性がある。
さらに、米国最高裁判所の最近の裁決は、場合によっては入手可能な特許保護範囲を縮小し、場合によっては特許所有者の権利を弱める。我々の将来の特許取得能力の不確実性の増加に加えて,このようなイベントの結合は,いったん特許を取得することの有効性と実行可能性に関する不確実性をもたらしている.米国議会、連邦裁判所、USPTOの将来の行動によると、特許を管理する法律·法規は予測不可能な方法で変化する可能性があり、私たちが新しい特許を獲得したり、私たちの既存の特許と私たちが将来獲得する可能性のある特許を強制的に執行する能力を弱める可能性がある。例えば、この場合、アソークです。分子病理学はMyriad Genetics,Inc.を訴える米国最高裁判所は,DNA分子に対するいくつかの主張は特許を申請できないと判断した。私たちは裁判所、アメリカ議会、またはUSPTOのこの決定と未来の判決が私たちの特許価値にどのように影響するか予測できない。他の管轄区域特許法の任意の類似した不利な変化は、私たちの業務、財務状況、運営結果、および将来性に大きな悪影響を及ぼす可能性もあります。
特許条項は候補製品に対する私たちの競争地位を十分に長く保護するのに十分ではないかもしれない。
特許の寿命は限られている。個別特許の条項は、特許が付与された国の特許法律条項に依存する。米国を含む大多数の国では、すべての維持費が適時に支払われれば、特許の自然失効時間は、通常、適用国における最初の非臨時出願日から20年である。しかしながら、特許によって提供される実際の保護は、国によって異なり、特許のタイプ、そのカバー範囲、規制に関連する延長の利用可能性、特定の国の法的救済の利用可能性、および特許の有効性および実行可能性を含む多くの要因に依存する。PTEおよびPTAを含む様々な延期は利用可能である可能性があるが、特許の有効期間およびその提供される保護は限られている。私たちの候補製品をカバーする特許を取得しても、特許有効期限が切れたら、私たちは模造薬を含む競争製品からの競争に直面するかもしれません。新製品候補製品の開発、テスト、監督審査に要する時間を考慮すると、私たちの候補製品を保護する特許は、私たちまたは私たちのパートナーがこれらの候補製品を商業化する前または近いうちに満期になる可能性がある。したがって、私たちが持っている特許と許可された特許の組み合わせは、他社が私たちと似ているか同じ製品を商業化することを排除するために十分な権利を提供してくれないかもしれない。
86
もし私たちが開発する可能性のある候補製品のPTEとデータ独占経営権を獲得しなければ、私たちの業務は実質的に損害を受ける可能性があります。
FDAが私たちが開発する可能性のある任意の候補製品の上場承認の時間、期限、および詳細によれば、私たちの1つ以上の米国特許は、1984年の“薬品価格競争および特許期限回復法”または“ハッジ·ワックスマン修正案”に従って限られたPTEを得る資格があるかもしれない。Hatch−Waxman修正案は,FDA規制審査過程で失われた特許期間の補償として,特許期間を最長5年に延長した。特許特許権の残り期間は、製品が承認された日から14年を超えてはならず、各製品は1つの特許しか延長できず、承認された薬物、その使用方法または製造方法に関する特許請求を延長することしかできない。しかしながら、個人的な技術移転を求めても、試験段階または規制審査中に職務調査を行うことができなかった場合、適用された最終期限内に出願を提出できなかった場合、関連する特許が満了する前に出願を提出できなかった場合、または適用要件を満たしていなかった他の任意の場合に承認を得ることができない可能性がある。しかも、特許保護の適用期間や範囲は私たちが要求しているものよりも短いかもしれない。もし私たちがPTEやこのような延期された期限が私たちが要求したよりも短いことができなければ、私たちの競争相手は私たちの特許が満期になった後に競争製品の承認を得るかもしれません。私たちの業務、財務状況、運営結果、および見通しは実質的な損害を受ける可能性があります。
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの商業と競争の地位は損なわれるだろう。
私たちの技術および製品候補のための特許出願に加えて、私たちは、特に特許保護が不適切であるか、または入手不可能であると考えられる場合に、当社の従業員、コンサルタント、および第三者と締結された秘密協定、秘密協定、および発明譲渡協定に依存して、独自の技術および商業秘密保護に依存する。
私たちの政策は、私たちの従業員、会社の協力者、外部科学協力者、CRO、契約製造業者、コンサルタント、コンサルタント、および他の第三者に、私たちとの雇用やコンサルティング関係の構築を開始する際に秘密協定を実行することを要求します。これらのプロトコルは、特定の場合がない限り、個人またはエンティティが私たちとの関係中に開発またはそれに開示するすべての私たちの業務または財務に関する機密情報を秘密にし、第三者に開示しないことを規定している。従業員の場合、合意は、個人的に構想された、私たちの現在または計画中の業務または研究開発に関連するもの、または通常の勤務時間内、私たちのオフィス内で行われる、または私たちの設備または独自の情報を使用して行われるすべての発明が、私たちの固有財産であることを規定している。コンサルタントおよび他の第三者の場合、プロトコルは、提供されたサービスに関連するすべての発明が私たちの固有財産であることを規定する。しかし、私たちは私たちが私たちの商業秘密またはノウハウおよびプロセスに可能性があるか、または接触したすべての当事者とそのような合意に到達したことを保証することはできない。さらに、知的財産権の譲渡は自動的に実行されない可能性があり、あるいは譲渡協定が違反される可能性があり、私たちは第三者にクレームを出すことを余儀なくされるか、あるいは彼らは私たちが私たちの知的財産権の所有権と考えていることを確認するために、私たちが提起したクレームに対抗する可能性がある。どちらも合意に違反し、私たちのビジネス秘密を含む私たちの固有の情報を開示する可能性があり、私たちはこのような違反について十分な救済措置を得ることができないかもしれない。強制執行側が商業秘密を不正に開示したり流用したりする主張は困難であり、高価で時間がかかり、結果は予測できない。
契約措置に加えて、物理的および技術的セキュリティ対策のような他の適切な予防措置を通じて、私たちの独自の情報の機密性を保護しようとしています。しかし、商業秘密と技術的ノウハウを保護することは難しいかもしれない。例えば、従業員またはアクセス許可権限を有する第三者が商業秘密を盗用する場合、これらの措置は、私たちの独自の情報に十分な保護を提供しない可能性がある。私たちの安全措置は、従業員やコンサルタントが私たちのビジネス秘密を盗用して競争相手に提供することを阻止できないかもしれませんが、私たちはこのような不正行為に対するいかなる救済措置も、私たちの利益を十分に保護するための十分な救済措置を提供できないかもしれません。また、商業秘密は、私たちが法的追跡権を得ることを阻止する方法で他の人によって独立して開発された可能性がある。私たちのビジネス秘密のような私たちの任意の機密または独自の情報が漏洩または流用されている場合、またはこれらの情報のいずれかが競争相手によって独立して開発されている場合、私たちの競争地位は損なわれる可能性がある。
また、米国国内外のいくつかの裁判所は商業秘密を保護することをあまり望まないか、または保護したくないことがある。もし私たちが第三者が私たちのどんな商業秘密を使用するのを阻止するために法廷に訴えることを選択すれば、私たちは巨額の費用を生むかもしれない。私たちが勝訴しても、このような種類の訴訟は私たちの時間と他の資源を消費するかもしれない。上記のいずれも、当社の業務、財務状況、経営結果、見通しに重大な悪影響を及ぼす可能性があります。
87
第三者は、将来的に、私たち、私たちの従業員、コンサルタント、またはコンサルタントが機密情報を誤って使用または開示したり、商業秘密を流用したりすると主張しているかもしれない。
バイオテクノロジーや製薬業界でよく見られるように、私たちが雇っている個人は、現在または以前、私たちの競争相手や潜在的なライバルを含む、大学、研究機関、または他のバイオテクノロジーや製薬会社に雇われている。さらに、我々は、研究機関、外部科学協力者、CRO、第三者メーカー、コンサルタント、コンサルタント、潜在的パートナー、および他の第三者を保護して、技術の潜在的な発展を評価するために、第三者の独自の地位を保護するために、秘密保持および秘密保持協定を定期的に締結する。 私たちと私たちの従業員、コンサルタント、およびコンサルタントが他人の独自の情報やノウハウを使用しないことを確実にするために努力していますが、私たちは、商業秘密または他の固有情報を含む第三者の知的財産権を意図的にまたは他の方法で誤って使用または開示したことが、将来的に告発される可能性があります このような個人の現職または前任雇用主は誰でもない。また、私たちは過去に、未来に告発される可能性があり、これらの個人は、その前の雇用主との競争禁止協定に違反している。そして、私たちはその中のいずれかの疑いに対抗するために訴訟を提起しなければならないかもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭的損害賠償を支払う以外に、私たちは貴重な知的財産権や人員を失う可能性がある。このようなクレームに対抗することに成功しても、訴訟は巨額の費用を招き、私たちの技術や管理者の正常な責任を分散させる可能性がある。さらに、公聴会、動議、または他の一時的な手続きまたは事態の発展の結果が公開される可能性があり、証券アナリストまたは投資家がこれらの結果がマイナスであると考える場合、このような見方は、私たちの普通株の価格に大きな悪影響を及ぼす可能性がある。このようなタイプの訴訟や訴訟は、私たちの運営損失を大幅に増加させ、開発活動に利用できる資源を減少させる可能性があり、このような訴訟や訴訟を十分に展開するのに十分な財政的または他の資源がないかもしれません。例えば、私たちのいくつかの競争相手は、彼らの財政資源がはるかに大きいので、私たちよりもこのような訴訟や法的手続きの費用を効果的に受けることができるかもしれない。いずれの場合も、知的財産権訴訟または他の知的財産権関連訴訟の開始および継続によって生じる不確実性は、市場での競争能力に悪影響を及ぼす可能性がある。
例えば、私たちはその機関と私たちの間の秘密協定に関する研究機関の手紙を受け取った。秘密保護協定は、私たちのいくつかのプロジェクトに関連する開発で評価されたいくつかの技術に関するものだ。我々は機密協定の条項に違反し,その機関の商業秘密や他の機密情報を流用し,不公平かつ欺瞞的な商業行為に従事し,我々の療法(BEAM−302を含む)を開発した際に不公正に富を得たと手紙で伝えられている。研究機関は、金銭損害賠償(わが社への価値分担の損害、および告発された故意違反に対する増加損害賠償を含む)、および違約が疑われる技術に関連するいくつかの進行中の特許権使用料および/またはマイルストーン支払い、および他の可能な救済措置を得る権利があると主張している。私たちはこの研究機関と和解交渉をしてきて、継続される予定だ。私たちがこのようなクレームについて和解できるかどうか、あるいは私たちがこれらのクレームに関連する任意の訴訟や訴訟で勝つかどうかを予測することはできない。また、どの訴訟も負の宣伝を招く可能性があり、その結果にかかわらず、重大な金銭的損失責任を負わせ、私たちの業務、財務状況、運営結果に実質的な悪影響を与える可能性がある。詳細については、本年度報告に他の表格10−Kに記載されている総合財務諸表付記6を参照されたい。
もし私たちの商標と商号が十分に保護されていなければ、私たちは興味のある市場で知名度を作ることができないかもしれません。私たちの業務は悪影響を受けるかもしれません。
私たちの登録または未登録商標または商号は、疑問、侵害、回避、または汎用商標として発表されるか、または他の商標を侵害していると認定される可能性がある。私たちはこれらの商標や商品名に対する私たちの権利を保護できないかもしれません。私たちは私たちが関心のある市場の潜在的なパートナーや顧客の中で知名度を確立するために必要です。時々、競争相手や他の第三者は、私たちと同様の商号や商標を採用して、ブランド表示を確立する能力を阻害し、市場の混乱を招く可能性があります。さらに、他の登録商標または商標の所有者は、我々の登録または未登録商標または商号の変異体を含む商号または商標侵害クレームを提出することができる。長期的には、私たちの商標や商号に基づいて名称を確立することができなければ、効果的に競争できない可能性があり、私たちの業務は悪影響を受ける可能性があります。商標、商業秘密、ドメイン名、著作権、または他の知的財産権に関連する専有権を実行または保護する努力は無効である可能性があり、大量のコストおよび資源移転を招き、私たちの業務、財務状況、運営結果、および成長の見通しに悪影響を及ぼす可能性があります。
知的財産権はすべての潜在的な脅威を解決できるとは限らない。
私たちの知的財産権が提供する未来の保護の程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できないか、あるいは競争優位性を維持することができるかもしれない。例えば:
88
これらの事件のいずれかが発生した場合、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
89
承認されれば、生物製品として許可され、規制された候補製品は、規制経路を簡略化することで承認された生体模倣薬の競争に直面する可能性がある。
2009年の生物製品価格競争および革新法、またはBPCIAは、患者保護および平価医療法案の一部であり、“医療および教育負担能力調整法”によって改正され、または総称してACAと呼ばれ、生物類似および交換可能な生物製品を承認する短い方法を確立する。規制経路はFDAのために生物類似生物製品を審査および承認する法律的権威を確立し、生物類似体と承認された生物製品との類似性に基づいて、それを“交換可能な”生物製剤として指定することを含む。BPCIAによれば、参照生物製品には12年のデータ独占権が付与され、この製品が初めて許可を得たときから、FDAは、参照製品が初めて許可を得た日から4年まで、参照生物製品に基づく生物類似または交換可能製品の申請を受け入れない。この12年間の排他的期間内に、別の会社は、BLAが参照製品、スポンサーのデータを返信しない限り、または生物学的に類似した申請の形態で出願を提出する限り、競合生物の承認を得ることができる。
BLAによりバイオ製品として開発されたどの候補製品も12年の専門期間を得る資格があるべきであると考えられる。しかしながら、国会の行動または他の理由により、このような排他性は短縮される可能性があり、またはFDAは候補製品を競合製品の参考製品とみなさず、これは予想よりも早く生物学的類似競争のための機会を創出する可能性がある。さらに、承認されると、生物学的類似体が、どの程度、非生物製品に類似した伝統的な模倣薬代替の方法で任意の参照製品を置換するかは、まだ発展中のいくつかの市場および規制要因に依存するであろう。それにもかかわらず、競争激化と価格設定圧力により、私たちの候補製品と類似した生物を承認することは、私たちの業務に実質的な悪影響を及ぼす可能性がある。
規制やその他の法律的コンプライアンスに関するリスク
遺伝子薬物、特に私たちが開発する可能性のある任意の新しい遺伝子薬物に対する規制要求は常に変化し、将来も変化し続けるかもしれない。
遺伝子薬物と細胞薬物に対する規制要求、特に私たちが開発する可能性のある任意の新しい遺伝子薬物製品は、すでに頻繁に変化し、将来的に変化し続ける可能性がある。FDAとEMAの発売許可を得た遺伝子薬物の数は限られていることが知られている。遺伝子医学領域のより成熟した製品の面でも、監督管理構造はまだ発展中である。例えば,FDAはCBER内に組織·高度療法室(前身は細胞,組織,遺伝子療法オフィス)を設置し,遺伝子薬物や関連製品の審査を統合し,細胞,組織,遺伝子療法諮問委員会を設置し,その審査についてCBERにアドバイスを提供している。NIHから組換えDNA研究資金を得た機関で行われる遺伝薬物臨床試験は,バイオテクノロジー活動オフィス組換えDNA諮問委員会(RAC)の審査を受ける可能性もあるが,NIHは,RACは標準監督機関で評価され異常リスクを構成できない場合にのみ臨床試験を公開審査すると発表した。
同じ状況は連合にも適用される。EMAの高級治療委員会(CAT)は高級治療薬物製品の品質、安全性と有効性を評価する責任がある。CATの役割は、CHMPがその最終意見を通過する前にCHMPに提出される候補遺伝子薬物の上場許可申請に関する意見草案を準備することである。EUでは,遺伝子薬物製品の開発と評価はEU関連ガイドラインを背景に考慮しなければならない。EMAは遺伝子薬物製品の開発とマーケティング許可に関する新しいガイドラインを発表し、これらの新しいガイドラインを遵守することを要求する可能性がある。したがって,遺伝子薬や細胞治療製品に適したプログラムや基準は,我々が開発可能な任意の候補製品に適用可能であるが,現在のところ確定していない。
これらの規制審査委員会や諮問グループおよびその発表された新しいガイドラインは、規制審査プロセスを延長する可能性があり、追加的な研究を求め、私たちの開発コストを増加させ、規制の立場や解釈の変化を招き、私たちが開発した任意の候補製品の承認や商業化を延期または阻止したり、重大な承認後の制限や制限を招いたりする可能性がある。私たちが開発可能な任意の候補製品を提案する時、私たちはこれらの規制や諮問グループと協議し、適用されるガイドラインを遵守することを要求されるだろう。もし私たちがこれをできなかったら、私たちはこのような候補製品の開発を延期したり停止したりすることを要求されるかもしれない。潜在的な製品を市場に出すために必要な規制承認を遅延させたり、監督管理の承認を得られなかったりする意外なコストは、業務を維持するのに十分な製品収入を生成する能力を低下させる可能性がある。
90
FDAは個別遺伝薬プログラムを継続できるかどうかを決定しているにもかかわらず,RAC公共審査プログラムを行うと臨床試験の開始が延期される可能性があり,たとえFDAが試験設計や詳細を審査して起動を承認したとしても。対照的に、RACが有利な審査を提供しても、または深い公共審査を免除しても、FDAはINDを臨床的保留状態に置くことができる。NIH助成機関を招いて臨床試験を行う場合,当機関のIBCとそのIRBは提案された臨床試験を審査し,試験の安全性を評価する必要がある。さらに、他の人が行う遺伝子薬物臨床試験の不利な発展は、FDAまたは他の監督機関が、私たちが開発する可能性のある任意の候補製品の承認要求を変更することをもたらす可能性がある。同様に、EMAは遺伝子薬物製品の開発とマーケティング許可に関する新しいガイドラインを発表し、これらの新しいガイドラインを遵守することを要求する可能性がある。
我々は最初に新しい技術を用いて疾患を治療する候補製品の決定と開発を求めているため,FDA,EMAあるいは他の規制機関は,臨床的意義のある結果を提供する臨床試験終点を提案するリスク増加を考慮しない可能性がある。これらの終点が臨床的意義があると考えられても,われわれは統計的意義のあるこれらの終点を実現できない可能性があり,特にわれわれが我々のプラットフォームを用いて対象としている多くの疾患は,T細胞急性リンパ球性白血病,グリコーゲン貯蔵障害,Stargardt病を含め,患者数が少なく,大型かつ厳しい臨床試験の開発が困難であるためである。また,あらかじめ指定された基準を達成しても,予測不可能な結果や,非主要端点の結果や他の関連データと一致しない可能性がある.FDAは製品のメリットとリスクもトレードオフし,FDAは安全性の観点から治療効果の結果を見る可能性があり,規制部門の承認を支持していないと考えられる。連合と他の国の他の規制機関はこのようなサイトとデータについて似たような論評をするかもしれない。私たちが開発する可能性のあるどの候補製品も新しい技術に基づいており、開発とその後の規制承認の時間とコストを予測することは困難である。現在、遺伝子編集治療製品はアメリカ或いはヨーロッパで承認されておらず、数量の限られた遺伝子治療製品だけが欧州委員会或いはFDAの発売許可或いは発売許可を得ている。その中のいくつかの製品は数年かけて登録を行い、その発売後の経験中の重大な問題を解決しなければならない。
他の遺伝子薬物または細胞療法製品の発売後の経験または臨床試験の不利な発展は、FDA、EMAおよび他の規制機関が、非ウイルス遺伝子薬物技術を使用する可能性のある製品の任意の候補製品の要求を開発または承認することを修正または承認する可能性があり、これら2つの場合のいずれも、私たちの業務に実質的な損害を与える可能性がある。そのほか、FDA、EMAと他の監督機関の臨床試験要求及びこれらの監督機関は候補製品の安全性と有効性を決定するための標準は、潜在製品のタイプ、複雑性、意外性と期待用途と市場によって大きく異なる。さらに、規制行動または個人訴訟は、私たちの研究計画または生成された製品の商業化に費用、遅延、または他の障害をもたらす可能性がある。
また,遺伝子医学,遺伝子テスト,遺伝子研究の倫理,社会,法律面の懸念は,追加の法規制や使用が禁止される過程を招く可能性がある。連邦と州機関、国会委員会、そして外国政府はバイオテクノロジーをさらに規制する意図を表明した。より厳格な規制や、私たちが開発する可能性のあるどの候補製品も安全ではないと主張したり、危険を構成したりして、私たちがどんな製品を商業化することを阻止するかもしれません。新しい政府要求を制定し、規制部門が私たちが開発している可能性のある任意の候補製品を承認することを延期または阻止するかもしれない。立法が変わるかどうか,条例,政策やガイドラインが変わるかどうか,機関や裁判所の解釈が変わるかどうか,あるいはこれらの変化の影響(あれば)が何になるかは予測できない。
私たちが臨床開発を通じて任意の候補製品を開発する際には、これらの規制や諮問グループと協議し、適用されるガイドラインを遵守することが求められる。これらの規制審査委員会および諮問グループおよびその発表された任意の新しいガイドラインは、規制審査プロセスを延長し、追加的な研究を要求し、私たちの開発コストを増加させ、規制の立場や解釈の変化を招き、私たちが開発した任意の候補製品の承認や商業化を延期または阻止したり、重大な承認後の制限または制限を招いたりする可能性がある。潜在的な製品を市場に出すために必要な規制承認を遅延または獲得できなかったり、監督管理の承認を得られなかったりする意外なコストは、十分な製品収入を生成する能力を低下させる可能性がある。
たとえ私たちが必要な臨床前研究と臨床試験を完成しても、上場審査過程は高価で、時間と不確定であり、私たちが開発する可能性のある任意の候補製品の商業化審査を獲得することを阻止するかもしれない。もし私たちが必要な規制承認を得たり遅延したりすることができなければ、私たちが開発する可能性のある候補製品を商業化したり、商業化を遅延させることができなくなり、私たちが収入を作る能力は深刻な損害を受けるだろう。
91
我々が開発する可能性のある任意の候補製品およびその開発および商業化に関連する活動は、設計、テスト、製造、記録保存、ラベル、貯蔵、承認、広告、販売促進、販売、輸入、輸出、流通を含み、米国FDA、EMAおよび他の規制機関、および他の国の類似機関によって全面的に規制されている。候補製品のマーケティング承認が得られなければ、指定された司法管轄区域で候補製品を商業化することはできないだろう。私たちはまだどのような管轄区域の規制機関からもどんな候補製品のマーケティングの承認を得ていない。上場承認を得るために必要な申請の提出と支援に必要な経験は限られており、この過程で第三者に協力していただく予定です。監督部門の承認を得るためには、各監督機関に広範な臨床前と臨床データと支持情報を提出し、各治療適応の安全性、純度と効力を確定する必要がある。規制機関の承認を得るためには,製品製造過程に関する大量の情報を提出し,関連規制機関が製造施設を検査する必要がある。我々が開発した任意の候補製品は効果がない可能性があり、中程度の効果のみである可能性があり、または不良または予期しない副作用、毒性、または市販承認または商業使用を阻止または制限することを阻止または制限する可能性があることが証明される可能性がある。
米国や海外でマーケティング承認を得る過程は高価であり、承認されるには何年もかかる可能性があり、関連する候補製品のタイプ、複雑性、新規性を含む様々な要素によって大きく異なる可能性がある。開発期間中の市場承認政策の変化、追加法規又は法規の変化、又は各提出製品申請に対する規制審査の変化は、出願の承認又は拒絶の遅延を招く可能性がある。FDAと他国の類似機関は承認過程において大きな自由裁量権を有しており、いかなる申請も拒否することができ、私たちのデータが承認を得るのに十分ではなく、追加の臨床前、臨床、あるいは他の研究を行う必要があると決定することもできる。そのほか、臨床前と臨床試験から得られたデータの異なる解釈は候補製品の発売許可を延期、制限或いは阻止する可能性がある。私たちが最終的に得たどの上場承認も限られているかもしれないし、制限されたり、承認された後の約束は、承認された薬物が商業的に実行できないようにする可能性がある。
さらに、小児科研究公平法またはPREAによれば、いくつかの生物学的製品のBLAまたは補足BLAは、すべての関連する小児科亜集団における生物製品の安全性および有効性を評価し、スポンサーがFDAの延期または免除を受けない限り、製品の安全に有効な各小児科亜集団の用量および投与をサポートしなければならない。延期は、小児科試験が完了する前に、製品または候補治療薬が成人で使用を許可する準備ができていることを発見するか、または小児科試験が開始される前に追加の安全性または有効性データを収集する必要があることを含むいくつかの理由があるかもしれない。EUの適用される立法はまた、スポンサーがEMA小児科委員会が許可した小児科調査計画に基づいて小児科群で臨床試験を行うか、免除されるか、この小児科委員会によるこれらの研究を延期することを要求する。私たちが米国やEU規制の承認を求めている任意の製品候補製品については、私たちが免除されたり、代替的に必要な研究や他の要求をタイムリーに完成させることができる保証はありません。これは、関連する名声の損害を招き、法執行行動の影響を受ける可能性があります。
もし私たちが承認を得る上で遅延に遭遇した場合、あるいは私たちが開発する可能性のある候補製品の承認を得ることができなければ、これらの候補製品の商業的な見通しが損なわれる可能性があり、私たちが収入を作る能力は実質的に損なわれるだろう。
外国の管轄区域でマーケティングの許可を得ることができなければ、私たちが開発する可能性のあるどの候補製品もこれらの管轄区で販売できなくなり、逆に収入を創出する能力を深刻に弱めることになる。
私たちがEUや他の外国司法管轄区域で開発する可能性のある任意の候補製品をマーケティングおよび販売するためには、私たちまたは私たちの第三者パートナーは単独のマーケティング承認(EUに対する単一承認)を得なければならず、多くの異なる法規要件を遵守しなければならない。承認手続きは国によって異なり、追加的なテストが含まれるかもしれない。承認を得るのに要する時間は、FDA承認を得る時間とは大きく異なる可能性がある。米国以外の規制承認手続きには、通常、FDA承認の取得に関するすべてのリスクが含まれる。また、米国以外の多くの国では、候補製品が精算承認された後、その候補製品が同国での販売を許可されることが求められている。私たちやこれらの第三者は(あれば)米国以外の規制機関の承認をタイムリーに得ることができないかもしれない。FDAの承認は、他国又は管轄区域の規制機関の承認を確保するものではなく、米国以外の1つの規制機関の承認も、他の国又は司法管区の規制機関又はFDAの承認を確保することができない。私たちは上場承認を申請できないかもしれませんし、どの司法管轄区でも私たちの薬品を商業化するために必要な承認を得ることができないかもしれません。これは私たちの収益能力を深刻に損なうことになります。
92
また、イギリスのEU離脱により、私たちはイギリスのマーケティング許可を得る上でより高いリスクに直面する可能性があり、一般にイギリスの離脱と呼ばれる。イギリスはこれ以上ヨーロッパ単一市場とEU関税同盟の一部ではない。2021年1月1日から、薬品と保健品監督局(MHRA)は国内法律に基づいてイングランド、スコットランド、ウェールズからなる大ブリテン(GB)の薬品と医療機器の監督を担当し始めたが、北アイルランド議定書の条項によると、北アイルランドは現在EU規則の制約を受けている。しかし、イギリスとEUは、イギリスの医療製品に関する規制を含む北アイルランド議定書下の既存の制度を根本的に変更するウィンザー枠組みに同意した。施行されると、ウィンザー枠組みの導入の変化は、MHRAがイギリス市場(すなわちイギリスと北アイルランド)に輸送されたすべての医療製品を承認することを担当し、EMAは北アイルランドへの医療製品の輸送を承認する上で何の役割も果たすことはできないだろう。イギリスの離脱やその他の理由により、いかなるマーケティング許可を得る上でのいかなる遅延も、いかなるマーケティング許可も得ることができず、イギリスで私たちの候補製品のための規制承認を求める努力を制限または延期させる可能性があり、これは私たちの業務に重大で実質的な損害を与える可能性がある。
また、外国の規制機関はその承認政策を変更し、新たな規定を制定する可能性がある。例えば、欧州委員会が2020年11月に開始した欧州薬品戦略イニシアティブを背景に、EU薬品立法は現在全面的な審査が行われている。欧州委員会は、医薬製品に関するいくつかの立法文書の改正に関する提案(規制データ保護の期限を短縮する可能性があり、迅速通路の資格を改訂するなど)。2023年4月26日に出版された。提案された改正は依然として欧州議会と欧州理事会の同意と採択を得る必要があるため,提案は採択前に重大な修正が行われる可能性があり,2026年初めまではないと予想される。しかし、長期的には、これらの改正は製薬業と私たちの業務に大きな影響を与える可能性がある。
どの規制機関も私たちの製品の販売を許可することは指標によって制限されるだろう。FDA規制に違反して私たちの製品を未承認の用途に使用する規定を遵守できなかったり、発見されたりした場合、刑事罰、巨額の罰金、または他の制裁、損害賠償を受ける可能性があります。
未承認用途の製品の普及に関する法規は複雑であり,FDA,EMA,MHRA,他の政府機関から実質的に解釈されている。FDAは2021年9月、医薬品の予期される用途を決定する際に機関が考慮すべき証拠タイプを記載した最終法規を公表した。それにもかかわらず,医師は承認されたラベルと一致しない方法で,ラベル外で患者に我々の製品を処方する可能性がある。私たちは私たちのどの販売とマーケティング行為も適用された法規に適合することを確実にするために、コンプライアンスと訓練計画を実施するつもりだ。これらの計画にもかかわらず、FDAや他の政府機関は、私たちのやり方が未承認の用途への私たちの製品の普及を禁止するようになっていると主張したり発見したりするかもしれない。私たちの従業員が会社の政策や適用された法規を遵守して、承認されていない用途のための製品を普及させるかどうかも確認できません。
ラベル外販売促進に規制制限があるにもかかわらず、FDAと他の監督管理機関は会社がある場合にその製品について真実、非誤解性と非販売促進の科学的な交流を行うことを許可する。例えば,FDAは2023年10月にガイドラインを発表し,未承認用途に関する科学的情報を医療保健提供者に配布する拘束力のない政策を管理することについて概説した。本ガイドライン草案は、このような通信が真実で、誤解性がなく、事実と偏らないことを要求し、医療保健提供者が許可されていない使用に関する情報の利点および欠点、ならびに有効性および実用性に必要なすべての情報を説明することを含む。また,FDAの最近の指導や,2023年の総合支出法案の一部として法律に署名した“承認前情報交換法”によると,会社は処方情報と一致する情報を普及させ,支払人の処方委員会メンバーと未承認薬物や承認済み薬物の未承認用途のデータについて能動的に会話することも可能である。私たちはこれらの議論に参加し、すべての適用された法律、法規指導、業界ベスト実践を遵守し、ヘルスケア提供者、支払人、他の顧客とコミュニケーションを取ることができるかもしれない。FDAの様々な法規、ガイドライン、政策、そして最近公布された立法をよく考慮して、我々の製品普及に関する制限を遵守することを確保する必要がある。
93
近年、大量の製薬と生物技術会社は様々な連邦と州監督管理、調査、検察と行政実体調査と調査の目標となり、これらの実体は司法省と各種アメリカ検事事務室、衛生·公衆サービス部監察長事務室、FDA、連邦貿易委員会または連邦貿易委員会、および各州総検察長事務室を含む未承認用途および他の販売行為のための製品の使用を促進することに関する。これらの調査容疑は、独占禁止法違反、FDCA違反、虚偽申告法、処方薬営業法および逆控除法の違反、および他の承認されていない用途の製品の販売促進、定価および連邦医療保険および/または医療補助精算に関連する行為に違反する疑いがあるなど、様々な連邦および州の法律および法規に違反している。これらの調査の多くは“りっぱな担い手“”虚偽申告法“による行動。虚偽請求法によれば、どの個人も政府を代表してクレームを提出することができ、個人または実体が虚偽クレームを提出したこと、または虚偽クレームが政府に支払いを要求することになると主張することができる。この人は1部持ってきたりっぱな担い手訴訟はどんな回復や和解から一杯のスプーンを分ける権利があるりっぱな担い手訴訟は、通常“密告者訴訟”とも呼ばれ、通常は現職または前任社員によって提起される。1つはりっぱな担い手訴訟を提起する時、政府はこの事件に介入して起訴するかどうかを決定しなければならない。もしそれが拒否すれば、個人は単独で起訴することができる。
FDAや他の政府機関が私たちに法を執行しているならりっぱな担い手許可されていない用途への製品の普及に関する禁止令に違反すれば、法令や会社の誠実な合意に同意するような巨額の民事または刑事罰金や損害賠償、その他の制裁に直面する可能性があり、これらの制裁に基づいて、私たちの活動は、適用される法律や法規の遵守を確保するために継続的な審査と監督を受けることになる。このような罰金、報酬、または他の制裁は、私たちの収入、業務、財務の見通し、そして名声に悪影響を及ぼすだろう。
私たちが将来発売許可を得たどの製品も発売後に市場から制限されたり撤退したりする可能性があります。もし私たちが規制要求を守っていない場合、あるいは承認された後にこのような製品の意外な問題に遭遇した場合、私たちは重大な処罰を受けるかもしれません。
私たちが発売許可を得た任意の製品、およびその製品の製造プロセス、承認後の研究と措置、ラベル、広告、販売促進活動などは、FDAと他の監督管理機関の持続的な要求と審査を受ける。これらの要求には、安全およびその他の発売後の情報と報告の提出、登録と上場要求、製造、品質管理、品質保証と記録と書類の対応する維持に関する要求、医師へのサンプルの配布と記録の保存に関する要求が含まれる。製品が上場承認されても、承認は、再生可能エネルギー管理システムを実施する要求を含む、その製品が発売可能な指定用途の制限または承認条件に制限される可能性がある。FDAはまた、製品の安全性或いは有効性を監視するために、高価な発売後の研究或いは臨床試験とモニタリングを要求する可能性がある。
FDA及び司法省を含む他の機関は、製品の製造、販売及び流通が承認の適応にのみ適用され、承認されたラベルの規定に適合することを確実にするために、製品の承認後のマーケティング及び販売促進活動を密接に規制し、監督する。FDAは、ラベル外使用に関するメーカーのコミュニケーションに厳格な制限を加えており、もし私たちが許可されていない製品を販売すれば、ラベル外マーケティングの警告や法執行行動を受ける可能性がある。“虚偽請求法”を含むFDCAおよび他の処方薬販売促進および広告に関する法規に違反することは、連邦および州医療詐欺および法律乱用および州消費者保護法違反の調査または告発を招く可能性がある。
さらに、市場承認を得る可能性のある任意の製品およびその製造業者または製造プロセスの以前に未知の有害事象または他の問題、または法規要件を遵守できなかったことが後に発見された場合、様々な結果が生じる可能性がある
94
最後に,新薬製品を開発·マーケティングする能力は,行われている挑戦FDAによるミフェプリストン承認訴訟の影響を受ける可能性がある。具体的には、2023年4月7日、アメリカテキサス州北区地区裁判所はFDAのミフェプリストンに対する許可を保留し、ミフェプリストンは1種の薬物製品であり、最初は2000年に承認され、その流通はREMS下で採用された各種条件の制約を受けた。この決定を下した際、地域裁判所は、米国の薬品の開発、承認、流通に悪影響を及ぼす可能性のある調査結果を下した。他の裁決では、地域裁判所は、原告が彼らの告発に勝つ可能性が高い、すなわちFDAがミフェプリストンを承認する際に独断的で気まぐれな行為を行った可能性が高く、そのラベルで決定された条件下での薬剤の使用が安全であるかどうかに関する証拠を十分に考慮していない。また,地域裁判所は連邦裁判所訴訟の長期的な要求を,原告がそのNDAを承認する決定についてFDAを提訴することを許可するか,あるいはREMSに基づいて要求を確立し,原告またはそのメンバーが被害を受ける程度,すなわちFDAの薬品承認決定が実際に原告に与えられた薬物による不良事件を受けた患者に看護を提供させることに基づいている。
2023年4月12日、米国第五巡回控訴裁判所は地域裁判所の裁決をある程度棚上げした。その後、2023年4月21日、米国最高裁判所は地域裁判所のすべての決定を棚上げにし、第5巡回控訴裁判所が地域裁判所の裁決の控訴を処理し、任意のまたは最高裁判所に提出した移審令申請を処理するのを待った。第五巡回控訴裁判所は2023年5月17日にこの事件に対して口頭討論を行い、2023年8月16日に裁決を下した。裁判所はミフェプリストンの市場からの除去を拒否し,FDAが2000年に最初に承認した挑戦は訴訟時効で禁止されていることを発見した。しかし裁判所は、原告は彼らの訴訟で勝つ可能性が高いと考えており、FDAが2016年と2021年にミフェプリストンの使用範囲を拡大することを許可した変化は独断的で気まぐれだと考えている。2023年9月8日、司法省とミフェプリストンメーカーは、米国最高裁判所に第5巡回控訴裁判所の裁決の再審査を要求する移審令の請願書を提出した。2023年12月13日,最高裁は控訴裁判所の裁決に関するこれらの移審令の請願書を承認した。
似たような制限はEUでの私たちの製品の承認にも適用される。販売許可の保持者は、医薬製品の製造、マーケティング、普及と販売に適用される一連の要求を守らなければならない。これらの問題は、EUの厳格な薬物警戒または安全報告規則を遵守し、許可後の研究および追加の監視義務を強制的に実施することができること、医薬製品の製造を許可すること、それに対して単独のメーカー許可証を取得しなければならないこと、および許可された薬品のマーケティングと普及を含むことであり、これらはEUで厳格に規制されている。EUの制約も受けています加盟国の法律。これらと他の連合の要求を守らないことはまた重大な処罰と制裁につながる可能性がある。
FDAが指定した迅速なチャネル、突破的、または再生医学高度療法は、実際には、より速い開発または規制審査または承認過程をもたらすことができない可能性があり、FDAが私たちが開発可能な任意の候補製品を承認することを保証することもできない。
FDAの快速チャンネル、突破性と再生医学高級療法或いはRMAT計画はある合格製品の開発を加速することを目的とし、これらの製品は深刻な疾病と状況の治療を目的としている。候補製品が深刻または生命に危険な疾患の治療に使用され、臨床前または臨床データがこの製品がこのような疾患が満たされていない医療需要を満たす潜在力があることを示す場合、スポンサーはFDA迅速チャネル認証を申請することができる。候補製品が深刻または生命に危険な疾患の治療を意図しており、予備臨床証拠が、候補製品が1つまたは複数の臨床的重要終点において既存の療法の実質的な改善を示す可能性があることを示す場合、候補製品は突破的療法として指定することができる。もし製品候補が治療、修正、逆転、または深刻な生命を脅かす疾患を治療、修正、または治癒するための再生医学療法であり、初歩的な臨床証拠が、候補製品がこのような疾患が満たされていない医療需要を解決する潜在力を有することを示す場合、候補製品はRMAT称号を得ることができる。高速チャネル、突破、および/またはRMAT認証を求めることができるが、このような認証に成功する保証はない。たとえ私たちがそのような認証を受けたとしても、私たちは従来のFDAプログラムよりも速い開発過程、審査、または承認を経験しないかもしれない。高速チャネル、突破、またはRMAT指定は、候補製品が市場承認を得るか、または任意の特定の時間範囲内で承認されることを保証することができない。また,FDAが我々の臨床開発プロジェクトのデータが高速チャネル,画期的またはRMAT指定をサポートしなくなったと考えた場合,その指定を取り消す可能性がある。高速チャネル、突破性、および/またはRMAT指定自体は、FDAの優先審査手順に適合することを保証することはできない。
95
FDAが指定した優先審査は、より速い規制審査や承認過程をもたらすことができない可能性があり、いずれにしても、FDAが我々が開発する可能性のある任意の候補製品を承認する保証はない。
FDAが、候補製品が重篤な疾患または状態の治療を意図していると判断し、承認された場合、そのような疾患または状態の治療、予防または診断の安全性または有効性の面で有意な改善を提供する場合、FDAは、候補製品を優先的に検討するように指定することができる。優先審査指定は、FDAがマーケティング申請を審査する目標が、出願提出日から6ヶ月であり、標準的な10ヶ月の審査期間ではないことを意味する。私たちは私たちの特定の候補製品を優先的に検討することを要求するかもしれない。FDAは候補製品優先審査地位を付与するか否かについて広範な裁量権を有しているため,特定の候補製品がこのような指定や地位を獲得する資格があると考えても,FDAはその資格を付与しないことに同意しない可能性がある。また、従来のFDAプログラムと比較して、優先審査指定は、必ずしもより速い規制審査プロセスを意味するわけではなく、承認面のいかなる利点も与えるとは限らない。FDAの優先審査を受けることは、6ヶ月の審査期間内または後に承認されることを保証しない。
私たちは私たちの候補製品のためにEUで良質な称号を求めるかもしれないが、私たちはそのような称号を受け取っても、そのような称号はより速い開発や規制審査や承認過程をもたらさないかもしれない。
連合で、私たちは未来に私たちのいくつかの候補製品のための良質な称号を求めるかもしれない。PRIMEはEMAの役割を強化し、科学と監督管理支持を強化し、開発を最適化し、未満足の医療需要を解決する潜在力を有する重大な公衆衛生利益の新薬の評価を加速することを目的とした自発的計画である。この計画は,EUで満足できる治療法がない疾患に対する薬物に集中しているか,あるいはそのような方法が存在しても,既存の治療法よりも大きな治療利点を提供することが可能である。Primeは、開発中でEUで許可を得ていない薬品に限られ、スポンサーは集中プログラムで初期上場許可申請を申請しようとしている。Primeとして受け入れられるためには、候補製品はその主要な公共健康利益と治療革新方面の資格基準に適合しなければならず、この基準は声明を実証できる情報に基づいている。Prime指定の利点は、マーケティング許可申請の前に継続的な支援を提供し、知識の蓄積を支援するCHMP調査委員を任命することと、重要な開発マイルストーンで早期対話および科学的提案を行うことと、製品の加速的な審査を行う可能性があることとを含み、これは、申請中に承認の程度に関する意見をより早く発表するために、審査時間を減少させることを意味する。PRIMEはスポンサーが同時にEMA科学提案と衛生技術評価提案を要求することができ、適時な市場進出を促進することができる。いずれの候補製品の良質な認証を得ても,従来のEMAプログラムと比較して,この認証は実質的に速い開発過程,審査または承認をもたらさない可能性がある。さらに、Prime称号を取得することは、EMAがマーケティング許可を付与する可能性を保証または増加させることを保証しない。
米国食品医薬品局、米国証券取引委員会および他の政府機関の資金不足は、政府の閉店やこれらの機関の運営の他の中断を含め、重要な指導部や他の人員の能力を雇用·保留し、新製品やサービスのタイムリーな開発や商業化を阻止するか、あるいはこれらの機関が私たちの業務運営に依存する可能性のある正常な業務機能を履行することを阻止することを阻害する可能性があり、これは私たちの業務に負の影響を与える可能性がある。
FDAの新製品の審査と承認能力は、政府予算と資金レベル、肝心な人員の雇用と維持及びユーザー費用の支払いを受ける能力、及び法定、監督と政策の変化を含む様々な要素の影響を受ける可能性がある。したがって、その機関の平均検討時間は近年変動している。FDAや他の機関の中断も、新製品候補製品が必要な政府機関によって審査および/または承認されるのに要する時間を遅らせる可能性があり、これは私たちの業務に悪影響を及ぼすだろう。また、政府が米国証券取引委員会や我々の業務に依存する可能性のある他の政府機関に提供する資金は、研究開発活動に資金を提供する機関を含め、政治プロセスの影響を受けており、政治プロセス自体が不安定で予測不可能である。
FDA、EMA、および他の機関の中断も、必要な政府機関の新薬の審査および/または承認に要する時間を遅らせる可能性があり、これは私たちの業務に悪影響を及ぼすだろう。例えば、近年、2018年や2019年を含め、米国政府は何度も閉店しており、米国食品·医薬品局や米国証券取引委員会などのある規制機関は、キー従業員を休暇させ、キー活動を停止せざるを得ない。また,新冠肺炎の大流行のような事件も中断する可能性がある。新冠肺炎の大流行期間中、一部の会社は完全な返信を受けたことを発表した。原因はアメリカ食品と薬物管理局がその応用に対する必要な検査を完成できないからである。将来的に類似した公衆衛生緊急事態が発生すれば、FDAは現在のペースを継続できない可能性があり、審査スケジュールが延長される可能性がある。米国以外で類似した状況に直面している規制当局は、類似した公衆衛生緊急事態に対応するために、同様の制限や他の政策措置をとる可能性があり、規制活動において遅延に遭遇する可能性がある。
緊急事態に関連した中断が発生すると、FDAが提出した規制文書をタイムリーに審査·処理する能力に深刻な影響を与える可能性があり、これは我々の業務に重大な悪影響を及ぼす可能性がある。将来的に緊急事態に関連する中断は、米国証券取引委員会のような他の政府機関にも影響を与える可能性があり、これは、私たちの公開申告文書の審査(必要があれば)および公開市場に参入する能力を遅らせることで、私たちの業務に影響を与える可能性もあります。
96
私たちは私たちの1つ以上の候補製品の孤立薬物独占経営権を得ることができないかもしれません。たとえ独占特許権を獲得しても、このような独占経営権はFDAやEMAが他の競争製品を承認することを阻止しない可能性があります。
孤児医薬品法によれば、FDAは、まれな疾患または疾患を治療するための医薬または生物学的製剤である場合、候補製品を孤児薬として指定することができる。EU EMAは孤児製品候補製品の承認に対しても似たような規制制度を持っている。一般に、孤児薬物名を有する製品がその後、その名称を有する適応の最初の発売許可を得た場合、製品は、一定期間内に市場排他期間を得る権利があり、これは、FDAまたはEMAがこの期間内に同じ孤児治療適応の別の候補製品の別のマーケティング申請を承認することを阻止するであろう。適用期間はアメリカでは7年、EUでは10年だ。もし製品が孤児薬物指定の基準に適合しなくなった場合、特にその製品の利益が十分に高く、市場排他性がもはや合理的でない場合、EUでの排他的期限は6年に短縮されることができる。
FDAが遺伝子治療において孤児薬に独占特許を与える基準は明確ではなく,進化しつつある。例えば、FDAは2021年9月に最終指導意見を発表し、現在、1つの遺伝子治療製品と孤児排他的目的製品のための別の“同じ”との見方を述べている。この指導の下で、2つの遺伝子治療製品間の遺伝子組換えまたはベクターが異なり、“微小な”差を反映しない場合、この2つの製品は孤児薬物の排他的目的のための異なる薬物とみなされる。FDAはケースに基づいて、同じウイルスクラスからの2つのベクターが同じかどうかを決定し、評価と同時に他の重要な特徴を考慮する可能性がある。さらに、FDAが私たちの候補製品のうちの1つに孤児薬物独占経営権を付与するために、機関は、米国で200,000人未満または米国で200,000人を超える影響を与える疾患または疾患の治療のための候補製品の使用を発見し、疾患または疾患に対する候補製品の開発および提供を合理的に期待することができないコストが、米国での販売から回収されるであろう。FDAは孤児薬物の排他的な条件や疾患がこの基準を満たしていないと結論するかもしれない。候補製品の孤立薬物排他性を獲得しても,この排他性は候補製品を競合から効果的に保護できない可能性があり,異なる候補製品が同じ条件で承認される可能性があるからである。FDAまたはEMAが、指定された要求に重大な欠陥があると判断した場合、または製造業者が稀な疾患または疾患を有する患者の需要を満たすのに十分な数の製品を保証できない場合、孤児薬の排他性を失う可能性もある。また,孤児薬物が承認された後であっても,FDAが後者の候補製品の方が臨床的に優れていると結論すれば,孤児排他性を有する製品よりも安全で有効であることが証明されているか,あるいは患者ケアに大きく貢献しているため,FDAはその後も同じ薬剤を同じ疾患に使用することを承認することができる。
2017年8月3日、国会でFDA 2017年再認可法案、略称FDARAが可決された。その他の事項以外に、FDARAはFDAが以前に存在した監督管理解釈を編纂し、薬品スポンサーに1種の孤児薬物の臨床優位性を証明することを要求し、この薬物は他の方面で以前許可された同じ稀な疾病を治療する薬物と同じであって、孤児薬物の排他性を獲得することができる。この新しい立法は以前の前例を覆し、即ち“孤児薬品法”はFDAに孤児排他期を認めることを明確に要求し、その臨床優勢にかかわらず。また、2020年12月に署名された総合立法によると、製品の臨床的優位性を示す要求は、2017年FDARA公布前に孤児薬物指定を受けたが、FDAの許可や許可を得ていない薬物や生物製品に適用される。
FDAは孤児薬物法とその規制と政策をさらに再評価するかもしれない。控訴裁判所の11人に対する裁決を考慮すると、この点は特に正しいかもしれないこれは…。2021年9月、巡回裁判所は、排他的範囲を決定するために、用語“同一疾患または状況”は、指定された“まれな疾患または状況”を意味し、この機関は、それを“適応または使用”と解釈することができないと判断した。したがって,裁判所は孤児薬物は“適応や使用”ではなく,指定された疾患や状況全体に排他的に適用されると結論した。この決定を覆すための立法提案があったが、それらはまだ法律になっていない。FDAは2023年1月23日、裁判所の命令範囲を超える事項において、FDAは、孤児薬の独占性と孤児薬の使用または適応のために許可された使用または適応とを束ねて、その既存の法規を適用し続けると発表した。FDAや国会が将来、いつ、あるいはどのように孤児の薬物法規や政策を変更するかどうかもわかりませんし、どのような変化が私たちの業務に影響を与える可能性があるかどうかもわかりません。FDAがその孤児の薬物法規や政策に変化する可能性があることによって、私たちの業務は悪影響を受ける可能性がある。
97
医療提供者、医師、第三者支払者との関係は、刑事制裁、民事処罰、契約損害、名声損害、利益および将来の収入の減少に直面する可能性がある反リベート、詐欺および乱用、反賄賂、および他の医療保健法律法規の制約を受けることになります。
医療保健提供者、医師、第三者支払人は、私たちが市場で承認された任意の候補製品を開発し、獲得する可能性があることを推薦し、処方する際に主な役割を果たしている。私たちの将来の第三者支払者や顧客との手配は、広く適用される詐欺や乱用、その他の医療法律や法規に直面する可能性があり、これらの法律と法規は、私たちがマーケティング、販売、流通を制限する可能性があり、私たちが上場許可を得た薬品の業務または財務手配と関係を制限するかもしれない。私たちが発売されている製品がある場合にのみ適用されるいくつかの法律と法規を含む、連邦および州医療保健法律および法規の制限を適用する
私たちが第三者の業務配置と適用される医療法律や法規に適合するように努力することは、多くのコストに関連することを確実にします。法律法規の広範性、ある法律法規に対する指導が限られていること、及び政府の法律法規が絶えず変化している解釈を考慮して、政府当局は結論を出す可能性があり、私たちの商業行為は医療保健法律法規に符合しない可能性がある。私たちの業務が上記の任意の法律または私たちに適用される任意の他の政府法規に違反していることが発見された場合、民事および刑事罰、損害賠償、罰金、連邦医療保険や医療補助などの政府医療計画から除外され、監禁、および私たちの業務を削減または再編することができ、これらはいずれも、私たちの業務、財務状況、運営結果、および見通しに悪影響を及ぼす可能性があります。
EUでは、医師の処方、推薦、裏書き、購入、供給、注文、または医薬製品の使用を誘導または奨励するために、医師に福祉または利点を提供することが禁止されている。医師への福祉や利点の提供も、英国の“2010年反賄賂法”のようなEU加盟国の反賄賂法の管轄を受けている。このような法律に違反することは巨額の罰金と監禁につながるかもしれない。
いくつかの連合会員国で医者に支払われた費用は公開されなければならない。また、医師との合意は、通常、医師の雇用主、その主管する専門組織、および/またはEU加盟国の監督当局に事前に通知し、承認しなければならない。これらの要件は、EU加盟国に適用される国家法律、業界規則、または専門行為規則に規定されている。このような要求を守らないことは、名声のリスク、公開非難、行政処罰、罰金、または監禁につながる可能性がある。
98
最近公布された将来の法律は、私たちと任意の未来のパートナーが私たちの候補製品のマーケティング承認を獲得し、それを商業化する難しさとコストを増加させ、私たちまたは彼らが獲得する可能性のある価格に影響を与えるかもしれない。
米国および一部の外国司法管轄地域では、医療システムに関する立法および法規の変更および提案された変更は、私たちが開発する可能性のある任意の候補製品の上場承認を阻止または遅延させ、承認後の活動を制限または規制し、私たちまたは任意の未来のパートナーがマーケティングの承認を得た任意の製品の収益性に影響を与える可能性がある。私たちは、現在の法律、および将来取られる可能性のある他の医療改革措置は、より厳しいカバー基準をもたらす可能性があり、私たちまたは未来のパートナーが受け取る可能性のある任意の承認された製品の価格に追加の下振れ圧力をもたらす可能性があると予想している。
2010年3月、アメリカ議会は2010年の患者保護と平価医療法案を公布し、PPAAと略称した。また、PACAが公布されて以来、他の立法改正も提案され、採択された。2011年8月、“2011年予算制御法案”(Budget Control Act Of 2011)などの法案は国会のための支出削減措置を制定した。赤字削減合同特別委員会の任務は、2013年から2021年までの間に少なくとも1.2兆ドルの赤字削減を提案することであるが、同委員会は必要な目標を達成できず、立法をいくつかの政府プロジェクトに自動的に削減することを触発した。これらの変化には、各年度に提供者に支払われる連邦医療保険総額が2%に達し、2013年4月に発効し、2031年まで続くことが含まれている。
2012年の“米国納税者救済法”は,いくつかの医療サービス提供者に支払う連邦医療保険を減少させ,政府が提供者に多額の金を取り戻す訴訟時効を3年から5年に延長した。これらの新しい法律は、連邦医療保険および他の医療資金のさらなる減少をもたらし、他の方法で、規制によって承認される可能性のある任意の候補製品の価格、またはそのような任意の候補製品の処方または使用頻度に影響を与える可能性がある。
PACAが公布されて以来、提供者に支払われる医療保険支払い総額を前期ごとに2%に削減することを含む他の立法変化も採択され、2013年4月に施行され、2031年まで続く。現在の立法により、医療保険支出の実際の減少幅は4%に達する可能性がある。総裁·バイデン氏は2022年12月に法律となる“総合支出法案”に署名し、医療保険計画の自動減額にいくつかの改正を行った。CAA第1001条は2010年の4%の法定現金支払法またはPAYGO自動減額を2024年末まで2年延期した。“2021年米国救援計画法案”の公布後、連邦医療保険計画の4%削減計画は2023年1月に発効する。CAAの医療補償タイトルは、2011年の連邦医療保険自動減額の2%予算制御法案を6ヶ月~2032年度に延長し、2030年度および2031年度の支払減少率を低減する第4163条を含む。
ACAが公布されて以来、この法律の条項を廃止し、代替するために、多くの法的挑戦と国会行動が続くだろう。例えば、2017年の税法の公布に伴い、国会は“個人強制令”を廃止した。この条項は、ほとんどのアメリカ人が最低レベルの医療保険を購入することを要求する条項が2019年に施行される。また、2021年6月、米国最高裁は原告が訴訟を起こす資格がないことが分かったため、ACA合憲性を問う訴訟を却下した。ACAに関する訴訟と立法は継続される可能性があり、結果は予測不可能で不確実である。
2021年1月、新たな行政命令は、連邦機関に、米国人の医療保険取得を制限するルールや他の政策を再検討し、この機会を保護し強化するための行動をとることを考慮するよう指示した。この命令によれば、連邦機関は、過去の疾患を有する人の保護の政策を破壊すること、医療補助およびPACAに基づいて、作業要件を含むカバー範囲または破壊計画のデモおよび免除を減少させる可能性があること、医療保険市場または他の医療保険市場を破壊する政策、連邦医療補助およびPPACAによる登録をより困難にする政策、および養育者への負担能力を含む保険または経済援助の負担能力を低減する政策を再検討するように指示される。
2021年12月13日,EUは衛生技術評価又はHTAに関する第2021/2282号条例を採択し,第2011/24/EU指令を改正した。この条例は2022年1月に施行されるが,2025年1月から適用され,その間に実施に関する準備と手順がとられる。適用されると、それは関連製品に従って段階的に施行されるだろう。この条例は、新しい医薬製品及びある高リスク医療機器を含むEU加盟国の衛生技術評価における協力を促進し、これらの領域で共同臨床評価を行うEUレベルの協力に基礎を提供することを目的としている。これは、EU加盟国がEU範囲内で汎用的なHTAツール、方法、およびプログラムを使用することを可能にし、患者に最大の潜在的影響を有する革新的な衛生技術の共同臨床評価、共同科学相談、開発者がHTA当局にアドバイスを求め、新興衛生技術および早期発見の将来性のある技術を決定し、他の分野で自発的な協力を継続することを含む4つの主要分野で協力することを可能にする。個別EU加盟国は、衛生技術の非臨床(例えば、経済、社会、倫理)の評価を引き続き担当し、定価と精算について決定する。
99
我々は,これらの医療改革,および将来とりうる他の医療改革措置は,連邦医療保険や他の医療資金のさらなる減少,より厳しいカバー基準,新たな支払い方法,および任意の承認された製品のために得られる価格および/または医師が市場に進出する可能性のある任意の承認された製品を管理することによって得られる補償レベルの追加的な下振れ圧力をもたらす可能性があると予想している。精算レベルの低下は私たちが受け取った価格や私たちの製品の処方や管理頻度に悪影響を及ぼす可能性があります。連邦医療保険や他の政府が計画している精算のいかなる減少も、個人支払者の支払いの同様の減少を招く可能性がある。したがって、これらの改革が発効すれば、市場で承認された候補製品の期待収入の開発に成功する可能性があり、我々の全体的な財務状況および候補製品の開発または商業化の能力に影響を与える可能性がある。
処方薬の米国や外国の司法管轄区域における価格はかなりの立法や行政行動の影響を受けており、許可を得ると、我々の製品の価格に影響を与える可能性がある。
アメリカでは、処方薬の価格が話題になってきた。アメリカ議会は最近数回の調査を行い、州と連邦立法を提案し、公布し、薬品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、連邦医療保険と医療補助下の薬品コストを下げることを目的とした。2020年には、総裁·トランプ氏が処方薬のコスト削減を目的としたいくつかの行政命令を発表し、これらの命令のいくつかが条例に盛り込まれている。これらの規定には、価格最恵国モデルが実施され、連邦医療保険B部分のある医師が管理する薬品に対する支払いを他の経済先進国が支払う最低価格とリンクさせ、2021年1月1日から発効する臨時最終規則が含まれている。しかし、この規則は全国的な初期禁止によって制限され、2021年12月29日、CMSはそれを廃止するための最終規則を発表した。CMSは、この規則の発表に伴い、それは価値をMedicare B部分の薬品支払いのすべての選択に組み入れ、そして受益者が根拠に基づく看護を獲得する機会を改善することを探索する。
また、2020年10月、HHSとFDAは、各州と他のエンティティが804条の輸入計画、すなわちSIPを制定し、いくつかの処方薬をカナダから米国に輸入することを許可する最終規則を発表した。この規定は米国薬物研究やメーカー協会(PhRMA)の訴訟で挑戦されたが,裁判所でPhRMAがHHSを起訴する資格がないことが発見された後,2023年2月に連邦地域裁判所に却下された。9つの州(コロラド州、フロリダ州、メイン州、ニューハンプシャー州、ニューメキシコ州、ノースダコタ州、テキサス州、バーモント州、ウィスコンシン州)はカナダからの麻薬の輸入を許可する法律が採択された。いくつかの州は第804条輸入計画提案を提出しており、FDAの承認を待っている。FDAは2023年1月5日にフロリダ州のカナダ薬物輸入計画を承認した。
また、2020年11月20日、HHSは薬品メーカーのD部分下でスポンサーの値下げを計画する安全港保護を廃止し、法律が値下げを要求しない限り、直接或いは薬局福祉マネージャーを通過する法規を決定した。この規定はまた、販売時点での値下げを反映するための新しい安全港を創出し、薬局福祉マネージャーと製造業者との間のいくつかの固定料金手配のための安全港を作成する。裁判所の命令により,上記安全港の除去と増加は延期され,最近の立法は同規則の実施を2026年1月1日に一時停止した。アイルランド共和軍はさらにこの規定の施行を2032年1月1日に延期した。
また、2020年10月、HHSとFDAは各州と他のエンティティがSIPを制定し、いくつかの処方薬をカナダから米国に輸入することを許可する最終規則を発表した。最終規則は現在行われている訴訟のテーマであるが、少なくとも6つの州(バーモント州、コロラド州、フロリダ州、メイン州、ニューメキシコ州、ニューハンプシャー州)がFDAの審査と承認のためにSIPsを開発することを目的としてカナダからの薬物の輸入を許可している。また、2020年11月20日、HHSは薬品メーカーのD部分下でスポンサーの値下げを計画する安全港保護を廃止し、法律が値下げを要求しない限り、直接或いは薬局福祉マネージャーを通過する法規を決定した。現在行われている訴訟により、インフラ投資·雇用法案は、この規則の実施を2026年1月1日に延期している。この規定はまた、販売所での値下げを反映するための新しい避難港を創出し、薬局福祉マネージャーとメーカーとの間のいくつかの固定費用手配のための新しい避難港を創出し、これらの計画の実施もインフラ投資と雇用法案によって2026年1月1日に延期された。
2021年9月,総裁·バイ登が署名した行政命令により,衛生·公衆サービス部は薬品価格を下げる計画を発表した。この計画の主な特徴は,(A)メーカーとの薬品価格交渉を支援することにより,薬品価格がすべての消費者や医療システム全体により負担と公平になること,(B)サプライチェーンの強化を支援し,生体模倣薬や後発薬を促進し,透明性を増加させる市場改革により,処方薬業界全体の競争を改善·促進すること,(C)公共·民間研究を支援し,市場インセンティブを確保することで価値と入手可能な新しい療法の発見を促進し,科学的革新を促進し,より良い医療保健と健康改善を促進することである。
100
最近、2022年8月16日に“アイルランド共和軍”が総裁·バイデンによって法律に署名された。新しい立法は連邦医療保険D部分に影響を与え、D部分は計画であり、連邦医療保険A部分または連邦医療保険B部分に加入する個人は毎月外来処方薬保険料を支払うことを選択することができる。他の事項を除いて、アイルランド共和軍はある薬品のメーカーに連邦医療保険との価格交渉(2026年から)を要求し、価格は交渉できるが上限があり、連邦医療保険B部分と連邦医療保険D部分に基づいてリベートを実施し、インフレを超える価格上昇を処罰し(2023年に初めて満了)、D部分のカバーギャップ割引計画の代わりに新しい割引計画を用いる(2025年から)。アイルランド共和軍は,衛生·公衆サービス部(HHS)秘書が最初の数年間,規制ではなく指導によってその多くの規定を実施することを許可した。
具体的には、価格交渉において、国会はMedicareが、競合する模倣薬または生体模倣薬を有さず、Medicare B部分およびD部分によって精算されるいくつかの高価な単一由来薬剤および生物製品のための低い価格を交渉することができる。CMSは、2026年からMedicare D部分によって支払われる10種類の高コスト薬剤の価格を交渉することができ、その後、2027年の15種類のD部分薬剤、2028年の15種類のB部分またはD部分薬剤、および2029年以降の20種類のB部分またはD部分薬剤の価格を交渉することができる。この規定は、少なくとも9年間承認された医薬品および許可13年の生物製品に適用されるが、単一のまれな疾患または疾患のための許可された医薬および生物製品には適用されない。それにもかかわらず、CMSは価格交渉でこれらの製品の最高価格を決定する可能性があるため、もし私たちの製品がMedicare価格交渉の対象であれば、私たちは完全に政府行動のリスクに直面するだろう。また、存在する可能性のあるリスクを考慮すると、金利協定のこれらの条項は、もし私たちの薬物製品が市場に発売されて9年後に価格を制定しなければ、医薬品の期待リターンや私たちの製品の特許のすべての価値を保護することができないというリスクをさらに増加させる可能性がある。
また、この立法は、製薬業者が提供する価格が法律で規定された交渉で達成された“最高公平価格”以下でない場合、または値上げ幅がインフレを超える場合、製薬業者は民事罰金と潜在的な消費税を受けることになると規定している。この立法はまた、メーカーに連邦医療保険D部分の価格上昇幅がインフレを超えた薬品にリベートを支払うことを要求している。新法律では,2024年の連邦医療保険の自己払い薬品コスト上限は年間4000ドルと規定されており,その後2025年から年間2000ドルが上限となっている。さらに、Medicare Part D処方薬計画に参加した個人については、彼らが計画の高いハードルや“悲劇的な時期”に達する前に要求された保証範囲がその初期の年間保証限度額を超えていれば、アイルランド共和軍は個人に関する法的リスクを増加させる可能性がある。最初の年間保証限度額を超え、悲劇的な時期を下回るサービスを必要とする個人は、壊滅的な時期に達するまで100%の処方費用を支払わなければならない。他にも、アイルランド共和軍には、共同保険と共同支払いコストの削減、低所得補助金計画の資格の拡大、年間自己負担費用に価格上限を設定することで、個人の財務負担を軽減するための多くの条項が含まれており、各規定は定価と報告書に潜在的な影響を与える可能性がある。
2023年6月6日,メルク社はHHSとCMSを提訴し,アイルランド共和軍の連邦医療保険に対する薬品価格交渉計画が憲法第5改正案に違反し,無償獲得となったと主張した。その後、他の一部の当事者は、アメリカ商会、百時美施貴宝会社、アメリカ薬物研究とメーカー、ノとノド会社、楊森製薬会社、ノワ製薬、アスリコンとブリンガー-インゲルハイム国際有限会社を含み、異なる裁判所にも訴訟を提起し、HHSとCMSに対して類似した憲法クレームを提出した。私たちはIreland共和軍のこのような条項と他の条項に関連した訴訟が継続され、結果的に予測可能で不確実だと予想する。
アメリカの薬品定価のやり方に対する行政、立法と法執行方面の興味もますます大きくなっている。アメリカ議会は調査を行い、大統領は行政命令を発表し、薬品定価の透明性を高め、連邦医療保険下の処方薬のコストを下げ、定価とメーカー患者計画との関係を審査し、政府計画の薬品精算方法を改革するための立法を提出し、公布した。例えば、総裁·バイ登政府は行政命令の中で、薬品価格を下げるための何らかの政策措置をとるつもりであることを示し、それに応じて衛生部は“高薬品価格に対応する総合計画”を発表し、その中で薬品価格改革の原則を概説し、国会が取ることができる様々な潜在的立法政策を列挙して薬品価格を低下させた。また、バイデン政府の2022年10月の行政命令に応答するため、衛生·公衆サービス部は2023年2月14日に報告を発表し、CMS革新センターによってテストされた3種類の新しいモデルを概説し、これらのモデルはそれらの薬物コストを下げ、獲得性を促進し、医療の質を高める能力に基づいて評価を行う。これらのモデルが将来の任意の医療改革措置で使用されるかどうかは不明である。また、2023年12月7日、バイデン政府は1980年ベハ·ドール法案下の進入権を使用することで処方薬の価格を制御するイニシアチブを発表した。2023年12月8日、米国国家標準·技術研究所は、権限行使を考慮した機関間指導枠組み草案を発表し、その中で初めて製品価格を機関が進行権を行使する際に使用できることを決定する要因とした。これまでデモの権利を行使したことはなかったが、新たな枠組みの下で、この権利が継続するかどうかは定かではない。
したがって,IRAがどのように実施されるかは不明であるが,いかなる連邦や州医療改革が我々にどのような影響を与えるかは確定できないが,このような変化は我々の活動に新たなあるいはより厳しい規制要求を加えたり,製品の精算を減少させたりする可能性があり,いずれも我々の業務,運営結果,財務状況に悪影響を及ぼす可能性がある。
101
州レベルでは、各州はますます積極的に、価格または患者の精算制限、割引、ある製品への参入の制限、マーケティングコストの開示および透明性措置を含む薬品および生物製品の価格設定を制御するための法規を立法と実施し、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。さらに,地域医療機関や個別病院では,どの薬品やサプライヤーが処方薬や他の医療計画に含まれるかを決定するために入札プログラムが使用されるようになってきている。これらの措置が承認されると、私たちの製品に対する最終的な需要を下げたり、私たちの製品の価格設定に圧力をかけるかもしれません。私たちは将来、より多くの州と連邦医療改革措置を取ることが予想され、そのいずれも連邦と州政府が医療製品やサービスのために支払う金額を制限する可能性があり、これは私たちの候補製品に対する需要の減少または追加の価格設定圧力を招く可能性がある。
EUでは、承認されれば、同様の政治、経済、規制発展が候補製品を利益的に商業化する能力に影響を与える可能性がある。米国やEU以外の市場では,精算や医療保険支払いシステムは国によって異なり,多くの国で特定製品や療法に価格上限が設定されている。いくつかの国、特にEU諸国では、処方薬の価格設定は政府によって統制されている。これらの国では、製品の発売許可を受けた後、政府当局との定価交渉にかなりの時間がかかる可能性がある。一部の国で精算或いは定価の承認を得るためには、私たちの候補製品のコスト効果を他の利用可能な治療法と比較する臨床試験を行う必要があるかもしれない。もし私たちの製品が精算を受けられない場合、あるいは範囲や金額が制限されている場合、あるいは定価が満足できないレベルに設定されている場合、私たちの業務は損害を受ける可能性があり、実質的かもしれません。
私たちの従業員、主要な調査者、コンサルタント、ビジネスパートナーは、規制基準と要求を遵守しないこと、およびインサイダー取引を含む不正行為または他の不正活動に従事する可能性があります。
私たちは従業員、コンサルタント、ビジネスパートナーの詐欺や他の不正行為のリスクに直面しており、臨床試験を開始すれば、私たちの主要な研究者も詐欺や他の不正行為のリスクに直面する。これらの当事者の不正行為は、FDA法規またはEUおよび他の司法管轄区域に適用される法規を故意に遵守しないこと、FDA、EMAおよび他の規制機関に正確な情報を提供すること、米国および海外の医療詐欺および法律法規を乱用し、財務情報またはデータを正確に報告すること、または不正な活動を開示することを含む可能性がある。特に、医療業界の販売、マーケティング、商業配置は、詐欺、不正行為、リベート、自己取引、その他の乱用行為を防止するための広範な法律法規によって制約されている。これらの法律法規は、広範な定価、割引、マーケティングと販売促進、販売手数料、顧客激励計画などの業務手配を制限または禁止している。このような不正行為はまた、臨床試験過程中あるいはFDA、EMA或いは他の監督機関との相互作用過程で得られた情報の不適切な使用に関連する可能性があり、これは規制制裁を招き、著者らの名声に深刻な損害を与える可能性がある。我々はすべての従業員に適用される行動基準を採択したが、常に従業員の不正行為を識別し、阻止できるわけではなく、このような行為を発見し防止するための予防措置は、未知または未管理のリスクや損失を効果的に制御できない可能性があり、またはこれらの法律や法規を遵守できないことによる政府の調査または他の行動や訴訟から私たちを保護することができるかもしれない。もし私たちにこのような行動を取って、私たちが自分の権利を弁護したり、維持することに成功しなかったら、これらの行動は、巨額の罰金や他の制裁を加えることを含む、私たちの業務、財務状況、運営結果、将来性に大きな影響を及ぼすかもしれない。
私たちが将来所有する可能性のある任意の国際業務を管理する法律法規は、米国以外でいくつかの候補製品を開発、製造、販売することを阻止し、コストの高いコンプライアンス計画の開発と実施を要求するかもしれない。
私たちが事業を展開しているアメリカ以外のすべての司法管轄区域では、私たちは多くの法律と法規の制約を受けている。国際ビジネス慣行コンプライアンス計画の作成、実施、維持のコストが高く、特に第三者に依存する必要がある場合には、このような計画の実行は困難である。
“反海外腐敗法”(FCPA)は、個人または企業が業務を獲得または保持するのを助けるために、任意の米国人または企業が、任意の外国人官僚、政党または候補者に直接的または間接的に支払い、提供、許可支払い、または任意の価値のあるものを提供することを禁止する。“海外腐敗防止法”はまた、米国に上場する証券会社にある会計条項を遵守することを要求しており、これらの条項は、企業(国際子会社を含む)のすべての取引の帳簿と記録を正確かつ公平に反映し、国際業務のために十分な内部会計制御システムを設計し、維持することを要求している。“海外腐敗防止法”の反賄賂条項は主に司法省によって執行される。米国証券取引委員会は“反海外腐敗法”における帳簿と記録条項の執行に関与している。
同様に、イギリスの“2010年反賄賂法”は、イギリスと関連のある会社や個人に対して域外効力を持っている。イギリスの反賄賂法は、公職者や個人や組織への誘惑を禁止している。“海外腐敗防止法”とイギリスの“反賄賂法”を遵守することは高価で困難であり、特に腐敗は公認問題である国である。また、海外腐敗防止法は製薬業に特別な挑戦をもたらしており、多くの国では病院が政府によって運営されているため、医師や他の病院従業員は外国人官僚とされている。臨床試験やその他の仕事に関連して病院に支払われた何らかの金は、政府関係者に支払われた不正金と考えられ、“海外腐敗防止法”の法執行行動につながった。
102
様々な法律、法規、および行政命令はまた、米国国外での使用および伝播を制限するか、または国家セキュリティ目的のために秘密にされた情報と、特定の製品およびこれらの製品に関連する技術データとを特定の非米国国民と共有する。私たちのアメリカ以外の拡張は、これらの法律を遵守するためにより多くの資源を投入することを要求し続けています。これらの法律は、アメリカ以外の場所で特定の薬剤や候補薬の開発、製造、販売を阻止するかもしれません。これは、私たちの成長潜在力を制限し、私たちの開発コストを増加させるかもしれません。国際ビジネス慣行を管理する法律を遵守しない場合、政府契約の資格停止または取り消しを含む重大な処罰を受ける可能性がある。“海外腐敗防止法”違反は重大な民事·刑事罰を招く可能性がある。“反海外腐敗法”による訴訟だけで、未解決のクレームが解決されるまで、米国政府とのビジネスを一時停止する権利を招く可能性がある。海外腐敗防止法違反の有罪判決は、政府請負業者の資格を長期的に廃止する可能性がある。私たちは国際ビジネス慣行法に基づいて私たちのいかなる義務も履行できなかったため、政府契約や関係の終了を招き、私たちの運営にマイナス影響を与え、私たちの名声と政府契約を得る能力を損なうだろう。米国証券取引委員会は、発行者が海外腐敗防止法の会計規定に違反したため、発行者の米国取引所での証券取引を一時停止または禁止する可能性もある。
私たちは、データプライバシーやセキュリティに関する厳しいプライバシー法、情報セキュリティ法律、法規、政策、契約義務の制約を受けており、このような法律、法規、政策、契約義務の変化は、私たちの業務に悪影響を及ぼす可能性があります。
私たちは、個人情報の収集、送信、記憶、使用に適したデータプライバシーおよび法律法規の保護に適した様々な制約を受けており、その中には、米国、EU、イギリス、および世界の他の国の全面的な規制制度を含む、個人情報のプライバシー、セキュリティ、および伝送に何らかの要求を加えることが含まれている。プライバシーとデータ保護の立法と規制構造は世界各地の司法管轄区域で発展し続けており、プライバシーやデータ保護問題にますます注目されており、これは私たちの業務に影響を与える可能性がある。これらの法律および法規を遵守しないことは、罰金、監禁会社の役人および大衆の非難、影響を受けた個人の損害クレーム、私たちの名声被害、および名誉損失を含む、私たちに対する法執行行動を引き起こす可能性があり、これらはいずれも、私たちの業務、財務状況、運営結果、または将来性に重大な悪影響を及ぼす可能性がある。さらに、これらの法律および法規は、サプライヤーおよび他のビジネスパートナーとの契約のコスト、および臨床試験または後続治療のための適切な患者を決定するコストを含む、私たちのビジネス活動に追加のコストをもたらす可能性がある。
もし私たちが個人が識別できる健康情報のプライバシーと安全を適切に保護できなければ、私たちは私たちの契約に違反していることが発見されるかもしれない。さらに、私たちが適用されるプライバシー法に従わない場合、私たちは民事と刑事罰、または私たちの業務に関連する他の法執行リスクに直面する可能性があります。このような潜在的な処罰に加えて、このような法執行活動は大量の内部資源を消費する可能性がある。また、州総検察長は民事訴訟を起こし、禁止令や損害賠償を求め、州住民のプライバシーを脅かす侵害行為に応える権利がある。私たちはこのような規定が私たちの業務にどのように解釈、実行されるかを決定することができない。法執行活動や潜在的な契約責任に関連するリスクに加えて、私たちは連邦や州レベルで変化する法律と法規を遵守するために努力している可能性が高く、私たちの政策、手続き、システムを絶えず修正する必要があります。
最近のデータプライバシー規制の変化の影響を扱い続けているが,新法規の発効と継続的な法的挑戦に伴い,データプライバシーは国内レベルでも国際的にも変化しており,変化するデータ保護ルールを遵守する努力は成功しない可能性がある.このような法律の解釈と適用は私たちの接近と一致しない可能性がある。私たちはこの変化する状況を理解して順応するために多くの資源を投入しなければならない。データ保護に関する法律を守らないことは、欧州経済区や他の地方データ保護当局が法執行行動をとるリスクに直面し、法律違反が発見されれば、重大な処罰を受ける可能性がある。同様に,米国連邦や州の個人情報のプライバシーやセキュリティに関する法律を守らなければ,このような法律の罰を受ける可能性がある.このようなデータ保護およびプライバシー法を遵守しない行為は、政府に罰金や命令を科す可能性があり、私たちのやり方、クレームまたは他の責任、規制調査および法執行行動、訴訟、および巨額の救済費用を変更することを要求し、いずれも私たちの業務に悪影響を及ぼす可能性がある。これらの法律に違反していると判断されなくても、政府のこれらの問題の調査には通常、大量の資源がかかり、負の宣伝が必要であり、これは私たちの業務、財務状況、運営結果、または将来性を損なう可能性がある。
103
ソーシャルメディアプラットフォームは私たちの業務に新しい危険と挑戦をもたらす。
ソーシャルメディアの持続的な拡張に伴い、それはまた私たちに新しい危険と挑戦をもたらしてくれる。ソーシャルメディアは、私たち、私たちの計画、そして私たちの候補製品が治療のために開発されている疾患に関する情報を交流するためにますます使用されている。製薬やバイオテクノロジー業界のソーシャルメディア実践が進化しており,不確実性と我々の業務に適用される法規を遵守しないリスクをもたらしている。例えば、患者は、ソーシャルメディアプラットフォームを使用して、製品または候補製品の有効性または不良体験についてコメントする可能性があり、これは、報告義務または他の結果をもたらす可能性がある。さらに、私たちのスタッフまたは他の人がメディアチャネルを介して意外にまたは意図的に非公開情報を開示することは、情報損失を招く可能性がある。さらに、任意のソーシャルメディアプラットフォーム上で、敏感な情報または私たち、私たちの製品、または私たちの候補製品に関する否定的または不正確な投稿またはコメントを不適切に開示するリスクがある。もしこのような事件が発生したり、私たちが適用された法規を遵守できなかった場合、私たちは責任を招き、制限的な規制行動に直面したり、私たちの業務に他の損害を与えたり、私たちの名声、ブランドイメージ、商業的名声に迅速で不可逆的な損害を与えることを含むかもしれません。
従業員事務·管理成長·情報技術に関するリスク
私たちの将来の成長は、私たちが事業や技術を識別して買収する能力にかかっているかもしれません。もし私たちがこれを成功させることができない場合、あるいは他の方法で任意の新しい業務や技術を私たちの業務に統合できなかったら、私たちの成長機会は限られているかもしれません。これは、重大な減価費用や他の不利な財務的結果を招く可能性があります。
私たちは私たちの業務戦略にマッチすると思う業務や技術の買収を求め続けています。しかし、将来の買収は、多くの運営と財務リスクをもたらすかもしれない
私たちは買収業務や技術を識別して実行し、現在のインフラに統合するのに十分な資源がないかもしれない。特に、私たちはより大きなバイオテクノロジー会社や他の競争相手と競争し、新たな協力と許可内の機会の構築に努力するかもしれない。これらの競争相手は私たちよりも多くの財務資源を得ることができ、新しい機会を発見して評価する上でより多くの専門知識を持っているかもしれない。また、私たちの候補製品と私たちが買収した会社との間に重複がある可能性があり、これは関係や他の約束の面で衝突し、統合された業務に不利になる可能性がある。さらに、新たな技術またはビジネスの支出と、その後これらの買収された技術またはトラフィックから生じる収入との間の時間(またはライセンス契約および/または戦略的協力に関連する収入確認時間)を取得するために使用されることは、異なる時期に当社の財務業績に変動をもたらす可能性がある。最後に、未完成の潜在的な買収機会に資源を投入したり、これらの努力の予想された利益を達成できなかったりすれば、重大な減価費用や他の不利な財務的結果が生じる可能性がある。
私たちの未来の成功は私たちがCEO総裁と他の重要な幹部を引き留めることができるかどうか、そして合格した人材を誘致、維持、激励できるかどうかにかかっている。
私たちは私たちの最高経営責任者John Evans、Giuseppe Ciaramella博士、私たちの総裁、そして私たちの管理と科学チームの他の主要なメンバーに非常に依存している。さん、ジアラメラ博士、その他の主要メンバーが“勝手”に雇われていることは、当社または彼らがいつでも彼らの雇用を終了できることを意味しています。私たちは私たちの役員や他の職員たちに“キーパーソン”保険を提供しない。これらの人たちの誰かを失ったサービスは、研究、開発、商業化目標の達成を阻害する可能性がある。
104
合格した科学、臨床、製造及び販売とマーケティング人員を募集と維持することも著者らの成功の鍵となる。私たちが最近発表したポートフォリオの優先順位と戦略的再編は、深い機関や技術知識を持つ人の流出を招くだろう。また,コスト増加,運営効率の低下,従業員の士気や生産性の低下,人員流動率の増加により,移行は我々の運営や従業員,サプライヤー,パートナーとの関係を乱す可能性がある。また、このような人事異動は、私たちの指導部の他のメンバーや私たちに残っている他の従業員への依存を増加させるかもしれません。彼らは契約上私たちに雇われ続ける義務がなく、いつでも離れることができます。どのような離職も特に破壊的である可能性があり、また、私たちが経験した追加人員の流れについては、トップレベルの人材に対する競争が非常に激しく、私たちは私たちの要求に合った候補者の探しと採用を延期するかもしれない。私たちの競争相手は、私たちに対する競争優位性を得るために、これらの移行と関連する潜在的な中断を利用することを求めるかもしれない。現在、私たちの産業には素質の高い人材が不足しており、このような状況は続くかもしれない。しかも、私たちがいる地域では、素質の高い人たちの不足が特に深刻だ。
多くの製薬とバイオテクノロジー会社の間の類似者に対する競争を受けて、私たちは受け入れ可能な条件でこれらの人員を吸引し、維持することができないかもしれない。私たちはまた、大学や研究機関から科学や臨床人を募集する競争に直面している。また、私たちは、科学と臨床コンサルタントを含むコンサルタントとコンサルタントに依存して、私たちの研究開発と商業化戦略の制定を助けてくれます。私たちの科学共同創始者を含め、私たちのコンサルタントやコンサルタントは、私たち以外の雇用主に雇われる可能性があり、他の実体との相談や相談契約に基づいて約束することができ、これは私たちが彼らを得る機会を制限するかもしれない。一部の幹部、重要な従業員、コンサルタント、コンサルタントのサービスを採用したり失うことは、私たちの研究、開発、商業化目標の進展を阻害し、私たちの業務、財務状況、運営結果、将来性に実質的な悪影響を与える可能性がある。
私たちは私たちの開発、規制、そして将来の販売とマーケティング能力を拡大することを望んでいますので、私たちは私たちの成長を管理する上で困難に直面するかもしれません。これは私たちの運営を乱すかもしれません。
私たちの製品ラインの成長と発展に伴い、私たちは私たちの従業員の数を増加させ、薬物開発、法規制事務、販売とマーケティング分野における私たちの業務範囲を拡大することが予想される。このような分野での私たちの予想される将来の成長を管理するためには、私たちの管理、運営、財務システムを継続して実施し、改善し、私たちの施設を拡大し、より多くの合格者を募集し、訓練し続けなければならない。私たちの財務資源が限られていることと、私たちの管理チームがこのような成長を期待している会社を管理する上での経験が限られているため、私たちが期待している業務拡張や採用やより多くの合格者を効率的に管理することができない可能性があります。また、我々が予想する業務実体の拡張は、巨額のコストを招く可能性があり、私たちの管理と業務発展資源を移転する可能性がある。成長を管理できないどんな状況も、私たちの業務計画の実行を延期したり、私たちの運営を妨害したりする可能性がある。
初期段階にあるバイオテクノロジー会社として、私たちは多くの治療分野と広範な疾病分野で新しいプラットフォームと候補製品を積極的に求めている。これらのすべての治療領域と疾病状態に適した候補製品の開発に成功し、そしてすべてのこれらの治療領域と疾病状態の監督と製造経路を十分に理解し、多くの領域で同時に実行するために、深い人材、資源と会社のプロセスが必要である。私たちの資源が限られているため、私たちはこれを効果的に管理し、私たちの業務を同時に実行し、拡大したり、より多くの合格者を募集したり訓練することができないかもしれない。これは私たちのインフラが弱く、操作ミス、法律や法規のコンプライアンスの失敗、ビジネス機会の喪失、従業員の流失、残りの従業員の生産性の低下を招く可能性がある。私たちの業務の実際の拡張は巨大なコストを招く可能性があり、他のプロジェクトから財務資源、例えば私たちの候補製品を開発することが可能です。私たちの経営陣が将来のどんな成長も効果的に管理できなければ、私たちの費用は予想以上に増加する可能性があり、私たちの収入を創出したり増加させる能力は低下する可能性があり、私たちは私たちの業務戦略を実施できないかもしれない。私たちの将来の財務業績と私たちの効果的な競争と私たちの候補製品を商業化する能力は、承認されれば、当社の将来の発展と拡張を効果的に管理する能力にある程度依存するだろう。
私たちの普通株に関するリスクは
私たちの普通株の市場価格は大きく変動する可能性があり、これは私たちの普通株の購入者に大きな損失をもたらし、私たちを証券集団訴訟に直面させる可能性がある。
私たちの株価は過去ずっと、将来も、大きな変動を受けていたかもしれない。総じて,株式市場,特にバイオ製薬会社の市場は,極端な変動を経験しており,この変動は特定の会社の経営業績とは無関係であることが多い。このような変動のため、初期購入価格あるいは初期購入価格以上で普通株を売ることができないかもしれません。私たちの普通株の市場価格は多くの要素の影響を受けるかもしれません
105
ある会社の証券市場価格にこのような変動が生じた後、同社に対して証券集団訴訟が提起されることが多い。私たちの株価の潜在的な変動性のため、私たちは未来に証券訴訟の目標になるかもしれない。証券訴訟は巨額のコストを招き、経営陣の注意と資源を私たちの業務から移す可能性がある。
財務報告書に対する適切かつ効果的な内部統制を確立し、維持することができなければ、私たちの経営業績と私たちの経営業務の能力が損なわれる可能性がある。
適切な内部財務·会計制御·プログラムを維持し、正確な財務諸表をタイムリーに作成できるようにすることは、高価で時間のかかる作業であり、常に再評価が必要である。財務報告の内部統制は、財務報告の信頼性を合理的に保証することを目的としたプログラムであり、公認された会計原則に基づいて財務諸表を作成する。上場企業の要求に適合するために、2002年のサバンズ·オクスリ法(SOX)第404条の要求に適合するために、我々の内部統制およびプログラムを記録、審査、改善するなどの行動をとり、経営陣が財務報告の内部統制の有効性を年間評価し、私たちの公認会計士事務所が年次報告とそのような評価の証明を提出することを要求している。私たちがこれらの措置を取ったにもかかわらず、私たちの内部統制の十分性を効果的に維持できないかもしれません。このような十分性を維持できなかったり、それによって正確な財務諸表がタイムリーに作成できなかったり、私たちの監査人がこのような十分な分岐を維持しているかどうかについて、私たちの運営コストを増加させ、私たちの業務を損なう可能性があります。また、投資家は、私たちの内部統制が不足しているか、あるいは正確な財務諸表をタイムリーに作成できないと考えており、これは私たちの普通株価格を損なう可能性があり、私たちのサービスを新老顧客に効果的にマーケティングし、販売することを困難にしている。
106
上場企業としては、引き続きコストを増加させることが予想されており、我々の経営陣は、新たなコンプライアンス措置やコーポレートガバナンス実践を実施するために多くの時間を投入する必要がある。
上場企業として、巨額の法律、会計、その他の費用を引き続き負担することが予想されている。2002年のサバンズ-オクスリ法案、ドッド-フランクウォール街改革と消費者保護法、ナスダック株式市場の上場要求、その他の適用された証券規則と法規は、有効な開示、財務制御、会社管理やり方の確立と維持を含む上場企業に対して様々な要求を提出した。私たちの経営陣と他の人たちはこのような要求に対する遵守を維持するために多くの時間を投じた。このような要求は私たちの法律と財政的コンプライアンス費用を増加させ、いくつかの活動をもっと時間と費用を増加させる。例えば、上場企業として、取締役と役員責任保険を維持する難しさとコストが高く、合格した取締役会のメンバーを引き付け、維持することが難しくなる可能性があります。これらの規則や条例は往々にして異なる解釈を持ち,多くの場合特殊性に欠けるため,規制機関や理事機関が新たな指導意見を提供するにつれて,実践における適用は時間とともに変化する可能性がある。これは遵守事項に関する持続的な不確実性と、開示と統治慣行を絶えず修正するために必要なより高いコストをもたらす可能性がある。
私たちは予測可能な未来に何の配当もないと予想している。投資家は彼らが彼らが購入した価格より高い価格で私たちの普通株を売らない限り、彼らの投資から見返りを得ないかもしれない。
あなたは私たちの普通株への投資に依存して配当収入を提供してはいけない。私たちは予測可能な未来に、私たちは普通株式保有者に何の配当も支払わないと予想している。代わりに、私たちは私たちの既存の業務を維持して拡大するためにどんな収益も維持する予定だ。さらに、任意の将来の信用スケジュールには、私たちの普通株が発表または支払い可能な配当金の金額を禁止または制限する条項が含まれている可能性がある。そのため、投資家は、投資リターンを実現する唯一の方法として、価格上昇後に普通株を売却することに依存しなければならない。
私たちが4回目に改訂した会社登録証明書、私たちの第2の改正と再記述の定款、およびデラウェア州法律の条項は逆買収効果がある可能性があり、他の会社が私たちを買収することを阻止するかもしれません。たとえ買収が私たちの株主に有利になり、私たちの株主が現在の経営陣を交換または更迭しようとするのを阻止することができます。
私たちが4回目に改訂した会社登録証明書またはわが社登録証明書、ならびに私たちの第2の改正と再記載の定款、私たちの定款およびデラウェア州法律に含まれる条項は、株主が有利と思われる可能性のある私たちの統制権の変更または私たちの経営陣の変更を阻止、延期、または阻止する可能性があり、あなたの株式からプレミアム取引を得ることができることを含む。当社の会社登録証明書および付例は、以下の条文を含む
これらの条項は単独でまたは一緒に敵意の買収、統制権の変更、または私たちの経営陣の変動を延期または阻止する可能性がある。これらの条項はまた、投資家が私たちの普通株に支払いたいかもしれない価格を制限し、それによって私たちの普通株の市場価格を下げることができる。
また、私たちはデラウェア州に登録して設立されたので、私たちはデラウェア州会社法第203条の規定によって管轄されています。この条項は、規定された方法で合併または合併が承認されない限り、私たちが発行した議決権のある株を15%以上保有している人が取引日後3年以内に私たちと合併または合併することを禁止しています。
107
私たちの会社の登録証明書、定款、あるいはデラウェア州法律の遅延または制御権の変更を阻止する条項は、私たちの株主が彼らが持っている普通株からプレミアムを得る機会を制限する可能性があり、また一部の投資家が私たちの普通株に支払いたい価格に影響を与える可能性があります。
我々の会社登録証明書および定款は、デラウェア州内の州または連邦裁判所が、私たちの株主が開始する可能性のある特定のタイプの訴訟および訴訟の独占フォーラムとして指定されており、これは、私たちまたは私たちの役員、上級管理者または従業員との紛争において有利な司法フォーラムを得ることができる株主の能力を制限することができる。
私たちの会社登録証明書は、限られた例外を除いて、デラウェア州内の州または連邦裁判所は、以下の態様の専属フォーラムとなる:(1)私たちを代表して提起された任意の派生訴訟または訴訟、(2)私たちの任意の取締役、上級管理職、または他の従業員の私たちまたは私たちの株主に対する受託責任に違反すると主張するいかなる訴訟も、(3)DGCL、私たちの会社登録証明書、または私たちの定款の任意の規定に基づいて、私たちにクレームを提起する任意の訴訟、(4)任意の解釈、適用、適用される。当社の登録証明書又は当社の定款の有効性を強制的に執行又は決定し、又は(5)内部事務原則によって管轄されていることを主張する他の任意の訴訟を執行又は決定する。また、私たちの付例は、私たちが書面で代替裁判所を選択することに同意しない限り、アメリカ連邦地域裁判所は、証券法に基づいて提起された任意の訴因を解決する独占的な裁判所になると規定している。当社の株式の権益を購入又はその他の方法で取得した者又は実体は、当社の上記会社の登録証明書及び附例の規定に了承し、同意したものとみなさなければならない。これらの裁判所条項の選択は、私たちまたは私たちの役員、役員、または他の従業員とのトラブルに有利だと考える株主のクレームを司法裁判所で提出する能力を制限する可能性があり、これは、私たちと私たちの役員、役員、および従業員に対するこのような訴訟を阻止する可能性があります。代替的に、裁判所が、私たちの会社登録証明書または添付例のこれらの条項が、1つまたは複数の特定のタイプの訴訟または法的手続きに適用されないことを発見した場合、または1つまたは複数の指定されたタイプの訴訟または法的手続きについて実行できない場合、私たちは、他の司法管轄区域において、そのような問題の解決に関連する追加費用を発生させる可能性があり、これは、私たちの業務および財務状況に悪影響を及ぼす可能性がある。例えば、デラウェア州衡平裁判所は最近、米国連邦地域裁判所が“証券法”に基づいて訴因を提出した苦情を解決する独占フォーラムであることを規定する条項は実行できないと判断した。しかし、この決定はデラウェア州最高裁判所によって再審され、最終的に覆されるかもしれない。
一般リスク因子
突発的な公衆衛生事件や流行病は私たちの業務に悪影響を及ぼすかもしれない。
突発的な公衆衛生事件や流行病は私たちの業務と財務業績に実質的な悪影響を与える可能性があり、私たちの候補製品の開発中断を招く可能性があります。このような突発的な公衆衛生事件が私たちの業務、運営結果、および将来の成長見通しに影響を与える可能性のある程度は、緊急事態の持続時間、範囲、および深刻さ、米国および他の国の旅行制限および社会的距離の存在および程度、企業閉鎖または業務中断、米国および他の国がとる行動の有効性、および治療介入の利用可能性を含む様々な要素に依存するであろう。
突発的な公衆衛生事件は私たちの臨床前と臨床活動の完成に遅延または他の方法で不利な影響を与える可能性があり、疫病の持続時間、未来の臨床試験の開始、および私たちの全体的な業務に基づいて、これらの要素は:
108
突発的な公衆衛生事件はまた、本節の“プロジェクト1 A”と題する多くの他のリスクを増加させる可能性がある。リスク要因は、“例えば、私たちが追加資金を調達する必要があることに関するリスク、私たちの四半期の財務業績の変動、および私たちが規制承認を得て維持する能力。
包括的な税制改革立法は私たちの商業と財政状況に悪影響を及ぼすかもしれない。
税法の最近または未来の変化は私たちの業務や財務状況に悪影響を及ぼすかもしれない。2017年12月22日、税法が法律に署名された。“CARE法案”で改正された“税法”には、会社税率を35%の最高限界税率から21%に下げる統一税率を含む会社税への大きな変化が含まれており、純営業損失の控除額を今年度の課税所得額の80%に制限し、2018年またはその後に生じる純営業損失に充てる。税法によると、2018年と今後数年間に発生したいかなる連邦純運営赤字は現在無期限に繰り越すことができるが、これ以上繰り越すことはできない。似たような規則と制限は州所得税提案に適用されるかもしれない。
税法に加えて、新冠肺炎の流行に対応する国会の一部として、2020年と2021年にCARE法案と2022年8月に法律に署名したアイルランド共和軍を含む税収条項を含む経済救済立法が公布され、いくつかの新しい税収条項が導入された。IRAは特に、上場企業がある株を買い戻すことに1%の消費税を課すことを含み、これは通常、上場企業(またはその一部の付属会社)が会社の株主から株を買収し、金銭や他の財産(会社自体の株を除く)と交換するのに適しているが、極めて最低限の例外がある。したがって、消費税は非伝統的な株の買い戻しのいくつかの取引に適用される可能性がある。税法やこのような追加立法の規制指導は継続されており、このような指導は最終的には、これらの法律が私たちの業務や財務状況に与える影響を増加または減少させる可能性がある。しかも、各州がどの程度税法とこの追加的な税金立法を遵守するかどうかはまだ確定されていない。
不安定な市場や経済状況は、私たちの業務、財務状況、株価に深刻な悪影響を及ぼす可能性がある。
過去数年間、世界の信用と金融市場は極端な変動と破壊を経験し、流動性と信用供給の深刻な減少、消費者自信の低下、経済成長の減速、失業率の上昇と経済安定の不確定性、世界の供給の中断、ロシアとウクライナ間の衝突、ロシアに対する関連制裁、インフレ率の上昇と金利の変化を含む。信用と金融市場のさらなる悪化と経済状況への自信が起こらない保証はない。私たちの全体的な業務戦略は、このような経済低迷、不安定なビジネス環境、または持続不可能で不安定な市場状況のいずれかの悪影響を受ける可能性がある。現在の株式や信用市場が悪化したり改善されていない場合、任意の必要な債務や株式融資をより困難にし、コストがより高く、希釈度が高くなる可能性がある。また、私たちの株価は下落する可能性があり、一部の原因は株式市場の変動と全体的な経済低迷である。
適切かつ有利な条件で必要な融資を得ることができなければ、私たちの成長戦略、財務業績、株価に実質的な悪影響を及ぼす可能性があり、私たちの1つまたは複数の候補製品の開発と商業化を延期、削減、または停止すること、または潜在的な許可内または買収を求めることを延期することを要求するかもしれない。さらに、我々の現在の1つまたは複数のサービスプロバイダ、製造業者、および他のパートナーは、経済的困難な時期を乗り切ることができない可能性があり、これは、運営目標を達成する能力に直接影響を与える可能性がある。
もし証券アナリストが私たちの業務に関する研究や報告を発表しなければ、あるいは彼らが私たちの株に対するマイナス評価を発表したら、私たちの株価は下落するかもしれません。
私たちの普通株の取引市場は、業界や金融アナリストが発表した私たちまたは私たちの業務に関する研究と報告にある程度依存している。これらのアナリストのうちの1人以上がわが社への報道を停止したり、私たちに関する報告書を定期的に発表できなかったりすれば、金融市場で可視度を失う可能性があり、逆に私たちの株価や取引量を低下させる可能性がある。また、もし私たちのアナリストが私たち、私たちのビジネスモデル、あるいは私たちの株式表現に否定的または誤った意見を発表した場合、あるいは私たちの経営業績が投資家グループの期待に達しなかった場合、一人以上のわが社を追跡したアナリストがわが社に対する彼らの提案を変える可能性があり、私たちの株価は下落する可能性があります。
109
私たちの内部コンピュータシステム、または私たちの第三者サプライヤー、協力者、または他の請負業者またはコンサルタントのシステムは、故障したり、セキュリティホールに遭遇したりする可能性があり、これは、私たちの製品開発計画を深刻に中断させ、私たちの業務に関連する敏感な情報を危険にさらし、または私たちの重要な情報へのアクセスを阻止し、それによって、私たちが責任を負うか、または他の方法で私たちの業務に悪影響を及ぼす可能性があります。
我々の内部コンピュータシステム、ならびに私たちの現在および未来の任意の第三者サプライヤー、協力者および他の請負業者またはコンサルタントのコンピュータシステムは、コンピュータウイルス、コンピュータハッカー、悪意のあるコード、従業員の窃盗または乱用、サービス拒否攻撃、複雑な民族国家および民族国家によって支持される参加者、許可されていないアクセス、自然災害、テロ、戦争、および電気通信および電気故障の破壊または中断を受けやすい。我々は、我々の情報セキュリティ計画および業務パートナーとの関連契約プロトコルによって、システム障害、事故、セキュリティホールの影響から情報技術システムを保護することを求めていますが、このような事件が発生し、当社の運営中断を招く場合、可能な個人データの損失を含む、当社の開発計画および業務運営中断を引き起こす可能性があります。例えば、未来の臨床試験中の臨床試験データの紛失は著者らの監督管理の審査作業の遅延を招く可能性があり、著者らのデータの回復或いは複製のコストを著しく増加させ、そして潜在的な個人データの損失に関連する義務とリスクを負担させる。もし私たちの情報システムやデータが深刻なネットワークセキュリティホールに遭遇した場合、米国または他の国の規制調査に関連する潜在的なコストに加えて、調査、救済、および取引相手およびデータ主体に違反を通知する可能性があることに関連するコストは巨大である可能性がある。しかも、私たちの救済努力は成功しないかもしれない。適切な技術およびネットワークセキュリティインフラを確立し、維持するために必要なリソースを割り当てて効率的に管理しなければ、取引エラー、サプライチェーンまたは製造中断、処理効率の低下、データ損失または知的財産権、または他の固有情報の損失または破損を含む深刻な業務中断を受ける可能性があります。
もしいかなる中断やセキュリティホールが私たちまたは私たちの第三者サプライヤー、協力者または他の請負業者またはコンサルタントのデータやアプリケーションが失われたり、破損したり、あるいは機密または独自の情報を適切に開示しない場合、私たちは訴訟暴露、処罰、罰金を含む責任を招く可能性があり、私たちは規制行動や調査の対象となる可能性があり、私たちの競争地位が損なわれる可能性があり、私たちの候補製品のさらなる開発と商業化は延期される可能性がある。以上のいずれも、当社の業務、財務状況、運営結果、または将来性に重大な悪影響を及ぼす可能性があります。
項目1 B。取消解析Dスタッフがコメントした。
ない。
プロジェクト1 C。ネットワークセキュリティです。
我々は、我々の全体的なリスク管理計画/情報技術機能に内蔵されたネットワークセキュリティリスクを評価、識別、管理する特定のプロセスを有しており、我々の情報資産および運営を内部および外部ネットワークの脅威から保護し、許可されていないアクセスや攻撃から従業員および臨床試験情報を保護し、ネットワークおよびシステムセキュリティを保護することを目的としている。これらのプロセスは、リスクを識別し、私たちのやり方を改善するために、物理、プログラム、および技術保障、応答計画、私たちのシステムの定期的なテスト、イベントシミュレーション、および私たちの政策およびプログラムの定例審査を含む。私たちはコンサルタント、独立プライバシー評価員、コンピュータセキュリティ会社とリスク管理会社、同業者会社、業界団体と管理専門家を招いて、私たちのネットワークセキュリティ監督を強化します。第三者サービスプロバイダと接触する前に、関連する抜け穴から当社の会社を保護するのを助けるために、彼らの内部リスク監視計画を考慮します。
我々は現在、ネットワークセキュリティ脅威からの既知のリスクが存在するとは考えておらず、これらのリスクは、合理的にわが社または当社の業務戦略、運営結果、または財務状況に重大な影響を与える可能性がある。
我々の取締役会の監査委員会はネットワークセキュリティリスクを直接監督し、このような監督に関する最新の状況を取締役会に提供する。監査委員会は、四半期ごとに管理層からネットワークセキュリティ問題に関する最新の状況を受け取り、更新中に重大な新しいネットワークセキュリティ脅威または事件に関する通知を受けた。
我々の情報セキュリティ副総裁は、全社のネットワークセキュリティ戦略、政策、標準とプロセスの運営監督を指導し、関連部門間で仕事を展開し、私たちおよび私たちの従業員と第三者サービスプロバイダがネットワークセキュリティリスクに対応する準備をしていることを評価し、助けてくれる。このIS副社長のサイバーセキュリティ訓練には、25年間のネットワークセキュリティプロジェクトの構築と維持の経験があり、国際情報システムセキュリティ認証連盟(ISC 2)から認証された情報システムセキュリティ専門家(CISSP)認証を取得し、ISACAからネットワークセキュリティ·インフラセキュリティ局(CISA)認証を取得した。
ネットワークの脅威を阻止し、発見するために、私たちは毎年アルバイトと臨時従業員を含むすべての従業員にデータ保護、ネットワーク安全とイベント応答と予防訓練とコンプライアンス計画を提供し、社会工学、ネット釣り、暗号保護、機密データ保護、資産使用と移動安全を含むタイムリーかつ関連するテーマをカバーし、従業員にすべての事件の重要性を直ちに報告するように教育している。我々はまた,技術に基づくツールを用いてネットワークセキュリティリスクを低減し,従業員ベースのネットワークセキュリティ計画を支援している.
110
プロジェクト2.ニュースオペラです。
2028年に満期になった賃貸契約によると、私たちはマサチューセッツ州ケンブリッジ市で約38,203平方フィートのオフィスと実験室空間をレンタルし、2034年に満期したレンタルによって、マサチューセッツ州ケンブリッジ市で約130,258平方フィートのオフィスと実験室空間を借りました。また,Alexandria Real Estate Equities,Inc.と賃貸契約を締結し,協定に基づき,ノースカロライナ州研究三角公園に広範な臨床プロジェクトを支援するために10万平方フィートの製造工場を建設した。レンタル契約は2022年12月にこの施設に入居して15周年に満了し、レンタル期間を2~5年延長することを選択することができます。私たちの施設は私たちの現在の需要を満たすのに十分で、必要な時に適切な追加空間を提供すると信じている。
項目3.法律法律手続き。
私たちは現在どんな実質的な法的手続きの当事者でもない。私たちは時々様々な法的手続きやクレームの影響を受けるかもしれません。これらの訴訟とクレームは私たちの正常な業務活動の過程で発生します。
4つ目:地雷の安全TYが披露する。
適用されません。
111
部分第2部:
項目5.登録者普通株式市場、関連株持株者は重要で発行者が株式証券を購入する。
市場情報
私たちの普通株は2020年2月6日からナスダック世界精選市場で公開取引され、取引コードはBEAMである。その前に、私たちの普通株は市場を公開しなかった。
所持者
2024年2月20日現在、私たちの普通株は約33人の登録保有者がいます。この数字には,その株式が街中の被指名者が所有する実益所有者は含まれていない.
配当をする
設立以来、私たちはどんな現金配当金も発表したり支払ったりしたことがない。私たちは将来の収益(あれば)を残し、私たちの業務の運営と拡張に資金を提供し、予測可能な未来に普通株式保有者に現金配当金を支払うことはないつもりです。
112
株式表現グラフ
1934年の“証券取引法”(改訂本)第18節または“取引法”については、以下の業績グラフおよび関連情報は、1933年の“取引法”(改正本)または“証券法”に基づいて提出された任意の将来の文書に、特に引用によってこのような文書に組み込まれない限り、引用によってこのような情報を“証券法”(改正本)または“証券法”に提出された任意の将来の文書に組み入れてはならない。
次の図は、私たちの普通株の2020年2月6日(私たちの初公募株の日)から2023年12月31日までの株主累積総リターンと同期(A)ナスダックバイオテクノロジー指数と(B)ナスダック総合指数の累積総リターンを比較した。この図は,2020年2月6日に我々の普通株,ナスダックバイオテクノロジー指数,ナスダック総合指数への投資を100ドルと仮定し,再投資配当金(あれば)を仮定している。このグラフは、一般向けの最初の発行価格ではなく、私たちの普通株の2020年2月6日の終値1株18.75ドルを私たちの普通株の初期価値として使用している。次の図に示す比較は履歴データに基づく.この図に含まれる株価表現は,必ずしも未来の株価表現を示唆しているとは限らない.

113
発行者または関連購入者が株式証券を購入する
2023年第4四半期に、私たちまたは任意の関連バイヤー、または私たちを代表して、または関連バイヤーを代表して行動する任意の1人当たりは、私たちの普通株式の任意の株式を購入しなかった。
第六項です[Re料理を出す]
114
項目7.経営陣の財務状況と経営結果の検討と分析国家統計局です。
以下、我々の財務状況と経営結果の検討と分析は、我々の連結財務諸表および本年度報告書の他の部分がForm 10-K形式で含まれるこれらの報告書に関する付記とともに読まなければならない。歴史財務情報を除いて、以下の議論と分析には、リスク、不確定性、および仮説に関連する前向きな陳述が含まれている。新聞の掲載を容易にするために、本明細書のいくつかの数字は四捨五入された。我々の実際の結果は,これらの前向き陳述で予想された結果と大きく異なる可能性があり,これは,本年度報告10−K表の第1 A項(リスク要因)で議論された要因を含む多くの要因の結果である。
2021年度に関する情報は、2022年12月31日現在の年次報告Form 10−Kの第120~126ページの第II部第7項の下で、2023年2月28日に米国証券取引委員会(“米国証券取引委員会”)に提出されている。
概要
私たちはバイオテクノロジー会社で、リードし、全面的に統合された精密遺伝子薬物プラットフォームの構築に力を入れている。私たちのビジョンは深刻な病気を患っている患者に生涯治療を提供することだ。このビジョンを実現するために、遺伝子編集と交付技術のセットと内部製造能力を含むプラットフォームを組み立てた。
我々の遺伝子編集技術キットは我々の独自の塩基編集技術を基礎としており、これはDNA二本鎖切断を招くことなく、ゲノム中の単一塩基に対する差別化精密遺伝薬を可能にする可能性がある。この方法は、標的配列上で正確で予測可能かつ効率的な遺伝結果を生成することを目的とする化学反応を使用する。著者らの独自の塩基編集プログラムは2つの主要な構成部分がある:(I)RNA結合を誘導するクラスター形成の規則間隔短回文反復配列或いはCRISPR蛋白質であり、それはCRISPRが確立したDNA標的能力を利用するが、改造された二本鎖切断を招くことはない;及び(Ii)塩基編集酵素、例えばデアミナーゼであり、それは必要な標的DNA塩基の化学修飾を実行する。この設計は、従来の遺伝子編集方法と比較して、より正確かつ効率的な編集を実現するのに役立ち、遺伝子編集の影響を著しく増加させる可能性があると信じている。私たちはこの2つの配送方法を求めています離体するそして体内にある術式は,組織型に依存する。塩基編集方法の優雅さは組織特定の送達方式と結合し、配向、高効率、正確かつ高度に汎用的な遺伝子編集システムに基礎を提供し、このシステムはいくつかの遺伝子を同時に遺伝子修正、遺伝子サイレンシングまたは遺伝子活性化、遺伝子修正および/または多重編集を行うことができるように設計されている。
我々の目標は,異なる遺伝子検証された編集目標に対して,広範で多様な基礎エディタの組合せ,および革新的なプラットフォームビジネスモデルを推進し,我々のプログラムのカバー範囲をより多くの患者に拡大することである.全体的に言えば、著者らはリードする正確な遺伝医学総合プラットフォームを構築することを求めており、これは広範な治療適用性と正確な遺伝医学領域を変える潜在力を持っているかもしれない。
製造業
高品質製造及び生産タイミングと技術ノウハウの制御に重要であるため、著者らはノースカロライナ州研究三角園区に100,000平方フィートの製造工場を設立し、広範な臨床プロジェクトを支持することを目的とした。この施設は2023年末にcGMP運営を開始し、私たちの製造を支援することを目指しています離体する 血液学と血液学における細胞治療計画体内にある 肝臓と肝臓介在性疾患に対する非ウイルス伝達計画は、潜在的な商業供給を支持するために規模を拡大する能力がある。われわれの予備臨床試験では,主に我々の内部製造能力と,遺伝子薬物に関して製造経験のあるCMOに依存することが予想される。この投資は私たちのポートフォリオと能力の価値、私たちが技術的に成功する可能性、そして私たちが患者に潜在的な生涯治療の速度を提供することを最大限に高めると信じています。
買収する
2021年2月には、Guide Treateutics,Inc.またはGuideを買収し、2021年2月19日までの10取引日における普通株の出来高加重平均価格に基づいて、通常の購入価格調整を含まない普通株の株式前払い総金額1.2億ドルを設定した。また、Guideの前株主とオプション所有者は、私たちの普通株で支払う1.00億ドルまでの追加技術マイルストーン支払いと2.200億ドルの製品マイルストーン支払いを受ける資格がある。
115
財務運営の概要
一般情報
私たちは2017年1月に設立され、2017年7月に運営を開始した。私たちの設立以来、私たちはほとんどの資源を私たちの基礎編集プラットフォームの構築と私たちのプログラムの組み合わせの発展を推進し、私たちの知的財産権を確立し、保護し、研究開発活動を行い、私たちの会社、業務計画を組織し、配備し、資金を調達し、これらの業務に一般的かつ行政的な支援を提供してきた。これまで、私たちは主に私たちの償還可能な転換可能な優先株を売却し、私たちの普通株の収益を発行し、協力と許可協定によって受け取った支払いによって、私たちの業務に資金を提供してきました。
私たちは初期段階にある会社で、私たちのほとんどのプロジェクトは臨床前または早期臨床開発段階にあります。これまで、私たちは製品販売から何の収入も得ておらず、予測可能な未来にも製品販売から収入を得ることはない。今まで、私たちの収入は主にパートナーとの許可と協力協定から来ていた。設立以来、私たちは重大な運営損失を受けた。2023年12月31日現在、2022年と2021年12月31日までの年間純損失はそれぞれ1.325億ドル、2.891億ドル、3兆706億ドルだった。2023年12月31日現在、私たちの累計赤字は12億ドルです。私たちが候補製品の臨床前と臨床開発を継続することに伴い、より多くの候補製品の臨床開発を推進する;ノースカロライナ州のcGMP施設を運営する;更に私たちの基礎編集プラットフォームを開発する;Guideを買収することによって得られたLNP技術を含む、私たちの基礎編集者に技術投資を提供し続ける;私たちがより多くの候補製品を発見と開発する過程で研究活動を展開する;私たちの知的財産権の組み合わせを維持、拡張、実行、保護し、そして研究開発、臨床、技術運営と商業者を募集し続ける。また、上場企業の運営に関連したコストが引き続き発生すると予想される。
このような予想された支出のために、私たちは私たちの持続的な運営を支援し、私たちの成長戦略を実施するために、より多くの資金を集める必要があるだろう。私たちが製品販売から相当な収入を得ることができるまで、もしあれば、株式発行、債務融資、協力、戦略連合、許可手配の組み合わせで私たちの運営に資金を提供する予定です。私たちは必要な時に割引条項や追加資金を調達できないか、このような他の合意に到達できないかもしれない。私たちは必要な時に資金を集めることができなくて、これは私たちの財務状況と私たちの業務戦略を実施する能力にマイナス影響を与えるだろう。私たちは私たちの運営を支援するためにこのような追加的な資本源を得ることができるという保証はないし、もし私たちがそのような資本を持っていれば、私たちはこれらの追加的な資本が私たちの短期的または長期的な需要を満たすのに十分であることを保証することはできない。
収入確認
2019年4月、我々はVerve Treateutics,Inc.またはVerveと、心血管疾患治療に専念する遺伝子編集の会社であるVerveプロトコルの協力および許可協定を達成した。2021年6月、私たちはApellis PharmPharmticals、Inc.またはApellisと研究協力プロトコル、すなわちApellisプロトコルを締結し、私たちのいくつかの塩基編集技術を使用して補体系によって駆動される疾患の新しい療法を発見することに重点を置いた。2021年10月、我々はSana Biotech,Inc.またはSanaとオプションおよびライセンス契約、またはSana協定を締結し、この協定に基づいて、私たちのCRISPR Cas 12 b技術にSanaの非独占的研究開発および商業権を付与し、ヌクレアーゼ編集をある程度実行する離体する遺伝子工学細胞治療計画です2021年12月にファイザーと4年間の研究協力協定を締結しました体内にある肝臓、筋肉と中枢神経系の3種類の稀な遺伝病に対する基礎編集プログラム。2022年9月、軌道治療会社または軌道会社と許可および研究協力協定、または軌道協定を締結し、軌道会社は非ウイルス送達およびRNA技術の推進に専念する新たなエンティティである。2023年10月、私たちはVerve Treateutics、Inc.またはVerveとの修正協力および許可プロトコルまたはVerveプロトコルに基づいて、Verveの心血管疾患基礎編集プログラムの選択権を含む、Verve Treateutics、Inc.またはVerveとの修正協力および許可プロトコルまたはVerveプロトコルに従って、Verveの共同開発および共同商業化を含む、礼来会社または礼来会社と譲渡および許可協定または礼来協定を締結した。
これまで、私たちは製品販売から何の収入も得ておらず、近い将来も何の収入も生じないと予想されています。次の年度まで 2023年12月31日,2022年12月31日,2021年12月31日に,それぞれ許可と連携協定から3.77億ドル,6,090万ドル,5,180万ドルの収入を確認した。
当社の収入確認ポリシーの詳細については、本年度報告Form 10−Kに含まれる監査総合財務諸表付記2および付記11を参照されたい。
研究開発費
研究·開発費用には、研究·開発活動を行うことによるコストが含まれている
116
私たちの外部研究開発費用は私たちの様々な臨床前と臨床プロジェクトを支持します。我々の内部研究開発費には,従業員に関する費用,施設関連費用,全体の研究開発支援のための他の間接研究開発費が含まれている。私たちは発生した費用に応じて研究と開発費用を支払う。私たちが将来受け取る研究·開発活動のための商品やサービスのための前金は前払い費用として記録されています。前払い額は福祉の消費に伴って支出される。
開発の初期段階では,我々の研究開発コストは通常製品プラットフォームと概念検証臨床前研究に用いられており,これらの研究は必ずしも特定の目標に割り当てることができるとは限らない。
われわれの計画の臨床前と臨床開発によるわれわれの計画の推進に伴い,われわれの研究·開発費用は大幅に増加することが予想される。
一般と行政費用
一般と行政費用は主に行政、知的財産権、業務発展と行政機能者の給料とその他の関連費用を含み、株式ベースの給与を含む。一般および行政費用には、知的財産権および会社の事務に関連する法律費用、会計、監査、税務およびコンサルティングサービスの専門費用、保険料、出張、および施設に関連する直接および分配費用、および他の運営コストも含まれる。
私たちは今後、私たちの増加した研究·開発活動を支援するために、私たちの一般的かつ行政的費用が増加すると予想する。また、上場企業として財務報告の制御を維持し、ナスダックと米国証券取引委員会が要求する会計、監査、法律、規制および税務関連サービスの遵守に関連するコスト、取締役および役員保険コスト、および投資家および広報コストを含む関連コストが引き続き発生することを予想している。
他の収入と支出
他の収入および支出には以下の項目が含まれている
117
行動の結果
2022年実績の検討および2021年実績との比較については,2022年12月31日までの財政年度Form 10−K年度報告第2部第7項“経営陣の財務状況と経営成果の検討と分析”を参照されたい。
2023年12月31日までと2022年12月31日までの年度比較
次の表は私たちの行動結果(千計)をまとめています
|
|
十二月三十一日までの年度 |
|
|
|
|
||||||
|
|
2023 |
|
|
2022 |
|
|
変わる |
|
|||
許可と協力収入 |
|
$ |
377,709 |
|
|
$ |
60,920 |
|
|
$ |
316,789 |
|
運営費用: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
|
437,381 |
|
|
|
311,594 |
|
|
|
125,787 |
|
一般と行政 |
|
|
116,813 |
|
|
|
87,805 |
|
|
|
29,008 |
|
総運営費 |
|
|
554,194 |
|
|
|
399,399 |
|
|
|
154,795 |
|
運営損失 |
|
|
(176,485 |
) |
|
|
(338,479 |
) |
|
|
161,994 |
|
その他の収入(支出): |
|
|
|
|
|
|
|
|
|
|||
派生負債の公正価値変動 |
|
|
7,500 |
|
|
|
23,900 |
|
|
|
(16,400 |
) |
非持株投資の公正価値変動 |
|
|
(18,592 |
) |
|
|
20,200 |
|
|
|
(38,792 |
) |
価格負債があって価値変動を公正にすることができます |
|
|
9,740 |
|
|
|
18,904 |
|
|
|
(9,164 |
) |
利息とその他の収入,純額 |
|
|
46,676 |
|
|
|
15,297 |
|
|
|
31,379 |
|
その他収入合計 |
|
|
45,324 |
|
|
|
78,301 |
|
|
|
(32,977 |
) |
所得税前純損失 |
|
|
(131,161 |
) |
|
|
(260,178 |
) |
|
|
129,017 |
|
所得税支給 |
|
|
(1,366 |
) |
|
|
(3,410 |
) |
|
|
2,044 |
|
権益法投資損失 |
|
|
— |
|
|
|
(25,500 |
) |
|
|
25,500 |
|
純損失 |
|
$ |
(132,527 |
) |
|
$ |
(289,088 |
) |
|
$ |
156,561 |
|
許可と協力収入
2023年12月31日までの事業年度では,許可·協力収入は約3.77億ドルであったが,2022年12月31日現在の事業年度では,許可·協力収入は約6,090万ドルと3兆168億ドル増加した。2023年に記録された許可および連携収入は、礼来社、ファイザー社、アペリス社、軌道会社、Verveプロトコルによって記録された収入です。2022年には、ファイザープロトコルに関連するライセンス収入6,090万ドルと、ApellisおよびVerveプロトコルに関連する収入を記録しました。収入の増加は,礼来社との取引(付記11参照)の2.164億ドルと,2022年と比較して2023年に提供されたサービスと比較して,ファイザーやアペリス取引に関する確認収入の増加を反映している。ファイザーとApellisの収入は払戻不可能な前金に関係しており,進捗の測定基準により,実際に発生した総コストを用いて総見積りコストと比較する方法であることを確認した。
研究開発費
2023年12月31日と2022年12月31日までの年度の研究開発費はそれぞれ4.374億ドルと3.116億ドルだった。次の表は、2023年12月31日と2022年12月31日までの研究開発費、およびこれらの項目の変化(ドル単位)をまとめています
|
|
十二月三十一日までの年度 |
|
|
|
|
||||||
|
|
2023 |
|
|
2022 |
|
|
変わる |
|
|||
外部研究開発費 |
|
$ |
154,178 |
|
|
$ |
111,831 |
|
|
$ |
42,347 |
|
従業員関連費用 |
|
|
109,878 |
|
|
|
89,547 |
|
|
|
20,331 |
|
施設やITに関する費用 |
|
|
69,339 |
|
|
|
54,048 |
|
|
|
15,291 |
|
株に基づく報酬費用 |
|
|
57,812 |
|
|
|
52,004 |
|
|
|
5,808 |
|
その他の費用 |
|
|
46,174 |
|
|
|
4,164 |
|
|
|
42,010 |
|
研究開発費総額 |
|
$ |
437,381 |
|
|
$ |
311,594 |
|
|
$ |
125,787 |
|
1兆258億ドル増加しました主な理由は以下の通りです
118
私たちがBEAM-101、BEAM-201、BEAM-302とBEAM-301の臨床試験を推進することに伴い、私たちの現在の研究計画を続け、新しい研究計画を開始し、私たちの候補製品の臨床前と臨床開発を継続し、任意の未来の臨床前研究を行い、患者を私たちの任意の候補製品に参加して臨床試験を行い始め、研究開発費用は引き続き増加する予定である。
一般と行政費用
2023年12月31日と2022年12月31日までの年度の一般·行政費はそれぞれ1兆168億ドルと8780万ドル。2900万ドル増加しました主な理由は以下の通りです
増加額は以下の項目の一部によって相殺される:
派生負債の公正価値変動
2023年12月31日までの1年間に、普通株価格の下落により、支払成功負債公正価値の変化に関する750万ドルの他の収入を記録しましたが、2022年12月31日現在の年間は2390万ドルです。成功した支払いの一部は2021年6月に支払われた;2023年12月31日現在、残りの成功支払債務はまだ返済されておらず、各報告期間で再評価が継続される。
非持株投資の公正価値変動
2023年12月31日と2022年12月31日までの年間で、VerveとPrime Medicine普通株投資に対する公正価値が変化したため、それぞれ1860万ドルの他の支出と2020万ドルの他の収入を記録した。
対価格負債の変動があります
2023年12月31日と2022年12月31日までの年間で、それぞれ970万ドルと1890万ドルの他の収入を記録しており、これらの収入は、プロジェクトスケジュールの更新と予想されるマイルストーンの実現可能性によるガイド技術や製品、または対価格負債の公正価値の変化に関連している。
利息とその他の収入,純額
2023年12月31日と2022年12月31日までの年度の利息とその他の収入(支出)純額はそれぞれ4670万ドルと1530万ドル。この成長は主に市場金利の上昇と私たちポートフォリオの成長が金利収入の増加を推進したためです。
所得税支給
2023年12月31日および2022年12月31日までに、それぞれ140万ドルおよび340万ドルの所得税支出を記録し、発生した課税収入が税務運営赤字繰越および税務相殺で完全に販売されていないことを反映している。私たちの課税収入には、2023年12月31日までに年度確認された礼来合意に関する収入と、2022年12月31日までにApellisやファイザーと協力して確認された収入が含まれており、この収入には、税務目的で2021年に延期されている。また、2017年の“減税と雇用法案”は、納税者に5年または15年以内に研究開発支出を資本化·償却することを要求しているため、課税所得額は帳簿支出よりも高い。
119
権益法投資損失
当社は2022年12月31日までに権益法投資損失2,550万ドルを記録しており,Orbaryが行っている研究開発や将来他の用途のない基礎差額に関連しており,この差額はこの投資日に直ちに計上されている。2022年12月31日まで、この投資はゼロに減記された。権益会計方法に関するより多くの情報は、当社の連結財務諸表付記2および付記11を参照してください。
流動資金と資本資源
2017年1月の設立以来、私たちは製品販売から何の収入も得ておらず、私たちの許可と協力協定のみから限られた収入を得ており、私たちの運営には大きな運営損失と負のキャッシュフローが生じています。予測可能な未来には,候補製品の臨床前·臨床開発の推進に伴い,巨額の費用と運営損失を招くことが予想される。
2021年4月、米国証券取引委員会または2021年棚にS-3表の形態で汎用自動棚登録声明を提出し、米国証券取引委員会に届出した後に発効する1つまたは複数の製品のうち不確定な数量の普通株式、優先株、債務証券、権証および/または単位を登録する。
2021年4月、私たちはJefferies LLCまたはJefferiesと市場またはATMでの販売協定または販売協定を締結し、この協定によると、私たちは時々当時の市場価格で私たちの普通株を発売し、販売する権利があり、総収益は30000万ドルに達する。私たちは、Jefferiesが販売契約に従って販売した任意の株の総販売収益の3.0%の最高3.0%の手数料をJefferiesに支払うことに同意する。2023年12月31日まで、私たちは販売契約に従って1株103.16ドルの平均価格で2,908,009株の普通株を売却し、総収益は3,000万ドルで、手数料と私たちが支払うべき発売費用を差し引く。
2021年7月と2023年5月に、吾らはJefferiesと販売協定を改正し、2023年5月10日に、吾らは総発行価格が8.0億ドルの普通株を追加できるように販売契約項目の総発売金額を増加させることを規定した。2023年12月31日まで、修正された販売契約に基づいて、1株51.93ドルの平均価格で10,860,992株の普通株を売却し、総収益は5.64億ドルで、手数料と発売前に支払うべき費用を差し引いた。
2023年10月、礼来社または礼来社と礼来契約を締結し、この協定によると、礼来社は、Verveとの改訂協力および許可協定またはVerveプロトコルに基づいて、Verveの心血管疾患基礎編集プログラムの選択権を共同開発し、共同商業化することを含むいくつかの資産および他の権利を獲得した。私たちは2億ドルの前金を受け取り、ある臨床、監督、そして連盟活動が完了した後、3億5千万ドルに達する潜在的な未来開発段階の支払いを受ける資格がある。礼来会社協定について、私たちは礼来会社と株式購入協定を締結し、礼来会社に2,004,811株の私たちの普通株を売却·発行することを規定しており、総購入価格は5,000万ドルです。私は2023年10月に株式購入協定の下で5,000万ドルを受け取り、2023年11月に前金2億ドルを受信したに等しい。私たちは約4,330万ドルの債務を蓄積しており、これらの債務は、礼によって合意された支払いに関連している可能性があり、ライセンス契約条項に対するこのような支払いの潜在的な適用性に関する議論が続いている。
2023年12月31日現在、私たちは12億ドルの現金、現金等価物、有価証券を持っている。
私たちは普通株の公正時価の増加に基づいてハーバードとブロド研究所に成功的な支払いを支払うことを要求された。満期の金額は現金や普通株の形で支払うことができ、私たちが自分で決めます。私たちはまたハーバード大学とブロイド大学にそれぞれ9000万ドルの追加費用を借りているかもしれない。
私たちはまだ私たちの候補製品を商業化していません。予測可能な未来に、私たちは私たちの候補製品を販売することから収入を得ないと予想されます。私たちは、私たちが計画した臨床前研究と臨床試験、私たちの商業規模cGMP製造施設の維持と運営、新製品開発、そして私たちの一般運営に資金を提供するために、より多くの資金を調達する必要があるかもしれないと予想しています。必要があれば、株式や債務融資など、様々な潜在的な源を通じて、あるいは会社協力やライセンス契約を通じて、より多くの資金を調達することを求めていきます。私たちは私たちの運営を支援するためにこのような追加的な資金源を得ることができるという保証はないし、もし私たちがこのような資金を持っていれば、私たちはこのような追加的な資金が私たちの需要を満たすのに十分であるという保証はない。
120
キャッシュフロー
次の表は私たちの現金源と用途(千計)をまとめています
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
経営活動提供の現金純額 |
|
$ |
(149,195 |
) |
|
$ |
22,527 |
|
|
$ |
(66,268 |
) |
投資活動提供の現金純額 |
|
|
71,840 |
|
|
|
(461,336 |
) |
|
|
(294,144 |
) |
融資活動提供の現金純額 |
|
|
276,448 |
|
|
|
111,590 |
|
|
|
756,141 |
|
現金、現金等価物、および限定的な現金純変化 |
|
$ |
199,093 |
|
|
$ |
(327,219 |
) |
|
$ |
395,729 |
|
経営活動
2023年12月31日までの1年間、経営活動で使用された現金純額は1.492億ドルで、礼来会社の取引で受け取った2.164億ドルの現金、計算すべき費用と他の負債の6770万ドルの増加、株式ベースの給与支出9860万ドル、非持株投資の公正価値の1860万ドルの減少、減価償却と償却費用2000万ドルの減少、経営リースROU資産の変化950万ドルを含む非現金プロジェクトが含まれている。
これらの現金源は、私たちの純損失1.325億ドル、繰延収入1.596億ドルの減少、経営賃貸負債の1020万ドルの減少、売掛金760万ドルの減少、その他の長期負債20万ドルの減少、前払い費用およびその他の流動資産の650万ドルの増加、協力売掛金10万ドルおよび非現金プロジェクト、2970万ドルの投資割引と保険償却、750万ドルの派生負債の公正価値の減少、970万ドルまたは対価格負債の公正価値の変化によって部分的に相殺された。
2022年12月31日現在、経営活動が提供する現金純額は2,250万ドルで、主にファイザー協定に関する売掛金3,000万ドル、経営賃貸負債の1,240万ドルの増加、売掛金の2,440万ドルの増加、および8,430万ドルの株式給与支出、2,550万ドルの権益法投資損失、1,410万ドルの減価償却と償却費用、840万ドルの経営賃貸純資産変動を含む非現金項目が含まれている。
これらの現金源は、私たちの純損失2.891億ドル、繰延収入3590万ドルの減少、2022年12月31日までの年度にApellisから徴収された第1周年支払純額2500万ドル、課税費用およびその他の負債の1690万ドルの減少、前払い費用およびその他の流動資産の780万ドルの増加、その他の長期負債の260万ドルの減少、および派生負債公正価値の2390万ドルの減少、非制御持分投資公正価値の20万ドルの増加を含む非現金プロジェクト。あるいは対価格負債のある公正価値変動は1890万ドル、償却投資割引と保険料は940万ドルである。
投資活動
2023年12月31日までの1年間に、投資活動が提供した現金7180万ドルは主に有価証券が満期になった純結果であり、3370万ドルの財産や設備購入を除いて、有価証券の購入部分はこの純結果を相殺した。
2022年12月31日までの1年間、投資活動のための現金は主に有価証券を購入した純結果であり、一部は4.124億ドルの有価証券満期日で相殺され、また4900万ドルの財産と設備が購入された。
融資活動
2023年12月31日までの年度,融資活動が提供する現金純額は2.764億ドルであり,主に2.366億ドルの株式発行収益(販売手数料控除),礼来合意発行株の収益3360万ドル,株式オプション行使収益610万ドル,および我々の従業員の株式購入計画に基づいて普通株を発行する収益300万ドルを含み,一部は設備融資負債純返済230万ドルと配当発行コスト60万ドルで相殺されている。
2022年12月31日までの1年間に、融資活動が提供する現金純額は、主に株式発行収益1.083億ドル、販売手数料を差し引いて、私たちの従業員株式購入計画に基づいて普通株収益310万ドル、株式オプション収益270万ドルを発行するが、230万ドルの設備融資債務の返済と20万ドルの株式発行コスト部分で相殺される。
121
資金需要
私たちが引き続き私たちのプロジェクトの組み合わせを進めるにつれて、私たちの運営費用は増加し、引き続き大幅に増加する予定です。
具体的には、以下のような状況に伴い、私たちの支出が増加するだろう
私たちは、2023年12月31日の現金、現金等価物、および有価証券が、添付の連結財務諸表の発表日から少なくとも今後12ヶ月間、現在および計画された運営費用および資本支出に資金を提供できるようになると予想している。私たちが基づいているこのような推定は不正確であることが証明されるかもしれないし、私たちは現在予想されているよりも早く私たちが利用できる資本資源を枯渇させるかもしれない。私たちのプロジェクト開発に関連する多くのリスクと不確実性のため、私たちの候補製品の研究や開発の完了に関連する増加した資本支出や運営費用を見積もることができません。
私たちの未来の支出需要は多くの要素に依存するだろう
122
我々の任意の候補製品の開発に関連するこれらまたは他の変数のいずれの結果も変化し、候補製品の開発に関連するコストおよび時間を著しく変化させることができる。また、私たちの運営計画は将来的に変化する可能性があり、運営需要とそのような運営計画に関連する資本要求を満たすために追加の資金が必要になるかもしれません。
これまで、相当な製品収入を生み出すことができれば、株式発行、債務融資、協力、戦略連合、許可手配の組み合わせで現金需要を満たす予定です。私たちは約束された外部資金源を持っていない。私たちは従来、私たちの資本需要を満たすために株式発行に依存していたが、将来は株式発行に依存するかもしれない。債務融資が可能であれば、追加債務を招く、資本支出を行う、または配当を宣言するなど、特定の行動をとる能力を制限または制限する契約を含むいくつかの合意が含まれる可能性がある。
もし私たちが追加的な協力、戦略連合、または第三者との許可手配を通じて資金を調達すれば、私たちは私たちの技術、未来の収入流、研究計画、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちは私たちに不利な条項で許可を与えなければならないかもしれない。もし私たちが必要な時に株式や債務融資を通じてより多くの資本を調達できない場合、私たちは私たちの製品開発を延期、制限、減少または終了する必要があるかもしれません。あるいは承認された場合、私たちは将来の商業化努力を延期、制限、減少、または終了する必要があるかもしれません。あるいは、私たちが自分で開発し、マーケティングしたいと思っていた候補製品を開発し、マーケティングする権利を与える必要があります。私たちは私たちの運営を支援するためにこのような追加的な資金源を得ることができるという保証はないし、もし私たちがこのような資金を持っていれば、私たちはこのような追加的な資金が私たちの需要を満たすのに十分であるという保証はない。
契約義務
私たちはキャンセルできない経営レンタル方式である資産をレンタルして、レンタル期間は2037年までです。レンタルは主に事務空間、実験室、製造空間、そして設備に関するものだ。2023年12月31日現在、これらのオフィスと実験室賃貸および設備レンタルの将来の最低承諾額は合計2兆514億ドルで、関連する公共エリア維持費や不動産税は含まれていません。
2021年5月、ハーバード許可協定と遠大許可協定に基づいて第1回成功支払い測定を行い、ハーバードと遠大学院への支払い成功支払いはそれぞれ1,500万ドルと1,500万ドルと算出した。私たちは普通株で各金額を支払うことを選択し、2021年6月にハーバード大学とブロイド研究所にそれぞれ174,825株の普通株を発行し、これらの債務を返済した。残りの支払成功義務は2023年12月31日まで返済されていない。私たちはまたハーバード大学と遠大学院にそれぞれ9000万ドルの追加的な支払いに借りがあるかもしれない。
私たちがある機関と締結した知的財産権許可協定によると、いくつかのマイルストーンと成功費用、非印税再許可収入費用、特許権使用料、許可維持費、特許維持費用の精算を義務的に支払うことができるかもしれません。私たちの知的財産権許可協定には、協定によって許可された知的財産権を使用した製品の開発に依存し、開発または規制承認マイルストーンの実現、および商業および成功支払いマイルストーンに依存する潜在的なマイルストーン支払いが含まれています。これらの額は未来の事件の発生状況によりますが、このような潜在的な債務の時間と可能性はまだ正確にはわかりません。
また、Guideの前株主とオプション所有者に1.00億ドルまでの追加技術マイルストーン支払いと2.2億ドルの製品マイルストーン支払いを支払うことに同意します。これらは私たちの普通株式価値で支払い、私たちの普通株の出来高加重平均価格を使用して、適用マイルストーンを受け取る日前の2取引日に終了した10日間の取引期間内です。これらの支払いは未来の事件の発生に依存しており、このような潜在的な債務の時間と可能性はまだ正確には知られていない。
また、私たちは正常な業務過程でCRO、CMO、他のサプライヤーと契約を締結し、私たちの研究開発活動や他の運営サービスや製品の実行に協力します。これらの契約は一般的に通知後に終了することが規定されているので、取り消すことができる契約です。
123
重要な会計政策と重大な判断と見積もり
私たちの経営陣は、私たちの財務状況と経営結果の議論と分析は、私たちの合併財務諸表に基づいており、私たちはアメリカ公認の会計原則に基づいて作成しています。これらの財務諸表を作成するには、私たちの財務諸表に報告されている資産、負債および費用金額、または資産および負債の開示に影響を与える推定、判断、および仮定を作成する必要があります。我々は,歴史的経験,既知の傾向や事件,および当時の状況で合理的と考えられる様々な他の要因に基づいて推定し,これらの要因の結果は資産や負債の帳簿価値の判断の基礎を構成しているが,これらの資産や負債の帳簿価値は他の源から容易に見られるものではない.私たちは持続的な基礎の上で私たちの推定と仮定を評価する。異なる仮定や条件の下で、私たちの実際の結果はこれらの推定とは異なる可能性がある。
我々の主な会計政策は、本年度報告書10-K表の総合財務諸表付記2により詳細に説明されているが、以下の会計政策は、財務諸表作成に使用する判断と推定に最も重要であると考えられる。
収入確認
最初に、契約がASC 606の範囲内にあるかどうかを確認します取引先と契約した収入またはASC 606または他の主題。ASC 606の範囲内で決定された契約の場合、収入は、顧客が約束された商品またはサービスの制御権を取得したときに確認される。確認された収入額は、これらの商品とサービスと交換するために、私たちが獲得する権利があると予想された対価格を反映している。このコア原則を実現するために,(I)顧客との契約の決定,(Ii)契約中の履行義務の決定,(Iii)取引価格の決定,(Iv)契約に取引価格を割り当てる履行義務,および(V)契約履行義務を履行する際に収入を確認する,の5つのステップを採用した.顧客の意図と支払い承諾に応じた対価格の能力を決定し、譲渡された商品とサービスのほぼすべての対価格を実質的に受け取ることができる場合にのみ、5段階モデルを契約に適用する。
契約で約束された履行義務は,顧客に移行する貨物やサービスによって決定され,これらの貨物とサービスは区別できるとともに,契約範囲内で異なる.1つの契約に複数の約束された貨物およびサービスが含まれている場合、私たちは、約束された貨物およびサービスが契約の文脈で区別できるかどうかを決定するために判断を使用する。これらの基準を満たしていなければ、約束された貨物とサービスは総合履行義務として入金されるだろう。
取引価格は、商品とサービスを顧客に移転するために、私たちが獲得する権利のある対価格によって決定されます。取引価格に可変対価格が含まれている場合には、可変対価格の性質に基づいて、期待値方法または最も可能な金額方法を用いて、取引価格に含まれるべき可変対価格金額を推定する。経営陣の判断により、契約項下の累積収入が将来的に大きく逆転しない可能性が高い場合には、可変対価格が取引価格に含まれる。可変考慮に対する制約の影響を含む任意の推定は、どのような変化があるかどうかを決定するために、各報告期間において評価される。取引価格の決定には重要な判断が必要であり,我々の各許可と連携プロトコルは,本年度報告Form 10−Kの連結財務諸表付記11においてより詳細な検討がある。
契約が単一履行義務を含む場合、取引価格全体をその単一履行義務に割り当てる。複数の履行義務を含む契約要求は、取引価格が可変でない限り、相対的に独立した販売価格に基づいて取引価格を個々の履行義務に割り当て、かつ、履行義務または単一履行義務の一部を構成する異なるサービスに完全に割り当てられる基準に適合する。受信した対価格を相対的に独立した販売価格に応じて単独の履行義務の間に割り当てる.独立販売価格の決定には重大な判断が必要であり、我々の各許可および連携プロトコルは、注釈11において詳細に議論される。
時間が経つにつれても、ある時点でも、私たちは義務を履行するつもりだ。以下の場合、収入は、時間とともに確認される:(I)顧客がエンティティ業績によって提供される利益を同時に受け取り、消費すること、(Ii)エンティティ業績が資産作成または強化時に顧客によって制御される資産を創造または強化するか、または(Iii)エンティティ業績は代替エンティティ用途の資産を生成しておらず、エンティティは、これまでに完了した業績支払いを強制的に実行する権利がある。エンティティが一定期間義務を履行していない場合、ある時点で、約束された貨物またはサービスの制御権を顧客に移すことによって、関連する履行義務を履行する。
サービス実行時間や連携プロトコルでの項目に関する外部コストの発生は,収入の特定時期の確認方式に影響を与える可能性がある.
知的財産権許可:我々の知的財産権許可が合意で決定された他の履行義務とは異なると判定された場合、許可が顧客に譲渡されたときに許可に割り当てられた対価格収入を確認し、顧客は許可を使用して利益を得ることができる。他の承諾と組み合わせたライセンスについては、合併履行義務が時間の経過とともに履行されるか、ある時点で履行されるかを決定するために、合併履行義務の性質を判断するために利用され、時間の経過とともに収入を確認するための進展を測定するための適切な方法が決定される。私たちは各報告期間の進捗測定基準を評価し、必要であれば、業績評価基準と関連するものを調整する
124
収入確認。我々は一般にこれまでに発生したコストを予想総コストと比較して収入を確認する.発生予定の内部と外部費用総額推定数とこれらの費用が発生予定の時間の変化は変動期間内に累積追跡調整であることを確認した。見積り費用総額の見積り数が変化したり,契約修正が履行義務の範囲を変更したりすると,その影響は大きい可能性がある.IPライセンスの収入確認の決定には重要な判断が必要であり,付記10と11で許可や連携プロトコルごとにより詳細に検討する.
マイルストーン支払い:開発または規制マイルストーン支払いを含む各スケジュールの開始時に、マイルストーンに到達する可能性を評価し、可能な金額方法を用いて取引価格に含まれる金額を推定します。将来大きな収入逆転が起こらない可能性が高い場合、関連するマイルストーン価値は取引価格に含まれるだろう。規制承認のような我々または被許可者の制御範囲内でない記念碑的支払いは、これらの承認を受ける前に実現可能とは考えられないため、管理職が収入が逆転することは不可能であると断言できないため、確認された収入が制限される。そして,取引価格は相対的に独立した販売価格で個々の履行義務に割り当てられ,そのため,収入を契約下の履行義務が履行された場合に確認する.その後の報告期間ごとに終了した場合には,このような発展マイルストーンを達成する可能性や任意の制限を再評価し,必要に応じて全体の取引価格の見積りを調整する.いずれの調整も累積追跡をもとに記録されており,調整期間中の収入や収益に影響を与える.今まで、私たちは私たちのどんな合意でも記念碑的な収入を確認していない。
商業マイルストーン支払い及び特許権使用料:販売に基づく特許権使用料(販売レベルに基づく記念碑的特許権使用料を含む)を含む手配については、ライセンスが特許権使用料に関連する主要項目とみなされている場合は、(I)関連販売が発生した場合、又は(Ii)特許権使用料に割り当てられた履行義務が履行された(又は部分的に履行されている)ときに収入を確認する。今まで、私たちは私たちのいかなる合意によって生成されたいかなる特許権使用料収入も確認していない。
私たちが義務を履行する必要がない場合、または義務履行期限が終了した後、貨物またはサービスの制御権を顧客に移した場合、金額は収入として確認される。通常、販売に基づくマイルストーンや印税を除いて、受領または満了したすべての金額は、許可および協力収入に分類される。販売に基づくマイルストーンおよび特許権使用料は、関連販売が発生したか、またはすべての特許権使用料分配の履行義務が履行された(または部分的に履行された)ときに、特許権使用料収入として確認される。
今後12ヶ月以内に確認されると予想される繰延収入は流動負債に分類される。
公正な価値計量--成功的な支払い
私たちは普通株の公正時価の増加に基づいてハーバードとブロド研究所に成功的な支払いを支払うことを要求された。満期の金額は現金や普通株の形で支払うことができ、私たちが自分で決めます。支払いに成功したのは会計基準コード815に従って派生ツールとして入金され、派生ツールおよびヘッジそして、初歩的に公正価値に従って入金し、相応して研究開発費用を計上した。負債は資産負債表ごとに市価で計算され、すべての価値変動は総合経営報告書で確認された利息とその他の収入(費用)とその他の全面赤字で確認されている。支払い義務の達成または満期の早い者まで、価値変動を公正に許可する負債を調整し続ける。支払いに成功した推定公正価値を決定するために、イベント発生確率、イベント発生時間、および支払い成功時の1株当たり価格を含むいくつかのキー変数に基づいて負債価値をモデル化するモンテカルロシミュレーションモデルを使用した。私たちの株価や変動性の大きな変化は負債の価値に大きな影響を与えるかもしれない。
計算と前払いの研究と開発コスト
財務諸表作成過程の一部として、私たちの費用と前払いの研究開発コストを見積もる必要があります。このプロセスは、未締結契約および調達注文を審査し、私たちの担当者とコミュニケーションして、私たちに代わって実行されるサービスを決定し、インボイスを受け取っていない場合、または他の方法で実際のコストを通知している場合に、実行されたサービスレベルおよびサービスによって生じる関連コストを推定することを含む。私たちのほとんどのサービスプロバイダは毎月私たちが提供してくれるサービスや契約マイルストーンに達した時に私たちに借金の領収書を発行してくれます。私たちは、当時私たちが知っている事実と状況に基づいて、財務諸表において、各貸借対照表の日付までの課税費用を推定します。計算すべき研究·開発費用の例としては,臨床前開発活動に関連するサプライヤーへの費用と,製品候補材料の開発,製造,流通に関するサプライヤーへの費用が予想される。
125
我々は,我々を代表して臨床前研究を行って管理している複数のサプライヤーとの契約に基づいて,受信したサービスと費用の見積もりに基づいて,臨床前研究に関する費用を計算した。これらの合意の財務条項は交渉が必要であり、契約によって異なり、支払いの不均衡を招く可能性がある。場合によっては、私たちのサプライヤーに支払われるお金は、提供されたサービスレベルを超え、事前支払い費用につながる可能性があります。サービス料を計算する際には、サービスを提供する時間帯と各時間内の労働支出レベルを見積もり、それに応じて調整します。
第七A項。数量化と高質市場リスクの開示について。
私たちは金利の変化と関連した市場リスクに直面している。2023年12月31日現在、私たちは現金、通貨市場基金、商業手形、会社手形、会社株式証券を含む12億ドルの現金、現金等価物、および有価証券を持っている。私たちの市場リスクに対する主な開口は金利感受性であり、金利感受性はアメリカの金利全体の水準変化の影響を受けており、特に私たちの投資は短期有価証券だからです。私たちのポートフォリオの存続期間が短く、私たちの投資リスクが低いため、即時10%の金利変動は私たちのポートフォリオの公平な市場価値に実質的な影響を与えないと信じています。私たちは満期まで私たちの投資を持つことができますので、私たちの経営業績やキャッシュフローは市場金利の変化が私たちのポートフォリオに与える影響を大きく受けないと予想しています。
私たちは現在、外貨為替レートの変化に関する重大な市場リスクに直面していません。しかし、私たちは確かにアメリカ以外のサプライヤーと契約を締結しており、これらのサプライヤーは外貨為替レートの変動の影響を受ける可能性があります。私たちは将来アメリカ以外のサプライヤーと追加の契約を締結するかもしれません。これは私たちの外貨両替リスクを増加させるかもしれません。
インフレは一般的に労働コストと研究開発、製造、開発コストを増加させることで私たちに影響を与える。インフレは私たちの財務諸表に実質的な影響を与えていないと信じています。これらの財務諸表は本年度報告の10-K表に含まれています。しかし、私たちの運営は未来にインフレの悪影響を受けるかもしれない。
プロジェクト8.財務諸表Sと補足データ。
本項目8の要求に基づいて提出された財務諸表は、本年度報告のテーブル10−Kの後に添付される。これらの財務諸表のインデックス項目15を参照してください展示品と財務諸表の付表本年度報告の表格10−K。
項目9.Accouとの変更と分岐会計と財務情報開示の専門。
ない。
第9条。制御するプログラムがあります
情報開示制御とプログラムの評価
2023年12月31日現在、我々の経営陣は、取引法第13 a-15(E)及び15 d-15(E)条の規定に適合する当社の開示制御及びプログラムの有効性を評価している。取引法規則13 a-15(E)および15 d-15(E)に定義されている用語“開示制御および手順”は、取引法に従って提出または提出された報告において開示を要求する会社の情報が米国証券取引委員会規則および表によって指定された期間にわたって記録、処理、集約および報告されることを保証するための会社の制御および他のプログラムを意味する。開示制御及び手続は、会社が“取引法”に基づいて提出又は提出された報告書において開示を要求する情報が蓄積され、その主要幹部及び主要財務官を含む会社経営者に伝達されることを保証することに限定されるものではないが、必要な開示に関する決定をタイムリーに行うために、同様の機能を果たす者の制御及び手続を適宜行う。経営陣は、どのような制御やプログラムが、どんなに設計や操作が良くても、その目標を実現するために合理的な保証を提供するしかないことを認識しており、管理部門は、可能な制御とプログラムのコスト-利益関係を評価する際にその判断を運用しなければならない。
2023年12月31日までの開示統制及び手続の評価によると、我々の最高経営責任者及び最高財務官は、その日までに、我々の開示制御及び手続が合理的な保証レベルで有効であると結論した。
126
財務報告の内部統制に関する経営陣の報告
私たちの経営陣は財務報告書の十分な内部統制を確立して維持する責任がある。“取引法”第13 a-15(F)及び15 d-15(F)条に基づく財務報告の内部統制の定義は、公認された会計原則に基づいて財務報告の信頼性及び外部目的の財務諸表作成のための合理的な保証を提供するために、我々の取締役会、管理層及び他の人員によって実施されるプログラムであり、公認された会計原則に基づいて財務報告の信頼性及び外部目的の財務諸表作成のための合理的な保証を提供することである
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。将来的にどのような有効性評価を行うかの予測には,条件の変化により制御が不十分になる可能性がある,あるいは政策やプログラムを遵守する程度が悪化する可能性があるというリスクがある.私たちは財務報告に対する私たちの内部統制を検討し続け、その有効性を向上させ、私たちのシステムが私たちの業務と共に発展することを確実にするために時々変更する可能性がある。
最高経営責任者や最高財務責任者を含む経営陣の監督と参加の下、トレデビル委員会後援組織委員会が発表した“内部統制-総合枠組み(2013)”の枠組みに基づいて、財務報告の内部統制の有効性を評価した。この評価に基づき、我々の上級管理職は、財務報告内部統制が2023年12月31日から有効であると結論した。
私たちの独立公認会計士事務所徳勤会計士事務所は私たちの財務報告書の内部統制に関する認証報告書を発表しました。以下を参照されたい。
財務報告の内部統制の変化
私たちは内部統制の効率性と効力を向上させることを絶えず求めている。これは私たちの会社全体の流れを改善させた。2023年12月31日までの1年間、財務報告の内部統制(“外国為替法案”第13 a-15(F)および15 d-15(F)規則の定義に基づく)に大きな影響を与えなかったか、または合理的に財務報告の内部統制に大きな影響を与える可能性のある変化はなかった。
127
独立公認会計士事務所報告
ビム治療会社の株主と取締役会に。
財務報告の内部統制については
当社は、以下の基準に基づいて、ビム治療会社及びその付属会社(“当社”)の2023年12月31日までの財務報告内部統制を審査しました内部統制--統合フレームワーク(2013)テレデビル委員会(COSO)が主催して組織委員会が発表した。2023年12月31日現在、当社はすべての重要な面で財務報告に対する有効な内部統制を維持しており、その根拠は内部統制--統合フレームワーク(2013)COSOから発表されます。
また、米国上場企業会計監督委員会(PCAOB)の基準に従って、当社の2023年12月31日現在および2023年12月31日現在の総合財務諸表および2024年2月27日までの報告書を監査し、このような財務諸表に対して保留のない意見を表明した。
意見の基礎
当社経営陣は、効果的な財務報告内部統制を維持し、添付されている“経営陣財務報告内部統制報告”に含まれる財務報告内部統制の有効性を評価する責任を負う。私たちの責任は私たちの監査に基づいて、会社の財務報告書の内部統制に意見を述べることです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務報告の有効な内部統制がすべての重要な面で維持されているかどうかを決定するために、合理的な保証を得るために監査を計画し、実行することを要求する。我々の監査には,財務報告の内部統制を理解すること,重大な弱点があるリスクを評価すること,評価されたリスクテストに基づいて内部統制の設計·運用有効性を評価すること,および状況下で必要と考えられる他のプロセスを実行することが含まれる。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
財務報告の内部統制の定義と限界
会社の財務報告に対する内部統制は、公認された会計原則に基づいて、財務報告の信頼性と外部目的の財務諸表の作成に合理的な保証を提供することを目的としたプログラムである。会社の財務報告に対する内部統制には、(1)合理的で詳細かつ正確かつ公平に会社の資産を反映した取引および処分に関する記録の保存、(2)公認された会計原則に従って財務諸表を作成するために必要な取引が記録されている合理的な保証を提供し、会社の収入および支出は会社の経営陣と取締役の許可のみに基づいて行われる。(三)財務諸表に重大な影響を及ぼす可能性のある不正買収、使用、または処分会社の資産を防止またはタイムリーに発見する合理的な保証を提供する。
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来的にどのような有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムの遵守度が悪化する可能性がある.
/s/徳勤法律事務所
ボストン、マサチューセッツ州
2024年2月27日
128
プロジェクト9 B。他にも情報です。
役員と上級社員の取引手配
次の表は、2023年12月31日までの四半期間、私たちの役員および上級管理者が採用または終了した各会社の証券売買取引スケジュール、すなわち、(1)ルール10 b 5-1(C)正面防御条件を満たすことを目的とした契約、命令または書面計画、または“ルール10 b 5-1取引スケジュール”、または(2)“非ルール10 b 5-1取引スケジュール”(定義S-Kルール408(C)項参照)を示す
名前.名前(タイトル) |
とった行動(訴訟日) |
取引手配の種別 |
取引手配の性質 |
取引手配の継続時間 |
証券総数量 |
養子縁組 ( |
販売する |
または、すべての取引が実行されずに完了または満了するより早い日。 |
…まで |
||
( |
養子縁組 ( |
販売する |
または、すべての取引が実行されずに完了または満了するより早い日。 |
…まで |
|
養子縁組 ( |
販売する |
または、すべての取引が実行されずに完了または満了するより早い日。 |
…まで |
プロジェクト9 Cです。秘密を明かすE検査を阻止する外国司法管轄区域について。
ない。
129
部分(三)
プロジェクト10.役員·役員職ICERSと会社管理。
第10条に要求される情報は、2024年年次総会に関する我々が米国証券取引委員会又は米国証券取引委員会に提出した最終委託書に含まれ、引用により本明細書に組み込まれる。
私たちは、私たちの最高経営責任者、最高財務官、最高会計官、財務総監、または同様の機能を実行する者を含む、私たちのすべての役員、高級管理者、および従業員に適用される書面による商業行為および道徳的規則を採択した。規則の最新コピーは、Investors.beamtex.comのウェブサイトの投資家部分で取得できます。私たちは米国証券取引委員会規則に基づいて開示されなければならない私たちのコードの任意の修正または免除を私たちのウェブサイトで開示するつもりだ。
第11項.実行五、補償する。
第11項に要求される情報は、2024年年次総会に関する取締役に提出された最終委託書の“役員報酬”および“米国証券取引委員会報酬”と題する部分に含まれ、引用によって本明細書に組み込まれる。
プロジェクト12.特定の受益者の保証所有権従業員と経営陣と関連した株主問題。
本第12項に要求される情報は、2024年年次総会に関する我々が米国証券取引委員会に提出した最終委託書における“特定の利益所有者及び管理層の担保所有権”及び“株式補償計画に従って発行された証券”の2節に含まれ、参照によって本明細書に組み込まれる。
第13条に要求される情報は、2024年年次総会に関する我々が取締役に提出した最終委託書の“特定の関係及び関連者取引”および“米国証券取引委員会独立性”の部分に含まれ、参照によって本明細書に組み込まれる。
第14項.元金口座TING料金とサービスです。
本第14項に要求される資料は、2024年株主総会について米国証券取引委員会に提出された最終委託書に当社が提出した“主要会計士費用及びサービス”と“監査委員会事前承認政策及びプログラム”の2節を含み、参考方式で本文に組み込まれる。
130
部分IV.IV
プロジェクト15.展示品、資金エーアールアイチェックリストです。
1.財務ファクトシート
ここに掲げる財務諸表のリストについては、参照されたい連結財務諸表索引本年度報告表格10−Kに記載されているF−1ページは,参照により本プロジェクトに組み込まれる。
2.財務諸表の添付表
財務諸表の添付表は、必須でないか、適用されないか、またはこれらの情報が連結財務諸表またはその付記に含まれているので省略されている。
3.展示品
展示品 番号をつける |
|
展示品説明 |
表 |
ファイル.ファイル 番号をつける |
日取り 保存する |
展示品 番号をつける |
保存済み ここから声明する |
|
|
|
|
|
|
|
|
2.1# |
|
BEAM治療会社,Galileo Merge Sub I,Inc.,Galileo Merge Sub II,LLC,Guide Treateutics,Inc.(以下,ガイドライン),株主代表サービス有限責任会社とそのガイドライン保有者間の合併協定と計画,期日は2021年2月22日 |
10-K |
001-39208 |
03/15/2021 |
2.1 |
|
|
|
|
|
|
|
|
|
3.1 |
|
BEAM治療会社登録証明書の4回目の改訂。 |
8-K |
001-39208 |
02/11/2020 |
3.1 |
|
|
|
|
|
|
|
|
|
3.2 |
|
BEAM治療会社の付例を2回目の改訂と再改訂。 |
10-K |
001-39208 |
2/28/2023 |
3.2 |
|
|
|
|
|
|
|
|
|
4.1 |
|
普通株式の証明書証明書サンプル |
S-1 |
333-233985 |
09/27/2019 |
4.1 |
|
|
|
|
|
|
|
|
|
4.2 |
|
ビム治療会社と投資家との間で2018年11月8日に締結された“投資家権利協定”の改訂と再署名 |
S-1 |
333-233985 |
09/27/2019 |
4.2 |
|
|
|
|
|
|
|
|
|
4.3 |
|
BEAM治療会社と各買い手間の購入プロトコルフォーマット、日付は2021年1月16日 |
8-K |
001-39208 |
01/19/2021 |
10.1 |
|
|
|
|
|
|
|
|
|
4.4 |
|
登録証券説明 |
S-3 |
333-254946 |
4/01/2021 |
4.11 |
|
|
|
|
|
|
|
|
|
10.1 |
|
Up 26 Landsdown,LLCとBeam Treateutics Inc.の間のレンタルは2018年2月21日 |
S-1 |
333-233985 |
09/27/2019 |
10.1 |
|
|
|
|
|
|
|
|
|
10.2 |
|
マサチューセッツ工科大学とBEAM治療会社間の賃貸契約は、2019年4月24日 |
S-1 |
333-233985 |
09/27/2019 |
10.2 |
|
|
|
|
|
|
|
|
|
10.3 |
|
BEAM治療会社とJefferies LLCとの間の販売協定は,2021年4月1日である。 |
8-K |
001-39208 |
04/01/2021 |
1.1 |
|
|
|
|
|
|
|
|
|
10.4# |
|
総裁とハーバード大学の院士とビーム治療会社との間の許可協定は2017年6月27日 |
S-1 |
333-233985 |
09/27/2019 |
10.4 |
|
|
|
|
|
|
|
|
|
10.5# |
|
ハーバード大学総裁院士とビーム治療会社許可協定第1号修正案、日付は2017年12月12日 |
10-K |
001-39208 |
03/15/2021 |
10.5 |
|
|
|
|
|
|
|
|
|
10.6# |
|
ハーバード大学総裁院士とBEAM治療会社許可協定第2号改正案,期日は2020年3月27日 |
10-K |
001-39208 |
03/15/21 |
10.6 |
|
|
|
|
|
|
|
|
|
10.7# |
|
BRoad Institute,Inc.とBlink Treateutics Inc.の間のライセンス契約は,2018年5月9日である |
S-1 |
333-233985 |
09/27/2019 |
10.5 |
|
|
|
|
|
|
|
|
|
131
10.8# |
|
ブロッド研究所とBlink治療会社との間のライセンス契約第1修正案、日付は2018年9月4日 |
10-K |
001-39208 |
03/15/2021 |
10.8 |
|
|
|
|
|
|
|
|
|
10.9# |
|
Editas Medicine,Inc.とBeam Treateutics Inc.の間のライセンス契約は,2018年5月9日である |
S-1 |
333-233985 |
09/27/2019 |
10.6 |
|
|
|
|
|
|
|
|
|
10.10# |
|
Beam治療会社,ブロード研究所,ハーバード学院総裁と研究員およびEditas Medicine,Inc.間の書簡合意は2018年9月26日であった |
10-K |
001-39208 |
03/15/2021 |
10.10 |
|
|
|
|
|
|
|
|
|
10.11 |
|
総裁とハーバード大学、ブロド研究所、ビーム治療会社の研究員との間の書簡合意は、2021年1月7日。 |
10-K |
001-39208 |
03/15/22021 |
10.11 |
|
|
|
|
|
|
|
|
|
10.12# |
|
Bio Palette Co.,Ltd.とBeam Treateutics Inc.の間のライセンス契約は,2019年3月27日である |
S-1 |
333-233985 |
09/27/2019 |
10.7 |
|
|
|
|
|
|
|
|
|
10.13+ |
|
BEAM治療会社2017年株式オプションと贈与計画 |
S-1/A |
333-233985 |
01/27/2020 |
10.8 |
|
|
|
|
|
|
|
|
|
10.14+ |
|
BEAM治療会社2017年株式オプションおよび贈与計画下の制限株式プロトコルフォーマット |
S-1 |
333-233985 |
09/27/2019 |
10.9 |
|
|
|
|
|
|
|
|
|
10.15+ |
|
BEAM治療会社2017年株式オプションと付与計画下の奨励株式オプション付与通知のフォーマット |
S-1 |
333-233985 |
09/27/2019 |
10.10 |
|
|
|
|
|
|
|
|
|
10.16+ |
|
BEAM治療会社2017年株式オプションと付与計画下の非限定株式オプション付与通知のフォーマット |
S-1 |
333-233985 |
09/27/2019 |
10.11 |
|
|
|
|
|
|
|
|
|
10.17+ |
|
ビム治療会社とその役員と上級管理職との間の賠償協定フォーマット |
S-1 |
333-233985 |
09/27/2019 |
10.12 |
|
|
|
|
|
|
|
|
|
10.18+ |
|
BEAM治療会社とジョン·エバンスが2021年6月9日に署名した書簡協定を改訂し再署名しました |
10-Q |
333-233985 |
08/10/2021 |
10.1 |
|
|
|
|
|
|
|
|
|
10.19+ |
|
ビム治療会社とジュゼッペ·シアラメラとの雇用協定の改正と再署名は2020年1月24日 |
S-1/A |
333-233985 |
01/27/2020 |
10.14 |
|
|
|
|
|
|
|
|
|
10.20+ |
|
BEAM治療会社とTerry-Ann Burrell間の書簡合意は2020年1月24日 |
S-1/A |
333-233985 |
01/27/2020 |
10.15 |
|
|
|
|
|
|
|
|
|
10.21+ |
|
BEAM治療会社2019年持分インセンティブ計画 |
S-1/A |
333-233985 |
01/27/2020 |
10.16 |
|
|
|
|
|
|
|
|
|
10.22+ |
|
BEAM治療会社2019年持分インセンティブ計画下のインセンティブ株式オプションプロトコルフォーマット |
S-1/A |
333-233985 |
01/27/2020 |
10.17 |
|
|
|
|
|
|
|
|
|
10.23+ |
|
BEAM治療会社2019年株式インセンティブ計画下の非法定株式オプション契約のフォーマット |
S-1/A |
333-233985 |
01/27/2020 |
10.18 |
|
|
|
|
|
|
|
|
|
10.24+ |
|
BEAM治療会社2019年持分インセンティブ計画下の非法定株式オプション協定(非従業員取締役)のフォーマット |
S-1/A |
333-233985 |
01/27/2020 |
10.19 |
|
|
|
|
|
|
|
|
|
10.25+ |
|
BEAM治療会社2019年持分インセンティブ計画下制限株式単位奨励協定のフォーマット |
10-K |
001-39208 |
3/15/2021 |
10.25 |
|
|
|
|
|
|
|
|
|
10.26+ |
|
BEAM治療会社2019年持分インセンティブ計画下制限株式奨励プロトコルのフォーマット |
10-K |
001-39208 |
03/15/2021 |
10.26 |
|
|
|
|
|
|
|
|
|
10.27+ |
|
BEAM治療会社2019年従業員株式購入計画の改訂と再発表 |
10-K |
001-39208 |
2/28/2022 |
10.27 |
|
|
|
|
|
|
|
|
|
10.28+ |
|
BEAM治療会社2019年現金奨励計画 |
S-1/A |
333-233985 |
01/27/2020 |
10.21 |
|
|
|
|
|
|
|
|
|
10.29+ |
|
ビム治療会社の非従業員役員報酬政策を改正し、再確認しました。日付は2024年1月22日です |
|
|
|
|
X |
|
|
|
|
|
|
|
|
132
10.30 |
|
BEAM治療会社とARE−NC地域第14号有限責任会社とのリース契約 |
10-Q |
001-39208 |
08/12/2020 |
10.1 |
|
|
|
|
|
|
|
|
|
10.31 |
|
2020年4月14日マサチューセッツ工科大学とBEAM治療会社との賃貸借契約の第1号改正案 |
10-K |
001-39208 |
02/28/2022 |
10.31 |
|
|
|
|
|
|
|
|
|
10.32 |
|
2020年11月17日マサチューセッツ工科大学とBEAM治療会社との間の賃貸借契約の改正案第2号 |
10-K |
001-39208 |
02/28/2022 |
10.32 |
|
|
|
|
|
|
|
|
|
10.33 |
|
2021年8月24日マサチューセッツ工科大学とBEAM治療会社との間の賃貸契約の改正案第3号 |
10-K |
001-39208 |
02/28/2022 |
10.33 |
|
|
|
|
|
|
|
|
|
10.34 |
|
BEAM治療会社とJefferies LLCとの間で2021年7月7日に署名された販売協定の改正案第1号 |
8-K |
001-39208 |
07/07/2021 |
1.1 |
|
|
|
|
|
|
|
|
|
10.35 |
|
2022年12月7日マサチューセッツ工科大学とBEAM治療会社間の賃貸契約第4号改正案 |
10-K |
001-39208 |
02/28/2023 |
10.35 |
|
|
|
|
|
|
|
|
|
10.36+ |
|
BEAM治療会社とChristine Bellonとの間の書簡合意は2020年1月24日 |
10-Q |
001-39208 |
5/10/2023 |
10.1 |
|
|
|
|
|
|
|
|
|
10.37+ |
|
BEAM治療会社とエイミー·サイモンの間の手紙の契約は2021年1月29日です |
10-Q |
001-39208 |
5/10/2023 |
10.2 |
|
|
|
|
|
|
|
|
|
10.38 |
|
BEAM治療会社とJefferies LLCとの間で2023年5月10日に署名された販売協定の改正案第2号 |
8-K |
001-39208 |
5/10/2023 |
1.1 |
|
|
|
|
|
|
|
|
|
10.39 |
|
BEAM治療会社とARE-NC地域第14号レンタルの第1修正案、期日は2022年6月23日 |
|
|
|
|
X |
|
|
|
|
|
|
|
|
10.40 |
|
BEAM治療会社とARE-NC地域間のレンタルの第2の修正案、番号14、LLC、日付2023年3月31日 |
|
|
|
|
X |
|
|
|
|
|
|
|
|
10.41 |
|
BEAM治療会社とARE-NC地域第14号レンタルの第3回改訂、日付は2024年1月1日 |
|
|
|
|
X |
|
|
|
|
|
|
|
|
21.1 |
|
ビム治療会社の子会社リストです。 |
10-K |
001-39208 |
2/28/2022 |
21.1 |
|
|
|
|
|
|
|
|
|
23.1 |
|
徳勤法律事務所が同意した |
|
|
|
|
X |
|
|
|
|
|
|
|
|
31.1 |
|
2002年のサバンズ·オキシリー法第302節で可決された1934年の証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて発行された首席執行幹事証明書。 |
|
|
|
|
X |
|
|
|
|
|
|
|
|
31.2 |
|
2002年サバンズ−オキシリー法第302節で可決された1934年証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて首席財務幹事が認証された。 |
|
|
|
|
X |
|
|
|
|
|
|
|
|
32.1* |
|
2002年にサバンズ·オクスリ法案第906節で可決された“米国法典”第18編1350条に基づいて発行された首席執行幹事証明書。 |
|
|
|
|
X |
|
|
|
|
|
|
|
|
32.2* |
|
2002年にサバンズ·オクスリ法案第906節で可決された“米国法典”第18編1350条による首席財務官の証明。 |
|
|
|
|
X |
|
|
|
|
|
|
|
|
97.1 |
|
払戻政策 |
|
|
|
|
X |
|
|
|
|
|
|
|
|
133
101.INS |
|
連結されたXBRLインスタンス文書-インスタンス文書は、そのXBRLタグがイントラネットXBRL文書に埋め込まれているので、対話データファイルには表示されない |
|
|
|
|
X |
|
|
|
|
|
|
|
|
101.書院 |
|
イントラネットXBRL分類拡張アーキテクチャ文書 |
|
|
|
|
X |
|
|
|
|
|
|
|
|
101.カール |
|
インラインXBRL分類拡張計算リンクライブラリ文書 |
|
|
|
|
X |
|
|
|
|
|
|
|
|
101.def |
|
インラインXBRL分類拡張Linkbase文書を定義する |
|
|
|
|
X |
|
|
|
|
|
|
|
|
101.介護会 |
|
XBRL分類拡張ラベルLinkbase文書を連結する |
|
|
|
|
X |
|
|
|
|
|
|
|
|
101.Pre |
|
インラインXBRL分類拡張プレゼンテーションLinkbaseドキュメント |
|
|
|
|
X |
|
|
|
|
|
|
|
|
104 |
|
表紙インタラクションデータファイル(イントラネットXBRL文書に埋め込む) |
X |
||||
登録者はそれらが材料ではないことを決定したので、登録者はそれらを個人または機密タイプとみなすので、この展示品の#部分は省略されている。
+管理契約または補償計画を示します。
*改正された1934年証券取引法第18条または取引法の場合、本証明書は提出されたとみなされないか、またはこの条項の責任によって制限されています。このような証明は、参照によってそのような出願が明示的に組み込まれない限り、改正された1933年の証券法または“取引法”に基づいて提出された任意の出願に引用によって組み込まれているとはみなされない。
項目16.表10-Kの概要
ない。
134
サイン
改正された1934年証券取引法第13又は15(D)節の要求に基づいて、登録者は、正式に許可された次の署名者がその代表を代表して本報告書に署名することを正式に手配した.
|
|
BEAM治療会社 |
|
|
|
|
|
日付:2024年2月27日 |
|
差出人: |
/S/ジョン·エバンズ |
|
|
|
ジョン·エバンス |
|
|
|
最高経営責任者 |
本報告書は、改正された1934年の証券取引法の要求に基づき、以下の登録者によって登録者として指定日に署名された。
名前.名前 |
|
タイトル |
|
日取り |
|
|
|
|
|
/S/ジョン·エバンズ |
|
取締役CEO兼最高経営責任者 |
|
2024年2月27日 |
ジョン·エバンス |
|
(首席行政主任) |
|
|
|
|
|
|
|
/S/テリー-アン·ベレル |
|
首席財務官兼財務主管 |
|
2024年2月27日 |
テリー·アン·ベレル |
|
(首席財務官と首席会計官) |
|
|
|
|
|
|
|
/S/クリスティーナ·ブロ |
|
役員.取締役 |
|
2024年2月27日 |
クリスティーナ·ブロ |
|
|
|
|
|
|
|
|
|
寄稿S/グレアム·クーパー |
|
役員.取締役 |
|
2024年2月27日 |
グレアム·クーパー |
|
|
|
|
|
|
|
|
|
/S/マーク·フィッシュマン |
|
役員.取締役 |
|
2024年2月27日 |
マーク·フィッシュマン医学博士 |
|
|
|
|
|
|
|
|
|
寄稿S/何超瓊 |
|
役員.取締役 |
|
2024年2月27日 |
Carole Ho医学博士 |
|
|
|
|
|
|
|
|
|
/S/ジョン·マラガノール |
|
役員.取締役 |
|
2024年2月27日 |
ジョン·マラガノ博士 |
|
|
|
|
|
|
|
|
|
/投稿S/Christi Shaw |
|
役員.取締役 |
|
2024年2月27日 |
クリスティ·ショー |
|
|
|
|
|
|
|
|
|
/S/キャサリン·ウォルシュ |
|
役員.取締役 |
|
2024年2月27日 |
キャサリン·ウォルシュ |
|
|
|
|
135
連結財務諸表索引
独立公認会計士事務所報告(PCAOB ID番号 |
F-2 |
合併貸借対照表 |
F-4 |
合併経営報告書その他全面的な損失 |
F-5 |
合併株主権益報告書 |
F-6 |
統合現金フロー表 |
F-7 |
連結財務諸表付記 |
F-9 |
F-1
独立公認会計士事務所報告
ビム治療会社の株主と取締役会に。
財務諸表のいくつかの見方
Beam治療会社とその子会社(“当社”)2023年12月31日現在と2022年12月31日までの連結貸借対照表、2023年12月31日現在の3年度の関連総合経営報告書とその他の全面赤字、株主権益と現金流量、および関連する付記(総称して“財務諸表”と呼ぶ)を監査した。これらの財務諸表は,すべての重要な点で当社の2023年12月31日と2022年12月31日までの財務状況,および2023年12月31日までの3年度の経営結果とキャッシュフローを公平に反映しており,米国公認の会計原則に適合していると考えられる。
我々はまた、米国上場企業会計監督委員会(PCAOB)の基準に基づき、以下の基準に基づいて、会社が2023年12月31日までの財務報告内部統制を監査した内部統制--統合フレームワーク(2013)テレデビル委員会は組織委員会が発表した報告書と2024年2月27日の報告書を後援し、会社の財務報告書の内部統制について保留のない意見を表明した。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
以下に述べる重要な監査事項とは、監査委員会に伝達または要求された財務諸表を当期監査する際に生じる事項であり、(1)財務諸表に対して大きな意味を有する勘定または開示に関するものであり、(2)特に挑戦的で主観的または複雑な判断に関するものである。重要監査事項の伝達は、財務諸表に対する私たちの全体的な意見をいかなる方法でも変えることはなく、次の重要な監査事項を伝達することによって、重要な監査事項又はそれに関連する勘定又は開示について個別の意見を提供することもない。
収入確認−財務諸表付記2と付記11を参照
重要な監査事項の説明
同社が確認した収入は2023年12月31日までの年間で3兆777億ドル。連結財務諸表付記2で述べたように、当社は開始時に契約がASC 606の範囲に属するか否かを判定する取引先と契約した収入またはASC 606または他の主題。ASC 606の範囲内で決定された契約の場合、収入は、顧客が約束された商品またはサービスの制御権を取得したときに確認される。確認された収入金額は、これらの商品やサービスと交換するために、会社が獲得する権利があると予想されている対価格を反映している。この核心原則を実現するために,会社は,(I)顧客との契約の決定,(Ii)契約中の履行義務の決定,(Iii)取引価格の決定,(Iv)契約に取引価格を割り当てる履行義務,および(V)契約履行義務を履行する際に収入を確認する,の5つのステップを採用している。一定期間内に収入を確認する業績義務については、企業は各報告期間内に進捗の測定基準を評価し、必要に応じて業績測定基準と関連収入確認を調整する。同社はこれまでに発生したコストを総見積もりコストと比較して収入を確認するのが一般的だ。総見積もりコストの変化が収入に与える影響は変化している間は累積ベースで確認した。総費用推定数が変化した場合、または契約修正が履行義務の範囲を変更した場合、必要な収入調整は実質的である可能性がある。
ASC 606によれば、監査会社は、契約履行義務(S)および取引価格の決定に対して複雑かつ判断する性質を有するため、監査会社が収入契約の会計処理に対してより大きな努力と高度な監査役判断力を必要とする。また、進捗測定基準を用いて確認された収入を監査することは特に挑戦的であり、進捗測定基準を決定するためには、契約規定の適用履行義務を履行する総コストを推定するために管理層が重大な判断を行う必要があるからである。
監査で重要な監査事項をどのように処理するか
F-2
私たちの収入に関する監査手続きは以下のものを含む
/s/ |
|
|
2024年2月27日 |
2018年以来、当社の監査役を務めてきました。
F-3
BEAM治療会社
統合された貸借対照表
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
有価証券 |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
流動資産総額 |
|
|
|
|
|
|
||
財産と設備、純額 |
|
|
|
|
|
|
||
制限現金 |
|
|
|
|
|
|
||
経営的リース使用権資産 |
|
|
|
|
|
|
||
その他の資産 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
負債と株主権益 |
|
|
|
|
|
|
||
流動負債: |
|
|
|
|
|
|
||
売掛金 |
|
$ |
|
|
$ |
|
||
費用とその他の流動負債を計算しなければならない |
|
|
|
|
|
|
||
派生負債 |
|
|
|
|
|
|
||
繰延収入の当期分 |
|
|
|
|
|
|
||
賃貸負債の当期分 |
|
|
|
|
|
|
||
流動負債総額 |
|
|
|
|
|
|
||
長期賃貸負債 |
|
|
|
|
|
|
||
対価格負債があります |
|
|
|
|
|
|
||
繰延収入の長期部分 |
|
|
|
|
|
|
||
その他負債 |
|
|
|
|
|
|
||
総負債 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
株主権益: |
|
|
|
|
|
|
||
優先株、$ |
|
|
|
|
|
|
||
普通株、$ |
|
|
|
|
|
|
||
追加実収資本 |
|
|
|
|
|
|
||
その他の総合収入を累計する |
|
|
|
|
|
( |
) |
|
赤字を累計する |
|
|
( |
) |
|
|
( |
) |
株主権益総額 |
|
|
|
|
|
|
||
総負債と株主権益 |
|
$ |
|
|
$ |
|
||
付記はこれらの連結財務諸表の構成要素である。
F-4
BEAM治療会社
合併運営報告書イギリス国家統計局その他全面的な損失
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
|
$ |
|
|
$ |
|
|
$ |
|
||||
運営費用: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
|
|
|
|
|
|
|
|
|||
一般と行政 |
|
|
|
|
|
|
|
|
|
|||
総運営費 |
|
|
|
|
|
|
|
|
|
|||
運営損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
その他の収入(支出): |
|
|
|
|
|
|
|
|
|
|||
派生負債の公正価値変動 |
|
|
|
|
|
|
|
|
( |
) |
||
非持株投資の公正価値変動 |
|
|
( |
) |
|
|
|
|
|
|
||
価格負債があって価値変動を公正にすることができます |
|
|
|
|
|
|
|
|
|
|||
利息とその他の収入,純額 |
|
|
|
|
|
|
|
|
( |
) |
||
その他収入合計 |
|
|
|
|
|
|
|
|
|
|||
所得税前純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
所得税支給 |
|
|
( |
) |
|
|
( |
) |
|
|
— |
|
権益法投資損失 |
|
|
— |
|
|
|
( |
) |
|
|
— |
|
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
有価証券の未実現収益 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
総合損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
普通株1株当たりの基本損失と償却後の純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
加重平均発行済み普通株式、基本普通株式、希釈後普通株 |
|
|
|
|
|
|
|
|
|
|||
付記はこれらの連結財務諸表の構成要素である。
F-5
BEAM治療会社
合併報告書株主権益について
(単位は千で、シェアは含まれていない)
|
|
普通株 |
|
|
その他の内容 |
|
|
積算 |
|
|
積算 |
|
|
合計する |
|
|||||||||
|
|
株 |
|
|
金額 |
|
|
資本 |
|
|
収入(損) |
|
|
赤字.赤字 |
|
|
権益 |
|
||||||
2020年12月31日残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
私募による普通株発行は、発行コストを差し引いて#ドルです |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
市場から普通株を発行し,発行コストを差し引くと#ドルになる |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
普通株式を発行して成功支払責任を支払う |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
普通株買い入れガイドラインを発行する |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
制限された普通株の帰属 |
|
|
|
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
||
株に基づく報酬 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
普通株式オプションの行使 |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
その他全面収益(赤字) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2021年12月31日の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
ESPPにより普通株を購入する |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
市場から普通株を発行し,発行コストを差し引くと#ドルになる |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
制限された普通株の帰属 |
|
|
|
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
||
株に基づく報酬 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
普通株式オプションの行使 |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
その他全面収益(赤字) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2022年12月31日の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
ESPPにより普通株を購入する |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
市場から普通株を発行し,発行コストを差し引くと#ドルになる |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
許可契約に関する未登録普通株の発行 |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
制限された普通株の帰属 |
|
|
|
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
||
株に基づく報酬 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
普通株式オプションの行使 |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
その他全面収益(赤字) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2023年12月31日の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||||
付記はこれらの連結財務諸表の構成要素である。
F-6
BEAM治療会社
合併状態現金流動額
(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
経営活動 |
|
|
|
|
|
|
|
|
|
|||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
純損失と経営活動への現金純額の調整: |
|
|
|
|
|
|
|
|
|
|||
権益法投資損失 |
|
|
— |
|
|
|
|
|
|
— |
|
|
減価償却および償却 |
|
|
|
|
|
|
|
|
|
|||
償却投資保険料 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
進行中の研究開発費 |
|
|
— |
|
|
|
— |
|
|
|
|
|
株に基づく報酬費用 |
|
|
|
|
|
|
|
|
|
|||
経営的リース使用権資産変更 |
|
|
|
|
|
|
|
|
|
|||
派生負債の公正価値変動 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
価格負債があって価値変動を公正にすることができます |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
非持株投資の公正価値変動 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
他にも |
|
|
— |
|
|
|
|
|
|
|
||
経営性資産と負債変動状況: |
|
|
|
|
|
|
|
|
|
|||
前払い費用と他の流動資産 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
その他長期資産 |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
売掛金 |
|
|
( |
) |
|
|
|
|
|
|
||
費用とその他の負債を計算すべきである |
|
|
|
|
|
( |
) |
|
|
|
||
リース負債を経営する |
|
|
( |
) |
|
|
|
|
|
|
||
協同して応収する |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
収入を繰り越す |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
その他長期負債 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
経営活動提供の現金純額 |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
投資活動 |
|
|
|
|
|
|
|
|
|
|||
財産と設備を購入する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
有価証券を購入する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
有価証券の満期日 |
|
|
|
|
|
|
|
|
|
|||
グイドから得た現金純額 |
|
|
— |
|
|
|
— |
|
|
|
|
|
投資活動提供の現金純額 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
融資活動 |
|
|
|
|
|
|
|
|
|
|||
普通株を発行して得られる収益(手数料を差し引く) |
|
|
|
|
|
|
|
|
|
|||
特別引き出し権に基づいて株を発行して得た金 |
|
|
|
|
|
|
|
|
— |
|
||
持分発行費用を支払う |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
設備融資,純額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
株式オプションを行使して得られる収益 |
|
|
|
|
|
|
|
|
|
|||
株を非公開で発行して得た金 |
|
|
|
|
|
— |
|
|
|
— |
|
|
融資活動提供の現金純額 |
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|||
現金、現金等価物、および限定的な現金純変化 |
|
|
|
|
|
( |
) |
|
|
|
||
現金、現金等価物、制限現金--期初 |
|
|
|
|
|
|
|
|
|
|||
現金、現金等価物、制限された現金--期末 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
付記はこれらの連結財務諸表の構成要素である。
F-7
BEAM治療会社
合併現金フロー表(継続)
(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
キャッシュフロー情報の追加開示: |
|
|
|
|
|
|
|
|
|
|||
利子を支払う現金 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|||
非現金投資と融資活動を追加開示します |
|
|
|
|
|
|
|
|
|
|||
売掛金·売掛金のうち財産·設備の増加 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
使用権資産の取得による経営リース負債 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
売掛金と売掛金における持分発行コスト |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
成功支払責任を解決するために発行された普通株の公正価値 |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
資産購入で負担しているまたは対価格負債がある |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
資産買収に関連して発行された権益ツールの公正価値 |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
Orbary Treeutics,Inc.に知的財産権の非現金貢献を提供する. |
|
$ |
— |
|
|
$ |
|
|
$ |
— |
|
|
付記はこれらの総合財務諸表の構成要素である
F-8
BEAM治療会社
合併後の注釈財務諸表
1. 業務の性質と列報根拠
組織する
BEAM治療会社、私たちはここで“会社”または“BEAM”と呼ばれ、バイオテクノロジー会社であり、リードし、完全に統合された精密遺伝薬物プラットフォームの構築に取り組んでいる。BEAMのビジョンは遺伝病を有する患者に生涯治療を提供することである。同社は2017年1月25日にデラウェア州会社として設立され、2017年7月に運営を開始した。その主な事務所はマサチューセッツ州ケンブリッジ市に設置されている。
流動資金と資本資源
設立以来、同社はその基礎編集プラットフォームの構築とそのプログラム組合せの開発の推進、その知的財産権の確立と保護、研究と開発活動の展開、契約製造組織との製造活動、臨床前研究とIND支援研究を含む研究と開発コスト、組織と会社の人員の配備、その施設と新施設の拡張、業務計画、資金調達、これらの業務に一般的かつ行政的支援を提供するために、そのほとんどの資源を使用してきた。その会社は内部製造能力を確立した。同社は生物技術業界の早期会社によく見られるリスクと不確定要素の影響を受け、これらのリスクと不確定要素は候補製品の成功研究、開発と製造に関連する技術リスク、競争相手の新技術革新の開発、肝心な人員への依存、ノウハウの保護、政府法規の遵守及び追加資本を獲得して運営を援助する能力を含むがこれらに限定されない。現在と未来のプロジェクトは広範な臨床前と臨床テスト及び商業化前の監督管理承認を含む大量の研究と開発努力が必要となる。このような努力は多くの追加資本、十分な人員、そしてインフラを必要とする。会社の製品開発努力が成功しても、会社がいつ(あれば)製品販売から相当な収入を得るかは定かではない。
2021年4月に、当社はJefferies、LLCまたはJefferiesと販売協定を締結し、この合意により、当社は時々現行の市場価格で当社の普通株の株式を発売および売却する権利があり、総収益総額は最高$に達する
当社は2021年7月および2023年5月にJefferiesと販売協定の改訂を締結し、販売契約下の総発売金額を増加させることを規定し、当社が2023年5月10日から発売総発行価格を追加$とすることができるようにした
2023年10月、会社は、Verve Treateutics,Inc.またはVerveとの修正協力および許可プロトコルまたはVerveプロトコルに従って、会社がVerveを共同開発および共同商業化する心血管疾患基礎編集プログラムの選択権を含む、会社と礼来会社または礼来会社と譲渡および許可協定または礼来協定を締結した。その会社は1ドルを受け取った
F-9
設立以来、同社は巨額の損失を発生し、累計損失#ドルを計上した
同社は、2023年12月31日までに、その現金、現金等価物、有価証券が達成されると予想している
2. S重要会計政策の概要
陳述の基礎
合併原則
添付されている総合財務諸表には、当社とその完全子会社の経営結果が含まれています。すべての会社間取引と残高は合併で販売された。
予算の使用
公認会計基準に従って財務諸表を作成することは、報告期間及び報告期間内の資産、負債、収入及び支出の報告金額、又は有資産及び負債の開示に影響を与えるために、管理層に推定と仮定を要求する。当社は過去の経験とその当時の状況で合理的と考えられる様々な要素に基づいて見積もりと仮説を立てています。これらの連結財務諸表に反映される重大な推定および仮定は、リース負債、研究および開発費用、普通株式公正価値、株式報酬、または対価負債、成功支払い、推定許可料、または負債を計算する際に使用される増分借入金金利を含むが、これらに限定されない収入確認に関するいくつかの判断。実際の結果はこれらの推定とは異なる可能性がある。
現金、現金等価物、制限された現金
現金および現金等価物には、小切手口座、通貨市場口座、および購入日の残り期限が3ヶ月以下のすべての高流動性投資が含まれる。限定現金とは,保証金として発行された信用状のための担保であり,これらの保証金は会社がその施設をレンタルすることに関係している。
以下の表では、会社合併貸借対照表に報告されている現金、現金等価物、および限定的な現金と、合併現金フロー表に表示されている総金額(千計)とを照合します
|
|
十二月三十一日 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
現金と現金等価物 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
制限現金 |
|
|
|
|
|
|
|
|
|
|||
現金総額、現金等価物、制限された現金 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
F-10
有価証券
その会社は有価証券を売却可能な証券に分類している。売却可能な証券には、商業手形、高級会社手形、米国国債、政府証券が含まれる。売却可能な証券は公正価値に従って勘定し、損益計上は累計他の総合(損失)収入を計上しておらず、株主権益の構成部分として、実現までとしている。購入時に生成される任意の割増または割引は、手形の有効期間内に償却および/または利息収入および/または支出に計上される。損益は特定の確認方法を用いて決定され,利息や他の収入(費用),純額に計上されている。
会社持分証券
同社は公正価値が確定しやすい株式証券投資を会社総合貸借対照表の有価証券に分類している。その会社の有価証券は公正価値に基づいて列報する。一般に、これらの証券の公正な価値は、同じ持分証券の見積もりに基づいている。同社は個人発行の会社持分証券の投資を持っており、これらの証券は株式証券投資とされているこの投資は簡単に決定できる公正な価値がなく、当社は可視市場取引或いは減値(あればある)調整後の権益証券コストに基づいてこの投資を推定し、いかなる収益を通じて発生した価値変動を記録する。当社はその権益証券の公正価値変動を他の収入(費用)、総合経営報告書純額、その他全面赤字に計上している。
信用リスクが集中する
保証と補償
デラウェア州の法律で許可されている場合、会社が会社との関係又は会社が担当している職によって発生した何らかの事件又は事件は、会社はその高級管理者、取締役、コンサルタント及び従業員に対して賠償を行う。2023年12月31日まで,2022年および2021年12月31日まで年度当社は、当該等の賠償責任により何の損害も受けていません
株式発行コスト
同社はその株式発行に直接関連する逓増法律、専門、会計、その他の第三者費用を他の非流動資産として資本化し、発行が完了するまで資本化している。取引完了後、これらのコストは発行による追加実収資本の減少として株主権益に計上される。2023年12月31日と2022年12月31日までいくつありますか
金融商品の公正価値
ASCテーマ820、公正価値計量あるいは、あるいは ASC 820は、公正価値計測のためのツールのための公正価値レベルを構築し、市場データに基づく仮説(観察可能な投入)と会社自身の仮説(観察不可能な投入)とを区別する。観察される投入は、独立したソースから得られた市場データに基づいて市場参加者が資産または負債価格を設定する際に使用される投入である。観察できない投入は、市場参加者が資産や負債の価格設定のために使用されるという会社の仮定を反映し、その時点で入手可能な最適な情報に基づいて制定される。ASC 820は、公正価値を、計量日の市場参加者間の秩序ある取引において資産を売却するか、または負債を移転して受信した価格として決定する。公正価値計量において市場参加者の仮説を考慮する基礎として、ASC 820は以下の項目を区別した三級価値階層構造を構築した
第1レベル-同じ資産または負債の活発な市場見積もり。
第2レベル-第1レベル以外の直接または間接的に観察可能な投入、例えば見積市場価格、金利、収益率曲線。
第3レベル--資産または負債の観察不可能な投入(すなわち、市場活動の支援が少ないか、またはない)。第3レベルの投入には、管理層自身が資産または負債のために価格を設定する際に市場参加者自身が使用する仮説(リスクの仮定を含む)が含まれる。
ある程度、推定値は市場で観察または観察できないモデルや投入に基づいており、公正価値の決定にはより多くの判断が必要である。そのため、当社が価値を公平に判断する際に下した判断は、第3級に分類されたツールに対する判断度が最も高く、公正価値レベル内の金融商品レベルは、公正価値計量に大きな意味を持つ任意の投入の中で最低レベルに基づいている。
F-11
2023年12月31日までおよび2022年12月31日までに採用した推定方法に変動はなかったそれは.当社は各報告期間の終了時に異なるレベル間の移行を評価します。
財産と設備
財産と設備はコストから減価償却累計を引いて申告する.減価償却費用は、資産ごとの推定耐用年数内に直線法で確認され、具体的には以下の通り
資産種別 |
|
使用寿命を見込む |
コンピュータ装置及びソフトウェア |
|
|
実験室設備と事務家具 |
|
|
賃借権改善 |
|
退職又は売却時には、資産を処分するコスト及び関連する減価償却が勘定から差し引かれ、それによって生じる収益又は損失のいずれも利息及びその他の収入(費用)に計上される。修理費と維持費は発生時に料金を記入します。
長期資産減価準備
当社はその長期資産(主に物件や設備および経営賃貸使用権資産を含む)の減値を評価し、事件や状況変化がそのような資産(あるいは資産グループ)の額面が回収できない可能性があることを示した場合、当社はその減値を評価する。保有·使用する資産の回収可能性は,資産の帳簿価値と資産予想による将来の未割引現金流量を比較することで測定した。当該等の資産は減値とみなされ、確認すべき減値は、当該資産の帳簿価値が当該資産の公正価値を超える金額で計量される。いくつありますか
独立した金融商品と派生商品
レンタルとレンタル料
当社は、レンタル開始日にレンタル者にレンタル金を支払う財務義務があり、レンタル期間内に対象資産を使用する権利を得るために、レンタルを使用権またはROUモードで会計処理することを認めている。
手配開始時に、当社は手配に存在する独特の事実と状況に基づいて、その手配が賃貸契約であるかどうかを決定する。レンタル期間が1年を超えるリースは貸借対照表で純収益資産および短期·長期賃貸負債であることが確認された(場合によっては)。ROU資産は会社が標的を使用する権利を代表する リース期間中の資産及びリース負債とは、リースにより生じるリース金の支払い義務である。同社は、レンタルスケジュールの期限を評価する際に、通常、初期レンタル期間のみを含む。それはまた選択を終了し、このような要素をレンタル支払いを決定することを考慮する。当社が継続することが合理的に確定されていない限り、レンタル期間には契約更新の選択は含まれていません。
レンタル負債及びそれに対応するROU資産は、期待される残りレンタル期間のリース支払い現在値に基づいて入金される。受け取った報酬などの項目については、ROU資産を何らかの調整する必要があるかもしれない。レンタル契約に隠されている金利は一般的に確定しにくい。そこで,当社はその逓増借入金利を利用して,類似経済環境下で同じ通貨,類似期限で担保方式で借金した固定金利を反映している。レンタルレンタルで支払われたレンタル料金はレンタル期間内に直線法で確認します。
ある経営賃貸(非レンタル部分)の毎月の基本賃貸料のほか、会社は運営費用を支払う必要がある。当社は実行可能な方便を選択し、すべての資産カテゴリの非レンタル構成要素とレンタル構成要素を結合することを許可した。変動賃貸支払は、総合貸借対照表に記載されているリース使用権資産やリース負債に計上されるのではなく、発生期間中に費用として反映される。
F-12
レンタル改善は唯一無二ではなく、レンタル終了時にレンタル者が保留します。しかし、当社の特定の不動産需要に適した空間として設計されている場合、当社がコスト超過に責任を負うと、当社はレンタル改善の会計所有者であり、関連コストは資本化される。
同社の不動産経営賃貸契約はリース期間全体で予定されている年間賃料上昇を規定している。当社はリース期間全体で予定賃貸料増加の影響を直線的に確認しています。所有者が提供するテナント改善手当(あれば)は、レンタル開始時に当該テナントに関するROU資産の減少として記録される。
資産買い入れ
当社は、資産を買収するコスト(取引コストを含む)に基づいて、業務合併とはみなされない資産買収を計量及び確認し、対価は相対公正価値方法で買収項目に分配する。営業権は資産買収で確認されていません。資産買収では、買収が行われている研究·開発に割り当てられ、他の将来の用途のコストがなく、買収日に研究·開発費用を計上する。
買収の際、当社は一つの取引を企業合併または資産買収に計上すべきかどうかを決定する。
対価格負債があります
会社は、特定の製品および技術的マイルストーンの実現に応じて、Guide治療会社またはGuideの前株主およびオプション所有者に普通株の形で記念碑的な支払いを支払うことを要求される可能性がある。これらの支払いはASC 480項目で課金されます負債と持分を区別するそれは.この等または有価負債は、確率に基づくモデルを適用することによって推定され、このモデル利用の投入は、主に市場では観察されないいくつかの製品および技術マイルストーンの実現状況および関連する時間スケジュールに基づく公正価値で帳簿に記載されている。購入日に最初に計量および記録された、または対価負債の推定公正価値は、第3レベル計量とみなされ、公正価値が変化することを示すイベントまたは状況が四半期または発生するたびに審査される。または、各報告期間の終了時に公正価値に従って入金され、公正価値変動が合併業務表に記入された他の収入(費用)および他の包括的損失を推定する対価負債がある。
公正価値を推定するのは確率調整により現金流量モデルを割引することであり、このモデルは技術と製品の発展と関連する重大な推定と仮定を含む。成功確率や確率が大きく変化していますマイルストーンを実現する時期とリンクすることは公正価値の計量の著しい向上或いは低下を招く。当社は、当該等の責任が早期に実現または満期になるまで、価値変動の公正性について負債を調整し続ける。
収入確認
開始時に、会社は、契約がASC 606または他の主題の範囲内にあるかどうかを判断する。ASC 606の範囲内で決定された契約の場合、収入は、顧客が約束された商品またはサービスの制御権を取得したときに確認される。確認された収入金額は、これらの商品やサービスと交換するために、会社が獲得する権利があると予想されている対価格を反映している。この核心原則を実現するために,会社は,(I)顧客との契約の決定,(Ii)契約中の履行義務の決定,(Iii)取引価格の決定,(Iv)契約に取引価格を割り当てる履行義務,および(V)契約履行義務を履行する際に収入を確認する,の5つのステップを採用している。会社が顧客の意図と支払い承諾の対価格の能力に基づいて、譲渡された商品およびサービスの実質的にすべての対価格を受け取る可能性があると決定した場合にのみ、会社は5段階モデルを契約に適用する。
契約で約束された履行義務は,顧客に移行する貨物やサービスによって決定され,これらの貨物とサービスは区別できるとともに,契約範囲内で異なる.1つの契約に複数の承諾された商品およびサービスが含まれている場合、会社は、承諾された商品とサービスが区別可能であるかどうかを決定するために、判断を運用しなければならない。これらの基準を満たしていなければ、約束された貨物とサービスは総合履行義務として入金されるだろう。
取引価格は、商品やサービスを顧客に移転することと引き換えに、会社が獲得する権利のある対価格によって決定される。取引価格に可変対価が含まれている場合、当社は可変対価の性質に基づいて、期待値法または可能金額法を用いて取引価格に含まれるべき可変対価金額を推定する。経営陣の判断により、契約項下の累積収入が将来的に大きく逆転しない可能性が高い場合には、可変対価格が取引価格に含まれる。可変考慮に対する制約の影響を含む任意の推定は、どのような変化があるかどうかを決定するために、各報告期間において評価される。取引価格を決定するには重大な判断が必要であり、付記11で会社の各許可および協力協定についてより詳細に検討する。
F-13
契約が単一履行義務を含む場合、取引価格全体をその単一履行義務に割り当てる。複数の履行義務を含む契約要求は、取引価格が可変でない限り、相対的に独立した販売価格に基づいて取引価格を個々の履行義務に割り当て、かつ、履行義務または単一履行義務の一部を構成する異なるサービスに完全に割り当てられる基準に適合する。独立した販売価格を決定するには重大な判断が必要であり、付記11では会社の各許可と協力協定について詳細に検討した。
その会社は一定期間、またはある時点で契約履行義務を履行する。以下の場合、収入は、時間とともに確認される:(I)顧客がエンティティ業績によって提供される利益を同時に受け取り、消費すること、(Ii)エンティティ業績が資産作成または強化時に顧客によって制御される資産を創造または強化するか、または(Iii)エンティティ業績は代替エンティティ用途の資産を生成しておらず、エンティティは、これまでに完了した業績支払いを強制的に実行する権利がある。エンティティが一定期間義務を履行していない場合、ある時点で、約束された貨物またはサービスの制御権を顧客に移すことによって、関連する履行義務を履行する。
知的財産権許可:会社の知的財産権許可が合意で決定された他の履行義務とは異なると判断された場合、許可が顧客に譲渡された場合、会社は許可に割り当てられた対価格収入を確認し、顧客は許可を使用して利益を得ることができる。他の承諾と組み合わせたライセンスについて、会社は、合併履行義務が時間の経過とともに履行されるか、ある時点で履行されるかを決定するために、判断を利用して、合併履行義務の性質を評価し、時間が経過するにつれて、収入を確認するための進展を測定するための適切な方法を決定する。当社は報告期間ごとに進捗指標を評価し、必要に応じて業績指標と関連収入確認を調整します。同社はこれまでに発生したコストを総見積もりコストと比較して収入を確認するのが一般的だ。総見積もりコストの変化が収入に与える影響は変化している間は累積ベースで確認した。総費用推定数が変化した場合、または契約修正が履行義務の範囲を変更した場合、必要な収入調整は実質的である可能性がある。知的財産権許可の収入確認を決定するには重要な判断が必要であり、付記10および11では、会社の各許可および協力協定についてより詳細な議論が行われている。
マイルストーン支払い:開発または規制マイルストーン支払いを含む各手配の開始時に、会社はマイルストーンに達する可能性を評価し、最も可能な金額法を用いて取引価格に含まれる金額を推定する。将来大きな収入逆転が起こらない可能性が高い場合、関連するマイルストーン価値は取引価格に含まれるだろう。規制承認のような会社または被許可者の制御範囲内でない記念碑的支払いは、これらの承認を受ける前に実現可能とは考えられないため、取引価格に含まれる対価格が制限される。そして、取引価格は相対的に独立した販売価格で個々の履行義務に割り当てられ、会社は契約項目の履行義務を履行する際に収入を確認する。その後各報告期間が終了した時点で,当社はこのような発展マイルストーンを達成する可能性や任意の制限を再評価し,必要に応じて全体の取引価格の見積りを調整する。いずれの調整も累積追跡をもとに記録されており,調整期間中の収入や収益に影響を与える.
商業マイルストーン支払いおよび特許権使用料:販売に基づく特許権使用料(販売レベルに基づくマイルストーン特許使用料を含む)を含む手配については、ライセンスが特許使用料に関連する主要項目とみなされている場合、企業は、(I)関連販売が発生した場合、または(Ii)特許権使用料に割り当てられた履行義務が履行された(または部分的に履行されている)ときに収入を確認する。現在まで、同社はそのいかなる合意によって生成されたいかなる特許権使用料収入も確認していない。
当社が残りの履行義務を必要としなくなった場合や、履行義務期間が終了した後、当該等の金額は、貨物又はサービスの制御権を顧客に移転する際に収入として確認される。通常、販売に基づくマイルストーンや印税を除いて、受領または満了したすべての金額は、許可および協力収入に分類される。販売に基づくマイルストーンおよび特許権使用料は、関連販売が発生したか、またはすべての特許権使用料分配の履行義務が履行された(または部分的に履行された)ときに、特許権使用料収入として確認される。
今後12ヶ月以内に確認されると予想される繰延収入は流動負債に分類される。
F-14
契約残高
会社が顧客が対価または満期支払いを支払う前に貨物またはサービスを顧客に譲渡する場合、会社は契約資産を確認するが、口座または他の入金として示されるいかなる金額も含まない。契約資産は、実体が顧客に譲渡する商品又はサービスの対価格権利である。契約負債または繰延収入は、主に会社が支払いを受けたが、関連履行義務を履行または完全に履行していない契約に関するものである。前払い金および費用は、受領または満了時に繰延収入と表記され、当社が当該等の手配下での責任を履行するまで、収入確認を将来の期間まで遅延させることが要求される可能性がある。会社ライセンス契約により生じる前金契約債務は、融資部分を代表するものではなく、支払は貨物又はサービスの譲渡に資金を提供するのではなく、許可を付与する技術は、会社が発生した研究及び開発費用を反映している。
2023年、2023年、2022年12月31日終了年度の繰延収入総額の変動状況は以下の通り(単位:千)
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
期初残高 |
|
$ |
|
|
$ |
|
||
ライセンス契約からの繰延収入の増加 |
|
|
— |
|
|
|
|
|
収入で確認した金額 |
|
|
( |
) |
|
|
( |
) |
期末残高 |
|
$ |
|
|
$ |
|
||
研究開発コスト
研究·開発コストは発生時に費用を計上する。研究開発コストには、賃金およびボーナス、株式ベースの給与、従業員福祉、施設コスト、実験室用品、減価償却、製造費用、臨床前費用、相談、および他の契約サービスが含まれる研究開発活動を行うことによって生じるコストが含まれる。許可された技術または知的財産権が技術的可能性に達しておらず、将来的に他の用途がない場合、発生時にいくつかの技術または知的財産権の許可を得るコストは、研究および開発費用に計上されるある研究開発活動のコストは個別手配の条項によって確認され、発生したコストモデルと異なる可能性があり、財務諸表に前払いまたは計算すべき研究開発コストとして反映される。
株に基づく報酬
同社の株式ベース報酬計画は、株式オプション、制限株式奨励、制限株式単位の付与を許可する。補助金は役員を含む従業員と非従業員に付与される。
当社はASCテーマ718に基づいて株式ベースの報酬を計算している報酬--株式報酬またはASC 718。ASC 718は、従業員、非従業員、および取締役に支払われたすべての株式を総合経営報告書で費用として確認し、その公正価値に基づいて他の全面的な損失を確認することを要求する。同社は、ブラック·スコアーズオプション定価モデルまたはブラック·スコルスオプション定価モデルを使用して、従業員および非従業員の株式オプションの公正価値を付与すると推定している。会社普通株の公正価値は、制限株式奨励と制限株式単位の公正価値を決定するために使用される。
株式ベースの報酬報酬は、サービスまたは業績に基づく帰属条件によって制限される。サービスを基礎とする帰属条件の従業員、取締役及び非従業員が獲得した奨励に関する補償支出は、付与日に応じて関連サービス期間(一般に帰属期限)の公正価値を直線基準で確認する。業績条件を達成する可能性のある範囲内で、奨励業績の帰属条件に関連する従業員の報酬支出は、授与日をもとに、必要なサービス期間の公正価値によって確認し、加速帰属法を採用する。
ブラック·スコアーズオプション価格設定モデルは、(I)予想株価変動、(Ii)予想報酬期間、(Iii)無リスク金利、および(Iv)期待配当を含む、いくつかの主観的仮定に基づく入力を必要とする。当社の予想変動率は代表的な上場企業の歴史変動率に基づいて計算され、これらの会社は製品開発段階と生命科学業界の重点を含む当社と類似した特徴を持ち、その株式上場期間の変動率に応じて重み付けを行う。履歴変動率は,期待期限の仮定に見合った一定期間に基づいて計算される.当社は“米国証券取引委員会従業員会計公報第107号”に規定されている簡略化方法を採用している株式支払従業員および非従業員に付与されたオプションの期待期間、すなわち、十分な履歴データが不足しているため、期待期間は、オプションの帰属期限および元の契約期間の算術平均値に等しい。無リスク金利は、満期日に関連奨励の期待期限に見合った米国債に基づく。期待配当収益率は
F-15
特許費用
特許出願の提出及び起訴に関するすべての特許に関する費用は、発生時に費用に計上される。支出回収の不確実性により、発生した金額は、添付された総合業務報告書およびその他の包括的損失のうち、一般的および行政費用に分類される。
可変利子実体
当社は、そのエンティティが可変利益エンティティであるか否か、またはVIEであるか否かを決定するために、財務的利益を有する各法人エンティティを審査する。エンティティがVIEである場合、当社は、一連の要因に基づいて、(I)どの当事者がVIEの経済パフォーマンスに最も影響を与える活動を指導する権利があるか、(Ii)任意の契約合意に従って、当事者の契約権利および責任、および(Iii)どちらが損失を負担するか、またはVIEから利益を得る義務があるかどうかを含む一連の要因に基づいてVIEの主な受益者であるかどうかを評価する。会社がVIEの主な受益者であると判断した場合、VIEの財務諸表をその連結財務諸表に統合する。当社がこれが合併VIEの主要な受益者ではないと判断した場合、またはVIEにおいて可変権益を有しなくなった場合、当社は決定を下した間にVIEの合併をキャンセルする。
所得税
当社は繰延税金資産と負債が当社の財務諸表と納税申告書に含まれている事件の予想される将来の税務結果であることを確認します。繰延税項資産と負債は財務諸表帳簿額面と現有資産及び負債及び赤字及び貸記繰越の税基との差額に基づいて決定し、予想差額を採用してその年に発効した制定税率を振り戻す。繰延税金資産が現金化できない可能性が高い場合は、推定準備金を差し引く。当社は審査の結果、税務状況をより維持する可能性があるかどうかを決定します。より可能性がある可能性がない場合、ハンドヘルドデバイスのいかなる利点に起因することができるかは確認されない。確認された税金割引は、より確認可能な閾値に適合する任意の税務ヘッドについて、事項解決後に達成される可能性が50%を超える最大金額として計算される。所得税引当金の一部として、同社は不確定な税収状況に関連した利息及び罰金を計上する。
総合損失
総合損失とは、企業株主権益が一定期間内に非所有者源の取引及びその他の事件と状況によって発生した変化である。総合損失には純損失および純損失以外に含まれない他の権益変動が含まれる。同社の他の全面赤字の唯一の要素は有価証券の未実現損益である。
1株当たり純損失
当社は2段階法で1株当たり純損失を計算しており、当社は参加証券の定義に合った株式を発行しているため。2級法は、発表または累積された配当金および未分配収益の参加権に基づいて、各種類の普通株および参株証券の1株当たり純損失を決定する。二段階法は、この期間中に得られる収入が、その期間のすべての収入が割り当てられているかのように、普通株主がそれぞれ配当を得る権利に基づいて、普通株式と参加証券との間で分配されることを要求する。
普通株株主が1株当たり基本純損失を占めるべき計算方法は、普通株株主が純損失を当期発行普通株の加重平均で割るべきである。普通株主が償却純損失を占めるべきであることは、普通株株主の純損失を調整して、希釈証券の潜在的な影響に基づいて未分配収益を再分配することによって計算される。普通株株主が希釈1株当たり純損失を占めるべき計算方法は、普通株株主は希釈純損失を当期発行普通株で割った加重平均を占めるべきであり、その中に普通株等価物の希釈影響を仮定する潜在希釈性普通株を含む。
1株当たりの純損失計算については、業績および市場帰属条件は可能な株式オプションおよび株式単位、潜在的希薄化証券、未帰属制限株および普通株は普通株等価物とみなされ、業績または市場帰属条件は適用帰属条件を満たすことができない株式オプションおよび株式単位は普通株等価物とみなされない。
したがって、当社が純損失を報告している間、このような損失はこのような参加証券に割り当てられていない。当社は普通株株主が純損失を占めるべき期間を報告し、普通株株主は希釈1株当たり純損失と普通株株主が基本的に1株当たり純損失を占めるべきであり、希釈性普通株の効果が逆薄であれば、希釈性普通株を発行したと仮定しないからである。会社報告2023年12月31日まで、2022年12月31日と2021年12月31日まで年度普通株主は純損失を占めるべき.
F-16
権益会計法
当社が自社が普通株または実質普通株に投資する実体の経営や財務政策に重大な影響を与えることができるが制御できない場合には、当社は権益会計方法を用いて関連投資活動を記録する。当社が大きな影響力を持っているかどうかを評価する際には、投資の性質や規模、当社が持つ参加権、例えば協力や他の業務関係があるかどうかなどを考慮します。
権益会計法により、当社の投資は最初にコストで総合貸借対照表に計上されます。権益法投資を計上する際に、当社は、被投資会社関連純資産の帳簿価値と公正価値との間に基準差があるか否かを当社が割合で評価する。通常、収益や損失を計算する際には、当社は対象資産や負債の推定利用可能年数で確認された基礎差を直線的に償却するが、将来他の用途のない研究開発やIPR&Dのベース差は含まれていない。基礎差は、知的財産権研究開発に関連し、投資先がASC 805で定義されていない企業の範囲内である企業が合併し会社がすべての基礎差を被投資先の特定の資産または負債に帰することができない場合、投資コストが投資先の資産および負債の割合公正価値を超える残りの部分は権益法営業権とみなされ、権益投資残高で確認され、その残高は会社のメモ口座で個別に追跡される。当社はその後、総合経営報告書と全面赤字報告書の中で権益損失法投資プロジェクトにおいて他の実体の収益または損失を計上すべきである。損失シェアが当社の投資の帳簿価値を超えた場合、当社は追加損失の確認を一時停止し、追加資金の提供を約束したり、被投資債務の保証を約束しない限り確認を継続します。
事件や環境変化がその権益法投資の帳簿価値が減値可能であることを示すたびに,当社はこのような投資の減値を評価し,被投資者の財務指標,製品および商業見通し,現金使用などの定性的および定量的要因を考慮する。権益法投資の価値低下が非一過性と決定されれば、損失は当期収益に計上され、投資を公正価値に減記する。
2023年12月31日会社の軌道会社への投資は権益会計法を用いて会計計算を行う。その会社は所有している
市場と地理情報を細分化する
業務部門は、エンティティの構成要素として定義され、そのエンティティに関する個別離散情報は、首席業務意思決定者または意思決定グループが、リソースをどのように割り当てるかを決定し、業績を評価する際に評価することができる。CODMは会社の最高経営責任者です。会社はその業務を管理しています
最近公表された会計声明
2023年11月、FASBはASU 2023-07を発表した細分化された報告(主題280)今回の更新における改訂は、単一の報告可能な支部を有するエンティティに対する新しい支部開示要求および他の開示要求を含む部分開示要求を拡大する。この更新は、2023年12月15日以降の会計年度と、2024年12月15日以降の会計年度の中期に適用されます。この基準を採用することは会社の総合財務諸表に実質的な影響を与えないと予想される。
FASBは2023年12月にASU 2023-09を発表しました所得税(話題740)。今回の更新における改正は、税率調整、支払いされた所得税、その他の開示に関する追加情報を含む所得税開示要求を拡大する。この更新は、2024年12月15日以降の年間期間に適用されます。この基準を採用することは会社の総合財務諸表に実質的な影響を与えないと予想される。
3. P不動産と設備、網
財産および装置には、以下のものが含まれる(千計)
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
賃借権改善 |
|
$ |
|
|
$ |
|
||
実験室装置 |
|
|
|
|
|
|
||
家具と固定装置 |
|
|
|
|
|
|
||
コンピュータ装置 |
|
|
|
|
|
|
||
建設中の工事 |
|
|
|
|
|
|
||
総資産と設備 |
|
|
|
|
|
|
||
減価償却累計を差し引く |
|
|
( |
) |
|
|
( |
) |
財産と設備、純額 |
|
$ |
|
|
$ |
|
||
F-17
下表は、発生した減価償却費用(千単位)をまとめた
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
減価償却費用 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
4. f金融商品の空気価値
会社が公正な価値によって恒常的に計量する金融商品は、現金等価物、有価証券、Verve Treateutics、Inc.およびPrime Medicine,Inc.の株式証券、またはPrime、または会社とGuideとの間で2021年2月23日に締結された合併協定および計画に関連するまたは対価負債、またはGuide合併協定、およびハーバードと遠大許可協定による成功的な支払い派生債務を含む。
以下の表は、公正価値階層構造内の会社金融資産と負債の公正価値を示している2023年12月31日(千):
|
|
携帯する |
|
|
公平である |
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
|||||
資産 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
現金等価物: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
貨幣市場基金 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|||
有価証券: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
商業手形 |
|
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|||
会社手形 |
|
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|||
アメリカ国債 |
|
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|||
アメリカ政府証券 |
|
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|||
会社持分証券 |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|||
総資産 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
負債.負債 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
支払いに成功した責任-ハーバード |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|||
支払い成功責任-遠大研究院 |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
対価格負債があります技術は |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
対価格負債がある製品は |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
総負債 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|||
F-18
以下の表は、公正価値システム内の2022年12月31日までの会社の金融資産と負債の公正価値(単位:千)を示している
|
|
携帯する |
|
|
公平である |
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
|||||
資産 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
現金等価物: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
貨幣市場基金 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|||
商業手形 |
|
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|||
会社手形 |
|
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|||
有価証券: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
商業手形 |
|
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|||
会社手形 |
|
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|||
アメリカ国債 |
|
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|||
アメリカ政府証券 |
|
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|||
有価証券に含まれる権益証券: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
会社持分証券 |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|||
総資産 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
負債.負債 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
支払いに成功した責任-ハーバード |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|||
支払い成功責任-遠大研究院 |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
対価格負債があります技術は |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
対価格負債がある製品は |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
総負債 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|||
現金等価物現金等価物に含まれる通貨市場基金は、アクティブ市場のオファー市場価格から推定されるので、公正価値レベルの第1レベルに分類される。商業手形および会社手形は、価格設定がアクティブ市場のオファーとは異なるので、公正価値レベルの第2レベルに分類され、報告日に直接または間接的に観察されることができ、公正価値は、モデルまたは他の推定方法を使用することによって決定される。
有価証券有価証券(会社持分証券を含まない)は、価格投入がアクティブな市場のオファーではないので、公正価値レベルの第2レベルに分類され、公正価値は、モデルまたは他の推定方法を使用して決定されることが報告日に直接または間接的に観察されることができる。
同社は2023年12月31日と2022年12月31日までの年間で、Verveの普通株を含むVerveへの投資を保有している。2023年12月31日までに会社は
2022年10月、Primeは普通株の初公募株を完成させた。2023年12月31日と2022年12月31日まで会社が持っています
以下の表は、保有する会社持分証券の公正価値変化による他の収入(費用)(単位:千):
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
その他の収入(費用) |
|
$ |
( |
) |
|
$ |
|
|
$ |
|
||
成功支払責任付記10でさらに議論されているように、当社は、当社Aシリーズ優先株の初期加重平均価値の指定倍数に基づいて、または初公開発売後、当社の普通株の時価を指定推定日にハーバードおよびブロイド研究所に支払う必要がある。ハーバードと遠大許可協定によると、会社は株式に基づく成功支払いの責任を公正価値で計算する。負債の支払いに成功した推定公正価値を決定するために、同社はモンテカルロシミュレーション方法を使用して、いくつかのキー変数に基づいて株価の将来の動向をモデル化する。
F-19
ハーバードとブロードカレッジの支払い成功負債の推定公正価値を計算する際に、以下の変数が格納されている
|
|
ハーバード |
|
|
ボッド·カレッジ |
|
||||||||||
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||||
普通株公正価値(1株当たり) |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
予想変動率 |
|
|
% |
|
|
% |
|
|
% |
|
|
% |
||||
所期期間(年) |
|
|
|
|
|
|
|
|
||||||||
予想変動率の計算は、自社の履歴変動率と、期待期限仮定にマッチした上場企業株の履歴変動率に類似した既存情報を用いて推定される。また、当社は、支払成功負債を算出する際に、推定計量日の推定数、時間、可能性を計上している。
次の表は、第3レベル投入に基づく成功支払負債の公正価値変化(千単位)を照合する
|
|
2023年12月31日までの年度 |
|
|||||||||
|
|
ハーバード |
|
|
ボッド·カレッジ |
|
|
合計する |
|
|||
2021年12月31日の残高 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
価値変動を公平に承諾する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
2022年12月31日の残高 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
価値変動を公平に承諾する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
2023年12月31日の残高 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
対価格負債があります-付記9でさらに議論されているように、ガイドライン合併プロトコルによれば、ガイドラインの前株主およびオプション所有者は、最大の追加$を取得する資格がある
価格負債の推定公正価値を計算または有する場合には、以下の変数が格納される
|
|
技術的マイルストーン |
|
|
製品の一里塚 |
|
|
||||||||||
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
|
||||
割引率 |
|
|
% |
|
|
% |
|
|
% |
|
|
% |
|
||||
実現の確率 |
|
|
|
|
|
|
|
|
|
||||||||
予想達成年 |
|
|
|
|
|
|
|
|
|
||||||||
次の表は、第3レベルの投入または対価格負債に基づく公正な価値変動をチェックした(千で計算)
|
|
|
|
|||||||||
|
|
技術的マイルストーン |
|
|
製品の一里塚 |
|
|
合計する |
|
|||
2021年12月31日の残高 |
|
$ |
|
|
$ |
|
|
|
|
|||
価値変動を公平に承諾する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
2022年12月31日の残高 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
価値変動を公平に承諾する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
2023年12月31日の残高 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
F-20
5. 有価証券
会社が持っている有価証券を下表にまとめた2023年12月31日(千):
|
|
原価を償却する |
|
|
毛収入 |
|
|
毛収入 |
|
|
公正価値 |
|
||||
商業手形 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
会社手形 |
|
|
|
|
|
|
|
|
— |
|
|
|
|
|||
アメリカ国債 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
アメリカ政府証券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
会社持分証券 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
合計する |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
次の表は、会社が2022年12月31日に保有する有価証券(単位:千)をまとめた
|
|
原価を償却する |
|
|
毛収入 |
|
|
毛収入 |
|
|
公正価値 |
|
||||
商業手形 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
会社手形 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
||
アメリカ国債 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
||
アメリカ政府証券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
会社持分証券 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
合計する |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
有価証券の償却コストは割増償却と満期割引増加に応じて調整される。2023年12月31日と2022年12月31日にその他の総合(赤字)収入を累計する残高には有価証券関連の活動のみが含まれる。いくつありますか
当社は信用品質の高い会社の債務証券を保有しており、どの債務証券の信用リスクにも大きな変化はないことが確認されている。すべての投資の契約期限は
6. 費用とその他の流動負債を計算しなければならない
計算すべき費用および他の流動負債には、以下の項目が含まれる(千で計算)
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
計算すべきか債務があるかは付記10を参照 |
|
$ |
|
|
$ |
— |
|
|
従業員補償と関連福祉 |
|
|
|
|
|
|
||
研究コスト |
|
|
|
|
|
|
||
プロセス開発と製造コスト |
|
|
|
|
|
|
||
専門費 |
|
|
|
|
|
|
||
設備融資コスト |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
合計する |
|
$ |
|
|
$ |
|
||
当社は当機関と当社との間の守秘協定に関する研究機関から手紙を受け取りました。秘密保護協定は、その会社がそのいくつかのプロジェクトに関連する開発を評価するいくつかの技術に関する。同社は機密協定の条項に違反し,同機関の商業秘密や他の機密情報を流用し,不公平や欺瞞的な商業行為に従事し,BEAM−302を含む治療法の開発において不公正に富を得たと手紙で伝えられている。研究機関は、金銭的損害賠償(会社への価値を分担する損害賠償、および故意違反の疑いによって増加した損害賠償を含む)、および違約の疑いのある技術に関連して行われている特定の特許権使用料および/またはマイルストーン支払い、ならびに他の可能な救済措置を得る権利があると主張している。誰も不満を提起せず、会社は研究機関とこの問題を議論し続けた。2023年12月31日までどんな損失額も確定できません。会社は#ドルを計算しなければなりません
F-21
7. 賃貸借証書
賃貸借契約を経営する
同社の経営賃貸契約は以下の通り
次の表は、経営リースコストと転貸収入(単位:千):をまとめています
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
リースコストを経営する |
|
$ |
|
|
$ |
|
|
$ |
|
|||
可変リースコスト |
|
|
|
|
|
|
|
|
|
|||
短期賃貸コスト |
|
|
|
|
|
|
|
|
|
|||
転貸収入 |
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
合計する |
|
$ |
|
|
$ |
|
|
$ |
|
|||
経営性賃貸のレンタル期間と割引率をまとめた表
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
加重平均残存賃貸年限(年) |
|
|
|
|
|
|||
加重平均割引率 |
|
|
% |
|
|
% |
||
次の表は、レンタル負債を計測する際に含まれるレンタル料金(千単位)をまとめています
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
レンタル経営のための経営キャッシュフロー |
|
$ |
|
|
$ |
|
|
$ |
|
|||
ROU資産の取得による経営リース負債 |
|
|
|
|
|
|
|
|
|
|||
はい当社の今後5年間の年間経営賃貸期限とその後の合計は、2023年12月31日までに以下のようになります(千計)
12月31日までの年度 |
|
金額 |
|
|
2024 |
|
$ |
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 |
|
|
|
|
2028 |
|
|
|
|
その後… |
|
|
|
|
未割引賃貸払い |
|
|
|
|
差し引く:推定利息 |
|
|
( |
) |
リース負債総額を経営する |
|
$ |
|
|
F-22
8.戦略的構造調整
2023年10月、会社は会社を再編して業務運営を簡素化するための最新のポートフォリオ優先事項と戦略計画を発表した。このポートフォリオの優先順位と戦略再編に伴い、会社は約
総合貸借対照表の課税費用とその他の流動負債に記録されている2023年12月31日現在の年間リストラの再編負債活動の詳細は以下の通り
|
|
金額 |
|
|
2022年12月31日残高再編成 |
|
$ |
— |
|
発生した費用 |
|
|
|
|
支払い |
|
|
( |
) |
2023年12月31日残高再編成 |
|
$ |
|
|
9.購入ガイド
開ける
さらに、Guideの元株主とオプション所有者は追加の最高$を取得する資格があります
当社は、すべての未来の技術および製品マイルストーン支払いがASC 480項目の下の負債に分類されることを決定したので、会社は案内合併合意日にこれらのマイルストーン支払いについて#ドルの公正価値の負債を記録した
F-23
取引価格の決定と割当ては以下のとおりである(千単位)
成約価格 |
|
|
|
|
|
発行済み権益ツールの公正価値 |
|
$ |
|
|
|
技術や製品や対価格負債があります |
|
|
|
|
|
取引コスト |
|
|
|
|
|
成約総価格 |
|
$ |
|
|
|
分配取引価格 |
|
|
|
|
|
現在行われている研究と開発 |
|
$ |
|
|
|
得られた現金 |
|
|
|
|
|
前払い費用と他の資産 |
|
|
|
|
|
財産と設備 |
|
|
|
|
|
集結した労働力 |
|
|
|
|
|
他の負債を負担する |
|
|
( |
) |
|
成約総価格 |
|
$ |
|
|
|
10. 許可証 協議
同社にはその研究や開発活動で使用されている技術に関する様々なライセンスプロトコルがある。ライセンス契約には、プリペイド、オプション費用、持続維持費、再許可料、印税ベースの支払い、マイルストーン支払い、成功ベースの支払い、および他の支払いが含まれる場合があります。オプション費用(適用される場合)は、行使時に、維持費、再許可料、その他の支払いが推定された満期金額または最終的に支払われた金額を発生した費用として記録することを確認する。派生ツールとして入金する必要がないか、または支払いが発生したことが確認された。当社のライセンススケジュールに基づいて対応する成功ベースの支払いは派生ツールであるため、成功ベースの支払いの公正価値は、経営報告書および全面損失表内の単独項目に変動して確認され、以下でさらに検討する。または債務および非特許権使用料転用許可料に含まれる経営および総合損失報告書に含まれる研究および開発費用の総額は#ドルである
当社のライセンス契約によると、再ライセンスに占めるべき価値や関連する再ライセンス費用は、契約要件に関する見積もりやその他の判断が必要となる可能性があり、これらの手配に基づいて支払われる最終金額の不確実性をもたらしている。満期の契約金額は計算すべきであり,ライセンス契約下の満期金額を解釈することに関する意外な場合があれば,会社は可能かつ推定可能な金額に対する負債を確認する。しかしながら、潜在的な支払い範囲内のどの金額も他の金額よりも良い推定値でない場合には、その範囲内の最低金額を計算すべきである。ある損失範囲内のある額がその範囲内の任意の他の額よりも良いような推定であれば、その額を計算すべきである。同社は、約#ドルを含む推定数を含む許可料負債を計上しなければならない
ハーバード大学許可協定
2017年6月、当社はいくつかの基礎編集技術について“ハーバード許可協定”を締結し、この合意に基づき、当社は、指定特許権の下で独占的、世界的に再許可可能な印税許可を取得し、許可された製品の開発及び商業化、及びいくつかの特許権の下の非独占的、世界的に再許可可能な印税の許可に基づいて、許可された製品を研究及び開発する。当社は、発展計画に基づいて、商業的に合理的な努力で特許製品を開発し、規制許可を得た任意の特許製品を商業市場に投入し、このような製品を市場に投入した後に監督管理許可を得た特許製品を販売し、監督管理許可を得た特許製品を合理的に公衆に提供することに同意する。ライセンス期間は、(I)ライセンス製品をカバーするライセンス特許の最終期限、(Ii)ライセンス製品に関連する排他的期限、または(Iii)ライセンス製品が初めて商業販売された後の特定の期限まで延長され、いずれか一方が特定の規定に従って早期に終了しない限り。
ハーバード許可協定に従って付与された権利の一部の対価格は、以下にさらに説明する成功した支払いを含む。
支払いに成功するハーバード許可協定によれば、ハーバードは、指定された推定日に達成された会社Aシリーズ優先株の初期加重平均価値の指定された倍数に基づいて、現金または会社株の形態で成功的な支払いを得る権利がある。Success支払いは$1から$1まで様々です
F-24
2021年5月に最初の支払い成功測定が行われ、ハーバードで不足している金額が$と算出されました
次の表は、ハーバード大学に対する会社の支払い成功責任(千単位)をまとめた
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
ハーバード大学の支払い成功責任 |
|
$ |
|
|
$ |
|
||
次の表は、ハーバード成功支払負債の公正価値変化による費用(千単位)をまとめたものである
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
ハーバード大学の支払い成功負債の公正価値変動 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
|
その他の支払い –
年間維持費は,適用年度の規定額に基づき,年次別に費用と記す。年間特許コストは発生時に費用を計上する。ハーバード製品マイルストーンが発生する可能性があると判断されると、支払うべき金額は研究と開発費用として記録される。当社はハーバード製品マイルストーン支払いのこの手配を継続的に監視します。
ハーバードライセンス協定に基づいて製品が商業化されている範囲では,会社はその支払い義務のある金額に応じて印税費用と再許可非印税支払い(場合によっては)を計上し,販売に応じて調整する。
広範な許可協定
2018年5月、遠大研究院とRNA編集プラットフォームを含むいくつかのRNA塩基編集技術について遠大な許可協定を締結した。遠大許可協定によると、遠大研究所は特定特許権の下で独占的かつ非独占的な世界範囲内で再許可可能な許可を付与し、許可製品を開発と商業化し、特定の特許権の下で研究と開発許可製品の非独占的、世界的範囲内の再許可可能な許可を付与する。協定によると、会社は開発計画に基づいて、商業的に合理的な努力で許可製品を開発し、規制部門の許可を得た任意の許可製品を商業市場に発売し、このような製品を市場に出した後、監督部門の許可を得た許可製品を販売し、監督部門の許可を得た許可製品を合理的に公衆に提供する。ライセンス期間は、(I)ライセンス製品をカバーするライセンス特許が最終的に満了した時間、(Ii)ライセンス製品に関連する規制排他期間、または(Iii)ライセンス製品が初めて商業販売された後の特定の期間まで延長され、いずれか一方が特定の規定に従って早期に終了しない限り。
遠大許可協定項の追加的な考慮事項は支払い成功を含めており,詳細は以下のとおりである.
F-25
支払いに成功する遠大許可協定によれば、遠大研究所は、成功した支払いを得る権利があり、現金または会社の普通株は、指定された推定日に達成されたAシリーズ優先株の初期加重平均価値の指定された倍数に基づいて決定される。Success支払いは$1から$1まで様々です
2021年5月、最初の成功支払い測定を行い、遠大研究所に対応する金額を#ドルと算出した
表には、遠大学院に対する会社の成功支払い責任(単位:千)をまとめた
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
遠大学院の支払い成功責任 |
|
$ |
|
|
$ |
|
||
表は、遠大研究所が負債公允価値変動に成功したことによる費用(千)をまとめた
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
遠大研究院の支払い成功責任の公正価値変動 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
|
その他の支払い –
年間維持費は,適用年度の規定額に基づき,年次別に費用と記す。年間特許費用は発生時に費用を計上する。遠大な製品マイルストーンが発生する可能性があると確定すると、支払うべき金額は研究と開発費用として記録される。同社は遠大な製品マイルストーンの支払い状況を継続的に監視している。
製品が遠大ライセンス契約に基づいて商業化されている限り、会社は特許権使用料費用を計算し、適用された場合には非特許権使用料支払いを再許可し、支払い義務のある金額を支払い、販売時に調整する。
Editasライセンス契約
F-26
以下のような場合はEditasに追加的な考慮事項を提供します 会社はEditas Researchライセンスの基礎となる3種類の知的財産権のいずれかのEditas開発と商業化ライセンスを取得するためにオプションを行使することを選択し、費用は1組15万ドルから中8桁まで様々であり、具体的にはオプション行使の時間に依存する。また、当社は、(I)Editasライセンス契約によって満了した開発、規制および商業マイルストーン支払い、およびいくつかの許可可能な収入支払い、および(Ii)Editasのある許可者が支払うべき一定の割合の年間保守費および特許料を含む、既存のEditasライセンス契約に従って支払う義務がある可能性がある知的財産権に関する何らかの支払いを返済しなければならない。さらに、任意の製品がEditas開発および商業化ライセンスに従って商業化される場合、会社は、EditasがEditas所有の特許によってカバーされているかどうかに応じて、Editasが任意の適用可能なEditasライセンシーに支払うべきそのような許可製品の販売に関連する印税を支払うことを要求されるであろう。
Editasが当社に付与するライセンス権と選択権は,Editasが基礎ライセンスプロトコル側の基礎ライセンスプロトコルとして付与する条項や条件の制約を受け,当該合意により,Editasは当社にライセンス権または選択権を付与し,当該等のライセンス内の権利を終了する。いずれか一方が合意条項に従って早期に終了しない限り、Editasライセンス契約は引き続き全面的に有効であり、(I)Editasの任意の適用機関の許可の下で最後に満了するライセンス使用料期限と、(Ii)当該製品がもはやその国によって許可されていないEditasが所有する特許の有効な権利要件によってカバーされる日(以下の列の両方の後の者を基準とする)が、ライセンス製品および国/地域に基づいて満了する。当社はEditasに90日間の書面通知を出した後、Editasライセンス契約を随時完全に終了またはグループごとに終了する権利があります。Editasライセンス契約が終了すると、Editasが当社のすべての権利および許可(オプションの行使およびそのような許可を得る権利を含む)を付与することは直ちに終了し、特許のセット中の特許はもはや許可された特許とみなされなくなる。Editasライセンス契約の満了または終了は、いずれの理由でも生じた義務または責任を免除しないか、または満了または終了前の期間の義務または責任に起因することができる。
会社が当該オプションを行使すれば,合意項下のオプション行使費用は研究·開発費に計上される。今まで、何のオプションも行使されていない。年間維持費は,適用年度の規定額に基づき,年次別に費用と記す。年間特許コストは発生時に費用を計上する。また、会社は指定されたマイルストーンに達したときに、いくつかの開発、規制、ビジネスマイルストーンの支払いをEditasに支払わなければならない。適用される範囲内で、再許可収入支払いは、会社毎に適用される許可証内に規定された義務に応じて支払われる金額を積算し、Editasに支払う金額とする。最後に,Editasライセンス契約により製品を商業化すると,会社は支払い義務のある金額に応じて特許権使用料を計算し,販売に応じて調整する。
11.連携とライセンスプロトコル
礼来会社
2023年10月、会社は、Verve Treateutics,Inc.またはVerveとの修正された協力および許可プロトコルまたはVerveプロトコルに従って、会社がVerveを共同開発および共同商業化する心血管疾患基礎編集プログラムの選択権を含むいくつかの資産および他の権利を取得した(Verveプロトコルに関する議論を参照)。当社はVerveプロトコルにより当社のVerve技術への独占再許可を当初許可していた礼来社に授与した。礼来社はまた、Verveプロトコルに従って支払われた任意の将来のマイルストーンまたは特許使用料を受信する権利と、代表を指定し、Verveと共に共同指導委員会に参加する権利と義務を取得した。その会社は1ドルを受け取った
しばらくの間
礼来会社協定について、当社は礼来社と株式購入契約を締結し、売却と発行を規定しています
当社は株式購入契約の下の対価を受け取りました$
F-27
礼来会社協定と株式購入協定は一括合意として同時に交渉され、合併契約として入金されている。当社はASC 505に従って礼来社に普通株を譲渡する手配の構成要素を担当している権益ASC 606の下で定義された顧客−仕入先関係を含み、契約と考えられる基準を満たすので、ASC 606の収入部分となる。当社は、まず、ASC 505の指針を適用して、発行された普通株式の公正価値を計測し、残りの対価格を手配されたASC 606部分に割り当てる。
契約開始時まで、ASC 606の総取引価格は$と決定された
当社の結論は,知的財産権の連携権と許可は同じ譲渡モデルと時間を持ち,礼来合意が発効した日に譲渡することである。礼来会社が研究開発サービスを要求する権利は協議の中の1つのオプション購入であり、実質的な権利を構成しない。合意の脈絡で、プレゼントを約束した他のすべての項目はどうでもいい。
同社は2023年10月のある時点で履行義務の収入を確認しており,履行義務に関するすべての要求が完了しているためである。ASC 606の規定に従って選択権を行使すれば、礼来社が会社の研究開発サービスを選択的に購入することに関するいかなる掛け値も単独の契約として入金される。2023年までに年度まで同社は$を確認した
軌道.軌道
2022年9月、同社は軌道治療会社または軌道会社と許可および研究協力協定、または軌道協定を締結した
会社が軌道プロトコルにより提供する許可とサービスの交換として,会社は軌道会社が制御するいくつかのRNA技術と非ウイルス伝達技術の非独占研究許可,および研究計画で概説した研究開発支援サービスを取得した。軌道会社はまた、軌道会社によって制御されるいくつかのRNA技術および非ウイルス送達技術の開発許可証を同社に授与した。開発ライセンスは遺伝子編集や調節分野では独占的であり,ワクチンやあるタンパク質療法以外のすべての分野では排他的ではない。その会社はまた受け取りました
この研究計画の期限は
当社はASC 606項の軌道プロトコルにより受信した対価格を計算し、取引先と契約した収入またはASC 606は、ASC 606の下で定義された顧客−仕入先関係を含み、契約と考えられる基準を満たすので、ASC 606である。契約開始時の総成約価格は#ドルに決定された
当社の結論は,研究·開発許可証は軌道協定における他の承諾と変わらないため,当社はライセンスと研究開発サービス,技術移転,委員会参加と材料譲渡を組み合わせることが義務であることを決定した。契約期間内に,同社は軌道履行義務に関する収入が一定期間満たされていることを確認した,すなわち
F-28
ビームは軌道会社の持株権を持っていない。ピムには軌道会社の活動を指揮する権限がないからだ。同社は軌道会社取締役会における非制御的代表と軌道会社における会社の持分を通じて軌道会社に大きな影響を与えている。そのため、当社は軌道会社の財務諸表を合併せず、権益会計方法を用いてその投資を計算する。2022年9月の締め切りまで、会社の軌道会社への投資の公正価値は#ドルです
ファイザー社
ファイザー協定が始まった時、同社は返金できない前金#ドルを得る権利がありました
協力期間内に、ファイザーは、特定の計画テーマである疾患を、予め定義された代替疾患で置換することを一度に選択することができる。協力の初期期限は
同社は、ASC 606によって定義された顧客-仕入先関係を含み、契約とみなされる基準に適合するので、ASC 606に従ってファイザー協定を計算する。
契約開始時の総成約価格は#ドルに決定された
当社の結論は,その基礎編集技術のライセンスは,独占開発権と商業化権利を含み,他の性能義務と区別できないため,当社はライセンスが他の研究開発サービスと組み合わせて性能義務を代表することを決定しており,ライセンスは前払い収入を確認していない。
F-29
契約義務ごとの販売価格は、会社が推定した独立販売価格またはESSPに基づいて決定されます。同社はファイザー協定に含まれるすべての履行義務のためにESSPを制定し,個々の履行義務を履行する総見積もりコストを決定することであり,定期的に独立して販売すればその製品の価格を決定することを目的としている。当社は相対独立販売価格法により独立販売価格を契約履行義務に割り当てています。
同社は契約期間内に入力法を用いて満たされているため、契約履行義務ごとの収入を確認している。会社が割り当てた出来高は$である
サナバイオテクノロジー
2021年10月、同社は、Sana Biotech,Inc.またはSanaと、特定の工学的細胞療法計画を開発し、商業化するために、SanaにそのCRISPR Cas 12 bヌクレアーゼシステムの非独占ライセンスを付与したSana契約を締結した。ライセンスに加えて、当社はサナ協定の要求に応じて、サナ協定の発効日後に予備技術譲渡を行い、サナに何らかの独自技術を提供している。この技術移転は2021年に発生した。許可証譲渡と初期技術移転が完了した後、SANAは自費で治療製品の開発と商業化を担当し、これらの製品は特定のCAR抗原標的或いは多能性幹細胞或いはPSC製品タイプを含まなければならない。
ライセンスの対価格として、同社は#ドルの前金を受け取りました
当社は、ASC 606によって定義された顧客-仕入先関係を含み、契約とみなされる基準に適合するため、ASC 606に従ってSANAプロトコルを計算する。契約開始時の総成約価格は#ドルに決定された
当社は、(I)当社の特許権及びノウハウに基づいてSanaに付与された非独占ライセンスと、(Ii)発効日後の初期技術譲渡とを含む単一履行義務を決定した。当社はさらに,選択権,置換権,遺伝的目標命名権にSANAの実質的な権利は付与されていないと結論した。会社は契約履行義務を1つしか確定していないため、取引価格を割り当てる必要はない。
当社は2021年12月31日までに年度内に確認します
F-30
アペリス製薬会社は
提携の一部として、同社は合計$を取得する資格がある
当社は、ASC 606によって定義された顧客-仕入先関係を含み、契約とみなされる基準に適合するため、ASC 606に従ってApellisプロトコルを計算する。
契約開始時の総成約価格は#ドルに決定された
同社の結論は、6つの基礎編集計画のそれぞれが研究開発サービス、許可証、代替権、ガバナンス参加と結合しており、区別できる重大な約束であり、“アペリス協定”の背景でも異なり、単独の義務履行を代表している。だから会社は確かに
契約義務ごとの販売価格は、会社が推定した独立販売価格またはESSPに基づいて決定されます。当社はApellisプロトコルに含まれるすべての履行責任のためにESSPを作成し、方法はすべての履行責任を履行する総見積もりコストを決定することであり、定期的にそのような物品を独立して販売する価格を決定することを目的としている。当社は相対独立販売価格法により独立販売価格を契約履行義務に割り当てています。
会社はすべての履行義務の収入を確認して、それが満足しているからです
良質な医療
2019年9月、会社はPrime Medicineと協力と許可協定を締結し、会社の創業者の一人によって開発された新しい遺伝子編集技術を研究·開発した。合意条項によると、同社は、ヒト疾患を治療するための遺伝子編集製品を開発し、商業化するために、Prime Medicineに、そのいくつかのCRISPR技術(Cas 12 bを含む)、伝達技術、および同社によって制御されるいくつかの他の技術の非独占的許可を付与した。Prime Medicineは、任意の単塩基変換変異の作成または修正、および鎌状細胞疾患の治療のための任意の編集のためのオリジナル遺伝子編集技術の開発および商業化のための独占的ライセンスを付与する。
F-31
Prime Medicine許可技術を使用するいかなる製品についても、会社はある臨床、監督、商業活動が完了した後、Prime Medicineに記念碑的な支払いを支払わなければならない。また、特定の国で開発され、Prime Medicine許可技術を使用した2つの製品の承認を求め、特定の国で承認された任意のこのような製品(S)を商業化するために、商業的に合理的な努力が求められている。Prime Medicineは,ある製品を共同開発·商業化し,その費用と収入を共有する権利があり,これらの製品が使用する技術は米国Prime Medicineから許可を得ている。本協定によれば、いずれも商業化製品の純売上高に応じて他方に特許使用料を支払うことが可能である。
さらに、同社は2021年3月までにPrime Medicineに非実質的な一時管理および起動サービスを提供しているが、2022年または2023年の間にこのようなサービスは提供されていない。
同社は2023年12月31日現在、同協定下の将来のマイルストーンや特許権使用料が認められる可能性が低いと判断した。
神韻
2019年4月、当社はVerveと、冠動脈疾患のリスク増加に関連する遺伝子を修正するための遺伝子編集戦略を検討するための協力·許可プロトコルであるVerveプロトコルを締結した。当社はVerveとVerveプロトコルを改訂した。改訂されたVerveプロトコル条項によると、当社はVerveにいくつかの基礎編集技術および改善された独占許可を付与し、VerveはVerveによって制御されたいくつかの独自技術および特許に基づいて当社の非独占許可、連携連携技術権益およびいくつかの交付技術の非独占許可を付与する。
VerveプロトコルについてVerveは会社を発表しました
同社は2020年12月31日までの年間でVerveのAシリーズ優先株も購入した。Verveは2021年6月、普通株の初公募を完了した。Verveの初公募株について、Verveが完成しました
経営陣は,Verveプロトコルに関する履行義務は,ライセンス技術に関する許可と改善の組合せであることを確認した.ウィヴィに約束された他のすべてのプロジェクトは合意の脈絡でどうでもいい。Verveが会社に発行した株式の公正価値は、固定前払い#ドルとみなされる
2023年10月、会社、Verve、および礼来社は、Verveプロトコルの下に存在するいくつかの権利および義務を、Opt-In製品を共同開発および共同商業化する権利および義務、マイルストーンおよび特許権使用料の支払いの財務権利、および特定の共同決定委員会に参加する権利を含むVerveプロトコルの下に存在するいくつかの権利および義務を礼来会社に譲渡することに同意する添付契約を締結した。同社は、権利の譲渡および許可はVerveプロトコルの修正であり、ASC 606の規定に従って、このプロトコルは既存の契約の一部とみなされると認定している。修正前に、会社は、契約開始およびその後の修正時に“Verveプロトコル”に関連する履行義務は明確ではなく、単一の履行義務であり、その契約の履行期間内に完了すると判断した。そこで,連携の契約期間(2019年4月から2038年)では,時間に基づく比例業績モデルを用いて前金を収入として確認し,許可収入と確認した.マイルストーン支払いと特許権使用料は可変対価格とされており、マイルストーンや特許使用料に達していないため、改正日までに制限されている。修正後,経営陣は,当社は手配中にVerveに大きな継続義務はなく,プロトコル下の余剰収入は修正後のある時点で確認すべきであると結論した。そこで、会社は残りの$を確認しました
2023年12月31日までの年度、会社は$を確認しました
F-32
12.普通株式
2021年1月に会社が発行して販売する
2021年4月に当社はJefferiesと販売協定を締結し、これにより、当社は現行の市価で株式を随時発売および売却する権利があり、総収益は最高$に達する
当社は2021年7月および2023年5月にJefferiesと販売協定の改訂を締結し、販売契約下の総発売金額を増加させることを規定し、当社が2023年5月10日から発売総発行価格を追加$とすることができるようにした
2021年5月,ハーバードとブロッド許可協定に基づいて第1回成功支払い測定を行い,ハーバードとブロダー研究所への支払い成功支払いを$と算出した
13.株式オプションおよび付与計画
2017年株式オプションと贈与計画
2017年6月、会社取締役会は、会社の普通株を発行または購入するために、会社従業員、高級管理者、取締役、コンサルタントおよび外部コンサルタントに合格奨励株式オプションおよび非制限株式オプション、制限株式またはその他の奨励を付与するBEAM治療会社2017年株式オプションおよび付与計画、すなわち2017年計画を採択した。
2017年計画は取締役会が管理しています。2017年計画により付与された株式オプションが満期になります
2019年インセンティブ計画
2019年10月、会社取締役会はBEAM治療会社2019年持分インセンティブ計画、すなわち2019年計画を採択し、IPO後、すべての持分ベースの奨励は2019年計画に基づいて付与された。2019年には、会社の従業員、上級管理職、取締役、コンサルタントおよび外部コンサルタントに合格および非適格株式オプション、株式付加価値権、制限および非制限株式および株式単位、業績奨励およびその他の株式ベースの奨励を付与する予定です。
2019年計画によると発行可能な会社普通株の最高株式数は最初は
2023年12月31日まで会社が所有しています
F-33
合併経営報告書とその他の総合損失のうち研究開発費と一般および行政費用と記載されている株式補償費用は以下の通り(単位:千):
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
研究開発 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
一般と行政 |
|
|
|
|
|
|
|
|
|
|||
株式に基づく報酬総支出 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
株式オプション
ブラック·スコアーズオプション定価モデルで使用される株式オプション定価モデルは以下のように仮定される
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
予想変動率 |
|
|
|
|
|
|
||||||
加重平均無リスク金利 |
|
|
% |
|
|
% |
|
|
% |
|||
期待配当収益率 |
|
|
% |
|
|
% |
|
|
% |
|||
予想期限(年単位) |
|
|
|
|
|
|
|
|
|
|||
次の表は、会社の株式奨励計画下のオプション活動の概要を提供します
|
|
番号をつける |
|
|
重みをつける |
|
|
重みをつける |
|
|
骨材 |
|
||||
2022年12月31日に返済されていません |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
授与する |
|
|
|
|
|
|
|
|
|
|
|
|
||||
鍛えられた |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
没収する |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
2023年12月31日現在の未返済債務 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
2023年12月31日から行使可能 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
同社は、いくつかの業績に基づく帰属基準を含む普通株株を購入するために、ある従業員に株式オプションを付与しており、これらの基準は、主に編集アプリケーションに関連するいくつかの発展マイルストーンの実現に関連しており、会社普通株の初回公募後の終値と関連している。業績状況が実現可能であると考えられた場合には、これらの業績ベース株式オプションに関する株式ベース報酬費用の確認を開始し、マイルストーンの将来結果に関する内在的リスクおよび不確実性を考慮した管理層の最適推定を使用する。あったことがある
2023年,2022年および2021年12月31日までに年度内に付与された加重平均授受日の1株当たりの株式公開価値は$である
2023年12月31日までに
制限株
当社は2023年まで、2023年、2022年および2021年12月31日までに制限された普通株式を発行し、制限株式単位のみで構成されています発行された制限的な普通株式は一般に一定期間内に至れり尽くせり
一般に、制限株式単位の所有者が当社と業務関係がなくなった場合、帰属していない制限株式単位はログアウトされる。
F-34
以下は同社の限定的な株式活動の概要である
|
|
株 |
|
|
重み付けの- |
|
||
2022年12月31日現在帰属していません |
|
|
|
|
$ |
|
||
発表されました |
|
|
|
|
|
|
||
既得 |
|
|
( |
) |
|
|
|
|
没収される |
|
|
( |
) |
|
|
|
|
2023年12月31日現在帰属していません |
|
|
|
|
$ |
|
||
2023年,2022年,2021年12月31日までの年度内帰属制限株式の公正価値合計はい$です
2023年12月31日には約$
2019年従業員株購入計画
当社はBlack−Scholesオプション推定モデルを用いて,付与日ESPP項における購入権の公正価値を推定した。予想変動率は,会社普通株のオプション期待寿命に対応した一定期間の履歴変動率から計算される。無リスク金利は、満期期限がオプションの期待寿命に似た証券を付与した場合の米国債収益率曲線に基づいている。期待寿命は,ESPPにより提供される贈与の購入期間をもとにしている。
当社は発売期間の費用を直線帰納法で記録しています。2023年12月31日までの年度ESPP関連株式報酬支出2022年、2021年は$
その会社は発行した
14.普通株主の1株当たり純損失
上述したように、会社が普通株株主が純損失を占めるべき期間を報告する場合、潜在的希薄化証券は、それらの影響が逆薄になるため、1株当たりの純損失の計算から除外されている。したがって、普通株株主が基本純損失を占めるべきであることと希釈後の1株当たり純損失を計算するための発行済み普通株加重平均は同じである
|
|
12月31日まで |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
無帰属制限株 |
|
|
|
|
|
|
|
|
|
|||
普通株購入の未償還オプション |
|
|
|
|
|
|
|
|
|
|||
ESPP |
|
|
|
|
|
|
|
|
|
|||
合計する |
|
|
|
|
|
|
|
|
|
|||
F-35
以下の表は、会社の普通株株主が1株当たり基本純損失と償却純損失を占めるべき計算方法(千では、株と1株当たりの金額を含まない)をまとめたものである
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
分子: |
|
|
|
|
|
|
|
|
|
|||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
分母: |
|
|
|
|
|
|
|
|
|
|||
加重平均発行済み普通株式、基本普通株式、希釈後普通株 |
|
|
|
|
|
|
|
|
|
|||
普通株1株当たりの基本損失と償却後の純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
15. 所得税
連邦法定所得税率を用いて計算された所得税費用と会社の有効所得税率との入金は以下のとおりである
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
連邦法定金利 |
|
|
% |
|
|
% |
|
|
% |
|||
連邦福祉を差し引いた州所得税 |
|
|
|
|
|
|
|
|
|
|||
研究開発税収控除 |
|
|
|
|
|
|
|
|
|
|||
控除不可/課税不可永久項目 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
“知的財産権と開発ガイドライン”調達 |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
評価免除額を変更する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
合計する |
|
|
- |
% |
|
|
- |
% |
|
|
% |
|
同社の繰延税金の構成要素は以下の通り(千計)
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
繰延税金資産: |
|
|
|
|
|
|
||
純営業損失が繰り越す |
|
$ |
|
|
$ |
|
||
研究開発税収控除 |
|
|
|
|
|
|
||
費用その他を計算する |
|
|
|
|
|
|
||
収入を繰り越す |
|
|
|
|
|
|
||
派生負債 |
|
|
|
|
|
|
||
株式オプション |
|
|
|
|
|
|
||
償却する |
|
|
|
|
|
|
||
資本化研究 |
|
|
|
|
|
|
||
リース責任 |
|
|
|
|
|
|
||
繰延税金資産総額 |
|
|
|
|
|
|
||
ROU資産 |
|
|
( |
) |
|
|
( |
) |
財産と設備 |
|
|
( |
) |
|
|
( |
) |
他にも |
|
|
( |
) |
|
|
( |
) |
減算:推定免税額 |
|
|
( |
) |
|
|
( |
) |
税金資産を繰延し,純額 |
|
$ |
|
|
$ |
|
||
当社が記録した当期税額は、2023年12月31日および2022年12月31日までに計上されています
F-36
また、改正された1986年の国税法第382条及び383条は、企業が“所有権変更”を経験した範囲で税収属性を利用する能力を制限している。“所有権変更”は、通常、3年間のスクロール試験期間内に、5%以上の株主間の所有権変化が50%を超える(価値で測定される)と定義される。もし会社が所有権変更を経験した場合、所有権変更前の税収属性(例えば純営業損失と一般営業税控除)を利用して所有権変更後の課税所得額或いは税金が年間制限されていることを相殺し、通常は会社の所有権変更前の権益価値と計算し、ある規定の調整を経て、アメリカ国税局が毎月公表した長期免税率を乗じる。同社は2023年12月31日までの第382節の研究を完了し、2017年6月、2018年12月、2021年12月に歴史所有権変更が発生したことを決定した。また、株式所有権の変化により、会社は将来的に所有権の変化を経験するかもしれない。
2023年12月31日と2022年12月31日まで、会社は
2023年12月31日と2022年12月31日まで、会社は
同社の設立以来のすべての納税年度はアメリカとマサチューセッツ州で所得税申告書を提出しました。課税開始年度
16.関連先取引
軌道.軌道
付記11で述べたように、当社は軌道会社取締役会における非持株代表及び当社の軌道会社における持分を通じて、軌道会社に重大な影響を与えるが、軌道会社を制御していない。同社と軌道会社も協力·許可協定の締約国であり、複数の共同取締役会メンバーを有している。
創始者
2023年12月31日まで,2022年および2021年12月31日まで年度同社は#ドルを支払いました
神韻
同社とVerveは協力·許可協定の締約国であり、2022年上半期に共通の取締役会メンバーを持つ2023年12月31日までに会社は
当社はVerveに無形金額のある材料を購入し、これらの材料はそれぞれ添付されている2022年12月31日と2021年12月31日までの年度の総合経営報告書とその他の全面赤字に研究·開発費用として記録されている。当社は2023年12月31日までVerveに材料を購入していません。
2021年10月に、当社は契約を締結し、この合意に基づき、Verveが転貸した
良質な医療
同社とPrime Medicineは協力と許可協定の双方であり、共通の創業者がいる2022年第3四半期まで共通の取締役会メンバーを持っています2023年12月31日までに会社は
17.従業員福祉
2018年、会社は#節の下で固定払込計画を構築しました
F-37