アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
本財政年度末まで
あるいは…。
1934年証券取引法第13条又は15(D)条に基づいて提出された |
手数料書類番号
(登録者の正確な氏名はその定款に記載)
(明またはその他の司法管轄権 会社や組織) |
(税務署の雇用主 識別番号) |
(主にオフィスアドレスを実行) |
(郵便番号) |
登録者の電話番号、市外局番を含む:(
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
|
取引 記号 |
|
登録された各取引所の名称 |
|
|
同法第12条(G)により登録された証券:なし
登録者が証券法第405条に規定する有名な経験豊富な発行者である場合は、再選択マークで表示してください。はい。☐
登録者がこの法第13又は15(D)条に従って報告書を提出する必要がないか否かを、再選択マークで示す。はい。☐
登録者が(1)過去12ヶ月以内(または登録者がそのような報告を提出する必要があるより短い期間)に1934年の証券取引法第13条または15(D)条に提出されたすべての報告書を再選択マークで示すかどうか、および(2)このような提出要求を過去90日以内に遵守してきた
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
|
☒ |
|
ファイルマネージャを加速する |
|
☐ |
|
非加速ファイルサーバ |
|
☐ |
|
規模の小さい報告会社 |
|
|
|
|
|
|
新興成長型会社 |
|
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に第240.10 D−1(B)条に従って受信されたインセンティブベースの補償に従って回復分析を行う必要があるかどうかを再選択マークで示す
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)。はい違います
登録者非関連会社が保有する投票権と無投票権普通株の総時価は、2023年6月30日現在のナスダック世界精選市場普通株の終値に基づいているはい$です
2024年2月12日現在、登録者が発行する普通株式数は
引用で編入された書類
登録者は,2023年12月31日までの財政年度終了後120日以内に,2024年株主総会に関する第14 A条に基づいて最終的な依頼書を提出する予定である。2024年このような最終依頼書の一部株主年次総会は、本明細書に記載された範囲内の本年度報告の表格10−Kの第3の部分を参照して組み込む。
カタログ表
|
|
ページ |
前向き陳述に関する特別説明 |
1 |
|
|
|
|
第1部 |
|
|
第1項。 |
業務.業務 |
3 |
第1 A項。 |
リスク要因 |
49 |
項目1 B。 |
未解決従業員意見 |
113 |
プロジェクト1 C。 |
ネットワーク·セキュリティ |
113 |
第二項です。 |
属性 |
114 |
第三項です。 |
法律訴訟 |
114 |
第四項です。 |
炭鉱安全情報開示 |
114 |
|
|
|
第II部 |
|
|
五番目です。 |
登録者普通株市場、関連株主事項及び発行者による株式証券の購入 |
115 |
第六項です。 |
保留されている |
116 |
第七項。 |
経営陣の財務状況と経営成果の検討と分析 |
117 |
第七A項。 |
市場リスクの定量的·定性的開示について |
131 |
第八項です。 |
財務諸表と補足データ |
131 |
第九項です。 |
会計と財務情報開示の変更と相違 |
131 |
第9条。 |
制御とプログラム |
131 |
プロジェクト9 B。 |
その他の情報 |
134 |
プロジェクト9 Cです。 |
検査妨害に関する外国司法管区の開示 |
134 |
|
|
|
第三部 |
|
|
第10項。 |
役員·幹部と会社の管理 |
135 |
第十一項。 |
役員報酬 |
135 |
第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
135 |
十三項。 |
特定の関係や関連取引、取締役の独立性 |
135 |
14項です。 |
チーフ会計士費用とサービス |
135 |
|
|
|
第4部 |
|
|
第十五項。 |
展示·財務諸表明細書 |
136 |
第十六項。 |
表格10-Kの概要 |
138 |
|
|
|
サイン |
139 |
|
連結財務諸表 |
F-1 |
|
私たちは時々私たちのウェブサイトやLinkedIn上のプロフィールwww.linkedin.com/Company/amylyxを使用して材料情報を配布するかもしれない。私たちの財務と他の重要な情報は通常、私たちのウェブサイトの投資家部分に公開され、www.amylyx.comで得ることができます。私たちは、私たちが他の方法で伝播していない重要な情報をこのサイトで発表するかもしれないので、投資家に私たちのサイトの投資家の部分を見ることを奨励します。我々のサイトやLinkedInページに含まれてアクセス可能な情報は,本Form 10-K年次報告には含まれず,その報告の一部を構成することもない.
FOに関する特別な説明RWARD-Look文
本年度報告におけるForm 10−K又は年次報告は、改正された1933年証券法第27 A節又は改正された証券法及び改正された1934年“証券取引法”第21 E節又は取引法における安全港条項に基づいて行われた前向きな陳述を含む。本年度報告では歴史的事実に関する陳述を除き,他のすべての陳述は前向き陳述である。場合によっては、前向きな陳述は、“予想”、“信じ”、“継続”、“可能”、“推定”、“予想”、“意図”、“可能”、“計画”、“潜在”、“予測”、“プロジェクト”、“すべき”、“目標”、“将”またはこれらの用語または他の同様の用語の否定語によって識別することができる。このような陳述は未来の結果や業績の保証ではなく、重大なリスクと不確実性に関するものだ。本年度報告における展望的陳述は、以下の明示的または暗示的な陳述を含むが、これらに限定されない
1
本年度報告中の任意の展望性陳述は私たちの未来の事件と私たちの未来の財務表現に対する現在の見方を反映し、既知と未知のリスク、不確定性とその他の要素に関連し、私たちの実際の結果、業績或いは成果はこれらの展望性陳述と明示或いは暗示する任意の未来の結果、業績或いは成果とは大きく異なるかもしれない。実際の結果が現在の予想と大きく異なる可能性がある要因には,第I部第1 A項“リスク要因”と本年度報告の他の部分に記載されている要因がある。このような不確実性を考慮して、あなたはこのような前向きな陳述に過度に依存してはいけない。法的要求がない限り、私たちは未来に新しい情報があっても、これらの前向きな陳述を任意の理由で更新または修正する義務がない。
私たちのすべての展望的な陳述は本年度報告書までの日に限られている。いずれの場合も、実際の結果は、これらの前向き情報とは大きく異なる可能性がある。私たちはそのような期待や展望的な陳述が正しいことが証明されることを保証できない。本年度報告書に言及されているか、または我々の他の開示または他の定期報告または他の文書に言及されているか、または米国証券取引委員会または米国証券取引委員会に提出された他の文書または文書に記載されている1つまたは複数のリスク要因またはリスクおよび不確定要因の発生または任意の重大な悪影響は、私たちの業務、見通し、財務状態、および運営結果に重大な悪影響を及ぼす可能性がある。法律に別の規定があることを除いて、私たちは、本年度報告日後に発生した実際の結果、計画、仮説、推定または予測の変化、またはそのような展望的表現に影響を与える他の状況を反映するために、そのような前向き表現の更新または修正を承諾または計画しない。本年度報告の後になされた任意の開示声明または開示は、本年度報告に含まれる任意の前向き陳述に修正または影響を与える場合、本年度報告に含まれるそのような陳述を修正または置換するものとみなされる。
私たちは時々、これらの市場の潜在規模および特定の疾患の推定発病率および流行率の推定を含む、私たちの業界、一般的な商業環境、および特定の疾患に関する市場の推定、予測、および他の情報を提供するかもしれない。推定、予測、予測、市場研究或いは類似方法に基づく情報自体は不確定要素の影響を受け、実際の事件、状況或いは数字は、実際の疾病罹患率と市場規模を含み、本年度報告に反映された情報とは大きく異なる可能性がある。他に明確な説明がない限り、我々は、市場研究会社および他の第三者、業界、医療および一般出版物、政府データおよび同様のソースによって準備された報告、研究調査、研究および類似データから、当業界、商業情報、市場データ、流行率情報および他のデータを取得し、場合によっては、私たち自身の仮説および分析を適用し、これらの仮説および分析は、将来的に不正確であることが証明される可能性がある。
商標
便宜上、本報告では、私たちの商標および商号に言及する際に使用および?記号は使用されていませんが、このような言及は、適用法に基づいて、これらの商標および商号に対する私たちの権利を最大限に主張しないと解釈されるべきではありません。
2
第1部
第1項公事です。
概要
Amylyx PharmPharmticals,Inc.(Amylyx,We,OurあるいはUsとも呼ばれる)は商業段階の生物技術会社であり、その使命は神経変性疾患による苦痛を終わらせることである。十数年来、著者らは筋萎縮性側索硬化症(ALS)と神経変性疾患の研究に力を入れ、これらの疾病の治療方法を変える上で重大な進展を得た。
2013年の設立以来、私たちは神経変性疾患患者の需要解決に専念する研究段階会社から、複数の適応開発プロジェクトを持つ商業企業に転換した。
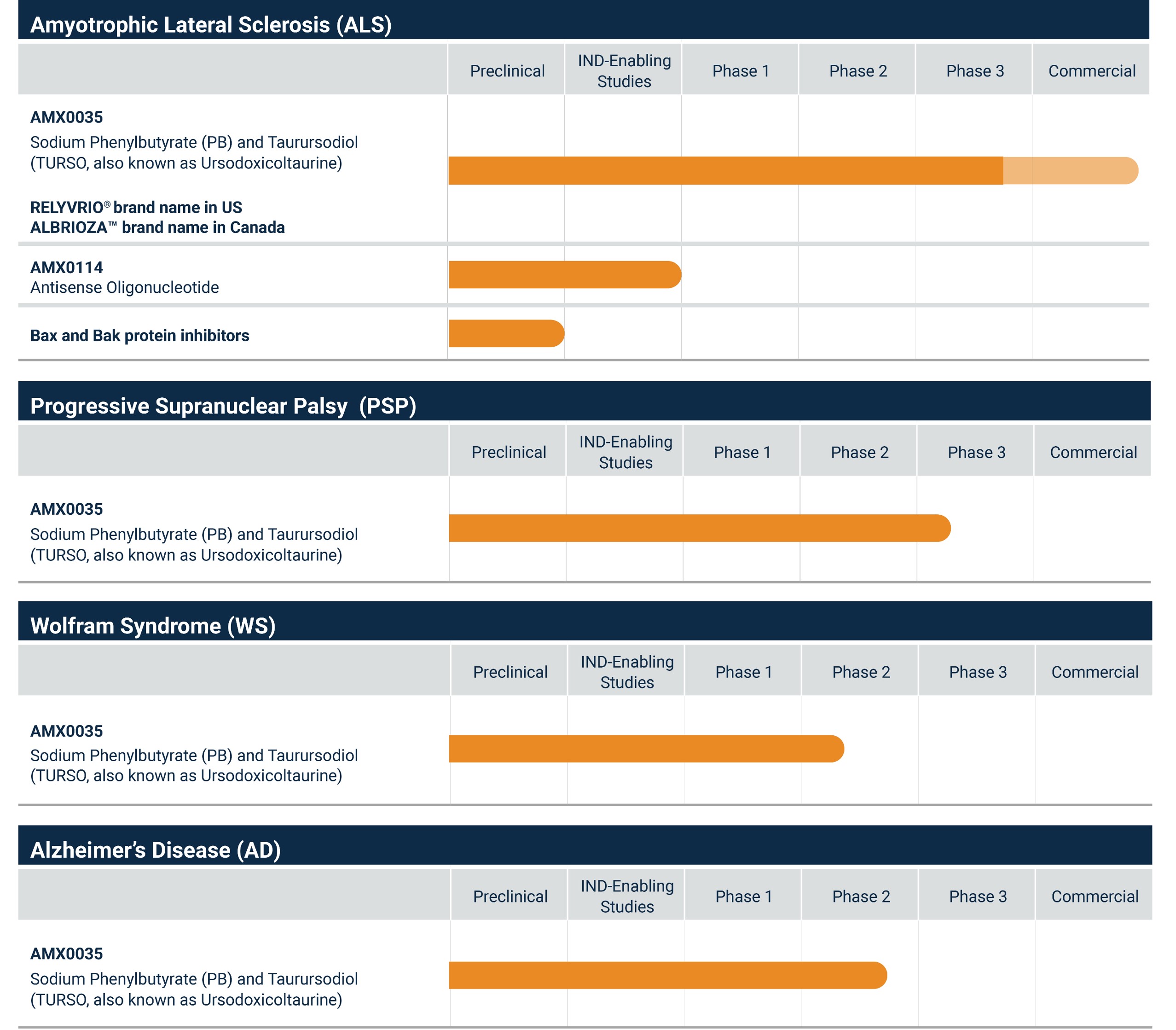
最初の商業製品AMX 0035(フェニルブタン酸ナトリウム)[鉛]タウリンとジオール[トルソ](米国ではRELYVRIOとも呼ばれ、カナダではALBRIOZAと呼ばれる)は私たちが知っている唯一のALS療法であり、同じ臨床試験で疾病の進展を遅らせることができ、機能独立を維持し、総生存期間を延長することを助けることが証明され、副作用の概況と経口投与の普遍的な耐性は良好である。AMX 0035は2022年10月にRELYVRIOとしてアメリカで商業発売され、2022年7月にカナダでALBRIOZA商業で発売された。RELYVRIOとALBRIOZAが発売されて以来、2023年12月31日までに、私たちはすでに4.03億ドルの製品純収入を生み出した。AMX 0035は,広く使用されているALS薬となる可能性があると信じ,症状管理を基準とした疾患から有意義な介入措置を有する疾患にALSを転換する機会を提供している。さらに、AMX 0035は、広範な神経変性疾患の治療パラダイムを変化させるために、単独でまたは他の療法と一緒に使用できることを意味する神経変性疾患の基礎療法になる可能性があると信じている。
我々はAMX 0035のメリットを世界の20万人以上のALS患者にもたらすことに取り組んでいる。私たちは、承認される可能性のある他の司法管轄区域でAMX 0035を商業化し、世界各地の重要な利害関係者と接触し、EUや日本を含むAMX 0035を獲得する機会を探るためのグローバルインフラを建設している。
我々は引き続きAMX 0035が筋萎縮性側索硬化症を治療する全世界フェニックス第3段階臨床試験に注目しており、これはアメリカとヨーロッパの臨床場所で行われた48週間の無作為、二重盲検、プラセボ対照試験であり、2024年第2四半期または前にTOPLINE結果を報告する予定である。フェニックスからのデータが支持されていれば,ALSの利点を証明した2つの臨床試験が初めてとなる。支持性の鳳凰データはAMX 0035の商業化発売とALS治療の転換をさらに加速させると信じている。
筋萎縮性側索硬化症に加えて,AMX 0035を用いた他の神経変性疾患の治療には強い科学的根拠があると考えられる。AMX 0035は、標的小胞体(ER)、圧力およびミトコンドリア機能障害によって神経変性を緩和または軽減することを意図しており、この2つの相互接続された中枢経路は神経変性をもたらす。我々独自のPBとTursoの組み合わせとそのそれぞれの作用機序は,いずれかの作用機序のみに対する治療よりも神経変性を予防するために,異常細胞死に協同作用することができると信じている。われわれは進行性核上性麻痺(PSP)とWolfram症候群(WS)におけるAMX 0035の効果を評価するために臨床試験を積極的に進めている。
我々は、ALSおよび他の神経変性疾患の他の潜在的治療法を決定するための研究に取り組んでおり、ALS患者の治療のためのAMX 0114、アンチセンスオリゴヌクレオチドも開発されている。私たちの現在のチャンネルは次の表のようになります。
3
4
私たちの会社とチームは
Amylyxは簡単で答えられていない質問に基づいています:何がニューロン死をもたらすのでしょうか?共同席最高経営責任者ジョシュ·コーエンとジャスティン·クレイは、11年前の2013年の進行性神経変性疾患による苦痛を終わらせるために世界的な会社を設立した。著者らは深い科学、臨床、商業と指導経験を持つチームを結成し、生物技術の専門知識を支持し、私たちの目標の実現を助けた。私たちの首席財務官James FratesはAlkermesの首席財務官として20年以上の経験を持っている。われわれの首席医療官Camille L.Bedrosianは,Ultragenyx,Alexion,ARIAD製薬会社で首席医療官を務めることを含め,製薬業界で成功した臨床開発と翻訳研究プロジェクトについて30年近くの経験を有している。私たちの最高技術運営官Tom HolmesはBiogen社で25年以上のサプライチェーン、薬品製造、プロジェクト管理の指導経験を持っている。我々の最高法務官兼総法律顧問Gina M.Mazzarielloは、Boehringer Inglheim USA,Inc.で指導職を担当することを含む医療保健業界で20年以上の企業と商業法律経験を持っている。私たちの首席人的資源官Linda arcaultは30年以上のリーダーシップと戦略ビジネスの鋭敏さをもたらし、最近彼女はSunovionで首席人的資源官を務めている。このチームは一連の独特な技能をもたらし、特にALSにおけるAMX 0035の成功した商業化を推進するのに適しており、同時にAMX 0035の他の適応を研究し、内部で新しい方法と化合物を探索し、有力な臨床医や研究者と協力することを含む、我々の導管を引き続き推進している。
私たちの戦略
私たちの使命はいつか神経変性疾患による苦痛を終わらせることだ。私たちがこの使命を達成するための戦略の重要な要素は
5
神経変性疾患
神経退行性変化の予防は現在最も重要な満足されていない医療需要の一つである。神経細胞の健康を保護する治療方法の発展は従来から独特な挑戦の制限を受けており、潜在生物学に対する不完全な理解と、臨床前研究で観察された活動を臨床試験結果に転化することが不足している。多くの神経変性疾患は症状を改善する治療選択しかなく、許可されていない有意義に疾病過程の治療方法を変える。疾患変更療法が承認された他の国では,疾患の進展をさらに緩和し,生存結果を改善するためにより多くの選択が必要であるが,依然として満足されていない。多くの神経変性疾患、特にALS、PSP、WSとADなどの進行性と深刻な疾患を解決するための新しい方法が依然として切実に必要である。
AMX 0035神経変性疾患治療の背景と理論的基礎
身体中の多くの他の細胞が定期的に死亡して健康機能の一部として置き換えられているのに対し,成熟したニューロンは通常細胞死に抵抗し,通常再生できない。多種のストレス因子が神経細胞の回復能力を超えて活性化された時にのみ、神経細胞死亡はトリガされ、このような状況は神経変性疾患によく見られる。大多数の神経退行性疾患は複雑な病理生理機序を有し、多数の経路が参与し、集結し、最終的に神経細胞の死亡を招く。ニューロン中のこれらの病理変化の大部分は小胞体やミトコンドリア機能障害に関与している可能性があり,後者は脂質やタンパク質の新陳代謝や分泌,カルシウム恒常性やエネルギー産生に影響する。この2種類の基本細胞構造の機能障害は多くの神経退行性疾患と関係があり、それらの神経細胞の健康と生存を維持する方面の核心作用を明らかにし、そしてAMX 0035に理論基礎を提供し、AMX 0035は小胞体とミトコンドリア機能を救い、そして神経細胞を保護と保存することを目的とした。
AMX 0035は二重未フォールディング蛋白反応或いはUPR-Baxアポトーシス阻害剤であり、2種類の小分子の特許経口固定用量の組み合わせである:PBはUPRを減少させる小分子シャペロンであり、UPRによる細胞死を防止する;Tursoは1種のBax阻害剤であり、アポトーシスによって細胞死を減少させる。
UPRの分解とbaxのミトコンドリア膜外へのシフトを抑制することにより、AMX 0035が様々な条件および圧力でニューロンの生存を維持することができることが、様々なモデルにおいて示されている体外培養神経変性モデル,小胞体ストレスモデル,ミトコンドリア機能障害モデル,酸化ストレスモデルと各種他疾患の特定モデル,および体内にあるALS、AD、多発性硬化症(MS)のモデル。
我々はAMX 0035を設計し,小胞体ストレスとミトコンドリア機能障害を同時に緩和することで神経細胞死を減少させた。PBはDJ-1というタンパク質を上方制御することによって小胞体ストレスを軽減することが証明されており、DJ-1は主要なシャペロン調節タンパク質であり、他のシャペロン蛋白質を募集し、小分子パートナーとして使用されている。Tursoは胆汁酸の一種であり,ミトコンドリア膜に組み込むことにより,Baxのミトコンドリア膜への転座を減少させ,ミトコンドリアの透過性を低下させ,細胞のアポトーシス閾値を向上させ,ミトコンドリアの生物エネルギー欠陥を回復する。我々の研究により,これらの鍵となる相互関連パスウェイに対するPBとTursoの組合せの特定の比率を決定し,神経細胞活性の向上における協同活性を示した体外培養それは.2022年、臨床前データによると、単独の化合物と比較して、PBとTursoの連合潜在的協同作用は、同業者評議の医学雑誌に発表された臨床と転化性神経病学年鑑それは.これらの散発性筋萎縮性側索硬化症成人と非筋萎縮性側索硬化症成人の初代皮膚線維芽細胞転写や代謝プロファイルに関するデータは,フェニルブタン酸ナトリウムやタウリン(タウリンとも呼ばれる)単独使用に比べて,PBとTursoの併用がALS関連経路に関与する遺伝子や代謝物に大きく有意に影響していることを示している。そして、最適な経口製剤としてAMX 0035を開発しました体内にある臨床的にもそうです
6
著者らの臨床前研究により、PBとTursoを併用することは、細胞培養と動物モデルにおいて神経退行性疾患と関連するいくつかの病理経路を抑制できることを表明した。例えば1つの体外培養神経変性モデルを構築するために,PBとTurso単独と併用酸化誘導ニューロン死や細胞生存防止の潜在能力を測定し,PrestoBlue試薬を用いて測定した。本実験では,ラット初代皮質ニューロンに過酸化水素を適用し,その濃度は約40%のニューロンを死滅させるのに十分であった。特定の用量のPBとTursoはそれぞれ一部の神経細胞を死亡から保護し、細胞生存率は約80%に達した。しかし,これらのラット初代皮質ニューロンをPBとTursoを特定の割合で併用投与した場合,酸化誘導ニューロン死はほぼ100%防止された。このようにした結果体外培養モデルを次の図に示す。
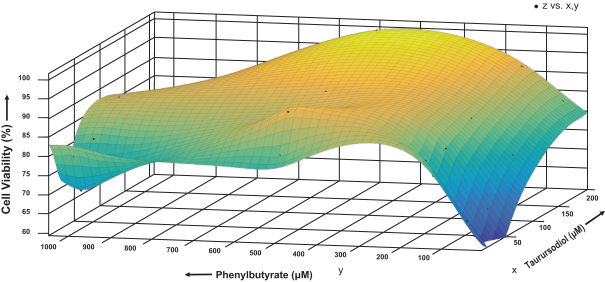
また,特定の割合のPBとTursoを用いた利点も観察された体外培養小胞体ストレス,ミトコンドリア機能障害,酸化ストレスのモデル,およびALS,AD,パーキンソン病,多発性硬化症,フリードリヒ運動失調,原発線粒体ミオパチーや各種他疾患の疾患特定モデルである。私たちはまた行いました体内にあるPBとTursoの併用モデルには,ALS,AD,MSのモデルが含まれており,また,学術グループではALS,AD,MS,パーキンソン病,ハンチントン病,PSP,多系統萎縮,X連鎖副腎白質ジストロフィー,様々な他のモデルにおいてTursoおよび/またはPBの単一療法治療研究が行われている。この証拠集団は,この組み合わせを用いた神経変性疾患の適応治療を支持し,他の疾患適応におけるAMX 0035の開発を促すと信じている。
AMX 0035筋萎縮性側索硬化症の治療
筋萎縮性側索硬化の概要
我々が最初に開発したAMX 0035は筋萎縮性側索硬化症を治療するためであり、これは非情な進行性と致命的な神経変性疾患である。ALSは脳や脊髄中の運動ニューロン死によるものであり,筋機能が悪化し,移動,発話,嚥下,食事ができず,呼吸が麻痺し,最終的に死亡する。疾患の後期には,一人で持続的なケアが必要となる可能性があり,車椅子が必要であり,コミュニケーション,摂食,呼吸のための機械的支援が必要である。ALSは依然として一般的に致命的であり,約半数の患者が症状出現後に死亡した中位数は3年未満である。
ALSはアメリカ食品と薬物管理局(FDA)とヨーロッパ薬品管理局(EMA)によって稀な疾患とされているが、それは全世界でよりよく見られる成人発病神経筋疾患の一つであると考えられている。世界で少なくとも20万人が筋萎縮性側索硬化症と診断されている。公開された情報源によると、アメリカには約30,000人のALS患者がいると推定されています。30,000人以上のALS患者がEUとイギリスに位置し、3,000人のALS患者がカナダにいると推定されています。
7
90%以上の患者に筋萎縮性側索硬化症の家族歴,すなわち散発性筋萎縮性側索硬化症はない。他の開発手法ではALSの遺伝例の解決が求められているが,AMX 0035は散発性であっても遺伝的であってもすべてのALS事例に対して行うことを目指している。筋萎縮性側索硬化症の多くは40歳から70歳の間であり,確定診断時の中位年齢は55歳であった。しかし,この疾患の症例は確かに2,30歳の人に発生している。
米国では,筋萎縮性側索硬化症と新たに診断された患者の医療費が高く,個々の障害マイルストーンの出現に伴い医療費が急速に増加する。筋萎縮性側索硬化症患者の看護は密集しており,医療専門チーム,特殊設備,日常活動協力が必要である。介護者はしばしば、予期した仕事を逃したり、介護を提供する雇用機会を放棄させられたりし、経済的圧力が増加する。このような疾患は患者の家族にも影響を与え、彼らは通常大部分の看護を提供し、これはしばしば24時間の看護を必要とする。介護者は筋萎縮性側索硬化症の疾患進展の要求に絶えず適応し,巨大な体力と精神的疲労,特に疾患の末期に必要である。
筋萎縮性側索硬化症と診断された患者の半数が症状出現後の中位数が3年未満であることを考慮すると,高い割合の患者が最近診断された。ALS患者の独立性を延長し、その身体機能喪失を緩和することができる療法であって、彼らの疾患と一緒に生活し続けることができる人の数を増加させる可能性がある。
筋萎縮性側索硬化症の重大な需要は満たされていない
筋萎縮性側索硬化症は多種の機序による異質性疾患であり、患者の異なる発病と遅延診断、筋肉機能の持続進展と喪失、及び生存時間の短縮を招く。
多種の発病経路に対して、疾病を改善でき、患者に機能と生存利益を提供できるALS療法に対して、重大な満足されていない需要がある。AMX 0035単独治療およびFDAによって承認された他の2つのALS治療薬と併用して、抗グルタミン酸作動薬リルゾールおよびフリーラジカルスカベンジャーエダラボンはALSの進行を調節できることが証明された。重要な臨床試験では,リルゾールはプラセボに比べて気管切開あるいは死亡時間が長く,エダラボンはプラセボに比べて機能保持時間が長い。しかし,依然としてALS療法が必要であり,機能を温存し,生存期間を延長することができ,患者がより長時間独立性を保つことができる。
ALSの多経路病理生理学のため、専門家たちは成功した治療は複数の重要な神経細胞死亡経路を同時に標的にする必要がある可能性があると一致している。これらの重要な経路の合流点に対して治療を行う強力な理論的基礎があり、小胞体とミトコンドリアに含まれ、AMX 0035のような複数の経路に対する治療は、新興のALS治療モデルと一致すると考えられる。
AMX 0035筋萎縮性側索硬化治療の臨床研究進展
我々はNealsのリーディングALS専門家からの意見を設計し,AMX 0035とプラセボの間の有意差を測定するために,我々の第2段階半人馬試験を設計した。この研究はまた、試験中に既存の承認された治療法リルゾールおよびエダラボンの使用を継続する選択肢を参加者に提供した。FDAは2017年9月にALS患者を治療する孤児薬としてAMX 0035を承認した。2019年12月、私たちは私たちの半人馬試験の積極的な背線結果を発表した。この実験は主なゴールに達しました“ニューイングランド医学雑誌”2020年9月と筋肉と神経雑誌2020年10月。EMAは2020年4月にAMX 0035孤児資格を与え、ALS患者の治療に用いられた。2021年第2四半期にカナダで新薬申請(NDS),2021年第4四半期に米国で新薬申請(NDA),2022年第1四半期に欧州でMAAを提出した。
AMX 0035は2022年6月、カナダ保健省がALBRIOZAを条件としてALSを治療する上場許可を取得し、2022年7月にカナダでALBRIOZAを商業的に発売した。2022年9月、AMX 0035はFDAの許可を得て成人ALSを治療するためのRELYVRIOを獲得し、私たちは2022年10月にアメリカでRELYVRIOを発売した。
8
2023年10月、EMAの人は、EU成人ALSの治療に使用されたAMX 0035のMAAに対する最初の否定的な意見を医薬品委員会(CHMP)で確認した。この決定は,2023年6月に採択された初歩的な否定意見に対するCHMPの正式な再審手続きが終了した後に行われた。2024年1月、欧州委員会はCHMPの否定的な意見の採択を確認した。我々は、2021年11月にMAAに提出する前に開始されたAMX 0035によるALS治療に焦点を当てた全世界フェニックス第3段階臨床試験の完了に引き続き焦点を当てている。もしフェニックスが支持を表明したら、私たちは可能な限り早く再び連合の承認を求める予定だ。
CentaurはALSでAMX 0035を用いた第2段階実験です
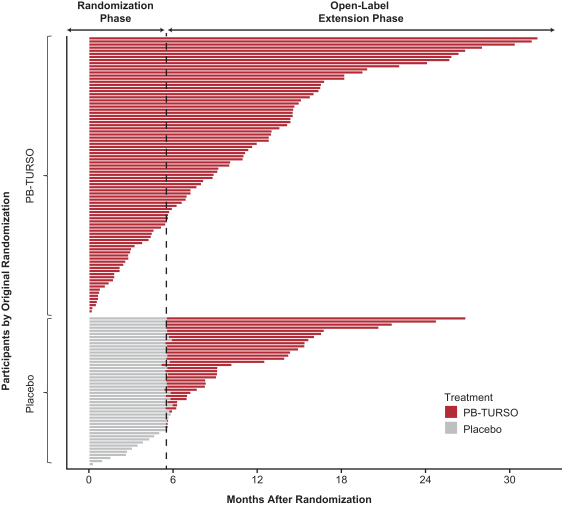
2020年9月と10月に,第2段階無作為,二重盲検,プラセボ対照のCentaur試験の詳細な結果を公表した。Centaur試験は25のNealsセンターで行い,ALSを有する成人患者を評価した。重要な組み入れ基準は改訂されたEl Ecore標準によって定義された明確なALSであり、この標準は少なくとも3つの定義された身体領域に各種の臨床バイタルサインと症状があり、上下運動神経細胞バイタルサインと定義され、症状の出現から18ケ月未満、肺活量が遅い、或いはSVCが60%より大きいことを要求する。これらの基準を選択するのは,同質で進展の速い患者群を選択し,治療効果を観察する可能性を潜在的に増加させるためである。参加者は、リルゾールおよび/またはエダラボン治療を含む、彼らの選択された看護基準を継続することを許可された。条件に適合した参加者(n=137)は、ランダムに2対1に分けてAMX 0035治療を受け、前の3週間に1日1つの香パック(各香パックは1グラムのTursoおよび3 g PBを含む)を与え、耐性がある場合、24週間の残りの治療中に1日2回、または一致するプラセボに用量を増加させる。2人の参加者は後続の治療効果評価を行わず、治療効果群(改良意向治療、またはMITT,n=135)にも組み入れられなかった。この2人の参加者は、セキュリティ集団(意図治療、またはITT、n=137)に組み込まれる。24週間の平行グループ化試験が終了した後、参加者は、すべての参加者が35ヶ月間追跡された開放ラベル拡張(OLE)試験に参加する資格があり、参加者および医師は依然として元の治療群を無視していた。Centaur実験を終えたランダム段階の参加者のうち,92%がOLEに参加することを選択した.♪the the the
9
OLEの最初の合意は2021年3月に完了した。PB−Turso組み合わせおよびプラセボを使用した患者のランダム化段階およびOLEにおける実際の治療持続時間を以下の図に示す
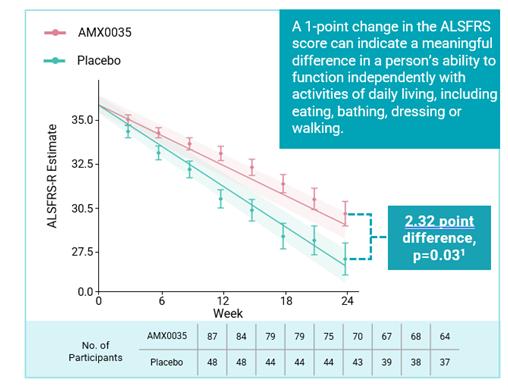
Centaur試験の主要な治療効果結果の測定基準はALSFRS-R総得点の低下率である。ALSFRS-R尺度はALSの臨床実践と臨床試験の中で最も広く使用されているALS分類標準である。それは患者の機能能力を測定し、4つの領域に分けられる:ボール(話す、よだれと嚥下を含む)、細かい運動(手書き、食べ物を切る/器を処理する、着衣と衛生を含む)、大運動(ベッドの上での寝返り、歩くことと階段を登ることを含む)と呼吸(呼吸困難、整形呼吸と呼吸機能不全を含む)。ALSFRS-R尺度の1点の低下は患者の独立性の深刻な制限を反映でき、ALSFRS-R尺度の2点の増加は以下の方面と関連する
Centaur試験はその主要な終点に達し,24週間以内にAMX 0035(n=87)をランダムに服用した参加者はプラセボ(n=48)を服用した参加者と比較して臨床低下率は統計的に有意に減少した(p値は0.03)。これらの結果は,24週間後にAMX 0035治療を受けた患者のALSFRS−Rでの得点がプラセボ治療を受けた患者より平均2.32点高く,25%差があることを示しており,次の図のようになる。Nealsが行ってスポンサーしたALS臨床医や研究者に対する調査では,ALSFRS−Rの減少がどの程度臨床的意義があると考えられるかを決定することを目的としており,ALSFRS−R総得点の差は20%以上である
10
調査を受けた臨床医や研究者の多くは臨床的意義があると考えられている。筋萎縮性側索硬化症の専門家が最近発表した文章でも,一点差が臨床的意義があると考えられるという観点を示している。
1.2人の参加者は、後続の治療効果評価を行わず、治療群にも含まれなかった(修正された治療意向n=135)。
疾病進展のない副次的な治療効果を測定した結果は肢体等長力正確テスト(ATLIS)を通じて測定した筋肉力の低下であり、SVC測定のテストと肺機能を通じて、両者はすべて予測値と肝心な研究イベントのパーセンテージとして表され、死亡、永久機械換気と入院を含む。神経フィラメントも生物学的指標として測定した。ATLIS上で測定したAMX 0035治療は上肢力の保護に統計学的意義があるが(p=0.042),下肢の測定は統計学的有意差に達していないことが示唆された(p=0.34)。この2つの指標の平均値は,ATLIS総得点と呼ばれ,AMX 0035(p=0.11)に傾いている。上大静脈容量の測定によると,AMX 0035治療を支持して肺機能を温存する傾向もあり,有意差は5.11%であったが,統計学的に有意差はなかった(p=0.076)。これらの効能データを下表にまとめた。また、24週間の無作為試験段階で、死亡、永久機械通気と入院事件を含む重要な研究事件に対してイベント時間分析を行った。Centaur試験に参加した患者は,6カ月のフォローアップが完了できると考えられる患者に限られているため,最初の24週間の無作為試験段階では,このような性質のイベントは少ないと予想される。その結果,24週間の無作為試験段階では,試験治療群と対照群の間に積極的な差が認められたが,統計学的有意差は認められなかった
11
この研究です。24週間の無作為研究段階では,試験治療群と対照群で観察された神経糸血漿レベルの低下速度に統計学的有意差は認められなかった。
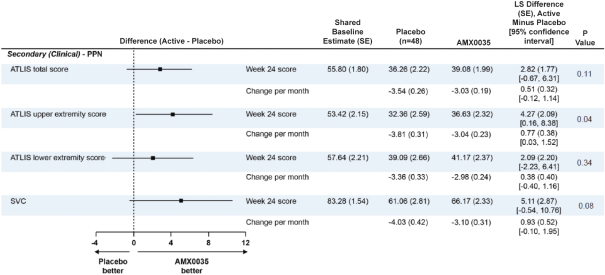
Centaur試験では,血漿中のリン酸化神経糸重鎖の含有量を測定した。この結果では,両群間に統計学的有意差はなかった。この結果の限界は,脳脊髄液ではなく血漿で測定されていることであり,この結果とALSとの最終的な相関は現場調査中である。
多くの参加者(77%)が研究に入る際または以前にリルゾールまたはエダラボン治療を受けており、プラセボ群でエダラボン治療を受けている割合(50%)がAMX 0035群(25%)よりも高いことに注意されたい。薬物の使用が結果に影響するかどうかを決定するために,あらかじめ指定された分析を行った。これらの分析により、ベースライン使用併用薬(リルゾールおよび/またはエダラボン)にかかわらず、AMX 0035のS効果が主要結果に与える影響は一致していることが分かった。
Centaur試験(ITT分析)でランダムに抽出した全被験者の総生存率(OS)を分析し,最初にAMX 0035をランダムに服用した患者(n=89)とプラセボをランダムに服用した患者(n=48)を比較した。この特殊な分析では、各参加者の生命状態は、2021年3月1日までの米国社会保障死亡指数などの源を使用し、OLEの服用を継続していなくても、研究薬物の中止、研究脱退、またはフォローアップを失った参加者の位置特定サービスによって測定されている。フォローアップ中、最初にAMX 0035をランダムに服用した患者は、最初にプラセボをランダムに服用した患者と比較して、死亡リスクが36%低下した(リスク比またはHRは0.64;95%信頼区間は0.42~1.00;p値は0.048)。平均生存期間は23.5
12
以下の図に示すように,AMX 0035を前にランダムに服用した群と,以前にプラセボをランダムに服用した群では18.7カ月(IQR 11.7~31.3カ月)はそれぞれ14.6~39.3カ月,18.7カ月であった。

AMX 0035の生存データについても3回の追加事後分析を行い,1回の追加事後バイオマーカー分析を行った。これらの分析には,秩序保持構造失効時間モデル(RPSF)と呼ばれる統計的方法を用いて交差治療の影響を調整する分析,Centaur試験で観察された生存率をALS自然病歴データベースから派生したヨーロッパALS治療ネットワーク生存予測モデルを用いた比較分析,Centaur治療群で観察された生存率とALS歴史臨床試験からの適応治療無邪気参加者の生存率を比較する分析,最後にCentaur参加者の血漿試料を用いた神経炎症性バイオマーカーの分析がある。
2022年5月、RPSF(登録商標)を使用した後期分析推定では、AMX 0035を最初にランダムに服用した参加者の平均生存期間は、最初にプラセボをランダムに服用した参加者よりも9.7カ月多かった。RPSF(登録商標)は、プラセボ交差を説明するために腫瘍学的によく使用される方法である。さらに、グループ分析において、AMX 0035をランダムに受け入れ、OLE段階に入り続けた参加者は、AMX 0035治療を受けていない参加者と比較して、彼らの平均生存期間が15ヶ月延長したことを示している。
ヨーロッパALS治癒ネットワーク(ENCALS)は1万人以上のALS患者のデータを収集し、これらのデータを用いてベースライン因子に基づく予後モデルを作成し、患者の生存時間を予測した。Amylyxはこのモデルの発起人と協力し,半人馬座の各参加者の治療無邪気な全体生存時間を予測した。このモデルを用いて生成した予測(治療無邪気)生存データをCentur研究で実際に観察された生存データと比較した。この特殊な分析では,AMX 0035をランダムに服用した参加者の平均生存期間は予測群と比較して9.9カ月延長した。
2023年10月、半人馬研究参加者の長期生存率と傾向スコアとが一致するAMX 0035無邪気傾向スコアが一致するAMX 0035を比較した一時的な分析が、同業者評議の医学ジャーナルに発表された臨床と転化性神経病学年鑑それは.この分析では,AMX 0035群の中位総生存期間の10.4カ月の延長が観察された。臨床試験開始時にAMX 0035治療を受けた参加者は、プラセボ服用を開始した参加者よりも6ヶ月前にAMX 0035の使用を開始し、服用時間が長く、より大きな生存利益が見られたことを意味する。
2023年12月、Centaur試験参加者の血漿試料を用いた神経炎症性バイオマーカーの特別な分析が発表された神経学神経外科精神医学誌はそれは.結果的には
13
筋萎縮性側索硬化症の2つの血漿神経炎症性バイオマーカーYKL−40(キチナーゼ−3様蛋白1とも呼ばれる)と全身炎症バイオマーカーC反応蛋白(CRP)の血漿濃度は24週間で有意に低下し,Centaur試験の参加者では12週前に低下が認められた。
共同レベル検定を含むCentaur試験データの感受性分析も行い,参加者の死亡によるデータ損失は,主要な機能結果を推定する際にばらつきがないことを示した。失われたデータと死亡や死亡の同値事象を説明するために感受性分析も行った。これらの感受性分析は予備分析と類似した結果を得た。併用を解釈するために設計された感受性分析では,併用を伴う分析を予備的に分析·修正した治療効果の大きさが一致した。
AMX 0035の全体的な耐性は良好であり、不良イベントの発生率はプラセボと基本的に類似している。AMX 0035を服用した参加者の97%(86/89)は有害事象を報告したが,プラセボ服用参加者の96%(46/48)は有害事象を報告しており,両群の有害事象の性質はほぼ類似していた。次の表はいずれの治療群の中で最もよく見られる副作用(5%以上)を示した。筋萎縮性側索硬化症の進行性神経変性変化の本質のため、これらの多くの副作用(例えば、筋肉無力、転倒、呼吸困難、疲労)は潜在的な筋萎縮性側索硬化症疾患に起因する可能性が高い。プラセボ治療を受けた患者と比較して、AMX 0035治療を受けた患者の中で発生したイベントの割合は5%以上であり、AMX 0035治療を受けた患者のイベントの発生頻度はより高く(2%以上)、これらのイベントは主に胃腸イベントであり、これらのイベントは深刻ではなく、強度の多くは比較的に軽く、治療3週間後の発生率は明らかに低下した。AMX 0035治療群とプラセボ群では,19%の患者と8%の患者が副作用で試験参加を中止した。
最もよく見られる副作用は下痢、腹痛、吐き気、上気道感染、便秘、筋肉無力、転倒、頭痛、眩暈とウイルス性上気道感染である。カナダ保健省もまた過度に興奮していることに気づいた。既知のTursoの安全性特徴と一致し,AMX 0035を服用した患者はプラセボを受けた患者よりも下痢や嘔気の発生率が高かった。対照的に、プラセボ治療を受けた患者の筋肉無力、転倒、便秘、頭痛の発生率はより高かった。観察された半人馬座試験の副作用を次の図に示す。
不良事件(AES)(1)いずれの治療群でも5%の患者が≥ |
||||||
MedDRAシステム臓器類第一選択用語 |
|
プラセボ+SOC |
|
AMX 0035+SOC |
|
総括する |
胃腸疾患 |
|
29 (60.4%) |
|
60 (67.4%) |
|
89 (65.0%) |
筋肉骨格と結合組織疾患 |
|
21 (43.8%) |
|
38 (42.7%) |
|
59 (43.1%) |
損傷、中毒、プログラム合併症 |
|
23 (47.9%) |
|
35 (39.3%) |
|
58 (42.3%) |
神経系疾患 |
|
19 (39.6%) |
|
33 (37.1%) |
|
52 (38.0%) |
感染と侵入 |
|
21 (43.8%) |
|
28 (31.5%) |
|
49 (35.8%) |
呼吸器、胸部、縦隔疾患 |
|
10 (20.8%) |
|
29 (32.6%) |
|
39 (28.5%) |
調べる |
|
10 (20.8%) |
|
26 (29.2%) |
|
36 (26.3%) |
全身性障害と投与部位の状況 |
|
13 (27.1%) |
|
20 (22.5%) |
|
33 (24.1%) |
皮膚と皮下組織疾患 |
|
8 (16.7%) |
|
16 (18.0%) |
|
24 (17.5%) |
精神障害 |
|
9 (18.8%) |
|
14 (15.7%) |
|
23 (16.8%) |
腎臓と泌尿系疾患 |
|
8 (16.7%) |
|
10 (11.2%) |
|
18 (13.1%) |
新陳代謝と栄養失調 |
|
4 (8.3%) |
|
10 (11.2%) |
|
14 (10.2%) |
心臓疾患 |
|
0 (0.0%) |
|
7 (7.9%) |
|
7 (5.1%) |
目の病気 |
|
1 (2.1%) |
|
5 (5.6%) |
|
6 (4.4%) |
14
プラセボ治療群(18.8%)と比較して、AMX 0035治療群(12.4%の患者)は名目上深刻な有害事象或いはSAEが発生する頻度が低い。この差は,AMX 0035治療群(3.4%の患者)と比較して,プラセボ治療群(8.3%の患者)の呼吸不全などの呼吸事象の発生率が高いことが大きい。筋萎縮性側索硬化症の疾患進展は通常呼吸不全を招き,筋萎縮性側索硬化症患者に最もよく見られる死亡原因である。Centaur実験で観察されたSAEを次の表にまとめる.
重大な有害事象(SAE)
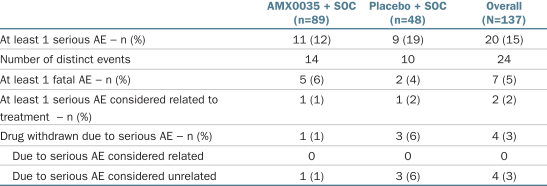
全体的に,この24週間の二重盲検研究では,計7名の患者が死亡し,そのうち2名(患者総数の4%)がプラセボを受け,5名(総患者の6%)がAMX 0035を受けた。すべての死亡例にAMX 0035が関与していると考えられている。筋萎縮性側索硬化症患者に最もよく見られる死亡原因と一致し,研究期間中の多くの死亡(7名中4名)は呼吸不全(各群2名)によるものであった。他の死亡原因(AMX 0035群では)には,抜管後の声門上と声門下吸入(誤嚥性肺炎による),憩室炎,転倒に続発する硬膜下血腫がある。死亡当量は気管切開術あるいは永久補助通気術,あるいはPAVと定義される。PAVは1日の非侵襲的機械換気が22時間を超え,1週間(7日)を超えると定義されている。24週間の研究では,プラセボ群では1名(2%の患者)とAMX 0035群で死亡に相当する事件(すなわち気管切開/PAV)を経験した患者は1人もいなかった。
2022年3月、特定の参加資格基準に適合するALS患者に適用するために、FDAが許可した米国EAPを開始することを発表した。EAPはRELYVRIOの米国での商業発売に伴い停止した。
AMX 0035はALS患者において機能的に統計学的に有意な候補を示した最初の候補であり,ALSFRS−Rであらかじめ指定された平均速度変化によって測定され,OSの長期分析で測定された2つの結果がALS患者にとって重要であると信じている。つまり,われわれのCentaur試験では患者の機能は統計的に有意に改善し,総生存率も統計的に有意に改善し,AMX 0035は全体的に耐性が良好であることが証明された。
筋萎縮性側索硬化におけるAMX 0035の臨床開発計画
我々のAMX 0035による筋萎縮性側索硬化症の治療の全世界フェニックス第3段階臨床試験は、筋萎縮性側索硬化症に対するAMX 0035の治療の安全性および有効性を評価するために、米国およびヨーロッパの臨床場所で行われた48週間の無作為、二重盲検、プラセボ対照試験である。この試験の登録作業は2022年3月に米国で完了し,欧州では2023年2月に完了した。我々は2023年2月にフェニックス試験の登録を完了し,664人が参加したと発表した。私たちは2024年第2四半期または以前にフェニックス試験のバックライン結果を報告する予定だ。
我々のフェニックス試験において、主要な終点は48週間以内の生存率とALSFRS-R総得点の進展及び48週間以内の安全性と耐性の総合測定基準である。我々のフェニックス試験の副次的な終点はSVC、ALSAQ-40(ALS患者の生活の質を専門的に評価するための主観的健康指標を提供するアンケート)、EQ 5 D-5 L(標準的な生活の質指標)、King‘sの低下(ALSの分期指標は中枢神経系或いはCNSの数量、罹患区域及び胃造口或いは非侵襲性人工呼吸の需要に基づく)、Mitos分期(標準方法を用いてALSFRS-R採点から予想される機能的分期指標)、無換気生存と長期生存である。フェニックス試験の鍵は基準に含まれています
15
臨床的に確定診断されたか、または臨床的に筋萎縮性側索硬化症に罹患している可能性がある筋萎縮性側索硬化症患者(2~4つの身体部位が筋萎縮性側索硬化症と一致)、55%、およびリルゾール/エダラボンの使用が許可されていることを含む。
48週間の試験を完了したヨーロッパ参加者は、登録を開放ラベル延期段階に選択することができる。この段階では、すべての参加者がAMX 0035を受け入れ、持続的な安全性および有効性措置を評価した。
私たちがこれまでに獲得したマーケティング承認は限られている可能性があるため、制限または承認後に要求される制限を受けて、これらの管轄区域で上場後の支援を提供する必要があるかもしれません。例えば,われわれが米国でRELYVRIOを承認した一部として,発売後にマウスやラットへの発癌研究,薬物−薬物相互作用研究,腎臓や肝臓損傷患者の研究が求められている。さらに、カナダにおけるAMX 0035のマーケティング許可の条件のうちの1つは、我々が行っているフェニックス試験および他の計画または行われている研究のデータを提供することである。フェニックス実験の結果は私たちの業務に実質的な影響を及ぼすかもしれない。
われわれはまた,ALS患者におけるAMX 0035の安全性と有効性をさらに評価し,実世界での状況を含めて検討する予定である。これらの連携研究には,単一センター体験や支払者データベース観察的研究などがある.
ALSにおけるAMX 0035の商業化
ALSにおけるAMX 0035の世界的なビジネス機会は,われわれが知られている1つ目であり,疾患の進行を緩和し,機能独立性を維持し,同一試験で総生存率を延長するのに役立つ唯一のALS療法であると信じている。AMX 0035は一般的に耐性が良好で、副作用はコントロールでき、経口は便利である。AMX 0035はALS患者の初回入院と永久機械換気の時間を減少させることを含む、臨床上意義のある終点に重大な影響があることが証明された。リリースから2023年12月31日まで、AMX 0035の販売から4.03億ドルの製品純収入を得ました。
AMX 0035は他の潜在的な適応の治療に使われています
著者らの疾病経路に対する広範な理解に基づいて、AMX 0035はAD、WS、パーキンソン病、ハンチントン病、PSP、多系統萎縮、原発性側索硬化症、虚血性脳卒中、多発性硬化症、Friedreich運動失調、Leigh症候群とLeber遺伝性視神経病変を含む多種の疾患に利益を提供できると信じている。
我々は,AMX 0035のSポテンシャルを支援するデータの強度,満たされていない需要の緊急性,これらの条件下での臨床試験の可能性,臨床開発活動の効率とビジネス潜在力に基づき,適応ごとにこれらを優先した。いくつかの適応については,同社がAMX 0035上で生成したデータを考慮すると,第3段階の安全性および有効性評価に直接入る可能性があり,経路の迅速な開発が可能である可能性が考えられる。患者に最もメリットをもたらす可能性が最も高いと考えられる適応と,最も早く市場に参入する適応を優先する。
AMX 0035進行性核上性麻痺治療の臨床研究進展
著者らは2023年12月にOrion試験を開始し、これはAMX 0035によるPSP治療の全世界の重要な3期試験である。Orionは世界的、無作為、二重盲検、プラセボ対照の3期臨床試験であり、AMX 0035がプラセボと比較した有効性と安全性を評価することを目的としている。約600名の参加者が米国,カナダ,EU,イギリス,日本の約100地点に登録され,これまでで最大規模のPSP臨床試験となることが予想される。
主要な治療効果の終点はベースラインから第52週までの疾病進展の変化を評価し、28項目の進行性核上性麻痺評価量表(PSPRS)の総得点で評価し、PSPRSはPSP臨床試験における確定と検証の終点である。
二次治療効果の終点は改良した10項目のPSPRS採点で測定した疾病進展、及び運動障害協会-統一パーキンソン病評価表の第二部分(MDS-UPDRS第二部分)から測定した日常生活活動の運動面である。探索的結果は日常生活能力、認知機能、生活の質、総生存率、脳領域体積、神経元損傷/炎症の液体バイオマーカーと介護者負担の変化を含む。
安全性および耐性は、緊急有害事象(TEAE)および重篤な有害事象(SAE)の治療頻度を評価することによって評価される。2025年か2026年には背線結果が発表される予定だ。参加者が完成する
16
52週間の無作為、プラセボ対照試験段階はOpen Labelへの登録を延期することを選択することができ、すべての参加者はAMX 0035を獲得し、最長でもう1年延長することができる。
オリオン座は全世界の主要な学術リーダー、PSP患者及びその介護者及び業界と組織協力設計と計画を提唱したものである。
PSPは1種の散発的、稀な成人発病の神経変性疾患であり、歩行とバランス、眼球運動、嚥下と言語に影響する。世界の10万人に7人がこの病気に感染していると報告されている。PSPを有する人の初回診断後の期待寿命は6~8年であり,PSPは通常中末期から始まり,時間の経過とともに急速に発展する。PSPの治療法は現在のところ承認されていない。
臨床前データはAMX 0035固定用量組み合わせ中の2つの小分子ベンゼン酪酸ナトリウムPBとタウリンジオールTursoによるPSP治療の潜在力を支持した。様々な体外培養実験中、フェニルブタン酸ナトリウムはパートナー蛋白を上昇と募集し、蛋白質の折り畳みを安定させ、小胞体ストレスと折り畳まれていない蛋白質反応を減少させ、これは細胞アポトーシスを招くことができ、圧力が圧倒的であれば。PSPの研究により、PSP中の未フォールディング蛋白反応は疾病影響領域で活性化され、遺伝的証拠はこの活性化は保護性反応ではなく、PSP発生の危険因子であることを表明した。Tursoは細胞死調節因子Baxの転座を減少させることでミトコンドリア膜を安定させ,ミトコンドリア機能とエネルギー産生を改善し,アポトーシス閾値を向上させる。PSPでは,PSP患者からの細胞質細胞系およびPSP患者からのニューロンはミトコンドリア機能障害を示した。
PSPは異常なtau包有体が特徴であることからtau病とも呼ばれる。他の神経退行性疾患と類似し、PSPの病理生理変化は多要素であり、いくつかの遺伝と環境要素はtau機能障害と凝集を招く可能性がある。遺伝子突然変異、小胞体ストレスと未フォールディング蛋白反応の活性化、ミトコンドリア機能障害と神経炎症を含む多種の経路はtau機能障害と凝集の原因と考えられている。AMX 0035によるAD治療の第2段階フライ馬試験の臨床前データおよびバイオマーカー分析によると、AMX 0035はtauおよび他の神経変性マーカーのレベルを有意に低下させることが証明された。
AMX 0035治療WSの臨床研究進展
我々は2020年11月にFDAがWSを治療する孤児薬としてAMX 0035を承認したと発表した。2023年3月,WSに対するAMX 0035治療の第2段階臨床試験の現場活性化を完了し,2023年4月に1人目の参加者が用量を受けたことを発表した。この試験は探索的な開放ラベル生物学的研究であり、AMX 0035の安全性と耐性の効果、及び内分泌、神経と眼科機能の各種指標を評価する。2024年試験のTOPLINE結果を予定している。
セントルイスワシントン大学医学院の研究者はAmylyxと協力し、臨床前データを発表し、WSを治療する新しい方法としてのAMX 0035の潜在力を探索した。これらのデータは同業者が評議したものです臨床研究雑誌WFS 1遺伝子の発病変異(WFS 1 c.1672 C>T,p.R 558 C)を確定し、更なる遺伝子型-表現型分析にプラットフォームを提供し、そしてWSにおけるAMX 0035の治療開発に初歩的な概念検証を提供した。この研究により、IPSC由来のWSモデルは臨床観察に関連する遺伝子型-表現型関係モデルを提供できることを表明した。AMX 0035に関する研究の要点は、以下のとおりである
WSは常染色体劣性遺伝性神経変性疾患であり、児童期に発症する糖尿病、視神経萎縮と神経変性を特徴とする。WSのよく見られる所見は,糖尿病,視神経萎縮,中枢性尿崩症,感音神経性難聴,神経原性膀胱,進行性神経機能障害である。WSの予後
17
貧困、この病気を患っている多くの人たちは深刻な神経障害で早世した。WSはまれで生命にかかわる小児科疾患であり,WS WFS 1遺伝子(あるいはWFS 1)の変異によるものと考えられ,一部の患者ではCDGSH鉄チオドメイン蛋白2 CISD 2遺伝子(あるいはCISD 2)の原因変異によるものである。WS用の薬剤は現在のところ承認されていない。
WSは小胞体ストレスの疾患のようである。WFS 1は重要なwolframin蛋白をコードし、産生し、この蛋白は小胞体制御過程に参与しているようである。WFS 1欠乏は慢性小胞体ストレスとUPRを引き起こす。WFS 1はまたUPR分子を負に調節して転写因子6(ATF 6)を活性化し,細胞死を招く。また,最近の研究では,WFS 1はミトコンドリア関連ER膜(MAM)を介して小胞体からミトコンドリアにカルシウムイオンを輸送し,ミトコンドリアの機能に影響することが示唆されている。
UPRを含むWSの中心経路を標的とするAMX 0035は、細胞モデルおよび患者由来細胞系モデルを含む様々なWSモデルにおいて有益な効果を示す。例えば、WSの背景で小胞体ストレス調節におけるAMX 0035の潜在的な作用をテストするために、PB、TursoおよびAMX 0035の効果が試験された体外培養野生型およびWFS 1欠陥のランゲルハンス島β細胞系モデル。これらの細胞のうち、PBまたはTURSO単独ではなく、対照群と比較してAMX 0035のみが、衣マイシン誘導性WFS 1欠損ランゲルハンス島β細胞株の細胞死を有意に阻止することができ、caspase 3/7活性測定(p=0.017)。また,PBとTursoの組合せも検討した体外培養ヒト患者由来の神経前駆細胞では,WFS 1に変異が存在し,WSを引き起こす。対照条件と比較して,PBとTurso単独応用では3種類の異なるヒト細胞株における細胞死を抑制することができ,PBやTurso単独処理と比較して,PBとTursoの併用は3つの単独の患者由来WS細胞株の細胞死レベルを著しく低下させ,これらの細胞は患者由来神経前駆細胞に分化した。したがって,WSの関連モデルでは,対照群やPBまたはTurso単独処置と比較してAMX 0035が細胞死を低下させる相乗効果が観察された。これらの理由から,AMX 0035は有望なWS臨床候補薬であると考えられる。
AMX 0035 AD治療の臨床研究進展
2021年に、私たちのPegasus第二段階試験の結果を完成し、報告し、末期軽度認知障害(MCI)または早期から中等度痴呆患者におけるAMX 0035の安全性、耐性および活動度を評価した。この試験の目的は,ADや他の神経変性疾患に関連するバイオマーカーデータを収集し,ADにおけるAMX 0035の開発を支援することである。
Pegasus試験はランダム、プラセボ対照の第2段階試験であり、95人の米国参加者が参加した。患者はランダムに3~2群に分けられ、それぞれAMX 0035、1つの香包(各香包は1グラムTursoと3 g PBを含む)の治療を受け、24週間以内に1日2回服用するか、または一致したプラセボを服用した。
Pegasus試験の首席研究員Steven Arnold博士は2021年第4四半期に開催されたアルツハイマー病臨床試験会議(CTAD)でPegasus試験の主な結果を示した。これらのTOPLINE結果によると、AMX 0035の全体的な耐性は良好であり、約80%の患者はAMX 0035 ARMの試験で用量を完成した。セキュリティ効果を図に示す.Centaur試験と同様に,AMX 0035を服用した患者のうちより高い割合の患者に胃腸不良が出現した。しかしながら、Pegasus試験では、SAEはAMX 0035に起因しなかった。
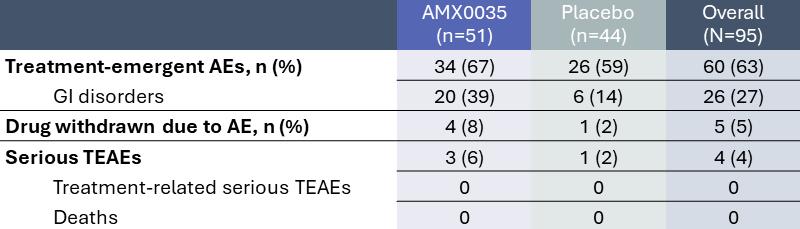
6ヶ月間のこの試験は、認知、機能、またはイメージングにおけるAMX 0035およびプラセボ腕の差を評価しなかった。
18
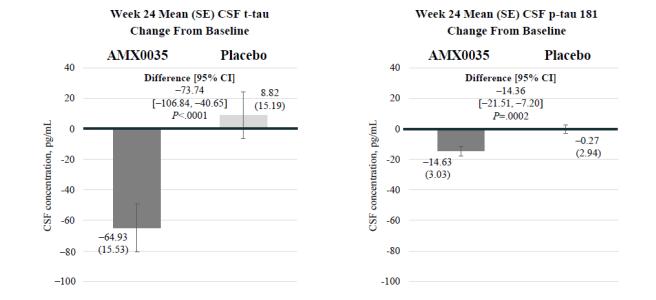
この試験の探索的目標は,すべてのボランティアの脳脊髄液中のアミロイド1−42,アミロイド1−40,総tau(t−tau),トレオニン181リン酸化tau(Ptau 181),神経元損傷マーカー,ミトコンドリア酸化還元と機能マーカーおよび神経炎症に対するAMX 0035治療の影響を測定することである。
機能磁気共鳴分析はまだ進行中であるが,本試験では各用量群間に治療効果終点の有意差は認められなかった(p>0.05)。重要な治療効果の結果を以下の図に示す
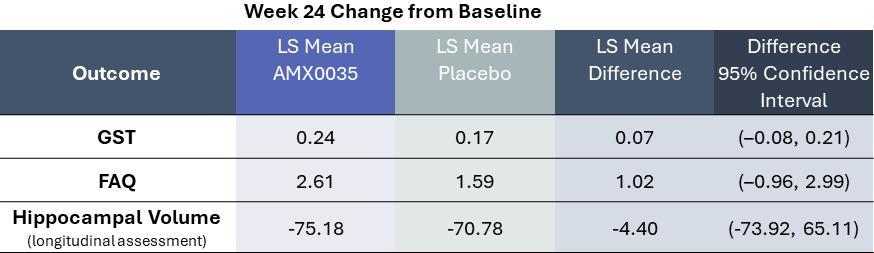
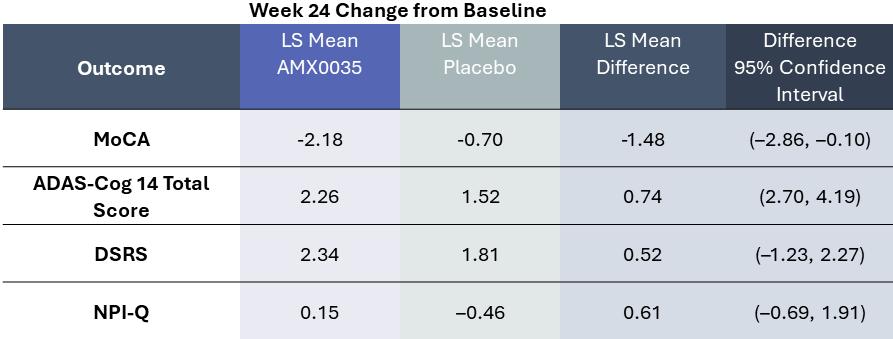
a海馬体の体積成分は標準的なADNI MRIアルゴリズムに基づいているが,統計解析計画に含まれる他のMRIアルゴリズムによる評価も行い,類似した結果が得られた。
19
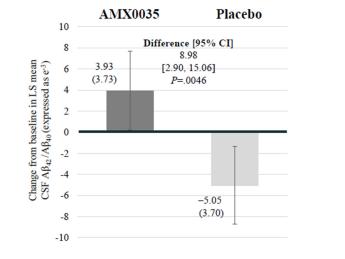
ADに興味のある複数のバイオマーカーへの有意な影響が試験的に観察された。脳脊髄液では,AMX 0035群のtau蛋白181の発現が有意に減少した(p
実験したバイオマーカーとイメージング結果は実質的に改善されたと信じており、AD進展に関連する神経変性経路に対するAMX 0035の影響を理解するために情報を提供し続ける。これらの結果を使用して、ADおよび他の潜在的適応における我々のAMX 0035の臨床開発戦略を評価し、参考を提供し続ける。我々は現在,これらのデータを評価し,科学コンサルタントとPegasus試験の結果を検討するとともに,われわれの臨床開発戦略においてADを治療するAMX 0035の潜在的な次のステップを開発することを考えている。
AMX 0035新剤形による筋萎縮性側索硬化治療の臨床研究進展
我々はAMX 0035の新しい味覚マスキング製剤を研究している。この提案法は新しい知的財産権を生成するかもしれない。
AMX 0114筋萎縮性側索硬化治療の臨床研究の進展
ALSの治癒には,疾患発症機序に関与する多くの細胞経路に対して併用する方法が必要であると考えられる。この仕事の一部として、軸索(ウォラー)変性経路の重要な貢献者であるCalain-2をコードする遺伝子に対するAMX 0114、アンチセンスオリゴヌクレオチドを研究している。
軸索変性はすでにALSと他の神経退行性疾患の臨床表現と発病機序の肝心な早期要素と考えられている。カルシウム依存性蛋白加水分解酵素Calain−2の活性化は軸索変性の重要な効果因子と考えられている。Calain−2のALS動物モデルにおける治療作用や,ALSで広く研究されているバイオマーカーであるCalain−2の切断神経糸における作用や,死後ALS組織におけるCalain−2とその切断産物レベルの上昇の発現は,ALSの発症機序に関与していると考えられている。
これまでに完成した臨床前研究により、AMX 0114は有効、用量依存と持続的なノックアウトを実現したことが明らかになったCAPn 2ヒト運動ニューロンのmRNA発現とcalain−2蛋白レベル。さらに、AMX 0114治療は、IPSC由来のヒト運動ニューロンの神経毒性損傷後の細胞外神経糸軽鎖(NFL)レベルを低下させ、AMX 0114治療は、ALS連鎖病原性TDP-43変異を有するIPSC由来のヒト運動ニューロンの生存率を増加させる。
AMX 0114は研究性新薬やINDにより研究が進んでおり,2024年にINDが提出され臨床に入る予定である。
20
筋萎縮性側索硬化症総合診断バイオマーカーの開発
筋萎縮性側索硬化症を有する人は約3分の1の経過をかけて診断を探した。筋萎縮性側索硬化症の診断遅延の重要な駆動要素の一つは信頼性が乏しく、有効なバイオマーカーが不足して診断を補助することである。早期診断を支援する技術は,ALSの看護と治療の推進やALS患者とその家族が長い診断過程で経験した重大な心理的ストレスを緩和するために重要である。
ALSの早期診断促進を目的とした新たな複合バイオマーカーの開発に努めている。バイオマーカーの性能を仮定した情報を提供し、候補バイオマーカーを決定し、後続の検証研究の設計に情報を提供するパイロット研究が行われている。
患者代弁者
筋萎縮性側索硬化症とその他の神経変性疾患の患者の提唱構造は巨大かつ複雑な総合ネットワークであり、国際、多区域と特定の国家レベルの集団をカバーしている。我々はすでにこれらの複雑なネットワークを介して国際レベルで信頼性と信頼できるパートナーシップを構築しており,現在我々の重点は米国,カナダ,ヨーロッパ,アジア太平洋地域のALS提唱団体である。
重要な提唱団体との協力は私たちの使命に重要です。これらの神経変性疾患を患っている人とその家族が私たちがしているすべての中心だからです。これは私たちの科学、データ、そして開発計画に対する透明なコミュニケーションと認識から始まる。これらの提唱団体が知ることを確保し,その有権者の質問に答えることができ,コミュニティに利益を与える機会や政策を適切に提唱することを求めている。
競争
生物技術と製薬業界の特徴は技術進歩が迅速で、競争が激しく、特許製品を強調することである。著者らは主要と専門製薬と生物技術会社、学術研究機構、政府機関及び公共と個人研究機関を含む多くの異なる源からの潜在的な競争に直面している。我々は、AMX 0035を含む任意の候補製品の開発および商業化に成功し、現在の療法および将来発売される可能性のある新しい療法と競合する可能性がある。私たちのすべての候補製品の成功に影響を与える重要な競争要素は、治療効果、安全性、用量、コスト、販売促進支援の有効性と知的財産権保護を含むと信じている。
私たちの多くの競争相手は、単独あるいはパートナーと協力しても、研究開発、臨床前試験、臨床試験、製造とマーケティング方面の財力と専門知識はすべて私たちよりずっと多い。将来の協力とM&Aは、より少ない数の競争相手に資源をさらに集中させる可能性がある。私たちの競争相手も私たちよりも早くFDAや他の規制機関からその製品の承認を得ることができ、これは私たちの競争相手が私たちが市場に入る前に強力な市場地位を確立したり、私たちの開発をもっと複雑にしたりする可能性がある。これらの競争相手はまた臨床試験のために類似の合格科学と管理人材バンク、場所と患者集団、及び著者らの計画のために補充或いは必要な技術を奪い合う可能性がある。
供給と製造
私たちは依存し、予測可能な未来には、商業供給のための、および我々の組織メンバーの指導の下で臨床試験のための、現在の良好な製造プロセスまたはcGMP要件に適合するAMX 0035およびAMX 0114を製造するために、第三者契約製造組織またはCMOに依存し続けることが予想される。AMX 0035については,第三者メーカーから製造·配布された原料薬PBとTursoの2種類の活性医薬成分を用いた。私たちは、これらの供給者の参照に関連する薬物マスターファイルを許可することを含む、これらの原料薬のための長期的、単一源の供給契約を締結した。著者らは既存のCMOで商業供給、臨床試験とその他の潜在的な需要のために単一源の原料薬の生産と包装手配を制定した。私たちはPatheon Inc.またはセマー飛世爾の子会社PatheonでAMX 0035原料薬を生産し、カナダのホイットビーに位置している。私たちは第三者製造能力を拡大しており、この能力はビジネスニーズをサポートし続けると信じており、AMX 0035製造2025年までをカバーする協定に署名しました。生産後,バルク薬品はその後イリノイ州ロックフォードに位置するPCI Pharma Servicesに1回と二次包装された。米国以外の市場に目を向けるにつれ、現地の市場ニーズを支援するために、より多くの製造·流通場所を増やす計画だ。また,リスクに基づく方法を利用して,必要に応じてより多くの製造場所を導入する。
21
私たちは製薬業界の技術運営担当者で構成されたチームを設立した。このチームは豊富な技術、製造、分析、品質、監督管理(cGMPを含む)とプロジェクト管理経験を持っており、私たちの第三者メーカーを監督し、品質と監督管理のコンプライアンスを維持することができる。また、同チームのメンバーは世界的に珍しい疾患製品の商業化と発売にも参加している。事業化が進むにつれて、私たちは私たちの技術運営チームを作り続ける予定だ。
私たちはまた、規制された業界と一致した品質管理システムを持っており、このシステムは、私たちのCMO監督を管理する標準的な操作政策とプログラムを概説している。
Patheonと製造合意に達しました
2019年11月、Patheonは、cGMP製造、品質管理、品質保証、安定性テスト、パッケージ、および関連サービスを提供するPatheon、Inc.またはPatheonとプライマリ製造サービス契約または製造契約を締結しました。私たちは“製造協定”に基づいてAMX 0035を含む予備製品協定に署名した。製造契約の初期期限は2025年12月に終了し、有効な製品契約が存在すれば自動的に更新され、継続期間は最後の有効な製品契約の終了時に終了する。AMX 0035をカバーする製品契約の初期期限は2025年12月に終了し、いずれか一方が審査しようとしないことを事前に通知しない限り、2年間の連続期限を自動的に更新する。
私たちは製造契約または任意の製品協定を終了することができます:任意の政府または監督機関がカナダ、EU、またはアメリカでAMX 0035を販売することを永久的に阻止する場合、またはもし私たちがこれ以上製造サービスを注文するつもりがなければ、もし私たちが製造サービスを注文しようとすれば、AMX 035‘S’が市場での販売を停止すれば、30日前に書面で通知することができます。Patheonは、製品契約期間内に12ヶ月連続で製品数がゼロになることを前提として、製造契約項目の任意の製品合意を終了することを30日前に書面で通知することができます。さらに、Patheonが不払いにより製造サービスを一時停止してから30日以内に全額支払いを受けていない場合、または場合によっては、製造契約または製品契約のいずれかの権利を譲渡する場合、Patheonは9ヶ月前に書面通知を出して、製造契約または任意の製品合意を終了することができる。また、いずれの当事者も、他方が治癒されていない実質的な違約を含む製造契約または任意の製品協定を終了することができ、他方が債務または破産を支払うことができない場合には、書面による通知を介して終了することができる。
CU Chemieと締結された供給契約
2019年10月に,吾らはCU Chemie Utikon,GmbHやCU Chemie(SEQENSグループの支社)と供給協定を締結し,これにより,CU ChemieはAMX 0035の製造のために原料薬PBを非独占的に吾等に供給することに同意した。この協定の初期期限は5年であり、早期終了しない限り、2年連続の期限を自動的に更新する。満了後、便宜上、いずれか一方は3ヶ月前に書面で合意を終了することを通知することができる。また、いずれか一方が他方が治癒していない実質的な違約の場合、または他方が倒産または破産した場合には、合意を終了することができる。
大陸間取引所と締結した供給協定
2023年8月、ICE S.p.A.またはICE(従来のProdotti Chimici e Alimentari S.p.A.)と商業供給契約を締結し、この協定に基づいて、ICEはAMX 0035を製造するためのタウリンデオキシコール酸原料薬を独占的に供給することに同意した。この協定はいくつかの最低購入量要件を含む。この協定の初期期限は2028年12月に終了し、共同指導委員会が更新を決定することができる。どちらか一方が借金を返済できない場合や破産した場合、または他方の違約が是正されていない場合には、合意を終了することができる。
知的財産権
PBおよびTurso分子は我々の特許ではないが、AMX 0035自体の固定用量の組み合わせを含むAMX 0035をカバーする特許および特許出願を有している。
知的財産権は私たちの分野と一般的な製薬産業で必須的だ。私たちのビジネスの成功は、AMX 0035とその用途、および任意の未来の製品の知的財産権を保護する能力にある程度依存します
22
候補者です。我々は、特に米国および外国の特許権を求め、維持し、擁護することによって、当社の業務およびAMX 0035の発展に重要なビジネス的意義を有するノウハウ、発明、および改善を保護し、強化することを求めている。
私たちはAMX 0035とAMX 0114の周りを含む、私たちの治療分野の知的財産権の組み合わせを積極的に確立している。私たちの現在の特許の組み合わせは8つの特許シリーズを含む。これら8つのシリーズのうち、我々は現在、AMX 0035を含む、我々の技術のための発行された特許および係属中の122個の特許出願を有している。現在、我々の特許組み合わせには、発行された米国特許6件、発行された外国特許58件、係属中の米国特許出願14件、係属中の外国特許出願44件が含まれている。我々が発行した特許および処理される出願は、フェニルブチレート化合物および胆汁酸(例えばTUDCA)の相対数をカバーしており、私たちが発表および未解決のいくつかのクレームは、これら2つの薬剤の特定の割合をカバーしている。
我々の最初の特許ファミリーは、Tursoおよび4−PBAを含む胆汁酸およびフェニルブチレート化合物の組成物、およびこれらの組成物を用いて神経変性疾患およびその関連原因を細胞レベルで治療する方法に関する。この一連は、4つの許可された米国特許および58個の許可された外国特許(私たちが許可した欧州特許が発効した国/地域の権利を含む)を含む。私たちが海外で特許を取得した司法管轄区域は、アルバニア、オーストリア、オーストラリア、ボスニア·ヘルツェゴビナ、ベルギー、ブルガリア、中国、クロアチア、キプロス、チェコ共和国、デンマーク、エストニア、EU、フィンランド、フランス、ドイツ、ギリシャ、香港、ハンガリー、アイルランド、アイスランド、イタリア、日本、リトアニア、ラトビア、マカオ、マケドニア、マルタ、モナコ、モンテネグロ、オランダ、ノルウェー、ポーランド、ポルトガル、ルーマニア、サンマリノ、セルビア、スロバキア、スロバキア、韓国、スペイン、スウェーデン、スイス、トルコ、イギリスを含む。私たちはまたオーストラリア、カナダ、EU、マカオ、日本、韓国、アメリカと他の司法管轄区で特許申請を待っています。この家族では,米国(米国特許番号11,071,742,2021年7月27日発行)とオーストラリアで発表された物質がクレームを構成している。これらの発行された特許およびその家族が発行した他の特許は、早ければ2033年12月に満期になる可能性がある。
我々の第2の特許シリーズは、フェニルブチレート化合物および胆汁酸(Tursoおよび4−PBAを含む)の特定の組成物およびその製造方法に関する。その家族は許可された2つのアメリカ特許を含む。私たちはアメリカ、EU、そして他の司法管轄区域でもこの一連の特許出願が審理されている。この一連の中で、米国、アルゼンチン、オーストラリア、ブラジル、カナダ、中国、EU、香港、イスラエル、日本、韓国、メキシコ、台湾が提出した申請で決定すべき事項クレームの構成がある。この家族が発行した特許と他の特許は早ければ2040年7月に満期になる可能性がある。
我々の第3の特許シリーズは、筋萎縮性側索硬化症を治療するための特別な症状および/またはフェニルブチレート化合物と胆汁酸(Tursoおよび4-PBAを含む)との組み合わせによって関連する有害事象を減少させる方法である。私たちはアメリカ、EU、そして他の司法管轄区域でこの一連の特許出願が審理されている。現在、私たちはこの家族で未解決の問題に対するクレームを持っていない。このシリーズはまだ特許が発行されていないにもかかわらず、このシリーズが発行する特許の有効期限は少なくとも2040年8月まで延長されると予想される。
我々の第4の特許シリーズは、フェニルブチレート化合物と胆汁酸(ベンゼン酪酸ナトリウムおよびTursoを含む)との組み合わせを用いてアルツハイマー病および他の進行性核上性麻痺を治療する方法を対象としている。私たちはアルゼンチン、台湾でこの一連の処理されている特許出願と、特許協力条約(PCT)出願を持っている。このシリーズはまだ特許が発行されていないにもかかわらず、シリーズが発行する特許の有効期限は少なくとも2042年11月まで延長されると予想される。
我々の第5の特許シリーズは,他の治療薬(チトクロームP 450を含む基質,治療指数の狭い薬物および有機陰イオントランスポーター1(OAT 1)の基質)を,フェニルブチレート化合物および胆汁酸(ベンゼン酪酸ナトリウムおよびTursoを含む)と組み合わせて使用する方法を対象としている。私たちはアメリカ、アルゼンチン、台湾でこの一連の特許出願が出願されており、特許協力条約(PCT)出願もある。このシリーズはまだ特許が発行されていないにもかかわらず、シリーズが発行する特許の有効期限は少なくとも2042年3月まで延長されると予想される。
我々の第6の特許シリーズは,トルソとフェニルブタン酸ナトリウム投与後の薬物動態学的特徴を対象としている。私たちはアメリカ、アルゼンチン、台湾でこの一連の特許出願が出願されており、特許協力条約(PCT)出願もある。このシリーズはまだ特許が発行されていないにもかかわらず、シリーズが発行する特許の有効期限は少なくとも2042年5月まで延長されると予想される。
我々の第7特許シリーズは、他の治療薬(胆塩外排出ポンプ(BSEP)の阻害剤を含む)を、フェニル酪酸化合物および胆汁酸(ベンゼン酪酸ナトリウムおよびTursoを含む)と組み合わせて使用する方法を対象としている。アメリカ、アルゼンチン、台湾でこの一連の特許出願が行われています
23
特許協力条約(PCT)出願。このシリーズはまだ特許が発行されていないにもかかわらず、シリーズが発行する特許の有効期限は少なくとも2042年5月まで延長されると予想される。
我々の8番目の特許ファミリーは,calain−2転写産物に対するオリゴヌクレオチドを対象としている。私たちは米国でこの一連の処理されている特許出願と,特許協力条約(PCT)出願を持っている。このシリーズはまだ特許が発行されていないにもかかわらず、シリーズが発行する特許の有効期限は少なくとも2043年5月まで延長されると予想される。
私たちは私たちの任意の未解決特許出願または私たちが将来提出する可能性のあるどの特許出願にも特許が付与されることを確実にすることはできない。さらに、私たちの既存の任意の特許または将来私たちに付与される可能性のある任意の特許が、私たちの商業製品を保護する上で商業的用途を持っているかどうかを確認することはできません。最後に、私たちは、私たちが付与した特許と、将来私たちに付与された任意の特許が、訴訟または行政手続きの後に有効および/または強制的に実行されると認定されたかどうかを決定することができない。
2021年1月、ブルチェティーニS.r.l.ライデルとケラー特許パートナーMBBはそれぞれ欧州特許庁に我々が発行した欧州特許EP 2978419に対する異議を提出した。より高いレベルでは、この特許は、胆汁酸およびフェニルブチレート化合物を使用して神経変性疾患(およびその原因または条件)を治療する様々な方法を必要とする。野党部門は2022年6月2日の口頭訴訟で欧州特許EP 2978419のライセンスを維持した。反対派区画の決定は最終決定となり、EP 2978419は変わらない。EP 2978419は、2023年4月26日から発効する統一特許裁判所の管轄権から脱退することを選択された。
個別特許の期限は特許を取得した国の法的期限に依存する。私たちが出願を提出したほとんどの国では,特許期間は最初の優先権を要求する非仮出願が提出された日から20年である。米国では、特許期限は、特許期限調整によって延長することができ、これは、特許権者が特許付与時の特許付与時の行政遅延による特許権者の損失を補償することができ、または、1つの特許が以前に提出された特許によって最終的に放棄された場合、特許期限が短縮される可能性がある。米国では、ハッジ·ワックスマン法によれば、FDAによって承認された薬物をカバーする特許期限も最大5年延長する資格がある可能性があり、FDA規制審査中に失われた特許期間を補うことを目的としている。特許期間延長の長さは、規制審査に要する時間長に基づいて算出される。“ハッジ·ワックスマン法”に基づいて延長された特許期間は,製品承認日から14年間を超えてはならず,承認された薬物に適用される特許を延長することしかできない。また、1つの特許は1回しか展示期間がないので、1つの特許が複数の製品に適用される場合は、1つの製品展示期間に基づくしかない。ヨーロッパおよび他のいくつかの外国司法管轄区域にも同様の規定があり、承認された薬物をカバーする特許の期限を延長する。RELYVRIOの米国での承認を経て、私たちはすでに私たちの製品および/またはその使用方法のいくつかの発行された米国特許のために特許期間を延長しました。
私たちはまた、商標、商業秘密、技術ノウハウ、持続的な技術革新、秘密保持協定、および発明譲渡協定に依存して、私たちの独自の地位を発展させ、維持しています。セキュリティプロトコルは、当社の独自の情報を保護することを目的としており、発明譲渡プロトコルは、当社の従業員、コンサルタント、または他の第三者によって開発された技術の所有権を付与することを目的としています。我々は,我々の場所の物理的セキュリティおよび我々の情報技術やITシステムの物理的および電子的セキュリティを維持することで,我々のデータやビジネス秘密の完全性とセキュリティを保護することを求めている.私たちは私たちの合意と安全措置に自信があるが、その中のどれも違反される可能性があり、私たちは十分な救済措置を持っていないかもしれない。しかも、私たちのビジネス秘密は競争相手に知られたり独立して発見されるかもしれない。
私たちのビジネスの成功は、他人の所有権を侵害することなく運営する能力と、他人が私たちの所有権を侵害することを防止する能力にもある程度依存しています。本年度報告では“リスク要因−我々の知的財産権に関するリスク”と題する項目1 Aが知的財産権に関するリスクについて全面的に検討した
私たちは、AMX 0035が有効で成功的に断言できると思う発行された特許は、AMX 0035のいくつかの場所およびいくつかの管轄区域の特許版図を検索しましたが、AMX 0035を商業化する能力を阻止することはできませんでした。
欧州特許EP 3016654名は“神経変性疾患の治療のためのタウリンデオキシコール酸(TUDCA)”であり、ブリュッセル社が所有している。この特許は哺乳動物ALSの治療におけるTursoの使用に関する。欧州特許庁(EPO)はEP 3016654の付与に反対意見を提出し、EPOにEP 3016654の撤回を要求した。欧州特許庁は2019年11月18日に初歩的な意見を発表し、少なくともEP 3016654の主な説は新規性に欠けていることを発見した。2021年6月11日,欧州特許庁反対部で口頭手続きが行われた。はい
24
口頭手続きが終わった後、野党はEP 3016654のすべてのクレームを撤回することを決定したと発表した。書面判決が発表された;しかし、ブルチェティーニは野党支部の決定を控訴委員会に上訴した。ブルーチェティーニの控訴は2022年6月7日に控訴の却下を求め、反対党支部がEP 3016654のすべてのクレームを撤回する決定を維持するために応答した。控訴委員会は、2023年5月24日に口頭尋問に出席することを要求する召喚状を出した。口頭審理は2024年6月5日に行われる予定だ。
政府の監督管理
FDAおよび州および地方司法管轄区および他の国および地域の同様の規制機関は、カナダおよびEU加盟国を含み、薬物の臨床開発、製造、マーケティング、流通に参加する会社に対して、例えば、我々が開発している会社のような要求を行う。これらの機関と他の連邦、州と地方実体は私たちの候補製品の研究開発、テスト、製造、品質管理、安全、有効性、ラベル、貯蔵、記録保存、承認、広告と販売促進、流通、承認後の監視と報告、サンプリングと輸出入などの方面に対して監督管理を行う。
アメリカ政府の薬品の監督管理
米国では,FDAは連邦食品,薬物と化粧品法案(FDCA)とその実施条例に基づいて薬品を規制している。規制の承認を得て、その後、適用される連邦、州、地方、外国の法規と条例を遵守する過程には、多くの時間と財政資源が必要だ。製品開発プロセス、承認プロセス、または承認後の任意の時間において、出願人が適用される米国の要求を遵守できない場合、FDAが係属中のNDAの承認を拒否する、承認の撤回、臨床棚上げの実施、警告状の発行、製品のリコール、製品の差し押さえ、生産または流通の完全または部分的な一時停止、禁止、罰金、政府契約の拒否、原状回復、返還、または民事または刑事罰のような様々な行政または司法制裁を受ける可能性がある。
FDAが米国で発売される前に必要とされるプログラムは、一般に以下のような態様を含む
臨床前研究
臨床前研究には、製品の化学、毒性、処方の実験室評価、および体外培養潜在的な安全性と有効性を評価するために動物研究を行っています臨床前研究の進行は連邦法規と要求の制約を受け、安全/毒理学研究の良好な実験室操作規程を含む。
25
INDスポンサーは,臨床前試験の結果を生産情報,分析データ,任意の利用可能な臨床データや文献,臨床研究計画などとともにFDAに提出し,INDの一部としなければならない。INDはFDAがヒトに研究製品の使用を許可する要求であり,ヒト臨床試験が開始される前に発効しなければならない。いくつかの前臨床試験、例えば生殖不良事象や発ガン性の動物試験は、IND提出後も継続する可能性がある。INDはFDAが受信した30日後に自動的に発効し、それ以前にFDAが1つまたは複数の提案された臨床試験に対して懸念または問題を提起しなければ、臨床試験を保留する。この場合,INDスポンサーやFDAは臨床試験開始前に未解決の問題を解決しなければならない。したがって,INDの提出はFDAが臨床試験の開始を許可しない可能性がある。
臨床試験
臨床試験は、CGCP要求に適合する合格した研究者の監督の下で、研究中の新薬をヒト被験者に投与することに関連し、すべての研究対象に任意の臨床試験に参加するためのインフォームドコンセントを書面で提供することを含む。臨床試験は,試験目標を詳細に説明し,安全性をモニタリングするためのパラメータと評価する有効性基準のシナリオで行った。INDの一部として,各臨床試験の案と任意の後続の案修正案をFDAに提出しなければならない。さらに、臨床試験に参加する各機関を代表するIRBは、その機関が任意の臨床試験計画を開始する前に、計画を審査し、承認しなければならない。IRBはまた、各臨床試験対象またはその法律代表に提供されなければならないインフォームドコンセントを審査および承認し、完成まで臨床試験を監視しなければならない。
ある臨床試験に関する情報は,そのwww.Clinicaltrials.govサイト上で公開されるために,特定の時間枠で米国国立衛生研究院(NIH)に提出されなければならない。臨床試験登録の一部として,臨床試験の製品,患者群,調査段階,研究場所や研究者,その他に関する情報が公開されている。スポンサーは試験完了後に臨床試験結果を開示する義務があるが,場合によっては結果の開示が試験完了日後2年に延期される可能性がある。対象の臨床研究や法律規定の研究結果を適時に登録できなかったことは民事罰金を招く可能性があり、また違反側が連邦政府の将来の支出を獲得することを阻止する。NIHのClinicalTrials.gov登録と報告要件に関する最終規則が2017年に発効し,NIHとFDAは最近,政府が要求に適合しない臨床試験スポンサーに対してこれらの要求を実行し始めることを希望していることを示している。
人体臨床試験は通常3つの連続段階に分けて行われ、この3つの段階は重なる可能性があり、合併する可能性もある
承認後試験は,4期臨床試験と呼ばれることがあり,最初の上場承認後に行われる可能性がある。これらの試験は,治療適応が予想される患者の治療から追加的な経験を得るために用いられている。場合によっては,FDAはNDA承認条件として4期臨床試験を強制的に要求する可能性がある。
臨床試験結果を詳細に説明する進捗報告は少なくとも毎年FDAに提出しなければならず,SAEが発生すればより頻繁に提出される。深刻かつ意外な疑わしい有害事象、他の研究または動物または動物からの発見については、FDAおよび調査者にIND安全報告書を提出しなければならない体外培養方案或いは研究者マニュアルに記載されたテストと比べ、人類被験者に重大なリスクがあるテスト、及び任意の臨床上重要な深刻な疑似副作用の発生率の増加を表明した。スポンサーは15日以内にINDセキュリティ報告書を提出し,スポンサーがその情報有資格報告を確定した後でなければならない。スポンサーは何か事故をFDAに通知しなければなりません
26
発起人が最初にメッセージを受け取った後の7つの暦内に致命的或いは生命に危害を及ぼす疑いの副作用が発生した。
第1段階、第2段階、および第3段階試験は、任意の指定された期限内に成功しないか、または全く成功しない可能性がある。また、FDAやスポンサーは、研究対象が受け入れられない健康リスクに直面していることを発見することを含む、随時様々な理由で臨床試験を一時停止または終了することができる。同様に、1つの臨床試験が委員会の要求に従って行われない場合、または薬剤が患者に予期せぬ深刻な傷害を与えた場合、IRBは、その所在機関の臨床試験の承認を一時停止または終了することができる。さらに、いくつかの臨床試験は、データ安全監視委員会または委員会と呼ばれる臨床試験スポンサーによって組織された独立した合格専門家グループによって監督される。このグループは,実験のあるデータへのアクセスにより,許可試験が指定されたチェックポイントで行えるかどうかを決定する.
臨床試験と同時に、会社は通常追加の動物研究を完成し、薬物化学と物理特性に関するより多くの情報を開発し、cGMPの要求に基づいて最終的に商業量産製品のプロセスを決定しなければならない。製造過程は一貫して高品質のロット製品を生産できる必要があり、また、会社は最終製品の特性、強度、品質、純度をテストする方法を開発しなければならない。また,適切な包装を選択·試験し,薬物が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
治療のための薬を研究する機会を広げる
使用を拡大することは、“同情的使用”と呼ばれることがあり、臨床試験以外に研究製品を使用し、比較可能または満足できる代替治療案がない場合には、重篤または直ちに生命を脅かす疾患または条件を有する患者を治療する。参入拡大に関する規則や条例は,研究療法の恩恵を受ける可能性のある患者が研究製品を獲得する機会を改善することを目的としている。FDAの法規は、個別患者(緊急時および非緊急時に治療された単一患者IND申請)、中規模の患者集団、および治療レジメンまたは治療INDに従って研究製品の使用を申請したより大きな集団のために、会社または治療医がINDの研究製品を治療目的で使用することを可能にする。
INDが患者または患者のグループを治療するために研究製品の使用を拡大する申請を審議するとき、スポンサーおよび治療医または調査者は、患者(S)が深刻または直ちに生命に危険な疾患または状態を有し、疾患または状態を診断、監視または治療するための類似または満足できる代替療法がなく、潜在的な患者利益は治療の潜在的リスクが合理的であり、潜在的リスクが治療すべき背景または条件で不合理ではないことを証明するすべての基準が適用される場合に適切であるかどうかを決定するであろう。要求された治療に対して、研究薬物の使用を拡大することは、製品の発売承認を支持する可能性のある臨床研究の起動、進行または完成、または他の方法で製品の潜在的開発を損害する可能性がある。
スポンサーはその薬品をより多くの人に提供する義務はないが、2016年に採択された“21世紀治療法”の要求に基づいて、スポンサーが獲得拡大の要請にどのように対応するかに関する政策があれば、この政策を公開しなければならない。スポンサーは、2期または3期試験の開始が早いとき、または研究薬または生物学的薬剤が突破的療法、迅速チャネル製品または再生医学高度療法として指定された後15日以内にこのような政策を開示しなければならない。
また、2018年5月30日には、“裁判権法案”が法律に署名された。他の事項に加えて、この法律は、ある患者に連邦フレームワークを提供し、彼らが第1段階の臨床試験を完了し、FDAの承認を得るために調査を行っているいくつかの研究製品を使用することを可能にする。場合によっては、条件を満たす患者は、臨床試験に参加することなく、FDA拡大参入計画に従ってFDA許可を得ることなく治療を求めることができる。“試用権法案”によると,メーカーはその研究製品を条件に適合した患者に提供する義務はない。
NDA提出と上場承認
必要な臨床試験が成功したと仮定すると、臨床前および臨床研究の結果は、製品の化学、製造、制御、および提案されたラベルなどに関する詳細な情報と共にFDAに提出され、NDAの一部として、製品を1つまたは複数の市場に使用することの承認を要求する
27
兆候があります。ほとんどの場合、秘密保持協定を提出するには高額な申請使用料を支払う必要がある。FDAは最初に秘密保持プロトコルの完全性を審査し,それの“届出”を受け入れる.FDAの手続きによると、当該機関は、NDAを受信してから60日の間、申請届出を受けるか否かを決定し、これは、当該機関が申請が十分に完全であり、実質的な審査を行うことができると判断した敷居に基づいている。現在発効している処方薬使用料法案(PDUFA)ガイドラインによると,FDAは標準NDA提出日から10カ月以内に,新たな分子実体を審査提出させて行動させることと,優先審査した新分子実体NDA提出日から6カ月以内を目標としている。したがって,NDAがFDAに提出された日から,この審査過程は通常それぞれ12カ月と8カ月を要する。FDAは、そのPDUFA規格または優先NDAの目標日を常に満たすわけではなく、審査プロセスは、FDAがより多くの情報を提供または明確にすることを要求することによって延長されることが多い。FDAは秘密保持プロトコルを審査し,他の事項に加えて,薬物がその期待用途に対して安全に有効であるかどうかを決定し(S),後者の決定は大量の証拠に基づいている。この発見は、2つの十分かつ良好な制御の研究によって確認することができ、または場合によっては、非常に説得力のある単一、大型、多中心、十分かつ良好な制御に基づく研究、または良好な研究および検証的証拠を十分かつ制御することから来る。FDAの法規とガイドラインはまた、まれで致命的な疾患を背景に不確実性に対してより大きな柔軟性と寛容度を有することを可能にしている。FDAはまた、製品の製造、加工、包装、または製品を保有する施設が、製品の持続的な安全、品質、および純度を確保するための基準に適合しているかどうかを評価する。
さらに、改正された2003年の“小児科研究公平法”によれば、いくつかの新薬または新薬補充剤は、すべての関連する小児科亜群において主張される適応の安全性および有効性を評価し、製品に対して安全かつ有効な各小児科亜群の投与および投与をサポートするのに十分なデータを含まなければならない。FDAは、成人のために製品が使用されるか、または小児科データ要件を完全にまたは部分的に免除するまで、申請者の要求に応じて、または小児科データの一部または全部の提出を延期することを許可することができる。薬物としてマーケティング申請を提出するスポンサーは、第2段階会議の終了後60日以内に予備小児科研究計画を提出しなければならず、そのような会議がない場合、第3段階または第2/3段階研究を開始する前に早期に予備小児科研究計画を提出しなければならない。最初の小児科研究計画は、研究目標および設計、年齢群、関連する終点および統計方法、またはそのような詳細な情報を含まない理由、および小児科研究データおよび支援情報の提供の要求を延期することを要求する任意の任意の要求を含む、スポンサー計画によって行われる1つまたは複数の小児科研究の大綱を含まなければならない。FDAとスポンサーは小児科研究計画について合意しなければならない。臨床前研究,早期臨床試験および/または他の臨床開発計画から収集したデータに基づいて小児科計画の変化を考慮する必要があれば,スポンサーは合意した初期小児科研究計画の修正案を随時提出することができる。PREAは孤児薬に指定された適応が付与された薬剤には適用されない。
FDAは、届出を受ける前に、提出後の最初の60日以内にすべてのNDAを予備審査して、それらが十分に完全であるかどうかを決定し、実質的な審査を行うことができる。FDAは秘密協定の申請を受け入れるのではなく、より多くの情報を提供することを要求するかもしれない。この場合、申請は追加情報と共に再提出されなければならない。再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。提出された申請が受け入れられると、FDAは深い実質的な審査を開始する。
FDAは,新薬や安全性や有効性の問題を提起する薬物の申請を諮問委員会に提出することができる。諮問委員会は,臨床医や他の科学専門家を含む独立した専門家からなるグループであり,審査,評価を担当し,申請を承認すべきかどうか,どのような条件で提案すべきかについて提案している。FDAは諮問委員会の提案に制限されていないが、それは決定を下す時にこれらの提案を慎重に考慮するだろう。
FDAはまた、薬物の利点がそのリスクよりも大きいことを保証し、薬物の安全な使用を確保するために、REMSを提出する必要があると判断した場合、リスク評価および緩和戦略の提出を要求することができる。RMSは、薬物ガイド、医師コミュニケーション計画、患者パッケージ挿入、および/または制限された配布方法、患者登録、または他のリスク最小化ツールのような安全な使用を保証する要素を含むことができる。FDAは具体的な状況に応じてREMSに対する要求および具体的なREMS条項を決定する。FDAがREMSが必要であると結論した場合,NDAのスポンサーは提案したREMSを提出しなければならない。必要であれば、FDAはREMSなしでNDAを承認しないだろう。
秘密協定を承認する前に、FDAは通常、製品を生産する1つまたは複数の施設を検査する。FDAは、製造プロセスおよび施設がcGMP要件に適合していると判断しない限り、申請を承認せず、製品が要求された範囲で一貫して生産されることを保証するのに十分である
28
規格です。さらに、NDAを承認する前に、FDAはCGCP要件に適合することを確実にするために、1つまたは複数の臨床試験地点を検査する可能性がある。
NDAとすべての関連情報を評価した後,諮問委員会のアドバイス(あれば)や製造施設や臨床試験地点に関する検査報告を含めて,FDAは承認状を発行する可能性があり,場合によっては完全な返信が発行される可能性がある。完全な返信状は、一般に、NDAの最終承認を保証するために満たされなければならない特定の条件の宣言を含み、FDAが出願を再検討するために追加の臨床または臨床前試験が必要とされる可能性がある。完全な返信が発行された場合、出願人は、秘密協定を再提出し、手紙で決定されたすべての不足点を解決するか、または出願を撤回することができる。この補足情報を提出しても、FDAは最終的にその申請が承認された規制基準を満たしていないと決定する可能性がある。もしこのような条件がFDAの満足を得たら、FDAは通常承認書を発行するだろう。この薬物の商業マーケティングを許可し、特定の適応の具体的な処方情報を提供する。
FDAが製品を承認しても、承認された製品の使用適応を制限することができ、製品ラベルに禁忌症、警告または予防措置を含むことを要求し、承認後に薬物の安全性をさらに評価するための第4段階の臨床試験を含む承認後の研究を要求することができ、製品の商業化後に製品を監視するための試験および監視計画を要求するか、または流通および使用制限またはREMS下の他のリスク管理メカニズムを含む他の条件を適用することが、製品の潜在的な市場および収益性に大きな影響を与える可能性がある。FDAは発売後の研究或いはモニタリング計画の結果に基づいて、製品の更なるマーケティングを阻止或いは制限することができる。承認後、新たな適応の追加、製造変更、および追加のラベル宣言など、承認された製品のいくつかのタイプの変更は、さらなるテスト要件およびFDAの審査および承認を受けるであろう。
孤児薬の指定と排他性
孤児医薬品法によれば、FDAは、(I)米国では200,000人未満、または(Ii)米国では200,000人を超え、米国でそのような疾患または状態の製品を開発および提供することに合理的な期待がなく、そのような疾患または状態のための製品を開発および提供するコストは、そのような製品の販売から回収される、まれな疾患または状態を治療するための医薬製品の孤児の称号を与えることができる。会社は秘密保持協定を提出する前に孤児薬物指定を申請しなければならない。この要求が承認された場合、FDAは、治療薬の識別情報およびその潜在的用途を開示するであろう。指定孤児薬は、規制審査と承認過程でいかなる利点も伝達されず、規制審査と承認過程の持続時間を短縮することもない。
孤児状態を有する製品が、そのような指定された疾患または条件を有するFDAの最初の承認を受けた場合、またはその指定されたまれな疾患または条件下で選択された適応または使用が得られた場合、製品は、孤児製品の独占経営権を得る権利がある。孤立製品排他性は、FDAが、いくつかの限られた場合に限り、同じ適応で同じ製品を販売するために、7年以内に他の出願を承認しない可能性があることを意味する。孤児薬に指定された薬物が最終的に市販承認された場合,その適応の範囲は指定された範囲よりも広く,排他性を得る資格がない可能性がある。場合によっては、孤児特許は、同じ適応を有する同じ有効成分の後続製品が、より良い治療効果または安全性に基づいて承認された製品よりも臨床的に優れていることが証明された場合、または患者ケアに重大な貢献をする場合、または孤児の薬物特許を有する会社が市場ニーズを満たすことができない場合を含む別の製品の承認を阻止しない。また、FDAは、これらの製品が異なる有効成分を含む限り、1つ以上の製品を同一の孤児適応または疾患に使用することを許可することができる。また,競合相手は孤児薬が排他的な適応を持つことで異なる製品の承認を得たり,同じ製品を獲得しているが孤児薬が排他的な異なる適応の承認を得たりする可能性がある。米国の立法者は最近、競争を促進するために“孤児薬物法案”の規制や立法改正が必要になる可能性も提案されている。これには立法の導入が含まれており、法律になれば、私たちの排他性を維持するための競争に直面している場合、AMX 0035は経済的に不可能であることをFDAに証明することが要求されるだろう。
開発と優先審査案を加速させる
FDAは新薬開発と審査を促進と加速するためのいくつかの計画を維持し、深刻或いは生命に危害を及ぼす疾病或いは条件の治療において満たされていない医療需要を満たす。これらの計画は、FDA標準の開発および審査手順よりも早く患者に提供するために、重要な新薬の開発または審査を加速させることを目的とした迅速なチャネル指定、画期的な治療指定、優先的な審査指定、および承認の加速を含む。
29
FDAはある標準に符合する新薬の審査過程を加速或いは促進することを目的としたFastTrack計画を持っている。具体的には、新薬が深刻な或いは生命に危害を及ぼす疾患を治療することを目的とし、臨床前或いは臨床データがこのような疾病が満たされていない医療需要を解決する可能性があることを示した場合、迅速なチャンネル指定を受ける資格がある。高速チャネル指定は製品にも,検討中の特定の適応にも適用可能である。スポンサーは、NDAの承認を得る前の任意の時間に、FDAに製品を高速チャネル状態に指定することを要求することができるが、NDA前の会議よりも遅くないことが好ましい。迅速チャネル指定は、スポンサーが臨床前および臨床開発中にFDAと相互作用するためにより多くの機会を提供し、さらに、マーケティング申請が提出されると、機関がスクロール審査を行うことも可能であり、これは、機関がスポンサーが完全な申請を提出する前に、一部のマーケティング申請を審査することができ、以下に説明する優先審査を行うことができることを意味する。
さらに、1つの薬剤が単独で、または1つまたは複数の他の薬剤または生物学的製品と組み合わせて、深刻または生命に危険な疾患を治療するために使用され、初期臨床証拠は、製品が1つまたは複数の臨床的に重要な終点で現在承認されている療法よりも実質的に改善されている可能性があることを示している場合、薬剤は突破的療法として指定される資格がある可能性がある。画期的な治療指定の利点には,迅速チャネル指定と同様の利点と,有効な薬物開発計画を確保するためのFDAの密な指導がある。製品が重篤または生命に危険な疾患を治療し、承認された場合、既存の治療法と比較して、安全性および有効性の面で顕著な改善を提供する場合、優先審査を受ける資格もある可能性がある。FDAがマーケティング申請を提出する際に,具体的な状況から他の既存療法と比較して,提案薬が疾患の治療,予防または診断における有意な改善を表すかどうかを決定する。FDAは、優先審査に指定された新薬出願を評価するために追加の資源を使用することを試み、審査を促進するために努力し、新規分子実体のNDAに対するFDAの行動の目標を提出日から10ヶ月から6ヶ月に短縮する。
1つの製品が深刻または生命に危害を及ぼす疾患または状態を治療する場合、通常、既存の療法よりも有意な利点を提供し、臨床的利益を合理的に予測する可能性のある代替終点への影響、または不可逆的な発病率または死亡率(IMM)よりも早く測定可能な臨床終点に対して、IMMまたは他の臨床的利益への影響を合理的に予測する可能性が高い場合、製品も加速承認を得る資格がある。承認を加速させる条件として,FDAはスポンサーに十分かつ制御された上場後検証性試験を要求することができ,2022年の食品·薬物総合改革法案(FDORA)によると,FDAは現在,このような試験を承認前または承認加速日後の特定の時間帯に行うことを適宜要求することが許されている。FDORAによれば、FDAはまた、検証試験が製品の予期される臨床的利益を検証することができない場合、承認の加速下で承認された薬物または適応の承認を撤回することができるように、手続きを加速させる権限を増加させる。さらに、機関から別の通知がない限り、FDAは、通常、承認を加速させることを考慮した製品の販売促進材料の事前承認を要求し、これは、製品の商業発売時間に悪影響を及ぼす可能性がある。
1つの製品がこれらの計画のうちの1つまたは複数に適合していても、FDAは、製品がもはや資格条件に適合していないことを後で決定することができ、またはFDAの審査または承認を決定する期間が短縮されないことができる。迅速チャネル指定、画期的な治療指定、および優先審査指定は、承認の基準を変更することはありませんが、開発や審査プロセスを加速させる可能性があります。加速的な承認を得た薬品はまた、伝統的な承認を得た薬品と同じ安全と有効性法定基準に符合しなければならない。
米国の非特許排他性
FDCAにおけるデータ排他性条項は、いくつかの後続申請の提出または承認を延期する可能性がある。FDCAはNCE守秘協定の承認を得た最初の申請者に5年間の米国内データ独占期間を提供した。FDAが以前に同じ活性部分を含む他の新薬を承認していなければ,薬物はNCEであり,活性部分は薬物物質の作用を担う分子やイオンである。排他期間内に、出願人が法定の権利参照承認に必要なすべてのデータを所有していないか、または所有していない場合、FDAは、薬剤の模倣薬のANDAまたは薬剤の別のバージョン505(B)(2)NDAの審査を受け入れない可能性がある。しかしながら、後続の出願が特許の無効または非侵害の証明を含む場合、4年後に提出することができる。
FDAのこれまでの立場は,固定用量組合せ製品中の活性部分の1つが以前に医薬製品で承認されていた場合,NCE排他性はこの組合せ製品に使用できないということであった。しかし、2014年10月、FDAはこの立場を変え、固定用量組合せ製品の申請に単一の新しい活性部分を有する医薬物質が含まれており、固定用量組合せ製品であっても以前に承認された活性部分を有する医薬物質が含まれていれば、5年間のNCE排他性を得る資格があるとする最終指導意見を発表した。
30
FDAが、出願人が行っているまたは後援する新しい臨床研究(バイオアベイラビリティ研究を除く)が承認申請に不可欠であると考えている場合、FDCAはまた、NDA、505(B)(2)NDAまたは既存のNDAの補充のために、既存の薬剤の新しい適応、用量または強度のような3年間の市場排他性を提供する。この3年間の専門期間は新しい臨床研究に関連する使用条件のみをカバーし、保護された臨床データを参考にしない後続の申請をFDAが許可することを禁止しない。5年と3年の排他性は完全な秘密協定の提出や承認を延期したり承認したりしないだろう。しかしながら、完全なセキュリティプロトコルを提出する出願人は、安全かつ有効であることを証明するために、必要なすべての臨床前研究および十分かつ良好に制御された臨床試験を参照する権利を行うか、または得ることを要求されるであろう。
小児科排他性は、米国が市場排他性を規制するもう一つのタイプである。小児科排他性が得られた場合、すべての製剤、剤形、および有効部分および特許条項の適応の既存の規制排他性期間は6ヶ月増加する。この6ヶ月間の専門権は、FDAによって発表されたこのような試験の“書面請求”に基づいて小児科試験を自発的に完成させることができ、小児科専門権を付与する際に、9ヶ月以上の期限があることが条件である。
承認後に要求する
FDAによって生産または流通を許可された薬品はFDAの普遍的かつ持続的な監督管理を受けなければならず、その中には記録保存、定期報告、製品サンプリングと流通、広告と販売促進、および製品の不良反応の報告に関連する要求が含まれている。承認後、承認された製品の大多数の変更は、新たな適応または他のラベル宣言を追加するなど、FDAの審査および承認を事前に受けなければならない。どの市場製品に対しても、持続的で毎年の使用料要求がある。
FDAはNDAを承認するための条件として、いくつかの承認後の要求を加えるかもしれない。例えば、FDAは、第4段階の臨床試験を含む上場後試験を要求し、製品の商業化後の安全性と有効性をさらに評価し、監視するために監視を行う可能性がある。
FDAの規定は,製品が特定の施設で生産され,cGMP規定に適合することを要求しており,この規定は他を除いて,品質管理と品質保証,記録やファイルの維持,cGMPとの任意の偏差を調査·是正する義務を要求している。また、医薬品メーカーおよび他の承認された薬品の生産·流通に参加するエンティティは、薬品、成分および成分を供給するエンティティを含み、FDAおよび州機関にその機関を登録し、FDAとこれらの州機関の定期的な抜き打ち検査を受けて、cGMP要求に適合することを確保しなければならない。また、2013年には、米国で流通されているいくつかの処方薬や生物製品を識別して追跡するための電子システムを構築することを目的とした“医薬品サプライチェーン安全法案”が公布された。DSCSAは薬品メーカー,卸売業者,流通業者に2023年11月終了の10年間に段階的と資源集約型の義務を負うことを求めている。FDAはDSCSAのいくつかの要求を満たすために、2023年11月から2024年11月までの1年安定期を設定し、貿易パートナーに相互運用可能なシステムとプロセスを引き続き構築し、検証させる。この法律の要求には、疑わしい製品に対して検疫と迅速な調査を行い、不正製品であるかどうかを決定すること、貿易相手およびFDAに任意の不正製品を通報すること、および製品追跡および追跡要求を遵守することが含まれる。製造プロセスの変更は厳しく規制されており,通常FDAが事前に承認して実施する必要がある。FDA法規はまた、cGMP要求との任意の偏差を調査および是正し、スポンサーおよびスポンサーが使用を決定する可能性のある任意の第三者製造業者に報告および文書要求を提出することを要求する。そのため、メーカーはcGMPコンプライアンスを維持するために、生産と品質管理に時間、お金、労力をかけ続けなければならない。
1つの薬剤が承認されると、規制要件および基準の遵守を維持することができない場合、または製品が発売された後に問題が発生した場合、FDAは、例えば、申請に含まれていない臨床経験に基づく新しい証拠、または申請が承認された後にFDAが得ることができる場合、承認された製品が、そのラベルに規定された、推奨または提案された使用条件下で主張または表示される効果を有することを示す実質的な証拠が乏しい場合がある。スポンサーも同様の理由で自発的にその承認された製品を市場から引き下げることができる。製品はその後、予期されない重症度または頻度の有害事象、または製造プロセス、または法規要件を遵守できなかったことを含む、以前に未知の問題を発見し、新しいセキュリティ情報を追加するために承認されたラベルの強制改訂をもたらす可能性がある;新しい安全リスクを評価するために発売後の研究または臨床試験を強制的に実施する;または強制的に流通する
31
またはREMS計画下の他の制限。承認またはマーケティングに関するこれらの制限は、製品の商業普及、流通、処方、または配布を制限する可能性がある。他の他の潜在的な結果には
FDAは市場に投入された製品のマーケティング、ラベル、広告、販売促進を厳格に規制している。製造業者および製造業者を代表する任意の第三者は、承認された適応に対してのみ、製品の承認されたラベルと一致する方法で薬物を普及させることができる。FDAや他の機関はラベル外用途の普及を禁止する法律法規を積極的に実行しており,ラベル外用途の普及が不適切であることが発見された会社は重大な責任を負う可能性がある。
アメリカの他の医療保険法は
医療保健提供者、医師、第三者支払人は、私たちが発売許可を得た薬品の推薦と処方に主な役割を果たす。第三者支払人、医療保健提供者と医師及び患者と他の第三者と製品の臨床研究、販売、マーケティングと普及及び関連活動について達成した手配は、承認されると、製薬業者を広範に適用される詐欺と濫用及びその他の医療保健法律と法規に直面させる可能性がある。米国では、これらの法律には、州および連邦反リベート、虚偽声明、医師透明性、および患者データプライバシーおよびセキュリティ法律法規が含まれているが、これらに限定されない
32
これらの法律の広汎性、および既存の法定例外および安全港の規制の狭いため、製薬業者のいくつかの商業活動は、1つまたは複数のそのような法律の挑戦を受ける可能性がある。商業計画が適用された医療保険法に適合することを確実にするための努力は多くのコストに関連する。政府および法執行当局は、医薬品製造業者の商業行為が、詐欺および乱用または他の医療保健法律および法規を適用する現在または未来の法規、法規または判例法に適合していないと結論するかもしれない。製薬メーカーに対してこのような訴訟を提起した場合、同社が自分の権利を弁護または維持することに成功しなかった場合、これらの訴訟は、重大な民事、刑事および行政処罰、損害賠償、返還、監禁、罰金、連邦医療保険、医療補助および他の連邦医療保健計画への参加から除外される可能性があり、もし私たちが誠実さと監督協定の制約を受けて、違反疑惑、契約損害、名声損害、利益および将来の収益減少および業務削減を解決するために、信頼と監督協定の制約を受けた場合、これらは製薬メーカーの業務運営能力および運営結果に悪影響を及ぼす可能性がある。また,いずれの薬品も米国以外での商業化は,上記の医療保健法や他の外国法の外国等価物に制約される可能性がある。
33
アメリカでは、州データ漏洩通知法、州と連邦健康情報プライバシー法、連邦と州消費者保護法を含む多くの連邦と州の法律法規が、健康に関連する個人情報と他の個人情報の収集、使用、開示と保護を管理している。例えば、カリフォルニアは最近“カリフォルニア消費者プライバシー法”(California Consumer Privacy Act、略称CCPA)を公布し、カリフォルニアの消費者のために新しいプライバシー権(法律で定義されているような)を創造し、消費者または家庭の個人データを処理するエンティティに対してより多くのプライバシーおよびセキュリティ義務を規定している。CCPAは、カバーする会社に、そのデータ収集、使用、および共有アプローチに関するいくつかの開示を消費者に提供し、影響を受けたカリフォルニア住民に、特定の個人情報販売または移転から撤退することを選択する方法を提供する。CCPAは2020年1月1日に施行され、カリフォルニア州総検察長は様々なバージョンの最終法規を提出した。2020年7月1日以来、カリフォルニア州総検察長は違反者に対して法執行行動をとる権利がある。また、カリフォルニアプライバシー法“カリフォルニアプライバシー権法案”は2020年11月3日にカリフォルニア州有権者によって可決され、2023年1月1日に施行された。CPRAは個人情報の処理と蓄積に追加的な義務を規定している.CPRAに関連する発展を引き続き監視し、CPRAコンプライアンスに関連する追加コストと費用を予想する。米国の他の州でも総合的なプライバシー立法(コロラド州、コネチカット州、ユタ州、バージニア州では別の法律が採択されている)の制定が考えられており、業界組織はこれらの分野で常に新基準を採用し、提唱している。CPRAによって改正されたCCPAは、HIPAA下のPHIのいくつかの活動に関連する例外的な状況を含んでいるが、これらの法律はまだ私たちに適用されないか、まだ施行されていないので、CCPA、CPRA、または他のこのような未来の法律、法規、および基準が私たちの業務に与える可能性があることを決定することはできない。
私たちが臨床試験を行うか、または私たちが行っているまたは未来の臨床試験で被験者を募集することを決定した場合、州や連邦法律または他の義務によって、追加のプライバシー制限を受ける可能性があります。私たちはまた、ヨーロッパの一般的なデータ保護条例を含む他の国/地域の法律に支配される可能性がある。
アメリカの現在と未来の医療改革法案は
国内でも海外でも,政府も個人も,支払側は医療コストを抑えるための複雑な方法を開発しているが,これらの方法は,遺伝子療法やまれな疾患に対する療法,例えば我々が開発しているような新しい技術に特化しているわけではない。米国や一部の外国司法管轄地域では、医療システムの立法や規制に多くの変化が生じており、製品販売の収益性に影響を与える可能性がある。特に、2010年には、2010年に“医療·教育和解法案”(または総称して“ACA”)によって改正された“患者保護·平価医療法案”が公布され、それ以外にも、生体製品を低コストの生体模倣薬の潜在的競争を受けるようにすること、多くのメーカーが医療補助薬品還付計画の下で不足している最低医療補助税還付を増加させること、医療補助薬品還付計画を医療補助管理に登録された介護組織を使用する個人の処方に拡大すること、メーカーにあるブランドの処方薬に新たな年会費と税金を徴収することを要求すること;連邦医療保険D部分の保険切欠き割引計画を作成し、この計画によると、メーカーは必ずその保証間隔期間内に、条件を満たす受益者に適用ブランド薬品交渉価格の70%の販売時点割引を提供し、メーカーの外来薬物として連邦医療保険D部分の保証の条件に組み入れなければならない;そして連邦政府の比較有効性研究の計画を増加するために激励を提供する。
公布以来、ACAのいくつかの方面に対して多くの司法、行政、行政と立法方面の挑戦を提出した。例えば,総裁·バイデンは複数の行政命令を発表し,処方薬コストの低減を図っている。いくつかの措置および他の提案された措置は発効するために追加の立法によって許可される必要があるかもしれないが、バイデン政府はこれらの措置を撤回または他の方法で変更する可能性があるが、バイデン政府と国会は薬品コストを制御するための新しい立法措置を求め続けると表明している。また、ACAが公布されて以来、米国は他の立法改正を提案し、採択した。2011年の予算統制法やその後の立法などは、年度ごとに提供者に支払う医療保険総額を2%削減することを含む国会支出削減策を作り、2031年まで有効となる。2012年の“米国納税者救済法”は,病院や癌治療センターを含むいくつかの医療サービス提供者に支払う連邦医療保険をさらに減少させ,政府が医療サービス提供者に支払う多額の支払いを取り戻す訴訟時効を3年から5年に延長した。2024年1月1日から、“2021年米国救援計画法案”は単一源と革新者多源薬物に対する法定医療補助薬品リベート上限を廃止し、現在この上限は薬品メーカーの平均価格の100%に設定されている。2010年の法定現金給付法案、2021年の米国救援計画法案、および後続立法による予算赤字推定が増加したため、これ以上の立法がない場合、2025年から提供者に支払われる医療保険金額はさらに減少する。これらの法律および法規は、医療保険や他の医療資金のさらなる減少を招き、規制承認を得る可能性のある任意の候補製品の価格に影響を与えるか、またはそのような候補製品を開発または使用する頻度に影響を与える可能性がある。
34
また、ACAが公布されて以来、米国は他の立法改正を提案し、採択した。2011年の予算統制法やその後の立法などは、年度ごとに提供者に支払う医療保険総額を2%削減することを含む国会支出削減策を作り、2031年まで有効となる。2012年の“米国納税者救済法”は,病院や癌治療センターを含むいくつかの医療サービス提供者に支払う連邦医療保険をさらに減少させ,政府が医療サービス提供者に支払う多額の支払いを取り戻す訴訟時効を3年から5年に延長した。2024年1月1日から、“2021年米国救援計画法案”は単一源と革新者多源薬物に対する法定医療補助薬品リベート上限を廃止し、現在この上限は薬品メーカーの平均価格の100%に設定されている。2010年の法定現金給付法案、2021年の米国救援計画法案、および後続立法による予算赤字推定が増加したため、これ以上の立法がない場合、2025年から提供者に支払われる医療保険金額はさらに減少する。これらの法律および法規は、医療保険や他の医療資金のさらなる減少を招き、規制承認を得る可能性のある任意の候補製品の価格に影響を与えるか、またはそのような候補製品を開発または使用する頻度に影響を与える可能性がある。
2022年“インフレ低減法案”(IRA)には、2025年から連邦医療保険D部分の受益者の自己支出上限を7,050ドルから2,000ドルに引き下げ、カバー格差を効果的に除去する条項が含まれている;連邦医療保険D部分下のいくつかの薬物に新しいメーカー財務責任を適用し、アメリカ政府がある高コスト薬物と生物製品の連邦医療保険B部分とD部分の価格上限について交渉することを可能にし、模造薬や生物類似競争が存在しない;会社にインフレよりも価格が速い薬品の連邦医療保険へのリベートを要求する;そして、HHSリベート規則の実施を2032年1月1日に延期し、この規則は薬局福祉マネージャーが受け取ることができる費用を制限する。また,IRAによると,孤児薬は連邦医療保険薬品価格交渉計画の影響を受けないが,孤児名があることを前提としており,唯一承認された適応はこの疾患や状況に対するものである。1つの製品が複数の孤児の称号または複数の承認の適応を得た場合、それは孤児薬物免除を受ける資格がない可能性がある。アイルランド共和軍の実施は現在行われている訴訟の影響を受けており,これらの訴訟はアイルランド共和軍の医療保険薬品価格交渉計画の合憲性を疑問視している。Ireland共和軍が私たちの業務と医療産業全体に及ぼす影響はまだ明確ではない。
カナダの承認プロセス
カナダでは,我々の小分子製品候補製品と我々の研究·開発活動は主に“食品·薬物法”とその下の規則·法規によって規制されており,これらの法規はカナダ保健省によって施行されている。その他の事項以外に、カナダ衛生部は薬品の研究、開発、テスト、承認、製造、包装、ラベル、貯蔵、記録保存、広告、販売促進、流通、マーケティング、承認後のモニタリング及び輸出入に対して監督管理を行う。カナダの法律によると、薬品審査手続きは製造施設に対して許可証を発行し、製品に対して厳格な制御の研究とテストを行うことを要求し、薬品の商業化販売を承認する前に、政府は実験結果を審査と承認する。監督管理機構は通常また任意の薬物製品の製造、非臨床開発と臨床開発においてそれぞれcGMP、良好な実験室実践或いはGLPと持続良好な臨床実践或いはCGCPなどの厳格かつ具体的な標準に従うことを要求している。カナダで規制の承認を得る過程と、その後適用される法規と条例を遵守するには、多くの時間と財力が必要だ。詳細は“リスク要因”を参照されたい
カナダで薬品審査を行うために必要な主な手順は以下のとおりである
非臨床安全性薬理学と毒理学研究
非臨床研究を行う体外培養そして、臨床研究および開発過程全体で投与する前に候補薬物安全性の証拠を提供するために、動物において薬物動態学、新陳代謝および可能な毒性効果を評価する。このような研究は適用された法律とプロスに基づいて行われる。
臨床試験
米国と類似した法規はカナダの臨床試験に適用される。カナダでは、臨床試験がカナダで行われると、研究倫理委員会(REBS)が臨床試験計画の審査·承認に用いられる。ヒト被験者への研究用新薬の投与に関する臨床試験については,カナダ衛生部に申請して承認を得なければならず,カナダの地点で試験を開始することができる。CGCPの要求によると,試験は合格した調査者の監督下で行われ,多くの場合医師である。臨床試験は,試験目標,試験手順,安全性モニタリングのためのパラメータ,評価すべき治療効果基準,統計分析計画などを詳細に説明する案に基づいて行った。被験者によって署名された案とインフォームドコンセントは,試験地点に属するREBによって審査·承認された。人間
35
新薬の臨床試験は、米国政府の規制を背景に上述したように、3つの連続した段階で行われるのが一般的である。カナダ保健省も、マーケティング申請を支援するために、外国の臨床試験データを受け入れている。また,ヒト臨床試験のための研究薬の製造はcGMP要求に拘束されている。
新薬が提出される
カナダでは,カナダ衛生部が同意すれば,第3段階臨床試験または早期試験を成功させた後,新薬を支援する会社は,この製品の薬理,化学,製造および制御に関連するすべての非臨床および臨床データおよび他の試験を収集し,NDSの一部としてカナダ衛生部に提出する。
カナダ衛生部は新薬を承認せず、cGMP-規範生産の品質体系に符合しない限り-満足でき、しかもNDSに含まれるデータは大量の証拠を提供し、この薬物が研究の適応と投与量の下で安全かつ有効であることを証明した。カナダ衛生部がNDSに十分な情報が含まれていることに満足すれば、新薬の発売許可を承認する可能性がある。カナダでは,新薬の発売認可はコンプライアンス通知,あるいはNOCと呼ばれている。
新しいデータリストに格納されたデータを生成するために必要なテスト,および新しいデータリストの承認プロセスは,膨大な時間,労力,財力を必要とし,完成までに数年かかる可能性がある.これは製品の効果、安全性、そして品質を確保するために必要だ。非臨床と臨床テストから得られたデータは常に決定的ではなく、異なる解釈の影響を受ける可能性があり、これは監督管理部門の承認を延期、制限或いは阻止する可能性がある。カナダ保健省はNDSをタイムリーに承認しないか、または全く承認しないかもしれない。カナダではNDSSは使用料を支払う必要があり、これらの費用は通常毎年増加し、インフレを反映している。
カナダ衛生部は深刻な、生命に危害を及ぼす或いは深刻な虚弱な疾病或いは状況を治療する新薬に対してNDS審査を行った後、条件のある発売許可を与える権利がある。条件に適合する通知(NOC/C)は、既存のデータに基づいて、(I)カナダで現在販売されていない薬物の疾患または疾患の有効な治療、予防または診断、または(Ii)治療効果を著しく向上させ、および/またはリスクを著しく低下させ、それにより、カナダで販売されている薬剤が十分に管理できない疾患または疾患のための既存の治療、予防または診断試薬を提供し、全体的な利益/リスクプロファイルを改善するために、承認されることができる。NOC/cを付与するとき、NOC/cを獲得した会社は、カナダ衛生部に対していくつかの約束をしなければならず、その中には、新薬の安全な使用および治療効果を支援するために、カナダ衛生部に確認性データを提供することを要求することが一般的に含まれている。
カナダ保健省が候補製品を承認しても、候補製品の承認適応を制限することができ、製品ラベルに禁忌症、警告または予防措置を含むことを要求し、承認後の薬物の安全性をさらに評価するための第4段階の臨床試験を含む承認後の研究を要求することができ、商業化された製品を監視するための試験および監視計画を要求し、または流通制限または他のリスク管理メカニズムを含む他の条件を適用する。
カナダ衛生部は発売後の研究或いはモニタリング計画の結果に基づいて、製品の更なるマーケティングを阻止或いは制限する可能性がある。承認後、新たな適応の追加、製造変更、および追加のラベル宣言など、承認された製品のいくつかのタイプの変更は、変更を実施する前に、さらなるテスト要件、通知、および審査および承認を受ける必要があります。さらに、新しいセキュリティ情報が発生した場合、追加のテストおよび/または規制通知が必要とされる可能性があり、またはカナダ保健省は、承認された適応の範囲または臨床使用の他の条件に影響を与えるために、製品ラベルの更新を要求する可能性がある。
EU承認手続き
EUの医薬製品に対する承認の流れはアメリカとほぼ同じである。それは満足できるように臨床前研究を完成させ、十分かつ制御された臨床試験を行い、各提案適応に対する製品の安全性と有効性を確定する必要がある。MAAの関係主管当局に提出することを要求し、これらの当局がマーケティング許可を付与し、その後、その製品は
36
EU英国のEU離脱後、医薬製品を英国市場に投入するためには、単独のマーケティング許可が必要である(北アイルランド議定書によると、EU規制枠組みは北アイルランドに引き続き適用され、EU集中認可は引き続き認められている)。2024年1月、新医薬製品の開発者は現在、薬品と保健製品監督機関(MHRA)の国際認可手続き(IRP)に基づいて申請を提出することができる。IRPは、MHRA指定参照規制機関のうちの1つによって承認された製品に開放され、米国とカナダを含む。
臨床試験許可
2014年4月,EUは2022年1月31日に臨床試験指令2001/20/ECに代わる新たな臨床試験条例(EU)第536/2014号を採択した。新法規の一時的な条項は,2025年1月31日までに行われているすべての臨床試験を新法規に移行しなければならないと規定している。この新しい規定はEUの臨床試験承認制度を徹底的に改革した。具体的には、EUの臨床試験の承認を簡略化し、簡略化することを目的として、すべての加盟国に直接適用される(これは、各加盟国で国家立法を実施する必要がないことを意味する)、EUの臨床試験の承認を簡略化することを目的としている。新条例の主な特徴は:臨床試験情報システム(CTIS)を通じて単一入口点を通じて申請プログラムを簡略化すること;申請のために単一文書を準備と提出すること、及び臨床試験発起人の報告プログラムを簡略化すること;臨床試験申請評価の統一プログラムは2つの部分に分けられる(第1部分は科学と医薬製品文書を含み、第2部分は国家と患者レベルの文書を含む)。第1部は、加盟国が作成した報告書を参照して臨床試験許可申請(関係加盟国)が提出されたEUのすべての加盟国またはEU加盟国の主管当局の協調審査によって評価される。二番目の部分は関連する各会員国によって個別的に評価される。臨床試験申請の評価には厳しい期限が設定されている。
EUのトップクラスの称号
2016年3月,EMAは適応候補の開発を促進するイニシアティブを開始したが,これらの適応はまれであり,現在のところ治療法はほとんどない。優先薬物計画或いはPrime計画は満たされていない医療需要領域の薬物開発を奨励し、重大な革新を代表する製品に加速評価を提供することを目的としており、これらの製品の中で、上場許可申請は集中プログラムを通じて行われる。条件に適合する製品は、満たされていない医療需要が存在する条件(EUには満足できる診断、予防または治療方法がない、または、ある場合、新薬は重大な治療利点をもたらす)でなければならず、それらは、新しい治療方法を導入することによって、または既存の方法を改善することによって、満たされていない医療需要を満たす潜在力を示さなければならない。中小企業の製品は大企業よりも早くPrime計画に参加する資格があるかもしれません。Primeの称号を持つ候補製品のスポンサーは多くのメリットを得ることができ、しかし限らず、早期にEMAと積極的な監督対話を行い、臨床試験設計とその他の開発計画要素を頻繁に討論し、及びファイルを提出した後にMAA評価を加速する。EMAのCHMPまたは高度治療委員会の専任連絡先および調査委員は、EMA委員会レベルで製品のより多くの理解を促進するために、Prime計画の早期に任命されることが重要である。会議を開始してこれらの関係を開始し、EMAを含む1つの多学科専門家チームは、全体発展と監督戦略に関する指導を提供する。
固定用量共同ガイドライン
FDAと同様に、EMAも固定用量組合せ製品の審査および承認問題を解決するためのガイドラインを発表した。このEMAの固定組合せ薬物臨床開発に関するガイドラインは2017年10月1日から施行された。任意の固定組合せ医薬製品の基本的な科学的要求は、この組み合わせの薬理と医学原理が合理的であることを証明し、そして証拠基礎を構築し、すべての活性物質の期待される治療効果(有効性及び/又は安全性)に対する関連貢献及び目標適応におけるこの組み合わせの積極的な利益-リスクを証明することである。EMAは、2つの活性成分の初期組み合わせに関連する製品の場合、初期治療のための固定組合せ医薬製品の臨床的有効性/安全性研究を支持する設計は、その基本原理に依存し、特に単一活性物質の使用と比較して、より良い治療効果またはより高い安全性を達成することを示している。単一療法が不十分、不適切、または道徳的に所望の治療効果を達成することが決定された場合には、最初の併用療法が合理的であることを容易に証明すべきである(例えば、 HIV)です
37
マーケティング許可
欧州経済地域(すなわち、EUおよびアイスランド、リヒテンシュタインおよびノルウェー)または欧州経済地域における製品のマーケティング許可を得るためには、出願人は、欧州経済区が管理する中央プログラムまたはEU加盟国主管当局が管理するプログラムのうちの1つ(分散プログラム、国家プログラムまたは相互承認手順)に従ってMAAを提出しなければならない。マーケティング許可はヨーロッパ経済地域に設立された申請者にのみ付与されることができる。(EC)第1901/2006号条例では、EUがマーケティング許可を得る前に、出願人は、EMAによって承認された小児科人口のすべてのサブクラスをカバーする小児科調査計画(PIP)に含まれるすべての措置を証明しなければならない。(1)特定の製品免除、(2)カテゴリ免除、または(3)PIPに含まれる1つまたは複数の措置を延期しなければならない(例えば、製品が子供に無効または安全でない可能性があるので、この製品は成人人口でのみ発生する疾患または状態である。または、この製品が小児科患者の既存の治療法と比較して有意な治療的利益がない場合)。PIPによる小児科臨床試験結果の発売許可を得た製品(結果陰性であっても)は,6カ月の補充保護証明書またはSPC延期を取得する資格がある(その製品のSPC申請を提出すると同時に,またはSPC満期2年前のいつでも延期申請を提出することが前提である)。
集中化手続きは、欧州委員会によって欧州経済地域全体で効果的な単一マーケティング許可が付与されることを規定している。(EC)第726/2004号条例によると、特定の製品には、いくつかのバイオテクノロジーによって生産された薬品、孤児薬品に指定された製品、高級治療薬が含まれる(EC)即遺伝子治療、体細胞治療および組織工学医薬製品)および新規活性物質を含む製品は、HIV、エイズ、癌、神経変性疾患、糖尿病、自己免疫および他の免疫機能障害、およびウイルス性疾患を含む特定の疾患の治療に使用可能であることを示している。他の疾患の治療のための新しい活性物質を含む製品、および高度な革新性または集中処理プロセスを有する公衆の健康に有利な製品については、集中処理プログラムが任意である。他の場合には、出願人の要求に応じて、集中手順を使用することもできる。私たちは私たちが開発している候補製品について集中化手続きが義務的になると予想する。
中央手続きの下で、CHMPは、製品の予備評価を担当し、既存のマーケティング許可の修正または拡張を評価するなど、いくつかの許可後および保守活動を担当する。中央手続によれば、出願人が“気候変動管理計画”の質問に答えるために補足資料または書面または口頭解釈を提供する場合、重大な影響評価を評価する最長期限は210日であり、タイマポーズを含まない。タイミングを停止することは、MAAを評価する時間フレームを210日を超えるように大幅に延長する可能性がある。特殊な場合、公衆衛生の観点、特に治療革新の観点から見ると、1種の医薬製品が重大な利益を持っている場合、CHMPは加速的な評価を与える可能性がある。CHMPがこの要求を受信した場合、210日の制限時間は150日(クロックポーズを含まない)に減少するが、CHMPが加速評価にもはや適していないと考える場合、中央プログラムの標準時限に回復する可能性がある。この期限終了時に,CHMPは医薬製品に関する販売許可を承認すべきかどうかについて科学的な意見を提供する。CHMPが肯定的な意見を与えた場合、EMAは、EMA提案を受信してから67日以内に発行されるマーケティング許可を付与する最終決定を下す支援ファイルと共にEU委員会に提供する。
欧州委員会はいわゆる“特別な場合のマーケティング許可”を承認するかもしれない。このような許可は、(I)関連製品の適応が非常にまれであり、申請者が全面的な証拠を提供することを合理的に期待できないこと、(Ii)現在の科学的知識状態では、包括的な情報を提供できないこと、または(Iii)そのような情報を収集することが公認された医学道徳原則に違反するため、通常の使用条件下で有効性および安全性に関する包括的なデータを提供できないことを証明することができる製品に適用される。したがって、特別な場合には、いくつかの特定の義務の制約の下でマーケティング許可を承認することができ、これらの義務は、以下を含むことができる
38
特別な場合、上場許可は年度見直し手続きでリスクと収益のバランスを再評価するために年間審査を受けなければならない。ライセンスの継続は年間再評価と関連があり、否定的な評価はマーケティング許可が一時停止または撤回される可能性がある。しかし、特殊な場合、医薬製品の発売許可の継続は“正常”販売許可と同じ規則に従う。したがって、特別な場合のマーケティング許可は最初は5年であり、その後のライセンスは、EMAがセキュリティ理由を決定しなければ、さらに5年間延長する価値がある限り、無期限に発効する。
集中認証プログラムとは異なり、分散マーケティング許可プログラムは、製品販売が存在する各EU加盟国の主管当局に個別に申請し、それによって単独で承認する必要がある。この申請は,中央プログラムを介して環境管理機関に提出して許可する申請と同様である。EUは加盟国が有効な申請を受けてから120日以内に評価草案と関連材料の草稿を作成することを参考にする。このようにして生成された評価報告書は関係EU加盟国に提出され、これらの加盟国は評価報告および関連材料を受け取ってから90日以内にその報告書および関連材料を承認するかどうかを決定しなければならない。もしEU加盟国が公衆健康の潜在的な深刻なリスクに対する懸念から評価報告書や関連材料を承認できない場合、論争内容はすべてのEU加盟国に対して拘束力を持つことを決定する欧州委員会に提出することができる。
相互承認手続きの基礎は、同じくEU加盟国の主管当局がEUの他の加盟国の主管当局によるある医薬製品の販売許可を受け入れることである。国家マーケティング許可の所有者は、EU加盟国の主管当局に申請を提出することができ、その主管部門に、他のEU加盟国の主管当局が提供するマーケティング許可を認めるように要求することができる。
条件付きマーケティング許可
欧州委員会はまた、包括的なマーケティング許可を申請するために必要な包括的な臨床データを得る前に、いわゆる“条件付きマーケティング許可”を承認することができる。深刻な衰弱または生命に危険な疾患(孤児薬として指定された薬剤を含む)または公衆衛生緊急時の候補製品の治療、予防または診断のための候補製品については、(I)候補製品のリスク-利益バランスが積極的である場合、(Ii)許可後に出願人が必要な包括的な臨床試験データを提供することができる可能性が高い、(Iii)この製品が満たされていない医療需要を満たし、(Iv)関連医薬製品が直ちに発売される公衆の健康に対する利点が、依然として追加のデータが必要であるという事実に固有のリスクを超える場合、そのような条件付きマーケティング許可を付与することができる。条件付き販売許可には,現在行われているあるいは新たな研究の完了や薬物警戒データの収集義務など,販売許可保持者が履行しなければならない具体的な義務が含まれていることができる。条件付きマーケティング許可の有効期間は1年であり、リスク−収益バランスが正のままであり、追加または修正された条件および/または特定の義務の必要性が評価された後、毎年更新することができる。上記の集中プログラムのスケジュールは、条件付きマーケティング許可申請の審査にもCHMPに適用される。上場許可所有者が規定の義務を履行し、完全なデータが薬品の利益がそのリスクよりも引き続き大きいことを確認すると、条件付きマーケティング許可は標準的な集中マーケティング許可に変換することができる(これ以上特定の義務の制約を受けない)。
EUの規制データ保護
EUでは、完全かつ独立したデータパケットによって承認された革新医薬製品はマーケティング許可を得た後、8年間のデータ独占経営権を獲得する資格があり、他の2年間の市場独占経営権を有する。データ排他性が付与された場合、EUが初めて参照製品を許可した日から8年以内に、これらの革新的製品の模倣薬または生物模倣薬の許可を付与した場合、出願人は、模倣薬または生物類似製品マーケティング許可を申請する際に、製品アーカイブに含まれる革新者の臨床前および臨床試験データを参照することができない。追加の2年間の市場排他期間内に、模造薬または生物に類似したMAAを提出し、革新者のデータを参照することができるが、市場排他性が満了するまで、いかなる模倣薬または生物に類似した薬品もEU市場に投入することができない。この10年の最初の8年間に、マーケティング許可保持者が1つまたは複数の新しい治療適応の許可を得た場合、10年全体の期間は最大11年に延長され、許可前の科学的評価では、これらの適応は既存の療法と比較して有意な臨床的利益をもたらすことができると考えられる。1つの化合物が革新的な医療製品とされていても、革新者は所定のデータ独占期間を獲得し、別の会社は
39
しかしながら、同社がMAAベースのマーケティング許可を取得し、完全かつ独立した薬物試験、臨床前試験、および臨床試験データパケットを有する場合、製品の別のバージョンを販売することもできる。
授権期間と継続期間
上場授権書の初期有効期限は原則として5年である。マーケティング許可は、5年後に、EMA(中央許可製品のための)または関連するEU加盟国の主管当局(国家許可製品のため)のリスク-収益バランスの再評価に基づいて更新することができる。そのため、上場授権書保持者は上場授権書が失効する少なくとも9ヶ月前に、マーケティング授権書を授与してから導入されたすべての変化を含むEMA或いは主管当局に品質、安全と効果に関する文書の総合バージョンを提供しなければならない。欧州委員会やEU加盟国の主管当局は、薬物警戒に関する正当な理由に基づいて、5年間の上場許可期間を継続することを決定することができる。その後最終的に更新されると、上場許可の有効期限は無期限となる。いかなる認可の後も、認可後3年以内に医薬品が実際にEU市場に投入されていない場合(集中手続きである場合)、またはEU加盟国の市場(国家によって許可された製品)が許可されていない場合、または製品が3年連続で市場から除去されている場合には、もはや有効ではない(いわゆる日没条項)。
孤児薬の指定と排他性
(EC)第847/2000号条例により施行された条例(EC)第141/2000号によれば、そのスポンサーが証明できることを前提として、欧州委員会により孤児医薬製品に指定することができる。(1)生命又は慢性衰弱に危険な疾患の診断、予防又は治療を目的としていること、(2)又は(I)申請を提出したとき、EUでの影響が万分の5を超えないこと、又は(Ii)EUにおける販売が十分な見返りをもたらす可能性がない場合、その開発に必要な投資が合理的であることを証明するためのインセンティブ措置がない場合、(3)EUから許可されていない満足できる診断、予防または治療方法、または、そのような方法があれば、製品は、その疾患の影響を受けている人に大きな利点を有するであろう。
許可を得ると、孤児薬品はすべてのEU加盟国で10年間の市場排他性を有する権利があり、研究プログラムの科学的援助、集中マーケティング許可プログラムによる許可、登録およびマーケティング許可費用の削減または廃止など、開発と監督審査過程で一連の他のメリットを得る権利がある。市場排他期間内に、以下の場合にのみ、同じ孤児適応を有する“類似医薬製品”の発売許可を承認することができる:(I)元孤児医薬製品の販売授権者は、第2の孤児医薬製品の許可に同意する;(Ii)原始孤児医薬製品の製造業者は、十分な数の製品を供給することができない;または(Iii)第2の製品は、元の孤児医薬製品よりも安全で、より効果的で、または臨床的に良いと決定される。類似医薬製品“の定義は、承認された孤児医薬製品に含まれる1つ以上の類似活性物質を含む医薬製品であり、同じ治療適応のためのものである。また、5年目の終了時に当該製品が指定された孤児の基準を満たしていないと判定された場合、例えば、最初の孤児医薬製品の利益が十分に高く、市場固有性を維持するのが合理的であることを証明するのに十分ではないため、市場専門期間を6年に短縮することができる。
営業許可を得た後の規制要件
もし医薬製品がEUで許可された場合、マーケティング許可の保持者は、医薬製品の製造、マーケティング、普及、販売に適した一連の要求を守らなければならない。これらの措置には
40
上記の連合規則は一般的にヨーロッパ経済地域に適用される。
EU規制枠組みの改革
欧州委員会は2023年4月に立法提案を提出し,実施されれば,EUの現在のすべての薬品(まれな疾患や児童を治療する薬品を含む)の規制枠組みに代わる。欧州委員会は立法提案を欧州議会と欧州理事会に提出して審査と承認を行った。2023年10月、欧州議会は立法提案に対して修正案を提出する報告案を公表し、欧州議会はこれを討論する。欧州委員会の立法提案が承認されると(修正または修正されない)、それらはEU法律として採択されるだろう。
ヨーロッパ経済圏とイギリスのデータ保護法規
個人健康データを含むヨーロッパ経済地域における個人の個人データを収集、使用、開示、移転、または他の方法で処理することは、EU一般データ保護条例またはEU GDPRの制約を受け、イギリスの個人個人データの同様の処理は、イギリス一般データ保護条例またはイギリスGDPRおよびイギリス2018年データ保護法によって制限される。本年度報告では、他の説明がない限り、GDPRとはEU GDPRとイギリスGDPRを指す。GDPRの範囲は広く,個人データを処理する会社に対して多くの要求がなされており,健康や他の敏感なデータの処理,個人データに関する個人の同意の取得,個人へのデータ処理活動に関する情報の提供,個人データの安全と機密性の保護のための保障措置の実施,データ漏洩に関する通知の提供,第三者処理者の採用時に何らかの措置をとるなどの要求がある。GDPRはまた、欧州経済圏/イギリス以外の国(米国を含む)への個人データの移転に厳しいルールを実施し、データ保護当局が2000万ユーロ(1,750万ポンド)や世界年収の4%に達する可能性のある罰金を含むGDPR違反行為に巨額の罰金を科すことを許可している。GDPRはまた、データ主体と消費者協会に個人訴訟権利を与え、監督当局に苦情を提出し、司法救済を求め、GDPR違反による損害について賠償を受けることができる。GDPRを守ることは厳格で時間のかかる過程となり、ビジネスコストを増加させたり、会社にそのビジネスやり方を変えて完全な遵守を確保したりする可能性がある。
イギリスの離脱とイギリスの規制枠組み
イギリスは2020年1月31日にEU(通称離脱)を正式に離脱し、EUはイギリスと貿易·協力協定を締結し、TCAと略称し、2021年1月1日から暫定的に適用され、2021年5月1日から正式に適用される。TCAには薬品に関する具体的な条項が含まれており,その中にはGMP相互承認,薬品の製造施設の検査,発表されたGMP文書が含まれているが,イギリスとEUを大規模に相互承認する薬品法規は規定されていない。現在、イギリスは“2012年人類薬品条例”(改正された)(北アイルランド議定書に基づき、EU規制枠組みは引き続き北アイルランドに適用されている)を採択し、医薬製品のマーケティング、普及、販売に関するEUの立法を実施している。そのため,新たなEU臨床試験規制を除いて,イギリスの規制制度はEUの規制制度とほぼ一致しているが,イギリスの規制制度はEUから独立しており,“薬物規制法”もイギリスとEUの薬剤業立法について相互に認める規定がないため,これらの制度は後日より大きく異なる可能性がある。しかしながら、TCAによると、EU薬品立法は卸売によって認められていないにもかかわらず、以下に述べるイギリスの薬品規制機関MHRAによって2024年1月1日から実施される新しい枠組みの下で、MHRAは、イギリスのマーケティング許可の申請を考慮する際に、EMA(およびいくつかの他の規制機関)がマーケティング許可を承認する決定を考慮すると表明している。
2023年2月27日、イギリス政府と欧州委員会は、北アイルランド議定書、すなわち“ウィンザー枠組み”と呼ばれる新たな手配の代わりに、原則的な政治的合意を発表した。この新しい枠組みはイギリスの医薬製品に対する規制を含む北アイルランド議定書下の既存制度を根本的に変えた。特にMHRAはすべての医療製品を承認する責任があります
41
イギリス市場(すなわちイギリスと北アイルランド)では、EMAは北アイルランドへの医薬製品の輸送を承認する上で何の役割も果たすことはできないだろう。MHRAは、イギリスで販売されているすべての医薬製品のイギリス範囲での単一マーケティング許可を付与し、製品がイギリス各地で単一パッケージおよび単一ライセンスで販売できるようにする。2023年3月24日、EU·イギリス合同委員会はウィンザー枠組みを承認したため、イギリス政府とEUは立法措置を制定し、法律にする。2023年6月9日,MHRAはウィンザーフレームワークの薬品について2025年1月1日から適用すると発表した。
MHRAは,患者に利益を与える新薬を優先的に獲得するプログラム,評価プログラムの加速,新製品やバイオテクノロジー製品の新たな評価経路を含む国家許可プログラムを変更している。すべての既存のEU中央ライセンス製品のマーケティングライセンスは、2021年1月1日にイギリスのマーケティングライセンスに自動変換(元)されます。2024年1月1日以来、マーケティング許可を承認する新しい枠組みが確立されており、この枠組みに基づいて、MHRAは新しいイギリスのマーケティング許可の申請を決定する際に、EMAおよびいくつかの他の規制機関がマーケティング許可を承認するための決定を考慮する可能性がある。イギリスでは、今発売前の許可された孤児の称号がない。代わりに,MHRAは対応するMAAと同時に孤児指定申請を審査する.これらの基準は基本的に同じであるが,イギリス市場のためにオーダーメイドされている,すなわちイギリス(EUではなく)という疾患の流行率は万分の5を超えてはならない。孤児の称号が与えられた場合、市場独占期間はその製品がイギリスで初めて承認された日から設定される。
製品の定価決定を承認する
連合では、様々な国の価格設定と補償プログラムの差が大きい。一部の国では、補償価格を合意した後にのみ、製品を販売することができると規定されている。いくつかの国は、特定の候補製品の費用対効果を現在利用可能な療法またはいわゆる衛生技術評価と比較して、精算または定価承認を得るために、追加の費用対効果評価を達成することを要求する可能性がある。例えば、EU加盟国は、その国の健康保険制度が精算を提供する製品範囲を制限し、人が使用する医療製品の価格を制御することを選択することができる。EU加盟国は製品の具体的な価格を承認することができ、その製品を市場に投入する会社の収益力を直接または間接的に制御する制度をとることもできる。他のEU加盟国は会社が自分の製品価格を固定することを許可したが、処方量を監視と制御し、医師に指導を発表して処方を制限した。最近,EUの多くの国が薬品要求の割引額を増加させ,各国が医療支出を管理しようとしているに伴い,これらの努力は継続する可能性があり,特にEUの多くの国が深刻な財政危機や債務危機を経験している場合である。全体的には,医療コスト,特に処方薬の下り圧力が大きくなっている。
そのため、新製品の参入にはますます高い壁が設けられている。政治、経済、規制面の事態は定価交渉をさらに複雑化させる可能性があり、補償を受けた後、定価交渉が継続される可能性がある。EUの各加盟国が使用する参考定価と平行貿易、すなわち低価格と高価なEU加盟国間の裁定は、価格をさらに下げることができる。薬品に対して価格規制や精算制限を実施することが保証されていない国は、これらの国で承認されれば、任意の製品に対して有利な精算と定価手配を許可する。
日本の承認の流れ
日本政府の薬品規制
日本の薬品規制枠組みは様々な法律法規に基づいており,主に“薬品と医療機器法”(略称“PMD法”)から構成されている。衛生、労働と福祉部は薬品と医療機器の開発、製造と商業化を監督する主要な監督機関であり、薬品と医療機器会社を指導して監督し、製品の品質と安全を確保する。薬品と医療機器管理局(“PMDA”)は衛生部と協力した監督管理機関であり、製品の安全性、有効性と品質を確保する。PMDAは薬品と医療機器の臨床試験と上場許可申請に対して科学的な審査を行うことを許可された;そしてその発売後の安全性をモニタリングした。薬品監督管理局はまた,薬品や医療機器による薬物副作用の患者への補償を担当している。
42
全体的に、国際協調会議(“ICH”)のメンバーとして、日本の薬品法規は基本的に米国やEUと類似している。非臨床研究を行うのは,新たな化学や生物物質の健康安全性を証明するためである。非臨床研究は経済協力と発展組織の要求を反映した日本の良好な実験室実践(GLP)の原則に従わなければならない。現在,日本とEUはGLPに対して相互承認の合意があり,生成されたEUの要求に応じたデータは日本当局に受け入れられる。アメリカと似たような合意はない。
日本での臨床試験はICHの良好な臨床実践ガイドラインやGCPに基づく日本の法規に従って行わなければならない。臨床試験のスポンサーが日本国内で成立していない場合は,日本国内でその守護者として実体を指定しなければならず,その実体はスポンサーを代表して行動することを許可されるべきである。スポンサーは臨床試験保険を購入し,業界協定に基づいて,試験による傷害のための共通の賠償政策を策定しなければならない。人体臨床研究が開始される前に、スポンサーは研究製品の安全性の評価を完成し、そして参加機関のIRBの同意を経て、事前に当局に臨床試験通知と臨床試験方案を提出しなければならない。当局が通知をコメントしない場合,発起人は臨床試験を継続することができる。試験案または提出された他の情報の任意の重大な変更は、IRBの承認を得て当局に通知しなければならない。臨床試験で使用される薬品はGMPで生産されなければならない。
日本で薬を販売するためには、私たちは規制部門の承認を得なければならない。研究薬物の規制承認を得るためには、私たちは新薬申請を提出しなければならない。新薬応用の評価は製品のリスク-効果バランスの評価に基づいており、評価の基礎は製品の品質、安全性と有効性に関する科学標準である。臨床データの数量と品質は承認決定の重要な決定要素である。海外で発生した臨床試験データはICHの提案に適合したパケットの一部として受け入れられている。通常,日本の参加者の中で臨床試験を行い,データが日本の人口に対して外挿可能であることを確保する必要がある。PMDAはデータ適合性審査、良好な臨床実践の現場検査及び現在の良好な製造実践に符合する監査と詳細データ審査を担当する。審査組織が審査を完了すると、この事項は専門家諮問委員会によって審議され、政府は委員会の積極的な提案に基づいて承認される。
孤児薬の指定と排他性
この製品が何らかの“難治性疾患”や患者規模が50,000名以下に制限されている患者を治療するために設計されていれば,高度な医療ニーズを満たしていることが証明されれば,孤児に指定された薬物製品を得ることができる可能性がある。孤児薬物に指定された薬品は,財政援助,研究費税収減免,申請と相談費減免,PMDAの指導と提案,優先審査,および薬品の再審期間を最長10年に延長する権利がある。この製品の薬品価格を計算する際には,新薬創設保険料も適用される。
開発と優先審査案を加速させる
標準審査制度の下で、承認過程全体に12ヶ月を要する。衛生部は特に重要で革新的な薬物のためにいくつかの加速開発または審査計画を導入した
新冠肺炎など公衆の健康に深刻な影響を与える可能性のある疾病伝播に必要な製品に対して特殊な審査制度を実施する。これは、限られた状況で使用され、他に利用可能な治療法がなく、特定の外国での使用が許可されているためである。迅速承認審査プログラム、GMP審査、国家認証、コンテナ、包装、ラベルなどの特殊な例外があります。
“医薬品経営許可証”
承認要求に加えて、適切な種類のライセンスを持たなければならず、メーカーは日本で商業流通を行うことができる。ライセンスを取得するためには、製造業者または販売業者は、少なくともいくつかの製造、マーケティング、品質、およびセキュリティスタッフを雇用しなければならない。薬物製造·販売業務の発行規定には,薬剤師1名を委任する一般薬物市場コンプライアンス主任など,品質制御を遵守する良好な品質規則と上場後の安全監査の良好な警戒規則がある。
43
製品のみの承認を持つ非日本会社は、自ら流通するのではなく、日本で適切なライセンス所持者を指定して商業的に製品を流通させることができる。ライセンスの有効期限は五年です。PMD法案は,他国で生産された薬品を日本に輸入·販売する際にマーケティング許可証を取得することを要求している。また,外国メーカーの製造場所ごとに認証を受け,日本で販売されている薬品製造場所となることも求められている。
規制排他性
上場許可保有者は新薬の発売後調査を行わなければならず、衛生部は上場承認後の特定の期間内に、再審査を通じて有効性と安全性を再確認しなければならず、一般的に少なくとも6年である。リスク管理計画の概念は再検討に含まれている。このような点で、模造薬の申請は再審が完了するまで提出できない。この間、ブランド製品は後発薬の影響を受けなかった。
いずれの薬物も,部長が特に延長が必要であると考えていれば,厚労省は再審期間を10年以下に延長することができる。一般に,新たな活性成分を含む薬剤は8年の再審期が得られ,新投与経路の薬剤は6年の再審期が得られる。この点で,小児科薬には具体的な期限はないが,再審期間は必要に応じて延長(10年以下)することができ,小児科投与量や用法再審に必要な臨床データを収集することができる。
知的財産権保護
特許期間は一般的に出願日から20年である。しかし、薬品、再生医学製品の安全を保障するなどの法律法規は実施できないため、特許期間は最長5年延長できる。医薬品承認に関する情報が記載されているOrange Bookもありますが、これはアメリカ式の特許関連ではありません。衛生部は後発薬を承認せず,原薬の物質特許や応用特許が満了するまで,有効成分の生産が可能となる。
定価決定
この製品は定価の承認を得て、医療保険の償還を申請することができます。承認を得て発売されると、薬品はまた良好な生産規範の基準に基づいて、発売後に定期的に安全と品質に対して警戒を維持する。日本では、国家健康保険システムは、どの薬品が精算を受ける資格があるかを規定し、厚生労働省がこの価格表の製品の価格を決定する薬品価格表を維持している。市場承認後、メーカーや販売業者はMHLWと補償価格の交渉を開始し、補償価格は通常60日から90日以内に決定される。歴史的には、日本は年2回予定の価格調整を行い、これをコスト低減の方法としているが、最近では、この2年間で追加調整が行われている。しかしながら、小児科使用に指定された革新的または有用と考えられる新製品、または孤児または小人口疾患のための新製品は、定価プレミアムを取得する資格がある可能性がある。政府はまた、可能であれば後発薬の使用を促進した。
世界の他の地域の規制
カナダ、EU、米国以外の他の国、例えば中東、アフリカ、ラテンアメリカ、アジア諸国では、臨床試験、製品許可、定価、精算の要求は国によって異なる。また,臨床試験はGCP要求および“ヘルシンキ宣言”からの適用法規要求と倫理原則に基づいて行わなければならない。
もし私たちが適用される外国監督管理要求を遵守できない場合、私たちは罰金、規制許可の一時停止または撤回、製品のリコール、製品の差し押さえ、経営制限、刑事起訴などに直面する可能性がある。
保証と精算を請け負う
新薬製品の商業化成功は政府衛生行政部門、私営健康保険会社とその他の組織によるこれらの薬物製品の清算程度にある程度依存する。米国では、個人健康保険会社や健康維持組織などの政府当局や第三者支払者が、どのような薬物製品のためにお金を払い、精算レベルを確立するかを決定している。政府と個人支払者の獲得可能性と精算範囲は大多数の患者が薬物を負担できる鍵である
44
製品です。薬品の販売は国内外の薬品コストが健康維持、管理保健、薬局福祉と類似の医療管理組織が支払う程度、あるいは政府衛生行政当局、個人健康保険会社と他の第三者支払人が精算する程度に大きく依存する。
アメリカの医療業界と他の地域の主な傾向の一つはコストコントロールだ。政府当局と第三者支払者は、特定の薬品のカバー範囲や精算金額を制限することでコストを抑制しようとしている。第三者決済者は、価格に挑戦し、医療の必要性を審査し、医療製品、療法、サービスの費用対効果を審査するとともに、それらの安全性と有効性を疑問視するようになっている。私たちの製品のための精算は特に難しいかもしれません。ブランド薬と医者の監督下で使用される薬物は往々にして価格が高いからです。私たちは、私たちの製品の医療の必要性と費用効果、FDAと外国の承認を得るのに必要なコストを証明するために、高価な薬物経済学研究を行う必要があるかもしれない。このような研究は私たちが開発した製品の遅延や不利なカバー範囲をもたらすかもしれない。私たちの候補製品は医学的に必要で費用効果があると思われないかもしれない。政府または他の第三者支払人から製品の保証と精算承認を得ることは時間がかかり、高価なプロセスであり、これは、各支払人に科学的、臨床的、費用効果的なデータを提供して、個々の支払人に基づいて私たちの製品を使用するために必要かもしれないが、保険と十分な精算を得ることは保証されないかもしれない。支払者が製品に保険を提供することを決定することは、十分な返済率を承認することを意味するものではない。また、支払者が1つの商品に保険を提供することを決定し、他の支払者もその商品に保険を提供することを保証することはできない。製品開発への投資の適切なリターンを実現するために、十分な第三者精算が得られない可能性があり、十分な価格レベルを維持することができる。精算が得られない場合や限られたレベルの精算のみが提供されなければ、私たちが開発に成功した任意の候補製品を商業化することに成功できないかもしれません。
多くの国では、国家衛生システムの一部として、薬品価格は異なる価格制御メカニズムの制約を受けている。一般的に、この制度での薬品価格はアメリカを大きく下回っています。他の国は会社が自分の薬品価格を固定することを許可していますが、会社の利益を監視してコントロールしています。そのため,米国以外の市場では,薬品の精算が米国より減少する可能性がある。
アメリカでは、新薬製品の精算に関する主な決定は通常衛生と公衆サービス部に属するCMSによって行われる。CMSは新薬製品がどの程度連邦医療保険の下でカバーと精算されるかを決定し、個人支払者はよくCMSに大きく従う。しかし、第三者支払者の間には統一された薬品保険と精算政策がなく、支払者間の薬品保険と精算レベルに大きな差がある可能性がある。第三者支払人の保険範囲と精算は多くの要素に依存する可能性があり、第三者支払人が薬品の使用が以下の条件を満たすかどうかの確定を含む
私たちが商業化しているどの製品にも保険や精算があることを確実にすることはできません。もし保険と精算があれば、清算レベルはいくらですか。カバー範囲はまた、FDAや同様の外国規制機関が製品を承認する目的よりも限られている可能性がある。将来の保険·精算は、事前に許可された要求や、米国や国際市場における孤児薬物製品の販売率の変化など、より多くの制限を受ける可能性がある。清算は私たちが規制部門の承認を受けた任意の製品の需要や価格に影響を及ぼすかもしれない。
MMAはMedicare Part D計画を創立し、Medicare受益者に自発的な処方薬の福祉を提供した。D部によると、連邦医療保険受益者は、個人実体が提供する処方薬計画に参加することができ、これらの計画は外来処方薬の保険を提供する。すべての連邦医療保険薬物計画は少なくとも連邦医療保険が設定した標準保険レベルを提供しなければならないが、D部分処方薬計画発起人はすべてのD部分薬物に費用を支払う必要はなく、各D部分処方薬計画は自分の薬物処方を開発し、それがどの薬物および被覆のレベルまたはレベルをカバーするかを決定することができる。しかしながら、D部分処方薬処方は、必ずしも各カテゴリまたはカテゴリのすべての薬剤を含むとは限らないにもかかわらず、各治療カテゴリおよびカバーされたD部分薬剤カテゴリの薬剤を含む必要がある。D部分の処方薬計画に使用されるどの処方も薬局と治療委員会が開発·審査しなければならない。政府が支払った費用
45
処方薬のいくつかの費用は私たちが発売承認された薬に対する需要を増加させるかもしれない。D部分の処方薬計画がカバーしているどの製品のいかなる交渉価格も私たちが入手可能な価格より低いかもしれません。また,MMAは連邦医療保険受益者の薬品福祉にのみ適用されるが,個人支払者は自分の支払率を設定する際に連邦医療保険カバー政策や支払制限に従うことが多い。MMAによる任意の支払い減少は、非政府支払者支払いの同様の減少をもたらす可能性がある。
MedicaidまたはMedicare Part B計画に従って連邦補償を獲得するか、または米国政府機関に直接販売する薬品の場合、製造業者は割引を340 B薬品定価計画に参加する資格のあるエンティティに拡大しなければならない。製品を与えるために必要な340 B割引は,メーカー平均価格(AMP)とメーカーから報告された医療補助返金金額から計算される。ACAは2010年から、現在の法律状況に応じて、これらの新しい条件に適合するエンティティ(児童病院を除く)は、孤児薬品340 B割引定価を取得する資格がないことにもかかわらず、340 B割引定価を取得する資格があるエンティティタイプを拡大した。必要な340 b割引は、製造業者の平均価格またはAMPおよび医療補助ポイントデータに基づいて決定されるため、Medicaid返却式およびAMP定義の上記の修正は、必要な340 b割引を増加させる可能性がある。2009年の“米国回復·再投資法案”は、同一疾患の異なる治療法の有効性を比較するために連邦政府に資金を提供した。この研究の計画は2012年にHHS,ヘルスケア研究,品質局,NIHによって公表され,研究状況と関連支出の報告が定期的に国会に提出される。比較有効性研究の結果は,公共または個人支払者の保証政策を許可するためのものではないが,いずれかのこのような薬物や治療しようとしている場合が試験のテーマであれば,研究が候補製品の販売にどのような影響を与えるかは不明である(あれば)。比較有効性研究では,競合他社の薬物にメリットがあり,候補製品の販売に悪影響を及ぼす可能性もある。もし第三者支払者が私たちの薬物が他の利用可能な療法と比較して費用効果があると思わない場合、彼らは承認後に私たちの薬物をその計画の下の福祉としてカバーしないかもしれない、または、もし彼らがそう思う場合、支払いレベルは利益に基づいて私たちの薬を販売するのに十分ではないかもしれない。
これらの法律および将来の州および連邦医療改革措置は将来的に採択される可能性があり、いずれも連邦医療保険および他の医療保険資金のさらなる減少をもたらし、他の方法で規制の承認を得る可能性のある任意の候補製品の価格、またはそのような候補製品の処方または使用頻度に影響を与える可能性がある。
アメリカ以外の多くの国では、薬品の定価は政府によってコントロールされている。例えば、連合では、様々な国の価格設定と補償プログラムの差が大きい。一部の国では、補償価格を合意した後にのみ、製品を販売することができると規定されている。一部の国は追加の費用対効果評価の完成を要求する可能性があり、特定の治療法の費用対効果を現在利用可能な治療法またはいわゆる衛生技術評価と比較して、精算または定価の承認を得る。他の国では会社が製品の価格設定を許可する可能性があるが,製品数を監視し,処方を制限するための指導意見を医師に発表する。各国が医療支出を管理しようとするにつれ,薬品価格や使用を抑える努力が続く可能性がある。
従業員と人的資本
2023年12月31日現在、384人のフルタイム従業員を抱えています。私たちの従業員チームのうち、151人の従業員が直接研究開発に従事し、残りは行政、商業、運営支援を提供している。私たちの従業員の中の一人も労働組合代表でもなく、集団交渉協定のカバー範囲もない。私たちは従業員と仲がいいと思います。
私たちの人的資本は、神経変性疾患による苦痛を終わらせる目標を達成するのを助けるために不可欠だ。私たちの人的資本資源の目標は、適用された場合、既存の従業員と新規従業員を識別、募集、維持、激励、統合することを含む。私たちの株式激励計画の主な目的は株に基づく報酬奨励と現金に基づく業績ボーナス奨励を授与することによって、選定された従業員、顧問と取締役を吸引、維持、激励することである。
利用可能な情報
我々は,Form 10−Kの年次報告,Form 10−Qの四半期報告,Form 8−Kの現在報告,依頼書,その他の情報を米国証券取引委員会に提出した。私たちがアメリカ証券取引委員会に提出した書類は、アメリカ証券取引委員会のウェブサイトwww.sec.govで調べることができます。ウェブサイトもありますhttp://www.amylyx.comです私たちはウェブサイトの投資家の一部で、私たちの年間報告書10-K表、四半期報告を含む、アメリカ証券取引委員会に提出または提出した書類を無料で提供します
46
表格10-Q、表格8-Kの最新報告、およびこれらの報告に対する任意の証拠および修正。我々は、このような材料を米国証券取引委員会に電子的にアーカイブしたり、そのような情報を提供したりした後、合理的で実行可能な範囲でこれらの情報をできるだけ早く提供する。私たちのウェブサイト上の他の情報は、本報告書または私たちがアメリカ証券取引委員会に提出した任意の他の報告書の一部ではない。
環境、社会、ガバナンス(ESG)
私たちがいつか神経変性疾患による苦痛を終わらせる使命を駆動する価値観は私たちのビジネスの核心だ。大胆さ、好奇心、参加度、責任感、真実性に対する私たちの約束は私たちを国際社会の責任ある一員にさせた。私たちは私たちが世界に与える影響、私たちがどのような点でよくやっているのか、そして私たちがどこで改善できるのかをもっとよく知るために努力している。2023年にはESG委員会を設立し、この仕事を指導し、成長過程で私たちの環境、社会、ガバナンスの旅を正規化するのを手伝ってくれました。その委員会は行政指導チームに報告し、職能を越えた従業員チームで構成されている。仮想会社として、私たちはサプライヤーやサプライヤーとの協力の重要性を認識しており、これらのサプライヤーのやり方は持続可能な発展への約束を示している。私たちの最近の急速な成長に伴い、私たちは現在、調達機能を実施し、新しいサプライヤーの持続可能な努力を評価するプロセスを導入している。
環境.環境
会社として、私たちは急速に発展している。私たちの多くの従業員は遠隔作業を選択しているが、私たちはすでに施設で回収計画や自動照明などの機能を実施している。Amylyxは、その契約製造、パッケージ、および供給パートナーが、現地の法律および法規に適合し、これらの計画を検討するための定期監査の能力を保持する同様のESG計画を維持すると予想している。それにもかかわらず、私たちはこの分野でやるべき仕事があり、私たちの環境影響を研究することが私たちの未来の仕事の一部になることを認める。
社交的である
人々に対する私たちの約束は私たちの核心的な価値観に現れ、私たちの行動に反映される。著者らは神経変性疾患患者の潜在的な治療方法の発見と開発に取り組んでいる。
私たちが注目している病気を患っている人は私たちの真の北であり、私たちは筋萎縮性側索硬化症や他の神経変性疾患コミュニティと密接に協力し続けている。これには,我々の臨床試験設計と材料募集に対する彼らの意見を求めることと,定期的にこれらの個人や看護者と接触し,多学科看護と承認された治療の障害を含む複数のテーマへのフィードバックを聞くことが含まれる。神経変性疾患コミュニティのニーズに耳を傾け、理解し続けることを確保するために、私たちは個人を私たちの全社会議で毎月彼らの観点を共有するように招待した。私たちはまた私たちの宣伝や他の外部材料でALS患者と彼らの介護者を紹介できることを誇りに思っています。我々が新たな候補薬を推進する過程では,すべての人が平等な機会を確保して臨床試験に参加すること,特に性別,年齢,社会経済的背景,皮膚色,人種などの人からなる臨床試験に取り組んでいる。
私たちの製品が承認され、商業的に使用されている地域では、教育や参入支援を含む一連のサービスを提供する専門チームを持っています。条件に合った患者に対して、私たちは彼らが負担できるように経済的支援計画を提供する。
有用な薬物を薬物を取得する資格のある多くの人に提供するためには,各国のルールに従い,規制手続きを通過することが望ましいと考えられる。しかし、場合によっては、承認されていなくても、あるいは研究中であっても、本来治療を得ることができない人に治療機会を提供することができる研究用薬剤を提供することができるかもしれない。
雇用主としては、組織の最上位層で異なる観点や背景を代表する多様性も重要である。私たちの7人の幹部のうち3人は女性だ。時間が経つにつれて、私たちがより多くのメンバーを増やすにつれて、私たちの取締役会は多様性を増やすことに取り組んでいます。現在、私たちの6人の取締役会のメンバーのうち、2人は女性で、1人は人種多様性だ。私たちはすでに私たちのウェブサイトで取締役会多様性行列を発表した。
私たちは職員たちの健康と福祉に関心を持っている。私たちは提供します
47
統治する
私たちの取締役会は会社の業務と管理を監督する責任があります。私たちの管理実践の一部として、私たちは高い基準の道徳基準に取り組んでおり、これは私たちの商業行為と道徳基準に反映され、私たちの役員、高級管理者、従業員、指定エージェントに適用される。このコードはわが社のサイトに公開されています。私たちには独立した会長がいて、私たちの6人の取締役会のメンバーのうち4人は独立している。私たちの監査委員会、指名委員会、会社管理委員会、報酬委員会はすべて独立した役員で構成されています。
48
第1 A項リスク要因です
我々の業務と見通しを評価する際には、本年度報告書や米国証券取引委員会に提出された他の文書に記載されている他の情報に加えて、以下のリスク要因を慎重に考慮すべきである。私たちの普通株に投資することは高い危険と関連がある。もし実際に以下のいかなるリスクと不確定要素が発生すれば、私たちの業務、将来性、財務状況と経営結果は重大な不利な影響を受ける可能性がある。以下に説明する危険は詳細ではなく、私たちが直面している唯一の危険でもない。新しいリスク要素は時々出現する可能性があり、いかなる要素或いは要素の組み合わせが私たちの業務、将来性、財務状況と運営結果に与える影響を予測できない。
リスク要因の概要
私たちの財務状況と資金需要に関連するリスク
AMX 0035または将来の候補製品の商業化に関するリスク
私たちの現在と未来の候補製品の発見と開発に関するリスク
49
私たちの第三者への依存に関するリスク
私たちの知的財産権に関するリスクは
私たちの業務運営、従業員事務、管理成長に関するリスク
私たちの普通株に関するリスクは
50
私たちの財務状況と資金需要に関連するリスク
私たちは過去に大きな損失を受けました。もし私たちが承認した製品から十分な収入を発生させて私たちの費用を支払うことができなければ、未来にもっと多くの損失が生じるかもしれません。
生物製薬製品開発への投資は非常に高い投機性があり、それは大量の前期資本支出を必要とし、重大なリスクが存在するため、即ち任意の潜在的な候補製品は十分な効果或いは受け入れ可能な安全状況を証明できず、監督部門の承認を得ることができず、商業上実行可能ではない。私たちはすでに大量の資源を投入して、私たちの製品開発の仕事とRELYVRIOとALBRIOZAの商業化において、RELYVRIOはすでにFDAの許可を得て、ALBRIOZAはカナダ衛生部の条件付きマーケティングの許可を得たが、私たちは限られた時期にアメリカとカナダの製品販売から収入を得ただけだ。私たちは引き続き臨床開発、商業化、より多くの司法管轄区域とより多くの適応の承認、持続的な運営に関する巨額の研究開発とその他の費用を発生させる。設立以来、著者らは著者らの大部分の財務資源と努力を研究開発に投入し、臨床前研究と臨床試験を含み、商業化と商業化活動のために準備した。私たちの財務状況と経営結果は、私たちの収入、支出、純収益(赤字)を含めて、四半期ごとに毎年大きく変動する可能性があります。したがって、今後の経営業績の指標として、いかなる四半期や年度の業績にも依存してはいけません。また、純損失と負のキャッシュフローは、将来的には私たちの株主権益や運営資本に悪影響を及ぼす可能性がある。2023年12月31日までの累計赤字は3.049億ドル。私たちは未来に再び重大な損失が出る可能性があり、私たちの財務業績はRELYVRIOのアメリカとALBRIOZAのカナダでの持続的に成功した商業販売に高度に依存するだろう。
私たちは次のような状況が発生すれば、私たちの費用が大幅に増加するかもしれないと予想している
私たちは、アメリカとカナダにおけるAMX 0035の商業化を支援するために、販売とマーケティング、流通、および製造能力を含むインフラを拡大し続けています。2023年12月31日現在、384人のフルタイム従業員を抱えています。
FDA、カナダ保健省、EMA、または他の規制機関が私たちが現在予想しているのではなく、臨床試験や他の研究を要求する場合、あるいは
51
我々の臨床試験またはAMX 0035または我々が開発する可能性のある任意の他の現在または将来の候補製品のための適切な製造スケジュールを確立するか、またはAMX 0035または任意の他の候補製品の開発を完了する上で、任意の遅延がある。
製品販売収入の歴史が限られており、長期的な持続可能な収益性を実現したり維持できない可能性があることを確認しました。
私たちが収入を創出し、利益を達成する能力は、米国におけるRELYVRIOとカナダのALBRIOZAの商業化を含む、製品開発を成功させ、商業化に必要な規制承認を得る能力に依存する。私たちは製品販売収入の能力が私たちの以下の成功に大きくかかっていることを確認した
アメリカのRELYVRIOとカナダのALBRIOZA以外に、私たちはまだ他の許可された製品を商業販売に発売していません。RELYVRIOやALBRIOZAの商業化には多大なコストがかかり続けると予想され,我々が開発している別の候補製品が商業販売を許可されても,どのような承認された候補製品の商業化にも巨額のコストが生じることが予想される。たとえ私たちがRELYVRIOおよびALBRIOZAの販売から収入を得始めたとしても、AMX 0035が他の管轄区域で承認されない限り、または私たちの現在または未来の候補製品の他の適応または他の製品が将来承認されない限り、長期的な持続可能な収益性を達成または維持することができない可能性がある。これらの活動に関連する不確実性とリスクのため、私たちは収入の時間と金額、将来の損失の程度、または利益を維持することができるかどうかを正確かつ正確に予測することができない。
私たちが利益を維持できなかったことは、私たちの普通株の市場価格を低くし、資金を調達し、業務を拡大し、製品を多様化し、あるいは運営を継続する能力を弱める可能性がある。もし私たちが過去のように損失を被ったら、投資家は何の投資リターンも得られず、すべての投資を失うかもしれない。
私たちの運営の歴史は限られていて、現在は一つの商業製品AMX 0035しかありません。アメリカではRELYVRIOと呼ばれ、カナダではALBRIOZAと呼ばれています。これは私たちの未来の生存の将来性を評価することを難しくするかもしれません。
ここ数年間、私たちはまだ臨床段階から商業段階へ移行する比較的早い段階にある。今まで、私たちの業務は主に組織、人員の配備と会社に資金を提供し、資金を調達し、臨床前研究と臨床試験を含む研究と開発活動を展開し、そして最近AMX 0035を準備し、それを商業化することに限られている。長期的な持続に基づいて相当な収入を創出したり、長期製品の商業化に成功するために必要な販売やマーケティング活動を行うことができることは証明されていません。2022年6月、AMX 0035はカナダ保健省から筋萎縮性側索硬化症を治療する条件のマーケティング許可を取得し、1つの条件は、フェニックス試験および他の計画におけるまたは行われている研究のデータを提供することである。
52
2022年9月、AMX 0035は成人ALSの治療のためにFDAの発売許可を得た。2024年1月、欧州委員会は、EUにおけるAMX 0035の条件付きマーケティング許可のMAAに対するCHMPの負の意見を採択することを確認した。我々は引き続き我々の全世界フェニックス3期臨床試験を完成させることに集中し、ALS患者におけるAMX 0035の治療効果と安全性に関するより多くのデータを提供する。もしフェニックスが支持を表明したら、私たちは私たちがこのような承認を得ることが保証されないにもかかわらず、できるだけ早くEUで承認を求める予定だ。
また、私たちがアメリカでRELYVRIOを承認した一部として、私たちは発売後、ALBRIOZAはカナダで承認されましたが、条件があり、満たさなければ、AMX 0035を商業化し続ける能力を弱める可能性があります。2022年9月7日のFDAの末梢と中枢神経系薬物諮問委員会のAMX 0035によるALS治療に関する第2回会議で、もし我々のフェニックス試験が成功しなければ、自発的に市場から製品を上下させることを含む患者に正しいことをすると表明した。鳳凰衛視のデータが利用可能な時、私たちは規制部門と協議するつもりだ。規制部門はまたAMX 0035を市場からリコールすることを要求するかもしれない。われわれが米国で承認したRELYVRIOの一部として,発売後にマウスやラットへの発癌性研究,ヒトボランティアにおける薬物−薬物相互作用研究,腎臓や肝臓障害の被験者での研究が求められている。これらの研究の結果は、フェニックス試験および任意の可能な脱退を含めて、私たちの業務に実質的な悪影響を及ぼす可能性がある。
また,我々はカナダ保健省が規定しているALBRIOZAによるALS治療のマーケティング許可のすべての条件を満たしていない可能性がある。もし私たちがそれをできなかったら、私たちはカナダ保健省が適用した追加条件の制約を受けて、あるいはALBRIOZAの商業化を止めなければならないかもしれません。これは私たちの利益見通しに影響を与えるかもしれません。
私たちの業務目標を達成する時、私たちは予見できない費用、困難、複雑な状況、遅延、および他の既知または未知の要素に遭遇する可能性がある。したがって、あなたは私たちの将来性を考慮して、コスト、不確実性、遅延、会社が初期の商業段階でよく遭遇する困難、特に私たちのような製薬会社を考慮しなければなりません。もし私たちがこのような活動についてもっと長い運営歴史を持っていれば、あなたは私たちの未来の成功や生存能力に対するどんな予測もそんなに正確ではないかもしれない。
私たちの四半期と年間経営業績は将来的に変動するかもしれません。したがって、私たちは研究アナリストや投資家の期待を達成できないかもしれません。これは私たちの株価の下落を招き、私たちの融資や融資能力にマイナス影響を与え、独立会社としての私たちの生存能力にマイナスの影響を与える可能性があります。
過去、私たちの財務状況と経営業績はそれぞれ異なり、未来は様々な要素のため、私たちの財務状況と経営業績は引き続き四半期と年度の間に変動し、その中の多くの要素は私たちがコントロールできない。これらの変動を引き起こす可能性のある当社の業務に関連する要因には、本年度報告に記載されている他の要因が含まれています
53
ここで言及した様々な要素と他の要素のため、私たちは私たちの前のどの四半期や年度の業績も私たちの未来の経営業績の指標として依存すべきではありません。
私たちの財務業績は四半期間と年度間に大きく変動する可能性がありますので、私たちの運営結果を期ごとに比較することは、私たちの将来の業績の良い指示ではないかもしれません。いずれの特定の四半期においても、私たちの経営業績は証券アナリストや投資家の予想を下回る可能性があり、これは私たちの株価を下落させる可能性がある。私たちが行っているまたは未来の1つまたは複数の臨床試験の意外な臨床試験の結果もまた、私たちの株価を下落させる可能性がある。
私たちは未来に私たちの財政的需要を満たし、私たちの業務目標を達成するために多くの追加資金を必要とするかもしれない。もし私たちが必要な時に資金を集めることができなければ、私たちは私たちの製品発見と開発活動や商業化努力を延期、減少、または廃止させることを余儀なくされるかもしれない。
設立以来、私たちの業務は大量の現金を消費した。私たちは引き続き大量の資金を費やし、監督管理の許可を得た司法管轄区域でAMX 0035を商業化し、より多くの適応とより多くの候補製品の臨床前と臨床開発においてAMX 0035の臨床開発を継続する予定である。AMX 0035または私たちが開発した任意の他の現在または未来の候補製品のために追加のマーケティング承認を得ることができない場合、AMX 0035および任意の他の現在または未来の候補製品を継続して開発し、私たちの運営に資金を提供するために、多くの追加の現金が必要になるかもしれません。また、我々の開発や商業化努力の過程で、他の予期せぬコストが発生する可能性がある。私たちが行っていることと期待される臨床試験の設計と結果は高い不確実性があるため、私たちが開発した任意の候補製品の開発と商業化に成功するために必要な実際の数量を合理的に見積もることはできない。
私たちの将来の資本需要は多くの要素に依存しています
54
私たちは許容可能な条件で追加資金を提供するかどうか、あるいは根本的にできないかどうかを確認することができない。ウクライナとイスラエルの持続的な衝突を含む地政学的不安定と衝突による様々なグローバル市場経済の不確定性による挑戦、世界の信用と金融市場は最近の間に深刻な変動と中断を経験し、流動性と信用供給の減少、消費者自信の低下、高インフレ率と高金利、および経済安定面の不確定性を含む。株式市場や信用市場が悪化すれば、任意の必要な債務や株式融資をより困難にし、コストをより高くしたり、希釈度を高くしたりする可能性がある。
私たちが約束した追加資本源はありません。もし私たちが追加資本(必要であれば)、十分な金額、または私たちが受け入れられる条項を集めることができない場合、私たちはAMX 0035または任意の他の現在または未来の候補製品または他の研究開発計画の開発または商業化を大幅に延期、削減、または停止しなければならないかもしれません。私たちは、他の場合が望ましいのではなく、AMX 0035および任意の他の現在または未来の候補製品のためのパートナーを探す必要があるかもしれないし、あまり有利でない条項でAMX 0035および任意の他の現在または未来の候補製品のためにパートナーを探すか、または不利な条項で放棄することができるかもしれませんが、そうでなければ、私たちは開発または商業化を求めます。上記のどの事件も、私たちの業務、将来性、財務状況と経営結果を深刻に損害し、私たちの普通株価格の下落を招く可能性があります。
私たちは、AMX 0035のアメリカとカナダの商業販売から得た収入と、私たちの既存の現金、現金等価物、および短期投資は、本年度の報告日後少なくとも12ヶ月の予想される運営と資本支出需要を満たすのに十分になると信じています。しかし、私たちの推定は間違っていることが証明されるかもしれないし、私たちは現在予想されているよりも早く私たちが利用できる資本資源を使用することができる。さらに、変化する状況-そのいくつかは私たちの制御を超えているかもしれない--私たちの資本消費速度は私たちの現在の予想よりも大きく速く、私たちは計画よりも早く追加資金を求める必要があるかもしれない。
追加資本の調達は私たちの株主に希釈し、私たちの運営を制限したり、私たちの技術や候補製品に対する権利を放棄することを要求するかもしれません。
私たちは私たちが計画した業務に関連した費用が引き続き増加すると予想している。私たちが大量の収入を持続的に生成し、製品販売から持続的な収益性を示すことができない限り、私たちは、公開または私募株式発行、特許使用料または債務融資、協力、許可手配、または他のソース、またはこれらの任意の組み合わせによって将来の現金需要に融資する必要があるかもしれない。また、有利な市場条件や戦略的考慮により、現在または将来の運営計画のために十分な資金があると考えても、追加の資本を求めることができる。
私たちが普通株、転換可能証券、または他の株式証券を売却することによって追加資本を調達する場合、あなたの所有権権益は希釈される可能性があり、これらの証券の条項は清算または他の特典および逆希釈保護を含む可能性があり、これは普通株主としての権利に悪影響を及ぼす可能性がある。さらに、債務融資(実行可能であれば)は、固定支払義務をもたらす可能性があり、追加債務を招く、資本支出を招く、留置権を作成する、株式を償還する、または配当を宣言するなど、私たちの具体的な行動をとる能力を制限する制限的な契約を含むいくつかの合意に関連する可能性があり、これらは、私たちの業務を展開する能力に悪影響を及ぼす可能性がある。融資獲得には、我々の経営陣が多くの時間と精力を投入する必要がある可能性もあり、彼らの注意を日常活動から移す可能性があり、我々の経営陣がAMX 0035や任意の将来の候補製品の開発や商業化を監督する能力に悪影響を及ぼす可能性がある。
もし私たちが第三者との協力やマーケティング、流通、または許可手配を通じてより多くの資金を調達すれば、私たちは私たちの技術、将来の収入源、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になる可能性のある条項でライセンスを授与しなければならないかもしれない。
税法の変化は私たちの業務と財政状況に悪影響を及ぼすかもしれない。
米国連邦、州、地方、国際所得税に関する規則は立法過程に参加する人員およびアメリカ国税局とアメリカ財務省の審査を受け続けている。税法の変化(これらの変化は追跡力を持つ可能性がある)は、私たちまたは私たちの普通株の所有者に悪影響を及ぼすかもしれない。近年、このような変化は多く発生しており、未来も変化し続けるかもしれない。将来の税法の変化は、私たちの業務、キャッシュフロー、財務状況、または運営結果に実質的な悪影響を及ぼす可能性があります。私たちは強く求めています
55
投資家は税法の潜在的な変化が私たちの普通株投資に与える影響について彼らの法律と税務顧問に相談します。
金融サービス業の不利な事態の発展に影響を与え、例えば流動資金、金融機関又は取引相手の違約又は不履行に関する実際の事件又は懸念は、我々の現在及び予想される業務運営及び我々の財務状況及び運営結果に悪影響を及ぼす可能性がある。
金融機関、取引相手または金融サービス業他社の限られた流動性、違約、不良業績または他の不利な事態の発展に影響を与える事件、またはそのような事件または他の同様のリスクへの懸念または噂は、過去に発生し、将来的に市場全体の流動性問題を引き起こす可能性がある。私たちは現在、このような事件を経験した金融機関とは投資していませんが、私たちと関係のあるどの金融機関も破産管理状態に置かれていれば、このような資金を得ることができないかもしれません。また、吾等と業務往来のあるいずれか一方が当該等の金融機関と締結した手形や貸借手配に基づいて資金を取得できなければ、当該等の当事者が吾等に債務を支払ったり、新たな商業手配を締結して吾等に追加金を支払うことを要求する能力が悪影響を受ける可能性がある。
インフレと金利の急速な上昇は、以前に発行された金利が現在の市場金利よりも低い国債の取引価値を低下させる。米国財務省、連邦預金保険会社、および連邦準備委員会は、このようなツールの売却による潜在的損失のリスクを低減するために、金融機関が保有するいくつかの政府証券を保証する金融機関に250億ドルまでの融資を提供する計画を発表しているが、金融機関の顧客引き出しの広範な需要または金融機関の即時流動性に対する他の流動性需要は、このような計画の能力を超える可能性がある。また、将来的に他の銀行や金融機関が閉鎖された場合、米国財務省、連邦預金保険会社、連邦準備委員会が未加入資金のルートを提供する保証はなく、タイムリーにそうする保証もない。
私たちの銀行関係を必要または適切に評価する必要があると思いますが、私たちは、私たちの現在と将来の業務運営に資金や資本化を提供するのに十分な資金源を得て、私たちの要素に影響を与える深刻な被害を受ける可能性があります。これらの要因は、私たちと直接財務的に手配されている金融機関、あるいは金融サービス業や経済全体に影響を与えています。他にも、これらの要因には、流動性の緊張または失敗、様々な金融、信用または流動資金協定または手配された義務を履行する能力、金融サービス業または金融市場の中断または不安定、または金融サービス業会社の将来性に対する懸念または否定的な予想が含まれる可能性がある。
さらに、米国または国際金融システムに対する投資家の懸念は、より高い金利またはコスト、より厳しい財務および運営契約、または信用および流動性源を得るための体系的な制限を含む、あまり有利ではない商業融資条項を招く可能性があり、それによって、私たちは融資を受けにくく、さらには融資を受けることができない。他のリスクに加えて、利用可能な資金または現金および流動資金源の減少は、運営費用、財務義務、または他の義務を履行する能力に悪影響を及ぼす可能性があり、私たちの財務および/または契約義務に違反したり、連邦または州賃金および労働時間法違反を招いたりする可能性がある。上記のいずれかの影響、または上記の要因または他の関連または同様の要因に起因する任意の他の影響は、我々の流動資金、私たちの現在および/または予想される業務運営、ならびに財務状態および運営結果に重大な悪影響を及ぼす可能性がある。
AMX 0035または将来の候補製品の商業化に関するリスク
私たちの販売とマーケティング経験は限られています。もし私たちがアメリカ、カナダ、または他の場所でAMX 0035または任意の他の現在または未来の候補製品を商業化することに成功できなければ、承認された場合、私たちは意味のある追加の製品収入を生成できないかもしれない。
AMX 0035は私たちが商業化した最初の製品です。われわれは現在専門チームを介してカナダでALBRIOZAを販売しており,米国では専門チームを通じてRELYVRIOを販売しており,ALSが比較的まれであること,我々が狙っている他の適応を考慮している。我々は、AMX 0035および任意の将来の候補製品(承認された場合)のマーケティング、販売、および流通のためのグローバルマーケティングおよび販売チームを確立し続けている。筋萎縮性側索硬化症の治療のためのAMX 0035を商業化し、PSP、WS、ADおよび他の適応を治療するためのAMX 0035を商業化し続けるか、または現在または将来承認される可能性のある任意の他の候補製品を商業化するためには、各地域に基づいてマーケティング、販売、流通、管理および他の能力を確立し、または第三者とこれらのサービスを提供しなければならないが、私たちは成功しないかもしれない。
56
私たち自身の販売とマーケティング能力の確立、および第三者とこれらのサービスを実行する計画を達成することはリスクに関連している。例えば、私たちは高価で時間のかかる機関であるアメリカの商業機関を募集して訓練しました。承認された場合、AMX 0035または任意の他の現在または将来の候補製品を商業化することを阻害する要素が含まれるかもしれません
もし私たちが第三者と販売、マーケティング、流通サービスの手配を達成すれば、私たちの製品収入またはこれらの収入流から得られる収益力は、私たちが自分たちが開発した任意の候補製品をマーケティングし、販売する場合よりも低いかもしれません。さらに、私たちは、第三者との販売およびマーケティングAMX 0035または現在または未来の任意の他の候補製品の配置を成功的に達成することができないか、または私たちに有利な条項でそうすることができないかもしれない。私たちはこれらの第三者に対して限られた制御権を持っているかもしれませんが、彼らのいずれかは、AMX 0035または任意の他の現在または未来の候補製品を効率的に販売およびマーケティングするために必要なリソースおよび注意を投入できないかもしれません。もし私たちが販売およびマーケティング能力を成功的に確立できなければ、私たち自身も第三者と協力しても、AMX 0035または他の現在または未来の候補製品を商業化することに成功しないかもしれない。
ALSおよび他の神経変性疾患医療界および支払者に、AMX 0035または任意の未来の候補製品の利点を理解させるために努力しており、これは大量の資源を必要とする可能性があり、決して成功しないかもしれない。AMX 0035または任意の将来の候補製品の複雑さおよび独自性、ならびに私たちの目標適応のために、これらの努力は、通常必要なリソースよりも多くのリソースを必要とする可能性がある。たとえAMX 0035または任意の未来の候補製品が任意の司法管轄区で承認されても、もし私たちが私たちの製品をうまくマーケティングできなければ、私たちはこのような製品から相当な収入を得ることができません。
マーケティング、製造、流通能力を拡大することができない場合、またはAMX 0035をマーケティングおよび販売するために第三者と合意することができない場合、またはマーケティング承認を得た他の現在または将来の候補製品を提供することができない場合、追加の製品収入を生成することができません。
私たちの開発活動が生み出す可能性のある任意の製品を商業化することに成功するためには、私たち自身も他の人とも協力して、マーケティング、薬物警戒、製造、流通能力を拡大し続ける必要があります。私たち自身のマーケティングと流通の発展は、今は、高価で時間がかかり続け、さらなる製品発表を延期する可能性があります。しかも、私たちは私たちがこの能力を成功的に発展させ続けることができるということを確信できない。私たちは、確立されたマーケティングおよび流通能力を利用するために、他のエンティティと任意の承認された候補製品について協力することができるが、もしあれば、有利な条項でこのような合意に到達できないかもしれない。将来のパートナーがAMX 0035または他の現在または将来の候補製品を商業化するのに十分な資源を投入していない場合、または必要な機能を自ら開発することができない場合、私たちは私たちの業務を維持するのに十分な製品収入を生成することができません。私たちは多くの会社と競争して、これらの会社は現在広範で、経験が豊富で、資金の豊富な販売、流通とマーケティング業務を持っていて、採用、採用、訓練とマーケティングと販売人員を維持します。私たちは、承認された場合、第三者がAMX 0035および任意の他の現在または将来の候補製品を販売およびマーケティングするのを助けることを探す過程でも競争に直面している。マーケティングおよび販売機能を実行するための内部チームまたは第三者の支援がなければ、これらのより成熟した会社との競争に成功することができないかもしれない。
ALS、PSP、WS、ADおよび他の神経変性疾患のためのAMX 0035は、現在開発または将来開発されている可能性のある任意の他の候補製品の市場は、私たちが予想しているよりも小さいかもしれない。
57
私たちは神経変性疾患の研究と製品開発に集中している。私たちの市場機会推定は、これらの疾患を有する私たちの人数の推定、私たちが承認した製品ラベルの潜在的範囲、AMX 0035または任意の他の現在または将来の候補製品から利益を得る可能性があるこれらの疾患患者のサブセット、様々な価格設定スキーム、および特定の国の珍しい疾患精算政策についての私たちの理解を含む様々な要因に基づいている。これらの推定は多くの仮定に基づいており,不正確であることが証明されている可能性があり,新しい研究はこれらの疾患の推定発症率や流行率を低下させる可能性がある。まれな適応の場合、市場機会を推定することは特に挑戦的である可能性があり、疫学データはしばしばより一般的な適応よりも限られているので、潜在的な患者集団を評価するための追加の仮定が必要となる可能性がある。私たちは引き続きRELYVRIOをアメリカ、ALBRIOZAをカナダで商業化し、そして他の司法管轄区でAMX 0035をマーケティングし始め、そしてもっと多くの市場動態に関する情報を理解し、そして監督機関とより多くの潜在的なマーケティング審査について接触することによって、私たちの製品の最初の潜在的な市場機会に対する見方は更に精密になる。例えば,米国の筋萎縮性側索硬化症市場は,米国では大きな割合の筋萎縮性側索硬化症患者がより大きな治療センター外で治療を受けており,これらの患者の識別や接触が困難になっているため,我々の最初の推定とは異なる可能性が認められた。さらに、我々は主に筋萎縮性側索硬化症の年間発病率に注目しており、これは、AMX 0035の初期市場機会が時間の経過とともに達成可能な総潜在的市場機会よりも小さい可能性があることを意味する。患者を識別できず、AMX 0035または任意の他の魅力的な市場機会の現在または将来の候補製品を商業化することに成功した場合、私たちの将来の製品収入は予想を下回る可能性があり、私たちの業務は影響を受ける可能性がある。
患者識別の努力は患者集団を解決する能力にも影響する。患者識別における努力が成功しなかったり、効果が期待されなかったりすれば、例えば、診断計画の不足、医療専門家の疾患意識の不足、大型治療センター以外で患者を識別および接触させることの困難さ、または他の原因により、私たちが探している機会全体を解決できない可能性がある。したがって、患者は識別および接触が困難である可能性があり、米国、カナダ、EUおよび他の地域の潜在的な患者数は予想を下回る可能性があり、または患者は他の方法で私たちの製品治療を受けることができない可能性があり、これらはすべて私たちの業務、財務状況、運営結果、および将来性に悪影響を及ぼすであろう。
AMX 0035は、商業的成功を継続するか、または利益を維持するために必要である医師、患者、第三者支払者、および医学界の他の人の市場に対する受容度を維持することができないかもしれない。
任意の適応を治療するためのAMX 0035または私たちの現在または未来の任意の他の候補製品が適切な規制機関のマーケティングおよび販売承認を得ても、医師、患者、第三者支払者、および医学界の他の人の十分な市場受容度を得ることができない可能性がある。医師は、AMX 0035または他の製品を患者の治療レジメンに追加することを望まない場合があり、またはAMX 0035またはそのような製品の患者の治療レジメンへの追加を停止する可能性がある。さらに、患者は、通常、医師が推奨しない限り、追加の治療を増加させたくない、彼らが現在受けている治療計画に適応する。さらに、保険および十分な精算が不足しているため、患者は、AMX 0035またはそのような他の製品を彼らの治療レジメンに追加することができない可能性がある。また,カナダ保健省,FDA,EMA,他の規制機関に我々候補製品の安全性と有効性を示すことができても,医学界の安全性や有効性に対する懸念は市場受け入れを阻害する可能性がある。特定の管轄区域のマーケティング制限によると、特に適用される衛生当局によって承認された特定のラベルに関連している場合には、医療専門家および患者を能動的に教育する能力が制限される可能性がある。
教育医療界および第三者支払者が、現在および将来の候補製品の利点を理解するための努力は、管理時間および財務資源を含む大量の資源を必要とし、成功しない可能性がある。AMX 0035または任意の他の現在または将来の候補製品が承認されたが、十分な市場受容度に達していない場合、私たちは顕著な収入を生じない可能性があり、利益を上げ続けることはできないかもしれない。AMX 0035および任意の他の将来の候補製品の市場受容度は、商業販売のために許可された場合、多くの要因に依存する
58
AMX 0035または私たちの現在または未来の任意の他の候補製品が市場承認または商業成功を得るために規制部門の承認を得られなかった場合、私たちのビジネスの見通しに悪影響を及ぼすだろう。
非タグはPBを用いてALSを治療し、PBは1種の模倣薬であり、いくつかの司法管轄区におけるTursoの潜在的な販売に加えて、AMX 0035ビジネス機会を減少または除去する可能性のある追加のリスクに直面させる。
TursoとPBの組合せとしてAMX 0035を開発·推進している.PBはすでにFDAと他の監督機関によっていくつかの尿素循環障害患者の治療に許可されている。
Tursoは、米国を含むいくつかの司法管轄区域でALS治療のための製剤を未承認の形で販売している。医療専門家がPBを処方してALSを治療する可能性があり、この組み合わせがAMX 0035の利点を複製できると信じているので、承認されていない、ラベル、または販売されていないALS治療のためのTurso商業製剤を得ることを提案している。EU Humanitas Mirasole spaが行ったALS患者のTursoの安全性と有効性を評価する第三段階の臨床試験が陽性結果を報告した場合、ある司法管轄区でもTursoがALS患者の患者誘導治療が出現する可能性がある。これらのやり方は医学界が推薦したものではなく、いかなる規制機関の許可も得られていないが、それらは米国のRELYVRIO、カナダのALBRIOZAおよびAMX 0035の販売(他の司法管轄区で許可された場合)、および/またはアメリカまたは海外の公衆のAMX 0035に対する見方に影響を与える可能性がある。
FDA、カナダ保健省、EMA、または他の同様の外国規制機関がAMX 0035の模倣バージョン、または私たちが現在または将来規制によって承認された任意の他の候補製品を承認した場合、またはこれらの機関がそのような製品の模倣バージョンを承認する前に適切な非特許専門期間を与えていない場合、このような製品の販売は悪影響を受ける可能性がある。
米国では、NDAが承認されると、そのカバーされた製品は、FDA出版物“治療同等性評価を有する承認された医薬製品”またはオレンジブックの“参照リスト薬物”となる。米国では、製造業者は簡略化された新薬申請やANDAを提出することによって、上場薬物の模倣薬バージョンの承認を求めることができる。ANDAを支持するためには、模倣薬メーカーは通常、その製品が参考発売薬と同じ有効成分(S)、剤形、強度、投与経路、適切なラベルを持っていることを証明しなければならず、後発薬バージョンは参考発売薬と生物学的同等性があり、これは体内での吸収速度と程度がある程度同じであることを意味する。後発薬の発売コストは参考発売薬物よりはるかに低い可能性があり、模造薬を生産する会社は通常比較的に低い価格でこれらの製品を提供することができる。また,多くの州では,ブランド薬を処方しても薬局レベルで治療上等価な後発薬の代替が許可または要求されている。そのため,後発薬を発売した後,任意のブランド製品や参考リスト薬の売上の大部分が後発薬に流出する可能性がある。
市販薬の任意の適用された非特許専有期間の満了を参照する前に、FDAは模倣薬のANDAを承認してはならない。FDCAは新しい化学実体或いはNCEを含む新薬に5年間の非特許専門期を提供した。本条項の場合、NCEは、FDAが以前に任意の他のNDAで承認された活性部分を含まない薬剤を意味する。活性部分は薬物物質の生理的あるいは薬理作用を担う分子またはイオンである。具体的には、このような排他性が付与されている場合、ANDAは、提出された文書に第4段落の証明、すなわち、列挙された薬物をカバーする特許が無効であるか、強制的に実行できないか、または模倣薬によって侵害されない限り、5年の満了前にFDAにANDAを提出することができない。この場合、出願人は、市販薬が承認されて4年後に出願を提出することができ、その模倣薬の発売を求めることができ、我々の製品が依然として特許保護を有していても、NDAまたは特許所持者が直ちに侵害訴訟を提起しない限り、FDAは裁判所が裁判所にいない限り、30ヶ月以内にANDAを承認することができない
59
後発医薬品製造業者に対する支援は以前に発表された。固定用量組合せ製品の場合,FDAは,組合せ製品が新たな活性部分を含む場合,その組合せ製品はNCE排他性(データ排他性とも呼ばれる)を獲得する資格があり,固定組み合わせであっても先に承認された活性部分を有する医薬物質を含むという立場である。
我々はすでにFDAからRELYVRIOのNCE独占経営権を取得しており、この独占経営権は2027年9月に満了する。また、私たちのカナダ保健省の条件付きマーケティング許可について、ALBRIOZAは8年間の市場排他期を提供する革新的な薬物登録に追加された。欧州の規制機関はFDAやカナダ衛生部からAMX 0035の排他性について異なる結論を出す可能性がある。
もし我々が開発したいかなる製品も5年間のNCE独占特許権を取得していない場合,FDAは承認日3年後にその製品の模造薬バージョンを承認することができるが,ANDA出願人に我々の製品がオレンジブックに記載されているいかなる特許も無効であるか,または侵害していないことを証明するように要求する。NDAまたは特許所持者が直ちに侵害訴訟を提起した場合、FDAは30ヶ月以内にANDAを最終的に承認することができず、裁判所が後発薬メーカーに有利な判決を事前に発表しない限り、最終的にANDAを承認することができない。薬物が以前に承認された活性部分を含む場合、薬剤に3年間の排他性を投与し、NDAは、生物学的利用度または生物学的同等性の研究を含まない1つ以上の新しい臨床研究の報告を含み、これらの研究は、出願人または出願人のために行われ、NDAを承認するために必要であるとFDAによって決定される。このような形式のデータ排他性は新臨床調査,あるいはNCI,排他性と呼ばれる.AMX 0035が将来の使用のために承認された場合、または現在および将来の候補がNCI独占特許のみが承認された場合、後発薬製造業者は、AMX 0035が承認された後に随時ANDAを提出し、3年の市場排他期間の終了後に、たとえ私たちの製品が特許保護を有していても、彼らの模倣薬製品の発売を求めることができる。
さらに、米国では、FDCAは、患者数が20万人未満または患者数が20万人を超える薬物を治療するための7年間の孤児薬物専門期間を提供しているが、このような薬物の米国販売において、そのような疾患または状態を治療する薬剤の開発および製造のコストを回収することができる合理的な期待はない。AMX 0035はALS治療のための孤児薬物指定が許可されており、AMX 0035(RELYVRIO)が2022年9月に成人ALSの治療のためにFDAによって許可されたことに伴い、この製品は孤児薬物の独占経営権を付与され、FDAはAMX 0035と同じ孤児適応を含む後発薬またはブランド製品を承認することができず、有効期間は7年であるが、いくつかの例外は除外されている。この期限はNCE専用期間と同時に実行される.
カナダのデータ保護制度は“革新薬物”に特許保護から独立した8年間の市場独占期を提供した。革新的薬物は、以前カナダ衛生部によって許可されていなかった医薬成分を含み、塩、エステル、エナンチオマー、溶媒酸塩または多形体などの以前に承認された医薬成分ではない変異体を意味する。ある薬物がカナダで“革新薬物”と認定された場合、データ保護期間の前6年以内に、模倣薬および製造業者は革新薬物との直接または間接比較に基づいてその製品の承認を求めることができず、カナダ衛生部は8年以内にコンプライアンス通知やNOCまたは上場承認を発表することができない。ALBIOZAの一つの成分(ウルソル酸)は革新薬物であるため、ALBIOZAは承認された後に革新薬物登録に添加される。ALBIOZAのデータ保護期間は2030年6月10日まで,すなわちそのNOC発表日から8年である。
カナダには承認されたまれな疾患の治療製品に孤児薬物排他性を提供する規制条項がない。
EUでは、革新的な医薬製品(小分子と生物医薬製品を含む)、新しい活性物質やNASと呼ばれることがあり、マーケティング許可を得た後、8年間のデータ独占権を獲得する資格があり、他の2年間の市場独占権がある。もしデータ排他性、模倣薬或いは生物類似薬を授与する申請者が模倣薬或いは生物類似薬の発売許可を申請する時、参考製品が初めてEUで許可を得た日から8年以内に、製品ファイルに含まれる革新者の臨床前と臨床試験データを参照してはならない。追加の2年間の市場排他期間内に、模倣薬または生物類似製品の発売許可を提出することができ、革新者のデータを参考にすることができるが、市場排他期間が満了するまで、いかなる模倣薬や生物類似製品も発売できない。この10年前の8年間に、マーケティング許可保持者が1つまたは複数の新しい治療適応の承認を得、これらの新しい治療適応が既存の治療法と比較して有意な臨床的利益をもたらす場合、この10年間の市場独占期間を11年に延長することができる。しかしながら、1つの革新的な医薬製品が所定のデータ独占期間を取得しても、別の会社が完全かつ独立した薬物試験、臨床前試験、および臨床試験データパケットを有するアプリケーションに基づくマーケティング許可を取得した場合、同社はその製品の別のバージョンを販売することができる。私たちはEUでAMX 0035のNAS地位を申請した。NASの地位にかかわらず、EUがマーケティング許可を与えた後も孤児資格を維持すれば、AMX 0035は孤児市場の独占経営権を獲得する資格があると予想される。EUの現行の孤児薬物制度
60
孤児薬物は10年間の市場独占期を有する権利があり,スポンサーが合意した小児科研究計画を遵守すれば,12年まで延長することができる。しかし、欧州委員会は2023年4月に立法提案を提出し、実施されれば、ある孤児薬の現在の排出期間を短縮する可能性がある。
AMX 0035または任意の未来の製品は、承認された場合、そのような製品の模造バージョンからの競争に直面する可能性があり、将来の収入、収益性、およびキャッシュフローに悪影響を与え、これらの候補製品から投資収益を得る能力を大きく制限する可能性がある。
AMX 0035および任意の他の現在または未来の候補製品は、承認された場合、上場後の制限、要求、または市場からの撤退を受ける可能性があり、もし私たちまたは彼らが規制要求を遵守できなかった場合、または私たちまたは彼らの製品が承認された後に予期せぬ問題に遭遇した場合、巨額の費用を招く可能性があり、私たちまたは任意の未来のパートナーは重大な処罰を受ける可能性がある。
AMX 0035または私たちまたは任意の未来のパートナーが規制の承認を得た任意の他の現在または将来の候補製品、およびそのような製品の製造プロセス、承認後の研究、ラベル、広告および販売促進活動などは、FDA、カナダ保健省、EMA、および他の適用可能な規制機関の継続的な要求および審査を受ける。これらの要求には、安全およびその他の発売後の情報と報告の提出、登録と上場要求、製造、品質管理、品質保証と記録と書類の対応する維持に関する要求、医師へのサンプルの配布と記録の保存に関する要求が含まれる。いくつかの商業処方薬製品については、製造業者およびサプライチェーンに関連する他の当事者は、流通チェーン要件を満たし、電子的、相互運用可能な製品追跡および追跡システムを構築し、偽、移転、盗難、故意に偽の製品または米国での流通に適していない他の製品をFDAに通報しなければならない。私たちと契約メーカーはまた、これらの要求および私たちが得る可能性のある製品承認条項の遵守状況を監視するために、使用料および規制機関の定期的な検査を受ける。製品候補が規制部門の承認を得ても、承認は、米国でのリスク評価および緩和策(REMS)の要求を含む、製品上場適応または用途の制限または承認条件の制限を受ける可能性がある。
FDA、カナダ衛生部、EMAとその他の監督機関もまた、製品の安全性或いは有効性を監視するために、高価な発売後の研究或いは臨床試験とモニタリングを要求する可能性がある。例えば,米国で承認されたRELYVRIOの一部として,発売後にマウスやラットへの発癌性研究,ヒトボランティアにおける薬物−薬物相互作用研究,腎臓や肝臓障害の被験者での研究が求められている。また、FDAおよび司法省を含む他の機関は、製品の製造、マーケティング、流通が承認の適応にのみ適用され、承認されたラベルの規定に適合することを確実にするために、製品の承認後のマーケティングおよび販売促進活動を密接に規制·監督する。規制当局はラベル外使用に関するメーカーのコミュニケーションに厳格な制限を加えた。しかしながら、会社は、一般に、製品承認されたラベルと一致する真で誤解されない情報を共有するかもしれない。もし私たちまたは任意の未来のパートナーがマーケティングAMX 0035または私たちまたは彼らが規制部門の承認された適応のみを得ている他の現在または未来の候補製品がなければ、もし私たちまたは彼らがそうしていると告発された場合、私たちまたは彼らはラベル外マーケティングの警告または法執行行動を受けるかもしれない。処方薬販売促進および広告に関する法律および法規に違反することは、虚偽申告法および任意の類似した外国法を含む連邦および州医療保健詐欺および乱用法律および州消費者保護法の違反に対する調査または告発を引き起こす可能性がある。EUでは、消費者に直接向けた処方薬の広告が禁止されている。EUの医療製品普及規則に違反した行為は行政措置、罰金、監禁の罰を受ける可能性がある。承認されれば、これらの法律は、私たちの製品を大衆に宣伝·普及させることをさらに制限または制限し、医療専門家との販売促進活動に制限を加える可能性がある。
カナダの発売後、米国と類似することが求められています。私たちの新薬提出または条件付きNDSが承認された後、カナダ衛生部はリスク管理計画やRMPの提出を要求しています。RMPの一部として,カナダ保健省は追加の臨床試験を要求する可能性がある。例えば、カナダにおけるAMX 0035(ALBRIOZA)の販売許可条件のうちの1つは、我々が行っているフェニックス試験および他の計画または行われている研究のデータを提供することである。市販されている薬品もまた標準的な薬物警戒活動を行わなければならない。製品サプライチェーンのいかなるラベルの変更や変更もカナダ保健省の承認を提出して実施する必要があります。私たちの広告は競争相手や医療提供者によって慎重に審査され、カナダ保健省や他の機関にクレームされる可能性があります。カナダでは、精算は複雑で、処方薬の処方リストを得るためには、公共と個人支払者に提出する必要がある。さらに、AMX 0035に関連する任意の特許がある場合、この製品は、特許薬品価格審査委員会またはPMPRBの価格によって規制される。
61
さらに、後に、我々の製品またはその製造業者または製造プロセスに、以前に未知の有害事象または他の問題が発生したり、法規要件を遵守できなかったりすることが発見され、様々な結果が生じる可能性がある
承認後の規制要件を遵守できない場合、私たちは規制機関によってAMX 0035または任意の将来承認された製品のマーケティング承認を撤回または制限される可能性があり、または私たちは自発的にそうするかもしれません。私たちはAMX 0035または任意の未来に承認された製品をマーケティングし、アメリカ、カナダまたは他の司法管轄地域でAMX 0035を開発またはより多くの適応のために開発する能力が制限される可能性があり、より多くの候補製品の承認を開発し、求めることは、私たちの収益性の達成または維持に悪影響を及ぼすかもしれません。そのため、承認後の規制要求を遵守するコストは、私たちの経営業績や財務状況にマイナス影響を与える可能性があります。
もし私たちが新しい地域でAMX 0035または任意の他の現在または未来の候補製品の保険および精算を得ることができない場合、AMX 0035または任意の他の現在または未来の候補製品を利益的に販売することは困難になるかもしれない。
AMX 0035および現在または将来の任意の他の候補製品の成功は、承認された場合、十分な保証範囲および第三者支払者の補償があるかどうかに依存する。AMX 0035および任意の他の現在または未来の候補製品は、その目標疾患を治療する新しい方法を表すので、我々は、AMX 0035および任意の他の現在または将来の候補製品、または私たちが開発する可能性のある任意の製品の保証範囲および精算範囲が利用可能であるか、またはAMX 0035および任意の他の現在または未来の製品からの潜在的収入を正確に推定することを保証することができない。もし私たちが十分な精算レベルを得ることができなければ、私たちがこのような候補製品をマーケティングして販売する能力は不利な影響を受けるだろう。私たちが開発する可能性のある任意の現在または将来の候補製品に関連するサービスの精算方法およびレベルも重要である(例えば、患者に対して私たちの候補製品を管理する)。このようなサービスの精算不足は、医師および支払者のボイコットを招き、AMX 0035または私たちが開発する可能性のある他の任意の現在または将来の候補製品をマーケティングまたは販売する能力に悪影響を及ぼす可能性がある。しかも、私たちは十分な価値を達成するために新しい精算モデルを開発する必要があるかもしれない。支払者はこのような新しいモデルを採用できないか望まない可能性があり,患者はこのモデルが負担する費用の一部を負担することができないかもしれない。もし私たちがこれらの新しいモデルが必要だと判断すれば、私たちはそれらを開発することに成功しなかったか、あるいは支払人がこれらのモデルを採用しなければ、私たちの業務、財務状況、運営結果、見通しは不利な影響を受けるかもしれない。引受範囲および補償に関するより多くの情報は、本年度報告書の“企業−政府規制−保険·補償”と題する部分を参照されたい。
自分の病状に医療サービスを提供する患者は通常,第三者支払者によってその治療に関連する費用の全部または一部を精算する。政府医療保健計画(例えばMedicareとMedicaid)および商業支払人(例えば、個人健康保険会社と健康維持組織)の十分なカバーと精算は新製品の受容度に重要である。政府当局と他の第三者支払人は、彼らがどのような薬物と治療と費用をカバーするかを決定する。第三者支払者の保証範囲と補償は多くの要素に依存するかもしれない。
62
米国では、第三者支払者の間に統一された製品保証や精算政策はない。したがって、政府や他の第三者支払人から製品の保証と精算承認を得ることは、時間がかかり、高価なプロセスであり、各支払人に科学的、臨床的、費用効果的なデータを提供して、個々の支払人に基づいて私たちの製品を使用することを支援する必要があるかもしれないが、第三者支払者から保険と十分な精算を受ける保証はないかもしれない。新たに承認された製品の保険カバー範囲や精算に関する不確実性が大きく,製品が発売後の条件や追加の臨床データ提供の要求の影響を受けると,この不確実性が悪化する可能性がある。アメリカでは、新薬精算に関する主な決定は通常医療保険と医療補助サービスセンター(CMS)によって行われ、CMSはアメリカ衛生と公衆サービス部の一つの機関であり、CMSは新薬が連邦医療保険の下でカバーと精算するかどうか、及びどの程度精算するかを決定する。個人支払者はCMSに大きく従う傾向がある。特定の製品の保険を受けても,それによる精算支払率は利益を実現したり維持したりするのに不十分である可能性があり,あるいは患者が受け入れられないと考える高い共済額が必要となる可能性がある。米国や国際市場では、将来の保険や精算は、事前に許可された要求など、より多くの制限を受ける可能性がある。孤児薬は通常費用分担が最も高いレベルに置かれ、かなりの割合で事前許可の要求を受けている。連合の清算機関はCMSよりもっと保守的かもしれない。
米国以外では、国際業務は通常、広範な政府価格制御および他の市場によって規制されており、私たちは、カナダ、EU、および他の国のコスト制御措置がますます重視され、AMX 0035などの医薬製品および私たちが開発する可能性のある他の任意の現在または将来の候補製品の価格設定と使用に圧力をかけ続けると信じている。AMX 0035または任意の未来の候補製品が発売後の条件によって承認された場合、公共および個人支払者に補償を求める際にも追加の挑戦に遭遇する可能性があります。多くの国、特にEU諸国では、国家衛生システムの一部として、医療製品の価格は異なる価格制御メカニズムの制約を受けている。これらの国では、製品の発売許可を受けた後、政府当局との定価交渉にかなりの時間がかかる可能性がある。例えば、カナダでは、省レベル当局との価格交渉には、合意された定価や精算料率を達成するのに18カ月以上かかる可能性がある。これらの交渉の前に、CADTHとINESSSは、薬物が衛生システムに提供する価値を評価するための審査を行った。特許薬品については,PMPRBは薬品の販売価格に管轄権を有しており,PMPRBの受け入れ可能価格の評価は支払者との交渉に影響する可能性がある。このような交渉はまた、補償を与える前に組合せ製品の追加的な研究および理由をもたらす可能性がある。そのため、特定の国/地域での製品のマーケティング承認を得ることができるかもしれませんが、その後、価格法規の制約を受けて、これらの法規は私たちの製品の商業発表を延期したり、阻止したりする可能性があり、長い間続くかもしれません。一部の国で精算或いは定価の承認を得るためには、私たちの候補製品のコスト効果を他の利用可能な治療法と比較する臨床試験を行う必要があるかもしれない。一般的に、この制度での製品価格はアメリカよりずっと低いです。他の国は会社が自分の製品価格を固定することを許可していますが、会社の利益を監視しています。追加の外国価格規制や定価規制の他の変化は、候補製品に受け取ることができる費用を制限するかもしれません。したがって、米国以外の市場では、AMX 0035および私たちが開発する可能性のある任意の他の現在または将来の候補製品の精算は、米国よりも減少する可能性があり、商業的に合理的な収入および利益を生成するのに十分ではないかもしれない。
さらに、第三者支払者は、候補製品を使用した後に必要な長期的な後続評価をカバーしない可能性があり、十分な補償を提供しない可能性がある。患者は、保険が提供されなければ、AMX 0035または任意の他の現在または未来の候補製品を使用することはあまり不可能であり、AMX 0035または任意の将来の候補製品の大きな一部のコストを支払うのに十分である。AMX 0035および任意の他の現在または将来の候補製品は、従来の療法よりも高い商品コストを有する可能性があり、長期的な後続の評価を必要とする可能性があるので、カバー範囲および販売率は、利益を維持するのに十分ではない可能性が高い可能性がある。カナダ保健省がAMX 0035によるALS治療の条件を承認した後,カナダの一部の提供者から積極的な回答を受けたが,保険カバー範囲や精算には大きな不確実性があった。第三者決済者がAMX 0035と任意の現在または未来の候補製品の保証と精算についてどのような決定を下すかを予測することは現在困難である。
さらに、米国、カナダ、EU、および他の外国司法管轄区域の政府および他の第三者支払者は、医療コストを制限または低減するためにますます努力しており、これらの組織は、新しい承認製品の保証範囲および精算レベルを制限する可能性があり、したがって、AMX 0035または任意の他の現在または将来の候補製品に十分な支払いを支払うことができないか、または十分な支払いを提供することができない可能性がある。アメリカでは、特殊薬物の価格決定に関する立法と法執行の関心が増加している。管理型ヘルスケアの傾向、健康維持組織の日々の影響力、コスト制御措置、および追加の立法変化により、AMX 0035または任意の他の現在または将来の候補製品の販売に関連する価格設定圧力に直面することが予想される。
63
行われている医療立法や規制改革措置は、私たちの業務や運営結果に実質的な悪影響を及ぼす可能性がある。
規制、法規、または既存の規制の解釈の変化は、(I)私たちの製造スケジュールを変更すること、(Ii)製品ラベルを追加または修正すること、(Iii)私たちの製品をリコールまたは停止すること、または(Iv)追加の記録保存要件を要求することなど、私たちの将来の業務に影響を与える可能性がある。このような変化を強制的に実施すれば、私たちの業務運営に悪影響を及ぼす可能性がある。もっと知りたいことがあれば、タイトルを参照してください“企業−政府規制−米国の現在と将来の医療改革立法この年間報告書にあります。
私たちは未来に取られる可能性のある計画を予測できない。政府、保険会社、医療組織、その他の医療サービスを管理する支払人は、医療コストの抑制または低減、および/または価格規制の実施に努力し続けており、悪影響を及ぼす可能性がある
これらの法律および将来採用される可能性のある州および連邦医療改革措置は、連邦医療保険および他の医療保険資金のさらなる減少をもたらす可能性があり、他の方法では、AMX 0035または規制の承認を得る可能性のある任意の他の現在または将来の候補製品のために得られる価格、または任意のそのような候補製品の処方または使用頻度に影響を与える可能性がある。連邦医療保険や他の政府が計画している精算のいかなる減少も、個人支払者の支払いの同様の減少を招く可能性がある。コスト抑制措置や他の医療改革を実施することは、収入の創出、収益性の維持、あるいは候補製品の商業化を阻止するかもしれない。
また,米国や海外の政府や第三者支払者が医療コストの制限や低減に力を入れており,これらの組織が新承認製品のカバー範囲や精算レベルを制限する可能性があるため,候補製品の支払いや十分な支払いを提供できない可能性がある。アメリカでは、特殊薬物の価格決定に関する立法と法執行の関心が増加している。具体的には、米国議会は最近数回の調査を行い、薬品定価の透明性を高め、連邦医療保険下の処方薬のコストを下げ、定価とメーカー患者計画との関係を審査し、政府計画の薬品精算方法を改革するための連邦と州立法を提出し、公布した。これらの改革努力が私たちの業務と医療業界全体に与える影響はまだ明らかではない。
将来的にはより多くの州および連邦医療改革措置が取られることが予想され、いずれも連邦および州政府が医療製品およびサービスのために支払う金額を制限する可能性があり、これはいくつかの薬品に対する需要の減少または追加の価格設定圧力を招く可能性がある。
その中のいくつかの措置および他の提案された措置は発効するために追加の許可を必要とする可能性があるが、国会とバイデン政府は薬品コストを制御するために新しい立法および/または行政措置を求め続けると表明している。
米国以外の政府は厳しい価格制御を実施する可能性があり、これがあれば私たちの収入に悪影響を及ぼす可能性がある。
一部の国では、カナダとあるEU加盟国を含め、処方薬の価格設定は政府によってある程度コントロールされている。他の国は処方薬の価格設定に対して似たような接近をするかもしれない。これらの国では、製品の規制承認を受けた後、政府当局との定価交渉にかなりの時間がかかる可能性がある。EUはEU加盟国に各種の選択を提供し、その国家健康保険制度がそれに補償を提供する薬品の範囲を制限し、人が使用する薬品の価格を制御する。EU加盟国は製品の具体的な価格を承認することができ、その製品を市場に投入する会社の収益力を直接または間接的に制御する制度をとることもできる。他のEU加盟国は会社が自分の薬品価格を固定することを許可したが、処方量をモニタリングと制御し、医師に指導を発表して処方を制限した。また、費用抑制措置の一部として、各国政府や他の利害関係者は価格や補償レベルにかなりの圧力をかける可能性がある。政治、経済、規制の発展の可能性
64
定価交渉をさらに複雑化させ、保険や補償を受けた後、定価交渉が継続される可能性がある。各国で使用されている参考定価と平行配分、あるいは低価格と高価な国の間で裁定を行うことで、価格をさらに下げることができる。いくつかの国では、AMX 0035または任意の他の現在または将来の候補製品を、他の利用可能な治療法と費用対効果比較して、精算または価格決定の承認を得るか、または維持する必要があるかもしれないが、これは時間も高価である。私たちはそのような価格と精算が私たちに受け入れられるという保証がない。第三者支払者や主管当局が割引を公表することは、公布国や他の国の価格や補償レベルにさらなる圧力を与える可能性がある。価格設定レベルが満足できない場合、または私たちの製品が精算または精算範囲または金額に制限されていない場合、私たちまたは私たちの戦略パートナーの販売収入およびAMX 0035または任意の他の現在または将来の候補製品のこれらの国/地域における潜在的な収益力は負の影響を受けるだろう。
医療提供者、医師、患者、第三者支払者との関係は、適用される反リベート、詐欺および乱用、および他の医療法律法規の制約を受ける可能性があり、これは、私たちを刑事制裁、民事処罰、契約損害、名声損害、および利益と将来の収入の減少に直面させるかもしれない。
米国や他の地域の医療提供者,医師,第三者支払者は薬品の推奨と処方に主な役割を果たしている。第三者支払者および顧客との手配は、製薬業者が医薬品研究、販売、マーケティング、流通の業務または財務手配および関係を制限する可能性があるが、連邦反バックル法規および連邦虚偽クレーム法案を含むが、連邦反バックル法規および連邦虚偽クレーム法案を含む広範に適用される詐欺および乱用、および他の医療法律および法規に直面する可能性がある。特に、医療製品やサービスの普及、販売およびマーケティング、および医療業界のいくつかの商業的配置は、詐欺、リベート、自己取引、および他の乱用行為を防止するための広範な法律によって制限されている。これらの法律法規は、幅広い価格設定、割引、マーケティングおよび販売促進、構造および手数料(S)、特定の顧客インセンティブ計画、および他のビジネススケジュールを制限または禁止する可能性があります。これらの法的拘束を受けた活動は,患者を募集して臨床試験を行う過程で得られた情報の不正使用に関するものである。もっと知りたいことがあれば、タイトルを参照してください“企業-政府規制-その他アメリカ医療保険法この年間報告書にあります。
米国では,患者が承認した製品を負担できるように支援するために,患者支援計画や条件に適合した患者の自己負担クーポン計画を含む第三者組織支援計画や患者支援計画を提供する。政府の法執行機関は、精算支援サービスを含む製薬会社の製品や患者支援計画にますます大きな興味を示しており、これらの計画のいくつかの調査は、重大な民事·刑事和解につながっている。さらに、少なくとも1つの保険会社は、そのネットワーク薬局が、保険会社が決定した特定の特殊薬物の共同支払い券を受け入れないように指示している。私たちの自己負担クーポン計画は保険会社の似たような行動の目標になるかもしれない。2014年9月、HHS監察長弁公室(OIG)は、適切な措置を講じず、Dの一部の受益者が共同支払いクーポンを使用することを排除し、連邦反リベート法規および/または民事罰金法律の制裁を受ける可能性があると警告する特別諮問公告を発表した。そこで,会社はこれらのD部分受益者を自己払いクーポンの使用から除外し,我々のAmylyxケアチームも同様である。保険会社の自己支払いクーポン政策の変更および/または新しい立法または規制行動の導入および公布は、これらの患者支援計画を制限または否定的に影響する可能性があり、これは、米国のRELYVRIOのような影響を受けた製品の使用をより少ない患者にもたらす可能性があり、したがって、私たちの販売、業務、および財務状態に実質的な悪影響を及ぼす可能性がある。
会社の資金支援を受ける第三者患者支援プロジェクトも政府や規制機関の審査強化の対象となっている。OIGはすでにガイドラインを策定しており,製薬業者が医療保険患者に自己援助を提供する慈善団体への寄付が合法であることを示している。しかし、患者支援プロジェクトへの寄付はいくつかの負の宣伝を受け、政府の多くの法執行行動の対象となった。なぜなら、他のコストの低い代替製品ではなく、ブランド薬品を普及させるために寄付金が乱用されたと告発されたからである。具体的には,近年,様々な連邦や州法により,政府が第三者患者援助計画の合法性を疑問視し,複数の和解を招いている。私たちは以前、時々独立した慈善基金会に慈善寄付金を提供し、経済的に困難な患者が保険料、共同支払い、共同保険の義務を履行することを支援してきた。もし私たちがそうすることを選択し、私たちまたは私たちのサプライヤーまたは寄付受給者が、慈善寄付またはこれらのプロジェクトを提供したり運営したりする際に関連する法律、法規、または変化する政府指導に従わなかったと考えられた場合、私たちは損害賠償、罰金、処罰または他の刑事、民事または行政制裁または法執行行動を受ける可能性がある。私たちは私たちのコンプライアンス統制、政策、そして手続きが私たちの従業員、業務パートナー、サプライヤー、または慈善基金の行為が私たちの管轄区域の法律や法規に違反することを防ぐのに十分であることを確実にすることはできない。法律を遵守しているかどうかにかかわらず政府の調査にはビジネスパートナーやサプライヤーや慈善財団の調査も含まれています
65
私たちの業務実践に影響を与え、私たちの名声を損ない、経営陣の注意をそらし、私たちの費用を増加させ、助けを必要とする患者に基礎的な支援を提供する可能性を減らすことができる。
医薬製品の流通はまた他の規定と条例を遵守しなければならず、許可されていない医薬製品の広範な記録保存、許可、貯蔵と安全要求を防止することを含む。我々の内部運営と将来の第三者の業務配置が適用される医療法律や法規に適合することを確保することは、多くのコストに及ぶ。政府当局は、私たちの業務やり方が現在または未来に適合していないと結論するかもしれないが、詐欺および乱用または他の医療保健法律および法規の適用に関する現行または将来の法規、機関指導または判例法に関連している。これらの法律の範囲も執行も不確定であり,現在の医療改革環境の急速な変化の影響を受けている。私たちの業務が上記のいずれかの法律または私たちに適用される可能性のある他の政府の法律および法規に違反していることが発見された場合、私たちは、民事、刑事および行政処罰、損害賠償、罰金、米国政府の援助から除外された医療計画(MedicareおよびMedicaidなど)や他の国または司法管轄区域の同様の計画の外、返還、監禁、契約損害、名声損害、利益減少、追加報告要件および監督を含む重大な処罰を受ける可能性があり、もし私たちが会社の誠実協定または同様の合意に制約された場合、これらの法律に違反した疑い、および私たちの業務の遅延、減少、終了、または再編を解決します。しかも、このような行動を防御するのは高価で時間がかかる可能性があり、大量の財政と人的資源が必要となる可能性がある。したがって、私たちが私たちに提起される可能性のあるいかなる訴訟も防ぐことに成功しても、私たちの業務は損害を受ける可能性がある。もし私たちがそれと業務を展開することを期待している任意の医師または他の提供者または実体が適用されない法律を遵守していないことが発見された場合、彼らは政府の援助された医療計画から除外され、監禁されたことを含む重大な刑事、民事または行政制裁を受ける可能性がある。上記のいずれかの場合が発生すると、我々の業務運営能力や運営結果に悪影響を及ぼす可能性がある。
もし私たちが医療補助薬品リベート計画または他の政府定価計画の下での報告と支払い義務を履行できなければ、私たちは追加の精算要求、処罰、制裁、罰金を受ける可能性があり、これは私たちの業務、財務状況、運営結果、成長の見通しに実質的な悪影響を及ぼすかもしれない。
私たちは医療補助薬品リベート計画、340 B計画、アメリカ退役軍人事務部、連邦供給スケジュールまたはFSS定価計画、およびTricare小売薬局計画に参加し、これらの計画は私たちにメーカーの平均価格を開示することを要求し、将来的にはいくつかの薬物の平均販売価格をMedicare計画に報告することを要求するかもしれない。定価と返却点計算は製品と計画によって異なり、非常に複雑で、通常私たち、政府あるいは監督機関と裁判所の解釈を受ける。さらに、これらの計画および政策(カバー範囲の拡大を含む)に関する規制および立法変化および司法判断は増加しており、私たちのコストおよびコンプライアンスの複雑さを増加させ続け、継続的に時間をかけて実施され、特にCMSまたは他の機関が実施中に取った方法に疑問を提起する場合、私たちの運営結果に実質的な悪影響を与える可能性がある。例えば、私たちのMedicaid定価データの場合、前の四半期の報告書が正しくなかったり、再計算価格データによって変化したことを認識した場合、私たちは通常、元のデータが満期になってから最大3年以内に修正されたデータを再提出する義務があります。このような再記述は私たちのコストを増加させ、過去数四半期の私たちの返却責任超過または未成年を招く可能性がある。価格再計算は、340 B計画に従って製品の最高価格を提供することにも影響を与える可能性があり、340 B計画に参加したエンティティが過去数四半期に価格再計算によって徴収した高すぎる費用の返還を義務化する可能性があります。
もし私たちが故意に政府に虚偽の価格や製品情報を提出したことが発見された場合、私たちの平均販売価格を報告する際に虚偽の陳述をしたことが発見された場合、必要な価格データを適時に提出しなかった場合、または340 B保証エンティティに徴収された費用が法定最高価格を超えていることが発見された場合、民事罰金を適用することができる。また,我々が340 B計画に参加するプロトコルや我々の医療補助薬品リベート協定は終了する可能性があり,この場合,連邦政府は医療補助や連邦医療保険D部分に基づいて保険を受けている外来薬に支払うことができない可能性がある。また、FSSやTricare小売薬局との手配で政府に過大な費用を徴収した場合、差額を政府に返金することを要求されます。必要な開示および/または識別契約の多収料金を行うことができなかったことは、FCAおよび他の法律法規に基づいて私たちに告発される可能性がある。政府の意外な返金、および政府の調査や法執行行動への対応は、高価で時間がかかり、私たちの業務、財務状況、運営結果、成長の見通しに実質的な悪影響を及ぼす可能性がある。
さらに、立法が導入される可能性があり、通過すれば、他の事項に加えて、340 B計画をより多くのカバーエンティティに拡大するか、または参加する製造業者に入院環境で使用される薬剤に340 Bの割引価格を提供することに同意することを要求するか、および将来の製造業者の平均価格または医療補助の定義の任意の追加的な変更を要求するであろう
66
返却金額は私たちの340億上限価格計算に影響を与え、私たちの運営結果にマイナス影響を与える可能性があります。また、ある製薬業者は340 B計画下の契約薬局手配に関する持続的な訴訟に巻き込まれた。例えば,2023年11月3日,米国サウスカロライナ州地方裁判所はGenesis Healthcare Inc.訴えBeceraらの事件について意見を述べた。これは340 B定価処方を取得する資格のある患者の範囲を拡大させる可能性がある。340 B計画に関するこの司法手続きおよび他の司法手続きの結果、および340 B計画に従って契約薬局を介してカバーエンティティに割引を提供する製造業者への潜在的な影響は、まだ確定していない。
グローバルなプライバシーやデータセキュリティ要件を遵守することは、追加的なコストと責任をもたらしたり、グローバルでデータを収集して処理する能力を抑制したりする可能性があり、これらの要求を守らないことは、私たちを巨額の罰金と処罰に直面させる可能性があり、これは、私たちの業務、財務状況、または運営結果に大きな悪影響を及ぼす可能性があります。
全世界の収集、使用、保護、共有、譲渡、その他の情報処理の規制枠組みは急速に変化しており、予見可能な未来には依然として不確定である可能性がある。世界的に、私たちが業務を展開しているほとんどの管轄区域は、自分のデータセキュリティとプライバシーの枠組みを構築しており、これらの枠組みを守らなければならない。例えば、ヨーロッパ経済地域における個人の個人データは、GDPRによって制限され、同様に、イギリスの個人の個人データの処理は、イギリスのGDPRおよびEU GDPRのGDPRによって制限される個人健康データを含む、収集、使用、開示、転送、または他の方法で処理される。GDPRの範囲は広く,個人データを処理する会社に対して多くの要求がなされており,健康や他の敏感なデータの処理に関する要求,個人データに関する個人の同意の取得,個人へのデータ処理活動に関する詳細な情報の提供,個人データの安全と秘密保護の保障措置の実施,データ漏洩の通知の提供,および第三者処理者を採用する際に何らかの措置を講じている。GDPRはまた、欧州経済圏/イギリス以外の国(米国を含む)への個人データの送信に厳しいルールを適用しており、欧州委員会やイギリス政府は、これらの国/地域は個人データに“十分な”保護を提供できないと考えているため、欧州経済地域に位置する臨床試験地点を、このような場所から十分なデータ保護レベルが不足していると考えられる国(例えば、米国)への個人データの移転に適用すべきである。個人データの欧州経済地域やイギリス以外への移行を禁止している(例えば、欧州委員会は標準契約条項やSCC、SCCを承認している。一方,イギリス国際データ転送プロトコル/付録,あるいはイギリスIDTA)はすでに到着している.SCCS/UK IDTAに依存してデータ転送を行う場合には、受信者が現地の法律によって制限されているかどうかを評価するための転送影響評価が要求される可能性もあり、これらの法律は、公共当局が個人データにアクセスすることを可能にする。EEA/UKデータ保護制度下の国際移転義務は、多くの努力とコストを必要とし、EEA/UK個人データをどこに転送するか、どのサービスプロバイダを利用してEEA/UK個人データを処理することができるかについて戦略的に考慮する必要があるかもしれない。データ保護法に基づいて個人データをヨーロッパ経済圏やイギリスから米国に移すことができない行為は、私たちの実験能力を阻害し、私たちの業務や財務状況に悪影響を及ぼす可能性があります。GDPRはまた、データ保護当局が不正に収集または使用した個人情報の廃棄を要求し、GDPR違反行為に巨額の罰金を科すことを許可し、罰金金額は世界の収入の4%または2000万ユーロ(イギリスのGDPRによると、金額は1,750万ユーロ)に達し、金額が大きい者を基準に、データ主体と消費者協会がデータ主体と消費者協会に訴訟を提起する私的な権利を与え、監督機関に苦情を提出し、司法救済を求め、GDPR違反による損害について賠償を受ける。英国はEU GDPR下の第3の国とされているにもかかわらず、欧州委員会は現在、英国がEU GDPRまたは十分な決定に基づいて十分な保護を提供していることを認める決定を発表しているため、欧州経済圏からの個人データの英国への移行は制限されていない。イギリス政府は、イギリスのデータ保護制度を改革するために、イギリスの立法手続きにデータ保護とデジタル情報法案を導入しており、もし通過すれば、イギリス法案の最終バージョンは、イギリスとヨーロッパ経済地域のデータ保護制度との類似性をさらに変更し、EU委員会のイギリスの十分性の決定を脅かす可能性があり、これは私たちの追加的なコンプライアンスコストを招き、私たちの全体的なリスクを増加させる可能性がある。イギリスのデータ保護法律や法規が中長期的にどのように発展するか、イギリスに出入りするデータ転送がどのように長期的に規制されるのかは不明だ。EU GDPRとEU GDPRの現在の義務はほぼ類似しているにもかかわらず,時間の経過とともにイギリスGDPRはEU GDPRとそれほど一致しなくなる可能性がある。また、EU加盟国はEU GDPRを補完する国家法律を採択しており、これはEU GDPRから部分的にずれている可能性があり、EU加盟国の主管当局のEU GDPR義務の解釈は国によって少し異なる可能性があるため、欧州経済圏はデータ保護法規の面で統一された法的環境では動作しないと予想される。EU GDPRと英国GDPRのそれぞれの条項と法執行が将来さらに分化する可能性は、追加の規制課題と不確実性をもたらしてくれた。イギリスの将来の法律法規とEUの法律法規との相互作用は解像度が不足しており、ヨーロッパ個人データ処理の法律リスク、不確実性、複雑性、コストを増加させる可能性があり、私たちのプライバシーとデータ安全コンプライアンス計画はイギリスとヨーロッパ経済区に対して異なるコンプライアンス措置を実施する必要があるかもしれない。
67
米国では、同様の法的要求が制定または提出されている。様々なデータ保護法が我々の活動に適用されており、州と連邦の2レベルの様々な法執行機関は、一般消費者保護法に基づいて会社のプライバシーやデータセキュリティ問題を審査することができる。連邦貿易委員会と州総検察長は消費者のプライバシーとデータ安全保護を積極的に検討している。州と連邦の二級も新しい法律を考慮している。例えば、カリフォルニア消費者プライバシー法は2020年1月1日に施行され、最近カリフォルニアプライバシー権法案によって改正された--GDPR創造のリスクや義務と類似したリスクと義務が生じている。この法案は、臨床試験の一部として収集されたいくつかの情報を免除しているにもかかわらず、企業が企業環境において収集した個人情報や、カリフォルニアに住む従業員、申請者、および退職者から収集された個人情報など、他の方法で扱うことが可能な他の個人情報に適用可能である。多くの州では似たような広範な消費者プライバシー法が採択され、バージニア州、コロラド州、コネチカット州の法律が施行された。また、他の多くの州や連邦政府は、広範な消費者プライバシー法の法案を制定することを検討している。
上記の要件および任意の他のデータプライバシーおよびデータセキュリティ法律法規を遵守することは、厳格かつ時間のかかるプロセスであり、大量のリソースを必要とし、私たちの技術、システムおよび実践、ならびに個人データを処理または送信する任意の第三者協力者、サービスプロバイダ、請負業者またはコンサルタントの技術、システムおよび実践を継続的に検討する必要がある。GDPRおよび他のいくつかのタイプの敏感なデータの保護を強化することに関連する法律または法規の変化、例えば、私たちの臨床試験からの医療データまたは他の個人情報は、私たちの業務慣行を変更し、追加のコンプライアンスメカニズムを確立する必要があるかもしれません。私たちの開発、規制、商業化活動を中断または延期し、私たちの業務コストを増加させ、政府の法執行行動、個人訴訟、および私たちに対する巨額の罰金および処罰を招き、私たちの業務、財務状況、または運営結果に重大な悪影響を及ぼす可能性があります。
人工知能によるリスクと挑戦は、私たちの機密情報、独自情報、個人データに対してセキュリティリスクを構成することを含む、私たちの業務に影響を与える可能性があります。
人工知能の開発と使用中の問題に加え、不確定な監督環境に加えて、私たちの業務運営に名声損害、責任或いはその他の不利な結果をもたらす可能性がある。多くの技術革新と同様に、人工知能によるリスクや挑戦は私たちの業務に影響を与える可能性がある。法律や情報セキュリティ審査を経た特定の用例に対しては,生成的人工知能ツールを用いて我々のシステムに統合する可能性がある.当社のサプライヤーは、その使用状況を開示することなく、生成人工知能ツールをその製品に統合する可能性があり、これらの生成人工知能ツールのプロバイダは、プライバシーおよびデータ保護に関する既存または迅速に変化する法規または業界基準を満たすことができず、私たちまたは私たちのプロバイダが十分なレベルのサービスおよび体験を維持する能力を抑制することができるかもしれない。もし私たち、私たちのサプライヤー、または私たちの第三者パートナーが生成的人工知能を使用して実際または予想される違反やプライバシーまたはセキュリティ事件を経験すれば、私たちは貴重な知的財産権と機密情報を失う可能性があり、私たちの名声と大衆の私たちのセキュリティ対策の有効性に対する見方が損なわれる可能性がある。また、世界各地の不良行為者は、人工知能を用いて、個人情報、機密情報、知的財産権の窃盗および乱用に関する不正活動を行うなど、ますます複雑な方法を使用している。これらの結果のいずれも、私たちの名声を損なう可能性があり、貴重な財産や情報損失を招き、私たちの業務に悪影響を及ぼす可能性があります。
私たちの現在と未来の候補製品の発見と開発に関するリスク
私たちは現在AMX 0035の成功に依存しています。これは私たちの最先端の候補製品です。もし私たちがAMX 0035の追加の規制承認を維持したり、それを商業化することに成功したり、あるいはそのような点で重大な遅延に遭遇した場合、私たちの業務は実質的な損害を受ける可能性があります。
私たちは現在1つの商業製品AMX 0035しかありません。アメリカではRELYVRIOで販売され、カナダでALBRIOZAで販売されています。私たちの現在の業務と未来の成功は、ALSに対するAMX 0035の監督管理の承認を維持し、商業化を継続する能力と、他の司法管轄区でALSとAMX 0035ならびにPSP、WS、ADおよび他の神経疾患のAMX 0114の開発、維持、および追加の規制承認を得て商業化に成功する能力に大きく依存しています。これまでわれわれが得たAMX 0035を支持する臨床試験データは限られており,ALS患者137名の臨床試験と95名のAD患者の臨床試験しか完了していなかった。われわれはALSにおけるAMX 0035の全世界3期臨床試験,AMX 0035のWSにおける2期臨床試験,およびAMX 0035のPSPにおける全世界3期臨床試験を行っており,将来的には他の適応や候補品についてより多くの臨床試験を行う予定である。われわれはALSにおいてもAMX 0114のINDイネーブル研究を行い,2024年に臨床試験を開始する予定である。
68
著者らはRELYVRIOによる成人ALSの治療に対するFDAの許可を得、カナダ衛生部のALBRIOZAによるALS治療のマーケティング許可を得たが、私たちの申請に対して否定的な意見を提出したため、私たちはまだEUのマーケティング許可を得ていない。もし私たちのフェニックス試験の結果が支持されたら、私たちはEUでALSの再治療のためにAMX 0035のMAAをできるだけ早く再提出する予定です。そこで,我々の努力の大部分をAMX 0035のさらなる開発と商業化に投入し,ALSや他の疾患の治療に投入している。AMX 0035は、私たちの初期および潜在的な追加適応の持続的な開発および追加の規制承認に、私たちのビジネスの将来の成功に重要です。私たちはALS、PSP、WS、AD、その他の適応の治療のために、十分な資金が必要であり、私たちのAMX 0035の臨床開発を登録し、完成させることに成功した。
AMX 0035の将来の規制および商業成功または他の現在または将来の候補製品は、以下のリスクを含むいくつかのリスクの影響を受ける
その中の多くのリスクは私たちがコントロールできないものであり、臨床開発に関連するリスク、監督提出過程、私たちの知的財産権に対する潜在的な脅威、及び任意の未来の協力者の製造、マーケティングと販売努力を含む。もし私たちが追加的な規制の承認を開発、獲得、維持できなければ、あるいは成功することができます
69
AMX 0035を開発する必要性を満たすためにAMX 0035を商業化するか、またはこれらの任意のリスクまたは他の理由で遅延に遭遇した場合、私たちの業務は実質的な損害を受ける可能性があります。
また,製薬業界で大量に開発されている薬物のうち,一部のみが規制部門に上場申請を提出しており,より少ない薬物が商業化承認されている。さらに、たとえ私たちがAMX 0035の任意の適応またはAMX 0114の規制承認を得たとしても、そのような承認は、製品を販売する私たちの適応または用途または患者集団によって制限される可能性がある。さらに、AMX 0035または任意の他の現在または将来の候補製品の神経疾患患者を適切に識別できない場合、AMX 0035またはマーケティング許可を得た任意の他の現在または将来の候補製品のすべての商業的潜在力を達成することができない可能性がある。したがって、私たちが必要な資金を得ることができても、私たちの開発活動に資金を提供し続けることができても、どの司法管区でもAMX 0035またはAMX 0114の開発または商業化に成功することは保証できません。もし私たちまたは私たちの未来のいかなるパートナーも、私たちの初期または潜在的な追加の適応のためにAMX 0035またはAMX 0114を開発、維持、または追加の規制承認を得ることができない場合、私たちは私たちの業務を継続するのに十分な収入を生成できないかもしれない。さらに,われわれはAMX 0035またはAMX 0114の任意の適応を開発している臨床試験では積極的な結果を示さなかったり,他の規制要件を満たしていなかったりして,他の適応におけるAMX 0035またはAMX 0114の開発に悪影響を及ぼす可能性がある。
監督管理の承認を遅延または拒否し、上場後の要求および上場後の義務を達成できない、または任意の司法管轄区で任意のマーケティング申請を再提出し、AMX 0035の追加データまたは情報を提供することを要求することは、AMX 0035の商業化を延期または一時停止し、私たちの収益能力、私たちの業務、および私たちの運営結果に悪影響を与え、私たちの運営を遅延または停止させる可能性がある。
薬品の研究、テスト、製造、ラベル、承認、販売、マーケティング、流通と発売後の義務はアメリカと他の国のFDA、カナダ衛生部、EMAとその他の監督管理機関の広範な監督管理を受けており、これらの監督管理規定は国によって異なる。2022年9月、著者らはFDAの許可を得てAMX 0035(RELYVRIO)を成人筋萎縮性側索硬化症の治療に応用し、著者らはアメリカでRELYVRIOを許可した一部として、発売後にマウスとラットに発ガン性研究、薬物-薬物相互作用研究、及び腎臓或いは肝臓損傷患者に対する研究を要求した。カナダ衛生部からALS治療のAMX 0035(ALBRIOZA)のマーケティング許可も得た。カナダで承認された条件の1つは,我々が行っているグローバルフェニックス試験や他の計画あるいは行われている研究のデータを提供することである。私たちはまた、EU規制機関でAMX 0035をALS治療のために承認することを求めているが、私たちの申請に否定的な意見を出した後、私たちはまだ欧州委員会のマーケティング許可を得ていない。フェニックスの結果が有利であれば,我々はできるだけ早くAMX 0035がEUでALS治療の承認を得ることを早急に求める予定であるが,EU委員会の承認を得ることに成功できないかもしれない。
FDA、カナダ保健省、EMA、または任意の他の外国規制機関は、様々な理由で、AMX 0035の上場承認を延期、制限、拒否、または撤回することができる
70
大量に開発されている薬物のうち,一部のみがFDA,カナダ衛生部,EMAあるいは他の規制承認手続きに成功し,商業化されている。
FDAまたは適用される外国規制機関もまた、AMX 0035の適応および/または患者数が私たちが最初に要求したものよりも限られていることを承認する可能性があるが、FDA、カナダ保健省、EMA、または任意の他の適用可能な外国規制機関は、AMX 0035の商業化に成功するために必要または望ましいラベルを承認しないかもしれない。適用可能な規制承認を得るか、または得ることができない遅延は、AMX 0035の商業化を延期または阻止し、私たちの業務および将来性に大きな悪影響を及ぼすだろう。
AMX 0035は1種の固定用量の組合せ薬物製品であり、ある監督管理機関は証明を要求する可能性があり、この組み合わせが期待される人の群れに対して安全かつ有効であることを証明する以外に、各成分が主張する効果に貢献していることを証明する必要がある。
FDAの組み合わせ規則によれば、FDAは、提案された医薬製品の各成分が主張された効果に貢献することが証明されなければ、固定用量の組み合わせ製品のためにNDAを提出または承認してはならず、各成分の用量(量、頻度、持続時間)は、予想される集団に対して安全に有効である。FDA連携規則に関するより多くの情報は、本年度報告書の“企業−政府規制−固定用量連合製品の共同規則”と題する部分を参照されたい。
私たちが規制承認を求める他の管轄区域では、EUのEMAと類似した規制機関は私たちに似たような要求を加えるかもしれない。本年度報告書の“企業-政府監督管理--固定用量組合せガイドライン”と題する節を参照する。EUでは,われわれの以前のMAAでは,AMX 0035,PB,Turso(TUDCAとも呼ばれる)の各成分の臨床効果を示すための臨床前データのみが提出されている。EMAが将来のMAAで結論を出すことは保証されず,われわれの臨床前データはこれらの目的に対して十分であるか,あるいはそれでもわれわれの臨床前研究結果はAMX 0035の各成分のALS治療に対する臨床効果を証明することはできない。私たちは、EUでの上場許可を得るために、固定用量の組み合わせに含まれる各成分のレベルでの貢献をサポートするために、臨床データを提供することを要求されるかもしれない。
FDAは、成人筋萎縮性側索硬化症を治療する固定用量組み合わせ製品としてAMX 0035(RELYVRIOと呼ばれる)を許可しているが、FDAおよび同様の外国の規制機関は、承認前に追求される可能性のある任意の他の適応を治療するために、AMX 0035または私たちが開発する可能性のある任意の他の固定用量組み合わせ製品の固定用量組み合わせ規則を満たすことを要求するかもしれない。
FDA、EMA、または他の同様の外国の規制機関が、分析研究のような1つまたは複数の臨床試験を行うことを要求する場合、このような臨床試験の設計、持続時間および範囲は、これらの機関および他の同様の外国の規制機関とさらに議論した後に決定されるであろう。したがって、私たちは、異なる司法管轄区域の固定用量組み合わせ製品に対するこれらの要求を満たすために、私たちが行う必要があるかもしれない任意のAMX 0035の将来の臨床試験の推定時間または範囲を正確に予測することができない。進行中の神経学的第三者データ、特にALSでは、我々の製品または他の製品に関するデータは、固定用量の組み合わせを含む規制決定に影響を与える可能性がある。
著者らは研究開発努力を神経変性疾患と中枢神経系疾患の治療に集中し、これは製品開発において非常に限られた成功を収めた領域である。
我々の研究と開発努力は神経変性疾患と中枢神経系疾患の解決に集中している。歴史的に見ると
71
製薬会社の神経変性疾患と中枢神経系疾患領域における努力は製品開発における成功は限られている。神経退行性疾患と中枢神経系療法の発展は独特な挑戦をもたらし、生物学に対する理解が不完全であり、薬物の脳への血液脳関門への制限が存在し、その後の臨床試験と投与量の選択においてよく臨床前研究結果の翻訳性が不足し、及び候補製品の影響が小さすぎて臨床試験中に選択した結果指標を使用できない可能性があり、或いは測定した結果が統計学的意義に達していない。筋萎縮性側索硬化症,AD,その他の神経変性疾患の患者では,承認された治療選択はほとんどない。私たちの将来の成功は、AMX 0035の成功した開発と商業化、および神経変性疾患および中枢神経系疾患の治療のための任意の他の現在または未来の候補製品に高度に依存する。神経変性疾患および中枢神経系疾患を開発および商業化するAMX 0035および任意の他の現在または未来の候補製品は、私たちが最適な用量を選択したことを保証し、適切な臨床試験を実行して有効性をテストすることを保証し、カナダ保健省、FDA、EMAおよび他の同様の外国監督管理機関の規制承認を得て維持することを含む多くの挑戦に直面するであろう。
FDA、カナダ衛生部、EMA、および他の類似外国機関の規制承認過程は長く、時間がかかり、本質的に予測不可能であり、もし私たちが最終的にAMX 0035または任意の他の現在または未来の候補製品の規制承認を維持または獲得できなければ、私たちの業務は深刻な損害を受けるだろう。もし私たちの世界第3段階フェニックス試験がALSの現在または追加のマーケティング許可を支持するのに十分でないことが発見された場合、FDAまたはカナダ保健省がRELYVRIOまたはALBRIOZAに対する以前の規制承認をそれぞれ制限または撤回することになる可能性があり、あるいは規制機関と協議した後、自発的に市場からRELYVRIOまたはALBRIOZAを撤回することを決定する可能性があり、そうでなければ、EUの規制承認を得ることに成功できないかもしれない。
FDA、カナダ衛生部、またはEMAの規制承認をそれぞれ得ておらず、私たちおよび任意の未来のパートナーは、米国、カナダ、またはEUで商業化、マーケティング、普及、または販売してはならない。他の管轄区域の規制当局もまた似たような要求を持っているかもしれない。FDA、カナダ衛生部、EMAと他の類似外国監督管理機関の承認を得るのに要する時間は予測不可能であり、通常臨床試験開始後数年を必要とし、そして多くの要素に依存し、これらの監督管理機関のかなり大きな適宜決定権を含む。また、承認政策、法規、あるいは承認を得るために必要な臨床データのタイプと数量は候補製品の臨床開発過程で変化する可能性があり、司法管轄区域によって異なる可能性がある。FDAはどんな承認でも実質的な有効性の証拠があるかどうかを確認する必要がある。この発見は、2つの十分かつ良好な制御の研究によって確認することができ、または場合によっては、非常に説得力のある単一、大型、多中心、十分かつ良好な制御に基づく研究、または良好な研究および検証的証拠を十分かつ制御することから来る。FDAの法規とガイドラインはまた、まれで致命的な疾患を背景に不確実性に対してより大きな柔軟性と寛容度を有することを可能にしている。
カナダ市場許可の条件の一つは、私たちが行っているグローバルフェニックス3期臨床試験のデータを提供することである。カナダ衛生部は著者らのフェニックス試験を受けたデータが条件を満たすことを保証できず、ALS治療の上場許可にALBRIOZAを許可する条件はない。もしカナダ衛生部がフェニックス試験のデータに満足していなければ、AMX 0035がカナダで販売を継続するために、検証性臨床試験の面で更なる約束をすることを要求することができる。さらに、これらのデータは、ALSを治療するためのAMX 0035のMAAをEUで再提出することをサポートしていない可能性がある。
諮問委員会が2022年9月7日に開催された第2回仮想会議で積極的な承認提案を行った後、FDAはRELYVRIOの承認を承認した。FDAはその後,成人筋萎縮性側索硬化症の治療におけるRELYVRIOの治療を承認したが,今回の会議と前回の諮問委員会会議で,FDAはあらかじめ指定された主な分析の統計モデルの選択と我々のマーケティングアプリケーションに含まれる生存結果の解釈可能性の懸念を提案した。他の規制当局は、筋萎縮性側索硬化症の治療のためのAMX 0035のマーケティングアプリケーションをサポートするために、私たちのデータを検討する際に、私たちのデータに対して同様の懸念を提起する可能性がある。フェニックスがサポートしていない場合、FDAはAMX 0035の承認を制限または撤回することができ、またはAMX 0035を市場から撤回することを求めることができる。もし私たちが規制承認を得る上で遅延に遭遇した場合、または私たちがこのような承認を得られなかった場合、AMX 0035の商業的見通しが損なわれる可能性があり、私たちが収入を創出するか、または追加的な承認を得る能力と、私たちの普通株の価値は深刻な損害を受けるだろう。
臨床試験は費用が高く,設計と実施が困難であり,完成には数年かかる可能性があり,結果自体は不確定である。もしあれば、どんな臨床試験も計画通りに行われるか、予定通りに完成することは保証できません。AMX 0114を含む、我々の初期および潜在的な追加適応または任意の将来の候補製品の臨床開発のためのAMX 0035の臨床開発は、臨床試験においてまたは広範な患者集団において有効性、有害事象の発生を証明することができなかったことを含む、任意の開発段階において固有の失敗リスクの影響を受けやすい
72
深刻なまたは医学的または商業的には受け入れられず、合意または適用される法規要件を遵守できなかったこと、ならびにFDA、カナダ保健省、EMA、または任意の他の同様の外国規制機関は、候補製品が開発または承認を継続できない可能性があると判断した。さらに、FDA、カナダ保健省、EMA、または任意の他の同様の外国規制機関が、現在予想されている基礎に基づいて臨床試験を行うことを要求する場合、または私たちの臨床試験またはAMX 0035が他の適応およびAMX 0114の開発に何らかの遅延が生じた場合、私たちの費用は増加する可能性がある。AMX 0035または任意の他の現在または将来の候補製品が有益な効果を有していても、我々の臨床試験の規模、持続時間、設計、測定、実施または分析を含む1つまたは複数の要因によって、臨床評価中にその効果が検出されない可能性がある。対照的に、同様の要因のため、我々の臨床試験は、AMX 0035または任意の他の現在または将来の候補製品の明らかな積極的効果が実際の積極的効果よりも大きい(あれば)ことを示す可能性がある。同様に、我々の臨床試験では、AMX 0035または任意の他の現在または将来の候補製品の毒性または耐性を検出することができないかもしれない、またはAMX 0035または任意の他の現在または未来の候補製品の毒性または耐性が悪いと誤って考えているかもしれないが、事実はそうではない。
AMX 0035およびAMX 0114は追加または初期の規制承認を得られない可能性があり、私たちの将来の任意の候補製品も規制承認を得ることができない可能性があり、原因は以下の理由を含む多くの理由がある
この長い承認過程と臨床試験結果の予測不可能性は、私たちが監督部門の承認を得ることができず、AMX 0035あるいは私たちが開発した任意の未来の候補製品を発売することができなくなり、これは私たちの業務、運営結果、将来性を深刻に損なうことになるかもしれない。現在承認されている神経変性疾患を治療するための薬剤の終点および試験設計が、AMX 0035およびAMX 0114を含む将来の承認において許容されることは保証されない。FDA、カナダ保健省、EMA、および他の同様の外国当局は、承認過程で大きな自由裁量権を持ち、私たちが開発した任意の候補製品がいつ、または規制されているかどうかを決定します。AMX 0035または任意の他の現在または将来の候補製品の過去または将来の臨床試験から収集されたデータが有望であると信じていても、これらのデータは、FDAまたは任意の他の規制機関の承認をサポートするのに十分ではない可能性がある。
FDAは、製品がその予期される用途に対して安全に有効であるかどうかを決定するためにセキュリティプロトコルを検討し(S)、後者の決定は、多くの証拠に基づく。この発見は、2つの十分かつ良好な制御の研究によって確認することができ、または場合によっては、非常に説得力のある単一、大型、多中心、十分かつ良好な制御に基づく研究、または良好な研究および検証的証拠を十分かつ制御することから来る。FDAはこの基準が満たされることに同意しないかもしれない。したがって、AMX 0035または任意の他の現在または将来の候補製品、FDAおよび他の規制機関、カナダ保健省およびEMAを含む保証はなく、私たちが計画している可能性のある追加の臨床試験を要求することはない。状況はそうかもしれません特にこのような規制機関は
73
上場承認を要請したり、該当機関の上場後の要求に応答したりするとともに、相互に相談したり、ALSに対するAMX 0035研究を行っていることをそれぞれの機関に通知する必要がある場合があります。2022年9月、我々はFDAの許可を得て、成人筋萎縮性側索硬化症の治療のためのAMX 0035を許可し、承認の一部として、発売後にマウスとラットに発癌研究、薬物-薬物相互作用研究、腎臓或いは肝臓損傷患者の研究を要求した。2022年7月、私たちはカナダ保健省からAMX 0035のマーケティング許可を取得し、ALSの治療に条件を付けた。承認の条件の一つは私たちのフェニックス試験のデータを提供することです。カナダ衛生部がわれわれフェニックス試験のデータを受けることは保証されず、ALSの治療にAMX 0035を許可する条件はない。通常,米国だけでなく,カナダやヨーロッパでは,発売承認は2つの3期臨床研究に基づいている。例えば、EMAおよび欧州委員会のCHMPは、半ヒト馬の臨床データが承認をサポートするのに十分であるかどうかに関連して、EUで成人ALSの治療を条件付きマーケティング許可を申請するAMX 0035に否定的な意見を提示する。さらに、別の規制機関は、我々の世界第3段階フェニックス試験は、ALSの追加的なマーケティング許可をサポートするのに十分ではなく、RELYVRIOおよびALBRIOZAの以前の規制承認をそれぞれ制限または撤回することをもたらす可能性があることを発見した。2022年9月7日の諮問委員会の第2回会議で、もし私たちのフェニックス試験が成功しなければ、私たちは自発的にこの製品を市場から出荷することを含む患者に正しい措置を取ると表明した。規制当局のこのような発見または自発的にAMX 0035を市場から撤退させる決定は、私たちの収入を創出し、利益を維持する能力を深刻に損なうだろう。
また、未来の任意の公衆衛生危機による妨害は、著者らが行っていると計画中の臨床前研究と臨床試験を開始、スクリーニング、登録、進行或いは完成する時に困難或いは遅延に遭遇する可能性を増加させる可能性がある。将来公衆衛生危機が発生した場合、病院資源の優先順位により、臨床サイトの起動と患者スクリーニングと登録が遅延する可能性がある。隔離が患者の行動を阻害したり,医療サービスを中断したりすると,調査者や患者は臨床試験案を遵守できない可能性がある。同様に,医療提供者として将来の高度な感染性や感染性疾患への接触が増加する可能性があり,逆にわれわれの臨床試験運営に悪影響を及ぼす可能性があるため,患者,首席調査者,現場スタッフを募集·維持する能力も限られている可能性がある。さらに、私たちは臨床試験場所のモニタリングのような重要な臨床試験活動の中断に遭遇する可能性があり、原因は連邦或いは州政府、雇用主と他の人為的な未来のいかなる高度な伝染性或いは伝染性疾病の発生の強要或いは提案の旅行、隔離或いは社会距離協定に関する制限である。将来の公衆衛生危機の結果として,進行中と計画中の臨床試験の予想スケジュールを実現する上で遅延に直面する可能性がある。
そのほか、監督管理部門は著者らの臨床或いは製造業務に対して検査を行う可能性があり、通常のモニタリング、生物研究モニタリングと審査前検査を含む。さらに、私たちが承認されても、規制機関は、AMX 0035または他の現在または未来の候補製品を承認することができ、その適応は、私たちが要求しているものよりも少ないか、または私たちの製品のために徴収しようとしている価格を承認しないかもしれないし、高価な発売後の臨床前研究および臨床試験の表現に基づいて承認されるかもしれないし、あるいは候補製品を承認する可能性のあるラベルは、候補製品の商業化に成功するために必要または必要なラベル声明を含まないかもしれない。上記のいずれの場合も、AMX 0035または任意の他の現在または将来の候補製品の商業的見通しに実質的な損害を与える可能性がある。
カナダでは,承認前のGMP検査はNDSに関連して行われない。逆に、カナダ衛生部は薬品機関許可証(DEL)によってこのサイトがGMPに符合するかどうかを決定する。DELSはカナダの会社のみが保有しており,同社はこの薬の記録のある輸入業者となっている。輸入時には,薬品と薬品の生産,検査,包装場所を薬品カタログに記載しなければならない。上場は公認された姉妹監督管理機関(例えばEMA或いはFDA)の検査報告に依存する。新冠肺炎が大流行したため,検査報告は現在最大3年前といえる。AMX 0035薬物製品の生産場所はカナダにあり、カナダ衛生部の定例検査を受けている。新冠肺炎が大流行したため,カナダのこれらの検査は現在遠隔で行われている。
我々は、AMX 0035または任意の他の現在または将来の候補製品の開発を完了する間に、予期しないコストまたは遅延が生じるか、または最終的には達成できない可能性がある。
監督部門の承認を得るために、AMX 0035と任意の他の現在或いは未来の候補製品を商業化するために、私たちは広範な臨床前研究と臨床試験を通じて、これらの候補製品が人体に対して安全かつ有効であることを証明しなければならない。臨床前や臨床試験は費用が高く,完成までに数年かかる可能性があり,その結果自体は不確定である。失敗は臨床試験中のいつでも発生する可能性があり、私たちの将来の臨床試験結果は成功しないかもしれません。これは任意の適用を満たすためにAMX 0035の追加法規の承認を得ることに影響を与えるかもしれません
74
発売後の条件または要求、または米国とカナダでAMX 0035を販売し続ける。規制部門はまたAMX 0035を市場からリコールすることを要求するかもしれない。これは、他の適応および任意の他の現在または将来の候補製品に対するAMX 0035開発計画に影響を与え、私たちの運営結果に影響を与える可能性がある。
著者らは臨床試験或いは臨床試験を完成する前の研究及び他の臨床試験を開始或いは完成する上で遅延に遭遇する可能性がある。私たちはまた、臨床試験中に多くの予見不可能な事件に遭遇する可能性があり、これらのイベントは、上場承認を得るか、またはAMX 0035または私たちが開発した任意の他の現在または将来の候補製品を商業化する能力を延期または阻止する可能性がある
監督機関、臨床試験を行っている機関のIRBs或いはデータ監視委員会は様々な原因で臨床試験を一時停止或いは中止する可能性があり、これらの要素は監督要求或いは著者らの臨床規程に従って臨床試験を行うことができなかったこと、FDA或いは他の監督機関の臨床試験操作或いは試験場所の検査による臨床休止の強制実施、予見できない安全問題或いは不良副作用、ある種の薬物の使用がメリットがあることを証明できなかったこと、政府法規或いは行政措置の変化或いは十分な資金が不足して臨床試験を継続することを含む。
ALSまたはADの治療のための早期のAMX 0035臨床試験または私たちが動物で行った任意の他の臨床試験または臨床前研究の結果の負または不確定な印象は、重複または追加の臨床前研究または臨床試験が要求される可能性があり、発売承認が延期される可能性があり、または他の適応の臨床前研究または臨床試験のAMX 0035の変更または遅延をもたらす可能性がある。私たちが行う可能性のある任意の臨床試験が、規制部門が私たちの初期または潜在的な追加適応、または任意の未来の候補製品へのAMX 0035の使用を許可するために、十分な有効性および安全性を証明するかどうかはわかりません。我々が行っているALS全世界3期フェニックス試験を含む後期臨床試験が、非常に強い統計的意義を有する有利な結果を生成しなければ、我々は、以前に発表されたALSに対するAMX 0035の規制承認(我々のFDA承認および私たちのマーケティングを含む)を得ることができるか、または維持することができる
75
カナダ保健省の許可)や潜在的な他の適応、または任意の未来の候補製品は、悪影響を受ける可能性がある。
我々はALS、PSP、WS、ADまたは潜在的な他の適応に対するAMX 0035の臨床試験の開始と完了に成功せず、その各構成要素を含むAMX 0035の有効性と安全性を証明することができず、これはAMX 0035の発売を獲得し、維持するために必要な規制承認であり、もし私たちの世界第3段階鳳凰試験が成功しなければ、私たちの業務を深刻に損害し、任意の適応のためのAMX 0035の開発とマーケティングを継続する能力を含む。もし私たちが検査や規制承認の獲得と維持に遅延があれば、私たちの候補製品開発コストも増加し、臨床試験を完成するために追加の資金を得る必要があるかもしれない。私たちの臨床試験が予定通りに開始または予定通りに完了することを保証することはできません。あるいは試験開始後に私たちの試験を再構成する必要はありません。重大な臨床試験の遅延または臨床試験の追加データの必要性は、AMX 0035または任意の他の現在または将来の候補製品を商業化する期限を独占的に所有するか、または私たちの競争相手が私たちの前に製品を市場に出すことを可能にし、このような候補製品を商業化することに成功する能力を弱める可能性があり、これは私たちの業務および運営結果を損なう可能性がある。さらに、臨床試験の遅延をもたらすか、または引き起こす要因の多くは、最終的にAMX 0035または任意の将来の候補製品の規制承認が拒否される可能性がある。
私たちが必要な臨床前研究と臨床試験を完成したとしても、マーケティング承認過程は高価で、時間がかかり、不確定であり、私たちまたは任意の未来のパートナーが私たちの初期または潜在的な他の適応を阻止し、私たちが開発した任意の未来の製品候補製品がAMX 0035の商業化の承認を得るか、または維持することができるかもしれない。
我々が開発可能な任意の候補製品およびその開発および商業化に関連する活動は、その設計、テスト、製造、安全性、有効性、記録保存、ラベル、貯蔵、承認、広告、販売促進、販売、流通を含み、米国および他の国/地域のカナダ保健省、FDA、EMA、および他の規制機関によって全面的に規制されている。候補製品のマーケティング承認を得られなければ、指定された司法管轄区域で候補製品を商業化することはできないだろう。これまで、私たちは規制承認と潜在的な商業化を求めるために多くの時間と資源を投入してきたが、私たちはアメリカでのみAMX 0035(RELYVRIO)の監督許可を得て、カナダでAMX 0035(ALBRIOZA)条件を持つマーケティング許可を得て、まだどの司法管轄区の監督機関からも他の規制許可を得ておらず、任意の候補製品をマーケティングするために、将来的に開発を求める可能性のある候補製品は永遠に規制されないかもしれない。マーケティング承認を得るために必要な申請を提出して支援した経験はなく、この過程で第三者CROまたは規制コンサルタントに依存して支援してくれます。監督部門の承認を得るためには、各監督機関に広範な臨床前と臨床データ及び支持情報を提出し、生物製剤候補薬物の安全性、純度、有効性と効力を確定する必要がある。監督管理の承認を得るためには、製品の製造過程に関する情報を関連監督機関に提出し、関連監督機関が製造施設を検査する必要がある。私たちが開発した任意の候補製品は効果がないかもしれません。中程度の効果しかないかもしれません。あるいは不良または意外な副作用、毒性、あるいは他の特徴があることが証明される可能性があり、上場承認を得ることを阻止したり、商業使用を阻止したり制限したりする可能性があります。
アメリカ、カナダ、EUと他の外国司法管轄区では、上場承認を得る過程コストが高く、追加の臨床試験が必要であれば、数年かかるかもしれないが、もし本当に承認されれば、関連する候補製品のタイプ、複雑性と新規性を含む様々な要素によって大きく変化するかもしれない。開発期間中の市場承認政策の変化、追加法規又は法規の変化、又は各提出製品申請に対する規制審査の変化は、出願の承認又は拒絶の遅延を招く可能性がある。FDA、カナダ保健省、EMAおよび他の国の類似機関は、承認過程において大きな自由裁量権を有しており、任意の申請の受け入れを拒否することができ、あるいは審査中に私たちのデータが承認を得るのに十分ではなく、追加の臨床前、臨床、または他の研究を行う必要があると決定した。例えば、AMX 0035に対する筋萎縮性側索硬化症の治療に対する我々のNDAを検討している間、FDAは、私たちの臨床前および臨床データに関する情報を明らかにすることを要求し、諮問委員会会議で、私たちの臨床データ解釈のいくつかの懸念に留意することを要求する。また、前臨床試験と臨床試験から得られたデータの異なる解釈は、候補製品の上場承認を遅延、制限、または阻止し、その承認のラベルを制限する可能性がある。例えば,種々の神経変性疾患モデルの臨床前研究が行われているが,FDAの観点では,RELYVRIOがALS患者に対して治療効果を発揮する機序は不明である。また,承認されたRELYVRIOタグでは,FDAは対照研究外で収集したデータの限界を考慮して,ランダム後の長期探索的生存分析を慎重に解釈すべきであることを指摘している。また,FDAは新薬や安全性や有効性に問題のある薬物の申請を諮問委員会に提出する権利がある。例えば、諮問委員会が2022年9月7日に開催した第2回仮想会議の後、FDAはRELYVRIOの承認を承認した。♪the the the
76
諮問委員会は2022年3月30日に初会議を行い,われわれの無作為対照2期Centaur試験とOLEのデータがAMX 0035がALS患者の治療に有効であることを確認したかどうかについて4回(賛成)と6回(反対)の投票を行った。諮問委員会第2回会議では,諮問委員会は,筋萎縮性側索硬化症の治療承認を支持するのに十分な有効な証拠があるかどうかを賛成7票,反対2票の結果で回答し,筋萎縮性側索硬化症が満たされていないニーズ,行われているグローバルフェニックス試験の状況,および筋萎縮性側索硬化症の深刻さを考慮した。今回の会議と諮問委員会の前回の会議で,FDAはあらかじめ指定された主な分析の統計モデルの選択と生存結果に対する解釈可能な関心を提出した。また、私たちがALBRIOZAをカナダで販売することを許可する条件の一つは、フェニックス試験のデータを提供することです。カナダ衛生部が我々のフェニックス試験のデータが条件を満たすことを受け入れることを保証することはできず、ALSまたはフェニックス試験の治療のためのALBRIOZAの上場許可を許可する条件はない。もしカナダ衛生部がフェニックス試験のデータに満足していなければ、AMX 0035がカナダで販売を継続するために、検証性臨床試験の面で更なる約束をすることを要求することができる。したがって、私たちは私たちが求めているマーケティング承認を維持できないかもしれません。私たちが最終的に得た任意のマーケティング承認は、任意の条件付き承認を含めて、拒否され、制限され、撤回されるか、または承認された製品を商業的に不可能にする可能性のある制限または承認された後に約束される可能性があります。
もし私たちが承認を得る上で遅延に遭遇した場合、または私たちがAMX 0035または私たちが開発する可能性のある任意の候補製品の承認を維持または獲得できなかった場合、これらの候補製品(AMX 0035を含む)のビジネス見通しが損なわれる可能性があり、私たちが収入を創出する能力は深刻な損害を受けるだろう。
早期臨床試験と臨床前研究の結果は未来の結果を予測できないかもしれない。著者らの臨床試験中の初期或いは初歩的なデータはこれらの試験の完了時或いは後期試験で得られた結果を示すことができないかもしれない。
臨床前研究の結果は臨床試験の結果を予測できない可能性があり,われわれが開始したいかなる早期臨床試験の結果も後期臨床試験の結果を予測できない可能性がある。また,臨床試験の初期データは,このような試験完了後の結果を反映できない可能性がある。我々の任意の臨床試験が最終的にAMX 0035または任意の他の現在または未来の候補製品のさらなる臨床開発に成功または支持することは保証されない。また,Centaur試験で見られた臨床結果は,われわれの全世界第3段階フェニックス臨床試験で重複しない可能性があり,これは,カナダで無条件にALBRIOZAの許可を得,米国でRELYVRIOの承認を維持し,EUで承認を求め,AMX 0035の開発を継続してより多くの適応を得る能力に重大な影響を与える可能性がある。臨床試験中の薬物や生物製品の失敗率が高かった。多くの製薬と生物科学技術業界の会社は臨床開発において重大な挫折に遭遇し、早期の研究で人を奮い立たせる結果を得ても、著者らの臨床開発中のいかなるこのような挫折は著者らの業務と経営業績に重大な不利な影響を与える可能性がある。
また,我々は過去に使用したことがあり,将来的には“オープンラベル”臨床試験設計を用いることも可能である。オープンタグ“臨床試験”とは、患者および研究者の両方が、患者が研究製品候補を受け入れているかどうかを知っているかどうか、または既存の承認薬またはプラセボを意味する。大多数の開放ラベル臨床試験は候補の研究製品のみをテストし、時々異なる用量レベルでテストを行う可能性がある。開放ラベル臨床試験は様々な制限を受けており,これらの制限は任意の治療効果を誇張する可能性があり,開放ラベル臨床試験中の患者が治療を受ける際に知られているからである。オープンラベル臨床試験は“患者偏見”の影響を受ける可能性があり,すなわち患者が症状が改善したと考えているのは,実験的治療を受けていることを意識しているだけである。また,オープンラベル臨床試験は,“調査者偏見”の影響を受ける可能性があり,すなわち臨床試験の生理結果を評価·審査する人は,どの患者が治療を受けているかを知り,その知識を知っている場合に治療群の情報をより有利に解釈することが可能である。プラセボまたは能動対照の制御された環境で研究を行う場合、開放ラベル試験の結果は、AMX 0035または任意の未来の候補製品の将来の臨床試験結果を予測できない可能性がある。
著者らが時々発表或いは公表した臨床試験の一時的な背線と初歩的なデータはより多くの患者データの出現に従って変化する可能性があり、そして監査と検証プログラムの影響を受け、これは最終データの重大な変化を招く可能性がある。
時々、私たちは臨床試験の一時的な背線または初歩的なデータを公表するかもしれない。著者らが完成する可能性のある臨床試験の中期データは、患者登録の継続とより多くの患者データの獲得に伴い、1つ以上の臨床結果が実質的に変化する可能性があるというリスクに直面している。初歩的な結果或いはベースラインの結果は依然として監査とチェック手続きを守らなければならず、これは最終データが著者らが以前公表した初歩的なデータと大きく異なる可能性がある。したがって、最終データを得る前に、中期と予備データを慎重に見なければならない。初期または中期データと最終データとの間の不利な差は、私たちの名声およびビジネスの将来性を深刻に損なう可能性があります。
77
臨床試験で患者を登録と保留することは高価で時間のかかる過程であり、私たちがコントロールできない多種の要素によってより困難あるいは不可能になる可能性がある。
患者登録は臨床試験時間スケジュールの重要な要素であり、臨床試験の時間スケジュールはある程度著者らが患者を募集して試験に参加する速度、及び必要なフォローアップ期間の完成状況に依存する。カナダ保健省、FDA、EMA、または他の同様の外国規制機関の要求に基づいて、これらの試験に参加するのに十分な数の合格者を見つけて募集することができない場合、AMX 0035または任意の他の現在または未来の候補製品の臨床試験を開始または継続することができないかもしれない。さらに、AMX 0035および任意の他の現在または将来の候補製品のいくつかの臨床試験は、患者数が比較的少ない適応に重点を置いている可能性があり、これは、条件に適合する患者の登録人数をさらに制限するか、または登録者数が予想よりも遅いことをもたらす可能性がある。われわれの臨床試験の資格基準が確立すると,利用可能な試験参加者をさらに制限する可能性がある。例えば,筋萎縮性側索硬化症を有する患者数は少なく,場合によっては正確に決定されていない場合がある。これらの疾患の実際の患者数が予想よりも少ない場合、我々は、AMX 0035または任意の他の現在または将来の候補製品の開発および承認を延期または阻止するために、患者を臨床試験に参加させる際に困難に遭遇する可能性がある。一度登録しても、十分な数の患者を残して、私たちのどんな実験もできないかもしれません。例えば、筋萎縮性側索硬化症患者は深刻な活動能力の問題、発病率とその他の合併症があり、これは歴史的に筋萎縮性側索硬化症試験中の保留を更に挑戦的にさせる。これらの挑戦は,われわれが将来臨床試験を行う可能性のある適応も含め,多くの他の神経変性疾患の適応にも存在する。過去にわれわれの臨床試験では,われわれのCentaur試験やCentaur Ole試験を含めて中止されたことがある。中断は現在或いは未来の試験中に発生する可能性があり、著者らの臨床試験の完成遅延を招く可能性があり、著者らが臨床試験中に更に多くの患者を募集する能力を影響し、そして著者らの臨床試験データの完全性に影響を与える。
患者の臨床試験における登録と保持は,患者群の大きさ,調査中の疾患の重症度,試験案の性質,候補製品の既存の安全性と有効性データ,競合療法の数や性質,行われている競合療法の臨床試験や同じ適応を獲得する競争療法を獲得する機会の拡大,患者と臨床場所の接近度,試験の資格基準,試験期間中に患者の能力を十分にモニタリングすること,研究している候補製品に対する臨床医や患者の潜在的優位性の見方,すべての現場訪問が完了する前に試験を終了するリスクを含む多くの要因に依存する。適時かつ費用効果を持って著者らの臨床試験を完成するために、選択可能な患者は限られており、これは著者らが対象とする神経疾患が少ないためである。
また,患者コミュニティとの関係構築努力は成功しない可能性があり,われわれの臨床試験における患者登録の遅延を招く可能性がある。
我々は、AMX 0035または任意の将来の候補製品の臨床試験において報告されるかもしれない任意の負の結果も、候補製品の他の臨床試験の募集および維持を困難にするか、または不可能にする可能性がある。計画中の患者の登録または保留の遅延または失敗は、コスト増加、計画遅延、または両方をもたらす可能性があり、これは、ALS、PSP、WS、ADおよび他の適応、ならびに任意の他の現在または将来の候補製品においてAMX 0035を開発する能力に有害な影響を与えるか、またはさらなる開発を不可能にする可能性がある。さらに、患者が私たちの臨床試験から退出した場合、予定された用量または後続のアクセスを逃したり、他の方法で臨床試験プログラムに従わなかった場合、公衆衛生流行病および関連疾患のためにも、私たちの臨床試験データの完全性が損なわれる可能性があり、あるいはカナダ保健省、FDA、EMAまたは他の規制機関に受け入れられない可能性があり、これは計画に適用される重大な挫折になるだろう。また,将来の臨床試験の適切かつタイムリーな進行を確保するためにCROや臨床試験サイトに依存する可能性があり,彼らのサービスについて合意しようとしているが,実際の表現を強制する能力は限られている。
候補製品の製造または処方を変更する方法は、追加のコストや遅延を招く可能性がある。
候補製品が臨床前研究から後期臨床試験までを通じて、潜在的な承認と商業化を得るために、製造方法や調合などの開発活動の様々な側面に伴い、過程の最適化と結果の最適化に努力するために変更されることが一般的である。これらの変化のいずれも、AMX 0035または任意の他の現在または将来の候補製品の表現が異なることをもたらし、進行中の臨床試験またはプロセスを変更して製造された材料を使用して行われる他の将来の臨床試験の結果に影響を与える可能性がある。例えば、欧州でAMX 0035を承認することを求める過程で、Centaur試験で評価された処方とは異なるAMX 0035製剤をサポートするデータを提出した。それらの臨床研究の商業処方の変更は、これら2つの異なる処方の生物利用度が比較可能性を有することがより多くの臨床データによって証明されるまで、規制当局が私たちの上場申請の承認を延期することを招く可能性があり、または臨床評価の以前の処方に回復することを要求する可能性がある。早期製造方法または処方下で生産された候補製品から得られた早期臨床データと計画中の商業データとを接続するために比較可能性テストを行う必要があるかどうか
78
制定過程で、規制部門はこの検査結果の説明に同意しないかもしれない。これは臨床試験の完成を遅らせる可能性があり、1つ以上の臨床試験を繰り返し、臨床試験コストを増加させ、AMX 0035または任意の他の現在または未来の候補製品の承認を延期し、私たちが販売を開始し、収入を創出する能力を脅かす必要がある。
規制の承認が得られた場合、AMX 0035または任意の将来の候補製品は、不良副作用をもたらす可能性があり、またはその規制承認を延期または阻止し、承認されたラベルの商業イメージを制限するか、または重大な負の結果をもたらす可能性がある他の特性をもたらす可能性がある。
AMX 0035または任意の将来の候補製品によって引き起こされる副作用は、私たちまたは規制機関の臨床試験の中断、延期、または停止をもたらす可能性があり、より厳しいラベル、またはFDA、カナダ保健省、EMA、または同様の外国の規制機関の遅延、拒否、または規制承認の撤回をもたらす可能性がある。著者らの臨床前研究或いは臨床試験の結果は副作用或いは予期しない特徴の高さと受け入れられない重症度と普遍性を示す可能性がある。これまで、AMX 0035の臨床試験において、AMX 0035の全体的な耐性は良好であり、最もよく見られる治療副作用は下痢、腹痛、吐き気、上気道感染、便秘、頭痛、疲労、蛋白尿と食欲低下を含む。また,AMX 0035服用時に味不良を体験することが報告されている。動物研究では、妊娠および授乳中にラットにAMX 0035を投与することは、すべての試験用量の子孫死亡率を増加させ、著者らの臨床試験試験の臨床用量よりも低いか、または類似している。しかしながら、我々の世界第3期フェニックス臨床試験または他の未来のALSまたは他の適応臨床試験においてAMX 0035の類似耐性が観察される保証はない。多くの最初に臨床或いは早期試験で所望の化合物を示した後、不良或いは予期しない副作用を引き起こすことが発見され、化合物の更なる発展を阻止した。
AMX 0035または任意の他の現在または将来の候補製品の開発中に受け入れられないまたは深刻な副作用が発生した場合、私たち、FDA、カナダ保健省、EMAまたは同様の外国の規制機関、IRBまたは私たちの試験を行う機関の独立道徳委員会、または独立した安全監視委員会は、私たちの臨床試験を一時停止または終了することができ、または規制機関は、臨床試験を停止するか、またはAMX 0035または任意の他の現在または将来の候補製品の任意のまたはすべての目標適応の承認を拒否するように命令することができる。薬物に関連すると考えられる治療に生じる副作用は、被験者の募集または被験者の試験を完了する能力に影響を与える可能性があり、または潜在的な製品責任クレームをもたらす可能性がある。1つの適応の臨床試験においてAMX 0035が出現する副作用は、他の適応におけるAMX 0035の臨床試験登録、規制承認、および商業化に悪影響を及ぼす可能性がある。さらに、他の当事者は、AMX 0035のコンポーネントまたは将来の候補製品に対して負の調査結果を行う可能性がある。例えば、Humanitas Mirasole SpaまたはHumanitasはEUで第3段階臨床試験を行っており、ALS患者に対するTursoの安全性および有効性を評価するために、Tursoの安全性に関するより多くの発見をもたらす可能性がある。サードパーティの任意の負の発見は、AMX 0035または私たちが開発する可能性のある他の候補製品の将来の承認またはラベルに影響を与える可能性がある。さらに、治療医療従事者は副作用を適切に認識したり、処理したりしない可能性がある。私たちはアビタスとは何の関係もない。彼らの第3段階臨床試験が成功し、TursoがFDAまたは任意の他の規制機関の承認を得た場合、Tursoは、私たちの知的財産権保護および私たちが所有または将来所有する可能性のある規制排他性がこのような商業化を阻止しない限り、AMX 0035と競合する商業化製品になる可能性がある。AMX 0035または任意の将来の候補製品の潜在的副作用を認識または管理するためのトレーニングが不足しており、患者の負傷または死亡を招く可能性がある。このようなどんな状況でも、私たちの業務、財政状況、そして見通しに大きな被害を及ぼす可能性がある。
われわれのAMX 0035の臨床試験では苦味がしばしば認められた。苦味自体は患者にとって安全なリスクとはならないが、それは患者のより多くの不コンプライアンスをもたらす可能性があり、これは、我々が行っている全世界3期フェニックス試験を含む、我々の臨床試験で観察されたAMX 0035の治療効果を低下させるか、またはその商業応用を制限するかもしれない。
また,AMX 0035の臨床試験は,丁寧に定義された同意が臨床試験に入った患者群で行った。したがって、我々の臨床試験または任意の将来のパートナーの臨床試験は、AMX 0035または任意の他の現在または将来の候補製品が、実際の積極的効果(あれば)を超える、または不良副作用を識別することができないことを示す可能性がある。
最後に,AMX 0035はTursoとPBの組合せである.PBはすでにFDAと他の監督管理機関によってある尿素循環障害の治療に許可され、Tursoはすでにイタリアで肝硬変疾患、例えば原発性胆汁性肝硬変の治療に許可されている。AMX 0035のうちの1つまたは複数の活性部分は、FDAまたは他の規制機関の承認を得ている可能性もある。AMX 0035が市場承認を得て管轄区域で商業化されても、適用される規制機関がPBまたはTursoまたはAMX 0035の任意の有効部分の承認を取り消す可能性がある(適用される場合)、または治療効果、製造、または供給の問題が可能であるというリスクに直面するであろう
79
PBまたはTursoまたはAMX 0035の任意の活性部分と共に現れる(適用される場合)。これは私たち自身の製品が市場から撤退されたり、商業的にあまり成功しないことを招くかもしれない。
増加しているAMX 0035アクセス権限の需要は、私たちの名声にマイナスの影響を与え、私たちの業務を損なう可能性があります。
われわれはALS,PSP,WS,AD,その他の潜在的な将来適応の治療にAMX 0035を開発しており,現在これらの適応の治療選択は限られているかない。個人や団体は会社を目指し,破壊的なソーシャルメディア活動により,重大な満足されていない医療ニーズを有する患者のための未承認薬物の獲得を要請することに関与している可能性がある。もし私たちがアクセス権限拡大政策の下でAMX 0035または任意の未来の製品候補製品へのアクセス権限を提供または提供しないことを決定した場合、私たちの名声はマイナスの影響を受ける可能性があり、私たちの業務は損なわれる可能性があります。
過去、メディアによる個別患者参入要求の拡大への関心は、ALSのキー療法の獲得を加速させる法案および以前の“試用権”法、例えば2017年の連邦試用権法案など、従来のEAPよりも早く未承認治療を可能にすることを目的とした地方および国家レベルの立法の制定と公布を招き、前者はALSに関連する研究および開発を支援することを目的としていた。この分野の急進主義と立法の可能な結果の一つは、私たちがFDAに提出したEAPを開始する必要があるか、または予想よりも早くAMX 0035または任意の未来の候補製品をより広く発売する必要があるということかもしれない。
さらに、一部の患者は、商業的承認を得る前に、慈悲使用、EAP、または薬物の権利を獲得しようと試みることによって薬物を獲得し、彼らは生命を脅かす疾患を有し、他のすべての利用可能な治療方法を使い切っている。これらの患者集団では、SAEのリスクが高く、これは、AMX 0035または将来の候補製品の安全状態に悪影響を及ぼす可能性があり、これは、重大な遅延をもたらすか、またはAMX 0035または将来の候補製品を商業化することに成功できない可能性があり、これは、私たちの業務に実質的な損害を与える可能性がある。私たちは、AMX 0035または将来の候補製品の規制承認および商業化に成功するために必要な制御された臨床試験を実行するために、将来的に、AMX 0035または将来の候補製品の規制承認および商業化に成功するために必要な制御された臨床試験を実行するために、私たちが開始した任意の未来の医薬品および/またはEAPを再構成または一時停止する必要があるかもしれず、これは、そのような計画の既存または潜在的参加者に関連する負の宣伝または他の中断を引き起こす可能性がある。
ソーシャルメディアプラットフォームの使用はますます多くなり、新たなリスクと挑戦をもたらしている。
ソーシャルメディアは、私たちの臨床開発活動やAMX 0035が治療のために開発されている疾患を交流するためにますます使用されており、私たちは、米国でのRELYVRIOとカナダのALBRIOZAの商業化努力、そして私たちが規制の承認を得ている他の任意の管轄区域を実現するために適切なソーシャルメディアを利用し続けるつもりです。バイオテクノロジーや製薬業界のソーシャルメディア実践は発展し続けており,このような使用に関する条例や管理ガイドラインも発展しているが,常に明確ではない。この変化は、不確実性と、我々の業務に適用される法規を遵守しないリスクをもたらし、私たちに対する規制行動、およびラベル外マーケティングまたは他の禁止された活動に関連する可能性のある訴訟、ならびにFDA、米国証券取引委員会、および他の規制機関のより厳しい審査をもたらす可能性がある。例えば、患者は、ソーシャルメディアチャネルを使用して、AMX 0035を使用した治療の経験をレビューすることができ、または進行中の盲目的臨床試験における彼らの経験、またはいわゆる有害事象を報告することができる。このような開示が発生すると、試用登録に悪影響を及ぼす可能性があり、適用される有害事象報告義務を監視して遵守することができない可能性があり、またはソーシャルメディアによる政治的および市場的圧力の下で、私たちの業務または公衆の合法的な利益を守ることができない可能性があります。これは、候補製品に対する私たちの発言が制限されているからです。いずれのSNSにおいても、敏感または機密情報または否定的または不正確な投稿またはコメントを不適切に開示するリスクがある。さらに、私たちはソーシャルメディアで、当社、管理職、AMX 0035、または未来の製品候補製品に関する攻撃に遭遇するかもしれません。もしこのような事件が発生したり、私たちが適用された法規を遵守できなかった場合、私たちは責任を負い、規制行動に直面したり、私たちの業務に他の損害を与える可能性があります。
より多くの適応を得るためにAMX 0035を開発し、商業化することができない場合、または他の現在または将来の候補製品を発見、開発、商業化することができなければ、私たちの業務を発展させることができない可能性があり、戦略目標を達成する能力は損なわれるだろう。
筋萎縮性側索硬化症を治療するAMX 0035の商業化が現在の主な重点であるにもかかわらず、我々の長期成長戦略の一部として、他の適応へのAMX 0035の応用を継続的に評価し、他の候補製品を開発することを計画している。我々は、AMX 0035または他の潜在的候補製品の内部機会を評価することを意図しており、神経変性疾患および中枢神経系または他の疾患を有する患者を治療するために、他の候補製品および商業製品の許可または買収を選択することも可能であり、これらの患者は、有意な未満足の医療需要および限られた治療を有する
80
選択します。これらの他の潜在的な候補製品は、商業販売の前に、臨床前研究、臨床試験およびFDA、カナダ衛生部、EMAおよび/または他の適用可能な外国規制機関の承認を含む追加的で時間のかかる開発作業が必要となるであろう。すべての候補製品は薬品開発固有の失敗リスクに直面しやすく、候補製品は十分な安全と有効が証明されない可能性があり、監督管理機関の許可を得られない可能性がある。さらに、承認されたこのような製品は、経済的に製造または生産され、商業化に成功したり、市場に広く受け入れられたり、他の商業代替製品よりも効果的であることを保証することはできません。
候補製品を決定する研究活動には、最終的に任意の候補製品が決定されたか否かにかかわらず、大量の技術、財政、人的資源が必要である。著者らの研究活動は最初に潜在的な候補製品の決定に希望を示す可能性があるが、多くの原因で臨床開発のための候補製品を生成できなかった
もし私たちがより多くの候補製品を発見し開発することに成功しなければ、私たちの成長潜在力と私たちの戦略目標を達成することは損なわれるかもしれない。
私たちは、より多くの候補製品または将来検討すべきAMX 0035の適応および修正を決定することによって、私たちのチャネルを拡大する努力は成功しないかもしれない。AMX 0035の特定の候補製品または適応または処方を追求するために限られたリソースがかかる可能性があり、AMX 0035を利用することは、より有利であるか、またはより成功する可能性のあるこれらの候補製品または適応または処方を利用することができないかもしれない。
私たちの財務と管理資源が限られているため、私たちはAMX 0035の具体的な適応と改良剤に集中している。したがって、AMX 0035がいくつかの適応において有害な副作用を有することが証明される可能性があることを含む、AMX 0035のためにより多くの臨床開発機会を作ることができない可能性があり、または他の特徴は、これらの追加の適応において発売承認および市場受け入れを得ることがあまり不可能であることを示している。
われわれは,WS,PSP,他の適応患者に対する臨床試験を含め,今後数年以内にAMX 0035を同時にいくつかの臨床試験を行う予定であり,どの適応を優先的に選択するかを決定することは困難である可能性がある。したがって、私たちは、私たちがより大きなビジネス潜在力や成功の可能性があると思う他の兆候を持つ機会を探すことを放棄または延期するかもしれない。さらに、AD患者のためのAMX 0035の探索計画、およびALSおよび他の神経変性疾患のための他の候補製品の評価を継続している。しかしながら、私たちは、他の潜在的な適応および候補製品ではなく、私たちの1つまたは複数の目標適応に重点を置くことができ、そのような開発努力は成功しない可能性があり、これは、AMX 0035および他の候補製品の臨床開発および承認を延期するであろう。また,AMX 0035の他の適応を決定する研究活動には大量の技術,財力,人的資源が必要である。化学関連,安定性関連,あるいはその他の理由により,これらの追加的な修飾の開発に成功できない可能性がある。我々は最近,筋萎縮性側索硬化症や他の神経変性疾患に対するCalain−2のアンチセンスオリゴヌクレオチドの開発を発表した。われわれは現在INDを有効にする研究によりAMX 0114を進めており,2024年に臨床に入る予定である。私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。AMX 0035またはAMX 0114または他の候補製品の特定の適応または処方の現在および将来の研究開発活動への支出は、いかなる商業的に可能な製品も生成しない可能性がある。
さらに、ライセンス内や買収開発段階の資産や計画を求めることができ、追加的なリスクをもたらす可能性がある。将来性のある候補製品を識別、選択、獲得するには大量の技術、財務、および
81
人的資源専門知識。このような努力は、可能な特定の候補製品を実際に獲得することを招くことはなく、何のメリットも生じることなく、私たちの管理職の時間と資源支出の分流をもたらす可能性がある。
例えば、最終的に製品の承認につながる計画を決定できなければ、最終的に投資収益を提供できない製品を評価、買収、開発するために大量の資本や他の資源がかかる可能性がある。
競合製品は、現在または将来の適応におけるAMX 0035のビジネス機会を減少または除去する可能性がある。もし私たちの競争相手が私たちよりも早く技術または候補製品を開発し、または彼らの技術または候補製品が私たちよりも効果的または安全であれば、私たちがAMX 0035を開発し、それを商業化することに成功する能力は不利な影響を受ける可能性がある。
筋萎縮性側索硬化症とアルツハイマー病を含む他の神経変性疾患を治療する臨床と商業前景の競争は激しく、そして迅速と重大な技術変化の影響を受ける。現在のAMX 0035適応については、AMX 0035の任意の将来の適応、または将来開発または商業化を求める可能性のある他の候補薬剤について、世界各地からの主要な製薬会社、専門製薬会社、バイオテクノロジー会社の競争に直面している。例えば、Humanitas Mirasole spaは現在EUで3期の臨床試験を行っており、ALS患者におけるTursoの安全性および有効性を評価し、承認されれば、AMX 0035の競争相手として商業化されている可能性がある。この研究が臨床的ゴールに達した場合、このような単一療法はFDA、EMA、および他の規制機関の承認を得る可能性があり、Tursoは、私たちの知的財産権保護および私たちが所有または未来に所有する可能性のあるいかなる規制排他性がこのような商業化を阻止しない限り、AMX 0035と競合する商業化製品になる可能性がある。また,大手製薬やバイオテクノロジー会社では現在薬物の販売·販売が行われているか,我々が求めている適応を治療するための候補薬が開発されている。潜在的な競争相手はまた学術機構、政府機関とその他の公共と個人研究組織を含み、これらの組織は研究を展開し、特許保護を求め、研究、開発、製造と商業化のための協力手配を確立する。
いくつかの大手製薬会社がFDAが承認したALS治療薬を販売している。これらの薬剤には、セノフィ−アンバンテ米国有限責任会社によって販売されているリルゾールと、三菱Tanabe Pharma America,Inc.で販売されているRadicavaがある。また、三菱Tanabe Pharma America,Inc.は、Mtpaと略称してRadicavaに代わる経口薬を開発している。2022年第1四半期、FDAはMTPAによるRadicavaの経口代替の優先審査申請を受け、2022年5月にRadicavaの経口代替申請を承認した。私たちの潜在的な競争相手は、Biogen,Inc.,UCB S.A.およびPTC Treateutics,Inc.,専門製薬および後発薬会社、学術機関、政府機関、研究機関などの製薬とバイオテクノロジー会社を含む。FDAは2022年7月にNDAを受け,tofersenの優先審査を承認し,tofersenはSOD 1 ALS患者を評価しているアンチセンス研究薬である。2023年4月、FDAはQALSODYの承認を加速した。
私たちの多くの競争相手は私たちよりもっと多くの財務資源を持っていて、市場で足場を固めて、研究開発、製造、臨床前と臨床テストの方面で専門知識を持って、監督管理の許可を得て、そして清算とマーケティングの許可を得た製品です。したがって、私たちの競争相手は私たちよりも治療の規制承認を得ることに成功し、幅広い市場で受け入れられるかもしれない。私たちが開発と商業化費用を回収する前に、私たちの競争相手の製品は、私たちが商業化する可能性のある任意の候補製品よりも有効であり、AMX 0035または任意の未来の候補製品を時代遅れにしたり、競争力を失ったりする可能性がある。AMX 0035が現在求められている適応のために承認された場合、一連の開発されている治療法と競合する可能性がある。さらに、私たちの競争相手は、AMX 0035または私たちが開発する可能性のある任意の未来の候補製品よりも効果的またはより安価であるかもしれない技術および医薬製品の開発、獲得に成功する可能性があり、これは、これらの候補製品を時代遅れにし、競争力を持たないかもしれない。
AMX 0035または任意の他の将来の候補製品が承認された後、私たちは、私たちの製品の有効性、安全性および耐性、私たちの製品の管理容易さ、これらの製品が監督管理によって承認された時間および範囲、製造、マーケティングおよび販売能力の利用可能性およびコスト、価格、精算範囲、および特許地位を含む多くの異なる要因に基づく競争に直面する可能性があります。既存および将来の競合製品は、私たちが商業化している任意の製品よりも効率的で、より安全で、より安価で、またはより効率的なマーケティングおよび販売を含む、より良い治療代替案を提供する可能性がある。私たちが候補製品を開発して商業化する費用を回収する前に、競争力のある製品は、私たちが商業化したどの製品も時代遅れになったり、競争力がないかもしれない。これらの競争相手はまた私たちの従業員を募集する可能性があり、これは私たちの専門レベルと業務計画を実行する能力にマイナスの影響を与える可能性がある。
82
さらに、私たちの競争相手は、私たちよりも早く特許保護、規制排他性、または規制承認を得て製品を商業化するかもしれません。これは、規制承認を受けた候補製品の将来の承認や販売に影響を与える可能性があります。RELYVRIOは米国とALBRIOZAのカナダの商業化で競争に直面することが予想され,承認されれば,どの将来の候補製品も競争に直面する。カナダ保健省、FDA、またはEMAがAMX 0035または任意の将来の候補製品の商業販売を承認した後、マーケティング能力および製造効率の面で競争を展開する。製品間の競争は、製品の効果と安全性、規制承認の時間と範囲、供給、マーケティングと販売能力の可用性、製品価格、政府と個人第三者支払者の清算範囲、規制排他性、特許地位に基づくと予想される。もし私たちの候補製品が規制部門の承認を得たが、市場で効果的に競争できなければ、私たちの収益性と財務状況は影響を受けるだろう。
製薬とバイオテクノロジー産業の合併と買収は、私たちの数の少ない競争相手により多くの資源を集中させる可能性がある。規模の小さい会社や他のスタートアップ会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配されている。これらの第三者は合格した科学と管理者を募集と維持し、臨床試験場を構築し、私たちの活動と相補的あるいは必要な技術を獲得する面で私たちと競争を展開している。
1つの管轄区域でAMX 0035または任意の将来の候補製品の規制承認を取得し、維持することは、他の管轄区域でこれらの候補製品の規制承認を得ることに成功することを意味するわけではない。
1つの管轄区域でAMX 0035および任意の将来の候補製品の規制承認を取得し、維持することは、任意の他の管轄区域で規制承認を得ることができるか、または維持することができる保証はなく、1つの管轄区域で監督管理承認を得ることができないか、または遅延することは、他の管轄区の監督管理過程に悪影響を及ぼす可能性がある。例えば、FDAはAMX 0035(RELYVRIO)を承認し、カナダ衛生部はAMX 0035(ALBRIOZA)の上場許可を許可したが、EUと他の外国司法管轄区の類似規制機関もこれらの国でのAMX 0035の製造、マーケティングと普及を許可しなければならない。承認手続きは司法管轄区域によって異なり、1つの管轄区域で行われる臨床試験は他の管轄区域の監督機関によって受け入れられない可能性があるので、米国、カナダ、またはEUとは異なるまたはそれ以上の要求および行政審査期間に関連する可能性がある。米国以外の多くの管轄区では、カナダとEUを含むいくつかの司法管轄区では、候補製品はまず精算許可を得なければならず、その後、その管轄区で販売許可を得ることができる。場合によっては、私たちが私たちの製品のために受け取る価格もまた承認されなければならない。
米国以外の管轄区域の監督管理機関は候補製品の承認に要求があり、私たちはこれらの管轄区が発売される前にこれらの要求を守らなければならず、これらの規制要求は国/地域によって大きく異なる可能性がある。他の規制の承認を得て、他の規制要求を遵守することは、私たちの重大な遅延、困難、コストを招く可能性があり、追加の臨床前研究または臨床試験が必要になる可能性があり、これは高価で時間がかかる可能性があり、私たちの製品がある国/地域で発売されることを延期または阻止する可能性がある。外国規制機関の承認過程はFDA承認に関連するすべてのリスクに関連する。アメリカとカナダを除いて、私たちはどの司法管轄区での販売を許可された候補製品もなく、私たちはカナダ以外の国際市場で監督管理の許可を受けた経験もありません。もし私たちが国際市場の規制要求を遵守できず、および/または適用されたマーケティング承認を得て維持できなければ、私たちの目標市場は減少し、AMX 0035または任意の将来の候補製品の市場潜在力を十分に発揮する能力は損なわれるだろう。
米国およびEUにおけるALSの治療および米国におけるWS治療のためのAMX 0035の孤児薬物指定を得ても、市場排他性を含む孤児の薬物状態に関する利点を得ることができないか、または維持することができない可能性がある。
米国やEUを含むいくつかの管轄区の規制機関は、患者数が比較的少ない薬物を孤児薬に指定する可能性がある。孤児医薬品法によると、医薬品がまれな疾患や疾患の治療を目的としている場合、FDAは、通常、米国では患者数が20万人未満、または米国では20万人を超えると定義されているが、米国の販売で薬物の開発コストを回収することは合理的に期待されていない。EUでは、診断に用いられる製品について孤児の称号を与えることができる。生命や慢性衰弱を脅かす疾患の予防や治療は,申請時にEU 10,000人あたり5人に影響を与えない。さらに、生命または長期的に衰弱している疾患の診断、予防または治療のための製品については、インセンティブ措置がなければ、EUにおける製品の販売が、製品を開発するための必要な投資が合理的であることを証明するのに十分でない可能性が高い場合には、指定を得ることができる。いずれの場合も,孤児に指定された申請者も証明しなければならない
83
このような疾患の満足できる診断、予防または治療を許可する方法はない(または、そのような方法が存在する場合、新製品は、既存の製品と比較して、影響を受けた人に大きな利点となるであろう)。
2017年9月、FDAはAMX 0035孤児薬物地位を付与し、米国のALS患者の治療のために使用し、2022年9月に成人ALSの治療のためにAMX 0035(RELYVRIO)を許可し、この製品は孤児薬物の独占経営権を付与され、FDAはAMX 0035と同じ孤児適応を含む後発薬またはブランド製品を許可できず、期限は7年であるが、いくつかの例外状況は除外されている。また、2020年6月、EMAはEU ALS患者の治療のためにAMX 0035孤児薬物地位を授与した。また,2020年11月に米国でWS患者のAMX 0035を治療する孤児薬物状態を獲得した。一般に、孤児の薬物名を有する薬物がその後、そのような名称を有する適応の最初の発売許可を得た場合、薬物は、市場排他期を有する権利がある可能性があり、これは、FDAまたはEMAがこの期間内に同じ薬物の別の上場許可申請を承認することを阻止するであろう。別の薬はAMX 0035の前に発売承認される可能性がある。適用期間は米国では7年,EUでは10年であり,候補製品がそれぞれの規制機関が合意した小児科調査計画に適合すれば,それぞれ6カ月と2年延長することができる。5年目の終了時に、1つの製品が孤児指定の基準を満たしていないことが証明された場合、またはその製品の利益が十分に高い場合、市場排他性がもはや合理的でない場合、EUの排他的期間は6年に短縮されることができる。EUでは、孤児販売排他的な10年間、EU加盟国の主管当局、欧州医薬品局、または欧州委員会は、許可された孤児製品に類似した医薬製品の申請またはマーケティング許可の付与を許可しない。類似医薬製品“の定義は、承認された孤児医薬製品に含まれる1つ以上の類似活性物質を含む医薬製品であり、同じ治療適応のためのものである。欧州委員会は施行されれば、場合によっては孤児マーケティング排他性の10年間の期間を短縮することができる立法を提案した。FDAまたはEMAが、指定された要求に重大な欠陥があると判断した場合、または製造業者が稀な疾患または疾患を有する患者の需要を満たすのに十分な数の薬剤を保証できない場合、孤児薬の排他性を失う可能性がある。さらに、1つの薬剤が孤児の独占独占権を付与され、承認された後であっても、FDAまたはEMAが、より安全で、より効果的であることが証明され、または患者ケアに重大な貢献をしていることが証明されているので、FDAまたはEMAが、その後、7年(またはEUでは10年)の排他的期間が満了する前に、同じ疾患の治療を承認することができると結論した。また、孤児指定製品が発売承認され、その適応範囲が指定された適応よりも大きいか、または異なる場合、その製品は孤児排他性を得る権利がない可能性がある。FDAはWSの治療のためにAMX 0035を承認したにもかかわらず、AMX 0035の修正または異なる適応の承認を得た場合、現在の孤児指定は排他性を提供しない可能性がある。
孤児薬物指定は、規制審査または承認過程においていかなる利点も伝達されず、規制審査または承認過程の持続時間を短縮することもない。また、どの候補製品の規制承認も撤回される可能性があり、他の候補製品は私たちの前に承認され、孤児薬の独占経営権を得る可能性があり、これは私たちの市場進出を阻止するかもしれない。
我々がAMX 0035の稀な薬排他性を獲得しても、この排他性は、異なる薬物が孤児薬排出期間が満了する前に同じ状況に対して承認されることができるので、競争から効果的に保護することができない可能性がある。例えば、AMX 0035が孤児薬物排他性を得る権利があっても、この排他性は、TursoがAMX 0035とは異なる医薬製品であると判断された場合、FDA、EMA、または他の規制機関がALSの単一療法としてTursoを承認することを阻止しない可能性がある。また,規制部門は,この単一療法が臨床的にわれわれの固定用量製品よりも優れており,孤児薬物排他的特許を付与されても承認される可能性がある。米国の立法者は最近、競争を促進するために“孤児薬物法案”の規制や立法改正が必要になる可能性も提案されている。これには立法の導入が含まれており、法律になれば、私たちの排他性を維持するための競争に直面している場合、AMX 0035は経済的に不可能であることをFDAに証明することが要求されるだろう。
AMX 0035を孤児薬として指定し,他の適応の治療に用いることが求められるかもしれない。これらの適応のターゲット患者群の発病率と流行率は、私たちの推定および第三者データに基づく。もしこのようなターゲット集団の市場機会が私たちが推定したより大きいなら、私たちは孤児薬の指定を得ることができないかもしれない。また、孤児薬物指定が与えられた場合、私たちは市場排他性を含む孤児薬物指定に関連するいかなる利点も維持できないかもしれない。
著者らは定期的に各種の第三者源と内部から発生した分析に基づいて、ターゲット患者群の発病率と流行率を推定した。私たちの推定は正確ではないかもしれないし、不正確なデータに基づいているかもしれない。上述したように、孤児医薬品法によれば、医薬がまれな疾患または疾患の治療を意図している場合、FDAはそれを孤児薬として指定することができ、米国では、まれな疾患または疾患は、患者数が20万人未満、または患者数として定義されるのが一般的である
84
米国では20万人を超えるが、合理的な期待はなく、薬物開発のコストは米国での販売から回収される。もし私たちの発症率や流行推定の将来の適応があれば、孤児薬の指定を求めることができるかもしれないし、孤児薬の指定を得ることができないかもしれない。
FDAがAMX 0035の他の適応の孤児薬物指定を承認しても,孤児指定適応よりもFDAの範囲の広い適応のマーケティング承認を求めると,米国での独占営業権が制限される可能性がある。さらに、孤児の薬物状態指定を最初に取得した任意の候補製品は、FDAが後に指定要求に重大な欠陥があると判断した場合、または製造業者が稀な疾患または疾患を有する患者の需要を満たすのに十分な数の製品を保証できない場合、そのような指定を失う可能性がある。さらに、他の人は、私たちが開発している製品と同じ疾患または疾患を治療する製品のために孤児薬物の地位を得ることができ、それにより、かなり長い間、そのような疾患または疾患を治療する市場で競争する能力を制限することができる。したがって、私たちの業務と展望は影響を受けるかもしれない。
私たちが開発する可能性のある候補製品の優先審査指定を求めることができるかもしれませんが、このような指定を受けない可能性があり、優先審査指定は、より速い開発や規制審査や承認過程につながらない可能性があります。
FDAが候補製品が重篤な疾患を治療する方法を提供すると判断し、承認された場合、製品は安全性または有効性の面で有意な改善を提供するであろう場合、FDAは候補製品を優先的に検討するように指定する可能性がある。優先審査指定は、FDA審査申請の目標が6ヶ月であり、標準的な10ヶ月の審査期間ではないことを意味する。例えば、筋萎縮性側索硬化症を治療するAMX 0035の優先審査を受け、将来的に任意の将来の候補製品の優先審査指定を要求する可能性があるが、AMX 0035または私たちが開発する可能性のある任意の他の候補製品の将来の適応がこの指定された基準に適合すると仮定することはできない。FDAは候補製品優先審査地位を付与するか否かについて広範な裁量権を有しているため,特定の候補製品がこのような指定や地位を獲得する資格があると考えても,FDAはその資格を付与しないことを決定する可能性がある。さらに、優先審査指定は、必ずしも開発または規制審査または承認過程が速いことを意味するわけではなく、必ずしもFDA標準審査および承認と比較して、承認において何らかの利点があることを意味するとは限らない。例えば,FDAは最初にALSを治療するAMX 0035のPDUFA日付を2022年6月29日に設定し,われわれNDAの審査スケジュールを2022年9月に延長した。FDAの優先審査を受けることは、6ヶ月の審査期間内に承認されることが保証されない、あるいは全くできない。
私たちは私たちが開発した候補製品のためにFDAの迅速なチャネル指定を求めるかもしれないが、私たちは成功しないかもしれない。もし私たちが成功すれば、この指定は実際にはより速い開発や規制審査や承認過程をもたらすことができないかもしれない。
私たちは私たちが開発した未来の候補製品のために高速チャネル認証を求めるかもしれない。1つの製品が重篤または生命に危険な疾患を治療するために使用され、臨床前または臨床データが、このような疾患が満たされていない医療要件を満たす可能性があることを示す場合、製品スポンサーは、迅速チャネル認証を申請することができる。FDAは幅広い裁量権を有しており,この称号が付与されているかどうか,したがって,ある特定の候補製品がこの称号を得る資格があると考えても,FDAがそれを付与することを決定することを保証することはできない。私たちが高速チャネル認証を受けたとしても、私たちは従来のFDAプログラムよりも速い開発過程、審査、または承認を経験しないかもしれない。FDAが我々の臨床開発活動からのデータが高速チャネル指定をサポートしなくなったと考えた場合,その指定を取り消す可能性がある。
私たちが開発した候補製品にFDA指定の突破療法を求めるかもしれませんが、成功しないかもしれません。私たちが成功すれば、指定はより速い開発や規制審査や承認過程をもたらすことができず、私たちの製品候補が上場承認される可能性も増加しないかもしれない。
私たちは私たちが開発したすべての候補製品のために突破療法の称号を求めることができる。画期的な治療法は、1つまたは複数の他の薬剤と単独でまたは1つまたは複数の他の薬剤と組み合わせて重篤または生命を脅かす疾患または状態を治療することを意図した薬剤として定義され、初歩的な臨床証拠は、1つまたは複数の臨床的に重要な終点において、現在承認されている療法よりも実質的に改善された効果、例えば臨床開発早期に観察された実質的な治療効果を示す可能性があることを示している。画期的な治療法として指定された薬物に対して,FDAと試験スポンサーとの相互作用やコミュニケーションは,臨床開発の最も有効な経路を決定するのに役立つとともに,無効対照案中の患者数を最小限にすることができる。FDAで画期的な治療法に指定されている薬物も加速承認と優先審査を受ける資格がある。
画期的療法に指定されたのはFDAの裁量権である。したがって,我々が開発した候補製品が画期的療法として指定された基準に適合していると考えても,FDAは同意せず,このような指定をしないことにする可能性がある。いずれにしても画期的な治療の称号を得た製品は
85
FDAの通常の手順に従って承認を考慮している薬剤と比較して、候補薬剤は、より速い開発プロセス、審査または承認をもたらすことができず、FDAの最終承認を保証することができない可能性がある。また,我々が開発したどの候補製品も画期的な療法となる資格があっても,FDAは後でその薬剤が資格条件を満たしていないことを決定し,その指定を取り消す可能性がある。
私たちまたは私たちの任意の未来のパートナーに対する製品責任訴訟は、私たちの資源と注意を移し、私たちが重大な責任を負い、AMX 0035または任意の他の現在または未来の候補製品の商業化を制限する可能性があります。
私たちは潜在的な製品責任と専門賠償リスクに直面しており、これらのリスクは医薬製品の研究、開発、製造、マーケティングと使用に固有のものである。私たちと任意の協力者は臨床試験でAMX 0035を使用し、AMX 0035のアメリカ、カナダ、その他の司法管轄区域での販売(承認されれば)は、将来私たちを責任クレームに直面させるかもしれない。私たちの臨床試験に参加した参加者、患者、医療提供者、製薬会社、私たちの協力者、または私たちの将来承認された任意の製品を使用、管理、または販売する他の人は、私たちまたは私たちのパートナーに製品責任クレームを出すかもしれません。もし私たちがこのようないかなるクレームに対しても自分を弁護することに成功できない場合、私たちは重大な責任を招くか、またはAMX 0035または任意の他の現在または未来の候補製品の商業化を制限することを要求されるかもしれない。事件がどうであっても最終的な結果がどうであろうと、賠償責任は次のようになるかもしれない
臨床試験過程は潜在的な副作用を識別と評価することを目的としているが、臨床開発は常に新薬の安全性と有効性を完全に表現するわけではなく、しかも監督管理部門の許可後であっても、薬物は常に予見できない副作用を示す可能性がある。もしAMX 0035が臨床試験期間中あるいは承認後に副作用を引き起こす場合、私たちは大きな責任に直面する可能性がある。医師および患者は、既知の潜在的副作用を決定するいかなる警告も遵守しない可能性があり、AMX 0035または将来の任意の候補製品を使用すべきでない患者。もし私たちが現在または未来の任意の候補製品が商業販売を許可されれば、私たちは消費者の私たちに対する見方と私たちの製品の安全と品質に強く依存するだろう。もし私たちが患者が私たちの製品や他の会社が販売している任意の類似製品を使用したり、誤用したりすることで、病気や他の悪影響に関連した負の宣伝を受けたら、私たちは悪影響を受けるかもしれない。
臨床試験責任を含む製品責任保険の総金額は500万ドルに達しているにもかかわらず、この保険は私たちが生じる可能性のある潜在的な責任を完全にカバーできないかもしれない。いかなる製品責任訴訟や他の訴訟の費用も、私たちに有利な問題を解決しても、巨大である可能性がある。私たちがアメリカ、カナダ、他の司法管轄区域でAMX 0035を商業化する時、承認された場合、あるいは規制部門の許可を得た他の現在または未来の候補製品があれば、私たちの保険カバー範囲を増加させる必要があるだろう。しかも、保険カバー範囲はますます高くなっている。許容可能なコストで十分な保険範囲を維持することができない場合、または潜在的な製品責任クレームに対して他の方法で保護を提供することができない場合、AMX 0035または任意の他の現在または将来の候補製品の開発および商業生産および販売を阻止または阻止することができ、これは、私たちの業務、財務状況、運営結果、および将来性を損なう可能性がある。
私たちまたは任意の未来のパートナーがAMX 0035または任意の他の現在または未来の候補製品の規制承認を得て維持しても、私たちの製品に対する承認条項と持続的な規制は、私たちの製品の生産とマーケティングの方法を制限するかもしれません。これは、私たちの収入を創出する能力を弱めるかもしれません。
86
規制の承認を受けると、承認された製品とそのメーカーや営業業者は、持続的な審査と広範な規制を受けることになる。したがって、私たちおよび任意の未来のパートナーは、AMX 0035または私たちまたは彼らが規制部門の承認を得た任意の他の現在または将来の候補製品に関する広告および販売促進要件を遵守しなければならない。処方薬に関する販売促進情報は様々な法律や法規によって制限されており,製品が承認されたラベル中の情報と一致しなければならない。したがって、私たちと未来のパートナーは、私たちが開発した未承認の適応や用途のためのいかなる製品も普及させることができないだろう。
さらに、承認された製品の製造業者およびその工場は、品質管理および製造手順がcGMPに適合することを保証することを含むFDA、カナダ衛生部、およびEMAの広範な要求を遵守しなければならず、その中には、品質管理および品質保証に関する要求、および対応する記録および文書保守および報告要件が含まれている。私たち、私たちの契約メーカー、任意の未来の協力者及びその契約メーカーはcGMPに適合しない可能性があり、FDA、カナダ衛生部、EMAの定期的な抜き打ち検査を受けて、cGMPに適合することを監督し、確保する。我々は規制適合性の検査と検証に努力しているが、FDA、カナダ衛生部、EMAまたは他の機関の監督管理検査では、私たちの1つまたは複数の第三者製造サプライヤーがcGMP規定に適合していないことが発見される可能性があり、これは第三者サプライヤーの閉鎖または薬品ロットまたはプロセスの無効を招く可能性がある。場合によっては、製品のリコールが必要または要求される可能性があり、これは私たちの薬品を供給してマーケティングする能力に大きな影響を与えるだろう。
したがって、私たちまたは任意の未来の協力者がAMX 0035または1つまたは複数の将来の候補製品の規制承認を得た任意の司法管轄区域では、私たち、任意の未来の協力者、および私たちと彼らの契約製造業者は、製造、生産、製品監視、および品質管理を含むすべてのコンプライアンス分野に時間、お金、エネルギーを投入し続けるだろう。
もし私たちと任意の未来のパートナーが承認後の規制要件を遵守できない場合、私たちと任意の未来のパートナーは、規制機関によるAMX 0035または任意の他の現在または未来の製品に対する規制承認を受ける可能性があり、私たちまたは任意の未来のパートナーが任意の未来の製品をマーケティングする能力が制限される可能性があり、これは、私たちが利益を達成または維持する能力に悪影響を及ぼす可能性がある。また、承認後の法規を遵守するコストは、私たちの経営業績や財務状況に悪影響を及ぼす可能性があります。
私たちが将来予想する任意の国際業務を管理する法律法規は、アメリカ以外で特定の製品を開発、製造、販売することを阻止し、コストの高いコンプライアンス計画の開発と実施を要求するかもしれません。
私たちはアメリカとカナダに業務を持っていて、EUを含む他の管轄区および他の潜在的な管轄区域で業務を展開したいと思っています。私たちは現在、あるいは各管轄区で業務を展開することを計画している多くの法律と法規を遵守するために追加の資源を投入しなければなりません。“反海外腐敗法”(FCPA)は、個人または企業が業務を獲得または保持するのを助けるために、任意の米国人または企業が、任意の外国人官僚、政党または候補者に直接的または間接的に支払い、提供、許可支払い、または任意の価値のあるものを提供することを禁止する。“海外腐敗防止法”はまた、米国に上場する証券会社にある会計条項を遵守することを要求しており、これらの条項は、企業(国際子会社を含む)のすべての取引の帳簿と記録を正確かつ公平に反映し、国際業務のために十分な内部会計制御システムを設計し、維持することを要求している。
“反海外腐敗法”を遵守することは高価で困難であり、特に腐敗は公認問題である国である。また、海外腐敗防止法は製薬業に特別な挑戦をもたらしており、多くの国では病院が政府によって運営されているため、医師や他の病院従業員は外国人官僚とされている。臨床試験やその他の仕事に関連して病院に支払われた何らかの金は、政府関係者に支払われた不正金と考えられ、“海外腐敗防止法”の法執行行動につながった。
輸出規制および貿易制裁法を含む様々な法律、法規、および行政命令は、米国国外での使用および伝播を制限したり、国家安全目的のための機密情報を特定の非米国国民と共有したり、特定の製品およびこれらの製品に関連する技術データを共有したりする。もし私たちがアメリカ以外での業務を拡大すれば、これらの法律を遵守するためにもっと多くの資源を投入する必要があります。これらの法律は、私たちがアメリカ以外で特定の製品や候補製品を開発、製造、販売することを阻止するかもしれません。これは、私たちの成長潜在力を制限し、私たちの開発コストを増加させるかもしれません。
87
国際ビジネス慣行に関する法律を遵守しなければ、重大な民事·刑事罰を受け、政府契約の資格を一時停止または廃止する可能性がある。米国証券取引委員会は、発行者が海外腐敗防止法の会計規定に違反したため、発行者の米国取引所での証券取引を一時停止または禁止する可能性もある。
もし私たちが環境、健康、安全の法律法規を守らなければ、罰金や処罰を受けたり、私たちの業務を損なう可能性のあるコストが発生するかもしれません。
私たちは多くの環境、健康と安全法律と法規の制約を受けて、それらの研究室の手続きと危険材料と廃棄物の処理、使用、貯蔵、処理と処理を管理する法律と法規を含む。時々、将来、私たちの業務は、化学品および生物学的材料を含む危険かつ可燃性材料の使用に関連する可能性があり、危険な廃棄物製品を生成する可能性もある。これらの材料や廃品を処分する契約を第三者と締結しても、これらの材料による汚染や傷害のリスクを完全に解消することはできません。私たちの危険な材料を使用または処分して汚染または損傷をもたらす場合、私たちはそれによるいかなる損害に責任を負う可能性があり、いかなる責任も私たちの資源範囲を超える可能性がある。民事や刑事罰金やこのような法律や法規を遵守しない罰に関する巨額の費用を招く可能性もある。
私たちは従業員の負傷によって生じる可能性のある費用と費用を支払うために労働者補償保険を維持していますが、この保険は潜在的な責任に対応するのに十分ではないかもしれません。しかし、私たちは私たちが提起した環境責任や有毒侵害請求に保険を提供することはできない。
また、現在または未来の環境、健康、安全法律法規を遵守するためには、巨額のコストが生じる可能性がある。環境法律法規は私たちの研究、開発、あるいは生産努力を損なうかもしれない。しかも、このような法律法規を遵守しないことは巨額の罰金、処罰、または他の制裁につながる可能性がある。
私たちの第三者への依存に関するリスク
私たちは協力関係の構築を求めるかもしれませんが、ビジネスの合理的な条件で協力関係を構築し、維持できなければ、私たちの開発と商業化計画を変えなければならないかもしれません。
AMX 0035および任意の現在または将来の候補製品および開発計画または活動の推進、ならびに米国およびカナダALBRIOZAにおけるRELYVRIOの商業化、および他の司法管轄地域および任意の将来の候補製品におけるAMX 0035の潜在的な商業化は、費用を支払うために多くの追加の現金を必要とする。AMX 0035または他の現在または将来の候補製品のいくつかの適応について、私たちは、他の製薬およびバイオテクノロジー会社と開発および潜在的な商業化の面で協力することを決定するかもしれない。可能な協力者には、大中型製薬会社、地域的、全国的な製薬会社、およびバイオテクノロジー会社が含まれるかもしれない。また、外国の規制機関による候補製品の規制承認を得ることができれば、国際バイオテクノロジーや製薬会社と協力して、これらの候補製品を商業化することができるかもしれない。
私たちは適切な協力者を探すことで激しい競争に直面している。私たちが協力について最終的な合意に到達するかどうかは、他に加えて、パートナーの資源と専門長の評価、提案された協力の条項と条件、提案されたパートナーのいくつかの要因の評価に依存する。これらの要因は、我々の候補製品と競合候補製品との潜在的な差異、臨床試験の設計または結果、カナダ保健省、FDA、EMAまたは他の同様の外国規制機関の承認を得る可能性、およびこのような承認の規制経路、候補製品の潜在的市場、製品の製造および患者への製品の提供のコストおよび複雑さ、および競争製品の潜在性を含む可能性がある。協力者はまた、代替候補製品または技術を考慮して、連携に利用可能な同様の指示を得ることと、我々の候補製品に対して、そのような連携が私たちとの連携よりも魅力的であるかどうかを考慮することができる。もし私たちが私たちの支出を増やし、私たち自身の開発や商業化活動に資金を提供することを選択すれば、私たちは追加の資本を得る必要があるかもしれないし、これらの資本は受け入れ可能な条件で私たちに提供できないかもしれないし、全く得られないかもしれない。もし私たちが十分な資金がなければ、私たちはAMX 0035または他の現在または未来の候補製品をさらに開発することができず、それらを市場に出して製品収入を生成することもできないかもしれない。
協力の交渉と記録は複雑で時間がかかる。また,最近では大手製薬会社間の業務合併数が大きく,将来の潜在的パートナー数の減少を招いている。私たちが将来達成する任意の協力合意は、私たちが潜在的に協力したり、他の方法で特定の候補製品を開発する能力を制限するかもしれない。私たちはタイムリーに基づいて受け入れ可能な条項で協力することができないかもしれないし、交渉することさえできないかもしれない。それができなければ削減しなければならないかもしれません
88
我々は、協力候補製品の開発を求め、その開発計画または私たちの1つまたは複数の他の開発計画または活動を減少または延期し、その潜在的な商業化を延期したり、任意の販売またはマーケティング活動の範囲を縮小したり、私たちの支出を増加させ、自費で開発または商業化活動を行う。
我々は、今後、AMX 0035または任意の他の現在または将来の候補製品の開発および商業化について第三者と協力する可能性があり、AMX 0035および他の現在または将来の候補製品についての見通しは、これらの協力の成功に大きく依存するであろう。
我々は、AMX 0035および任意の他の現在または将来の候補製品を協力によって開発し、商業化することができる。例えば、私たちは、サニーブルック研究所とのパートナー関係のように、AMX 0035の商業化を促進するために、または神経変性疾患を治療する新薬候補薬を決定するために、1つまたは複数の第三者との様々な流通、協力、および他のマーケティングスケジュールを利用することができる。私たちの流通、開発、販売、マーケティング、許可、またはより広範な協力手配において可能なパートナーは、大中型製薬会社、地域的、全国的な製薬会社、およびバイオテクノロジー会社を含む。もし私たちがそのような協力に参加すれば、私たちの協力者がAMX 0035または任意の他の現在または将来の候補製品の開発または商業化に特化したリソースの数および時間を限られた制御することができるかもしれない。私たちがこれらの計画から収入を創出する能力は、任意の未来の協力者がこれらの手配の中で彼らに割り当てられた機能を成功的に履行する能力に依存するだろう。さらに、任意の将来の協力者は、合意された条項の満了前または後に研究または開発プロジェクトを放棄し、資金義務を含む適用された合意を終了する権利があるかもしれない。
AMX 0035および任意の他の現在または将来の候補製品に関する協働は、以下のような多くのリスクをもたらす
89
協調プロトコルは、最も効率的な方法で、または候補製品の開発または商業化を引き起こさない可能性がある。もし私たちの未来の任意のパートナーが業務統合に参加すれば、それは私たちが許可した任意の候補製品の開発または商業化を延期、減少、または終了することを決定するかもしれない。
私たちは第三者に依存して臨床試験を支援してくれます。それらのパフォーマンスが満足できない場合、私たちは規制部門の承認を得ることができないか、またはAMX 0035または任意の他の現在または未来の候補製品を正常に商業化することができないか、またはそのような承認または商業化が延期される可能性があり、私たちの業務は実質的な損害を受ける可能性がある。
我々はCRO,臨床データ管理組織,医療機関,臨床研究者など第三者による臨床試験を継続する予定であり,これらの第三者による我々が開発した任意の将来の候補製品の臨床試験を行う予定である。場合によっては、このような第三者のいずれかが私たちとの雇用関係を終わらせることができる。私たちは代替計画に到達できないかもしれないし、商業的に合理的な条項でそうすることができないかもしれない。しかも、新しいCROが仕事を始める時、自然な過渡期がある。そのため、遅延が生じる可能性があり、これは予想される臨床開発スケジュールを満たす能力に負の影響を与え、私たちの業務、財務状況、将来性を損なう可能性がある。臨床試験は多数の臨床サイト、サプライヤーとその他の第三者に関連し、著者らはこれらのサプライヤーに依存して適切な研究実施、統計分析とランダム化を確保する。彼らがこれらの活動で犯したいかなるミスや偏差も、監督機関の臨床試験結果に対する有用性と解釈可能性に影響を与える可能性がある。例えば、2022年3月30日の諮問委員会会議でFDAはいくつかの懸念に注目しており、FDAから見ると、これらの懸念はCentaur実験結果の解釈可能性に影響を与えている。臨床試験に偏りが生じることがあり,その中で案や標準操作手順が完璧に実行されず,是正措置がとられている。これらの事件は低リスクであると考えられるかもしれないが,リスクに対する私たちの見方や適切な是正行動は規制機関の見方とは異なる可能性がある。研究過程中に方案や標準操作手順を逸脱することは負の規制意見と結果を招く可能性がある。
また,これらの第三者の臨床開発活動への依存は,これらの活動の制御を制限しているにもかかわらず,個々の試験が適用案,法律,法規要求,科学的基準に従って行われることを確実にする責任がある。さらに、FDA、EMA、およびEU加盟国の主管当局は、データおよび報告の結果が信頼性かつ正確であることを保証し、試験参加者の権利、完全性、および機密性を保護するために、臨床試験結果を行い、記録し、報告する良好な臨床実践またはGCPを遵守するように要求されている。FDAは,試験スポンサー,主要研究者,臨床試験地点,IRBsを定期的に検査することでこれらのGCPを実行している。もし私たちまたは私たちの第三者請負者が適用されたGCPに従わなかった場合、私たちの臨床試験で生成された臨床データは信頼できないと考えられるかもしれませんが、FDA、EMA、または他の規制機関は、追加の適応、または他の現在または未来の候補製品を含む追加の臨床試験を承認する前に、規制承認プロセスを延期することを要求するかもしれません。検査後、FDA、EMA、あるいは他の規制機関が私たちの任意の臨床試験がGCPに適合しているかどうかを確認することはできません。例えば,われわれの臨床試験サイトや研究者は過去と将来に案のずれが生じる可能性があり,われわれの臨床試験結果の全体的な解釈に影響を与える可能性がある。我々はまた,ある臨床試験を一定の時間範囲で登録し,完成した臨床試験の結果をClinicalTrials.govのような政府後援のデータベースに発表することも求められている。そうしないと罰金、否定的な宣伝、そして民事と刑事制裁につながるかもしれない。
また,我々を代表して臨床試験を行っている第三者は我々の従業員ではなく,これらの請負業者との合意によって得られる救済措置を除いて,我々が行っている開発活動に十分な時間,スキル,資源を投入しているかどうかを制御することはできない。これらの請負業者はまた、私たちの競争相手を含む他の商業実体と関係がある可能性があり、彼らはまた、これらのエンティティのための臨床試験または他の薬物開発活動を行っている可能性があり、これは、彼らが適切な時間を私たちの臨床活動に投入することを阻害するかもしれない。これらの第三者が臨床研究者を含む場合、法規要件または私たちが宣言した規程に従って契約義務の履行に成功し、予期される期間内に私たちの臨床試験を完了または実施することができなかった場合、私たちは、AMX 0035または任意の他の現在または将来の候補製品の規制承認に必要な臨床データを得ることができないか、または遅延する可能性がある。このような状況が発生した場合、私たちは、AMX 0035または任意の他の現在または将来の候補製品を商業化することに成功し、私たちの努力を遅らせることができないか、または延期することができるだろう。この場合、私たちの財務業績とAMX 0035、または私たちが開発を求めている任意の他の現在または将来の候補製品のビジネス見通しは損なわれる可能性があり、私たちのコストは増加する可能性があり、私たちが収入を生成する能力は遅延、損傷、または担保償還権を失う可能性がある。
また、隔離、現地避難、および同様の政府命令、またはこのような命令、閉鎖または他の業務運営行為の制限が発生する可能性があるとの見方は、感染症に関連しており、例えば政府が新冠肺炎の大流行中に講じた措置は、私たちCROの人員に影響を与える可能性があり、これは私たちの臨床スケジュールを混乱させる可能性があり、これは私たちの業務、将来性、財務状況、および運営結果に実質的な悪影響を及ぼす可能性がある。
90
私たちはまた他の第三者に私たちの臨床試験のための薬品の貯蔵と配布に依存している。我々のディーラーのいかなる業績ミスも、AMX 0035または任意の他の現在または将来の候補製品の臨床開発または規制承認、またはそれによって生成された製品の商業化を延期し、追加の損失をもたらし、潜在的な製品収入を奪う可能性がある。
CGMPに準拠するAMX 0035を生産するために第三者を使用することは、時間的に、または許容可能なコストで十分な数のcGMPに適合するAMX 0035または必要な数のそのような材料を得ることができないリスクを増加させる可能性がある。
私たちは臨床または商業バッチAMX 0035を生産または運営する製造施設を持っていないが、現在私たちはそのような資源と能力が不足している。したがって、我々は現在、AMX 0035中の活性医薬成分または原料薬を第三者に生産および供給し、適用される法律、法規および標準に従ってAMX 0035の混合および包装を行うことに依存している。我々の現在の戦略は、AMX 0035のすべての製造と、任意の現在または未来の候補製品を第三者にアウトソーシングすることである。
著者らは現在第三者メーカーを招いてAMX 0035の原料薬とAMX 0035の最終薬物製品調合を提供し、著者らの臨床試験及び参入と商業供給の拡大に用い、著者らは単独の第三者を招いて完成した臨床材料を混合と包装した。私たちは異なるサプライヤーの薬物物質の比較可能性と、異なるサプライヤーの安定性データを証明することができなければならない。私たちは現在、私たちのAPIを供給するためにメーカーに依存しており、他の製造業者は別の種類を供給する。いくつかの潜在的な代替メーカーがAMX 0035の各原料薬を製造することができると信じているが、そのような任意の代替製品を決定および同定する際に、追加のコストおよび遅延が生じる可能性がある。さらに、地政学的事件または世界的衛生危機が、AMX 0035および任意の他の現在または未来の製品および候補製品のために十分な供給を得る能力にどの程度影響を与える可能性があるかは、このような事件による経済的挑戦が世界経済およびサプライチェーンなど多くの要素に影響を与え続けるかどうかに依存する。私たちは私たちが満足できる条件で必要な供給スケジュールをタイムリーに得ることができるか、あるいは根本的に保証できないという保証はない。必要に応じてこれらの配置を確保できない場合、AMX 0035または任意の他の現在または将来の候補製品の開発を完了するか、または(承認された場合)それを商業化する能力に大きな悪影響を及ぼす可能性がある。私たちは第三者製造業者と商業供給合意に到達できないかもしれないし、受け入れ可能な条項でそうすることができないかもしれない。AMX 0035のビジネス規模や配合を拡大することは困難かもしれませんし、製造コストが目を引くほど高いかもしれません。
たとえ第三者製造業者と手配を確立し、維持することができても、第三者製造業者に依存することは、追加的なリスクをもたらす
もし私たちが私たちの重要な製造関係を維持しなければ、あるいは私たちのどの契約メーカーもその義務を履行できなければ、私たちは代替メーカーを見つけたり、私たち自身の製造能力を発展させることができないかもしれません。これは、アメリカとカナダでAMX 0035を商業化する能力に悪影響を与え、規制機関が私たちの製品を承認する能力を延期または弱めるかもしれません。もし私たちが代替メーカーを見つけたら、私たちは私たちに有利な条項と条件で彼らと合意できないかもしれないし、新しい施設がFDA、カナダ保健省、EMA、および他の外国規制機関の資格と登録を得る前に、大きな遅延があるかもしれない。
91
メーカーのいかなる変化も生産プロセスとプロセスの変化に関連する可能性があり、これは私たちが臨床試験で使用した以前の臨床供給と任意の新しいメーカーの供給との間の過渡的な研究を要求するかもしれない。さらに、任意の新しい製造プロセスが、以前FDA、カナダ保健省、EMA、または他の規制機関に提出された仕様に基づいて、私たちの候補製品を生産するかどうかを検証する必要があります。臨床用品の比較可能性の証明には成功しない可能性があり,追加の臨床試験が必要かもしれない。
場合によっては、AMX 0035または任意の他の現在または将来の製品または候補製品を製造するために必要な技術スキルは、元の契約製造業者固有または独自である可能性があり、困難に遭遇する可能性があり、またはそのようなスキルを予備または代替サプライヤーに譲渡することを禁止する契約制限が存在する可能性があり、またはそのようなスキルを全く譲渡できない可能性がある。さらに、契約製造業者は、AMX 0035または契約製造業者が独立して所有する任意の他の現在または将来の製品候補製品の製造に関連する技術を所有または取得することができる。これは、別の契約製造業者にAMX 0035または任意の他の現在または将来の候補製品を生産させるために、契約製造業者への依存を増加させるか、または契約製造業者からライセンスを取得することを要求するであろう。AMX 0035が私たちの任意の初期または潜在的な追加適応または任意の未来の候補製品のために使用された場合、任意の規制機関の承認を得た場合、私たちは、第三者との契約製造業者の配置を使用して、これらの製品の商業生産を行う予定である。このプロセスは困難で時間がかかり、cGMPで運営されるAMX 0035または任意の他の現在または将来の候補製品を生産する能力のある契約製造業者の数が限られているので、製造施設に入る競争に直面する可能性がある。したがって、私たちは第三者製造業者と満足できる条項について合意できない可能性があり、これは私たちの商業化を延期するかもしれない。
私たちまたは私たちの第三者製造業者が適用された法規を遵守できなかったことは、臨床棚上げ、罰金、禁止、民事処罰、遅延、一時停止または承認撤回、AMX 0035または任意の他の現在または未来の候補製品、運営制限、および刑事起訴を含む制裁を実施する可能性があり、いずれもAMX 0035または任意の他の現在または未来の候補製品の供給に深刻な影響を与える可能性がある。我々の契約製造業者は、AMX 0035または任意の他の現在または将来の候補製品を生産するための施設は、FDA、カナダ保健省、EMA、およびいくつかの他の外国規制機関によって評価されなければならない。私たちは私たちの契約製造パートナーの製造過程をコントロールせず、cGMPを守るために彼らに完全に依存している。もし私たちの契約メーカーが私たちの規格やFDA、カナダ保健省、EMA、または他の機関の厳格な規制要件に合った材料を生産することに成功しなければ、私たちはこれらの工場で生産された製品が規制部門の承認を得ることを確保および/または維持できないかもしれません。しかも、私たちの契約製造業者が十分な品質管理、品質保証、合格者の能力を維持することを制御することはできません。FDA、カナダ保健省、EMA、または他の同様の外国規制機関が、AMX 0035または任意の他の現在または将来の候補製品を製造するためにこれらの施設を不足しているか、または承認していないことを発見した場合、または将来的にそのような承認を撤回した場合、代替製造施設を探す必要があるかもしれません。これは、規制機関の承認またはマーケティングAMX 0035または任意の他の現在または未来の候補製品を開発、獲得する能力に深刻な影響を与えます(承認された場合)。また、契約メーカーの交換を要求された場合、新たな契約製造業者が品質基準およびすべての適用規制に適合する施設やプログラムを持っているかどうかを確認することを要求され、さらなるコストや遅延を招く可能性があります。契約製造業者は、製造または品質管理の問題に直面し、薬品生産や輸送遅延を招く可能性があり、あるいは請負者が適用されるcGMP要求を遵守できない可能性がある。CGMP要件または他のFDA、カナダ保健省、EMA、および同様の外国法規要件を遵守しないいかなる行為も、私たちの臨床研究活動およびAMX 0035または任意の他の現在または未来の候補製品を開発し、私たちの製品をマーケティングする能力に悪影響を及ぼす可能性がある。
FDA、カナダ保健省、EMA、その他の外国規制機関はメーカーに製造施設の登録を要求している。FDA,カナダ衛生部,EMA,対応する外国規制機関もこれらの施設を検査し,cGMPに適合しているかどうかを確認する。契約製造業者は、製造または品質管理の問題に直面し、薬品生産や輸送遅延を招く可能性があり、あるいは請負者が適用されるcGMP要求を遵守できない可能性がある。CGMP要件または他のFDA、カナダ保健省、EMA、および同様の外国法規要件に準拠できなかったいかなる行為も、私たちの臨床研究活動およびAMX 0035または任意の将来の候補製品を開発し、承認された後に私たちの製品を販売する能力に悪影響を及ぼす可能性がある。
AMX 0035の任意の第三者製造業者または任意の他の現在または将来の候補製品が、そのような候補製品の生産規模を拡大することができず、および/またはその製造製品の歩留まりを向上させることができない場合、私たちは製品を製造するコストが増加する可能性があり、商業化が遅れる可能性がある。
臨床試験の需要を満たすのに十分な数を生産するために、AMX 0035の米国およびカナダにおける拡張参入および商業化、およびその後、AMX 0035の他の司法管轄区域での任意の商業化が承認されれば、
92
または私たちが開発する可能性のある他の任意の現在または将来の候補製品は、当社の第三者製造業者は、製品の品質を維持しながら生産を増加させ、その製造プロセスを最適化することを要求されるであろう。より大規模な生産への移行は難しいことが証明されるかもしれない。さらに、我々の第三者製造業者が、AMX 0035または任意の他の現在または将来の候補製品の製品生産量を向上させるために、その製造プロセスを最適化することができない場合、または製品の品質を維持し、cGMPを遵守しながら、そのような候補製品の数を増加させることができない場合、臨床試験、参入拡大、または市場需要を満たすことができない可能性があり、これは、私たちの利益を創出する能力を低下させ、私たちの業務および運営結果に大きな悪影響を及ぼす可能性がある。
我々は、AMX 0035または将来の候補製品を開発および商業化するために、第三者からの活性成分のライセンスを維持する必要があるかもしれないが、これは、私たちの開発コストを増加させ、このような候補製品を商業化する能力を遅らせる可能性がある。
任意のAMX 0035または任意の他の現在または将来の候補製品において原料薬を使用することを決定した場合、これらの製品は、1つまたは複数の第三者の特許である場合、これらの第三者からの活性成分のライセンスを維持する必要があるであろう。臨床試験を支援するための臨床前毒理学研究を行う前に、これらの活性成分にアクセスまたは継続する権利が得られなければ、代替活性成分にアクセスまたは開発することによって、これらの計画された代替候補製品を開発する必要があり、開発コストの増加およびこれらの候補製品の商業化遅延を招く可能性がある。商業的に合理的な条項で所望の活性成分への持続的なアクセス権を獲得したり、適切な代替活性成分を開発できなければ、これらの計画の候補製品を商業化することができない可能性がある。
私たちの知的財産権に関するリスクは
私たちのビジネス成功は私たちが知的財産権とノウハウを保護する能力にかかっている。
私たちのビジネス成功は、特許候補製品AMX 0035および任意の将来の特許製品候補を介して、米国および他の国/地域で特許、商標および商業秘密によって知的財産権保護を取得し、維持する能力に大きく依存する。もし私たちが私たちの知的財産権を十分に保護しなければ、競争相手は私たちが持つ可能性のあるいかなる競争優位性を侵食、否定、または占有する可能性があり、これは私たちの業務と持続的な収益性を損なう可能性がある。私たちの独自の地位を保護するために、私たちは特許出願を提出し、AMX 0035または私たちの業務に重要な任意の他の現在または将来の候補製品に関連する他の特許出願を米国または海外で提出することができ、私たちはまた特許または他の人が提出した特許出願を許可または購入することができる。特許出願過程は高価で時間がかかる。私たちは必要または望ましいすべての特許出願を合理的な費用またはタイムリーに提出して起訴することができないかもしれない。
もし私たちが獲得した特許保護範囲が十分でなければ、私たちは他の人たちが私たちと似ているか同じ技術や製品を開発して商業化することを防ぐことができないかもしれない。私たちが市場で競争に成功するために必要な特許保護の程度は、場合によっては入手できないかもしれないし、深刻に制限されている可能性があり、私たちの権利を十分に保護できないかもしれないし、どんな競争優位性を獲得したり維持したりすることを許可しているかもしれません。私たちは、私たちのいかなる特許も持っていることを保証することができない、または私たちの任意の係属中の特許出願が発行された特許に成熟していることは、私たちの独自療法を保護するのに十分な範囲、または他の方法でいかなる競争優位性を提供するのに十分な権利要件を含むだろう。他の当事者は、我々の方法に関連しているか、または我々の方法と競合する可能性のある技術を開発しており、特許出願が出願されているか、または提出されている可能性があり、同じ化合物、処方または方法を要求するか、または要求によって我々の特許地位を支配する可能性のある標的を要求することによって、我々の特許出願と重複または衝突する可能性のある特許請求の特許を取得している可能性がある。また、外国の法律はアメリカの法律のように私たちの権利を保護しないかもしれません。また、特許のライフサイクルは限られています。アメリカでは、特許の自然失効期間は一般的に申請後20年だ。様々な延期があるかもしれない;しかし、特許の有効期限とその提供される保護は限られている。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちの特許組み合わせは、他の会社がAMX 0035のような製品または任意の他の現在または将来の候補製品を商業化することを阻止するのに十分かつ継続的な特許保護を提供することができないかもしれない。代替組み合わせまたはTursoが単一医薬製品として開発され、承認を求める可能性のある適応のために承認され、我々の特許請求の範囲を超える場合、AMX 0035の適合性および商業的成功は実質的な損害を受ける可能性がある。
挑戦されていなくても、私たちが持っている特許と未解決の特許出願は、発表されても、私たちに意味のある保護を提供してくれないかもしれませんし、競争相手が私たちの特許声明を迂回して設計することを阻止することもできません
93
類似または代替療法を非侵害的に開発する。例えば、第三者は、私たちの候補製品と同様の利点を提供するが、私たちの特許保護または許可権の範囲内ではない競争療法を開発することができる。AMX 0035または任意の他の現在または将来の候補製品に関する特許および特許出願によって提供される特許保護が十分に広くないか出願されている場合、そのような競争を阻害するのに十分ではない場合、候補製品を商業化することに成功する能力は負の影響を受ける可能性があり、これは私たちの業務を損なうであろう。
私たちまたは任意の未来のパートナー、協力者、または許可された人は、開発および商業化活動中に行われた発明の特許可能な態様を識別できない可能性があり、そうでなければ遅すぎて、特許保護を得ることができない。したがって、私たちは私たちの特許地位を強化する予想された潜在的な機会を逃すかもしれない。
適切な優先権請求、発明権、特許請求の範囲、または特許期限調整要求のような形態的欠陥が、我々の特許または特許出願の準備または提出中に存在するか、または将来生じる可能性がある。もし私たちまたは私たちのパートナー、協力者、または許可者が、現在であっても将来であっても、そのような特許および他の知的財産権を確立、維持または保護することができなかった場合、そのような権利は減少またはキャンセルされる可能性がある。もし私たちのパートナー、協力者、または許可者が任意の特許権を起訴、維持または実行する上で私たちと完全に協力しないか、または同意しない場合、そのような特許権は損害を受ける可能性がある。私たちの特許または特許出願が形態、準備、起訴または実行において重大な欠陥がある場合、そのような特許は無効および/または強制的に実行できない可能性があり、そのような出願は有効で強制的に実行可能な特許を決して生成しない可能性がある。これらの結果のいずれも、第三者の競争を阻止する能力を弱める可能性があり、第三者競争は私たちの業務に悪影響を及ぼす可能性がある。
バイオテクノロジーと製薬会社の特許状況には不確実性がある。さらに、薬物化合物特許権の決定は、通常、現在の法律および知的財産権の背景、既存の法的前例、および個人の法律の解釈に依存する複雑な法律および事実の問題に関連する。そのため、中国の特許権の発行、範囲、有効性、実行可能性と商業価値はすべて不確定性がある。
係属中の特許出願は、特許がこのような出願から発行されるまで、そのような出願において要求される技術を実施する第三者に対して強制的に実行することはできない。他の特許性要件を満たすと仮定すると,現在,最初に特許出願を提出した者は通常特許を取得する権利がある。しかし、2013年3月16日までに、米国では、最初に発明者がこの特許を取得した。科学文献中の発見発表は往々にして実際の発見に遅れており、アメリカと他の司法管轄区の特許出願は出願後18ヶ月まで公表され、時には全く公表されないこともある。したがって、私たちは、私たちが私たちの特許または係属中の特許出願に要求された最初の発明をした人であるか、または私たちがこれらの発明のために特許保護を申請した最初の人であることを確認することができない。第三者が私たちの特許で要求された発明について以前の特許出願を提出した場合、または2013年3月15日またはそれ以前に提出された出願である場合、そのような第三者は、最初に我々が出願した特許請求の範囲がカバーする任意の主題の人であるかを決定するために、米国で干渉手順を開始することができる。第三者が2013年3月15日以降にこのような以前の出願を提出した場合、そのような第三者は、私たちの発明が彼らの発明から来たかどうかを決定するために、米国で派生プログラムを開始することができる。
さらに、特許の発行は、その発明性、範囲、有効性、または実行可能性に対して決定的ではないため、我々の特許は、米国および海外の裁判所または特許庁で挑戦される可能性がある。さらに、私たちは、私たちの特許および特許出願に関連するすべての潜在的に関連する先行技術を開示したと信じているが、私たちが各関連する司法管轄区域でこのようなすべての先行技術を発見または開示したことは保証されない。科学文献中の発見発表は往々にして実際の発見より遅れており、米国と他の司法管轄区の特許出願は通常出願後18ヶ月後に公表され、場合によっては全く公表されない場合もある。
したがって、私たちは、私たちが最初に私たちの特許または係属中の特許出願で主張された発明を提出したのか、最初にそのような発明のために特許保護されたのかを正確に知ることができない。そのような従来技術が存在する場合、それは、特許無効を宣言するために使用されてもよく、または係属中の特許出願からの特許の発行を阻止することができる。例えば、このような特許出願は、第三者が既存技術を米国特許商標局または世界各地の他の特許庁に提出する必要がある場合がある。代替または補足として、私たちは、許可後審査手続き、反対意見、派生手続き、一方的再審、当事者再審、補充審査、または地域裁判所、米国または様々な外国特許庁における妨害訴訟または挑戦に参加することができ、私たちが権利を有する挑戦的な国および地域特許または特許出願を含む、私たちの業務に依存する特許を含む。このような任意の挑戦において不利な裁決を下すことは、特許の損失または特許または特許出願の特許請求の範囲が全部または部分的に縮小され、無効または実行できない、または特許出願が拒否されること、または特許または特許出願の1つまたは複数の特許請求の範囲の喪失または縮小をもたらす可能性があり、いずれも私たちが阻止する能力を制限する可能性がある
94
他の会社は、同様の技術および製品を使用または商業化したり、我々の技術および製品の特許保護期間を制限したりしてはならない。
未解決および将来の特許出願は、私たちの業務の全部または一部を保護する特許の発行、または他社が競合製品を商業化することを効果的に阻止する特許を発行することにつながる可能性があります。競争相手はまた私たちの特許を迂回して設計することができる。米国や他の国の特許法や特許法解釈の変化は,我々の特許の価値を低下させたり,我々の特許保護の範囲を縮小したりする可能性がある。また、外国の法律は、米国の法律のように私たちの権利を保護しないかもしれない。例えば、欧州特許庁、中国、日本のような重要な商業市場をカバーする司法管轄区を含む異なる司法管轄区の特許法は、米国の法律よりも人体治療方法の特許性の制限が大きい。このような状況が発生すれば、私たちの創造能力に実質的な悪影響を及ぼす可能性がある。
特許出願中には多くのリスクおよび不確実性が存在し、私たちまたは私たちの任意の未来の開発パートナーが、特許を取得して保護することによって、AMX 0035または任意の他の現在または未来の候補製品を保護することに成功することを保証することはできない。これらのリスクと不確実性には
私たちがすでに取得または許可している可能性のある発行された特許は、私たちに何の意味のある保護も提供してくれないかもしれないし、競争相手が私たちと競争することを阻止したり、他の方法で私たちにどんな競争優位性を提供してくれたりしないかもしれない。私たちの競争相手は、非侵害的に類似または代替技術または製品を開発することによって、私たちの特許を迂回することができるかもしれない。私たちの競争相手も承認を求めて、彼ら自身の製品を販売して、私たちの製品と似ているか、あるいは他の面で私たちの製品と競争するかもしれません。代替的に、私たちの競争相手は、私たちが所有または許可している特許が無効であるか、強制的に実行できないか、または侵害されていないと主張するANDAをFDAに提出することができ、それにより、任意の承認された製品の模倣薬バージョンの販売を求めることができる。この場合、私たちは訴訟を提起することによって特許侵害を告発することを含む、私たちの特許を擁護したり維持したりする必要があるかもしれない。このような訴訟のいずれにおいても、裁判所または他の管轄権を有する機関は、私たちの特許が無効または強制執行できない、または私たちの競争相手が私たちの特許を侵害していないと認定する可能性がある。したがって、私たちが効果的かつ強制的に実行可能な特許を持っていても、これらの特許は、私たちのビジネス目標を達成するのに十分な競争製品またはプロセスを保護することができない可能性がある。
さらに、私たちは私たちのビジネス秘密と独自の非特許を保護する技術的ノウハウに依存している。第三者とのセキュリティ協定の締結や、従業員、コンサルタント、協力者、サプライヤー、およびコンサルタントとの秘密情報および発明譲渡協定の締結を含む、当社の商業秘密および非特許ノウハウを保護する措置をとっているにもかかわらず、このようなすべてのプロトコルが正式に実行されていることは保証されておらず、第三者がこの情報を取得する可能性があり、または独立して情報または同様の情報を取得する可能性がある。我々の業務に関連する可能性のある技術は,このようなセキュリティや発明譲渡プロトコルの一方ではない人によって独立して開発されるであろう.私たちの技術知識や取引を無許可で開示したり使用することを防ぐことはできないかもしれません
95
コンサルタント、協力者、サプライヤー、コンサルタント、元従業員、そして現従業員の秘密。さらに、もし私たちの秘密協定の当事者がこれらの合意の条項に違反したり、違反したりした場合、私たちはそのような違反や違反を補うための十分な救済措置がないかもしれません。私たちはそのような違反や違反によって私たちの商業秘密を失うかもしれません。そうでなければ、私たちの商業秘密は私たちの競争相手に知られたり独立したりするかもしれない。また,我々のビジネス秘密を保護するための措置が不十分であると考えられれば,第三者が我々のビジネス秘密を流用することに対抗する十分な追跡権がない可能性がある.もしこのような事件が発生した場合、または私たちが私たちの商業秘密やノウハウの保護を失った場合、私たちの業務は損なわれる可能性がある。
私たちの知的財産権と私たちの独自技術を保護することは難しくて高価で、私たちはそれらの保護を保障できないかもしれない。
私たちのビジネス成功は、私たちの特許候補製品AMX 0035の特許保護および商業秘密保護の取得および維持にある程度依存し、これらの特許を潜在的な第三者挑戦から保護することに成功するであろう。私たちの候補製品を第三者から無許可に製造、使用、販売、販売、または輸入から保護する能力は、これらの活動をカバーする有効かつ実行可能な特許の下で私たちが所有する権利の程度に依存する。
製薬、生物技術とその他の生命科学会社の特許地位は非常に不確定である可能性があり、複雑な法律と事実問題に関連し、これらの問題の重要な法律原則はまだ解決されておらず、近年すでに多くの訴訟のテーマになっている。米国や他の国の特許法や特許法解釈の変化は,我々の知的財産権の価値を低下させる可能性がある。過去10年間、米国連邦裁判所はますます訴訟で製薬とバイオテクノロジー特許の無効を宣言し、これらの訴訟は往々にして特許法の変化に対する解釈に基づいている。さらに、特許出願または特許請求の範囲が特許可能性に適合するすべての要件を決定することは、法律適用および判例に基づく主観的決定である。米国特許商標局または米国裁判所または他の事実審査員または対応する外国国家特許庁または裁判所が、権利要件が特許可能性のすべての要件に適合しているかどうかについて最終裁決を下すことは保証されない。したがって、私たちは、私たちが所有する特許または特許出願において許容または実行される可能性のある特許請求の範囲の広さを予測することができない。
私たちは、私たちの既存技術出版物や特許文献、または特許として発行されることを含む、私たちの任意の特許出願が出願可能な特許であることを保証することはできない。私たちはまた、私たちの未解決および将来の特許出願が提起される可能性のある任意のクレームの範囲を保証することもできず、いかなる潜在的な第三者が米国または外国の司法管轄区域における私たちの特許および特許出願の特許性、有効性、または実行可能な任意の訴訟の結果に挑戦する可能性があることを保証することもできない。このような挑戦は、成功すれば、私たちの製品および現在または未来の候補製品の特許保護を制限することができ、および/または私たちの業務に実質的な損害を与える可能性がある。
訴訟中の挑戦に加えて、第三者は、米国における私たちの特許の有効性を疑問視するために、ライセンス後審査および当事者間審査手続きを使用することができ、一部の第三者は、競合他社が発行した特許の一部または全部の権利要件をキャンセルするためにこれらの手続きを利用してきた。2013年3月16日以降に出願された特許については,第三者は特許発表後9ヶ月の窓口内でライセンス後審査出願を提出することができる。特許の有効出願日が2013年3月16日までであれば,特許発表後ただちに当事者間の審査出願を提出することができる。有効提出日が2013年3月16日以降である特許については、付与後審査出願の提出から9ヶ月の期限満了後に当事者間の審査出願を提出することができる。ライセンス後の再審手続きは任意の無効な理由で提起することができ、当事者間の再審手続きは、公表された既存技術および特許に基づいて無効な疑問を提起することしかできない。米国特許商標局のこれらの対抗性訴訟は、米国連邦裁判所訴訟における米国特許の有効性を推定することなく特許主張を審査し、米国連邦裁判所訴訟で使用されているよりも低い立証責任を使用する。したがって、競争相手または第三者は、USPTO許可後の審査または当事者間審査手続きにおいて、米国連邦裁判所の訴訟で無効ではなく、米国特許を無効にしやすいと考えられる。もし私たちのどの特許もこのようなUSPTO訴訟で第三者の挑戦を受けた場合、私たちがその特許を守ることに成功する保証はありません。これは、疑問視されている特許権を失う可能性があります。
EUでは,第三者は欧州特許庁に反対意見を提出することで,我々の特許の有効性に挑戦することができる。野党委員会の不利な裁決は欧州特許の範囲を縮小または無効にする可能性がある。もし私たちのどのヨーロッパ特許もこのような反対手続きで第三者の挑戦を受けたら、私たちがその特許を守ることに成功する保証はありません。これは私たちが疑問視されている特許権を失う可能性があります。
96
法的手段は限られた保護しか提供できず、私たちの権利を十分に保護できないか、あるいは私たちの競争優位性を獲得または維持することができるかもしれないので、未来の私たちの所有権の保護の程度は不確定だ。例えば:
さらに、AMX 0035または任意の他の現在または将来の候補製品の特許保護を取得して維持することができない場合、またはそのような特許保護が満了した場合、後続の適応の製品または候補製品をさらに開発することによって、私たちの製品組み合わせを拡張することは、もはやコスト効果を有さない可能性がある。
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの技術的価値は実質的な悪影響を受ける可能性があり、私たちの業務は損害を受けるだろう。
特許に加えて、私たちは、特に特許保護が適切でないか、または入手不可能であると考えられる場合に、商業秘密によって私たちの特許候補製品を保護することができる。ビジネスの秘密は保護するのが難しい。従業員、コンサルタント、科学コンサルタント、請負業者、協力者と秘密協定や発明譲渡協定を締結することで、私たちの機密独自情報をある程度保護することを求めています。このような協定は私たちの固有の情報を保護することを目的としている。しかし、私たちはすべての関係者とこのような合意を締結したことを確認することはできず、私たちの商業秘密や他の機密固有情報が開示されないか、あるいは競争相手が私たちの商業秘密を取得したり、実質的に同等の情報や技術を独立して開発しないかを決定することもできない。例えば、私たちの従業員、コンサルタント、請負業者、外部科学協力者、および他のコンサルタントは、競争相手に私たちの情報を意図的にまたは意図的に開示することができないかもしれない。第三者エンティティに我々のいかなる商業秘密を不正に取得して使用することを強制するかは高価で時間がかかり、結果的に予測不可能であり、私たちはこのような違反に対して十分な救済措置を得ることができないかもしれない。私たちはまた、私たちの場所の物理的セキュリティと私たちのITシステムの物理的および電子的セキュリティを維持することで、私たちの機密固有情報の完全性と機密性を保護しようとしていますが、これらのセキュリティ対策は破壊される可能性があります。しかも、アメリカ以外の裁判所は商業秘密を保護することをあまり望まないことがある。また,我々の競争相手は同等の知識,方法,ノウハウを自主的に開発することができる.商業秘密保護されたノウハウは、このような同等の技術が商業秘密保護された技術の後に発明されたとしても、独立して開発された同等の技術の特許よりも優先されていないことに留意されたい。もし私たちの任意の機密固有情報が競争相手によって合法的に取得または独立して開発された場合、私たちはその競争相手がその技術または情報を使用して私たちと競争することを阻止する権利がなく、これは私たちの競争地位を損なう可能性がある。
特許条項は製品に対する私たちの競争地位を十分な時間で保護するのに十分ではないかもしれない。
特許の寿命は限られている。米国では、すべての維持費が適時に支払われる場合、特許の自然失効時間は、通常、米国で最初の非臨時出願日から20年である。米国特許の特許期間は、特許期限調整によって延長することができ、これは、特許権者が米国特許商標局の特許付与時の行政遅延による損失を補償することができ、または、1つの特許がより早く提出された特許によって最終的に放棄された場合、短縮することができる。
様々な延期があるかもしれないが、特許の有効期限とその提供される保護は限られている。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。
米国では,1984年の“薬品価格競争と特許期限回復法”は,特許所有者が長い規制承認過程により実行可能な特許期限を失ったことを補償するために,通常の満期後に最大5年間延長することを許可している。PTEは承認された適応に限定される(または任意の他の適応
97
延期期間中に承認を得る)。私たちはアメリカでPTEを申請する予定です。私たちが特許を起訴している他の国も同様の延期がある可能性があり、私たちも同様にこのような延期を申請する予定です。
このような特許期間の延長は保証されているわけではなく、多くの要求を満たす必要がある。私たちは、適用期間内に出願できなかったこと、関連する特許が満了する前に出願できなかったこと、または多くの適用要件のいずれかを他の方法で満たすことができなかったため、延期を得ることができない可能性がある。さらに、適用当局は、米国のFDAおよびUSPTO、および他の国/地域の任意の同等の規制機関を含み、このような延期が利用可能かどうかの評価に同意しない可能性があり、私たちの特許延期の承認を拒否するか、または私たちが要求したよりも限られた延期を承認する可能性がある。このような状況が発生すれば、私たちの競争相手は私たちの特許が満期になった後、私たちの臨床と臨床前データを参考にすることで競争製品の承認を得て、他の場合よりも早く彼らの製品を発売するかもしれない。このような状況が発生すれば、私たちの創造能力に実質的な悪影響を及ぼすかもしれない。
米国や他の管轄地域の特許法解釈の変化は,特許の全体的な価値を低下させ,製品を保護する能力を弱める可能性がある。
米国議会は特許性基準を確立する法律の採択を担当している。どんな法律と同じように、仕事を実行することは連邦機関と連邦裁判所に彼らの法律の解釈に基づいている。特許基準の解釈は,米国特許商標局内部および最高裁判所を含む各連邦裁判所の間に大きな差がある可能性がある。最近、最高裁はいくつかの特許案件に対して裁決を下し、全体的に特許を申請できる発明タイプを制限している。また,特許性基準の解釈については,最高裁はこれらの問題を果敢に解決していない。最高裁の明確な指導がないため,米国特許商標局は特許法や標準を解釈する際にますます保守的になっている。
我々の将来の特許取得能力に対する不確実性の増加に加え,米国の法的環境も特許価値に関する不確実性をもたらしている。国会のいかなる行動、連邦下級裁判所、最高裁判所の将来の裁決、および米国特許商標局の解釈によると、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、新しい特許を獲得したり、既存の特許および将来獲得可能な特許を強制的に執行する能力を弱める可能性がある。
私たちは世界各地で私たちの知的財産権を強制的に施行できないかもしれない。
世界のすべての国で私たちの候補製品申請、起訴、強制執行、および特許保護の費用は目を引くほど高く、そして私たちのアメリカ以外のいくつかの国での知的財産権はアメリカほど広くないかもしれない。いくつかの国では特許性に対する要求が異なるかもしれない。特に発展途上国では、したがって、私たちが確かに特許保護を求めている国であっても、どの特許も私たちの製品に関連している保証はない。
また、私たちが知的財産権を保護し、実行する能力は、外国の知的財産法の意外な変化の悪影響を受ける可能性がある。また、米国やEU以外のいくつかの国の法律は知的財産権の保護の程度は米国やEUの法律よりも劣る。ある外国司法管轄区では、多くの会社が知的財産権の保護と保護に重大な問題に直面している。これは私たちが私たちの特許を侵害したり、私たちの他の知的財産権を流用することを防ぐことを難しくするかもしれない。例えば、多くの外国国には強制許可法があり、これらの法律によると、特許権者は第三者に許可を付与しなければならない。したがって、私たちは、米国およびEU以外のいくつかの国/地域で第三者が私たちの発明を実施したり、米国または他の司法管轄区で私たちの発明で作られた製品を販売したり、輸入したりすることを阻止することができないかもしれない。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの技術を使用して自分の製品を開発して販売することができ、また、もし私たちが特許を実行して侵害活動を阻止する能力が不足していれば、競争相手は他の侵害製品を私たちが特許保護を持っている地域に輸出するかもしれない。これらの製品は私たちの製品と競争するかもしれませんが、私たちの特許や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。
私たちの特許権訴訟を実行することは、成功するか否かにかかわらず、巨額のコストを招き、私たちの努力と資源を私たちの業務の他の側面から移転する可能性があります。さらに、このような訴訟は、私たちの特許が無効または強制執行できないリスクに直面するか、または無効と狭く解釈され、私たちの係属中の特許出願を発行できないリスクに直面させ、第三者が私たちにクレームを提起する可能性があるかもしれない。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。また、製品の主要市場で私たちの知的財産権を保護しようとしていますが、私たちが同様の努力を開始または維持できることを確実にすることはできません
98
もし承認されたら、私たちはすべての管轄区域で私たちの製品を販売することを望むかもしれない。したがって、このような国で知的財産権を保護するための私たちの努力は十分ではないかもしれない。
他の人は、発明権に挑戦したり、私たちの知的財産権の所有権を要求したりする可能性があり、これは訴訟に直面させ、その将来性に重大な悪影響を及ぼす可能性がある。
第三者または元従業員または協力者は、私たちまたは私たちのライセンシーの1つまたは複数の特許または他の独自または知的財産権の発明権または所有権権益を要求することができる。第三者は、影響を受けた製品または製品の臨床試験、製造、およびマーケティングを禁止するために、金銭賠償および/または禁止を求める法的訴訟を提起することができる。私たちは現在、第三者が私たちの特許や他の知的財産権の在庫や所有権について提起したいかなるクレームや主張も知らないが、第三者がこのような特許や知的財産権のクレームや利益を主張しない保証はない。もし私たちがこのようなクレームを弁護することができなかった場合、金銭的損害賠償を支払うことに加えて、AMX 0035または任意の他の現在または未来の候補製品に対して非常に重要な知的財産の独占所有権または使用権のような貴重な知的財産権を失う可能性がある。また、結果にかかわらず、私たちがどんな訴訟に巻き込まれれば、私たちの大量の資源を消費し、私たちの技術や管理者の多くの気晴らしを招く可能性があります。
もし私たちが第三者の知的財産権侵害で起訴された場合、このような訴訟は費用が高く、時間がかかり、AMX 0035または任意の他の現在または未来の候補製品の開発または商業化を阻止または延期する可能性がある。
当社のビジネス成功は、第三者の知的財産権および他の独自の権利を侵害することなく、当社の候補製品を開発、製造、マーケティング、販売する能力にある程度依存しています。しかし、私たちの研究、開発、および商業化活動は、第三者が所有または制御している特許または他の知的財産権を侵害または他の方法で侵害される可能性がある。第三者は、米国および非米国によって発行された特許、ならびに化合物、化合物の製造方法、および/または我々が開発しているAMX 0035または任意の他の現在または将来の候補製品の疾患適応を治療するための方法に関連する係属中の特許出願を有する可能性がある。任意の第三者特許または特許出願がAMX 0035または任意の他の現在または将来の候補製品またはそれらの使用または製造方法をカバーしていることが発見された場合、私たちは、許可を得ずに、そのような候補製品を計画的に製造または販売することができない可能性があり、この許可は、商業的に合理的な条項で提供できないか、または全く得られない可能性がある。
バイオテクノロジーや製薬業界に大量の知的財産権訴訟があり、私たちは、米国または海外での特許侵害訴訟を含む、私たちの候補製品に関連する知的財産権訴訟または他の対抗性訴訟の一方になるか、または脅かされる可能性がある。AMX 0035または任意の他の現在または将来の候補製品の組成、使用または製造に関連する材料、調製、製造方法または治療方法の第三者特許または特許出願が存在する可能性がある。私たちは、私たちの候補特許薬物AMX 0035に関連する特許および特許出願を定期的に検索しているが、関連特許の識別、特許請求の範囲、または関連特許の満了を含むが、関連特許の識別、特許請求の範囲、または関連特許の満了を含むが、AMX 0035または任意の他の現在または将来の製品の任意の司法管轄区域における商業化に関連するまたは必要なすべての特許および係属中の出願がAMX 0035または任意の他の現在または将来の製品と識別されていることを保証することはできない。特許出願は、発行するのに数年の時間を要する可能性があるので、現在処理されている特許出願が存在する可能性があり、これらの出願は、AMX 0035または任意の他の現在または将来の候補製品が侵害された発行された特許を告発する可能性がある。さらに、第三者は将来的に特許を取得し、私たちの技術を使ってこれらの特許を侵害したと主張するかもしれない。したがって、第三者は現在存在するまたは未来に出現する知的財産権に基づいて私たちに権利侵害を請求するかもしれない。知的財産権訴訟の結果は不確定要因の影響を受け,これらの不確定要因は事前に十分に定量化できない。製薬およびバイオテクノロジー産業は大量の特許を生成しており、私たちを含む業界参加者は、どの特許が様々なタイプの製品または使用または製造方法をカバーしているのかを常に明確にしていないかもしれない。特許提供の保護範囲は裁判所の解釈に依存し、解釈はいつも一致しているわけではない。もし私たちが特許侵害を起訴された場合、私たちは私たちの候補製品、製品、または方法が関連特許の特許主張を侵害していないか、または特許主張が無効または実行不可能であることを証明する必要があり、私たちはそれができないかもしれない。無効性を証明することは難しい。例えば,米国では,証明無効は,発行された特許享受の有効性の推定を覆すために,明確で納得できる証拠を提示する必要がある.私たちがこれらの訴訟で勝訴しても、私たちは巨額のコストを招く可能性があり、私たちの経営陣や科学者の時間と注意力はこれらの訴訟に移される可能性があり、これは私たちの業務と経営業績を深刻に損なう可能性があります。さらに、私たちにクレームを出した当事者は、彼らがより多くの資源を持っているので、私たちよりも複雑な特許訴訟の費用を効果的に維持することができ、私たちはこれらの訴訟を成功させるのに十分な資源がないかもしれない。
99
もし私たちが第三者の知的財産権を侵害していることが発見された場合、裁判所の命令を含めて、権利侵害の候補製品や製品の開発、製造、または商業化を停止させられる可能性がある。代替的に、権利侵害技術を使用して、権利侵害候補製品または製品の開発、製造、または販売を継続するために、第三者から許可を得る必要があるかもしれない。もし私たちが影響を受けた製品を製造または販売し続けるために許可を要求された場合、巨額の印税を支払うか、私たちに付与された特許交差許可を支払う必要があるかもしれない。しかし、このようなライセンスが受け入れ可能な条項で提供されることを保証することはできません。最終的に、私たちは特許侵害や他の知的財産権侵害の疑いで製品の商業化を阻止されたり、いくつかの方面の商業運営を停止させられたりする可能性がある。また、知的財産権訴訟の結果は不確定要素の影響を受け、これらの不確定要素は証人の振る舞いや信頼性、いかなる敵の身分も含めて事前に十分に定量化することができない。知的財産権事件では特に、これらの事件は専門家が合理的に同意しない可能性のある技術的事実に関する専門家の証言に依存するかもしれない。しかも、私たちは商業的に合理的な条項や必要な許可証を得ることができないかもしれない。たとえ私たちが許可を得ることができても、それは非排他的で、私たちの競争相手が私たちに許可された同じ技術にアクセスできるようにすることができる;あるいは、あるいはさらに、それは商業市場での私たちの成功した競争の能力を阻害または破壊する条項を含むことができる。さらに、もし私たちが故意に特許を侵害したことが発見された場合、私たちは3倍の損害賠償と弁護士費を含む重大な金銭損害に責任があると判断されるかもしれない。権利侵害の発見は、AMX 0035または任意の他の現在または将来の候補製品を商業化することを阻止するか、またはいくつかの業務運営を停止させることを阻止するかもしれません。これは、私たちの業務を損なう可能性があります。我々が第三者の機密情報や商業秘密を盗用したと主張することは,我々の業務に類似した負の影響を与える可能性がある.さらに、知的財産権訴訟や行政訴訟に関連する大量の開示要求により、私たちのいくつかの機密情報が開示によって漏洩される可能性がある。さらに、任意の訴訟の開始および継続によって生じる任意の不確実性は、追加資金を調達する能力に重大な悪影響を及ぼすか、または私たちの業務、運営結果、財務状況、および将来性に重大な悪影響を及ぼす可能性がある。
私たちは、私たちの従業員や私たちが彼らの知的財産権を流用したと主張したり、私たち自身の知的財産権の所有権を要求したりする第三者のクレームを受けるかもしれない。
私たちの多くの現職および元従業員、および私たちの許可側の現職および前職の従業員は、競争相手または潜在的な競争相手である可能性のある会社を含む、大学または他のバイオテクノロジーまたは製薬会社に雇われていた。その中の一部の従業員は、私たちの上級管理職のメンバーを含み、これまでの仕事に関連する専有権、秘密、およびスポーツ禁止協定、または同様の協定に署名しているかもしれない。私たちは、私たちの従業員が私たちのために働いているときに他人の固有情報やノウハウを使用しないことを保証するために努力しているが、私たちまたはこれらの従業員は、商業秘密または他の固有情報を含む、そのような第三者の知的財産権の使用または開示を告発される可能性がある。このようなクレームを弁護するために訴訟を提起する必要があるかもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭的損失を支払うことに加えて、私たちは損害を受けたり、重要な知的財産権や人員の仕事の成果を失ったりする可能性があり、これは私たちの技術商業化を阻害または阻止する可能性があり、逆に私たちのビジネス開発努力に大きな影響を与える可能性がある。このような知的財産権は第三者に付与することができ、私たちの技術や製品を商業化するためには第三者から許可を得る必要があるかもしれない。そのような許可は商業的に合理的な条項や根本的に存在しないかもしれない。このようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣の注意を分散させる可能性がある。
また、私たちは通常、知的財産権開発に参加する可能性のある従業員、コンサルタント、請負業者に、このような知的財産権を譲渡する協定に署名することを要求していますが、実際に私たちが自分の知的財産権を開発しているすべての側とこのような合意を実行することができない可能性があり、このような知的財産権の所有権について私たちにクレームを出したり、私たちにクレームをつけたりする可能性があります。もし私たちがこのようなクレームを起訴したり、弁護することができなければ、金銭的損害賠償を支払う以外に、私たちは貴重な知的財産権を失う可能性がある。このようなクレームの起訴や抗弁に成功しても、訴訟は巨額の費用を招き、私たちの上級管理者や科学者の注意を分散させる可能性がある。
もし私たちの商標と商号が十分に保護されていなければ、私たちは私たちの商標で知名度を確立できないかもしれません。私たちの業務は不利な影響を受けるかもしれません。
私たちの商標または商号は、挑戦、侵害、回避、または汎用商標として発表されるか、または他の商標を侵害していると判断される可能性がある。私たちの商標は登録と一般法の保護に依存している。私たちはこれらの商標や商品名に対する私たちの権利を保護できないかもしれないし、これらの名前の使用を停止させることができないかもしれません。私たちは関心のある市場で潜在的なパートナーや顧客の名前の承認を得るために必要です。商標登録過程で、私たちは拒否を受けるかもしれない。私たちはこのような拒否に答える機会があるにもかかわらず、私たちはできないかもしれない
100
この拒絶を克服する。また,米国特許商標局や多くの外国司法管轄区の類似機関では,第三者は係属中の商標出願に反対し,登録商標の取り消しを求める機会がある。私たちの商標に反対またはキャンセル訴訟を提起するかもしれないが、私たちの商標は継続できないかもしれない。また、私たちが米国で私たちの製品のために使用するどの名称も、登録されているかどうか、または商標として登録されているかどうかにかかわらず、FDAの承認を受けなければならない。FDAは、通常、他の製品名と混同される可能性がある可能性を含む、提案された製品名を検討する。FDAが私たちが提案した任意の製品名に反対する場合、私たちは、適用可能な商標法に適合し、第三者の既存の権利を侵害せず、FDAによって受け入れられる利用可能な代替名を決定するために多くの追加資源を必要とするかもしれない。もし私たちが私たちの商標と商号に基づいて名称を確立することができなければ、私たちは効果的に競争できない可能性があり、私たちの業務は不利な影響を受けるかもしれない。
知的財産権は私たちの業務が直面しているすべての潜在的な脅威を解決できるとは限らない。
私たちの知的財産権が提供する将来の保護の程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できないか、あるいは競争優位性を維持することができるかもしれない。以下の例は例示的である
私たちは第三者に依存して研究開発と製造を行い、これは私たちのビジネス秘密を共有することを要求し、これは私たちのビジネス秘密が流用されたり漏洩したりする可能性が増加し、従業員や第三者と締結された秘密保護協定は、ビジネス秘密の漏洩を十分に防止し、他の固有情報を保護できない可能性がある。
私たちは、独自の商業秘密または機密ノウハウおよび非特許ノウハウが、私たちのビジネスに非常に重要であると考えている。私たちは、特に特許保護の価値が限られていると思う場合、商業秘密または機密技術によって私たちの技術を保護するかもしれない。私たちは第三者に依存して研究と開発を行い、将来的に私たちの特許候補製品AMX 0035および任意の他の現在または未来の候補製品の製造は第三者に依存すると予想される。私たちはまた、第三者と協力してAMX 0035および任意の現在または未来の候補製品を開発したい。上記の協力の結果として、私たちは時々私たちの協力者とビジネス秘密を共有しなければならない。
商業秘密や機密ノウハウを機密として維持することは難しいかもしれない。このような情報を競争相手から開示したり盗んだりしないようにするために、私たちの政策は私たちの従業員、コンサルタント、請負業者と
101
コンサルタントは、独自の情報の研究または開示を開始する前に、(適用可能であれば)秘密協定および材料譲渡プロトコル、コンサルティングプロトコル、または他の同様の合意を締結する。これらの協定は、一般に、私たちの商業秘密を含む、第三者が私たちの機密情報を使用または開示する権利を制限する。しかしながら、現従業員または元従業員、コンサルタント、請負業者、およびコンサルタントは、競争相手に私たちの機密情報を意図的にまたは意図的に開示することができず、機密情報を無許可に開示することができない場合に十分な救済措置を提供できない可能性がある。商業秘密および他の機密情報を共有する必要は、そのような商業秘密が私たちの競争相手に知られ、意図せずに他の人の技術に組み込まれるか、または開示されるか、またはこれらの合意に違反して使用されるリスクを増加させる。私たちの独自の地位が私たちのノウハウおよびビジネス秘密にある程度基づいていることを考慮すると、競争相手は、私たちのビジネス秘密または他の許可されていない使用または開示が私たちの競争地位を損なうことを発見し、私たちの業務および運営結果に悪影響を及ぼす可能性があります。第三者が商業秘密または機密技術を不法に入手して使用することを強制することは高価で、時間がかかり、予測できない。また,セキュリティプロトコルの実行可能性は管轄によって異なる可能性がある.
さらに、これらの合意は、一般に、私たちの合意がいくつかの限られた発行権を含む可能性があるにもかかわらず、私たちのコンサルタント、従業員、第三者請負業者、およびコンサルタントが、私たちのビジネス秘密に関連する可能性のあるデータを発行する能力を制限します。私たちは私たちのビジネス秘密を保護しようと努力しているにもかかわらず、私たちの競争相手は、私たちと第三者との合意、独立開発、または私たちの任意の第三者協力者が情報を発表することによって、私たちのビジネス秘密を発見するかもしれない。競争相手は私たちのビジネス秘密が私たちの競争地位を損なうことを発見し、私たちの業務に悪影響を及ぼすだろう。
私たちは第三者から知的財産権を取得する必要があるかもしれませんが、このような許可は得られないかもしれませんし、商業的に合理的な条項では得られないかもしれません。
第三者は、将来の他の候補製品の開発に非常に重要又は必要な特許権を含む知的財産権を保有することができる。私たちは、私たちの現在および未来の候補製品を商業化するために、1つまたは複数の第三者の特許または独自技術を使用する必要があるかもしれない。このような知的財産権を直接得ることができない場合や、必要に応じて、または商業的に合理的な条件下でそのような第三者からそのような知的財産権の許可を得ることができなければ、他の将来の候補製品を商業化する能力を遅らせる可能性が高い。
他の場所で説明されている私たちの知的財産権に関するリスクは、私たちが許可を得る可能性のある知的財産権にも適用され、私たちまたは私たちのライセンシーがこれらの権利を獲得、維持、擁護、実行できなかった場合、私たちの業務に悪影響を及ぼす可能性がある。場合によっては、私たちが許可した特許の起訴、保守、または強制執行を制御できない可能性があり、そのような特許の起訴、保守、および弁護過程に投入する十分な能力がない可能性があり、将来の潜在的な許可者は、許可された特許を取得、維持、弁護、および強制執行するために必要または適切なステップを取ることができないかもしれない。
私たちの業務運営、従業員事務、管理成長に関するリスク
私たちは現在、経済不確定と資本市場が混乱している時期にあり、地政学的不安定、持続的な軍事衝突(ロシアとウクライナの間の衝突、イスラエルの衝突を含む)、高インフレと金利上昇は、私たちの業務、財務状況、運営結果に大きな悪影響を与えている。
米国と世界市場は最近、持続的なロシア-ウクライナ紛争、ロシアに対する制裁の影響、最近のイスラエルとガザ地区の衝突など、経済的不確実性による変動と混乱を経験している。2022年2月、ロシア軍はウクライナへの全面的な軍事侵入を開始した。現在の軍事衝突の持続時間と影響は極めて予測しにくいが、ウクライナ衝突はすでに市場混乱を招き、商品価格、信用と資本市場の大幅な変動、及びサプライチェーンの中断を含み、これは全世界のインフレ率が過去最高を記録した。また、イスラエルとガザ地区の現在の武力衝突により、世界市場は最近の襲撃により、イスラエルが米国が指定した外国のテロ組織ハマスに宣戦布告したため、より多くの混乱を経験する可能性がある。私たちはインフレ、ウクライナ、イスラエルの情勢、および世界的な資本市場を監視し続け、AMX 0035または任意の他の現在または未来の候補製品を生産するために依存するサプライチェーンへの影響を含む、我々の業務に対する潜在的な影響を評価している。
これまで、我々の業務は、上記のような事件、地政学的緊張、あるいは記録的なインフレの実質的な影響を受けていないにもかかわらず、私たちの業務が短期的かつ長期的にどの程度影響を受けるか、あるいはこれらの問題がどのような方法で私たちの業務に影響を与える可能性があるかは予測できない。ウクライナとイスラエルの紛争の範囲と持続時間は
102
地政学的緊張、記録的なインフレ、それに伴う市場混乱は予測できないが、巨大かもしれない。このような中断はまた私たちが直面している他の危険の影響を増幅させるかもしれない。
米国食品医薬品局、米国証券取引委員会および他の政府機関の資金不足は、政府の閉店やこれらの機関の運営の他の中断を含め、重要な指導部や他の人員の能力を雇用·保留し、新製品やサービスのタイムリーな開発や商業化を阻止するか、あるいはこれらの機関が私たちの業務運営に依存する可能性のある正常な業務機能を履行することを阻止することを阻害する可能性があり、これは私たちの業務に負の影響を与える可能性がある。
FDAの新製品の審査と承認能力は、政府予算と資金レベル、肝心な人員の雇用と維持及びユーザー費用の支払いを受ける能力、及び法定、監督と政策の変化を含む様々な要素の影響を受ける可能性がある。したがって、その機関の平均検討時間は近年変動している。また、政府が米国証券取引委員会や我々の業務に依存する可能性のある他の政府機関に提供する資金は、研究開発活動に資金を提供する機関を含め、政治プロセスの影響を受けており、政治プロセス自体が不安定で予測不可能である。
FDAや他の機関の中断も、新製品候補製品が必要な政府機関によって審査および/または承認されるのに要する時間を遅らせる可能性があり、これは私たちの業務に悪影響を及ぼすだろう。政府が長期的に停止すれば、FDAが私たちが提出した規制文書を適時に審査して処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。また、将来的に政府の閉店は、適切な資本化と事業継続のために、私たちが公開市場に進出し、必要な資本を得る能力に影響を与える可能性がある。
私たちは私たちの幹部、主要な顧問、そして他の人たちに深刻に依存して、彼らのサービスを失うことは私たちの業務に深刻な損害を与えるだろう。
私たちの成功は、現在の行政官、主要顧問、他の人たちのサービスを採用し、保留する能力があるかどうかにかかっており、そうし続ける可能性が高い。私たちは私たちの現職幹部と雇用協定を締結したが、彼らはいつでも私たちとの雇用関係を終わらせることができる。彼らのサービスを失うことは私たちが研究、開発、そして商業化目標を達成するのを阻害するかもしれない。
生物技術と製薬業界における私たちの競争力は私たちが高い素質の管理、科学と医療人員を誘致し、維持できるかどうかにかかっている。近年、私たちの業界の管理職の流出率は高い。例えば、2024年2月、私たちの当時の首席人力資源官デブラ·カンナはリンダ·アーセノールに置き換えられ、私たちの現在の首席人的資源官となりました。2023年12月、私たちの当時の世界臨床研究開発担当兼首席医療官パトリック·Yeramian医学博士はCamille L.Bedrosian医学博士に置き換えられ、私たちの現在の首席医療官となった。また、2023年12月、私たち当時の首席商務官マーガレット·オリンガーは会社を退社しました。幹部や他の重要な従業員を交換することは困難かもしれませんし、私たちの業界では開発に成功し、監督部門の承認を得て製品を商業化するために必要な技能と経験を持っている個人の数が限られているので、長い時間がかかるかもしれません。
この限られた人材バンクから募集する競争は非常に激しく、多くの製薬と生物技術会社の間の類似人員に対する競争を考慮して、私たちは受け入れ可能な条件でこれらの追加の肝心な従業員を募集、訓練、維持或いは激励することができないかもしれない。私たちはまた、大学や研究機関から科学や臨床人を募集する競争に直面している。
私たちはコンサルタントとコンサルタントに依存して、科学と臨床コンサルタントを含めて、私たちの研究開発と商業化戦略の制定を助けてくれます。私たちのコンサルタントとコンサルタントは他のエンティティに雇われる可能性があり、これらのエンティティとの相談やコンサルティング契約に基づいて約束することができ、これは私たちの彼らに対する利用可能性を制限するかもしれない。もし私たちが高い素質の人材を引き付け、維持することができなければ、AMX 0035または任意の他の現在または未来の候補製品を開発し、商業化する能力は制限されるだろう。
私たちは限られた職員だけが私たちの業務を管理して運営する。
2023年12月31日現在、384人のフルタイム従業員を抱えています。私たちはAMX 0035の開発と商業化に集中して、現金使用を最適化し、効率的な方法で私たちの業務を管理し、運営することを要求します。AMX 0035を開発および商業化するのに十分な人員を雇用および/または維持することができ、または私たちの業務を運営し、および/または私たちが本来達成しようとしていたすべての目標を達成することができることを保証することはできません。
103
私たちの従業員、独立請負業者、コンサルタント、協力者、CROは規制基準と要求を守らないことを含む不当な行為や他の不当な活動に従事する可能性があり、これは私たちが重大な責任を負い、私たちの名声を損なう可能性があります。
私たちは、従業員、独立請負業者、コンサルタント、協力者、およびCROが詐欺または他の不正活動に従事する可能性があるというリスクに直面している。これらの当事者の不適切な行為は、故意、無謀、および/または不注意な行為を含むことができ、または以下の規定に違反する不正な活動を開示することができる
これらの法律に拘束された活動はまた、不適切なマーケティング、使用或いは歪曲臨床試験過程で得られた情報を含み、著者らの臨床前研究或いは臨床試験において虚偽データを作成したり、製品材料を不法に流用したりすることは、規制制裁と私たちの名声を深刻に損なう可能性がある。不正行為を識別し、阻止することは常に可能ではなく、このような行為を発見し、防止するための予防措置は、未知または未管理のリスクまたは損失を効果的に制御することができないか、またはこのような法律、基準または法規に準拠していないことによる政府の調査または他の行動または訴訟から私たちを保護することができない可能性がある。さらに、誰かや政府がこのような詐欺や他の不正行為を告発する可能性があり、起こらなくてもリスクに直面している。もし私たちにこのような訴訟を提起した場合、私たちは私たちの権利を弁護したり、維持することに成功しませんでした。これらの訴訟は、民事、刑事と行政処罰、損害賠償、罰金、返還、誠実な監督と報告義務の適用、Medicare、Medicaidおよび他の連邦医療保健計画から除外される可能性があり、契約損害、名声損害、利益および将来の収益の減少、および私たちの業務の能力および運営結果に実質的な悪影響を及ぼす可能性があります。
私たちは私たちの組織を拡大することを望んでいるので、私たちは私たちの成長を管理する上で困難に直面するかもしれないし、これは私たちの運営を混乱させるかもしれない。
私たちは現在、私たちの従業員の数と業務範囲を引き続き大幅に増加させ、特に規制事務と販売、マーケティング、流通の分野で、私たちの上場企業の運営を支援する予定です。これらの成長活動を管理するためには、私たちの管理、運営、財務システムを継続して実施し、改善し、私たちの施設を拡大し、より多くの合格者を募集し、訓練し続けなければならない。私たちの経営陣はこのような成長活動を管理するために多くのエネルギーを投入しなければならないかもしれない。しかも、私たちが予想している成長は私たちがその国の違う地理的地域に移転する必要があるかもしれない。私たちの財務資源が限られていることと、私たちの管理チームがこのような成長を期待している会社を管理する上での限られた経験のため、私たちは私たちの業務の拡張や移転を効果的に管理し、肝心な従業員を維持したり、より多くの合格者を探したり、採用したりすることができないかもしれません。私たちは私たちの業務の拡張や移転を効果的に管理することができず、私たちのインフラが弱く、操作ミス、ビジネス機会の喪失、従業員の流失、余剰従業員の生産性の低下を招く可能性があります。私たちの予想成長はまた、大量の資本支出を必要とし、より多くの候補製品を開発するような他のプロジェクトから財務資源を移転する可能性がある。私たちの予想成長を効果的に管理できなければ、私たちの費用は予想よりも増加する可能性があり、私たちの収益能力は低下する可能性があり、AMX 0035や他の現在または未来の候補製品の成功した商業化を含む当社の業務戦略を実施できないかもしれません。
大流行、流行病、あるいは伝染病の爆発は、私たちの臨床前研究、臨床試験、私たちが依存している第三者、私たちのサプライチェーン、資金を集める能力、私たちが通常の業務を行う能力、そして私たちの財務業績を含む、私たちの業務に実質的な悪影響を及ぼすかもしれない。
私たちは公衆衛生危機と関連した危険に直面している。例えば、2020年から2022年まで、著者らは著者らの臨床前と臨床試験活動の変化、例えばいくつかの非現場仕事の手配と非現場評価の実行を含む新冠肺炎の大流行のいくつかの影響を経験した。私たちが将来的に他の影響に遭遇しないことは保証されません例えば、さらなる登録の延期や一時停止を余儀なくされ、私たちのサプライチェーンの潜在的な中断を経験して、
104
将来の臨床試験で患者を募集するか、または予想される時間範囲内で我々の研究の完全な統合を実現することができる点で、困難または追加コストに直面しているか、または全くできない。
将来の大流行または同様の中断が、患者登録または治療、またはAMX 0035および任意の他の現在または将来の候補製品の開発に生じる任意の負の影響は、臨床試験活動のコストの高い遅延をもたらす可能性があり、これは、規制部門の承認を得て、AMX 0035および任意の他の現在または将来の候補製品を商業化する能力に悪影響を及ぼす可能性があり、承認されれば、私たちの運営費用を増加させ、これは、私たちの財務業績に大きな悪影響を及ぼす可能性がある。新冠肺炎の疫病とその他の全世界のマクロ経済要素も公開株式市場の大幅な変動を招き、アメリカと世界経済に妨害をもたらし、未来のいかなる疫病或いは類似の破壊は更なる市場混乱を招く可能性がある。このような変動性と経済混乱の激化は、私たちがより有利な条件で資金を調達することを困難にするかもしれないし、根本的にそうではない。私たちまたは私たちと接触している第三者が業務中断に遭遇した場合、現在計画されている方法やスケジュールに従って業務を展開する能力は大きな負の影響を受ける可能性があり、これは私たちの業務および私たちの運営結果や財務状況に大きな悪影響を及ぼす可能性があります。将来の任意の大流行または同様の破壊が私たちの業務および財務業績に悪影響を及ぼす場合、本“リスク要因”の節で説明した多くの他のリスク、例えば、私たちの臨床試験の時間および完了、および将来の融資を得る能力に関連するリスクを増加させる可能性もあります。
私たちの普通株に関するリスクは
私たちの株価は変動するかもしれません。あなたはあなたの投資の全部または一部を失うかもしれません。
私たちの普通株の取引価格は大きく変動する可能性があり、様々な要素の影響を受けて大幅に変動する可能性があり、その中のいくつかの要素は取引量が限られていることを含む制御できない。例えば、2022年1月7日、すなわち私たちの株がナスダック世界ベスト市場で取引された初日から、2023年12月31日まで、私たちの株は41.93ドルと低価格6.51ドル/株の範囲で取引されてきた。“リスク要因”の節や年報の他の部分で議論されている要因のほかに、これらの要素には、以下のような要素が含まれる
105
また,株式市場,特に製薬会社は,極端な価格や出来高変動を経験しており,これらの変動はこれらの会社の経営業績に関係なくあるいは比例しないことが多い。私たちの実際の経営業績にかかわらず、広範な市場と業界要素は私たちの普通株の市場価格にマイナス影響を与える可能性がある。
不安定な市場、経済、政治、地理的条件は、私たちの業務、財務状況、株価に深刻な悪影響を及ぼす可能性がある。
広く報道されているように、世界の信用と金融市場は過去数年間、流動性と信用供給の深刻な減少、消費者自信の低下、インフレ率と金利上昇、および最近のウクライナとイスラエルの衝突と関連した状況を含む極端な変動と破壊を経験している。信用と金融市場のさらなる悪化と経済状況への自信が起こらない保証はない。私たちの全体的な業務戦略は、このような経済低迷、不安定なビジネス環境、または持続不可能で不安定な市場状況のいずれかの悪影響を受ける可能性がある。私たちの業務はウクライナやイスラエルの衝突などの地政学的事件による変動の影響も受ける可能性がある。現在の株式や信用市場が悪化したり改善されていない場合、任意の必要な債務や株式融資をより困難にし、コストがより高く、希釈度が高くなる可能性がある。また、私たちの株価は下落する可能性があり、一部の原因は株式市場の変動と全体的な経済低迷である。
106
適切かつ有利な条件で必要な融資を得ることができなければ、私たちの成長戦略、財務業績、株価に実質的な悪影響を及ぼす可能性があり、私たちの1つまたは複数の候補製品の開発と商業化を延期、削減、または停止すること、または潜在的な許可内または買収を求めることを延期することを要求するかもしれない。さらに、私たちの現在の1つまたは複数のサービスプロバイダ、製造業者、および他のパートナーは、経済的に困難な時期を乗り切ることができない可能性があり、これは、私たちが時間通りに、予算で運営目標を達成する能力に直接影響を与える可能性がある。
私たちはもう新興成長型企業ではなく、新興成長型会社に適用されるコンプライアンス要件はもう私たちには適用されません。
私たちは2012年にJumpstart Our Business Startups ActやJOBS Actで定義された新興成長型企業の資格を満たしていないので、私たちは新興成長型企業に適用されるいくつかのコンプライアンス要件の免除に依存する権利はなくなりました。したがって、いくつかの猶予期間の規定の下で、私たちは今:
私たちはこれ以上雇用法案に関連した費用節約を利用することができない。また、非新興成長型企業に適用される追加要求が、他の業務に対する私たちの経営陣や人員の関心を移した場合、私たちの業務、財務状況、運営結果に大きな悪影響を及ぼす可能性があります。増加したコストは私たちの純収益を減少させ、あるいは私たちの純損失を増加させ、他の業務分野でコストを下げることを要求するかもしれない。私たちは、これらの要求に応答して生成される可能性のある追加コストの金額や時間を予測または推定することができない。また、非新興成長型企業としての義務を履行できなければ、普通株退市、罰金、制裁、その他の規制行動に直面する可能性があり、民事訴訟に直面する可能性もある。
私たちの総流通株の大部分は市場に売却される可能性があり、これは私たちの業務が良好であっても、私たちの普通株の市場価格を大幅に低下させる可能性がある。
公開市場で私たちの大量の普通株を売ることはいつでも起こる可能性がある。これらの売却、あるいは市場で大量の株式保有者が株を売却しようとしているとの見方は、我々の普通株の市場価格を低下させる可能性がある。2024年2月12日現在、我々が発行した普通株は67,782,139株であり、私たちの関連会社が保有していない限り、直ちに公開市場で制限されずに転売することができる。また、約1,180万株の自社普通株を保有する株主は、特定の条件に適合する場合には、その株式に関する登録声明を提出したり、自分又は他の株主に提出する可能性のある登録声明にその株を組み込むことを要求する。
私たちは予測可能な未来に私たちの株にどんな現金配当金も支払わないと予想している。そのため、株主は資本付加価値(あれば)に依存して投資リターンを得ることができる。
私たちは私たちの株の現金配当金を発表したり支払ったりしたことがない。私たちは現在、私たちの未来のすべての収益を保留する予定で、もしあれば、私たちの業務の運営、発展、成長に資金を提供します。しかも、未来のどんな債務や信用協定の条項も私たちが配当金を支払うことを阻止するかもしれない。したがって、私たちの普通株の資本付加価値は(もしあれば)あなたが予測可能な未来に唯一の収益源になるだろう。
私たちの普通株の所有権は私たちの既存の幹部、役員、主要株主に集中しており、新しい投資家が重大な会社の意思決定に影響を与えることを阻止する可能性がある。
私たちの役員と取締役は、私たちが発行した普通株の5%以上を持つ株主に加えて、私たちの普通株の大きな部分を持っています。したがって,これらの株主が共同行動することは,我々株主に承認されたすべての事項や我々の管理や事務に大きな影響を与えることができる.例えば、これらの株主が共同行動を選択した場合、彼らは取締役の選挙および私たちのすべてまたはほとんどの資産の任意の合併、合併、または売却の承認に大きな影響を与える可能性がある。これは私たちの株主がこれが彼らの最適な利益に合っていると思うかもしれないので、私たちの普通株に対する能動的な買収提案や要約を阻止または阻止する可能性がある。
107
デラウェア州の法律および私たちが改正して再記載した会社登録証明書、または当社の会社登録証明書、ならびに改正および再記載された定款または私たちの定款の条項は、合併、要約買収または代理権競争を困難にし、私たちの普通株の取引価格を低くする可能性がある。
会社登録証明書及び定款の規定は、株主がその株式によって割増される可能性のある取引を含む、実際又は潜在的な支配権変更又は管理層変更に係る取引を遅延又は阻止することができ、又は我々の株主がその最適な利益に適合すると考えられる取引を含むことができる。したがって、このような規定は私たちの普通株の価格に悪影響を及ぼすかもしれない。他の事項を除いて、私たちの会社の証明書と定款:
これらの条項の改正は、私たちの取締役会が優先株株を発行し、任意の権利、優遇、特権を指定する能力を除いて、私たちが当時発行した普通株の少なくとも66-2/3%の保有者の承認を得る必要がある。
さらに、デラウェア州会社として、私たちはデラウェア州会社法第203条の制約を受けている。これらの規定は、大株主、特に私たちが発行した議決権株の15%以上を有する株主が、一定期間内に私たちと合併または合併することを禁止することができる。デラウェア州の会社は宅配便で本条項を脱退することができます
108
元会社登録証明書における規定、又は株主が承認した会社登録証明書又は定款の改訂。しかし、私たちはこの条項から脱退することを選択しなかった。
当社の登録証明書、定款、およびデラウェア州法律のこれらおよび他の条項は、株主または潜在的な買収者が私たちの取締役会に対する支配権を得ることを困難にするか、またはわが社に関連する合併、要約買収、または代理権競争を延期または阻害することを含む、当時の取締役会の反対行動を開始する可能性があります。これらの条項の存在は、私たちの普通株の価格に悪影響を与え、会社の取引で価値を実現する機会を制限する可能性があります。
もし私たちが財務報告書に対して適切かつ有効な内部統制を維持できなければ、私たちが正確かつ適時に財務諸表を作成する能力は損害を受ける可能性があり、これは私たちの経営業績、投資家の私たちに対する見方を損なう可能性があり、それによって私たちの普通株の価値を損なう可能性がある。
サバンズ·オキシリー法404条または404条によると、私たちの経営陣は、財務報告書の内部統制に対する私たちの内部統制の有効性を毎年評価して報告し、財務報告の内部統制に対する私たちの任意の重大な弱点を発見しなければならない。JOBS法案で定義されている新興成長型会社の資格を満たしておらず,大型加速申告会社となっているため,404(B)条の監査人認証要求などの要求も遵守しなければならない。このような認証報告書を準備し、私たちが以前実施していなかった報告書要求を遵守するコストを準備し、私たちの費用と大量の管理時間を増加させ続ける。投資家たちは追加的なコンプライアンスコストのため、私たちの普通株がそんなに魅力的ではないことを発見するかもしれない。したがって、一部の投資家が私たちの普通株の吸引力が低下していることを発見すれば、私たちの普通株はそれほど活発ではない取引市場がある可能性があり、私たちの株価はもっと変動するかもしれない。
経営陣と私たちの独立公認会計士事務所は、財務報告の内部統制に達成しなければならない基準を評価するルールが複雑で、大量の文書、テスト、可能な救済措置が必要です。私たちと私たちの独立公認会計士事務所の財務報告の内部統制の評価については、情報技術を含め、追加の財務·管理制御、報告システムおよびプログラムを実施し、追加の会計·財務担当者を招聘するシステムをアップグレードする必要があるかもしれません。
必要な新しい制御措置や改善された制御措置を実施できなかったり、実行中に遭遇した困難は、私たちの報告義務を履行できない可能性があります。さらに、私たちまたは私たちの独立公認会計士事務所が第404条に基づいて行った任意のテストは、財務報告の内部統制に存在する欠陥を明らかにすることができ、これらの欠陥は、重大な弱点と考えられているか、または私たちの財務諸表を前向きまたは追跡的に変更する必要がある場合があり、またはさらなる関心または改善が必要な他の分野を発見することができる。悪い内部統制はまた、投資家が私たちが報告した財務情報に自信を失う可能性があり、これは私たちの普通株の取引価格にマイナス影響を与える可能性がある。内部統制の欠陥はまた私たちが未来に財政的業績を再説明することにつながるかもしれない。私たちは株主または他の第三者訴訟の対象となる可能性があり、米国証券取引委員会、ナスダック世界ベスト市場または他の規制機関の調査は、追加の財務および管理資源を必要とし、罰金、停止、損害賠償の支払い、または他の救済措置を招く可能性がある。さらに、第404条の監査人認証条項の遵守を遅延させることは、短い転売登録の資格の廃止、米国証券取引委員会の行動、および私たちの普通株が一時停止または退市されることを含む様々な行政制裁を受ける可能性があり、これは、私たちの普通株の取引価格を低下させ、私たちの業務を損なう可能性がある。
私たちの規約では、デラウェア州衡平裁判所とアメリカ連邦地域裁判所は、私たちと株主との間のほとんどの紛争の独占法廷となり、これは、私たちまたは私たちの役員、役員、または従業員との紛争において有利な司法フォーラムを獲得する私たちの株主の能力を制限するかもしれません。
私たちの別例では、法律で許可された最大範囲内で、裁判所が被告とされる不可欠な当事者に対して個人管轄権を有する場合、デラウェア州衡平裁判所は、デラウェア州成文法または一般法に基づいて提起された以下のタイプの訴訟または手続の独占裁判所である
109
この規定は、取引法で規定されている義務または責任を執行するための訴訟には適用されない。また、証券法第22条は、連邦裁判所と州裁判所は、このようなすべての“証券法”訴訟に対して同時に管轄権を持っていると規定している。したがって、州裁判所と連邦裁判所はこのようなクレームを受理する管轄権を持っている。複数の司法管轄区で訴訟を提起せざるを得ないことや、異なる裁判所が不一致や逆の裁決を下す脅威などの考慮要因を避けるために、わが社の登録証明書はさらに、米国連邦地域裁判所は、証券法に基づいて提出された任意の訴因を解決する独占的なフォーラムとなると規定している。デラウェア州裁判所は、このような選択の裁判所条項が事実上有効であることを決定しているが、株主は依然として専属裁判所条項が指定された場所以外の場所でクレームを出すことを求めることができる。このような状況の中で、当社の登録証明書の独占フォーラム条項の有効性と実行可能性を強く主張する予定です。これは、他の法ドメインでこのような訴訟を解決することに関連する多くの追加費用を必要とする可能性があり、これらの規定がこれらの他の法ドメインの裁判所によって実行されることを保証することはできない。
これらの排他的フォーラム条項は、司法フォーラムにおいて、私たちまたは私たちの役員、役員、または他の従業員との紛争に有利であると考える株主のクレームを提出する能力を制限する可能性があり、これは、私たちと私たちの役員、役員、および他の従業員に対する訴訟を阻止するかもしれない。もし裁判所がわが社の登録証明書のいずれかの排他的な裁判所条項が訴訟で適用されないか、または実行できないことを発見した場合、私たちは他の管轄区域での紛争解決に関連するさらなる重大な追加費用が生じる可能性があり、これらすべては私たちの業務を深刻に損なう可能性がある。
将来的に私たちの株主の所有率をさらに希釈し、私たちの株式インセンティブ計画に基づいて、私たちの株主の所有率をさらに希釈し、株価を下落させる可能性があることを含む、私たちの普通株を売却して発行する権利、または普通株を購入する権利。
将来的には、臨床試験、商業化努力(任意のAMX 0035または任意の他の現在または将来の候補製品のマーケティング承認を得ることができれば)、研究および開発活動、上場企業の運営に関連するコストなど、私たちの計画中の業務を継続するために多くの追加資本が必要になると予想される。資本を調達するために、私たちは1回または複数回の取引で時々決定された価格および方法で普通株、転換可能証券、または他の株式証券を販売することができる。もし私たちが普通株、転換可能証券、または他の株式証券を販売する場合、投資家はその後の売却によって深刻に希釈される可能性がある。このような売却はまた、私たちの既存株主の実質的な希釈をもたらす可能性があり、新しい投資家は、私たちの公開発行で販売された普通株を含む、私たちの普通株式保有者よりも優先的な権利、優遇、および特権を得ることができる。
私たちの2022年株式オプションとインセンティブ計画または2022年計画によると、私たちの経営陣は、従業員、取締役、コンサルタントに株式オプションを付与することを許可されています。また、2022年計画によると、発行のために予約された普通株式数は、2023年1月1日から2032年1月1日(2032年1月1日を含む)まで、前年の12月31日に発行された普通株式総数の5.0%または取締役会が決定したより少ない数の普通株式を自動的に増加させる。また、我々のESPPによると、2023年1月1日(2032年1月1日まで)から、発行のために予約された普通株式数は、例年の1月1日に自動的に増加し、(I)自動増持日前のカレンダー月最終日に発行された普通株式総数の1.0%と、(Ii)1,210,000株とに増幅され、任意の増資日前に、増加した普通株式数が第(I)及び(Ii)項に規定された金額よりも少なくなることを決定することが可能である。私たちの取締役会が毎年将来付与可能な株式数を増加させないことを選択しない限り、私たちの株主は追加的な希釈を経験する可能性があり、これは私たちの株価を下落させる可能性がある。
一般リスク因子
上場企業として、私たちの運営コストは高く、私たちの経営陣はコンプライアンスイニシアチブに多くの時間を投入する必要があります。
上場企業として、私たちは大量で持続的な法律、会計、その他の費用を発生させ、特に今は新興成長型会社ではなくなりました。私たちは取引法の報告要求を受けて、私たちの業務と財務状況に関する年間、四半期、現在の報告書をアメリカ証券取引委員会に提出することを要求しています。また、サバンズ-オクスリ法案および米国証券取引委員会とナスダック世界精選市場が後にサバンズ-オクスリ法案の条項を実施するために採択された規則は、有効な情報開示と財務制御の維持を要求し、コーポレートガバナンスのやり方を変更することを含む上場企業に重大な要求を出した。さらに、2010年7月、ドッド·フランクウォールストリート改革と消費者保護法案、あるいはドッド·フランク法案と呼ばれ、
110
制定する。テレス·フランク法案には重要な会社管理と役員報酬に関する条項があり、米国証券取引委員会はこれらの分野で“報酬発言権”や代理アクセスのような追加的な規則を取ることを要求している。株主急進主義、現在の政治環境、および現在の高度な政府介入と規制改革は、大量の新しい法規と開示義務を招く可能性があり、これは追加のコンプライアンスコストを招き、現在予見できない方法で業務を運営する方法に影響を与える可能性がある。
また、私たちはもはや新興成長型企業ではないため、上場企業の様々な報告要求に適した特定の免除を再利用しない可能性がある。その報告書の要求の増加は私たちのコンプライアンス負担をさらに増加させるだろう。上場企業に適用されるルールや法規を遵守するために、巨額のコストが発生し続けると予想しています。これらの要求が私たちの経営陣と従業員の注意を他の業務から移すと、それらは私たちの業務、財務状況、運営結果に実質的な悪影響を及ぼす可能性がある。増加したコストは私たちの純収益を減少させ、あるいは私たちの純損失を増加させ、他の業務分野のコストを下げたり、私たちの製品やサービスの価格を向上させることを要求するかもしれません。私たちは、これらの要求に応答して生成される可能性のある追加コストの金額や時間を予測または推定することができない。これらの要求の影響は、私たちの取締役会、取締役会委員会、または役員に参加することをより難しくし、合格した人を引き付け、維持することを可能にするかもしれない。
当社の電気通信またはITシステム、または当社の協力者、CRO、サードパーティ物流プロバイダ、販売業者または他の請負業者またはコンサルタントのネットワーク攻撃または他の障害は、情報の盗難、データ破損、および当社のビジネス運営の深刻な中断をもたらす可能性があります。
私たち、私たちの協力者、当社のCRO、サードパーティ物流プロバイダ、流通業者、および他の請負業者およびコンサルタントは、ITシステムおよびネットワークを使用して、私たちのビジネス活動に関連する電子情報を処理し、送信し、保存します。デジタル技術使用の増加に伴い、第三者が盗難または推定された証拠、コンピュータマルウェア、ウイルス、社会工学、迷惑メール、または他の手段を使用して従業員アカウントにアクセスすること、およびコンピュータシステムおよびネットワークへの意図的な攻撃および不正アクセスを試みることを含む、ネットワークイベントの頻度および複雑性が一般的に増加する。ネットワーク攻撃はまた、支払いまたは情報が予期されない受信者に送信されるようなネットワーク釣りまたは電子メール詐欺を含むことができる。これらの脅威は、私たち、私たちのパートナー、私たちのCRO、第三者物流プロバイダ、流通業者、および他の請負業者およびコンサルタントのシステムおよびネットワークのセキュリティ、ならびに私たちのデータの機密性、可用性、および完全性に危険をもたらします。私たちがサイバー攻撃を防ぐことに成功したり、その影響を軽減することに成功する保証はない。同様に、私たちの協力者、CRO、第三者物流提供者、流通業者、および他の請負業者およびコンサルタントが、彼らのシステムに格納されている、または彼らがアクセスする権利のある私たちの臨床および他のデータを保護することに成功することは保証されません。いかなるネットワーク攻撃、データ漏洩、セキュリティ事件またはデータ破壊、乱用または損失は、適用される米国と国際プライバシー、データ保護およびその他の法律に違反する可能性があり、私たちにアメリカ連邦、州と地方監督管理エンティティおよび国際規制エンティティの訴訟と政府の調査と訴訟を受け、それによって重大な民事および/または刑事責任に直面させる。さらに、私たちの一般責任保険および会社リスク計画は、私たちが直面しているすべての潜在的なクレームを含まない可能性があり、私たちが適用する可能性のあるすべての責任を賠償するのに十分ではなく、私たちの業務や将来性に実質的な悪影響を及ぼす可能性があります。例えば、AMX 0035または私たちの将来の候補製品の完了、進行中、または将来の臨床試験の臨床試験データの損失は、我々の開発および規制承認作業の遅延をもたらす可能性があり、データを回復または複製するコストを著しく増加させる可能性がある。さらに、ネットワーク攻撃または他のデータセキュリティホールまたは事件によって名声損害または訴訟または不利に直面する規制行動を受ける可能性があり、さらなるデータ保護または救済措置の実施、罰金および処罰または他の責任、および既存および将来の業務の損失を含む名声損害および重大な追加費用を招く可能性がある。
私たちは純営業損失と研究開発控除を使って将来の課税収入を相殺する能力が一定の制限を受ける可能性があります。
2023年12月31日現在、6980万ドルのアメリカ連邦純運営損失があり、無期限に繰り越しています。2017年の減税や雇用法案(TCJA)の規定により、これらのNOL繰り越しの年間使用量が制限される可能性がある。2023年12月31日まで、私たちは680万ドルのアメリカ連邦研究開発税収の繰越免除があります。私たちはまたアメリカ州NOLの繰延納税資産と研究開発税収の繰延980万ドルを記録しました。これらの米国連邦研究開発税控除と米国州繰越がそれぞれ2042年と2035年に使用されなければ、満期が始まる可能性がある。すべてのNOLと研究開発税収の繰越免除の使用は私たちがアメリカ連邦と州課税収入を生成することを条件としている。
所有権変更は2016年12月31日と2023年12月31日までの年度に発生した。一般的に、1986年に改正された国内税法第382節および383節、またはIRC、および州法の該当条項によると、所有権変更を経験した会社は、変更前の純資産または税収控除繰り越しを利用して将来の課税所得額を相殺する能力が制限される。これらの目的のために、所有権変更は、一般に、会社の株式の少なくとも5%を所有する1つまたは複数の株主または株主集団の総株式所有権で発生する
111
指定されたテスト期間内に、株式の保有量はその最低持株割合より50ポイント以上増加した。未来の私たちの株式所有権の変化は、その多くは私たちがコントロールできるものではなく、IRC第382条と383条による所有権の変化を招く可能性がある。私たちの既存の連邦と州NOLおよび研究開発税収の繰越免除はこれらの未来の所有権変更の制限を受けるかもしれない。したがって、私たちはこのような転換の実質的な部分を利用できないかもしれない。以下に述べるように、私たちは私たちのすべてのアメリカ繰延税金資産に対して全額推定準備金を維持します。私たちは短期的にすべてまたは一部の推定手当を支給するかもしれないが、推定手当の支給および支給の正確な時間と金額は、依然として私たちの利益水準、収入の増加、臨床プロジェクトの進展、将来の収益性への期待などの要素の影響を受ける。
私たちは証券集団訴訟の影響を受けており、将来的にはより多くの証券集団訴訟の影響を受ける可能性がある。
従来、証券集団訴訟は、ある会社の証券市場価格が下落した後に提起されることが多かった。このリスクは製薬会社が近年大幅な株価変動を経験しているため、私たちに特に関連している。このような訴訟は巨額のコストを招き、経営陣の注意力や資源を移転させる可能性があり、これは私たちの業務を損なう可能性がある。詳しくは“プロジェクト3.--法的訴訟”を参照されたい
もし私たちがナスダックの持続的な上場の要求を満たすことができなければ、私たちの普通株はカードを取られるかもしれない。
もし私たちがナスダックの全世界の精選市場の持続的な上場要求、例えば会社の管理要求や最低終値要求を満たすことができなければ、ナスダックは措置を取って、私たちの普通株を撤退させるかもしれない。このような退市は、私たちの普通株の価格にマイナス影響を与える可能性があり、私たちの普通株の売却または購入を希望する時に私たちの普通株を売却または購入する能力を弱める可能性があります。退市事件では、上場要求を遵守するための私たちのいかなる行動も、私たちの普通株の再上場を許可し、市場価格を安定させたり、私たちの普通株の流動性を高めたり、私たちの普通株が最低入札価格要求以下に下落することを防止したり、将来ナスダック世界の精選市場の上場要求に合わないことを防止することを保証することはできません。
もし証券アナリストが私たちの株に対する否定的な評価を発表すれば、私たちの株価は下落するかもしれない。
私たちの普通株の取引市場は、業界または証券アナリストが発表した私たちまたは私たちの業務に関する研究と報告にある程度依存している。もし私たちの一人以上のアナリストが私たちの会社に否定的な意見を発表することができれば、私たちの株価は下落するかもしれません。もしこれらのアナリストのうちの1人以上が私たちの研究報告を停止したり、私たちに関する報告書を定期的に発表できなかったりすれば、私たちは金融市場で可視性を失う可能性があり、これは逆に私たちの株価や取引量を低下させる可能性がある。
112
項目1 B未解決の従業員のコメント。
ない。
プロジェクト1 Cネットワークセキュリティです。
リスク管理と戦略
私たちは一般市場と私たちの産業で、サイバーセキュリティ脅威の数と深刻さが増加していることを認識している。これらの脅威に対応するために,我々はネットワークセキュリティリスク管理戦略を堅持し,我々の業務が直面しているネットワークセキュリティリスクを識別,評価,管理することを目的としている.我々のネットワークセキュリティリスク管理戦略は、様々な政策と構成要素を含み、ネットワークセキュリティ評価、イベント応答計画、私たちの主要なサプライヤーのセキュリティ実践の評価、および私たち従業員に対するネットワークセキュリティ意識訓練を含む。また、警報および監視ツールを含む第三者技術およびセキュリティツールおよび解決策を利用して、ネットワークセキュリティ計画をサポートします。
我々は第三者を招いて毎年ネットワークセキュリティリスク評価を行い,この評価は国家標準と技術研究所(NIST)ネットワークセキュリティフレームワークが情報を提供する.私たちはすでに私たちのITセキュリティチームのためのプロセスを構築し、ネットワークセキュリティリスク登録簿を介して既知のITセキュリティリスクと私たちの救済努力を追跡し、定量化した。ITセキュリティチームは定期的に会議を行い,組織全体のフィードバックと我々NISTが提供する年間ネットワークセキュリティリスク評価に含まれる結果に基づいて,ネットワークセキュリティリスク登録簿を審査·更新する.ITセキュリティチームは少なくとも毎年実行リーダーチームと取締役会に調査結果を報告している。
我々は、入社前に主要なソフトウェアサプライヤーのセキュリティ慣行を審査し、評価するプロセスを構築しており、ネットワークセキュリティアンケートおよびセキュリティ監査報告および認証に対するサプライヤーの回答(状況に応じて決定される)を含む。私たちの流れには、データ保護保障を維持するためにデータを処理する主要サプライヤーの契約要件も含まれています。
私たちは職員たちに入社中に提供する安全意識訓練プログラムを提供する。私たちはまた年間を通じてインターネット釣り訓練を含む追加的な義務訓練を提供する。
私たちは私たちの業務に関連した多くのネットワークセキュリティの危険に直面している。このようなリスクは、私たちの業務戦略、運営結果、または財務状況に大きな影響を与えていないにもかかわらず、私たちの業務戦略、運営結果、または財務状況に大きな影響を与える可能性は低いと考えられていますが、これまで、私たちおよび第三者サプライヤーの情報システムに関連する脅威やセキュリティ事件にしばしば遭遇してきました。我々が直面しているネットワークセキュリティリスクに関するより多くの情報は、項目1 A-リスク要因の“当社の電気通信またはITシステム、または私たちの協力者、CRO、第三者物流プロバイダ、販売業者または他の請負業者またはコンサルタントのネットワーク攻撃または他の障害、情報の盗難、データ破損、および業務運営の深刻な中断をもたらす可能性がある”と題するリスク要因を参照してください。
ネットワークセキュリティリスクを管理する
私たちの取締役会はネットワークセキュリティリスクの全面的な監督を担当し、少なくとも毎年私たちの監査委員会と執行指導チームの関連メンバーがサイバーセキュリティプロセスの鍵となる更新を通報してくれます。
私たちの監査委員会と私たちの実行指導チームのメンバーは四半期ごとに私たちのグローバル情報技術責任者および私たちのITセキュリティチームの他のメンバーと定期的に会い、新たに出現したサイバーセキュリティ脅威構造、私たちのサイバーセキュリティプロセスの重大な発展、そして私たちのネットワークセキュリティリスク評価のようなネットワークセキュリティ問題を討論します。
我々のITセキュリティチームは全世界の情報セキュリティ、管理とアーキテクチャ担当者(“グローバルISGA担当者”)が指導し、組織の日常情報セキュリティ戦略を管理と指導し、私たちのネットワークセキュリティツール、制御と戦略を監督し、組織の資産、ネットワークとデータを保護することを含む。グローバルISGA担当者は我々のグローバル情報技術担当者に仕事を報告した。グローバルISGA担当者は定期的にグローバル情報技術担当者にネットワークセキュリティリスク,プロジェクト,イニシアティブを報告し,後者は定期的に上記事項について実行管理層と監査委員会に報告する。
このグローバルISGA担当者は認証情報システムセキュリティ専門家証明書を持ち,約20年間のITセキュリティ管理経験を持っている.ITセキュリティチームは外部からサポートされています
113
必要に応じてネットワークサポート、セキュリティ運営、他のIT分野にホストサービスを提供するサプライヤー。私たちのITセキュリティチームはまた、個人データの保護に関するネットワークセキュリティイニシアティブと戦略を調整するために、当社の企業データ保護計画を監督する当社のグローバルプライバシー委員会と定期的に面会しています。
第二項です財産です。
2023年12月31日現在、私たちの主要物件の詳細は以下の通りです
属性説明 |
|
位置 |
|
正方形 |
|
|
属性 |
|
初期借約 |
|
事務スペース |
|
マサチューセッツ州カンブリッジ |
|
|
8,850 |
|
|
レンタルする |
|
2026年10月 |
事務スペース |
|
マサチューセッツ州カンブリッジ |
|
|
24,400 |
|
|
レンタルする |
|
2025年7月 |
私たちの施設は私たちの現在の需要を満たすのに十分であり、必要であれば、私たちは適切な追加または代替空間を提供するだろうと信じている。
第三項です法律訴訟。
2024年2月9日、米国ニューヨーク南区地方裁判所は、私たちの会社と私たちの一部の現職および元官僚(ShihはAmylyx製薬会社らの事件を訴えた。案件番号1:24-CV-00988(“クレーム”)。施正栄の起訴状は、取引所法案第10(B)節及びその公布された第10 b-5条に違反したことを告発し、第20条(A)条に基づいて特定の現職及び前任者を告発し、告発された統制者であることを告発した。施正栄の起訴状によると、被告はRELYVRIOの商業結果と将来性について重大な虚偽と誤解性陳述をしたという。施正栄の訴状は、指定されていない損害賠償、利息、費用及び弁護士費、並びに裁判所が適切と考えている他の指定されていない救済を要求する。その会社は施正栄の苦情を強力に弁護しようとしている。
第四項です炭鉱の安全情報開示。
適用されません。
114
第II部
五番目です。登録者普通株、関連株主事項及び発行者が株式証券を購入する市場。
市場情報
我々の普通株は2022年1月7日にナスダック世界ベスト市場で取引を開始し、コードは“AMLX”である。その前に、私たちの普通株は市場を公開しなかった。
記録保持者
2024年2月12日現在、私たちは約21人の普通株式保有者がいる。一部の株式は“街道”名義で保有されているため、当該等の株式の実益所有者の数は知られていないか、または上記の数字に含まれている。この数の登録所有者は、その株式が他のエンティティが信託形式で保有する可能性のある株主も含まれていない。
配当政策
私たちは普通株の配当金を発表したり支払ったりしたことがなく、私たちは予測可能な未来にも私たちの普通株の配当金を発表したり支払わないと予想している。私たちは現在、私たちの未来の収益を維持するつもりで、もしあれば、私たちの業務の発展と成長に資金を提供します。
株式補償計画に基づいて発行された証券
表格10-K第5項に要求される株式報酬計画に関する資料は、本年度報告第3部第12項に組み込まれる。
発行者および関連購入者が株式証券を購入する
本年報で述べた期間、吾らは何の登録株も購入していない。
未登録証券販売
2023年12月31日までの年間で、未登録証券を発行または販売していません。
115
株式表現グラフ
次の図は我々の普通株の各時期における表現とナスダック総合指数とナスダックバイオテクノロジー指数の表現を比較した。この図は,2022年1月7日の終値後に我々の普通株およびナスダック総合指数とナスダックバイオテクノロジー指数への投資を100ドルと仮定し,再投資配当金(あれば)を仮定している。次の図に示す株価表現は,必ずしも将来の株価表現を示唆しているとは限らない.このグラフは“募集材料”ではなく、米国証券取引委員会の“届出”とはみなされず、証券法または取引法に従って提出された任意の文書に引用的に組み込まれることもなく、本文書の日付の前または後に作成されたものであっても、そのような文書中の任意の一般的な合併言語も考慮されない。
第六項です[保留されている]
116
第七項。経営陣の財務状況と経営結果の検討と分析。
以下の情報は,本年度報告における他の地方の総合財務情報とその付記とともに読まなければならない。
本議論および本年度報告の他の部分は、私たちの計画、目標、期待、および意図の陳述のようなリスクおよび不確実性に関する前向きな陳述を含む。本年度報告“リスク要因”の一部で述べられた要因を含む多くの要因の影響により、我々の実際の結果は、以下の議論および分析における前向き陳述に記載されているか、または示唆された結果とは大きく異なる可能性がある.
概要
Amylyx製薬会社は商業段階の生物技術会社であり、その使命は神経変性疾患による苦痛を終わらせることである。十数年来、著者らはずっと筋萎縮性側索硬化症と神経退行性疾患を研究し、そしてこれらの疾病の治療方法を変える方面で重大な進展を得た。
2013年の設立以来、私たちは神経変性疾患患者の需要解決に専念する研究段階会社から、複数の適応開発プロジェクトを持つ商業企業に転換した。
最初の商業製品AMX 0035(フェニルブタン酸ナトリウム)[鉛]タウリンとジオール[トルソ](米国ではRELYVRIOとも呼ばれ、カナダではALBRIOZAと呼ばれる)は私たちが知っている唯一のALS療法であり、同じ臨床試験で疾病の進展を遅らせることができ、機能独立を維持し、総生存期間を延長することを助けることが証明され、副作用の概況と経口投与の普遍的な耐性は良好である。AMX 0035は2022年10月にRELYVRIOとしてアメリカで商業発売され、2022年7月にカナダでALBRIOZA商業で発売された。RELYVRIOとALBRIOZAが発売されて以来、2023年12月31日までに、私たちはすでに4.03億ドルの製品純収入を生み出した。AMX 0035は,広く使用されているALS薬となる可能性があると信じ,症状管理を基準とした疾患から有意義な介入措置を有する疾患にALSを転換する機会を提供している。さらに、AMX 0035は、広範な神経変性疾患の治療パラダイムを変化させるために、単独でまたは他の療法と一緒に使用できることを意味する神経変性疾患の基礎療法になる可能性があると信じている。
我々はAMX 0035のメリットを世界の20万人以上のALS患者にもたらすことに取り組んでいる。私たちは、承認される可能性のある他の司法管轄区域でAMX 0035を商業化し、世界各地の重要な利害関係者と接触し、AMX 0035を獲得する機会を探るためのグローバルインフラを建設している。
我々は引き続きAMX 0035が筋萎縮性側索硬化症を治療する全世界フェニックス第3段階臨床試験に注目しており、これはアメリカとヨーロッパの臨床場所で行われた48週間の無作為、二重盲検、プラセボ対照試験であり、2024年第2四半期または前にTOPLINE結果を報告する予定である。フェニックスからのデータが支持されていれば,ALSの利点を証明した2つの臨床試験が初めてとなる。支持性の鳳凰データはAMX 0035の商業化発売とALS治療の転換をさらに加速させると信じている。
筋萎縮性側索硬化症に加えて,AMX 0035を用いた他の神経変性疾患の治療には強い科学的根拠があると考えられる。AMX 0035は小胞体ストレスとミトコンドリア機能障害を標的とすることによって神経変性を緩和または軽減することを目的としており、この2つの相互接続された中枢経路は神経変性を引き起こす。我々独自のPBとTursoの組み合わせとそのそれぞれの作用機序は,いずれかの作用機序のみに対する治療よりも神経変性を予防するために,異常細胞死に協同作用することができると信じている。われわれはPSPおよびWSにおけるAMX 0035の効果を評価するために臨床試験を積極的に進めている。
設立以来、私たちはほとんどの努力を研究開発、商業化前、商業化活動に投入し、管理と技術者の募集、資金の調達、臨床前研究と臨床試験のための材料の生産、インフラの建設を含めてこれらの活動を支援してきた。2023年12月31日現在、私たちは主に普通株の公開発行、優先株の私的売却、転換可能な手形、および最近それぞれ米国とカナダでRELYVRIOとALBRIOZAを販売する収入を通じて、私たちの運営に資金を提供しています。
2023年までに運営損失が発生し、2023年12月31日までに累計3.049億ドルの赤字が発生しました。これらの損失は主に研究や開発活動に関する費用によるものである
117
そして私たちの運営に関連する販売、一般、行政コスト。私たちは、私たちが承認した製品を製品販売、マーケティング、製造、販売に関する巨額の商業化費用が発生すると予想している。私たちは重大な損失を招くかもしれません。私たちの財務結果は、私たちがアメリカで成功したRELYVRIOの商業化に高度に依存します。私たちは引き続き巨額の費用を発生します。私たちは臨床前と臨床開発を通じてAMX 0035と他の現在または未来の候補製品を推進し、より多くの試験を確立し、開始し、より多くの臨床、科学、管理、行政の人員を募集し、規制部門の承認を求め、任意の承認された候補製品を商業化するからです。これまで、私たちのCROネットワークと他のコンサルタントの助けを借りて、私たちは主に内部でAMX 0035とAMX 0114を開発しました。これは研究開発支出の増加を招いたが、開発および製造過程を通じてAMX 0035およびAMX 0114を効率的に管理することができるようにした。
我々はまた、重大な法律、会計、投資家関係、その他の費用を含む上場企業の運営に関する追加コストを引き続き発生させる予定だ。したがって、私たちは私たちの持続的な運営を支援し、私たちの成長戦略を実施するために多くの追加資金が必要かもしれない。利益を維持するために製品販売から十分な収入を生み出すことができる前に、他社との潜在的な協力、特許使用料融資、または他の戦略取引を含む、株式、債務融資、または他の資本源を売却することで、当社の運営に資金を提供する予定です。私たちは必要な時に資金を集めることができず、これは私たちの財務状況や業務戦略を実行する能力に悪影響を及ぼす可能性がある。私たちは私たちの現在の業務計画が達成されることを保証することもできないし、必要であれば、私たちが受け入れられる条件で追加資金を提供するか、または全く保証できないという保証もない。
2023年12月31日現在、私たちは3億714億ドルの現金、現金等価物、短期投資を持っている。私たちは、AMX 0035がアメリカとカナダの商業販売で得た収入と、2023年12月31日までの既存の現金、現金等価物、および短期投資は、今年度の報告日から少なくとも1年間の予想される運営と資本支出需要を満たすのに十分であると信じている。私たちは間違っていることが証明される可能性があるという仮定に基づいてこの推定をして、私たちは予想よりも早く利用可能な資本資源を枯渇させるかもしれない。
マクロ経済要因の影響
AMX 0035および任意の将来の候補製品の開発は、任意の流行病または災害によって妨害され、将来的に実質的な悪影響を受ける可能性がある。また、ウクライナとイスラエルの持続的な衝突、および世界的な流行病や他の公衆衛生事件による経済的挑戦のような政治的不安定と衝突は、米国、ヨーロッパ、中東を含む様々な世界市場の経済不確実性を招き、商品価格の大幅な変動、信用と資本市場の不安定、およびサプライチェーンの中断を含む市場混乱を招き、これらは世界的に記録的なインフレを招いた。これらのグローバル経済状況がグローバル経済や資本市場にさらなる悪影響を与え、特にこのような状況が継続的または悪化すれば、我々の業務、財務状況および経営業績に実質的かつ不利な影響を与える可能性がある。
これまで、我々の業務は、これらのグローバル経済や地政学的状況の実質的な影響を受けていなかったにもかかわらず、私たちの業務が短期的かつ長期的にどの程度影響を受けるか、あるいはこのような不安定がどのような方法で私たちの業務や運営結果に影響を与える可能性があるかは予測できない。これらの市場の混乱の程度と持続時間は予測できないが、巨大かもしれないが、巨大かもしれない。このような中断のいずれもまた、本報告書に記載されている他のリスクの影響を拡大する可能性がある。
世界経済の不確実性による様々なリスクに関するより多くの情報は、本年度報告の“リスク要因”と題する部分を読んでください。
私たちの運営結果の構成要素は
製品収入、純額
AMX 0035は2022年6月、カナダ保健省がALBRIOZAを条件としてALSを治療するマーケティング許可を取得し、2022年7月にカナダ国内でALBRIOZAの商業販売を開始した。2022年9月、AMX 0035はFDAの承認を得てALSを治療するRELYVRIOとして、2022年10月にアメリカでRELYVRIOを発売した。この間に確認された製品収入純額は主にALBRIOZAとRELYVRIOがそれぞれカナダと米国で販売されている単位に関係している。
118
運営費
販売コスト
販売コストには、主にRELYVRIO、ALBRIOZAの製造に関連するコスト、およびいくつかの期間のコストが含まれる
世界のマクロ経済状況により、私たちのグローバルサプライチェーンネットワークはいくつかの中断と変動が発生する可能性があり、将来私たちは原材料と包装供給の中断、出荷遅延及び関連するコスト上昇に遭遇する可能性がある。
研究と開発費
研究と開発費用には主にAMX 0035研究と開発に関連するコストが含まれている。私たちは発生した費用に応じて研究と開発費用を支払う。これらの費用には
私たちが将来受け取る研究·開発活動のための商品やサービスのための前金は前払い費用として記録されています。これらの金額は、貨物が関連サービスを交付または提供する際に費用として確認されるか、または貨物が提供または提供されるサービスがもはや予期されなくなるまで確認される。
AMX 0035では,我々のいくつかの間接研究および開発費用は適応ごとに追跡されていない。従業員コストや施設(減価償却や他の間接コストを含む)を特定の適応に割り当てないが,これらのコストは複数の適応に配置されているため,単独では分類されていない。著者らは内部資源を用いて研究と発見を監督し、著者らの臨床前開発、技術開発、製造と臨床開発活動を管理する。これらの従業員は複数の適応にまたがって働いているため,適応別にコストを追跡しない。
研究開発活動は私たちのビジネスモデルの核心だ。AMX 0035のような臨床開発後期段階にある候補製品は、通常、後期臨床試験の規模および持続時間の増加および関連する製品製造費用に起因するAMX 0114のような臨床開発初期段階の製品よりも高い開発コストを有する。私たちは、最近と未来に私たちが計画した臨床開発活動のため、私たちの研究と開発費用は引き続き増加し、アメリカ、カナダ、その他任意のAMX 0035を承認する司法管轄区の商業化活動に資金を提供することを予想しています。この時点では正確には
119
AMX 0035および任意の将来の候補製品の臨床開発を完了するために必要な努力の性質、時間、およびコストを推定または理解する。以下の要因により,われわれの臨床開発コストは大きく異なる可能性がある
製品開発および商業化に関連する多くのリスクおよび不確実性のため、AMX 0035および任意の他の現在または将来の候補製品の開発および商業化は、非常に高い不確実性を有する:
120
AMX 0035または任意の将来の候補製品の開発に関連するこれらの変数のいずれかの結果の変化は、我々の候補製品開発に関連するコストおよび時間に大きな影響を与える可能性がある。適用されれば、私たちはAMX 0035または任意の将来の候補製品の規制承認を得るか、または維持することができないかもしれない。
販売、一般、行政費用
販売、一般と行政費用は主に行政、財務、販売、マーケティングと行政機能者の給料と関連費用を含む。販売、一般および行政費用は、特許および会社事務に関連する法律費用、会計、監査、税務および行政相談サービスの専門費用、会社保険費用、行政出張費用、販売およびマーケティング費用、情報技術、独立慈善基金への慈善寄付、施設に関連する費用および他の運営費用も含む。AMX 0035の持続的な研究と開発、AMX 0035の商業化を支援するために従業員をさらに増加させることに伴い、私たちの販売、一般、および管理費用は今後も増加すると予想される。また、より多くの会計、監査、法律、規制、コンプライアンス、および役員と役員の保険コスト、および上場企業に関連する投資家や広報費用が発生することを予想しています。我々はすでにカナダにおけるALBRIOZAのALS治療の発売許可とRELYVRIOの米国での成人ALS治療のマーケティング許可を得た。
その他の収入、純額
利子収入
利息収入には主に私たちの短期投資のプレミアム償却と割引の増加と、私たちの現金、現金等価物、短期投資から稼いだ利息収入が含まれています。
その他の費用、純額
他の費用を除いて、純額は主に外国為替取引の実現されたと実現されていない純損失を含む。
所得税
所得税は貸借対照法を用いて決定される。繰延税項資産及び負債とは、財務諸表の帳簿額面と資産及び負債の計税基準との間の一時的な差異による将来の税項結果を指し、税項属性については、差異反転が予想される年間発効の制定税率を用いて繰り越す税項を指す。私たちの繰延税金資産の現金化は未来の課税収入の発生に依存しており、その金額と時間はまだ確定していない。既存の証拠の量に基づいて、繰延税金資産の一部または全部が現金化できない可能性が高い場合、推定免税額が提供される。2023年の間に、私たちの推定手当の一部は逆転され、今年度のアメリカ連邦と州課税所得額で達成された金額に関連している。経営陣の利用可能な証拠(運営中に大きな損失を受けた歴史を含む)のすべての評価によると、2023年12月31日現在、私たちはすべての米国繰延税金資産に対して全額評価準備金を維持し続けている。私たちのすべての既存の証拠に対する評価はまた、これらの製品販売による実際と予測収入を含む監督管理機関のALBRIOZAとRELYVRIOの許可と発展を考慮することを含む。私たちの製品の商業化はまだ初期段階にあることを考慮して、私たちは未来の販売の時間と数量を決定しない。私たちは短期的に残りの推定免税額の全部または一部を発行するかもしれませんが、推定免税額の発行と、発行の正確な時間と金額は、以下のような要素の影響を受けなければなりません
121
他の面では、私たちの利益水準、収入増加、臨床プロジェクトの進展、そして将来の収益性に対する期待。
2023年12月31日と2022年12月31日までに、それぞれ約6980万ドルと2.032億ドルのNOL繰り越しと、それぞれ約1兆246億ドルと1億641億ドルの州NOL繰り越しを持ち、これらの繰り越しは将来の課税所得額を減らすために使用できる。2023年12月31日現在、すべての米国連邦NOLは無期限繰り越しとなる。繰り越した1兆246億ドルの州純運営損失のうち、8280万ドルはマサチューセッツ州と関係があり、2035年に満期になる。2023年12月31日と2022年12月31日までに、それぞれ680万ドルと460万ドルの連邦税収免除、160万ドルと120万ドルの州税免除を受けた。繰越税金控除は2035年からの異なる日に満期になるだろう。
122
経営成果
2023年12月31日までと2022年12月31日までの年度比較
次の表は、2023年12月31日までと2022年12月31日までの年間経営結果をまとめています
|
|
十二月三十一日までの年度 |
|
|||||||||||||
|
|
2023 |
|
|
2022 |
|
|
$Change |
|
|
変更率 |
|
||||
|
|
(単位:千) |
|
|
|
|
||||||||||
製品収入、純額 |
|
$ |
380,786 |
|
|
$ |
22,230 |
|
|
$ |
358,556 |
|
|
|
1613 |
% |
運営費用: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
販売コスト |
|
|
25,441 |
|
|
|
2,993 |
|
|
|
22,448 |
|
|
|
750 |
% |
研究開発 |
|
|
128,187 |
|
|
|
93,450 |
|
|
|
34,737 |
|
|
|
37 |
% |
販売、一般、行政 |
|
|
188,356 |
|
|
|
127,128 |
|
|
|
61,228 |
|
|
|
48 |
% |
総運営費 |
|
|
341,984 |
|
|
|
223,571 |
|
|
|
118,413 |
|
|
|
53 |
% |
営業収入(赤字) |
|
|
38,802 |
|
|
|
(201,341 |
) |
|
|
240,143 |
|
|
|
(119 |
)% |
他の収入、純額: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
利子収入 |
|
|
16,155 |
|
|
|
4,291 |
|
|
|
11,864 |
|
|
|
276 |
% |
その他の費用、純額 |
|
|
(660 |
) |
|
|
(551 |
) |
|
|
(109 |
) |
|
|
20 |
% |
その他の収入合計,純額 |
|
|
15,495 |
|
|
|
3,740 |
|
|
|
11,755 |
|
|
|
314 |
% |
所得税前収入 |
|
|
54,297 |
|
|
|
(197,601 |
) |
|
|
251,898 |
|
|
|
-127 |
% |
所得税支給 |
|
|
5,026 |
|
|
|
774 |
|
|
|
4,252 |
|
|
|
549 |
% |
純収益(赤字) |
|
$ |
49,271 |
|
|
$ |
(198,375 |
) |
|
$ |
247,646 |
|
|
|
(125 |
)% |
*NM--意味がない
製品収入、純額
我々は2022年7月にカナダでALBRIOZAの販売を開始し、2022年10月に米国でRELYVRIOの販売を開始した。2023年12月31日と2022年12月31日までの年間で、それぞれ約3億808億ドルと2220万ドルの製品収入純額を記録した。この増加は主にRELYVRIOとALBRIOZAの2023年通年の販売と関係があるが、大部分は2022年第4四半期に販売されている。我々の収入確認政策のさらなる検討については、本年度報告に含まれる総合財務諸表付記2、重要会計政策の概要を参照されたい。
販売コスト
2023年12月31日までの1年間の販売コストは2540万ドルだったが、2022年12月31日までの年間販売コストは300万ドルだった。その間、販売コストには、私たちの市場製品RELYVRIOとALBRIOZAの調達、製造、流通のコストが含まれています。さらに、販売コストには、私たちの一時訪問または患者支援計画を介して特定の患者に無料で提供されているマーケティング製品を製造するコストが含まれています。患者に無料で提供される薬物製品は製品収入に含まれず,純額である。規制部門の承認前の製品製造に関する支出コストの政策によると、2023年12月31日と2022年12月31日までの年間で収入として確認された単位のいくつかのコスト、すなわちそれぞれ約1,120万ドルおよび340万ドルが、規制部門の承認を得る前に支出されているため、これらの期間の販売コストには含まれていない。私たちはこれらの在庫を消費するにつれて、販売コストが増加し、毛金利が下がると予想しています。2024年第2四半期に残りの商業化前の在庫を製品販売に使用する予定です。
123
研究と開発費
次の表は、2023年12月31日と2022年12月31日までの年間研究開発費をまとめています
|
|
十二月三十一日までの年度 |
|
|||||||||||||
|
|
2023 |
|
|
2022 |
|
|
$Change |
|
|
変更率 |
|
||||
|
|
(単位:千) |
|
|
|
|
|
|
|
|||||||
AMX 0035-ALS |
|
$ |
64,987 |
|
|
$ |
60,649 |
|
|
$ |
4,338 |
|
|
|
7 |
% |
AMX 0035-PSP |
|
|
5,495 |
|
|
|
93 |
|
|
|
5,402 |
|
|
|
5,809 |
% |
給与明細と人事関係の |
|
|
44,734 |
|
|
|
28,501 |
|
|
|
16,233 |
|
|
|
57 |
% |
他にも |
|
|
12,971 |
|
|
|
4,207 |
|
|
|
8,764 |
|
|
|
208 |
% |
|
|
$ |
128,187 |
|
|
$ |
93,450 |
|
|
$ |
34,737 |
|
|
|
37 |
% |
2023年12月31日までの年間研究開発費は1億282億ドルだったが、2022年12月31日までの年間は9350万ドルだった。この間,われわれの研究開発費の大部分はAMX 0035の開発と臨床試験に関与していた。3,470万ドルの増加は、主に、賃金および人員関連費用の1620万ドルの増加によるものであり、株ベースの報酬が420万ドル増加し、PSP治療のためのAMX 0035の支出が540万ドル増加し、筋萎縮性側索硬化症の治療のためのAMX 0035の支出が430万ドル増加し、他のすべてのコストが880万ドル増加したためである。賃金や人事に関する費用増加の要因は,研究·開発を支援する従業員数の増加である。PSPを治療するためのAMX 0035の支出が増加したのは、主にオリオン3期試験の開始をサポートする費用によるものである。筋萎縮性側索硬化症を治療するAMX 0035の支出増加は、主に臨床前開発活動の増加によるものであり、開放ラベル延長段階、および他のコストの増加を含む、我々の全世界第三段階フェニックス試験に関連するコストと関連している。今後しばらくは他の適応にAMX 0035の研究と開発が増加することが予想される。
販売、一般、行政費用
2023年12月31日までの1年間の販売、一般、行政費用は1兆884億ドルだったが、2022年12月31日までの年間は1億271億ドルだった。6120万ドル増加した主な理由は、株式ベースの報酬が1120万ドル増加し、コンサルティングおよび専門サービスが1610万ドル増加し、その他の費用が1490万ドル増加したことを含む賃金および人事関連費用が3020万ドル増加したことだ。賃金や人事関連コストの増加は、主にビジネス、一般、行政機能の面でより多くの人員を採用して私たちの成長を支援することと、EUでの商業化準備活動によるものである。コンサルティングや専門サービスやその他の費用の増加は,主に商業活動,上場企業運営,その他の費用の支出増加によるものである。
その他の収入、純額
利子収入
2023年12月31日までの年度の利息収入は1620万ドルであるのに対し、2022年12月31日までの年度の利息収入は430万ドルである。この成長は、主に有利な金利とより高い短期投資および現金等価物残高によるものであり、これは、2022年以降に発行された収益とAMX 0035の売却現金収入によって推進される。
所得税支給
2023年12月31日と2022年12月31日までの年度に、それぞれ500万ドルと80万ドルの所得税支出を記録した。2023年12月31日までの年間所得税の計上には、交付部分の推定免除額が含まれており、今年度の米国連邦と州の課税所得額で実現される予定の金額に関連している。本年度の米国連邦と州の課税収入はTCJA税法の変化の大きな影響を受け,この税法は2022年1月1日から施行され,IRC第174条に基づいてすべての研究や実験コストの資本化と償却を要求している。
124
流動性と資本資源
流動資金源
2022年下半期、私たちは承認された医薬製品RELYVRIO(カナダでALBRIOZAと呼ばれる)を販売することで収入を生成し始めた。今まで、私たちは主に私たちが承認した製品を販売し、普通株を売却し、発行し、転換可能な優先株と転換可能な手形の収入を通じて、私たちの運営に資金を提供してきました。2023年12月31日現在、私たちは3億714億ドルの現金、現金等価物、短期投資を持っている。
設立から2023年12月31日まで、私たちはすでに6.693億ドルの総収益を集め、発行コストを差し引くと、主に普通株の発行、転換可能な優先株、転換可能な手形、授与協定から来ている。私たちの現在の運営計画と仮定によると、私たちは、アメリカとカナダでAMX 0035商業販売から得た収入と、私たちの既存の現金、現金等価物、および短期投資は、本年度報告を提出した日後少なくとも12ヶ月の予想運営と資本支出需要を満たすのに十分であると信じている。私たちは間違っていることが証明される可能性があるという仮定に基づいてこれらの推定をして、私たちは期待よりも早く利用可能な資本資源を利用することができる。
資本資源
私たちは私たちが行っている活動に関連する費用が増加することを予想し、特に私たちはAMX 0035と他の現在または未来の候補製品の臨床前活動、製造、臨床試験を推進し、カナダでのALBRIOZAとアメリカのRELYVRIOの商業化計画を実行し、AMX 0035の他の管轄区での商業発売の準備をしている(承認されれば)。また、重大な法律、会計、投資家関係、その他の費用を含む上場企業の運営に関連する追加コストを予想しています。JOBS法案で定義されている新興成長型企業の資格に適合していないため、現在は大規模加速申告会社とみなされているため、404(B)条の監査役認証要件および削減報告要件を含む新興成長型会社に適用される特定のコンプライアンス要件の免除に依存する権利はなくなっている。私たちの支出も増えますなぜなら私たちは
125
候補製品やプロジェクトの研究,開発,商業化に関する多くのリスクと不確実性のため,我々の運営資金需要の正確な金額を見積もることはできない。私たちの将来の資金需要は多くの要素に依存し、それによって大幅に増加するかもしれない
利益を維持するのに十分な製品収入を生み出すことができる前に、株式発行、債務融資、政府または他の第三者資金、マーケティングおよび流通手配、その他の協力、戦略連合、許可手配によって、私たちの現金需要に融資することができるかもしれません。ある程度、私たちは普通株、転換可能証券、または他の株式証券を売却することで追加資本を調達し、現在の所有権権益は希釈される。もし私たちが第三者との協力やマーケティング、流通、または許可手配を通じてより多くの資金を調達すれば、私たちは私たちの技術、将来の収入源、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になる可能性のある条項でライセンスを授与しなければならないかもしれない。さらに、債務融資(実行可能であれば)は、固定支払義務をもたらす可能性があり、追加債務を招く、資本支出を招く、留置権を作成する、株式を償還する、または配当を宣言するなど、私たちの具体的な行動をとる能力を制限する制限的な契約を含むいくつかの合意に関連する可能性があり、これらは、私たちの業務を展開する能力に悪影響を及ぼす可能性がある。もし私たちが必要な時に追加資金を集めることができなければ、私たちは私たちの製品開発や将来の商業化努力を延期、制限、減少または終了することを要求されるかもしれません。あるいは私たちが自分で開発し、マーケティングすることをより望む候補製品の権利を与えることができます。
126
キャッシュフロー
2023年12月31日までと2022年12月31日までの年度比較
次の表は、2023年12月31日と2022年12月31日までの年間の現金源と用途をまとめています
|
|
十二月三十一日までの年度 |
|
|||||||||||||
|
|
2023 |
|
|
2022 |
|
|
$Change |
|
|
変更率 |
|
||||
|
|
(単位:千) |
|
|
|
|
||||||||||
経営活動提供の現金純額 |
|
$ |
11,919 |
|
|
$ |
(179,871 |
) |
|
$ |
191,790 |
|
|
|
(107 |
)% |
投資活動提供の現金純額 |
|
|
92,053 |
|
|
|
(238,988 |
) |
|
|
331,041 |
|
|
|
(139 |
)% |
融資活動が提供する現金純額 |
|
|
3,543 |
|
|
|
431,789 |
|
|
|
(428,246 |
) |
|
|
(99 |
)% |
現金·現金等価物に対する為替レート変動の影響 |
|
|
160 |
|
|
|
(65 |
) |
|
|
225 |
|
|
|
(346 |
)% |
現金、現金等価物、および限定的な現金の純増加 |
|
$ |
107,675 |
|
|
$ |
12,865 |
|
|
$ |
94,810 |
|
|
|
737 |
% |
経営活動
2023年12月31日までの1年間、経営活動は、主に4930万ドルの純収益、3720万ドルの非現金株式報酬支出、110万ドルの減価償却費用から1,190万ドルの現金を提供したが、私たちの運営資産と負債に使用された純現金は6570万ドル増加し、990万ドルの投資プレミアムと割引償却によって相殺された。
私たちの営業資産と負債に使用される現金純額は主に売掛金の2,160万ドルの増加、売掛金の1,590万ドルの増加、および経営リース使用権資産の180万ドルの減少を含む。これは在庫増加7 310万ドル、売掛金2 470万ドルの増加、前払い費用およびその他の流動資産の480万ドルの増加、および経営リース負債の200万ドルの減少によって相殺される。
2022年12月31日までの年間で、経営活動は1億799億ドルの現金を使用しており、主な理由は、私たちの純損失1.984億ドルと、210万ドルの投資割増と割引償却であるが、2170万ドルの非現金株式給与支出、50万ドルの減価償却費用、および私たちの運営資産と負債に使用されている純現金の160万ドルの増加によって相殺されている。
私たちが経営している資産や負債に使用されている現金純額には、主に計算費用の2610万ドルの増加と繰延レンタル料が含まれています。これは、私たちの増加を支持する外部研究·開発支出の増加、売掛金の190万ドルの増加、短期投資からの受取利息の50万ドルの減少によるものです。売掛金は1530万ドル増加し、在庫は980万ドル増加し、他の資産は50万ドル増加し、前払い費用とその他の流動資産は520万ドル増加し、この増加を相殺した。
投資活動
2023年12月31日までの年間で、投資活動が提供する現金純額が9210万ドルであったのは、期間満了の3.941億ドルの投資が短期投資を購入した3.008億ドルと不動産や設備を購入した120万ドルで相殺されたためである。
2022年12月31日までの年間で、投資活動のための現金純額は2.39億ドルで、うち250万ドルは財産と設備の購入、4.159億ドルは短期投資の購入に使用され、同期満期投資1億794億ドルの影響を相殺した。
融資活動
2023年12月31日までの年間で、融資活動が提供する現金純額は350万ドル。この額には700万ドルの株式オプション行使の収益が含まれており、330万ドルの株式奨励源泉徴収税と10万ドルの繰延発行コストによって相殺されている。
127
2022年12月31日までの年間で、融資活動が提供する現金純額は4.318億ドル。この金額には、私たちの初公募株(IPO)の2.09億ドルの収益(引受業者の割引と手数料を差し引く)、2022年に後続発行された収益2.316億ドル(引受業者の割引と手数料を差し引く)、株式オプションを行使する収益220万ドルが含まれ、280万ドルの繰延発行コストで相殺されている。
購入承諾s
私たちは正常な業務過程で契約製造組織と原材料調達と製造サービスについて合意しました。2023年12月31日現在、私たちはこれらの原材料調達と製造サービスに関する合意に基づいて約1.95億ドルを約束し、2028年までに支払う予定だ。
重要な会計政策と重大な判断と見積もり
私たちの総合財務諸表はアメリカ公認会計基準に基づいて作成されました。我々の連結財務諸表および関連開示を作成する際には、総合財務諸表における資産、負債、コストおよび費用に影響を及ぼす報告金額、または資産および負債の開示に影響を及ぼす推定および判断を行う必要がある。我々は,歴史的経験,既知の傾向や事件,および当時の状況で合理的と考えられる様々な他の要因に基づいて推定し,これらの要因の結果は資産や負債の帳簿価値の判断の基礎を構成しているが,これらの資産や負債の帳簿価値は他の源から容易に見られるものではない.私たちは持続的な基礎の上で私たちの推定と仮定を評価する。異なる仮定や条件の下で、私たちの実際の結果はこれらの推定とは異なる可能性がある。
我々の主な会計政策は、本年度報告書末の総合財務諸表付記2により詳細に記載されているが、以下の会計政策は、総合財務諸表の作成に使用される判断と推定に最も重要であると信じている。
収入確認
私たちの収入は会計政策が報告書の結果に大きな影響を与えることを確認し、いくつかの推定に依存している。収入は、ASCトピック606の次の5ステップモデルに従って確認される−取引先と契約した収入または主題606。収入もまた、以下に説明するいくつかの総額が純額またはGTN調整に関連する可変対価格によって減少する。これらのGTN調整は、歴史的経験、支払者チャネルの組み合わせ(例えば、MedicareまたはMedicaid)、適用計画下の現在の契約価格、未発行請求書のクレームおよび処理時間遅延、および流通チャネル内の在庫レベルを考慮した後に行われる重大な推定および判断に関する。推定数は期間ごとに評価を行い,必要に応じて情報や実際の経験を修正するように調整する.主題606によれば、顧客が私たちの製品に対する制御権を取得すると、ある時点(納入時)に発生する製品販売収入を確認する。製品収入は適用されたGTN調整後の純額を差し引いて割引と手当を含めています。お客様の支払いは通常領収書が発行された日から30日以内に支払わなければなりません。
これらの情報が利用可能な場合には,実際の活動に関する情報を含む新たな情報に基づいて我々のGTN推定を調整する.これまで,GTNの実際の活動は我々の推定と実質的な差はなかった。GTN調整の以下のカテゴリは、外部ソースから取得された重大な推定、判断、および情報に関する。
プロバイダはストレージ容量に応じて料金と割引を使用します
私たちはアメリカ退役軍人事務部などの政府実体と他の各方面(340 B薬品定価計画下の保証実体を含む)と共に計画に参加し、この計画によると、製品の定価は卸売業者価格よりも低く参加実体に拡張される。これらのエンティティは卸を通じてより低い計画価格で製品を購入し、卸売業者は私たちに彼らの調達コストとより低い計画価格との差額を受け取ってくれます。製品収入および売掛金から、販売による未処理引当請求に起因する推定金額を差し引く。
顧客に現金割引を提供して、彼らが適時に支払うことを奨励します。製品の収入と売掛金は販売時に推定された現金割引金額を引いて、割引は通常お客様が負担します
128
一ヶ月以内です。
支払人は返却します
私たちは州政府医療補助計画や他の条件を満たす連邦と州政府計画に参加し、参加する州と地方政府実体に割引とリベートを提供することを要求した。これらの計画を通じて提供されたすべての割引とリベートは私たちの医療補助リベート項目に含まれています。私たちのリベート計算は私たちがこのようなリベートの影響を受ける収入の大きさを推定することを要求する。我々の返却ポイント項目は,関連収入が確認された同期間に記録されており,製品収入の減少を招いている。未払いまたは未開のリベートの推定金額を負債として列記する。
返却点と割引はアメリカで管理されている医療機関に提供し、処方薬計画を管理し、Medicare Part D薬品福祉をカバーするMedicare Advantage処方薬計画を提供する。未払いまたは未開請求書のリベートと割引の推定金額を負債として列記する。
その他の奨励、返品、割引、調整
GTNの他の調整には、条件を満たす商業保険患者に経済的援助を提供し、支払者の要求に応じて処方薬を支払い共同で支払うことを目的とした自発的な患者支援計画、例えば私たちの自己援助計画が含まれている。援助対象費用の共同支払いの計算は、各報告期間の収入として確認されたクレーム推定数と、私たちが受信することが予想される製品に関する各クレームの費用に基づく。この等は、関連収入を確認する同一期間の入金を調整し、製品収入の減少及び流動負債の構築を招き、当該流動負債は支出及び他の流動負債の一部として総合貸借対照表に計上される。
既存製品の推定製品返品量は定量的と定性的情報を用いて決定され、これらの情報には返品の予想経験、予測された需要、流通ルートにおける在庫レベル、製品日付と有効期限、製品が生産停止されたかどうかなどが含まれているがこれらに限定されない。同社はこれまでわずかな返品を受けており、今後しばらくの製品返品はわずかになると信じている。
研究と開発費用を計算すべきである
連結財務諸表作成過程の一部として、我々が計上すべき研究·開発費用を見積もる必要がある。このプロセスは、未締結契約および調達注文を審査し、私たちの担当者とコミュニケーションして、私たちに代わって実行されるサービスを決定し、インボイスを受け取っていない場合、または他の方法で実際のコストを通知している場合に、実行されたサービスレベルおよびサービスによって生じる関連コストを推定することを含む。私たちのほとんどのサービスプロバイダは予定のスケジュールに従って、あるいは契約マイルストーンに達した時に私たちに借金の領収書を発行しますが、前金が必要なものもあります。私たちは、当時私たちが知っている事実と状況に基づいて、連結財務諸表において、各貸借対照表の日付までの課税費用を推定します。我々は定期的にサービスプロバイダとこれらの推定の正確性を確認し,必要に応じて調整する.
計算すべき研究および開発費用の推定は、CRO、CMOおよび他の第三者サービスプロバイダから受信されたタイムリーかつ正確な報告にある程度依存する。計算すべき研究および開発費用の推定例には、以下の者に支払われる費用が含まれる
私たちは、私たちを代表して臨床前研究と臨床試験を行って管理する複数のCMOとCROのオファーと契約に基づいて、受信したサービスと費用の努力を推定し、これらの見積もりと契約に基づいて臨床前研究と臨床試験に関連する費用を計算する。これらの合意の財務条項は交渉が必要であり、契約によって異なり、支払いの不均衡を招く可能性がある。場合によっては、私たちのサプライヤーに支払われるお金は、提供されたサービスレベルを超え、事前支払い費用につながる可能性があります。サービス料を計算する際に、サービスを提供する時間帯と各時間の努力度を見積もります。実際の時間を計算すれば
129
サービスパフォーマンスや努力程度が見積もり値と異なる場合は、それに応じて料金や前払い費用の金額を調整します。実際に発生した金額と実質的に異なることはないと予想されるが、提供されたサービスの実際の状態および時間に対する提供サービスの状態および時間の理解が異なる可能性があり、報告された金額が任意の特定の時期に高すぎたり、過小になったりする可能性がある。これまで,我々が計算した研究や開発費の先行推定には何の大きな調整もなかった.
所得税
私たちは貸借対照法を使用して所得税を計算する。繰延税項資産及び負債とは、財務諸表の帳簿額面と資産及び負債の計税基準との間の一時的な差異による将来の税項結果を指し、税項属性については、差異反転が予想される年間発効の制定税率を用いて繰り越す税項を指す。私たちの繰延税金資産の現金化は未来の課税収入の発生に依存しており、その金額と時間はまだ確定していない。既存の証拠の量に基づいて、繰延税金資産の一部または全部が現金化できない可能性が高い場合、推定免税額が提供される。2023年12月31日現在、経営陣の利用可能なすべての証拠(運営中に大きな損失を受けた歴史を含む)の評価に基づいて、私たちのすべてのアメリカ連邦と州繰延税金資産に対して全額推定手当を維持しています。我々のすべての既存の証拠の評価には、2023年にALBRIOZAとRELYVRIOを売却したことによる収入も考慮されている。私たちの製品発売の初期段階を考慮して、私たちは未来の販売の時間と金額を確定しません。これらの販売は持続的な収益力をもたらし、私たちの繰延税金資産の回収可能性に十分な積極的で客観的な証拠を提供します。私たちは短期的にすべてまたは一部の推定手当を支給するかもしれないが、推定手当の支給および支給の正確な時間と金額は、依然として私たちの利益水準、収入の増加、臨床プロジェクトの進展、将来の収益性への期待などの要素の影響を受ける。私たちは地域税務機関の所得税監査と調整を受けるかもしれない。不確定な税務状況の性質は経営陣の重大な判断に依存し、大きな変化が生じる可能性がある。我々は,内部専門知識と第三者専門家の支援を用いて,不確定な税収頭寸と関連する累積確率を評価した.より多くの情報を得るにつれて、修正されて改善されることが予想される。予想と最終決済の間に差が生じる可能性があり、追加の税金が発生する。
在庫品価格計算
コストまたは推定可能な純価値の中の低い者で在庫を推定する。私たちは先進的な先出しに基づいて在庫コストを決定し、材料と製造費用に関する金額を含む。在庫の消費または販売予想が12ヶ月を超える場合、在庫を長期在庫に分類します。我々は,報告期間ごとに資本化在庫の回収可能能力を評価し,任意の過剰および古い在庫を初回確認減値期間の推定可変動純値に減記した。この等減算費用が発生した場合は,販売コストに記入する.在庫コストが実現可能かどうかを決定するには、我々の製品の将来の需要、予想される将来の売上高、手元の貨物の残賞味期限、私たちの現在と未来の戦略計画など、管理層が見積もりを行う必要がある。
私たちの製品に対する実際の需要が低下したり、実際の市場状況が経営陣が予測しているほど有利でなければ、追加的に在庫を減記する必要があるかもしれません。これは総合経営報告書に販売コストと記入されます。しかも、私たちの製品は製造過程全体にわたって厳格な品質管理と監視を受けています。もしあるロットあるいは単位の製品が品質規格を満たしていなければ、販売コストを記録し、いかなる滞売在庫をその推定した可変現金値に減記する。
在庫減記を推定するための仮定は合理的であると考えられるが,これらの仮定の将来の重大な変化や将来のイベントや市場状況の変化が異なる見積りをもたらす可能性は保証されない.
最近発表された会計公告
最近発表された我々の財務状況及び経営結果に影響を与える可能性のある会計声明の記述は、我々の総合財務諸表の付記2に開示されている。
130
第七A項。市場リスクに関する定量的で定性的な開示。
市場リスクとは、我々の業務または既存または予想される金融取引に関連する市場変化に起因する可能性のある潜在的損失を意味する。我々は正常な業務過程で様々な市場リスクに直面しており,これらのリスクは以下のとおりである.
金利リスク
私たちは金利の変化と関連した市場リスクに直面している。2023年12月31日現在、私たちは3億714億ドルの現金、現金等価物、短期投資を持っている。私たちの現金等価物は主に銀行預金と通貨市場共同基金に投資される。私たちの市場リスクに対する主な開口は金利感受性であり、金利敏感性はアメリカの金利全体の水準変化の影響を受けている。私たちの投資は金利リスクの影響を受けており、市場金利が上昇すれば、私たちの投資は値下がりする可能性がある。私たちのポートフォリオの持続時間が長く、私たちの投資リスクが低いため、金利が直ちに100ベーシスポイント変化することは、私たちのポートフォリオの公平な市場価値に実質的な影響を与えないと思います。私たちは満期まで私たちの投資を持つことができますので、私たちの経営業績やキャッシュフローは市場金利の変化が私たちの投資に与える影響に大きな影響を受けないと予想しています。
外貨と貨幣の両替リスク
私たちは現在、外貨為替レートの変化に関する重大な市場リスクに直面していません。しかし、私たちはすでにアメリカ以外のサプライヤーと契約を締結し続ける可能性があります。そのため、私たちの業務は将来的に外貨為替レートの変動の影響を受ける可能性があります。また、貸借対照表の日には、海外子会社の資産と負債をそれぞれの機能通貨から適切なレートでドルに変換する。為替変動によるこれらの資産と負債の帳簿価値変動は、我々の総合貸借対照表の累計その他の全面収益(損失)に計上される。損益表勘定は年内の毎月平均為替レートで換算します。
インフレリスク
インフレは、2023年12月31日と2022年12月31日までの12ヶ月間、我々の業務、財務状況または運営結果に実質的な影響を与えないと考えられる。しかし、インフレは、適格な人材を誘致し、維持するための私たちの労働コストに影響を与え続ける可能性がある。
プロジェクト8それは.財務諸表と補足データ。
私たちの総合財務諸表は、私たちの独立公認会計士事務所の報告とともに、本年度報告のF-1ページから始まります。
プロジェクト9それは.会計や財務開示における会計士との変更と食い違い。
ない。
第9条。制御とプログラムです
情報開示制御とプログラムの評価
取引法の下でルール13 a-15(E)および15 d-15(E)で定義されているように、取引法に従って提出または提出された報告書に開示されなければならない情報が、米国証券取引委員会規則および表で指定された期間内に記録、処理、集約および報告され、これらの情報が、必要な開示に関する決定をタイムリーに行うために、我々の経営陣に蓄積され、伝達されることを目的としている。
我々の経営陣は、最高経営責任者と財務責任者の参加の下、2023年12月31日までの開示統制と手続きの有効性を評価した。対に基づいて
131
我々の開示制御及び手続は、2023年12月31日現在、我々の最高経営責任者及び最高財務官は、その日までに、我々の開示制御及び手続は合理的な保証レベルで有効であると結論した。
経営陣財務報告内部統制年次報告書
我々の経営陣は、取引法第13 a-15(F)および15 d-15(F)条で定義されているように、財務報告の十分な内部統制の確立と維持を担当している。財務報告の内部統制は、財務報告の信頼性を合理的に保証することを目的としたプログラムであり、公認された会計原則に基づいて外部目的の財務諸表を作成する。私たちの財務報告書に対する内部統制は以下の政策と手続きを含む
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来的にどのような有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムの遵守度が悪化する可能性がある.
当社の最高経営責任者と最高財務責任者の監督の下、トレデビル委員会後援組織委員会が発表した2013年の“内部統制-総合枠組み”における財務報告の有効な内部統制基準に基づいて、当社の財務報告の内部統制の有効性を評価した。このような評価に基づき、我々の経営陣は、2023年12月31日現在、財務報告に対して有効な内部統制を維持していると結論した。
財務報告の内部統制の変化
2023年12月31日まで、財務報告の内部統制に大きな影響を与えたり、合理的に財務報告の内部統制に大きな影響を与える可能性のある変化はありません。
132
独立公認会計士事務所報告
Amylyx製薬会社の株主と取締役会に。
財務報告の内部統制については
Amylyx製薬会社とその子会社(“当社”)の2023年12月31日までの財務報告内部統制を監査しました内部統制--統合フレームワーク(2013)テレデビル委員会(COSO)が主催して組織委員会が発表した。2023年12月31日現在、当社はすべての重要な面で財務報告に対する有効な内部統制を維持しており、その根拠は内部統制--統合フレームワーク(2013)COSOから発表されます。
また、米国上場企業会計監督委員会(PCAOB)の基準に従って、当社の2023年12月31日までおよび2023年12月31日までの年度の総合財務諸表および2024年2月22日までの報告を監査し、このような財務諸表に対して保留のない意見を表明した。
意見の基礎
当社経営陣は、効果的な財務報告内部統制を維持し、付随する経営陣財務報告内部統制年次報告に含まれる財務報告内部統制の有効性の評価を担当する。私たちの責任は私たちの監査に基づいて、会社の財務報告書の内部統制に意見を述べることです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務報告の有効な内部統制がすべての重要な面で維持されているかどうかを決定するために、合理的な保証を得るために監査を計画し、実行することを要求する。我々の監査には,財務報告の内部統制を理解すること,重大な弱点があるリスクを評価すること,評価されたリスクテストに基づいて内部統制の設計·運用有効性を評価すること,および状況下で必要と考えられる他のプロセスを実行することが含まれる。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
財務報告の内部統制の定義と限界
会社の財務報告に対する内部統制は、公認された会計原則に基づいて、財務報告の信頼性と外部目的の財務諸表の作成に合理的な保証を提供することを目的としたプログラムである。会社の財務報告に対する内部統制には、(1)合理的で詳細かつ正確かつ公平に会社の資産を反映した取引および処分に関する記録の保存、(2)公認された会計原則に従って財務諸表を作成するために必要な取引が記録されている合理的な保証を提供し、会社の収入および支出は会社の経営陣と取締役の許可のみに基づいて行われる。(三)財務諸表に重大な影響を及ぼす可能性のある不正買収、使用、または処分会社の資産を防止またはタイムリーに発見する合理的な保証を提供する。
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来的にどのような有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムの遵守度が悪化する可能性がある.
/s/徳勤法律事務所
ボストン、マサチューセッツ州
2024年2月22日
133
プロジェクト9 B。他の情報。
次の3ヶ月以内に2023年12月31日、以下の上級管理者または取締役(規定規則16 a-1(F)参照)は、適用計画の条項に基づいて、以下の普通株売却の取引計画を採択した。これらの計画は、取引法規第10 b 5-1(C)(1)の平権弁護条件を満たすことを目的としている
違います
プロジェクト9 Cそれは.検査を妨害する外国司法管轄区域を開示する。
適用されません。
134
第三部
第10項それは.役員、幹部、会社が管理する。
本第10項で要求される情報は、2024年株主総会に関する我々が米国証券取引委員会に提出した最終委託書の第1号提案であるコーポレートガバナンス及び役員部分に含まれ、参照により本明細書に組み込まれる。
プロジェクト11それは.役員報酬。
第11項に要求される情報は、2024年年次総会の最終委託書に関する取締役に提出された役員報酬と、米国証券取引委員会報酬部分(“報酬及び業績”というタイトルの下の情報を含まない)とを含み、参照により本明細書に組み込まれる。
プロジェクト12それは.いくつかの実益所有者の保証所有権及び管理層及び関連株主事項。
第12項に要求される情報は、2024年年次総会の最終委託書に関する米国証券取引委員会に提出されたいくつかの利益所有者の保証所有権および管理部分を含み、参照によって本明細書に組み込まれる。
第13項それは.特定の関係と関連取引、そして役員の独立性。
第13項で要求される情報は、2024年年次総会の最終委託書に関する我々が米国証券取引委員会に提出したいくつかの関係および関連先取引および会社管理部分を含み、参照によって本明細書に組み込まれる。
プロジェクト14それは.チーフ会計士料金とサービス料です。
私たちの独立会計士事務所は
第14項に要求される情報は、2024年年次総会に関する我々が米国証券取引委員会に提出した最終委託書に関する第2号提案部分に含まれ、引用により本明細書に組み込まれる。
135
第4部
プロジェクト15それは.展示品、財務諸表明細書。
本報告書に記載されている総合財務諸表一覧については、本年度報告F−1ページの総合財務諸表インデックスを参照し、本プロジェクトに参照して組み込まれている。
展示品 番号をつける |
|
説明する |
|
|
|
3.1 |
|
Amylyx PharmPharmticals,Inc.の4回目の改訂および再登録証明書(登録者を参照して2022年1月11日に証券取引委員会に提出された8−Kフォームの現在の報告書の添付ファイル3.1に組み込まれる)。 |
|
|
|
3.2 |
|
Amylyx製薬会社の規約は、2回目の改正と再改訂(登録者が2022年1月11日に証券取引委員会に提出した8−K表の現在の報告書の添付ファイル3.2を引用して編入された)。 |
|
|
|
4.1 |
|
普通株式証明書サンプル(2022年1月3日に米国証券取引委員会に提出された登録者登録説明書S−1/A表(文書番号333−261703)添付ファイル4.1参照)。 |
|
|
|
4.2 |
|
第2の改正および再署名された投資家権利協定は、2021年7月1日であり、登録者と協議者との間の合意(参照登録者が2021年12月16日に米国証券取引委員会に提出されたS-1表登録声明(文書番号333-261703)添付ファイル4.2によって成立する)。 |
|
|
|
4.3 |
|
証券説明(登録者により2022年3月31日に証券取引委員会に提出された10−K表添付ファイル4.3により法団として成立)。 |
|
|
|
10.1# |
|
2015年株式オプション及びインセンティブ計画及びその奨励協定のフォーマット(2022年1月3日に米国証券取引委員会に提出された登録者登録説明書S−1/A表(文書番号333−261703)添付ファイル10.1参照)。 |
|
|
|
10.2# |
|
2022年株式オプション及びインセンティブ計画及びその奨励協定のフォーマット(2022年1月3日に米国証券取引委員会に提出された登録者登録説明書S−1/A表(文書番号333−261703)添付ファイル10.2合併を参照)。 |
|
|
|
10.3# |
|
非従業員役員報酬政策(参照登録者が2023年3月21日に米国証券取引委員会に提出した8−K表の現在報告書の添付ファイル10.1により成立)。 |
|
|
|
10.4# |
|
役員現金インセンティブボーナス計画(2022年1月3日に米国証券取引委員会に提出された登録者登録説明書S−1/A表(文書番号333−261703)添付ファイル10.4参照)。 |
|
|
|
10.5# |
|
2022年従業員株式購入計画(2022年1月3日に米国証券取引委員会に提出された登録者登録説明書S-1/A表(文書番号333-261703)添付ファイル10.3参照)。 |
|
|
|
10.6# |
|
リース契約は,日付が2018年10月23日であり,登録者とBullfinch Square Limited Partnership(登録者が2021年12月16日に米国証券取引委員会に提出したS-1表登録声明(文書番号333-261703)添付ファイル10.6登録成立)によって改訂された. |
|
|
|
10.7# |
|
登録者とジョシュ·コーエンとの間の雇用契約表(参照登録者は2022年1月3日に米国証券取引委員会に提出された登録説明書S−1/A表(文書番号333−261703)添付ファイル10.7が法団として成立する)。 |
|
|
|
10.8# |
|
登録者とJustin Kleeとの間の雇用契約表(登録者が2022年1月3日に米国証券取引委員会に提出した登録者登録説明書S−1/A表(文書番号333−261703)添付ファイル10.8が法団として成立する)。 |
|
|
|
10.9# |
|
登録者とジェームズ·フレッツとの間の雇用契約表(登録者が2022年1月3日に米国証券取引委員会に提出した登録者登録説明書S−1/A表(書類番号333−261703)添付ファイル10.9が法団として成立する)。 |
|
|
|
136
10.10# |
|
登録者とマーガレット·オリンガーとの間の雇用契約表(登録者は2022年1月3日に米国証券取引委員会に提出された登録説明書S−1/A表(文書番号333−261703)添付ファイル10.10を法団に登録する)。 |
|
|
|
10.11# |
|
登録者とパトリック·D·エラミアン医学博士との間の雇用協議表(2022年1月3日に米国証券取引委員会に提出された登録者登録声明S−1/A表(文書番号333−261703)添付ファイル10.11を参照して編入)。 |
|
|
|
10.12# |
|
当社とパトリックYeramianとの間の雇用協定改正案は、2022年12月1日から施行される(登録者は2022年12月6日に米国証券取引委員会に提出された8-K表現在報告の添付ファイル10.1が法団として設立される)。 |
|
|
|
10.13#* |
|
当社とパトリック·Yeramianとの間の雇用協定改正案は、2023年11月27日から発効します。 |
|
|
|
10.14# |
|
登録者とGina Mazarielloとの間の雇用合意表(登録者が2023年3月13日に米国証券取引委員会に提出したForm 10−K年度報告の添付ファイル10.18が法団として設立された)。 |
|
|
|
10.15#* |
|
登録者とCamille Bedrosianの間の雇用協定形式。 |
|
|
|
10.16# |
|
上級者弁済協議表(参照登録者が2021年12月16日に証券取引委員会に提出した登録声明S−1表(文書番号333−261703)の添付ファイル10.13が法団として設立された)。 |
|
|
|
10.17#* |
|
登録者とマーガレット·オリンガーが2023年12月31日に締結した別居協定。 |
|
|
|
10.18 |
|
総製造サービス協定は、登録者とPatheon Inc.によって署名され、日付は2019年11月12日である(2021年12月16日に米国証券取引委員会に提出された登録者登録説明書S−1表(文書番号333−261703)添付ファイル10.14を参照して編入)。 |
|
|
|
10.19 |
|
登録者とPatheon Inc.との間で2019年11月12日に署名された2019年1月18日の製品協定の第1改正案によると、2019年11月12日の製品協定(登録者を引用して2023年5月11日に米国証券取引委員会に提出された10-Q表四半期報告書の添付ファイル10.2を統合した)。 |
|
|
|
10.20 |
|
第2修正案は、日付が2023年3月20日、製品協定、日付が2019年11月12日であり、登録者とPatheon Inc.との間の日付が2019年11月12日である製造サービス総合意(登録者が2023年5月11日に米国証券取引委員会に提出された10-Q表四半期報告書の添付ファイル10.3を引用して統合されたことにより)、2021年1月18日の改正改正案第1号を参照して改正される。 |
|
|
|
10.21 |
|
登録者がCU Chemie Utikon GmbHと締結した供給契約は、2019年10月29日(登録者が2021年12月16日に米国証券取引委員会に提出されたS-1表登録説明書添付ファイル10.15(文書番号333-261703))である。 |
|
|
|
10.22 |
|
第1修正案は、2023年1月1日に施行され、登録者とCU Chemie Uentikon GmbHとの間の供給協定により、2019年10月29日となる(登録者は2023年5月11日に米国証券取引委員会に提出された10-Q表四半期報告書の添付ファイル10.4が法団として設立される)。 |
|
|
|
10.23 |
|
登録者はICE S.p.A.(前身はProdotti Chimici e Alimentari S.p.A.)と締結され,2021年7月26日に署名され,2019年12月9日の研究,開発·供給協定および日付が2021年7月26日の改訂書である(2021年12月16日に米国証券取引委員会に提出された登録者登録説明書S−1表(第333−261703号文書)添付ファイル10.16を参照して統合される)。 |
|
|
|
10.24 |
|
商業供給契約は,2023年8月8日に登録者とICE S.p.A.(前身はProdotti Chimici e Alimentari S.p.A.)によって締結され,ICE S.p.A.の間で締結される。(登録者が2023年8月10日に証券取引委員会に提出したForm 10-Q四半期報告の添付ファイル10.1を参照)。 |
|
|
|
21.1* |
|
登録者子会社リスト。 |
|
|
|
23.1* |
|
独立公認会計士事務所徳勤会計士事務所の同意を得ました。 |
|
|
|
31.1* |
|
2002年サバンズ-オキシリー法第302節に基づいて可決された1934年“証券取引法”第13 a-14(A)及び15 d-14(A)条に基づいて認証連席首席執行幹事。 |
|
|
|
31.2* |
|
2002年サバンズ-オキシリー法第302節に基づいて可決された1934年“証券取引法”第13 a-14(A)及び15 d-14(A)条に基づいて認証連席首席執行幹事。 |
|
|
|
31.3* |
|
2002年サバンズ−オキシリー法第302節で可決された1934年証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて首席財務幹事が認証された。 |
|
|
|
137
32.1+ |
|
2002年の“サバンズ-オックススリー法案”906節で採択された“アメリカ法典”第18編1350節連合最高経営責任者証明書による。 |
|
|
|
32.2+ |
|
2002年の“サバンズ-オックススリー法案”906節で採択された“アメリカ法典”第18編1350節連合最高経営責任者証明書による。 |
|
|
|
32.3+ |
|
2002年の“サバンズ-オックススリー法案”906節で採択された“米国法典”第18編1350条による首席財務官の認証。 |
|
|
|
97.1* |
|
賠償追討政策 |
|
|
|
101.INS* |
|
連結されたXBRLインスタンス文書−インスタンス文書は、XBRLタグがイントラネットXBRL文書に埋め込まれているので、相互作用データファイルには表示されない。 |
|
|
|
101.Sch* |
|
Linkbase文書を組み込んだインラインXBRL分類拡張アーキテクチャ |
|
|
|
104 |
|
表紙インタラクションデータファイル(イントラネットXBRL文書に埋め込む) |
*アーカイブをお送りします。
+ 手紙で提供する。 改正された1934年の証券取引法第18条の規定によると、本証明書は提出されたものとみなされず、当該条項の責任にも拘束されない。このような証明は、引用によってそのような出願が明示的に組み込まれない限り、改正された1933年の証券法に基づいて提出されたいかなる出願にも組み込まれているとはみなされない。
#は、管理契約または任意の補償計画、契約、またはスケジュールを表します。
S-K法規第601(B)(10)項によれば、本展示品の部分(星番号で示す)は省略されている。
必要または適用されないので、必要な資料が連結財務諸表またはその付記に含まれているので、いかなる財務諸表も提出されていない。
プロジェクト16それは.表格10-Kの概要
ない。
138
サイン
改正された1934年証券取引法第13又は15(D)節の要求に基づいて、登録者は、正式に許可された次の署名者がその代表を代表して本報告書に署名することを正式に手配した.
|
|
AMYLYX製薬会社 |
|
|
|
|
|
日付:2024年2月22日 |
|
差出人: |
/S/ジョシュア·B·コーエン |
|
|
|
ジョシュア·B·コーエン |
|
|
|
合同最高経営責任者 |
|
|
|
|
日付:2024年2月22日 |
|
差出人: |
/S/ジャスティン·B·クレイ |
|
|
|
ジャスティン·B·クレイ |
|
|
|
合同最高経営責任者 |
本報告書は、改正された1934年の証券取引法の要求に基づき、以下の登録者によって登録者として指定日に署名された。
名前.名前 |
|
タイトル |
|
日取り |
|
|
|
|
|
/S/ジョシュア·B·コーエン |
|
合同CEO兼取締役(最高経営責任者) |
|
2024年2月22日 |
ジョシュア·B·コーエン |
|
|
|
|
|
|
|
|
|
/S/ジャスティン·B·クレイ |
|
合同CEO兼取締役(最高経営責任者) |
|
2024年2月22日 |
ジャスティン·B·クレイ |
|
|
|
|
|
|
|
|
|
/S/ジェームズ·M·フレディス |
|
首席財務官(首席財務官と首席会計官) |
|
2024年2月22日 |
ジェームズ·M·フレディス |
|
|
|
|
|
|
|
|
|
/S/ジョージ·マクレーン·ミルン |
|
役員.取締役 |
|
2024年2月22日 |
ジョージ·マクレーン·ミルンです博士号。 |
|
|
|
|
|
|
|
|
|
/S/ポール·フォンダン |
|
役員.取締役 |
|
2024年2月22日 |
ポール·フォンダンM.S.,M.B.A. |
|
|
|
|
|
|
|
|
|
/S/ダフニー·クミ |
|
役員.取締役 |
|
2024年2月22日 |
ダフニー·クミ |
|
|
|
|
|
|
|
|
|
/投稿S/カレン·フェルストン |
|
役員.取締役 |
|
2024年2月22日 |
カレン·フェルストン |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
139
Amylyx製薬会社
連結財務諸表索引
独立公認会計士事務所レポート(PCAOB監査人ID:34) |
F-2 |
合併貸借対照表 |
F-4 |
連結業務報告書 |
F-5 |
総合総合収益表(損益表)) |
F-6 |
転換可能優先株と株主権益連結報告書(損失)を償還可能) |
F-7 |
統合現金フロー表 |
F-8 |
連結財務諸表付記 |
F-9 |
F-1
“独立報告書”公認会計士事務所
株主に敬意を表する Amylyx製薬会社の取締役会です
財務諸表のいくつかの見方
添付されているAmylyx製薬会社とその子会社(“当社”)の2023年12月31日及び2022年12月31日の総合貸借対照表、2023年12月31日までの各年度の関連総合経営表、全面収益(赤字)、償還可能転換可能優先株及び株主権益(損失)及び現金流量、及び関連付記(総称して“財務諸表”と呼ぶ)を監査した。これらの財務諸表は,すべての重要な点で当社の2023年12月31日と2022年12月31日までの財務状況,および2023年12月31日までの3年度の経営結果とキャッシュフローを公平に反映しており,米国公認の会計原則に適合していると考えられる。
我々はまた、米国上場企業会計監督委員会(PCAOB)の基準に基づき、以下の基準に基づいて、会社が2023年12月31日までの財務報告内部統制を監査した内部統制--統合フレームワーク(2013)テレデビル委員会は組織委員会が発表した報告書と2024年2月22日の報告書を後援し、会社の財務報告書の内部統制について保留のない意見を表明した。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
以下に述べる重要な監査事項とは、財務諸表を当期監査する際に生じる事項であり、監査委員会に伝達または要求され、(1)財務諸表に対して大きな意味を有する勘定または開示に関し、(2)特に挑戦的、主観的、または複雑な判断に関するものである。重要監査事項の伝達は、財務諸表に対する私たちの全体的な意見をいかなる方法でも変えることはなく、次の重要な監査事項を伝達することによって、重要な監査事項又はそれに関連する勘定又は開示について個別の意見を提供することもない。
総額対純額(“GTN”)調整に関する可変考慮事項−財務諸表付記2と3を参照−
重要な監査事項の説明
製品販売収入は販売純価格(取引価格)で入金され、その中にはいくつかのGTN調整に関する可変対価格推定が含まれている。GTN調整の構成要素は、貿易割引と手当、製品返品、第三者支払者リベート、および会社、その顧客と支払人との間の製品販売に関する契約で提供される他の手当を含む。これらのGTN調整は、関連販売について請求された金額に基づいて、または関連販売について請求する金額に基づいており、売掛金減少(顧客に支払うべき場合)または流動負債(顧客以外に支払うべき場合)に分類される。貿易割引と割引、仕入先返金と返金は総合貸借対照表に売掛金の減少額として記入されています。政府リベートやその他のリベートは総合貸借対照表に課税費用の一部として入金される。
GTNのいくつかの調整は、重要な管理仮説と判断の使用に関する。これらの重要な仮定および判断は、歴史的経験、支払者チャネルの組み合わせ(例えば、MedicareまたはMedicaid)、適用計画下の現在の契約価格、未発行請求書のクレームおよび処理時間遅延、および流通チャネルにおける在庫レベルの考慮を含む。
F-2
関連する複雑さを考慮して、我々は経営陣の重大な仮定の評価を重要な監査事項として決定した。監査のような重大な仮定は特に主観的判断と監査努力に関するものだ。
監査で重要な監査事項をどのように処理するか
GTN調整に関連する監査プログラムには以下のことが含まれています
/s/徳勤法律事務所
ボストン、マサチューセッツ州
2024年2月22日
2020年以来、当社の監査役を務めてきました。
F-3
AMYLYX製薬会社
合併残高シーツ
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
短期投資 |
|
|
|
|
|
|
||
売掛金純額 |
|
|
|
|
|
|
||
棚卸しをする |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
流動資産総額 |
|
|
|
|
|
|
||
財産と設備、純額 |
|
|
|
|
|
|
||
制限現金等価物 |
|
|
|
|
|
|
||
経営的リース使用権資産 |
|
|
|
|
|
|
||
長期在庫 |
|
|
|
|
|
— |
|
|
その他の資産 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
負債と株主権益 |
|
|
|
|
|
|
||
流動負債: |
|
|
|
|
|
|
||
売掛金 |
|
$ |
|
|
$ |
|
||
費用を計算する |
|
|
|
|
|
|
||
賃貸負債を経営し、今期の部分 |
|
|
|
|
|
|
||
流動負債総額 |
|
|
|
|
|
|
||
賃貸負債を経営し,当期分を差し引く |
|
|
|
|
|
|
||
総負債 |
|
|
|
|
|
|
||
*(付記18) |
|
|
|
|
|
|
||
株主権益: |
|
|
|
|
|
|
||
優先株、$ |
|
|
|
|
|
|
||
普通株、$ |
|
|
|
|
|
|
||
追加実収資本 |
|
|
|
|
|
|
||
赤字を累計する |
|
|
( |
) |
|
|
( |
) |
その他の総合収益を累計する |
|
|
|
|
|
( |
) |
|
株主権益総額 |
|
|
|
|
|
|
||
総負債、償還可能な転換可能優先株、株主権益 |
|
$ |
|
|
$ |
|
||
付記はこれらの連結財務諸表の構成要素である。
F-4
AMYLYX製薬会社
統合された運営説明書
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
収入: |
|
|
|
|
|
|
|
|
|
|||
|
$ |
|
|
$ |
|
|
$ |
— |
|
|||
奨学金収入 |
|
|
— |
|
|
|
— |
|
|
|
|
|
総収入 |
|
|
|
|
|
|
|
|
|
|||
運営費用: |
|
|
|
|
|
|
|
|
|
|||
販売コスト |
|
|
|
|
|
|
|
|
— |
|
||
研究開発 |
|
|
|
|
|
|
|
|
|
|||
販売、一般、行政 |
|
|
|
|
|
|
|
|
|
|||
総運営費 |
|
|
|
|
|
|
|
|
|
|||
営業収入(赤字) |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
その他の収入(費用)、純額: |
|
|
|
|
|
|
|
|
|
|||
利子収入 |
|
|
|
|
|
|
|
|
|
|||
転換可能手形は価値変動を公正に許容する |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
その他の費用、純額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
その他の収入を合計して純額 |
|
|
|
|
|
|
|
|
( |
) |
||
所得税前収入 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
所得税支給 |
|
|
|
|
|
|
|
|
— |
|
||
純収益(赤字) |
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
1株当たり純収益 |
|
|
|
|
|
|
|
|
|
|||
基本的な情報 |
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
薄めにする |
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
1株当たり純利益(損失)を計算するための加重平均株式 |
|
|
|
|
|
|
|
|
|
|||
基本的な情報 |
|
|
|
|
|
|
|
|
|
|||
薄めにする |
|
|
|
|
|
|
|
|
|
|||
付記はこれらの連結財務諸表の構成要素である。
F-5
AMYLYX製薬会社
統合された総合報告書収入(損)
(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
純収益(赤字) |
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
その他の全面収益(損失): |
|
|
|
|
|
|
|
|
|
|||
外貨換算収益 |
|
|
|
|
|
( |
) |
|
|
|
||
保有投資は純収益を実現していない |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
その他全面収益(赤字) |
|
|
|
|
|
( |
) |
|
|
|
||
総合収益(赤字) |
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
付記はこれらの連結財務諸表の構成要素である。
F-6
AMYLYX製薬会社
総合償還可能報告書転換可能優先株株主の権益と
(単位:千、共有データを除く)
|
|
償還可能である |
|
|
|
普通株 |
|
|
その他の内容 |
|
|
積算 |
|
|
積算 |
|
|
合計する |
|
||||||||||||||
|
|
株 |
|
|
金額 |
|
|
|
株 |
|
|
金額 |
|
|
資本 |
|
|
収入(損) |
|
|
赤字.赤字 |
|
|
権益(赤字) |
|
||||||||
2021年1月1日現在の残高 |
|
|
|
|
$ |
|
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
( |
) |
|||||
C-1シリーズの償還可能な転換可能優先株を発行し、発行コストを差し引く$ |
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
||
変換可能手形と課税利息をC-2シリーズの転換可能優先株に変換し、発行コスト#ドルを差し引く |
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
||
株式オプション行使時に普通株を発行する |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
その他総合損失 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
純損失 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2021年12月31日現在の残高 |
|
|
|
|
$ |
|
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
||||||
初公開時に優先株を普通株に転換する |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
初公開時に普通株を発行し、発行コストを差し引く$ |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
後続発行時に普通株を発行し、発行コストを差し引いて#ドル |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
株式オプション行使時に普通株を発行する |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
その他総合損失 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
純損失 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2022年12月31日現在の残高 |
|
|
— |
|
|
$ |
— |
|
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
株式オプション行使時に普通株を発行する |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
RSUに帰属するときに普通株式を発行する |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
その他総合収益 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
純収入 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
||
2023年12月31日現在の残高 |
|
|
— |
|
|
$ |
— |
|
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||||
付記はこれらの連結財務諸表の構成要素である。
F-7
AMYLYX製薬会社
統合された現金フロー表
(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
経営活動提供のキャッシュフロー: |
|
|
|
|
|
|
|
|
|
|||
純収益(赤字) |
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
純収益(損失)と業務活動で使用される現金純額を調整する: |
|
|
|
|
|
|
|
|
|
|||
株に基づく報酬費用 |
|
|
|
|
|
|
|
|
|
|||
減価償却費用 |
|
|
|
|
|
|
|
|
|
|||
償却投資保険料 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
転換可能手形は価値変動を公正に許容する |
|
|
— |
|
|
|
— |
|
|
|
|
|
経営性資産と負債変動状況: |
|
|
|
|
|
|
|
|
|
|||
売掛金純額 |
|
|
( |
) |
|
|
( |
) |
|
|
— |
|
棚卸しをする |
|
|
( |
) |
|
|
( |
) |
|
|
— |
|
受取利息 |
|
|
|
|
|
|
|
|
( |
) |
||
前払い費用と他の流動資産 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
経営的リース使用権資産 |
|
|
|
|
|
|
|
|
— |
|
||
その他の資産 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
売掛金 |
|
|
|
|
|
|
|
|
|
|||
費用と繰延レンタル料を計算しなければならない |
|
|
|
|
|
|
|
|
|
|||
リース負債を経営する |
|
|
( |
) |
|
|
( |
) |
|
|
— |
|
受取利息と受取利息の関係者 |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
経営活動提供の現金純額 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
投資活動によって提供されるキャッシュフロー: |
|
|
|
|
|
|
|
|
|
|||
財産と設備を購入する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
短期投資を購入する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
短期投資満期収益 |
|
|
|
|
|
|
|
|
|
|||
投資活動提供の現金純額 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
資金調達活動が提供するキャッシュフロー: |
|
|
|
|
|
|
|
|
|
|||
購買力平価ローンの返済と収益 |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
初めて公募して得た収益 |
|
|
— |
|
|
|
|
|
|
— |
|
|
増発所得収益 |
|
|
— |
|
|
|
|
|
|
— |
|
|
初公募株払いのコスト |
|
|
— |
|
|
|
( |
) |
|
|
— |
|
後続サービス料金はお支払いいただきました |
|
|
( |
) |
|
|
( |
) |
|
|
— |
|
転換可能な手形を発行して関係者が得た金 |
|
|
— |
|
|
|
— |
|
|
|
|
|
転換手形を発行して得られた金は発行コストを差し引く |
|
|
— |
|
|
|
— |
|
|
|
|
|
変換可能チケットに関する発行コスト |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
C-1シリーズの償還可能な転換優先株を発行して得られた金 |
|
|
— |
|
|
|
— |
|
|
|
|
|
C-1シリーズ償還可能な転換可能優先株の発行に関する発行コスト |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
株式オプションを行使して得られる収益 |
|
|
|
|
|
|
|
|
|
|||
株奨励前納税 |
|
|
( |
) |
|
|
— |
|
|
|
— |
|
繰延発売費を支払う |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
融資活動が提供する現金純額 |
|
|
|
|
|
|
|
|
|
|||
現金,現金等価物および限定的現金等価物に及ぼす為替レート変動の影響 |
|
|
|
|
|
( |
) |
|
|
|
||
現金、現金等価物、および制限的現金等価物の純増加 |
|
|
|
|
|
|
|
|
|
|||
期初現金、現金等価物、および限定的現金等価物 |
|
|
|
|
|
|
|
|
|
|||
期末現金、現金等価物、および制限現金等価物 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
キャッシュフロー情報の追加開示: |
|
|
|
|
|
|
|
|
|
|||
変換可能手形と課税利息をC-2系列償還可能優先株に変換する |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
短期投資の未実現収益 |
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
課税費用に含まれる株式奨励源泉徴収 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
売掛金に掲げる財産と設備の購入 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
売掛金と売掛金に含まれる繰延発売コスト |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
ASC 842を採用した場合の使用権資産と負債 |
|
$ |
— |
|
|
$ |
|
|
$ |
— |
|
|
賃貸負債と引き換えに使用権資産 |
|
$ |
— |
|
|
$ |
|
|
$ |
— |
|
|
繰延発行コストの株式への移転 |
|
$ |
— |
|
|
$ |
|
|
$ |
— |
|
|
後続発売コストは売掛金と売掛金に計上される |
|
$ |
— |
|
|
$ |
|
|
$ |
— |
|
|
初公開時に優先株を普通株に転換する |
|
$ |
— |
|
|
$ |
|
|
$ |
— |
|
|
納めた所得税 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
||
付記はこれらの連結財務諸表の構成要素である。
F-8
AMYLYX製薬会社
総合財務備考報告書
1.ビジネスの性質
Amylyx製薬会社及びその完全子会社Amylyx或いは同社は商業段階の生物技術会社であり、その使命は神経変性疾患による苦痛を終わらせることである。同社は筋萎縮性側索硬化症(ALS)をその第1の適応として求めており,AMX 0035の世界的なALSの開発と潜在的商業化に専念している。AMX 0035はアメリカ食品医薬品局(FDA)が許可し、RELYVRIOの名前で発売された®米国では成人筋萎縮性側索硬化症の治療に使用されている。AMX 0035はカナダ保健省の条件付き承認を受け、ALBIOZAで販売されているカナダでは筋萎縮性側索硬化症を治療している。同社は、ALS患者におけるAMX 0035の有効性および安全性に関するより多くのデータを提供し、他の神経変性疾患のためにAMX 0035を開発しているグローバルフェニックスの第3段階臨床試験の完了に引き続き集中している。AMX 0035は小胞体、ストレスとミトコンドリア機能障害に対して、この2つの相互接続された中枢経路は神経退化を招く可能性がある。同社はさらにAMX 0035が進行性核上性麻痺(PSP)やWolfram症候群(WS)を含むERやミトコンドリアストレスに関連する疾患を検討している。2023年4月、同社はHELIOS試験の第1段階参加者にAMX 0035治療WSの第2段階試験を服用した。2023年12月、同社は、PSPを治療するAMX 0035の世界的に重要な3期試験であるOrion試験の最初の参加者のために薬物を服用した。同社はまた、アンチセンスオリゴヌクレオチドAMX 0114、Calain-2を標的とし、軸索変性疾患の重要な蛋白質などを含む神経変性疾患を治療する候補薬物をもっと多く発売した。
リスクと不確実性
同社は生物技術業界会社によく見られるリスクと不確定要素の影響を受け、臨床前研究と臨床試験の結果、市場受容度及びその許可された製品ALBRIOZAとRELYVRIOの商業化に成功したが、ALBRIOZAは2022年6月にカナダで条件付きマーケティング許可を獲得し、RELYVRIOは2022年9月にアメリカでFDAの許可を得て、監督管理の審査過程中に時間困難或いは遅延が出現する可能性があり、競争相手は新しい技術革新を開発し、肝心な人への依存、ノウハウの保護、政府法規に符合し、追加資本を獲得して運営に資金を提供する能力、新冠肺炎などの世界衛生危機による経済挑戦や地政学的不安定や衝突による世界各市場の経済不確実性に関するリスクがある。同社およびその請負業者は、AMX 0035を製造するための原材料およびバルク医薬物質、ならびに任意の追加または未来の候補製品を欧州およびカナダから輸入するなど、その研究開発および商業活動に重要なプロジェクトの供給中断に遭遇する可能性がある。
2.主な会計政策の概要
新聞の掲載と合併の基礎-添付されている総合財務諸表は米国公認会計原則または公認会計原則に従って作成され、その中には当社及びその完全子会社の勘定が含まれている。すべての会社間の残高と取引はすでに合併中に販売されている。本説明で言及された任意の適用指針は、財務会計基準委員会(FASB)の“会計基準編纂”(ASC)および“会計基準更新”(ASU)における権威GAAPを意味する。
予算を使って-公認会計原則に基づいて連結財務諸表を作成する際には、財務諸表日の資産及び負債報告金額及び報告期間内に報告された費用金額に影響を与えるために、会社管理層が推定及び仮定を行う必要がある。実際の結果はこれらの推定とは異なる可能性がある。経営陣は、財務諸表作成に用いる見積もりや仮定を作成する際に、適切な財務会計政策を選択する際に多くの要因を考慮している。経営陣はこの過程で重要な判断をしなければならない。経営陣の試算過程は往々にして一連の潜在的な合理的な試算を生成する可能性があり、管理職はその合理的な推定範囲内の額を選択しなければならない。その他を除いて,見積数は,総額と純額の調整,在庫の回収可能性,製品発売のための在庫,課税費用,ストックオプション評価,繰延税金資産の見積準備と研究開発費などの分野で用いられている。
F-9
収入確認-2022年6月、AMX 0035はカナダ衛生部がALBRIOZAを条件としてALSを治療する上場許可を得て、会社は2022年7月にカナダでALBRIOZAを発売した。2022年9月、AMX 0035はFDAの許可を得て成人筋萎縮性側索硬化症を治療するRELYVRIOとして、同社は2022年10月にアメリカでRELYVRIOを発売した。
同社は卸売業者、専門薬局と専門流通業者あるいは顧客と合意し、ALBRIOZA、RELYVRIOと将来承認された製品を流通する。ASC主題606-によると-取引先と契約した収入または主題606は、顧客が約束された商品またはサービスの制御権を取得すると、収入が確認され、その額は、これらの商品またはサービスと交換するために、エンティティが取得する権利があることを望む対価格を反映する。
(I)顧客との契約を決定するステップ(S),(Ii)契約中の履行義務を決定する,(Iii)取引価格を決定する,(Iv)契約に取引価格を割り当てる履行義務,および(V)会社が契約義務を履行した場合に収入を確認する,の5つのステップを実行する。企業は、企業が顧客に譲渡する商品またはサービスと交換するために、企業が獲得する権利が予想される対価格を受け取る可能性が高いことを含む、主題606で定義された契約に適合する手配にのみ5ステップモデルを適用する。契約開始時に、契約が主題606の範囲内にあると判断されると、会社は、各契約において承諾された商品又はサービスを評価し、それらが義務を履行していると判断し、各承諾された商品又はサービスが異なるか否かを評価する。そして、会社は、履行義務を履行する際にそれぞれの履行義務に割り当てられた取引価格の金額が収入であることを確認する。
製品収入、純額
その会社はその顧客にその承認された製品を販売する。これらの顧客はその後、私たちの製品を専門薬局プロバイダ、専門流通業者、ヘルスケア提供業者、いくつかの医療センターまたは病院、および患者に転売する。顧客との合意に加え、同社は専門薬局、ヘルスケア提供者、支払人と合意し、我々の製品を購入するために政府が規定しているおよび/または私的に協議したリベートを提供する。同社の顧客識別プロセスは、契約や法的要因、誰が会社の製品をコントロールし、在庫リスクを担っているかなど、多くの要因を考慮している。同社は、収入を確認するために適切な顧客を決定するために、顧客ごとにこれらの要因を評価している。場合によっては、会社は第三者物流プロバイダを使用して会社の製品を顧客に渡す可能性がありますが、会社は第三者物流プロバイダが私たちの代理であると判断したので、会社は顧客に渡す際に収入を確認します。これらの要素の変化やこれらの要素の仮定は私たちの収入確認に影響を与えるかもしれません
顧客がわが製品に対する統制権を取得すると、会社は製品販売収入を確認し、これはある時点(納入時)に発生する。製品収入は,適用されたGTN調整後の純額を差し引くと,以下のようになる。
製品販売に関連する顧客に税金を徴収し、政府当局に送金すべきであれば、これらの税金は収入から除外される。もし会社が確認すべき資産の予想償却期限が
GTN調整
製品販売収入は販売純価格(取引価格)で入金され、その中にはいくつかのGTN調整に関する可変対価格推定が含まれている。GTN調整の構成要素には、貿易割引と手当、製品返品、第三者支払人のリベート、および会社、その顧客と支払人の間で私たちの製品販売に関する契約で提供される他の手当が含まれている。これらのGTN調整は、以下に説明するように、稼いだ金額に基づいて、または関連販売において請求され、売掛金が減少する(その金額が顧客に支払われるべきである場合)または流動負債(その金額が顧客以外の方に支払われるべきである場合)に分類される。これらの推定は、主題606における期待値方法に従って、歴史的経験、支払人チャネルの組み合わせ(例えば、連邦医療保険または医療補助)、適用計画下の現在の契約価格、未発行請求書のクレームおよび処理時間遅延、および流通チャネルにおける在庫レベルのような関連要因に対して確率的重み付けを行う一連の可能な結果を考慮する。場合によっては、会社は、主題606の中で最も可能性のある方法を採用する。♪the the the
F-10
期待値手法を用いるか最も可能であるかは,GTN調整のタイプと,どの方法が得られることが期待される対価格額をより良く予測するかに依存する.全体的に、これらのGTN調整は、約束された商品またはサービスを顧客に移転するために、会社が獲得する権利があると予想される対価格金額を取引価格に反映している。
取引価格に含まれる可変対価金額は制限されることができ、契約項で確認された累積収入金額が今後の期間に大きな逆転が生じない可能性が高い場合にのみ、純販売価格に含まれる。最終的に受け取った実際の対価格金額は私たちの見積もりとは違うかもしれません。もし未来の実際の結果が私たちの推定と異なる場合、会社はこれらの推定値を調整し、これは製品収入、純額、および既知の差異期間の収益に影響を与えるだろう。
貿易割引と手当
流通サービスおよび流通業者が提供してくれるいくつかのデータについて、会社は通常、顧客に即時支払い割引および支払い費用を提供し、これらのデータは、私たちの契約において明確に規定され、関連製品の収入確認中に収入減少として記録される。お客様の支払いは通常、即時支払い割引を考慮することなく、領収書日の30日後のカレンダー日に満了しなければなりません。
製品を返品する
業界慣例によると、会社は製品の納期に応じて限られた返品権利を顧客に提供するのが一般的であり、この納期は契約に規定された特定の期間内に失効する。また、我々の有限返品権利政策は、お客様が製品の出荷ミスや出荷中に破損したり、製品が公式薬品リコールによって返品された場合に合格返品を行うことを許可しています。
当社は、当社の顧客が返金可能な製品売上高を推定し、この推定を、関連製品収入期間の収入減少と、合併貸借対照表における純売掛金の減少を確認するために記録しています。同社は現在、返品の歴史的経験、予測された需要、流通ルートにおける在庫レベル、製品の日付と有効期限、製品が生産中止になったかどうかなどを含むが、返品を推定するために定量的かつ定性的な情報を使用している。これまで、同社は実質的ではない返品を受けており、今後しばらくの間に製品の返品はわずかになると信じている。
プロバイダはストレージ容量に応じて料金と割引を使用します
プロバイダへの費用および割引とは、当社から製品を直接購入した顧客に直接請求する価格よりも低い価格で合格したヘルスケア提供者に製品を販売することを契約が承諾したことによる推定義務である。顧客は彼らが製品に支払った価格と合格医療提供者の最終販売価格との差額に基づいて会社に費用を徴収する。これらのGTN調整は,関連収入が確認された同時期に決定され,製品収入と売掛金純額が減少した。GTNによる記憶容量使用課金の調整には,顧客がまだ申請していないポイントが含まれているが,各報告期間末に流通チャネル在庫に保持されている機器にポイントを発行する予定であり,これらの単位は合格したヘルスケア提供者や,顧客が申請したがポイントの課金を発行していないと予想される.
支払人は返却します
当社は、ある政府や個人支払人組織と、主に政府と商業健康保険会社と契約を結び、私たちの製品の使用に関するリベートを支払います。州医療補助計画と連邦医療保険によると、同社は割引義務を履行しなければならない。当該等GTNは、関連収入を確認する同一期間内に入金することを調整し、製品収入の減少及び流動負債を構築し、当該負債は総合貸借対照表の計上支出及びその他の流動負債に計上する。連邦医療保険については,処方薬カバーギャップのうち連邦医療保険D部分計画により追加責任を負う患者数も推定されている。会社のこれらのリベートに対する負債には、前の四半期にまだ支払われていない、または領収書を受け取っていない請求書、クレーム推定数が含まれている
F-11
各報告期間終了時に、収入が確認されたが流通チャネル在庫に残っている製品については、将来的にクレームが出ると予想される。
他のインセンティブ
同社が提供する他のインセンティブは、支払者が要求する処方薬の共同支払いを有する条件に適合する商業保険患者に財政的援助を提供することを目的とした自発的な患者支援計画、例えば、その共同支払い援助計画を含む。自己援助課税費用の計算は、報告期間ごとに収入が確認された製品に関するクレーム推定数と、企業が受信する予定の各クレームのコストに基づいている。この等は、関連収入を確認する同一期間の入金を調整し、製品収入の減少及び流動負債の構築を招き、当該流動負債は支出及び他の流動負債の一部として総合貸借対照表に計上される。
全面的な損失-総合損失には、純損失と、株主との取引や経済事件以外の取引や経済事件による株主権益(赤字)の他の変化がある。総合損失は純損失と他の総合(損失)収益からなる。その他総合(赤字)収入には有価証券と外貨換算の未実現損益が含まれる。
現金と現金等価物-
制限された現金等価物- 制限された現金等価物は#ドルを含む
売掛金、純額- 同社の売掛金には、顧客の製品販売に関する売掛金が含まれており、標準的な支払い条件がある。同社は期限を過ぎた売掛金を分析し、顧客が支払うことができない可能性のある予想信用損失のために売掛金を準備した。回収できないと判断された金額は、作成された準備金からログアウトします。2023年12月31日と2022年12月31日までしかし、会社の顧客の信用状況は良好とされており、期待される信用損失は実質的ではない。
短期投資-短期投資は米国国庫券と手形、会社債務証券、商業手形、機関債券からなり、期限は貸借対照表日から1年未満である。その会社はそのすべての短期投資を販売可能に分類した。そのため、これらの投資は公正価値によって入金され、公正価値はオファーされた市場価格によって決定される。売却可能な証券の未実現損益は他の累計総合損失の単独構成要素として計上される。短期投資のコストは割増の償却と満期前割引の増加によって調整される。このような償却と付加価値は利息収入に計上される.実現した損益は他の費用、純額に計上される。同社は貸借対照表の日に非一時的減値の短期投資を評価している。非一時的と判断された公正価値低下(あれば)も他の純収益に計上されている。
非一時的な価値低下の短期投資を評価する際に、当社が考慮する要因は、価値低下が元のコストに占める割合がどれだけ大きいか、投資の時価がその元のコストよりも低い時間がどれだけ長いか、および当社は、公正な価値および全体的な市場状況の任意の予想回復を達成するために、投資を十分な時間保持する能力と意図があることを含む。2023年12月31日と2022年12月31日まで短期投資には減価費用はありません。
信用リスク集中-会社を集中的な信用リスクに直面させる可能性のある金融商品には、主に現金と現金等価物、短期投資、売掛金純額が含まれる。当社は高い信用品質を持つと考えられる金融機関に現金を預けています。当社はこのような口座でいかなる損失を被っていないし、商業銀行関係に関連する正常な信用リスクではなく、いかなる異常な信用リスクに直面しているとも思わない。
当社の売掛金は、純額が顧客が当社に不足している金額を表しています。Amylyxはその顧客に対して継続的な信用評価を行い、通常担保を必要としない。その会社はそのリスクを監視し
F-12
必要に応じて不良債権を記録して準備する
公正価値計測-総合貸借対照表では,公正価値で常時記録されている資産と負債は,その公正価値を計測するための投入に関する判断レベルに基づいて分類される.公正価値は、計量日市場参加者間の秩序ある取引において、資産または負債が元金または最も有利な市場で負債を移転するために徴収または支払いされる交換価格として定義される。公正価値を計量するための推定技術は,観察可能な投入を最大限に利用し,観察不可能な投入を最大限に減少させなければならない。“公正価値計量に関する権威指針”は公正価値計量の開示のために三級公正価値等級を確立し、具体的には以下の通りである
ある程度、推定値は市場で観察または観察できないモデルや投入に基づいており、公正価値の決定にはより多くの判断が必要である。そのため、当社が価値を公平に判断する際に下した判断は、第3級に分類されたツールに対する判断度が最も高く、公正価値レベル内の金融商品レベルは、公正価値計量に大きな意味を持つ任意の投入の中で最低レベルに基づいている。
同社の金融商品には、現金、現金等価物、制限された現金等価物、短期投資、売掛金、純額、売掛金、売掛金が含まれている。当社の短期投資は公正価値に基づいて計上され、上記公允価値等級の第1級及び第2級投入に基づいて決定される。当社の2021年手形(定義付記8参照)は、上記公正価値階層における第3級投入に基づいて決定される公正価値に基づいて入金される。残りの金融商品は,そのそれぞれの台帳に記載されており,その等資産や負債の短期的な性質のため,当該等の帳簿額面はおおむね公正価値である.
在庫-当社はその在庫をコストまたは推定可処分純価値の低い者で推定しています。同社は先進的な先出しの原則に従って在庫コストを決定し、その中には材料や製造間接費用に関する金額が含まれている。在庫の消費または販売予想がその12ヶ月の正常運営周期を超えた場合、会社は在庫を長期在庫に分類する。当社は報告期間ごとに資本化在庫の回収可能性を評価し,任意の過剰と古い在庫を減値期間中の推定可変価値を初めて確認するまで減記した。この等減算費用が発生した場合は,販売コストに記入する.在庫コストが現金化できるかどうかを判断するには経営陣が試算する必要がある。実際の市場状況が経営陣が予想していたほど有利でなければ、追加減記在庫が必要となる可能性があり、合併経営報告書に販売コストとして記録されることになる。
経営陣の判断により、将来の商業化が可能であり、将来の経済効果が期待できると考えられた場合、会社は規制部門の承認後に会社製品に関する在庫コストを資本化する。規制機関から候補製品の承認を受ける前に得られたライブラリの存在が発生した場合には研究·開発費用を計上する。臨床または商業製品生産に利用可能な在庫は最初に資本化され、その後、まだ開発中の薬物の生産を決定する際に研究および開発費用として支出される。
財産と設備、純資産-属性設備を減価償却累計額を差し引いたコストで列記する。財産や設備の減価償却は直線法で計算され、計算された時間はそれぞれの資産の推定耐用年数となる。資産寿命の改善や延長のないメンテナンスやメンテナンスは、発生時に費用を計上する。資産を売却または廃棄する際には,コストおよび減価償却は総合貸借対照表から差し引かれる
F-13
そしてこれによって生じる任意の収益または損失は、実現された期間の総合経営報告書に反映される
|
|
使用寿命を見込む |
|
||
家具と固定装置 |
|
|
コンピュータハードウェアとソフトウェア |
|
|
|
長期資産減価-事件や環境変化が資産の帳簿価値を回収できない可能性があることを示した場合、当社は資産の潜在的減値を評価する。回収能力は,資産の帳簿価値と資産予想による予想将来の未割引純現金フローを比較することで測定した。当該等の資産が減値とみなされる場合、確認すべき減値は、資産の帳簿価値がその公正価値を超える金額で計量される。その会社は所有している
研究と開発-研究·開発費用には、研究·開発活動に直接起因することができる費用が含まれている。研究と開発に関する支出は発生した期間内に支出される。将来の研究および開発活動のための貨物およびサービスの払い戻し不可能な前金は、支払い時に支出するのではなく、活動が行われたときまたは貨物を受信したときに支出される。また、研究開発に関連する賃金や福祉、施設や管理費用、用品やその他の関連コストも研究開発費に含まれている。
販売とマーケティングコスト-販売とマーケティング費用は主に販売とマーケティング担当者の給料と福祉、専門と相談費、行政出張費用、マーケティングと広告費用、例えばマーケティング文献、販売促進活動、会議とシンポジウム、ブランド普及などを含む。販売及び市場普及及び広告コストは、発生した時に販売、一般及び行政費用に計上し、添付されている総合経営報告書に計上する。当社は広告費用を当社の商業製品の普及に関する費用と見なしています。2023年12月31日と2022年12月31日までの年間広告コストは$
特許関連コスト-特許出願の提出及び起訴に関する特許関連費用は、支出回収の不確実性により発生した費用に計上される。発生した金額は添付されている総合経営報告書で販売、一般、行政費用に分類される。
株の報酬に基づいて株式の報酬に基づいて、総合経営報告書において、付与された日の必要なサービス期間内の公正価値によって確認され、このサービス期間は、通常、対応する報酬の帰属期間に等しい。没収は発生状況に応じて計算されます。一般に,当社が発行する株式オプション報酬はサービス性の帰属条件のみを持ち,これらの奨励の費用を直線法を用いて記録する.同社が株式に基づく報酬費用を分類する方式は、受賞者の給料やサービスプロバイダのコストを分類する方式と同じだ。
各制限された普通株奨励の公正価値は、付与された日に会社普通株の同一日の公正価値に基づいて推定される。
各オプション付与の公正価値は、付与された日にBlack-Scholesオプション定価モデルを使用して推定され、このモデルは、予想される株価変動、予想される付与期限、無リスク金利、および予期される配当を含むいくつかの主観的仮定に基づいて入力を必要とする。当社は上場同業者の歴史的変動率に基づいて期待株価変動率を推定しています。同社の株式オプションの期待期限は、“簡略化”方法で決定され、“普通”オプション資格に適合する株を奨励するために使用される。無リスク金利は、奨励付与時に発効した米国債収益率曲線を参考にして決定され、期間は奨励の予想期限にほぼ等しい。当社は普通株について現金配当金を支払ったことがなく、予測可能な未来にはいかなる現金配当金も支払わないと予想されているため、予想される配当収益率はない。当社の株価は授出日の終値を基準としています。初公募前に、当社の普通株は公開市場がないため、普通株の推定公正価値は当社取締役会が各購入株式権の授与日、管理層の意見、その普通株を考慮した第三者推定値、及び当社取締役会が関連していると考えられ、最近の第三者推定日から当社の上場日までの間にすでに変化する可能性のある他の客観及び主観的要素の評価によって決定された
F-14
グラントです。これらの第三者評価は、米国公認会計士協会会計·評価ガイドラインで概説された指導に基づいて行われている補償として発行された個人持株会社株式証券の推定値.
事件があったり-当社には通常の業務活動で発生したものや負債が時々あるかもしれません。損失が可能になり、合理的に推定できるようになった場合、会社は損失を計上しなければならない。損失の合理的な推定が範囲であり、その範囲内のいずれの金額もより良い推定でない場合、その範囲の最低額は、会社の総合貸借対照表に負債として記録される。当社はその判断が合理的であると考えられるが起こり得ない損失や損失を考慮すべきではないが、合理的に可能な損失範囲を開示している。いくつありますか
賃貸借証書-会社は財務会計基準ASC 842を採用しています賃貸借契約あるいは…。 ASC 842,2022年1月1日。ASC 842は、(I)エンティティが満了または既存の契約がレンタルであるかどうかまたはレンタルを含むかどうかを再評価する必要がないことと、(Ii)エンティティが満了または既存のレンタルのレンタル分類を再評価する必要がないことと、(Iii)エンティティが任意の既存のレンタルの任意の初期直接コストを再評価する必要がないこととを含む、当社が実用的な便宜的なセットを選択することを可能にする。もう1つの実際の方便は,テナントが賃貸借契約の延長または終了および対象資産の購入を考慮した場合,会社がリース期間を事後に決定することができることである.当社はこの実用的な方便を使用することを選択しており、ASC 842を実施する際には事後諸葛亮の方法は選択されていない。
当社はオフィスをレンタルし、時々他のレンタル契約を締結して業務を展開することができます。当社は契約開始時に1つの手配にレンタルが含まれているかどうかを決定します。当社のリース手配ごとに、当社は、当社がレンタル期間内に対象資産を使用する権利、及びリース負債を代表して、当社に代わってリース支払いの義務を支払う使用権資産を記録している。経営リース使用権資産および経営リース負債は、レンタル開始日にレンタル期間内に残り将来最低賃貸支払いの正味現在価値を確認します。当社の賃貸借契約に隠されている金利は簡単には特定できませんが、賃貸支払い純現在値を計算するための加重平均割引率を設定する際には、当社は市場源(対象資産別に定められた類似期限の信用素相若会社の金利を含む)に基づいて借金金利を増加させて推定し、賃貸支払いを割引します。当社が経営しているリースのレンタル料金はレンタル期間内の直線ベースで確認し、可変レンタルコストは発生した費用を計上します。同社は2023年12月31日と2022年12月31日まで融資リースを行っていない。
当社は実際の便宜策を選択し、確認及び計量要求を短期賃貸契約、すなわち借約開始日が1年以下のいずれの賃貸契約にも適用しない。レンタルは会社に追加のメンテナンスと他の費用を支払うことを要求するかもしれません。これらの費用は一般的に非レンタルコンポーネントと呼ばれます。当社はすでに実行可能な方便を選択し、レンタルと非レンタル構成部分を結合した。レンタルにレンタル期間の延長の選択権が含まれている場合、当社は、そのオプションがその初期リース期間評価に行使されるとは仮定せず、リース開始日に存在する経済的要因の評価に基づいて、当社が継続期間を合理的に把握していない限り、継続期間を合理的に把握する。
ASC 842を採用する前に、会社はレンタルが開始されるたびにレンタルプロトコルを評価して、レンタルが経営的賃貸か資本賃貸かを決定するASC 840、レンタル(ASC 840)それは.ASC 840の4つのテスト基準のいずれかが満たされると、レンタルは資本リースとなる資格がある。賃貸契約に契約更新選択権、テナント改善手当、賃料祝祭日または賃貸料上昇条項が含まれている場合、当社は繰延賃貸資産または負債に計上し、このような繰延賃貸資産または負債は、賃貸料支出と将来の最低賃貸支払いの支払いとの差額に相当する。経営リースに関する賃貸料支出は,リース期間ごとの経営報告書で直線的に確認されている。
所得税-同社は貸借対照法を用いて所得税を計算している。繰延税項資産及び負債とは、財務諸表の帳簿金額と資産及び負債の計税基準との間の一時的な差異及び差異が年間発効すると予想される税率繰越の損失を逆転させる将来の税務結果を指す。繰延税金資産を期待して現金化された金額に減らすための推定準備を設ける。同社はまた、“より可能性が高い”場合にのみ、その地位が持続可能であり、その技術的優位性に基づいて、不確定な税収状況の税収利益を確認している。所得税引当金の一部として、同社は不確定な税収状況に関連した利息及び罰金を計上する。現在まで、同社では所得税に関する重大な利息や罰金は発生していない。
F-15
既存の証拠の量に基づいて、繰延税金資産の一部または全部が現金化できない可能性が高い場合、推定免税額が提供される。2023年12月31日現在、経営陣の利用可能なすべての証拠(運営中に大きな損失を受けた歴史を含む)の評価に基づいて、私たちのすべてのアメリカ連邦と州繰延税金資産に対して全額推定手当を維持しています。私たちのすべての利用可能な証拠の評価はまた、2023年のこれらの製品の販売による収入を含む規制部門のALBRIOZAとRELYVRIOの承認を考慮することを含むそれは.私たちの製品発売の初期段階を考慮すると、将来の販売時間と数量は確定できません。私たちは短期的にすべてまたは一部の推定手当を支給するかもしれないが、推定手当の支給および支給の正確な時間と金額は、依然として私たちの利益水準、収入の増加、臨床プロジェクトの進展、将来の収益性への期待などの要素の影響を受ける。
市場情報を細分化します経営支部は、収入および支出を生成する可能性があり、1人以上の首席運営意思決定者によって定期的に評価される離散財務情報を取得して、支部に割り当てられた資源について決定し、その業績を評価することができる業務活動に従事する構成要素として定義される。同社はその連合席最高経営責任者が最高経営決定者、すなわちCODMであることを決定した。CODMは総合経営結果を審査し、会社の全体戦略と目標に基づいて、資源又は資本を特定の化合物又はプロジェクトに割り当てることを決定する。会社の業務全体は単一の管理チームによって管理されており、このチームは連合席最高経営責任者に仕事を報告している。同社には神経変性疾患の治療法を研究·開発する業務部門がある。2023年12月31日まで及び2022年12月31日まで年度しかし、同社のすべての長期資産は米国内で保有されている。
1株当たり純収益-当社は1株当たり純収益(損失)を計算する際に2段階法を採用しており、当社は参加証券の定義に合った株を発行しているからです。2級法は、発表または累積された配当金および未分配収益の参加権に基づいて、各種類の普通株および参株証券の1株当たり純収益(損失)を決定する。二段階法は、この期間中に得られる収入が、その期間のすべての収入が割り当てられているかのように、普通株主がそれぞれ配当を得る権利に基づいて、普通株式と参加証券との間で分配されることを要求する。
1株当たり基本純収益(損失)の計算方法は、普通株株主が占めるべき純収益(損失)を当期発行普通株の加重平均で割ったものである。希釈純収益(損失)は,普通株主が純収益(損失)を占めるべきであることを調整し,希釈証券の潜在的影響に応じて未分配収益を再分配することで計算される。普通株株主は1株当たりの純利益(損失)を占めるべき計算方法は普通株株主が償却純収益(損失)を当期に発行された普通株で割る加重平均であり、潜在的な希釈性普通株を含む。この計算については、株式オプション、転換可能手形、償還可能優先株は潜在的な希釈性普通株とみなされている。
当社の償還可能な転換可能優先株は、契約に当該等の株式の保有者に配当に参加する権利を付与しているが、契約上は当該等の株式の所有者に自社の損失を分担することを要求していない。したがって,会社が普通株株主が純損失を占めるべきであることを報告している間,このような損失はこのような参加証券に分配されない。会社が普通株株主が純損失を占めるべき期間を報告する時、普通株株主は希釈1株当たり純損失と普通株株主は基本的に1株当たり純損失を占めるべきであり、希釈性普通株の影響が反希薄であれば、希釈性普通株を発行したと仮定しないからである。
最近の会計公告
未採用の新会計公告
FASBは2023年12月にASU 2023-09を発表しました所得税(特集740):所得税の改善開示所得税開示の透明性および意思決定の有用性を向上させるために、またはASU 2023-09。ASU 2023-09は、2024年12月15日以降の年間期間を予想に基づいて発効します。早期採用と遡及応用を可能にする。同社は現在、このASUの連結財務諸表や関連開示への影響を評価している。
2023年11月、FASBはASU 2023-07号を発表した分部報告(テーマ280):改善可能報告分部開示またはASU 2023-09は、その報告可能な部門の中期および年間の重大な支出情報を開示することを公共エンティティに要求する。ASU 2023−07は2025年5月31日までの年度から会社に対して発効している。同社は現在、このASUの連結財務諸表や関連開示への影響を評価している。
F-16
最近採用された会計公告
2016年6月、FASBはASU第2016-13号を発表した金融商品·信用損失(主題326)金融商品信用損失の測定またはASU 2016-13。ASU 2016-13の条項は、現在使用されている発生した損失方法の代わりに期待損失方法を使用するために、低減モデルを修正し、信用損失推定値を提供するために、より広範な合理的かつサポート可能な情報を考慮することを要求する。売却可能な債務証券に関連する信用損失は、直接証券に減記するのではなく、信用損失準備金によって記録される。当社はASU 2016-13を採用し、2023年1月1日から発効し、その連結財務諸表や関連開示に実質的な影響はありません。
同社は2022年1月1日から改正遡及移行法を採用し,ASC 842の要求を採択した。比較期間は再記述されていない.本基準は、リース資産のエンティティが、貸借対照表上のこれらのリースによって生成された権利および義務の資産および負債を確認することを要求する。同社は利用可能な一括実際の便宜策を選択し、これまでの手配が借約であるかどうか、あるいはその賃貸借契約の分類および初期直接コストの処理を含む会計結論を再評価する必要がないようにした。同社はすでに会計政策選択を行い、初期期限が12ヶ月以下の賃貸契約を貸借対照表から除外した。ASC 842を発行することは、レンタルスケジュールに関連する財務報告の透明性および比較可能性を増加させるためである。従来のGAAPまたはASC 840とASC 842との主な違いは、テナントがASC 840によって経営的リースに分類されたリースに対して使用権リース資産とリース負債を確認することであった。2022年1月1日現在、会社が記録した使用権資産は#ドル
3.製品収入純額
これまで,同社の製品収入の唯一の源はRELYVRIOの販売であり,カナダではALBRIOZAと呼ばれてきた。歴史的経験、支払者チャネルの組み合わせ(例えば、MedicareまたはMedicaid)、適用計画下の現在の契約価格、未発行請求書のクレームおよび処理時間遅延、および流通チャネルにおける在庫レベルを考慮すると、GTN調整を推定する際には重大な判断が必要である
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
|
|
(単位:千) |
|
|||||||||
製品収入、毛収入 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
||
GTN調整 |
|
|
( |
) |
|
|
( |
) |
|
|
— |
|
製品収入、純額 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
||
GTN調整の活動と期末準備金残高は以下のとおりである
|
|
記憶容量に応じて料金と現金割引をご利用ください |
|
|
医療補助と医療保険のリベート |
|
|
その他のリベート、返品、割引、調整 |
|
|
合計する |
|
||||
|
|
(単位:千) |
|
|||||||||||||
2021年12月31日までの期末残高 |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
本年度の販売に関する準備 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
前期販売に関する調整 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
貸方と支払い |
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2022年12月31日までの期末残高 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
本年度の販売に関する準備 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
前期販売に関する調整 |
|
|
( |
) |
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
貸方と支払い |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
2023年12月31日までの期末残高 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
含まれているGTN調整の期末準備金残高には,契約承諾が自社から製品を直接購入した顧客に直接受け取る価格よりも低い価格で合格ヘルスケア提供者に製品を販売することによる返金,顧客へのタイムリーな支払いの割引,製品返品の推定が含まれている。引き落とし、割引、返品は売掛金の減少額とし、純額は連結貸借対照表に記入する。また,GTN調整の期末準備金残高には,医療補助と医療保険リベート,その他が含まれている
F-17
戻ってきて自発的な患者支援計画下の義務,顧客に支払うべき費用のために用いられる。医療補助と医療保険リベート,その他リベート,費用は課税費用の一部として合併貸借対照表に記録されている。
4.短期投資
その会社は、購入時に有価証券の適切な分類を決定し、各貸借対照表の日付にそのような指定を再評価する。当社はASC 320に基づき、2023年12月31日および2022年12月31日に発行されたすべての有価証券を“販売可能”に分類している投資--債務と持分証券それは.同社は公正価値記録によって売却可能な証券を記録し、未実現収益と損失を他の累積総合収益(損失)の単独構成部分として計上している。いくつありますか
同社は債務証券を売却できるコストを調整し、割増と満期時の割引を増加させる。このような償却と付加価値は利息収入に計上される.証券売却のコストは具体的な識別方法によって決定される。その会社は証券を売ることができる利息と配当金に利子収入を計上するように分類される。当社の証券売却に係る課税利息は,添付の簡明総合貸借対照表における前払い費用及びその他の流動資産に記載されており,総額は$
以下に12カ月までに赤字を達成していない売却可能証券の概要を示す2023年12月31日および2022年12月31日(単位:千):
|
|
2023年12月31日 |
|
|
2022年12月31日 |
|
||||||||||
|
|
公正価値 |
|
|
未実現損失 |
|
|
公正価値 |
|
|
未実現損失 |
|
||||
国庫券 |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
$ |
( |
) |
|
国庫券 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
( |
) |
|
会社債務証券 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
( |
) |
|
機構債券 |
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
未実現損失状態にある売却可能証券総額 |
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
||
2023年12月31日現在、会社の証券組合は
2023年1月1日までに、会社は貸借対照表の日に非一時的減値の短期投資を評価した。非一時的と判断された公正価値の低下(あれば)も他の純収益に含まれる。非一時的な価値下落の短期投資を評価する際に、当社が考慮する要因は、価値下落が元のコストに占める割合がどれだけ大きいか、投資の時価がその元のコストよりも低い時間がどのくらいあるか、および当社は、公正な価値および全体的な市場状況の任意の予想回復を達成するために、投資を十分に長い時間保持する能力および意図があることを含む。同社は、2022年12月31日現在、非一時的減値の投資を持っていないことを決定した。
販売可能な短期投資に分類されるのは、以下のことを含む
2023年12月31日 |
|
償却する |
|
|
実現していない |
|
|
実現していない |
|
|
公平である |
|
||||
|
|
(単位:千) |
|
|||||||||||||
国庫券 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|||
機構債券 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
||
短期投資総額 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
F-18
2022年12月31日 |
|
償却する |
|
|
実現していない |
|
|
実現していない |
|
|
公平である |
|
||||
|
|
(単位:千) |
|
|||||||||||||
国庫券 |
|
$ |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
|
||
国庫券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
商業手形 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
会社債務証券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
機構債券 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
短期投資総額 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
5.在庫
在庫には以下の内容が含まれている
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
|
|
(単位:千) |
|
|||||
原料.原料 |
|
$ |
|
|
$ |
|
||
Oracle Work in Process |
|
|
|
|
|
|
||
完成品 |
|
|
|
|
|
|
||
総在庫 |
|
$ |
|
|
$ |
|
||
経営陣の判断により、将来の商業化が可能であり、将来の経済効果が期待できると考えられた場合、会社は規制部門の承認後に会社製品に関する在庫コストを資本化する。2023年12月31日までに会社は$
6.財産と設備、純額
財産と設備、純額は:
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
|
|
(単位:千) |
|
|||||
家具と固定装置 |
|
$ |
|
|
$ |
|
||
コンピュータハードウェアとソフトウェア |
|
|
|
|
|
|
||
賃借権改善 |
|
|
|
|
|
|
||
建設中の工事 |
|
|
|
|
|
|
||
総資産と設備 |
|
|
|
|
|
|
||
減算:減価償却累計 |
|
|
( |
) |
|
|
( |
) |
財産と設備の合計 |
|
$ |
|
|
$ |
|
||
F-19
7.課税料金
計算すべき費用には以下が含まれている
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
|
|
(単位:千) |
|
|||||
外部研究と開発に対応すべきである |
|
$ |
|
|
$ |
|
||
福祉と奨励的報酬を計算すべきである |
|
|
|
|
|
|
||
応算製造業 |
|
|
|
|
|
|
||
相談料やその他の専門費を計算します |
|
|
|
|
|
|
||
リベートと自己援助を計算しなければならない |
|
|
|
|
|
|
||
課税税 |
|
|
|
|
|
|
||
その他の課税費用 |
|
|
|
|
|
|
||
費用総額を計算する |
|
$ |
|
|
$ |
|
||
8.変換可能なチケット
2021年債券を発行する
2021年1月、当社はドルを発行します
当社は公正価値選択権項の2021年手形の資格に適合し、それを計算することを選択したため、潜在的な内蔵派生製品の特徴の分析を迂回した。当社は、公正価値オプションの方が2021年債の基本的な経済状況を反映すると信じている。したがって,2021年に発行された手形は発行時に公正価値で入金され,これはこれらの手形の元本金額#ドルに等しいと決定される
2021年紙幣の両替
2021年7月、会社は融資取引を完了し、C-1シリーズの償還可能な転換可能優先株を発行した。今回の融資取引の完成により、2021年手形はその元の条項に従って自動的にC-2シリーズの償還可能な転換可能優先株に変換された(C-1シリーズの償還可能な転換可能な優先株、即ち“Cシリーズ優先株”と一緒に)。Cシリーズ優先株の公正価値は#ドルに決定された
変換可能チケット関連先
いくつありますか
F-20
9それは.公正価値計量
以下の表は、同社が公正価値に応じて恒常的に計量する金融資産と負債の情報を示し、このような公正価値を決定するための公正価値レベルを示す
|
|
2023年12月31日 |
|
|
|
|
||||||||||
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
|
合計する |
|
||||
|
|
(単位:千) |
|
|||||||||||||
資産: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
現金等価物 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
短期投資: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
国庫券 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
機構債券 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
短期投資総額 |
|
|
|
|
|
|
|
|
— |
|
|
|
|
|||
制限現金等価物 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
金融資産総額 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|||
|
|
2022年12月31日 |
|
|
|
|
||||||||||
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
|
合計する |
|
||||
|
|
(単位:千) |
|
|||||||||||||
資産: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
現金等価物 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|||
短期投資: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
国庫券 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
国庫券 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
商業手形 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
会社債務証券 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
機構債券 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
短期投資総額 |
|
|
|
|
|
|
|
|
— |
|
|
|
|
|||
制限現金等価物 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
金融資産総額 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|||
短期投資の価値評価
当社はその通貨市場基金、国庫券および国庫券を公正価値システム下の1級資産に分類しており、これらの資産はアクティブ市場で同じ資産の見積市場価格で評価されており、評価調整を必要としないためである。同社は、取引情報、仲介人または取引業者のオファー、入札、要約、またはこれらのデータソースの組み合わせを含む可能性がある第三者定価サービスによって各貸借対照表の日付で得られた情報を使用して、観察可能な市場入力を使用して推定されるので、その商業手形、会社債務証券、および機関債券を公正価値レベルの2次資産に分類する。
その会社はやった
いくつありますか
10.レンタル証書
同社は、2026年10月まで、取消不可能な経営リースでオフィス施設をレンタルしている
F-21
ASC 842に要するレンタル費用の構成要素を以下に示す2023年12月31日および2022年12月31日:
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
|
|
(単位:千) |
|
|||||
レンタル料 |
|
|
|
|
|
|
||
リースコストを経営する |
|
$ |
|
|
$ |
|
||
総賃貸コスト |
|
$ |
|
|
$ |
|
||
賃貸負債は、賃貸手配による残りの賃貸支払いの現在値を計算することで計量される。私どものレンタルに隠されている金利は確定しにくいため、当社は推定した逓増借款金利を用いて、残りの賃貸支払いの現在値を計算するための割引率を決定します。逓増借款金利とは、当社が支払わなければならない金利であり、経済環境下の賃貸年期のような賃貸支払いに担保方式で借入することである。逓増借款金利は開始日の情報に基づいて決定される。当社は最近対外借金をしていないため、増額借款は吾らの当社の信用格付けに対する理解に基づいて定められた仮定金利であり、担保借入を反映するように調整されている。
同社の賃貸借には継続オプションが含まれており、賃貸契約をさらに数年延長することができる。当社はこのような継続選択権を合理的に行使することはできないため、賃貸条項を決定する際にはそのような選択権は考慮されず、関連する潜在的な追加支払いも賃貸支払いに含まれていない。当社は、各レンタル構成要素とその関連する非レンタル構成要素を単一賃貸構成要素として会計処理することを選択し、リース構成要素の間にのみすべての契約対価格を割り当てることを選択した。当社は、非レンタル部分(公共エリアメンテナンスなど)が実際に発生したコストに応じて賃貸料とは別に支払われている既存の純賃貸を有しているため、経営賃貸使用権資産や賃貸負債には含まれておらず、発生期間を費用として反映している。
以下の表は、同社の総合貸借対照表における経営リース列報の状況をまとめたものである
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
|
|
(単位:千) |
|
|||||
資産 |
|
|
|
|
|
|
||
経営的リース使用権資産 |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
負債.負債 |
|
|
|
|
|
|
||
経営賃貸使用権負債流動 |
|
$ |
|
|
$ |
|
||
経営的リース使用権負債は当期分を差し引く |
|
|
|
|
|
|
||
リース負債総額を経営する |
|
$ |
|
|
$ |
|
||
当社は、2023年12月31日および2022年12月31日までの期間内に、以下の経営リースについて現金を支払います$
|
|
自分から |
|
|
2024 |
|
$ |
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 |
|
|
— |
|
2028 |
|
|
— |
|
未割引賃貸支払総額 |
|
|
|
|
差し引く:推定利息 |
|
|
( |
) |
リース負債総額を経営する |
|
$ |
|
|
加重平均残存期間は、2023年12月31日および2022年12月31日まで
F-22
11.転換可能優先株式償還可能
2021年7月1日、会社は許可された会社登録証明書を修正しました
2021年7月、当社は融資取引を完了し、その取引において、会社が発行した
同社の償還可能な転換可能優先株には以下の内容が含まれる
|
|
2021年12月31日 |
|
|||||||||||||||||
|
|
(千ドル) |
|
|||||||||||||||||
|
|
優先して優先する |
|
|
優先株 |
|
|
携帯する |
|
|
清算する |
|
|
普通株 |
|
|||||
Aシリーズ優先株 |
|
|
|
|
|
|
|
$ |
|
|
$ |
|
|
|
|
|||||
Bシリーズ優先株 |
|
|
|
|
|
|
|
$ |
|
|
$ |
|
|
|
|
|||||
C-1シリーズ優先株 |
|
|
|
|
|
|
|
$ |
|
|
$ |
|
|
|
|
|||||
C-2シリーズ優先株 |
|
|
|
|
|
|
|
$ |
|
|
$ |
|
|
|
|
|||||
|
|
|
|
|
|
|
|
$ |
|
|
$ |
|
|
|
|
|||||
2022年1月、会社初公募が完了した後、会社が発行したすべての優先株を普通株に転換する。いくつありますか
12.株主資本(損失))
普通株-普通株ごとに所有者に権利を持たせる
同社は発行のために普通株式を予約しており、以下の事項に関連している
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
普通株式を許可する |
|
|
|
|
|
|
||
発行済みと発行済み普通株式 |
|
|
|
|
|
|
||
将来発行のための普通株式の許可と予約: |
|
|
|
|
|
|
||
株式オプション行使のために保留された普通株 |
|
|
|
|
|
|
||
未帰属限定株式単位のための普通株 |
|
|
|
|
|
|
||
将来の株式に基づく奨励保留の普通株を発行する |
|
|
|
|
|
|
||
将来の発行のために予約された普通株式の総数を許可して |
|
|
|
|
|
|
||
将来発行可能な非備蓄普通株 |
|
|
|
|
|
|
||
2022年1月、会社は初公募株を完成し、発行と販売
F-23
2022年10月、当社は後続公開を完了し、発行しました
13.株式オプションおよび贈与計画
株式インセンティブ計画-2022年1月、会社の取締役会が採択され、会社の株主は2022年の株式オプションとインセンティブ計画を承認し、あるいは2022年計画と呼ばれ、この計画は2022年1月5日に発効する
当初、2022年計画の規定に基づいて調整した後、2022年計画により発行可能な会社普通株の総株式数は
一カレンダー年度内に、2022年計画又はその他の方法で任意の個人非従業員取締役に付与された会社の普通株式の最高数に加えて、当該カレンダー年度内に当該非従業員取締役に当社の取締役会に勤務している会社に支払われるいかなる現金費用も、$を超えない
2015年計画に基づいて付与されたすべてのオプションと奨励は会社普通株で構成されている。2022年1月6日までに
インセンティブ計画-2023年7月、会社の取締役会はAmylyx PharmPharmticals,Inc.2023激励計画を通過し、現在会社に雇われていない高素質の潜在幹部と従業員を誘致し、彼らに会社の独自の権益を提供するために株式奨励を授与した。当社はすでに予約しました
従業員の株購入計画-2022年1月、会社取締役会は2022年従業員の株式購入計画を採択し、その後、会社の株主の承認を得た。ESPPは最初に最大合計の発行を保留して許可しました
通常のオプション情報
F-24
同社はブラック·スコアーズオプション定価モデルを用いて株式オプション奨励の付与日における公正価値を推定し、以下の加重平均仮定を採用した
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
授権価格 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
無リスク金利 |
|
|
% |
|
|
% |
|
|
% |
|||
予想期限(年単位) |
|
|
|
|
|
|
|
|
|
|||
予想変動率 |
|
|
% |
|
|
% |
|
|
% |
|||
配当率 |
|
|
% |
|
|
% |
|
|
% |
|||
2023年、2022年および2021年12月31日までに年度内に付与された1株当たりの加重平均授受日の株式購入の公正価値は$
のオプション活動の概要
|
|
量 |
|
|
重み付けの- |
|
|
重み付けの- |
|
|
骨材 |
|
||||
2022年12月31日に返済されていません |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
授与する |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
鍛えられた |
|
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
キャンセルまたは没収 |
|
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
2023年12月31日現在の未返済債務 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2023年12月31日に行使できます |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2023年12月31日に帰属していません |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
株式オプションの総内的価値は、会社普通株公正価値よりも低い価格を行使するオプションの行権価格と会社普通株公正価値との差額として計算される。2023年,2022年および2021年12月31日までに年度内に行使されたオプションの内的価値の合計は$
2023年,2022年および2021年12月31日までの年度内に帰属する株式オプションの総公平価値は$
制限株式単位活動
本年度までの限定株式単位活動概要2023年12月31日、具体的な状況は以下の通り
|
|
株式数 |
|
|
加重平均付与日公正価値 |
|
||
2022年12月31日現在帰属していません |
|
|
|
|
$ |
|
||
授与する |
|
|
|
|
$ |
|
||
既得 |
|
|
( |
) |
|
$ |
|
|
没収される |
|
|
( |
) |
|
$ |
|
|
2023年12月31日現在帰属していません |
|
|
|
|
$ |
|
||
株の報酬に基づいて
F-25
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
|
|
(単位:千) |
|
|||||||||
研究開発費 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
販売、一般、行政費用 |
|
|
|
|
|
|
|
|
|
|||
株に基づく報酬総額 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
会社は株式報酬費を資本化した$
次の表は、報酬タイプ別に株ベースの報酬をまとめています
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
|
|
(単位:千) |
|
|||||||||
株式オプション |
|
$ |
|
|
$ |
|
|
$ |
|
|||
制限株式単位 |
|
|
|
|
|
|
|
|
— |
|
||
株式に基づく報酬総支出 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
下表はこれまでの未確認株式報酬支出をまとめたものである2023年12月31日、報酬タイプおよびその費用が確認される予定の加重平均期間。未確認株による補償費用総額は、実際の没収が発生した場合に応じて調整されます。
|
|
2023年12月31日まで |
|
|||||
|
|
未確認費用 |
|
|
加重平均識別期間 |
|
||
|
|
(単位:千) |
|
|
(単位:年) |
|
||
株式オプション |
|
$ |
|
|
|
|
||
制限株式単位 |
|
$ |
|
|
|
|
||
14.所得税
所得税準備金控除前の純損失の構成は以下のとおりである
|
|
現在までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
|
|
(単位:千) |
|
|||||||||
アメリカです。 |
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
アメリカではない |
|
|
|
|
|
|
|
|
( |
) |
||
所得税前収入 |
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
所得税の規定は以下のとおりである
|
|
現在までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
|
|
(単位:千) |
|
|||||||||
現行の所得税支給 |
|
|
|
|
|
|
|
|
|
|||
アメリカ-連邦 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
アメリカ-州 |
|
|
|
|
|
— |
|
|
|
— |
|
|
アメリカではない |
|
|
|
|
|
|
|
|
— |
|
||
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
||
繰延所得税の準備 |
|
|
|
|
|
|
|
|
|
|||
アメリカではない |
|
$ |
( |
) |
|
$ |
— |
|
|
$ |
— |
|
所得税支給 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
||
F-26
当社の有効所得税率と米国法定連邦所得税率
|
|
現在までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
アメリカの法定税率で課税する |
|
|
% |
|
|
% |
|
|
% |
|||
州所得税割引 |
|
|
% |
|
|
% |
|
|
% |
|||
研究開発税収控除 |
|
|
( |
)% |
|
|
% |
|
|
% |
||
役員報酬 |
|
|
% |
|
|
( |
)% |
|
|
— |
% |
|
不確定税収状況 |
|
|
% |
|
|
( |
)% |
|
|
( |
)% |
|
推定免税額 |
|
|
( |
)% |
|
|
( |
)% |
|
|
( |
)% |
他にも |
|
|
% |
|
|
( |
)% |
|
|
( |
)% |
|
有効所得税率 |
|
|
% |
|
|
( |
)% |
|
|
% |
||
繰延税金資産と負債は以下の通り
|
|
現在までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
|
|
(単位:千) |
|
|||||
繰延税金資産: |
|
|
|
|
|
|
||
連邦純営業損失繰越 |
|
$ |
|
|
$ |
|
||
国有純営業損失を繰り越す |
|
|
|
|
|
|
||
資本化研究開発コスト |
|
|
|
|
|
|
||
在庫品 |
|
|
|
|
|
|
||
税金控除 |
|
|
|
|
|
|
||
株に基づく報酬 |
|
|
|
|
|
|
||
応算項目その他 |
|
|
|
|
|
|
||
繰延税金資産総額 |
|
$ |
|
|
$ |
|
||
推定免税額 |
|
|
( |
) |
|
|
( |
) |
繰延税項目純資産総額 |
|
$ |
|
|
$ |
|
||
繰延税金負債: |
|
|
|
|
|
|
||
他にも |
|
|
( |
) |
|
|
( |
) |
繰延税金負債総額 |
|
$ |
( |
) |
|
$ |
( |
) |
繰延税項目純資産 |
|
$ |
|
|
$ |
— |
|
|
当社は確立された推定免税額を定期的に再評価し、すべての正と負の証拠をトレードオフする。2023年、会社は評価準備を再評価し、2023年12月31日までの3年間の累積損失、および最近の監督管理機関が許可したALBRIOZAとRELYVRIO、2023年の利益と正キャッシュフロー、および前年のアメリカ連邦と州NOLの一部および研究開発税収の繰越を実現することを含む負の証拠を考慮した。マイナスとプラスの証拠を評価した後、同社は2023年12月31日までの繰延税項目純資産は引き続き完全な推定値を維持すべきだと結論した。短期的には推定手当の全部または一部が支給される可能性がある。推定手当の支給及び支給の正確な時間と金額は依然として利益レベル、収入の増加、臨床プロジェクトの進展と未来の利益能力に対する期待などの要素の影響を受ける。
2023年12月31日と2022年12月31日現在、同社の連邦NOL赤字繰越は約$
IRC第382条及び383条の規定により、NOL及び研究開発税収控除繰越の使用はかなりの年間制限を受ける可能性がある。所有権変更は2016年12月31日までの年度と
F-27
2023年。これらの所有権変動は会社のNOL繰越と研究開発税収控除の全体能力に影響を与えないが、毎年将来の課税収入を相殺するために使用できる金額を制限する可能性がある。
下表は会社の年度までの見積計上の繰り越し状況を反映している2023年12月31日、2022年、2021年:
|
|
現在までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
|
|
(単位:千) |
|
|||||||||
年初の評価免税額 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
所得税計上の増加 |
|
|
( |
) |
|
|
|
|
|
|
||
年末評価免税額 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
本年度に収録された推定免税額の減少は,主に2023年の税引き前利益による課税収入と,研究開発費の資本化により増加した課税収入に関連している。
米国会計基準第740条の規定によると、同社は、連結財務諸表における納税申告の一頭寸の利益を確認する敷居を“より可能性がある”と定義して税務機関の支援を受けている所得税における不確実性を会計処理している。税金優遇は最終移民が達成される可能性が50%を超える最大優遇によって測定されるt.
|
|
現在までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
|
|
(単位:千) |
|
|||||||||
期初残高 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
数年前の税務頭寸に関する増加(減少) |
|
|
|
|
|
( |
) |
|
|
— |
|
|
今年度の税収頭寸に関する増加 |
|
|
|
|
|
|
|
|
|
|||
期末残高 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
当社は現在税務機関が審査できるすべての課税年度の申告表内で採用されているか、あるいは取る税務立場について検討しています。すべての不確定な税収割引が確認されれば、確認後の実際の税率に影響を与えるが、会社の推定手当の変化によって相殺され、実際の税率にも影響を与える
同社は異なる司法管轄区でアメリカ連邦、外国、州所得税申告書を提出した。制限の状況は管轄区域によって異なる。現在連邦や州の監査や行われている検討はない。
15. 従業員福祉計画
当社は1つを維持するこれは条件に合ったアメリカ人従業員に税金優遇に基づいて退職貯蓄の機会を提供する。計画参加者は適用されたIRC年度制限に応じて条件に合った報酬を延期することができる。この年度までに2022年12月31日、会社は避難港貢献を提供したf
1株当たり純収益
1株当たり純収益
1株あたりの基本収益の算出方法は,純収益(損失)を当期発行普通株の加重平均で割ったものである。希釈後の1株当たり収益は合併加重平均値から計算されます
F-28
普通株式および潜在的希薄化株式の数は、行使を想定して行使された従業員株式オプションおよび非帰属制限株式単位を含む。希釈後の1株当たり収益を計算する際には、会社は在庫株方法を採用している。
1株当たり収益を計算する際に使用する分子と分母の要約は以下のとおりである(単位:千,1株当たりおよび1株当たりデータは含まれていない:
|
|
十二月三十一日 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
分子: |
|
|
|
|
|
|
|
|
|
|||
純収益(赤字) |
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
分母: |
|
|
|
|
|
|
|
|
|
|||
1株当たりの基本純利益(損失)を計算するための加重平均株式 |
|
|
|
|
|
|
|
|
|
|||
従業員株式オプションと制限株式単位の希釈効果 |
|
|
|
|
|
— |
|
|
|
— |
|
|
加重平均株式数は,1株当たり減額後の純収益(損失)の計算に用いられる |
|
|
|
|
|
|
|
|
|
|||
1株当たり純収益 |
|
|
|
|
|
|
|
|
|
|||
基本的な情報 |
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
薄めにする |
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
同社は2022年12月31日と2021年12月31日までの12カ月間の純損失を報告したため、1株当たりの基本純損失は希釈後の純損失と同じだった。これらの証券は、2022年12月31日と2021年12月31日までの12ヶ月間に逆希釈影響を与えるため、すべての株式オプションおよび制限株式単位は希釈加重平均流通株の計算から除外される.
|
|
十二月三十一日 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
普通株購入オプション |
|
|
|
|
|
|
|
|
|
|||
制限株式単位 |
|
|
|
|
|
|
|
|
— |
|
||
転換可能優先株を償還する |
|
|
— |
|
|
|
— |
|
|
|
|
|
除外された普通株式総等価物 |
|
|
|
|
|
|
|
|
|
|||
17.関連するパーティ取引
転換可能な手形
2021年債券の発行については、当社は合わせて$を発行しております
仕入先協定
通常業務中、当社は当社の関連先の基準を満たす側に関連する実体から材料、用品、またはサービスを購入することができます。これらの取引は四半期ごとに審査されており、これまでこれらの取引は会社の総合財務諸表にとって重要ではなかった。
18.支払いの引受およびまたは事項
法律訴訟-当社は2023年12月31日現在、いかなる重大な法的手続きの一方でもありません。報告日ごとに、当社は潜在損失金額又は潜在損失範囲が権威的な案内処理又は事項の規定によって可能かつ合理的に推定されているかどうかを評価する。当社はその法的訴訟に関する費用が発生したことを確認した。
特許権使用料の支払い-中間にある2016年8月と2019年2月、会社はALS協会、ALS発見治癒基金、アルツハイマー病薬物発見基金、アルツハイマー病協会と
F-29
癒すアルツハイマー病基金や贈与者です協定の条項によると,同社は全部で$を獲得した
会社、筋萎縮性側索硬化症協会、筋萎縮性側索硬化症発見治癒のそれぞれの贈与協定の条項によると、当社は授与者1人当たりに総額等しい支払いを要求されます
会社、アルツハイマー薬発見基金、アルツハイマー協会とアルツハイマー治療基金の間のそれぞれの贈与協定の条項によると、会社は最高額を$に支払う
調達承諾-当社は通常業務中に契約製造組織と原材料調達と製造サービスについて合意しています。同社は2023年12月31日までに約束した$
19それは.後続事件
2024年2月9日、米国ニューヨーク南区地方裁判所は、同社とその一部の現職および前任上級管理者(ShihはAmylyx製薬会社らの事件を訴えた。案件番号1:24-CV-00988(“Shihクレーム”)。施正栄の起訴状は、取引所法案第10(B)節及びその公布された第10 b-5条に違反したことを告発し、第20条(A)条に基づいて特定の現職及び前任者を告発し、告発された統制者であることを告発した。施正栄の起訴状によると、被告はRELYVRIOの商業結果と将来性について重大な虚偽と誤解性陳述をしたという。施正栄の訴状は、指定されていない損害賠償、利息、費用及び弁護士費、並びに裁判所が適切と考えている他の指定されていない救済を要求する。その会社は施正栄の苦情を強力に弁護しようとしている。現在,これらのクレームの影響を見積もることはできない(あれば).
F-30