アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
本財政年度末まで
あるいは…。
1934年証券取引法第13条又は15(D)条に基づいて提出された |
手数料書類番号
(登録者の正確な氏名はその定款に記載)
(国やその他の管轄区域 会社または組織の名称) |
(税務署の雇用主 識別番号) |
(主にオフィスアドレスを実行) |
(郵便番号) |
登録者の電話番号、市外局番を含む:(
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
|
取引コード |
|
登録された各取引所の名称 |
|
|
同法第12条(G)に基づいて登録された証券:ありません
登録者が証券法第405条に規定する有名な経験豊富な発行者である場合は、再選択マークで表示してくださいはい、そうです ☐
登録者がこの法第13又は15(D)条に従って報告書を提出する必要がないか否かを、再選択マークで示すはい、そうです ☐
登録者が(1)過去12ヶ月以内(または登録者がそのような報告を提出する必要があるより短い期間)に1934年の証券取引法第13条または15(D)条に提出されたすべての報告書を再選択マークで示すかどうか、および(2)このような提出要求を過去90日以内に遵守してきた
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
大型加速ファイルサーバ |
|
☐ |
|
ファイルマネージャを加速する |
|
☐ |
|
|
|
|
|
|
|
|
☒ |
|
規模の小さい報告会社 |
|
||
|
|
|
|
|
|
|
新興成長型会社 |
|
|
|
|
|
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に第240.10 D−1(B)条に従って受信されたインセンティブベースの補償に従って回復分析を行う必要があるかどうかを再選択マークで示す☐
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)はい、そうです
2023年6月30日現在,登録者の非関連会社が保有する投票権と無投票権普通株の総時価は約$
2024年3月4日現在、登録者の発行済み普通株式数は
引用で編入された書類
本報告の第3部が開示を要求するいくつかの情報は、引用的に登録者の最終委託声明に組み込まれる2024年次株主総会では,依頼書は本報告でカバーした財政年度終了後120日以内に提出される。
カタログ表
|
|
ページ |
第1部 |
|
|
第1項。 |
業務.業務 |
1 |
第1 A項。 |
リスク要因 |
56 |
項目1 B。 |
未解決従業員意見 |
113 |
プロジェクト1 C。 |
ネットワーク·セキュリティ |
113 |
第二項です。 |
属性 |
114 |
第三項です。 |
法律訴訟 |
114 |
第四項です。 |
炭鉱安全情報開示 |
114 |
|
|
|
第II部 |
|
|
五番目です。 |
登録者普通株市場、関連株主事項及び発行者による株式証券の購入 |
115 |
第六項です。 |
[保留されている] |
115 |
第七項。 |
経営陣の財務状況と経営成果の検討と分析 |
116 |
第七A項。 |
市場リスクの定量的·定性的開示について |
133 |
第八項です。 |
財務諸表と補足データ |
134 |
第九項です。 |
会計と財務情報開示の変更と相違 |
134 |
第9条。 |
制御とプログラム |
134 |
プロジェクト9 B。 |
その他の情報 |
135 |
プロジェクト9 C |
検査妨害に関する外国司法管区の開示 |
135 |
|
|
|
第三部 |
|
|
第10項。 |
役員·幹部と会社の管理 |
136 |
第十一項。 |
役員報酬 |
136 |
第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
136 |
十三項。 |
特定の関係や関連取引、取締役の独立性 |
136 |
14項です。 |
最高料金とサービス |
136 |
|
|
|
第4部 |
|
|
第十五項。 |
展示·財務諸表明細書 |
137 |
第十六項。 |
表格10-Kの概要 |
169 |
i
前向き陳述に関する特別説明
本年度報告書には前向きな陳述が含まれている。本年度報告における歴史的事実に関する陳述を除くすべての陳述は、以下の陳述を含む前向き陳述である
“信じる”、“可能”、“将”、“推定”、“継続”、“予想”、“設計”、“意図”、“予想”、“可能”、“計画”、“潜在”、“予測”、“求める”、“すべき”、“できる”、またはこれらの言葉の否定バージョン、および同様の表現は、前向き陳述を識別することを意図している。これらの展望的な陳述は、私たちの現在の未来の事件と傾向の予想と予測に基づいており、これらの事件と傾向は、私たちの財務状況、運営結果、戦略、短期と長期業務運営、および目標と財務需要に影響を与える可能性があると考えられる。
これらの展望的陳述は、“リスク要因”の節で述べたリスク、不確実性、および仮説を含むいくつかのリスク、不確実性、および仮説の影響を受ける。しかも、私たちの運営環境は競争が激しく、変化が迅速だ。新しいリスクが時々発生する。私たちの経営陣はすべてのリスクを予測することはできませんし、すべての要素が私たちの業務に与える影響を評価することもできません。あるいは任意の要素や要素の組み合わせは、実際の結果が私たちが含まれる可能性のある任意の前向きな陳述の結果と大きく異なる程度をもたらす可能性があります
II
製造します。これらのリスク、不確定性と仮定を考慮して、本年度報告で議論された前向き事件と状況は発生しない可能性があり、実際の結果は展望性陳述中の予想或いは示唆の結果と大きく異なる可能性がある。
あなたは未来の事件の予測として前向きな陳述に依存してはいけない。私たちは展望性陳述に反映された予想は合理的であると考えているが、私たちは展望性陳述に反映された未来の結果、進展、発見、活動レベル、業績或いは事件と状況が実現或いは発生することを保証できない。さらに、法的規定を除いて、私たちまたは他の誰もが展望的陳述の正確性と完全性に責任を負わない。私たちは、これらの陳述が実際の結果または私たちが予想している変化と一致するように、本年度報告日の後に任意の理由で任意の前向き陳述を公開更新する義務はありません。
あなたは本年度報告書と私たちが本年度報告書で参考にしてアメリカ証券取引委員会に提出した文書を読んで、私たちの将来の実績、活動レベル、業績、事件、状況が私たちの予想と大きく異なるかもしれません。
私たちの業務に関するリスクの概要
以下は,我々の証券投資に投機的あるいはリスクを持たせる要因の概要である.この結論は私たちが直面しているすべての危険を解決していない。本リスクファクター要約でまとめたリスクおよび我々が直面している他のリスクに関する他の議論は,“リスクファクター”というタイトルの部分で見つけることができ,よく考慮すべきである。
三、三、
四
パー?パーT I
項目1.B役に立つ。
我々は臨床段階の細胞·遺伝子治療会社であり,癌やまれな疾患の患者に新たな治療法を提供している。我々のコア独自プラットフォームに基づいて、我々の非ウイルスiggyBac DNA送達システム、Cas-Clover部位固有遺伝子編集システム、およびナノ粒子およびAAVに基づく遺伝子送達技術を含む、様々な適応の広範な候補製品の組み合わせを発見し、開発している。
著者らの核心プラットフォーム技術は単独で使用することができ、結合して使用することもでき、多種の細胞と遺伝子治療方式に応用することができ、そして著者らの候補製品の組み合わせを設計することができ、現在の世代の細胞と遺伝子治療技術の主要な制限を克服することを目的としている。
細胞療法
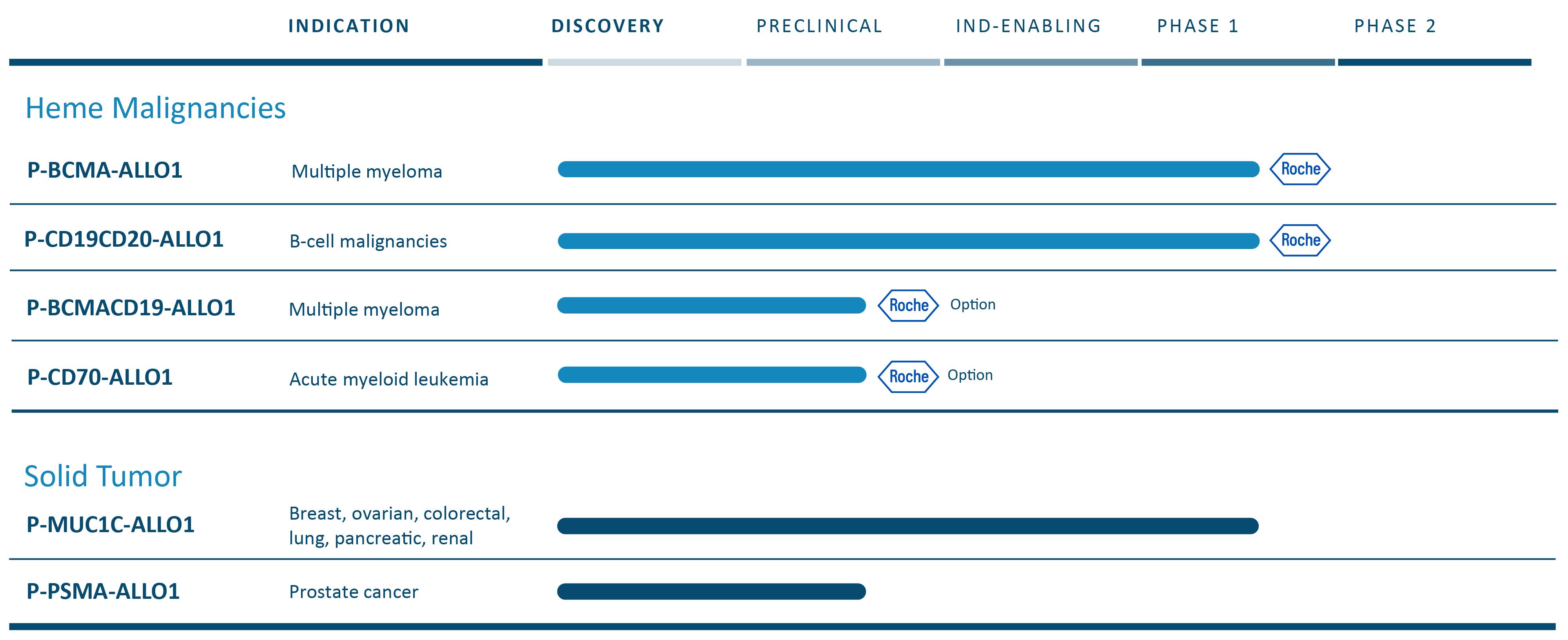
細胞治療において、私たちの技術は、幹細胞記憶T細胞、あるいはT細胞を含む工学細胞を用いて候補製品を作ることができると信じています供給チェーン管理細胞は、患者の体内に移植し、潜在的に持続的な持続的な反応を駆動することができる。我々のキメラ抗原受容体T細胞、またはCAR-T、治療組み合わせは現在同種異体または既製の候補製品を含む。この業界では、異体CAR-T細胞製品は自己製品よりも早く開発され、一部の原因はその生産過程において遺伝子編集技術が必要であるが、この方法はいつでも利用可能で、品質がずっと高い既製製品であるため、この分野の次の重大な進歩になる可能性がある。私たちはすでに血液学的および固形腫瘍適応に私たちの自己プログラムの知識を使用して私たちの同種プログラムを助けている。われわれは広範なパイプラインを進めており,血液や固形腫瘍腫瘍適応の臨床段階では多くのCAR−T候補製品がある。
私たちの内部は固形腫瘍細胞治療に集中している。P-MUC 1 C-ALLO 1は現在第1段階試験にあり、乳癌、卵巣癌、および他の上皮由来の癌を含む多種の固形腫瘍を治療する可能性がある。そのほか、著者らは他のいくつかの実体腫瘍同種異体計画があり、期待される研究新薬(IND)に書類を提出しており、P-PSMA-ALLO 1、転移性去勢耐性前立腺癌、或いはmCRPCの治療のために開発されている臨床前段階計画である。このプログラムは我々の自己バージョンP−PSMA−101の研究結果を用いており,第1段階研究で38名の患者を治療しており,これらの発見が異遺伝子計画に有用であると信じている。著者らはまた固形腫瘍と細胞治療における他の方法のいくつかの計画を探索しており、現在臨床前段階であり、その標的はまだ開示されておらず、及び他の組み合わせは、潜在的なCAR-TとT細胞受容体(TCR)を含む二重製品を含む。
戦略的パートナーシップ
2022年8月、私たちはF.Hoffmann-La Roche LtdとHoffmann-La Roche Inc.,または総称してRocheと呼ばれ、パートナー関係を構築することを発表し、この2つのパートナーの中で、彼らは私たちの主要な血液学的適応を許可または選択した。プリペイドライセンスに含まれ、ロー氏は、P−BCMA−ALLO 1およびP−CD 19 CD 20−ALLO 1を許可するか、またはそれぞれ1つのTier 1計画を許可する。P−BCMA−ALLO 1は現在1期試験であり,再発性/難治性多発性骨髄腫患者のために開発されている。P-CD 19 CD 20-ALLO 1はB細胞血液学的徴候を治療するために開発した計画であり、著者らはIND文書を提出した後、著者らは2023年6月にFDAの承認を得て、著者らは1期の臨床試験を開始できるようにした。この2つの許可計画に加えて,羅氏はP-CD 70-ALLO 1とP-BCMACD 19-ALLO 1を許可するか,2つのTier 2計画をそれぞれ許可することができる.P-CD 70-ALLO 1は臨床前段階の方案であり、血液学的適応の治療に開発されている。P-BCMACD 19-ALLO 1は1種の臨床前両標的方案であり、多発性骨髄腫の治療に開発されている。Tier 1とTier 2計画以外に、著者らは羅氏会社と研究協力を達成し、その中で羅氏会社は著者らのある知的財産権の下で独占的な許可を持ち、最大6種類の血液学適応の同種異体CAR-T細胞治療製品、或いは各製品の1つの協力計画を開発、製造と商業化することができる。
1
我々が羅氏と締結した協力·許可協定や羅氏協力協定によると、羅氏は私たちに1.1億ドルを前払いした。ロ氏の第2レベル計画オプションの行使、協力計画の指定、許可製品の選択権の行使(以下に定義する)、商業許可、および第1レベル計画、オプションの第2レベル計画および協力計画によって特定の開発、規制、および純売上マイルストーン事件を実現する製品によると、特定の精算、費用、マイルストーン支払いを取得する資格があり、合計60億ドルに達し、(I)第1レベル計画15億ドル、(Ii)第2レベル計画11億ドル、(Iii)協力計画29億ドル。(Iv)特許製品の4.15億ドルです我々はまた,製品ごとに,Tier 1計画,オプションTier 2計画と連携計画による製品純売上高の中位から下位2桁,および許可製品の中央桁から中央値の階層印税支払いを許可する権利があり,いずれの場合も,いくつかの通常の減免と補償の制限を受けている.使用料は、その製品をカバーするライセンス特許がその国で満期になるまで、またはその製品が同国で初めて商業販売された日から10年まで製品および国/地域によって支払われる。
細胞治療管
遺伝子治療
遺伝子治療において,我々の技術は,長期的で安定した遺伝子発現を提供し,時間の経過とともに減弱することなく,単一療法の治癒につながる可能性がある新たな治療法を創出する可能性があると信じている。われわれの独自遺伝子工学技術は,従来の遺伝子療法の一過性の限局性を解決し,肝臓ガイド遺伝子療法から明らかな優位性を提供する可能性があると信じている。さらに私たちは複数のことを追求する可能性があると信じています体内にあるそして離体する広範な細胞タイプや組織で非肝臓ガイドの遺伝子治療を行う。
私たちは内部に集中しています体内にある遺伝子療法です。P-FVIII-101、iggyBac技術をナノ粒子送達システムと組み合わせてRNAおよびDNAを同時に送達する非ウイルス方法を含む、我々の先行プロジェクト。P-FVIII-101は血友病Aの治療に開発されている。また、P-OTC-101は肝臓ガイドの遺伝子療法であり、iggyBac技術をAAVとナノ粒子と組み合わせて治療に使用している体内にあるオルニチンアミノトランスフェラーゼ欠乏症の治療。OTCDは通常致命的或いは病的な尿素循環疾患であり、オルニチントランスアミノメチル酵素(OTC)遺伝子の先天性突然変異によって引き起こされ、高度に満足されていない医療需要を持っている。FDAは2023年7月、孤児薬としてP-OTC-101を承認した。我々は,非ウイルスナノ粒子送達システムの混合送達RNAとAAVを用いてDNAを送達するP−OTC−101計画を開発しており,この計画のための最新のスケジュールを策定している。
2
遺伝子治療管
私たちの独自の細胞と遺伝子工学プラットフォーム技術は
私たちはすでに独自の遺伝子工学技術を開発し、広範な実用価値を持っている。著者らの技術プラットフォームの広さと深さは主に3種類に分けられる:(1)遺伝子挿入、(2)遺伝子編集と(3)遺伝子交付、追加のCAR-Tツールによって支持される。
3
遺伝子挿入:iggyBac DNA送達システム
DNAトランスポゾンは,切り取りや貼付機構によりプラスミドから染色体に効率的に移動する遺伝子エレメントである。CAR-T製造に含まれる遺伝子転移方法としてDNAトランスポゾンが使用されている。GiggyBac DNA伝達システムは著者らの独自の非ウイルス遺伝子工学技術であり、高効率なSuper iggyBacトランスポナーゼを用いて治療性遺伝子組換えDNAをゲノムに添加することができ、このトランスポナーゼは遺伝子改造された超活性酵素であり、非常に効率的に豚Bacトランスポゾンを転座することができる。PiggyBacは,効率的かつ正確なシフトや多様な異なる製品属性を実現できると信じている.
4
次の図はブタというBac DNA送達システムを示しています
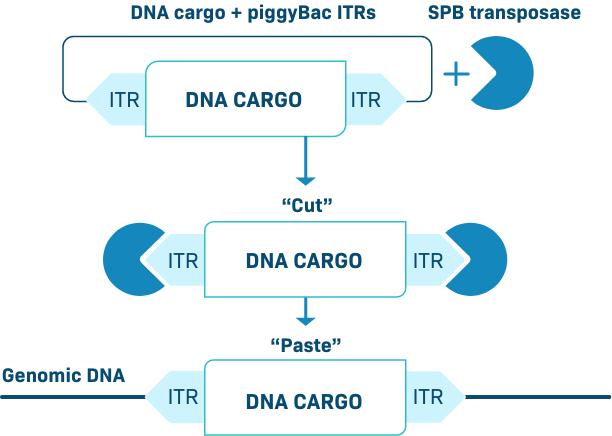
IggyBac DNAトランスポゾン遺伝子組換えの貨物輸送領域内でコードされる治療用遺伝子の両側非翻訳逆末端反復配列(ITRs)は、トランス座酵素がこれらの配列を特異的に認識して、治療用トランスジェニック貨物をゲノム中の特定の配列(TTAAヌクレオチド)に効率的に安定的に統合する。トランスポナーゼは、タンパク質として、またはDNAまたはRNAの形態で細胞にコードされてもよい。
PiggyBacプラットフォームは我々のコア技術であり,CAR−Tや我々が開発している他の候補遺伝子治療製品の開発に用いられている。私たちのiggyBac DNA送達システムは様々な差別化された製品属性を実現できると信じています
5
ブタBacトランスポゾンは優先的に治療用遺伝子組換えをT細胞を含む早期記憶T細胞に転座する供給チェーン管理細胞です。レトロウイルスの遺伝子組換え伝達方法、例えばレンチウイルスとγ逆転写ウイルスは、遺伝子組換えを早期記憶T細胞に伝達する上で有効ではないと考えられる。これは重要な差別化要因であり、高い割合のTを持つCAR-T製品を製造することができます供給チェーン管理細胞は理想的な特徴を与えます
慢性ウイルスやγ逆転写ウイルスなどのCAR−T製造に一般的に用いられるウイルスの遺伝子負荷は約10−20 kbまたはkb程度に制限されているが,iggyBacは200 kbを超える負荷を証明しており,複数の有用な遺伝子の転移を可能にしている。著者らが現在CAR-T候補製品中のすべてのCAR-T細胞が1つのCAR分子遺伝子、1つの安全スイッチ遺伝子と1つの選択遺伝子を携帯することに伴い、iggyBacの超大型キャリア能力は複数の遺伝子を著者らの候補製品に統合することを可能にし、耐性と効力を更に高めることができる。貨物輸送能力はまた、二重および他のマルチCAR-T候補製品を作成するために、複数のCAR-Tコード遺伝子および/またはTCR遺伝子を包装することを可能にする。
PiggyBac ITRsと他のコンポーネントは強力な絶縁体として機能し、安定な遺伝子組換え発現を確保し、腫瘍発生のリスクを低下させる。PiggyBacはレンチウイルスと比較して低い遺伝子内領域統合を示し,有害変異を引き起こす可能性が低いことを意味している。
また,PiggyBacはウイルスベクターのGMP生産に比べて良好な製造仕様やGMP材料の生産コストがはるかに低く,GMP生産時間もはるかに短いと予想される。
次の図は、CAR−T候補製品を作成するためのGiggyBacトランスポゾン遺伝子組換え方法を示す

精密特異的遺伝子編集−CAS−クローバー定点遺伝子編集技術−
我々は,IIS制限エンドヌクレアーゼClo051の一部からなる独自のホモ二量体ヌクレアーゼシステムClo051を用いた遺伝子編集技術を開発した。この酵素のゲノム切断は二量体化に厳密に依存し,完全な二量体系となり,正確な位置特異性を与える。CAS−CloverはCRISPR(クラスター状,規則的間隔の短い回文重複)関連蛋白9,あるいはCas 9を用いており,この酵素は永久的に改変されており,DNA(dCas 9と呼ぶ)を切断できない。DCas 9が適切な誘導RNA(GRNA)に結合する場合,dCas 9はDNA結合蛋白の役割のみを果たす。CAS-CLOVERは第一世代CRISPRシステムの利点(設計が簡単、コストが低く、多重化能力)と独自の相同二量体ヌクレアーゼシステムの利点(精確特異性)を結合した。T細胞の応用には,Cas−Cloverが重要であり,静止したT細胞でよく動作しており,生産過程での成熟や疲労を回避し,T細胞の保存を助けることができる供給チェーン管理表現型です。
6
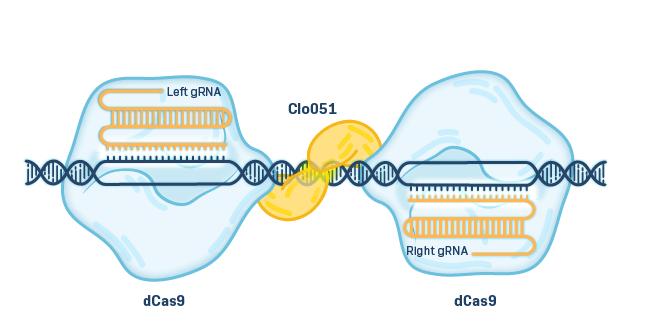
最も広く用いられている遺伝子編集プラットフォームはCRISPRと関連タンパク質Cas 9である。この遺伝子編集技術は細菌から自然に産生されるウイルス防御機構に由来する。その動作原理は,Cas 9酵素を誘導RNAに結合させ,Cas 9酵素を特定のDNA配列に配向させ,二本鎖DNAを切断することである。DNAが切断されると,細胞は自然に発生するDNA修復機構を用いて切断された末端を再接続する。
CRISPR/Cas 9技術は、一般に標的DNA部位と類似しているが完全に同じではない予期されない位置で追加的な切断を行うことを意味する不必要な脱標的切断をもたらすことが証明されている。このような目標から外れた切断はゲノムDNAの永久的な突然変異を招く可能性があり、これは無意識に有害な突然変異と腫瘍の発生を招く可能性があり、それによって細胞と遺伝子治療薬物の製造に重大な安全問題を産生する。
細胞および遺伝子治療のために流行している別の部位特異的遺伝子編集プラットフォームは、転写活性化子様エフェクターヌクレアーゼ、またはTALENである。これらはTAL DNA結合ドメインとDNA切断ドメイン(通常はFokI)を融合させることで構築されており,FokIは独自のホモ二量体として機能しており,2つの半部位がまったく同じ位置とまったく同じ時間に集積しなければ切断できないことを意味している。2つの半位を必要とするため,このタイプのシステムは全二量体系と呼ばれることがある。
TALEN技術は通常、CRISPR/Cas 9よりもはるかに高い忠実度でDNA中の特定の位置を切断することができるが、相対的に労働集約型および建造コストが高い。概念的に類似しており、亜鉛指ヌクレアーゼ、或いはZFN、技術は1種の遺伝子編集技術であり、1種類のDNA結合蛋白からなり、DNA二本鎖を切断させるために用いられる。TALEN技術のように、ZFNは、特定のために必要な編集に必要な配列を作成することによって使用するために、より多くの準備および作業を必要とする。Talen,ZFNともに細胞を活性化して編集する必要があり,静止したT細胞ではうまく動作しないため,高い割合のT細胞を保持することができない供給チェーン管理CAR-T表現型。
もう一つの新しい遺伝子編集技術は塩基編集と呼ばれている。塩基編集はCRISPR系のコンポーネントを用いて他の酵素とともに,二本鎖DNA切断を起こさずに直接細胞DNAやRNAに点変異を装着した。DNA塩基編集は、塩基デアミナーゼに融合した触媒不活性化ヌクレアーゼを含み、場合によってはDNAグリコシル酵素阻害剤をさらに含む。塩基編集技術はある程度の不必要な標的外突然変異が生じることが知られているが、具体的な程度は不明であり、同種異体CAR-Tに対して安全問題を引き起こす可能性があり、製品が多くの患者に服用する可能性があるからである。
7
遺伝子伝達技術:ナノテクノロジー、体内外電気穿孔技術とAAV
非ウイルス遺伝子挿入のためのPiggyBacプラットフォームと遺伝子編集のためのCas-Cloverプラットフォームのほかに、RNA、DNA、タンパク質を細胞に伝達できるように遺伝子伝達のためのプラットフォーム技術を開発しました離体するそして体内にある様々な応用に適している。これらの技術にはナノ粒子技術AAV技術離体するそして体内にある電気穿孔技術と方法です。PiggyBacとCas-Cloverは潜在的な実用価値を持つため,アプリケーションによって異なる配送方式が必要であると予想される.
同種CAR-T製品候補ではT細胞を編集します離体する電気穿孔技術を使用して、治療用トランスジェニックを細胞ゲノムに安定的に挿入するために必要なiggyBacアセンブリを提供する。Cas−Cloverを電気穿孔によりT細胞に導入し,同種反応を除去するために細胞を編集した。
いくつかの肝臓ガイド遺伝子治療計画では、AAV技術と脂質ナノ粒子(LNPs)を使用してiggyBacを肝臓に輸送します体内にあるそれは.様々な送達目標を達成するために、様々な異なるナノ粒子組成物を開発した。これらのナノ粒子は一般に2つに分類され,重合体とLNPsである。重合体は新規ブロック共重合体からなる単成分粒子であり,タンパク質などの大きな複雑分子を輸送することを目的としている。LNPsは既知と新たな脂質からなる多成分ナノ粒子であり,mRNAやDNAを含む核酸の輸送を目指している。私たちはPiggyBacとCas-Clover技術を提供するためにこれらのナノ粒子の概念を評価している。
私たちのナノ粒子プラットフォームの長期目標はAAVへの需要をなくすことです体内にあるナノ粒子を用いて私たちの技術を細胞に伝えることで遺伝子治療を行う。私たちは、私たちの完全な非ウイルス送達技術を使用して、P-FVIII-101プロジェクトを指名し、より多くのプロジェクトをサポートするために、独自のナノテクノロジープラットフォームを積極的に成熟させています。
細胞療法
早期世代CAR−T療法の限界を解決する
早期世代のCAR−T療法はすでに大きな潜在力を示しているにもかかわらず,いくつかの限界も存在する。早期と現在のCAR−T療法の多くはウイルスによる製造を用いて生産されている。ウイルスによる製造に関するいくつかの固有問題は,他のCAR−T療法の潜在力を制限していると考えられる。T細胞工学は通常ウイルス形質導入によって実現され、これはウイルスを用いて外来DNAを細胞に導入する過程であり、最も明らかなのはレトロウイルス、例えばγ逆転写ウイルス或いはレンチウイルスである。
これらのウイルスベクターは広く最適化されているにもかかわらず、挿入輪郭に対する安全懸念、限られた遺伝子負荷量および最終製品の不良特性を含む限界がますます明らかになっている。我々は我々固有の非ウイルスiggyBac DNA送達システムを用いてCAR分子遺伝子をT細胞に送達した。非ウイルス法を用いた最大の利点は,高パーセンテージT細胞からなるCAR−T製品を生成できることである供給チェーン管理細胞です。これは治療がより一致して持続的な反応を引き起こし、毒性がより小さい可能性があると信じている。さらに、私たちの非ウイルス的方法は、より低い製造コストとより短い製造時間を有すると信じている。われわれは,自己CAR−T製品と同様に優れ,さらには優れている可能性があり,既製のコストは自己療法の一部のみである健康ドナー由来の同種異体や既製のCAR−T療法も開発した。
細胞タイプの問題である幹細胞記憶
T供給チェーン管理細胞は細胞治療の理想的な細胞と考えられているが、それらは移植、長寿、自己更新と多能性の潜在力を持っているため、1波また1波のより多くの分化した細胞を作ることができるからである。T細胞には一方向成熟経路があります供給チェーン管理細胞は中央記憶T細胞またはT細胞にセンチメートルそしてエフェクターにT細胞、またはTを記憶するエム!最後にTには電信為替細胞です。T細胞の成熟と分化に伴い、その核心機能と能力は変化し、その効力と持続性に影響する。私たちの方法は私たちの候補製品に高い割合の分化の少ないT細胞を利用することであり、持続性を増加させ、早期世代CAR-T製品のいくつかの重要な制限を緩和することを目標としている。私たちも高いTを持つ製品を作ると信じています供給チェーン管理これが固形腫瘍の臨床効果でこのように成功した理由かもしれません供給チェーン管理細胞は腫瘍を攻撃するために一連の細胞を移植して産生することができる。概念的にはより成熟したより多くのエフェクター細胞を含む製品は薬のようなものです
8
T含有量の高い製品供給チェーン管理細胞はプロドラッグのようなものです“The T”供給チェーン管理細胞は腫瘍細胞を殺さず、体内に移植し、より多くの分化した細胞を作って殺傷する。
次の図はこの一方向T細胞成熟経路をT細胞から説明しています供給チェーン管理細胞からTへ電信為替セル:
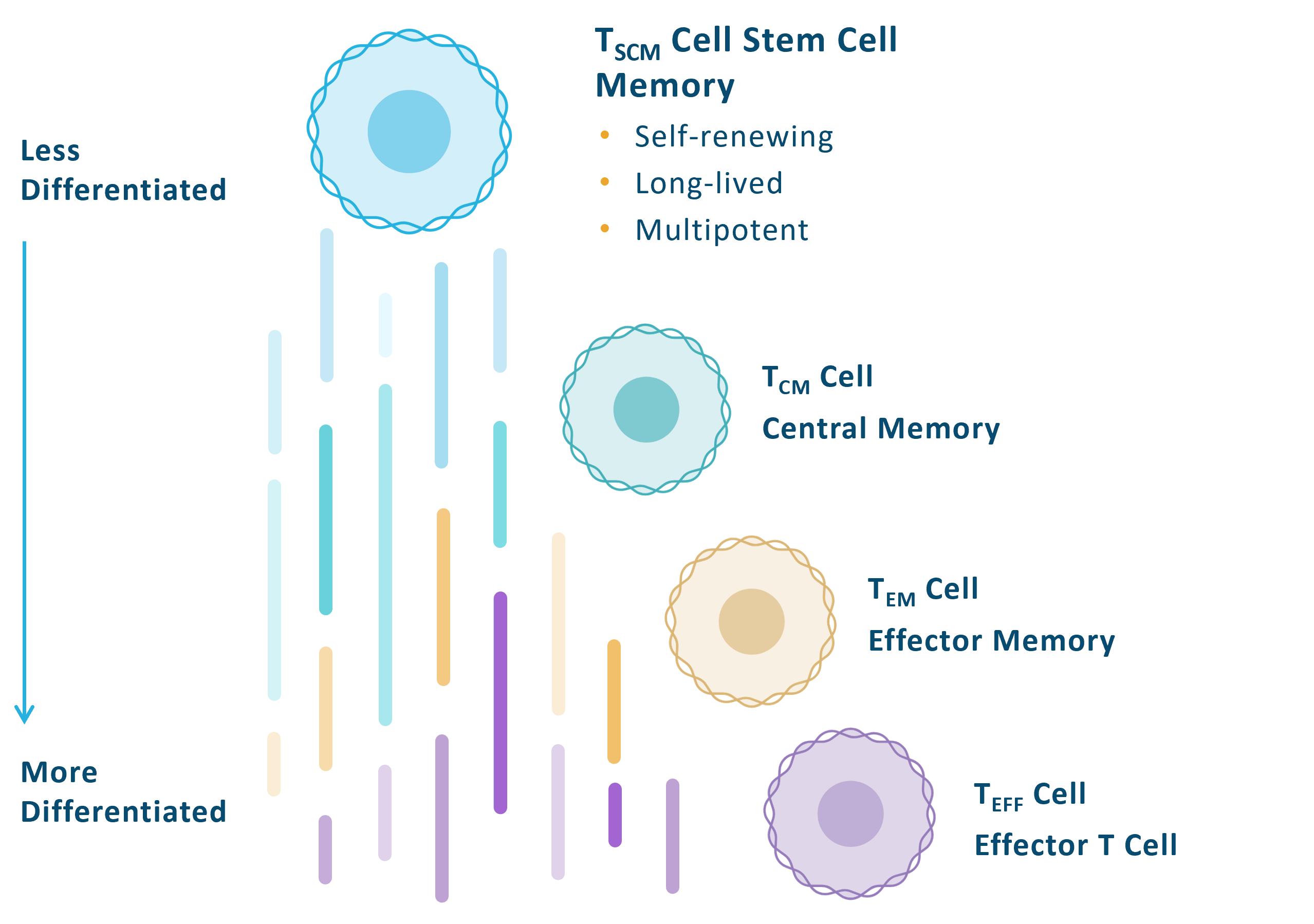
これまでの自己候補製品の臨床データによると,T細胞パーセンテージの間には強い相関が認められた供給チェーン管理製品候補と最適な臨床反応の面で。私たち自身の経験を除いて、ますます多くの証拠とTを認識しています供給チェーン管理臨床治療効果と関連している。
遺伝子編集
遺伝子編集ツールは、ある細胞表面分子の発現を除去するために広く使用されており、これは、ドナー細胞の患者に対する潜在的反応性を回避するために使用される可能性があり、これは、移植片対宿主病(GvHD)、および患者細胞のCAR-T産物に対する反応性をもたらし、この反応は、宿主対移植片と呼ばれる。同種異体CAR−T製品を作成する際には,活性化されたT細胞が成熟経路を起動するため,静止T細胞を効率的に編集できる遺伝子編集ツールを用いる必要があると考えられる。T細胞が成熟すると欲しいT細胞を失い始めます供給チェーン管理そのため疲れ果て、最終製品の効果があまり効果的ではない。
他の多くの遺伝子編集技術とは異なり,Cas-Cloverの方法を用いて静止T細胞を効率的に編集することができ,非常に理想的なT細胞を保持することができる供給チェーン管理製品組成中の同種異体候補製品は、著者らのCAR-T方法の重要な構成部分である。私たちのすべての同種異体候補製品の目標は、同じ製品の自己バージョンに匹敵する外形あるいはそれ以上の製品を作ることであり、私たちが最初に多発性骨髄腫を治療した完全同種異体製品候補製品P-BCMA-ALLO 1の場合、私たちの治療効果基準は他のBCMA標的計画と対照する。
9
コスト、規模、カバー範囲
早期のCAR−Tの市場進出の勢いは強いにもかかわらず、これまで商業採用は比較的緩やかであった。CAR−Tの広範な採用には2つの主な障害があると考えられる。第一の障害物はコストだ。これらの療法自体は数十万ドルを要する可能性があり,早期世代CAR−T療法のまれに生じる実質的な毒性を管理することは多大な追加コストをもたらす可能性がある。二番目の障害物は毒性自体だ。副作用コントロールには若干の進展が得られているが,多くの患者にとってリスクは依然として大きく,より便利なコミュニティ病院や外来輸液センターに比べて,これらの早期世代のCAR−T製品は集中治療室を有する大型病院や治療センターでしか使用できない。
我々の方法は,これらの障害を克服し,CAR−T療法の潜在力を放出することができると信じている。より高い割合のTの組み合わせは供給チェーン管理製品と改善可能な耐性は私たちを学術医学センターを超えて、これらの製品のカバー範囲を拡大させるかもしれない。われわれの最初の臨床試験P−BCMA−101では,FDAとの検討を経て完全外来に投与されており,同様にわれわれのP−BCMA−ALLO 1とP−MUC 1 C−ALLO 1計画も外来投与の許可を得ている。外来CAR−T投与量はすでにP−MUC 1 C−ALLO 1とP−BCMA−ALLO 1臨床試験で行われている。外来投与はカバー範囲を拡大し、コストを下げると信じている。また,我々のブースター分子技術は,単一健康ドナーの単一製造運転から数百剤の可能性のある同種異体CAR−T候補製品を生産できるため,我々の同種異体製造プロセスの規模を拡大することができる。これはCAR−T療法の製造コストを大きく低下させ,腫瘍学における伝統的な生物療法のレベルに達し,既製品を即座に使用できるようにした。
血液系腫瘍におけるCAR−Tの役割
早期世代のCAR−T療法は,血液系悪性腫瘍で印象的な反応を得ることが証明されており,再発や/あるいは従来の標準療法に無効な前治療患者においても同様である。いくつかの適応の応答率はすべての以前に報告された治療法よりはるかに高く,一部の患者は治癒される可能性がある。しかし、このような成果にもかかわらず、安全と費用の面で大きな挑戦が残っている。また,多くの患者がCAR−T治療後に再発し,全体的に反応時間が悪いため,反応持続時間をさらに改善することができると考えられる。
早期世代CAR-T療法の主要な制限は潜在的な深刻な毒性であり、最も明らかなのはCRSと神経毒性であり、この2つの毒性のいずれも致命的である可能性がある。現在のCAR−T療法はICUを設置した大型医療センターで行われているため,CAR−T治療を受けているすべての患者にICUを保持し,これらの重篤な毒性反応に遭遇しないようにすることができる。また,CAR−Tに関連する毒性を処理するコストは治療自体のコストを超えることが多い。他のCAR−T候補にとって、将来的には、ウイルスベースの製造の性質による大きなコスト、製造、および商業拡張性の課題に直面している。これらの問題は現在のCAR-T製品の商業範囲を大きく制限している。反応持続時間が短いにはいくつかの潜在的な原因があり、通常は体内のCAR-T細胞の除去と腫瘍細胞上のCAR-T標的の発現喪失、いわゆる抗原脱出の2つに分類される。
安全問題
不幸にも,初期世代CAR−T法の最初の印象的な反応に対する興奮は,潜在的な生命を脅かす毒性に薄まっており,最も明らかなのはCRSと神経毒性である。神経毒性の典型的な臨床症状は頭痛、意識不明、精神錯乱、言語障害とてんかん発作を含む。人々のこれらの毒性に対する理解がますます多くなるにつれ、現在人々はそれらが異なる分子機序によって引き起こされる可能性があることを認識している。しかし,両者ともT細胞に根ざした反応は,本質的に速すぎて強すぎる。患者のCAR−T細胞や他の免疫細胞はサイトカインや他の分子を放出し,免疫低下を開始し,治療を回避あるいは成功させなければ致命的である可能性がある。
T供給チェーン管理成熟したT細胞と比べ、細胞はより少ない細胞毒性応答分子を発現し、細胞毒能力を徐々に分化と発展させると考えられている。私たちはTを信じて供給チェーン管理細胞表現型はCAR-Tのより制御可能な拡張と腫瘍細胞に対するより漸進的な殺傷を招く可能性があり、それによって毒性の重症度を軽減する
10
例えばCRSおよび神経毒性は、外来で完全に使用可能なCAR−T製品をもたらす。
我々のCAR−T候補製品の第2の安全機能は,製造過程でCAR正極電池を積極的に選択することである。薬剤耐性遺伝子は他の細胞治療にも用いられ,遺伝子修飾細胞を選択·精製し,遺伝子治療の効率を向上させる機序として用いられている。著者らの候補製品はヒトジヒドロ葉酸還元酵素(DHFR)遺伝子の変異体を発現することを目的としている。このDHFR遺伝子変異体を含む細胞は薬物メトトレキサート(MTX)に対して軽微な耐性を有する。他の薬剤耐性策略と比較して、DHFRの優勢はMTXが遺伝毒性がなく、分裂細胞を優先的に殺すことである。重要なのはこの遺伝子-薬物の組み合わせは以前に許可されていました離体する比較的低濃度のメトトレキサート遺伝子組換えT細胞のスクリーニング。
また,遺伝子修飾CAR陽性細胞が豊富に行われている離体するしたがって,我々のCAR−T製品は基本的に100%CAR陽性である。これは競争相手の製品と異なり、これらの製品は順方向選択を利用せず、通常は大量のCAR陰性細胞を含み、これらの細胞は癌細胞を殺すことはできないが、体外で人工的に活性化および拡張することができ、CRSおよび/または神経毒性を引き起こす可能性がある。したがって,Tの高い割合に加えて,積極的な選択が別のメカニズムであると考えられる供給チェーン管理細胞、これは我々のCAR-T候補製品が有意に大きな治療指数を有することをもたらす可能性がある。
各CAR-T細胞は安定してゲノムに統合された遺伝子組換えを持っていることから、CAR-T製造過程中の遺伝子組換え伝達部分は有害な突然変異を産生し、細胞を制御されない方法で拡張させ、これは細胞自身が癌になる可能性がある。また,ウイルス製造の場合,トランスジェニックの一部としてCAR−T細胞に組み込まれたいくつかのウイルス成分,例えばトランスジェニックの長末端反復配列やLTrSは,細胞中の既存の遺伝子を活性化し,細胞癌化を引き起こす可能性があり,この過程を発癌と呼ぶ。
レンチウイルス製CAR−T製品を受け,クローン性拡張を認めた例がある。クローン性増幅は単一T細胞が増殖優位を与えられ,患者のすべてのCAR陽性細胞の大多数に成長することを意味する。この場合,クローン増幅はレンチウイルスが増殖に重要な遺伝子を挿入することによるものである。私たちのCAR-T候補製品は私たち独自のPiggyBac技術を使用します。PiggyBacは遺伝子内領域との低統合を示しており,有害な変異を引き起こす可能性が低いことを意味している。さらに、レトロウイルスと異なり、iggyBacはLTR配列を含まず、ITRsと他の構成要素であり、それらは強力な絶縁体として機能し、安定したトランスジェニック発現を増強し、腫瘍発生のリスクを低下させる。
私たちは、追加のセキュリティメカニズムとして、各候補製品にセルラーセキュリティスイッチを含む。CRSや神経毒性はいずれも過剰に活発なT細胞反応に関与していると考えられている。したがって、CAR-T細胞の数を減少させるために適時に介入することは大多数の不良イベントを管理する有効な方法であるべきである。理想的な関与技術は滴定可能な技術であり,すべてのCAR−T細胞を除去することなく,いくつかの細胞を残して治療効果を発揮し続けると考えられる。
商業拡張性
初期世代CAR-T製品のもう1つの課題は、それらの商業的拡張性である。定義によると、自己CAR-T製品は個人化された製品である。これらの生産コストは通常も高く,特にウイルスベースの製造方法を用いた場合である.私たちの非ウイルス方法iggyBacは、GMP核酸、DNA、RNAを使用しているので、従来のCAR-T法よりも効果的でコスト的であると信じており、GMPウイルスよりも生産が速く、安価である。我々はさらに製造プロセスを最適化し、磁気ビーズおよびサイトカインを含むウイルスベースの方法に関連する高価な材料を除去した。
CRSおよび神経毒性などの深刻かつ潜在的な致命的毒性をもたらすCAR−T製品は、三次ケア病院での投与が要求され、そこで、医師はこれらの毒性の治療を熟知し、集中治療室に入ることを選択することができる。これらの重篤な毒性の可能性は,コミュニティ病院や外来輸液センターで使用されている可能性は排除されている。われわれの用量増加P−BCMA−101 1期臨床試験では,CRSやCRSにより集中治療室に入院せざるを得ない患者はいないことが知られている
11
神経毒性です。これらの結果から,FDAと検討したところ,完全外来に基づいて投与することができた。P−BCMA−ALLO 1臨床試験で外来CAR−T投与量も行った。P−CD 19 CD 20−ALLO 1計画の初歩的な結果を評価したところ,これまでの試験と同様の安全反応を継続すれば,外来用量も継続する予定である。
治療効果の挑戦:CAR-T細胞を除去する
なぜCAR−T細胞が投与後に患者から除去されるのかについては多くの解釈があるが,主な解釈は,他のCAR−T製品の多くのT細胞がT細胞を含めてより成熟して短いことであると考えられる電信為替細胞です。すべてのT細胞が同じではなく,主に早期記憶T細胞,特にT細胞からなる製品を開発する能力を信じている供給チェーン管理細胞は,反応持続時間と耐性を増加させる鍵である。我々の非ウイルス型PiggyBac製造方法は、高い割合で所望のT細胞を有するCAR−T製品を製造することができる既知の商業的に実行可能な唯一の方法である供給チェーン管理細胞と私たちの技術の効率性です
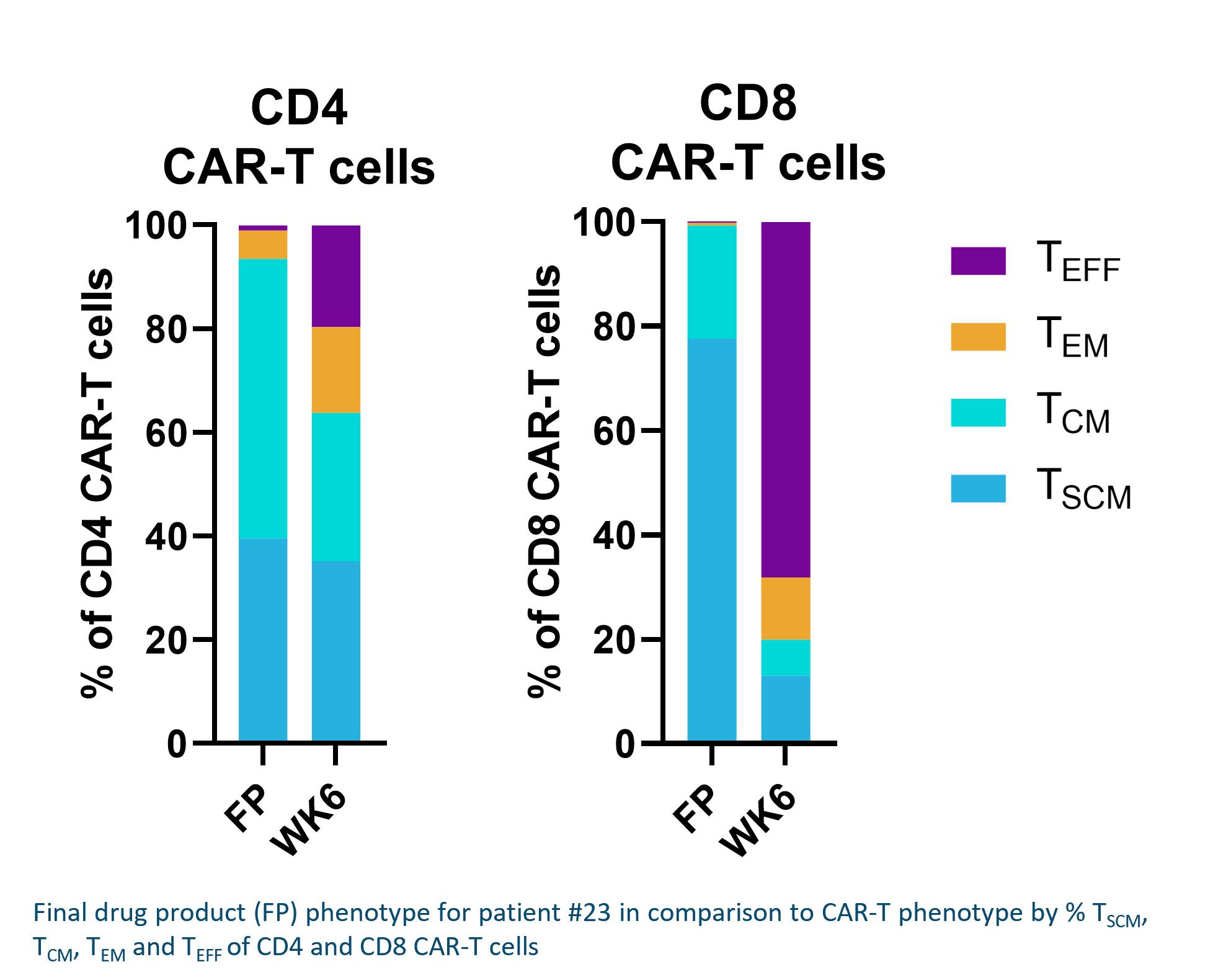
PiggyBac DNA送達システムがCARを含むトランスジェニックをT細胞に優先的に送達する能力をテストするために供給チェーン管理T細胞について、著者らは臨床前実験を行い、T細胞を異なる亜型に分け、その後、それぞれこれらの亜群を最適化されたiggyBac製造技術或いは最適化されたレンチウイルスプロセスに置き、各最終製品亜群における転座或いは形質導入細胞のパーセンテージを測定した。次の図に示すように,piggyBacは転座(遺伝子組換えを含むpiggyBac過程を伝達する)に非常に有効である供給チェーン管理T細胞におけるレンチウイルスの形質導入(レンチウイルスがCARトランスジェニックを含む過程を伝達する)は相対的に無効である供給チェーン管理細胞です。CD 4+T細胞(T補助細胞とも呼ばれる)とCD 8+T細胞(細胞毒性T細胞とも呼ばれる)を測定し,相互作用と考えられる2つのT細胞亜群を代表し,免疫機能とT細胞反応に重要な役割を果たしている。
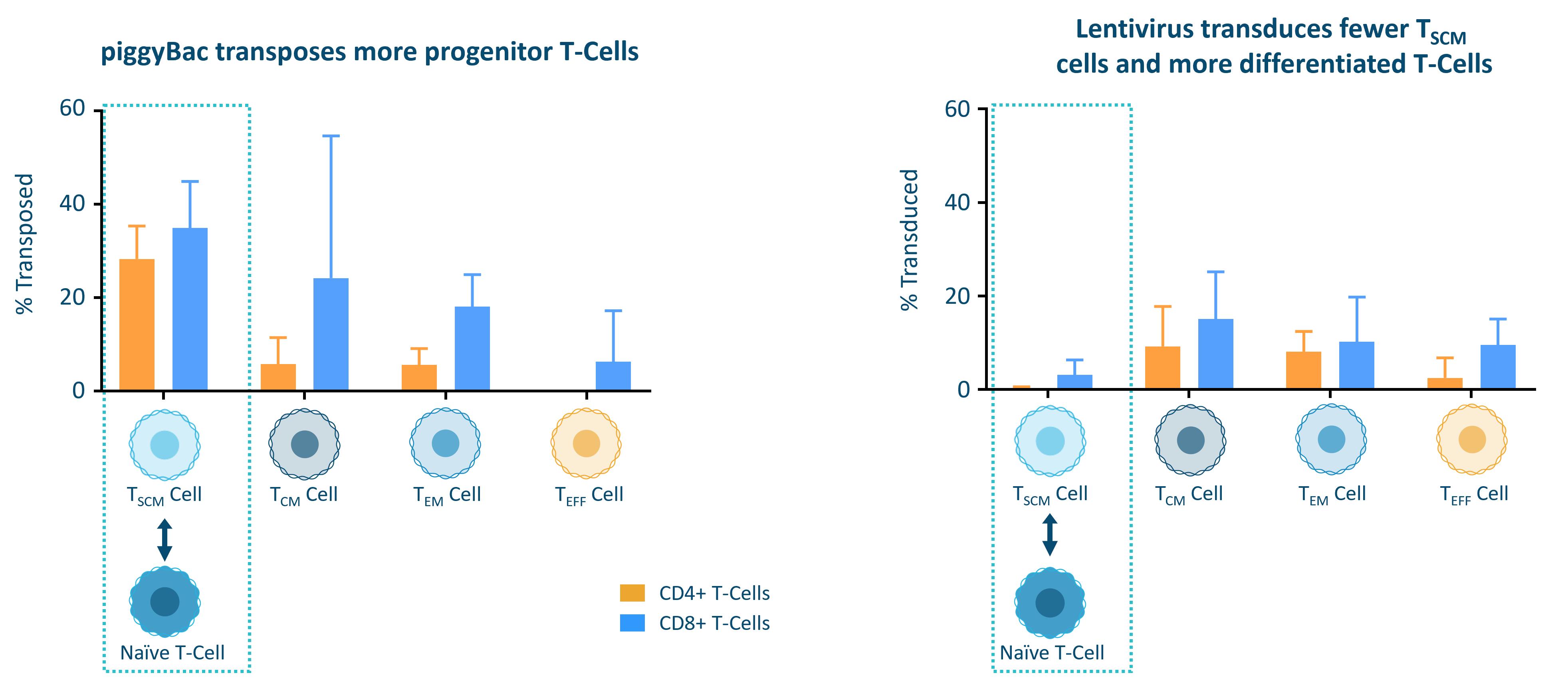
T細胞の一方向成熟経路から,遺伝子工学的手法を用いてT細胞を優先的に修正すると考えられる供給チェーン管理細胞は高い割合のTを持つ最終製品を作るために不可欠である供給チェーン管理細胞です。製造過程で遺伝子改変工程が完了すると,改変されていない細胞を除去する陽性選択工程を実行する。最後に、T細胞の自己更新に有利な条件下で残りの細胞を活性化し、拡張します供給チェーン管理分化していない細胞は,産物中のT細胞の割合が高い供給チェーン管理細胞はT細胞の割合が相対的に低い患者材料から始めても供給チェーン管理細胞です。私たちの非ウイルス豚Bac DNA伝達システムは通常T細胞を産生します供給チェーン管理細胞百分率は80%に達している。我々のiggyBac製造方法をレンチウイルスによる製造方法と比較し,レンチウイルス製造方法は異なる媒体(Aim V,Thermo Fisher Science),異なるT細胞刺激(Dyal/Thermo Fisher Science由来CD 3/CD 28ビーズ)とベクター統合用ウイルス(レンチウイルス)を用いた。1つのドナーの細胞に対する予備実験では,分類後のT細胞亜群を1回iggyBac過程を行い,3つのドナーの細胞を用いたレンチウイルス過程と比較した。初期記憶コンポーネント、または組み合わせT供給チェーン管理Tとセンチメートル細胞には通常
12
90%以上の細胞が私たちの製品候補です注目すべきは2019年12月に25%以上のT細胞を含む修飾T細胞製品をカバーする米国特許を取得しました供給チェーン管理細胞です。
CAR-T開発分野の他社もT-Tの割合を増加させようとしている供給チェーン管理製造工程では,小分子阻害剤薬や各種サイトカインを添加し,培養時間を減少させ,選別方法により早期T細胞を物理的に濃縮することを含む代替方法によりその製品に細胞を添加する。しかし、これらの方法には固有の問題があり、これらの問題は、Tの高いパーセントを有する最終製品候補を作成することに成功する能力を制限すると考えられる供給チェーン管理細胞です。
私たち自身の臨床データでも、他の人が発表·提出したデータでも、T細胞の割合が高い供給チェーン管理CAR−T製品中の細胞は臨床反応と相関することが証明されており,我々のCAR−T候補製品には高い割合のT細胞が含まれている供給チェーン管理細胞です。私たちの目標は、私たちの候補製品が応答の効力と耐久性を含む他のCAR-T製品の多くの側面での制限を克服することだ。
より成熟したT細胞はT細胞に比べて寿命が短いです供給チェーン管理細胞は、持続的に存在できないため、患者の体内から除去でき、製品の治療効果が良くない。CAR−Tが早期に失われた原因の1つは,T細胞表面に相互作用するCAR結合分子が存在することである。これはCAR分子の架橋と緊張性シグナルと呼ばれる現象を招き,この現象ではCAR−T細胞はほぼ常に刺激され活性化される。緊張性シグナルは早期に効力を失い、拡張不良と細胞死を招き、T細胞枯渇と呼ばれる。重鎖のみの抗体断片や厳選された単鎖断片可変抗体などの結合分子を用いて,架橋や緊張性シグナルのリスクを最大限に低減する。
効果的課題:抗原脱出と抗体
いくつかのCAR-T製品はすでにいわゆる抗原脱出による治療効果を失うことが証明され、これはCAR-T療法の選択的圧力により、腫瘍細胞上のCAR-T標的の発現が失われ或いは急激に減少し、CAR-Tからそれらを殺す能力を脱出する腫瘍細胞の拡張を招くことである。抗原脱出を避けるために,発現が減少すると考えられる標的の選択に集中するように努力する。例えば,BCMAは細胞増殖に重要であることから,CAR−T治療後に腫瘍細胞に失われる可能性は低いと考えられる。
もう1つの抗原脱出防止方法は,同じCAR−T製品を用いて癌細胞上で複数の標的を追跡することである。癌細胞は単一標的ではなく、複数の標的の発現を同時に低下または失う可能性が大幅に低下する。ウイルスベクターの遺伝子輸送能力はかなり限られているが,iggyBacは20倍を超える遺伝輸送能力を輸送できることを証明しており,複数のCAR分子遺伝子の同時転移を許可している。2つ以上のCARやTCRを同一のT細胞に含めることで,iggyBacの大きな遺伝子貨物容量をさらに抗原脱出問題を解決できると信じている。我々のP−CD 19 CD 20−ALLO 1計画は,CD 19とCD 20を同時に対象とした初の同種異体二重CAR−T計画であると考えられる。この計画は,他の自己CAR−T試験で見られた潜在的なCD 19抗原脱出を克服することを目的としている。私たちは最近この計画の第一段階臨床試験を開始した。
固形腫瘍におけるCAR−T
効能挑戦
悪性血液疾患の治療におけるT細胞の持続性の標準的な懸念に加え,CAR−T製品を用いて固形腫瘍を治療する際には,この問題を悪化させる因子がある。これまで、大多数の早期に生成したCAR-T製品は固形腫瘍において顕著な反応を示さず、このような比較的に悪い治療効果に対して多くの潜在的な解釈がある。まず,CAR−T細胞は固形腫瘍細胞に接触しにくい可能性がある。いくつかの疾患では、急性リンパ球性白血病のように、腫瘍細胞はCAR-T細胞に近づきやすい。しかし,多くの固形腫瘍では,CAR−T細胞を腫瘍に近づけることが困難である可能性が多い。次に、固形腫瘍細胞はあるチェックポイント遺伝子の発現を変化させ、T細胞の殺傷に抵抗力を発生させる可能性がある。第三に,多くの固形腫瘍の中心は非常に低酸素,あるいは酸素濃度が低く,この環境はT細胞機能に不利であると考えられている。
13
固形腫瘍におけるCAR-Tの不良治療効果も少数の例外があり、特に多形性神経膠芽腫と肝細胞癌において、CAR-Tの治療は固形腫瘍の中で完全な緩和或いはCRを招く。これらのまれな症例では,CAR−T製品の治療を複数回受けている。固形腫瘍細胞に対するCAR−T細胞の殺傷作用は血液腫瘍細胞ほど有効ではないにもかかわらず,CAR−Tを複数回投与することでこれを克服することができ,より成熟したT細胞を大量に死滅させることができる。患者を治療するための無限数の細胞があれば、この方法はもっと実行可能になるだろう。しかし、初期世代のCAR-T製品を製造するのは相対的に時間と高価であり、しかも最終製品は限られた数量の細胞から構成されているため、この方法は多くの患者にとって非現実的である。
高比率のT細胞からなるP-MUC 1 C-ALLO 1を含む固形腫瘍候補製品供給チェーン管理細胞はT細胞を含む各T細胞サブタイプに移植、自己更新、成熟することができると考えられます電信為替細胞、これらの細胞は深層反応が可能になるまで腫瘍を攻撃し続けることができる。したがって、著者らのCAR-T候補製品は一回の投与を通じて固形腫瘍に対する高い応答率を実現する潜在力があると信じている。われわれの最初の固形腫瘍プログラムP−PSMA−101の早期臨床結果では,有望な奏効率が認められた。2022年2月17日に報告によると、上位14名の患者の中で、71%の患者はPSAが低下し、その中の36%の患者のPSA低下は50%を超えた。また,1名の患者はPSMA PETと他のマーカーにより腫瘍がほぼ完全に消失したことを証明した。
安全問題
我々の解決策は,CRSに関連するCAR−T毒性問題や血液腫瘍に関連する神経毒性問題を解決するために用いられ,固形腫瘍にも適している。しかし、CAR-T製品が固形腫瘍の治療に使用される場合、追加の毒性問題がある。血液腫瘍と比べ、固形腫瘍は通常より少ない独特な表面標的を持ち、これらの標的は健康細胞に発現しないため、標的を選択する時にもっと慎重にしなければならず、標的上/腫瘍外毒性を回避し、CAR-T細胞が健康細胞上の予想される標的を識別し、その細胞を殺す時にこのような毒性が発生する。MUC 1−Cのような癌細胞に過剰発現する標的を選択し,癌化形式の標的に結合する上でより有効であると考えられる結合分子を用いることでこのリスクを解決しようとしている。
われわれのP−PSMA−101試験では,われわれは研究早期に臨床放置を経験し,患者の死亡を評価し,P−PSMA−101の治療に関与している可能性があるが,一部は患者の不コンプライアンス事件によるものである。プロトコル修正後,臨床保留は解除され,それ以来,治療に関連する可能性のあるより多くの患者の死亡を経験することなく,試験中により多くの患者に投与量を提供した。2022年2月17日に米国臨床腫瘍学会泌尿生殖系癌シンポジウム(ASCO-GU)で報告された報告によると、我々は57%の患者にCRSが観察され、そのうちの14%の患者が3級以上のレベルを経験し、14%の評価可能な患者に免疫効果細胞関連神経毒性症候群(ICANS)が観察された。私たちは2022年に私たちの候補製品P-PSMA-ALLO 1の同種バージョンに集中するためにこの計画を終えた。
著者らが固形腫瘍CAR-T導管を拡張することに伴い、著者らは固形腫瘍細胞特有の標的を識別することは更に困難になると予想している。そこで我々は,標的の組み合わせが存在するか否かに応じてCAR−T細胞が腫瘍細胞を死滅させるように指導する複雑なシステムを開発している。例えば,表面に標的Aも標的Bもある腫瘍細胞のみを殺すCAR−Tを開発できると信じているが,表面に標的Aや標的Bがある正常な細胞を単独で殺すことはない。
1つの関連する戦略は、標的AとB(癌細胞および正常細胞に存在する可能性がある)を発現する時にのみ細胞を殺すCAR-Tを開発することであるが、標的Cを発現しない(正常細胞にのみ存在する可能性がある)。これらの戦略はいずれも2つ以上のCAR分子が同一のCAR−T細胞表面に共発現する必要がある。その巨大な遺伝貨物容量により,iggyBac DNA伝達システムはこれらの方法を可能にすると信じている。対照的に、ウイルスに基づく方法は通常、2つの全長を超える自動車分子を伝達することができない。
単一のブタトランスポゾンを用いて4つの全長CAR分子遺伝子を発現するCAR-T細胞を生産できることを証明しました各遺伝子は異なる標的性と他の2つの遺伝子を持っています
14
製造業では(左図)。さらに、発現すると、すべてのCAR分子が標的を発現する対応する細胞株に対して特定の殺傷作用を行うことが証明された(右図)
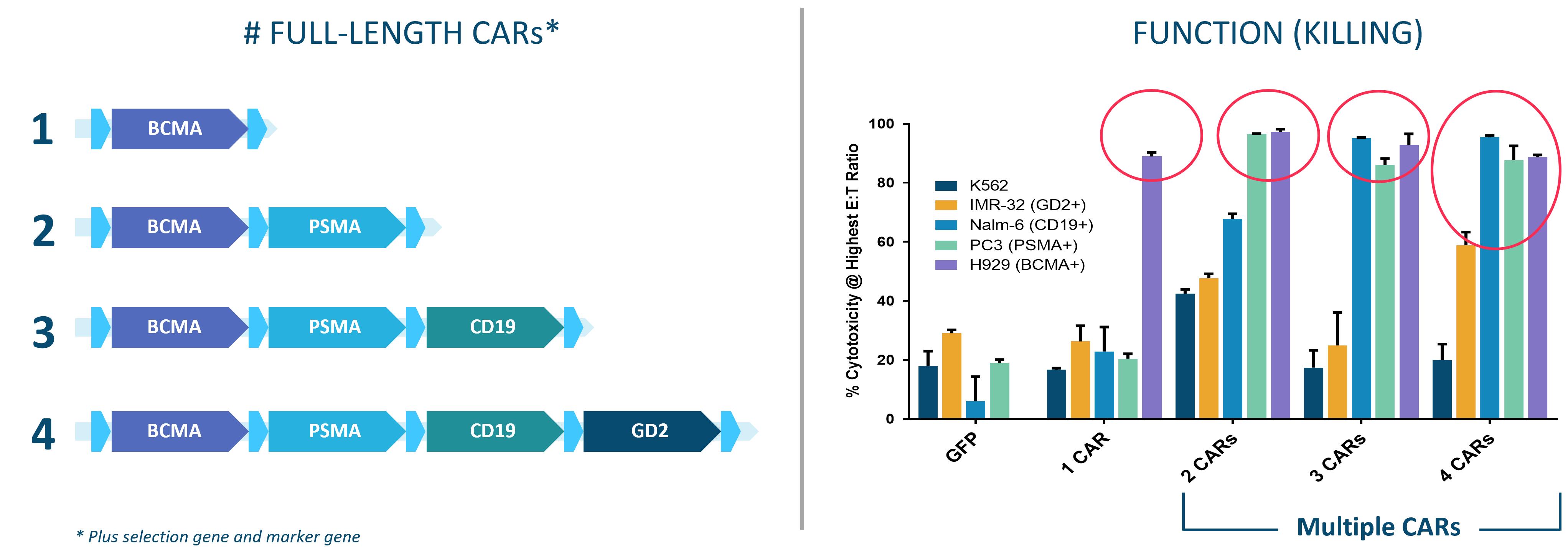
報告されたヒト腫瘍細胞がルシフェラーゼ遺伝子を発現するように遺伝子改変されている、報告に基づく殺傷試験によって特異的殺傷が評価される。これらの腫瘍細胞はCAR−T細胞とインビトロで10:1の効果標的比(10:1)で24時間共培養した。CARR-T細胞発現全長CARTyrinの異なる組み合わせ:(1)BCMA CARTyrin,(2)BCMA CARTyrinとPSMA CARTyrin,(3)BCMA CARTyrin,PSMA CARTyrinとCD 19 scFv-Car,あるいは(4)BCMA CARTyrin,PSMA CARTyrin,CD 19 scFv-CARとGD 2 scFv-CAR。細胞毒性パーセンテージは、フルオレセイン基質を添加し、発光シグナルを読み取ることによって細胞毒性(特異的溶解)を評価し、CAR−T細胞を含む腫瘍細胞に対する単独の腫瘍細胞の発光を計数することによって計算される。個々の単独CARはその相同抗原に対して細胞毒性を示し,他の3つの全長CARが存在する場合でも同様である。
固形腫瘍を治療する別の方法は、癌細胞内部にのみ癌関連タンパク質を発現するTCRの変異体を発現することであり、CAR分子は細胞表面の標的のみを認識することである。TCR戦略とCAR戦略を組み合わせて用いることができ,iggyBac製造法を用いることにより同一細胞表面にCARとTCR分子の組み合わせを発現させることができると信じている。
商業拡張性
私たちの方法は血液腫瘍のすべての商業的で拡張性の利点も固形腫瘍にも適用できると信じています。
異遺伝子や既製のCAR−T療法
効能挑戦
同種または既製CAR−T製品の目標は、単一ドナーまたは細胞株から大量のCAR−Tを創出することである。成功した同種CAR-T製品は、特定の適応を有する任意の患者の治療に既製品として使用することができ、それにより、製造に関連するコストを大幅に低減することができる。しかしながら、1つの異体製品が類似の自己製品と同じ活性を達成するために高用量または複数回の用量を必要とする場合、異体製品の多くの潜在的なコスト節約利点は達成できないであろう。
遺伝子編集ツールは、ある細胞表面分子の発現を除去するために広く使用されており、これは、ドナー細胞の患者に対する潜在的な反応性を回避するために使用される可能性があり、これは、GvHD、およびCAR-T産物に対する患者細胞の反応性をもたらす可能性があり、この反応は、宿主対移植片と呼ばれる。同種異体CAR−Tを作成する際には,静止T細胞を効率的に編集できる遺伝子編集ツールを用いることが必要であると考えられる
15
T細胞を活性化する産物として成熟経路を起動する。T細胞が成熟すると欲しいT細胞を失い始めます供給チェーン管理そのため疲れ果て、最終製品の効果があまり効果的ではない。
多くの他の遺伝子編集技術と異なり,Cas−Cloverは静止したT細胞を効率的に編集することができ,非常に理想的なT細胞を保持することができる供給チェーン管理製品組成中の同種異体候補製品は、著者らのCAR-T方法の重要な構成部分である。私たちのすべての同種異体候補製品の目標は同じ製品の自己バージョンに匹敵するかそれ以上の外形ができる製品を作ることである;私たちの最初の多発性骨髄腫を治療する完全同種異体候補製品P-BCMA-ALLO 1の場合、私たちの治療効果基準は他のBCMA標的計画と対照する。
安全問題
CRSや神経毒性に関する標準的な懸念のほかに,同種製品に関する追加的な安全性懸念がある。上述したように、同種異体製品は2つの形態の同種異体反応を引き起こすことができる:移植片対宿主病と宿主対移植片。宿主の移植片に対する懸念は、それが同種異体CAR-T細胞の早期除去を招く可能性があるためだけであり、前に議論したすべての製品の持続性の差に関連する治療効果の挑戦を招くが、それは安全問題を引き起こさない。
しかしながら、GvHDは深刻で潜在的な致命的な疾患、すなわちCAR−T細胞が患者の健康細胞を殺している。内因性TCRはGvHD予防のために除去すべき分子であることが示唆された。この分子がほぼ100%のCAR−T細胞で完全に除去できなければ,GvHDが問題となる可能性がある。我々の効率的なCas−Clover技術および後続の精製工程は,少なくとも99%の細胞TCR発現を除去しており,このレベルはGvHD予防に必要なレベルよりも安全であると考えられる。
同種異体製品の1つの利点は、単一ドナーまたは細胞株が多くの用量を産生することができることである。しかしながら、1つの潜在的な欠点は、製造中に生じる任意の有害変異が、自己製品ではなく、多くの患者に投与される用量に存在する可能性があることであり、自己製品において、このようなリスクは個人患者に限定されるからである。そのため、不必要な脱標的突然変異を最大限に減少或いは完全に防止することは特に重要である。CRISPRのようないくつかの遺伝子編集技術は、望ましくない突然変異を産生する可能性があることが知られている。前臨床試験では,われわれのCas−Coverer技術は正確な部位特異性を示しており,標的外変異を引き起こす傾向はないかほとんどない。われわれ自身の臨床前データとこれまでに発表された他の全二量体CRISPRシステムに関する結果から,Cas−Cloverは利用可能な最も特異的な遺伝子編集法であると考えられる。
商業拡張性
完全同種異体のCAR-T製品は、製造過程において時間とコストを著しく節約する可能性を提供し、それによって各薬剤のコストを大幅に低下させ、患者の可及性を増加させた。それにもかかわらず、製造プロセスは、固定用量の同種異体製品を産生するために、単一のドナーまたは細胞系材料上で動作しなければならない。ウイルスに基づく製造方法の生産過程において,最も高価な部分の1つはウイルス自体である.この生産システムはGMP DNAとRNAのみを用いており,GMPウイルスを必要としない。これは同種異体CAR−T製品を背景にしても、候補製品の生産コストを大幅に低下させると信じている。また,PiggyBacの開発や製造時間がウイルスよりも短いことは,人々が製品概念からGMP材料に早く移行できることを意味している.例えば、2年もたたないうちに、私たちはP-BCMA-101を製品概念から臨床試験で使用された最初の患者に移し、これらの経験を適用して、未来の製品候補のスケジュールを満たすか、または超えることができると信じている。
TCRの遺伝子改変は先に議論したGvHDを避けるために必要であり,T細胞を産生し,製造過程で増幅が困難である可能性がある。多くの自己CAR−T製造戦略では、TCRは通常、T細胞刺激の重要な受容体として使用される。しかし,同種異体戦略では,TCRをノックアウトするどの単一成分も工学T細胞表面にTCR複合体全体を失わせ,製造過程におけるCD 3抗体に対する反応性を著しく低下させる。TCRおよび他の遺伝子修正を除去するこれらの結果は、一般に“対立遺伝子税”と呼ばれる。TCR統合体を次の図に示す。
16
私たちはT細胞のパーセンテージを維持し、潜在的に増加させながら、この問題を克服することができる独自のプロモーター分子を開発した供給チェーン管理最終製品中の細胞ですBooster分子は、T細胞のRNAベース技術を製造過程で導入し、T細胞表面の受容体を瞬時に発現させ、細胞が抗体ベースの活性化分子に反応し、細胞を著しく拡張させることができ、細胞の成熟や枯渇を招くことなく、細胞を著しく拡張させる。独自のプロモーター分子を用いることで細胞の膨張と収量を増強し,単回生産細胞数をブースター分子を使用しない場合の5倍以上とした(下図参照)。
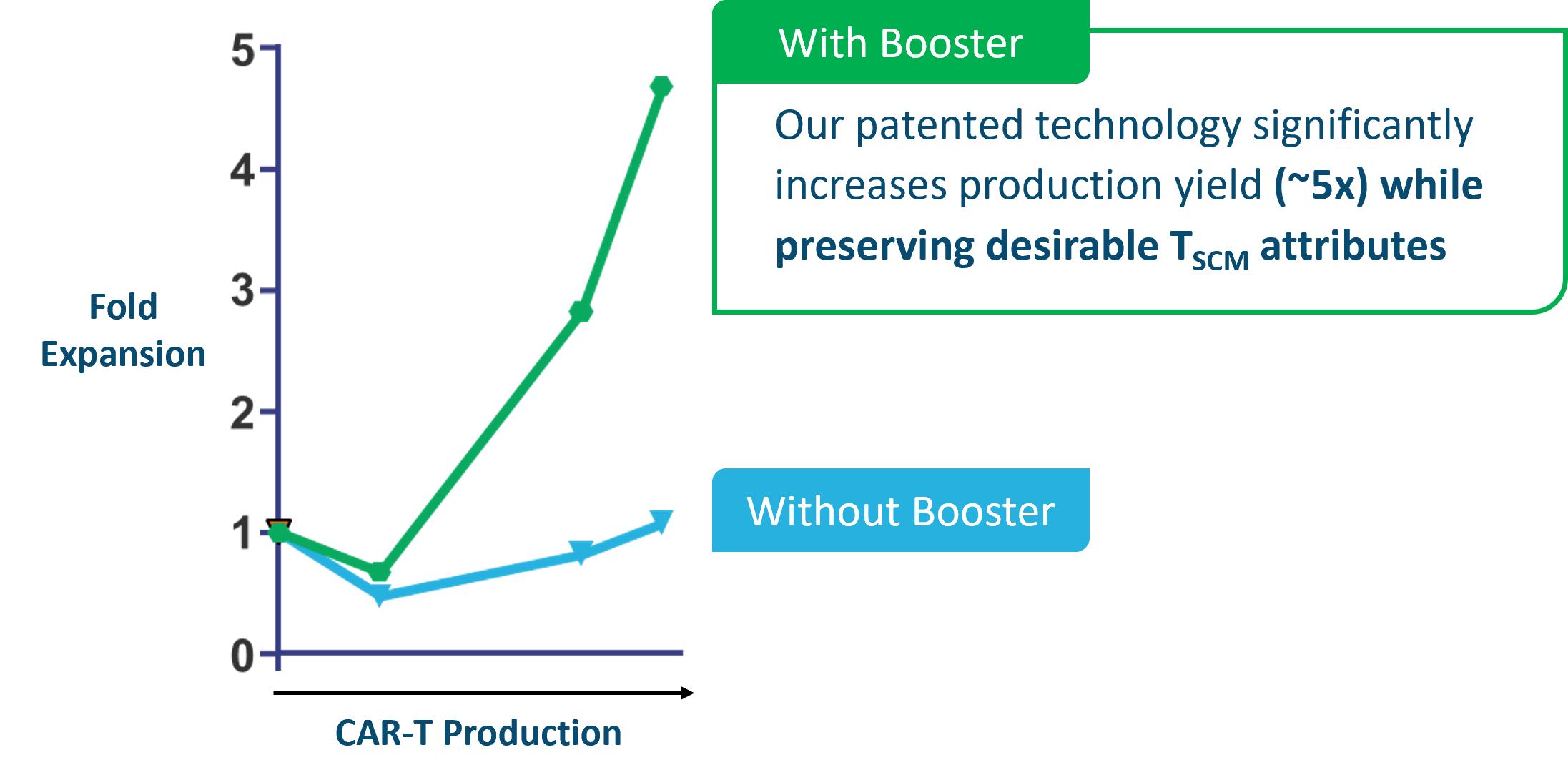
17
私たちは、完全に同種異体の候補製品を作ることができ、適用すれば、対応する自己製品に相当するプロファイルを維持することができるが、十分な用量を作ることができ、一度の生産から数百人の患者を治療することができると信じている。
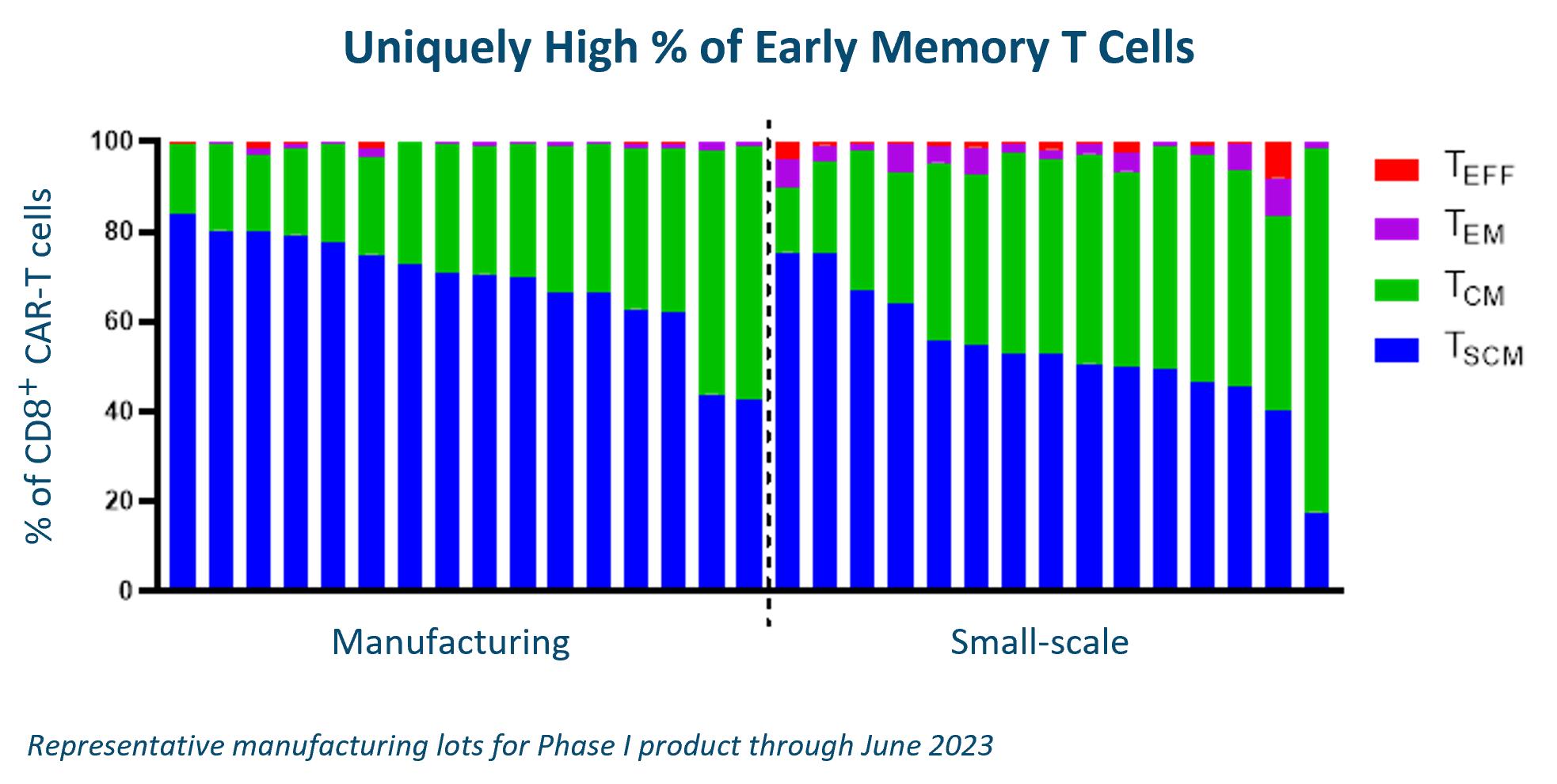
製造過程の1つの鍵はTを保存することだと思います供給チェーン管理表現型です。以下に示すように,小規模とGMP生産ロットでは,いずれも一定割合の早期記憶T細胞を保持することができた。
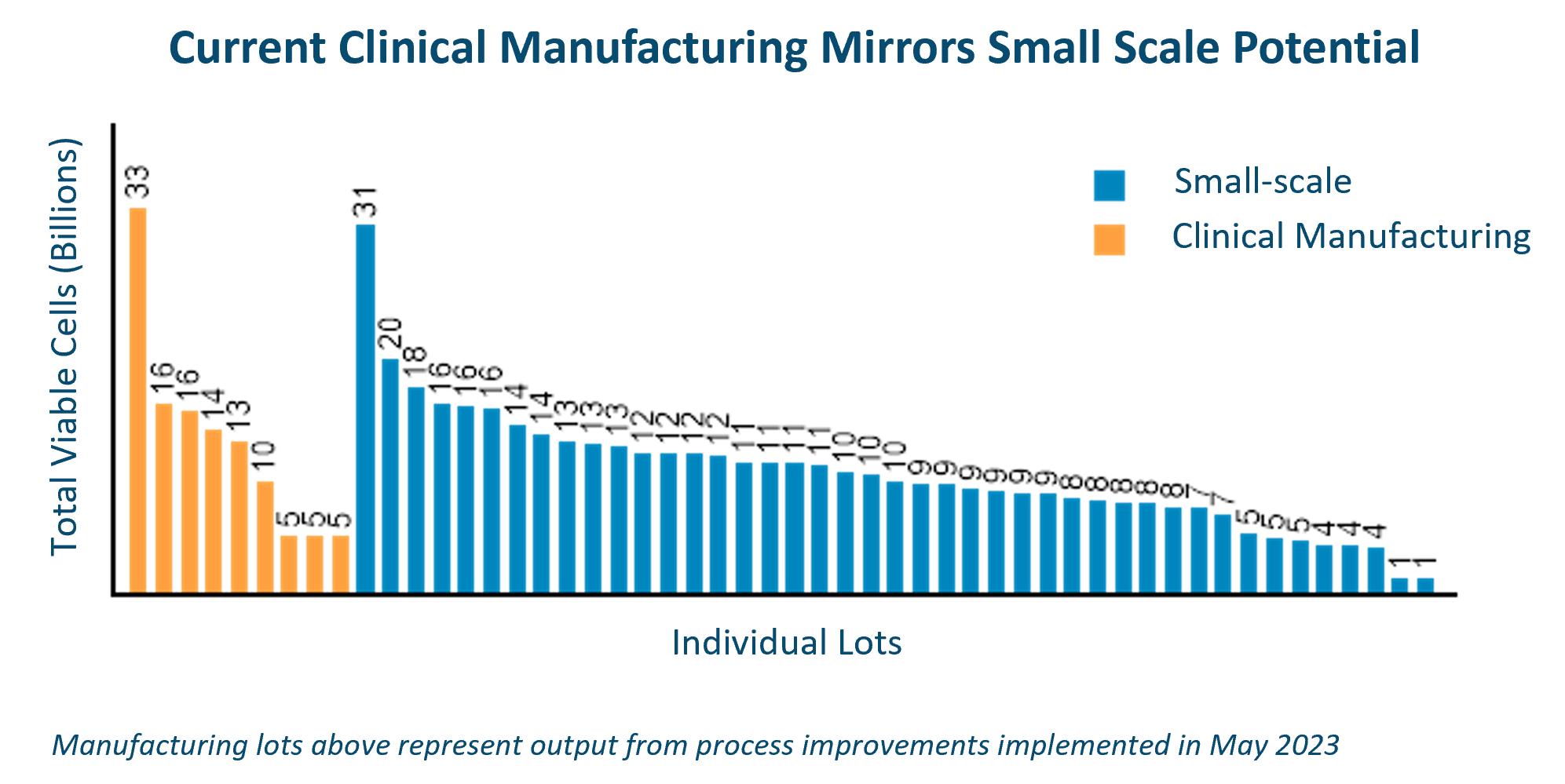
私たちが私たちの製品を開発するにつれて、私たちは商業成功の鍵は同種異体製品を大規模化する能力になると信じています。私たちの独自の推進剤分子のため、より高い生産量で私たちの製品を生産することができ、その結果、用量によって白血球を1つ使用し、1回当たり最大100剤を生産することができた。
18
二重CAR−T同種移植候補案
IggyBacの非常に大きなキャリア能力は、ウイルスベースの技術と比較して、より大きなまたはより多くの治療用トランスジェニックを含むことを可能にする。我々が2つ以上の機能的に整ったCARおよび/またはTCR分子をT細胞に組み込む能力は重要な競争優位である可能性があると信じている。二重特定または直列結合を使用してこの問題を解決しようと試みるいくつかの競合他社とは異なり、2つ以上の全自動車またはTCR分子を含むことがより有効な方法である可能性が考えられる。
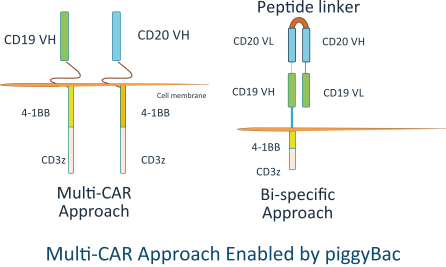
同種異体CAR−T
以下の表は、現在のCAR-T候補製品の組み合わせをまとめたものである
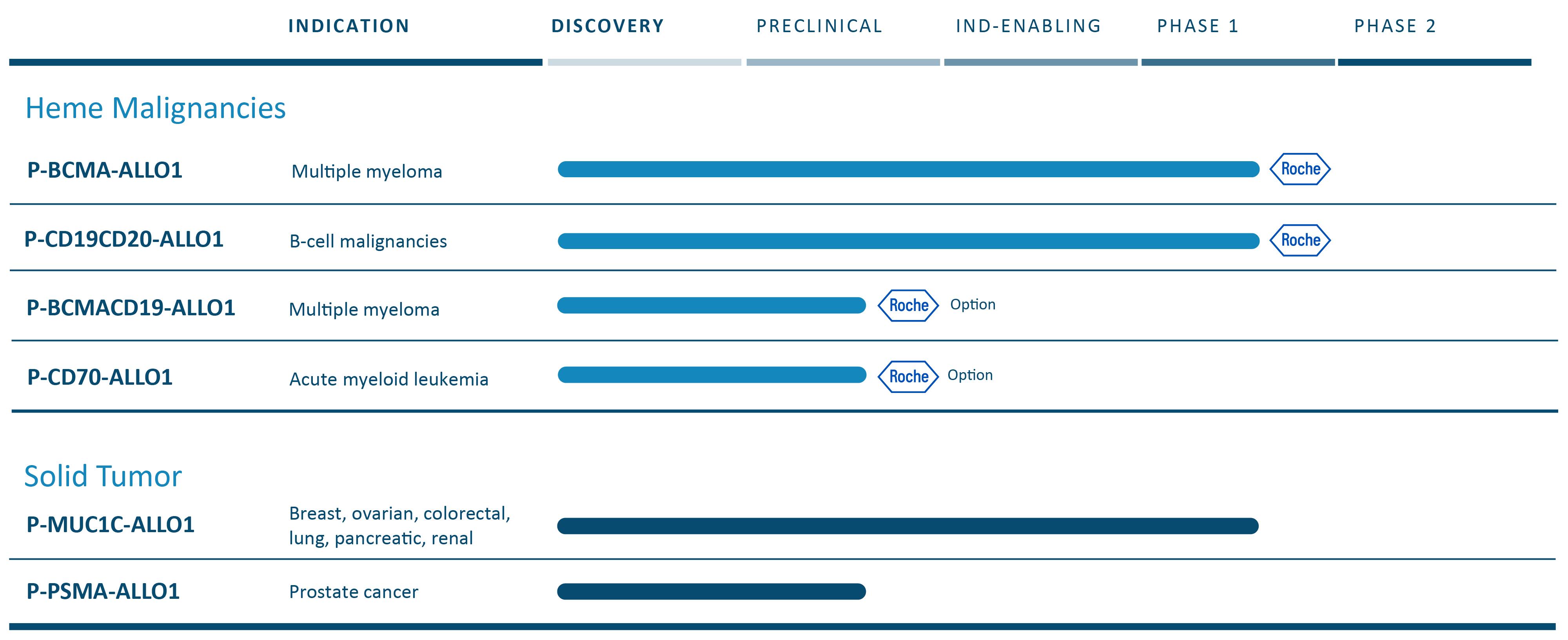
我々の完全同種異体CAR−T候補は,健康ドナーからの特徴の良い細胞を出発材料として開発し,一次生産運転から潜在的な数百名の患者を治療できることを目標としている。投与量は凍結保存され,将来の既製品使用に備えて治療センターに貯蔵されている。
19
P-MUC 1 C-ALLO 1:多種の固形腫瘍の適応症
概要
P-MUC 1 C-ALLO 1は完全同種異体CAR-T候補製品であり、多種の固形腫瘍適応を治療する潜在力がある。標的分子MUC 1-Cは腫瘍選択的グリコシル化切断産物であり、大部分の上皮性腫瘍に高度に発現している。我々のP-BCMA-ALLO 1とP-PSMA-101計画を利用した学習のためにP-MUC 1 C-ALLO 1を設計した.著者らは現在P-MUC 1 C-ALLO 1の1期臨床試験を評価しており、2022年12月に開催されたヨーロッパ医学腫瘍学免疫腫瘍学2022年年会(ESMO I-O)でこの計画の初歩的な早期臨床データ更新を共有した。2024年4月の米国癌研究協会(AACR)年次総会で、より高いリンパ消費と細胞拡張との関連性、2024年下半期に適切なフォーラムで発表されたより煩雑な臨床更新に重点を置いたこのプロジェクトに関する要約を共有する予定である。
当社独自のPiggyBac DNA送達システムを用いて、高比率のT細胞を含む高純度P−MUC 1 C−ALLO 1製品を製造した供給チェーン管理これらの細胞は固形腫瘍を治療するCAR−T療法の開発の鍵である可能性が考えられる。我々はGvHDと宿主の移植片に対する同種反応を減少または除去するために、我々の独自のCas-Cloverプラットフォームを用いてT細胞に対して遺伝子工学を行った。
目標指示
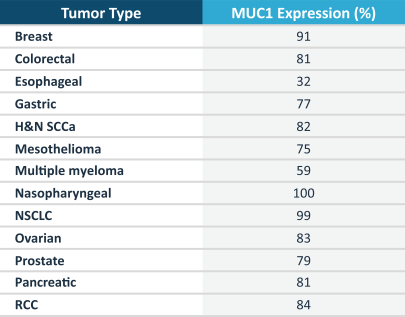
さらに評価し,後にP−MUC 1 C−ALLO 1がMUC 1−C発現を生じる適応における最初に発展した臨床適応を決定する予定である。癌の約90%は上皮組織に由来し、これらの癌のうちのかなりの割合のヒトは、乳癌、結腸直腸癌、肺癌、卵巣癌、膵臓癌および腎臓癌などの一般的な癌を含むMUC 1-Cを発現する。
臨床発展戦略
著者らは現在、一期臨床試験でP-MUC 1 C-ALLO 1を評価している。P-MUC 1 C-ALLO 1は、私たちの他のプロジェクトの経験的教訓を利用することを目的としています。現在の第1段階レジメンは、RECISTによって測定することができ、標準的な看護治療を受ける資格がない、または標準的な看護治療を受ける資格がない180人までの末期または転移性上皮原性癌を有する成人被験者の登録を可能にする。標準的な3+3用量-漸増設計を採用し、患者は単一回と複数回(循環)投与の4つの分枝を通じて登録することができ、いくつかの異なるリンパ除去方案を使用し、各方案は最大7つの用量増加行列がある。コホート研究開始標準の3日間シクロホスファミド(300 mg/m)2/日)およびフルダラビン(30 mg/m2/日)細胞注入前にリンパ濾過レジメンを投与した。他にも
20
リンパ浄化化学療法(シクロホスファミド500 mg/m)を大量に服用した患者2/d×3日、フルダラビン30 mg/m2/日×3日またはシクロホスファミド1000 mg/m2/d×3日、フルダラビン30 mg/m2/日×3日)。各アーム計画のP-MUC 1 C-ALLO 1用量の増加範囲は0.75~15 x 106細胞/kg。治療を受けた患者は安全性、耐性と腫瘍反応の一連の測定を受け、そして最後の剤P-MUC 1 C-ALLO 1後に15年間のフォローアップを行う。
この段階の臨床試験の主要な目標は最大耐容量、即ちMTDを決定し、全体の安全性と耐性を評価し、及び初歩的な治療効果と疾病反応を含む。他の探索的終点は、MUC 1−Cの腫瘍発現の評価と、P−MUC 1 C−ALLO 1との反応および拡張動態との相関を含むであろう。
2022年12月、著者らはESMO-IOで初歩的な早期臨床データを公表した。2022年11月14日までに、この研究はすでに7名の上皮由来癌患者に薬物を服用し、食道癌、結腸直腸癌、乳癌、膵臓癌と前立腺癌を含み、その中の4名の患者の治療効果は評価できる。これまで、乳癌患者は1人しか薬物治療を受けていなかった;このHR+、HER 2-乳癌を有する患者は、以前に4つの一連の治療を受け、0.75 x 10の投与量で部分的な緩和を得た6細胞/kg。他の2名の大量治療を受けた胃腸腫瘍(結腸直腸癌と膵臓癌)患者は0.75 x 10の用量で病状が安定している6細胞/kgおよび2 x 106細胞/kg。これらの早期臨床データに基づいて、P-MUC 1 C-ALLO 1は安全かつ耐性が良好であり、DLTS、CRS、GVHDまたはICANはない。
P−PSMA−ALLO 1:転移性去勢抵抗型前立腺癌
概要
P−PSMA−ALLO 1は我々の自己バージョンプログラムP−PSMA−101の学習を用いて開発されており,第1段階臨床試験で評価され,第1段階臨床試験では38名の患者が用量治療を受け,予備臨床結果は2022年2月にASCO−GUで公表された。
P−PSMA−ALLO 1はPSMAを発現する細胞を標的とし,PSMAはmCRPC細胞に高発現する。PSMAは葉酸摂取に関与しており,PSMAを発現する腫瘍細胞に増殖優位であると考えられている。また,腫瘍細胞がアンドロゲンに依存しなくなるにつれてPSMAレベルも増加し,前立腺癌進展のマーカーである。したがって,PSMAは抗原脱出が起こりにくい可能性が考えられる。
目標指示
前立腺癌は全世界で4番目によく見られる癌であり、アメリカ男性の癌死亡の第二の原因でもあり、65歳以上の男性の発病率は約60%である。米国だけで約310万人の男性が前立腺癌を患っており,毎年約4万の新しいmCRPC症例が推定されている。
前立腺癌の治療パターンは患者の年齢や診断時の他の潜在的な健康状態によって異なる。早期前立腺癌の治療選択は能動モニタリング、放射線治療、凍結治療、ホルモン治療と手術治療を含む。 転移性疾患患者の例であり,ホルモン感受性型疾患と去勢抵抗型前立腺癌の間にさらに分岐している。
早期前立腺癌患者の5年生存率は100%に近いにもかかわらず、mCRPCに対する需要は依然として高く、5年生存率は約30%しかない。P-PSMA-ALLO 1は
21
臨床で成功し、承認を得ることは、mCRPC患者の生存率と生活の質を著しく高めることができる。
P−BCMA−ALLO 1:多発性骨髄腫
概要
P-BCMA-ALLO 1は羅氏社と協力して開発した多発性骨髄腫を治療する完全同種異体CAR-T候補製品である。著者らは現在P-BCMA-ALLO 1の1期臨床試験を評価しており、著者らは2023年12月のアメリカ血液学会(ASH)年会で早期安全性と初歩的な治療効果結果を公表した。2024年4月のAACR年次総会でBCMA難治性患者の最新データを共有する予定である。私たちは2024年下半期の科学会議でこのプロジェクトの追加臨床更新を共有する予定で、これは羅氏との調整にかかっている。
P-BCMA-ALLO 1は私たちの最初の健康ドナー細胞からの完全同種異体CAR-T候補製品であり、既製の治療法として無関係な多発性骨髄腫患者に使用される可能性がある。我々は,GvHDと宿主の移植片に対する同種反応を減少または除去するために,我々の独自のCas−Clover遺伝子編集ツールを用いてT細胞を遺伝子工学した。CAS-Cloverは静止T細胞を有効に編集することを目的とし、そしてすでに精確な特異性を示し、それによって望ましくない標的外突然変異を制限し、そして耐性を高めることに役立つ。P−BCMA−ALLO 1は単鎖VH BCMA結合剤も含み,臨床前データに基づく結合剤はわれわれP−BCMA−101計画における結合剤よりも優れていると考えられる。
目標指示
多発性骨髄腫は1種の致命的な血液癌形式であり、異常形質細胞から発展し、形質細胞は1種の免疫細胞であり、通常抗体を分泌して感染に対抗する。多発性骨髄腫の根本的な原因はまだ不明であるが、それは異常な形質細胞を産生することによって患者に影響し、これらの細胞は高レベルの抗体或いは抗体断片を分泌し、腎臓と他の器官の機能障害を招き、最終的に致命的である。また,血液中の異常形質細胞の過剰生産や,骨髄や軟部組織における形質細胞腫と呼ばれる腫瘍腫瘍を引き起こす。
アメリカには約16万人の多発性骨髄腫患者があり、毎年3.5万例近くが追加され、死亡は1.3万人近くである。それは男性では女性よりもよく見られ、通常高齢者に影響を与え、平均診断年齢は約70歳である。すでにいくつかの多発性骨髄腫を治療する新薬が承認されたにもかかわらず、大多数の患者にとって、それは依然として不治の病である。現在多発性骨髄腫の治療モードはプロテアソーム阻害剤(PI)、免疫調節薬物(IMiDS)と自己幹細胞移植から始まる。大多数の患者はこれらの薬物に対して薬剤耐性と/或いは再発を産生し、再発/難治性患者に対する高度に満足されていない治療需要を引き起こす。プロテアソーム阻害剤とIMiD失敗後,患者は通常モノクロナル抗体,異なるPIとIMiDあるいはキメラ抗原受容体T細胞(CAR−T)の治療を受ける。多くの患者は最終的に緩和ケアに移行する。治療を行わなければ,多くの多発性骨髄腫患者は確定診断後1年目に死亡した。現在の治療案で治療を受けている患者の約半数は,確定診断後5年間生存している。P−BCMA−ALLO 1が臨床的に成功すれば,再発/難治性多発性骨髄腫患者の生存率と生活の質を著しく向上させることができると信じている。
臨床発展戦略
第一段階の臨床試験の主要な目標は安全性と任意の用量制限毒性(DLT)を評価し、そしてP-BCMA-ALLO 1の単回投与による再発及び/又は常規治療が無効な多発性骨髄腫成人患者のMTDを決定することである。また、著者らは国際骨髄腫ワーキンググループ(IMWG)の標準を用いて抗骨髄腫反応活性を評価している。
22
我々の最初のポイントは、プロテアソーム阻害剤、IMiDおよび抗CD 38治療、および/またはプロテアソーム阻害剤、IMiDおよび抗CD 38治療を含む少なくとも3つの以前の治療を受けた再発性/難治性多発性骨髄腫患者を募集することである。
この試験は開放ラベルの投与量増加試験であり、現在の方案は最大235名の再発/難治性多発性骨髄腫を有する成年被験者の登録を許可し、各列は最大7回の投与量が増加し、標準の3+3用量増加設計を使用する。P−BCMA−ALLO 1 CAR−T細胞を受ける前に,条件リンパ浄化化学療法レジメンを受けた。標準案には300 mg/mの投与量が含まれています2シクロホスファミドと30 mg/m2フルダラビンは毎日静脈点滴し、3日間連続し、その後2日にP-BCMA-ALLO 1を単回静脈点滴した。被験者はさらに高用量化学療法(シクロホスファミド500 mg/m)を含む代替リンパ浄化レジメンを受けた2/d×3日、フルダラビン30 mg/m2/日×3日またはシクロホスファミド1000 mg/m2/d×3日、フルダラビン30 mg/m2/日×3日)。各アーム計画のP-BCMA-ALLO 1用量増加範囲は0.75~15 x 106細胞/kg。治療を受けた患者は安全性、耐性と腫瘍反応の一連の測定を受け、そして最後の剤P-BCMA-ALLO 1後に15年間のフォローアップを行う。
同社は最近2023年12月に開催された第65回アメリカ血液学会(ASH)年会と博覧会で積極的な早期安全性と初歩的な治療効果データを共有した。
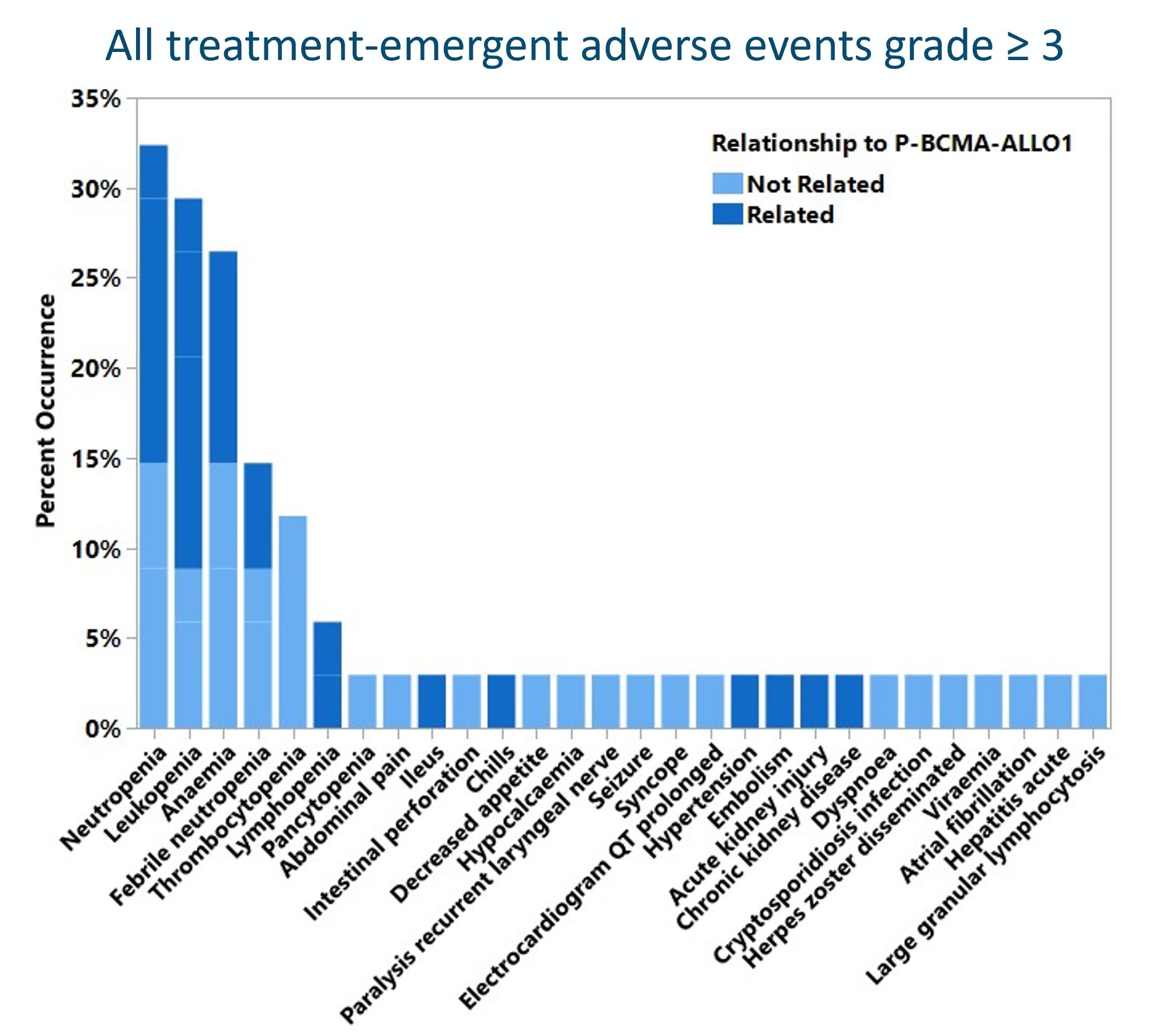
会議で強調されたデータによると,P−BCMA−ALLO 1は耐性良好な既製療法であり,良好な新出現の安全性を有し,架橋化学療法や他の抗骨髄腫架橋療法を用いずに,意図的に治療した群の100%の患者に提供した。移植片対宿主病や用量制限毒性は発生しておらず,CRSと神経毒性の発生率は低い(いずれも2級)。
23
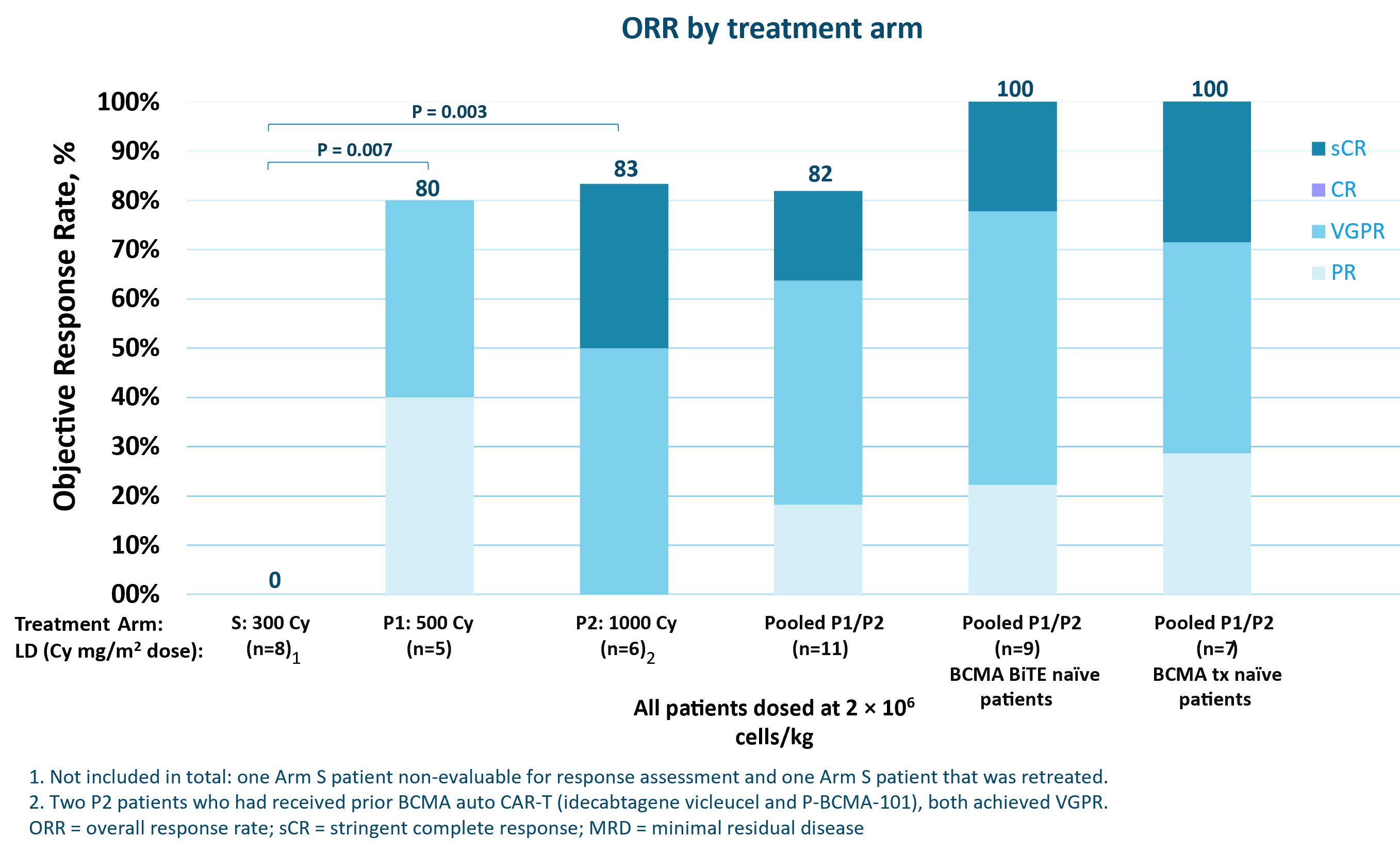
データによると、82%の総有効率(OOR)と深い臨床反応は、MRD陰性患者の厳格な完全反応(SCR)、十分なリンパ浄化を受けた重度前処理患者からの既製の同種異体BCMA標的CAR-Tを含む。これらの患者のうち,これまでBCMA標的二重特異性T細胞結合抗体治療を受けていなかった患者は100%のORRであった。研究登録からリンパ除去化学療法開始までの中位時間は1日,P−BCMA−ALLO 1投与までの中位時間は7日であった。2023年10月23日現在のデータ締め切り時には,反応のある患者9名中8名が臨床反応中であった。
24
初歩的なデータによると異遺伝子T細胞供給チェーン管理豊富なCAR-T細胞が骨髄に輸送され、細胞傷害効果T細胞に分化し、治療後少なくとも6週間持続することは、細胞が腫瘍関連部位に持続的に存在するという仮説を支持する。
P-CD 19 CD 20-ALLO 1: B細胞悪性腫瘍
概要
P-CD 19 CD 20-ALLO 1は同種異体、既製のCAR-T製品であり、羅氏会社と協力し、B細胞白血病とリンパ腫の臨床前開発に応用されている。CD 19とCD 20を同時に対象とすることにより,早期CD 19 CAR−T製品で観察された抗原脱出のいくつかの問題を克服する可能性があると信じている。
臨床発展戦略
IND申請を提出した後,2023年6月にFDAの承認を得て,最近臨床試験を開始することができた。この第一段階、マルチセンター、開放ラベル、用量増加研究は、いくつかの再発/難治性B細胞悪性腫瘍を有する成人被験者約70名を募集と治療し、最大7回の用量増加行列、範囲は0.75から15×10までである6細胞/kgは、標準的な3+3用量逓増設計を採用した。P−CD 19 CD 20−ALLO 1 CAR−T細胞を受ける前に,条件リンパ浄化化学療法レジメンを受ける2シクロホスファミドと30 mg/m2フルダラビンは毎日静脈点滴し、3日間連続し、その後2日にP-CD 19 CD 20-ALLO 1を単回静脈点滴した。被験者はまた、交互リンパ浄化レジメンおよびより高い用量の化学療法(シクロホスファミド750 mg/m)を受けることができる2/d×3日、フルダラビン30 mg/m2/日×3日またはシクロホスファミド1000 mg/m2/d×3日、フルダラビン30 mg/m2/日)。
P-BCMACD 19-ALLO 1それは.P-BCMACD 19-ALLO 1は多発性骨髄腫の臨床前に開発された同種異体、既製のCAR-T候補製品である。P-BCMACD 19-ALLO 1は2つの機能が揃ったCAR分子を含み、BCMA或いはCD 19を発現する細胞を標的とすることができる。すでに発表された多発性骨髄腫患者のCD 19候補治療に対する研究によると、ある患者では、CD 19を発現する可能性があるがBCMAを発現しない骨髄腫幹細胞が存在する可能性があるため、BCMAおよびCD 19を同時に標的化することは、BCMA単独標的化よりも有効である可能性が考えられる。また,CD 19を含めて抗薬抗体反応を防ぐことができ,この反応はある患者のBCMA治療のみの有効性を短縮する可能性がある。我々がP−BCMA−ALLO 1計画から学んだ知識を適用することにより,完全に同種異体であるようにこの候補製品を開発している。P−BCMA−ALLO 1ステージ1臨床試験で観察された初歩的な結果を分析した後,IND申請を提出する予定である。羅氏はこの計画許可証を取得する独占的な選択権を持っている。
P-CD 70-ALLO 1それは.P-CD 70-ALLO 1はCD 70を標的とし、CD 70はすでにこの領域で公認されている標的になっている。我々はこの候補製品を開発しており,他の同種異体計画から学んだ知識を適用することで,完全に同種異体としている。羅氏はこの計画許可証を取得する独占的な選択権を持っている。
我々は,我々の早期研究の結果を最もよく利用し,さらなるパイプライン開発に情報を提供するために,我々の最初とこれからの臨床計画を戦略的に設計した。私たちはいくつかの臨床前計画があり、私たちの様々な目標を代表する第二世代あるいは第三世代計画を目的とし、私たちのプラットフォームの異なる機能を利用して他の適応を探索している。
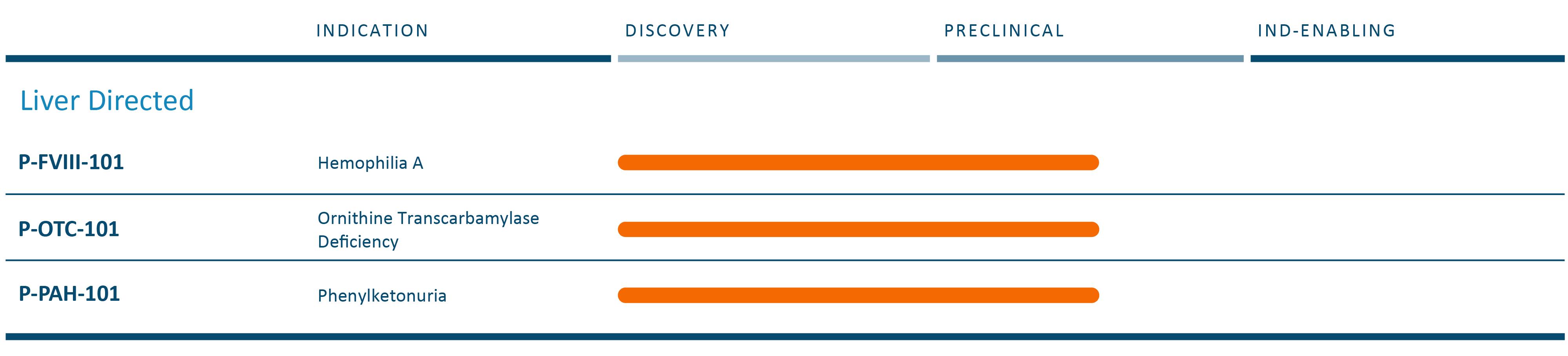
肝臓ガイド遺伝子治療
この概念は体内にある遺伝子療法は20世紀70年代初めのSに興り、最初の人体試験は1980年に始まった。しかし、早期の臨床失敗はこの領域の発展を阻害し、関連する資金と進展は過去10年まで緩慢であった。この10年間、遺伝子療法は拡張され、より多くの受け入れられてきた。期限が切れる
25
いくつかの臨床上の成功や関連する資金やM&A活動により,この分野は現在新たな治療開発の主な焦点となっている。興味と発展が新たに現れたにもかかわらず体内にある遺伝子治療は重大な挑戦に直面している。
現在の多くの遺伝子療法の主な限界の1つは,これらの療法が通常一過性であるため,狭い適応範囲に限られていることである。これらの制限は多くの要素によって駆動され、これらの要素はAAVを伝達治療性遺伝子組換えの標準方法として使用することと関係がある。まず、特定のAAVカプシドは、インビボで複数の細胞タイプに効果的に感染するために使用することができるが、AAVは、治療用トランスジェニックに適応するために遺伝子治療用途において除去されたウイルスのrep遺伝子なしにゲノムに組み込まれることは通常ない。統合の不足は治療性遺伝子組換えの低発現レベルを招き、通常時間の経過とともに低下する。細胞分裂時、発現は最終的に失われるため、急速に分裂した組織でAAV媒介遺伝子治療、例えば小児科肝臓を使用することは困難または不可能である。不幸にも、小児科肝臓は多くの単遺伝子先天性代謝ミスを治療するために標的を必要とする組織であり、特に多くのより深刻な影響を受けている患者である。次に、AAVの負荷量は相対的に小さく、これはその治療により大きな治療性遺伝子組換えを必要とし、潜在疾患の適応を是正する能力を制限する可能性がある。比較的小さいキャリア量も付加的な特徴の添加を制限し、例えばより大きな組織専用促進剤、絶縁体または安全スイッチを提供する。第三に、一部の患者では、AAV自体が予め存在する抗体と免疫原性を有することができる。また,AAVによる療法は通常抗体による免疫反応を起こし,繰り返し投与は非常に挑戦的である。最後に,前世代のAAV療法は臨床効果を産生するのに十分な遺伝子を伝達するのに比較的高用量のウイルスが必要であり,AAV自体に関する安全問題が生じている。
我々の技術は,いくつかの重要な点から他のAAV手法の欠点を解決することを目的としている.まず,われわれのiggyBac技術をAAVと組み合わせることにより,治療用遺伝子組換えをDNAに統合し,患者DNAの安定した部分となり,急速に分裂した細胞においても同様の治療法を作ることができると信じている。これは単一治療治癒の可能性を招き,治療が主に小児科肝臓の適応に現れた場合でも同様である。次に,iggyBacはDNAへの統合が非常に効率的であり,比較的低い用量でも安定かつ高レベルの治療的遺伝子組換え発現が生じ,現在AAV技術のみでは治療できない適応に有効な活性を許容している可能性が考えられる。また,PiggyBacとAAVの併用は,AAVのみを用いた技術に比べてウイルス量がはるかに低い場合に有効である可能性があるため,AAV自体によるいくつかの毒性リスクを軽減する。
私たちはまた、私たちのGiggyBac技術を私たちのナノ粒子技術と組み合わせて、AAVに対する需要を完全に除去するために治療的遺伝子組換えを提供する。これはウイルス関連の毒性を完全に回避し,より大きな遺伝子や重複投与を伝達することができ,治療可能な適応数をさらに拡大することになる。
私たちの技術プラットフォームは開発を支援していますが体内にある遺伝子治療は広範な応用の中で、著者らは最初の努力を肝臓ガイド遺伝子治療に集中しており、この方面で、著者らは将来性の高い臨床前データを持っており、そして私たちは早期世代の遺伝子治療より顕著な競争優位があると信じている。我々の技術はAAV技術だけでは解決できない適応や患者群問題を解決する可能性があると信じている。場合によっては,我々のiggyBac技術をAAVやナノ粒子輸送と組み合わせることにより,これらの一過性療法を単一治療,一生持続的な反応に変えることが可能であると信じている。
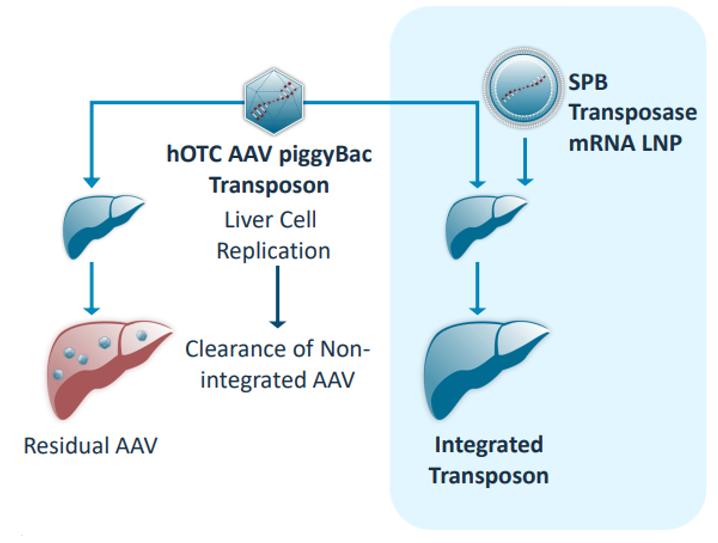
いずれのAAVベースシステムも、AAV ITR(AAV+PiggyBacトランスポゾン)に50塩基対まで小さくすることができるPiggyBac ITRを簡単に追加することによって、PiggyBac-AAVベクターに変換することができる。このベクターはiggyBacトランスポナーゼなしで標準AAVベクターと同様の性能を示し,後者は第2のAAV(AAV+iggyBacトランスポナーゼ)で伝達できると予想される。治療用遺伝子の代替として増強緑色蛍光蛋白(EGFP)レポーター遺伝子を用い,AAV+iggyBacトランスポゾン(トランスポナーゼなし)を動物に注入したところ,マウス肝臓に低レベルのEGFP発現が認められた(左下図)。他の標準的なAAV療法と同様に,上体(非統合)AAVの発現レベルが低いため,時間の経過とともに発現レベルが減弱し,特に細胞分裂時には低下する。しかし,AAV+PiggyBacトランスポゾンとAAV+トランスポゾンを共注射したところ,右下図に示すように,多くの肝細胞に高レベル,安定した発現が認められた。この場合,iggyBacトランスポナーゼは遺伝子組換えをトランスポゾンから抽出し,安定してゲノムに統合する。細胞が分裂するにつれて彼らは統合された
26
治療用遺伝子組換えなので、すべての子孫細胞はそれを永久的に発現している。この戦略は,OTCD,シトルリン血症I型,進行性家族性肝内胆汁うっ滞症III型の3つの異なる重症先天性肝臓遺伝病のマウスモデルに用いられており,各症例の単一治療の潜在力を示している。
我々の遺伝子治療計画の目標の1つは,ナノ粒子を介して我々の遺伝子工学技術を伝達し,AAVの限界によるAAVの使用ニーズを解消することである。臨床前作業では,iggyBacトランスポゾン(DNA)とiggyBacトランスポナーゼ(RNA)を動物モデルに導入した積極的な結果が見られ,肝臓のすべての領域で有意な統合と遺伝子組換え発現が実現された。次の図は実験を示し、この実験では、1匹の若年マウスに異なるナノ粒子を構成するiggyBacトランスポゾン(DNA)とiggyBacトランスポナーゼ(RNA)を併用し、肝臓におけるレポーター遺伝子の発現レベルを測定し、7ケ月間持続した。これらのデータは初歩的であるが,我々のナノ粒子送達iggyBacの目標に向かって重要な一歩を踏み出していることを表している可能性があり,従来の遺伝子療法と比較した大きな進歩を表していると信じている。
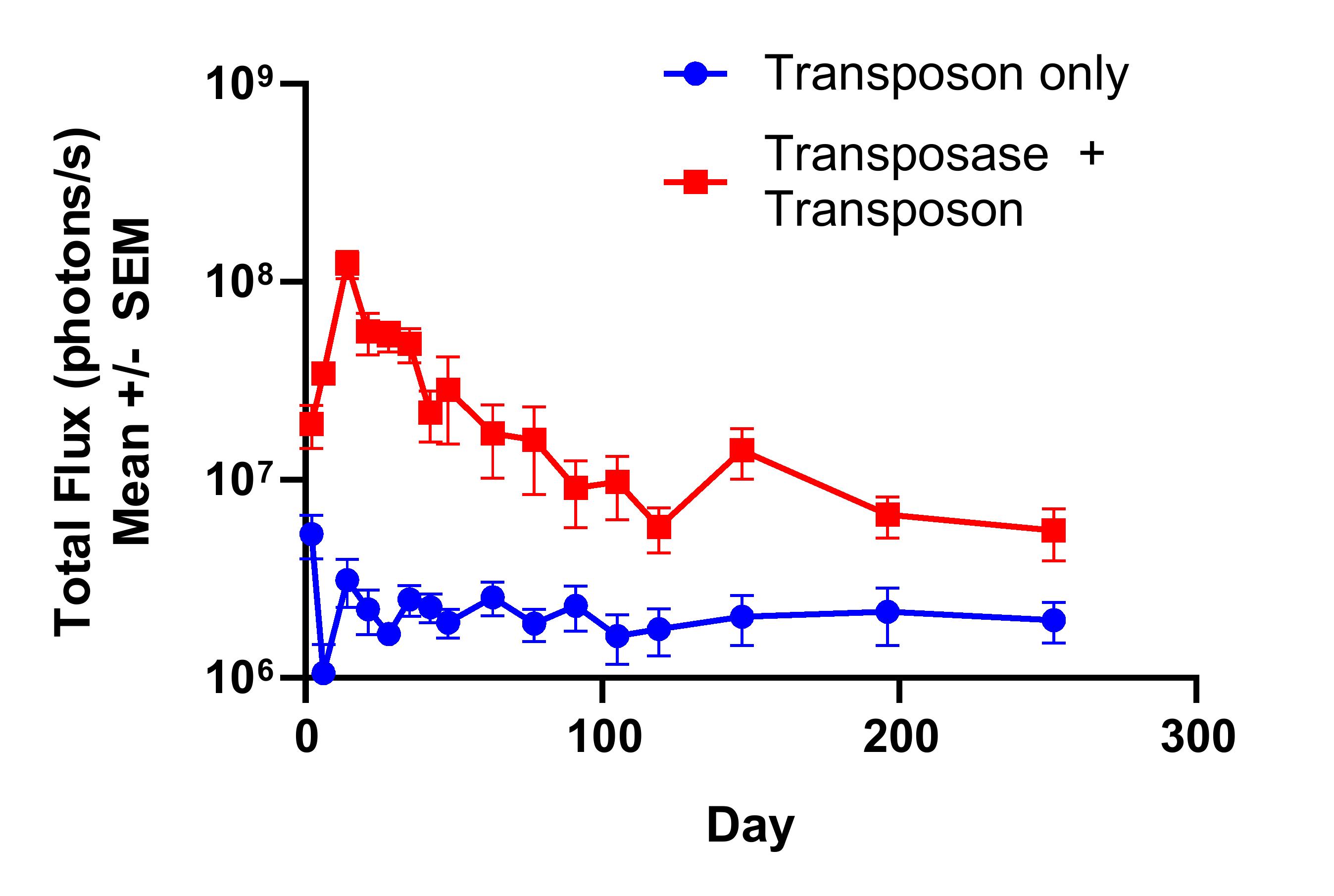
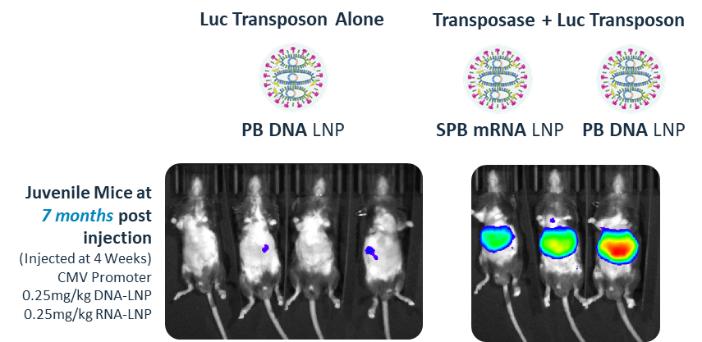
27
私たちの遺伝子治療計画は
遺伝子治療
次の表は現在の遺伝子治療候補の組み合わせをまとめました
我々の最終目標は,我々のナノ粒子技術でAAVを完全に置換し,将来の遺伝子治療における製品開発をAAVの制限から脱却させることである。
P−FVIII−101
概要
P-FVIII-101は肝臓ガイド遺伝子療法であり、私たち固有のiggyBac DNA送達システムを私たち固有の非ウイルスナノ粒子技術と組み合わせて、体内にある血友病Aを治療する。
私たちは、このプロジェクトに私たちの完全な非ウイルス伝達システムを配備することを選択し、AAVや他のウイルスベースの技術に依存しない。
目標指示
血友病Aは凝固因子VIII産生不足による出血性疾患であり,満足されていない需要が高い。疾病の重症度は軽微から深刻まで様々であり、第八因子レベルは疾病の重症度と関係がある。
臨床前データ
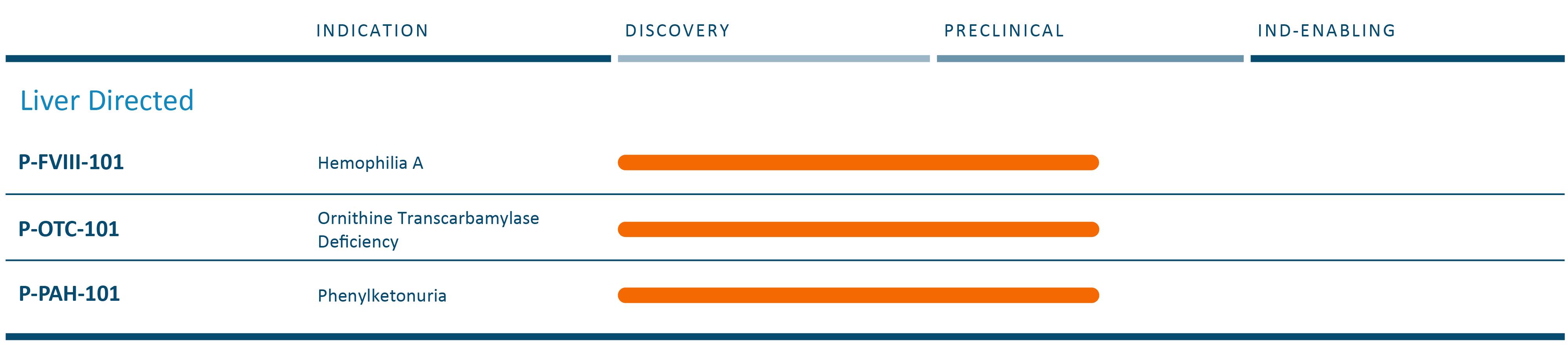
我々の臨床前データは、我々のP-FVIII-101潜在的製品候補のナノ粒子を使用して送達され、凝固因子VIII欠乏を若年マウスモデルにおいて機能正常化を提供することが予想されるレベルまで是正することができることを示している。著者らは2023年12月のアメリカ血液学会(ASH)年会でこの計画の臨床前データを公表し、P-FVIII-101が単剤後に(約50%)ヒト凝固因子VIII活性に達することを示し、単剤と繰り返し用量後に成人マウスに12ケ月持続する治療性凝固因子VIII活性を提供し、用量滴定の可能性を示した。これらのデータ支持は、我々の非ウイルス送達システムにおいて、治療用トランスジェニック発現カセットを肝細胞のゲノムに安定的に統合することができ、一貫性および持続的な治療活性を提供することができる。
概要
P-OTC-101は体内にある肝臓ガイド遺伝子治療は深刻な早発性OTCDを治療する候補薬物であり、この疾患は単一治療、一生持続的な反応を実現する可能性があると信じている。私たちは私たちの方法を信じて
28
生命早期の患者に対して治療を行うことができ、伝統的なAAVによる遺伝子療法と比較して重要な利点を提供し、後者は新生児と青少年に有効である可能性が低い。私たちは独自のiggyBac DNA送達システムと肝臓ガイドAAVまたはナノ粒子を組み合わせたことを評価しています体内にあるOTCDの治療。OTCDは通常致命的或いは病的な尿素循環系疾患であり、OTC遺伝子の先天性突然変異によって引き起こされ、高度に満足されていない医療需要を持っている。私たちは2023年7月にFDAからこの計画の孤児薬物指定を取得した。
目標指示
OTCDは稀な遺伝性疾患であり、OTC酵素が完全または部分的に欠乏していることを特徴とする。OTCは酵素であり,体内の窒素を分解·除去する過程に作用し,この過程を尿素循環と呼ぶ。OTC酵素の欠乏は,窒素が血液中にアンモニア(高アンモニア血症)として過剰に蓄積する。過剰なアンモニアは神経毒素であり,血液を介して中枢神経系に入り,傾眠,嘔吐,怒りやすなどの症状を招き,より重篤な場合には筋張力低下,てんかん発作,肝臓腫大,呼吸困難,死亡をきたす。この病気の深刻な形は赤ちゃんに影響を与え、通常は新しく生まれた男性です。この疾患の軽い形は乳児期の遅い子供に影響を与える。より深刻な非処方薬は高度に満たされていない医療需要を含む。
われわれの臨床前研究では,iggyBacとAAVとLNPsを組み合わせた方法により,治療性ヒトOTC遺伝子組換えが新生OTC欠損マウスの肝臓で安定かつ高レベルで発現していることが証明された。対照的に,同じヒトOTC遺伝子組換えを含む従来のAAV遺伝子治療を受けたマウス(iggyBac媒介統合を使用しない)は,無視できるヒトOTC発現を示した。
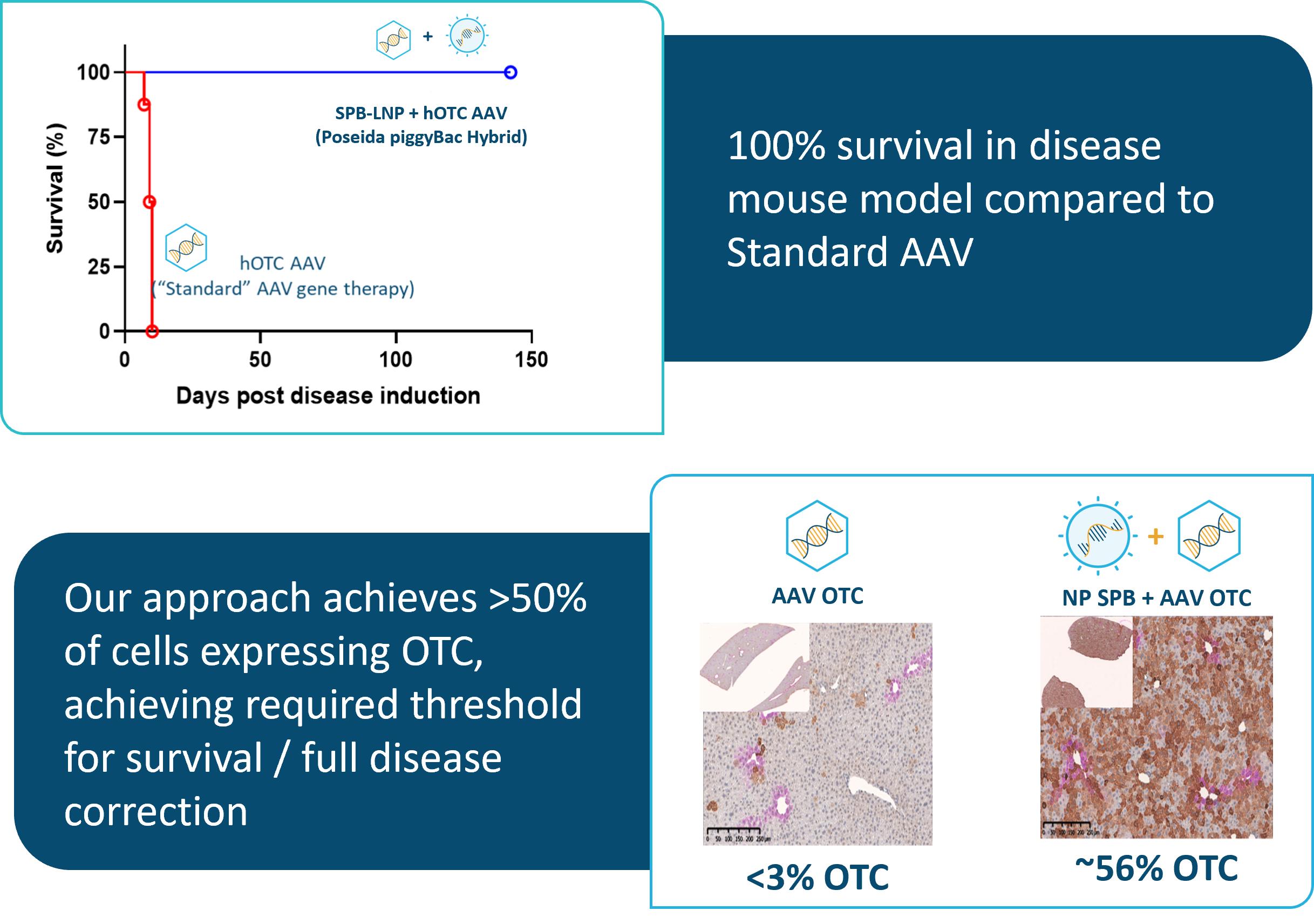
29
別の単独研究では、我々のヒトOTC発現カセットをコードするiggyBacトランスポゾンを含む3つのユニークなAAVカプシドが、OTCDのマウスモデルにおいて評価された。すべての3つのカプシドは、持続的なヒトOTC mRNA転写および対応するOTCタンパク質の肝臓内での活性を示した。これらのデータは,iggyBacが様々な既存の臨床的に成熟したAAVカプシドに容易に統合できることを示している。また,非処方薬活性の急激な増加は,標準的なAAV単独療法と比較してPiggyBac−OTCの用量低下の可能性と,依然として単一治療,持続反応を実現できる能力を顕著に示しており,標準AAV療法と比較して余分なコストと耐性の利点がある。
P-PAH-101
概要
P-PAH-101は肝臓ガイド遺伝子療法であり、iggyBac技術と我々のナノ粒子送達技術を組み合わせて治療に使用されている体内にあるフェニルアセトン尿症の治療、あるいはPKU。私たちは独自のiggyBac DNA送達システムと肝臓ガイドAAVとナノ粒子を組み合わせたことを評価しています体内にある北京大学の治療。
目標指示
PKUは代謝異常であり、正常な情況下でフェニルアラニンをチロシンに変換する酵素欠陥によって引き起こされる。これはフェニルアラニンの蓄積を招き、フェニルアラニンは脳に有害であり、色素沈着の減少、及び成長と神経予後不良を招く。食事蛋白質の制限は看護の基準であるが、厳格な一生堅持が必要であり、これは多くの患者、特に年齢の大きい児童にとって挑戦的である可能性がある。
臨床前データ
著者らの臨床前データは、典型的なフェニルアセトン尿症マウスモデルにおいて、血清フェニルアラニン、1つの重要なPKUバイオマーカーが、正常レベルまで分解できることを示した。もう一つの単独研究において、著者らは治療性フェニルアラニンヒドロキシ酵素或いはPAH遺伝子組換えのマウス単一治療後の肝臓全体における発現を証明した。著者らのデータは、生命早期に治療を受けた後、高治療性PAH蛋白発現と広範な肝細胞分布を維持できることは著者らのiggyBacプラットフォームの統合機序によるものであることを表明した。
私たちの戦略
我々の使命は,治癒能を有する次世代細胞と遺伝子療法の開発である。
我々の広範な遺伝子工学プラットフォーム技術を用いて新しい細胞や遺伝子治療製品を開発·商業化し,様々な適応の中で高度に満足されていない医療ニーズの患者を治療する予定である。現在の製品ラインには腫瘍適応のための同種CAR-T製品と
30
子豚Bac+AAVと子豚Bac+ナノ粒子製品は孤児遺伝病肝臓ガイド遺伝子治療方案の候補とすることができる。私たちは以下の戦略で私たちの使命を実現する予定です
血液悪性腫瘍に対する同種異体CAR−T療法を迅速に開発·商業化した。われわれは,現在市販されているCAR−T療法の後方勤務,コスト,安全制限を解決するために,再発/難治性多発性骨髄腫患者に適した候補品であるP−BCMA−ALLO 1を開発している。我々は,現在市販されているCAR−T療法の後方勤務,コスト,有効性,安全性制限を解決するために,再発/難治性B細胞悪性腫瘍患者に適した候補品であるP−CD 19 CD 20−ALLO 1を開発している。時間が経つにつれて、早期の治療シリーズで私たちの候補製品を開発する予定です。我々のCAR−T候補製品の観察および予想される安全プロファイルは、コミュニティ腫瘍診療所を含む外来環境での通常の投与を可能にすべきである。
著者らのプラットフォーム技術の実力と広さを利用して、固形腫瘍に対する同種異体CAR-T療法を開発した。著者らのプラットフォーム技術は固形腫瘍の治療におけるCAR-Tの歴史的制限を解決することを目的としており、これらの制限はこれらの適応に臨床影響を与えるために必要な製品持続性の不足によるものである。固形腫瘍を治療する候補薬剤としてP−MUC 1 C−ALLO 1を進めており、CARおよびTCR法がより多く使用される可能性のある製品の評価を継続していく。
我々のプラットフォーム技術を用いて肝臓ガイドの遺伝子治療計画を行った。我々の先行遺伝子治療候補製品P−OTC−101とP−PAH−101は我々のiggyBac技術を用いてAAVとナノ粒子と結合し,孤児遺伝病に対して単一療法の開発を目指している。また、P−FVIII−101は、ナノ粒子をベースとした我々の体内にある遺伝子療法は,AAV技術の需要に代わっている。耐性を向上させ,コストを低減し,再投与を許可し,AAVが有効に解決できない兆候を解決することにより,遺伝子治療のナノ粒子伝達は,大型治療性遺伝子組換えを伝達する必要がある疾患の是正を含むAAV伝達に対する重大な進歩である可能性が考えられる。我々と我々の将来の潜在的パートナー(あれば)は我々の候補遺伝子治療製品の開発を計画しており,承認されれば商業化される。
我々の技術と能力を利用して同種異体多車-T製品を開発した。著者らのダブルCAR同種異体製品候補はB細胞悪性腫瘍のためのP-CD 19 CD 20-ALLO 1と多発性骨髄腫のためのP-BCMACD 19-ALLO 1を含む。これらの多車計画は,我々のiggyBacプラットフォームが他の技術では容易に実現できない候補製品を支援できる能力を顕著に示していると信じている.我々は引き続き多車候補製品の開発を計画しており,次世代CAR−T療法を代表する可能性があると信じている。
戦略的パートナーシップと構造を評価して価値を創造し、私たちのプラットフォーム技術を革新し、開発し続ける.我々のプラットフォーム技術は高度に差別化され、広範な治療方式と適応の中で多くの候補製品を創造することができる。そこで、私たちは私たちのカバー範囲を拡大し、私たちの使命を追求する過程で追加的な価値を創出するために、2つの重要な協力を行った。2021年10月から2023年7月まで、武田協力協定に従って武田と協力し、遺伝子治療努力をさらに拡大していきます。2022年8月,われわれは羅氏協力協定を発表し,血液学的適応の範囲内でわれわれの同種輸血管をさらに開発した。2023年8月、私たちはアストラスの戦略投資を発表した。我々の技術の広さを考慮すると,細胞療法による自己免疫疾患の治療など,我々の技術を開発し,戦略的パートナーシップを評価する分野があると信じている。
仲間関係
羅氏
2022年8月、私たちは羅氏とパートナー関係を構築することを発表し、彼らは私たちの血液学先導適応を許可または選択した。プリペイドライセンスに含まれ、ロー氏は、P−BCMA−ALLO 1およびP−CD 19 CD 20−ALLO 1を許可するか、またはそれぞれ1つのTier 1計画を許可する。P−BCMA−ALLO 1は現在第1段階試験であり,再発/難治性多発性骨髄腫患者のために開発されており,われわれの最初の自己プログラムP−BCMA−101の経験を用いている。P-CD 19 CD 20-ALLO 1はB細胞血液学的指標を治療するために開発された計画であり、著者らは最近この計画の一期試験を開始した。この2つの許可計画に加えて,羅氏はP-CD 70-ALLO 1とP-BCMACD 19-ALLO 1を許可するか,2つのTier 2計画をそれぞれ許可することができる.P−CD 70−ALLO 1は開発中の血液学的適応に対する臨床前段階計画である。P-BCMACD 19-ALLO 1は1種の臨床前両標的方案であり、多発性骨髄腫の治療に開発されている。Tier 1とTier 2計画のほかに、羅氏会社と研究協力を達成しました。その中で、羅氏会社は私たちのいくつかの知的プロジェクトの下で独占的な許可を持っています
31
最大6種類の血液学的適応の同種異体CAR-T細胞治療製品を開発、製造と商業化した。
羅氏の協力協定によると、羅氏は私たちに1.1億ドルを前払いした。ロ氏の第2レベル計画オプションの行使、協力計画の指定とライセンス製品の商業許可の選択権の行使、第1レベル計画、オプション第2レベル計画と協力計画に基づいて特定の開発、監督、純売上マイルストーン事件を実現する製品などによると、(I)第1レベル計画15億ドル、(Ii)第2レベル計画11億ドル、(Iii)協力計画29億ドルを含むいくつかの精算、費用、マイルストーン支払いを獲得する資格がある。(Iv)特許製品の4.15億ドルです我々はまた,製品ごとに,Tier 1計画,オプションTier 2計画と連携計画による製品純売上高の中位から下位2桁,および許可製品の中央桁から中央値の階層印税支払いを許可する権利があり,いずれの場合も,いくつかの通常の減免と補償の制限を受けている.使用料は、その製品をカバーするライセンス特許がその国で満期になるまで、またはその製品が同国で初めて商業販売された日から10年まで製品および国/地域によって支払われる。
2023年11月7日から、羅氏協力協定は、(I)羅氏が私たちに支払ったいくつかの既存の製造関連費用を再分配して、私たちの既存の各第1段階計画のために新しい製造プロセスの開発および移転費用を追加するために再分配され、(Ii)羅氏が各第1級計画のために私たちに支払ったいくつかの開発マイルストーンの支払いの金額を再分配するように改正された。改正案では、1500万ドルのマイルストーン費用を払えば、既存の2年期研究計画をさらに18カ月延長できることも規定されている。その他の点では、ロ氏協力協定下のすべての履行義務と支払い条件は変わらない。
武田さん
2021年10月には、武田薬品米国社または武田社と協力·許可協定、すなわち武田協力協定を締結し、この協定に基づき、4500万ドルの前金および合意期間全体の研究開発費を取得した。
2023年5月31日、武田から書面通知を受け、武田は武田協力協定を中止することを選択し、2023年7月30日から発効、または終了日とする。終了日まで、双方はそれぞれ武田協力協議での責任を果たしていたが、終了日には、吾らの武田協力協議での独占責任は終了した。また、武田の免許を付与し、武田協力協定に基づいて研究活動を行う許可証を付与した。終了後,P-FVIII-101およびP-PAH-101を含むすべての以前に許可されたプログラムを開発するすべての権利が付与される.遺伝子治療の分野で新たな戦略的協力を求めることができるかもしれないが、これまで武田協力協定に含まれていた計画の一部または全部と、現在このような戦略的協力の約束または合意に達していないにもかかわらず、可能な追加の内部計画が含まれている可能性がある。
潜在的な他の計画とパートナーの機会
我々は我々のプラットフォーム技術を利用して現在CAR-Tと肝臓ガイド遺伝子治療製品の候補製品を開発しているが、著者らの技術は広範な細胞と遺伝子治療方式と疾病に広範な適用性がある。現在のチャネルを除いて、著者らと著者らの協力者は臨床前データを持っており、これらのデータは様々な方法で組み合わせた場合の技術プラットフォームの将来の潜在的な応用を示している。我々は将来的にこれらのツールを使用して、腫瘍学以外の適応、例えば自己免疫疾患、伝染病、アレルギー関連疾患、甚だしきに至っては神経変性疾患を解決するためにT細胞ベースの製品を創造するかもしれない。CAR-Tも幹細胞移植の代替と非清髄性プレコンディショニング方案とすることができる。我々の技術は,多能性幹細胞,ナチュラルキラー細胞,造血幹細胞,B細胞,肝細胞,筋肉,および多くの他の細胞を誘導することを含む他のタイプの細胞や組織にも適用可能であり,将来的に様々な適応で治療を行う他の方法が可能となる。最後にCas-Clover技術を直接使うことができます体内にある他の遺伝子編集会社が採用している方法と同様です
32
私たちのチームは
われわれは経験豊富な管理チームが指導し,治癒能を有する次世代細胞や遺伝子療法の開発に揺るぎなく取り組んでいる。私たちのCEO Mark J.Gergen,J.D.は医療·生命科学会社で25年以上の経験を持ち,最近では2022年2月にCEOに移行し,最近では2024年1月1日にCEOに任命された。Gergenさんは2018年初めに当社に加入する前に、Amylin PharmPharmticals、Mirati Treeutics、Halozyme Treeuticsを含む複数の成功したバイオテクノロジー会社の実行管理チームのメンバーでした。私たちの最高経営責任者であるKristin Yarema博士は、本名社長で、2022年4月から細胞療法会社のCEOを務め、CEOを務め、医療·生命科学会社で20年以上の経験を持ち、2022年4月にわが社に入社する前に、Yarema博士はヤダラ生物療法会社、安進会社、ノワール社を含む多くの成功したバイオテクノロジーや製薬会社の実行管理チームのメンバーである。同管理チームは2023年12月31日現在、330人の従業員の支持を得ており、そのうち165人が高度な学位を有し、そのうち91人が博士号および/または医学博士号を有し、多くの人が薬物発見と開発の面で豊富な経験を持っている。
私たちのCAR-T製造技術は
我々のCAR−T候補製品は健康なドナーT細胞からなり,これらのT細胞は遺伝子工学を経てCAR分子や他の遺伝子を発現することができる。登録された病院で標準的な白血球分離プログラムによりPBMCを採取し,このプログラムの直後に白血球分離細胞を製造場所に搬送した。
CAR−T候補製品の製造は、陽性選択によりCD 4陽性およびCD 8陽性T細胞を単離することを含む。その後、電気穿孔トランスポゾン遺伝子組換え(CAR分子遺伝子、DHFR正選択遺伝子と安全スイッチ遺伝子をコード)、スーパーブタBacトランスポナーゼRNA(iggyBacトランスポゾン遺伝子組換えを動員する酵素)、Cas-Coverer遺伝子編集システムをコードするmRNA及び2つの同種異体拒絶反応に関連する異なる遺伝子に対する誘導RNA、及び増強分子をコードするmRNAである。その後,メトトレキサートによりCAR陽性のT細胞を選択し,これらの細胞を増幅し,さらに精製してトランスジェニック細胞を得た。
最後の製品は袋に入れて超低温保存されます。製品が管理のために発表された後、凍結保存された候補製品は、登録研究センターの薬局または適用された細胞治療機関に宅配便で搬送され、そこで管理時間に格納される。
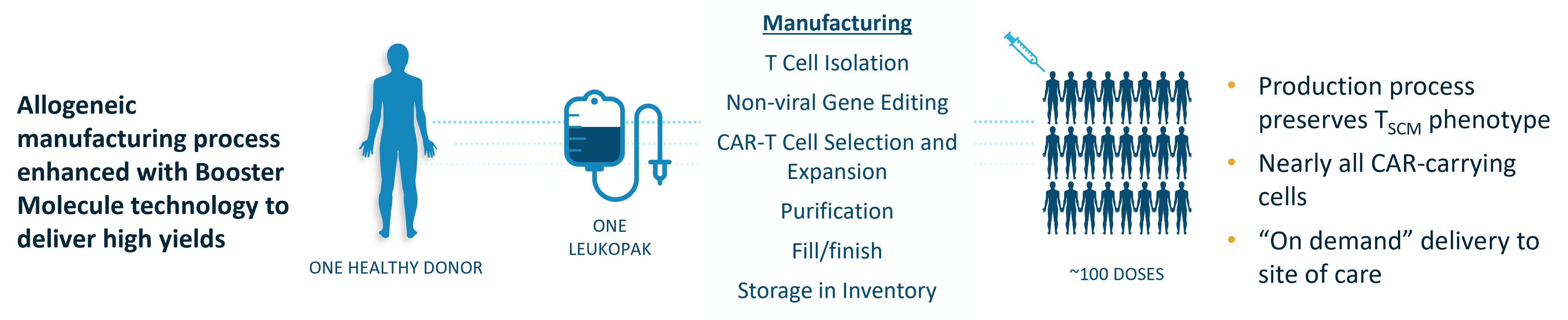
CAR-T代行
私たちはサンディエゴに内部臨床GMP生産工場を持っていて、私たちの本部に隣接して、第一段階と第二段階の臨床試験のために臨床前材料と臨床用品を開発と生産します。我々は2021年第3四半期にGMPの生産を開始し,最初にこの工場を用いて我々のP−MUC 1 C−ALLO 1計画を生産した。私たちは今、P-BCMA-ALLO 1とP-CD 19 CD 20-ALLO 1を含む他の候補製品を追加しました。これは私たちの臨床製造の唯一の源です。
私たちは以前、複数の第三者契約メーカーと協力して私たちの候補製品を生産した。P−BCMA−ALLO 1の製造には,われわれは以前薬明カント社と協力し,彼らから臨床供給を得ていた。2022年第4四半期、将来の製品源となる当社の内部臨床工場製造施設でP-BCMA-ALLO 1を生産する許可を得ました
33
前へ行ってください。私たちの他の候補製品について、私たちは様々な第三者製造業者の臨床供給を評価している。私たちは様々なサプライヤーと協力して原材料を提供してくれました現在私たちのメディアはStemcell、DNAコンポーネントはAldevronからです私たちは私たちが契約製造業者と供給者との関係が良いと信じている。将来、私たちはまた任意の承認された候補製品のための1つ以上の商業製造施設を建設することができるかもしれない。
商業化計画
私たちは内部製品候補製品と計画を発見する世界的な権利を持っている。私たちは私たちの候補製品に対して重要な開発と商業化の権利を保持するつもりで、マーケティングの承認を得たら、アメリカと他の地域で単独で、またはパートナーと私たちの候補製品を商業化することができます。私たちは現在、販売、マーケティング、あるいは商業製品の流通能力がなく、会社として製品をマーケティングした経験もありません。私たちの候補製品のさらなる発展に伴い、私たちは時間の経過とともにアメリカに必要なインフラと能力を構築する予定で、他の地域も含まれるかもしれません。臨床データ、アドレス指定可能な患者集団の規模、商業インフラの規模及び製造需要は著者らの商業化計画に影響或いは変更する可能性がある。
競争
生物技術業界、特にCAR-Tと遺伝子治療科学は、新技術と特許製品の開発の激しいと迅速な変化の競争を特徴とする。我々はCAR-Tと遺伝子療法における特許方法と科学専門知識が私たちに競争優位を提供してくれると信じているが、私たちはより規模が大きく、資金が豊富な製薬会社、学術と研究機関を含む多くの異なる源からの潜在的な競争に直面している。もし私たちの競争相手が私たちが開発する可能性のあるどの製品よりも効果的で、安全で、副作用が少ない、より便利で、コストの低い製品を開発し、商業化すれば、私たちのビジネス機会は減少または消失するかもしれない。私たちのプロジェクトの成功に影響を与える重要な競争要素は、それらの有効性、安全性、利便性、コストかもしれない。
現在,我々が最初に選択した適応の既存療法や/または新療法の商業化に努力している他の組織もある。もしこれらの努力が成功し、彼らの候補製品が私たちの前に承認されたり発売されたりすれば、彼らは私たちの候補製品を採用するハードルを増加させるかもしれない。
CAR−T候補製品は臨床試験において良好な臨床効果を有するため,先進的なT細胞療法や他のタイプの腫瘍療法を開発した他の組織と直接競争することが予想される。これにはAdaptimmune治療会社、allgene社、Arcell社、Astellas製薬会社、Autolus社、百時美施貴宝社、Cariou生物科学社、Cellectis S.A.,ヤンソン製薬会社、Juno治療会社(ケルキン社に買収され、現在は百時美施貴宝社)、亘喜生物社(アスリーカンに買収されている)、Kite製薬会社(ジリッド科学社の一社)、伝説的生物、ノワ製薬、武田社が含まれるCAR−T分野の会社が含まれる。
いくつかの小さなバイオテクノロジー会社や大きな製薬会社が免疫療法や遺伝子療法をさらに求めている。私たちはまた、安進、アスリカン、百時美施貴宝会社、F.Hoffman-La Roche AG、グラクソ史克、メルク社、ファイザーなどの会社から提供された非細胞治療の競争に直面している。私たちの多くの競争相手は、単独でも、彼らとのパートナーとも、より多くの資金、技術、および他の資源、例えば、より多くの研究開発者および/または研究開発、製造、臨床試験、および臨床試験を行う上でより強い専門知識を持っている。
最近の承認やM&A活動も多くの会社の設立を刺激しており,これらの会社は現在遺伝子治療技術や適応を求めている。状況は急速に変化しており,これらの会社は数が多すぎて発売できないが,Alnylam製薬会社,Astellas社,Beam治療会社,BioMarin製薬会社,Bluebird Bio,Cellectis,CRISPR治療会社,Editas Medicines社,F.Hoffman−La Roche AG(買収したSpark治療会社),Generation Bio,Inc.,Intellia治療会社,
34
LogicBio治療社,Moderna,ノワ製薬(買収されたAveXis社),Passage Bio社,Sangamo治療社,Sarepta治療社,Ultragenyx社である。
また、規模が小さいかスタートアップ段階にある会社は、より成熟した会社と協力することで私たちと競争する可能性があります。技術の商業適用性の進歩やこれらの企業に投資する資本の増加により、競争はさらに激化する可能性がある。製薬、生物技術と遺伝子治療業界の合併と買収は一般的であり、より多くの資源が私たちの少数の競争相手に集中する可能性がある。著者らの競争相手はまた合格した科学と管理人員を募集と維持し、臨床試験場所と臨床試験患者登録を確立する方面で著者らと競争している。
知的財産権
知的財産権は私たちの分野と全体的なバイオテクノロジー分野で必須的だ。我々は、特許権を求め、維持し、守ることにより、内部開発、第三者から取得された特許権であっても、第三者から取得された特許権であっても、我々の業務発展に重要なビジネス的意義を有するノウハウ、発明、改善を保護し、強化することを求める。孤児薬の指定,開発·審査の加速,データ独占性,市場独占性,特許期限延長などによる規制保護も求めている。
我々の知的財産権は、(1)プラットフォーム技術に対する特許権、(2)我々の製品で使用されるコアコンポーネントに対する特許権、(3)特定の製品に対する特許権、(4)治療適応治療方法に対する特許権、(5)コアコンポーネントおよびプラットフォーム技術使用方法に対する特許権、および(6)革新的製造プロセスをカバーする特許権を含む多層的な保護を提供することを目的としている。私たちはまた私たちの業務発展に必須的かもしれない商業秘密に依存している。
私たちの現在の階層特許権は、私たちの次世代技術特許の開発と申請の努力に加えて、実質的な知的財産権保護を提供してくれると信じています。
我々は、米国および国際的にP−MUC 1 C−ALLO 1、P−PSMA−ALLO 1、P−BCMA−ALLO 1、P−CD 19 CD 20−ALLO 1および我々の他の候補製品、我々の細胞治療候補製品、ならびに我々の遺伝子治療候補製品P−FVIII−101、P−OTC−101、およびP−PAH−101のために特許保護を出願している。しかし、バイオテクノロジーにおける特許や他の知的財産権分野は絶えず発展している分野であり、多くのリスクと不確実性を持っている。
上記のプラットフォーム技術およびコアコンポーネントについて(例えば、T供給チェーン管理組成物および製造方法、トランスジェニックHSC製造方法、誘導式安全スイッチ、PiggyBac DNA伝達システム、Cas-Clive遺伝子編集技術、免疫細胞増幅を増強するブースター分子、鎧策略およびナノ粒子伝達方法)知的財産権は主に会社の所有または会社が獲得した知的財産権からなる。我々は、現在および新しい候補製品および新しいプラットフォームおよびコア技術をサポートするために、より多くの特許出願を提出する予定である。私たちのビジネス成功は、私たちの現在および未来の候補製品の獲得と維持、およびこれらの製品を開発および製造するための方法の特許保護および商業秘密保護にある程度依存し、これらの特許を第三者の挑戦から保護し、他人の独占権を侵害することなく運営されるであろう。私たちが第三者が私たちの製品を製造、使用、販売、提供、または輸入することを阻止する能力は、これらの活動をカバーする効果的かつ強制的に実行可能な特許または商業秘密の下で当社が所有する権利の程度に依存する。私たちは、私たちの任意の未解決特許出願または未来に提出された任意の特許出願に特許が付与されるかどうかを決定することができず、また、私たちの任意の既存特許または将来私たちに付与される可能性のある任意の特許が、私たちの候補製品、発見計画、およびプロセスを保護する上で商業的用途を有することを保証することはできない。このリスクおよび私たちの知的財産権に関連するより包括的なリスクについては、“リスク要因-私たちの知的財産権に関連するリスク”というタイトルの部分を参照してください
個別特許の期限は特許を取得した国の法的期限に依存する。私たちが出願したほとんどの国では,米国を含め,特許期間は非臨時特許出願が提出された最初の日から20年である。アメリカでは特許は特許の有効期限を延長することができる
35
期限調整は,特許権者が米国特許商標局の特許審査及び付与時の行政遅延による補償,又は1つの特許が直前に提出された特許により最終的に放棄された場合又は特許権者側の遅延により短縮されることができる。米国では,FDAが承認した薬物をカバーする特許期限も特許期間を延長する資格があり,FDA規制審査中に失われた特許期限の補償として特許期限の回復を可能にしている。ハッジ-ワックスマン法は特許期間を特許満了後最大5年間延長することを許可している。特許期間の延長の長さは,薬物が規制審査を受ける時間の長さと関係がある。1つの特許の残り期間は、製品が承認された日から14年を超えてはならず、承認された薬物に適した特許を延長することしかできず、承認された薬物、その使用方法、またはその製造方法に関する請求項を延長することしかできない。欧州や他の外国司法管区にも同様の規定があり、承認された薬物をカバーする特許の期限を延長する。将来、私たちの製品がFDAの承認を得たら、私たちはこれらの製品の特許出願のために特許期間を延長する予定です。私たちは、特許を取得できる任意の管轄区域で、私たちが発行した任意の特許のために特許期間の延長を求める予定ですが、米国のFDAを含む適用当局が保証することはできません。このような延長を承認すべきかどうか、承認されれば、そのような延長の長さの評価に同意します。我々の知的財産権に関連するリスクに関するより多くの情報は、“リスク要因-私たちの知的財産権に関連するリスク”というタイトルの部分を参照してください
場合によっては、一時特許出願として、米国特許商標局に直接特許出願を提出する。仮特許出願は、米国でより低コストの初特許出願を提供するためのものである。該当する非臨時特許出願は,仮出願日の後12ヶ月以内に提出されなければならない。対応する非仮出願の利点は、特許出願の優先日(S)が早い仮出願日(S)であり、最終的に発行される特許の特許期間が遅い非仮出願日から計算されることである。この制度は,優先権日を早期に取得することができ,優先権年度内に特許出願(S)に材料を増加させ,特許期間の開始を延期し,起訴費用を延期することができ,出願を審査しないことを決定した場合に有用である可能性がある。我々は,我々の仮特許出願に関連する非仮特許出願をタイムリーに提出する予定であるが,このような特許出願がいかなる競争優位を提供してくれる特許の発行につながるかどうかは予測できない.
我々は、米国非仮出願及び特許協力条約又はPCT出願を提出し、これらの出願は、より早く提出された仮出願の優先日の利益を有することを要求する(適用される場合)。PCT制度は、特許出願の最初の優先日の後12ヶ月以内に単一出願を提出することを許可し、後でPCTに提出された国際特許出願に従って国家特許出願を行うことができる全152個のPCT加盟国を指定する。PCT検索機関は、特許性検索を実行し、申請料を生成する前に外国国出願の成功機会を評価するために使用することができる拘束力のない特許性意見を発表する。PCT出願は特許として発行されていないが、出願人が国家段階出願を介して任意の加盟国で保護を求めることを可能にしている。特許出願の第1の優先権の日から2年半の期限が終了したとき、PCTのどの加盟国も直接国家出願を通過することができ、または場合によっては欧州特許機関のような地域特許組織を介して単独の特許出願を出願することができる。PCTシステムは費用を遅延させ、国/地域特許出願の成功機会の限られた評価を可能にし、出願が出願の最初の2年半以内に放棄された場合に大量の節約を実現した。
すべての特許出願について、私たちは具体的な状況に基づいて特許請求戦略を決定するつもりだ。私たちはいつも弁護士の提案と私たちのビジネスモデルと需要を考慮するつもりだ。私たちが提出した特許は、これらのアプリケーションが戦略的価値を有することを前提として、私たちの独自技術および任意の製品のすべての有用なアプリケーションおよび既存技術および製品のために発見されたすべての新しいアプリケーションおよび/または用途に対する保護権利を含む。我々は、既存の特許庁規則及び条例の場合、我々のプロセス及び組成が最大のカバー範囲及び価値を得ることを確実にするために、特許出願の数及びタイプ、並びに係属中及び発表された特許請求の範囲を絶えず再評価する。また、特許訴訟中には、私たちの知的財産権および業務ニーズを満たすためにクレームが修正される可能性がある。
特許保護を得る能力およびこのような保護の程度は、従来技術の範囲、発明の新規性および非顕著性、および特許法実施要件を満たす能力を含む多くの要因に依存することを認識している。さらに、特許出願において要求されるカバー範囲は、特許発行前に大幅に縮小することができ、その範囲は、再解釈またはさらに解釈することができる
36
特許が発行された後でさえ変化した。したがって、私たちは私たちの未来の任意の候補製品や私たちの技術プラットフォームのために十分な特許保護を獲得または維持することができないかもしれない。私たちが現在求めている特許出願が任意の特定の司法管轄区域で特許として発行されるかどうか、または任意の発行された特許の権利主張が競争相手の影響を受けないように十分な特許保護を提供するかどうかを予測することはできない。私たちが持っているどんな特許も第三者によって挑戦され、回避され、無効に発表される可能性がある。
特許保護に加えて、私たちは商標登録、商業秘密、ノウハウ、他の独自情報、および持続的な技術革新に依存して、私たちの競争的地位を発展させ、維持しています。私たちは、私たちの業務において特許保護から保護されているか、または特許保護に適していないと考えられる側面を保護し、保護し、独自の情報の機密性を維持することを求めている。私たちは、当社の従業員やコンサルタントと契約を締結することを含む、当社の独自情報およびビジネス秘密を保護する措置をとっていますが、第三者は、実質的に同じ独自の情報および技術を独立して開発したり、他の方法で私たちのビジネス秘密を取得したり、当社の技術を開示したりすることができます。したがって、私たちは私たちの商業秘密を意味的に保護することができないかもしれない。私たちの政策は、私たちの従業員、コンサルタント、外部科学協力者、協賛研究者、および他のコンサルタントに、私たちとの雇用や相談関係を開始する際に秘密協定を実行することを要求します。これらの合意は、個人と私たちとの関係中に開発または開示された当社の業務または財務に関するすべての機密情報は、特定の場合を除いて第三者に開示されてはならないことを規定している。私たちと従業員の合意はまた、従業員が私たちに雇われた過程で構想されたすべての発明または従業員が私たちの機密情報を使用することによって生成されたすべての発明が私たちの固有財産であることを規定している。しかしながら、このようなセキュリティプロトコルおよび発明譲渡プロトコルは違反される可能性があり、このような違反に対応するための十分な救済措置がない可能性がある。しかも、私たちのビジネス秘密は競争相手に知られたり独立して発見されるかもしれない。私たちのコンサルタント、請負業者、または協力者が、私たちのために働いているときに他人が所有している知的財産権を使用する場合、関連またはそれによって生じる商業秘密、ノウハウ、および発明の権利について議論が生じる可能性がある。我々の知的財産権に関連するリスクに関するより多くの情報は、“リスク要因-私たちの知的財産権に関連するリスク”というタイトルの部分を参照してください
私たちのようなバイオテクノロジー会社の特許地位は通常不確実であり、複雑な法律、科学、および事実の問題に関連している。私たちのビジネス成功はまた第三者の独占権を侵害しないことにある程度依存するだろう。いかなる第三者特許の発行が、私たちの開発や商業戦略、あるいは私たちの製品やプロセスを変更し、ライセンスを取得したり、いくつかの活動を停止したりすることを要求するかどうかはまだ確定されていません。私たちは、任意のライセンス契約に違反したり、将来の製品を開発または商業化するために必要な独自の権利の許可を得ることができず、私たちに実質的な悪影響を及ぼす可能性があります。もし第三者が米国で準備して提出した特許出願も権利の技術を要求する場合、私たちは発明の優先権を決定するために、米国特許商標局の干渉または派生プログラムに参加しなければならないかもしれない。より多くの情報については、“リスク要因-私たちの知的財産権に関連するリスク”というタイトルの節を参照してください
市場排他性を拡大することができる場合、我々の戦略は、現在または予想される開発プラットフォーム、技術、および/または候補製品のコア要素に関連することができるかもしれない追加の知的財産権を得ることである。
会社が持っている知的財産権
複数のファイルは、2022年6月に国家段階に入る2020年12月に提出された公表されたPCT出願を含むP-MUC 1 C-ALLO 1をカバーする。米国以外のいくつかの国では、主要市場国の大多数を含め、国段階の申請が待っている。これらの申請から発行された事項構成クレームは2040年まで満期になりません。
P-BCMA-ALLO 1は、2020年6月に国家段階に入る2018年12月に提出された公表されたPCT出願を含む複数のファイルでカバーされている。米国以外のいくつかの国では、主要市場国の大多数を含め、国段階の申請が待っている。本出願から発行された事項からなるクレームは2038年までに満期になりません。
P-CD 19 CD 20-ALLO 1は、2つの公表されたPCT出願を含み、それぞれ2022年4月に提出され、2023年10月に国家段階に入る複数の届出ファイルによってカバーされる。いくつかの国段階の申請が待っています
37
アメリカ以外の国は、ほとんどの主要市場国を含む。本出願が発行した事項組成請求書は2042年までに満了しない。
我々のP−PSMA−ALLO 1,P−BCMACD 19−ALLO 1,P−CD 70−ALLO 1,他の細胞治療計画はまだ早い開発段階にあり,我々の計画は特定の知的財産権カバー範囲が開発されている。
各候補製品のコアコンポーネントは、ScFv結合タンパク質(P-MUC 1 C-ALLO 1)または重鎖抗体断片結合タンパク質(P-BCMA-ALLO 1)、免疫細胞増幅を増強する増強分子(現在のすべての同種異体製品)、早期記憶T細胞(T細胞を含む)に対して、会社が所有するプラットフォームアプリケーションによって保護されている供給チェーン管理)およびその製造方法(P-MUC 1 C-ALLO 1,P-BCMA-ALLO 1)、癌治療のための方法(全製品)、ブタBac転座システム(全製品)、誘導可能な安全スイッチ(全製品)、製品製造のための修飾細胞の同時選択および増幅を促進するためのマーカー遺伝子、および三価トランスポゾン構造のための自己切断ペプチド(全製品)。P−CD 19 CD 20−ALLO 1では,重鎖のみを含む抗体断片結合剤が使用されており,会社所有ではなく,我々の許可下で他の人を排除する独占的な権利によって保護されている。注目すべきは2019年12月に25%以上のT細胞を含む修飾T細胞製品をカバーする米国特許を取得しました供給チェーン管理細胞、特許期間は2037年に満期になる。これらのトランスジェニックT細胞を製造するための製造方法および細胞培養液に関する米国特許を発行しました供給チェーン管理特許期限が2037年に満期になる細胞。私たちはまた、私たちのCas-Clover特定部位遺伝子編集システムを保護するために米国で発行されたComposal of Matter特許を持っており、このシステムの特許期間は2037年に満了する。私たちはまた米国でComposal Of Matter特許を発表し、私たちのiggyBac DNA送達システムを保護し、このシステムの特許期限は2030年に満了する。
我々の遺伝子治療計画は,P−FVIII−101,P−OTC−101,P−PAH−101を含む。複数の文書は、2022年9月に国家段階に入る2021年3月に提出された公表されたPCT出願を含むP−OTC−101を含む。米国以外のいくつかの国では、主要市場国の大多数を含め、国段階の申請が待っている。これらの申請から発行された事項構成クレームは2041年まで満期にならない。また、2022年2月に提出された公表されたPCT出願と、2022年3月に提出された公表されたPCT出願とを含む複数の交付技術出願があり、それぞれ2023年8月と2023年9月に国家段階に入る。これらの申請から発行された事項構成クレームは2042年までに満了しない。最後に、私たちは2023年1月に提出された未解決の仮出願を2つ持っており、この2つの申請は2024年1月に非臨時出願に転換される。これらの申請から発行された事項構成クレームは2044年まで満期にならない。
得られた知的財産権
Transposagen BiopPharmticals,Inc.またはTransposagen,Inc.から剥離した製品として,我々は最初にGiggyBac転座システムや方法に関する知的財産権を獲得した.この買収には、次世代遺伝子編集システムや使用方法に関する知的財産権も含まれている。
Vindo NanoBioTechnology,LLC(前身はVindo NanoBioTechnology,Inc.)を買収した2016年10月。この取引の一部として、ポリマーナノ粒子組成物および例えば遺伝子治療技術を提供するための方法に関連する知的財産権を得た。
協力協定
羅氏協力協定
2022年7月、私たちは羅氏と協力協定を締結し、協定に基づいて、羅氏に授与した:(I)私たちの特定の知的財産権に基づいて、世界的に独占的に許可し、私たちの既存のP-BCMA-ALLO 1およびP-CD 19 CD 20-ALLO 1計画から同種CAR-T細胞治療製品を開発、製造、商業化する、または各計画、Tier 1計画;(Ii)既存のP-BCMACD 19-ALLO 1およびP-CD 70-ALLO 1計画または各Tier 2計画における同種CAR-T細胞治療製品を開発、製造および商業化するために、我々の特定の知的財産権に基づいて独占的なグローバルライセンスを取得し、(Iii)我々の特定の知的財産権の独占的許可に基づいて、ロー氏によって指定された最大6つの協力計画の中から同種CAR-T細胞治療製品を開発、製造、商業化する;(Iv)いくつかの限られた知的財産権の下で非独占商業許可を選択する
38
あるロー氏特許細胞療法製品を開発、製造と商業化する権利があり、羅氏が確定した最大3つの固形腫瘍標的に使用され、可能かもしれない;及び(V)著者らの既存の2つの血液悪性腫瘍早期計画の第一の約束権。
Tier 1計画ごとに,第1段階用量逓増臨床試験により開発活動を実行し,羅氏はこのような活動を実行する際に生じるいくつかの費用の特定の割合を精算する義務があり,各Tier 1計画には最大1つの指定された精算上限がある。第一段階の線量を超えて増加する第一級計画活動について、羅氏はこの計画によって発生したすべての費用を精算する義務がある。Tier 2計画ごとに,IND支援研究の開発候補を選択するか,あるいはロ氏がオプション維持費を選択して支払う場合には,第1段階用量逓増臨床試験を完了することで研究·開発活動を行う。また,羅氏が独占的に選択権を行使するTier 2計画ごとに,羅氏は我々に選択権使用料を支払う義務がある.Tier 1プランとTier 2プランごとに,羅氏への技術移転が完了するまで製造活動を実行する.
双方は2年間の初歩的な研究計画を行い、臨床前に特定の数量の同種異体CAR-T細胞療法に関連する合意された次世代治療概念を探索とテストする。羅氏が選択して費用を支払う場合、双方はその後、18ヶ月間の第2の研究計画を行い、この計画によれば、双方は初期期間で行われている既存の仕事を延長することができ、および/または同種異体CAR-T療法に関連する特定の数の追加的に合意された次世代治療概念を探索および臨床前に試験することができる。ロー氏はこの2つの研究計画の中から最大6つのヘム悪性腫瘍に対する同種異体CAR−T計画を指定する可能性があり,各計画についてIND活動を促進する開発候補,あるいは各協力計画を選択して研究·開発活動を行う。各連携計画を指定する際には,羅氏は指定費用を支払う義務がある.私たちが協力計画の主導的な最適化活動を完了した後、羅氏はその計画を羅氏に移行し、私たちに支払いまたは終了することを選択することができる。代替的に、限られた数の協力計画の場合、羅氏は、第1段階用量漸増臨床試験を完了することによって、いくつかの追加の開発および製造活動を行うことを選択することができ、この場合、羅氏は、いくつかのマイルストーンを支払い、そのような開発および製造活動に関連する特定のパーセントのコストを精算する。各連携計画については,羅氏への技術移転が完了するまで製造活動を実行する.
羅氏の協力協定によると、羅氏は私たちに1.1億ドルの前金を支払った。ロ氏の第2レベル計画オプションの行使、協力計画の指定とライセンス製品の商業許可の選択権の行使、第1レベル計画、オプション第2レベル計画と協力計画に基づいて特定の開発、監督、純売上マイルストーン事件を実現する製品などによると、(I)第1レベル計画15億ドル、(Ii)第2レベル計画11億ドル、(Iii)協力計画29億ドルを含むいくつかの精算、費用、マイルストーン支払いを獲得する資格がある。(Iv)特許製品の4.15億ドルです
我々はまた,製品ごとに,Tier 1計画,オプションTier 2計画と連携計画による製品純売上高の中位から下位2桁,および許可製品の中央桁から中央値の階層印税支払いを許可する権利があり,いずれの場合も,いくつかの通常の減免と補償の制限を受けている.使用料は、その製品をカバーするライセンス特許がその国で満期になるまで、またはその製品が同国で初めて商業販売された日から10年まで製品および国/地域によって支払われる。
ロ氏協力協定は、2022年9月に改正された1976年の“ハート-スコット-ロディノ反トラスト改正法”に基づく適用待機期間の満了または終了後に発効し、残りの特許権使用料や他の支払い義務がなくなるまで製品や国/地域で行われ続ける。ロ氏協力協定には、重大な違約や破産に対する条項、羅氏の便宜のための標準的な終了条項が含まれている。これらのうちのいくつかの終了権利は、特定の製品またはライセンスおよびロ氏協力協定全体に対して行使することができる。
2023年11月7日から、羅氏協力協定は、(I)羅氏が私たちに支払ったいくつかの既存の製造関連費用を再分配して、私たちの既存の各第1段階計画のために新しい製造プロセスの開発および移転費用を追加するために再分配され、(Ii)羅氏が各第1級計画のために私たちに支払ったいくつかの開発マイルストーンの支払いの金額を再分配するように改正された。修正案には
39
既存の2年間の研究プログラムを提供し、さらに18ヶ月の能力を延長し、1500万ドルのマイルストーン費用を支払う。その他の点では、ロ氏協力協定下のすべての履行義務と支払い条件は変わらない。
武田協力協定
2021年10月には、武田薬品米国社または武田社と協力·許可協定、すなわち武田協力協定を締結し、この協定に基づき、4500万ドルの前払金と協定期間中の研究開発精算金を取得した。
2023年5月31日、武田から書面通知を受け、武田は武田協力協定を中止することを選択し、2023年7月30日から発効、または終了日とする。終了日まで、双方はそれぞれ武田協力協議での責任を果たしていたが、終了日には、吾らの武田協力協議での独占責任は終了した。また、武田の免許を付与し、武田協力協定に基づいて研究活動を行う許可証を付与した。2023年12月31日までの1年間に、これまでに繰延された890万ドルの収入を確認し、これらの業績義務を履行する必要がないことを確認した。終了後,P-FVIII-101およびP-PAH-101を含むすべての以前に許可されたプログラムを開発するすべての権利が付与される.遺伝子治療の分野で新たな戦略的協力を求めることができるかもしれないが、これまで武田協力協定に含まれていた計画の一部または全部と、現在このような戦略的協力の約束または合意に達していないにもかかわらず、可能な追加の内部計画が含まれている可能性がある。
許可内合意
2017年4月にTeneoBio,Inc.(安進の子会社)と締結されたビジネスライセンス契約
2017年4月27日、私たちは、TeneoBio、Inc.またはTeneoBioと商業許可協定、または2017年TeneoBio協定を締結し、この協定に基づいて、TeneoBioが提供するヒト疾患を治療するための特定の重鎖配列のみを治療するためのCAR分子(非自然産生重鎖抗体断片のみを含むCAR)を使用して開発することができる、TeneoBio、Inc.またはTeneoBioとビジネスライセンス契約を締結した。我々は我々のP−BCMA−ALLO 1候補製品にこれらのライセンスを使用した。
2017年のTeneoBio協定によると、私たちは2017年のTeneoBioプロトコルで許可された抗体を選択することで、TeneoBioに50万ドルの前払い費用を支払った。私たちは、任意の同種製品のいくつかの臨床および規制マイルストーンが初めて達成されたときにTeneoBioに合計2050万ドルを支払い、任意の自己製品のある臨床および規制マイルストーンが初めて達成されたときにTeneoBioに合計2050万ドルを支払うことを要求された。個々の製品と国ごとに、すべての特許製品の純売上高の低い桁の割合で特許使用料を支払う義務があります。
2017年のTeneoBio協定は、すべての国/地域で特許を許可する有効な権利主張が最後に満了した時点で終了します。私たちはまた60日前にTeneoBioに書面で通知して、2017年のTeneoBio合意をいつでも終了することができます。一方が違約書面通知を受けてから90日以内に救済されなかった場合、または他方が破産した場合、どちらも2017年のTeneoBio合意を終了することができます。
2018年8月にTeneoBio,Inc.(安進の子会社)とビジネスライセンス契約を締結しました
2018年8月3日,我々はTeneoBioとCAR−T細胞療法のためのTeneoBioのヒト重鎖抗体の開発と使用のための商業許可合意,すなわち2018年TeneoBio合意を達成した。2018年のTeneoBio協定の条項によると、TeneoBioから特定の数の標的を研究、開発、商業化する独占的な権利を得ることを選択することができる。
2018年のTeneoBio協定によると、私たちはTeneoBioに400万ドルの前金を支払った。私たちは、(1)特定の目標の独占経営権を選択し、TeneoBioが一定期間、特定の目標を第三者に権限を付与することを制限する特定の目標の独占経営権を選択し、(3)各目標に対してビジネス選択権を行使するため、低いから6桁の範囲の追加費用を支払うことを要求された
40
目標です。私たちは各製品のいくつかの臨床と規制マイルストーンが初めて実現された時、TeneoBioに合計3100万ドルを支払うことを要求された。どの特許製品の純売上高に対しても製品や国/地域に低い一桁パーセントの印税を支払う義務があります。場合によっては、特許使用料が低下する可能性がある。
2018年のTeneoBio協定は、すべての国/地域の特許を許可する有効な権利主張が最後に満了した時点で終了します。私たちはまた、1つ以上の目標に関連した2018年のTeneoBio合意をいつでも終了するために、60日前に書面で通知することができます。一方が違約書面通知を受けてから90日以内に救済されなかった場合、または他方が破産した場合、どちらも2018年TeneoBio合意を終了することができます。
2019年10月にXyone Treateutics,Inc.(Genus Oncology,LLCの利益継承者)と許可合意に達した
2019年10月24日、Xyone TreeuticsまたはXyoneとライセンス契約またはXyoneプロトコルを締結しました。Xyoneプロトコルによれば、私たちはXyoneに150万ドルの前払い費用を支払い、Xyoneは、Xyone制御のいくつかの特許下の独占的、再許可可能なグローバル許可、およびXyone制御のいくつかの独自で再許可可能なグローバルライセンスを取得するために、2020年4月に追加150万ドルで行使される選択権を付与し、MUC 1に対する抗体を含む自動車細胞およびその誘導体を含む医薬製品、またはXyone制御のいくつかの特許および独自技術における非独占的、再許可可能なグローバルライセンスを研究、開発および商業化治療に伴う診断のために研究、開発および商業化する。人間の病気と状況を予防し緩和する。Xyoneプロトコルによって付与された許可は、上流許可者によって保持されているいくつかの権利および米国政府の権利によって制限される。上流ライセンス側が保持する権利は,上流ライセンス側がライセンス分野で教育,教育,その他の非商業的研究活動を展開する能力,その他の学術,政府または非営利組織がライセンス分野で非商業的研究活動を展開する能力に限られており,我々の計画や候補製品を追求する能力を制限していない。我々のP−MUC 1 C−ALLO 1製品候補では,結合剤としてMUC 1に対するXyone抗体またはその誘導体を用いた。
Xyoneプロトコルによると、Xyoneが許可した任意の製品とセット診断が初めてある臨床、監督、販売マイルストーンを実現した場合、Xyoneに合計7100万ドルを支払わなければならない。Xyoneライセンス製品と関連診断製品の純売上高の中央桁パーセントに低い等級版税を製品や国/地域に応じて支払う義務がありますが、いくつかの慣例の減免を遵守しなければなりません。
Xyoneプロトコルは、製品および国/地域に基づいて決定され、(1)Xyoneライセンス製品をカバーするライセンス特許内の最後の満了の有効な権利要件、(2)Xyoneライセンス製品の同国での規制排他性満了、および(3)Xyoneライセンス製品が同国で初めて商業販売されてから10年である最終期限の使用料期限に満了する。私たちはまた30日前にXyoneに書面で通知した場合にいつでもXyone合意を終了することができます。いずれも違約書面通知を受けてから90日以内に是正されなかった重大な違約行為は、Xyoneライセンス契約を終了することができます。Xyoneもまた、私たちが破産した後、またはXyoneライセンス製品のIND承認後20ヶ月以内にXyoneライセンス製品の第1段階臨床試験を開始できなかった場合、直ちにXyoneプロトコルを終了する権利がある。
HMGUとのライセンス契約を修正し再署名しました
2021年3月12日、我々はHelmholtz-Zentum München-Deutsches Forschungszentrum für Gesundheit and UmWelt GmbH(HMGUと略す)と改訂され再説明された特許許可協定またはHMGU許可協定を締結し、この協定に基づいて、ヒト医薬製品を含むいくつかの研究、開発、製造および商業化されたHMGUが所有する特定の特許出願および特許が主張する製品およびサービスのグローバル独占権利を取得した。我々は,これらのライセンスをCas−Clover遺伝子編集技術に使用し,P−BCMA−ALLO 1,P−MUC 1 C−ALLO 1,P−CD 19 CD 20−ALLO 1,および我々が計画した他の同種計画を含む。
41
HMGU許可プロトコルにより,支払い当日の10,000ユーロに相当する11,506ドルの前払い料金をHMGUに支払った.我々はHMGUの年間保守費の支払いを求められ,同年満期の印税に記入された。私たちはまた、最初に許可された製品(Clo051は治療薬の一部である)のいくつかの臨床および規制マイルストーンが初めて実現されたときに、HMGUに合計170万ユーロを支払い、最初に許可された製品(Clo051は治療薬の一部ではない)のいくつかの臨床および規制マイルストーンの第1の段階で、90万ユーロまでの総額をHMGUに支払うことを要求された。ライセンス製品またはライセンスサービスに従って、年間純売上高の低い桁数パーセントの範囲の印税をライセンスサービスおよび国/地域に支払う義務があり、印税料率は、治療用途のために許可製品またはライセンスサービスが使用されるか、Clo051が治療剤の一部であるか、または治療剤を製造するためのものであるかどうかに依存する。私たちは現在私たちの遺伝子工学技術の一部としてClo051を使用して、私たちの候補製品を生成しています。
HMGUライセンス契約は、最終印税期限が満了した時点で終了し、印税期限は、ライセンス製品および国·地域ごとのライセンス製品に基づいて決定される。我々もカレンダー年度終了前3カ月以内に書面通知を出し,HMGUライセンス契約を終了する権利がある.いずれも違反書面通知を受けてから6週間以内に是正されなかった重大な違約行為は、“HMGU許可協定”を終了することができる。もし私たちが破産したら、HMGU許可協定は自動的に終了するだろう。
政府の監督管理
FDAと連邦、州と地方の各レベル及び国外の他の監督管理機関は著者らが開発している生物製品の研究、開発、テスト、製造、品質管理、輸出入、安全、有効性、ラベル、包装、貯蔵、流通、記録保存、承認、広告、販売促進、マーケティング、承認後の監視と承認後の報告などの方面に対して広範な監督管理を行った。私たちは、第三者請負業者と共に、私たちが研究したい、または承認を求めることができるかもしれない国/地域規制機関の様々な臨床前、臨床、および商業承認要件を満たすことが要求されるだろう。
FDAがバイオ製品候補製品が米国で発売される前に必要とされるプログラムは、一般に以下のような態様を含む
42
臨床前と臨床発展
候補製品の第1回臨床試験を開始する前に,INDをFDAに提出しなければならない。INDはFDAが人類の研究を許可した新薬製品の要求である。IND提出の中心焦点は臨床研究の全体的な研究計画と方案(S)である。INDはまた、製品の毒性学、薬物動態学、薬理学および薬効学的特徴を評価する動物およびインビトロ研究結果、化学、製造および制御情報、および研究製品の使用を支援するための任意の利用可能なヒトデータまたは文献を含む。INDはヒト臨床試験が始まる前に発効しなければならない。INDはFDAが30日以内に提案された臨床試験に対して安全懸念または問題を提起しない限り、FDA受信後30日以内に自動的に発効する。この場合,INDは臨床的に放置される可能性があり,INDスポンサーやFDAは臨床試験が開始される前に未解決の問題や問題を解決しなければならない。したがって,INDの提出はFDA認可による臨床試験の開始につながる可能性があり,そうでない可能性もある。米国で臨床試験が開始される前にFDAにINDを提出するほか、組換えあるいは核酸分子の合成に関連するいくつかのヒト臨床試験はFDAの監督と他の臨床試験法規の制約を受け、NIHガイドラインに規定されている地方レベルの監督を受けている。具体的には,NIHのガイドラインによると,ヒト遺伝子転移試験の監督にはIBCによる評価と評価があり,IBCは地方機関委員会であり,組換えや合成核酸分子を用いた研究の審査·監督を担当している。IBCは、研究の安全性を評価し、公衆の健康または環境に対する任意の潜在的リスクを決定し、このような審査は、臨床試験開始前のいくつかの遅延をもたらす可能性がある。NIHガイドラインは強制的ではないが,関連研究がNIH組換えや合成核酸分子研究助成を受けた機関で行われているか,あるいはその助成によって行われていない限り,多くの会社や他のNIHガイドラインに拘束されていない機関は自発的にこれらのガイドラインに従っている。
臨床試験は、GCPに従って合格した研究者の監督の下でヒト被験者に研究製品を服用することを含み、すべての研究被験者に任意の臨床研究への参加についてインフォームドコンセントを提供することを含む。臨床試験は,研究目標を詳細に説明し,安全性をモニタリングするためのパラメータと評価する有効性基準のシナリオで行った。製品開発中に行われる各後続の臨床試験および後続の任意のレジメン修正は、既存のINDに個別に提出されなければならない。また,臨床試験を推奨する各地点の独立IRBは,その地点で臨床試験を開始する前に任意の臨床試験の計画とそのインフォームドコンセントを審査·承認しなければならず,完成まで研究を監視しなければならない。規制当局、IRB、またはスポンサーは、対象が受け入れられない健康リスクに直面していることを発見すること、または試験がその目標を達成する可能性が低いことを含む、様々な理由で臨床試験を随時一時停止することができる。いくつかの研究はまた、臨床研究スポンサーによって組織された独立した合格専門家グループの監視を含み、このグループは、研究のいくつかのデータへのアクセスに基づいて、研究が指定されたチェックポイントで行うことができるかどうかを許可するデータ安全監視委員会と呼ばれ、被験者に受け入れられない安全リスクまたは他の理由があると判定された場合、治療効果を示さない場合、臨床試験を停止する可能性がある。現在行われている臨床研究や臨床研究結果を公的登録機関に報告することに関する要求もある。
BLAの承認を得るために,ヒト臨床試験は通常3つの重複可能な連続段階に分けて行われる。
43
場合によっては、FDAは、製品が承認された後に追加の臨床試験を自発的に実施して、製品に関するより多くの情報を得ることを要求する可能性がある。このようないわゆる第4段階研究は“法案”を承認する条件かもしれない。臨床試験と同時に、会社は追加の動物研究を完成させ、候補製品の生物学的特徴に関する追加情報を開発することができ、cGMP要求に基づいて商業量産製品の過程を最終的に決定しなければならない。製造過程は高品質の候補製品ロットを持続的に生産できる必要があり、特に最終製品の特性、強度、品質と純度をテストする方法を開発しなければならない、あるいは生物製品に対しては、安全、純度、効力の試験方法を開発しなければならない。また,適切な包装を選択·テストし,候補製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
BLA提出と審査
すべての適用された法規要件に基づいて必要なすべてのテストが成功したと仮定すると,製品開発,非臨床研究,臨床試験の結果はBLAの一部としてFDAに提出され,その製品を1つまたは複数の適応の市場に使用することの承認が求められる。BLAは,否定や曖昧な結果や積極的な発見,製品の化学,製造,制御,アドバイスのラベルなどに関する詳細な情報を含む関連する臨床前および臨床研究から得られたすべての関連データを含まなければならない。BLAの提出には、免除または免除が適用されない限り、FDAに相当な申請使用料を支払う必要があり、承認されたBLAのスポンサーは、年間計画費を支払う必要がある。
BLAが提出されると,FDAは提出申請を受けてから10カ月以内に審査基準申請を行うか,または申請が優先審査資格を満たしている場合には,FDAが提出申請を受けてから6カ月以内に審査基準申請を行うことを目標としている。標準審査および優先審査では、FDAがより多くの情報を提供することを要求するか、または明確にすることを要求する要件は、しばしば審査プロセスを大幅に延長する。FDAは、製品が安全で、純粋かつ有効であるかどうか、およびその製造、加工、包装、または保持されている施設が、製品の持続的な安全、純度および効力を保証するための基準に適合しているかどうかを決定するためにBLAを審査する。FDAは諮問委員会を招集し,審査申請について臨床的知見を提供する可能性がある。BLAを承認する前に、FDAは通常、製品を生産する1つまたは複数の施設を検査する。FDAは、製造プロセスおよび施設がcGMP要件に適合していることを決定し、要求された仕様の下で製品が一貫して生産されることを保証するのに十分でない限り、申請を承認しないであろう。さらに、BLAを承認する前に、FDAは、GCPに適合することを確実にするために、通常、1つまたは複数の治療場所を検査する。FDAが申請、製造プロセス、または製造施設が受け入れられないと判断した場合、それは、提出された文書に不足点を列挙し、追加の試験または情報の提供を要求することが多い。任意の要求された補足情報が提出されたにもかかわらず、FDAは最終的に、その申請が承認された規制基準を満たしていないと決定する可能性がある。
FDAがBLAを評価し、研究製品および/またはその薬物を生産する製造施設を検査した後、FDAは承認状または完全な返信を発行することができる。承認書は、製品の商業マーケティングを許可し、特定の適応に関する具体的な処方情報を提供する。完全な返信は、FDAがBLAで発見されたすべての欠陥を記述するが、FDAが申請をサポートするデータが承認をサポートするのに不十分であると判断した場合、FDAは、必要な検査、試験提出された製品バッチ、および/または提案されたラベルを最初に検討することなく、完全な返信を発行する可能性がある。完全な返信を発行する際に、FDAは、BLAがより多くの情報の提供または明確化を要求することを含む、BLAが承認された条件であるように、申請者がとる可能性のある行動を提案する可能性がある。適用される規制基準を満たしていない場合、FDAは、製品の安全性または有効性を監視するために、追加の試験または情報を要求し、および/または上場後の試験および監視を要求するBLAの承認を延期または拒否することができる。
1つの製品が規制部門の承認を受けた場合、このような承認は特定の適応が付与され、製品が発売される可能性のある指定用途の制限をもたらす可能性がある。例えば、FDAは、製品の利点を保証するために、リスク評価および緩和策を有するBLA、またはREMSを承認する可能性がある
44
リスクを超えていますREMSは、製品に関連する既知または潜在的に深刻なリスクを管理し、そのような薬剤の安全な使用を管理することによって、患者がこれらの薬剤を継続的に得ることを可能にするための安全戦略であり、制限された分配方法、患者登録、および他のリスク最小化ツールのような薬物ガイドライン、医師のコミュニケーション計画、または安全使用を保証する要素を含むことができる。FDAはまた,提案されたラベルを変更したり,適切な制御や仕様を作成したりすることを条件に承認することも可能である.承認されると、発売前と上場後の要求に対する遵守が保たれていない場合、あるいは製品が市場に進出した後に問題が発生した場合、FDAは製品承認を撤回する可能性がある。FDAは、製品の商業化後の安全性と有効性をさらに評価し、監視するために、1つまたは複数の第4段階上場後の研究および監視を要求することができ、これらの発売後の研究の結果に基づいて製品のさらなる販売を制限する可能性がある。
開発と審査計画を加速する
FDAは合格した候補製品に一連の迅速な開発と審査計画を提供した。高速チャネル計画は、特定の基準に適合した新製品の審査プロセスを加速または促進することを目的としている。具体的には、新製品が深刻または生命に危険な疾患や状況を治療し、その疾患または状況が満たされていない医療需要を満たす潜在力を示す場合、迅速なチャネル指定を受ける資格がある。高速チャネル指定は,製品と研究中の特定の適応の組合せに適している.高速チャネル製品のスポンサーは,製品開発期間中に審査チームと頻繁にやり取りする機会があり,BLAを提出すると,優先審査を受ける資格がある可能性がある。高速チャネル製品もスクロール審査を行う資格がある可能性があり、この場合、FDAは、完全な出願を提出する前にBLAの審査部分をスクロールして考慮することができ、スポンサーがBLA部分を提出するスケジュールを提供した場合、FDAはBLAの部分を受け入れることに同意し、スケジュールが許容可能であると判断し、スポンサーはBLAの第1の部分を提出する際に任意の必要な使用料を支払うことができる。
深刻または生命に危害を及ぼす疾患または状態を治療することを目的とした製品も、その開発と審査を加速するために、突破的な治療指定を受ける資格がある可能性がある。初歩的な臨床証拠が、1つの製品が1つまたは複数の臨床的に重要な終点で既存の治療法よりも実質的な改善を示す可能性があることを示す場合、例えば、臨床開発早期に観察された実質的な治療効果が示された場合、製品は画期的な治療指定を得ることができる。この指定には、高速チャネル計画のすべての機能と、第1段階で開始されたより密集したFDA相互作用および指導と、高度な管理者の参加を含む製品開発および審査を加速する組織約束とが含まれる。
迅速なチャネル指定および/または画期的な治療指定を有する製品を含むFDA承認された生物学的製品に提出された任意のマーケティング申請は、優先審査および加速承認のようなFDAの審査および承認プロセスを加速することを意図した他のタイプの計画の資格に適合する可能性がある。市場で販売されている製品と比較して、製品が重篤な疾患や状況の治療、診断または予防において著しく改善される可能性がある場合、優先審査を受ける資格がある。新分子実体を含む製品について、優先審査指定は、FDAが60日申請後6ヶ月以内に上場申請に行動することを目標としていることを意味する(標準審査は10カ月)。
さらに、深刻または生命を脅かす疾患または状態の治療における安全性および有効性について研究された製品は、臨床的利益を合理的に予測する可能性のある代替終点に有効であるか、または不可逆的な発病率または死亡率よりも早く測定することができる臨床終点に有効であると判断され、病状の重症度、希少性または流行率、および代替治療を利用可能または不足し、不可逆的な発症率または死亡率または他の臨床的利益への影響を合理的に予測することを考慮すると、加速承認を得ることができる。承認を加速する条件として、FDAは通常スポンサーに十分かつ良好な制御を行う上場後の臨床研究を要求し、不可逆的な発病率或いは死亡率或いは他の臨床利益に対する期待影響を検証と記述する。また、FDAは現在、承認を加速させる条件として宣伝材料を事前承認することを求めており、製品商業発売の時期に悪影響を及ぼす可能性がある。
(1)細胞療法、治療用組織工学製品、ヒト細胞および組織製品、またはそのような治療法または製品を使用する任意の組み合わせ製品として定義されるRMATの資格に適合する、再生医学高度療法、またはRMATと呼ばれる、以下の基準に適合する任意の薬剤の効率的な開発計画および検討を加速することを目的とする、(2)その目的は、治療である
45
深刻な或いは生命に危害を及ぼす疾患或いは状況を修正、逆転或いは治癒する;及び(3)初歩的な臨床証拠により、この薬物はこのような疾病或いは状況が満たされていない医療需要を解決する可能性があることを表明した。RMAT指定は,画期的な治療指定と同様に,FDAとのより頻繁な会議,候補製品の開発計画の検討,審査や優先審査の資格のスクロールなど,潜在的な利点を提供する。承認が得られると、適切なとき、FDAは、臨床証拠、臨床研究、患者登録、または電子健康記録のような他の真の証拠源を提出することによって、より大きな検証データセットを収集することによって、または承認前にすべての治療を受けた患者を承認後に監視することによって、加速承認の下で承認後の要求を満たすことを可能にすることができる。
迅速チャネル指定、画期的な治療指定、優先審査、加速承認、およびRMAT指定は、承認の基準を変更することはありませんが、開発または承認プロセスを加速する可能性があります。
孤児薬名
孤児医薬品法によれば、FDAは、米国では20万人未満に影響を与えるか、または米国では20万人を超える影響を与える稀な疾患または疾患を治療するための薬剤または生物に孤児の称号を付与することができるが、米国では、そのような疾患または疾患を治療する薬剤または生物薬剤を米国で開発および提供するコストが、米国での薬剤または生物学的薬剤の販売から回収されることを合理的に予想することができない。BLAを提出する前に,指定孤児薬を申請しなければならない。FDAが孤児薬物指定を承認した後、FDAは、治療薬の模倣薬識別情報およびその潜在的孤児の使用を開示する。孤児薬物の指定は、規制審査または承認過程においていかなる利点も伝達されず、規制審査または承認過程の持続時間を短縮することもない。
孤児薬物指定を有する製品がその後、このような指定された疾患に対するFDAの最初の承認を得た場合、この製品は、孤児薬物の独占的承認(または排他性)を得る権利があり、これは、FDAが完全なBLAを含む他の出願を承認しない可能性があり、限られた場合、例えば孤児薬物に対する排他性を有する製品に対する臨床的利点を示さない限り、7年以内に同じ生物適応を販売する可能性があることを意味する。孤児薬物排他性は、FDAが同じ疾患または状態に対する異なる薬物または生物学的製剤、または異なる疾患または状態に対する同じ薬物または生物学的製剤を承認することを妨げるものではない。孤児薬物を指定する他の利点は、いくつかの研究の税金相殺およびBLA申請料の免除を含む。
指定された孤児薬物が孤児が指定された適応よりも広い用途で承認された場合,孤児薬物の排他性を得ることはできない。さらに、FDAが指定された要求に重大な欠陥があると後に判断した場合、または製造業者がこのような稀な疾患または疾患患者の需要を満たすのに十分な数の製品を保証できない場合、米国での独占営業権を失う可能性がある。
承認後に要求する
我々は、FDAによって製造または流通を許可されたどの製品も、記録保存、有害事象報告、定期報告、製品サンプリングおよび流通、ならびに製品広告および販売促進に関連する要件を含むFDAによって普遍的かつ持続的に規制されている。承認後、承認された製品の大多数の変更は、新たな適応または他のラベル宣言を追加するなど、FDAの事前審査および承認を経なければならない。継続的なユーザ料金要求もあり,この要求に基づいて,FDAは承認されたBLAで決定された製品ごとの年間計画費用を評価する.生物製造業者およびその下請け業者は、FDAおよびある州機関に彼らの機関を登録し、FDAおよび特定の州機関の定期的な抜き打ち検査を受けて、cGMPのコンプライアンスを理解しなければならない。これは、私たちと私たちの第三者製造業者にいくつかの手続きと文書要求を加えている。製造プロセスの変更は厳しく規制されており,変更の重要性により,FDAが事前に承認して実施する必要がある可能性がある。FDAの規定はまた、cGMPから外れた場合の調査と是正を要求し、任意の第三者メーカーとの報告を要求しています
46
使用することになるかもしれません。そのため、メーカーは生産と品質管理の分野で時間、お金、精力をかけ続け、cGMPやその他の法規遵守性を維持しなければならない。
規制要件や基準が守られていない場合、あるいは製品発売後に問題が発生した場合、FDAは承認を撤回する可能性がある。その後、製品には、予期されない深刻度または頻度の不良事象、または生産プロセス、または規制要件を遵守できなかったことを含む、以前に未知の問題が存在することが発見され、新しいセキュリティ情報を追加するために承認されたラベルの改訂を招く可能性がある;新しい安全リスクを評価するために発売後研究または臨床研究を実施すること、またはREMS計画に従って流通制限または他の制限を実施することが可能である。他の他の潜在的な結果には
FDAは生物製品のマーケティング、ラベル、広告、販売促進を厳格に規制している。1社はFDAが承認したラベルの規定に基づいて、安全性と有効性、純度、効力に関する声明しか提出できない。FDAと他の機関は非ラベル用途の普及を禁止する法律法規を積極的に施行している。これらの要求を守らないことは、否定的な宣伝、警告状、改正広告、および潜在的な民事と刑事罰を招く可能性がある。医師は、製品ラベルに記載されていない使用のための合法的に入手された製品の処方、および我々が試験およびFDAによって承認された用途とは異なる使用を行うかもしれない。このようなラベル外の使用は医学専門科でよく見られる。医師は,異なる場合,このような非ラベル使用が多くの患者の最適な治療法であると考えるかもしれない。FDAは医者が治療を選択する時の行動を規範化しない。しかし、FDAは製品ラベルの外使用問題に対する製造業者のコミュニケーションを制限した。
生体模倣薬と参考製品の排他性
2009年の“生物製品価格競争と革新法”(BPCIA)は、FDAによって承認された参考生物製品と類似または交換可能な生物製品のための短い承認経路を作成した。
生物類似性とは生物製品と参照製品が安全性、純度と効力の面で臨床的に意義のある差異がないことを指し、これは分析研究、動物研究と臨床研究によって証明できる。互換性は、製品が基準製品生物と類似していることを必要とし、この製品は、任意の所与の患者において、参照製品と同じ臨床結果を生成することが期待できることを証明しなければならず、複数回投与された製品の場合、以前の投与後、生物および参照生物は、安全リスクを増加させることなく、または参照生物の独占的使用と比較して治療効果のリスクを低下させることなく、交互にまたは交換することができる。生物製品がもっと大きく、よくもっと複雑な構造に関連する複雑性、及びこのような製品を製造する技術は、FDAがまだ制定している簡略化審査経路の実施に対して重大な障害を構成した。
47
BPCIAによると,生物類似製品の申請は,参考製品が初めてFDA許可を得た4年後にFDAに提出されなければならない。また,FDAによる生物類似製品の承認は,参考製品が初めて許可された日から12年後に発効する可能性がある。この12年間の独占期間内に、FDAが競合製品の完全なBLAを承認した場合、出願人自身の臨床前データと、その製品の安全性、純度および有効性を証明するために、十分かつ良好に制御された臨床試験からのデータとを含み、別の会社は、参照製品の競合バージョンを販売する可能性がある。BPCIAはまた、交換可能な製品として承認された生物模倣薬のためのいくつかの排他的期限を設けている。この節では,FDAが“交換可能”と考えている製品が本当に州薬剤法に管轄されている薬局に取って代わられるかどうかは不明である。
生物製品は米国でも小児科市場の排他性を得ることができる。小児科専有権が付与された場合、既存の専有期間と特許条項を6ヶ月増加させる。この6カ月間の排他性は,他の排他的保護や特許期間終了時から,FDAが発表したこのような研究の“書面請求”によって小児科研究を自発的に完成させることができる。
他のアメリカの医療保険法やコンプライアンスの要求は
米国では、FDA以外にも、連邦医療保険·医療補助サービスセンター(CMS)、米国衛生·公衆サービス部(HHS)の他の部門(例えば、総監察長事務室や衛生資源·サービス管理局)、司法省または司法省、司法省内の個別連邦検事室、州および地方政府を含む様々な連邦、州、地方当局によって規制されている可能性がある。例えば、販売、マーケティング、および科学/教育補助計画は、“社会保障法”、“虚偽請求法”、“健康保険携行性および責任法案”または“HIPAA”のプライバシーおよび安全条項、ならびに改正された同様の州法の詐欺および乱用条項を遵守しなければならない可能性がある。
他の事項に加えて、連邦反リベート法規は、任意の個人またはエンティティが、購入、レンタル、注文または手配、購入、注文または手配購入、レンタルまたは注文として、Medicare、Medicaid、または他の連邦医療計画に従って全部または部分的に精算することができる任意の物品またはサービスの見返りとして、故意に、または故意に現金または実物で直接または間接的に、公開または隠蔽的に提供、支払い、請求または任意の報酬を受けることを禁止する。報酬という単語は価値のあるものを含むと広く解釈されている。連邦反リベート法規は、治療製品製造業者と処方者および購入者との間の配置に適用されると解釈される。いくつかの法的例外と規制避難所がいくつかの一般的な活動を保護することは起訴されない。例外や安全港の範囲は狭く,処方,購入または推奨の報酬を誘導するために告発される可能性があるやり方に関連しており,例外や安全港の資格を満たさなければ審査される可能性がある。特定の適用された法定例外や安全港を規制するすべての要求を満たすことができなかったことは、連邦反リベート法規に基づいて、このような行為自体が不法であることを意味するわけではない。代わりに、そのすべての事実と状況の累積審査に基づいて、この手配の合法性を逐案的に評価する。私たちのやり方は、私たちと医者の手配を含めて、すべての場合、法定例外や安全港保護を規制するすべての基準を満たしていないかもしれない。
また、連邦“反リベート法令”下の意図基準は、2010年に“保健·教育和解法案”によって改正された“患者保護·平価医療法案”によって改正され、または総称してACAと呼ばれ、より厳しい基準を達成し、個人または実体がその法令またはその法令に違反する具体的な意図を実際に知る必要がなくなり、違反行為を実施することができる。また,ACAは連邦虚偽クレーム法案(FCA)の目的により,連邦反リベート法規違反による物品やサービスのクレームが虚偽や詐欺的クレームを構成することを含む判例法を編纂した。
FCAを含む連邦虚偽請求および民事罰金法は、一般市民が民事訴訟によって強制的に実行することができ、任意の個人または実体が虚偽または詐欺的クレームの提出を意図的にまたは提出することを禁止し、連邦医療保健計画(MedicareおよびMedicaidを含む)に支払いまたは承認するために、または虚偽記録または陳述を作成、使用または作成または使用して、連邦政府に虚偽または詐欺的クレーム材料を提供することを禁止する。クレームには、米国政府に提出された金銭または財産に対する“任意の請求または要求”が含まれている。例えば、歴史的には、製薬や他の医療保険会社は、顧客に無料製品を提供した疑いでこれらの法律の起訴を受けている
48
顧客がこの製品のために連邦計画に請求書を発行することを期待する。他の会社も起訴されました。これらの会社のマーケティング製品は未承認、ラベル外の用途に使用されているため、通常は精算されず、虚偽の声明を提出することになりました。
HIPAAは、他の事項に加えて、故意および故意に計画を実行または実行しようとすることを禁止し、虚偽または詐欺的な口実、陳述または約束の方法で任意の医療福祉計画(プライベート第三者支払者を含む)が所有または管理または保管している任意の金銭または財産を詐欺または取得し、医療違反行為の刑事調査を意図的に阻害し、悪巧み、計画または装置、重大な事実、または医療福祉、プロジェクトまたはサービスの提供または支払いに関連する任意の重大な虚偽、架空または詐欺的陳述を故意に妨害し、故意に詐欺、計画または装置、重大な事実、または医療福祉、プロジェクトまたはサービスの提供または支払いに関連する任意の重大な虚偽、架空または詐欺的な陳述を行うことを禁止する追加の連邦刑法を制定する。連邦反リベート法規と同様に、ACAはHIPAA下のある医療詐欺法規の意図標準を改正し、個人或いは実体がこの法規を実際に理解する必要がなく、或いはこの法規に違反する特定の意図があれば違反を実施できるようにした。
さらに、多くの州は、連邦医療補助および他の州計画に従って精算されるプロジェクトやサービス、またはいくつかの州では、支払者が誰であるかにかかわらず、類似した通常より禁止された詐欺や法律または法規の乱用を持っている。しかも、私たちの製品が外国で販売されていれば、私たちは似たような外国の法律に制約されるかもしれない。
私たちは連邦政府と私たちが業務を展開している州のデータプライバシーと安全規制によって制限されるかもしれない。HIPAAは“経済と臨床健康情報技術法案”(HITECH)及びその実施条例の改正後、ある医療保健提供者、医療情報交換所と健康計画(保証実体と呼ばれる)、独立請負業者或いは保証実体を代表する代理人に対して、保証実体(業務パートナーと呼ぶ)及びその保証下請け業者を代表してサービスを提供する際に、単独で識別可能な健康情報のプライバシー、安全と伝送を受信或いは取得することに関連する要求を提出した。このうち,HITECHはHIPAAのプライバシーやセキュリティ基準をビジネスパートナーに直接適用する.HITECHはまた4つの新しい民事罰金等級を作成し、HIPAAを改訂し、民事と刑事処罰を商業パートナーに直接適用し、州総検察長に新しい権力を与え、連邦裁判所に民事訴訟を提起し、損害賠償または禁制令を要求してHIPAAを実行し、連邦民事訴訟の提起に関連する弁護士費と費用を求めることができる。また,多くの州の法律は特定の場合に健康情報のプライバシーやセキュリティを管理しており,その多くの法律は互いに大きく異なり,HIPAAに先制されておらず,HIPAAよりも禁止的な効果があり,コンプライアンス作業を複雑化している可能性がある.
また、多くの製薬業者は平均販売価格と最適価格のようないくつかの価格報告指標を計算し、政府に報告しなければならない。また、これらの薬品の価格は、政府の医療計画や個人支払者が要求する強制的な割引やリベート、および将来的には米国よりも販売価格が低い国からの薬品の輸入を制限している法律を緩和することによって低下する可能性がある。将来の連邦医療保険カバー範囲と精算政策がどのように私たちの製品に応用されるかを予測することは困難であり、しかも異なる連邦医療保健計画下のカバー範囲と精算政策は常に一致しているわけではない。連邦医療保険の販売率は連邦医療保険計画に加えられた予算制限を反映している可能性もある。
さらに、ACA内の連邦医師支払い法案または日光法案およびその実施条例は、連邦医療保険、医療補助または児童健康保険計画(いくつかの例外)に従って支払うことができる薬品、器具、生物および医療用品を定義するいくつかの製造業者に、医師(医師、歯医者、検眼師、足科医および脊椎マッサージ師を含む定義)、他の保健専門家(例えば、医師アシスタントおよび勤務看護師)および教育病院に支払いまたは分配されたいくつかの支払いまたは他の価値移転に関する情報、ならびに医師およびその直系親族が所有する所有権および投資権益に関する情報を毎年CMSに報告することを要求する。また、多くの州では支払いや他の価値移転の報告も管理されており、その多くは互いに大きく異なり、往々にして先制されておらず、“陽光法案”よりも尻込み的な効果が生じ、遵守努力をさらに複雑化させる可能性がある。
製品を商業的に流通させるためには、ある州に薬品や生物製品のメーカーや卸売業者を登録することを要求する州の法律を守らなければならない
49
これらのメーカーや流通業者がその州に営業場所がなくても、製品を同州のメーカーと流通業者に輸送する。一部の州はまた、メーカーと流通業者が流通チェーン中で製品の系統を確立することを要求しており、いくつかの州はメーカーと他の州に流通チェーン中の製品の流れを追跡し追跡できる新しい技術を採用することを要求している。いくつかの州はすでに立法を公布し、製薬と生物技術会社にマーケティングコンプライアンス計画を確立し、州政府に定期報告を提出し、販売、マーケティング、定価、臨床試験およびその他の活動を定期的に公開し、および/またはその販売代表を登録し、薬局および他の医療保健実体が製薬および生物技術会社にいくつかの医師処方データを販売およびマーケティングのために提供することを禁止し、いくつかの他の販売およびマーケティング行為を禁止する。私たちのすべての活動は連邦と州消費者保護と不正競争法によって制限されるかもしれない。
第三者との業務配置が適用される医療法律や法規に適合することを確保することは、費用の高い努力である。もし私たちの業務が上記の任意の連邦および州医療保健法律または任意の他の現在または将来私たちに適用される政府法規に違反していることが発見された場合、私たちは民事、刑事および/または行政処罰、損害賠償、罰金、返還、個人監禁、MedicareとMedicaid、禁止、個人通報者が政府名義で提起した個人訴訟、または政府契約、契約損害、名声損害、行政負担、利益減少、および未来の収入のような政府計画から除外されることを含む重大な処罰を受ける可能性があります。私たちがこれらの法律を遵守していないという疑惑や、私たちの業務の縮小または再編を解決するために、会社の誠実な合意または他の合意の制約を受けている場合、追加の報告義務および監督を負う必要があり、これらはいずれも、私たちの業務運営能力および私たちの運営結果に悪影響を及ぼす可能性があります。
保証範囲·定価·精算
私たちが規制部門の承認を得る可能性のある任意の候補製品のカバー範囲と精算状態には、重大な不確実性がある。米国や海外市場では、規制部門の承認を得て商業販売を行う任意の製品の販売は、第三者支払者がこのような製品に保険を提供する程度にある程度依存し、十分な補償レベルを確立する。米国では,第三者支払者には連邦や州医療計画,個人管理のヘルスケア提供者,医療保険会社,その他の組織が含まれている。カバー範囲と政府医療計画(米国の連邦医療保険や医療補助のような)および商業支払者からの十分な補償は、新製品の受容度に重要である。同様に、セット診断テストは単独の保証と精算が必要であり、しかもそのセットの薬品或いは生物製品の保証と精算は含まれていない。
第三者支払人は彼らがどのような治療費用を支払い、精算レベルを確立するかを決定する。第三者支払人の治療性薬物の使用に対する決定を含む第三者支払人の保証範囲と精算は多くの要素に依存する可能性がある
私たちは商業化されたどの製品も精算されることを確実にすることはできません。保険と精算があれば、精算レベルが十分かどうかも確認できません。カバー範囲はまた、FDAや同様の外国規制機関が製品を承認する目的よりも限られている可能性がある。限られたカバー範囲と不足した精算は、規制承認を受けた任意の製品に対する需要を減らしたり、その価格を低下させたりする可能性がある。
第三者決済者は、価格に挑戦し、医療の必要性を審査し、医療製品、療法、サービスの費用対効果を審査するとともに、それらの安全性と有効性を疑問視するようになっている。私たちの製品のために精算するのは特に難しいかもしれません。高い価格を伴うからです
50
ブランド薬と医師の監督下で使用される薬を使用する。私たちは、私たちの製品の医療の必要性と費用効果、FDAの承認を得るのに必要なコストを証明するために、高価な薬物経済学的研究を行う必要があるかもしれない。私たちの候補製品は医学的に必要で費用効果があると思われないかもしれない。第三者支払人から製品の保証と精算承認を得ることは時間がかかり、高価な過程であり、これは各支払人に科学的、臨床的、費用効果的なデータを提供して、個々の支払人に私たちの製品を使用するために必要かもしれないが、保証と十分な精算を得ることは保証されないかもしれない。第三者支払者が製品に保険を提供することを決定することは、十分な返済率を承認することを意味するものではない。また、米国では、第三者支払者は保険や精算に対して統一的な政策を持っていない。第三者支払者が自己の保険·精算政策を設定する際には,通常連邦医療保険引受政策や支払制限に依存するが,独自の方法や承認の流れもある。したがって、第三者支払者は、1つの商品に保険を提供することを決定し、他の支払者もその商品に保険を提供することを保証することはできない。十分な第三者支払者の精算が得られない可能性があり、製品開発への投資の適切なリターンを実現するために十分な価格レベルを維持することができる。精算が得られない場合や限られたレベルの精算のみが提供されなければ、私たちが開発に成功した任意の候補製品を商業化することに成功できないかもしれません。
私たちのいくつかの製品は承認されると、医者が管理することができる。現在適用されているアメリカの法律によると、ある非自己投与された製品(注射可能な薬物を含む)は、連邦医療保険B部分を通じて連邦医療保険の保険を受ける資格がある可能性がある。連邦医療保険B部分は元の連邦医療保険の一部であり、老人と障害者に医療福祉を提供し、受益者の健康状態を治療するために必要な外来サービスと用品をカバーし、ある医薬製品を含む。メーカーの合格薬品或いは生物製品が連邦医療保険B部分精算を獲得する条件の一つとして、メーカーは医療補助薬品リベート計画と340 B薬品定価計画を含む他の政府医療保健計画に参加しなければならない。医療補助薬品還付計画は製薬業者が衛生と公衆サービス部部長と締結し、有効に全国的な税金還付協定を締結することを要求し、各州が連邦マッチング資金を獲得する条件として、メーカーが医療補助患者に提供する外来薬物に用いられる。340 B薬品定価計画によると、製造業者は割引をこの計画に参加するエンティティに拡大しなければならない。
他の国もまた違う価格設定と精算プログラムを持っている。EUでは,政府はその定価と精算規則や国家医療システムの制御により薬品の価格に影響を与え,これらのシステムは消費者が支払う薬品コストの大部分に資金を提供する。いくつかの法域はプラスリストとネガティブリスト制度を実行し、補償価格を合意した後にのみ、製品を販売することができる。精算または定価の承認を得るために、その中のいくつかの国は臨床試験の完成を要求する可能性があり、特定の候補製品のコスト効果を現在利用可能な治療法と比較する。他の会員国たちは会社が自分の薬品価格を固定することを許可するが、会社の利益を監視する。医療コストの下振れ圧力が大きくなる。そのため、新製品の参入にはますます高い壁が設けられている。また、一部の国では、低価格市場からの国境を越えた輸入が一国国内の定価に商業圧力をかけている。
政府や第三者支払者が保険と十分な補償を提供できない場合、規制部門の承認を得て商業販売を行う任意の候補製品の適正性が影響を受ける可能性がある。また,管理式医療への重視,健康維持組織の日増しに増加している影響力,米国の追加立法変化は医療定価の圧力を増加させており,この圧力は増加し続けることが予想される。医療費は一般的に上昇し,特に処方薬,医療機器,外科手術プログラムなどの治療費上昇の下振れ圧力が非常に大きくなっている。保証政策と第三者精算料率は随時変化する可能性がある。規制部門の承認を得た1つまたは複数の製品が有利な引受·精算状態を獲得しても、将来的にはあまり有利ではない引受政策や精算料率が実施される可能性がある。
医療改革
米国や一部の外国司法管轄区では、医療保健システムに関するいくつかの立法と規制の変化、提案された変化が継続され、これらの変化は候補製品の上場承認を阻止または延期し、承認後の活動を制限または規範化し、マーケティングの許可を得た候補製品を収益的に販売する能力に影響を与える可能性がある。アメリカの政策立案者や支払者の中で
51
他の地域では,医療システムの変革を促進することに非常に興味があり,医療コストの抑制,質の向上,および/または参入拡大を既定の目標としている。米国では、製薬業はこれらの努力の重点であり、重大な立法計画の大きな影響を受けてきた。
例えば、ACAは政府と民間保険会社の医療融資とサービス提供を大きく変えた。ACAは他の事項を除いて:(1)ある吸入、輸液、点滴、移植または注射に対して、小売コミュニティ薬局で通常配布されていない薬品と生物製品について、メーカーの医療補助薬品リベート計画下のリベートを計算する新しい方法を導入した;(2)医療補助薬品リベート計画下でメーカーの最低医療補助リベートを増加した;(3)ブランド処方薬メーカーが連邦政府に支払わなければならないブランド処方薬費用を確立した;(4)計画中に新しい実体を増加させることにより、340 B薬品定価計画に参加する資格のある実体リストを拡大した。(5)新しい連邦医療保険D部分引受切欠き割引計画を構築し、メーカーは現在、保証間隔期間内に条件を満たす受益者に適用ブランド薬品交渉価格の70%の販売時点割引を提供し、メーカーの外来薬として連邦医療保険D部分の条件に入れることに同意しなければならない;(6)メーカーの医療補助バックオフ責任を連邦医療補助管理保健組織に参加する個人に分配された保険薬物に拡大する。(7)各州がより多くの個人に医療補助を提供することを可能にし、連邦貧困レベル133%未満の個人に新たな強制資格カテゴリを増加させることによって、メーカーの医療補助リベート責任を潜在的に増加させることを含む医療補助計画の資格基準を拡大するステップと、(8)後続のバイオ製品のためのライセンスフレームを作成するステップと、(9)CMSに医療保険および医療補助革新センターを設立し、革新的な支払いおよびサービス交付モードをテストして、処方薬支出を含む可能性がある医療保険および医療補助支出を低減するステップと、(10)患者を中心とした新たな結果研究所を作成し,優先順位を監督·決定し,臨床的有効性の比較検討を行った。
ACAのいくつかの側面は法律と政治的な挑戦を受けている。トランプ総裁は、ACA規定のいくつかの要求を延期、回避、または緩和するために、いくつかの行政命令と他の指令に署名した。同時に、国会はACAの全部または一部を廃止または廃止し、代替する立法を審議した。国会ではまだ全面的な廃止立法は成立していないが、ACAの下にある税収実施に影響を与えるいくつかの法案が署名されて法律となっている。2017年12月には、2017年の減税·雇用法案が公布され、2019年1月1日から個人がACA規定を維持できなかった医療保険に対する税収処罰が廃止され、一般に“個人強制要求”と呼ばれている。また、2020年に連邦支出計画は永久に廃止され、2020年1月1日から“平価医療法案”は、高コスト雇用主が後援する医療保険と医療機器税に“キャデラック”税を徴収することを規定し、2021年1月1日から医療保険会社税も廃止された。また,2018年に両党予算法案,あるいはBBAなどが改正され,ACAは2019年1月1日から施行され,多くの連邦医療保険薬物計画におけるカバーギャップを埋めるために,通常“ドーナツ穴”と呼ばれている
2021年6月17日、米国最高裁は、ACAは全体的に違憲であり、“個人権限”が国会で廃止されたため、プログラム理由に基づく挑戦を却下した。したがって、ACAは現在の形態で継続的に有効であるだろう。また,米国最高裁判所が裁決を下す前に,総裁·バイデンは2021年1月28日にACA市場による医療保険の取得を目的とした特殊な保険加入期間を開始する行政命令を発表した。行政命令はまた、特定の政府機関に、医療補助モデルプロジェクトおよび免除計画の再審査、および医療補助またはACAによる医療保険カバー範囲の獲得による不必要な障害をもたらす政策を含む医療補助モデルプロジェクトおよび免除計画の再審査および見直し、医療補助またはACAによる医療保険のカバー範囲を制限する既存の政策およびルールを再検討するように指示する。ACAは未来に司法や国会で挑戦される可能性がある。2022年8月16日、総裁·バイ登は2022年インフレ削減法案に署名し、個人が平価医療法案市場で医療保険を購入する強化補助金を2025年に延長した。2025年からアイルランド共和軍は,受益者の最大自己負担コストと新たに構築されたメーカー割引計画を著しく低減することにより,連邦医療保険D部分計画下の“ドーナツ脆弱性”を解消した。このような挑戦やバイデン政府の医療改革措置がACAと我々の業務にどのように影響するかは不明である。
52
ACAが公布されて以来、他の立法改正も提案され、採択された。2011年8月、オバマ総裁は、2013年4月1日から施行された医療保険提供者への医療保険支払総額の最高2%削減を含む“2011年予算制御法案”に署名し、その後の立法改正により、国会がさらに行動しない限り2032年まで有効となる。2013年1月、“2012年米国納税者救済法”が法律に署名され、病院を含むいくつかの医療サービス提供者への医療保険支払いが減少し、政府が提供者に多額の支払いを取り戻す訴訟時効が3年から5年に延長された。2015年の連邦医療保険アクセスとチップ再認可法案によると、新計画の拡大は、医師の業績計画に対する連邦医療保険支払い計画のような我々の業務にも影響を与える可能性があり、品質支払い計画とも呼ばれる。
また、米国は特殊薬品の価格設定に対する立法と法執行への興味もますます大きくなっている。具体的には、米国議会は最近数回の調査を行い、薬品定価の透明性を高め、連邦医療保険下の処方薬のコストを下げ、定価とメーカー患者計画との関係を審査し、政府計画の薬品精算方法を改革するための連邦と州立法を提出し、公布した。2021年7月、バイデン政府は“米国経済における競争を促進する”という行政命令を発表し、その中には処方薬に対する条項が複数ある。バイデン行政命令への対応として,2021年9月9日,HHSは高薬価に対応した総合計画を発表し,薬品定価改革の原則を概説し,国会がとりうる様々な潜在的立法政策を示し,これらの原則を推進した。また、アイルランド共和軍(IRA)は、他の事項に加えて、(I)HHSが連邦医療保険(Medicare)がカバーするいくつかの高支出、単一由来の薬物および生物製品の価格について交渉し、法律で規定されているこのような薬物と生物製品との交渉に等しくない“最高公平価格”の価格を提供することによって、薬品メーカーに民事と潜在的な消費税を科し、(Ii)連邦医療保険B部分またはD部分にカバーされているいくつかの薬品および生物製品にリベートを徴収し、インフレを超える価格上昇を処罰するように指示する。アイルランド共和軍は衛生と公共サービス部が最初の数年に規制ではなく指導によってその多くの規定を実施することを許可した。このような規定は2023年度から段階的に施行される。2023年8月29日、HHSは、連邦医療保険薬品価格交渉計画が現在法的挑戦を受けているにもかかわらず、価格交渉を受ける上位10種類の薬物のリストを発表した。これらの計画の実施に伴い,HHSは指導意見を発表·更新し続けている。アイルランド共和軍がどのように実施されるかは不明であるが,製薬業に大きな影響を与える可能性がある。また、バイデン政府の2022年10月の行政命令に応答するため、衛生·公衆サービス部は2023年2月14日に報告を発表し、CMS革新センターによってテストされた3種類の新しいモデルを概説し、これらのモデルはそれらの薬物コストを下げ、獲得性を促進し、医療の質を高める能力に基づいて評価を行う。これらのモデルが将来の任意の医療改革措置で使用されるかどうかは不明である。また、2023年12月7日、バイデン政府はベハ-ドール法案下の入場権利を使用することで処方薬の価格を制御するイニシアチブを発表した。2023年12月8日、米国国家標準·技術研究所は、権限行使を考慮した機関間指導枠組み草案を発表し、その中で初めて製品価格を機関が進行権を行使する際に使用できることを決定する要因とした。これまでデモの権利を行使したことはなかったが、新たな枠組みの下で、この権利が継続するかどうかは定かではない。州レベルでは、立法機関は、価格や患者の精算制限、割引、特定の製品への参入の制限、マーケティングコストの開示と透明性措置を含む薬品および生物製品の価格設定を制御するための法規を立法および実施することが増えており、場合によっては、他の国からの輸入と大量購入も奨励されている。例えば、FDAは2024年1月5日、特定の州医療計画のためにカナダから特定の薬剤を輸入するフロリダ州第804条輸入計画(SIP)の提案を承認した。この計画がどのように実施されるか,どのような薬剤が選択されるか,その計画が米国やカナダで法的挑戦を受けるかどうかは不明である。他の州もFDAの審査を待っているSIP提案を提出した。このような承認された輸入計画が一旦実施されると、これらの計画がカバーする製品の薬品価格をより低くする可能性がある。
“反海外腐敗法”
“海外腐敗防止法”は、いかなる米国人個人または企業が、任意の外国人官僚、政党または候補者に直接または間接的に支払い、支払いを提供または許可するか、または任意の価値のあるものを提供することを禁止し、個人または企業が業務を獲得または保持するのを助けるために、外国の実体の任意の行為または決定に影響を与えることを目的としている。海外腐敗防止法は証券を持つ会社にも要求されています
53
米国に上場し、会計規定を遵守するために、企業(国際子会社を含む)のすべての取引の帳簿や記録を正確かつ公平に反映し、国際業務のために適切な内部会計制御システムを設計·維持することを要求する。
法規を付加する
これらの規定に加えて,環境保全や有害物質に関する州や連邦法は,“職業安全と健康法”,“資源節約と回収法”,“有毒物質制御法”を含めて,我々の業務に影響を与える。これらの法律と他の法律は私たちの様々な生物、化学、放射性物質の使用、処理と処理、これらの物質は私たちの行動、そして私たちの行動によって生成された廃棄物を規範化している。もし私たちの運営が環境汚染を招いたり、個人を危険物質に曝露させたりすれば、損害賠償と政府罰金の責任を負う可能性があります。私たちは、私たちが適用される環境法律を実質的に遵守し、これらの法律を遵守し続けることは、私たちの業務に実質的な悪影響を与えないと信じている。しかし、私たちはこのような法律の変化が私たちの未来の運営にどのように影響するか予測できない。
その他の規則
私たちはまた多くの連邦、州、地方法律の制約を受けており、これらの法律は安全作業条件、製造実践、環境保護、火災危険制御と危険或いは潜在的危険物質の処分に関連している。私たちが今または未来にこのような法律と法規を遵守することは大きな費用をもたらすかもしれない。
人的資本資源
私たちのチームは2023年12月31日までに330人の従業員に発展し、そのうち165人が高度な学位を持ち、うち91人が博士号および/または医学学位を持っている。私たちのすべての従業員はアメリカで雇われている。私たちの高い素質と経験豊富なチームは研究、臨床、製造、監督及び一般と行政機能の科学者、医者と専門家を含み、これは私たちの成功に重要である。我々の従業員のうち,278人が研究·開発活動に従事し,52人が一般·行政活動に従事している。私たちの従業員の中の一人も労働組合代表でもなく、集団交渉協定のカバー範囲もない。私たちはまたアルバイトを利用して私たちの業務需要に柔軟性を提供する。
2023年、私たちのチームは16人の従業員を増やした。私たちは2024年に引き続き従業員を増やす予定で、臨床と臨床前研究·開発における専門知識と能力を拡大することに重点を置いている。
多様性公平性包括性
私たちは多様化と包括的な作業環境を促進することを誇りに思います。このような環境では、チームメンバーは彼らの潜在力を十分に発揮し、彼ら独自の視点に貢献して協力を推進する能力があると感じています。私たちは多様化、公平と包容の職場を構築することに努力し、同時に一流のチームを構築して医学の未来を変える。
2023年12月31日現在、従業員の自己報告によると、私たちの全従業員の約46%が女性で、約54%が少数民族や少数民族集団のメンバーだ。
私たちは労働力チーム全体での私たちの多様性と包括的な計画を測定して強化することに集中し続けるつもりだ。私たちは性別、性別、民族、人種、宗教、あるいは他の保護された特質にかかわらず、最も合格した従業員を募集し、私たちの政策は職場差別に関するすべての適用法律を遵守することだ。
採用·発展·留用
著者らの戦略の成功実行は各級の高技能従業員の多元化チームを吸引、維持、激励することに依存する。2023年、私たちのチームは私たちの現在の最高経営責任者を含めて16人の従業員を増やした。
私たちは一般的に基本給と短期的と長期的なインセンティブの組み合わせを含む包括的で競争力のある総給与プログラムを私たちの永久従業員のために作成した。他には
54
私たちの従業員に競争力のある給料を提供し、従業員は私たちの計画の進歩による潜在的な経済収益を共有すべきだと思います。そのため、私たちは通常、初回採用時とその後、毎年永久従業員に株式オプションと制限株式単位を付与し、従業員の株式購入計画への参加を許可し、会社および/または個人目標の実現に応じて永久従業員に年間ボーナスを支払う。私たちの休暇計画には有給休暇、休暇、病気休暇、その他の有給休暇と無給休暇が含まれています。私たちの健康および健康計画には、医療、歯科、視力ケア、一部の費用がPoseida、会社が支払う生命保険、401(K)貯蓄計画退職貯蓄の均等支払い、従業員援助計画(EAP)、および他の福祉支払いが含まれている。
私たちは、持続的な学習と発展、訓練とその他の資源も私たちの従業員を維持し、学習と指導文化を創造する重要な構成要素であると信じている。私たちは私たちの従業員がオンライン技能課程、リーダーシップシンポジウム、EAPによる職業相談、その他の専門発展計画と非公式指導を含む各種の学習と発展資源を参加し、利用することを奨励する。
従業員のコミュニケーションと敬業度
私たちは、私たちの従業員が彼らの仕事が私たちの全体戦略にどのように貢献しているのかを知っている時、彼らのパフォーマンスが最も良いということを認識している。これを実現するために,様々なチャネルを用いた公開と直接のコミュニケーション,市庁会議,全社範囲の書面コミュニケーション,およびわが社内ネットワークへの掲示を強調した.
我々は従業員の士気が強いことを確保するために努力し、従業員の関心の重要な領域を確定し、従業員のフィードバックを求め、従業員の流出率を監視することによって、従業員の労働環境と文化に対する理解を強化することを目的とした従業員敬業度調査を実施して評価を行った。敬業度調査の結果は,従業員の敬業度の向上と従業員体験の改善を目的とした計画と流れの実施に用いられた。
企業情報
私たちは2014年12月にデラウェア州で登録設立された。私たちの主な実行事務室はカリフォルニア州サンディエゴにあり、郵便番号:92121、ドンセンター通り9390 Towne Centre Drive 9390 Suit 200、私たちの電話番号は(8587793100)です。私たちの会社のサイトの住所はwww.pose ida.comです。本年度報告に含まれるまたは当社のサイトから取得可能な情報は、本年度報告の一部ではなく、本報告には当社サイトアドレスが含まれており、非アクティブテキストとしてのみ参照されている。
新興成長型会社
私たちは2012年のJumpStart Our Business Startups Actで定義されているように“新興成長型会社”です。私たちは、(1)財政年度の最終日まで、(A)2020年7月に初公募(IPO)完了5周年後、(B)私たちの年間総収入が少なくとも12.35億ドル、または(C)取引法の報告書を12ヶ月間遵守していることを意味し、前年6月30日現在、非関連会社が保有する普通株式の時価が7000万ドルを超えることを意味する新興成長型会社である。そして(2)前3年の間に10億ドルを超える転換不能債券を発行した日。我々は本年度報告で2012年のJumpStart Our Business Startups Actを“JOBS Act”と呼び,言及した“新興成長型会社”はJOBS Actにおける意味と同じである。
55
第1 A項。国際ロータリーSK因子です。
私たちの普通株への投資は投機的で、高い危険を扱っている。我々の普通株を購入、保有または売却するか否かを決定する前に、以下に述べるリスク、および本年度報告Form 10-Kに含まれる他の情報、ならびに我々の合併財務諸表および関連付記、ならびに“経営陣の財務状況および経営結果の検討および分析”の節の情報をよく考慮しなければならない。以下のいずれかのリスクが発生すれば、私たちの業務、財務状況、経営業績、将来の成長見通しは重大な悪影響を受ける可能性がある。この場合、私たちの普通株の市場価格は下落する可能性があり、あなたは投資の全部または一部を失うかもしれない。このForm 10-K年次報告書には、リスクと不確実性要因に関する前向きな陳述も含まれている。以下に説明するリスクを含む様々な要因のため、私たちの実際の結果は、前向き陳述で予想される結果とは大きく異なる可能性がある。“前向きな陳述に関する特別な説明”というタイトルの章を参照してください
私たちの限られた経営歴史、財務状況、資本要求に関するリスク
私たちは臨床段階の細胞と遺伝子治療会社で、運営の歴史は限られている。成立以来、純損失が発生しており、予測可能な未来には、引き続き重大な損失を受けることが予想される。私たちは製品販売から何の収入も得られなかったし、永遠に利益を上げないかもしれない。
私たちは臨床段階の細胞と遺伝子治療会社で、運営の歴史が限られていて、これまでの私たちの業務の成功を評価することが難しくなり、私たちの将来の生存能力を評価することも難しいかもしれません。今まで、私たちの業務は組織と配備の会社、業務計画、資金の調達、私たちの知的財産権の組み合わせの確立と保護、私たちのプラットフォーム技術の開発、潜在的な候補製品の決定及び研究開発と製造活動に限られており、私たちの候補製品の臨床前研究と臨床試験を含む。私たちのすべての候補製品は初期開発段階にあり、商業販売のために承認された製品はありません。私たちは製品販売から何の収入も得たことがなく、運営を開始してから毎年純損失が出ています2023年12月31日と2022年12月31日までの年間で、それぞれ1億234億ドルと6400万ドルの純損失を出した2023年12月31日までの累計赤字は5.943億ドルだった。もしあれば、規制部門の承認と商業化のための製品候補製品が準備されるまでにも数年かかると予想される。今後数年と予見可能な未来には,臨床開発による我々の候補製品の推進に伴い,ますます多くの運営損失を招くことが予想される。私たちのこれまでの損失は、予想された将来の損失に加え、私たちの株主権益や運営資本に悪影響を与え続けるだろう。
利益を実現し、維持するためには、巨大な市場潜在力を持つ1つ以上の製品を開発し、最終的に商業化しなければならない。これは私たちの候補製品の臨床前研究と臨床試験を完成させ、これらの候補製品のためにマーケティング許可を得て、製造、マーケティングと販売を獲得し、任意の発売後の要求を満たす製品を含む一連の挑戦的な活動で成功することを要求するだろう。私たちはこのような活動で決して成功しないかもしれません。たとえ私たちが私たちの1つ以上の候補製品を商業化することに成功しても、私たちは利益を達成するために十分な大きさまたは十分な収入を生成しないかもしれません。また、比較的若い企業として、予期せぬ費用、困難、合併症、遅延などの既知と未知の挑戦に直面する可能性がある。もし私たちが確実に利益を達成すれば、私たちは四半期や年度の収益性を維持または向上させることができないかもしれません。私たちは引き続き多くの研究開発と他の支出を生み出して、より多くの候補製品を開発し、マーケティングします。もし私たちが実現して利益を維持できなければ、会社の価値を下げ、資金を調達し、研究開発努力を維持し、業務を拡大し、あるいは運営を継続する能力を弱める可能性がある。わが社の価値の低下はあなたの投資損失の全部または一部を招く可能性もあります。
私たちは私たちの候補製品の開発と商業化を達成するために多くの追加資金を得る必要があるだろう。もし私たちが必要な時にこの資金を集めることができなければ、私たちは私たちの製品開発計画や他の業務を延期、減少またはキャンセルさせることを余儀なくされるかもしれない。
設立以来、私たちは大量の現金を使って私たちの運営に資金を提供し、今後数年で私たちの支出は大幅に増加すると予想される。バイオ製薬候補製品の開発は資本集約型である。著者らの候補製品が臨床前研究と臨床試験に入り、進展を得るに伴い、著者らは大量の追加資金が必要であり、著者らの臨床、監督管理、品質と製造能力を拡大する。はい
56
また、いずれかの候補製品が市場承認されれば、マーケティング、販売、製造、流通に関連した巨額の商業化費用が発生すると予想される。
2023年12月31日現在、私たちは2.122億ドルの現金、現金等価物、短期投資を持っている。私たちの現在の運営計画によると、私たちは私たちの既存の現金、現金等価物、および短期投資が少なくとも今後12ヶ月以内に私たちの運営に資金を提供できると信じている。しかし、私たちの現在の現金、現金等価物、および短期投資は、規制機関の承認によって任意の候補製品に資金を提供するのに十分ではなく、私たちの候補製品の開発と商業化を達成するために多くの追加資本を集める必要があるだろう。
追加の資本は、株式発行および/または債務融資によって、または他の潜在的な流動性源から得ることができ、その中には、我々の1つまたは複数の研究プロジェクトまたは特許の組み合わせの新しいまたは既存の協力、許可、または他の商業プロトコルを含むことができる。必要であれば、私たちは受け入れ可能な条件で十分な資金を得ることができないかもしれないし、全くないかもしれない。私たちがより多くの資金を得る能力は、いくつかの国と地域の内乱と政治的動揺、世界の経済状況が悪化する可能性があること、および公衆衛生危機が米国と世界各地の信用と金融市場に与える破壊と変動の影響を受ける可能性がある。もし私たちが必要な時や魅力的な条件下で資金を集めることができなければ、私たちは私たちの研究開発計画や他の業務を延期、減少、またはキャンセルすることを余儀なくされるだろう。もしこのような事件が発生したら、私たちが行動目標を達成する能力は実質的で不利な影響を受けるだろう。私たちの将来の資本需要と利用可能な資金の十分性は、“リスク要因”に記載されている要素を含む多くの要素に依存するだろう。これらの要素が私たちに与える深刻さと直接的な影響によって、私たちは私たちの運営要求、条項が私たちに有利、あるいは根本的にできないために追加的な融資を得ることができないかもしれない。
私たちが基づいているこれらの推定は、不正確であることが証明されたり、商業意思決定によって調整される必要がある可能性があり、私たちは現在予想されているよりも早く私たちの利用可能な資本資源を枯渇させるかもしれない。私たちの将来の資本需要は多くの要素に依存します
私たちは長年製品販売から収入を得ないと予想されているので、もしあれば、私たちの持続的な運営と予想される費用増加に関連した大量の追加資金を得る必要があります。私たちの候補製品を販売することから相当な収入を得ることができる前に、もしあれば、株式発行、債務融資、あるいは他の資本源を通じて私たちの現金需要を満たすことを望んでいます
57
潜在的な寄付金、協力、許可、または他の似たような計画。また、有利な市場条件や戦略的考慮により、現在または将来の運営計画を実行するのに十分な資金があると考えても、追加の資本を求めることができる。資本市場における金利と経済インフレの変化は、私たちが将来獲得できる融資の獲得可能性、金額、タイプに影響を与える可能性がある。2021年8月13日私たちは支配権発行協定を締結しましたSM販売プロトコル,あるいはCantor Fitzgerald&Co.やCantorと締結された販売プロトコルは,時々“市場発売”計画により普通株を売却し,その計画の総発行価格は8,500万ドルと高く,Cantorはこの計画により販売代理を担当する。販売契約に基づいて証券を売却できるという要求を満たし続ける保証はありません。もし私たちが要求を満たしていれば、有利な条件で十分な資金を集めることができます。もし私たちが必要な時や魅力的な条件下で資金を集めることができなければ、私たちは私たちの研究開発計画や将来の商業化努力を延期、減少または廃止することを余儀なくされるだろう。
私たちの融資協定の条項は私たちの運営と財政的柔軟性を制限する。もし私たちが債務融資を通じて追加資本を調達すれば、どんな新しい債務の条項も私たちが業務を運営する能力をさらに制限するかもしれない。
2023年12月31日現在、オックスフォード金融有限責任会社(Oxford Finance LLC)と合意した融資と保証協定によると、私たちは6,000万ドルの未返済定期融資を持っている。このローンの保証は留置権であり、私たちのほとんどの個人財産、権利、資産をカバーしており、知的財産は含まれていない。融資協定は私たちに適用される慣用的な肯定と否定の条約と違約事件を含む。これらの肯定的な条約には、政府の承認、いくつかの財務報告書の提出、保険カバー範囲の維持、在庫の良好かつ販売可能な状態の維持、重大な知的財産権の保護を要求する条約が含まれている。負の条約には、私たちが担保を譲渡すること、追加債務を招くこと、合併または買収を行うこと、現金配当金を支払うこと、または他の分配を行うこと、投資を行うこと、留置権を設立すること、資産を売却すること、二次債務を支払うことの制限が含まれ、いずれの場合も、いくつかの例外的な状況によって制限される。融資協定の制限条項は、私たちが私たちまたは私たちの株主が有益だと思うビジネス機会を追求できないかもしれない。さらに、他の違約トリガ要因では、オックスフォード大学は、融資合意定義の重大な不利な変化と考えられる任意の事件が発生したときに違約を宣言することができる。もし私たちが融資協定の下で違約すれば、オックスフォード大学は私たちのすべての返済義務を加速させ、私たちの質権の資産を制御するかもしれないが、これは私たちが合意の中で私たちにあまり有利ではない条項を再交渉するか、または直ちに運営を停止する必要があるかもしれない。さらに、もし私たちが清算された場合、オックスフォード大学が返済を得る権利は、私たちの普通株式所有者が清算から任意の収益を得る権利よりも優先されるだろう。オックスフォードの違約事件に対するいかなる声明も私たちの業務と見通しを深刻に損なう可能性があり、私たちの普通株の価格下落を招く可能性がある。もし私たちが追加的な債務融資を調達すれば、このような追加債務の条項は私たちの運営と財政的柔軟性をさらに制限するかもしれない。
我々の候補製品の発見,開発,規制承認に関するリスク
私たちの候補製品は初期開発段階にあり、私たちの候補製品の人体上の歴史が限られていることをテストするために臨床試験を行っています。
我々の開発はまだ初期段階であり,これまで我々の業務の大部分は我々のプラットフォーム技術の開発,製造能力の構築,薬物発見や臨床前研究に限られてきた。2021年11月、私たちは私たちのP-BCMA-101計画の臨床開発を段階的に終わらせることを決定し、これは私たちの最初の候補製品が人体でテストを行った。2022年11月、私たちは私たちの最初の固形腫瘍臨床試験であるP-PSMA-101計画の臨床開発を段階的に終了することを決定したと発表した。われわれは2021年末にP−BCMA−ALLO 1とP−MUC 1 C−ALLO 1の1期臨床試験を開始し,最近2023年末にP−CD 19 CD 20−ALLO 1の1期臨床試験を開始した。そのため、私たちのインフラは限られており、会社として臨床試験を行った経験と監督の相互作用は限られており、私たちの臨床試験が時間通りに完成するかどうかは確定できず、私たちが計画した臨床試験は時間通りにスタートするかどうか、私たちの計画の開発計画はFDAや他の類似した外国の監督機関に受け入れられるかどうか、あるいは承認されれば、これらの候補製品は商業化に成功することができる。
58
私たちの候補製品の開発はまだ初期段階にあるので、最終的に製品販売から相当な収入を得る能力は多くの要素に依存します
もし私たちがこのような要求のうちの1つ以上をタイムリーに達成できなければ、私たちは重大な遅延に遭遇したり、私たちの候補製品を商業化することに成功できない可能性があり、これは私たちの業務に深刻な損害を与えるだろう。
私たちの候補製品は新しい技術に基づいており、これにより、候補製品開発の時間、結果、コスト、及び規制承認を得る可能性を予測することは困難である。
私たちは私たちのプラットフォーム技術を使った候補製品の開発に集中します。私たちの将来の成功はこの方法の成功開発にかかっています。一般に,CAR−Tも遺伝子編集も新興分野であり,我々の手法は特にどの重要な時間帯でも広範なテストを受けていない。特に,CAR−T製品ではTの割合が高いと考えられるが,供給チェーン管理細胞は早期世代CAR-T製品が直面しているいくつかの挑戦を克服することができるかもしれないが、修正された細胞が体内に長期的に存在する可能性があることを含む、これらの細胞の割合を増加させることが予想される利点を生じるかどうか、または時間の経過とともに予見できない負の結果をもたらすかどうかを決定することはできない。私たちはまだ、私たちのプラットフォーム技術に基づく任意の候補製品が臨床試験中または後に市場承認を得た場合の有効性および安全性を成功的に証明することもできず、私たちのプラットフォーム技術を使用することは決して適切な製品を生成しないかもしれない。私たちはまた、持続可能で反復可能で拡張可能な製造プロセスの開発、またはそのプロセスを商業パートナーに移転するか、または私たち自身の商業製造能力を確立する上で遅延に遭遇する可能性があり、これは、私たちがタイムリーに、または利益的に臨床試験を完了することを阻止するか、または任意の製品を商業化することを阻止するかもしれない。
そのほか、FDA、ヨーロッパ薬品管理局(European Medicines Agency、EMAと略称する)とその他の監督管理機関の臨床試験要求及びこれらの監督管理機関は候補製品の安全性と有効性を確定する標準に用いられ、潜在製品のタイプ、複雑性、意外性及び期待用途と市場によって大きく異なる。我々のような新製品候補製品の規制承認プロセスは、他のより有名で広く研究されている医薬品または他の製品候補製品と比較して、より高価であり、時間もかかる可能性がある。CAR-Tと遺伝子治療製品は近年進展しているが、一部の製品しかない
59
アメリカや他の市場では、私たちの候補製品が規制部門の承認を得るのにどのくらいの時間またはどのくらいのコストがかかるかを決定することは難しい。
また、遺伝子編集業界は急速に発展しており、私たちの競争相手は新しい技術を導入し、私たちの技術を時代遅れにしたり、魅力を低下させたりする可能性がある。私たちの候補製品の開発サイクルのどの時点でも新しい技術が現れる可能性があります。競合他社が代替技術を使用または開発するにつれて、このような技術の任意の故障は、私たちの計画に悪影響を及ぼす可能性がある。例えば、いくつかの研究により、CRISPR-Cas 9方法を用いた遺伝子編集は編集細胞自体の癌化のリスクを増加させる可能性があり、2021年10月、臨床意義が未知の染色体異常を発見し、著者らの競争相手がTALEN方法を使用する計画は全面的に臨床保留になった。FDAは2023年11月,CAR−T療法の二次性悪性腫瘍のリスクを調査していると発表した。我々の非ウイルスCas-Clover遺伝子編集方法がその中のいくつかの問題を回避する可能性があると信じているにもかかわらず、私たちの方法は類似したリスクに関連しているかもしれないし、他の遺伝子編集技術やCAR-T療法が直面している問題は、私たちの技術および候補製品に否定的な見方を与えたり、審査を強化したりするかもしれない。
遺伝子編集技術を用いて作られた製品や遺伝子治療治療に関連する製品に対する規制要求は常に変化し,将来も変化し続ける可能性がある。1つの規制機関の承認は、いかなる他の規制機関が何を承認する必要があるかを代表しない可能性があり、遺伝子治療製品と遺伝子編集技術を用いて作られた他の製品を監督する責任者との間には多くの重複があり、時には不整合である。例えば、国家衛生研究院(NIH)の組換えDNA分子に関する研究ガイドラインによると、ヒト遺伝子転移試験の監督には、機関生物安全委員会(IBC)による評価と評価が含まれており、IBCは地方機関委員会であり、当該機関の組換えまたは合成核酸分子を用いた研究の審査·監督を担当している。IBCは、研究の安全性を評価し、公衆の健康または環境に対する任意の潜在的リスクを決定し、このような審査は、臨床試験開始前のいくつかの遅延をもたらす可能性がある。NIHガイドラインは強制的ではないが,関連研究がNIH組換えや合成核酸分子研究助成を受けた機関で行われているか,あるいはその助成によって行われていない限り,多くの会社や他のNIHガイドラインに拘束されていない機関は自発的にこれらのガイドラインに従っている。NIHを介して候補製品提出案を審査する必要がない可能性があっても,政府規制機関を除いて候補製品の臨床試験を行う各機関のIBCとIRBの適用,あるいは適切な中央IRBであれば提案された臨床試験を審査·承認する必要がある。
さらに、遺伝子治療製品またはゲノム編集技術を使用して作られた製品(例えば、CRISPR-Cas 9技術を適用して開発された製品)に対する他の人の臨床試験における不利な発展、または遺伝子編集分野に対する公衆の不利な見方は、FDAおよび他の規制機関が、私たちが開発する可能性のある任意の候補製品の承認要件を修正すること、または遺伝子編集技術を使用する製品を制限することをもたらす可能性があり、両方とも、私たちの業務に実質的な損害を与える可能性がある。さらに、規制行動または個人訴訟は、私たちの研究計画または現在または将来の候補製品の開発または商業化に費用、遅延、または他の障害をもたらす可能性がある。
健康ドナーT細胞から設計された同種異体CAR−T候補製品も開発されており,ある癌を有する任意の患者への応用を目指している。CAR-T候補製品の同種異体バージョンは実証されていない開発領域であり、ドナーT細胞の品質の変異性及び患者の外国ドナー細胞に対する潜在免疫反応を理解と解決することを含む定量化が困難な特殊なリスクに直面しており、これは最終的に安全性、有効性と著者らが信頼性と一致した方法で製品を生産する能力に影響する可能性がある。例えば,P−BCMA−ALLO 1に関するFDAからの我々INDのフィードバックに応答するために,特定の同種候補製品特有の検出放出基準の更新が求められている。実施は私たちの臨床スケジュールに影響を与えていないが、それまたは同様の規制要件が将来的に影響を与えないことは保証されず、どのような遅延も、私たちの業務、財務状況、運営結果、および将来の成長見通しに実質的な悪影響を及ぼす可能性がある。
60
私たちの業務は私たちの主要候補製品の成功に強く依存している。もし私たちが臨床開発を進め、承認され、私たちの主要な候補製品を商業化することに成功し、承認適応を有する患者の治療に使用できなければ、私たちの業務は深刻な損害を受ける。
私たちの業務と将来の成功は、私たちが臨床開発を進める能力にかかっており、規制部門の承認を得て、私たちの先行候補製品を商業化することに成功しています。我々の3つの同種CAR-T候補製品P-BCMA-ALLO 1、P-MUC 1 C-ALLO 1およびP-CD 19 CD 20-ALLO 1は、安全性、有効性または耐久性の理由から、候補製品を開発する能力を阻害し、公衆および投資家の私たちの同種細胞治療計画導管の実行可能性に対する意見を含む、これらの候補製品の失敗または他の同種細胞治療計画の失敗に属するため、我々の候補製品を開発する能力を阻害する可能性がある。私たちのすべての候補製品は、私たちの主要な候補製品を含めて、より多くの臨床と非臨床開発、複数の司法管轄区域の監督審査と承認、大量の投資、引き続き十分な臨床と最終商業製造能力、そして重大なマーケティング努力を獲得して、製品販売から任意の収入を得ることができる。また、私たちの他の候補製品は、私たちのリード候補製品と同様の技術に基づいているので、任意のリード候補製品が追加のセキュリティ問題、効能問題、製造問題、開発遅延、規制問題、または他の問題に遭遇した場合、私たちの開発計画と業務は深刻な損害を受けることになります。
私たちの候補製品は開発中または承認後に深刻な有害事象、不良副作用、または他の予期しない特性が発見される可能性があり、これは私たちの臨床開発計画の中断を招く可能性があり、規制機関は私たちの候補製品の承認を拒否し、マーケティング承認後に発見されれば、マーケティング許可を取り消したり、私たちの候補製品の使用制限を取り消したりして、その候補製品の商業潜在力を制限する可能性がある。
これまで,限られた数の癌患者のみで候補製品をテストしてきたが,これらの臨床試験参加者の多くは服薬後に限られた時間しか観察していなかった。私たちが私たちの候補製品を開発し続け、他の候補製品の臨床試験を開始するにつれて、深刻な副作用、疾患の再発、または予期しない特徴が出現する可能性があり、それらの候補製品を放棄するか、またはそれらの開発をより狭い用途またはサブグループに制限することにつながり、これらの人々の中で、リスク効果の観点から、SAEまたは不良副作用または他の特徴は、それほど一般的ではなく、それほど深刻ではない、またはより容易に受け入れられ、または治療効果がより顕著または持続的である。例えば、CAR-T製品の臨床試験で観察される1つの重大なリスクは、サイトカイン放出症候群、すなわちCRSに発展し、場合によっては神経毒性および患者の死亡を引き起こすことである。本文書が提出された日まで、我々の同種遺伝子計画の臨床試験で観察されたCRSまたは神経毒性例は比較的限られているにもかかわらず、我々の既存の試験または将来のCAR−T計画のより高い用量で、これらまたは他の有害事象のより高い比率が観察される可能性がある。臨床試験でより多くのまたは重篤なCRS症例が観察されたり、その重症度に応じて他の副作用や他の予期しない発見が認められたりすると、私たちの試験は延期または停止する可能性があり、私たちの開発計画は完全に停止する可能性があります。2020年8月,われわれはP−PSMA−101試験が臨床的に放置され,1人の患者の死亡状況を評価することを発表し,P−PSMA−101の治療に関与している可能性がある。2020年11月,我々はFDAがこの事件の調査および患者のコンプライアンスと安全性を向上させるための提案案改正案に基づき,臨床棚上げを中止し,試験を再開したと発表した。同様の患者の死亡または他の試験の一時停止または終了を必要とする他の有害事象を観察することができ、これは、このような計画の重大な挫折を表す可能性がある。
著者らの候補製品が最初に早期臨床試験で希望を示したとしても、生物製品の副作用は往々にしてそれらがより大きく、より長く、より広範な臨床試験でテストを行った後にのみ検出され、場合によっては、承認後に患者に商業規模の製品を提供した後にのみ検出できる。場合によっては、重篤な不良または予期せぬ副作用が候補品または他の因子によって引き起こされるか、特に他の疾患を有し、他の薬剤を服用している可能性がある腫瘍学的対象において決定することが困難である。開発中または承認後に深刻な不良または予期しない副作用が発見され、私たちの候補製品によると決定された場合、候補製品を使用した治療の利点が各潜在的患者へのリスクよりも大きいことを確実にするために、リスク評価および緩和策を策定する必要があるかもしれないが、医療従事者とのコミュニケーション計画、患者教育、広範な患者監視または分配システムおよびプロセスが含まれている可能性があり、これらのシステムおよびプロセスは高度に制御され、制限されており、コストは業界基準よりも高い。製品に関連する副作用も可能です
61
潜在的な製品責任クレームを招く。このようなどんな状況でも、私たちの業務、財政状況、そして見通しに大きな被害を及ぼす可能性がある。
さらに、もし私たちの1つ以上の候補製品が発売許可を得て、私たちまたは他の人が後にこのような製品による不良副作用を発見した場合、多くの潜在的な重大な負の結果を招く可能性がある
否定的な世論や遺伝子編集に関する遺伝子研究や療法の規制審査の増加は、私たちの候補製品に対する大衆の見方を損なう可能性があり、あるいは私たちの業務を展開したり、規制機関が私たちの候補製品を承認してくれる能力に悪影響を及ぼす可能性がある。
私たちが使っている遺伝子編集技術は珍しいです公衆の認知は遺伝子編集が安全でないという説の影響を受ける可能性があるが,遺伝子編集を含む製品は公衆や医学界の受け入れを得られない可能性がある。これは私たちの製造努力や遺伝子編集技術が検討されたり、安全ではないとみなされる可能性がある。私たちの成功は私たちの目標疾患を専門的に研究する医師に依存し、承認されれば、私たちの候補製品を既存の、よりよく知っている治療法として代替または補充することができ、より多くの臨床データが利用可能になるかもしれない。遺伝子編集に対するいかなる否定的な見方の増加も、より少ない医師が私たちの治療法を開発することをもたらすか、または患者が私たちの治療方法を使用したり、私たちの候補製品の臨床試験に参加する意欲を低下させる可能性がある。
さらに、遺伝子編集および細胞治療技術の新規性を考慮して、政府は、これらの技術の制御を維持し、またはその使用を制限するために、輸入、輸出または他の態様を制限する可能性がある。米国または国際的には、ますます多くの負の世論またはより厳しい政府法規が、私たちの業務または財務状況に悪影響を与え、私たちの候補製品の開発および商業化、またはそのような候補製品の需要を遅延または損害する可能性がある。
臨床開発は長く、高価で不確実な過程だ。臨床前研究と早期臨床試験の結果は常に未来の結果を予測するわけではない。われわれが臨床試験に入ったどの候補製品も今後の臨床試験で有利な結果が得られない可能性があり,あれば上場承認も得られない。
薬物と生物製品の開発リスクは極めて大きい。開発過程に入った候補製品のうち、一部だけが市場承認を得た。規制部門の許可を得て私たちの候補製品を販売する前に、私たちは広範な臨床試験を行い、候補製品の人体上の安全性と有効性を証明しなければならない。臨床試験費用は高く,完成までに数年かかる可能性があり,その結果も確定していない。
我々の候補製品や他の製品の臨床前研究や早期臨床試験の結果は,同じあるいは類似した作用機序を有する製品であっても,後期臨床試験の結果を予測できない可能性がある。特に,候補製品は人体試験において予見できない安全性や有効性を示す問題は珍しくなく,臨床前動物モデルで良好な結果が得られたにもかかわらず。2020年8月、著者らはP-PSMA-101試験が臨床保留され、患者の死亡を評価することを発表した。この臨床は2020年11月に解除されました
62
患者のコンプライアンスおよび安全性を向上させるための議定書改正案が実施され、その中には、導入および排除基準の改正、およびモニタリングおよび実験室検査の頻度が含まれている。さらに、主に著者らの最初の臨床試験P-BCMA-101においていくつかの患者の抗薬物抗体が観察されたため、私たちは、30日前により小さい周期で薬物を投与し、事前適応レジメンにリツキシマブを添加して、任意の抗体反応を潜在的に阻害するような追加の用量戦略を探索した。これらの抗薬物抗体が候補産物を中和した場合、P-BCMA-101または抗薬物抗体中和候補産物の任意の他の候補産物の活性が制限される可能性がある。ある程度、私たちは、以前に評価された第1の段階キューよりも限られたデータに基づく可能性がある任意の臨床試験において、これらの他の用量戦略のうちの1つを選択して進める。P−BCMA−101,P−PSMA−101とわれわれの現在の臨床試験を除いて,われわれの候補製品はヒトで試験されていない。我々は最近,我々の最初の3つの同種異体CAR−T候補製品P−BCMA−ALLO 1,P−MUC 1 C−ALLO 1とP−CD 19 CD 20−ALLO 1の臨床試験を開始した。我々はすでにP−BCMA−ALLO 1の開発に我々の自己P−BCMA−101製品候補から学んだ知識を適用しているが,これらの知識が異遺伝子プログラムに適用されているかどうか,あるいは臨床試験では意外な結果に遭遇しないことは確認できない。著者らの候補製品の臨床前と臨床テストの未来の結果もあまり確定していない。著者らはCAR-Tと遺伝子治療開発及び関連プラットフォーム技術の方法に対して意外性と相対的にテストされていない性質を持っているからである。一般的に、臨床試験失敗は多種の要素によるものである可能性があり、研究設計、用量選択、患者登録標準及び良好な安全性或いは有効性特徴を証明できなかった。そのため、臨床試験の失敗はテストのどの段階でも発生する可能性がある。早期の試験で良好な結果が得られたにもかかわらず、治療効果や不良な安全性状況が乏しいため、生物製薬業界のいくつかの会社は臨床試験の推進において挫折した。
もし私たちの臨床試験結果が確定しなければ、あるいは私たちの候補製品に関連する安全問題や有害事象であれば、
著者らの候補腫瘍学製品の治療は化学療法と清髄治療を含み、これは副作用或いは不良事件を招く可能性があり、これらの副作用或いは不良事件は著者らの候補製品と関係がないが、依然として著者らの臨床試験の成功に影響を与える可能性がある。しかも、私たちの候補製品は他の不良事件を招くかもしれない。重篤な患者を著者らの臨床試験に入れることはこれらの患者が使用する可能性のある他の治療法或いは薬物によって死亡或いはその他の不良医療事件を招く可能性がある。上述したように、これらの事件は、私たちが規制部門の承認を得ることを阻止したり、私たちの候補製品に対する市場の受け入れ度を獲得したり、製品を商業化する能力を弱めることができます。私たちのすべての候補製品は私たちのプラットフォーム技術に由来しているので、私たちの候補製品の臨床的失敗はまた、私たちの他の候補製品が似たような故障を経験する実際または予想される可能性を増加させるかもしれない。
63
私たちの臨床試験は大きな遅延に遭遇するかもしれない。
もしあれば、どんな臨床試験も計画通りに行われるか、予定通りに完成することは保証できません。例えば、私たちがいくつかの臨床を完成する前に開発し、提出し、INDS下で行われた許可を得る前に、私たちが計画している肝臓指向遺伝子治療候補薬の第一段階の臨床試験を開始することはできない。われわれは2021年8月にFDAがP−BCMA−ALLO 1のINDを承認し,2021年12月にP−MUC 1 C−ALLO 1のINDを承認し,2023年7月にP−CD 19 CD 20−ALLO 1のINDを発表したが,臨床サイトに依存して患者を募集し続けた。われわれは2020年8月に,われわれのP−PSMA−101試験は臨床放置され,1人の患者の死亡を評価することを発表した。2020年11月、我々はFDAがこの事件の調査と患者のコンプライアンスと安全性を高めるための提案案修正案に基づいて、臨床保留を取り消したことを発表した。私たちは裁判を再開することができるが、他の裁判で似たような棚上げは裁判の最終完了を延期するかもしれない。成功を妨げたり、速やかに臨床開発を完了したりする可能性がある他のイベントには、
64
臨床前と臨床開発を成功させることができないいかなる状況も、私たちの追加コストを招き、あるいは私たちの資金を調達し、製品販売から収入を獲得し、協力手配を達成または維持する能力を弱める可能性がある。例えば、いくつかの臨床試験サービスプロトコルは、患者登録によって変化しない費用に基づく。したがって,臨床試験の登録速度が鈍化すれば,試験に関する何らかの費用は減少しないため,試験完了総コストが増加する。また、私たちの候補製品を製造変更すれば、修正された候補製品をより早いバージョンに関連付けるために追加の研究を行う必要があるかもしれません。臨床試験遅延は、候補製品を商業化する独占的な権利を持つ可能性のある任意の期限を短縮することもできますし、私たちの競争相手が私たちの前に製品を市場に出すことを可能にすることは、候補製品を商業化することに成功する能力を弱める可能性があり、私たちの業務と運営結果を損なう可能性があります。
もし私たちが臨床試験の患者登録中に遅延や困難に遭遇したら、必要な監督許可を受けることは遅延あるいは阻止する可能性があります.
FDA、EMA、または任意の他の同様の規制機関の要求に応じて、または必要に応じて所与の試験に適切な統計データを提供することができない場合、私たちまたは私たちの協力者は、私たちが決定または開発した任意の候補製品のために臨床試験を開始または継続することができず、これらの試験に参加するのに十分な数の合格者を募集することができないかもしれない。私たちが計画しているいくつかの珍しい病気には、登録は特に挑戦的かもしれない。さらに、患者が細胞治療、遺伝子治療または遺伝子編集領域に関連する有害事象の負の宣伝、患者集団のような競争的臨床試験、競合製品の臨床試験または他の理由で私たちの試験に参加したくない場合、私たちが開発する可能性のある任意の候補製品の募集患者、研究を行い、監督管理の承認を得るスケジュールが遅れる可能性がある。また,我々の競争相手のいくつかは現在も将来も候補製品に対する臨床試験を行っており,これらの候補製品は我々が開発中で将来開発可能な候補製品と同様の適応を有しており,本来われわれの臨床試験に参加する資格のある患者は競争相手の候補製品の臨床試験に参加することができる。
臨床試験患者の入選はまた、調査中の疾患の重症度を含む他の要素の影響を受ける;患者集団の大きさと患者の流れを決定する;試験方案の設計;入選患者が試験獲得が完了する前に退出するリスク;調査中の疾患に対する承認薬物の有効性;患者のインフォームドコンセントを獲得し、維持する能力;関連試験の資格と排除基準;著者らは治療期間と治療後に患者の能力を十分に監視する;および潜在患者、特に患者数が限られた疾患の臨床試験場所の隣接と利用可能性。
私たちの臨床試験の登録遅延は、私たちが開発する可能性のある任意の候補製品の開発コストを増加させる可能性があり、これはわが社の価値を低下させ、追加融資を得る能力を制限する。もし私たちまたは私たちの協力者が計画通りに臨床試験を行うのに十分な数の患者を募集することが困難な場合、私たちは進行中または計画中の臨床試験を延期、制限または終了する必要があるかもしれないが、いずれも私たちの業務、財務状況、運営結果、および見通しに悪影響を及ぼすだろう。
著者らの臨床試験中の中期、背線と初歩データは更に多くの患者データの出現に従って変化する可能性があり、そして監査と検証プログラムの影響を受け、これは最終データの実質的な変化を招く可能性がある。
著者らは時々著者らの臨床前研究と臨床試験の初歩的、中期或いは背線データを公開する可能性があり、これらのデータは当時利用可能なデータの初歩的な分析に基づいており、結果と関連する発見と結論は患者登録と治療の継続及びより多くの患者データの獲得に従って変化する可能性がある。以前の初期データまたは中間データと将来の中期または最終データとの間の不利な差は、私たちのビジネスの将来性を深刻に損なう可能性があります。臨床前研究や臨床試験完了後にTOPLINEデータを公表することも可能であり,特定の研究や試験に関するデータをより全面的に審査した後,これらのデータが変化する可能性がある。私たちはまた、私たちのデータ分析の一部として、すべてのデータを全面的かつ詳細に評価する機会がないか、または受け取る機会がないかもしれないという仮説、推定、計算、および結論を出した。したがって、追加のデータが受信され、十分に評価されると、私たちの報告書の中期、ベースライン、または予備結果は、同じ研究の将来の結果とは異なるかもしれないし、または異なる結論または考慮要因が、これらの結果を合格させる可能性がある。裏線データもまだ監査と確認手続きを受けなければならず、これは最終的な結果を招く可能性がある
65
データは私たちが以前に発表した初期データと実質的に違う。したがって、最終データが利用可能になる前に、中間データ、バックラインデータ、および予備データは慎重に見られなければならない。
さらに、規制機関を含む他の人は、私たちの仮定、推定、計算、結論または分析を受け入れないか、またはデータの重要性を異なる方法で解釈またはトレードオフする可能性があり、これは、特定の計画の価値、特定の候補製品または製品の承認または商業化、およびわが社の全体的な状況に影響を与える可能性がある。さらに、開示された特定の研究または臨床試験に関する情報を選択することは、一般に、広範な情報に基づいており、あなたまたは他の人は、私たちが開示に含めるために重要な情報または他の適切な情報として決定することに同意しない可能性がある。
私たちは最終的に私たちの候補製品が孤児薬として指定された潜在的な利点を受け取ったり認識したりしないかもしれない。
私たちは私たちの特定の候補製品のために孤児薬物の称号を求めるかもしれない。FDAは孤児を米国で20万人未満の患者を治療する稀な疾患または20万人を超える影響を与えるように指定したが、米国では治療薬の開発およびマーケティングのコストを合理的に予想する薬剤はなかった。我々は以前、再発/難治性多発性骨髄腫の治療のためのP−BCMA−101の孤児薬物名を取得したが、我々が申請すれば、将来的にP−BCMA−ALLO 1または任意の他の候補製品のこの名称を受信しないかもしれない。米国では,孤児薬を指定することで一方が臨床試験費用,税収割引,申請費の減免のために贈与資金を提供する機会が得られるような財政インセンティブを得る権利がある。FDAが孤児薬物指定を承認した後、FDAは、この薬剤の模倣薬識別情報およびその潜在的孤児用途を開示する。しかし、孤児薬物を指定することは、候補製品の開発時間や監督審査時間を短縮することも、監督審査や承認過程において候補者にいかなる利点をもたらすこともない。
さらに、1つの製品が孤児指定の適応を有するFDAの最初の承認を得た場合、この製品は、孤児薬物の排他性を得る権利があり、これは、FDAが、限定された場合、例えば孤児に対して排他的な製品に対する臨床的優位性を示すか、または製造業者が孤児患者集団に対して十分な製品数を保証することができない限り、同じ適応の同一の薬剤を販売するために、7年以内に他の申請を許可することができないことを意味する。我々または我々の協力者が孤立指定適応よりも広い適応の承認を求めると,米国での独占営業権を得ることができない可能性もあり,FDAが後に指定要求に重大な欠陥があると判断すれば,独占営業権を失う可能性がある。孤児薬の称号を得ても,医薬品開発に関する不確実性により,特定の孤児適応の上場承認を得た最初の会社ではない可能性がある。また,候補製品の孤立薬物排他性を獲得しても,この排他性は競合から製品を効率的に保護できない可能性があり,異なる薬物が同じ条件で承認される可能性があるからである。
私たちは私たちのいくつかの候補製品のために再生医学高度療法やRMAT認証を求めることができるかもしれないが、承認されても、このような認証はより速い開発や規制審査や承認過程を招くことはなく、私たちの候補製品が発売承認される可能性を増加させることはない。
FDAは2017年,21世紀治療法案の実施の一部としてRMATの指定を確立した。以下の条件に適合する研究薬物は、RMATとして指定される資格がある:(1)再生医学療法の定義に適合し、再生医学療法の定義は、細胞療法、治療用組織工学製品、ヒト細胞および組織製品、またはこのような治療または製品を使用する任意の組み合わせ製品であり、(2)深刻な疾患または状態を治療、修正、逆転または治癒することを目的としている;および(3)予備臨床証拠は、研究薬物がこのような疾患または状態を解決する可能性のある満たされていない医療需要を解決する可能性があることを示す。私たちは以前に再発/難治性多発性骨髄腫の治療のためのP-BCMA-101のRMAT称号を獲得したが、もし私たちが申請すれば、私たちは未来に任意の他の候補製品のこの称号を獲得しないかもしれない。RMATの指定は、FDAとより頻繁に会議を開いて候補製品の開発計画を検討することと、BLASおよび優先審査をスクロール審査する資格があることとを含む潜在的な利点を提供する。RMAT資格が付与された候補製品は、長期臨床的利益を合理的に予測する可能性のある代替物または中間終点に基づいて、または臨床試験を適切に拡大し、加速承認を得ることを含む、大量の場所から得られたデータに依存する資格がある可能性もある。RMAT指定の候補製品
66
FDAの判断によれば、加速された承認を得た患者は、臨床証拠、臨床研究、患者登録または他の真の証拠源(例えば、電子健康記録)を提出することによって、より大きな検証的データセットを収集することによって、または治療を承認する前に、そのような治療を受けたすべての患者を承認後に監視することによって、承認後の要求を満たすことができる。
RMAT指定は、製品承認の基準を変更することなく、そのような指定またはそのような指定された資格が審査または承認を加速させる保証もなく、承認された指示がRMAT指定によってカバーされる指示範囲よりも狭くならない保証もない。また,臨床データの出現に伴い資格基準を満たさなくなると,RMATの指定が取り消される可能性がある。
私たちの候補製品は商業化されるために広範な規制要求を満たさなければならず、どの規制承認にも、大量の追加開発費用を必要としたり、製品を商業化することに成功した能力を制限する制限や条件が含まれている可能性がある。
著者らの候補製品の臨床開発、製造、ラベル、貯蔵、記録保存、広告、販売促進、輸出入、マーケティングと流通はすべてアメリカFDAと国外市場の類似外国監督管理機関の広範な監督管理を受けている。アメリカでは、FDAの規制承認を受けるまで、私たちの候補製品の販売は許可されていません。監督管理の承認を得る過程は費用が高く、通常臨床試験開始後数年後に必要であり、しかも関連する候補製品のタイプ、複雑性と意外性及び目標適応と患者群によって大きく異なる可能性がある。候補製品の臨床開発に時間と費用が投入されているにもかかわらず、監督管理部門の承認は永遠に保証されない。
これまで、私たちは、BLAまたは他のマーケティング許可申請をFDAに提出したり、比較可能な外国規制機関に同様の薬物承認申請を提出したりしていない。加速審査要求データは、候補薬物の代替終点への影響は合理的に臨床利益を予測する可能性があり、あるいは臨床終点への影響は不可逆的な発病率或いは死亡率の影響よりもっと早く測定することができ、この影響は合理的に不可逆的な発病率或いは死亡率或いは他の臨床利益を予測する可能性があり、同時に病状の重症度、希少性或いは流行率及び代替治療の獲得可能性或いは不足を考慮する。特に,FDAは我々の候補製品に対して治療のための何らかの適応を設計する治療法を承認しており,候補製品を開発する際により多くの薬剤がこれらの適応に承認される可能性があるため,BLA提出時に候補製品の承認を加速できるかどうかを予測することは困難である。
承認された候補製品を米国または海外で商業化する前に、私たちまたは将来の潜在的パートナーは、十分かつ良好に制御された臨床試験によって大量の証拠を提供し、FDAまたは同様の外国の規制機関に、これらの候補製品がその予期される用途に対して安全かつ有効であることを満足させなければならない。私たちの候補製品の臨床前または臨床データが有望であると信じていても、これらのデータはFDAおよび同様の外国規制機関の承認を支持するのに十分ではないかもしれない。特に、新技術を使用した候補製品の識別と開発を求めているため、FDAや他の規制機関は、上場を承認する前に安全研究やモニタリングの強化を含む追加の要求を加えるリスクが増加する可能性がある。また、特定のカテゴリー製品の中のより多くの候補製品が臨床開発を通じて監督管理審査と承認に入ることに伴い、監督管理機関が必要とする可能性のある臨床データの数量とタイプは増加或いは変化する可能性がある。
FDAまたは同様の外国の規制機関は、多くの理由で、承認候補製品を延期、制限、または拒否することができる
67
海外市場については、審査手続きは国によって異なり、上記のリスクに加え、追加の製品テスト、行政審査期限、価格主管部門との合意に及ぶ可能性がある。また、ある上場薬品の安全性に対する疑問を引き起こす事件は、FDAと類似の外国監督管理機関が安全性、有効性或いは他の監督管理に基づいて新製品を審査することを考慮する時にもっと慎重であり、監督管理の承認を得る重大な遅延を招く可能性がある。
私たちが最終的に臨床試験を完了し、私たちの候補製品を商業化する承認を得ても、FDAまたは同様の外国の規制機関は、第4段階の臨床試験を含む高価な追加の臨床試験の表現に基づいて承認される可能性があり、および/またはREMSを実施する。FDAなどの外国規制機関も、適応や患者数が私たちが最初に要求したよりも限られた候補製品を承認する可能性があり、製品の商業化に成功することが必要または望ましいと考えるラベルを承認しない可能性もある。私たちの製品のメーカーやメーカーの施設も、品質管理や品質保証に関する要求、それに応じた記録やファイルのメンテナンスを含むcGMP法規の遵守が求められています。また、規制当局は、これらの製造施設を承認してから、我々の製品を生産するために使用されなければなりません。これらの施設は、cGMP法規に適合することを確実にするために、FDAや他の同様の外国規制機関の継続的な審査および定期検査を受けることになります。
適用可能な規制承認を得るか得られないかのいずれの遅延も、候補製品の商業化を延期または阻止し、我々の業務および将来性に実質的な悪影響を及ぼすであろう。
68
たとえ私たちの候補製品が規制機関の承認を得ても、私たちは持続的な義務と持続的な規制審査の制約を受けることになり、これは多くの追加費用を招く可能性がある。また、私たちの候補製品が承認されれば、ラベルや他の制限や市場撤退を受ける可能性があり、規制要求を遵守していない場合や私たちの製品が予期せぬ問題に遭遇した場合、私たちは罰を受けるかもしれません。
FDA、EMA、または任意の他の同様の規制機関が私たちの任意の候補製品を承認する場合、製品の製造プロセス、ラベル、包装、流通、有害事象報告、貯蔵、広告、販売促進および記録は、広範かつ持続的な規制要件の制約を受ける。これらの要件には,我々が承認後に行った任意の臨床試験に対して安全および他の上場後の情報や報告,登録要件の提出,cGMPやGCPの遵守を継続することが含まれている。その後、予想されなかった重症度または頻度の不良事件、または私たちの臨床製造施設、第三者製造業者または製造プロセス、または規制要求を遵守できなかったことを含む以前の未知の製品問題が発見され、他を除いて、以下のようになる可能性がある
さらに、もし私たちの候補製品が承認されれば、私たちの製品ラベル、広告、販売促進は規制要求と持続的な規制審査を受けるだろう。FDAは生物製薬製品を販売促進する可能性のある声明を厳格に規制している。特に、製品は、当該製品が承認されたラベルに反映されるように、FDA承認されていない用途に使用されてはならない。
政府は違法の疑いのあるいかなる調査にも対応するために多くの時間と資源を必要とする可能性があり、否定的な宣伝が生じる可能性がある。上記のいずれかの事件や処罰が発生すると、私たちまたは私たちの協力者が私たちの候補製品を商業化する能力を抑制し、私たちの業務、財務状況、および運営結果を損なう可能性があります。
さらに、FDA、EMA、および他の同様の規制機関の政策は変化する可能性があり、私たちの候補製品に対する規制承認を阻止、制限、または延期するための追加の政府法規が公布される可能性がある。もし私たちが既存の要求の変化に適応できない場合、あるいは新しい要求や政策を採用することができない場合、または私たちがコンプライアンスを維持できない場合、私たちは私たちが得る可能性のあるマーケティング承認を失う可能性があり、これは私たちの業務、将来性、利益を達成または維持する能力に悪影響を及ぼすだろう。
米国や海外の将来の立法や行政や行政行動によって生じる可能性のある政府規制の可能性、性質、程度も予測できない。私たちが既存の要求の変化に緩やかに適応できない場合、あるいは新しい要求を採用することができない場合、またはコンプライアンスを維持できない場合、得られたマーケティング承認が失われる可能性があり、利益を達成したり維持することができない可能性があります。
資金不足や世界的な健康問題によるFDAや他の政府機関の中断は、重要な指導部や他の人員の採用、保留または配置の能力を阻害する可能性があり、または新たなまたは修正された製品がタイムリーに開発、承認または商業化されることを他の方法で阻止することは、私たちの業務に悪影響を及ぼす可能性がある。
FDAが新製品を審査·承認する能力は、政府予算と資金レベル、法律、法規と政策の変化、FDAの採用と保留の能力を含む様々な要素の影響を受ける可能性がある
69
キーパーソンは、ユーザ料金の支払い、およびFDAが日常的な機能を履行する能力に影響を与える可能性のある他のイベントを受け入れる。したがって、その機関の平均検討時間は近年変動している。また,他の研究開発活動を援助する政府機関への政府の援助は政治過程の影響を受けており,この過程は本質的に不安定で予測不可能である。FDAおよび他の機関の中断は、必要な政府機関によって新生物製品が審査および/または承認されるのに要する時間を遅らせる可能性もあり、これは私たちの業務に悪影響を及ぼすだろう。例えば、過去数年間、2018年12月22日から35日間を含めて、米国政府は何度も閉鎖されており、FDAのようないくつかの規制機関は、FDAのキー従業員を休暇させ、キー活動を停止しなければならない。
また、全世界の新冠肺炎流行に対応するため、アメリカ食品と薬物管理局は2020年の間に製造施設と製品の大部分の国内外の検査を数ヶ月延期し、リスクに基づいて検査を回復し、遠隔監視方法を採用した。新冠肺炎疫病に対して、アメリカ以外の監督管理機関も類似した制限と政策措置を取った。政府が長期的に停止している場合、または世界的な健康問題がFDAまたは他の規制機関の定期検査、審査または他の規制活動を阻害する場合、FDAまたは他の監督管理機関が私たちの規制提出を適時に審査し、処理する能力に深刻な影響を及ぼす可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。
私たちは、より利益的または成功可能性の高い候補製品または適応を利用することなく、特定の候補製品または適応を追求するために限られたリソースを費やす可能性がある。
私たちの財務と管理資源が限られているため、私たちは私たちの研究計画を優先し、私たちの発見と開発は選定された候補製品と適応に重点を置く必要がある。潜在的候補製品の広さと我々のプラットフォーム技術を用いて追求できると信じている兆候により,我々の研究·開発活動の優先順位を正確に決定することが我々にとって特に重要である.したがって、私たちは他の候補製品を探す機会を放棄または延期するか、または後により大きな商業潜在力を有することが証明された他の兆候を探す機会を放棄または延期するかもしれない。私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。私たちの現在と未来の研究開発計画および特定の適応候補製品への支出はいかなる商業的に実行可能な製品も発生しないかもしれない。特定の候補製品の商業的潜在力や目標市場を正確に評価していなければ、協力、許可、または他の印税手配によって候補製品に価値のある権利を放棄することも可能であり、この場合、私たちは候補製品の独占的な開発と商業化の権利を維持した方が有利である。
私たちが未来にもっと多くの候補製品を発見したり発見したりする努力は成功しないかもしれない。
我々の研究計画は当初、潜在的な候補製品の決定に希望を示す可能性があるが、様々な原因で臨床開発のための候補製品を生成できなかった
新製品候補製品を確定する研究プロジェクトには大量の技術、財力と人的資源が必要である。臨床前と臨床開発のために適切な他の候補を見つけることができなければ、治療製品の開発と商業化に成功する機会は限られるだろう。
70
製造、商業化、第三者依存に関するリスク
著者らは第三者に依存して臨床試験を行い、そしていくつかの研究と臨床前研究を行った。これらの第三者が契約の責務を満足に履行できない場合、または予期された期限内に完了できなかった場合、私たちの開発計画は、コストを延期または増加させる可能性があり、いずれも、私たちの業務および将来性に悪影響を及ぼす可能性がある。
私たちは自分で前臨床試験や臨床試験のすべての側面を行うことができない。したがって,我々が行っている臨床試験や我々の候補製品の任意の将来の臨床試験を第三者に依存して行い続ける予定である。具体的には,CRO,臨床研究者,コンサルタントはこれらの試験の進行とその後のデータ収集と分析に重要な役割を果たしている。しかし、私たちは彼らの活動のすべての側面を統制できないだろう。しかし、私たちは私たちのすべての実験が適用された合意と法律、法規、そして科学的基準に従って行われ、私たちのCROと他の第三者への依存が私たちの規制責任を免除しないことを確実にする責任がある。私たちと私たちのCROは、FDA、欧州経済圏加盟国の主管当局、および同様の外国規制機関によって、私たちの臨床開発におけるすべての候補製品に対して実行される法規およびガイドラインであるGCP要求を遵守しなければならない。監督管理機関は定期的に試験スポンサー、臨床試験研究者、臨床試験地点を検査することによって、これらのGCP要求を実行する。もし私たちまたは私たちの任意のCROまたは臨床試験サイトが適用されたGCP要件を遵守できなかった場合、私たちの臨床試験で生成されたデータは信頼できないと考えられる可能性があり、FDAまたは同様の外国の規制機関は、私たちの上場申請を承認する前に追加の臨床試験を行うことを要求するかもしれない。また,われわれの臨床試験はcGMP法規により生産された製品を用いて行わなければならない。もし私たちがこれらの規定を守らなければ、私たちに臨床試験を停止および/または繰り返すことを要求するかもしれません。これは上場承認過程を延期します。
私たちが依存しているどのようなCRO、臨床試験研究者、または他の第三者が、私たちの開発活動に十分な時間および資源を投入するか、または契約の要求に従ってタスクを実行することは保証されない。これらの第三者のいずれかが予想される最終期限内に完了できなかった場合、私たちの臨床プログラムを遵守したり、法規要件を満たしたり、他の方法で目標を達成しなかったり、私たちとの協力を終了したりすることができない場合、私たちの開発計画のスケジュールは延長または延期される可能性があり、または私たちの開発活動は一時停止または終了する可能性がある。私たちの任意の臨床試験サイトが任意の理由で終了した場合、私たちは、これらの被験者を別の合格した臨床試験サイトに移すことができない限り、そのような臨床試験に参加した被験者の後続情報を失う可能性がある。また、私たちの臨床試験の臨床試験調査員は時々私たちの科学顧問や顧問を担当し、このようなサービスによって現金や株式補償を受ける可能性がある。これらの関係および任意の関連賠償が感知または実際の利益の衝突をもたらし、またはFDAまたは任意の同様の外国の規制機関が、財務関係が試験の解釈に影響を及ぼす可能性があると結論した場合、適用される臨床試験場所で生成されたデータの完全性が問われる可能性があり、臨床試験自体の効用が脅かされる可能性があり、これは、FDAまたは任意の同様の外国規制機関によって提出された任意のマーケティング申請が延期または拒否される可能性がある。そのような遅延や拒否は私たちが候補製品を商業化することを防ぐことができる。
さらに、これらの第三者は、他のエンティティと関係がある可能性もあり、その中のいくつかは私たちの競争相手である可能性がある。これらの第三者が規制要件や私たちが規定した規程に従ってその契約責任を成功的に履行し、予想される期限内に私たちの臨床試験を完了または行うことができない場合、私たちは候補製品のマーケティング承認を得ることができないか、遅延する可能性があり、私たちの製品の商業化に成功する努力を遅らせることができないか、または延期することができません。
私たちは臨床製造工場を運営しており、私たちのすべてのCAR-T候補製品のために臨床前と臨床材料を開発と製造しており、これには大量の資源が必要である。もし著者らの臨床製造施設が成功に運営できなかった場合、重大な遅延を招く可能性があり、著者らの研究開発努力(臨床試験を含む)及び著者らのCAR-T候補製品の将来の商業可能性(承認されれば)に不利な影響を与える。
私たちの臨床製造施設は検証され、合格され、全面的に運営されている。我々は引き続き外部CMOから原材料を調達するが,外部CMOからわれわれの臨床製造施設に移行しており,われわれの臨床製造施設はP−BCMA−ALLO 1,P−MUC 1 C−ALLO 1,P−CD 19 CD 20−ALLO 1を含む臨床試験の臨床材料の唯一の供給源となることが予想される。この単一の供給源依存はCAR-T製品の数量不足のリスクを増加させます
71
許容可能なコストまたは品質の候補者では、これが承認されれば、我々の開発または商業化の努力を阻止または損害する可能性がある。もし私たちの臨床製造施設で十分な臨床前または臨床材料を生産できなければ、外部CMOとの契約を余儀なくされるかもしれませんが、もし本当にそうすれば、商業的に合理的な条項ではできないかもしれません。商業的に合理的な条項があっても,われわれの臨床製造施設から外部CMOへのどの製造移行も非常に時間がかかる可能性があり,合格した代替品の数が限られているため,多くの努力と専門知識が必要である。場合によっては、我々のCAR−T候補製品を製造するために必要な技術的スキルまたは技術は、唯一無二または独自である可能性があり、私たちは、そのようなスキルまたは技術を別のCMOに移すことが困難である可能性があり、実行可能な代替案が存在しない可能性がある。もし私たちが私たちの臨床製造施設で生産できなかった場合、あるいは適用された規格に従って直ちにCMOから十分な臨床材料を得ることができなかった場合、私たちの研究と開発は、臨床試験、私たちのCAR-T候補製品の将来の商業可能性(承認されれば)、ならびに私たちの業務、財務状況、運営結果、および成長の見通しは重大な不利な影響を受ける可能性がある。
私たちまたは私たちは、臨床前試験および臨床試験のためのいくつかの候補製品を製造し、供給する第三者は、品質と数量の両方が満足できる候補製品の供給を確立または維持できない可能性がある。
私たちの研究室で生産された製品の数は相対的に少なく、私たちの研究計画の評価に使われている。私たちは引き続き第三者に依存して、私たちのいくつかの候補製品を生産して臨床前と臨床テストを行い、もし私たちの任意の候補製品が承認されれば、私たちはこれらの第三者に依存して商業生産を行うことができるかもしれない。私たちの現在の製造スケジュールは限られており、予測可能な未来に、私たちのすべての候補製品は単一ソースのサプライヤーのみによって提供されると予想される。このような依存は、承認されれば、十分な数の候補製品や製品がない、または許容可能なコストまたは品質で十分な数の製品を得ることができなくなり、私たちの開発または商業化努力を延期、阻止、または損害する可能性があるというリスクを増加させる。
さらに、臨床試験または商業販売のための治療薬の準備に参加するすべての実体は、私たち自身と既存の候補製品契約メーカーを含めて、広く規制されている。商業販売または臨床試験のための完成治療製品の使用が許可された成分は、cGMP要求に従って生産されなければならない。これらの規定は、調査製品及び承認販売された製品の品質を制御及び確保するために、記録保存、並びに品質システムの実施及び実行を含む生産プロセス及びプログラムを管理する。生産過程の不良制御は汚染物質の導入を招く可能性があり、あるいは私たちの候補製品の性能や安定性に意外な変化をもたらす可能性があり、これらの変化は最終製品テストでは検出できない可能性がある。我々または私たちの契約製造業者は、BLAをサポートするすべての必要なファイルをタイムリーに提供しなければならず、FDAがその施設検査計画によって実行されるFDAの良好な実験室動作仕様およびcGMP規定を遵守しなければならない。似たような外国の規制機関は似たような要求を遵守することを要求するかもしれない。我々の施設及び品質システム、並びに我々の第三者契約メーカーの施設及び品質システムは、適用される法規に適合するために、承認前の検査を通過しなければならず、私たちの候補製品上場承認の条件としなければならない。私たちは契約メーカーの生産活動をコントロールせず、契約メーカーがcGMP法規を遵守することに完全に依存している。
もし私たちのどの製造業者も、そのような要求を遵守できなかった場合、または品質、時間、または他の側面での私たちの義務を履行できなかった場合、または私たちのコンポーネントまたは他の材料の供給が他の理由で制限または中断された場合、私たちは自分で材料を製造することを余儀なくされるかもしれませんが、私たちは能力や資源がないか、または他の第三者と合意することができず、私たちは商業的に合理的な条項でそうすることができないかもしれません。特に、私たちのメーカーのどの交換にも大量の努力と専門知識が必要であり、合格した交換数は限られている可能性があるからです。場合によっては、我々の候補製品を製造するために必要な技術的スキルまたは技術は、元の製造業者固有または独自である可能性があり、そのようなスキルまたは技術を他の第三者に譲渡することは困難である可能性があり、実行可能な代替案が存在しない可能性がある。また、私たちのいくつかの候補製品と私たち自身の独自の方法は、会社以外で生産または実施されたことがないので、これらの候補製品や方法のための新しい第三者製造計画を構築しようとすれば、私たちの開発計画は遅延に遭遇する可能性があります。これらの要素は、私たちのこれらの製造業者への依存を増加させ、あるいは他の第三者が私たちの候補製品を生産するために、これらのメーカーからライセンスを取得することを要求するだろう。もし…
72
どんな理由でも、私たちはメーカーを要求したり、自発的に交換したりして、新しいメーカーの施設やプログラムが品質基準およびすべての適用される法規やガイドラインに適合しているかどうかを確認するように要求されます。新メーカーの検証に関する遅延は、タイムリーまたは予算内で候補製品を開発する能力に悪影響を及ぼす可能性があります。
私たちまたは第三者が私たちの製造要件を実行できなかった場合、またはcGMP製造基準を遵守しない場合、様々な態様で私たちのビジネスに悪影響を及ぼす可能性があります
遺伝子工学製品を製造することは複雑で、私たちまたは私たちの第三者メーカーは生産中に困難に直面するかもしれない。もし私たちまたは私たちの任意の第三者メーカーがそのような困難に遭遇した場合、承認されれば、臨床試験に候補製品を提供したり、患者に製品を提供する能力が延期または阻止される可能性がある。
遺伝子工学製品の製造は複雑であり,革新的な技術を用いて生細胞を処理する必要があるかもしれない。これらの製品を製造するには,この目的のために設計·検証するための施設が必要であり,複雑な品質保証と品質制御プログラムが必要である。製造過程中の任意の場所の微小な偏差は、充填、ラベル、包装、貯蔵と輸送及び品質管理とテストを含み、すべてロット故障、製品のリコール或いは変質を招く可能性がある。生産過程が変化した時、著者らは臨床前と臨床データを提供することを要求される可能性があり、このような変化前後の製品の比較性、強度、品質、純度或いは効力を示す。製造施設で微生物、ウイルス、または他の汚染が発見された場合、これらの施設は、汚染を調査および修復するために長い時間閉鎖する必要がある可能性があり、臨床試験を延期し、私たちの業務に悪影響を及ぼす可能性がある。バイオ由来成分の使用はまた、感染またはアレルギー反応、または可能な汚染のために製品施設を閉鎖することを含む危害疑惑を引き起こす可能性がある。
そのほか、臨床試験或いは商業規模の大規模生産はまだ関連リスクが存在し、その中にコスト超過、技術拡大の潜在問題、技術再現性、安定性問題、良好な生産実践に符合し、ロット一致性と原材料の適時可獲得性などが含まれる。我々の任意の候補製品が市場承認を得ても、当社または我々のメーカーがFDAまたは他の同様の外国規制機関が許容可能な規格に従って承認された製品を生産して、その製品の潜在的な商業投入または将来の潜在的需要を満たすのに十分な数の製品を生産することができる保証はない。もし私たちや私たちのメーカーが臨床試験や商業化のために十分な数を生産できなければ、私たちの開発と商業化の努力が損なわれ、私たちの業務、財務状況、運営結果、成長の見通しに悪影響を及ぼすだろう。
候補製品の製造方法を変更することは追加的なコストや遅延を招く可能性がある。
候補製品が前臨床試験から後期臨床試験まで、そして発売承認と商業化に伴い、開発計画の各方面、例えば製造方法は、この過程でよく変化し、生産量の最適化、生産ロットの最適化、最大限にコストを下げ、そして一致した品質と結果を実現するために努力する。このような変化はこのような期待された目標を達成できない可能性がある。これらの変化のいずれも私たちの候補製品の表現が異なり、計画中の臨床結果に影響を与える可能性があります
73
改変された材料を用いた試験や他の将来の臨床試験。これは臨床試験の完成を遅らせる可能性があり、移行臨床試験或いは1つ以上の臨床試験を繰り返し、臨床試験コストを増加させ、著者らの候補製品の承認を延期し、そして著者らの候補製品を商業化し、収入を創造する能力を脅かす。
いかなる承認された製品も医師、患者、病院、癌治療センター、医療保健支払人と医学界の他の人が商業成功に必要な市場受容度を達成できない可能性がある。
もし私たちのすべての候補製品が市場の承認を得たら、それらはまだ医者、患者、医療保険支払人と医学界の他の人の十分な市場受容度を得ることができないかもしれない。例えば,現在の癌治療法,例えば化学療法や放射線治療は,医学界では成熟しており,医師はこれらの治療法に依存し続けている可能性がある。私たちの候補製品の多くは、現在限られているか、または承認されていない製品のメカニズムを対象としており、これは、医師、患者、支払人の採用が遅い速度をもたらす可能性がある。もし私たちの候補製品が十分な受容度に達していなければ、私たちは著しい製品収入が生じないかもしれません。私たちは利益を上げることができないかもしれません。もし私たちの候補製品が商業販売に使用されることが許可されれば、市場の受け入れ度は多くの要素に依存するだろう
不利な価格設定法規や第三者保険と精算政策のため、私たちの候補製品を商業化することに成功できないかもしれません。これは私たちの候補製品を利益的に販売することを難しくするかもしれません。
政府または他の第三者支払人から製品の保証と精算承認を得ることは時間がかかり、高価な過程であり、結果は不確定であり、これは支払人に私たちの製品使用を支援する科学的、臨床的、費用効果的なデータを提供する必要があるかもしれない。新たに承認された製品については、このような保険や精算を得る上で大きな遅延が生じる可能性があり、保険は利用できない可能性があり、あるいはFDAや同様の外国規制機関がこの製品を承認する目的よりも限られている可能性がある。さらに、保険および精算を受ける資格があることは、製品がすべての場合に支払いを受けることを意味するのではなく、研究、開発、知的財産権、製造、販売、および流通費用を含む、私たちのコストをカバーするレートで支払うことを意味するわけではない。新製品の臨時精算水準(適用すれば)も私たちのコストを支払うのに十分ではない可能性があり、恒久的にならない可能性があります。販売率は製品の使用や臨床環境によって異なる可能性があり,すでに低コスト製品のために設定された精算レベルに基づいている可能性があり,他のサービスの既存支払いにも組み込まれている可能性がある。製品の正味価格は、政府医療計画または個人支払者が要求する強制的な割引またはリベート、将来の任意の薬品価格を制限する法律、および将来的に米国価格よりも低い価格で製品が販売される国からの輸入を制限する法律の任意の緩和によって低下する可能性がある。
新たに承認された製品の保険カバーと精算に関する不確実性が大きい。米国では、第三者支払者は保険や精算に統一された政策を持っていない。第三者支払者は精算戦略を設定する際には通常Medicare保証政策と支払い制限に依存するが、Medicare保証と精算確定以外にも、彼ら自身の方法と承認の流れがある。したがって、第三者支払者は、1つの商品に保険を提供することを決定し、他の支払者もその商品に保険を提供することを保証することはできない。
74
第三者支払者の製品使用状況の決定を含む第三者支払人の保証範囲と精算は多くの要素に依存する可能性がある
私たちは商業化されたどの製品も精算できることを確実にすることはできません。保険と精算があれば、精算レベルはいくらですか。私たちは政府の資金援助と個人支払人から私たちが開発してくれた任意の承認された製品から保険と十分な販売率を迅速に得ることができません。これは私たちの経営業績、製品の商業化に必要な資金を調達する能力、そして私たちの全体的な財務状況に実質的な悪影響を及ぼすかもしれません。
精算は私たちが市場で承認されたどんな製品の需要と価格に影響を及ぼすかもしれない。第三者支払者が特定の製品のために保険を受けていると仮定すると,それによる精算支払率が十分に高くない可能性があり,あるいは患者が受け入れられないと考えて受け入れられないほど高い共同支払いが必要となる可能性がある。自分の病態を治療するために処方薬を服用している患者とその処方を行う医師は,通常第三者支払者に依存してこれらの薬物に関する費用の全部または一部を精算する。患者は保険を提供しなければ、私たちの製品を使用することはあまりできません。私たちの製品の全部または大部分のコストを支払うのに十分な費用を精算するのに十分です。そのため、保証範囲と十分な精算は新製品の受容度に重要である。カバー範囲の決定は臨床および経済基準に依存する可能性があり、より成熟またはより低コストの治療代替製品が利用可能になった場合、またはその後に利用可能である場合、これらの基準は新製品に不利である。
医師の監督下で管理されている製品については、このような薬物が高い価格に関連することが多いため、保険および適切な補償を得ることは特に困難である可能性がある。しかも、製品自体が単独で精算されるかもしれないし、できないかもしれない。逆に、病院や主管医は、私たちの製品を使った治療や手続きを提供することで精算されるかもしれません。また、医療保険·医療補助サービスセンター(CMS)は、連邦医療保険医料金表や病院外来予想支払制度を含む医療保健提供者への精算のための精算制度を時々改正し、医療保険支払いを減少させる可能性がある。
私たちは、管理型ヘルスケアの傾向、ヘルスケア組織の日々の影響力、および追加の法的変化により、私たちの任意の候補製品の販売に関する定価圧力に直面することを予想しています。全体的に,医療コストの下り圧力は非常に大きくなり,特に処方薬,医療機器,外科手術プログラム,その他の治療が行われている。そのため、新製品の商業化成功にますます高い壁が設けられている。さらに、任意の将来の政府コスト制御や他の医療改革措置を実施することにより、私たちが受け取る可能性のある任意の承認された製品の価格に追加の下振れ圧力を与える可能性がある。
また、私たちまたは私たちの協力者は、私たちの候補製品と一緒に使用するセット診断テストを開発するかもしれません。私たちまたは私たちの協力者は、私たちの製品候補者のために求められるかもしれない保険と精算を除いて、これらのテストのために単独で保険と精算を受けることを要求されるだろう。私たちはまだ候補製品のためのセットの診断テストを開発していませんが、もし私たちが開発すれば、私たちの候補製品に適用されるのと同じ理由で、私たちが保険と十分な補償を受ける能力には大きな不確実性があります。
米国以外では、多くの国が製品の販売価格が承認されてから発売されることを要求しているが、定価審査期間はマーケティングや製品の許可を得てからのみ開始される。その中のいくつかの国で精算或いは定価の承認を得るために、私たちは臨床試験を行い、私たちの候補製品の費用効果を他の利用可能な治療法と比較する必要があるかもしれない。一部の外国市場では、処方薬の定価は初歩的な承認を得た後も、政府の持続的なコントロールを受けている。そのため、ある候補製品の特定の国/地域でのマーケティング承認を得ることができますが、その後
75
価格規制の制約を受けて、これらの規制は私たちの製品の商業発表を延期し、私たちがその国で製品を販売することによって生じる収入(あれば)に悪影響を及ぼすかもしれません。不利な価格設定制限は、これらの候補製品が市場承認を得ても、1つ以上の候補製品への投資を回収する能力を阻害する可能性がある。
私たちは承認されたバイオ製品候補製品が予想以上に早く競争に直面する可能性があることを求めるつもりだ。
2010年3月23日に法律として署名された“患者保護および平価医療法案”、または総称して2010年3月23日に法律となる“平価医療法案”に署名された“平価医療法案”は、“2009年生物製品価格競争および革新法案”、またはBPCIAという副題を含み、FDA許可の参考生物製品生物と類似または交換可能な生物製品のための短い承認経路を作成する。BPCIAによると,生物類似製品の申請は,参考製品が初めてFDA許可を得た4年後にFDAに提出されなければならない。また,FDAによる生物類似製品の承認は,参考製品が初めて許可された日から12年後に発効する可能性がある。この12年間の独占期間内に、FDAが競合製品の完全なBLAを承認した場合、スポンサー自身の臨床前データおよび十分かつ良好に制御された臨床試験からのデータを含み、その製品の安全性、純度および有効性を証明するために、別の会社は依然としてこの参照製品の競合バージョンを販売する可能性がある。
BLAによりバイオ製品として承認されたいずれの我々の候補製品も12年の専門期間を得る資格があるべきであると考えられる。しかしながら、国会の行動または他の理由により、この排他性は短縮される可能性があり、またはFDAは、私たちの製品候補製品を競合製品の参考製品とみなさず、予想よりも早く後発薬競争の機会を創出する可能性がある。BPCIAの他の面では,そのいくつかがBPCIAの排他的条項に影響を与える可能性があり,最近の訴訟のテーマでもある.さらに、承認されると、生物類似体が私たちのいずれかの参考製品をどの程度置換するかは、非生物製品の伝統的な模造薬代替と類似しており、これはまだ発展中のいくつかの市場および規制要因に依存するかどうかは不明である。
もしすべての承認された製品が私たちが予想していたより早く生物類似競争を受けたら、私たちは巨大な価格設定圧力に直面し、私たちのビジネス機会は制限されるだろう。
もし私たちのすべての候補製品の市場機会が私たちが思っているより小さいなら、私たちの収入は不利な影響を受けるかもしれません。私たちの業務は影響を受けるかもしれません。
私たちは最初に癌治療法の開発に集中した。我々の候補製品治療から利益を得る可能性のある潜在的な患者集団の予測は推定に基づいている。もし私たちのいかなる推定も正確でなければ、私たちの任意の候補製品の市場機会は著しく減少し、私たちの業務に悪影響を及ぼす可能性がある。
私たちの第三者への依存は、私たちのビジネス秘密を共有することを要求し、これは、競争相手がこれらの秘密や私たちのビジネス秘密が流用または開示されていることを発見する可能性を増加させる。
私たちは第三者に依存して私たちの候補製品を開発して製造するので、私たちは彼らとビジネス秘密を共有しなければならない。独自の情報の研究または開示を開始する前に、当社のコンサルタント、従業員、第三者請負業者およびコンサルタントと秘密協定、材料譲渡協定、コンサルティング協定、または他の同様の合意を締結することによって、当社のノウハウを部分的に保護することを求めています。これらの協定は、一般に、私たちの商業秘密を含む、第三者が私たちの機密情報を使用または開示する権利を制限する。第三者と協力する際に契約条項が採用されているにもかかわらず、商業秘密および他の機密情報を共有する必要は、そのような商業秘密が私たちの競争相手に知られ、無意識に他の人の技術に組み込まれているか、またはこれらの合意に違反した場合に開示または使用されるリスクを増加させる。私たちの独自の地位が私たちのノウハウおよびビジネス秘密にある程度基づいていることを考慮すると、競争相手は、私たちのビジネス秘密または他の許可されていない使用または開示を独立して発見することは、私たちの競争地位を損なうことになり、私たちのビジネスに大きな悪影響を及ぼす可能性があります。
76
さらに、これらの合意は、一般に、私たちの合意がいくつかの限られた発行権を含む可能性があるにもかかわらず、私たちのコンサルタント、従業員、第三者請負業者、およびコンサルタントが、私たちのビジネス秘密に関連する可能性のあるデータを発行する能力を制限します。例えば、私たちが協力する可能性のある任意の学術機関は、そのような協力によって生成されたデータを発表する権利を付与されるかもしれません。任意の共同研究開発プロジェクトは、私たちの研究開発または同様の合意の条項に従って商業秘密を共有することを要求するかもしれません。私たちは私たちのビジネス秘密を保護しようと努力しているにもかかわらず、私たちの競争相手は、私たちと第三者との合意に違反したり、独立して開発したり、私たちの任意の第三者協力者が情報を発表することによって、私たちのビジネス秘密を発見するかもしれない。競争相手は私たちのビジネス秘密が私たちの競争地位を損なうことを発見し、私たちの業務に悪影響を及ぼすだろう。
もし私たちの候補製品がマーケティングおよび商業化のために承認され、販売およびマーケティング能力を確立することができない場合、または第三者と合意して私たちの候補製品を販売およびマーケティングすることができなければ、私たちの候補製品が承認された場合、私たちはそれを商業化することに成功できないだろう。
私たちは販売、マーケティング、流通能力や経験がありません。私たちが販売とマーケティング責任を保留する任意の承認された製品を商業的に成功させるためには、高価で時間がかかるか、これらの機能を他の第三者にアウトソーシングする販売とマーケティング組織を構築しなければならない。将来、私たちのいくつかの候補製品が承認されれば、私たちのいくつかの候補製品を販売したり、私たちの協力者と一緒に販売活動に参加したりするために、重要な販売·マーケティングインフラを構築することを選択するかもしれません。
私たち自身の販売とマーケティング能力の確立、および第三者とこれらのサービスを実行する計画を達成することはリスクに関連している。例えば、販売チームの採用と訓練は高価で時間がかかり、どんな製品の発表も延期される可能性がある。販売チームを募集し、マーケティング能力を確立する候補製品の商業発表が何らかの理由で延期または発生していない場合、これらの商業化費用を早期または不必要に発生させる。これは費用が高いかもしれません。もし私たちが私たちの販売とマーケティング担当者を維持したり再配置できなければ、私たちの投資は損失します。
将来の製品の商業化を阻害する可能性があります
もし私たちが第三者と販売、マーケティング、流通サービスの手配を達成すれば、私たちの製品収入またはこれらの製品収入は私たち自身が開発した任意の製品をマーケティングし、販売する場合よりも低いかもしれません。さらに、私たちは第三者と私たちの候補製品の販売とマーケティングの手配を成功的に達成できないかもしれないし、私たちに有利な条項でそうすることができないかもしれません。第三者のマーケティングや流通手配を達成する際に、私たちが得た任意の収入は、第三者の努力に依存し、これらの第三者が十分な販売および流通能力を確立するか、または必要なリソースを投入し、任意の将来の製品を効果的に販売およびマーケティングすることを保証することはできません。もし私たちが販売とマーケティング能力を確立することに成功できなければ、私たち自身も第三者と協力しても、私たちの候補製品を商業化することに成功しません。
たとえ私たちの候補製品がFDAの承認を得ても、私たちは決してアメリカ以外で承認されたり、そのような製品を商業化したりすることはありません。これは、私たちがそのすべての市場潜在力を達成する能力を制限するだろう。
米国以外の市場で任意の製品を販売するためには、安全性と有効性に関する他国の多くの規制要件を確立し、遵守しなければならない。一国で行われる臨床試験は、他の国の規制部門に受け入れられない可能性があり、一国で規制の承認を得ることができる
77
他のどの国でも規制の承認を受けることを意味するのではない。承認手続きは国によって異なり、追加の製品テストと検証、および追加の行政審査期限が含まれる可能性があります。外国の監督管理機関の承認を求めることは著者らの重大な遅延、困難とコストを招く可能性があり、追加の臨床前研究或いは臨床試験を必要とする可能性があり、これは高価で時間がかかるだろう。各国の規制要求には大きな違いがある可能性があり、私たちの製品がこれらの国で発売されることを延期または阻止する可能性がある。これらと他の規制要件を満たすことは高価で、時間がかかり、不確定であり、予期しない遅延が生じる可能性がある。また、私たちはどの国でも規制承認を得ることができず、他の国の規制承認過程を延期したり、マイナス影響を与える可能性がある。私たちは国際市場を含めてどの司法管轄区でも候補製品の販売を承認しておらず、国際市場で規制承認を受けた経験もありません。もし私たちが国際市場の規制要求を守らない場合、あるいは必要な承認を得て維持できなければ、私たちが製品全体の市場潜在力を実現する能力は損なわれるだろう。
私たちのライセンス内の他の戦略的合意に関連するリスク
私たちは現在、いくつかの許可内協定に署名しており、これらの合意に基づいて、私たちは、私たちのいくつかのプラットフォーム技術および最終候補製品を使用、開発、製造、および/または商業化する権利を得ている。もし私たちがこれらの合意の下の義務に違反したら、私たちは損害賠償金の支払いを要求されるかもしれません。これらの技術に対する私たちの権利を失ったり、両方を持っていたりすることは、私たちの業務と将来性に悪影響を及ぼすでしょう。
私たちは開発と商業化活動の勤勉義務、あるマイルストーンを実現する支払い義務、製品販売の特許権使用料、消極的な契約、その他の実質的な義務を含む様々な義務を負わせるライセンスと他の戦略的合意にある程度依存している。例えば、P-BCMA-ALLO 1、P-CD 19 CD 20-ALLO 1、P-PSMA-ALLO 1については、TeneoBio,Inc.(安進の子会社)またはTeneoBio、P-MUC 1 C-ALLO 1とのプロトコルに従って重鎖のみの接着剤を許可しており、Xyone Treateutics,Inc.(Genus Oncology,LLCの利益継承者)またはXyoneとのプロトコルにより、追加の二輪車計画および他の同種遺伝子臨床計画を許可し、TeneoBioとのプロトコルにより、接着剤の許可を継続することが可能である。P-BCMA-ALLO 1、P-MUC 1 C-ALLO 1、P-CD 19 CD 20-ALLO 1、および将来の同種製品を製造するためのCas-Clover遺伝子編集技術について、私たちはHelmholtz-Zentum München-Deutsches Forschungszentrum für Gesundeit und UmWelt GmbHとの合意に基づいていくつかの知的財産権を付与した。もし私たちがライセンス契約の義務を履行できなかったり、許可されていない方法で私たちに許可された知的財産権を使用できなかった場合、私たちは損害賠償金の支払いを要求される可能性があり、私たちの許可者は許可を終了する権利があるかもしれない。私たちのライセンス契約が終了した場合、私たちは、そのような製品と一緒にテストまたは承認された製品を開発、製造、マーケティング、または販売プロトコルでカバーすることができないかもしれません。このような状況は、任意のこのようなプロトコルに従って開発された候補製品の価値に実質的な悪影響を及ぼす可能性がある。
さらに、知的財産権または技術を第三者または第三者に許可するプロトコルは複雑であり、そのようなプロトコルのいくつかは様々な解釈の影響を受ける可能性がある。可能性のある契約解釈の相違の解決は、関連する知的財産権または技術に対する私たちの権利と考えられる範囲を縮小するか、または関連協定の下での私たちの財務または他の義務を増加させる可能性があり、いずれも、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。また、我々が許可している知的財産権をめぐる紛争が、商業的に許容可能な条項で現在の許可手配の能力を維持していることを阻害したり、損害を与えたりすれば、影響を受けた候補製品の開発に成功し、商業化することができない可能性がある。
現在または将来のライセンシーがライセンス条項を遵守できない場合、ライセンシーが侵害第三者に対してライセンス特許を強制的に実行することができない場合、ライセンス特許または他の権利が無効または強制執行できないことが発見された場合、または許容可能な条項で必要なライセンスを締結できない場合、私たちの業務も影響を受けるであろう。さらに、私たちの許可者は私たちに許可されていない知的財産権を所有したりコントロールしたりする可能性がありますので、その是非にかかわらず、私たちは私たちが許可者の権利を侵害しているか、または他の方法で侵害しているというクレームを受ける可能性があります。
また、将来の製品の販売にどのくらいの印税義務が支払われるかは確定できませんが、金額は大きいかもしれません。私たちの将来の印税義務の額は、成功した製品で使用する技術と知的財産権にかかっています
78
もしあれば商業化していますしたがって、私たちが製品を開発して商業化することに成功しても、私たちは利益を達成したり維持することができないかもしれない。
もし私たちが必要な第三者知的財産権を得ることに成功したり、私たちの既存の知的財産権を維持することができなければ、関連する研究プロジェクトや候補製品の開発を放棄しなければならないかもしれません。私たちの業務、財務状況、運営結果、見通しは影響を受ける可能性があります。
私たちの協力者は、私たちの候補製品を開発または商業化するのに十分な資源を投入していないかもしれないし、開発または商業化努力に失敗する可能性があり、これは、私たちがいくつかの候補製品を開発または商業化する能力および私たちの財務状況および経営業績に悪影響を及ぼす可能性がある。
私たちと羅氏との協力については、私たちのどの第三者との任意の他の協力計画も、私たちの協力者が私たちの候補製品の開発や商業化に取り組んでいる資源の数と時間に限られた制御を持っている可能性がある。例えば,武田は最近,腺関連ウイルス(AAV),遺伝子治療,まれな血液学から内部戦略転換を行い,武田との協力を中止した。また,羅氏と協力して最大10個の同種異体CAR−T細胞治療計画を開発する予定であるが,羅氏はこのような計画を2つしか指定しておらず,羅氏が羅氏協力プロトコルに基づいてより多くの細胞治療計画を選択開発することは保証されていない。もし羅氏が協力協定の下で協力可能な目標または計画よりも少ない最大数を追求することを決定した場合、羅氏の協力協定に従って受け取る可能性のある潜在的な支払いを制限し、私たちの開発スケジュールを延期したり、他の方法で私たちの業務に悪影響を与えることになる。一般的に、私たちがこのスケジュールから収入を得る能力は、私たちの協力者がこのスケジュールで彼らに割り当てられた機能を成功的に履行する能力と、他の側面で彼らの契約義務を遵守する能力に依存する。
私たちの既存または未来のどの協力も最終的には成功しない可能性があり、これは私たちの業務、運営結果、財務状況、および成長の見通しに否定的な影響を与えるかもしれない。さらに、このような協力または他の手配のいずれかの条項は、私たちに不利であることが証明されるか、または不利とみなされる可能性があり、これは、私たちの普通株の取引価格に負の影響を与える可能性がある。場合によっては、私たちは協力中の製品または候補製品または研究計画の持続的な開発または製造を担当することができ、パートナーから得られた支払いは、この製品の開発または製造のコストを支払うのに十分ではないかもしれない。たとえば,ロ氏協力協定によると,羅氏は我々がP-CD 19 CD 20-ALLO 1に関する開発活動を実行する際に生じる特定の割合の費用を補償する義務があるが,我々は残高を担当し,羅氏は我々の金額に最高上限があることを補償する義務がある.
私たちと私たちの協力者との間には、例えば、臨床データの解釈、マイルストーンの実現、開発責任または費用の区分、開発計画、財務条項の解釈、または協力中に開発された知的財産権の所有権に関する衝突が生じる可能性がある。もしそのような衝突があれば、協力者は自分の利益のために行動するかもしれないが、これは私たちの最良の利益に反するかもしれない。私たちとパートナーとの間のどのような相違も、私たちの候補製品の開発や商業化を遅延または阻止する可能性がある。
さらに、現在と未来の第三者とのいかなる協力も、以下の追加のリスクに直面しており、これらのリスクの発生は、私たちの協力計画を失敗させる可能性があります
79
私たちは買収、許可、または戦略連合のメリットを意識していないかもしれませんが、私たちはより大きなビジネス機会やより成功をもたらす可能性のある計画に参加または利用できません。
私たちの業務は私たちが研究プロジェクトや製品を識別、開発、商業化する能力にかかっている。我々のビジネス戦略の重要な要素の1つは、我々の非ウイルスiggyBac DNA送達システム、Cas-Clover部位特定遺伝子編集システム、およびナノ粒子およびAAVに基づく遺伝子送達技術を含む、我々のコア独自プラットフォームに基づいて、より多くのプログラムを発見し、開発することである。内部研究開発努力に加えて、我々は、例えば羅氏との協力、他の戦略協力を探索して、新たなプロジェクトを発見するために、戦略協力によってこれを実現することを求めている。また、複数のライセンス側とライセンス内合意を締結し、将来的には第三者との買収や追加のライセンス手配を求める可能性があり、既存の技術や候補製品を補完または強化すると信じています。
これらの取引は、未知の債務を負担すること、私たちの業務を中断すること、私たちの管理層の時間と注意を移動させることを含む多くの運営および財務リスクをもたらす可能性があり、協力または買収された製品、候補製品または技術を管理するために、取引対価格またはコストを支払うために大量の債務を生成または希釈し、予想よりも高い開発または製造コスト、予想以上の人員および他の資源約束、予想よりも高い、予想よりも高い
80
協力、買収または統合コスト、資産減価または営業権または減価費用、償却費用の増加、任意の買収業務の業務および人員の協力または合併を促進する困難およびコスト、任意の買収業務の主要なサプライヤー、メーカーまたは顧客との関係は、管理層および所有権の変化によって減少し、任意の買収業務を維持できない重要な従業員。したがって、買収が合意または戦略的パートナーシップに達する可能性があれば、これらの取引を私たちの既存の業務や会社文化と組み合わせることに成功できなければ、あるいは公衆衛生危機が私たちや取引相手の運営に大きな悪影響を与える場合、スケジュールを延期したり、他の方法で私たちの業務に悪影響を与える可能性があり、このような取引のメリットを実現できないかもしれません。さらに、私たちの資源が限られているため、特定のタイプの治療の開発を追求し、援助すること、または特定のタイプの癌の治療を追求し、援助することを選択しなければならないため、私たちは、いくつかの計画または製品、または後により大きな商業的潜在力を有することが証明された適応の機会の追求を放棄または延期する可能性がある。私たちの計画の潜在市場の推定は不正確かもしれませんが、特定の計画のビジネス潜在力を正確に評価していなければ、私たちは戦略的協力、許可、または他の手配を通じてその計画の貴重な権利を放棄するかもしれませんが、この場合、その計画の独占的な開発と商業化の権利を維持することは私たちに有利です。あるいは,内部資源を1つのプロジェクトに割り当てることができ,そのプロジェクトでは,協調スケジュールを達成した方が有利である.これらの事件のいずれかが発生した場合、特定の候補製品の開発作業を放棄または延期すること、または潜在的な成功計画を開発することができないことを余儀なくされる可能性がある。
私たちは将来的に私たちの候補製品についてより多くの協力を形成することを望んでいるかもしれないが、そうすることができないかもしれないし、このような取引の潜在的なメリットを実現できない可能性があり、これは私たちの開発と商業化計画を変更または延期させる可能性がある。
私たちの候補製品の開発と潜在的な商業化は費用を支払うために多くの追加資本を必要とするだろう。将来、私たちは他の生物製薬会社と協力して、いくつかの候補製品を開発し、アメリカ以外の地域やいくつかの適応を含めて商業化することを決定するかもしれない。適切な協力者を探すことで、私たちは激しい競争に直面するだろう。私たちの候補製品のための戦略的パートナーシップや他の代替手配を構築する努力は成功しないかもしれない。彼らは協力努力の開発段階が早すぎると思われるかもしれないので、第三者は私たちの候補製品が安全性と有効性を示すために必要な潜在力を持っていないと思うかもしれない。第三者協力は、通常、適用候補製品に対して将来成功する制御権の一部または全部を第三者に譲ることを要求する。私たちが協力して最終合意に到達する能力は、協力者の資源と専門知識の評価、協力の条項と条件、提案された協力者の私たちの技術、候補製品、および市場機会の評価に依存するだろう。協力者はまた、同様の協力可能な指示を得るための代替候補製品または技術を考慮することができ、そのような連携が、私たちと私たちとの連携よりも私たちの候補製品に魅力的であるかどうかを考慮することができる。いかなる許可協定によれば、私たちはまた制限される可能性があり、特定の条項や潜在的な協力者と協定を締結することはできない。
協力の交渉と記録は複雑で時間がかかる。また,最近では大手製薬会社間の大量の業務合併により将来の潜在パートナー数が減少し,合併後の会社の戦略も変化している。したがって、私たちはタイムリーで受け入れ可能な条件で協力を交渉することができず、交渉することさえできないかもしれない。もし私たちがそれができない場合、私たちはいくつかの候補製品の開発を減らし、私たちの1つまたは複数の他の開発計画を減少または延期し、潜在的な商業化を延期したり、特定の候補製品の任意の計画販売やマーケティング活動の範囲を縮小したり、私たちの支出を増加させ、自費で開発、製造、または商業化活動を行わなければならないかもしれない。もし私たちが私たちの支出を増やし、私たち自身の開発、製造、商業化活動に資金を提供することを選択すれば、私たちは追加の資本を得る必要があるかもしれないし、これらの資本は受け入れ可能な条件で私たちに提供できないかもしれないし、全く得られないかもしれない。もし私たちが十分な資金がなければ、私たちは私たちの候補製品をさらに開発したり、それらを市場に出して製品収入を作ることができないかもしれない。
私たちの候補製品はまた、効率的かつ効率的に動作するために特定のコンポーネントを必要とする可能性があり、これらのコンポーネントの権利は他の人によって保持されている可能性がある。私たちは、私たちが決定した第三者から任意の成分、使用方法、プロセス、または他の第三者知的財産権の内部許可を得ることができないかもしれない。私たちはこのようなライセンスのいずれかを合理的な費用または合理的な条項で得ることができないかもしれないが、これは私たちの業務を損なうだろう。たとえ我々が許可を得ることができても,それは非排他的である可能性があり,我々の競争相手が同じ技術を得ることができる
81
私たちに権限を与えてくれた。この場合、私たちは代替技術を開発または許可するために多くの時間と資源を必要とするかもしれない。
私たちの産業や商業運営に関わるリスクは
私たちは私たちのキーパーソンに高度に依存していて、私たちが高い素質の人材を誘致し、維持することに成功できなければ、私たちの業務戦略を成功的に実施することができないかもしれません。
私たちが競争の激しい生物技術と製薬業界の中で競争できるかどうかは、私たちが高い素質の管理、科学と医療人員を引き付けることができるかどうかにかかっている。私たちは私たちの管理、科学、そして医療者たちに非常に依存している。私たちの役員、他の重要な従業員、他の科学や医療コンサルタントのサービスを失うこと、そして適切な代替者を見つけることができず、製品開発の遅延を招き、私たちの業務を損なう可能性があります。
私たちのほとんどの業務はサンディエゴの施設で行われている。この地域は多くの他の生物製薬会社と多くの学術·研究機関の本部である。私たちの市場は技術者に対する競争が非常に激しく、私たちが受け入れられる条件で高い素質の人員を採用し、維持する能力を制限する可能性があり、甚だしきに至っては根本的にはできない。
価値のある従業員をわが社に引き付けるために、給料と現金激励のほか、株式オプションと時間とともに付与されたRSUを提供しています。時間の経過とともに、従業員に対する株式オプションおよびRSUの価値は、当社の制御範囲を超えており、他社が提供するより利益のあるオファーを相殺するにはいつでも不十分である可能性がある。私たちは価値のある従業員を引き留めるために努力しているにもかかわらず、私たちの管理、科学、開発チームのメンバーは短時間で私たちとの雇用関係を終了するかもしれない。例えば、2022年には、私たちの幹部2人が辞任と退職の通知を提供した。私たちは私たちのいくつかの重要な従業員と雇用協定を持っているが、これらの雇用協定は自由に雇用できることを規定しており、これは私たちのどの従業員も通知するかどうかにかかわらず、いつでも私たちの職場を離れることができることを意味する。私たちはどんな幹部の生命保険証にも“キーパーソン”保険証を提供しないつもりだ。私たちの成功はまた私たちが引き続き高技能の初級、中級と高級管理者及び初級、中級と高級科学と医療人員を吸引、維持、激励できるかどうかにかかっている。近年、生物技術業界の求人市場競争は日々激しくなっているため、著者らは正常レベルより高い人員流動率を経験し、もし私たちが現有の従業員を維持し、新しい従業員を招いて自然減員の影響に対応できなければ、私たちの運営は不利な影響を受ける可能性がある。
私たちは激しい競争に直面しています。これは他の人が私たちよりも早く製品を発見し、開発したり、商業化したり、あるいは私たちよりも製品のマーケティングに成功する可能性があります。
新製品の開発と商業化競争は激しい。我々は製薬,バイオテクノロジー,その他関連市場で競争を展開しており,これらの市場では癌治療のための免疫療法や遺伝性遺伝疾患のための遺伝子療法が開発されている。もし私たちの競争相手が開発し商業化した製品が私たちが開発する可能性のあるいかなる製品よりも安全で、より効果的で、副作用が少なく、より深刻ではなく、より便利で、あるいは私たちが開発する可能性のあるいかなる製品も時代遅れまたは競争力がなければ、私たちのビジネス機会は減少または消滅するかもしれない。私たちの競争相手も私たちよりも早く製品の発売承認を得るかもしれません。これは私たちの競争相手が市場に入る前に強力な市場地位を築くことにつながるかもしれません。また,腫瘍や遺伝疾患分野の新薬や療法の急増に伴い,新技術の出現に伴い,ますます激しい競争に直面することが予想される。もし私たちが技術変革の最前線にいられなければ、私たちは効果的に競争できないかもしれない。我々が開発と商業化に成功した任意の候補製品は,既存の療法や将来出現する可能性のある新しい療法と競争するであろう。バイオテクノロジーと製薬業界の高度な競争性質と迅速な技術変化は、私たちの候補製品や私たちの技術を時代遅れにし、競争力を低下させたり、経済的ではないかもしれない。
私たちの候補製品のいくつかと同じカテゴリーに属する他の製品は承認されたか、またはさらに開発されています。特定の種類の生物製薬製品の中でより多くの候補製品が臨床開発を通じて監督審査と承認を行うことに伴い、必要とされる可能性のある臨床データの数量とタイプ
82
規制部門が要求することは増加したり変化したりするかもしれない。そのため、私たちはこのような候補製品に対する臨床試験結果は、これらの製品と候補製品と競争する或いはより有利なリスク効果の概況を表示する必要があるかもしれないし、上場承認を得ることができ、あるいは承認されれば、商業化に有利な製品ラベルを表示する必要があるかもしれない。リスク収益プロファイルがこれらの製品または候補製品と競争力がない場合、私たちが開発した製品は商業的に実行できない可能性があり、利益を上げることができない場合、または割引された価格設定や精算を実現できない可能性があります。このような状況で、私たちの未来の製品収入と財政状況は実質的な悪影響を受けるだろう。
具体的には,Adaptimmune治療会社,allgene社,Arcell社,Astellas製薬会社,Autolus社,百時美施貴宝社,Cariou生物科学社,Cellectis S.A.,ヤンソン製薬会社,Juno治療会社(Celgene社に買収され,現在はBristol−Meyers Squibb社),亘喜生物社(アスリーカンに買収),Kite製薬会社(吉利科学社の一社),伝説的生物,ノワ製薬,武田社など,様々なCAR−T療法の方法が求められている。多くの小さなバイオテクノロジー会社や大きな製薬会社が免疫療法や遺伝子療法をさらに求めている。私たちはまた、安進、アスリーカン、ビム治療会社、百時美施貴宝社、F.ホフマン-ラロ社、世代生物会社、グラクソ·スミスクライン、メルク社、PassageBio社、ファイザーなどから提供された非細胞または他の遺伝子療法の競争に直面している。私たちの多くの競争相手は、単独でもパートナーとも、より多くの財務、技術および他の資源、例えばより多くの研究開発者および/または研究開発、製造、臨床前テスト、臨床試験を行う上でより強い専門知識を持っている。
製薬とバイオテクノロジー産業の合併と買収は、私たちの数の少ない競争相手により多くの資源を集中させる可能性がある。規模の小さい会社や他のスタートアップ会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配されている。これらの第三者は、合格した科学·管理者を募集し、維持し、臨床試験場および臨床試験の被験者登録を確立し、私たちの計画を補完したり、私たちの計画のために必要な技術を獲得する上で私たちと競争している。
私たちのすべての計画の成功に影響を与える重要な競争要素は、それらの有効性、安全性、利便性、精算の利用可能性である可能性がある。もし私たちが開発、商業化に成功し、競争相手よりも高い精算レベルを実現できなければ、私たちは彼らと競争できなくなり、私たちの業務は実質的に損害を受けるだろう。
私たちは潜在的な製品責任に直面しています。もし私たちにクレームが成功すれば、大量の責任とコストを招くかもしれません。もし私たちの候補製品を使用して患者を傷つけたり、患者を傷つけたと考えられたりすれば、このようなダメージが私たちの候補製品と関係がなくても、私たちの規制承認が撤回されたり、他の方法で負の影響を受ける可能性があり、私たちは費用の高さと破壊的な製品責任のクレームを受ける可能性がある。
臨床試験で私たちの候補製品を使用して市場の承認を得たどの製品を販売しても製品責任クレームのリスクに直面させます。消費者、医療提供者、製薬会社、または他の販売または他の方法で私たちの製品に接触した人は、私たちに製品責任を請求するかもしれません。私たちの候補製品は不良事件を引き起こすかもしれない。もし私たちが製品責任クレームに成功的に対抗できなければ、私たちは大量の責任とコストを招くかもしれない。さらに、製品責任クレームは、是非曲直や最終結果にかかわらず、以下のようになる可能性がある
83
私たちは製品責任保険を保証して、事故ごとに1,000万ドル賠償して、総限度額は1,000万ドルです。私たちは現在の臨床計画によると、私たちの製品責任保険範囲は十分だと信じています。しかし、私たちは責任損失から私たちを守るために、合理的なコストあるいは十分な金額で保険範囲を維持することができないかもしれません。もし私たちが候補製品の発売許可を得たら、私たちは私たちの保険カバー範囲を拡大し、商業製品の販売を含めるつもりです;しかし、私たちは商業的に合理的な条項や十分な金額で製品責任保険を得ることができないかもしれません。私たちの保険証書にも様々な例外があるかもしれません。私たちは製品責任クレームの影響を受けるかもしれません。私たちは保険範囲を持っていません。薬物や治療に思わぬ悪影響を与えた集団訴訟では,多額の判決が下されることがある。成功した製品責任クレームや一連の私たちに対するクレームは私たちの株価を下落させる可能性があり、私たちの保険範囲を超えていると判断すれば、私たちの運営と業務結果に悪影響を及ぼす可能性があります。
私たちの候補製品は癌および他の疾患患者に対して通常すでに深刻かつ末期の疾患段階にあり、既知のものもあれば未知の重大な事前存在と潜在的に生命を脅かす健康リスクもある。治療中、患者は死亡を含む有害事象を受ける可能性があり、原因は、私たちの第1段階P−PSMA−101試験で発生した患者死亡のような、我々の候補製品に関連する可能性がある。このような事件は、怪我をした患者に巨額の費用を支払うこと、遅延、負の影響、または規制部門が私たちの製品を販売することを許可または維持する機会を得ること、または私たちの商業化努力を一時停止または放棄することを要求する費用の高い訴訟に直面する可能性がある。有害事象が我々の製品に関係しているとは考えられない場合でも,この場合の調査は時間や不確実性がある可能性がある.これらの調査は、他の国/地域での規制承認プロセスを延期したり、製品候補者の獲得や維持に影響を与えたり制限したりする規制承認タイプを遅らせるための販売努力を中断する可能性がある。これらの要因により、製品責任クレームが弁護に成功しても、我々の業務、財務状況又は運営結果に実質的な悪影響を及ぼす可能性がある。
私たちは私たちの開発、規制、運営能力を拡大することが予想されますので、私たちは管理成長に困難に直面する可能性があり、これは私たちの運営を混乱させるかもしれません。
2023年12月31日まで、私たちは330人の従業員を持っている。私たちの研究開発計画の推進に伴い、私たちは更に私たちの従業員の数量と業務範囲を増加させる必要があるかもしれません。特に臨床開発、製造、品質、規制事務、もし私たちの任意の候補製品がマーケティングの許可、販売、マーケティングと流通を獲得すれば、未来の成長を管理するためには
私たちの将来の財務業績および私たちの候補製品を開発、製造、商業化する能力は、将来のいかなる成長の能力を効果的に管理するかにある程度依存し、私たちの経営陣はまた、これらの成長活動を大量の時間を投入して管理するために、財務および他の資源および比例しない多くの注意を日常活動から移行させなければならないかもしれない。
もし私たちが新入社員を雇用し、私たちのコンサルタントや請負業者チームを拡大することで、私たちの組織を効果的に拡大することができなければ、私たちの候補製品をさらに開発し、商業化するために必要な任務を実行することができない可能性があり、私たちの研究、開発、商業化の目標を達成できないかもしれない。
私たちまたは私たちが依存している第三者は地震、火災、または他の自然災害の悪影響を受けるかもしれない。
私たちの本部、主要な研究施設と臨床製造施設はカリフォルニア州のサンディエゴにあり、そこは過去に深刻な地震と火災を経験したことがある。もしこのような地震、火災、他の自然災害、テロ、そして似たような私たちがコントロールできない意外な事件が、私たちがすべてまたは重大なものを使用することを阻止したら
84
私たちの本部や研究施設の一部では、私たちは難しいかもしれませんし、場合によっては長い間私たちの業務を継続することはできません。我々には災害復旧や業務継続計画がなく、当社の内部または第三者サービスプロバイダの災害復旧や業務連続性計画の欠落や性質が限られているために巨額の費用が発生する可能性があり、業務に大きな悪影響を及ぼす可能性があります。また、私たちのサプライチェーンに不可欠な各当事者は単一の場所で運営されており、自然災害や他の突発的、予見不可能、深刻な不良事件におけるそれらの脆弱性を増加させている。このような事件がわれわれのサプライチェーンに影響を及ぼすと,われわれの臨床試験を行う能力,われわれの開発計画,業務,財務状況あるいは運営結果に実質的な悪影響を及ぼす可能性がある。
私たちの純営業損失の繰越と他の税務属性を使用する能力は限られているかもしれません。
私たちは歴史的に大きな損失を受けて、近い将来の利益を期待していませんし、私たちは永遠に利益を達成しないかもしれません。2018年1月1日までに開始された課税年度未使用の米国連邦純営業損失、またはNOLは、将来の課税収入を相殺するために繰り越すことができ、あれば、これらの未使用のNOLが満期になるまで。現行法によると、2017年12月31日以降の課税年度に発生した米国連邦NOLは無期限に繰り越すことができるが、2020年12月31日以降の課税年度ではこのような米国連邦NOLの控除額は課税収入の80%に制限されている。各州がどの程度連邦税法を遵守するかはまだ確定されていない。
2023年12月31日まで、私たちは2.853億ドルのアメリカ連邦NOLがあり、現行法により無期限に繰り越すことができます。2023年12月31日現在、私たちは合計約4690万ドルのアメリカ連邦孤児薬物信用と研究開発信用を持っています。私たちのNOL繰越と研究開発相殺はアメリカと州税務当局の審査と可能な調整を受けます。
また、改正された1986年の国内税法第382条および383条、またはこの法規および州法律の対応条項によると、ある会社が“所有権変更”を経験した場合、これは通常、3年間の間にその持分所有権の変化が50ポイントを超える(価値で計算される)と定義され、同社は変更前の純資産の繰越、研究開発控除、およびいくつかの他の税収属性を使用して変更後の収入または税金を相殺する能力が制限される可能性がある。これは、将来の課税収入や納税義務を相殺するために毎年使用できるNOL、研究開発控除、または他の適用可能な税金属性の金額を制限することができるかもしれません。その後、NOL、R&D相殺や他の適用税収属性の使用における所有権変更や米国の税収ルールの変更は今後数年間の制限にさらに影響を与える可能性がある。さらに、州レベルでは、NOLの使用を一時停止または制限する時期がある可能性があり、これは州の課税税を加速または永久的に増加させる可能性がある。したがって、課税収入の純額を稼ぐと、私たちの純営業損失の全部または大部分の繰越と他の税務属性を使うことができないかもしれません。これは私たちの将来の納税義務を増加させ、私たちの将来のキャッシュフローに悪影響を及ぼす可能性があります。
医療法や施行条例の変化、医療政策の変化は、現在予測できない方法で私たちの業務に影響を与える可能性があり、私たちの業務や運営結果に大きな悪影響を及ぼす可能性があります。
米国や一部の外国司法管轄地域では、医療システムに関するいくつかの立法や法規の変化、提案された変化が継続されており、これらの変化は、候補製品の上場承認を阻止または延期し、承認後の活動を制限または規範化し、マーケティング承認を得た候補製品を収益的に販売する能力に影響を与える可能性がある。米国やEUを含む他の地方の政策立案者や支払者の中には,医療コストを抑え,質の向上および/または参入を拡大するために医療システムの改革を推進することに大きな興味がある。米国では、製薬業はこれらの努力の重点であり、重大な立法計画の大きな影響を受けてきた。
“平価医療法案”は、政府と民間保険会社が医療保健に資金を提供する方式を大きく変え、米国の製薬業に大きな影響を与えた。その他の事項を除いて、“平価医療法案”:(1)吸入、輸液、点滴、インプラントまたは注射、通常小売コミュニティ薬局で配布されていないいくつかの薬物および生物製品に対して、メーカーが医療補助薬物リベート計画の下で不足しているリベートを計算する新しい方法を導入し、(2)医療補助薬物リベート計画の下でメーカーの最低医療補助リベートを増加させる;(3)ブランド処方を確立する
85
ブランド処方薬の薬品メーカーは連邦政府に支払わなければならない薬品費用;(4)計画中に新しい実体を増加させることによって、340 B薬品定価計画に参加する資格のある実体をカバーするリストを拡大した;(5)新しい連邦医療保険D部分カバーギャップ割引計画を構築し、メーカーは現在そのカバー間隔内に条件を満たす受益者に適用ブランド薬品交渉価格の70%の販売時点割引を提供することに同意しなければならず、メーカーの外来薬物として連邦医療保険D部分の条件に組み入れられている。(6)メーカーの医療補助税還付責任を、医療補助管理保健組織に参加する個人に割り当てられた対象薬物に拡大すること、(7)各州がより多くの個人に医療補助を提供することを可能にし、連邦貧困レベル133%未満の収入を有する個人に新たな強制資格種別を増加させることによって、メーカーの医療補助税還付責任を潜在的に増加させることを含む医療補助計画の資格基準を拡大すること、(8)後続生物製品のためのライセンスフレームを作成すること、(9)CMSに医療保険および医療補助革新センターを設立し、革新的な支払いおよびサービス交付モードをテストして、処方薬支出を含む可能性がある連邦医療保険および医療補助支出を低減すること。(10)患者を中心とした新たな結果研究所を作成し,優先順位を監督·決定し,臨床的有効性の比較検討を行った。
“平価医療法案”のいくつかの面で行政、司法、国会の挑戦を受けている。例えば、2021年6月17日、米国最高裁は、“個人強制令”が国会で廃止されたため、“平価医療法案”全体が違憲だと弁明する手続き理由に基づく挑戦を却下した。また、米国最高裁判所が2021年1月28日に裁決を下す前に、バイデン総裁は、平価医療法案市場による医療保険の獲得を目的とした特殊な加入期間を開始する行政命令を発表した。行政命令はまた、特定の政府機関に、医療補助モデルプロジェクトおよび免除計画の再検討、医療補助または“平価医療法案”による医療保険獲得に不必要な障害をもたらす政策を含む医療補助モデルプロジェクトおよび免除計画の再審査および見直し、医療補助または“平価医療法案”の見直しを指示する。“平価医療法案”は将来的に司法や国会の挑戦を受ける可能性がある。2022年8月16日、総裁·バイ登は2022年インフレ削減法案に署名し、個人が平価医療法案市場で医療保険を購入する強化補助金を2025年に延長した。2025年からアイルランド共和軍は,受益者の最大自己負担コストと新たに構築されたメーカー割引計画を著しく低減することにより,連邦医療保険D部分計画下の“ドーナツ脆弱性”を解消した。バイデン政府のいかなる追加医療改革措置が“平価医療法案”や我々の業務や財務状況にどのように影響するかは不明である。
“平価医療法”が公布されて以来,他の立法改正も提案され,採択された。これらの変化には、2013年に開始された2011年予算制御法案によると、提供者に支払われる医療保険支払い総額を2%削減することが含まれており、インフラ投資·雇用法案、2023年総合支出法案を含む法案の立法改正により、追加の国会行動がとられない限り、2032年まで有効となる。2013年1月、“2012年米国納税者救済法”が法律に署名され、病院を含むいくつかの医療サービス提供者への医療保険支払いが減少し、政府が提供者に多額の支払いを取り戻す訴訟時効が3年から5年に延長された。我々の業務に影響を与える可能性のある他の変化は、2015年の“連邦医療保険アクセスとチップ再許可法案”に基づいて新しい計画を拡大することを含み、例えば医師業績計画に対する連邦医療保険支払いは、品質支払い計画とも呼ばれる。2019年11月、CMSは最終ルールを発表し、最終的に品質支払い計画の変化を決定した。現在,品質支払い計画の導入が連邦医療保険計画下の医師全体の精算にどのように影響するかは不明である。連邦医療保険や他の政府が計画している精算のいかなる減少も、個人支払者の支払いの同様の減少を招く可能性がある。また、新しい法律は、連邦医療保険や他の医療資金のさらなる減少を招く可能性があり、これは、顧客が私たちの製品の需要や負担能力、私たちの財務運営結果に大きな悪影響を及ぼす可能性があります。
そのほか、政府は最近薬品メーカーがその上場製品の価格設定の方式に対して更に厳格な審査を行い、国会が数回の調査を行い、連邦と州立法を提出し、公布し、これらの立法は製品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、政府の薬品に対する精算方法を改革することを目的としている。連邦レベルでは、トランプ政権はいくつかの手段を用いて、連邦予算提案、行政命令、政策措置を含む薬品定価改革を提出または実施している。2021年7月バイデン政府は“促進”という行政命令を発表しました
86
アメリカ経済“は、処方薬に対する多くの規定を持っている。バイデンの行政命令に応えるため、2021年9月9日、アメリカ衛生·公衆サービス部(HHSと略称する)は高薬価に対応する総合計画を発表し、その中で薬品定価改革の原則を概説し、国会が取ることができる各種の潜在立法政策を列挙し、これらの原則を推進した。また、アイルランド共和軍(IRA)は、他の事項に加えて、(I)HHSが連邦医療保険(Medicare)がカバーするいくつかの高支出、単一由来の薬物および生物製品の価格について交渉し、法律で規定されているこのような薬物と生物製品との交渉に等しくない“最高公平価格”の価格を提供することによって、薬品メーカーに民事と潜在的な消費税を科し、(Ii)連邦医療保険B部分またはD部分にカバーされているいくつかの薬品および生物製品にリベートを徴収し、インフレを超える価格上昇を処罰するように指示する。アイルランド共和軍は衛生と公共サービス部が最初の数年に規制ではなく指導によってその多くの規定を実施することを許可した。このような規定は2023年度から段階的に施行される。2023年8月29日、HHSは、連邦医療保険薬品価格交渉計画が現在法的挑戦を受けているにもかかわらず、価格交渉を受ける上位10種類の薬物のリストを発表した。これらの計画の実施に伴い,HHSは指導意見を発表·更新し続けている。アイルランド共和軍がどのように実施されるかは不明であるが,製薬業に大きな影響を与える可能性がある。また、バイデン政府の2022年10月の行政命令に応答するため、衛生·公衆サービス部は2023年2月14日に報告を発表し、CMS革新センターによってテストされた3種類の新しいモデルを概説し、これらのモデルはそれらの薬物コストを下げ、獲得性を促進し、医療の質を高める能力に基づいて評価を行う。これらのモデルが将来の任意の医療改革措置で使用されるかどうかは不明である。また、2023年12月7日、バイデン政府はベハ-ドール法案下の入場権利を使用することで処方薬の価格を制御するイニシアチブを発表した。2023年12月8日、米国国家標準·技術研究所は、権限行使を考慮した機関間指導枠組み草案を発表し、その中で初めて製品価格を機関が進行権を行使する際に使用できることを決定する要因とした。これまでデモの権利を行使したことはなかったが、新たな枠組みの下で、この権利が継続するかどうかは定かではない。
州レベルでは、立法機関は、価格または患者の精算制限、割引、特定の製品参入の制限、マーケティングコスト開示および透明性措置を含む、医薬品および生物製品の価格設定を制御するための法規を立法および実施することが増えており、場合によっては、他の国からの輸入および大量購入を奨励することを目的としている。例えば、FDAは2024年1月5日、特定の州医療計画のためにカナダから特定の薬剤を輸入するフロリダ州第804条輸入計画(SIP)の提案を承認した。この計画がどのように実施されるか,どのような薬剤が選択されるか,その計画が米国やカナダで法的挑戦を受けるかどうかは不明である。他の州もFDAの審査を待っているSIP提案を提出した。このような承認された輸入計画が一旦実施されると、これらの計画がカバーする製品の薬品価格をより低くする可能性がある。
将来的に採用される可能性のあるこれらや他の医療改革措置は、より厳しいカバー基準とより低い精算を招き、私たちが受け取った任意の承認された製品の価格に追加の下振れ圧力をもたらす可能性があると予想される。連邦医療保険や他の政府援助計画のいかなる精算減少も、個人支払者の支払いの同様の減少を招く可能性がある。コスト抑制措置や他の医療改革を実施することは、上場承認を得た後に収入を創出し、利益を実現し、あるいは私たちの薬物を商業化することを阻止する可能性がある。
EUでは、私たちが規制承認を受けた任意の候補製品の保証範囲と精算状態は、EU加盟国の国家法律によって規定されている。EU加盟国の要求事項は違うかもしれない。また、国家レベルでは、製薬会社と医療専門家との支払いに関する透明性に関する法律が公布されている。
私たちは適用される詐欺と乱用、透明性、政府価格報告書、そして他の医療保険の法律と規制によって制限されている。もし私たちがこのような法律を遵守できないか、または完全に遵守できなければ、私たちは巨額の処罰に直面するかもしれない。
医療提供者と第三者支払者は、私たちが将来開発する可能性のある任意の候補製品と、マーケティングの許可を得た任意の候補製品の推薦および処方において主な役割を果たすだろう。現在と未来の臨床研究者、第三者支払者、医療保健提供者と顧客との手配は、私たちの製品の研究、マーケティング、販売、流通の業務または財務配置と関係に影響を与える可能性がある広範に適用される詐欺と乱用、その他の医療法律と法規に直面させます。私たちの運営能力に影響を与える可能性のある法律は含まれているが、これらに限定されない
87
88
私たちはまた、市場活動や消費者を損なう可能性のある活動を広く規制している連邦および州消費者保護および不正競争法の制約を受ける可能性がある。私たちは、私たちの候補製品の使用に影響を与える可能性のある条項(承認されれば)を含む株式オプション条項を含む諮問·科学顧問委員会と医師や他のヘルスケア提供者と合意している。これらの法律の複雑かつ深遠な性質のため、規制機関はこれらの取引を再編成または終了しなければならない禁止措置と見なすかもしれない。そうでなければ、私たちは他の重大な処罰を受けるかもしれない。もし規制機関が私たちとサプライヤーとの財務関係を適用法違反と解釈し、これらのサプライヤーが私たちの候補製品の注文と使用に影響を与える可能性があれば、私たちは不利な影響を受ける可能性がある。
連邦と州法執行機関は引き続き医療会社と医療提供者との相互作用を審査し、医療業界の重大な調査、起訴、有罪判決、和解を招いた。調査への対応に時間と資源がかかる可能性があり、経営陣の業務への関心を分散させる可能性がある。このような調査や和解は、私たちのコストを増加させるか、または他の方法で私たちの業務に悪影響を及ぼす可能性がある。
私たちが第三者の業務手配と適用される医療法律や法規に適合することを確保することは、コストが高くなる可能性があります。もし私たちの運営がこれらの法律のいずれかまたは任意の他の現在または将来、私たちの政府の法律および法規に適用される可能性があることが発見された場合、私たちは重大な民事、刑事および行政処罰、損害賠償、罰金、返還、監禁、連邦医療保険や医療補助、契約損害、名声損害、利益減少および将来の収益減少、追加の報告義務および監督(これらの法律違反に関する告発を解決するために当社の誠実な合意または他の合意に拘束された場合)、および私たちの業務の削減または再編は、いずれも私たちの運営を深刻に混乱させる可能性がある。もし私たちがそれと業務を展開することを期待している任意の医師または他の医療提供者または実体が適用されない法律に適合していないことが発見された場合、彼らは政府の援助された医療計画から除外されることを含む重大な刑事、民事または行政制裁を受ける可能性がある。
私たちの知的財産権に関するリスクは
もし私たちが私たちのプラットフォーム技術や候補製品のために十分な知的財産権保護を得ることができなければ、あるいは知的財産権保護の範囲が十分でなければ、私たちの競争相手は私たちと類似または同じ製品を開発し、商業化する可能性があり、私たちの製品を商業化することに成功する能力は悪影響を受ける可能性がある。
私たちの成功は、私たちがアメリカや他の国で私たちのプラットフォーム技術と候補製品に関する特許保護を獲得し、維持する能力に大きく依存する。私たちが守ることを求めているのは
89
米国や海外で我々の新たな発見や技術に関連する特許地位を出願することにより,これらの発見や技術は我々の業務に非常に重要である。我々の未解決および将来の特許出願は、私たちの候補製品またはその予期される用途を保護する特許の発行、または他社が競合技術、製品または候補製品を商業化することを効果的に阻止する特許をもたらすことができないかもしれない。
特許の取得および実行は高価で時間がかかり、必要または必要なすべての特許出願を合理的なコストまたはタイムリーに提出して起訴することができない場合があり、または我々の特許出願に基づいて発行される可能性のある特許を維持および/または実行することができる。我々は,特許保護を得るのが遅くなるまで,我々の研究や開発結果の特許可能性を確認できない可能性もある.私たちは、私たちの従業員、会社の協力者、外部科学協力者、契約研究組織、契約メーカー、コンサルタント、コンサルタント、その他の第三者などと、私たちの研究開発成果を得る権利のある特許に関する当事者と秘密保持および秘密協定を締結したが、これらの当事者のいずれかは、これらの合意に違反し、特許出願を提出する前にこれらの結果を開示し、特許保護を求める能力を危険にさらす可能性がある。
物質構成である生物および医薬製品の特許、例えば自動車ベースの候補製品は、このような特許によって提供される保護が任意の使用方法とは無関係であるため、これらのタイプの製品に強力な知的財産権保護を提供することが多い。私たちの未決特許出願中の私たちの候補製品の組成に関連する権利要件が、米国特許商標局または外国特許庁によって出願可能特許とみなされるかどうか、または私たちが発行した任意の特許における権利要件が、米国または外国裁判所によって有効かつ強制的に実行可能であるとみなされるかどうかを決定することはできない。使用方法特許は、特定の方法を使用する製品を保護する。このようなタイプの特許は、製品が特許方法の範囲を超えていることを示すために、競合他社が我々の製品と同じ製品を製造および販売することを阻止しない。また,競争相手が我々の目標適応に対して彼らの製品を積極的に普及させなくても,医師はラベルの外にこれらの製品を処方する可能性がある。ラベル外の処方は使用方法の特許侵害を侵害または助長する可能性があるにもかかわらず,このような行為は一般的であり,このような侵害行為の予防や起訴は困難である。
バイオ製薬会社の特許地位は通常高度に不確定であり、複雑な法律や事実問題に関連しており、近年多くの訴訟のテーマとなっており、最高裁の裁決を含む裁判所の裁決を招き、将来の特許権を執行する能力の不確実性を増加させている。しかも、外国の法律はアメリカの法律のように私たちの権利を保護しないかもしれないし、その逆も同様である。
さらに、我々は、私たちの候補製品またはその予想用途に関連する可能性のあるすべての第三者知的財産権を知らないかもしれないので、これらの第三者知的財産権が私たち自身の特許および特許出願の特許性に及ぼす影響、およびこれらの第三者知的財産権が私たちの運営自由に与える影響は、非常に不確実である。米国および他の管轄地域では、特許出願は通常、出願18ヶ月後に公表され、場合によっては全く公表されない場合もある。したがって、私たちは、私たちが最初に私たちの特許または係属中の特許出願で主張された発明を提出したのか、最初にそのような発明のために特許保護されたのかを正確に知ることができない。したがって,我々の特許権の発行,範囲,有効性,実行可能性,商業的価値は高い不確実性を持っている.私たちの特許または係属中の特許出願は、米国および海外の裁判所または特許庁で挑戦される可能性がある。例えば、第三者が既存技術の発行前に米国特許商標局に提出したり、ライセンス後の審査手続き、反対意見、派生、再審に参加したりする必要があるかもしれません各方面間米国又は他の場所で我々の特許権又は他の者の特許権に挑戦する審査手続。このような任意の挑戦において不利な裁決を下すことは、排他性喪失または特許主張の全部または部分的な縮小、無効または実行不能を招き、それにより、他人が類似または同じ技術および製品を使用することを阻止したり、それを商業化する能力を制限したり、私たちの技術および製品の特許保護期間を制限したりする可能性がある。さらに、新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。私たちの候補製品に対する特許保護を獲得または維持できなかったことは、私たちの業務、財務状況、運営結果、および将来性に重大な悪影響を及ぼす可能性があります。
90
もし私たちが第三者の知的財産権侵害を起訴されれば、このような訴訟は費用が高く時間がかかり、私たちの候補製品の開発や商業化を阻止または延期する可能性がある。
私たちのビジネス成功は、第三者の知的財産権および他の独自の権利を侵害することなく、私たちの候補製品を開発、製造、マーケティング、販売する能力にある程度依存する。第三者は私たちが彼らの知的財産権を侵害したり流用したと主張するかもしれない。例えば、2019年初めに、私たちは第三者から受け取った材料を不正に使用すると主張し、当社のCAR-T製品でセキュリティスイッチを使用することに関連するいくつかの特許を侵害すると信じていると主張する第三者から手紙を受け取りました。私たちは第三者のいかなる材料を不正に使用するかを否定し、特許が侵害されないか、無効であるか、または両方が併存すると信じているが、第三者がその容疑を主張するかどうか、または続く訴訟を予測することはできない。正当な理由の有無にかかわらず、知的財産権クレームに関連する訴訟や他の法的手続きは予測不可能であり、通常コストが高く、時間がかかり、解決策が私たちに有利であっても、私たちの技術や管理者の正常な役割を分散させることを含む、私たちのコア業務から大量の資源を移転する可能性がある。また、公聴会、動議、または他の一時的な手続きや事態の発展の結果が公表される可能性があり、証券アナリストや投資家がこれらの結果がマイナスだと思う場合、私たち普通株の市場価格に大きな悪影響を及ぼす可能性がある。このような訴訟または訴訟は、私たちの運営損失を大幅に増加させ、開発活動または任意の将来の販売、マーケティング、または流通活動に使用することができるリソースを減少させる可能性がある。私たちはそのような訴訟や法的手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らがより多くの財政資源とより成熟して発展した知的財産権の組み合わせを持っているので、このような訴訟や法的手続きの費用を私たちよりも効率的に負担するかもしれない。特許訴訟または他の訴訟の開始と継続によって生じる不確実性は、市場での競争能力に重大な悪影響を及ぼす可能性がある。
バイオテクノロジーや製薬業界には大量の知的財産権訴訟があり、私たちは私たちの候補製品に関連する知的財産権訴訟や他の対抗性訴訟の当事者になったり、これらの訴訟や訴訟の脅威にさらされたりする可能性がある。私たちはあなたに、私たちの候補製品と私たちが開発する可能性のある他のノウハウが第三者が所有する既存または未来の特許を侵害しないということを保証することはできません。第三者は既存または未来の知的財産権に基づいて私たちに権利侵害を請求するかもしれない。私たちはすでに発行された特許、および第三者、例えば私たちが候補製品分野の競争相手を開発していることを知らないかもしれません。私たちの候補製品の成分、配合、製造方法、または使用方法、または処理を含む私たちの候補製品が私たちの権利を侵害していると主張するかもしれません。私たちは第三者が所有する特許を知っているが、これらの特許は、私たちの候補製品や私たちが開発する可能性のある他のノウハウとは関係なく、私たちの候補製品に侵害される可能性もあると考えられる。さらに、特許出願が発行されるまでに数年かかる可能性があるため、現在処理されている特許出願が存在する可能性があり、これらの出願は、私たちの候補製品が発行された特許を侵害する可能性がある。私たちは米国と海外の競争相手であり、その多くはより多くの資源を持っており、特許の組み合わせおよび競争技術に大量の投資を行っており、それらはすでに特許を出願または獲得しているかもしれないし、将来的に特許を申請して獲得する可能性があり、これらの特許は、私たちの候補製品の製造、使用、販売の能力を阻止、制限、または他の方法で妨害するだろう。製薬およびバイオテクノロジー産業はかなり多くの特許を生み出しており、私たちを含む業界参加者は、どの特許が様々なタイプの製品または使用方法をカバーしているのかを常に明確にしていないかもしれない。特許のカバー面は裁判所の解釈にかかっており、解釈は常に統一されているわけではない。もし私たちが特許侵害で起訴された場合、私たちは私たちの候補製品、製品、または方法が関連特許の特許主張を侵害していないか、または特許主張が無効または実行不可能であることを証明する必要があり、私たちはそれができないかもしれない。その無効性を証明することは難しいかもしれない。例えば、米国では、法廷で無効を証明するには、発行された特許の有効性の推定を覆すために明確で納得できる証拠を提示する必要があり、管轄権を有する裁判所が、そのような米国特許の権利要件の無効を宣言することは保証されない。私たちがこれらの訴訟で勝訴しても、私たちは巨額のコストを生む可能性があり、私たちの経営陣や科学者の時間と注意力はこれらの訴訟に移される可能性があり、これは私たちの業務や運営に実質的な悪影響を及ぼす可能性がある。しかも、私たちはこのような行動を円満に達成するための十分な資源がないかもしれない。
もし私たちが第三者の知的財産権を侵害していることが発見された場合、裁判所の命令を含めて、権利侵害候補製品または製品の開発、製造、または商業化を余儀なくされる可能性がある。あるいは権利侵害技術を使用するためには第三者から許可を得る必要があるかもしれません
91
権利侵害候補製品の開発、製造、販売を継続している。しかし、私たちは商業的に合理的な条項や必要な許可証を得ることができないかもしれない。我々が許可を得ることができても,非排他的である可能性があり,我々の競争相手が我々に許可された同じ技術にアクセスできるようにする.さらに、もし私たちが故意に特許を侵害したことが発見された場合、私たちは3倍の損害賠償と弁護士費を含む金銭損害賠償責任を負われる可能性がある。権利侵害の発見は、候補製品を商業化したり、一部の業務運営を停止させたりすることを阻止し、私たちの技術者や経営陣の時間と注意を移し、開発遅延を招く可能性があり、および/または、費用対効果に基づいて不可能である可能性があり、いずれも私たちの業務に実質的な損害を与える可能性があります。もし私たちに対する侵害クレームが成功した場合、故意に侵害した3倍の損害賠償と弁護士費、特許権使用料および他の費用の支払い、私たちの侵害薬物の再設計、または第三者から1つ以上の許可証を取得することを含む巨額の金銭損害賠償を支払わなければならないかもしれないし、多くの時間とお金の支出が必要である可能性がある。我々が第三者の機密情報や商業秘密を盗用したと主張することは,我々の業務に類似した負の影響を与える可能性がある.
関連する第三者特許を識別できないか、または第三者特許の関連性、範囲、または満了時間を誤って解釈することができない可能性があり、これは、私たちが製品を開発およびマーケティングする能力に悪影響を及ぼす可能性がある。
私たちは、関連特許の識別、特許請求の範囲、または関連特許の満了を含む、私たちのいかなる特許検索または分析も保証することはできず、私たちはまた、私たちの候補製品の任意の司法管轄区域での商業化に関連する、または必要な、米国および海外のすべての第三者特許および係属中の特許および係属中の出願を識別したことを保証することはできない。
特許請求の範囲は、法律の解釈、特許における書面開示、および特許の起訴履歴に依存する。特許または保留出願の関連性または範囲の解釈は正しくない可能性があり、これは私たちの製品を販売する能力に悪影響を及ぼす可能性がある。私たちの製品が第三者特許のカバー範囲内にないか、または第三者の保留出願が関連範囲のクレームを出すかどうかを誤って予測する可能性がある。私たちが関連する特許の満期日の決定は、アメリカまたは海外のどのようなものでも正しくないかもしれません。これは、私たちの候補製品を開発し、マーケティングする能力に悪影響を及ぼす可能性があります。私たちは関連特許を識別して正確に解釈することができず、私たちが製品を開発し、マーケティングする能力に悪影響を及ぼすかもしれない。
もし私たちが第三者に知的財産権を許可する合意の義務を履行できなかった場合、あるいは私たちとライセンス者との業務関係が妨害された場合、私たちは私たちの業務に非常に重要な許可権を失う可能性があります。
私たちは、私たちの業務に非常に重要な知的財産権ライセンス協定の多くの締約国であり、将来的により多くのライセンス契約を締結したいと思っています。私たちの既存の許可協定は、私たちが未来の許可協定が私たちに様々な勤勉さ、マイルストーン支払い、特許権使用料、その他の義務を負担することを要求すると予想している。公衆衛生危機による私たちの業務運営への影響、または破産の影響を含めて、これらの合意下の義務を履行できなかった場合、許可側は許可を終了する権利がある可能性があり、この場合、許可がカバーする製品を販売することができません。
私たちは第三者から許可を得て、私たちの研究を進めたり、私たちの候補製品の商業化を許可する必要があるかもしれません。私たちはよくそうします。私たちはもしあれば、合理的な費用または合理的な条項でこのような許可証のいずれかを得ることができないかもしれない。この場合、私たちは代替技術を開発または許可するためにかなりの時間と資源を必要とするかもしれない。もし私たちがそれができなければ、影響を受けた候補製品を開発したり商業化することができないかもしれません。これは私たちの業務を深刻に損なう可能性があります。現在の候補製品または将来の製品に対して強制的に執行される可能性のある第三者特許が存在しないことを保証することはできません。それによって、私たちの販売を禁止したり、私たちの販売については、第三者に印税および/または他の形態の賠償を支払う義務があります。
多くの場合、私たちが許可した技術の特許訴訟は完全に許可者によって統制されている。公衆衛生危機が私たちの許可者の業務運営に及ぼす影響を含む、私たちの許可された独自の知的財産権の特許または他の保護を彼らから得ることができなければ、私たちは私たちの知的財産権の権利またはこれらの権利に対する私たちの排他性を失う可能性があり、これらの特許と出願は私たちの業務と私たちのものに適合しないかもしれない
92
競争相手は知的財産権を利用して競争相手の製品をマーケティングすることができる。場合によっては、私たちは許可技術によって作られた特許の起訴を統制する。もし私たちがこのような起訴に関連したいかなる義務にも違反したら、私たちは私たちの許可パートナーに重大な責任を負うかもしれない。知的財産権許可は私たちの業務に重要であり、複雑な法律、商業と科学問題に関連し、そして私たちの業界の科学発見の迅速な発展によって複雑になった。ライセンス契約によると、知的財産権に関する論争が発生する可能性があります
私たちが許可している知的財産権をめぐる紛争が許容可能な条項で現在の許可手配の能力を維持していることを阻害したり、損害を与えたりすれば、影響を受けた候補製品の開発に成功し、商業化することができない可能性がある。私たちは通常、私たちがここで説明した私たちが持っている知的財産権のように、私たちが許可した知的財産権保護に関するすべてのリスクに直面している。もし私たちまたは私たちの許可者がこの知的財産権を十分に保護できなければ、私たちの製品商業化能力は影響を受ける可能性がある。
将来的には、私たちは第三者技術の追加許可を得る必要があるかもしれません。これらの許可は私たちに提供できないかもしれないし、商業的に不合理な条項でしか得られないかもしれません。これは、私たちがより高価で不利な方法で私たちの業務を運営することにつながるかもしれません。
私たちは現在私たちの候補製品と他の独自技術をカバーする知的財産権を持っている。他の製薬会社や学術機関も、我々の業務に関連する可能性のある特許出願を既に又は計画して提出している可能性がある。時々、これらの第三者特許の侵害を避けるために、他の第三者から技術的許可を得て、私たちの候補製品をさらに開発したり、商業化したりする必要があるかもしれません。もし私たちが私たちの候補製品を製造、使用、または販売するために必要な任意のこのような特許を含む任意の第三者技術の許可を要求された場合、私たちは商業的に合理的な条項でそのような許可を得ることができないか、またはそのような許可を得ることができないかもしれない。私たちの任意の候補製品を開発または商業化するために必要ないかなる第三者ライセンスも取得できないことは、私たちの業務と運営を深刻に損なう可能性があり、関連する努力を放棄する可能性があります。
第三者知的財産権の許可または買収は競争分野であり、より多くの老舗企業は、魅力的または必要と考えられる第三者知的財産権許可または買収戦略をとることができる。これらの老舗会社はその規模、資本資源及び更に強い臨床開発と商業化能力のため、私たちより競争優位を持っているかもしれない。しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。
さらに、私たちが所有し許可しているいくつかの特許または特許出願または将来の特許は、第三者と共同で所有されているか、または共同で所有されている可能性がある。もし私たちがこのような特許または特許出願における第三者共同所有者の権利の独占的な許可を得ることができない場合、これらの共通所有者は、私たちの競争相手を含む他の第三者に権利を許可することができ、私たちの競争相手は競争製品および技術を販売することができる。さらに、私たちは、第三者に対してそのような特許を強制的に実行するために、私たちの特許のどのような共通所有者の協力も必要とするかもしれませんが、そのような協力は私たちに提供されないかもしれません。さらに、私たちが持っている特許および許可中の特許は、1つまたは複数の第三者の権利によって保持されるかもしれない。上記のいずれも、我々の競争地位、業務、財務状況、経営結果、および見通しに重大な悪影響を及ぼす可能性がある。
93
私たちは、私たちの係属中の特許出願に記載され、要求された発明に関連する特許が付与されるか、または私たちの特許出願に基づく特許が挑戦および無効および/または実行不可能であることを保証することはできない。
私たちのポートフォリオには未解決のアメリカと外国特許出願があります。しかし、私たちは予測できません
私たちは、私たちの係属中の特許出願における私たちの候補製品および/または技術のための権利要件が、米国特許商標局または外国特許庁によって出願可能特許とみなされることを決定することはできない。そのような特許出願がライセンス特許として発行されることは保証されない。我々の発明の特許可能性を決定する一態様は、“従来技術”の範囲および内容、すなわち保護を要求する発明の優先日の前に、関連分野の技術者がかつてまたは入手可能と考えられていた情報に依存する。私たちが知らない既存技術が存在する可能性があり、これらの技術は、私たちの特許請求の範囲の特許可能性に影響を与える可能性があり、または、発表された場合、特許請求の有効性または実行可能性に影響を及ぼす可能性がある。特許が確かに我々の特許出願に基づいて発行されていても、第三者はその有効性、実行可能性、または範囲に疑問を提起する可能性があり、これは、このような特許が縮小され、無効にされ、または実行できない可能性がある。さらに、それらが挑戦されていなくても、私たちの製品の組み合わせにおける特許は、第三者が関連技術を実践することを排除したり、私たちの権利要件をめぐる他の人の設計を阻止するのに十分ではない可能性がある。もし私たちの候補製品における知的財産権の地位の広さや実力が脅かされれば、会社が私たちと協力して開発することを阻止し、候補製品を商業化する能力を脅かすかもしれない。訴訟や行政訴訟では、私たちが発行した任意の特許における権利要件が米国または他の国の裁判所によって有効とみなされるかどうかを判断することはできません。
知的財産権は必ずしも私たちの競争優位に対するすべての潜在的な脅威を解決できるとは限らない。
私たちの知的財産権が提供する未来の保護の程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できないか、あるいは競争優位性を維持することができるかもしれない。例えば:
94
このような事件が発生した場合、私たちの業務、運営結果、そして将来性を深刻に損なう可能性がある。
私たちは私たちの特許や他の知的財産権を保護したり強制したりする訴訟に巻き込まれるかもしれないが、これは高価で時間がかかり、成功しないかもしれない。
競争相手または他の第三者は、私たちの特許、商標、著作権、または他の知的財産権を侵害するかもしれない。権利侵害や不正使用に対抗するために、費用がかかり、時間がかかる可能性があり、私たちの管理者や科学者の時間と注意力を分散させる権利侵害請求を要求される可能性があります。私たちの係属中の特許出願は、特許がこのような出願から発行されるまで、そのような出願において要求される技術を実施する第三者に対して強制的に実行することはできない。私たちが認定された侵害者に対するいかなるクレームも、私たちが彼らの特許を侵害したと主張し、また、私たちの特許が無効または強制執行できない、または両方を持っていると主張するように、これらの当事者に反クレームを促す可能性がある。米国の特許訴訟では,被告が無効および/または実行不可能と主張する反訴が一般的である。有効性を疑問視する理由は、新規性の欠如、明らかな、実施できない、または書面記述の不足を含む、いくつかの法定要件のいずれかを満たしていないと言われているからかもしれない。主張を実行できない理由は,特許訴訟に関連する者が米国特許商標局に関連情報を隠蔽したり,起訴中に誤った陳述をしたりすることである可能性がある.法的に無効と実行不可能と断言された後の結果は予測できない。いかなる特許侵害訴訟においても、裁判所は私たちの特許の全部または一部が無効または強制的に執行できないと判断する可能性があり、私たちは他の当事者が論争のある発明を使用することを阻止する権利がない。もう1つのリスクは,これらの特許の有効性が支持されても,裁判所が特許権利請求を偏狭に解釈したり,我々の特許請求が発明を含まないことを理由に,相手が関連する発明の使用を阻止する権利がないと判断したり,米国法第35編271(E)(1)条に基づいて,相手が我々の特許技術を使用して特許侵害の安全港に属すると判断することである.我々の特許に関連する訴訟または訴訟における不利な結果は、これらの当事者または他の競争相手に対して私たちの特許を主張する能力を制限し、第三者が類似または競合製品を製造および販売する能力を制限または排除する可能性がある。これらの状況のいずれも、私たちの競争業務の地位、業務の見通し、および財務状況に悪影響を及ぼす可能性があります。同様に、私たちが商標侵害クレームを主張する場合、裁判所は、私たちが主張する商標が無効または強制執行できないと判断するか、または商標侵害を主張する側が関連商標に対する優先権を有することを主張するかもしれない。この場合、私たちは最終的にこのような商標の使用を中止することを余儀なくされるかもしれない。
95
侵害行為が成立したと認定しても,裁判所はさらなる侵害活動に禁止令を付与せず,金銭賠償のみを決定する可能性があり,十分な救済措置ではない可能性もある。さらに、知的財産権訴訟は大量の開示を必要とするため、私たちのいくつかの機密情報は訴訟中に開示によって漏洩される可能性がある。聴聞、動議、または他の一時的手続き、または事態発展の結果を公開することもできる。もし証券アナリストや投資家がこれらの結果がマイナスだと思っていれば、私たち普通株の株価に実質的な悪影響を及ぼす可能性がある。しかも、私たちはこのような侵害請求を提起して追跡するのに十分な財政的または他の資源を持っていることを保証することはできません。これらのクレームは通常数年続いています。たとえ私たちが最終的に勝訴しても、このような訴訟の金銭的代価と私たちの経営陣と科学者の注意の移動は、私たちが訴訟から得たいかなる利益を超えるかもしれない。
訴訟の費用と不確実性のため、私たちは第三者に対して私たちの知的財産権を強制的に実行できないかもしれない。
訴訟の費用と不確実性のため、第三者が私たちが発表した特許を侵害しても、私たちの未解決または将来の特許出願または他の知的財産権によって発行される可能性のある任意の特許は、そのようなクレームや訴訟を提起して実行するリスク調整コストが高すぎるか、わが社または私たちの株主の最適な利益に適合していないか、または他の方法で特定の第三者に私たちの知的財産権を強制的に実行することは非現実的または望ましくない可能性があると結論するかもしれません。より多くの財力とより成熟して発展した知的財産の組み合わせを持っているので、私たちの競争相手または他の第三者は、複雑な特許訴訟または訴訟の費用を私たちよりも効果的に負担することができる。この場合、私たちは、状況を簡単に監視するか、または他の非訴訟の行動または解決策を開始または求めることで、より慎重なやり方を決定するかもしれない。また、訴訟に関連する不確実性は、私たちの臨床試験を継続し、私たちの内部研究計画を継続し、必要な技術または他の候補製品の許可を得るために必要な資金を調達し、あるいは候補製品を市場に出す能力を支援するために、必要な資金を調達することに影響を与える可能性がある。
私たちは、私たちの従業員、コンサルタント、または独立請負業者が第三者の機密情報を誤って使用または開示することに関するクレームを受けるかもしれない。
私たちは将来、私たちまたは私たちの従業員が無意識に、または他の方法で前の雇用主または競争相手のいわゆる商業秘密または他の機密情報を使用または漏洩したという疑惑を受けるかもしれない。私たちの従業員およびコンサルタントが、私たちのために働いているときに他人の知的財産権、独自の情報、技術的ノウハウ、または商業秘密を使用しないことを確実にするために努力しているにもかかわらず、私たちは、従業員が彼らの競業禁止または競業禁止協定の条項に違反しているか、または私たちまたはこれらの個人が、元の雇用主または競争相手のいわゆる商業秘密または他の固有情報を意図せず、または他の方法で使用または開示しているという疑惑を受ける可能性がある。
私たちは訴訟を通じて自分を弁護するかもしれませんが、私たちが勝訴しても、訴訟は巨額のコストを招き、経営陣の注意を分散させる可能性があります。もし私たちがこれらのクレームに対する抗弁に失敗した場合、私たちに金銭損害賠償を要求することに加えて、裁判所は、これらの技術または機能が以前の雇用主の商業秘密または他の固有情報を含むか、または由来することを前提として、私たちの候補製品に重要な技術または機能の使用を禁止することができる。さらに、このような訴訟またはその脅威は、私たちの名声、戦略連合を形成したり、協力者に私たちの権利を譲渡したり、科学コンサルタントと接触したり、従業員やコンサルタントを雇う能力に悪影響を及ぼす可能性があり、すべては私たちの業務、運営結果、および財務状況に悪影響を及ぼすだろう。このようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣の注意を分散させる可能性がある。
私たちは世界各地で私たちの知的財産権を保護できないかもしれない。
特許は国や地域の影響を持っており、世界各地で私たちのすべての候補製品の出願、起訴、弁護特許の費用は目を引くほど高いだろう。したがって、私たちはアメリカ以外のいくつかの国の知的財産権に米国の知的財産権が広くないかもしれません。私たちは第三者がアメリカ以外のすべての国で私たちの発明を実施することを阻止できないかもしれません。あるいは第三者がアメリカや他の司法管轄区で私たちの発明を使用して製造した製品を販売または輸入することを阻止できないかもしれません。競争相手は特許保護を受けていない管轄区で私たちの技術を使って自分の製品を開発するかもしれません
96
私たちは他の侵害製品や技術を私たちが特許保護を持っている地域に輸出するかもしれないが、執行権はアメリカほど強くない。これらの製品は私たちの製品と競争するかもしれませんが、私たちの特許や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。さらに、特定の国の法制度、特に特定の発展途上国の法制度は、特許や他の知的財産権保護、特に薬品や生物製品に関連する特許保護の強制執行に賛成しておらず、これは、私たちの特許の侵害を阻止したり、私たちの専有権を侵害する方法で競争製品を販売することを困難にする可能性がある。外国の管轄区域で私たちの特許権を強制的に執行する訴訟手続きは、巨額のコストを招き、私たちの努力と注意を私たちの業務の他の側面から移転させる可能性があり、私たちの特許が無効または狭義に解釈されるリスクに直面する可能性があり、私たちの特許出願を発表できないリスクに直面させ、第三者からのクレームを引き起こす可能性がある。私たちは私たちが始めたどのような訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。同様に、私たちのビジネス秘密が外国司法管轄区域で漏洩されれば、世界各地の競争相手は私たちの独自の情報を得ることができ、私たちは満足できる追跡権を持っていないかもしれない。そのような開示は私たちの業務に実質的な悪影響を及ぼすかもしれない。また、私たちが知的財産権を保護し、実行する能力は、外国の知的財産法の意外な変化の悪影響を受ける可能性がある。また,中国やインドを含むいくつかの発展途上国には強制許可法があり,これらの法律により特許権者は第三者に許可を強制的に付与される可能性がある。これらの国/地域では、特許が侵害された場合、または私たちまたは私たちのライセンシーが第三者にライセンスを付与することを余儀なくされた場合、私たちおよび私たちのライセンス者は限られた救済措置を受ける可能性があり、これらの特許の価値を大幅に低下させる可能性がある。また、多くの国は政府機関や政府請負業者に対する特許の実行可能性を制限している。これは私たちの潜在的な収入機会を制限するかもしれない。したがって、私たちが世界各地で私たちの知的財産権を実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネスメリットを得るのに十分ではないかもしれません。
米国や他の管轄区特許法の変化は、特許の全体的な価値を低下させ、私たちの候補製品を保護する能力を弱める可能性がある。
他の生物製薬会社と同様に、私たちの成功は知的財産権、特に特許に大きく依存している。生物製薬業界で特許を獲得と実施することは技術上の複雑性と法律上の複雑性にも関連するため、コストが高く、時間がかかり、しかも内在的な不確定性を持っている。米国特許法または特許法解釈の変化は不確実性やコストを増加させる可能性があり、私たちの発明、取得、維持、および実行能力を弱める可能性があり、より広く言えば、私たちの知的財産権の価値に影響を与えたり、私たちが所有し許可している特許の範囲を縮小したりする可能性がある。2011年9月16日に法律となった“Leahy-Smith America発明法”や“Leahy-Smith Act”に署名することを含む米国と他の国の最近の特許改革立法は、私たちの特許出願をめぐる起訴や、私たちが発表した特許の実行または保護の不確実性とコストを増加させる可能性がある。“ライシー·スミス法案”は米国特許法を多くの重大な改正を行った。これらの条項は、特許出願起訴方式に影響を与える条項を含み、既存技術を再定義し、競争相手に特許の有効性に挑戦するために、より効果的かつ費用効果的な方法を提供する。これらの措置には、第三者が特許訴訟中に米国特許商標局に以前の技術を提出することを可能にすることと、特許の有効性を攻撃するために、ライセンス後審査を含む米国特許商標局によって管理される認可後手続きとが含まれる各方面間審査と派生手続き。2013年3月以降、“ライシー·スミス法案”によれば、米国は第1の発明者から出願制に移行し、このような制度の下で、他の法定要件が満たされたと仮定すると、第1の特許出願を提出した発明者は、第三者が第1の発明によって要求された発明であるか否かにかかわらず、発明の特許を取得する権利がある。したがって、2013年3月以降であるが、同じ発明を含む出願を提出する前に米国特許商標局に特許出願を提出した第三者は、当該第三者が発明を行う前にこの発明を作成したとしても、我々の発明をカバーする特許を付与することができる。これは私たちが発明から特許出願までの提出時間を認識することを要求するだろう。米国およびほとんどの他の国/地域の特許出願は、提出後または発行前の一定期間秘密であるため、(1)私たちの候補製品および私たちが開発する可能性のある他の独自技術に関連する特許出願を提出するか、または(2)私たちまたは私たちのライセンス者の特許または特許出願に要求される任意の発明を発明することが最初であることは確認できない。私たちが効果的かつ強制的に実行可能な特許を持っている場合であっても、他方が私たちの出願日前にその発明を商業に使用していることを証明することができる場合、または他方が強制許可から利益を得ることができる場合、私たちは、他の人が要求された発明を実践することを排除することができない。しかし、Leahy-Smith法案およびその実施は、私たちの特許出願をめぐる起訴および私たちが発行した特許の実行または保護の不確実性およびコストを増加させる可能性があり、これらのすべては、私たちの業務、財務状況、運営結果、および見通しに重大な悪影響を及ぼす可能性がある。
97
また、薬品研究開発と商業化における企業の特許地位は特に不確定である。近年、米国最高裁判所はいくつかの特許事件に対して裁決を下し、場合によっては利用可能な特許保護範囲を縮小するか、場合によっては特許所有者の権利を弱めるかを決定している。米国議会、米国裁判所、米国特許商標局、および他の国の関連立法機関の将来の行動によれば、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、それによって、私たちが新しい特許を獲得したり、私たちの既存の特許と将来獲得可能な特許を実行する能力を弱める可能性がある。例えば2013年の事件ではアソークです。分子病理学V。 万得遺伝株式会社米国最高裁判所は,DNA分子に対するいくつかの主張は特許を申請できないと判断した。私たちが所有または許可したいかなる特許もこの決定によって無効と認定されるとは信じていないが、裁判所、米国議会、またはUSPTOの将来の裁決がどのように私たちの特許価値に影響を与えるかを予測することはできない。
特許保護の獲得と維持は、政府特許機関が提出した様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要件に適合しない場合、私たちの特許保護は減少またはキャンセルされる可能性がある。
特許および/または特許出願の有効期間内に、定期維持費、継続費、年会費、および特許および/または特許出願に関する様々な他の政府費用を、いくつかの段階に分けて米国特許商標局および外国特許代理機関に支払わなければならない。米国特許商標局および各種外国政府特許機関はまた、特許出願中にいくつかのプログラム、文書、費用支払い、およびその他の同様の規定を遵守することを要求している。多くの場合、公共健康危機が私たちまたは私たちの特許維持サプライヤーに与える影響による意外なミスを含むことは、滞納金を支払うことによって、または規則を適用する他の方法で治癒することができるが、場合によっては、規定を遵守しないことは、特許または特許出願が放棄または失効され、関連する管轄区域の特許権の一部または全部を喪失させる可能性がある。特許または特許出願の放棄または失効をもたらす可能性のある規定を遵守しないイベントは、規定された期限内に公式行動に応答できなかったこと、費用を支払わなかったこと、および適切に合法化されず、正式な文書を提出することができなかったことを含むが、これらに限定されない。もし私たちが私たちの候補製品をカバーする特許と特許出願を維持できなければ、私たちの競争的地位は不利な影響を受けるだろう。
私たちは私たちの商業秘密の機密性を保護できなければ、私たちの商業と競争の地位が損なわれるかもしれない追跡と実行が困難な商業秘密とノウハウに依存するかもしれない。
私たちのいくつかの候補技術と製品のための特許を求める以外に、私たちは、特許を取得していないノウハウ、技術、および他の独自の情報を含む商業秘密に依存して、私たちの競争地位を維持することができる。私たちの候補製品の要素は、その製造および製造プロセスを含み、特許がカバーされていない独自技術、情報または技術に関連する可能性があり、したがって、これらの態様については、商業秘密および技術を私たちの主要な知的財産権と見なすことができる。私たちの従業員、私たちの施設を共有する第三者従業員、または研究、臨床試験または製造活動を行う第三者コンサルタントおよびサプライヤーの任意の意図的または意図的な開示、または第三者による私たちのビジネス秘密または独自の情報の流用(例えば、ネットワークセキュリティホールを介して)は、競争相手が私たちの技術的成果をコピーまたは超えることを可能にし、それによって市場における私たちの競争地位を侵食する可能性がある。私たちは私たちの候補製品の開発と製造において第三者に依存したいので、私たちは時々彼らとビジネス秘密を共有しなければならない。私たちの第三者への依存は、私たちのビジネス秘密を共有することを要求し、これは、競争相手がこれらの秘密や私たちのビジネス秘密が流用または開示されていることを発見する可能性を増加させる。
ビジネス秘密と技術的ノウハウを保護することは難しいかもしれない。私たちは従業員に秘密条項を含む書面雇用協定を締結し、彼らが雇用中に発生した任意の発明を私たちに譲渡する義務があることを要求します。私たちは、双方の財産、潜在的な商業秘密、ノウハウ、情報を保護するための守秘義務および知的財産義務を含む、私たちの共有施設の任意の第三者と書面協定を締結します。私たちはさらに、当社の協力者、外部科学協力者、契約研究組織、契約製造業者、コンサルタント、コンサルタント、および他の第三者のような、アクセス権限を取得した当事者と秘密および秘密協定を締結することで、私たちの潜在的なビジネス秘密、ノウハウ、および情報を保護することを求めています。私たちのコンサルタント、請負業者、および外部科学協力者の場合、これらの合意は、一般に発明譲渡義務を含む。私たちは私たちが可能であるか、または私たちの商業秘密またはノウハウとプロセスに接触したすべての当事者とこのような合意に到達したことを保証することができない。私たちは確信できません
98
ビジネス秘密および他の機密固有情報を漏洩することはなく、または競合他社は、我々の商業秘密または独立開発と実質的に同じ情報および技術を他の方法で取得することはない。このような努力にもかかわらず、どちらも合意に違反し、私たちのビジネス秘密を含む私たちの固有の情報を漏洩する可能性があり、私たちは十分な救済措置を得ることができないかもしれない。強制執行側が商業秘密を不正に開示したり流用したりする主張は困難であり、高価で時間がかかり、結果は予測できない。しかも、米国国内外のいくつかの裁判所は商業秘密をあまり望んでいないか、または保護したくない。もし私たちの任意の商業秘密が競争相手または他の第三者によって合法的に取得または独立して開発された場合、私たちは彼らがその技術や情報を使用して私たちと競争することを阻止する権利がないだろう。もし私たちの任意の商業秘密が競争相手または他の第三者に漏洩された場合、または競争相手または他の第三者によって独立して開発された場合、私たちの競争地位は損なわれるだろう。
私たちは私たちの特許と他の知的財産権の発明権または所有権のクレームに挑戦する対象になるかもしれない。
私たちは、元従業員、協力者、または他の第三者が、発明者または共同発明者として、私たちの特許または他の知的財産権において権益を有するクレームの制約を受ける可能性がある。特許出願上適切な発明者が示されていないことは、その上で発行された特許が強制的に実行できない可能性がある。発明権紛争は、発明者として指定された異なる個人の貢献に関する相互矛盾した意見、外国国民が特許標的開発に参加する外国法律の影響、我々の候補製品の開発に参加する第三者の義務衝突、または潜在的な共同発明の共同所有権に関する問題によるものである可能性がある。訴訟は、これらおよび他の挑戦在庫および/または所有権のクレームを解決するために必要である可能性がある。代替または追加として、私たちはこのような知的財産権上の私たちの権利範囲を明確にするために協定を締結することができる。もし私たちがこのようなクレームを弁護することができなければ、金銭損害賠償を支払うことに加えて、貴重な知的財産権、例えば貴重な知的財産権の独占所有権や使用権を失う可能性がある。このような結果は私たちの業務に実質的な悪影響を及ぼすかもしれない。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。
私たちの許可者は、第三者コンサルタントまたは協力者または第三者(例えば、米国政府)からの資金に依存する可能性があり、したがって、私たちの許可者は、私たちが許可を得た特許の唯一および独占所有者ではありません。もし他の第三者が私たちが特許を許可する所有権または他の権利を持っている場合、彼らはこれらの特許を私たちの競争相手に許可し、私たちの競争相手はそれと競争する製品と技術を販売することができるかもしれない。これは私たちの競争地位、業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼすかもしれない。
また、私たちの政策は、知的財産権の概念や開発に参加する可能性のある私たちの従業員と請負業者が合意に署名し、このような知的財産権を私たちに譲渡することを要求しているにもかかわらず、実際に私たちが自分の知的財産権と見なしているすべての当事者とこのような合意を実行することができないかもしれない。知的財産権の譲渡は自動的に実行されない可能性があり、あるいは譲渡協定が違反される可能性があり、私たちは第三者にクレームをつけさせられたり、私たちが私たちの知的財産権の所有権だと思うことを決定するために、私たちが提起する可能性のあるクレームを弁護したりすることができます。このようなクレームは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。
特許条項は候補製品に対する私たちの競争地位を十分に長く保護するのに十分ではないかもしれない。
特許権の期限は限られています。米国では、すべての維持費が適時に支払われる場合、特許の自然失効時間は、通常、その最初の有効出願日から20年後である。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。私たちの候補製品をカバーする特許を取得しても、製品の特許有効期限が切れると、私たちは生物類似薬や模倣薬からの競争に直面する可能性がある。したがって、私たちの特許の組み合わせは、他の会社が私たちと似ているか同じ候補製品を商業化することを排除するために、私たちに十分な権利を提供していないかもしれない。米国では、特許が発行された後、USPTOによるいくつかの遅延に従って特許の寿命を延長することができるが、このような増加は、特許出願人が特許訴訟中にもたらすいくつかの遅延に基づいて減少または除去することができる。特許期間の延長
99
規制に基づく遅延はアメリカで利用可能かもしれない。しかし、各上場承認は1つの特許しか延長できず、1つの特許は単一製品に対して1回しか延長できない。また,特許期間延長期間の保護範囲は,特許請求の範囲のすべてまで延長されるのではなく,承認された製品範囲に限定される。外国司法管轄区域が特許期間延長のような法律の違いを管理することは大きく異なり、1つの特許家族から複数の特許を取得する能力を管理する法律も同様である。さらに、試験段階または規制審査中に職務調査を行うことができず、適用の締め切り内に出願を提出することができず、関連する特許が満了する前に出願を提出することができなかった場合、または他の方法で適用の要求を満たすことができなかった場合、延期されることはないかもしれない。もし私たちが特許期間の延長や回復を得ることができない場合、あるいはこのような延長の期限が私たちが要求しているよりも短い場合、私たちの製品を独占販売する権利がある期間は短縮され、私たちの競争相手は私たちの特許が満期になった後に競争製品の承認を得る可能性があり、私たちの臨床と臨床前のデータを参考にすることで、私たちの開発と臨床試験への投資を利用することができ、他の場合よりも早く彼らの製品を発売することで、私たちの収入は減少する可能性があり、実質的である可能性がある。
もし私たちの商標と商号が十分に保護されていなければ、私たちは興味のある市場で知名度を作ることができないかもしれません。私たちの業務は悪影響を受けるかもしれません。
私たちの現在または未来の商標または商号は、挑戦、侵害、回避、または汎用または記述商標として宣言されるか、または他の商標を侵害していると判断される可能性がある。私たちはこれらの商標や商品名に対する私たちの権利を保護できないかもしれないし、これらの名前の使用を停止させることができないかもしれません。私たちは関心のある市場で潜在的なパートナーや顧客の名前の承認を得るために必要です。商標登録過程で、私たちは米国特許商標局または他の外国司法管轄区から私たちの出願の拒絶を受けることができる。私たちはこのような拒否に答える機会があるだろうが、私たちはこの拒否を克服できないかもしれない。また,米国特許商標局や多くの外国司法管轄区の類似機関では,第三者は係属中の商標出願に反対し,登録商標の取り消しを求める機会がある。私たちの商標に反対またはキャンセル訴訟を提起するかもしれないが、私たちの商標は継続できないかもしれない。もし私たちが私たちの商標と商号に基づいて名称を確立することができなければ、私たちは効果的に競争できない可能性があり、私たちの業務は不利な影響を受けるかもしれない。私たちは私たちの商標と商品名を流通業者のような第三者に許可するかもしれない。これらのライセンス契約は、私たちの商標や商号をどのように使用するかに指針を提供する可能性がありますが、ライセンシーは、これらの合意に違反したり、私たちの商標およびビジネス番号を乱用したりすることで、私たちの権利を危険にさらしたり、私たちの商標や商号に関連する商標の名誉を弱める可能性があります。
また、私たちは、私たちが登録したかどうかにかかわらず、私たちの候補製品と共に使用されるどんな名前もFDAの承認を受けなければならないと提案しています。ヨーロッパにも似たような要求がある。FDAは、通常、他の製品名と混同される可能性がある可能性を含む、提案された製品名を検討する。FDA(または外国司法管轄区の同等の行政機関)が私たちが提案した任意の独自製品名に反対する場合、適用商標法の資格に適合し、第三者の既存の権利を侵害せず、FDAのために受け入れられる適切な代替名を決定するために、多くの追加資源が必要となる可能性がある。さらに、多くの国では、商標登録を所有し維持することは、高級商標所有者がその後に提起した侵害クレームに対して十分な弁護を提供できない可能性がある。時々、競争相手や他の第三者は、私たちと同様の商号や商標を採用して、ブランド表示を確立する能力を阻害し、市場の混乱を招く可能性があります。さらに、他の登録商標または商標の所有者は、我々の登録または未登録商標または商号の変異体を含む商号または商標侵害クレームを提出することができる。もし私たちが商標侵害クレームを主張する場合、裁判所は私たちが主張する商標が無効または強制執行できない、または私たちが商標侵害を主張する側が関連商標に対する優先権を持っていると判断する可能性がある。この場合、私たちは最終的にこのような商標の使用を中止することを余儀なくされるかもしれない。
100
私たちの普通株に関するリスクは
私たちの経営業績にかかわらず、私たちの普通株の市場価格はずっと変動し続けているかもしれません。あるいは低下する可能性があります。あなたはあなたのすべてまたは一部の投資を失うかもしれません。
私たちの普通株の市場価格は多くの要素によって大幅に変動する可能性があります。その中の多くの要素は私たちがコントロールできないことです
また、株式市場、特にナスダック世界市場は、極端な価格と出来高変動を経験し、多くの免疫腫瘍や遺伝子治療会社の株式証券の市場価格に影響を与え続けている。その中の多くの会社の株価変動はその経営業績に関係ないか比例しないが、私たちは過去にも私たちの経営業績とは無関係か比例しない変動を経験したことがある。2023年1月1日から2024年3月4日まで、私たちの普通株の終値は1株1.62ドルから8.73ドルの間です。過去、市場変動期間中、株式市場は証券集団訴訟を起こした。私たちが証券訴訟に巻き込まれれば、私たちに巨額の費用を負担させ、資源や経営陣の私たちの業務への関心を移転させ、私たちの業務に悪影響を及ぼすかもしれません。
私たちの主要株主と経営陣は私たちのかなりの割合の株を持っており、株主の承認が待たれる事項に大きな制御を加えることができるだろう。
2024年3月4日現在、私たちの役員、取締役、5%の株主とその付属会社の実益は、私たちの約56%の議決権付き株を持っています。したがって、このような株主たちは彼らの所有権地位を通じて私たちに影響を与えることができる。この株主たちは株主の承認を必要とするすべての事項を決定することができるかもしれない。例えば、これらの株主が共同で行動することは、取締役選挙を制御し、私たちの組織文書を修正し、任意の合併、資産売却、または他の重大な会社取引を承認することができるかもしれない。これは私たちの普通株に対する能動的な買収提案や要約を阻止または阻止する可能性がありますが、これらの提案や要約は私たちの株主としての最良の利益に合致していると考えられるかもしれません。
101
もし私たちが将来有効な内部統制システムを維持できなければ、私たちの財務状況や運営結果を正確かつタイムリーに報告することができないかもしれません。これは投資家の信頼に悪影響を与え、私たちの普通株の価値に影響を与えるかもしれません。
私たちは“取引所法案”、“サバンズ-オキシリー法案”、“ナスダック株式市場規則と条例”の報告要件を遵守しなければならない。“サバンズ-オキシリー法”(Sarbanes-Oxley Act)は、財務報告書に対して効率的な開示制御と手続きおよび内部統制を維持することを要求する。
重大な欠陥は財務報告内部統制の欠陥または欠陥の組み合わせであり、私たちの年度または中期連結財務諸表の重大なミス報告は合理的な可能性があり、適時に防止または発見できないようにする。私たちは将来、私たちの内部財務と会計制御とプログラムシステムにおける重大な弱点を発見するかもしれません。これは、私たちの連結財務諸表に重大なミスを招く可能性があります。私たちの財政報告書に対する内部統制はすべてのミスと詐欺を阻止したり発見したりしないだろう。設計および動作がどんなに良好であっても、絶対的な保証ではなく、合理的な保証しか提供できず、保証制御システムの目標が実現される制御システム。すべての制御システムの固有の限界により,どの制御評価も誤りや不正による誤り陳述が発生しないことは絶対に保証されず,すべての制御問題や不正が発見されることは絶対に保証されない.
もし私たちがサバンズ-オキシリー法案404条の要求を直ちに守ることができない場合、あるいは財務報告書に対して適切かつ効果的な内部統制を維持できない場合、私たちはタイムリーで正確な財務諸表を作成できないかもしれない。このような状況が発生すれば、私たちの投資家は私たちが報告した財務情報に自信を失う可能性があり、私たちの株式の市場価格は下落する可能性があり、私たちはアメリカ証券取引委員会や他の規制機関の制裁や調査を受けるかもしれない。
一般リスク因子
上場企業として、私たちの運営コストは引き続き大幅に増加し、私たちの経営陣は新しいコンプライアンスを実施するために多くの時間を投入する必要があります。
上場企業として、私たちはすでに大量の法律、会計、その他の費用を発生させ続けており、これらの費用は私たちが民間会社として発生していない。また、サバンズ-オクスリ法案や、米国証券取引委員会がその後に実施したルールや、ナスダックグローバル精選市場が上場企業に提出した様々な要求を守らなければならない。2010年7月、“ドッド·フランクウォール街改革·消費者保護法案”または“ドッド·フランク法案”が公布された。テレス·フランク法案には重要な会社管理と役員報酬に関する条項があり、米国証券取引委員会はこれらの分野で“報酬発言権”や代理アクセスのような追加的な規則を取ることを要求している。“新興成長型会社”としては、2012年のJumpStart Our Business Startups Actで定義されているように、より長い時間と初公募(IPO)完了から最大5年間で多くの要求を実施することが許可されています。私たちはこの新しい立法を継続して利用しようとしていますが、私たちがまだ“新興成長型会社”であることは保証されず、予算や計画よりも早くこれらの要求を実施することが要求され、予期しない費用が発生する可能性があります。株主急進主義、現在の政治環境、および現在の高度な政府介入と規制改革は、大量の新しい法規と開示義務を招く可能性があり、これは追加のコンプライアンスコストを招き、現在予見できない方法で業務を運営する方法に影響を与える可能性がある。私たちの経営陣と他の人たちはこのようなコンプライアンス計画を実施するために多くの時間を投入する必要があるだろう。しかも、このような規則と規制は私たちの法律と財務コンプライアンスコストを増加させ、いくつかの活動をより時間とコストを高くするだろう。例えば、これらの規則や規定は、取締役や上級管理者責任保険をより難しく、より高価にすることが予想され、現在のこのような保険レベルを維持するために多くのコストが必要になるかもしれません。上場企業として私たちに課せられた要求を守るために、毎年約400万~500万ドルの追加費用が発生すると思います。
私たちの従業員、主要な調査者、コンサルタント、ビジネスパートナーは、規制基準と要求を遵守しないこと、およびインサイダー取引を含む不正行為または他の不正活動に従事する可能性があります。
私たちは従業員、主な調査者、コンサルタント、ビジネスパートナーの詐欺や他の不適切な行為のリスクに直面している。これらの当事者の不正行為には、FDAと非米国規制機関の規定を故意に遵守せず、FDAと非米国規制機関に正確な情報を提供することが含まれる可能性がある
102
米国や海外の医療詐欺や法律法規の乱用を遵守し、財務情報やデータを正確に報告したり、不正な活動を開示したりする。特に、医療業界の販売、マーケティング、商業配置は、詐欺、不正行為、リベート、自己取引、その他の乱用行為を防止するための広範な法律法規によって制約されている。これらの法律法規は、広範な価格設定、割引、マーケティングおよび販売促進、販売手数料、顧客インセンティブ計画、および他のビジネス計画を制限または禁止する可能性があります。このような不正行為はまた臨床試験過程で得られた情報を不当に使用する可能性があり、これは監督部門の制裁を招き、著者らの名声に深刻な損害を与える可能性がある。従業員の不正行為を常に識別し、阻止することができるわけではなく、このような行為を発見し、防止するための予防措置は、未知または未管理のリスクや損失を効果的に制御することができないか、またはこれらの法律や法規を遵守できないことによる政府の調査または他の行動または訴訟から私たちを保護することができないかもしれない。私たちにこのような訴訟を提起すれば、これらの訴訟は、連邦医療保険や医療補助、契約損害、名声損害、利益減少および将来の収益減少、追加の報告義務および監督(会社の誠実な合意または他の合意の制約を受けた場合)を含む、重大な民事、刑事および行政処罰、損害賠償、罰金、監禁、連邦医療保険および医療補助、契約損害、名声損害、利益減少および将来の収益減少、追加の報告義務および監督(会社の誠実な合意または他の合意の制約を受けた場合)を適用して、これらの法律違反に関する疑惑を解決し、私たちの業務を削減または再構成することを含む、私たちの業務に大きな影響を与えるかもしれない。
もし私たちの情報技術システムまたはデータまたは私たちが依存している第三者のシステムまたはデータが破壊された場合、私たちは規制調査または行動を含むが、制限されないが、訴訟、罰金と処罰、私たちの業務運営の中断、私たちの候補製品の開発計画の実質的な破壊、名声損害、収入または利益の損失、顧客または販売の損失、および他の不利な結果を含むこのような損害を経験する可能性がある。
私たちはますます情報技術システム、インフラ、データに依存して私たちの業務を運営している。通常のビジネスプロセスでは、我々は、個人データ(例えば、健康関連データ)、知的財産権、および商業秘密(総称して“敏感情報”)を含む個人データ(例えば、健康関連データ)、知的財産権、および商業秘密(総称して“敏感情報”と総称する)を収集、受信、記憶、処理、生成、使用、送信、開示、アクセス可能、保護、処置、送信および共有(総称して“処理”と呼ぶ)する。重要なのは、私たちはこのような機密情報の機密性と完全性を維持するために、安全な方法でそうしなければならないということだ。我々はまた,我々の運営要素を第三者に外注するため,我々の機密情報にアクセスできる第三者請負者を多く管理している.このような第三者のサイバーセキュリティアプローチを監視する能力は限られており、これらの第三者には十分な情報セキュリティ対策が整っていない可能性がある。
ネットワーク攻撃、インターネットベースの悪意のある活動、オンラインおよびオフライン詐欺、および他の同様の活動は、私たちの敏感な情報および情報技術システム、ならびに私たちが依存する第三者システムの機密性、完全性、および可用性を脅かす。このような脅威は一般的で、増加し続けており、ますます気づきにくくなっている。このような脅威は様々な源から来ている。伝統的なコンピュータ“ハッカー”のほかに、行為者、人員(例えば窃盗や乱用)、複雑な民族国家、民族国家によって支持される行為者も現在、攻撃に参加している。一部の脅威行為者は現在、地政学的な理由で軍事衝突と防御活動を組み合わせた民族国家行為者を含むサイバー攻撃に従事し続けることが予想されている。戦争と他の重大な衝突の間、私たちと私たちが依存している第三者は、報復ネットワーク攻撃を含むこれらの攻撃のリスクが増加しやすいかもしれません。これは、私たちのシステムと行動を実質的に混乱させるかもしれません。
私たちと私たちが依存している第三者は、社会工学攻撃(深さ偽装による偽造を含む、ウイルスおよびワームのような)、マルウェア(高度な持続的な脅威侵入の結果を含む)、サービス拒否攻撃、証拠充填、人員不正行為またはエラー、恐喝ソフトウェア攻撃、サプライチェーン攻撃、ソフトウェア脆弱性、サーバ障害、ソフトウェアまたはハードウェア障害、データまたは他の情報技術資産損失、広告ソフトウェア、電気通信障害、地震、火災、洪水など、様々な変化する脅威を受ける可能性があります。人工知能の強化や協力的な攻撃、他の似たような脅威。組織犯罪脅威行為者、民族国家、民族国家によって支持される行為者によって実施される攻撃を含む恐喝ソフトウェア攻撃は、ますます一般的かつ深刻になっており、私たちの運営の深刻な中断、製品またはサービスを提供する能力、データおよび収入損失、名声損害、および資金移転を招く可能性がある。恐喝支払いは恐喝ソフトウェア攻撃の否定的な影響を軽減するかもしれないが、例えば、適用された法律または法規によってそのような支払いが禁止されているため、私たちはそのような支払いを望んでいないか、または支払うことができないかもしれない。同様にサプライチェーン攻撃は
103
私たちは、私たちのサプライチェーンまたは私たちの第三者パートナーのサプライチェーン内の第三者およびインフラが損なわれていないことを保証することはできませんし、利用可能な欠陥や脆弱性を含まない保証はありません。これらの欠陥や脆弱性は、私たちの情報技術システムや私たちと私たちのサービスをサポートする第三者情報技術システムを破壊または中断させる可能性があります。将来的または過去のビジネス取引(例えば、買収または統合)は、我々のシステムが、買収または統合エンティティのシステムおよび技術に存在する脆弱性の負の影響を受ける可能性があるので、より多くのネットワークセキュリティリスクおよび脆弱性に直面する可能性がある。また,これらの買収や統合されたエンティティを職務調査する際に発見されなかったセキュリティ問題が発見される可能性があり,会社を我々の情報技術環境やセキュリティ計画に統合することは困難である可能性がある.
以前に決定されたまたは同様の脅威は、セキュリティイベントまたは他の中断をもたらす可能性がある。セキュリティイベントまたは他の中断は、許可されていない、不法または意外に取得、修正、廃棄、損失、変更、暗号化、開示、またはデータへのアクセスをもたらす可能性があり、または私たち(および私たちが依存する第三者)がサービスを提供する能力を乱す可能性がある。このような事件が発生したり、発生したと考えられたりすると、我々の製品開発計画や業務運営の実質的な中断を招く可能性がある。これらの脅威は,我々のシステムのセキュリティ,セキュリティおよび我々のデータの可用性と完全性にリスクとなり,これらのリスクは我々のシステムに依存して業務を行う第三者にも適用可能である.もし私たちの第三者サービス提供者がセキュリティ事件や他の中断に遭遇したら、私たちはまた悪い結果に直面するかもしれない。第三者サービス提供者がプライバシーや安全に関する義務を履行できなかった場合、損害賠償を受ける権利があるかもしれませんが、どの賠償も私たちの損害を補うのに十分ではないかもしれません。あるいはそのような賠償を取り戻すことができないかもしれません。
私たちは、セキュリティイベントを防止し、私たちの情報システム(例えば、私たちのハードウェアおよび/またはソフトウェア、私たちが依存する第三者のハードウェアおよび/またはソフトウェアを含む)における脆弱性を検出、緩和、および修復するために、大量のリソースを費やしたり、私たちのビジネス活動(私たちの臨床試験活動を含む)を修正したりする可能性があります。特定のデータプライバシーおよびセキュリティ義務は、私たちの情報技術システムおよびデータを保護するために、特定のセキュリティ対策、業界標準、または合理的なセキュリティ対策を実施し、維持することを要求する可能性があります。セキュリティ対策が実施されているにもかかわらず、これらの措置の規模と複雑さと、私たちと私たちが依存する第三者が保存している機密情報がますます多くなっていることを考慮して、これらの措置が有効であることは保証されない。私たちは、脆弱性を利用するためのこのような脅威と技術が常に変化し、しばしば複雑であるため、私たちの情報技術システムにおけるすべての脆弱性をタイムリーに発見して修復することができないかもしれない。したがって、このような脆弱性はセキュリティ事件が発生した後に検出されるかもしれない。私たちは私たちの情報技術システムの抜け穴を認識して修復しようと努力しているにもかかわらず、私たちの努力は成功しないかもしれない。さらに、私たちはこのような決定された抜け穴を解決するための救済措置を制定して配置することに遅延があるかもしれない。このような抜け穴は私たちの業務に実質的な危険をもたらし、利用され、セキュリティ事件を招く可能性がある。
私たちは私たちのデータ保護努力と情報技術への投資がセキュリティ事件の発生を防ぐことができるかどうかを確認することができない。このような事件に遭遇した場合、適用されるデータプライバシーおよびセキュリティ義務は、影響を受けた個人、顧客、規制機関、および投資家を含むセキュリティイベントを関連する利害関係者に通知することを要求する可能性があります。このような開示は費用が高く、そのような要求を開示または遵守しないことは悪い結果をもたらすかもしれない。もし私たち(または私たちが依存する第三者)がセキュリティ事件を経験した場合、またはセキュリティ事件を経験したと考えられる場合、私たちは、政府の法執行行動(例えば、調査、罰金、処罰、監査、および検査)、追加の報告要件および/または監視、データ(個人データを含む)の処理の制限、訴訟(カテゴリクレームを含む)、賠償義務、負の宣伝、名声損害、管理層の注意の移動、金銭支出、私たちの運営中断(データ利用可能性を含む)、財務損失、および他の同様の損害に遭遇する可能性がある。セキュリティ事件とそれに伴う結果は、私たちの候補製品の開発遅延を招き、顧客が私たちの製品やサービスの使用を停止し、新しい顧客が私たちの製品やサービスを使用することを阻止し、私たちの業務の成長と運営能力にマイナスの影響を与える可能性がある。
私たちの契約には責任制限が含まれていないかもしれませんが、あっても、私たちの契約における責任制限は、私たちのデータプライバシーとセキュリティ義務に関連する責任、損害、またはクレームから私たちを保護するのに十分である保証はありません。私たちの保険範囲が私たちのプライバシーと安全慣行によって生じる責任から私たちを保護または軽減するのに十分か、または私たちを保護するのに十分かどうかは確認できません。私たちはこのような保険が商業的に合理的な条項や根本的に存在しない、あるいはそのような保険が未来のクレームを支払うと判断することはできません。
104
私たちが業務を拡大し、顧客群を拡大し、独自かつ敏感なデータを処理、保存、転送することによって、私たちのリスクが増加する可能性があります。
私たちまたは私たちの顧客に不利な税金法律または法規の変化は、私たちの業務、キャッシュフロー、財務状況、または経営結果に重大な悪影響を及ぼす可能性があります。
新しい収入、販売、使用、または他の税金法律、法規、規則、法規または条例はいつでも公布される可能性があり、これは私たちの業務運営および財務業績に悪影響を及ぼす可能性があります。さらに、既存の税金法律、法規、規則、法規または条例は、私たちに解釈、変更、修正、または適用される可能性がある。例えば、2017年に公布された非公式名称は減税·雇用法案、コロナウイルス援助、救済·経済安全法案、アイルランド共和軍が米国税法に多くの重大な改正を行った。国税局や他の税務機関の将来のこのような立法の指導は私たちに影響を与える可能性があり、このような立法のいくつかの側面は将来の立法で廃止または修正される可能性がある。また,各州がどの程度連邦税法を遵守するかは不明である。将来の税改正立法は、私たちの繰延税金資産の価値に実質的な影響を与える可能性があり、大量の一次費用を招く可能性があり、将来のアメリカでの私たちの税金支出を増加させる可能性がある。
2022年1月1日から、減税·雇用法案は、発生年度に税収目的の研究開発費を差し引く選択肢を廃止し、納税者に5年以内に米国内で行われた研究活動のためにこれらの費用を資本化し、15年以内に米国国外で行われた研究活動のためにこれらの費用を償却することを求めている。廃止や資本化要求を数年後に延期する立法提案があるが、この規定が廃止されたり、他の方法で改正される保証はない。国税局や他の税務機関の将来のこのような立法の指導は私たちに影響を与える可能性があり、このような立法のいくつかの側面は将来の立法で廃止または修正される可能性がある。
私たちは厳格で変化しているアメリカと外国の法律、法規と規則、契約義務、業界基準、政策、その他のデータプライバシーとセキュリティに関する義務を受けています。私たちが実際にまたは健康およびデータ保護義務を遵守できなかったと考えられることは、規制調査または行動(民事または刑事罰を含む可能性がある)、個人訴訟(集団訴訟を含む可能性がある)、および大規模な仲裁要求、罰金および処罰、業務運営中断、名声損害、収入または利益損失および/または負の宣伝を招き、私たちの経営業績および業務に負の影響を与える可能性がある。
私たちは、個人データおよび他の敏感なデータ(私たちが収集した臨床試験に関連する試験参加者の健康データを含む)、独自および機密の商業データ、商業秘密、知的財産権、および敏感な第三者データを処理する。私たちのデータ処理活動は私たちに多くのデータプライバシーとセキュリティ義務を負わせた。したがって、私たちおよび任意の潜在的な協力者は、様々な法律、法規、ガイドライン、業界基準、外部および内部プライバシーおよびセキュリティポリシー、契約、およびデータプライバシーおよびセキュリティに関する義務、または私たちを管理し、私たちを代表して個人データを処理する他の義務など、多くの連邦、州、および外国データプライバシーおよび保護義務の制約を受ける可能性があります。
米国、ヨーロッパ諸国、私たちが業務を展開している他の国では、データプライバシーや情報セキュリティが大きな問題となっている。プライバシーやセキュリティ問題に対する法律や規制の枠組みが急速に変化しており,コンプライアンスコストや責任の開放が増加することが予想される。米国には、連邦健康情報プライバシー法、違反通知法、健康情報プライバシー法、個人データプライバシー法、連邦および州消費者保護法(例えば、連邦貿易委員会法第5条)、および他の同様の法律(例えば、盗聴および録音法)を含む多くの連邦および州法律があり、健康関連情報および他の個人情報の収集、使用、開示および保護を管理する法律は、私たちの運営または私たちの協力者の運営に適用される可能性がある。過去数年間、米国の多くの州-カリフォルニア州、バージニア州、コロラド州、コネチカット州、ユタ州を含めて全面的なプライバシー法が公布され、プライバシー通知に具体的な開示を提供し、その個人データに関するいくつかの権利を住民に提供することを含む、カバーされた企業にいくつかの義務が加えられた。適用可能であれば、そのような権利は、特定の個人データにアクセス、訂正または削除する権利、および指向性広告、分析、および自動決定のような特定のデータ処理活動から退出する権利を選択する権利を含むことができる。このような権利の行使は私たちの業務と製品とサービスを提供する能力に影響を及ぼすかもしれない。いくつかの州はまた、データプライバシーの影響など、敏感な情報を含むいくつかの個人データの処理により厳しい要求を提出している
105
評価する。この州の法律は規定を守らない行為に法的罰金を科すことを許可している。例えば、2020年に“カリフォルニアプライバシー権法案”(CCPA)によって改正された2018年“カリフォルニア消費者プライバシー法”(California Consumer Privacy Act)は、消費者、企業代表、および従業員の個人情報に適用され、プライバシー通知において具体的な開示を提供し、個人が特定のプライバシー権を行使する要求を尊重することを企業に要求する。CCPAは、故意違反行為ごとに最高7500ドルの民事罰金を科すことができ、特定のデータ漏洩の影響を受けた個人訴訟当事者が重大な法定損害賠償を取り戻すことを許可すると規定している。CCPAはいくつかの臨床試験で処理されたデータを免除したが、CCPAはコンプライアンスコストを増加させ、著者らが維持しているカリフォルニア住民の他の個人データに関する潜在的な責任を増加させた。また,近年連邦,州,地方の各レベルでも同様のデータプライバシーやセキュリティ法律が提案されており,今後より多くの州が類似した法律を通過することが予想され,コンプライアンス作業をさらに複雑化させ,我々と我々が依存する第三者の法的リスクやコンプライアンスコストを増加させることが予想される.これらの法律および/または新しいまたは改正されたデータプライバシー法の制約を受けた場合、私たちの法執行行動に対するリスクは増加する可能性があり、私たちは規制の枠組みの下での義務の制約を受ける可能性があり、私たちに訴訟を提起する可能性のある個人または実体の数が増加する可能性があり(個人訴訟を通じて権利を通過する個人を含む)、また、私たちのコンプライアンス努力をさらに複雑化させる可能性があるからである。私たちは消費者の健康データのプライバシーを管理する新しい法律によって制限されるかもしれない。例えば、ワシントンの“私の健康私のデータ法案”(MHMD)は、消費者の健康データを広く定義し、消費者の健康データの処理に制限を加え(同意に厳しい要求を加えることを含む)、消費者にその健康データに関するいくつかの権利を提供し、個人が違法行為を起訴することを可能にする個人訴訟権利を作成する。他の州は似たような法律を考慮して通過する可能性がある。
また,我々は第三者(臨床試験データを取得した研究機関を含む)から健康情報を取得する可能性があり,これらの情報はHIPAA(HITECH改訂)のプライバシーやセキュリティ要求に制約されており,このプロトコルは個人が識別可能な健康情報のプライバシー,セキュリティ,送信に具体的な要求をしている.もし私たちがHIPAAに違反したら、私たちは重大な処罰を受けるかもしれない。さらに、プライバシー権擁護者や業界団体が提案され、将来的には法律や契約で守らなければならない基準が提案される可能性がある。さらに、個人データを処理するためには、様々なプライバシー法や他の義務によって、何らかの同意を得る必要があるかもしれません。例えば、チャットロボットおよびセッション再放送プロバイダを含む様々な方法によって第三者から消費者情報を取得する場合、または第三者マーケティング画素を介して、私たちのいくつかのデータ処理アプローチは、盗聴法律の挑戦を受ける可能性がある。このような接近はますます多くの集団訴訟原告の挑戦を受けるかもしれない。私たちはこれらのやり方について同意を得ることができないか、あるいは同意を得ることができず、集団訴訟と大規模な仲裁要求を含む不良な結果を招く可能性がある。
米国以外では,我々が業務を行っているほとんどの管轄区域で,健康に関する情報や他の個人情報にも適用可能な独自のデータセキュリティやプライバシーの法的枠組みが構築されている.例えば,EUの一般データ保護条例(EU GDPR)やイギリスのGDPR(イギリスGDPR)は個人データの処理に厳しい要求をしている。例えば、EU GDPRによれば、政府規制機関は、データ処理に対して一時的または最終的な禁止を実施し、EU GDPRに基づいて最高2000万ユーロの罰金を科すことができ、英国GDPRに基づいて最高1750万ポンドの罰金を科すか、場合によっては世界の年収の4%に罰金を科し、両者は大きな額を基準にして、または法的権限によってその利益を表すデータ主体または消費者保護組織カテゴリの個人データを処理することに関する個人訴訟を行うことができる。EUデータ保護構造の不安定な性質は、内部コンプライアンスの巨額の運営コストとわが業務のリスクを招く可能性がある。
通常の業務の過程で、私たちは個人データをヨーロッパや他の司法管轄区域からアメリカや他の国に移すことができる。ある司法管轄区域はすでにデータ現地化法律と国境を越えた個人データ移転法を制定した。例えば、ヨーロッパや他の司法管轄区域では、データの現地化や他の国への個人データの移転を制限する法律が制定されている。特に,欧州経済圏(European Economic Area,EEAと略す)やイギリスは,米国や他のプライバシー法が不足していると考えられている国への個人データの移転に大きな制限を与えている.他の司法管轄区域はそのデータ現地化と国境を越えたデータ転送法に対して類似の厳格な解釈を行う可能性がある。現在、欧州経済区の標準契約条項、イギリスの国際データ転送プロトコル/付録、EU-米国データプライバシーの枠組み(自己認証コンプライアンスへの移行を許可し、この枠組みに参加する関連米国組織)のような個人データを法に基づいてヨーロッパ経済区およびイギリスから米国に移転するための様々なメカニズムがあるが、これらのメカニズムは法的挑戦を受けており、これらの措置を満たしたり依存したりして個人データを合法的に米国に転送できる保証はない。ヨーロッパ経済圏やイギリスや他の地域から個人データを移動させる合法的な方法がなければ
106
もし私たちが米国司法管轄区に業務を移した場合、またはコンプライアンス移転の要求が煩雑すぎる場合、私たちは、私たちの業務中断または降格、高いコストで業務の一部または全部またはデータ処理活動を他の司法管轄区(例えば、ヨーロッパ)に移す必要があり、より多くの規制行動に直面し、巨額の罰金と処罰、データを転送できない、パートナー、サプライヤーおよび他の第三者との協力、私たちの業務を処理または移転するために必要な個人データの処理または移転を禁止する禁止を含む深刻な不利な結果に直面する可能性があります。また,個人データをヨーロッパ経済区やイギリスから他の司法管轄区,特に米国に移転した会社は,規制機関,個人訴訟当事者,維権団体のより厳しい審査を受けることになる。一部の欧州規制機関は、これらの会社がGDPRのクロスボーダーデータ転送制限に違反している疑いがあるため、一部の企業に外部へのデータ転送の一時停止または永久停止を命じている。
私たちはまた、データプライバシーやセキュリティに関する契約義務の制約を受けており、これらの義務を守る努力は成功しないかもしれません。例えば、GDPRおよびCCPAのようないくつかのプライバシー法は、私たちのクライアントがそのサービスプロバイダに特定の契約制限を適用することを要求する。我々は、データプライバシーおよびセキュリティに関連するいくつかの認証または自律原則を遵守するなど、プライバシーポリシー、マーケティング材料、および他の声明を発表します。もしこのような政策、材料、または声明が私たちの接近に欠陥があることが発見された場合、透明性の欠如、詐欺性、不公平または不実であれば、私たちは規制機関の調査、法執行行動、または他の不利な結果を受けるかもしれない。
データプライバシーやセキュリティ(および個人データプライバシー期待)に関する義務は急速に変化しており,ますます厳しくなり,規制の不確実性をもたらしている.さらに、これらの義務は、法ドメイン間で不一致または衝突する可能性がある異なる適用および解釈される可能性がある。これらの義務を準備し、遵守するには、私たちのサービス、情報技術、システム、やり方、および個人データを処理する任意の第三者を代表するサービス、情報技術、システム、およびやり方を変更する必要があるかもしれません。適用されるすべてのデータプライバシーおよびセキュリティ義務を遵守しようと努力しているにもかかわらず、私たちはそれをできなかった場合があります(またはできなかったとみなされる)。さらに、私たちが努力したにもかかわらず、私たちが依存している人たちや第三者がこのような義務を履行できない可能性があり、これは私たちのコンプライアンス状態に影響を及ぼすかもしれない。例えば、第三者加工業者が適用される法律、法規、または契約義務を遵守できないことは、私たちの業務を運営できないこと、政府の実体または他の人が私たちに提起した訴訟を含む悪影響をもたらす可能性がある。米国および国際データ保護法律および法規を遵守していないか、または遵守されていないと考えられることは、政府の法執行行動(民事または刑事罰調査、罰金、監査および検査を含む可能性がある)、個人訴訟(カテゴリに関連するクレームを含む)および大規模仲裁要件、違反報告要件、追加の報告要件および/または監督、個人データの処理を禁止し、個人データを廃棄または使用しない命令、および/または負の宣伝をもたらし、私たちの経営業績および業務に負の影響を与える可能性がある。特に,原告は集団クレームや大規模仲裁要求を含むプライバシーに関するクレームを積極的に会社に提起してきた.その中のいくつかのクレームは毎回違反した上で法定損害賠償を取り戻すことを許可し、実行可能であれば、巨大な法定損害賠償をもたらす可能性があり、具体的にはデータ量と違反数量に依存する。また,我々または我々の潜在的協力者が情報を取得する臨床試験対象,およびこれらの情報を共有する提供者は,我々の情報の使用や開示能力を契約的に制限する可能性がある。私たちがプライバシー権を侵害していると主張し、データ保護法を遵守できなかったり、私たちの契約義務に違反したりして、私たちが責任を負わなくても、弁護の費用が高く、時間がかかり、否定的な宣伝を招き、私たちの業務を損なう可能性があります。このようなイベントは、顧客流出、業務運営中断または中断(関連する臨床試験を含む)、個人データを処理できない、またはいくつかの司法管轄区域で運営できない、時間および資源をかけて任意のクレームまたは照会を正当化すること、または私たちの業務モデルまたは運営に重大な変化が生じることを含むが、これらに限定されないが、これらのようなイベントは、私たちの名声、業務または財務状態に重大な悪影響を及ぼす可能性がある。
ソーシャルメディアプラットフォームは私たちの業務に新しいリスクと挑戦をもたらします.
ソーシャルメディアの持続的な拡張に伴い、それはまた私たちに新しい危険と挑戦をもたらしてくれる。ソーシャルメディアは、私たち、私たちの計画、そして私たちの候補製品が治療のために開発されている疾患に関する情報を交流するためにますます使用されている。製薬やバイオテクノロジー業界のソーシャルメディア実践が進化しており,不確実性と我々の業務に適用される法規を遵守しないリスクをもたらしている。例えば、患者は、ソーシャルメディアプラットフォームを使用して、製品または候補製品の有効性または不良体験についてコメントする可能性があり、これは、報告義務または他の結果をもたらす可能性がある。さらに、私たちのスタッフや他の人がメディアチャネルを介して意外または意図的に非公開情報を開示することは、おそらく
107
情報が失われる。さらに、任意のソーシャルメディアプラットフォーム上で、敏感な情報または私たち、私たちの製品、または私たちの候補製品に関する否定的または不正確な投稿またはコメントを不適切に開示するリスクがある。もしこのような事件が発生したり、私たちが適用された法規を遵守できなかった場合、私たちは責任を招き、制限的な規制行動に直面したり、私たちの業務に他の損害を与えたり、私たちの名声、ブランドイメージ、商業的名声に迅速で不可逆的な損害を与えることを含むかもしれません。
私たちはアメリカと外国のいくつかの反腐敗、反マネーロンダリング、輸出規制、制裁、その他の貿易法律と法規の制約を受けている。私たちは違反によって深刻な結果に直面するかもしれない。
米国および外国の反腐敗、反マネーロンダリング、輸出規制、制裁および他の貿易法律法規、または総称して貿易法と呼ばれ、会社およびその従業員、代理人、CRO、法律顧問、会計士、コンサルタント、請負業者および他のパートナーの許可、承諾、提供、提供、誘致、直接または間接的に公共または民間部門の受取人の腐敗または不当な支払いまたは任意の他の価値のあるものを受け入れることを禁止する。貿易法違反は、大量の刑事罰金と民事処罰、監禁、貿易特権の喪失、資格取り消し、税収の再評価、契約違反と詐欺訴訟、名声損害、その他の結果を招く可能性がある。私たちは政府機関や政府付属病院、大学、その他の組織の役人や従業員と直接または間接的な相互作用を持っている。私たちはまた、時間が経つにつれて、アメリカ以外での私たちの活動が増加すると予想している。我々は、第三者に依存した研究、臨床前研究および臨床試験、および/または必要な許可、許可証、特許登録、および他の市場承認を得ることが予想される。私たちは、私たちがそのような活動を明確に許可していなくても、私たちの人員、代理、またはパートナーの腐敗や他の不法活動に責任を負うことを要求されるかもしれない。
もし私たちが環境、健康、安全の法律法規を守らなければ、私たちは罰金や罰金を科されたり、コストを発生したりする可能性があり、これは私たちの業務の成功に実質的な悪影響を及ぼすかもしれない。
私たちおよび私たちと施設を共有する第三者は、実験室の手続きおよび処理、使用、貯蔵、処理、危険材料および廃棄物の管理を含む多くの環境、健康および安全な法律と法規を遵守しなければならない。私たちのすべての行動は化学品、生物学的、そして放射性材料を含む危険で燃えやすい材料の使用に関するものだ。私たちのすべての業務は危険な廃棄物製品を生成するだろう。私たちは一般的に第三者と契約を結び、このような材料と廃棄物を処理する。私たちはこのような材料が汚染や傷害をもたらす危険を除去することができない。もし私たちが私たちと施設を共有している第三者が危険材料を使用して汚染や損傷をもたらした場合、私たちはそれによるいかなる損害に責任を負うかもしれません。どんな責任も私たちの資源範囲を超えている可能性があります。私たちはまた民事や刑事罰金と処罰に関連した巨額の費用を発生させるかもしれない。
危険材料の使用による従業員の負傷により生じる可能性のあるコストや支出を支払うために労働者補償保険を維持しているが、潜在的な責任を支払うのに十分ではない可能性がある。私たちは私たちが生物、危険または放射性物質を貯蔵したり処分したりすることによって、私たちが提起した環境責任や有毒侵害に対して保険を維持することはできません。
また、現在または未来の環境、健康、安全法律法規を遵守するためには、巨額のコストが生じる可能性がある。このような現行または未来の法律法規は私たちの研究と開発を損なうかもしれない。このような法律法規を遵守しないことはまた巨額の罰金、処罰、または他の制裁につながる可能性がある。
不利で不安定な市場と経済状況は、私たちの業務、財務状況、および株価に深刻な悪影響を及ぼす可能性がある。
我々の経営業績は、米国と世界経済の全体的な状況、米国と世界の金融市場、および不利な地政学的およびマクロ経済発展の悪影響を受ける可能性がある。部品不足や関連するサプライチェーンの挑戦、公衆衛生危機などの地政学的事態の発展、ウクライナとロシア間の衝突と関連制裁、銀行の倒産、上昇するインフレ率、中央銀行当局がこのようなインフレをコントロールする措置など、多くの要因により、米国と世界の市場と経済状況は引き続き妨害と変動を受けることになる。私たちの業務、財務状況、あるいは経営結果に影響を与える可能性のある一般的な商業と経済状況は、経済成長、債務と株式資本市場の変動、世界金融市場の流動性、そして私たちの流動性を獲得することを含む
108
アメリカの銀行システム内では、信用の獲得性とコスト、投資家と消費者の自信、そして私たち、私たちのメーカーと私たちのサプライヤーがいる経済の実力。
また、ロシアがウクライナに侵攻した後、世界各地の金融市場は変動を経験した。侵入への対応として、米国、連合王国、EUは他の国とともに、ロシア、ロシア銀行、一部のロシア個人に対して新たな重大な制裁と輸出規制を実施し、将来的に追加の制裁を実施するか、またはさらなる懲罰行動をとる可能性がある。ロシアに対する制裁(および将来実施される可能性のある懲罰的措置)およびロシアが実施する反措置、およびウクライナとロシア間の持続的な軍事衝突および関連制裁(これらの影響が周辺地域に拡大する可能性が予想される)のすべての経済的および社会的影響は依然として不確定である;しかし、紛争および関連制裁は、欧州と世界の貿易、商業、価格安定、信用供給、サプライチェーンの連続性、流動性を得る機会の減少を招き、世界市場に大きな不確実性をもたらし続ける可能性がある。特に、継続的なロシア-ウクライナ紛争や関連制裁は、欧州や他の先進経済体の生活コストを急速に上昇させている(主により高いエネルギー価格によって推進されている)。しかも、経済が疲弊したり下落したりすることは、私たちのサプライヤーと製造業者に圧力を与えるかもしれない。したがって、私たちの業務と業務結果は、特にそれがより多くの国、さらなる経済制裁、またはより広い軍事衝突に関連する程度にアップグレードすれば、ウクライナとロシアとの間の持続的な衝突と関連する制裁の悪影響を受ける可能性がある。
将来的に私たちの株式を売却して発行したり、私たちの株式を購入する権利を含めて、私たちの株式インセンティブ計画によると、私たちの株主の所有権の割合が希釈され、私たちの株価が下落する可能性があります。
未来には私たちが計画している業務を継続するための追加的な資金が必要になるだろう。ある程度、私たちは株式証券を発行することで追加資本を調達し、私たちの株主は深刻な希釈を経験するかもしれない。私たちは1回または複数回の取引で、私たちが時々決定した価格および方法で普通株、転換可能証券、または他の株式証券を販売することができる。もし私たちが1つ以上の取引で普通株、転換可能証券、または他の株式証券を販売すれば、投資家はその後の売却によって深刻に希釈されるかもしれない。これらの売却はまた、私たちの既存株主の実質的な希釈をもたらす可能性があり、新しい投資家は私たちの既存株主よりも優れた権利を得る可能性がある。
私たちの2020年の持分インセンティブ計画または2020年計画によると、私たちの経営陣は、私たちの従業員、取締役、コンサルタントに株式オプションおよびその他の持分ベースの奨励を付与することを許可されています。我々の2020計画によると、発行のために予約された普通株式数は、2021年1月1日から2030年1月1日まで毎年1月1日に自動的に増加し、金額は(I)毎回の自動増資日前のカレンダー月最終日の私たちの普通株式総流通株数の5%に相当するか、または(Ii)我々の取締役会が適用される1月1日までに決定したより少ない数の普通株式に相当する。もし私たちの取締役会が毎年最大額で将来付与可能な株式数を増加させることを選択すれば、私たちの株主は追加的な希釈を経験する可能性があり、これは私たちの株価を下落させる可能性がある。
私たちは証券集団訴訟の影響を受けるかもしれない。
従来、証券集団訴訟は、ある会社の証券市場価格が下落した後に提起されることが多かった。このリスクは製薬会社が近年大幅な株価変動を経験しているため、私たちに特に関連している。私たちがこのような訴訟に直面すれば、巨額のコストを招き、経営陣の注意と資源を移転させる可能性があり、これは私たちの業務を損なう可能性があります。
証券や業界アナリストが私たちの株に不利または誤った意見を発表すれば、私たちの株価や取引量は低下する可能性がある。
私たちの普通株の取引市場は、業界や証券アナリストが発表した私たちまたは私たちの業務に関する研究と報告の影響を受ける可能性があります。もし私たちのどのアナリストが私たち、私たちのビジネスモデル、私たちの知的財産権、あるいは私たちの株式表現に否定的または誤った意見を発表した場合、あるいは臨床試験と運営結果がアナリストの期待に達していなければ、私たちの普通株の取引価格は否定的な影響を受けるだろう。これらのアナリストのうちの1人以上が私たちの報告書を停止したり、私たちの報告書を定期的に発表できなかった場合、私たちの普通株に対する需要が減少する可能性があり、これは私たちの普通株価格と取引量を低下させる可能性がある。
109
私たちの開示統制と手続きはすべてのミスや詐欺を阻止したり検出できないかもしれない。
私たちは“取引所法案”の定期報告書の要求事項を守らなければならない。我々の開示制御および手続きは、取引所法案に基づいて提出または提出された報告書に開示されなければならない情報が蓄積され、管理層に伝達され、米国証券取引委員会規則および表に指定された期間内に記録、処理、まとめ、報告されなければならないことを合理的に確保するために設計されている。任意の開示制御およびプログラムまたは内部制御およびプログラムは、発想および動作がどのように完全であっても、絶対的な保証ではなく、合理的な保証を提供することしかできず、制御システムの目標が達成されることを確保することしかできないと信じている。
これらの固有の限界は,意思決定過程における判断が誤りである可能性があり,簡単な誤りや誤りによって故障が発生する可能性があるという現実を含む.例えば、私たちの役員または役員は、意図せずに新しい関係または手配を開示できなかった可能性があり、関連するいかなる取引も開示できなかった。さらに、ある人の個人的な行動、2人以上の結託、または無許可超越制御は、制御を回避することができる。したがって,我々の制御システム固有の制限により,誤りや詐欺による誤り陳述が発生し,発見されない可能性がある.
将来の財務会計基準や慣行の変化は、不利で予期せぬ収入変動を招き、我々が報告した経営業績に悪影響を及ぼす可能性がある。
将来の財務会計基準の変化は、不利で予期せぬ収入変動を招き、私たちが報告した財務状況や経営結果に影響を与える可能性がある。米国の財務会計基準は絶えず審査されており、過去には新たな声明と声明の異なる解釈がしばしば出現し、今後も再び出現することが予想される。したがって、私たちは私たちの会計政策を変えることを要求されるかもしれない。これらの変化は、私たちの財務状況や経営結果に影響を与えたり、このような財務状況や経営結果を報告する方法に影響を与える可能性があります。我々は、持続的な発展の基準を遵守するために資源を投入する予定であり、このような投資は一般的かつ行政費用の増加を招き、管理時間と注意をビジネス活動からコンプライアンス活動に移す可能性がある。
私たちは“新興成長型企業”であり、新興成長型企業に適用される報告要件を下げることが投資家に対する私たちの普通株の吸引力を低下させるかどうかを決定することはできません。
我々はJOBS法案で定義されている“新興成長型企業”であり、他の非新興成長型企業に適用される上場企業の報告書を利用して免除を要求する予定である
また、“新興成長型企業”として、JOBS法案は、このような声明が民間企業に適用されるまで、このような声明が民間企業に適用されるまで、上場企業に適用される新たなまたは改正された会計声明の採用を延期することを許可しており、その後、この免除を利用しないことを撤回することができない。雇用法案に基づいて、私たちはこの延長された過渡期を使用することを選択した。したがって、我々の総合財務諸表は発行者の財務諸表と比べものにならない可能性があり、後者は上場企業に適用される新しい会計基準や改正会計基準の発効日を守らなければならず、私たちの財務状況を他の上場企業の財務諸表と比較することをより困難にする可能性がある。
110
私たちは投資家が私たちがこのような免除に依存して私たちの普通株の吸引力が低下することを発見するかどうか予測できない。したがって、一部の投資家が私たちの普通株の吸引力が低下していることを発見すれば、私たちの普通株はそれほど活発ではない取引市場がある可能性があり、私たちの株価はもっと変動するかもしれない。私たちは私たちがこれ以上新興成長型会社ではないまで、このような報告書免除を利用するかもしれない。私たちは、(1)財政年度の最終日まで、(A)私たちの初公募(IPO)完了5周年後、(B)私たちの年間総収入が少なくとも12.35億ドル、または(C)前年6月30日まで、非関連会社が保有する私たちの普通株の時価が7億ドルを超え、(2)前3年間に10億ドルを超える転換不能債券を発行したことを意味する新興成長型会社である。
デラウェア州の法律と私たちが改訂して再記載した会社証明書および改正と再記述の定款中の条項は合併、要約買収あるいは代理権競争を困難にし、それによって私たちの普通株の取引価格を下げる可能性があります。
我々のデラウェア州会社としての地位とデラウェア州一般会社法の反買収条項は、利益関連株主が利益株主になってから3年以内に当該株主と業務合併を行うことを禁止しているため、制御権変更が既存の株主に有利になるため、制御権の変更を阻止、延期、または阻止する可能性がある。また、当社が改訂·再記述した会社登録証明書および改訂·再記述の定款には、以下の内容を含む当社の買収をより困難にする可能性のある条項が含まれています
さらに、デラウェア州会社として、私たちはデラウェア州会社法第203条の制約を受けている。これらの規定は、大株主、特に私たちが発行した議決権株の15%以上を有する株主が、一定期間内に私たちと合併または合併することを禁止することができる。デラウェア州会社は、その元の会社の登録証明書に明文で規定したり、その会社の登録証明書またはその株主の承認を受けた定款を修正することによって、この規定から脱退することを選択することができる。しかし、私たちはこの条項から脱退することを選択しなかった。
当社の会社登録証明書の改訂と再発行、改正と再記述の定款およびデラウェア州法律のこれらおよびその他の条項は、株主または潜在的な買収者がより入手しにくいかもしれません
111
私たちの取締役会は、当社の合併、買収要約、代理権競争を延期または阻害することを含む、当時の取締役会が反対する行動を開始しました。これらの条項の存在は、私たちの普通株の価格に悪影響を与え、会社の取引で価値を実現する機会を制限する可能性があります。
私たちが改訂して再記述した会社登録証明書は、デラウェア州の州裁判所を指定するか、または、デラウェア州内に州裁判所が管轄権を持っていなければ、デラウェア州地域の連邦裁判所は、私たちの株主が起こしうるいくつかのタイプの訴訟と訴訟の唯一のおよび独占フォーラムとして、わが社と私たちの役員、上級管理者、従業員に対する訴訟を阻止するかもしれない。
私たちが改正して再記載した会社登録証明書は、法的に許容される最大範囲で、私たちが書面で代替裁判所を選択することに同意しない限り、デラウェア州衡平裁判所(または、衡平裁判所に管轄権がない場合、デラウェア州連邦地域裁判所)は、デラウェア州成文法または一般法の下で以下のタイプの訴訟または手続きの唯一かつ独占的な裁判所になる。(1)私たちが提起した任意の派生訴訟または訴訟を代表する。(2)当社の現職または前任取締役、上級職員または他の従業員が当社または当社の株主に対して信頼された責任を有する任意の訴訟または手続に違反すると主張する、(3)“デラウェア州会社法”、我々が改正および再記載した会社登録証明書または当社の改正および再記載された付例の任意の条文に基づいて、当社または当社の任意の現職または前任取締役、上級職員または他の従業員に対してクレームを提出する任意の訴訟または手続、(4)解釈、適用、強制執行、または当社の改正および再記載された会社登録証明書または当社の改正および再記載の添付例の有効性を決定する任意の訴訟または手続;(5)デラウェア州会社法がデラウェア州衡平裁判所に管轄権を与える任意の訴訟または手続、および(6)内部事務原則によって管轄されている、私たちまたは私たちの任意の役員、高級職員または他の従業員のクレームに対する任意の訴訟。
この規定は、取引法で規定されている義務または責任を執行するための訴訟には適用されない。また、証券法第22条は、連邦裁判所と州裁判所は、このようなすべての“証券法”訴訟に対して同時に管轄権を持っていると規定している。したがって、州裁判所と連邦裁判所はこのようなクレームを受理する管轄権を持っている。複数の司法管轄区でクレームを提訴せざるを得ないことや、異なる裁判所が不一致や逆の裁決を下す脅威などの考慮要因を回避するために、我々が改訂·再記述した会社登録証明書は、アメリカ合衆国連邦地域裁判所が証券法に基づいて提起された任意の訴因を解決するための独占的なフォーラムとなることをさらに規定している。デラウェア州裁判所は、このような選択の裁判所条項が事実上有効であることを決定しているが、株主は依然として専属裁判所条項が指定された場所以外の場所でクレームを出すことを求めることができる。このような状況の中で、私たちは、私たちが改訂して再記載した会社登録証明書の独占フォーラム条項の有効性と実行可能性を強く主張する予定です。これは、他の法ドメインでこのような訴訟を解決することに関連する多くの追加費用を必要とする可能性があり、これらの規定がこれらの他の法ドメインの裁判所によって実行されることを保証することはできない。
これらの排他的フォーラム条項は、司法フォーラムにおいて、私たちまたは私たちの役員、役員、または他の従業員との紛争に有利であると考える株主のクレームを提出する能力を制限する可能性があり、これは、私たちと私たちの役員、役員、および他の従業員に対する訴訟を阻止するかもしれない。もし裁判所が私たちが改正して再記載した会社登録証明書のいずれかの独占的な裁判所条項が訴訟で適用されないか、または実行できないことが発見された場合、私たちは他の管轄区域での論争の解決に関連するさらなる重大な追加費用を発生させる可能性があり、これらのすべては私たちの業務を深刻に損なう可能性がある。
112
項目1 B。取消解析Dスタッフがコメントした。
ない。
プロジェクト1 C。ネットワークセキュリティです。
リスク管理と戦略
我々は、当社の重要なコンピュータネットワーク、第三者ホストサービス、通信システム、ハードウェアおよびソフトウェア、当社のキーデータ(知的財産権を含む)、および独自の戦略的または競争的性質を有する機密情報(“情報システムおよびデータ”)からなるネットワークセキュリティ脅威の重大なリスクを識別、評価、管理するために、様々な情報セキュリティプロセスを実施し、維持している。
我々の企業リスク管理委員会は、会社のセキュリティ·法律チームおよび第三者サービスプロバイダとともに、会社のネットワークセキュリティ脅威やリスクの識別、評価、管理を支援しています。また、企業リスク管理委員会、セキュリティチーム、法律チーム、会社の第三者サービスプロバイダは、手動および自動化ツールの使用、ネットワークセキュリティ脅威を識別する報告およびサービスの購読、脅威環境のスキャン、私たちおよび業界のリスク状況の評価、私たちに報告された脅威の評価、制御評価、脅威および脆弱性評価、および専門家第三者組織によるシミュレーション浸透テストおよびデスクトップイベント応答演習に参加し、ネットワークセキュリティ脅威からのリスクを識別および評価するために、様々な方法を使用することによって、私たちの脅威環境および会社のリスク状況を監視し、評価する。
我々は、ネットワークセキュリティ脅威が私たちの情報システムおよびデータに及ぼす重大なリスクを管理および低減するために、様々な技術、物理的および組織的措置、プロセス、基準、およびポリシーを実施し、維持し、例えば、イベント応答計画およびポリシーを維持し、定期的なリスク評価を行うこと、セキュリティ基準と認証を実施すること、データを暗号化すること、ネットワークセキュリティ制御を維持すること、データを隔離すること、物理セキュリティ制御を維持すること、システムとデータ監視ツール、定期従業員安全訓練およびシミュレーションテスト、浸透テスト、およびネットワークセキュリティ保険を維持することを目的とする。
ネットワークセキュリティ脅威の重大なリスクの評価と管理は、会社全体のリスク管理プロセスに統合されている。我々のネットワークセキュリティリスク管理枠組みは,国家標準と技術研究所のリスク評価ガイドラインに基づいている。例えば、ネットワークセキュリティリスクは、所定の確率および潜在的影響行列に従って評価される。そして,これらのリスクはリスク処理計画とともに企業リスク登録簿に記録され,企業リスク管理委員会の承認に供される。
私たちは、法律顧問、ネットワークセキュリティコンサルタント、ネットワークセキュリティソフトウェアプロバイダ、ホスト·ネットワーク·セキュリティサービスプロバイダ、および浸透試験会社を含む専門サービス会社のような、第三者サービスプロバイダを使用して、ネットワークセキュリティ脅威からの重大なリスクを時々識別し、評価し、管理するのを助ける。
我々は、アプリケーションプロバイダ、ホスト会社、契約研究機関、契約製造機関、流通業者、サプライチェーンリソースなど、第三者サービスプロバイダを使用して、業務全体で様々な機能を実行する。提供されたサービスの性質、関連する情報システムおよびデータの感度、およびプロバイダの識別に基づいて、当社のプロバイダ管理プロセスは、プロバイダに関連するネットワークセキュリティリスクの決定を支援し、ネットワークセキュリティに関連する契約義務をプロバイダに課すことを目的とした異なるレベルの評価を含む可能性がある。
企業に重大な影響を及ぼす可能性のあるネットワークセキュリティ脅威のリスクおよび起こりうる方法の説明については、本年度報告Form 10-Kの一般的なリスク要因項目のリスク要因を参照してください。“私たちの情報技術システムまたはデータまたは私たちが依存する第三者のシステムまたはデータが破壊された場合、規制調査または行動に限定されないが、訴訟、罰金および罰金、私たちの業務運営の中断、私たちの候補製品の開発計画の重大な中断、名声損害、収入または利益損失、顧客または販売損失を含むこのような損害によって生じる不利な結果を経験する可能性があります。他の不利な結果もあります
113
統治する
我々の取締役会は、その一般的な監督機能の一部として、会社のネットワークセキュリティリスク管理を担当しています。監査委員会は、ネットワークセキュリティ脅威からのリスクを監督し、緩和することを含む、会社のネットワークセキュリティリスク管理プロセスを監督する。
我々のネットワークセキュリティリスク評価と管理プロセスは、IT副総裁、IT運営部取締役、総法律顧問を含むいくつかの会社経営陣によって実施·維持されている。
我々のIT副総裁とIT運営の取締役は適切な人員募集を担当し,ネットワークセキュリティリスク考慮を会社全体のリスク管理戦略に取り入れ,関係者に重要な優先事項を伝えるのを支援している。我々のIT副総裁は、ネットワークセキュリティイベントの準備、ネットワークセキュリティプロセスの承認、セキュリティ評価および他のセキュリティ関連報告の審査を支援する責任があります。
我々のネットワークセキュリティイベント応答プロセスと政策は、状況に応じていくつかのネットワークセキュリティイベントを管理層メンバーに報告することを目的としている。私たちのセキュリティチームと総法律顧問は、会社のイベント応答チームと協力して、通知を受けたサイバーセキュリティイベントを緩和し、修復するのを助けます。また、会社のイベント応答プロセスおよびポリシーには、いくつかのネットワークセキュリティイベントについて取締役会に報告することが含まれている。
取締役会は、企業リスク管理委員会とIT副総裁から、会社の重大なネットワークセキュリティ脅威とリスク、およびこれらの脅威やリスクに対応するために会社が実施したプログラムに関する報告を定期的に受けている。取締役会はまた、ネットワークセキュリティ脅威、リスク、緩和に関連する様々な報告書、要約、またはプレゼンテーションを閲覧することができる。
プロジェクト2.専門家ペルテスです。
私たちは現在カリフォルニア州サンディエゴで約110,000平方フィートの製造、オフィス、および実験室空間をレンタルしており、87,000平方フィートの賃貸契約は2029年12月31日に満了し、その中には臨床製造施設が含まれており、私たちのオフィスと実験室空間に隣接している。残りの2.3万平方フィートのオフィスと実験室空間は転貸状態にあり、2025年12月31日に満期になる。私たちは私たちの既存のレンタル空間が予測可能な未来の施設需要を満たすのに十分であると信じています。私たちが必要かもしれないどんな追加空間も商業的に合理的な条項で提供されます。
項目3.法律法律手続き。
私たちは現在どんな実質的な法的手続きの当事者でもない。時々、私たちは正常な業務過程で発生した法的訴訟に巻き込まれるかもしれない。結果にかかわらず、訴訟は弁護と和解費用、管理資源の移転、負の宣伝、名声損害などの要素によって私たちに不利な影響を与える可能性がある。
プロジェクト4.地雷安全安全に開示する。
適用されません。
114
パー?パーT II
項目5.登録者普通株式市場、関連株持株者は重要で発行者が株式証券を購入する。
市場情報
私たちの普通株は、一株当たり0.0001ドルの価値があります。ナスダック全世界の精選市場で取引して、コードは“PSTX”です
記録保持者
2024年3月4日現在、私たちの普通株には約64人の登録株主がいます。一部の株式は“街道”名義で保有されているため、当該等の株式の実益所有者の数は知られていないか、または上記の数字に含まれている。
配当政策
私たちは私たちの普通株式に対するどんな配当金も発表したり支払ったりしたことがない。私たちは現在、すべての利用可能な資金と任意の将来の収益(もしあれば)を維持し、私たちの業務の発展と拡張に資金を提供するつもりで、予測可能な未来にはいかなる現金配当も支払わないと予想しています。将来配当金を派遣するかどうかは私たちの取締役会が適宜決定し、適用される法律に依存し、そして私たちの経営結果、財務状況、契約制限と資本要求などの要素に依存する。さらに、私たちがオックスフォード大学と達成した私たちの債務を管理する融資協定は、私たちが現金配当金を申告して支払う能力を制限した。
株式補償計画に基づいて発行された証券
本プロジェクトが提供を要求した情報は,2024年株主総会のために作成した最終依頼書を参考にした。第3部第12項“特定の利益を受けるすべての人と管理されている保証所有権”を参照
最近売られている未登録証券
ない。
収益の使用
ない。
発行者および関連購入者が株式証券を購入する
ない。
第六項です[保留されている]
115
項目7.経営陣の以下の問題の議論と分析財務状況と経営実績。
以下、当社の財務状況と経営結果の検討と分析は、当社の連結財務諸表と本年度報告書10-K表の他の部分に関する付記と一緒に読まなければなりません。今回の議論は、特に、本年度報告Form 10−Kにおける“前向き陳述に関する特別な説明”で述べたように、我々の将来の経営結果や財務状況、業務戦略および将来の経営の管理計画および目標に関する情報、特にリスクおよび不確実性に関する前向きな陳述を含む。あなたは本年度報告書の“リスク要因”の項目の開示を読むべきであり、討論は私たちの実際の結果がこれらの前向きな陳述で予想された結果と大きく異なる重要な要素をもたらす可能性がある。
概要
我々は臨床段階の細胞·遺伝子治療会社であり,癌やまれな疾患の患者に新たな治療法を提供している。我々は2014年12月に登録設立され,その後Transposagenから剥離され,同社は2003年から遺伝子工学技術を開発してきた。設立以来、私たちの業務の重点は私たちの会社を組織と配備し、業務計画を制定し、資金を調達し、許可を獲得し、知的財産権を開発と獲得し、そして私たちの知的財産権の組み合わせの確立と保護、著者らの遺伝子工学技術の開発、潜在的な候補製品の確定及び研究開発と製造活動を展開することであり、著者らの候補製品の臨床前研究と臨床試験、及び戦略取引に従事することを含む。私たちは候補製品の販売が許可されていないし、製品販売から何の収入も得られていない。
私たちは主に株式売却、債務融資、戦略協力を通じて私たちの業務に資金を提供します。設立以来,普通株を公開発売することで3.054億ドルの総収益を調達し,償還可能な転換可能な優先株を売却することで3.343億ドルの総収益を調達し,我々の融資合意により6000万ドルの借金総収益を獲得し,カリフォルニア再生医学研究所(CIRM)から合計2380万ドルの贈与資金を獲得した。私たちはまたいくつかの戦略的合意に到達した。2021年第4四半期に武田と協力協定を結び、4500万ドルの前払いを受けた。2022年第3四半期、私たちは羅氏と協力合意に達し、1.1億ドルの前払いを受けた。2023年第3四半期、私たちはAstellasと証券購入協定と戦略権利書協定を締結し、5000万ドルを一度に受け取りました。
2023年12月31日現在、私たちは現金、現金等価物、短期投資2.122億ドルを持っている。私たちは候補製品の販売が許可されていないし、製品販売から何の収入も得られていない。設立以来、重大な運営損失が発生しており、予測可能な未来には引き続き重大な運営損失を被ることが予想される2023年12月31日と2022年12月31日までの年度の純損失はそれぞれ1億234億ドルと6400万ドルだった。2023年12月31日までの累計赤字は5.943億ドルだった。
我々のコア独自プラットフォームに基づいて、我々の非ウイルスiggyBac DNA送達システム、Cas-Clover部位固有遺伝子編集システム、およびナノ粒子およびAAVに基づく遺伝子送達技術を含む、様々な適応の広範な候補製品の組み合わせを発見し、開発している。著者らの核心プラットフォーム技術は単独で使用することができ、結合して使用することもでき、多種の細胞と遺伝子治療方式に応用することができ、そして著者らの候補製品の組み合わせを設計することができ、現在の世代の細胞と遺伝子治療技術の主要な制限を克服することを目的としている。
細胞治療において,われわれの技術は工学的細胞を有する候補製品を作り出すことができ,これらの細胞を患者に移植し,持続的な反応を駆動し,単一治療治癒を引き起こす能力があると信じている。私たちのCAR-T治療組み合わせは同種または既製の候補製品を含む。われわれは広範なパイプラインを進めており,固形腫瘍と血液腫瘍学的適応の臨床段階では多くのCAR−T候補製品がある。遺伝子治療において,我々の技術は,長期的で安定した遺伝子発現を提供し,時間の経過とともに減弱することなく,単一療法の治癒につながる可能性がある新たな治療法を創出する可能性があると信じている。
116
以下の表に現在細胞治療に用いられている候補製品の組み合わせをまとめた
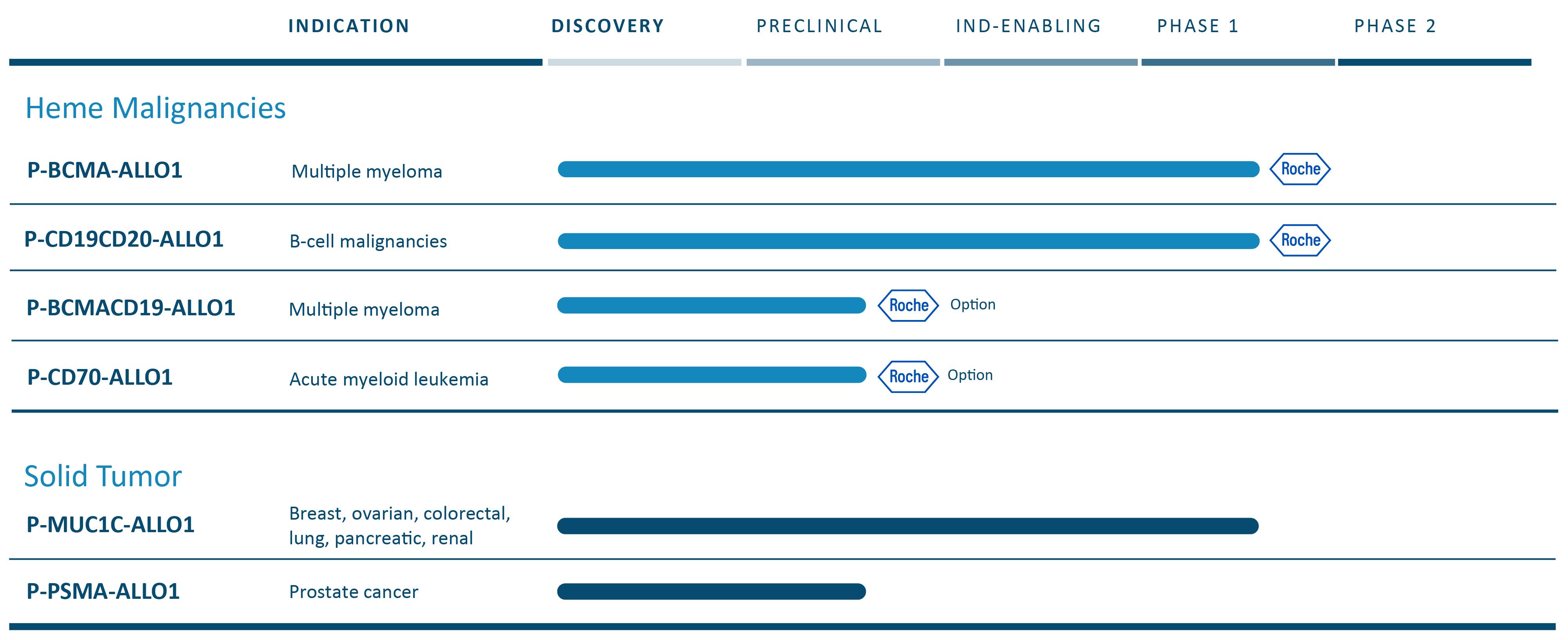
私たちは私たちの非ウイルスiggyBac DNA送達システムを使ってこれらの候補製品を生産した。我々の完全同種異体CAR−T候補は,健康ドナーからの特徴の良い細胞を出発材料として開発し,一次生産運転から潜在的な数百名の患者を治療できることを目標としている。投与量は凍結保存され,将来の既製品使用に備えて治療センターに貯蔵されている。さらに、我々の候補同種製品は、同種反応性を低減または除去するために、我々独自のCas-Clover部位固有遺伝子編集システムを使用し、拡張性のブースター分子技術を製造するために使用される。
私たちの最先端の内部固形腫瘍計画は
私たちが羅氏と協力した最先端の血液学プロジェクトは
117
次の表は現在の遺伝子治療分野の候補製品の組み合わせをまとめています
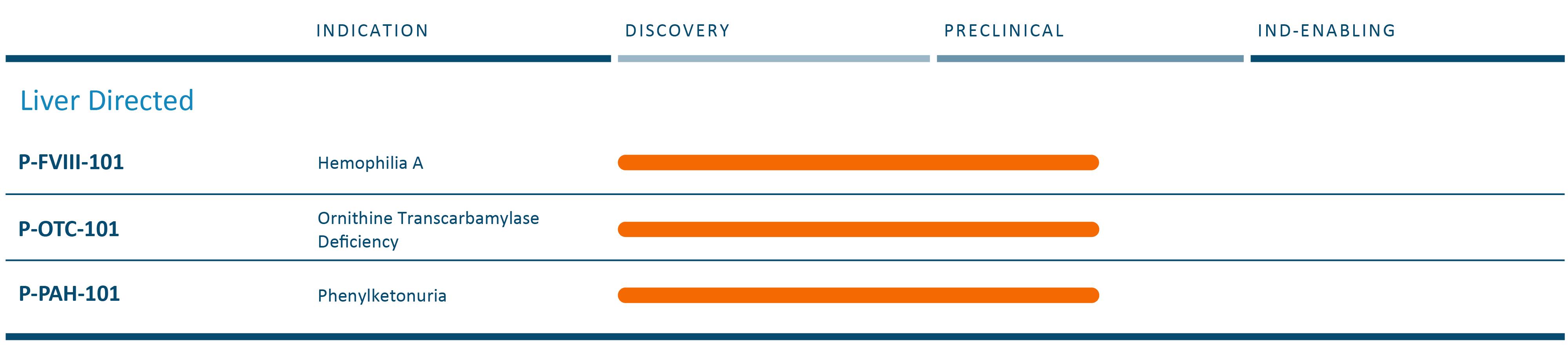
我々の候補遺伝子治療製品は,従来のAAV遺伝子治療の主な制限を克服するために,我々のiggyBac技術を用いてAAVとともに開発された。我々の方法は,統合と長期的に安定した発現をもたらすことができ,潜在的な用量はAAV技術よりもはるかに低いため,コストや耐性の利点ももたらすと信じている。我々の最終目標は,我々の非ウイルスナノ粒子技術でAAVを完全に置換し,将来の遺伝子治療製品開発をAAVの制限から脱却させることである。
私たちの最先端の遺伝子治療計画は
118
予測可能な未来には、P-MUC 1 C-ALLO 1を含む候補製品の開発を継続し、規制部門の承認を求め、任意の承認された製品を商業化し始めるにつれて、費用や損失が大幅に増加すると予想される。我々が開発している候補製品の数を拡大し,これらの候補製品の臨床開発を求めるにつれ,開発コストは全体的に上昇することが予想される。これらの増加した開発コストの一部を相殺するために,羅氏との協力には計画補償が含まれている.我々は,研究開発活動の増加,従業員数の増加,ナスダック上場規則の遵守,米国証券取引委員会の要求および上場企業としての運営継続に伴い,一般的かつ行政費用が増加すると予想している。われわれの純損失は四半期間と年度間に大きく変動する可能性があり,これはわれわれの臨床試験の時間と他の研究·開発活動への支出にかかっている。
私たちは現在私たちの内部の臨床GMP製造工場から私たちの候補製品を調達しています。私たちはまた様々なサプライヤーと協力して、媒体、DNA、RNAコンポーネントを含む私たちの製造原材料を提供します。将来、私たちはまたFDAが承認した任意の候補製品のための1つ以上の商業製造施設を建設するかもしれない。
協力協定
羅氏協力協定
2022年7月、私たちは羅氏と協力協定を締結し、協定に基づいて、羅氏に授与した:(I)私たちの特定の知的財産権に基づいて、世界的に独占的に許可し、私たちの既存のP-BCMA-ALLO 1およびP-CD 19 CD 20-ALLO 1計画から同種CAR-T細胞治療製品を開発、製造、商業化する、または各計画、Tier 1計画;(Ii)我々の既存のP-BCMACD 19-ALLO 1およびP-CD 70-ALLO 1計画または各Tier 2計画から同種CAR-T細胞治療製品を開発、製造、商業化するために、我々のいくつかの知的財産権に基づいて独占的なグローバル許可の独占的選択権を獲得し、(Iii)我々の特定の知的財産権の独占的許可に基づいて、ロー氏が指定した最大6つの協力計画から同種異体CAR-T細胞治療製品を開発、製造、および商業化する。(Iv)いくつかの限られた知的財産権の下で非独占的商業許可の選択権を獲得して、いくつかの羅氏固有細胞療法製品を開発、製造および商業化し、羅氏が決定した最大3つの固形腫瘍標的、もしかしたら製品が可能かもしれない;および(V)私たちの2つの血液悪性腫瘍早期既存計画に対する優先的な契約権。
Tier 1計画ごとに,第1段階用量逓増臨床試験により開発活動を実行し,羅氏はこのような活動を実行する際に生じるいくつかの費用の特定の割合を精算する義務があり,各Tier 1計画には最大1つの指定された精算上限がある。第一段階の線量を超えて増加する第一級計画活動について、羅氏はこの計画によって発生したすべての費用を精算する義務がある。Tier 2計画ごとに,IND支援研究の開発候補を選択するか,あるいはロ氏がオプション維持費を選択して支払う場合には,第1段階用量逓増臨床試験を完了することで研究·開発活動を行う。また,羅氏が独占的に選択権を行使するTier 2計画ごとに,羅氏は我々に選択権使用料を支払う義務がある.Tier 1プランとTier 2プランごとに,羅氏への技術移転が完了するまで製造活動を実行する.
双方は2年間の初歩的な研究計画を行い、臨床前に特定の数量の同種異体CAR-T細胞療法に関連する合意された次世代治療概念を探索とテストする。羅氏が選択して費用を支払う場合、双方はその後、18ヶ月間の第2の研究計画を行い、この計画によれば、双方は初期期間で行われている既存の仕事を延長することができ、および/または同種異体CAR-T療法に関連する特定の数の追加的に合意された次世代治療概念を探索および臨床前に試験することができる。ロー氏はこの2つの研究計画の中から最大6つのヘム悪性腫瘍に対する同種異体CAR−T計画を指定する可能性があり,各計画についてIND活動を促進する開発候補,あるいは各協力計画を選択して研究·開発活動を行う。Vt.に基づいて
119
羅氏は指定された協力計画ごとに指定された費用を支払う義務がある。私たちが協力計画の主導的な最適化活動を完了した後、羅氏はその計画を羅氏に移行し、私たちに支払いまたは終了することを選択することができる。代替的に、限られた数の協力計画の場合、羅氏は、第1段階用量漸増臨床試験を完了することによって、いくつかの追加の開発および製造活動を行うことを選択することができ、この場合、羅氏は、いくつかのマイルストーンを支払い、そのような開発および製造活動に関連する特定のパーセントのコストを精算する。各連携計画については,羅氏への技術移転が完了するまで製造活動を実行する.
羅氏の協力協定によると、羅氏は私たちに1.1億ドルの前金を支払った。ロ氏の第2レベル計画オプションの行使、協力計画の指定とライセンス製品の商業許可の選択権の行使、第1レベル計画、オプション第2レベル計画と協力計画に基づいて特定の開発、監督、純売上マイルストーン事件を実現する製品などによると、(I)第1レベル計画15億ドル、(Ii)第2レベル計画11億ドル、(Iii)協力計画29億ドルを含むいくつかの精算、費用、マイルストーン支払いを獲得する資格がある。(Iv)特許製品の4.15億ドルです
我々はまた,製品ごとに,Tier 1計画,オプションTier 2計画と連携計画による製品純売上高の中位から下位2桁,および許可製品の中央桁から中央値の階層印税支払いを許可する権利があり,いずれの場合も,いくつかの通常の減免と補償の制限を受けている.使用料は、その製品をカバーするライセンス特許がその国で満期になるまで、またはその製品が同国で初めて商業販売された日から10年まで製品および国/地域によって支払われる。
ロ氏協力協定は、2022年9月に改正された1976年の“ハート-スコット-ロディノ反トラスト改正法”に基づく適用待機期間の満了または終了後に発効し、残りの特許権使用料や他の支払い義務がなくなるまで製品や国/地域で行われ続ける。ロ氏協力協定には、重大な違約や破産に対する条項、羅氏の便宜のための標準的な終了条項が含まれている。これらのうちのいくつかの終了権利は、特定の製品またはライセンスおよびロ氏協力協定全体に対して行使することができる。
2023年11月7日から、羅氏協力協定は、(I)羅氏が私たちに支払ったいくつかの既存の製造関連費用を再分配して、私たちの既存の各第1段階計画のために新しい製造プロセスの開発および移転費用を追加するために再分配され、(Ii)羅氏が各第1級計画のために私たちに支払ったいくつかの開発マイルストーンの支払いの金額を再分配するように改正された。改正案では、1500万ドルのマイルストーン費用を払えば、既存の2年期研究計画をさらに18カ月延長できることも規定されている。その他の点では、ロ氏協力協定下のすべての履行義務と支払い条件は変わらない。
武田協力協定
2021年10月には、武田薬品米国社または武田社と協力·許可協定、すなわち武田協力協定を締結し、この協定に基づき、4500万ドルの前払金と協定期間中の研究開発精算金を取得した。
2023年5月31日、武田から書面通知を受け、武田は武田協力協定を中止することを選択し、2023年7月30日から発効、または終了日とする。終了日まで、双方はそれぞれ武田協力協議での責任を果たしていたが、終了日には、吾らの武田協力協議での独占責任は終了した。また、武田の免許を付与し、武田協力協定に基づいて研究活動を行う許可証を付与した。2023年12月31日までの1年間に、これまでに繰延された890万ドルの収入を確認し、これらの業績義務を履行する必要がないことを確認した。終了後,P-FVIII-101およびP-PAH-101を含むすべての以前に許可されたプログラムを開発するすべての権利が付与される.遺伝子治療の分野で新たな戦略的協力を求めることができるかもしれないが、これまで武田協力協定に含まれていた計画の一部または全部と、現在このような戦略的協力の約束または合意に達していないにもかかわらず、可能な追加の内部計画が含まれている可能性がある。
120
許可内合意
以下に我々の鍵許可プロトコルの概要を示す.これらのライセンス契約と我々の他のライセンスプロトコルの詳細については、本年度報告における“ライセンス経営プロトコル”と題する部分と我々の合併財務諸表の付記12を参照されたい。
CIRM贈与基金
2018年、私たちはCIRMから400万ドルの報酬を得て、私たちはP-PSMA-101臨床前研究をサポートするためにすべての収益を受けました。この報酬の条項には、プロジェクトが商業化された後、贈与金の返済を選択するか、印税義務に変換することができるオプションが含まれている。贈与契約の条項によると、私たちは収益を受け取った時に負債として記録します。2023年第3四半期には,P−PSMA−101計画に関する追加臨床や他の開発活動を行わないことにしたが,P−PSMA−101計画に関連する金額の返済義務はなく,それぞれの負債確認を廃止し,2023年12月31日までの年度中にこのような金額を他の収入に記録した。
私たちの運営結果の構成要素は
収入.収入
協力収入
協力収入には,羅氏や武田との連携·ライセンス契約で確認した収入が含まれており,このようなパートナーに契約書を納入して成果を渡す時間とパターンを反映している。
121
運営費
研究と開発
研究と開発費用は主に私たちの研究と開発活動による外部と内部コストを含み、私たちのプラットフォーム技術の開発、私たちの薬物発見努力、私たちの候補製品開発を含む。
外部コストには:
内部コストには
私たちは発生した費用に応じて研究と開発費用を支払う。外部費用は,我々のサービス供給者が提供してくれた情報に基づいて特定のタスクを達成する進捗を評価したり,報告日ごとのサービス量の推定を行って確認したりする.技術許可のための前金とマイルストーン支払いは臨床段階計画に関連しており,費用が発生している間は研究·開発支出としている。私たちが将来受け取る研究·開発活動のための商品またはサービスのための前払いは、前払い費用または他の長期資産として記録される。これらの金額は、関連する商品が提供されたり、サービスが提供される時に支出される。
いつでも、私たちは複数の研究プロジェクトを行っている。われわれは計画された段階で外部コストを追跡し,臨床段階の計画についてはコストを1つずつ計画的に追跡し,早期段階の仕事については臨床前計画を含めてこれらの総コストを追跡した。私たちの内部資源、従業員、およびインフラは、私たちの臨床製造施設を含み、どの計画とも直接バンドルされず、一般的に複数の計画に配置されている。したがって、私たちは特定の計画の内部費用を追跡しない。
臨床開発後期段階にある候補製品は通常,臨床開発早期段階の候補製品よりも高い開発コストを有しており,これは主にCRO活動や製造費用によるものである。われわれは,近い将来,われわれが計画している臨床前と臨床開発活動により,われわれが行っているP−MUIC−ALLO 1による上皮由来固形腫瘍患者の第1段階試験,P−BCMA−ALLO 1治療再発/難治性多発性骨髄腫患者の第1段階試験,B細胞悪性腫瘍患者に対するP−CD 19 CD 20−ALLO 1第1段階試験およびその他の臨床試験を含む,われわれの研究·開発費用が大幅に増加することを予想している
122
私たちが候補薬のチャンネルを拡大するにつれて、このような計画は始まる予定だ。私たちは私たちの任意の候補製品の臨床前と臨床開発を完成するために必要な努力の性質、時間とコストを正確に推定或いは知ることができない。以下の要因により,我々の開発コストは大きく異なる可能性がある
これらの変数のいずれかの結果の変化は、我々の任意の候補製品の開発のために、各候補製品の開発に関連するコスト構造およびスケジュールを著しく変更する可能性がある。私たちのどんな候補製品も規制部門の承認を得ることができないかもしれない。臨床試験や前臨床研究から思わぬ結果が得られるかもしれない。
一般と行政
一般と行政費用は主に行政、財務と行政機能者の賃金と関連費用を含み、株式報酬を含む。一般および行政費用には、施設に関する直接分担費用や法律、特許、相談、投資家と公共関係、会計や監査サービスの専門費用も含まれている。私たちは、私たちの候補製品(P-MUC 1 C-ALLO 1、P-BCMA-ALLO 1、P-CD 19 CD 20-ALLO 1を含む)の持続的な研究活動と開発を支援するための従業員の増加に伴い、任意の承認された製品を商業化し始め、私たちの一般的かつ行政的費用は将来的に増加すると予想される。
その他の収入(費用)
利子支出
利息支出には、我々の融資協議項における未返済借入金の利息支出及び債務割引と債務発行コストの償却が含まれています。利上げの環境を考慮すると、市場金利を反映するために金利支出が徐々に増加することが予想される。
その他の収入、純額
その他の収入、純額には利息収入と私たちの核心業務とは関係のない雑収入と費用が含まれています。利息収入は私たちの証券を売って稼ぐことができる利息で構成されています。
123
経営成果
2023年12月31日までと2022年12月31日までの年度比較
次の表は私たちの行動結果(千計)をまとめています
|
|
十二月三十一日までの年度 |
|
|
|
|
||||||
|
|
2023 |
|
|
2022 |
|
|
変わる |
|
|||
収入: |
|
|
|
|
|
|
|
|
|
|||
協力収入 |
|
$ |
64,703 |
|
|
$ |
130,492 |
|
|
$ |
(65,789 |
) |
総収入 |
|
|
64,703 |
|
|
|
130,492 |
|
|
|
(65,789 |
) |
運営費用: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
|
156,772 |
|
|
|
152,899 |
|
|
|
3,873 |
|
一般と行政 |
|
|
37,435 |
|
|
|
37,539 |
|
|
|
(104 |
) |
総運営費 |
|
|
194,207 |
|
|
|
190,438 |
|
|
|
3,769 |
|
運営損失 |
|
|
(129,504 |
) |
|
|
(59,946 |
) |
|
|
(69,558 |
) |
その他の収入(支出): |
|
|
|
|
|
|
|
|
|
|||
利子支出 |
|
|
(8,671 |
) |
|
|
(6,370 |
) |
|
|
(2,301 |
) |
その他の収入、純額 |
|
|
14,852 |
|
|
|
2,858 |
|
|
|
11,994 |
|
所得税前損失 |
|
|
(123,323 |
) |
|
|
(63,458 |
) |
|
|
(59,865 |
) |
所得税費用 |
|
|
(107 |
) |
|
|
(544 |
) |
|
|
437 |
|
純損失 |
|
$ |
(123,430 |
) |
|
$ |
(64,002 |
) |
|
$ |
(59,428 |
) |
協力収入
2023年12月31日までの1年間の協力収入は6470万ドルであったのに対し,2022年同期は1兆305億ドルであった。6,580万ドル減少したのは,主に羅氏協力協定による許可と研究サービス確認の収入が7,110万ドル減少したためであり,これは主にこの合意により2022年に付与された初期許可に関する収入の確認に関連している。協力収入の全体的な減少は、武田協力協定で実行された研究サービスで確認された530万ドルの収入増加によって相殺された。この成長は主に武田協力協定が2023年第3四半期に終了したことで確認された890万ドル以前の繰延収入である。
124
研究と開発費
次の表に私たちの研究開発費(単位:千)をまとめます
|
|
十二月三十一日までの年度 |
|
|
|
|
||||||
|
|
2023 |
|
|
2022 |
|
|
変わる |
|
|||
外部コスト: |
|
|
|
|
|
|
|
|
|
|||
臨床段階計画: |
|
|
|
|
|
|
|
|
|
|||
同種異体計画: |
|
|
|
|
|
|
|
|
|
|||
* |
|
$ |
11,626 |
|
|
$ |
10,910 |
|
|
$ |
716 |
|
*P-MUC 1 C-ALLO 1 |
|
|
7,699 |
|
|
|
5,244 |
|
|
|
2,455 |
|
*-CD 19 CD 20-ALLO 1 |
|
|
1,907 |
|
|
|
— |
|
|
|
1,907 |
|
完全同種移植計画 |
|
|
21,232 |
|
|
|
16,154 |
|
|
|
5,078 |
|
自己プログラム: |
|
|
|
|
|
|
|
|
|
|||
*P-BCMA-101 |
|
|
59 |
|
|
|
10,256 |
|
|
|
(10,197 |
) |
*P-PSMA-101 |
|
|
2,285 |
|
|
|
19,529 |
|
|
|
(17,244 |
) |
自己プログラム総数 |
|
|
2,344 |
|
|
|
29,785 |
|
|
|
(27,441 |
) |
臨床分期計画の総数 |
|
|
23,576 |
|
|
|
45,939 |
|
|
|
(22,363 |
) |
臨床前段階計画とその他の未分配費用 |
|
|
43,947 |
|
|
|
30,715 |
|
|
|
13,232 |
|
内部コスト: |
|
|
|
|
|
|
|
|
|
|||
人員 |
|
|
72,848 |
|
|
|
61,341 |
|
|
|
11,507 |
|
施設やその他 |
|
|
16,401 |
|
|
|
14,904 |
|
|
|
1,497 |
|
研究開発費総額 |
|
$ |
156,772 |
|
|
$ |
152,899 |
|
|
$ |
3,873 |
|
2023年12月31日までの1年間の研究開発費は1兆568億ドルだったが、2022年12月31日までの年間の研究開発費は1兆5229億ドルだった。研究開発費が390万ドル増加した要因は,研究協力活動の増加により,臨床前段階計画やその他の未分配費用が1320万ドル増加し,人員費が1150万ドル増加したこと,従業員数の増加により増加した株による報酬支出270万ドル,施設やその他の費用に関する内部コストが150万ドル増加したが,臨床段階計画は2240万ドル減少したためである。臨床段階計画は2,240万ドル減少したが,これは主にわれわれの自己計画に関連する臨床開発活動の減少により,異体計画の増加によって相殺され,これは主に全体登録とわれわれの第三項同種異体臨床試験P−CD 19 CD 20−ALLO 1の開始によるものである。
一般と行政費用
2023年12月31日までの1年間は、一般·行政費は3740万ドルだったが、2022年12月31日までの年間は3750万ドルだった。一般的および行政支出が10万ドル減少したのは、主に保険料が150万ドル減少したが、人事費の120万ドル増加に相殺されたのは、主に前執行主席の辞任に関する一次支出による株式報酬支出の増加によるものである。
利子支出
2023年12月31日までの年間の利息支出は870万ドルであるが、2022年12月31日までの年度の利息支出は640万ドルであり、オックスフォード金融有限責任会社(Oxford Finance LLC)との定期融資で元金残高を返済していない利息が含まれている。利息支出が230万ドル増加したのは、主に金利上昇によるものだ。
その他の収入、純額
2023年12月31日までの1年間、その他の収入純額は1,490万ドルだったが、2022年12月31日までの年間は290万ドルだった。1200万ドル増加の主な原因は利息収入が730万ドル増加することで、金利上昇と利用可能な投資残高の増加、支払い延期の核販売が原因だ
125
CIRMの贈与負債は2023年12月31日までの1年間で400万ドルであり,返済しようとしなくなった贈与に関連しており,P−PSMA−101のための追加的な臨床や他の開発活動は行わないことにしたからである。
流動性と資本資源
2014年の設立以来、私たちは深刻な運営損失と運営キャッシュフローを負に受け、主に私たちの能力に頼って株式と債務融資および戦略協力を通じて私たちの運営に資金を提供してきた。2023年12月31日と2022年12月31日までの年間純損失はそれぞれ1兆234億ドルと6400万ドルだった運営されているマイナスキャッシュフローから9220万ドル2680万ドルですそれは.少なくとも今後数年以内に、運営は純損失とマイナスキャッシュフローを継続すると予想される。2023年12月31日までの累計赤字は5.943億ドルだった。
今まで、私たちの業務は主に会社の組織と人員の配備、業務計画、資金の調達、許可と知的財産権の獲得及び私たちの知的財産権の組合せの確立と保護、著者らの遺伝子工学技術の開発、潜在的な候補製品の確定及び研究開発と製造活動に集中し、著者らの候補製品の臨床前研究と臨床試験、及び戦略取引に従事することを含む。
私たちの現金の主な用途は私たちの運営費用に資金を提供することであり、主に研究と開発支出、主に私たちの臨床前と臨床段階計画に関連する賃金と外部コスト、およびより小さい程度の一般と行政支出を含む。運営費に資金を提供するための現金は、これらの費用を支払う時間の影響を受けており、これは私たちの未払い売掛金や売掛金の変化に反映されている。
私たちはまだ私たちの候補製品を商業化していません。もしあれば、数年以内に候補製品の販売から収入が発生しないと予想しています。私たちは主に株式売却、債務融資、戦略協力を通じて私たちの業務に資金を提供します。設立以来,普通株の公開発売により3.054億ドルの総収益を集め,償還可能な優先株を売却することで3.343億ドルの総収益を調達し,我々の融資合意により6000万ドルの借入総収益を獲得し,CIRMから合計2380万ドルの贈与資金を獲得した。2021年第4四半期に武田と協力協定を結び、4500万ドルの前払いを受けた。2022年第3四半期、私たちは羅氏と協力合意に達し、1.1億ドルの前払いを受けた。2023年第3四半期、私たちはAstellasと証券購入協定と戦略権利書協定を締結し、5000万ドルを一度に受け取りました。
私たちは、2023年12月31日まで、私たちの現金、現金等価物、および短期投資2.122億ドルが、少なくとも本10-K年度報告書に含まれる財務諸表が発表された日から12ヶ月以内に、私たちの運営に資金を提供するのに十分であると予想している。長期的には、私たちは私たちの持続的な運営を支援し、私たちの成長戦略を実施するために追加の資金が必要になるだろう。
P-MUC 1 C-ALLO 1または他の候補製品の開発に成功し、規制部門の承認を得なければ、少なくとも今後数年以内には承認されない限り、製品販売からいかなる収入も得られないと予想される。もし私たちの候補製品が規制部門の承認を得たら、製品販売、マーケティング、製造、流通活動に関連した巨額の商業化費用が発生すると予想される。したがって、相当な製品収入を生成することができる前に、株式発行、債務融資、または他の資本源(潜在的な贈与、協力、許可、または他の同様の手配を含む)によって、私たちの運営に資金を提供する予定です。しかし、私たちは、もしあれば、タイムリーに、または優遇条件で追加資金を得たり、そのような他の計画を達成することができないかもしれない。特に、最近の金融機関の倒産や流動性レベルの影響、最近や予想されている金利や経済インフレの変化を含む公衆衛生危機、銀行業界の現在の金融状況を考慮すると、私たちの業務を支援するためのこれらの追加資金源を得ることができる保証はありません、あるいはこれらの資金があれば、これらの追加資金が私たちの需要を満たすのに十分であるという保証はありません。私たちが必要な時に資金を調達できなかったり、このような他の計画を達成することができなかったことは、私たちの財務状況にマイナスの影響を与え、私たちの研究を延期、減少、または終了させる可能性があります
126
開発計画やその他の業務、あるいは私たちが自分で開発·マーケティングしたい候補製品を開発·マーケティングする権利を付与します。
融資協定
2017年、私たちはオックスフォードと融資と保証協定を締結し、その後、未返済残高総額3,000万ドルを改正した。
2022年2月、私たちはオックスフォード大学と新しい融資と保証協定、すなわち2022年の融資協定を締結した。2022年ローン協定の条項によると、私たちは6,000万ドルの定期融資を借り入れ、その一部は改訂されたローン協定下の未返済残高の返済に使われている。2022年ローン協定によると、最初の利息期限は2025年4月1日で、その後23ヶ月等額で元金と適用利息が支払われる。2022年9月、2022年ローン協定で定義された適格株式事件を実現し、受取利息のみの期限を2026年4月1日に延長し、その後毎月11件の元本と適用利息を支払う。そのため、2022年ローン協定下のすべての未返済金額は2027年2月1日に満期になる。
ロンドン銀行の同業解体が2023年6月30日に発表を停止したため、私たちは現在、保証付き隔夜融資金利(“SOFR”)を用いて私たちの借金の受取利息金額を計算しています。SOFRは隔夜の現金借り換えコストを測る指標であり、米国国債を担保として、直接観察可能な米国財務省が支持する買い戻し取引に基づいている。
2022年ローン協議での未返済残高は、浮利年利で7.83%に相当するプラス(A)30日期ドルLIBOR金利と(B)0.11%の大きな者が利息を計上します。2022年ローン協定には、2023年6月30日にLIBORを段階的に廃止する際に、LIBORの代わりに代替基準金利を使用する条項が含まれている。2023年7月1日から、2022年ローン協議下の未返済残高は変動年利で利息を計上し、金利は(A)7.94%及び(B)(I)計上月直前の1ヶ月の営業日の1ヶ月CME期限SOFR、(Ii)0.10%及び(Iii)7.83%の和に相当する。2023年12月31日現在、私たちの定期ローンの適用金利は13.27%です。
私たちは2022年のローン合意下の未返済債務をいつでも500万ドルの増分で返済する権利があり、早期返済の罰を受けることはない。改訂された融資協定によると、(I)新満期日、(Ii)新ローンの早期支払い或いは(Iii)新ローン前払いの両者のうち早い者は、7.5%の最終支払い費用を支払わなければならない。
経営賃貸契約
2018年10月、私たちはカリフォルニア州サンディエゴで研究開発と行政活動のための施設賃貸協定を締結した。レンタル期間は2019年4月1日から2029年12月31日まで満了する。2019年10月、私たちは既存物件を拡張するための賃貸契約改正を締結した。新規物件の賃貸期間は2020年7月29日から始まり、2029年12月31日まで満了する。
2019年7月、私たちはカリフォルニア州サンディエゴの工場のレンタル契約を締結しました。この工場は改造されて良好な製造実践基準に適合し、私たちの早期臨床試験の製造に使用されました。レンタル期間は2020年6月26日から始まり、2029年12月31日まで満了する。
2021年10月、カリフォルニア州サンディエゴに研究·行政活動のための約23,000平方フィートの施設を設立する分譲協定を締結した。レンタル期間は2022年3月から始まり、2025年12月31日に満了する。
2023年12月31日まで、私たちはカリフォルニア州サンディエゴで約110,000平方フィートの製造、実験室、およびオフィス空間の運営リースを有し、87,000平方フィートの賃貸契約は2029年12月31日に満了し、オフィスおよび実験室空間に隣接する臨床製造施設を含む。レンタル協定には2つのオプションが含まれており、各オプションは期間を5年間延長する。
127
契約義務と約束
次の表は、2023年12月31日までの契約義務と約束(単位:千)をまとめています
|
|
期限どおりの支払い |
|
|||||||||||||
|
|
合計する |
|
|
少ないです |
|
|
1~3年 |
|
|
4~5年 |
|
||||
賃貸承諾額を経営する |
|
$ |
34,012 |
|
|
$ |
6,226 |
|
|
$ |
16,787 |
|
|
$ |
10,999 |
|
債務義務 |
|
|
86,721 |
|
|
|
8,095 |
|
|
|
78,626 |
|
|
|
— |
|
合計する |
|
$ |
120,733 |
|
|
$ |
14,321 |
|
|
$ |
95,413 |
|
|
$ |
10,999 |
|
上の表に記載されている契約義務と引受以外に、私たちに必要な現金の大部分は給与、雇用福祉、求人、従業員の留任と訓練費用を含む人員費用と関係があります。私たちは予測可能な未来に、私たちが引き続き従業員数を増やすにつれて、私たちの人員費用総額が増加すると予想している。
そのほか、著者らは正常業務過程中に契約研究機関、CMOとその他の第三者と臨床前研究、臨床試験及びテストと製造サービスについて契約を締結した。このような契約には最低購入約束は含まれておらず、私たちは事前に書面で契約をキャンセルすることを通知することができる。キャンセル時に支払われるべき金額には、提供されたサービスの支払いまたは発生した費用が含まれており、当社サービスプロバイダのキャンセル不可義務を含めて、最長でキャンセル日から1年となります。これらの支払いの金額と時間が分からないので、上の表にはこれらの支払いは含まれていません。
いくつかのライセンス契約も締結されており,これらの合意により,指定された臨床前,臨床,規制マイルストーン,および特許権使用料の支払い時に合計のマイルストーン支払いが義務付けられている。これらのライセンス契約の下での将来の支払いは、これらのライセンス契約下の支払い義務が、例えば、指定されたマイルストーンを達成したり、製品販売を生成したりするような未来のイベントに依存するので、上の表には含まれていません。私たちはそれらが評価可能で実現可能な時、このような記念碑的な支払いを記録する。これらのマイルストーンの実現や将来の製品販売が生じるタイミングや可能性を見積もるには重大な判断が必要であり,不確実性の影響を受ける。2023年12月31日現在、これらのマイルストーンを実現したり、将来の製品販売が発生する時間や可能性を見積もることはできません。上記の“ライセンス中のビジネスプロトコル”と題するセクションを参照してください。
キャッシュフロー
次の表は、現金および現金等価物の主な出所と用途を示しています(千で計算)
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
経営活動用の現金 |
|
$ |
(92,167 |
) |
|
$ |
(26,772 |
) |
投資活動によって提供される現金 |
|
|
39,134 |
|
|
|
(203,334 |
) |
融資活動で提供された現金 |
|
|
16,127 |
|
|
|
105,159 |
|
現金と現金等価物の純減少 |
|
$ |
(36,906 |
) |
|
$ |
(124,947 |
) |
2023年12月31日までの1年間に、経営活動は9220万ドルの現金を使用し、主な理由は、私たちの純損失1.234億ドル、1710万ドルの非現金プロジェクトによって相殺され、私たちの運営資産と負債の変化によって提供された現金純額1410万ドルだった。非現金費用には主に2,310万ドルの株式ベースの補償、560万ドルの減価償却費用、80万ドルの発行済み定期債務の割引増加が含まれ、840万ドルの投資割引増加と400万ドルの繰延CIRM贈与負債の解約によって相殺される。2023年12月31日までに、当社の運営資産および負債変動が提供する現金純額は、主にAstellas戦略権利関数によって受信された前払いに関する繰延収入が670万ドル増加し、負債が500万ドル増加し、経営賃貸使用権資産が340万ドル減少し、前払い支出およびその他の流動資産が160万ドル減少し、売掛金が100万ドル増加したが、経営リース負債によって370万ドル分相殺された。
128
2022年12月31日までの1年間に、経営活動は2680万ドルの現金を使用したが、主な理由は、私たちの純損失6400万ドル、2350万ドルの非現金プロジェクトによって相殺され、私たちの運営資産と負債の変化によって提供された現金純額1370万ドルだった。非現金費用は主に1890万ドルの株式補償、520万ドルの減価償却費用、80万ドルの発行済み定期債務の割引増加、180万ドルの投資割引増加によって相殺される。2022年12月31日までの年度、我々の経営資産と負債変化が提供する現金純額は、主に武田協力協定に基づいて受信した前払い関連の繰延収入が2,730万ドル増加し、経営賃貸使用権資産が460万ドル減少し、負債が140万ドル増加し、他の長期資産が60万ドル減少し、前払い費用および他の流動資産が50万ドル減少し、一部が売掛金の910万ドル増加と売掛金670万ドル減少によって相殺される。経営リース負債は490万ドル減少した。
投資活動によって提供される現金
2023年12月31日までの年間で、投資活動が提供する現金純額は3910万ドルで、その中には2.8億ドルの短期投資満期収益が含まれており、一部は2.378億ドルの短期投資購入および310万ドルの不動産·設備購入によって相殺されている。
2022年12月31日までの年間で、投資活動のための現金純額は2.033億ドルで、2.944億ドルの短期投資購入と、390万ドルの不動産と設備購入が含まれており、一部は9500万ドルの短期投資満期収益によって相殺されている。
私たちが短期投資を購入して売るタイミングは私たちの利用可能な現金残高と既存投資の満期日にかかっています。会社のオフィススペースを除いて、研究開発や製造活動の拡大に伴い、設備調達に関するすべての時期の財産や設備の購入を行っています。
融資活動で提供された現金
2023年12月31日までの年間で,融資活動が提供する現金純額は1,610万ドルであり,私募普通株から得られた1,440万ドルを含み,我々のESPPと株式オプション購入による130万ドル,Cantorとの販売協定により我々の普通株を定期的に発行·販売して得られた90万ドルは,株式純額決済配当金奨励に関する50万ドルの税金によって部分的に相殺された。
2022年12月31日までの年間で、融資活動が提供する現金純額は1.052億ドルで、普通株を公開発売した7530万ドルの純収益、2022年の融資協定の2860万ドルの収益、債務発行コストと返済改定後の融資合意を差し引いた純額、および我々のESPPによる株式オプションの購入と行使の130万ドルの収益が含まれている。
重要な会計政策と重大な判断と見積もり
経営陣による我々の財務状況や経営結果の検討·分析は、米国で一般的に受け入れられている会計原則に基づいて作成されている我々の財務諸表に基づいている。これらの財務諸表を作成する際には、報告中に報告された資産および負債の報告金額、財務報告日のまたは負債に関する開示、ならびに報告の費用および他の収入の報告金額に影響を与えるために、推定および判断を行う必要がある。著者らは絶えず著者らの推定と判断を評価し、その中で最も重要なのは収入確認、臨床前と臨床研究の計算すべき費用及び株に基づく報酬コストに関する推定と判断である。私たちの見積もりと判断は歴史的経験と他の私たちが当時の状況で合理的な要素だと思っています。状況の変化とより多くの情報の理解に伴い、異なる結果が出る可能性がある。
私たちの主な会計政策は私たちの総合財務諸表付記2により詳細に説明されていますが、以下の会計政策と見積もりは私たちの総合財務諸表の作成に最も重要だと思います。次の議論は私たちの最も重要な会計問題を解決すると信じています
129
政策は我々の財務状況と経営結果を記述するために最も重要な政策であり、管理層が最も困難で、最も主観的で、最も複雑な判断を行う必要がある。
収入確認
今まで、私たちの収入は主に協力と許可協定から来ていた。私たちの協力および許可協定には、知的財産権許可および研究開発サービスを含む複数の要素が含まれている可能性がある。これらの手配によると、私たちが受け取った考慮事項には、前金、研究開発資金、コスト補償、研究開発、規制、商業マイルストーン支払い、特許使用料が含まれる可能性があります。
会計基準コード化主題606を適用します取引先と契約した収入(“ASC 606”)、財務会計基準委員会(“FASB”)によって発行され、顧客との契約を示す。ASC 606によれば、収入は、顧客が約束された商品またはサービスの制御権を取得したときに確認される。確認された収入金額は私たちがこのようなサービスの価格を獲得する権利があると予想していることを反映している。我々は,異なる性能義務を評価するために,個人連携や許可プロトコルを背景にこれらの性能義務の性質を分析した.基本的な活動の性質に応じて総合経営報告書に適切な分類を行うために、顧客との契約を評価します。顧客との取引記録は我々の総合経営報告書に記載されており、提携関係の特徴に応じて、毛額または純額で記録されている。
ASC 606の範囲内で配置された収入確認を決定するために、(I)顧客との契約を決定するステップと、(Ii)契約中の履行義務を決定するステップと、(Iii)可変対価格(あれば)を含む取引価格を決定するステップと、(Iv)契約中の履行義務に取引価格を割り当てるステップと、(V)エンティティが履行義務を履行する場合(または履行義務として)収入を確認するステップと、の5つのステップを実行する。顧客の商品やサービスに移行するために獲得する権利のある対価格を受け取る可能性が高い場合にのみ、5段階モデルを契約に適用します。
顧客の履歴支払い経験や(新規顧客に対する)顧客に関する信用や財務情報などの要因に基づいた判断を用いて顧客の支払い能力や意図を決定する。約束された貨物やサービスが個別的な義務であるかどうかを決定するには重大な判断が必要だ。このような判断の変化は、収入確認期間の大きな変化を招く可能性がある。私たちは私たちの全体価格と割引目標に基づいて、サービスタイプ、一時間当たりの市場料率の推定、及び研究、開発或いは臨床試験の段階を考慮して、独立した販売価格を決定します。取引価格を確定する過程は重大な判断に関連し、収入の推定、市場規模と開発リスク、及び顧客との手配を交渉する際に考慮する他の要素など、複数の要素を考慮することが含まれる。私たちは顧客に商品とサービスを譲渡するために獲得する権利が期待される価格に基づいて取引価格を決定します。取引価格を決定する際には,我々の判断により,契約項での累積収入が将来大きく逆転しない可能性が高い場合には,適用範囲内で任意の可変要因を考慮する.ASC 606による知的財産権許可に関する特許権使用料例外によれば、取引価格には、将来私たちの顧客から受信した特許権使用料支払いは含まれていない。我々の連携·許可プロトコルには、顧客に支払う対価格も含まれておらず、重要な融資部分も含まれていない。
契約義務を履行する
以下は私たちが収入を創出する主な商品とサービスの説明だ。
知的財産権許可証
私たちは独自技術と開発と商業化権利を含む私たちの知的財産権を許可することで収入を生成する。これらのライセンスは、顧客に期限に基づくライセンスを提供し、私たちの内部で発見された特定の治療適応のためのプラットフォーム技術をさらに研究、開発し、商業化する。機能性知的財産権許可を顧客に譲渡する際に、契約中の他の履行義務とは異なると判定された許可について、機能性知的財産権許可に割り当てられた対価格は、払戻不可能な前払い対価格の形で確認される。ライセンスが他の商品又はサービスと合併して義務を履行する場合、収入は一定期間内に私たちの
130
進展を測定する方法であり、この方法では、私たちは合併履行義務を履行した。私たちは報告期間ごとに進捗指標を評価し、必要に応じて業績指標と関連収入確認を調整する。私たちの許可協定は一般的にキャンセルできる。顧客は通知を受けた後に契約を終了する権利がある。お客様が契約に違反し、契約条項に従って救済されていない場合にのみ、私たちは契約を終了する権利があります。
物質的権利
顧客によって適宜行使可能な追加の商品またはサービスを含む権利の配置は、一般に代替方法と考えられる。これらのオプションが顧客に実質的な権利(すなわち、無料または割引によって提供されるオプション商品またはサービス)を提供しているかどうかを評価し、そうであれば、それらが義務を履行しているとみなされるかどうかを評価する。重大な権利の確認は、割引金額およびオプションが行使される可能性に関する仮定を含む、オプション行使価格に対する基礎許可の価値の決定に関する判断が必要である。会計目的のため、物質的権利の行使は契約改正とみなされる。任意の物質権利に割り当てられた金額は、関連する将来の貨物またはサービス譲渡時またはオプション満了時に収入として確認される。
研究と開発サービス
我々は,ライセンス知的財産権に関する研究開発サービスを顧客に提供することで収入を創出している.私たちが顧客に提供するサービスは主に科学研究活動を含む。これらのサービスに関する収入は,債務履行の総見積もりコストに基づいて,使用コスト比入力法が時間の経過とともに確認されている.
多重履行義務を持つ契約
私たちの顧客との協力および許可協定には、複数の約束された商品やサービスが含まれている可能性がある。取引価格は相対的に独立した販売価格で単独の履行義務に割り当てられる。私たちは私たちの全体価格と割引目標に基づいて、サービスタイプ、1時間当たりの市場料率の推定、及び開発と臨床試験の段階を考慮して、独立販売価格を決定します。
ASC 606は、契約の取引価格を決定し、その金額を割り当てるべき履行義務を決定した後、相対的に独立した販売価格で義務履行毎に価格を割り当てることを要求する。ASC 606において、相対的に独立した販売価格は、エンティティが約束された商品またはサービスをクライアントに個別に販売する価格として定義される。同じ履行義務の他の観察可能な取引を単独で販売していなければ、各履行義務の独立販売価格を推定します。我々の製品やサービスの独立販売価格が直接観察できない場合、入手可能な関連情報を用いて独立販売価格を決定し、適切な推定方法を採用して、基礎知的財産権許可の特徴と機能、持続的な研究活動に関連する経済潜在力を含むが、基礎知的財産権許可の特徴と機能を含む市場状況と特定の実体の要素を考慮する。独立販売価格を決定するための重要な仮定には、開発スケジュール、人員コストの販売率、割引率、および技術と規制が成功する確率も含まれる可能性がある。
研究と開発費用を計算すべきである
連結財務諸表作成過程の一部として、我々が計上すべき研究·開発費用を見積もる必要がある。このプロセスは、未締結契約および調達注文を審査し、私たちの適用者とコミュニケーションして、私たちを代表して実行されるサービスを決定し、インボイスを受け取っていない場合、または他の方法で実際のコストを通知している場合に、実行されたサービスレベルおよびサービスによって生じる関連コストを推定することを含む。私たちのほとんどのサービス提供者は私たちにサービス料を滞納する領収書を発行してくれますが、前金が必要なものもあります。私たちは、私たちが当時知っていた事実と状況に基づいて、私たちの連結財務諸表において、各貸借対照表の日付までの課税費用を推定します。私たちは
131
定期的にサービスプロバイダと推定の正確性を確認し,必要に応じて調整する.検討·開発費用を算定すべき例としては,臨床前開発活動に関する費用,プロセス開発·拡大活動に関するCMO費用,臨床試験に関連する臨床試験材料の生産,臨床試験に関する契約研究組織をサプライヤーに支払うことが想定される。
材料及びサービスを提供する複数のCMO及び契約研究組織とのオファー及び契約に基づいて、受信したサービス及び費用の努力を推定し、契約研究及び製造に関連する費用を記録する。これらの合意の財務条項は交渉が必要であり、契約によって異なり、支払いの不均衡を招く可能性がある。場合によっては、私たちのサプライヤーに支払われるお金は、提供されたサービスレベルを超え、事前支払い費用につながる可能性があります。サービス料を計算する際に、サービスを提供する時間帯と各時間の努力度を見積もります。サービス実行の実際の時間または努力の程度が推定値と異なる場合、それに応じて計算すべき費用または前払い費用を調整する。実際に発生した金額と実質的に異なることはないと予想されるが、提供されたサービスの実際の状態および時間に対する提供サービスの状態および時間の理解が異なる可能性があり、報告された金額が任意の特定の時期に高すぎたり、過小になったりする可能性がある。これまで,我々が計算した研究や開発費の先行推定には何の大きな調整もなかった.
株に基づく報酬
ブラック·スコアーズオプション定価モデルを用いて、付与された日の公正価値に基づいて、従業員、非従業員、取締役の株式奨励を測定した。これらの賠償金の補償費用は必要なサービス期間内に確認され、サービス期間は通常各賠償金の授権期間である。私たちは直線法を用いてサービスの付与条件に基づく奨励費用を記録した。没収行為は発生時に確認します。
ブラック·スコアーズオプション定価モデルは、株式ベースの報酬およびESPPに従って購入可能な株の公正価値を決定するために主観的仮定を使用することを必要とする。これらの仮説には
2023年12月31日現在、従業員の株式オプションに関する未確認株式報酬支出は2410万ドルで、約2.3年の加重平均期間に支出として確認される予定だ。2023年12月31日現在,すべての未償還株式オプションの内在価値は約170万ドルであり,そのうち約80万ドルは既得オプションに関係しており,約90万ドルは非既得オプションと関係がある。
“雇用法案”
私たちは改正された1933年証券法第2(A)節で定義されたように、あるいは2012年にJumpStart Our Business Startups ActまたはJOBS Actによって改正された証券法のような新興成長型会社である。“雇用法案”
132
我々のような“新興成長型企業”が、上場企業に適用される新たなまたは改訂された会計基準を遵守するために、延長された過渡期を利用することを可能にする。私たちは、私たち(I)が新興成長型会社または(Ii)が“雇用法案”に規定されている移行期間の延長を明確かつ撤回できない日(早い者を基準とする)まで、雇用法案に規定されている延長移行期間を使用することを選択した。したがって、我々の連結財務諸表は、上場企業の発効日までに新たなまたは改訂された会計声明を遵守している会社と比較できない可能性がある。JOBS法案はまた、改正された2002年の“サバンズ-オキシリー法案”第404条の監査人認証要件の免除、当社の登録声明、定期報告、委託書における役員報酬に関する広範でない開示義務、役員報酬についての非拘束性相談投票の要求を免除し、株主承認までに承認されなかった金パラシュート支払いを免除することを含む、他の非新興成長型企業の上場企業に適用される様々な報告要件のいくつかの免除を利用することを可能にしている。私たちはまた未来の届出文書に他の低減された報告要件を利用することを選択することができる。したがって,我々の株主は重要と思われる何らかの情報を得ることができない可能性があり,我々が株主に提供する情報は他の公開報告会社が提供する情報とは異なる可能性があり,それと比較することもできない.
私たちは、(1)本年度の最終日、私たちの年収が12.35億ドルを超える、(2)“大型加速申告会社”の資格に適合した日まで、(1)関連会社が少なくとも7.00億ドルの株式証券を保有している、(3)前の3年間で10億ドルを超える転換不可能債務証券を発行し、(4)2025年12月31日まで発生する新興成長型会社である。
1934年の“証券取引法”によると、私たちも規模の小さい報告会社です。私たちがもう新興成長型会社ではなくても、私たちは規模の小さい報告会社であり続けるかもしれない。私たちは、規模の小さい報告会社が入手可能ないくつかの比例して開示された情報を利用して、(I)私たちの非関連会社が保有する投票権と非投票権普通株が第2四半期の最終営業日に2.5億ドル未満である限り、または(Ii)最近終了した会計年度の年収が1.00億ドル未満であり、私たちの非関連会社が保有する投票権および非投票権普通株が第2期の最終営業日に7.00億ドルを下回る限り、これらの比例開示された情報を利用することができる。
最近の会計公告
最近発表された我々の財務状況、経営業績又はキャッシュフローに影響を与える可能性のある会計声明の記述は、本年度報告に含まれる総合財務諸表の付記2に開示される。
第七A項。数量化と高質市場リスクの開示について。
金利リスク
2023年12月31日現在、私たちは現金、現金等価物、短期投資2.122億ドルを持っている。現金は金融機関の預金に含まれている。金利収入は金利の総水準の変化に敏感です。しかし、これらの投資の性質により、金利がどの期間にわたって10%の変化が生じても、我々の総合財務諸表に実質的な影響を与えないと仮定する。
2023年12月31日現在、吾らは2022年の融資協議の下で6,000万ドルが借金を返済しておらず、変動金利で利息を計上しており、金利は(A)7.94%と(B)(I)の利息月直前の1ヶ月の営業日の1ヶ月のCME期限SOFR、(Ii)0.10%と(Iii)7.83%の和に等しい。金利が上記のいずれの期間に10%の変動が発生しても、我々の総合財務諸表に実質的な影響を与えないと仮定する。
外貨両替リスク
これまで、外貨取引損益は私たちの連結財務諸表にとって重要ではなく、私たちも正式な外貨ヘッジ計画はありませんでした。私たちの費用は普通ドルで計算されます。しかし、私たちは限られた数の外国の供給者たちと契約を結んだ
133
ヨーロッパとカナダに位置し、未来に外国のサプライヤーと契約するかもしれない。私たちの業務は将来外貨為替レートの変動の影響を受けるかもしれません。為替レートが上記のいずれの期間に10%の変動が発生しても我々の総合財務諸表に実質的な影響を与えないと仮定する。
インフレの影響
インフレは一般的に私たちの労働コストを増加させることで私たちに影響を及ぼす。私たちはインフレが私たちの総合財務諸表に実質的な影響を及ぼすとは思わない。
プロジェクト8.財務諸表Sと補足データ。
本プロジェクトに必要な財務諸表と補足データは、本年度報告シート10-K第4部第15項に列挙された各ページに掲載されています。
項目9.Accoとの変更と分岐“会計と財務開示”誌。
ない。
第9条。制御するプログラムがあります
情報開示制御とプログラムの評価
私たちは、1934年に改正された証券取引法または“取引法”の下の規則13 a-15(E)および15 d-15(E)で定義されているように、開示制御および手続きの維持を担当する。開示制御およびプログラムは、取引法に基づいて提出または提出された報告書において開示を要求する情報が、米国証券取引委員会規則および表に指定された期間内に記録、処理、集約および報告されることを保証するための制御および他の手続きである。開示制御及び手続は、取引所法案に基づいて提出又は提出された報告書において開示を要求する情報が蓄積されていることを確保し、開示すべき情報を速やかに決定するために、我々の経営陣に適宜伝達されることを目的としているが、制御及び手続に限定されない。
我々の経営陣の評価によると、最高経営責任者と最高財務官は、2023年12月31日に我々の開示統制と手続きが発効したと結論した。
財務報告の内部統制に関する経営陣の報告
我々の経営陣は、取引法第13 a-15(F)および15 d-15(F)条で定義されているように、財務報告の十分な内部統制の確立と維持を担当している。我々は財務報告に対して内部統制を維持し、財務報告の信頼性に合理的な保証を提供し、公認された会計原則に基づいて外部目的の財務諸表を作成することを目的としている。
財務報告の内部統制はその固有の限界のため、財務報告目標の実現に絶対的な保証を提供することができない。財務報告の内部統制は人の勤勉さとコンプライアンスに関わる過程であり、人のミスによって判断ミスや故障が発生しやすい。財務報告に対する内部統制も談合や不当な管理を凌駕することで回避することができる。発想や動作がどんなに良くても、絶対的な保証ではなく、合理的な保証しか提供できず、制御システムの目標が実現されることを確保する制御システム。また,制御システムの設計は,資源制約が存在し,そのコストに対する制御の利点を考慮しなければならないという事実を反映しなければならない.すべての制御システムの固有の限界により,どの制御評価もわが社内のすべての制御問題や不正事件(あれば)が発見されていることを絶対に保証することはできない.このような制限のため、財務報告書の内部統制は重大な誤報をタイムリーに防止したり発見できない可能性がある。しかし、このような固有の制限は財務報告手続きの既知の特徴だ。したがって、(除去ではないにもかかわらず)このリスクを低減するために、プロセス中に保障措置を設計することが可能である。
134
私たちの最高経営責任者と最高財務責任者の監督の下で、私たちの経営陣は、トレデビル委員会後援組織委員会が発表した“内部統制-総合枠組み”(2013年の枠組み)で提案された基準に基づいて、私たちの財務報告の内部統制の有効性を評価した。この評価に基づき、我々の経営陣は、2023年12月31日現在、財務報告の内部統制が合理的な保証水準で有効であると結論した。
公認会計士事務所認証報告
本年度報告には、雇用法案が“新興成長型会社”のために設立された免除による財務報告の内部統制に関するわが公認会計士事務所の証明報告は含まれていない。
財務報告の内部統制の変化
2023年12月31日までの四半期内に、財務報告の内部統制(“取引法”第13 a-15(F)および15 d-15(F)規則の定義による)に大きな影響を与えなかったか、または合理的に財務報告の内部統制に大きな影響を与える可能性のある変化はなかった。
プロジェクト9 B。他にも情報です。
ない。
プロジェクト9 Cです。開示する検査を阻止した外国司法管轄区のUREについて。
適用されません。
135
部分(三)
プロジェクト10.役員·役員職ICERSと会社管理。
本プロジェクトに要求される情報(以下に述べるを除く)は、本10−K年度報告書に含まれる財政年度終了後120日以内に米国証券取引委員会に提出される依頼書を参考にして取締役に組み込まれ、タイトルは“提言1:取締役選挙”、“取締役が著名人と現職取締役に言及された情報”、“取締役会及び会社統治に関する情報”、“執行役員”である
私たちは、私たちの最高経営責任者、最高財務責任者、および最高会計官を含む、私たちのすべての役員、高級管理者、および従業員に適用される書面ビジネス行動と道徳基準を採択しました。ビジネス行為と道徳基準の最新コピーは、当サイトのコーポレート·ガバナンス部分で取得することができます。サイトはwww.pose ida.comです。私たちは、米国証券取引委員会規則に基づいて開示される必要がある私たちの商業行為および道徳基準の任意の修正または免除を私たちのウェブサイトで開示するつもりだ。
プロジェクト11.行政官イー補償します。
本プロジェクトに要求される情報は、本年度報告がカバーする財政年度終了後120日以内に米国証券取引委員会に提出される2024年年次総会に関する我々の委託書を参考にして統合され、この年次報告はForm 10−K形式で“役員報酬”と題する部分で行われる
プロジェクト12.特定の受益者の保証所有権従業員と経営陣と関連した株主問題。
本プロジェクトに要求される情報は、本年度報告書に含まれる10-Kフォームの“特定の利益所有者および経営陣の保証所有権”という節の10-Kフォームに含まれる財政年度終了後120日以内に米国証券取引委員会に提出される2024年株主年次総会に関する私たちの委託書を参照することによって統合される
本プロジェクトが要求する情報は本稿に含まれ,本年度報告がカバーする10−K表に含まれる財政年度終了後120日以内に米国証券取引委員会に提出される2024年年次総会に関する我々の依頼書を参考にし,タイトルは“関係者との取引と賠償”と“取締役会と会社のガバナンスに関する情報”である
第14項.元金口座TING料金とサービスです。
本プロジェクトに要求される情報はここに含まれており,本年度報告に含まれる10−K表に含まれる財政年度終了後120日以内に米国証券取引委員会に提出される2024年年次総会に関する我々の依頼書を参考にして,“提案2:独立公認会計士事務所の選択を承認する”というタイトルである
136
部分IV.IV
プロジェクト15.物証、資金請求書の明細書です。
本年度報告書の一部として、以下の財務諸表と財務諸表を提出した
連結財務諸表索引
独立公認会計士事務所レポート(PCAOB ID番号) |
|
138 |
|
|
|
独立公認会計士事務所レポート(PCAOB ID番号) |
|
139 |
|
|
|
2023年12月31日と2022年12月31日までの連結貸借対照表 |
|
140 |
|
|
|
2023年12月31日と2022年12月31日までの総合経営報告書と全面赤字 |
|
141 |
|
|
|
2023年12月31日までと2022年12月31日まで年度株主権益変動表 |
|
142 |
|
|
|
2023年12月31日と2022年12月31日までの統合現金フロー表 |
|
143 |
|
|
|
連結財務諸表付記 |
|
144 |
陳列品
添付されている展示品インデックスに記載されている展示品は,10-K表年次報告の一部としてアーカイブや統合を行っており,参考にしている.
137
“独立地域裁判所報告書”英国王立会計士事務所
ポセイダ治療会社の株主と取締役会に。
財務諸表のいくつかの見方
添付されているポセイダ治療会社とその子会社(当社)の2023年12月31日現在の総合貸借対照表と、2023年12月31日現在の関連総合経営報告書と全面赤字、株主権益変動と現金流量、および関連付記(総称して“総合財務諸表”と呼ぶ)を監査した。総合財務諸表は,すべての重要な点において,会社の2023年12月31日の財務状況と,2023年12月31日までの年度の経営結果とキャッシュフローを公平に反映しており,米国公認会計原則に適合していると考えられる。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査することを求められておらず、私たちも招聘されて監査を行っていません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性について意見を述べるためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
/s/
2023年以来、私たちは会社の監査役を務めてきた。
2024年3月7日
138
独立公認会計士事務所報告
ポセイダ治療会社の取締役会と株主に。
財務諸表のいくつかの見方
添付のポセイダ治療会社とその子会社(“当社”)2022年12月31日現在の総合貸借対照表と、同年度までの株主権益とキャッシュフロー変化に関する総合経営と全面赤字報告書(総称して“総合財務諸表”と呼ぶ)を監査した。総合財務諸表は,すべての重要な面で当社の2022年12月31日までの財務状況と,同年度までの経営結果とキャッシュフローを公平に反映しており,米国公認の会計原則に適合していると考えられる。
意見の基礎
これらの連結財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の総合財務諸表に意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従ってこれらの連結財務諸表を監査した。これらの基準は、連結財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。
我々の監査には、連結財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであっても詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれる。これらの手続きは、連結財務諸表中の金額および開示に関する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、合併財務諸表の全体列報の評価も含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
物質的重点
統合財務諸表付記1に記載されているように、同社は将来の運営に資金を提供するための追加資金を必要とする。付記1では,経営陣のイベントや状況の評価およびこの問題を軽減する計画について説明した。
/s/
2023年3月9日
私たちは2015年から2023年まで当社の監査役を務めています。
139
ポセイダ治療会社
合併B割当書
(単位は千で、シェアは含まれていない)
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
短期投資 |
|
|
|
|
|
|
||
売掛金 |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
流動資産総額 |
|
|
|
|
|
|
||
財産と設備、純額 |
|
|
|
|
|
|
||
経営的リース使用権資産 |
|
|
|
|
|
|
||
無形資産、純額 |
|
|
|
|
|
|
||
商誉 |
|
|
|
|
|
|
||
その他長期資産 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
負債と株主権益 |
|
|
|
|
|
|
||
流動負債: |
|
|
|
|
|
|
||
売掛金 |
|
$ |
|
|
$ |
|
||
費用とその他の負債を計算すべきである |
|
|
|
|
|
|
||
賃貸負債を経営し、流動 |
|
|
|
|
|
|
||
収入を繰延し,当期 |
|
|
|
|
|
|
||
流動負債総額 |
|
|
|
|
|
|
||
定期債務 |
|
|
|
|
|
|
||
繰延CIRM贈与負債 |
|
|
— |
|
|
|
|
|
収入を繰延し、流動ではない |
|
|
|
|
|
|
||
非流動経営賃貸負債 |
|
|
|
|
|
|
||
その他長期負債 |
|
|
|
|
|
|
||
総負債 |
|
|
|
|
|
|
||
*(注12) |
|
|
|
|
|
|
||
株主権益: |
|
|
|
|
|
|
||
普通株、$ |
|
|
|
|
|
|
||
追加実収資本 |
|
|
|
|
|
|
||
その他の総合収益を累計する |
|
|
|
|
|
( |
) |
|
赤字を累計する |
|
|
( |
) |
|
|
( |
) |
株主権益総額 |
|
|
|
|
|
|
||
総負債と株主権益 |
|
$ |
|
|
$ |
|
||
付記はこれらの財務諸表の不可分の一部だ。
140
ポセイダ治療会社
業務所合併報告書損失と総合損失
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
収入: |
|
|
|
|
|
|
||
協力収入 |
|
$ |
|
|
$ |
|
||
総収入 |
|
|
|
|
|
|
||
運営費用: |
|
|
|
|
|
|
||
研究開発 |
|
|
|
|
|
|
||
一般と行政 |
|
|
|
|
|
|
||
総運営費 |
|
|
|
|
|
|
||
運営損失 |
|
|
( |
) |
|
|
( |
) |
その他の収入(支出): |
|
|
|
|
|
|
||
利子支出 |
|
|
( |
) |
|
|
( |
) |
その他の収入、純額 |
|
|
|
|
|
|
||
所得税前純損失 |
|
|
( |
) |
|
|
( |
) |
所得税費用 |
|
|
( |
) |
|
|
( |
) |
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
|
|
|
|
|
|
||
その他の全面的な収入(費用): |
|
|
|
|
|
|
||
短期投資の未実現収益 |
|
|
|
|
|
( |
) |
|
総合損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
|
|
|
|
|
|
||
普通株主は1株当たり基本損失と希釈して1株当たり純損失を占めるべきである |
|
$ |
( |
) |
|
$ |
( |
) |
加重-流通株、基本株と希釈株の平均 |
|
|
|
|
|
|
||
付記はこれらの財務諸表の不可分の一部だ。
141
ポセイダ治療会社
年合併変動表株主権益
(単位は千で、シェアは含まれていない)
|
|
ごく普通である |
|
|
その他の内容 |
|
|
積算 |
|
|
積算 |
|
|
合計する |
|
|||||||||
|
|
株 |
|
|
金額 |
|
|
資本 |
|
|
収入(損) |
|
|
赤字.赤字 |
|
|
権益 |
|
||||||
2021年12月31日の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
|
|
|||||
従業員株補償計画下普通株の発行 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
公募から普通株を発行し,発行コストを差し引いて$とする |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
証券売却可能な未実現損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2022年12月31日の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
従業員株補償計画下普通株の発行 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
ATMで普通株を発行し,発行コストを差し引く |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
方向性増発で普通株を発行する |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
証券売却可能な未実現収益 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2023年12月31日の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||||
付記はこれらの財務諸表の不可分の一部だ。
142
ポセイダ治療会社
合併状態キャッシュフロープロジェクト
(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
経営活動: |
|
|
|
|
|
|
||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
純損失と経営活動で使用される現金純額の調整: |
|
|
|
|
|
|
||
減価償却および償却費用 |
|
|
|
|
|
|
||
財産と設備処分損失 |
|
|
|
|
|
|
||
株に基づく報酬 |
|
|
|
|
|
|
||
核販売が延期されているCIRM贈与負債 |
|
|
( |
) |
|
|
— |
|
発行済み定期債務の割引が増加する |
|
|
|
|
|
|
||
投資証券増額純額 |
|
|
( |
) |
|
|
( |
) |
経営性資産と負債変動状況: |
|
|
|
|
|
|
||
売掛金 |
|
|
|
|
|
( |
) |
|
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
経営的リース使用権資産 |
|
|
|
|
|
|
||
その他長期資産 |
|
|
( |
) |
|
|
|
|
売掛金 |
|
|
|
|
|
( |
) |
|
費用とその他の負債を計算すべきである |
|
|
|
|
|
|
||
リース負債を経営する |
|
|
( |
) |
|
|
( |
) |
収入を繰り越す |
|
|
|
|
|
|
||
経営活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
投資活動: |
|
|
|
|
|
|
||
財産と設備を購入する |
|
|
( |
) |
|
|
( |
) |
財産と設備を売却して得た収益 |
|
|
— |
|
|
|
|
|
短期投資を購入する |
|
|
( |
) |
|
|
( |
) |
短期投資満期収益 |
|
|
|
|
|
|
||
投資活動提供の現金純額 |
|
|
|
|
|
( |
) |
|
融資活動: |
|
|
|
|
|
|
||
従業員株補償計画に基づいて普通株で得られた金を発行する |
|
|
|
|
|
|
||
株式奨励金の株式純額決済に関する税額を支払う |
|
|
( |
) |
|
|
— |
|
ATMで普通株を発行する収益は,発行コストを差し引く |
|
|
|
|
|
— |
|
|
普通株に私募して得た収益 |
|
|
|
|
|
— |
|
|
普通株を公開発行して得られる収益は,発行コストを差し引く |
|
|
— |
|
|
|
|
|
定期債務収益 |
|
|
— |
|
|
|
|
|
債務発行コストを支払う |
|
|
— |
|
|
|
( |
) |
融資活動が提供する現金純額 |
|
|
|
|
|
|
||
現金と現金等価物の純減少 |
|
|
( |
) |
|
|
( |
) |
期初現金及び現金等価物 |
|
|
|
|
|
|
||
期末現金および現金等価物 |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
非現金経営、投資、融資活動: |
|
|
|
|
|
|
||
支払すべき帳簿に掲げる財産と設備の購入と |
|
$ |
|
|
$ |
|
||
経営性リース負債と引き換えに使用権資産 |
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
||
キャッシュフロー情報の追加開示: |
|
|
|
|
|
|
||
支払の利子 |
|
$ |
|
|
$ |
|
||
付記はこれらの財務諸表の不可分の一部だ。
143
ポセイダ治療会社
連結財務諸表付記
違います。TE 1.ビジネスの性質と届出根拠
運営の性質
ポセイダ治療会社(“会社”または“ポセイダ”)は、癌およびまれな疾患の患者に新たな治療方法を提供する臨床段階の細胞および遺伝子治療会社である。同社は、その非ウイルス型iggyBac DNA送達システム、Cas-Clover部位固有遺伝子編集システム、およびナノ粒子およびAAVに基づく遺伝子送達技術を含む、そのコア独自プラットフォームに基づく様々な適応の広範な候補製品の組み合わせを発見し、開発している。
同社は生物技術業界の発展段階の会社によく見られるリスクと不確定要素の影響を受け、競争相手の新技術革新の開発、肝心な者への依存、ノウハウの保護、政府法規の遵守及び追加資本を獲得して運営を援助する能力を含むが、これらに限定されない。現在開発されている候補製品は、広範な臨床前と臨床試験及び商業化前の監督管理承認を含む大量の追加的な研究と開発作業が必要となる。このような努力は多くの追加資本、十分な人員とインフラ、そして広範囲なコンプライアンス報告能力を必要とする。同社の治療開発努力が成功しても,同社がいつ(あれば)製品販売から相当な収入を得るかは定かではない。
流動性と資本資源
設立以来、同社の運営は純損失と負のキャッシュフローを経験し、主に株式と債務融資および戦略協力を通じてその運営に資金を提供する能力に依存している当社は2023年12月31日および2022年12月31日までに純損失を計上した$
会社は、2023年12月31日まで、その現金、現金等価物と短期投資を予定している$
列報と合併の基礎
添付されている連結財務諸表は、同社の財務状況、経営結果と現金流量を反映し、米国公認の会計原則(“GAAP”)に適合し、その中にポセイダ治療会社及びその全額子会社の勘定を含む。すべての会社間取引と残高は無効になりました。
リスクと不確実性
全世界中東紛争、ロシアのウクライナへの侵入、米国、NATO、その他の国がすでに取っているか、あるいは将来取られる可能性のある報復措置などの事件は、世界の安全懸念を引き起こし、地域衝突を招く可能性がある。そうでなければ、
144
地域性これらのリスクのいずれかまたは全部が会社のサプライチェーンを混乱させる可能性があり、会社が実施しており、将来的に会社の候補製品に対する臨床試験を行う能力に悪影響を及ぼす可能性がある。このような持続的あるいは将来的な衝突が最終的に会社業務に与える影響の程度は高度に不確実であり,現時点では把握的に予測することはできない.
付記2.主要会計政策の概要
予算の使用
公認会計原則に従って連結財務諸表を作成する場合、会社は連結財務諸表と付記中の報告金額に影響を与える推定と仮定を行う必要がある。当社は、収入、課税支出、研究開発費、株式報酬支出、繰延税項目の評価免除額に関する推定数字を含むが、これらに限定されない推定数字を評価し続けている。当社は過去の経験その他その時点で部下が合理的と考えている特定の市場あるいは関連仮定に基づいて推定しています。実際の結果は,これらの推定や仮定とは異なる可能性がある.
市場情報を細分化する
同社の独占業務には,高度に満たされていない医療ニーズの患者のための治療薬の開発が含まれている。そこで当社は
公正価値計量
特定の金融商品は公正な価値で入金することを要求する。現金のような他の金融商品は、コストで入金され、公正な価値に近い。現金等価物と短期投資は販売可能な証券からなり、公正価値に基づいて入金される。また、売掛金の満期日が短いため、帳簿金額は公正価値に近い。その変動金利は市場金利に近いため、当社の定期債務の帳簿価値はその公正価値に近い。
業務リスク集中
同社は依存しており、引き続き少数のサプライヤーに依存して物資や材料を開発計画していると予想される。このような製造サービスの重大な中断はこのような計画に悪影響を及ぼすかもしれない。同社の収入は2人の顧客との協力と許可協定から来ている。
賃貸借証書
当社は会計基準コードテーマ842に基づいてレンタルを会計処理している賃貸借証書(“ASC 842”)。当社は契約開始時に一つの手配がレンタルかどうかを確定します。契約譲渡が一定期間内に確定した財産、工場または設備の使用権を制御して対価格と交換する場合、レンタルは存在する。賃貸の定義には、(1)契約に確定された資産が土地または減価償却可能資産(すなわち、不動産、建屋および設備)であること、および(2)会社が決定された資産の使用を制御する権利があることの2つの条件がある。
当社はテナントの経営リースに経営賃貸使用権(“ROU”)資産、経営賃貸負債、流動及び経営賃貸負債、総合貸借対照表中の非流動資産を計上している。賃貸負債の最初とその後はレンタル開始日未払い賃貸支払いの現在値で計測します。
145
ASC 842は、テナントがレンタルに隠れた金利を用いて未払いのリース支払いを割引することを要求し、その金利を容易に決定できない場合には、その逓増借入金利を用いて割引を行う。当社の賃貸借契約に隠されている金利はあまり知られていませんので、増額借入金金利は発効日に得られた資料に基づいてレンタル支払いの現在値を決定します。当社の賃貸の逓増借入金利とは、担保に類似条項の下の賃貸支払いに等しい金額を借り入れるのに必要な金利のことです。
当社のすべての賃貸借契約の期限には借約の取消不能期限が含まれています。当社のリース期間が賃貸契約の延長または終了の選択権の影響を受け、その選択権を行使すると合理的に決定された場合、賃貸支払いはリース資産または負債の計量に計上される。
当社は、すべてのレンタル期間が12ヶ月以下の短期賃貸契約を確認しない純収益資産と賃貸負債を選択しました。当社はその短期賃貸に関する賃貸支払いがレンタル期間内の直線法支出であることを確認した。いくつありますか
信用リスクが集中する
会社をかなり集中的な信用リスクに直面させる可能性のある金融商品には、主に現金、現金等価物、短期投資が含まれる。当社の連邦保険金融機関での預金は連邦保険限度額を超えています。当社は当該等の口座に何の損失も生じておらず、経営陣は当社が当該等の預金及び投資を保有する預金機関の財務状況により重大な信用リスクに直面することはないと信じている.
現金と現金等価物
当社は、購入日から90日以下の原始最終満期日に購入したすべての高流動性投資を現金等価物とみなしている。現金及び現金等価物は、金融機関の預金及び有価証券を含む。現金等価物は公正価値によって報告される。
短期投資
残り満期日が3カ月を超える投資は、総合貸借対照表では短期投資に分類され、主に米国財務省と他の政府機関債務からなる。企業のポートフォリオ全体が現在の業務に利用可能であると考えられているため、会社はすべての投資を売却および流動資産に分類し、満期日が現在の総合貸借対照表の日付より1年以上遅れている可能性がある場合であっても、管理層が必要に応じてこれらの証券を売却する収益を使用してその業務に資金を提供しようとしていることを反映しているそれは.債務証券は公正価値に基づいて入金され、損益を実現せずに他の全面収益(損失)を計上し、株主権益の構成部分として、実現までとしている。購入時に生成される任意の割増または割引は、手形の有効期間内に償却され、および/または利息収入および/または費用が増加される。公正価値のいかなる調整も当社が“非一時的”と考えている投資価値の低下を反映していれば、当社は経営報告書と全面赤字に費用を計上することで、投資を公正価値に減少させる。本報告で述べた間、このような調整を行う必要はない。
商業権その他無形資産
目に見えない資産は業務合併の一部として買収され、買収日に公平な価値で資本化されている。無期限に建設研究開発(“IPR&D”)は償却する必要はないが、毎年減値テストを行ったり、減値指標がある場合にはより頻繁にテストを行ったりする。同社は第4四半期に毎年その無期限知的財産権研究開発の減値状況をテストした。無限寿命知的財産権の研究開発に対して減値テストを行う時、当社はまず定性要素を評価して、事件或いは状況の存在がその公正価値がその帳簿価値よりもその帳簿価値よりも低い可能性があるかどうかを確定することができるかどうかを確定することができ、あるいは当社は数量化減値分析を行い、無限寿命知的財産権研究開発の公正価値を確定し、定性評価を行わないことができる。会社が考えている定性的要因
146
含まれています予想、重大な負の業界或いは経済傾向及び資産使用方面の重大な変化或いは計画変化に関連する業務の表現は明らかに良くない。もし当社がまず品質要素を評価し、無限寿命知的財産権研究開発の公正価値がその帳簿価値よりも少ない可能性が高いと判断した場合、当社はその後、無限寿命知的財産権研究開発の公正価値を決定する。いずれの方法でも、無限寿命知的財産権研究開発の公正価値がその帳簿価値より少ない場合、公正価値と帳簿価値との差額について減値費用を確認する。あったことがある
第4四半期に、またはイベントまたは環境変化が減値が発生する可能性があることを示す場合、同社は毎年その営業権を減値テストを行う。減値が存在する場合、減値損失は、資産または資産グループの帳簿価値が推定資産の公正価値を超えることに基づいて計量される。減値は,買収業務の業績悪化,開発作業の不利な結果,適用法律や法規の不利な変化,様々なその他の状況による可能性がある。商誉帳簿価値の回収可能性を評価する際には、会社は推定された将来の現金流量およびその他の要素を仮定して、買収資産の公正価値を決定しなければならない。戦略や市場状況の変化は将来のこれらの判断に大きな影響を与える可能性があり,記録された残高を調整する必要がある.いくつありますか
財産と設備
財産と設備はコストによって申告し、減価償却或いは償却は直線法を採用し、その推定耐用年数は以下の通りである
資産分類 |
|
使用寿命を見込む |
|
|
実験室装置 |
|
|
|
|
賃借権改善 |
|
|
||
コンピュータ装置及びソフトウェア |
|
|
|
|
家具と固定装置 |
|
|
|
|
メンテナンスとメンテナンス費用は発生時に費用を計上します。資産が抹消またはその他の方法で処分された場合、コストおよび減価償却は、会社の総合貸借対照表から除外され、それによって生じる任意の収益または損失は、会社の総合経営報告書および全面赤字に反映される。
イベントや環境変化がある資産の帳簿金額が回収できない可能性があることを示すたびに,物件や設備の減価状況を審査する。回収能力は,帳簿金額と資産予想による将来の未割引純現金流量を比較することで測定した。当該等の資産は減値とみなされ、確認すべき減値は、当該資産の帳簿金額が当該資産による予想割引将来のキャッシュフロー純額の金額を超えて計量される。もうあります
収入確認
これまで、同社の収入は主に協力と許可協定から来ていた。同社の協力·許可協定には、知的財産権許可および研究·開発サービスを含む複数の要素が含まれている可能性がある。これらの手配によると、会社が受け取った対価格には、前金、研究開発資金、コスト補償、研究開発、監督管理、商業マイルストーン支払い、特許権使用料が含まれる可能性がある。
当社は会計基準コード特別テーマ606を適用している取引先と契約した収入(“ASC 606”)は、顧客との契約を示すために、財務会計基準委員会(“FASB”)によって発行される。ASC 606によれば、収入は、顧客が約束された商品またはサービスの制御権を取得したときに確認される。確認された収入金額は、会社が#年に受け取る権利があると予想された対価格を反映している
147
これらのサービスを交換します。同社は、個人協力やライセンス契約を背景に、異なる履行義務を評価するために、これらの履行義務の性質を分析している。当社は、総合経営報告書に適切な分類を行うために、基本活動の性質に基づいて顧客との契約を評価します。顧客との取引記録は、会社の総合経営報告書に記載されており、提携関係の特徴に応じて、毛数または純額で記録されている。
ASC 606の範囲内に配置された収入確認を決定するために、当社は、(I)顧客との契約を決定するステップと、(Ii)契約中の履行義務を決定するステップと、(Iii)可変対価格(あれば)を含む取引価格を決定するステップと、(Iv)契約中の履行義務に取引価格を割り当てるステップと、(V)エンティティが契約義務を履行する際に収入を確認するステップと、の5つのステップを実行する。会社が顧客に譲渡された商品やサービスと交換するために獲得する権利のある対価格を受け取る可能性がある場合にのみ、会社は5ステップモードを契約に適用する。
同社は、顧客の履歴支払い経験や(新規顧客に対する)顧客に関する信用や財務情報などの要因に基づいた判断を用いて顧客の支払い能力や意図を決定する。約束された貨物やサービスが個別的な義務であるかどうかを決定するには重大な判断が必要だ。このような判断の変化は、収入確認期間の大きな変化を招く可能性がある。同社はその全体定価と割引目標に基づき,サービスタイプ,1時間あたりの市場料率の推定および研究,開発あるいは臨床試験の段階を考慮して,独立した販売価格を決定している。取引価格を確定する過程は重大な判断に関連し、収入の推定、市場規模と開発リスク、及び顧客との手配を交渉する際に考慮する他の要素など、複数の要素を考慮することが含まれる。同社は、商品やサービスを顧客に移転することと引き換えに、その予想される取引価格に基づいて取引価格を決定する。取引価格を決定する際に、会社の判断により、契約項での累積収入が将来大きく逆転しない可能性が高い場合には、適用範囲内で、いかなる可変考慮要素も考慮する。ASC 606による知的財産権許可に関する特許権使用料例外によれば、取引価格には、将来私たちの顧客から受信した特許権使用料支払いは含まれていない。その会社の協力と許可協定には
契約義務を履行する
以下に同社が収入を生み出した主な商品とサービスについて述べる。
知的財産権許可証
同社は、知的財産権(ノウハウ、開発権、商業化権利を含む)を許可することで収入を得ている。これらのライセンスは、社内で発見された特定の治療適応のためのプラットフォーム技術をさらに研究、開発し、商業化するための期限に基づくライセンスを顧客に提供する。会社が機能性知的財産権許可を顧客に譲渡する場合、契約中の他の履行義務とは異なると判断された許可については、会社が払戻不可能な前払い対価格の形で受信した対価格はその時点で確認される。ライセンスが他の商品やサービスと統合された場合
物質的権利
顧客によって適宜行使可能な追加の商品またはサービスを含む権利の配置は、一般に代替方法と考えられる。同社は、これらのオプションが顧客に実質的な権利(すなわち、無料または割引によって提供されるオプション商品またはサービス)を提供しているかどうかを評価し、そうである場合、それらが義務を履行しているとみなされているかどうかを評価する。物質権利の確認には物質権利の価値決定に関する判断が必要である
148
オプション価格に対する標的許可には,割引額とオプションが行使される可能性に関する仮定が含まれる.会計目的のため、物質的権利の行使は契約改正とみなされる。任意の物質権利に割り当てられた金額は、関連する将来の貨物またはサービス譲渡時またはオプション満了時に収入として確認される。
研究と開発サービス
同社はその顧客にライセンス知的財産権に関する研究·開発サービスを提供することで収入を得ている。会社が顧客に提供するサービスは主に科学研究活動を含む。これらのサービスに関する収入は,債務履行の総見積もりコストに基づいて,使用コスト比入力法が時間の経過とともに確認されている.
多重履行義務を持つ契約
会社と顧客との協力およびライセンス契約には、複数の約束された商品またはサービスが含まれている場合があります。約束された貨物とサービスの特徴に基づいて、会社はそれらが単独であるか合併の履行義務であるかを分析する。取引価格は相対的に独立した販売価格で単独の履行義務に割り当てられる。同社はその全体定価と割引目標に基づき,サービスタイプ,1時間あたりの市場料率の推定および開発と臨床試験の段階を考慮して,独立した販売価格を決定している。
ASC 606は、当社に、契約の取引価格を決定し、その金額を分配すべき履行義務を決定した後、相対的に独立した販売価格で履行義務毎に価格を割り当てるように要求する。ASC 606において、相対的に独立した販売価格は、エンティティが約束された商品またはサービスをクライアントに個別に販売する価格として定義される。当社が同じ履行義務の他に観察可能な取引を単独で販売していなければ、当社は契約義務ごとの独立販売価格を推定しています。会社の製品やサービスの独立販売価格を直接観察できない場合、会社は既存の関連情報を用いて独立販売価格を決定し、適切な推定方法を採用して、基礎知的財産権許可の特徴と機能、持続的な研究活動に関連する経済潜在力を含むが、基礎知的財産権許可の特徴と機能を含む市場状況と特定の実体の要素を考慮する。独立販売価格を決定するための重要な仮定には、開発スケジュール、人員コストの販売率、割引率、および技術と規制が成功する確率も含まれる可能性がある。
可変考慮事項
会社が顧客と締結する契約には、一般に、(I)研究、開発、および規制マイルストーン支払い、これらの特定のマイルストーンを達成する際にこれらの支払いを得る権利がある、(Ii)特許知的財産権に関連する一次的な販売ベースの支払いおよび販売ベースの使用料の2つの可変価格が含まれる。
もし一つの手配に研究、開発、または規制マイルストーンの支払いが含まれている場合、当社はマイルストーンが達成可能であると考えられているかどうかを評価し、最も可能な金額法を用いて取引価格に含まれる金額を推定する。大きな収入逆転が起こらない可能性が高い場合、関連するマイルストーン価値は取引価格に含まれるだろう。会社の統制範囲内でない記念碑的支払いは通常承認を受けるまでは実現できないと考えられる。
ライセンス知的財産権下の製品販売に基づく印税は印税例外項に計上される。同社は、ライセンス知的財産権の下で販売された特許使用料に基づく収入と、販売の発生または履行義務が履行または部分的に履行された場合の遅い時間に一度に支払うことを確認する。
ASC 606が知的財産権を許可する特許権使用料例外によれば、当社は、販売の発生または履行義務が満たされるか、または部分的に満たされるまで、販売に基づく特許権使用料およびマイルストーンに関連する可変金額のいかなる収入も確認しない。将来に関連した収入は
149
販売に基づく特許権使用料およびマイルストーンは、未履行業績義務に関する将来予想確認の推定収入には含まれていない。
収入の分類
同社は以下の地域で運営している
契約資産と契約負債
その会社は契約条項に基づいて顧客から支払いを受けた。売掛金は対価格権が無条件になったときに入金される。研究開発サービスについては、会社は通常、サービスを提供する際に毎月または四半期ごとに顧客に請求書を送信する
契約の獲得と履行のコスト
その会社は普通そうします
売掛金と信用損失の準備
売掛金には主に顧客が支払うサービス料と契約条項に応じて支払う金が含まれています。売掛金は対価格権が無条件になったときに入金される。研究開発サービスについては、会社は通常、サービスを提供する際に毎月または四半期ごとに顧客に請求書を送信する
研究と開発
研究開発費には、会社が研究開発プロジェクトに従事している科学研究者の人力、材料、設備、分配された施設コストが含まれる。研究·開発コストは発生時に運営費を計上する。
技術許可のための前金とマイルストーン支払いは,発生期間中に研究·開発支出とした。将来的に受信される研究および開発活動のための貨物またはサービスの前払いは、前払い費用または他の長期資産と表記される。前金は、関連貨物の交付またはサービス提供時に料金を計上します。
研究と製造契約コストと対策プロジェクト
同社は様々な研究開発と製造協定を締結した。これらの合意は通常キャンセル可能であり、関連支払いは相応の費用が発生したときに入金される。同社はこれまでに発生した見積もりコストの計上項目を記録している。計算すべき負債の十分性を評価する際に、当社は、事件の段階または完了状況、受信した請求書および契約コストを含む研究研究または臨床試験および製造活動の進捗状況を分析する。いずれの報告期間終了時の当計残高を決定する際には,重大な判断と見積もりを行う。実際の結果は会社の見積もりとは違うかもしれません。同社の歴史計算制見積もりは実際のコストと実質的な差はありません。
150
特許費用
特許出願の提出及び起訴に関するすべての特許に関する費用は、支出回収状況が不確定であることによる費用に基づいて計上される。発生した金額は一般費用と行政費用に分類される。
株に基づく報酬
会社員およびコンサルタントに付与された株式オプションおよび制限株式単位(“RSU”)に関する株式報酬および2020年従業員株式購入計画(“ESPP”)奨励は、付与日に報酬の公正価値で算出される。公正価値は、必要なサービス期間中に総合財務諸表において株式ベースの報酬支出として確認され、サービス期間は一般に各報酬の帰属期間である。サービスによる報酬に関する報酬は、授与日から授権期間内に直線的に確認され、通常は至れり尽くせり
当社はブラック·スコアーズ推定値モデルを用いて日株オプション奨励とESPPによって購入可能な株の公正価値を推定しています。オプション定価モデルを用いて各株式奨励の公正価値を決定する場合、会社のいくつかの変数に対する仮定の影響を受け、これらの変数は付与された日の普通株の公正価値、奨励の期待期限、奨励期間の予想株価変動、無リスク金利と配当率を含む。
総合収益(赤字)
全面収益(損失)は、短期投資の未実現収益と損失を含む一定期間内に非所有者由来の取引や他の事件や状況による権益変化と定義されている。総合収益(損失)は,列報の全期間の総合業務報告書と全面収益(損失)表に反映されている。
1株当たり純損失
1株あたりの基本純損失の計算方法は,純損失を当期発行済み普通株の加重平均で割ったものであり,普通株等価物は考慮しない。1株当たり純損失は普通株1株当たりの純損失とほぼ同じであり,全期間の純損失状況により,潜在的希薄化証券の影響は逆薄であるためである。
所得税
繰延税項資産及び負債は、資産及び負債の帳簿額とそのそれぞれの税額に基づく財務諸表の帳簿金額との差額と、差額を予想して返送した年度に発効した制定税率に適用された純営業損失及び貸出金繰り越しとの差額に基づいて決定される。繰延税金資産と負債の変動を所得税に計上する準備。当社は、その繰延税金資産が将来の課税収入から回収される可能性を評価し、既存の証拠の重みに基づいて、繰延税金資産のすべてまたは一部が現金化できない可能性があると考え、所得税費用を計上することで推定値を設定している。予想未来の課税オーバー額及び慎重かつ実行可能な税務計画策略を考慮して、繰延税金資産を回収する潜在力を評価する。
♪the the the会社は連結財務諸表で確認された所得税の不確実性を計算し、2ステップ法を用いて確認すべき税収割引額を決定した。まず納税状況は
151
評価する税務機関が外部審査を行った後にその税金を維持する可能性を確認するために。税務状況がより継続する可能性があると考えられる場合、税務状況は、連結財務諸表で確認された利益金額を決定するために評価される。確認可能な利益額は、最終和解時に50%以上の実現可能性がある最大額である。所得税の準備には、それによって生じる税収準備金または未確認の税収割引の影響が適切であると考えられ、関連する純利息および罰金が含まれる。
新興成長型会社の地位
当社は2012年のJumpStart Our Business Startups Act(“JOBS法案”)で定義されている新興成長型会社である。“雇用法案”によると、新興成長型企業は、これらの基準が民間企業に適用されるまで、“雇用法案”公布後に発表された新たなまたは改正された会計基準の採用を延期することができる。当社は、(I)新興成長型会社または(Ii)が“雇用法案”に規定されている延長移行期間から脱退することを明確かつ撤回できなくなるまで、この延長移行期間を使用することを選択した。したがって、これらの連結財務諸表は、上場企業の発効日までに新たなまたは改訂された会計声明を遵守している会社と比較できない可能性がある。
最近採用された会計公告
FASBは2016年6月、2016-13年度会計基準更新(ASU)を発表した金融商品信用損失の計量ASC 326が確立されました金融商品--信用損失それは.このASU及びその後の修正案は、金融機関及び他の組織が保有する融資及び他の金融商品の信用損失をタイムリーに記録し、財務報告を改善することを要求する。ASU 2016-13は、報告日に保有される金融資産のすべての予期される信用損失を、歴史的経験、現在の状況、および合理的かつサポート可能な予測に基づいて測定することを必要とする。当社は2023年1月1日にASU 2016-13を採用しています。この基準を採用することは会社の総合財務諸表や開示に影響を与えない。
最近発表された会計公告
2020年3月にFASBはASU 2020-04を発表しました参考為替レート改革(テーマ848):参考為替レート改革の促進が財務報告に及ぼす影響2021年1月にASU 2021-01を発表しました参考為替レート改革:範囲それは.これらの更新は、米国公認会計原則を契約、契約保証関係、および他の参考為替レート改革の影響を受ける取引に適用するために、いくつかの基準が満たされれば、オプションの便宜的な措置と例外を提供する。オプションのガイドラインを提供することは、為替レート改革を参考にした潜在的な会計負担を軽減するためだ。この指導意見は有効であり、遅くとも2022年12月31日に採択されることができる。FASBは2022年12月、為替レート改革(テーマ848)を参考に、テーマ848の日没日を延期し、テーマ848での臨時会計規則を2022年12月31日から2024年12月31日に延長するASU 2022-06を発表した。会社は現在、新たな会計基準の採用が会社の財務状況や経営結果に影響を与える可能性があることを評価しているが、会社の総合財務諸表や開示に実質的な影響を与えないと予想される。
2023年11月、FASBはASU 2023-07を発表した分部報告書(主題280):報告可能な分部開示の改善この基準は、主に重大な部門費用に関する開示を強化することによって、公共実体に関する報告可能な部門の開示を改善する。ASU 2023−07は,2023年12月15日以降の財政年度と2024年12月15日以降の財政年度内の移行期間で有効であり,早期採用を許可している。会社は現在、新たな会計基準の採用が会社の財務状況や経営結果に影響を与える可能性があることを評価しているが、会社の総合財務諸表や開示に実質的な影響を与えないと予想される。
FASBは2023年12月にASU 2023-09を発表しました所得税(特集740):所得税開示の改善. ASU 2023-09は、報告エンティティの有効税率入金に関する分類情報および所得税が支払われた情報を提供することを要求する。ASU 2023-09は公共エンティティに有効で、毎年
152
周期.周期2024年12月15日以降から、早期養子縁組が許可される。同社は現在、この指導が連結財務諸表や開示に及ぼす影響を評価している。
付記3.ある貸借対照表の構成要素
前払い費用と他の流動資産
前払い料金および他の流動資産には、以下のものが含まれている(千計)
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
契約研究サービス |
|
$ |
|
|
$ |
|
||
前払い保険 |
|
|
|
|
|
|
||
賃料を前払いする |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
前払い費用とその他の流動資産総額 |
|
$ |
|
|
$ |
|
||
財産と設備、純額
財産と設備、純額は以下の部分からなる(千計)
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
実験室装置 |
|
$ |
|
|
$ |
|
||
賃借権改善 |
|
|
|
|
|
|
||
コンピュータ装置及びソフトウェア |
|
|
|
|
|
|
||
家具と固定装置 |
|
|
|
|
|
|
||
総資産と設備 |
|
|
|
|
|
|
||
減算:減価償却累計と償却 |
|
|
( |
) |
|
|
( |
) |
財産と設備の合計 |
|
$ |
|
|
$ |
|
||
財産や設備に関する減価償却と償却費用は$
費用とその他の負債を計算すべきである
計算すべき費用とその他の負債は以下のように構成される(千計)
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
契約研究サービス |
|
$ |
|
|
$ |
|
||
給与明細及び関連費用 |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
計算しなければならない費用とその他の負債総額 |
|
$ |
|
|
$ |
|
||
153
注4.金融商品
次の表は証券を売ることができる分担コストと公正価値をまとめた(単位:千):
|
|
成熟性 |
|
償却する |
|
|
実現していない |
|
|
実現していない |
|
|
公正価値 |
|
||||
2023年12月31日: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|||
アメリカ政府機関証券と国債 |
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
||||
合計する |
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
2022年12月31日: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|||
アメリカ政府機関証券と国債 |
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
||||
合計する |
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
当社はそのすべての短期投資を販売可能に分類しており、このような証券は満期前に売却して管理戦略を実施する必要がある可能性があるため、このような証券は公正価値に基づいて入金される。2023年12月31日まで、2022年12月31日までに保有する売却可能債務証券の残り満期日はいずれも1年を超えていない。証券売却可能な未実現損益は全面損失に計上されている。2023年12月31日までに
当社は、各報告期間終了時にその投資保有量を審査し、期待される信用損失モデルを用いて未実現損失を評価し、未実現損失が信用損失によるものか他の要因によるものかを決定する。当社も、当社が満期前に関連証券を売却しようとしていることや、当社がその償却基準を回収する前にその証券を売却することを要求される可能性が高いかどうかなど、様々な要因を用いて投資保有量の減値を評価しています。2023年12月31日まで及び2022年12月31日まで年度その会社はできました
付記5.公正価値計量
公正価値は、計量日の市場参加者間の秩序ある取引において、資産または負債が元金または最も有利な市場で負債を移動させるために課金または支払いされる交換価格(退出価格)として定義される。適用される会計基準は公正価値を計量する際に使用する投入に既定の階層構造を提供し、使用可能な時に最も観察可能な投入を使用することを要求することによって、観察可能な投入を最大限に利用し、使用が観察できない投入を最大限に減少させる。観察される投入は,市場参加者が資産や負債を評価する際に使用する投入であり,当社以外のソースから得られた市場データに基づいて策定されている。観察できない投入は、市場参加者が資産や負債を評価するために使用される要素に対する会社の仮定を反映した投入である。公正な価値を測定するために3つのレベルの投資があります
154
同社はその通貨市場基金と米国国債を公正価値レベルの1級資産に分類しており、これらの基金と米国債の推定値は活発な市場の見積もりに基づいており、推定調整を行わない。
以下の表は、同社が公正価値で恒常的に計測している金融資産と負債の推定レベル(千単位)をまとめたものである
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
|||
2023年12月31日: |
|
|
|
|
|
|
|
|
|
|||
資産: |
|
|
|
|
|
|
|
|
|
|||
貨幣市場基金 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
アメリカ政府機関証券と国債 |
|
|
|
|
|
— |
|
|
|
— |
|
|
合計する |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
|
|
|
|
|
|
|
|
|
|
|||
2022年12月31日: |
|
|
|
|
|
|
|
|
|
|||
資産: |
|
|
|
|
|
|
|
|
|
|||
通貨市場基金とアメリカ政府機関国債 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
アメリカ政府機関証券と国債 |
|
|
|
|
|
— |
|
|
|
— |
|
|
合計する |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
注6.戦略投資
アステラス
協議条項
2023年8月4日(“署名日”)、当社はAstellas US,LLC(“Astellas”)と一連の合意(総称して“Astellasプロトコル”)を締結しており、詳細は以下の通りである。
証券購入と登録権協定
証券購入協定(“証券購入協定”)に基づき,当社はAstellasへの私募方式での発行および売却合算に同意した
当社もAstellasと登録権協定(“登録権協定”)を締結しており、この合意によると、当社はAstellasの登録に同意していますこれは…。締め切りの翌日です。
戦略権利書簡協定
署名日には、当社もAstellasと戦略権利書簡協定(“Astellas Strategic Rights Letter”)を締結し、これにより、会社は、(I)Astellasの自社取締役会、当社取締役会のいずれの委員会および当社科学顧問委員会のオブザーバー席を付与することに同意し、(Ii)18ヶ月の間、Astellasの制御権変更に関する通知権を付与する(Astellas Strategic Rights Letter参照)。(3)締め切りから締め切り12ヶ月周年日までの期間(“排他期”)は,(1)募集,故意の奨励,交渉,あるいは他の方法で締結してはならない善意の任意の第三者との計画取引(以下の定義)、(2)会社がP-MUC 1 C-ALLO 1に関連する任意の機密情報へのアクセスを提供し、P-MUC 1 C-ALLO 1は、複数の固形腫瘍適応のための会社の完全同種異体CAR-T製品(以下、“計画”と略す)であり、それは、知っている場合に計画取引を促進することを目的としており、または(3)計画取引について任意の意向書、契約または他の約束を締結することを目的としている(“計画取引”は、独占的または共同独占的許可または共同販売促進または共同マーケティング手配または商業販売権利の付与である)。世界中で使用するために、本計画の1つまたは複数の製品を普及またはマーケティングすること)。(Iv)向
155
Astellas(1)会社が受け取ったら善意の第三者が提出した統制権変更取引提案が、当該提案が会社取締役会に拒絶されない限り、又は(2)会社取締役会が承認した統制権変更手続を開始し、(3)会社が受領した場合善意の第三者による計画取引の提案は、その提案が会社取締役会によって拒否されない限り(“計画取引提案”)、または(4)排他期間終了後、計画取引開始について第三者と会社取締役会が承認した計画取引の流れについて実質的に検討する(“計画過程”)。(X)計画取引提案に関する通知については、Astellasは、計画取引提案よりも会社に有利な競争的提案を全体的に優先的に提供する権利があり、それにより、特定の時間内に可能な計画取引を独占的に交渉する権利があり、(Y)計画過程は、会社が任意の第三者と有意な実質的な議論を行う前に、Astellasは、指定された時間内に交渉計画取引を優先的に提出する権利を有するべきであり、いずれの場合も、Astellas戦略権利レターに規定された手続きおよび条件に適合しなければならない。
Astellas戦略権利書によってAstellasに付与された権利の一部の対価格として、Astellasは会社に#ドルを一度に支払った
Astellas戦略的権利レターは、次の時間の中で最も早く発生しなければならない:(I)閉鎖日の18ヶ月の記念日、(Ii)Astellasは以下の期間を有する
収入確認
証券購入プロトコルおよび登録権プロトコルは、ASC 606の範囲に属さず、株式発行入金として、発行済み株式の公正価値は、取引価格の範囲を超えているとみなされる。株式発行のさらなる詳細については、付記10を参照されたい。Astellas Strategic Rights Letterによって付与された権利は、プロトコルがクライアントとの契約を表すので、ASC 606に従って評価される。これらの権利はAstellasにいかなる支配権も譲渡しないと考えられているが、Astellasに将来の権利を与え、前金の全部または一部として$を与える
会社は発行日の見積もり公正価値に基づいて普通株発行金額を#ドルに計上しています
“会社”ができた
注7.連携とライセンス契約
羅氏
協議条項
はい2022年7月、会社はF.Hoffmann-La Roche LtdとHoffmann-La Roche Inc.(総称して“羅氏”と呼ぶ)と協力と許可協定(“羅氏協力協定”)を締結し、この協定に基づいて、会社は羅氏に:(I)ある会社の知的財産権の下での独占的なグローバル許可を授与した
156
物業既存の各P−BCMA−ALLO 1およびP−CD 19 CD 20−ALLO 1計画に基づいて、同種CAR−T細胞治療製品(各計画は“第1レベル計画”)の開発、製造および商業化を計画し、(Ii)既存の各P−BCMACD 19−ALLO 1およびP−CD 70−ALLO 1計画の同種CAR−T細胞治療製品(各計画は“第2レベル計画”)を開発、製造および商業化するために、ある企業の知的財産権に基づいて世界的に独占的に許可されている。(Iii)ある企業の知的財産権の下での独占ライセンスは、開発、製造および商業化から
Tier 1計画ごとに、会社は第一段階用量逓増臨床試験による開発活動を行い、羅氏は会社がこのような活動を実行する中で発生したいくつかの費用の特定の割合を返済する義務があり、各Tier 1計画は指定された補償上限に達することができる。第一段階の線量を超えて増加する第一級計画活動について、羅氏はこの計画によって発生したすべての費用を精算する義務がある。Tier 2計画ごとに,同社はINDを有効にするための研究開発候補を選択するか,あるいは羅氏がオプション維持費を選択して支払う場合には,第1段階用量逓増臨床試験を完了することで研究·開発活動を行う。また,羅氏が独占的に選択権を行使するTier 2計画ごとに,羅氏は選択権使用料を支払う義務がある.Tier 1計画とTier 2計画ごとに,社は羅氏への技術移転が完了するまで製造活動を実行する.
双方は初歩的な
ロ氏の協力協定に基づいて羅氏に与えられた権利の代償として、会社は前払#ドルを受け取った
♪the the the会社はまた,製品ごとにTier 1計画,オプションTier 2計画,連携計画の製品純売上高の中位から下位2桁および特許製品の下位から中央値の分級使用料支払いを行う権利があり,いずれの場合も何らかの条件によって制限されている
157
習慣.習慣減少と補償。特許使用料は、その製品がその国または地域でのライセンス特許が遅くとも満了するまで、製品および国/地域によって支払われる
ロ氏協力協定は、特許権使用料または他の支払い義務がなくなるまで、製品および国/地域ごとに有効化される。ロ氏協力協定には、重大な違約や破産に対する条項、羅氏の便宜のための標準的な終了条項が含まれている。これらのうちのいくつかの終了権利は、特定の製品またはライセンスおよびロ氏協力協定全体に対して行使することができる。
2023年11月7日から、羅氏協力協定は、(I)羅氏が私たちに支払ったいくつかの既存の製造関連費用を再分配して、私たちの既存の各第1段階計画のために新しい製造プロセスの開発および移転費用を追加するために再分配され、(Ii)羅氏が各第1級計画のために私たちに支払ったいくつかの開発マイルストーンの支払いの金額を再分配するように改正された。修正案は既存の
収入確認
契約開始時に会社は確定した
取引価格を決定するために、同社は羅氏協力協定中に受け取ったすべての支払いを評価した。いくつかのマイルストーンおよび追加料金は、可能な金額法に基づく初期取引価格に含まれない可変対価格とみなされる。各報告期間内に、不確定イベントが解決された場合、または他の状況が変化した場合、会社は取引価格を再評価する。同社は、羅氏協力協定開始時の取引価格は#ドルであることを確認した
ロ氏協力協定が発効した日から,Tier 1計画ライセンスに関する履行義務が履行されている。他のすべての履行義務は、提供される基本サービスが実際に発生した費用が推定総費用の割合に基づいて決定されるため、比例して確認される。当社は,コストに基づく入力法がこれらの履行義務を履行するモデルを最もリアルに記述していると考えている。達成される見積もりコストを修正するためのいかなる累積影響も
158
会社の履行義務は、変化と金額が合理的に見積もることができる期間に記録されます。この方法は会社に重大な判断を用いて、将来の支出を見積もることを要求する。会社の推定または判断が協力過程で変化した場合、それらは、会社が現在および将来の間に確認した収入の時間および金額に影響を与える可能性がある。
武田さん
2021年10月、当社は武田薬品米国社(“武田”)と協力及び許可協定(“武田協力協定”)を締結し、これにより、当社はPiggyBac、Cas-Clover、生分解性DNA及びRNAナノ粒子輸送技術及びその他の独自遺伝子工学プラットフォームを含む当社のいくつかのプラットフォーム技術の下でのグローバル独占許可を授与し、ある適応の遺伝子治療製品を研究、開発、製造及び商業化した。武田協力協定によると、会社は前払金#ドルを受け取った
当社は2023年5月31日、2023年7月30日(“終了日”)に発効する武田社から武田協力協定の中止を選択した旨の書面通知を受けた。終了日まで、双方はそれぞれの協力合意の下での責任を履行したが、終了日には、当社は協力協定の下での独占的責任を終了した。なお、当社が武田に付与したライセンス及び武田は、提携協定により当社が研究活動を行うライセンスを付与して終了します。同社は$を確認した
収入を繰り越して勘定する
当社のAstellas戦略権利関数への加入は戦略投資であるが,余剰金額を確認するために収入として決定されているため,次の表にはこれらのプロトコルの残り繰延収入への参考が含まれている。1つの契約資産$
プロトコルに関する繰延収入期末残高残高の入金状況は以下の通り(千で計算)
|
|
羅氏協力協定 |
|
|
武田協力協定 |
|
|
アスタラス戦略権利手紙 |
|
|
合計する |
|
||||
2021年12月31日現在の残高 |
|
$ |
— |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
||
領収書の受け取り/発行金額 |
|
|
|
|
|
|
|
|
— |
|
|
|
|
|||
収入が確認された |
|
|
( |
) |
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
2022年12月31日現在の残高 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|||
領収書の受け取り/発行金額 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
収入が確認された |
|
|
( |
) |
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
2023年12月31日現在の残高 |
|
$ |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
|||
注8.カリフォルニア再生医学研究所賞
同社はカリフォルニア再生医学研究所(“CIRM”)から資金を得ており,ある内部計画の開発に用いられている。助成条項によると、CIRMと同社は共に特定のプロジェクトを援助しており、これらのプロジェクトによると、資金は一部を奨励すると決定された発展マイルストーンの中で提供される。同社は関連プロジェクトの将来の潜在収入をCIRMと共有する義務がある。CIRMからの将来の収入の割合は、受け取った報酬金額および収入が製品販売からかライセンス料によって生成されるかに依存する。会社がCIRMに支払うことを要求される可能性がある最高収入共有金額は
159
至れり尽くせり収入を共有し、会社は任意の奨励をローンに転換する権利があり、会社は当日あるいは前にこのような選択権を行使しなければならない米国食品医薬品局(FDA)は、同社の上場許可申請を受けてから数営業日以内に同社に通知した。会社がどんな報酬を融資に転換する権利を行使すれば、それは義務があるだろうこのような選択をする平日。CIRM対応の返済金額はご褒美からローンに移行する時間によって異なります
2018年9月、同社は#ドルの奨励金を獲得した
注9.定期債務
2017年に、当社は融資及び担保協定を締結しました オックスフォード金融有限責任会社(“オックスフォード”)と締結され、その後改訂された(“改訂融資協定”))によると、会社の借入金総額は#ドルとなる
当社は米国会計基準470号特集“債務”に基づき、この修正を債務改正としているが、改正は実質的とは考えられないからである。
2022年ローン契約下の未返済残高は最初に変動年利で計算され、金利は
160
率のです
当社は“2022年ローン協議”の項目の未返済債務を随時返済する権利があり、債務増加を$とする
当社は定期融資で得られた金を完全に運営資金需要に利用し,その一般業務運営に資金を提供することができる。当社が“2022年融資協定”に基づいて負う義務は、そのほとんどの既存と将来の資産(知的財産権を除く)の優先担保権益を担保としている。また、同社は2022年の融資協定の許可を得ない限り、その知的財産権資産を担保しないことにも同意した。“2022年ローン協定”によると、まだ返済されていない金があるが、当社は財産、業務合併或いは買収に関する契約、その他の慣用契約を含む複数の肯定及び制限的契約を遵守しなければならない。同社はまた、配当金を発表するか、#ドルを超える株式を他の分配または支払いすることを制限されている
付記10.株主権益
授権株
当社が2020年7月に初の公募を完了したことを受けて、当社はその会社の登録証明書を修正してライセンスを付与します
株式承認証
改訂された融資協定によると、当社は2017年にOxford(I)に株式承認証を発行して購入した
普通株を売る
2022年8月、当社は対を完成しました
当社は2023年8月4日にAstellasと証券購入協定を締結し、これにより、当社は私募でAstellasへの発行及び販売を合算することに同意しました
161
市場施設では
2021年8月に当社は制御持分発行を行っているSMCantor Fitzgerald&Co.(“Cantor”)と販売協定(“販売契約”)を締結し,時々“市価発売”計画により普通株を販売する予定であり,総発行価格は最高$に達する
普通株
未来発行の普通株を確保する
将来の発行のために予約された普通株式には以下が含まれる2023年12月31日:
発行済みおよび未償還株式オプション |
|
|
|
|
発行済みおよび未発行の限定株式単位 |
|
|
|
|
未来のためのオプションと奨励付与を許可する |
|
|
|
|
従業員の下で将来の発行を許可する |
|
|
|
|
合計する |
|
|
|
注11.株ベースの報酬
持分激励計画
2020年7月、会社取締役会と株主は“2020年株式インセンティブ計画”(略称“2020年計画”)を承認した。2020年計画によると、会社は会社の現従業員、高級管理者、取締役またはコンサルタントに株式オプション、株式付加価値権、制限株式、制限株式単位、その他の株式または現金に基づく奨励を付与することができる。合計する
2023年12月31日までに
162
2022年2月、会社取締役会は“2022年インセンティブ計画”(略称“インセンティブ計画”)を承認した。インセンティブ計画によると、会社は当社の以前従業員又は非従業員取締役でない個人に非法定株式オプション、株式付加権、制限性株式奨励、制限株式単位奨励、業績奨励及びその他の奨励を付与することができ、当社の就職への激励とすることができる。2022年2月に承認されたインセンティブ計画によると、最初に発行された普通株の最高数は
株式オプション
以下に当社の今年度までの株式オプション活動の概要を示す2023年12月31日:
|
|
株 |
|
|
重みをつける |
|
|
重みをつける |
|
|
骨材 |
|
||||
2023年1月1日の残高 |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
授与する |
|
|
|
|
|
|
|
|
|
|
|
|
||||
鍛えられた |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
没収/キャンセルされる |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
2023年12月31日の残高 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
帰属していると予想されているオプション |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
現在までに行使可能なオプションに帰属した |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2023年12月31日までおよび2022年12月31日まで年度授権権の加重平均授受日公正価値は$
2023年12月31日現在の株式オプションに関する未確認報酬コスト総額は$
付与されたオプションの公正価値は,付与された日にBlack-Scholesオプション定価モデルを用いて推定される.付与されないオプションに関連して発生したとみなされる株式ベースの補償費用の払込を没収する。同社が従業員、非従業員、取締役に付与するオプションの公正価値を決定するための仮定は以下の通りである
|
|
十二月三十一日までの年度 |
||
|
|
2023 |
|
2022 |
無リスク金利 |
|
|
||
予想変動率 |
|
|
||
所期期間(年) |
|
|
||
配当率 |
|
— |
|
— |
163
無リスク金利·無リスク金利は、付与時に有効な米国財務省のゼロ金利に基づいて発行され、期限は期待オプション期限に対応する。
予想変動率−予想変動率は、株式オプション付与の期待期間と等しい期間における上場バイオテクノロジー会社の平均変動率に基づいて推定される。比較可能な会社は、類似した規模、ライフサイクル、または専門分野の段階に基づいて選択される。
所期期限·予想期間は、株式ベースの報酬が突出すると予想される期間を表す。オプション付与の期待期間は、行使パターンおよび付与後の雇用行為の終了に関する十分な履歴データがない場合に使用される簡略化方法を用いて決定される。簡略化方法は、期待期間を株式報酬の帰属日と契約期間との間の中点とする。
配当を期待する·会社は、その普通株に配当金を支払ったこともなく、その普通株に配当金を支払う計画もない。そこで同社が使用している予想配当収益率は
限定株単位
以下に会社の今年度までの限定株式単位(“RSU”)活動の概要を示す2023年12月31日:
|
|
株 |
|
|
重みをつける |
|
||
2023年1月1日の残高 |
|
|
|
|
$ |
|
||
授与する |
|
|
|
|
|
|
||
既得 |
|
|
( |
) |
|
|
|
|
没収/キャンセルされる |
|
|
( |
) |
|
|
|
|
2023年12月31日の残高 |
|
|
|
|
$ |
|
||
RSU奨励は株式奨励であり、付与されると、保有者に会社普通株の株式を交付する。従業員と非従業員役員に授与されるRSUは最大で授与されます
2020年従業員株購入計画
2020年7月、会社の取締役会と株主は、会社が株式を初公開した日から発効する“2020年従業員株購入計画”を承認した。合計する
164
同社はブラック·スコアーズ定価モデルを用いて、発行日ごとにESPPによって発行された購入権の公正価値を推定している
|
|
十二月三十一日までの年度 |
||
|
|
2023 |
|
2022 |
無リスク金利 |
|
|
||
予想変動率 |
|
|
||
所期期間(年) |
|
|
||
配当率 |
|
— |
|
— |
同社は、株式オプション、RSU、ESPPに関する株式ベースの報酬支出総額を、添付の総合経営報告書と包括収益(損失)の以下の費用種別(千単位)に記録している
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
研究開発 |
|
$ |
|
|
$ |
|
||
一般と行政 |
|
|
|
|
|
|
||
株式に基づく報酬総支出 |
|
$ |
|
|
$ |
|
||
付記12.支払いの引受及び又は事項
賃貸借契約を経営する
2018年10月、当社は、カリフォルニア州サンディエゴにある施設を研究開発·行政活動に使用する賃貸契約を締結しました。レンタル期間は#年から始まります
2019年7月、当社はカリフォルニア州サンディエゴの工場について賃貸契約を締結し、この工場は改造を経て良好な製造実践基準に適合し、早期臨床試験の製造に用いられた。レンタル期間は#年から始まります
2023年12月31日現在、同社が所有する運営リースには
2021年10月、当社はカリフォルニア州サンディエゴにある施設について分譲契約を締結し、この施設は約
2023年12月31日及び2022年12月31日まで、当社は確認しました$
165
賃貸借契約。2023年12月31日までの経営リースの加重平均残存期間と加重平均割引率は
自分から2023年12月31日、賃貸負債満期日は以下の通り(単位:千):
十二月三十一日までの年度 |
|
|
|
|
2024 |
|
$ |
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 |
|
|
|
|
2028 |
|
|
|
|
その後… |
|
|
|
|
将来のレンタル支払総額 |
|
|
|
|
利子を推定する |
|
|
( |
) |
賃貸負債残高合計 |
|
|
|
|
賃貸負債の当期分を差し引く |
|
|
|
|
賃貸負債、当期分を差し引く |
|
$ |
|
|
賠償協定
通常の業務過程において、会社は、ある事項について、供給者、レンタル者、契約研究機関、業務パートナー、および他の当事者に、このような合意または第三者による知的財産権侵害クレームに違反することによる損失を含むが、これらに限定されない異なる範囲および条項の賠償を提供することができる。また、当社はすでにその取締役会メンバー及びいくつかの行政人員と補償協定を締結し、当社は取締役又は高級管理者としての身分或いはサービスとして発生する可能性のあるいくつかの責任について賠償を行わなければならないと規定している。これらの賠償協定によると、会社が将来支払う必要がある可能性のある最大潜在金額は多くの場合無制限である。同社はこのような賠償によりいかなる物質コストも発生しておらず、賠償要求があることも知られていない。
2017年4月にTeneoBio,Inc.(安進の子会社)と締結されたビジネスライセンス契約
当社は2017年4月にTeneoBio,Inc.(“TeneoBio”)と商業許可協定(“2017 TeneoBioプロトコル”)を締結し,これにより当社は世界的な独占使用権を獲得し,TeneoBioがヒト疾患の治療に提供するいくつかの重鎖配列を含むCAR分子を含む同種異体T細胞を使用·開発した。
2018年8月にTeneoBio,Inc.(安進の子会社)とビジネスライセンス契約を締結しました
2018年8月,当社はTeneoBioとビジネスライセンスプロトコル(“2018年TeneoBioプロトコル”)を締結し,このプロトコルにより,当社はTeneoBioのいくつかの標的を独占的に研究,開発および商業化し,TeneoBioのヒト重鎖抗体をCAR−T細胞療法に開発および使用した。
2019年10月にXyone Treateutics,Inc.(Genus Oncology,LLCの利益継承者)と許可合意に達した
2019年10月に,当社はXyoneとライセンス契約(“Xyoneプロトコル”)を締結し,これにより,当社はXyone制御のいくつかの特許項におけるグローバル独占許可およびXyone制御のいくつかのノウハウ下の非独占グローバルライセンスを取得し,MUC 1抗体を標的とした自動車細胞とその誘導体を含む医薬製品やXyoneライセンス製品,およびXyone制御のいくつかの特許およびノウハウの下での非独占グローバルライセンスを研究,開発および商業化し,ヒト疾患や疾患の治療,予防および緩和に伴う診断薬を研究,開発および商業化した。
166
法律や事項がある
正常な業務過程において、当社は第三者の当社に対するクレームに直面する可能性があります。当社は、現在、まだいかなる訴訟、訴訟の脅威、または断言または断言されていないクレームは未定であり、これらの訴訟またはクレームは個別または全体的に会社の経営業績または財務状況に重大な悪影響を及ぼす可能性があると信じている。
注13.所得税
営業税前損失の構成要素具体的には以下のとおりである(千単位)
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
アメリカ国内では |
|
$ |
( |
) |
|
$ |
( |
) |
所得税前純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
所得税支給以下の構成からなる(千単位):
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
現在: |
|
|
|
|
|
|
||
連邦制 |
|
$ |
|
|
$ |
|
||
状態.状態 |
|
|
|
|
|
|
||
総当期に準備する |
|
|
|
|
|
|
||
延期: |
|
|
|
|
|
|
||
連邦制 |
|
|
— |
|
|
|
— |
|
状態.状態 |
|
|
— |
|
|
|
— |
|
繰延準備金総額 |
|
|
— |
|
|
|
— |
|
支出総額 |
|
$ |
|
|
$ |
|
||
2022年から2017年の減税·雇用法案は、納税者に研究開発支出の資本化と償却を要求している
以下の差(千計)のため、所得税は、税前収入に適用される米国連邦法定所得税率を適用することによって決定される所得税金額とは異なる
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
連邦法定金利 |
|
$ |
( |
) |
|
$ |
( |
) |
以下の側面の税金影響を調整する |
|
|
|
|
|
|
||
税金控除 |
|
|
( |
) |
|
|
( |
) |
州税、純額 |
|
|
( |
) |
|
|
( |
) |
未確認税収割引 |
|
|
|
|
|
|
||
株に基づく報酬 |
|
|
|
|
|
|
||
その他、純額 |
|
|
|
|
|
|
||
評価免除額を変更する |
|
|
|
|
|
|
||
合計する |
|
$ |
|
|
$ |
|
||
167
繰延税金資産と負債の重要な構成要素は、以下の項目(千計)を含む
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
繰延税金資産: |
|
|
|
|
|
|
||
純営業損失 |
|
$ |
|
|
$ |
|
||
所得税の繰越免除 |
|
|
|
|
|
|
||
研究開発資本化 |
|
|
|
|
|
|
||
収入を繰り越す |
|
|
|
|
|
|
||
賃貸負債 |
|
|
|
|
|
|
||
償却する |
|
|
|
|
|
|
||
費用を計算する |
|
|
|
|
|
|
||
補助金収入 |
|
|
— |
|
|
|
|
|
その他、純額 |
|
|
|
|
|
|
||
繰延税金資産総額 |
|
|
|
|
|
|
||
繰延税金負債: |
|
|
|
|
|
|
||
使用権資産 |
|
|
( |
) |
|
|
( |
) |
減価償却 |
|
|
( |
) |
|
|
( |
) |
あさって無期限に生きている無形資産 |
|
|
( |
) |
|
|
( |
) |
繰延税金負債総額 |
|
|
( |
) |
|
|
( |
) |
推定免税額 |
|
|
( |
) |
|
|
( |
) |
繰延税金純負債 |
|
$ |
( |
) |
|
$ |
( |
) |
繰延税金資産の現金化は、当社が今後数年間、繰延税金資産に関連する関連司法管轄区で十分な収入を生み出す能力に依存する可能性がある。同社は、繰延税金負債の予定沖販売、将来の課税収入、税務計画戦略、および最近の財務業績を含む、利用可能なすべてのプラスおよび負の証拠を考慮している。推定免税額$
無期限無形資産に関連する繰延税金負債は、これらの繰延税金負債の償却が無期限とされているため、繰延税金資産の実現を支援する収入源と見なすことはできない。しかし、当社は同一管轄区域内に無限損失繰越期間を有する無期限資産を有しており、適切な性質を有しているため、無期限無形資産に関する繰延税金負債は、繰延税金資産の課税収入源を実現することをサポートしており、両者とも不確定な償却または満期期があるためである。
当社は2023年12月31日までに国家純営業損失を繰り越します$
2023年12月31日まで、同社は連邦孤児薬物相殺と研究開発控除及び州研究開発税控除を持っている$
また、国税法第382節の規定により、純営業損失と研究開発税収控除繰越の使用が年間制限されている。第382条により決定された将来の所有権変更は、繰越純営業損失の使用をさらに制限する可能性がある。評価準備の存在により、これらの税金属性に関する繰延税金資産の将来の変化は、当社の実際の税率に影響を与えない。
その会社はアメリカと各州の管轄区域で税金を払わなければならない。2023年12月31日現在、会社の2012年から現在までの納税年度は連邦と他州税務機関の審査を受ける
168
未使用の純営業損失と研究開発税収を繰り越して免除する。当社が税務属性の繰越を持っている場合、その属性が発生した納税年度は依然として国税局または国家税務機関の審査を経て今後一定期間調整することができます。同社は現在、米国国税局や州や地方税務機関の審査を受けていない。
自分から2023年12月31日、当社は未確認の税金割引があります$
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
年初残高 |
|
$ |
|
|
$ |
|
||
今年度はポストが増える |
|
|
|
|
|
|
||
前年度はポストが増えた |
|
|
|
|
|
( |
) |
|
年末残高 |
|
$ |
|
|
$ |
|
||
このような認められていない税金優遇は今後12ヶ月以内に変わらないと予想される。まとに命中する$
注14.従業員福祉計画
国税法第401条(K)によると、会社は固定納付退職貯蓄計画を持っている。この計画は、最低年齢とサービス要件に適合するすべての従業員をカバーし、参加者が年度を一部延期することを可能にしているそれは.その計画に対する会社の貢献は取締役会が適宜決定することができる。当社の今年度末までの総供出2023年12月31日と2022年12月31日$
付記15.1株当たり純損失
1株当たり基本純損失(収益)の計算方法は、当期普通株株主が占めるべき純損失(収益)を当期発行普通株の加重平均で割る。1株当たり純損失(収益)は、株式オプションの行使やESPPによって購入された株、および行使可能な発行済株式証など、普通株を発行可能な追加償却を反映している。
同社の潜在的希薄化証券は、ESPPから普通株、普通株オプション、普通株を購入する引受権証を含み、1株当たりの希釈純損失の計算から除外されており、1株当たりの純損失を減少させるからである。したがって、普通株株主が1株当たり基本純損失を占めるべきであることと希釈後の1株当たり純損失を計算するための発行済み普通株加重平均株式数は同じである。
計算期間中に普通株式株主が1株当たりの純損失を占めるべきである場合、会社は以下の潜在普通株を含まない。これらの潜在普通株は期末ごとの発行済み金額に基づいて記載されており、これらの株式を計上することは逆償却効果が生じるからである
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
未償還株式オプションとRSU |
|
|
|
|
|
|
||
普通株購入引受権証 |
|
|
|
|
|
|
||
ESPP株 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
項目16.表10-Kの概要。
ない。
169
展示品索引
展示品番号 |
|
説明する |
|
|
|
|
|
|
2.1^ |
|
登録者、エルメス合併第一子会社、エルメス合併第二子会社、有限責任会社、Vindo NanoBioTechnology,Inc.とChristopher Youngの株主代表間の合併再編協定及び計画は、2016年10月10日に改訂された(2020年6月19日に米国証券取引委員会に提出された登録者登録説明書S-1表(文書番号333-239321)添付ファイル2.1を参照して編入)。 |
|
|
|
3.1 |
|
改訂および再改訂された登録者登録証明書(2020年7月14日に米国証券取引委員会に提出された登録者が現在報告している8−Kテーブル(ファイル番号001−39376)の添付ファイル3.1を参照して編入)。 |
|
|
|
3.2 |
|
登録者規約(登録者が現在報告している8−K表の添付ファイル3.2(書類番号001−39376)を引用して改訂·再改訂し、2020年7月14日に米国証券取引委員会に提出する)。 |
|
|
|
4.1 |
|
登録者普通株式証明書フォーマット(S−1登録者登録説明書添付ファイル4.1(フレット番号333−239321を参照して、2020年6月19日に米国証券取引委員会に届出)。 |
|
|
|
4.2^ |
|
登録者及びその一部株主は、2020年6月24日に登録者及びその一部株主の間で改正及び再署名された投資家権利協定(改訂された登録者登録説明書S−1表添付ファイル4.2を参照して編入され、2020年7月6日に米国証券取引委員会の第333−239321号文書に提出される)。 |
|
|
|
4.3 |
|
オックスフォード金融有限責任会社に発行された引受権証表は、2017年7月25日(2020年6月19日に米国証券取引委員会に提出された登録者登録声明S-1表(書類番号333-239321)添付ファイル4.3を参照して編入されます)。 |
|
|
|
4.4 |
|
2018年8月13日にオックスフォード金融有限責任会社に発行された引受権証表(2020年6月19日に米国証券取引委員会に提出された登録者登録声明S-1表(文書番号333-239321)添付ファイル4.4を参照して編入)。 |
|
|
|
4.5 |
|
2019年2月11日にオックスフォード金融有限公司に発行された引受権証表(2020年6月19日に米国証券取引委員会に提出された登録者登録声明S-1表(書類番号333-239321)添付ファイル4.5を参照して編入)。 |
|
|
|
4.6 |
|
普通株説明(登録者年次報告表を引用することにより10−K(アーカイブ番号0)添付ファイル4.6に組み込む01-39376)、2021年3月11日に米国証券取引委員会に提出)。 |
|
|
|
4.7 |
|
登録者と安仕達米国有限責任会社が2023年8月4日に締結した登録権協定(添付ファイル10.2を参照して登録者に組み込まれ、2023年8月4日に米国証券取引委員会の現在の8−K表報告書(文書番号001−39376)に提出される)。 |
|
|
|
10.1+ |
|
賠償協定表は、登録者及びその役員と上級職員との間で記入される(参照登録者が2020年6月19日に米国証券取引委員会に提出された登録者登録説明書S−1表(書類番号333−239321)添付ファイル10.1に編入)。 |
|
|
|
10.2+ |
|
改訂されたポセイダ治療会社の2015年株式インセンティブ計画及びその下のオプション付与通知、オプション協定及び行使通知のフォーマット(登録者登録声明の添付ファイル10.2を参照して改訂S-1表(第333-239321号文書)に組み込まれ、この表は2020年7月6日に米国証券取引委員会に提出される)。 |
|
|
|
10.3+ |
|
ポセイダ治療会社2020年株式インセンティブ計画及びその下の株式オプション付与通知、オプション協定及び行使通知のフォーマット(添付ファイル10.3を参照して改訂された登録者登録説明書S-1表(第333-239321号文書)に組み込まれ、この表は2020年7月6日に米国証券取引委員会に提出される)。 |
|
|
|
10.4+ |
|
ポセイディダ治療会社2020年株式インセンティブ計画(添付ファイル10.2を参照して登録者に編入され、2021年11月9日に米国証券取引委員会の10-Q表四半期報告書(ファイル番号001-39376)に提出された)下の制限株式単位奨励通知および制限株式単位奨励協定の表による。 |
|
|
|
10.5+ |
|
ポセイダ治療会社2020年従業員株購入計画(2020年7月6日に米国証券取引委員会に提出された登録者登録声明の改訂S-1表(文書番号333-239321)の添付ファイル10.4)。 |
170
|
|
|
10.6+ |
|
Poseida Treateutics,Inc.制御計画および参加プロトコルの終了および変更の形態(2020年7月6日に米国証券取引委員会に提出された登録者登録声明に修正されたS-1表(文書番号333-239321)添付ファイル10.5を参照して組み込む)。 |
|
|
|
10.7+ |
|
非米国株式オプション付与通知、株式オプション協定、およびポセディア治療会社2020年株式インセンティブ計画(参照登録者が2023年3月9日に米国証券取引委員会に提出した10-K表年次報告書(文書番号001-39376)第10.7条に基づく行使通知。 |
|
|
|
10.8+ |
|
ポセイダ治療会社2020年株式インセンティブ計画下の非米国限定株式単位奨励通知および制限株式単位奨励協定の表(添付ファイル10.8を参照して登録者に組み込まれ、2023年3月9日に米国証券取引委員会に提出されたForm 10−K年間報告書(文書番号001−39376))。 |
|
|
|
10.9+^ |
|
登録者とEric Ostertagによって2023年2月1日に署名された“移行·諮問協定”(添付ファイル10.9を参照して登録者に編入された登録者は、2023年3月9日に米国証券取引委員会の10−Kフォーム年次報告書(文書番号001−39376)に提出される)。 |
|
|
|
10.10+^ |
|
登録者とマーク·グガンとの間で2024年1月1日に締結された第2次改正および再署名された幹部雇用協定(添付ファイル10.3を参照して登録者に組み込まれ、2024年1月4日に米国証券取引委員会の現在の8−K表報告書(第001−39376号文書)に提出される)。 |
|
|
|
10.11+ |
|
改訂および再署名された参加協定は、登録者およびMark Gergenによって2022年2月1日に署名される(添付ファイル10.2を参照して登録者が2022年2月1日に米国証券取引委員会に提出された8−Kフォームの現在の報告書(ファイル番号001−39376)に組み込まれる)。 |
|
|
|
10.12+^ |
|
登録者とJohanna Myletとの間の招待状は,2015年6月8日(2020年6月19日に米国証券取引委員会に提出された登録者登録声明S−1表(文書番号333−239321)添付ファイル10.10を参照して編入)である。 |
|
|
|
10.13+^ |
|
登録者とHarry Leonhardtとの間の招待状は,日付が2020年7月1日である(2023年3月9日参照により米国証券取引委員会の登録者10−K年次報告書(文書番号001−39376)の添付ファイル10.13への編入)。 |
|
|
|
10.14+^ |
|
登録者とブレント·ワーナー社との間に発行された招待状は,2022年1月6日(添付ファイル10.14を参照して登録者に編入された2023年3月9日に米国証券取引委員会に提出された10−K表年次報告(書類番号001−39376)に記載されている). |
|
|
|
10.15+^ |
|
登録者とKristin Yarema博士との間の役員採用協定は、2024年1月1日である(添付ファイル10.1を参照して登録者に組み込まれることにより、2024年1月4日に米国証券取引委員会の現在の8−K表報告書(文書番号001−39376)に提出される)。 |
|
|
|
10.16+ |
|
登録者およびKristin Yarema博士によって2024年1月1日に署名された改訂および再署名された参加協定(添付ファイル10.2を参照して登録者に組み込まれることにより、2024年1月4日に米国証券取引委員会の現在の8−K表報告書(文書番号001−39376)に提出される)。 |
|
|
|
10.17+ |
|
ポセイダ治療会社は、2023年11月9日に米国証券取引委員会に提出された非従業員役員報酬政策(登録者の10-Q四半期報告書(ファイル番号001-39376)を参照することによって)の添付ファイル10.1を改訂し、再実施した。 |
|
|
|
10.18+ |
|
ポセイダ治療会社は、2022年誘導計画(登録者登録声明S-8表(文書番号333-276948)添付ファイル99.4を参照して編入し、2024年2月8日に米国証券取引委員会に提出することにより、2022年2月8日に改訂·再起動した。 |
|
|
|
10.19+ |
|
ポセイディダ治療会社の2022年誘導計画によると、通知、株式オプション協定、行権通知、制限株式単位付与通知および奨励協定を付与する表(2022年2月18日に米国証券取引委員会に提出された登録者登録声明の99.5号文書S−8(文書番号333−262869)を参照して編入される)。 |
|
|
|
10.20* |
|
登録者がTeneoBio,Inc.と締結した商業許可協定は、2017年4月27日から発効する(2020年6月19日に米国証券取引委員会に提出された登録者登録説明書S−1表(文書番号333−239321)添付ファイル10.13を参照して編入)。 |
|
|
|
10.21* |
|
登録者が天諾生物株式会社と締結した商業許可協定は、2018年8月3日から施行される(2020年6月19日に米国証券取引委員会に提出された登録者登録説明書S−1号書類添付ファイル10.14がこの協定に組み込まれることを参照)。 |
|
|
|
171
10.22* |
|
登録者とGenus Oncology,LLCとの間のライセンス契約は、2019年10月24日に発効する(2020年6月19日に米国証券取引委員会に提出された登録者登録説明書S−1表(文書番号333−239321)の添付ファイル10.16を参照して編入)。 |
|
|
|
10.23^ |
|
融資および保証協定は、登録者、Vindo NanoBioTechnology、LLCおよびオックスフォード金融有限責任会社によって署名され、日付は2022年2月22日である(2022年3月10日に米国証券取引委員会に提出された登録者10−K年度報告書の添付ファイル10.23(文書番号001−39376)を参照して組み込まれる)。 |
|
|
|
10.24^ |
|
“融資·担保協定の同意及び第1改正案”は、登録者と登録者との間で署名される。Vindo NanoBioTechnology,LLCおよびOxford Finance LLCは,2023年7月19日(登録者を参照して2023年8月8日に米国証券取引委員会に提出された10−Q表四半期報告(ファイル番号001−39376)の添付ファイル10.1によって編入された)。 |
|
|
|
10.25 |
|
登録者とBMR 9360-9390都市中心有限責任会社との間で締結された賃貸契約は、2018年10月1日であり、2019年10月4日から2020年3月11日まで改訂される(2020年6月19日に米国証券取引委員会に提出された登録者S-1表登録声明(書類番号333-239321)添付ファイル10.18を参照して編入)。 |
|
|
|
10.26 |
|
登録者とビマーウェイ·イーストガイットショッピングセンター株式会社との間で2019年7月12日に締結された賃貸契約(2020年6月19日に米国証券取引委員会に提出された登録者登録説明書S-1表(書類番号333-239321)の添付ファイル10.19)。 |
|
|
|
10.27 |
|
被支配持分発行SM登録者とCantor Fitzgerald&Co.との間の販売合意は、2021年8月13日(2021年8月13日に米国証券取引委員会に提出された登録者登録声明S-3表(文書番号333-258804)添付ファイル1.2を参照して編入される)。 |
|
|
|
10.28* |
|
登録者とHelmholtz-Zentum München-Deutsches Forschungszentum für Gesundheit and UmWelt GmbHとの間で改訂および再署名された許可協定は、2021年3月12日に署名される(合併時参照登録者は、2021年5月11日に米国証券取引委員会の10-Q表四半期報告添付ファイル10.1(文書番号001-39376)に提出される。 |
|
|
|
10.29* |
|
登録者、F.Hoffmann-La Roche Ltd.とHoffmann-La Roche Inc.との間の協力および許可協定は、2022年7月30日(登録者を参照することによって2022年11月10日に米国証券取引委員会に提出された10-Q表四半期報告(文書番号001−393376)添付ファイル10.1に組み込まれる)。 |
|
|
|
10.30^* |
|
登録者F.Hoffmann-La Roche LtdとHoffmann-La Roche Inc.が2023年11月7日に署名した協力および許可協定第1修正案。 |
|
|
|
10.31 |
|
証券購入協定は、登録者と安仕達米国有限責任会社によって2023年8月4日に署名される(添付ファイル10.1を参照して登録者に組み込まれる2023年8月4日に米国証券取引委員会に提出された現在の8−K表報告(文書番号001−39376)。 |
|
|
|
10.32^ |
|
登録者と安泰来米有限責任会社との間で2023年8月4日に締結された戦略権利書簡協定(登録者は2023年8月4日に米国証券取引委員会の8−K表に現在報告されている添付ファイル10.3(文書番号001−39376))に提出される。 |
|
|
|
23.1 |
|
独立公認会計士事務所安永法律事務所同意。 |
|
|
|
23.2 |
|
独立公認会計士事務所普華永道有限責任会社は同意した。 |
|
|
|
24.1 |
|
授権書(本文書の署名ページを参照)。 |
|
|
|
31.1 |
|
2002年のサバンズ·オキシリー法第302節で可決された1934年の証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて発行された首席執行幹事証明書。 |
|
|
|
31.2 |
|
2002年サバンズ−オキシリー法第302節で可決された1934年証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて首席財務幹事が認証された。 |
|
|
|
32.1 |
|
2002年にサバンズ·オクスリ法案第906条で可決された“米国法典”第18編1350条に規定されている最高経営責任者証明書。 |
|
|
|
32.2 |
|
2002年の“サバンズ-オックススリー法案”906節で採択された“米国法典”第18編1350条による首席財務官の認証。 |
|
|
|
97.1 |
|
ポセイダ治療会社です奨励的補償政策です |
|
|
|
172
101.INS |
|
XBRLタグがイントラネットXBRL文書に埋め込まれているので、連結されたXBRLインスタンス文書−インスタンス文書は、対話データファイルには表示されない |
|
|
|
101.書院 |
|
Linkbase文書を組み込んだインラインXBRL分類拡張アーキテクチャ |
|
|
|
104 |
|
表紙インタラクションデータファイル(イントラネットXBRL文書に埋め込む) |
^ S−K条例第601(A)(5)項によれば、一部の展示品及び付表は省略されている。登録者は、米国証券取引委員会の要求に応じて、任意の漏れた展示品又はスケジュールのコピーを補充提供することを約束する。
+ 契約または補償計画を管理すること。
* S-K法規第601(B)(10)項によれば、本展示品のいくつかの部分は、登録者がこの情報が実質的ではないと判断したので、星番号で表記された方法で省略されており、開示されている場合、登録者に競争損害を与える可能性がある。
173
標札すきま
改正された1934年“証券取引法”第13又は15(D)節の要求に基づいて、登録者は、カリフォルニア州サンディエゴの次の署名者が代表して本報告に署名することを正式に手配した.
|
|
POSEIDA治療会社 |
|
|
|
|
|
日付:2024年3月7日 |
|
差出人: |
寄稿S/クリスチャン·アレマ |
|
|
|
クリスチャン·アレマ博士 |
|
|
|
最高経営責任者 |
以下の署名の各人は、Kristin Yarema,Ph.D.及びJohanna Mylet,並びに彼らの各々の真及び合法的な事実代理人及び代理人を構成して任命し、十分な代替及び再代理の権限を有し、任意及び全ての身分で、その人の氏名、場所及び代理として、本報告の任意及び全ての修正案(発効後の修正案を含む)に署名し、そのすべての証拠物及びその他の関連文書とともに証券取引委員会に提出し、上記事実代理人及び代理人を付与する。一方、彼らの各々は、これに関連するすべての必要かつ必要なことを行い、実行するための完全な権力および権限を有しており、その人が可能であるか、または自ら行うことができるすべての意図および目的に完全に沿って、本明細書で上述したすべての事実代理人および代理人、または彼らのいずれか、または彼らの代替者または代替者を承認し、確認することは、本条例によることを合法的にまたは手配することができる。
本報告書は、改正された1934年の証券取引法の要求に基づき、以下の登録者によって登録者として指定日に署名された。
名前.名前 |
|
タイトル |
|
日取り |
|
|
|
|
|
寄稿S/クリスチャン·アレマ |
|
取締役最高経営責任者総裁 (首席行政主任) |
|
2024年3月7日 |
クリスチャン·アレマ博士 |
|
|
|
|
|
|
|
|
|
/S/ジョンナ·M·マイレット |
|
首席財務官 (首席財務会計官) |
|
2024年3月7日 |
ヨナ·M·マレットC.P.A. |
|
|
|
|
|
|
|
|
|
/S/マーク·J·グガン |
|
取締役執行主席兼最高経営責任者 |
|
2024年3月7日 |
マーク·J·グガンJ.D |
|
|
|
|
|
|
|
|
|
/S/ラファエロ·アルマード |
|
役員.取締役 |
|
2024年3月7日 |
ラファエル·アマド医学博士 |
|
|
|
|
|
|
|
|
|
/S/チャールズ·M·ボーム |
|
役員.取締役 |
|
2024年3月7日 |
チャールズ·M·ボーム医学博士 |
|
|
|
|
|
|
|
|
|
/S/シンシア·コリンズ |
|
役員.取締役 |
|
2024年3月7日 |
シンシア·コリンズ商工管理修士 |
|
|
|
|
|
|
|
|
|
寄稿S/ルーク·康寧 |
|
役員.取締役 |
|
2024年3月7日 |
ルーク·康寧 |
|
|
|
|
|
|
|
|
|
/S/Marcea B.Lloyd |
|
役員.取締役 |
|
2024年3月7日 |
Marcea B.Lloyd、J.D |
|
|
|
|
|
|
|
|
|
/S/ジョン·P·シュミット |
|
役員.取締役 |
|
2024年3月7日 |
ジョン·P·シュミット商工管理修士 |
|
|
|
|
174