オクフィル製薬会社
表格10-K
表格10-K
カタログ
|
第1部
|
7 | ||
|
第1項。
|
商売人
|
7 | |
|
第1 A項。
|
リスク要因
|
44 | |
|
項目1 B。
|
未解決従業員意見
|
83 | |
|
プロジェクト1 C。
|
ネットワーク·セキュリティ
|
83 | |
|
第二項です。
|
特性
|
84
|
|
|
第三項です。
|
法律手続き
|
84 | |
|
第四項です。
|
炭鉱安全情報開示
|
84 | |
|
|
|||
|
第II部
|
|||
|
五番目です。
|
登録者普通株市場、関連株主事項、発行者による株式証券の購入
|
85 | |
|
第六項です。
|
[保留されている]
|
85 | |
|
第七項。
|
経営陣の財務状況と経営成果の検討と分析
|
85 | |
|
第七A項。
|
市場リスクの定量的·定性的開示について
|
99
|
|
|
第八項です。
|
財務諸表と補足データ
|
100 | |
|
第九項です。
|
会計·財務開示面の変化と会計士との相違
|
100 | |
|
第9条。
|
制御とプログラム
|
100 | |
|
プロジェクト9 B。
|
その他の情報
|
101 | |
|
プロジェクト9 Cです。
|
検査妨害に関する外国司法管区の開示
|
101 | |
|
第三部
|
|||
|
第10項。
|
役員、行政、会社の管理
|
102 | |
|
第十一項。
|
役員報酬
|
102 | |
|
第十二項。
|
ある実益所有者の担保所有権及び経営陣及び株主に関する事項
|
102
|
|
|
十三項。
|
特定の関係や関連取引、取締役の独立性
|
102 | |
|
14項です。
|
チーフ会計士費用とサービス
|
102 | |
|
|
|||
|
第4部
|
|||
|
|
|||
|
第十五項。
|
展示品と財務諸表の付表
|
103 | |
|
第十六項。
|
表格10-Kの概要
|
108 | |
|
サイン
|
|||
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
本Form 10-K年次報告では、他に説明がない限り、言及されている“私たち”、“私たち”、“Ocuphire”または“会社”とは、Ocuphire Pharma,Inc.を意味する。私たちの財務諸表は、米国公認会計原則(“米国公認会計原則”)に基づいて作成されている。
前向きに陳述する
この10-K表年次報告書には、1933年証券法(“証券法”)第27 A節(“証券法”)、
及び改正された1934年“証券取引法”(“取引法”)第21 E節の安全港条項に適合する前向きな陳述が含まれている。これらの展望性表現は私たち、私たちの業務の将来性と私たちの経営結果と関係があり、
の多くの要素と事件構成のあるリスクと不確定要素の影響を受け、これらの要素と事件は私たちの実際の業務、経営の見通しと経営結果はこのような展望性表現の予想と大きく異なるかもしれない。このような差異をもたらすか、または促進する可能性のある要因は、本10−K表の年次報告書に“リスク要因”のタイトルで記載されている要因を含むが、これらに限定されない。読者に、これらの前向き陳述に過度に依存しないように注意することができ、これらの陳述は、本報告の発表日からのみ発表される。
場合によっては、“予想”、“信じ”、“継続”、“可能”、“推定”、“予想”、“予定”、“可能”、“進行中”、“計画”、“潜在”、“予測”、“項目”、“すべきである”という言葉によって、前向き陳述を識別することができる。すべての前向き表現がこれらの語を含むわけではないが、“将”、“将”または
といった用語または他の類似用語の否定。私たちはその後起こる可能性のある事件や状況を反映するためにどんな前向きな陳述も修正する義務はない。私たちは、本報告および私たちが米国証券取引委員会(“米国証券取引委員会”)に提出した他の報告書で行われた様々な開示を慎重に検討し、考慮することを読者に促し、これらの報告書は、関心のある当事者に、我々の業務に影響を与える可能性のあるリスクおよび要因の提案を提供する。
リスク要因をまとめる
私たちの業務は“第1 A項”に記載されているように、多くのリスクの影響を受ける。本年度報告書の“リスク要因”。他の要因以外にも、主な要因および不確実性要因は、以下の通りである
| • |
私たちは私たちの製品ラインと私たち(または私たちの現在または未来の戦略パートナー)の成功に大きく依存している。Ocuphireおよび/またはViatrisは、PSの臨床開発を決して完了せず、市場の承認を得ることができないか、またはPSを単独または小用量ピロカ品(LDP)、APX 3330、または私たちの任意の他の候補製品としての補助療法の商業化に成功させる可能性がある。さらに、私たち(または私たちの戦略パートナー)がAPX 3330やPSを十分に開発して商業化できなかった場合、私たちの業務は深刻な被害を受ける可能性があります。
|
| • |
以前の臨床試験結果は未来の結果を予測できない可能性があり、著者らの現在と計画中の臨床試験結果はFDA或いは非アメリカ監督管理機関の要求に符合しない可能性がある。
|
| • |
もし私たちが臨床試験の患者登録、私たちが臨床試験を行い、完成する能力、そして私たちが必要な監督管理の承認を求める能力に遅延或いは困難があれば、
は延期或いは阻止される可能性がある。
|
| • |
法規要求やFDAガイドラインの変化、あるいは著者らの臨床試験中に発生した意外な事件は、臨床試験方案の変化或いは追加の臨床試験要求を招く可能性があり、これは私たちのコスト増加或いはその開発スケジュールを延期させる可能性がある。
|
| • |
私たちまたは他の人たちは、競争相手の製品と比較して、私たちの候補製品が十分な治療効果または十分な効果を欠いているか、またはそれらが以前に発見されなかった不良副作用を招き、規制部門の承認または商業化を延期または阻止する可能性があることを発見するかもしれない。
|
| • |
私たちの候補製品がアメリカでマーケティング承認を得ても、私たちは決して規制部門の承認を得ず、アメリカ以外のところでこれらの候補製品をマーケティングすることができるかもしれません。
|
| • |
私たちは激しい競争と迅速な技術変革に直面しており、これは他の人が私たちよりも早く、あるいはより成功的に製品を発見、開発、商業化することを招くかもしれない。
|
3
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| • |
私たちは現在、APX 3330が承認された場合、販売およびマーケティング能力を確立するか、または第三者販売、マーケティング、およびAPX 3330を招聘することで困難に直面する可能性がある販売またはマーケティングインフラを持っていません。
|
| • |
私たちの将来の商業成功は私たちの候補製品が承認されれば、医者、患者、第三者支払人と医学界の他の人の中で著しい市場受容度を得ることにかかっている。
|
| • |
FDAまたは同様の外国規制機関が上場承認された私たちの候補製品の模造薬バージョンを承認した場合、またはこれらの機関が私たちの製品の模造薬を承認する前に候補製品の適切な独占特許期間を与えない場合、私たちの製品の販売は悪影響を受ける可能性がある。
|
| • |
私たちはどんな製品の販売からも何の収入も生じていません。予測可能な未来に損失が出ることを予想して、永遠に実現したり、利益を維持したりすることができません。
|
| • |
私たちの比較的短い経営歴史は、投資家が私たちのこれまでの業務を評価し、私たちの将来の生存能力を評価することを困難にするかもしれない。
|
| • |
私たちは未来に多くの追加資本が必要になるだろう。追加資本を得ることができない場合、または許容可能な条項で追加資本を得ることができない場合(金融サービス業の変化、私たちの財務業績、または他の理由でも)、私たちは運営を延期、減少、または停止しなければならない。
|
| • |
追加資本の調達は私たちの株主に希釈し、私たちの運営を制限したり、私たちの技術や候補製品に対する権利を放棄することを要求するかもしれません。
|
| • |
世界経済と社会の不安定または不利なグローバル経済状況は、私たちの収入、財務状況、または運営結果に悪影響を及ぼす可能性がある。
|
| • |
私たちの従業員や私たちの代表は、適用される規制基準や要求に違反することを含む不適切な行為や他の不適切な活動に従事する可能性があり、これは私たちの業務を深刻に損なう可能性があります。
|
| • |
私たちまたは私たちのサプライヤーやメーカーに対する製品責任訴訟は、私たちが重大な責任を負うことになり、私たちが開発する可能性のある任意の候補製品の商業化を制限することができます。
|
| • |
連邦立法や州や地方政府の行動は、外国の薬品を米国に再輸入することを許可する可能性があり、薬品が外国で低い価格で販売されている場合、これは私たちの経営業績に悪影響を及ぼす可能性がある。
|
| • |
私たちは第三者に依存して臨床前と臨床試験を行い、他の任務を実行してくれた。これらの第三者がその契約責任を成功的に履行できない場合、予想される期限までに完了するか、またはbrを遵守することができなければ、規制部門の候補製品の承認を得ることができないか、または商業化することができず、私たちの業務が損なわれる可能性がある。
|
| • |
私たちは第三者に完全に依存して原料薬を供給して製造し、私たちの候補製品の非臨床薬品供給と臨床薬品供給を調合と包装し、生産過程で薬品物質と製品の分析テストを行い、私たちは第三者に依存して私たちの現在と未来の候補製品の商業供給を生産してテストするつもりだ。
|
4
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| • |
我々は、候補製品の開発または販売について許可スケジュール(例えば、Viatrisライセンス合意(以下のように定義)を達成し、将来的にAPX 3330を含む他の戦略的同盟を形成または求めるか、または許可スケジュールを達成することが可能である。もし私たちが有利な条件でこれらの連合を設立したり維持したりできなければ、私たちの業務は損害を受ける可能性があり、私たちは開発、製造、商業化計画を変更しなければならないかもしれない。
|
| • |
もし私たちが買収、許可、または戦略的協力を行えば、これは私たちの資本要求を増加させ、私たちの株主を希釈し、私たちに債務を発生させたり、負債を抱えたり、私たちを他のリスクに直面させたりする可能性がある。
|
| • |
もし私たちが候補製品のために十分な特許保護を得ることができなければ、私たちの競争相手は私たちと似ているか、または同じ製品や技術を開発し、商業化するかもしれません。これは、私たちが開発する可能性のある任意の候補製品を商業化する能力、私たちの業務、運営結果、財務状況、および見通しに悪影響を及ぼすでしょう。
|
| • |
もし私たちが“ハッジ-ワックスマン法”や同様の外国立法に基づいていなければ、特許期間を延長し、私たちの候補製品のデータ独占権を得ることで保護されなければ、私たちの業務は実質的な損害を受ける可能性がある。
|
| • |
私たちは世界各地で私たちの知的財産権を保護したり実践することができないかもしれない。
|
| • |
私たちの特許保護の獲得と維持は、政府機関が提出した様々な手続き、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
|
| • |
私たちは私たちのキーパーソンに依存して、私たちが高い素質の人材を誘致し、維持することに成功できなければ、私たちのビジネス戦略を成功的に実施することができないかもしれない。
|
| • |
私たちと私たちの協力者にとって、国際運営に関連する様々なリスクが私たちの業務に悪影響を及ぼす可能性があります。
|
| • |
ネットワークイベント、ネットワークセキュリティホール、サービス中断、またはデータ破損を含むシステム障害または計画外イベントが発生した場合、私たちの業務および運営は影響を受けます。
|
| • |
私たちは現在、私たちの株式信用限度額の手配と関連した潜在的な発行を必要とする相当な数の株を持っている。私たちのELOCに基づいて株式を発行または売却することは、発行済み株の数を大幅に増加させ、私たちの証券保有者の株式希釈につながります。これは普通株の市場価格を大幅に下げるかもしれない。
|
| • |
私たちは予測可能な未来に何の現金配当金も支払わないと予想している。
|
| • |
ナスダック資本市場の持続的な上場基準を満たさなければ、私たちの普通株は銘柄を取られるかもしれません。もし退市すれば、私たちの普通株と私たちの普通株の流動性はbrの影響を受けるだろう。
|
| • |
私たちの普通株の市場価格は大きく変動するかもしれない。
|
| • |
私たちは証券訴訟の影響を受けるかもしれませんが、これは高価で、経営陣の注意をそらすかもしれません。
|
5
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
業界と市場データ
本年度報告では,医療機器や保健業界に関する情報,統計データ,推定を引用した。私たちは業界と一般出版物、市場研究会社の報告、その他のソースを含む様々な第三者ソースからこの情報を取得した。この情報は多くの仮定と制限を含み,我々はこの
情報の正確性や完全性を独立に検証していない.いくつかのデータおよび他の情報もまた、我々の研究、内部調査の検討、業界内で議論されている一般的な情報、および第三者ソースからの管理層の善意の推定に基づいている。私たちはこのような外部情報源と推定が信頼できると信じているが、独立して確認されていない。“第1 A項”に記載されている要因を含む様々な要因により、我々が経営している業界は高度な不確実性、変化、リスクの影響を受けている。リスク要因ですこれらの要因や他の要因は,本年度報告や他の出版物で表現された結果とは大きく異なる結果を招く可能性がある。
6
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
第1部
| 第1項。 |
商売人
|
概要
Ocuphire製薬会社は臨床段階の生物製薬会社であり、新しい治療法の開発に専念し、網膜と屈光性眼症患者の満足されていない需要を治療するために使用されている。
同社の主要候補網膜製品APX 3330はRef−1(還元酸化効果因子−1蛋白)の小分子阻害物である。REF-1はHIF-1αとNF-κBなどの転写因子の調節因子であり、REF-1を抑制することは血管内皮増殖因子と炎症性サイトカインのレベルを下げることができ、これらのサイトカインは眼血管新生と炎症に重要な役割を果たす。APX 3330は1種の経口錠剤であり、毎日1回或いは2回服用し、糖尿病網膜症(DR)の治療に用いられる。DR或いは糖尿病黄斑水腫被験者に対する第二段階の研究はすでに完成し、結果は2023年1月に公表された。2023年10月,同社は米国食品医薬品局(FDA)と第2段階終了(EPOP 2)会議を行い,将来の臨床試験へのAPX 3330の登録終点を支援することで合意した。同社は2024年2月にFDAに特別案評価(“SPA”)を提出し、臨床試験案と統計分析計画について合意し、FDAとSPA合意を達成する際に研究設計パラメータと期待時間の詳細を共有することを求めた。
DRは約1000万人の糖尿病患者に影響を与えており,2050年には1400万人以上の米国人に影響すると予想される。糖尿病網膜症は非増殖性糖尿病網膜症(NPDR)或いは増殖性糖尿病網膜症(“PDR”)に分けられ、前者は早期糖尿病であり、病状は軽微或いは存在せず、後者は糖尿病眼病の比較的に末期であり、高度な失明症状が出現することができる。約800万のDR患者がNPDRを有しており,治療を行わなければPDRに進展する可能性がある。この疾患は視力喪失を招くリスクがあるにもかかわらず、90%を超えるNPDR患者は現在、眼看護専門家の観察以外に、彼らが視力を脅かす合併症が出現するまで、いかなる治療コースも受けていない。これは様々な要因によるものであり,現在承認されているこの疾患を含めた治療法には頻繁な眼部注射による治療負担が必要である。APX 3330は経口錠剤として、アメリカの約800万NPDR患者の早期非侵襲性治療になる潜在力がある。
同社はまたAPX 2009とAPX 2014の許可を得ており、この2つの製品はAPX 3330の第2世代類似製品である。このRef-1阻害剤ファミリーの独特な作用機序は血管新生と炎症を減少させることができ、これは老年性黄斑変性、地理萎縮と非眼科疾患などの他の網膜疾患の治療に役立つかもしれない。
2022年11月、会社はFamyGen Life Science,Inc.(“Famy”)(2023年1月にViatris,
Inc.(“Viatris”)に買収され、現在Viatrisと改名した)は許可と協力協定(“Viatris許可協定”)を締結し、これにより、会社はViatrisの開発、製造、輸入、輸出、その屈折製品候補フェントラミン眼科溶液とそれを商業化する独占許可を授与した。PSは毎日1回のメタンスルホン酸フェントラミン点眼液の調合であり、瞳孔直径の縮小と視力向上を目的としている。2023年9月、PSは食品医薬品局によって薬物による散瞳の治療のために許可され、商標はRYZUMVI
である。PSは現在老眼(年齢に関連する近視力不明瞭)を治療する第三段階の臨床試験にある。Vega−2は老眼に対する第3段階研究が主な終点に達しており,我々の開発·ビジネスパートナーのViatrisは2024年上半期に第3段階の研究を継続する予定である。角膜屈折術後に暗い(中視または低)光線条件下で視力が低下した患者でも,PSは第3段階であった。2023年12月5日、同社は、角膜屈折手術後に暗い(中視または弱光)条件下で視力低下を治療するPSの第3段階試験であるLynx-2に関するFDAの合意を受け、我々の開発および商業パートナーViatrisは、2024年上半期にこの適応の第3段階開発を継続する予定である。
Viatrisライセンス契約によると、Ocuphireは、RYZUMVIの秘密協定承認を得るために、3500万ドルの前払い現金と1000万ドルのライセンス契約マイルストーン支払いを受け取りました。また、Ocuphireは、ある指定された規制や純売上のマイルストーンに達した後、合計1.2億ドルにのぼる潜在的な追加支払いを受ける資格がある。Ocuphireはまた、PSの米国と米国以外の年間純売上高に基づく階層印税(印税の範囲は米国の年間純売上高の2桁低い数百点から米国以外の年間純売上高パーセント
)を獲得する。Viatrisライセンス契約は合意条項によって終了するまで満期になりません。
7
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
企業の歴史
2018年2月、Ocuphire Pharma,Inc.(Rexahnと合併する前に、“Private Ocuphire”)が成立し、その後、2018年4月にOcularis Pharma,LLC(メタンスルホン酸フェントラミン眼科ソリューションの原始革新者)と合併し、2020年1月にApexian PharmPharmticals,Inc.からAPX 3330を含むRef-1阻害剤計画のいくつかの権利を獲得した(“Apexian再許可プロトコル”参照)。
2020年11月、Private OcuphireはRexahn PharmPharmticals,Inc.(“Rexahn”)に対する逆合併(“合併”)を完了し、Rexahnは薬物開発業務を停止した上場企業であり、同時に普通株と引受権証の発行により2000万ドルを調達し、普通株を購入するために使用された。合併については、RexahnはOcuphire Pharma,Inc.と改名し、その後上場会社として、Raxahnは以前Private Ocuphireが経営していた業務を経営した。
Ocuphireは2022年11月にViatrisライセンス契約を締結し、PS製品をViatrisに許可しました。
Ocuphireの多くの従業員、取締役、コンサルタント、コンサルタントはLumifyのような承認された製品を含むPSと他の眼科薬の開発に参加している® ボストン社のマーケティングZirganによって®ボストン社とデュレイゾ社がマーケティングしています®ノワール社のマーケティングUpneqによって®RVL PharmPharmticals(RVL製薬社)で販売されている®RocklatanのAlconマーケティングによって®Alcon、Vyzultaがマーケティングしています® ボストン社とシードラがマーケティングしています® ボストン株式会社(CEQUA)で販売® 太陽製薬工業株式会社が販売し、Thea Pharma Inc.が販売するIyuzehとDextzaで販売しています® この管理チームはGeorge Magrath最高経営責任者(“CEO”)がリードし、George Magrath、修士、医学博士、MBAが指導し、製薬会社の経営及び多くの治療分野で発見、開発、商業化治療において豊富な経験を持っている。
戦略.戦略
Ocuphireの目標は有力な生物製薬会社を構築し、患者の発見、開発、商業化、および/または一流療法の許可証を発行し、医師と支払人に魅力的な解決策を提供することである。Ocuphireがその目標を実現する戦略の鍵となる要素は以下のとおりである
| • |
私たちの製品の臨床開発を推進する。
|
APX 3330計画に対して,会社は2023年10月に米国食品·薬物管理局(FDA)と第2段階終了(EPOP 2)会議を行い,APX 3330の将来の臨床試験の登録終点への進出を支援することで合意した。同社は2024年2月にFDAにSPAを提出し,FDAとSPA合意に達した際に研究設計,コスト,スケジュールについてさらなる指導を提供する。
PSについては,Ocuphireは2022年11月にViatris許可協定に署名し,この合意によりViatrisはPSの開発と商業化の独占的権利を持つ.Viatris許可協定によると,OcuphireはViatrisと協力して米国で開発活動を継続し,このような予算開発活動をViatrisが精算する。PSは2023年9月にFDAにより薬物による散瞳の治療に許可され,ブランドはRYZUMVIであり,老眼と角膜屈折手術後に暗い(中間または低)光線条件下で視力低下の3期試験が進められている。Viatrisは米国以外の地域でPS開発と商業化努力を行う独占的な権利を持っている。
8
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| • |
知的財産権の組み合わせを維持し、拡大するそれは.Ocuphireは、Ref-1阻害剤計画の世界的に独占的な再許可、そのすべての眼科および糖尿病適応の候補製品APX 3330、ならびにAPX 2009およびAPX 2014を含むRef-1導管候補の組成物および使用方法を有する。
|
Ocuphireは米国と他の司法管轄区域でこの知的財産権保護を拡大し、拡大するより多くの機会を探索し続けている。Ocuphireは、その候補製品APX 3330、そのすべての眼科および糖尿病適応のためのRef-1阻害剤計画のグローバル独占的再許可、ならびにAPX 2009およびAPX 2014を含むRef-1導管候補の組成物および使用方法を有する。Ocuphireは米国と他の司法管轄区域でこの知的財産権保護を拡大し、拡大するより多くの機会を探索し続けている。
| • |
APX 3330とPSの価値を最大化します。Ocuphireは、1つまたは複数のパートナーを探し、米国国内外でAPX 3330を開発し、商業化する可能性がある。
|
現在FDAで承認されているRYZUMVIとPSは,角膜屈折手術後の暗い(中視または低)光線条件下での老眼と視力低下が承認された場合,Viatris Eye看護部門がViatris許可協定に基づいて米国と主要な非米国市場で商業化する。
| • |
内部許可と買収機会の評価それは.Ocuphireのチームは臨床段階の資産を識別と許可或いは買収する資格があり、そして絶えず
はそのルートの拡大と多様化の機会を評価している。
|
Ocuphireは、多様な適応のためにAPX 3330を開発し続けており、独自に開発および商業化しているか、またはAPX 3330の開発および商業パートナーを求めている可能性がある。Ocuphireは,そのパートナーであるViatrisと複数の適応PSの開発を継続している。Ocuphireは、この2つの計画は、(1)今まで有望な臨床データ、(2)2種類の小分子末期臨床候補薬物、(3)便利な投与経路とスケジュール、(4)第一線または補助治療の潜在力、および(5)巨大な商業潜在力を含む、類似した潜在的優位性を示すと考えている。以下の図1にOcuphireの現在の候補製品開発プロセスとその目標指標と期待マイルストーンをまとめる
図1:Ocuphireパイプ:候補製品と適応パイプライン
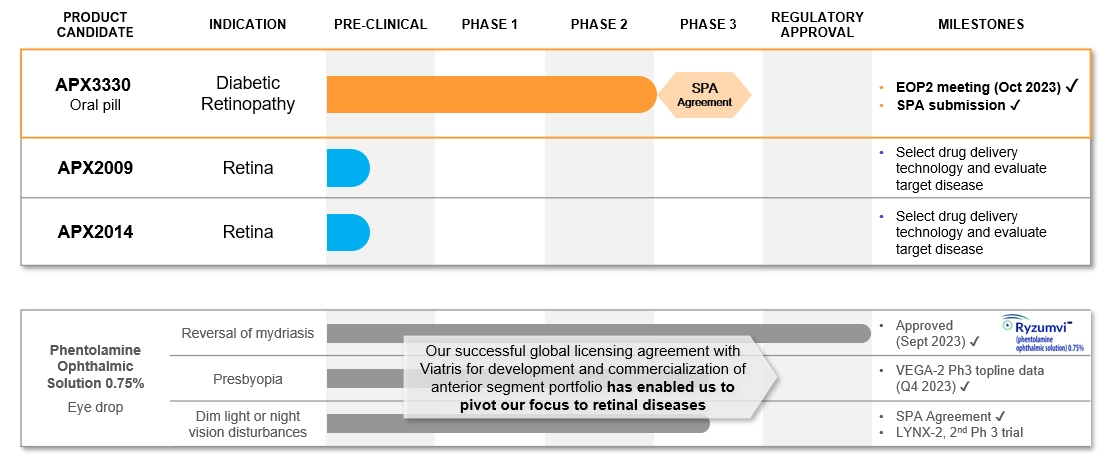
注:RYMZUMVIと0.75%PS(フェントラミン眼薬液)は1%PMOS(メタンスルホン酸フェントラミン眼薬液)と同様であった。本文書では2つの名前のPSを一般的に引用しているが,この2つの名前は記述に区別がない.
9
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
PSは2023年9月にFDAにより薬物による散瞳の治療に許可され,ブランドはRYZUMVIであり,角膜屈折手術後の暗い(中視または低)光線条件下での老眼や視力低下の治療に第3段階試験が行われている。Ocuphireは,角膜屈折術後の暗い(中視または低)光線条件下での老眼と視力低下の治療のためのPSに対する補足NDAを提出する予定であり,APX 3330の将来のNDAへの前進を推進している。
眼科疾患市場概況
網膜症市場
網膜損傷は失明を招く主要な原因の一つであり、世界各地の高齢化と更に多くの糖尿病人口の増加に伴い、網膜損傷は依然として増加し続けている。糖尿病は20−74歳の成人が失明する主な原因である。国家眼科研究所のデータによると、アメリカだけで1000万人を超える患者が糖尿病網膜症を患っており、これは糖尿病の合併症であり、慢性高血糖レベルは網膜血管損傷を招く。また750,000名の患者は糖尿病黄斑水腫(“DME”)を患っており、これは糖尿病網膜症の最もよく見られる合併症の一つであり、黄斑は損傷血管滲出の液体による腫脹である。DRとDMEの疾病進展はすべて血管異常増殖に関連し、血管内皮増殖因子シグナルと炎症反応を介している。OcuphireのAPX 3330経口錠剤は最近DRの第二段階の臨床試験を完成し、そして1種の斬新な二重作用機序
でこの巨大なDR市場の需要を満たす可能性があり、この二重作用機序は血管新生の調節剤VEGFと炎症を抑制できる。また、アメリカでは100万人以上の患者がwAMDを患っている。これらの網膜と脈絡膜血管疾患は黄斑に損害を与え、深刻で永久的な視力喪失を招く主要な原因である。現在、市場ではいくつかの薬物がLucentisを含む抗血管内皮増殖因子治療のために指定されている®(Ranibizumab)、遺伝子テーク社が販売しているモノクロナル抗体およびEYLEA®Regeneron製薬会社が販売している組換え融合タンパク質であり、深刻な形態のDMEおよびwAMDなどの網膜疾患を治療する標準的な看護になっている(AfLibercept)。これらの注射薬は生物製剤であり,眼科医のオフィスで治療を行っている。2020年には,LucentisとEYLEAの全適応の世界年間売上高は合計130億ドルを超えた(Lucentisは35億ドル,EYLEAは100億ドルを超えた)。
眼前節病市場
米国では約1億例の拡大手術があり,高齢化や糖尿病人口の増加に伴いこの数字はさらに上昇することが予想され,より頻繁な眼科検査やbr手術が必要である。何百万人ものアメリカ人たちも様々な屈折不正を患っている。
老眼はこのような屈折不正であり,40歳以上の患者によく見られ,物体を近距離で観察する能力が低下する。この状況は1.2億人を超えるアメリカ人に影響を与え、通常は近距離物体に焦点を合わせるために老眼鏡および/またはコンタクトレンズが必要である。
また,GlobalDataのデータによると,米国では約3800万人の患者がLASIK,夜間近視,円錐角膜,眼科手術あるいは自然老化過程による光線の暗さや暗視障害に悩まされている。スマートフォンの画面を使いすぎるため、若者の視力障害も世界的な傾向である。
APX 3330およびPSの概要
APX 3330
APX 3330は1種の小分子であり、特異的に脱プリン/脱ピリミジン核酸エンドヌクレアーゼ1/酸化還元因子-1(APE-1/Ref-1、Ref-1と略称する)、Ref-1は1種の二機能蛋白であり、細胞シグナル伝達の重要な転写因子の調節に参与する。REF-1は炎症、血管新生(血管形成)と酸化還元(Redox)シグナルを調節し、及び神経細胞の正常機能に重要なDNA修復である。DNA修復ではなく酸化還元活性を抑制することによって、臨床前研究により、APX 3330はいくつかの重要な血管新生促進と炎症促進転写因子、例えば核因子-κBと低酸素誘導因子-1α及び下流標的血管内皮増殖因子を調節することによって、血管新生と炎症を減少させることを表明した。これらの転写因子は多種の網膜と脈絡膜血管疾患の病理生理過程に参与し、糖尿病網膜症(DR)、糖尿病黄斑水腫(DME)、湿性老年性黄斑変性(WAMD)と地理萎縮(GA)を含む。
10
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
Ocuphire候補製品APX 3330の主な特性は、
| • |
最初の経口療法になる可能性がある。頻繁な硝子体内注射抗血管内皮増殖因子に関連する眼部合併症と比べ、もし承認されれば、毎日2回APX 3330を内服することは大量の網膜疾患患者にとって便利な新しい予防的治療方案或いは補助治療方案であるかもしれない。
|
| • |
上流目標は二つの有効な経路に関連している。APX 3330は、様々な網膜疾患を引き起こすことが知られている2つの確認された細胞シグナル経路(血管新生および炎症)を調節することを目的としている。また,APX 3330の作用機序は現在の抗血管内皮増殖因子療法の上流で著明な作用があり,抗血管内皮増殖因子療法を補充し,受診や硝子体内注射の頻度を減少させる可能性が示唆された。
|
| • |
良好な耐性の特徴。12個の完成した1期と2期の臨床試験では,APX 3330耐性が良好であった。副作用(“副作用”)は多少見られ、しかも軽微であり、一過性掻痒が最もよく見られる。
|
| • |
体系的な管理の潜在的な利点。APX 3330は全身投与として両眼(両眼)網膜血管疾患の治療が期待できる。
|
| • |
拡張可能な製造プロセスを有する口腔錠剤。APX 3330は良好な安定性を有する経口投与錠剤の調合であり、その活性薬物成分は小分子であり、標準化、拡張可能と低コストの製造技術優勢を持っている。
|
Ocuphireは最初にAPX 3330をDR適応の第一線の治療とし,DME,wAMD,GAなどの他の網膜適応としての単一療法や補助療法として臨床的利益を得る機会を探索することが可能である
| • |
ドラッカー!これは20~74歳の成人の視力喪失の主な原因であり、これは血液中のグルコースの慢性的な上昇による網膜細胞損傷である。br}網膜の重要なオピニオンリーダーのフィードバックは、経口薬物によるDR進展の緩和は有用な治療方法であり、背景DRがあり、視覚機能が良好な患者に適していることを示している。
|
| • |
ジメチルエーテルこれはDRの最もよく見られる合併症の一つであり、血管漏出は網膜黄斑腫脹と視力喪失を招く。
|
| • |
WAMD慢性眼疾患であり、視力中央部の視覚歪みを招き、異常血管は液体や血液を黄斑に浸透させ、黄斑は眼の中で中央と色覚に重要な部分である。
|
| • |
ガリウム.ガリウム老年性黄斑変性(AMD)の高級形式は、進行性と不可逆性の視力喪失を招く。
|
PS(0.75%フェントラミン点眼液)
PSは2022年にViatrisの許可を得,1日1回の無菌·無防腐剤の目薬製剤であり,メタンスルホン酸フェントラミンを含み,アドレナリン神経系に作用し平滑筋収縮を抑制する可逆的,非選択的α−1とα−2アドレナリン拮抗薬である。Ocuphireは2022年11月に505(B)(2)経路に従ってNDAをFDAに提出し,2023年9月にPSはRYZUMVIブランドでFDAの薬物誘導のための散瞳の治療許可を得た。RYZUMVIは薬物による瞳孔拡張を逆転させることができ,この拡張は光に対する感度を増加させ,焦点を合わせることができず,読書,作業,運転を困難にする。
11
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
Viatrisは引き続きRYZUMVIを商業化し,薬物による散瞳の治療に用いる。OcuphireとViatrisはViatrisライセンスプロトコルに基づいて以下の他の適応にPSを求めている:
| • |
老眼、目の水晶体は弾性を失い、近距離物体に焦点を合わせる能力に影響を与える。老眼は通常40歳以降に発生し,多くの患者が老眼鏡を使用するのは近くの物体を読んだり見たりするためである。ワイトニーTM2021年10月に承認され、2023年10月に承認されたQLOSIは、老眼の治療のための2つの承認された目薬のみである。
|
| • |
角膜屈折術後は暗い(中間または低)光線条件下で視力が低下し,瞳孔が暗い光で大きく開いた場合,角膜の周辺欠陥(収差)散乱光の場合である。患者はグレア、ハロー、星嵐とコントラスト感度が低下する。暗い(中間)光条件下での視力低下は新たな適応であり,承認されていない治療法である。
|
APX 3330型S標的指標
糖尿病網膜症
糖尿病網膜症の概要
DRは糖尿病による眼病であり、アメリカでは1000万人以上の患者が影響を受け、慢性血糖レベルの上昇は網膜の血管を損害する。20~74歳の成人では,視力喪失の要因となっている。DRには主に2つのタイプがある:
| • |
非増殖性DR、またはNPDRと呼ぶ.NPDRはDRの早期段階であり,治療を行わなければ高血糖値に曝露し続けると,時間の経過とともにより重篤なDRに進展する可能性がある。
|
| • |
増殖性DR、またはPDRと呼ばれる.PDRはNPDRよりも高度なDR段階である.その特徴は網膜新生血管であり、治療しなければ、永久的な損傷と失明を招く。
|
NPDRとPDRの治療法は異なる。NPDRでは、治療は通常、生活様式の観察、変化、およびNPDR進展をもたらす高血糖を治療する。そのほか、現在の治療モードは医者が早期DR/NPDR患者を待って監視し、そして増殖期DR或いはDMEに進展した患者に抗血管内皮増殖因子或いはステロイド注射治療或いはレーザー治療を保留することである。糖尿病網膜症臨床研究ネットワークのS方案試験において、Lucentisは糖尿病網膜症患者の治療においてレーザー治療に劣らないことを発見した。また,2018年,Regeneronのパノラマ実験では,EYLEA®中重度から重度のNPDR患者は疾病の進展を逆転した。
糖尿病網膜症の既存の治療法の限界
DR(特にNPDR)では,近年抗血管内皮増殖因子の治療薬が承認されているにもかかわらず,実際には疾患に症状がなく,患者は注射やレーザー治療を受けたくないため,注射剤の使用は第一選択の治療法ではない。
末期糖尿病網膜症において、硝子体内注射血管内皮増殖因子阻害剤は全世界範囲で承認された;しかし、これらの治療法は糖尿病網膜症に関連する潜在血管問題の完全な解決策を提供することは少ない。これらの治療薬物はいくつかの患者に対して成功したが、かなりの患者は薬剤耐性と難治性である。また,硝子体内注射では出血や眼内感染を含む重篤な副作用が出現する可能性があり,LucentisやEYLEAも動脈中血栓のリスク増加に関与している。また,硝子体内注射は眼科医によく通院する必要があり,通常は4週間に1回であり,開発中の抗血管内皮増殖因子療法br}も注射間隔(8−12週)の延長に努めている。
12
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
また,網膜疾患は最初または時間の経過とともに二国間であったため,一方の眼のみを治療し,他方の眼を残して治療しなかった。
APX 3330の災害復旧の機会
血管内皮増殖因子レベルの異常上昇を特徴とするほか、最近の科学文献報告によると、糖尿病眼疾患は血管内皮増殖因子とは関係のない炎症成分があることが明らかになった。br}は炎症と酸欠シグナル(血管内皮増殖因子の産生)が糖尿病網膜症の血管漏出と新生血管形成に重要な作用を発揮するため、炎症促進と酸欠シグナルを同時に抑制する治療は有望な治療策略を提供する。Ref-1(炎症と免疫反応に関連するタンパク質)を標的とするAPX 3330‘Sはこの二重作用機序(あるいはMOA)を利用して血管内皮増殖因子の発生を減少させ、血管内皮増殖因子の数を減少させ、同時に炎症損傷を防止する可能性がある。APX 3330のMOAと伝統的な抗血管内皮増殖因子療法の違いは、それは上昇した血管内皮増殖因子レベルを中和することができず、血管内皮増殖因子レベルを正常な動態平衡レベルに回復させ、それによって糖尿病眼病の早期進展或いは悪化を予防する理想的な治療方案になることである。
これはDR治療に対する反応を改善する可能性があり、末期網膜疾患(DME,wAMD)の侵襲的治療間の持続時間を延長する可能性がある。また、潜在的な1日2回の経口投与の候補製品として、疾病の早期より便利な選択となる可能性があり、特にDRにとっては硝子体内注射よりも便利である可能性があり、後者は患者に負担を与え、白内障形成、眼圧上昇、眼内感染と網膜剥離を含む顕著な副作用を有する。また,APX 3330は全身療法として両眼治療の潜在力を提供し,良好な安全性
を維持している。
潜在的な他の兆候は
糖尿病黄斑浮腫
糖尿病黄斑浮腫の概要
DMEはDRのよく見られる合併症であり、糖尿病網膜症の悪化、黄斑腫脹により、損傷した血管から液体が滲出する。それは糖尿病患者が失明に至る最もよく見られる原因の一つであり、約75万名の患者に影響を与える。ジメチルエーテルは視覚中心がぼやけ、波形の直線、暗く見えたり色が落ちたり、盲点に見える可能性がある。DMEの発病機序は血管漏出、網膜虚血及び血管増殖増殖因子と炎症メディエーターの放出に関連する。
DMEでは,コルチコステロイドや抗血管内皮増殖因子薬が血管漏出,炎症,低酸素/血管新生の治療に用いられている。DME合併DRに進展した患者では,抗血管内皮増殖因子薬が第一線の治療であり,次いでコルチコステロイドである。LucentisはDMEの治療のために許可され、用量レジメンは約4週間ごとに0.3 mg注射された。同様にEYLEAは®承認された用量レジメンは約4週間ごとに2.0 mgを注射する。
DMEでは、硝子体内血管内皮増殖因子阻害剤は全世界的に承認されているが、これらの療法はDMEに関連する潜在的血管問題の完全な解決策を提供することは少ない。これらの治療薬はいくつかの患者に対して成功しているが、かなりの割合の患者は薬剤耐性と難治性である。また,硝子体内注射では出血や眼内感染を含む重篤な副作用が出現する可能性があり,LucentisやEYLEAも動脈中血栓のリスク増加に関与している。また,硝子体内注射は眼科医によく通院する必要があり,通常は4週間に1回であり,開発中の抗血管内皮増殖因子療法br}も注射間隔(8−12週)の延長に努めている。
また,網膜疾患は最初または時間の経過とともに二国間であったため,一方の眼のみを治療し,他方の眼を残して治療しなかった。
13
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
湿性AMD
年齢関連性黄斑変性(AMD)はよく見られる眼病であり、アメリカでは1100万人が罹患し、全世界で1.96億人が罹患しており、その大部分は55歳以上の人である。これは進行性疾患であり、網膜の中央部分に影響を与え、黄斑と呼ばれ、これは目の中の鮮鋭度、中央視覚、色感知を担当する領域である。WAMDはAMDの高度な形態であり、網膜下新生血管と液体漏出を特徴とする。アメリカとEUでは、50歳以上の患者の深刻な視力喪失を招く要因となっている。WAMDはAMD症例総数の10%しか占めていないが,AMDに関連する重篤な視力喪失の90%を招いている。未治療或いは治療不足のwAMDは更なる血管漏出、黄斑内液体を招き、最終的に瘢痕組織を形成し、これは永久性視力喪失、甚だしきに至っては失明を招く可能性があり、これは未治療或いは治療不足の間に発生した瘢痕と網膜変形の結果である。深刻なDRとDMEと類似し、現在wAMDの治療は硝子体内注射を含み、主にLucentisとEYLEAである。これらの治療法の限界は,上で“DRとDMEの既存治療法の限界”と題する章で述べられている。APX 3330に基づくRef-1を標的とするMOAはVEGFの過剰発現と炎症を減少させることができ、wAMDにおいて潜在的な応用の将来性を有する。さらに、注射可能なwAMD市場に入るために、Ocuphireは、APX 3330およびその第2世代類似体APX 2009およびAPX 2014の硝子体内または徐放製剤の効用を考慮している。APX 2009とAPX 2014のデータは,APX 3330(“薬理学·実験治療学雑誌”に発表)に比べてRef−1目標に対する奏効率が向上していることを示している。
ガリウム.ガリウム
地理的萎縮(GA)はAMDの高度な形式であり、進行性と不可逆的な視力喪失を招く。先進国では,AMDは65歳以上の高齢者の恒久的視力喪失の要因であり,AMDのリスクは加齢とともに増加する。発表された研究によると,米国では約100万人がGAに罹患しており,世界では500万人がGAを患っている。GAを有する人では,光受容器は光感受性細胞であり,黄斑悪化,黄斑は網膜の中心部であり,中央視覚と色知覚を担当している。この被害は小さな斑点から始まり,大きなプラークに成長する。黄斑の細胞死に伴い、この人は視力を失い始めた。早期AMDを有する人は、読書または暗視の問題に気づくかもしれない。最終的に,疾患が末期まで進行すると,視野中心の恒久的な盲点(暗点)が進展する。GAの原因は炎症や,多くの環境や遺伝的リスク因子を含む多因子と考えられる。人体免疫システムの重要な構成部分として、補体下落の失調は重要な役割を果たしている。低下補体の過剰活性化は健康細胞の破壊を招き,GAを含む多くの疾患の発生や発展を招く。SYFOVRE®IZERVAYと®FDAが承認した唯一のGA治療薬であり,毎月または1カ月ごとに硝子体内注射が必要である。
候補製品
APX 3330
APX 3330(E 3330)は1種の経口製剤であり、毎日2回投与し、網膜と脈絡膜血管疾患に関連する多数の経路、例えばDRとDMEに対して、治療を行わなければ、永久性視力喪失と最終失明に進展する可能性がある。機序研究と以前の臨床経験により、APX 3330は非常に有望な候補薬物であり、DRとDMEからこれらの疾病の治療の有効性と安全性の臨床評価に応用できることを表明した。
動物モデルや体外の臨床前薬理学的研究では,APX 3330は経口投与,全身投与でも硝子体内注射により直接眼に入り,APX 3330は網膜の血管新生や炎症を減少させることが証明されている。ヒトにおいて、APX 3330は複数の1期と2期試験において良好な臨床耐性を有することが証明され、10%未満の患者のみが吐き気や下痢などの軽微な自己制限性副作用が出現した。また、APX 3330の経口投与量は人体の血流濃度に達し、網膜に影響を与えるマウスのレベルよりも高いことが示唆された。
Ocuphireは最初に中等度から重度のNPDR適応を求めた。さらに,Ocuphireは,他の網膜適応(例えば,DME,WAMDおよびGAまたは他の非眼科適応)としての単一療法または補助療法として臨床的利益を得る機会を探索する可能性がある。第2世代候補薬剤APX 2009およびAPX 2014は、眼科および非眼科適応のための硝子体内、経口、または他の注射経路も考慮することができる。
14
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
APX 3330の作用機序
APX 3330は1種の選択性小分子であり、二重機能を有するデオキシリボ核酸エンドヌクレアーゼ1/酸化還元効果因子-1(APE 1/Ref-1)蛋白に作用し、Ref-1と略称する。この蛋白は酸化還元シグナルやDNA修復に関与している。APX 3330は分子がDNA修復を行う能力に影響を与えることなく酸化還元機能を選択的に抑制するため,正常な細胞機能は不変である。また、HIF-1αとNF-κBのようないくつかの重要な転写因子を同時に低下させることによって、APX 3330を妨害するREF-1活性は血管新生と炎症を阻止することができる(参照)以下の図2は直感的な説明を提供する酸欠誘導因子-1αは血管新生に重要な蛋白血管内皮増殖因子の発現を調節し、核因子-κBは炎症過程中の上流調節蛋白、例えば腫瘍壊死因子αとケモカインである。
DR/DMEの発生は網膜血管漏出、網膜血流不足及び血管新生増殖因子と炎症メディエーターの放出に関連する。HIF-1αとNF-κBの下流標的はこれらの疾病特徴の肝心な媒質であり、現在糖尿病眼病とWAMD治療の標的でもある。APX 3330はVEGF蛋白の作用を抑制するのではなく、臨床前モデルでその形成を抑制できることが証明された;これはAPX 3330と現在承認或いは開発されているDR薬物(例えばLucentisとEYLEA)との重要な潜在的な違いである。Sがこの2つの転写因子活性を抑制する潜在的能力は,抗血管内皮増殖因子やステロイドを頻繁に硝子体内注射する必要を軽減する可能性がある。
APX 3330は異常血管新生と炎症を減少させる二重機序を有する。APX 3330はRef−1下流のチャネルを遮断した。低酸素誘導因子-1αを遮断することは血管内皮増殖因子シグナル伝達を減少させ、核因子-κBを遮断することは血管内皮増殖因子、腫瘍壊死因子-αなどの炎症性サイトカインの産生を調節できる。逆に,抗血管内皮増殖因子薬は血管内皮増殖因子の作用のみを抑制する(では)以下の図2は直感的な説明を提供する).
15
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
図2:検証された疾患経路におけるAPX 3330の二重作用機序
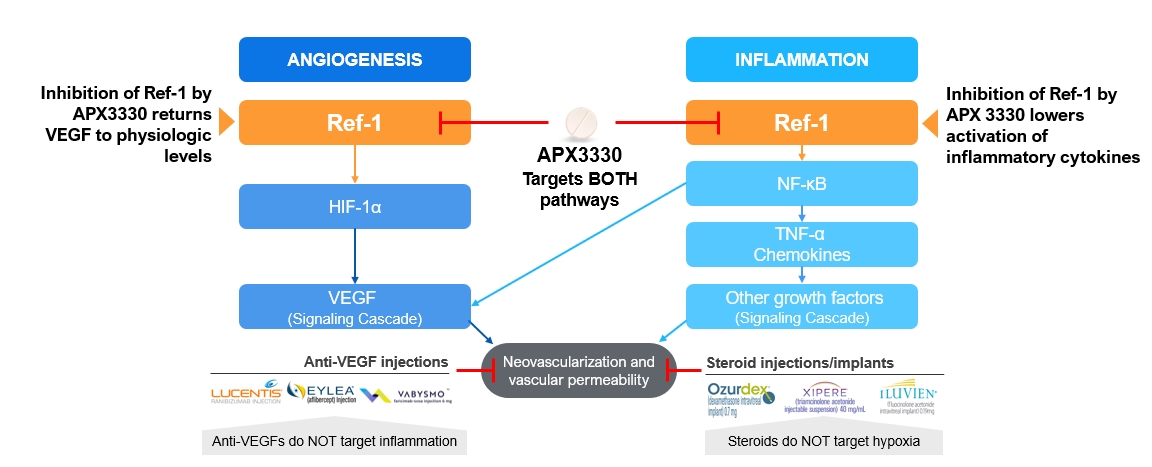
注:イーリー®RegeneronとLucentisの登録商標です®羅氏/遺伝子泰克の登録商標であり、VABYSMOは羅氏集団の登録商標である® Allergan,XIPEREの登録商標です® Clearside Biomedical,Inc.,ILUVIENの登録商標です® Alimera Sciences Inc.の登録商標です。
APX 3330臨床経験まとめ
APX 3330はすでに375人を超える健康ボランティアまたは肝炎、癌または糖尿病網膜症患者において研究されており、その中で270人を超える人が候補製品を平均75日以上服用し、1日あたり720 mgまでの用量である。この12個の1期と2期非眼科と眼科臨床試験において,安全性と有効性,Ref−1分子標的への影響および薬効学的特徴に関する臨床データを収集した。APX 3330は良好な安全性、耐性と生物学的治療効果を示した。
この10試験でAPX 3330が予測可能なPKを示すことが認められ,非臨床研究で得られたPKデータと一致した。2つの研究では,食事はAPX 3330のPKに影響しないことが分かった。APX 3330はすでに600 mg/日までの用量で耐性が良好であることが証明された。
5つの第1段階試験(CLN_0001、2、3、4および8)では、プラセボまたはAPX 3330治療を受けた75名の患者のうち、5名の患者が急性毒性反応(1日120 mg、180 mgまたは240 mg用量の軽度下痢)を出現した。APX 3330治療を受けた279名の5つの2期試験では,40名(14%)に副作用が認められ,その多くは軽微であった。成人固形腫瘍患者の第一段階試験APX_CLN_0011において、患者は毎日720 mgまでのAPX 3330用量を受けた。1日720 mgを服用した6名中2名に自発可逆的なびまん性皮疹が出現した。毎日600ミリグラムの用量を受けた患者は急性毒性の兆候がない。APX_CLN_0011 1期腫瘍学試験の19名の患者のうち、4名の患者の暴露時間は6ケ月を超え、3名の患者(600 mg/日)の暴露時間は300日を超え、明らかな薬物関連副作用はなかった。
DR/DME患者におけるAPX 3330の2 b期試験(ZETA-1)(完了)
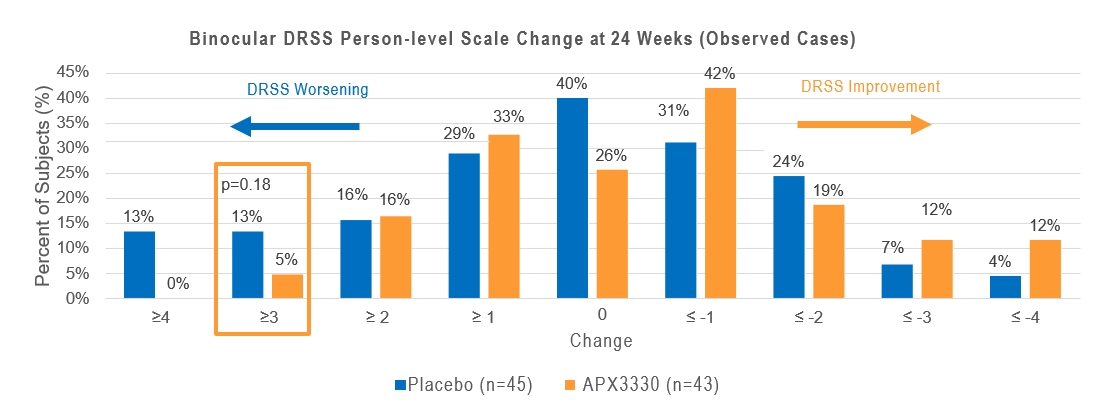
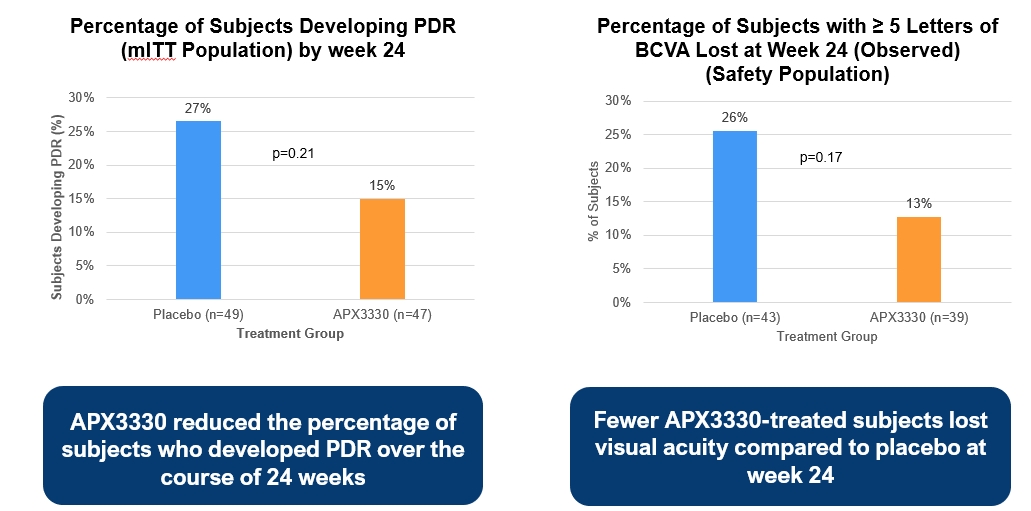
2022年8月、Ocuphireは2 b期双マスキング、無作為、プラセボ対照の多中心DRとDME患者試験であり、2021年4月に開始したZeta-1を完成させた。103人のDR被験者が入選し、その90%がNPDR(ベースラインDRSSが47または53)を有し、平均ベースラインCSTが270ミクロンであった。この研究は、中重度NPDRから軽度PDR、および中心視力喪失のないDME患者を含む1日600 mgのAPX 3330によるDR患者の治療効果を評価した。主な終点は研究眼の24週目、糖尿病網膜症早期治療研究において2級改善した患者のパーセンテージである。複数の時点の重要な二次終点は、単眼および両眼DRSS、CSTおよびBCVAの変化を含む。AEモニタリング、臨床実験室評価、眼圧とバイタルサイン評価を通じて患者の安全性を評価した。Zeta-1試験は研究眼の主要な終点に達しなかった;しかし、この試験は両眼を評価する時に糖尿病網膜症進展を予防する臨床意義の潜在力があることを証明した。硝子体内注射治療単眼とは異なり,全身薬物で両眼を治療するため,両眼の反応を考慮する必要がある。APX 3330治療を受けた患者のDR重症度がプラセボと比較して悪化する可能性は低い。ZETA−1試験では,プラセボ群の13%の患者がAPX 3330群の5%(p=0.18)と比較して,ベースライン両眼レベル47,53または61(図3)の場合,両眼ヒトレベル尺度は24週でベースラインレベルより3段階悪化した。追加の治療効果の終点は方向性においてDR進展の緩和と視力保護におけるAPX 3330の生物効果の支持に有利である。APX 3330の視力は安定しており、APX 3330によって治療された患者は、プラセボと比較して5つ以上の遠隔視力文字を失った患者の方が少ない(13%は26%、p=0.17)(図3)。
16
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
図3:24週におけるDR、PDR、および視力喪失進行の被験者の割合
APX 3330は良好な安全性を示した。緊急治療の重篤な副作用では,14例が研究薬物とは無関係と考えられ,11例はプラセボ群,3例はAPX 3330群であった。各群の2人の被験者がAEにより退出し,プラセボ対象の1人のOU DMEの悪化が治療に関与していると考えられた。全体的に、64名の被験者(29名のAPX 3330、35名のプラセボ)は211例の副作用(91例のAPX 3330、120例のプラセボ)があった。そのうち31例のみが麻薬に関与していると考えられた(APX 3330,17例のプラセボ)。すべての治療に関連する副作用は軽度または中等度であった。これらの治療は、眼科検査の任意の他の特徴または全身安全性の任意の評価に悪影響を与えない(図4).
17
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
図4:ZETA-1セキュリティの概要
APX 3330-10段階1および2試験-EISAI(APX CLN 0001-0010)
衛材株式会社の協賛の下で、日本は10項目の臨床試験を行い、対象は健康ボランティアと慢性肝炎患者(即ちアルコール性C型、B型肝炎)を含み、腫瘍壊死因子-α遮断薬の開発を目的としている。彼らが臨床試験を行ったところ,APX 3330の分子標的は確認されておらず,Ref−1蛋白であることも知られていない。
この10試験でAPX 3330が予測可能な薬物動態を示すことが発見され,非臨床研究で得られた薬物動態データと一致した。また,600 mg/日以下では明らかな急性毒性はなかった。APX 3330は良好な耐性を有することが証明されている。また、2つの研究で、食事は候補製品の薬物動態学に影響しないことが分かった。安全耐性br測定ではバイタルサインは変化せず,臨床実験室値も変化しなかった。観察された唯一の有害事象は下痢と皮疹であり,いずれの場合も5%未満で軽微であった。
APX 3330臨床発展計画
APX 3330の場合、APX 3330の網膜脈絡膜血管疾患の治療に関するINDの申請は、2018年12月に食品医薬品局眼科部門に提出され、発効している(IND 142152)。APX 3330はまた,米国食品·薬物管理局腫瘍科と膵癌治療のIND(IND 125360)を締結した。APX 3330はすでに12項目の試験(6項目の1期試験と6項目の2期試験)を完成し、主に肝疾患、固形腫瘍と糖尿病網膜症患者に関連している。OcuphireはFDAとEP 2会議を開催し、2023年10月に会議結果を共有し、Dr.のシステム薬物登録の主な終点を確認することを含む登録計画に関する合意を述べた。同社はFDAにSPAを提出した。
APX 3330の潜在的な臨床計画:
APX 3330の潜在的臨床計画は、DR/DME患者における2023年1月に報告されたAPX 3330の第2段階の安全性、耐性および有効性の結果、および2023年10月にFDAと開催されたEPOP 2会議の結果に基づいて、登録終点を確認し、最終的にAPX 3330をDR第一線療法の将来の試験設計として決定し、また、慢性安全曝露試験およびNDA提出前に必要な任意のさらなる動物毒理学研究を定義する。OcuphireはDME,WAMD,GAなどの他の網膜適応としての単一療法や補助療法の臨床的利益機会を探索する可能性がある。
18
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
PS
PSの作用機序
PSは毎日1回のメタンスルホン酸フェントラミン無菌点眼液の調合であり、瞳孔直径の縮小と視力向上を目的としている。PSの有効薬物成分であるメタンスルホン酸フェノールトラミンは1種の非選択性α-1とα-2アドレナリン拮抗薬であり、虹彩平滑筋の活性化を抑制し、瞳孔直径を縮小できる。フェントラミンと異なり、コリン作動薬(例えばピロカルピン、カルバコールとアキシロジン)は虹彩括約筋に作用して瞳孔直径を縮小し、そして毛様体筋接触の副作用と関連し、例えば頭痛、眉痛と網膜剥離、高度近視患者に使用は限られている。PS
は現有の目薬の多くの特性を持ち、便利な投与経路とコスト効果の高い製造技術、及び毎日1回の投与の潜在的な優勢を含む。
PSは相対的に非選択的なα1とα2アドレナリン拮抗薬である。瞳孔の拡張は主に瞳孔を囲む放射状虹彩拡張器筋によって制御され,これらの筋はα−1アドレナリン受容体によって活性化される。フェントラミンは虹彩拡張器筋肉上のこれらの受容体に可逆的に結合し,瞳孔径を縮小する。フェントラミンはα-1アドレナリン作動薬の散瞳作用を直接拮抗することができ、間接的にムチン拮抗剤の虹彩括約筋に対する散瞳作用を逆転させることができる。Alpha-1拮抗薬は受容体と結合し、瞳孔反応を抑制し、乱視を減少させる(図5Br}
メタンスルホン酸フェノールトラミンはFDAが承認した2種類の注射用薬物RegitineとOraVerseの有効成分であり,前述したとおりである。
薬物によって引き起こされる散瞳適応の治療において、散瞳は、αアゴニスト(例えば、フェニレフリン)を用いて虹彩拡張器筋肉を刺激することによって、またはM受容体アンタゴニスト(例えば、トピカルミン)を用いて虹彩括約筋を遮断することによって達成される。PSは,α−1アゴニストを直接拮抗するか,あるいはM受容体遮断薬の散瞳効果を間接的に拮抗させることにより,自然逆転前に散瞳を加速させることができる。
老眼患者では,レンズが形状(調節)や近傍物体に焦点を合わせることができない光線を克服するために,瞳孔径を小さいサイズに小さくすることで,光線が直線に近い方向にのみ眼に入ることを可能にし,焦点深度を増加させる(“ピンホール効果”)。Ocuphireは,PSを用いて虹彩拡張器筋をリラックスさせ,ピロカなどのマウス塩基作動薬を用いて虹彩括約筋を収縮させることにより,2 mmから3 mmの最適瞳孔径を達成できるとしている。これは、レンズ調整なしに最適な焦点深度および視覚に近い解像度を生成することができる。
最後に,光線がぼやけた視力障害に対して,PSを用いた適度な屈折効果が暗視困難を緩和する可能性があり,その大部分は角膜周辺に存在する欠陥や収差によるものであることを提案した。したがって,瞳孔を小さな直径に縮小することでこれらの欠陥の影響を減少または除去することができ,小さい瞳孔は
焦点が外れた網膜への異常光線を遮断することが知られているからである.
19
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
図5:PSの動作機構
PS臨床経験まとめ
PSはすでに研究者が発起·スポンサーした12の1期,2期,3期の臨床試験で評価されている。すべての試験において,700名以上の成人患者が少なくとも1剤のフェントラミン点眼液を接触させた。第二段階と第三段階試験の臨床試験データは,米国眼科学会(AAO),視力·眼科研究協会(ARVO)あるいは米国白内障·屈折手術学会(ASCRS)の年次会議で公表された。VEGA−2実験の結果は将来の医学会議で発表される可能性がある。
VEGAプログラムによるPS治療の老眼適応
PS老眼:VEGA-2第3段階試験(完了)
Vega-2(NYXP-301)は二重盲検、ランダム、プラセボ対照の多中心試験であり、PS、プラセボと補助的LDPを賦形剤(プラセボ)と比較した。約330人の被験者はランダムに4つの治療群に分けられ、2段階に分けられた。
この研究のデータは将来の医学会議で発表されるかもしれません
第2段階織姫-1試験(完了)
VEGA-1(NYXP-201)はダブルマスキング、ランダム、プラセボ対照の多中心試験であり、PSとLDPと賦形剤(プラセボ)眼液による老眼患者の治療の比較に用いられる。計150名の患者をランダムに4つの治療群に分け、各群3:2:2:3。この研究の主要な治療効果は終点に達した。この研究のデータは2021年と2022年のいくつかの医学会議で公表された。
Lynx計画−角膜屈折術後の暗(中視または低)条件下での視力低下PSの適応
PS:第3段階Lynx-1実験(完了済み)
LYNX-1(NYXDLD-301)は、3期二重マスキング、ランダム、プラセボ対照のマルチセンター研究であり、アメリカの複数の地点に弱光視力障害を有する患者に対してPSとプラセボ眼溶液の比較を行った。この実験では、弱光条件下で視力障害が出現した患者145名がランダムにPS群またはプラセボ群に分類された。各被験者は、PSまたはプラセボ眼液治療を1:1の割合でランダムに受け、虹彩色(明暗虹彩)で階層化した。各眼は寝る前または寝る前に自分で投与し,1日1回,14日間持続する。
20
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
8日目(主終点)にMITT群でPS治療を受けた被験者では,プラセボ治療群と比較し(それぞれ13%と3%;p=0.0459),PS治療を受けた被験者では,研究眼の15文字はmLCVA
でベースラインより有意に改善した。この研究では,PS治療を受けた被験者は15日目にプラセボ治療群と比較し(それぞれ21%および3%;p=0.0042),PS治療を受けた被験者の研究眼の15文字はmLCVAにおいてもベースラインより有意に改善した(21%比3%;p=0.0042)。
PS非臨床毒理学研究
包括的非臨床毒性計画の一部として,Ocuphireはウサギとビーグル犬においてメタンスルホン酸フェノールトラミン薬剤の単回と反復用量毒性研究を行った。PSに関連する眼部病理変化は認められなかった。
文献にメタンスルホン酸フェントラミンの非臨床情報(薬理特性、一般と生殖毒理学)及びその他の承認されたフェントラミン製品と調合を述べ、RYZUMVI、OraverseとRegitineの市場申請審査過程においてFDAによって審査を行った。
PS臨床発展計画
OcuphireはViatrisと協力してPSを開発している。PSについては、新薬(IND)の研究申請は2011年7月に食品と薬物管理局眼科部門に提出され、発効した(IND
70499)。Ocuphireは2022年11月に薬物性散瞳を治療するためのPSのNDAを提出し,2023年9月に承認された。PSはすでに12試験(3項目1期試験,5項目2期試験,4項目3期試験)を完了しており,主に若年·年配の健常ボランティアで行い,薬物による散瞳,老眼,暗光線障害,緑内障患者を逆転させている。2023年12月、Ocuphireは、角膜屈折手術後に暗いbr(中間または低)光線条件下で視力が低下したPSを治療するためのFDAとのSPAプロトコルを取得した。
PS潜在臨床計画
OcuphireとそのパートナーViatris,Vega-3(NYXP-302)試験は約545名の老眼患者においてダブルマスク、無作為、プラセボ対照の多中心試験を行う予定である。第2の登録試験は、Vega-2と同様のPSの有効性および安全性を評価し、類似した主要および重要な補助端末および分析を含み、迅速な反応を評価し、合計48週間延長することができる。私たちのbr開発およびビジネスパートナーViatrisは、2024年上半期に第3段階の開発を継続する予定である。
第1段階第3段階試験の積極的な結果によると、Lynx−1、OcuphireおよびそのパートナーであるViatrisは、角膜屈折手術後に暗い(中視または低)光線条件下で視力が低下したbrを治療するために、200人までの被験者において同様のPS 2回目の登録試験を計画している。私たちの開発とビジネスパートナーのヴィアヤリスは2024年上半期に第三段階の開発を続ける予定です。
将来の許可と買収の機会
Ocuphireは絶えず科学的価値、特許保護、監督管理経路と商業機会に基づいて候補製品を評価している。
販売とマーケティング
同社は一連の眼科薬物会社とAPX 3330の開発と商業化について討論し、共同開発、流通、許可或いは合併とbr}買収を含む。Ocuphireは,商業化前計画の一部として,主要オピニオンリーダーとの接触や,網膜,眼科,検眼医学や業界会議での知名度と知名度の向上を含めた市場開発活動を開始している。
21
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
Ocuphireは2022年11月、米国および米国以外の市場(アジアの特定の国を含まない)ですべてのPS適応を開発し、商業化するライセンス契約をViatrisと締結した。
製造業
APX 3330、PSと将来開発される他の候補製品について、Ocuphireの契約メーカーは現在、その原料薬と薬物製品を生産し、Ocuphireの臨床前研究と臨床試験に使用し、信頼性と重複可能な合成技術とよく見られる製造技術を採用している。
Viatris許可プロトコルにより,OcuphireはPSのビジネス製造責任をViatrisに移そうとしている.Ocuphireには長期合意は何もないが,Ocuphireやその開発パートナーは薬物物質や薬物製品のこのような手配を適宜確保する予定であり,現在複数のメーカーとの調達注文が使用されている。OcuphireはNDA規制報告書をFDAに提出しており,選定されたメーカーが原料薬br物質や薬品を提供する資格があるようにしている。Ocuphireは引き続き契約メーカーおよび潜在的なパートナーに依存して商業ロットの薬物
物質や薬物製品を生産し続ける予定であり,適用された規制機関の承認を得て発売されれば。Ocuphireは製造施設を所有したり運営したりしておらず,現在のところ何の製造施設も建設する計画はない。
APX 3330
APX 3330は、既存の原材料と伝統的な化学プロセスから合成された小分子薬物の経口製剤である。APX 3330薬剤はすでに新しい形式に最適化された。APX 3330薬物物質と薬物製品のプロセスと分析開発を完了し,cGMP法規の要求に基づいて生産を拡大している。以前、APX 3330医薬品メーカーは、60 mgおよび120ミリグラムの用量強度の錠剤のcGMP生産活動を支持するための薬物開発を行い、後者は以前の臨床試験に使用されてきた。Ocuphireはすでに薬物製品を再調製し、用量強度brを300 mgに向上させ、1日1~2回の服用を容易にした。Ocuphireはまた、未来の大量のAPX 3330の薬剤と薬物製品に対して追加の安定性研究を行い、新しい強度、製剤とbrの有効期限を決定し、監督管理と商業段階を支持することを計画している。
PS
PSの特許配合は,無菌,無防腐剤,等張,緩衝の水溶液であり,メタンスルホン酸フェントラミン,マンニトール,酢酸ナトリウムを含む。メシル酸フェノールトラミンは1種の小分子薬物であり、信頼性と反復可能な合成技術によって既製の原料から製造することができる。Ocuphireは現在イタリアのサプライヤーからPSの活性医薬成分を獲得しており,現在第2の源の開発に取り組んでいるが,Viatrisと協力して長期商業製造戦略を決定している。臨床試験に使用したすべての薬品物質であるメタンスルホン酸フェントラミンとPS薬品は現行の良好な生産規範(CGMP)に従って生産され、サプライヤーは有効薬物主文書(DMF)にFDAにプロセスを登録した。PSは従来使い捨てボトルに包装されており,蓋は第1段階と第2段階の臨床試験の容器閉鎖システムとしていた。Ocuphireはコンテナ閉鎖システムをPSの業界標準のための使い捨て無防腐剤吹込密封(BFS)容器に変換しており,先行する米国メーカーが充填している。BFS容器中のPS点眼液はFDAによって薬物−デバイス組合せ製品に分類された。現在の製造技術はすでに商業生産能力に拡大している。PSは冷蔵少なくとも2年間の5℃
で安定性を示した。Ocuphireは、有効期間を決定し、規制提出および商業生産を支援するために、大量の薬物メタンスルホン酸フェントラミンおよびPSの医薬製品に対して追加の安定性研究を行っている。最終的な世界市場を供給し,単一施設への依存を回避するために,OcuphireとそのパートナーであるViatrisは,薬物物質や薬物製品の第2源製造施設を構築する可能性を評価している。
22
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
Apexian分割許可プロトコル
2020年1月21日、OcuphireはApexianと再許可協定を締結し、この協定によると、OcuphireはAPX 3330候補製品と第二世代候補製品に関連する特許および他の知的財産権を獲得し、APX 3330に関連するいくつかの研究報告、製造と分析記録、データ、ノウハウ、技術および他の独自情報を含むApexianが衛材から取得した知的財産権を含む。この知的財産権はRef−1阻害剤計画を構成し,眼科や糖尿病に関連する疾患を治療するための治療アプリケーションの開発に重点を置いている。Ref−1阻害剤計画における先導化合物はAPX 3330であり,OcuphireはDRとDMEおよび潜在的なwAMDを治療する経口投与錠剤として開発しようとしている。参照してください“Ocuphire経営陣の財務状況と運営結果の検討と分析
契約義務と承諾−Apexian分許可協定“Apexian再許可プロトコルの詳細を理解します。
知的財産権
APX 3330
Ocuphireが所有するAPX 3330およびその関連化合物の特許付与は、2023年12月31日現在、9つの米国特許および2つの出願されている米国非一時特許出願、ならびにヨーロッパ、日本、カナダ、中国およびオーストラリアで発行されている特許、ならびにヨーロッパ、日本、カナダ、韓国で出願されている特許出願を含む。このライセンスは、眼科および糖尿病適応のヒト健康用途分野においてカバーされる主題特許および特許出願のためのAPX 3330および関連物質化合物組成物の使用および商業化のためのものである。
許可された米国特許第9,040,505号は、2030年に満了する予定のAPX 3330のような糖尿病網膜症および他の疾患を治療する方法を要求する。欧州、日本、オーストラリア、カナダはすでに対応する特許
を発行しており、これらの特許計画は2028年に満期になり、関連する米国特許出願が審理中であり、その治療法は特許として発表されれば2028年に満期になると主張している。係属中の米国特許出願第18/341,077号および欧州、日本、カナダ、韓国の係属中の出願は、APX 3330、APX 2009、またはAPX 2014を使用してwAMDおよび他の疾患を治療する方法を必要とする。これらの未解決特許出願に基づく特許が付与されれば,2039年に満了する。米国や一部の外国諸国は,政府の規制審査過程で失われた新薬特許期間を補償するために,特許期間を最大5年間延長することを許可している。米国特許
9,040,505に丸5年間の特許期間を延長する資格があれば,米国特許9,040,505の有効期限は2035年に満了する。米国特許9,040,505は、完全な5年間の特許期間を延長する資格があるかどうかは、FDAがこの新薬を承認する日にある程度依存する。
Ocuphireはまた、2030年に満了する予定であるAPX 2014を用いて特定の網膜疾患を治療する方法に対する米国特許を許可している。Ocuphireはまた、APE 1/REF-1阻害剤(例えば、APX 3330)および第2の治療剤を含む併用療法組成物に対するbrの特許出願を米国およびカナダで許可し、このような併用療法を用いて網膜疾患および/または他の適応を治療する方法が米国およびカナダで懸案されている。これらの出願に基づく特許が付与された場合、2038年に満期になる。欧州、日本、カナダでは、ニューロン感受性を低下させ、および/または他の適応を治療するために、単一療法または併用療法においてAPE 1/REF−1阻害剤(例えば、APX 3330)を使用するための特許出願が承認されており、これらの出願に基づいて付与された特許は、2038年に満了する。同じ特許ファミリーには、APX 3330を併用療法の一部として使用して炎症および疼痛を治療する方法に対する特許が含まれている。
OcuphireがAPX 3330デリバティブを有する米国特許は、9,089,605件、9,193,700件、9,877,936件、10,154,973件、11,160,770件、11,648,226件、および11,723,886件を含む。これらの米国特許計画は、2029年から2032年までの満了を計画している。OcuphireがAPX 3330のデリバティブに付与した外国特許には、欧州、日本、オーストラリア、中国、カナダの特許が含まれており、これらの特許計画は2028年から2039年の間に満了する予定である。
Ocuphireは、Ocuphireが許可された特許および特許出願に加えて、2023年12月31日までに、APX 3330を用いて糖尿病網膜疾患を治療する方法に関する米国、ヨーロッパ、日本、オーストラリア、カナダ、および他の国で出願中の特許出願を有する。上記特許出願に基づいて承認された場合、Ocuphireは2042年に満了する。また、Ocuphireは、APX 3330塩およびエステルに関する未解決の国際特許出願を有している。APX 3330塩またはエステルを含む組成物、ならびにAPX 3330塩およびエステルの治療用途。上記特許出願に付与された特許は、2043年に満了する。Ocuphireはまた、糖尿病網膜疾患患者のAPX 3330を使用する他の治療方法に対する係属中の米国仮特許出願と、新世代分子APX 2009またはAPX 2014を使用する他の治療方法に対する米国仮特許出願とを有しており、これらの一時特許出願に基づいて提出された任意の特許が承認された場合、2044年に満了する。
23
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
PS
Ocuphireの特許権には,様々な形態のメタンスルホン酸フェントラミン,メタンスルホン酸フェントラミンを含む製剤,メタンスルホン酸フェントラミンの使用方法およびメタンスルホン酸フェントラミンを製造する方法の特許および特許出願がある。Ocuphireは主に発明特許および特許出願、Ocuphire製品名の商標保護、およびOcuphireが適切と考える商業秘密保護によってその知的財産権を保護する。Ocuphireはすべての適応のPSのすべてのグローバル権利を持っているが,Viatris許可プロトコルにより,PSのある権利の許可権はPSを超えている.
OcuphireのPS関連特許資産には、2023年12月31日現在、米国特許12件、出願中の米国非仮特許9件、オーストラリア、カナダ、欧州、日本、メキシコで取得された特許、オーストラリア、カナダ、欧州、日本、中国、その他の国で出願されている特許が含まれている。
Ocuphireの米国特許第9,795,560,10,278,918,10,772,829,11,090,261および11,844,858ならびにオーストラリア、ヨーロッパおよび日本の特許は、メタンスルホン酸フェントラミン水性製剤を含む物質組成請求項を含み、2034年に満了する。同じ特許ファミリーにおいて、Ocuphireは、メタンスルホン酸フェントラミン水性製剤を要求する係属中の米国特許出願もあり、この出願によれば、承認された場合、特許は2034年に満了する。特許および特許出願は、PS製品の現在の臨床処方をカバーする。
Ocuphireの米国特許番号9,089,560;9,789,088;11,000,509および11,717,510は、2034年に満了する予定であるメタンスルホン酸フェントラミンを使用して視覚性能を改善する方法に対する請求項を含む。オーストラリア、カナダ、ヨーロッパ、日本はすでに相応の特許を発行しており、これらの特許は2034年に満期になる予定だ。同一特許ファミリーにおいて,Ocuphireには未解決の米国特許出願が1つあり,メタンスルホン酸フェントラミンのような視覚性能を改善する方法も要求されており,これにより,この係属中の出願に基づく特許は2034年に満了する。特許および特許出願は、PS製品の現在の臨床処方の使用をカバーする。
Ocuphireは米国,オーストラリア,カナダ,中国,ヨーロッパ,日本でメタンスルホン酸フェノールトラミンを用いた緑内障や他の医学疾患の治療に対する特許出願が出願されている。これらの未決出願に基づく特許が承認されれば,2039年に満期となる。
Ocuphireの米国特許第10,993,932号は、補助ピロカル品を有するメタンスルホン酸フェノールタールを用いて老眼を治療する方法に対する請求項を含み、2039年に満了する予定である。Ocuphireの米国特許第11,400,077号は、メタンスルホン酸フェノールタールを用いて散瞳を治療する方法に対する請求項を含み、2039年に満了する予定である。米国特許10,993,932および11,400,077と同じ特許ファミリーに属し、Ocuphireには老眼の治療が可能であると主張されている4つの係属中の米国特許出願があり、他の2つの米国特許は散瞳の治療が可能であると主張している。オーストラリア,カナダ,中国,ヨーロッパ,日本,その他の国の対応特許出願が承認されているため,これらの未解決の米国と外国特許出願に基づく特許が承認されれば,2039年に満了する。
Ocuphireの米国特許第11,566,005号は、物質成分として高純度メタンスルホン酸フェントラミン、フェントラミンからメタンスルホン酸フェントラミンを製造する方法、および被験者の虹彩平滑筋収縮を抑制し、視覚コントラスト感度または視力を向上させ、暗または暗視障害を治療し、薬物性誘導散瞳を治療または逆転させ、老眼を治療する方法を含む。米国、ヨーロッパ、日本および他の外国では、対応する特許が出願されている。上記の特許出願が承認された場合、2042年に満了することが予想される。オプリーには、高純度メタンスルホン酸フェントラミンの製造方法およびメタンスルホン酸フェントラミン含有組成物に関する特許出願が中国出願中である。この中国のbr出願について付与された特許は2041年に満期となり,また,Ocuphireにはメタンスルホン酸フェントラミン結晶形態とその用途に対する特許出願が欧州で決定されている。この欧州特許出願に付与された特許は2043年に満期になる。Ocuphireは、米国、ヨーロッパ、日本および他の国においても、散瞳および緑内障を治療する他の方法に対する特許出願が行われており、この出願によれば、上記特許出願に基づく任意の外国特許が承認された場合、2042年に満了する。
24
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
Ocuphireは2025年に満期になる予定でメキシコにも特許を有しており,眼科用処方を所有していると主張している。
OcuphireはすでにアメリカでNYXOLのために商標保護を登録しました®そして処理中のRYZUMVI米国連邦商標出願。
より多くの特許条項情報
背景として,特許期間は通常,非仮出願を提出した日から20年である。アメリカでは、特許の有効期限はいくつかの方法で延長されることができる。まず,特許期限調整(PTA)は,特許権者による米国特許商標局の特許付与時の行政遅延による損失を補償する。第二に、場合によっては、FDA規制審査期間により実際に失われた期間の一部を再獲得するために、1984年の“医薬品価格競争及び特許期限回復法”(“ハッジ·ワックスマン法案”と略す)の規定に基づいて、特許期間(PTE)の延長を許可することができる。医薬品承認のこの回復期は5年を超えてはならず,回復期を含む特許総期限はFDA承認後14年を超えてはならない。承認された薬物に適用される特許(S)のみPTEを取得する資格があり,延期出願は特許満了前に提出され,上場承認後60日以内に提出されなければならない。米国では、“ハッジ·ワックスマン法案”は特許保護から独立しており、新化学エンティティ(NCE)秘密保持協定の承認を得た最初の申請者に5年間の米国内の非特許データ独占期間を提供している。この条項によると、“ハッジ-ワックスマン法案”によると、APX 3330は、FDAがAPX 3330活性成分を含む他のbr薬を承認していないため、NCEとみなされているため、5年間にわたるデータおよび市場排他性を得る資格がある可能性がある。
ヨーロッパでは、データ排他性指令により、製薬会社は競争リスクがなく、11年に及ぶ彼らの製品を販売する可能性がある。日本では、日本の“薬品法”によると、必要な研究期間の再審の長さによると、市場許可所持者が市場に参入するのに10年に及ぶ可能性がある。また、政府が新薬の販売許可を求める期間に適応するように特許期間の延長を許可している国では、ある特許の期限を最大5年間延長することができ、政府が新薬の販売許可を求めるのにかかる時間に適応することができる。
Ocuphireはまた書面協定を通じてその固有の情報を保護する。Ocuphireの従業員、コンサルタント、請負業者、パートナー、および他のコンサルタントは、雇用または採用を開始する際に、秘密協定および譲渡発明協定に署名しなければならない。また,OcuphireはOcuphire
機密情報を取得可能な外部当事者と書面セキュリティプロトコルを締結することで独自の情報を保護する.
競争
製薬業界の内部競争は激しい。OcuphireはRYZUMVIおよび現在の候補製品APX 3330およびPSは適応ごとに良好な開発地位にあると考えているが,Ocuphireまたはその開発パートナーはブランドや後発薬会社および現在開発中の製品からの競争に直面するであろう。その中の多くの会社は薬物開発、研究開発と商業化の面で明らかに多くの財力と人力資源及び経験を持っている。これらの競争相手は合格した科学と管理人員を募集と維持し、臨床試験場を構築し、臨床試験患者を募集し、製品、候補製品或いはその他のOcuphire計画を補充する技術の面でOcuphireと競争を展開する。小さな会社や他の早い段階にある会社が大手成熟会社と協力することを選択すれば、それらも重要な競争相手になる可能性がある。
25
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
政府の監督管理と製品審査
その他の事項以外に、アメリカ連邦、州と地方各級及び欧州連合(EU)を含む他の国家と司法管轄区の政府当局は薬品の研究、開発、テスト、製造、品質管理、審査、包装、貯蔵、記録保存、ラベル、広告、販売促進、流通、マーケティング、審査後の監視と報告及び輸出入などの方面に対して広範な監督管理を行った。米国や他の国や司法管轄区で規制承認を得る流れと、その後適用される法規や法規および他の規制機関の遵守には、多大な時間と財力が必要である。
EMAは独立した管理委員会によって管理され、EUの薬品の評価、監督、安全監視を担当する分散機関である。薬品と医療製品監督機関(MHRA)はイギリス(UK)の薬品、医療機器、血液成分を監督し、イギリスで離脱した後、その機能はEU EMAに類似している。日本医薬品·医療機器庁の機能は米国のFDAに類似しており,独立した行政機関である。国家医療製品管理局は中国が薬品と医療機器を規範化する機関(前身は中国食品薬品監督管理局)である。
アメリカの薬品審査と承認
米国では,FDAは連邦食品,薬物と化粧品法案(FDCA)とその実施条例に基づいて薬品を規制している。製品開発プロセス、承認プロセス、または承認後の任意の時間にFDCAおよび他の適用法律の適用要件を遵守することができない場合、FDAが係属中の出願の承認拒否、承認撤回、臨床保留の実施、警告状および他のタイプの手紙の発行、製品リコール、製品差し押さえ、生産または流通の完全または一部の一時停止、禁止、禁止、br}罰金、政府契約の拒否、原状回復、利益の返還、利益の完全または部分的な一時停止、禁止、br}の罰金、政府契約の拒否、原状回復、利益の返還など、申請者および/またはスポンサーに様々な行政または司法制裁を受ける可能性がある。あるいはFDAと司法省または他の政府実体によって提起された民事または刑事調査および処罰。
米国での新薬製品の販売および流通の承認を求める出願人は、通常、以下の義務を履行しなければならない
| • |
“動物福祉法”とFDAの良好な実験室規範に基づいて臨床前実験室テスト、動物研究と調合研究を完成した
|
| • |
FDAにINDを提出するには、ヒト臨床試験が開始される前に発効しなければならない
|
| • |
各臨床試験を開始する前に、各臨床場所を代表する独立した機関によって委員会またはIRBによって承認される
|
| • |
各提案適応に対する推奨薬物製品の安全性と有効性を決定するために、良好な臨床実践或いはGCP及び他の適用法規に基づいて十分かつ制御された人体臨床試験を行う
|
| • |
薬品と薬品の製造、包装、ラベルと流通はFDAの良好な製造規範(GMP)規定に符合し、GLP非臨床研究とGCP臨床研究に応用して候補薬物を研究する
|
| • |
商業製品の使用および包含のための製品ラベル、包装挿入ページ、および処方情報を作成する;
|
| • |
食品医薬品局に秘密保持協定を作成して提出します
|
| • |
適切な場合、または適用される場合、製品は、FDA諮問委員会によって審査される
|
| • |
現在の良好な製造実践またはcGMP要件に適合する状況を評価し、施設、方法、および製品の特性、強度、品質、および純度を維持するのに十分な施設、方法、および制御を保証するために、FDAによる製品またはその構成要素の1つまたは複数の製造施設の1つまたは複数の検査を満足的に完了させる
|
26
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| • |
GCPおよび臨床データの完全性を保証するために、FDAの臨床試験場所に対する監査を満足的に完了させる
|
| • |
使用料(適用例)を支払い、FDAがNDAを承認することを保証する
|
| • |
リスク評価および緩和策、またはREMS、ならびにFDA要件の承認後の研究を含む、承認された任意の要件を遵守する。
|
臨床前研究
臨床前研究は、製造された医薬物質または活性医薬成分および製剤医薬または医薬製品の純度および安定性の実験室評価、および体外培養そして体内にある動物実験、薬物の安全性と活性を評価し、人類の初歩的な試験に応用し、治療使用の理論基礎を確立する。臨床前研究の進行はGLP法規を含む連邦法規と要求された制約を受ける。臨床前試験の結果は,生産情報,分析データ,任意の利用可能な臨床データや文献や臨床試験計画などとともに,INDの一部としてFDAに提出される。いくつかの長期的な臨床前試験、例えば長期反復用量毒理学研究は、IND提出後に引き続き行われる可能性がある。
会社は通常長期の臨床前テスト、例えば長期反復用量毒理学研究を完成しなければならず、また研究製品の化学と物理特性に関する追加情報を開発し、そしてcGMP要求に基づいて最終的に商業大量生産製品の技術を確定しなければならない。製造過程は一貫して高品質の候補製品ロットを生産できる必要があり,また,メーカーは最終製品の特性,強度,品質,純度を試験する方法を開発しなければならない。また,適切な包装を選択·テストし,候補製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
INDとIRBプロセス
INDはFDCAの免除であり、未承認の薬物の州間商業での輸送が許可され、臨床研究に応用されている。IND申請を支援するためには,申請者
は臨床試験ごとに1つの案を提出しなければならず,どの後続の案修正もINDの一部としてFDAに提出しなければならない。また,前臨床試験の結果は,生産情報,分析データ,任意の利用可能な臨床データや文献,臨床試験計画などとともにINDの一部としてFDAに提出されている。INDは、この30日間の間にFDAが懸念または問題を提起し、臨床的保留を実施しない限り、申請後30日に発効する。
臨床保留はFDAがスポンサーに発表した命令であり,提案された臨床研究の延期や進行中の研究の一時停止が要求されている。一部の臨床保留はIND要求を遅延或いは一時停止する一部の臨床仕事のみである。例えば、特定のプロトコルまたはプロトコルの一部が継続されることは許可されず、他のプロトコルはそうすることができる。臨床保留或いは一部の臨床保留を実施した後30日を超えない後、FDAはスポンサーに棚上げ根拠に関する書面解釈を提供する。臨床棚上げや一部の臨床放置を発表した後,FDAがスポンサー調査が継続可能であることを通知した後にのみ,調査を回復することができる。FDAは、スポンサーによって提供された情報に基づいて、上述した欠陥が修正されたかどうかを決定するか、またはFDAを満足させる、すなわち調査を継続することができる。FDAはまた臨床試験開始後に試験の臨床休止或いは一部の臨床休止を行うことができる。
27
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
スポンサーは選択できるが,必要ではなく,IND下で外国臨床試験を行う。IND下で海外臨床試験を行う場合,放棄しない限り,FDAのすべてのIND要求を満たさなければならない。国外の臨床試験がINDの下で行われない場合、スポンサーは試験がFDAのいくつかの法規要求に符合することを保証しなければならず、試験をINDまたは上場承認申請の支持として使用しなければならず、このような試験はGCPに従って行われなければならず、独立道徳委員会またはIECによる審査と承認を含み、患者のインフォームドコンセントを得なければならない。最終規則中のGCP要求は臨床研究の倫理基準とデータ完全性基準を含む。FDAの規定は非IND外国の臨床研究に参加する人類患者の保護、及び結果データの質と完全性の確保を助けることを目的としている。これらはまた,IND研究に匹敵する方法で非IND外国学習が行われることを確保するのに役立つ.
また,臨床試験に参加する各機関を代表するIRBは,臨床試験が開始される前に任意の臨床試験の計画を審査·承認しなければならず,IRBは試験を継続的に監視しなければならない。IRBは試験患者に提供する試験案やインフォームドコンセント情報などを審査·承認しなければならない。IRBの運営はFDAの規定に適合しなければならない。臨床試験がIRBの要求に従って行われない場合、あるいは候補製品が患者に与える意外な深刻な傷害に関連している場合、IRB
は、その機関またはその代表機関の臨床試験の承認を一時停止または終了することができる。
さらに、いくつかの試験は、データ安全監視委員会または委員会と呼ばれる試験スポンサーによって組織された独立した合格専門家グループによって監督される。このグループは,グループ保守のみの実験に利用可能なデータへのアクセス権限に基づいて,指定されたチェックポイントで許可を提供できるかどうかを実験する.参加者または患者が受け入れられない健康リスクに直面していると判定された場合、臨床試験の任意の段階で開発を一時停止または終了することができる。Ocuphireは、変化するビジネス目標および/または競合環境に応じて、一時停止または終了の他の理由を提示する可能性がある。
いくつかの臨床試験に関する情報は,そのClinicalTrials.govサイト上で公開伝播するために,特定の時間枠で米国国立衛生研究院(NIH)に提出されなければならない。
NDAを支持するヒト臨床試験
臨床試験は、GCP要求に従って合格した研究者の監督の下でヒト患者または健康ボランティアに研究製品を服用することを含み、すべての研究患者に任意の臨床試験に参加する前に書面でインフォームドコンセントを提供することを含む。臨床試験は書面試験案に基づいて行い,その中に組み入れと排除基準,試験目標,研究参加者に対する試験,安全性モニタリングのためのパラメータ,評価を行う有効性基準を詳細に説明した。
ヒト臨床試験は通常3つの連続段階で行われるが,これらの段階は重なる可能性がある。
| • |
ステップ1それは.この薬剤は、最初に健康なヒト対象、または癌、標的疾患または状態のようないくつかの適応の患者に導入され、その安全性、用量耐性、吸収、代謝、分布、排泄を試験し、可能であれば、その有効性の早期兆候を得、最適な用量を決定する。
|
| • |
第二段階それは.この薬物は限られた患者集団に適用され、可能な副作用と安全リスクを決定し、特定の目標疾患に対するこの製品の治療効果を初歩的に評価し、用量耐性と最適な用量を決定する。
|
| • |
第3段階それは.この薬物は制御された良好な臨床試験においてより多くの患者集団に応用され、通常は地理的に分散した臨床試験地点であり、十分なデータを生成して承認のために製品の有効性と安全性を統計的に評価し、製品の全体的なリスク-利益概況を確立し、製品のラベルに十分な情報を提供する。
|
INDでの活動および状態を詳細に説明する報告書は,少なくとも毎年FDAに提出されなければならない。さらに、以下のいずれかについては、IND安全報告書をFDAに提出しなければならない:
深刻かつ意外な疑わしい副作用;他の研究または動物または体外培養この薬物に暴露された人体は重大なリスクのテストが存在することを表明した;及び方案或いは研究者マニュアルに記載されているのと比較して、任意の臨床上重要な深刻な副作用の疑いの発生率の増加。第1段階、第2段階、および第3段階の臨床試験は、任意の指定された時間内に成功しないか、または全く成功しない可能性がある。さらに、FDAまたはスポンサーは、研究患者が受け入れられない健康リスクに直面していることを発見することを含む、様々な原因に基づいて臨床試験を一時停止または終了することができる。同様に、臨床試験がIRBの要求に従って行われない場合、または薬物が患者に意外な深刻な傷害を与えた場合、IRBは、その機関またはその代表機関の臨床試験の承認を一時停止または終了することができる。FDAは、通常、GCPに適合し、提出された臨床データの完全性を保証するために、1つまたは複数の臨床サイトを検査する。
28
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
スポンサーは臨床試験の設計について特別案評価(SPA)合意を達成することができる。FDAのSPAプログラムは、FDAがある臨床或いは動物研究の提案設計と規模を評価することを可能にし、候補製品の治療効果を決定するための主要な基礎を形成するための臨床試験を含むことによって、FDAの薬物と生物製品に対する審査と承認を促進することを目的としている。臨床試験スポンサーの具体的な要求に基づいて、FDAはこの方案を評価し、スポンサーの方案設計及び科学と監督管理要求に関する質問に答える。FDAの目標は、要請を受けてから45日以内にSPA審査を完了することだ。FDAは、規制部門が研究適応の有効性について製品を承認することを支持するために、試験方案設計の特定の要素(例えば、基準、用量選択、終点および/または計画分析に入る)が受け入れられるかどうかを最終的に評価する。FDAとスポンサー間のSPAに関するすべてのコミュニケーションは、SPAレターまたはスポンサーとFDAとの間の会議記録に明確に記録されなければならない。
FDAはSPAに同意するかもしれないが、SPA協定は製品が承認されることを保証しない。FDAがSPAプロセスで検討されたプロトコルで提案された設計、実行、および分析に同意したとしても、FDAは、場合によってはそのプロトコルを破棄または変更することができる。特に,SPA協定締結時に認識されていない公衆衛生問題,製品の安全性や有効性に関する他の新たな科学的問題,スポンサーが合意した試験案を遵守できなかった場合,あるいはスポンサーがSPA変更要求で提供した関連データ,仮説や情報,あるいは虚偽や漏れが発見された場合には,SPAプロトコルはFDAに対して拘束力がない。
また、SPAプロトコルが最終的に決定された後であっても、SPAプロトコルを修正することができ、このような修正は、上述した場合でなければ、FDAおよびスポンサーが書面でこのプロトコルを修正することに同意しない限り、FDA審査部門に拘束力があるとみなされる。一般的に、そのような修正は研究を改善するためのものだ。FDAは,SPAプロトコルの条項やSPAプロトコルのテーマである任意の研究のデータや結果を解釈するうえで大きな自由度と裁量権を保持している.さらに、FDAがSPAに従ってそのプロトコルを撤回または変更したり、臨床試験から収集されたデータを私たちとは異なる解釈をしたりする場合、FDAは、このデータが規制承認申請をサポートするのに十分ではないと考える可能性がある.
臨床試験と同時に,会社は通常追加の動物研究を完成させなければならず,薬物化学や物理特性に関する追加情報
を開発して,cGMP要求に応じて商業的に量産される製品のプロセスを最終的に決定しなければならない。製造プロセスは一貫して高品質の候補薬物ロットを生産することができなければならず、その中で最終薬物の識別、強度、品質、純度と効力をテストするための方法を開発しなければならない。また,適切な包装を選択·試験し,候補薬物が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
アメリカ食品医薬品局に秘密保持協定を提出します
成功に必要な臨床試験や他の要求,臨床前研究や臨床試験の結果,製品の化学,製造,制御,提案されたラベルなどに関する詳細な情報がセキュリティプロトコルの一部としてFDAに提出され,医薬製品の1つまたは複数の適応への使用の承認が求められると仮定する。連邦法によると、ほとんどのNDAの提出には申請使用料を支払う必要がある。承認されたNDAのスポンサーは処方薬計画の年間費用も支払う必要がある。一部の費用については、孤児に指定された薬物の申請料例外、および特定の小企業の免除のようないくつかの例外および免除がある。*FDAは、NDAを受信してから60日以内に予備審査を行い、74社によってスポンサーに通知されるこれは…。FDAが提出材料を受信した翌日に、申請が十分に完全であるかどうかを決定し、実質的な審査を可能にする。FDAはNDAの届出を受けるのではなく、より多くの情報を提供することを要求することができ、
スポンサーは届出拒否通知を受信する。この場合、申請は追加情報と共に再提出されなければならない。再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。提出された申請が受け入れられると、FDAは深い実質的な審査を開始する。FDAはNDA審査中のいくつかの業績目標に同意した。多くの標準出願の審査目標は提出日から10カ月以内に審査を行うことであるが,“優先審査”製品については,審査目標は提出後6カ月以内に審査を行うことである。FDAは、最初の提出後にFDAによって発見された突出した欠陥
を解決するために、出願人によって提供された新しい情報を考慮して、または明確にするために、審査手続きを延長することができる。
29
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
セキュリティ協定を承認する前に、FDAは、通常、製品を生産しているか、または生産する1つまたは複数の施設を検査する。これらの承認前検査(“PAI”)は、薬品成分製造(例えば、活性薬物成分)、完成薬品製造および制御検査実験室を含むNDA提出に関連するすべての施設をカバーすることができる。FDAは、製造プロセスおよび施設がcGMP要件に適合していることを決定し、商業規模で要求された規格に従って製品を生産することを保証するのに十分でない限り、申請を承認しないであろう。さらに、NDAを承認する前に、FDAは、GCPに適合することを確実にするために、通常、1つまたは複数の臨床場所を検査する。
また,承認の条件として,FDAは申請者にリスク評価と緩和策(“REMS”)の策定を要求する可能性がある。REMSは、製品の
収益が潜在リスクよりも大きいことを保証するために、リスク最小化戦略を使用する。REMSは、薬物ガイドライン、医療専門家の医師コミュニケーション計画、および安全使用を確保する要素、またはETASUを含むことができる。ETASUは、処方または調剤に限定されないが、特殊なトレーニングまたは認証、特定の場合にのみ調剤、特殊な監視、および患者登録簿の使用を含むことができる。FDAが製品の使用に関連する深刻なリスクを認識した場合、それは承認または承認後にREMSを要求する可能性がある。REMSへの要求は製品の潜在的な市場と収益力に大きな影響を与える可能性がある。
FDAは新薬の申請を諮問委員会に提出したり、なぜこのような推薦がないのかを説明することができる。通常,諮問委員会は臨床医や他の科学専門家を含む独立した専門家からなるグループであり,申請を審査,評価し,申請を承認すべきかどうか,どのような条件でアドバイスを提供すべきかを担当する。FDAは諮問委員会の提案に制限されていないが、それは決定を下す時にこれらの提案を慎重に考慮するだろう。
迅速チャネル、画期的な治療、優先的な指定の検討
FDAは、特定の製品を指定して迅速な審査を行う権利があり、これらの製品が、深刻または生命に危険な疾患または状態の治療において満たされていない医療ニーズを解決することを目的としている場合、迅速チャネル指定、突破的治療指定、および優先審査指定と呼ばれる。
特に、FDAは、深刻なまたは生命に危険な疾患または状態を治療するために1つまたは複数の他の製品と単独でまたは組み合わせて使用することが意図されている場合、迅速な検討のための製品を指定することができ、そのような疾患または状態を満たす満たされていない医療需要の可能性を示す。Fast Track製品については,スポンサーがFDAとより多くのインタラクションを行う可能性があり,FDAは申請完了前にFast Track製品申請の
部分の審査を開始する可能性がある.スポンサーはまた、残りの情報を提出するスケジュールを提供しなければならず、FDAはこのスケジュールを承認しなければならず、スポンサーは適用された使用料を支払わなければならない。
しかし、FDAがFast Track申請を審査する期間目標は、提出申請の最後の部分まで開始される。また,FDAが臨床試験中に出現したデータがFast Track指定を支持しなくなったと考えると,FDAはこの指定を撤回する可能性がある。
第二に、1つの製品が単独で、または1つまたは複数の他の製品と組み合わせて、深刻なまたは生命を脅かす疾患または病状の治療のために使用され、予備臨床証拠が、製品が1つまたは複数の臨床的に重要な終点で既存の治療法よりも実質的に改善されている可能性があることを示している場合、例えば、臨床開発早期に観察された顕著な治療効果が示されている場合、製品は突破的療法として指定することができる。FDAは、開発過程全体でスポンサーとの会議を行うこと、製品スポンサーに開発と承認に関する提案をタイムリーに提供すること、より多くの上級者を審査過程に参加させること、および審査チームに学際的なプロジェクト担当者を割り当てることを含む画期的な治療法に対して何らかの行動をとる可能性がある。
30
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
第3に、FDAは、製品が重篤な疾患を治療し、承認された場合、安全性または有効性の面で顕著な改善を提供する製品を優先的に検討することができる。FDAはケース別に,他の既存療法と比較して推奨されている製品が有意な改善を示しているかどうかを確認した。顕著な改善は,ある疾患の治療の有効性の増加,治療を制限する産物反応の除去あるいは大幅な減少,記録されている患者のコンプライアンスの向上が重篤な結果の改善を招く可能性があること,および新亜群の安全性と有効性の証拠である可能性が示唆された。優先的に指定された目的は、全体的な関心とリソースをこのような申請の評価に誘導し、FDAがマーケティング申請に行動する目標を10ヶ月から6ヶ月に短縮することである。
FDAの秘密保持協定に関する決定
FDAによるNDAの評価および付帯情報によると、製造施設の検査結果を含めて、FDAは承認状または完全な返信を発行する可能性がある。承認書は、製品の商業マーケティングを許可し、特定の適応に関する具体的な処方情報を提供する。完全な応答文は、一般に、提出中の不足点を概説し、FDAが出願を再検討するために、多くの追加のテストまたは情報を必要とする可能性がある。NDAを再提出する際に、これらの不足点がFDAによって満足的に解決された場合、FDAは承認状を発行する可能性がある。FDAは、含まれる情報タイプに依存して、そのような再提出を2または6ヶ月以内に検討することを約束している。これらの追加情報を提出しても、FDAは最終的にその申請が承認された規制基準を満たしていないと決定する可能性がある。
FDAが製品を承認する場合、製品承認の適応を制限する可能性があり、製品ラベルに禁忌症、警告または予防措置を含むことを要求し、承認後の薬物の安全性をさらに評価するための第4段階の臨床試験を含む承認後の研究を要求することを要求し、製品の商業化後に製品を監視するための試験および監視計画を要求するか、またはREMSを含む他の条件を適用するか、REMSを含む他の条件を適用することは、製品の潜在的な市場および収益性に大きな影響を与える可能性がある。FDAは発売後の研究或いはモニタリング計画の結果に基づいて、製品の更なるマーケティングを阻止或いは制限することができる。承認後、承認された製品の多くのタイプの変更、例えば、新しい適応の追加、製造変更、および追加のラベル宣言など、さらなるテスト要件およびFDA
審査および承認を受けなければなりません。
承認後に要求する
FDAによって生産または流通を許可された薬品は、FDAの普遍的かつ持続的な監督管理を受けなければならず、その中には、記録保存、定期報告(例えば、前3年の年間報告および四半期安全報告)、製品サンプリングと流通、広告および販売促進、および製品不良反応の報告に関連する要求が含まれている。承認された後、新しい適応や他のラベル宣言を追加するなど、承認製品の多くの変更は、FDAの審査と承認を事前に受ける必要があります。いずれの市販製品に対しても、継続的な使用料要求、及び臨床データを有する補充アプリケーションの新規アプリケーション料金を提供する。
また,薬品メーカーや他の生産·流通承認薬品に参加する実体は,FDAや州政府機関にその機関を登録し,FDAやこれらの州機関からcGMP要求に対する定期的な抜き打ち検査を受けなければならない。製造プロセスの変更は厳しく規制されており,通常FDAが事前に承認して実施する必要がある。FDAはまた、cGMPとのいかなる偏差も調査·是正し、スポンサーやスポンサーが使用を決定する可能性のある任意の第三者メーカーに報告および文書要求を行うことを要求している。そのため、メーカーはcGMPコンプライアンスを維持するために、生産と品質管理に時間、お金、労力をかけ続けなければならない。
承認後、監督管理の要求と基準を満たしていない場合、あるいは製品の発売後に問題が発生した場合、FDAは承認を撤回する可能性がある。
後に製品に以前に未知の問題があることを発見し、意外な深刻さや頻度の不良事件、あるいは製造技術、あるいは規制要求を遵守できなかった場合、新しい安全情報を追加するためにbr承認のラベルを改訂することを招く可能性がある;発売後の研究または臨床試験を実施して新しい安全リスクを評価するか、あるいはREMS計画に従って流通またはその他の制限を実施する。他の事項を除いて、他の潜在的な結果は以下のことを含む
31
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| • |
製品の販売や製造を制限し、市場から製品を完全に撤回したり、製品をリコールしたりする
|
| • |
承認後の臨床試験には罰金、警告状、一時停止を科す
|
| • |
FDAが承認すべきNDAまたは承認されたNDAの補充剤を承認することを拒否するか、または製品ライセンス承認を一時停止または停止または停止すること;
|
| • |
製品を差し押さえたり、差し押さえたり、製品の輸出入を許可することを拒否したりする
|
| • |
民事または刑事処罰を禁令または適用する。
|
FDAは市場に投入された製品のマーケティング、ラベル、広告、販売促進を厳格に規制している。承認された適応に基づいて承認されたラベルの規定に基づいて薬物を普及させるしかない。すべての宣伝材料は、最初の使用前にFDAに提出されなければなりません。FDAや他の機関は、ラベル外用途の普及を禁止する法律法規を積極的に実行しており、ラベル外用途の普及に不適切が発見された会社は重大な責任を負う可能性があります。
そのほか、処方薬製品の流通は“処方薬販売法”(PDMA)の制約を受け、この法案は連邦一級で薬品サンプルの流通を監督し、各州の薬品サンプル流通業者に対する登録と監督管理に最低基準を設定した。PDMAも州法も処方薬製品サンプルの配布を制限し,配布におけるbrの責任を確保することが求められている。
第五百五十五条第二項新開発区
多くの新薬製品のNDAは2つの十分かつ良好な制御に基づく臨床試験であり,これらの試験は新薬の安全性と有効性を提案する大量の証拠を含まなければならない。これらの出願はFDCA第505(B)(1)条に基づいて提出された。しかしながら、FDCA第505(B)(2)条によれば、FDAは、代替タイプのNDAを承認することを許可される。このようなタイプの出願は、出願人が同様の製品に対するFDAの安全性および有効性の以前の発見、または発表された文献に部分的に依存することを可能にする。具体的には、第505条(B)(2)条は、薬物が安全に有効であるか否かを示す医薬品の秘密協定に適用され、出願によって承認された出願人に依存しており、“出願人又は出願人のために行われているのではなく、出願人が調査を行っている者又はそのために調査を行っている者から参考又は使用権を得ていない”とされている
したがって,第505条(B)(2)条の認可FDAは,非出願人が開発したセキュリティ及び有効性データに基づいてNDAをある程度承認する。第505条(B)(2)条は、FDAが新しいまたは改善された処方または以前に承認された製品を承認するための別の可能性のあるより迅速な方法を提供することができる。第505条(B)(2)の出願人がFDAの以前の承認に依存して科学的に適切であると判断できる場合、出願人は、新しい製品のいくつかの臨床前研究または臨床試験の必要性を除去することができる。FDAはまた、承認された製品からの変更をサポートするために、追加の研究または測定を行うことを企業に要求する可能性がある。次に、FDAは、参照製品のすべてまたは一部が承認されたラベル適応および第505(B)(2)節の出願人が求めた任意の新しい適応について新薬候補を承認することができる。
後発薬の略新薬申請
1984年、FDCAに対するHatch-Waxman修正案の採択に伴い、国会はFDAが法規NDA(Br)条項に従って以前に承認した薬物と同じ模倣薬を承認することを許可した。後発薬の承認を得るためには,申請者はこの機関に短い新薬申請,あるいはANDAを提出しなければならない。このような応用を支持するために、後発薬メーカーは以前に機密協定によって承認された薬品による臨床前と臨床
テストに依存することができ、参考発売薬物或いはRLDと呼ばれる。
32
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
具体的には,ANDAを承認するためには,FDAは通常,後発薬が有効成分,投与経路,剤形,使用条件,薬物強度においてRLDと同様であることを発見しなければならない。FDAはまた、模倣薬と革新薬が生物学的同等性を有することを確定しなければならない。この法規によると,模倣薬は生物的にRLDと同等でなければならない。
ANDAが承認された後,FDAはその出版物“承認された治療同等性評価を有する医薬製品”(オレンジブックとも呼ばれる)で,この後発薬がRLDと治療的に同等であるかどうかを指摘している。臨床医と薬剤師は通常,同量の後発薬を治療することはRLDを完全に代替できると考えられている。また、ある州の法律と多くの医療保険計画の実施により、FDAが指定した治療同等性は、処方臨床医や患者が知らない場合、あるいはその同意を得ない場合に後発薬の代替を招くことが多い。
Hatch−Waxman修正案によれば、FDAは、RLDの任意の適用可能な非特許固有期間が満了するまでANDAを承認しない可能性がある。FDCAは新しい化学実体を含む新薬に5年間の非特許データ排他性を提供した。本条項の場合、新しい化学物質またはNCEは、FDAが任意の他のNDAで以前に承認された活性部分を含まない薬剤を意味する。活性部分は薬物物質の生理的あるいは薬理作用を担う分子またはイオンである。このようなNCE排他性が付与されている場合、ANDAは、提出された文書に第4項の証明が添付されていない限り、5年の満了前にFDAに出願を提出することができず、この場合、出願人は、元の製品の承認後4年以内に出願を提出することができる。
FDCAはまた、NDAが1つまたは複数の新しい臨床研究(バイオアベイラビリティまたは生物学的同等性研究を除く)の報告を含み、
が出願人または出願人のために行われ、承認出願に重要である場合、3年間の排他性があると規定している。この3年間の専門期間は、新たな臨床研究を行う使用条件(S)に適用され、通常、新しい剤形、投与経路、組み合わせまたは適応など、以前に承認された医薬製品の変化を保護する。新たな臨床研究の法定要求を満たしていれば,先に承認された活性部分を含む医薬製品は3年間の独占経営権を得ることになる。5年間のNCE排他性とは異なり,3年間の排他的裁決は,FDAがオリジナル薬物製品の承認を受けた日からその薬物の後発薬を承認するANDAを求めることを阻止しなかった。
Hatch-Waxman特許認証と30ヶ月の滞在
NDAまたはその付録が承認された後、NDAスポンサーは、出願人の製品または承認された製品使用方法をカバーすることを必要とする各特許をFDAにリストしなければならない。NDAスポンサーに列挙された各特許は、オレンジブックに発表される。ANDA出願人がFDAに出願する場合,出願人はオレンジブックに記載されている参照製品に関する任意の特許をFDAに証明しなければならないが,ANDA出願人が承認を求めていない使用方法の特許は除外される。第505条(B)(2)条の出願人が承認された製品の検討に依存する場合,出願人は,ANDA出願人と同程度であるオレンジマニュアルに記載されている任意の特許をFDAに証明しなければならない。
具体的には、出願人は、各特許証明について、(1)必要な特許情報がまだ提出されていないこと、(2)特許が満了していること、(3)記載されている特許が満了していないが、特定の日に満了し、特許が満了した後に承認を求めること、または(4)に記載された特許が無効であり、強制的に実行できないか、または新製品の侵害を受けないこと、について証明しなければならない。
新製品が承認された製品の上場特許又はそのような特許を侵害しないか、又は強制的に実行できない認証を第4項認証と呼ぶ。出願人が列挙された特許に異議を唱えていない場合、または特許使用方法の承認を求めていないことを示す場合、ANDAまたは505(B)(2)出願は、参照製品を要求するすべての特許が期限切れになるまで承認されないであろう(出願人が承認を求めていない適応に関連する使用方法特許は除く)。
33
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
ANDA出願人が第4項の認証をFDAに提供した場合、FDAがANDA届出を受けた後、出願人はまた、NDA及び特許所有者に第4項の認証の通知を送信しなければならない。そして、NDA及び特許所有者は、第4項の認証の通知に対して特許侵害訴訟を提起することができる。第4項の認証を受けてから45日以内に特許侵害訴訟を提起すると、第4項の通知、特許満了又は侵害事件においてANDA出願人に有利な裁決を受けてから30ヶ月まで、FDAがANDAを承認することは自動的に阻止される。
第505条(B)(2)の出願人が承認された製品の研究に依存する場合,出願人はオレンジブックに記載されているいずれかの承認された製品の特許をFDAに証明しなければならず,証明の程度はANDA出願人と同じである。したがって、オレンジブックに記載された参照製品の任意の非特許(例えば、新しい化学物質の承認を得る排他性)が期限切れになり、第4項の認証およびその後の特許侵害訴訟の場合には、訴訟和解または侵害事件において条項505(B)(2)出願人に有利な裁決まで、条項505(B)(2)秘密協定の承認が保留されるまで、参照製品のすべての列挙された特許が満了するまで放置することができる。
505(B)(2)および米国NCEデータ独占性
米国では、“ハッジ·ワックスマン法”は、505(B)(2)の申請によって承認された第1の申請者に、規制部門の新たな適応、用量、または既存の薬剤の強度のような規制部門の承認を求める3年間の米国内非特許データ独占期間を提供する。この3年間の専門権は,新たな臨床研究に関連する使用条件のみをカバーしており,原活性物質を含む薬物のANDAのFDA承認は禁止されていない。この条項によれば、老眼、散瞳または角膜屈折手術後に暗い(中視または低)光条件下で視力が低下したPSを治療するために使用されるPSは、“ハッジ·ワックスマン法”に基づいて3年間のデータ独占権を得ることができる。
米国では、“ハッジ·ワックスマン法案”は、新しい化学物質を含む新薬に5年間の非特許データ排他期を提供している。本条項の場合、新しい化学物質またはNCEは、FDAが任意の他のNDAで以前に承認された活性部分を含まない薬剤を意味する。活性部分は薬物物質の生理的あるいは薬理作用を担う分子またはイオンである。このようなNCE排他性が付与された場合、ANDAは、提出された書類に第4項の証明が添付されていない限り、5年の満了前にFDAに提出することができず、この場合、出願人は、元の製品の承認から4年後に出願を提出することができる。
小児科研究と排他性
2003年の“小児科研究公平法”によれば、セキュリティプロトコルまたはその付録は、すべての関連する小児科亜群において医薬製品が主張する適応の安全性および有効性を評価するのに十分なデータを含み、製品に対して安全かつ有効な各小児科亜群の用量および投与をサポートしなければならない。2012年の食品·薬物管理局安全·革新法案(FDASIA)の公布に伴い、スポンサーはデータを評価する前に小児科試験計画を提出しなければならない。これらの計画は、試験目標および設計、任意の延期または免除請求、ならびに法規要件の他の情報を含む、出願人計画によって行われる提案された小児科試験または研究の概要を含まなければならない。そして,申請者,FDA,FDAの内部審査委員会は提出された情報を審査し,相互に協議し,最終計画について合意しなければならない。FDA
または申請者は、いつでも計画の修正を要求することができる。
FDAは、成人のために製品が使用されるか、または小児科データ要件を完全にまたは部分的に免除するまで、申請者の要求に応じて、または小児科データの一部または全部の提出を延期することを許可することができる。延期要求および延期要求に関する他の要求および手続きは、“連邦延期審査法”に記載されている。法規が別途要求されない限り、小児科データ要求は孤児として指定された製品には適用されない。
34
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
小児科排他性は米国のもう一つの非特許マーケティング排他性であり、付与された場合、任意の特許または規制排他性(孤児排他性を含む)の期間をさらに6ヶ月延長することが規定される。NDAスポンサーから提出された小児科データがこのようなデータに対するFDAの書面要求に公平に応答すれば,この6カ月の排他性を与えることができる。これらのデータは、この製品が研究されている小児科群において有効であることを示す必要はなく、逆に、臨床試験がFDAの要求に公平に応答していると考えられる場合、追加的な保護が付与される。要求された小児科研究報告が法定期限内にFDAに提出され、FDAによって受け入れられた場合、製品の最新の法定または規制独占または特許期間は6ヶ月延長される。これは特許期間の延長ではないが、FDAが別の出願を承認できない規制期間を効果的に延長する。
孤児薬の指定と排他性
孤児医薬品法によれば、1つの医薬製品が、まれな疾患または疾患の治療を意図している場合(一般に、米国で影響を受けている個人が200,000人未満であることを意味する場合、または米国で疾患または疾患を治療するための医薬製品の開発および製造のコストが製品の販売から回収されることが合理的に期待できない場合)、FDAは、医薬製品を“孤児薬物”として指定することができる。会社は秘密協定を提出する前に孤立製品指定を申請しなければならない。この要求が承認された場合、FDAは、治療薬の識別情報およびその潜在的用途を開示するであろう。孤立製品指定は、規制審査や承認過程で何のメリットも伝達されず、持続時間を短縮することもない。
孤児状態を有する製品が、そのような指定された疾患または状態を有するFDAの最初の承認を得た場合、製品は、通常、孤児薬物排他性を得るであろう。孤児薬物
排他性とは,FDAがある限り限られた場合を除いて,7年以内に同一製品の同一適応を承認できない他の任意の出願を意味する。競争相手は,この孤立製品が排他的な
指示によって異なる製品の承認を得る可能性があり,同一製品であるが異なる指示によって承認される可能性がある.孤立製品に指定された薬物や薬物製品が最終的に上場承認され,その適応範囲がその孤立製品申請で指定された範囲を超えていれば,独占経営権を得る権利がない可能性がある。
特許期限の回復と延長
“ハッジ·ワックスマン法”によると、新薬製品を有する特許は、製品開発およびFDA規制審査中に失われた特許期間が最長5年間の特許回復を可能にする限られた特許期間延長を得る資格がある可能性があると主張している。承認された回復期は、通常、IND発効日とNDA提出日との間の時間の半分であり、NDA提出日と最終承認日との間の時間を加える。特許期間回復は特許の残存期間の延長には利用できず,製品承認日から合計14年を超える。承認された医薬製品に適用される特許は1つのみ延期する資格があり、延期出願は関連特許が満了する前に提出されなければならない。承認を求める複数の薬物を含む特許は、そのうちの1つが承認された場合にのみ延期される。米国特許商標局は、FDAと協議した後、任意の特許期限の延長または回復の出願を審査および承認する。Ocuphireは,任意の米国特許に関連する任意の特許期間を延長することを保証することはできず,獲得すれば,その任意の候補製品に関する延長期限を保証することはできない。
EUによる薬品の審査と承認
アメリカ国外で任意の製品をマーケティングするために、会社はまた他の国/地区と司法管轄区域の品質、安全性と有効性及び製品に対する臨床試験、マーケティング許可、商業販売と流通などの方面の多くの異なる監督管理要求を守らなければならない。製品がFDAの承認を得るかどうかにかかわらず、Ocuphireは相応の外国の監督管理機関の必要な許可を得て、それからこれらの国或いは司法管轄区で臨床試験或いは製品マーケティングを開始することができる。承認の流れは最終的に国と司法管轄区域によって異なり、追加の製品テストと追加の行政審査期間に及ぶ可能性がある。他の国や管轄地域で承認を得るのに要する時間は、FDAの承認を得るのに要する時間とは異なり、さらに長くなる可能性がある。1つの国/地域または司法管轄区で規制承認を得ることは、別の国または管轄区域で規制承認を得ることは保証できないが、1つの国/地域または司法管轄区で監督管理の承認を得ることができなかったり、遅延したりすることは、他の国または司法管轄区の監督管理プロセスに悪影響を及ぼす可能性がある。
35
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
EU薬品審査手続き
欧州臨床試験指令によると、EUはすでに加盟国の国家立法を通じて臨床試験承認制度を実施した。この制度によれば,申請者は臨床試験を行うEU加盟国の主管国当局の承認を得なければならない。また,出願人は主管倫理委員会が賛成の意見を発表した後にのみ臨床試験を開始することができる。臨床試験申請はヨーロッパ臨床試験指令と加盟国の相応国家法律で規定されている支持情報を持つ研究用薬品ファイルを添付し、適用された指導文書の中でさらに詳細に説明しなければならない。
EU規制制度に基づいて製品の上場承認を得るためには,出願人は集中または分散したプログラムに従ってマーケティング許可申請またはMAA
プログラムを提出しなければならない。中央手続きは欧州委員会によってすべての欧州連合加盟国に有効な単一マーケティング許可を付与することを規定している。特定の製品の場合、集中手順は強制的であり、特定のバイオテクノロジープロセスによって製造された薬品、孤児薬品として指定された製品、高級治療製品、および特定の疾患を治療するための新しい活性物質を含む製品を含む。他の疾患を治療するための新しい活性物質を含む製品、および高度に革新的な製品または患者に有利な集中プロセスについては、集中プロセスが任意である可能性がある。
集中プログラムにより、ヨーロッパ薬品管理局(EMA)に設置された人用薬品委員会(CHMP)が製品の初歩的な評価を担当している。CHMPはまた、既存のマーケティング許可の修正または拡張を評価するなど、いくつかの許可後および保守活動を担当する。欧州連合の中央手続きによると、重大な影響評価の最長期限は210日であり、タイミングポーズは含まれておらず、申請者は“気候変動管理計画”の質問に答えるために、補充資料または書面または口頭解釈を提供しなければならない。特殊な場合、公衆衛生の観点、特に治療革新の観点から見れば、1種の医薬製品は重大な価値があり、CHMPは加速評価を与える可能性がある。この場合,環境保護局は150日以内にCHMPの意見を出すことを確保している。
複数のEU加盟国で製品を販売することを希望する申請者は、分散したプログラムを使用することができ、これらの国の製品は、以前にどの欧州連合加盟国でもマーケティング承認を受けたことがない。分権手続きは、出願人によって指定された1つの加盟国(参照加盟国と呼ばれる)による出願の評価を、1つまたは複数の他のまたは関連加盟国によって承認することを規定する。この手続きによれば、出願人は、製品特性概要草案、ラベル、および包装チラシ草案を含む、同じロールおよび関連材料に基づいて、参照加盟国および関連加盟国に出願を提出する。会員国が有効な申請を受けてから210日以内に評価報告書の草稿と関連材料の草稿を作成することを参考にする。各関係加盟国は、加盟国の評価報告及び関連材料を参考にした日から90日以内に、評価報告及び関連材料を承認するか否かを決定しなければならない。
もし加盟国が公衆の健康に深刻な危害を及ぼす可能性があることを理由に評価報告と関連材料を承認できない場合、論争点は紛争解決メカニズムの制約を受け、最終的にEU委員会に提出される可能性があり、その決定はすべての加盟国に対して拘束力がある。
この枠組みの中で,メーカーは指令2001/83/EC第10条(3)項に基づいて混合薬用製品の承認を求めることができる。混合応用は参考製品からの情報とデータ、及び適切な臨床前試験と臨床試験からの新しいデータにある程度依存する。提案された製品が模倣薬の厳密な定義に適合していない場合、またはバイオアベイラビリティ研究が生物学的同等性を証明するために使用できない場合、または模倣薬の活性物質(S)、治療適応、強度、薬形または投与経路が参照薬と比較して変化する場合には、そのような出願が必要である。この場合、試験および試験の結果は、2003/63/EC号命令によって改訂された2001/83/EC号命令添付ファイルに要求されるデータ含有量基準に適合しなければならない。
参照製品がこのプログラムを通じて発売されることが許可された場合、混合医薬製品アプリケーションは自動的に集中プログラムに入ることができる。参照製品が分散プロセスによって許可された場合、申請者が医薬製品が重大な治療、科学的または技術革新を構成していることを証明する場合、または医薬製品のコミュニティ許可がコミュニティ一級患者の利益に適合することを承認する場合、集中的な手順の下での審議のための混合出願を受け入れることができる。
36
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
EU臨床試験が承認された
EUで臨床試験を行う要求は、良好な臨床実践或いはGCPを含み、臨床試験指令2001/20/ECとGCP指令2005/28/ECに規定されている。改正された第2001/20/EC号指令と第2005/28/EC号指令に基づき、EU加盟国の国家立法によりEU臨床試験承認制度が実施された。この制度によると、試験を計画している各EU加盟国の主管国当局の承認を得なければならない。そのためには,研究用医薬品アーカイブやIMPDと,2001/20/EC号指令と2005/28/EC号指令および他の適用可能なガイドライン文書に規定されているさらなる支援情報が必要なCTAを提出しなければならない。また,主管倫理委員会がその国の臨床試験申請に積極的な意見を発表した後にのみ,臨床試験を開始することができる。
2014年4月,EUは新たな臨床試験条例(EU)第536/2014号を採択した。新しい“臨床試験条例”は“臨床試験指令”の代わりに、EUの現有の医療製品の臨床試験法規に対して徹底的な改革を行い、新しい臨床試験許可調整プログラムを含み、これは薬品発売許可の相互認識プログラムを想起させ、スポンサーが臨床試験結果を公表する義務を増加させた。臨床試験条例の施行は延期された。“臨床試験指令”は2022年末に新しい“臨床試験条例”に代替される可能性がある。新しい臨床試験条例はEUの臨床試験の審査を簡略化と簡素化することを目的としている。この条例の主な特徴は、単一の入口点であるEUポータルによって申請手続きが簡略化されたこと、申請のために準備されて提出された単一の文書、および簡略化された報告手続きによって、スポンサーが各機関および異なる加盟国にそれぞれ実質的に同じ情報を提出する必要がないようにすること、臨床試験申請評価の統一的な手順が2つの部分に分けられること(第1の部分はすべての関連加盟国によって共同評価され、第2の部分は各関連加盟国によって個別に評価される)である。臨床試験申請評価の最終期限を厳格に規定し,倫理委員会は関係加盟国の国家法律に基づいているが,“臨床試験条例”で規定されている全体的な時間範囲で評価手続きに参加している。
授権期間と継続期間
マーケティング許可は原則として5年間の有効期間であり、マーケティング許可は5年後にEMAまたはライセンス加盟国の主管当局によるリスク-収益バランスの再評価に基づいて更新することができる。このため、マーケティング許可所有者は、マーケティング許可を付与してから導入されたすべての変化を含む品質、セキュリティ、および有効性に関するファイルの統合バージョンをEMAまたは主管当局に提供しなければならず、少なくとも6ヶ月前にマーケティング許可が無効になる前に。一旦更新されると、上場許可の有効期限は無期限であり、欧州委員会又は主管当局が薬物警戒に関する正当な理由に基づいて5年間の継続を決定しない限り、5年間の継続が決定される。認可が失効した後3年以内に薬品を実際にEU市場(集中手続きであれば)または加盟国市場のいかなる許可も許可されていない(いわゆる日没条項)。
EUにおけるデータと市場排他性
EUでは、新しい化学実体はマーケティング許可を得た後に8年間のデータ独占経営権を獲得する資格があり、他の2年間の市場独占経営権を持っている。このようなデータ排他性が付与された場合、EUの規制機関は、8年以内にイノベーターのデータを参照して非特許(簡略化)出願を評価することができず、その後、非特許マーケティング許可を提出することができ、イノベーターのデータ
は参照することができるが、2年以内に承認を得ることはできない。この10年前の8年間に、マーケティング許可保持者が1つまたは複数の新しい治療適応の許可を得、許可前の科学的評価において、これらの適応が既存の療法と比較して有意な臨床的利益をもたらすと考えられる場合、10年全体の期間は最大11年に延長される。1つの化合物が新しい化学実体であると考えられ、スポンサーが所定のデータ独占期間を得ることができても、別の会社はその製品の別のバージョンを販売することができ、その会社が薬物試験、臨床前試験および臨床試験の完全なデータベースを含む完全なMAAを完成できることを前提とし、その製品の発売許可を得ることができる。
37
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
孤児薬の指定と排他性
第141/2000条に規定されている薬物は、その発起人が証明できることを条件として、その発起人が、生命又は慢性衰弱にかかわる疾患の診断、予防又は治療に使用され、その影響がヨーロッパ共同体の万分の5以下の人、又は生命にかかわる疾患の診断、予防又は治療を目的としていることを証明することができる。ヨーロッパ共同体には深刻な虚弱または深刻な慢性疾患が存在し、インセンティブがなければ、その薬剤の欧州共同体での販売は、必要な投資が合理的であることを証明するために十分な見返りをもたらす可能性が低い。上記の2つの場合のいずれについても、申請者は、欧州共同体が好ましい診断、予防または治療に関連する方法が許可されていないことを証明しなければならない、または、そのような方法が存在する場合、薬物は、その疾患の影響を受けている人に大きな利益をもたらすであろう。
規制847/2000は欧州連合で孤児薬を指定する基準と手続きを規定している。具体的には、孤児製品に指定された申請は、製品発売承認申請を提出する前の任意の時間に提出することができる。孤児薬の発売許可は10年間の市場排他期をもたらすだろう。しかし、5年目の終了時に、製品が指定された孤児薬の基準を満たしていないと判定された場合、例えば、製品の利益が十分に高く、市場排他性が合理的であることを証明するのに不十分である場合、この期間は6年に短縮されることができる。非常に特殊な場合にのみ、マーケティング許可所有者の同意を得る、十分な数量の製品を供給できない、類似医薬製品が“臨床関連優位性”を示す、あるいは委員会が孤児薬品を審査した後、加盟国が市場独占期5年目に提出した要求(認定基準がもはや適用されない場合)など、市場独占経営権を撤回することができる。第141/2000条例により孤児薬に指定された医薬製品は,孤児薬の研究,開発,供給を支援するために,欧州共同体と加盟国から提供される奨励を受ける資格がある。
営業許可後の規制要件
米国と同様に、医薬製品の販売許可保持者とメーカーは、生産と販売許可を承認する前と後に、欧州薬品管理局とEU個別加盟国主管当局の全面的な監督管理を受けている。例えば、医薬製品のEU上場許可保持者はEU薬物警戒立法及びその関連法規とガイドラインを守らなければならず、これらの法規とガイドラインは薬物警戒或いは評価と医療製品の安全性のモニタリングに対して多くの要求を提出した。EUの医薬製品の製造過程も厳格に監督管理されており、監督管理機関は規定に合わないと思う製造施設を閉鎖する可能性がある。製造には製造許可が必要であり、製造許可保持者は適用されるEU法律に規定されている各種要求を遵守しなければならず、医薬製品と活性薬物成分を製造する際にEU cGMP標準を遵守することを含む。
EUでは、承認された製品の広告および販売促進は、医薬製品販売促進、臨床医師との相互作用、誤解性、および広告および不公平な商業行為を比較するEU加盟国の法律によって制限される。さらに、個別のEU加盟国が採択した他の立法は、医薬製品の広告や販売促進に適用される可能性がある。これらの法律は、医薬製品に関連する宣伝材料および広告が、主管当局によって承認された製品特性要約またはSmPCに適合することを要求する。SmPCに適合しない医薬製品の普及はラベル外の普及を構成していると考えられ,EUでは禁止されている。
38
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
医療改革
アメリカの医療保健業界は、製薬業界を含み、厳格な監督管理を受け、常に重大な変化が発生している。連邦または州政府が米国内での医療保健の提供または資金調達方式を変更するためのいかなる重大な努力もOcuphireの業務に実質的な影響を与える可能性がある。現在、“医療·教育和解法案”(“ACA”と総称する)によって改正された2010年の“患者保護·平価医療法案”は、医療業界に影響を与え、引き続き大きな影響を与える先駆的な立法である。ACAは未加入個人の医療保険カバー範囲
を拡大するとともに,医療サービスの総コストを抑えることを目的としている。ACAは立法、行政命令、そして司法挑戦の改革を受けてきた。例えば、2017年の減税および雇用法案には、ACAが1年の全部または一部で合格医療保険を維持できなかった個人に対する税金ベースの分担責任支払いを廃止する条項が含まれており、これは一般に“個人強制”と呼ばれている。また、2020年総合支出法案は、ACAがある高コスト雇用主が支援する医療保険に徴収する“キャデラック”税と、非免除医療機器に徴収される医療機器消費税を完全に廃止し、健康保険会社税を廃止した。2018年の両党予算法案(“BBA”)はACAを改正し、Medicare Dに参加する一部の製薬メーカーが不足している販売時点割引を50%から70%に引き上げ、Medicare薬物計画のカバーギャップを縮小し、一般に“ドーナツ穴”と呼ばれている。インフレ削減法案(IRA)によると、このカバーギャップは2025年1月1日から解消される。IRAはまた、Medicare Part D受益者が初期保証段階にある時にブランド、生物製品と生物類似製品の交渉価格の10%を支払い、Medicare Part D保証の壊滅的な段階で交渉価格の20%を支払うことを製薬業者に要求した。2021年6月17日、米国最高裁判所はいくつかの州がACAに対して提出した最新の司法挑戦を却下したが、この法律の合憲性を具体的に裁くことはなかった。最高裁の将来の行動,その他のこのような訴訟,バイデン政府の医療改革措置がACAにどのように影響するかは不明である。
ACAにおけるOcuphireの潜在的候補薬に影響を与える条項は以下の通りである
| • |
特定の政府医療計画におけるこれらのエンティティの市場シェアに基づいて、孤児適応のために許可された特定の製品の販売には適用されないにもかかわらず、指定されたブランドの処方薬および生物製剤を製造または輸入する任意のエンティティに徴収される差し引くことのできない特別な費用
|
| • |
医療補助計画の資格基準を拡大し、他を除いて、各州が連邦貧困レベル133%以下の収入を有するある個人に医療補助を提供することを許可し、それによってメーカーの医療補助返点責任を潜在的に増加させる
|
| • |
ブランドと模倣薬の最低税金還付を高めることによって、外来処方薬の価格を計算と報告するための医療補助薬品還付の定義を修正し、税金還付責任を連邦医療保険優勢計画に参加する個人の処方に拡大し、医療補助薬品リベート計画(MDRP)下でのメーカーのリベート責任を拡大した;br}はMDRP下でのメーカーのリベートは吸入、輸液、点滴、インプラント或いは注射の薬物に対して計算される新しい方法を解決した
|
| • |
340 B薬品割引計画に適合する実体タイプを拡大した
|
| • |
患者を中心とした新しい結果研究所は、監督、優先事項を決定し、臨床治療効果の比較研究を行い、このような研究に資金を提供する
|
| • |
連邦医療保険と医療補助サービスセンター(“CMS”)内に連邦医療保険と医療補助革新センターを設立し、革新的な支払いとサービス交付モデルをテストして、連邦医療保険と医療補助のbr支出を低減し、処方薬支出を含む可能性がある。
|
ACAを廃止し、代替する可能性のある条項を含む、より多くの立法変化があるかもしれない。将来的にACA条項を廃止し、代替する任意の潜在的立法の時間および範囲は多くの点で非常に不確実であるが、研究ベースの製薬部門に不利ないくつかのACA条項もACAカバー範囲の拡大に伴い廃止される可能性がある。
39
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
ACAが公布されて以来、アメリカはまた他の立法改正を提案し、採択した。2011年8月には、他の事項を除いて、“2011年予算制御法案”が国会支出削減措置を制定した。赤字削減合同特別委員会の任務は、2013年から2021年までの間に少なくとも1.2兆ドルの赤字削減を提案することであるが、同委員会は必要な目標を達成できず、いくつかの政府プロジェクトの立法の自動削減を触発した。これには、2013年4月に施行された各年度に提供者に支払われる連邦医療保険総額が2%減少することが含まれており、後続の立法改正により、追加の国会行動が行われない限り、2032年度の自動減額令の前6ヶ月以内に有効に維持される(一時停止、その後一時的に減少し、新冠肺炎大流行期間中に2022年7月1日に満了する)。
また、処方薬や生物製品価格の上昇を受けて、米国政府は薬品定価のやり方の審査を強化した。このような審査はいくつかの国会調査を招き、連邦と州立法を提出し、公布し、製品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、br政府計画製品の精算方法を改革することを目的とした。例えば、2021年3月11日、総裁·バイデンは2021年の米国救援計画法案に署名し、ACAによって2021年と2022年の補助金を取得する資格のある個人への保険税免除援助を一時的に増加させ、保険料控除を受ける資格がある税収控除に本来適用されていた400%の連邦貧困レベル制限を撤廃した。アイルランド共和軍はこの増加した税金免除援助と400%の連邦貧困制限を2025年に延長した。
州レベルでは、立法機関は、価格または患者の精算制限、割引、いくつかの製品参入の制限、およびマーケティングコスト開示および透明性措置を含む、医薬品および生物製品の価格設定を制御するための法規を立法し、実施しており、場合によっては、他の国からの輸入および大量購入を奨励することを目的としている。例えば、いくつかの州の法律要件brは、関連法規によって決定されたいくつかの価格設定のハードルを超える値上げおよび新製品発表状況を州機関および/または商業調達業者に開示する。その中のいくつかの法律は政府職員たちがまだ明確にしていない曖昧な要求を含む。法律とその実施の不明確性を考慮して、私たちの報告行為は関連連邦と州の法律法規の処罰条項によって制限される可能性がある。一部の州はまた、消費者と支払者に負担能力に挑戦する可能性のある高コスト処方薬製品を決定し、このような製品をコスト審査し、場合によってはこのような製品に支払い上限を適用する処方薬負担能力委員会を設立した。
ACAの全部または一部を修正または廃止するか、または新しい医療立法を実施することを含む政策変化は、医療システムに重大な変化をもたらす可能性があり、これは、収入を創出し、利益を達成することができ、または私たちの薬品を商業化することを阻止するかもしれない。私たちは将来、より多くの州と連邦医療改革措置を取ることが予想され、いずれも連邦と州政府が医療製品およびサービスのために支払う金額を制限する可能性があり、これは私たちの候補製品の需要の減少や定価の低下、あるいは追加の価格設定圧力をもたらす可能性がある。
医療詐欺と乱用およびコンプライアンス法律
他にも医療に関する詐欺や濫用やコンプライアンス法があり,Ocuphireなどの製薬会社の運営方式が広く規範化され,薬品に関する活動が規範化されている。このような法律法規は施行されるために行政指導が必要かもしれない。規定を遵守しないことは、巨額の罰金および/または処罰を含む可能性がある法律および/または行政行動に直面する可能性があり、違反活動の停止を命じること、刑事告発、警告状、製品のリコールまたは差し押さえ、製品の承認遅延、政府の精算計画または契約から除外され、適用司法管轄区域での業務が制限される可能性がある。
連邦と州の医療法律と法規には
| • |
個人および実体がインフォームドコンセントおよび故意の場合に現金または実物で直接または間接的に報酬brを請求、提供、受け入れ、または提供することを禁止し、個人の転転または購入を誘導または奨励し、任意の商品またはサービスを注文または推薦することができ、これらの商品またはサービスは、連邦医療保険および医療補助などの連邦医療計画に従って全部または部分的に支払いを行うことができる刑法である連邦反控除法規。個人や実体は、法規や法規違反の具体的な意図を実際に知る必要がなく、違反行為を実施することができる。連邦反リベート法違反は、重大な民事罰金と刑事罰金を招き、監禁と監禁は政府医療計画から除外される可能性がある
|
40
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| • |
連邦民事虚偽請求法は、民事通報者または準訴訟によって強制的に執行され、個人または実体に重大な民事処罰、3倍の損害賠償、および政府医療計画から除外される可能性があるbr個人または実体を故意に連邦政府に提出するか、または連邦政府に虚偽または詐欺的な支払い請求を提出させることをもたらすか、または虚偽記録または陳述を連邦政府への支払いの義務に実質的な影響を与えるか、または連邦政府への支払いの義務を故意かつ不当に回避、減少または隠蔽することができる。また,連邦反リベート法規違反は連邦民事虚偽クレーム法案に基づいて責任を負う根拠とすることができる.連邦民事虚偽請求法案のように、連邦政府に虚偽、架空または詐欺的なクレームを提出または提出した人に刑事責任を課す連邦刑事虚偽クレーム法案もある
|
| • |
連邦民事通貨罰法は、以下の活動に従事するエンティティに実質的な民事罰金を科すことを許可する:(1)インフォームドコンセント、br、または要求通りに提供されていないサービスまたは他の任意の方法で虚偽または詐欺のサービスのクレームを引き起こすことをもたらす;(2)連邦医療保健計画から除外された個人またはエンティティと契約を締結して、連邦医療計画によって精算可能な項目またはサービスを提供する;(3)連邦反リベート法規に違反する。(四)既知の過払いの払戻を申告していない者
|
| • |
連邦支払い透明性追跡および報告要求は、Medicare、Medicaidまたは児童健康保険計画などに従って支払われた薬品、装置、生物製品、および医療用品のいくつかのメーカーが、米国衛生公衆サービス部(HHS)内のCMSに毎年追跡して追跡し、米国に登録医(医師、歯科医師、検眼士として定義され、米国衛生公衆サービス部(HHS)内のCMSに報告することを要求する)連邦医師支払い陽光法案と呼ばれることを要求する。足科医師と脊椎マッサージ師)、医師アシスタント、勤務看護師、臨床看護師専門家、麻酔科医アシスタント、登録看護師麻酔科医、登録助産師と教育病院、及び医師及びその直系親族が持つ所有権と投資権益。すべての支払い、価値譲渡および所有権、または投資利益に必要な情報をタイムリーかつ正確かつ完全に提出できなかった場合、民事罰金を招く可能性がある
|
| • |
1996年“連邦健康保険携行性と責任法案”(HIPAA)は、詐欺の任意の医療福祉計画(任意の第三者支払人を含む)の計画を実行または実行しようとする行為に刑事と民事責任を適用し、医療福祉計画を故意かつ故意に流用または窃取し、医療保健違法行為に対する刑事調査を故意に阻害し、および重大な事実を故意に偽造、隠蔽または隠蔽し、あるいは任意の重大な虚偽、架空または詐欺的な陳述または陳述、または医療福祉、プロジェクトまたはサービスに関連する虚偽陳述を行う。連邦反リベート法規と同様に、個人や実体は法規や法規違反の特定の意図を実際に知る必要がなく、違反を実施することができる
|
| • |
2009年に“衛生情報技術促進経済·臨床衛生法”改正された“HIPAA”は、他のほか、共同保健取引における情報電子交換、及び個人が健康情報を識別できるプライバシーと安全に関する基準の統一的な基準を採用することが規定されている。これらの基準は、このような情報を保護するために行政、物理、および技術保障措置を要求する。また、多くの州では、健康情報のプライバシーやセキュリティ問題を解決するための同様の法律が公布されており、いくつかの法律はHIPAAよりも厳しい。このような法律を遵守しないことは重大な民事と刑事処罰をもたらす可能性がある
|
41
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| • |
州法律は、処方薬価格詐欺を禁止すること、または国が“高コスト”と考えているいくつかの薬品に支払い上限を設定すること、および値上げに理由を提供する情報を含むいくつかの価格情報を報告することを要求する
|
| • |
同様の州および外国の法律、例えば州反リベートおよび虚偽請求法は、非政府第三者支払者(個人保険会社を含む)によって精算される医療項目またはサービスの販売またはマーケティング手配およびクレームに関連することに適用される可能性があり、一部の州法律は、製薬会社に製薬業界の自発的コンプライアンスガイドラインおよび連邦政府が発行した関連コンプライアンスガイドラインを遵守することを要求し、また、医薬品メーカーに医師および他の医療保健提供者への支払いまたはマーケティング支出に関する情報を報告することを要求する。
|
| • |
また、承認されれば、私たちの製品は連邦医療保険(Medicare)の保険を受ける資格がある可能性があり、連邦医療保険は高齢者と障害者に医療福祉を提供する連邦医療計画であると予想されます。具体的には、私たちの製品は主に連邦医療保険Dによって部分的に精算され、この部分は連邦医療保険受益者に外来処方薬福祉を提供する予定です。連邦医療保険D部分は,個人保険計画と連邦政府との契約手配に基づいてこの計画によって実施されている。個人健康保険で提供される薬品保険と同様に、D部分は処方を計画し、使用制御(例えば事前許可、手順治療、数量制限)を実施し、薬品メーカーと割引を協議する。だからこそ,D部分計画がカバーする処方薬リストは計画によって異なる。しかしながら、制限された例外を除いて、法規制は、個人計画が、いくつかの治療カテゴリおよびカテゴリの薬物または生物学的製品をカバーし、各独自の治療カテゴリまたはカテゴリに少なくとも2つの薬剤を含むことを必要とする。私たちの製品はまた他の政府計画の下でカバーと精算を得ることができます。以下に議論する計画を含む
|
| • |
MDRPは製薬業者が衛生と公衆サービス部部長と国家税金還付協定を締結し、発効することを要求し、各州が医療補助患者に提供するメーカー外来薬物の連邦マッチング資金を獲得する条件としている。MDRPによると,メーカーは外来で使用する製品数(一部の例外を除く)のために州ごとの医療補助計画にリベートを支払わなければならず,これらの製品は医療補助受益者に配布され,州医療補助計画によって支払われなければならない。MDRP返却点は,公定式,州報告の利用率データ,定価データを用いて計算され,これらのデータはメーカーが月と四半期ごとに計算してCMSに報告する。これらのデータは、AMPと、単一ソースおよびイノベーティブ多源製品の場合の各薬剤の最適価格とを含む。
|
| • |
340 B医薬品定価計画は、参加する製造業者が、法定定義された保証エンティティに、製造業者が外来医薬品の340 B“最高価格”を超えない費用を請求することに同意することを要求する。br}これら340 B保証エンティティは、連邦合格健康センター、ライアン·ホワイトHIV/エイズ計画受給者、いくつかのタイプの病院および専門診療所、および低所得患者に不比例なサービスを提供するいくつかの病院を含む、特定の連邦称号または特定の連邦計画援助を受ける医療組織を含む。ACAは340 B計画を拡大し、ある児童病院、ある独立した癌病院、肝心な通路病院、ある農村転院センターとある唯一のコミュニティ病院も含め、各病院はACAによって定義されている。340 B上限価格は、MDRPによって計算された保険外来薬のAMPおよびリベート金額に基づく法定式を使用して計算され、一般に、MDRPに拘束された製品も340 B上限価格計算および割引要求によって制約される。Medicaid AMP
の定義やMedicaid返却金額の任意の変更も,我々の製品の340 B最高価格計算に影響を与える可能性があり,我々の運営結果に悪影響を与える可能性がある.
|
42
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| • |
連邦法律はさらに、ある会社がMDRPとMedicare Part B計画の下で連邦資金を使用してその製品を支払う資格があり、ある連邦機関と被贈与者によって購入され、退役軍人事務部(“VA”)連邦供給スケジュール(“FSS”)定価計画に参加しなければならないことを要求した。参加するためには、メーカーは退役軍人管理局と任意の保険薬品についてFSS契約とその他の協定を締結しなければならない。これらの合意によると、製造業者は、1992年の“退役軍人医療保健法案”(VHCA)603節に規定された連邦法定最高価格(FCP)である四大連邦機関である退役軍人管理局、国防総省(DoD)、公衆衛生サービス(インド衛生サービスを含む)、および沿岸警備隊にこのような製品を提供しなければならない。FCPは加重平均
非連邦平均メーカー価格(“非FAMP”)に基づいており、メーカーは四半期ごとと毎年退役軍人管理局にこの価格を報告することを要求されている。
|
| • |
連邦医療計画下の価格報告や返金支払い義務を遵守できなかった行為は、私たちの財務業績にマイナスの影響を与える可能性がある。もし私たちが知らずに政府に虚偽の価格情報を提出したことが発見された場合、私たちの平均販売価格を報告する際に虚偽の陳述をしたことが発見された場合、または必要な価格データをタイムリーに提出しなければ、民事罰金を適用することができる。このような行為はまた、他の連邦法(例えば、“虚偽申告法”)によって規定される他の潜在的責任に根拠を提供することができる。
|
医療費精算
医療保健提供者と第三者支払人は監督管理の許可を得た薬品を推薦と発行する上で主要な役割を果たしている。もし私たちの潜在的な候補薬物が監督部門の承認を得たら、製品の商業化の成功は第三者支払人から得られ、十分な精算レベルを維持することにある程度依存する可能性があり、これらの支払人は商業健康保険会社、管理型医療保健組織、およびMedicareとMedicaidのようなアメリカの政府医療計画を含む。しかし,米国の医療業界や他の地域では,コスト抑制が増加傾向にある。
最近、政府機関は薬品メーカーとディーラーがどのようにその製品の価格を設定するかについてより厳格な審査を行った。これは,国会の調査や他の提案·公布の立法を招き,(I)製品定価の透明性の向上,(Ii)薬品や他の医療製品のカバー範囲や精算の制限,および(Iii)医療システム全体での政府医療計画の精算改革を目指している。提案された改革措置は米国議会が立法を通過して発効する必要があるが、総裁·バイ登政府と米国議会はそれぞれ、薬品コストを抑制するために新たな立法および/または行政措置を求め続けると表明している。
例えば、2021年3月11日、総裁·バイデンは“2021年米国救援計画法案”に署名し、その条項には、ACAにおける製薬業者のMDRPによる還付責任を制限する条項の日没が含まれている。ACAによると、メーカーのリベート責任は以前カバーされていた外来薬の平均メーカー価格の100%に制限されている。2024年1月1日から、メーカーのMDRPリベート責任はもはや
に上限がなく、これはメーカーが支払うMDRPリベートがある保険外来薬を販売する際に得られるリベートよりも多くなる可能性がある。それは.また、2022年10月14日、バイデン総裁はアメリカ人の処方薬コストの低減に関する行政命令を発表し、衛生·公衆サービス部部長にCMS革新センターテストのために新しい医療支払いと交付モードを選択するかどうかを考慮することを指示し、そして連邦医療保険と医療補助計画に参加する受益者が革新的な薬物療法を獲得することを促進した。2023年2月14日、衛生·公衆サービス部は、CMS革新センターによるテストを含む2022年10月14日の行政命令に応答する報告書を発表した。具体的には、(1)D部分スポンサーが“高価値br}薬品リスト”のモデルを構築することを許可し、いくつかのよく見られる模倣薬の最高共付額を2ドルに設定することに関する。(2)CMS、製造業者と州医療補助機関との間にパートナー関係を確立し、いくつかの細胞および遺伝子治療薬の結果に基づく多州合意を生成する医療補助に重点を置いたモデル、および(3)連邦医療保険B部分承認計画薬の支払い金額を加速させるモデルを調整して、新しい療法の開発を推進する。
43
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
また、2022年8月16日、総裁·バイデンは“アイルランド共和軍”に署名し、法律にした。IRAには、(I)2025年からMedicare Part Dの受益者の自己負担費用に2,000ドルの上限を設定すること、(Ii)Medicare Part D内のすべての薬品に対して新たな製造業者割引義務を実施すること、(Iii)米国政府がCMSと価格交渉過程を行った後、Medicare Part BおよびDがカバーする固定数の薬品および生物製品のための“最高公平価格”を確立することを可能にする条項が含まれている。(4)企業にインフレよりも薬品価格の上昇のために医療保険にリベートを支払うことを要求し、CMSは2023年6月30日に、2023年から2024年の間に行われた第1回価格交渉の要求とパラメータを詳細に説明し、2026年に発効した“最高公平価格”条項に拘束された製品を実施するステップを採用し、2023年8月29日に、価格交渉を受けた10種類の薬品の予備リストを発表した。2023年11月17日、特定の製造業者を決定する方法
が、適用製品の割引がMedicare Part Dメーカー割引計画によって要求される段階段階よりも低くなることを決定する方法について概説し、2023年12月14日に、2024年1月1日から2024年3月31日までの間にIRAのインフレリベート条項に基づいて共通保険料率を調整した48種類のMedicare Part B製品を含むリストを発表する。これらの薬品の価格設定措置がどのようにもっと広範な製薬業界に影響するかはまだ観察が必要である。Ireland共和軍が私たちの業務と医療産業全体に及ぼす影響はまだ明確ではない。
さらに、米国個別州も、精算制限、割引、特定の製品参入の制限、マーケティングコスト開示および透明性措置を含む薬品の価格設定を制御するための法規を法律および実施することによってますます積極的になっており、場合によっては、他の国からの輸入および大量調達を奨励するメカニズムも含まれている。
人的資本資源
Ocuphireは2023年12月31日までに14名のフルタイム従業員を有し,任務は以下のとおりである:3名が臨床研究開発活動に従事し,そのうち1名が博士号を有し,br}5名が研究開発活動および業務開発と財務に従事し,6名が財務,人的資源,行政支援に従事している。Ocuphireは引き続き専門家コンサルタントと契約組織を利用して日常業務の実行を支援する予定である.Ocuphireのすべての従業員は労働組合代表もなく、集団交渉合意のカバー範囲もない。
Ocuphireはそれが従業員と良い関係を維持していると思っている。
利用可能な情報
その会社のインターネットアドレスはWwwwwo.ocupire.comそれは.我々は、米国証券取引委員会に電子的にアーカイブまたは米国証券取引委員会に文書を提出した後、合理的で実行可能な範囲内で、我々の年間報告書(Form
10-K)、Form 10-Q四半期報告、Form 8-K現在の報告、および1934年証券取引法第13(A)または15(D)節に基づいて提出または提出された報告修正案を、我々の投資家関係サイトir.ocupire.com/米国証券取引委員会を通じてできるだけ早く提供する。我々のサイトに含まれる情報は本報告の一部ではなく,参考に本報告にも組み込まれていない.
| 第1 A項。 |
リスク要因
|
私たちの証券に投資するのは高いリスクがある。投資の前に、あなたは以下のリスクと不確実性と今年度の報告書の他の情報を慎重に考慮しなければならない。本文で述べたいかなるリスクと不確定性も、私たちの業務、経営結果、財務状況に重大な悪影響を与える可能性があり、更に私たちの証券の取引価格或いは価値に重大な不利な影響を与える可能性がある。私たちが現在知らないことや私たちが現在把握している情報に基づいてどうでもいいと思う他のリスクも私たちに大きな悪影響を及ぼす可能性がある。したがって、あなたは投資の全部または一部を失うかもしれない。
44
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
私たちの候補製品の商業化と開発に関するリスク
私たちは製品ラインの成功に大きく依存しています。私たち(または私たちの戦略パートナー)がRYZUMVIの商業化やAPX 3330やPSの開発および商業化を十分に実現できなければ、私たちの業務は深刻な被害を受けるだろう。
著者らの業務はAPX 3330とフェントラミン目薬“PS”の成功した臨床開発、監督管理の承認と商業化に依存している。Viatrisは我々の戦略パートナーであり、FDAが承認したRYZUMVIの商業化およびPSのさらなる開発と商業化を担当している(FDAが承認されれば)。APX 300はまだ臨床開発段階にある。私たちはAPX 3330、RYZUMVI、PSの開発に多くの精力と財力を投入しており、私たち(または私たちの戦略パートナー)は将来的にAPX 3330、RYZUMVI、PSの開発と商業化に多くのエネルギーと財力を投入したい。私たちやViatrisはRYZUMVIや私たちの候補製品の開発に成功して商業化できないという重大なリスクが残っている。我々はAPX 3330がいつあるいは人体に有効あるいは安全であることが証明されるかどうかを正確に予測することができず、それが発売許可を得るかどうかを正確に予測することもできない。私たちが製品収入を創出する能力は、RYZUMVIの持続的な商業化と、APX 3330およびPS(総称して私たちの候補製品と呼ぶ)のマーケティング承認および商業化に大きく依存する。
薬品の研究、テスト、製造、ラベル、承認、販売、マーケティングと流通はFDAと他の監督管理機関の広範な監督管理を受けており、これらの監督管理機関はアメリカと他の国/地区に異なる監督管理規定が存在する可能性がある。FDAまたは任意の外国国/地域の機密協定の承認を得るまで、これらの国/地域から必要なbr承認を得るまで、米国で私たちの候補製品を販売することは許可されていません。特定の適応候補製品の商業化販売の規制承認を得る前に,この目標適応に適用する候補製品
が安全かつ有効であることを非臨床試験と臨床試験により証明しなければならない。この過程は長年の時間を要する可能性があり、その後、同時に上場後の研究と監督を行う可能性があり、これは私たちがこれまで株式と債務融資を通じて調達した収益ではなく、大量の資源が必要になるだろう。米国で大量に開発されている薬物のうち、一部の薬剤だけがFDA規制承認プロセスを成功させ、商業化されている。したがって、私たちの候補製品の開発とFDA承認を完了することができても、候補製品が承認または商業化され、市場に広く受け入れられているか、または他の商業代替製品よりも有効であることを保証することはできない。私たちがより多くの候補製品の開発と商業化に成功しなかったり、RYZUMVIを商業化し続けることに成功しなければ、私たちのビジネス機会は限られるだろう。APX 3330、RYZUMVI、PSの成功は以下のいくつかの要素の影響を受ける可能性がある
| • |
RYZUMVIの打ち上げ遅延または広範な商業化は困難に直面している(現在、2024年上半期に打ち上げ予定)
|
| • |
生産または臨床試験中、または生産試験または臨床試験の遅延、終了、または多くの予見不可能な事象による;
|
| • |
非臨床的および臨床的研究から候補製品に不利な結果を得た
|
| • |
臨床試験のコストは予想以上だった
|
| • |
患者或いは医学研究者は著者らの臨床試験方案及び参加したい患者の数に従うことを望むかどうか
|
| • |
私たちの候補製品の申請を遅延させ、規制機関に適用されたマーケティングおよび秘密協定の承認を受けた
|
| • |
他の政府または規制の遅延および規制要件、政策およびガイドラインの変化は、規制の承認を得るために追加の臨床試験を行うか、または多くの追加の資源を使用する必要があるかもしれない
|
45
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| • |
RYZUMVIと当社の候補製品の商業数量を第三者メーカーと手配し、適用された規制機関から当社の製造プロセスおよび第三者メーカー施設の規制承認を得る問題
|
| • |
販売、マーケティング、および流通能力を確立し、RYZUMVIおよび私たちの候補製品の商業販売を開始し、承認されれば、単独でも他社とも協力する
|
| • |
RYZUMVIと私たちの製品候補製品に対する患者、医学界、および第三者支払人の受容度;
|
| • |
既存の看護基準を含む他の療法と効果的に競争する;
|
| • |
承認後、RYZUMVIおよび私たちの候補製品の持続的に許容可能なセキュリティ状態を維持する
|
| • |
第三者支払者から保険と適切な補償を受けて維持する
|
| • |
特許と商業秘密保護と規制排他性を獲得して維持する
|
| • |
RYZUMVIと私たちの候補製品に関連する知的財産権の組み合わせにおける私たちの権利を保護し、
|
| • |
私たちは私たちの候補製品に関するより多くのデータの要求を満たすことができる。
|
| • |
さらに、Apexian許可プロトコルによると、Ocuphireは、眼科および糖尿病疾患のためにいくつかの化合物を使用する権利がある。他の非眼科適応では,Ocuphireはこれらの化合物の進行を制御しない。
|
ViatrisはRYZUMVIとPSを世界の主要市場で商業化する独占的なグローバル権利を持っている。Viatrisはこれらの製品を適時に開発したり、商業化したりすることができず、私たちの業務と経営業績に大きな悪影響を与えるだろう。
私たちはViatrisが世界のキー市場でRYZUMVIとPSを独占的に商業化する権利を与えた。また、Viatrisには、米国以外でRYZUMVIとPSを開発する独占的な権利とライセンスを付与しています。次のような要因により,Viatrisとの協力は成功しない可能性がある
| • |
Viatrisは私たちの製品をタイムリーにまたは費用効率的に作ることができないかもしれません
|
| • |
ViatrisはViatrisライセンス契約の下での義務をタイムリーに履行できないかもしれない
|
| • |
Viatrisは私たちの製品を効果的に商業化できないかもしれません
|
| • |
Viatrisは、RYZUMVIまたはPSを米国以外の1つまたは複数の適切な当事者に再許可することができない場合がある
|
| • |
私たちとViatrisとの間の契約紛争や他の相違は、私たちの製品の開発、製造、再許可と商業化、ライセンス契約の解釈、および所有権に関する紛争を含みます。Viatrisは、Ocuphireに90日間の通知を出した後、RYZUMVIとPSのために米国で新たな開発パートナーを選択する可能性がある。
|
46
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
上記のいずれの条項も、Viatris許可協定に従って受け取る資格のある任意の支払いの可能性と時間に悪影響を及ぼす可能性があります。会社はViatrisに依存して私たちの製品の商業化と販売を推進するだろう。ViatrisがViatrisライセンス契約の義務を履行しなければ、これは私たちの業務、運営結果、将来性に大きな悪影響を与え、私たちの株価を下落させる可能性があります
以前の臨床試験結果は未来の結果を予測できない可能性があり、私たちの現在と計画中の臨床試験結果はFDAあるいは非アメリカ監督機関の要求に符合しない可能性がある。
本年度報告で他の場所で検討されたAPX 3330とPSの先行非臨床研究と臨床試験の結果は,必ずしも将来の非臨床研究や臨床試験の結果を予測するとは限らない可能性がある。我々の現在の開発スケジュールに基づいて我々が計画した候補製品臨床試験を完成させることができても,我々が以前候補製品に対して行った臨床試験の結果は将来の試験で複製されない可能性がある。製薬やバイオテクノロジー業界の多くの企業(私たちよりも多くの資源や経験を持つ会社を含む)が早期開発に積極的な成果をあげた後、後期臨床試験で大きな挫折を経験し、類似した挫折に直面しないことは確認できない。これらの挫折の原因には,以前に報告されていない有害事象(“副作用”)を含む臨床試験実施期間中の非臨床発見や臨床試験における安全性や有効性観察がある。そのほか、非臨床と臨床データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの製品
候補製品は非臨床研究と臨床試験で満足できると思っているが、依然としてFDAの承認を得られなかった。もし私たちが私たちの任意の候補製品の臨床試験で十分な有効性と安全性
を反映するのに十分な結果を提供できなければ、私たちの候補製品の開発スケジュール、規制承認と商業化の見通し、およびOcuphireの業務と財務の見通しは不利な影響を受けるだろう。また,Ocuphireの候補製品は追加の第3段階登録試験でそれぞれの主要端末に達していても承認されない可能性がある.FDA或いは非アメリカ監督機関は著者らの試験設計或いは著者らの非臨床研究と臨床試験データの解釈に同意しないかもしれない。さらに、これらの規制当局のいずれも、臨床登録試験案に関するコメントまたは提案を審査して提供した後であっても、候補製品に対する承認要求を変更することができ、この案は、FDAまたは他の規制機関の承認をもたらす可能性がある。
例えば、角膜屈折術後に暗い(中視または低)光線条件下で視力が低下したPSの第3段階研究についてFDAと合意し、APX 3330の承認を支援するためのSPAプロトコルを求める予定であるが、FDAは最終的に承認を得るためにより多くの研究を必要とする可能性がある。
報告によると、FDAのSPAプロセスは、FDAがある臨床或いは動物研究の提案設計と規模を評価することを可能にし、候補製品の治療効果を決定するための主要な基礎を形成するための臨床試験を含むことによって、FDAの薬物と生物製品に対する審査と承認を促進することを目的としている。臨床試験スポンサーの具体的な要求に基づいて、FDAはこの方案を評価し、スポンサーの方案設計及び科学と監督要求に関する質問に答える。FDAの目標は、要請を受けてから45日以内にSPA審査を完了することだ。FDAは、規制部門が研究適応の有効性についてこの製品を承認することを支持するために、試験方案設計の特定の要素を最終的に評価し、例えば、基準、用量選択、終点、および/または計画分析が許容できるかどうかを分析する。FDAとスポンサー間のSPAに関するすべてのコミュニケーションは,SPAレターやスポンサーとFDA間の議事録に明確に記録されなければならない.
FDAがSPAに同意するかもしれないにもかかわらず、SPA協定は製品が承認されることを保証しないと言った。FDAがSPAプロセスで検討されたプロトコルで提案された設計、実行、および分析に同意しても、FDAはそのプロトコルを破棄または変更することができる場合がある。特に,SPA協定締結時に認識されていない公衆衛生問題,製品の安全性や有効性に関する他の新たな科学的問題,スポンサーが合意した試験案を遵守できなかった場合,あるいはスポンサーがSPA変更要求で提供した関連データ,仮定や情報,あるいは虚偽やbrが漏れていることが発見された場合,SPAプロトコルはFDAに対して拘束力を持たない。
47
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
また、SPAプロトコルが最終的に決定された後であっても、上述した場合でない限り、このような修正は、FDAおよびスポンサーが書面でこのプロトコルを修正することに同意すれば、FDA審査部門に拘束力があるとみなされる可能性がある。一般的に、そのような修正は研究を改善するためのものだ。FDAは,SPAプロトコルの条項やSPAプロトコルのテーマである任意の研究のデータや結果を解釈するうえで大きな自由度と裁量権を保持している.さらに、FDAがSPAに従ってそのプロトコルを撤回または変更したり、臨床試験から収集されたデータを我々とは異なる解釈をしたりする場合、FDAは、このデータが規制承認申請をサポートするのに十分ではないと考える可能性がある。
さらに、これらの規制機関は、私たちが要求しているものよりも少ないか、またはbrの高価な発売後の臨床試験の表現によって承認される可能性がある当社の候補製品を承認することもできます。任意の目標適応のための任意の候補製品の商業販売の規制承認を得る前に、我々は、非臨床研究および良好な臨床研究において収集された大量の証拠によって、候補製品が目標適応に使用されることが安全かつ有効であることを証明し、米国の承認について、FDAはこれに満足しなければならない。FDAまたは非アメリカ規制機関が私たちが計画した臨床試験が私たちの任意の適応候補製品を承認するための基礎となるのに十分だと考えることは保証できません。FDAと非アメリカ規制機関は、私たちの臨床試験結果の評価と、結果が私たちの候補製品の安全かつ有効性を証明するかどうかを評価する上で広範な裁量権を持っています。もし私たちが承認前に計画したものではなく、候補製品の臨床試験を要求されたら、私たちは大量の追加資金を必要とするかもしれませんし、このような結果や他の臨床試験の結果が承認されるのに十分であることを保証することはできません。また,我々の現在と計画されている非臨床·臨床試験がFDAや非米国規制機関の要求に適合していなければ,我々の業務は実質的な損害を受ける可能性がある。
患者の臨床試験への参加を募集する上で遅延や困難に遭遇すれば,これらの臨床試験を行って完成する能力,必要な規制承認を求め,獲得する能力は,延期または阻止される可能性がある。
FDAや米国国外の同様の規制機関の要求に応じて、これらの試験に参加するのに十分な数の合格者を見つけることができなければ、私たちまたは未来の協力者は、私たちの候補製品の臨床試験を開始または継続できないかもしれない。患者登録は様々な要素の影響を受ける
| • |
調査中の病気の重症度は
|
| • |
研究中の疾患のための薬物の獲得可能性および有効性が承認された
|
| • |
試験に関する資格基準と探訪スケジュール
|
| • |
他社と競争して条件に合った患者は、同じ適応または患者群の候補製品を治療するための臨床試験を行う
|
| • |
私たちが臨床試験に支払う費用は
|
| • |
研究を受けた製品候補製品のリスクと収益を感知する
|
| • |
臨床試験への参加を促進するために努力しています
|
| • |
医者の患者は治療法を変え
|
| • |
治療中および治療後に患者の能力を十分に監視する;
|
48
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| • |
潜在的患者のための臨床試験場の距離および利用可能性を提供する
|
| • |
患者が任意の突発公共衛生事件の中で臨床試験に参与する能力。
|
私たちの臨床試験に参加するのに十分な数の患者を募集することができない、あるいは私たちの試験が完了した後に十分な登録人数を維持することは、重大な遅延を招き、あるいは1つ以上の臨床試験を完全に放棄する必要があるかもしれない。私たちの臨床試験の登録遅延は私たちの候補製品の開発コストを増加させ、私たちの株価を下落させるかもしれません。
私たちまたは他の人たちは、私たちの候補製品が競争相手の製品と比較して十分な治療効果が不足しているか、またはそれらが以前に発見されなかった副作用brをもたらし、規制部門の承認または商業化を延期または阻止する可能性があることを発見するかもしれない。
APX 3330,PSともに比較的少ない患者群で試験されているため,1日720 mgと0.75%フェントラミン眼液(これは1.0%メタンスルホン酸フェントラミン眼液と同じ)を毎日服用し,持続時間が限られていることから,これまでの臨床試験ではAPX 3330やPSの有意な正面効果が実際の正面効果よりも大きいか,あるいはその開発が進むにつれて,追加的かつ予見不可能な副作用が観察される可能性がある。APX 3330またはPSが十分な治療効果が不足していることが発見された場合、またはそれらが不良副作用(以前に私たちが完成していない臨床試験で決定されなかった副作用を含む)を引き起こす場合、私たちまたは監督機関の臨床試験の中断、延期または停止を招く可能性があり、FDAまたは他の非米国規制機関が任意またはすべての標的適応の規制承認を拒否する可能性がある。
もし私たちの候補製品が十分な治療効果が不足していることが発見された場合、あるいはそれらが以前に発見されなかった副作用を招いた場合、このような候補製品の商業化を阻止し、販売から収入を得ることができるかもしれない。また、もし私たちの候補製品と私たちまたは他の人たちが後で市場の承認を得たら:
| • |
有効性が低いことや候補製品による副作用が発見されました
|
| • |
規制部門は製品の承認を撤回することができる
|
| • |
私たちは製品をリコールし、製品の投与方法を変更し、追加の臨床試験を行うか、または製品のラベルまたは流通(REMSを含む)を変更することを要求されるかもしれない
|
| • |
製品の販売または製造プロセスに追加的な制限を加えることができる
|
| • |
私たちは罰金、禁止、あるいは民事または刑事処罰を受けるかもしれない
|
| • |
私たちは起訴され、患者への傷害に責任を負うかもしれない
|
| • |
製品は競争力を低下させ、売上が低下する可能性がある
|
| • |
私たちの名声は臨床医と患者で普遍的に損なわれるかもしれない。
|
これらのイベントのいずれかまたは組み合わせは、影響を受けた候補製品に対する市場の許容度を達成または維持することを阻止または維持することができ、または候補製品を商業化するコストおよび支出を大幅に増加させる可能性があり、これは、逆に、販売候補製品から相当な収入または任意の収入を得ることを延期または阻止する可能性がある。
49
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
法規要求やFDAガイドラインの変化、あるいは著者らの臨床試験中に発生した意外な事件は、臨床試験方案の変化或いは追加の臨床試験要求を招く可能性があり、これは私たちのコストを増加させ、或いは開発スケジュールを延期する可能性がある。
法規要求やFDAガイドラインの変化、あるいは著者らの臨床試験中に発生した意外な事件は、著者らが臨床試験方案を修正する必要があるかもしれない、あるいはFDAは追加の臨床試験要求を強制的に実施する可能性がある。著者らの臨床試験方案の修正はFDAとIRBsに再提出して審査と承認を行う必要があり、臨床試験のコスト、時間或いは成功達成に不利な影響を与える可能性がある。もし私たちが遅延に遭遇したり、任意の試験を終了したり、あるいは追加の臨床試験を要求されたら、私たちの候補製品のビジネスの将来性が損なわれる可能性があり、私たちが製品の収入を作る能力が延期されるかもしれない。
私たちの候補製品に対する規制機関の任意の計画適応の承認を得られなかった場合、あるいはより多くの候補製品を開発できなかった場合、私たちのビジネス機会は制限されるだろう。
われわれは最初にわれわれの目標適応候補製品DR,逆転薬物誘導散瞳の開発,老眼の治療および角膜屈折術後の暗(中視または低)照明条件下での視力低下に焦点を当てた。RYZUMVIは薬物性散瞳の治療のために承認されている。しかし、承認された後、私たちの候補製品に対する規制部門の任意の他の適応の承認を得ることができるか、または私たちの候補製品を商業化することに成功することを保証することはできません。私たちの1つまたは複数の目標または他の適応候補製品が規制部門の承認または商業化に成功しなければ、私たちのビジネス機会は制限されるだろう。
私たちが規制部門から私たちの候補製品の承認を得たり、商業化に成功したりしても、それらは持続的な規制審査と批判を受けるだろう。この継続的な検討と批判は規制の承認を失う可能性がある。
私たちは他の獲得性もしかしたら候補製品の臨床開発を続けるかもしれない。開発、監督機関の他の候補製品に対する承認を得て、それを商業化して
は私たちが完成した株式と債務融資の純収益のほかに、大量の追加資金が必要であり、しかも薬物製品開発固有の失敗リスクが発生しやすい。私たちは開発過程で他の候補製品を進めることに成功することを保証することはできません。
我々の薬物研究と発見能力は限られており、第三者から候補製品を買収し、追加資本を調達し、あるいは資本の資源を移転して、私たちの候補製品パイプラインを拡大する必要があるかもしれない。
私たちの現在の薬物研究と発見能力は限られている。したがって、候補製品ラインの外に私たちのルートを拡張するには、第三者から可能かもしれない候補製品を購入したり、追加資本を調達したり、資本資源を移転してこのような拡張に資金を提供する必要があるかもしれません。私たちは将来性のあるかもしれない候補製品の買収を求める時に激しい競争に直面し、追加資本を調達できないかもしれない、あるいは会社の他の分野から資本資源を移転する可能性があり、資金減少によって実質的な結果に直面する可能性がある。私たちの多くの競争相手は非臨床試験と臨床試験、監督許可を獲得し、医薬製品の製造とマーケティングにおいて明らかに多くの財務資源とより豊富な経験を持っている可能性があり、そのため、潜在的許可者にとっては、私たちよりも魅力的かもしれない。もし私たちが他の有望な候補製品を得ることができなければ、より多くの資本を集めたり、資本資源を移転したりすることができなければ、私たちの候補製品ルートを拡大できないかもしれません。
もし私たちが他の候補製品を得ることができるならば、そのような許可協定は私たちに様々な義務を課す可能性があり、私たちの許可者は、重大な違約が発生した場合、または場合によっては勝手にその下の許可を終了する権利があるかもしれない。将来のライセンスの終了は、私たちが知的財産権を使用する権利を失う可能性があり、これは、将来の候補製品を開発し、それを商業化する能力に悪影響を与え(承認されれば)、私たちの競争業務の地位と業務の将来性を損なう可能性があります。
50
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
我々は限られた資源をかけて特定の
適応を追求する可能性があり,より利益や成功確率が大きい可能性のある適応を利用することはできない.
私たちの財務と管理資源が限られているため、私たちは現在、私たちの候補製品のための特定の適応を決定する開発計画に集中しています。したがって、
私たちは、他の適応を探す機会を放棄または延期するか、または後に、より大きな商業的潜在的な他の潜在的候補製品を証明する可能性がある。潜在市場の規模を変化させたり、正確に予測できなかったりするため、私たちの資源配分決定は、実行可能な商業製品や利益のある市場機会を利用できない可能性がある。特定の適応または将来の候補製品に対する私たちの研究開発計画への支出は、いかなる商業的に実行可能な製品も生じないかもしれない。もし私たちが候補製品の商業潜在力や目標市場を正確に評価しなければ、私たちはその候補製品の承認や市場承認を得ることができない可能性があり、私たちの業務と財務業績は損なわれるだろう。
私たちの財務状況と追加資本需要に関連するリスク
私たちはどの製品の販売からも何の収入も得ておらず、予測可能な未来に損失が出ることが予想され、永遠に実現したり、利益を維持したりすることはできないかもしれない。
私たちが唯一商業化販売を許可された製品はRYZUMVIであり、RYZUMVIは2024年上半期に私たちの商業化パートナーViatrisによって発売されると予想されます。私たちの候補製品が1つ以上の管轄区域の商業化に必要な規制承認を得ない限り、追加の製品
収入は発生しないと予想されます。私たちが収入を作る能力は私たちの能力を含む多くの要素にかかっています
| • |
RYZUMVIの成功と広範な商業化
|
| • |
私たちの候補製品から有利な結果を得て、これらの適応の追加臨床試験を成功させることを含む、その計画適応の非臨床および臨床開発を完成させる
|
| • |
2つの候補製品の申請を監督部門に提出し、直ちに米国と外国のマーケティング許可を得た
|
| • |
第三者と商業的に実行可能な供給および製造関係を確立し、維持することができ、これらの第三者は、臨床開発を支援するために十分な製品およびサービスを数量および品質で提供することができ、RYZUMVIおよび我々が開発した候補製品に対する市場の需要を満たすことができる
|
| • |
私たちの候補製品を単独でまたは製薬パートナーと米国または他の市場で効果的にマーケティングおよび販売するための販売およびマーケティング能力を確立する
|
| • |
競争製品や技術や市場発展の問題を解決しています
|
| • |
RYZUMVIおよび我々が開発した候補製品は、政府および第三者支払者が顧客および患者に提供する保険および十分な補償を得る
|
| • |
RYZUMVIと私たちの候補製品を市場に承認させます。
|
また、2023年12月31日までの累計赤字は8150万ドルです。私たちは主に私募による約束手形と転換可能な手形を発行し、上場企業になった後に普通株式と引受証を発行し、最近Viatris許可協定に基づいて受け取った費用とマイルストーン支払いによって私たちの業務に資金を提供しています。私たちはほとんどの財政資源と努力を私たちの候補製品の臨床開発に投入した。私たちの1つ以上の候補製品が追加の規制承認を得たと仮定しても、APX 3330は数年かかると予想される[PSと]商業化の準備ができているかもしれませんが、私たちの候補製品は市場の承認や商業成功を得ることができないかもしれません。私たちは製品収入が発生した直後に利益を達成できない可能性があり(あれば)、持続資金なしで運営を継続できない可能性がある。
51
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
私たちの候補製品から利益を得て利益を維持するためには、市場潜在力のある製品を開発し、最終的に商業化しなければならない。これは私たちが一連の挑戦的な活動の中で成功することを要求し、非臨床試験と臨床試験を完成し、候補製品の監督管理許可、製造、マーケティングと販売を獲得し、いかなる規制承認を得、任意の
発売後の要求を満たす薬物を含む。私たちはこのような活動に関連した巨額の費用が発生すると予想している。私たちはほとんどこのような活動の初期段階にいる。私たちはこのような活動で決して成功しないかもしれないし、たとえ私たちが成功しても、私たちは利益を達成するのに十分な大きさや十分な収入を生むことができないかもしれない。
もし私たちの候補製品が確かに利益を達成したら、私たちは年間収益性を維持したり向上させることができないかもしれない。私たちは候補製品から利益を得たり、利益を維持することができず、私たちの価値を下げ、資金を調達し、研究開発努力を維持し、業務を拡大し、あるいは運営を継続する能力を弱める可能性がある。
私たちの比較的短い経営歴史は、投資家が私たちの業務のこれまでの成功度を評価することを困難にし、私たちの未来の生存能力を評価することも困難になるかもしれない。
私たちは臨床段階の会社で、これまで、私たちの業務は組織と当社を配備した会社、業務計画、資金調達、私たちの候補製品の開発に限られていました。私たちは、ビジネス規模の製品を生産したり、製品の商業化に成功するために必要な販売やマーケティング活動を展開する能力があることを証明していません。
しかも、投資家が私たちの業務と将来性を評価するための運営履歴はありません。私たちのような臨床段階の会社への投資は、固有に多くのリスクの影響を受けている。これらのリスクおよび困難は、(A)累積損失、(B)経営履歴の長い会社に比べて、業務戦略を策定および評価する時間が比較的限られていることによる不確実性、(C)将来の製品の販売開始に必要な法規を遵守すること、(D)臨床、製造、分析実験室作業、非臨床、監督、商業化、または他の活動に依存することを含む正確な財務計画上の挑戦を含む。(E)企業に資金を提供すること、および(F)本明細書に記載された他のリスク要因の挑戦を迎えること。私たちは投資家が私たちの業績を評価するための経営履歴がありません;したがって、私たちは新しい業務の設立と発展に関連するすべてのリスクを受けています。持続可能または利益のあるビジネスを実現するために、予定されたスケジュールに従って私たちの計画を実現できる保証はありません。
金融サービス業に影響を与える不利な事態は、私たちの現在と予想されている業務運営および私たちの財務状況や運営結果に悪影響を及ぼす可能性があります。
私たちが必要か適切だと思う方法に基づいて私たちの銀行関係を評価しているにもかかわらず、私たちが現在および予想されている将来の業務運営に資金または資本化を提供するのに十分な資金源や他の信用手配の機会を得ることは、私たちと直接手配されている金融機関または金融サービス業全体または全体の経済に影響を与える深刻な被害を受ける可能性があります。これらの要因は、流動性制限または失敗、様々な金融、信用または流動性プロトコルまたは手配に従って義務を履行する能力、金融サービス業または金融市場の中断または不安定、または金融サービス業会社の将来性に対する懸念または負の予想を含むことができる。これらの要因は、我々と財務や業務関係にある金融機関や金融サービス業界会社に関連する可能性があるが、金融市場や一般金融サービス業界に関連する要因も含まれている可能性がある。1つまたは複数のこれらの要因に関連するイベントまたは問題の結果は、私たちの現在および予想されるビジネス運営、ならびに私たちの財務状態および運営結果に生じる様々な重大かつ不利な影響を含むことができる。これらは、以下を含むことができるが、これらに限定されない
52
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| • |
預金または他の金融資産または未加入預金または他の金融資産の損失の取得を遅延させる;
|
| • |
既存の循環クレジット手配または他の運営資金源を得ることができず、および/または払い戻し、展示期間または満期日の延長、または新しいクレジット手配または他の運営資金源に入ることができない
|
| • |
潜在的または実際に契約義務に違反して、私たちに手紙または信用または他の信用支援手配を維持することを要求する;または
|
| • |
現金管理スケジュールの終了および/または遅延獲得または実際の損失現金管理スケジュールによって制約された資金。
|
さらに、米国または国際金融システムに対する投資家の懸念は、より高い金利またはコスト、より厳しい財務および運営契約、または信用および流動性源を得るための体系的な制限を含む、あまり有利ではない商業融資条項を招く可能性があり、それにより、私たちがより受け入れ可能な条項や受け入れ可能な条項で融資を受けないようにする。利用可能な資金または現金および流動資金源の任意の減少は、他のリスクに加えて、運営費用、財務義務、または他の義務を履行する能力に悪影響を及ぼす可能性があり、私たちの財務および/または契約義務の違反、または連邦またはbr州賃金および労働法違反をもたらす可能性がある。上記の任意の影響、または上記要因または他の関連または同様の要因に起因する任意の他の影響は、我々の流動資金および私たちの現在および/または予想される業務運営および財務状態および運営結果に重大な悪影響を及ぼす可能性がある。また、マクロ経済や金融サービス業のいずれのさらなる悪化も、私たちと業務往来のある当事者との損失や違約を招き、さらに、私たちの現在および/または予想される業務運営および運営結果および財務状況に重大な悪影響を及ぼす可能性がある。例えば、私たちと業務を展開している側は、期限が切れたときに支払うことができず、私たちとの合意に基づいて違約、破産、または破産を宣言する可能性があります。私たちのいかなる取引相手のいかなる破産や資本も債務不履行、または満期時に支払うことができなかったり、いかなる重大な関係を失っても、私たちの重大な損失を招き、私たちの業務に重大な悪影響を及ぼす可能性がある。
私たちは未来に多くの追加資本が必要になるだろう。もし追加的な資金がなければ、私たちは運営を延期、減少、または停止しなければならないだろう。
私たちは私たちの候補製品と運営のさらなる開発に資金を提供し続けるために追加の資金を調達する必要があるだろう。私たちの将来の資本需要は大きく、多くの要素に依存するかもしれない
| • |
私たちの候補製品を研究と開発し、著者らの非臨床研究と臨床試験の範囲、規模、進捗、結果とコストを開始し、完成させる
|
| • |
私たちの候補製品を米国および他の国/地域でさらなる市場承認を得るための努力のコスト、時間、および結果は、私たちの候補製品のための準備とNDAのFDAへの提出、および他の国/地域のFDA関連要求および規制要件を満たすことを含む
|
| • |
私たちが開発または獲得した任意の他の候補製品の数量および特徴(あれば)
|
| • |
私たちが有利な条件で協力を確立し維持する能力があれば
|
| • |
もし私たちの製品候補が発売承認されたら、商業販売からの収入(もしあれば)
|
53
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| • |
マーケティング承認を得た場合、私たちの候補製品を商業化することに関連するコストは、販売およびマーケティング能力の開発、またはマーケティングおよび販売のための戦略的協力のためのコストおよび時間を含む
|
| • |
私たちの候補製品や商業化に成功した製品を作るコストは
|
| • |
特許主張の提出、起訴、執行、規制届出提出のコストなど、一般企業活動に関連するコスト。
|
変化する環境は私たちの現在の予想よりも資本消費の速度を大きく速めるかもしれない。いかなる臨床試験の結果も高度に不確定であるため、著者らは著者らの候補製品の開発、監督管理許可と商業化に成功した実際の金額を合理的に推定することができない。私たちが必要な時、追加的な融資を受けることができないかもしれないし、私たちに有利な条項で融資を受けることができないかもしれない。また、有利な市場条件や戦略的考慮により、現在または将来の運営計画を実行するのに十分な資金があると考えても、追加の資本を求めることができる。もし私たちが十分な資金をタイムリーに得ることができなければ、私たちは戦略パートナーが見つからない限り、私たちの候補製品を開発し続けたり、私たちの候補製品を商業化することができないかもしれない。
54
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
世界経済と社会の不安定または不利なグローバル経済状況は、私たちの収入、財務状況、または運営結果に悪影響を及ぼす可能性がある。
グローバル経済の健康、特に株式と信用市場の健康、そして私たちの社会構造の安定は、私たちの業務と経営業績に影響を与えている。例えば、現在の欧州と中東の衝突、中国不動産などの分野の負の傾向、および講じられた対応は、株式や信用市場に悪影響を及ぼす可能性がある。もし株と信用市場が不利なら、私たちは必要な時や優遇条件でより多くの資金を集めることができないかもしれない。私たちのサプライヤーと開発パートナーは、財務的困難に遭遇したり、お金を借りてその運営に資金を提供できない可能性があり、これは、彼らが私たちの製品を購入したり、適時に支払う能力に悪影響を及ぼす可能性があります(もしあれば)。国際貿易紛争を含め、疲弊したり低下したりする経済的または政治的中断は、私たちのメーカーやサプライヤーに圧力を与え、供給中断を招く可能性があり、あるいは私たちの顧客に潜在的な製品の支払いを延期させる可能性があります。また、最近のサプライチェーン中断や労働力不足や持続的なインフレのような不利な経済状況は、私たちのサプライヤーがメーカーに材料やコンポーネントを提供する能力に悪影響を与え続ける可能性があり、これは私たちの業務に負の影響を与える可能性があります。これらの経済状況は私たちの将来の業務活動を正確に予測して計画することを難しくしている。
また、世界経済の全般的な減速は、景気後退、または特定の地域や業界の減速、米国との貿易パートナーとの貿易緊張の激化、インフレや信用市場の引き締めを含み、いずれも私たちの業務、財務状況、流動性に悪影響を及ぼす可能性がある。不利な世界経済状況は、時々私たちの経営の業界や市場の著しい減速を招いたり、悪化させたりして、私たちの業務や経営業績に悪影響を与えています。マクロ経済の疲弊と不確定性もまた、私たちが収入、毛金利と支出を正確に予測することを困難にし、債務融資或いは再融資の難度を増加させる可能性がある。
上記のいずれも私たちの業務を深刻に損なう可能性があり、私たちは政治的または経済的気候と金融市場の状況が私たちの業務のすべての方法を深刻に損なう可能性があることを予見できない。
追加資本の調達は私たちの株主に希釈して、私たちの運営を制限したり、私たちの技術や権利を放棄することを要求するかもしれません 製品
候補製品。
これまで、相当な製品収入を生み出すことができれば、株式と債務融資、潜在的な戦略的協力と許可手配によって、私たちの現金需要を満たすことが予想されています。私たちは約束された外部資金源を持っていない。債務融資または優先持分融資(利用可能な場合)が関与する可能性のあるプロトコルは、追加債務を生成する、資本支出を行うか、または配当を宣言するなど、我々が特定の行動をとる能力を制限または制限する契約を含む。したがって,より多くの資本を調達することは実現できない可能性があり,必要であっても,より多くの資本を調達することが可能であれば,理想的な条項ではできない可能性がある.もし私たちが第三者との戦略的協力やマーケティング、流通、または許可手配を通じて資金を調達する場合、私たちは、私たちの技術、将来の収入源、研究計画、または候補製品に対する貴重な権利を放棄しなければならないか、または私たちに不利になる可能性のある条項でライセンスを付与しなければならないかもしれない。もし私たちが必要な時にもっと多くの資金を集めることができなければ、私たちは私たちの製品開発や将来の商業化努力を延期、制限、減少または終了することを要求されるかもしれません。あるいは私たちが自分で開発し、マーケティングすることをより望んでいた候補製品の権利を与えることができます。これは私たちの普通株の価値を下げるかもしれない。
55
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
政府の規制に関連するリスク
私たちの候補製品がアメリカでマーケティング承認を得ても、私たちは決して規制部門の承認を得ず、アメリカ以外のところでこれらの候補製品をマーケティングすることができるかもしれません。
アメリカ以外にも、私たちは規制部門の承認を求めて、ヨーロッパ、日本、カナダ、オーストラリアで私たちの候補製品を販売し、他の市場で販売する可能性があります。もし私たちが未来に他の候補製品を求めるなら、私たちはアメリカ以外の規制機関がこのような候補製品を承認することを求めるかもしれない。しかし、米国以外の市場で任意の製品を販売するためには、これらの他の国/地域の多くの
および異なる安全性、有効性、および他の法規制要件を確立し、遵守しなければならない。承認手続きは国によって異なり、追加の製品候補テストと追加の行政審査期限が含まれる可能性があります。他の国/地域で承認を得るのに要する時間は、FDA承認を得るのに要する時間とは異なる可能性がある。他の国/地域の上場承認プロセスには、上述したFDAの米国での承認に関するすべてのリスクとその他のリスクが含まれる可能性がある。特に、米国以外の多くの国では、製品が商業化される前に定価と精算承認を受けなければならない。この承認を得ることは、これらの国/地域で製品が市場に出す時間を大幅に遅延させる可能性がある。一方の国の上場承認は、他の国の上場承認を確保することはできないが、一方の国の上場承認の失敗や遅延は、他国の規制プロセスに悪影響を及ぼす可能性がある。他の国/地域でマーケティング承認を得ることができなかったか、またはそのような承認を得る上でいかなる遅延や他の挫折が生じても、これらの海外市場で私たちの候補製品をマーケティングする能力を弱めることになる。このような減価はいずれも私たちの潜在市場の規模を縮小し、私たちの業務、運営結果、将来性に悪影響を及ぼす可能性がある。
私たちが私たちの候補製品のためにさらなるマーケティング許可を得ても、これらの候補製品は発売後、義務、制限、あるいは市場からの撤退制限を受ける可能性があり、もし私たちが規制要求を遵守できなかったり、承認後に予期しない製品問題に遭遇した場合、私たちは重大な処罰を受けるかもしれません。
私たちまたは私たちの未来のパートナーが将来的に上場承認された任意の候補製品、およびその薬剤の製造プロセス、承認後の研究および措置、ラベル、広告、販売促進活動などは、FDAおよび他の規制機関の持続的な要求および審査を受けるだろう。これらの要求には、安全とその他の発売後の情報と報告の提出、登録と上場要求、記録と書類の製造、品質管理、品質保証と相応の維持に関連する要求、医師へのサンプルの配布と記録の保存に関する要求が含まれる。候補製品の発売が承認されても、承認は、REMSの実施の要件を含む、医薬品が発売される可能性のある指定用途の制限または承認条件によって制限される可能性があり、これは、制限された流通システムの要件を含む可能性がある。
FDAはまた、候補製品の安全性或いは有効性を監視するために、高価な発売後の研究或いは臨床試験とモニタリングを要求する可能性がある。FDAと司法省を含む他の機関は、薬品の生産、販売および流通が承認の適応にのみ適用され、承認されたラベルの規定に適合することを確保するために、薬品承認後のマーケティングおよび販売促進活動を密接に監督·監督する。FDAは、ラベル外使用に関する製造業者のコミュニケーションに厳しい制限を加えており、もし私たちまたは任意の未来のパートナーが承認された適応のみを獲得した候補製品を販売していない場合、私たちまたはパートナーはラベル外販売促進によって警告または法執行行動を受ける可能性がある。処方薬の普及や広告に関する虚偽声明法を含む“連邦食品、医薬品および化粧品法”およびその他の法規に違反することは、連邦または州医療詐欺および法律乱用および州消費者保護法違反を調査または告発する可能性がある
さらに、私たちの候補製品、私たちの製造業者、または製造プロセスには、以前に未知の上級エンジニアまたは他の問題が存在したり、法規要件を遵守できなかったりして、様々な結果が生じる可能性があることが分かった
| • |
患者が私たちの薬を服用している訴訟は
|
| • |
このような薬物、製造業者、または製造プロセスの制限;
|
| • |
薬のラベルやマーケティングの制限
|
| • |
薬の配布や使用の制限
|
| • |
発売後の研究や臨床試験が求められている
|
56
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| • |
警告状や見出しのない手紙
|
| • |
薬を市場から引き揚げ
|
| • |
私たちが提出した保留申請または承認された申請を承認する補充申請を拒否する;
|
| • |
医療専門家に製品リコールまたは公開通知または医療製品安全警報を発すること
|
| • |
罰金、返還、または利益または収入の返還;
|
| • |
上場承認の一時停止または撤回
|
| • |
潜在的な協力者との関係を破壊し
|
| • |
不利なニュース記事と私たちの名声への損害
|
| • |
麻薬の輸入や輸出を拒否しています
|
| • |
製品を検収する
|
| • |
民事または刑事処罰を禁令または適用する。
|
立法改革や私たちの業務に影響を与える規制環境の変化は、私たちの候補製品の上場承認を得る難しさやコストを増加させるか、あるいは他の方法で候補製品の価格設定や商業実行可能性に影響を与える可能性があります。
米国や一部の外国司法管轄地域では、医療システムに関する立法や法規の変更および提案された変更は、候補製品の上場承認を阻止または延期し、承認後の活動を制限または規範化し、私たちまたは未来のパートナーが私たちまたは彼らが発売許可を得た任意の薬物を販売する能力に影響を与える可能性がある。現在の法律および将来取られる可能性のある他の医療改革措置は、より厳しいカバー基準をもたらす可能性があり、私たちまたは未来のパートナーが任意の承認されたbr薬物の価格に下振れ圧力を与える可能性があると予想される。
例えば、2010年3月、米国議会は“患者保護·平価医療法案”(“ACA”)と“医療·教育調整法案”、あるいは“医療改革法案”を公布し、医療補助の拡大と個人医療保険の実施により医療カバー範囲を拡大し、政府のヘルスケア計画下の薬品のカバー範囲の変更や精算範囲を含む医療カバー範囲を拡大した。
連邦や州政府関係者や立法者も、薬品輸入に関する立法を含む薬品価格や支払いを規制する措置の実施に努めている。最近、公衆と政府は薬品定価に対してかなりの審査を行い、人々が考えている薬品コストが高すぎることを解決する提案を提出した。最近は州立法も薬品コスト問題の解決に努力しており、通常薬品コストの透明性の向上或いは薬品価格の制限に重点を置いている。一般的な立法コスト制御措置はまた私たちの製品候補製品の精算に影響を及ぼす可能性がある。改正された“予算制御法案”は、2013年に医療サービス提供者に支払われた医療保険(ただし医療補助ではない)を2%減少させ、国会がさらに行動しない限り、2027年まで有効となる。Medicare、Medicaid、または実施可能な他の公共援助または補助医療計画に影響を与える重大な支出削減、および/または私たちに徴収される可能性のある任意の重大な税金または費用は、運営結果に悪影響を及ぼす可能性がある。連邦や州レベルで新しい立法が採用されれば、販売が許可されれば、私たちの現在または未来の製品に対する需要や定価に影響を与える可能性がある。しかし,医療改革法案や他の連邦や州改革努力の最終的な内容,時間,影響を予測することはできない。連邦や州医療改革が私たちの将来の業務や財務業績に悪影響を与えないことは保証されない。
57
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
司法と議会はACAのいくつかの側面に挑戦と修正案を提出し、今後もACAにより多くの挑戦と修正案を提出し、それを廃止し、代替する努力を予定している。さらに、ACAが公布されて以来、他の立法改正も提案され、採択された。これらの新しい法律は、連邦医療保険および他の医療資金のさらなる減少をもたらし、そうでなければ、マーケティング承認を得る可能性のある任意の候補製品の価格に影響を与える可能性がある。連邦医療保険や他の政府援助計画の精算を減らすことは、個人支払者の支払いのような減少を招く可能性がある。また、最近政府は、メーカーがそのマーケティング製品に価格を設定する方法について審査を強化している。コスト制御措置や他の医療改革を実施することは、収入を創出し、利益を実現し、あるいは私たちの薬品を商業化することを阻止することができるかもしれない。
また,2021年3月11日,総裁·バイデンは2021年米国救援計画法案に署名し,2024年1月1日から単一源と革新者多由来薬の法定医療補助薬品還付上限を廃止した。また,国会では,連邦医療保険D部分でカバーされている処方薬の費用に上限を設定し,2024年から年度自己負担上限を2000ドルに設定し,他の医療改革イニシアティブの一部として,より多くの医療改革措置が検討されている。承認後の要求を拡大し、医薬品の販売や販売促進活動を制限するための立法·規制提案がなされている。私たちは
が追加の立法変更を公布するかどうか、あるいはFDAの法規、ガイドライン、解釈が変更されるかどうか、あるいはそのような変更が候補製品の上場承認にどのような影響を与える可能性があるかどうかを決定することができません(ある場合)。また、米国議会のFDA承認過程に対するより厳格な審査は、上場承認を著しく延期したり阻止したり、あるいは私たちまたは未来の協力者がより厳しい薬物ラベルと上場後テストとその他の要求を受ける可能性がある。最近、総裁·バイデンは2022年8月に連邦医療保険受益者が使用する製品価格の上昇がインフレよりも速い場合に連邦医療保険にリベートを支払うようにメーカーに要求することを含む2022年インフレ削減法案に署名した。
私たちと医療保健提供者と第三者支払者との関係は、適用される詐欺や乱用、その他の医療法律法規の影響を受け、これは私たちを刑事制裁、民事処罰、契約損害、名声損害、利益減少、将来の収入減少などの処罰と結果に直面させる可能性がある。
医療サービス提供者および第三者支払者は、マーケティング承認された任意の候補製品を推薦および処方する際に主な役割を果たすであろう。私たちの現在と将来の第三者支払者や顧客との手配は、マーケティング、販売、流通、マーケティングの承認を得た候補製品の業務または財務的配置および関係を制限する可能性がある幅広い適用される詐欺や乱用、および他の医療法令に直面する可能性がある。適用される連邦と州医療法律法規に規定されている制限と義務には以下のものが含まれている。適用可能なbr法のより詳細な情報については,“第I項,第1項−業務−医療詐欺と濫用及びコンプライアンス法律·法規”と題する部分を参照されたい。ある州と外国の法律はまた異なる方法で健康情報のプライバシーやセキュリティ
を管理しており,HIPAAは通常先制されず,コンプライアンス作業を複雑化させている。
我々の現在と将来の第三者との業務配置が適用される医療法律や法規に適合することを確保するために努力することは、多くのコストに関連する。政府当局は、私たちの業務実践は、現在または将来、詐欺および乱用または他の医療保健法律および法規の適用に関する現行または将来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。もし私たちの業務がこれらの法律または任意の他の私たちに適用される可能性のある任意の政府法規に違反していることが発見された場合、私たちは重大な民事、刑事と行政処罰、損害賠償、罰金、政府援助の医療計画から除外され、MedicareおよびMedicaidのようなbr、返還、個人監禁、契約損害、名声損害、利益減少、および将来の収入減少、ならびに私たちの業務の削減または再編に直面する可能性があります。もし私たちがそれと業務を展開することを期待している任意の医師または他の提供者または実体が適用されない法律に適合していないことが発見された場合、彼らは政府の援助された医療計画から除外されることを含む刑事、民事、および行政処罰を受ける可能性がある。このような操作に対してbrを保護するには、コストが高く、時間がかかる可能性があり、大量の財力および人的資源が必要となる可能性がある。したがって、私たちが私たちに提起されるかもしれないどんな訴訟も防ぐことに成功しても、私たちの業務は損害を受ける可能性がある。
58
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
私たちはアメリカとある外国の輸出入規制、制裁、禁輸、反腐敗法、反マネーロンダリング法律法規の制約を受けている。このような法律基準を遵守することは国内と国際市場での私たちの競争能力を弱めるかもしれない。私たちは違反によって刑事責任と他の深刻な結果に直面するかもしれないし、これは私たちの業務を損なうかもしれない。
私たちは輸出規制と輸入法律法規に支配されており、“アメリカ輸出管理条例”、“アメリカ税関条例”、“アメリカ財務省外国資産規制事務室で実施されている各種経済·貿易制裁条例”、改正された“1977年米国反海外腐敗法”、“アメリカ法典”第18編201条に掲載されている米国国内賄賂法規、“米国旅行法”、“米国愛国者法”、そして私たちが活動している国の他の州と国の反賄賂と反マネーロンダリング法。腐敗防止法は広く解釈され、会社およびその従業員、代理人、請負業者、および他のパートナーの許可、約束、提供、直接的または間接的に公共または民間部門の受給者に不当なお金または任意の他の価値のあるものを支払うことを禁止する。私たちはアメリカ国外で第三者を招いて臨床試験を行い、商業化段階に入った後、私たちの製品を海外に販売し、および/または必要な許可、許可証、特許登録、その他の規制承認を得る可能性がある。私たちは政府機関や政府付属病院、大学、その他の組織の役人や従業員と直接または間接的な相互作用を持っている。私たちは、明確な権限やbrが実際にそのような活動を理解していなくても、従業員、代理、請負業者、および他のパートナーの腐敗や他の不正活動に責任を負わなければならないかもしれない。上記の法律法規に違反するいかなる行為も、重大な民事と刑事罰金と処罰、監禁、輸出入特権の喪失、資格取り消し、税金の再評価、契約違反と詐欺訴訟、名誉損害、その他の結果を招く可能性がある。
私たちの従業員または代表は、適用される規制基準や要求に違反することを含む不当な行為または他の不適切な活動に従事する可能性があり、これは私たちの業務を深刻に損なう可能性がある。
私たちは従業員詐欺や他の不適切な行為の危険に直面している。従業員の不正行為には、以下のような義務を故意に履行しないことが含まれる可能性がある
| • |
FDAと適用される非米国規制機関の規定を遵守する
|
| • |
FDAと適用される非米国規制機関に正確な情報を提供する
|
| • |
アメリカと海外の医療詐欺と法律法規の乱用
|
| • |
財務情報やデータを正確に報告する;または
|
| • |
許可されていない活動を私たちに開示する。
|
特に、医療業界の販売、マーケティング、業務配置は、詐欺、不正行為、リベート、自己取引、その他の乱用行為を防止するための広範な法律法規によって制約されている。これらの法律法規は、広範な定価、割引、マーケティングと販売促進、販売手数料、顧客激励計画などの業務手配を制限または禁止している。従業員の不正行為はまた、不適切な使用(取引を含む)が臨床試験中に得られた情報に関連する可能性があり、これは規制制裁を招き、私たちの名声を深刻に損なう可能性がある。従業員の不正行為を常に識別し、阻止することができるわけではなく、私たちがこのような行為を検出し、防止するための予防措置(従業員コンプライアンス訓練を含む)は、未知または未管理のリスクまたは損失を効果的に制御することができないか、またはこれらの法律または法規を遵守できないことによる他のbr行動または訴訟から私たちを保護することができないかもしれない。もし私たちにこのような訴訟を提起した場合、私たちは私たちの権利を弁護したり、維持することに成功しませんでした。これらの訴訟は、重大な民事、刑事および行政処罰、損害賠償、罰金、MedicareおよびMedicaidのような政府援助から除外された医療計画(例えば、MedicareおよびMedicaid)から除外された、返還、個人監禁、契約損害、名声損害、利益減少および将来収益、ならびに私たちの業務を削減または再構築することを含む、私たちの業務に重大な影響を与える可能性があります。
59
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
FDAと他の規制機関は非ラベル使用の普及を禁止する法律法規を積極的に実行している。もしラベルの外使用を不当に普及させることが発見されたら、私たちは重大な責任を負うかもしれない。
FDAや他の規制機関は、処方薬の販売促進を行う可能性のある声明を厳格に規制している。特に、製品は、製品承認ラベルに反映されるように、FDAまたは他の規制機関によって承認されていない使用に使用されてはならない。もし私たちの候補製品がある適応のマーケティング承認を得たら、医師はまだ承認されたラベルと一致しない方法で彼らのbr患者にこのような製品を処方するかもしれない。もし私たちがこのようなラベル外の使用を普及させることを発見されたら、私たちは重大な責任を負うかもしれない。連邦政府は不当な販売促進会社に巨額の民事と刑事罰金を科し、いくつかの会社がラベル外販売促進に従事することを禁止している。FDAはまた、企業に同意法令または永久禁止を締結し、これらの法令または永久禁止に基づいて、特定の販売促進行為を変更または制限することを要求する。もし私たちが私たちの候補製品の普及を成功的に管理できなければ、承認されれば、私たちは重大な責任を負う可能性があり、これは私たちの業務や財務状況に悪影響を及ぼすだろう。
米国税法や州税法の変化、例えば純営業損失の繰越やいくつかの他の税務属性を使用する能力の変化に影響を与え、私たちの財務状況や経営結果に悪影響を及ぼす可能性があり、これらの変化に対して会計を調整する必要がある可能性があるリスクが生じる。
私たちは歴史的に大きな損失を受けて、近い将来の利益を期待していませんし、私たちは永遠に利益を達成しないかもしれません。2018年1月1日までに開始された課税年度未使用の連邦純営業損失またはNOL
は、このような未使用のNOLが満期になるまで、将来の課税収入(ある場合)を相殺するために繰り越すことができます。現行法によると、2017年12月31日以降の課税年度に発生した連邦NOLは無期限に繰り越すことができるが、このような連邦NOLの控除額は課税収入の80%に制限されている。各州がどの程度連邦税法を遵守するかはまだ確定されていない。
また、改正後の1986年の国税法第382条及び383条又は同法規によれば、ある会社が“所有権変更”を経験した場合、通常、ある株主が3年間の間にその持分所有権の変化が50ポイントを超える(価値で計算される)と定義され、同社は変更前のNOL及び他の変更前の税収属性(例えば、税収控除を検討する)を用いて、その変更後の収入又は税収を相殺する能力が制限される可能性がある。私たちは過去に所有権変更を経験したことがありますが、将来は株式所有権の後続変動によって所有権変動を経験する可能性があります(その中のいくつかの変動は私たちのbr制御範囲内ではありません)。したがって,純課税所得額を得ると,変動前のNOLを用いてこのような課税収入を相殺する能力が制限される。州税法の類似条項は、累積州税br属性の使用を制限することにも適用可能です。さらに、州レベルでは、NOLの使用を一時停止または制限する時期がある可能性があり、これは州の課税税を加速または永久的に増加させる可能性がある。したがって、私たちが利益を達成しても、私たちのNOLや他の税金属性の大きな部分を使用できないかもしれません。これは、私たちの将来のキャッシュフローや運営結果に悪影響を及ぼすかもしれません。
米国や州税法変更における他の変更の会計処理は複雑であり、変更は今期や将来期間に影響を与える可能性がある。米国証券取引委員会の指導と一致して、私たちのbr合併財務諸表は、現在の税収法律法規に対する私たちの税収影響の推定を反映している。
60
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
私たちの候補製品の商業化に関するリスクは
私たちは激しい競争と迅速な技術変革に直面しており、これは他の人が私たちよりも早く、あるいはより成功的に製品を発見、開発、商業化することを招くかもしれない。
新薬製品の開発と商業化競争は激しい。私たちの候補製品が承認されれば、競争に直面し、世界各地の主要な製薬会社、専門製薬会社、バイオテクノロジー会社、大学および他の研究機関、および政府機関からの任意の将来の候補製品の競争に直面することが予想される。眼科治療市場は競争が激しく、活力に満ちている。私たちの成功は私たちが計画した適応のために市場シェアを得る能力があるかどうかにある程度かかっているだろう。
他の製薬会社は、同じ適応に対して治療法を開発する可能性があり、承認されれば、これらの適応はRYZUMVIまたは私たちの候補製品と競合し、私たちの許可内の特許、係属中の特許出願、または他の固有の権利を侵害することはなく、これは私たちの業務および運営結果に悪影響を及ぼす可能性がある。
私たちの競争相手は私たちが開発している製品よりも効果的で、安全で、より便利で、あるいはコストが低い製品を開発するかもしれません。あるいは私たちの候補製品を時代遅れにしたり、競争力がないかもしれません。我々の競争相手も既存の技術方法の進歩や新しい方法あるいは異なる方法の開発のために私たちの技術を時代遅れにし、それによって薬物発見過程における私たちの優位性を潜在的に除去する可能性がある。私たちの競争相手も私たちよりも早くFDAや他の規制機関のその製品のマーケティング承認を得ることができ、これは私たちの競争相手が私たちが市場に入る前に強力な市場地位を築くことができるようになるかもしれない。競争に関するより多くの情報は,本年度報告の“項目1,業務”に掲載されている。
私たちと比べ、私たちの多くの競争相手は研究開発、製造、非臨床テスト、臨床試験を行い、監督管理の許可とマーケティング許可を得た製品の面でもっと高い知名度、財力と専門知識を持っている。製薬·バイオテクノロジー業界の合併·買収は、私たちの数の少ない競争相手により多くの資源を集中させる可能性があります。規模の小さい会社や他の早期会社も、特に大手や成熟企業との協力によって重要な競争相手であることが証明される可能性があります。これらの会社は合格した科学と管理者を採用、採用と維持し、契約サービス提供者、メーカーと顧問を採用し、臨床試験場を設立し、患者を募集して臨床試験を行い、戦略取引を達成し、著者らの計画と相補的或いは必要な技術を獲得する方面で私たちと競争を展開している。
私たちは現在、販売やマーケティング能力を確立したり、第三者販売、マーケティング、流通APX 3330を招聘したりする上で、困難に直面している可能性があります。
私たちには販売やマーケティングインフラがありません。現在、薬品を販売、販売、流通する能力もありません。販売およびマーケティングの責務を保持する任意の承認された製品を商業的に成功させるためには、販売およびマーケティング組織を構築するか、またはこれらの機能の一部または全部を他の第三者にアウトソーシングしなければならない。Viatrisライセンスプロトコルには、RYZUMVIおよびPSの商業化(承認された場合)が含まれていますが、APX 3330に対する同様のプロトコルはありません。
私たちは自分の販売とマーケティング能力を確立し、第三者とこれらのサービスを提供する手配を達成することには、リスクがあります。例えば、販売員の募集と訓練は高価で時間がかかり、これはどんな製品の発表も延期される可能性がある。販売チームを募集し、マーケティング能力を確立した候補製品の商業発表が何らかの理由で遅延または発生していない場合、商業化費用のコストを早期または不必要に発生させる。これは費用が高いかもしれません。もし私たちが私たちの販売とマーケティング担当者を維持したり再配置できなければ、私たちの投資は損失します。
61
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
候補製品の商業化を阻害する可能性がある要素は
| • |
十分な数の有効な販売およびマーケティング担当者を募集および保持することができない、または第三者と流通契約を締結することができない
|
| • |
販売員は医師に触れたり、十分な数の医師に対して私たちの製品の利益に関する訓練を行うことができません
|
| • |
販売員が提供する相補的な製品が不足しており、より広い製品ラインを持つ会社と比較して競争的に不利になる可能性がある
|
| • |
独立した販売およびマーケティング組織を構築することに関連する予測不可能なコストおよび費用;
|
| • |
第三者支払者と政府機関から十分な保険と補償を受けることができない。
|
| • |
もし私たちが第三者と合意して販売、マーケティング、流通サービスを実行すれば、私たちの製品収入またはこれらの製品収入が私たちにもたらす収益力は私たち自身が開発した製品をマーケティングして販売することよりも低いかもしれません。さらに、私たちは、第三者との販売およびマーケティングの任意の候補製品の配置を成功的に達成できないかもしれないし、私たちに有利なbr条項に従ってそうすることができないかもしれません。私たちはこのような第三者に対して支配権がほとんどない可能性が高く、彼らのいずれも必要な資源と注意力を投入して薬を効果的に販売し、マーケティングすることができないかもしれない。もし私たちが販売とbrのマーケティング能力を確立することに成功できなければ、私たち自身も第三者と協力しても、私たちの候補製品を商業化することに成功しません。
|
私たちの将来のビジネス成功はRYZUMVIと私たちの候補製品(承認されれば)が医師、患者、第三者支払者、医学界の他の人の間で顕著な市場受容度を得ることにかかっている。
RYZUMVIと我々の候補製品は,市場承認を得ても,医師,患者,医療支払者あるいは医学界の他の人の十分な市場受容度を得ることができない可能性がある。もしRYZUMVIと私たちの候補製品が十分な受容度に達していなければ、著しい製品収入が生じないかもしれませんし、利益を上げることができないかもしれません。RYZUMVIと私たちの候補製品の市場受容度
商業販売のために許可された場合、多くの要素に依存します
| • |
代替療法と比較した治療効果と潜在的優位性
|
| • |
競争力のある価格で私たちの製品を売ることができます
|
| • |
対象患者群が新たな療法を試みる意欲と,医師がこれらの治療法を処方する意欲
|
| • |
私たちの製品を他の薬と一緒に使用する制限はありません
|
| • |
私たちの製品と患者が服用している他の薬との相互作用
|
| • |
あるタイプの患者は私たちの製品を服用できません
|
| • |
患者を治療する能力を示し、任意の適用可能な規制機関が要求すれば、他の利用可能な治療方法と比較して、目標適応を承認する能力がある
|
| • |
承認された適応の他の治療法と比較して,投与は比較的便利で容易である
|
62
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| • |
どんな副作用の流行率や重症度も
|
| • |
FDA承認タグに含まれる制限または警告;
|
| • |
承認されたか、または近い将来に商業的に使用される予定の代替療法の供給状況
|
| • |
私たちの販売とマーケティング戦略の有効性
|
| • |
私たちはマーケティングを通じて知名度向上に努めています
|
| • |
代替療法が提唱される可能性のある様々な疾患の研究,治療,予防に関与する組織のガイドラインと提言;
|
| • |
私たちは十分な第三者保険と十分な補償を得ることができる
|
| • |
第三者保険がない場合、患者が自分で費用を支払うことを希望するかどうか
|
| • |
医師または患者は、より有効、より安全、またはより便利である可能性があっても、既存の治療方法を変更したくないかもしれない。
|
私たちはまだ私たちの製品を何も売っていません。私たちは市場が私たちの製品に対して十分な需要を持っていることを投資家に保証することはできない。市場に我々の製品を受け入れさせるためには、業界参加者の知名度と需要を高めるために、大量のマーケティング努力と資金支出が必要となる。私たちは私たちが提供する製品に対するいかなる需要度を決定するために独立した市場研究を行っていないし、市場が私たちが生産してくれた製品やサービスに十分な興味を持っていることを保証することはできない。私たちが計画して提供する製品に対する需要不足は私たちに実質的な悪影響を及ぼすだろう。
FDAまたは同様の外国規制機関がRYZUMVIの模倣薬または私たちが発売承認された候補製品を承認した場合、またはこれらの機関が私たちの製品の模倣薬を承認する前に候補製品brの適切な排除期間を与えない場合、私たちの製品の販売は悪影響を受ける可能性がある。
NDAが承認されると、そのカバーされた製品は、FDA出版物“承認された治療同等性評価を有する医薬製品”における“参照リスト薬物”となる。製造業者は、米国で簡略化された新薬申請(“ANDA”)を提出することによって、市販薬の模倣薬バージョンの承認を参照することができる。ANDAを支援するためには,後発薬メーカーは臨床研究を必要としない。逆に、出願人
は、通常、その製品の有効成分(S)、剤形、強度、投与経路、使用条件またはラベルが参考発売薬(“RLD”)と同じであり、模倣薬がRLDと生物学的同等性を有することを証明しなければならず、これは体内での吸収速度と程度が同じであることを意味する。後発薬の市場へのコストはRLDよりはるかに低い可能性があり、模倣薬を生産する会社は通常、これらの製品をより低い価格で提供することができる。そのため,後発薬を発売した後,どのブランド製品やRLDの売上の大部分が後発薬に流出する可能性がある。
FDC法案は新化学実体(“NCE”)を含む新薬で5年間の非特許専有期間を規定している。具体的には、このような排他性が付与されている場合、ANDA
は、提出された材料に第4段落の証明が添付されていない限り、5年の満了前にFDAに出願を提出することができない。この場合、出願人は、RLD承認4年後に出願を提出することができる。FDAがその候補製品中の活性成分をNCEと見なすかどうかは不明であるため,それらが承認されれば,それらに5年間のNCE排他性
を与える。もし私たちが開発したどの製品もNCE 5年の独占経営権を獲得しなければ、私たちはまだ3年間の独占経営権を得る資格があります。私たちの候補製品は後発薬からの競争に直面し、これは私たちの将来の収入、収益力、およびキャッシュフローに悪影響を与え、私たちがこのような候補製品から投資収益を得る能力を大きく制限するかもしれない。
63
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
私たち(または私たちのパートナー)がRYZUMVIと私たちの候補製品を商業化することができても、私たちの収益性は第三者の清算やり方に大きく依存する可能性が高く、これらのやり方が不利であれば、私たちの業務を損なうことになります。
私たち(または私たちのパートナー)がRYZUMVIと私たちの候補製品の商業化に成功する能力は、政府衛生行政部門、個人健康保険会社、および他の組織がどの程度保険と十分な補償を提供するかにある程度依存するだろう。政府当局と第三者支払者、例えば個人健康保険会社や健康維持組織は、どの薬物のために費用を支払い、精算レベルを確立するかを決定する。政府当局と第三者支払者は,ある薬物のカバー範囲や精算金額を制限することでコストを抑制しようとしている。ますます多くの第三者支払者がbr製薬会社に価格に基づいて所定の割引を提供することを要求し、医療製品の料金に挑戦している。私たちは私たちが商業化した任意の候補製品が利用可能かどうかを確認することができず、利用可能であれば、精算レベルが十分かどうかを決定することもできない。我々の候補商品のために保険を受けていると仮定すると(第三者支払者の承認を得た場合),それによる精算支払率が十分でない可能性があり,あるいは患者が受け入れられない共同支払い
を発見する必要がある可能性がある.その病態を治療するために処方薬を服用している患者およびその処方医は、通常、その処方薬に関連する費用の全部または一部を第三者支払者に依存して精算する。承認された場合、患者は、保険を提供しなければ、候補製品を使用することができず、その製品の全部または大部分のコストを支払うのに十分な費用を精算する。したがって,カバー範囲と十分なbr精算は新製品の受容度に重要である。清算が得られない場合、あるいは限られたレベルに限られていれば、市場で承認された任意の候補製品を商業化することに成功できないかもしれません。また、米国と国際的な薬品の価格設定や参入政策が変化し、候補製品の商業的可能性にマイナスの影響を与える可能性があります。提案された政策変化は,連邦医療保険が薬品メーカーと交渉する可能性を含め,承認されれば候補製品の価格設定能力を制限する可能性がある。新たに承認された薬物の精算には大きな遅延がある可能性があり,カバー範囲は候補製品がFDAや米国以外の同様の規制機関の承認を得る目的よりも限られている可能性がある。さらに、精算を受ける資格があるということは、どの製品もすべての場合に支払われることを意味するのではなく、brの研究開発、製造、販売、流通を含む私たちのコストをカバーするレートで支払うことを意味するわけではない。新製品の臨時精算水準(適用すれば)も私たちのコストを支払うのに十分ではない可能性があり、恒久的にならない可能性があります。精算料率は製品の使用や臨床環境によって異なる可能性があり,低コスト薬品のために設定された精算レベルに基づく可能性があり,他のサービスの既存支払いにも組み込まれる可能性がある。製品の正味価格は、以下のように低下することができる:
政府医療計画または個人支払者が要求する強制割引またはリベート、および将来の医薬品からの輸入を制限する任意の法律の緩和は、これらの国/地域では、医薬品の販売価格が米国
よりも低い可能性がある。第三者支払者が自分の精算政策を設定する際には,通常連邦医療保険カバー政策と支払制限に依存する。しかし,米国の第三者支払者では,薬品の保証や精算に統一された政策要求はない。そのため、薬品の保証範囲と精算範囲は支払人によって異なる。したがって、保証範囲を決定する過程は通常時間も高価であり、これは各支払者にそれぞれ私たちの製品を使用した科学的および臨床的支援を提供することが要求される。保証範囲と適切な補償が一貫して適用されるか、または最初に得られることは保証されない。政府援助や個人支払者から私たちが開発した承認された製品の保証範囲と利益の支払率を迅速に得ることができなければ、私たちの経営業績、製品の商業化に必要な資金を調達する能力、および私たちの全体的な財務状況に悪影響を及ぼす可能性がある。
私たちまたは私たちのサプライヤーやメーカーに対する製品責任訴訟は、私たちが重大な責任を負うことになり、私たちが開発する可能性のある任意の候補製品の商業化を制限することができます。
私たちは私たちの候補製品が人体臨床試験でテストを行うことに関連する固有の製品責任リスクに直面しています。もし私たちが開発する可能性のある任意の製品を商業的に販売すれば、私たちはより大きなリスクに直面します。製品テスト、製造、マーケティング、またはbrの販売中に、患者、ヘルスケア提供者、または他の販売、または他の方法で私たちの候補製品に接触した者は、製品責任クレームを私たちに提示する可能性がある。例えば、私たちはある候補製品が被害を与えたと告発されるかもしれないし、その製品は他の点では適していない。任意のそのような製品責任クレームは、製造または設計欠陥の告発、アルコールまたは他の薬剤との相互作用、不注意、または保証違反など、製品固有の危険を警告できなかったことを含むことができる。州消費者保護法によると、クレームも主張することができる。もし私たちが私たちの候補製品に被害を与えるクレームを自己弁護することに成功できなければ、私たちは巨額の責任を招くかもしれない。是非曲直や最終結果にかかわらず、賠償責任は
64
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| • |
私たちが開発している候補製品の需要を減らすことができます
|
| • |
私たちの名声とメディアの深刻な否定的な関心を損なう
|
| • |
臨床試験参加者の脱退
|
| • |
製品ラベルへのFDAの警告を増加させた
|
| • |
関連訴訟の巨額の抗弁費用
|
| • |
実験参加者や患者に多額の報酬を与え
|
| • |
私たちの主な業務に対する管理職の関心を分散させる
|
| • |
収入損失
|
| • |
私たちが開発可能な候補品を商業化することはできません
|
| • |
規制当局が調査を行い
|
| • |
後発薬製品に適した製品責任訴訟制限を利用できず,危険や欠陥とされている製品への責任開放が増加する可能性がある。
|
私たちの製品責任および/または臨床試験保険カバー範囲は、私たちが生じる可能性のあるすべての責任をカバーするのに十分ではないかもしれません。私たちが臨床試験を拡大するにつれて、候補製品の商業化に成功すれば、私たちの保険カバー範囲を増やす必要があるかもしれません。保険範囲はますます高くなっており、私たちは商業的に合理的な条項や十分な金額で製品責任保険を得ることができず、出現する可能性のある責任を満たすことができないかもしれない。
同様に、私たちは、私たちの製造業者およびサプライヤーから購入された製品に関連する係属中または脅威の訴訟または他のクレームの当事者であるか、または他の方法でこれに責任を負うことができる。我々のサプライヤーに製品責任保険の加入を要求しようとしているにもかかわらず,このような保険や保険の十分性が得られるかどうかは不明である。このような事件およびクレームは、困難および複雑な事実および法的問題を引き起こす可能性があり、各特定の事件またはクレームの事実および状況、訴訟を提起する管轄権、および適用される法律の違いを含むが、これらに限定されない多くの不確実性および複雑さの影響を受ける可能性がある。このような訴訟は、特にこのような事項が保険範囲内でない場合、予想される準備金を超える追加費用およびリスクをもたらす可能性がある。未解決の法的問題や他のクレームが解決された後、既定の準備金を超える費用が発生する可能性があります。将来の製品責任訴訟およびクレーム、安全警報、または製品リコールは、その最終結果にかかわらず、業務および名声、および顧客および戦略パートナーの能力を吸引し、保持することに大きな悪影響を及ぼす可能性があります。もし私たちがこのような否定的な宣伝に直面すれば、業務、収益性、そして成長の見通しが影響を受けるかもしれない。
65
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
もし私たちまたは私たちの第三者製造業者が環境や健康と安全の法律法規を遵守できなかった場合、私たちは罰金や処罰を受けたり、コストを発生したりする可能性があり、これは私たちの業務の成功に悪影響を及ぼす可能性があります。
私たちの研究と開発活動は、私たち自身と私たちの第三者メーカーによる潜在的危険物質(化学および生物材料を含む)の制御使用に関するものです。私たちのメーカーはアメリカと海外の連邦、州、現地の法律法規の制約を受け、実験室プログラムおよび医療と危険材料の使用、製造、貯蔵、処理と処置を管理しています。私たちのメーカーがこれらの材料を使用、処理、貯蔵、処分する手続きは法律で規定されている基準に適合していると信じていますが、医療や危険材料による汚染や傷害のリスクを除去することはできません。このような汚染や傷害のため、私たちは責任を負うかもしれません。あるいは連邦、州、市、地方当局はこれらの材料の使用を制限し、私たちの業務運営を中断するかもしれません。もし事故が発生した場合、私たちは損害賠償責任を要求されるか、罰金を科される可能性があります。このような責任や罰金は私たちの資源範囲を超える可能性があります。私たちは医療や危険材料による責任に保険をかけない。我々は従業員のために危険材料の使用により負傷する可能性のあるコストと支出のために労働者補償保険を維持しているが、この保険は潜在的な責任を負うのに十分ではないかもしれない。適用される環境、健康と安全法律、およびbr法規を遵守することはコストが高く、現在または未来の環境法規は私たちの研究開発と生産努力を損なう可能性があり、それによって私たちの業務、将来性、財務状況あるいは運営結果を損なう可能性がある。
私たちの第三者への依存に関するリスクは
私たちは第三者に依存して私たちの非臨床と臨床試験を行い、他の任務を実行してくれます。これらの第三者が契約の責務をうまく履行できない場合、予想される締め切りまでに仕事を完了したり、規制要求を遵守したりすることができなければ、規制部門の私たちの候補製品の承認を得ることができないか、商業化できない可能性があり、私たちの業務は損なわれる可能性があります。
著者らは第三者CROと他の第三者に依存して、管理、監視、その他の方法で著者らの非臨床研究と臨床試験を実行する。将来的にはCRO、臨床データ管理組織、医療機関と臨床研究者などの第三者に依存して、著者らの非臨床研究と臨床試験を行うことが予想される。私たちは他の多くの会社とこのような第三者の資源を争っている。
そこで,これらの非臨床研究と臨床試験の進行,時間と完成および非臨床研究や臨床試験により開発されたデータの管理を限られた制御を行う。私たちは過去に経験したことがありますが、未来も経験したことがありますが、私たちが依存している第三者の業績に影響を与える事件によるスケジュール中断を経験したことがあります。外部の当事者とのコミュニケーションも挑戦的である可能性があり、ミスや協調活動の困難を招く可能性がある。また、サプライチェーンや運営中の他の意外な自然イベントや中断は、第三者が私たちに対する義務を履行する能力に影響を与える可能性がある。外部は人員配置の困難に直面する可能性がある
| • |
契約義務を履行しない者
|
| • |
規制コンプライアンスの問題に直面しています
|
| • |
所有権や経営陣の変更を経験します
|
| • |
優先順位が変化したり、財務的苦境に陥ったりする
|
| • |
他の実体との関係を構築し、その中のいくつかは私たちの競争相手かもしれない。
|
これらの因子は第三者の臨床試験の意思や能力に悪影響を及ぼす可能性があり,われわれの制御範囲を超える意外なコスト増加に直面する可能性がある。
66
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
これらの第三者に依存した研究や開発活動は,これらの活動に対する我々の制御を減少させるが,これは我々の責任や要求を軽減することはない.例えば、FDAは私たちが臨床試験記録と臨床試験結果を報告する標準を遵守することを要求し、一般に良好な臨床実践(GCP)と呼ばれ、データと報告の結果の信頼性と正確性を保証し、そして臨床試験参加者の権利、完全性と機密性を保護する。
任意のCROの作業の即時性または品質の問題は、任意のこのようなCROとの関係を終了し、代替サービスプロバイダを使用することを求めることをもたらす可能性がある。このような変更をすることは、コストが高いか、または私たちの臨床試験を延期する可能性があり、契約制限は、そのような変化を困難または不可能にする可能性がある。臨床試験を行っているCROを交換しなければならない場合,類似したサービスを提供する別のCROが見つかるまで,臨床試験を一時停止しなければならない可能性がある。代替組織を探すのに要する時間は、候補製品の商業化遅延を招く可能性があり、あるいは失われたデータを複製する際に巨額の費用を発生させる可能性があります。私たちが依存しているいかなるCROも他の場所では提供できないサービスを提供するとは信じていませんが、受け入れ可能な方法やbr}許容可能なコストで臨床試験を行うことができる代替組織を見つけることは困難かもしれません。私たちの臨床試験のいかなる遅延も完成できないことは、候補製品のための規制承認を得る能力に深刻な影響を与える可能性があり、候補製品を商業化することを阻止し、それによって販売収入を創出する能力を制限または阻止することができる。
さらに、臨床試験に関連する要求は継続的に発展し、これは、追加の監視、より大きなコスト、および/または遅延を必要とする可能性がある。FDAは2023年、CROに追加的な要求を行う可能性があるインフォームドコンセントおよびGCPに関連するガイダンスファイルを発行した。
FDAは2023年8月,過去の指導意見の代わりにガイドライン“インフォームドコンセント,IRBs,臨床調査員,スポンサーに対する指導意見”を発表し,最終的にインフォームドコンセントに関するガイドライン案を決定した。さらに、FDAは、2023年12月に、臨床調査がヒト被験者に構成されるリスクが最低リスクを超えず、ヒト被験者の権利、安全、および福祉を保護するための適切な保障措置を含む場合、例外的にインフォームドコンセント要求を遵守しないことを可能にする、最低リスク臨床研究に対する機関審査委員会のインフォームドコンセント免除または変更の最終規則を発表した。これらの指導文書は絶えず変化するインフォームドコンセント要求を提出しており,臨床試験における患者の募集や保留に影響する可能性がある。患者募集と維持への影響は臨床試験を阻害或いは延期する可能性があり、これはコストを増加させ、臨床計画を延期する可能性がある。
そのほか、2023年6月、FDAはガイドライン草案E 6(R 3)の良好な臨床実践(GCP)を発表し、ICH加盟国と地区の臨床試験データ標準を統一することを目的とした。データ要求の変更はFDA或いは類似の外国の監督管理機関が臨床前研究或いは臨床試験のデータに同意しない可能性があり、更なる研究が必要かもしれない。
私たちは第三者に完全に依存して原料薬を供給して製造し、私たちの候補製品の非臨床薬品供給と臨床薬品供給を調合と包装し、生産過程で薬品物質と製品の分析テストを行い、私たちは第三者に依存して私たちの現在と未来の候補製品の商業供給を生産してテストするつもりだ。
我々は現時点ではなく,我々のbr非臨床研究や臨床試験使用のために,我々の臨床薬物供給のインフラや能力を内部生産するつもりもない。私たちは内部資源と能力が不足していて、臨床や商業規模でどんな候補製品も生産できない。薬品の生産過程は複雑で、監督管理が厳しく、そして多種のリスクに直面している。例えば、私たちの契約製造業者は、候補製品の生産および活性医薬成分(または薬物)および最終薬物製品の分析試験を行うための施設であり、適用される規制機関に秘密協定または関連する外国規制を提出しなければならない場合には、FDAおよび他の同様の外国規制機関によって検査を行わなければならない。また、薬品或いは製品の生産は汚染、設備故障、設備設置或いは操作が不適切、サプライヤー或いはオペレータのミスにより製品損失を招きやすい。また、候補製品を製造する製造施設は、設備故障、労働力不足、自然災害、電力故障、または他の要因の悪影響を受ける可能性がある。全世界のサプライチェーン不足などの要素により、製造スケジュールは材料不足、施工遅延とサプライチェーン挑戦の負の影響を受ける可能性がある。
67
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
また,眼科製品製造に関連する需要が変化する可能性があり,現在の製造プロセスを修正する必要があるかもしれない。FDAは2023年12月、眼科外用薬製品の品質考慮因子である改訂されたガイドライン草案を発表し、その中で眼科医薬製品が眼内と眼周囲外用に使用される品質考慮因子を重点的に紹介した。更新された品質考慮因子は新しい要求への適応の遅延を招く可能性があり、製造に関連するコストを増加させる可能性もある。
我々は契約メーカーと分析実験室の製造と試験過程を制御せず、彼らが活性薬物物質と完成品の製造と良好な実験室規範(GLP)の現在の良好な製造規範(CGMP)を遵守することに完全に依存している。私たちの契約メーカーや分析実験室が、私たちの規格やFDAまたは適用される外国規制機関の厳しい規制要件に適合する材料の製造と試験に成功しなければ、私たちの製品に対する規制承認を確保および/または維持することができません。しかも、私たちの契約製造業者と分析実験室が十分な品質管理、品質保証、合格者の能力を維持することはできません。これらの材料や製品の生産と試験の規制要求を満たすことができなければ、私たちの契約メーカーと分析実験室施設の規制許可に影響を与える可能性がある。FDAまたは同様の外国規制機関が候補製品を製造および試験するためのこれらの施設を承認しない場合、または将来的にbrの承認を撤回する場合、代替の製造および試験施設を探す必要があるかもしれないが、これは、私たちが規制機関の承認を得たり、候補製品を販売する能力に悪影響を及ぼすだろう。さらに、私たちのすべての契約製造業者および分析実験室は他の会社と契約し、これらの会社のために材料または製品を供給および/または製造および/または試験し、これにより、私たちのメーカーはそのような材料および製品を生産する際に規制および調達リスクに直面する。可能な範囲で、私たちは複数の供給者たちを決定しようと努力した。しかし,単一ソースからしか入手できない原材料もあるし,1つのサプライヤーのみを決定しており,複数のソースが存在する場合でも同様である.
我々は、非臨床および臨床試験のためのAPX 3330およびPSを製造および試験するために、米国および海外の第三者製造者および試験実験室に依存し、将来的にAPX 3330、PSおよび任意の他の候補製品のためにこのようにし続ける予定である。もし私たちの第三者製造業者および分析実験室が薬物および/または薬物
製品を商業ベースで供給または試験できない場合、私たちは候補製品の生産および販売に成功しないかもしれない(承認されれば)、または私たちはそうすることを延期するかもしれない。例えば、私たちは現在イタリアのPS薬剤サプライヤーとインドのAPX 3330薬剤メーカーに依存している。これらの薬物物質の生産に何らかの遅延または問題が生じた場合、またはこれらの医薬物質、開発およびPS、私たちの候補製品の承認および潜在的な商業投与から遅延または他の悪影響が生じる場合。我々は、製品規格(同一性、強度、品質および効力)を比較することによって、現在の薬物物質および/または医薬製品と、以前に完成した非臨床試験および臨床試験で使用された薬物物質および/または医薬製品との等価性を証明する。もし私たちがこのような等価性を証明できなければ、私たちは私たちの候補製品に対して追加のbr}非臨床試験および/または臨床試験を行う必要があるかもしれない。これらや他の潜在的な問題から,米国メーカーとAPX 3330やPSの活性薬物成分
との追加供給源を確立する可能性を模索している。これらの追加的な源を確立し、彼らの製造技術の鑑定と彼らの製品の等価性を証明することを含み、費用が膨大で、時間がかかり、実現が困難である可能性があり、そして私たちの研究開発活動を延期する可能性がある。たとえ私たちが生産を異なる第三者に移すことができても、どの移転も高価で時間がかかる可能性があり、特に新しい工場は必要な規制要求を遵守する必要があり、私たちはその工場で生産されたどの製品を使用または販売する前にFDAの承認が必要だからだ。もし私たちがどのメーカーも交換しなければならないなら、私たちの研究開発活動は似たようなサービスを提供する別のメーカーを見つけるまで一時停止しなければならないかもしれない。代替組織を探すのにかかる時間は、候補製品の開発や商業化の遅れを招く可能性がある。
68
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
我々は、候補製品の開発または販売について許可スケジュール(例えば、Viatris許可合意)を達成し、将来的に他の戦略連合を形成したり、または許可スケジュールを達成したりすることが可能である。もし私たちが有利な条件でこれらのbr連合を設立または維持できなければ、私たちの業務は損害を受ける可能性がある。
私たちは、他の戦略連合を結成したり、求めたり、合弁企業や協力を作成したり、第三者と他の許可取り決めを達成したりすることが可能であり、これらの手配は、Viatrisライセンス協定のような候補製品に関する私たちの開発および商業化努力を補完または強化すると信じています。これらの関係のいずれも、非日常的な費用や他の費用の発生、短期的および長期的な支出を増加させること、または既存の株主を希釈する証券を発行することを要求することができ、これは私たちの管理や業務を混乱させる可能性がある。私たちの可能なパートナーは大中型、地域的、または全国的な製薬会社とバイオテクノロジー会社を含む。もし私たちがどの第三者ともこのような計画を達成すれば、候補製品の開発や商業化のための私たちの協力者の資源の数と時間を限られた制御を行う可能性が高い。
私たちがこのような計画から収入を創出する能力は、私たちの協力者がこれらの手配の中で彼らに割り当てられた機能を成功的に履行する能力に依存するだろう。戦略的取引やライセンスの後、このような取引の合理的な収入または特定の純収入を証明することが達成されるかどうかは確認できません。候補製品に関する協力は私たちに次のようなリスクをもたらすだろう
| • |
協力者は彼らがこれらの協力の努力と資源に適用することを決定する上で大きな自由裁量を持っている
|
| • |
協力者は期待通りに義務を履行していないかもしれない
|
| • |
協力者は、開発および商業化を行ってはならない、または臨床試験結果、協力者の戦略的重点または利用可能な資金の変化または外部要因(例えば、資源の移転または競争優先事項を作成する買収)に基づいて、開発または商業化計画を継続または更新しないことを選択することができる
|
| • |
協力者は臨床試験を延期し、臨床試験計画に資金不足を提供し、臨床試験を停止或いは候補製品を放棄し、新しい臨床試験を繰り返し或いは行うことができ、或いは臨床試験候補製品の新しい調合を要求することができる
|
| • |
協力者は、第三者開発と直接または間接的に私たちの候補製品と競合する製品を独立して開発または間接的に開発することができ、協力者が競争力のある製品がより開発に成功する可能性があると思う場合、または私たちよりも魅力的な条項で商業化することができる
|
| • |
1つまたは複数の候補製品に対してマーケティングおよび流通権利を有する協力者は、マーケティングまたは流通の任意のそのような候補製品のために十分なリソースを使用してはならない
|
| • |
協力者は、私たちの知的財産権を正しく維持したり守ったりすることができないかもしれないし、何らかの方法で私たちの固有の情報を使用して、訴訟を招いて、私たちの固有の情報を危険にさらしたり、無効にしたり、私たちを訴訟に直面させたりすることができる
|
| • |
協力者は第三者の知的財産権を侵害する可能性があり、これは私たちを訴訟と潜在的な責任に直面させるかもしれない
|
| • |
私たちと協力者の間で紛争が発生する可能性があり、私たちの候補製品の研究、開発または商業化の遅延または終了を招き、あるいは訴訟や仲裁に経営陣の関心と資源を分散させることができる
|
| • |
私たちの協力で発見された場合、私たちはコントロール権の変更を経験すれば、いくつかの価値のある権利を失うかもしれない
|
| • |
協力は終了される可能性があり、このような終了は、適用可能な候補製品をさらに開発または商業化するための追加の資金需要を生成する可能性がある
|
69
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| • |
協力者は私たちの発見を理解し、これらの知識を利用して未来に私たちと競争するかもしれない
|
| • |
協力者の非臨床または臨床研究の結果は、他の開発プロジェクトを損害または損害する可能性がある
|
| • |
異なる協力者の間に衝突が存在する可能性があり、これは、これらの協力および潜在的な他の協力に負の影響を与える可能性がある
|
| • |
私たちの協力の数量と性質は、私たちの未来の潜在的な協力者や買収者の魅力に悪影響を及ぼすかもしれない
|
| • |
協力協定は、最も効果的な方法で、または私たちの候補製品の開発や商業化を招くことがないかもしれない。もし私たちの現在または未来のパートナーが業務統合に参加する場合、このような協力の下で私たちの製品開発または商業化計画を追求し、重視し続けることは、延期、減少、または終了する可能性がある
|
| • |
協力者たちは必要なマーケティング承認を得ることができないかもしれない。
|
将来の協力パートナーが上記のいずれの理由で候補製品を開発できなかったり、有効に商業化できなかった場合、これらの候補製品は販売が許可されない可能性があり、承認されれば、候補製品の販売が制限される可能性があり、これは私たちの運営業績や財務状況に悪影響を及ぼすだろう。
もし私たちが合理的なビジネス条項でAPX 3330のために新しい協力関係を構築できなければ、私たちは私たちの開発、製造、商業化計画を変えなければならないかもしれない。
私たちは開発、製造、商業化計画のための協力者を誘致する上で激しい競争に直面している。我々はすでにViatrisとRYZUMVIとPSの開発と商業化について協力してきた。APX 3330の連携について最終的な合意を達成するかどうかは,提案した協力者の資源,専門知識の評価,および関連する候補製品に関する複数の要因の評価,および提案する連携の条項や条件に依存する.これらの要因は、臨床試験の設計または結果、FDAまたは米国国外の同様の規制機関が承認する可能性、候補製品の潜在的市場、そのような候補製品の製造および患者への配送のコストおよび複雑性、競争製品の潜在性、技術所有権の不確実性
を含むことができ、挑戦の是非を考慮せずにこのような所有権に挑戦する場合、存在する可能性のある不確実性、および一般的な業界および市場状況を含むことができる。協力者はまた,類似した連携を得るために代替製品
候補製品や技術を考慮することができ,そのような連携が我々の連携よりも魅力的である可能性があるかどうかを考えることができる.私たちは商業的に合理的なbr条項でこれらの合意を締結できないかもしれないし、根本的にはできないかもしれない。
将来の商業協力の潜在的収入の大部分は、規制マイルストーンの支払いを達成するか、または私たちの候補製品を販売する際に支払われるべき印税(承認された場合)のような支払いを含むか、またはある可能性がある。私たちがこれらの協力の下で得る可能性のあるマイルストーンと印税収入は、私たちの協力者が私たちの候補製品を開発、発売、マーケティング、販売する能力に依存するだろう(承認されれば)。また、協力者は、我々の候補製品に関する協力開発された製品を商業化することで、第三者と合意することを決定することができ、受け取るマイルストーンや印税収入(あれば)を減少させる可能性がある。
既存の協力協定によれば、潜在的なパートナーと特定の条項について未来の合意を締結することができないように制限される可能性もあります。連携は複雑で時間のかかる
交渉と記録である.また,最近では大手製薬会社間の業務合併数が大きく,将来の潜在的パートナー数の減少を招いている。
70
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
私たちはタイムリーで受け入れ可能な条件で協力を交渉することができないかもしれないし、交渉できないかもしれない。もし私たちがそれができなければ、私たちは私たちが協力を求めているbr候補製品の開発を削減し、私たちの開発計画や私たちの1つ以上の他の開発計画の開発計画を減らしたり、私たちの潜在的な商業化を延期したり、どんな販売やマーケティング活動の範囲を縮小したり、私たちの支出を増やし、自費で開発または商業化活動を行わなければならないかもしれない。もし私たちが私たちの支出を増やして私たち自身の開発や商業化活動に資金を提供することを選択すれば、私たちは追加の資本を得る必要があるかもしれません。これらの資本は受け入れ可能な条項で私たちに提供できないかもしれません。もし私たちが十分な資金がなければ、私たちは私たちの候補製品をさらに開発することができないかもしれないし、私たちを市場に出して製品収入を生むこともできないかもしれない。
もし私たちが買収、許可、または戦略協力に従事すれば、これは私たちの資本要求を増加させ、私たちの株主を希釈し、私たちに債務を発生させたり、負債を負担したり、他のリスクに直面させたりする可能性がある。
我々は、相補的製品、知的財産権、技術または業務の許可または買収を含む様々な買収および戦略的パートナーシップに従事することができる。買収や戦略的パートナーシップは多くのリスクをもたらす可能性があります
| • |
業務費と現金需要が増加した
|
| • |
借金や負債を抱えています
|
| • |
私たちの株式証券を発行することで、私たちの株主が希釈されるだろう
|
| • |
買収された会社の業務、知的財産権、製品、候補製品を吸収し、新しい人員の統合に関する困難を含む
|
| • |
経営陣の関心は、既存の候補製品と、このような買収や戦略的パートナーシップを求める取り組みから移っている
|
| • |
重要な従業員の保留、キーパーソンの流出、そして私たちがキー業務関係を維持する能力の不確実性
|
| • |
そのような取引の他方に関連するリスクおよび不確実性は、その当事者およびその既存製品または候補製品の将来性および規制承認を含む;
|
| • |
私たちは得られた知的財産権、技術、および/または製品から私たちの目標を達成するのに十分な収入を得ることができず、関連する取引と維持コストを相殺することさえできない。
|
また、このような取引を行えば、大量の使い捨て費用や無形資産の買収が生じる可能性があり、将来的に巨額の償却費用につながる可能性がある。
私たちの知的財産権に関するリスクは
もし私たちが候補製品のために十分な特許保護を得ることができなければ、私たちの競争相手は私たちと似たような製品や技術を開発し、商業化する可能性があり、これは私たちが開発する可能性のある任意の候補製品を商業化する能力、私たちの業務、運営結果、財務状況、見通しに悪影響を及ぼすだろう。
私たちと私たちの許可側は、私たちの新技術や候補製品に関連する特許出願をアメリカと海外に提出することで、私たちの独自の地位を保護しようとしています。
私たちの未定および将来の特許出願は、私たちの技術または製品の全部または一部を保護するために、または他社が競争相手の技術および製品を商業化することを効果的に阻止するために、特許の発行を招くことができないかもしれない。特に、任意の特許出願の起訴中に、その出願に基づく任意の特許の発行は、我々が提案する権利要件の特許性をサポートするために、追加の臨床前または臨床データ
を生成する能力があるかどうかに依存する可能性がある。私たちは十分な追加データをタイムリーに生成できないかもしれないし、全く生成できないかもしれない。
71
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
特許起訴過程は高価で時間がかかり、私たちと私たちの未来の許可者、被許可者、または協力パートナーは、すべての必要または理想的な特許出願を合理的なコストで、または適時に準備、提出、起訴することができないかもしれない。私たちまたは任意の未来のライセンシー、ライセンシー、または協力パートナーは、特許保護を得る前に、開発および商業化活動中に行われる発明の特許可能な態様を決定することができない可能性がある。私たちおよび私たちのライセンシーの特許出願は、そのような出願が特許を発行するまで、次いで、発行された特許要件がその技術の範囲をカバーする限り、そのような出願において要求される技術を実施する第三者に対して強制的に実行することはできない。
私たちのいかなる特許が成熟しているか、または私たちの任意の未解決特許出願が発行された特許に成熟することを保証することはできません。その中には、私たちの製品を保護するのに十分な特許請求の候補製品が含まれています。他社は、我々の方法に関連するか、または我々の方法と競合する可能性のある技術を開発し、特許出願が提出されたか、または提出された可能性があり、我々の特許出願と重複または衝突する特許を受信したか、または受信した可能性があり、
は、例えば、同じ化合物、方法または処方を要求することによって、または要求によって、我々が所有する特許またはライセンス内の特許の主題を支配することができる。バイオテクノロジーおよび製薬会社の特許地位は、我々の特許地位を含み、複雑な法律および事実の問題に関連しているため、私たちが得ることが可能な任意の特許請求の発行、範囲、有効性、および実行可能性は確定的に予測できない。特許が発行されると、疑問視され、実行不可能、無効、または回避される可能性がある。米国の特許や特許出願も妨害プログラムの影響を受ける可能性があります一方的再試験、あるいは各方面間
は地方裁判所で訴訟手続き、補充審査、質疑を再審査する。特許は、異なる国および地域の特許庁で反対、許可された後に審査または同様の手続きを受ける可能性がある。これらの訴訟は、特許喪失または特許出願が拒否されたか、または特許または特許出願の1つまたは複数の特許請求の範囲を喪失または縮小させる可能性がある。また、このような妨害、再審、反対、支出後の審査、各方面間検討、補充検査、または撤回手続きは費用がかかるかもしれないし、時間がかかるかもしれない。したがって、私たちが所有または独占的に許可されている可能性のある任意の特許は、
競争相手に対していかなる保護も提供できないかもしれない。さらに、介入手続きにおける不利な決定は、第三者が我々が求める特許権を取得することをもたらす可能性があり、これは、逆に、私たちの候補製品を開発、マーケティング、または他の方法で商業化する能力に影響を与える可能性がある。
また,特許の発行は有効と推定されているが,その有効性や実行可能性は決定的ではなく,十分な独自保護や類似製品を持つ競争相手に対する競争優位性を提供できない可能性がある.競争相手はまた私たちの特許を迂回して設計することができる。他の当事者は、より効果的な技術、設計、または方法のために開発され、特許保護を得ることができる。私たちは、コンサルタント、サプライヤー、元従業員、または現職の従業員が許可されていない開示または任意の技術知識または商業機密を使用することを阻止できないかもしれない。一部の国の法律はアメリカの法律のように専有権を保護しておらず、私たちはこれらの国で私たちの専有権を保護することは重大な問題に直面する可能性がある。このような事態が発生すれば、我々の販売に実質的な悪影響を及ぼす可能性がある。
私たちが特許権を行使する能力は私たちが侵害行為を検出する能力にかかっている。その製品で使用されているコンポーネントを宣伝しない侵害者を発見することは困難である.
また,競争相手や潜在的なライバルの製品で侵害証拠を得ることは困難または不可能である可能性がある.私たちの特許権を強制的に執行または擁護するための訴訟(もしあれば)は、私たちが勝訴しても、費用が高く時間がかかる可能性があり、経営陣とキーパーソンの私たちの業務運営に対する注意を移すことができます。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれません。もし私たちが勝てば、得られた損害賠償や他の救済措置は商業的な意味がないかもしれません。
さらに、私たちの特許を強制的に実行または保護する手続きは、私たちの特許を無効、実行不可能、または狭い解釈に直面させる危険性があるかもしれない。このような訴訟はまた、私たちの1つまたは複数の特許の一部または全部のクレームが無効であるか、または強制的に実行できないことを含む、第三者からのクレームを引き起こす可能性がある。もし任意の訴訟において、裁判所が私たちの候補製品をカバーする特許が無効または強制執行できないと発表すれば、私たちの財務状況と運営結果は不利な影響を受けるだろう。また、裁判所が第三者が持っている有効で強制的に実行可能な特許が私たちの候補製品をカバーしていることが発見された場合、私たちの財務状況やbr}運営結果も悪影響を受けるだろう。
72
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
未来の私たちの所有権の保護の程度は不確定で、私たちは保証できない
| • |
私たちの任意の特許、または私たちの任意の未解決特許出願は、発行された場合、私たちの候補製品を保護するのに十分な範囲を有する権利要件を含むであろう
|
| • |
私たちの任意の未解決特許出願は発行された特許をもたらすだろう
|
| • |
承認されれば、関連特許が満期になる前に候補製品の商業化に成功することができる
|
| • |
私たちはすべての特許と出願されている特許をカバーする最初の発明です
|
| • |
私たちはこれらの発明の最初の特許申請者です
|
| • |
他の会社は私たちの特許を侵害しない類似または代替技術を開発しません
|
| • |
私たちのどんな特許も効果的で強制的に施行されるだろう
|
| • |
私たちに与えられたいかなる特許も、私たちの商業的に実行可能な製品に独占市場の基礎を提供し、いかなる競争優位性を提供してくれるか、あるいは第三者の挑戦を受けないだろう
|
| • |
私たちは、特許を個別に出願できる他の独自技術または製品候補を開発する
|
| • |
私たちの商業活動や製品は他人の特許を侵害しないだろう。
|
特許の寿命は限られている。特許の自然失効期間は、一般的にその発効出願日から20年後である。様々な延期があるかもしれない;しかし、特許の有効期限とその提供される保護は限られている。候補製品の特許出願と規制承認との間に長い時間があることを考慮すると、私たちは特許保護された候補製品を販売する時間を列挙することができ、私たちの特許は承認される前に期限切れになる可能性がある。もし私たちの候補製品に特許保護がなければ、私たちは私たちの候補製品からの模造バージョンの競争を受けやすいかもしれません。これは私たちの候補製品の収益性に影響を与えるかもしれません。
さらに、私たちの特許保護の獲得と維持は、政府機関によって提出された様々な手続き、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。米国特許商標局および各種外国政府特許機関は、特許出願中にいくつかのプログラム、文書、費用支払い、または他のbr規定を遵守することを要求する。さらに、発行された特許の定期維持費および年会費は、特許有効期間内にいくつかの段階で米国特許商標局および外国特許代理機関に支払われなければならない。適用規則によれば、多くの場合、滞納金を支払うことによって、または他の方法でミスを訂正することができるが、場合によっては、規則を遵守しないことは、特許または特許出願の放棄または失効をもたらす可能性があり、関連する司法管轄区域の特許権の一部または全部の喪失をもたらす可能性がある。この場合、私たちの競争相手が市場に参入する可能性があり、これは私たちの業務に悪影響を及ぼすだろう。
我々は,特許保護を得るのが遅くなるまで,我々の研究開発成果で特許を申請できることを確認できない可能性もある.米国で特許を維持することは高価なプロセスであり、海外で特許や特許出願を維持するコストはより高い。したがって、私たちと私たちの許可側はこのような特許を維持することができず、私たちの製品の組み合わせの権利を減らすことができないかもしれません。製薬、バイオテクノロジー、医療機器会社の特許地位は通常高度に不確定であり、複雑な法律や事実問題に関連しており、近年多くの訴訟のテーマとなっています。したがって,我々と我々ライセンシーの特許権の発行,範囲,有効性,実行可能性,商業的価値は非常に不確定である.我々と我々の許可者との係属中および将来の特許出願は、我々の技術または製品を保護する特許の発行、または他社が競争技術および製品を商業化することを効果的に阻止する特許をもたらすことができないかもしれない。
73
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
もし私たちが“ハッジ·ワックスマン法”や同様の外国立法に基づいていなければ、特許期間の延長と私たちの候補製品のデータ独占権を得ることで保護されなければ、私たちの業務は深刻な損害を受ける可能性がある。
米国特許のうちの1つは、1984年の“医薬品価格競争および特許期限回復法”(“ハッジ·ワックスマン法案”と略す)に従って特許期間を回復する資格がある場合、我々または他の候補製品の時間、規制審査持続時間、およびFDAが上場を許可した日(ある場合)に基づいている可能性がある。ハッジ·ワックスマン法は、FDA規制審査期間中の製品の補償として、特許回復期間または特許期間を延長することを規定している。特許期間延長の期限は,規制審査にかかる時間に基づいて計算される。将来、私たちは、私たちのRYZUMVIまたは他の候補製品に関連する1つまたは複数の特許
のために特許期間の延長を求める計画があるかもしれない。しかし,適用の締め切り内に出願できなかったこと,関連特許が承認される前に満期になったこと,適用要求を満たしていなかったことなどから延期が得られなかった可能性がある.さらに、適用される期間または提供される特許保護範囲は、私たちが要求するものよりも短いか、または短くなる可能性がある。もし私たちが特許期間の延長を得ることができない場合、あるいはどのような延長の期間が私たちが要求したよりも短い場合、私たちの収入は減少し、実質的な損失をもたらす可能性がある。
米国特許法の変化は特許の全体的な価値を低下させ、候補製品を保護する能力を弱める可能性がある。
2011年、米国は“米国発明法”(“AIA”)により範囲の広い特許改革立法を公布した。AIAによる重要な変化の1つは、2013年3月16日から、米国が同一発明を主張する異なる当事者が2つ以上の特許出願を提出する際に、どちらの特許を付与すべきかを決定するために、“先出願”制度に移行することである。その日の後であるが、したがって特許を得ることができる前に、発明が第三者によってなされる前にこの発明がなされていても、米国特許商標局に特許出願を提出する第三者がある。これは私たちが発明から特許出願を提出するまでの時間を知ることを要求するだろうが、状況は私たちの発明に関する特許出願を迅速に提出することを阻止するかもしれない。AIAが導入した他のいくつかの変化には,特許権者が特許侵害訴訟を提起できる範囲を制限することと,USPTOで任意の発行された特許に挑戦する機会を第三者に提供することが含まれている。これは2013年3月16日までに発表された特許であっても、私たちのすべてのアメリカ特許に適用される。USPTO手続きの証拠基準は、米国連邦裁判所が特許請求の無効を宣言するために必要な証拠基準よりも低いため、第三者がUSPTO手続きにおいてUSPTOの権利請求を無効にするのに十分な証拠を提供する可能性があり、たとえ地域裁判所訴訟で同じ証拠が初めて提出された場合でも、権利請求を無効にするのに十分ではない。したがって,第三者はUSPTO手続きを使用して我々の特許主張を無効にしようと試みる可能性があり,地域裁判所訴訟で第三者が被告として最初に疑問を提起されれば,我々の特許主張は無効にならない.AIAおよびその実施は、我々の特許出願をめぐる起訴および当社が発行する特許の実行または保護の不確実性およびコストを増加させる可能性がある。また、米国最高裁は近年、分子病理学協会がMyriad Genetics、Inc.(Myriad I)、Mayo Collaborative Servicesがプロメテウス実験室会社、
とAlice Corporation Ptyを訴えるなど、いくつかの特許事件における訴えを訴えている。特許保護の範囲を縮小したり、特許権者の権利を弱体化させたりする場合もある。私たちの将来の特許取得能力に対する不確実性を増加させるほか、これらのイベントの組み合わせは、一旦特許を取得する価値に関する不確実性をもたらす。米国議会、連邦裁判所、および米国特許商標局の決定によると、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、それにより、私たちが新しい特許を獲得したり、私たちの既存の特許と私たちが将来獲得する可能性のある特許を強制的に執行する能力を弱める。
74
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
私たちは世界各地で私たちの知的財産権を保護したり実践することができないかもしれない。
我々が特許保護を受けていない司法管轄区では,競争相手は我々の知的財産権を利用して独自の製品を開発し,さらに特許保護を持っているが米国よりも特許を実行することが困難な地域に他の侵害製品を輸出する可能性がある.競争相手の製品は、私たちが発行または付与していない特許、または私たちが発行または付与した特許声明または他の知的財産権が、これらの司法管轄区域での競争相手の活動を阻止するのに十分ではない司法管轄区で私たちの候補製品と競争することができないかもしれない。ある国、特にある発展途上国の法律制度は特許の強制執行を困難にし、これらの国は他のタイプの知的財産権保護、特に薬品に関連する知的財産権保護を認めない可能性がある。これは私たちが特定の管轄区域で私たちの特許の侵害を防止したり、私たちの独占権を侵害する方法で競争製品を販売することを困難にするかもしれない。外国の管轄区域で私たちの特許権を強制的に執行する訴訟は巨額のコストを招く可能性があり、私たちの努力と注意を私たちの業務の他の側面からbrに移します。ある司法管轄区の法律は知的財産権の保護程度はアメリカの法律に及ばず、多くの会社はこのような司法管轄区の保護とこのような権利を守ることに大きな困難に直面している。もし私たちまたは任意の未来の許可者が、これらの管轄区域内の私たちの業務に重要な知的財産権を保護することが困難になったり、効果的な保護から排除されたりすれば、これらの権利の価値は低下する可能性があり、私たちはこれらの管轄区域内の他の人からの追加的な競争に直面するかもしれない。多くの国に強制許可法があり,これらの法律により特許権者は第三者に強制的に許可を付与される可能性がある。また、多くの国は政府機関や政府請負業者に対する特許の実行可能性を制限している。これらの国では,特許権者の救済措置は限られている可能性があり,このような特許の価値を大幅に低下させる可能性がある。もし私たちまたは任意の許可者が私たちの業務に関連する任意の特許について第三者に許可を与えることを余儀なくされた場合、関連する司法管轄区における私たちの競争地位が損なわれる可能性があり、私たちの業務と経営結果は不利な影響を受ける可能性がある。
私たちは私たちの特許と他の知的財産権を保護したり強制したりする訴訟に巻き込まれるかもしれないが、これは高価で時間がかかり、成功しないかもしれない。
競争相手は私たちの特許、私たちの許可パートナーの特許、または他の知的財産権を侵害するかもしれない。権利侵害または不正使用に反撃するために、私たちは高価で時間がかかるかもしれないbr侵害クレームの提出を要求されるかもしれない。さらに、侵害訴訟では、裁判所は、私たちの特許が無効または強制執行できないと判断したり、私たちの特許が関連技術をカバーしていないことを理由に、相手がその技術を使用することを阻止することを拒否することができる。どんな訴訟手続きの不利な結果も、私たちの1つ以上の特許を無効または狭義に解釈されるリスクに直面させる可能性がある。さらに、知的財産権訴訟は大量の発見を必要とするため、私たちのいくつかの機密情報は、このような訴訟中に開示によって漏洩される可能性がある。さらに、このような侵害クレームを提起し、追及するのに十分な財政的または他の資源があることは保証されず、これらのクレームは通常数年継続して解決される。
訴訟手続きは失敗する可能性があり、成功しても費用が高く、私たちの経営陣や他の従業員の注意を分散させる可能性がある。私たちは単独で、または私たちの協力者と共に、私たちの固有の権利の盗用を防ぐことができないかもしれません。特に法律では、米国のようにこれらの権利を十分に保護できないかもしれません。
さらに、公聴会、動議、または他の一時的な手続き、または事態の発展の結果が発表される可能性がある。もし証券アナリストや投資家がこれらの結果が否定的だと思うなら、私たちは私たちの普通株価格に大きな悪影響を及ぼすかもしれない。
75
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
第三者は、私たちが彼らの知的財産権を侵害したと主張する法的訴訟を提起する可能性があり、その結果は不確実であり、私たちの業務成功に悪影響を及ぼす可能性がある。
私たちのビジネス成功は、第三者の独自の権利および知的財産権を侵害することなく、私たちの候補製品を開発、製造、マーケティング、販売し、当社の独自技術を使用する私たちと私たちの協力者の能力に依存します。バイオテクノロジーと製薬産業の特徴は特許と他の知的財産権に関する広範な訴訟だ。私たちは未来に私たちの薬品や技術に関連する知的財産権に関する対抗手続きや訴訟の一方になったり、妨害や派生手続き、許可後の審査、各方面間米国特許商標局の前の審査または他の手続き、または外国司法管轄区域の他の同様の手続。第三者は既存の特許または将来付与される可能性のある特許に基づいて私たちに侵害請求をするかもしれない。もし私たちが第三者の知的財産権を侵害していることが発見された場合、私たちは第三者からライセンスを取得して、私たちの薬品や技術の開発とマーケティングを継続することを要求される可能性があります。しかし、私たちは商業的に合理的な条項や必要な許可証を得ることができないかもしれない。たとえ我々が許可を得ることができても,我々の競争相手や他の第三者が我々に許可してくれた同じ技術
にアクセスできるように,非独占的であることができる.私たちは裁判所の命令を含めて、権利侵害技術や薬物の開発を停止し、商業化することを余儀なくされるかもしれない。さらに、故意の侵害が発見された場合、私たちは重大な金銭的損害に責任を負う必要があり、3倍の損害賠償と弁護士費が含まれる可能性がある。権利侵害の発見は、候補製品を商業化することを阻止したり、いくつかの業務運営を停止させたりする可能性があり、これは私たちの業務を損なう可能性があります。あるいは、私たちは私たちの権利侵害製品を再設計する必要があるかもしれないし、それは不可能かもしれないし、大量の時間とお金の支出が必要かもしれない。第三者機密情報や商業秘密を盗用した疑いは,我々の業務に類似した負の影響を与える可能性がある.特許または他の独占権に関連する任意の訴訟または他の手続きが私たちにもたらすコストは、私たちに有利であっても、巨額である可能性があり、大量のコストをもたらす可能性があり、私たちの経営陣や他の従業員の気晴らしを招く可能性がある。私たちのいくつかの競争相手は、彼らがより多くの資源を持っているので、私たちよりも複雑な特許訴訟の費用を効果的に受けるかもしれない。特許訴訟や他の訴訟の開始と継続による不確実性は、私たちの研究開発を延期し、私たちが運営を継続する能力を制限する可能性がある。
私たちは私たちの職員たちや私たちが以前の雇用主の知的財産権を間違って流用したことで損害賠償を受けるかもしれない。
私たちの従業員とコンサルタントは以前、私たちの競争相手や潜在的な競争相手を含む他のバイオテクノロジーや製薬会社に雇われていた。私たちは現在私たちに対するクレームが解決されていることを知らないにもかかわらず、私たちはこれらの従業員たちまたは私たちが意図していない、または他の方法で私たちのbr従業員の元雇用主の商業機密または他の固有の情報や知的財産権を使用または漏洩したと告発される可能性がある。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。これらのクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣の注意を分散させる可能性がある。もし私たちがこのようなクレームを弁護できなければ、お金のクレームを支払う以外に、私たちは貴重な知的財産権や人員を失う可能性があります。キーパーソンや彼らの仕事の成果の流出は、私たちの候補製品を開発したり商業化したりする能力を弱めるかもしれない。
もし私たちがビジネス秘密や他の固有情報の漏洩を十分に防ぐことができなければ、私たちが追求する可能性のあるどの製品の価値も大幅に縮小する可能性がある。
私たちの政策は、知的財産権開発に参加する可能性のある従業員と請負業者に、このような知的財産権を譲渡する協定を実行することを要求していますが、私たち
は、実際に私たちが自分と見なしている知的財産権を開発している当事者とこのような合意を実行できない可能性があります。
私たちと彼らの譲渡協定は自動的に実行されないかもしれないし、違反される可能性があります。私たちは第三者にクレームを出すことを余儀なくされたり、私たちが私たちの知的財産権の所有権とみなされていることを決定するために、私たちが提起したクレームを弁護する可能性があります。私たちはビジネス秘密、技術ノウハウ、持続的な技術革新に依存して、私たちの競争地位を発展させ、維持することができる。しかし、商業秘密は保護することが難しい。私たちは、従業員、コンサルタント、外部科学協力者、協賛研究者、契約製造業者、サプライヤー、および他のコンサルタントと締結された秘密協定にある程度依存して、私たちのビジネス秘密および他の独自の情報を保護します。これらの
プロトコルは、機密情報の漏洩を効果的に防止できない可能性があり、機密情報を不正に漏洩させた場合に適切な救済措置を提供できない可能性がある。さらに、これらの
プロトコルは、可能またはアクセスされた商業機密の各々と実行されていることを保証することはできない。一方が合意に違反し、私たちのビジネス秘密を含めて個人情報を漏らした場合、私たちは十分な救済措置を得ることができないかもしれません。br}一方が商業秘密を不正に開示したり、流用したりするクレームを実行することは困難で、高価で時間がかかり、結果は予測できません。しかも、米国国内外のいくつかの裁判所は商業秘密をあまり望んでいないし、保護したくない。もし私たちのいかなる商業秘密が競争相手によって合法的に取得または独立して開発された場合、私たちは彼らまたは彼らがそのような商業秘密を漏らした人がその技術や情報を使用して私たちと競争することを阻止する権利がないだろう。もし私たちの任意の商業秘密が競争相手または他の第三者に漏洩された場合、または競争相手または他の第三者によって独立して開発された場合、私たちの競争地位は損なわれるだろう。
76
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
私たちの商標保護を獲得し、維持することは、米国特許商標局および他の外国政府機関の承認に依存し、第三者は、他の方法で私たちの商標権を弱めることに挑戦、侵害、またはbrする可能性がある。
私たちはすでにアメリカで“RYZUMVI”商標の登録を取得した。私たちはまだ他の候補製品の商標をどの管轄区域にも登録していません(
これ以上使用されていない“Nyxol”を除いて)。私たちが商標の登録を確保して維持しなければ、第三者に対してこれらの登録を実行する際に他の場合よりも多くの困難に直面する可能性があり、これは私たちの業務に影響を与える可能性があります。私たちが候補製品のために商標出願を提出する場合、これらの出願は登録が許可されない可能性があり、登録商標は取得、維持、または強制的に実行されない可能性がある。アメリカと外国の管轄区域の商標登録手続きで、私たちは拒否を受けるかもしれない。私たちはこのような拒否に答える機会があるが、このような拒否を克服できないかもしれない。また、米国特許商標局および多くの外国司法管轄区の類似機関は、第三者
が未解決の商標出願に反対し、登録商標のキャンセルを求める機会を得ることを可能にする。私たちの商標に反対したり取り消したりするかもしれませんが、私たちの商標は継続できないかもしれません。さらに、米国の将来の候補製品で使用される任意の固有名詞は、登録または出願が商標として登録されているか否かにかかわらず、FDAの承認を受けなければならないことを計画している。FDAは、一般に、他の薬剤名と混同される可能性を評価することを含む、提案された薬物名を検討する。FDAが任意の候補製品の任意の提案された特許薬物名に反対する場合、私たちは、適用商標法の資格に適合し、第三者の既存の権利を侵害せず、FDAのために受け入れられる適切な代替特許薬剤名を決定するために、多くの追加のリソースをかかる可能性がある。もし私たちが私たちの任意の商標を登録した場合、私たちの商標または商号は挑戦、侵害、回避、汎用商標として発表されるか、または他の商標が侵害されたと認定される可能性がある。私たちはこれらの商標や商品名に対する私たちの権利を保護できないかもしれないし、これらの名前の使用を停止させることを余儀なくされる可能性があります。私たちは、関心のある市場で潜在的なパートナーや顧客の名前の承認を得るためにこれらの名前が必要です。もし私たちが私たちの商標と商号に基づいて名称を確立することができなければ、私たちは効果的に競争できない可能性があり、私たちの業務は不利な影響を受けるかもしれない
我々は、第三者との協力、許可手配、合弁企業、戦略連合、またはパートナー関係を行う可能性があるが、これらの協力または協力は、商業的に可能性のある製品の開発や、相当な将来の収入をもたらすことはないかもしれない。
私たちは未来に特定の許可や他の協力協定を締結するかもしれない。このような合意は、様々な職務調査、マイルストーン支払い、特許権使用料、保険、または他の義務を私たちに強要するかもしれない。もし私たちがこのような義務を履行できなかった場合、私たちの許可者またはパートナーは関連合意を終了する権利があるかもしれません。この場合、このようなライセンス知的財産権に含まれる製品
を開発またはマーケティングすることができません。さらに、ライセンス契約によると、知的財産権に関する紛争が生じる可能性があります
| • |
ライセンス契約に従って付与された権利範囲および解釈に関連する他の問題;
|
| • |
私たちの候補製品、技術、およびプロセスは、ライセンス契約に拘束されていないライセンス側の知的財産権をどの程度侵害しているか
|
| • |
私たちの協力開発関係に基づいて、特許と他の権利を再許可する
|
| • |
私たちのライセンス契約の下での義務と、どのような活動がこれらの職務義務を満たしていますか
|
77
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| • |
知的財産権を共同で創造または使用することによって生成された発明およびノウハウのリストおよび所有権;
|
| • |
特許技術発明の優先権。
|
さらに、第三者から知的財産権または技術許可を得るプロトコルは複雑であり、このようなプロトコルのいくつかの条項は、様々な解釈の影響を受ける可能性がある。起こりうる任意の契約解釈の相違の解決は、関連する知的財産権または技術に対する私たちの権利と考えられる範囲を縮小するか、または関連する合意の下での私たちの財務的義務または他の義務を増加させる可能性があり、いずれも、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。また、我々が許可している知的財産権をめぐる紛争が、商業的に許容可能な条項で許可スケジュールを維持する能力を阻害したり、弱体化したりすれば、影響を受けた候補製品の開発に成功し、商業化することができない可能性があり、これは、私たちの業務、財務状況、運営結果、および将来性に大きな悪影響を及ぼす可能性がある。さらに、私たちは、任意の将来の許可者の準備、届出、起訴、および維持活動が適用された法律および法規を遵守するか、または効果的かつ強制的に実行可能な特許および他の知的財産権を生成するかどうかを決定することはできない。
我々は、Apexian PharmPharmticals,Inc.が開発しているAPX 3330候補製品(“Apexian”)の候補製品のための第三者(例えば、Apexian PharmPharmticals,Inc.が開発しているAPX 3330候補製品)と、我々の他の候補製品
から得られた再許可の知的財産権とに依存しており、今回の許可下での権利を終了、減少、または失うことは、私たちの業務を損なうことになる。
我々はApexianと再許可プロトコル(改訂され、“Apexian再許可プロトコル”と呼ばれる)を締結し、Apexianが所有するAPX 3330候補製品と第二世代候補製品に関連する内許可特許と他の知的財産権を付与し、Apexianは衛材株式会社(“衛材”)から内許可を得た知的財産権を付与し、APX 3330に関するいくつかの研究報告、製造と分析記録、データ、ノウハウ、技術およびその他の
独自情報を含む。将来、私たちはAPX 3330または他の候補製品のために同じまたは同様の性質の追加の再許可プロトコルを締結するかもしれない。“Apexian再許可協定”のような再許可プロトコルによって付与される権利は、私たちの様々なマイルストーン支払い、特許権使用料、保険または他の義務によって制限される可能性があり、場合によっては、業務を停止する場合、合意に従って満期金を支払うことができない場合、書面通知を受けた後の特定の期間内に合意を修正できなかった重大な違約行為、または特定の開発およびビジネススケジュールを満たすことができなかったことを含む場合がある。Apexian従属許可プロトコルのような従属許可プロトコルを終了することは、同じ特典条項で私たちに提供できない可能性があり、または全く提供できない可能性がある新しいまたは回復されたプロトコルを交渉しなければならない可能性があり、これは、APX 3330およびbr}第2世代資産を開発することができないことを意味するかもしれない。私たちは、再許可プロトコル(Apexian再許可プロトコルを含む)に従って許可された技術をカバーする特許および特許出願の準備、届出、起訴、および維持を完全に制御するわけではない。
再許可プロトコルによれば、インディアナ大学研究·技術会社(“IURTC”)、すなわちApexianに許可され、私たちに再許可された特許の所有者は、そのような特許を制御するすべての
起訴および維持の権利を保持する。したがって、私たちはこのような特許と特許出願が私たちの業務の最適な利益に合った方法で準備、提出、起訴、そして維持されるということを常に決定することはできない。私たちは、ライセンス特許の起訴と維持について私たちの意見を考慮し、費用を負担することに同意する権利があるにもかかわらず、IURTCがそのような特許を起訴および維持できなかった場合、または起訴活動の制御によってこれらの特許または特許出願の権利を失った場合、私たちが許可した権利は減少または廃止される可能性があり、そのような許可権利の対象となる任意の候補製品を開発および商業化する権利は悪影響を受ける可能性がある。同様の権利減少または終了は、将来の再許可プロトコルでも発生する可能性がある。また,ApexianがIURTCとのライセンスプロトコルに違反し,60日間の救済期間内にその違反を修正できなかった場合,IURTCはApexianとのライセンスプロトコルを終了する可能性があり,この場合,我々の許可も終了し,Apexianとのライセンスプロトコルでのすべての権利を失うことになる.
78
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
Apexian再許可プロトコルは,Apexianがこのような許可プロトコルの終了を防ぐためにApexianとIURTCの許可プロトコルに違反していることを我々と連携して修復しなければならないことを規定しているが,このような努力がApexianとIURTC間の許可プロトコルの終了を阻止することに成功する保証はない.同様に、Apexianが衛材との許可協定に違反し、60日間の治療期間内にこのようなbr違反を是正できなかった場合、衛材はApexianとの許可協定を終了する可能性があり、この場合、私たちの許可下での再許可権利も終了する。我々は衛材とApexian間の許可プロトコル
の下では何の実質的な義務もないが,Apexianには一定の守秘と支払い義務があり,これらの義務を履行しなければ衛材許可協定に違反する可能性がある.
ApexianとIURTCのライセンス契約によると、もし私たちがIURTCとのライセンス契約に違反するいかなる行為もApexianのせいにしない場合、特に私たちの支払い、報告、監査、賠償義務に違反することを含む、Apexianがこのようなライセンス契約に違反し、終了した原因とみなされる。
候補製品と承認された製品を取得する権利を買収することで拡張は成功しない可能性がある。
私たちは未来に他の製品、候補製品、または技術の権利を得るかもしれない。私たちのビジネスの将来の成長は、承認されたbr製品、他の候補製品、または技術の権利を得る能力があるかどうかにある程度依存するかもしれません。しかし、私たちは第三者からそのような製品、候補製品、または技術の権利を得ることができないかもしれない。医薬製品の買収は競争の激しい分野であり、多くの成熟した会社も私たちが魅力的と思われる製品、候補製品、あるいは技術の戦略を許可したり買収したりすることを求めている。これらの老舗会社は私たちより競争優位を持っているかもしれません。それらの規模、現金資源及びより強い臨床開発と商業化能力のためです。しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。私たちはまた、私たちの投資が適切なリターンを得るための条項で関連製品、候補製品、または技術のbr権利を得ることができないかもしれない。さらに、私たちは私たちの重点分野で適切な製品、候補製品、または技術を決定することができないかもしれない。もし私たちが適切な製品、候補製品、または技術の権利を得ることに成功できなければ、私たちがこの戦略要素を追求する能力は損なわれる可能性がある。
私たちの従業員の事務と管理成長に関連するリスク
私たちは私たちのキーパーソンに依存して、私たちが高い素質の人材を誘致し、維持することに成功できなければ、私たちは私たちのビジネス戦略を成功的に実施できないかもしれません。
私たちはジョージ·マグラス、医学博士、工商管理修士、修士、最高経営責任者、取締役会のメンバーを含む、私たちの管理、科学と医療者に高度に依存している。私たちは役員と雇用協定を締結したが、どの従業員も私たちとの雇用関係を終わらせることができる。予見可能な未来に、私たちの任意の幹部、他の重要な従業員あるいはコンサルタント、あるいは他の科学と医学コンサルタントのサービスを失うことは、私たちの研究、開発、商業化目標の実現を阻害するかもしれない。もし私たちがキーパーソンを維持できず、質の高い後継者を雇うことができなければ、財務目標を達成し、私たちの業務を維持または拡大するなど、重要な目標を達成できないかもしれない。私たちは科学と臨床コンサルタントを含むコンサルタントとコンサルタントに依存して、私たちの開発と商業化戦略の制定を助けてくれます。私たちのコンサルタントとbrコンサルタントは、私たち以外の雇用主に雇われる可能性があり、他のエンティティと締結されたコンサルティングまたはコンサルティング契約に基づいて約束することができ、これは、私たちに対する彼らの利用可能性を制限するかもしれない。合格した科学者と商業者を募集して維持することもまた私たちの成功の鍵になるだろう。多くの製薬とバイオテクノロジー会社の間の類似者に対する競争を受けて、私たちは受け入れ可能な条件でこれらの人員を吸引し、維持することができないかもしれない。私たちは大学や研究機関から科学者を募集する競争も経験した。臨床試験で成功できなかったことは、合格した科学者を採用と維持することをもっと挑戦的にする可能性もある。
79
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
私たちは、会社の多くの会社の機能(販売、マーケティング、流通チームを含む)の開発と拡張が必要になると予想されていますので、このような開発や拡張を管理することが困難になる可能性があり、運営を混乱させる可能性があります。
2024年3月1日現在、17人のフルタイム従業員がおり、候補製品の臨床開発に伴い、従業員数と業務範囲を増やすことが予想されています。私たちが予想している発展と拡張を管理するためには、私たちの管理、運営、財務システムを引き続き実施し、改善し、私たちの施設を拡大し、より多くの合格者を募集し、訓練し続けなければなりません。また,我々の経営陣は,我々の日常活動から不比例な注意を移し,これらの開発活動を管理するために多くの時間を投入する必要があるかもしれない.私たちの資源が限られているので、私たちは私たちの業務の拡張や採用とより多くの合格者を効率的に管理することができないかもしれません。これは、私たちのインフラが弱く、操作ミス、業務機会の喪失、従業員の流失、または残りの従業員の作業効率の低下を招く可能性があります。私たちの業務の実際の拡張は大きなコストを招く可能性があり、候補製品の開発など、他のプロジェクトから財源を移転することが可能です。もし私たちの経営陣が私たちの予想した発展と拡張を効果的に管理できなければ、私たちの費用は予想よりも多く増加するかもしれません。私たちの収入を創出したり増加させる能力は低下する可能性があり、私たちは私たちの業務戦略を実施できないかもしれません。私たちの将来の財務業績と候補製品を商業化し、効果的に競争する能力は、私たちが将来の発展と拡張を効果的に管理する能力にある程度依存するだろう。
私たちと私たちの協力者にとって、国際運営に関連する様々なリスクが私たちの業務に悪影響を及ぼす可能性があります。
私たちの米国での業務に加えて、将来的に国際業務を展開する可能性があり、不利になる可能性のある規制、定価、精算、法律、政治、税収、労働力条件を含むこのようなグローバル業務に関連するリスクに直面する可能性があり、これは私たちの業務を損なう可能性がある。私たちはアメリカ以外で臨床試験を行う予定です。私たちは国際ビジネスに関連する多くのリスクに直面しています
| • |
私たちの候補製品の違いや予期せぬ規制要件に適合しています
|
| • |
異なる医療実践および慣習は、承認された場合、私たちの候補製品または任意の他の承認された製品の市場での受容度に影響を与える
|
| • |
言語障害;
|
| • |
契約紛争が発生した場合、外国の法律によって管轄される契約条項の解釈
|
| • |
外国人業務員の配置と管理が困難であり、第三者に依存したビジネスやその他の活動を制御することができない
|
| • |
労働騒乱がアメリカよりも一般的な国では労働力の不確実性
|
| • |
1977年の“反海外腐敗法”や同様の外国法規に基づいて負う可能性のある責任
|
| • |
海外の原材料供給や製造能力に影響を与える事件による生産不足
|
| • |
外国政府の税収、法規、許可要件
|
| • |
米国と外国政府の関税、貿易制限、価格と外国為替規制、その他の規制要件
|
80
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| • |
インフレ、自然災害、戦争、テロ事件、特に外国の政治的不安定を含む経済的疲弊
|
| • |
通貨レートの変動は、業務費用の増加と収入の減少を招く可能性がある
|
| • |
税務、雇用、移民、労働法律、法規、および従業員の海外居住や旅行の制限を遵守する
|
| • |
外交と貿易関係の変化;
|
| • |
私たちの契約や知的財産権を実行する上での挑戦は、特に米国のように知的財産権を尊重し、保護しない外国である。
|
もし私たちがこれらのリスクに遭遇すれば、私たちの非アメリカ司法管轄区での販売は損害を受ける可能性があり、私たちの運営結果は影響を受け、私たちの名声と業務の将来性はマイナスの影響を受けるだろう。
ネットワークイベント、ネットワークセキュリティホール、サービス中断、またはデータ破損を含むシステム障害または計画外イベントが発生した場合、私たちの業務および運営は影響を受けます。
セキュリティ対策が実施されているにもかかわらず、私たちの内部コンピュータシステムおよび私たちの現在および未来の請負業者およびコンサルタントのコンピュータシステムは、コンピュータウイルス、許可されていないアクセス、自然災害、テロ、戦争、および電気通信および電気故障の破壊を受けやすい。2021年3月、私たちは商業電子メール流出の被害者になった。この詐欺は私たちに何の損失も与えなかった。このような事件が再発して我々の運営が中断されると,我々の開発計画や業務運営の実質的な中断を招く可能性がある.例えば、完成した或いは未来の臨床試験中の臨床試験データの損失は私たちの監督管理の承認作業の遅延を招く可能性があり、そして私たちのデータの回復或いは複製のコストを著しく増加させる。もしどんな中断やセキュリティホールが私たちのデータやアプリケーションを紛失したり破損したり、あるいは機密または独自の情報を不適切に漏洩させた場合、私たちは責任を招く可能性があり、私たちの候補製品のさらなる開発および商業化は延期される可能性がある。私たちは大量の資源を使って、私たちの業務活動と実践を根本的に変えたり、私たちの臨床試験活動或いは情報技術を修正して、セキュリティホールを防止し、実際或いは潜在的な
脆弱性を緩和、検出、修復するために努力する必要があるかもしれない。また、私たちの情報技術インフラをネットワークイベント、ネットワークセキュリティホール、サービス中断またはデータ破損の影響から保護できなかったことは、私たちの運営を深刻に混乱させ、私たちの業務、運営実績、または財務報告の内部統制の有効性に悪影響を及ぼす可能性があります。また、洪水、火災、爆発、竜巻、地震、極端な天気条件、医療流行病、電力不足、電気通信故障、ネットワークセキュリティ事件、ネットワークセキュリティホール、サービス中断或いはデータ破損、他の自然或いは人為事故或いは事件或いは流行病のような計画外事件は、私たちがbr施設を十分に利用できなくなり、私たちの業務運営能力に悪影響を与え、特に日常生活において、その財務と運営状況に重大なマイナス影響を与える可能性がある。これらの施設を使用できないと
コストが増加し,我々候補製品の開発遅延や業務運営が中断する可能性がある.
私たちの普通株式所有権に関連するリスク
私たちは現在相当の数の普通株を持っていて、私たちの株式信用限度額手配に関連する株を発行するかもしれません。私たちのELOCに基づいてbr株を発行または販売することは、発行済み株の数を大幅に増加させ、私たちの証券保有者の株式を希釈することになります。これは普通株の市場価格を大幅に下げるかもしれない。
私たちは未来に発行される可能性のある相当な数の普通株式を持っている。
81
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| • |
私たちの株式信用限度額あるいはELOCの手配によると、リンカーン公園資本基金有限責任会社246,792株の普通株を発行しました。
|
| • |
我々のELOCプロトコルによれば,ELOCプロトコルの36カ月の期限内にリンカーンパーク資本有限責任会社に50,000,000ドルまでの普通株を売却することができ,購入プロトコルに規定されているいくつかの条件が満たされてからのみ開始することができる.
|
普通株が私たちのELOCスケジュールに従って発行または販売されていれば、私たちの証券保有者は希釈される可能性があります。これらの追加証券の発行は、我々の証券の市場価格に悪影響を及ぼす可能性がある。
私たちは予測可能な未来に何の現金配当金も支払わないと予想している。
現在の予想は、私たちは将来の収益を維持し、もしあれば、私たちの業務の発展と成長に資金を提供することだ。したがって、予測可能な未来には、私たち普通株の資本付加価値(あれば)が投資家の唯一の収益源となる。
ナスダック資本市場の持続的な上場基準を満たさなければ、私たちの普通株は銘柄を取られるかもしれません。もし退市すれば、私たちの普通株の流動性は影響を受けるだろう。
私たちの普通株がナスダックで上場し続けるかどうかは一連の上場基準を守り続けるかどうかにかかっています。私たちがこのような
基準を守り続けるという保証はない。ナスダックからの退市は、株式証券を公開または私的に売却することで追加融資を調達する能力に悪影響を与え、投資家が私たちの証券を取引する能力に著しく影響を与え、私たちの普通株の価値と流動性に悪影響を及ぼす。退市はまた、私たちの戦略選択と潜在的な取引相手に対する吸引力を制限し、従業員の自信を失う可能性があり、機関投資家あるいは業務発展機会に対する興味を失う可能性があるなど、他の負の結果を生じる可能性がある。さらに、我々の普通株がナスダック資本市場から撤退し、取引価格が1株当たり5.00ドル以下に維持されている場合、私たちの普通株の取引は、取引法が公布した特定の規則の要求を受ける可能性もあり、これらの規則は、経営者が“細価格株”と定義されている株式(一般に、非国家証券取引所上場またはナスダックからオファーされた任意の株式証券、その市場価格が1株当たり5.00ドル未満であるが、いくつかの例外を除く)に関連する任意の取引において追加的に情報を開示することを要求する可能性がある。
私たちの普通株の市場価格は大きく変動するかもしれない。
私たちの普通株の市場価格は様々な要素によって大幅に変動する可能性があります。その中のいくつかの要素は私たちがコントロールできません
| • |
私たちや私たちの競争相手は新製品や製品の強化を発表します
|
| • |
私たちは許可者や他の戦略的パートナーとの関係の変化;
|
| • |
知的財産権と規制承認の発展について
|
| • |
私たちは競争相手とは経営結果が違います
|
| • |
ロックプロトコルの解除により、私たちの普通株は大量に販売された
|
| • |
臨床試験結果の公表
|
| • |
希釈可能な融資を発表しました
|
| • |
証券アナリストの利益予想や提案を変えること
|
82
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| • |
医療支払い制度の構造を変え
|
| • |
新冠肺炎の大流行を含む製薬と生物技術業界の発展と市場状況
|
| • |
APX 3330、PS、または私たちが開発する可能性のある任意の他の候補製品の臨床試験結果。
|
また、全体的に、株式市場、特にバイオテクノロジー会社の市場は、極端な価格や出来高変動を経験している。この変動により、投資家は彼らの証券を売却して利益を得ることができない可能性がある。持続的な市場変動は私たちの普通株価格の極端な変動を招く可能性があり、これは私たちの経営業績に関係なく、あるいは比例しない可能性があり、これは私たちの普通株の価値を低下させ、私たちの普通株購入者の重大な損失を招く可能性がある。
私たちは証券訴訟の影響を受けるかもしれませんが、これは高価で、経営陣の注意をそらすかもしれません。
我々普通株の市場価格は変動する可能性があり、過去に株式市場価格の変動を経験した会社は証券集団訴訟の影響を受ける。私たちは未来にこのような訴訟の目標になるかもしれない。私たちに対する証券訴訟は巨額の費用を招く可能性があり、私たちの経営陣の関心を他の業務に移すことは、私たちの業務を深刻に損なう可能性があります。
| 項目1 B。 |
未解決従業員意見
|
適用されません。
| プロジェクト1 C。 |
ネットワーク·セキュリティ
|
リスク管理と戦略
同社は、ネットワークセキュリティ脅威からのリスクを識別、評価、管理、監視するためのプロセスを含むネットワークセキュリティリスク管理計画を採用している。我々は、当社のネットワークセキュリティ意識とリスク管理文化を促進するために、ネットワークセキュリティリスク管理をより広範なリスク管理の枠組みに統合してきた。これらの流れは、会社データおよびシステムのセキュリティ、セキュリティ、完全性および可用性の内部および外部脅威、および会社の運営の他の重大なリスクを評価し、少なくとも年に1回、会社のシステムまたは運営が大きく変化したときに評価することと、決定されたリスクに応答することとを含む。同社のネットワークセキュリティとリスク管理計画は,米国国家標準·技術研究所(NIST)の枠組みに基づいている。我々のリスク管理プロセスの一部として,会社は外部サプライヤーを招いて定期的な安全評価を行っている。当社の第三者リスク管理計画の一部として、当社のクラウド·プロバイダおよび他の第三者に関連するリスクを含むベンダーのネットワークセキュリティリスクを評価します。
サイバーセキュリティの脅威
本報告日現在,我々の業務戦略,運営結果,財務状況に大きな影響を与えると考えられるネットワークセキュリティ脅威や事件によるリスクは何も発見されていない.私たちは努力を続けているにもかかわらず、私たちのネットワークセキュリティ保障措置が、私たちまたは私たちの第三者サービスプロバイダの情報技術システムが破壊されたり、崩壊したりすることを防ぐ保証はありません。特に、ネットワークセキュリティ脅威が発展し、脅威行為者が複雑になっている場合には、特にネットワークセキュリティ脅威が発展し、脅威行為者が複雑になることを保証できません。より多くの情報については、第1 Aのリスク要因を参照してください“システム障害や計画外イベント(ネットワークイベント、ネットワークセキュリティホール、サービス中断、またはデータ破損を含む)が発生した場合、私たちの業務および運営は影響を受けます.”
83
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
統治する
ネットワークセキュリティリスク管理計画は、ネットワークセキュリティ事件の予防、検査、緩和と救済を含み、会社の財務部門が指導し、財務部門の高級副総裁と財務部門の高級取締役を含む。この2人はいずれも我々のネットワークセキュリティと情報技術プロジェクトを監督した経験があり,これまでのポストで類似した監督機能を担当していた.私たちは情報技術コンサルタントの提案と専門知識を提供して、絶えず発展する業界標準を監視し、私たちの適用政策の遵守状況を監視しています。財務総監上級副総裁は少なくとも毎年会社監査委員会にネットワークセキュリティ問題を報告し、会社のシステムや運営に重大な変化が発生した場合、毎回の取締役会で材料更新を共有します。財務総監の上級副総裁はまた、状況に応じて会社の最高経営責任者や他の上級管理職に仕事を報告した。これらの報告は、会社が会社のネットワークセキュリティ政策を遵守する全体的な評価を含む可能性があり、リスク評価、リスク管理と制御決定、サービスプロバイダの手配、テスト結果、セキュリティイベントと応答、政策とプログラムの変更と更新提案などのテーマを含むことができる。また,我々のネットワークセキュリティ計画に対する任意の外部
審査結果は上級管理職と監査委員会に報告される.
| 第二項です。 |
特性
|
私たちの本社は現在ミシガン州ファミントン山にあります。2024年12月31日に満期になった賃貸契約によると、私たちは約1,600平方フィートのレンタルオフィス空間を持っています。ビジネスの拡張に伴い、私たちは私たちの既存の空間やより多くの空間と施設を拡張することができるかもしれません。私たちは将来的に商業的に合理的な条項で適切な追加的かつ代替空間を提供すると信じています。
| 第三項です。 |
法律手続き
|
私たちは日常業務の過程で時々訴訟とクレームの影響を受ける。訴訟及びクレームの結果は正確には予測できないが、本出願の日まで、私たちはいかなるクレーム又は訴訟の当事者であるとは信じておらず、これらのクレーム又は訴訟の結果が私たちに不利であれば、これらの結果が私たちの業務又は財務状況に重大な悪影響を及ぼすことが予想される理由がある。結果にかかわらず,弁護や和解コスト,管理資源分流などにより,訴訟は我々に悪影響を与える可能性がある。
| 第四項です。 |
炭鉱安全情報開示
|
適用されません。
84
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
第II部
| 五番目です。 |
登録者普通株市場、関連株主事項、発行者による株式証券の購入
|
市場情報
私たちの普通株はナスダック資本市場で取引され、コードはOCKUPです。
所持者
2024年3月5日現在、私たちの普通株は約75人の登録保有者がいます。登録された所有者数は,我々の譲渡代理帳簿に登録されている実際の所有者数に基づいて計算され,“街名”の株式保有者や預託信託会社によって維持されている証券倉位リストに記載されている個人,組合,協会,会社またはその他の実体は反映されていない.
配当政策
設立以来、私たちは普通株について何の現金配当金も支払ったことがなく、予測可能な未来にもいかなる現金配当金も支払わないだろう。私たちは現在、私たちの収益を保留する予定で、もしあれば、私たちの業務拡張に資金を提供します。
最近売られている未登録証券
ない。
| 第六項です。 |
[保留されている]
|
適用されません。
|
第七項。
|
経営陣の財務状況と経営成果の検討と分析
|
私たちの財務状況と経営結果の議論と分析、私たちの財務諸表と本年度報告書の他の部分に含まれる以下の説明を読むべきです。本議論および分析に含まれるいくつかの情報または本年度報告の他の部分に記載された情報は、我々の業務および関連融資の計画および戦略に関する情報を含み、リスクおよび不確実性に関する前向きな陳述を含む。本年度報告の“リスク要因”の一部に列挙された要素を含む多くの要素の影響により、我々の実際の結果は、以下の議論と分析に含まれる前向き陳述に記述または示唆された結果とは大きく異なる可能性がある。
概要
我々は臨床段階の生物製薬会社であり,網膜や屈光性眼疾患患者が満足していない需要を治療する新しい療法の開発に専念している。
APX 3330
我々の主要な候補網膜製品APX 3330はRef-1(還元酸化効果因子-1蛋白)の小分子阻害物である。REF−1はHIF−1αやNF−kBなどの転写因子の調節因子である。Ref-1を抑制することは血管内皮増殖因子(VEGF)と炎症性サイトカインのレベルを下げることができ、この2種類のサイトカインは眼血管新生と炎症において重要な役割を果たすことが知られている。APX 3330は開発中の経口錠剤であり、毎日1~2回服用し、糖尿病網膜症(DR)の治療に用いられる。
85
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
DRは約1000万人の糖尿病患者に影響を与えており,2050年には1400万人以上の米国人に影響すると予想される。糖尿病網膜症は非増殖性糖尿病網膜症(“NPDR”)或いは増殖性糖尿病網膜症(“PDR”)に分けられ、前者は早期症状であり、症状は軽微或いは存在せず、後者は糖尿病眼疾患の比較的に末期段階であり、高度な失明症状を有する。約800万のDR患者がNPDRを有しており,治療を行わなければPDRに進展する可能性がある。このような疾患に関連する視力喪失のリスクがあるにもかかわらず,現在90%を超えるNPDR患者は眼看護専門家の観察以外に,視力を脅かす合併症が出現するまで何の治療も受けていない。APX 3330は経口錠剤として、アメリカ800万NPDR患者の早期非侵襲的治療法になる可能性がある。
2023年1月,103名の被験者(600ミリグラムAPX 3330を1日51名服用)のDR患者で行ったZeta−1第2段階試験のトップクラスの治療効果と安全性の結果,中,重度NPDRと軽度PDR,中心視力喪失のない糖尿病黄斑浮腫患者を含めて報告した。毎日APX 3330を服用することはプラセボと比較して研究の主要な終点に達していないにもかかわらず、すなわち眼が24週に糖尿病網膜症早期治療研究(“”)糖尿病網膜症重症度尺度(“DRSS”)の2段階改善患者のパーセンテージ(br})を研究したが、プラセボと比較したが、両眼DRSSヒト尺度でDRS三段階改善の治療効果が見られた。DR進展を防止或いは緩和することは視力を脅かす合併症であり、例えばPDRは臨床意義のある終点である。APX 3330は糖尿病患者においても良好な安全性と耐性を示した。2023年10月,米国食品医薬品局(FDA)と第2段階末期会議を開催し,両目DRSS人スケールで登録終点が3ステップ悪化することで合意した。APX 3330はZeta−1試験で良好な安全性と耐性を示した。Ocuphireは2024年2月に臨床試験案と統計分析計画の合意を求めるための特別案評価(SPA)をFDAに提出したまた,FDAとSPA合意を達成する際に研究設計パラメータと期待時間の詳細を共有する.
また、APX 3330の第2世代類似製品であるAPX 2009とAPX 2014を許可しました。このファミリーRef-1阻害剤は血管新生と炎症を減少させる独特な作用機序
は老年性黄斑変性、地理萎縮と非眼科疾患のような他の網膜疾患の治療に役立つかもしれない。
我々は現在、APX 3330およびその第2世代類似体(APX 2009およびAPX 2014)の局所投与経路と、その網膜治療パイプライン拡張の一部である全身(経口)経路とを評価している。
RYZUMVIとフェントラミン眼液0.75%(PS)
2022年11月に私たちは許可と協力協定(“Viatrisライセンス契約Br}FamyGen生命科学社(2023年1月にViatris,Inc.に買収)これにより、Viatris独占ライセンスを授与して、開発、製造、輸入、輸出、商業化(I)私たちの屈折製品候補製品ですフェントラミン液0.75%、本名ニコランチン(“PS”)と呼ばれ,(A)薬物による散瞳の逆転の治療,(B)角膜屈折性手術後の中視(低)光条件下での視力低下,および(C)老眼,および(Ii)PSと小用量ピロカルピンによる老眼の治療に用いられる(要するに、“PS製品”)アジアのある国と司法管轄区域を除いて(“ヴィアヤリス領土”)。2023年9月、PSはFDAによって治療薬による散瞳のために承認され、ブランド名はRYZUMVIであり、Viatris許可協定に従って支払われた1000万ドルの記念碑的支払いを引き起こした。
Viatrisライセンス契約の条項によると、OcuphireはViatrisと協力するアメリカでPS製品を開発するそれは.Viatrisは開発に関する予算コストを精算するPS製品FDAの承認により。ヴィアットリースが開発を担当しますPS製品アメリカ以外のヴィアトリス地域のbr個の国と司法管轄区にあります。
PSは毎日1回のメタンスルホン酸フェントラミン点眼液の調合であり、瞳孔直径の縮小と視力向上を目的としている。PSは、薬物性散瞳(“RM”)(散瞳)、老眼(年齢に関連する近視ボケ)の治療など、様々な適応に用いることができる屈折参差性角膜屈折術後の中視(弱)光条件下での視力低下手術をします。PSはすでに合計1100人以上の研究参加者(うち650人以上がPS治療を受けている)の12の臨床試験(3つの第1段階,5つの第2段階,4つの第3段階)で研究を行い,3つの目標屈折適応に有望な臨床データを示した。
86
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
逆転薬物性散瞳,老眼,弱光干渉におけるPSの複数の晩期臨床試験の正面データ
を報告した。老眼に対するVega−2段階3研究は主な終点に達しており,我々の開発とビジネスパートナーのViatrisは2024年上半期に段階3開発を継続する予定である角膜屈折術後中視光(弱光)下視力低下私たちはLynx-2に関するFDAの特別なプロトコル評価プロトコルを獲得することは,PSの第3段階実験である.私たちの開発とビジネスパートナーのViatrisは、2024年上半期にこの段階の開発を継続する予定です。
戦略的展望
著者らは引き続き多くの資産を買収する機会を探索し、既存のAPX 3330、APX 2009とAPX 2014網膜適応を他の網膜適応に拡張し、APX 3330の全世界のキー市場における後期開発、監督管理準備と商業化のために戦略パートナーを探すつもりである。これまで私たちの主な活動は、研究開発活動を行い、商業·財務計画を実行し、人員を募集し、資金を調達することだった。私たちは1つの製品RYZUMVIのみが販売を許可されており、Viatrisの売上に応じて印税が発生する可能性があり、私たちは許可と協力収入
を除いて、FDAや他の規制機関が許可しない限り、大量の収入が持続的に発生しないことを予想し、APX 3330やPSを他の適応に使用することに成功した。これまで、私たちが相当な製品収入を持続的に生み出すことができれば、私たちは株式と債務融資、協力、戦略連合、許可手配を通じて私たちの現金需要を満たすことができると予想しています。
2023年12月31日現在,我々は主に合計6,330万ドルの総収益の株式融資により我々の運営に資金を提供しており,そのうち2,115万ドルはRexahn PharmPharmticals,Inc.(“Rexahn”)の合併(“合併”)に関連しており,私募による転換可能手形の発行,合計毛収入純現金850万ドルである。また,合計4500万ドルのライセンス料とマイルストーン支払い,開発に関する費用精算を受けており,これらはViatrisライセンス契約に関連している。
2023年12月31日までの年度の純損失は1,000万ドルですが、2022年12月31日までの年度の純収益は1,790万ドルです。2023年12月31日現在、私たちの累計赤字は8150万ドルです。さらに私たちは費用が増えると予想していますなぜなら私たちは
| • |
APX 3330、PS、および将来のパイプラインの任意の他の候補製品の臨床試験を継続した
|
| • |
APX 3330、APX 2009、およびAPX 2014、PS、ならびに私たちの将来のパイプラインにおける任意の他の候補製品の非臨床研究を継続した
|
| • |
私たちが決定し、許可し、買収した他の候補製品を開発する
|
| • |
臨床試験に成功した候補製品のために監督部門の承認を求める
|
| • |
私たちの候補品を作ってくれました
|
| • |
私たちの知的財産権の組み合わせを維持し、拡大し、保護する
|
| • |
私たちの業務計画を実行するために、臨床、科学、運営、財務者を含むより多くの従業員を招聘する
|
| • |
私たちの製品開発と潜在的な将来の商業化努力を支援するために、運営、財務、および管理情報システムおよび人員を増加させる
|
| • |
上場企業として運営しています
|
| • |
私たち自身またはパートナーと一緒に販売、マーケティング、流通インフラを構築し、規制の承認を受ける可能性のある任意の製品を商業化します。
|
87
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
私たちの純収入(損失)は、私たちの非臨床研究、臨床試験、他の研究開発活動の支出(およびその精算)の時間、およびViatris許可協定または私たちが締結した任意の他の許可および協力協定から受け取った潜在的なマイルストーン支払いおよび稼いだ収入、およびApexian再許可協定に従って時々支払われる潜在的な支払いを必要とする可能性がある四半期間および年間にわたって大幅に変動する可能性がある。
最新の発展動向
臨床マイルストーン
APX 3330
2023年1月、我々は、糖尿病網膜症患者におけるAPX 3330の2 b期試験であるZeta-1のトップクラスの治療効果および安全性結果を発表した。Zeta−1試験では,APX 3330は良好な安全性と耐性を示し,両眼DRSSスケールでDR悪化を緩和または防止する効果を示した。FDAは私たちのEP 2会議でこれが承認可能な登録の終点だということに同意した。
PS
2023年1月、PSおよびPSおよびLDPのみの老眼適応をサポートするために、2段階3登録試験の最初のVega-2段階3キー試験の開始を発表した。Vega-2第3段階研究は主な終点に達しており,我々の開発·ビジネスパートナーViatrisは2024年上半期に第3段階の開発を継続する予定である。
法規更新
2023年9月、FDAはRYZUMVIブランド下のPSをRM治療のために承認したと発表した。この承認に関連して、Viatris許可協定によると、私たちは1,000万ドルのマイルストーン支払いを受けた。
2023年11月,FDAと開催されたEPOP 2会議の結果を発表し,APX 3330の登録終点の推進を支援することで合意した。Ocuphireは2024年2月にFDAにSPAを提出し,臨床試験案と統計分析計画の合意を求め,FDAとSPA合意時に研究設計パラメータと期待時間の詳細を共有する。
2024年1月,われわれはFDAの臨床試験プログラムに関する合意を受け,Lynx−2段階3試験の統計分析の評価を計画し,br勧告の治療暗(中間)光線条件下での視力低下の適応を評価することを発表した屈折性角膜手術後それは.我々の開発·ビジネスパートナーViatrisは2024年上半期に第3段階開発を継続する予定である。
CEOが交代する
2023年4月19日、当社は元総裁兼当社最高経営責任者のミナ·スッチの採用を終了し、リチャード·ロジャースを当社の臨時総裁兼最高経営責任者に任命した。2023年6月8日、当社はSoochさんと別居と解除協定を締結した。
当社は2023年11月1日、George Magrath,M.D.,M.B.A.,M.S.をCEOと取締役会メンバーに任命することを発表した。この任命により、臨時総裁と最高経営責任者を務めたリチャード·ロジャースが辞任し、取締役会に残り続けた。
リンカーンパーク資本基金有限責任会社(“リンカーンパーク”)との購入協定
88
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
2023年8月10日、Ocuphireは購入契約の30ヶ月以内に、Ocuphireは唯一の権利を有するが、リンカーン公園は購入契約の30ヶ月の期間内に常に最大5,000万ドルの普通株を購入し、額面0.0001ドル(“普通株”)を随時購入するようにリンカーン公園と普通株購入契約(“購入契約”)を締結した。購入契約を締結するとともに、Ocuphireもリンカーン公園と登録権協定(“登録権協定”)を締結することにより、吾らは登録声明に基づいて購入契約に基づいてリンカーン公園に発行された普通株“br}株式の転売を登録することに同意した。購入契約に署名した後、吾らはリンカーン公園に246,792株の普通株を発行し、購入契約に基づいて当社の普通株を購入することを承諾した代償として発行した。リンカーン公園は私たちの普通株を直接または間接的に空売りしたり、反対したりしないことに同意した。
財務運営の概要
許可と協力収入
これまで,許可と連携収入は,許可譲渡に関連した一度に払戻不可能な支払い,追加のマイルストーン支払い,Viatris許可合意によって得られた費用精算から来ており,Biosense Global LLC(“Biosense”)やProcessa PharmPharmticals,Inc.(“Processa”)とRexahn RX−3117薬物化合物に関するライセンス合意から小さい程度であった。Viatrisライセンス契約に関連する研究開発サービスの精算を取得する際に収入を確認し、Viatrisとの合意から潜在的なマイルストーンと特許使用料の支払いの追加収入を得ることが可能であると予想されます,
Biosense,Processa,または将来締結される他のライセンス契約であるが,以下の説明の理由により,マイルストーンや印税支払いを稼ぐために必要な販売レベルは非常に不確定である.
これまで、上記のライセンスおよび協調収入に加えて、パートナーのViatrisがRYZUMVIを商業化したり、規制の承認を得たりしない限り、相当な収入は生じないと予想され、RM以外の適応の商業化にAPX 3330またはPSの使用が開始された。APX 3330、PS、または私たちが将来追求する可能性のある他の候補製品の開発を適時に完了できない場合、あるいは規制部門の承認を得ることができない場合、私たちが相当な収入を創出する能力は影響を受けるだろう。
運営費
Ocuphireの運営費は,一般と行政費用および研究開発費の2つに分類される。
一般と行政費用
一般費用および行政費用には、主に、研究や開発活動と直接関係のない職能者のための賃金、福祉、株式ベースの報酬費用が含まれる。その他の重大なコストには、取締役および上級管理者の保険および他の財産および責任リスク、知的財産および会社の事務に関連する法律費用、会計および税務サービスの専門費用、商業コンサルタントが提供する他のサービスおよび法律和解が含まれる。
研究と開発費
今まで私たちは研究開発費相関のある主にAPX 3330とPSの臨床段階発展を対象としている。研究開発費には、研究開発活動を行うことによるコスト、研究開発従業員およびコンサルタントの報酬、福祉および株式報酬コスト、非臨床研究および臨床試験に関連するコスト、監督活動、臨床活動を支援する製造活動、許可費、非合法特許コスト、特定の研究開発を行う外部サービスプロバイダに支払う費用、および管理費用の分配が含まれる。
89
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
Viatrisライセンスプロトコルによると,PS開発に関する予算研究と開発費用はViatrisが全額精算する.ただし,すべての研究·開発費用は,PSに関する費用を含めて発生した費用として費用を計上し,第三者で発生した費用は契約作業を実行する際に費用を計上する.我々は,
研究やプロジェクトの状態を監視し,我々の外部サービスプロバイダから伝票を受信することで,サービス提供によるコストを積算する.実際の費用が知っている時、私たちは計算すべきプロジェクトを調整する。研究開発活動は私たちのビジネスモデルの核心だ。
APX 3330やPSの臨床開発後期段階での開発コストは,早期開発段階のコストよりも高くなることが予想され,これは主に後期臨床試験や関連非臨床研究の規模と持続時間の増加によるものである。私たちは今後数年で私たちの研究開発費が増加すると予想している。しかしながら、APX 3330、PSおよび他の候補製品を完成させる現在または未来の非臨床計画および臨床試験の持続時間、コストおよびタイミングを決定することは困難である。
融資コスト
融資コストには株式限度額融資による発行コストが含まれており、リンカーン公園は以下でさらに検討する。
利子支出
利子支出…からなる短期ローンに関する元本利息コスト(保険証券融資に関する)はこの間返済されておらず、2022年5月に全額返済されている。
派生負債の公正価値変動
派生負債の公正価値変動…からなる私たちの株式限度額融資に関連する派生負債の公正価値変化
は、株式限度額融資未償還期間中である。また、Rexahn承認株式証に関する権利証負債の公正価値変化は返済されていないが、本プロジェクトにも含まれている。
その他の収入,純額
その他の収入(支出)は、純額には、現金及び現金等価物に投資して稼いだ利息、株式投資の実現及び未実現収益(損失)及び贈与金及び他の出所に関する精算(発生時)が含まれる。さらに、他の収入(支出)純額には、前Rexahn株主と締結された、または価値のある権利協定(“CVRプロトコル”)によって支払われた吾らによって支払われたお金も含まれる。
所得税支給
所得税の準備には、米国の連邦および州所得税、ならびに財務報告のための資産および負債の帳簿金額と所得税のための金額との間の一時的な差を反映する純税影響の繰延所得税と関連する推定免税額の変化が含まれる。現在、将来の課税収入の不確定性及びその他の現金化能力或いは余剰繰延税項純資産に影響する関連要素を考慮して、2023年12月31日と2022年12月31日までの繰延税項純資産はすでに全額推定値を計上して準備している。
90
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
経営成果
表に示した期間の運営結果(千単位)をまとめた
|
この年度までに
十二月三十一日
|
||||||||||||
|
2023
|
2022
|
変わる
|
||||||||||
|
許可と協力収入
|
$
|
19,049
|
$
|
39,850
|
$
|
(20,801
|
)
|
|||||
|
運営費用:
|
||||||||||||
|
一般と行政
|
11,959
|
7,269
|
4,690
|
|||||||||
|
研究開発
|
17,653
|
14,355
|
3,298
|
|||||||||
|
総運営費
|
29,612
|
21,624
|
7,988
|
|||||||||
|
営業収入(赤字)
|
(10,563
|
)
|
18,226
|
(28,789
|
)
|
|||||||
|
融資コスト
|
(1,328
|
)
|
—
|
(1,328
|
)
|
|||||||
|
利子支出
|
—
|
(9
|
)
|
9
|
||||||||
|
派生負債の公正価値変動
|
80
|
—
|
80
|
|||||||||
|
その他の収入,純額
|
1,837
|
(14
|
)
|
1,851
|
||||||||
|
所得税前収入
|
(9,974
|
)
|
18,203
|
(28,177
|
)
|
|||||||
|
所得税支給
|
(12
|
)
|
(315
|
)
|
303
|
|||||||
|
純収益
|
$
|
(9,986
|
)
|
$
|
17,888
|
$
|
(27,874
|
)
|
||||
2023年12月31日までと2022年12月31日までの年次比較
許可と協力収入
許可·協力収入は2023年12月31日現在で1,900万ドルであるのに対し、2022年12月31日現在の事業年度は3,980万ドルである。2023年の収入はFDAがPSを承認した100ドルの記念碑的支払いから来ていますブランド名はRYZUMVI薬剤による散瞳の治療に使用される。1,000万ドルのマイルストーン支払い前に、Viatris許可プロトコルに関連する初期取引総価格に含まれるので、会社によって制限される。最後に,2023年に確認された収入残高は,Viatrisライセンスプロトコルに関する研究開発サービスの産出に関係している.2022年の例年の収入は、主に開発、製造、輸入、輸出、商業化の永久再許可許可の譲渡と関係がある第4四半期のViatrisライセンス契約から来ていますPS製品また,研究開発サービスからの補償も少ない.
一般と行政
2023年12月31日までの年度の一般·行政費は1200万ドルであるのに対し,2022年12月31日までの年度は730万ドルである。470万ドルの増加は、元最高経営責任者の退職に関する解散費120万ドル、株式報酬140万ドル、その他の人事関連コスト30万ドル、専門サービス80万ドル、法律支援60万ドル、業務発展活動およびその他のコスト純額20万ドルによるものである。一般および行政費用には、2023年12月31日と2022年12月31日までの年間で、それぞれ240万ドルと110万ドルの株式ベースの報酬支出が含まれている。
研究と開発
以下の表は、列挙期間中の我々の研究と開発費用の構成要素(千単位)を説明する
|
この年度までに
|
||||||||||||
|
十二月三十一日
|
||||||||||||
|
2023
|
2022
|
変わる
|
||||||||||
|
外部コスト:
|
||||||||||||
|
0.75%フェントラミン液(PS)
|
$
|
9,983
|
$
|
8,962
|
$
|
1,021
|
||||||
|
APX 3330
|
4,818
|
3,047
|
1,771
|
|||||||||
|
未分配
|
678
|
647
|
31
|
|||||||||
|
総外コスト
|
15,479
|
12,656
|
2,823
|
|||||||||
|
内部コスト:
|
||||||||||||
|
従業員関連費用
|
2,148
|
1,637
|
511
|
|||||||||
|
施設、用品、その他
|
26
|
62
|
(36
|
)
|
||||||||
|
内部総コスト
|
2,174
|
1,699
|
475
|
|||||||||
|
研究開発費総額
|
$
|
17,653
|
$
|
14,355
|
$
|
3,298
|
||||||
91
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
2023年12月31日までの年間の研究開発費は1,770万ドルですが、2022年12月31日までの年間は1,440万ドルです。330万ドルの増加は主にPS Vega−2試験の臨床コストが40万ドル増加し,APX 3330とPSの製造·毒性活動が約250万ドル増加し,株による報酬を含む賃金コストが70万ドル増加し,その他の運営費が30万ドル増加したが,規制活動の60万ドル減少によって相殺された。Viatrisライセンスプロトコルによると,PS
製品開発に関する予算研究開発費はViatrisが全額精算する。研究開発費には、2023年12月31日と2022年12月31日までの年間で、それぞれ110万ドルと70万ドルの株式報酬支出が含まれている。
融資コスト
2023年12月31日までの年間融資コストは130万ドルで、リンカーン公園との株式投資額融資に帰属する発行コストが含まれています。2022年12月31日までの年間融資コストは何もありません。
利子支出
2022年12月31日までの年間の利息支出は9,000ドルで、その中には短期融資(保険融資に関する)に関する元本利息が含まれている2023年12月31日までの年間、私たちは何の利息も支出していない。
派生負債の公正価値変動
派生負債の公正価値変動は、権益限度額融資、すなわち2023年12月31日までの年度収益80,000ドルに起因し、私たちの普通株式公正価値変動と権益限度額融資の下で各割引層が発行可能な普通株式潜在株式数に起因する。最後に、tRexahn株式証に関する権利証負債の公正価値変動も本プロジェクトに含まれているが,2023年12月31日と2022年12月31日までの年間で最低水準である。負債に分類された最後のRexahn株式承認証は2023年4月に満期になり、行使されなかった。
その他の収入,純額
Ocuphireは2023年12月31日までの年間で他の収入があり,純額は180万ドルであり,主に我々の現金や手元現金等価物に関する利息収入に関係している。
2022年12月31日までの年間で、私たちは170,000ドルの短期投資の未実現純損失と実現された通貨損失約3,000ドルによって、主に現金と現金等価物に関する159,000ドルの利息収入によって相殺される他の費用純額14,000ドルがあります。
所得税支給
所得税支出には,2023年12月31日と2022年12月31日までの年度までの米国連邦と州所得税が含まれており,金額はそれぞれ12,000ドルと315,000ドルであり,br}は純営業損失の繰越と研究控除後の課税所得額の純額から生じている。
92
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
流動性と資本資源
資本資源
2023年12月31日まで、私たちの主要な流動性源は5050万ドルの現金と現金等価物を含む。2023年末に手元の現金は私たちの業務に資金を提供するのに十分だと信じています本出願の日から少なくとも12か月以内それは.2023年12月31日現在、私たちの現金と現金等価物は、主に2つの大型金融機関の現金預金と現金等価物投資に投資されています。
歴史資本資源
私たちが業務に資金を提供する主な現金源は、各種株式6,330万ドルの発行と転換可能手形850万ドルの発行であり、Ocuphire変換可能手形の本チケット(“Ocuphire変換可能手形”)を含む。また,我々は2022年第4四半期に3,500万ドルの一度に現金払戻不可能な支払いを受け取り,2023年第4四半期に1,000万ドルのマイルストーン支払いを受け取り,2022年第4四半期以来開発に関するコスト精算を受けており,これらはViatris許可協定に関連している.
リンカーン公園購入契約
2023年8月10日私たちは普通株購入協定リンカーン公園と“リンカーン·パーク”株式限度額融資(“購入契約”)それは.購入契約は、購入契約に規定されている条項と条件を満たした場合、唯一の権利がありますが、リンカーン公園に購入契約の30ヶ月以内に最大5000万ドルの会社普通株を時々購入するように指示する義務はありません。購入契約を締結するとともに、吾らも登録権利協定を締結し、この合意に基づき、吾らは登録声明に基づいて、購入契約に基づいてリンカーン公園に発行された普通株株式の転売を登録することに同意した。購入契約に署名した後、吾らはリンカーン公園に246,792株の当社普通株を発行し、購入契約に基づいて当社の普通株を購入することを承諾した代償として発行した。リンカーン公園は、私たちの普通株を直接または間接的に空売りまたはヘッジすることに同意しました。上記の約束株を除いて、購入合意によると、2023年12月31日までに、合計1,300,000株の普通株が売却され、総収益は450万ドルです。2023年第3四半期までは、購入契約に基づいて普通株を売却しなかった。
市場で計画する
2024年1月10日、私たちは証券法に基づいてS-3保留登録表を提出した。この表は2024年1月23日にアメリカ証券取引委員会によって発効が発表され、この表によると、当社は時々1.75億ドルの証券の発売を自ら決定することができる2021年3月11日、私たちはJones Trading Institution Services LLC(“Jones Trading”)
と販売契約を締結し、この協定によると、私たちは時々任意に、またはJones Tradingを代理および/または依頼者として、総発行価格が4,000万ドルに達する普通株(“ATM”)を発売または販売することができる。ATM機が成立して以来、2023年12月31日までに6,045,316株の普通株が売却され、総収益は2,270万ドルだった。
登録された直売製品
2021年6月4日,我々はA.G.P./Alliance Global Partners(“AGP”)と配給エージェント合意に達した.配給代理契約の条項によると、AGPは2021年6月8日に合計3,076,923株の自社普通株および引受権証を売却し、1株4.875ドルの発行価格および0.5株のRDO承認株式証の発行価格で1,538,461株の自社普通株(“RDO株式承認証”)を購入し、総収益は1,500,000ドルであり、AGPの費用および関連発売支出を差し引いて1,100,000ドルである。購入契約には、Ocuphireの習慣陳述、保証と合意、成約の習慣条件、Ocuphireの賠償義務、当事者の他の義務、終了条項が含まれる。
93
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
RDO株式証の行使価格は1株6.09ドルで、2021年6月8日の初期発行日に行使でき、初期行使日後5年で満期になる。限られた例外を除いて、RDO株式証所有者は、所有者およびその関連会社がその権利を行使した後に4.99%(または発行日前に所有者を選択した場合、9.99%を超える)を超える発行済み普通株を所有することを前提としているRDO株式証の任意の部分を行使する権利がない。しかし、事前に通知された場合、所有者は利益所有権制限を増加または減少させることができ、
また、いずれの場合も利益所有権制限は9.99%を超えてはならないことが条件である。2023年12月31日まで、まだ1,538,461件のRDO株式証明書が決済されていない。証券の発売は,S-3表における我々の有効保留登録
宣言に基づいて行った.
M&A前融資
証券購入協定
2020年6月17日、Private Ocuphire、Rexahn及びいくつかの投資家は証券購入協定を締結し、この協定は2020年6月29日に改訂及び再記述された(改訂及び再記述を経て、“証券購入協定”と呼ばれる)。証券購入協定によると、合併完了時には、投資家はプライベートOcuphire取締役が投資した300,000ドルとレクサーンの取締役を含む2,115万ドルの現金を投資する(“合併前融資”)。合併前融資により、(I)Private Ocuphireは投資家にPrivate Ocuphire普通株(“初期株”)を発行して売却し、合併中の交換比率によって合計1,249,996株普通株(“転換後の初期株”)に変換し、(Ii)は投資家の利益であり、Private Ocuphireの追加普通株(“追加株式”)を第三者信託に入金する。合併中の交換比率に基づいて合計3,749,992株の普通株式(“変換後追加株式”)に変換され、この変換後の追加株式は2020年11月19日に引渡し(またはbr可能株式となる)を投資家に発行し、(Iii)吾らは合併完了後の第10取引日に投資家1人当たり(X)Aシリーズ承認株式証を発行することに同意し、(A)投資家が購入した転換初期株式、(B)転換済み追加株式が交付されたか、または投資家に交付可能な合計に相当する。証券購入プロトコルに記載されている交付のいかなる制限にも影響を与えることなく、(C)投資家に発行されるBシリーズ株式承認証に係る普通株初期数(ありあれば)および(Y)普通株株式を購入する追加株式証明書。
免除協定
2021年2月3日から、合併前に融資に投資した各投資家(“所有者”)は、当社と放棄合意(総称して“放棄合意”と呼ぶ)を締結した。
放棄合意に基づいて、所有者とOcuphireは、ある権利を放棄することに同意し、最終的にAシリーズ権証とBシリーズ承認株式証の行使価格と数量を決定し、ある融資制限を解除し、ある漏れ合意の期間を延長し、ある所有者の場合、株式証に関連するある株式登録権を付与する。
免除協定はAシリーズ株式承認証に記載されている全額ラチェット逆希釈条項を永久的に免除することを規定している(ある逆希釈条項は以前にAシリーズ株式承認証の負債会計処理を招いたため)。免除協定の発効日に、Aシリーズ株式承認証は株式に再分類される。
免除協定によると、すべてのBシリーズ株式承認証は、すべての所有者に関連する株式総数を1,708,335株に固定する。
第1回株式承認証
Aシリーズ権証は2020年11月19日に発行され、初期行使価格は1株4.4795ドルで、発行時に直ちに行使することができ、期限は
発行日から5年となる。Aシリーズ株式承認証は合計5,665,838株の普通株式を行使することができる(その中に掲載されているいかなる行使制限にも影響を与えない)。2023年12月31日までに、5,665,838件のAシリーズ権証が返済されていません。
94
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
発行時、Aシリーズ株式承認証にはいくつかの条項が含まれており、Ocuphireがその時の行使価格よりも低い価格で任意の普通株を発行または売却する場合、または合意に達して任意の普通株を発行または売却する場合、初期行使価格の引き下げを招く可能性があり、株式証関連株式数
を上昇させる可能性がある。免除協定の条項によると、これらの
条項はもはや有効ではない。
Bシリーズ株式承認証
Bシリーズ株式承認証の発行価格は0.0001ドルであり、発行時に行使することができ、(I)予約日(上述したように)または(Ii)投資家のBシリーズ株式証全数行使の日(その中に記載された行使制限が発効しない)の翌日に満了する。Bシリーズ株式承認証は最初に合計665,836株の普通株式
を行使することができ(その中に記載されているいかなる行使制限にも影響を与えない)、最終的に免除協定に署名した後に1,708,335株の普通株を行使することができる。2023年12月31日まで、Bシリーズ株式承認証はすべて決済されなかった。
発行時に、Bシリーズ株式承認証にはいくつかの条項が含まれており、これらの条項は、45取引日のリセット期間内の普通株のドル出来高加重平均価格に依存する追加のBシリーズ株式承認証の発行をもたらす可能性がある。合意を放棄する条項によると、このような規定はこれ以上有効ではない。
Ocuphire変換可能手形
2018年5月から2020年3月まで、Ocuphire変換可能手形を発行し、総収益は850万ドルで、Ocuphire変換可能手形の本チケットを含む。
Ocuphire変換可能手形は2020年3月10日に最終成約した。Ocuphire転換債券の年利率は8%である。2020年11月4日、合併の完了に伴い、Ocuphireのすべての発行済み手形は977,128株のOcuphire普通株に変換された。
キャッシュフロー
次の表は、示す期間のキャッシュフロー(千単位)をまとめた
|
この年度までに
十二月三十一日
|
||||||||
|
2023
|
2022
|
|||||||
|
経営活動が提供する現金純額
|
$
|
(1,112
|
)
|
$
|
14,314
|
|||
|
投資活動のための現金純額
|
—
|
—
|
||||||
|
融資活動が提供する現金純額
|
8,979
|
3,786
|
||||||
|
現金と現金等価物の純増加
|
$
|
7,867
|
$
|
18,100
|
||||
経営活動のキャッシュフロー
2023年12月31日までの1年間に,運営活動に使用された現金110万ドルは純損失1000万ドルにより,一部は480万ドルの非現金運営費用
で相殺され,Ocuphire運営資産と負債変化による約410万ドルの純現金源で相殺された。非現金支出には、主に350万ドルの株式ベースの報酬、120万ドルの株式限度額融資に関連する非現金融資コスト、20万ドルが融資活動の発行コストに再分類されるが、派生負債10万ドルの公正価値収益によって相殺される。営業資産と負債の変化は,主にViatrisライセンス契約に関連する売掛金や契約資産が250万ドル減少し,次いで売掛金や売掛金が120万ドル増加したことと,Ocuphireの運営費変動に関する前払い費用が40万ドル減少したためである。
95
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
2022年12月31日までの1年間で,経営活動が提供した現金は1,430万ドル,純収益は1,790万ドルであり,一部は200万ドルの非現金運営支出によって相殺され,Ocuphire運営資産と負債変化による約560万ドルの現金純使用によって相殺された。非現金支出には主に180万ドルの株式報酬と20万ドルの短期投資が含まれており、純損失を達成していない。営業資産と負債の変化は,主にViatrisライセンス契約に関する売掛金と契約資産が490万ドル
増加し,次いで我々の売掛金と売掛金の減少,Ocuphireの運営費用変動に関する前払い費用が合計70万ドル増加したためである。
投資活動によるキャッシュフロー
報告期間内に投資活動はなかった.
融資活動によるキャッシュフロー
2023年12月31日までの年間で,融資活動が提供する現金純額は900万ドルであり,主に購入プロトコルとATMから受け取った収益を含み,発行コスト
を差し引くと,それぞれ430万ドルと460万ドルである。
2022年12月31日までの1年間に、融資活動が提供する現金純額は380万ドルで、その中には主にATMから受け取った収益、発行コストを差し引いた430万ドルが含まれ、一部は50万ドルの短期ローン支払いで相殺されている。
流動資金と資本資源要求
2023年12月31日まで、私たちは5050万ドルの現金と現金同等物を持っている。これまで,許可と連携の収入は,一度に払い戻し不可能な3500万ドルからであり,記念碑的な1000万ドルの支払いと,Viatrisライセンス契約により得られた精算と予想精算費用,Biosense Global LLC(“Biosense”)やProcessa製薬会社(“Processa”)とRexahn RX−3117医薬化合物とのライセンス合意であった。Viatrisライセンス契約に関連する研究開発サービスの精算時に収入を確認し、Viatris、Biosense、Processa、または将来締結された他のライセンス契約の将来の潜在的マイルストーンおよび特許権使用料支払いから追加収入を得る可能性があると予想されていますが、以下の説明の理由で、マイルストーンや特許使用料支払いを稼ぐために必要なbrの販売レベルは極めて不確定です。
これまで,上記の許可や協調収入以外には,我々のパートナーViatris
が商業化されない限り,大量の収入は生じないと予想されているRYZUMVIまたは、規制部門の承認を得て、RM以外の適応のためのAPX 3330またはPSの商業化を開始する。APX 3330、PS、または将来追求する可能性のある任意の他の候補製品の開発がタイムリーに完了できなかった場合、または規制部門のこのような候補製品の承認を得ることができなかった場合、私たちが相当な収入を創出する能力は影響を受けるだろう。
また、2023年8月10日に、私たちはリンカーン公園と購入契約を締結し、その中で、私たちは唯一の権利があるが、義務はありません。リンカーン公園は購入契約の30ヶ月以内に、5000万ドル以下の普通株を時々購入するように指示しました。購入契約を締結するのはATMを補充するためです。購入契約を締結すると同時に、リンカーン公園と登録権利協定を締結し、この協定に基づいて、購入契約に従ってリンカーン公園に発行された普通株の転売を登録したか、または可能性があることに同意した。私たちは2023年8月11日に米国証券取引委員会に目論見書付録(文書番号333-252715)を提出した。購入契約の条項によると、販売価格が1株0.25ドル以下であれば、普通株をリンカーン公園に売ることができません。したがって、私たちが購入契約期間内にこの施設を完全に使用することは保証されない。
96
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
もし私たちが株式または転換可能な債務証券を売却することによって追加資本を調達する場合、私たちの株主の所有権権益は希釈され、これらの証券の条項は、清算、株式承認証、または普通株主としての権利に悪影響を及ぼす他の特典を含む可能性がある。債務融資が可能であれば、関連する可能性のあるプロトコルは、追加債務を招く、資本支出を行う、または配当を宣言するなど、特定の行動をとる能力を制限または制限する契約を含む。もし私たちが将来製薬パートナーとの協力、戦略連合、または許可手配を通じてより多くの資金を調達すれば、私たちは私たちの技術、未来の収入流の貴重な権利を放棄しなければならないかもしれない、または私たちに不利になる可能性のある条項で許可証を授与しなければならないかもしれない。もし私たちが株式や債務融資を通じて、あるいは必要な時に協力、戦略連盟、許可手配を通じてより多くの資金を調達することができない場合、私たちは私たちの製品開発、将来の商業化努力を延期、制限、または中止する必要があるかもしれません。あるいは私たちの候補製品を開発してマーケティングする権利を与えて、そうでなければ、私たちは私たちの製品を自分で開発し、マーケティングすることを望んでいます。
将来の資本需要
Viatrisライセンスプロトコルによると,PS開発に関する予算研究と開発費用はViatrisが全額精算する.APX 3330の開発は多くの不確実性の影響を受けており、私たちが基づいているこれらの推定は、現在の予想とは大きく異なることが証明され、現金資源の使用速度が現在の予想よりも速い可能性がある。また、臨床試験において早期候補製品と試験候補製品を推進する過程はコストが高く、これらの臨床試験の進展時間も確定していない。私たちが成功的にbr収益に移行できるかどうかは、私たちのコスト構造をサポートするのに十分な製品販売レベルを達成することにかかっている。私たちは私たちが永遠に利益を上げたり、経営活動から正のキャッシュフローを生成することを保証できない。
契約義務と約束
施設レンタル
私たちはキャンセルできないレンタル施設を経営しています。このレンタルは2024年12月31日に満了します(改訂されました)、基本レンタル料は月3,000ドルです。
Apexian分割許可プロトコル
2020年1月21日,我々はApexian再許可協定を締結し,この協定に基づき,独占的な世界特許と他の知的財産権を取得し,眼科疾患や糖尿病関連疾患の治療に関連するRef−1阻害剤計画を構成した。Ref-1阻害剤計画中の先導化合物はAPX 3330であり、現在開発中であり、NPDR患者DRを治療する経口投与錠剤である。Ref-1阻害剤(例えばAPX 3330、APX 2009およびAPX 2014)が血管新生および炎症を減少させる作用機序は、糖尿病性黄斑浮腫(DME)、湿性老年性黄斑変性(WAMD)および地理的萎縮(GA)および非眼科適応などの他の網膜疾患の治療に役立つ可能性がある。Ocuphireは現在,網膜や非眼科適応へのチューブ拡張の一部として,全身(経口)経路を除く局所投与経路
を評価している
Apexian再許可プロトコルについては,Apexianとそのいくつかの連属会社に843,751株の普通株を発行した.
私たちはApexian分割許可プロトコルに従って最初の眼科適応と最初の糖尿病適応のための一次マイルストーン支払いに同意した。これらのマイルストーン支払い
は、(I)特定の開発および規制マイルストーンに対する支払い(米国で第1の第2段階試験(この試験が主要終点に達した場合)および第1の第3段階キー試験を完了し、FDAによる化合物の最初の新薬申請に対する規制承認を提出し、得られた)、合計1,100万ドルに達し、(Ii)合計2,000万ドルに達する特定の販売マイルストーン支払いを含み、各純販売マイルストーン支払い
は、このようなマイルストーンが初めて達成されたときに1回支払われる。
また,Apexian再ライセンスプロトコルで特許がカバーしている製品の純売上高の1桁パーセントに相当する使用料を支払うことにも同意した.本年度報告日まで、マイルストーンや特許使用料支払いはトリガされていません。
97
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
他の約束
正常な運営過程において、著者らはサプライヤーとキャンセル可能な調達承諾を締結し、各種の肝心な研究、臨床と製造サービスに応用した。このような計画がカバーする調達約束は私たちの研究と開発によって変化するかもしれない。
他の資金需要
上述したように、私たちのいくつかの現金需要は、私たちが行っているAPX 3330の研究と開発の資金と関係があり、私たちの知的財産権ライセンスの下の任意の潜在的なマイルストーンと印税義務を含む。私たちが将来行う可能性のある設計、開発、臨床前および臨床活動の議論については、本年度報告の“第1部-業務-APX 3330の潜在的臨床計画-PS潜在臨床計画-将来の許可および買収機会-製造-Apexian再許可プロトコル-米国医薬品の審査および承認”を参照されたい。このような活動に必要な予想される現金支出が含まれている。
私たちの今後12ヶ月以内の他の現金需要には、売掛金、課税費用、調達約束、その他の流動負債が含まれています。我々の他の12ヶ月を超える様々な契約義務および約束された現金需要には、2024年までの第三者サービスプロバイダとの運営リースおよび契約協定が含まれている可能性がある。本年報“第II部第8項-財務諸表及び補足データ”内の付記3-承諾及び又は事項を参照して、将来の支払い時間のリース義務及びライセンス契約のさらなる詳細を知ることができる。
私たちは、私たちが商業販売から十分な収入を生むまで、手元の現金、未来の株式と債務融資、Viatris許可協定での精算支払い、潜在的なマイルストーンと特許使用料支払い、および任意の未来の協力および許可協定を通じて、商業販売から十分な収入が生じるまで、費用を支払いたいと思っています。
重要な会計政策と試算
私たちの財務諸表はアメリカ公認会計基準に従って作成されています。これらの会計原則は、財務諸表日までの資産及び負債報告金額、並びに列報期間中の収入及び費用報告金額に影響を及ぼす可能性がある推定及び判断を要求する。私たちが依存している推定と判断は、私たちがこれらの推定と判断を下した時に得られた情報に合理的に基づいていると信じている。もしこれらの見積もりと実際の結果の間に大きな違いがあれば、私たちの財務業績は影響を受けるだろう。以下では、我々のより重要な見積もりと判断を反映した会計政策を紹介し、これらの会計政策は、我々の報告書の財務業績を全面的に理解し、評価するために最も重要であると考えられる。
我々の重要会計政策は、本年報第2部第8項-財務諸表と補足データの付記1-会社記述と重要会計政策要約で検討した。私たちは、以下の会計政策と見積もりが、私たちの報告書の財務結果を全面的に理解し評価するために最も重要だと思います。これらの推定は、本質的に不確実な問題に関連するので、最も困難、最も主観的、または最も複雑な判断を行う必要がある。私たちは私たちの取締役会の監査委員会とこのような重要な会計政策と推定と関連した開示を検討した。私たちはこれまで何の大きな変化もしていませんが、以下に述べる分野の会計方法が将来大きく変わる可能性も考えられません。
98
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
許可と協力収入
我々は、財務会計基準委員会(FASB)会計基準編纂(ASC)606の規定に従って許可および連携収入を会計処理する取引先と契約した収入それは.この指導意見は収入をどのように確認するかを決定するための統一的なモデルを提供する。私たちは収入確認に関する許可と連携協定
を締結しました。我々は,まず契約の独立価格に基づいて契約の取引価格を契約下の履行義務ごとに割り当て,許可と連携収入を確認する.履行義務ごとの独立価格
は,その公正価値をもとに,キャッシュフロー手法を採用し,確率評価に基づく項目ごとの予想グローバル純利益,内部予測による予測,業界データ,同一業界他指導会社からの情報,その他の関連要因を含む様々な仮定を考慮した。私たちは将来的に私たちの既存のbrライセンスや協力収入に関する重大な可変価格調整はないと予想しています。ライセンスおよび連携収入決定に関する議論は、本年度報告書“第2部、第8項-財務諸表および補足データ”
における注釈9-ライセンスおよび連携プロトコルを参照されたい。
株に基づく報酬
私たちはASC 718“報酬-株式報酬”の規定に従って株式報酬を計算します。したがって,授受権益ツールに関する補償コストは,授出日に再計測されないBlack-Scholesモデルの公正価値で確認される.持分道具の没収が発生した時、私たちは記録するつもりだ。日公許可価値の付与と当社の株式ベースの報酬の適用に関する議論は、本年度報告第2部第8項-財務諸表および補足データにおける付記6-株式ベースの報酬を参照されたい。
所得税資産負債
将来の課税所得額の不確実性と、私たちの残りの繰延税項目の純資産現金化能力に影響する他の要素を考慮して、私たちは私たちの繰延税項目の純資産に全額推定を提供しました。詳細は,本年度報告“第2部第8項−財務諸表及び補足データ”に11−所得税が付記されている。
事件があったり
私たちは通常の業務過程でいくつかの許可協定に関連する義務を含む多くの意外な状況に遭遇するだろう。詳細は、本年度報告“第2部第8項--財務諸表及び補足データ”に付記されている3−引受金及び又は事項を参照されたい。
最近の会計公告
財務会計基準委員会や他の基準制定機関は時々新しい会計声明を発表するだろう。適用された場合、私たちは指定された発効日に基づいてこのような新しい基準を採用するつもりだ。本年報に掲載されている財務諸表付記に別途開示されていない限り、最近発表されたいかなるまだ発効していない基準(S)の影響は、当社の財務状況やbr}経営業績に実質的な影響を与えないと信じています。最近発表された会計基準(S)についてより深く議論するには、本年度報告第2部第8項-財務諸表および補足データの付記1“会社説明および重大会計政策概要”を参照されたい。
| 第七A項。 |
市場リスクの定量的·定性的開示について
|
適用されません。
99
カタログ表
オクフィル製薬会社
表格10-K
表格10-K
| 第八項です。 |
財務諸表と補足データ
|
本プロジェクトに要求される情報は、本年度報告に含まれ、F−1ページから始まり、参照されて本明細書に組み込まれる。
| 第九項です。 |
会計·財務開示面の変化と会計士との相違
|
ない。
| 第9条。 |
制御とプログラム
|
情報開示制御とプログラムの評価
改正された1934年証券取引法第13 a-15条または取引法第13 a-15条または取引法の要求に基づいて、最高経営責任者および最高財務官、我々の最高経営責任者および最高財務官の指示の下、本年度報告が包含されるまでの期間終了時規則13 a-15(E)または15 d-15(E)で定義された開示制御プログラムおよびプログラムを評価した。この評価によると、我々の最高経営責任者および最高財務官は、本年度報告がカバーする期間が終了するまで、我々の開示制御および手続きが有効であると結論した。
経営陣財務報告内部統制年次報告書
我々の経営陣は、財務報告書の信頼性を合理的に保証し、米国公認会計原則に基づいて外部目的で財務諸表を作成するために、財務報告書の十分な内部統制を確立し、維持する責任がある。財務報告の内部統制には、(I)米国公認会計原則に基づいて財務諸表を作成するために必要な取引を記録するために必要な取引を記録するための合理的な保証を提供し、米国公認会計原則に基づいて財務諸表を作成し、私たちの収入と支出は私たちの経営陣と取締役会の許可のみに基づいて行われるための合理的な保証と手続きが含まれる。(Iii)財務諸表に重大な影響を与える可能性のある不正買収、使用、または処分について、財務諸表に重大な影響を与える可能性のある不正買収、使用または処分について合理的な保証を提供することを防止またはbr}する。
私たちの経営陣は、CEOやCEOを含め、財務報告に対する内部統制がすべてのエラーやすべての詐欺を防止または発見できないことを認識しています。設計や動作がどんなに良くても、絶対的な保証ではなく、合理的な保証しか提供できず、保証制御システムの目標が実現される制御システム。制御システムの設計は,資源制約が存在し,
かつ制御の利点がそのコストに対して考慮されなければならないという事実を反映しなければならない.さらに、すべての制御システムの固有の限界のため、どの制御評価も、誤りまたは不正による誤った陳述が発生しないこと、またはある場合には、すべての制御問題および不正事例が検出されないことを絶対に保証することはできない。任意の制御システムの設計は、将来のイベント可能性のいくつかの仮定にある程度基づいており、任意の設計
がすべての潜在的な未来条件でその目標を成功的に達成することを保証することはできない。
経営陣は、最高経営責任者と財務責任者の参加の下、2023年12月31日現在、すなわち我々の財政年度終了時の財務報告の内部統制を評価した。経営陣は、トレデビル委員会後援組織委員会が発表した“内部統制--総合枠組み(2013年)”で決定された基準に基づいて評価を行った。この評価によると、経営陣は、財務報告に対する会社の内部統制は2023年12月31日から発効すると結論した。
財務報告の内部統制の変化
2023年12月31日までの四半期内に、財務報告の内部統制(“外国為替法案”規則13 a-15(F)および15 d-15(F)の定義に基づく)には何の変化もなく、これらの変化は私たちの財務報告の内部統制に大きな影響を与えたり、合理的に大きな影響を与える可能性がある。
100
カタログ表
オクフィル製薬会社
表格10-K
表格10-K