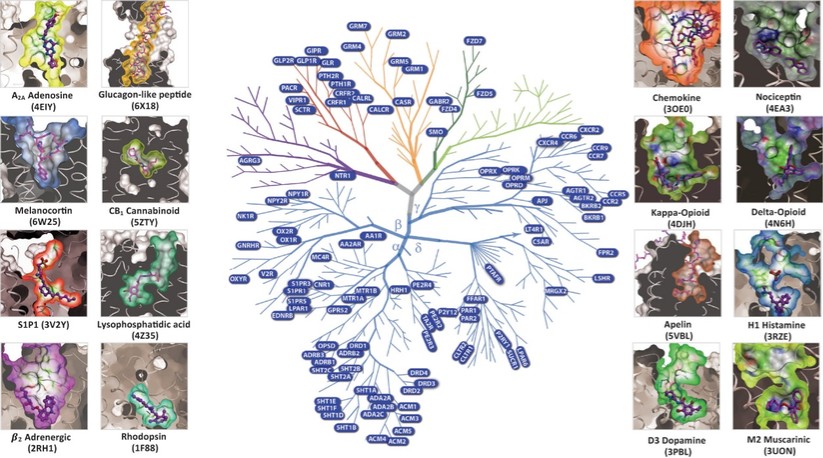
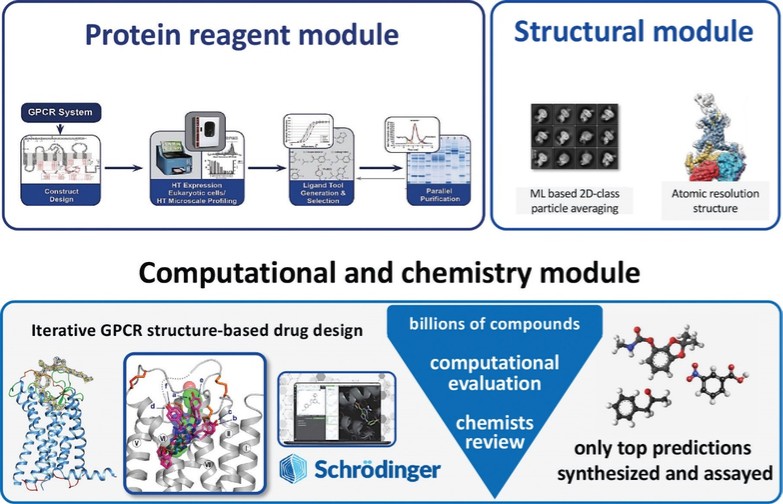
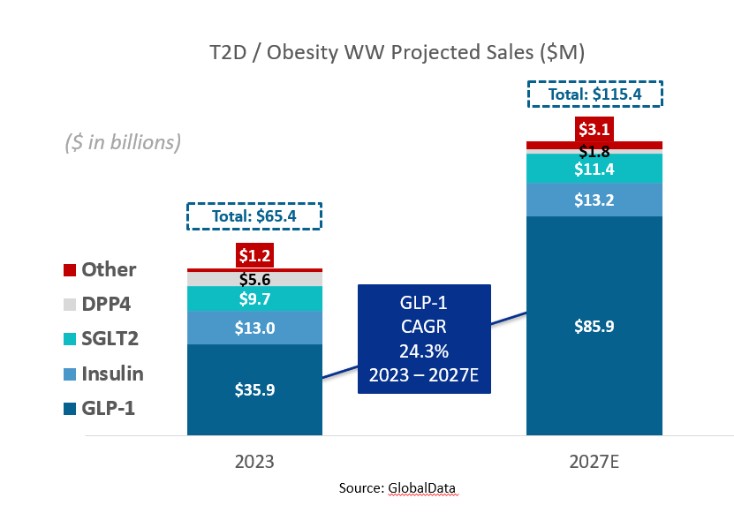
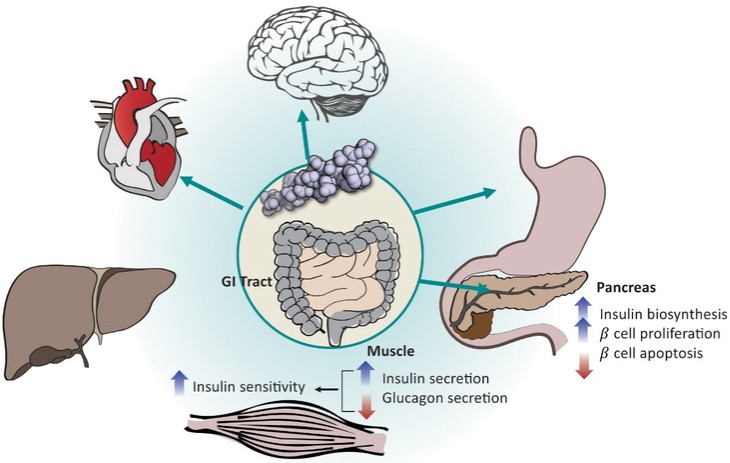
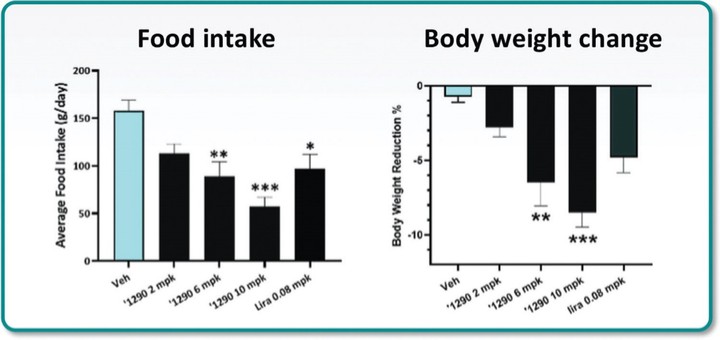
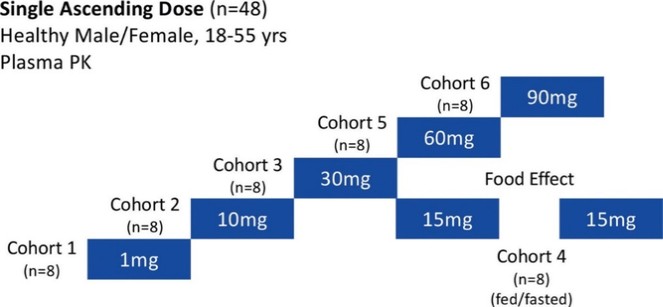
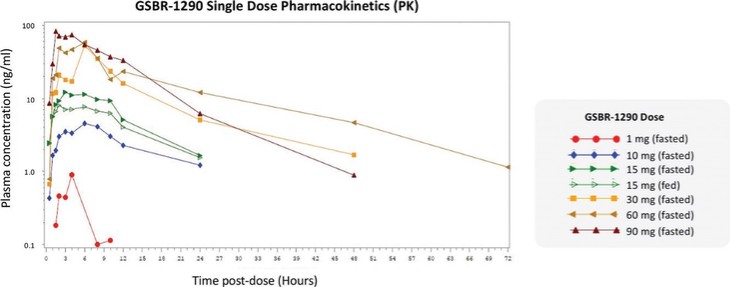
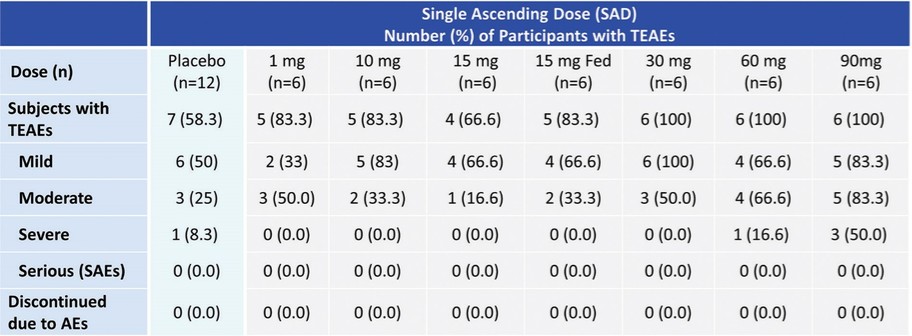
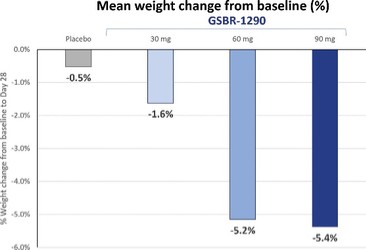
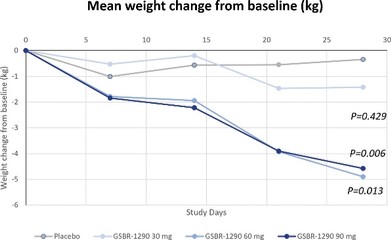
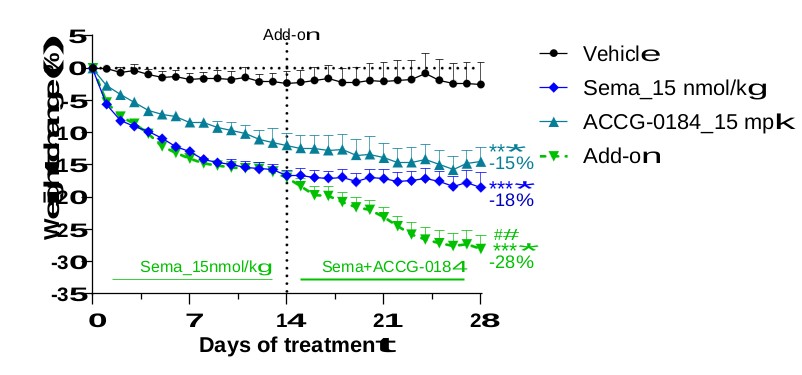
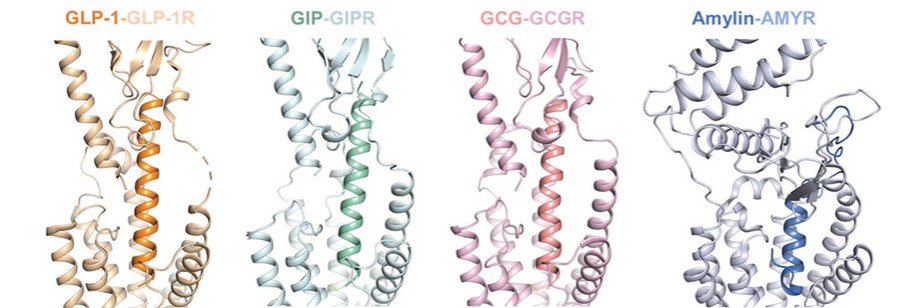
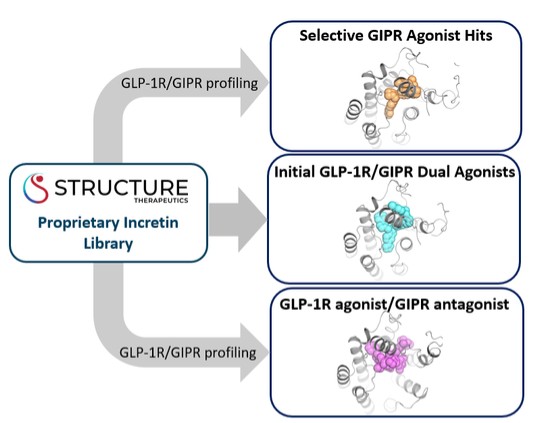
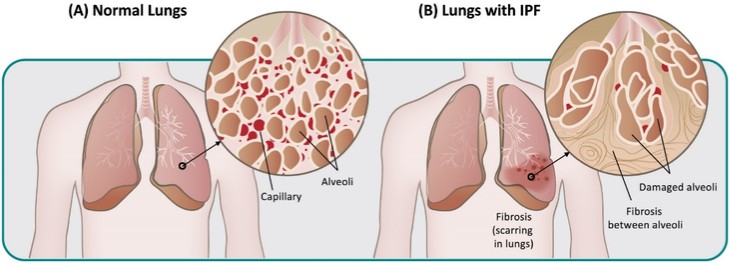
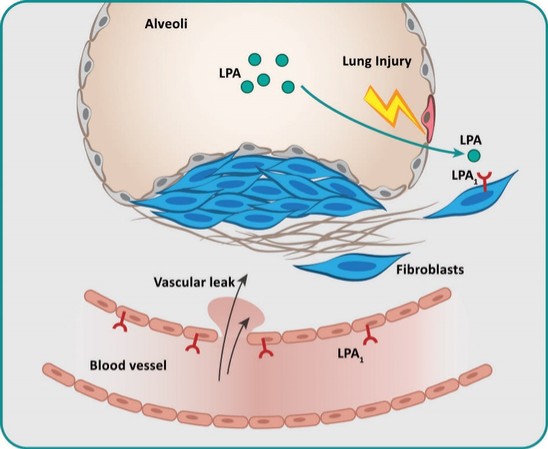
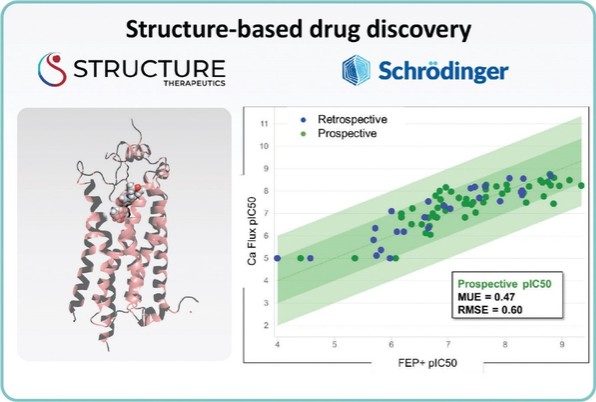
アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表10-K
☒ | 1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
本財政年度末まで十二月三十一日, 2023
あるいは…。
☐ | 1934年証券取引法第13又は15(D)節に提出された移行報告書 |
移行期になります至れり尽くせり
依頼書類番号:001-41608
構造治療会社です。
(登録者の正確な氏名はその定款に記載)
ケイマン諸島 | | 98-1480821 |
(法団又は組織の他の司法管区として設立された国) | | (国際税務局雇用主身分証明書番号) |
601 Gateway大通り, 900番のスイートルーム 南サンフランシスコ, カリフォルニア州 | | 94080 |
(主にオフィスアドレスを実行) | | (郵便番号) |
登録者の電話番号、市外局番を含む:(650) 457-1978
同法第12条(B)に基づいて登録された証券:
| | | | |
| | 各取引所名 | | |
クラスごとのタイトル | | 取引コード | | いつ登録しましたか |
米国預託株式(ADS)は、1株当たり3株の普通株に相当し、1株当たり額面0.0001ドル | | GPCR型 | | ナスダック世界市場 |
普通株は、1株当たり0.0001ドルの価値があります* | | | | ナスダック世界市場* |
* 違います。Tは取引に用いられるが,米国預託株式の登録に関する場合に限られる
同法第12条(G)により登録された証券:なし
登録者が証券法規則405で定義されている経験豊富な発行者である場合は、再選択マークで示してください。
はい、そうです☐ 違います。 ☒
登録者がこの法第13節または第15節(D)節に基づいて報告を提出する必要がないかどうかを再選択マークで示す。
はい、そうです☐ 違います。 ☒
登録者が(1)過去12ヶ月以内(または登録者がそのような報告の提出を要求されたより短い期間)に、1934年の証券取引法第13または15(D)節に提出されたすべての報告を提出したかどうか、および(2)このような提出要求を過去90日以内に遵守してきたかどうかを、再選択マークで示すはい、そうです ☒違います☐
登録者が電子的に提出されたか否かをチェックマークで示す;過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間内に)、S−Tルール405(本章232.0405節)に従って提出された各相互作用データファイルを要求するはい、そうです ☒違います☐
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
| | | | | | | | |
大型加速ファイルサーバ☐ | | ファイルマネージャを加速する☐ | | 非加速ファイルサーバ ☒ | | 規模の小さい報告会社☒ | | 新興成長型会社☒ |
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する☐
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる☐
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する☐
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に第240.10 D−1(B)条に従って受信されたインセンティブベースの補償に従って回復分析を行う必要があるかどうかを再選択マークで示す☐
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)。はい、そうです☐違います☒
2023年6月30日現在,すなわち登録者が最近完成した第2財期の最終営業日であり,登録者の非関連会社が保有する登録者普通株の総時価は約$である501.7ナスダック世界市場における米国預託証券代表の登録者普通株の終値によると、米国預託株式あたり41.57ドル。非連属会社が保有する投票権付き株式の時価を決定する際には、登録者の普通株には、取締役や上級職員1人当たり実益が所有する普通株、および登録者1人当たり10%以上発行されている者は含まれていない。他の目的に対して,このような関連地位の決定は必ずしも決定的な決定であるとは限らない.
2024年2月29日現在、登録者の発行済み普通株式数は、1株当たり額面0.0001ドルである139,794,124このうち129,882,333株の普通株は米国預託証明書の形で保有されている。
引用で編入された書類
登録者は,登録者が2023年12月31日までの財政年度後120日以内に第14 A条に基づいて証券取引委員会に提出した2024年株主総会最終依頼書の一部の内容を,本年度報告の第3部Form 10−Kに引用により組み込む予定である。