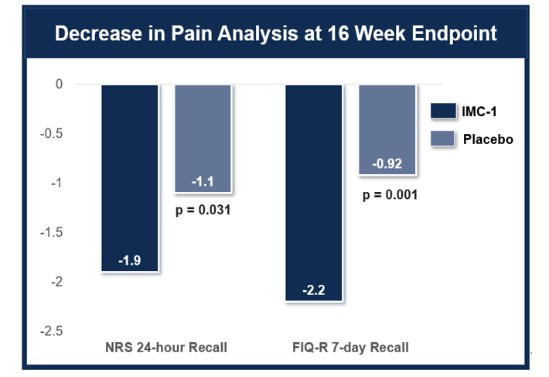
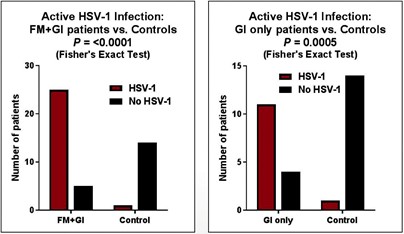
米国
証券取引委員会
ワシントンD.C. 20549
フォーム10-K/A
修正第1号
☒ | 1934年の証券取引法のセクション13または15 (d) に基づく年次報告書 |
終了した会計年度について 12 月 31 日, 2023
または
☐ | 1934年の証券取引法第13条または第15条 (d) に基づく移行報告書 |
コミッションファイル番号: 001-39811
ビリオス・セラピューティクス株式会社
(憲章に明記されている登録者の正確な名前)
| |
デラウェア州 | 85-4314201 |
(法人または組織のその他の管轄区域の州) | (IRS雇用者識別番号) |
| |
44 ミルトンアベニュー, アルファレッタ, ガスの | 30009 |
(主要執行機関の住所) | (郵便番号) |
登録者の電話番号 (市外局番を含む): (866) 620-8655
同法第12条 (b) に従って登録された証券:
| | | | |
| | | | 各取引所の名前 |
各クラスのタイトル | | トレーディングシンボル | | どちらに登録したか |
普通株式、1株あたり額面0.0001ドル | | ビリ | | ナスダックキャピタルマーケット |
法のセクション12 (g) に従って登録された証券:なし
登録者が証券法第405条で定義されている有名なベテラン発行体である場合は、チェックマークで示してください。
はい ☐ いいえ ⌧
登録者が法の第13条または第15(d)条に従って報告を提出する必要がない場合は、チェックマークで示してください。
はい ☐ いいえ ⌧
登録者が、(1) 1934年の証券取引法第13条または第15条 (d) で提出が義務付けられているすべての報告書を過去12か月間(または登録者がそのような報告を提出する必要があったほど短い期間)に提出したかどうか、および(2)過去90日間にそのような申告要件の対象であったかどうかをチェックマークで示してください。 はい ⌧いいえ ☐
登録者が電子的に提出したかどうか、過去12か月間(または登録者がそのようなファイルを提出する必要があったほど短い期間)に、規則S-T(この章の§232.0405)の規則405に従って提出する必要があったすべてのインタラクティブデータファイルをチェックマークで示してください。はい ⌧いいえ ☐
登録者が大規模な加速申告者、加速申告者、非加速申告者、小規模な報告会社、または新興成長企業のいずれであるかをチェックマークで示してください。取引法規則12b-2の「大規模加速申告者」、「加速申告者」、「小規模報告会社」、および「新興成長企業」の定義を参照してください。
| | | |
大型加速フィルター☐ | アクセラレーテッド・ファイラー☐ | 非加速ファイラー ⌧ | 小規模な報告会社☒ |
| | | 新興成長企業☒ |
新興成長企業の場合は、登録者が取引法第13条 (a) に従って規定された新規または改訂された財務会計基準を遵守するために延長された移行期間を使用しないことを選択したかどうかをチェックマークで示してください。 ☐
登録者が、監査報告書を作成または発行した登録公認会計士事務所が、サーベンス・オクスリー法(15 U.S.C. 7262 (b))のセクション404(b)に基づく財務報告に対する内部統制の有効性に関する報告書を提出し、経営陣による評価を証明したかどうかをチェックマークで示してください。 ☐
証券が同法第12条(b)に従って登録されている場合は、申告に含まれる登録者の財務諸表に、以前に発行された財務諸表の誤りの訂正が反映されているかどうかをチェックマークで示してください。 ☐
これらの誤りの訂正のいずれかが、セクション240.10D-1(b)に従って、関連する回復期間中に登録者の執行役員が受け取ったインセンティブベースの報酬の回収分析を必要とした修正であるかどうかをチェックマークで示してください。 ☐
登録者がシェル会社(同法第12b-2条に定義)であるかどうかをチェックマークで示してください。はい ☐いいえ ⌧
登録者が最近終了した第2四半期の最終営業日である2023年6月30日の時点で、登録者の非関連会社が保有する登録者の普通株式の時価総額は$でした24,371,988.18ナスダック・キャピタル・マーケットで報告された終値に基づいています。
2024年2月26日現在の登録者の普通株式の発行済み株式数は 19,257,937.
参照により組み込まれた文書
第III部には、2023年12月31日に終了した年度から120日以内に、規則14Aに従って2024年定時株主総会に関連して登録者が証券取引委員会に提出する最終的な委任勧誘状(「委任勧誘状」)からの参照により特定の情報が組み込まれています。ただし、そのような期間内にそのような委任勧誘状が提出されない場合、そのような情報はこの年次報告書の修正に含まれますフォーム10-Kは、この120日以内に提出してください.