アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
本財政年度末まで
あるいは…。
手数料書類番号
(登録者の正確な氏名はその定款に記載)
(明またはその他の司法管轄権 会社や組織) |
(税務署の雇用主 |
(主にオフィスアドレスを実行) |
(郵便番号) |
登録者の電話番号、市外局番を含む:(
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
|
取引 記号 |
|
登録された各取引所の名称 |
|
|
|||
|
|
|
|
(ナスダック世界市場) |
同法第12条(G)に基づいて登録された証券:ありません
登録者が証券法第405条に規定する有名な経験豊富な発行者である場合は、再選択マークで表示してください
登録者がこの法第13又は15(D)条に従って報告書を提出する必要がないか否かを、再選択マークで示すはい、そうです ☐
登録者が(1)過去12ヶ月以内(または登録者がそのような報告を提出する必要があるより短い期間)に1934年の証券取引法第13条または15(D)条に提出されたすべての報告書を再選択マークで示すかどうか、および(2)このような提出要求を過去90日以内に遵守してきた
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
|
☒ |
|
ファイルマネージャを加速する |
|
☐ |
|
|
|
|
|
|||
非加速ファイルサーバ |
|
☐ |
|
規模の小さい報告会社 |
|
|
|
|
|
|
|
|
|
新興成長型会社 |
|
|
|
|
|
|
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する☐
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に第240.10 D−1(B)条に従って受信されたインセンティブベースの補償に従って回復分析を行う必要があるかどうかを再選択マークで示す☐
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)はい、そうです ☐ 違います。
ナスダック世界市場普通株2023年6月30日の終値によると、登録者の非関連会社が保有する投票権と無投票権普通株の総時価は約1ドルである
登録者が発行された普通株式の数2024年2月26日は
引用で編入された書類
登録者の2024年株主総会に関する最終委託書部分は,本報告日後に米国証券取引委員会に提出され,参考として本報告の第3部に組み込まれる。このような依頼書は,登録者が2023年12月31日までの財政年度終了後120日以内に米国証券取引委員会に提出される.
監査役事務所ID: |
監査役の名前: |
監査役位置: |
キセノン製薬会社です。
表格10-K
2023年12月31日までの財政年度
カタログ表
|
|
|
|
|
|
|
|
|
ページ |
第1部 |
|
1 |
||
第1項。 |
|
業務.業務 |
|
3 |
第1 A項。 |
|
リスク要因 |
|
28 |
項目1 B。 |
|
未解決従業員意見 |
|
66 |
プロジェクト1 C。 |
|
ネットワーク·セキュリティ |
|
67 |
第二項です。 |
|
属性 |
|
67 |
第三項です。 |
|
法律訴訟 |
|
68 |
第四項です。 |
|
炭鉱安全情報開示 |
|
68 |
|
|
|
|
|
第II部 |
|
69 |
||
五番目です。 |
|
登録者普通株,関連株主事項及び発行者が株式証券を購入する市場 |
|
69 |
第六項です。 |
|
[保留されている] |
|
70 |
第七項。 |
|
経営陣の財務状況と経営成果の検討と分析 |
|
71 |
第七A項。 |
|
市場リスクの定量的·定性的開示について |
|
80 |
第八項です。 |
|
財務諸表と補足データ |
|
81 |
第九項です。 |
|
会計と財務情報開示の変更と相違 |
|
102 |
第9条。 |
|
制御とプログラム |
|
102 |
プロジェクト9 B。 |
|
その他の情報 |
|
103 |
プロジェクト9 Cです。 |
|
検査妨害に関する外国司法管区の開示 |
|
103 |
|
|
|
|
|
第三部 |
|
104 |
||
第10項。 |
|
役員·幹部と会社の管理 |
|
104 |
第十一項。 |
|
役員報酬 |
|
104 |
第十二項。 |
|
ある実益所有者の担保所有権及び経営陣及び株主に関する事項 |
|
104 |
十三項。 |
|
特定の関係や関連取引、取締役の独立性 |
|
104 |
14項です。 |
|
チーフ会計士費用とサービス |
|
104 |
|
|
|
|
|
第4部 |
|
105 |
||
第十五項。 |
|
展示·財務諸表明細書 |
|
105 |
第十六項。 |
|
表格10-Kの概要 |
|
107 |
|
|
|
|
|
サイン |
|
108 |
||
i
パー?パーT I
前向きに陳述する
適用法によれば、本年度報告書にForm 10−K形式で含まれるいくつかの陳述が前向き陳述を構成する可能性がある。“すべき”、“許可する”、“意図する”、“可能”、“信じる”、“計画”、“可能な結果”、“継続する予定”、“継続する”、“予想する”、“推定する”、“プロジェクト”または同様の表現、またはそのような言葉またはフレーズの否定は、“前向きな陳述”を識別することを目的とする。未来の予想を議論し、将来の運営結果や財務状況の予測を含む、または他の“前向き”情報を述べているので、これらの声明をよく読むべきだ。これらの陳述は、私たちの未来の計画、目標、期待、意図、および財政的表現、およびこれらの陳述が根拠とする仮定と関連がある。これらの展望的な陳述は含まれているが、これらに限定されない
1
これらの展望性陳述はいくつかのリスクと不確定性の影響を受け、これらのリスクと不確定性は実際の結果と展望性陳述で予想されるものと大きく異なることを招く可能性がある。このような差をもたらす可能性のある要因には、本報告の第1の部分1 A項である“リスク要因”および本報告の他の部分で議論された要因が含まれるが、これらに限定されない。前向きな陳述は、私たちの経営陣の信念と仮定と、私たちの経営陣が現在把握している情報に基づいている。本報告のすべての声明と同様に、これらの声明は締め切りのみを説明しており、将来の事態の推移に応じてこれらの声明を更新または修正する義務はありません。本報告では,“我々”,“キセノン”,“会社”とはキセノン製薬会社とその子会社を指す。他の説明がない限り、本報告書のすべてのドルの金額はドルで表される。
また、“私たちが信じている”という声明と類似した声明は、関連テーマに対する私たちの信念と意見を反映している。これらの陳述は,本年度報告10−K表までの日に我々に提供された情報に基づいており,これらの情報がこのような陳述の合理的な基礎を構成していると考えられるが,このような情報は限られているか不完全である可能性があり,我々の陳述は,入手可能なすべての関連情報を徹底的に調査または検討していることを示していると解釈されてはならない.このような陳述は本質的に不確実であり、投資家たちにこのような陳述に過度に依存しないように想起させる。
このForm 10-K年次報告書には、XenonロゴおよびXenonの他の商標またはサービスマークを含む当社の商標および登録商標が含まれています。本年度報告でForm 10−K形式で出現する他の商標,商号またはサービスマークは,それぞれの所有者に属する。
リスク要因の概要
我々の業務は、本報告のタイトルが“リスク要因”と題する部分的に強調されたリスクおよび不確定要因を含む多くのリスクおよび不確定要因の影響を受ける。以下は私たちが直面している主なリスクの概要です
2
我々のリスク要因は,本報告の日までにこのような状況が存在しないことを保証しているわけではなく,このようなリスクや状況がすべてまたは部分的に発生していないと肯定的に解釈されるべきでもない。
プロジェクト1.ビジネス
私たちは神経科学に専念する生物製薬会社で、神経や精神障害患者の生活改善に取り組んでいます。我々は、てんかんやうつ病を含む高度に満たされていない医療ニーズ分野を解決するための新しい製品ラインを進めている。私たちの特許候補品に加えて、私たちはNeurocrine Biosciences、Inc.またはNeurocrine Biosciencesを含む学術および産業パートナーと協力している。
私たちの戦略
我々の目標は、一連の神経科学疾患を発見、開発、商業化する革新的な治療法を完全に統合し、利益を得るバイオ製薬会社を設立することである。
私たちの戦略の主な構成要素は
3
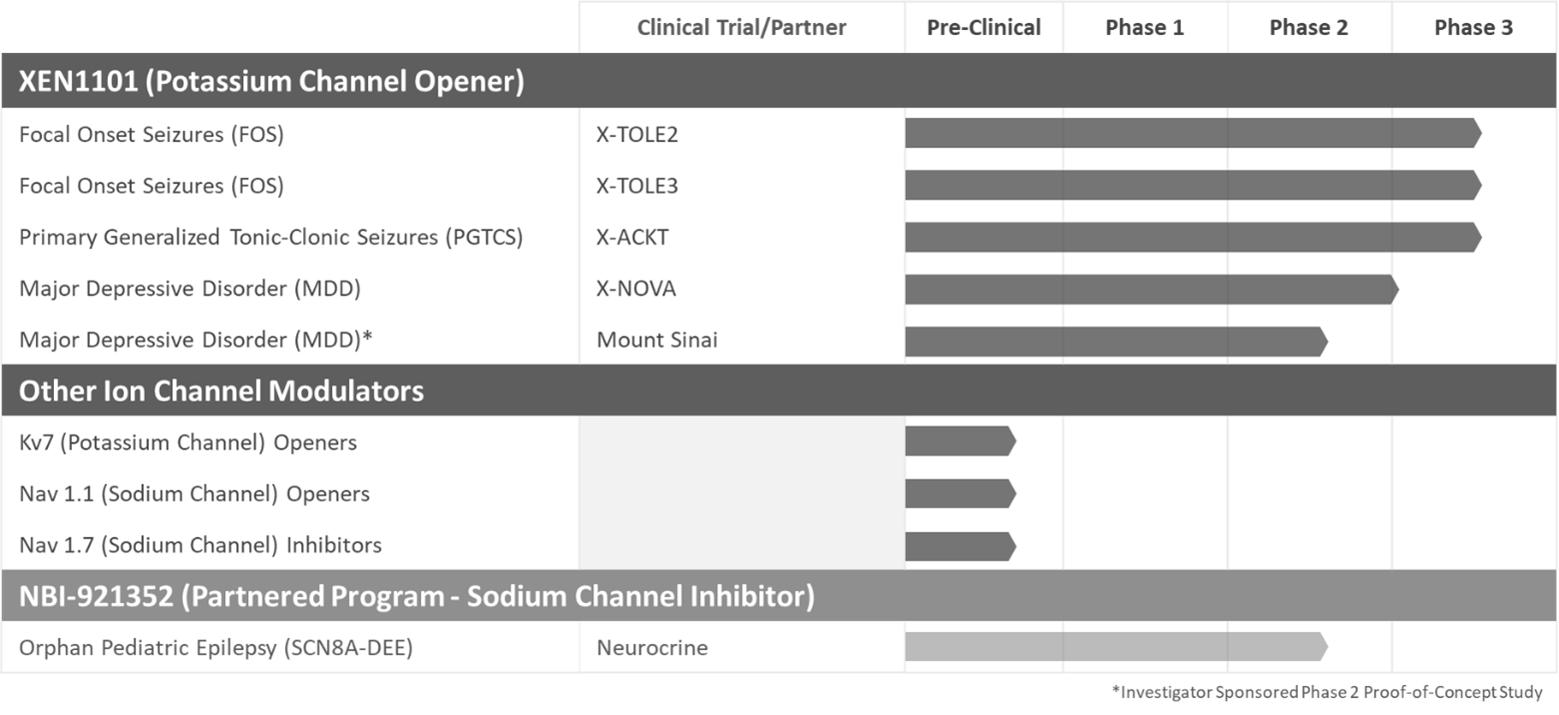
私たちのパイプは
私たちの候補製品
XEN 1101
XEN 1101は新型、有効なKV 7カリウムチャネル開口薬であり、てんかん、重篤な抑うつ障害或いはMDD、及び潜在的な他の神経疾患の治療に開発されている。
XEN 1101はてんかんを治療する
我々のXEN 1101 3期てんかん計画は,X−TOLE 2とX−TOLE 3と呼ばれる2つの同じ3期臨床試験を含み,この2つの試験は2 b期X−Tole臨床試験の後に設計された。これらの多中心、無作為、二重盲検、プラセボ対照試験は15 mg或いは25ミリグラムXEN 1101を食物と一緒に服用する補助治療の臨床有効性、安全性と耐性を評価しており、各研究は約360名の局所てんかん発作(FOS)患者がいる。主要な治療効果の終点はXEN 1101がプラセボと比べ、ベースラインから二重盲検期(DBP)までの毎月のてんかん発作頻度の中央値パーセンテージ変化(MPC)である。XENONはX−TOLE 2の患者登録が2024年末から2025年初めに完了すると予想している。
XEN 1101治療てんかん(原発性全身性強直−クローヌス発作)
著者らの3期X-Ackt臨床試験は原発性全般性強直クローヌス発作(PGTCS)の追加てんかん徴候に対する潜在的な監督管理提出を支持することを目的とした。この多中心、ランダム、二重盲検、プラセボ対照研究は約160名のPGTCS患者における補助治療として25ミリグラムXEN 1101の臨床有効性、安全性と耐性を評価している。プラセボと比較して、主要な治療効果の終点はベースラインからXEN 1101までのDBPの毎月PGTCS頻度におけるMPCである。
てんかん用XEN 1101(オープンタグ拡張)
X-TOLE 2、X-TOLE 3あるいはX-Ackt中のDBPを完成させた後、条件を満たす患者は開放ラベル拡張或いはOLE研究に入ることができ、最長3年に達する。また,進行中のX−Toleステージ2 b OLEは5年から7年に延長されており,XEN 1101のために重要な長期データを生成し続けている。
4
XEN 1101てんかん治療の臨床効果まとめ
第一段階:健康な被験者で行われた第一段階の研究により、XEN 1101は検査された用量において全体的な耐性が良好であり、その薬物動態学的特徴は滴定を必要とすることなく、毎日1回の食物投与計画を支持し、これはすべての第二段階と第三段階試験で使用されている。著者らは1つの第1段階の臨床試験を完成し、XEN 1101の健康被験者における単回上昇用量(SAD)と複数の上昇用量(MAD)の安全性、耐性と薬物動態学的特徴を評価した。XEN 1101の第1段階の結果は、交差食品効果行列(n=10)を含む5~30 mg(n=34、プラセボ=8)の6つのSAD行列からのデータを含み、単一用量は20 mgである。MAD結果は、3つのキューを含み、1日1回の用量が15~25 mg(n=18、プラセボ=6)であり、2つのキューを含み、15 mgは、それぞれ絶食および摂食状態で7日および10日を評価し、1つのキュー25 mgは、10日を超える食事状態を評価する。多くの副作用(AEs)は軽度あるいは中等度であり、自発的に緩和し、抗てんかん薬(ASM)と一致する。深刻な有害事象、死亡または臨床的に有意に遅延した心室再分極または実験室結果はなかった。第一段階の結果は、XEN 1101は検査された用量で一般的な耐性が良好であることを示した(単回用量は最大30 mg、複数回の用量は1日最大25 mg)。
ステップ1 b:XEN 1101‘Sが皮質興奮性を調節する能力と効力を評価するための1 b期経頭蓋磁気刺激(TMS)研究のデータにより、目標中枢組織に活性が存在し、そして著者らの2 b期の臨床試験の用量選択に参考を提供した。この1 b期二重盲検、プラセボ対照、ランダム交差TMS研究は20名の健康男性被験者を含む。すべての被験者について2時間および4時間でTMS測定を行い、XEN 1101が示す吸収段階の延長のため、一部の被験者について6時間で追加のTMS評価時点を増加させた。対象は、最初にXEN 1101またはプラセボの20 mg用量にランダムに分配され、その後、1週間の溶出期後に、別の群の治療群に交差した。XEN 1101は皮質脊髄興奮性を低下させ、濃度依存の安静運動閾値(RMT)の上昇を示し、RMTはTMS-EMGの重要な指標である。RMTはXEN 1101血中濃度に比例し,平均4.9±0.7%(P)増加した
2 b期X-Tole臨床試験:2021年10月、著者らは2 b期X-Tole臨床試験のTOPLINE結果を発表し、この試験は無作為、二重盲検、プラセボ対照の多中心研究であり、10 mg、20 mgまたは25 mg XEN 1101を1日1回の補助治療として成人局所てんかん患者における臨床治療効果、安全性と耐性を評価することを目的とした。この研究は、安全な集団中の325人のランダムに治療を受けた被験者と、治療を意図した集団の323人の被験者を改良して治療効果分析を行うこととを含む。被験者の平均年齢は40.8±13.3歳,8.9%,40.6%または50.5%の被験者は全研究過程で1,2または3種類の安定した背景ASMをそれぞれオンおよび継続し,研究に入るまで6回のASMの中央値を通過しなかった。すべての研究グループの中位ベースラインてんかん発作頻度は月約13.5回のてんかん発作であった。二重盲検期を終えた285名の被験者のうち、96.5%の被験者がXEN 1101の長期安全性、耐性および有効性を評価するためにOLEに入った。
DBPにおけるX−Tole治療効果の結果まとめ:X-Tole試験はその主要な治療効果の終点に達し、XEN 1101はプラセボ(単調用量と反応;pを示した
5
|
XEN 1101 25 mg (N=112) |
XEN 1101 20 mg (N=51) |
XEN 1101 10 mg (N=46) |
プラセボ (N=114) |
毎月局所発作頻度はベースラインの中央値より減少している |
52.8% (p |
46.4% (p |
33.2% (p=0.035) |
18.2% |
病気の患者少なくとも50%は-毎月焦点性てんかん発作頻度がベースラインよりも減少 |
54.5% (p |
43.1% (p |
28.3% (p=0.037) |
14.9% |
病気の患者少なくとも75%は-毎月焦点性てんかん発作頻度がベースラインよりも減少 |
29.5% |
29.4% |
8.7% |
6.1% |
病気の患者100% 毎月局所てんかん発作頻度はベースラインより減少している |
6.3% |
7.8% |
2.2% |
1.8% |
X-Toleは、以前に失敗したASMの数、研究に伴うASM、およびベースラインてんかん負担から定義される治療が困難な患者集団を含むので、異なるベースライン特徴を有する患者において追加的なサブ分析が行われた。以下の表は25 mg用量群内のてんかん発作減少の中央値パーセンテージのグループ分け分析を概説し、ベースライン時に疾病の重症度が低い患者のてんかん発作減少の幅が明らかに増加したことを示した
|
XEN 1101 25 mg |
プラセボ |
X-TOLEの全体 |
52.8% (N=112) |
18.2% (n=114) |
前に故障したASM>6 |
43.2% (n=45) |
14.2% (n=47) |
前に故障したASM 6 |
58.3% (n=67) |
20.5% (n=67) |
ASM=3を伴う |
51.3% (n=54) |
20.4% (n=56) |
随伴ASM 2 |
59.7% (n=58) |
14.4% (n=58) |
毎月ベースライン押収量>8.5回 |
50.8% (n=83) |
18.2% (n=84) |
ベースライン押収量毎月8.5ドルです |
70.6% (n=29) |
18.8% (n=30) |
また,全発作亜型のてんかん発作減少の解析では,25 mg用量群で全身性強直間代発作をきたす‘タイプ4’焦点性発作毎月焦点性発作頻度の中央値百分率が86.9%(n=23)減少した。二重盲検期内にベースライン月焦点性てんかん発作回数に達する時間を行ったイベント時間分析では、XEN 1101と比較して、XEN 1101はプラセボと比較して、てんかん再発率は明らかに低下し、用量依存関係を呈した。
てんかん発作の著明な減少は全体状況の著明な改善に関与しており,この点は医師が臨床グローバル変化印象(CGI−C)を用いて評価し,被験者がXEN 1101 25 mg群の患者のグローバル変化印象(PGI−C)尺度を用いて自己報告したものを以下の表に示す
|
XEN 1101 25 mg (N=112) |
プラセボ (N=114) |
CGI-C(一部の患者の病状は明らかに好転または明らかに好転) |
46.4% (p |
22.8% |
PGI-C(一部の患者の病状は明らかに好転または明らかに好転) |
42.9% (p=0.001) |
21.9% |
XEN 1101 25 mg群のCGI-CとPGI-Cは統計学的意義があり、XEN 1101 20 mg群のPGI-Cは統計学的意義があったが、CGI-CのXEN 1101 20 mg群とXEN 1101 10 mg群のCGI-CとPGI-Cはすべてプラセボより改善したが、統計学的有意差はなかった。
6
DBP X-TOLEセキュリティ結果のまとめ:XEN 1101はDBPで耐性が良好であり,そのAEsは他のASMと一致した。プラセボ群と比較して治療群の緊急有害事象(TEAE)の発生率は高く,プラセボ群では62.3%の患者,XEN 1101 10ミリグラム群で67.4%,XEN 1101 20ミリグラム群で68.6%の患者およびXEN 1101 25ミリグラム群で85.1%の患者が少なくとも1回のTEAEを経験した。すべての治療群において、TEAEが5%以上の原因は神経系疾患、精神疾患、全身疾患、胃腸疾患、眼疾患と感染である--大多数は中枢神経系と関係があり、重症度は軽度或いは中度であり、治療期の早期に発生する。すべてのXEN 1101用量群(n=211)の中で、最もよく見られるTEAEはめまい(n=52、24.6%)、傾眠(n=33、15.6%)、疲労(n=23、10.9%)および頭痛(n=21、10.0%)であった。プラセボを含む異なる用量群のめまい被験者は、プラセボ群8名の被験者(7.0%)、10ミリグラム群の3名の被験者(6.5%)、20ミリグラム群の13名の被験者(25.5%)、25ミリグラム群の36名の被験者(31.6%)であった。この研究のすべての4つの分枝において、緊急治療深刻な有害事象(SAE)の発生率は類似しており、プラセボ群の2.6%の患者、XEN 1101 10ミリグラム群の4.3%の患者、XEN 1101 20ミリグラム群の3.9%の患者、およびXEN 1101 25ミリグラム群の2.6%の患者が少なくとも1つの治療-緊急SAEを経験した。プラセボ群では3.5%,XEN 1101 10ミリグラム群では2.2%,XEN 1101 20ミリグラム群では13.7%,XEN 1101 25ミリグラム群では15.8%の被験者でAEによる治療中断があった。積極的な治療群では尿貯留TEAEが2例あり,そのうちの1例は投与量の減少が必要であり,2名とも服薬を継続し,他の変化や関与はなかったと報告されている。米国泌尿系協会症状指数の全体あるいは個別項目の平均差によると,尿滞留を示す証拠はない。安静や立位テストによるバイタルサインは,心血管側の懸念信号はなく,健康診断や神経検査では安全面の懸念信号はなく,心電図,安全実験室や尿分析でも懸念信号は認められなかった。体重変化は大きくなく,平均(SD)変化:プラセボ群で0.2 kg(2.4),10ミリグラム群で0.6 kg(2.3),20ミリグラム群で1.6 kg(2.2),25ミリグラム群で1.9 kg(2.9)であった。
X−Toleデータおよび一時開放ラベル拡張(OLE)データの他のPOST特設子分析:2022年と2023年には,XEN 1101段階2 b X−Tole臨床試験データと進行中のX−Tole OLE中期データの他のパケット分析を提供した。
X-Toleデータの他のサブ分析により、XEN 1101の迅速な効果は有効な治療性用量の使用開始と関係があることが分かった。プラセボと比較して,全用量の1週間以内にてんかん発作の中央値頻度は統計学的に有意に低下した。XEN 1101は1週目から急速に効果を発揮し,ベースラインと比較して週発作頻度の中央値は39.1%(P)減少した
OLE中期データの分析により、XEN 1101は全体的に依然として耐性が良好であり、毎日1回20 mgの用量で長期治療効果を産生し、分析締め切り2023年9月5日まで、OLE中の24ケ月の保留率は60%であった。18~30カ月のOLE研究期間中、てんかん発作頻度は、二重盲期ベースラインと比較して毎月持続的に減少し(78%~95%の中央値百分率変化)、ベースラインで1~2つのASMを受けた患者は、3つのASMを受けた患者よりも高い減少が認められた。≥3カ月、6カ月、および12カ月連続治療した患者のうち、37.5%、22.2%および14.9%の患者がOLEでてんかん発作の緩和を得た。連続3カ月,6カ月,12カ月の患者では,それぞれ56.4%,34.5%,23.6%の患者がOLEで少なくとも24カ月の治療を受けていた。OLE治療を受けた24ケ月後、てんかん患者の生活の質-31(QOLIE-31)の臨床的重要な改善はすべての患者(n=162)のてんかん懸念、社会機能と薬物効果のサブスケールで見られ、てんかんなし群(n=39)の改善は更に大きかった。また,QOLIE−31により測定された生活の質の改善は,最初に1年目に報告され,X−Tole OLEの翌年に維持または改善された。新たな安全シグナルは認められず,これまで解析されてきた臨床データでは,XEN 1101はOLEで良好な耐性を保ち続けており,その副作用は先の結果と一致し,他の抗てんかん薬の副作用と一致していることが示唆された。引き続き患者に顕著な利益を提供する潜在力に基づいて、著者らはすでにX-Tole OLEを5年から7年に延長した。
てんかんとてんかん発作タイプについて
てんかんは1種の慢性神経系疾患であり、その特徴は反復発作、理由と予測不可能な発作である。個人的には理由のないてんかんが2回(あるいは理由のないてんかんが繰り返される可能性がある)が2回あり,これらの発作は既知や可逆的な医学的状況によるものでなければてんかんと診断される。てんかん発作は一般的に2種類に分けられる:焦点性発作或いはFOS発作と全身性発作。FOSはてんかん患者に最もよく見られるてんかん発作タイプである。FOSは脳内に限局しており,局所にも脳全体にも拡散することができ,これは通常二次性全身性てんかんに分類される。FOSは米国てんかん発作の約60%を占め,FOS患者総数は約180万人であった。全身性てんかん発作は,脳両側あるいは大脳両側の細胞群に同時に関与する。この用語は原発全般性強直クローヌス発作、或いはPGTCSと呼ばれ、欠神発作と無張力発作を含む。米国では全身性発作がてんかん発作の約30%,約90万人を占め,その多くがPGTCSを経験している。米国で残る10%のてんかん発作の特徴は未知の発作であり,てんかん発作の開始が未知のときに発生する。より多くの情報が得られるに伴い,原因不明の発作は後で焦点性発作や全身性発作と診断される可能性がある。
7
米国ではてんかん発作の治療に多くのASMが用いられているが,PGTCSに用いられる薬剤は少ない。現在、FOS或いはPGTCS患者個体に対する治療の重点はてんかん発作頻度の減少であり、最終的な目標はてんかん発作の緩和である。早期治療は通常単一療法から始まり,多剤投与を増やして残存てんかん負担のある患者を管理する。多種の治療方案が選択可能であるにもかかわらず、50%もの患者は管理が悪いと考えられ、最初の治療経路は追加の治療方案が必要である。管理の悪い患者に対して、医師はますます補助治療の補充機序としててんかん発作を制御することに助けを求めている。FOSやPGTCSに対して新たな,より効果的かつ耐性のある治療が必要であり,これらの治療法は効果が速く,独自の機序は多薬併用において重要であり,服用しやすく(例えば,1日1回),持続すると考えられる。我々の市場研究によると、XEN 1101が承認されれば、FOSとPGTCSを解決するための納得できる価値主張を提供できると信じています。
重度抑うつ障害(MDD)用XEN 1101
2023年11月、著者らは168名の中重度うつ病(MDD)患者の1日1回の食物と共に10 mgと20 mg XEN 1101を服用する臨床有効性、安全性と耐性を評価した無作為、二重盲検、プラセボ対照、第二段階概念検証X-Nova臨床試験のTOPLINE結果を報告した。主な目的はモンゴメリ-オズバーグ抑うつ評価表(MADRS)の6週間以内の分数変化を用いて、中重度MDD患者の抑うつ症状の改善におけるXEN 1101とプラセボの治療効果を評価することである。
我々は、MDD後期XEN 1101臨床計画の開始を支援するために、2024年4月に米国食品医薬品局(FDA)と共に“第2段階終了”会議に参加する予定であり、その中には3つの第3段階臨床試験が含まれ、第1の第3段階研究は2024年下半期に開始される予定である。私たちはまたXEN 1101の未来の発展の他の潜在的な指標を評価している。
DBPにおけるX-Nova効果データの概要:研究の主な終点は6週間におけるMADRSの変化であり,プラセボ群の平均低下幅は13.90,XEN 1101 10ミリグラム群とXEN 1101 20ミリグラム群の平均低下幅はそれぞれ15.61と16.94であった。プラセボとXEN 11020 mg群の間には,有意な用量反応と臨床的に有意であったが統計学的有意差は認められなかった(p=0.135)。6週目にハミルトン抑うつ尺度またはHAM−D 17のあらかじめ指定された終点は統計学的有意差に達し,プラセボ群とXEN 11020 mg群の平均低下幅はそれぞれ10.18%と13.26%(p=0.042)であった。スナイズ−ハミルトン快楽尺度(Shaps)変化の肝心な副次終点では,6週目に快感欠損を測定した肝心な副次終点で統計学的有意差が得られ,プラセボ群では5.3,XEN 11020ミリグラム群では7.77(p=0.046)減少した。1週目のMADRSは統計学的有意差があり,プラセボ群は平均4.88%,XEN 11020 mg群は平均7.54%(p=0.047)低下し,比較的早い治療効果を示した。プラセボと比較して,医師が臨床世界的改善印象(CGI−I)を用いて評価したところ,XEN 1101 20ミリグラム群のうつ病症状が少なくとも軽微に改善したとの報告は統計学的に有意であった(p=0.004)。
DBPにおけるX-Novaセキュリティおよび耐性データの概要:XEN 1101耐性は良好であり、すべての治療薬物が報告した不良イベントの発生率は類似している。XEN 1101 20 mg群の最もよく見られるTEAEは眩暈(17.9%)、傾眠(10.7%)、頭痛(8.9%)と注意障害(8.9%)を含み、プラセボ群は眩暈(7.3%)、傾眠(1.8%)、頭痛(12.7%)と注意障害(0%)を報告した。XEN 1101 20ミリグラム群の中に3名の患者(5.4%)がいたが、プラセボ群は2名の患者(3.6%)であった。2つのXEN 1101治療群では重篤な有害事象やSAEは報告されず,プラセボ群では2名の患者(3.6%)が緊急SAEの治療を経験した。XEN 1101は著明な体重増加に関係なく,明らかな性機能障害も報告されていない。
研究者主導のXEN 1101のMDDにおける第2段階概念検証研究
著者らはまた、シナイ山のイカン医学院と協力し、進行中のXEN 1101の第二段階概念検証、ランダム、平行対照、プラセボ対照の多点研究を支持し、約60名の被験者のMDDの治療に用いた。この研究の主な目的はXEN 1101の脳奨励回路への影響を研究し、機能磁気共鳴画像(FMRI)による両側腹側線条体活動の変化を評価することである。第2の目標は,うつ病や快感欠損の臨床測定におけるXEN 1101とプラセボの効果をMADRSとShapsを用いてそれぞれテストすることである。
8
重度抑うつ障害(MDD)について
MDDはよく見られる慢性神経疾患であり、その特徴は情緒が低下し、喜びを感じることができず、罪悪感と無駄を感じ、精力不足、及びその他の2週間或いはそれ以上の時間持続する感情と身体症状であり、そして社会、職業、教育或いはその他の重要な機能を損害する。MDDは高度に流行し、治療が困難な疾患である。米国国立衛生研究院のデータによると、毎年7.8%のアメリカ成人(2100万人)がMDDを患っており、そのうちの約3分の2の人が抑うつに関連する深刻な損害を患っていると推定されている。国家精神衛生研究所が援助した抑うつ緩和の序列治療代替方案或いはSTAR*D試験の結果により、確定診断と治療患者の3分の2近くが第一線の治療の十分な治療反応を経験しておらず、これらの最初の失敗も二線治療を通過できなかったことは、新しい作用機序を有する新しい抗うつ薬の必要性を明らかにした。
XEN 1101に関する知的財産権
私たちはXEN 1101をカバーする知的財産権の組み合わせを保護し、拡大するための包括的な戦略を制定した。重要なことは、2021年には、(1)XEN 1101医薬物質および関連する医薬組成物の独特の結晶形態、およびそれらの調製および使用方法、(2)食事時または食事に近い食事時にXEN 1101を経口投与する様々な方法を含む2つの米国特許を付与されている。これらの米国特許はそれぞれ2040年と2039年に満了する予定であり,特許期間は延長されない。私たちの知的財産権の組み合わせのより詳細な説明については、私たちの候補製品ラインを含めて、次の“-知的財産権”を参照してください。
新たなルート機会
薬物発見に関する我々の専門知識から,我々の努力はイオンチャネル標的の識別に集中しており,新たな調節剤が重大な治療進歩を代表している可能性が考えられる。私たちのチャネルの拡張は、私たちの内部研究努力と、他の外部候補製品の買収または獲得の許可から来るかもしれません。最近の重点はKV 7,Nav1.1,Nav1.7に対する内部開発候補製品である。これらの臨床前候補薬物が臨床開発に入ることに伴い、追加の更新を提供する。
私たちの協力プロジェクトは
臨床分期選択性ナトリウムチャネル遮断薬NBI−921352によるてんかん治療
2019年12月、我々はNeurocrine Biosciencesと許可と協力協定を締結し、てんかんの治療法を開発した。Neurocrine BiosciencesはXEN 901(現在NBI−921352と呼ばれる)の独占的許可を有し,XEN 901は臨床段階選択的Nav1.6ナトリウムチャネル阻害剤であり,選択的Nav1.6阻害剤およびデュアルNav1.2/1.6阻害剤を含む開発のための臨床前化合物の独占的許可を有する。この協定はまた、2022年6月に完了したより多くの新型Nav1.6およびNav1.2/1.6阻害剤を発見、識別し、開発するための長年の研究協力を含む。合意条項によると、私たちはいくつかの臨床、規制、商業マイルストーン支払い、そして将来の販売特許権使用料を得ることができる。Neurocrine Biosciencesと達成されたこの合意条項のより詳細な説明については、以下の“-協力、商業、および許可協定”を参照されたい。
NBI−921352は、SCN 8 A発育性およびてんかん性脳症またはSCN 8 A−DIEを有する小児患者の治療のために開発されている。第2段階臨床試験は、SCN 8 A−DEEを有する小児患者(年齢2歳から21歳まで)におけるNBI−921352の使用を評価している。Neurocrine Biosciencesは,SCN 8 A−DIEにおけるNBI−921352の孤児薬およびまれな小児科疾患に対する米国食品医薬品局の指定を得ている。
2023年11月,Neurocrine Biosciencesは,成人FOS患者におけるNBI−921352の第2段階臨床試験を評価することはてんかん発作頻度の有意な減少を証明できなかったと報告しており,FOSにおけるNBI−921352のさらなる開発は計画されていない。
協力、ビジネス、ライセンス契約
Neurocrine Biosciences,Inc.と許可と協力協定に署名する。
2019年12月、2021年1月及び2022年2月の改訂後、吾らはNeurocrine Biosciencesと許可及び協力協定或いは協力協定を締結し、協力関係を確立し、協力協定に基づき、双方は著者らの臨床候補薬物XEN 901を含むナトリウムチャネル阻害剤を識別、研究及び開発し、現在NBI-921352、及びNeurocrine Biosciencesは協力協定に記載された条項と条件に基づいて独占的に開発及び商業化されたいくつかの臨床前候補薬物(“DTC”)及び研究化合物と呼ばれる。
9
Neurocrine Biosciencesは、これらの化合物を世界的に研究、開発および商業化するための、任意のヒト疾患または障害、状態、状態および/または疾患の治療、治癒、診断、予測または予防のための独自、印税、および再許可可能な許可を有しているが、協力協定に規定されているいくつかの例外は除外されている。我々はまた、特定の知的財産権の非排他性、非特許権使用料、再許可可能なライセンスをNeurocrine Biosciencesに付与し、精選NAV阻害剤(以下で定義する)として識別するための化合物をスクリーニングし、協力協定において明確に排除されたいくつかの化合物または排除された化合物を研究するために使用する。
協力合意期間内に、排除された化合物を除いて、協力合意の条項によると、我々または私たちそれぞれのどの関連会社も、その主要な作用機序である化合物を直接または間接的に研究、開発、製造し、選択NAV阻害剤と呼ばれる電位依存性ナトリウムチャネルNav1.2およびNav1.6を抑制する。
すべての当事者は、開発および研究計画の下で活動を行うことによって生じるすべての費用を個別に担当しているが、Neurocrine Biosciencesは、NBI-921352開発および研究活動について、特定の従業員および私たちによって発生した自己負担費用を精算し、特定のDTCに関連するいくつかの開発活動については、Neurocrine Biosciencesが精算することを約束することができることを前提としている。
Neurocrine Biosciences社は,開発計画で規定されている活動を除いて,化合物や化合物を含む任意の医薬製品のすべての開発と製造を独自に担当しており,費用と費用は完全にNeurocrine Biosciencesが負担しているが,共同出資案(以下の定義)を遵守しなければならない。私たちは、開発計画の下で指定された流行閾値または主要適応を満たすか、またはそれを超える第1の適応において、ある製品の開発に共通資金を提供し、協力協定または共同援助オプションの条項に基づいて、そのような製品の米国での純売上を計算し、純売上高で計算された1桁の中央値パーセントの印税増加を得る権利がある。もし私たちが共同出資選択権を行使すれば、双方はNeurocrine Biosciencesが適応に適用されるこのような製品の開発によって生じるすべての合理的で記録されたコストと費用を平均的に分担するが、このような製品を開発して米国以外の規制機関が承認するためのコストと費用のみを除外する。2023年12月31日現在、この選択権を行使していない。
Neurocrine Biosciencesは私たちに3000万ドルの現金支払いを含む5000万ドルの前払いを支払った。前払い金の残りについては,提携合意を締結するとともに,双方が株式購入プロトコル(定義は後述)を締結することにより,吾らはNeurocrine Biosciencesに株式を発行·売却し(後述),総購入価格は2,000万ドルである。
欧州規制機関がNBI-921352の成人局所てんかん発作の臨床試験申請を承認したことに基づいて、2021年9月、合計1,000万ドルの記念碑的支払いを受け、現金450万ドル、株式投資550万ドルであった。2022年1月には、675万ドルの現金支払いと825万ドルの株式投資を含む1500万ドルの記念碑的支払いを受け、FDAがSCN 8 A-DeE小児科臨床試験研究者を拡大する案修正案を受け入れたことに基づく。
協力協定はまた、Neurocrine Biosciencesが私たちに支払った潜在的な総開発と規制マイルストーン支払いは、NBI-921352製品について、最大3.25億ドルに達し、他の化合物ごとに、最大2.475億ドル、最大で他の3種類の化合物に達することを規定している。各化合物は、いくつかの純販売目標が達成された後にNeurocrine Biosciencesから最大4種類の化合物を支払うNBI-921352製品を含む最高1.5億ドルの販売マイルストーンを含む。
Neurocrine Biosciencesの製品および国/地域に対する印税の支払い義務は、(I)(A)双方が研究期間またはその後の指定された期間内に提出する共通特許権または(B)協力協定で指定された特許権(いずれの場合もこのような製品をカバーする)の最後の有効主張が満了したとき、(Ii)当該製品が当該国で初めて商業販売されてから10年、および(Iii)当該製品の国家/地域における規制または特許使用料の排他的期限が満了する期間の中で遅くとも満了する。特許権の満了または後発薬の発売後の平均純売上高がある割合低下することを含む、特許権使用料の支払いは、特定の場合に減少する可能性がある。事前に終了しない限り、協力協定の期限は、その製品の国/地域における印税期間が満了するまで、製品および国/地域に基づいて継続される。
10
Neurocrine Biosciencesは、(I)Neurocrine BiosciencesがNbI-921352製品について、Neurocrine Biosciencesがその商業的に合理的な努力を使用してNbI-921352製品の第2段階臨床試験を完了するまで、少なくとも90日間の書面通知を提供することによって、またはいかなる理由もなく、NbI-921352製品の第2段階臨床試験を完了することができる;(I)Neurocrine Biosciencesがその商業的に合理的な努力を使用してDTC製品の第1段階臨床試験を完了するまで、DTC製品について;そして(Iii)Neurocrine Biosciencesがその商業的に合理的な努力を使用してこの2つの臨床試験を完成する前に、協力プロトコル全体について。特定の条件を満たす場合、いずれか一方は、重大な違約の全部または一部が発生した場合に連携プロトコルを終了することができる。もしNeurocrine Biosciencesが私たちが治癒していない材料の違約のために協力協定を終了する権利がある場合、Neurocrine Biosciencesは終了の代わりにNeurocrine Biosciencesが私たちのすべての後続支払いを半減することを選択することができる。
協力協定が任意の理由で終了した後、私たちはNeurocrine Bioscionsに付与されたすべてのライセンスおよび他の権利は終了すべきであるが、終了が1つ以上の製品または国についてのみ適用される場合、そのような終了は、終了された製品または国にのみ適用されるであろう。場合によっては、Neurocrine Biosciencesは、終了した国/地域で研究、開発、および商業化が終了した製品の開発、製造、または商業化によって終了するために、Neurocrine Biosciencesによって実際に終了した製品の開発、製造、または商業化によって使用されるいくつかのNeurocrine Bioscienceに合理的に必要な知的財産権の許可を与えることに同意している。有効日の終了前に第2段階の臨床試験が完了していない任意の終了製品については、このような許可は特許使用料を免除し、そうでなければ、終了発効日に応じて製品の開発段階を適用し、低い桁数パーセントから1桁パーセントまでの使用料を負担する。
株式購入協定
2019年12月2日、吾らはNeurocrine Biosciencesと株式購入契約を締結し、この合意に基づき、吾らはNeurocrine Biosciencesに1,408,847株の普通株を私募方式で発行·販売し、総購入価格は2,000万ドル、または1株当たり14.196ドルであった。買収価格は我々普通株2019年11月29日の終値より20%割増します。
我々は2021年9月と2022年1月にそれぞれNeurocrine Biosciencesと追加のSPA協定を締結し,これにより,我々の株式275,337株および258,986株をそれぞれ私募方式でNeurocrine Biosciencesに発行および売却し,総購入価格はそれぞれ550,000ドル(1株19.9755ドル)および8,250,000ドル(1株31.855ドル)であった。買い取り価格は公告30日前の出来高加重平均価格より15%割増した。
第一注文製薬会社と締結された資産購入契約。
2017年4月、私たちはFirst Order PharmPharmticals、Inc.またはFirst Orderと資産購入協定を締結し、この合意に基づいて、XEN 1101(以前は1 OP 2198と呼ばれていた)のすべての権利を獲得した。第一注文前に博世健康会社の間接子会社Valeant PharmPharmticalsルクセンブルクS.a.r.l.およびValeant PharmPharmticalsアイルランド有限会社博世健康から1 OP 2198を買収し、潜在的なマイルストーンと特許権使用料の支払いを含むいくつかの義務を負った。
2018年9月、私たちは、3960万ドルまでの潜在的な臨床開発、規制、販売ベースのマイルストーン、および商業販売の上位数%の特許権使用料を含め、3960万ドルまでの潜在的な臨床開発、規制、販売に基づくマイルストーン、および商業販売の上位数%の特許権使用料を購入する協定に調印しました。
2020年8月には、協定のいくつかの定義を修正し、特定のマイルストーンの支払いスケジュールを修正するための資産購入協定修正案に署名した。修正案を施行した後、私たちは最初の注文に30万ドルを支払った。2023年2月には、臨床およびその他のマイルストーンを実現するために140万ドルが支払われた。私たちはまだ未来に支払われる可能性のある600万ドルまでの規制マイルストーンに責任がある。最初の注文には印税義務がありません。
知的財産権
私たちのビジネス戦略の一部として、私たちの業務に重要であると考えられるノウハウを保護するために努力しています。私たちの候補製品とその使用方法および製造プロセスをカバーするための特許保護、および私たちの業務に重要な他の発明を求めて維持することを含みます。我々は、成分、使用方法、治療および患者選択、調製および製造プロセスに対する特許出願を提出することによって、私たちが開発している候補製品および将来の製品の中で創造または決定された知的財産権を拡大していく予定である。私たちはまた、ビジネス秘密、内部知識、技術革新、および第三者との合意によって、私たちの競争地位を発展、維持、保護している。私たちの競争力はこの戦略の成功にかかっているだろう。
2023年12月31日現在、我々は、16件の米国特許および56件の外国司法管轄区の特許(欧州特許国家検証を含まない)、370件以上の係属特許出願を有し、共同所有している。
11
XEN 1101については、2023年12月31日現在、4つの米国特許、海外司法管轄区における29件の特許(欧州特許国家検証を含まない)および200件以上の出願中の特許を有している。発行された特許、およびこれらの出願から発行された任意の特許は、2028年から2044年の間に満了することが予想される(何の延期もない)。
NBI−921352(前身はXEN 901)については,2023年12月31日現在,4つの米国特許,12件の海外司法管轄区の特許(欧州特許国検証を除く),18件の出願されている特許出願を有しているか,共同で所有している。発行された特許、およびこれらの出願から発行された任意の特許は、2037年から2044年までの間に満了することが予想される(何の延期もない)。我々のNav1.6および/またはNav1.2の選択的阻害剤(NBI-921352を含まない)については、2023年12月31日現在、米国特許6件、外国管轄区における15件の特許(欧州特許国検証を除く)、および90件以上の係属特許出願を有している。発行された特許およびこれらの出願によって発行された任意の特許は、2037年から2039年の間に満了することが予想される(何の延期もない)。Neurocrine Biosciencesとの私たちの協力によると、Neurocrine Biosciencesは、私たちが評価する権利があるにもかかわらず、NBI-921352と他の選択的Nav1.6阻害剤とダブルNav1.2/1.6阻害剤特許との組み合わせの起訴、維持、および他の関連事項を制御している。
我々の開発プロジェクトについては,KV 7(XEN 1101を除く),Nav1.1,Nav1.7に関する目標が含まれており,2023年12月31日現在,米国発行特許1件と未解決特許20件以上を有している.発行された特許、およびこれらの出願から発行された任意の特許は、2036年から2044年までの間に満了することが予想される(何の延期もない)。
競争
生物技術と製薬業界の競争は激しく、その特徴は技術進歩が迅速で、独自製品に対する重視度が高いことである。私たちは私たちの技術、開発経験、科学知識と薬物発見方法が私たちにいくつかの優勢を提供していると信じているが、著者らは発見と候補製品開発の方面で製薬と生物技術会社、学術機関と政府機関、公共と個人研究機関を含む多くの異なる方法と源からの潜在的な競争に直面している。私たちまたは私たちのパートナーが開発および商業化に成功した任意の候補製品または製品は、既存製品および将来発売される可能性のある新製品と競争するだろう。
私たちまたは私たちの協力者と比較して、私たちは競争しているか、あるいは将来競争する可能性のある多くの会社は、研究開発、製造、臨床前テスト、臨床試験を行い、監督管理許可とマーケティング承認を得た製品について、より多くの財務資源と専門知識を持っている。製薬とバイオテクノロジー産業の合併と買収は、私たちの数の少ない競争相手により多くの資源を集中させる可能性がある。規模が小さいか早い段階にある会社も重要な競争相手であることが証明される可能性があり、特に大手·成熟会社との協力により手配されている。
もし私たちの競争相手が私たちが開発する可能性のある任意の製品よりも安全で、より効果的で、副作用が少なく、より便利で、より安価な製品や療法を開発し、商業化すれば、私たちのビジネス機会は減少または消失するかもしれない。私たちの競争相手も私たちよりも早くFDA、欧州医薬品局、EMA、あるいは他の外国規制機関からその製品の承認を得る可能性があり、これは私たちの競争相手が市場に入る前に強力な市場地位を確立することをもたらすかもしれない。また、我々の競争能力は、多くの場合、保険会社や他の第三者支払者の影響を受ける可能性がある。
製品市場以外に、私たちの競争相手は合格した科学と管理者を募集と維持し、臨床試験場を設立し、臨床試験患者を募集し、著者らの計画と相補的或いは必要な技術を獲得することで私たちと競争を展開する。
私たちのすべての候補製品の成功に影響を与える重要な競争要素は、承認されれば、それらの効果、安全性、利便性、価格、代替製品の有効性、競争レベルとカバー範囲の可用性、および政府と他の第三者支払人の十分な補償である可能性が高い。我々が臨床開発している候補製品は,市場や現在開発されている様々な療法や薬物と競合する可能性がある。
12
私たちの1つ以上の特許または協力候補製品がてんかん治療のために承認された場合、他のASMやお互いと競争する可能性が予想される。これらの現在よく使われているASMはブレバシタン、カルバマゼピン、メトロニダス、クロロバザン、エチミアミン、ガバブジン、ラモトリアジン、レボエチルシタン、オカルバゼピン、パラパタン、フェニトイン、トピラメートとバルプロ酸塩を含む。Bioaven株式会社、Cerevel治療ホールディングス、Equilibre生物製薬会社、ジョンソン革新薬会社、Neurona治療会社、Praxis Precision Medicines社、Rapport治療会社、SK生命科学社、Supernus製薬会社、志盟生物製薬会社が開発している候補製品を含む他の製品と競合する可能性のあるASMが開発されています。もし私たちの1つ以上の候補特許製品がMDDの治療に承認されれば、他の抗うつ薬や米国預託株式と競争する可能性が予想されます。MDDを有する患者は、一般に、選択的セロトニン再取り込み阻害剤またはSSRI、ベンゾジアゼピン薬、セロトニン/ノルアドレナリン再取り込み阻害剤またはSNRI、ノルアドレナリンおよびドーパミン再取り込み阻害剤、またはNDRI、N-メチル-D-アスパラギン酸またはN-メチル-D-アスパラギン酸、受容体アゴニストおよび非定型抗精神病薬を含む、様々な米国預託株式の治療を受ける。現在処方されている抗うつ薬には,ベンゾジアゼピン類,ブレシプラゾール,アンフェタミン,アンフェタミン/右メサフェン,カリラジン,シタロプラム,度ロキセチン,エスピラン,エスピラム,フルオキセチン,ケタミン,セルトラリン,トラゾロン,三環系薬物,ベンラファキシン,ベラシドン,ボルチキセチンがある。いくつかの会社がMDD治療の候補製品を開発しており,エバービー社,Bioaven株式会社,細胞内治療会社,ジョンソン革新薬会社,Neumora治療会社,Relmada治療会社,Sage治療会社,住友製薬米国社を含むことが知られている。
政府の監督管理
我々は薬物としてFDAと米国以外の同等の規制機関によって規制されている小分子候補製品を開発している。FDA内部では、薬物評価·研究センター(CDER)が薬物の規制を担当している。薬品は“連邦食品、薬物と化粧品法”あるいは“食品、薬物と化粧品法”及びその他の連邦、省、州、地方と外国法規の規制を受けている。その他の事項を除いて、“食品と薬物規制法”と相応の法規は薬品のテスト、製造、安全、効果、ラベル、包装、貯蔵、記録保存、流通、輸入、輸出、報告、広告、その他の販売促進活動を管理する。試験的新薬申請またはINDは、薬物臨床試験開始前にFDAの承認を得なければならず、このような候補製品の各臨床研究案は、米国が開始する前にFDAと機関審査委員会(IRB)の審査を通過しなければならない。米国で薬物が発売される前にもFDAの承認を得なければならない。規制承認を得る過程とその後、適切な連邦、省、州、地方、外国の法律法規を遵守する過程には多大な時間と財力が必要であり、必要な規制承認を得ることができない可能性がある。製品開発プロセス、承認プロセス又は承認後のいずれかにおいて、適用される米国の規制要件を遵守しない場合、出願人及び/又はスポンサーは、様々な行政又は司法制裁を受ける可能性がある。他の行動に加えて、これらの制裁は、FDAが係属中の申請の承認の拒否、承認の撤回、臨床一時停止の実施、警告または無タイトルの手紙の発行、他のタイプの法執行関連手紙、製品のリコール、製品の差し押さえ、再標識または再包装、生産または流通の完全または部分的な一時停止、禁止、罰金、政府契約の拒否、原状回復、利益の返還、またはFDAと司法省または他の政府エンティティによって提起された民事または刑事調査および処罰を含むことができる。
アメリカの薬物開発プロセスは
FDAが米国で医薬品を発売する前に必要なプログラムは通常、以下のいくつかの態様を含む
13
NDAを支持するために必要なデータは、2つの異なる発展段階で生成される:臨床前と臨床。新しい化学実体の場合、臨床前開発段階は通常、活性成分の合成、調合の開発と製造技術の決定、純度と安定性の評価、および実験室で非ヒト毒理学、薬理学と薬物代謝研究を行い、その後の臨床試験を支持する。臨床前試験の進行はGLPと米国農務省の動物福祉法を含む連邦法規に適合しなければならない。スポンサーは,臨床前試験の結果を,生産情報,分析データ,任意の利用可能な臨床データや文献,提案された臨床案とともにFDAに提出し,INDの一部としなければならない。INDはFDAが研究薬物製品の使用をヒトに許可する要求である。INDが提出した文書の中心的な焦点は人体試験の全体調査計画と案である(S)。INDはFDAが提案された臨床試験に対して懸念または問題を提起しなければ、FDAが受信後30日以内に自動的に発効し、30日以内にINDを臨床保留状態に置かなければならない。この場合,INDスポンサーやFDAは臨床試験開始前に未解決の問題や問題を解決しなければならない。IND提出後、生殖AEsや発ガン性の動物試験など、いくつかの長期的な臨床前試験が継続される可能性がある。FDAはまた,臨床試験の前または期間のいつでも,安全考慮または規定を遵守しない理由で,候補薬物の臨床保留を実施することも可能である。したがって,IND提出はFDAが臨床試験の開始を許可することを保証していない,あるいは開始すると試験の一時停止や終了を招く可能性があるという問題は生じない。
FDAがINDを受けていれば,この薬物はヒト臨床試験で検討し,安全かつ有効であるかどうかを確認することができる。臨床開発段階はGCPにより合格調査者の監督下で患者を含むヒト被験者に薬物製品を服用することに関連し、GCPは臨床試験、記録データ、臨床試験結果を報告するための基準を確立し、すべての研究被験者に任意の臨床試験に参加するインフォームドコンセントを書面で提供することを要求することを含む。
ヒト臨床研究は通常3つの連続した段階で行われ、これらの段階は重複または合併する可能性がある:
承認後の臨床研究は,4期臨床研究と呼ばれることがあり,最初の上場承認後に行うことができる。これらの臨床研究は,治療適応が予想される患者の治療から追加的な経験を得るためのものであり,特に長期安全なフォローアップのためである。場合によっては,FDAはNDAを承認する条件として4期臨床試験を強制的に実行することができる。臨床開発のすべての段階において、監督管理機関はすべての臨床活動、臨床データと臨床研究調査人員に対して広範なモニタリングと監査を行うことを要求している。
臨床研究と同時に、会社は通常追加の動物研究を完成し、薬物物理特性に関する追加情報を開発し、GMP要求に基づいて最終的に商業量産製品のプロセスを決定しなければならない。製造過程は一貫して高品質の候補製品ロットを生産できなければならず、他の要求以外に、スポンサーは最終薬物の品質、特性、強度と純度を確保する方法を制定しなければならない。また,適切な包装を選択·試験し,薬物がその標識された棚期に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
さらに、災害や突発的な公衆衛生事件のため、対象者の保護を支援するために、追加の臨床試験政策やプログラムを制定して実施する必要があるかもしれない。例えば、FDAは、災害および公衆衛生突発事件による重大な中断中の臨床試験に関する指導意見を発表し、これらの事件の影響を受ける臨床試験スポンサーのいくつかの考慮事項を記載して、試験参加者の安全を確保し、GCPコンプライアンスを維持し、試験完全性のリスクを最小限に低減する。災害と突発公共衛生事件による重大な中断により、私たちは現在或いは未来の指導と監督要求に基づいて、著者らの臨床試験或いは業務運営を更に調整する必要があるかもしれない。
14
アメリカの審査と承認の流れ
薬物の臨床研究を完了した後、FDAのNDAに対する承認を得なければならず、その後、この薬物の商業マーケティングを開始することができる。NDAは、製品開発、実験室と動物研究、人体研究、製品製造と成分の情報、提案されたラベル、その他の関連情報を含む必要がある。さらに、小児科研究公平法またはPREAによれば、NDAまたはNDAの補充は、すべての関連する小児科亜群において主張される適応の安全性および有効性を評価し、各安全で有効な小児科亜群に対する製品の用量および投与をサポートするためのデータを含まなければならない。FDAはデータの提出を延期することを許可するか、またはすべてまたは部分的な免除を与えることができる。規制が別途要求されない限り、PREAは孤児の称号が付与されたいかなる適応薬にも適用されない。テストや承認過程には多大な時間と労力が必要であり,FDAがNDAの届出を受ける保証はなく,届出してもどの承認もタイムリーに承認される保証はない.
改正された処方薬使用料法案(PDUFA)によると、各NDAは相当な使用料を伴わなければならない。PDUFAは薬品に年間製品費,処方薬を生産するための施設に年間施設費を徴収している。場合によっては、小企業が初めて出願した出願料を免除することを含む、費用を免除または減免することができる。また,孤児薬として指定された製品については,NDAに対して使用料を評価せず,非孤児適応も含まれていない限りである。
出願が提出されてから60日以内に、FDAは、機関が提出を受け入れる前に実質的に完了したかどうかを決定するために、NDAを審査する。FDAは、それが不完全であるか、または提出時に適切に審査できないと考えられる任意のマーケティング申請の提出を拒否することができ、追加の臨床データを含む追加の情報の提供を要求することができる。この場合、秘密協定と追加情報を再提出しなければならない。再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。提出された申請が受け入れられると,FDAはNDAの深い実質的な審査を開始する。FDAは、提案された製品がその予期される用途に対して安全に有効であるかどうか、およびGMPに従って生産されるかどうかを決定するために、申請を審査する。FDAは、新製品の申請または安全性または有効性の問題が生じる製品の申請を諮問委員会に提出することができ、一般に、申請を承認すべきかどうか、どのような条件下で承認すべきかを審査、評価および提案するための臨床医および他の専門家を含むグループである。FDAは諮問委員会の提案に制限されていないが、それは決定を下す時にこれらの提案を慎重に考慮するだろう。製品承認中に、FDAはまた、製品の安全な使用を保証するために、リスク評価および緩和策、またはREMSが必要であるかどうかを決定する。FDAがREMSが必要であると考える場合、NDAのスポンサーは提案されたREMSを提出しなければならない;必要であれば、FDAはREMSのない出願を承認しないであろう。
関連データおよび情報が提出されたにもかかわらず、FDAは最終的にNDAがその承認の規制基準を満たしていないことを決定し、承認を拒否する可能性がある。臨床研究から得られたデータは常に決定的ではなく,FDAのデータ解釈は我々の同じデータに対する解釈とは異なる可能性がある。機関が上場申請を承認しないことを決定した場合、FDAは、FDAによって決定された出願中のすべての特定の欠陥が一般的に記述される完全な返信を発行する。決定された欠陥は軽微である可能性があり(例えば、ラベル変更が必要である)、重大である可能性もある(例えば、追加の臨床研究が必要である)。さらに、完全な返信状は、出願人がとり得る、申請を承認条件に置くための提案行動を含むことができる。完全な返信が発行された場合、出願人は、秘密協定を再提出し、手紙で決定されたすべての不足点を解決するか、または出願を撤回することができる。
1つの製品が規制部門の承認を得た場合、承認は臨床試験で研究された特定の疾患および用量に限定され、そうでなければ使用適応が制限される可能性があり、これは製品の商業的価値を制限する可能性がある。さらに、FDAは、いくつかの禁忌症、警告、または予防措置を製品ラベルに含めることを要求する可能性がある。FDAは、REMS要求に従って製品流通、処方または調剤に制限および条件を適用することができ、または他の方法で任意の承認範囲を制限することができる。薬品審査は規制基準を満たしていないか、最初の発売後に問題が発生したため撤回される可能性がある。
FDAがPDUFAに同意した業績目標の1つは、申請提出日から10ヶ月以内に90%の標準新分子実体(NME)NDAの審査を完了し、提出日から6ヶ月以内に90%優先NME NDAの審査を完了し、審査決定を行うことである。FDAは常にPDUFAの目標日を達成するわけではなく,その審査目標は時々変わる可能性がある。PDUFA目標日の最後の3ヶ月以内に、FDA要求(またはアプリケーションスポンサーが他の方法で提供する)が、以前に提出されていない、またはFDA審査されていない新しいデータまたは新しい情報(例えば、新しい重大な臨床安全性または有効性研究報告、提案されたREMS)、または係属中の出願に以前に提出された研究の新しい分析または重大な再分析を行う場合、審査プロセスおよびPDUFA目標日を3ヶ月延長することができる。
15
承認後に要求する
FDAは承認後も引き続き薬品に対して厳格かつ広範な監督管理を行い、特にGMP方面である。私たちは依存し、私たちが商業化する可能性のある任意の製品の臨床的および商業的ロットを生産するために第三者に依存し続けることが予想される。私たちの製品のメーカーは、品質管理と品質保証、記録とファイルのメンテナンスを含むGMP法規に適用される要求を遵守することを要求されます。他の薬品メーカーの承認後要求に適用され、報告は配布製品の安全性、有効性或いは品質に影響を与える可能性のあるGMP偏差、記録保存要求、不良反応の報告、最新の安全と治療効果情報の報告、及び電子記録と署名要求を遵守することを含む。
また、FDAの広告および販売促進要件、例えば、消費者向け直接広告に関連する要件、製品承認ラベルに記載されていない、または他の態様で製品承認ラベルと一致しない製品が、患者集団において製品を普及させるための要求、および業界スポンサーの科学的および教育活動のために使用されることを禁止しなければならない。以前未知の問題が発見されたり、適用された規制要求を遵守できなかったりすると、製品の販売を制限したり、その製品を市場から引き揚げたりし、民事または刑事制裁を受ける可能性がある。製品開発プロセス、承認プロセスまたは承認後の任意の時間に適用される米国の要求を遵守できなかった場合、出願人または製造業者は、行政または司法民事または刑事制裁および負の宣伝を受ける可能性がある。FDAの制裁には、承認保留申請の拒否、承認撤回、臨床封印、警告または無名手紙、製品リコール、製品差し押さえ、生産または流通の完全または部分的な一時停止、禁止、罰金、政府契約の拒否、強制要求の是正広告、または医師とのコミュニケーション、取り締まり、原状回復、利益の返還または民事または刑事罰が含まれる可能性がある。機関や司法法執行行動は私たちに実質的な悪影響を及ぼすかもしれない。
医薬品メーカーと承認薬品の生産と流通に参加する他のエンティティは、FDAとある州機関にその機関を登録し、FDAとある州機関の定期的な抜き打ち検査を受けて、GMPと他の法律を遵守することを保証しなければならない。そのため、メーカーはGMPコンプライアンスを維持するために、生産と品質管理に時間、お金、労力をかけ続けなければならない。承認後に製品が発見された問題は、製品を市場から撤回することを含む、製品、製造業者、または承認された機密協定保持者の制限をもたらす可能性がある。さらに、製造プロセスまたは施設の変更は、通常、新たな適応および追加のラベル宣言を追加するような承認された製品の他のタイプの変更を実施するためにFDAの承認を得る必要があり、FDAのさらなる審査および承認を受ける必要がある。
迅速チャネル指定、優先審査、画期的な治療指定、承認の加速
FDAには、迅速なチャネル指定、優先審査、画期的な治療指定、および承認の加速を含む様々な計画があり、これらの計画は、薬物開発およびFDA審査のプロセスを加速または簡略化することを目的としている。1つの薬剤がこれらの計画のうちの1つまたは複数に適合していても、FDAは後で、その薬剤がもはや資格条件を満たしていないと決定する可能性がある。一般に,これらの計画に適合する薬剤は,重篤または生命に危険な疾患を治療する薬剤,満たされていない医療ニーズを解決する可能性のある薬剤,既存の治療法よりも意義のある薬剤である。これらの経路は、FDAがNDAを審査するのに要する時間を低減することができるが、製品がFDAの承認を得ることを保証することはできない。
迅速なチャネル指定を受ける資格があるために、FDAは、スポンサーの要求に基づいて、深刻または生命に危険な疾患または条件を治療することを目的とした薬剤を決定しなければならないが、有効な治療方法はなく、このような疾患または条件を満たす可能性のある満たされていない医療需要を証明しなければならない。クイックチャネル計画によれば、候補薬剤のスポンサーは、候補薬剤のIND提出と同時にまたは後に、FDAに特定の適応の製品を迅速チャネル製品として指定することを要求することができる。FDAはスポンサーからの要請を受けて60日以内に指定決定を迅速に行わなければならない。他の利点に加えて、プロキシ端末を使用してFDAとより大きなインタラクションを行うことができれば、FDAは、申請が完了する前に、Fast Track製品のNDA部分の審査を開始する可能性がある。申請者が余剰情報を提出するスケジュールを提供し、申請者が適用された使用料を支払うことができれば、スクロール審査を行うことができる。しかしながら、FDAが高速チャネル申請の期間目標を検討することは、NDAの最後の部分が提出されるまで開始される。高速通路薬はまた加速承認と優先審査を受ける資格があるかもしれない。また,FDAが高速チャネルの指定が臨床試験中に出現したデータの支持を得なくなったと考えた場合,その指定を撤回する可能性がある。
FDAは,治療に大きな進展を遂げた薬剤を優先的に検討したり,適切な治療法がない場合に治療を提供したりする可能性がある。優先審査は、現在のPDUFAガイドライン下の標準審査10ヶ月ではなく、FDA審査申請の目標が6ヶ月であることを意味する。この6ヶ月および10ヶ月の審査期間は、受信日ではなく、新しい分子実体の“提出”日から計算され、これは、通常、提出日から審査および決定を行うために、約2ヶ月のタイムラインを増加させる。高速チャネル指定を受ける資格のある製品の多くは、優先審査を受けるのに適していると考えられる可能性もある。
16
米国議会が2012年に公布した新たな“食品·薬物管理局安全·革新法案”(FDASIA)の規定によると、スポンサーは候補薬物を“画期的療法”に指定することを申請することができ、通常は薬物の第2段階試験終了時である。画期的な治療法は、1つまたは複数の他の薬剤と単独でまたは1つまたは複数の他の薬剤と組み合わせて重篤または生命を脅かす疾患または状態を治療することを意図した薬剤として定義され、初歩的な臨床証拠は、1つまたは複数の臨床的に重要な終点において、臨床開発早期に観察される実質的な治療効果のような既存の療法よりも有意な改善を示す可能性があることを示す。画期的な治療法に指定された薬物も加速承認を得る資格がある。画期的な治療法については,FDAは薬物開発計画の密かつ早期の指導など,承認申請の開発や審査を加速させるための何らかの行動をとる可能性がある。
FDASIAはまた、FDAの加速承認条例の編纂と拡張を行い、この条例によると、FDAは深刻または生命を脅かす疾患のための薬物の使用を許可することができ、この薬物は合理的に臨床利益を予測する可能性のある代替終点に基づいて、あるいは不可逆的な発病率または死亡率よりも早く測定できる中間臨床終点に基づいて、不可逆的な発病率または死亡率または他の臨床利益への影響を合理的に予測することができ、それによって既存の治療よりも意義のある治療利益を提供する。代替終点はマーカーであり,それ自体は臨床的利益を評価していないが,臨床的利益を予測できると考えられる。この決定は,疾患や状況の重症度,希少性や流行率,代替治療法の有無を考慮している。承認された条件として、FDAは、不可逆的な発症率または死亡率または他の臨床終点に対する予期される効果を検証および説明するために、加速承認された薬物のスポンサーに第4段階または発売後の研究を要求することができ、加速中止手順を必要とする可能性がある。加速法規によって承認された候補薬物のすべての宣伝材料はFDAの事前審査を経なければならない。
1つの製品がこれらの計画のうちの1つまたは複数に適合していても、FDAは、製品がもはや資格条件に適合していないことを後で決定することができ、またはFDAの審査または承認を決定する期間が短縮されないことができる。また,迅速チャネル指定,優先審査,加速承認,突破的治療指定は承認の基準を変更することはなく,最終的に開発や承認過程を加速させない可能性もある。
孤児薬名
孤児医薬品法によれば、FDAは、まれな疾患または疾患を治療するための薬剤を孤児として指定することができ、このような疾患または疾患は、通常、米国では20万人未満に影響し、米国では20万人を超え、このような疾患または疾患を治療する薬剤を米国で開発および生産するコストは、製品の販売から回収されることが合理的に予想されていない。秘密保持協定を提出する前に、指定孤児薬を申請しなければならない。FDAが孤児薬物指定を承認した後、FDAは、治療薬の識別およびその潜在的な孤児の使用を開示する。指定孤児薬は、規制審査と承認過程でいかなる利点も伝達されず、規制審査と承認過程の持続時間を短縮することもない。
まれな小児科疾患を有する患者の50%以上が19歳以下であり、米国で影響を受けている患者が200,000人未満である場合、孤児医薬製品もまれな小児科疾患またはRPD称号を取得する資格がある。製品の承認時には、RPD称号を有する製品のスポンサーは、他の製品の後続マーケティング申請を得るための優先審査クーポンに交換することができる優先審査クーポンを取得する。このような証明書は別の会社に移すことができる。
孤児によって指定された候補製品がその後、孤児によって指定された疾患または条件を有する薬剤に対するFDAの最初の承認を得た場合、この製品は、孤児の薬物排他性を得る権利があり、これは、FDAが、限定された場合、例えば、孤児に対して排他的な製品に対する臨床的利点を示さない限り、7年以内に同じ適応について同じ薬剤を販売する任意の他の出願を許可しないことを意味する。しかし,孤児薬物に対して排他的な同一適応に対しては,競争相手は異なる製品の承認を得ることが可能であるか,あるいは同一製品に対して孤児薬物に対して排他的な異なる適応を有する場合には,競争者が承認される可能性がある。競合他社がFDAで定義された同じ孤児適応の同じ製品の承認を得た場合、または我々の候補製品が同じ孤児適応または疾患のために競合相手の製品に含まれると決定された場合、孤立薬物排他性は、7年以内に候補製品が承認されることを阻止する可能性もある。孤児薬物に指定された候補品が発売承認された場合,その適応範囲は指定されたものよりも広く,孤児薬物排他性を得る資格がない可能性がある。EUの孤児薬物の地位は似ているが、10年に及ぶ排他性を含む異なる利点を持っている。
17
はいCatalyst Pharms,Inc.Becera事件を訴える、1299“連邦判例編”第14巻(第11巡回法廷)2021年)、裁判所はFDAの長期的な立場に同意しない、すなわち孤児薬物排他性は、条件に適合する疾患内の承認用途または適応にのみ適用され、疾患全体または状況におけるすべての用途または適応には適用されない。特に,巡回裁判所は,Catalyst薬物の孤児薬物排他性は,Catalystの薬物が成人LEMSの治療にのみ許可されていたにもかかわらず,Catalystの薬物が同一孤児指定疾患であるLambert−Eaton筋無力症候群(LEMS)のすべての用途または適応のための別の薬剤の使用をFDAが承認したことを排他的に阻止したと考えている。したがって、裁判所はFDAに児童LEMSのための薬物の承認を保留するように命令した。この決定は孤児薬の専有権の適用に不確実性をもたらす。FDAは2023年1月24日、Catalystにおける裁判所の命令を遵守しているが、FDAは、Catalyst命令の範囲外の事項に法規の長期解釈を適用し続けることを意図している--すなわち、機関は、孤児薬物独占の範囲を薬物が承認された用途または適応と一緒にバンドルし続けることを明らかにし、これにより、他のスポンサーが同じ孤児指定疾患または条件下で承認されていない新しい用途または適応のために承認されることを可能にする。将来の訴訟、立法、機関決定と行政行動が孤児薬物の排他性の範囲にどのように影響するかはまだ不明である。
制御物質規制
米国制御物質法案、あるいはCSAは、登録、安全、記録保存、報告、貯蔵、配布、その他の薬品監督管理局(DEA)が管理する要求を規定している。麻薬·犯罪問題事務室は、違法な商業ルートへの損失や移転を防止するために、規制された物質を規制する操作者と、これらの物質を製造·包装するための設備や原材料を担当する。DEAは制御物質を付表I,II,III,IVまたはV物質として管理している。定義によると、別表1物質は乱用の可能性が高く、既定の医療用途がなく、米国で販売または販売されてはならない。医薬品は付表II、III、IVまたはVとすることができ、その中で付表II物質は最も乱用リスクが高いと考えられ、付表V物質はこのような物質の中で相対的に乱用リスクが最も低い物質と考えられている。規制された物質を製造、流通、配布、輸入または輸出するいかなる施設も年次登録を行わなければならない。登録は特定の場所、活動、そして制御物質スケジュールに対して行われる。例えば、輸入と製造は個別に登録する必要があり、登録ごとにどのような制御物質の付表が許可されているかを明確にする。
DEAは通常、登録を発行する前に施設を検査してその安全対策を検討する。安全要求は制御物質別表によって異なり,最も厳しい要求は付表1と付表2に適用される。必要な安全対策には,従業員の背景調査,ケージ,監視カメラ,在庫台帳などによる在庫の実物制御がある。すべての制御物質の処理記録を保存し,定期的にDEAに報告を提出しなければならない。また、どんな制御物質が盗まれたり、紛失されたかを報告し、任意の制御物質を廃棄する許可を得なければならない。しかも、特別な許可と通知要求は輸出入に適用される。
DEAはある規制対象物質を処理する登録機関を定期的に検査している。適用された要求、特に損失や移転を遵守できなかったことは、法執行行動を招く可能性があり、私たちの業務、運営結果、財務状況に重大な悪影響を及ぼす可能性がある。DEAは民事処罰を求め、必要な登録の更新を拒否したり、制限、一時停止、またはこれらの登録の手続きを開始したりすることができる。場合によっては、違反は刑事訴訟を引き起こす可能性がある。個別の州も制御物質を規制しています。私たちと契約メーカーは許可、記録保存、安全を含む州政府のこれらの製品の流通に対する規制を受けます。
制御物質もいくつかの国際薬物規制条約に基づいて規制されている。これらの条約は国連国家麻酔薬委員会によって施行された。米国はこれらの条約の署名国であるため、その法律と法規を国際要求に適合させなければならず、これらの要求には通常、許可、記録保存、報告要求が含まれている。これらの製品分類に関する国際条約のいかなる変化も、米国や他の国のこれらの物質に対する規制に影響を与える可能性がある。また,上場承認や制御物質分類手続きは国によって異なり,追加のテストや行政審査期限が関与する可能性があり,我々の候補製品が我々が開発した国で制御対象物質に分類された成分が含まれていれば複雑になる可能性があり,このような候補製品が商業化前に適用される制御対象物質法律に制約される可能性がある。外国の制御物質に対する規制は米国DEAと州政府の規制と大きく異なる可能性がある。他国での上場承認や制御物質分類の取得に要する時間は、米国でFDA承認やDEA分類を取得するのに要する時間とは異なり、さらに長い可能性がある。
18
米国特許期間延長と市場排他性
FDAが私たちの候補製品の使用を許可した時間、期限、および詳細によると、私たちのいくつかの米国特許は、1984年の“薬品価格競争および特許期限回復法”(一般に“ハッジ-ワックスマン法案”と呼ばれる)に基づいて限られた特許期間を延長する資格がある可能性がある。ハッジ·ワックスマン法は、製品開発やFDA規制審査の過程で失われた特許期間の補償として、特許期間を最大5年間延長することを許可している。特許期間の延長は,特許の残存期間を製品承認日から合計14年間延長することはできない.承認製品に適用される特許は1つのみ延期する資格があり,特許期間を延長する出願はFDAの承認を受けてから60日以内に提出されなければならない。米国特許商標局はFDAと協議し,任意の特許期限延長の出願を審査し,承認する。
ハッジ−ワックスマン法によると,新しい化学実体をその有効成分として含む薬物製品は,5年間の市場排他性を得る権利がある。FDAが以前に同じ活性部分を含む任意の他の新薬を承認していない場合、医薬製品は、薬物がエステル、塩または他の非共有結合誘導体である分子をもたらす分子またはイオンを含む医薬物質の活性を担当する分子またはイオンであると定義される新しい化学物質である。この排他的期間内に、FDAは、以前に承認された活性部分を含む別の会社によって提出された簡略化された新薬出願またはANDAまたは505(B)(2)NDAを承認しない可能性がある。しかしながら、ANDAまたは505(B)(2)NDAが特許無効または非侵害の証明を含む場合、4年後に提出することができる。
活性成分は以前FDAが承認した薬物製品であり,スポンサーが新たな臨床研究を要求され,新たな適応,用量,強度あるいは剤形を支持すれば,3年間の市場排他性を得る権利がある。この3年間の専門権は、新しい臨床研究に関連する使用条件のみをカバーしており、一般的な事項として、FDAがANDAまたは505(B)(2)NDAを元に修正されていない薬物の模倣薬バージョンへの使用を許可することは禁止されておらず、このような申請の承認は早くても丸5年の市場専門権の満了後に発効する可能性がある。
薬物製品は米国で小児科市場の独占経営権を獲得することもでき、承認されれば、既存の市場独占経営権とオレンジ図書に列挙された任意の特許期間を6ヶ月延長する(S)。他の専門期間および/または特許期間終了(S)から6カ月の専門権は,FDAが発表した小児科研究“書面請求”に基づいて小児科研究を完了した後にタイムリー,自発的,取り決めに基づいて付与することができ,データがその薬物が研究している小児科群に有効であることを示していなくてもよい。
法規を付加する
薬品承認後の製造、販売、普及とその他の活動は、FDAの監督を受ける以外に、多くの監督管理機関の監督を受けて、アメリカの医療保険と医療補助サービスセンター、衛生と公衆サービス部の他の部門、制御物質薬品監督管理局、消費財安全委員会、連邦貿易委員会、職業安全と健康管理局、環境保護局及び州と地方政府を含む。米国では,製薬業者の活動は連邦や州の法律に拘束されており,これらの法律は医療業界の“詐欺や乱用”を防止することを目的としている。これらの法律は、一般に、製造業者と医療サービス業者または医療業界の他の参加者との間の財務的相互作用を制限し、および/または、そのような相互作用を政府および公衆に開示することを要求する。その中の多くの法律法規の要求が曖昧で、あるいは行政指導が必要である。製薬業者はまた、連邦医療計画(例えば、医療補助)の保険を得るために、政府医療計画に基づいて、または特定の政府および個人購入者に割引またはリベートを提供することを要求されている。このような計画に参加するには特定の薬品の価格を追跡して報告する必要があるかもしれない。そのような価格を正確に報告しなければ、製造業者は罰金と他の処罰を受けるだろう。いずれの制御物質の処理も米国の“制御物質法”と“制御物質輸出法”に適合しなければならない。薬品はアメリカの“毒物予防包装法”に適用される児童保護包装要求に符合しなければならない。製造、販売、販売促進、その他の活動はまた、連邦と州消費者保護および不正競争法によって制限される可能性がある。
規制要求を遵守できなかったことは、製造業者を可能な法律や規制行動に直面させる。場合によっては、適用される規制要件を満たさないことは、刑事起訴、罰金またはその他の処罰、禁止、リコールまたは薬品の差し押さえ、生産の完全または部分的な一時停止、製品の承認の拒否または撤回、政府医療計画から除外されるか、または政府契約を含む会社の供給契約の締結を許可することを拒否する可能性がある。さらに、1つの会社がFDAおよび他の要求を遵守していても、製品の安全性または有効性に関する新しい情報は、FDAが製品承認を修正または撤回することをもたらす可能性がある。私たちが販売している未来の製品の販売を禁止または制限または撤回することは、不利な方法で私たちの業務に大きな影響を与えるかもしれません。
規制、法規、または既存の規制の解釈の変化は、(I)私たちの製造スケジュールを変更すること、(Ii)製品ラベルを追加または修正すること、(Iii)私たちの製品をリコールまたは停止すること、または(Iv)追加の記録保存要件を要求することなど、私たちの将来の業務に影響を与える可能性がある。このような変化を強制的に実施すれば、私たちの業務運営に悪影響を及ぼす可能性がある。
19
世界反腐敗法
米国“海外腐敗防止法”及びカナダ“外国公職者腐敗法”、“米国旅行法”、“OECD反賄賂条約”第18章米国法典第201条、及び我々が適用する他の任意の適用される国内又は海外反腐敗又は反賄賂法は、会社及び個人が業務を取得又は保留し、公式身分で働く者を取得又は保留することを禁止している。いかなる外国の政府職員、政府職員、政党または政治候補者に支払いを提案し、任意の価値のあるものを支払うことを提案して、業務を獲得または保留しようとしているか、または他の方法で公的身分で働く人に影響を与えることは、不法である。米国の“海外腐敗防止法”、カナダ“外国公職者腐敗法”、その他の適用される反腐敗·反賄賂法によると、我々の第三者代理の行為に責任を負うことも求められる可能性がある。これらの法律を遵守しないことは、私たちを調査、制裁、和解、起訴、他の法執行行動、利益返還、巨額の罰金、損害賠償、他の民事と刑事罰または禁止、特定の人との契約の一時停止または取り消し、輸出特権の喪失、通報者の苦情、名声損害、不利なメディア報道、およびその他の付随的な結果に直面する可能性がある。いかなる調査、行動、または制裁、または前述した他の損害は、私たちの業務、経営業績、および財務状況に実質的な負の影響を及ぼす可能性がある。
アメリカ以外の政府規制
米国の法規以外にも、研究、開発、テスト、製造、品質管理、制御物質、承認、ラベル、包装、貯蔵、記録保存、販売促進、広告、流通、承認後の監視と報告、マーケティングと薬品の輸出入、精算要求を含む他の司法管轄区域の様々な法規を遵守する。一般的に、新薬が発売される前に、その品質、安全性、有効性を証明するデータを大量に取得し、各規制機関の特定のフォーマットに組織し、審査を提出し、監督機関の承認を得なければならない。私たちがFDAの候補製品の承認を得るかどうかにかかわらず、私たちは外国の規制機関がこれらの国で臨床研究を始めたり、その候補製品をマーケティングしたりする前に必要な承認を得なければならない。米国以外のある国にも類似したプログラムがあり,ヒト臨床研究を開始する前に臨床研究申請を提出することが求められており,INDに似ている。
臨床研究、製品許可、カバー範囲、定価と精算を指導する要求と手続きは国によって異なる。いずれの場合も,臨床研究はGCPおよび“ヘルシンキ宣言”からの適用法規要求と倫理原則に基づいて行われている。EU第536/2014号臨床試験法規、あるいはCTRと呼ばれ、2022年1月31日に発効し、臨床試験指令の代わりに、EUの臨床薬物試験提出、評価とモニタリングのプログラムを統一し、それによって現在の臨床試験の許可と表現標準の規則を簡略化した。CTRは臨床試験スポンサーに臨床試験を行うEU加盟国ごとの国家主管当局(NCA)の承認を求める。また,スポンサーは現地研究倫理委員会(REC)が積極的な意見を発表した後のみ,特定の研究地点で臨床試験を開始することができる。以下に述べる移行手配によると、スポンサーは臨床試験情報システム(CTI)と呼ばれるEU中央臨床試験ポータルサイトを通じて臨床試験許可或いはCTAの申請を提出する。NCA(スポンサーによって選択された報告書提出EU加盟国)が率先して検証および評価申請を行い、その中で臨床試験を行う他の関連加盟国と相談および調整を行う。申請が拒否された場合、申請は修正され、CTISによって再提出されることができる。関連加盟国は限られた状況で“選択脱退”の承認を宣言し、臨床試験がこの加盟国で行われることを阻止することができる。CTRは3年間の過渡期が予想される。2023年1月31日までに,すべての新しいCTA申請はCTIで提出され,CTRに基づいて提出されなければならない。2025年1月31日からその後,臨床試験指令(現在CTRに置換されている)によって承認されたいずれかのまだ行われている試験は,CTRに適合してCTIに記録される必要がある。
イギリスでは、人用薬品の臨床試験は主に“2004年人用薬品(臨床試験)条例”(改正)によって管轄されている。EUと同様に,イギリスで臨床試験が開始される前に,薬品や保健製品規制機関(MHRA)のCTA,RECの積極的な意見を得なければならない。
20
EUの監督管理制度によると、研究薬物の監督管理の承認を得るためには、上場許可申請、すなわちMAAを提出しなければならない。2種類のマーケティング許可があります
連合王国については,2024年末までに,“大ブリテンおよび北アイルランド連合王国の欧州連合と欧州原子力共同体からの離脱に関する協定”に掲載された“北アイルランド議定書”に基づき,集中化されたMAは北アイルランドの医療製品の商業化に有効な基礎を提供し続けている。しかし、集中化されたMAはもはやイギリスの医薬製品の商業化に有効な基礎を提供しなくなった。ウィンザー·フレームワーク(EU委員会とイギリス政府がイギリスの離脱後の薬品を含む貨物の流れの制限を是正するために発表した政治的声明)によると、2025年1月1日から、イギリス市場のすべての新しい医薬製品はMHRAの許可を得、イギリスで販売しようとしているすべての医薬製品に単一のイギリス範囲内のMAをイギリス許可局に代表して、単一パッケージで北アイルランドを含むイギリス各地で販売できるようにするが、イギリスパッケージには明確で読みやすい“UK Only”を持ってこそイギリス市場への進出が許可されなければならない。
EU離脱以来,MHRAは患者に利益を与える新薬を優先的に獲得するプログラム,加速評価プログラム,新製品とバイオテクノロジー製品の新たな評価経路を含む国家許可プログラムを変更した。司法管轄区域間ではEU薬品立法の卸売承認はなく、EU MAも英国医薬製品の商業化に有効な基礎を自動的に提供することはない。2024年1月1日から、会社はMHRAに新しい国際認可手続き(IRP)に基づいて外国司法管轄区(EUを含む)が受け入れ可能な参考監督管理機関によって付与されたMAを認めることを要求することができる。IRPは,MHRAが信頼されている規制機関の専門知識や意思決定を考慮し,IRP申請を的確に評価することを可能にし,提供された証拠が十分でないと考えられる場合に申請を拒否する権限を保持することを可能にする。
米国が機密協定を提出するための申請はEU要求の申請と類似しているが、他の要求に加えて、特定の国に対する文書要求がある点で異なる。薬物が商業化される前に、規制部門のこの薬物の精算承認を得る必要がある。
21
連合はまたデータと市場排他性に機会を提供する。例えば、EUでは、マーケティング許可を得た後、品質、臨床前試験結果、および臨床試験データからなる完全な独立したデータパケットから承認された革新医薬製品(小分子および生物医薬製品を含む)に基づいて、MA付与後8年間のデータ独占権を取得し、2年間のマーケティング独占権を追加的に取得する。承認された場合、データ排他性は、模倣薬または生物類似申請者が模倣薬または生物類似MAを申請する際に、データ排除期間が満了するまで、革新者の臨床前と臨床試験データを交差引用することを防止する。模倣薬や生物類似製品が承認されても、丸10年の専門期間が終わる前にしか発売できない。この10年の最初の8年間に、MA保持者が1つまたは複数の新しい治療適応の許可を得た場合、許可前の科学的評価では、これらの適応は、現在承認されている療法と比較して有意な臨床的利益をもたらすと決定された場合、10年全体の期間を最大11年に延長することができる。イギリス国内法は同じ規制データとマーケティング排他的公式に従っている。しかし、製品がEU規制機関によって新しい化学実体や新しい活性物質とみなされることは保証されず、製品はデータ排他性を得る資格がない可能性がある。
EUで孤児の称号を獲得した製品は10年間の市場排他性を得ることができ、マーケティング許可を与える時も孤児の称号を維持することを前提としている。10年間の孤児市場専門期間内に、EUのいかなる監督機関も同じ適応の類似医薬製品の申請を受け入れて許可を得てはならない。孤児製品は合意された小児科調査計画に基づいて小児科研究を完成させることもでき,EUでは追加2年間の市場排他性を得ることができ,データが小児科適応の承認を招くことはない。いかなる補充保護証明書も孤児の症状に関する小児科研究によって延期してはならない。
EUが“孤児医薬製品”を指定する基準は、原則として米国と類似しているが、全く同じではない。(EC)141/2000号条例第3条によると、生命または長期的に衰弱した疾患の診断、予防または治療に医薬製品が使用されている場合は、孤児として指定することができる;(2)申請した場合、(A)このような疾患は、EUで10,000人を超えないうちの5人に影響を与えるか、または(B)当該製品に孤児身分によるメリットがなければ、EUでは十分な見返りが生じず、投資が合理的であることを証明するのに十分ではない。および(3)このような疾患を満足できる診断、予防または治療する方法がEU市場で販売されていないこと、またはそのような方法がある場合、製品は、(EC)847/2000法規によって定義されているこのような疾患の影響を受けている人に大きな利点を有するであろう。販売許可を付与する際には、10年間の孤児市場独占期間から利益を得るために、指定孤児の基準を再評価して確認しなければならない。MHRAはイギリスに特定された基準について同様の評価を行った。欧州医薬品局では,MA申請を提出する前に,孤児薬の指定を要求しなければならない。EMAの孤児医薬品委員会(COMP)は、MAの付与時に承認された孤児指定を再評価して、指定保持基準に適合し続けることを確実にするように要求されている。そうでなければ、孤児の指定は撤回されることができる。対照的に、MHRAは医薬製品の開発過程で孤児の称号を与えない。逆に,MHRAはMA付与時に基準を満たすかどうかを決定する.
ヨーロッパ医薬品局とイギリスでは、孤児薬品は費用を下げたり費用を免除したりするなどの経済奨励を受ける資格があり、マーケティング許可を得た後、承認された治療適応の10年間の市場独占経営権を得る権利がある。孤児薬物指定が承認された場合、出願人は、上場許可申請の費用減免を受けるが、上場許可を提出したときにその指定が待っている場合は、そうではない。孤児薬物の指定は、監督審査と承認過程においていかなる利点も伝達されず、規制審査と承認過程の持続時間を短縮することもない。
5年目の終了時に、製品が指定された孤児の基準を満たしていないと判定された場合、例えば、製品の利益が十分に高く、市場排他性を維持するのが合理的であることを証明するのに十分でない場合、10年間の市場排他性は6年に減少することができる。さらに、以下の場合、いつでも同じ治療適応の類似医薬製品にマーケティング許可を与えることができる
類似医薬製品“の定義は、承認された孤児医薬製品に含まれる1つ以上の類似活性物質を含む医薬製品であり、同じ治療適応のためのものである。
22
欧州医薬品局では,新医薬製品を開発する会社は,欧州薬品管理局の小児科委員会(“PDCO”)と小児科研究計画(“PIP”)について合意しなければならず,免除が適用されない限り,この計画に基づいて小児科臨床試験を行わなければならない。PIPは,MAが求められている薬物の小児科適応を支援するために,データ発生時間とアドバイスの措置を規定している。PDCOは、成人における製品の有効性および安全性を証明するのに十分なデータがあるまで、PIPの一部または全ての措置の実施を延期する義務を許可することができる。さらに、小児科臨床試験データが不要または不適切に提供される場合、PDCOは、子供に無効または安全でない可能性があるので、これらのデータを提供する義務を免除することができ、この製品は、治療のために使用される疾患または状態が成人集団でのみ発生することが予想される、または小児科患者の既存の治療に対して有意な治療利益がない場合である。MAが付与された製品は,PIPによる小児科臨床試験の結果(結果陰性であっても),6カ月の補充保護証明書の延期(承認時に有効であれば)を得る資格がある。孤児薬品の場合、孤児市場の独占権を2年間延長することができる。この小児科奨励は特定の条件の制約を受け,PIPに適合したデータを開発·提出する際に自動的に獲得されない。現地の法律要求に基づき,イギリスのMHRAはEEAと類似した方法を採用し,小児科人口のための医療製品の開発を促進している。
EU以外の他の国、例えばカナダや東欧、ラテンアメリカまたはアジアの国では、臨床研究、製品と機関の許可、カバー範囲、データ保護、定価と精算の要求は国によって異なる。繰り返しますが,すべての場合,臨床研究はGCPおよび“ヘルシンキ宣言”からの適用法規要求と倫理原則に基づいて行われています。
もし私たちが適用される外国監督管理要求を遵守できない場合、私たちは罰金、規制許可の一時停止または撤回、製品のリコール、輸出入できない、製品の差し押さえ、経営制限、刑事起訴などに直面する可能性がある。
薬品の保証範囲,定価と精算
私たちが規制部門の承認を得た任意の候補製品のカバー範囲と精算状態には、重大な不確実性がある。米国や他国の市場では、規制部門の承認を得て商業販売を行う任意の製品の販売は、第三者支払者が保険及び十分な補償を提供するか否かにある程度依存する。第三者支払者には、MedicareまたはMedicaidなどの政府プロジェクト、個人健康保険および管理医療計画などの個人支払者、および他の組織が含まれる。これらの第三者支払者は製品の保証や精算を拒否したり制限したりする可能性がある。
米国では,支払先によって薬品のカバー範囲や精算方式が大きく異なる可能性がある。1つの第三者支払人は、ある特定の薬品又はサービスを保証することを決定し、他の支払者も当該製品に保険を提供することを確保することができず、又は適切な販売率で保険を提供することができる。第三者支払者は、カバー範囲を制限すること(例えば、承認リスト上の特定の医薬製品または処方に限定され、FDAによって承認された特定の適応のすべての医薬製品を含まない場合がある)、使用を制御すること(例えば、特定の患者に保険を提供する前に、新しいまたは革新的な薬物療法の事前承認または事前許可を要求すること)、および薬品の精算金額を制限することによってコストを制御しようと試みることができる。
薬品コストは引き続き政府と第三者支払人の深い興味を引き起こした。管理式ヘルスケアの傾向,管理式ヘルスケア組織のますますの影響力,追加的な立法提案により,製薬業は定価圧力に直面することが予想される。第三者決済者は、価格、医療製品およびサービスの医療必要性および費用効果、ならびにそれらの安全性および有効性を検討することにますます挑戦している。私たちは、私たちの製品の医療の必要性と費用効果、FDAの承認を得るのに必要なコストを証明するために、高価な薬物経済学的研究を行う必要があるかもしれない。私たちの候補製品は医学的に必要で費用効果があると思われないかもしれない。支払人が薬品に保険を提供することを決定することは適切な販売率を承認することを意味しない。製品開発への投資の適切な見返りを実現するために、十分な価格レベルを維持することができる十分な第三者精算がないかもしれない。
将来どのようなコスト制御措置が取られるか、または他の方法で実施されるかは予測できないが、新しい措置または任意の提案された措置の発表は、候補製品のために十分な価格および利益運営を得る能力に重大な悪影響を及ぼす可能性がある。
国際市場では,精算や医療保険支払い制度は国によって異なり,多くの国で特定製品や治療法に価格上限が設定されている。私たちの製品が医学的に合理的であることは保証されず、特定の適応が必要であり、私たちの製品が第三者支払者によって費用効果があると考えられている保証はなく、私たちの保険範囲や精算レベルが十分であることは保証されず、第三者支払者の精算政策が利益を得て製品を販売する能力に悪影響を与えないことも保証されない。
23
また、多くの外国の国では、薬品の提案価格は必ず承認されなければならず、合法的に発売されることができる。各国の薬品定価と精算に対する要求は大きく異なる。例えば、EUは、その加盟国に様々な選択を提供し、その国の健康保険制度が補償を提供する医療製品の範囲を制限し、人が使用する医療製品の価格を制御する。加盟国は医薬製品の具体的な価格を承認することができ、医薬製品を市場に投入する会社の収益力に対して直接或いは間接的に制御制度をとることもできる。薬品に対して価格制御や精算制限を実行する国が私たちのいかなる製品にも有利な精算と定価手配を許可することは保証されません。歴史的に見ると、EUで発売された製品はアメリカの価格構造に従わず、通常価格ははるかに低くなることが多い。
医療改革
米国や他の管轄地域では、医療システムの立法や規制面のいくつかの変化が、私たちの将来の運営結果に影響を与える可能性がある。特に、アメリカ連邦と州レベルはすでにいくつかの措置を取り続け、全体的に医療コスト、特に薬品コストの低減を求めている。
例えば、米国では、改正された“患者保護と平価医療法案”(Patient Protection and Affordable Care Act,PPACAと略称する)は2010年3月に公布された全面的な法律であり、医療補助の拡大と個人医療保険のカバー範囲の実施を通じて医療保険のカバー範囲を拡大し、その中に政府医療保健計画下の薬品のカバー範囲の変更と精算範囲を含む。PPACA以外にも,広く行われている医療改革努力があり,その中のいくつかの重点は薬品の価格や支払いを規制することである。薬品の定価と支払い改革はトランプ政権の重点であり、バイデン政権の重点でもある。例えば、2021年に公布された連邦立法は、2024年1月1日に施行された医療補助薬品税還付計画の法定上限を廃止した。もう一つの例として、2022年の“インフレ低減法案”(IRA)には様々な薬品価格交渉、インフレリベート、定価条項が含まれているが、実施日はそれぞれ異なる。
アイルランド共和軍は連邦政府がある高価な単一源の連邦医療保険薬物のために最高公平価格を交渉することを許可し、薬品価格交渉要求を守らないメーカーに罰と消費税を加え、すべての連邦医療保険B部分とD部分の薬物(限られた例外の場合)が薬品価格の増加がインフレより速い場合にインフレリベートを獲得することを要求し、連邦医療保険D部分を再設計して受益者の自己処方薬コストなどの変化を減少させる。Ireland共和軍が施行中であるため、私たちの業務とより広い製薬業への影響はまだ不確定だ。もう一つの例は、2022年、アイルランド共和軍が公布した後、バイデン政府はHHS報告書が連邦医療保険と医療補助革新センター(CMMI)を利用して連邦医療保険と医療補助受益者の薬品コストを下げる新しいモデルをテストする方法を指示する行政命令を発表した。この報告は2023年に発表され,CMMIが現在開発中の様々なモデルを提案し,薬物コストの低減,獲得性の促進と看護の質の向上を目指している。このような変化や他の変化は私たちの製品の市場状況に影響を及ぼすかもしれない。私たちは議会、機関、そして他の機関が薬品価格と政府価格報告書を引き続き検討すると予想している。
州レベルでは、立法機関は、価格または患者の精算制限、割引、特定の製品参入の制限、マーケティングコスト開示および透明性措置を含む、医薬品および生物製品の価格設定を制御するための法規を立法および実施することが増えており、場合によっては、他の国からの輸入および大量購入を奨励することを目的としている。例えば、FDAは、2024年1月5日に、特定の州医療計画のためにカナダから特定の薬剤を輸入するフロリダ州804条輸入計画(SIP)の提案を承認した。この計画がどのように実施されるか,どのような薬剤が選択されるか,その計画が米国やカナダで法的挑戦を受けるかどうかは不明である。他の州もFDAの審査を待っているSIP提案を提出した。このような承認された輸入計画が一旦実施されると、これらの計画がカバーする製品の薬品価格をより低くする可能性がある。また、多くの州はすでに州薬品価格透明性と報告法を公布し、もし私たちが製品が発売されれば、私たちのコンプライアンス負担を大幅に増加させ、このような州法律に基づいてより大きな責任を負わせるかもしれない。
医療改革の努力はすでに検討され、法的挑戦を受け続ける可能性がある。例えば,PACAでは税収改革立法が公布され,2019年から強制医療保険範囲を維持していない個人への税収処罰が廃止され,2021年には米国最高裁はいくつかの州がACAに対する最新の司法挑戦を却下したが,PPACAの合憲性を具体的に裁くことはなかった。別の例として、連邦反減税法規下の法規改正は、製薬業者が薬局福祉マネージャーおよび健康計画に提供する従来のMedicare Part D割引の保護を廃止する。裁判所の命令により更迭が延期され、最近の立法はこの規則の施行を2032年1月に一時停止した。もう一つの例として,アイルランド共和軍の薬品価格交渉計画は,複数の製薬業者や業界団体による訴訟で挑戦されている。
すでにまたは将来的にとりうる医療改革措置は、より厳しいカバー基準をもたらす可能性があり、承認された任意の製品の価格に追加の下振れ圧力をもたらし、将来の任意の収入創出を深刻に損なう可能性があると予想される。連邦医療保険や他の政府が計画している精算のいかなる減少も、個人支払者の支払いの同様の減少を招く可能性がある。どんな新しい法律と法規の変化を遵守することは時間がかかり、費用がかかる可能性があり、それによって私たちの業務に実質的な悪影響を与える可能性がある。
24
承認後の要求を拡大し、医薬品の販売や販売促進活動を制限するための立法·規制提案がなされている。私たちは、より多くの立法変化が公布されるかどうか、あるいはFDAの法規、ガイドライン、解釈が変わるかどうか、あるいはこれらの変化が私たちの候補製品の規制承認(あれば)にどのような影響を与える可能性があるかどうかを決定することはできない。また、FDA承認過程に対する国会のより厳しい審査は、規制部門の承認を著しく延期または阻止する可能性があり、より厳しい製品ラベルや上場後テストなどの要求の影響を受ける可能性がある。
しかも、他の国も違う価格設定と補償プログラムを持っている。ヨーロッパ共同体では,各国政府はその定価と精算規則や国家医療システムの制御により医薬製品の価格に影響を与え,これらのシステムは消費者にこれらの製品の大部分のコストを支払っている。いくつかの法域はプラスリストとネガティブリスト制度を実行し、補償価格を合意した後にのみ、製品を市場で販売することができる。その中のいくつかの国は、精算或いは定価の承認を得る条件として、臨床試験を完成し、特定の製品のコスト効果を当時利用可能な他の治療法と比較することを要求する可能性がある。他の会員国たちは会社が自分の薬品価格を固定することを許可するが、会社の利益を監視する。全体的には,医療コスト,特に処方薬の下り圧力が非常に大きくなっている。そのため、新製品の参入にはますます高い壁が設けられている。また、一部の国では、低価格市場からの国境を越えた輸入が一国国内の定価に商業圧力をかけている。
2021年5月1日、EUとイギリスの貿易·協力協定(TCA)が発効した。TCAは薬品に関する具体的な規定を含み,その中には相互承認GMP検査の結果が含まれている。申請者とMA保有者は,EU法規として提出された支援情報として,MHRAがEU/欧州経済区以外に位置する地点に発行したGMP証明書を提出することができる。しかし,TCAはイギリスとEUの薬品法規が大規模に相互承認されないと予想している。現在、イギリスの規制制度はEUの規制制度とほぼ一致している。しかし、欧州委員会が提出した薬品監督制度の全面的な改革の提案のような提案された立法改革を考慮すると、これらの制度は将来的に食い違いが生じる可能性がある。
他の医療法とコンプライアンス要件
アメリカでは、製薬業者は多くの他の連邦、州と地方の法律を遵守しなければならない。これらの法律は詐欺と乱用を防止すること、医療保健業界の他の人との相互作用の透明性を高めること、薬品の定価を監督し、個人情報のプライバシーを保護することを目的としている。これらの法律は、米国司法省および司法省、米国衛生·公衆サービス部(HHS)内の個別の米国検事室、HHSの各部門を含むが、医療保険および医療補助サービスセンター(CMS)、監察長事務室、州薬局委員会を含むが、これらに限定されない異なる連邦および州法執行機関によって実行される。
私たちは、リベート法および虚偽請求法を含む、医療保健の“詐欺および乱用”に関連する様々な連邦および州法律の制約を受ける可能性があり、これらの法律は、連邦医療保険計画(連邦医療保険および医療補助計画を含む)、または場合によっては、商業健康計画など、第三者支払者によって精算可能な製品の過去および将来の販売活動に関するものである。逆バックル法は、一般に、購入、処方、または特定の薬物の使用を含む任意の価値のあるものを製造するために、製薬業者が誘致、提供、受け入れ、または支払うことを禁止する。虚偽申告法は、一般に、任意の虚偽または詐欺的な精算薬品またはサービス支払い請求を第三者支払者に提出するか、または提出することを禁止する。これらの法律の具体的な規定はそれぞれ異なるにもかかわらず、それらの範囲は通常広く、これらの法律は特定の業界で実践されている法規、指導、または裁判所判決に適用されていないかもしれない。したがって、私たちの接近はこのような法律の挑戦を受ける可能性がある。
連邦政府と各州も製品を販売する薬品メーカーの販売とマーケティングのやり方を規範化するために法律と法規を公布した。法律と法規は一般にメーカーと医療保健提供者との間の財務的相互作用を制限する;メーカーにいくつかのコンプライアンス標準を採用することを要求する;政府と公衆に財務的相互作用を開示することを要求する;マーケティング支出または定価情報の開示を要求する;薬品の価格設定を規範化し、および/または薬品販売代表の登録を要求する。その中の多くの法律法規の要求が曖昧で、あるいは行政指導が必要である。法律およびその実施の曖昧性を考慮して、将来の任意の活動(私たちが連邦医療計画の候補製品の承認および/または補償を得たら)が挑戦される可能性がある。
医療補助薬品リベート計画を含む連邦法律は、製薬業者が何らかの計算された製品価格を政府に報告すること、または政府当局または個人実体に何らかの割引またはリベートを提供することを政府に要求し、通常、政府医療計画下の精算条件とする。これらの法律は複雑であり、報告された価格を正確に計算できなかったり、適切な価格およびリベートを提供したりすることは、製造業者を処罰および他の制裁に直面させる可能性がある。
薬品と生物製品の流通は広範な記録保存、許可、貯蔵と安全要求を含む追加の要求と条例を遵守し、許可されていない薬品の販売を防止しなければならない。
25
連邦と州消費者保護と不正競争法律法規は市場活動を広く規制しており、これらの活動は消費者を損なう可能性があり、製薬業者の活動に適用される可能性がある。
私たちが個人の身分情報を運営し、取得したり、保存したりする各管轄区では、データプライバシーとセキュリティ法律の制約を受ける可能性があります。多くのアメリカ連邦と州法律は個人情報の収集、使用、開示と保存を規範化している。複数の外国の国でも、個人情報の収集、使用、開示、保存を管理する法律が制定されている。世界的に、私たちの業務に影響を及ぼす可能性のあるプライバシーやデータ保護問題がますます注目されている。
私たちの活動が適用された医療法律と法規に適合することを確実にする努力は巨額のコストに関連するだろう。法律法規の広範性、ある法律法規に対する指導が限られていること、および政府の法律法規が絶えず変化している解釈を考慮すると、政府当局は結論を出す可能性があり、私たちの商業実践はこれらの法律に適合していないかもしれない。もし私たちの運営がこれらの法律または任意の他の私たちに適用される可能性のあるすべての政府法規に違反していることが発見された場合、私たちは重大な民事、刑事、行政処罰、損害賠償、罰金、連邦医療保険や医療補助のような政府援助の医療計画から除外され、私たちの業務の削減や再編を受ける可能性がある。さらに、このような行動を防御するには費用がかかり、時間がかかる可能性があり、多くの人的資源が必要となる可能性がある。したがって、私たちが私たちに提起される可能性のあるいかなる訴訟も防ぐことに成功しても、私たちの業務は損害を受ける可能性がある。
我々の製品が海外で販売されている場合、または米国以外のサプライヤーまたは独立請負業者と契約を締結した場合、同様の外国の法律および法規の制約を受ける可能性があり、例えば、安全監視、反腐敗/反賄賂法、リベート法、医療詐欺および乱用法律、および会社のコンプライアンス計画を実施し、医療保健専門家に価値を支払うか、または移転する場合を含む適用された承認後の要求が含まれる可能性がある。現在何の問題もあることはわかりませんが、医療保険法を適用した訴訟や、このような行動が私たちの業務に影響を与えるかどうかは予測できません。しかし、このような行動またはクレームを弁護するコスト、および適用される任意の制裁は、私たちの業務または財務状況に実質的な悪影響を及ぼす可能性がある。
環境問題
私たちの運営には危険材料(生体材料を含む)が必要であり、これは様々な連邦、省、地方環境、安全法律法規の制約を受けている。現行の監督管理構造下のいくつかの規定は厳格な責任を規定し、一方の当事者に潜在的な責任を負うことを要求し、過失や不注意を考慮しない。環境汚染や個人接触危険物質が発生すると,我々や他者の業務運営による損害や罰金の負担が要求される可能性がある。私たちは、私たちが適用される環境法律を実質的に遵守し、これらの法律を遵守し続けることは、私たちの業務に実質的な悪影響を与えないと信じている。しかし、私たちは法律の変化や新しい規制の発展が私たちの業務運営やコンプライアンスコストにどのように影響するのか予測できない。
人力資本
我々の取締役会と経営陣は、長期的な企業価値の創造は、我々従業員を含む多くの利害関係者の利益や懸念を考慮することで進められていることを認識している。2023年12月31日現在、私たちは251人のフルタイムとアルバイトの永久従業員を含む259人の従業員を持っており、そのうち179人はカナダに位置し、72人はアメリカに位置している。私たちの従業員のうち、190人が主に研究開発に従事しており、そのうち78人が博士や医学博士(または同等の学歴)の学位を持ち、69人が一般的で行政や商業活動に従事している。私たちの職員たちの中で労働組合が代表する人は一人もいない。私たちは何の停止も経験しておらず、私たちは私たちが従業員と仲がいいと思う。
生物技術と製薬領域の合格人材に対する競争は非常に激しいため、各級の合格従業員を誘致と維持することは著者らの業務に重要である。従業員の士気が強いことを確保し、従業員の尊敬度調査を行い、重点分野を決定し、従業員の流出率を監視する努力を続けている。2023年12月31日現在、わが社の離職率は過去数年間、業界市場平均を下回ってきた。
我々は、高素質の人員を誘致し、維持し、優秀な業績を奨励·奨励するために、全面的かつ競争力のある報酬·福祉計画を構築している。我々の従業員に競争力のある賃金を提供するほか、従業員は会社および/または個人目標の実現に応じて、永久従業員に年間ボーナスを支給することで、私たちの計画を進めることで生じる潜在的な財務収益を共有すべきだと考えます。私たち従業員の利益を私たちの株主の利益と一致させるために、私たちは最初の採用時もその後も毎年、すべての永久従業員に株式オプションを付与します。私たちの休暇計画には有給休暇、個人休暇、病気休暇、障害休暇、その他の有給休暇と無給休暇が含まれています。私たちの健康と健康計画には、医療、歯科、視力ケア、退職貯蓄、従業員援助計画、柔軟な勤務時間、その他の福祉が含まれています。
高学歴の従業員を持つバイオ製薬会社として、私たちの従業員は業界の進歩に続いて、彼らのキャリアの中で成長し続けなければならないと信じている。会議への出席や授業料の精算など、様々な内部研修や外部専門の機会を通じて、従業員のさらなる発展を支援しています。
26
私たちは会社の各レベルの多様性、公平性、包摂性、獲得性に取り組んでおり、これらの重要な問題を推進するための共同経営陣/従業員委員会を設立しています。私たちは私たちの労働チーム全体で私たちの多様性と包括的な計画を測定して拡大することに集中し続けるつもりだ。私たちは性別、性別、民族、人種、宗教、あるいは他の保護された特質を問わず、最も合格した従業員を募集し、私たちの政策は職場差別に関するすべての適用法律を遵守することだ。
製造業
我々は現在依存しており,引き続き第三者契約メーカーに依存して我々の製品を生産(または十分な数の製造に必要な材料)を生産していく予定であり,臨床前試験や臨床試験のために,我々の製品の商業生産に用いる予定である。同様に、私たちは協力者たちに直接またはCMOを通じて彼らに許可された候補製品を製造することに依存するかもしれない。したがって、私たちは内部でどんな製造施設を開発したり、関係者を招いたりしなかった。
これまでに、私たちは複数の第三者メーカーとサプライヤーから私たちの候補製品の材料を獲得しました。私たちは私たちの候補製品を作るために必要なすべての材料が一つ以上の供給源から得られると信じている。しかし、候補製品ごとに製造プロセスが異なり、関連する候補製品のタイプに応じて、十分な供給を探すことがより困難になる可能性がある。私たちの候補製品は通常信頼性があり、再使用可能な合成技術によって生産することができ、原料、補助剤と包装アセンブリはどこでも得ることができる。
企業情報
私たちは1996年11月5日にブリティッシュコロンビア州で“商業会社法”(ブリティッシュコロンビア省)の前身に基づいて設立され、名称は“Xenon BioResearch Inc.”である。2000年5月17日、カナダ商業会社法(CBCA)第187条によると、私たちはブリティッシュコロンビア州から引き続き連邦管轄範囲に入り、同時に私たちの名称を“Xenon Genetics Inc.”と改称した。2000年7月10日,ブリティッシュコロンビア州で省外会社に登録され,“キセノン製薬会社”と改称された。2004年8月24日。2023年12月31日現在、Xenon PharmPharmticals USA Inc.の完全子会社、Xenon Pharmticals USA Inc.が、2016年12月2日にデラウェア州に登録設立された。私たちの主な実行事務室はカナダブリティッシュコロンビア州バーナビGilmore Way 3650にあります。郵便番号:V 5 G 4 W 8、私たちの電話番号は(604)484-3300です。私たちはブリティッシュコロンビア州、エバータ州、オンタリオ州の報告発行元ですが、私たちの株はどの公認のカナダ証券取引所にも上場していません。私たちの普通株はナスダックの世界市場で取引され、コードは“XENE”です
そこでもっと多くの情報を見つけることができます
私たちは、これらの資料を電子的にアメリカ証券取引委員会(アメリカ証券取引委員会)に提出または提出した後、合理的で実行可能な範囲内で、私たちの投資家関係サイトを通じて、私たちの年間報告、四半期報告、現在の報告、依頼書、およびこれらの報告のすべての修正をできるだけ早く無料で提供します。これらの報告書はカナダブリティッシュコロンビア州バーナビーのXenon製薬会社投資家関係部3650 Gilmore Way,V 5 G 4 W 8,電子メール:Investors@xenon-pharma.comにも無料で連絡することができる。我々のサイトとその含まれたり統合された情報は、本Form 10-K年次報告に含めるつもりはありません。米国証券取引委員会は、報告書、依頼書、および情報宣言と、www.sec.gov上で電子的に提出または提供された報告書に関する他の情報とを含むウェブサイトを維持している。キセノンに関するより多くの情報はSEDARサイトwww.sedar.comでも入手可能である。
27
第1 A項。リスク要因
本報告に含まれる他の情報に加えて、本報告書の“財務状況および経営結果の検討および分析”というタイトルの部分、および私たちの財務諸表および関連付記を含むリスク要因をよく考慮しなければなりません。以下のリスク要因に記載されている任意の事件および本報告書の他の場所に記載されているリスクが発生した場合、我々の業務、経営業績、および財務状況は深刻な損害を受ける可能性がある。Form 10-Kに関するこの報告書はまた、リスクと不確実性に関する前向きな陳述を含む。本報告の以下および他の部分に説明される要因のため、我々の実際の結果は、前向き陳述で予想される結果とは大きく異なる可能性がある。我々のリスク要因は,本報告の日までにこのような状況が存在しないことを保証しているわけではなく,このようなリスクや状況がすべてまたは部分的に発生していないと肯定的に解釈されるべきでもない。
私たちの財務状況と資本要求に関連するリスク
設立以来、私たちは大きな被害を受けており、予測可能な未来には、引き続き大きな被害を受けると予想されています。
生物製薬製品開発への投資は非常に高い投機性があり、それは大量の資本支出を必要とし、そして重大なリスクが存在するため、即ち候補製品は十分な治療効果或いは受け入れ可能な安全性を証明できない可能性があり、監督管理部門の許可を得ることができず、商業上実行可能ではない。これまで、商業販売が許可されている製品は何もありませんし、製品販売から何の収入も得られていません。私たちの臨床開発と持続的な運営に関連する大量の研究開発やその他の費用が発生し続けます。したがって、私たちは利益を上げず、設立以来各時期に損失を出した。設立以来、著者らはほとんどの財務資源と努力を臨床前研究、研究薬物の製造と臨床試験を含む研究と開発に投入した。私たちの財務状況と経営結果は、純損失を含めて、四半期ごとに毎年大きく変動する可能性があります。したがって、今後の経営業績の指標として、いかなる四半期や年度の業績にも依存してはいけません。私たちは予測可能な未来に持続的な収益性がないと予想する。2023年12月31日,2022年,2021年12月31日までの年度の純損失はそれぞれ1.824億ドル,1.254億ドル,7890万ドルであり,2023年12月31日までの累計赤字は6.651億ドルであり,我々の研究開発計画に関する費用および我々の運営に関する一般的かつ行政コストが原因である。私たちは予測可能な未来に引き続き大きな損失を受けることが予想され、私たちが私たちの候補製品を研究·開発し、規制部門の承認を求めるにつれて、これらの損失は増加するだろう。
私たちは予測可能な未来に巨額の費用が発生し、運営損失が増加すると予想している
様々な理由で、米国食品医薬品局、FDA、欧州医薬品局またはEMAまたは他の規制機関が、承認後の約束を含む現在の予想に基づいて臨床および他の研究を行うことを要求する場合、または私たちの臨床試験を支援するための適切な製造計画が確立され、私たちの任意の候補製品の開発または商業化に遅延が生じた場合を含む、私たちの費用は予想を超える可能性がある。私たちのこれまでの損失は、予想された将来の損失に加え、私たちの株主権益や運営資本に悪影響を与え続けるだろう。
28
私たちは製品販売から何の収入も得ませんし、永遠に利益を上げないかもしれません.
私たちが収入を創出し、利益を達成する能力は、私たち単独またはパートナーと私たちの候補製品の開発を成功させ、必要な規制承認を得る能力にかかっている。成功した商業化は、臨床試験で安全性と有効性を証明し、マーケティングを含む規制を獲得し、これらの候補製品を承認し、私たちまたは私たちの既存または未来の任意のパートナーが規制の承認を得る可能性のある製品を製造、マーケティング、販売し、任意の上場後の要求を満たし、個人保険や政府支払者から私たちの製品の精算を得ることを含む多くの重要なマイルストーンを実現する必要があるだろう。これらの活動に関連する不確実性とリスクのため、私たちは収入の時間と金額、さらなる損失の程度、または私たちがいつ利益を達成することが可能かどうかを正確かつ正確に予測することができない。私たちと私たちの既存または未来の協力者たちは決してこのような活動で成功しないかもしれないし、たとえ私たちが成功しても、私たちは利益を達成するのに十分な収入を生むことができないかもしれない。たとえ私たちが確実に利益を達成したとしても、私たちは四半期や年度の収益性を維持したり向上させることができないかもしれない。さらに、FDA、EMA、または他の規制機関が現在予想されている基礎の上で臨床試験を行うことを要求する場合、または私たちの臨床試験を完了したり、私たちの任意の候補製品の開発に遅延が生じた場合、私たちの費用は増加する可能性があります。
もし私たちの候補製品が臨床試験で失敗したり、規制部門の承認を得なかったりすれば、あるいは私たちの未来のどの製品も、一旦承認されれば、市場承認や十分な市場シェアを得ることができなければ、私たちは決して利益を上げないかもしれない。利益を達成し、このような状態を維持するために十分な収入を生み出すことができなければ、私たちの財務状況と経営業績はマイナスの影響を受け、私たちの普通株の市場価格は不利な影響を受ける可能性がある。
私たちは全くなければ受け入れられる条件で提供できないかもしれない追加資金を調達する必要があるだろう。必要なときに資金を得ることができない場合、私たちの製品発見と開発計画や商業化努力、または他の運営を延期、制限、または終了させることができないかもしれません。
設立以来、私たちは私たちの資源の大部分を私たちの臨床前と臨床候補製品の発見と開発に使用してきた。私たちは引き続き大量の資源を費やして、私たちの現在と未来の計画の臨床前と臨床開発を継続することが予想される。もし私たちが開発した候補製品のマーケティング承認を得ることができれば、これらの候補製品を発売して商業化するために多くの追加資金が必要になります。このような発売と商業化はパートナーの責任ではありません。また,我々の開発努力の過程で他の予期しないコストが発生する可能性がある.著者らの計画と期待される臨床試験の設計と結果は高度に不確定であるため、私たちが開発に成功した任意の候補製品の開発と商業化に必要な実際の数量を合理的に見積もることができない。
私たちの未来の資本需要は多くの要素に依存していますが、これらに限定されません
29
我々の研究と開発計画およびプロジェクトの進展に関する時間予想によると、本報告日までに、私たちの既存の現金と現金等価物および有価証券は、少なくとも今後12ヶ月の運営費用と資本支出需要に資金を提供できるようになると予想される。
現在知られていない多くの要因により、私たちの運営計画は変化する可能性があり、私たちは計画よりも早く追加資金を求める必要があるかもしれない。将来の資金調達は追加的な挑戦をもたらすかもしれないし、未来には十分な金額や私たちが受け入れられる条項が得られないかもしれない(もしあれば)。もし私たちが受け入れられる条件で十分な追加資本を調達できなければ、私たちは私たちの製品開発計画や商業化計画を大幅に延期、減少、または中止しなければならないかもしれない。
私たちは、特定の候補製品または適応を追求するために限られたリソースを割り当てる可能性があり、より利益的またはより成功する可能性の高い他の候補製品または適応を利用することができない。
私たちの財務と管理資源が限られているので、私たちは限られた数の研究プロジェクトと候補製品に集中している。したがって、私たちは、後により大きな商業潜在力を有することが証明された他の兆候における他の候補製品または私たちの現在の候補製品を求める機会を放棄または延期することができるかもしれない。私たちの資源分配決定は私たちが実行可能な商業薬や利益のある市場機会を利用できないかもしれない。私たちの現在と未来の研究開発計画と特定の適応候補製品への支出はいかなる商業的に実行可能な薬物も発生しないかもしれない。もし私たちが特定の候補製品の商業的潜在力や目標市場を正確に評価しなければ、私たちは独占開発権と商業化権利を保留することが私たちにより有利な場合、協力、許可、または他の配置を通じてその候補製品に貴重な権利を放棄するかもしれない。
追加資本の調達は、私たちの既存の株主を希釈し、私たちの運営を制限したり、私たちの技術や候補製品に対する権利を放棄することを要求するかもしれません。
私たちは私たちが計画した運営に関連した費用が増加すると予想している。私たちが任意の承認された候補製品から大量の収入を生み出すことができない限り、私たちは公開または私募株式発行、債務融資、特許使用料に基づく融資、協力、許可手配、または上記の任意の組み合わせによって、私たちの将来の現金需要に資金を提供すると予想される。また、有利な市場条件や戦略的考慮により、現在または将来の運営計画のために十分な資金があると考えても、追加の資本を求めることができる。
私たちが達成した任意の融資計画の条項は、私たちの株主の持株や権利に悪影響を及ぼす可能性があり、私たちが追加証券(株式または債務を問わず)、またはそのような証券を発行する可能性は、私たちの普通株の市場価格の下落を招く可能性がある。追加的な株式や転換可能な証券の売却はまた私たちのすべての株主の権利を希釈するだろう。
歴史的に、私たちはまた債務を通じて私たちの運営に資金を提供する。将来発生するいかなる債務も固定支払義務の増加を招き、制限的な契約を強制的に実施する可能性がある。このような条約には、私たちが追加債務を発生させる能力の制限、私たちが知的財産権を獲得し、販売する能力の制限、および私たちが業務を展開する能力に悪影響を及ぼす可能性のある他の運営制限が含まれている可能性がある。
私たちはまた、協力またはマーケティング、流通または許可手配、または第三者との特許権使用料に基づく融資によって資金を求めることを要求される可能性があり、私たちは、私たちの技術、将来の収入源または候補製品の貴重な権利を放棄しなければならないかもしれない、または私たちに不利になる可能性のある条項で許可を付与しなければならないかもしれない。もし私たちが必要な時にもっと多くの資金を集めることができなければ、私たちは私たちの製品発見と開発計画、商業化努力を延期、減少、または中止することを要求されるかもしれません。あるいは私たちはもともと自分で開発とマーケティングを望んでいた候補製品を授与する権利を与えます。また、いかなる追加的な拠出努力も、私たちの管理職の彼らの日常活動に対する関心を移す可能性があり、これは私たちの候補製品を開発し、商業化する能力に悪影響を及ぼすかもしれない。
私たちは為替変動に関連するリスクの影響を受けて、これらのリスクは私たちの経営業績に影響を与えるかもしれません.
2023年12月31日まで、私たちは約3%の現金と現金等価物と有価証券を元建てで価格を計算します。私たちのカナダでの業務はカナダドルで計算された巨額の費用を発生させた。私たちは現在私たちのカナダリング支出のために外貨ヘッジ手配を行っていません。そのため、外貨変動は私たちの収益に不利な影響を与える可能性があります。しかし、将来、為替レートのリッジ活動に従事して、為替レート変動の影響を軽減するために努力するかもしれません。私たちが施行したどんなヘッジ技術も効果がないかもしれない。もし私たちのヘッジ活動が有効でなければ、通貨レートの変化は私たちの普通株の市場価格にもっと大きな影響を与えるかもしれない。
30
私たちのビジネスや産業に関するリスクは
私たちと私たちの協力者は私たちの候補製品市場で激しい競争に直面しており、これは他の人たちが私たちの前に製品を発見、開発したり、商業化したり、あるいは私たちまたは私たちの協力者よりもこれに成功する可能性がある。
生物技術と製薬業界の特徴は技術進歩が迅速で、競争が激しく、そして独自製品を高度に重視していることである。著者らは薬物発見と製品開発において、主要な製薬、専門製薬と生物技術会社、学術機関、政府機関及び公共と民間研究機関を含む多くの異なる方法と源からの潜在的な競争に直面している。私たちまたは私たちのパートナーが開発と商業化に成功した任意の候補製品は、既存製品および将来発売される可能性のある任意の新製品と競争するだろう。
私たちのすべての候補製品が承認された場合、その成功に影響を与える重要な競争要素は、それらの治療効果、安全性および/または耐性、管理の利便性および簡易性、価格、代替製品の潜在的優位性、後発薬競争レベル、および政府および他の第三者支払者が保険および十分な補償を提供するかどうかである可能性がある。
私たちまたは私たちの協力者と比較して、私たちは競争しているか、あるいは将来競争する可能性のある多くの会社は、研究開発、製造、臨床前テスト、臨床試験を行い、監督管理許可とマーケティング承認を得た製品について、より多くの財務資源と専門知識を持っている。製薬とバイオテクノロジー産業の合併と買収は、私たちの数の少ない競争相手により多くの資源を集中させる可能性がある。規模が小さいか早い段階にある会社も重要な競争相手であることが証明される可能性があり、特に大手·成熟会社との協力により手配されている。
もし私たちの競争相手が私たちが開発する可能性のある任意の製品よりも安全で、より効果的で、副作用が少なく、より便利で、より安価な製品や療法を開発し、商業化すれば、私たちのビジネス機会は減少または消失するかもしれない。私たちの競争相手も私たちよりも早くFDA、EMA、または他の外国の規制機関からその製品の承認を得ることができ、これは私たちの競争相手が市場に入る前に強力な市場地位を確立することができるかもしれない。また、私たちの競争能力は、保険会社や他の第三者支払者による決定の影響を受ける可能性があります。
我々の1つ以上の特許または協力製品がてんかん治療のために承認された場合、他の抗てんかん薬やASMまたは互いに競合する可能性が予想される。さらに、我々の1つまたは複数の特許製品が重篤な抑うつ障害の治療のために承認された場合、他の抗うつ薬または米国預託株式と競合する可能性が予想される。
私たちは発売された特許製品もなく、第二段階の臨床試験後の臨床開発も完了しておらず、将来の候補製品の開発と任意の結果製品を独立して商業化する能力を評価することは困難である。
ある会社として、私たちはこれまで3期の臨床試験と関連する規制要求を完成した経験がなく、新薬申請或いはNDA或いは同等の提出、あるいは製品の商業化を含む。私たちは、第2段階以降に独立して繰り返し臨床開発を行う能力があることを証明しておらず、規制部門の承認を得て、薬物や医薬製品を登録·商業的に製造したり、第三者代表者を配置したりして、治療製品を商業化している。もし私たちのビジネス戦略を実行し、候補製品を開発し、独立して商業化するなら、私たちはこれらの能力を育成する必要があるだろう。私たちが開発した独立プロジェクトの業務計画を実行するためには、成功する必要があります
もし私たちがこれらの目標を成功的に達成できなければ、私たちは未来の候補製品を独立して開発し、商業化することができず、そうする潜在的な優位性を実現できないかもしれない。
31
私たちがより多くの候補製品を発見、開発、商業化することに成功しなければ、事業を拡大し、戦略目標を達成する能力が損なわれる可能性がある。
我々は、我々の内部研究から、または他の製品または技術を買収または許可することによって、候補製品を決定する製品開発チャネルを構築している。私たちの内部発見作業と潜在的買収が可能かもしれない機会の評価には、可能な候補製品が見つかったかどうかにかかわらず、大量の技術、財務、人的資源が必要だ。
我々の内部研究から、あるいは他の候補製品や技術を買収または許可することによって、臨床開発および商業化に適したより多くの候補製品を決定することができなければ、将来的に製品収入を得ることができない可能性があり、これは私たちの財務状況に大きな損害を与え、私たちの普通株の市場価格に悪影響を及ぼす可能性がある。
もし私たちの幹部とキーパーソンを引き付けることができなければ、私たちは私たちの候補製品の開発に成功できないかもしれません。私たちの協力協定で規定された義務を履行することができず、私たちの臨床試験を行うことができず、私たちの候補製品を商業化することもできません。
私たちの成功部分は私たちが引き続き高い素質の管理、臨床と科学者を吸引、維持と激励する能力にかかっている。近年、私たちの業界の管理職の流出率は高い。幹部や他の重要な従業員を交換することは困難かもしれませんし、私たちの業界では開発に成功し、監督部門の承認を得て製品を商業化するために必要な技能と経験を持っている個人の数が限られているので、長い時間がかかるかもしれません。
私たちは私たちの幹部に高い依存を持っています,イアン·モティマーさん,私たちの社長やCEOを含めて。もし私たちの一人以上の幹部がサービスを失ったら、私たちの候補製品の開発成功を大幅に遅延させるかもしれません。
また、臨床開発活動を拡大し、ビジネス能力を発展させる際には、独立した商業化努力を支援する販売インフラを含むより多くの人員を募集する必要があります。多くの製薬会社とバイオテクノロジー会社が似たようなスキルを持つ人を争っているため、受け入れ可能な条件で人材を引き付けることができないかもしれない。役員や重要な従業員を募集したり失うことができないサービスは、私たちの研究、開発、商業化目標の進展を阻害する可能性があります。
私たちの従業員、協力者、および他の人たちは、法律と法規の基準と要求を守らないことを含む不正行為や他の不正活動に従事する可能性があり、これは私たちに重大な責任を与え、私たちの名声を損なうかもしれない。
私たちは、従業員、協力者、サプライヤー、調査現場スタッフ、コンサルタント、ビジネスパートナー、その他の人員の詐欺または他の不正行為のリスクに直面しています。これらの当事者の不適切な行為は、故意、無謀、および/または不注意な行為を含むことができ、または以下の規定に違反する不正な活動を開示することができる
特に、医療業界の販売、マーケティング、商業配置は、詐欺、不正行為、リベート、自己取引、その他の乱用行為を防止するための広範な法律法規によって制約されている。しかも、私たちは適用される外国、連邦、州データプライバシーと安全法律の制約を受けている。より多くの情報については、“リスク要因-私たちは、プライバシー、データ保護、および情報セキュリティに関連して変化するグローバルな法律および法規に支配されており、これは、多くのコンプライアンスコストを発生させることを要求するかもしれませんが、私たちがこのような法律および法規を遵守できないか、または遵守できないと考えられていることは、私たちの業務および運営を損なう可能性があります”を参照してください
32
様々な法律法規は、広範な定価、割引、マーケティングおよび販売促進、販売手数料、顧客インセンティブ計画、およびその他の業務スケジュールを制限または禁止する可能性があります。いかなる不正行為も臨床試験過程で得られた情報の不適切な使用或いは虚偽陳述、或いは著者らの臨床前研究或いは臨床試験において虚偽データを作成することに関連する可能性があり、これは監督部門の制裁を招き、著者らの名声に深刻な損害を与える可能性がある。私たちは、私たちすべての従業員、高級管理者、役員、代理人、および代表(コンサルタントを含む)に適した行動基準を採択しましたが、常に不正行為を識別し、阻止することができるわけではありません。私たちが不正行為を発見し、防止するための予防措置は、未知または未管理のリスクや損失を効果的に制御できないか、または政府の調査または他の行動またはクレーム、要求または訴訟から私たちを保護することができない可能性があります。これらの訴訟は、実際にまたはこれらの法律および法規を遵守できないと言われていることによるものです。さらに、誰かや政府がこのような詐欺や他の不正行為を告発する可能性があり、起こらなくてもリスクに直面している。もし私たちにこのような訴訟を提起した場合、私たちは自分を弁護し、有利な和解を達成し、または他の方法で私たちの権利を維持することに成功できなかった場合、これらの行動は、民事、刑事と行政処罰、損害賠償、罰金、返還、誠実な監督と報告義務の適用、連邦医療保険、医療補助および他の連邦医療保健計画から除外される可能性があり、契約損害、名声損害、利益および将来の収益の減少、および私たちの業務の縮小を含むかもしれない。さらに、このような行動を防御するには費用がかかり、時間がかかる可能性があり、大量の財政と人的資源が必要となる可能性がある。したがって、私たちが私たちに提起される可能性のあるいかなる訴訟も防ぐことに成功しても、私たちの業務は損害を受ける可能性がある。
私たちはリストラと事業拡大の成功を含めて私たちの成長を管理することに困難に直面するかもしれない。
我々の業務戦略には,継続開発と,開発に成功した場合に選定された候補製品を商業化することがある.この戦略を実行するためには、規制、販売、製造、サプライチェーン、マーケティングインフラを構築し、私たちの開発能力を拡大したり、第三者と契約を結んだりして、これらの能力とインフラを提供してくれる必要があります。この目標を達成するためには、競争が激しさを増し、インフレが激化している市場で適切な条件で従業員を補償し、私たちの管理、運営、財務制度を引き続き実施し、改善する必要がある。私たちの業務の拡大に伴い、様々な戦略パートナー、サプライヤー、他の第三者とのより多くの関係を管理する必要があると予想されます。
今後の成長は、より多くの従業員を特定、採用、維持、インセンティブ、統合を含む管理職メンバーにより多くの責任を負わせるだろう。また、私たちの経営陣は、私たちの日常活動から不比例な注意を移し、これらの成長活動を大量に管理する必要があるかもしれない。私たちは私たちの業務の拡張を効果的に管理できないかもしれません。これは私たちの業務の疲弊を招き、操作ミス、ビジネス機会の喪失、従業員の流失、余剰従業員の生産性の低下を招く可能性があります。私たちの予想成長は大量の資本支出を必要とし、既存およびより多くの候補製品を開発するなど、他のプロジェクトから財政資源を移転する可能性がある。もし私たちが私たちの成長を効果的に管理できなければ、私たちの支出は予想よりも増加するかもしれないし、私たちの収入を創出して成長する能力は低下するかもしれないし、私たちは私たちの業務戦略を実施できないかもしれない。私たちの将来の財務業績と私たちが候補製品を商業化し、効果的に競争する能力は、私たちが未来のどんな成長を効果的に管理する能力にある程度依存するだろう。
私たちはプライバシー、データ保護、情報セキュリティに関連して変化する世界的な法律と法規に支配されており、これは大量のコンプライアンスコストを発生させることを要求するかもしれませんが、私たちがこのような法律や法規を遵守できないことは、私たちの業務と運営を損なう可能性があります。
通常の業務過程で、私たちは個人データおよび他の敏感な情報を処理し、私たちの独自および機密の商業データ、商業秘密、知的財産権、臨床試験に関連する試験参加者が収集したデータ、および他の敏感なデータを含む。私たちのデータ処理活動は、様々な法律、法規、ガイドライン、業界基準、外部および内部プライバシーおよびセキュリティポリシー、契約、および私たちを代表して個人データを処理する他の義務など、多くのデータプライバシーおよびセキュリティ義務を負担することを要求しています。
米国では、連邦、州、地方政府はデータ漏洩通知法、個人データプライバシー法、消費者保護法を含む多くのデータプライバシーとセキュリティ法律を制定している。例えば、2009年に“経済·臨床健康情報技術法案”(HITECH)によって改正された米国連邦1996年“健康保険携帯性と責任法案”(HIPAA)は、個人が健康情報を識別できるプライバシー、安全、および伝送に具体的な要求を提出し、これらの要求は、私たちが相互作用する多くのアメリカの医療保健提供者、例えば私たちのアメリカの臨床試験場所に適用される。州レベルでは、カリフォルニアプライバシー権法案の改正と補充を経た2018年カリフォルニア消費者プライバシー法、またはCCPAと呼ばれ、この法案を適用する企業に義務が課せられている。CCPAは規定を守らない行為に法定罰金を科すことを許可する。CCPAはいくつかの臨床試験で処理されたデータを免除しているが、私たちの業務と運営に適用される範囲内で、CCPAはコンプライアンスコストと潜在的な責任を増加させ、私たちが維持しているカリフォルニア住民に関する他の個人情報に関連する可能性がある。他の州でもデータプライバシー法が公布された。近年,連邦,州,地方の各レベルでより多くのデータプライバシーやセキュリティ法律が提案されており,コンプライアンス作業をさらに複雑化させる可能性がある.
33
米国以外では,EUの一般データ保護条例(EU GDPR)とイギリスのGDPR(イギリスGDPR)が個人データの処理に厳しい要求をしている。例えば、EU GDPRによると、政府規制機関は、データ処理に対して一時的または最終禁止を実施し、最大2000万ユーロまたは世界の年収4%に達する罰金を、金額が大きい者を基準とする可能性がある。しかも、個人は私たちがその個人データを処理することに関する訴訟を提起することができる。いくつかの外国司法管轄区は、データローカライズ法および国境を越えた個人データ転送法を公布しており、これは、EUまたはEUからの個人データの転送または受信など、司法管轄区域にわたる情報の転送をより困難にする可能性がある。他の管轄区域ではデータプライバシー法が公布され、改正され続けており、データプライバシー分野の複雑さを増加させている。
私たちは適用されるすべてのデータプライバシーとセキュリティ義務を遵守しようと努力しているにもかかわらず、これらの義務はますます厳格な方法で急速に変化しており、どのように遵守するかにいくつかの不確実性をもたらし、私たちの政策ややり方の修正を要求するかもしれません。これは費用が高く、経営陣や技術者の注意をそらす可能性があります。さらに、私たちは時々遵守できなかったり、遵守できなかったと思われたりして、重大な結果に直面する可能性がある。これらの結果は、政府の法執行行動、調査、および他の手続き、追加の報告要件および/または監視、個人データの処理の禁止、個人データの廃棄または使用の命令、および会社の役人の監禁を含むことができるが、これらに限定されない。このような事件は、私たちの臨床試験を含む、私たちの業務運営の中断または中断、私たちの臨床試験を含む、私たちの名声、業務、または財務状態に重大な悪影響を及ぼす可能性があります。個人データを処理できない、または特定の司法管轄区域で運営されていること、私たちの製品を開発すること、またはそれを商業化する能力が限られていること、任意のクレームまたは調査のために時間と資源をかけて弁護すること、否定的な宣伝、または私たちの業務の修正または再構成。
ネットワークセキュリティホール、システム障害、またはサードパーティ請負業者またはプロバイダによって所有される情報を含む当社のシステムおよび/または情報のような実際または感知された情報セキュリティイベントが発生した場合、私たちのトラフィックおよび運営は、他の被害を受ける可能性がある。
我々は、内部情報技術システムおよびネットワーク、ならびに第三者プロバイダおよび請負業者のシステムおよびネットワークに依存して、当社の業務活動に関する情報を取得、送信、記憶、処理する。私たちが業務を効率的に管理する能力は、私たちと私たちの第三者請負業者とサプライヤーの技術システムの安全性、信頼性、十分性に依存します。ネットワーク釣り、商業電子メール漏洩、社会工学、恐喝ソフトウェアまたは他のマルウェア、または私たちに代わって維持または他の方法で処理されたデータの任意のセキュリティホール、セキュリティイベントまたは他の破壊、紛失または許可されていない使用または他の処理を含む、当社のシステムおよび/またはデータの機密性、完全性または可用性に悪影響を及ぼすいかなるイベントであっても、知的財産権損失または商業機密が流用される可能性があり、私たちの業務および運営を中断し、増加したコストに直面し、問題を解決するために時間と資源をかけることを要求し、私たちは個人的なクレーム、要求および訴訟、規制調査、および他の訴訟手続きの影響を受ける可能性がある。罰金、処罰、その他の責任があり、私たちの業務に実質的な悪影響を及ぼす。さらに、私たちの候補製品が完成したか、または行われている臨床試験における前臨床データまたは臨床試験データの損失、変更、または他の破損または他の利用不可能は、私たちの開発と規制承認作業の遅延を招き、データの回復または複製のコストを著しく増加させる可能性がある。任意のネットワーク攻撃、セキュリティホールまたは事件、または私たちの名義で維持または他の方法で処理されたデータの他の破壊、失われたまたは許可されていない処理、または任意のこのような事件が発生したとの見方は、適用される米国および国際プライバシー、データ保護、情報セキュリティおよび他の法律法規に違反し、私たちの名声を損なう可能性があり、米国連邦、州および現地規制エンティティおよび国際規制エンティティの訴訟および政府調査および訴訟に直面させ、実質的な民事および/または刑事訴訟および責任に直面させる可能性がある。さらに、プライバシー、データ保護、および情報セキュリティに関連するさらなる措置を実施することによって、実際または予期されるセキュリティホールまたはイベントまたは他の状況に対応するために、多くの追加費用を招く可能性がある。
これまで、私たちは、ネットワーク攻撃や他の情報セキュリティイベントが私たちの業務、財務状態、または運営に実質的な影響を与えたことは経験していませんが、攻撃技術の頻繁な変化、およびそのような攻撃の数および複雑さの増加により、私たちの業務、財務状態、または運営は将来的に悪影響を受ける可能性があります。また,計算が分散している性質は,モバイルデバイスを一般的に使用して機密情報にアクセスすることや,遠隔データセンターにホストされるクラウドベースのアプリケーションの広範な使用を含み,セキュリティホールや事故のリスクを増加させる.私たちと私たちの第三者サプライヤーと請負業者の遠隔作業者の数が増加しているため、これらのリスクは増加する可能性があります。ネットワーク脅威の持続的な発展に伴い、私たちは、私たちの保護措置を修正したり強化したり、情報セキュリティホール、脅威、および事件を調査し、修復するために、多くの追加資源を必要とするかもしれません。我々は階層的セキュリティ対策を実施しているが,我々の計算機システムおよび我々の第三者契約製造業者(CMO),契約研究機関(CRO)とそのサプライヤーや請負業者が使用する外部システムやサービスは,これらのイベントの影響を受ける可能性があり,ネットワーク攻撃の防止やその影響の軽減に成功する保証はない.私たちの責任保険は、セキュリティホール、サイバー攻撃、および他の関連違反に対する私たちのクレームをカバーするのに十分ではないかもしれません。
34
国際業務に関連した様々なリスクは私たちの業務に実質的な損害を与える可能性があります。
私たちは、私たちが臨床試験を行っているアメリカ以外の国を含む、アメリカ以外で運営·運営を計画している各司法管轄区域の多くの法律法規を遵守するために、より多くの資源を投入しなければならない。私たちは重大な国境を越えて国際活動をしているので、私たちは国際業務に関連するリスクに直面します
もし私たちが国境を越えて国際活動に関連したこれらのリスクをうまく管理できなければ、私たちの業務は実質的な損害を受けるかもしれない。
もし私たちが受動的な外国投資会社と説明されれば、私たちの普通株のアメリカ保有者は不利な税金結果を受けるかもしれない。
一般に、任意の納税年度において、私たちの総収入の75%以上が受動的収入である場合、または平均資産率の少なくとも50%(適用される財務省法規に基づいて決定され、これは、私たちの普通株の時価に部分的に依存し、変化する可能性がある)が受動的収入を生成するために使用される場合、米国連邦所得税目的のための受動的外国投資会社、またはPFICとして説明される。私たちの総収入と総資産によると、2023年12月31日までの納税年度はPFICとみなされ、その後の納税年度はPFICとみなされる可能性がある。我々のPFICとしての地位は毎年事実に基づいて決定されており,本課税年度や将来納税年度のPFIC地位については何の保証も提供できない。
もし私たちがどの年にもPFICなら、私たちの普通株のアメリカ保有者は不利な税金結果を受けるかもしれない。非会社アメリカ株主が私たちの普通株を売却して実現した収益は資本利益課税ではなく普通収入として課税され、私たちの普通配当金に適用される優遇税率は失われます。利息費用はまた、すべての米国保有者が達成した収益と配当の税金に追加されるだろう。アメリカの所有者は彼らの特定の状況について彼ら自身の税務顧問に相談しなければならない。
35
アメリカの保有者は合格した選挙基金選挙をタイムリーに行うことで、このような不利な税金結果を避けることができる。PFICの毛収入または資産テストに達する年ごとに、当選した米国の保有者は、私たちの純一般収入と純資本利益(あれば)に比例して計上することを要求される。私たちがアメリカの所有者に私たちの純一般収入と純資本利益を比例的に提供することを約束した時だけ、アメリカの所有者は合格した選挙基金選挙を行うことができる。要求に応じて、私たちは私たちのアメリカ人所有者に必要な情報を提供して、彼らが合格した選挙基金選挙を行い、私たちがPFICの毎年の一般収益と純資本利益の割合だと思う割合を比例的に報告します。アメリカの保有者は今回の選択と関連する報告をして、彼ら自身の税務顧問に相談することを要求しなければならない。
米国の保有者は時価ベースの選挙をタイムリーに行うことで不利な税収結果を軽減することもできる。一般に,PFICの毛収入や資産テストに適合する毎年,当選した米国の保有者は,その納税年度の毛収入に普通株価値の増加を計上し,その毛収入からその納税年度におけるその株の価値の減少を差し引く。我々の普通株が適格な取引所(ナスダック世界市場またはナスダックを含む)で定期的に取引されている場合にのみ、時価ベースの選択を行い、維持することが可能である。我々の普通株が定期的に合格した取引所で取引を行うかどうかは,我々の制御範囲を部分的に超えている事実に基づいた年次決定である.したがって、PFICと記述されていれば、米国の保有者は、不利な税収結果を軽減するために、時価建ての選挙を行う資格がない可能性がある。アメリカの保有者はこの選挙が可能かどうかについて自分の税務顧問に相談しなければならない。
さらに、もし私たちがPFIC(または私たちのPFIC地位が不確定)になった場合、一部のアメリカ投資家が私たちの普通株を購入することを阻止する可能性があり、これは私たちの普通株の市場価格に悪影響を及ぼす可能性がある。
私たちの純営業損失の繰越と他の税務属性を使用する能力は限られているかもしれません。
私たちは重大なカナダ連邦純営業赤字の繰越があり、カナダ連邦投資税収の繰越免除と省級投資税収の繰越免除があり、それらは満期になる可能性があり、未使用で、未来の所得税負債を相殺することができない。カナダと米国連邦、省、州、地方所得税に関するルールは、立法過程に参加する人員およびカナダ税務局、国税局、米国財務省の審査を受け続けている。税法の変更または既存の法律解釈の変更(これらの変更はトレーサビリティを有する可能性がある)、純営業損失および税収控除に関する変更を含むことは、私たちまたは私たち普通株の保有者に悪影響を及ぼす可能性がある。近年、このような変化は多く発生しており、未来も変化し続けるかもしれない。将来の税法の変化は、私たちの業務、キャッシュフロー、財務状況、または運営結果に実質的な悪影響を及ぼす可能性があります。
私たちは私たちの組織や業務がある管轄区に所得税を支払うかもしれません。これは私たちの将来の収入を減らすことになります.
私たちはカナダやアメリカ以外の管轄区に所得税を納める可能性があり、もしこのような管轄区域の法律に基づいて、私たちはそこで貿易や業務を行っているとみなされたり、そこからと思われる収入を稼いだりして、免除を受ける資格がありません。私たちが納税する必要がないと思っている司法管轄区では、これらの管轄区の税務機関が1つ以上の納税年度を審査しないことを確認することはできません。税務審査は通常複雑であり、税務機関は私たちが報告した項目の処理に同意しない可能性があり、その結果、私たちの経営業績や財務状況に重大な悪影響を及ぼす可能性がある。
買収や他の戦略取引は、私たちの業務を混乱させ、私たちの株主の株式希釈を招き、他の方法で私たちの業務を損なう可能性があります。
我々は、他の業務、製品または技術の買収、戦略連合、ライセンス取引、または相補業務への投資を求めるなど、継続的に行われる様々な戦略取引を積極的に評価する。これらの取引のいずれも私たちの財務状況と経営業績に重大な影響を与える可能性があり、私たちを多くのリスクに直面させます
36
これらのリスクに加えて、外国買収は、異なる文化や言語を越えた業務統合に関するリスク、通貨リスク、および特定の国に関連する特定の経済、政治、規制リスクを含む独自のリスクにも関連する。
しかも、どんな戦略連合や買収の期待的な利益も達成できないかもしれない。将来の買収または処分は、私たちの株式証券の潜在的希釈発行、債務、または負債または償却費用または営業権の償却をもたらす可能性があり、これらはいずれも私たちの財務状況を損なう可能性がある。私たちは将来の買収の数量、時間、規模を予測することができず、どのような取引が私たちの経営業績に影響を与える可能性も予測できない。
私たちの現在と未来のアメリカおよび他の地域での業務は、連邦および州の反リベート、詐欺と乱用、虚偽声明、透明性、医療情報プライバシーと安全、および他の医療保健法律および法規の制約を直接または間接的に適用され、これは、私たちを刑事制裁、民事処罰、契約損害、名声損害、行政負担、利益および将来の収入の減少に直面させる可能性がある。
アメリカや他の地域の医療提供者や第三者支払者は、マーケティングの承認を得た任意の候補製品を推薦し、処方する上で主な役割を果たしています。ヘルスケア提供者、第三者支払者、患者、医療業界内の他の当事者との合意は、マーケティング、販売、流通がマーケティングの承認を得た任意の製品の業務または財務配置および関係を制限する可能性がある広範に適用される詐欺および乱用、および他の医療法律法規に直面する可能性があります。適用される医療およびデータプライバシー法律には、以下の制限が含まれており、いくつかの制限は、上場製品を持っている場合にのみ適用されます
37
私たちの活動が適用された医療法律と法規に適合することを確実にする努力は巨額のコストに関連するだろう。法律法規の広範性、ある法律法規に対する指導が限られていること、および政府の法律法規が絶えず変化している解釈を考慮すると、政府当局は結論を出す可能性があり、私たちの商業実践はこれらの法律に適合していないかもしれない。もし私たちの運営がこれらの法律または任意の他の私たちに適用される可能性のあるすべての政府法規に違反していることが発見された場合、私たちは重大な民事、刑事、行政処罰、損害賠償、罰金、連邦医療保険や医療補助のような政府援助の医療計画から除外され、私たちの業務の削減や再編を受ける可能性がある。さらに、このような行動を防御するには費用がかかり、時間がかかる可能性があり、多くの人的資源が必要となる可能性がある。したがって、私たちが私たちに提起される可能性のあるいかなる訴訟も防ぐことに成功しても、私たちの業務は損害を受ける可能性がある。
もし私たちが環境、健康、安全の法律法規を守らなければ、私たちは罰金や罰金を科されたり、コストを発生したりする可能性があり、これは私たちの業務の成功に実質的な悪影響を及ぼすかもしれない。
我々の研究·開発活動は,潜在的有害生物材料および危険材料,化学品および通常分子や細胞生物学に用いられる様々な放射性化合物の制御使用に関する。例えば、培養に細胞を使用し、少量の放射性同位体を使用することが多い。これらの材料の使用、保存、処理、または処置による予期しない汚染または傷害のリスクを、最新の許可および訓練計画を維持することで完全に除去することはできません。汚染や傷害が発生した場合、私たちはそれによる損害に責任を負う可能性があり、どんな責任も私たちの資源範囲を超える可能性がある。私たちは現在このような材料の使用によって引き起こされたいくつかのクレームのために保険をかけている。しかし、もし私たちが合理的な費用と十分な保険範囲で私たちの保険範囲を維持できなければ、私たちの保険は出現する可能性のあるいかなる責任もカバーしないかもしれない。私たちはカナダ連邦、省と地方の法律法規の制約を受け、アメリカおよび/または外国のこれらの材料と指定廃棄物製品の使用、貯蔵、運搬と処分に関する法律法規の制約を受ける可能性がある。これらの材料の使用に関する規定を遵守することは費用がかかる可能性があり、私たちがこれらの規定を遵守しなければ、私たちの運営と収益性に実質的な悪影響を及ぼす可能性がある。
私たちまたは私たちが依存している第三者は、地震や他の自然災害の悪影響を受ける可能性があり、私たちの業務の連続性や災害復旧計画は、深刻な災害から私たちを十分に保護できないかもしれません。
私たちの本社はカナダブリティッシュコロンビア州のバーナビーにあります。私たちは地震などの自然災害の影響を受けやすく、これらの自然災害は私たちの行動を乱すかもしれない。自然災害、停電、火災、または他の事件が発生した場合、本社の全部または大部分を使用することができなくなり、私たちのCMOの製造施設のような重要なインフラを破損したり、他の方法で運営を中断したりすることは難しいかもしれません。場合によっては、長い間私たちの業務を継続することはできません。地震や他の自然災害の保険を購入しましたが、発生する可能性のあるすべての損失を補償するのに十分な業務中断保険を購入しないかもしれません。深刻な災害や同様の事件が発生した場合、我々の既存の災害復旧および業務連続計画は十分ではない可能性がある。私たちは自然災害や地震によって多くの費用が発生する可能性があり、これは私たちの業務に実質的な悪影響を及ぼすかもしれない。しかも、私たちはサンプルや他の価値のあるデータを失うかもしれない。上記のいずれの状況の発生も私たちの業務に重大な悪影響を及ぼす可能性がある。
私たちの候補製品の発見、開発、商業化に関するリスク
私たちの業務はXEN 1101の成功開発に大きく依存しています。もし私たちが規制部門のXEN 1101の承認を得られず、それを商業化することに成功すれば、私たちの業務は実質的な損害を受ける可能性があります.
私たちはまだ商業販売を許可されていない製品で、大量の精力と財力を投入して、著者らの臨床段階の候補製品XEN 1101を開発して、てんかん、MDDと潜在的な他の神経疾患の治療に応用している。私たちの将来の事業成功はXEN 1101の持続的な開発と最終規制承認にかかっている。著者らはXEN 1101てんかん3期臨床試験と任意の他の未来の3期臨床試験を成功的に登録と完成する必要がある。XEN 1101の将来の規制と商業成功はいくつかのリスクの影響を受ける
38
その中の多くのリスクは私たちがコントロールできないものであり、臨床開発に関連するリスク、監督提出過程、私たちの知的財産権に対する潜在的な脅威、及び任意の未来の協力者の製造、マーケティングと販売努力を含む。もし私たちまたは任意のパートナーが私たちの初期または潜在的な他の適応のために開発し、規制部門の承認を得たり、XEN 1101の商業化に成功したりすることができない場合、または私たちがこれらのいかなるリスクや他の理由で遅延に遭遇した場合、私たちの業務は実質的な損害を受ける可能性がある。
また,製薬業で開発されている大量の薬物のうち,一部の薬物のみがFDAに秘密保持協定を提出しており,より少ない薬物が商業的に承認されている。さらに、XEN 1101の任意の適応の規制によって承認されたとしても、そのような承認は、XEN 1101を販売する我々の適応または用途または患者集団によって制限される可能性がある。したがって、私たちが開発計画に資金を提供し続けるために必要な資金を得ることができても、XEN 1101の開発に成功したり、商業化したりすることを保証することはできません。
私たちの薬物発見方法は確認されておらず、商業的価値のある製品を開発できるかどうかもわかりません。
我々の薬物発見方法は、再現性或いは費用効果の方法で候補製品の発見と人類疾患を安全かつ有効に治療する商業的に実行可能な製品の開発を招くことができない可能性がある。
著者らの薬物発見仕事は最初に他の候補製品を決定する上で希望を示す可能性があるが、臨床開発或いは商業化に利用可能な実行可能な製品候補製品を生成することができない。このような故障が発生する原因は多くあり、任意の候補製品がさらなる研究後に深刻なまたは予期しない副作用または他の特徴を有することが証明される可能性があり、それが安全である可能性が高いか、または適用可能な規制基準に適合していないこと、および/または許容可能なコストで商業的に量産できないこと、または根本的に不可能であることを示す。
我々の発見活動が薬物発見の新たな標的を決定できなかった場合,あるいはこれらの標的がヒト疾患の治療に適していないことが証明された場合,あるいはこれらの標的に対する特異性と選択性を有する候補製品を開発できなければ,可能な製品を開発することはできない。もし私たちが実行可能な製品を開発して商業化しなければ、私たちは商業的に成功しないだろう。
臨床前研究及び/或いは早期臨床試験の結果は後期臨床試験の結果を予測できない可能性があり、著者らの臨床試験結果は監督管理の要求に符合しない可能性があり、著者らは監督管理の承認を得る上で遅延或いは思わぬ困難に遭遇する可能性がある。
臨床前研究の結果は,われわれ,われわれのCROによるものであっても,我々がそれから許可を得たり,候補製品を獲得したりした他の第三者によるものであっても,臨床試験の結果を予測できない可能性がある。そのほか、各種の原因により、臨床前結果はよく異なる研究で比較することが困難であり、実験方案と技術、人員、設備とその他の要素の差異を含み、これは臨床前結果の信頼性と臨床試験結果の予測性を低下させる可能性がある。また,第三者が発表した臨床データや症例報告やわれわれ候補製品の早期臨床試験データは後期臨床試験の結果を予測できない可能性がある。早期研究結果の解釈には慎重が必要であり、これらの研究は通常規模が比較的に小さく、ある患者の反応は臨床意義があることを表明した。より多くの患者を募集した臨床試験後期結果は,期待される安全性や有効性結果を示すことができないか,あるいは同一候補製品の早期試験結果と一致しない可能性がある。その後の臨床試験結果は早期の臨床試験を複製できない可能性があり、原因は多種あり、試験設計の差異、異なる試験終点(或いは探索性研究中に試験終点が不足)、患者群、患者数、患者選択標準、試験持続時間、薬物投与量と調合及び早期研究中に統計能力が不足している。研究中の疾病或いは障害が既定の臨床終点が不足し、有効な治療効果測定基準が乏しい場合、これらの不確定性は増加し、以前はいかなる薬物開発に対する疾患もなかった場合のように、候補製品が新しい機序を対象としている。
39
また、私たちの候補製品は承認されないかもしれません。たとえそれらが私たちの第三段階の臨床試験で主要な終点に達していても。FDA、EMAあるいは外国の監督管理機関は私たちの試験設計と臨床前研究と臨床試験データの解釈に同意しないかもしれない、あるいは追加のデータが必要である。さらに、これらの規制機関のいずれも、重要な臨床試験案に関するコメントまたは提案を審査して提供した後であっても、候補製品に対する承認要求を変更することができ、成功すれば、FDA、EMA、または他の外国規制機関が承認を申請するための基礎となる可能性がある。例えば、FDAは、私たちが計画したセキュリティ協定の実質的な審査を拒否するか、または私たちのデータを審査した後に結論を出す可能性があり、私たちの申請は規制部門の承認を得るのに十分ではないと考えることができる。FDAが私たちが計画したNDAを承認しなければ、追加の臨床、非臨床、または生産研究を要求し、その後、私たちの申請を再検討するかもしれない。FDAまたは他の規制機関が要求するこれらまたは任意の他の研究の範囲によれば、NDAまたは同等の出願の承認は著しく遅れる可能性があり、または利用可能なリソースよりも多くのリソースを必要とする可能性がある。さらに、適用される規制機関は、私たちの候補製品を承認することも可能であり、その適応や人数は、私たちが要求しているものよりも狭いかもしれないし、高価な上場後の約束の履行状況に応じて承認される可能性がある。
著者らは時々発表或いは公表した臨床試験の一時、初期、“主要”と初歩的なデータはより多くの患者データの獲得に従って変化する可能性があり、そして監査と検証プログラムの制限を受け、これは最終データの重大な変化を招く可能性がある。
著者らは時々著者らの臨床前研究と臨床試験の初歩的或いは主要なデータを公開開示する可能性があり、これらのデータは当時使用可能なデータの初歩的な分析に基づいており、結果と関連する発見と結論は特定の臨床前研究或いは臨床試験の関連データに対してより全面的な審査を行った後に変化する可能性がある。私たちはまた、私たちのデータ分析の一部として、すべてのデータを全面的かつ詳細に評価する機会がないか、または受け取る機会がないかもしれないという仮説、推定、計算、および結論を出した。したがって、他のデータが受信され、十分に評価されると、私たちの報告の主要または予備結果は、同じ研究または実験の将来の結果とは異なる可能性があり、または異なる結論または考慮要因が、これらの結果を合格させる可能性がある。最も重要なデータもまだ監査と確認手続きを受けなければならないが、これは最終データが以前に公表された予備データと大きく異なる可能性がある。したがって、最終データが利用可能になる前に、トップラインデータは慎重に表示されなければならない。
さらに、規制機関を含む他の人は、私たちの仮定、推定、計算、結論または分析を受け入れないか、またはデータの重要性を異なる方法で解釈またはトレードオフする可能性があり、これは、特定の計画の価値、特定の候補製品または製品の承認または商業化に影響を与え、私たちの業務の成功に実質的な悪影響を及ぼす可能性がある。さらに、開示された特定の研究または臨床試験に関する情報を選択することは、通常、広範な情報に基づいており、あなたまたは他の人は、私たちが決定した重要な情報または他の適切な情報が私たちの開示に含まれることに同意しない可能性がある。もし私たちが報告した中期、主または予備データが実際の結果と異なる場合、または規制機関を含む他の人が結論に同意しない場合、私たちが承認を得て私たちの候補製品を商業化する能力が損なわれる可能性があり、これは私たちの業務、運営結果、将来性、または財務状況を損なう可能性がある。さらに、私たちまたは私たちの競争相手が中期、営業、または予備データを開示することは、私たちの普通株式価格の変動を招くかもしれない。
臨床試験は、私たちまたは私たちの協力者の候補製品の臨床開発のどの段階での安全性と有効性を十分に証明できないかもしれない。私たちまたは私たちのパートナーの任意の候補製品の開発を終了することは、私たちの業務と私たちの普通株の市場価格に実質的な損害を与えるかもしれません。
XEN 1101とNBI-921352(私たちの協力者Neurocrine Biosciencesによって開発された)を含む私たちの協力者の候補臨床製品と、私たちの臨床前化合物を含む臨床開発の候補製品に入る予定であり、異なる開発段階にあり、商業化前に大量の臨床開発、テスト、および規制承認が必要になる。
私たちの候補製品の商業化販売が規制される前に、私たちまたは私たちのパートナーは、各目標適応における各候補製品の使用が安全かつ有効であることを、長く、複雑で高価な臨床前試験および臨床試験によって証明しなければならない。臨床試験では,いつでも失敗する可能性がある。臨床試験は目標適応研究に対する候補製品の安全性と有効性を証明できないことが多い。臨床試験を開始した候補製品の多くは製品として承認されなかった。生物製薬業界のいくつかの会社は高級臨床試験で重大な挫折を受け、早期の試験で良好な結果を得たが、治療効果或いは副作用が乏しいためである。任意の候補製品の安全性と有効性試験以外に、臨床試験失敗は多種の要素によるものである可能性があり、試験設計、用量選択、統計分析計画、プラセボ効果、患者登録標準、患者コンプライアンスと試験実行中の欠陥を含む。試験と研究から得られたデータは異なる解読の影響を受けやすく、規制機関は私たちのように私たちのデータを有利に解読しない可能性があり、これは規制部門の承認を延期、制限、あるいは阻止する可能性がある。これらの原因による臨床試験の失敗は私たちの業務と私たちの普通株の市場価格に実質的な損害を与える可能性があります。
40
私たちと私たちの協力者のいくつかの候補製品について、私たちと私たちの協力者は、いくつかの疾患や障害に対する治療方法の開発を求めており、これらの疾患または障害の臨床経験は比較的限られており、臨床試験は新しい終点と測定方法または主観的な患者フィードバックを使用する可能性があり、これはこれらの臨床試験の複雑さを増加させ、監督部門の承認を遅らせる可能性がある。私たちまたは私たちの協力者の臨床試験の陰性または非決定性結果は、追加の臨床前試験または臨床試験を決定または要求すること、または候補製品の継続開発の終了を決定することをもたらす可能性がある。例えば,2021年10月に成人局所てんかん患者におけるXEN 1101の2 b期X−Tole臨床試験のTOPLINEデータを発表した。また,2023年11月にXEN 1101のMDD患者における2期X−Nova臨床試験のTOPLINEデータを発表した。著者らが行っているXEN 1101てんかん3期臨床試験或いは任意の他の未来の3期臨床試験は十分な治療効果と安全性結果を証明することは保証できず、著者らがXEN 1101の監督管理の許可を得ることができることを保証することはできない。上記のいずれの結果も、私たちの業務、候補製品、および将来の見通しに重大な悪影響を及ぼすだろう。
もし私たちまたは私たちのパートナーの候補製品が臨床試験で安全かつ有効であることが証明されなければ、これらの候補製品は規制部門の承認または商業化に成功することができないだろう。また,われわれまたはわれわれの協力者が臨床試験においてわれわれまたはわれわれの協力者が開発している候補臨床製品のいずれの適応においても積極的な結果を示すことができなければ,他の適応の開発に悪影響を及ぼす可能性がある。この場合、私たちは他の化合物を開発し、関連する臨床前テストと臨床試験を行う必要があり、追加の融資を求める可能性があり、これらはすべて私たちの業務、成長の見通し、経営業績、財務状況、経営業績に重大な悪影響を与える。
私たちまたは私たちの協力者は、患者を私たちの臨床試験に参加することが困難であることを発見することができ、これは私たちの候補製品の臨床試験の成功を延期または阻止することができるかもしれない。
私たちまたは私たちの協力者は、十分な数の患者を識別、募集、募集できないかもしれない、あるいは研究において多様性を実現するために必要または希望する特徴を有する患者は、直ちに臨床試験を完了できないか、または全く達成できない。患者の臨床試験への参加に影響する要素は以下のとおりである
私たちと私たちの協力者の臨床試験は、私たちの試験に参加することを選択する可能性のある患者のいくつかが競争相手による試験に参加することを選択する可能性があるので、私たちの候補製品と同じ治療分野の製品を他の臨床試験と争います。合格臨床研究者の数が限られているため、著者らはいくつかの競争相手が使用した同じ臨床試験地点で著者らのいくつかの臨床試験を行うことが予想され、これは著者らがこれらの臨床試験地点で臨床試験を行うことができる患者数を減少させる。
私たちと私たちの協力者は私たちの臨床試験のために十分な数の患者を募集することができません。これは重大な遅延を招き、あるいは1つ以上の臨床試験を完全に放棄する必要があるかもしれません。患者募集の遅延はコスト増加を招く可能性があり、計画された臨床試験の時間や結果に影響を与え、候補製品の開発·承認過程に影響を与え、製品販売の開始と収入創出に必要な規制承認を求める能力を危険にさらす可能性があり、いずれも当社の価値低下を招き、必要に応じて追加融資を受ける能力を制限する可能性がある。
41
われわれの成功はまた,われわれの臨床試験地点を管理する者の集団表現,貢献,専門知識に依存している。バイオ製薬や関連サービス業界では,合格人材に対する競争が非常に激しく,特に高等教育の学位を持つ人たちである。生物製薬サービス業界が直面している人員の流動と労働力不足の増加は、著者らが臨床試験を実行することに依存する第三者に負の影響を与える可能性がある。十分な人員支援のある試験地点の選択を求めているが,その業界の人員流動やより広範な労働市場動態が我々の試験地点に悪影響を与えないことは確認できない。もし私たちのサイトがこれらの要素の負の影響を受けたら、私たちは適時に臨床試験を登録する能力が阻害される可能性があり、私たちの業務、開発スケジュールと財務状況に負の影響を与える可能性がある。
私たちまたは私たちの協力者は、私たちまたは私たちの協力者の候補製品の開発および商業化を完成または最終的に完了できない場合、意外なコストや遅延が生じる可能性がある。
私たちの任意の候補製品を商業化するために必要な規制承認を得るためには、私たちまたは私たちの協力者は、広範な臨床前研究と臨床試験を通じて、私たちまたは私たちの協力者の候補製品が人体に対して安全かつ有効であることを証明しなければならない。私たちまたは私たちの協力者は、私たちまたは私たちの協力者の臨床試験または臨床試験を完了する前の研究、および他の臨床試験または臨床前研究の開始または完了において遅延に遭遇する可能性があり、これは、規制機関がIND下での臨床試験の実施を許可していないか、または私たちが臨床試験を開始するために必要な任意の臨床試験支出または同様の承認を承認しないか、または延期することを含む。私たちまたは私たちの協力者はまた、臨床試験中に多くの予見できない事件に遭遇する可能性があり、これらのイベントは、私たちまたは私たちの協力者が候補製品の開発を完了するのを遅延または阻止するか、または市場の承認を得るか、または私たちまたは私たちの協力者が開発した候補製品を商業化するかもしれない
42
これらのリスクおよび不確実性は、私たちまたはパートナーの任意の臨床計画に影響を与える可能性があり、上記の任意の臨床、規制または運営イベントは、私たちまたは私たちのパートナー計画の臨床および規制活動を変更する可能性がある。著者らの臨床試験における患者の募集と保留の挑戦は、著者らのXEN 1101てんかん3期臨床試験において、流行病、地政学的事件、あるいは任意の他の原因によっても、試験をさらに延期するか、あるいは試験中断を招く可能性がある。
任意の3期または他の重要な臨床試験の結果は、上場承認を支持するのに十分ではないかもしれない。これらの臨床試験は非常に時間がかかり、通常数百から数千人の患者に関連する。患者を識別する挑戦により,臨床試験も長い可能性がある。患者が認識に成功しても,てんかん臨床試験のベースラインてんかん負担を含めてスクリーニング基準を通過できない可能性があり,試験に参加できない。私たちの試験に参加する患者を識別、スクリーニング、および/または募集することに関連するいかなる挑戦も、私たちの臨床試験の完了に要する時間を延長するか、または目標登録者数を達成し、私たちの臨床試験を完了するためにより多くのサイトを開始する必要があり、これは、私たちの操作コストを増加させ、および/または私たちの規制承認時間を遅らせる可能性がある。さらに、FDA、EMA、または他の外国規制機関がキーテスト基準または主要終点の選択に私たちまたは私たちの協力者を同意しない場合、または主要終点の結果が実験治療を受けていない対照群患者と比較して信頼できないか、または私たちの統計分析に定説がない場合、規制機関は、その管轄地域で私たちの候補製品を承認することを拒否する可能性がある。FDA、EMA、または他の外国規制機関も、これらの任意の候補製品を承認する条件として、追加の臨床試験を要求する可能性がある。
もし臨床試験が私たち、私たちの協力者、試験を行っている機関のIRBs、この試験のための任意のデータ安全監視委員会、またはFDA、EMA、または他の外国の規制機関によって一時停止または終了された場合、私たちまたは私たちの協力者も遅延に遭遇する可能性がある。このような主管部門は一連の要素のために臨床試験を一時停止或いは中止する可能性があり、これらの要素は監督管理要求或いは著者らの臨床規程に従って臨床試験を行うことができなかったこと、FDA、EMA或いは他の外国の監督機関による臨床試験操作或いは試験場所の検査による臨床一時停止、候補製品の製造問題、予見できない安全問題或いは不良副作用、ある種の薬物の使用が治療効果があることを証明できなかったこと、政府法規或いは行政措置の変化或いは十分な資金が不足して臨床試験を継続することを含む。また,別の会社の候補製品は我々の化合物種別と同じであるため,試験や他の臨床データによるセキュリティ問題により遅延が生じる可能性がある。
また,適用される法規要件やガイドラインが変化する可能性があり,これらの変化や他の目標を反映するために臨床試験案を修正する必要がある可能性があり,これらの目標は我々の全体的な発展戦略に重要な科学的情報を生成する可能性がある。方案の修正過程は常にいくつかの審査機関の審査と承認が必要であり、監督機構と科学、監督と倫理委員会及びIRBsを含み、これは臨床試験の適時な完成に影響する可能性がある。さらに、これらの案修正案は、審査機関によって提出されないか、または全く受け入れられない可能性があり、臨床試験のコスト、時間、または成功達成に影響を及ぼす可能性がある。
私たちまたは私たちのパートナーが候補製品の任意の臨床試験の完了または終了を遅延または終了すれば、候補製品のビジネス見通しが損なわれる可能性があり、特許保護された製品の商業化の独占的権利を有する可能性のある期間が短縮される可能性があり、私たちまたはパートナーが製品販売を開始し、製品から製品収入を創出する能力が延期されるかもしれない。そのほか、臨床試験を完成するいかなる遅延も著者らのコストを増加し、著者らの候補製品の開発と承認過程を緩和し、そして最終的に臨床試験と候補製品の開発を中止する可能性がある。このようなどんな状況でも、私たちの業務、財政状況、そして見通しに大きな被害を及ぼす可能性がある。さらに、臨床試験の開始または完了遅延をもたらす多くの要因は、最終的には、私たちまたは私たちの協力者の候補製品が規制部門の承認を得られない可能性もある。
規制の承認が得られた場合、私たちの候補製品は、不良副作用や、その規制承認を遅延または阻止し、承認されたラベルの商業イメージを制限したり、重大な負の結果をもたらす可能性がある特性をもたらす可能性がある。
私たちの任意の候補製品による副作用は、私たちまたは規制機関の臨床試験の中断、延期、または停止を招き、より厳しいラベルをもたらすか、またはFDA、EMAまたは同様の外国の規制機関の規制承認を延期または拒否する可能性がある。
われわれの臨床試験結果は副作用や予期せぬ特徴の重症度と流行度を示す可能性がある。例えば,われわれのX−ToleおよびX−Nova臨床試験における副作用は通常軽微または中等度であるが,進行中の3期てんかんや他の臨床試験や他の将来の臨床試験でXEN 1101類似の耐性が認められる保証はない。多くの最初に臨床あるいは早期試験で所望の化合物を示した後、不良或いは予期しない副作用を引き起こすことが発見され、それによってこの化合物のさらなる発展を阻止した。
43
もし私たちの候補製品が開発中に受け入れられない副作用が発生した場合、私たち、FDA、EMAまたは同様の外国の規制機関、IRBs、または私たちの試験を行う機関の独立倫理委員会は、私たちの臨床試験を一時停止、制限または終了することができ、または独立した安全監視委員会は、私たちの試験を一時停止、制限または終了することを提案することができ、またはFDA、EMAまたは同様の外国規制機関は、臨床試験を停止するか、または私たちの製品候補製品の任意またはすべての目標適応を承認することを拒否するように命令することができる。薬物関連の治療に出現すると考えられる副作用は,臨床試験被験者の募集を延期したり,われわれの臨床試験に参加した被験者の臨床試験への参加を中止したりする可能性がある。さらに、治療医療従事者は、これらの副作用を適切に認識または処置していない可能性がある。私たちは、私たちの臨床試験および任意の候補製品が商業化された時の副作用を理解するために、私たちの候補製品を使用する医療従事者を訓練する必要があるかもしれない。私たちの候補製品の潜在的な副作用を認識したり管理したりする上で訓練が不足し、私たちの候補製品を服用している患者にダメージを与える可能性があります。このような状況のいずれも私たちの業務、財務状況、および見通しに悪影響を及ぼす可能性がある。
また,われわれの候補製品の臨床試験は,丁寧に定義された臨床試験に同意した患者群で行った。したがって,われわれの臨床試験では,候補製品の有意なプラス効果が実際のプラス効果(あれば)よりも大きいか,あるいは不良副作用を認識できない可能性がある。
候補製品の製造または処方を変更する方法は、追加のコストや遅延を招く可能性がある。
候補製品の開発が前臨床試験から後期臨床試験まで承認と商業化に向かうことに伴い、開発計画の各方面、例えば製造方法と調合は、開発過程中に常に変化し、製品、技術と結果の最適化に努力し、特許保護を拡大し、或いは異なる人々に対して。例えば、我々は、Neurocrine Biosciencesのライセンスに含まれるNBI-921352のための小児科製剤を開発した。これらのいずれの変化も、小児において同じ薬物曝露状態を提供せず、および/または成人において観察される同じ処方または他の処方とは異なる副作用をもたらす可能性がある候補製品の表現を異なるものとする可能性がある。新配合性能の意外な変化は,計画中の臨床試験や変更後の材料を用いた他の将来の臨床試験の結果に影響する可能性がある。これは臨床試験の完了を遅らせる可能性があり、追加の移行臨床試験を行うか、または1つ以上の臨床試験を繰り返し、臨床試験コストを増加させ、および/または私たちの候補製品の承認を延期または危険にさらし、および/または私たちまたは私たちの協力者が製品販売を開始し、収入を創出する能力を脅かす必要がある。
FDA、EMAと他の外国司法管轄区域の監督管理機関の監督管理審査過程は長く、時間がかかり、しかも本質的に予測できない。もし私たちまたは私たちの協力者が規制部門の私たちの候補製品に対する承認をタイムリーに得ることができなければ、あるいは根本的にできなければ、私たちの業務は実質的な損害を受けるかもしれない。
規制審査過程はコストが高く、FDA、EMA或いは他の司法管轄区の他の外国監督管理機関が任意の製品の販売を許可するのに要する時間は不確定であり、数年かかるかもしれない。規制承認を受けるかどうかは予測不可能であり、規制当局のかなりの自由裁量を含む多くの要因に依存する。承認政策、法規或いは承認を得るために必要な臨床前と臨床前データのタイプと数量は候補製品の臨床開発過程で変化する可能性があり、そして司法管轄区域によって異なる可能性がある。そのほか、臨床前と臨床データはよく異なる解釈と分析の影響を受けやすく、臨床前研究が有望な結果と臨床試験の成功を示しても、著者らはFDA、EMA或いは他の司法管轄区の他の外国監督管理機関が私たちのように結果を解釈することを保証できず、著者らが候補製品を提出して承認に供する前に、もっと多くの試験、製造に関連する研究或いは非臨床研究が必要であるかもしれない。多くの会社は彼らの候補製品が臨床前研究と臨床試験で満足できると思っているが、依然としてその製品のマーケティング許可を得られなかった。もし私たちの研究および試験結果がFDA、EMA、または他の司法管轄区域の他の外国規制機関を満足させてマーケティング申請を支援することができない場合、私たちの候補製品の承認は大幅に延期されるかもしれません。あるいは、私たちの候補製品の潜在的な承認を支援するために追加的な試験を行うために多くの追加資源が必要かもしれません。私たちの既存の候補製品や未来の任意の候補製品も、私たちがそのような承認を求めるのに時間と資源を費やしても、規制部門の承認を永遠に得られない可能性がある。
私たちの候補製品は多くの理由で規制部門の承認を得ることができないかもしれません
44
私たちまたは私たちの協力者が特定の製品の承認を得たとしても、規制機関は、臨床試験を含む高価な承認後に約束された表現に基づいて製品を承認することができ、またはそのラベルが製品の商業化に必要または必要なラベル宣言を含まないことを承認するかもしれない。
また、FDA、EMA、あるいは他の外国監督機関の臨床試験に関する政策が変わる可能性があり、追加の政府法規が公布される可能性がある。例えば,EUの臨床試験に関する規制構造が最近変化している。EU臨床試験条例、あるいはCTRは、2014年4月に採択され、EU臨床試験指令を廃止し、2022年1月31日に発効した。CTRの実施はまた臨床試験情報システム(CTIS)を実施することを含み、これは新しい臨床試験門戸とデータベースであり、欧州薬品管理局(EMA)が欧州委員会とEU加盟国と協力して維持する。CTRの目標は、EU全体で試験を行う一致ルール、一致したデータ基準および有害事象リスト、および許可状態に関する一致情報を含む。EUで行われた各臨床試験の進行と結果の情報が公開される。CTRはEU加盟国が国家レベルで臨床試験を管理するいくつかの側面を許可する。臨床試験を計画しているEU加盟国がCTISの採用や国家レベルでの他の規制改革の実施が遅い場合、あるいはCTISシステムおよび/またはプロセスが技術的な問題に遭遇した場合、私たちの臨床試験はEU加盟国で遅延する可能性があり、私たちのコストが増加する可能性がある。イギリスの臨床試験に適用される主要な立法はイギリス人用薬物(臨床試験)法規2004であり、それはEU臨床試験指令を国内法律に転換する。イギリスではすでに単一申請が同時に薬品と保健品監督機関と研究倫理委員会によって審査されることを可能にする総合研究応用システムが実施されている。イギリスでの臨床試験に関する要求や義務はEUの立場と大きく一致している。異なる司法管轄区域の法規要求の変化を遵守することは追加のコストを招く可能性があり、著者らの臨床開発計画を延期し、或いは私たちに更に大きな責任を負わせる可能性があり、もし私たちが既存の要求の変化に適応できない或いは新しい要求或いは臨床試験を管理する政策を採用すれば、著者らの開発計画、著者らのXEN 1101 3期てんかん臨床試験を含むことは影響を受ける可能性がある。
また,特定の疾患や障害の承認を求める治療法がある可能性があるため,規制部門の承認を得るためには,我々が開発したこれらの疾患や障害を治療するための候補製品が安全かつ有効であるだけでなく,既存製品と比較する必要がある可能性があることを臨床試験で証明する必要がある可能性があり,規制部門の承認や十分な補償を得ることが困難になる可能性がある。
45
私たちが一つの管轄区から私たちの候補製品の承認を得て維持しても、私たちは他の管轄区域で私たちの候補製品の承認を決して得られないかもしれません。これは私たちの市場機会を制限し、私たちの業務に悪影響を及ぼすでしょう。
私たちが承認した製品の販売(ある場合)は、私たちが規制承認を受けた国/地域マーケティング承認の規制要件によって制約され、私たちの候補製品を北米、EU、他の国/地域で商業化するために、私たち自身またはパートナーと規制承認を求める予定です。一国で行われる臨床試験は他の国の規制機関に受け入れられない可能性があり、一国の規制承認は他のどの国でも承認されることは確保できず、一国で規制承認を得ることができなかったり、遅延したりすることは、他の国の規制承認過程に悪影響を及ぼす可能性がある。例えば、米国におけるFDAの承認は、他国または管轄地域の規制機関の承認を確保しておらず、外国規制機関の承認は、FDA、EMAまたは他の国の規制機関の承認を確保していない。審査手続きは司法管轄区域によって異なり、長くて高価である可能性があり、関連する要求と行政審査期限はアメリカと異なり、追加の臨床前研究或いは臨床試験を含む米国の要求と行政審査期限を超える可能性がある。私たちの候補製品が承認されても、特定の管轄区域の規制機関は、任意の製品の規制承認を撤回する可能性がある。私たちは国際市場で規制の承認を受けた経験がない。もし私たちまたはパートナーが規制要件を遵守できなかったり、必要な承認を得られずに維持できなかったりすれば、私たちの目標市場は減少し、候補製品のすべての市場潜在力を達成する能力は損なわれるだろう。
米国およびヨーロッパではSCN 8 A−DeEの治療のためのNBI−921352の孤児薬物指定が承認されているにもかかわらず,このような指定から何の価値も達成できない可能性がある。
我々のパートナーであるNeurocrine Biosciencesによって開発されたNBI-921352は、米国食品医薬品局の孤児薬物の称号を獲得し、欧州委員会がSCN 8 A-DeEを治療する孤児薬物の称号を獲得した。現在,孤児薬物指定は米国とEUでそれぞれ7年と10年の市場排他性を提供しており,1つの製品がこのような孤児適応のために最初に承認された製品である場合がある。EUでは、孤児薬物に対して、有効かつ完全な小児科調査計画(PIP)は、スポンサーが孤児薬物指定を審査する際に付与された10年間のマーケティング排他性に基づいて2年間のマーケティング排他性を延長する資格を有することができる。しかし,孤児薬物市場排他性は承認中に特定された適応以外の他の適応には触れず,他のタイプの薬物がこれらの同じ適応で孤児指定や承認を得ることも阻止しない。また、孤児薬物が承認された後であっても、FDAが新薬が臨床的に孤児製品より優れていると結論した場合、或いは市場不足が出現した場合、FDAはその後類似化学構造を有する薬物を同じ条件に使用することができると結論した。孤児薬物指定は、規制審査または承認中に薬物にいかなる利点も提供せず、可能な費用減免に加えて、このような指定も薬物が市販承認される可能性を増加させることはない。
はいCatalyst Pharms,Inc.Becera事件を訴える、1299“連邦判例編”第14巻(第11巡回法廷)2021年)には,裁判所はFDAの長期的な立場,すなわち孤児薬物排他性が条件を満たす疾患内の承認用途または適応にのみ適用されることに同意しない。FDAは2023年1月24日、FDAがCatalystに対する裁判所の命令を遵守しているが、FDAはCatalyst命令の範囲外に規制の長期的な解釈を適用し続けることを意図していることを明らかにし、FDAは、同じ孤児指定疾患または条件の下で、他のスポンサーが未承認の新しい用途または適応のために承認された新しい用途または適応のために、孤児薬物の独占的適用の範囲を薬物が承認された用途または適応と一緒にバンドルし続けることを可能にすることを明らかにした。将来の訴訟、立法、機関決定と行政行動が孤児薬物の排他性の範囲にどのように影響するかはまだ不明である。
EUでは、薬物が最初の指定基準を満たさなくなった場合、または上場許可所有者が2回目の孤児薬の申請に同意した場合、または十分な薬物を提供できない場合、または第2の申請者がその薬物が最初の孤児薬よりも臨床的に優れていることを証明した場合、孤児の排他性は6年に減少する可能性がある。
46
FDAはSCN 8 A-DeEのまれな小児科疾患またはRPDの治療のためにNBI-921352を許可しているが、私たちはこのような指定からいかなる価値も達成できないかもしれない。
NBI−921352は,我々の協力者Neurocrine Biosciencesによって開発され,SCN 8 A−DeEの治療のためのRPD指定が得られた。FDAは“まれな小児科疾患”を米国では20万人未満の疾患と定義しており,主に18歳以下の人である。FDAのRPD優先審査クーポン計画によれば、RPDを治療するためのNDAまたはバイオ製品ライセンス申請BLAが承認されると、そのような出願の発起人は、後続のNDAまたはBLAを取得するための優先審査クーポンを取得するために使用することができる優先審査クーポンを取得する資格がある。Neurocrine BiosciencesがRPD優先審査券を受信することは保証されず、優先審査券の使用が後続のマーケティング申請のより速い審査または承認をもたらす保証もない。Neurocrine BiosciencesがSCN 8 A-DIEでNBI-921352の承認を得て、このような優先審査証明書を取得する資格があっても、この計画は製品候補を承認する際にもはや有効ではない可能性がある。また,優先査読クーポン券を第三者に無料で販売または譲渡することができるにもかかわらず,我々や我々の任意の協力者が優先査読クーポンを第三者に販売する保証はない場合には,任意の価値を実現することができる.また、議会はFDAの許可を2024年9月30日にRPDを指定し、RPD優先審査証明書を2026年9月30日に付与することを延長した。RPD指定は、規制審査または承認中に薬物にいかなる利点も提供せず、可能な費用減免および優先審査証明書を除いて、このような指定は、薬物が市販承認される可能性を増加させることはない。
私たちに製品責任訴訟を提起すれば、私たちは私たちの限られた製品責任保険のカバー範囲を超えた大量の責任を負うことができ、私たちの現在と未来の製品の商業化を制限することが要求されるかもしれません。
私たちの候補製品の臨床テストのため、私たちは固有の製品責任リスクに直面しています。もし私たちが任意の候補製品を商業化すれば、私たちはもっと大きなリスクに直面します。例えば、私たちの任意の候補製品が、他の療法と組み合わせて開発された任意の製品を含む場合、製品テスト、製造、マーケティング、または販売中にダメージを与えたり、不適切であることが発見された場合、私たちは起訴される可能性があります。このような製品責任クレームは、製造欠陥、設計欠陥、製品固有の危険について警告、不注意、厳格な責任と保証違反の告発を含む可能性がある。クレームはまた州や省の消費者保護法によって提起されることができる。もし私たちが製品責任クレームで自分自身を弁護することに成功できなければ、私たちは重大な責任を招いたり、私たちの候補製品の商業化を制限することを要求されるかもしれません。成功的な防御であっても、多くの財政的で管理的な資源が必要だ。私たちは補償の第三者も責任を負う可能性があるということに同意する。事件がどうであっても最終的な結果がどうであろうと、賠償責任は次のようになるかもしれない
私たちが現在保険を受けている製品責任保険の保険金額は現在の臨床計画と比較して適切です。しかし、私たちは責任による損失から私たちを保護するために、合理的なコストあるいは十分な金額で保険範囲を維持することができないかもしれません。もし私たちが候補製品の発売許可を得たら、私たちは私たちの保険カバー範囲を拡大し、商業製品の販売を含めるつもりです;しかし、私たちは商業的に合理的な条項や十分な金額で製品責任保険を得ることができないかもしれません。薬物や治療に思わぬ悪影響を与えた集団訴訟では,多額の判決が下されることがある。成功した製品責任クレームや一連の私たちに対するクレームは、私たちの普通株の市場価格を下落させる可能性があり、もし私たちの保険範囲を超えていると判断すれば、私たちの将来の運営と業務業績に悪影響を及ぼすかもしれません。
47
私たちの候補製品が対象とするいくつかの疾患または障害の患者は、通常、深刻かつ末期の疾患段階にあり、既知および未知の重大な事前存在および潜在的に生命を脅かす場合がある。治療中,我々の候補製品に関与している可能性があるため,患者は過去および未来に死亡を含む有害事象を受ける可能性がある。このような事件は、怪我をした患者に大量の資金を支払うこと、遅延、負の影響、または規制部門がこれらの候補製品のマーケティングを許可または維持する機会を得るか、または私たちの商業化努力を一時停止または放棄することを要求する費用の高い訴訟に直面する可能性がある。有害事象が我々の製品に関係しているとは考えられない場合でも,この場合の調査は時間や不確実性がある可能性がある.これらの調査は、他の国/地域での規制承認プロセスを延期したり、製品候補者の獲得や維持に影響を与えたり制限したりする規制承認タイプを遅らせるための販売努力を中断する可能性がある。これらの要因により、製品責任クレームが弁護に成功しても、我々の業務、財務状況又は運営結果に実質的な悪影響を及ぼす可能性がある。
もし私たちが自分の販売、マーケティング、流通能力を確立できなかったり、これらの目的のために合意したりすることができなければ、私たちはいかなる未来の製品を独立して商業化することに成功できないかもしれない。
私たちは販売やマーケティングインフラを持っていません。会社として、販売、マーケティング、流通経験がありません。私たちの戦略は、私たち自身の商業インフラを構築することを含み、ある商業市場で将来の製品を選択的に商業化することは、高価で時間がかかるだろう。XEN 1101、および/または特定の商業市場を含むいくつかの製品の場合、ビジネスパートナーを時々評価します。場合によっては、このような製品の将来の開発や商業化に参加することが私たちの業務を促進すると考えていれば、参加する権利の保留を求めることができるかもしれません。私たちは私たちがこのようなビジネスパートナーシップを成功的に達成するかどうか、あるいは完成すれば、このようなパートナーシップが成功するかどうかを確信できない。
米国での内部販売、流通、マーケティング能力を発展させるためには、私たちの任意の候補製品が承認されることを確認する前に投入する必要がある大量の財務および管理資源を投入しなければならない。会社として、私たちは以前、生物製薬製品のマーケティング、販売、流通の面で経験がなく、重大なリスクに関連するビジネス組織の設立と管理を行ってきた。私たちが自分で販売、マーケティング、流通機能を実行することを決定した任意の未来の製品に対して、私たちは多くの追加のリスクに直面するかもしれません
適切な場合、私たちは、販売、マーケティング、および流通能力を有する契約販売者、流通パートナー、または協力者を利用して、私たちの候補製品の商業化または独立商業化を支援することを選択することができる。もし私たちが第三者と合意して、製品に販売、マーケティング、流通サービスを提供すれば、私たちがそれによって生成した収入またはこれらの収入から得られた利益は、私たち自身が販売、マーケティング、および流通している場合よりも低いかもしれません。さらに、私たちは第三者との販売、マーケティング、流通に成功できないかもしれません。あるいは私たちに有利な条項でそうすることができないかもしれません。私たちはこれらの第三者に対して支配権がほとんどない可能性が高く、これらの第三者のいずれかは、私たちの現在または任意の未来の製品を効果的に販売、マーケティング、流通するために必要な資源と注意を投入することができないかもしれない。
48
私たちの任意の候補製品を商業化する規制承認を得ても、私たちは持続的な規制義務と持続的な規制審査の制約を受けることになり、多くの追加費用と遅延を招く可能性がある。
私たちまたは既存または未来のパートナーが規制の承認を受けた任意の私たちの候補製品、およびそのような製品の製造プロセス、承認後の研究、ラベル、広告、および販売促進活動などは、FDAおよび他の規制機関の持続的な要求および審査を受けるだろう。これらの要求には、安全およびその他の発売後の情報と報告の提出、登録と上場要求、製造、品質管理、品質保証と記録と書類の対応する維持に関する要求、医師へのサンプルの配布と記録の保存に関する要求が含まれる。私たちと私たちの契約製造業者はまた、FDAや他の規制機関の使用料や定期検査を受けて、これらの要求と私たちが入手可能な任意の製品承認条項の遵守状況を監視します。また,我々の候補製品は,1970年の“制御物質法”下の付表分類(あるいは米国以外の類似立法下の付表分類)を得る可能性がある。これは追加的な複雑性をもたらし、製造、サプライチェーン、許可、輸出入、および流通における遅延および制限をもたらす可能性がある。
製品が承認されても、FDAまたは他の適用可能な規制機関(場合によっては)は、製品ラベル上で広く予防措置および警告をとることを要求するか、または臨床試験または重いリスク管理活動を含む臨床試験または重いリスク管理活動、またはREMSを含む臨床試験または重いリスク管理活動を含む、製品ラベル上で広範な予防措置および警告を要求することを制限する可能性があり、承認条件として、薬剤の利点が潜在リスクよりも大きいことを保証することを助ける。REMSは、薬物ガイドライン、医療専門家のコミュニケーション計画、および安全な使用を確保する要素、またはETASUを含むことができる。ETASUは、処方または調剤のための特殊トレーニングまたは認証、特定の場合にのみ調剤、特殊モニタリング、および患者登録簿の使用を含むことができるが、これらに限定されない。REMSに対する要求はこの薬物の潜在的な市場と収益力に重大な影響を与える可能性がある。
任意の承認された製品について、私たちまたは私たちの協力者は、製品の製造プロセス、ラベル、包装、系列化、流通、有害事象報告、貯蔵、広告、販売促進および記録などに関する広範な法規および要求を引き続き遵守することを保証する必要がある。これらの要求は、安全性および他の承認後の情報および報告の提出、および我々または私たちの協力者が承認後に必要とする任意の臨床試験について、現在の良好な生産実践(CGMP)、良好な流通実践(GDP)、および現在の良好な臨床実践(CGCP)を継続的に遵守することを含む。
承認後に製品に以前に未知の問題が存在することが発見され、予想されていない深刻度または頻度の不良事象、または私たちの製品または第三者製造業者または製造プロセスの他の問題、または規制要件を遵守できなかった場合、他に加えて、ラベルまたはマーケティング、撤回、同意法令、臨床保有、承認後の要求または制限、リコール、罰金、警告状、禁止、処罰、連邦医療保健計画の排除、差し押さえおよび/または拘束などの結果および不良行為の制限を招く可能性がある。上記のいずれの状況の発生も、我々の業務や経営結果に重大な悪影響を及ぼす可能性がある。
また,処方薬は承認されたラベルに基づいて,承認された適応に対してしか普及しない。FDA,EMA,その他の機関はラベル外使用を禁止する法律法規を積極的に実行しており,ラベル外使用を不当に普及させていることが発見された会社は重大な責任を負う可能性がある。しかし、医師はその独立した医学判断に基づいて、ラベル外で使用される合法的に使用可能な製品のために処方することができる。FDAは医師が治療を選択する行為を規範化していないが、FDA、EMAと他の外国の監督管理機関は確かにラベル外でその製品を使用するメーカーのコミュニケーションを制限している。
私たちが制御物質を含むか、または対象とされる候補製品を開発し、商業化する場合、私たちまたは私たちのCRO、CMO、および他の請負業者は、制御対象物質の法律や法規を遵守できなければ、私たちの業務運営結果や私たちの財務状況に悪影響を及ぼす可能性があります。
私たちは将来、複数の司法管轄区域(例えば、アメリカ、カナダ、EU)で制御物質とみなされる候補製品を開発する可能性があり、これにより、私たちは規制活動(貯蔵、製造、研究、臨床試験、輸入、輸出などを含む)に従事する各適用司法管轄区域で追加の制御物質規制要求に直面することになるだろう。例えば、必要な登録の取得と維持は、私たちの制御物質候補製品の輸入、製造、または流通の遅延を招き、臨床試験の予想スケジュールを延長する可能性がある。
規制された物質や規制された物質は麻薬取締庁が改正案に基づいて規制される。DEAは化合物を付表I,II,III,IVまたはV類として管理している。米国で使用が許可されている薬品は別表II,III,IVまたはVとされている可能性があり,付表IIは乱用または依存の可能性が最も高い物質と考えられ,付表V物質はこのような物質の中で乱用の相対的リスクが最も低い物質と考えられている。
49
DEAのスケジュール決定は、1つの物質または1つの物質に対するFDAの特定の処方の承認に依存する。このスケジュールの決定は、適切なスケジュールに関するFDAの承認およびFDAの提案に依存し、承認および任意の潜在的な再スケジュールプロセスの遅延をもたらす可能性がある。DEAが有利なスケジューリング決定を行う保証はない.連邦レベルが付表II,III,IVまたはV制御物質に属する物質も,州法や法規および類似した外国制御物質条例(適用すれば)によって付表決定する必要がある可能性がある。FDAの承認を得た場合、いくつかの制御物質に関する承認後の活動はDEAの規制を受け、施設の登録と検査、製造、貯蔵、流通、医師処方プログラムなどに関するDEA規定を含む。さらに、我々の請負業者(例えば、我々のCROおよびCMO)が開発および/または商業化の過程でCSA(例えば、適用される)を遵守できない場合、私たちの業務、財務状況、および運営結果に大きな悪影響を及ぼす可能性がある。
米国個別州や米国以外の国でも制御物質法律法規が制定されている。これらの法律と法規は、常に連邦法律を反映するとは限らない州制御物質法律を含み、私たちの候補製品を単独で手配するかもしれない。異なる管轄区域で異なる制御された物質要求を遵守することは、私たちの運営コストを増加させ、追加的な責任を負わせるかもしれない。
私たちの候補製品が市場によって承認されても、候補製品に制御された物質が存在することは、私たちの現在または未来の候補製品に対する否定的な宣伝や大衆的な印象をもたらす可能性がある。
もし私たちが制御された物質によって規制された候補製品が商業販売のために承認された場合、制御された物質または他の制御された物質に対する公衆の否定的な宣伝または認知は、市場の受け入れまたは消費者が私たちの候補製品の認知に負の影響を与える可能性がある。臨床医や患者が制御物質を含む新しい療法を試みたくなければ,限られた採用に直面している可能性がある。疾病に関連するいかなる不良宣伝、あるいは患者が私たちまたは他社の流通の類似療法を使用または誤用したことによる他の悪影響は、私たちの業務、将来性、財務状況、および運営結果に重大な悪影響を及ぼす可能性がある。
将来の有害事象および候補製品中に存在する制御された物質の研究はまた、より厳しい政府規制、より厳しいラベル要件、および私たちの候補製品をテストまたは承認する際に生じる可能性のある規制遅延をもたらす可能性がある。どんなより厳しい審査も、私たちの候補製品のための規制承認のコストを延期または増加させる可能性がある。
私たちが開発したすべての候補製品は不利な第三者の保証と清算のやり方と定価法規の制約を受けるかもしれません。
私たちまたは私たちのパートナーが任意の製品を商業化することに成功しているかどうかは、政府医療計画(例えば、MedicareおよびMedicaid)および個人第三者支払人(例えば、個人健康保険会社、管理保健計画および他の組織)によるこれらの製品および関連治療の保証および清算の程度にある程度依存するであろう。政府当局と個人第三者支払人は、彼らがどの薬に支払うかを決定し、清算水準を確立する。アメリカの医療産業の主な傾向の一つはコスト統制だ。政府当局と第三者支払者は,特定の薬物のカバー範囲や精算金額を制限することでコストを抑制しようとしている。私たちまたは私たちの協力者が商業化したどの製品も保険と精算を受けることができることを保証することはできません。精算することができれば、精算することもできます。さらに、保証範囲および精算は、私たちまたは協力者がマーケティング承認を得た任意の候補製品の需要または価格に影響を与える可能性があります。保険や精算が得られない場合、あるいは精算が限られたレベルに限られている場合、私たちまたは私たちの協力者は、マーケティングの承認を得た候補製品を商業化することに成功しない可能性があります。
第三者支払者のカバー範囲や新承認製品の精算に関する不確実性が大きい。米国では、保証範囲と精算範囲は第三者支払人によって異なる。第三者支払者は、ある候補商品に保険を提供することを決定し、他の支払者もその候補商品に保険を提供することを保証することはできない。したがって、カバー範囲を決定するプロセスは、しばしば時間がかかり、高価である。支払人が精算を決定する際に考慮する要素は、製品が(I)その健康計画下での保険福祉、(Ii)安全、有効、および医学的に必要であるかどうか、(Iii)特定の患者に適しているか、(Iv)費用効果、および(V)試験的でも研究的でもないことに基づく。私たちは各第三者支払者に私たちの製品を使用した科学的かつ臨床的支援を単独で提供することを要求されるかもしれないが、保険と適切な精算を一貫的に適用すること、または最初に十分な精算を得ることは保証されない。
連邦政府と州政府が処方薬の定価を下げる措置を含む追加の医療コスト制御措置を実施することに伴い、私たちの製品が承認された場合、個人または公共支払者が保証を受け、保険を受けた場合、精算金額が十分かどうか、あるいは他の市場製品と競争するかどうかを決定することはできない。連邦と州政府および医療計画の行動は薬品定価と医療保健コストに追加の下振れ圧力をもたらす可能性があり、承認されれば、これは私たちの製品のカバー範囲と清算、私たちの収入、および私たちが他の市場製品と競争し、研究開発コストを回収する能力にマイナス影響を与える可能性がある。
50
さらに、政府医療計画または個人支払者が要求する強制的な割引またはリベート、および将来、米国より低い価格で販売される可能性のある国/地域からの輸入薬品の法的規制が緩和されることによって、薬品の正味価格が低下する可能性がある。許容可能な精算レベルを獲得し、維持するためには、合理的かつ慣例的な共同支払いレベルでの患者の参入を確保するために、価格から割引またはリベートを提供しなければならないか、または他の不利な定価修正を実施しなければならない可能性がある。私たちは私たちが商業化されたどの候補製品も精算できることを確実にすることはできない。もし精算できるなら、精算のレベルもそうだ。
米国以外では,治療薬の商業化は通常広範な政府価格制御や他の市場規制の制約を受けており,欧州,カナダ,その他の国ではコスト制御の取り組みが重視されており,候補製品などの治療薬の定価や使用に圧力を与え続けていると考えられる。多くの国、特にEU諸国では、国家衛生システムの一部として、医療製品価格は異なる価格制御メカニズムの制約を受けている。これらの国では、製品が発売承認された後、政府当局との定価交渉にはかなりの時間がかかるかもしれない。いくつかの国で精算または定価の承認を得るために、私たちあるいは私たちの協力者は臨床試験を行う必要があるかもしれません。私たちの候補製品の費用効果を他の利用可能な治療法と比較します。一般的に、この制度での製品価格はアメリカよりずっと低いです。他の国は会社が自分で価格を設定することを許可していますが、会社の利益を監視しています。追加の外国価格規制や定価規制の他の変化は、私たちまたは私たちの協力者が私たちの候補製品に受け取ることができる費用を制限するかもしれません。そのため、米国以外の市場では、米国に比べてわが製品の精算が減少する可能性があり、商業的に合理的な収入や利益を生み出すには不十分である可能性がある。
私たちまたは私たちの協力者のいくつかのターゲット患者集団は、SCN 8 A-DIEのような孤児またはマイナー適応である可能性がある。小さい患者群を治療するための治療法を商業的に実行できるようにするためには、このような治療法の定価、カバー範囲、精算金額は相対的に高く、数量不足の問題を解決する必要がある。したがって、私たちまたは私たちの協力者は、より小さい潜在市場規模を占める任意の承認製品に対して定価、カバー、精算戦略を実施する必要があるかもしれない。もし私たちまたは私たちの協力者が、私たちまたは私たちの協力者の現在および第三者支払者または政府からの任意の未来の製品のために保証範囲と十分な精算を確立または維持することができない場合、これらの製品の採用および販売収入は悪影響を受け、ひいてはマーケティングまたはこれらの製品を販売する能力に悪影響を及ぼす可能性がある。
医療やその他の改革は、私たちまたは私たちの協力者が開発した任意の製品を商業化する難しさとコストを増加させ、入手可能な価格に影響を与えるかもしれません。
米国や一部の外国司法管区は、医療システムを変更するための一連の立法·規制提案を検討または公布しており、このような製品が販売を許可されると、任意の製品の販売収益性に影響を与える可能性がある。米国や他地域の政策立案者や支払者の中では,医療システムの改革を推進することに大きな興味があり,医療コストの抑制,質の向上および/または参入拡大を既定の目標としている。米国では、製薬業はこれらの努力の重点であり、重大な立法計画の大きな影響を受けてきた。
例えば、2010年に“医療·教育和解法案”により改正された“患者保護·平価医療法案”は、総称してPPACAと呼ばれるものであり、その中に含まれる措置は、政府や民間保険会社が医療に資金を提供する方法を著しく変更している。ACA以外にも,広く行われている医療改革努力があり,その中のいくつかの重点は薬品の価格や支払いを規制することである。薬品の定価と支払い改革はトランプ政権の重点であり、バイデン政権の重点でもある。例えば、2021年に公布された連邦立法は、2024年1月1日に施行された医療補助薬品税還付計画の法定上限を廃止した。もう一つの例として、2022年の“インフレ低減法案”(IRA)には様々な薬品価格交渉、インフレリベート、定価条項が含まれているが、実施日はそれぞれ異なる。アイルランド共和軍は連邦政府がある高価な単一源の連邦医療保険薬物のために最高公平価格を交渉することを許可し、薬品価格交渉要求を守らないメーカーに罰と消費税を加え、すべての連邦医療保険B部分とD部分の薬物(限られた例外の場合)が薬品価格の増加がインフレより速い場合にインフレリベートを獲得することを要求し、連邦医療保険D部分を再設計して受益者の自己処方薬コストなどの変化を減少させる。Ireland共和軍が施行中であるため、私たちの業務とより広い製薬業への影響はまだ不確定だ。もう一つの例は、2022年、アイルランド共和軍が公布した後、バイデン政府はHHS報告書が連邦医療保険と医療補助革新センター(CMMI)を利用して連邦医療保険と医療補助受益者の薬品コストを下げる新しいモデルをテストする方法を指示する行政命令を発表した。この報告は2023年に発表され,CMMIが現在開発中の様々なモデルを提案し,薬物コストの低減,獲得性の促進と看護の質の向上を目指している。このような変化や他の変化は私たちの製品の市場状況に影響を及ぼすかもしれない。私たちは議会、機関、そして他の機関が薬品価格と政府価格報告書を引き続き検討すると予想している。
また、いくつかの州はすでに州薬品価格透明性と報告法を公布し、もし私たちが製品が発売されれば、私たちのコンプライアンス負担を大幅に増加させ、このような州法律に基づいてより大きな責任を負わせるかもしれない。これらの措置や実施されている他の医療改革措置は,我々の業務に実質的な悪影響を及ぼす可能性がある。
51
医療改革の努力はすでに検討され、法的挑戦を受け続ける可能性がある。例えば,PACAでは税収改革立法が公布され,2019年から強制医療保険範囲を維持していない個人への税収処罰が廃止され,2021年には米国最高裁はいくつかの州がACAに対する最新の司法挑戦を却下したが,PPACAの合憲性を具体的に裁くことはなかった。別の例として、連邦反減税法規下の法規改正は、製薬業者が薬局福祉マネージャーおよび健康計画に提供する従来のMedicare Part D割引の保護を廃止する。裁判所の命令により更迭が延期され、最近の立法はこの規則の施行を2032年1月に一時停止した。もう一つの例として,アイルランド共和軍の薬品価格交渉計画は,複数の製薬業者や業界団体による訴訟で挑戦されている。
私たちはアメリカ連邦や州医療立法の将来の行方を予測できません。法律や規制枠組みのさらなる変化は、私たちの将来の収入創出能力を低下させたり、私たちのコストを増加させたりする可能性があり、どちらも私たちの業務、財務状況、および運営結果に実質的な悪影響を及ぼす可能性があります。政府および他の第三者支払者は、医療コストの制御または低減および/または価格制御の実施に努力し続けており、承認されれば、候補製品の需要および利益を達成または維持する能力に悪影響を及ぼす可能性がある。
EUでは、同様の政治、経済、規制発展は、私たちまたは私たちの協力者が私たちの現在または任意の未来の製品を利益的に商業化する能力に影響を及ぼすかもしれない。価格およびコスト制御措置に対する持続的な圧力に加えて、EUまたは加盟国レベルの立法発展は、著しい追加的な要求や障害を招く可能性があり、これは私たちの運営コストを増加させる可能性がある。国際市場では,精算や医療保険支払い制度は国によって異なり,多くの国で特定製品や治療法に価格上限が設定されている。私たちの将来の製品は、あれば第三者支払者に医学的に合理的だと思われないかもしれませんが、特定の適応が必要、あるいは割に合わないです。このような製品は十分な精算レベルが得られない可能性があり、第三者支払者の精算政策は、私たちまたは協力者が未来の製品を販売する収益性に悪影響を及ぼす可能性がある。
承認後の要求を拡大し、医薬品の販売や販売促進活動を制限するための立法·規制提案がなされている。私たちは、より多くの立法変化が公布されるかどうか、あるいはFDAの法規、ガイドライン、解釈が変わるかどうか、あるいはこれらの変化が私たちの候補製品の上場承認にどのような影響を与える可能性があるかどうかを決定することはできない。また、米国議会のFDA承認過程に対するより厳格な審査は、上場承認を著しく延期または阻止する可能性があり、より厳しい製品ラベルと承認後のテストとその他の要求の影響を受ける可能性がある。
また、他のより広範な立法改革も採択されており、これは私たちの製品の商業成功に悪影響を与え、私たちの製品の商業成功を阻止する可能性があります。例えば、改正2011年の予算制御法により、2013年に医療保険(医療補助ではない)提供者に支払われるお金が減少し、国会が追加行動を取らない限り、2032年まで有効になる。Medicare、Medicaid、または実施可能な他の公共援助または補助医療計画に影響を与える重大な支出削減、および/または私たちに徴収される可能性のある任意の重大な税金または費用は、私たちの運営結果に悪影響を及ぼす可能性がある。
米国や他の管轄区域の将来の立法や行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することはできない。もし私たちまたは私たちの協力者が既存の要求の変化に緩やかに適応できない場合、あるいは新しい要求や政策を採用することができない場合、または私たちまたは私たちの協力者が法規遵守を維持できない場合、私たちの候補製品は獲得された可能性のあるいかなるマーケティング承認も失う可能性があり、私たちは利益を達成したり維持することができない可能性があり、これは私たちの業務に悪影響を及ぼすだろう。
私たちの第三者への依存に関するリスク
私たちの協力製品と候補製品の成功した開発と商業化の将来性は、私たちの協力者の研究、開発、マーケティングの努力にかかっています。
私たちは、私たちの協力者が私たちの計画に投入する可能性のある資源、時間、エネルギーを制御することができず、そのような計画に関する情報やこれらの計画によって生成された情報に限らずアクセスすることができない。私たちはNeurocrine Biosciencesを含む私たちのパートナーに依存し、私たちが彼らとそれぞれ達成した合意に基づいて、候補製品の研究および臨床開発に資金を提供し、任意の臨床開発を行い、そのような1つまたは複数のそのような製品または候補製品を承認、マーケティング、商業化することに成功した。そのような成功は重大な不確実性の影響を受けるだろう。
協力に成功した収入からの能力は、様々な要因の影響を受ける可能性があることを確認した
52
もし私たちの協力者が私たちの期待した方法で彼らの責任を表現したり、適時に履行しなかったりすれば、臨床開発、規制承認、商業化努力は延期され、終了され、または商業的に成功しないかもしれない。私たちと私たちの協力者の間で衝突が起こるかもしれない。もし私たちの1つまたは複数の協力合意が終了した場合、私たちは任意の終了した製品または候補製品の責任を自費で負担するか、または新しいパートナーを探す必要があるかもしれない。この場合、私たちは、1つ以上の独立したプロジェクトの規模と範囲を制限すること、または私たちの支出を増加させ、受け入れられないかもしれない条項や根本的に得られない追加資金を求めることが要求される可能性があり、私たちの業務は実質的で不利な影響を受ける可能性がある。
私たちは新しい協力を成功させたり、既存の連合を維持することができないかもしれませんが、これは私たちが候補製品を開発し、製品を商業化する能力に悪影響を及ぼすかもしれません。
通常の過程で、私たちは他のバイオテクノロジーや製薬会社と接触し、潜在的な内部許可、外部許可、連合、および他の戦略的取引について議論する。私たちの候補製品と開発計画の進歩、そして私たちの現在と未来の候補製品の潜在的な商業化は、費用を支払うために多くの追加の現金が必要になるだろう。しかも、いくつかの管轄区域では、パートナーは私たちの候補製品の市場潜在力を私たちよりもよく認識するかもしれない。これらの理由やその他の理由で、より多くの製薬やバイオテクノロジー会社と開発と潜在的な商業化に協力することにしたかもしれません。
私たちは適切な協力者を探す上で激しい競争に直面しており、交渉過程は時間がかかり複雑だ。さらに、私たちは、私たちの研究開発ルートが不足している可能性があるので、現在または未来の候補製品のための他の協力または他の代替計画を確立することに成功できないかもしれません。私たちの現在または未来の候補製品は、協力努力の開発段階が早すぎると考えられるかもしれません。および/または第三者は、私たちの候補製品が安全性および有効性を示す必要な潜在力が不足していると考えるかもしれません。私たちが協力を確立するために成功したとしても、私たちが合意した条項は私たちに不利になる可能性があり、例えば、候補製品の開発や承認が延期または承認された製品販売に失望した場合、私たちはそのような協力を維持できないかもしれない。
もし私たちの既存の任意の協力合意が終了した場合、または他の製品協力に参加することが私たちの最良の利益に合致すると判断した場合、私たちは達成できなかった、達成を延期し、またはそのような協力を維持できなかった
53
私たちは私たちの候補製品を生産するために第三者に依存しており、これは私たちが必要な時に、または許容可能なコストで十分な数の候補製品、原材料、原料薬、または医薬製品がないリスクを増加させるかもしれない。
私たちは臨床や商業ロット候補製品を生産するための製造施設を持ったり運営したりしていません。私たちはそうする資源と能力が足りません。したがって、私たちは現在、私たちの候補製品中の活性医薬成分または原料薬、ならびに私たちの臨床試験と臨床前研究および臨床試験用品の包装、ラベル、流通のためのすべての候補製品の最終医薬製品の調合を製造し、供給するために第三者に依存している。私たちの現在の戦略は私たちのすべての候補製品の製造を第三者にアウトソーシングすることだ。
さらに、私たちは、私たちのパートナーに依存して、直接またはCMO生産によって彼らに許可された候補製品、またはCMOと協力して、私たちの候補製品を生産するのに十分な数の材料を生産して、臨床前試験および臨床試験のために使用し、私たちの製品の商業生産に使用する予定です。もし私たちまたは私たちの協力者がそのような第三者の製造源を手配できない場合、あるいは商業的に合理的な条項でそうすることができなければ、私たちまたは私たちの協力者は十分な候補製品を成功的に生産できないかもしれない、あるいは私たちまたは私たちの協力者はそうすることを延期するかもしれない。このような失敗や重大な遅延は私たちの臨床試験を延期し、私たちの業務に実質的な損害を与える可能性がある。バイオ製薬製品の製造は複雑であり、先進的な製造技術とプロセス制御の開発を含む大量の専門知識と資本投資が必要である。汚染、設備故障或いは設備設置或いは操作が不適切であるため、サプライヤー或いはオペレータのミス、生産量の汚染と不一致、製品特性の多変性及び生産過程の困難により、私たちの候補製品の製造過程は製品損失の影響を受けやすい。正常製造プロセスとの微小な偏差でも生産量の低下、製品欠陥、その他の供給中断を招く可能性がある。私たちの候補製品または私たちの候補製品を製造する第三者製造施設で微生物、ウイルス、または他の汚染が発見された場合、そのような製造施設は、汚染を調査および修復するために長い間閉鎖される必要があるかもしれない。私たちの候補製品の生産運営に影響を与える不利な発展は、承認されれば、出荷遅延、在庫不足、ロット故障、製品の撤回やリコール、あるいは私たちの製品供給の他の中断を招く可能性があります。また、在庫を解約し、規格に適合しない製品に他の費用や支出を発生させ、コストの高い救済努力を行ったり、よりコストの高い製造代替案を求めたりしなければならない可能性もある。
第三者製造商会に依存することは、私たち自身が候補製品を製造すれば、これらの第三者に依存してコンプライアンスおよび品質管理および保証、量産、第三者が制御できない要因(私たちの製品仕様に従って私たちの候補製品を合成および生産できなかったことを含む)によって製造協定に違反する可能性、業界統合の影響(このような第三者に関連する業務統合を含む)、および第三者がコストが高いか、または私たちに損害を与える時間に合意を終了または終了する可能性を含む、これらの第三者に依存するリスクをもたらすであろう。いくつかの潜在的な代替メーカーが私たちの候補製品を生産できると信じていますが、そのような代替製品を決定して同定する際に、追加のコストと遅延が生じる可能性があります。
また、私たちは通常、原材料、原料薬と薬品とサービスを購入注文の形で注文し、どの商業メーカーとも長期専用生産能力或いは最低供給手配を締結していない。私たちは私たちが満足できる条件で必要な供給スケジュールをタイムリーに得ることができるか、あるいは根本的に保証できないという保証はない。必要に応じてこれらの手配を確保できなければ、候補製品の開発を完了したり、それを商業化したり(承認されれば)能力に実質的な悪影響を及ぼす可能性がある。私たちは第三者製造業者と商業供給合意に到達できないかもしれないし、受け入れ可能な条件でそうすることができないかもしれない。私たちの候補製品の商業数量と配合を拡大することは困難かもしれませんし、製造コストは目を引くほど高いかもしれません。
54
さらに、FDA、EMA、および他の外国規制機関は、私たちの候補製品がcGMPおよび同様の外国基準で生産されなければならないことを要求している。医薬品メーカー及びその下請け業者は、販売申請を提出する際にその施設及び/又は製品を登録することを要求され、その後、毎年FDA、EMA及び他の外国監督管理機関に登録される。それらはまたFDA、EMA、他の外国規制機関の事前承認検査と定期抜き打ち検査を受けている。その後、製品または私たちまたは私たちの協力者が使用する製造または実験室施設に問題があることが発見された場合、製品のリコール、製造の一時停止、輸入禁止、製品の差し押さえ、または自発的に薬物を市場から引き下げることを含む、製品またはその製造または実験室施設の制限を引き起こす可能性がある。私たちまたは私たちのパートナーの第三者製造業者は、cGMPに準拠できなかったか、または十分な数の候補製品をタイムリーに配送できなかったことで、私たちの任意の候補製品が遅延したり、規制部門の承認を得られなかったりする可能性があります。
第三者製造業者以外に、私たちは他の第三者に依存して、貯蔵、監視、ラベル、包装、大量の薬品と薬品を輸送する。もし私たちがそのような第三者源を手配できない場合、あるいは商業的に合理的な条項でそうすることができなければ、私たちは十分な候補製品を提供することに成功できないかもしれないし、私たちはそれを延期するかもしれない。そのような失敗や重大な遅延は私たちの業務に実質的な被害を及ぼすかもしれない。
もし私たちの候補製品のいかなる第三者メーカーも、私たちの候補製品の生産規模を拡大することができず、および/またはその製造製品の良率を向上させることができなければ、私たちの製品を製造するコストが増加する可能性があり、商業化が遅れる可能性がある。
臨床試験の需要を満たすのに十分な数を生産するために、承認されれば、私たちの候補製品はその後商業化され、私たちの第三者メーカーは製品の品質を維持しながら生産量を増加させ、製造プロセスを最適化することを要求されるだろう。より大規模な生産への移行は難しいことが証明されるかもしれない。また、私たちの第三者メーカーが、私たちの候補製品の生産量を向上させるためにその製造プロセスを最適化できない場合、または製品の品質を維持しながら私たちの候補製品の生産量を増加させることができない場合、登録および検証要求および臨床試験の需要や市場需要を満たすことができない可能性があり、規制部門の承認を延期し、利益を創出する能力を低下させ、私たちの業務や運営結果に大きな悪影響を及ぼす可能性がある。
私たちは第三者に依存して臨床前研究と臨床試験を行う。これらの第三者が、適用される法律法規の遵守や予想される期限までに完了することを含む、彼らの契約義務を成功的に履行できない場合、私たちの業務は深刻な損害を受ける可能性があります。
私たちは、学術機関、CRO、病院、診療所、および他の第三者パートナーを含む可能性があり、私たちの現在および未来の候補製品の臨床前および臨床研究を監視、支持、実施、および/または監視するために、私たちによってコントロールされていない実体に依存する。したがって,我々自身がこれらの実験を行うのに比べて,これらの研究の時間やコストおよび被験者を募集する能力の制御は少ない.例えば,研究者が協賛する第二段階概念検証臨床試験は,シナイ山イカン医学院の学術協力者と協力して行われており,MDDや快感障害におけるXEN 1101の役割を検査している。
もし私たちが受け入れ可能な条項でこれらの第三者と合意を維持することができない場合、またはそのような約束が早期に終了した場合、私たちは直ちに患者を募集したり、他の方法で予想される方法で実験を行うことができないかもしれない。また,これらの第三者が我々の研究に十分な時間と資源を投入することや,我々の候補製品に関する臨床試験情報を維持することを含む,我々の契約要件や法規の要求に応じてタスクを実行することは保証されない.これらの第三者が予想される最終期限内に、任意の規制情報をタイムリーに送信することができず、合意を遵守できなかった場合、または規制要求または彼らと達成された合意に従って行動できなかった場合、または彼らが不適格な方法で、またはその活動または彼らが取得したデータの品質または正確性を損なう方法で動作した場合、私たちの将来の候補製品の臨床試験は延長または延期され、追加のコストが生じる可能性があり、または私たちのデータはFDA、EMA、または他の外国の規制機関によって拒否される可能性がある。
結局、私たちは私たちのすべての臨床試験が適用された方案、法律、法規、科学的基準に従って行われることを保証する責任があり、私たちの第三者への依存は私たちの規制責任を免除しない。
55
我々のCROおよびCMOは、現在の良好な実験室実践、またはFDA、欧州経済圏加盟国の主管当局および同様の外国規制機関が臨床開発製品に対して実行しているcGLP、CGCPおよびcGMP法規およびガイドラインを遵守しなければならない。監督管理部門は定期的に臨床試験スポンサー、主要な研究者、臨床試験場、製造施設、非臨床試験施設とその他の請負業者を検査することによって、これらの規定を実行する。もし私たちまたは私たちの任意のCROまたはCMOがこれらの適用された法規を遵守できなかった場合、私たちの非臨床研究および臨床試験で生成された臨床データは信頼できないと考えられるかもしれません。私たちのマーケティング申請は提出が延期されるかもしれません。あるいはFDA、EMA、または他の外国の規制機関は、私たちのマーケティング申請を承認する前に追加の臨床試験を行うことを要求するかもしれません。検査後、FDA、EMAあるいは他の外国の監督管理機関は著者らの任意の臨床試験が失敗したか、または適用されたCGCP法規を遵守できなかったことを決定することができる。また,われわれの臨床試験はFDA,EMA,他の外国規制機関が施行したcGMP法規下で生産された製品を用いて行わなければならず,われわれの臨床試験には大量の試験対象が必要である可能性がある。私たちはcGLP、CGCP、cGMP法規を遵守できず、臨床試験を繰り返したり、より多くのロットの薬物を生産する必要があるかもしれません。これは規制承認過程を延期し、私たちのコストを増加させます。さらに、私たちの任意のCROまたはCMOが連邦または州の詐欺および乱用または虚偽請求法律または医療プライバシーおよび安全法律に違反している場合、またはこのような状況が発生したと断言または報告した場合、私たちの業務は影響を受ける可能性がある。
私たちの任意の臨床試験サイトが任意の理由で終了すれば、これらの患者の看護を別の合格した臨床試験サイトに移すことができない限り、進行中の臨床試験に登録されている患者の後続情報を失う可能性がある。さらに、私たちが私たちのいかなるCROまたはCMOとの関係が終了した場合、私たちはCROまたはCMOの代わりに商業的に合理的な条項で合意できないか、または全く合意できないかもしれない。
CRO、CMO、または他のサプライヤーを交換または増加させることは、大量のコストを伴う可能性があり、大量の管理時間および重点を必要とする可能性があります。また、新しいCRO、CMO、またはサプライヤーが作業を開始すると、自然な過渡期がある。したがって,遅延が生じる可能性があり,期待される臨床開発スケジュールを満たす能力に実質的な影響を与える可能性がある。もし私たちが代替供給計画を求めることを要求された場合、それによって生じる遅延と適切な代替案が見つからない可能性があり、私たちの業務に実質的で不利な影響を与える可能性がある。
知的財産権に関するリスク
私たちは私たちの1つまたは複数の候補製品または未来の製品のために十分な特許保護を獲得または維持することができないかもしれない。
私たちのビジネス成功は、私たちの候補製品と未来の製品、それらのそれぞれのコンポーネント、調合、製造方法および治療方法の特許、商標および商業秘密保護、および第三者の挑戦を成功させる能力を獲得し、維持する能力があるかどうかにある程度依存するだろう。私たちは、私たちの候補製品および将来の製品のために、私たちの戦略的重点分野および重要な市場で特許保護を強化し、終了した開発計画、分野、または戦略的重要性の低い市場に関連する既存の特許または特許出願を放棄する可能性があるために、日常業務中に私たちのグローバル特許の組み合わせを評価する。特許は、私たちが現在保留している特許出願に関連していないかもしれませんが、発行された特許は、後で無効または強制的に実行できないことが発見される可能性があり、私たちの候補製品や任意の未来の製品を十分に保護できないと解釈されたり、他の方法で私たちにどんな競争優位性を提供することもできません。バイオテクノロジーや製薬会社の特許地位は通常,複雑な法律や事実を考慮しているため,法律変化の影響を受ける可能性が高い。また,米国特許商標局(USPTO)や外国特許庁が特許を発行する際に適用される基準は,常に統一的または予測可能ではなく,法的変化の影響を受ける可能性もある。したがって、私たちは決定されたことのない特許出願で特許を発行することができないかもしれないし、私たちは異なる管轄区域で異なる範囲の特許請求を提起するかもしれない。したがって、将来の私たちの未来の製品やノウハウ(あれば)の保護の程度や、私たちの候補製品や未来の製品や他のノウハウについて十分な知的財産権保護を得ることができなかったことは、私たちの業務や利益を達成する能力に大きな悪影響を及ぼす可能性があります。
特許および/または特許出願の定期維持費、継続費、年会費および様々な他の政府費用は、特許および/または特許出願の有効期間内にいくつかの段階で米国特許商標局および米国以外の様々な政府特許庁に支払われる。米国特許商標局および外国政府特許庁は、特許出願および保守過程において、いくつかのプログラム、文書、費用支払い、およびその他の同様の規定を遵守することを要求する。私たちは私たちが所有または共同所有している特許と特許出願を遵守するのを助けるために、信頼性の良い法律事務所と他の専門家を招いて、私たちが許可を超えた特許と特許出願のコンプライアンスを達成するために協力者に依存している。
56
私たちの知的財産権は必ずしも私たちに競争優位性を提供するとは限らない。
私たちの知的財産権が提供する将来の保護の程度は不確定であり、知的財産権には限界があり、私たちの業務を十分に保護できないかもしれないし、競争優位性を維持することが許されないかもしれない。以下の例は説明のみである
上記のような私たちの競争優位に対するどんな脅威も私たちの業務に実質的な悪影響を及ぼす可能性がある。
私たちは世界各地で私たちの知的財産権を保護できないかもしれない。
世界のすべての国で候補製品の申請、起訴、強制執行、特許保護の費用は目を引くほど高く、私たちの米国以外のいくつかの国の知的財産権は米国の知的財産権を広く持っていないかもしれない。また、いくつかの外国の法律は米国の法律のように知的財産権を保護していない。そのため、第三者が米国以外のすべての国で私たちの発明を実践したり、米国または他の司法管轄区域内で米国または他の司法管轄区に販売、販売、使用、製造、輸入することを阻止することができないかもしれない。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの発明を使用して彼ら自身の製品を開発することができ、私たちの特許保護を持っている地域に他の侵害製品を輸出することもできますが、法執行力はアメリカほど強くありません。これらの製品は私たちの未来の製品と競争する可能性があり、私たちの特許や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。
いくつかの国では、多くの会社が知的財産権の保護、実行、保護に重大な問題に直面している。いくつかの国の法律制度、特に特定の発展途上国の法律制度は、特許、商業秘密、および他の知的財産権の実行、特にバイオテクノロジーおよび医薬製品に関連する特許を支持しておらず、これは、私たちの特許の侵害を阻止したり、私たちの専有権を侵害する方法で競争製品を販売することを困難にする可能性がある。
57
海外で私たちの特許権を強制的に執行する訴訟は巨額のコストを招く可能性があり、私たちの努力と注意を私たちの業務の他の側面から移し、私たちの特許は無効または狭義に解釈されるリスクに直面する可能性があり、私たちの特許出願は特許として発行されないリスクに直面し、第三者が私たちにクレームを提起する可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。したがって、私たちが世界各地で私たちの知的財産権の努力を実行することは、私たちの知的財産権から顕著な商業的優位性を得るのに十分ではないかもしれない.
私たちは私たちの特許を強制的に執行する人かもしれない特許の訴訟に巻き込まれるかもしれません。これは高価で、時間がかかり、成功していないかもしれません。挑戦されれば、これらの特許は無効であることが発見されるか、または強制的に実行できないかもしれません.
私たちまたは私たちの許可者は、私たちの候補製品および独自技術に関連する特許および他の知的財産権保護を得るための措置を取っているにもかかわらず、私たちのどの知的財産権も挑戦または無効にされる可能性がある。
競争相手は私たちの特許や私たちの許可側の特許を侵害するかもしれない。権利侵害や無許可の使用を阻止するために、私たちは権利侵害請求を要求されるかもしれません。これは高価で時間がかかるかもしれません。さらに、侵害訴訟では、裁判所は、訴訟中の特許を無効または強制執行できないと判断することができる。この場合、第三者は、ライセンス料や印税を支払うことなく、我々の技術を使用することができる可能性がある。我々の特許の有効性が支持されても,裁判所は我々の特許が相手の活動をカバーしていないことを理由に,相手が論争のある技術の使用を阻止することを拒否する可能性がある.さらに、第三者は、私たちの特許に対する私たちの権利、または私たちの特許の範囲または有効性に肯定的に挑戦することができる。任意の訴訟またはライセンス後の訴訟における不利な結果は、より多くの特許が無効が宣言されるか、強制的に実行できないか、または狭い解釈されるリスクに直面する可能性があり、私たちの任意の特許出願が発行された特許を生成できないリスクに直面させる可能性がある。私たちの知的財産権を実行するためのどんな努力も費用が高く、私たちの科学と管理者の努力を分散させることができるかもしれない。
例えば、私たちが第三者に対して法的訴訟を提起して、私たちの候補製品または未来の製品をカバーする特許を強制的に執行しようとする場合、被告は私たちの特許を無効および/または実行できないと抗弁または反訴することができる。米国や一部の外国の特許訴訟では,被告の抗弁や反訴が無効および/または実行不可能と主張することはありふれている。有効性を疑問視する理由は、新規性の欠如、明らかな特許不合格、優先権権利要件の喪失、書面記載の欠如、または実施の欠如など、いくつかの法定要件のいずれかを満たすことができなかったと言われている可能性がある。強制執行できないと主張する理由は、特許訴訟に関連する者が、米国特許商標局または開示義務の存在する適用外国人同業者に重要な情報を隠蔽するか、または起訴中に誤った陳述をすることである可能性がある。例えば、派生訴訟、権利訴訟、一方的再審、当事者間の審査、付与後審査または異議訴訟など、第三者によって開始された、または米国特許商標局または任意の外国特許当局によって開始された行政訴訟は、私たちの特許または私たちの許可者の発明権、所有権、権利要件の範囲または有効性に挑戦するために使用される可能性がある。これらの訴訟のいずれの不利な結果も、関連技術の使用を停止することを要求するか、または勝利者から許可権利を得ようとすることを要求するかもしれない。もし勝利者が商業的に合理的な条項で私たちに許可を提供しなければ、何か許可を提供すれば、私たちの業務は損害を受ける可能性があります。訴訟や行政訴訟は失敗する可能性があり、成功しても巨額のコストを招き、我々の経営陣や他の従業員の注意を分散させる可能性がある。
例えば、私たちの特許または私たちのライセンシーの特許の有効性に関する挑戦は、私たちおよび特許審査員が起訴中に知らない以前の技術が存在する可能性がある。被告が無効および/または強制執行できない法的主張に勝った場合、私たちは候補製品または将来の製品の少なくとも一部またはすべての特許保護を失うだろう。別の例として,我々の特許訴訟は誠実で善意の義務に従って行われていると考えても,訴訟当事者や米国特許商標局自体はこの点に基づいて我々の特許に疑問を提起することができる.このような挑戦の後の結果は予測できない。挑戦者が勝利しなくても、私たちの特許主張は、挑戦者および他の人に対してこのような権利主張を実行する能力を制限すると解釈される可能性がある。このような挑戦を弁護するコスト、特に外国でのコスト、およびそれによる任意の特許保護の喪失は、私たちの1つまたは複数の候補製品および私たちの業務に実質的な悪影響を及ぼす可能性がある。
私たちのいくつかの候補製品と未来の製品の特許保護と特許起訴は、第三者が特許を維持し、無効クレームを弁護する能力にかかっている。
私たちの候補製品または任意の未来の製品に関連するいくつかの特許は、ライセンサー、再ライセンシーが可能な人を含む、将来的には私たちの協力者によって制御される可能性がある。このような手配によれば、私たちは取った行動について私たちの協力者と協議し、起訴と実行の予備権利を持つ権利があるかもしれないが、私たちは過去に、将来的に私たちのポートフォリオにおける特許および特許出願の権利を起訴し、維持し、権利侵害者のためにそのような特許を主張する能力を放棄する権利があるかもしれない。例えば、XEN 901(現在NBI-921352と呼ばれる)の特許の組み合わせに関連する現在の権利、他の選択的Nav1.6阻害剤、およびデュアルNav1.2/1.6阻害剤は、Neurocrine Biosciencesにのみ許可され、Neurocrine Bioscienceは、製品侵害に関連する任意の訴訟を提起および制御する優先権を有する。
58
現在または将来、私たちの候補製品または将来の製品に関連する特許を提出、起訴、実行および/または擁護する権利のあるパートナーが、私たちの任意の候補製品をカバーする特許保護を適切に起訴および維持することができない場合、または私たちの任意の候補製品または将来の製品に関連する特許が侵害者によって主張された特許、または無効または強制的に実行できない主張を弁護し、そのようなカバー範囲に悪影響を与える場合、私たちは、そのような候補製品または将来の製品を開発および商業化する能力が悪影響を受ける可能性があり、競争相手の製造、使用、輸入、販売および/または販売競合製品を阻止することができないかもしれない。
我々の候補製品または将来の製品の販売、要約販売、輸入、製造または使用が第三者の特許または他の知的財産権を侵害していると主張することは、訴訟を回避しても、解決のために多くの時間とお金を必要とする可能性があり、私たちの候補製品の開発または商業化を阻止または延期する可能性がある。
私たちのビジネス成功は、他人の知的財産権や他の独占権を侵害することなく、私たちの候補製品の開発と、私たちの未来の製品を商業化する能力にある程度依存する。第三者は私たちまたは私たちの協力者が彼らの知的財産権を侵害、流用している、または他の方法で侵害していると主張するかもしれない。このような第三者は、既存の知的財産権に基づいている可能性があり、将来出現する知的財産権に基づく可能性がある賠償に同意する他の当事者に訴訟を提起する可能性があります。
私たちの候補製品は、発表時に第三者が保有する関連特許または特許出願がカバーされる可能性があり、私たちの候補製品をカバーすると断言される可能性のある関連特許または第三者が保有する特許出願を識別することもできない可能性がある。例えば、2000年11月29日までに提出された米国出願と、その日の後に提出された米国国外で提出されていないいくつかの出願は、特許発行前に秘密にされていた。米国および他のいくつかの司法管轄区の他の特許出願は、優先権を要求する最初の出願の約18ヶ月後に発行され、このような最も早い出願日は、一般に優先権日と呼ばれる。さらに、科学や特許文献に発表された発見は、実際の発見に遅れていることが多い。したがって、私たちまたは私たちの協力者が最初の発明または最初に私たちの候補製品またはその用途について特許出願を提出した人であるか、または私たちの候補製品が現在発表されているか、または未来に発表される特許を侵害しないかを決定することはできない。さらに、処理されるべき特許出願および公表された特許は、現在または将来の製品(あれば)またはその用途をカバーするために、後に修正することができるが、いくつかの制限された制限を受ける。
特許のカバー面は裁判所の解釈にかかっており、解釈は常に統一されているわけではない。もし私たちが特許侵害で起訴された場合、私たちは私たちの候補製品、私たちの未来の製品、または使用方法が関連特許の特許主張を侵害していないか、あるいは特許主張が無効または実行不可能であることを証明する必要があり、私たちはそれができないかもしれない。その無効性を証明することは難しいかもしれない。私たちがこれらの訴訟で勝訴しても、私たちは巨額のコストを生む可能性があり、私たちの経営陣や科学者の時間と注意力はこれらの訴訟に移される可能性があり、これは私たちの業務や運営に実質的な悪影響を及ぼす可能性がある。しかも、私たちはこのような行動を円満に達成するための十分な資源がないかもしれない。
我々は、派生手続、権利手続き、一方的再審、当事者間の審査、付与後審査、または反対手続のような行政訴訟手続を米国特許商標局または任意の外国特許機関に請求することによって、第三者特許請求の実行可能性または有効性に挑戦することを選択することができる。このような行政訴訟は費用が高く、私たちの時間や他の資源を消費するかもしれない。このような行政訴訟の費用は高いかもしれないし、私たちの時間や他の資源を消費するかもしれない。もし私たちが米国特許商標局、ヨーロッパ特許庁、または他の外国特許庁で有利な結果を得ることができなかった場合、私たちの候補製品または将来の製品が私たちの特許を侵害している可能性があると主張する第三者の訴訟に直面する可能性がある。
結果にかかわらず、特許侵害や他の知的財産権侵害の疑いを弁護するには費用が高く時間がかかる可能性がある。したがって、私たちが最終的に勝訴したり、早い段階で和解が成立したりしても、このような訴訟や脅威訴訟は私たちに予期しない多くの費用をもたらすかもしれない。また,訴訟や脅威訴訟は,我々の管理チームの時間や注意力に大きな要求を与え,会社の他の業務への追求を分散させる可能性がある.したがって、販売、使用、製造、販売、提供または当社の候補製品または将来の製品の侵害、流用、または他の方法で第三者知的財産権に違反すると主張することは、私たちの業務に重大な悪影響を及ぼす可能性があります。
第三者は私たちよりも複雑な知的財産権訴訟や訴訟の費用を効率的に負担することができる。また,訴訟に関する不確実性は,臨床試験に必要な資金を調達し,我々の内部研究計画を継続し,必要な技術の許可を得たり,候補製品を市場に出すのに役立つ戦略的協力を達成する能力に実質的な悪影響を与える可能性がある。
59
知的財産権訴訟の不利な結果は、私たちの研究開発活動および/または特定の製品を商業化する能力を制限するかもしれない。
未来のどんな知的財産権訴訟と他の行政訴訟は私たちの人員の追加費用と気晴らしにつながるだろう。知的財産権訴訟には不確実性が避けられず、私たちに不利な事件に弱さや欠陥があっても、私たちは敗訴する可能性がある。このような訴訟または訴訟における不利な結果は、私たちまたは任意の未来の戦略的パートナーを私たちの独自の地位を失い、私たちに重大な責任を負わせるか、または商業的に許容可能な条項では得られない可能性のあるライセンスを求めることを要求するかもしれません。全くなければ、それぞれが私たちの業務に実質的な悪影響を及ぼす可能性があります。もし私たちが第三者の知的財産権を侵害していることが発見された場合、裁判所の命令を含めて、権利侵害候補製品または将来の製品の開発、製造または商業化を余儀なくされる可能性がある。代替的に、権利侵害技術を使用して、権利侵害候補製品または将来の製品の開発、製造、または販売を継続するために、第三者から許可を得る必要があるかもしれない。
例えば、第三者が私たちに彼らの知的財産権を主張することに成功した場合、私たちは私たちの技術のいくつかの側面の使用を禁止されたり、ある未来の製品の開発と商業化が禁止されるかもしれない。あるいは、私たちは、故意に特許を侵害していることが発見されたら、原告に3倍もの損害賠償金と弁護士費を含む巨額の損害賠償金を支払うことを要求される可能性がある。別の選択として、私たちは、私たちの研究開発計画を継続したり、任意の結果製品を販売したりするために、知的財産権所有者からライセンスを取得する必要があるかもしれない。私たちはまた、第三者の知的財産権の侵害または他の方法での侵害を回避するために、私たちの候補製品または未来の製品を修正または再設計することを要求される可能性がある。これは技術的にも商業的にも不可能である可能性があり、これらの製品の競争力を低下させるか、またはこれらの製品の市場進出を延期または阻止する可能性がある。上記のいずれも、私たちの研究開発活動を制限し、1つ以上の候補製品を商業化する能力、または両方を制限することができる。
もし私たちが第三者から許可を求めることを選択したり要求された場合、私たちは許可料や印税、または両方を支払うことを要求されるかもしれないが、これは相当な費用になるかもしれない。このようなライセンスは許容可能な条項で提供されないかもしれないし、全く提供されないかもしれない。たとえ私たちまたは任意の未来の協力者が許可を得ることができても、これらの権利は非排他的である可能性があり、これは私たちの競争相手が同じ知的財産権を獲得することをもたらす可能性がある。最終的に、私たちは、実際に訴訟の結果、許容可能な条項で許可を得ることができないので、製品を商業化することを阻止されたり、裁判所によって、または他の方法で私たちの業務運営のいくつかまたは全ての態様を強制的に停止させることができません。また、私たちは知的財産権侵害のクレームによる重大な金銭的損失に責任があると判断されるかもしれない。将来的には、第三者から許可要項や許可要求を受ける可能性があり、知的財産権の侵害やライセンス料の滞納を主張しており、このようなクレームに法的根拠がなくても、このようなクレームを回避または解決することができない可能性がある。
Neurocrine Biosciencesまたは他の協力者が様々な場合、第三者によって制御される知的財産権の権利を許可または他の方法で取得する場合、例えば、第三者知的財産権がない場合、製品が国/地域で開発または商業化できない場合、または製品を開発または商業化するためにそのような第三者の権利を獲得することが決定された場合には有用である場合、我々の協力協定によれば、彼らは製品および場合によっては国/地域の方法で私たちに支払うお金を減らす資格がある。上記のどのような事件も私たちの業務を深刻に損なう可能性がある。
もし私たちがいかなる合意に違反しても、この合意に基づいて、私たちは第三者から私たちの候補製品や技術の使用、開発、商業化の権利を獲得し、私たちは私たちの業務に非常に重要な許可権を失うかもしれません。
既存または将来の許可および他の合意によれば、私たちは、開発および商業化義務、および潜在的なマイルストーン支払いおよび他の義務などの職務遂行義務を含む様々な義務の制約を受けているか、または制限されている可能性がある。もし私たちがこのような義務を履行できなかった場合、または他の方法で私たちの許可協定に違反した場合、私たちの許可パートナーは、適用の許可を全部または部分的に終了する権利があるか、または独占的許可を非独占的許可に変換する権利があるかもしれない。一般に、このようなライセンスの損失またはそれによって生じる任意のライセンス排他性は、私たちの業務、将来性、財務状態、および運営結果に重大な損害を与える可能性がある。
60
もし私たちが商業秘密と他の固有情報の不正流出を防ぐことができなければ、私たちの競争地位は損なわれるかもしれない。
特許に加えて、私たちは、私たちの競争地位を維持するために、ビジネス秘密、技術ノウハウ、および私たちの発見プラットフォーム、ビジネス戦略、および候補製品に関する独自の情報に依存しています。私たちの研究開発活動と商業活動の過程で、私たちはしばしば秘密保護協定によって私たちの固有の情報を保護する。例えば、我々が実験室、製造、臨床前開発または臨床開発製品またはサービスのサプライヤーまたは潜在的な戦略パートナーと話した場合、このようなセキュリティプロトコルが使用される。また、私たちのすべての従業員とコンサルタントは、わが社に参加する際に秘密保護協定と発明譲渡協定に署名しなければなりません。私たちの従業員、コンサルタント、請負業者、業務パートナー、または外部科学協力者は、私たちの商業秘密情報を故意にまたは無意識に漏らし、これらの秘密協定に違反したり、私たちの商業秘密が流用されたりする可能性があります。私たちの協力者はまたデータを発表する権利があるかもしれないが、私たちは発表前に特許保護を申請できないかもしれない。競争相手はこのような情報を利用するかもしれないし、私たちの競争地位は損なわれるだろう。さらに、私たちの従業員、コンサルタント、または請負業者が、私たちのために働いているときに他人が所有している知的財産権を使用する場合、関連するまたはそれによって生じるノウハウおよび発明の権利について議論が生じる可能性がある。第三者が私たちのいかなる商業秘密を不法に取得して使用することを強要するかは高価で時間がかかり、結果は予測できない。また、米国以外の裁判所は米国の裁判所のように商業秘密を保護したくないことがある。また,我々の競争相手は同等の知識,方法,ノウハウを自主的に開発することができる.もし私たちが私たちのノウハウや他の機密情報を秘密にすることができなければ、私たちが特許保護を受けたり、私たちの商業秘密情報を保護する能力が脅かされ、これは私たちの競争的地位に悪影響を及ぼすだろう。
さらに、私たちまたは私たちの許可者は、元従業員、協力者または他の第三者が、発明者または共同発明者として、私たちの所有または許可内の特許、商業秘密、または他の知的財産権において権益を有するというクレームを受ける可能性がある。例えば、私たちまたは私たちの許可者は、私たちの候補製品の開発に参加する従業員、コンサルタント、または他の人の義務衝突によって在庫紛争が生じる可能性があります。私たちの在庫または私たちまたは私たちの許可者が私たちの所有または認可内の特許、商業秘密、または他の知的財産権の所有権に挑戦するこれらおよび他のクレームに対抗するために、訴訟を提起する必要があるかもしれない。もし私たちまたは私たちの許可者がこのようないかなるクレームも弁護できなかった場合、金銭損害賠償を支払うことに加えて、私たちの候補製品に非常に重要な知的財産権の独占所有権や使用権のような貴重な知的財産権を失う可能性があります。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。上記のいずれも、当社の業務、財務状況、経営結果、見通しに重大な悪影響を及ぼす可能性があります。
米国特許法または他の国/地域法律の変化は、私たちの特許出願をめぐる起訴および私たちの特許の実行または保護の不確実性およびコストを増加させ、私たちの候補製品および未来の製品を保護する能力を弱める可能性がある。
私たちの成功は知的財産権、特に特許に大きく依存する。製薬と生物製薬会社の特許地位は非常に不確定である可能性があり、複雑な法律と事実問題に関連し、その重要な法律原則はまだ解決されていない。これまで,米国や他の国は,これらの分野の特許によって許容される権利要件の広さについて一致した政策を策定していない。しかも、国会や他の外国立法機関は私たちに不利な特許改革立法を通過するかもしれない。例えば,最近では特許法をどのように解釈するかが変化しており,米国特許商標局も国会も最近特許制度の大きな改革を行っている。米国最高裁判所のいくつかの裁決は現在、最高裁がいくつかの特許に対して明らかに否定的な傾向を示している。これらの決定の傾向や,USPTOが実施している特許性要求の変化は,我々の製品の特許を取得,維持および/または実行することがますます困難になる可能性がある。将来の特許法解釈の変化や特許法が法律の変化として制定される可能性があることを正確に予測することはできない。これらの変化は、私たちの特許、私たちの特許を得る能力、私たちの特許出願を起訴し、私たちの特許および/または私たちの協力者の特許およびアプリケーションを実行するコストに実質的な影響を与えるかもしれない。米国以外のこれらの分野の特許状況にも不確実性がある。米国や他の国の特許法や特許法解釈の変化は,我々の知的財産権の価値を低下させたり,我々の特許保護の範囲を縮小したりする可能性がある。したがって、私たちは、私たちが所有しているか、または私たちがライセンスを持っている特許または第三者特許において許容または強制される可能性のある権利要件の広さを予測することができない。例えば、欧州特許出願は、特許発行後に単一特許になることを選択することができ、単一特許裁判所(UPC)の管轄を受ける。単一特許の選択とUPCの作成はヨーロッパ特許実践の重大な変化である。UPCは新しい裁判所制度であるため、裁判所はほとんど前例がなく、UPCにおける任意の訴訟の不確実性を増加させている。
61
知的財産権訴訟や行政訴訟は不利な宣伝を招き、私たちの名声を損なう可能性があり、私たちの普通株の市場価格の下落を招く可能性がある。
任意の知的財産権訴訟または行政訴訟において、訴訟または手続の提起および聴聞結果、動議裁決および訴訟または手続の他の臨時裁決を公表することができる。もし証券アナリストや投資家がこれらの声明が否定的だと思うなら、私たちの既存製品、計画、または知的財産権の知覚価値は低下する可能性がある。したがって、私たちの普通株の市場価格は下がるかもしれない。このような声明はまた私たちの名声や私たちの未来の製品の市場を損なう可能性があり、これは私たちの業務に実質的な悪影響を及ぼすかもしれない。
もし私たちが候補製品の特許期間を延長することで米国の“ハッジ·ワックスマン法案”や同様の外国立法の保護を得なければ、私たちの業務は実質的な損害を受ける可能性がある。
FDAによる我々の候補製品の上場承認の時間、期限、詳細(ある場合)によると、“ハッジ·ワックスマン法案”によると、1つ以上の米国特許は、限られた特許期間を延長する資格がある可能性がある。しかし,適用の最終期限内に出願を提出できなかったこと,関連特許の満了前に出願を提出できなかったことや適用の要求を満たしていなかったことなどにより延期が得られなかった可能性がある.さらに、適用される期間または提供される特許保護範囲は、5年未満であってもよく、その数字が5年未満であれば、私たちの要求よりも少なくてもよい。
もし私たちが特許期間の延長を得ることができない場合、あるいはこのような延長の持続時間が私たちが要求したものよりも短い場合、私たちは私たちの製品を独占販売する権利があり、私たちの競争相手は私たちの特許が満期になった後に競争製品の承認を得るかもしれません。私たちの収入は大幅に減少するかもしれません。
もし私たちの商標が十分に保護されていなければ、私たちは興味のある市場で知名度を作ることができないかもしれません。これは私たちの業務に悪影響を及ぼすかもしれません。
私たちの現在と未来の商標は、私たちの会社名Xenonを含めて、私たちが運営·運営を計画している各市場に商標を登録していません。当社の会社名又は製品名の商標出願は登録が許可されていない可能性があり、当社の登録商標は維持又は強制執行されない可能性があります。商標登録過程で、私たちは拒否を受けるかもしれないが、私たちの反応はこのような拒否を克服できないかもしれない。第三者はまた、その製品にXenon名を使用して商標を登録しようと試みる可能性があり、このような使用を阻止することに成功しない可能性があります。また,米国特許商標局や多くの外国同様の特許庁では,第三者は係属中の商標出願に反対し,登録商標のキャンセルを求める機会がある。私たちの商標はこのような訴訟で生きられないかもしれない。もし私たちが私たちの商標のために登録されていなければ、私たちは第三者に対してこれらの登録を実行する時に他の場合よりも大きな困難に直面するかもしれない。
さらに、米国の候補製品で使用される任意の名称は、商標として登録または登録申請されているか否かにかかわらず、FDAの承認を受けなければならないことを提案する。ヨーロッパと他の国も似たような要求を持っている。FDA(または外国の同等管理機関)が私たちが提案した任意の製品名に反対する場合、私たちは、例えば、適用される商標法に従って、第三者の既存の権利を侵害せず、FDAのために受け入れられる適切な代替名を決定する必要があるかもしれない。これは追加料金がかかるかもしれません。
さらに、他の登録商標の所有者は、私たちが登録または未登録商標の変形を含む潜在的な商標侵害クレームを提起する可能性がある。もし私たちが商標侵害クレームを主張する場合、裁判所は私たちが主張する商標が無効または強制執行できない、または私たちが商標侵害を主張する側が関連商標に対する優先権を持っていると判断する可能性がある。この場合、私たちは最終的にこのような商標の使用を中止することを余儀なくされるかもしれない。もし私たちが私たちの商標に基づいて名称識別を確立できなければ、私たちは効果的に競争できないかもしれないし、私たちの業務は不利な影響を受けるかもしれない。
私たちの普通株式所有権に関連するリスク
私たちの普通株の市場価格は変動する可能性があり、私たちの普通株を購入した人は大きな損失を受けるかもしれません。
私たちの普通株はナスダックに上場して、取引コードは“XENE”です。私たちの普通株の市場価格は過去に変動しており、未来も変動するかもしれない。このような変動のため、投資家は私たちの普通株に投資する時に損失を受ける可能性がある。私たちの普通株の市場価格は以下の要素を含む多くの要素の影響を受ける可能性がある
62
また、株式市場全体、特にナスダックやバイオ製薬業界は、時々変動を経験しているが、この変動は往々にして対象会社の経営業績とは無関係である。例えば、新冠肺炎の大流行と上昇し続けるインフレと金利は著しい変動を招いている。これらの広範な市場と業界の変動は、私たちの経営業績にかかわらず、私たちの普通株の市場価格に悪影響を及ぼすかもしれない。最近のいくつかの場合、1株の市場価格が変動しており、当該株の保有者は株式を発行する会社に対して証券集団訴訟を起こしている。もし私たちのいかなる株主が私たちを提訴すれば、訴訟の弁護と処置は費用が高く、私たちの経営陣の時間と注意を分散させ、私たちの経営業績を損なう可能性があります。
将来的に私たちの普通株を売却して発行したり、普通株に変換したり、普通株に交換可能な証券は、私たちの株主を希釈し、私たちの普通株の市場価格を下落させる可能性があります。
私たちの普通株を大量に売却したり、これらの売却が発生する可能性があると考えたりするため、私たちの普通株の市場価格は下がる可能性があります。これらの売却、あるいはこれらの売却が起こりうる可能性は、将来的に私たちが適切だと思う時間と価格で株式証券を売却することをより困難にする可能性もある。
63
私たちの持分インセンティブ計画によると、私たちの報酬委員会(またはそのサブセットまたは許可)は、私たちの従業員、役員、およびコンサルタントに株式ベースのインセンティブを付与する権利がある。私たちの株式ベースの補償計画によると、将来的に株式オプションの付与と普通株の発行はすべての株主の希薄化を招き、私たちの普通株の市場価格に悪影響を及ぼす可能性がある。
さらに、将来的には、融資、協力協定、買収、訴訟和解、従業員手配、または他の関連する追加の普通株、優先株、または他の普通株に変換可能な株式または債務証券を発行する可能性がある。私たちはまた私たちが時々発行する事前融資承認株式証を行使する時に追加の普通株を発行することができる。私たちとJefferiesおよびStifelとの販売協定によると、私たちの“市場”株式発行計画に基づくいかなる発行も含めて、私たちの既存株主の株式を大幅に希釈し、私たちの普通株の市場価格の下落を招く可能性がある。
私たちはカナダの会社法と証券法の管轄を受けており、場合によってはこれらの法律が株主に与える影響はデラウェア州の会社法や米国証券法とは異なる。
私たちは“カナダ商業会社法”(CBCA)や他の関連法律の管轄を受けており、これらの法律の株主権利への影響は、米国司法管轄区域の法律によって管轄されている会社の権利とは異なる可能性があり、私たちの条項や定款とともに、要約買収、代理競争または他の方法で当社に対する支配権を得ることを遅延、延期、または阻止する効果がある可能性があり、あるいは買収側がこの場合提供したい価格に影響を与える可能性がある。CBCAとデラウェア州一般会社法(DGCL)との間の大きな違いは、(I)重大な会社取引(例えば、合併と合併、他の非常会社取引、または私たちの定款の改正)については、CBCAは一般に株主の3分の2の多数票を必要とし、DGCLは一般に多数票のみを必要とするが、(Ii)CBCAによると、5%以上の株式を保有し、株主総会で投票する権利を有する保持者は、株主特別会議の開催を要求することができ、DGCLにはこのような権利はない。また、我々の取締役会は、私たちの管理チームメンバーに責任を持って命令し、CBCAのいくつかの条項および私たちの定款や細則は、株主が取締役会メンバーを交換する難しさを増やすことで、現在の経営陣の任意の試みを変更または罷免することを挫折または阻止する可能性があります。これらの条項のいくつかは
私たちの条項、定款、CBCA、あるいは任意の適用されるカナダ証券法の遅延または制御権の変更を阻止する条項は、私たちの株主がその普通株からプレミアムを得る機会を制限する可能性があり、また、一部の投資家が私たちの普通株に支払いたい価格に影響を与え、それによって私たちの普通株の市場価格を下げる可能性がある。
アメリカの民事責任は私たち、私たちの役員、あるいは私たちの管理者に強制的に執行されないかもしれません。
私たちはCBCAによって管理されています。私たちの主な業務場所はカナダブリティッシュコロンビア州にあります。私たちの多くの役員や上級管理者はアメリカ国外に住んでいて、彼らの全部または大部分の資産と私たちの資産の全部または大部分はアメリカ国外にあります。そのため、投資家はアメリカ国内で私たちと私たちのいくつかの役員と上級管理者に訴訟手続きを送達することが難しいかもしれません。あるいは、アメリカ連邦証券法またはアメリカの任意の他の法律に基づく民事責任条項の訴訟を含む、アメリカの裁判所で私たちまたはこれらの人々に対する判決を実行することができます。米国連邦証券法または米国の任意の他の法律の民事責任条項のみに基づく権利は、カナダ裁判所(ブリティッシュコロンビア州裁判所を含む)で提起された元の訴訟または米国裁判所で得られた強制執行判決の訴訟で強制的に執行できない可能性がある。
64
私たちは証券集団訴訟の危険に直面している。
証券集団訴訟は、ある会社の証券市場価格が下落した後に提起されることが多い。このリスクは、バイオテクノロジー会社が近年大幅な株価変動を経験しているため、特に私たちと関連がある。私たちがこのような訴訟に直面すれば、巨額のコストを招き、経営陣の注意と資源を移転させる可能性があり、これは私たちの業務を損なう可能性があります。さらに、バイオテクノロジー会社に対する訴訟の増加は、取締役や上級管理者責任保険を獲得することをより困難かつ高価にする可能性があり、同じまたは同様の保険を得るために、低減された保険限度額および保証範囲を受け入れること、または同じまたは同様の保険を得るためにより高い費用を生成することを要求される可能性がある。
私たちの経営陣は現金の使用に広範な裁量権を持っており、私たちは私たちの現金を有効に使用できないかもしれません。これは私たちの運営結果に悪影響を及ぼすかもしれません。
私たちの経営陣は私たちの現金資源を運用する上で幅広い裁量権を持っている。株主は私たちの決定に同意しないかもしれません。現金資源の使用は私たちの経営結果を改善したり、私たちの普通株の価値を高めたりしないかもしれません。もし私たちがこれらの資金を有効に利用できなければ、私たちの業務に実質的な悪影響を与え、私たちの候補製品の開発を延期し、私たちの普通株の市場価格を下落させるかもしれない。さらに、それらを使用する前に、それらは大量の収入が生じないか、または値下がりする可能性のある投資に投資される可能性がある。
私たちは予測可能な未来に私たちの普通株に現金配当金を支払わないと予想している。
私たちは現在予測可能な未来に私たちの普通株にどんな現金配当金も支払うつもりはない。私たちは現在、私たちの未来のすべての収益を維持するつもりで、もしあれば、私たちの業務の成長と発展に資金を提供します。したがって、私たち普通株の資本増加(あれば)は、投資家が予測可能な未来に唯一の収益源である可能性がある。
アナリストが発表した報告書は、私たちの実際の結果とは異なる報告書での予測を含め、私たちの普通株の価格や取引量に悪影響を及ぼす可能性がある。
私たちの普通株の取引市場は、業界または証券アナリストが発表した私たちまたは私たちの業務に関する研究と報告にある程度依存している。私たちの一人以上のアナリストを追跡して私たちの会社に否定的な意見を発表すれば、私たちの普通株の株価は下落するかもしれません。もし一人以上のアナリストが私たちの研究報告を停止したり、私たちに関する報告書を定期的に発表できなかった場合、私たちは金融市場での可視性を失う可能性があり、これは逆に私たちの普通株の価格や取引量を低下させる可能性がある。
私たちの未償還事前融資権証明書は市場を公開していない。
私たちの未償還事前融資権証は公開取引市場を持っておらず、私たちは市場が発展しないと予想している。また、私たちは、発行されていない事前融資権証をナスダックまたは他の任意の国の証券取引所または国家公認の取引システムに上場するつもりはない。活発な取引市場がなければ、未発行の予融資権証の流動性が制限される。
65
一般リスク因子
不安定な市場と経済状況は私たちの業務や財務状況に深刻な悪影響を及ぼす可能性がある.
世界の信用と金融市場は時々極端な破壊を経験し、新冠肺炎の大流行と関係のある破壊を含み、その特徴は市場変動の激化、インフレ率の上昇、消費者の自信の低下、経済成長の低下、失業率の上昇、経済安定の不確定である。同様に、現在ウクライナとロシアの間および中東の衝突、および最近の世界の銀行業の失敗は、資本市場の変動をもたらし、さらなる世界経済結果が生じると予想される。流動性が限られている、違約、不良表現、その他の影響を及ぼす金融機関や私たちと業務往来のある当事者の不利な事態の発展、あるいはこれらのようなリスクに対する見方は、過去および未来に市場全体の流動性問題を招く可能性がある。例えば、2023年3月には、シリコンバレー銀行が閉鎖されて破産管理手続きに入り、その後、より多くの金融機関が破産管理手続きに入る。米国政府または他の管轄区の政府が将来他の金融機関が倒産した場合に介入することは保証されず、未保険資金を得る方法を提供することも、米国政府または他の管轄区の政府が直ちにそうすることを保証することはできない。信用や金融市場に再びこのような混乱が生じ、経済状況への自信が悪化すれば、我々の業務は悪影響を受ける可能性がある。株式市場や信用市場が将来的に大幅に悪化すれば、大流行、政治的動揺や戦争、あるいは世界の銀行業のさらなる不安定を含め、任意の必要な株式や債務融資をより困難にし、コストが高く、希釈度が高くなる可能性がある。適時かつ有利な条件で必要な融資を得ることができなければ、私たちの成長戦略、財務業績、および私たちの普通株の市場価格に重大な悪影響を及ぼす可能性があり、開発や商業化計画の延期または放棄を要求することができるかもしれない。さらに、この場合、私たちの現在の1つまたは複数のパートナー、サービスプロバイダ、製造業者、および他のパートナーは、生存できないか、または私たちへの約束を履行できない可能性があり、これは、時間通りおよび予算で運営目標を達成する能力に直接影響を与える可能性がある。
企業統治やその他の事項に関する法律、法規、投資家駆動の基準により、巨額のコストを招き続けることが予想されています.
“ドッド·フランクウォール街改革と消費者保護法”、2002年の“サバンズ·オックススリー法案”、“カナダ証券法”の条項、適用されたカナダ証券法、米国証券取引委員会、ナスダック、カナダ会社、適用されたカナダ証券監督管理機関が採択または提案したルールを含む上場企業の法律·法規に影響を与え、これらのルールの影響を評価し、その要求に応じているため、引き続き大きなコンプライアンスコストをもたらすであろう。
上場企業に適用される各種報告書やその他の要求を遵守するにもかなりの時間と経営陣の注意が必要である。将来、私たちの財務報告の内部統制の評価を要求に応じて発表できない場合、あるいは私たちまたは私たちの独立公認会計士事務所が、私たちの財務報告の内部統制が無効であると判断しなければ、この欠陥は私たちの業務や財務業績に悪影響を及ぼす可能性があり、私たちの普通株式価格はマイナスの影響を受ける可能性があります。新しい規則は、取締役や上級者責任保険を含むいくつかのタイプの保険をより難しくまたは高価にする可能性があり、私たちは、現在の保険範囲と同じか似たような保険を得るために、低減された保険限度額と保険範囲を受け入れさせられたり、より高い費用を発生させたりする可能性があります。これらの事件の影響はまた、私たちの取締役会や取締役会委員会に在任し、私たちの役員を務めることを引き付け、維持することを難しくするかもしれない。私たちは、このような規則および規則を遵守するために私たちが発生する可能性のある総コストまたはそのようなコストの時間を予測または推定することはできない。
また,米国証券取引委員会と適用されるカナダ証券規制機関は,最近の環境,社会,ガバナンスにおけるルール策定作業を含む様々なルール策定作業を定期的に行っている。様々な組織もこのようなESGテーマにおける会社の表現を測定しており,これらの評価の結果が広く宣伝されている.このような評価で良好な会社に投資するために専門的に投資するファンドはますます人気があり、主要機関投資家は、このようなESG措置の投資決定に対する重要性を公開している。ESG事項に関する追加のルールが採用されている場合、または投資家がESG事項への関心を高め続ける場合、私たちは、遵守しようと努力している間に、より高いコストが生じる可能性があり、規制機関またはそのような投資家が、私たちの努力が十分であると思うかどうかを決定することはできない。
項目1 B未解決従業員意見
ない。
66
プロジェクト1 C。ネットワークがあります安全だ
我々は、内部情報技術システムおよびネットワーク、ならびに第三者プロバイダおよび請負業者のシステムおよびネットワークに依存して、当社の業務活動に関する情報を取得、送信、記憶、処理する。私たちが業務を効率的に管理する能力は、私たちと私たちの第三者請負業者とサプライヤーの技術システムの安全性、信頼性、十分性に依存します。そこで,ネットワークセキュリティ脅威からのリスクの評価,識別,管理を目的とした情報セキュリティ計画を実施した.
私たちは少なくとも年に一回、ネットワークセキュリティと技術的リスクに関するリスク評価を行っている。我々のネットワークセキュリティリスク管理計画は,米国国家標準と技術研究所(NIST)が発表した基準を含む業界基準に基づいて策定されている。この計画のハイライトは:
我々のネットワークセキュリティ計画によって決定されたリスクは、潜在的な影響および発生の可能性を決定し、それに応じて緩和計画を策定して実施するために評価される。
取締役会監査委員会はネットワークセキュリティリスクの監視に対して最も重要な責任を持っている。監査委員会は、リスク管理、技術、金融を含む異なる専門知識を持つ取締役会メンバーで構成され、ネットワークセキュリティリスクを効果的に監督できるようにしている。経営陣の監督は、IT指導委員会、最高財務官(CFO)、最高法務官を含む実行管理層のサブセット、および我々の情報システム上級副総裁上級副総裁を含む機能に関する専門知識によって実行される。我々の上級副社長ISはIT指導委員会の主要メンバーであり、我々のネットワークセキュリティリスクの評価、監視、管理を担当している。彼は情報技術戦略や運営において20年以上の経験を持ち、背景には複数の会社でIT幹部を務めた豊富な経験が含まれている。上級副総裁、情報システム、首席財務官は、ネットワークセキュリティリスク評価、新たに出現した脅威および業界基準の変更、およびイベント報告および修復(あれば)に関する総合的な報告書を少なくとも毎年監査委員会に提出する。
上級副社長、IS、潜在的な脆弱性を含む、私たちの情報システムの定期的な監視プロセスを監視します。ネットワークセキュリティイベントが発生した場合、IT組織および指定された実行管理層メンバーは、既定のネットワークセキュリティイベント応答計画に従うべきである。この計画には、直ちに行動して脅威を抑制し、影響を軽減し、重要性を評価することが含まれており、将来のリスクを低減するために、是正措置の回顧と決定が求められている。
私たちは、2023年12月31日までの財政年度内に、以前に発見されたネットワーク攻撃や他の情報セキュリティ事件が、私たちの業務、財務状況、または運営に実質的な影響を与えないことを経験していませんが、未来にそのようなリスクや将来の重大な違反行為の大きな影響を受けないことを保証することはできません。これらのリスクの議論については、“プロジェクト1 A-リスク要因-当社のビジネスおよび業界に関連するリスク-当社のビジネスおよび運営は、第三者請負業者またはサプライヤーによって所有されている情報を含むネットワークセキュリティホール、システム障害、または当社のシステムおよび/または情報の他の被害など、実際または感知された情報セキュリティイベントにおいて影響を受ける可能性がある”を参照されたい
項目2.Pサーカス.サーカス
私たちの本社はブリティッシュコロンビア州のバーナビーにあり、現在私たちはそこで約53,023平方フィートの事務と実験室空間を占めています。レンタル期間は2032年6月に満期になります。私たちは現在毎月合計約145,353ドルの基本賃貸料、不動産税、公共地域維持費、管理費を支払い、大家は約67,932ドルの保証金を持っている。
私たちはマサチューセッツ州のニダムで約17,057平方フィートのオフィス空間を占めています。レンタル期間は2027年11月に満期になります。私たちが現在毎月支払っている基本賃貸料総額は約63,964ドルで、大家さんが持っている保証金は約187,627ドルです。
67
私たちの既存の施設は私たちの短期的な業務需要を満たすのに十分で、必要であれば、商業的に合理的な条項に従って追加の空間を提供すると信じています。
第3項それは.法律訴訟
私たちは時々法的手続きに巻き込まれたり、私たちの正常な業務過程でクレームを受けるかもしれない。当社は現在、いかなる法的手続きにも関与していませんが、当社の経営陣は、当社に不利であると判断すれば、当社の業務、財務状況、経営業績またはキャッシュフローに重大な悪影響を及ぼすことが合理的に予想されると考えています。結果にかかわらず,弁護や和解コスト,管理資源分流などにより,訴訟は我々に悪影響を与える可能性がある。
プロジェクト4それは.炭鉱安全情報開示
適用されません。
68
第II部
第5項それは.登録者普通株,関連株主事項及び発行者が株式証券を購入する市場
市場情報
私たちの普通株は2014年11月5日からナスダック全世界市場で取引され、取引コードはXENEである。2024年2月26日、私たちの普通株の最終売却価格は1株49.13ドルだった。
所持者
2024年2月26日現在、私たちの普通株式には約82人の登録所有者がいます。実株主数はこの記録保持者の数を超えており,実益所有者であるがその普通株はブローカーや他の被指名者が街頭名義で保有する株主が含まれている.
配当をする
私たちは私たちの普通株式または任意の他の証券の任意の現金配当金を発表したり支払ったりしたことがない。私たちは現在、予測可能な未来にすべての利用可能な資金と任意の未来収益(あれば)を維持し、私たちの業務運営のために、予測可能な未来に現金配当金を支払うことは期待されていないと予想している。将来的に現金配当金を派遣することは、適用法律に基づいて取締役会が適宜決定し、私たちの財務状況、経営業績、現在と予想される現金需要、既存または当時に存在する債務ツールの要求、および取締役会が関連すると考えている他の要素を含む様々な要素に依存する。
カナダの源泉徴収税率は25%(カナダが署名した任意の適用可能な所得税条約または条約の規定に基づいて減免されなければならない)であり、普通配当金の支払いまたは貸記、または所得税法(カナダ)によってカナダ住民または非カナダ住民の普通株式所有者の総金額に支払われるとみなされる。カナダの源泉徴収税は、私たちまたは私たちの支払い代理人がそうでなければ支払うべき配当金の金額から直接差し引かれ、カナダの係に送金されます。カナダ-米国税条約(1980)または条約によると、非カナダ住民株主は配当の実益所有者であり、条約の全利益を享受する資格があり、我々普通株が非カナダ住民に支払う配当に適用される源泉徴収税率は一般的に15%に低下し、非カナダ住民所有者が実益が少なくとも10%の議決権を有する株式を有する会社である場合、適用される源泉徴収税率は5%に低下する。条約についてアメリカ住民であるすべての人が条約のメリットを享受する資格があるわけではない。場合によっては、財政的に透明な実体(有限責任会社を含む)によって額を獲得した人は、条約に規定された福祉を受ける権利がない可能性がある。カナダは、これらの条約に基づいて利益を申請する能力を含む、カナダの多くの税金条約(ただし、条約に影響を与えない)に影響を与える税ベース侵食および利益移転を防止するための税収条約に関する措置を実行する多国間条約の署名国である。カナダではない住民は彼ら自身の税務顧問に相談して、適用された所得税条約や条約に基づいて減免を受ける権利があることを決定しなければならない。
株式補償計画に基づいて発行された証券
本プロジェクトが提供を要求した情報は,2023年12月31日までの財政年度終了後120日以内に提出される米国証券取引委員会に提出された2024年年次総会依頼書を参考にしている。
株式表現グラフ
本業績グラフは、改正された1934年“証券取引法”(以下、“取引法”という。)第18節の規定に基づいて提出されたものとみなされてはならず、また、当該出願書類に特に明確な説明が引用されていない限り、改正された1933年“証券法”又は“取引法”に基づいてXenon製薬会社が提出したいかなる出願書類にも引用されてはならない。
次の図は,2018年12月31日から2023年12月31日までの,(I)我々の普通株,(Ii)ナスダックバイオテクノロジー指数と(Iii)ナスダック総合指数に100ドルの現金を投資した総累積リターンの比較を示している。グラフ中の比較は米国証券取引委員会が必要としているものであり,我々の普通株の将来可能性を予測または指示するための表現ではない.この図は、すべての配当金が再投資されたと仮定している(これまで、私たちは何の配当も発表していない)。
69
発行者は株式証券を買い戻す
ない。
イットm 6. [保留されている]
70
項目7.経営陣の以下の問題の議論と分析財務状況及び経営実績
以下の議論および分析、ならびに本年度報告書の他の部分に含まれる総合財務諸表および付記を読まなければなりません。以下の議論には、リスクと不確定要素に関する前向きな陳述が含まれている。我々の実際の結果は、任意の前向き陳述において明示的または示唆された結果とは異なる可能性があり、これは、タイトル第I部分第1 A項である“リスク要因”に列挙された要因を含む様々な要因の結果である。議論全体において、文脈が別に説明や暗示がない限り、用語“キセノン”、“私たち”はキセノン製薬会社およびその子会社を意味する。
概要
我々は神経科学に専念する生物製薬会社であり,革新的な療法を発見·開発し,それを商業化し,神経や精神障害患者の生活を改善することに取り組んでいる。我々は、てんかんやうつ病を含む高度に満たされていない医療ニーズ分野を解決するための新しい製品ラインを進めている。
XEN 1101
XEN 1101は新型、有効なKV 7カリウムチャネル開口薬であり、てんかん、重篤な抑うつ障害或いはMDD、及び潜在的な他の神経疾患の治療に開発されている。
XEN 1101はてんかんを治療する
我々のXEN 1101 3期てんかん計画は、2 b期X−Tole臨床試験の後に設計されたX−TOLE 2とX−TOLE 3と呼ばれる2つの同じ3期臨床試験を含む。これらの多中心、無作為、二重盲検、プラセボ対照試験は毎日1回15 mg或いは25ミリグラムXEN 1101を補助治療としての臨床治療効果、安全性と耐性を評価しており、各研究は約360名の局所てんかん発作(FOS)患者がある。主要な治療効果の終点はXEN 1101がプラセボと比べ、ベースラインから二重盲検期(DBP)までの毎月のてんかん発作頻度の中央値パーセンテージ変化(MPC)である。X−TOLE 2の患者登録は2024年末から2025年初めに完了すると予想されている。
XEN 1101治療てんかん(原発性全身性強直−クローヌス発作)
著者らの3期X-Ackt臨床試験は原発性全般性強直クローヌス発作(PGTCS)の追加てんかん徴候に対する潜在的な監督管理提出を支持することを目的とした。この多中心、ランダム、二重盲検、プラセボ対照研究は約160名のPGTCS患者に対する補助治療として25ミリグラムXEN 1101の臨床治療効果、安全性と耐性を評価している。プラセボと比較して、主要な治療効果の終点はベースラインからXEN 1101までのDBPの毎月PGTCS頻度におけるMPCである。
てんかん用XEN 1101(オープンタグ拡張)
X-TOLE 2、X-TOLE 3あるいはX-Ackt中のDBPを完成させた後、条件を満たす患者は開放ラベル拡張或いはOLE研究に入ることができ、最長3年に達する。また,進行中のX−Toleステージ2 b OLEは5年から7年に延長されており,XEN 1101のために重要な長期データを生成し続けている。
大うつ病を治療するXEN 1101
2023年11月、著者らは168名の中重度MDD患者が1日に1回食物と共に10 mgと20 mg XEN 1101を服用する臨床有効性、安全性と耐性を評価した第2段階概念検証X-Nova臨床試験のエキサイティングなTOPLINE結果を報告した。研究の主な終点は6週目におけるモンゴメリ-オズバーグ抑うつ評価量表(MADRS)の変化である。プラセボとXEN 11020 mg群の間に明らかな用量反応と臨床的に有意であったが統計学的有意差は認められなかった(p=0.135)。他の二次治療効果の終点に、ハミルトン抑うつ尺度(HAM-D 17)、快感を測定するSnaith-Hamilton悦尺度(Shaps)と1週目のMADRSを含み、すべて統計学的意義があり、治療効果の効果が比較的に早いことを表明した。XEN 1101の全体的な耐性は良好であり、すべての治療単位で報告された全体的な不良イベントの発生率及び類似の薬剤停止率はすべて類似している。2つのXEN 1101治療群では重篤な有害事象は報告されておらず,XEN 1101は有意な体重増加に関係なく,参加者も有意な性機能障害を報告していない。著者らはMDDにおけるXEN 1101の末期臨床開発を積極的に計画しており、2024年に第三段階臨床計画を開始する予定である。私たちはまたXEN 1101の未来の発展の他の潜在的な指標を評価している。
また,我々は西奈山イカン医学院と協力して,研究者が支援しているXEN 1101の第2段階概念検証,ランダム,平行試験,プラセボ対照の多地点研究を支援し,約60名の被験者のMDDを治療している。TOPLINE X−NOVA結果のその他の情報については、“最新の進展”を参照されたい。
71
他ルート商機
我々は,我々の広範なイオンチャネルの専門知識と薬物発見能力を利用して,検証された薬物目標を決定し,新たな候補製品を開発し続けている。最近の重点はKV 7,Nav1.1,Nav1.7に対する内部開発候補製品である。
協力計画:NBI-921352
我々はNeurocrine Biosciencesと協力しててんかんの治療法を開発している。Neurocrine BiosciencesはXEN 901の独占的な許可を有し,XEN 901は現在NBI−921352と呼ばれ,選択的Nav1.6ナトリウムチャネル阻害剤である。2023年11月、Neurocrine Biosciencesは、成人FOS患者におけるNBI-921352の第2段階臨床試験は、てんかん発作頻度の有意な減少を証明できなかったと報告した。Neurocrine生物科学社は,FOSにおいてNBI−921352をさらに開発する計画はないと指導している。現在,2歳から21歳のSCN 8 A発育性とてんかん性脳症を有する患者へのNBI−921352の応用を評価する第2段階臨床試験が行われている。
私たちは主に株式証券を売却することで、2023年11月に公開された普通株と事前資本権証から約3.24億ドルの純収益を獲得し、私たちの特許所有者と協力者から資金を得ること、債務融資を含む私たちの業務に資金を提供する。2023年12月31日までの年度では何の収入も確認されていないが,2022年12月31日と2021年12月31日までの年度では,連携協定からの収入はそれぞれ940万ドルと1840万ドルであった。今まで、私たちは何の製品も販売を許可されていませんし、製品販売から何の収入も得ていません。製品開発に成功し,規制機関の候補製品の承認を得るまで製品販売から収入を得ることはないと予想され,数年(あれば)かかると予想され,その結果には大きな不確実性がある。
私たちは引き続き私たちの候補製品を開発し、予測可能な未来に運営に資金を提供するために追加の資金を必要とするだろう。成立以来、私たちは毎年純損失を出しており、予測可能な未来にも純損失が続くことが予想される。2023年12月31日現在、2022年12月31日と2021年12月31日までの純損失はそれぞれ1.824億ドル、1.254億ドル、7890万ドルだった。2023年12月31日現在、私たちの累計赤字は6.651億ドルです。我々のほとんどの純損失は,我々の研究や開発計画に関するコストと我々の運営に関する一般的かつ行政的コストによるものである.私たちは特に私たちの運営費用が大幅に増加すると予想しています
財務運営の概要
収入.収入
今まで、私たちの収入は主に協力と許可協定から来ていて、私たちは製品販売から何の収入も得ていません。候補製品の開発努力が成功し、規制部門の承認を得られれば、将来的に製品販売から収入を得ることができるかもしれない。私たちは私たちの候補製品の商業化と販売からどの程度収入を得るか、いつ、あるいはどの程度収入を得るか予測できない。私たちのどんな候補製品も規制部門の承認を得ることができないかもしれない。
私たちはまた、Neurocrine Biosciencesとの許可および協力協定、またはNeurocrineとの協力、本年度報告Form 10-Kの他の部分の連結財務諸表の“業務-協力、商業および許可協定”および“付記11”に記載された、私たちの任意の候補製品または知的財産権との許可または協力によって生じる支払いによって収入を生成する可能性がある。私たちはNeurocrineが協力して未来のマイルストーンや特許使用料支払いの時間を保証することもできないし、私たちがこのような支払いを受けることを保証することもできない。
72
次の表は、2023年12月31日、2022年、2021年12月31日までに年度確認された収入の概要(単位:千):
|
十二月三十一日までの年度 |
|
|||||||
|
2023 |
|
2022 |
|
2021 |
|
|||
神経分泌生物科学: |
|
|
|
|
|
|
|||
取引価格の確認 |
$ |
— |
|
$ |
372 |
|
$ |
3,715 |
|
研究と開発サービス |
|
— |
|
|
1,938 |
|
|
6,452 |
|
一里塚払い |
|
— |
|
|
7,124 |
|
|
5,270 |
|
Pacira BioSciences: |
|
|
|
|
|
|
|||
一里塚払い |
|
— |
|
|
— |
|
|
3,000 |
|
*総収入 |
$ |
— |
|
$ |
9,434 |
|
$ |
18,437 |
|
Neurocrine協力条項によると、私たちは2019年12月に3,000万ドルの前払い現金と2,000万ドルの普通株式投資を受け取りました。この手配の全体的な取引価格は計量されて何らかの履行義務に割り当てられ,収入はこれらの履行義務が完了したときに確認される.2021年9月、規制機関は欧州規制機関によるNBI-921352成人局所てんかん発作の臨床試験申請を承認し、合計1,000万ドルの記念碑的支払いを受け、現金450万ドル、普通株投資550万ドルであった。また,2022年1月,米国食品医薬品局(FDA)の承認によりSCN 8 A−DIE研究者を2歳から11歳までの研究対象に拡大し,合計1,500万ドルのマイルストーン支払いを獲得し,現金形式は675万ドル,普通株式投資は825万ドルであった。いずれの場合も、株式投資は発行日に公正価値で計量され、それにより生じる現金支払割増が収入として確認される。サービスを提供する際には,サービスを研究·開発して公平な市場価値で収入として確認する.この研究協力は2022年6月に完了した。
2021年12月31日までの年間で300万ドルの収入が確認され,Pacira BioSciences,Inc.が達成したPCRX 301の開発と商業化の世界的権利協定に関連しており,FDAが新薬研究申請を承認した100万ドルのマイルストーンと1 b期の臨床試験開始の200万ドルのマイルストーンを含む。2023年12月31日および2022年12月31日までの年間では,Pacira合意に関する収入は確認されていない。2022年11月、Pacira BioSciencesはPCRX-301の臨床開発を追求しない戦略決定を下した。
運営費
次の表は、2023年12月31日、2022年12月31日、2021年12月31日までの年間運営費(単位:千)をまとめたものです
|
|
十二月三十一日までの年度 |
|
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|
|||
研究開発 |
|
$ |
167,512 |
|
|
$ |
105,767 |
|
|
$ |
75,463 |
|
|
一般と行政 |
|
|
46,542 |
|
|
|
32,810 |
|
|
|
21,967 |
|
|
総運営費 |
|
$ |
214,054 |
|
|
$ |
138,577 |
|
|
$ |
97,430 |
|
|
研究と開発費
研究および開発費用とは、任意の取得または許可された候補製品または技術、および私たちのパートナー候補製品を支援するコストを含む、我々の特許候補製品および我々の薬物発見努力を開発することによって生じるコストである。
研究·開発費用には、研究·開発活動を行うために発生するコストが含まれる
73
プロジェクトに特化した費用は,われわれの臨床開発候補に直接起因するコストを反映しており,そのために多くの費用が生じている。すべての残りの研究と開発費用は臨床前,発見,その他の計画費用に反映されている。所与の時間に、私たちはいくつかの肯定的な早期研究と薬物発見計画を持っている。私たちの人員とインフラは通常複数のプロジェクトに配置されており、任意の単独の内部早期研究や薬物発見計画と直接関連していない。したがって、特定のプロジェクトに基づいて内部早期研究および内部薬物発見計画の財務情報を維持することはない。
私たちのすべての研究開発費は発生した費用で計算されます。私たちはサービスを提供する前に研究開発サービスのために支払ったお金を私たちの総合貸借対照表に前払い資産として記録し、サービスを提供する際に費用を計上します。いくつかの開発活動のコストは,我々のプロバイダと第三者サービスプロバイダが提供してくれた情報やデータを用いて特定のタスクを達成する進捗を評価することによって確認される.
我々の計画が開発の後期段階に入るにつれて,臨床試験を継続し,我々の内部薬物発見計画を臨床前開発に進め,我々の早期研究を継続することにより,我々の将来の研究·開発費用は大幅に増加することが予想される。製造への投資を含めて,我々の候補製品の開発に関する研究·開発活動に投資し続けているからである。臨床開発後期にある候補製品は通常,臨床開発早期段階の候補製品よりも高い開発コストを有しており,これは主に後期臨床試験の規模,範囲,持続時間が増加しているためである。
臨床開発スケジュール、規制承認の可能性および商業化と関連コストは不確定であり、見積もりが困難であり、大きな差がある可能性がある。必要な臨床研究を行い、監督部門の許可を得る過程は高価で時間がかかり、著者らの候補製品の開発成功も非常に不確定である。したがって、私たちは、私たちの任意の候補製品の臨床前および臨床開発を完成させるために必要な性質、時間、およびコストを正確に推定または知ることができず、私たちがいつ、およびどの程度の候補製品の商業化および販売から収入を得ることができるか、または利益を達成できるかを知ることができない。
一般と行政費用
一般及び行政支出は主に人事関連の支出を含み、行政、財務、法律、業務発展、商業及び行政機能に従事する従業員の賃金、福祉及び株式給与、保険料、監査、税務及び法律サービスの専門費用、知的財産権の維持及び申請に関連する費用、商業化のために準備するために発生した費用、及び研究開発費に計上されていない分配施設関連及び情報科学技術費用を含む。
継続的な研究活動や候補製品の開発を支援するための経営活動の拡大に伴い,事業化に備えて将来の一般的かつ管理費が増加することが予想される。また、増加した会計、監査、法律、規制、コンプライアンスと役員と役員の保険コスト、および上場企業の運営に関連する投資家や広報費用を発生させていきます。
その他の収入(費用)
利息収入利息収入には私たちの現金と投資残高から稼いだ収入が含まれています。私たちは私たちの金利収入が変動し続けると予想しています。これは私たちの現金と投資残高と金利にかかっています。
証券取引の未実現公正価値収益(損失)取引証券は公正価値で入金される。取引証券の未実現公正価値収益(損失)は期内投資の市場定価変化と関係がある。取引証券の未実現公正価値収益(損失)は引き続き変動し、これは私たちの投資残高と市場収益率に依存すると予想される。
為替損益。純為替損益には、為替変動が我々の通貨資産や負債に与える影響による損益が含まれており、これらの資産や負債はドル以外の通貨(主にカナダドル)で価格を計算している。私たちは引き続き多額の費用をカナダドルで支払い、外貨変動に関連するリスクの影響を受け続ける。
重要な会計政策と重大な判断と見積もり
私たちの経営陣は私たちの財務状況と経営結果の討論と分析は私たちの連結財務諸表に基づいています。これらの報告書はアメリカ公認会計原則あるいはアメリカ公認会計原則に従って作成されています。総合財務諸表を作成する際には、報告期間内に報告された資産および負債額および発生した収入および費用に影響を与える推定と仮定を行う必要がある。我々は,我々の歴史的経験,既知の傾向,当時の状況では合理的と考えられる様々な他の要因に基づいて推定し,これらの要因の結果は資産や負債の帳簿価値の判断の基礎を構成しているが,これらの資産や負債の帳簿価値は他の源からは明らかではない。異なる仮定または条件では、実際の結果は、これらの推定値とは異なる可能性がある。
74
我々の歴史と将来の業績を十分に理解し評価する上で、最も重要な会計政策は、収入確認、研究開発コスト、株式に基づく報酬であると考えられる。より多くの情報を知る必要がある場合は、本年度報告書10-K表の他の部分に記載されている連結財務諸表の“付記3”を参照してください
収入確認:
私たちが受け取った収入の規模と性質のため、収入確認は重要な会計推定である。
私たちの主な収入源は払い戻しできない前金、研究開発サービス資金、マイルストーン支払い、許可と協力協定下の印税です。
お客様に商品やサービスを提供する契約では、1つ以上の履行義務があり、それぞれの履行義務が異なるかどうかを確認するために評価しなければなりません。そして、契約項の下の対価格はそれぞれの相対独立販売価格に応じて異なる履行義務の間に分配される。各成果の推定独立販売価格は、納入可能な成果が定期的に独立して販売される販売価格の最適な推定値を反映し、他人に販売する際に商品やサービスの市場価格を参照して決定されるか、または独立販売価格なしに調整された市場評価方法によって決定される。私たちは一般に、義務履行の見積もり期間内または潜在的利益が顧客に移転している間に払い戻しできない前払いからの収入を確認します。私たちは報告期間ごとに進捗指標を評価し、必要に応じて業績指標と関連収入確認を調整する。
関連商品やサービスの制御権が我々の顧客に転送された場合,それぞれの異なる履行義務に割り当てられた対価格が収入として確認される.我々は,ライセンシーが提供する研究·開発サービスの対価格を代表して現行の市場価格と一致するレートでこのような活動を行う際に確認した。確認された累計収入が大きく逆転しない可能性が高いことが確認された場合,リスク実質的業績マイルストーン(販売ベースのマイルストーンを含む)に関する対価格が収入として確認された。その後の各報告期間の終了時に、これらのマイルストーンを実現する可能性を再評価し、必要に応じて全体の取引価格の推定を調整します。知的財産権許可に関連する販売ベースの使用料は、収入基準において特定の例外があり、すなわち、顧客のその後の販売または使用が発生する前に、対価格は取引価格に含まれず、収入で確認される。
研究開発コスト:
研究開発コストは重要な会計政策であり、コストが大きいため、しかもサプライヤーの業績割合を推定し、第三者の計算と前払いの研究開発費用を計算する必要がある。
我々は,臨床前研究と臨床試験および製造活動を含む第三者サービスプロバイダによる研究と開発活動の見積りコストを計上した。サード·パーティ·サービス·プロバイダは、通常、適切な計算すべき項目を決定するために、比例業績の推定値を提供する。計算すべき項目の十分性を決定する際には,提供されるサービスレベル,研究進捗状況(活動の段階や完了状況を含む),および契約コストに基づいて進捗状況を分析する.これらの手配に基づいて関連サービスを受信する前に第三者に支払われるお金は、サービスを提供する前に前払い料金と表記される。私たちの歴史的前払いと計算された見積もりは実際のコストと実質的な差がない。
株式ベースの報酬:
株式ベースの報酬は、株式ベースの報酬費用の計算には大量の仮定と多くの仮定が必要であるため、重要な会計推定である。
私たちは私たちの株式オプション計画に基づいて従業員、コンサルタント、役員、上級管理者に株式オプションを付与します。給与費用は公正価値法で入金される。我々は、オプションの期待寿命および株式の期待変動率を含むいくつかの仮定を推定することを要求するBlack-Scholesオプション定価モデルを用いて、オプションの公正価値を計算する。予想変動率は,期待期限仮定に見合った一定時間に基づいて計算された我々普通株の履歴変動率に基づいている。私たちの株式オプションの期待期限は私たちの既存の履歴データを利用して決定されました。没収が発生した時に確認します。私たちは直線法を使用してオプションの帰属中に株式オプションの公正価値を償却する。
75
経営成果
2023年、2022年、2021年12月31日終了年度比較
次の表は、2023年、2023年、2022年、2021年12月31日までの年間業務結果とこれらの項目の変化(千計)をまとめています
|
|
年.年 一段落した 12月 31, |
|
|
変わる |
|
変わる |
|
|||||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|
増加/(減少) |
|
増加/(減少) |
|
|||||
収入.収入 |
|
$ |
— |
|
|
$ |
9,434 |
|
|
$ |
18,437 |
|
|
$ |
(9,434 |
) |
$ |
(9,003 |
) |
研究開発費 |
|
|
167,512 |
|
|
|
105,767 |
|
|
|
75,463 |
|
|
|
61,745 |
|
|
30,304 |
|
一般と行政費用 |
|
|
46,542 |
|
|
|
32,810 |
|
|
|
21,967 |
|
|
|
13,732 |
|
|
10,843 |
|
その他: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
利子収入 |
|
|
27,620 |
|
|
|
8,713 |
|
|
|
466 |
|
|
|
18,907 |
|
|
8,247 |
|
取引証券の未実現公正価値収益(損失) |
|
|
3,550 |
|
|
|
(2,934 |
) |
|
|
(719 |
) |
|
|
6,484 |
|
|
(2,215 |
) |
為替損益 |
|
|
199 |
|
|
|
(1,891 |
) |
|
|
358 |
|
|
|
2,090 |
|
|
(2,249 |
) |
所得税前損失 |
|
$ |
(182,685 |
) |
|
$ |
(125,255 |
) |
|
$ |
(78,888 |
) |
|
$ |
(57,430 |
) |
$ |
(46,367 |
) |
収入.収入
2023年12月31日までの1年間で、2022年同期に比べて940万ドル減少した。減少の主な原因はNeurocrine協力である;前金に関するすべての業績義務は2022年3月に完成し、収入が40万ドル減少し、私たちが協力した研究部分は2022年6月に完成し、研究開発サービス収入を190万ドル減少させた。また,我々は2022年にNeurocrine協力で710万ドルのマイルストーンを確認したが,2023年にはこの協力でマイルストーンを確認しなかった.
2022年12月31日までの1年間で、2021年同期に比べて900万ドル減少した。減少の主な原因はNeurocrine協力である;前金に関するすべての業績義務は2022年3月に完成し、収入が330万ドル減少し、私たちが協力した研究部分は2022年6月に完成し、研究開発サービス収入を450万ドル減少させた。また、私たちは2021年に私たちのPacira合意に基づいて300万ドルのマイルストーンを確認したが、2022年にはこの合意は何のマイルストーンも認めなかった。
研究と開発費
次の表は,2023年12月31日まで,2022年と2021年までの研究と開発費およびこれらの項目の変化(千単位)をまとめたものである
|
|
年.年 一段落した 12月 31, |
|
|
変わる |
|
変わる |
|
|||||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|
増加/(減少) |
|
増加/(減少) |
|
|||||
直接外部コスト: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
XEN 1101 |
|
$ |
89,303 |
|
|
$ |
37,367 |
|
|
$ |
21,664 |
|
|
$ |
51,936 |
|
$ |
15,703 |
|
XEN 496 |
|
|
5,152 |
|
|
|
14,765 |
|
|
|
13,017 |
|
|
|
(9,613 |
) |
|
1,748 |
|
臨床前発見その他の計画は |
|
|
15,552 |
|
|
|
12,711 |
|
|
|
13,342 |
|
|
|
2,841 |
|
|
(631 |
) |
間接コスト: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
関係者(株ベースを含む) |
|
|
45,981 |
|
|
|
32,447 |
|
|
|
21,225 |
|
|
|
13,534 |
|
|
11,222 |
|
施設や他の未分配の研究と |
|
|
11,524 |
|
|
|
8,477 |
|
|
|
6,215 |
|
|
|
3,047 |
|
|
2,262 |
|
研究開発費 |
|
$ |
167,512 |
|
|
$ |
105,767 |
|
|
$ |
75,463 |
|
|
$ |
61,745 |
|
$ |
30,304 |
|
76
2022年同期と比較して、2023年12月31日までの1年間で、研究開発費は6170万ドル増加した。この増加は、主に我々のXEN 1101計画および人員関連コストによるものであるが、これは、後期開発を支援する従業員数の増加と、より高い公正価値で付与されたオプション数の増加による株式ベースの報酬支出であるが、XEN 496の支出減少分はこの増加を相殺している。XEN 1101の増加は著者らの第三段階てんかん臨床試験が現場起動と患者登録を行っており、現在と未来の臨床試験と潜在的なNDA申請の製造活動の増加、及び著者らのX-Nova第二段階MDD臨床試験の完成を支持しているためである。XEN 496の減少は、EPIK臨床試験および開放ラベル拡張を支持する外部コストの低下によるものであり、これは、XEN 496の臨床開発を追求しないことに決定したためである。
2021年同期と比較して、2022年12月31日までの1年間で、研究開発費は3030万ドル増加した。この増加は、主に我々のXEN 1101計画および人員関連コストによるものであり、これは、後期開発を支援する従業員数の増加と、より高い公正価値で付与されたオプション数の増加による株式ベースの報酬支出である。XEN 1101の増加は、我々のてんかん3期臨床試験の開始に関連する費用、X-Tole開放ラベル拡張をサポートする持続コスト、および私たちのX-Nova 2期MDD臨床試験を含む。
一般と行政費用
表は,2023年12月31日,2022年,2021年12月31日終了年度までの一般費用と行政費用およびこれらの項目の変動状況(千計)をまとめたものである
|
|
年.年 一段落した 12月 31, |
|
|
変わる |
|
変わる |
|
|||||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|
増加/(減少) |
|
増加/(減少) |
|
|||||
関係者(株ベースを含む) |
|
$ |
32,166 |
|
|
$ |
22,814 |
|
|
$ |
14,123 |
|
|
$ |
9,352 |
|
$ |
8,691 |
|
専門と相談料 |
|
|
8,760 |
|
|
|
5,189 |
|
|
|
4,367 |
|
|
|
3,571 |
|
|
822 |
|
他にも |
|
|
5,616 |
|
|
|
4,807 |
|
|
|
3,477 |
|
|
|
809 |
|
|
1,330 |
|
一般と行政費用 |
|
$ |
46,542 |
|
|
$ |
32,810 |
|
|
$ |
21,967 |
|
|
$ |
13,732 |
|
$ |
10,843 |
|
2023年12月31日までの1年間で、2022年同期に比べて一般·行政費が1370万ドル増加した。成長は主に,我々の拡大する研究開発活動を支援するために増加した従業員数による人事関連コストと,より高い公正価値で付与されたオプション数の増加や専門や相談費の増加による株式ベースの報酬支出である。
2022年12月31日までの1年間で、2021年同期に比べて一般·行政費が1080万ドル増加した。成長は主に,我々の拡大する研究開発活動を支援するために増加した従業員数による人事関連コストと,より高い公正価値で付与されたオプション数の増加や求人費用,保険料および専門·相談費の増加による株式ベースの報酬支出である。
その他の収入
次の表は、2023年12月31日現在、2022年12月31日現在、2021年12月31日までの他の収入およびこれらの項目の変化(千単位)をまとめています
|
|
年.年 一段落した 12月 31, |
|
|
変わる |
|
変わる |
|
|||||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|
増加/(減少) |
|
増加/(減少) |
|
|||||
利子収入 |
|
$ |
27,620 |
|
|
$ |
8,713 |
|
|
$ |
466 |
|
|
$ |
18,907 |
|
$ |
8,247 |
|
取引証券の未実現公正価値収益(損失) |
|
|
3,550 |
|
|
|
(2,934 |
) |
|
|
(719 |
) |
|
|
6,484 |
|
|
(2,215 |
) |
為替損益 |
|
|
199 |
|
|
|
(1,891 |
) |
|
|
358 |
|
|
|
2,090 |
|
|
(2,249 |
) |
その他の収入 |
|
$ |
31,369 |
|
|
$ |
3,888 |
|
|
$ |
105 |
|
|
$ |
27,481 |
|
$ |
3,783 |
|
2023年12月31日までの1年間で、2022年同期に比べて他の収入は2750万ドル増加した。この増加は主に利息収入が1890万ドル増加したことによるものであり、これは有価証券残高の増加および投資市場収益の増加によるものである。また,取引証券の市場収益率の変化により,取引証券の未実現公正価値収益は650万ドル増加したが,取引証券残高の減少分で相殺された.カナダドル価値の変動やカナダドル建ての現金と現金等価物残高の減少により,為替収益(損失)も210万ドル変化した。
2021年同期と比較して、2022年12月31日までの1年間で、他の収入は380万ドル増加した。これは主に有価証券残高が増加し、利息収入が820万ドル増加したためだ
77
投資の市場収益率を高めることができますカナダドル建ての現金と現金等価物残高の増加及びカナダドル価値の低下により、外国為替損失は220万ドル増加し、取引証券公正価値の未実現損失は220万ドル増加し、取引証券残高の増加と投資市場収益率が増加し、部分的にこの増加を相殺した。
流動性と資本資源
流動資金源
これまで、私たちは主に株式証券の売却、協力と許可協定から得られた資金、債務融資を通じて、私たちの運営に資金を提供してきました。初公募株から2023年12月31日まで、主に株式証券の発行により合計13億ドルを超える現金純収益を集め、2023年11月に公募株で普通株と事前融資の引受権証を売却して得られた約3.24億ドルの純収益を含む。2023年12月31日現在、9.309億ドルの現金と現金等価物と有価証券を持っています.
私たちの協力者たちが彼らとの合意に基づいてマイルストーン支払いのいかなる義務を支払うかを除いて、私たちは外部資金源を約束していない。私たちが相当な製品収入を生み出すことができる前に、もしあれば、私たちは協力協定と株式または債務融資の組み合わせで私たちの現金需要を満たす予定だ。
私たちは2020年8月にJefferies LLC、Stifel、Nicolaus&Company、InCorporation、またはStifelと2022年3月に改正された“市場で”の株式発行販売協定を締結し、この合意に基づいて、私たちは時々私たちの普通株を売るかもしれない。2020年8月6日に米国証券取引委員会に提出された目論見書補編によると、2021年1月に合計73.3万株の普通株を売却し、手数料と取引費用を差し引いた収益は1,070万ドルとなった。2022年3月1日に米国証券取引委員会に提出された新募集説明書の補編によると、2020年8月の現金自動支払機の代わりに、2.5億ドルの普通株や、2022年3月のATMを時々販売する可能性がある。2023年12月31日現在、2022年3月のATMにより、合計855,685株の普通株が売却され、手数料と取引費用を差し引いた収益は2950万ドルだった。
資金需要
設立以来、私たちは重大な運営損失が発生した。2023年12月31日現在、私たちの累計赤字は6.651億ドルです。私たちは引き続き収入を超える大量の支出が発生する見通しで、今後数年で運営赤字が発生すると予想される。私たちの純損失は四半期ごとと毎年大きく変動するかもしれません。私たちは予測可能な未来に、私たちが私たちの候補製品の研究と臨床前と臨床開発を続けるにつれて、私たちの現在と潜在的な候補製品の研究範囲を拡大する;私たちの候補製品のためにより多くの臨床前、臨床あるいは他の研究を開始する;臨床試験と商業化生産のための薬品の供給と薬品;臨床研究を成功させた任意の候補製品のための規制とマーケティング承認を求める;より多くの人を採用し、保留する;より多くの候補製品を識別と検証する;おそらく他の候補製品と技術を獲得する;第1注文製薬会社および他の第三者への支払いを含むが、これらに限定されないが、我々の許可または他の合意に従ってマイルストーン支払いまたは他の支払いを行うこと、当社の知的財産権の組み合わせを維持、保護および拡大すること、販売、マーケティング、流通および他の商業インフラを確立し、マーケティング承認を得る可能性のある任意の製品を商業化すること、追加のインフラを作成し、私たちの運営および私たちの製品開発および計画における将来の商業化努力を支援するための追加コストを生成すること、および上記の任意の遅延または問題を経験すること。
私たちの将来の資本需要は予測が難しく、多くの要素に依存しています
78
我々の研究と開発計画およびプロジェクトの進展に関する時間予想によると、本報告日までに、私たちの既存の現金と現金等価物および有価証券は、少なくとも今後12ヶ月の運営費用と資本支出需要に資金を提供できるようになると予想される。しかし、私たちの推定と仮定は間違っていることが証明される可能性があり、私たちの既存の資本資源が私たちのすべての予想された研究開発努力と将来の商業化努力を行うのに十分であるという保証はありません。また,臨床試験で候補薬物を試験する過程はコストが高く,これらの試験の進展時間は依然として不確定である。また、インフレは私たちの労働コストと研究開発費を増加させ、資本資源の使用に影響を与える可能性がある。私たちの長期資金需要には、以下に説明する契約約束を含む運営、資本、および製造支出が含まれるだろう。我々の候補製品の開発と商業化に関連する固有のリスクと不確実性のため、長期的に期待されている臨床前研究と臨床試験に関連する資本流出と運営支出を見積もることができない。
契約承諾
2017年4月、資産購入契約により、第一注文製薬会社または第一注文会社からXEN 1101(前身は1 OP 2198)を買収した。2020年8月には、第1注文と資産購入協定を修正し、合意のいくつかの定義を修正し、いくつかのマイルストーンの支払いスケジュールを修正した。2023年12月31日まで、私たちはこのようなマイルストーンの進展に基づいて200万ドルを支払った。未来の最初の注文に対する潜在的な支払いは600万ドルまでの規制マイルストーンを含む。最初の注文には印税義務がありません。
私たちはブリティッシュコロンビア州のバーナビーに研究実験室とオフィススペースの運営リースがあり、マサチューセッツ州のニダムにオフィススペースの運営レンタルがあります。賃貸借契約条項はそれぞれ2032年6月と2027年11月に満期となる。2023年12月31日現在、賃貸義務を経営する将来の賃貸支払いに関する金額は合計1250万ドルであり、そのうち170万ドルは今後12ヶ月以内に支払う予定です。
キャッシュフロー
次の表に2023年12月31日,2022年12月31日,2021年12月31日までの年間キャッシュフローの概要(単位:千)を示す
|
|
十二月三十一日までの年度 |
|
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|
|||
経営活動のための現金純額 |
|
$ |
(145,327 |
) |
|
$ |
(98,430 |
) |
|
$ |
(69,502 |
) |
|
投資活動のための現金純額 |
|
|
(117,170 |
) |
|
|
(296,003 |
) |
|
|
(246,770 |
) |
|
融資活動が提供する現金純額 |
|
|
353,522 |
|
|
|
278,471 |
|
|
|
447,543 |
|
|
経営活動
2023年12月31日までの1年間、経営活動に用いられた純現金総額は1兆453億ドルだったが、2022年同期は9840万ドルだった。この増加は主に研究開発および一般·行政費の増加と,2023年に確認されなかった収入によるものである。利子収入の増加および経営資産と負債の変動部分はこの増加を相殺した。
2022年12月31日までの1年間、経営活動に用いられた純現金総額は9840万ドルだったが、2021年同期は6950万ドルだった。増加の要因は,研究と開発および一般·行政費用の増加,Neurocrine連携やPacira協定に関する確認収入の減少である。利子収入の増加および経営資産と負債の変動部分はこの増加を相殺した。
投資活動
2023年12月31日までの1年間、投資活動に用いられた純現金総額は1兆172億ドルだったが、2022年同期は2.96億ドルだった。この変化は主に購入した有価証券の償還増加を差し引いたものである。不動産購入、建屋、設備の増加分はこの増加を相殺した。
79
2022年12月31日までの1年間で、投資活動に用いられた純現金総額は2.96億ドルだったが、2021年同期は2兆468億ドルだった。この変化は主に償還要因を差し引いた有価証券購入量の増加によるものである。
融資活動
2023年12月31日までの1年間で、融資活動が提供した純現金総額は3兆535億ドルだったが、2022年同期は2兆785億ドルだった。この増加は主に2023年の3.535億ドルの純収益と関係があるが、2022年に普通株と事前融資株式証を発行する純収益は2兆778億ドルである。
2022年12月31日までの1年間で、融資活動が提供した純現金総額は2兆785億ドルだったが、2021年同期は4億475億ドルだった。減少の要因は、2022年の純収益が2兆778億ドルだったのに対し、2021年に普通株と予備資金権証を発行した純収益は4億473億ドルだったことだ。
関係者取引
当社関連側取引の説明については、“特定関係と関連取引と取締役独立性”を参照されたい
流通株データ
2024年2月26日まで、私たちは75,432,482株の普通株と発行され、発行された、発行された事前融資権証を持って、2,173,081株の普通株を追加購入することができ、発行された株式オプションは8,860,105株の普通株を追加購入することができ、発行された株式証明書は40,000株の普通株を追加購入することができる。
第七A項。定量と合格IVE市場リスクに関する開示
私たちは通貨為替レートと金利の変化を含む様々な市場リスクに直面している。市場リスクとは,市場金利や価格の不利な変化による潜在的損失である。
外貨リスク
2023年12月31日現在、我々はドル建ての現金および現金等価物と有価証券9.071億ドル、カナダドル建ての現金および現金等価物と有価証券を3,150万カナダドルとしている。
私たちはドル以外の通貨での取引、特にカナダドルでの取引を行っているので、外貨為替リスクの影響をある程度受けています。私たちはまたカナダドル建ての現金と現金等価物、売掛金、売掛金を持っています。
外貨為替レートの変化は私たちに大きな外貨収益や損失をもたらすことができる。私たちは現在、私たちのリスクの開放をヘッジしないため、保有カナダドルの将来収益や損失のリスクを負担している。私たちは最近より多くの外貨変動を経験しましたが、ドルのカナダドルに対する相対的な価値が直ちに上昇または10%低下することが私たちの経営業績に実質的な影響を与えるとは思いません。
金利感度
2023年12月31日現在、私たちは9.309億ドルの現金と現金等価物、および有価証券を持っています。私たちの金利敏感性は主に私たちの現金と現金等価物と有価証券に起因する。金利の不利な変化は100ベーシスポイントまたは1%で、2023年12月31日現在の我々の有価証券の公正価値を約570万ドル減少させる。私たちは投機目的のために投資することもなく、派生金融商品を使用して金利リスクを管理しないつもりだ。
インフレリスク
インフレは一般的に私たちの労働コストと研究開発費を増加させることで私たちに影響を及ぼすだろう。 私たちは最近いくつかの時期の運営費用が増加していますが、一部の原因は最近のインフレの上昇だと思いますが、インフレが2023年12月31日までの年度の業務、財務状況、あるいは運営結果に実質的な影響を与えるとは思いません。しかし、インフレにより、今後しばらく運営費用が増加し続ける可能性があります。
80
項目8.財務諸表と補足データ
キセノン製薬会社です。
連結財務諸表索引
2023年12月31日までの年度
|
|
索引.索引 |
|
|
|
独立公認会計士事務所報告 |
|
82 |
|
|
|
2023年12月31日と2022年12月31日までの連結貸借対照表 |
|
84 |
|
|
|
2023年,2022年と2021年12月31日までの総合経営報告書と全面赤字報告書 |
|
85 |
|
|
|
2023年まで、2022年、2021年12月31日まで年度株主権益総合レポート |
|
86 |
|
|
|
12月31日に年間連結キャッシュフロー表を終了し、 2023年、2022年、2021年 |
|
87 |
|
|
|
連結財務諸表付記 |
|
88 |
81
独立公認会計士事務所報告
株主や取締役会に
キセノン製薬会社:
連結財務諸表に対するいくつかの見方
Xenon製薬会社(当社)の2023年12月31日現在と2022年12月31日までの連結貸借対照表、2023年12月31日までの3年間の年間ごとの関連総合経営報告書と全面赤字、株主権益と現金流量、および関連付記(総称して連結財務諸表と呼ぶ)を監査しました。総合財務諸表は、すべての重要な点で、会社の2023年12月31日まで、2023年12月31日と2022年12月31日までの財務状況と、2023年12月31日までの3年間の運営結果とキャッシュフローを公平に反映しており、米国公認会計原則に適合していると考えられる。
我々はまた、米国上場企業会計監督委員会(PCAOB)の基準に基づき、以下の基準に基づいて、会社が2023年12月31日までの財務報告内部統制を監査した内部統制--統合フレームワーク(2013)トレデビル委員会後援組織委員会が発表した報告書と、2024年2月29日の私たちの報告書は、社内財務報告の内部統制の有効性について保留されていない意見を表明した。
意見の基礎
これらの連結財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいてこのような連結財務諸表に意見を発表することだ。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、連結財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。我々の監査には、連結財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであっても詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれる。これらの手続きは、連結財務諸表中の金額および開示に関する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、合併財務諸表の全体列報の評価も含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
重要な監査事項とは、合併財務諸表を当期監査する際に発生した、監査委員会に伝達または要求された事項である:(1)総合財務諸表に対して重大な意義を有する勘定または開示に関連する;(2)私たちが特に挑戦的で、主観的または複雑な判断に関連する。私たちは重要な監査事項が存在しないと確信する。
/s/ピマウェイ法律事務所
フランチャイズ専門会計士
1999年以来、当社の監査役を務めてきました。
カナダバンクーバー
2024年2月29日
82
独立公認会計士事務所報告
株主や取締役会に
キセノン製薬会社:
財務報告の内部統制については
我々はS(会社)の2023年12月31日までの財務報告内部統制を監査し、根拠の基準は内部統制--統合フレームワーク(2013)テレデビル委員会が主催して委員会が発表された。2023年12月31日現在、当社はすべての重要な面で財務報告に対する有効な内部統制を維持しており、その根拠は内部制御--統合フレームワーク (2013)テレデビル委員会が主催して委員会が発表された。
また、米国上場企業会計監督委員会(PCAOB)の基準に従って、当社の2023年12月31日まで、2023年12月31日と2022年12月31日までの総合貸借対照表、2023年12月31日までの3年間の各年度の関連総合経営報告書と全面赤字、株主権益と現金流量および関連付記(総称して総合財務諸表と呼ぶ)を監査し、2024年2月29日の報告でこのなどの総合財務諸表に対して留保のない意見を表明した。
意見の基礎
当社経営陣は、効果的な財務報告内部統制を維持し、付随する経営陣財務報告内部統制年次報告に含まれる財務報告内部統制の有効性の評価を担当する。私たちの責任は私たちの監査に基づいて、会社の財務報告書の内部統制に意見を述べることです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務報告の有効な内部統制がすべての重要な面で維持されているかどうかを決定するために、合理的な保証を得るために監査を計画し、実行することを要求する。我々の財務報告の内部統制の監査には、財務報告の内部統制を理解すること、重大な弱点があるリスクを評価すること、評価されたリスクテストに基づいて内部統制の設計と運営有効性を評価することが含まれる。私たちの監査はまた、私たちがこのような状況で必要だと思う他の手続きを実行することを含む。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
財務報告の内部統制の定義と限界
会社の財務報告に対する内部統制は、公認された会計原則に基づいて、財務報告の信頼性と外部目的の財務諸表の作成に合理的な保証を提供することを目的としたプログラムである。会社の財務報告に対する内部統制には、(1)合理的で詳細かつ正確かつ公平に会社の資産を反映した取引および処分に関する記録の保存、(2)公認された会計原則に従って財務諸表を作成するために必要な取引が記録されている合理的な保証を提供し、会社の収入および支出は会社の経営陣と取締役の許可のみに基づいて行われる。(三)財務諸表に重大な影響を及ぼす可能性のある不正買収、使用、または処分会社の資産を防止またはタイムリーに発見する合理的な保証を提供する。
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来的にどのような有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムの遵守度が悪化する可能性がある.
/s/ピマウェイ法律事務所
フランチャイズ専門会計士
カナダバンクーバー
2024年2月29日
83
キセノン製薬会社です。
合併貸借対照表
(株式金額を除いて、千ドル単位)
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
|
||
|
|
2023 |
|
|
2022 |
|
|
||
資産 |
|
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
|
||
有価証券(付記6) |
|
|
|
|
|
|
|
||
売掛金 |
|
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
||
長期有価証券(付記6) |
|
|
|
|
|
|
|
||
経営性賃貸使用権資産、純額(付記8) |
|
|
|
|
|
|
|
||
財産·工場·設備,純額(付記7) |
|
|
|
|
|
|
|
||
繰延税金資産(付記13) |
|
|
|
|
|
|
|
||
前払い費用、長期 |
|
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
|
||
|
|
|
|
|
|
|
|
||
負債と株主権益 |
|
|
|
|
|
|
|
||
流動負債: |
|
|
|
|
|
|
|
||
売掛金と売掛金(付記9) |
|
$ |
|
|
$ |
|
|
||
賃貸負債を経営する(付記8) |
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
||
長期経営賃貸負債(付記8) |
|
|
|
|
|
|
|
||
総負債 |
|
$ |
|
|
$ |
|
|
||
|
|
|
|
|
|
|
|
||
株主権益: |
|
|
|
|
|
|
|
||
普通株額面価値発行·発行の株式 |
|
$ |
|
|
$ |
|
|
||
追加実収資本 |
|
|
|
|
|
|
|
||
赤字を累計する |
|
|
( |
) |
|
|
( |
) |
|
その他の総合損失を累計する |
|
|
( |
) |
|
|
( |
) |
|
株主権益総額 |
|
$ |
|
|
$ |
|
|
||
総負債と株主権益 |
|
$ |
|
|
$ |
|
|
||
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|||
付記はこれらの連結財務諸表の構成要素である。
84
キセノン製薬会社です。
合併経営報告書と全面赤字
(1株当たりおよび1株当たりの金額を除く、千ドルで表す)
|
|
十二月三十一日までの年度 |
|
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|
|||
収入(付記11) |
|
$ |
— |
|
|
$ |
|
|
$ |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|||
運営費用: |
|
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
|
|
|
|
|
|
|
|
|
|||
一般と行政 |
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|||
運営損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
その他の収入(支出): |
|
|
|
|
|
|
|
|
|
|
|||
利子収入 |
|
|
|
|
|
|
|
|
|
|
|||
取引証券の未実現公正価値収益(損失) |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
|
為替損益 |
|
|
|
|
|
( |
) |
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|||
所得税前損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
所得税払戻(別注13) |
|
|
|
|
|
( |
) |
|
|
|
|
||
純損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
優先株株主は純損失を占めるべきだ |
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
|
普通株主は純損失を占めなければならない |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|||
その他の全面収益(損失): |
|
|
|
|
|
|
|
|
|
|
|||
証券売却可能な未実現収益(赤字)(付記6) |
|
$ |
|
|
$ |
( |
) |
|
$ |
— |
|
|
|
総合損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|||
普通株1株当たり純損失(付記4): |
|
|
|
|
|
|
|
|
|
|
|||
基本的希釈の |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
加重平均発行済み普通株式(注4): |
|
|
|
|
|
|
|
|
|
|
|||
基本的希釈の |
|
|
|
|
|
|
|
|
|
|
|||
付記はこれらの連結財務諸表の構成要素である。
85
キセノン製薬会社.
(株式金額を除いて、千ドル単位)
|
|
オープンカー |
|
|
普通株 |
|
|
その他の内容 |
|
|
積算 |
|
|
その他を累計する |
|
|
合計する |
|
||||||||||||||
|
|
株 |
|
|
金額 |
|
|
株 |
|
|
金額 |
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
締め切りの残高 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||||
今年度の純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
普通株式と |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
株に基づく報酬 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
演習に基づいて発表する |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
|
|||
締め切りの残高 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||||
今年度の純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
普通株式と |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
優先株の転換 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
||
株に基づく報酬 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
演習に基づいて発表する |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
|
|||
その他全面赤字(付記6) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
締め切りの残高 |
|
|
— |
|
|
$ |
— |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
今年度の純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
普通株式と |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
事前出資株式証の転換 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
||
株に基づく報酬 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
演習に基づいて発表する |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
|
|||
その他全面収益(付記6) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
||
締め切りの残高 |
|
|
— |
|
|
$ |
— |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
付記はこれらの連結財務諸表の構成要素である。
86
キセノン製薬会社です。
合併状態キャッシュフロープロジェクト
(単位:千ドル)
|
|
十二月三十一日までの年度 |
|
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|
|||
経営活動: |
|
|
|
|
|
|
|
|
|
|
|||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
現金を扱っていないもの: |
|
|
|
|
|
|
|
|
|
|
|||
減価償却 |
|
|
|
|
|
|
|
|
|
|
|||
税金を繰延する |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
株に基づく報酬 |
|
|
|
|
|
|
|
|
|
|
|||
為替損失を実現しない |
|
|
( |
) |
|
|
|
|
|
|
|
||
証券取引の未実現公正価値(収益)損失 |
|
|
( |
) |
|
|
|
|
|
|
|
||
経営性資産と負債変動状況: |
|
|
|
|
|
|
|
|
|
|
|||
売掛金 |
|
|
|
|
|
|
|
|
( |
) |
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
|
売掛金と売掛金 |
|
|
|
|
|
|
|
|
|
|
|||
収入を繰り越す |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
経営活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|||
投資活動: |
|
|
|
|
|
|
|
|
|
|
|||
家屋·工場·設備を購入する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
有価証券を購入する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
有価証券収益 |
|
|
|
|
|
|
|
|
|
|
|||
投資活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|||
融資活動: |
|
|
|
|
|
|
|
|
|
|
|||
普通株式と事前融資権証を発行する |
|
|
|
|
|
|
|
|
|
|
|||
株式オプションの行使により普通株を発行する |
|
|
|
|
|
|
|
|
|
|
|||
融資活動が提供する現金純額 |
|
|
|
|
|
|
|
|
|
|
|||
現金および現金等価物に及ぼす為替レート変動の影響 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
|
現金と現金等価物を増やす(減らす) |
|
|
|
|
|
( |
) |
|
|
|
|
||
現金と現金等価物、年明け |
|
|
|
|
|
|
|
|
|
|
|||
現金と現金等価物、年末 |
|
$ |
|
|
$ |
|
|
$ |
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|||
補足開示: |
|
|
|
|
|
|
|
|
|
|
|||
賃貸経営のための現金 |
|
$ |
|
|
$ |
|
|
$ |
|
|
|||
レンタル奨励金で受け取った現金 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
非現金取引の追加開示: |
|
|
|
|
|
|
|
|
|
|
|||
無現金ベースで行使された株式オプションの公正価値 |
|
|
|
|
|
|
|
|
|
|
|||
事前資金権証の公正価値を行使する |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
帳簿に掲げる財産·工場·設備の購入 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
新経営権と引き換えに得られた使用権資産 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
賃貸負債と売掛金の経営に関する増加 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
経営的リース使用権資産と経営的リース負債が増加 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
付記はこれらの連結財務諸表の構成要素である。
87
キセノン製薬会社です。
連結財務諸表付記
(1株当たりおよび1株当たりの金額を除く、千ドルで表す)
Xenon製薬会社(“会社”)は神経科学に専念する生物製薬会社であり、1996年に“商業会社法”(ブリティッシュコロンビア州)の前身登録により設立され、2000年に“カナダ商業会社法”に基づいて連邦政府に登録され続け、神経·精神疾患患者の生活改善に取り組んでいる。
設立以来、同社は重大な経営赤字を発生させた。2023年12月31日現在、同社の累積損失は$
企業が相当な製品収入を生み出すことができる前に、経営陣は協力協定、株式、債務融資の組み合わせで会社の現金需要に資金を提供する予定だ研究·開発活動の継続とその製品の将来の商業化は、必要に応じて同社が追加資金を調達することに成功する能力に依存する。将来の研究開発計画の結果を予測することもできないし、同社が将来これらの計画に資金を提供し続ける能力も予測できない。
同等の総合財務諸表は米ドルで列報され、米国公認会計原則(“米国公認会計原則”)に基づいて作成されている。
2023年12月31日現在、会社は完全子会社を持っているXenon製薬会社は
当該等の総合財務諸表には、当社及びその全額付属会社の勘定が含まれている。すべての会社間取引と残高は合併時に出荷されました。
米国公認会計原則に基づいて総合財務諸表を作成する際には、管理層は、財務諸表日の資産及び負債額及び又は有資産及び負債の開示、及び報告期間内の収入及び支出の報告金額に影響を与える推定と仮定を行わなければならない。推定数の重要な分野には、履行義務を完了する推定時間と、株式ベースの報酬の決定とを含む収入確認に限定されるものではない。これらの見積もりと仮定は、会社が合理的だと思う歴史的かつ前向きな要素を考慮している。推定と仮定は四半期ごとに検討される。会計推定数のすべての改訂は、推定カウントを修正する期間および影響を受けた任意の将来の期間で確認される。
現金等価物は高流動性の投資であり、買収時に現金に変換しやすく、期限は3ヶ月以下である。現金等価物はコスト加算利息で入金されます。
有価証券とは、元の満期日が3ヶ月を超え、固定金利で利上げされる債務証券のことである。2022年7月1日から、同社はその有価証券を取引可能証券または売却可能証券に分類した。有価証券は公正価値に基づいて勘定する.
取引性証券に分類された有価証券の公正価値損益は総合経営報告書で入金される。これらの証券は、会社が処罰を受けることなく、今後12ヶ月以内にこれらの証券を現金に変換する意欲と能力があるため、流動資産に分類される。
88
売却可能な有価証券に分類された未実現公正価値損益は,株主権益における他の全面収益(損失)によって計上される。販売可能な証券の公正価値が償却コストベースよりも低い場合、価値低下が信用損失に起因するかどうかを決定するために評価を行う。信用損失の公正価値の減少は直接総合経営報告書に計上し、相応の信用損失支出を計上することができるが、公正価値が余剰コスト基礎より低い金額を超えてはならない。信用品質がその後改善された場合、控除限度額は以前に記録された信用損失の最高限度額に戻る。当社が減価された売却可能な証券を販売しようとしている場合、あるいは当社が余剰コスト基準を回収する前にその証券を売却しなければならない可能性が高い場合、全体の公正価値調整は直ちに総合経営報告書で確認され、対応する信用損失準備を考慮することなく確認される。売却可能な証券の実現損益と信用損失(ある場合)は、具体的な確認方法に基づいて利子収入を計上する。販売可能な証券も、割増償却および満期日割引増加に応じて調整され、このような償却および割引増加は利息収入に含まれる。残りの満期日が1年以上の売却可能証券は非流動資産に分類される.
国内で開発された知的財産権特許の確立と維持に発生するコストは、発生した期間内に計上される。
物件、工場及び設備はコストから減価償却累計及び/又は累積減価損失(あればある)を引いて帳簿に記入する。修理とメンテナンス費用は発生した期間内に計上されます。
財産、工場と設備はその推定使用年数内に直線法で以下の比率で償却した
資産 |
|
料率率 |
研究設備 |
|
|
オフィス家具と設備 |
|
|
コンピュータ装置 |
|
|
賃借権改善 |
|
同社はその長期資産の減価指標を監視している。このような指標があれば,当社は影響を受けた資産の回収可能度を評価し,その等資産の帳簿価値が当該等資産の未割引将来のキャッシュフローの総和よりも少ないかどうかを決定する方法である.当該等資産が回収できないことが発見された場合、当社は、当該等資産の帳簿価値と当該等資産の公正価値とを比較して、当該等減価の金額を計量するが、当該等公正価値は、当該等資産に関する予想将来の現金流量の現在値に基づいて決定されるのが一般的である
レンタルを運営するレンタルに分類されたレンタルは、レンタル期間内の最低賃貸支払いの現在値で入金され、レンタル者の暗黙的な金利が容易に確定できない場合は、レンタルに隠れているレンタル者の金利や会社の逓増借款金利で割引を行う。リース期間は、当社が継続権の行使または終了オプションの行使を合理的に決定する継続期間および終了オプションがカバーするすべての期間を含む。レンタル負債、初期直接コスト、および任意のレンタルインセンティブ支払いを含む、対応する使用権資産が確認されました。賃貸負債はリース金を支払う際に減記し、使用権資産はレンタル期間内に減価償却する。運営リース支出は賃貸期間内に直線原則で確認され、賃貸負債は利息及び使用権資産減価償却を含め、変動期間中の指数に基づく可変賃貸支払い変動に応じて調整される。レンタル期間が12ヶ月以下の短期経営リースのリース支払いは、レンタル期間内に直線的に計算される。同社は、そのレンタルプロトコルに埋め込まれていない非レンタル要素を分離することを選択した。
89
会社を深刻な集中信用リスクに直面させる可能性のある金融商品には、主に現金と現金等価物および有価証券が含まれる。同社の投資は、資本の保存と流動性の維持を目的とした高い信用格付けを有する投資級証券に限られている。現金と現金等価物はカナダとアメリカの主要金融機関に保管されており、連邦保険の限度額を超える場合がある。当社は商業銀行関係に関する標準信用リスクを超えるとは考えていない。
Neurocrine Biosciences,Inc.(“Neurocrine Biosciences”)が占める
当社は公正な価値に応じていくつかの金融商品その他の項目を計量します。
公正価値を確定するため、当社は公正価値階層構造を採用して金融資産と負債の公正価値を計量する。この階層構造は、公平な価値を評価するための推定技術の入力を、レベル1(最高優先度)、レベル2、レベル3(最低優先度)の3つのレベルに分類する。
資産と負債は,公正価値計測に重要な意味を持つ最低投入レベルによって分類される。評価投入の観測可能性の変化は公正価値レベル中のある証券のレベルの再分類を招く可能性がある。売掛金、売掛金及び売掛金の額面は、当該等の手形の性質及び短期的な性質により公正価値に近い。当社の現金および現金等価物および有価証券は公正価値で恒常的に計量され,採用した公正価値階層レベルは付記5に記載されている。
同社は,顧客への承諾した貨物やサービスの譲渡により得られる収入額を5ステップモデルで確認する:(1)顧客との契約を決定する(S),(2)契約中の履行義務を決定する,(3)取引価格を決定する,(4)契約に取引価格を割り当てる履行義務,および(5)契約履行義務を履行する際に収入を確認する。
協力協定は、知的財産権またはライセンス、および研究開発サービスを含む様々な権利および/またはサービスを会社に提供することを要求することができる。このような協力協定によると、同社は通常、払い戻し不可能な前払い、研究開発サービス資金、マイルストーン支払い、特許権使用料を取得する資格がある。
当社が1つ以上の義務を履行してその顧客に貨物またはサービスを提供する契約では、各履行義務は、(I)顧客が単独でまたは他の既製資源と共に貨物またはサービスから利益を得ることができるかどうか、(Ii)貨物またはサービスを契約内の他の約束とは別に識別することができるかどうかによって評価される。そして、契約項の下の対価格はそれぞれの相対独立販売価格に応じて異なる履行義務の間に分配される。各納入可能成果の推定独立販売価格は、納入可能成果の定期独立販売時の販売価格の自社の最適な推定を反映し、他人に販売する際の商品やサービスの市場価格に基づいて決定され、独立販売価格がなければ、調整後の市場評価方法により決定される。
90
関連商品やサービスの制御権がクライアントに移行した場合,それぞれの異なる履行義務に割り当てられた対価格が収入として確認される.当社は,被許可者が提供する研究·開発サービスの対価格を代表して,現行の市場料率と一致したレートでこのような活動を行う際に確認した。リスクのある実質的な業績マイルストーン(販売ベースのマイルストーンを含む)に関する対価格は、確認された累計収入が大きく逆転しない可能性が高い場合には、可能金額法を用いて収入として確認される。知的財産権許可に関連する販売ベースの使用料は、収入基準において特定の例外があり、すなわち、顧客のその後の販売または使用が発生する前に、対価格は取引価格に含まれず、収入で確認される。
研究と開発コストは発生した期間内に支出される。
会社は、付記10 cに記載されている株式オプション計画に基づいて、従業員、コンサルタント、取締役、上級管理者に株式オプションを付与する。
従業員株補償支出は、授与日に奨励の推定公正価値に基づいて計量し、必要なサービス期間の費用を確認し、実際の没収を差し引いた後、追加実収資本はそれに応じて増加する。株式ベースの報酬支出は、報酬の必要なサービス期間全体にわたって直線的に償却され、これは通常、奨励の授権期間である。株式オプション行使で受け取ったどの対価格も配当金に記入する。
当社とその子会社の機能通貨と報告通貨はドルです。ドル以外の通貨建ての通貨資産と負債を貸借対照表日の現行為替レートでドルに再計量する。ドル以外の通貨で購入した非貨幣的資産と負債を取引日ごとの歴史的為替レートに換算する。
収入と費用取引にはイーは取引日の大まかな為替レートで換算されます。為替損益を換算して総合経営表と総合収益(赤字)為替損益を計上する。
繰延所得税は、資産及び負債の帳簿金額及びそれぞれの課税基礎と純営業損失及び信用繰越との差額による将来の税務結果について確認する。繰延所得税資産と負債は、定められた税率で計量され、これらの仮差額と繰越を予定する年度の課税所得額に適用される予定です。税率変動が繰延所得税資産と負債に及ぼす影響は,公布日を含む期間の総合経営と全面収益(赤字)表で確認された。繰延所得税資産の現金化が“可能性が高い”確認基準を満たしていない場合には、推定準備を提供する。
経営部門は企業の構成要素として定義され、その独立した離散情報は、首席経営意思決定者あるいは意思決定グループが資源をどのように分配するかを決定し、業績を評価する際に評価することができる。会社は以下の点でその運営·管理業務を確認します
91
新しい会計声明は、財務会計基準委員会または会社が指定された発効日から採用された他の基準作成機関によって時々発表される。当社は最近発表された会計声明を評価しており、初歩的な評価によると、どの声明も当社の財務諸表に大きな影響を与えないと考えられています。
普通株1株当たりの基本純収入(損失)は証券参加に必要な2段階法を用いて計算され、その中には系列1優先株を1つの単独カテゴリとして含む。転換優先株は保有者が普通株と同等の基礎の上で会社の配当と損益に参加する権利を持たせることができる。そこで,未分配収益(損失)は,期間内に発行されたカテゴリごとの加重平均株式に応じて普通株と参加優先株に分配される.2022年3月には
基本と希釈後の1株当たりの純利益(損失)を計算する際に使用する普通株の加重平均は、いつでも名義現金で対価格行使できるため、期間内に発行された加重平均事前融資権証を含む。
在庫株方法は、会社の株式オプションと引受権証の希薄化効果を計算するために用いられる。この方法によれば、1株当たりの普通株式償却純収益(損失)を計算するための普通株式増分数は、仮想発行された普通株数と、仮想収益を用いて購入した普通株数との差額である。
折算法は会社が転換可能な優先株の削減効果を計算するために用いられる。IF-転換法により、優先株の配当(適用すれば)が普通株株主の占有収益に加算され、優先株と実収配当は期末に適用される株価で転換されたと仮定する。効果が希釈性である場合にのみ,IF変換方法を適用する.
公正価値を決定するための公正価値等級現金および現金等価物および有価証券は、以下のものを含む
|
|
2023年12月31日 |
|
|
2022年12月31日 |
|
||||||||||||||||||||||||||
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
|
合計する |
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
|
合計する |
|
||||||||
現金と現金等価物 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
現金と貨幣市場基金 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||||
有価証券 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
担保投資証明書 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
アメリカ国債 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
アメリカ政府証券 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||||
商業手形 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||||
会社債務証券 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||||
合計する |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
||||||
同社の米国政府証券、商業手形、会社債務証券の公正価値は、独立定価源から得られた価格に基づいている。価格設定サービスからの有効なオファーの証券は、主に同様の資産の観察可能な価格設定または他の市場で観察可能な投入に基づくので、第2レベルに反映される。これらの価格設定サービスによって使用される典型的な情報は、報告された取引、基準収益率、発行者利益差、入札、キャッシュフロー見積もりまたは推定、早期返済利差、および違約率を含むが、これらに限定されない。
当社は2023年12月31日と2022年12月31日まで、3級に分類された証券を何も持っていません。
92
2023年12月31日までその会社は$を持っている
|
|
2023年12月31日 |
|
|
2022年12月31日 |
|
||||||||||||||||||
|
|
償却する |
|
|
未達成収益 |
|
|
公平である |
|
|
償却する |
|
|
実現していない |
|
|
公平である |
|
||||||
契約満期日は0から1年: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
担保投資証明書 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|||||
アメリカ国債 |
|
|
|
|
|
( |
) |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
||||
アメリカ政府証券 |
|
|
|
|
|
( |
) |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
||||
商業手形 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
|||||
会社債務証券 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||||
契約期間は1~3年: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
アメリカ国債 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||||
アメリカ政府証券 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
( |
) |
|
|
|
||
会社債務証券 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||||
合計する |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||||
これらの有価証券の信用損失や減価準備は確認されていないが、これらの証券は高信用品質の投資級証券であるため、会社は売却しようとせず、予想回復前に売却することも要求されず、公正価値低下の要因は金利の変化である。
不動産、工場、設備には、
|
|
十二月三十一日 |
|
||||
|
|
2023 |
|
2022 |
|
||
研究設備 |
|
$ |
|
$ |
|
||
オフィス家具と設備 |
|
|
|
|
|
||
コンピュータ装置 |
|
|
|
|
|
||
賃借権改善 |
|
|
|
|
|
||
減算:減価償却累計と償却 |
|
|
( |
) |
|
( |
) |
帳簿純価値 |
|
$ |
|
$ |
|
||
93
経営リースのコスト構成は以下のように同年度までである2023年12月31日、2022年、2021年:
|
|
十二月三十一日までの年度 |
|
|||||||
|
|
2023 |
|
2022 |
|
2021 |
|
|||
レンタル料 |
|
|
|
|
|
|
|
|||
レンタル費用を経営する |
|
$ |
|
$ |
|
$ |
|
|||
可変レンタル費用(1) |
|
|
|
|
|
|
|
|||
レンタル期間と割引率 |
|
|
|
|
|
|
|
|||
加重平均残存賃貸年限(年) |
|
|
|
|
|
|
|
|||
加重平均割引率 |
|
|
% |
|
% |
|
% |
|||
12月31日までの年度: |
|
|
|
|
2024 |
|
$ |
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 |
|
|
|
|
2028 |
|
|
|
|
2029年以降 |
|
|
|
|
将来の最低賃貸支払い総額 |
|
$ |
|
|
差し引く:推定利息 |
|
|
( |
) |
賃貸負債現在価値 |
|
$ |
|
|
売掛金および売掛金には以下の内容が含まれる
|
|
十二月三十一日 |
|
||||
|
|
2023 |
|
2022 |
|
||
貿易応払い |
|
$ |
|
$ |
|
||
従業員の報酬、福祉、関連プロジェクト |
|
|
|
|
|
||
相談と契約研究 |
|
|
|
|
|
||
専門費 |
|
|
|
|
|
||
他にも |
|
|
|
|
|
||
合計する |
|
$ |
|
$ |
|
||
2020年8月、会社はJefferies LLC(“Jefferies”)とStifel、Nicolaus&Company,Inc.(“Stifel”)と2022年3月に改訂された“市場で”の株式発行販売協定を締結したこれにより、当社は時々普通株を販売することができます。2021年1月、当社は販売しました
94
2021年3月,当社は引受公開を完了した
2021年9月、Neurocrine Biosciencesと2019年12月に締結され、2021年1月に改訂された許可及び協力協定(“Neurocrine協力協定”)について、当社は株式購入協定(“SPA”)に署名し、これにより当社が発行した
2021年10月、当社は引受公開を完了しました
2022年6月、当社は引受公開を完了しました
2023年11月、会社が完成引受の公開発行
同社の法定株式は無額面無限数量の普通株と優先株からなる。
当社には,(I)あらかじめ存在する株式オプション計画(“株式オプション計画の改訂および再編成”),(Ii)が2020年6月および2022年6月に改訂および再記述された2014年株式インセンティブ計画(“2014計画”),および(Iii)2019年持分インセンティブ計画(“2019年インセンティブ計画”)の3つの持分インセンティブ計画がある。
改正·再編された株式オプション計画は、会社が初公開する前に、取締役、上級管理者、従業員、コンサルタントに普通株を購入する株式オプションを付与することが規定されている。改訂及び再編成された株式オプション計画に基づいて付与された株式オプション
2014年6月、当社の株主は2014年計画を承認し、2020年6月と2022年6月に改訂され、改訂された2014年株式インセンティブ計画(“改訂後に再策定された2014年計画”)に置き換えられた。
95
2019年9月、会社取締役会は“2019年インセンティブ計画”を採択し、“2019年インセンティブ計画”調整規定を満たすことを前提として、保留します
当社の株主は、2020年6月及び2022年6月に改訂された改訂及び改訂された2014年計画を承認し、当社の2014年計画のいくつかの条文を改訂する。改訂及び再予約された2014年計画は、引き続き当社取締役、高級管理者、従業員及びコンサルタントに株式ベースの報酬奨励、及び発行制限株式、限定株式単位、株式付加価値権及び業績株式の授与を許可する。改訂と再確認された2014年計画によると、付与されたオプションは一般に階層に基づいて付与される
次の表は、この期間の株式オプション活動をまとめています
|
|
量 |
|
|
重みをつける |
|
|
骨材 |
|
|||
|
|
オプション |
|
|
値段(ドル)(1) |
|
|
固有の 価値がある |
|
|||
優秀で2020年12月31日 |
|
|
|
|
|
|
|
|
|
|||
授与する |
|
|
|
|
|
|
|
|
|
|||
鍛えられた(2) |
|
|
( |
) |
|
|
|
|
|
|
||
没収、キャンセル、または期限切れ |
|
|
( |
) |
|
|
|
|
|
|
||
未返済、2021年12月31日 |
|
|
|
|
|
|
|
|
|
|||
授与する |
|
|
|
|
|
|
|
|
|
|||
鍛えられた(2) |
|
|
( |
) |
|
|
|
|
|
|
||
没収、キャンセル、または期限切れ |
|
|
( |
) |
|
|
|
|
|
|
||
未返済、2022年12月31日 |
|
|
|
|
|
|
|
|
|
|||
授与する |
|
|
|
|
|
|
|
|
|
|||
鍛えられた(2) |
|
|
( |
) |
|
|
|
|
|
|
||
没収、キャンセル、または期限切れ |
|
|
( |
) |
|
|
|
|
|
|
||
未返済、2023年12月31日 |
|
|
|
|
|
|
|
|
|
|||
行使可能、2023年12月31日 |
|
|
|
|
|
|
|
|
|
|||
2023年12月31日未償還および行使可能な株式オプションの加重平均残存契約期間は
96
当社の今年度までの非既得株式オプション活動及び関連資料の概要2023年12月31日の状況は以下の通り
|
|
量 |
|
|
加重平均 |
|
||
非既得権益者、2023年1月1日 |
|
|
|
|
|
|
||
授与する |
|
|
|
|
|
|
||
既得 |
|
|
( |
) |
|
|
|
|
没収またはキャンセルされる |
|
|
( |
) |
|
|
|
|
非既得権益者、2023年12月31日 |
|
|
|
|
|
|
||
2023年12月31日まで年度内に帰属するオプションの公正価値合計はい$です
株式オプションの付与日における公正価値は,ブラック−スコアーズオプション定価モデルを用いて推定され,このモデルは複数の主観投入を必要とする。オプションの無リスク金利は、付与日に発効した米国債収益率曲線に基づいており、期限はオプションの予想期限と似ている。予想変動率は、予想期限仮定に見合った一定期間に基づいて算出された当社普通株の履歴変動率である。予想寿命は会社の履歴データに基づいていると仮定する。配当率は当社が現金配当金を派遣したことがなく、現在現金配当金を派遣するつもりはないことに基づいている。没収行為は発生時に確認します。
加重平均オプション価格は以下のように仮定される
|
|
十二月三十一日までの年度 |
|
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|
|||
平均無リスク金利 |
|
|
% |
|
|
% |
|
|
% |
|
|||
予想変動率 |
|
|
% |
|
|
% |
|
|
% |
|
|||
平均予想期限(年) |
|
|
|
|
|
|
|
|
|
|
|||
期待配当収益率 |
|
|
% |
|
|
% |
|
|
% |
|
|||
付与オプションの加重平均公正価値 |
|
$ |
|
|
$ |
|
|
$ |
|
|
|||
株式ベースの報酬費用は、連結経営報告書と総合収益(損失)で以下のように分類される
|
|
十二月三十一日までの年度 |
|
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|
|||
研究開発費 |
|
$ |
|
|
$ |
|
|
$ |
|
|
|||
一般と行政費用 |
|
|
|
|
|
|
|
|
|
|
|||
|
|
$ |
|
|
$ |
|
|
$ |
|
|
|||
2023年12月31日まで非既得株式オプションに関する未確認株式報酬支出は#ドルである
2018年3月、当社はBVFと交換協定を締結し、この合意に基づき、当社はBVFに発行します
当社の資産が株主間で清算、解散または清算または他の方法で分配された場合、第1シリーズ優先株は普通株と同等の地位を有し、第1シリーズ優先株の保有者は転換後に単一カテゴリである普通株と一緒に投票する権利があるが、いくつかの制限によって制限される。
シリーズ1優先株は現金決済ができず、償還機能もないため、シリーズ1優先株はASC 480項の下ですべて権益として入金され、主契約の転換特徴と分岐していない。
BVFは2018年12月31日までの年間で
97
下表は年末までの資本権証活動をまとめたものである2023年12月31日、2022年、2021年:
|
|
発行日 |
|
|
|
|
||||||||||||||
|
|
2021年3月 |
|
|
2021年10月 |
|
|
2022年6月 |
|
|
2023年12月 |
|
|
合計する |
|
|||||
優秀で2020年12月31日 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
発表されました |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
鍛えられた |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
未返済、2021年12月31日 |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
発表されました |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
鍛えられた |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
未返済、2022年12月31日 |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
発表されました |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
||
鍛えられた |
|
|
( |
) |
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
未返済、2023年12月31日 |
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
2021年3月、2021年10月、2022年6月と2023年12月に完成した包売公開について、会社は予資権証を発行し、同値数の普通株を購入し、価格は1ドルとなる
事前資金権証は発行日から予資権証がすべて行使された日から、所持者が適宜行使することができる
予融資権証は株式分類条件を満たしているため、予融資権証を発行して得られた金は#ドルである
2018年8月購入権証
当社は、ASC 606に従って、払戻不可能な事前支払いを確認することを含む、付記3 Jに記載された5ステップモードで各協調プロトコルを評価した。当社では、責任履行の見積もり期間や関連利益を顧客に移している間に、払い戻し不可能な前払いからの収入を確認するのが一般的です。払戻不可能な許可料が未交付の履行義務とは独立して顧客に価値があれば,交付時に確認する。当社は報告期間ごとに進捗指標を評価し、必要に応じて業績指標と関連収入確認を調整します。
98
会社の協力協定における研究および開発マイルストーンには、以下のタイプのイベントが含まれる可能性がある
規制されたマイルストーン支払いには、以下のタイプのイベントが含まれる場合があります
商業化マイルストーン支払いは、予め指定された閾値に達した年間製品販売によってトリガされた支払いを含むことができる。
同社は、マイルストーンが達成可能とされているかどうかを決定し、可能な金額法を用いて取引価格に含まれる金額を推定するために、研究開発と販売マイルストーン支払いを含む各手配を評価する。会社の統制範囲内でないマイルストーン支払いは実現可能とは考えられません。大きな収入逆転が起こらない可能性が高い場合、関連するマイルストーン価値は取引価格に含まれるだろう。そして、取引価格は相対的に独立した販売価格で個々の履行義務に割り当てられ、会社は契約項目の履行義務を履行する際に収入を確認する。その後の各報告期間終了時に、当社は当該等のマイルストーンを実現する可能性を再評価し、必要に応じて全体の取引価格の推定を調整する。
2023年12月31日までの年間収入は以下の通り2022年と2021年:
|
十二月三十一日までの年度 |
|
|||||||
|
2023 |
|
2022 |
|
2021 |
|
|||
神経分泌生物科学: |
|
|
|
|
|
|
|||
取引価格の確認 |
$ |
— |
|
$ |
|
$ |
|
||
研究と開発サービス |
|
— |
|
|
|
|
|
||
一里塚払い |
|
— |
|
|
|
|
|
||
Pacira BioSciences: |
|
|
|
|
|
|
|||
一里塚払い |
|
— |
|
|
— |
|
|
|
|
*総収入 |
$ |
— |
|
$ |
|
$ |
|
||
2019年12月、会社はNeurocrine BiosciencesとNeurocrine協力協定を締結した。このプロトコルによれば、同社はXEN 901(現在NBI-921352と呼ばれる)に独占許可を付与し、特定の臨床前化合物に開発のための独占許可(“DTC”)を付与する。この協定はまた、より多くの新型Nav1.6およびNav1.2/1.6阻害剤(“研究化合物”)を発見、識別、開発するための2年間の研究協力を含む。同社とNeurocrine Biosciencesは,(A)臨床前開発研究化合物の共同研究協力(“研究計画”)を発見·決定し,2022年6月に完成,(B)共同指導委員会が選定したNBI−921352と2つのDTCの共同開発計画(“予備開発計画”)の2つの協力計画の実施に協力している。
協定調印時、Neurocrine Biosciencesは同社に#ドルの費用を前払いした
同社は特許製品に関する商業前と商業マイルストーンの支払いを得る資格があり、総額は最高$に達する
99
その会社は共同出資する権利がある
このプロトコルには,(I)関連技術とノウハウ譲渡を有するNBI-921352の独占ライセンス,(Ii)関連ノウハウ譲渡を有するDTCの独占ライセンス,(Iii)研究計画下の研究化合物と研究サービスのライセンス,(Iv)NBI-921352の初期開発計画下での開発サービス,および(V)DTCの初期開発計画下での開発サービスが含まれる.Neurocrine Biosciencesは、同社の独自の専門知識が市場で容易に得られないため、このような許可自体や業界でよく見られる他の資源から利益を得ることができないため、研究化合物および研究計画下の研究サービスの許可は単一の履行義務と考えられる。研究複合体の早期開発段階に鑑み,業績義務と関連収入は研究サービスの業績に完全に関連している。
プロトコルを実行する際には,取引価格は#ドルからなる
この手配により,当社は履行責任(Iii)および(Iv)によるいくつかのフルタイム同値および外部コストを支払うために資金を獲得する権利がある。Neurocrine Bioscionsを代表して履行義務(3)および(4)項目の下のサービスを履行することに関連する手配対価格は、Neurocrine Bioscions要求に依存してサービスを提供することに依存するので、初期取引価格割り当てから除外され、これらのサービスの価格は、推定公正価値に基づいて計算される。
総成約価格は$
2021年9月、欧州規制機関がNBI-921352の成人局所てんかん発作の臨床試験申請を承認したことに基づいて、同社は記念碑的な支払い総額#ドルを受け取った
202年1月、米国食品医薬品局(FDA)からNBI-921352への完全IND受け入れを受けたことにより、同社は合計1ドルの記念碑的支払いを受けた
2022年12月31日と2021年12月31日までの年間で、会社確認収入は
2019年9月、当社は、2021年11月にPacira BioSciencesによって買収されたFlexion Treateutics Inc.(“Flexion”)と合意し、この合意に基づいて、Flexionは、会社が所有または制御する特定の法規文書、知的財産権、報告、データ、およびすべての数のXEN 402、すなわちPCRX-301を含むXEN 402および関連化合物(総称して“XEN 402”)に関連するすべての権利を取得する。
FDAは2021年12月31日までの1年間にPCRX−301の最初の研究新薬申請を承認し,1 b期の臨床試験を開始し,記念碑的な支払い$を生成した
100
2017年4月、当社は資産購入協定に基づいて第1注文からXEN 1101(前身は1 OP 2198)を買収した。2020年8月、当社と第1注文は資産購入協定を改訂し、協定のいくつかの定義を修正し、いくつかのマイルストーンの支払いスケジュールを修正した。同社は2023年12月31日までに支払いを完了した
同社はすでに、業界慣行の賠償条項を含む第三者と許可·研究協定を締結している。これらの賠償条項は、一般に、第三者のクレームまたはこれらの取引による損害によって生じたいくつかの損害および費用を会社に賠償することを要求する。
未来の潜在的賠償の最高金額は無限だ;しかし、会社は現在商業と製品責任保険を持っている。この保険は会社のリスク開放を制限し、将来支払う任意の金額の一部を回収できるようにしている。歴史的に見ると、当社は当該等の合意に基づいていかなる賠償金も支払われておらず、当社は当該等の賠償義務の公正価値が極めて低いと信じている。したがって、列報のいずれの期間においても、当社は当該等の債務に関するいかなる負債も確認していない。
所得税の還付は、予想されるカナダ連邦と省の法定所得税税率を適用することで計算される金額とは異なる
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
カナダ連邦とカナダでの計算回収 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
評価免除額を変更する |
|
|
|
|
|
|
|
|
|
|||
得られた税収控除 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
税金属性の有効期限が切れた/使用済み |
|
|
|
|
|
|
|
|
|
|||
差し引かれない支出 |
|
|
|
|
|
|
|
|
|
|||
他にも |
|
|
|
|
|
|
|
|
( |
) |
||
所得税支出(回収) |
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
2023年12月31日,2022年,2021年12月31日までの年度の所得税支出(回収)は,同社の米国における完全子会社であるキセノン米国製薬会社の業務からである。
繰延所得税資産および負債は、財務諸表で確認された資産と負債額と所得税との一時的な差によるものである。会社の繰延所得税の純資産の重要な構成部分は以下の通りである
|
|
十二月三十一日 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
科学研究と実験発展池 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
税金控除 |
|
|
|
|
|
|
|
|
|
|||
非資本損失 |
|
|
|
|
|
|
|
|
|
|||
減価償却可能資産 |
|
|
|
|
|
|
|
|
|
|||
融資費を繰延する |
|
|
|
|
|
|
|
|
|
|||
株に基づく報酬 |
|
|
|
|
|
|
|
|
|
|||
他にも |
|
|
|
|
|
|
|
|
|
|||
減価免税額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
所得税純資産を繰延する |
|
$ |
|
|
$ |
|
|
$ |
|
|||
101
繰延所得税資産の現金化は,将来の期間に十分な課税収入が生じるかどうかに依存し,これらの期間では一時的な差が逆転することが期待される。推定免税額は季節ごとに検討され、“最も可能性が高い”基準の評価が変われば、推定免税額はそれに応じて調整される。当社はこのような資産の現金化が“可能性が高い”という基準を満たしていないと評価しているため、カナダの繰延所得税資産は引き続き全額推定手当を適用している。2023年12月31日現在と2022年12月31日までに総合貸借対照表に記録されている繰延所得税資産は、財務諸表で確認された資産および負債額とXenon米国製薬会社の業務に関する所得税純額との一時的な差によるものである。
2023年12月31日同社は、科学研究と実験開発支出を減税#ドルに申請する人がいない
2023年12月31日その会社は$を持っています
2023年12月31日会社は非資本損失があり、不確定な税務状況を差し引いて税務目的に繰り越し、来年度の課税所得額約$を減らすために用いることができる
投資税控除と損失繰越は異なる年度に満期になる.
2023年12月31日当社が税務状況を確定していないため確認されていない税収割引総額は$
同社はカナダと米国で所得税申告書を提出し、同社はこの2つの管轄区が課税すべきだと考えている。当社では課税対象とされていないため所得税申告書を提出していない司法管轄区では、当該管轄区の税務機関が1つまたは複数の納税年度を審査しないことは特定できません。また、所得税申告書を提出した各管轄区の訴訟時効は一般的に審査期限が制限されているにもかかわらず、赤字繰越のため、審査時効期間は赤字繰越使用後数年で満了するのが一般的である。税務機関が当社が主張している税務控除及び税金還付について定例監査を行う以外、当社はいかなる税務管轄区域で現在行われているいかなる他の重大な所得税審査も知りません。課税年度から
項目9.Accouとの変更と分岐会計と財務情報開示の専門家
ない。
第9条。制御とプログラム
開示制御とプログラムの評価それは.我々の経営陣は、最高経営責任者と財務責任者の参加の下、2023年12月31日までの開示統制と手続きの有効性を評価した。取引法規則13 a-15(E)および15 d-15(E)に定義されている用語“開示制御および手順”は、取引法に従って提出または提出された報告において開示を要求する会社の情報が米国証券取引委員会規則および表によって指定された期間にわたって記録、処理、集約および報告されることを保証するための会社の制御および他のプログラムを意味する。開示制御及び手続は、取引所法に基づいて提出又は提出された報告書に開示を要求する情報が蓄積されていることを確保し、その主要幹部及び主要財務官を含む会社管理層に適宜伝達し、開示要求に関する決定を直ちに行うことを目的としているが、制御及び手続に限定されない。
経営陣は、どのような制御やプログラムが、どんなに設計や操作が良くても、その目標を実現するために合理的な保証を提供するしかないことを認識しており、管理部門は、可能な制御とプログラムのコスト-利益関係を評価する際にその判断を運用しなければならない。我々の2023年12月31日までの開示制御およびプログラムの評価によると、我々の最高経営責任者および最高財務官は、この日までに、私たちの開示制御およびプログラムは設計および運営において有効であり、合理的な保証レベルにあると結論した。
102
経営陣財務報告書内部統制年次報告書。我々の経営陣は、我々のCEO及び最高財務官の参加の下で、我々の財務報告に対する十分な内部統制の確立及び維持を担当しており、この用語は、1934年の証券取引法規則13 a−15(F)及び規則15 d−15(F)で定義されている。我々の財務報告に対する内部統制は公認された会計原則に基づいて、財務報告の信頼性と外部目的の財務諸表の作成に合理的な保証を提供する過程である。私たちの財務報告書に対する内部統制は以下の政策と手続きを含む
いかなる財務報告内部制御制度の有効性は、私たちの内部制御制度を含めて、内在的に制限されており、設計、実施、運営と制御とプログラムを評価する時に判断力を行使し、不正行為を完全に除去できないことを含む。したがって、どのような財務報告書の内部統制制度も、私たちの制度を含めて、設計と運営がどんなに良くても、絶対的な保証ではなく、合理的な保証しか提供できない。また,将来的にどのような有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムの遵守度が悪化する可能性がある.経営陣は、財務報告書の内部統制に対する2023年12月31日までの我々の有効性を評価した。評価にあたっては、経営陣は、財務報告書の内部統制に対する我々の有効性を評価するために、トレデビル委員会後援組織委員会(COSO)が“内部統制-総合枠組み(2013)”で提案した基準を使用した。これらの基準を用いた評価によると、経営陣は、財務報告に対する内部統制は2023年12月31日から有効であると結論した。
独立公認会計士事務所認証報告。2023年12月31日現在、財務報告の内部統制の有効性は、独立公認会計士事務所ピマウェイ有限責任会社が監査しており、このことは、本年度報告書10−K表の他の部分の報告で説明されている。
財務報告書の内部統制の変化。2023年12月31日までの3ヶ月間、取引所法案規則13 a-15(D)および15 d-15(D)の要求に基づいて行われた評価によると、財務報告の内部統制には何の変化もなく、財務報告の内部統制に大きな影響を与えたり、合理的な可能性が財務報告の内部統制に大きな影響を与えたりしている。
プロジェクト9 B。その他の情報
私たちは私たちのすべての役員、上級管理職、そして従業員に適用される書面行動基準を持っている。私たちの行動基準の最新バージョンは、会社のウェブサイトの“投資家”の部分で取得することができます。URLは:Http://www.xenon-pharma.comSEDARではWwwv.sedar.comそれは.私たちのウェブサイトの“投資家”欄で、私たちの行動基準の改正または役員と役員の免除を発表しますHttp://www.xenon-pharma.com.
2023年12月31日までの3ヶ月間、役員又は第16条の役員はいない
プロジェクト9 Cです検査妨害に関する外国司法管区の開示
適用されません。
103
部分(三)
プロジェクト10.取締役、上級管理者、および企業管理
Form 10−K第10項に要求される情報は,2023年12月31日までの財政年度終了後120日以内に提出される米国証券取引委員会に提出された2024年年次総会依頼書を参考にして組み込まれている。
プロジェクト11.役員報酬
Form 10−K第11項に要求される情報は,2023年12月31日までの財政年度終了後120日以内に提出される米国証券取引委員会に提出された2024年年次総会依頼書を参考にして組み込まれている。
プロジェクト12.特定の実益所有者の保証所有権及び管理職及び株主に関する事項
Form 10-K第12項に要求される情報は、2023年12月31日までの財政年度終了後120日以内に提出される米国証券取引委員会に提出された2024年年次総会依頼書を参照して組み込まれている。
第13項:特定の関係及び関連取引、並びに取締役独立性
Form 10−K第13項に要求される情報は,2023年12月31日までの財政年度終了後120日以内に提出される米国証券取引委員会に提出された2024年年次総会依頼書を参考にして組み込まれている。
プロジェクト14.主な会計費用とサービス
Form 10−K第14項に要求される情報は,2023年12月31日までの財政年度終了後120日以内に提出される米国証券取引委員会に提出された2024年年次総会依頼書を参考にして組み込まれている。
104
部分IV.IV
項目15.物証、財務諸表付表
(A)(1)財務諸表--項目8に掲げる財務諸表を、本年度報告シート10-Kの一部として提出する。
(A)(2)財務諸表明細書--すべての明細書が省略されているのは、これらの明細書が適用または不要であること、または本年度報告のリスト10-K第8項に記載されている連結財務諸表または付記に規定されている資料が含まれているためである。
(A)(3)展示品-S-K法規第601号に記載されている展示品は、以下(B)段落に掲げる。
(B)展示品-次の表に記載されている展示品は、添付アーカイブを添付するか、または米国証券取引委員会に以前に届出された展示品を参照して統合される。
展示品 |
|
書類説明 |
|
引用で編入する |
||||||
|
|
表 |
|
書類番号. |
|
展示品 |
|
提出日 |
||
|
|
|
|
|
|
|
|
|
|
|
3.1 |
|
会社の定款。 |
|
10-Q |
|
001-36687 |
|
3.1 |
|
2014年12月15日 |
|
|
|
|
|
|
|
|
|
|
|
3.1A |
|
会社定款改正案は、系列1優先株を設立する。 |
|
8-K |
|
001-36687 |
|
3.1 |
|
2018年3月28日 |
|
|
|
|
|
|
|
|
|
|
|
3.2 |
|
会社の付例を改訂及び再発注する. |
|
10-Q |
|
001-36687 |
|
3.2 |
|
2014年12月15日 |
|
|
|
|
|
|
|
|
|
|
|
4.1 |
|
普通株式証明書フォーマット。 |
|
S-1/A |
|
333-198666 |
|
4.1 |
|
2014年10月6日 |
|
|
|
|
|
|
|
|
|
|
|
4.2 |
|
Xenon製薬会社とシリコンバレー銀行の間で株を購入する権利証は、2018年8月3日。 |
|
8-K |
|
001-36687 |
|
4.1 |
|
2018年8月7日 |
|
|
|
|
|
|
|
|
|
|
|
4.3 |
|
証券説明。 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.4 |
|
あらかじめ出資して株式証の書式を承認する. |
|
8-K |
|
001-36687 |
|
4.1 |
|
2021年10月6日 |
|
|
|
|
|
|
|
|
|
|
|
4.5 |
|
資金を前払いして株式証明書を発行する. |
|
8-K |
|
001-36687 |
|
4.1 |
|
2022年6月23日 |
|
|
|
|
|
|
|
|
|
|
|
4.6 |
|
資金を前払いして株式証明書を発行する. |
|
8-K |
|
001-36687 |
|
4.1 |
|
2023年11月30日 |
|
|
|
|
|
|
|
|
|
|
|
10.1# |
|
修正された株式オプション計画とそのオプション協定のフォーマット。 |
|
S-1/A |
|
333-198666 |
|
10.7 |
|
2014年10月6日 |
|
|
|
|
|
|
|
|
|
|
|
10.2# |
|
2014年の株式インセンティブ計画及びそれに使用された株式オプション協定形式を改訂し、再改訂する。 |
|
8-K |
|
001-36687 |
|
10.1 |
|
2022年6月2日 |
|
|
|
|
|
|
|
|
|
|
|
10.3# |
|
役員は役員と賠償協議形式を協議した。 |
|
S-1/A |
|
333-198666 |
|
10.15 |
|
2014年10月6日 |
|
|
|
|
|
|
|
|
|
|
|
10.4 |
|
資産購入協定は、2017年4月25日に、会社と第一注文製薬会社が署名した。 |
|
10-Q |
|
001-36687 |
|
10.2 |
|
2017年8月3日 |
|
|
|
|
|
|
|
|
|
|
|
10.5 |
|
マイルストーンと特許権使用料買収協定は,2018年9月7日にXenon製薬会社,Valeant製薬アイルランド有限会社とValeant製薬ルクセンブルクS.a.r.l.が署名した。 |
|
8-K |
|
001-36687 |
|
10.1 |
|
2018年9月11日 |
|
|
|
|
|
|
|
|
|
|
|
10.6# |
|
当社とRobin Sherringtonとの間で2019年3月19日に改訂され、再署名された雇用協定。 |
|
10-K |
|
001-36687 |
|
10.24 |
|
2020年3月9日 |
|
|
|
|
|
|
|
|
|
|
|
10.7# |
|
当社がChristopher Von Seggernと締結した雇用協定は,2020年7月17日である。 |
|
10-Q |
|
001-36687 |
|
10.3 |
|
2020年11月5日 |
|
|
|
|
|
|
|
|
|
|
|
10.8# |
|
会社がイアン·モティマーと結んだ雇用契約は、2021年1月13日。 |
|
8-K |
|
001-36687 |
|
10.2 |
|
2021年1月14日 |
|
|
|
|
|
|
|
|
|
|
|
10.9# |
|
当社がシェリー·オーリンと締結した雇用契約は、2021年1月13日となっている。 |
|
8-K |
|
001-36687 |
|
10.4 |
|
2021年1月14日 |
|
|
|
|
|
|
|
|
|
|
|
105
展示品 |
|
書類説明 |
|
引用で編入する |
||||||
|
|
表 |
|
書類番号. |
|
展示品 |
|
提出日 |
||
|
|
|
|
|
|
|
|
|
|
|
10.10# |
|
当社がクリストファー·ケニーと締結した雇用契約は、2021年8月18日となっている。 |
|
10-Q |
|
001-36687 |
|
10.3 |
|
2021年11月10日 |
|
|
|
|
|
|
|
|
|
|
|
10.11# |
|
2019年株式インセンティブ計画及び関連形式の株式オプション協定. |
|
8-K |
|
001-36687 |
|
10.1 |
|
2019年9月10日 |
|
|
|
|
|
|
|
|
|
|
|
10.12 |
|
許可と協力協定は、2019年12月2日にXenon PharmPharmticals Inc.とNeurocrine Biosciences,Inc.によって署名された。 |
|
8-K |
|
001-36687 |
|
10.1 |
|
2019年12月2日 |
|
|
|
|
|
|
|
|
|
|
|
10.13 |
|
Xenon製薬会社とNeurocrine Biosciences,Inc.との間の株式購入契約は,2019年12月2日である。 |
|
8-K |
|
001-36687 |
|
10.2 |
|
2019年12月2日 |
|
|
|
|
|
|
|
|
|
|
|
10.14 |
|
当社と第一注文製薬会社との間で2020年8月4日に署名された資産購入協定の第1号改正案。 |
|
10-Q |
|
001-36687 |
|
10.2 |
|
2020年8月6日 |
|
|
|
|
|
|
|
|
|
|
|
10.15 |
|
2020年8月6日までの市場持分発行販売協定は,Xenon製薬会社,Jefferies LLCとStifel,Nicolaus&Company,Inc.が署名した。 |
|
8-K |
|
001-36687 |
|
1.1 |
|
2020年8月6日 |
|
|
|
|
|
|
|
|
|
|
|
10.16 |
|
Xenon製薬会社,Jefferies LLCとStifel,Nicolaus&Company,Inc.が2022年3月1日に署名した市場持分発行販売協定の第1号改正案。 |
|
8-K |
|
001-36687 |
|
1.1 |
|
2022年3月1日 |
|
|
|
|
|
|
|
|
|
|
|
10.17 |
|
Xenon PharmPharmticals Inc.とNeurocrine Biosciences,Inc.が2019年12月2日に署名した許可及び協力協定の第1号修正案は,2021年1月13日である。 |
|
8-K |
|
001-36687 |
|
10.1 |
|
2021年1月14日 |
|
|
|
|
|
|
|
|
|
|
|
10.18 |
|
Xenon製薬会社、遺伝子技術会社とF.Hoffmann-La Roche Ltd.の間で調印された終了協定は、2021年8月6日である。 |
|
10-Q |
|
001-36687 |
|
10.1 |
|
2021年8月11日 |
|
|
|
|
|
|
|
|
|
|
|
10.19 |
|
Xenon製薬会社とNeurocrine Biosciences,Inc.との間の株式購入契約は,2021年9月8日である. |
|
8-K |
|
001-36687 |
|
10.1 |
|
2021年9月8日 |
|
|
|
|
|
|
|
|
|
|
|
10.20 |
|
当社は赤石企業有限公司と締結し、2021年11月24日に発効した賃貸契約。 |
|
8-K |
|
001-36687 |
|
10.1 |
|
2021年12月1日 |
|
|
|
|
|
|
|
|
|
|
|
10.21 |
|
Xenon製薬会社とNeurocrine生物科学社との間の株式購入協定は,2022年1月11日である。 |
|
8-K |
|
001-36687 |
|
10.1 |
|
2022年1月12日 |
|
|
|
|
|
|
|
|
|
|
|
10.22 |
|
Xenon PharmPharmticals Inc.とNeurocrine Biosciences,Inc.が2019年12月2日に署名した許可及び協力協定の修正案第2号は,2022年2月25日である。 |
|
10-K |
|
001-36687 |
|
10.30 |
|
2022年3月1日 |
|
|
|
|
|
|
|
|
|
|
|
10.23 |
|
Redstone企業有限公司とXenon製薬会社との間の改築とレンタル改正協定同意書が、2022年5月19日に発効した。 |
|
10-Q |
|
001-36687 |
|
10.1 |
|
2022年8月9日 |
|
|
|
|
|
|
|
|
|
|
|
10.24# |
|
会社とアンドレア·ディファビオとの雇用協定は、2022年11月7日となっている。 |
|
10-Q |
|
001-36687 |
|
10.1 |
|
2022年11月8日 |
|
|
|
|
|
|
|
|
|
|
|
10.25# |
|
役員は報酬計画を奨励する。 |
|
8-K |
|
001-36687 |
|
10.1 |
|
2023年3月14日 |
|
|
|
|
|
|
|
|
|
|
|
106
展示品 |
|
書類説明 |
|
引用で編入する |
||||||
|
|
表 |
|
書類番号. |
|
展示品 |
|
提出日 |
||
|
|
|
|
|
|
|
|
|
|
|
10.26 |
|
Redstone Enterprise Ltd.とXenon PharmPharmticals Inc.の間の改装合意同意書は,2023年3月27日に発効した。 |
|
10-Q |
|
001-36687 |
|
10.1 |
|
2023年8月9日 |
|
|
|
|
|
|
|
|
|
|
|
10.27 |
|
Redstone Enterprise Ltd.とXenon PharmPharmticals Inc.の間の改装合意同意書は,2023年8月16日に発効した。 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.28# |
|
当社がジェームズ·エンプフィールドと締結した雇用契約は、2023年12月15日となっている。 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
21.1 |
|
当社の子会社リストです。 |
|
10-K |
|
001-36687 |
|
21.1 |
|
2017年3月8日 |
|
|
|
|
|
|
|
|
|
|
|
23.1 |
|
独立公認会計士事務所ピマウェイ会計士事務所が同意します。 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
24.1 |
|
授権書(署名ページに掲載)。 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
31.1 |
|
細則13 a-14(A)/15 d-14(A)特等実行幹事の証明書 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
31.2 |
|
細則13 a-14(A)/15 d-14(A)首席財務幹事の証明 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
32.1* |
|
第一百五十条首席行政官の証明書 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
32.2* |
|
第一百五十条首席財務主任の証明 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
97 |
|
還付政策は、2023年10月2日から施行されます |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.INS |
|
XBRLタグがイントラネットXBRL文書に埋め込まれているので、連結されたXBRLインスタンス文書−インスタンス文書は、対話データファイルには表示されない |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.書院 |
|
インラインXBRL分類拡張アーキテクチャ文書と埋め込まれたLinkbase文書 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
104 |
|
表紙インタラクションデータファイル(イントラネットXBRL文書に埋め込む) |
|
|
|
|
|
|
|
|
本展示品のいくつかの部分に対して、秘密保護待遇が与えられた。見落とした部分は米国証券取引委員会(Securities And Exchange Commission)に単独で提出されている。
S-K法規第601(B)(10)項の規定により、本展示品の一部の内容は省略されており、それらは秘密であり、実質的ではない。
#は、管理契約または補償計画を示します。
*本10-K表年次報告に添付されている証拠物32.1および32.2に添付されている証明は、証券取引委員会に提出されたものとはみなされず、参照によってXenon製薬会社が1933年の“証券法”(改訂本)または1934年の“証券取引法”(改訂本)に従って提出されたいずれの文書にも、当該文書が本リスト10-Kの日付の前または後に提出されたものであっても、その文書に含まれる任意の一般的な登録言語にかかわらず、適用されてはならない。
第十六項表格10-Kの概要
適用されません。
107
標札すきま
1934年“証券取引法”第13又は15(D)節の要求に基づいて、登録者は、正式に許可された以下の署名者がその代表を代表して本報告に署名することを正式に手配した。
日付:2024年2月29日 |
|
キセノン製薬会社です。 |
||
|
|
差出人: |
|
/S/イアン·モティマー |
|
|
|
|
イアン·モティマー |
|
|
|
|
社長と最高経営責任者 |
授権依頼書
以下の署名のすべての人は、Simon Pimstone、Ian MortimerおよびSherry Alinを構成し、任命し、それぞれ彼または彼女の真の合法的な事実弁護士および代理人として、他の人なしに彼または彼女の名義、位置、および代替の身分で行動する十分な権力を有する(彼または彼女が取締役および/またはXenon製薬会社の上級職員として機能することを含む)。本報告書の任意およびすべての修正および補足、ならびに本報告書に関連する任意およびすべての他の必要または付随文書に署名し、すべての証拠品およびこれに関連するすべての他の文書と共に委員会に提出する。
本報告は、1934年の証券取引法の要求に基づき、以下の登録者によって登録者として指定日に署名された。
サイン |
|
タイトル |
|
日取り |
/S/イアン·モティマー イアン·モティマー |
|
取締役CEO社長(最高経営責任者) |
|
2024年2月29日 |
投稿S/シェリー·オーリン |
|
首席財務官(首席財務·会計幹事) |
|
2024年2月29日 |
シェリー·オーリン |
||||
寄稿S/シモン·ピムストーン |
|
取締役会議長 |
|
2024年2月29日 |
サイモン·ピムストーン |
|
|
||
/S/ムハンマド·アザブ |
|
役員.取締役 |
|
2024年2月29日 |
モハメド·アザブ |
|
|
||
/S/ジリアン·カノン |
|
役員.取締役 |
|
2024年2月29日 |
ジリアン·カノン |
|
|
||
寄稿S/スティーブン·ガンノン |
|
役員.取締役 |
|
2024年2月29日 |
スティーブン·ガンノン |
|
|
||
/投稿S/エリザベス·ガロファロ |
|
役員.取締役 |
|
2024年2月29日 |
エリザベス·ガロファロ |
|
|
||
/投稿S/ジャスティン·ゴヴァー |
|
役員.取締役 |
|
2024年2月29日 |
ジャスティン·ゴファー |
|
|
||
/S/パトリック·マッチャド |
|
役員.取締役 |
|
2024年2月29日 |
パトリック·マチャド |
|
|
||
/S/ゲイリー·パトゥ |
|
役員.取締役 |
|
2024年2月29日 |
ゲイリー·パトゥー |
|
|
||
/S/ドーン·スヴォロノス |
|
役員.取締役 |
|
2024年2月29日 |
ドーン·スヴォロノス |
|
|
108