VRNA-20231231誤り2023会計年度0001657312P 2 Y前払い費用前払い料金には以下の料金が含まれています(単位:千):
| | | | | | | | | | | | | | |
| | 十二月三十一日 |
| | 2023 | | 2022 |
| 臨床試験やその他の開発費 | | $ | 59 | | | $ | 38 | |
| 保険 | | 3,558 | | | 2,027 | |
| 他にも | | — | | | 434 | |
| 前払い費用総額 | | $ | 3,617 | | | $ | 2,499 | |
前払い料金には以下の料金が含まれています(単位:千):
| | | | | | | | | | | | | | |
| | 十二月三十一日 |
| | 2023 | | 2022 |
| 臨床試験やその他の開発費 | | $ | 59 | | | $ | 38 | |
| 保険 | | 3,558 | | | 2,027 | |
| 他にも | | — | | | 434 | |
| 前払い費用総額 | | $ | 3,617 | | | $ | 2,499 | |
2022年5月2日、残りのすべての引受権証が満期になりました。2023年12月31日及び2022年12月31日までの年度内に、持分証の行使或いは没収は何もない。
2016年、会社は新投資家と既存投資家に31,115,926単位を発行し、配給価格は1単位あたり1.4365 GBだった。各単位は普通株と株式承認証を含む。2022年5月2日までに、権証所持者は1株当たり1.7238 GBの発行価格で0.4%の普通株を承認することができる。権利証所有者は、無現金でその権利を行使することを選択することができ、権証所有者は、ゼロ対価格で行使可能な権証をより少ない数の権証と交換することを選択することができる。減少した株式引受証数は,株式承認証の株価と行使価格を考慮した式から計算される.
取引後、権利証が非上場証券に対して行使可能である場合、権証所持者は、対象証券を交付するのではなく、現金支払いを要求することができる。そのため、株式承認証はアメリカ会計基準第480号“負債と権益を区別する”項の下の負債とされている。株式承認証は公正価値によって計量し、公正価値レベルの中で第三級に分類され、その変動は総合経営報告書と全面損失中の他の収入/(支出)に計上される。
2021年12月31日まで、31,003,155件の株式承認証がまだ発行されておらず、投資家は合計最大12,401,262株の普通株を引受する権利がある。
2021年12月31日まで、このような権証には内在的な価値がない。
2023年12月31日から2022年12月31日までの年間で,推定技術や公正価値計測レベル間の移行は変化しなかった。公正価値は、2021年12月31日から2022年5月2日までの間に変化しない。株式承認証の推定値にはブラック·スコルスモデルを用い、使用した仮定を以下の表に示す
| | | | | | | | |
| | 十二月三十一日 |
| | 2022 |
| |
| ポンド単位の行権価格 | £ | 1.7238 | |
| 無リスク金利 | | 0.07 | % |
| 行使期限を見込む | | 0.33 |
| 年化変動率 | | 51.6 | % |
| 配当率 | — | % |
| 権利証の計算価値は千ドル単位で | $ | — | |
次の表に株式承認証価値の変動状況(千計)を示す
| | | | | | | | |
| | 十二月三十一日 |
| | 2022 |
| 一月一日 | | $ | 2,246 | |
| | |
| 公正価値調整 | | (2,246) | |
| | |
| | |
| 十二月三十一日 | | $ | — | |
| | | | | | | | |
| | 十二月三十一日 |
| | 2022 |
| |
| ポンド単位の行権価格 | £ | 1.7238 | |
| 無リスク金利 | | 0.07 | % |
| 行使期限を見込む | | 0.33 |
| 年化変動率 | | 51.6 | % |
| 配当率 | — | % |
| 権利証の計算価値は千ドル単位で | $ | — | |
次の表に株式承認証価値の変動状況(千計)を示す
| | | | | | | | |
| | 十二月三十一日 |
| | 2022 |
| 一月一日 | | $ | 2,246 | |
| | |
| 公正価値調整 | | (2,246) | |
| | |
| | |
| 十二月三十一日 | | $ | — | |
アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表10-K
☒1934年証券取引法第13条又は15条に基づいて提出された年次報告
本財政年度末まで十二月三十一日, 2023
あるいは…
☐1934年証券取引法第13条又は15条に基づいて提出された移行報告
_から_への過渡期
依頼書類番号:001-38067
ヴェローナ製薬会社
(登録者の正確な氏名はその定款に記載) | | | | | | | | | | | |
| イギリス.イギリス | | | 98-1489389 |
| (登録設立又は組織の国又はその他の管轄区域) | | | (国際税務局雇用主身分証明書番号) |
| | | |
ロンドンのリバーサイドをもう3つください ロンドン.ロンドンSE 1 2 REイギリス.イギリス | | | 適用されない |
(主にオフィスアドレスを実行) | | | (郵便番号) |
登録者の電話番号、市外局番を含む:+44203283 4200
同法第12条(B)に基づいて登録された証券:
| | | | | | | | |
| クラスごとのタイトル | 取引記号 | 登録された各取引所の名称 |
| 普通株、1株当たり0.05 GB* | VRNA | ナスダック(Tmall Stock Market LLC)ナスダック世界市場) |
*普通株式は米国預託株式(1株当たり8株普通株に相当)に代表され、規則12 A-8に基づいて改正された1934年の証券取引法第12条(A)条の影響を受けない。
同法第12条(G)に基づいて登録された証券:
ありません
登録者が証券法第405条に規定する有名な経験豊富な発行者である場合は、再選択マークで表示してくださいはい、そうです☒ありません。☐
登録者がこの法第13又は15(D)条に従って報告書を提出する必要がないか否かを、再選択マークで示す。はい。☐違います。 ☒
登録者が(1)過去12ヶ月以内(または登録者がそのような報告を提出する必要があるより短い期間)に1934年の証券取引法第13条または15(D)条に提出されたすべての報告書を再選択マークで示すかどうか、および(2)このような提出要求を過去90日以内に遵守してきたはい、そうです☒ありません。☐
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示すはい、そうです☒ありません。☐
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
| | | | | | | | | | | | | | |
| 大型加速ファイルサーバ | ☒ | | 加速されたファイルマネージャプロファイル | ☐ |
| 非加速ファイルサーバ | ☐ | | 小型報告会社 | ☒ |
| | | 新興成長型会社 | ☐ |
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる☒
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する☐
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に第240.10 D−1(B)条に従って受信されたインセンティブベースの補償に従って回復分析を行う必要があるかどうかを再選択マークで示す
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)。はい、違います☒
非関連会社が保有する登録者が投票権と無投票権を有する普通株の総時価は約#ドルである1.52023年6月30日まで、つまり登録者が最近完成した第2四半期の最終営業日である。本開示の目的のためにのみ、登録者の役員、取締役、および特定の株主がその日に保有する株式は、そのような者または実体が登録者の連属会社とみなされる可能性があるので、除外される
2024年2月23日現在登録者は646,524,958普通株は、1株当たり額面0.05 GB、発行済みであり、すべて米国預託株式形式で保有すれば、80,815,620株米国預託株式を代表し、1株当たり8(8)株普通株に相当する。
引用で編入された書類
登録者は,登録者2024年年度株主総会に関連する最終委託書の内容の一部を第14 A条に基づいて証券取引委員会に提出する予定であり,本稿で述べた範囲内で本年度報告のForm 10−K第3部を参照して組み込む予定である。
一般情報
本年度報告で言及した“ヴェローナ”,“会社”,“グループ”,“我々”はいずれもヴェローナ製薬会社とその合併子会社を指す。本年度報告では、米国証券取引委員会を“米国証券取引委員会”、1933年に証券法が改正されて“証券法”、1934年に証券取引法が改正されて“取引法”と呼ばれている
商標、商標名、サービスマーク
本年度報告書には、他の組織の財産に属する商標、商標名、およびサービスマークが含まれる場合がある。便宜上、本年度報告で言及された商標および商標名は使用および記号を使用していないが、これらの参照は、適用法律に基づいて私たちの権利を最大限に主張しないこと、または適用されたすべての人がこれらの商標および商標名に対する権利を主張しないことを意味するわけではない。
前向き陳述に関する警告説明
本年度報告書は展望的な陳述を構成する陳述を含む。場合によっては、前向き表現は、すべての前向き表現がこれらの語を含むわけではないが、“可能”、“可能”、“会議”、“すべき”、“予想”、“計画”、“予想”、“可能”、“意図”、“目標”、“プロジェクト”、“想定”、“信じ”、“推定”、“予測”、“潜在的”または“継続”またはこれらの用語の否定または他の同様の表現によって識別することができる。本年度報告に含まれる歴史的事実の陳述を除いて、本年度報告に含まれるすべての陳述は、私たちの将来の運営の結果および財務状況、業務戦略および将来の運営の計画および管理目標、イソフェンまたは任意の他の候補製品の開発に関する陳述を含むが、我々の臨床試験および潜在的な規制承認に関するデータの予想開始、時間、進展および利用可能性に関する陳述、およびイソフェンタニルに適用される予想法規、研究開発コスト、成功のタイミングと可能性、潜在的な協力、私たちの特許組み合わせの持続時間、費用、将来の収入、資本要求、債務超過義務の推定、および追加融資に対する私たちの需要を含む。私たちは、2023年の定期融資とイギリスの税金免除からの現金収入で得られる資金と、私たちの現金と現金等価物が運営に資金を提供するのに十分かどうかを予想している
本年度報告書の展望的陳述は予測のみであり、主に私たちの現在の未来の事件と財務傾向の予想と予測に基づいており、これらの事件と財務傾向は私たちの業務、財務状況、運営結果に影響を与える可能性があると考えられる。これらの前向き表現は、本年度報告が発表された日までの状況のみについて、本年度報告“リスク要因”および“経営層の財務状況および経営結果の議論と分析”および米国証券取引委員会に提出された他の報告書の他の章に記載された重要な要素を含む、多くの既知および未知のリスク、不確実性および仮定の影響を受ける。
展望性陳述自体はリスクと不確実性の影響を受けるため、その中のいくつかのリスクと不確定性は予測できないか定量化されており、いくつかは私たちが制御できないので、あなたは未来の事件の予測としてこれらの展望的陳述に依存してはならない。著者らの展望性陳述に反映された事件と状況は実現できない或いは発生できない可能性があり、実際の結果は展望性陳述中の予測結果と大きく異なる可能性がある。しかも、私たちは持続的な環境で運営している。法的要件が適用されない限り、私たちは、任意の新しい情報、未来のイベント、状況の変化、または他の理由による、本明細書に含まれる任意の前向きな陳述を公開または修正するつもりはありません。我々は,本年度報告に含まれる前向き陳述を証券法第27 A節と取引法第21 E節の前向き陳述に関する安全港条項に盛り込む予定である。
本年度報告には,業界出版物から得られた市場データと業界予測が含まれている。このようなデータは多くの仮定と制限と関連があり、あなたにこのような推定を過度に重視しないように想起させる。私たちはまだ第三者情報を独立して確認していない。本年報に掲載されている市場地位,市場機会,市場規模情報はほぼ信頼できると信じているが,このような情報は本質的に正確ではない.
リスク要因をまとめる
私たちの業務は、第1部1 A項で述べたリスクと不確実性を含む多くのリスクと不確実性に直面している。本年度報告書の“リスク要因”。私たちのアメリカ預託証明書に投資する時、あなたはこのような危険と不確実性を慎重に考慮しなければならない。私たちの業務に影響を与える主なリスクと不確定要素は以下の通りです
•私たちの経営の歴史は限られていて、どんな製品収入も生まれたことがありません
•承認されれば、アンテフェンタニルおよび任意の将来の候補製品の開発および商業化を達成するための追加の資金が必要になるかもしれません。または承認された場合、アンテフェンタリンの他の製剤または標的適応を開発および商業化するために追加の資金が必要になるかもしれません
•2023年4.0億ドルの定期融資項目の下敷きは、いくつかの臨床および規制マイルストーンおよび他の指定された条件の実現に依存する。もし私たちがこのような条件を満たすことができなければ、私たちは代替資金源を探す必要があるだろう
•私たちの税率の変化、いくつかの税金控除または減免を得ることができない、または追加の税金負担または評価に直面することは、私たちの収益力に影響を与える可能性があり、税務機関の監査は、前のいくつかの時期の追加納税をもたらす可能性がある
•私たちはアンテファニンの成功に頼っていますこれが私たちが開発している唯一の候補品です
•私たちは私たちの候補製品の開発と商業化の過程で追加のコストが発生したり、遅延が発生したりすることができないかもしれない
•エンゼフェンタンは深刻な不良、不良、あるいは受け入れられない副作用がある可能性があり、上場承認を延期または阻止する可能性がある
•他の適応を得るために患者を臨床試験に組み込むことができない場合や募集速度が予想より遅い場合、私たちの研究や開発は悪影響を受ける可能性があります
•私たちは高価で破壊的な責任クレームに直面するかもしれません。臨床テストでも商業段階でも、私たちの製品責任保険はこのようなクレームのすべての損害を含まないかもしれません
•監督管理の審査過程は長く、時間がかかり、本質的に予測できず、もし私たちが最終的に監督部門のアンテフェンに対する承認を得られなければ、私たちの業務は実質的な損害を受けるだろう
•制定と将来の立法は、アンテファンドリンの上場承認と商業化の難しさとコストを増加させ、私たちが制定する可能性のある価格に影響を与える可能性がある
•私たちの業務運営と調査者、医療専門家、コンサルタント、第三者支払者、顧客との現在と将来の関係は、適用される医療規制法の制約を受け、処罰される可能性があります
•私たちがいる業界の競争は激しく、変化が迅速で、これは他の人が私たちよりも早く、あるいはより成功的に製品を発見、開発、または商業化することにつながるかもしれない
•私たちは依存し、そして引き続き第三者に依存して、独立した臨床研究者と臨床研究機関を含めて、著者らの臨床前研究と臨床試験を行う予定である
•Nuance Pharmaとの協力と許可協定は私たちの業務に非常に重要だ。Nuance Pharmaがアンテフェンタリンを含む製品を大中華区で開発および販売できない場合、もし私たちまたはNuance PharmaがNuanceプロトコルを十分に履行できなかった場合、または私たちまたはNuance PharmaがNuanceプロトコルを終了した場合、私たちのビジネスは悪影響を受けるだろう
•もし私たちがアンテファンドリンのために新しい戦略関係を築くことができなければ、私たちの業務、研究開発、商業化の将来性は不利な影響を受ける可能性がある
•私たちは依存し、引き続き第三者メーカーとサプライヤーに依存して活性医薬成分アンチフェンとそれから誘導された製剤医薬製品を生産することが予想される。このような第三者への依存は私たちの研究開発計画の進展やアンテフェンタニルの開発を損なう可能性があります
•私たちの薬品の販売、マーケティング、精算、流通はすべて第三者に依存し、これらの第三者が職責を十分に履行できなければ、私たちの業務に悪影響を及ぼすと予想されています
•私たちと私たちの製造業者、サプライヤー、および他の重要な第三者のネットワークセキュリティリスク管理計画およびプロセスは、私たちのシステム、ネットワーク、および機密情報を効果的に保護できないかもしれません
•私たちは特許と他の知的財産権に依存してアンテファンドリンを保護し、その実行、弁護、メンテナンスは挑戦的でコストがあるかもしれない
•私たちは関連する第三者特許を識別していないかもしれないし、第三者特許の関連性、範囲、または失効を誤って解釈する可能性があり、これは私たちがエステファン塩基を開発、製造、販売する能力に悪影響を及ぼす可能性がある
•私たちは、法廷で疑問が提起された場合、発行された特許が無効または実行不可能と認定される可能性があるアンテファンをカバーする特許を保護または強制執行する訴訟に巻き込まれる可能性がある
•私たちの将来の成長と競争能力は、私たちがキーパーソンを維持し、より多くの合格者を募集する能力にかかっている
•私たちは私たちの開発、規制、販売とマーケティング能力を拡大することを望んでいます。したがって、私たちは私たちの成長を管理する上で困難に直面する可能性があり、これは私たちの運営を乱すかもしれません
•私たちのアメリカ預託株式の価格は変動する可能性があり、私たちがコントロールできない要素によって変動するかもしれません
•上場企業として、米国での運営コストは引き続き増加し、私たちの上級管理職は、新たなコンプライアンス措置やコーポレートガバナンス実践を実施するために多くの時間を投入する必要がある。
| | | | | | | | | | | |
| カタログ表 |
| | | ページ |
| 第1部 | | | |
第1項。 | 業務.業務 | | 1 |
第1 A項。 | リスク要因 | | 32 |
項目1 B。 | 未解決従業員意見 | | 77 |
プロジェクト1 C。 | ネットワーク·セキュリティ | | 77 |
第二項です。 | 属性 | | 79 |
第三項です。 | 法律訴訟 | | 79 |
第四項です。 | 炭鉱安全情報開示 | | 79 |
| 第II部 | | | |
五番目です。 | 登録者普通株市場、関連株主事項及び発行者による株式証券の購入 | | 80 |
第六項です。 | [保留されている] | | 80 |
第七項。 | 経営陣の財務状況と経営成果の検討と分析 | | 80 |
第七A項。 | 市場リスクの定量的·定性的開示について | | |
第八項です。 | 財務諸表と補足データ | | 90 |
第九項です。 | 会計と財務情報開示の変更と相違 | | 90 |
第9条。 | 制御とプログラム | | 91 |
プロジェクト9 B。 | その他の情報 | | 92 |
プロジェクト9 Cです。 | 検査妨害に関する外国司法管区の開示 | | 92 |
| 第三部 | | | |
第10項。 | 役員·幹部と会社の管理 | | 93 |
第十一項。 | 役員報酬 | | 93 |
第十二項。 | 特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 | | 93 |
十三項。 | 特定の関係や関連取引、取締役の独立性 | | 93 |
14項です。 | 最高料金とサービス | | 93 |
| 第IV部 | | | |
第十五項。 | 展示·財務諸表明細書 | | 94 |
第十六項。 | 表格10-Kの概要 | | 95 |
サイン | | | 96 |
第1項:商業銀行業務
概要
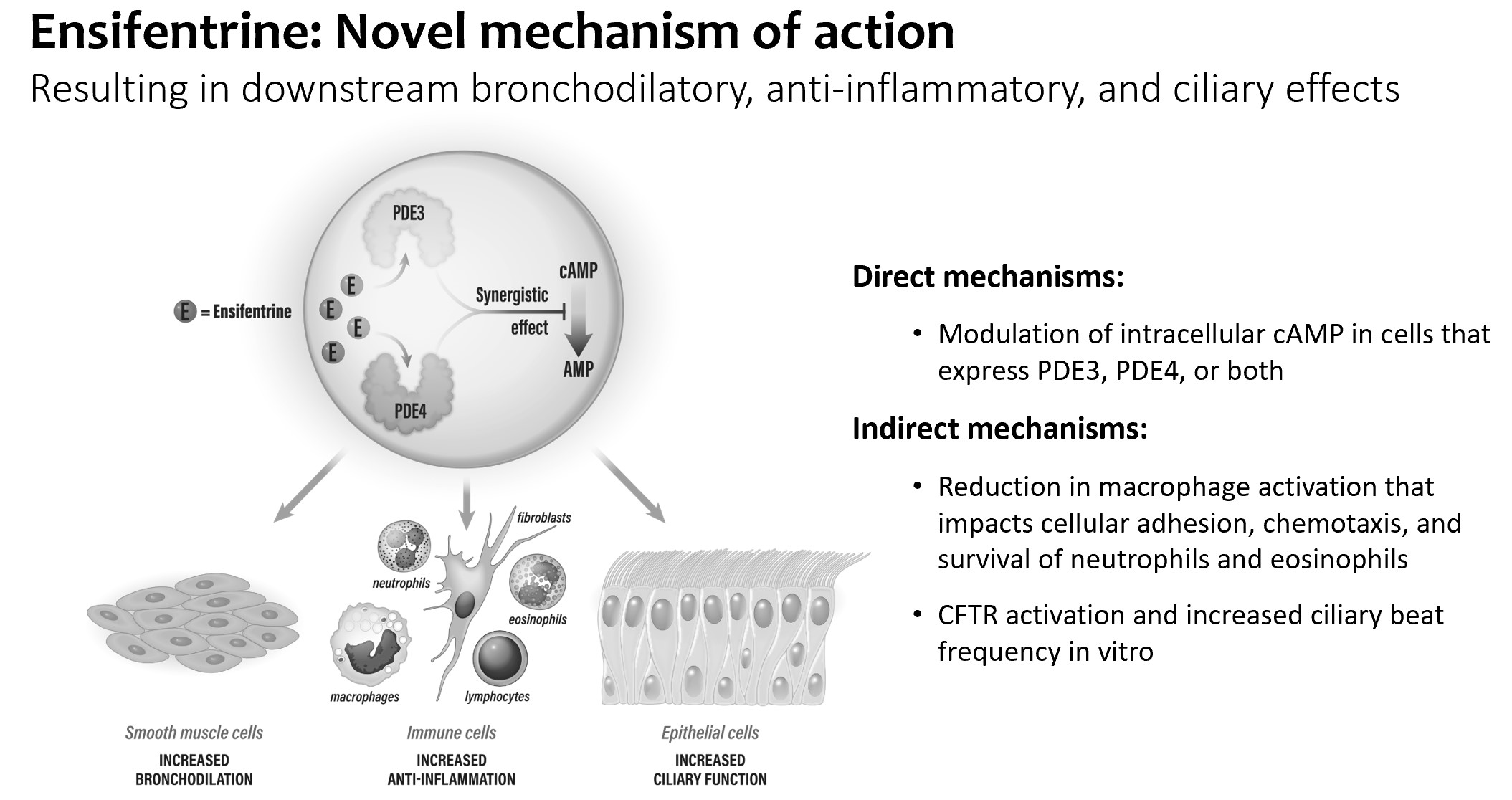
著者らは臨床段階の生物製薬会社であり、革新療法の開発に専念し、それを商業化し、重大な医療需要を満たしていない呼吸器疾患の慢性治療に応用している。我々の候補製品ensfentineは研究中の一流の吸入性、選択的ホスホジエステラーゼ3と4酵素(“PDE 3”と“PDE 4”)の二重阻害剤であり、1つの分子に気管支拡張剤と非ステロイド抗炎症活性を結合した
最初、著者らは吸入型アンチフェンタニルを開発しており、慢性閉塞性肺疾患(COPD)の維持治療に応用されており、COPDはよく見られる、慢性的、進行性、生命を脅かす呼吸器疾患であり、治愈できない。承認されれば、アンテフェンタニルは20数年来初めて新しい作用モードでCOPD維持治療に応用する吸入性療法になることが期待される。
2023年8月,米国食品医薬品局(FDA)はCOPD維持治療のための新薬申請(NDA)の承認を求める審査を受け,処方薬使用料法案(PDUFA)の目標行動日を2024年6月26日とした。FDAは,現在この申請を検討するために諮問委員会会議を開催する予定はないと述べている。
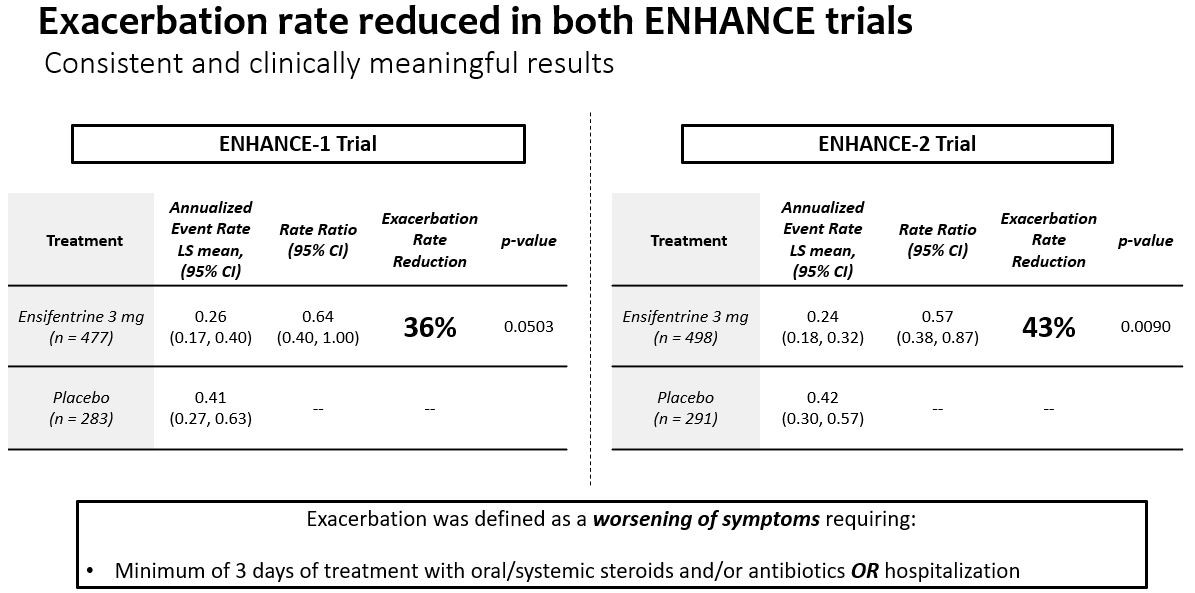
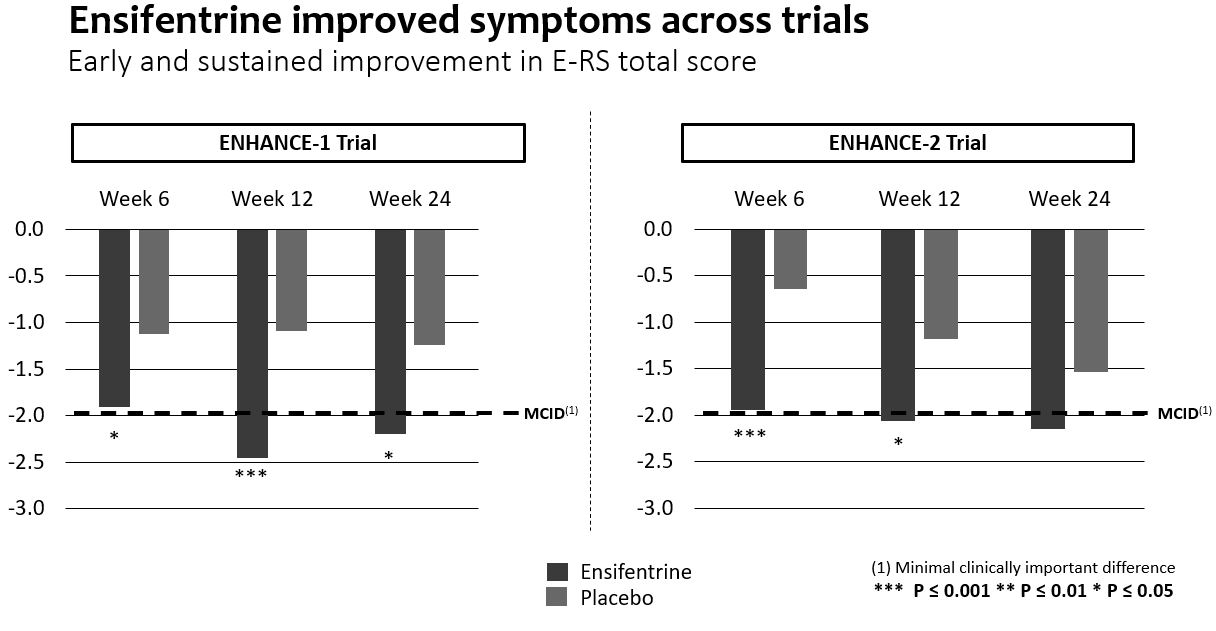
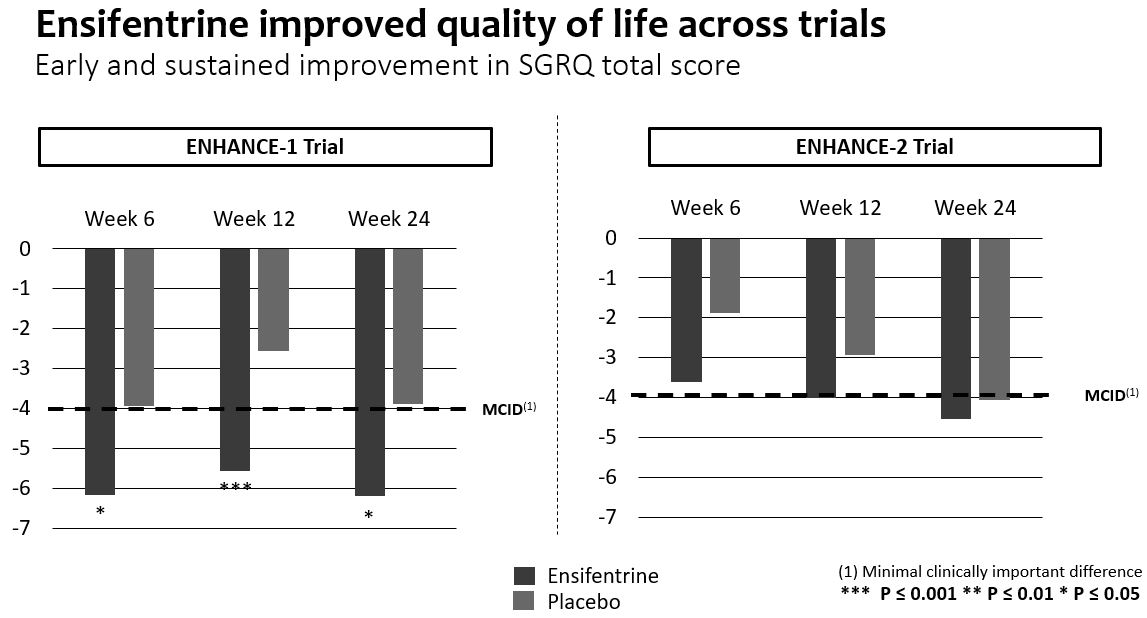
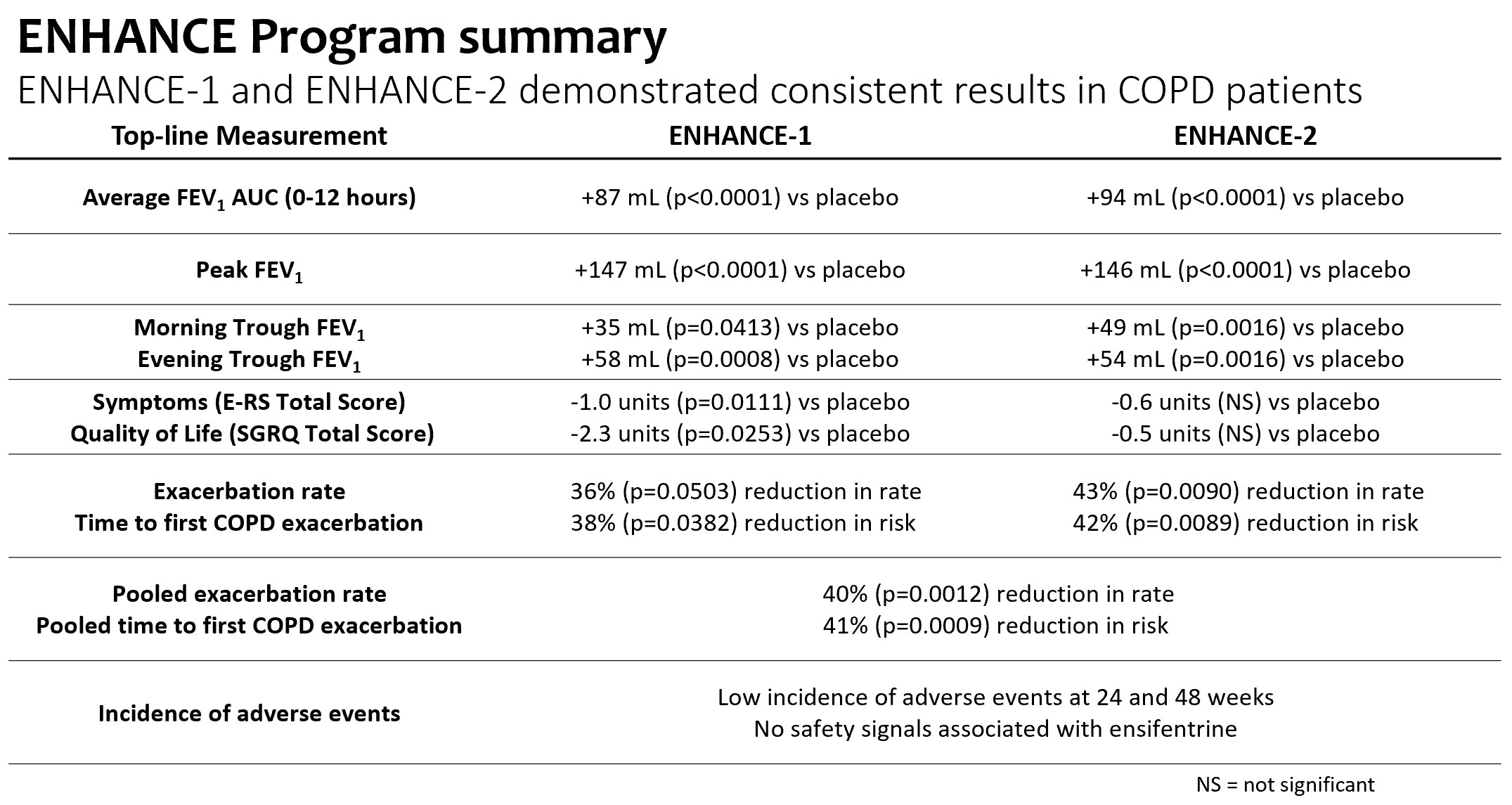
著者らが成功した第三段階Ensifentine(“新型吸入型COPD霧化療法として”)計画の結果に基づいて、著者らは承認されれば、アンテフェンタニルはCOPDの治療モードを変える可能性があると信じている。EnsiftreineはEnhance−1とEnhance−2試験ともに主要な終点に達しており,肺機能測定に統計的有意かつ臨床的に有意な改善が示唆された。肝心な副次的な終点で、アンテフェンタニルは症状と生活の質の早期と持続的な改善を示した。また,他の終点データでは,Enhance−1とEnhance−2では,イソフェントラリンがCOPD悪化の確率とリスクを有意に低下させた。エンセフェンタニルは両試験とも耐性が良好であった。
2023年に私たちは国際科学会議で強化試験のデータの追加分析を提出し、これらのデータは同業者評議の出版物に発表された
•2023年5月,我々は2023年米国胸科学会国際会議(ATS)で12編の要約と1回のシンポジウムを提出し,病状悪化,救急薬の使用,医療保健の利用を含む増強試験の拡張の事前指定と事後分析を検討した。新たな革新薬を強調するために温存した臨床試験シンポジウムの一部として,増強試験結果の概要を紹介した。つまり,すべての臨床関連亜群と評価の集約分析において,アンテフェンタニルは肺機能の改善と悪化確率とリスクの低下を含む高度に一致した結果を示した。その他の重要な分析により、アンテフェンタニルはSGRQ*サブドメインを含む症状と生活の質指標を改善し、救急薬物の使用と医療保健の利用を減少した。また,拡張した安全性分析では,アンテフェンタニルは良好な耐性を示した
*セントジョージ呼吸アンケートは効果的な患者報告結果ツールです
•これらの要約はATSサイトおよび“アメリカ呼吸と危篤看護医学雑誌”(AJRCCM)に発表された
•2023年6月、拡張結果がAJRCCMに発表された
•2023年9月、著者らは2023年ERS国際大会でEnhance-1 24週の悪化データの分析を提出し、このデータはイソプロピルアドレナリン治療は中度と重度COPD悪化の発生率とリスクを明らかに低下させたことを表明した。この要約は“ヨーロッパ呼吸雑誌”に発表されました
•2023年10月、著者らは2023年胸科年会でEnhance-1とEnhance-2のまとめとグループ分け後の分析に関する4回の報告を発表し、病状の悪化、肺機能、症状と生活の質の終点及び日常救援薬物の使用に関するデータをカバーした。最近の増悪歴にかかわらず,アンテフェンタニル治療はCOPD悪化の比率とリスクを大きく低下させ,耐性は良好であった。また、グループ分け分析により、アンテフェン洗浄治療は肺機能、症状と生活の質の指標を改善し、背景治療を考慮することなく、悪化の確率とリスクを低下させ、日常救急薬物の使用を減少させることができることを示した。これらのデータは“胸科年会”のオンライン副刊に発表された。また、胸部では、著者らは疾病意識運動を開始し、どれだけのCOPD患者が自分の病状を討論しにくいことを強調した。
私たちは依然としてアメリカでのアンチフェンニンの商業化に集中すると同時に、私たちは霧化吸入による慢性閉塞性肺疾患患者の維持治療のために、アンチフェンニンと長時間作用性ムスカリン拮抗薬(“LAMA”)グリピラメートとの固定用量と組み合わせた製剤を開発している。開発活動を追跡して実行可能なことを確認する
著者らは2024年下半期にFDAに研究用新薬申請(IND)を提出し、そして承認を得た場合に第二段階の臨床試験を開始し、固定用量連合製剤のCOPD患者に対する安全性と有効性を評価する予定である。
また2024年下半期には、霧化吸入アンチフェンによる非嚢胞性線維性気管支拡張(NCFBE)患者の有効性と安全性を評価するための第2段階の臨床試験を開始する予定であるが、FDAの承認が必要である
第二段階臨床試験では,アンテファニンは慢性閉塞性肺疾患,喘息,嚢胞性線維化(“CF”)患者で陽性結果を示した。慢性閉塞性肺疾患を治療する第二段階試験では,他の2種類の製剤:乾燥粉吸入器(“DPI”)と加圧計量吸入器(“PMDI”)を評価した。エンセフィン亭はCOPD試験において積極的な2期データを示し、これらの調合のすべてが提供された時
嚢胞性線維化や喘息のためのアンテファタリンの開発やアンテフェントーリンの他の配合は管路拡張とライフサイクル機会,米国以外での協力の潜在力を提供していると信じている。
承認されれば,吸入型アンテフェンタニルをCOPDの維持治療に応用して米国(“米国”)で商業化する予定である。アンテフェンは薬物装置の組合せとして管理されていないと考えられるが,患者は既製の標準噴霧噴霧器を用いてアンテフェンタリンを服用している。米国以外では,これらの地域で製品の開発や商業化に専門知識や経験を持つ会社にイソフェンタニルの許可証を発行する予定である。そのため、著者らは上海専門製薬会社のニュース製薬有限会社(“ニュース製薬”)と戦略協力を達成し、大中国地区でアンテフェンタニルを開発と商業化した。Nuanceは2023年、その重要な3期試験で第1の被験者を募集し、中国がアンテフェントーリンを用いた慢性閉塞性肺疾患の維持治療を評価した。
慢性閉塞性肺疾患及びその治療現状の概要
COPDはよく見られる、進行性、生命を脅かす呼吸器疾患であり、治愈できない。それは肺機能喪失を招き、虚弱な呼吸困難、入院、死亡を招く。慢性閉塞性肺疾患は日常生活に重大な影響を与える。患者は起床,入浴,食事,歩行などの基本活動に苦慮している。慢性閉塞性肺疾患の全世界の計画によると、全世界で、慢性閉塞性肺疾患は約3.92億人に影響しており、第三の死亡原因である。
COPD薬物治療の目標は症状を軽減し、悪化の数量と重症度(通常は症状のアップグレード)を減少することによって患者の生活の質を改善し、そして患者の機能能力を改善することである。
約40年間、COPDの治療はFDAと欧州委員会がヨーロッパ薬品管理局(EMA)の意見に基づいて使用を許可した3種類の吸入療法に主導されてきた:抗毒マウス、β-アゴニストと吸入性コルチコステロイド(ICSS)。COPD患者はよく気管支拡張剤の治療を受け、LAMAと長時間作用β-アゴニスト(“LABA”)を含み、気道収縮を緩和し、呼吸を更に容易にする。また,悪化のリスクのある患者はICSSを処方されて彼らを予防する可能性がある
あるCOPD患者はPDE 4阻害剤ロフルラスト(Daliresp)内服治療を用いた®)は、重篤な慢性気管支炎患者の悪化リスクの低下を示している。しかし、PDE 4内服治療は全身暴露を招き、吐き気、嘔吐、下痢、腹痛、食欲不振と体重減少などの不良胃腸副作用と関係がある。
アメリカに約860万のCOPD患者は単独或いは連合してLAMA、LABA或いはICS治療を受け、COPDの重症度にかかわらず。これらの薬剤を服用し,早期に二重(LAMA/LABA)と三連(LAMA/LABA/ICS)療法を使用したにもかかわらず,多くの患者が衰弱症状を継続していた。Phreesia 2022年12月の研究によると,49%の患者が毎月24日を超える症状が持続している。この負担は新たな吸入療法に重要な機会を残し,これらの療法は3つの主要な治療種別に基づいて追加的な利点を増加させている。これらの患者の肺機能と症状を改善し、病状の悪化を減少させ、全体の生活の質を高めるために、新しい治療方案が切実に必要である。
エンシフィトリン
エンセフィン亭は1種の研究中の、一流の、吸入性、小分子と選択性PDE 3とPDE 4二重阻害剤である。この二重抑制は,単一化合物において気管支拡張剤や非ステロイド抗炎症剤として機能することができる。重要なことは,アンテフェンタニルの治療概況は既存の分類の気管支拡張剤と抗炎症治療とは異なることである。米国やヨーロッパ,あるいはFDAまたは欧州委員会が承認した呼吸器疾患を治療する他の単一化合物では,気管支拡張剤および消炎剤の役割を同時に果たす化合物はないことが知られている。開発と承認に成功すれば,吸入型アンチフェンタニルは20年以上ぶりにCOPDを治療する新しい薬剤となり,LAMA,LABA,ICSを含む既存の吸入療法に添加できる唯一の気管支拡張剤となる可能性がある。
セキュリティ構成ファイル
これまで約3000名の被験者を対象とした臨床試験では,エンセフェンタン耐性は良好であった。また,健常ボランティアの包括的QT研究では,アンチフェンタニルはQT間隔を延長したり,他の心臓伝導パラメータに影響したりしなかった。それは吸入を通じて直接肺に入り、肺部のイソフェンに対する暴露を最大限に増加させ、同時に全身暴露を最大限に減少させる。この特徴はPDE 4阻害剤の経口投与に関連する胃腸機能障害などの全身副作用を最大限に減少させる。また,非臨床試験では,PDE 3とPDE 4に対するアンチフェンの選択性が他の酵素や受容体よりも高く,脱標的効果を最大限に減少させることができると考えられる。
差別化プロファイル
PDE 3とPDE 4を選択的に抑制することによって、アンチフェンは呼吸器疾患の3つの重要な機序:気管支拡張、炎症と粘液繊毛除去に影響する。エンシフェントレリンは平滑筋細胞と炎症細胞中のcAMPとcGMPのレベルを増加させ、それによって気管支拡張と抗炎症作用を達成することを目的としている。エンゼファトリンは嚢胞性線維化膜貫通コンダクタンス調節因子(CFTR)を刺激することも証明されており,気道上皮細胞におけるイオンチャネルの一種である。CFTR蛋白の突然変異はイオンチャネル機能が不良あるいは機能しないことを招き、CFを招く。CFTR機能障害は慢性閉塞性肺疾患においても潜在的な重要性を有している。CFTR刺激は肺内電解質バランスを改善し,粘液を薄くし,粘液繊毛除去を促進し,肺機能を改善し,肺感染を減少させる可能性がある
いくつかの臨床前研究において、PDE 3とPDE 4の二重抑制はPDE単独による気道平滑筋の収縮抑制と炎症メディエーターの放出抑制より増強或いは協同作用を示した。これらの増強作用は,COPD,NCFBE,喘息,CFを含む呼吸器疾患の治療におけるアンチフェンタニルの有効性を増加させる可能性が考えられる
著者らは、肺機能、COPD症状と生活の質を改善する方面において、アンテフェンタニルはCOPD治療中に満足されていない大量の需要を解決する潜在力があると信じている。
アンテフィタリンの研究進展は
アンチフェンタニルによる慢性閉塞性肺疾患治療の臨床研究進展
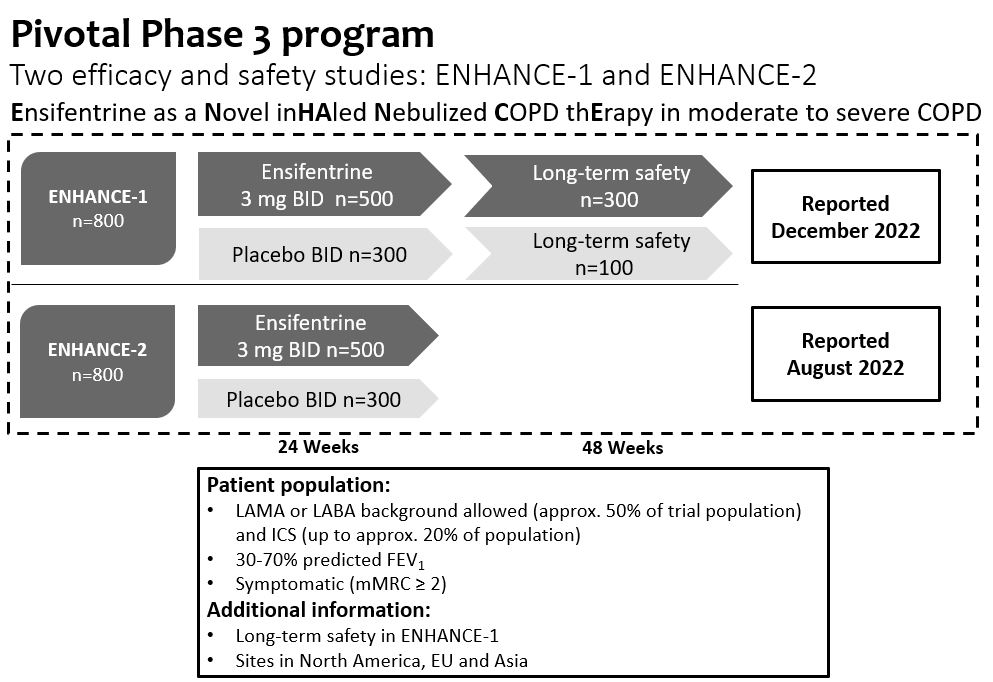
第3段階増強計画
2つのランダム、二重盲検、プラセボ対照の3期試験において、Ensifentineは成功に主要な終点、即ちEnhance-1とEnhance-2に達し、中から重度COPD患者までの肺機能指標は統計的に顕著かつ臨床的に意義のある改善を示した。両試験とも症状と生活の質指標の改善を示し,Enhance−1で統計学的有意差を達成した。この2つの試験の中で、エンゼフェンタニンは中重度COPD悪化の発生率とリスクを大幅に低下させた。エンセフェンタニルは両試験とも耐性が良好であった。
増強試験は,単一療法としてアンチフェンタニルを評価し,単一の気管支拡張剤に添加し,約50%の被験者がLAMAまたはLABAを受けたことを目的とした。また,約20%の被験者が仲間のLAMAやLABAとともにICSSを受けた。
各試験は主に米国とヨーロッパに位置する地点で約800名の被験者を募集し,合計約1600名の被験者を募集した。この2つの試験は24週間を超える有効性と安全性データの重複証拠を提供したが,Enhance−1は48週間で約400人の被験者の長期安全性を評価した。
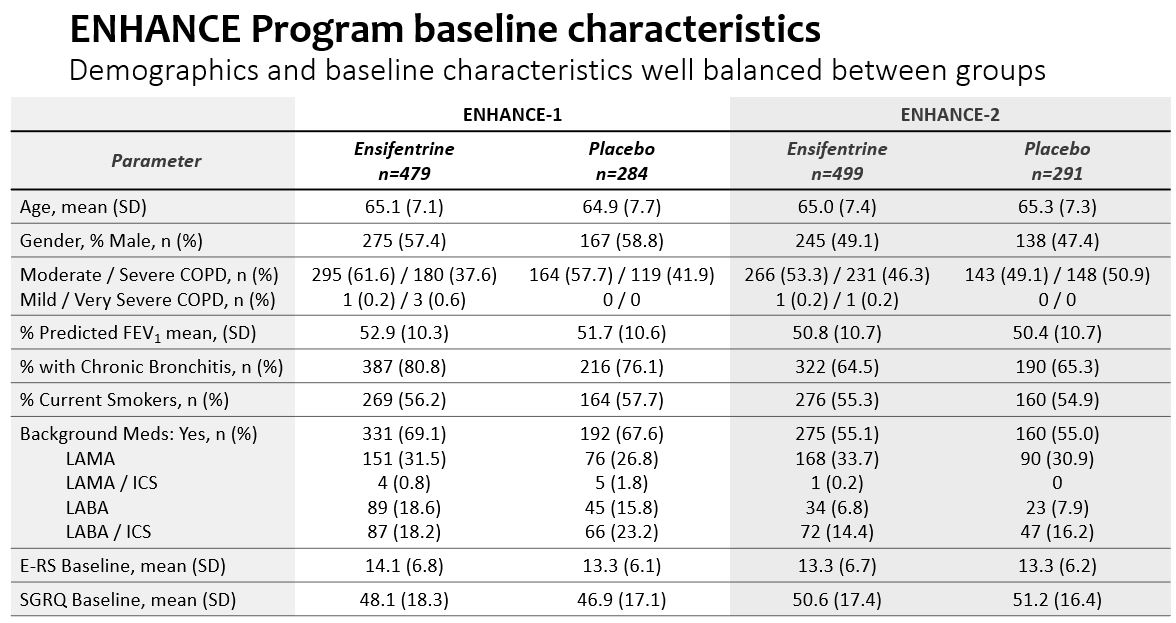
両試験において,被験者の人口統計学的特徴と疾患特徴は両治療群の間で良好なバランスが得られた。
•Enhance−1では,約69%の被験者が背景COPD治療を受けており,LAMAかLABAであった。また,約20%の被験者がLAMAやLABAを伴うICSを受けた。
•Enhance−2では,約55%の被験者が背景COPD治療を受けており,LAMAかLABAであった。また,約15%の被験者がLAMAやLABAを伴うICSを受けた。
それぞれ2022年8月と12月にEnhance−2とEnhance−1の正面営業結果を報告した。2つの試験の中で、エンゼフェンタニンは主要な終点に成功し、中から重度COPD患者の肺機能測定において統計的に顕著かつ臨床的に意義のある改善を示した。両試験とも症状と生活の質指標の改善を示し,Enhance−1で統計学的有意差を達成した。エンセフェンタンは中から重度COPD悪化の発生率とリスクを大幅に低下させ、2つの試験において非常に良い耐性を得た。
ハイライト
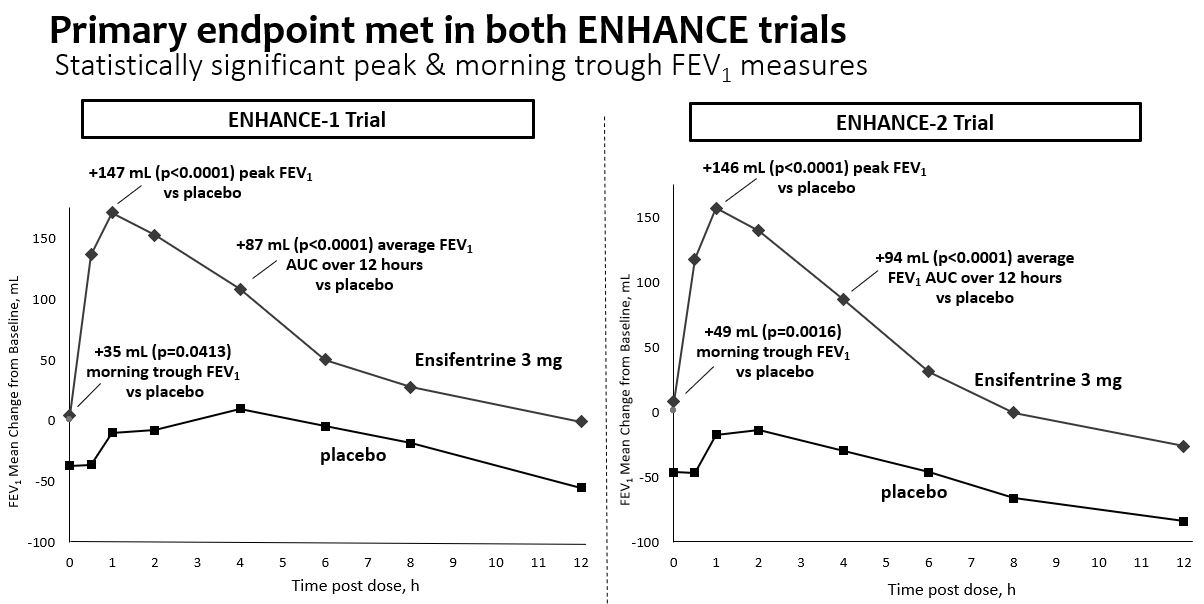
メインエンドポイント(FEV)に適合1 AUC 0-12時間)
•プラセボは修正され、平均FEVはベースラインより変化した112週投与後0~12時間の曲線下面積87 ml(P
•性別,年齢,喫煙状況,COPD重症度,背景薬物,ICS使用,慢性気管支炎,FEVなどすべてのサブ群において,アンチフェンタニルと一致した改善を示した1可逆性と地理的地域です
肺機能を評価する副次的な終点は
•プラセボ修正、FEVピーク増加1147ミリリットル(p
•プラセボ訂正、朝谷FEV増加1Enhance-1群とEnhance-2群は12週にそれぞれ35 mL(p=0.0413)と49 mL(p=0.0016)であり、1日2回の投与レジメンを支持した。
重量増加率とリスク低減
•プラセボを服用した患者と比較して,プラセボを服用した被験者は24週間以内に重度慢性閉塞性肺疾患が悪化する割合が36%(p=0.0503)低下し,Enhance−1を服用した被験者は43%(p=0.0090)減少した。
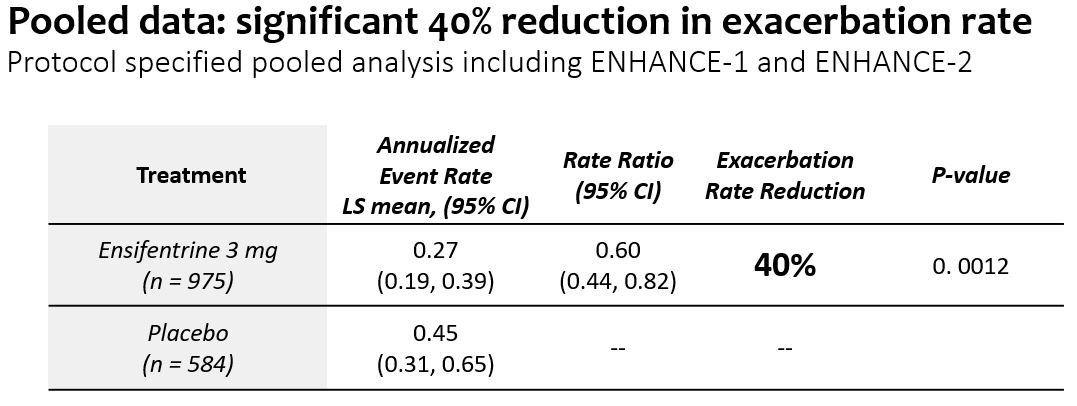
•Enhance−1とEnhance−2の合併増悪データでは,アンテフェンタニルはプラセボ服用患者と比較して24週間で中等度から重度の慢性閉塞性肺疾患の増悪発生率を40%低下させた(p=0.0012)
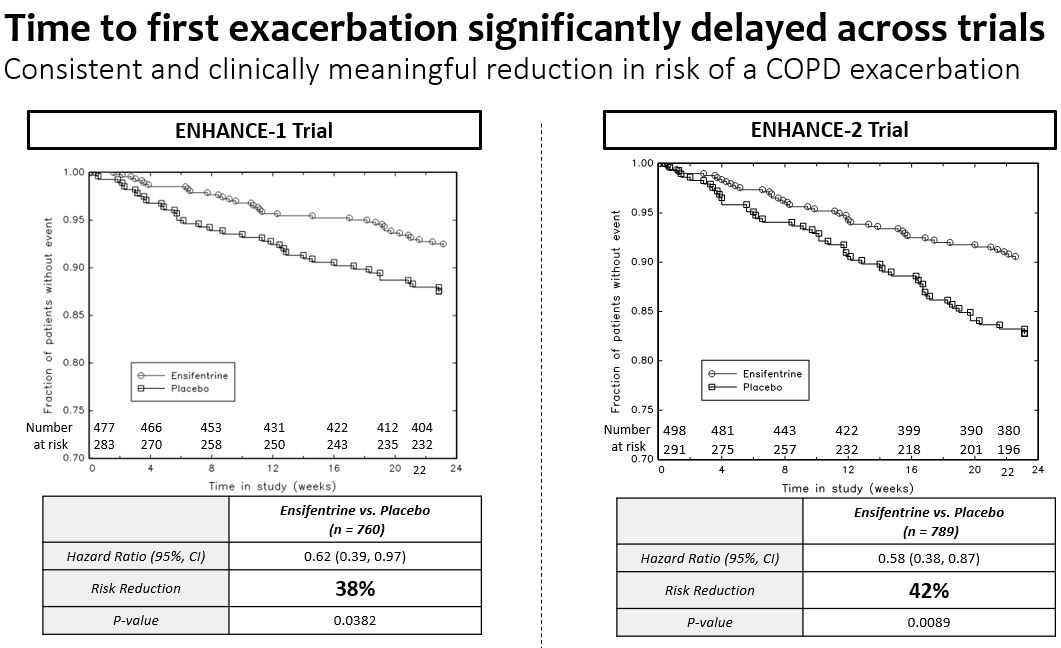
•プラセボを服用したEnhance−1群とEnhance−2群はプラセボと比較してそれぞれ38%(p=0.0382)と42%(p=0.0089)の中等度/重度悪化リスクを低下させた。
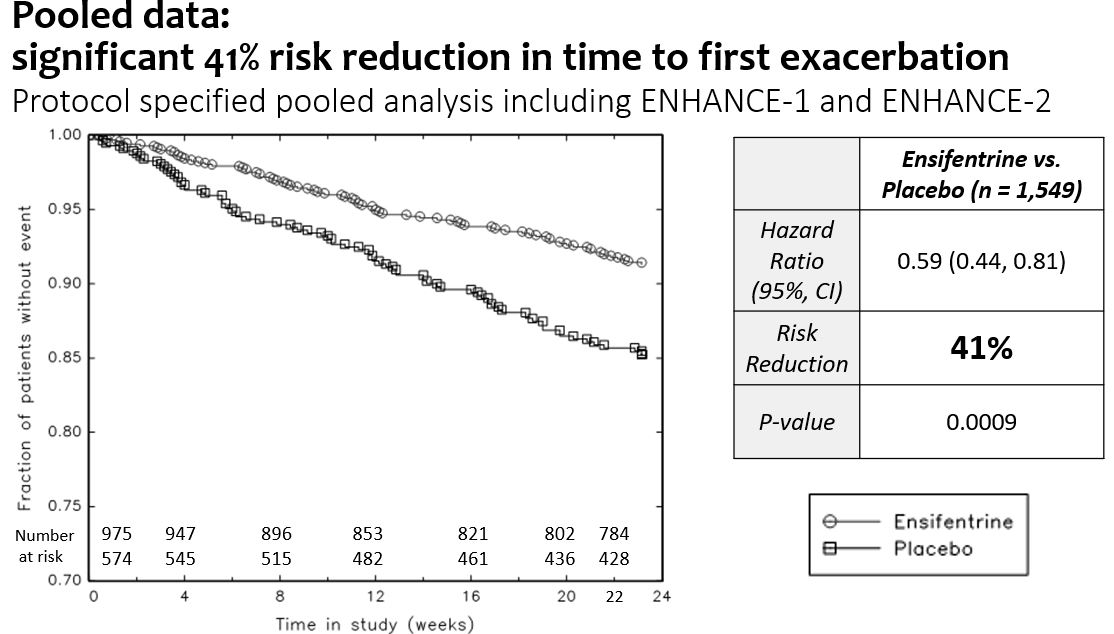
•Enhance−1とEnhance−2の合併悪化データでは,初回悪化の時間で比較してプラセボと比較して,イソフェントラリンは中等度/重度悪化のリスク41%(p=0.0009)を有意に低下させた。
慢性閉塞性肺疾患症状と生活の質(“生活の質”)
•Enhance−1試験では,E−RS*総スコアで測定された毎日の症状は,プラセボ服用24週間でプラセボより有意に改善し,ベースラインから−2単位以上の最小臨床的重要差(“MCID”)であった。6,12および24週目では,プラセボと比較して症状の改善は早期と持続であり,統計学的有意であった。Enhance−2でも類似した改善が認められたが,プラセボ群では時間の経過とともに改善が認められたため,統計学的有意差は得られなかった。
•Enhance−1試験では,SGRQ*総得点で測定した生活の質は,24週でプラセボと比較してベースラインから−4単位以上のMCIDに向上し,統計学的に改善した。6、12と24週目には、プラセボと比較して、生活の質の改善は早期と持続であり、統計学的意義がある。Enhance−2では,SGRQ*総得点で測定した生活の質は12週と24週でもベースラインから−4単位以上のMCIDに改善し,測定ごとに値がプラセボを上回ったが,プラセボ群の時間経過による改善が認められたため,統計学的有意差は得られなかった。
*呼吸症状を評価するE-RSおよびセントジョージ呼吸アンケートSGRQは、検証された患者報告結果ツールである
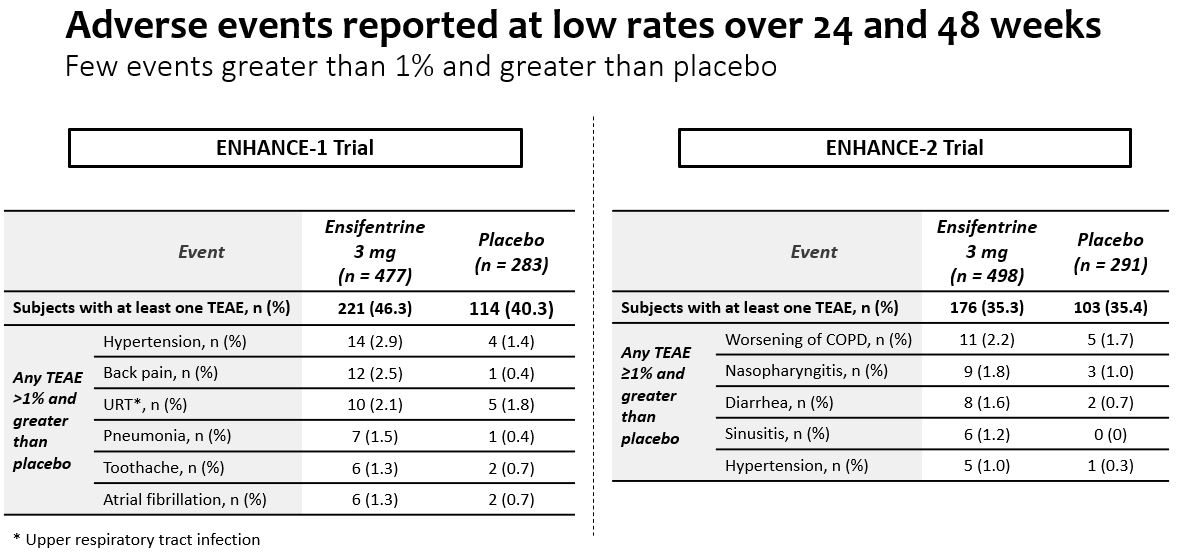
良好な安全状況
•エンシフェンタニル耐性は良好であり,1%を超える被験者で発生する有害事象は少なく,24週と48週でプラセボを上回った。
承認されれば,アンテフェンタニルはCOPDの治療パターンを変化させる可能性があると信じている。臨床試験の全データ,特にEnhance計画の主要な結果は,肺機能,症状,生活の質指標と病態緩解の改善を含み,一致した安全性結果に加え,われわれの信念を支持している。
レシピ.レシピ
我々はすでに3種類の最も広く使用されている吸入器:噴霧器、DPI、PMDIのためにアンテフェンタニルの処方を開発した。アンテフェンタリンの霧化配合は、独自の装置ではなく、標準噴射式噴霧器に適して設計されている。ネブライザーを介してCOPD薬物を輸送することは非常に重要であり,ほとんどの年齢と器用な成人がこの薬物を使用できるため,ピーク吸気流量にかかわらず,手持ち吸入器装置やピーク吸気流量の低い患者の操作が困難な患者に利点を提供した。DPIとpMDI手持ち式吸入器フォーマットは相対的に携帯性と便利であり、重要な輸送メカニズムでもある。
我々は引き続きアンテフェンタリン霧化製剤の開発に集中しているが、pMDIとDPI製剤の開発は新しい潜在的適応、調合の組み合わせ、協力を含むより多くのライフサイクル機会を提供すると信じている。2021年2月,われわれは中重度COPD患者にpMDIアンテフェンタニルを使用した第二段階多用量試験の陽性結果を報告した。PMDIから提供されたエンシフェントーリンはすべての原発と二次肺機能終点を満たす。プラセボと比べ、肺機能の改善は用量順に行われ、ピークと12時間の投与間隔期間中に統計学的意義があり、そしてpMDIの1日2回の投与によるCOPDの治療を支持した。本研究の単剤部分のデータは2020年3月に報告されている。
著者らはすでにCOPD第二段階試験においてすべての3種類の調合を用いて概念を証明することに成功した。また,第2段階試験のデータは3種類のレシピとも一致していた.すべての3種類の剤形は肺機能と作用持続時間の面で統計的に顕著かつ臨床的に意義のある改善を示し、毎日2回の用量とプラセボに類似した安全概況を支持した
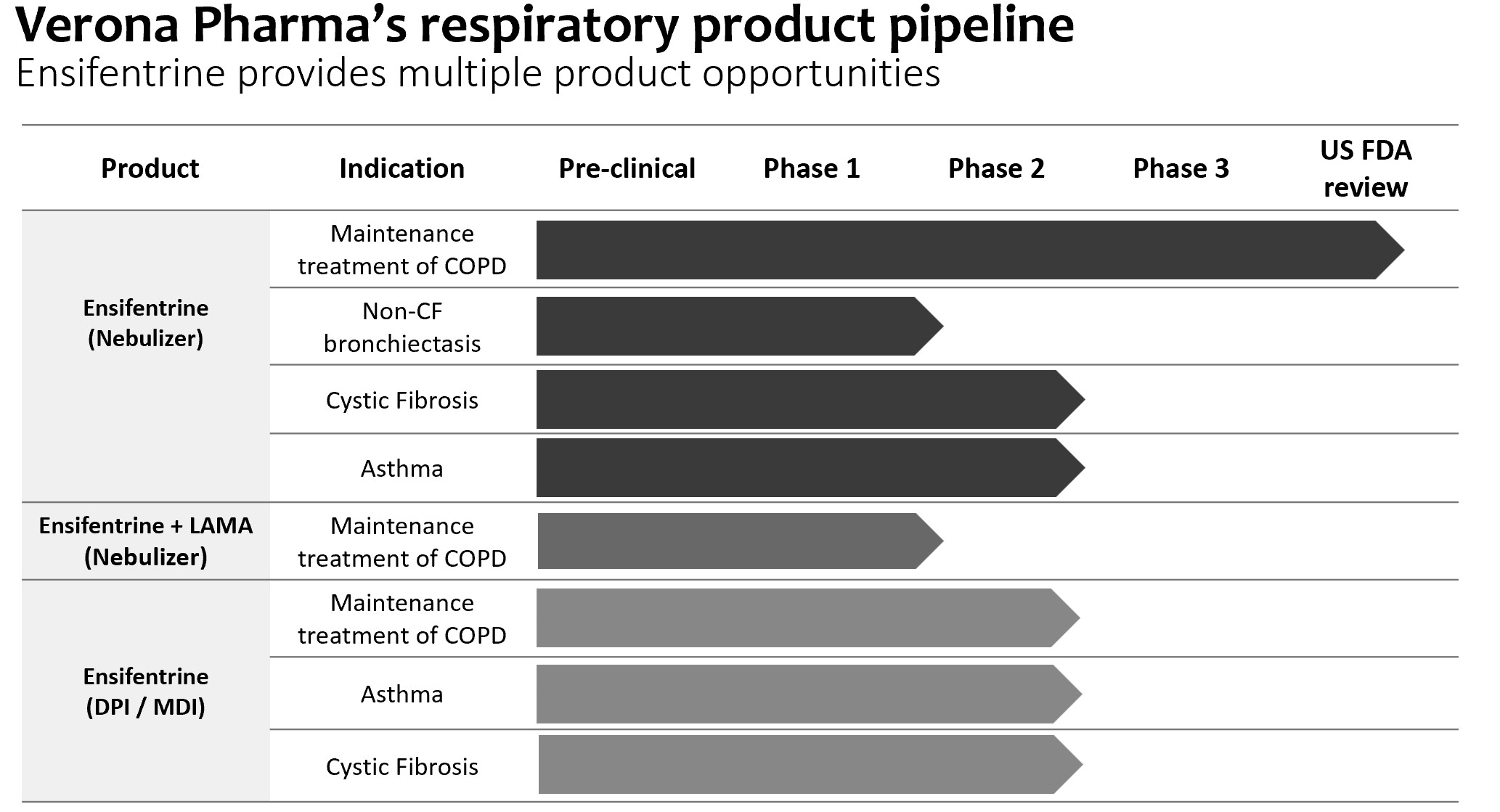
パイプ.パイプ
次の表に私たちの開発計画をまとめました。
計画的臨床開発活動
エンシフィタリン/ラマ固定用量併用投与
固定用量連合療法、例えばLABA/LAMA、LABA/ICSとLABA/LAMA/ICSは通常COPDの治療に応用され、著者らの市場研究により、霧化固定用量連合療法に対する需要はまだ満たされていない。アンテファニンとLAMAの結合はCOPD患者に最初の霧化固定用量の組み合わせを提供でき、二重機序を通じて気管支拡張を提供し、PDEを抑制することによって非ステロイド抗炎症作用を発揮する可能性があると信じている。著者らは、噴霧吸入によるCOPD患者の維持治療のためのアンテファニンおよびグリピラメートを含む固定用量の併用製剤を開発している。私たちはアメリカを含む複数の管轄区で特許出願を提出した。
もし実行可能な調合を開発すれば、著者らは2024年下半期にFDAにIND申請を提出する予定であり、もし実行が許可されれば、第二段階の臨床試験を開始し、固定用量の組み合わせ処方のCOPD患者に対する安全性と有効性を評価する。
非嚢胞性線維性気管支拡張症
NCFBEは1種の慢性肺疾患であり、持続的な咳、痰多産と頻繁な呼吸器感染を特徴とし、病状が深刻であるほど、病状は深刻である。米国では50万人もの成人がこの疾患を患っており,現在のところ専門的な治療法は承認されていない。医師は現在気管支拡張剤,抗生物質,ステロイド,粘液希釈剤,手術を使用している。
COPD患者に観察された臨床結果から,肺機能の改善や咳や痰の症状を含め,アンチフェンは潜在的にNCFBEを治療する有効な方法である可能性が信じられた。2024年下半期に第二段階臨床試験を開始する予定であり,FDAが許可すればNCFBE患者に対するスプレー剤の治療効果と安全性を評価する。
アンテフェンタニルの潜在的付加適応症
嚢胞性線維症と喘息
COPDとNCFBE以外に、著者らはアンチフェンタニルは他の呼吸器疾患においても潜在的な応用があると信じており、CFと喘息を含み、これは導管拡張に機会を提供し、そしてアメリカ以外の協力に潜在力を提供した。
Cfは進行性、致命的な遺伝病であり、治癒できず、死亡年齢の中央値は46歳である。この場合の特徴は,ドロドロした粘液が体の多くの器官を損なうことである。これは反復と持続性の肺感染を招き,頻繁な病態悪化や入院治療を招く。その他の症状は栄養不良、便秘と下痢を含み、一部の成人は糖尿病、関節炎と肝臓問題が出現する。
Cfはアメリカとヨーロッパで最もよく見られる致命的な遺伝病である。嚢胞性線維化財団のデータによると,90以上の国や地域では,米国では約4万人がCFと診断され,世界では10.5万人がCFと診断され,毎年約1000例の新症例が診断されている。アメリカとヨーロッパの監督管理機関はCFは稀な或いは孤児疾患であると考え、激励措置を提供して有効な新しい治療法の開発を奨励する。Cf患者は毎日多くの薬物治療、頻繁な病状悪化と入院治療に耐えなければならない。最終的に,選定した患者は肺移植を受ける。
1つの2 a期の臨床試験において、単用量霧化吸入アンチフェンタニルは慢性閉塞性肺疾患患者の肺機能を改善できる。また,臨床前研究では,アンチフェンは嚢胞性線維化膜貫通コンダクタンス調節器(CFTR)を活性化し,粘液粘度の低下や粘液繊毛除去の改善に有用であった。これらのデータは潜在的な治療法としてのアンチフェンの継続的な発展を支持していると信じている。
喘息はよく見られる慢性炎症性肺疾患であり、散発的な呼吸困難を招く。この疾患は気道狭窄や腫脹を招き,呼吸困難,喘息,咳,胸部圧迫感などの症状をきたす。アレルゲンや刺激物などのトリガーに曝露すると喘息発作を引き起こす。
喘息発作の重症度と頻度はそれぞれ異なる。世界保健機関の推定によると、全世界で2.6億人以上が喘息を患っており、それは児童の中で最もよく見られる慢性病である。米国では成人喘息患者の約60%が定期的に服薬しているにもかかわらず,喘息はコントロールされていない。治癒方法はないが,症状はトリガー要因の回避や既定の維持療法により予防でき,気管支拡張剤,ICS,抗IgE薬,ロイコトリエン阻害剤を含む
エンセフィン亭は喘息の2 a期臨床試験において潜在力を示した。この実験のデータは2019年10月に同誌に発表された肺薬理と治療Sは、アンテフェンタニルは肺機能の改善に対して用量依存性があり、現在の救急薬物である大量のサルブタモール霧化吸入に相当することを証明した。重要なことは,アンテフェンタニル耐性が良好であり,サルブタモールを受けた患者よりも全身反応を経験することである。
私たちのチーム
われわれの専門家チームは呼吸療法の開発と商業化において数十年の経験を持ち,以下のCOPD療法:Advairを含む®Anoro Ellipta®;Breo®フロヴェント®笛の形®Incruse Ellipta®SerEvent®Symbiort®Tudorza Pressair®バントリンと®.
製造業
我々には製造施設がなく、現在の良好な製造規範(“cGMP”)に適合するアンチフェン臨床試験材料および任意の未来候補製品、ならびに商業数量のアンチフェンおよび任意の未来候補製品を第三者契約製造組織(“CMO”)に依存して供給し続けることが予想される
将来的には他のCMOと契約する可能性があるが,現在1つのCMOがプロポフォール薬剤の生産を担当しており,1つのCMOが各製剤のプロポフォールの生産を担当している。
私たちは現在すべてのCMOが商業規模の製造能力を持っている。正常な業務過程では,イソニアジド系薬物物質や薬物製品の製造プロセスを他のCMOに譲渡し,臨床や商業用品を生産することができると考えられる。
商業化する
2023年の間、私たちは引き続き私たちの商業能力と発売準備を建設して、承認可能なアンテフェンタリンの準備をします。主な商業化前活動には,経験豊富な幹部の増加,疾患意識活動の開始,われわれの患者支援と流通戦略の整備と実施,およびわれわれのイソニアジド投与材料の開発開始が含まれており,これらは広範な市場研究の支持を得ている。
著者らは従業員数を79人まで大幅に拡大し、医療事務、コンプライアンス、製造、金融とIT方面の重要な指導職を増加させ、マーケティング、市場参入と商業運営におけるビジネスチームを深化させた。これらの任命は医療事務副主任高級副総裁、コンプライアンス副主任総裁と薬物警戒副主任総裁を含む。
また,“言わずと知れた慢性閉塞性肺疾患”という疾患キャンペーンを開始した。この運動は,どれだけの患者が依然として日常生活に影響を与える持続的な症状を有しているかを明らかにしている。この活動は医療専門家がさらに彼らの患者に慢性閉塞性肺疾患にどのように対応するかを聞くことを奨励する。
未来を展望すると、著者らは引き続き著者らの上場戦略を推進し、定価、流通と患者支持サービス、HCPと患者参加計画、及び引き続き著者らの疾病宣伝活動を含む多くの重要な策略を決定する。
アメリカです
アメリカでは、承認されれば、私たちは自分で霧化フェンタニル林を商業化しようとしている。現在米国の慢性閉塞性肺疾患の維持的治療は約100億ドルの売上を生み出している。米国では約860万人の患者が慢性閉塞性肺疾患の慢性維持治療を受けている。これらの患者は単独或いは連合してすべてのCOPD重症度のLAMA、LABAとICS製品の治療を受けた。これらの治療法にもかかわらず,約50%の患者が毎月24日を超える症状が報告されている。この負担は重大であり、COPD患者を治療する新しいと新しい行動機序の必要性を明らかにした。これらの患者たちは彼らの肺機能と症状の治療を改善するのを助ける必要がある。まだ症状のある患者の数以外に、COPDはアメリカの医療システムに巨大な負担をもたらし、直接と間接コストは約500億ドルである。
われわれが米国のヘルスケア提供者や支払人と行った市場調査によると,COPDの重症度や治療法にかかわらず,アンテフェンタニルはすべての症状のある患者の補充療法として広く採用されると信じている。LAMA,LABA/ICS,LAMA/LABAあるいは三連療法を受けている既存患者では,アンチフェンの大部分の用途が付加療法となる。これは,これらの患者の肺機能,症状,生活の質の改善に役立つ新たな治療法が切実に必要であるためである。われわれの市場研究では,多くのイソフェンの使用が最初に肺科医から始まることが示唆されている。この集中的な処方薬の基礎により,約100名の代表からなる現場販売チームが潜在的なイソフェンタニル機会に触れることが予想される
国際的に
COPDは全世界の約3.92億人を影響し、多くの患者はまだ確定診断されていない。アジア、ヨーロッパ、ラテンアメリカを含む米国以外の戦略は、これらの地域におけるアンテファンドリンのさらなる開発と商業化を支援するために、有力会社とパートナーシップを構築することである。
2021年6月、私たちは上海専門製薬会社Nuance Pharmaと戦略協力を達成することを通じて、この戦略を実行し、潜在価値は2.19億ドルに達し、大中国地区でアンテフェンタニルを開発と商業化した。協定条項によると、私たちはNuance Pharmaに中国大区でのアンテフィン亭の開発と商業化の独占的権利を付与する。その見返りに、私たちは2021年6月9日まで、Nuance Pharmaの親会社Nuance Biotechで、2,500万ドルの現金と1,500万ドルの株式を含む合計4,000万ドルの前払いを受けた。私たちは1.79億ドルに達する更なるマイルストーン支払いを得る資格があり、これらの支払いはある臨床、監督と商業マイルストーンを実現する時にトリガしたものであり、大中国地区の純売上高の2桁分級特許権使用料である。
Nuance Pharmaは大中国の臨床開発と商業化に関連するすべてのコストを担当している。この地域におけるアンテファタリンの臨床開発が我々の世界的な開発と商業化戦略と一致することを確実にするための共同指導委員会が設立された。2023年4月、Nuance Pharmaは、中国大陸における中国の慢性閉塞性肺疾患維持治療の治療効果を評価する第1の被験者をその重要な3期試験で募集したと発表した。Nuance Pharmaは2023年3月に健康ボランティアの中でアンテフェンタニルの第1段階試験を開始した。これらの研究は中国新薬評価センターが中国大陸でのアンチフェンによる慢性閉塞性肺疾患治療の第一段階と第三段階の研究を許可した後に行われた
競争
製薬業の特徴は技術が迅速に進歩し、競争が激しく、特許薬物を高度に重視していることである。著者らは主要な製薬、専門製薬と生物技術会社、学術機関、政府機関及び公共と個人研究を含む多くの異なる源からの潜在的な競争に直面している
機構です。開発に成功し商業化すれば,アンテフェンタニルは既存療法や将来出現する可能性のある新しい療法と競争する。
エンシフェントーリンは独特で一流の候補治療薬物であり、単一分子中に気管支拡張剤と非ステロイド抗炎症特性を同時に有する。米国やヨーロッパでは,他のダブルPDE 3やPDE 4阻害剤は市販されておらず,臨床開発中の薬剤もないことが知られている。われわれの市場研究によると,病態の重症度にかかわらず,アンテフェンタニルは患者範囲で使用されることが予想される。主にすべての既存の治療種別(LAMA,LABA,ICS)の有症状患者の補充治療に用いられることが予想される。一部の医療提供者は,アンテフェンタニルの臨床状況に応じて単一療法として使用することを示している
そのため、もし承認されれば、霧化アンチフェンの独特な特性は霧化と手持ち吸入器調合、DPIとPMDIを含むすべての許可されたCOPD療法と競争できると信じている。また,アンテフェンタニルの作用機序は既存療法の補完であることから,これらの治療以外にも利用可能であると考えられる。
現在COPD維持治療のための噴霧製品が承認されている中で、アンチフェンタニルのアメリカ市場における潜在的な競争相手はLABA(Brovana)であると考えられている®Performomistと®(ユペリー)とラマは®).
COPD市場のDPI/pMDI維持治療において、アンテフェンタニルが現在最も近い潜在競争相手はセビコンである®SPIRIVA社のアスリコンにより販売されている長時間作用性β2アゴニスト気管支拡張剤とICSとの組み合わせ®Boehringer Inelheim GmbH,Advairによって販売されている長時間作用性抗毒マウス強気管支拡張剤®グラクソ·スミスクラインによって販売されている長時間作用性β2−アゴニストとICSとの組み合わせ®グラクソ·スミスクラインとAnoroによって販売されている長時間作用性β2−アゴニスト気管支拡張剤とICSとの組み合わせ®グラクソ·スミスクラインによって販売されている長時間作用性β2アゴニスト気管支拡張剤と、長時間作用性抗毒マウス強気管支拡張剤との組み合わせ。グラクソ·スミスクラインが開発したラマ、LABA、ICSの三連療法®アメリカとEUで承認されていますアスリコンにも三聯療法の組み合わせ製品(LAMA/LABA/ICS),Breztri Aeroballがあります®米国は2020年7月に同法案を批准し、EUは2020年12月に同協定を承認し、中国は2019年12月に同協定を承認した。またChiesiの三連療法組合せ製品Trim弓は®2017年にEUで承認され、現在米国では第3段階試験にある。
臨床開発においてCOPDの悪化を予防するための他の潜在療法は生物製剤の注射を含む。セノフィの抗IL 4 Dupixent®すでに第三段階計画を完成し、アメリカでCOPD補充生物製品許可証の申請を提出した。アスリカンの抗IL 33,トゾラックモノクロナル抗体,グラクソ·スミスクラインの抗IL 5,Nucala®,およびChiesiのPDE 4阻害剤であるテミラストは,3期試験を行っている。いくつかの抗炎症薬や気管支拡張剤がCOPD治療の第二段階臨床試験を行っていることが知られている。
知的財産権
私たちは、内部開発でも第三者から許可を得ても、特許権を求め、維持し、擁護することを含む、私たちの業務に重要と考えられるノウハウ、発明、改善の保護と強化に努めています。私たちの政策は、米国や米国以外の司法管轄地域で当社のノウハウ、発明、改善、プラットフォーム、製品候補に関する特許保護を求め、獲得することを求めることであり、これらは私たちの業務の発展と実施に非常に重要です
2023年12月31日現在、我々の特許組み合わせは、発行された11件の米国特許、7つの係属中の米国特許出願(4つの米国仮特許出願を含む)、発行された外国特許79件、および係属中の外国出願75件(国際PCT出願6件を含む)を含む。これらの特許および特許出願は、イソフェントラリンの結晶形態、イソフェントリリンおよびいくつかの呼吸器薬の組成物、特定の塩のイソフェントラリン、嚢胞性線維症および非嚢胞性線維症気管支拡張の治療、およびいくつかの他の呼吸器疾患のいくつかの態様を治療するためのイソフェントリン、および2044年までの予期される有効期間を有するイソフェントラリンを調製するためのいくつかの呼吸可能な製剤に対する請求項を含む
私たちは“Verona Pharma”をアメリカといくつかの他の主要な司法管轄区域の商標に登録した。承認されれば、私たちはまた米国でアンチリンの潜在的商標を登録することを申請した
個別特許の展示期間は、特許出願の提出日又は特許発行日及び特許を取得した国の特許法律期限に応じて定められる。一般に,米国で定期的に提出された出願のために発行された特許有効期間は20年であり,最も早く有効な非臨時出願の日から計算される。さらに、場合によっては、特許期間を延長して、米国特許商標局(USPTO)が特許の発行を遅延させた部分時間と、FDA規制審査期間によって実際に損失した部分期間とを再取得することができる。しかしながら、FDAの構成要素については、回復期間は5年を超えることはできず、回復期限を含む総特許期間はFDA承認後14年を超えてはならない。外国特許の有効期限は適用される現地法の規定によって異なるが,通常は最も早い有効出願日から20年である。しかし、特許提供の実際の保護は製品によって異なり、国によって異なり、多くの要素に依存している
特許のタイプ、そのカバー範囲、規制に関連する延長の利用可能性、特定の国の法的救済措置の可用性、および特許の有効性および実行可能性を含む
また、私たちはビジネス秘密と技術ノウハウ、持続的な技術革新によって、私たちの競争地位を発展させ、維持しています。私たちは、私たちの専門情報を保護し、私たちの協力者、従業員、コンサルタントとの秘密協定、および私たち従業員との発明譲渡協定の一部を使用することを求めています。また、私たちの協力者と選定されたコンサルタントと秘密協定や発明譲渡協定を締結しました。これらのプロトコルは、我々の独自の情報を保護し、発明譲渡プロトコルの場合、第三者との関係によって開発された技術の所有権を付与することを目的としている。このような合意は違反されるかもしれないし、私たちはどんな違反にも対応する十分な救済策がないかもしれない。しかも、私たちのビジネス秘密は競争相手に知られたり独立して発見されるかもしれない。もし、私たちの協力者、従業員、およびコンサルタントが、私たちのために働いているときに他人が所有している知的財産権を使用する場合、関連またはそれによって生じるノウハウおよび発明の権利について議論される可能性がある
私たちのビジネス成功はまた第三者の独占権を侵害しないことにある程度依存するだろう。いかなる第三者特許を発行することが、私たちの開発や商業戦略、あるいは私たちの薬物やプロセスを変更し、許可を得たり、いくつかの活動を停止することを要求するかどうかはまだ確定されていません。私たちは、私たちの将来の薬物の開発または商業化のために、私たちが必要とする可能性のある独占権許可を得ることができなかったり、私たちの将来の薬物を開発または商業化することができません。もし第三者が2013年3月16日までに米国で特許出願を準備して提出し、私たちが権利を持つ技術を持っていると主張した場合、私たちは発明の優先権を決定するために、米国特許商標局の介入手続きに参加しなければならないかもしれない。詳細は“項目1 A”を参照されたい。リスク要因-知的財産権と情報技術に関するリスク
Ligand(前身Vernalis)とのライセンスプロトコル
2000年2月にLigand UK Development Limited(前Vernalis Development Limited)(“Ligand”)と譲渡および許可プロトコルを締結し,Ligand UK Development Limited(前Vernalis Development Limited)(“Ligand”)は2018年10月からLigand PharmPharmticals,Inc.の完全子会社であった。2022年3月24日、私たちはLigandと合意し、譲渡と許可協定を修正した。私たちは譲渡と許可協定および修正協定を総称してLigandプロトコルと呼ぶ。Ligandプロトコルによれば、Ligandは、アンテフェンおよび関連化合物に関連するいくつかの特許および特許出願のすべての権利を、私たち、またはLigand特許に譲渡している。Ligandの事前同意なしに、私たちはLigand特許を第三者にさらに譲渡することはできない。Ligandはまた、ヒトまたは動物のアレルギー性または炎症性疾患を治療するためのイソプロピルアドレナリンの計画IPを含むLigand特許を用いて開発されたPDE阻害剤、Ligandノウハウおよび特定の化合物の実物在庫に基づいて、ヒトまたは動物のアレルギー性または炎症性疾患を治療するためのイソプロピルアドレナリンの計画IPを含む、いくつかのLigand独自技術に基づいて、我々に独占的、世界的に印税許可を付与する。Ligand協定によると、私たちはLigand特許を維持し、商業的に合理的で勤勉な努力を使用して製品を開発し、商業化しなければならない
2022年3月に、吾らはLigandと改訂協定(“改訂”)を締結し、これによりLigandプロトコルを改訂して、Ligandプロトコルのいくつかの曖昧な言葉を明らかにした。改正により、吾らは、改訂日から5営業日以内にLigandに2,000,000ドルおよび(Ii)を吾等または許可者に分けて初の商業販売イソフィン亭に支払うことに同意し、その金額は現金で支払うか、あるいは吾らが同等の価値の会社持分を発行することを適宜決定して支払い、この金額は、吾らの米国預託株式のナスダック世界市場における出来高加重平均価格に基づいて決定され、この10(10)の取引日および当該マイルストーン事件発生前10取引日に含まれる。
私たちは2022年3月にLigandに200万ドルを支払い、実行時に支払われた200万ドルを販売、一般、管理費用として総合運営報告書に計上した。この支払いは契約修正に関係しているからだ。
Ligand協定はいずれか一方がその条項に基づいて早期に終了しない限り、2042年3月24日に満了する。どちらも他方の破産や債務不履行、あるいは他方の未治癒の重大な違約行為により“Ligand合意”を中止することができ、合意の終了を求める一方が英国高裁の最終判決を得なければならないことが条件であり、他方が“Ligand協定”に規定されている義務に深刻に違反していると発表した。私たちはLigand協定を終わらせるために90日前に書面で通知することができる。もし私たちがLigandに私たちがLigand特許を放棄したり、任意のLigand特許が失効することを許可することを通知すれば、LigandはLigandプロトコルを終了するかもしれない。Ligandプロトコルが終了した後,任意のプログラムIPの使用を停止し,Ligand特許とその任意の改良をLigandに譲渡しなければならないが,我々の任意の分割許可者は,その分許可者が許可したプログラムIP部分についてLigandと直接ライセンス契約を締結する権利がなければならない.
政府の監督管理
FDAと州と地方司法管轄区及びその他の国の類似監督機関は臨床開発、製造、マーケティングと流通に参与する会社に対して大量と重い要求を提出した
私たちが開発している薬のようにこれらの機関と他の連邦、州、地方と外国実体は私たちの候補製品の研究開発、テスト、製造、品質管理、安全、有効性、ラベル、貯蔵、記録保存、承認、広告と販売促進、流通、承認後の監視と報告、サンプリングと輸出入などの面で監督管理を行う。
FDA薬品承認プロセス
米国では,FDAは“連邦食品,薬物と化粧品法”(“FDCA”)及びその実施条例に基づいて薬品を規制している。規制の承認を得て、その後、適用される連邦、州、地方、外国の法規と条例を遵守する過程には、多くの時間と財政資源が必要だ。製品開発プロセス、承認プロセス、または承認後のいずれかの場合、出願人が適用される米国の要求を遵守できない場合、FDAが係属中の新薬申請の提出を拒否するか、承認を拒否するか(“NDA”)、承認の撤回、臨床棚上げの実施、警告状の発行、製品のリコール、製品の差し押さえ、生産または流通の完全または部分的な一時停止、禁止、罰金、政府契約の拒否、原状回復、返品または民事または刑事罰のような様々な行政または司法制裁を受ける可能性がある。
FDAが米国で発売される前に必要とされるプログラムは、一般に以下のような態様を含む
•非臨床実験室テスト、動物研究を完成し、その中のいくつかはFDAの良好な実験室規範(“GLP”)に従って行い、調合研究を行わなければならない
•米国がヒト臨床試験を開始する前に発効しなければならないINDをFDAに提出する
•各臨床試験が開始される前に、各臨床サイトの独立した機関審査委員会(“IRB”)または倫理委員会の承認;
•良好な臨床実践(“GCP”)に基づいて、各適応に対する薬物製品の安全性と有効性を決定するために、十分かつ制御された人体臨床試験を行うことが要求されている
•NDAはすべての重要な試験が完了した後にFDAに提出される
•FDAが要求した場合、FDA諮問委員会の審査が完了する
•現在の良好な製造規範(“cGMP”)要求に対するコンプライアンスを評価し、GCPのコンプライアンスを評価するために、製品を製造するための1つまたは複数の製造施設の検査を良好に完了させ、施設、方法、および薬物の特性、強度、品質および純度を維持するのに十分な制御を確保し、GCPのコンプライアンスを評価するために選択された臨床研究場所を検査することができる
•FDAは、米国で使用される特定の適応の製品の商業マーケティングを可能にするために、NDAおよび米国処方情報を審査および承認する。
非臨床研究
非臨床研究は製品の化学、毒性と調合に対する実験室評価、及び潜在的安全性と有効性を評価する動物研究を含む。INDスポンサーは,INDの一部として,非臨床試験の結果を製造情報,分析データ,任意の利用可能な臨床データや文献などとともにFDAに提出しなければならない。INDはFDAが米国食品·薬物管理局に提出した許可申請であり,州間商業で輸送し,ヒトへの研究薬物製品の使用を要求している。INDはFDAが受信した30日後に自動的に発効し、それ以前にFDAが1つまたは複数の提案された臨床試験に対して懸念または問題を提起しなければ、臨床試験を保留する。この場合,INDスポンサーやFDAは臨床試験開始前に未解決の問題を解決しなければならない。したがって,INDの提出はFDAが臨床試験の開始を許可しない可能性がある
臨床試験
臨床試験は、GCP要求に適合する合格した研究者の監督の下で、すべての研究対象または法律代表に任意の臨床試験に参加することを要求するインフォームドコンセントを書面で提供することを含む、ヒト対象者に研究薬を服用することを含む。臨床試験は,試験の目標や終点,安全性をモニタリングするためのパラメータおよび評価する有効性基準を詳細に説明するシナリオで行った。INDの一部として,各臨床試験の案と任意の後続の案修正案をFDAに提出しなければならない。INDが活発な状態にある場合には、前回の進展報告以来行われた臨床試験と非臨床研究の結果の進展報告をまとめ、他の情報を除いて、少なくとも毎年fdaにind安全報告を提出しなければならず、深刻かつ意外な疑わしい有害事象を理解するために、fdaと調査者に書面のind安全報告を提出しなければならず、他の研究結果は、同じまたは類似の薬物に曝露されたヒトに対する重大なリスク、動物または体外試験結果がヒトに重大なリスクがあること、および任意の臨床上の重大なリスクを示すことを示している
重要なことは,案や研究者マニュアルに記載されているものと比較して,重篤な副作用の発生率が増加していることである。
また,臨床試験に参加する各機関のIRBは,その機関が臨床試験を開始する前に任意の臨床試験の計画を審査·承認しなければならない。規制当局、IRB、またはスポンサーは、対象が受け入れられない健康リスクに直面していることを発見すること、または試験がその目標を達成する可能性が低いことを含む、様々な理由で臨床試験を随時一時停止することができる。いくつかの研究はまた、臨床研究スポンサーによって組織された独立した合格専門家グループの監督を含み、このグループは、データ安全監視委員会と呼ばれ、委員会はデータを審査し、指定されたチェックポイントで進めることができるかどうかの研究を提案する。もしそれが受け入れられない安全リスクや他の理由があると判断した場合、治療効果が証明されていなければ、臨床試験を停止する可能性がある。ある臨床試験に関する情報は,そのwww.Clinicaltrials.govサイト上で公開されるために,特定の時間枠で米国国立衛生研究院(NIH)に提出されなければならない
人体臨床試験は通常3段階に分けて行われ、この3つの段階は重なる可能性があり、合併する可能性もある
•第1段階:候補薬剤は、最初に健康なヒト対象または標的疾患または状態を有する患者に導入され、安全性、用量耐性、吸収、代謝、分布、排泄が試験され、可能であれば、その有効性の早期兆候が得られる
•第二段階:候補薬物は、可能な副作用と安全リスクを決定するために限られた患者集団に使用され、特定の標的疾患に対するこの製品の治療効果を初歩的に評価し、用量耐性および最適用量を決定する
•第三段階:良好に制御された臨床試験において、候補薬剤は、一般に地理的に分散された臨床試験場所に投与され、承認のために製品の有効性および安全性を統計的に評価するのに十分なデータを生成し、製品の全体的なリスク-利益プロファイルを確立し、製品のラベルに十分な情報を提供する
承認後試験は,第4段階研究と呼ばれることがあり,最初の上場承認後に行われる可能性がある。これらの試験は,治療適応が予想される患者の治療から追加的な経験を得るために用いられている。場合によっては,FDAはNDAを承認する条件として4期臨床試験を強制的に実行することができる。
新薬開発期間中、スポンサーはいつかFDAと会う機会がある。これらの要件は、INDを提出する前、第2段階の終了時、および秘密協定の提出前にある可能性がある。他の時間に会議を開催することを要求することができます。これらの会議は,スポンサーにこれまで収集してきたデータに関する情報を共有する機会を提供し,FDAにアドバイスを提供し,スポンサーとFDAに次の段階の開発について合意することができる
臨床試験と同時に、会社は通常追加の動物研究を完成し、薬物化学と物理特性に関する追加情報を開発し、cGMPの要求に基づいて最終的に商業量産製品のプロセスを決定しなければならない。製造過程は一貫して高品質の候補製品ロットを生産できる必要があり、また、メーカーは最終薬物の身分、強度、品質、純度をテストする方法を開発しなければならない。また,適切な包装を選択·テストし,候補製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない
上場承認
成功に必要な臨床試験,臨床前研究や臨床試験の結果,製品の化学,製造,制御,提案されたラベルなどに関する詳細な情報を想定し,NDAの一部としてFDAに提出し,その製品を1つまたは複数の適応に使用することの承認を求めた。ほとんどの場合、秘密保持協定を提出するには高額な申請使用料を支払う必要がある。そのほか、小児科研究公平法(PREA)はスポンサーに大多数の薬物、新しい有効成分、新しい適応、新しい剤形、新しい投与方案或いは新しい投与経路に対して小児科臨床試験を行うことを要求している。PREAによれば、元のNDAおよびいくつかのサプリメントは、スポンサーが延期または免除を受けていない限り、小児科評価を含まなければならない。要求された評価は、すべての関連する小児科亜群において適応の安全性および有効性を主張する製品を評価し、その安全に有効であると考えられる各小児科亜群の用量および投与を支持しなければならない。スポンサーまたはFDAは、小児科亜群の一部または全部の小児科臨床試験の延期を要求することができる。延期は、小児科臨床試験が完了する前に、成人で使用を許可する準備ができていることを発見するか、または小児科臨床試験が開始される前に追加の安全性または有効性データを収集する必要があることを含むいくつかの理由があるかもしれない。FDAは、必要な評価を提出できなかった、延期された最新の状況を維持し、または小児科処方承認要求を提出できなかった任意のスポンサーに、規定に適合しない手紙を送信しなければならない。
FDAは、届出を受ける前に、提出後の最初の60日以内にすべてのNDAを予備審査して、それらが十分に完全であるかどうかを決定し、実質的な審査を行うことができる。FDAはより多くの情報を提供することを要求するかもしれません
秘密保護協定の申請を受けるのではありませんこの場合、申請は追加情報と共に再提出されなければならない。再提出された申請はFDAが届出を受ける前にも審査を行わなければならない
提出された申請が受け入れられると、FDAは深い実質的な審査を開始する。FDAは、薬物が安全かつ有効であるかどうか、および薬物を製造、加工、包装、または保有する施設が、製品の持続的な安全、品質、および純度を確保するための基準に適合しているかどうかを決定するために、秘密保持プロトコルを検討する。現在発効している“処方薬使用料法案”ガイドラインによると,FDAは標準NDA提出日から10カ月以内に,新たな分子実体に提出された材料を審査して行動させることを目標としている。この審査には通常12カ月の時間が必要であり,NDAがFDAに提出された日から計算すると,FDAは申請提出後約2カ月で“届出”決定を下すためである。
FDAは新薬の申請を諮問委員会に提出するかもしれない。諮問委員会は,臨床医や他の科学専門家を含む独立した専門家からなるグループであり,申請を審査,評価し,申請を承認すべきかどうか,どのような条件でアドバイスを提供すべきかを担当する。FDAは諮問委員会の提案に制限されていないが、それは決定を下す時にこれらの提案を慎重に考慮するだろう
秘密協定を承認する前に、FDAは通常、製品を生産する1つまたは複数の施設を検査する。FDAは、製造プロセスおよび施設がcGMP要件に適合していることを決定し、要求された仕様の下で製品が一貫して生産されることを保証するのに十分でない限り、申請を承認しないであろう。さらに、NDAを承認する前に、FDAは、GCP要件に適合することを確実にするために、1つまたは複数の臨床試験地点を検査する可能性がある。
NDAとすべての関連情報を評価した後,諮問委員会のアドバイス(あれば)や製造施設や臨床試験地点に関する検査報告を含めて,FDAは承認状を発行する可能性があり,場合によっては完全な返信が発行される可能性がある。完全な応答は、一般に、再提出されたNDAが最終的な承認を得ることを保証するために満たされなければならない特定の条件を含む声明を含み、FDAが出願を再検討するために追加の臨床または臨床前試験が必要とされる可能性がある。この補足情報を提出しても、FDAは最終的にその申請が承認された規制基準を満たしていないと決定する可能性がある。もしこのような条件がFDAの満足を得たら、FDAは通常承認書を発行するだろう。この薬物の商業マーケティングを許可し、特定の適応の具体的な処方情報を提供する
1つの製品が規制部門の承認を受けた場合、このような承認は特定の適応が付与され、製品が発売される可能性のある指定用途の制限をもたらす可能性がある。例えば、FDAは、製品の利点がそのリスクよりも大きいことを確実にするために、リスク評価および緩和戦略(“REMS”)を有するNDAを承認する可能性がある。REMSは、既知または潜在的な薬物に関連する深刻なリスクを管理し、薬物の安全な使用を管理することによって、患者がそのような薬物を継続的に得ることができるようにするための安全戦略であり、制限された分配方法、患者登録、および他のリスク最小化ツールのような薬物ガイドライン、医師のコミュニケーション計画、または安全使用を保証する要素を含むことができる。FDAはまた,提案されたラベルを変更したり,適切な制御や仕様を作成したりすることを条件に承認することも可能である.FDAはまた、製品の商業化後の安全性と有効性をさらに評価し、監視するために、1つまたは複数の第4段階上場後の研究とモニタリングを要求する可能性があり、これらの発売後の研究の結果に基づいて製品のさらなる販売を制限する可能性がある
開発と審査計画を加速する
FDAは合格した候補製品に一連の迅速な開発と審査計画を提供した。例えば、迅速チャネル計画は、深刻なまたは生命を脅かす疾患または状態を治療するための製品の審査プロセスを加速または促進し、疾患または状態を解決する満たされていない医療需要の潜在力を示すことを意図している。高速チャネルは,候補製品と研究中の特定の適応に適した組合せを指定する.高速チャネル候補製品のスポンサーは,製品開発期間中に適用されるFDA審査チームとより頻繁なインタラクションを行う機会があり,機密協定が提出されると,申請は優先審査を受ける資格がある可能性がある.高速チャネル候補製品のセキュリティプロトコルは、スクロール審査を行う資格がある可能性もあり、この場合、FDAは、完全な出願を提出する前に、セキュリティプロトコルを考慮する審査部分をスクロールさせることができ、スポンサーがセキュリティプロトコル部分を提出するスケジュールを提供した場合、FDAは、セキュリティプロトコルの部分を受け入れることに同意し、スケジュールが許容可能であると判断し、スポンサーは、セキュリティプロトコルの第1の部分を提出する際に任意の必要な使用料を支払うことができる
重篤または生命に危険な疾患や状況を治療しようとする候補品も,その開発や審査を加速するための画期的な療法指定を受ける資格がある可能性がある。予備臨床証拠が、候補製品が単独で、または1つまたは複数の他の薬物または生物学的製品と組み合わせて使用されることを示す場合、候補製品は、1つまたは複数の臨床的重要終点において、例えば、臨床開発早期に観察される実質的な治療効果を示す可能性がある場合、候補製品は、画期的な治療指定を得ることができる。この指定は、高速チャネル計画のすべての機能と、第1段階で開始されたより密集したFDA相互作用および指導と、高度管理者の参加を含む候補製品開発および審査を加速する組織約束とを含む。
迅速なチャネル指定および/または画期的な治療指定を有する候補製品を含むFDA承認を提出する任意の薬物マーケティング申請は、優先審査のようなFDAの他のタイプの計画に参加する資格がある可能性がある。候補製品が、深刻なまたは生命を脅かす疾患または状態の治療を意図しており、承認された場合、そのような疾患または状態の既存の代替品と比較して、安全性または有効性の点で有意な改善を提供する場合、NDAは、優先審査を受ける資格がある。新しい分子実体NDAについて、優先審査指定は、FDAの目標が、提出日60日後6ヶ月以内に上場申請に行動することであることを意味する(標準審査では10カ月)
さらに、臨床研究に適用される設計によれば、深刻または生命に危害を及ぼす疾患または状況の治療において安全性と有効性研究を行う候補製品は加速承認を得る可能性があり、前提は、この製品が代替終点に対して有効であることを決定することであり、この代替終点は合理的に臨床利益を予測することができるか、または臨床終点において不可逆的な発病率または死亡率よりも早く測定することができ、この終点は不可逆的な発病率または死亡率または他の臨床利益を合理的に予測する可能性が高く、同時に病状の深刻性、希少性または盛行度、および代替治療の利用可能性または不足を考慮することである。承認を加速する条件として、FDAは、通常、不可逆的な発症率または死亡率または他の臨床的利益に対する予想される影響を検証および説明するために、スポンサーに十分かつ制御された検証的臨床研究を要求し、承認加速の前にそのような検証的研究を行うことを要求する可能性がある。スポンサーが必要な研究を適時に行うことができなかった場合、あるいはこのような研究が予測の臨床的利益を検証できなかった場合、加速承認を得た製品は迅速な脱退プログラムの影響を受ける可能性がある。また、FDAは現在、承認を加速させる条件として宣伝材料を事前承認することを求めており、製品商業発売の時期に悪影響を及ぼす可能性がある
迅速チャネル指定、画期的な治療指定、優先審査、および承認の加速は、承認の基準を変更することはありませんが、開発または承認プロセスを加速させる可能性があります。製品候補が1つまたは複数の計画の条件に適合していても、FDAは、その製品がもはや資格条件を満たしていないと後で決定するか、またはFDAの審査または承認を決定する期間が短縮されない可能性がある。
孤児薬の指定と排他性
孤児医薬品法によれば、FDAは、まれな疾患または疾患を治療するための薬剤を孤児として指定することができる。まれな疾患または疾患の定義は、患者数が米国で20万人未満であるか、または米国での患者数が20万人を超え、米国での薬剤または生物学的薬剤の開発および提供が合理的に予想されていないコストが、米国での薬剤または生物学的薬剤の販売から回収されることである。秘密保持協定を提出する前に、指定孤児薬を申請しなければならない。FDAが孤児薬物指定を承認した後、FDAは、治療薬の模倣薬識別情報およびその潜在的孤児の使用を開示する。
孤児薬物指定を有する製品が、その後、そのような指定された疾患または疾患の特定の有効成分を有するFDAの最初の承認を得た場合、この製品は、孤児製品の排他性を得る権利があり、これは、FDAが完全なNDAを含む他の出願を承認しない可能性があり、限られた場合を除いて、同じ疾患または疾患の同じ薬剤が7年以内に販売される可能性があることを意味する。例えば、孤児薬物排他性を有する製品に対する臨床的優位性を示すか、またはFDAは、指定された薬物の疾患または状態を有する患者の需要を満たすために十分な数の孤児薬を得ることができることを証明していないことを発見する。孤児薬物排他性は、FDAが同じ疾患または状態に対する異なる薬物、または異なる疾患または状態に対する同じ薬物を承認することを阻止しない。孤児薬を指定する他の利点は、いくつかの研究の税金控除とNDA申請使用料の免除を含む。
指定された孤児薬物が孤児として指定された疾患または状況よりも広い使用として承認された場合、孤児薬物排他性が得られない可能性がある。さらに、FDAが後に指定要求に重大な欠陥があると判断した場合、または上述したように、第2の出願人が、その製品が孤児排他性を有する承認製品よりも臨床的に優れていることを証明する場合、または製品を承認する製造業者が、まれな疾患または疾患患者の需要を満たすのに十分な数の製品を保証できない場合、米国における孤児薬の独占営業権を失う可能性がある
承認後に要求する
FDAによって生産または流通を許可された薬品はFDAの普遍的かつ持続的な監督管理を受けなければならず、その中には記録保存、定期報告、製品サンプリングと流通、広告と販売促進、および製品の不良反応の報告に関連する要求が含まれている。承認後、承認された製品の大多数の変更は、新たな適応または他のラベル宣言を追加するなど、FDAの審査および承認を事前に受けなければならない。また,どの上場製品に対しても継続的な年間使用料要求があり,この要求に基づき,NDA申請者はNDAが承認した処方薬製品ごとに相当な“計画費”を支払わなければならない
また、医薬品メーカーや他の生産·流通承認薬品に参加する実体は、FDAと国家機関にその機関を登録し、米国食品医薬品局(FDA)の定期的な抜き打ち検査を受けなければならない
FDAとこのような州機関はcGMP要求を遵守する。製造プロセスの変更は厳しく規制されており,通常FDAが事前に承認して実施する必要がある。FDA法規はまた、cGMP要求との任意の偏差を調査および是正し、スポンサーおよびスポンサーが使用を決定する可能性のある任意の第三者製造業者に報告および文書要求を提出することを要求する。そのため、メーカーはcGMPコンプライアンスを維持するために、生産や品質管理の分野で時間、お金、労力をかけ続けなければならない
承認された場合、規制要求や基準の遵守が維持されていない場合、または製品発売後に問題が発生した場合、FDAは承認を撤回する可能性がある。その後、製品には、予期されない深刻度または頻度の不良事象、または生産プロセス、または法規要件を遵守できなかったことを含む、以前に未知の問題が存在することが発見され、新しいセキュリティ情報を追加するために承認されたラベルの強制改訂をもたらす可能性がある;新しい安全リスクを評価するために発売後研究または臨床試験を実施すること、またはREMS計画に従って流通または他の制限を実施することが可能である。他の他の潜在的な結果には
•製品の販売や製造を制限し、市場から製品を完全に撤回したり、製品をリコールしたりする
•承認後の臨床試験には罰金、警告状、一時停止を科す
•FDAが承認すべきNDAまたは承認されたNDAの補充剤の承認を拒否するか、または製品承認を一時停止または撤回すること;
•製品を差し押さえたり、差し押さえたり、製品の輸出入を許可することを拒否したりする
•民事または刑事処罰を禁令または適用する
FDAは市場に投入された製品のマーケティング、ラベル、広告、販売促進を厳格に規制している。承認の適応と承認のラベルの規定に基づいてのみ薬物を普及させることができる。FDAや他の機関はラベル外用途の普及を禁止する法律法規を積極的に実行しており,ラベル外用途の普及が不適切であることが発見された会社は重大な責任を負う可能性がある。これらの要求を守らないことは、否定的な宣伝、警告状、改正広告、および潜在的な民事と刑事罰を招く可能性がある。
薬品マーケティング排他性
FDCAによって許可された市場排他性条項は、いくつかのマーケティング申請の提出または承認を延期する可能性がある。例えば、FDCAは、新しい化学エンティティ秘密協定の承認を得た最初の出願人に、5年間の米国内の非特許データ排他期間を提供する。FDAが以前に同じ活性部分を含む他の新薬を承認していなければ,薬物は新しい化学実体であり,活性部分は薬物物質の作用を担う分子やイオンである。排他的期間内に、FDAは、出願人が合法的な参照権を有していない場合、または承認されていない場合、または他の会社が505(B)(2)条(a“505(B)(2)NDA”)に従って提出された簡略化された新薬出願(“ANDA”)またはNDA(a“505(B)(2)NDA”)を承認しないか、または承認するために必要なすべてのデータを参照する場合、元の革新薬と同じ適応のために使用されるか、または他の適応のために使用される可能性がある。しかしながら、出願がイノベーターNDA所有者がFDAに記載された特許のうちの1つを含む特許が無効または未侵害証明である場合、4年後に出願することができる
FDAが、出願人が行っているまたは後援する新しい臨床研究(バイオアベイラビリティ研究を除く)が、既存の薬剤の新しい適応、用量または強度のような承認出願に不可欠であると考えている場合、FDCAは、NDAに3年間の非特許排他性、または既存のNDAの補充を提供することもできる。この3年間の排他性は、この薬剤が新しい臨床研究に基づいて承認された修正のみを含み、FDAがANDAまたは505(B)(2)NDAを許可することは禁止されておらず、元の適応または使用条件の有効成分を含む薬剤のために使用される。5年と3年の排他性は完全な秘密協定の提出や承認を延期したり承認したりしないだろう。しかしながら、完全なセキュリティプロトコルを提出する出願人は、安全かつ有効であることを証明するために、任意の臨床前研究および十分かつ良好に制御された臨床試験を参照するために必要な権利を行うか、または得ることを要求されるであろう。
小児科専門権は米国で利用可能な別のタイプのマーケティング専門権である。小児科排他性規定は,スポンサーがFDAの“書面請求”に応じて児童に臨床試験を行う場合,既存の排他性または利用可能な特許期間の別の期間に追加6カ月の市場排他性を付加することを規定している。書面申請の発表はスポンサーに述べた臨床試験を要求することはなく,FDAの小児科専門権の承認もFDAに行われた研究に基づく小児科使用情報を含むラベルの承認も要求されない。
外国監督管理
アメリカ国外で任意の医薬製品をマーケティングするために、各司法管轄区域はGLP、良好な臨床規範(GCP)と良好な製造規範(GMP)を遵守し、臨床試験を開始し、その後新しい薬物の発売許可を得て、しかも各国が異なることを含む類似の監督管理要求がある。
各司法管轄区域はその臨床試験申請と上場許可申請の評価にこれらの規定を適用する。外国規制承認過程には、上述したFDA承認に関連するすべてのリスクと、具体的な国に対する追加法規が含まれている。外国規制承認過程には、上述したFDA承認に関連するすべてのリスクと、具体的な国に対する追加法規が含まれている。私たちの製品がFDAの承認を得ているかどうかにかかわらず、私たちはこれらの国で臨床試験を開始したり、販売したりするために、外国の比較可能な規制機関の承認を得なければならない。他の国や管轄地域で承認を得るのに要する時間は、FDAの承認を得るのに要する時間とは異なり、さらに長くなる可能性がある。一方の国または管轄区域の規制承認は、他の国または管轄区域の規制承認を確保することはできない。また、1つの国または管轄区域で監督管理の承認を得ることができなかったり、遅延したりすることは、他の国または司法管轄区の監督プロセスにマイナス影響を与える可能性がある。適用される外国の監督管理要求を守らなければ、他のほかに、罰金、監督管理の一時停止または撤回、製品のリコール、製品の差し押さえ、経営制限、刑事起訴などの処罰を受ける可能性がある。
非臨床研究と臨床試験
アメリカと類似して、欧州連合(“EU”)の非臨床と臨床研究の各段階は厳格な監督管理によって制御されている。
非臨床研究を行うのは,新生物物質の健康や環境安全性を証明するためである。非臨床研究(薬物-毒理学)は、EU指令2004/10/ECに規定されているGLP原則を遵守しなければならない(特定の特定の医薬製品に他の理由がない限り、例えば放射性ラベルのための放射性薬物前駆体)。特に、体外と体内の非臨床研究は必ずGLP原則に従って計画、実行、モニタリング、記録、報告とアーカイブを行い、GLP原則は組織過程の品質体系と非臨床研究の条件として一連の規則と標準を定義した。このようなプロス基準は経済協力と開発組織の要求を反映する。
EUでは,医療製品の臨床試験はEUと国家法規および国際人用薬品技術要求調整理事会(“ICH”)のGCPに関するガイドラインおよび“ヘルシンキ宣言”からの適用法規要求と倫理原則に従って行わなければならない。臨床試験の発起人がEU内で成立していない場合、それはEU実体をその法定代表者として指定しなければならない。スポンサーは臨床試験保険証書を購入しなければならず、大多数のEU加盟国では、スポンサーは臨床試験で負傷したいかなる研究対象にも“非のない”賠償を提供する責任がある。
EUの臨床試験に関連する規制構造は最近変化した。EU臨床試験条例(“CTR”)は2014年4月に採択され、EU臨床試験指令が廃止され、2022年1月31日に施行された。指示とは異なり、CTRはすべてのEU加盟国に直接適用され、加盟国がそれをさらに国家法律として実施する必要はない。CTRは臨床試験情報システムを通じてEU全体の臨床試験の評価と監督過程を著しく調整し、このシステムは集中したEU門戸とデータベースを含む。
EU臨床試験指令は,臨床試験を行う各加盟国で主管する国家衛生当局と独立した倫理委員会に単独の臨床試験申請(“CTA”)を提出することを要求しているが,FDAやIRBのように,CTRは集中的な手続きを導入し,多センター試験の申請を提出することのみを要求している。CTRは、スポンサーが各会員国の主管当局と道徳委員会に文書を提出することを可能にし、各会員国が決定を下すことを可能にする。他の事項以外に、CTAは試験方案のコピーと被調査薬品の生産と品質情報を含む調査薬品ファイルを含まなければならない。CTAの評価手続きも統一されており、すべての関連加盟国による共同評価を含み、道徳基準を含む各加盟国が個別にその領土に関する具体的な要求を評価する。各会員国の決定は集中されたEUポータルサイトを通じてスポンサーに伝達される。CTAが承認されると,臨床研究開発は継続可能である
CTRは3年間の過渡期が予想される。進行中の臨床試験と新たな臨床試験がCTRによってどの程度制御されるかはそれぞれ異なる。2022年1月31日までにEU臨床試験指令に基づいて申請を提出した臨床試験、または(Ii)が2022年1月31日から2023年1月31日までの間、かつスポンサーがEU臨床試験指令に適用される臨床試験を選択し、2025年1月31日までこの指令の管轄を受ける。この日以降,すべての臨床試験(行われている臨床試験を含む)はCTR条項に拘束される。
臨床試験で使用される薬品はGMPで生産されなければならない。他の国と連合の範囲の規制要件も適用される可能性がある。
マーケティング許可
EUでは、医薬製品はマーケティング許可(MA)を得た後にのみ市場に投入されることができる。EU規制制度によると、規制機関による候補製品の承認を得るためには、M&A申請(MAA)を提出しなければならない。このようにする過程は,他を除いて医薬製品の性質に依存する。2つのタイプのMAがあります
•“集中MA”は欧州委員会が欧州薬品管理局人用薬品委員会(“CHMP”)の意見に基づき、集中プログラムを通じて発行され、EU全体で有効である。特定のタイプの医療製品、例えば、(I)バイオテクノロジープロセスで抽出された薬剤、(Ii)高度治療医薬製品(“ATMP”)(例えば、遺伝子治療、体細胞治療および組織工学製品)、(Iii)孤児指定医薬製品、および(Iv)HIV/エイズ、癌、神経変性疾患、糖尿病、自己免疫疾患および他の免疫機能障害およびウイルス疾患などの特定の疾患を治療するための新しい活性物質を含む製品は、集中プログラムを実行しなければならない。EUで許可されていない新しい活性物質を含む製品、または重大な治療、科学的または技術革新、またはEUの公衆衛生上の利益に適合する製品については、集中手順がオプションである。
•“国家MA”はEU加盟国の主管当局によって発行され、それぞれの領土のみをカバーし、集中プログラムの強制範囲に属さない製品候補を使用することができる。1つの製品がEU加盟国で販売されることが許可されている場合、その国MAは、相互認識手順によって別の加盟国で承認を得ることができる。この製品が申請時にどの加盟国でも国MAを取得していない場合、分散手続きによって各加盟国で同時に承認を得ることができる。分権手続きに基づいて、MAを求める各加盟国の主管当局に同じ書類を提出し、申請者はそのうちの1つを参考加盟国として選択する。
上記の手順により、市場参入を付与するために、欧州市場管理局又はEU加盟国主管当局は、製品の品質、安全性及び有効性に関する科学的基準に基づいて、製品のリスク−利益バランスを評価する。MAの初期期限は5年である.この5年後、リスク-収益バランスを再評価した上で許可を更新することができる
中央プログラムによると,環境評価機関が重大な影響評価を評価する最長時限は210日であり,クロックポーズは含まれていない。特別な場合、CHMPは、150日以下(クロックポーズを含まない)でMAAを加速評価する可能性がある。満たされていない医療需要に対して、公衆の健康に大きな影響を与えることが期待される革新的な製品は、米国の画期的な治療指定と同様のインセンティブを提供する一連の加速開発および審査計画、例えば優先薬物(Prime)計画を得る資格がある可能性がある。EMAは2016年3月、満たされていない医療需要に対する薬物開発の支援を強化するためのPrime計画という自発的な計画を開始した。その基礎は、有望な薬剤を開発している会社との相互作用と早期対話を増加させ、彼らの製品開発計画を最適化し、より早期に患者に接触するのを助けるために、彼らの評価を加速させることである。Prime指定を受けた製品開発者は加速評価を受ける資格が期待されるが,これは保証ではない。Primeの称号を持つ候補製品のスポンサーは多くのメリットを得ることができ、しかし限らず、早期にEMAと積極的な監督対話を行い、臨床試験設計とその他の開発計画要素を頻繁に討論し、及びファイルを提出した後にMAA評価を加速する。CHMPの専任連絡先と調査委員は、EMA委員会レベルで製品のより多くの理解を促進するために、Prime計画の早期に任命されたことが重要である。最初の会議はこれらの関係を開始し、EMAの多学科専門家チームを含み、全体的な発展と監督戦略に関する指導を提供した。
データとマーケティングの排他性
EUでは、ライセンス発売された新製品(すなわち、参考製品)は、通常、MAで8年間のデータ独占および追加2年間の市場独占を得る。承認された場合、データ固有期間は、模倣薬または生物類似薬の申請者がEUで模倣薬または生物類似薬を申請することを防止することになり、参照製品がEUで初めて許可された日から8年以内に、参照製品プロファイルに含まれる臨床前および臨床試験データに依存する。市場排他期は、参考製品がEUで最初の許可を得た10年後まで、成功した模倣薬または生物類似申請者がその製品をEUで商業化することを禁止している。MA保持者が10年の最初の8年間に1つまたは複数の新しい治療適応の許可を得た場合、10年間の市場専門期間は最大11年に延長されることができる。許可前の科学的評価では、これらの適応は既存の治療法と比較して有意な臨床的利益をもたらすと考えられる。しかし、一つの製品がEU規制機関によって新たな活性物質とみなされる保証はなく、製品はデータ排他性を得る資格がない可能性がある。
小児科発展
EEAでは,新医薬製品のMAAは小児科群で行われた研究結果を含まなければならず,EMA小児科委員会(PDCO)と合意された小児科調査計画(PIP)に適合している。PIPセット
MAが求められている薬物の小児科適応をサポートするためのデータ生成の時間および推奨措置を決定する。PDCOは、成人における製品の有効性および安全性を証明するのに十分なデータがあるまで、PIPの一部または全ての措置の実施を延期する義務を許可することができる。さらに、小児臨床試験データを必要としないか、または提供するのに適していない場合、PDCOは、子供に無効または安全でない可能性があるので、これらのデータを提供する義務を免除することができ、この製品は、治療のために使用される疾患または状態が成人集団でのみ発生することが予想される場合、または小児科患者の既存の治療に対して有意な治療利益がない場合。著者らはすでにCOPD小児科データの免除を受けた。
孤児医薬製品
EUの“孤児薬品”の認定基準は原則的にアメリカと似ている。スポンサーが、(1)生命または慢性衰弱にかかわる疾患の診断、予防または治療を目的としている場合、(2)または(A)申請時に、EUにおけるそのような疾患の影響が10,000人中5人以下であること、または(B)孤児身分によるメリットがなければ、EUで十分な見返りを生じることなく、投資が合理的であることを証明することができる場合、(3)EUに市販されているような疾患の診断、予防または治療に満足できる方法がないか、またはそのような方法が存在する場合、製品は、そのような疾患の影響を受けている人に大きな利点を有するであろう。
MAAを提出する前に、孤児としての指定を要請しなければならない。EUが指定した孤児は一方に費用の削減や免除、礼賓援助、集中手続きへの進入などのインセンティブを得る権利がある。MAを承認した後、孤児医薬製品は、承認された治療適応の10年間の市場排他性を得る権利があり、これは、EU規制機関が別のMAを受け入れることができないこと、またはMAを承認すること、または同じ適応の類似製品のMAを10年間延長する申請を受け入れることを意味する。合意されたPIPにも適合する孤児薬品については,市場専門期間を2年間延長した。いかなる補充保護証明書も孤児の症状に関する小児科研究によって延期してはならない。孤児指定は、規制審査および承認過程においていかなる利点も伝達されず、規制審査および承認過程の継続時間を短縮することもない。
5年目の終了時に、製品が市場排他性を維持するのに不十分であることを証明するのに不十分であることを証明することを含む、指定された孤児の基準に適合しないと決定された場合、または疾患の流行率が限界値を超えている場合、孤児専門期間は6年に短縮されることができる。さらに、以下の場合、いつでも同じ適応の類似製品のためにMAを承認することができる:(1)第2の出願人は、その製品が類似しているが、より安全で、より効果的で、または臨床的により良いことを証明することができる、(2)出願人は、第2の申請孤児薬に同意するか、または(3)出願人は、十分な孤児薬を提供することができない。
承認後に要求する
アメリカと同様に、MA保有者と医薬製品メーカーはすべてヨーロッパ薬品管理局、欧州委員会及び/又はEU加盟国主管監督機関の全面的な監督管理を受けている。MAの所持者は薬物警戒制度を設立·維持し,合資格の薬物警戒者(“QPPV”)を委任し,この制度の設立と維持を担当し,医療製品の安全概況や新たに出現した安全問題を監督しなければならない。主な義務は深刻な副作用の疑いの報告の加速と定期的な安全更新報告(“PSURs”)の提出を含む。
すべての新しいMAAは、企業が実施するリスク管理システムを記述し、製品に関連するリスクを防止または最大限に低減するための措置を記録するリスク管理計画(RMP)を含まなければならない。規制当局はまた、特定の義務を金融管理専門家の条件として規定することができる。このようなリスク最小化措置または認可後の義務は、追加の安全監視、PSURsのより頻繁な提出、または追加の臨床試験または認可後の安全性研究を含む可能性がある。
医薬製品の広告と販売促進も医薬製品の販売促進、医師との相互作用、誤解性と比較広告及び不公平な商業行為に関する法律の制約を受けている。製品のすべての広告および販売促進活動は、承認された製品特性要約と一致しなければならないため、すべてのラベル外の販売促進活動は禁止されます。連合はまた消費者に直接向けた処方薬の広告を禁止する。薬品広告や販売促進の一般的な要求はEU指令に基づいて制定されているが、詳細は加盟国ごとの法規によって管理されており、各国が異なる可能性がある。
上記のEU規則は、27のEU加盟国にノルウェー、リヒテンシュタイン、アイスランドを加えた欧州経済圏(EEA)に一般的に適用されている。
私たちまたは私たちの任意の第三者パートナーは、サプライヤー、製造業者、および流通業者を含み、EUおよびEU加盟国が臨床試験、製造承認、医薬製品のMAおよびそのような製品のマーケティングに適用される法律を遵守することができず、MA付与の前後、医薬製品の製造、法定健康保険、賄賂および反腐敗または他の適用可能な法規要件にかかわらず、行政、民事または
刑事罰。これらの処罰は、臨床試験またはMAの承認の遅延または拒否、製品の撤回およびリコール、製品の差し押さえ、一時停止、マーケティング許可の撤回または変更、生産の完全または部分的な一時停止、流通、製造または臨床試験、経営制限、禁止、免許取り消し、罰金、および刑事罰を含むことができる。
イギリスの離脱とイギリスの規制枠組み
2021年1月1日の離脱移行期間が終了して以来、イギリス(イングランド、スコットランド、ウェールズ)はEU法律の直接的な制約を受けていないが、アイルランド/北アイルランド議定書の条項によると、EU法律は北アイルランドに一般的に適用されている。二次立法によってイギリス法律に転換されたEU法律は依然としてイギリスに適用されるが、EU CTRなどの新しい立法はイギリスには適用されない。
“2021年医薬品·医療機器法”によると、国務長官または“適切な当局”は、医療製品や医療機器分野の既存の法規を改正または補充する権利がある。これにより,人間の薬物,臨床試験,医療機器分野の規制格差や将来の変化を解決する上で柔軟性を許すことを目的として,今後二次立法で新たなルールを導入することが可能となる。
2021年1月1日から、薬品と保健品監督局(“MHRA”)はイギリスSの独立した薬品と医療機器監督機関になった。北アイルランド議定書の結果として、北アイルランドはイングランド、ウェールズ、スコットランドとは異なる規則、GBが適用されるだろう;全体的に、北アイルランドはEUの規制制度に従うが、その国家主管機関はMHRAであるだろう
臨床試験に関連するイギリスの規制枠組みは、既存のEU立法(二次立法によってイギリス法で実施される)に由来する。2022年1月17日、MHRAはイギリスの臨床試験立法の再制定に関する8週間の諮問を開始し、臨床試験の審査を簡略化し、革新を促進し、臨床試験の透明性を高め、リスクの割合を高め、そして患者と公衆の臨床試験への参加を促進することを目的とした。MHRAは2023年3月21日に諮問に応じ,立法を早期に改正することを確認した。イギリス政府が提出した最終法律テキストは最終的にイギリスの臨床試験枠組みとCTRの一致或いは乖離の程度を決定する
MHRAは、150日間の評価およびスクロール審査プログラムを含む、患者に利益を得る新薬を優先的に得るプログラムを含む、国家許可プログラムを変更している。中央許可製品のためのすべての既存のEU MAは、MA所有者が離脱を選択しない限り、GBでのみ有効であり、無料で、2021年1月1日にイギリスMAに自動的に変換またはキャンセルされる。集中プログラムを用いて欧州経済区全体で効果的なM&Aを獲得するためには,欧州経済区に会社を設立しなければならない。したがって,イギリスが離脱して以来,まずヨーロッパ経済区実体が構築されていない場合,イギリスに設立された会社はEU集中プログラムを使用することはできず,欧州経済区実体はいかなる集中式MAを持たなければならない。英国で製品を商業化するイギリスMAを得るためには、申請者はイギリスで設立されなければならず、イギリスで製品を商業化するためには、イギリスの国家認可プログラムのうちの1つまたはイギリス離脱後に残りの国際協力プログラムのうちの1つに従わなければならない。2024年1月1日から、新しい国際認可フレームワークがすでに到着しており、この枠組みに基づいて、MHRAは新しいGB MAの申請を決定する際に、EMAとある他の規制機関によるMAの承認決定を考慮する。
MA前の孤児の称号はありません。逆に、MHRAは、対応するMA申請を審査しながら孤児指定申請を審査する。これらの基準は基本的に同じであるが,すべて市場のためにオーダーメイドされている,すなわちEUではなくイギリスのこのような疾患の流行率は万分の5を超えてはならない。孤児の称号が与えられた場合、市場独占期間はこの製品がGBで初めて承認された日から設定される。
他の医療保険法
FDAによる薬品と生物製品マーケティングの制限以外に、アメリカの他の連邦と州医療監督法律は製薬業界の商業行為を制限し、その中には州と連邦の反リベート、虚偽声明と医師支払い、および薬品定価透明性法律が含まれているがこれらに限定されない。外国の管轄区域にも似たような法律がある。
他の事項に加えて、米国連邦反バックオフ法規は、任意の個人またはエンティティが、購入、レンタル、注文、手配または推奨購入、レンタル、またはMedicare、Medicaidまたは他の連邦医療保健計画に従ってすべてまたは部分的に精算することができる任意の商品、施設、物品またはサービスの購入、レンタル、または注文を全部または部分的に精算することができる任意の商品、施設、物品またはサービスの購入、レンタル、または注文を誘導または補償するために、意図的または意図的または間接的に、公開的または間接的に提供、支払い、請求、受け入れ、または任意の報酬を提供することを禁止する。“報酬”という言葉は価値のあるものを含むと広く解釈されている。逆リベート法規は、薬品メーカーと処方者、購入者、処方管理人と受益者との間の手配に適用されると解釈される。いくつかの法定例外状況と規制安全港保護のいくつかの一般的な活動は起訴されないが、例外状況と安全港の範囲は狭い。報酬に関する接近は、処方、購入、または推薦を誘導することを目的としていると告発される可能性があり、もしそうであれば、審査されるかもしれない
法的または規制の例外や避難港の要求を満たしていない。特定の適用された法定例外や安全港を規制するすべての要求を満たすことができなかったことは、米国連邦反リベート法規に基づいて、このような行為自体が不法であることを意味するわけではない。代わりに、そのすべての事実と状況の累積審査に基づいて、この手配の合法性を逐案的に評価する。いくつかの裁判所は、この法規の意図要求を、報酬の手配に関連する任意の目的が連邦医療カバーの業務への移行を誘導することである場合、法規が違反されたと解釈する
また,個人や実体は,法規や法規違反の具体的な意図を実際に知る必要がなく,違反行為を実施することができる
任意の個人または実体が意図的に連邦政府に提出するか、または虚偽、架空または詐欺的な支払いまたは承認クレームの提出を引き起こすことを禁止し、連邦政府への虚偽または詐欺的クレームに関連する虚偽記録または陳述の作成、使用、または作成または使用をもたらし、または米国連邦政府への金銭支払い義務を回避、減少または隠蔽するための虚偽陳述を意図的に行うことを禁止する民事虚偽請求法案を含む連邦虚偽クレーム法律。クレームには、米国政府に提出された金銭または財産に対する“任意の請求または要求”が含まれている。民事虚偽申告法に基づく訴訟は総検事長が提起することができ、個人が政府名義で訴訟を提起することもできる。民事虚偽請求法違反は非常に深刻な罰金と3倍の損害賠償を招く可能性がある。これらの法律によると、いくつかの製薬会社や他のヘルスケア会社が起訴されており、顧客に製品を無料で提供した疑いがあり、顧客がその製品の連邦計画に料金を徴収することを期待している。これらの会社のマーケティング製品が未承認またはラベル外の用途のために使用され、虚偽の声明を提出することになったため、他の会社も起訴された。また、連邦民事虚偽請求法案については、米国連邦反ダンピング法案違反による物品やサービスを含むクレームが虚偽または詐欺的クレームを構成している。さらに、民事罰金法規は、連邦健康計画にクレームを提起することが決定されたまたは原因となった任意の者に処罰を加え、その人は、クレームに従って提供されていない項目またはサービスまたは虚偽または詐欺的な項目またはサービスであることを知っているか、または知っているべきである。多くの州でも同様の詐欺や法律や法規の乱用があり、医療補助や他の州で計画されている精算プロジェクトやサービスに適用されたり、いくつかの州では支払者にかかわらず適用されています。
1996年の連邦健康保険携行性と責任法案、あるいはHIPAAは、個人第三者支払人、医療福祉計画を故意に流用または盗み取り、医療保健違法行為に対する刑事調査を故意に妨害し、故意に偽造、隠蔽または隠蔽または隠蔽し、あるいは医療福祉、プロジェクトまたはサービスの提供または支払いに関連する重大な虚偽、架空または詐欺的陳述を行うことを含む追加の連邦刑法を制定し、故意に詐欺の任意の医療福祉計画を知りながら実行または実行しようとする計画を禁止している。アメリカ連邦反リベート法規と類似して、個人或いは実体は実際にこの法規或いはこの法規に違反する具体的な意図を理解する必要がなく、違反を実施することができる
また,最近では連邦や州政府が医師や他の医療提供者に支払う費用の規制を強化する傾向が見られている。その他の事項以外に、“医師報酬陽光法案”は、医師(定義は医師、歯科医師、検眼師、足科医師と脊椎マッサージ師を含む)、ある非医師従事者(医師アシスタント、勤務看護師、臨床看護師専門医、勤務看護師麻酔科医、麻酔学アシスタントと勤務看護師助産師を含む)と教育病院に何らかの支払いと“価値移転”を提供し、医師及びその直系親族が持つ所有権と投資権益を規定し、メーカーの年間報告要求をカバーする。すべての支払い、価値譲渡および所有権または投資利益のために必要な情報をタイムリーかつ正確かつ完全に提出できなかった場合、重大な民事罰金を招き、“失敗を承知でいる”ために追加処罰を受ける可能性がある。保険引受メーカーは、その後の例年の90日目までに報告書を提出しなければならない。さらに、ある州は、製薬業の自発的コンプライアンスガイドラインおよび連邦政府が発行した関連コンプライアンスガイドラインを遵守し、医師および他の医療保健専門家および実体に提供されるプレゼント、補償および他の報酬または価値項目のマーケティング慣行および/または追跡および報告に制限を加えるコンプライアンス計画の実施を要求する
このような任意の法律または任意の他の適用される政府法規に違反することは、損害賠償、罰金、連邦医療保険および医療補助を含む連邦医療計画から除外される可能性、返還および企業誠実協定を含む重大な刑事、民事および行政処罰をもたらす可能性があり、これらの合意は、他に加えて、これらの法律を遵守しないことに関する疑惑を解決するために、会社に厳しい運営および監督要求を加える。このような会社の行政職員たちと従業員たちはまた似たような制裁と処罰、そして監禁を受けることができる。実際と潜在的な和解額が巨大であることから,政府当局は引き続き大量の資源を投入し,医療提供者やメーカーが適用される詐欺や法律の乱用を調査する予定である。また、類似した国や外国の法律法規の範囲は上記の規定の範囲よりも広い可能性があり、支払者にかかわらず適用可能である。これらの法律と法規は大きく異なる可能性があり、コンプライアンス作業をさらに複雑化させる可能性がある。例えばEUでは多くのEU加盟国が具体的な
贈与法規は,医療製品の商業行為,特に医療専門家や組織に対してさらに制限されている。また、最近では、医療専門家や実体への支払いや価値移転の規制を強化する傾向があり、多くのEU加盟国は国家サンシャイン法案を可決し、製薬会社に対して米国の要求(通常は年に1回)のような報告や透明性要件を実施している。いくつかの国はまた、商業コンプライアンス計画を実施することを要求するか、またはマーケティング支出および価格設定情報の開示を要求する。このような任意の法律または任意の他の適用可能な政府法規に違反することは、重大な行政、民事および刑事罰、損害賠償、罰金、返還、追加の報告義務および監督を含むが、これらの法律を遵守しない、削減または再運営、政府医療計画から除外され、監禁された疑惑を解決するために、会社の誠実な合意または他の合意の制約を受ける場合、処罰を招く可能性がある。
保証と精算を請け負う
著者らが監督管理の許可を得た任意の薬品或いは生物製品のカバー範囲と精算状態に対して、重大な不確定性が存在する。米国や他国の市場では,自分の病状に応じて処方治療を受けている患者や処方サービスを提供する提供者は,通常第三者支払者に依存してすべてまたは一部の関連医療費を精算している。患者は保険を提供しなければ、私たちの製品を使用することはあまりできません。私たちの製品の大きなコストを支払うのに十分な費用を精算するのに十分です。したがって、規制部門の承認を得て商業販売を行う任意の製品の販売は、保証範囲の可用性と第三者支払者の十分な補償にある程度依存する。第三者支払者には、政府当局、医療計画の管理、個人健康保険会社、その他の組織が含まれる
米国では、第三者支払い者が医薬品または生物製品に保険を提供するかどうかを決定するプロセスは、一般に、そのような製品の価格を設定すること、または保険が承認された後に支払者が製品に支払うべき支払率を決定するプロセスから分離される。第三者支払者は、承認リスト上の特定の製品に保証範囲を制限することができ、処方表とも呼ばれ、FDAによって承認された特定の適応のすべての製品を含まない可能性がある。第三者支払人は私たちの候補製品を保証しないことを決定し、承認されると、医師の私たちの製品に対する使用を減少させ、私たちの販売、運営結果、財務状況に実質的な悪影響を与える可能性があります。また,第三者支払者が薬品や生物製品に保険を提供することを決定することは,適切な返済率を承認することを意味するものではない。製品開発への投資の適切な見返りを実現するために、十分な価格レベルを維持することができる十分な第三者精算がないかもしれない。また、製品の保証範囲と精算範囲は支払人によって異なる。第三者支払者は、ある特定の医療製品又はサービスを保証することを決定し、他の支払者も当該医療製品又はサービスに保険を提供するか、又は適切な販売率で保険を提供することを確保することができない。したがって、保証範囲の決定過程は、各支払人にそれぞれ私たちの製品を使用する科学的かつ臨床的支援を提供する必要があり、これは時間のかかる過程になる
国際市場では、精算や医療支払いシステムは国によって異なる。EUでは,政府はその定価と精算規則や国家医療システムの制御により製品の価格に影響を与え,これらのシステムは消費者にこれらの製品の大部分のコストを支払っている。加盟国はその国の医療保険制度が精算を提供する薬品の範囲を自由に制限し、人が使用する薬品の価格と精算レベルを制御することができる。一部の司法管轄区域はプラスリストとネガティブリスト制度を実施しており、この制度の下で、製品は政府が価格を精算することに同意した後にのみ販売することができる。会員国は、医薬製品の具体的な価格または補償レベルを承認することができ、または医薬製品を市場に投入する責任を負う会社の利益能力に対して、数量に基づく手配、上限、および参考定価メカニズムを含む直接または間接的な制御制度をとることができる。精算または定価の承認を得るために、その中のいくつかの国は臨床試験の完成を要求する可能性があり、特定の候補製品のコスト効果を現在利用可能な治療法と比較する。他の会員国たちは会社が自分の薬品価格を固定することを許可するが、会社の利益を監視する。薬品に対して価格制御や精算制限を実行する国が私たちのいかなる製品にも有利な精算と定価手配を許可することは保証されません。医療コスト、特に処方薬の下振れ圧力は非常に大きくなっている。そのため、新製品の参入にはますます高い壁が設けられている。また、一部の国では、低価格市場からの国境を越えた輸入が一国国内の定価に商業圧力をかけている
医療コストを抑えることはすでに連邦、州と外国政府の優先事項になり、薬品或いは生物製品の価格はずっとこの努力の重点である。第三者支払者は安全性と有効性を疑問視するほか、医療製品やサービスの価格に挑戦し、医療の必要性を審査し、薬品や生物製品、医療機器と医療サービスの費用効果を審査することが増えている。もしこれらの第三者決済者が私たちの製品が他の利用可能な療法と比較して費用効果があると思わない場合、彼らはFDA承認後に私たちの製品をカバーしないかもしれないし、もし彼らがそうした場合、支払いレベルは私たちに利益的な方法で製品を販売させるのに十分ではないかもしれない
医療改革
アメリカの医療業界と他の地域の主な傾向の一つはコストコントロールだ。政府当局や他の第三者支払者は,特定の医療製品のカバー範囲や精算金額を制限することでコストを抑制しようとしている。例えば、2010年3月に“保健と教育和解法案”によって改正された“患者保護と負担できる医療費用法案”が公布され、その中で、他を除いて、医療補助薬品還付計画の下で大多数のメーカーが不足している最低医療補助税金還付を向上させた;吸入、注入、点滴、移植または注射の薬品計算メーカーに対して医療補助薬品還付計画の下で不足している払い戻しを行う新しい方法を採用し、医療補助薬品還付計画を医療補助管理に参加する看護計画を使用する個人の処方に拡大し、ある連邦医療保険D部分の受益者に対して強制割引を実施し、メーカーが連邦医療保険D部分でカバーする外来薬の条件とする。
ACAのいくつかの側面は公布以来、司法、行政、そして国会の挑戦を受けてきた。2021年6月17日、米国最高裁はいくつかの州がACAに対して提出した最新の司法挑戦を却下したが、ACAの合憲性を具体的に裁くことはなかった。最高裁が裁決を下す前に,総裁·バイデンはACA市場による医療保険の取得を目的とした2021年2月15日から2021年8月15日までの特殊保険期間を開始する行政命令を発表した。行政命令はまた、特定の政府機関に、医療の獲得を制限する既存の政策や規則を審査し、再考するよう指示した。
また、2011年8月2日には、提供者に支払う医療保険の全面的な削減を含む支出削減措置を制定するための2011年予算制御法が公布され、この法案は2013年4月1日に施行され、この法規の後続立法改正により、国会がさらなる行動を取らない限り、2020年5月1日から2022年3月31日まで一時的に一時停止される2032年まで有効となる。2013年1月2日、病院、画像センター、がん治療センターを含むいくつかの提供者に支払う医療保険をさらに減らし、政府が提供者に多額の支払いを取り戻す訴訟時効を3年から5年に延長する“2012年米国納税者救済法”が法律に署名された。また,2021年3月,2021年に米国救援計画法案が署名されて法律となり,2024年1月1日から法定の医療補助薬品還付上限が廃止された。これまで,還付の上限は薬品メーカーの平均価格の100%であった。
最近、政府はメーカーがその販売製品に価格を設定する方式をより厳格に審査し、最近の国会で数回の調査を行い、製品の価格設定の透明性の向上、価格設定の審査とメーカー患者計画との関係、政府計画の薬品精算方法を改革するための立法を提出し、公布した。2022年8月16日、2022年インフレ削減法案(IRA)が署名され法律となった。他の事項を除いて、アイルランド共和軍はある薬品のメーカーに連邦医療保険との価格交渉を要求し(2026年から)、連邦医療保険B部分と連邦医療保険D部分に基づいてリベートを徴収し、インフレを超える価格上昇を罰し(初めて2023年に満期)、D部分のカバーギャップ割引計画の代わりに新しい割引計画を採用する(2025年から)。アイルランド共和軍は,衛生·公衆サービス部(HHS)秘書が最初の数年間,規制ではなく指導によってその多くの規定を実施することを許可した。2023年8月29日、衛生·公衆サービス部は価格交渉を受ける上位10種類の薬品リストを公表した。HHSはMedicare薬品価格交渉計画が現在法的挑戦を受けているにもかかわらず、IRA実施の指導意見を発表し、引き続き発表する。アイルランド共和軍の製薬産業への影響はまだ完全には確定されていないが、それは重大かもしれない。
将来的には、より多くの州、連邦、外国の医療改革措置が取られると予想され、いずれも連邦と州政府が医療製品やサービスのために支払う金額を制限する可能性があり、これは私たちの製品に対する需要の減少を招き、承認されると、追加の価格上昇を招く可能性がある。特に,連邦医療保険B部分はアンテフェンタニルの精算に重要な役割を果たすことが予想される。承認されれば,連邦医療保険Bによる製品の一部精算方式の変化がアンテフェンタニルの全体的な保証範囲に影響する可能性がある。連邦医療保険や他の政府援助計画のいかなる精算減少も、個人支払者の支払いの同様の減少を招く可能性がある。
類似した政治、経済、規制発展がEUで発生しており、製薬会社がその製品を商業化する能力に影響を与える可能性がある
2021年12月13日,衛生技術評価に関する第2021/2282号条例が可決され,第2011/24/EU号指令が改正された。この規定は2022年1月に施行されたが、2025年1月から適用され、その間に実施に関する準備と手順が取られるだけである。適用されると、それは関連製品に従って段階的に施行されるだろう。この条例はEU加盟国の新医薬製品を含む衛生技術の評価における協力を促進し、これらの領域の共同臨床評価にEUレベルの協力基礎を提供することを目的としている。EU加盟国がEU範囲内で汎用的なHTAツール、方法、プログラムを使用することを可能にし、患者に最大の潜在的影響を有する革新的な衛生技術の共同臨床評価、共同科学相談、開発者がHTA当局にアドバイスを求め、新興衛生技術および早期発見の将来性のある技術を決定し、自発的に継続することを含む4つの主要分野で協力することができる
他の分野の協力。個別EU加盟国は、衛生技術の非臨床(例えば、経済、社会、倫理)の評価を引き続き担当し、定価と精算について決定する
データプライバシーとセキュリティ法
多くの州、連邦、および外国の法律、法規、標準は、健康に関連する個人情報および他の個人情報の収集、使用、アクセス、秘密およびセキュリティを規定しており、現在または将来的には、私たちの運営または私たちのパートナーの運営に適用される可能性がある。アメリカでは、データ漏洩通知法、健康情報プライバシーと安全法、および消費者保護法律と法規を含む多くの連邦と州の法律法規が、健康に関連する個人情報と他の個人情報の収集、使用、開示と保護を規範化している。また、いくつかの外国法は、健康に関するデータを含む個人データのプライバシーおよびセキュリティを管理する。プライバシーとセキュリティ法律、法規、その他の義務は絶えず変化し、互いに衝突し、コンプライアンス作業を複雑化させ、調査、訴訟あるいは行動を招き、重大な民事および/または刑事罰およびデータ処理の制限を招く可能性がある。
法規を付加する
これらの規定に加えて,環境保全と有害物質に関する州や連邦法は,“職業安全と健康法”,“資源保護·回収法”,“有毒物質制御法”を含め,我々の業務に影響を及ぼす。これらの法律や他の法律は,様々な生物,化学,放射性物質の使用,処理,処分を管理しており,これらの物質は作業,作業から発生する廃棄物に用いられている。もし私たちの運営が環境汚染を招いたり、個人を危険物質に曝露させたりすれば、損害賠償と政府罰金の責任を負う可能性があります。他の特定の国も同様の法律を採択し、似たような義務を規定している。
アメリカの“海外腐敗防止法”
米国の“反海外腐敗法”(FCPA)は、海外業務を獲得または保留したり、公的な身分で働いている人に影響を与えるために、米国の会社や個人が何らかの活動に従事することを禁止している。いかなる外国の政府職員、政府職員、政党または政治候補者に支払いを提案し、任意の価値のあるものを支払うことを提案して、業務を獲得または保留しようとしているか、または他の方法で公的身分で働く人に影響を与えることは、不法である。FCPAの範囲は多くの国のある医療専門家との相互作用を含む。他の国も同様の法律を採択し、似たような義務を規定している。
従業員
2023年12月31日現在、私たちは79人のフルタイム従業員がいる。私たちの従業員はみんな集団交渉合意の側でもなく、労働組合や労働組合の代表でもない。私たちは私たちが従業員と仲がいいと思う。
情報を付加する
我々は2005年2月にイングランドとウェールズ法によりISIS Resources plcとして成立した。2006年9月、私たちはカナダに登録設立された民間会社Rhinophma Limitedを買収し、私たちの名前をVerona Pharma plcに変更した。私たちの主な事務所は3 More London Riverside、London、SE 12、2 RE、UKにあります
我々は、Form 10-K年間報告、Form 10-Q四半期報告、Form 8-K現在の報告、およびこれらの報告の任意の修正を含む米国証券取引委員会に提出された文書を公開し、これらの材料を米国証券取引委員会または米国証券取引委員会にこのような材料を電子的にアーカイブした後、合理的で実行可能な範囲でできるだけ早く我々のウェブサイトwww.veronapharma.comの“投資家”の一部を介して米国証券取引委員会を無料で提供する。当社サイトに含まれているか、または本サイトで取得可能な情報は、本年度報告の一部を構成していません。米国証券取引委員会はまた、ヴェローナ製薬会社を含む米国証券取引委員会に電子的に提出された発行者に関する報告書、依頼書、その他の情報を含むウェブサイトをwww.sec.gov上に保持している。
第1 A項。リスク要因
私たちのアメリカ預託証明書に投資することは高度な危険と関連がある。私たちの総合財務諸表と関連付記、および“経営陣の財務状況と経営業績の検討と分析”を含む、以下のリスクおよび本年度報告書の他の情報をよく考慮しなければなりません。次のいずれの事件や事態の発生も、当社の業務、財務状況、経営業績、成長見通しに悪影響を及ぼす可能性があります。この場合、私たちのアメリカ預託証明書の市場価格は下落する可能性があり、あなたは投資の全部または一部を損失する可能性があります
私たちのビジネスや産業に関するリスクは
私たちの経営の歴史は限られていて、どんな製品収入も発生したことがありません。
私たちは臨床段階の生物製薬会社で、運営の歴史は限られており、設立以来ずっと重大な運営損失を受けている。2023年12月31日と2022年12月31日の会計年度までの純損失はそれぞれ5440万ドルと6870万ドルだった。2023年12月31日現在、私たちの累計赤字は3億884億ドルです。私たちの損失は主に私たちの唯一の候補製品であるアンテファインの研究と開発費用と、私たちが業務インフラを建設する際に生じる一般的かつ行政コストによるものです。予測可能な未来には,我々の研究と開発努力の拡大に伴い,他の配合や他の適応における我々の臨床開発を進め,様々な配合や様々な適応の中で規制部門の承認を得て商業化することを求め,重大な運営損失を招き続ける可能性がある。私たちの費用は大幅に増加すると予想されています
•非嚢胞性線維気管支拡張症(NCFBE)、嚢胞性線維症(CF)、喘息または他の適応の治療に用いられる臨床試験を開始し、実施した
•COPDまたは他の適応の治療のための、固定用量の組み合わせを含む他の活性成分との組み合わせを含む、他の製剤において起動および他の臨床試験を行うこと;
•臨床薬理学的研究を開始し実施しました
•より多くの呼吸器候補製品の発見と開発または許可を求めています
•臨床前研究を行い、アンテフェンタリンおよび他の潜在的な未来候補製品を支持する
•臨床および商業的に供給されるアンテフェンタリン活性医薬成分およびそれから誘導される製剤医薬製品を開発すること
•規制部門のアンテファンドリンの承認を求めた
•販売、マーケティング、運営、精算、流通インフラを含むアンテフェンタニルの潜在的な商業化を支援するための商業インフラを発展させ、承認されれば、アンテフェンタニル林を商業化するための製造能力を拡大する
•私たちの知的財産権の組み合わせを維持し、拡大し、保護する
•私たちが許可した技術と製品の操作の自由を確保し、維持し、または獲得すること
•私たちの製品開発および潜在的な将来の商業化努力を支援する人員を含む、臨床、科学、運営、財務および管理情報システムおよび人員を増加させる;
•アメリカ、イギリス(“UK”)および他の場所での可能性のあるビジネスを拡大します。
もし私たちがいかなる遅延或いは上述のいかなる問題に遭遇した場合、失敗した臨床前研究或いは臨床試験、複雑な結果、安全問題或いは監督管理挑戦を含むが、これらに限定されないが、著者らの費用も大幅に増加する可能性がある。
著者らはほとんどの財政資源と努力をアメリカのCOPD維持治療のための噴霧式アンテフェンタニルの研究開発、臨床前研究と臨床試験及び商業化に投入した。他製剤や他の適応のためのアンテフェンタリンの開発を継続し,他地域で商業化している。
利益を達成して維持するためには、相当な収入を生み出すために、開発と最終的な商業化に成功しなければならない。これは、他の処方と他の適応におけるアンチフェンの臨床試験を完成させ、より多くの候補製品を発見し、開発し、監督部門のアンチフェンに対する承認を得、未来に以下の条件を満たす任意の候補製品を含む、一連の挑戦的な活動で成功することを要求する
臨床試験に成功し、製造、商業とマーケティング能力を確立し、最終的に監督管理の許可を得る可能性のある任意の製品を流通と販売する。私たちはただその中のいくつかの活動の初期段階にいるだけだ。私たちはこのような活動で決して成功しないかもしれないし、たとえ私たちが成功しても、私たちは利益を達成するのに十分な収入を生むことができないかもしれない。
医薬品開発に関連する多くのリスクや不確実性のため、費用を増加させる時間や金額、あるいはいつ、または利益を達成できるかどうかを正確に予測することはできない。FDA、欧州医薬品局(“EMA”)または他の規制機関が現在予想外の研究を行うことを要求した場合、または臨床試験の完了またはアンテフェンタニルまたは任意の他の候補製品の開発に遅延が生じた場合、私たちの費用は増加する可能性があり、収入はさらに遅れる可能性がある。
たとえ私たちが製品印税や製品販売を生み出したとしても、私たちは四半期や年度の利益を達成したり維持したりしないかもしれない。私たちが持続的に利益を得ることができなかったことは、私たちのアメリカ預託証明書の市場価格を低くし、資金を調達し、業務を拡大し、私たちの製品を多様化し、あるいは運営を継続する能力を弱める可能性がある。私たちのアメリカ預託証券市場価格の下落はまた私たちのアメリカ預託株式保有者が彼らの全部または一部の投資を損失させる可能性があります。
承認されれば、任意の将来の候補製品の開発および商業化、または他の製剤または目標適応の開発および商業化を達成するために追加の資金が必要となるだろう。もし私たちが必要な時に資金を集めることができない場合、あるいは現金と現金等価物を維持する任意の金融機関の失敗が未保険資金を獲得することを阻止または遅延した場合、私たちは私たちの製品開発計画や商業化努力の延期、減少、またはキャンセルを余儀なくされる可能性がある。
われわれが行っている計画や活動に関する費用が増加することが予想され,特に臨床試験やアンチフェンを商業化しようとしている場合や,他の製剤や他の適応にアンチフェンを開発·準備しようとしている場合である。また,アンチフェンや他の任意の候補製品に対する規制部門の承認を得た場合,製品位置決め研究,製品製造,医療事務,マーケティング,販売,流通などの活動に関連した巨額の商業化費用が発生すると予想される。また、米国で上場企業としての運営やナスダック世界市場(すなわちナスダック)での上場維持に関する持続的なコストが発生することが予想される。したがって、私たちは私たちの持続的な業務と関連した多くの追加資金を得る必要があるだろう。もし私たちが必要な時や魅力的な条件下で資金を集めることができなければ、私たちは私たちの研究開発計画や将来の商業化努力を延期、減少、または廃止することを余儀なくされるかもしれない。
もし私たちが監督機関のアメリカでのアンチフェンタリンのCOPD治療の許可を得たら、私たちは現有の現金資源と2023年の定期融資項目の下で獲得する予定の追加資金は少なくとも2026年末までの計画運営費用と資本支出需要に資金を提供することができ、アンテフェンタリンの商業発売を含むことができる。2023年の定期ローンでの将来の前払いは、特定の規制およびビジネスマイルストーン、および他の特定の条件の実現に依存します。私たちは不正確であることが証明される可能性があるという仮定に基づいてこの推定を行い、私たちは現在予想されているよりも早く私たちが利用できる資本資源を使用することができる。しかも、多くの未知の要素のため、私たちの運営計画は変化する可能性がある。他の要素を除いて、このような要素は私たちが現在計画されているより早く追加資金を求める必要があるかもしれない。また、有利な市場条件や戦略的考慮により、現在または将来の運営計画を実行するのに十分な資金があると考えても、追加の資本を求めることができる。私たちはほとんどの現金と現金等価物をアメリカの主要金融機関と国際金融機関の口座に預けており、これらの機関での預金は保険限度額を超えています。市場状況はこのような機関の生存能力に影響を及ぼすかもしれない。もし私たちが現金と現金同等物を維持しているどの金融機関が倒産すれば、私たちが未保険の資金をタイムリーにまたは根本的に得ることができる保証はない。このような資金を得ることができない場合や遅延されたどんな状況も、私たちの業務と財務状況に悪影響を及ぼす可能性がある。
私たちの将来の資本需要は多くの要素に依存します
•監督管理部門は監督管理部門が要求する可能性のある任意の発売後の研究を含むアンテフェンタニルのコスト、時間と結果を提出と審査し、もし監督部門の許可を得たら;
•もし監督部門の許可を得た場合、アンテフェンタリンによる慢性閉塞性肺疾患の商業定位を治療するのに必要な任意の他の研究のコスト、進捗、および結果を支持する
•NCFBE、CFBE、喘息または他の適応、または固定用量組み合わせ製品を含む他の製剤を治療するための任意の臨床試験のコスト、進行および結果;
•臨床的および商業的に供給されるアンテフェンタリン活性成分および誘導製剤医薬製品の製造コスト;
•その他の適応の臨床前開発、実験室試験と臨床試験の範囲、進展、結果と臨床試験、およびCOPDと潜在的なNCFBE、CF、喘息および他の呼吸器疾患を維持するためのアンチフェンを維持するためのDPIとPMDI製剤または固定用量のアンチフェン併用製剤の範囲、進展、結果とコスト;
•アンテフェンタリンの将来の潜在的な商業化活動のコスト、タイミング、結果、生産、マーケティング、販売、流通を含む
•特許出願を準備し、提出し、起訴し、私たちの知的財産権を維持し、実行すること、および知的財産権侵害に関する第三者の請求を含む任意の知的財産権に関するクレームを弁護するコストと時間
•アンテフェンタリンの商業販売から収入を得る時間と額
•アンテフェンタニルの販売価格と十分な第三者保険と精算があるかどうか
•競争の技術と市場発展の影響
•私たちは現在、このような取引の約束や合意を達成していないにもかかわらず、アンテフェンに関する許可または協力計画を達成することを含む、企業、製品、および技術の程度に買収または投資している。
いかなる追加的な拠出努力も、私たちの管理職の彼らの日常活動に対する注意を移すかもしれないが、これは私たちがアンテフェンを開発し、商業化する能力に悪影響を及ぼすかもしれない。しかも、私たちは未来の融資が十分な金額または私たちが受け入れられる条項で提供されることを保証できない。さらに、いかなる融資条項も、私たちの業務、私たちの株主の持株または権利、または私たちの普通株式またはアメリカ預託証明書の価値に悪影響を及ぼす可能性がある。
すぐに資金を得ることができなければ、プロポフォールに関連する研究開発計画や商業化努力の大幅な削減、延期、停止を要求される可能性があり、私たちの業務を拡大することができない、あるいは必要に応じて私たちのビジネスチャンスを利用することができなくなり、私たちの業務を損害し、運営を停止する可能性があります。
私たちはアンテファニンの成功に完全に依存していて、これは私たちが開発している唯一の候補品だ。アンテフェンタニルのどの適応も規制部門の承認を得ることは保証されておらず,商業化される前に必要である。もし私たちおよびそれと開発および商業化協定を締結した可能性のあるいかなる協力者もアンテフェンを商業化できない場合、あるいは商業化に大きな遅延がある場合、私たちの創造能力や財務状況は不利な影響を受けるだろう。
私たちは現在どんな製品の販売からも何の収入も得ていません。私たちは永遠に適切な製品を開発したり商業化することができないかもしれません。私たちはほとんどの精力と財力を投入してアンテフェンタニルを開発していますが、今のところ開発中の候補製品は何もありません。私たちが特許使用料と製品収入を生み出す能力は、アンテファンドリンの成功した商業化に大きく依存し、承認されれば、これは決して起こらないかもしれない。私たちが製品販売から任意の収入を得る前に、Ensifentineは規制部門の承認、製造供給の調達、商業化、大量の追加投資、重大なマーケティング努力が必要になるだろう。FDA、欧州委員会、または同様の外国規制機関の規制承認を得るまで、私たちは米国、ヨーロッパ、または他の国でアンチフェンまたは任意の候補製品をマーケティングまたは普及させることは許可されておらず、私たちはアンテフェンまたは任意の未来の製品候補に対する規制承認を決して得ないかもしれない。FDAは2023年8月、COPD維持治療のための承認を求めるNDAを受け入れ、処方薬使用料法案(PDUFA)の目標行動日を2024年6月26日に指定したが、承認されることは保証されないか、またはアンテフェンタニルの商業化に必要または望ましいラベル声明と共に承認されるであろう。また、私たちはまだ環境管理専門家に上場許可申請(“MAA”)を提出したり、他の規制機関に同様の申請を提出していない。アンテファンドリンの成功は、次のような要素を含む多くの要素に依存するだろう
•適切な規制機関を満足させるために、アンテフェンタニルの標的適応としての治療法が安全かつ有効であることを証明できないかもしれない
•適用される規制機関は、追加の臨床前または臨床試験を要求するかもしれません。これは私たちのコストを増加させ、私たちの開発期間を延長します
•アンテフェンの臨床試験結果は関連監督部門が上場を許可した統計或いは臨床意義レベルに符合しない可能性がある
•適用される規制機関は、私たちの臨床試験の数、設計、規模、進行、または実施に同意しないかもしれない
•私たちは臨床試験を行う契約研究機関(“CRO”)を招聘して、私たちのコントロールできない行動をとる可能性があり、私たちの臨床試験に重大な悪影響を与える
•適切な規制機関は、臨床前研究および臨床試験からのデータが、アンチフェンの臨床的および他の利益がその安全リスクを超えることを証明するのに十分であるか、またはデータの解釈に同意しない可能性があるかもしれない
•私たちは関連する規制機関が受け入れられる非臨床安全の概要を証明することができる
•意外な操作或いは臨床問題は臨床研究結果の完成或いは解釈を妨げる可能性がある
•予期しない製造問題、製品性能の問題、または安定性の問題は、遅延または他の方法で私たちの臨床開発計画の進捗に悪影響を及ぼす可能性がある
•米国食品薬品監督管理局または他の監督機関が、私たちの候補製品の製造施設や臨床場所に対してマーケティング応用に関する検査を行う必要があると判断した場合、このような監督管理機関は、戦争とテロ、例えばヨーロッパと中東の持続的な衝突を含む地政学的衝突によるか、旅行制限、例えば新冠肺炎疫病期間中に実施される制限を行うことができない
•良好な臨床実践(“GCP”)適合性問題、不当行為或いはその他の原因のため、適用される監督管理機関は著者らの臨床試験地点で生成したデータを受け入れない可能性がある
•私たちのNDAが諮問委員会によって検討された場合、FDAは諮問委員会会議を適時に手配することが困難である可能性があり、または諮問委員会は、承認の条件として、承認されたラベルまたは流通および使用制限を制限するために、追加の臨床前研究または臨床試験を要求することを提案することができるかもしれない
•適用される規制当局は、承認の条件として、リスク評価および緩和戦略(“REMS”)または同様のリスク管理措置の制定を要求することができる
•適用される規制機関は、私たちの第三者メーカーの製造プロセスや施設に欠陥があることを発見するかもしれない
•適用される規制部門は、その承認政策を変更したり、新しい規定を取ることができる
•もし私たちが異臭品を他の人に許可すれば、これらの当事者がイソフェント品の臨床試験を完成させ、監督管理の承認を得て、それを商業化するための努力を示している
•私たちの臨床試験では、アンテフェンタニルの商業的可能性を制限したり、アンテフェンタニルの商業化が不可能になったりすることが発見されるかもしれません
•もし私たちがアンテフェンタニルの協力協定に基づいて権利を保留すれば、私たちはアンテフェンタニルの臨床研究と臨床試験を完成させ、市場の承認を得て、アンテフェンタリンの商業製造能力と商業化の努力を確立する
•承認されれば、患者、医学界、および第三者支払人のアンテフェンタニルに対する受け入れは有効に他の治療法と競争し、承認を得て、私たちの知的財産権とクレームを獲得、維持、実行、保護した後、許容可能な安全性を維持し続ける。
これらの要素のいずれかの不利な結果は、私たちが重大な遅延を経験したり、アンテフェンタニルを商業化することに成功しなかったりする可能性がある
私たちはアンテフェンあるいは任意の未来の候補製品が臨床試験で成功するか、あるいは監督部門の承認を得るかどうかを決定することはできない。また、アンテフェンタニルあるいは任意の未来の候補製品は臨床試験で成功しても、監督部門の許可を得られない可能性がある。もし私たちが監督部門のアンテファンドリンや未来の候補製品の承認を得なければ、私たちは運営を続けることができないかもしれない。私たちが規制部門の承認を得て、アンフェタミンまたは任意の未来の候補製品を生産し、販売することに成功したとしても、私たちの収入は、私たちが規制部門の承認を得て商業権を持つ地域の市場規模にある程度依存するだろう。もし私たちが狙っている患者亜群市場が私たちが予想しているほど大きくなければ、承認されれば、このような製品の販売から大量の収入が発生しないかもしれません。
私たちは規制部門がアメリカでアンテファンドリンを商業化することを承認するための秘密保持協定を提出した。私たちは将来的に規制部門の承認を求め、アンテファンドリンをEUと他の国で商業化するかもしれない。多くの国の規制承認範囲は似ているが、複数の国で単独の監督管理許可を得るためには、これらの国の安全性と有効性及び臨床試験と商業販売の管理、定価と流通などの面での多くと異なる監督管理要求を遵守する必要があり、これらの管轄区域で成功するかどうかを予測することはできない。
私たちの限られた経営の歴史は、投資家が私たちの業務のこれまでの成功度を評価することを困難にし、私たちの未来の生存能力を評価することも困難になるかもしれない。
2005年の設立以来、著者らはほとんどの資源をプロポフォールの開発、私たちの知的財産権の組み合わせの確立、私たちのサプライチェーンの発展、私たちの業務の計画、資金の調達及びこれらの業務に一般と行政支持を提供することに投入した。著者らはすでに異なる剤形と異なる適応のアンテフェンタニルの複数の1期と2期の臨床試験、およびCOPDを維持治療するための2つの登録のための3期臨床試験を完成した。規制部門の承認を得たり、商業規模の製品を製造したり、第三者代表がそうしたり、成功した製品の商業化に必要な販売やマーケティング活動を行うことには成功していません。また、我々は利益を上げておらず、設立以来毎年赤字が発生しており、様々な要因により、財務状況や経営業績は四半期ごとや毎年大幅に変動し続け、その多くの要因はコントロールできないと予想される。したがって、投資家が私たちの未来の成功や生存能力に対するいかなる予測も、私たちがより長い運営歴史を持っている時のように正確ではないかもしれない。
私たちの信用手配の条項は私たちの経営と財政的柔軟性を制限し、私たちの既存と未来の債務は私たちの業務運営能力に悪影響を及ぼすかもしれない
2023年12月、Verona Pharma,Inc.はオックスフォード金融有限責任会社(“オックスフォード”)と担保代理およびオックスフォードとHercules Capital,Inc.(総称して“融資者”と呼ぶ)によって管理されるいくつかの基金として定期融資手配(“融資協定”)を締結し、この合意により、総額4.0億ドルに達する定期融資手配を5回に分けて提供することができ、2023年の定期融資と呼ぶことができる。ローン契約が終了した時、吾らは最初の5,000,000ドル(“A期ローン”)を受け取った。2023年の定期融資項目の立て替え金ごとに変動年利(“基本金利”)で累算して利息を計算し、この金利は、(A)(I)1ヶ月シカゴ商品取引所(CME)期限SOFR(ローン契約を定義する)基準金利の大きい者は、利息が発生する月の直前の月の最後の営業日であり、(Ii)5.34%プラス(B)5.85%である。しかし、条件は、(I)いずれの場合も、定期Aローンの基本金利は、(X)11.19%を下回ってはならないこと、(Y)他の各立て替え金の基本金利は、この立て替え融資日の直前の営業日の基本金利を下回ってはならないこと、(Ii)A期融資の自己清算から2023年12月31日までの間の基本金利は11.19%とし、(Iii)各立て替え金の基本金利は、このような立て替え融資日ごとに適用される基本金利に基づいて2.00%増加してはならないことである。
私たちの未返済債務は、2023年の定期ローンでの借金以外に発生した任意の追加債務を含み、私たちの他の財務義務と契約約束に加えて、重大な不利な結果が生じる可能性があります
•利息および元金の支払いに現金資源の一部を使用し、運営資本、資本支出、候補製品開発、および他の一般企業用途に利用可能な資金を削減することが求められている
•全体的な経済、産業、市場の不利な変化の影響を受けやすいようにしています
•私たちが特定の企業の行動を取ったり、さらなる債務や株式融資を得る能力を低下させるかもしれない制限的な条約に制限されている
•ビジネスと競争する業界の変化を計画したり対応したりする際の柔軟性を制限します
•私たちは債務がより少ないか債務超過がより良い競争相手を選択するのと比較して競争劣勢にある。
私たちは私たちの現在と未来の債務超過義務を私たちの当時の現金と現金同等物で返済するつもりだ。しかし、私たちは十分な資金を持っていないかもしれないし、追加の融資を手配することができないかもしれないし、融資協定や任意の他の債務ツールの下で満期になった金額を支払うことができないかもしれない。具体的な行動をとったり回避したりする契約を含め、融資合意に基づいて現在と将来の債務義務を履行できないことは、違約事件を招く可能性があるため、貸主はすべての満期金額を加速させる可能性がある。ローン契約の下の満期金額が違約事件によって加速された場合、十分な資金や可能性がないかもしれません
私たちの債務を返済するための追加融資を手配することができず、現在の業務戦略を実行しています。さらに、私たちの貸手は、そのような債務を担保する任意の担保上でその担保権益を強制的に実行することを求めることができる
また、私たちが清算された場合、貸手が償還を得る権利は、私たちの米国預託株式(“米国預託株式”)保有者または私たちの株主が清算から任意の収益を得る権利よりも優先される。融資者の違約事件に対するいかなる声明も、私たちの業務と将来性を深刻に損なう可能性があり、私たちのアメリカ預託証明書価格の下落を招く可能性がある。また、融資協定下の契約、私たちの資産を担保とし、私たちの知的財産権に対する負の質権は、私たちが追加債務融資を受ける能力を制限する可能性がある。もし私たちが追加的な債務融資を調達すれば、このような追加債務の条項は私たちの運営と財政的柔軟性をさらに制限するかもしれない。
追加資本を調達することは、私たちの所有者に希釈をもたらし、私たちの運営を制限するか、または私たちの技術または候補製品に対する権利を放棄することを要求するかもしれない。
これまで、私たちが相当な製品収入を生み出すことができれば、私たちは証券発行、債務融資、許可、協力協定、研究支出の組み合わせによって私たちの現金需要を満たす予定です。もし私たちが証券発行を通じて資金を調達すれば、私たちのアメリカ預託株式保有者と株主の所有権権益は希釈され、これらの証券の条項には清算や他の特典が含まれる可能性があり、これらの保有者が私たちのアメリカ預託証券所有者としての権利に悪影響を及ぼす可能性がある。債務融資は、実行可能であれば、固定支払義務をもたらす可能性があり、私たちは、例えば、追加債務の発生、取得、販売、おそらく知的財産権の発行、資本支出、配当金、または他の経営制限を発表する能力の制限のようないくつかの限定的な条約に同意することを要求される可能性がある。もし私たちが協力または許可協定を通じてより多くの資金を調達すれば、私たちは私たちの技術、将来の収入源、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になる可能性のある条項で許可を付与しなければならないかもしれない。さらに、私たちはまた、より早い段階で協力者や他の人との手配を通じて資金を求めることを要求されるかもしれない。そうでなければ望ましい。もし私たちが研究支出を通じて資金を調達すれば、私たちはいくつかの要求によって制限されるかもしれません。これは私たちが資金を使用する能力を制限したり、研究開発情報を共有することを要求するかもしれません。上記のいずれかまたは他の方法で追加資本を調達することは、我々の業務および米国預託株式保有者および株主の持株または権利に悪影響を及ぼす可能性があり、我々の米国預託証券の市場価格を低下させる可能性がある。
私たちの業務は国際業務に関連する経済、政治、規制、その他のリスクの影響を受ける可能性がある。
イギリスに本社を置き、ナスダックに上場している会社として、私たちの業務は国際業務の展開に関するリスクに直面しています。私たちの多くのサプライヤーと協力と臨床試験関係はイギリスとアメリカ以外に位置している。したがって、私たちの将来の業績は様々な要素の影響を受けるかもしれない
•インフレ、特に非アメリカ経済と市場の政治的不安定を含む経済的疲弊
•米国以外の国は薬品審査に対する異なる規制要求を持っている
•異なる法ドメインは、これらのドメインで動作する自由を保障、維持、または獲得する上で異なる問題をもたらす可能性がある
•知的財産権の保護を減らすことができます
•非アメリカの法律法規を守ることに困難があります
•アメリカ以外の規制と税関、関税、貿易障壁の変化
•ユーロの非アメリカ通貨為替レートの変化と通貨規制
•特定の国や地域の政治経済環境の変化
•米国または非米国政府の貿易保護措置、輸出入許可要求、またはその他の制限行動
•ある非アメリカ市場では異なる精算制度と価格規制が行われています
•税法変更による否定的な結果
•外国に住んだり旅行したりする従業員は税収、雇用、移民、労働法を遵守する
•労働騒乱がアメリカよりも一般的な国では労働力の不確実性
•異なる労使関係を含む人員配置と国際業務の管理に関する困難
•海外の原材料の供給や製造能力に影響を与える事件による生産不足;
•地政学的行動による業務中断は、ヨーロッパと中東の持続的な衝突、あるいは地震、台風、洪水、火災などの自然災害、あるいは新冠肺炎のような公共衛生事件の突発的な公共衛生事件を含む。
為替レートの変動は私たちの経営結果と財務状況に大きな影響を及ぼすかもしれない。
私たちの本社はイギリスにありますが、私たちの財務諸表はドル建てで、私たちの多くの業務活動はアメリカとイギリス以外のパートナーと行われています。これらの取引は別の通貨で価格を計算するかもしれません。したがって、私たちの業務と私たちのアメリカ預託証明書の価格はポンドとドルの為替レート変動の影響を受けるだけでなく、他の国の通貨為替レートの変動の影響を受ける可能性があり、これは私たちの運営業績やキャッシュフローに大きな影響を与える可能性があります。現在、私たちは為替レートのヘッジスケジュールを持っていません。
開発,臨床試験,規制承認に関するリスク
臨床薬物開発と規制審査は長くて高価な過程に関連し、結果は不確定である。私たちは、最終的に私たちの候補製品の開発と規制承認を完了できない場合、追加のコストが発生したり、遅延が発生する可能性があります。
臨床薬物開発は長く高価な過程であり、不確定なスケジュールと不確定な結果を持っている。もしアンテフェンタニルの臨床試験が延長或いは延期された場合、あるいは後期臨床試験が監督機関に要求された安全性と有効性を示さなかった場合、私たち或いは著者らの協力者は必要な監督管理の許可を得ることができず、適時にアンテフェンタニルを商業化することができず、甚だしきに至っては商業化を実現できないかもしれない。
アンテフェンタリンの発売と販売に必要な監督管理許可を得るために、著者ら或いはアンチフェントーリンの任意の協力者は広範な臨床前研究と臨床試験を通じてアンチフェンタリンが人体内で安全かつ有効であることを証明しなければならない。臨床試験費用は高価であり,完成まで数年かかる可能性があり,その結果自体も確定していない。臨床試験では,いつでも失敗する可能性がある。臨床前研究と早期臨床試験の結果は後期臨床試験の結果を予測できないかもしれない。臨床試験後期段階の候補製品は期待された安全性と有効性特徴を示すことができない可能性があり、すでに臨床前研究と初歩的な臨床試験を通じて進展を得たにもかかわらず。監督管理機関の結果に対する解釈は著者らと異なる可能性があり、製品が臨床開発段階にある時、時間の経過とともに変化する可能性が予想される
生物製薬業界のいくつかの会社は高級臨床試験で重大な挫折を受け、早期の試験で良好な結果を得たが、治療効果或いは副作用が乏しいためである。FDAは私たちに追加の臨床前研究或いは臨床試験を要求するかもしれないし、これらの研究或いは臨床試験は成功しないかもしれないし、監督機関に成功とみなされないかもしれない。我々の唯一の候補であるアンチフェンタニルについては,異なる剤形および異なる適応に対するアンチフェンタニルに対する複数の1期および2期臨床試験,および慢性閉塞性肺疾患の2つの登録を維持するための3期臨床試験を完了した。これらの研究結果から,COPDの維持治療へのアンテフェンタニルの承認を求め,FDAは2023年8月にわれわれのNDA審査を受け,PDUFAの目標行動日を2024年6月26日に指定したNDAを提出した。FDAの届出通信と2023年11月の中期通信はそれぞれ2つの審査問題について初歩的な通知を出し、これらの問題は申請に含まれるいくつかの二次データ(例えば谷底)と探索性データ(例えば病状の悪化)がそれぞれ有利な利益-リスク概況或いは治療効果発見をサポートするためにどの程度使用できるかに関連する。FDAはこの2つの手紙で、これらの論評は初歩的であり、審査の情報の最終決定を反映していないと指摘している
もし私たちが他の地域で霧化アンチフェンをCOPD維持治療の商業化に応用することを望むなら、これらの地区の監督機関は追加の臨床前研究或いは臨床試験を要求する可能性があり、もし私たちが他の製剤或いは他の適応のアンチフェンニンを商業化したいなら、私たちは更なる臨床研究を要求される。
われわれは臨床試験の遅延に遭遇する可能性があり,計画中の臨床試験が時間どおりに開始されるかどうか,再設計が必要かどうか,時間どおりに患者を募集するかどうか,あるいは予定通りに完成するかどうかは分からない。我々の臨床試験は延期される可能性があり、一時停止または終了される可能性があり、またはこれらの試験のデータの有効性が影響を受ける可能性があり、原因は多くある
•臨床試験の開始または継続を支援するために、十分な臨床前、毒理学または他の体内または体外データを生成することができない
•投与量および頻度を含む、臨床試験の設計または実施の遅延または実施についての規制プロトコルを得ることができなかった
•裁判を開始することを遅延させたり、監督部門の許可を得られなかった
•遅延または予期されるCROおよび臨床試験地点と受け入れ可能な条項と合意できなかったか、その条項は広範な交渉を必要とする可能性があり、異なるCROと試験地点の間に有意差が存在する可能性がある
•CROは、科目募集、データ収集、データ監視、実験室サンプル管理、プログラミングおよび分析、または他の活動における契約義務を履行することができない
•機関審査委員会(“IRB”)、または道徳委員会の承認または各場所の正面的な意見を得ることができなかったか、または遅延されたか、または得られなかった
•適切な患者を遅延させたり、試験に参加できなかったりする
•患者に試験を完成させなかったか、あるいは治療後のフォローアップを行った
•臨床サイトが試験方案から外れたり、試験から退出したり、深刻な不正行為または不正行為がある場合;
•新たな臨床試験場所の増加を延期し
•アンテファンドリンの二重盲検を達成したり維持したりすることはできません
•アンテフェンタニルと相応の薬品を生産する過程で発生した意外な技術問題
•医薬品の性能および/または安定性の可変性;
•アンテファンドリンの商業可能性の発見を低減する可能性がある
•臨床試験に十分な量のアンテフェンタリンを生産することはできません
•アンテフェンタリンの品質または安定性は、許容可能な安全性または有効性基準よりも低い
•臨床試験ではアンテファンが権利侵害を主張し、禁止された第三者行為が私たちの進展を妨害した
•欧州や中東で続く紛争や、地震、台風、洪水、火災などの自然災害による業務中断など、地政学的行動には、戦争やテロが含まれている
•米国や他の国政府が実施している貿易制裁は、ある国(例えばロシア)に臨床試験費用を支払う能力に影響を与えている
•もし私たちまたは私たちの協力者が参加者が受け入れられない健康リスクに直面していることを発見すれば、安全または耐性の問題は、私たちまたは私たちの協力者が(場合によっては)試験を一時停止または終了させることをもたらす
•規制要件、政策、指針の変化
•患者とボランティアの臨床試験における保持率は予想より低かった
•私たちの第三者研究請負業者は、法規の要求を遵守できなかったか、または私たちに対する契約義務をタイムリーに履行できなかったか、または全く遵守しなかった
•特定の実験で評価しようとしているサブグループを特定することは困難であり、登録を延期する可能性がある国もある。
臨床試験が我々、そのような試験を行っている機関のIRBs、そのような試験のためのデータ審査委員会またはデータ安全監視委員会またはFDAまたは他の規制機関によって一時停止または終了された場合、遅延に遭遇する可能性がある。このような主管部門は多種の要素のために臨床試験を一時停止或いは中止する可能性があり、これらの要素は監督管理要求或いは著者らの臨床規程に従って臨床試験を行っていない、FDA或いは他の監督機関の臨床試験操作或いは試験場所の検査による臨床一時停止、予見できない安全問題或いは副作用、ある種の薬物を使用するメリットを証明できなかった、著者らの臨床試験は十分な有効性と安全性を証明できなかった、政府法規或いは行政措置の変化或いは十分な資金が不足して臨床試験を継続することを含む。
また,われわれの臨床試験の首席研究員は時々私たちの科学コンサルタントやコンサルタントを務め,このようなサービスに関する報酬を得る可能性がある。場合によっては、私たちはFDAまたは他の規制機関にいくつかの関係を報告することを要求されるかもしれない。FDAまたは他の規制機関は結論を出すかもしれないが、私たちと主要な研究者との財務関係は利益の衝突をもたらしたり、他の方法でこの研究の解釈に影響を与えたりする。したがって,FDAや他の規制機関は,適用された臨床試験地点で発生するデータの完全性を疑問視する可能性があり,臨床試験自体の効用が脅かされる可能性がある。これは、FDAまたは他の規制機関が私たちの上場申請の承認を遅延または拒否することを招き、最終的にアンテフェンタニルの上場承認が拒否される可能性がある。
任意の適応の任意の臨床試験の完了を遅延させる場合、または任意の他の候補製品の臨床試験を完了するか、または任意の臨床試験または任意の他の候補製品を終了する場合、これらの候補製品の商業的将来性が損なわれる可能性があり、私たちが製品収入を創出する能力は延期されるであろう。また、臨床試験の完成のいかなる遅延も私たちのコストを増加させ、開発と承認過程を緩和し、私たちの製品販売と収入を創造する能力(あれば)を危険にさらす。重大な臨床試験遅延は、私たちの競争相手が私たちよりも先に製品を市場に出したり、候補製品を商業化する独占的な権利を持っている時間を短縮し、候補製品を商業化する能力を弱める可能性もあります。さらに、臨床試験の開始または完了遅延をもたらす多くの要因は、最終的には、監督部門がアンテフェンタニルまたは任意の他の候補製品の承認を拒否する可能性もある。
臨床試験はFDAの法律法規,EU規則や条例,その他の適用される規制機関の法律要求,法規やガイドラインに基づいて行われ,これらの政府機関や臨床試験を行う医療機関のIRBs(あるいは他の倫理委員会)の監督を受けなければならない。また,臨床試験は現在の良好な生産規範(“cGMP”)や類似した国外要求や他の法規に基づいて生産されたアンテフェンタニルを使用しなければならない。また,われわれはCROと臨床試験地点に依存してわれわれの臨床試験の適切かつタイムリーな進行を確保し,彼らが約束した活動に合意しているが,彼らの実際の表現への影響は限られている。われわれはわれわれの協力者および医療機関とCROに依存してGCP要求に応じた臨床試験を行っている。我々の協力者やCROがわれわれの臨床試験のために参加者を募集できなかった場合,GCP基準に従って研究できなかった場合,あるいは完全登録の実現を含めて試験実行中に長い時間遅延した場合,コスト増加,計画遅延,あるいは両方の影響を受ける可能性がある。また、EUとアメリカ以外の国で行われた臨床試験は、輸送コストの増加、追加の規制要求、および非EUと非アメリカCROの参加により、私たちをさらなる遅延と費用に直面させ、FDAやEMAの知らない臨床研究者に関連するリスク、および異なる診断、スクリーニング、医療基準に直面させる可能性がある。
また,FDAや他の規制機関の臨床試験に関する政策が変わる可能性があり,追加の政府法規が公布される可能性がある。例えば,EUの臨床試験に関する規制構造が最近変化している。EU臨床試験条例(CTR)は2014年4月に採択され、EU臨床試験指令が廃止され、2022年1月31日に施行された。EU臨床試験指令は,臨床試験を行う各加盟国で主管する国家衛生当局と独立した倫理委員会に単独の臨床試験申請(“CTA”)を提出することを要求しているが,CTRは集中的な手続きを導入し,多センター試験の申請のみを要求している。CTRは、スポンサーが各会員国の主管当局と道徳委員会に文書を提出することを可能にし、各会員国が決定を下すことを可能にする。CTAの評価手続きも統一されており、すべての関連加盟国による共同評価を含み、道徳基準を含む各加盟国が個別にその領土に関する具体的な要求を評価する。各会員国の決定は集中されたEUポータルサイトを通じてスポンサーに伝達される。CTAが承認されると,臨床研究開発は継続可能である。CTRは3年間の過渡期が予想される。進行中の臨床試験と新たな臨床試験がCTRによってどの程度制御されるかはそれぞれ異なる。2022年1月31日までにEU臨床試験指令に基づいて申請を提出した臨床試験、または(Ii)が2022年1月31日から2023年1月31日までの間、かつスポンサーがEU臨床試験指令に適用される臨床試験を選択し、2025年1月31日までこの指令の管轄を受ける。この日以降,すべての臨床試験(行われている臨床試験を含む)はCTR条項に拘束される。我々と我々の第三者サービスプロバイダ(例えばCRO)がCTR要求を遵守することは、我々の開発計画に影響を与える可能性がある
イギリスがどの程度その規制をEUと統合することを求めているのかは不明だ。臨床試験に関連するイギリスの規制枠組みは、既存のEU立法(二次立法によってイギリス法で実施される)に由来する。
2022年1月17日、イギリス薬品と保健品監督局(MHRA)はイギリスの臨床試験立法の再制定について8週間のコンサルティングを開始し、臨床試験の審査を簡略化し、革新を促進し、臨床試験の透明性を高め、リスクの割合を高め、そして患者と公衆の臨床試験への参与を促進することを目的とした。それによって生じる立法変化は密接に注目され、イギリスの臨床試験の枠組みと(EU)CTRとの一致或いは乖離の程度を決定する。アイルランド/北アイルランドに関する議定書の条項によると,(EU)CTRにおける研究用医薬品と補助薬品の製造·輸入に関する規定は北アイルランドに適用される。英国政府はその法規をEUが採用した新しい方法と密接に一致させないことを決定しており,他の国に比べてイギリスでの臨床試験のコストに影響する可能性がある。
もし私たちが既存の要求の変化にゆっくりあるいは適応できない場合、あるいは新しい要求を採用したり、臨床試験を管理する政策を採用すれば、私たちの発展計画は影響を受ける可能性がある。
エンシフェンタンは深刻な不良、不良或いは受け入れられない副作用がある可能性があり、上場承認を延期或いは阻止する可能性がある。もしアンチフェンを開発する過程で、または承認後にこのような副作用が発見された場合、私たちはアンチフェンの開発を放棄する必要があるかもしれず、任意の承認されたラベルの商業イメージが制限される可能性があり、あるいは上場承認後に他の重大な負の結果に直面する可能性がある。
アンテフェン洗浄によって引き起こされる可能性のある不良副作用は、私たちまたは規制機関の臨床試験の中断、延期、または停止を招く可能性があり、より厳格なラベルまたはFDAまたは他の同様の外国機関の規制承認遅延または拒否を招く可能性がある。臨床試験を行っている間、患者は彼らの研究医に疾病、傷害、不快感を含む彼らの健康変化を報告した。一般に、研究されている候補製品がこれらの状況をもたらしているかどうかを決定することは不可能だ。私たちがより大きく、より長く、より広い臨床試験で私たちの候補製品を試験するとき、またはこれらの候補製品の使用がより広くなるにつれて(規制部門の許可を得た場合)、患者は、以前の試験で観察された疾患、傷害、不快感、および他の有害事象、および以前の試験で発生しなかったか、または検出されなかったことを報告する。多くの場合,研究製品が大規模臨床試験で試験を行った後,あるいは承認後に患者にビジネス規模の製品を提供した後にのみ,副作用を検出することができる場合がある。われわれはすでに20項目以上のアンテフェンタニルの1期、2期、3期の臨床試験を完成した。これらの試験において、一部の患者は尿路感染、背部痛と高血圧を含む軽微から中度の副作用が出現した。
われわれの将来の臨床試験結果は不良副作用の重症度と普遍性を示す可能性があり,受け入れられない。この場合、私たちの実験は一時停止または終了される可能性があり、FDAまたは他の同様の外国規制機関は、任意またはすべての標的適応のアンテフェンのさらなる開発を停止または拒否するように命令することができる。薬物に関連する副作用は、患者の募集または患者の試験完了能力に影響を与える可能性があり、あるいは潜在的な製品責任クレームを招く可能性がある。また、もしアンテフェンタニンが発売許可を得た場合、私たちまたは他の人は後にアンテフェンタニルによる不良または受け入れられない副作用を発見し、いくつかの潜在的な重大な負の結果を引き起こす可能性がある
•規制当局はこのような製品の承認を撤回し、アンチフェンを市場から撤去することを要求するかもしれない
•規制当局は、医師および薬局にラベル宣言、具体的な警告、禁忌症、または現場警報を追加することを要求するかもしれない
•規制当局は、患者に配布するために、このような副作用のリスクを概説するための薬物ガイドラインを必要とするかもしれないが、アンテフェンタニルの利点がそのリスクよりも大きいことを確実にするために、REMS計画または同様のリスク管理措置を実施する
•私たちは投与方法を変更し、追加の臨床試験を行うか、またはアンテフェンタニルのラベルを変更することを要求されるかもしれない
•私たちがどのようにアンテフェンタニルを普及させるかは制限されるかもしれません
•アンテフェンタリンの販売量は大幅に減少する可能性がある
•私たちは訴訟や製品責任クレームの影響を受ける可能性があります
•私たちの名声は損なわれるかもしれない。
これらの事件のいずれも、アンテフェンに対する市場の受容度を達成または維持することを阻止または維持することができ、または商業化コストおよび費用を大幅に増加させる可能性があり、これは、逆に、アンテフィンの販売から相当な収入を得ることを延期または阻止する可能性がある。
私たちは、固定用量の組み合わせを含み、および/またはNCFBE、CF、喘息または他の呼吸器疾患を含む様々な適応のために、異なる処方においてアンテフェンタニルを開発することに成功できないかもしれない。
われわれの戦略の一部は,NCFBE,CFと喘息,固定用量組み合わせ,MDI,DPIを含む他の製剤の開発を継続することである。我々のこれまでの研究と開発は,アンテフェンタニルがNCFBE,CF,喘息を治療する潜在力を有することを示しているにもかかわらず,これらの適応や任意の他の疾患でアンテフェンタニルを開発できないか,あるいは開発が成功しない可能性がある。さらに、他の疾患におけるアンテフェンタニルの潜在的用途は、我々が開始する予定の任意の臨床研究のために患者を募集することが困難であること、または有害な副作用が出現する可能性があること、または市場承認および市場受け入れを示す可能性のある他の特徴を含む臨床開発に適していない可能性がある。多様な適応や製剤のアンテフェンタニルの商業化に成功し、開始しなければ、今後の時期に製品収入を得ることが困難に直面し、財務状況を深刻に損なう可能性がある。
私たちの臨床試験では、私たちは患者の登録に依存している。臨床試験で患者を募集できない場合や,募集速度が予想より遅い場合には,我々の研究や開発に悪影響を受ける可能性がある。
アンテフェンタリンの臨床試験を成功と適時に完成するには、十分な数の候補患者を募集する必要がある。患者登録時間が予想より長いことや,患者引き揚げや他の外部要因により,試験が遅延の影響を受ける可能性がある。患者の入選は多くの要素に依存し、患者群の規模と性質、調査中の疾病の重症度、試験の資格基準、患者と臨床場所の接近度、臨床方案の設計、患者の同意を獲得と維持する能力、入選患者が試験から退出するリスク、競争的臨床試験の可用性、臨床試験によって調査されている適応によって承認された新薬の可用性、および臨床医師と患者が研究している薬物の他の利用可能な治療法に対する潜在的な優位性に対する見方を含む。これらの要素は私たちが十分な患者を募集して、適時かつ費用効果的に著者らの臨床試験を完成させることを困難にするかもしれない。予想以上の患者数も臨床試験への参加を停止する可能性がある。任意のアンテフェンタニルまたは他の候補製品の臨床試験の完成を延期することは私たちのコストを増加させ、私たちのアンチフェンの開発と承認を遅くし、そして私たちの製品販売の開始と収入を創出する能力を遅延または危険にさらす可能性がある。また、臨床試験の開始或いは完成遅延を招くいくつかの要素は、最終的に監督部門がアンテフェンタニルの承認を拒否する可能性もある。
私たちは高価で破壊的な責任クレームに直面するかもしれません。臨床テストでも商業段階でも、私たちの製品責任保険はこのようなクレームのすべての損害を含まないかもしれません。
私たちは潜在的な製品責任と専門賠償リスクに直面しており、これらのリスクは医薬製品の研究、開発、製造、マーケティングと使用に固有のものである。現在、私たちはまだ商業販売のための製品を承認されていません;しかし、私たちとどの協力者も臨床試験で現在および未来に使用されているアンチフェン洗浄、および未来に販売されるアンチフェン洗浄は、承認されれば、私たちを責任クレームに直面させるかもしれません。これらのクレームは、この製品を使用する患者、医療提供者、製薬会社、私たちの協力者、またはアンテフェンを販売する他の人によって提起される可能性がある。私たちに対するいかなるクレームも、その是非にかかわらず、弁護することが困難である可能性があり、コストも高く、アンテフェンタニル林の市場やアンテフェンタニル林の任意の商業化の将来性に悪影響を及ぼす可能性がある。また、事件がどうであっても、最終的な結果がどうであろうと、賠償責任は次のようになるかもしれない
•アンテファンドリンの需要が減少した
•私たちの名声を損なう
•臨床試験参加者の脱退
•関連訴訟の弁護費用
•経営陣の時間と資源を移転する
•実験参加者や患者に多額の報酬を与え
•調査、製品のリコールまたは撤回、またはラベル、マーケティング、または販売促進制限を規制する
•収入の損失
•アンテファンドリンを商業化したり普及させたりすることはできない。
臨床試験過程は潜在的な副作用を識別と評価することを目的としているが、監督部門の許可後であっても、薬物は常に予測できない副作用を示す可能性がある。もしアンテフェンタニンが臨床試験期間中あるいは承認後に副作用を引き起こすならば、私たちは重大な責任を負うかもしれない。医師および患者は、既知の潜在的副作用を決定するいかなる警告も遵守しない可能性があり、アンテフェンタニルを使用すべきでない患者。
私たちは引き続きアンテファンのために製品責任保険に加入していますが、私たちの責任は保険範囲を超えている可能性があります。もし私たちがアンテフェンタリンの発売許可を得たら、私たちは私たちの保険範囲を拡大して、商業製品の販売を含めるつもりです。しかし、私たちは合理的なコストで保険範囲を維持できないかもしれないし、起こりうるいかなる責任にも対応するのに十分な保険範囲を得ることができないかもしれない。もし成功した製品責任クレームまたは一連のクレームが未保険の負債または保険を超えた負債によって私たちに提出された場合、私たちの資産はこのようなクレームを支払うのに十分ではない可能性があり、私たちの業務運営は損害を受ける可能性がある。
FDA、EMAと類似の外国監督管理機関の監督管理審査過程は長く、時間がかかり、しかも本質的に予測不可能であり、もし私たちが最終的にアンテフェンタニルの監督管理許可を得ることができなければ、私たちの業務は実質的に損害を受けるだろう。
FDA、欧州委員会、および類似の外国規制機関の承認を得るのに要する時間は予測できないが、通常は臨床試験開始後数年後に必要であり、監督機関のかなり大きな裁量権を含む多くの要素に依存する。また、承認政策、法規、あるいは承認を得るために必要な臨床データのタイプと数量は候補製品の臨床開発過程で変化する可能性があり、司法管轄区域によって異なる可能性がある。私たちはまだ規制部門のアンチフェンの承認を得ていないし、私たちが将来開発する可能性のあるどの候補製品も規制部門の承認を得られないかもしれない。
承認された候補製品を米国または海外で商業化する前に、私たちまたは私たちの協力者は、厳格に制御された臨床試験によって大量の証拠を提供し、FDAまたは外国の規制機関に、その予想される用途に対して安全かつ有効であることを満足させなければならない。非臨床研究と臨床試験の結果は異なる方法で解釈できる。私たちの候補製品の非臨床的または臨床的データが有望だと信じていても、これらのデータはFDAおよび他の規制機関の承認を支持するのに十分ではないかもしれない。FDAや外国の規制機関はまた、承認前または承認後に追加の臨床前研究または臨床試験を行うことを要求するか、または私たちの臨床開発計画の内容に反対する可能性がある。
Ensifentineは、以下のような理由で、多くの理由で規制許可を得ることができないかもしれない
•FDA、EMAあるいは類似の外国監督機関に証明できないかもしれないが、その提案された適応に対して、キフェントーリンは安全かつ有効であり、必要な統計学的意義がある
•アンテファニンの利益が安全リスクよりも大きいことは証明できないかもしれない
•FDA、EMA、または同様の外国の規制機関は、臨床前研究または臨床試験データの解釈に同意しないかもしれない、またはデータが受け入れられないことを発見する可能性がある
•FDA、EMA、または同様の外国の規制機関は、第3段階の臨床試験で評価された1つまたは複数の用量または二重盲検を実施する方法は受け入れられないことを発見するかもしれない
•様々な理由で、アンテファンドリン臨床試験から収集されたデータは、米国でのNDAの提出または承認、EUでのMAAの提出、または他の国で規制された承認を得る他の同様の提出をサポートするのに十分ではない可能性がある
•FDAまたは同様の外国の規制機関は、私たちと臨床および商業用品契約を締結する第三者メーカーの製造プロセスまたは施設を承認できない可能性がある
•FDAまたは類似の監督機関は、著者らの臨床研究に参加する臨床サイトまたはCROのGCPが適合または受け入れられないことを発見し、臨床データが承認を支持するのに不十分である可能性がある
•FDAなどの外国の監督管理機関の承認政策や法規は大きく変化する可能性があり、私たちの臨床データは承認を得るのに十分ではない
•FDA、EMA、または同様の外国の規制機関は、私たちの臨床試験の設計または実施に同意しないかもしれない
•FDA、EMA、または同様の外国規制機関は、私たちが提案した製品仕様および性能特徴に同意しない可能性があります。
この長い承認過程と未来の臨床試験結果の予測不可能性は、市場でのアンテフェンタニルの監督管理許可を得ることができない可能性がある。FDA、EMA、および他の規制機関は承認過程でかなりの自由裁量権を持ち、いつ、またはアンテフェンタニルの監督管理許可を得るかどうかを決定する。アンテフェン臨床試験から収集されたデータが有望であると信じていても、これらのデータはFDA、欧州委員会、または任意の他の規制機関の承認を支持するのに十分ではないかもしれない。例えば,FDAは2023年8月にCOPD維持治療のためのNDAの承認を求める審査を受け,PDUFAの目標行動日を2024年6月26日に指定した。NDA届出通信と2023年11月の周期中間審査はそれぞれ2つの審査問題について初歩的な通知を出し、これらの審査問題は申請に含まれるいくつかの二次データ(例えば谷底)と探索性データ(例えば病状の悪化)がそれぞれ有利な利益-リスク概況或いは治療効果の発見を支持するためにどの程度使用できるかに関連する。FDAはこの2つの手紙で、これらの論評は初歩的であり、審査の情報の最終決定を反映していないと指摘している。
また、たとえ私たちが任意の管轄区域の承認を得ても、規制機関は私たちが要求しているよりも少ないまたは限られた適応を承認するかもしれないし、私たちが徴収しようとしているアンテフェンの価格を承認しないかもしれないし、高価な発売後の臨床試験の表現によって承認されるかもしれないし、承認されたラベルにはアンテフェンの商業化に必要または所望のラベル声明が含まれていないかもしれない。上記のいずれの場合も、アンテフェンタリンのビジネス見通しに実質的な損害を与える可能性がある。
さらに、FDAと外国規制機関は彼らの承認政策を変更し、新しい規定を制定する可能性がある。例えば、欧州委員会が2020年11月に開始した欧州薬品戦略イニシアティブを背景に、EU薬品立法は現在全面的な審査が行われている。欧州委員会は、医薬製品に関するいくつかの立法文書の改正に関する提案(規制データ保護の期限を短縮する可能性があり、迅速通路の資格を改訂するなど)。2023年4月26日に出版された。提案された改正は依然として欧州議会と欧州理事会の同意と採択を得る必要があるため,提案は採択前に重大な修正が行われる可能性があり,2026年初めまではないと予想される。しかし,長期的には,今回の改正はバイオ製薬業界や我々の業務に大きな影響を与える可能性がある。
資金不足や世界的な健康問題によるFDAや他の政府機関の中断は、重要な指導部や他の人員の採用、保留、配置の能力を阻害する可能性があり、あるいは新製品や修正された製品のタイムリーまたは開発、承認、商業化を他の方法で阻止することは、私たちの業務にマイナスの影響を与える可能性がある。
FDAおよび同様の外国規制機関が新製品を審査·承認する能力は、政府予算および資金レベル、法定、規制および政策変化、FDAまたは外国規制機関のキーパーソンの雇用および保留、ユーザー費用支払いを受け入れる能力、およびFDAまたは外国監督機関が通常の機能を履行する能力に影響を与える可能性のある他の事件を含む様々な要素の影響を受ける可能性がある。したがって、FDAと外国規制機関の平均審査時間は近年変動している。また,他の研究開発活動を援助する政府機関への政府の援助は政治過程の影響を受けており,この過程は本質的に不安定で予測不可能である。アムステルダムへの移転後のEMAおよびそれに伴う人員変動のようなFDAおよび他の機関の中断は、新薬または承認または承認された薬物の修正が必要な政府機関による審査および/または承認に要する時間を遅らせる可能性もあり、これは私たちの業務に悪影響を及ぼす。例えば、ここ数年間、米国政府は何度か閉鎖されており、FDAのようないくつかの規制機関は、FDAのキー従業員を休暇にし、キー活動を停止しなければならない。
また、新冠肺炎の流行に対応するため、アメリカ食品薬品監督管理局は国内外の異なる場所の製造施設の大部分の検査を延期した。FDAは標準的な検査操作を回復したにもかかわらず、ウイルスのいかなる巻き返しや新しい変種の出現も、さらなる検査関連または行政遅延をもたらす可能性がある。もし政府が長期的に停止した場合、あるいは世界的な健康問題がFDAや他の規制機関が定期的な検査、審査、または他の規制活動を行うことを阻止した場合、それは
これは、FDAまたは他の規制機関が、私たちが提出した規制文書をタイムリーに審査して処理する能力に深刻な影響を与え、私たちの業務に大きな悪影響を及ぼす可能性がある。
規制部門の承認を得ても、継続的な義務と持続的な規制審査の制約を受けることになり、多くの追加費用を招く可能性がある。また,承認されれば,アンチフェンはラベルや他の制限や市場撤退を受ける可能性があり,規制要求を遵守できなかった場合やアンチフェン洗浄の意外な問題に遭遇した場合,処罰される可能性がある。
FDAまたは同様の外国規制機関がアンチフェンの製造過程、ラベル、包装、流通、不良事件報告、貯蔵、広告、販売促進および記録保存を承認した場合、広範かつ持続的な規制要求を受けるであろう。これらの要件には,年間使用料の支払い,安全および他の発売後の情報や報告の提出,施設登録や医薬品の発売,承認後に行われた任意の臨床試験においてcGMPや同様の海外産プロポフォールやGCP要求を継続することが含まれており,いずれも巨額の費用を招く可能性があり,プロポフォールを商業化する能力を制限している。さらに、私たちが得る可能性のあるアンテフェンタニルの任意の承認は、特定の年齢層の使用制限、警告、予防措置または禁忌症に関連する重大な制限を含む可能性があり、重い承認後の研究またはリスク管理要件を含む可能性がある。例えば、FDAは、私たちの候補製品を承認するためにREMSを必要とすることができ、これは、制限された分配方法、患者登録、および他のリスク最小化ツールのような薬物ガイドライン、医師トレーニングおよびコミュニケーション計画、または安全な使用を保証する他の要素を必要とする可能性がある
私たちと私たちの契約製造業者はまた、FDAと他の規制機関の定期的な検査を受けて、これらの要求と私たちが得る可能性のある製品承認条項の遵守状況を監視する。私たちまたは規制機関が、予想されていない深刻度または頻度の不良事象、または製品の製造施設に問題があるような以前に未知の問題があることを発見した場合、規制機関は、製品のリコールを要求するか、または市場から製品を撤回するか、または生産を一時停止することを含む、製品、製造施設、または私たちに制限を加えることができる。さらに、FDAや他の類似した外国規制要求を守らなければ、私たちの会社は行政または司法制裁を受ける可能性があります
•製品の承認を遅延または拒否します
•私たちの臨床試験を行う能力の制限は、進行中または計画中の試験の全部または一部の臨床保留を含む
•製品、製造業者、または製造プロセスの制限;
•警告状や見出しのない手紙
•民事と刑事罰
•禁令
•規制承認の一時停止または撤回;
•輸入製品の差し押さえ、差し押さえ、または禁止;
•自発的または強制的な製品のリコールと宣伝要求
•生産を停止しています
•費用の高い新しい製造要件を含む運営に制限を加える。
これらのいずれかの事件や処罰の発生は,プロポフォールの商業化や創収能力を抑制する可能性があり,大量の時間と資源をかけて対応する必要があり,負の宣伝が生じる可能性がある。
さらに、FDAおよび他の規制機関の政策は変わる可能性があり、私たちの候補製品に対する規制承認を阻止、制限、または延期するための追加の政府法規が公布されるかもしれない。米国や海外の将来の立法や行政または行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することはできない。もし私たちが既存の要求の変化に緩やかに適応できない場合、あるいは新しい要求や政策を採用することができない場合、あるいは規制コンプライアンスを維持できない場合、私たちは法執行行動の影響を受ける可能性があり、利益を達成したり維持したりすることができないかもしれない。
FDAや他の外国規制機関は、ラベル外使用の普及を禁止する法律や法規を積極的に実行しており、これらの法律に違反していることが発見されれば、重大な責任を招く可能性がある。
もしアンテフェンタニルがどんな適応にも許可されていれば,私たちはアンテフェンタニルのラベル外用途を不正に普及させたことが発見され,大きな責任を負う可能性がある。FDAや他の規制機関は、承認されれば、処方製品(例えば、私たちの候補製品)に対する販売促進主張を厳格に規制することができる。特に、製品は、製品が承認されたラベルに反映されるように、FDAまたは他の規制機関によって承認されていない使用に使用されてはならない。もし私たちが候補製品のマーケティング承認を得たら、医者は承認されたラベルと一致しない方法で患者に処方するかもしれない。もし私たちがこのようなラベル外の使用を普及させることを発見されたら、私たちは重大な責任を負うかもしれない。米連邦政府は、ラベル外使用の不当な普及の疑いがある会社に巨額の民事と刑事罰金を科し、いくつかの会社がラベル外販売促進に従事することを禁止している。FDAはまた、企業に同意法令または永久禁止を締結し、これらの法令または永久禁止に基づいて、特定の販売促進行為を変更または制限することを要求する。もし私たちがアンテファンドリンの普及を成功的に管理できなければ、承認されれば、私たちは重大な責任を負うかもしれませんが、これは私たちの業務や財務状況に実質的な悪影響を及ぼすでしょう。
ヨーロッパでは、非ラベル使用自体はEU薬品立法の監督管理を受けず、薬品に対する厳格な監督管理は医療実践における薬品の使用と区別がある。ラベル外使用は国家法規を遵守すべきであり、EU加盟国によって異なる可能性がある(S)。
米国やEUなどの主要な薬品市場でアンテフェンタニルの発売許可を得ても、他の主要市場で承認されたり、商業化されたりすることは決してなく、すべての市場の潜在力を実現する能力を制限するだろう。
1つの国または地域で任意の製品を販売するためには、その国または地域の安全性および有効性に関する多くのおよび異なる規制要件を確立し、遵守しなければならない。一国で行われる臨床試験は、他の国の規制部門に受け入れられない可能性があり、一国の規制承認は、他のどの国でも規制承認を受けることを意味するものではない。承認手続きは国によって異なり、追加の製品テストと検証、および追加の行政審査期限が含まれる可能性があります。すべての主要な市場で監督管理の承認を求めることは著者らの重大な遅延、困難とコストを招く可能性があり、追加の臨床前研究或いは臨床試験が必要である可能性があり、これは高価で時間がかかるだろう。各国の規制要求には大きな違いがある可能性があり、これらの国へのイソフェン導入を延期または阻止する可能性がある。これらと他の規制要件を満たすことは高価で、時間がかかり、不確定であり、予期しない遅延が生じる可能性がある。また、私たちはどの国でも規制承認を得ることができず、他の国の規制承認過程を延期したり、マイナス影響を与える可能性がある。私たちは現在、どの司法管轄区でも候補製品の販売が許可されておらず、EU、アメリカ、あるいは他の国際市場でも、国際市場で監督管理の承認を受けた経験がない。もし私たちが国際市場の規制要求を遵守できなかったり、必要な承認を得られなかったりすれば、私たちの目標市場は減少し、私たちはアンテフェンタリン市場の潜在力を十分に発揮する能力が影響を受けるだろう。
私たちの従業員と独立請負業者は、最高調査者、CRO、コンサルタント、サプライヤー、および協力パートナーを含み、法規基準および要求を遵守しないことを含む不適切な行為または他の不適切な活動に従事する可能性があります。
私たちは、首席調査者、CRO、コンサルタント、サプライヤー、およびパートナーが詐欺または他の不正活動に従事する可能性があるリスクを含む、私たちの従業員と独立請負業者に直面している。これらの当事者の不正行為は、(I)FDA、EUおよび他の同様の規制機関およびEUの法律および法規に違反し、これらの機関への真の、完全かつ正確な情報の報告を要求する法律、(Ii)製造基準、(Iii)連邦および州データプライバシー、セキュリティ、詐欺および乱用、ならびに米国および海外の他の医療保健法律法規、または(Iv)真実、完全かつ正確な財務情報およびデータの報告を要求する法律を含む、意図的、無謀または不注意な行為または不正な活動を含む可能性がある。具体的には、医療業界の販売、マーケティング、および商業配置は、詐欺、不正行為、リベート、自己取引、および他の乱用行為を防止するための広範な法律法規によって制限されている。これらの法律法規は、広範な価格設定、割引、マーケティングおよび販売促進、販売手数料、顧客インセンティブ計画、および他のビジネス計画を制限または禁止する可能性があります。これらの法律制約を受けた活動はまた、不適切な使用或いは歪曲臨床試験過程で得られた情報に関連する可能性があり、著者らの臨床前研究或いは臨床試験中に虚偽のデータを作成し、或いは薬物製品を不法に流用し、これは規制制裁を招き、著者らの名声に深刻な損害を与える可能性がある。従業員や他の第三者の不正行為を常に識別し、阻止することができるわけではなく、このような活動を発見し、防止するための予防措置は、未知または管理不可能なリスクや損失を効果的に制御することができないか、またはそのような法律や法規を遵守できないことによる政府調査または他の行動または訴訟から私たちを保護することができない可能性がある。さらに、誰かや政府がこのような詐欺や他の不正行為を告発する可能性があり、起こらなくてもリスクに直面している。もし私たちにこのような行動を取って、私たちが自分の権利を弁護したり、維持したりすることに成功しなかったら、これらの行動は
私たちの業務および経営結果は、重大な民事、刑事および行政処罰、損害賠償、金銭罰金、引き渡し、他の司法管轄区域から排除される可能性のあるMedicare、Medicaid、および他の米国連邦医療計画または医療計画の参加、誠実な監督と報告義務を含み、違反疑惑、個人監禁、他の制裁、契約損害、名声損害、利益減少および将来の収益の減少、および私たちの業務の削減を解決する。
私たちが時々発表したり公表したりする臨床試験の一時、“トップ”あるいは初歩的なデータは、より多くの患者データの獲得に伴い変化し、監査と検証手続きの影響を受ける可能性があり、これは最終データの実質的な変化を招く可能性がある。
私たちは時々私たちの臨床試験の中期、主要または予備データを公開するかもしれないが、これらのデータは当時利用可能なデータの初歩的な分析に基づいており、結果および関連する発見および結論は、特定の研究または試験に関連するデータをより全面的に検討した後に変化する可能性がある。私たちはまた、私たちのデータ分析の一部として、すべてのデータを全面的かつ詳細に評価する機会がないか、または受け取る機会がないかもしれないという仮説、推定、計算、および結論を出した。したがって、より多くのデータが受信され、十分に評価されると、私たちの報告の主要または予備結果は、同じ研究の将来の結果とは異なる可能性があり、または異なる結論または考慮要因が、これらの結果を合格させる可能性がある。トップラインデータ或いは初歩データは依然として監査と確認手続きを受ける必要があり、これは最終データが以前公表されたトップラインデータ或いは初歩データと大きく異なる可能性がある。したがって、最終データが利用可能になる前に、トップラインおよび予備データは慎重に見られなければならない。
私たちはまた時々私たちの臨床前研究と臨床試験の中期データを開示することができる。著者らが完成する可能性のある臨床試験の中期データは、患者登録の継続とより多くの患者データの獲得に伴い、1つ以上の臨床結果が実質的に変化する可能性があるというリスクに直面している。中間データと最終データの間の不利な違いは、私たちのビジネスの見通しを深刻に損なう可能性があります
さらに、規制機関を含む他の人は、私たちの仮定、推定、計算、結論または分析を受け入れないか、またはデータの重要性を異なる方法で解釈またはトレードオフする可能性があり、これは、特定の計画の価値、特定の候補製品または製品の承認または商業化、およびわが社の全体的な状況に影響を与える可能性がある。さらに、開示された特定の研究または臨床試験に関する情報を選択することは、通常、広範な情報に基づいており、あなたまたは他の人は、私たちが決定した重要な情報または他の適切な情報が私たちの開示に含まれることに同意しない可能性がある。
もし私たちが報告した中期、トップライン、または予備データが実際の結果と異なる場合、または規制機関を含む他の人が結論に同意しない場合、私たちが承認を得て私たちの候補製品を商業化する能力が損なわれる可能性があり、これは私たちの業務、経営業績、将来性、または財務状況を損なう可能性がある。
医療保健法やその他の法律コンプライアンス事項に関するリスク
制定と将来の立法は、アンテファンドリンの上場承認と商業化の難しさとコストを増加させ、私たちが制定する可能性のある価格に影響を与える可能性がある。
米国、EU、その他の外国司法管轄地域では、医療システムの複数の立法や規制改革、提案された改革が継続されることが予想されており、将来の運営結果に影響を与える可能性がある。特に,米国連邦や州レベルでは,医療コストの低減と医療の質の向上を図る取り組みが継続されている。例えば、2010年3月には、政府や民間保険会社がヘルスケアのために資金調達する方式を大きく変えた“医療·教育調整法”改正“患者保護·平価医療法案”が公布された。ACAの条項の中で、製薬とバイオテクノロジー産業にとって最も重要な条項は:
•特定のブランドの処方薬および生物製剤を製造または輸入する任意のエンティティが支払うべき控除できない年間費用は、特定の政府医療計画におけるそれらの市場シェアに基づいてこれらのエンティティ間で分担されるべきである
•新しい連邦医療保険D部分保証切欠き割引計画は、メーカーはその保証間隔期間内に条件を満たす受益者にブランド薬品交渉価格を適用する販売時点割引を提供することに同意しなければならず、メーカーの外来薬物として連邦医療保険D部分で保険を受ける条件である
•医療補助薬品リベート計画によると、メーカーが支払わなければならない法定最低リベートはそれぞれブランドと後発薬メーカーの平均価格の23.1%と13.0%に引き上げられた
•医療補助薬品バックル計画の下での製造業者の吸入、注入、点滴、インプラントまたは注射薬のバックルを計算するための新しい方法
•メーカーの医療補助税還付責任を医療補助管理保健組織に参加する個人に配布する保険薬品に拡大した
•医療補助計画の資格基準を拡大し、他を除いて、各州が連邦貧困レベル133%以下の収入のある個人に医療補助を提供することを許可し、それによってメーカーの医療補助リベート責任を潜在的に増加させる
•患者を中心とした新しい結果研究所は、監督、優先事項を決定し、臨床治療効果の比較研究を行い、このような研究に資金を提供する
•連邦医療保険·医療補助サービスセンター(CMS)に医療保険·医療補助革新センターを設立し、革新的な支払い·サービス交付モデルをテストし、連邦医療保険·医療補助支出を低減し、処方薬支出を含む可能性がある。
ACAのいくつかの側面は公布以来、司法、行政、そして国会の挑戦を受けてきた。2021年6月17日、米国最高裁はいくつかの州がACAに対して提出した最新の司法挑戦を却下したが、ACAの合憲性を具体的に裁くことはなかった。最高裁が裁決を下す前に,総裁·バイデンはACA市場による医療保険の取得を目的とした2021年2月15日から2021年8月15日までの特殊保険期間を開始する行政命令を発表した。行政命令はまた、特定の政府機関に、医療の獲得を制限する既存の政策や規則を審査し、再考するよう指示した。
また、ACAが公布されて以来、米国は他の立法改正を提案し、採択した。例えば、他の事項を除いて、2011年の予算制御法は、医療保険提供者の医療保険支払総額を減少させ、その後の法規の立法改正により、国会がさらに行動しない限り、2020年5月1日から2022年3月31日まで支払いを停止することを除いて2032年まで有効となる。2013年1月2日、病院、画像センター、がん治療センターを含むいくつかの提供者に支払う医療保険をさらに減らし、政府が提供者に多額の支払いを取り戻す訴訟時効を3年から5年に延長する“2012年米国納税者救済法”が法律に署名された。また,2021年に米国救援計画法案が法律に署名され,2024年1月1日から法定医療補助薬品還付上限が廃止された。これまで,還付の上限は薬品メーカーの平均価格の100%であった。これらの法律および将来公布される任意の法律は、連邦医療保険および他の医療保険資金のさらなる減少をもたらす可能性があり、これは、私たちの顧客および私たちの財務運営に実質的な悪影響を及ぼす可能性がある。
また、支払い方法は医療立法と規制措置の影響を受ける可能性がある。例えば、CMSは、バンドル支払いモードのような新しい支払いおよび配信モードを開発することができる。また、最近政府はメーカーが製品を販売するための価格設定方式の審査を強化している。最近は2022年8月16日に“2022年インフレ削減法案”(IRA)が署名されて法律となっている。他の事項を除いて、IRAはある薬品のメーカーが連邦医療保険と価格交渉を要求し(2026年から)、価格は交渉できるが上限がある;連邦医療保険B部分と連邦医療保険D部分によるリベート徴収は引き続きインフレを超える価格上昇を罰し、D部分のカバーギャップ割引計画の代わりに新しい割引計画を採用する(2025年から)。アイルランド共和軍は,衛生·公衆サービス部(HHS)長官が最初の数年間,規制ではなく指導によって多くの規定を実施することを許可した。これらの計画の実施に伴い,HHSは指導意見を発表·更新し続けている。2023年8月29日、HHSは、連邦医療保険薬品価格交渉計画が現在法的挑戦を受けているにもかかわらず、価格交渉を受ける上位10種類の薬物のリストを発表した。アイルランド共和軍の製薬業に対する影響はまだ完全には確定されていないが、重大な可能性が高い。
また、バイデン政府は2022年10月14日に追加の行政命令を発表し、HHSに90日以内に報告書を提出するよう指示し、連邦医療保険と医療補助革新センターをさらに利用して連邦医療保険と医療補助受益者の薬品コストを下げる新しいモデルをテストすることを説明した。この行政命令への応答として,2023年2月14日,HHSはCMS革新センターでテストされた3種類の新しいモデルについて概説し,これらのモデルは薬物コストを低減し,獲得性を促進し,医療の質を向上させる能力に基づいて評価する。これらのモデルが将来の任意の医療改革措置で使用されるかどうかは不明である
将来的にはより多くの米国連邦医療改革措置がとられることが予想され,いずれも米国連邦政府が医療製品やサービスのために支払う金額を制限する可能性があり,プロポフォールの需要減少や追加的な定価圧力を招く可能性がある。
米国の個別州も、価格や患者の精算制限、割引、ある製品への参入の制限、マーケティングコストの開示と透明性措置を含む、薬品と生物製品の定価を制御するための法規を立法と実施することをますます積極的に実施しており、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。法律で規定されている第三者支払者の支払金額の価格制御またはその他の制限は、私たちの業務、運営結果、財務状況、見通しを損なう可能性があります。また,地域医療当局や個別病院では,どの薬品やサプライヤーが処方薬や他の医療計画に含まれるかを決定するために入札プログラムが使用されるようになってきている。これはアンテフェンタリンに対する最終的な需要を下げるかもしれないし、私たちの製品の価格設定に圧力を与えるかもしれない。
EUでは、似たような政治的、経済的、規制的な発展が、承認されれば、私たちが利益を得てアンテフィンを商業化する能力に影響を及ぼすかもしれない。価格およびコスト制御措置に対する持続的な圧力に加えて、EUまたは加盟国レベルの立法発展は、著しい追加的な要求や障害を招く可能性があり、これは私たちの運営コストを増加させる可能性がある。EUで保健サービスを提供することは、保健サービスの確立と運営、薬品の定価と精算を含み、ほぼ完全に国家の法律と政策の問題であり、EUの法律と政策の問題ではない。この点で,各国政府と保健サービス提供者は,保健および製品定価や補償を提供する上で異なる優先順位や方法を持っている。しかし、全体的に、大多数のEU加盟国の医療予算制限は、関連医療サービス提供者の薬品定価と精算の制限を招いた。さらに、EUや国が製品の開発·販売を希望する人に増加している規制負担に加えて、これは、アンテフェンの上場承認を阻止または延期し、承認後の活動を制限または規制し、承認すればアンテフェンを商業化する能力に影響を与える可能性がある。国際市場では,精算や医療保険支払い制度は国によって異なり,多くの国で特定製品や治療法に価格上限が設定されている。
2021年12月13日,衛生技術評価に関する第2021/2282号条例が可決され,第2011/24/EU号指令が改正された。この規定は2022年1月に施行されたが、2025年1月から適用され、その間に実施に関する準備と手順が取られるだけである。適用されると、それは関連製品に従って段階的に施行されるだろう
この条例はEU加盟国の新医薬製品を含む衛生技術の評価における協力を促進し、これらの領域の共同臨床評価にEUレベルの協力基礎を提供することを目的としている。これは、EU加盟国がEU範囲内で汎用的なHTAツール、方法、およびプログラムを使用することを可能にし、患者に最大の潜在的影響を有する革新的な衛生技術の共同臨床評価、共同科学相談、開発者がHTA当局にアドバイスを求め、新興衛生技術を決定すること、および将来性のある技術を早期に決定することを含む4つの主要分野で協力することができる
他の分野で自発的な協力を続けている。個別EU加盟国は、衛生技術の非臨床(例えば、経済、社会、倫理)の評価を引き続き担当し、定価と精算について決定する。
米国や海外の将来の立法や行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することはできない。もし私たちまたは私たちの協力者が既存の要求の変化に緩やかに適応できない場合、または新しい要求や政策を採用することができない場合、または私たちまたは私たちの協力者が規制適合性を維持できない場合、アンテファニンは入手された可能性のあるいかなる規制承認も失う可能性があり、私たちは利益を達成または維持できないかもしれない。
我々の業務運営および調査者,医療専門家,コンサルタント,第三者支払者,顧客との現在と将来の関係は,適用される医療規制法の制約を受け,処罰される可能性がある。
私たちの業務運営および調査者、医療専門家、コンサルタント、第三者支払者、顧客との現在と将来の手配は、広範に適用される詐欺や乱用、その他の医療に関する法律や法規に直面する可能性があります。これらの法律は、私たちがどのように研究、マーケティング、販売、流通アンテフィンをどのように研究するか(承認されれば)を含む、私たちが業務を展開する業務または財務的配置および関係を制限するかもしれない。これらの法律には
•米国連邦反リベート法規は、他の事項に加えて、個人または実体が個人の推薦または購入、レンタル、注文または推薦の任意の商品、施設、物品またはサービスを誘導または奨励するために、個人の推薦または購入、レンタル、注文または推薦または任意の商品、施設、物品またはサービスを誘導または奨励するために、任意の報酬(任意のリベート、賄賂または何らかのリベートを含む)を提供することを禁止し、これらの商品、施設、物品またはサービスは、米国連邦および州医療保険および医療補助計画に従って全部または部分的に支払うことができる。個人またはエンティティは、法規または法規違反の具体的な意図を実際に知る必要がなく、違反を実施することができる
•民事虚偽請求法案を含む米国連邦虚偽クレームおよび民事罰金法律であって、米国連邦政府に故意に虚偽または詐欺的な支払いまたは承認請求を提出するか、虚偽または詐欺的クレームの作成または使用を意図的に作成、使用または誘導する虚偽記録または報告書を含む、または米国連邦政府への支払いの義務を回避、減少または隠蔽するための虚偽陳述を意図的に行う個人またはエンティティに対して、民事告発者または刑事訴訟を含む刑事および民事処罰を適用することを含む、米国連邦虚偽クレームおよび民事罰金法律。また、政府は、米国連邦反リベート法規に違反して発生した物品やサービスを含むクレームは、“虚偽請求法”については、虚偽または詐欺的クレームを構成していると断言できる
•1996年の米国連邦医療保険携帯および責任法案、またはHIPAAは、他にも、詐欺の任意の医療福祉計画を意図的かつ故意に実行または実行しようとする計画、または医療福祉、プロジェクトまたはサービスの提供または支払いに関連する重大な事実を故意および故意に偽造、隠蔽または隠蔽または隠蔽し、または任意の重大な虚偽陳述を行い、刑事および民事責任を適用する;米国連邦反リベート法令と同様に、個人または実体は、その法令またはその法令に違反する具体的な意図を実際に知る必要がなく、違法行為を犯すことができる
•FDCAは、他に加えて、医薬品、生物製品、および医療機器に偽または誤ったブランドを貼り付けることを禁止している
•米国連邦立法は、ACAおよびその実施条例の一部として、Medicare、Medicaidまたは児童健康保険計画によって精算可能ないくつかの薬品、設備、生物製品および医療用品の製造業者に毎年、医師(医師、歯科医師、視光師、足科医師および脊椎マッサージ師を含むと定義される)、いくつかの非内科従事者(医師アシスタント、勤務看護師、臨床看護師、登録看護師麻酔科助手、麻酔学アシスタントおよび登録看護師助産師)への何らかの支払いおよびその他の価値移転に関する情報を政府に報告する医師支払い陽光法案と呼ばれる。これらの医師とその直系親族が所有している所有権と投資権益
•州反リベートおよび虚偽クレーム法律は、研究、流通、販売およびマーケティングスケジュール、および任意の第三者支払人(個人保険会社を含む)の精算に関連する医療項目またはサービスに関するクレームを含むが、製薬会社に製薬業界の自発的コンプライアンスガイドラインおよび米国連邦政府によって発行された関連コンプライアンスガイドラインを遵守することを含むが、製薬会社に製薬業界の自発的コンプライアンスガイドラインおよび米国連邦政府によって発行された関連するコンプライアンスガイドラインを含むが、これらに限定されない、我々のビジネス実践に適用される可能性がある州の法律および法規を含む、同様の州法律および法規
そうでなければ、医療提供者および他の潜在的な紹介源に支払う可能性のあるお金を制限することと、医療専門家および実体に提供されるプレゼントおよび他の報酬および価値項目を追跡することを必要とする価格設定およびマーケティング情報に関連する報告書を医薬品製造業者に提出することを要求する州法律法規と、
•EUでは、製薬会社と医療専門家と医療機関との相互作用も、EUレベルでもEU個別加盟国でも、厳格な法律、法規、業界自律行動規則、医師職業行為規則の制約を受けている。EUでは、処方、推薦、裏書き、購入、供給、注文、または薬品の使用を誘導または奨励するために、医師に福祉または利点を提供することが禁止されている。医療専門家や協会との関係は厳しい反プレゼント法規や反賄賂法律に制約されており,これらの法律の範囲はEU諸国で異なる。また、国のサンシャイン法案は、保健専門家や協会に提供される価値移転状況を製薬会社に定期的(例えば、毎年)に報告·公表することを要求する可能性がある。このような要求を守らないことは、名声のリスク、公開非難、行政処罰、罰金、または監禁につながる可能性がある。
我々の内部運営と第三者の業務配置とが適用される医療法律や法規に適合することを確保し、大量のコストに関連する。政府当局は、私たちの業務やり方が現在または未来に適合していないと結論するかもしれないが、詐欺および乱用または他の医療保健法律および法規の適用に関する現行または将来の法規、機関指導または判例法に関連している。私たちの運営が上記の任意の法律または任意の他の私たちに適用される可能性のある政府の法律および法規に違反していることが発見された場合、私たちは、民事、刑事および行政処罰、損害賠償、罰金、米国政府によって援助された医療計画(例えば、MedicareおよびMedicaid)や他の国または司法管轄区域の同様の計画の外、会社誠実協定、または他の合意を含む重大な処罰を受ける可能性があり、これらの法律に違反した疑い、返還、個人監禁、契約損害、名声損害、利益減少、および私たちの業務の削減または再編を解決する。もし私たちがそれと業務を行うことを期待している任意の医師や他の提供者や実体が適用されない法律を遵守していないことが発見された場合、彼らは政府の援助された医療計画や監禁から除外されることを含む刑事、民事または行政制裁を受ける可能性があり、これは私たちの業務運営能力に影響を及ぼす可能性がある。さらに、このような行動を防御するには費用がかかり、時間がかかる可能性があり、多くの人的資源が必要となる可能性がある。したがって、私たちが私たちに提起される可能性のあるいかなる訴訟も防ぐことに成功しても、私たちの業務は損害を受ける可能性がある。
実際または予想されるように、適用されるデータ保護、プライバシーおよびセキュリティ法律、法規、基準、およびその他の要求を遵守できないことは、私たちの業務、運営結果、および財務状況に悪影響を及ぼす可能性があります。
世界的なデータ保護構造は急速に変化しており、私たちは多くの州、連邦および外国の法律、要求および法規によって制限されているかもしれないが、これらの法律、要求、および法規は、個人情報の収集、使用、開示、保持および安全を管理しており、例えば、私たちが収集する可能性のある臨床試験に関連する情報を管理している。予測可能な未来には、実施基準および法執行実践は依然として不確定である可能性があり、私たちはまだ未来の法律、法規、基準、またはその要求に対する見方が私たちの業務に与える影響を決定することができない。このような変化は私たちの業務に不確実性をもたらす可能性があり、私たちがある司法管轄区域で業務を展開し、あるいは個人情報を収集、保存、移転、使用、共有する能力に影響を与え、私たちの契約でより重い義務を受ける必要があり、私たちが責任を負うか、または追加コストをかける必要がある。このような法律、法規、そして基準を遵守する費用は高く、未来に増加するかもしれない。私たちが連邦、州、または外国の法律または法規、私たちの内部政策と手続き、または私たちの個人情報を処理する契約を遵守できなかったか、または遵守できなかったと思われることは、否定的な宣伝、政府の調査と法執行行動、第三者のクレームと私たちの名声の損傷を招く可能性があり、いずれも私たちの業務、運営結果、および財務状況に実質的な悪影響を及ぼす可能性がある。
私たちの運営と業務の増加に伴い、私たちは新しいまたは追加のデータ保護法律と法規の制約または影響を受け、規制機関のより厳しい審査や関心に直面する可能性がある。米国では、2009年に“経済·臨床健康情報技術法案”により改正されたHIPAAとその下で実施された法規、あるいは総称してHIPAAと呼ばれ、他の事項に加えて、個人が健康情報を識別できるプライバシー、安全、伝送、違反報告に関するいくつかの基準が規定されている。多くの医療提供者は,我々がそれから患者の健康情報を取得する研究機関を含め,HIPAAが公布したプライバシーと安全法規の制約を受けている。我々は現在HIPAA下の保証実体やビジネスパートナーとして行動しているとは思わないため,HIPAAの要求や処罰を直接受けることはない.しかしながら、事実および状況に基づいて、私たちが知らずにHIPAAによってカバーされているヘルスケア提供者または研究機関から個人識別可能な健康情報を受信し、その医療提供者または研究機関が個人識別可能な健康情報の開示に関するHIPAAの要求を満たしていない場合、重大な刑事罰に直面する可能性がある
いくつかの州でも同様のプライバシーおよびセキュリティ法律が採択されており、その中のいくつかはHIPAAよりも厳しい可能性がある。これらの法律と法規は、異なる裁判所や他の政府当局によって解釈され、私たちおよび未来の顧客と戦略パートナーに潜在的な複雑なコンプライアンス問題をもたらすだろう。例えば、カリフォルニア州は2020年1月1日に施行される“カリフォルニア消費者プライバシー法”(CCPA)を公布した。CCPAは、他にも、カバーする会社のためのデータプライバシー義務を制定し、彼らの情報へのアクセスおよび削除、特定の情報共有からの離脱を選択する権利、および彼らの個人情報がどのように使用されるかに関する詳細な情報を受信する権利を含むプライバシー権をカリフォルニアの消費者に提供する。CCPAはまた、特定のデータ漏洩のために法定損害賠償を有する個人訴権を作成し、それにより、データ漏洩に関連するリスクを潜在的に増加させる。法律は健康に関する情報に対して限られた例外を規定しているにもかかわらず,状況に応じて規範化したり,個人情報の処理に影響を与えたりする可能性がある。また、カリフォルニアプライバシー権法案(CPRA)は2023年1月1日に正式に施行され、CCPAの重大な改正が行われた。それは、カバーされた企業に、追加の消費者権利手続き、データ使用の制限、より高いリスクデータの新しい監査要件、および敏感なデータからの退出を選択するいくつかの用途を含む追加のデータ保護義務を課す。それはまた、実質的な法規の発行を許可し、プライバシーと情報セキュリティ法執行の強化につながる可能性がある新しいカリフォルニア州データ保護機関を作成した。追加的なコンプライアンス投資と潜在的なビジネスプロセスの変更が必要になる可能性があります。他の州でも同様の法律が可決され、州や連邦レベルで提案され続けており、これは米国がより厳しいプライバシー立法に傾いている傾向を反映している。このような法律の公布は相互衝突の要求があり、コンプライアンスを挑戦的にする可能性がある。もし私たちがHIPAA、CCPA、CPRA、または他の国内プライバシーおよびデータ保護法律の制約または影響を受けた場合、これらの法律の要求を遵守できなかったために負う任意の責任は、私たちの財務状況に悪影響を及ぼす可能性があります。
私たちはまた、“一般データ保護条例”(GDPR)を含む、EUと欧州経済圏のデータプライバシーとセキュリティに関する異なる法律と法規の制約を受けている。GDPRは2018年5月に発効し,欧州経済圏内の個人データの処理に厳しい要求を出した。GDPRは,ヨーロッパ経済域内の個人が健康に関する個人データや他の個人データを処理する能力に対して,このような個人データの使用,収集,分析,移転(国境を越えた移動を含む)に関する義務を厳格に規定している。GDPRを遵守しなければならない企業は、より強力なデータ保護要件の規制法執行、および不正があれば2000万ユーロまたは不適合会社の世界年収の4%までの罰金が科される可能性があるコンプライアンス義務およびリスクに直面しなければならない。罰金に加えて、GDPR違反は、規制調査、名声被害、データ処理活動の停止/変更の命令、執行通知、評価通知(強制監査のための)、および/または民事クレーム(集団訴訟を含む)を引き起こす可能性がある。他の要件では、GDPR規制は、GDPRによって制約された個人データを、このような個人データに対して十分な保護を提供することが発見されていない第3国に移すことで、米国、欧州経済地域と米国との間の既存の移動機構の有効性および持続性はまだ不確定である。欧州連合裁判所(以下“CJEU”)の判例法は,標準契約条項のみに依存する欧州委員会が適切な個人データ転送機構としての標準契約形式を承認することは,必ずしもすべての場合で十分であるとは限らず,移転はケースベースで評価しなければならないと指摘している。2023年7月10日、欧州委員会は、DPFをDPF自己認証による米国エンティティのGDPF移行機構として有効にし、DPFをDPF自己認証の米国エンティティのGDPF移行機構として有効にする新たなEU-米国データプライバシー枠組み(DPF)に関する十分な決定を採択した。私たちは、国際個人データ移転に関する既存の法律の複雑さと不確実性が引き続き存在すると予想する。特に,DPFの十分性決定が挑戦され,米国やより広い他の管轄地域への国際移転は規制機関の強化審査を受け続けることが予想される。したがって、いくつかの業務上の変更を行わなければならない可能性があり、規定された期限内に既存のデータ転送のために改訂された標準契約条項および他の関連文書を実行しなければならない。
これに関連して,2021年初め以来,連合王国の欧州経済圏と欧州連合の離脱,および過渡期の終了に伴い,会社は連合王国の国家法律に組み込まれたGDPRとGDPRを遵守しなければならず,後者はそれぞれ最高1,750万ポンドまたは世界売上4%の大きな罰金を科す権利がある。2023年10月12日、イギリスによるDPFの延期が発効し(イギリス政府の承認を得て)、イギリスからDPFによる自己認証に基づく米国エンティティにデータを転送する仕組みとなる。私たちが他の国や司法管轄区域に拡張し続けるにつれて、私たちは追加の法律と法規の制約を受けるかもしれません。これらの法律と法規は私たちが業務を展開する方法に影響を与えるかもしれません
適用されるデータ保護の法律および法規を遵守することは、私たちがデータを収集、使用、開示する能力を制限し、または場合によってはいくつかの司法管轄区域で運営される能力に影響を与える契約でより重い義務を負うことを要求するかもしれない。もし私たちまたは私たちの協力者と第三者プロバイダが適用されるデータ保護法律と法規を遵守できなければ、政府が法執行行動を取ることになる可能性があります(可能性がある)
民事又は刑事罰)、個人訴訟及び/又は負の宣伝は、我々の経営業績及び業務に負の影響を与える可能性がある。また,我々または我々の潜在的協力者が情報を取得する臨床試験対象,およびこれらの情報を共有している提供者は,このような情報の使用や開示能力を契約的に制限する可能性がある。私たちがプライバシー権を侵害し、データ保護法を遵守できなかったこと、または私たちの契約義務に違反したと主張することは、私たちが責任を負わないと認定されても、弁護の費用が高く、時間がかかる可能性があり、マイナスの宣伝を招く可能性があり、私たちの業務、財務状況、運営結果、および将来性に実質的な悪影響を及ぼす可能性がある。
環境持続可能性と社会的イニシアティブへの日々の関心は、私たちのコストを増加させ、私たちの名声を損ない、私たちの財務業績に悪影響を及ぼす可能性がある
投資家、環境保護活動家、メディア、政府、非政府組織は、様々な環境、社会、その他の持続可能な発展問題に対する国民の関心が高まっている。私たちは圧力に直面する可能性があり、持続可能性に関連する具体的なリスク緩和戦略の設計と実施を含む、私たちの持続可能性に影響を与える事項に関する約束を行うことが求められる。私たちの業務に影響を与える環境、社会、その他の持続可能な開発問題を効果的に解決したり、関連する持続可能な開発目標を策定して実現できなければ、私たちの名声や財務業績は影響を受ける可能性があります。また、私たちの持続可能な開発目標を達成し、これらの目標の達成度を測定するためには、コストが増加する場合があり、これは私たちの業務や財務状況に悪影響を及ぼす可能性があります。
また,環境,社会,その他の持続可能な問題への重視は,新たな報告要件を含む新たな法律や条例の採択につながる可能性がある。もし私たちが新しい法律、法規、または報告要件を遵守できなければ、私たちの名声と業務は不利な影響を受けるかもしれない。
私たちは環境、健康、安全法律法規の制約を受けており、私たちは環境コンプライアンスや救済活動に関連する責任と巨額の費用に直面する可能性がある。
私たちの下請け業務は、私たちの研究、開発、テストと製造活動を含めて、多くの環境、健康と安全法律法規の制約を受けています。他の事項に加えて、これらの法律と条例は、危険材料および生物材料の制御使用、処理、放出および処置および登録、例えば化学溶媒、ヒト細胞、発癌化合物、変異原性化合物、生殖、実験室プログラムおよび血液伝播病原体に接触することに対して毒性作用を有する化合物を管理する。もし私たちがこのような法律と法規を守らなければ、私たちは民事や刑事罰金、処罰、または他の制裁に関連する巨額のコストを招くかもしれない。
我々と同様の活動をしている他社と同様に,危険や生体材料の放出や接触に関する責任を含む現在および歴史的活動に固有の環境責任リスクに直面している。環境、健康、そして安全の法律法規はもっと厳しくなっている。私たちは将来の環境コンプライアンスや救済活動に多くの費用を発生させることを要求されるかもしれませんが、この場合、私たちの生産と開発作業は中断または遅延する可能性があります。
私たちは反腐敗法、そして輸出規制法、税関法、規制法、その他私たちの業務を管理する法律を守らなければならない。もし私たちがこのような法律を守らなければ、私たちは民事または刑事処罰、他の救済措置、そして法的費用を受けるかもしれない。
私たちの業務は、イギリスの“2010年収賄法”、米国の“海外腐敗防止法”、および私たちが業務を展開し、将来業務を展開する可能性のある国/地域に適用される他の反腐敗法律を含む反腐敗法を遵守しなければならない。“収賄法”、“海外腐敗防止法”およびその他の法律は、一般に、業務を獲得または保留し、またはいくつかの他の商業的利益を得るために、私たち、私たちの職員および中間者に賄賂を与え、賄賂されたり、政府関係者または他の人に他の禁止されたお金を支払うことを禁止している。私たちは将来的に“収賄法”や“海外腐敗防止法”に違反する可能性のある高リスク司法管轄区域で業務を展開する可能性があり、私たちは第三者との協力や関係に参加する可能性があり、これらの第三者の行為は私たちに“反収賄法”、“海外腐敗防止法”あるいは現地反腐敗法に規定された責任を負わせる可能性がある。さらに、私たちは将来の規制要求の性質、範囲、または影響を予測することができず、私たちのいかなる国際業務もこれらの要求によって制約される可能性があり、既存の法律が管理または解釈される可能性がある方法を予測することもできない。
私たちはまた、適用される輸出規制法規、国および人員に対する経済制裁、税関要件および通貨両替法規、または総称して貿易規制法を含む、任意の国際業務を管理する他の法律および法規、イギリスとアメリカ政府およびEU当局によって管理される法規を含む。特に,ロシアでは,ロシアとウクライナとの間の持続的な衝突や,それによる米国や他の国政府による制裁により,臨床場所に費用を支払うか,ロシアで臨床試験を行う能力が影響を受ける可能性のあるリスクが増加するため,我々の第3段階増強臨床計画に関連する臨床試験地点を招聘した。
私たちが貿易統制法を含む“反収賄法”、“海外腐敗防止法”または他の法律要件を含むすべての適用された反腐敗法律を遵守することを完全かつ効果的に保証することはできない。もし私たちが“反収賄法”、“反海外腐敗法”および他の腐敗防止法または貿易統制法を守らなければ、私たちは刑事と民事処罰、返還と他の制裁、救済措置、法的費用を受ける可能性がある。イギリス、アメリカ、または他の当局が“反収賄法”、“海外腐敗防止法”、他の腐敗防止法または貿易規制法に違反する可能性があることを調査した場合、最終的にこれらの法律に違反していないと判断されても、コストが高く、時間がかかり、大量の人的資源が必要となり、私たちの名声を損なう可能性がある。
私たちは私たちの内部統制制度を構築し、改善し、発見されたどんな弱点も修正するために努力するつもりだ。しかし、私たちの政策と手続きが常に遵守されることは保証されないし、私たちの1人以上の従業員、コンサルタント、代理人、または協力者が適用法に違反していることを効果的に発見し、防止することはできないので、私たちは罰金、処罰、または起訴を受けるかもしれない。
商業化に関連するリスク
私たちがいる業界の競争は激しく、変化が迅速で、これは他の人が私たちよりも早く、あるいはより成功的に製品を発見、開発、または商業化することにつながるかもしれない。
バイオ製薬や製薬業界は競争が激しく、重大かつ迅速な技術変革の影響を受けやすい。私たちの成功は、費用効果に基づいて新製品のマーケティング承認を発見、開発、獲得し、これらの製品を成功させる能力に大きく依存している。もしアンテフェンタリンがどんな用途でも使用されることが承認されれば、ヨーロッパ、アメリカ、その他の司法管轄区域の大型、完全に統合された製薬会社、専門製薬会社と生物製薬会社、学術機関、政府機関、その他の民間および公共研究機関を含む様々な企業からの激しい競争に直面するだろう。これらの組織は私たちよりもっと多くの資源を持って、類似の研究を行い、特許保護を求め、そしてアンテフェンタニルと競争する可能性のある製品の研究、開発、製造とマーケティングのための協力手配を確立するかもしれない。
市販されているCOPD,喘息,CF,NCFBEを治療する製品の数に鑑み,アンテフェンタニルがこれらの適応に使用されることが承認されれば,激しい競争に直面することが予想される。ブリンガー·インゲルハイム,グラクソ·スミスクライン,アスリーカン,ノワール,Vertex,Viatris,Theravance,GIlead,Genentechを含む会社は現在,慢性閉塞性肺疾患,慢性閉塞性肺疾患,喘息の治療薬を市場に有しており,将来的には新たな会社がこれらの市場に参入することが予想される。現在NCFBEの治療法は米国やEUでは発売されていないが,後期臨床開発段階にある製品のいくつかは将来承認される可能性がある。任意の適応のアンテフェンタニルの開発と商業化に成功すれば,既存の療法や将来起こりうる新しい療法と競争するであろう。生物製薬と製薬業界の高度な競争性質と迅速な技術変化はイソフェンを時代遅れにし、競争力を低下或いは経済的ではないかもしれない。私たちの競争相手は
•私たちよりも大きな知名度、財務、製造、マーケティング、薬物開発、技術と人的資源を持っており、将来的にバイオ製薬と製薬業界の合併と買収はより多くの資源を私たちの競争相手に集中させる可能性がある
•より安全で、より効率的で、より安価で、より便利に、または管理が容易で、または副作用が少ないまたはより軽い製品を開発し、商業化すること
•規制部門の承認をより早く得ることができます
•私たちの製品と技術をカバーする優れた独自の地位を確立する
•より効果的な販売、マーケティング、および流通方法を実施すること
•より優位な戦略的同盟を形成する。
規模の小さい会社や他のスタートアップ会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配されている。これらの第三者は合格した科学と管理者を募集と維持し、臨床試験場と臨床試験の患者登録を確立し、そして私たちの計画と相補的或いは必要な技術を獲得する上で私たちと競争している。さらに、私たちが持っている可能性のあるどの協力者も、アンテフェンタリンと競争する製品のマーケティングと販売を決定することができる。もし私たちの競争相手がアンテフェンタニルよりも効果的で、副作用が少ない、またはより深刻ではなく、より便利で、あるいはより安い製品を開発し、商業化すれば、私たちのビジネス機会は減少または消失するかもしれない。私たちの競争相手も私たちよりも早く彼らの候補製品のためにFDAや他の規制機関の承認を得ることができ、これは私たちの競争相手が私たちが市場に入る前に彼らの市場地位を確立したり強化したりすることができるかもしれない。
アンテフェンタニルの商業化の成功は、政府当局と健康保険会社がアンチフェン純のために適切な保険範囲、精算レベルと定価政策を制定する程度にある程度依存する。十分なアンテフェンタニル保険や精算を獲得または維持できなければ、承認されれば、私たちのアンテフェンタニルを販売する能力を制限し、収入を創出する能力を低下させる可能性がある。
承認されれば,政府医療計画(例えばMedicareやMedicaid),個人健康保険会社や他の第三者支払者が提供する保険や精算の可用性と十分性は,多くの患者にとってアンテフェンタニルなどの処方薬を負担できる鍵となる。私たちが政府当局、個人健康保険会社、他の組織が受け入れられるカバー範囲と精算レベルを達成できるかどうかは、私たちがアンテファンを商業化する能力に影響を与えることになる。仮に第三者支払者がアンテファンドリンの保険を獲得したとすると,それによる精算支払率が十分でない可能性があるか,あるいは患者が受け入れられないほど高いと考えられる共同支払いが必要となる可能性がある。また,医師の監督下で管理されている薬品や生物製品では,保険や適切な補償を得ることは特に困難である可能性があり,このような製品は価格が高いことが多いからである。私たちは、アメリカ、EU、あるいは他の場所の保険と精算が、アンテフェンタニルまたは私たちが開発する可能性のある任意の製品に適用され、将来的に入手可能ないかなる精算も減少またはキャンセルされる可能性があることを保証することはできない。
第三者支払者はますます薬品とサービスの価格に挑戦するようになり、多くの第三者支払者は同等の後発薬あるいはより安い治療法が利用可能な場合、特定の薬物への保険と精算を拒否する可能性がある。第三者支払者はアンテフェンタニルが代替可能であると考え,患者に安い製品のみを精算することを提案する可能性がある。たとえ私たちがアンチフェンタニルの治療効果の改善或いは投与の利便性を示しても、現有の薬物の定価は私たちのアンテフェンタニルに対する料金を制限するかもしれない。これらの支払者は、特定の製品の精算状態を拒否または撤回したり、新製品や既存市場製品の価格を低すぎるレベルに設定したりする可能性があり、アンテファニンへの投資から適切なリターンを実現することができない。精算が得られない場合や限られたレベルに限られていれば、アンテフェンタニルを商業化することに成功できないかもしれないし、満足できる財務的見返りが得られない可能性もある。
新たに承認された製品の保険カバーと精算に関する不確実性が大きい。米国では,連邦医療保険や医療補助計画のような第三者支払者,個人や政府支払者を含め,新薬や生物製品のカバー範囲を決定する上で重要な役割を果たしている。連邦医療保険や医療補助計画は,個人支払者や他の政府支払者が薬品や生物製品の保険·精算政策をどのように策定するかのモデルとして利用されるようになってきている。一部の第三者支払者は、新しいまたは革新的な設備または薬物療法の保証範囲を事前に承認し、その後、このような治療法を使用する医療提供者に精算する必要があるかもしれない。第三者決済者がアンテフェンタニルのカバー範囲や補償についてどのような決定を下すかは予測が困難である。
精算状態を取得して維持するのに時間もかかりお金もかかる。米国の第三者支払者には、統一された製品保証や精算政策はない。そのため、製品の保証範囲と精算範囲は支払人によって異なる。そのため、保証範囲の決定過程はしばしば時間と高価な過程であり、それぞれの支払人にアンチフェンを使用する科学と臨床支持を提供する必要があり、保証範囲と適切な補償が一致した応用を得るか、あるいは最初に補償を受けることを保証することができない。また、精算に関する規則や条例は常に変化しており、場合によっては短時間で通知されており、これらの規則や条例が変わる可能性があると考えられる。具体的には,アンテファニンはMedicare Part BやMedicare Advantage計画の下で精算され,これらの計画下での製品精算方式が変化する可能性があり,これらの変化がアンテフェンタニルの将来の全体カバー範囲に影響する可能性が考えられる
米国以外では、国際業務は通常、広範な政府価格制御と他の市場規制を受けており、ヨーロッパや他の国のコスト制御措置の日々の重視は、アンテフェンタニルの定価と使用に圧力を与え続けていると考えられる。多くの国では、国家衛生システムの一部として、医療製品の価格は異なる価格制御メカニズムの制約を受けている。他の国は会社が自ら医療製品を価格設定することを許可しているが、会社の利益を監督してコントロールしている。外国の追加価格規制や定価法規の他の変化は、私たちがアンテフェンタリンに対して受け取ることができる金額を制限するかもしれません。そのため,米国以外の市場では,米国に比べてアンテフェンタニルの精算が減少する可能性があり,商業的に合理的な収入や利益を生み出すには不十分である可能性がある。
また、米国と国外の政府と第三者支払人が医療コストの制限或いは低減を強化することは、これらの組織が新承認製品のカバー範囲と精算レベルを制限する可能性があるため、彼らは十分なアンテフェンタニル支払いをカバー或いは提供できない可能性がある。管理式ヘルスケアの傾向,ヘルスケア組織の日増しに増加する影響力,追加的な立法変化により,アンテフェンタニル販売に関する価格設定圧力に直面することが予想される。全体的に,医療コストの下振れ圧力は非常に大きくなり,特に処方薬,外科手術,その他の治療が行われている。そのため、新製品の参入にはますます高い壁が設けられている。
また、1種類の薬品がEUでマーケティング許可を得たとしても、適時あるいは根本的にその製品の精算を保証することはできない
Ensifentineは市場に受け入れられないかもしれないが、この場合、私たちが製品収入を生み出す能力は影響を受けるだろう。
FDAまたは任意の他の規制機関がアンテフェンタニルの発売を許可したとしても、私たちが独自に開発したものであっても、パートナーと協力して開発したものであっても、医師、医療保健提供者、患者、または医学界はアンテフェントンを受け入れたり使用したりしない可能性がある。もしアンテフェンタリンが十分な受容度に達していなければ、私たちは著しい製品収入やいかなる運営利益も生じないかもしれない。アンテフェンタリンに対する市場の受け入れ度は様々な要素に依存するだろう
•市場が発売されるタイミング
•競争製品の数量と臨床概要
•アンテフェンタニルの臨床適応を承認しました
•私たちは受け入れられる安全性と有効性の証拠を提供する能力;
•副作用の流行率や重症度は
•比較的便利で頻繁で管理が容易である
•費用対効果
•マーケティング、販売、流通支援
•健康維持組織および他の公共および個人保険会社から十分な保険、補償、および十分な支払いを受ける;
•代替治療法と比較した他の潜在的優位性。
もしアンテフェンタニルが市場に認められなければ,われわれの創収能力に悪影響を及ぼす。アンテフィンドリンが市場に認められても、市場は十分ではなく、私たちに相当な収入を与えるのには十分ではないかもしれない。
私たちは現在、販売、マーケティング、運営、流通、精算インフラを含む私たちの商業能力とインフラを発展させています。もし私たち自身があるいは協力を通じて商業能力とインフラを成功的に発展させることができなければ、販売、マーケティング、運営、流通、精算能力を含めて、私たちはアンテフェンタニルを商業化することに成功できないかもしれない。
私たちは販売、マーケティングと運営、流通と精算能力とインフラを発展させています。私たちは今まで薬品をマーケティング、販売あるいは流通したことがありません。販売、マーケティング、運営、流通と精算を含む商業能力とインフラを確立し、アンテフェンタリンを商業化するための技術専門長と支援流通能力を持っていることは、高価で時間がかかる。この費用の一部または全部はアンテファンドリンを承認する前に発生した。また,十分な規模や狙っている医療市場で十分な専門知識を持つ販売チームを採用することはできないかもしれない。私たち自身または協力を通じて内部販売、マーケティング、および流通能力を発展させる任意の失敗または遅延は、アンテフェンタニルの商業化に悪影響を及ぼすだろう。
マーケティング、販売、流通について合意した協力協定について言えば、承認されれば、私たちの製品収入は私たちが直接販売したり、販売したりする製品収入よりも低いかもしれません。さらに、私たちが得たどんな収入もこれらの第三者協力者の努力に全部または部分的に依存し、これらの努力は成功しないかもしれないし、一般的には私たちの統制範囲内ではないかもしれない。もし私たちが受け入れ可能な条件やこれらの計画を全く達成できなければ、私たちはアンテフェンタニルを商業化することに成功できないかもしれない。もし私たちが単独でまたは1つ以上の第三者と協力することでアンテフェンタニルを商業化することに成功できなければ、私たちの未来の製品収入は影響を受け、私たちは重大な追加損失を受けるかもしれない。
私たちの第三者への依存に関するリスク
我々は依存し,独立した臨床研究者とCROを含めて第三者に依存し続け,われわれの臨床前研究と臨床試験を行う予定である。もしこれらの第三者が彼らの契約義務を成功的に履行できなかったり、予想された最終期限までに完成しなかった場合、私たちは規制部門のアンテフェンの承認を得られなかったり、商業化されたりすることができず、私たちの業務は実質的に損害を受ける可能性がある。
著者らは独立した臨床研究者とCROを含む第三者に依存し、引き続き依存し、著者らの臨床前研究と臨床試験を行い、そして著者らが行っている臨床前と臨床計画のモニタリングと管理データを提供してきた。著者らはこれらの方面に依存して、著者らの臨床前研究と臨床試験を実行し、そして彼らの活動のいくつかの方面だけを制御する。しかし、私たちは私たちのすべての研究と実験が適用された合意と法律、法規、そして科学的基準に従って行われ、私たちのこれらの第三者への依存が私たちの規制責任を免除しないことを確実にする責任がある。我々と我々の第三者請負業者およびCROは、FDAが実行する法規およびガイドライン、および私たちの臨床開発におけるすべての製品に対する同様の外国規制機関であるGCP要求の遵守を要求されている。規制機関は,試験スポンサー,主要調査者,試験地点を定期的に検査することでこれらのGCP要求を実行する。もし私たちの任意のCROを十分に監視できなかった場合、または私たちまたは私たちの任意のCROが適用されたGCP要件を遵守できなかった場合、私たちの臨床試験で生成された臨床データは信頼できないと考えられる可能性があり、FDA、EMAまたは同様の外国の規制機関は、私たちの上場申請を承認する前に追加の臨床試験を行うことを要求するかもしれない。私たちまたは私たちのCROまたは私たちの臨床試験に関連するサービスを提供する他の第三者の監督検査を保証することができない場合、これらの規制機関は、私たちの任意の臨床試験がGCP規定に適合していると判断するであろう。また,われわれの臨床試験は適用されたcGMPや類似した海外法規により生産された製品を用いて行わなければならない。私たちがこれらの規定を遵守しないことは、私たちが臨床試験を繰り返す必要があるかもしれないが、これは規制部門の承認過程を遅らせるだろう。
また,これらの研究者やCROは我々の従業員ではなく,契約を除いて,時間を含めてアンテフェンタニルや臨床試験のための資源数を制御することはできない。もし独立した研究者やCROがアンテフェンタニルを開発するのに十分な資源を投入していない場合、あるいは彼らの表現が基準に達していない場合、アンテフェンタニルの承認と商業化の将来性を延期または損害する可能性がある。また、第三者サービス提供者を使用して、これらの側に独自の情報を開示することを要求することは、これらの情報が流用されるリスクを増加させる可能性がある。
私たちの既存と未来のCROは、治癒されていない重大な違約事件が発生したときに私たちとの合意を終了する権利があるか、または可能性がある。また、私たちの臨床試験に参加した被験者の安全が合意を終了する必要があることを合理的に証明できれば、債権者の利益のために一般的に譲渡するか、または清算されれば、私たちのCROのいくつかは私たちとの合意を終了することができる。
もし私たちがこれらの第三者CROとの任意の関係が終了すれば、私たちは代替CROと合意したり、商業的に合理的な条項でそうすることができないかもしれない。CROを交換したり増加させたりすることは追加的なコストに関連し、管理職の時間と労力を必要とする。しかも、新しいCROが仕事を始める時、自然な過渡期がある。したがって,遅延が生じる可能性があり,期待される臨床開発スケジュールを満たす能力に実質的な影響を与える可能性がある。さらに、もし私たちのCROがその契約の義務または義務を成功裏に履行できなかった場合、あるいは予想された期限内に達成できなかった場合、または彼らが得た臨床データの品質または正確性が私たちの臨床方案、規制要求または他の原因を遵守できなかった場合、私たちの臨床試験は延長、遅延または終了される可能性があり、私たちは監督部門のアンテフェンの承認を得ることができないか、または商業化できない可能性がある。したがって、私たちの運営結果とアンテファタリンのビジネス見通しは損なわれ、私たちのコストは増加する可能性があり、私たちの収入を創出する能力は延期されるかもしれない。
Nuance Pharmaとの協力と許可協定は私たちの業務に非常に重要だ。Nuance Pharmaがアンテフェンタリンを含む製品を大中華区で開発および販売できない場合、もし私たちまたはNuance PharmaがNuanceプロトコルを十分に履行できなかった場合、または私たちまたはNuance PharmaがNuanceプロトコルを終了した場合、私たちの業務は不利な影響を受けるだろう。
吾らとNuance Pharmaは2021年6月9日に発効した協力及び許可協定(“Nuance協定”)を締結し、この合意に基づいて、吾らはNuance Pharmaが中国大区(中国、台湾、香港及びマカオ)でアンテフェンを含む製品(“Nuance許可製品”)を独占的に開発及び販売する権利を付与した。
Nuanceプロトコルは、双方が事前に終了しない限り、各司法管轄区域および各製品に基づいて、当該司法管轄区域内の製品の印税支払い義務が満了するまで継続する。どちらももう一方が治癒していない重大な違約や破産によりNuanceプロトコルを終了することができる。Nuance Pharmaはまた、90日前の書面通知後にNuanceプロトコルを任意に終了することができる。
Nuance協定を中止することは、大中華区の中国でNuance許可製品を開発と商業化する能力に重大な挫折を招く可能性がある。適切な代替協力または許可協定は交渉にかなりの時間を必要とし、私たちにあまり有利ではない条項で行われるかもしれない。また,Nuanceプロトコルによると,Nuance Pharmaは大中華区におけるNuance許可製品の中国での臨床開発と商業化に関するすべてのコストを負担することに同意した。Nuanceプロトコルが終了すれば、他の適切なパートナーを見つけるか否かにかかわらず、大中国地域におけるNuanceライセンス製品の臨床開発および商業化を支援するための追加融資を求める必要がある可能性があり、これは私たちの業務に大きな悪影響を及ぼす可能性がある。
Nuanceプロトコルによると、私たちはNuance Pharmaに依存してNuanceライセンス製品の開発に成功し、商業化した。Nuance Pharmaと共同指導委員会を設立して、Nuanceライセンス製品の大中華地区における臨床開発と商業化の全体的な進行を監督·調整しているが、Nuance Pharmaの開発と商業化のすべての側面を制御しておらず、Nuanceプロトコルの下で決定されたNuanceライセンス製品開発のリソースに割り当てることも制御していない。私たちの利益とNuance Pharmaの利益は時々違いや衝突が発生するかもしれないし、あるいは私たちはNuance Pharmaの努力レベルや資源分配に同意しないかもしれない。Nuance Pharmaは、我々とは異なる方法で、連携中に開発されている計画を内部優先順位付けすることができ、またはNuance許可製品を効率的または最適化するために十分なリソースを割り当てていない場合があり、または商業化する可能性がある。これらの事件が発生すれば、Nuanceライセンス製品の商業化から収入を得る能力が低下し、私たちの業務は悪影響を受けるだろう。また、Nuanceプロトコルによると、私たちはNuance Pharmaの中国大区での開発と商業化活動にイソニアジド薬物製品を提供する責任があり、もし私たちの供給価格が高すぎる場合、Nuance Pharmaが大中国地域でこの薬品を販売する価格は競争力がない可能性があり、これはNuance PharmaがNuance許可製品の商業化に成功した能力とNuance合意による私たちのリターンに重大な悪影響を及ぼす可能性がある。また、Nuance Pharma臨床開発活動の安全性および/または有効性データは様々な原因で私たちのデータと異なる可能性があり、私たちの臨床開発と商業化活動に影響を与える可能性があり、私たちがアメリカと他の国/地域の監督機関がアンテフェンタニルを承認する能力を獲得することを含む。
もし私たちがアンテファン林のために新しい戦略関係を築くことができなければ、私たちの業務、研究開発、商業化の将来性は不利な影響を受ける可能性がある。
私たちのアンテフェンタリン開発計画とアンテフェントーリンの潜在的な商業化には費用を支払うために多くの追加の現金が必要になるだろう。そこで私たちは製薬や生物製薬会社と協力して潜在的な商業化を開発することにしたかもしれません
アンテファニン。例えば、COPDおよび潜在的喘息および他の呼吸器疾患の維持治療のためのパートナーを探すことができる。
私たちは適切な協力者を探すことで激しい競争に直面している。協力の交渉と記録は複雑で時間がかかる。既存と未来の協力協定によると、私たちはまた制限される可能性があり、他の潜在的な協力者と特定の条項の合意を締結することはできません。私たちは受け入れ可能な条件で協力を交渉することができないかもしれないし、根本的にできないかもしれない。このような状況が発生した場合、アンテファニンの開発を削減し、その開発計画を減少または延期し、潜在的な商業化を延期したり、販売やマーケティング活動の範囲を縮小したり、私たちの支出を増加させたり、自費で開発または商業化活動を行わなければならない可能性がある。もし私たちが私たちの支出を増やし、私たち自身の開発や商業化活動に資金を提供することを選択すれば、私たちは追加の資本を得る必要があるかもしれないし、これらの資本は受け入れ可能な条件で私たちに提供できないかもしれないし、全く得られないかもしれない。もし私たちが十分な資金がなければ、私たちはイソニアジドを市場に出して製品収入を作ることができないだろう。もし私たちが協力合意に達したら、私たちは以下のリスクに直面するかもしれません。そのいずれも、私たちがイソフェンを開発し、商業化する能力に悪影響を及ぼす可能性があります
•アンテファンドリン開発に協力者が投入した資源の数と時間をコントロールできないかもしれません
•協力者は経済的困難に直面するかもしれません
•私たちはマーケティング、流通、知的財産権などの重要な権利を放棄することを要求されるかもしれない
•協力者は、我々の競争相手を含む第三者と協力して競合製品を開発または開発することができる
•協力者の臨床開発活動の安全性および/または有効性データは私たちのデータと衝突する可能性があり、潜在的に私たちの世界の臨床開発と商業化活動に影響を与える可能性がある
•協力者は私たちに対する守秘義務に違反し、機密情報と材料を不正に使用または漏洩する可能性がある
•業務合併または協力者の業務戦略の重大な変化は、任意の手配の下で義務を履行する私たちの意志に悪影響を及ぼす可能性がある
•私たちまたは協力者は、合意によって規定された義務を十分に履行できない可能性があり、および/または合意が議論される可能性がある
•私たちは、アンテファンをカバーする特許を保護または強制的に執行する訴訟、または高価で時間がかかり、成功していないかもしれない私たちの協力条項に関連する訴訟に巻き込まれるかもしれない
•Ligand PharmPharmticals、Inc.またはLigandとの合意での支払い義務を履行した後、協力して利益を得るのに十分な資金を提供できないかもしれません。Ligandは2018年10月にVernalis Development LimitedまたはVernalisを買収しました。
著者らは現在、第三者メーカーとサプライヤーに依存して、活性薬物成分であるアントトーリン及びその派生製剤製品を生産している。これらの第三者への依存は,我々の研究開発計画の進展やアンテフェンタニルの開発を損なう可能性がある。さらに、承認された場合、私たちは第三者生産アンテファニンの商業供給に依存する予定であり、これらの第三者がFDAまたは同様の規制機関の必要な承認を得られなかった場合、十分な量の製品をタイムリーに提供できなかった場合、許容可能な品質レベルまたは価格で製品を提供することができなかった場合、または他の方法で彼らの私たちまたは他の当事者への義務を達成できなかった場合、商業化は阻止され、延期され、または利益が低下する可能性がある。
私たちはアンテフェンタリンとその派生調合品を生産する施設がない。対照的に、承認された場合、私たちは、cGMPまたはGMP級臨床試験材料および商業数量のアンテファニンおよびその由来製剤製品を供給する第三者契約製造組織(CMO)に依存し、引き続き依存すると予想される。将来的には他のCMOと契約する可能性があるが,現在1つのCMOがプロポフォール薬剤の生産を担当しており,1つのCMOが各製剤のプロポフォールの生産を担当している。アンチフェンおよびその由来製剤製品を製造するための施設は、FDAの承認を得なければならず、検査は、我々がFDAに機密協定を提出した後に行われ、米国国外の類似した外国規制機関の承認を得る。私たちはスポンサーによる生産活動の監督を提供しているが、私たちは私たちのCMOの生産過程を直接制御することもなく、基本的に私たちのCMOがcGMPと海外のアンテフェンタリンの生産およびその派生調合に対する類似の要求に適合しているかどうかに依存する
製品です。CMOが我々の規範およびFDAまたは外国規制機関の規制要件に適合する材料を成功的に生産できない場合、それは、その製造施設におけるアンテフェンタニルおよびその由来製剤製品の生産の規制承認を確保または維持することができないであろう。また,CMOが十分な品質管理,品質保証,合格者の能力を維持することはほとんど直接制御されていない。FDAまたは同様の外国の規制機関が、エフェントーリンおよびその派生製剤を生産するためにこれらの施設を承認しない場合、または将来的にそのような承認を撤回する場合、私たちは代替製造施設を探す必要があるかもしれません。これは、私たちの開発計画を延期し、規制承認を得たり、エフェントーリンおよびその派生製剤製品を販売する能力に深刻な影響を与えます(承認されれば)。さらに、これらの法律、法規、および基準を達成し、維持することができなかった場合は、アンテフェンタニルおよびその由来製剤製品の生産を一時停止し、または承認された製品が撤回される可能性があるというリスクに直面しなければならない可能性がある。さらに、第三者プロバイダは、私たちがコントロールできない要素のために、彼らと私たちとの間の既存の合意に違反するかもしれない。彼らはまた、自分の財務的困難またはビジネス優先事項のために、契約の更新を終了または拒否する可能性があり、この場合、私たちにとってコストが高いか不便である。もし私たちが十分な代替品や他の許容可能な解決策を見つけることができなければ、私たちの臨床試験は延期されるかもしれないし、私たちの商業活動は損害を受ける可能性がある。また、私たちは、私たちのサプライヤー、CMO、および他の第三者に依存して、イソフェントーリンおよびその派生製剤製品を製造、貯蔵、流通することは、私たちがイソフェントーリンおよびその派生製剤製品に製造欠陥が存在するリスクに直面する可能性があり、私たちがこれらの欠陥を予防、検出、または制御する能力が限られていることを意味する。
我々は、CMOに依存して第三者サプライヤーからアンテフェンタニル及びその由来製剤製品を生産するために必要な材料を購入し、アンチフェンタニルの吸入及び噴霧装置を提供することに依存し、引き続き依存する。私たちは、CMOまたはその第三者サプライヤーがこれらの物資を調達して配送するプロセスや時間、またはそのような物資の品質または数量を直接制御することもないだろう。これらの供給は時々中断される可能性があり、中断すれば、合理的な時間範囲内で許容可能なコストまたは品質で代替供給を得ることができるか、または根本的に不可能であることを保証することはできない。私たちはアンテフェンの原料と臨床試験のための薬物送達設備(例えば噴霧器)を生産するための供給者の数が限られていて、私たちの臨床試験が中断する可能性があることを防止するために代替サプライヤーを評価する必要があり、承認されれば、最終的には商業販売に使用される。著者らは一般的に臨床試験を開始しないが、著者らは私たちの手元に十分なイソフェント品供給を得て臨床試験を完成することができると信じない限り、しかし、著者らのCMO或いはその第三者サプライヤーが行っている臨床試験中のイソフェントロントの供給、イソフェント品の生産に必要な原材料成分或いは輸送装置のいかなる重大な遅延も、私たちの臨床試験、製品テストと監督部門のイソフェント品に対する承認を大幅に延期する可能性がある。もし私たちのCMO、彼らの第三者供給、あるいは私たちが監督部門の許可を得た後にこれらの供給を購入できなければ、アンテフィンの商業発売が延期されたり、供給不足が発生したりすることは、私たちがアンテフィンを販売することから収入を得る能力を弱めるだろう。また、これらの供給品のコストと費用の増加は、私たちが経済的に効率的な方法でアンテフェンタニルを生産する能力を弱める可能性がある。また,CMOは労働力制限を経験しており,アンテフェンタニルを製造·輸送する能力に影響を及ぼす可能性がある。
私たちはCMOと第三者サプライヤーが契約義務を履行する際に他人の所有権を遵守し、尊重することに依存し、引き続き依存するだろう。CMOまたは第三者サプライヤーが私たちにサービスを提供している間に適切なライセンスを取得できなかった場合、または他の方法で第三者の固有の権利を侵害した場合、CMOまたは第三者サプライヤーの代わりを探し、または侵害クレームに対して抗弁しなければならない可能性があり、承認された場合、これら2つの場合は、私たちが監督管理の承認を得ること、または管理許可を得ること、またはそれらの任意の派生製剤製品を販売する能力に深刻な影響を与える。
知的財産権や情報技術に関するリスク
私たちは特許と他の知的財産権に依存してアンテフェンタリンを保護し、その実行、弁護、メンテナンスは挑戦的でコストがあるかもしれない。これらの権利を十分に実行または保護できなければ、私たちの競争能力を損なう可能性があり、私たちの業務を損なう可能性がある。
我々の商業的成功は、アンチフェンの特許および他の形態の知的財産権の取得および維持にある程度依存し、アンチフェンの配合、アンチフェンの多形体、塩類および類似体、アンチフェンを製造するための方法、噴霧器、DPI、PMDIなどの異なる吸入器のための最終医薬製品の製造方法、およびアンチフェンニン単独または他の既存製品との併用による呼吸器疾患の治療方法、またはそのような権利の許可を得るためのものである。これらの特許及び特許出願は,ある司法管区の関係当局の譲渡及び特許出願の登録が承認されたが,いかなる保証もない
追加的な登録はタイムリーに施行されるか、完全に施行されないだろう。十分な特許および他の知的財産権を保護または取得、維持または延長することができない場合、エステファン塩基の開発および販売能力に悪影響を及ぼす可能性がある。
特許訴訟プロセスは高価で時間がかかり、私たちまたは私たちのライセンシー、ライセンシーまたは協力者は、すべての必要または望ましい特許出願を合理的なコストで、またはタイムリーに、またはすべての司法管轄区域で準備、提出および起訴することができない可能性がある。私たちまたは私たちのライセンシー、ライセンシーまたは協力者も、特許保護を受ける前に、開発および商業化活動中に行われた発明の出願可能な特許の態様を決定することができない可能性がある。さらに、私たちが将来加入する可能性のある任意の内部許可の条項によれば、場合によっては、特許出願の準備、提出および起訴を制御する権利がないか、または第三者から内部許可を得る技術を含む特許を維持する権利がない可能性がある。したがって、このような特許および出願は、私たちの業務の最適な利益に合った方法で起訴され、強制されてはならない。さらに、私たちと現在または未来のライセンシー、ライセンシーまたは協力者の特許権の発行、範囲、有効性、実行可能性、および商業的価値は非常に不確実である。私たちと私たちの許可者たちの未解決および将来の特許出願は、私たちを保護する技術または製品の全部または一部の特許を発行すること、または他社が競争技術および製品を商業化することを効果的に阻止することにつながる可能性があります。特許審査プロセスは、私たちまたは私たちのライセンシー、ライセンシーまたは協力者が、私たちまたは私たちのライセンシー、ライセンシーまたは提携者の係属中および将来の特許出願の特許請求範囲を縮小することを要求することができ、これは、入手可能な特許保護範囲を制限する可能性がある。私たちは私たちの特許と特許出願に関連するすべての潜在的に関連する既存技術が見つかったことを保証することはできない。そのような従来技術が存在する場合、それは、特許を無効にするか、または係属中の特許出願から特許を発行することを阻止することができる。特許が確実に発行されても、このような特許がプロポフォールをカバーしていても、第三者は、反対、干渉、再審査、許可後審査、当事者間の審査、法廷または特許庁で提起された無効または派生訴訟、または同様の手続きを開始することができ、このような特許の有効性、実行可能性または範囲を疑問視することができ、これは、特許請求の範囲の縮小または無効をもたらす可能性がある。私たちおよび私たちのライセンシー、ライセンシーまたは協力者の特許出願は、特許が他の出願から発行されるまで、発行された特許要件が技術の範囲をカバーしない限り、これらの出願に必要な技術を実施する第三者に対して強制的に実行することはできない。
特許出願は出願後しばらくは秘密であるため、一部の特許出願は発行前には依然として秘密であり、私たちまたは私たちの許可者がアンテビリン関連の特許出願を最初に提出した会社であることは確認できない。さらに、第三者が2013年3月15日または以前にこのような特許出願、すなわち米国特許出願制度が先行発明から先行出願基準に変更された日を提出した場合、そのような第三者は、誰が私たちが出願した特許請求の範囲によってカバーされる任意の主題を最初に発明したかを決定するために、干渉手順を開始することができる。第三者が2013年3月15日以降にこのような出願を提出した場合、そのような第三者は、私たちの発明が彼らの発明から来たかどうかを決定するために派生プログラムを開始することができる。私たちが効果的で強制的に実行可能な特許を持っている場合であっても、他の当事者が彼らが私たちの出願日前にその発明を商業に使用していることを証明することができれば、または他方が強制許可から利益を得ることができれば、他の人が私たちの発明を実践することを排除することはできない。
私たちは、関連する第三者特許を識別しないか、または第三者特許の関連性、範囲、または満了時間を誤って解釈する可能性があり、これは、エステファン塩基を開発、製造および販売する能力に悪影響を及ぼす可能性がある。
私たちは、関連特許の識別、特許請求の範囲、または関連特許の満了を含むが、関連特許の範囲または関連特許の満了を含むが、私たちまたは私たちの許可者の任意の特許検索または分析を保証することはできず、私たちは、任意の司法管区のアンテファンドリンの商業化に関連する、または必要な、米国および海外の各第三者特許および係属中の特許および係属中の出願を識別したことを確実にすることはできない。例えば、2000年11月29日までに提出された米国出願と、その日以降に提出されたいくつかの米国出願は、特許発行前に米国国外では提出されないが、依然として秘密にされるであろう。米国および他の地方の特許出願は、優先権を要求する最初の出願の約18ヶ月後に発行され、このような最も早い出願日は、一般に優先権日と呼ばれる。したがって、アンテフェンタリンに関する特許出願は、私たちが知らずに他の人によって提出されたかもしれない。さらに、いくつかの制限の下で、開示された係属中の特許出願は、後に修正することができ、修正された方法は、イソフェントレントまたはイソフェントレントの使用を含むことができる。特許請求の範囲は、法律の解釈、特許における書面開示、および特許の起訴履歴に依存する。特許や係属出願の関連性や範囲の解釈は正しくない可能性があり,これはイソフェンタニルを販売する能力に悪影響を及ぼす可能性がある。アンテファニンが第三者特許のカバー範囲内にないか、または第三者の保留出願が関連範囲の特許要件を提示するかどうかを誤って予測する可能性がある。米国や海外のいずれかに関連する特許の満期日の決定は正しくない可能性があり,これはわれわれのイソフェンタニルの開発·販売能力に悪影響を及ぼす可能性がある。もし私たちが関連特許を識別して正確に解釈できなければ、私たちがエフィチリンを開発·販売する能力に悪影響を及ぼすかもしれない。
もし私たちが関連特許を識別して正確に解釈できなければ、私たちは侵害請求を受けるかもしれない。私たちは私たちがこのような侵害請求を成功的に解決したり、他の方法で解決できるという保証はない。もし私たちがこのような紛争で失敗すれば、損害賠償金の支払いを余儀なくされるほか、アンテフェンタニルの商業化を一時的または永久的に禁止される可能性がある。可能であれば、私たちは第三者の知的財産権を侵害しないように、アンテファンドリンの再設計を余儀なくされるかもしれない。これらのすべての事件は、私たちが最終的に勝っても、私たちが大量の財務と管理資源を移転する必要があるかもしれません。そうでなければ、私たちは私たちの業務に投入することができます。
私たちは、アンテファンドリンをカバーする特許を保護または強制執行する訴訟に巻き込まれるかもしれないが、これは高価で、時間がかかり、成功していない可能性があり、法廷で疑問が提起された場合、発行された特許が無効または実行不可能であることが発見される可能性がある。
私たちの競争的地位を保護するためには、私たちが私たちに所有または権限を与えてくれた任意の特許または他の知的財産権を強制的に実行または保護するために、時々訴訟に訴える必要があるかもしれないし、第三者特許または他の知的財産権の範囲または有効性を決定または疑問視する必要があるかもしれない。知的財産権の実行は困難で、予測不可能で、時間がかかり、費用がかかるため、私たちは私たちの権利を実行できないかもしれない-この場合、私たちの競争相手は、私たちにいかなる許可料も支払うことなく、私たちの技術の使用を許可されるかもしれない。しかしながら、さらに、私たちの特許に関連する訴訟は、私たちの1つまたは複数の特許が無効(逐一請求に基づいて、全部または部分的に無効)と認定されるか、または強制的に実行できないリスクがある。このような不利な裁判所判決は、第三者がアンテビリンを商業化し、私たちに支払うのではなく、私たちと直接競争することを可能にするかもしれない。もし私たちが知的財産権を許可すれば、私たちの合意は私たちのライセンシーに第三者侵害請求を統制したり、有効性挑戦を正当化することができるかもしれない。したがって、これらの特許および特許出願は、私たちの業務の最適な利益に適合した方法で強制的に実行または弁護されないかもしれない。
もし私たちが第三者に対して私たちの製品の一つをカバーする特許を強制的に執行するために法的訴訟を提起すれば、被告は私たちの特許を無効または強制執行できないと反訴することができる。米国やヨーロッパの特許訴訟では,被告が無効または実行不可能と主張する反訴が一般的である。有効性を疑問視する理由は、新規性の欠如、明らかな、または実施できないなど、いくつかの法定要件のいずれかを満たすことができなかったと言われている可能性がある。実行不可能な断言の理由は,特許起訴に関連する者が起訴中に米国特許商標局に関連情報を隠蔽したり,誤った声明を発表したりしたためかもしれない.特許訴訟の間、法律が無効と主張した後の結果は予測できない。例えば、有効性の問題については、私たちと特許審査員が起訴中に知らない無効な以前の技術がないことを確認することはできない。もし被告が無効または強制執行できない法的主張で勝訴した場合、私たちはアンテフェンタリンに関する特許保護の少なくとも一部、さらにはすべてを失うだろう。もし競争相手が私たちの特許や他の知的財産権を侵害することなく私たちが保護された技術を中心に設計すれば、特許や他の知的財産権も私たちの技術を保護しないだろう。
さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。聴聞、動議、または他の一時的手続き、または事態発展の結果を公開することもできる。証券アナリスト、業界コメンテーター、または投資家がこれらの結果がマイナスだと考える場合、私たちのアメリカ預託証明書の価格に悪影響を及ぼす可能性がある。
第三者は私たちが彼らの知的財産権を侵害したことを告発する法的訴訟を提起する可能性があり、その結果は不確定であり、私たちの業務成功にマイナスの影響を与える可能性がある。
私たちのビジネス成功は、第三者の特許および独自の権利を侵害したり、実際に侵害されたり、流用したり、または他の方法で第三者の特許および固有の権利を侵害することなく、私たちの能力、および私たちの将来のパートナーの開発、製造、マーケティング、および私たちの候補製品を販売する能力にある程度依存する。バイオテクノロジーおよび製薬業界では、米国特許商標局および対応する外国特許庁に提起された特許侵害訴訟、妨害、反対および再審手続きを含む特許及び他の知的財産権に関する訴訟や他の手続が多い。私たちはその中で運営される各市場が頻繁で広範な特許と他の知的財産権訴訟に直面していることを計画している。また、バイオ製薬や製薬業界を含む知的財産権に依存する業界の多くは、競争相手に対する優位性を得るための手段として知的財産権訴訟を利用している。われわれがプロポフォールを開発している分野には,第三者が所有する米国や外国から発行された特許や係属中の特許出願が多く存在する。一部のクレーム者は私たちよりもはるかに多くの資源を持っている可能性があり、私たちよりも複雑な知的財産訴訟費用をより大きく長時間受けることができるかもしれない。また,特許権の強制執行による特許料の抽出と和解にのみ重点を置いた特許持株会社が我々を対象とする可能性がある。バイオテクノロジーや製薬業界の拡大に伴い
また,発行された特許が多ければ多いほど,アンチフェンが第三者の知的財産権侵害疑惑を受ける可能性が高いリスクが大きくなる。
将来的には、私たちは、妨害または派生訴訟、認可後審査、および米国特許商標局で行われる当事当事者審査、または他の司法管轄区域の同様の対抗訴訟または訴訟を含む、アンチフェン亭および任意の将来の候補製品に関連する知的財産権に関する対抗訴訟または訴訟に参加または関与することを脅かす可能性がある。同様に、私たちまたは私たちのライセンシーまたは協力者は、第三者に対して、例えば、第三者によって制御される知的財産権の有効性または範囲に挑戦するような訴訟または訴訟を提起することができる。第三者は、既存の特許または将来付与される可能性のある特許に基づいて、その是非曲直にかかわらず、侵害請求を私たちに提起するかもしれない。第三者が彼らの特許権を強制的または他の方法で主張するために私たちと訴訟を行うことを選択する可能性があるリスクがある。このような主張に法的根拠がないと考えても、管轄権のある裁判所は、これらの第三者特許が有効で、強制的に実行可能であり、侵害されていると判断することができ、そのような特許の所有者は、適用された特許によって許可されたか、またはこれらの特許が満了するまで、最終的に無効または実行不可能と判定されない限り、候補製品を商業化することを阻止することができるかもしれない。同様に、管轄権のある裁判所が、私たちの組成物、処方または治療、予防または使用方法のあらゆる面をカバーするために、任意の第三者特許を保有している場合、そのような特許の所有者は、許可証を取得しなければ、またはその特許が満了するまで、または最終的に無効または実行不可能と判定されない限り、適用可能な候補製品を開発し、それを商業化する能力を阻止することができるかもしれない。このような許可は,合理的な条項で提供されない可能性があるか,提供されないか,あるいは非排他的である可能性があり,我々の競合他社が我々に許可された同じ技術にアクセスできるようにする.
もし私たちがこのような紛争で敗訴した場合、裁判所が私たちが故意に特定の知的財産権を侵害していることを発見した場合、特許事件で3倍の損害賠償金を得る可能性がある損害賠償金の支払いを余儀なくされる可能性がある。私たちまたは私たちの許可者は、アンテフェンを商業化することを一時的または永久的に禁止されているか、または関連する知的財産権を使用する米国および/または他の管轄区域での私たちの製品の販売、合併、製造、または使用を禁止することができる。可能であれば、私たちはまた、第三者の知的財産権を侵害しないようにアンテファンを再設計することを余儀なくされるかもしれません。これは、私たちの重大なコストや遅延を招き、あるいは技術的に再設計することができないかもしれません。これらのすべての事件は、私たちが最終的に勝っても、私たちが大量の財務と管理資源を移転する必要があるかもしれません。そうでなければ、私たちは私たちの業務に投入することができます。
さらに、私たちまたは私たちのライセンシーまたは協力者の特許および特許出願によって提供される保護の広さまたは強度が脅かされている場合、会社が現在または将来の候補製品を許可、開発、または商業化するために、私たちと協力することを阻止するかもしれない。
私たちは私たちの特許と他の知的財産権発明権のクレームに疑問を受けるかもしれない。
私たちは現在、私たちの特許の発明権や私たちの知的財産権の所有権に疑問を提起するクレームを受けていませんが、私たちは将来、元従業員、協力者、または他の第三者が発明者または共同発明者として私たちの特許または他の知的財産権において権利を持っているというクレームを受ける可能性があります。私たちの政策は、知的財産権の概念や開発に参加する可能性のある私たちの従業員と請負業者が合意に署名し、このような知的財産権を私たちに譲渡することを要求していますが、実際に私たちが自分の知的財産権と見なしているすべての当事者とこのような合意を実行することはできないかもしれません。例えば、知的財産権譲渡は自動的に実行されない可能性があり、または譲渡協定が違反される可能性があり、または私たちの候補製品の開発に参加するコンサルタントや他の人の義務衝突によって在庫紛争が生じる可能性があります。訴訟はこれらと他の挑戦在庫のクレームに対抗するために必要かもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭損害賠償を支払うことに加えて、貴重な知的財産権、例えば貴重な知的財産権の独占所有権や使用権を失う可能性がある。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。
知的財産権訴訟は私たちに大量の資源を費やし、私たちの人員の正常な義務に対する注意を分散させるかもしれない。
解決策が私たちに有利であっても、知的財産権クレームに関連する訴訟や他の法的手続きは、巨額の費用を発生させ、私たちの技術や管理者の正常な責任を分散させる可能性があります。さらに、公聴会、動議、または他の一時的な手続きまたは事態の発展の結果が公表される可能性があり、証券アナリストまたは投資家がこれらの結果が否定的であると考える場合、このような見方は、我々の米国預託証明書の価格に大きな悪影響を及ぼす可能性がある。このような訴訟や訴訟は、私たちの運営損失を大幅に増加させ、開発活動に利用できる資源を減少させる可能性がある。私たちはそのような訴訟や法的手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちの競争相手の中にはこのような訴訟や訴訟の費用をより効果的に受けるかもしれません
彼らはより大きな財政資源を持っている。特許訴訟または他の訴訟の開始と継続によって生じる不確実性は、市場での競争能力に悪影響を及ぼす可能性がある。
もし私たちが既存と未来に第三者と達成した任意の知的財産権許可やローン協定で規定された義務を履行できなければ、私たちは私たちの業務に非常に重要な権利を失うかもしれない。
私たちはLigandとライセンス契約に署名し、この合意に基づいて、いくつかの知的財産権の許可を得て、私たちの業務に関連するいくつかの特許および特許出願を割り当てられた。私たちは未来に他の許可協定を締結するかもしれない。私たちは未来のどんな許可協定も、様々な職務調査、マイルストーン支払い、特許権使用料、保険、その他の義務を私たちに強要すると予想している。我々は最近、Oxford Finance LLCおよびHercules Capital,Inc.と定期融資協定を締結した。定期融資は、Verona Pharma,Inc.およびVerona Pharma plcのほとんどの資産(知的財産権を除く)の留置権を保証するものであり、(I)任意の定期融資の融資日は、融資プロトコルによって抽出された未返済定期融資の元金総額が5000万ドルを超えることを前提としており、(Ii)Verona Pharma,Inc.またはVerona Pharma Plcの融資合意で定義された使用料融資の前に定義されている。Verona Pharma,Inc.またはVerona Pharma plcはまたオックスフォードとHercules社の知的財産権に否定的な約束をした。融資協定のさらなる説明については、“リスク要因-私たちの商業·工業に関連するリスク”の節を参照されたい。これらの合意によれば、いかなる治癒されていない重大な違反も、これらの合意下の特許権および他の知的財産権を行使する権利を失う可能性があり、アンチフェンまたは任意の将来の候補製品の開発および商業化への私たちの努力を危うくする可能性がある。私たちがLigandと合意した合意によると、Ligandの同意を得ず、私たちはいかなる譲渡された特許を放棄したり、いかなる譲渡された特許の失効を許可したり、無理な遅延や抑留を許可してはならない。もし私たちがそのような同意をタイムリーにまたは全く得られず、これらの譲渡された特許権が失効または放棄された場合、私たちとLigandとの合意は完全に終了するかもしれない。例えば、もし私たちが商業的な理由で譲渡された特許を商業的重要性が大きくない国で失効させることにした場合、Ligandは同意を提供せず、このような特許権が失効すれば、アンテフェンをカバーするすべての知的財産権を複数の市場で失う可能性がある。さらに、私たちの未来の許可者は、私たちに許可されていない知的財産権を持っているか、またはコントロールしているかもしれないので、その是非にかかわらず、私たちは私たちが許可者の権利を侵害しているか、または他の方法で侵害しているというクレームを受けるかもしれない。
私たちはアンチフェンの必要な権利を維持することに成功したり、買収やライセンス内で私たちの業務に重要な他の知的財産権を得ることができないかもしれない。
私たちは現在、特許、特許出願、およびノウハウを含むアンテファンドリンに関連する知的財産権を持っており、私たちの成功はこれらの権利を維持することにかかっている可能性が高い。私たちの手続きは第三者が持っている独占権を使用する必要があるかもしれないので、私たちの業務の成長は、私たちがこれらの専有権を取得、許可、維持、または使用する能力にある程度依存するかもしれない。さらに、アンテフェンは効果的に機能するために特定の配合物を必要とする可能性があり、これらの製剤の権利は他の人によって保持されている可能性がある。私たちは、任意の組成物、使用方法、プロセス、または他の第三者知的財産権を取得または許可することができないかもしれませんが、これらの知的財産権はアンチフェンに必要であると考えられます。第三者知的財産権の許可·買収は競争分野であり、一部のより成熟した企業も魅力的であると考えられる第三者知的財産権許可または買収戦略をとっている。これらの老舗会社は私たちより競争優位を持っているかもしれません。それらの規模、現金資源及びより強い臨床開発と商業化能力のためです。
しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。私たちはまた、私たちの投資を適切な見返りを得るための条項に従うことができないか、第三者の知的財産権をタイムリーに許可したり、取得することができないかもしれない。私たちが関心のある知的財産権の許可を得ることができても、私たちは独占的な権利を得ることができない可能性があり、この場合、他の人たちは同じ権利を使用して私たちと競争するかもしれない。もし私たちが受け入れ可能な条項でアンテファニンや開発計画の開発に必要な第三者知的財産権の許可を得ることに成功できなければ、アンテファニンやその開発計画の開発を放棄しなければならないかもしれない。
私たちは提案された製品名に対するFDAの承認を得る必要があり、これに関連するどんな失敗や遅延も、私たちのビジネスに悪影響を及ぼす可能性があります。
私たちは、米国特許商標局から正式な商標登録を受けたかどうかにかかわらず、候補製品のために使用される任意の固有名をFDAの承認を得る必要があると考えている。FDAが提案した製品名を審査する際には,他の製品名との混同による医療ミスの可能性も考慮されており,提案された名称が空想しすぎたり,誤って独自の有効性や成分を示唆したり,製品の効果を誇張したり,リスクを最小限に抑えたり,製品適応を拡大したり,実証されていない優位性を示唆したりすることも考えられる。FDAと協力して、アンテフェンタリンのために適切なブランド名を決定しています
これはFDAが受け入れられることだ。もし私たちが受け入れられる名前を決定する上で遅延に遭遇すれば、この遅延はアンテフェンタニルの発売に不利な影響を与える可能性があり、もし承認すれば、製品製造を延期するいくつかの要素を含む
FDAが私たちが提案したどんな製品名にも反対すれば、私たちは私たちの候補製品のために代替名を採用することを要求されるかもしれない。代替名称を採用すれば、候補製品の任意の既存商標出願の利点を失う可能性があり、適用商標法の資格に適合し、第三者の既存の権利を侵害せず、FDAのために受け入れられる適切な製品名を識別するために多くの追加資源が必要となる可能性がある。私たちは新しい商標のための成功したブランド表示をタイムリーにまたは根本的に確立できないかもしれないが、これは候補製品を商業化する能力を制限するだろう
もし私たちの商標と商号が十分に保護されていなければ、私たちは私たちの興味のある市場で知名度を確立することができず、私たちの競争地位は不利な影響を受けるかもしれない。
私たちはすでにいくつかの地域に商標を登録し、他の地域での商標登録を申請し、私たちの業務や提案された医薬製品に潜在的な商標名を提供している。私たちは私たちに非常に重要だと思う地域で私たちの商標名のために商標保護を受けることができないかもしれない。もし私たちが商標を登録すれば、私たちの商標出願は商標登録手続きで拒否されるかもしれない。私たちはこのような拒否に答える機会があるにもかかわらず、私たちはこの拒否を克服できないかもしれない。さらに、我々の任意の商標または商号は、登録または未登録にかかわらず、疑問、反対、侵害、キャンセル、回避、または汎用商標として宣言されるか、または他の商標の侵害と判定される可能性がある。私たちはこれらの商標と商品名に対する私たちの権利を保護できないかもしれません。私たちは私たちが関心のある市場で潜在的な協力者や顧客の知名度を確立するために必要です。長期的には、私たちの商標や商号に基づいて名称を確立することができなければ、効果的に競争できない可能性があり、私たちの業務は悪影響を受ける可能性があります。他のエンティティが異なる司法管轄区域で私たちの商標と類似した商標を使用したり、私たちの商標に対して優先的な権利を持っている場合、私たちが世界各地で現在の商標を使用することを妨害する可能性があります。
もし私たちがハッジ·ワックスマン修正案や同様の非米国立法の保護を受けなければ、アンテファンや他の任意の候補製品をカバーする特許期間を延長すれば、私たちの効果的な競争能力は損なわれる可能性がある。
特許の寿命は限られている。米国では、すべての維持費が適時に支払われる場合、特許の自然失効時間は、通常、米国で最初の非臨時出願日から20年である。アンテフェン浄化物質からなる発行特許は2020年に満了し、私たちの他の発行された特許は2031年から2041年までであり、このような特許の任意の利用可能な特許の延期に応じて決定される。もし我々の未決特許出願が特許発行された場合,それによって生成された特許は2031年から2044年の間に満期になると予想される.様々な延期があるかもしれないが、特許の有効期限とその提供される保護は限られている。アンテフェンタリンをカバーする特許を取得しても、製品の特許有効期限が満了すると、模造薬を含む競争薬の競争に直面する可能性がある。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちが持っている特許と許可された特許の組み合わせは、他社が私たちと似ているか同じ製品を商業化することを排除するために十分な権利を提供してくれないかもしれない。
FDAがアンテファニンの発売を承認した時間、期限、条件によると、私たちの1つ以上の米国特許は、1984年の“薬品価格競争と特許期限回復法”(Hatch-Waxman修正案と略称する)とEUの同様の立法に従って限られた特許期間の延長を受ける資格がある可能性がある。Hatch-Waxman修正案は、製品開発およびFDA規制審査中に失われた有効特許期間の補償として、承認された製品をカバーする特許を最大5年間延長することを可能にする。しかし、適用の最終期限内に出願を提出できなかった場合、関連特許の満了前に出願を提出できなかった場合、または適用の要求を満たしていなかった場合、延期されない可能性がある。しかも、延期の長さは私たちが要求したものより短いかもしれない。もし私たちが特許期間の延長を得ることができない場合、あるいはこのような延長の期限が私たちが要求したものよりも短い場合、私たちはその製品に対して特許権を行使することができる期限が短縮され、競争相手はより早く承認され、競争製品市場に入るかもしれません。したがって、私たちは適用製品からの収入が減少する可能性があり、大幅に減少するかもしれない。
私たちは特定の特許に対して限られた地域保護しか有しておらず、特定の管轄区域で困難に直面している可能性があり、これはこれらの管轄区域における私たちの知的財産権の価値を弱めるかもしれない。
私たちは一般的にイギリス知的財産権局で私たちの最初の特許出願を提出するか、または優先的に申請する。特許協力条約(PCT)に基づく国際出願は、通常、優先権出願後12ヶ月以内に提出される。PCTの出願によると、国および地域特許出願は他の管轄区域に提出される可能性があり、候補製品はこれらの管轄区で上場または製造される可能性があると考えられる。今まではまだ申請していません
このような保護を提供することが可能なすべての国および地域司法管轄区域内でアンテフェンタニルの特許保護が行われている。世界のすべての国で特許を申請、起訴、保護する費用は目を引くほど高く、私たちのアメリカ以外のいくつかの国での知的財産権はアメリカほど広くないかもしれない。しかも、私たちは付与される前に国と地域の特許出願を放棄することに決定するかもしれない。各国または地域特許の付与手続は、特定の法ドメインにおいて、出願が関連特許庁によって拒絶され、他の法ドメインによって承認される可能性がある独立した手続である可能性がある。例えば,他の国と異なり,中国の方が特許性に対する要求が高く,特に主張されている薬物の医療用途の詳細な説明が求められている。さらに、模倣薬製造業者または他の競争相手は、私たちまたは私たちのライセンシーの特許の範囲、有効性、または実行可能性に疑問を提起し、私たちまたは私たちのライセンシーに複雑で長く、高価な訴訟、または他の訴訟を要求するかもしれない。後発薬メーカーは私たちの製品の模造薬バージョンを開発し、承認を求めて発売することができる。同様に一般的な場合は、国によって同じ候補製品または技術の特許保護範囲が異なる可能性があることである。
競争相手は、私たちが特許保護を受けていない司法管轄区域で、私たちまたは私たちのライセンシーまたは協力者の技術を使用して彼ら自身の製品を開発することができ、また、他の侵害製品を私たちまたは私たちのライセンシーまたは協力者が特許保護を持っている地域に輸出することもできるが、法執行力はアメリカに及ばない。これらの製品は私たちの候補製品と競争する可能性がありますが、私たちと私たちの許可者または協力者の特許または他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。
いくつかの司法管轄区の法律は知的財産権の保護程度はアメリカとEUの法律或いは規則に及ばず、多くの会社はこれらの司法管轄区の保護とこのような権利を保護する時に重大な困難に直面している。特定の国の法律制度、特に特定の発展途上国の法律制度は、特許、商業秘密、および他の知的財産権保護の実行を支持しておらず、これは、私たちの特許の侵害を阻止したり、私たちの専有権を侵害する方法で競争製品を販売することを困難にするかもしれない。外国の管轄区域で私たちの特許権の訴訟を強制的に執行することは、成功するかどうかにかかわらず、巨額のコストを招く可能性があり、業務の他の方面への私たちの努力と注意力を移転させ、私たちの特許を無効または狭義に解釈されるリスクに直面させる可能性があり、私たちの特許出願を発表できないリスクに直面させ、第三者が私たちにクレームを提起する可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。したがって、私たちが世界各地で私たちの知的財産権を実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネスメリットを得るのに十分ではないかもしれません。また、私たちが予想している重要な市場で私たちの知的財産権を保護するつもりですが、候補製品をマーケティングしたいかもしれないすべての司法管轄区域で同様の努力を開始したり維持したりできることを保証することはできません。したがって、これらの国で私たちの知的財産権を保護するための努力が不十分である可能性があり、これは、すべての予想される重要な海外市場で私たちの候補製品を商業化することに成功した能力に悪影響を及ぼす可能性がある。私たちまたは私たちの許可者が知的財産権の保護に困難に直面したり、他の理由でこれらの管轄地域の業務に重要な知的財産権を効果的に保護できない場合、これらの権利の価値は低下する可能性があり、私たちはこれらの管轄区域の他の人からの追加的な競争に直面する可能性がある。
一部の国には強制許可法があり,これらの法律により特許権者は第三者に許可を強制的に付与される可能性がある。さらに、いくつかの国は、政府機関または政府請負業者に対する特許の実行可能性を制限している。これらの国では,特許権者の救済措置は限られている可能性があり,このような特許の価値を大幅に低下させる可能性がある。もし私たちまたは私たちのいかなる許可者も私たちの業務に関連する任意の特許の許可を第三者に付与することを余儀なくされた場合、私たちの競争地位は損なわれる可能性があります。
知的財産権は必ずしも私たちの競争優位に対するすべての潜在的な脅威を解決できるとは限らない。
私たちの知的財産権が提供する将来の保護の程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できないか、あるいは競争優位性を維持することができるかもしれない。以下の例は例示的である
•他の人は、私たちの候補製品と同じまたは同様の化合物を製造することができるかもしれないが、これらの化合物は、私たちが所有または独占的に許可された特許請求の範囲内ではない
•第三者の特許は私たちの候補製品を開発または商業化する能力を弱めるかもしれない
•私たちまたは私たちのライセンシーまたは任意の未来の戦略的パートナーは、私たちが所有または独占的に許可された発行特許または係属特許出願によってカバーされた発明を実践する最初の概念または減少した会社ではないかもしれない
•私たちまたは私たちのライセンシーまたは任意の未来の協力者は、私たちのいくつかの発明をカバーする特許出願を最初に提出した人ではないかもしれない
•他の人は私たちの知的財産権を侵害することなく、類似または代替技術を開発したり、私たちの任意の技術を複製したりすることができる
•私たちが処理している特許出願は発行された特許を生成しない可能性がある
•私たちが所有または独占的に許可された発行された特許は、競争優位性を提供してくれないかもしれないし、競争相手の法的挑戦のために無効または実行不可能と認定される可能性がある
•私たちの競争相手は特許権のない国で研究や開発活動を行い、これらの活動から学んだ情報を利用して競争力のある製品を開発し、私たちの主要な商業市場で販売するかもしれません
•私たちの候補製品または技術を使用して製造またはテストを行ってくれる第三者は、適切な許可を得ることなく他人の知的財産権を使用することができる
•私たちは他の特許を申請できる技術を開発しないかもしれない。
特許法または特許法の変更は、特許の全体的な価値を低下させ、イソフェンまたは任意の将来の候補製品を保護する能力を弱める可能性がある。
他の生物製薬会社と同様に、私たちの成功は知的財産権、特に特許に大きく依存している。生物製薬業界で特許を取得し、実施することは技術複雑性と法律複雑性にも関連する。したがって、生物製薬特許を取得して実行することは高価で、時間と固有の不確実性だ。また、2011年9月16日に採択された米国発明法やAIAは、米国特許制度の大きな変化を招いた。
AIAによる重要な変化の1つは,2013年3月16日から,米国が同一発明を要求する異なる当事者が2つ以上の特許出願を提出する際に,どちらが特許を付与されるべきかを決定する“先着案”制度に転換したことである。したがって、その日の後であるが、私たちの前に米国特許商標局に特許出願を提出した第三者は、たとえ第三者が発明を行う前に発明をしていても、私たちの発明をカバーする特許を付与することができる。これは私たちが発明から特許出願を提出するまでの時間を知ることを要求するが、状況は私たちの発明に関する特許出願を迅速に提出することを阻止するかもしれない。
AIAが導入した他のいくつかの変化は、特許権者が特許侵害訴訟を提起できる範囲を制限することと、USPTOで任意の発行された特許に挑戦する機会を第三者に提供することとを含む。これは2013年3月16日までに発表された特許であっても、私たちのすべてのアメリカ特許に適用される。USPTO訴訟における証拠基準は、米国連邦裁判所が特許請求の無効を宣言するために必要な証拠基準よりも低いため、第三者は、USPTO手続きにおいて、USPTOが権利要求を無効とするのに十分な証拠を提供する可能性があり、同じ証拠が最初に地域裁判所訴訟で提出された場合には、権利請求を無効にするのに十分ではない。
したがって,第三者は米国特許商標局の手続きを用いて我々の特許主張の無効を宣言しようとする可能性があり,第三者が地域裁判所訴訟で最初に被告として疑問を提起すれば,我々の特許主張は無効ではない.友邦保険が我々の業務運営にどのような影響を与えるかは不明である(あれば)。しかしながら、AIAおよびその実施は、私たちまたは私たちのライセンシーまたはパートナーの特許出願をめぐる起訴、ならびに私たちまたは私たちのライセンシーまたはパートナーの発行された特許の実行または弁護の不確実性およびコストを増加させる可能性がある。
さらに、米国最高裁判所は近年、いくつかの特許事件に対して裁決を下しているか、場合によっては利用可能な特許保護範囲を縮小するか、場合によっては特許所有者の権利を弱めるかを決定している。我々の将来の特許取得能力に関する不確実性の増加に加えて,このようなイベントの結合は,いったん特許を取得する価値に関する不確実性をもたらしている.国会、連邦裁判所、および米国特許商標局の決定によると、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、新しい特許を獲得したり、既存の特許および将来獲得可能な特許を強制的に執行する能力を弱める可能性がある。同様に、近年、欧州特許法の複雑性と不確実性も増加している。しかも、欧州特許制度は起訴中に許可された修正タイプの面で相対的に厳しい。これらの法律法規を遵守することは、私たちが将来私たちの業務に重要かもしれない新しい特許を得る能力を制限するかもしれない。
最終的に,統一特許と統一特許裁判所(UPC)制度は2023年6月1日に欧州で実施された。この新しい制度はヨーロッパの競争相手に対抗するために特許権を保護し実行する能力に不確実性をもたらすかもしれない。UPCによれば、デフォルトの場合、すべての欧州特許は、欧州特許パッケージを承認する前に発表された特許を含み、自動的にUPCの管轄に属する。UPCは私たちの競争相手に
新しいフォーラムは私たちの欧州特許を集中的に撤回し、競争相手が汎ヨーロッパ禁止を獲得する可能性を許可する。UPCが認める特許権の範囲と提供される特許救済措置の力を知るのに数年かかるだろう。EU特許セットによると、私たちは裁判所が存在する7年前に私たちの特許をUPCから離脱することを選択する権利があるだろうが、そうすることは私たちが新しい統一裁判所の利点を達成することを阻止するかもしれない。
従業員や他の人と締結された秘密保持協定は、ビジネス秘密の漏洩を防ぐのに十分ではない可能性があり、他の固有情報を保護することもできない可能性がある。
私たちは、独自のビジネス秘密、秘密のノウハウ、および非特許のノウハウが私たちのビジネスに重要であると考えています。私たちは、特に特許保護が価値が限られていると考えられる場合、商業秘密または機密技術によって私たちの技術を保護するかもしれない。しかし、商業秘密と機密技術は機密として維持することは難しい。
このような情報を競争相手から開示されたり盗んだりしないようにするために、私たちの政策は、私たちの従業員、コンサルタント、請負業者、コンサルタントが私たちと秘密協定を締結することを要求することです。私たちはまた、私たちの情報技術システムの実体と電子セキュリティを維持するために、私たちの資料、商業秘密、そしてノウハウの完全性とセキュリティを保障するために努力しています。許可されていない使用と開示を監視することは困難であり、私たちはまた私たちのノウハウを保護するために私たちが取った段階が有効かどうか分からない。私たちは私たちの商業秘密と他の固有と機密情報が漏洩されないという保証もなく、競争相手が他の方法で私たちの商業秘密を取得しないという保証もない。しかしながら、現従業員または元従業員、コンサルタント、請負業者、およびコンサルタントは、競争相手に私たちの機密情報を意図的にまたは意図的に開示することができず、機密情報を無許可に開示することができない場合に十分な救済措置を提供できない可能性がある。第三者に商業秘密および/または機密技術の不法取得および/または使用を強制的に要求することは、高価で、時間的であり、予測不可能である。秘密保持プロトコルの実行可能性は管轄地域によって異なる.さらに、もし競争相手が私たちの任意の商業秘密を合法的に取得または独立して開発すれば、私たちはその競争相手がその技術や情報を使用して私たちと競争することを阻止する権利がなく、これは私たちの競争地位を損なう可能性がある。また,我々のビジネス秘密を守るための手順が不十分であると考えられれば,第三者による商業秘密の流用に対抗する十分な追跡権がない可能性がある.
ビジネス秘密および秘密を取得または維持できなかったノウハウ貿易保護は、私たちの競争的地位に悪影響を及ぼす可能性がある。また、我々の競争相手は、実質的に同等の独自情報を独立して開発することができ、さらにはこれらの情報について特許保護を申請することができる。もしこのような特許保護に成功すれば、私たちの競争相手は私たちの商業秘密および/または機密技術の使用を制限するかもしれない。
私たちは、私たちの従業員や私たちが彼らの知的財産権を流用したと主張したり、私たち自身の知的財産権の所有権を要求したりする第三者のクレームを受けるかもしれない。
私たちの多くの従業員は、私たちの高級管理者を含めて、私たちの競争相手や潜在的な競争相手を含む大学や他の生物製薬会社に雇われていた。その中の一部の従業員は以前の仕事に関連した所有権、秘密とスポーツ禁止協定に署名した。私たちは、私たちの従業員が私たちのために働いているときに他人の固有情報またはノウハウを使用しないことを保証するために努力しているが、私たちまたはこれらの従業員は、商業秘密または他の固有情報を含む任意のそのような従業員の前雇用主の機密情報または知的財産を使用または開示している疑いを受ける可能性がある。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。
もし私たちがこのようなクレームを起訴または弁護しなければ、金銭損害賠償の支払いに加えて、私たちは貴重な知的財産権や人員を失ったり、損害を受けたりする可能性がある。このような知的財産権は第三者に付与することができ、私たちの技術や製品を商業化するためには第三者から許可を得る必要があるかもしれない。そのような許可は商業的に合理的な条項や根本的に存在しないかもしれない。このようなクレームの起訴や抗弁に成功しても、訴訟は巨額のコストを招き、経営陣の注意を分散させる可能性がある。
私たちの特許保護の獲得と維持は、政府特許機関によって提出された様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
発行された特許の定期維持費および年会費は、特許有効期間内にいくつかの段階に分けて米国特許商標局および外国特許代理機関に支払われなければならない。米国特許商標局および各種外国政府特許機関は、特許出願過程において、いくつかのプログラム、文書、費用支払い、およびその他の同様の規定を遵守することを要求する。多くの場合,不注意は滞納金の支払いやルールを適用する他の方法で救済できるが,規定を遵守しない場合もある
特許又は特許出願が放棄又は失効され,管轄区域に関する特許権の一部又は全部が失われた。特許または特許出願が放棄または失効される可能性のある規定を遵守しないイベントは、規定された期限内に公式行動に応答することができなかったこと、費用を支払わなかったこと、および適切に合法化されず、正式な文書を提出することを含む。もし私たちまたは私たちの許可者や協力パートナーが私たちの候補製品をカバーする特許や特許出願を維持できなかった場合、私たちの競争相手は市場に参入する可能性があり、これは私たちの競争地位を損ない、任意の候補製品を商業化することに成功する能力を弱める可能性がある。
私たちの情報技術システムと私たちが臨床前と臨床試験を行うために使用し、あるいは他の方法で協力するための製造業者、サプライヤー、その他の第三者のシステムは故障したり、セキュリティホールが発生したりする可能性があり、これは私たちの運営を分散させ、私たちの研究開発作業を遅延させ、私たちの業務、運営、財務表現に不利な影響を与える可能性がある
私たちの正常な業務過程において、私たちと私たちは、私たちの臨床試験被験者と従業員の知的財産権、臨床試験データ、独自の業務情報および個人識別情報(総称して“秘密情報”と呼ぶ)を含む、前臨床試験および臨床試験または他の方法で協力し、敏感なデータを収集し、保存するための製造業者、サプライヤーおよび第三者、ならびに私たちおよび第三者ネットワークにおいて。秘密情報のセキュリティ処理、維持、そして伝送は私たちの運営に必須的だ。当社の情報技術および他の内部インフラシステムは、会社のファイアウォール、サーバ、レンタル回線とインターネット接続、および臨床前および臨床試験または他の方法で協力するための製造業者、サプライヤー、および他の第三者のシステムを含み、系統的な障害のリスクに直面しており、これは私たちの運営を乱す可能性があります。これらの情報技術や他の内部インフラシステムの可用性に重大な中断が生じた場合,我々の協力中断や開発作業の遅延を招く可能性がある
さらに、我々の情報技術システムおよび我々の第三者サービスプロバイダ、戦略パートナーおよび他の請負業者またはコンサルタントのシステムは、コンピュータウイルス、マルウェア(例えば、恐喝ソフトウェア)、誤った構成、“エラー”または他の脆弱性、自然災害、テロ、戦争、電気通信および電気故障、ハッカー、ネットワーク攻撃、ネットワーク釣り攻撃および他の社会工学計画、悪意のコード、従業員の窃盗または乱用、人為的エラー、詐欺、サービス拒否攻撃またはサービス降格、複雑な民族国家および民族国家によってサポートされる行為者、または本組織内部システムにアクセスする権利を有する者、または本組織内部システムにアクセスする権利のある者の不正アクセスまたは使用の損害、攻撃または中断を受けやすい。情報技術システムへの攻撃は頻度,持続性,複雑性,強度の面で増加しており,動機や専門長の異なる複雑で組織的な団体や個人によって実施されている.持続的な混合作業環境のため、私たちはインターネット技術への依存と私たちの遠隔作業の従業員の数のために、サイバー犯罪者の抜け穴を利用するためのより多くの機会を作るかもしれないので、より多くのネットワークセキュリティリスクに直面する可能性がある。さらに、不正アクセスまたはシステム破壊または撹乱のための技術はしばしば変化し、通常、ターゲットに対して攻撃が開始されるまで識別されるため、これらの技術を予測したり、十分な予防措置を実施することができない可能性がある。私たちはまたセキュリティホールに遭遇する可能性があり、このような抜け穴は長い間発見されないかもしれない。発見されても、攻撃者が法医学証拠の検出を回避し、検出を回避し、除去し、または混同するためのツールおよび技術を使用するために、事件または違反行為を十分に調査または修復することができない可能性がある。私たちと私たちの製造業者、サプライヤー、および他の重要な第三者のネットワークセキュリティリスク管理計画およびプロセスは、ポリシー、制御、またはプログラムを含めて、私たちのシステム、ネットワーク、および機密情報を完全に実施、遵守、または効果的に保護することも保証されません。
私たちは、私たちの重要な第三者(例えば、協力者)とセキュリティ対策を実施しているにもかかわらず、私たちの情報技術およびインフラは、ハッカーまたは内部非行者の攻撃、人為的ミス、技術的脆弱性、違反、または他の中断による侵入を受けやすいかもしれない。私たちと私たちの特定のサービス提供者たちは時々ネットワーク攻撃とセキュリティ事件の影響を受ける。私たちが知っている限り、私たちは今まで何の重大なセキュリティホールも経験していませんが、どのような脆弱性も私たちのネットワークを危険にさらし、そこに格納された機密情報がアクセス、公開、紛失、または盗まれる可能性があります。このようなアクセス、開示、あるいは他の情報損失は、法律クレームや訴訟、個人データのプライバシーを保護する法的責任、監督管理処罰、私たちの運営を乱し、私たちの名声を損なう可能性があり、人々が私たちと私たちの臨床試験を行う能力に自信を失うことを招き、これは私たちの名声に不利な影響を与え、私たちの候補製品の臨床開発を延期する可能性がある。我々または第三者システムまたは機密情報の利用可能性、完全性またはセキュリティへのいかなる悪影響も、法的クレームまたは訴訟(例えば、集団訴訟)、規制調査および法執行行動、罰金および処罰、既存または将来の顧客の負の名声の影響を失うこと、および/または重大なイベント応答、システム回復または修復、および未来をもたらす可能性がある
コンプライアンスコスト。いかなる損失、費用、または債務は、任意のまたはすべての適用保険証の保証範囲内にない場合があり、またはその保証範囲を超える可能性がある。
従業員の事務と管理成長に関するリスク
私たちの未来の成長と競争能力は私たちが重要な人員を維持し、より多くの合格者を募集する能力にかかっている。
私たちの成功は私たちの肝心な管理者、科学と技術者の貢献に依存して、彼らの中の多くの人は私たちに助けを提供して、アンテフェンタリンと関連技術の方面で豊富な経験を持っています。私たちの主な管理職には、CEO David、最高財務責任者マーク·ハーン、最高医療官クレア·ポール、キャサリン·リカルド、キャロライン·ディアス最高経営責任者、クリストファー·マーティン、タラ·ロット最高開発官が含まれています。重要な人員の流出は私たちの商業化と研究開発活動を延期するかもしれない。そのほか、生物製薬と製薬分野の合格人材に対する競争は非常に激しく、私たちの未来の成功は私たちが高技能科学、技術と管理従業員を誘致、維持と激励する能力にかかっている。私たちは他の会社、大学、公共、民間研究機関、そして他の組織からの人員競争に直面している。もし私たちの将来の採用と維持努力が成功しなければ、私たちの製品候補開発目標を実現し、追加資本を調達し、私たちの業務戦略を実施することは困難かもしれません。
私たちは私たちの開発、規制、商業、販売、マーケティング、精算、流通能力を拡大することが予想されますので、私たちは成長を管理する上で困難に直面する可能性があり、これは私たちの運営を混乱させるかもしれません。
私たちは、特に商業運営と販売、マーケティング、精算、流通の分野で、私たちの従業員数と業務範囲が大幅に増加することを予想しています。私たちが予想している将来の成長を管理するためには、私たちの管理、運営、財務システムを継続して実施し、改善し、私たちの施設を拡大し、より多くの合格者を募集し、訓練し続けなければならない。私たちの財務資源が限られていることと、私たちの管理チームがこのような成長を期待している会社を管理する上での経験が限られているため、私たちの業務の拡張を効果的に管理したり、より多くの合格者を募集したりすることができないかもしれません。私たちの業務の拡張は巨大なコストを招き、私たちの管理と業務発展資源を移転する可能性があります。成長を管理できないどんな状況も、私たちの業務計画の実行を延期したり、私たちの運営を妨害したりする可能性がある。
アメリカの預託証明書に関連するリスクは
我々の普通株式(米国預託証明書に代表される普通株を含む)を持ついくつかの株主、取締役会メンバー、上級管理職は私たちに重大な支配権を行使することができるかもしれない。
当社の株主総会に出席する人数に応じて、当該等株主は単独又は集団投票にかかわらず、当該等の株主総会が決定した結果を決定又は顕著に影響することができる。50%を超える株主または株主団体の出席を制御し、私たちの株主総会で投票するには、取締役会のメンバーの任命、私たちの資本構造に関連するいくつかの決定、およびいくつかの重大な会社取引の承認を含む、簡単な多数の採択を必要とする任意の株主決議を制御することができる。他の結果を除いて、このような所有権集中は制御権変更を延期または阻止する効果がある可能性があるため、私たちのアメリカ預託証明書と普通株の市場価格にマイナスの影響を与える可能性がある。
私たちは予測可能な未来に私たちのアメリカ預託証明書や普通株に現金配当金を支払わないと予想されているので、資本付加価値(あれば)は私たちのアメリカ預託株式保有者と株主の唯一の収益源になり、彼らは決して投資から見返りを得ないかもしれない。
英国の現行法律によると、会社の累計実現利益は、その累積達成損失(非合併ベース)を超えてこそ配当金を支払うことができる。したがって、配当金を発行する前に、私たちは分配可能な利益を持っていなければならない。私たちは過去に私たちの普通株に配当金を支払ったことがない。私たちは収益を維持して、私たちの業務のために、予測可能な未来に現金配当金を支払わないつもりです。したがって、私たちのアメリカ預託証明書または普通株の資本増価(あれば)は、私たちのアメリカ預託株式保有者と株主の予測可能な未来における唯一の収益源となり、もし彼らが購入時の価格以上の価格で保有するアメリカ預託証明書または普通株を売却できなければ、彼らの投資は損失を受けるだろう。現金配当金を求める投資家は私たちのアメリカ預託証明書や普通株を購入してはいけません。
私たちのアメリカ預託証明書保持者は、私たちの普通株式保有者と同じ投票権を持っていないかもしれないし、彼らの投票権を行使するために投票材料をタイムリーに受け取ることができないかもしれない。
私たちのアメリカ預託証明書所持者は私たちのアメリカ預託証明書が証明した普通株に関する投票権を個人名義で行使することはできません。我々の米国預託証明書所持者は,その代表として預託管理人を指定し,その米国預託証明書に代表される普通株に付随する投票権を行使している.私たちの米国預託証明書保持者は、管理機関に投票を指示するために投票材料をタイムリーに受け取ることができない可能性があり、彼らまたはブローカー、取引業者、または他の第三者によってその米国預託証明書を持っている人は投票権を行使する機会がないかもしれない。さらに、保管人は、いかなる採決指示、いかなる採決方法、または任意のそのような採決を実行できなかった効果に対しても責任を負わないであろう。したがって、もし私たちのアメリカ預託証明書の所持者が要求通りに投票しなければ、彼らは投票権を行使できないかもしれないし、追徴権もないかもしれない。しかも、私たちのアメリカ預託証明書の保有者は株主総会を開くことができないだろう。
もし私たちのアメリカ預託証明書所有者に普通株を提供することが不法または非現実的であれば、私たちのアメリカ預託証明書所有者は、私たちのアメリカ預託証明書によって代表される私たちの普通株の分配またはそのいかなる価値も得ることができない。
私たちアメリカ預託証券の受託者は、私たちの普通株式または他の預金証券から受け取った現金配当金または他の分配を、私たちのアメリカ預託証明書所持者に支払うことに同意し、その費用と費用を差し引いた。私たちのアメリカ預託証明書の保有者は、そのアメリカ預託証明書に代表される私たちの普通株式数の割合でこれらの分配を受けます。しかし、預託機関と締結された預金協定に規定されている制限により、我々の米国預託証明書保持者に流通を提供することは不法または非現実的である可能性がある。私たちは私たちのアメリカ預託証明書、普通株式、権利、または他のものを私たちのアメリカ預託証明書所有者に配布することを可能にする他の行動を取る義務はない。これは、もし私たちのアメリカ預託証明書所有者に分配を提供することが不法または非現実的であれば、彼らは私たちの普通株式に対する私たちの分配や彼らから得られたいかなる価値も受けないかもしれないということを意味する。このような制限は私たちのアメリカ預託証明書の価値に大きな悪影響を及ぼすかもしれない。
私どものアメリカ預託証明書保持者はそのアメリカ預託証明書を譲渡する際に制限される可能性があります。
アメリカ預託証明書は預かり人の帳簿に譲渡することができます。しかし,保管人は,職務遂行に関与していると考えられる場合には,その譲渡帳簿を随時あるいは随時閉鎖することができる。さらに、私たちの帳簿または委託者の帳簿が閉鎖されたとき、または私たちまたは係の人が、法律または任意の政府または政府機関の任意の要求から、またはホスト契約の任意の規定に基づいて、またはホスト契約の条項に従った任意の他の理由で、任意の場合、受託者は、米国預託証明書の交付、譲渡または登録を拒否することができる。これらの譲渡制限は私たちのアメリカ預託証明書の価値に実質的な悪影響を及ぼすかもしれない。
私たちの株主の権利はアメリカの会社の株主に通常提供される権利とは異なるかもしれない。
私たちはイギリスの法律登録に基づいて設立された。普通株式所有者の権利、及びアメリカ預託証明書所有者のいくつかの権利は、2006年の会社法の規定、及び著者らの組織定款を含むイギリスの法律によって管轄されている。これらの権利はいくつかの重要な点で典型的なアメリカ会社の株主権利とは異なる。したがって、私たちの普通株式またはアメリカ預託証明書の投資家は、彼らがアメリカ会社に投資した場合と同じ保護または権利を有していないかもしれない。これは私たちのアメリカ預託証明書のこのような投資家に対する魅力を低下させ、それによって私たちのアメリカ預託証明書の価値を損なうかもしれない。
アメリカの民事責任に対するクレームは私たちに強制的に施行できないかもしれない。
私たちはイギリスの法律登録に基づいて設立された。私たちのほとんどの資産はアメリカ以外に位置している。私たちのほとんどの上級管理職と取締役会はアメリカ以外に住んでいる。したがって、投資家は、米国内でそのような人々に法的手続き文書を送達することができないか、または米国連邦証券法民事責任条項に基づく判決を含む、米国裁判所で得られた彼らまたは私たちに不利な判決を実行することができないかもしれない。
米国と連合王国には現在、民商事判決(仲裁裁決を除く)の承認と執行を規定する条約はない。したがって、米国裁判所による最終支払い判決は、米国証券法に完全に基づいているか否かにかかわらず、自動的にイギリスで認められたり強制的に執行されることはない。また、イギリス裁判所が米国または米国のいずれの州の証券法に基づいてイギリスで我々または我々の役員や上級管理職に対して提起した元の訴訟を受理するかどうかにも、不確実性がある。米国裁判所で得られたいかなる最終的かつ決定的な金銭判決も,米国裁判所では連合王国裁判所が訴訟自体の原因とされ,一般法により債務として訴訟が提起されるため,何らかの要求を満たせば,これらの問題を再審する必要はない。米国証券法民事責任条項に基づく判決が,これらの要求に適合しているかどうかは,このような法律に基づいて金銭損害賠償が処罰となるかどうかを含む,このような裁決を下す裁判所の問題である。もしイギリスの裁判所がアメリカの判決に基づいて支払うべき金額について判決を下したら、イギリスの判決は
この目的のために一般的に使用可能な方法によって強制的に実行することができる。このような方法は一般的にイギリスの裁判所が強制執行の方法を適宜規定することを可能にする。
したがって、米国の投資家は、米国連邦証券法による判決を含む、私たちまたは私たちの上級管理職、取締役会、または本明細書で言及したいくつかの専門家(彼らはイギリスまたはアメリカ以外の国/地域の住民)に対して、米国裁判所で得られた民商事判決を実行することができないかもしれない。
もし私たちが効果的な財務報告内部統制制度を維持できなければ、私たちは私たちの財務結果を正確に報告したり、不正を防止することができないかもしれない。したがって、株主は私たちの財務や他の公開報告に自信を失う可能性があり、これは私たちの業務と私たちのアメリカ預託証明書の取引価格を損なうことになる。
財務報告に対する効果的な内部統制は、信頼できる財務報告を提供するために必要であり、適切な開示制御や手順とともに詐欺を防止することを目的としている。必要な新しい制御措置や改善された制御措置を実施できなかったり、実行中に遭遇した困難は、私たちの報告義務を履行できない可能性があります。さらに、第404条に基づいて実施される任意のテスト、または私たちの独立公認会計士事務所がその後に行う任意のテストは、財務報告の内部統制における私たちの欠陥を明らかにすることができ、これらの欠陥は、重大な弱点と考えられるか、または私たちの財務諸表を前向きまたは追跡的に変更する必要がある場合があり、またはさらなる関心または改善が必要な他の分野を発見することができる。内部統制不足は、投資家が私たちが報告した財務情報に自信を失う可能性もあり、これは私たちのアメリカ預託証明書の取引価格に負の影響を与える可能性がある。
税収に関するリスク
私たちの税率の変化、いくつかの税金控除または減免を得ることができない、または追加の税金負債や評価に直面することは、私たちの収益力に影響を与える可能性があり、税務機関の監査は、前のいくつかの時期の追加税金支払いをもたらす可能性がある。
新しい収入、販売用途、または他の税金法律、法規、規則、法規または条例は、いつでも公布されるか、または私たちに不利な解釈、変更、修正、または適用される可能性があり、これらはいずれも、私たちの業務運営および財務業績に悪影響を及ぼす可能性があります。私たちは今のところ、これらの変化が起こるかどうかを予測できません。そうすれば、私たちの業務に最終的な影響を与えます。これらの変化が関連する不確実性の結果を含む負の影響を与える場合、これらの変化は、私たちの業務、財務状況、運営結果、およびキャッシュフローに実質的な悪影響を及ぼす可能性がある
著者らは各種の適応と交付方法のためにイソフェンタリンを開発することを含むが、研究と開発活動を展開し、そのため、著者らは現在イギリスでイギリス税務と税関総署(HMRC)、中小企業の研究開発減免或いは中小企業の研究開発減免に利益を得ており、この措置は現在イギリスの会社税に減免を提供している。
全体的に、中小企業研究開発猶予には、(A)資格に適合した中小企業がその年間利益から合資格支出の186%を差し引くことを可能にすること、イギリス会社税のための(86%の追加控除に通常の100%の控除を加えることを可能にすることによって)、または中小企業研究開発費の追加控除を活用するためにイギリスの会社税が十分な利益を有していない場合、将来の課税利益を相殺するために超過した部分(“返還可能損失”)を繰返すことができる2つの要素がある。あるいは、現在このような上納可能な損失の10%に相当する税収控除を現金で申請することができるか、または中小企業が税収控除を開発することができる。
HMRCが制定した基準によると、私たちの研究開発活動に関連する支出の一部は、臨床試験、製造、コンサルタントと給料及び関連コストを含むが運営に限定されず、中小企業の研究開発追加控除を受ける資格がある。HMRCの基準によると、私たちがこれによって発生した返還可能な損失は現在中小企業の研究開発税収控除を受ける資格がある。
2023年12月31日と2022年12月31日までの年次財務諸表に、それぞれ230万ドルと860万ドルの中小企業研究開発税収控除を記録した。HMRCの基準によると、2024年12月31日までの会計年度にこれらの中小企業の研究開発税控除を受ける予定だ。
イギリスS中小企業研究開発救済制度の変化は私たちの財務状況に悪影響を及ぼす可能性がある。2023年秋の声明では、英国政府は単一のR&D減免制度を導入し、現行のRDECと中小企業R&D減免計画を組み合わせることを確認した。立法草案によると、提案された信用限度額は条件を満たす支出の20%であり、信用限度額自体はイギリス会社税を納めなければならない。したがって、相殺は、名目税率にもかかわらず、イギリスの会社税の適用税率(現在の主要税率は25%)によって減少する
これは赤字会社に適用され、新たな研究開発減免制度の下で19%の低い税率に設定される。そのため、提案された制度とイギリス会社税の現行税率によると、イギリス会社税の主要税率の影響を受けた利益企業は実際に条件に合った支出の15%の控除を受け、赤字企業は16.2%の控除を受ける。提案された立法には、研究開発減免の制限も含まれており、ある会社が研究開発活動を第三者に請け負ったり、外部から提供された労働者に報酬を支払う場合、納税者はイギリスで働いている場合にしか減免を申請することができない。英国以外で唯一許可されている支出は、地理的、環境または社会的条件がイギリスに存在しないか、複製できないために必要な活動になると提案されている。提案立法には、研究開発活動を第三者に分配するための新たな規定も含まれている
また、2024年4月1日以降に開始される会計期間において、研究開発密集損失の中小企業計画のハードル(おおむね、合資格の研究開発支出が総支出に占める割合)は30%となることを提案している。そのため、資格に符合する研究開発費が総支出の30%以上を占める赤字中小企業は、86%の増額減額と14.5%の返済可能な信用を申請することができる。
新しいイギリスの研究開発税収減免制度は、2024年4月1日以降に開始される会計期間に適用されることを提案している。新制度の立法はまだ決定されていないので、私たちの財務状況への影響を完全に理解することはできませんが、計画の提案変化および/または任意のさらなる変化は私たちの財務状況、経営業績、あるいはキャッシュフローに重大な悪影響を及ぼす可能性があります
もし私たちが受動的な外国投資会社に分類されれば、アメリカの保有者に不利なアメリカ連邦所得税の結果をもたらすだろう。
我々の収入と資産構成および2023年12月31日までの納税年度の資産価値から,2023年12月31日までの納税年度には,米国連邦所得税目的に採用されている受動外国投資会社(“PFIC”)であると考えられる。しかし、過去のいかなる納税年度、2024年12月31日までの納税年度、または将来のいかなる納税年度にもPFICの地位に関する保証を提供することはできません。もし私たちが任意の課税年度に米国株主(以下のように定義する)に分類された場合、(I)私たちの普通株式または米国預託証明書を売却する任意の収益の全部または一部を一般収入とすること、(Ii)このような収益に繰延利息費用を適用し、特定の配当金を受け取ること、および(Iii)特定の報告要件を遵守することを含む、我々の普通株式または米国預託証明書を保有する任意の課税年度に分類される場合。私たちは私たちが上記の申告と納税義務を履行するために必要かもしれない情報をどのアメリカの所有者にも提供する保証はありません。
いずれの課税年度においても、(I)その総収入の75%以上が受動的収入からなる場合、または(Ii)その資産の平均四半期価値の50%以上が受動的収入を生成するか、または受動的収入を生成するために保有する資産からなる場合、一般に受動的外国投資会社またはPFICとみなされる。これらのテストの場合、受動的収入には、配当金、利息、投資財産の売却または交換の収益、ならびにいくつかのレンタル料および特許使用料が含まれる。また、上記の計算については、他の会社の株式の少なくとも25%を直接または間接的に所有している非米国会社は、同社の資産および収入に比例して分配されたシェアを直接保有し、受け入れているとみなされる。私たちが個人投資委員会の決定であるかどうかについては、毎年多くの事実に基づいて決定されており、採用されている原則や方法は場合によっては不明であり、異なる解釈がある可能性がある。収入テストの下で、私たちのPFICとしての地位は私たちの収入構成に依存し、これは私たちが行っている取引と私たちの会社の構造にかかっています。私たちの収入と資産の構成はまた私たちがどの発行で調達した現金支出の影響を受けている。すべてのアメリカの所有者は自分の税務顧問に相談すべきで、私たちがPFICであれば、アメリカの税収に悪影響を与えるかもしれません。
“米国所有者”とは、米国連邦所得税について、私たちの普通株式または米国預託証明書の実益所有者であり、米国市民または個人住民の所有者であり、米国内または米国、その任意の州またはコロンビア特区の法律に従って作成または組織された会社または他の会社に納税すべき実体であり、その収入はその出所にかかわらず米国連邦所得税を納めなければならない遺産である。または(I)米国裁判所によって監督され、そのすべての実質的な決定が1つまたは複数の“米国人”(“法典”第7701(A)(30)条に示される)によって支配される信託、または(Ii)米国人とみなされる有効な選挙を有する
もし私たちがアメリカの保有者が私たちの普通株式またはアメリカ預託証明書を持っている任意の年度がPFICに分類された場合、アメリカの保有者が私たちの普通株式またはアメリカ預託証明書を保有した後のすべての年間は、上記のPFICテストに適合し続けるかどうかにかかわらず、私たちがPFICでない限り、アメリカの保有者は特定の選択をすることができない。
米国の保有者が少なくとも私たちの普通株式または米国預託証明書の10%を所有しているとみなされた場合、その保有者は不利な米国連邦所得税の結果の影響を受ける可能性がある。
米国の保有者(上記で定義したように)が、私たちの普通株式または米国預託証明書の少なくとも10%の価値または投票権を直接、間接的または建設的に所有しているとみなされる場合、米国所有者は、私たちのグループ内の“制御された外国企業”または“フルオロクロロカーボン”(ある場合)の“米国株主”と見なすことができる。私たちのグループは1つ以上のアメリカ子会社を含んでいるので、私たちのいくつかの非アメリカ子会社は、私たちがフルオロカーボンとみなされているかどうかにかかわらず、フッ化炭素と見なすことができる。フッ化炭素の米国株主は、米国の課税収入に比例して分配された“F分部収入”、“世界無形低税収入”およびこのようなフルオロ塩化炭素の米国財産への投資を毎年報告することを要求される可能性があり、このようなフルオロ塩化炭素がどのような分配されているかにかかわらず。フッ化炭素については、米国の株主である個人は、米国会社に属する米国株主に何らかの減税や外国税収控除を与えることは一般的に許されない。これらの報告義務を守らないと米国の株主に巨額の罰金を科される可能性があり、その株主が年度を報告すべき米国連邦所得税申告書に関する訴訟時効を阻止する可能性がある。私たちは私たちの投資家が私たちまたは私たちの任意の非米国子会社がCFCsとみなされているかどうか、あるいはその投資家がこのようなCFCsの米国の株主とみなされているかどうかを決定するのを助ける保証はありません。また、このリスク要因に記載されている報告や納税義務を遵守するために必要な情報を米国の株主に提供する保証はありません。アメリカの保有者は彼らの税務顧問に問い合わせ、これらの規則が私たちの普通株式またはアメリカ預託証明書への投資における潜在的な応用を理解しなければならない。
一般リスク
私たちのアメリカ預託証明書の価格は変動する可能性があり、私たちがコントロールできない要素によって変動するかもしれません。
発売された新興生物製薬や薬物発見と開発会社の取引市場は常に変動性が大きく,将来的には高度な波動性を保つ可能性がある。私たちのアメリカ預託証明書の市場価格は様々な要素によって大きく変動する可能性があります
•臨床試験の陽性または陰性結果または遅延;
•私たちの競争相手ビジネスの発展は
•イソフェンの開発や商業化に関する協力や戦略関係の達成を遅延させたり、私たちに有利と思われない条項で協力や戦略関係を達成したり、
•私たちや競争相手の技術革新やビジネス製品の導入
•政府の規則の変化
•特許及び訴訟事項を含む特許権の発展について
•アンテファンドリンの商業的価値や安全性への関心は
•資金調達や他の会社の取引
•研究報告書や証券、業界アナリスト、コメンテーターのコメントを発表する
•製薬業界や経済全体の一般的な市場状況
•私たちの重要な科学や高度な管理者を失いました
•私たち、私たちの上級管理職または取締役会のメンバー、および私たちのアメリカ預託証明書の重要な所有者が私たちのアメリカ預託証明書を売却します
•他の事件と要素、その多くは私たちがコントロールできない。
これらおよび他の市場と業界要素は、私たちのアメリカ預託証明書の市場価格と需要を大幅に変動させる可能性があり、私たちの実際の経営業績にかかわらず、これは投資家が彼らのアメリカ預託証明書を随時販売することを制限または阻止し、他の方法で私たちのアメリカ預託証明書の流動性に負の影響を与える可能性がある。また,株式市場,特にバイオ製薬会社は,極端な価格や出来高変動を経験しており,これらの変動はこれらの会社の経営業績に関係なくあるいは比例しないことが多い。従来、1株の市場価格が変動した場合、その株の保有者は発行者に対して証券集団訴訟を起こすことがあった。もし私たちのアメリカ預託証明書所持者が私たちにこのような訴訟を起こしたら、私たちは巨額の訴訟弁護費用が生じるかもしれません。私たちの上級管理職の注意は私たちの業務運営から移っていきます。訴訟のどんな不利な判決もまた私たちに重大な責任を負わせるかもしれない。
私たちの大量のアメリカ預託証明書または普通株の将来の販売または将来の販売の可能性は、私たちのアメリカ預託証明書の価格に悪影響を及ぼすかもしれません。
将来的に私たちのアメリカ預託証明書や普通株を大量に売却したり、このような売却が発生すると考えられたりして、私たちのアメリカ預託証明書の市場価格を低下させる可能性があります。アメリカでは私たちのアメリカ預託証明書と私たちの役員、上級管理者、関連株主が保有する普通株が制限されています。これらの株主が公開市場で大量の普通株や米国預託証券を売却したり、市場でこのような売却が発生する可能性があると考えられた場合、我々の米国預託証券の市場価格および将来的に株式証券を発行することで資金を調達する能力は悪影響を受ける可能性がある。
不安定な市場と経済状況は、私たちの業務や財務状況、私たちのアメリカ預託証明書の価格に深刻な悪影響を及ぼす可能性があります。信用と金融市場を含む世界経済は最近、流動性と信用供給の深刻な減少、金利とインフレ率の上昇、消費者自信の低下、経済成長の低下、失業率の上昇、経済安定の不確定を含む極端な変動と破壊を経験している。株式や信用市場が悪化し続けている場合、あるいはイギリスや米国が衰退に入った場合、任意の必要な債務や株式融資をタイムリーまたは有利な条件で得ることが難しくなり、コストがより高くまたは希釈度が高くなる可能性がある。さらに、我々の1つまたは複数のCRO、プロバイダ、または他の第三者プロバイダは、経済低迷または衰退の中で生き残ることができない可能性がある。したがって、私たちの業務、経営業績、私たちのアメリカ預託証明書の価格は不利な影響を受ける可能性があります。
証券や業界アナリストやコメンテーターが私たちの業務に関する不正確または不利な研究報告を発表した場合、私たちのアメリカ預託証明書と普通株の価格および私たちの取引量は低下する可能性があります。
私たちアメリカの預託証明書と普通株の取引市場は、証券、業界アナリスト、あるいはコメンテーターが発表した私たちまたは私たちの業務に関する研究と報告にある程度依存しています。もし私たちの1人以上のアナリストが私たちのアメリカ預託証明書の格付けを引き下げたと報道された場合、または彼らまたは他の業界の評論家が私たちの業務の不正確または不利な研究や論評を発表した場合、私たちのアメリカ預託証明書と普通株の価格は下落するかもしれない。これらのアナリストのうちの1人以上が私たちの報告書を停止したり、私たちに関する報告書を定期的に発表できなかった場合、私たちのアメリカ預託証明書に対する需要が減少する可能性があり、これは私たちのアメリカ預託証明書と普通株の価格および取引量を低下させる可能性がある。
米国上場企業としては、コストの増加が予想されており、我々の上級管理職は、新たなコンプライアンス措置やコーポレートガバナンス実践を実施するために多くの時間を投入する必要がある。
米国上場企業として、2023年12月31日までの大規模加速申告機関への移行に関する費用を含め、米国上場企業になるまで発生していなかった大量の法律、会計、その他の費用が発生し続けることが予想されています。2002年のサバンズ-オクスリ法案、ドッド-フランクウォール街改革と消費者保護法、ナスダックの上場要求、その他の適用された証券規則と条例は、有効な開示、財務制御、会社管理やり方の確立と維持を含む非米国上場企業に対して様々な要求を提出した。私たちの上級管理職たちと他の人たちはこのようなコンプライアンス計画を実施するために多くの時間を投入し続ける必要があるだろう。しかも、このような規則と法規は引き続き私たちの法律と財務コンプライアンスコストを増加させ、いくつかの活動をより時間的で高価にするだろう。
これらの規則や条例は往々にして異なる解釈を持ち,多くの場合特殊性に欠けるため,規制機関や理事機関が新たな指導意見を提供するにつれて,実践における適用は時間とともに変化する可能性がある。これは遵守事項に関する持続的な不確実性と、開示と統治慣行を絶えず修正するために必要なより高いコストをもたらす可能性がある。
2002年“サバンズ·オキシリー法”第404条又は第404条によれば、我々は、財務報告の内部統制に関する上級経営者の報告、及び我々の独立公認会計士事務所が発行した財務報告の内部統制に関する証明報告書を提出しなければならない。第404(B)節の遵守を準備して維持するために、財務報告に対する内部統制を記録して評価するプロセスを実施した。この点で、我々は、財務報告の内部制御の十分性を評価して記録するために、外部コンサルティング人を招聘し、詳細な作業計画を実行し、適切にステップを取って制御プログラムを改善し、制御措置がファイルのように機能しているかどうかをテストすることによって検証し、財務報告内部制御のための持続的な報告および改善プログラムを実施する必要があり、高価で挑戦的である。私たちが努力したにもかかわらず、私たちは第404条で要求されたように、財務報告書の内部統制に有効であるという結論を出すことができない可能性がある。1つまたは複数の重大な弱点を発見すれば、財務諸表の信頼性に対する自信を失った金融市場の不良反応を招く可能性がある。
業務中断は私たちの運営に悪影響を及ぼすかもしれない。
私たちの業務は火災、悪天候条件、停電、電気通信故障、テロ活動、公衆衛生危機と大流行疾患(例えば新冠肺炎)の妨害、そして他の私たちがコントロールできない天災人災や事件を受ける可能性がある。私たちの工場はいつも悪天候を経験する地域にあります。私たちは重大な竜巻、洪水、火災、地震、停電、テロ活動、公衆衛生危機、大流行疾病或いはその他の災害に対して私たちの業務と財務業績の潜在的な結果を系統的に分析しておらず、これらの災害のための回復計画も立てていない。また、発生する可能性のある業務中断による実際の損失を補償するのに十分な保険を提供していません。私たちによるいかなる損失や損害も私たちの業務を損なう可能性があります。このような業務中断の発生は、私たちの運営と財務状況を深刻に損害し、私たちのコストと支出を増加させる可能性がある。
項目1 B。未解決従業員意見
ない。
プロジェクト1 C。ネットワーク·セキュリティ
ネットワークセキュリティリスク管理と戦略
我々は,我々のキーシステムと情報の機密性,完全性,可用性を保護するためのネットワークセキュリティリスク管理計画を策定し実施した.
私たちはアメリカ国家標準と技術研究院のネットワークセキュリティ枠組み(“NIST CSF”)に基づいて私たちの計画を設計し、評価します。これは私たちが特定の技術標準、規範、または要求を満たすことを意味するわけではありませんが、私たちはNIST CSFをガイドラインとして使用して、私たちの業務に関連するネットワークセキュリティリスクの識別、評価、管理を助けてくれます。
我々のネットワークセキュリティリスク管理計画は、私たちの全体企業リスク管理計画に統合され、企業リスク管理計画全体に適用される汎用的な方法、報告ルート、管理プロセスを共有し、他の法律、コンプライアンス、戦略、運営、金融リスク分野に適用される。
私たちのネットワークセキュリティリスク管理プログラムは
•リスク評価は、我々のキーシステム、情報、製品、サービス、およびより広範な企業IT環境が直面する重大なネットワークセキュリティリスクの識別を支援することを目的としている
•情報技術チームは,(1)我々のネットワークセキュリティリスク評価過程,(2)我々のセキュリティ制御,および(3)ネットワークセキュリティイベントに対する我々の反応を主に管理する
•外部コンサルタントおよびサービスプロバイダを適宜使用して、私たちのセキュリティ制御の様々な態様を評価、テスト、または他の方法で支援する
•私たちの従業員、請負業者、イベント応答者、および高度管理者のためのネットワークセキュリティ意識トレーニングを行います
•ネットワークセキュリティイベントに対応するプログラムを含み、より広範な企業業務連続性計画と一致するネットワークセキュリティイベント対応計画、および
•当社のキーシステムおよび情報にアクセスできるサービスプロバイダ、プロバイダ、およびプロバイダのための第三者リスク管理プロセスを提供します。
私たちは、私たちの運営、業務戦略、運営結果、または財務状況を含む、既知のネットワークセキュリティ脅威(これまでの任意のネットワークセキュリティイベントを含む)から、私たちに重大な影響を与えるか、または合理的に私たちに重大な影響を与える可能性があるリスクを決定していません。より多くの情報については、“リスク要因--情報技術システム障害、ネットワーク攻撃、またはネットワークセキュリティ欠陥が発生した場合、私たちの業務および運営が影響を受ける可能性がある”というタイトルの節を参照してください
サイバーセキュリティ·ガバナンス
本委員会は,ネットワークセキュリティリスクがそのリスク監督機能の一部であると考え,ネットワークセキュリティや他の情報技術リスクの監視を監査委員会(委員会)に依頼した.その委員会は管理職たちが私たちのネットワークセキュリティリスク管理計画を実施することを監督する
委員会は四半期ごとに私たちのネットワークセキュリティリスクに関する管理職の報告書を受け取った。また,管理層は,必要に応じて任意の重大なネットワークセキュリティイベントおよび任意の影響の小さいイベントの最新状況を委員会に通報する
委員会は、ネットワークセキュリティに関する活動を含む取締役会全員にその活動を報告した。取締役会の全メンバーはまた、我々のネットワークリスク管理計画に関する経営陣のブリーフィングを聴取した。取締役会メンバーは、副総裁、デジタル·情報技術、内部セキュリティ者または外部専門家からネットワークセキュリティテーマに関する講演を聴取し、上場企業に影響を与えるテーマに関する取締役会の継続教育の一部である。
我々の管理チームは、デジタル·情報技術副総裁兼首席財務官総裁を含み、ネットワークセキュリティ脅威による我々の重大なリスクの評価と管理を担当している。このチームは、私たちの全体ネットワークセキュリティリスク管理計画に主な責任があり、私たちの内部ネットワークセキュリティ担当者と私たちが招聘した外部ネットワークセキュリティコンサルタントを監視します。我々の管理チームは,上場製薬会社を数十年間管理した経験があり,その関連する情報技術やネットワークセキュリティリスク管理プロジェクトを含む。
私たちの管理チームは、内部セキュリティ担当者のプレゼンテーション、政府、公共またはプライベートソース(私たちが招聘した外部コンサルタントを含む)から得られた脅威情報および他の情報、およびIT環境に配備されたセキュリティツールによって生成された警報および報告を含む様々な手段によって、ネットワークセキュリティリスクおよびイベントを予防、検出、緩和および修復する作業を監視する
プロジェクト2.建築と不動産
私たちの会社はイギリスロンドンロンドンリバー3号のレンタルオフィススペースに本部を置いています。これらのオフィスのレンタル契約は2025年第1四半期に満期になります。私たちはまたジョージア州サバンナ商業団地33号Suite 300、郵便番号31405、2025年第4四半期満期;ノースカロライナ州ローリー市六フォーク路8529 Six Forks Road、Suite 400、郵便番号27615、2027年第4四半期満期。私たちはこのような施設が私たちの現在と最近の必要性を満たすのに十分だと信じている。
項目3.法的訴訟を起こす
時々、私たちは正常な業務過程で発生した法的訴訟に巻き込まれるかもしれない。私たちは現在、実質的な法的手続きの影響を受けていない。
プロジェクト4.炭鉱の安全状況の開示
適用されません。
第II部
項目5.登録者普通株市場、関連株主事項及び発行者が株式証券を購入する市場
市場情報と保有者
2020年10月30日までに、我々の普通株はロンドン証券取引所AIM Marketで取引され、取引コードは“VRP”である。私たちは2020年10月30日にAIMでの普通株の参入を廃止し、私たちの普通株は現在公開取引をしていません。我々の米国預託株式(ADS)は2017年4月27日からナスダック全世界市場で公開取引され、取引コードはVRNAである。
1株あたりの米国預託株式はヴェローナ製薬会社の普通株8株に相当する。
2024年2月27日現在、私たちは普通議決権株の登録所有者423人がいます。私たちの99.9%の投票権のある普通株はアメリカ預託株式の形で保有されている。私たちの0.1%の普通議決権株式残高は非上場議決権普通株として保有しています。私たちはまだ48,088,896株の未上場で投票権のない普通株を持っている。
配当をする
私たちは私たちの普通株式に対するどんな配当金も発表したり支払ったりしたことがない。私たちは、私たちの将来のすべての収益を維持し、私たちの業務の運営と拡張のために、予測可能な未来に現金配当金を支払わないと予想している。
最近売られている未登録証券
ない。
プロジェクト6.パートナーシップ[保留されている]
プロジェクト7.財務管理部門の財務状況と経営成果の検討分析
当社の財務状況および経営結果に関する以下の議論および分析、および連結財務諸表とこれらのレポートとの関連付記を読むべきです。これらのレポートは、本年度報告の後のForm 10-Kに含まれています。歴史財務情報以外に、以下の討論は展望性陳述を含み、著者らがリスクと不確定要素に関連する計画、推定、信念と期待を反映している。私たちの実際の結果と事件の時間は、このような前向きな陳述で議論されているのとは大きく違うかもしれない。これらの差異を引き起こすかまたは促進する可能性のある重要な要素は、本年度報告における表格10−Kに関する以下および他の部分について議論される要素、特に第1の部分1 A項で議論される要素を含む。“リスク要因”と“前向き陳述に関する戒め”と題した章
本プロジェクト7では,2023年12月31日までと2022年12月31日までの経営実績,および2023年12月31日までと2022年12月31日までの年度との比較を検討する。私たちの2021年度の具体的な議論と分析、および2022年と2021年の財務業績の前年比比較は、2023年3月7日に米国証券取引委員会に提出された2022年12月31日までの会計年度10-K表における財務状況と運営結果の検討および分析の第2部第7項に位置する。
概要
我々は臨床段階の生物製薬会社であり,革新的な治療法の開発に専念し,それを商業化し,重大な満足されていない医療ニーズを持つ慢性呼吸器疾患の治療に用いている。著者らの候補製品ensfentineは研究中の一流の吸入性、選択性、小分子とホスホジエステラーゼ3と4酵素(“PDE 3”と“PDE 4”)の二重阻害剤であり、1種の化合物に気管支拡張剤と非ステロイド抗炎症活性を結合した。
最初、著者らは吸入型アンチフェンタニルを開発しており、慢性閉塞性肺疾患(COPD)の維持治療に応用されており、COPDはよく見られる、慢性的、進行性、生命を脅かす呼吸器疾患であり、治愈できない。開発に成功し、承認されれば、アンテフェンタニルは20数年来初めて新しい作用モードでCOPD維持治療に応用する吸入性療法になることが期待される。
2023年8月,米国食品医薬品局(FDA)はCOPD維持治療のための新薬申請(NDA)の承認を求める審査を受け,処方薬使用料法案(PDUFA)の目標行動日を2024年6月26日とした。FDAは,現在この申請を検討するために諮問委員会会議を開催する予定はないと述べている。
著者らが成功した第三段階Ensifentine(“新型吸入型COPD霧化療法として”)計画の結果に基づいて、著者らは承認されれば、アンテフェンタニルはCOPDの治療モードを変える可能性があると信じている。EnsiftreineはEnhance−1とEnhance−2試験ともに主要な終点に達しており,肺機能測定に統計的有意かつ臨床的に有意な改善が示唆された。また,他の終点データでは,Enhance−1とEnhance−2では,イソフェントラリンがCOPD悪化の確率とリスクを有意に低下させた。エンセフェンタニルは両試験とも耐性が良好であった。
私たちは最近国際科学会議でEnhance試験データの追加分析を提出した
•2023年10月、私たちは2023年胸科年会でEnhance-1とEnhance-2の集合とサブグループ分析に関する4回の報告を発表し、病状の悪化、肺機能、症状と生活の質の終点、および日常薬物使用に関するデータをカバーした。これらのデータは“胸科年会”のオンライン副刊に発表された
•同様に胸部年会で、著者らは疾病意識を高める活動を開始し、日常生活に重大な影響を与える症状を受けても、多くのCOPD患者は医療保健提供者にその症状の真実程度或いは重症度を十分に開示することが困難であることを強調した。この活動は医療保健提供者が患者がどのようにCOPDに対応しているかを理解することを奨励することを目的としている
•2023年9月,我々は2023年ERS国際大会でEnhance−1 24週悪化データの分析を紹介した。要約は同業者評議の出版物に発表されました“ヨーロッパ呼吸雑誌”です
承認されれば,吸入型アンテフェンタニルをCOPDの維持治療に応用して米国(“米国”)で商業化する予定である。患者は既製の標準噴霧噴霧器を使用してアンテフェンタリンを服用するため、薬剤装置の組み合わせとは考えられない。米国以外では,これらの地域で製品を開発·商業化する専門知識や経験を持つ会社にイソフェンタニルの許可証を発行する予定である。そのため、著者らは上海専門製薬会社のニュース製薬有限会社(“ニュース製薬”)と戦略協力を達成し、大中国地区でアンテフェンタニルを開発と商業化した。
第二段階臨床試験では,アンテファニンは慢性閉塞性肺疾患,喘息,嚢胞性線維化(“CF”)患者で陽性結果を示した。慢性閉塞性肺疾患を治療する第二段階試験では,他の2種類の製剤:乾燥粉吸入器(“DPI”)と加圧計量吸入器(“PMDI”)を評価した。
成立以来,経常赤字と運営キャッシュフローが負となっており,2023年12月31日現在の累計赤字は3.884億ドルであった。私たちの候補製品が規制部門の承認を得て商業利益を達成する前に、運営中に追加の損失と負のキャッシュフローを招くことが予想される。
私たちは私たちが行っている活動に関連した巨額の費用を予想しています
•販売、マーケティング、流通インフラを構築し、私たちの製造および他の化学、製造、制御活動を通じて生産を商業規模に向上させ、規制の承認を受ける可能性のある任意の製品を潜在的に商業化する
•DPIとPMDI製剤の臨床開発、および他の製剤の研究と開発、ならびに固定用量のイソフェンと長時間作用性ムスカリン拮抗薬の組み合わせを継続した
•非慢性閉塞性肺疾患、急性COPD、慢性閉塞性肺疾患または任意の他の適応の治療のための更なる臨床試験を開始し、行う
•アンテフェンタリンの他の潜在的適応に関する臨床前研究を開始し、推進した
•より多くの候補品を発見し開発することです
•臨床試験に成功した候補品のために規制の承認を求めています
•私たちの知的財産権の組み合わせを維持し、拡大し、保護する
•より多くの呼吸器候補製品の発見と開発または許可を求めています
•我々の製品開発と将来の潜在的な商業化努力を支援し、米国上場企業としての継続的な運営を支援する人員を含む、臨床、科学、運営、財務および管理情報システムおよび人員を増加させる
•上記のいずれの場合にも、研究失敗、結果複雑、安全問題、または他の規制課題を含むが、これらに限定されない任意の遅延、または任意の問題に遭遇する。
2023年12月27日、私たちは担保代理であるオックスフォード金融有限責任会社(オックスフォード)と、オックスフォードとHercules Capital,Inc.が管理するいくつかの基金と400.0ドルまでの定期融資計画(“2023年定期融資”)を締結した。取引完了時、5,000万ドルは最大4つの合計3.5億ドルの追加前払いによって資金を提供するが、いくつかの規制と商業マイルストーンを満たす必要がある。2023年の定期ローンは、オックスフォード金融ルクセンブルクS.A.R.L.で既存の1.5億ドルのローンを代替しています。付記5-債務を参照してください我々の連結財務諸表と本年度報告におけるその他の部分との関連付記もっと詳しい情報を知ります。
私たちは信じています私たちが2023年12月31日までの現金と現金等価物、および2023年の定期融資項目で得られる予定の資金は、少なくとも2026年末までの計画運営費と資本支出需要に資金を提供することができ、米国で計画されているアンテフェンタリン(承認されれば)を含む資金を提供することができる。2023年の定期ローンでの残りの前払いは、いくつかの臨床および規制マイルストーンの実現および他の指定された条件に依存します。より多くの情報については、“流動性と資本資源”を参照されたい。
重要な合意
配基協定
2004年にRhinophmaを買収し,Ligand UK Development Limited(“Ligand”)(前身はVernalis Development Limited)の債務を担った。私たちは譲渡と許可協定をLigandプロトコルと呼ぶ。
Ligandは,エステファン塩基および関連化合物に関連するある特許および特許出願のすべての権利(“Ligand特許”)と,Ligand特許,Ligand技術およびある化合物を用いた実物在庫開発,Ligand特許,Ligand技術およびある化合物の実物在庫を用いて開発された製品(“Ligand許可製品”)の独占的,世界的な特許使用料許可を譲渡してくれた。
私たちは、いかなる規制機関によるLigandライセンス製品の商業化の初承認を得た場合に500万GBの記念碑的支払いを支払い、すべてのLigandライセンス製品の将来の販売実績に応じて低い1桁の印税を支払うことと、任意の子許可側から受け取った任意のLigand特許とLigandノウハウの対価格の中央値の20%に相当する部分を支払う義務がある。支払うべき特許権使用料は将来の販売実績に基づいているため、支払うべき金額は無制限である。
各事項または問題を解決する際に、Ligandプロトコルに関連するまたは対価格支払い(または対応)を料金として記録する。
2022年3月に、吾らはLigandと改訂協定(“改訂”)を締結し、これによりLigandプロトコルを改訂して、Ligandプロトコルのいくつかの曖昧な言葉を明らかにした。修正案によると:
·修正案の日から5営業日以内にLigandに200万ドルを支払うことに同意し、(Ii)私たちまたは二次許可者が初めてイソニアジドを商業販売したときにLigandに1,500万ドルを支払い、この金額を現金で支払うか、またはナスダック世界市場における米国預託株式の出来高加重平均価格に基づいて適宜決定します。この等価会社の持分は、このマイルストーン事件を含めて10(10)の取引日に基づいて決定されます
·いずれか一方がその条項によって早期に終了しない限り、Ligandプロトコルは2042年3月24日に満了する
·*“Ligandプロトコル”の終了後、修正案で定義されたように、任意の二次ライセンス者は、修正案によって定義されたように、二次被許可者レベルの許可された番組IP部分についてLigandと直接許可プロトコルを締結する権利がある
·マイルストーン支払いを許可することは現金で支払うことができるか、私たちの適宜決定に応じて、Ligandに同値な会社株を発行することで支払うことができる;
·どちらもLigand合意を終了させる権利があり、一方が英国高裁の最終判決を得なければならず、他方がLigand合意下の義務に実質的に違反していることを宣言しなければならないことが条件である。
改正案を実行する際に支払われた200万ドルを総合経営報告書の販売、一般と行政費用、全面赤字に計上します。この支払いは契約改正に関係しているからです。
微妙な協議
吾らはNuance Pharmaと協力及び許可協定(“Nuance協定”)を締結し、2021年6月9日(“Nuance発効日”)から発効し、この協定に基づいて、吾らはNuance Pharmaが大中華中国(中国、台湾、香港及びマカオ)に独自に開発及び商業化する権利を付与した。その見返りに、私たちは合計4,000万ドルの無条件対価格権利を得て、その中には2,500万ドルの現金と、発効日までのNuance Pharma親会社Nuance Biotechで1,500万ドルの株式が含まれている。私たちは将来1.79億ドルに達する記念碑的支払いを得る資格があり、この支払いはある臨床、監督管理と商業マイルストーンの実現及び大中国地区の純売上高の2桁分級特許権使用料によって触発される。収入が大きく逆転しない可能性が高い時、私たちはこのようなマイルストーンを認識するだろう。
2023年12月31日現在、1500万ドルの持分は、本年度報告の他の部分を含む総合貸借対照表に株式と記載されている。株式はコストによって入金され、私たちはいつでも確定できる公正な価値のない株式投資に計量代替案を使用することを選択したからである。私たちは四半期ごとにこの投資の減価指標を評価するつもりだ。私たちは四半期ごとにこの投資の減価指標を評価する。2023年12月31日までの年間で、投資公正価値に大きな影響を与える可能性のある事件や状況変化は確認されていません。
Nuance製薬会社は中国大区におけるアンテフェンタリンの臨床開発と商業化に関するすべての費用を担当する。2022年8月、Nuance Pharmaは中国薬物評価センターの許可を得て、中国大陸でアンテフェントーリンによる慢性閉塞性肺疾患治療の一期と三期研究を開始した。Nuance Pharmaは2023年3月に健康ボランティアの中でアンテフェンタニルの第1段階試験を開始した。2023年4月、Nuance Pharmaは、その重要な3期臨床試験において、第1の被験者のために薬物を服用し、この試験は、中国大陸部におけるアンチフェンの慢性閉塞性肺疾患維持治療の治療効果を評価した。私たちとNuance Pharmaの間には、このような臨床開発と商業化の全体的な進行を監督し、調整するための共同指導委員会が設立されている。共同指導委員会を利用して,大中華区におけるアンチフェンの臨床開発がわれわれ全体のグローバル開発と商業化戦略と一致することを確保することを支援する予定である。
Nuance協定の条項によると、大中華区で中国が最初の新薬申請を提出することが予想される3ヶ月前の任意の時間に、(I)第三者が私たちと協力して開発および/または商業化することに興味がある場合、アンテフェンは、世界的にも、少なくとも米国またはヨーロッパをカバーする地域でも、または(Ii)私たちが制御権変更を発生した場合、Nuance Pharmaに付与されたライセンスおよびすべての関連資産を独占的に選択する権利を有する。双方が同意した価格は、(I)Nuance Pharmaがプロトコルに従って現金形式で私たちに支払ったすべての以前の金額と、(Ii)Nuance PharmaがNuanceプロトコルに従ってensftreineの開発および商業化について生成および支払いしたすべての開発および規制コストに、いくつかのマイルストーンの達成に依存する1桁の範囲を乗じたものに等しいが、特定の最高金額によって制限される。
Nuanceプロトコルは、双方が事前に終了しない限り、各司法管轄区域および各製品に基づいて、当該司法管轄区域内の製品の印税支払い義務が満了するまで継続する。どちらももう一方が治癒していない重大な違約や破産によりNuanceプロトコルを終了することができる。Nuance Pharmaはまた、90日前の書面通知後にNuanceプロトコルを任意に終了することができる
私たちは買い戻し選択権を審査し、第三者を条件としているため、実際にそれを行使する能力がないと判断したので、契約はASC 606に従って入金された。
2022年4月13日、私たちはNuance Pharmaとアンチフェン塩基の生産と供給の合意(“Nuance供給協定”)を締結した。Nuanceの生産と供給は独自の個別的な履行義務であり、その対価格はNuanceによって注文される数量に応じて変化すると確信している。ライセンス製品の製造と供給によって得られた収入は、Nuanceに供給されたときに確認される。私たちはこの協定の下での製造と供給で依頼者を担当することを確認した。元金として、私たちは毛数で関連収入を確認します。2022年12月31日までの1年間に,Nuance Pharma臨床供給アンテフェンタニルに関連する50万ドルを確認した。
Nuanceプロトコルに関するより多くの情報は、本年度報告書の他の部分に含まれる付記6-当社の総合財務諸表の重要なプロトコルおよび関連付記を参照されたい。
肝心な会計見積もり
我々の経営陣の財務状況と経営結果の議論と分析は、米国公認会計原則(“米国公認会計原則”)に基づいて作成された我々の総合財務諸表に基づいている。これらの財務諸表を作成する際には、資産および負債の報告金額、貸借対照表の日付までの、または負債開示および報告期間内に報告された費用金額に影響を与える推定、判断、および仮定を行う必要がある。アメリカ公認会計原則によると、私たちは私たちの推定と判断を継続的に評価するつもりだ。
我々の主な会計政策は,本年度報告その他の部分の総合財務諸表付記により詳細に記述されているが,以下の会計政策が総合財務諸表を作成する際に用いる判断と見積もりが最も重要であると考えられる。
研究開発コスト
研究と発展(“R&D”)コストは発生時に総合経営報告書及び全面損失を計上した。私たちは、サプライヤーとコンサルタントとの契約および私たちの研究開発に関連する臨床現場合意の義務による費用を推定することを要求された。これらの契約の財務条件は交渉を経なければならず、契約と契約の間に差があり、支払流量がこれらの契約に基づいて材料やサービスを提供する期限と一致しない可能性がある。我々の目標は,これらの費用をサービスや努力の支出期限に合わせることで,我々の財務諸表に適切な臨床試験費用を反映させることである。これらの費用は,試験の進展と他の開発活動の進展から計算され,これらの進展は患者の進展と試験の各方面の時間によって測定された。著者らはまた、関係者および外部サービスプロバイダと臨床試験の進展状況または完成した他のサービスを検討することによって、前払いおよび推定数を決定した。臨床試験中に,実際の結果がその推定結果と異なれば,臨床試験費用認知率を調整する可能性がある。私たちは、当時知られていた事実と状況に基づいて、財務諸表において、貸借対照表毎の日付までの前払いおよび計上費用を推定する。実際に発生した金額と実質的に差がないと予想されるにもかかわらず、提供されたサービスの実際の状態および時間に対する提供サービスの状態および時間の理解が異なる可能性があり、私たちが報告する任意の特定の時期の金額が高すぎるか、または低すぎる可能性がある。著者らの臨床試験前払いと費用は契約研究組織からの研究募集と他の第三者サプライヤーが展開した活動を適時かつ正確に報告し、契約研究組織からの任意の変更注文を適時に処理することに依存する。2023年12月31日と2022年12月31日までの臨床試験やその他の開発コストに関する課税費用はそれぞれ70万ドルと1230万ドルである。
業務成果の構成部分
私たちの費用は大幅に増加すると予想されています
•非嚢胞性線維気管支拡張症(NCFBE)、嚢胞性線維症(CF)、喘息または他の適応の治療に用いられる臨床試験を開始し、実施した
•COPDまたは他の適応の治療のための、固定用量の組み合わせを含む他の活性成分との組み合わせを含む、他の製剤において起動および他の臨床試験を行うこと;
•臨床薬理学的研究を開始し実施しました
•より多くの呼吸器候補製品の発見と開発または許可を求めています
•臨床前研究を行い、アンテフェンタリンおよび他の潜在的な未来候補製品を支持する
•臨床および商業的に供給されるアンテフェンタリン活性医薬成分およびそれから誘導される製剤医薬製品を開発すること
•規制部門のアンテファンドリンの承認を求めた
•販売、マーケティング、運営、精算、流通インフラを含むアンテフェンタニルの潜在的な商業化を支援するための商業インフラを発展させ、承認されれば、アンテフェンタニル林を商業化するための製造能力を拡大する
•私たちの知的財産権の組み合わせを維持し、拡大し、保護する
•私たちが許可した技術と製品の操作の自由を確保し、維持し、または獲得すること
•私たちの製品開発および潜在的な将来の商業化努力を支援する人員を含む、臨床、科学、運営、財務および管理情報システムおよび人員を増加させる;
•アメリカやイギリスでのビジネスを拡大しています他の場所にあるかもしれません
今まで、私たちはどんな製品を販売しても収入を得ていない。これまで、すべての収入はNuanceプロトコルによって受信された前払いと供給のアンテファンドリンから来ていた。
将来的には、私たちの製品(私たち自身の販売チームでも第三者販売チームでも)、ライセンス料、マイルストーン支払い、アンテファニンや他の潜在的製品との戦略的協力に関する印税を販売することで収入を得る予定です。私たちは私たちが発生したどんな収入も四半期の変化によって変動すると予想している。もし私たちまたは私たちの戦略パートナーがアンテファニンの開発を適時に完了したり、監督部門の承認を得られなかったり、あるいは私たちが自分の販売チームを発展させたり、1つ以上の戦略パートナーが承認した製品を商業化することができなかった場合、私たちの将来の収入を創出する能力および私たちの財務状況と運営結果は大きな悪影響を受けるだろう。
運営費
研究開発コスト
研究開発コストには、賃金と人事関連コスト、およびアンチフェン研究開発活動の第三者コストが含まれる。関係者のコストには、私たちの株式オプション計画に関連する株式ベースの報酬費用が含まれています。第三者コストの中で最大の部分は臨床試験,および臨床用品と関連開発の製造,および臨床前研究である。研究·開発コストは発生時に費用を計上する。
第三段階増強計画はすでに研究と分析が完了しているため、他の投与方法や適応に新しい化合物を添加したり、アンテフェンタニルをさらに開発したりするまで、2024年上半期よりも前年同期よりも研究コストが低下することが予想される。研究や開発の性質により,期待コストは本質的に不確実であり,我々の現在の予想とは大きく異なる可能性がある.
販売、一般、行政費用
販売、一般及び行政コストには、賃金及び人事関連コスト(株式ベースの給与を含む)、上場会社の運営に関する支出(専門費用、保険及び商業関連コストを含む)、その他の運営費が含まれる。
私たちは、私たちのビジネス運営を発展させ、潜在的な発表に備えて、ビジネスコストが大幅に増加し、規制部門の承認を得ることに成功すれば、販売者、マーケティング、その他の発表に関連するコストが生じると予想している。市場知識の発展と商業化計画の整備に伴い、予想コストは現在の予想と大きく異なる可能性がある。
その他収入/(支出)
その他の収入/(支出)は利息収入と支出、現金と現金等価物と課税すべき外貨変動及びイギリスの研究開発税収控除(“R&D税収控除”)の推進を受けている。
私たちはイギリスの中小企業が税金減免計画を開発することに参加した。税金控除は条件に合った研究開発支出のパーセンテージで計算され、イギリス政府が現金で支払ってくれた。2022年と2023年の財政年度に関する入金金額は2024年に受け取る予定です。
税収
私たちはアメリカとイギリスで会社税を払わなければなりません。私たちは設立以来赤字が発生したため、イギリスの会社税を納めていません。私たちの総合経営と全面赤字報告書の所得税は私たちのアメリカでの経営活動が税収に与える影響を代表しており、これらの活動は会社間のサービス手配に基づいて課税収入を生み出しています。
イギリスの損失は未来の課税利益を相殺するために無期限に繰り越すことができるが、各種の使用基準と制限の制限を受けなければならない。毎年相殺できる金額は500万GBとイギリスの課税利益の50%に制限されている。
2023年と2022年12月31日までの年間経営実績
次の表は,2023年12月31日と2022年12月31日までの年度経営報告書(単位:千)を示している
| | | | | | | | | | | | | | | | | | | | |
| | 十二月三十一日までの年度 | | |
| | 2023 | | 2022 | | 分散.分散 |
| 収入.収入 | | $ | — | | | $ | 458 | | | $ | (458) | |
| 販売コスト | | — | | | (346) | | | 346 | |
| 毛利 | | — | | | 112 | | | (112) | |
| 運営費用: | | | | | | |
| 研究と開発(付記11) | | 17,216 | | | 49,283 | | | (32,067) | |
| 販売、一般、行政 | | 50,353 | | | 26,579 | | | 23,774 | |
| 総運営費 | | 67,569 | | | 75,862 | | | (8,293) | |
| 営業損失 | | (67,569) | | | (75,750) | | | 8,181 | |
| その他の収入/(支出): | | | | | | |
| 研究開発税収控除 | | 1,104 | | 9,634 | | (8,530) | |
| 債務返済損失 | | — | | (815) | | 815 | |
| 利子収入 | | 12,761 | | 2,821 | | 9,940 | |
| 利子支出 | | (2,057) | | (521) | | (1,536) | |
| 為替損益 | | 1,866 | | | (3,817) | | | 5,683 | |
| その他の収入合計,純額 | | 13,674 | | | 7,302 | | | 6,372 | |
| 所得税前損失 | | (53,895) | | | (68,448) | | | 14,553 | |
| 所得税費用 | | (474) | | | (253) | | | (221) | |
| 純損失 | | $ | (54,369) | | | $ | (68,701) | | | $ | 14,332 | |
収入.収入
2022年12月31日までの1年間の収入は50万ドルであり,Nuance Pharmaへの臨床供給材料の販売に関与している。
販売コスト
2022年12月31日までの1年間,Nuance Pharmaに生産販売されている臨床供給材料に関する販売コストは30万ドルであった。
研究開発コスト
2023年12月31日までの1年間、研究開発コストは1720万ドルだったが、2022年12月31日までの1年間、研究開発コストは4930万ドルと3210万ドル減少した。この減少は主に臨床試験や他の開発コストが3,270万ドル減少したためであり,これは第3段階増強計画で発生するコストが少なく,2023年に研究進行と分析が完了したが,2022年には当時行われていた研究進行に関するコストが大きいためである。2023年の臨床試験および他の開発コストには、第3段階増強計画サプライヤーの最終財務入金に関連する220万ドルのクレジットの影響も含まれる。臨床試験およびその他の開発費用の削減には、付記11--引受およびまたは事項が記載されているように、2022年12月31日終了年度の仕入先問題の解決に関連する150万ドルの費用が含まれている私たちの総合財務諸表と本年度報告書の他の部分に含まれる付記。 .
販売、一般、行政費用
2023年12月31日までの1年間、販売、一般、行政コストは5040万ドルだったが、2022年12月31日までの年間は2660万ドルと2380万ドル増加した。この増加は主に人員関係の費用が1,560万ドル増加したためであり,その中に株式ベースの報酬を含めて970万ドル増加したためである
ビジネススタート、市場普及、市場開発費用、出張、その他の企業コスト。これらの増加は、Ligand UK Development Limitedの譲渡と許可協定の修正に関連する200万ドルの非日常的な費用によって部分的に相殺され、この費用は2022年3月31日までの3ヶ月間に発生した。
その他収入/(支出)
2023年12月31日までの年度のその他の収入/(支出)は1370万ドルだったが、2022年12月31日までの年度は730万ドルと640万ドル増加した。この増加は主に高い平均現金残高と高い金利が990万ドルの利息収入を増加させたことと、ポンドが強くなったことに関連して570万ドル増加し、ポンドは2022年に軟調になったためである。この部分が研究開発税収控除の850万ドル減少によって相殺されたのは、2022年と比較して、第3段階増強計画の活動が2023年に減少したことと、サプライヤーの最終的な入金免除の影響によるものである。
キャッシュフロー
次の表は、2023年12月31日と2022年12月31日までの年間キャッシュフロー(単位:千)をまとめています
| | | | | | | | | | | | | | | | | | | | |
| | 十二月三十一日までの年度 | | |
| | 2023 | | 2022 | | 分散.分散 |
| 年初現金および現金等価物 | | $ | 227,827 | | | $ | 148,380 | | | $ | 79,447 | |
| 経営活動のための現金純額 | | (50,222) | | | (59,862) | | | 9,640 | |
| 投資活動のための現金純額 | | — | | | (29) | | | 29 | |
| 融資活動が提供する現金純額 | | 92,869 | | | 140,818 | | | (47,949) | |
| 現金および現金等価物に及ぼす為替レート変動の影響 | | 1,298 | | (1,480) | | 2,778 |
| 年末現金および現金等価物 | | $ | 271,772 | | | $ | 227,827 | | | $ | 43,945 | |
経営活動
2023年12月31日までの年間では、経営活動用の現金純額は5020万ドルだったが、2022年12月31日までの年度は5990万ドルと960万ドル減少したそれは.経営活動に使用される現金の減少は主に臨床試験や他の開発コストの減少によるものであるが,2023年12月31日までの12カ月間の売掛金と2022年12月31日現在の総合貸借対照表における未計費用残高に関する支払部分によって相殺されている。この部分は関係者の費用と情報技術や商業インフラに基づいて建設され、計画中の商業発射に備えている。また現在までに2022年12月31日、2021年研究開発税控除の支払いを受けましたが、2023年12月31日現在、2022年研究開発税収控除870万ドルは受け取っていません。
融資活動
融資活動が提供する現金が減少した要因は,株式発行から受け取った収益が8,340万ドル減少したが,債務ツール発行に関する純収益が3,480万ドル増加し,この減少を部分的に相殺したことである。
流動資金と資本資源
私たちは現在どんな承認された製品もなく、製品販売から何の収入も得たことがない。これまで、私たちは主に株式証券(株式承認証を含む)、私たちの定期融資によって手配された借金、およびNuanceプロトコルに従って受け取った前金によって、私たちの業務に資金を提供してきました。より多くの情報については、“重要な合意”を参照されたい。
成立以来、2023年12月31日現在と2022年12月31日現在の純損失を含む経常赤字が発生し、それぞれ5440万ドルと6870万ドルだった。また、2023年12月31日現在、私たちの累計赤字は3億884億ドルです。予測可能な未来には,我々の研究·開発努力を拡大し,他の配合や他の適応における臨床開発を進め,様々な配合や適応の中で規制部門の承認を得て商業化することが求められているため,重大な運営損失を受け続ける可能性がある。
賃貸と私たちの定期ローンの手配以外に、私たちは信用限度額や保証などの持続的な重大な財務約束を持っておらず、今後5年間の流動性に影響を与えると予想される。
2023年融資と資本取引
•第2期ローン前払い金の下で1,000万ドルを受け取りましたオックスフォード金融ルクセンブルク社と締結された融資と保証契約は、総金額が1.5億ドルに達した(“オックスフォード定期融資”);
•2021年3月に締結した市場発売計画(“2021年ATM計画”)によって20,321,384株の普通株(2,540,173株の米国預託証券に相当)が販売され、平均価格は1株約2.88ドル(米国預託株式23.08ドルに相当)であり、発行コストを差し引いて純収益約5,690万ドルを調達した
•2021年のATM計画の代わりに、Jefferies LLC(“Jefferies”)との公開市場販売協定で、私たちの普通株を米国預託証明書の形で販売し、総収益は2億ドルに達した。
•入力されましたある条項と条件に基づいて、2023年の定期融資に5,000万ドルの定期融資前払いと、総額3.5億ドルに達する追加定期融資前払いを提供する。得られたお金の一部はオックスフォード定期ローンによって借りられた未済債務を全額返済するために使用される。
私たちの2023年の定期融資は私たちにいくつかの金融契約を遵守することを要求し、私たちはこれらのすべての契約を守った2023年12月31日。
付記5--債務を参照我々の連結財務諸表と本年度報告におけるその他の部分との関連付記2023年の定期融資に関するより多くの情報、およびATM計画に関するより多くの情報は、注1ビジネス運営の組織および説明を参照されたい。
資金需要
私たちは、2023年12月31日までの現金と現金等価物に、2023年の定期融資項目で利用可能と予想される追加資金を加えることで、少なくとも2026年末までに、COPD維持治療のための噴霧式アンテファンドリンを米国で発売する予定を含む、計画された運営費および資本支出需要に資金を提供することができると信じています。2023年の定期融資項目の下での将来の前払いは、特定の規制と商業マイルストーンの実現および他の指定された条件に依存します。
私たちはアンテフェンタリンを商業化し、私たちのDPIおよびpMDI製剤の臨床開発を継続し、アンチフェンタリンまたはアンテフェントーリンと一緒の他の製剤を研究および開発するために追加の資金が必要かもしれない。さらに、他の適応のための臨床前または臨床研究の開始または実施、またはより多くの候補製品の発見または許可および開発が求められる可能性がある。私たちは公共または個人融資、債務融資、協力または許可協定、および他の手配を通じて追加資金を求める必要があるかもしれない。しかし、私たちは私たちが受け入れ可能な条件で追加的な資本を得ることに成功するか、または根本的に保証されないという保証はない。
もし私たちが株式または転換可能な債務証券を売却することで追加資本を調達する場合、私たちの株主と米国預託株式保有者の所有権権益は希釈され、これらの証券の条項は清算または他の特典を含む可能性があり、保有者が株主または米国預託株式保有者としての権利に悪影響を及ぼす可能性がある。任意の将来の債務融資または優先株融資は、利用可能であれば、私たちの資産と将来の収入流の担保権益を含む合意に関連する可能性があり、追加債務を招く、資本支出を行う、または配当を宣言するなど、私たちの具体的な行動をとる能力を制限または制限する契約を制限し、承認株式証を発行する必要がある可能性があり、これは私たちの証券保有者の所有権利益を希釈する可能性がある。
もし私たちが第三者との協力、戦略連合、または許可手配を通じてより多くの資金を調達すれば、私たちは私たちの技術、将来の収入源、研究計画、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になる可能性のある条項で許可を与えなければならないかもしれない。もし私たちが必要な時に株式や債務融資を通じてより多くの資金を調達できない場合、私たちは私たちの製品開発計画や将来の商業化努力を延期、制限、減少または終了することを要求されるかもしれません。あるいは、私たちは自分で開発とマーケティングをより望んでいた候補製品の権利を与えます。
私たちの未来のアンチフェンまたは任意の未来の候補製品に対する資本要求は、多くの要素に依存するだろう
•アンテフェンタリンまたは任意の未来の候補製品の臨床前試験および臨床試験の進展、時間および完成状況、および私たちはより多くのアンテフェンタニル臨床試験の可能性を要求されるかもしれない
•私たちは許可と開発の潜在的な新製品候補数を決定した
•私たちの組織をアンチフェンまたは任意の未来の候補製品の研究、開発、および潜在的な商業化を可能にするために必要な規模に発展させるのにかかるコスト
•特許出願の提出、特許の維持および実施、または第三者が提出した特許請求または侵害行為に対する抗弁の費用;
•私たちのために開発されたアンテファンまたは任意の未来の候補製品が規制承認を得るために関連する時間とコスト、および私たちは規制要件の変化またはアンテビリンまたは任意の未来の候補製品に関する不利な結果によって遭遇する可能性のある遅延;
•任意の許可またはマイルストーン費用は、将来の発展、アンチフェン、または任意の未来の製品候補を支払わなければならないかもしれない
•承認されたような予想されるアンテフェンまたは任意の将来の候補製品の商業化に関連する販売およびマーケティング活動、ならびに効果的な販売およびマーケティング組織を確立することに関連する費用;
•私たちは、もしあれば、未来のアンチフェンまたは任意の未来の候補製品(承認された場合)から得られた収入を、特許権使用料の形で直接または販売することができる。
私たちの商業収入(あれば)は、2024年下半期に商業化されると予想される製品の販売から来ます。したがって、私たちは私たちの業務目標を達成するために多くの追加資金を得る必要があるかもしれない。
最近の会計声明
未定と最近採択された会計声明の議論については、付記2“列報基礎と重要会計政策概要”を参照s私たちの総合財務諸表は本年度報告書の他の部分に含まれている。
第七A項。市場リスクの定量的·定性的開示について
私たちが小さい報告会社の会計年度後の第1四半期のForm 10-Q四半期報告ではなく、本プロジェクト7 Aに要求された情報の提供を要求されていません
項目8.連結財務諸表と補足データ
本項目8の規定により提出しなければならない財務諸表は、本年度報告書の後に添付される。これらの財務諸表のインデックスは、本年度報告書第4部第15項に記載されている。
第九項会計基準と会計及び財務開示における会計担当者の変更及び相違
取締役会監査·リスク委員会は2023年12月14日、普華永道会計士事務所(“普華永道”)を解散し、会社が2024年12月31日までの財政年度及び2024年12月31日までの財政年度の総合財務諸表を審査するために、会社が2024年12月31日までの財政年度及び2024年12月31日までの財政年度の総合財務諸表を審査することを許可し、会社株主が2024年株主周年総会で安永を会社の独立監査師(“株主委任”)に委任することを条件とした。株主委任により、安永は当社の現独立原子力数師の普華永道に代わり、普華永道は株主指名を受けて再選されておらず、その独立コア数師の任期は当社の2024年の株主総会後に終了する予定だ。
2022年及び2021年12月31日まで及び12月31日までの年度の総合財務諸表に関する報告書は、不利な意見又は免責声明を含まず、不確実性、監査範囲又は会計原則に対していかなる保留又は修正も行われていない。
(I)当社と普華永道は、2022年及び2021年12月31日までの財政年度内、及び2023年12月14日までの次の移行期間内に、会計原則又は実務、財務諸表開示又は監査範囲又は手順等の事項に“食い違い”はなく(S−K条例第304(A)(1)(Iv)項参照)であり、普華永道を満足させることができない場合には、当該等の年次に関する財務諸表報告に言及することになる。(Ii)S-K条例第304(A)(1)(V)項に記載されているように、“報告可能なイベント”はない。
当社は、2023年12月18日に米国証券取引委員会に提出された現在の8-K表報告書に開示されている内容のコピーを普華永道に提供し、普華永道に米国証券取引委員会に書簡を提出し、本文に含まれる陳述に同意するかどうかを声明することを要求する
2022年12月31日と2021年12月31日までの最近の2つの会計年度、および2023年1月1日から2023年12月14日までのその後の移行期間内に、会社または会社を代表する誰も、(I)完了または予定されている特定の取引に会計原則を適用するか、または会社の総合財務諸表で提出される可能性のある監査意見タイプについて安永と協議しておらず、また、会社が会計、監査または財務報告問題について決定した重要な要素ではないと考えられる書面報告または口頭提案を会社に提供していない。又は(Ii)が“不一致”(S−K条例第304(A)(1)(Iv)項及びその関連指示を参照)又は“報告すべき事象”(S−K条例第304(A)(1)(V)項に記載されているような)に属する任意の事項。
第9条。制御とプログラム
制御とプログラムを開示する
制御とプログラムの有効性の制限
我々の開示制御およびプログラムを設計·評価する際に、管理層は、任意の制御およびプログラムは、設計および動作がどんなに良好であっても、予想される制御目標を達成するために合理的な保証を提供することしかできないことを認識している。また、開示制御およびプログラムの設計は、管理層に、そのコストに対する可能な制御およびプログラムの利益を評価する際に判断することが要求されるリソース制限が存在するという事実を反映しなければならない。
情報開示制御とプログラムの評価
我々の経営陣は、最高経営責任者及び最高財務官の参加の下、本年度報告書に記載されている期間が終了するまでの開示制御及び手順(“取引所法”第13 a−15(E)及び15 d−15(E)条に定義されている)の有効性を評価した。このような評価に基づき、我々の最高経営責任者及び最高財務責任者は、2023年12月31日まで、我々の開示統制及び手続が合理的な保証水準で有効であると結論した。
経営陣財務報告内部統制年次報告書
我々の経営陣は、我々のCEOおよび最高財務官を含み、我々の財務報告に対する十分な内部統制の確立·維持を担当しており、この用語は、“取引法”の下のルール13 a-15(F)および15 d-15(F)で定義されている。
我々の経営陣は,テレデビル委員会後援組織委員会が発表した“内部統制−総合枠組み(2013)”で確立された基準に基づき,2023年12月31日までの財務報告内部統制の有効性を評価した。
この評価に基づき、我々の経営陣は、2023年12月31日現在、財務報告の内部統制に有効であると結論した。
公認会計士事務所認証報告
我々の独立公認会計士事務所普華永道会計士事務所は、2023年12月31日までの財務報告内部統制の有効性を監査しており、このことは本年度報告のF−2ページForm 10−Kで説明している。
財務報告の内部統制の変化
財務報告書の内部統制(取引法第13 a-15(F)および15 d-15(F)条で定義されているように)は変化しておらず、これは、取引法第13 a-15(D)または15 d-15(D)条による経営陣の評価で決定された
2023年12月31日までの四半期内に発生する我々の財務報告の内部統制に重大な影響を与えるか、または合理的に大きな影響を及ぼす可能性が高い“外貨法案”である。
プロジェクト9 B.報告書およびその他の資料
ない。
項目9 C:検査を阻止する外国司法管区の開示について
適用されません。
第三部
プロジェクト10.役員、役員、および企業管理
道徳的規則
我々の取締役会は、我々の最高経営責任者、最高財務官、最高会計官または財務総監、または同様の機能を実行する者を含む、すべての上級管理者、取締役および従業員に適用される書面による商業行為および道徳基準を採択している。私たちはすでに私たちのウェブサイトwww.veronapharma.comの“会社管理”の下の“投資家”の部分に最新の“商業行為と道徳基準”を掲示した。我々は、Form 8-K第5.05項のビジネス行為および道徳基準条項の修正または免除に関する開示要件、および開示役員および役員免除に関するナスダックの要求を満たす予定であり、このような情報を私たちのサイトに公開する方法であり、アドレスおよび位置は上記のように指定されている。当社のウェブサイトに掲載されている資料は参考に本年報に組み込まれていません。
本プロジェクトに必要な残りの資料は,2024年株主総会のために作成した最終依頼書に含まれ,この依頼書を参考に本明細書に組み込まれる。
プロジェクト11.役員報酬
本プロジェクトに要求される資料は,2024年株主総会のために作成された最終依頼書に含まれ,この依頼書を引用することにより本明細書に組み込まれる。
プロジェクト12.特定の実益所有者の保証所有権及び管理職及び株主に関する事項
本プロジェクトに要求される資料は,2024年株主総会のために作成された最終依頼書に含まれ,この依頼書を引用することにより本明細書に組み込まれる。
第13項:特定の関係及び関連取引、並びに取締役独立性
本プロジェクトに要求される資料は,2024年株主総会のために作成された最終依頼書に含まれ,この依頼書を引用することにより本明細書に組み込まれる。
プロジェクト14.総会計士費用とサービス料
本プロジェクトに要求される資料は,2024年株主総会のために作成された最終依頼書に含まれ,この依頼書を引用することにより本明細書に組み込まれる。
第4部
プロジェクト15.証拠品および財務諸表の添付表s
(A)(1)財務諸表
以下の財務諸表および独立公認会計士事務所報告書を本年度報告の一部として提出する
| | | | | | | | |
独立公認会計士事務所レポート(PCAOB ID:876) | | F-2 |
2023年12月31日と2022年12月31日までの連結貸借対照表 | | F-4 |
2023年12月31日と2022年12月31日までの総合経営報告書と全面赤字 | | F-5 |
2023年12月31日と2022年12月31日までの総合株主権益変動表 | | F-6 |
2023年12月31日と2022年12月31日までの統合現金フロー表 | | F-7 |
連結財務諸表付記 | | F-8 |
(A)(2)財務諸表付表。
これらは、適用されない、不要、または必要な資料が財務諸表またはその付記に列挙されているので、すべての財務諸表の添付表は省略される。
(A)(3)展示品。
以下に本年度報告の一部として提出された証拠品リストを示す.
| | | | | | | | | | | | | | | | | | | | | | | |
| | | 指定された文書を引用して統合する | |
| | | | | | | |
| 展示品番号 | 展示品説明 | 表 | 書類番号. | 証拠品番号: | 提出日 | 同封のアーカイブ/提供 | |
| 1.1 | 公開市場販売協定SM日付は2021年3月19日でVerona Pharma PlcとJefferies LLCの間で改訂された | S-3 ASR | 333-270339 | 1.2 | 3/7/2023 | | |
| 3.1 | 改正された現行の有効な定款 | 6-K | 001-38067 | 1 | 12/30/2020 | | |
| 4.1 | 預金協定 | 20-F | 001-38067 | 2.1 | | 2/27/2018 | | |
| 4.2 | 米国預託証明書フォーマット(添付ファイル4.1に添付) | 20-F | 001-38067 | 2.2 | | 2/27/2018 | | |
| 4.3 | 別表Aに指名された投資家ごとに発行する引受権証表 | F-1 | 333-217124 | 4.3 | | 4/3/2017 | | |
| 4.4 | Nplus 1 Singer LLPに株式承認証を発行する | F-1 | 333-217124 | 4.4 | | 4/3/2017 | | |
| 4.5 | 証券説明書 | 10-K | 001-38067 | 4.5 | | 2/29/2024 | * | |
| 10.1 | ヴェローナ製薬会社とその投資家の間で2016年7月29日に締結された“登録権協定” | F-1 | 333-217124 | 10.1 | | 4/3/2017 | | |
| 10.2 | Verona Pharma plcと投資家の間で2020年7月16日に署名された登録権協定 | 6-K | 001-38067 | 2 | | 7/22/2020 | | |
| 10.3.1† | Vernalis Development LimitedとRhinophma Limitedが締結した知的財産権譲渡と許可協定は,Verona Pharma plcの前身として2005年2月7日である | F-1 | 333-217124 | 10.2 | | 4/3/2017 | | |
| 10.3.2 | Verona Pharma plcとLigand UK Development Limitedが2022年3月23日に署名した改訂協定 | 8-K | 001-38067 | 10.1 | | 3/30/2022 | | |
| 10.4.3 | ヴェローナ製薬会社とRegus Management(イギリス)との間の継続レンタル契約制限日:2017年9月16日#1 | 20-F | 001-38067 | 4.3.3 | 2/27/2020 | | |
| 10.4.4 | ヴェローナ製薬会社とRegus Management(イギリス)との間の継続レンタル契約制限日:2017年9月16日#2 | 20-F | 001-38067 | 4.3.4 | 2/27/2020 | | |
| 10.4.5 | ヴェローナ製薬会社とRegus Management(イギリス)との間の継続レンタル契約制限日:2017年9月16日#3 | 20-F | 001-38067 | 4.3.5 | 2/27/2020 | | |
| 10.4.6 | ヴェローナ製薬会社とレッグス管理グループ有限責任会社が2019年7月16日に締結した更新リース契約 | 20-F | 001-38067 | 4.3.6 | 2/27/2020 | | |
| 10.4.7 | ヴェローナ製薬会社とRegus Management(イギリス)との間の継続レンタル契約制限日:2021年11月9日 | 10-K | 001-38067 | 10.4.7 | 3/7/2022 | | |
| 10.4.8 | ヴェローナ製薬会社とRegus Management(イギリス)との間の継続レンタル契約制限日:2021年12月7日 | 10-K | 001-38067 | 10.4.8 | 3/7/2022 | | |
| 10.4.9 | Verona Pharma,Inc.とBrier Creek Office#4,LLC間のリースプロトコルは,2020年3月6日である | 10-K | 001-38067 | 10.4.9 | 3/7/2022 | | |
| 10.5 | 転貸契約は,日付は2023年8月28日であり,Verona Pharma,Inc.とInsightSoftware LLCの間で締結されている | 8-K | 001-38067 | 10.1 | | 9/1/2023 | | |
10.6# | EMIオプションプラン | F-1 | 333-217124 | 10.4 | | 4/3/2017 | | |
10.7# | 未承認の改訂された株式購入計画 | F-1 | 333-217124 | 10.5 | 4/3/2017 | | |
10.8# | ヴェローナ製薬会社が2017年に2回目の改訂と再策定したインセンティブ奨励計画 | 8-K | 001-38067 | 10.1 | | 5/1/2023 | | |
10.9# | ヴェローナ製薬会社とDavid製薬会社との雇用協定は、2020年1月28日となっている。D。 | 20-F | 001-38067 | 4.7 | | 2/27/2020 | | |
10.10# | ヴェローナ製薬会社とキャサリン·リカルドが2019年12月21日に署名した雇用協定 | 20-F | 001-38067 | 4.8 | | 3/19/2019 | | |
| 10.11# | Verona Pharma plcとClaire Pollの間の雇用契約は、2016年10月1日となっている | F-1 | 333-217124 | 10.9 | | 4/3/2017 | | |
| 10.12# | ヴェローナ製薬会社とマーク·ハーンとの雇用協定は、2020年2月1日となっている | F-1 | 333-247928 | 10.12 | | 8/17/2020 | | |
| 10.13# | 取締役会のメンバーの合意形式 | F-1/A | 333-217124 | 10.11.1 | 4/18/2017 | | |
| 10.14# | 行政員による協議の形式 | F-1/A | 333-217124 | 10.11.2 | 4/18/2017 | | |
| 10.15# | サービス福祉計画における従業員の変更を抑える | 8-K | 001-39067 | 10.1 | 8/11/2021 | | |
| 10.16 | Verona Pharma plcに関する関係プロトコルは,2016年7月29日,Verona Pharma plc,OrbiMed Private Investments VI,LPとNPlus 1 Singer Consulting LLPからなる | F-1 | 333-217124 | 10.12 | 4/3/2017 | | |
| 10.17 | Verona Pharma plcに関する関係プロトコルは,2016年7月29日にVerona Pharma plc,Abingworth BioVentures VI LPとNPlus 1 Singer Consulting LLPおよびそれらの間で署名された | F-1 | 333-217124 | 10.13 | 4/3/2017 | | |
| 10.18 | Verona Pharma plcに関する関係プロトコルは,2016年7月29日にVerona Pharma plc,Vivo Ventures Fund VII,L.P.,Vivo Ventures VII Affiliates Fund,L.P.,Vivo Ventures Fund VI,L.P.,Vivo Ventures VI Affiliates Fund,L.P.とNPlus 1 Singer Consulting LLPによって締結された | F-1 | 333-217124 | 10.14 | 4/3/2017 | | |
| 10.19 | Verona Pharma plcに関する関係プロトコルは,2016年7月29日にVerona Pharma plc,Vivo Ventures Fund VII,L.P.,Vivo Ventures VII Affiliates Fund,L.P.,Vivo Ventures Fund VI,L.P.,Vivo Ventures VI Affiliates Fund,L.P.とNPlus 1 Singer Consulting LLPによって締結された | 6-K | 001-38067 | 1 | 7/22/2020 | | |
10.20# | 董事非執行役員招聘状表 | 10-K | 001-38067 | 10.2 | 2/25/2021 | | |
10.21† | Verona Pharma plc,Nuance Pharma LimitedとNuance(Shanghai)Pharma Co Ltd間の協力および許可プロトコルは,2021年6月9日から発効する | 10-Q | 001-38067 | 10.1 | 8/5/2021 | | |
10.22† | 融資と保証契約は,期日は2023年12月27日であり,Verona Pharma,Inc.,Oxford Finance LLCが担保エージェントと貸手として他の貸手と締結されている | 8-K | 001-38067 | 10.1 | 1/2/2024 | | |
| 10.23† | Ritedose社とVerona Pharma社が2023年12月20日に締結した商業供給契約 | 10-K | 001-38067 | 10.23 | 2/29/2024 | * | |
| 21.1 | ヴェローナ製薬会社の子会社リスト | | | | | * | |
| 16.1 | 普華永道有限責任会社からの手紙は、期日は2023年12月14日。 | 8-K | 001-38067 | 16.1 | 12/18/2023 | | |
| 23.1 | 独立公認会計士事務所普華永道法律事務所同意 | | | | | * | |
| 31.1 | 細則13 a-14(A)/15 d-14(A)首席実行幹事の証明 | | | | | * | |
| 31.2 | 細則13 a-14(A)/15 d-14(A)首席財務官の証明 | | | | | * | |
| 32.1 | 第1350条行政総裁の証明 | | | | | ** | |
| 32.2 | 第一百五十条首席財務官の証明 | | | | | ** | |
| 97# | 誤って判決された賠償を追討する政策 | | | | | * | |
| 101.INS | XBRLインスタンスドキュメントを連結する | | | | | * | |
| 101.書院 | イントラネットXBRL分類拡張アーキテクチャ文書 | | | | | * | |
| 101.カール | インラインXBRL分類拡張計算リンクライブラリ文書 | | | | | * | |
| 101.介護会 | XBRL分類拡張ラベルLinkbase文書を連結する | | | | | * | |
| 101.Pre | インラインXBRL分類拡張プレゼンテーションLinkbaseドキュメント | | | | | * | |
| 101.def | インラインXBRL分類拡張Linkbase文書を定義する | | | | | * | |
| 104 | 表紙相互データファイル(添付ファイル101に含まれるイントラネットXBRLのフォーマット) | | | | | * | |
*アーカイブをお送りします。
**関数で提供されます。
#は、管理契約または補償計画を示します。
法規S−K第601(B)(10)項によれば、本展示品の鍔部分(星番号で示す)は省略されている。このような漏れた情報は実質的ではなく、登録者は通常、そのような情報を個人または機密と見なしている。また、S−K条例第601(A)(5)項によれば、本展示品の付表及び添付ファイルは省略されている。
項目16.表格10-Kの概要
ありません
サイン
1934年“証券取引法”第13又は15(D)節の要求に基づいて、登録者は、正式に許可された以下の署名者がその代表を代表して本報告に署名することを正式に手配した。
| | | | | | | | | | | |
| | ヴェローナ製薬会社 |
| | | |
| 日付:2024年2月29日 | | 差出人: | /S/David·ザカドリー |
| | | 製薬会社のDavid·ザカドリーですD。 |
| | | 社長と最高経営責任者 |
本報告は、1934年の証券取引法の要求に基づき、以下の登録者によって登録者として指定日に署名された。
| | | | | | | | | | | | | | |
| | | | |
/S/David·ザカドリー | | 社長と最高経営責任者 (首席行政官) | | 2024年2月29日 |
製薬会社のDavid·ザカドリーですD。 | | | | |
| | | | |
/S/マーク·W·ハーン | | 首席財務官 (首席財務会計官) | | 2024年2月29日 |
マーク·W·ハーン | | | | |
| | | | |
/S/David·エブスワース博士 | | 取締役会議長 | | 2024年2月29日 |
デヴィッド·エブス博士です | | | | |
| | | | |
/S/クリスティーナ·アッカーマン | | 役員.取締役 | | 2024年2月29日 |
クリスティーナ·アッカーマン | | | | |
| | | | |
/S/マイケル·オズウィック | | 役員.取締役 | | 2024年2月29日 |
マイケル·オストウィック | | | | |
| | | | |
/S/ジェームズ·ブレイディ | | 役員.取締役 | | 2024年2月29日 |
ジェームズ·ブレイディ | | | | |
| | | | |
/S/ケン·カンニングアン医学博士 | | 役員.取締役 | | 2024年2月29日 |
ケン·カンニンアン医学博士です | | | | |
| | | | |
/S/リサ·デシャン | | 役員.取締役 | | 2024年2月29日 |
リサ·デシャン | | | | |
| | | | |
/S/マーティン·エドワーズ医学博士 | | 役員.取締役 | | 2024年2月29日 |
マーティン·エドワーズ医学博士 | | | | |
| | | | |
著者S/Mahendra Shah,Ph.D. | | 役員.取締役 | | 2024年2月29日 |
Mahendra Shah博士 | | | | |
| | | | |
/投稿S/ヴィカス·シンハ | | 役員.取締役 | | 2024年2月29日 |
ヴィカス·シンハ | | | | |
| | | | |
S/アンダース·ウルマン医学博士 | | 役員.取締役 | | 2024年2月29日 |
アンダース·ウルマン医学博士 | | | | |
索引.索引
| | | | | | | | |
独立公認会計士事務所レポート(PCAOB ID: 876) | | F-2 |
2023年12月31日と2022年12月31日までの連結貸借対照表 | | F-4 |
2023年12月31日と2022年12月31日までの総合経営報告書と全面赤字 | | F-5 |
2023年12月31日と2022年12月31日までの総合株主権益変動表 | | F-6 |
2023年12月31日と2022年12月31日までの統合現金フロー表 | | F-7 |
連結財務諸表付記 | | F-8 |
独立公認会計士事務所報告
ヴェローナ製薬有限公司の取締役会と株主へ
財務諸表と財務報告の内部統制に関するいくつかの見方
本監査人はVerona Pharma plc及びその付属会社(“貴社”)の二零二三年十二月三十一日及び二零二年十二月三十一日の総合貸借対照表、及び当該年度までの関連総合運営及び全面損益表、株主権益及びキャッシュフロー表を審査し、関連付記(総称して“総合財務諸表”と呼ぶ)を含む。テレデビル委員会(COSO)協賛組織委員会が発表した“内部統制-総合枠組み(2013)”で確立された基準に基づき、2023年12月31日までの財務報告内部統制を監査した
これらの総合財務諸表は,当社の2023年12月31日と2022年12月31日までの財務状況,および当該日までの経営業績とキャッシュフローを各重大な面で公平に反映しており,米国公認の会計原則に適合していると考えられる。また,COSOが発表した“内部制御−総合枠組み(2013)”で確立された基準に基づき,2023年12月31日現在,会社はすべての実質的な面で財務報告に対して有効な内部統制を実施していると考えられる。
意見の基礎
当社経営陣は、このような総合財務諸表の作成、有効な財務報告内部統制の維持、財務報告内部統制の有効性の評価を担当しており、9 A項の経営陣の財務報告内部統制年次報告書に含まれている。私たちの責任は、私たちの監査に基づいて、会社の合併財務諸表と会社が財務報告の内部統制に対して意見を述べることです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、連結財務諸表に重大なミスがないかどうか、エラーによるものであっても詐欺であっても、すべての重大な点で財務報告に対する有効な内部統制が維持されているかどうかを決定するために、合理的な保証を得るために監査を計画し、実行することを要求する。
我々の連結財務諸表の監査には、連結財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであっても詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれる。これらの手続きは、連結財務諸表中の金額および開示に関する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、合併財務諸表の全体列報の評価も含まれています。我々の財務報告の内部統制の監査には、財務報告の内部統制を理解すること、重大な弱点があるリスクを評価すること、評価されたリスクテストに基づいて内部統制の設計と運営有効性を評価することが含まれる。私たちの監査はまた、私たちがこのような状況で必要だと思う他の手続きを実行することを含む。私たちは私たちの監査が私たちの意見に合理的な基礎を提供すると信じている。
財務報告の内部統制の定義と限界
会社の財務報告に対する内部統制は、公認された会計原則に基づいて、財務報告の信頼性と外部目的の財務諸表の作成に合理的な保証を提供することを目的としたプログラムである。会社の財務報告に対する内部統制は、(1)合理的で詳細かつ正確かつ公平に会社の資産を反映した取引および処分の記録を維持することに関連している、(2)公認された会計原則に従って財務諸表を作成するために取引が必要とされている合理的な保証を提供し、会社の収入および支出は会社の経営陣および取締役の許可のみに基づいて行われる、という政策と手続きを含む。および(Iii)財務諸表に重大な影響を与える可能性のある不正買収、使用または処分会社の資産を防止またはタイムリーに発見することについて合理的な保証を提供する。
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。さらに、将来のどのような有効性評価の予測も制御される可能性がある
条件の変化により不十分であったり,政策やプログラムを遵守する程度が悪化したりする可能性がある.
重要な監査事項
以下に述べる重要な監査事項は、監査委員会に監査委員会に伝達または要求を伝達することを指し、(I)総合財務諸表に対して重大な意義を有する勘定または開示に関連し、(Ii)私たちが特に挑戦的、主観的または複雑な判断を有する当期総合財務諸表監査によって生じる事項に関するものである。重要監査事項の伝達は、総合財務諸表に対する私たちの全体的な意見をいかなる方法でも変えることはなく、以下の重要な監査事項を伝達することによって、重要な監査事項又はそれに関連する勘定又は開示について個別の意見を提供することもない。
定期融資手配の修正を計算する
総合財務諸表付記1、2及び5に記載されているように、2023年12月27日(“2023年発効日”)に、当社は2023年発効日に総額5,000,000,000ドルの定期融資パッド金(“A期融資”)と、いくつかの条項および条件制限を受けた4件の追加定期融資パッド金を含む、最大4,000,000,000ドルまでの定期融資手配(“2023年定期融資”または“融資協定”)を締結した。2023年の定期融資は、会社の既存の1億5千万ドルの融資に代わった。当社はA期ローンから純収益を得ており、一部は会社が前の2000万ドルの定期融資によって借りた既存の未返済債務所によって全額返済されている。債務が修正され、新しい債務ツールと古い債務ツールの条項が“大きく異なる”場合、債務を返済すると見なすことができる。経営陣の融資協議会計に対する評価によると、管理層はASC 470-50に基づいてA期融資の一部を修正会計処理している債務修正と返済”.
我々は、定期融資手配の会計修正に関する実行手続きが重要な監査事項であることを主な考慮要因と認定している:(I)この事項は重大な取引を代表し、(Ii)監査人は、会社の定期融資手配の修正および清算評価会計に関する手続きを実行し、監査証拠を評価する上での高度な努力である。
この問題を処理することは、統合財務諸表に対する私たちの全体的な意見を形成するための実行手順および評価監査証拠に関するものである。これらの手続きには、債務改正に関連する重大な取引の管理層会計制御措置の有効性をテストすることが含まれる。他にも、これらの手続きは、(1)新しい定期融資に対する管理層の会計評価、特に適用される会計基準に基づいてその修正および補償の評価を評価するステップと、(2)連結財務諸表に開示される十分性を評価するステップと、を含む
/s/ 普華永道会計士事務所
レイデン、イギリス
2024年2月29日
2015年以来、当社の監査役を務めてきました。
ヴェローナ製薬会社
合併貸借対照表
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
| | | | | | | | | | | | | | |
| | 十二月三十一日 |
| | 2023 | | 2022 |
| 資産 |
| 流動資産: | | | | |
| 現金と現金等価物 | | $ | 271,772 | | | $ | 227,827 | |
| | | | |
| 前払い費用 | | 3,617 | | | 2,499 | |
| 課税優遇 | | 10,954 | | | 9,282 | |
| | | | |
| その他流動資産 | | 3,365 | | | 3,388 | |
| 流動資産総額 | | 289,708 | | | 242,996 | |
| | | | |
| 非流動資産: | | | | |
| 家具と設備、網具 | | 24 | | | 73 | |
| 商誉 | | 545 | | | 545 | |
| 株権 | | 15,000 | | | 15,000 | |
| 使用権資産 | | 2,847 | | | 854 | |
| | | | |
| 非流動資産総額: | | 18,416 | | | 16,472 | |
| 総資産 | | $ | 308,124 | | | $ | 259,468 | |
| | | | |
| 負債と株主権益 |
| 流動負債: | | | | |
| 売掛金 | | $ | 3,492 | | | $ | 2,910 | |
| 費用を計算する | | 3,585 | | | 13,752 | |
| 流動経営賃貸負債 | | 1,180 | | | 675 | |
| 課税税金を納める | | — | | | 283 | |
| その他流動負債 | | 435 | | | 1,409 | |
| 流動負債総額 | | 8,692 | | | 19,029 | |
| | | | |
| 非流動負債: | | | | |
| | | | |
| 定期ローン | | 48,374 | | | 9,768 | |
| 非流動経営賃貸負債 | | 1,775 | | | 205 | |
| 非流動負債総額 | | 50,149 | | | 9,973 | |
| 総負債 | | 58,841 | | | 29,002 | |
| | | | |
| 引受金とその他の事項 | | | | |
| | | | |
| 株主権益 | | | | |
普通国標0.05額面株:667,659,630そして631,338,246発表されました643,536,094そして606,301,054未返済は、それぞれ2023年12月31日と2022年12月31日です | | 42,771 | | | 40,526 | |
| 追加実収資本 | | 601,063 | | | 529,187 | |
| 国庫保有普通株 | | (1,517) | | | (1,549) | |
| その他の総合損失を累計する | | (4,601) | | | (4,601) | |
| 赤字を累計する | | (388,433) | | | (333,097) | |
| 株主権益総額 | | 249,283 | | | 230,466 | |
| 総負債と株主権益 | | $ | 308,124 | | | $ | 259,468 | |
付記はこれらの連結財務諸表の構成要素である。
ヴェローナ製薬会社
合併経営報告書と全面赤字
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
| | | | | | | | | | | | | | | | | |
| | 十二月三十一日までの年度 | | | |
| | 2023 | | 2022 | | | |
| 収入.収入 | | $ | — | | | $ | 458 | | | | |
| 販売コスト | | — | | | (346) | | | | |
| 毛利 | | — | | | 112 | | | | |
| 運営費用: | | | | | | | |
研究と開発(付記11) | | 17,216 | | | 49,283 | | | | |
| 販売、一般、行政 | | 50,353 | | | 26,579 | | | | |
| 総運営費 | | 67,569 | | | 75,862 | | | | |
| 営業損失 | | (67,569) | | | (75,750) | | | | |
| その他の収入/(支出): | | | | | | | |
| 研究開発税収控除 | | 1,104 | | 9,634 | | | |
| 債務返済損失 | | — | | (815) | | | |
| 利子収入 | | 12,761 | | 2,821 | | | |
| 利子支出 | | (2,057) | | (521) | | | |
| 為替損益 | | 1,866 | | | (3,817) | | | | |
| その他の収入合計,純額 | | 13,674 | | | 7,302 | | | | |
| 所得税前損失 | | (53,895) | | | (68,448) | | | | |
所得税費用 | | (474) | | | (253) | | | | |
| 純損失 | | $ | (54,369) | | | $ | (68,701) | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| 普通株1株当たりの損失--基本損失と赤字 | | $ | (0.09) | | | $ | (0.13) | | | | |
| | | | | | | |
| 加重平均流通株−基本と希釈 | | 634,142,660 | | | 529,071,526 | | | | |
付記はこれらの連結財務諸表の構成要素である。
ヴェローナ製薬会社
合併株主権益報告書
(共有データを除く単位は千)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | 普通株 | | 追加実収資本 | | 国庫保有普通株 | | その他の総合損失を累計する | | 赤字を累計する | | 株主権益総額 |
| | 番号をつける | | 金額 | | | | | |
| 2022年1月1日の残高 | | 489,177,550 | | | $ | 31,855 | | | $ | 385,070 | | | $ | (603) | | | $ | (4,601) | | | $ | (263,716) | | | $ | 148,005 | |
| 純損失 | | — | | | — | | | — | | | — | | | — | | | (68,701) | | | (68,701) | |
| 普通株発行は発行コストを差し引く | | 114,080,000 | | | 6,918 | | | 133,279 | | | — | | | — | | | — | | | 140,197 | |
| 市況協定に基づいて普通株を発行する | | 80,696 | | | 5 | | | 62 | | | — | | | — | | | — | | | 67 | |
| 国庫に普通株を発行する | | 28,000,000 | | | 1,748 | | | — | | | (1,748) | | | — | | | — | | | — | |
| 帰属制限株式単位 | | — | | | — | | | — | | | 680 | | | — | | | (680) | | | — | |
| 行使された購入権 | | — | | | — | | | 1,250 | | | 122 | | | — | | | — | | | 1,372 | |
| 株式ベースの報酬 | | — | | | — | | | 14,121 | | | — | | | — | | | — | | | 14,121 | |
| 既得株式奨励で税金を源泉徴収した普通株 | | — | | | — | | | (4,723) | | | — | | | — | | | — | | | (4,723) | |
| 株式決済は株式の報酬に基づいて現金決済に再分類される | | — | | | — | | | 128 | | | — | | | — | | | — | | | 128 | |
| 2022年12月31日の残高 | | 631,338,246 | | | $ | 40,526 | | | $ | 529,187 | | | $ | (1,549) | | | $ | (4,601) | | | $ | (333,097) | | | $ | 230,466 | |
| 純損失 | | — | | | — | | | — | | | — | | | — | | | (54,369) | | | (54,369) | |
| | | | | | | | | | | | | | |
| 市況協定に基づいて普通株を発行する | | 20,321,384 | | | 1,227 | | | 55,682 | | | — | | | — | | | — | | | 56,909 | |
| 国庫に普通株を発行する | | 16,000,000 | | | 1,018 | | | — | | | (1,018) | | | — | | | — | | | — | |
| 帰属制限株式単位 | | — | | | — | | | — | | | 967 | | | — | | | (967) | | | — | |
| 行使された購入権 | | — | | | — | | | 1,866 | | | 83 | | | — | | | — | | | 1,949 | |
| 株式ベースの報酬 | | — | | | — | | | 19,012 | | | — | | | — | | | — | | | 19,012 | |
| 既得株式奨励で税金を源泉徴収した普通株 | | — | | | — | | | (4,389) | | | — | | | — | | | — | | | (4,389) | |
| 株式決済は株式の報酬に基づいて現金決済に再分類される | | — | | | — | | | (295) | | | — | | | — | | | — | | | (295) | |
| 2023年12月31日の残高 | | 667,659,630 | | | $ | 42,771 | | | $ | 601,063 | | | $ | (1,517) | | | $ | (4,601) | | | $ | (388,433) | | | $ | 249,283 | |
付記はこれらの連結財務諸表の構成要素である。
| | | | | | | | | | | | | | | |
| | 十二月三十一日までの年度 |
| | 2023 | | 2022 | |
| 経営活動: | | | | | |
| 純損失: | | $ | (54,369) | | | $ | (68,701) | | |
| 純収入と業務活動で使用される現金純額を調整する: | | | | | |
| 外国為替(収益)/損失 | | (1,866) | | | 3,817 | | |
債務発行原価償却 | | 116 | | | 80 | | |
| 債務償還割増の増加 | | 106 | | | 108 | | |
| 債務返済損失 | | — | | | 815 | | |
| | | | | |
| | | | | |
| 株式ベースの報酬 | | 19,012 | | | 14,121 | | |
| 減価償却および償却 | | 677 | | | 636 | | |
| | | | | |
| 経営性資産と負債変動状況: | | | | | |
| | | | | |
| | | | | |
| 前払い費用 | | (1,118) | | | 1,538 | | |
| 課税優遇 | | (1,104) | | | 3,964 | | |
| その他流動資産 | | 777 | | | (1,325) | | |
| | | | | |
| 売掛金 | | 486 | | | (7,146) | | |
| 費用を計算する | | (10,351) | | | (8,504) | | |
| リース負債を経営する | | (591) | | | (597) | | |
所得税 | | (1,037) | | | 136 | | |
| その他流動負債 | | (960) | | | 1,196 | | |
| 経営活動のための現金純額 | | (50,222) | | | (59,862) | | |
| 投資活動によるキャッシュフロー: | | | | | |
| 家具と設備を買う | | — | | | (29) | | |
| | | | | |
| 投資活動のための現金純額 | | — | | | (29) | | |
| 資金調達活動のキャッシュフロー: | | | | | |
| 普通株式を発行して得た金 | | 56,909 | | | 149,797 | | |
| 普通株の発行に関する発売料金をお支払いください | | — | | | (9,533) | | |
オックスフォードの定期ローンの収益は | | 9,996 | | | 10,000 | | |
2023年の定期ローン収益、オックスフォード定期ローンの返済と発生した債務発行コストを差し引いた純額 | | 28,712 | | | — | | |
| 債務発行コストを支払う | | (12) | | | (245) | | |
SVBの定期ローンを返済する | | — | | | (5,000) | | |
SVB定期ローン返済コスト | | — | | | (850) | | |
| 株式奨励金から源泉徴収税を支払う | | (4,685) | | | (4,723) | | |
| 持分を行使して得た金 | | 1,949 | | | 1,372 | | |
| | | | | |
融資活動が提供する現金純額 | | 92,869 | | | 140,818 | | |
| 現金および現金等価物に及ぼす為替レート変動の影響 | | 1,298 | | | (1,480) | | |
| 現金と現金等価物の純変化 | | 43,945 | | | 79,447 | | |
| 年初現金および現金等価物 | | 227,827 | | | 148,380 | | |
| 年末現金および現金等価物 | | $ | 271,772 | | | $ | 227,827 | | |
| キャッシュフロー情報の追加開示: | | | | | |
| 納めた所得税 | | $ | 1,245 | | | $ | 120 | | |
| 支払の利子 | | $ | 2,006 | | | $ | 348 | | |
付記はこれらの連結財務諸表の構成要素である。
注1-業務運営の組織と記述
ヴェローナ製薬会社はイギリスに登録して設立して登録した。ヴェローナ製薬会社は1つはVerona Pharma,Inc.これはデラウェア州の会社です(Verona Pharma plcとともに“会社”と呼ばれています)。登録事務所の住所はイギリスカーディフ中央広場1番地、CF 10 1 FSです。
同社は臨床段階の生物製薬グループであり,重大に満たされていない医療ニーズを持つ呼吸器疾患の革新的な治療法の開発と商業化に専念している。会社の米国預託株式(“米国預託株式”)はナスダック世界市場(“ナスダック”)に上場し、取引コードは“VRNA”である
2023年8月、米国食品医薬品局(FDA)は、同社が慢性閉塞性肺疾患(COPD)維持治療のための新薬申請(NDA)の承認を求め、処方薬使用料法案(PDUFA)の目標行動日を2024年6月26日とした。FDAは,現在この申請を検討するために諮問委員会会議を開催する予定はないと述べている。同社は2024年に可能な商業発売に備えており、秘密保持協定の承認が待たれている。
2023年6月に秘密保持協定を提出すると同時に、会社は#ドルを支払った3.2FDAに10万PDUFA申請料を支払う。当社は、FDAの承認を受け、2023年12月31日までの3ヶ月以内に返金することを、小企業にこの申請料を免除することを要求している。
流動性
会社は設立以来経営に経常赤字とマイナスキャッシュフローが生じ,累計損失#ドル388.42023年12月31日現在、1億2千万ドル。同社は、その製品が規制部門の承認を得て商業利益(あれば)を実現する前に、運営に追加の損失と負のキャッシュフローが生じると予想している。
同社は、2023年12月31日までの現金および現金等価物は、発行日から少なくとも今後12カ月の運営費および資本支出需要を支払うのに十分であると予想している。
2022年8月、会社はより大規模な公募株を完成させた14,260,000ADS、各ADS代表8人当社の普通株、額面GB0.051株当たりの価格は$です10.50アメリカ預託株式によると、その中には引受業者がその購入追加を全面的に行使します1,860,000アメリカ預かり証明書です。はい発行された総純収益は$です140.2引受割引と手数料、支払うべき発売費用を差し引いた後、100万ドルとなります。
当社は2022年12月31日までに販売します80,696普通株(普通株に相当)10,087米国預託証明書)2021年3月に締結された市場発売計画(“2021年ATM計画”)によると、平均価格は約$0.861株あたり(同じ$に等しい6.86アメリカ預託株式によると,合計純収益ドルを調達する0.1発行コストを差し引いて100万ドルです。2022年12月31日まで$99.22021年のATM計画によると、売却可能な米国預託証券形式の普通株は1億8千万株。
当社は2023年12月31日までに販売します20,321,384普通株(普通株に相当)2,540,1732021年ATM計画では,平均価格は約$である2.881株あたり(同じ$に等しい23.08アメリカ預託株式によると,合計純収益ドルを調達する56.9発行コストを差し引いた百万ドルです
2023年3月、S表3の登録声明により、会社はジェフリー有限責任会社(“ジェフリ”)との公開市場販売協定で2021年のATM計画に代わり、米国預託証明書の形で普通株を売却し、総収益は最高$に達する200.0ジェフリーが販売代理を務める“市場で”株式発行計画(“2023年現金自動支払機計画”)により、不定期に2億5千万ドルの資金を提供する。ジェフリーにはガンダムを獲得する権利がある3.0毛収入の%です
当社は2023年12月に最高$までの定期融資取り決めを締結した(“2023年定期ローン”)400.01000万ドル、オックスフォード金融有限責任会社(オックスフォード)が担保代理として、オックスフォードとHercules Capital,Inc.が管理するいくつかの基金50.0億ドルの資金は最高から四つ合計追加前払い$350.01,000,000ドルは、会社が特定の規制とビジネスマイルストーンを達成することに依存します。2023年の定期融資は会社の既存の$に取って代わる150.0オックスフォード金融ルクセンブルクS.A.R.L.との100万ユーロローン。詳細は付記5-債務を参照されたい。
同社の商業収入(あれば)は、2024年下半期に発売される予定の製品から販売される。また、当社は時々ライセンス外取引を行う可能性がありますが、当社が後日このような取引を確保できる保証はありません。したがって、会社はその業務を実現するために多くの追加資金を得る必要があるかもしれない
目標は、臨床および規制活動をさらに推進し、関連コストの導入に資金を提供することと、承認された場合にアンテフェンタニルを商業化するための効果的な販売·マーケティング組織を作成することである。このような資金は、公的または個人融資、債務融資、協力または許可手配、または他の手配によって得られる必要がある。しかし、同社が受け入れ可能な条件で追加資本を得ることに成功するか、または全く保証されない保証はない。
注2-主要会計政策の列報根拠と概要
列報と合併の基礎
連結財務諸表には、Verona Pharma plcおよびその完全子会社Verona Pharma,Inc.の口座が含まれています。会社間のすべての残高および取引はログアウトされました
総合財務諸表は米国公認会計原則(“米国公認会計原則”)に基づいて作成され、以下の会計政策を踏襲してきた。
予算の使用
米国公認会計原則に基づいて連結財務諸表を作成することは、連結財務諸表の日付の資産、負債、又は有資産と負債の開示及び報告期間内に報告された費用金額に影響を与えるために、管理層に推定と仮定を要求する。これらの総合財務諸表に反映される重大な推定および仮定は、研究開発費の計算および前払い、ならびに株式ベースの報酬の公正な価値を含むが、これらに限定されない。状況、事実、経験の変化に応じて、見積数を定期的に審査する。推定された変化は、それらが知られている期間に記録される。実際の結果は会社の見積もりとは違うかもしれません。
企業合併
当社は買収方法を用いて企業合併を計算します。買収子会社の譲渡対価は,譲渡資産の公正価値,被買収側の前所有者に対する負債,および当社が発行した株式である.移転された対価格には、価格設定によって生成された任意の資産または負債の公正な価値が含まれる。買収コストは、買収した純資産に占めるシェアを識別できる公正価値の一部を営業権に計上することを超える
企業合併で取得した確認可能資産及び負担された負債及び又は有負債は、最初に買収日の公正価値に応じて計量される。買収に関するコストは発生時に費用を計上して行政費用に計上する。
現金と現金等価物
当社は買収時の原始満期日が90日以下のすべての高流動性投資を現金等価物と見なしている。現金および現金等価物には、銀行に随時待機する預金と、米国および英国政府債券に投資される通貨市場基金と、高格付け機関からの流動性証券が含まれる。
株権
Nuanceプロトコルの一部として,当社はNuance Pharma親会社Nuance Biotechの持分を獲得した(付記6−重大合意参照)。Nuance Biotechの証券は公開取引されていないため、株式の公正価値は容易に確定できない。したがって、会社はASC 321-10-35-2の指導に従い、公正価値を用いて代替案を計量し、Nuance Biotech株の最後に取引所が示す価値とみなされる証券をコスト別に計量するが、減値しなければならない。推定値は、Nuance Biotechが同じまたは類似した投資の順序取引において観察可能な任意の価格変化に基づいて調整されるか、または減少指標が存在するか否かに基づいて調整される。
家具と設備、網具
家具と設備はオフィス家具、コンピュータ設備及びレンタル改善を含み、コストから減価償却累計額を差し引いた。家具や設備の減価償却は一般に予想可能な経済年限内で直線的に計算される二つ至れり尽くせり5年それは.リース改善減価償却は、資産の経済寿命またはリース期間内に比較的短い時間で計上される。
商誉
営業権は鼻咽頭癌の買収と関連する商業的名声を含む。営業権は償却しないが、定期的に減価テストを行う。
長期資産減価準備
当社は毎年またはイベントや環境変化が資産の帳簿価値を完全に回収できない可能性がある場合には,長期資産の減価審査を行う。
債務と債務発行コスト
新たな債務ツールを発行する際には、当社が確認した負債は、受信した収益に等しく、債務と共に発行された他のツール、取引の他の要素又は債務ツール自体の特徴を差し引いた任意の収益分配を行う。収益は一般に債務の利息と元金支払いの現在値に近い。
借り手が当社の財務困難に関連する経済的又は法的理由から、考慮しない特許権を当社に提供する場合、関連融資は問題債務再編に分類される。再構成が問題債務再構成を構成しない場合は、終了または修正と見なすべきかどうかを考慮するために評価する
債務が修正され、新しい債務ツールと古い債務ツールの条項が“大きく異なる”(ASC 470-50“債務修正および弁済”の債務修正ガイドラインで定義されているように)場合、債務を弁済と見なすことができる。
当社の債務ツールに関連する債務発行コストは、総合貸借対照表の定期融資において関連債務帳簿金額の直接減価として計上され、これらのコストは実際の利息法を用いて繰延され、関連債務のそれぞれの条項に従って利息支出に償却される。
収入確認
同社の収入には、同社のアンテファンの戦略合意の開発と商業化の収入が含まれている。協定の条項は、払い戻し不可能な前払い費用、マイルストーンに基づく支払い、および最終的に商業化された製品からの収入を含むことができる。このような合意は一般的に固定的な対価もあれば、可変対価もある。払い戻し不可能な前払い費用は固定されていると考えられ、マイルストーン支払いおよび商業化製品の収入は可変考慮要素として決定される。
ASC主題606の範囲内で合意に規定された義務を履行する際に確認すべき適切な収入額が決定された場合、会社は、(1)契約で約束された貨物またはサービスを決定するステップと、(2)契約中に異なるかどうかを含む契約義務であるかどうかを決定するステップと、(3)可変対価格の制限を含む取引価格を測定するステップと、(4)推定販売価格に基づいて取引価格を履行義務に割り当てるステップと、(5)会社が各履行義務を履行する場合(または履行義務として)収入を確認するステップとを実行する
履行義務は、契約において独自の商品またはサービスを顧客に譲渡する承諾であり、ASCテーマ606における計算単位である。会社の履行義務は、知的財産権(ライセンス、特許、および開発および規制データを含む)および製造および供給を含むことができる。経営陣はいつ業績義務を履行し、いつ収入を確認したかを判断しなければならない。
当社は、契約義務ごとに係る約束商品又はサービスの推定相対独立販売価格に基づいて、契約義務毎に総取引価格を割り当てる
知的財産権ライセンス取り決めについては、販売水準に基づくマイルストーン支払いを含む販売ベースの使用料を含み、ライセンスが使用料に関連する主要項目とみなされ、当社は、(I)関連販売が発生した場合、又は(Ii)分配された使用料の履行義務が履行された場合に、使用料収入及び販売に基づくマイルストーンを確認する。会社の知的財産権の権利が手配中に決定された他の履行義務とは異なると判定された場合、会社は、権利が顧客に譲渡されたときにその権利に割り当てられた払戻不可能な前払い費用の収入を確認し、顧客はその権利を使用して利益を得ることができる。
手配開始時に,会社は開発マイルストーンが実現可能であると考えられているかどうかを評価し,最も可能性のあるものを使用する
数量法。大きな収入逆転が起こらない可能性が高い場合、関連するマイルストーン価値は取引価格に含まれるだろう。規制当局の承認など、会社の統制範囲内でない記念碑的支払いは、これらの承認を受ける前に実現可能であるとは考えられない。
研究開発コスト
研究と開発(“R&D”)コストは発生時に費用を計上する。研究開発費には、予め承認された製造コスト、臨床研究機関および契約メーカーとの契約を含む、従業員の給与、株式ベースの給与および福祉、および会社の研究開発活動に関連する他のコストが含まれる。同社は,サプライヤーやコンサルタントとの契約およびその研究開発に関連する臨床場合意に基づいて負担する義務による費用を推定しなければならない。これらの契約の財務条項は契約によって異なる可能性があり、支払流量が当該等の契約に基づいて当社に材料やサービスを提供する期限と一致しない可能性があります。同社の目標は,これらの費用をサービスや努力の支出期限に合わせることで,その総合財務諸表に適切な臨床試験費用を反映させることである。会社は試験や他の開発活動の進捗状況に基づいてこれらの費用を会計処理している。患者の進行および進行を測定するための試験の様々な態様の時間に関する仮定を判断する。同社は関係者や外部サービスプロバイダと臨床試験あるいは完成した他のサービスの進展状況を検討することにより、前払いと見積数を確定した。臨床試験中に,実際の結果がその推定結果と異なれば,同社はその臨床試験費用確認比率を調整する。同社は、当時既知の事実と状況に基づいて、その連結財務諸表において、貸借対照表毎の日付までの前払い及び計上費用を推定する。当社は、実際に発生した金額と実質的な差はないと予想しているが、提供されたサービスの実態や時間に対する提供サービスの状態や時間の理解が異なる可能性があり、当社が報告した任意の特定の時期の金額が高すぎたり低すぎたりする可能性がある。会社の臨床試験前払いと費用は契約研究機関の研究募集状況と他の第三者サプライヤーが展開する活動を適時かつ正確に報告し、契約研究機関の任意の変更注文を適時に処理することに依存する。
株式ベースの報酬
当社には株式ベースの報酬計画があり、この計画によると、株式オプション、制限株式単位(“RSU”)および業績制限株式単位(“PRSU”)を含む様々な株式ベースの奨励を付与することができる。株式オプションとRSUの公正価値はマイルストーンやサービス条件に制限され、階層帰属方法を用いて、直線原則で補償費用として確認され、没収行為は発生時に確認される。
PRSUの公正価値は、いくつかの性能およびサービス条件によって制限され、性能条件が生じる可能性があると決定されると、階層的帰属方法を使用して残りのサービス期間内に確認される。
当社は公正価値に基づく方法を採用し、従業員が株式を取得するすべての手配のための報酬を決定する。株式オプションの公正価値は、付与された日にBlack-Scholes推定モデルを用いて推定され、このモデルは、期待変動率、期待配当、期待期間、および無リスク金利の仮定を用いて推定される。予想変動率は、当社普通株のオプション期待期間内の履歴変動率に基づいている。オプションを付与する期待期限は、帰属期限と契約期間との和の平均値として計算される簡略化された方法を用いて得られる。歴史的に見ると、無リスク金利は適切なイギリス政府債券収益率に基づいてきた。同社は2020年10月30日にその普通株をAIMから退市した後、米国債収益率を使用した。
RSUとPRSUの公正価値は、会社普通株の付与日の終値から計算される。
使用された想定の詳細は付記7-株式報酬に記載されている 連結財務諸表。
他の収入はイギリスの研究開発税控除です
研究開発税収控除は、イギリスで受け取るべき研究開発税収控除に関連する。広範な研究と開発活動を展開する会社として、会社はイギリスの研究開発中小企業(SME)計画の制約を受けている。資格に適合した支出には,主に研究者の雇用コスト,消耗品,関連するライセンス下請け契約コストの一部,研究プロジェクトの一部として生じるいくつかの内部間接費用が含まれており,この研究項目には収入がない
中小企業計画に関連する税収控除は、現金形式で受信され、贈与収入に類似しているため、合併経営および全面赤字報告書に他の収入記録として記録されている。
所得税
当社は、米国会計基準第740号“所得税”(以下、“米国会計基準740”という。)の規定に従って所得税を計算する。ASC 740は、負債法の使用、すなわち繰延税金資産および負債口座残高が、財務報告と資産および負債の税ベースとの間の差に基づいて決定され、公布された税率および差異の予想が逆転したときに施行される法律を使用して計量されることを規定する。
必要があれば、当社は繰延税金資産をその推定可能な価値に減らすための推定手当を提供します。ASC 740は、税務状態が不確定な会計問題を解決するための単一のモデルを確立する。ASC 740は、税務状況が財務諸表において確認される前に達成される必要がある最低確認閾値を規定することにより、所得税の会計処理を明らかにした。その会社には不確定な税務状況はない。
総合損失
当社は米国会計基準220号“損益表-報告全面収益”に基づいて全面赤字を計算した。全面赤字は期間内の株主権益のすべての変動を指すが、株主投資や株主への分配による変動は除外する。
細分化市場報告
経営部門は企業の構成要素として定義され、その独立した離散情報は、首席経営意思決定者あるいは意思決定グループが資源をどのように分配するかを決定し、業績を評価する際に評価することができる。その会社は所有している1つは経営と報告部門、薬品開発。
報告通貨と機能通貨
連結財務諸表はドルで報告され、ドルも会社子会社の本位貨幣である。外貨による取引を取引当日の為替レートで再計量することを会社の機能通貨とする。これらの取引によって生成された任意の通貨資産および負債は、貸借対照表の日または決済時の為替レートによって我々の機能通貨として再計量される。これにより生じた損益は我々の総合経営報告書に為替損益を計上した。
国庫株
2020年12月31日までの年度内に、当社は従業員及び元従業員が株式を買収したり、従業員及び元従業員の利益のために株式を買収することを協力するための信託基金を設立した。会社は2023年12月31日と2022年12月31日までの年間で発行した16.01000万株普通株式(相当)2.0(百万人の米国預託証明書)28.01000万ポンドです3.5(百万株米国預託証券)従業員に株式奨励を付与する際に発行された期待株式を支払うために、当該信託にそれぞれ支払われる。
当社は信託を間接的に制御する能力を持っています。受託者は信託契約に従って行動しなければなりませんし、当社は株式の発行を制御して奨励金を支払わなければなりません。したがって、信託基金は会社の総合財務諸表に組み込まれる。信託を発行したが、株式奨励帰属株式を満たすために従業員に発行されていない株式は、国庫が保有する普通株として総合貸借対照表に計上される。
金融商品の公正価値
アメリカ公認会計原則は公正価値を定義し、そして会社に公正価値を計量し、三級方法を用いて公正価値計量を開示するための枠組みを構築することを要求した。これらのレベルは、アクティブ市場のオファーのような観察可能な入力として定義されるレベル1と、直接または間接的に観察可能なアクティブ市場のオファー以外の入力として定義されるレベル2と、市場データが少ないかないか、したがって、エンティティが自分の仮定を作成する必要がある観察不可能な入力として定義されるレベル3と、を含む。
私たちの金融商品には、現金等価物、株式、他の資産、支払すべきお金、計算費用、および他の負債が含まれています。これらのツールの公正価値推定は、関連する市場情報に基づいて特定の時点で行われる。これらの見積りは主観的である可能性があり,不確定要因や重大な判断事項に関与しているため,正確には決定できない.他の手形の額面は短期的な性質に属するため,その公正価値を代表するとみなされる.
信用リスクが集中する
会社を集中的な信用リスクに直面させる可能性のある金融商品は主に現金と現金等価物、銀行預金といくつかの売掛金を含む
同社は高格付け金融機関と高格付け通貨市場基金で現金と現金等価物を持っている。私たちのこれらの機関での預金は保険限度額を超える可能性がありますが、当社はこれらの口座で重大な信用損失を経験していませんし、当社がこれらのツールでいかなる重大な信用リスクに直面しているとも思いません。
リース会計
会社は最初から賃貸契約かどうかを確定しています。純収益資産および負債は、開始日にレンタル期間内の賃貸支払いの現在値によって確認される。そのため、当社は開始時に固定と確定可能な支払いのみを考慮しています。
当社の賃貸借契約は暗黙的な金利を提供していないため、当社は賃貸支払いの現在値を計算する際に借入金金利を増加させることを決定した。ROU資産は、開始前に支払われた任意のレンタル支払いをさらに含み、受信された任意のレンタルインセンティブを差し引いて記録される
同社のレンタル条項には、レンタルの延長または終了のオプションが含まれている場合があります。会社がこれらの選択権を行使すると合理的に判断された場合、レンタルは負債として確認され、対応するROU資産も確認される。
経営リースには、会社総合貸借対照表における使用権資産および流動および非流動経営リース負債が含まれています。
最近採用された会計基準
FASBは2016年6月、ASU 2016-13、金融商品-信用損失(主題326)-金融商品信用損失の測定を発表した。本指針は,現行の発生済み損失減値方法の代わりになっている.
このモードによれば、初回確認時および各報告期間において、エンティティは、歴史的経験、現在の条件、および合理的かつサポート可能な予測による金融商品の使用期間内に予想されるクレジット損失の現在の推定を反映する準備金を確認しなければならない。このガイドラインでは,採用期間開始までの留保報酬の累積効果調整を要求し,修正の遡及移行方法を採用している.この更新は2023年1月1日に会社に発効し、この更新の採用は会社の財務諸表や関連開示に実質的な影響を与えていない。
最近発表された未採用の会計基準
FASBは2023年12月、申告エンティティの有効税率入金に関する分類情報および支払われた所得税に関する情報の提供を要求する米国会計基準委員会第2023-09号“所得税改善開示”を発表した。この基準は、より詳細な所得税開示を提供することによって、投資家に利益を得ることを目的としており、これは資本分配決定に役立つだろう。本ASUにおける改正案は、2025年1月1日から毎年発効し、予想に基づいて適用され、この基準を遡及適用することを選択することができる。早期養子縁組を許可する。このASUは会社の総合貸借対照表や総合経営報告書や全面赤字に影響を与えない。その会社は現在、その所得税開示への影響を評価している。
2023年11月、FASBは米国会計基準委員会第2023-07号、“報告可能支部開示に対する改善”を発表し、主に重大支部費用の開示を強化することによって、報告可能支部の開示要求を改善した。さらに、改訂は、中間開示要求を強化し、1つのエンティティが複数の部分損益を開示することができる状況を明らかにし、1つの報告可能な支部のみのエンティティに新たな支部開示要求を提供し、他の開示要件を含む。改訂の目的は投資家が実体の全体的な業績をよりよく理解し、潜在的な未来のキャッシュフローを評価することである。本ASUにおける改正は,2024年1月1日からの年次期間と2025年1月1日からの移行期間に適用され,提出されたすべての期間にさかのぼって適用されなければならない。このASUは会社の総合貸借対照表や総合経営報告書や全面赤字に影響を与えない。その会社は現在、その部門に開示された影響を評価している。
注3-物件賃貸
同社の使用権資産(“ROU”)は、ロンドン、ジョージア州、ノースカロライナ州で借りたオフィススペースに関連しており、レンタル期間はそれぞれ2025年、2025年、2024年、2027年である。
当社は2023年12月31日までにノースカロライナ州にオフィスビル賃貸手配を締結し、既存のロンドン賃貸契約を延長した。この2つの合意の結果として、同社は賃貸負債と対応するROU資産#ドルを確認した2.71000万ドルです。また、賃貸負債の経営と引き換えに得られた補完非現金純資産は#ドルである2.71000万ドルです。
当社は2022年12月31日までにジョージア州にオフィスビル賃貸手配を締結し、既存のロンドン賃貸契約を延長し、レンタル負債とそれに応じたROU資産が#ドルであることを確認した0.71000万ドルです。
賃貸負債を計算するために、会社は加重平均割引率を採用しています11%和4それぞれ2023年12月31日までおよび2022年12月31日までの年度の%である。2023年12月31日と2022年12月31日までの加重平均残余レンタル期間s 3.3数年前発送する1.5それぞれ数年です。
2023年12月31日までの残り賃貸期間の最低年次支払いは以下の通り(千単位)
| | | | | | | | |
| 2024 | | $ | 1,175 | |
| 2025 | | 842 | |
| 2026 | | 788 | |
| 2027 | | 784 | |
| 将来の最低賃貸支払い総額 | | $ | 3,589 | |
| 差し引く:推定利息 | | (634) | |
| リース負債総額を経営する | | $ | 2,955 | |
販売、一般、行政費を含めた経営リース費用の総額は $0.72023年12月31日までの1年間で
注4-費用を計算する
計算すべき費用には、以下の項目が含まれる
| | | | | | | | | | | | | | |
| | 十二月三十一日 |
| | 2023 | | 2022 |
| 臨床試験やその他の開発費 | | $ | 752 | | | $ | 12,314 | |
| 専門費用、上場および一般会社費 | | 2,039 | | | 1,364 | |
| 人員関係のコスト | | 794 | | | 74 | |
| 費用総額を計算する | | $ | 3,585 | | | $ | 13,752 | |
注5-債務
2020年11月に当社は最高可達$を締結しました30.0#ドルの前払いを含む2000万ドル(“SVB定期ローン”)5.0取引完了時に100万ドルの資金を得ました10.01000万ドルと300万ドルです15.0600万ドルは、いくつかの臨床開発マイルストーンと他の指定された条件の達成に依存する
2022年10月14日(“2022年発効日”)に、当社はオックスフォード金融ルクセンブルク有限公司と融資と保証契約を締結し、総金額は最高$に達します150.02000万ポンドですオックスフォード定期ローンは#ドルの総額の初期定期ローンの前払いを提供する10.02022年発効日に資金を提供する(“オックスフォードA期融資”)が最も多い四つ追加の定期ローンの前払金、総額は#ドルです140.01000万ドルは、いくつかの臨床と規制発展マイルストーンの実現と他の指定された条件に依存する。オックスフォード定期ローンの収益は一般企業と運営資本用途に使用され、オックスフォードA期ローンの収益の一部はSVB定期融資項目下の既存の未返済債務の全額返済に使用される。2023年3月24日、同社は受け取りました10.0第2期ローン前払金(“オックスフォードB期ローン”)。
当社は2023年12月27日(“2023年発効日”)に最高$を締結しました400.02000万ユーロ(“2023年定期ローン”または“ローン契約”)には、合計の定期ローン前払いが含まれています
金額:$50.02023年の発効日に資金(“A期ローン”)と4つの追加の定期ローンの前払を提供しますが、以下に説明するいくつかの条項と条件を遵守しなければなりません。金額は#ドルです100.02000万ドル(“B期ローン”)、$75.02000万ドル(“定期C期ローン”),$75.02000万ドル(D期ローン)とドル100.02000万ユーロ(“E期ローン”)。2023年の定期融資は、担保代理であるデラウェア州有限責任会社オックスフォード金融有限責任会社(“オックスフォード”)と締結され、オックスフォードとHercules Capital,Inc.によって管理されるいくつかの基金(“貸主”と総称される)である。2023年の定期融資の純収益は、一般企業や運営資本用途に利用される
同社はA期ローンから純収益#ドルを獲得した28.4主に期限Aローン収益#ドルで構成されています50.01百万ドル、オックスフォード定期ローンによって会社が借りた既存の未返済債務#ドル部分を全額返済します20.0融資契約に関連した百万ドル、貸手、第三者費用1.41000万ドル、利息は$0.21000万ドルです。
当社の融資協定に対する会計評価、及び関連するオックスフォード実体及び2023年定期ローンとオックスフォード定期ローンの条項によると、当社はオックスフォードに関連するA期ローン部分に対して改訂会計処理を適用しているそのため、オックスフォード定期融資が2023年の発効日の未償却債務発行コストで改訂された場合、2023年の定期融資項目の未返済金額を含む損益は記録されていない。また、純収益に含まれるいくつかの費用は、適用される指導意見とER ASC 470です。
Hercules Capital,Inc.に関連する条項Aローン部分は新規債務を発行し、ASC 470に基づいて適用される会計処理を計上している。
慣例的な条項と条件を満たした場合、B期ローンは、会社が米国食品医薬品局の新薬申請を受けた日から2024年9月15日までの間((I)会社の承認を受けた日から30日以内)に提供される。通例条項と条件(B期融資の事前借用を含む)を満たす場合、C期融資は、(I)2025年9月15日と(Ii)2025年9月30日までに、指定された純売上高マイルストーンを実現する次の2つの中で遅い項目で提供される。通例条項と条件(C期融資の事前借用を含む)を満たす場合、D期融資は、(I)2026年2月15日と(Ii)2026年3月31日までに、指定された純売上高マイルストーンを実現するため、以下の両者のうち遅い期間で利用可能となる。慣例条項と条件(2028年6月1日までのD期ローンの借り入れを含む)によると、貸主は自ら決定し、当社の要求に応じてE期ローンを提供することができる。
2023年の定期ローンは2028年12月1日に満期になる。(A)(I)利息が発生する月の直前の月の最後の営業日の1ヶ月前のシカゴ商品取引所期限SOFR(ローンプロトコルの定義参照)基準金利のうち大きい者に等しい貸付契約項目の下の各立て替え金は、変動年利率(“基本金利”)で計算されなければならない5.34%、追加(B)5.85%です。上記の規定があるにもかかわらず、(I)いずれの場合も、A期ローンの基本金利(X)は下回ってはならない11.19%及び(Y)2023年定期貸出1件当たりの基本金利は、2023年定期貸出資金調達日直前の営業日の基本金利を下回っており、(Ii)発効日から2023年12月31日(2023年12月31日を含む)までのA期ローンの基本金利は11.19%および(Iii)2023年の定期融資当たりの基本金利は、増加してはならない2.002023年の定期融資ごとの融資日は、適用される基本金利より2%高い。2023年の定期ローンでは、2028年6月1日直前の支払日まで毎月利息のみを支払うことになっています。その後、償却支払いは月額の元金分割払いに毎月の利息を加算した方式で支払います
返済時(満期、スピードアップ、早期返済、その他の方法を問わず)、借り手は貸手に最後の金を支払わなければならない。金額は 2.50%から3.50その割合は2023年の定期融資総額前払いは、2023年の定期融資の返済時期(“最終支払い”)に依存する。借り手は2023年の定期ローンを全額前払いすることができますあるいは部分的に借り手(I)十日前(10)日前にオックスフォード大学と貸金人に書面で通知し、(Ii)前払の日に支払う(A)すべての未返済元金に応算と未払い利息を加え、(B)前払い費用#2.002025年12月27日までに支払う場合、2023年の定期ローンの割合を前払いする1.502025年12月27日以降から2026年12月27日までに支払う場合、2023年の定期ローンの%を前払いする1.002026年12月27日以降に支払われる2023年定期融資、(C)最終支払いおよび(D)期限満了に応じて対応する他のすべての金(ある場合)、期限を超えた金の違約率利息を含む。違約事件中に返済されなかった金額は、必要な融資者(ローン合意を定義する)の要求に応じて支払われ、以下の追加金利で利息を計算しなければならない5.00年利率;及び(Iii)2023年定期ローンの任意の部分早期返済の額面は#の整数倍とする5.01000万ドルです。
2023年の定期融資は、当社のほとんどの資産(知的財産権を除く)の留置権を担保とし、知的財産権留置権は、(I)任意の2023年の定期融資の融資日(融資協定により抽出された2023年の未償還定期融資の元金総額が$を超える)の早い日に付与されることが条件となる50.0及び(Ii)借主又は当社が締結許可使用料融資(定義は融資協定を参照)の前である。同社はまたオックスフォード大学と融資者にその知的財産権に関する否定的な質権を提供した。
融資協定には、2つの金融チノを含む慣用的な陳述と保証、契約違反事件が含まれている:(I)2025年7月1日から、借り手はアメリカで一定レベルの現金を維持しなければならないが、オックスフォードを受益者とする制御協定を守らなければならない;しかし、会社の当時の時価が少なくとも#ドルであれば、この流動資金契約はいかなる所与の時間にも適用されない3.0(Ii)2025年9月30日から、借り手と当社は、アンテフェンタリンの四半期過去6ヶ月の製品純収入を維持しなければならない。条件は、この収入契約がいつでも免除されることである:(X)借り手と会社のこの四半期内の毎月最終日の無制限現金残高は、その日未返済2023年の定期融資元金総額の積を1.25以上乗じたものである。(Y)(1)借入者と当社の当該四半期内の毎月最終日の無制限現金残高が0.5以上の日に返済されていない2023年の定期融資元金総額、および(2)本四半期の毎月最終取引日前5取引日の1日当たりの米国預託株式平均額に、自社発行および発行済み米国預託株式総数を乗じて少なくとも$1.5または(Z)本四半期の毎月最終取引日までの5取引日以内に、当社米国預託株式の1日あたりVWAPの平均値に、自社発行済みおよび発行済み米国預託株式総数を乗じて、少なくとも$とする3.01000億ドルですローン協定には、費用返済、オックスフォード大学と貸主の利益のための賠償権利などの他の慣行条項も含まれている。
2023年12月31日現在、金利は約11年利率と2023年の定期ローンの帳簿価値と推定公正価値の間に大きな差はない。
2023年12月31日現在、2023年12月31日までの2023年定期融資に関する将来元金支払い(定期料金終了を除く)は以下の通り(千単位)
| | | | | | | | |
| 2024 | | $ | — | |
| 2025 | | — | |
| 2026 | | — | |
| 2027 | | — | |
2028 | | 50,000 | |
合計する | | $ | 50,000 | |
注6-重要な合意
配基協定
当社は2004年にRhinophmaを買収し,Ligand UK Development Limited(“Ligand”)(前身はVernalis Development Limited)の有無を担っている。その会社は譲渡と許可協定をLigand協定と呼ぶ。
Ligandは,アルギニンや関連化合物に関連するある特許および特許出願に対するすべての権利(“Ligand特許”)と,あるLigandノウハウに基づいてLigand特許,Ligandノウハウおよびある化合物の実物在庫を利用して開発された製品(“Ligandライセンス製品”)の独占的,世界的な特許使用料許可を当社に譲渡した。
いかなる規制機関によるLigandライセンス製品の商業化の最初の承認を得た場合、会社は500万GBの記念碑的支払いを支払う義務があり、すべてのLigandライセンス製品の将来の販売実績に基づいて低いビット数の使用料を支払うことと、任意の子許可人から受け取った任意のLigand特許およびLigandノウハウの対価格の20%に相当する部分を支払うことが義務付けられている。支払うべき特許権使用料は将来の販売実績に基づいているため、支払うべき金額は無制限である。
各事項または事項を解決する際に、会社は、Ligandプロトコルに関連するまたは対価格支払い(または支払い)を費用として記録する。
当社は2022年3月にLigandと改訂協定(“改訂”)を締結し、これによりLigandプロトコルを改訂し、Ligandプロトコルのいくつかの曖昧な言葉を明らかにする。修正案によると:
•同社はLigandに(I)$を支払うことに同意した2.0改訂日から5営業日以内に15.0当社または分割許可者が初めてアンテファニンを商業的に販売する際には、その金額を現金で支払うか、または当社の適宜決定の下で、10年間のナスダック世界市場における当社の米国預託株式の出来高加重平均価格に基づいて同値な会社の株式を発行することにより、同値な会社の株式を発行する10)には、このような記念碑的イベントおよび以前の取引日が含まれます
•Ligand協定は、いずれか一方がその条項に基づいて早期に終了しない限り、2042年3月24日に満了する
•Ligandプロトコルの終了後、修正案の定義のような任意の二次ライセンス者は、修正案の定義のような二次ライセンシーが再許可する番組IPの一部についてLigandと直接ライセンスプロトコルを締結する権利がある
•マイルストーン支払いは現金で支払うか、会社の適宜決定の下で、Ligandに同値な会社株を発行することで支払うことができる
•いずれもLigand合意を終了する権利は,他方がLigand合意下の義務に実質的に違反していることを宣言する英国高裁の最終判決を得ることを条件としている。
その会社は$を占めている2.0改正案を実行する際には、支払いは契約改正に関係するため、総合経営報告書では販売、一般、行政費用として100万ドル、全面赤字を支払う。
微妙な協議
当社はNuance Pharma Limited(“Nuance Pharma”)と協力及び許可協定(“Nuance合意”)を締結し、2021年6月9日(“Nuance発効日”)から発効し、これにより、当社はNuance Pharmaに大中華中国(中国、台湾、香港及びマカオ)にアンテファンを独占開発及び商業化する権利を付与した。その見返りに、同社は無条件の対価格権を獲得し、総額は#ドルとなった40.0100万ドルです25.02000万ドルの現金と株式、#ドル15.0Nuance発効日まで,Nuance Pharmaの親会社Nuance Biotechはドルを持っている。同社は将来的に$までの記念碑的支払いを受ける資格がある179.0ある臨床、監督管理と商業マイルストーン、及び製品の純売上高のパーセンテージによって等級付けされた2桁の特許権使用料を実現したため、大中華区中国で2.5億ドルをトリガした。収入が大きく逆転しない可能性が高い場合、会社はこのようなマイルストーンを確認するだろう。
2023年12月31日までにドル15.01,000,000,000株の資本は、総合貸借対照表に持分と記載されている。当社は計量代替案の使用を選択したため,持分はコストで入金する
確定しやすい公正な価値のない株式投資への。同社は四半期ごとにこの投資の減価指標を評価している。当社は2023年12月31日までの年間で、投資公平価値に大きな影響を与える可能性のある事件や状況変化は確認されていません。
Nuance協定の条項によると、大中華区中国で最初の新薬申請の3ヶ月前の任意の時間に提出されることが予想され、(I)第三者が当社と協力して開発および/または商業化アンチリンを開発することに興味がある場合、世界的にも少なくとも米国またはヨーロッパをカバーする地域でも、または(Ii)当社が制御権変更を生じた場合、当社はNuance Pharmaに付与されたライセンスおよびすべての関連資産の買い戻しの独占的選択権を有することになる。双方が同意した価格は、(I)Nuance Pharmaがプロトコルに従って現金形式で会社に支払ったすべての以前の金額と、(Ii)Nuance PharmaがNuanceプロトコルに従ってensftreineの開発および商業化について生成および支払いしたすべての開発および規制コストに、いくつかのマイルストーンの達成に依存する1桁の範囲を乗じたものに等しいが、特定の最高金額によって制限される。
Nuanceプロトコルは、双方が事前に終了しない限り、各司法管轄区域および各製品に基づいて、当該司法管轄区域内の製品の印税支払い義務が満了するまで継続する。どちらももう一方が治癒していない重大な違約や破産によりNuanceプロトコルを終了することができる。Nuance Pharmaはまた、90日前の書面通知後にNuanceプロトコルを任意に終了することができる
同社は買い戻し選択権を審査し,その選択権が第三者を条件としているため,会社は実際にその選択権を行使する能力がないため,この契約はASC 606で入金されていると判断した。
同社は2022年4月13日、Nuance Pharmaとアンテフェンタニルを生産·供給する協定(“Nuance供給協定”)を正式に締結した。同社は,Nuanceの生産と供給は異なる単独の履行義務であり,受け取る対価格はNuanceが注文した数量に応じて変化することができると認定している。ライセンス製品の製造と供給によって得られた収入は、Nuanceに供給されたときに確認される。同社は、同協定の下での製造·供給において依頼者を務めることを決定した。当社は依頼人として、毛数で関連収入を確認します。2022年12月31日までに、当社は確認します0.590万ドルの収入はNuance Pharma臨床供給アンテフェンタニルと関連がある。
注7-株式ベースの報酬
当社は従業員のために各種株式インセンティブ計画を実施し、株式奨励を行使する際に普通株や米国預託証明書を発行する。
当社は、従業員及び取締役に付与された株式オプション及びRSUに関する株式ベースの報酬支出を記録している。従業員の個人機能の性質に応じて、費用は研究開発と販売、一般と行政コストに計上され、関連年度の費用配分を代表する。従業員に株式補償を発行するコストは総合経営及び全面赤字報告書内で確認し、帰属期間に相応して権益を増加させる。
オプションは前日終値を付与する行権価格で発行され,通常一定期間以内に付与される1つは至れり尽くせり4年すべてのオプションの契約期間は10年.
以下の表に株式ベースの給与の研究開発と販売、一般と行政コストの分配状況(千単位)を示す
| | | | | | | | | | | | | | |
| | 十二月三十一日 |
| | 2023 | | 2022 |
| 研究開発 | | $ | 4,228 | | | $ | 5,420 | |
販売、一般、行政 | | 14,784 | | | 8,701 | |
株式ベースの総報酬 | | $ | 19,012 | | | $ | 14,121 | |
百代オプション計画と初公募先行権計画
我々の取締役会はそれぞれ2006年9月18日と2012年7月24日に百代オプション計画と初公募株式前オプション計画を採択した。これらの計画により発行可能な株式総数は,現在発行されているオプションの数である114,000普通株、あるいは14,250EMIオプション計画用ADSSと1,320,000普通株、あるいは165,000株式先行権計画の米国預託証明書を初公開する。
違います。2017年のインセンティブ·インセンティブ計画が採択されて以来、この2つの計画に基づいてより多くの報酬が授与され、違います。このような規定に基づいて、もっと多くの賞が授与されるだろう。
2017年度インセンティブ·インセンティブ·プログラム
2017年度奨励計画は、当社の取締役会を通過し、会社の一部の役員および従業員に株式ベースの報酬を支給することを目的として2017年4月26日に施行されました。それは、会社役員、高級管理者、従業員、非従業員取締役に株式オプション、RSU、その他の株式ベースの奨励を付与することを規定している。
株式オプション活動
購入株式数、1株当たりの株式購入権の加重平均授出日の公正価値及び加重平均行権価格はすべて1株当たりの普通株基準に従って以下のように示した。その会社がナスダック世界市場に上場しているアメリカの預託証明書はそれぞれ8人普通株です。
次の表は、株式オプション活動を示し、すべての3つの計画の未償還オプションを含む:
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | 株式オプションの数 | | 加重平均行権値 (1) | | 加重平均残契約期間(年) | | 内在的価値を合計する |
2022年1月1日に返済されていません | | 12,695,200 | | | $ | 1.38 | | | 6.5 | | |
| 授与する | | 9,024,000 | | | 0.90 | | | | | |
| 没収される | | (620,016) | | | 1.04 | | | | | |
| | | | | | | | |
| 鍛えられた | | (1,822,688) | | | 0.75 | | | | | |
2022年12月31日に返済されていません | | 19,276,496 | | | $ | 1.22 | | | 7.2 | | $ | 39,412 | |
| 授与する | | 7,376,000 | | | 2.46 | | | | | |
| 没収される | | (464,680) | | | 0.89 | | | | | |
| 期限が切れる | | (240,000) | | | 3.07 | | | | | |
| 鍛えられた | | (1,258,192) | | | 1.55 | | | | | |
2023年12月31日現在の未返済債務 | | 24,689,624 | | | $ | 1.56 | | | 7.4 | | $ | 24,022 | |
2023年12月31日に行使できます | | 12,904,640 | | | $ | 1.30 | | | 5.8 | | $ | 15,574 | |
(1) 行権価格は普通株の同値価格に関係しており,1で計算される8人アメリカ預託株式価格のエイチです。
以下、12月31日までの年間株式オプション行使活動に関する内在的価値と現金収入総額をまとめた
| | | | | | | | | | | | | | |
(千ドル) | | 2023 | | 2022 |
| 株式オプション行使の合計内的価値 | | $ | 1,861 | | | $ | 2,413 | |
| 株式オプションを行使した現金収入 | | $ | 1,949 | | | $ | 1,372 | |
株式オプションの公正価値を決定する
ブラック·スコアーズオプション定価モデルを用いて推定した持分補償オプションの公正価値総額は#ドルであった13.22023年12月31日までの年度内に付与されたオプション5.92022年12月31日までの年度内に付与された手形。コストはオプションの帰属期間内に階層帰属法を用いて直線的に償却される.以下では、2023年と2022年に付与された株式オプションのブラック·スコアーズ推定値に用いられるものとする。
予想変動率
変動率は,オプションの期待寿命と一致する一定期間の会社株価の過去日平均値を用いて計算した。
普通株主公正価値
二零二零年十月にAIMを退市する前に、普通株の公正価値は当社株の授出日前日のAIMでの終値に基づいて計算された。その後、公正価値は、先日の夜にナスダックで取引された米国預託証明書の終値を付与したものに基づいていた。
無リスク金利
無リスク金利は、2020年10月20日までAIMから退市するまで、付与時関連期限の英国政府債券利回りに基づいてきた。その後、適切な米国債収益率が使用された。
所期期限
当社には、その予想期間を推定するのに十分な歴史がないため、当社は、米国証券取引委員会の職員会計公告107等の簡略化された方法でオプションの期待期間を推定する
110簡略化方法に従って計算された期待期間は、類似した契約条項を有するすべての株式オプションに適用される。この方法を使用して、予想期間は、付与された株式オプションの帰属期間および契約期間の平均値に基づいて決定される。
配当を期待する
期待していた配当金がありません。
適用年度に付与された株式購入権に適用した重み付き平均仮定の概要は以下のとおりである
| | | | | | | | | | | | | | |
| | 十二月三十一日 |
| | 2023 | | 2022 |
| 無リスク金利 | | 3.40% - 4.69% | | 2.09% - 4.20% |
| 期待寿命、年数 | | 5-7 | | 5-7 |
| 予想変動率 | | 80.64% - 87.26% | | 82.50% - 84.27% |
| 期待配当収益率 | | — | % | | — | % |
| 付与日公正価値(1株当たり) | | $1.69 - $3.27 | | $0.34 - $1.33 |
制限株式単位活動
次の表にRSU活動を示す:
| | | | | | | | | | | | | | | | | | | | |
| | RSU数 | | 加重平均授権日公正価値 | | 加重平均残契約期間(年) |
2022年1月1日に返済されていません | | 38,347,352 | | | $ | 0.97 | | | 1.2 |
| 授与する | | 12,877,864 | | | 1.07 | | | |
| 没収される | | (1,006,264) | | | 1.03 | | | |
| 既得 | | (15,676,608) | | | 0.96 | | | |
2022年12月31日に返済されていません | | 34,542,344 | | | $ | 1.01 | | | 1.2 |
| 授与する | | 3,596,872 | | | 1.66 | | | |
| 没収される | | (303,648) | | | 1.10 | | | |
| 既得 | | (18,332,944) | | | 0.99 | | | |
2023年12月31日現在の未返済債務 | | 19,502,624 | | | $ | 1.14 | | | 1.2 |
2023年12月31日と2022年12月31日までの年間で,帰属するRSUの内在的価値は$である41.51000万ドルと300万ドルです14.32億5千万ドルと2億5千万ドルです
2023年12月31日現在、付与されているが確認されていない株式オプションとRSUに関する総報酬コストは$20.01000万ドルです。このコストは加重平均残りの期間内に費用として償却される1.9年限は、その後の没収状況に応じて調整される。
業績限定株式単位(“PRSU”)
同社は2023年にPRSUの配布を開始した。PRSUは、いくつかの業績目標を達成した後に付与を開始し、サービスを継続する必要があります。履行条件が発生する可能性があると判定されると、PRSUの公正価値は、階層的ホーム方法を使用して残りのサービス期間内に確認されるであろう。業績条件の存在により、適用される会計の枠組みではまだ可能とされておらず、当社は2023年にPRSUの補償費用を確認していない
当社の2023年12月31日までの年間PRSU活動の概要は以下の通りです
| | | | | | | | | | | | | | | | | | | | |
| | PRSU数 | | 加重平均授権日公正価値 | | 内在的価値を合計する |
2023年1月1日現在返済されていない | | — | | | $ | — | | | |
| 授与する | | 10,790,144 | | | 1.66 | | | |
| 没収される | | (60,000) | | | 1.66 | | | |
2023年12月31日現在の未返済債務 | | 10,730,144 | | | $ | 1.66 | | | $ | 26,665 | |
2023年12月31日現在、非既得PRSUに関する未確認の総補償コストは#ドル17.82000万ドル加重平均期間で確認されます加重平均期間は1年性能条件に達すると.
注8-福祉計画
同社は米国では従業員と執行役員のために401(K)固定払込退職計画を維持し、イギリスではその従業員と執行役員のために固定払込計画を維持している。当該等計画の資産は当社の資産と独立して管理する基金で別々に保有しています。
退職計画コストとは、当社が同年度内に当該計画に支払うべき供出金を指す。2023年12月31日までと2022年12月31日までの固定拠出料は#ドル0.61000万ドルと300万ドルです0.32億5千万ドルと2億5千万ドルです
注9-税収
Verona Pharma plcはイギリスで運営され,Verona Pharma,Inc.は米国で運営されており,これらの国や地域では所得税を払わなければならない。2023年12月31日までの1年間、イギリスの法人税税率は23.5%、米国連邦所得税税率は21%である。
所得税前利益/損失の構成は以下のとおりである(単位:千):
| | | | | | | | | | | | | | |
| | 十二月三十一日 |
| | 2023 | | 2022 |
| アメリカです | | $ | (7,429) | | | $ | (3,868) | |
| イギリス.イギリス | | 61,324 | | | 72,316 | |
| 合計する | | $ | 53,895 | | | $ | 68,448 | |
所得税費用の構成は以下のとおりである(千計)
| | | | | | | | | | | | | | |
| | 十二月三十一日 |
| | 2023 | | 2022 |
| アメリカです | | $ | 474 | | | $ | 253 | |
| イギリス.イギリス | | — | | | — | |
当期税費総額 | | $ | 474 | | | $ | 253 | |
| | | | |
| アメリカです | | $ | — | | | $ | — | |
| イギリス.イギリス | | — | | | — | |
| 繰延税金総額 | | — | | | — | |
所得税総支出 | | $ | 474 | | | $ | 253 | |
英国の法定所得税税率と我々の有効所得税税率との入金は以下のとおりである(百分率で表す)
| | | | | | | | | | | | | | |
| | 十二月三十一日 |
| | 2023 | | 2022 |
| イギリス税率 | | 23.5 | % | | 19.0 | % |
| 差し引かれない費用 | | (8.0) | % | | (1.8) | % |
| 研究開発激励 | | (4.1) | % | | (8.0) | % |
| 行使された購入権 | | 5.4 | % | | 2.1 | % |
| 繰延税額推定免税額変動 | | (18.1) | % | | (11.6) | % |
| その他の違い | | 0.4 | % | | (0.1) | % |
| 有効所得税率 | | (0.9) | % | | (0.4) | % |
会社の繰延税金資産と負債の構成要素は以下の通り(千で計算)
| | | | | | | | | | | | | | |
| | 十二月三十一日 |
| | 2023 | | 2022 |
| 繰延税金負債: | | | | |
あるいは負債がある (1) | | $ | (53,851) | | | $ | (34,565) | |
| | | | |
| 繰延税金負債総額 | | (53,851) | | | (34,565) | |
| | | | |
| 繰延税金資産: | | | | |
| 純営業損失 | | 54,012 | | | 38,893 | |
知的財産権研究開発資産 (1) | | 47,793 | | | 32,700 | |
| 未来可行権株 | | 7,548 | | | 11,964 | |
| 他にも | | 21 | | | (516) | |
| 繰延税金資産総額 | | 109,374 | | | 83,041 | |
| 減算:推定免税額 | | (55,523) | | | (48,476) | |
| 繰延税金資産は,推定準備後の純額を差し引く | | $ | — | | | $ | — | |
| | | | |
| 繰延税額推定免税額の変動 |
| | | | |
| 1月1日の推定免税額 | | $ | 48,476 | | | $ | 30,252 | |
| 税率の変化 | | (851) | | | — | |
評価免税額を引き上げる | | 7,898 | | | 18,224 | |
| | | | |
| 12月31日の推定免税額 | | $ | 55,523 | | | $ | 48,476 | |
(1) これは知的財産権研究開発資産と負担する或いは負債のある税基及び財務報告基数の違いと関係があり、アメリカ公認会計原則によると、財務報告基数はゼロである。
管理職はすでに累計税項損失及び未来の課税損失に対する予測を検討し、それらが実現する可能性が低いことを確定した。したがって、繰延税金資産について評価免除額が提供された。
同社の英国における純営業損失(NOL)は2023年12月31日と2022年12月31日現在#ドルである216.01000万ドルと300万ドルです155.62億5千万ドルと2億5千万ドルですNOLは将来の課税利益を相殺するために無期限繰り越しが可能であるが,これは年間500万GBの免税額に限られ,その後繰り越し可能な損失でカバーできる利益は50%に制限される。
同社は英国と米国でそれぞれ所得税申告書を提出している。2022年12月31日(2022年12月31日を含む)までの全年度、すべての必要な所得税申告書が完成した。同社は2022年12月31日までの年度の研究開発税インセンティブ申請を現在HMRCの定例審査を受けている。今回の審査のため、クレーム額の実質的な調整は行われない予定です違います。利息または罰金は、総合経営報告書と全面赤字または総合貸借対照表で確認されます。2023年12月31日現在、会社には不確定な税務頭寸はありません。
付記10-1株当たり純損失
1株当たりの純損失は普通株で計算する.その会社がナスダック世界市場に上場しているアメリカの預託証明書はそれぞれ8人普通株です次の表は、2023年と2022年の基本1株当たり収益と希釈後の1株当たり収益の計算方法(単位は千、1株および1株当たりの金額は含まない)を示している
| | | | | | | | | | | | | | |
| | 十二月三十一日 |
| | 2023 | | 2022 |
| 分子: | | | | |
| 純損失 | | $ | (54,369) | | | $ | (68,701) | |
| 普通株主が獲得できる純損失−基本損失と希薄損失− | | $ | (54,369) | | | $ | (68,701) | |
| 分母: | | | | |
| 加重平均流通株−基本と希釈 | | 634,142,660 | | | 529,071,526 | |
| 1株当たり純損失--基本損失と赤字 | | $ | (0.09) | | | $ | (0.13) | |
2023年12月31日および2022年12月31日までの年間で、54.9百万ドルと53.8希釈後の普通株当たり収益を計算する際には,それぞれ1.8億ユーロが計上されておらず,逆希釈となるからである。
注11-引受金とその他の事項
当社の累計最高リスクは2023年3月31日までの3ヶ月間$となっています6.9300万ドルはサプライヤーの件と関連があり、$のいくつかの領収書があります1.5同じサプライヤーに支払われた勘定は2000万ドルだった。この二つのプロジェクトはいずれも2023年6月に#ドルで決済された2.11000万ドルです。これは#ドルの純輸出につながった6.32023年6月30日までの3ヶ月間の純売上は300万ドルでした1.52023年12月31日までの年度の総合経営·全面赤字報告書における研究·開発コストは4.6億ドル。
付記12-関係者取引その他株主事項
2023年12月31日と2022年12月31日までの年間では,関連先取引はなかった。