MRSN-2023123100014428362023会計年度誤りHttp://Fasb.org/us-GAAP/2023#その他負債現在Http://Fasb.org/us-GAAP/2023#その他負債現在Http://Fasb.org/us-GAAP/2023#その他の負債は現在ではありませんHttp://Fasb.org/us-GAAP/2023#その他の負債は現在ではありません00014428362023-01-012023-12-3100014428362023-06-30ISO 4217:ドル00014428362024-02-23Xbrli:共有00014428362023-12-3100014428362022-12-31ISO 4217:ドルXbrli:共有00014428362022-01-012022-12-3100014428362021-01-012021-12-310001442836アメリカ-アメリカ公認会計基準:普通株式メンバー2020-12-310001442836US-GAAP:AdditionalPaidInCapitalMembers2020-12-310001442836アメリカ公認会計原則:他の総合収入メンバーを累計2020-12-310001442836アメリカ-公認会計基準:前払いメンバーを保留2020-12-3100014428362020-12-310001442836Mrsn:AtMarketEquityOfferingProgramメンバ2021-01-012021-12-310001442836Mrsn:AtMarketEquityOfferingProgramメンバアメリカ-アメリカ公認会計基準:普通株式メンバー2021-01-012021-12-310001442836Mrsn:AtMarketEquityOfferingProgramメンバUS-GAAP:AdditionalPaidInCapitalMembers2021-01-012021-12-310001442836アメリカ-アメリカ公認会計基準:普通株式メンバー2021-01-012021-12-310001442836US-GAAP:AdditionalPaidInCapitalMembers2021-01-012021-12-310001442836アメリカ-公認会計基準:前払いメンバーを保留2021-01-012021-12-310001442836アメリカ-アメリカ公認会計基準:普通株式メンバー2021-12-310001442836US-GAAP:AdditionalPaidInCapitalMembers2021-12-310001442836アメリカ公認会計原則:他の総合収入メンバーを累計2021-12-310001442836アメリカ-公認会計基準:前払いメンバーを保留2021-12-3100014428362021-12-310001442836Mrsn:AtMarketEquityOfferingProgramメンバ2022-01-012022-12-310001442836Mrsn:AtMarketEquityOfferingProgramメンバアメリカ-アメリカ公認会計基準:普通株式メンバー2022-01-012022-12-310001442836Mrsn:AtMarketEquityOfferingProgramメンバUS-GAAP:AdditionalPaidInCapitalMembers2022-01-012022-12-310001442836アメリカ-アメリカ公認会計基準:普通株式メンバー2022-01-012022-12-310001442836US-GAAP:AdditionalPaidInCapitalMembers2022-01-012022-12-310001442836アメリカ公認会計原則:他の総合収入メンバーを累計2022-01-012022-12-310001442836アメリカ-公認会計基準:前払いメンバーを保留2022-01-012022-12-310001442836アメリカ-アメリカ公認会計基準:普通株式メンバー2022-12-310001442836US-GAAP:AdditionalPaidInCapitalMembers2022-12-310001442836アメリカ公認会計原則:他の総合収入メンバーを累計2022-12-310001442836アメリカ-公認会計基準:前払いメンバーを保留2022-12-310001442836Mrsn:AtMarketEquityOfferingProgramメンバ2023-01-012023-12-310001442836Mrsn:AtMarketEquityOfferingProgramメンバアメリカ-アメリカ公認会計基準:普通株式メンバー2023-01-012023-12-310001442836Mrsn:AtMarketEquityOfferingProgramメンバUS-GAAP:AdditionalPaidInCapitalMembers2023-01-012023-12-310001442836アメリカ-アメリカ公認会計基準:普通株式メンバー2023-01-012023-12-310001442836US-GAAP:AdditionalPaidInCapitalMembers2023-01-012023-12-310001442836アメリカ公認会計原則:他の総合収入メンバーを累計2023-01-012023-12-310001442836アメリカ-公認会計基準:前払いメンバーを保留2023-01-012023-12-310001442836アメリカ-アメリカ公認会計基準:普通株式メンバー2023-12-310001442836US-GAAP:AdditionalPaidInCapitalMembers2023-12-310001442836アメリカ公認会計原則:他の総合収入メンバーを累計2023-12-310001442836アメリカ-公認会計基準:前払いメンバーを保留2023-12-31Mrsn:候補者Xbrli:純0001442836Mrsn:ComputerEquipmentOfficeEquipmentAndSoftwareMembers2023-12-310001442836Mrsn:実験室装置のメンバー2023-12-310001442836Mrsn:GSKプロトコルのメンバー2022-08-012022-08-310001442836Mrsn:GSKプロトコルのメンバー2022-08-310001442836MRSN:ビジネス開発と規制メンバーMrsn:GSKプロトコルのメンバー2022-08-310001442836MRSN:早期臨床発展メンバーMrsn:GSKプロトコルのメンバー2022-08-310001442836Mrsn:GSKプロトコルのメンバー2022-08-06Mrsn:パフォーマンス無効化0001442836Mrsn:GSKプロトコルのメンバー2023-01-012023-12-310001442836Mrsn:GSKプロトコルのメンバー2022-01-012022-12-310001442836Mrsn:GSKプロトコルのメンバー2023-12-310001442836Mrsn:GSKプロトコルのメンバー2022-12-310001442836ジョンソンとジョンソンプロトコルのメンバーです2022-02-28MRSN:目標0001442836ジョンソンとジョンソンプロトコルのメンバーです2022-02-012022-02-28MRSN:ライセンス0001442836ジョンソンとジョンソンプロトコルのメンバーですMrsn:開発と規制のメンバー2022-02-280001442836Mrsn:ビジネスメンバージョンソンとジョンソンプロトコルのメンバーです2022-02-280001442836ジョンソンとジョンソンプロトコルのメンバーです2023-12-310001442836ジョンソンとジョンソンプロトコルのメンバーです2023-01-012023-12-310001442836ジョンソンとジョンソンプロトコルのメンバーです2022-01-012022-12-310001442836ジョンソンとジョンソンプロトコルのメンバーです2022-12-310001442836Mrsn:A 2022 MerckKGaAプロトコルメンバー2022-12-310001442836Mrsn:A 2022 MerckKGaAプロトコルメンバー2022-12-012022-12-310001442836Mrsn:開発と規制のメンバーMrsn:A 2022 MerckKGaAプロトコルメンバー2022-12-310001442836Mrsn:ビジネスメンバーMrsn:A 2022 MerckKGaAプロトコルメンバー2022-12-310001442836Mrsn:A 2022 MerckKGaAプロトコルメンバー2023-01-012023-12-310001442836Mrsn:A 2022 MerckKGaAプロトコルメンバー2022-01-012022-12-310001442836Mrsn:A 2022 MerckKGaAプロトコルメンバー2023-12-310001442836Mrsn:A 20142018協力·ビジネスライセンス契約およびプロビジョニング·プロトコルMerckKgaaのメンバー2023-01-012023-12-310001442836Mrsn:A 2014連携·ビジネスライセンス契約MerckKgaaのメンバー2023-09-300001442836Mrsn:A 20142018協力·ビジネスライセンス契約およびプロビジョニング·プロトコルMerckKgaaのメンバー2023-12-310001442836Mrsn:A 20142018協力·ビジネスライセンス契約およびプロビジョニング·プロトコルMerckKgaaのメンバー2022-12-310001442836MRSN:生物科学のメンバーとしての協働とビジネスライセンス契約Mrsn:制限サービスメンバー2022-01-012022-12-310001442836MRSN:生物科学のメンバーとしての協働とビジネスライセンス契約Mrsn:制限サービスメンバー2021-01-012021-12-310001442836MRSN:生物科学のメンバーとしての協働とビジネスライセンス契約Mrsn:制限サービスメンバー2023-01-012023-12-310001442836MRSN:生物科学のメンバーとしての協働とビジネスライセンス契約MRSN:発展の一里塚的成果メンバー2023-01-012023-12-310001442836アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定する2023-12-310001442836アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2023-12-310001442836アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2023-12-310001442836アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2023-12-310001442836アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2023-12-310001442836アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2023-12-310001442836アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2023-12-310001442836アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2023-12-310001442836アメリカ-GAAP:公正価値は再帰的メンバーを測定する2023-12-310001442836アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2023-12-310001442836アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー2023-12-310001442836アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2023-12-310001442836アメリカ-公認会計基準:アメリカ政府機関債務証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2023-12-310001442836アメリカ-公認会計基準:アメリカ政府機関債務証券メンバーアメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2023-12-310001442836アメリカ-公認会計基準:アメリカ政府機関債務証券メンバーアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2023-12-310001442836アメリカ-公認会計基準:アメリカ政府機関債務証券メンバーアメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2023-12-310001442836アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001442836アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001442836アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001442836アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001442836アメリカ-公認会計基準:アメリカ政府機関債務証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001442836アメリカ-公認会計基準:アメリカ政府機関債務証券メンバーアメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001442836アメリカ-公認会計基準:アメリカ政府機関債務証券メンバーアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001442836アメリカ-公認会計基準:アメリカ政府機関債務証券メンバーアメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001442836アメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001442836アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001442836アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー2022-12-310001442836アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001442836アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001442836アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001442836アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001442836アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001442836アメリカ-公認会計基準:アメリカ証券メンバー2023-12-310001442836アメリカ-公認会計基準:アメリカ政府機関債務証券メンバー2023-12-310001442836アメリカ-公認会計基準:アメリカ証券メンバー2022-12-310001442836アメリカ-公認会計基準:アメリカ政府機関債務証券メンバー2022-12-31Mrsn:安全0001442836Mrsn:実験室装置のメンバー2022-12-310001442836アメリカ-公認会計基準:リース改善メンバー2023-12-310001442836アメリカ-公認会計基準:リース改善メンバー2022-12-310001442836Mrsn:ComputerEquipmentOfficeEquipmentAndSoftwareMembers2022-12-310001442836MRSN:ExistingCreditFacilityMemberUS-GAAP:LineOfCreditMember2019-05-082019-05-080001442836MRSN:ExistingCreditFacilityMemberSRT:最小メンバ数US-GAAP:LineOfCreditMember2019-05-080001442836アメリカ-公認会計基準:良質料率メンバーMRSN:ExistingCreditFacilityMemberSRT:最小メンバ数US-GAAP:LineOfCreditMember2019-05-082019-05-080001442836US-GAAP:LineOfCreditMemberMrsn:AmendedCreditFacilityMember2020-08-282020-08-280001442836SRT:最小メンバ数US-GAAP:LineOfCreditMemberMrsn:AmendedCreditFacilityMember2020-08-280001442836アメリカ-公認会計基準:良質料率メンバーSRT:最小メンバ数US-GAAP:LineOfCreditMemberMrsn:AmendedCreditFacilityMember2020-08-282020-08-280001442836MRSN:NewCreditFacilityMemberUS-GAAP:LineOfCreditMember2021-10-292021-10-290001442836US-GAAP:LineOfCreditMemberMrsn:AmendedCreditFacilityMember2021-10-292021-10-290001442836MRSN:NewCreditFacilityMemberSRT:最小メンバ数US-GAAP:LineOfCreditMember2021-10-290001442836MRSN:NewCreditFacilityMemberアメリカ-公認会計基準:良質料率メンバーSRT:最小メンバ数US-GAAP:LineOfCreditMember2021-10-292021-10-290001442836MRSN:NewCreditFacilityMemberMrsn:前金2023年10月以降から12026年10月までUS-GAAP:LineOfCreditMember2021-10-292021-10-290001442836MRSN:NewCreditFacilityMember2023-01-012023-12-310001442836MRSN:NewCreditFacilityMember2022-01-012022-12-310001442836MRSN:ExistingCreditFacilityMember2021-01-012021-12-310001442836MRSN:ExistingCreditFacilityMember2023-01-012023-12-310001442836MRSN:ExistingCreditFacilityMember2022-01-012022-12-310001442836Mrsn:AtMarketEquityOfferingProgram 2020 ATMMembers2020-05-310001442836アメリカ-アメリカ公認会計基準:普通株式メンバーMrsn:AtMarketEquityOfferingProgram 2020 ATMMembers2021-01-012021-12-310001442836Mrsn:AtMarketEquityOfferingProgram 2020 ATMMembers2021-01-012021-12-310001442836アメリカ-アメリカ公認会計基準:普通株式メンバーMrsn:AtMarketEquityOfferingProgram 2020 ATMMembers2022-01-012022-12-310001442836Mrsn:AtMarketEquityOfferingProgram 2020 ATMMembers2022-01-012022-12-310001442836Mrsn:AtMarketEquityOfferingProgram 2022年2月メンバー2022-02-280001442836Mrsn:AtMarketEquityOfferingProgram 2022年2月メンバーアメリカ-アメリカ公認会計基準:普通株式メンバー2022-01-012022-12-310001442836Mrsn:AtMarketEquityOfferingProgram 2022年2月メンバー2022-01-012022-12-310001442836Mrsn:AtMarketEquityOfferingProgram 2022年2月メンバーアメリカ-アメリカ公認会計基準:普通株式メンバー2023-01-012023-12-310001442836Mrsn:AtMarketEquityOfferingProgram 2022年2月メンバー2023-01-012023-12-310001442836Mrsn:AtMarketEquityOfferingProgram 2022年11月ATMMembers2022-11-300001442836アメリカ-アメリカ公認会計基準:普通株式メンバーMrsn:AtMarketEquityOfferingProgram 2022年11月ATMMembers2023-01-012023-12-310001442836Mrsn:AtMarketEquityOfferingProgram 2022年11月ATMMembers2023-01-012023-12-310001442836アメリカ-アメリカ公認会計基準:普通株式メンバーMrsn:AtMarketEquityOfferingProgram 2022年11月ATMMembers2023-12-3100014428362013-12-3100014428362013-01-012013-12-3100014428362022-06-0900014428362022-06-08Mrsn:投票0001442836Mrsn:WarrantsAndShareBasedPaymentsのメンバー2023-12-310001442836Mrsn:WarrantsAndShareBasedPaymentsのメンバー2022-12-310001442836米国-公認会計基準:給与共有基礎報酬メンバーの延期米国-公認会計基準:従業員株式オプションメンバー2023-12-310001442836米国-公認会計基準:給与共有基礎報酬メンバーの延期米国-公認会計基準:従業員株式オプションメンバー2022-12-310001442836米国-公認会計基準:給与共有基礎報酬メンバーの延期米国-GAAP:制限株式単位RSUメンバー2023-12-310001442836米国-公認会計基準:給与共有基礎報酬メンバーの延期米国-GAAP:制限株式単位RSUメンバー2022-12-310001442836アメリカ公認会計基準:保証メンバー2023-12-310001442836アメリカ公認会計基準:保証メンバー2022-12-310001442836Mrsn:StockIncentivePlan 2017メンバー2017-06-012017-06-300001442836Mrsn:StockIncentivePlan 2017メンバー2023-01-012023-01-010001442836Mrsn:StockIncentivePlan 2017メンバー2023-01-012023-12-310001442836Mrsn:StockIncentivePlan 2017メンバー2023-12-310001442836Mrsn:StockIncentivePlan 2017メンバー米国-公認会計基準:従業員株式オプションメンバー2023-01-012023-12-310001442836Mrsn:取締役会のメンバーMrsn:StockIncentivePlan 2017メンバー米国-公認会計基準:従業員株式オプションメンバー2023-01-012023-12-310001442836SRT:役員メンバーMrsn:StockIncentivePlan 2017メンバー米国-公認会計基準:従業員株式オプションメンバー2023-01-012023-12-310001442836SRT:最大メンバ数Mrsn:StockIncentivePlan 2017メンバー2023-01-012023-12-310001442836Mrsn:報酬プログラムのメンバー2022-02-280001442836Mrsn:報酬プログラムのメンバー2023-01-012023-12-310001442836Mrsn:報酬プログラムのメンバー2023-12-310001442836MRSN:PreorToInducementAwardProgramメンバー2023-12-310001442836SRT:最小メンバ数米国-GAAP:制限株式単位RSUメンバー2023-01-012023-12-310001442836SRT:最大メンバ数米国-GAAP:制限株式単位RSUメンバー2023-01-012023-12-310001442836SRT:役員メンバー米国-GAAP:制限株式単位RSUメンバー2023-01-012023-12-310001442836米国-GAAP:制限株式単位RSUメンバー2022-12-310001442836米国-GAAP:制限株式単位RSUメンバー2022-01-012022-12-310001442836米国-GAAP:制限株式単位RSUメンバー2023-01-012023-12-310001442836米国-GAAP:制限株式単位RSUメンバー2023-12-310001442836米国-GAAP:制限株式単位RSUメンバー2021-01-012021-12-310001442836アメリカ公認会計基準:従業員ストックメンバーMrsn:A 2017 ESPPMメンバー2017-12-310001442836アメリカ公認会計基準:従業員ストックメンバーMrsn:A 2017 ESPPMメンバーアメリカ-アメリカ公認会計基準:普通株式メンバー2017-01-012017-12-310001442836SRT:最大メンバ数アメリカ公認会計基準:従業員ストックメンバーMrsn:A 2017 ESPPMメンバー2017-12-310001442836アメリカ公認会計基準:従業員ストックメンバーMrsn:A 2017 ESPPMメンバー2023-01-012023-01-010001442836アメリカ公認会計基準:従業員ストックメンバーMrsn:A 2017 ESPPMメンバー2023-01-012023-12-310001442836アメリカ公認会計基準:従業員ストックメンバーMrsn:A 2017 ESPPMメンバー2022-01-012022-12-310001442836アメリカ公認会計基準:従業員ストックメンバーMrsn:A 2017 ESPPMメンバー2023-12-310001442836米国-公認会計基準:従業員株式オプションメンバー2023-01-012023-12-310001442836米国-公認会計基準:従業員株式オプションメンバー2022-01-012022-12-310001442836米国-公認会計基準:従業員株式オプションメンバー2021-01-012021-12-310001442836米国-公認会計基準:制限された株式メンバー2023-01-012023-12-310001442836米国-公認会計基準:制限された株式メンバー2022-01-012022-12-310001442836米国-公認会計基準:制限された株式メンバー2021-01-012021-12-310001442836アメリカ公認会計基準:従業員ストックメンバー2023-01-012023-12-310001442836アメリカ公認会計基準:従業員ストックメンバー2022-01-012022-12-310001442836アメリカ公認会計基準:従業員ストックメンバー2021-01-012021-12-310001442836米国-公認会計基準:研究·開発費メンバー2023-01-012023-12-310001442836米国-公認会計基準:研究·開発費メンバー2022-01-012022-12-310001442836米国-公認会計基準:研究·開発費メンバー2021-01-012021-12-310001442836アメリカ-公認会計基準:一般と行政費用メンバー2023-01-012023-12-310001442836アメリカ-公認会計基準:一般と行政費用メンバー2022-01-012022-12-310001442836アメリカ-公認会計基準:一般と行政費用メンバー2021-01-012021-12-310001442836米国-公認会計基準:従業員株式オプションメンバー2023-12-310001442836米国-公認会計基準:従業員株式オプションメンバー2023-01-012023-12-310001442836米国-公認会計基準:従業員株式オプションメンバー2022-01-012022-12-310001442836米国-公認会計基準:従業員株式オプションメンバー2021-01-012021-12-310001442836米国-GAAP:制限株式単位RSUメンバー2023-01-012023-12-310001442836米国-GAAP:制限株式単位RSUメンバー2022-01-012022-12-310001442836米国-GAAP:制限株式単位RSUメンバー2021-01-012021-12-310001442836アメリカ公認会計基準:保証メンバー2023-01-012023-12-310001442836アメリカ公認会計基準:保証メンバー2022-01-012022-12-310001442836アメリカ公認会計基準:保証メンバー2021-01-012021-12-310001442836Mr.Office SpaceCambridgeのメンバー2023-12-310001442836Mrsn:Office SpaceExpansionCambridgeメンバー2021-04-050001442836SRT:最小メンバ数2023-12-310001442836SRT:最大メンバ数2023-12-310001442836米国-GAAP:国内/地域メンバー2023-12-310001442836アメリカ-公認会計基準:州と地方法律法規のメンバー2023-12-310001442836米国-GAAP:国内/地域メンバーアメリカ-公認会計基準:研究メンバー2023-12-310001442836アメリカ-公認会計基準:州と地方法律法規のメンバーアメリカ-公認会計基準:研究メンバー2023-12-31Mrsn:管轄権Mrsn:監査Mrsn:時間0001442836Mrsn:ライセンスプロトコルのアップグレード支払いメンバー2023-01-012023-12-310001442836Mrsn:ライセンスプロトコルのアップグレード支払いメンバー2022-01-012022-12-310001442836Mrsn:ライセンスプロトコルのアップグレード支払いメンバー2021-01-012021-12-310001442836Mrsn:ライセンス契約マイルストーン支払いメンバー2023-01-012023-12-310001442836Mrsn:ライセンス契約マイルストーン支払いメンバー2022-01-012022-12-310001442836Mrsn:ライセンス契約マイルストーン支払いメンバー2021-01-012021-12-3100014428362023-07-260001442836米国-GAAP:従業員サービスメンバー2023-12-310001442836US-GAAP:契約終了メンバー2023-12-310001442836米国-GAAP:従業員サービスメンバー2023-01-012023-12-310001442836US-GAAP:契約終了メンバー2023-01-012023-12-310001442836米国-GAAP:従業員サービスメンバー2022-12-310001442836US-GAAP:契約終了メンバー2022-12-310001442836Mrsn:MartinHuberMembers2023-01-012023-12-310001442836Mrsn:MartinHuberMembers2023-10-012023-12-3100014428362023-10-012023-12-31 アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表:10-K
(マーク1)
☒1934年証券取引法第13又は15(D)項の規定により年次報告書を提出する
本財政年度末まで十二月三十一日, 2023.
あるいは…。
☐1934年証券取引法第13項又は15(D)項に基づいて提出された移行報告
日本から日本への過渡期には、日本から日本への過渡期、日本から日本へ、中国から日本への移行期がある
手数料書類番号 001-38129
Mersana治療会社
(登録者の正確な氏名はその定款に記載) | | | | | | | | |
| デラウェア州 | | 04-3562403 |
| (法団または組織の州またはその他の管轄区域) | | (国際税務局雇用主身分証明書番号) |
| | |
記念大通り840号ケンブリッジ大学, 体積量 | | 02139 |
| (主な行政事務室住所) | | (郵便番号) |
登録者の電話番号は市外局番を含んでいます(617) 498-0020
同法第12(B)項に基づいて登録された証券: | | | | | | | | |
| クラスごとのタイトル | 取引コード | 登録された各取引所の名称 |
| 普通株、額面0.0001ドル | MRSN | ナスダック世界ベスト市場 |
同法第12(G)項により登録された証券:
ありません
登録者が証券法第405条規則で定義された有名な経験豊富な発行者であれば、登録者が有名発行者であるか否かをチェックマークで表記する☐ 違います。 ☒
登録者が当該法案の第13節又は第15節(D)節に基づいて報告書を提出する必要がない場合は,複選マークで示してください☐ 違います。 ☒
再選択マークは、登録者が(1)過去12ヶ月以内に(または登録者がそのような報告の提出を要求されたより短い期間内に)1934年の証券取引法第13節または第15(D)節に提出を要求したすべての報告書を提出したかどうか、および(2)過去90日以内に、登録者がそのような報告書を提出したかどうかを示すはい、そうです ☒いいえ、違います☐
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示すはい、そうです ☒いいえ、違います☐
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
| | | | | | | | | | | |
| 大型加速ファイルサーバ | ☐ | ファイルマネージャを加速する | ☐ |
| 非加速ファイルサーバ | ☒ | 規模の小さい報告会社 | ☒ |
| | 新興成長型会社 | ☐ |
新興成長型企業である場合、登録者が、取引法第13(A)節に従って提供された任意の新しいまたは改正された財務会計基準を遵守するために、延長された移行期間を使用しないことを選択したか否かを再選択マークで示す☐
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オキシリー法案”(米国連邦法典第15編7262(B)条)第404(B)条に基づいて財務報告の内部統制の有効性を評価したことを証明し、その評価は、その監査報告を作成または発表した公認会計士事務所によって行われる☐
証券が法第12条(B)に基づいて登録されている場合は,届出に含まれる登録者の財務諸表が,以前に発表された財務諸表の誤り訂正を反映しているか否か☐
これらのエラーのより真ん中に登録者の任意の幹部が関連回復中に§240.10 D−1(B)に従って受信されたインセンティブベースの報酬に基づいて回復分析を行う必要があるかどうかを再選択マークで示す☐
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)☐いいえ、違います☒
2023年6月30日現在,すなわち登録者が最近完成した第2四半期の最終営業日,非関連会社が保有している登録者普通株の総時価は$である363,809,750この日までにナスダック世界ベスト市場での株式の最新報告販売価格に基づいている。
2024年2月23日現在登録者は121,303,007普通株は、1株当たり額面0.0001ドル、発行された。
引用で編入された書類
登録者が2023年12月31日までの財政年度終了後120日以内に2024年株主総会に提出された登録者最終委託書の内容の一部は、本年度報告のForm 10−K第3部に引用的に組み込まれ、その範囲は本明細書に記載された範囲内である。
カタログ | | | | | | | | | | | |
| | ページ |
前向き陳述に関する特別説明 | | 2 |
| | |
リスク要因の概要 | | 3 |
| | | |
| 第1部 | | |
| | | |
第1項。 | 商売人 | | 5 |
| | | |
第1 A項。 | リスク要因 | | 50 |
| | | |
項目1 B。 | 未解決従業員意見 | | 102 |
| | | |
プロジェクト1 C。 | ネットワーク·セキュリティ | | 103 |
| | | |
第二項です。 | 特性 | | 105 |
| | | |
第三項です。 | 法律手続き | | 105 |
| | | |
第四項です。 | 炭鉱安全情報開示 | | 105 |
| | | |
| 第II部 | | |
| | | |
五番目です。 | 登録者普通株市場、関連株主事項、発行者による株式証券の購入 | | 106 |
| | | |
第六項です。 | [保留されている] | | 107 |
| | | |
第七項。 | 経営陣の財務状況と経営成果の検討と分析 | | 107 |
| | | |
第七A項。 | 市場リスクの定量的·定性的開示について | | 119 |
| | | |
第八項です。 | 財務諸表と補足データ | | 120 |
| | | |
第九項です。 | 会計·財務開示面の変化と会計士との相違 | | 157 |
| | | |
第9条。 | 制御とプログラム | | 157 |
| | | |
プロジェクト9 B。 | その他の情報 | | 158 |
| | | |
プロジェクト9 Cです。 | 検査妨害に関する外国司法管区の開示 | | 158 |
| | | |
| 第三部 | | |
| | | |
第10項。 | 役員、行政、会社の管理 | | 159 |
| | | |
プロジェクト11 | 役員報酬 | | 159 |
| | | |
第十二項。 | ある実益所有者の担保所有権及び経営陣及び株主に関する事項 | | 159 |
| | | |
十三項。 | 特定の関係や関連取引、取締役の独立性 | | 159 |
| | | |
14項です。 | チーフ会計士費用とサービス | | 159 |
| | | |
| 第4部 | | |
| | | |
第十五項。 | 展示表と財務諸表の付表 | | 160 |
| | | |
第十六項。 | 表格10-Kの概要 | | 163 |
| | |
サイン | | 164 |
Mersanaへの引用
本Form 10−K年度報告では、文意が別に指摘されているほか、“会社”、“Mersana”、“私たち”はいずれもMersana治療会社とその合併子会社を指し、“私たちの取締役会”はMersana治療会社の取締役会を指す。
前向き陳述に関する特別説明
このForm 10-K年間報告書は前向きな陳述を含んでいる。前向きな陳述は歴史的事実でもなく、未来の業績の保証でもない。逆に、それらは私たちの現在の私たちの業務の未来、未来の計画と戦略、私たちの臨床結果と他の未来の状況に対する信念、期待、仮説に基づいている。“目標”、“予想”、“信じる”、“考慮”、“継続”、“可能”、“見積もり”、“期待”、“目標”、“予定”、“可能”、“進行中”、“計画”、“可能”、“潜在”、“予測”、“プロジェクト”、“求める”、“すべき”、“目標”、“目標”、“意志”、“将”またはこれらの用語の否定または他の同様の表現は、すべての前向き陳述がこれらの識別語を含むわけではないが、前向き陳述を識別することを意図している。
これらの前向きな陳述には、他に加えて、以下の態様に関する陳述が含まれる
•私たちの現在と未来の研究開発活動、臨床前研究と臨床試験の開始、コスト、時間、進捗と結果、私たちのXMT-1660とXMT-2056の第一段階臨床試験を含む
•私たちの既存の戦略的協力の潜在的な利点と私たちがより多くの戦略的協力に入る能力
•私たちのXMT-1660およびXMT-2056在庫は、私たちが行っている臨床試験および計画の生産運用の結果をサポートするのに十分であるかどうか
•私たちは私たち自身の候補製品とパートナーの候補製品を生産するために必要なDolasyntenと免疫合成プラットフォーム材料の在庫が十分であるかどうか
•私たちの候補製品のために規制承認のタイミングと能力を獲得し、維持する
•私たちは他の候補製品を迅速かつ効率的に識別し、開発し、私たちの既存または未来の抗体薬物結合プラットフォームを革新することができる
•私たちは候補製品を進出させ臨床試験を成功させることができます
•癌適応患者のニーズは満たされていません
•私たちの知的財産権の地位には、私たちのビジネス秘密に関する地位が含まれている
•私たちの戦略的ポイントは
•私たちは支出、未来の収入、資本需要、私たちの現在と予想される現金資源の十分性、そして追加融資の需要に対する私たちの推定値を推定する。
私たちは私たちの展望声明で開示された計画、意図、または予想を実際に達成できないかもしれません。あなたは私たちの展望的声明に過度に依存してはいけません。実際の結果または事件は、私たちが前向きな陳述で開示した計画、意図、および予想とは大きく異なるかもしれない。私たちはこの10-K表の年次報告書の警告声明に重要な要素を含んでおり、特に“リスク要因”の部分では、これらの要素は実際の結果や事件をもたらす可能性があり、私たちが行った前向き声明とは大きく異なると考えられる。私たちの展望的な陳述は、私たちが未来に行う可能性のあるいかなる買収、合併、処置、合弁企業、あるいは投資の潜在的な影響を反映していない。
本稿に含まれる前向き陳述は,本年度報告10-K表までの日の観点を代表している 私たちは法律の要求がなければ、新しい情報、未来の事件、または他の理由でも、いかなる前向きな陳述も更新する義務はない。私たちはその後に発生した事件と事態の発展が私たちの観点を変化させると予想している。したがって、あなたはこのような前向きな陳述に依存して、本年度報告書10-K表の日付までの私たちの任意の日付の観点を代表してはいけません。
リスク要因の概要
私たちの業務は異なる程度の危険と不確実性に直面している。投資家は、以下の概要のリスク及び不確実性、並びに第1部第1 A項で議論されたリスク及び不確実性を考慮しなければならないリスク要因本年度報告の表格10−K
私たちの業務は以下の主なリスクと不確実性の影響を受けている
•私たちが臨床試験で評価している候補製品の数は限られている。現在または将来の任意の候補製品が臨床開発に失敗した場合、私たちの業務に悪影響を及ぼす可能性があり、同じプラットフォーム技術に基づく他の候補製品の開発を停止することが要求される可能性がある。
•私たちは私たちの目標を達成するために大量の追加資金を必要とし、必要な時に必要な資本を得ることができなければ、私たちの製品開発や商業化努力を延期、制限、減少、または中止させることができるかもしれない。
•私たちは設立以来純損失が発生しており、商業販売のための製品は何も承認されていません。将来的には大きな運営損失を受け続けることが予想されます。
•私たちは臨床開発の初期段階にいる。私たちは二つの候補製品、XMT-1660とXMT-2056があり、第一段階の臨床開発にあり、私たちはまだこの二つの候補製品の臨床試験を完成していません。
•私たちは私たちにいくつかの肯定的で否定的な条約を遵守し、私たちの経営と財政的柔軟性に制限を加えることを要求する信用計画を持っている。
•私たちは激しい競争に直面しており、これは他の人たちが私たちよりも早く、あるいは成功的に製品を発見、開発、商業化することにつながるかもしれない。
•薬物発見と開発は複雑で、時間と高価な過程であり、リスクと高い失敗率に満ちている。私たちは新しい抗体-薬物結合体やADC製品の成功と適時な開発を保証することができない。
•私たちは私たちの候補製品が規制部門の承認を受けるか、あるいは臨床試験の結果が有利になるという保証はない。
•もし私たちが高級管理者と肝心な科学者を引き付けることができなければ、私たちは私たちの候補製品の開発に成功し、私たちの臨床試験を行い、そして私たちの候補製品を商業化することができないかもしれない。
•私たちの活動は、ヘルスケア提供者、第三者支払者、患者、政府関係者との交流を含み、医療、反腐敗、データプライバシーと安全、および消費者保護法律に関する広範な規制を受け続けている。適用法を遵守しないことは、巨額の罰金、契約損害、名声損害、収入減少、および私たちの業務の削減または再編を招く可能性があります。
•私たちは特許と他の知的財産権に依存して私たちの技術を保護する。私たちは自分の知的財産権を保護できないかもしれないし、私たちは他人の知的財産権を侵害するために責任を負わなければならないかもしれない。
•不利なグローバル経済または地政学的条件は、私たちの業務、財務状況、または経営結果に悪影響を及ぼす可能性がある。
業界データ
このForm 10-K年度報告書には、業界および市場データが含まれている可能性があり、私たち自身の内部推定と研究、業界および一般出版物および研究、調査、および第三者による研究からこれらのデータを得ることができる。業界出版物、研究、および調査は、そのような情報の正確性または完全性を保証しないにもかかわらず、信頼できると考えられるソースから得られる一般的な声明である。私たちはこれらの研究と出版物が信頼できると信じているが、私たちはまだ第三者源からの市場と業界データを独立して確認していない。
商標に関する説明
私たちは様々な商標登録と申請、そして私たちの名前と会社標識を含む未登録商標を持っています。本報告に登場する他社のすべての他の商号、商標、サービスマークは、それぞれの所有者の財産である。便宜上、本報告における商標および商号は、ラベルまたは記号を有しない可能性があるが、そのような言及は、そのそれぞれの所有者が、適用法に従ってその権利を最大限に主張しないいかなる指示もないと解釈されるべきではない。私たちは、他の会社との関係、または私たちの裏書きやスポンサーを暗示するために、他の会社の商標や商品名を使用したり展示したりするつもりはありません。
第I部
第1項:商業銀行業務
概要
著者らは臨床段階の生物製薬会社であり、抗体-薬物結合体或いはADCの開発に専念し、重大な需要を満たしていない癌患者に臨床上意義のあるメリットを提供する。著者らは数十年の業界経験を利用して、DolasynThenと免疫合成の2つの独自と差別化のADCプラットフォームを開発した。DolasynThenは我々の細胞毒性ADCプラットフォームであり、サイトに特定の同質ADCを生成することを目的としている。次に、Dolasynnは、特定の標的に対して薬物対抗体比(DAR)を最適化することを可能にし、用量制限性の深刻な好中球減少症、末梢神経病変、および眼毒性を回避するために、臨床的に証明された独自の黄金色ペイロードを利用する。免疫合成は著者らの独自のSTIN(インターフェロン遺伝子刺激物)アゴニストプラットフォームであり、系統的に管理するADCを産生し、抗原を発現する腫瘍細胞と腫瘍常駐免疫細胞の中でSTINGシグナルを局部的に活性化し、先天性免疫刺激の抗腫瘍潜在力を放出することを目的としている。私たちはこれらのプラットフォームを利用して私たちの会社とパートナーのためにADC候補製品を生成しています。これらの製品は現在の看護基準を改善する潜在力があると信じています
私たちの2つの臨床段階候補製品はXMT-1660とXMT-2056である。XMT-1660はB 7-H 4を目標としたDolasynten ADCであり、そのDARは正確、目標最適化され、6であり、著者らは一期臨床試験を行っており、現在乳癌、子宮内膜癌、卵巣癌を含む様々な腫瘍患者を募集している。XMT-2056はシステム投与された免疫融合ADCであり、新型のヒト表皮増殖因子受容体2(HER 2)エピトープに対して、DARは8であり、著者らはHER 2発現の末期或いは再発固形腫瘍患者に対して第一段階の臨床試験を行っており、乳癌、胃癌、結腸直腸癌と非小細胞肺癌を含む。また、我々は楊森生物技術会社(あるいはジョンソン)およびメルクKGaA、ドイツダムシュタットまたはメルクKGaAの付属会社Ares Trading S.A.と戦略協力関係を構築し、それぞれ私たちのDolasyntenと免疫合成プラットフォームを利用して他のADC候補製品を発見、開発、商業化することに集中した。また、グラクソ·スミスクライン知的財産権(第4号)有限公司(GSK)に独占選択権を付与し、XMT-2056の共同開発と商業化の独占グローバルライセンスを取得した
私たちは大手製薬会社からの特定のADC経験を含む幅広い経験と関連経験を持つ管理チームを結成し、これらの会社はバイエル株式会社、Centocor Inc.,星座製薬会社、Cubist製薬会社、F.Hoffmann-La Roche Ltd.,グラクソ史克、メルク社、ミレニアム製薬会社、Momenta製薬会社、セノフィ社、Sunovion製薬会社、Tesaro社、Vertex製薬会社を含む。私たちは取締役会と科学顧問委員会の支持を得て、これらの委員会は薬物発見、開発と商業化、業務発展と上場会社管理の面で補充経験を提供した。私たちは高度に差別化されたプラットフォーム、候補製品、協力者とチームは私たちが癌に抵抗する患者のために生活を変えるADCを発見と開発することができると信じている。
私たちの現在の販売ルートは以下の通りです
私たちの戦略
現在承認されているADCは、ある患者集団に実質的な利点を提供しており、より多くの候補製品が開発されているにもかかわらず、顕著なプラットフォームとペイロード制限は、この治療カテゴリがそのすべての潜在力を達成することを阻害していると考えられる。著者らは、一連の癌患者のための著しく改善された安全性と有効性を有するADCを作成するために、新しいプラットフォームとペイロードの開発に焦点を当てている。私たちは以下の戦略目標に照らして実行することが私たちの目標達成に役立つと信じている
•私たちのADCプラットフォームを利用して革新を続けています我々の2つの独自ADCプラットフォーム,DolasynThenと免疫合成は,それぞれ有効な細胞毒性と免疫刺激ADCの開発に利用できると信じている。これらのプラットフォームは用量制限プラットフォームの毒性を減少し、有効負荷抵抗メカニズムを回避し、新しいペイロード代替方案を提供することによって、現在のADCの肝心な制限を解決することを目的としている。持続的なプラットフォーム、ペイロードと候補製品の革新は私たちをADCのリーダーにする可能性があると信じており、私たちはすでに内部研究と発見者からなる核心チームを構築し、新しいと改善されたADC設計方法を求めている。これらの努力を通じて、私たちは機会を発見し、利用して、わが社がADC分野で更に頭角を現し、強力で差別化された候補製品ルートを維持することを渇望しています
•XMT-1660の開発を進めている。XMT-1660はB 7-H 4向けDolasynten ADCである。著者らはXMT-1660がB 7-H 4発現の一連の癌患者が満足していない需要を解決する潜在力があると信じている。著者らは各種腫瘍患者におけるXMT-1660の第一段階臨床試験を引き続き推進し、乳癌、子宮内膜癌と卵巣癌を含む。 著者らは現在、XMT-1660の安全性と耐性を評価し、臨床開発の後期段階で更なる研究を行うために、患者の投与量増加と埋め戻しキューに参加する患者を募集している。著者らは2024年第2四半期に試験中に腫瘍特異的拡張キューを起動する予定であり、2024年に初期用量のアップグレードと埋め戻しキューデータを共有する予定である。
•XMT−2056の開発を進めている。XMT-2056は全身投与された免疫合成ADCであり、HER 2の新しいエピトープを標的とし、標的に依存する方法で腫瘍常駐免疫細胞と抗原を発現する腫瘍細胞においてSTINGシグナルを局所的に活性化することを目的としている。この方法はHER 2の高または低腫瘍患者を単一療法として標準看護薬と併用治療できる可能性があると信じている。2023年第4四半期、米国食品医薬品局(FDA)は、以前治療されたHER 2を発現する末期または再発固形腫瘍患者に対して、XMT-2056臨床試験を一時停止することを決定したと発表した。私たちは試験を再開しており、2024年に早期試験の用量アップ分を予定している
•有力な組織と連携する。著者らは、著者らのADCプラットフォームと候補製品は現有と未来の協力者によって利用され、全世界の多くの患者群の重大な未満足需要を満たすことができると信じている。著者らはすでにジョンソンとメルクKGaAと戦略研究開発協力関係を構築し、それぞれ著者らのDolasynThenと免疫合成プラットフォームを利用して、いくつかの精選したADC候補製品を研究、開発と商業化した。グラクソ·スミスクラインに独占選択権を付与し、グローバル独占許可を得て、XMT-2056を共同開発し、商業化した
ADC背景と既存の制限
ADCは現在検証された成熟した腫瘍学的治療法であり,現在11製品がFDAに承認され,100製品を超える臨床試験が行われている。Leerink Partnersの2024年1月の報告によると、2022年のADC薬物の世界収入は66億ドルに達し、2030年には420億ドルを超えると予測されている。
細胞毒性腫瘍学ADCは伝統的に1種のモノクロナル抗体が化学結合物を介して化学療法“ペイロード”或いは細胞殺傷剤に付着或いは結合する。この抗体は,腫瘍細胞上で健康組織に対して過剰に発現する選択抗原に対する標的能を提供し,腫瘍への優先的,標的化のための機会を提供している ADCが抗原に結合すると,ADCは腫瘍細胞によって内化され,ペイロードはコネキシンの切断や抗体の分解によって放出される。十分な数の細胞毒ペイロードが標的細胞内化されると、腫瘍細胞の死亡を招く。いくつかのADCはペイロードを利用して、抗体から放出されると、抗原に依存しない方式で自由に細胞膜を通過することができ、これは異なる抗原が発現する腫瘍に対する治療効果を増強する可能性がある。 この現象は “傍観者効果”と呼ばれ、衝撃を受けた腫瘍細胞であれば積極的な影響を与える可能性がある しかし健康な細胞が影響を受けると否定的な結果が生じる。
通常、ADC開発者は健康組織に非常に限られた腫瘍細胞に高発現する標的抗原を識別し、腫瘍細胞を死滅させ、健康細胞を殺し、標的毒性を引き起こすことを避けることを求めている。腫瘍細胞に伝達される有効負荷量は,ADCの抗原への結合とその後の内化に関与していることから,腫瘍全体で非常に高く一致(あるいは均一)な抗原発現が奏効率を増加させる可能性が考えられている。
ADCで使用される化学リンカーは、ボディサイクルにおいて早期または制御されずにペイロードを放出することが深刻な非標的毒性をもたらす可能性があるので、ペイロードとボディサイクル中の抗体との間に安定した接続を提供しなければならない。標的腫瘍細胞内化ADCの場合、一般に、腫瘍に対する迅速で効率的な殺傷を促進するために、リンカー切断または抗体分解によって抗体からペイロードを放出する必要がある
ADCのための接続部は、切断可能または切断不可能の2つに分類される。一般に、切断可能なリンカーは循環中に安定であり、腫瘍に発見された酵素によって分解されるように、腫瘍に到達したときに選択的に切断される。逆に,切断不可能なリンカーは抗体の分解に依存してペイロードを放出する。切断不可能なリンカーでは,放出されたリンカーペイロードは依然として抗体の断片に付着しており,細胞の透過性や傍観者効果を制限することができる。使用された接続体−ペイロードの組み合わせの溶解度も、得られたADCの性質に大きな影響を与える
第1世代ADCで使用される多くのリンカーおよびペイロードの水溶解度は非常に低く、凝集のためにDARを3~4つに制限し、DARがこの制限を超えるまで増加すると、不良な薬物様性能をもたらす。また,アダプターペイロードが抗体に結合する部位や方式もADCの安定性や性能に影響を与え,付着部位ごとに周囲の微小環境が異なる可能性があり,アダプターペイロードの性質に影響する可能性がある。 多くの第1世代ADCは、ランダムまたはランダム結合の方法を利用し、これは、これらのプラットフォームが異なるADC集団を生成することを意味し、抗体上の異なる結合部位に付着した高および低DARサブ集団を含む可能性がある。このようなADCの異質性はすでに次善の薬物動態学、低下した治療効果と耐性及び狭窄の治療指数に役立つことが証明された
最近,ADC開発者は特定の部位の共役手法を用いて,ADCの均質性を向上させることを目指してきた。この方法により、リンカー-ペイロードの組み合わせは、ランダムではなく正確で所定の方法で抗体に接続され、より大きなADC同質性および属性の一貫性を生成する。
ADCプラットフォーム毒性制限
同じADCプラットフォームに基づく複数の候補製品が標的外毒性が出現した場合、それは抗原非依存性と考えられ、私たちは“プラットフォーム毒性”と呼ぶ 多くのADCプラットフォームは公認されたプラットフォーム毒性制限があり、治療効果と耐性を影響し、それらがどの抗原に対してであろうと。第一世代ADCは主にチューブリン阻害剤のペイロードを含み、それは微小管の細胞分裂における作用を妨害し、迅速に分裂した腫瘍細胞を殺し、同時に正常組織を保留する。複数のADCはこれらのプラットフォームの使用が許可されているが、患者は治療過程中に深刻な治療関連不良事象、あるいは重症好中球減少症、末梢神経病変および眼毒性などのTRAEを経験することが多い。これらのタイプのプラットフォーム関連有害事象は、しばしば用量を制限し、少なくとも部分的には、健康組織のペイロードに対する非特異的摂取によるものと考えられる
最近のADCプラットフォームはトポイソメラーゼ-1あるいはTopo-1阻害剤のペイロードを利用して、Topo-1酵素のDNA複製と転写における作用を抑制し、腫瘍細胞を殺し、同時に正常組織を保留する。Topo−1阻害剤ペイロードを用いてADC治療を受けた患者は,好中球減少症,貧血,血小板減少症,白血球減少症,間質性肺疾患などの重篤なTRAEを経験し,標的は何であっても報告されている。これらの毒性は,これらの正常細胞型の維持におけるTopo−1酵素の作用に関与していると考えられ,これらのADC標的抗原とも無関係と考えられている
プラットフォーム毒性を含む重篤なTRAEは通常ADCの最大耐性量を決定しているため,これらの有害事象の発生率や重症度を低下させることができる革新的なADCはより有効な単一療法であり,他の療法と広く併用できる可能性が考えられる。
ペイロード抵抗制限
癌耐性の1つのよく見られる機序は多剤耐性ポンプ(MDR)の上昇であり、例えばP-糖タンパク質であり、それは能動的に薬物を癌細胞からポンプし、それらの生存を助けることができる。例えば、多剤耐性ポンプは、癌細胞をAdcetris(ブツキシマブ)とKadcyla(アドトリマブ抗エンタンシン)に耐性を産生させることができ、いずれもfdaが承認したホジキンリンパ腫および乳癌の治療のための第一世代adcであることが証明されている.
新たに出現した臨床データは,Topo−1阻害剤ペイロードを有するADCに耐性を有することが今日一般的であることも示唆されている。これは,少なくとも癌細胞がトポイソメラーゼ−2,あるいはTOPO−2を上昇させたためと考えられ,この酵素はTOPO−1酵素への細胞の依存を置換し減少させることができる。したがって,新たに出現した臨床データが示すように,患者がTOPO−1ペイロードADCを用いて予備治療を行った後,疾患進行後に別の異なるTOPO−1ベースのADCを受けると,患者の臨床利益持続時間が大幅に短縮される可能性がある これまで、アメリカ食品と薬物管理局は2種類のTOPO-1ペイロードを持つADC:ENHERTU(FAM-トラツズマ単抗DERXTECAN-NXKI)とTRODELVY(SASITUZUMAB GOGITECAN-HZIY)を許可し、それぞれが乳癌の治療に応用されている。
検証されたペイロード代替案に欠けている
現在腫瘍学的適応の治療に許可されているADCは細胞毒性ペイロードを搭載しており,直接細胞殺傷作用機序により癌細胞を死滅させることを目指している。ますます多くの会社は免疫刺激剤とタンパク質分解剤のような他のペイロードの方向性送達を実現するためにADCモードの優勢を探索しているが、今までこのようなADCは許可されていない。
我々のADCプラットフォームと革新は
私たちの革新的な努力は現在承認されているADCの制限を克服することに集中している。具体的には,プラットフォーム毒性を最小限に抑え,ペイロード抵抗機構を回避し,ADC領域を細胞毒性方法の外に拡張した新しいペイロード代替案を提供することに注力している。
ペイロード、DAR、位置および共役方法、および同質性など、多くの異なる要因が、ADCの性能に影響を与える可能性があることが知られている。これらの因子の最適な組み合わせは、異なる抗体または標的抗原について異なる可能性がある。いくつかのADC開発者が使用する“一刀両断”方法とは異なり、著者らは斬新かつ差別化されたDolasynThenと免疫合成プラットフォームを設計し、所与の目標に対してこれらの特性を最適化することができ、これはADCが単一療法として使用することも、他の看護標準と組み合わせて使用することもできるかもしれないと信じている
私たちのADC手法の重要な違いは、私たちがADCで使用しているペイロードを取り囲む独自のモジュール式足場を使用していることだと信じています。私たちのステントは、電荷や水の溶解性の増強などの正確な物理化学的性質を提供し、薬物動態を最適化するために、私たちのペイロードの特性を正確にバランスさせることができ、それによって、ペイロードを標的細胞により効率的に輸送し、私たちのADCの治療指数を潜在的に拡大する機会を得ることを目的としている。著者らのステントのキーモジュールは、所与の目標の正確なDARを実現し、水の溶解度と全体的な電荷バランスを調整するために、特定の部位の結合を可能にするように設計されている
我々の第1世代ADCプラットフォームDolaflexinは、高DAR ADC適用の平均負荷特性をバランスさせるために、本質的に異質な天然ポリマー誘導ステントを使用した。我々の次世代プラットフォームDolasynThenとその後の免疫合成を開発する際に、私たちは完全に合成された、同質で正確にバランスのとれたモジュール化ステント方法を設計した。私たちのDolasynThenと免疫合成ステントはモジュール化され、総合的に定義されているため、それらは1組の設計目標による最適化を可能にしていると信じている。各プラットフォームステントは数年来構造-活性関係に対して臨床前探索を行った結果であり、最適な特徴を決定し、特定の標的の変調領域に一致するように微調整される可能性がある
DolasynThen次世代細胞毒性プラットフォームは
私たちのDolasyntenプラットフォームの開発は私たちのDolaflexinプラットフォームを改善するためであり、より広いADC分野で私たちをさらに目立たせるためでもある。Dolaflexinが使用するステントの性質とそのランダム結合方式のため、Dolaflexin ADCは内在的な異質性を有し、高いDARと低DARを有するサブ集団を含む異なる特性を有するサブ個体群から構成される。臨床前データに基づき,低いDARドラフィシンア群と比較して,高DARドラフィシンア群は有意に低下した治療効果と健康組織への有効負荷輸送をもたらす可能性があり,検討した臨床前モデルにおける有効性や耐性低下に関与していると考えられる。
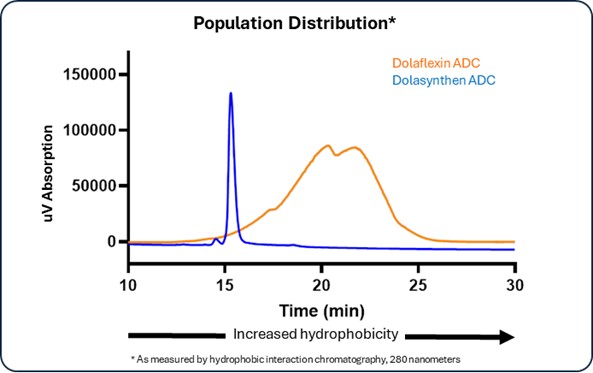
著者らは、DARと他の重要な特性を精確に制御できる同質ADCはドラファキシンと他の既存のADCプラットフォームと比較して独特な治療効果、耐性と治療指数優勢をもたらす可能性があると仮定した。これによって私たちはDolasyntenを開発した。DolasynThenは正確に定義された全合成足場および細胞毒性ペイロードを有し、DARを変更することによって特定の目標のためにADCをカスタマイズし、特定の目標のためにADCをカスタマイズするために水溶性および他の特性を最適化することを可能にする Dolasynten ADCの同質性を確保するために、Dolasynten足場は、正確に定義された一貫した特性を有する完全に均一なADCを作成するために、特定の部位の方法で抗体生物学的に結合される。図1はDolasynten ADC(単峰,鋭峰)とDolaflexin ADC(広いピーク)の異質性を示しており,両ADCとも同じ抗体で作成されており,これは疎水性相互作用クロマトグラフィーにより観察される。
図1
次に,DolasynnはDolaflexinと同様の固有Auistatin抗チューブリンペイロードを用いた。Auistatin専用のペイロードは体外培養そして体内にある臨床前研究は細胞毒性薬物を細胞内にロックすることによって傍観者効果を制御し、腫瘍の中で短時間の抗原非依存性拡散を許可する。薬物が隣接細胞に拡散するにつれて,ペイロードは依然として効率的に代謝されるが,細胞膜を通過できないように設計されている。私たちはこのような“制御された傍観者効果”が安全性と有効性を高める可能性があると信じている。われわれがいくつかの早期候補製品に対して行った複数の臨床試験では,他の第三者プラットフォームによるADC試験でよく報告される重篤な好中球減少症,末梢神経病変,眼毒性はわれわれのauristatin抗チューブリンペイロードではあまり見られないことが観察された。さらに、低透過性形態のペイロードは、PGPのような薬剤耐性ポンプの基質ではなく、このような抵抗機構を回避することができる可能性がある。最後に,我々のAuristatinペイロードはチューブリンを対象としているため,Topo−1やTopo−2の発現に依存せず,Topo−1阻害剤に関連する薬剤耐性機序を回避できる可能性が考えられる。
臨床前研究では,われわれのペイロードにより免疫原性細胞が死亡し,樹状細胞活性化により免疫系を刺激することが観察された。これと一致して,臨床前データは,われわれのペイロードがPD−1阻害剤などの免疫腫瘍学的薬物と相乗作用を有する可能性が示唆された。
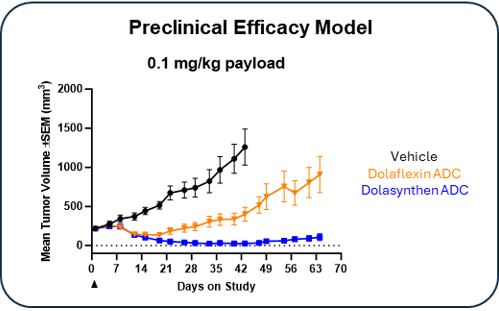
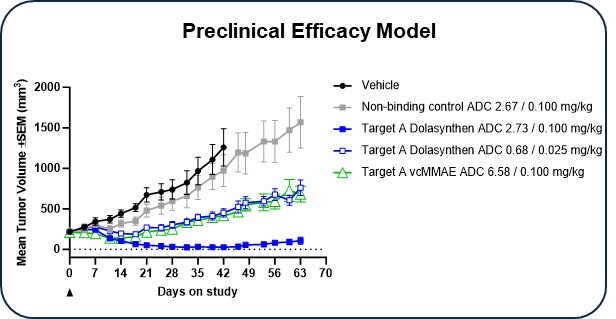
我々はすでに臨床前データを生成し,DolasynThenを用いて開発したADCの特性と性能を比較した 第1世代第三者プラットフォームであるDolaflexinおよびvcMMAEは、承認された複数のADC薬の開発に使用されている。これらのデータは,第一世代プラットフォームを用いて開発されたADCと比較して,DolasynThenが治療効果と薬物動態を改善する潜在力を有することを示していると信じている。図2に示すように,患者由来の腫瘍異種移植モデルでは,Dolaflexin ADCと比較してDolasynten ADCの用量後に腫瘍体積が長時間短縮され,両者とも同じ抗体を用いて作成され,同じ有効用量で投与された効果の向上が認められた。
図2
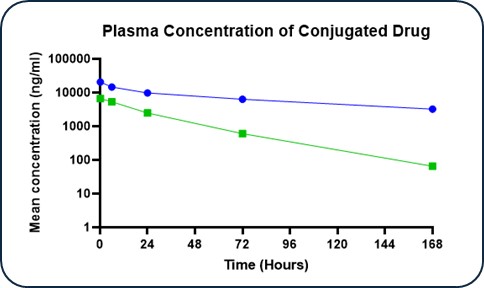
また,図3に示すように,より長い半減期とより大きな曲線下面積を含む改善された薬物動態が観察され,緑色で示されるvcMMAE ADCよりも青色で示されるDolasynThen ADCの半減期が長く,曲線下の面積が大きく,両者とも同じ抗体を用いて作成された。 この前臨床研究では、各ADCの用量は同じ抗体用量であった
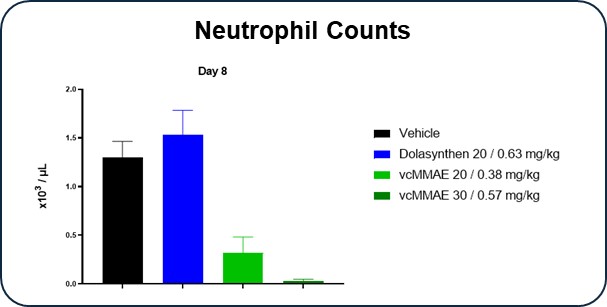
図3
図4に示すように,vcMMAE ADCは緑色で好中球あるいは白血球減少を示すのに対し,Dolasynten ADCは青色で示されており,臨床前モデルで直接比較したところ,同様の減少は経験していないことが観察された。 これらの臨床前データはvcMMAE ADCの投与量と関係があり、いくつかのvcMMAE ADC服用後に深刻な好中球減少が出現した不良事件の臨床報告と一致している。
図4
最後に、図5に示すように、患者由来の腫瘍異種移植モデルでは、同等のペイロード用量のvcMMAE ADC用量と比較して、平均腫瘍体積で測定して、Dolasynten ADC用量を使用することが、より良い治療効果をもたらすことが観察された。また,Dolasynten ADCを用いた用量はvcMMAE ADCを用いた用量と同様に,抗体用量とペイロード量がそれぞれ10倍と4倍低下することが観察された。
図5
著者らの主要なDolasynten候補製品XMT-1660はB 7-H 4に対するADCであり、著者らは各種の腫瘍患者に対して第一段階臨床試験を行っており、乳癌、子宮内膜癌と卵巣癌を含む。私たちは私たちのDolasyntenプラットフォームを通じて私たちの独自パイプラインのために追求した新しい目標を評価している。また、ジョンソンとの協力と許可協定に基づいて、最大3つの目標に適した新しいDolasynten ADCの発見と開発に専念し、仕事を進めていきます。
免疫合成-免疫刺激ADCプラットフォーム
免疫合成は著者らの新型免疫刺激ADCプラットフォームであり、先天性免疫システムに対する標的刺激を実現することによって、ADCに伝統的な細胞毒性ペイロードの交付を超えさせることを目的としている
STINGアゴニストは臨床前に抗癌治療法としての巨大な潜在力を有することが証明された。各種刺激剤候補製品はすでに第三者による臨床研究が行われており、全身投与あるいは腫瘍内注射により行われている。全身免疫活性化に関連する安全リスクのため、全身疼痛作動薬は通常亜治療性臨床用量に限定される。同時に,腫瘍内投与刺激剤も限られた治療効果を示しているが,これは腫瘍の可及性が非現実的であり,腫瘍に接触するためには重複した侵襲的手術が必要であり,刺激剤が腫瘍外に拡散する可能性があるためである。
著者らは、著者らの免疫合成プラットフォームを利用して作成したADCは、抗体を通じて新型の痛み作動薬のペイロードを伝達することを目標とし、全身或いは腫瘍内遊離(非結合)刺激剤の注射による伝達、有効性と耐性挑戦を解決する潜在力があると信じている。
我々が免疫合成プラットフォームで使用する独自刺激剤ペイロードは、その固有免疫活性化作用の伝達と定位を制御するために、非常に低い細胞透過性を有するように設計されている。著者らの臨床前データは、免疫合成ADCの抗腫瘍活性が、腫瘍常駐免疫細胞および腫瘍細胞における抗原依存性送達およびその後活性化されたSTING経路によって駆動されることを示している。Toll様受容体或いはTLRアゴニストなどの他の天然免疫経路と比べ、この2種類の細胞タイプのSTING経路の活性化は抗腫瘍活性を増強する潜在力を提供し、これらの経路はすでに免疫細胞のみを活性化し、腫瘍細胞を活性化しないことが証明された
我々はXMT-2056の第1段階臨床試験を再起動しており、これは我々のリードする免疫合成ADCであり、HER 2の新しいエピトープを目標としており、これまでFDAは2023年第4四半期にこの試験の臨床休止を解除した。私たちはまた免疫合成の新しい目標を評価しており、現在特許研究が行われている。また,我々はメルクKGaAとの連携やライセンスプロトコルに基づいて作業を進めており,2つの目標までの新たな免疫合成ADCの発見に専念している。
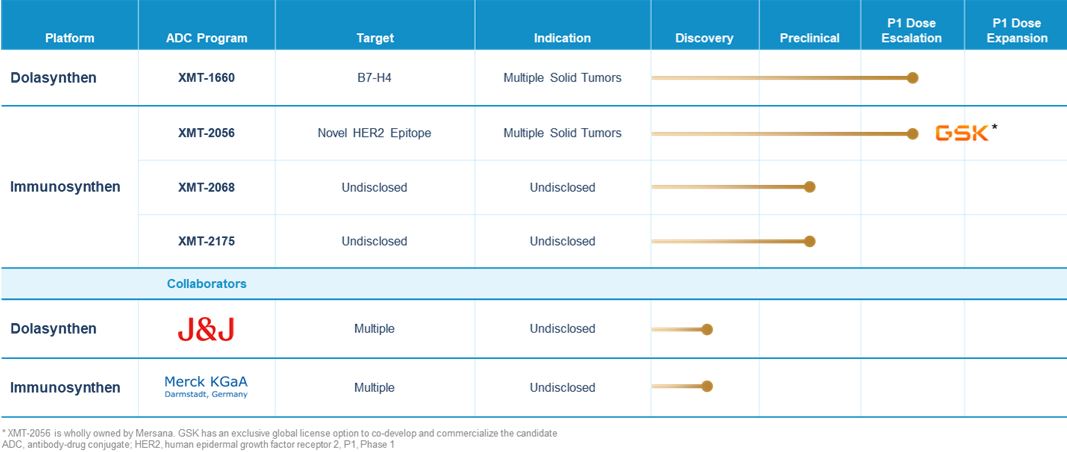
私たちの候補製品
我々は我々のプラットフォームを利用して強力な候補製品パイプラインを開発しており,これらの製品は臨床的意義のある癌療法となる可能性がある。私たちのパイプライン戦略はすでに生物検証(ADCまたは他の方式による)の目標に集中しており、これらの目標の中で、私たちのプラットフォームの優勢は臨床的に優れた治療効果をもたらす可能性があり、私たちは競争力のある、迅速に発売された発展戦略を追求することによって、一流または一流の地位を実現する潜在力がある
私たちは私たちの主要Dolasynten ADC XMT-1660と私たちの主要免疫合成ADC XMT-2056の第一段階臨床試験を進めている。私たちは2つの初期段階の臨床前候補薬があり、私たちはXMT-2068とXMT-2175と呼ばれ、それらは私たちの免疫合成プラットフォームを利用している。また,我々の協力者は異なる開発段階で複数のADC候補製品を持っている
XMT-1660:私たちがリードするDolasynThen ADC候補
XMT−1660はB 7−H 4に対するDolasynten ADCであり,正確で目標最適化されたDARは6であり,われわれは乳癌,子宮内膜癌,卵巣癌を含む様々な腫瘍を有する患者を募集している。B 7-H 4はCD 28/B 7細胞表面蛋白ファミリーのメンバーであり、抗腫瘍免疫を抑制することにより腫瘍の発生を促進し、多種の腫瘍タイプの陰性予後の指標とする。報告によると、それの正常な人体組織における発現は限られているが、多種の高度な需要が満たされていない腫瘍タイプの中ですべて高度に発現し、乳癌、子宮内膜癌と卵巣癌を含む
また,B 7−H 4の腫瘍における発現はプログラム死リガンド1やPD−L 1(B 7−H 1とも呼ばれる)の発現と極めて小さく重なっており,抗腫瘍免疫を抑制する機能冗長性を反映している可能性がある。そのため、B 7-H 4陽性或いはB 7-H 4+の腫瘍に対して、PD-L 1発現が欠損或いは低発現し、PD-(L)1免疫検査点阻害剤は無効である可能性があり、B 7-H 4標的薬物は有効な治療選択である可能性がある。重複しない発現プロファイルはまた、B 7-H 4とPD-(L)1の連合標的治療のために、腫瘍がこれらの免疫抑制蛋白の間で切り替えることによって治療から脱出する潜在的な利益を防止するために理論的基礎を提供する可能性がある。
我々はDolasynten ADCを用いてB 7-H 4を標的とし、良好な臨床前治療効果と耐性データを産生し、DARは正確に2と6であり、DARが約12のB 7-H 4 Dolaflexin ADCと比較した。これらの臨床前データから、XMT-1660にDX 6変異体を選択した。XMT−1660を目指したB 7−H 4は,需要の高さが満たされていない地域で重要な発展機会を提供していると考えられる
われわれは,乳癌,子宮内膜癌,卵巣癌を含む各種腫瘍患者におけるXMT−1660の一期開放ラベル臨床試験を行っている。 試験用量増加部分の主要な終点は最大許容用量を決定し、XMT-1660治療の安全性と耐性を評価することである。著者らは現在患者の投与量増加行列に参加する患者を募集しており、59 mg/平方メートルの用量まで増加している。本年度報告のセル10−Kまで,最大耐容量は決定されていない。引き続き投与量を増加させる以外に、著者らは臨床開発の後期段階で更なる研究を行うために、投与量とスケジュールを最適化するために、患者を埋め戻しキューに入れた。これまでトラツズマブやSavituzumab GOGITECANなどのTOPO−1抗癌剤治療を受けてきた乳癌患者の医療ニーズが満たされていないことや,TOPO−1耐性に関する新たな臨床データから,われわれが行っている第1段階臨床試験に参加する患者を募集している。著者らは2024年第2四半期に試験中に腫瘍特異的拡張キューを起動する予定であり、2024年に初期用量のアップグレードと埋め戻しキューデータを共有する予定である。
B 7-H 4-発現すぐに興味のある癌
各種の癌はすべてB 7-H 4、三陰性乳癌或いはTNBC、エストロゲン受容体/ホルモン受容体陽性乳癌、或いはER+/HR+BCを発現し、子宮内膜癌と卵巣癌は報告の中で最もよくこの抗原を過剰発現する癌である。われわれはXMT−1660の第1段階臨床試験でこれらの適応に参加する患者を募集している。
•TNBC:米国国立癌研究所(NCI)のデータによると,2023年に米国では約297,790例の新たな乳癌症例と43,170例の関連死亡があった。TNBCは乳癌症例の約12%を占めている。治療の特徴は単一薬物化学療法,次いでTopo−1阻害剤ADCである。免疫治療やPARP阻害剤もこれらの患者の一部に作用する可能性がある。XMT-1660は、進行性または転移性TNBCを有する成人患者の治療のためのFast Track製品としてFDAによって指定されており、これらの患者は、転移性環境において少なくとも1つの以前の化学療法を受けている
•ER+/HR+BC:米国癌協会のデータによると、米国297,790例の新規乳癌症例の中で、ER+/HR+BCは最もよく見られる亜型であり、症例の約73%を占めている。HR+の患者では、主要な治療選択はアロマターゼ阻害剤を含む内分泌治療である。この場合に使用される他の標的薬としては、CDK 4/6阻害剤、PI 3 K阻害剤、および最近のTopo−1阻害剤ADCが含まれる
•卵巣癌:NCIのデータによると、2023年にアメリカでは約1977例の新しい卵巣癌症例と13270例の関連死亡があり、アメリカ婦人科癌死亡の最もよく見られる原因となった。標準治療は、白金含有化学療法を繰り返し、その後、PARP阻害剤またはベバシズマブを用いて、患者が白金化学療法に耐性を生じるまで観察または維持することを含む。そして,これらの患者は単剤非白金系化学療法を受ける資格があるか,あるいは高葉酸受容体α発現が決定されれば,抗チューブリンADCミビツキシマブソラフタンシン治療を受ける。
•子宮内膜癌:NCIのデータによると、2023年にアメリカでは約66,200例の新しい子宮内膜/子宮癌症例と13,030例の関連死亡があり、アメリカで最もよく見られる婦人科悪性腫瘍になった。手術とシステム治療は末期子宮内膜癌治療の主流であり、マイクロサテライト不安定/ミスマッチ修復欠陥(MSI/dMMR)状態指導下のシステム治療である。免疫療法と化学療法或いはチロシンキナーゼ阻害剤の結合は標準的な治療方案であり、その後の治療選択は限られている
これらすべての癌タイプの高い再発率や新たに出現したTopo−1阻害剤ADC治療に対する耐性の証拠を考慮すると,XMT−1660などの新たな治療レジメンへの需要は有意に満たされておらず,特に再発の場合には有意に満たされていないと考えられる
XMT-2056:私たちの先行する免疫合成ADC候補
XMT-2056は全身投与された免疫合成STINGアゴニストADC(DAR 8)であり、HER 2の新しいエピトープを標的とし、トラツズマブ或いはpertuzumabの標的とは異なり、そして腫瘍常駐免疫細胞と腫瘍細胞中でSTINGシグナルを局所的に活性化し、HER 2の高或いは低腫瘍患者の単独治療に潜在的な可能性を提供し、そして標準看護薬物と連合治療することを目的としている
HER 2は1種類のシグナル分子ファミリーであり、多種の癌細胞表面に高度に優先的に発現し、腫瘍細胞の成長を促進する作用がある。HER 2蛋白過剰発現は多くの癌でよく記録されており、乳癌、胃癌、膀胱癌、肺癌、食道癌、結腸直腸癌、子宮内膜癌、卵巣癌、唾液腺癌、膵臓癌、子宮頸癌と他の癌を含み、HER 2の過剰発現の発生率は適応によって異なるにもかかわらず
臨床前研究では,XMT−2056がHER 2依存的に腫瘍貯留免疫細胞と腫瘍細胞におけるSTING経路の標的活性化により抗腫瘍活性を示すことが観察された。臨床前環境において,XMT−2056が承認された薬物(例えば抗PD−1抗体やトラツズマブジロテカン)と併用した場合,抗腫瘍活性が増強されることも観察された。
著者らは2023年1月にXMT-2056の多中心第一段階開放ラベル試験を開始し、対象は以前にHER 2を発現する末期/再発固形腫瘍を治療した患者であり、乳癌、胃癌、結腸直腸癌と非小細胞肺癌を含む。この試験は、最大許容用量または推奨される第2段階用量を決定し、XMT-2056治療の安全性および耐性を評価することを目的としている。試験設計は、用量漸増キューから用量レベルを除去する腫瘍タイプ特定のキューにおける治療を評価するための厳選された濃縮キューを含む。選りすぐりの濃縮行列にはHER 2+とHER 2−低乳癌の2つの乳癌濃縮行列が含まれると予想される。私たちは、推奨される第2段階用量を決定するために、用量増加および濃縮キューのデータを使用することを計画している。
2023年3月、我々は、XMT−2056に関連すると考えられるレベル5(致命的)深刻な有害事象(SAE)のために自発的に試験を一時停止したことをFDAに通知した後、FDAがXMT−2056のこの第1段階試験を臨床的に保留したことを発表した。SAEは第1段階の試験用量増加部分に初期用量レベルで登録された第2の患者に発生した。著者らはこの試験における上位2人の患者のサイトカインと他の臨床データを回顧し、免疫活性化のマーカーはXMT-2056がヒトにおいて、我々が以前臨床前研究で観察したよりも有効な先天性免疫刺激剤であることを示した
2023年10月31日、FDAは、XMT-2056第1段階臨床試験の臨床制限を解除したと発表した。私たちはすでに私たちの第1段階用量逓増設計において開始用量を低下させ、この試験を再起動している。私たちは2024年にこの第1段階試験の用量アップグレードを推進する予定だ。
HER 2-直接興味のある癌を表現する
多くの癌はHER 2を発現しているが、著者らは乳癌、胃癌、結腸直腸癌と非小細胞肺癌(NSCLC)を含む各種腫瘍患者を募集し、我々のXMT-2056第一段階臨床試験に参加する予定である。
•乳がん:NCIのデータによると,米国では297,790例の新たな乳癌症例のうち,約15−20%の腫瘍がHER 2+と考えられている。これらの患者は通常HER 2標的薬物治療を受け、例えばトラツズマブとpertuzumabなどである。トラツズマブderuxtecanはTOPO-1阻害剤ADCであり、最近すでに再発転移性HER 2+環境下の看護治療標準になり、そしてますます早期治療に応用されている。
•胃癌:胃癌NCIのデータによると,2023年に米国では約26,500例の新たな胃(胃)癌症例と11,130例の関連死亡があり,そのうち15−20%がHER 2+と考えられている。胃切除手術、化学療法と放射線治療は最初の疾病の主要な治療方法である。トラツズマブ、pertuzumabあるいはトラツズマブderuxtecanなどの免疫療法および/またはHER 2標的薬を使用することもできる。XMT−2056は2022年にFDAにより胃癌治療孤児薬として承認された。
•結腸直腸癌:NCIのデータによると,2023年に米国では約153,020例の新たな結腸直腸癌症例と52,550例の関連死亡があり,そのうち5%の腫瘍がHER 2+と考えられている。結腸直腸手術、化学療法と放射線治療は最初の疾病の主要な治療方法である。トラツズマブまたはpertuzumabなどの免疫療法および/またはHER 2標的剤を使用することもできる。
•NSCLC:NCIのデータによると,2023年に米国では約238,340例の新たな肺癌症例と127,070例の関連死亡があり,そのうち80%~85%が非小細胞肺癌と推定され,そのうち15%-20%の腫瘍がHER 2+と考えられている。肺切除手術、化学療法と免疫治療は最初の疾病の主要な治療方法である。放射線治療とある標的治療は、トラツズマブ、pertuzumab或いはトラツズマブderuxtecanを含み、疾病の後期と後期に応用されている。
これらすべての癌タイプの高再発率とTopo−1阻害剤ADC耐性機序の新たなエビデンスを考慮すると,XMT−2056などの新たな治療レジメンへの需要は有意に満たされておらず,特に再発の場合には有意に満たされていないと考えられる
生産停止候補製品
2023年7月、我々は、Upivitimab rilsodotin、またはUpRiとも呼ばれるXMT-1536の開発を停止し、これは、第1世代Dolaflexinプラットフォームを用いて開発されたナトリウム依存リン酸輸送体NaPi 2 bに対するADCである。この決定はUpliftのTOPLINEデータに基づいて行われ,Upliftは単腕臨床試験であり,1~4種類の先行治療案を受けた白金耐性卵巣癌患者を募集した。NaPi 2 b陽性群の中で、研究者が評価した客観応答率は15.6%であり、この試験はその主要な終点に達しておらず、標準看護単薬化学療法12%客観応答率の95%信頼区間下限を排除できなかった。NaPi 2 b陽性群で評価した応答持続期間は7.4カ月であった。上昇期において、治療に関連する最もよく見られる有害事象は一過性AST上昇、吐き気、血小板数低下(血小板減少を含む)と無力を含む。レベル3以上の眼毒性のない患者では,レベル3末梢神経病変と好中球減少の発生率は1%未満であった。9.7%の患者が肺炎を発症しており,NaPi 2 bの肺内II型肺胞細胞への発現による標的毒性であると推察される。5.6%の患者は救急3級以上の出血不良事件が発生し、5例の死亡病例を含む。著者らのすべてのUpRiとXMT-1592臨床データ(以下に議論するXMT-1592)および著者らは異なるステントとプラットフォーム上で同じ抗体とペイロードを使用する計画の臨床前データを深く分析した後、著者らは深刻な出血リスクはヘテロポリフレキシン混合物中の高DAR亜群の非特異的伝達による内皮損傷によるものであると仮定した。多くの患者に血小板数の低下を認めた。血小板のこの低下は血小板消費と活性化のマーカーであり,この内皮損傷の潜在的な指標でもあると推察される
2022年5月には、UpRiと同じペイロードを持つNaPi 2 bを目標としたDolasynten ADCであるXMT-1592の開発も停止した。XMT-1592はすでに卵巣癌と非小細胞肺癌患者の第一段階用量増加試験において研究を行った。著者らは31名の患者を募集して用量増加を行った後にこの試験を中止し、評価可能な卵巣癌患者(n=13)の2つの最高用量レベルでの全体客観応答率は31%であり、NaPi 2 b発現の選択を考慮しないことを観察した。また,UpRiで観察されたプラットフォーム毒性の理解から,この停止した第一段階試験のデータから,UpRiに比べてXMT−1592の非標的毒性が低下していることが示唆された。UpRiと比較して,XMT−1592を用いた患者では疲労,嘔気,一過性AST上昇の頻度と重症度が低かった。さらに、XMT−1592を使用した患者は、治療に関連する血小板減少有害事象(血小板減少を含む)または出血事象を報告していない。隆起試験の場合と同様に,XMT−1592のこの段階試験で肺炎が報告されている。この毒性は肺内II型肺胞細胞NaPi 2 bの発現に関与する標的毒性であると仮定した
著者らは2024年3月にヨーロッパ婦人科腫瘍学会或いはESGO大会でUpliftとXMT-1592 1期試験の臨床データを公表する予定である
戦略的協力
私たちは業務発展をわが社全体の戦略の中心的な柱としており、私たちのプラットフォームと候補製品は多様なタイプの潜在的な戦略協力を考えることができるようにしています。我々のADCプラットフォームは、多くのターゲットに広い適用性を有しており、パートナーが選択された数のターゲットに独自抗体を提供することを考慮することができ、私たちは私たちのプラットフォームを使用して新しいADC候補を発見することができると信じている。これらの協力プロトコルによれば、私たちは私たちのADCプラットフォーム(S)を任意の改善する権利を持っている。例えば,我々はジョンソンとDolasynThenに集中した発見協力に入り,メルクKGaAと免疫合成に焦点を当てた発見協力に入った。プラットフォームに基づく協力は私たちのプラットフォームの潜在力を利用して、短期資本を提供し、そして潜在的に患者に重要な新しい治療選択をもたらすことを助けることができると信じている。
私たちはまた、パートナーがいくつかの臨床前、臨床的、および/または商業的責任を負うことができるように、内部開発されたADC候補製品を持っている。例えば、我々はグラクソ·スミスクラインに独占選択権を付与し、グローバル独占許可を得て、XMT-2056を共同開発して商業化した。グラクソ·スミスクラインは今までこの選択権を行使しなかった。製品を重点とした協力は短期資金を提供することができ、臨床前、臨床或いは商業開発努力を推進し、拡大することができ、私たちが独立して完成した努力を超えて、患者に新しい治療選択をもたらす可能性があると信じている。
2022メルクKGaA協働
2022年12月、私たちはメルクKGaAと協力·商業許可協定、すなわち2022年メルクKGaA協定を締結した。2022年メルクKGaAプロトコルによれば、私たちは、マークKGaAプロトコルの発効後の一定期間内に選択された最大2つの特定の標的抗原または指定された標的に対する免疫合成ADCを使用することを可能にするために、マークKGaA独占ライセンスを付与する。メルクKGaAは2022年のメルクKGaAプロトコルに従って最初の指定されたターゲットを選択した。
2022年のメルクKGaA協定の条項によると、双方は最大2つの研究プロジェクトを展開する。各研究計画は、指定されたターゲットのための許可ADCの研究新薬申請またはIND(または海外等価物)、または各メルクKGaA許可製品が提出されるまで、選択されたターゲットの免疫合成ADCに関連する活動(各ADCは、2022年のメルクKGaAプロトコルに従って開発された許可ADC)に関連し、または定義された研究期間がより早く満了するまで、または定義された研究期間がより早く満了するまで、および関連する活動を含むであろう。すべての研究計画は双方が合意した研究計画に従うだろう。各指定されたターゲットについて、メルクKGaAは、指定されたターゲットに対して最大指定された数の抗体を提供することを担当し、私たちは、許可されたADCを作成するために、私たちの免疫合成プラットフォームを使用してこれらの抗体を結合することを担当する。すべての側は研究計画の下で自分の費用を担当するだろう。さらに、許可されたADCのいくつかの化学、製造および制御開発、およびいくつかの製造活動を担当し、いくつかの臨床前研究および臨床試験のための許可されたADCの薬物物質の製造を含み、それぞれの場合、費用はMerck KGaAによって負担され、いくつかの費用はMerck KGaAによって前払いされる。上記の規定を除いて、メルクKGaAは単独で責任を負う体外培養そして体内にある許可された任意のADCの特徴、他の臨床前作業、およびそれによって生成された任意のメルクKGaA許可製品に関連するすべての臨床開発および潜在的な商業化活動。
2022年メルクKGaA協定の条項によると、私たちは2023年2月に3000万ドルの前金を受け取った。メルクKGaAは私たちに研究プロジェクトのある開発と監督マイルストーンを支払い、ある発見マイルストーン、ある臨床試験の開始、及び監督部門がある地区のMerck KGaA許可製品を許可し、もしMerck KGaAがこの2つの指定目標に対するMerck KGaA許可製品を監督管理許可に提出すれば、総額は2億ドルに達する。
メルクKGaA許可製品の商業化により商業販売が発生すれば、メルクKGaA許可製品は、指定された目標を適用したメルクKGaA許可製品が指定された総売上の敷居に達した後、計画ごとにビジネスマイルストーンを支払い、この2つの指定目標に対するMerck KGaA許可製品がMerck KGaAによって商業化されれば、合計6億ドルに達する。また、将来的にメルクKGaAライセンス製品の純売上高で2桁から低い2桁の割合で階層印税を得る資格があります。
Merck KGaAは各国/地域と各Merck KGaA許可製品に対して引き続き印税義務を負担し、(I)このMerck KGaA許可製品がこの国である知的財産権保護を受けなくなった日、(Ii)このMerck KGaA許可製品が同国で初めて商業販売10周年の日、および(Iii)このMerck KGaA許可製品のこの国/地区でのマーケティング或いはデータ独占経営権が満期になった日まで。
2022年のメルクKGaA協定の条項によると、いくつかの例外的な場合を除いて、合意された期間内に、私たちは、指定されたターゲットのために、私たちの免疫合成プラットフォームを利用した他のADCを自らまたは第三者によって研究、開発、製造、または商業化しない。我々とメルクKGaAは共同研究委員会、共同製造委員会、共同知的財産権委員会を設立し、2022年のメルクKGaA協定による活動の調整を担当する。
各当事者は、いくつかの条件に適合する場合に、“2022年メルクKGaAプロトコル”の下でそれを再許可する権利を有する。事前に終了しない限り、“2022年メルクKGaA協定”は、最後のメルクKGaA許可製品の最後の満了の印税期限が満了するまで有効であり、またはメルクKGaAがMak KGaA許可製品を前払いしていない場合、最後の満了した研究計画が満了した時点で継続的に発効する。メルクKGaAは、私たちに特定の通知を出した後、その都合の良い場合、“2022年メルクKGaAプロトコル”を完全にまたは指定された目標で終了することができる。私たちまたはメルクKGaAは、他方の破産またはいくつかの未治癒違反によって、2022年のメルクKGaA協定を終了することができる。もしマークKGaAが私たちの未治癒の重大な違約行為のために2022年のメルクKGaA協定を終了する権利があれば、2022年のメルクKGaA協定を終了するのではなく、メルクKGaAは特定の罰金を引用することを選択することができ、私たちが適用する重大な違約行為の唯一と排他的な救済措置として、このような未治癒の重大な違約後に私たちに支払う可能性のある1つ以上の未来支払いに影響を与える。もしメルクKGaAが相応の研究計画を進めなければ、私たちは指定された目標に関する2022年のメルクKGaAプロトコルを終了するかもしれない。さらに、メルクKGaAまたはその任意の従属許可者または付属会社が、私たちのいくつかの特許の有効性、実行可能性に異議を唱えた場合(いくつかの例外を除いて)、2022年のメルクKGaAプロトコルを終了することができる。
グラクソ·スミスクラインと協力して
2022年8月、著者らはGSKと協力、選択及び許可協定を締結し、GSKに独占的な選択権を提供し、全世界の独占許可を取得し、XMT-2056を含む製品を共同開発及び商業化し、もしかしたら製品が可能かもしれず、GSKにXMT-2056を交付する第一段階単薬臨床試験において、乳癌患者の投与量の増加と濃縮完了後の特定の時間内或いは選択期間内に行使することができる。グラクソ·スミスクラインの選択権行使には、1976年にハート-スコット-ロディノ反独占改善法案や高速鉄道によって承認される必要があるかもしれない。グラクソ·スミスクラインが適用される任意の高速鉄道許可後にオプションを行使したり、GSKオプションを行使した場合、グラクソ·スミスクラインは私たちに9,000,000ドルのオプション行使金を支払う義務がある。
我々は我々のXMT-2056計画に関する研究や開発活動をリードし,もしあれば,合意された時間内にビジネス的に合理的な努力を用いてオプションパケットを生成する義務がある.GSKオプションを実施する前に,XMT−2056計画に関する製造,研究,早期臨床開発活動のコストを担当する。
グラクソ·スミスクラインが選択権を行使した後、もしあれば、グラクソ·スミスクラインはXMT-2056の生産を選択する可能性があり、私たちとグラクソ·スミスクラインは双方が策定する米国とEUの許可製品を承認するための共同開発計画に基づいてXMT-2056を共同開発し、GSKは大部分の開発活動とコストを担当する。グラクソ·スミスクラインは、米国とEU以外の承認を得るためだけのすべての開発コストを担当する。GSKプロトコルに規定されているいくつかの例外を除いて,このようなコスト分担スケジュールにより,米国とEUに重点を置いた開発コストにおける総シェア上限を固定額,すなわちMersana開発コスト上限とする.GSKプロトコルに規定されているいくつかの制限に適合する場合には、GSKが要求するいくつかの許可製品の後期臨床試験の開発コストを分担しないことを選択することもでき、開発コストを分担しない任意のこのような後期臨床試験のデータが、許可製品が米国またはEUで何らかのマーケティング承認を得ることにつながるか、または購入支払いとみなされる場合には、これらの費用を分担しないことを選択することも可能である。Mersana開発コストの上限を超える任意の開発コストは、購入された支払いとみなされる任意の金額を含めて、GSKが負担します私たちが利益シェア選択権を行使するまで(以下のように定義する))である。Mersana開発コストの上限を超える開発コストは、最優遇金利と特定の保証金に等しい変動金利で計上され、後で私たちが返済したり、将来の規制や販売マイルストーンや満期になる可能性のある特許使用料の支払いを相殺したりします。もし私たちが利益シェア選択権を行使すれば、Mersana開発コスト上限は適用されなくなり、当時返済されていなかった残りの部分に計算すべき利息を支払わなければならず、米国とEUに重点を置いた開発コストをより多く分担し続ける。
グラクソ·スミスクラインオプションを行使した後、もしあれば、私たちはグラクソ·スミスクラインからいくつかの後期臨床データおよび他のデータおよび情報を受け取った後の特定の期間内に、任意の特許製品の特定のシェアを受け入れる(または負担する)米国利益(または損失)、または利益シェア選挙を選択する権利があるだろう。また、もし私たちが利益分額選挙を行使すれば、私たちは同時にアメリカで任意の許可製品を共同で普及させることを選択することもできます。協調推進計画は、GSKプロトコルの残りの部分が依然として有効であるにもかかわらず、他方に何らかの違反が発生した場合、またはGSKがMersanaのいくつかの特定の制御権が変化した場合にプロトコルを終了する任意の一方によって終了することができる。また,Mersanaのある特定の制御権が変更された場合,GSKはこのような制御権変更後にGSKプロトコルによって起動される開発活動の実行を禁止することができる.
私たちはグラクソ·スミスクラインから1億ドルの前金を受け取った。早期臨床開発マイルストーンを満たすことで最高3000万ドルを得る資格があり,これらのマイルストーンはGSKオプション行使前に発生する可能性がある。GSK引受権の行使により、もし私たちの利益部数を選挙させることができなければ、私たちは5.92億ドルの追加未来の臨床開発と規制マイルストーン支払い、6.52億ドルに達する商業マイルストーン支払い、および世界の特許製品販売の2桁の等級版税(承認されれば)を得る資格があり、最高20%に達するが、慣例の減免を守らなければならない。もし私たちが利益分額選挙を行使すれば、上記の規制と商業マイルストーン金額の代わりに、減少した監督管理と商業マイルストーン支払いを獲得し、米国以外の販売の特許使用料を下げる資格があります。また、私たちが利益分額選挙を行使するか否かにかかわらず、グラクソ·スミスクラインは、指定された第三者にいくつかの記念碑的な支払いや特許権使用料を支払う責任があり、現在、これらの第三者とXMT-2056計画に関する合意を締結しています。
グラクソ·スミスクラインの各国および各ライセンス製品に対する印税義務は、(I)当該ライセンス製品が当該国が特定の知的財産権によって保護されなくなった日、(Ii)当該ライセンス製品が当該国で初めて商業販売されて12周年の日、および(Iii)当該ライセンス製品が当該国/地域の排他的規制が満了する日まで継続される。
GSKプロトコルの条項によれば、いくつかの例外を除いて、一定期間にわたって、我々およびGSKは、(A)刺激剤に結合したADCを含むか、または含むか、および(B)HER 2に対する他の製品または化合物を直接または第三者によって開発または商業化しない。さらに、我々は、STINGアゴニスト以外のペイロードに結合し、HER 2に配向された将来のADCに対するグラクソの優先交渉権を付与した。GSKオプション実施後,我々とGSKは共同指導委員会,共同開発委員会,共同製造委員会,共同商業化委員会,金融ワーキンググループを設立し,GSKプロトコルでのすべての活動の調整を担当し,GSKは多くの問題に対して最終決定権を持つが,いくつか列挙された例外は除外する.
グラクソ·スミスクラインがその選択権を行使しない場合、GSKプロトコルはオプション期限終了時に終了する。グラクソ·スミスクラインがその選択権を行使したが、吾らは契約側が当該高速鉄道承認文についてそれぞれの適用文書の最終日を提出した後の指定時間内に高速鉄道承認文を取得できなければ、GSKプロトコルを終了する権利がある。GSK購入権を行使する場合、GSKプロトコルは、GSKプロトコルに従って当該等の許可製品についてその国での支払い義務期間が満了するまで、いずれか一方がGSKプロトコルの条項に従って早期に終了しない限り、ライセンス製品および国/地域に従って有効に継続する。私たちまたはグラクソ·スミスクラインは、相手が借金を返済できないために“GSKプロトコル”を終了することができ、どちらも他方のいくつかの是正されていない違反によって“GSKプロトコル”を終了することができる。GSKプロトコルを終了することに加えて、私たちがいくつかの未治癒の重大な違約事件が発生した場合、GSKは法的または衡平法で利用可能な他の契約救済措置に加えて、このような未治癒の重大な違約事件後に当社に支払う可能性のある1つまたは複数の将来の支払いに影響を与えるために、指定された罰金を一度に選択する。GSKまたはその任意の二次ライセンスまたは付属会社がそのような特許の有効性、実行可能性に疑問を提起した場合、私たちは、GSKのいくつかの特許の許可を終了する可能性がある。便宜上、グラクソ·スミスクラインは私たちに知らせた後、グラクソ·スミスクラインとの合意を終了することができる。
きょうせい協力
2022年2月、私たちはジョンソンと研究協力と許可協定を締結した。我々は2023年7月14日と2023年9月25日に改訂された本協定を“ジョンソン協定”と呼ぶ。ジョンソン·プロトコルによると、私たちはジョンソンに独占的な許可を与え、私たちの独自のDolasyntenプラットフォームと他の技術開発、製造、商業化を使用して、ジョンソンによって選択された最大3つの標的に対する抗体-薬物結合体を使用することができる。我々の役割は,ジョンソンから提供された抗体による生物共役活動を行い,ジョンソンのためのADCを創出することである。 また,ジョンソンが開発により進展したADCのために何らかの化学,製造,制御開発と早期製造活動を行い,臨床薬物の製造を含め,費用はジョンソンが負担する。これらの限られた製造に加えて,ジョンソンはジョンソンプロトコルに基づいて開発したADCのさらなる開発,製造および商業化を担当し,任意の必要な規制承認を取得し,費用はジョンソンが支払う。
ジョンソン協定の条項によると、私たちは4,000万ドルの前払いを受けた。ジョンソンはまた、ある発見マイルストーン、ある臨床試験の開始及びある地区のある許可製品の監督許可を含むいくつかの開発と監督管理マイルストーンを研究プロジェクトに支払い、もしジョンソンがすべての三つの目標に対するADCを提出すれば、合計5.01億ドルに達する。ジョンソン開発のADCが商業化されれば、適用対象に基づくすべてのADCが指定された総販売限界に達したときに各プロジェクトのいくつかのビジネスマイルストーンを取得する資格があり、ジョンソンがすべての3つの目標のADCを商業化すれば、合計で約5.3億ドルに達する。また、ADCの将来の純売上高の中央値-1桁から低い-2桁の割合で階層印税を得る資格がある。
ジョンソン·プロトコルは、最後のADCの最後の特許使用料の期限が満了するまで、事前に終了しない限り有効になるだろう。特許権使用料期間とは、ADC毎および国/地域に基づいて、ADCの同国での初の商業販売開始から最近、以下の時間が発生するまでの期間である:(A)ADCに関連する最後の特許権使用料を負担する特許主張が国/地域で満了した日、(B)ADCがその国で規制排他的に満了した日、および(C)ADCがその国で初めて商業販売10周年を迎えた日である。ある国/地域のADCの使用料期間が満了した後、ジョンソンの許可は、当該国で開発、製造、商業化、または他の方法で利用するために、関連するプラットフォーム技術および任意の共同技術における権益の下で再許可を付与する権利がある永久的、撤回不可能、非独占的、全額支払いおよび免除される権利および許可となる。
2014年メルクKGaA協力
2014年6月、私たちはドイツDarmstadtのMerck KGaAと協力と商業許可協定、あるいは2014年のMerck KGaA協定を締結し、私たちのDolaflexinプラットフォームを利用してADC候補製品を開発と商業化し、最大6種類の標的抗原に応用できる。マクKGaAは標的抗原に対する抗体の産生を担当し,これらの抗体を用いてDolaflexin ADC候補を産生することを担当している。メルクKGaAは、これらのADC候補製品のさらなる開発および商業化を担当する独占的権利を有し、担当する。2018年5月、我々はドイツのダムシュタットのメルクKGaAと、IND研究および臨床試験に使用可能な材料を供給する2018年メルクKGaA供給協定を締結した。2023年12月15日、私たちはメルクKGaAと共同で、2014年のメルクKGaA協定と2018年のメルク供給協定を終了することに同意しました
アザナ生物科学が協力して
2012年3月、アザナ生物科学有限責任会社(遠藤製薬有限会社委託)やアザナ生物科学会社と協力協定を締結した。この合意の条項に基づき,我々はアザナの新しい抗体を用いて,我々の多楽欣プラットフォームのコンポーネントを用いて新しいADCを開発した。アサナ生物科学は任意のADC製品の製品開発、製造、商業化を担当している。
“Synaffixビジネスライセンスプロトコル”
2019年1月、私たちはSynaffix B.V.またはSynaffixと商業許可協定を締結し、2021年11月にこの合意を修正して再説明し、Synaffixとの関係を拡大し、2022年2月にジョンソンとの協力について再修正した。修正して再記述したプロトコルをSynaffixライセンスと呼ぶ.Synaffixライセンスにより,ターゲットに対するADCを開発,製造,商業化する権利があり,Synaffix独自のサイト特定共役技術を用いて,最大12個のターゲットに利用可能である.私たちは私たちの開発計画と協力に関する5つの目標を許可し、私たちは最大6つの追加的な目標を許可する権利がある。Synaffixライセンスに関連する680万ドルの費用を支払いました。その中には、400万ドルの予約料と許可料、180万ドルのマイルストーン支払い、100万ドルが将来の予約費および許可料、および将来の潜在開発マイルストーンの一部に使用される可能性があります。私たちは開発、規制、そして商業マイルストーンに4800万~1.32億ドルを支払う義務があるだろう。
ライセンスターゲットに対するADC製品が商業販売を開始する場合、もしあれば、Synaffixにそれぞれの製品の純売上高の1桁パーセント以下の階層印税を支払う必要がある。Synaffixライセンスは、製品のSynaffixライセンスの下で許可された特許のうちの最後の有効な権利要件が満了するまで、各国/地域および個々のライセンス製品に基づいて有効に維持される。各国/地域の各許可製品のSynaffixライセンスが満了した後、私たちがその国/地域で私たちに付与したこのような製品のライセンスは、全額支払いおよび永久的になります。私たちは製品を全部または一つずつ許可するSynaffix許可をいつでも終了することができる。どちらも,他方が合意の重要な条項に違反していることや,他方が破産に関するイベントが発生したことが原因で,所定の通知や救済期間内にSynaffixライセンスを終了することができる.
製造業
我々は所有したり経営したりせず,現在の良好な製造実践やcGMP,標準に適合した製造施設を構築する計画もない。私たちは現在、規制承認や商業製造を通じて私たちの活動を支援し、現在の協力の下での製造義務を支援するために、外部契約製造機関(CMO)に依存して製品製造を継続していく予定です。我々は薬物開発と製造経験のある者が我々のCMOとの関係を担当している。将来的には、これらのCMOを用いて私たちの製品の商業供給を生産したいと思っていますが、これらのCMOは生産規模を増加させる必要があるかもしれません。私たちは現在使用しているCMOが商業製造のために生産規模を拡大できないように、合格した代替サプライヤーがいません。Dolasyntenと免疫合成の製造過程は入手しやすい原料に触れ,化学/製薬生産分野で前例のあるユニット操作を用いた。現在、XMT-1660とXMT-2056のサプライチェーンにはいくつかの共通のサプライヤーがあり、私たちの現在の知っている限り、私たちはこれらのサプライヤーを使用することができると信じています。または商業化目的を達成するために、商業的に合理的な条項で他のサプライヤーを決定し、それと契約を締結することができます。
政府の監督管理
薬品と生物製品の研究、開発、テスト、製造、品質管理、包装、ラベル、貯蔵、記録保存、流通、輸入、輸出、販売促進、広告、マーケティング、販売、定価と精算は、米国と他の国と外国司法管轄区(EUを含む)政府当局によって広く規制されている。米国および外国や管轄区域で規制承認を受ける手続きや、適用される法規や条例、その他の規制要求を遵守するには、承認前も承認後も、多くの時間と財力が必要となる。生物製品の開発、承認とマーケティングに適用する監督管理要求は変化する可能性があり、法規と行政指導はよく機関によって私たちの業務に重大な影響を与える可能性がある方法で修正または再解釈される。
アメリカ政府の生物製品の規制は
米国では,FDAは“公衆衛生サービス法”(Public Health Service Act,PHSAと略す)に基づいて生物製品に許可証を発行し,“食品,薬物と化粧品法”(Food,Drug and Cosmetic Act,略称FDCA)に基づいてこのような製品を規制している。このような製品の臨床開発計画の開始と管理を担当し、その規制承認を担当する会社、機関、または組織は、一般にスポンサーと呼ばれる。 米国での新生物の販売と流通の承認を求めるスポンサーは、以下の各ステップを満足的に達成しなければならない
•良好な実験室実践又はGLP、法規又は他の適用法規に基づいて臨床前実験室試験、動物研究及び調合研究を完成する
•臨床方案を設計し、FDAにINDを提出し、この方案は人体臨床試験が開始される前に発効し、いくつかの変更をした時に更新しなければならない
•各臨床試験を開始する前に、各臨床試験場所を代表する独立機関審査委員会または倫理委員会によって承認される
•適用されたIND法規、良好な臨床実践或いはGCP及び他の臨床試験に関連する法規に基づいて十分かつ制御された人体臨床試験を行い、各提案適応に対する研究製品の安全性、有効性と純度を評価する
•アプリケーション使用料の支払いを含む1つまたは複数の推奨適応の上場承認を申請するBLAを作成してFDAに提出するステップと、
•適用される場合、BLAはFDA諮問委員会によって検討される
•生産生物の1つまたは複数の製造施設に対するFDAの1回または複数回の検査は、cGMP要件に適合する状況を評価して、施設、方法、および製品の特性、強度、品質、および純度を維持するのに十分な制御を保証するために満足的に完了する
•GCPに適合し、BLAをサポートするために提出された臨床データの完全性を保証するために、FDAの臨床試験場所に対する任意の監査を満足に完了させること;
•BLAに対するFDAの審査および承認は、リスク評価および緩和策(REMS)の実施の潜在的要件、およびFDAが要求する任意の承認後の臨床試験を含む承認後の追加的な要求を受ける可能性がある。
臨床前研究
スポンサーが潜在的な治療価値を有する候補製品のテストを開始する前に、候補製品は臨床前試験段階に入る。臨床前テストは製品の化学、調合と安定性の実験室評価、及びその他の候補製品の毒性を評価する研究を含む。これらの研究は一般にINDを支援する研究と呼ばれる.臨床前試験の実施及び試験に用いる化合物の配合は、GLP規制を含む連邦法規及び要件に適合しなければならない基準と米国農務省の動物福祉法(適用される場合)それは.臨床前試験の結果は,生産情報や分析データとともにINDの一部としてFDAに提出された。いくつかの長期的な臨床前試験、例えば生殖不良事件と発ガン性の動物試験、及び長期毒性研究は、IND提出後も継続する可能性がある。
INDとIRBプロセス
INDはFDCAの免除であり、未承認候補製品が州間商業で臨床研究のために輸送されることを許可し、FDAにこのような研究製品をヒトに使用することを許可することを要求する。承認されたBLAに属さない候補製品を州間輸送·管理する前に、INDの安全を確保しなければならない。INDの申請を支援するためには、スポンサーは各臨床試験にプログラムを提出しなければならず、任意の後続のプログラム修正はINDの一部としてFDAに提出されなければならない。INDは、FDAが受信した後30日以内に自動的に有効になり、それ以前にFDAが提案された臨床試験の製品または問題を提起しなければならない。この場合、INDスポンサーおよびFDAは、臨床試験が開始される前に、任意の懸案された懸念を解決しなければならない。したがって,INDの提出はFDAが臨床試験の開始を許可しない可能性がある。
IND下での臨床試験開始後,FDAもこの試験を臨床放置あるいは一部の臨床放置を実施することができる。臨床保留はFDAがスポンサーに発表した命令であり,提案された臨床研究の延期や進行中の研究の一時停止が要求されている。一部の臨床保留はIND要求の一部の臨床仕事を遅延或いは一時停止することである。例えば、一部の臨床的保留は、特定のプロトコルまたはプロトコルの一部が継続できないことを宣言する可能性があり、プロトコルの他の部分または他のプロトコルはそうすることができる。臨床保留或いは一部の臨床保留を実施した後30日を超えない後、FDAはスポンサーに棚上げ根拠に関する書面解釈を提供する。臨床保留あるいは一部の臨床保留を発表した後、FDAがスポンサー調査が継続可能であることを通知した後にのみ、臨床調査を回復することができる。FDAは、スポンサーによって提供された情報に基づいて、調査が継続または再開可能であることを決定し、これらの情報は、上述した欠陥を修正するか、または他の方法でFDAを満足させるであろう。臨床試験被験者に安全問題の製造問題をもたらす可能性があるため,臨床一時停止を強制する場合がある
上述したIND要件に加えて、臨床試験に参加する各機関を代表するIRBは、その機関が臨床試験を開始する前に、任意の臨床試験の計画を審査および承認しなければならず、IRBは少なくとも毎年継続的な審査および再承認試験を行わなければならない。IRBはFDAの規定に従って動作しなければならず,試験対象に提供された臨床試験案とインフォームドコンセント情報を審査·承認し,完成まで試験を監督しなければならない。臨床試験がIRBの要求に従って行われない場合、または候補製品が患者の予期しない深刻な損傷に関連している場合、IRBは、その機関またはその代表機関の臨床試験の承認を一時停止または終了することができる
さらに、いくつかの試験は、データ安全監視委員会、またはDSMBと呼ばれる試験スポンサーによって組織された合格専門家からなる独立したグループによって監視される。このグループは,試験の利用可能なデータの審査により,認可試験が指定されたチェックポイントで進めることができるかどうか,DSMBのみがこの試験にアクセスできるかどうかを検討している。Dsmbが参加者または患者が受け入れられない健康リスクに直面していると判断した場合、臨床試験の任意の段階で開発を一時停止または終了する可能性がある
拡張的アクセス
使用を拡大することは、“同情的使用”と呼ばれることがあり、臨床試験以外に研究性新製品を使用し、比較可能または満足できる代替治療案がない場合には、重篤または直ちに生命に危害を及ぼす疾患または条件を有する患者を治療する。参入拡大に関する規則や条例は,研究療法の恩恵を受ける可能性のある患者が研究製品を獲得する機会を改善することを目的としている。FDAの法規は、個別患者(緊急時および非緊急時に治療された単一患者IND申請)、中規模の患者集団、および治療レジメンまたは治療INDに従って研究製品の使用を申請したより大きな集団のために、会社または治療医がINDの研究製品を治療目的で使用することを可能にする。
INDが患者または患者のグループを治療するために研究製品の使用を拡大する申請を審議するとき、スポンサーおよび治療医または調査者は、患者(S)が深刻または直ちに生命に危険な疾患または状態を有し、疾患または状態を診断、監視または治療するための類似または満足できる代替療法がなく、潜在的な患者利益は治療の潜在的リスクが合理的であり、潜在的リスクが治療すべき背景または条件で不合理ではないことを証明するすべての基準が適用される場合に適切であるかどうかを決定するであろう。研究製品の要求される治療の拡大使用は、製品の発売承認を支持する可能性のある臨床研究の開始、進行または完了、または他の方法で製品の潜在的開発を損害する可能性がある
スポンサーは、取得を拡大するためにその研究製品を提供する義務はないが、“21世紀治療法案”または2016年に成立した“治療法案”におけるFDCAの修正案の要求に基づいて、開発中の重篤な疾患や疾患を治療する候補製品の拡大取得要求にどのように対応するかに関する政策がある場合には、この政策を公開しなければならない。スポンサーは、カバーされた研究製品の第2段階または第3段階試験の開始が早いとき、または研究製品がFDAによって画期的な治療法、高速チャネル製品、または再生医学高度療法として指定された15日後に、そのような政策を開示することを要求されている
また、2018年5月30日には、“裁判権法案”が法律に署名された。他の事項に加えて、この法律はある患者に連邦フレームワークを提供し、彼らが第一段階の臨床試験を完成し、FDAの許可を得た研究用新製品を得るために調査を行っていることを許可した。場合によっては、条件を満たす患者は、臨床試験に参加することなく、FDA拡大参入計画に従ってFDA許可を得ることなく治療を求めることができる。“試用権利法案”によると、製造業者はその製品を条件に適合する患者に提供する義務はないが、製造業者は内部政策を策定し、その政策に基づいて患者の要求に応答しなければならない
人体臨床試験
臨床試験は、GCP要求に適合した場合に、合格した研究者の監督の下で、すべての研究対象に任意の臨床試験に参加する前に書面でインフォームドコンセントを提供することを含む、ヒト被験者の研究製品候補を行うことに関する。臨床試験は書面による臨床試験案に基づいて行い,その中で試験の目標,組み入れと排除基準,安全性モニタリングのためのパラメータ,評価すべき有効性基準を詳細に説明した。各レジメンおよびその後のレジメンのいずれの実質的な修正もINDの一部としてFDAに提出されなければならず,臨床試験状態を詳細に説明する進捗報告は毎年FDAに提出されなければならない。FDAはすでに、ライセンススポンサーが臨床試験を行ういくつかの責任を契約研究組織、あるいはCROに移すことを規定している。
人体臨床試験は通常3つの連続段階に分けて行われるが、これらの段階は重複あるいは組み合わせられる可能性がある。承認された後、追加的な実験が必要になるかもしれない
ステップ1臨床試験は、最初に限られた集団で行われ、これらの集団は、健常人または患者における副作用、用量耐性、吸収、代謝、分布、排泄および薬効学を含む候補製品の安全性を試験するために、健康なボランティアまたは標的疾患を有する対象である可能性がある。第一段階臨床試験期間中に、候補製品の薬物動態学と薬理作用に関する情報を得ることができ、良好かつ科学的に有効な第二段階臨床試験を設計制御することができる
第二段階臨床試験は通常限られた患者集団で行われ、可能な副作用と安全リスクを決定し、特定の標的適応に対する候補製品の治療効果を評価し、用量耐性と最適用量を決定する。スポンサーは、より規模が大きく、コストの高い3期臨床試験を開始する前に情報を得るために、複数の2期臨床試験を行うことができる。第二段階の臨床試験は通常良好な制御と密接なモニタリングを受けている
第3段階第二段階の臨床試験が候補製品の一定量の範囲が潜在的に有効であり、許容可能な安全性を有することを証明した場合、臨床試験は引き続き行われる。第三段階の臨床試験はより多くの患者群を用いて行われ、更に投与量を評価し、臨床治療効果の実質的な証拠を提供し、そして複数の地理的に分散した臨床試験地点で拡大と多様化した患者群に対して更なる安全性テストを行う。コントロールが良好で、統計的に穏健な3期臨床試験を設計して、監督機関が使用するデータを提供して、承認するかどうか、そして承認すれば、どのように新しい生物製品を適切に標識するかを決定することができる。この3期臨床試験は“肝心な”試験と呼ばれる
1つの臨床試験は複数の段階の要素を結合する可能性があるが、FDAは通常、候補製品の発売承認を支持するために複数の第3段階試験を必要とする。ある会社が臨床試験を特定の段階に指定することは、この試験がこの段階のFDA要求を満たすのに十分であるとは限らない。なぜなら、FDAに案とデータを提出し、FDAによって審査する前に、この決定を下すことができないからである。さらに、上述したように、キー試験は、候補製品の安全性および有効性に対するFDAの評価要件を満たすと考えられ、規制承認を支援するために、他のキー試験または非キー試験と共に使用することができる。一般的に、重要な試験は3期試験であるが、設計が臨床利益の良好な制御と信頼できる評価を提供すれば、特に医療需要を満たしていない領域では、それらは2期試験である可能性がある。
場合によっては、FDAは候補製品のBLAを承認する可能性があるが、承認後の候補製品の安全性および有効性をさらに評価するために、スポンサーに追加の臨床試験を行うことが要求される。この試験は通常最初の上場承認後に行われるため、承認後または発売後の臨床試験と呼ばれる。これらの試験は、予想される治療群のより多くの患者の治療から追加の経験を得るためのものである。場合によっては、FDAは、承認条例に従って承認された製品を加速する場合など、承認後の臨床試験を強制的に実行することができ、臨床的利益を検証することができる。強制的な4期臨床試験での職務調査ができなかったことは,FDAが製品の承認を撤回する可能性がある。2022年12月、食品·薬物総合改革法案(FDORA)の成立に伴い、国会は、各新薬または生物製品の第3段階臨床試験または任意の他の“重要な研究”のための多様な行動計画の制定と提出をスポンサーに要求した。これらの計画はより多くの異なる患者群がFDA監督製品の後期臨床試験に参加することを奨励することを目的としている。具体的には,行動計画には,スポンサーの募集目標,これらの目標の基本原理,スポンサーがこれらの目標をどのように実現しようとしているかの解釈が含まれなければならない。このような要求に加えて、立法はFDAに多様な行動計画に関する新しいガイドラインを発表するように指示した。FDAは2024年1月、臨床試験で人種や民族データを収集する政策を示した指導草案を発表した。FDAは2024年1月、臨床試験で人種や民族データを収集する政策を示した指導草案を発表した。
2022年3月、FDAは“拡張コホート:最初のヒト臨床試験のために腫瘍薬物と生物製品の開発を加速するための”と題する最終ガイドラインを発表し、その中で開発者がどのように腫瘍学生物製品開発の初期段階(すなわち最初の人体臨床試験)で通常シームレス試験設計と呼ばれる適応試験設計を利用し、伝統的な3段階の試験を拡張行列試験と呼ばれる連続試験に圧縮するかについて概説した。個人拡張キュー設計をサポートする情報はINDアプリケーションに含まれ,FDAによって評価される.コホート試験を拡大することは,製品開発の効率を向上させ,開発コストや時間を削減する可能性がある
2023年6月、FDAはガイドライン草案を発表し、GCPに対する提案を更新し、臨床試験の設計と現代化を目指した。これらの更新は,より効率的な臨床試験の道を開き,医療製品の開発を促進することを目的としている。このガイドライン草案は国際調整理事会(ICH)が最近更新したE 6(R 3)ガイドライン草案から採択されたものであり、このガイドライン草案は迅速に発展した技術と方法革新を臨床試験企業に組み込むことを目的としている。また,FDAはガイドライン草案を発表し,分散臨床試験の実施について概説した。
最後に,臨床試験のスポンサーは,米国国立衛生研究院(NIH)が維持している公共登録センター(Clinicaltrials.gov)に登録し,何らかの臨床試験情報を開示することを求められている。特に,臨床試験登録の一部として,臨床試験の製品,患者群,調査段階,臨床試験地点と調査者,その他に関する情報が公開されている。米国国立衛生研究院の臨床試験登録と報告要求に関する最終規則は2017年に発効した。米国衛生·公衆サービス部(HHS)の長期遅延により、FDAは従来これらの報告要求を実行していなかったが、過去2年間にFDAはいくつかの自発的是正行動の事前通知と規定を遵守しないいくつかの通知を発表した。これらの違反通知は民事罰金を招くことはないが,FDCAの規定により,Clinicaltrials.govに臨床試験情報を要求通りに提出できなかったことは禁止されており,違反行為は毎日10,000ドルまでの民事罰金を受け続ける可能性がある。
臨床開発計画中のFDAとの相互作用
INDが承認され臨床試験を開始した後,スポンサーはFDAとの相互作用を継続する。臨床試験結果を詳細に説明する進捗報告は毎年IND発効60日以内に提出しなければならず,重篤な有害事象が発生すれば,より頻繁に進捗報告を提出する。このような報告書は安全更新報告書やDSURの開発を含まなければならない。さらに、以下の場合のうちの1つは、深刻かつ予期しない疑わしい副作用;他の試験または動物または体外培養この製品に接触する人体は重大なリスクのテストが存在することを表明した;及び方案或いは研究者マニュアルに列挙された情況と比べ、臨床上深刻な不良反応の発生が疑われるいかなる重要な増加である。第1段階、第2段階、および第3段階の臨床試験は、任意の指定された時間内に成功しないか、または全く成功しない可能性がある。FDAは、通常、GCPおよび提出された臨床データの完全性を保証するために、1つまたは複数の臨床場所を検査する。
また,スポンサーは臨床開発計画のあるときにFDAと会う機会がある。具体的には,スポンサーはIND提出前(IND前会議),2期臨床試験終了時(EP 2会議),BLA提出前(BLA前会議)にFDAと会うことができる。他の時間に会議を開催することを要求することもできる。スポンサーとFDAの間には5種類の会議がある。Aクラス会議は,もともと停滞していた製品開発計画を継続したり,重要なセキュリティ問題を解決したりするために必要な会議である.クラスB会議には,IND前会議とBLA前会議,EOP 2会議のような段階終了会議がある.クラスC会議は、製品開発および審査に関するA会議またはクラスB会議以外の任意の会議であり、例えば、提案使用時の製品承認の主な根拠として使用されていなかったバイオマーカーを新たな代替終点として使用することに関する早期協議を促進する会議を含む。D類会議は一連の狭い問題に重点を置いており、2つの重点議題を超えてはならず、3つの学科或いは分部を超える投入を要求すべきではない。最後に,インタラクティブ会議は,研究製品の早期開発において独自の挑戦を提示する新製品と開発計画を目指している.FDAは、議事録および諮問書簡で伝達された応答は、スポンサーに対する提案および/または提案のみを構成するため、スポンサーは、そのような提案および/または提案の制約を受けないと述べている。しかし,実践的には,スポンサーがFDAの提案に沿って臨床計画を設計していないことは,その計画を大きな失敗リスクに直面させる可能性がある。
FDAが承認したアメリカの海外臨床研究をサポートします
われわれの臨床開発計画については,米国以外の地点で試験を行う可能性がある。 ある国外の臨床研究がIND下で行われる時、放棄しない限り、すべてのIND要求を満たさなければならない。外国の臨床研究がINDの下で行われていない場合、スポンサーは、この研究をINDまたは上場承認申請の支持として使用するために、この研究がFDAのいくつかの法規要件に適合することを確実にしなければならない。具体的には,独立倫理委員会やIECの審査·承認を受けること,被験者のインフォームドコンセントを求めて受け入れることなど,GCPに従って行わなければならない。GCPは臨床研究の倫理とデータ完全性基準を含むことが要求される。FDAの規定は、非IND外国臨床研究に参加したヒト被験者の保護、及び結果データの質と完全性を確保することを目的としている。これらはまた、非IND外国研究がIND研究に匹敵する方法で行われることを確保するのに役立つ
FDAは米国国外で行われた臨床試験の研究データを受けて米国の承認を支持しており,何らかの条件によって制限される可能性もあり,まったく受け入れられない可能性もある。外国の臨床試験からのデータが米国での上場承認の唯一の根拠となることを意図している場合、FDAは通常、(I)データが米国の人口および米国の医療実践に適用されない限り、(I)データが米国の人口および米国の医療実践に適用されない限り、(Ii)試験は公認能力を有する臨床研究者がGCP規定に基づいて行うことができること、および(Iii)データはFDAによる現場検査を必要としないこと、またはFDAはこのような検査を行う必要があると考えており、FDAは現場検査や他の適切な方法でデータを検証することができる。
また,海外の研究データが承認の唯一の根拠としようとしなくても,FDAはこれらのデータを上場承認申請の支援として受け入れず,研究設計が良好でGCP要求に適合しない限り,FDAは必要と考えた場合に現場検査により研究データを検証することができる。多くの外国の規制機関もまた似たような承認要求を持っている。また、このような外国裁判は、裁判を行う外国司法管轄区に適用される現地法によって管轄されている。
製造やその他の規制要件
臨床試験と同時に、スポンサーは通常、追加の動物安全性研究を完成し、候補製品の化学的および物理的特性に関する追加情報を開発し、cGMP要求に基づいて商業ロット候補製品を生産するプロセスを最終的に決定する。製造過程は一貫して高品質の候補製品ロットを生産することができなければならず、他の標準以外に、スポンサーは完成品の特性、強度、品質と純度をテストする方法を制定しなければならない。また,適切な包装を選択·テストし,候補製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
具体的には,FDAの規定では,医薬品は特定の承認された施設で生産され,cGMPに適合しなければならないことが求められている。CGMP条例には、人員、建物および施設、設備、部品および製品容器および閉鎖的な制御、生産およびプロセス制御、包装およびラベル制御、保有および分配、実験室制御、記録および報告、ならびに返品または回収された製品に関する要件が含まれる。承認された医薬品の生産および流通に参加するメーカーおよび他のエンティティは、FDAおよびいくつかの州機関にその機関を登録し、cGMPおよび他の要求を遵守することを確実にするために、FDAの定期的な抜き打ち検査を受けなければならない。2022年12月に公布された“大流行病予防法”は、1種の薬物または生物が米国に輸入または提供される前に、米国国外の別の機関でさらなる製造、製造、繁殖、複合または加工を行っても、外国の薬品製造機関は登録と発売要求を守らなければならないことを明らかにした
検査は“リスクに基づくスケジュール”に従わなければならないが、これはいくつかの機関がより頻繁に検査されることを招く可能性がある。メーカーはまた、その工場に関する電子または実物記録の提供を要求しなければならないかもしれない。FDAの検査の延期、拒否、制限、または拒否は、製品が偽とみなされる可能性がある。承認された製品の製造プロセス,仕様や容器閉鎖システムの変更は厳しく規制されており,通常はFDAの承認を得て実施する必要がある。FDAの規定はまた、cGMPから外れたいかなる状況の調査と是正を要求し、スポンサーと生産承認製品の生産に参加する任意の第三者メーカーに対して報告と文書要求を提出することを要求している
製造業者や他の製品の製造と流通に参加する人たちもまたFDAと特定の州機関に彼らの工場を登録しなければならない。国内でも海外の製造企業でも,最初に生産過程に参加する際には,FDAに登録して追加的な情報を提供しなければならない。登録されていない工場で製造または輸入されたどの製品も、外国でも国内でも、FDCAの下に誤ったブランドが貼られているとみなされている。これらの生産施設は,cGMPや他の法律に適合することを確保するために,政府当局の定期的な抜き打ち検査を受ける可能性がある。製造施設が製品が承認されたときに適用される法規および要件に実質的に適合していない場合、その施設からの製品の出荷を禁止し、および/または以前に出荷された製品のリコールを禁止する警告状または禁止令を発行することが含まれる規制法執行行動をとることができる。
小児科試験
小児科研究公平法またはPREAによれば、アプリケーションおよびいくつかのタイプのサプリメントは、すべての関連する小児科亜集団において主張される適応の安全性および有効性を評価し、各安全で有効な小児科亜集団に対する製品の用量および投与をサポートするのに十分なデータを含まなければならない。スポンサーは、第2段階会議終了後60日以内に、またはスポンサーとFDAとの間で合意しなければならない場合に、初歩的な小児科研究計画またはPSPを提出しなければならない。これらの計画は、スポンサー計画によって提案された1つまたは複数の小児科臨床試験の大綱を含まなければならない。試験目標および設計、年齢グループ、関連終点および統計方法、またはそのような詳細な情報を含まない理由、および小児科試験データの提供要件および支援情報の要求を小児科評価または全部または部分的に免除することを延期することを含む。スポンサーとFDAは最終計画について合意しなければならない。非臨床研究,早期臨床試験および/または他の臨床開発計画から収集したデータに基づいて小児科計画の変化を考慮する必要があれば,スポンサーは合意した初期PSPに対する修正案を随時提出することができる
深刻な或いは生命に危害を及ぼす疾患或いは状況を治療することを目的とした研究製品に対して、FDAはスポンサーの要求に応じて会議を開催し、初期PSPの準備或いは討論を討論し、小児科評価を延期或いは放棄しなければならない。また、FDAは開発過程の早期に会議を開催し、PSPをスポンサーと議論し、FDAは深刻または生命に危険な疾患よりも遅くない第1段階の会議が終了する前に、FDAがPSPを受信してから90日後にスポンサーと面会しなければならない。
FDAは、成人のために製品が使用されるか、または小児科データ要件を完全にまたは部分的に免除するまで、スポンサーの要求に能動的にまたは対応することができ、承認は、小児科データの一部または全部の提出を延期することができる。延期は、小児科試験が完了する前に、製品または候補治療薬が成人で使用を許可する準備ができていることを発見するか、または小児科試験が開始される前に追加の安全性または有効性データを収集する必要があることを含むいくつかの理由があるかもしれない。FDAは、PREA要求を提出できなかった小児科評価、延期または延期を求めることができなかった、または必要な小児科処方の承認を要求できなかったスポンサーにPREA不適合書簡を送信することを要求されている。2023年5月、FDAは新しいガイドライン草案を発表し、さらにPREA下の小児科研究要求を述べた。
FDAがPREAにおいてこの法定免除を乱用すると考えられる行為を制限する措置をとっているにもかかわらず、小児科データ要件は、法規が別途要求されない限り、孤児として指定された製品には適用されない。さらに、2017年にFDA再認可法案によって改正されたFDCA第505 B条は、2020年8月18日または後に新しい活性成分のために提出された任意の元のNDAまたはBLAが、この要求を免除または延期しない限り、分子標的小児癌調査に関する報告を含まなければならず、申請された薬剤が、(1)成人癌の治療のために使用される場合、および(2)FDAの指導によれば、秘書が小児癌の成長または進行に密接に関連すると考えられる分子標的を含む必要がある。FDAはまた、児童人口中の疾病罹患率が比較的に低いため、PREA要求を免除する疾病リストを保留した
審査計画を速める
FDAはいくつかの方法で申請の検討を加速させることを許可された。高速チャネル計画によれば、候補製品のスポンサーは、IND提出と同時にまたは後に、FDAに特定の適応の製品を高速チャネル製品として指定することを要求することができる。もし候補製品が深刻な或いは生命に危害を及ぼす疾患を治療することを目的とし、このような疾病が満たされていない医療需要を満たす潜在力を示す場合、迅速なチャンネル認証を受ける資格がある。高速チャネルは,候補製品と研究中の特定の適応に適した組合せを指定する.他の利点に加えて、FDAとより大きな相互作用が可能であれば、FDAは、申請が完了する前にFast Track申請の部分の審査を開始する可能性があり、この過程をローリング審査と呼ぶ
迅速チャネル計画に従って、画期的な治療指定、優先審査、および承認の加速など、FDAの開発および審査を加速するための他のタイプの計画に参加する資格がある可能性があるFDA上場候補製品を提出することを含む
•画期的な治療指定画期的な治療計画の資格を得るためには、候補製品は深刻な或いは生命に危害を及ぼす疾患或いは状況の治療に使用されなければならず、初歩的な臨床証拠は、これらの候補製品が1つ以上の臨床顕著な終点で既存の治療法よりも著しく改善されている可能性があることを示しなければならない。FDAは突破的な候補治療製品のスポンサーが高効率開発計画に関する深い指導を得ることを確保し、高級管理者と経験豊富な従業員が積極的、協力と学際的な審査と転動審査に深く参与することを確保する
•優先的に審査する。候補製品が重篤な疾患を治療した場合、優先審査を受ける資格があり、承認されれば、市販されている製品と比較して、治療、診断または予防の安全性または有効性の重大な改善となる。FDAの目標は、標準審査の10ヶ月間ではなく、優先審査申請の審査を6ヶ月以内に完了させることである
•承認を速める研究された生物学的製品は、重篤または生命に危険な疾患の治療における安全性および有効性、ならびに既存の治療法よりも意義のある治療効果を提供する生物学的製品がより迅速に承認される可能性がある。承認を加速することは、候補製品が十分かつ良好に制御された臨床試験に基づいて、候補製品が臨床利益を合理的に予測する可能性のある代替終点に影響を与えるか、または生存または不可逆的な発病率または死亡率または他の臨床利益ではなく、臨床終点への影響を決定することができ、病状の重症度、希少性および流行度、ならびに代替治療の利用可能性または不足を考慮することを意味する。承認の一つの条件として,FDAは承認を加速させたバイオ製品候補のスポンサーに十分かつ良好に制御された上場後臨床試験を要求する可能性がある。また、FDAは現在、承認を加速する条件として宣伝材料を事前承認することを要求している
FDORAの2022年12月の採択に伴い、国会は薬品や生物製品の承認を加速させるためのいくつかの条項を改正した。具体的には、新しい立法認可FDAは、スポンサーが加速承認を得る前に検証的臨床試験を行うことを要求し、加速承認された製品のスポンサーが、研究が完了するまで承認後の研究の進展報告を6ヶ月ごとにFDAに提出することを要求し、検証試験が製品の臨床的利益を検証できなかった後、加速手順を使用して新薬申請またはBLAの加速承認を撤回する。また,FDORAは,承認加速後にこのような研究を要求しないことを決定した場合には,そのサイト上で“なぜ承認後研究に適していないのか,あるいは不要な理由”を公表することを求めている
2023年3月、FDAは指導意見草案を発表し、現在承認を加速する構想と方法を概説した。FDAによると、癌の深刻性と生命に危害を及ぼす性質のため、加速承認経路は通常腫瘍薬物の審査に用いられる。単一アーム試験は通常加速承認を支持するために使用されるが、無作為対照試験はより信頼性の高い治療効果と安全性評価を提供し、利用可能な治療との直接比較を可能にするため、第一選択方法である。そのため、FDAは実験データの設計、進行と分析の考慮要素を概説し、これらの試験は腫瘍治療薬物の加速承認を支持することを目的としている。このガイドラインは現在草案形式にすぎず,最終的に決定されても法的拘束力はないが,スポンサーはFDAのガイドラインを密接に遵守し,彼らの研究製品が加速的な承認を得る資格を確保するのが一般的である。
•再生性先進療法。2016年12月に採択された“21世紀治療法”に伴い,再生性先進療法に指定された製品の審査と承認がFDAに認可された。製品が深刻なまたは生命を脅かす疾患または状態を治療、修正、逆転または治癒することを目的とした再生医学療法であり、予備臨床証拠が、候補製品がそのような疾患または状態の満たされていない医療需要を満たす可能性があることを示す場合、製品はこの称号を得る資格がある。再生性高度治療指定の利点は、開発および審査を加速するためのFDAとの早期相互作用、画期的な治療の利点、潜在的な優先審査資格、および代替または中間終点に基づく加速承認を含む
これらの加速項目はいずれも承認の基準を変更していないが,各項目は候補製品の開発や承認過程の管理を加速させるのに役立つ可能性がある.
BLASの提出とアーカイブ
必要な臨床試験,臨床前研究および臨床試験の結果,および製品の化学,製造,制御,安全更新,特許情報,乱用情報および提案されたラベルに関する情報が,出願の一部としてFDAに提出され,候補製品を1つまたは複数の適応に押し出すことの承認を要求すると仮定する。上場承認を支持するために、提出したデータは生物製品の安全性、効力と純度を決定し、FDAを満足させるために、品質と数量で十分でなければならない。処方薬ユーザ料金法案(PDUFA)によるBLA提出に要する費用は高く(例えば,2024連邦財政年度では約4,048,695ドル),承認されたBLAのスポンサーは年間計画費用を支払う必要があり,現在は2024連邦財政年度に合格処方薬製品416,734ドルに設定されている。これらの費用は、通常、年に1回調整され、場合によっては、出願人が審査のために最初の人間治療申請を提出する小規模企業を含む免除および免除がある可能性がある。
FDAは、すべての出願を受信してから60日以内にすべての出願を予備審査し、実質的な審査のためにスポンサー申請が十分に完全であるかどうかをその時または前に通知しなければならない。関連部分では、FDAの法規は、FDAがすべての関連情報およびデータを受信する前に、申請は提出されたとみなされてはならないと規定している。FDAが出願がこの基準を満たしていないと判断した場合、出願人に提出拒否またはRTF決定を発行する。一般に、技術移転フレームワークは、実質的かつ意味的な審査を排除するために、情報または必要な情報の部分を明らかに見落としているような行政の不完全さに基づいている。FDAは申請を受け入れるのではなく、より多くの情報を提供することを要求するかもしれない。この場合、申請は追加情報と共に再提出されなければならない。再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。
出願が受け入れられた後、FDAは出願の深い実質的な審査を開始した。FDAは、提案された製品がその予期される用途に対して安全に有効であるかどうか、許容可能な純度プロファイルを有するかどうか、および製品がcGMPに従って製造されているかどうかを決定するために出願を審査する。FDAがPDUFAで合意した目標と政策によると、FDAは10ヶ月間、新しい分子実体としての標準出願の予備審査を完了し、“優先審査”を有する出願については6ヶ月の期間がある。FDAは、新しい情報を考慮するために、審査プロセスをさらに3ヶ月延長することができ、または出願人が明確な説明を提供する場合には、FDAが最初の提出後に発見した係属中の欠陥を解決することができる。これらの審査目標にもかかわらず,FDAによる申請の審査がPDUFA目標日を超えることは珍しくない。
出願を審査する際には,FDAは通常,申請者に情報要求を提出し,回答の最終期限を設定する.FDAはまた,新製品の製造施設の承認前検査を行い,製造プロセスや施設がcGMPに適合しているかどうかを確認する。FDAは、製造プロセスおよび施設がcGMP要件に適合していることを決定し、要求された仕様の下で製品が一貫して生産されることを保証するのに十分でない限り、この製品を承認しないであろう
FDAはまた、INDおよびGCP要件に適合することを保証し、FDAに提出された臨床データの完全性を保証するために、スポンサーおよび1つまたは複数の臨床試験場所を検査することができる。FDORAの成立に伴い,国会ではFDAに提出された臨床·非臨床研究の準備,進行や分析に関わる施設や研究記録や研究過程に関与している他の人の検査が許可されることが明らかになり,FDAが検査を行う権限が明らかになった。
さらに、FDAは、安全性または有効性の問題を提起する新製品候補出願を含む申請を、審査、評価、および提案を行って、申請を承認すべきかどうか、およびどのような条件で承認すべきかを決定するために諮問委員会に提出することができる。通常,諮問委員会は臨床医や他の科学専門家を含む独立した専門家からなるグループであり,申請を審査,評価し,申請を承認すべきかどうか,どのような条件でアドバイスを提供すべきかを担当する。FDAは諮問委員会の提案に制限されていないが、最終承認決定を下す際にこれらの提案を考慮する
FDAはまた、製品の利益がそのリスクよりも大きいことを保証し、製品の安全な使用を保証するために、REMSを提出する必要があると判断した場合、REMSの提出を要求することができる。REMSは、制限された分配方法、患者登録、または他のリスク最小化ツールのような薬物ガイドライン、医師コミュニケーション計画、評価計画、および/または安全使用を保証する要素を含むことができる。FDAは具体的な状況に応じてREMSに対する要求および具体的なREMS条項を決定する。FDAがREMSが必要であると考えている場合、申請されたスポンサーは提案されたREMSを提出しなければならず、FDAはREMSのない出願を承認しないであろう。
BLASの決定について
申請およびすべての関連情報を評価した後、諮問委員会のアドバイス(ある場合)および製造施設および臨床試験地点の検査報告を含めて、FDAは完全な返信またはCRLまたは承認書を発行する。この決定を達成するために、FDAは、提案製品の期待利益が患者に対する潜在的リスクよりも大きいことを決定しなければならない。この評価の根拠は:潜在疾病の深刻性と現有の治療法が患者の医療需要を満たす程度;発売前の臨床試験証拠はどのようにこの製品の発売後の環境中の実際の使用状況の不確定性を推定するか;及びリスク管理ツールが特定のリスクを管理する必要があるかどうかを含む。
CRLは,申請の審査周期が完了したことを示しており,申請は現在の形で承認されない.CRLは、通常、提出中の不足点を列挙し、FDAが申請を再検討するために、大量の追加のテストまたは情報を必要とする可能性がある。CRLは、追加の臨床または他のデータ、追加の重要な第3段階臨床試験(S)、および/または臨床試験、臨床前研究または生産に関連する他の重要で時間のかかる要件を必要とする可能性がある。CRLが発行された場合、出願人は、FDAが決定した欠陥に1年間応答することができ、FDAは、出願が撤回されたと考えるか、または申請者が6ヶ月の応答期間を追加的に延長することを適宜承認することができる。FDAは、発行されたCRLの再提出を2ヶ月または6ヶ月以内に検討することを約束しており、具体的には、含まれる情報のタイプに依存する。しかしながら、この補足情報を提出しても、FDAは最終的に、その申請が承認された規制基準を満たしていないと決定する可能性がある。
一方,その製品の商業マーケティングを承認し,特定の適応に関する具体的な処方情報を提供する。すなわち、承認は、FDA承認のラベルに記述された使用条件(例えば、患者数、適応)に限定される。さらに、解決すべき特定のリスク(S)に応じて、FDAは、承認後に製品の安全性をさらに評価するための承認後試験(4期臨床試験を含む)を要求するために、製品ラベルに禁忌症、警告または予防措置を含むことを要求することができ、試験および監視計画は、製品の商業化後に製品を監視すること、または販売および使用制限、または製品の潜在的な市場および利益に大きな影響を与える可能性のあるREMS下の他のリスク管理メカニズムを含む他の条件を適用することを要求することができる。FDAは発売後の試験或いはモニタリング計画の結果に基づいて、製品の更なるマーケティングを阻止或いは制限することができる。承認後、新たな適応の追加、製造変更、および追加のラベル宣言など、承認された製品のいくつかのタイプの変更は、さらなるテスト要件およびFDAの審査および承認を受けることになる。
承認した後要求する
新しい処方薬が承認された後、製造業者、承認された製品および製品の製造場所は、監視および保存活動の監視および記録、製品に関連する不良経験および製品問題、製品サンプリングおよび流通、製造および販売促進、および広告を含むFDAによって一般的かつ持続的に規制される。医師は、許可されていない用途または患者集団のために合法的に利用可能な製品(すなわち“ラベル外使用”)を発行することができるが、製造業者はそのような用途を販売または普及させてはならない。FDAや他の機関はラベル外用途の普及を禁止する法律法規を積極的に実行しており,ラベル外用途の普及が不適切であることが発見された会社は重大な責任を負う可能性がある。FDAは2021年9月、生物学的に予想される用途を決定する際に機関が考慮すべき証拠のタイプを記載した最終法規を公表した。
非常に具体的で狭い条件下では、製造業者が、科学または医学定期刊行物情報の配信のようなラベル外情報に関する非販売促進、非誤解的伝播に従事することを可能にすることができる。また,2022年12月の承認前情報交換法の成立に伴い,承認されていない製品のスポンサーは,製品承認後に患者の接触を加速させるために,開発中の製品に関する何らかの情報を支払者に能動的に伝達することができる。これまで,このような通信はFDAの指導の下で許可されていたが,新たな立法はスポンサーの保護を明確にしており,これらのスポンサーは未承認の製品用途を含む開発中の製品に関するいくつかの情報を支払者に伝えてきた。また,FDAは2023年10月にガイドラインを発表し,FDA管理が未承認用途の科学情報をヘルスケア提供者に配布する拘束力のない政策について概説した。 本ガイドライン草案は、このような通信が真実で、誤解性がなく、事実と偏らないことを要求し、医療保健提供者が許可されていない使用に関する情報の利点および欠点、ならびに有効性および実用性に必要なすべての情報を説明することを含む。
ある会社がラベル外の使用を促進することが発見された場合、それはFDA、司法省、または衛生·公衆サービス部監察長事務室、および州当局の行政および司法執行を受ける可能性がある。これは、民事および刑事罰金、企業の製品の宣伝または流通の方法を実質的に制限する協定、および不利な公共関係および名声の損害を含む一連の重大な商業的影響を及ぼす可能性のある一連の処罰を企業に受ける可能性がある。連邦政府は、不当な販売促進の疑いのある会社に対して巨額の民事と刑事罰金を科し、企業に同意法令または永久禁止令を締結し、これらの法令または永久禁止に基づいて、特定の販売促進行為を変更または制限することを要求する。
さらに、製品に適応、ラベル、または生産プロセスまたは施設の変化を含む修正がある場合、スポンサーは、新しい申請またはサプリメントに対するFDAの承認を得るために提出を要求される可能性があり、これは、スポンサーが追加のデータを開発すること、または追加の臨床前研究および臨床試験を行うことを必要とする可能性がある。FDAが新しい適応を承認することを確保する過程は、元の適応を承認する過程と類似しており、新適応における製品の安全性および有効性を証明するために、十分かつ制御された臨床試験データを提出する必要がある。このような実験を行っても,FDAはタイムリーに使用するためにラベル適応のいかなる拡張も承認しないか,あるいは全く承認しない可能性がある。継続的な年間ユーザー料金要求もあり、現在はある製品の計画費用として評価されている。
また、規制要件や基準を遵守していない場合、あるいは製品発売後に問題が発生した場合、FDAは承認を撤回する可能性がある。その後、製品に以前未知の問題が存在し、意外な深刻性或いは頻度の不良事件、或いは生産技術、或いは監督要求を遵守できなかったことを発見し、許可されたラベルを強制的に改訂して新しい安全情報を追加し、発売後の臨床試験要求を強制的に実施して新しい安全リスクを評価するか、あるいはREMS計画に従って流通或いはその他の制限を実施することを招く可能性がある
他の他の潜在的な結果には
•製品の販売や製造を制限し、市場から製品を完全に撤回したり、製品をリコールしたりする
•セキュリティ警報、親愛なるヘルスケア提供者への手紙、プレスリリース、または製品に関する警告または他のセキュリティ情報を含む他の通信;
•宣伝材料を強制的に修正し、ラベルを貼り付け、訂正情報を発表する
•承認後の臨床試験の罰金、警告状、無見出し状、または実行に関連する他の手紙または臨床保留;
•FDAは、承認されるべき出願または承認された出願の追加申請を拒否するか、または製品承認を一時停止または撤回する
•製品の差し押さえ、差し押さえ、あるいは製品の輸出入を許可しないことを拒否した
•民事又は刑事処罰の禁止又は適用;及び
•法令、会社誠実協定、資格取り消し、または連邦医療計画から除外することに同意します。
生物製品を管理する規制の排他性
ある生物製品がBLAの承認を得た後にFDAによって発売されると、この製品はあるタイプの市場とデータ排他性を有する権利がある可能性があり、FDAが特定の期間内に競合製品を承認することを禁止する。2010年3月、米国は2009年の生物製品価格競争および革新法案、またはBPCIAを含む患者保護および平価医療法案を公布した。BPCIAはPHSAを改訂し、FDAが許可した参考生物製品の生物と類似或いは交換可能な生物製品のために短い承認経路を作成した。FDAはこれまでにいくつかの生物類似製品と交換可能な生物類似製品を承認してきた。
BPCIAによれば、製造業者は、以前に承認された生物製品“生物類似”の製品の生産を要求する申請を提出することができ、この製品は“参考製品”と呼ばれる。FDAに生物類似製品を承認させるためには、参考製品と提案された生物類似製品が安全性、純度と効力の面で臨床的に意義のある差がないことを発見しなければならない。生物類似スポンサーは、分析研究、動物研究、および1つまたは複数の臨床試験のデータに基づいて、その製品が参考製品の生物と類似していることを証明して、1つまたは複数の適切な使用条件下での安全性、純度、および効力を証明することができる
FDAが生物類似製品を参照製品と交換することを許可するためには、この機関は、製品が参照製品と生物的に類似していることを発見するだけでなく、安全リスクを増加させることなく、または参照生物製品の独占的使用に対して治療効果のリスクを低下させることができる場合に、2つの製品が交換できるように、参照製品と同じ臨床結果を生成することができることを発見しなければならない。FDAの承認後、基準製品の衛生保健提供者の介入を開くことなく、交換可能な生物類似体で参照製品を代替することができる。交換可能な生物類似製品が承認された後、FDAは、第1の交換可能な生物類似製品が初めて商業化販売されてから1年後まで、任意の第2の生物類似製品に交換地位を付与してはならない。議会はFDORAを通じて、FDAは、これらの製品がこのような製品が基準製品と交換可能な最初の日に承認される限り、複数の第1の交換可能な生物類似生物製品を承認することができると明らかにした
参考生物製品には12年の独占経営権が付与されており、この製品が初めて許可を得た日から計算すると、FDAはこの参考生物製品に基づく生物類似または交換可能製品の申請を受け入れず、この参考製品が初めて許可を得た日から4年になる。しかしながら、1つの製品が独占特許を取得する資格がある参考製品と考えられていても、FDAが製品の完全なBLAを承認する場合、製品は、スポンサー自身の臨床前データと、その製品の安全性、純度、および効力を証明するために十分かつ制御された臨床試験データとを含み、別の会社は、製品の競合バージョンを販売することができる。最近,政府は12年の参考製品専門期間の短縮を提案したが,これまで公布されていない。同時に、BPCIAが成立して以来、多くの州で法律や法律改正案が可決され、生物類似製品に関連する薬局のやり方が解決された。
孤児薬の指定と排他性
米国の孤児薬物指定は,スポンサーにまれな疾患や疾患の治療のための製品の開発を奨励することを目的としている。米国では、法律は、まれな疾患または疾患を、米国で20万人未満または米国で20万人を超える影響を及ぼす疾患と定義しており、米国での製品の販売から疾患または疾患を治療する製品の開発および提供のコストを回収することができるという合理的な期待はない。
FDAが承認すれば、孤児薬物指定は会社が製品が承認された日から7年以内に税金控除と潜在的な市場排他性を得る資格がある。孤児製品に指定された出願は、当該製品の発売を承認する申請を提出する前のいつでも提出することができる。1つの製品が受け入れ可能な機密要求に基づいてFDA孤児製品開発事務所から孤児薬物指定を受けると、その製品は孤児となる。そして、その製品はどんな他の製品のように審査と承認手続きを通過しなければならない。
スポンサーは、以前承認されていなかった製品を孤児薬として指定したり、すでに発売されている製品のために新たな孤児適応を申請することを要求することができる。さらに、1つの製品が承認された孤児薬と他の態様で同じ製品である場合、製品が信頼された仮定を提示することができる場合、すなわち、その製品が第1の承認された製品よりも臨床的に優れている可能性がある場合、製品の発起人は、同じ稀な疾患または疾患の後続製品に対する孤児薬物名を求めることができ、取得することができる。複数のスポンサーは、同じ製品のために同じまれな疾患または疾患の孤児薬物指定を得ることができるが、孤児薬物指定を求める各スポンサーは、完全な指定申請を提出しなければならない。
孤児として指定された製品が、このような指定された疾患または条件を有するFDAの最初の承認を受けた場合、またはまれな疾患または条件下での特定の適応または使用として指定された場合、製品は、通常、孤児薬物排他性を得る。孤立薬物排他性とは、いくつかの限られた場合を除いて、FDAが同じ疾患または状態のための別のスポンサーの同一の製品のマーケティング申請を7年以内に承認しない可能性があることを意味する。孤児薬として指定された製品が最終的に発売承認された場合,その適応範囲は孤児薬物申請で指定された範囲よりも広く,排他性を得る資格がない可能性がある。
市場独占期間はFDAが上場申請を承認した日から始まり,当該製品の治療に用いられる疾患や状況の指定にのみ適用される。場合によっては、孤児薬物排他性は、孤児薬物排他性を有する会社が市場需要を満たすことができない場合、またはその後の製品が、より良い治療効果または安全性に基づいて臨床的に承認された製品よりも優れていることが証明された場合、または患者ケアに重大な貢献をすることを含む、別の製品の承認を阻止しない。トランプ総裁が2020年12月27日に署名した総合的な立法によると、製品は臨床的優位性を示す必要があり、2017年のFDCA改正案が公布される前に孤児薬物指定を受けたがFDAの承認を得ていない薬物製品に適用されなければならない。
2021年9月,第11巡回控訴裁判所は,市場排他性の範囲を決定するために,法規中の“同一疾患または状況”という言葉は指定された“まれな疾患または状況”を指すと判断し,FDAはそれを“適応または使用”と解釈することはできない。したがって,裁判所は孤児薬物は“適応や使用”ではなく,指定された疾患や状況全体に排他的に適用されると結論した。この決定を覆すための立法提案があったが、それらはまだ法律になっていない。FDAは2023年1月23日、裁判所の命令範囲を超える事項において、FDAは、孤児薬の独占性と孤児薬の使用または適応のために許可された使用または適応とを束ねて、その既存の法規を適用し続けると発表した。
小児科排他性
小児科専有権は米国では非特許マーケティング専有権であり、承認された場合、6ヶ月の独占権を追加することができる。生物製品については、6ヶ月の期間は、任意の既存の規制排他性に付加することができるが、いかなる特許条項にも付加されない。小児科専属の条件は、FDAが小児科集団における新製品の使用に関連する情報がその集団の健康に利益をもたらす可能性があることを決定すること、FDAが小児科臨床試験の書面請求を提出すること、およびスポンサーが法定時間の枠組み内で要求された臨床試験を実行し、報告することに同意することを含む。スポンサーから提出された小児科データがこのようなデータに対するFDAの書面要求に公平に応答すれば,この6カ月の排他性を付与することができる。これらのデータは、研究された小児科群においてこの製品が有効であることを示す必要はない。要求された小児科研究報告が法定期限内にFDAに提出され、FDAの受け入れを受ける場合、製品の法定または規制独占期間または特許期間にかかわらず6ヶ月延長される。これは特許期間の延長ではないが、FDAが別の出願を承認できない規制期間を効果的に延長している。
特許期限の回復と延長
米国では,ハッジ·ワックスマン法案によると,新製品,その使用方法または製造方法を有する特許は,製品開発やFDA規制審査中に失われた特許期間を最大5年間延長する資格がある可能性があると主張している。出願延期された特許が承認されたと仮定すると、製品をカバーする特許の回復期は、通常、人間のINDの発効日と“商業行為法”の提出日との間の時間の半分に関連し、出願提出日と最終承認日との間の時間を加える。特許期限回復は特許の残存期間を延長するために使用できず,製品が米国で承認された日から合計14年を超える。承認された製品に適用される特許は1つのみ延期する資格があり,延期出願は延期された特許が満了する前に提出されなければならない。複数の製品をカバーする特許は、そのうちの1つの承認に関連して延期することしかできない。米国特許商標局は,FDAと協議した後,任意の特許期限延長の出願を審査·承認する。
随伴診断
FDAは2014年8月に最終指導意見を発表し、承認治療製品への適用と体外培養診断を伴う。このガイドラインによれば、新しい生物学的製品のための、セットされた診断装置およびその対応する治療装置は、製品ラベルにおいて示される治療のためのFDAの承認または承認を同時に得るべきである。キット診断装置の承認または許可は、装置が十分に評価され、ターゲット集団において十分な性能特徴を有することを保証するであろう。FDAは2016年7月、治療製品のスポンサーと体外培養製品の共同開発に関する問題に関するキット診断装置。
2014年のガイドラインはまた、候補生物製品の臨床試験において治療決定を行うためのキット診断装置は、装置が承認または承認された予期される用途に使用されない限り、研究装置とみなされるであろうと説明している。患者選択のような重要な治療決定を行うために使用される場合、FDAの調査デバイス免除またはIDE法規に従って、診断デバイスは、一般に重大なリスクデバイスとみなされる。したがって、診断デバイスのスポンサーはIDEの規定を遵守することを要求されます。ガイドラインによると、診断装置と製品がそれぞれの承認を支援するために一緒に研究しなければならない場合、IDE法規とIND法規の要求を同時に満たすことができれば、この2つの製品は同じ調査研究で研究することができる。指導意見は,研究計画やテーマの詳細に応じて,スポンサーが単独でIND申請を提出したり,INDとIDE申請を同時に提出したりすることができる。
2020年4月,FDAは追加のガイドラインを発表し,適切な多様な生物腫瘍学製品の指定用途を支援するために,マーカーセット診断装置の開発と考慮事項を述べた。本ガイドラインは,診断に伴うラベルに関する既存の政策に基づいている。FDAはその2014年のガイドラインで、診断に伴う診断が特定のカテゴリーの治療製品と共に使用するのに適していると結論するのに十分な証拠があれば、診断に伴う期待用途/使用適応は、特定の製品ではなく、特定の治療製品群と命名すべきであることを示している。2020年ガイドラインは、2014年ガイドライン中の政策声明を拡張し、セット診断開発者は、彼らのテストを開発することができるかどうかを決定する際に、一連の要因を考慮することを提案するか、または、単一の治療製品(S)をリストするのではなく、特定のグループのための腫瘍学治療製品のようなより広いラベル宣言をサポートするために、承認されたセット診断のラベルを追加修正することができる。
FDCAのもとでは体外培養診断は,随伴診断を含め,医療機器として規制されている。アメリカでは、医療機器の設計と開発、臨床前と臨床試験、発売前の承認或いは承認、登録と上場、製造、ラベル、貯蔵、広告と販売促進、販売と流通、輸出と輸入及び発売後の監督などの方面はFDCA及びその実施条例と他の連邦と州法規と条例の管轄を受けている。免除が適用されない限り、診断テストは、商業配信の前に、事前に市場に通知して、またはFDAの承認を得る必要があります。FDAの前に要求されます体外培養随伴診断は、候補製品に反応して発売前承認またはPMAを得る患者を選択することを目的とし、同時に治療製品候補を承認することを目的としている。臨床および臨床前データの収集、およびFDAおよびFDAへの審査の提出を含むPMAプロセスは、数年またはそれ以上を要するかもしれない。これには厳格な上場前審査が含まれており、その間、スポンサーは設備の安全性と有効性の合理的な保証、設備設計、製造、ラベルなどに関する設備とその部品の情報を準備してFDAに提供しなければならない。PMA申請は申請費を支払う必要があり、2024年の連邦財政年度の申請料は483,560ドル、小企業の申請料は120,890ドルである。
医療適合性
米国では、バイオ製薬メーカーおよびその製品は、医療業界の詐欺や乱用を防止するための法律など、連邦や州レベルで広く規制されている。医療保健提供者と第三者支払人は推薦と処方が発売承認された薬品の面で主要な役割を果たしている。提供者、コンサルタント、第三者支払者および顧客との間の配置は、広く適用される可能性のある詐欺および乱用、リベート、虚偽請求法律、医療提供者への支払いの報告、および患者のプライバシー法律法規および他の、我々の業務および/または財務的手配を制限する可能性のある医療法令の制約を受ける可能性がある。私たちが発売されている製品がある場合にのみ適用されるいくつかの法律と法規を含む、連邦および州医療保健法律および法規の制限を適用する
•連邦虚偽声明、虚偽声明、および民事罰金法律は、他の者に加えて、政府資金を支払うために虚偽声明を故意に提出または提出させることを禁止するか、または虚偽クレームを得るために虚偽声明を作成または誘導することを禁止する
•連邦医療計画逆控除法は、他に加えて、個人が直接または間接的に提供、請求、受け入れ、または報酬を提供することを禁止し、連邦医療保険と医療補助などの連邦医療保健計画によって支払うことができる商品やサービスを個人に紹介または購入または注文させることを誘導する
•1996年の連邦健康保険携行性および責任法案、またはHIPAAは、医療提供者および他のエンティティのプライバシー保護に適用されることに加えて、詐欺の任意の医療福祉計画の実行または医療事項に関する虚偽陳述を禁止する
•連邦法律は、製薬業者に、特定の計算された製品価格を政府に報告するか、または政府当局または個人実体に、通常、政府医療計画下の精算条件として何らかの割引またはリベートを提供することを要求している
•製薬および医療機器会社に、特定の医療保健提供者との特定の財務的相互作用を監視し、公衆に再開示するために、医師およびその直系親族が所有する所有権および投資権益を再開示するために、医療および医療補助サービスセンター(CMS)に報告することを要求する連邦公開支払い(または連邦“陽光”法)
•市場活動を広く規制し、消費者の活動を損なう可能性がある連邦消費者保護法と不正競争法
•州逆リベートおよび虚偽申告法、州法律は、製薬会社が特定のコンプライアンス基準を遵守することを要求し、製薬会社と医療保健提供者との間の財務的相互作用を制限すること、または製薬会社に医療提供者への支払いまたはマーケティング支出に関する情報を報告することを要求すること、およびプライバシー、安全、および場合によっては健康情報に違反する州法律を要求することを含む同様の州法律および法規であり、多くの法律は、しばしばHIPAAによって先制されず、コンプライアンス努力を複雑化させることが多い、および
•“海外腐敗防止法”のような賄賂および腐敗を禁止する法律および法規は、米国企業およびその従業員および代理人が、外国政府官僚、国際公共機関または外国政府の所有または付属実体の従業員、外国公職候補者およびその外国政党または官僚への権限、約束、腐敗または不当な支払いまたは任意の他の価値のあるものを提供または提供することを禁止することを含む。
これらの法律に違反する行為は、連邦医療保険および医療補助のような連邦医療保険および州医療計画から除外される場合も含まれる刑事および/または民事制裁を受けることになる。コンプライアンスが時間がかかって費用がかかることを確実にする。EUや他の司法管轄地域にも同様の医療法律や法規があり、医療提供者との相互作用やその支払いの報告要求、個人情報のプライバシーや安全を管理する法律が含まれている。
医療改革
アメリカの医療業界と他の地域の主な傾向の一つはコストコントロールだ。過去数年間に、連邦と州政府は薬品の定価、医療製品のカバー範囲の制限と精算及びアメリカ医療保健システムのその他の変化についていくつかの提案を提出した。
2010年3月、米国議会は、2010年に“医療·教育調整法案”によって改正され、または総称して“PPACA”と呼ばれる“患者保護·平価医療法案”を公布し、これに加えて、政府医療計画下の薬品のカバー範囲や支払い方法の変更を含む。PACAが公布されて以来、他の立法改正も提案され、採択された。2011年8月、“2011年予算制御法案”(Budget Control Act Of 2011)などの法案は国会のための支出削減措置を制定した。赤字削減合同特別委員会の任務は、2013年から2021年までの間に少なくとも1.2兆ドルの赤字削減を提案することであるが、同委員会は必要な目標を達成できず、立法をいくつかの政府プロジェクトに自動的に削減することを触発した。これらの変化には、各年度に提供者に支払われる連邦医療保険総額が2%に達し、2013年4月に発効し、2031年まで続くことが含まれている
PACAが公布されて以来、この法律の条項を廃止し、代替するために、多くの法的挑戦や国会行動が続くだろう。例えば、2017年12月22日にトランプ総裁が署名した“2017年減税·雇用法案”や“税法”の公布に伴い、米議会は“個人強制令”を廃止した。この条項は、ほとんどのアメリカ人が最低レベルの医療保険を購入することを要求する条項が2019年に施行される。2021年6月17日、米国最高裁はいくつかの州がPPACAに提出した最新の司法挑戦を却下したが、PPACAの合憲性を具体的に裁くことはなかった。PACAをめぐる訴訟と立法は継続される可能性があり、結果は予測不可能で不確実だ。
トランプ政権はまた、PPACAに基づいて権力および責任を持つ連邦機関にPPACAの実施を放棄、延期、承認免除または延期を指示する任意の条項を破壊または延期する行政行動をとっており、これらの条項は、各州、個人、医療保健提供者、医療保険会社または薬品または医療機器メーカーに財政的または規制的負担をもたらす。しかし、2021年1月28日、総裁·バイデンはこれらの命令を撤回し、医療の獲得を制限するルールや他の政策を再検討し、その獲得を保護し強化するための行動を検討するように連邦機関に指示する新しい行政命令を発表した。この命令によると、連邦機関は、新冠肺炎に関連する合併症を含む以前の疾患を有する人の保護の政策を弱めること、連邦医療補助および“全米政治行動計画”によるデモおよび免除は、仕事要件を含むカバー範囲または破壊計画を減少させる可能性のある政策、健康保険市場または他の医療保険市場を破壊する政策、連邦医療補助および“全米政治行動計画”への参加の難しさを増加させる政策、および扶養者への負担能力を含む保険または経済援助の負担能力を低減する政策を含む再検討を指示されるであろう。
薬品価格
アメリカでも処方薬の価格はずっと話題になっています。アメリカ議会は最近数回の調査を行い、州と連邦立法を提案し、公布し、薬品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、連邦医療保険と医療補助下の薬品コストを下げることを目的とした。2020年には、総裁·トランプ氏が処方薬のコスト削減を目的としたいくつかの行政命令を発表し、これらの命令のいくつかが条例に盛り込まれている。これらの規定には、価格最恵国モデルを実施し、連邦医療保険B部分のある医師が管理する薬品に対する支払いを他の経済先進国が支払う最低価格とリンクさせ、2021年1月1日から発効する臨時最終規則が含まれている。しかし、この規則は全国的な初期禁止によって制限され、2021年12月29日、CMSはそれを廃止するための最終規則を発表した。CMSは、この規則の発表に伴い、それは価値をMedicare B部分の薬品支払いのすべての選択に組み入れ、そして受益者が根拠に基づく看護を獲得する機会を改善することを探索する。
また、2020年10月、HHSとFDAは、各州と他のエンティティが804条の輸入計画、すなわちSIPを制定し、いくつかの処方薬をカナダから米国に輸入することを許可する最終規則を発表した。この規定は米国薬物研究やメーカー協会(PhRMA)の訴訟で挑戦されたが,裁判所でPhRMAがHHSを起訴する資格がないことが発見された後,2023年2月に連邦地域裁判所に却下された。いくつかの州はカナダからの麻薬の輸入を許可する法律を採択した。いくつかの州は第804条輸入計画提案を提出しており、FDAの承認を待っている。FDAは2023年1月5日にフロリダ州のカナダ薬物輸入計画を承認した。
また、2020年11月20日、HHSは薬品メーカーのD部分下でスポンサーの値下げを計画する安全港保護を廃止し、法律が値下げを要求しない限り、直接或いは薬局福祉マネージャーを通過する法規を決定した。この規定はまた、販売時点での値下げを反映するための新しい避難港を作成し、薬局福祉マネージャーと製造業者との間のいくつかの固定料金手配のための避難港を作成する。裁判所の命令により,上記安全港の除去と増加は延期され,最近の立法は同規則の実施を2026年1月1日に一時停止した。2022年のインフレ削減法案、あるいはアイルランド共和軍と呼ばれ、さらにこの規定の実施を2032年1月1日に延期する。
2021年7月9日、総裁·バイ登は14063号行政命令に署名し、その中で薬品価格などの問題に重点を置いた。この命令はHHSに45日以内に1つの計画を制定し、処方薬の過度な定価を打撃し、国内の薬品サプライチェーンを強化し、連邦政府がこのような薬品に支払う価格を下げ、繰り返し出現した価格詐欺問題を解決するよう指示した。2021年9月9日、HHSは薬品値下げ計画を発表した。この計画の主な特徴は,(A)メーカーとの薬品価格交渉を支援することにより,薬品価格がすべての消費者や医療システム全体により負担と公平になること,(B)サプライチェーンの強化を支援し,生体模倣薬の促進と透明性を増加させる市場変化を支援することにより,処方薬業界全体の競争を改善·促進すること,および(C)公共および民間研究を支援することにより,市場インセンティブを確保して価値と入手可能な新しい療法を促進し,科学的革新を促進し,より良い医療保健と健康改善を促進することである
2022年8月16日、総裁·バイデンは2022年の“インフレ低減法案”であるアイルランド共和軍に署名した。新しい立法は連邦医療保険D部分に影響を与え、D部分は計画であり、連邦医療保険A部分または連邦医療保険B部分に加入する個人は毎月外来処方薬保険料を支払うことを選択することができる。他の事項を除いて、アイルランド共和軍はある薬品のメーカーに2026年から連邦医療保険と価格交渉を行うことを要求し、価格は上限がある場合に交渉することができ、連邦医療保険B部分と連邦医療保険D部分に基づいてリベートを実施し、2023年に初めて満期になったインフレを超える価格上昇を処罰し、2025年からD部分のカバーギャップ割引計画の代わりに新しい割引計画を採用する。アイルランド共和軍は衛生·公衆サービス部長官が最初の数年に規制ではなく指導によってその多くの規定を実施することを許可した
具体的には、価格交渉において、国会は、Medicareが、競合する模倣薬または生体模倣薬を有さず、Medicare B部分およびD部分によって精算されるいくつかの高価な単一由来薬剤および生物製品のためのより低い価格を交渉することができる。CMSは、2026年からMedicare D部分によって支払われる10種類の高コスト薬剤の価格を交渉することができ、その後、2027年の15種類の追加D部分薬剤、2028年の15種類の追加B部分またはD部分薬剤、および2029年以降の20種類の追加B部分またはD部分薬剤の価格を交渉することができる。この規定は、少なくとも9年間承認された医薬品および許可13年の生物製品に適用されるが、単一のまれな疾患または疾患のための許可された医薬および生物製品には適用されない。また、この立法は、製薬業者が提供する価格が法律で規定された交渉で達成された“最高公平価格”以下でない場合、または値上げ幅がインフレを超える場合、製薬業者は民事罰金と潜在的な消費税を受けることになると規定している。この立法はまた、メーカーに連邦医療保険D部分の価格上昇幅がインフレを超えた薬品にリベートを支払うことを要求している。新法律では,2024年の連邦医療保険の自己払い薬品コスト上限は年間4000ドルと規定されており,その後2025年から年間2000ドルが上限となっている。
2023年6月6日、メルク社はHHSとCMSを提訴し、アイルランド共和軍の連邦医療保険に対する薬品価格交渉計画が憲法第5改正案に違反した無償徴収を構成したと主張した。その後、米国商会、百時美施貴宝会社、アメリカ製薬研究とメーカー協会、PhRMA、アステラス、ノとノッド、ヤンソン製薬、ノワール、アスリカン、バーリンガー-インゲルハイムを含む他のいくつかの機関も異なる裁判所に訴訟を提起し、HHSとCMSに対して類似した憲法クレームを提出した。私たちはIreland共和軍のこのような条項と他の条項に関連した訴訟が継続され、結果的に予測可能で不確実だと予想する。
州レベルでは、各州はますます積極的に、価格または患者の精算制限、割引、ある製品への参入の制限、マーケティングコストの開示および透明性措置を含む薬品および生物製品の価格設定を制御するための法規を立法と実施し、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。例えば、いくつかの州は、健康担体、薬局福祉マネージャー、卸売流通業者を含む医薬品製造業者およびサプライチェーン内の他のエンティティに、薬品の価格設定に関する情報を開示することを要求する。さらに、地域医療機関および個別病院は、どの薬品およびどのサプライヤーが彼らの処方薬および他の医療計画に含まれるかを決定するために入札プログラムを使用するようになってきている。これらの措置が承認されると、私たちの製品に対する最終的な需要を下げたり、私たちの製品の価格設定に圧力をかけるかもしれません。私たちは将来、より多くの州と連邦医療改革措置を取ることが予想され、そのいずれも連邦と州政府が医療製品やサービスのために支払う金額を制限する可能性があり、これは私たちの候補製品に対する需要の減少または追加の価格設定圧力を招く可能性がある。
連邦と州データプライバシー法
複数のプライバシーやデータセキュリティ法律が私たちのビジネス活動に影響を与える可能性があり、アメリカや私たちが実験や将来業務を展開する可能性のある他の国/地域で行われています。このような法律は変化しており、私たちの義務と未来の規制リスクを増加させるかもしれない。一般に、ヘルスケア業界では、HIPAA、HHSに基づいて、保護された健康情報(PHI)のプライバシーおよびセキュリティを保護するための法規が発行されており、これらの情報は、特定の医療提供者、健康計画、および医療情報交換所を含むカバーされたエンティティによって使用または開示されている。HIPAAはまた,医療取引で使用されるデータ内容,コードとフォーマットの標準化,医療計画と提供者の識別子の標準化を規範化している。HIPAAはまた、保護された健康情報を取得した保証エンティティの商業パートナーに対して、保証エンティティまたは代表的な保証エンティティにサービスを提供する際に何らかの義務を課す。HIPAAは場合によっては私たちに適用される可能性があり、私たちのビジネスパートナーにも適用される可能性があり、その方法は私たちと彼らの関係に影響を与える可能性があります。私たちの臨床試験は、プライバシーに関連する具体的な条項も含む共通のルールによって規制されている。連邦プライバシー法規のほかに、多くの州の法律が健康情報のセキュリティと安全性を管理しており、これらの法律は私たちの業務に適用される可能性がある。州総検察長は、HIPAA違反行為に対して連邦民事·刑事処罰を行う可能性があるほか、連邦裁判所に民事訴訟を提起し、損害賠償や禁令を求めてHIPAAを執行し、連邦民事訴訟に関連する弁護士費と費用を求める権利がある。また、州総検察長(個人原告とともに)は、HIPAAのプライバシーや安全ルール違反の疑いで禁止令や損害賠償を求める民事訴訟を起こしている。州総検事長はまた州のプライバシーと安全法を執行する権利がある。未来にはまたプライバシーと安全に関する新しい法律と規制が採択されるかもしれない。
州レベルでは、カリフォルニア州で米国初のGDPRのような法律と呼ばれる法律が公布された。カリフォルニア消費者プライバシー法案、またはCCPAと呼ばれ、消費者のための新しいプライバシー権(法律で広く定義されているように)を創出し、消費者または家庭の個人データを処理するエンティティにより多くのプライバシーおよびセキュリティ義務を課す。CCPAは2020年1月1日に施行され、カバーする会社がカリフォルニアの消費者に新たな開示情報を提供することを要求し、これらの消費者に特定の個人情報の販売から撤退することを選択し、データ漏洩に対して新たな訴訟理由をとることを可能にする。また、2023年1月1日から、カリフォルニアプライバシー権法案(CPRA)は、ある敏感な個人情報に対する消費者の権利を拡大することを含むCCPAを大きく改正した。CPRAはまた、CCPAおよびCPRAを実施し実行する権限を付与される新しい国家機関を作成した。CCPAやCPRAは我々の業務活動に影響を与える可能性があり,これをどのように解釈するかに依存し,我々の業務はネットワークの脅威を受けやすいだけでなく,個人データや個人が識別できる健康情報に関する変化する規制環境の影響を受けることを示している。このような規定は私たちのいくつかの商業活動に適用されるかもしれない
カリフォルニア州を除いて、他の11州でもCCPAとCPRAのような全面的なプライバシー法が採択された。このような法律は施行されたか、2026年末までに施行されるだろう。これらの法律は,CCPAやCPRAと同様に,個人情報の処理に関する義務と,“敏感な”データを処理する特殊な義務を規定しており,健康データが含まれている場合もある。このような法律のいくつかの規定は私たちの商業活動に適用されるかもしれない。2025年以降に施行される包括的なプライバシー法が2024年の立法会議中に採択されたことを強く検討している州もある。他の州も将来的には似たような法律を考慮し、国会でも連邦プライバシー法の成立について議論されてきた。私たちの業務の健康情報に影響を及ぼすかもしれないいくつかの州の専門的な規制もある。例えば、ワシントン州は2023年に“私の健康私のデータ法案”を可決し、この法案はHIPAA規則によって規制されていない健康情報を専門的に規制し、この法律はまた個人訴訟権を有し、これは関連するコンプライアンスリスクをさらに増加させる。コネチカット州やネバダ州でも同様の法律が成立して消費者健康データを規制しており,より多くの州では2024年にこのような立法が制定されることが考えられている。これらの法律は、我々の研究対象の決定、業務パートナーとの関係、および最終的に私たちの製品のマーケティングおよび流通(承認されれば)を含む当社の業務活動に影響を与える可能性があります。
これらの法律の汎用性、およびこれらの法律によって提供される法定例外および規制安全港の範囲が狭いため、私たちの現在または未来のいくつかの業務活動は、いくつかの臨床研究、販売およびマーケティング実践、および特定のプロジェクトおよびサービスを私たちの顧客に提供することを含み、1つまたは複数のこのようなプライバシーおよびデータセキュリティ法律の挑戦を受ける可能性がある。ますます高いコンプライアンス環境、および複数の司法管轄区域の異なるプライバシーコンプライアンスおよび/または報告要件を遵守するために強力かつ安全なシステムを確立し、維持する必要があり、医療会社がそのうちの1つまたは複数の要件を完全に遵守できない可能性を増加させる可能性がある。私たちの運営が、我々のプライバシーまたはデータセキュリティに適用される上記の任意の法律または法規に違反していることが発見された場合、私たちは、潜在的な重大な刑事、民事および行政処罰、損害賠償、罰金、契約損害、名声損害、利益減少および将来の収益減少、追加の報告要件および/または規制を含む罰を受ける可能性があり、これらの法律違反に関する告発を解決するために、法令または同様の合意に同意する制約を受けた場合、いずれも私たちの業務運営能力および私たちの運営結果に悪影響を及ぼす可能性があります。ある程度、私たちが開発する可能性のある任意の候補製品は、承認されると、海外で販売され、私たちは似たような外国法の制約を受けるかもしれない。
EUの医療製品の承認と規制
アメリカの法規のほかに、私たちは様々な外国法規の制約を受けて、これらの法規は私たちの製品のアメリカ以外の臨床試験と商業販売と流通を管理しています。私たちがFDAの候補製品の承認を得るかどうかにかかわらず、私たちは、これらの国や地域で臨床試験や製品の販売を開始するために、外国または経済地域の比較可能な規制機関の承認、例えば27の加盟国のEUを取得しなければならない。連合では、私たちの候補製品もまた広範囲な規制要求を受ける可能性がある。アメリカと同様に、医薬製品は主管監督機関のマーケティング許可を得た後にのみ発売されることができる。アメリカと類似して、EUの臨床前と臨床研究の各段階は重要な監督管理によって制御されている。
EUと欧州経済区(EEA)が統一的な薬品監督規則を適用する以外に、臨床試験、製品許可、定価と精算の審査プロセスと要求は国家と司法管轄区の間で大きく異なり、追加のテストと追加の行政審査期限に関連する可能性がある。他の国や管轄地域で承認を得るのに要する時間は、FDAの承認を得るのに要する時間とは異なり、さらに長くなる可能性がある。1つの国または管轄区域で規制承認を得ることは、他の国または管轄区域で規制承認を得ることを保証することはできないが、1つの国または管轄区域で規制承認を得ることができなかったか、または遅延して監督管理許可を得ることは、他の国または司法管轄区の規制手続きに悪影響を及ぼす可能性がある。
非臨床研究
非臨床研究を行うのは,新たな化学や生物物質の健康や環境安全性を証明するためである。非臨床(薬物-毒性)研究は、EU指令2004/10/ECに規定されている良好な実験室規範(GLP)原則を遵守しなければならない(特定の医薬製品、例えば放射性ラベルのための放射性薬物前駆体については、別の正当な理由がない限り)。特に、体外と体内の非臨床研究は必ずGLP原則に従って計画、実行、モニタリング、記録、報告とアーカイブを行い、GLP原則は組織過程の品質体系と非臨床研究の条件として一連の規則と標準を定義した。このようなプロス基準は経済協力と開発組織の要求を反映する。
臨床試験
2022年1月31日,新しい臨床試験条例(EU)第536/2014号,あるいはCTRは,従来の臨床試験指令2001/20/ECに代わってEUで発効した。新しい規定はEUの臨床試験の許可、進行と透明性を簡略化し、簡素化することを目的としている。新しい臨床試験承認調整手続きによると、1つ以上の欧州連合加盟国またはEU加盟国で行われる臨床試験の発起人は、承認申請を提出するだけでよい。提出された材料は臨床試験情報システムを介して提出され、これはEMAが監督する新しい臨床試験ポータルサイトであり、臨床試験スポンサー、EU加盟国の主管当局と公衆に使用することができる
手続きの簡略化に加えて,CTRには,申請のための書類のセットと,簡略化された臨床試験スポンサー報告プログラムと,臨床試験申請評価の統一プログラムが含まれており,このプログラムは2つに分けられている。第1部は、臨床試験許可申請が提出されたすべてのEU加盟国の主管当局によって評価される。二番目の部分は関連されたすべての連合会員国によって個別的に評価される。臨床試験申請の評価には厳しい期限が設定されている。評価手続きにおける道徳管理委員会の役割は、EU加盟国に関する国家法律によって引き続き管轄されるだろう。しかし、全体的に関連するスケジュールは臨床試験条例によって定義されるだろう。
CTRは、スポンサーが事前に臨床試験を行うEU加盟国主管国当局の承認を得なければならないという、以前に存在した要求を変えていない。もし臨床試験が異なるEU加盟国で行われた場合、これらのEU加盟国の主管当局は臨床試験の承認を提供しなければならない。また,スポンサーは適用された倫理委員会が賛成の意見を発表した後にのみ,特定の臨床地点で臨床試験を開始することができる。
CTRは3年間の過渡期が予想される。進行中の臨床試験と新たな臨床試験がCTRによってどの程度制御されるかはそれぞれ異なる。(I)2022年1月31日までに“臨床試験指令”に基づいて申請を提出した臨床試験、または(Ii)2022年1月31日から2023年1月31日までの間、かつスポンサーが“臨床試験指令”を適用する臨床試験を選択し、2025年1月31日までこの指令によって管轄されている。この日以降,すべての臨床試験(行われている臨床試験を含む)はCTR条項に拘束される。
ある臨床試験を行う締約国は,米国のようにEUの臨床試験登録所でEUの臨床試験情報を公表しなければならない。
EUのマーケティング許可
マーケティング許可申請、またはMAASは、製品によって1つ以上のEU加盟国で許可された相互承認または分散手順であるにもかかわらず、いわゆる集中または国家認可プログラムに従って提出することができる。
集中化プログラムは、欧州薬品管理局(EMA)の良好な意見に基づいて、すべてのEU加盟国およびEEAに属するアイスランド、リヒテンシュタイン、ノルウェーで有効である単一マーケティング許可を付与することを規定している。特定のバイオテクノロジーによって生産された医薬、孤児医薬製品として指定された製品、高度な治療薬(例えば、遺伝子治療、体細胞治療または組織工学薬)、およびHIV/エイズ、癌、糖尿病、神経変性疾患または自己免疫疾患、ならびに他の免疫機能障害およびウイルス疾患などの特定の疾患を治療するための新しい活性物質を含む製品については、集中的な手順が実行されなければならない。重大な治療、科学的または技術的革新を代表する製品、またはその許可が公衆の健康に有利になる製品については、集中手順がオプションである。中央プログラムによると,環境保護局がMAAを評価する最長期限は210日であり,クロックポーズは含まれておらず,スポンサーは人間用薬品委員会やCHMPからの質問に答えるために追加の書面や口頭情報を提供する。CHMPは特殊な状況下で加速評価を承認することができ、特に治療革新の角度から見ると、1種の医薬製品が重大な公衆衛生利益を有することが予想される場合。加速評価プログラムによる重大な影響評価の期限は150日であり,停止クロックは含まれていない.
いくつかのEU諸国では、他の2つの可能な方法が医薬製品を許可することができ、これらの経路は集中プログラム範囲に属さない研究用医薬製品に使用することができる
•分散プログラムそれは.分散プログラムを使用して、スポンサーは、どのEU諸国でも許可されていない薬品を1つ以上のEU諸国で同時に許可することができ、集中プログラムの強制範囲に属さない。発起人は一つの会員国を参考会員国として選択し、先頭に立って申請を科学的に評価することができる。
•相互認識プログラムそれは.相互認識手続きでは、1つの薬剤は、まずEU加盟国(参照加盟国として)でその国の国家手続きによって許可される。その後、他のEU諸国にさらなるマーケティング許可を段階的に求めることができ、関連国は、加盟国が発行した元の国家マーケティング許可の有効性を参照することに同意することができる。
上記の手順により、販売許可を承認する前に、欧州経済区管理局又は欧州経済区加盟国主管当局は、製品の品質、安全性及び有効性に関する科学的基準に基づいて、製品のリスク−利益バランスを評価する。
条件付き承認
特別な場合、EU立法(第14条-a条例(EC)第726/2004号(条例(EU)2019/5及び条例(EC)第507/2006号の人用薬品に関する条件付きマーケティング許可改正)は、スポンサーが包括MAを申請するために必要な全面臨床データを取得する前に条件付きMAを取得することを可能にする。以下の場合、候補製品(孤児薬として指定された薬剤を含む)に条件付き承認を与えることができる:(1)候補製品は、深刻な衰弱または生命に危険な疾患の治療、予防または医学診断のためのもの、(2)候補製品は、患者が満たされていない医療需要を満たすことを目的としている、(3)包括的な臨床データを提出する前に、医薬品の即時発売に関するメリットが依然として補充データを必要とするという事実に固有のリスクを超えることを前提として、MAを承認することができる。(4)候補製品のリスク−収益バランスが積極的であり,(5)スポンサーが必要な包括的な臨床試験データを提供できる可能性が高い。条件付きMAは,進行中または新たな臨床試験の完了および薬物警戒データの収集に関する義務を含むMA保持者が履行しなければならない具体的な義務を含むことができる。条件付きMAの有効期間は1年であり,リスク−収益バランスが正のままであり,条件や特定の義務を付加または修正する必要があるかどうかを評価した後,毎年更新することができる。上記の集中プログラムのスケジュールは,CHMPによる条件付きM&A申請の審査にも適用可能である.
特殊な事情
“特別な場合”では、出願人が、製品が許可され、特定の手順に従った後であっても、正常な使用条件下での有効性及び安全性に関する包括的なデータを提供することができず、MAを付与することができる。特に期待される適応は非常にまれであり,現在の科学的知識状態では網羅的な情報を提供することが不可能である場合や,データを生成することが一般的に受け入れられている倫理原則に違反する可能性がある場合がある.このMAは、重篤な疾患または満たされていない医療需要のために承認されるべき医薬製品を保持し、出願人は、MAに付与するために必要な合法的な完全データセットを有さないので、条件付きMAに近い。しかし,条件付きMAと異なり,申請者は失われたデータを提供する必要もなく,提供する必要もない.“特殊な場合”のMAは最終的に承認されているが,毎年薬品のリスク−収益バランスが審査されており,リスク−収益比が有利でなければMAは撤回される。これらのプログラムにより,MAを付与する前に,EMAまたは加盟国主管当局は,製品の品質,安全性,有効性に関する科学的基準に基づいて,製品のリスク−利益バランスを評価する。MAの初期期間は条件付きMAを除いて5年である.この5年後、リスク-収益バランスを再評価した上で許可を更新することができる
小児科試験
EUのマーケティング許可を得る前に、スポンサーは、EMAがPIPに含まれる1つまたは複数の措置について特定の製品の免除、カテゴリ免除、または延期を許可しない限り、EMA承認に適合する小児科人口のすべてのサブグループをカバーする小児科調査計画(PIP)に含まれるすべての措置を証明しなければならない。(EC)第1901/2006号“小児科条例”と呼ばれる、すべての販売許可手続のそれぞれの要件が規定されている。会社が認可された薬物のために新たな適応,薬物形式あるいは投与経路を増加させようとした場合にも,この要求は適用される。EMAの小児科委員会(PDCO)はある薬物の延期を承認する可能性があり、会社が成人に対する有効性と安全性を証明する十分な情報があるまで、会社が児童薬物の開発を延期することを許可する。以下の場合、PDCOはまた、免除を与えることができる:(A)製品は、子供の集団の一部または全部に無効または非安全である可能性があり、(B)この疾患または状態は、成人集団においてのみ発生するか、または(C)この製品は、小児集団の既存の治療に対して有意な治療利益を有さない。マーケティング許可申請を提出する前に、または既存のマーケティング許可を修正する前に、EMAは、会社が各関連PIPに列挙された合意された研究および措置を実際に遵守していると判断する。
素数ラベル
2016年3月,EMAは適応候補の開発を促進するイニシアティブを開始したが,これらの適応はまれであり,現在のところ治療法はほとんどない。優先薬品計画、あるいはPrimeは、満たされていない医療需要分野の製品開発を奨励し、中央プログラムの下で審査された重大な革新を代表する製品に対して加速評価を提供することを目的としている。中小企業の製品は大企業よりも早くPrime計画に参加する資格があるかもしれません。Primeの称号を持つ候補製品のスポンサーは多くのメリットを得ることができ、しかし限らず、早期にEMAと積極的な監督対話を行い、臨床試験設計とその他の開発計画要素を頻繁に討論し、及びファイルを提出した後にマーケティング許可申請の評価を加速する。重要なのは,CHMPや高度治療委員会の専門機関連絡先や調査委員がPrime計画の早期に任命され,EMA委員会レベルで製品のより多くの理解を促進することである。会議を開始してこれらの関係を起動し、EMAを含む多学科専門家チームは、スポンサーに全体的な発展と監督管理戦略方面の指導を提供した。
授権期間と継続期間
マーケティング許可は原則として5年間有効であり、マーケティング許可は5年後に、EMAまたはライセンス加盟国の主管当局によるリスク-収益バランスの再評価に基づいて更新することができる。そのため、上場授権書保持者は上場授権書が失効する少なくとも9ヶ月前に、マーケティング授権書を授与してから導入されたすべての変化を含むEMA或いは主管当局に品質、安全と効果に関する文書の総合バージョンを提供しなければならない。一旦更新されると、上場許可の有効期限は無期限であり、欧州委員会または主管当局が薬物警戒に関する正当な理由に基づいて5年間の継続を決定しない限り、5年間の継続が決定される。いかなる許可も、許可が失効してから3年以内に製品をEU市場(集中手続きであれば)または許可加盟国の市場に実際に投入しなかった(いわゆる日没条項)。
上場認可後の規制要件
米国と同様に、医薬製品の販売許可保持者とメーカーは、生産と販売許可を承認する前と後に、欧州薬品管理局とEU個別加盟国主管当局の全面的な監督管理を受けている。例えば、EU薬品の上場許可を持っている人はEU薬物警戒立法とその関連法規とガイドラインを守らなければならず、これらの法規とガイドラインは薬物警戒或いは評価と医療製品の安全性のモニタリングに対して多くの要求を提出している。EUの医薬製品の製造過程も厳格に監督管理されており、監督管理機関は規定に合わないと思う製造施設を閉鎖する可能性がある。製造には製造許可が必要であり、製造許可保持者は適用されるEU法律に規定されている各種要求を遵守しなければならず、医薬製品と活性薬物成分を製造する際にEU cGMP標準を遵守することを含む。
EUでは、承認された製品の広告および販売促進は、医薬品販売促進、臨床医師との相互作用、誤解性および比較広告、および不公平な商業行為に関するEU加盟国の法律によって制限される。さらに、個別のEU加盟国が採択した他の立法は、医薬製品の広告や販売促進に適用される可能性がある。これらの法律は,医薬製品に関連する宣伝材料や広告が主管当局が承認した製品特性要約に適合することを求めている。SmPCに適合しない医薬製品の普及はラベル外の普及を構成していると考えられ,EUでは禁止されている
監督管理排他性
欧州連合では、許可されて発売された新製品(すなわち参考製品)は、8年間のデータ独占権と追加2年間の市場独占権を得る資格がある。データ専門期間は後発薬スポンサーがEUが後発薬の発売許可を申請する時に参考製品ファイルに含まれる臨床前と臨床試験データに依存し、参考製品が欧州連合が初めて許可を得た日から8年以内である。市場排他期は、参考製品が欧州連合で最初の許可を得た10年後まで、成功した後発薬スポンサーが欧州連合でその製品を商業化することを禁止している。10年の最初の8年間に、マーケティング許可保持者が1つまたは複数の新しい治療適応の許可を得た場合、許可前の科学的評価期間中に、これらの適応は既存の療法と比較して有意な臨床的利益をもたらすことができると考えられる場合、10年間の市場専門期間は最大11年に延長することができる。
孤児薬の指定と排他性
EUが指定した孤児薬品の基準は原則的にアメリカと似ている。条例(EC)141/2000第3条によれば、以下の場合、医薬品は、孤児として指定することができる:(1)生命または慢性衰弱にかかわる疾患の診断、予防または治療のための、(2)申請時に、そのような疾患がEUで10,000人以下の5人に影響を与えるか、または(B)孤児身分のメリットがない場合、当該製品は、投資が合理的であることを証明するためにEUで十分な見返りを生じない、(3)満足できる診断、予防または治療方法がEUで販売されることを許可していない、またはそのような方法が存在する場合、その製品はこのような状況の影響を受けた人たちに大きな利益を与えるだろう。条例(EC)847/2000の“顕著な利益”という言葉の定義は臨床に関連する優勢或いは患者看護に対する重大な貢献を意味する。
孤児医薬製品は費用の低減や費用の免除などの財政奨励を受ける資格があり、マーケティング許可を得た後、承認された治療適応の10年間の市場排他性を得る権利がある。この10年間の市場排他期間内に、欧州薬品管理局または欧州薬品管理局加盟国の主管当局は、同じ適応の類似医薬製品のマーケティング許可申請を受け入れることができない。類似医薬製品の定義は、許可された孤児医薬製品に含まれる1つ以上の類似活性物質を含む医薬製品であり、同じ治療適応のためのことを目的とする。上場許可を申請する前に、孤児指定申請を提出しなければならない。孤児が孤児として指定された場合、スポンサーはマーケティング許可申請の費用を減免するが、マーケティング許可の提出時にその指定が待っている場合は、そうではない。孤児指定は、規制審査および承認過程においていかなる利点も伝達されず、規制審査および承認過程の継続時間を短縮することもない。
5年目の終了時に、製品が指定された孤児の基準を満たしていないと判断された場合、例えば、製品の利益が十分に高く、市場排他性を維持するのが合理的であることを証明するのに十分でない場合、欧州連合の10年間の市場排他性は6年に減少することができる。さらに、以下の場合、(1)第2のスポンサーは、その製品が類似しているが、より安全で、より効果的であり、または臨床的により良いことを証明することができる、(2)スポンサーが2回目の孤児薬の申請に同意すること、または(3)スポンサーが十分な孤児薬を提供できないことを、同じ適応の類似製品の発売を随時承認することができる。
小児科排他性
スポンサーがすべてのEU加盟国のマーケティング許可を取得した場合、または欧州委員会が集中手続きで付与されたマーケティング許可を取得し、小児科集団に対する試験結果が製品情報に含まれている場合、否定的であっても、補充保護証明書またはSPCの期間を延長することによって追加6ヶ月の合格特許保護を得る資格があるか、またはマーケティング許可保持者によって規制市場占有権を10年から11年に延長することを選択する資格がある。
特許期限を延長する
EUはまた,補完保護証明書(SPC)による特許期間の延長を規定している。SPC獲得のルールと要求は米国と類似している。SPCは当初の満期日以降に特許の有効期限を最大5年間延長することができ、製品に最大15年間の市場独占経営権を提供することができる。場合によっては、小児科専門権を取得すれば、これらの期間はさらに6ヶ月延長することができる。SPCはEU全体で入手できるにもかかわらず、スポンサーは各国に基づいて申請しなければならない。EU以外のいくつかの他の外国司法管轄区域にも同様の特許期間延長権が存在する。
処方薬の精算と定価
連合では、様々な国の価格設定と補償プログラムの差が大きい。一部の国では、補償価格を合意した後にのみ、製品を販売することができると規定されている。いくつかの国は、特定の候補製品のコスト効果を現在利用可能な治療法またはいわゆる衛生技術評価と比較して、精算または価格設定の承認を得るために、追加の研究を達成することを要求するかもしれない。例えば、欧州連合は、その国の健康保険制度が精算を提供する製品範囲を制限し、人が使用する医療製品の価格を制御するために、EU加盟国に様々な選択を提供している。EU加盟国は製品の具体的な価格を承認することができ、製品を市場に投入する会社の収益力を直接または間接的に制御する制度をとることもできる。他のEU加盟国は会社が自分の製品価格を固定することを許可したが、処方量を監視と制御し、医師に指導を発表して処方を制限した。最近,欧州連合の多くの国で医薬品割引が増加し,各国が保健支出の管理を試みているに伴い,特に欧州連合の多くの国が深刻な財政危機や債務危機を経験した場合,これらの努力が継続される可能性がある。全体的には,医療コスト,特に処方薬の下り圧力が大きくなっている。そのため、新製品の参入にはますます高い壁が設けられている。政治、経済、規制面の事態は定価交渉をさらに複雑化させる可能性があり、補償を受けた後、定価交渉が継続される可能性がある。各加盟国が使用する参考定価と平行貿易、すなわち低価格と高価なEU加盟国間の裁定は、さらに価格を下げることができる。薬品に対して価格規制や精算制限を実施することが保証されていない国は、これらの国で承認されれば、任意の製品に対して有利な精算と定価手配を許可する。
セット診断設備の承認
EUでは,診断などに伴う医療機器は,EU医療機器条例(EU)2017/745)添付ファイルIで詳細に説明されている一般的な安全と性能要件やSPR,あるいは2021年5月26日に施行され,従来適用されていたEU医療機器指令(理事会指令93/42/EEC)のMDRに置き換えられなければならない。SPRとセット医療機器への適用の付加的な要求に適合することは,医療機器にCE適合性マーカーを貼り付けることができる前提条件であり,これらのマークがなければ,医療機器は市販や販売できない。SPRに適合することを証明するためには,メーカーは医療機器のタイプとその分類によって異なる合格評価プログラムを受けなければならない。MDRは、EU全体にわたって統一的、透明、予測可能かつ持続可能な医療機器規制枠組みを構築することを目的としている。
また,EU規制当局は2022年5月に施行された新たな体外診断法規(IVDR)(EU)2017/746を採択した。新法規は“体外診断指令”(IVDD)98/79/ECに取って代わった。その体外診断医療機器の合格評価を通知機関に申請したいメーカーは,要求を満たし,新たなより厳しい法規を遵守するために,2022年5月までにその技術文書を更新しなければならない。その他の事項以外に、この条例は設備を市場に投入することに関する規則を強化し、設備が発売された後に監督を強化した;メーカーの市場投入設備の品質、性能と安全に対する追跡責任を明確に規定した;唯一の識別番号を通じて、サプライチェーン全体の医療設備のエンドユーザー或いは患者に対する追跡可能性を高めた;中央データベースを構築し、患者、医療保健専門家と公衆にEUの既存製品に関する全面的な情報を提供した。そして、インプラントのようないくつかの高リスクデバイスの評価ルールを強化し、これらのデバイスは、市場に投入される前に専門家の追加的な検査を受ける必要がある可能性がある。
IVDRは2022年5月に施行された。しかし,2021年にはEU加盟国,衛生機関,経済経営者がIVDRを適用する準備ができていないことが明らかになった。したがって、欧州委員会はIVDR規則を段階的にまたは交互に導入することを提案した。現在の過渡期は2025年5月26日の高リスク体外診断(IVDS)から2027年5月26日の低リスクIVDまでである。衛生機関で製造·使用される設備に関するいくつかの規定は,2028年5月26日から適用されなければならない。これらの移行期間は、以前の法的枠組み(特にIVDD)に従って発行された証明書または適合性宣言によってカバーされる装置のいわゆる“従来の装置”にのみ適用される。
一般資料保障規程
ヨーロッパや他の国では、重要なプライバシーとデータセキュリティ法が適用されている。個人健康データを含む欧州経済地域内の個人に関する個人データを収集、使用、開示、移転、または他の方法で処理することは、2018年5月25日に施行されたEU“一般データ保護条例”(GDPR)によって制限される。GDPRの範囲は広く,個人データを処理する会社に多くの要求があり,健康や他の敏感なデータを扱う会社に対してより高い要求が出されており,たとえば多くの場合,企業にはこのようなデータを処理する前に,敏感な個人データに関する個人の同意を得なければならないことが要求される.GDPRがGDPRの範囲内に属する個人データを処理する会社に課す義務は、個人にデータ処理活動に関する情報を提供すること、個人データのセキュリティおよび機密性を保護するための保障措置を実施すること、データ保護幹事を任命すること、データ違反イベントについて通知を提供すること、および第三者プロセッサを採用する際に何らかの措置をとることである。
GDPRはまた、米国を含む欧州経済圏以外の国への個人データの移転に厳しいルールを実施し、データ保護当局が2000万ユーロや世界の年収の4%に達する可能性のある罰金を含む、GDPR違反行為に巨額の罰金を科すことを許可している。GDPRはまた、データ主体と消費者協会に個人訴訟権利を与え、監督当局に苦情を提出し、司法救済を求め、GDPR違反による損害について賠償を受けることができる。GDPRを守ることは厳格で時間のかかる過程であり,ビジネスコストを増加させたり,会社にそのビジネスやり方を変更して完全な遵守を確保したりする可能性がある
2020年7月、欧州連合裁判所(Court of the European Union、略称CJEU)は、個人データを欧州経済地域から米国に移転するための合法化メカニズムの一つである欧州連合-米国プライバシー遮蔽枠組みの無効を宣言した。CJEUが決定した後,2022年10月,総裁·バイデンはEU−米国プライバシーの盾に代わるEU−米国データプライバシー枠組みを実施する行政命令に署名した。EUは2022年12月にEU-米国データプライバシーフレームワークによる十分性決定を開始し,欧州委員会は2023年7月に十分性決定を採択した.十分な決定は、EU-米国データプライバシーフレームワークを自ら認証する米国社が、EUから米国にデータを送信するための有効なデータ転送機構とすることを可能にする。しかし、いくつかのプライバシー提唱団体は、EU-米国のデータプライバシーの枠組みに挑戦すると表明している。これらの挑戦が成功すれば、EU-米国のデータプライバシーの枠組みに影響を与えるだけでなく、標準契約条項や他のデータ伝送機構の生存能力もさらに制限される可能性がある。この問題をめぐる不確実性は私たちの業務に影響を及ぼす可能性がある。
2016年6月23日、イギリスの有権者はEU離脱、いわゆる離脱に賛成票を投じた。連合王国のEU離脱後,2018年にイギリスデータ保護法はイギリスで行われた個人データ処理に適用され,GDPRが規定している義務と平行した義務が含まれている。英国の2018年の“データ保護法”は“施行”と“GDPR”の補完であり、2018年5月23日に王立承認を受け、現在イギリスで施行されているが、“GDPR”に基づいて欧州経済地域からイギリスにデータを移行することが合法であるかどうかは不明であるが、これらの移行は現在欧州委員会の十分な決定によって許可されているが、これらの移行は現在、欧州委員会の十分な決定によって許可されている。英国政府は、EU 27カ国と欧州経済圏加盟国のすべてがデータ保護において十分であり、連合王国からEU/欧州経済区へのデータが影響を受けないことを確保することを決定した。また、欧州委員会の最近の決定は、連合王国が欧州連合から連合王国にデータを移すのに“ほぼ十分”と考えているようだが、この決定は今後再評価される可能性がある。英国と米国はまた、EU-米国データプライバシーの枠組みに類似した機能を有する米英データブリッジの構築に同意し、会社がイギリスから米国にデータを送信するための追加の法的メカニズムを提供した。スイスは英国に加えて、スイス-米国のデータプライバシーの枠組みに関する十分な決定を承認している(スイスの米国へのデータ移転では、EU-米国のデータプライバシーの枠組みと米英データブリッジのように動作している)。このような開発のどんな変化や更新も私たちの業務に影響を及ぼす可能性がある。
イギリスの離脱とイギリスの規制枠組み
イギリスは2020年1月31日にEUから離脱した。EUと英国は、2021年1月1日に臨時発効し、2021年5月1日に発効する貿易·協力協定における新たなパートナーシップについて合意した。この協定は主に自由貿易に注目し,医療製品を含む商品貿易に関税や割当を徴収しないことを確保している。その後、EUと連合王国は2つの独立した市場を形成し、2つの異なる規制と法制度によって管理される。そのため、この協定は貨物貿易障壁を最大限に減らすことを図るとともに、連合王国が単一市場の一部ではないため、国境検査は避けられないことを認めている。2021年1月1日から、薬品と保健品規制機関(MHRA)はイギリスの薬品と医療機器の監督を担当し始め、国内法によると、イギリスはイングランド、スコットランド、ウェールズを含み、北アイルランドは北アイルランド議定書によってEU規則の制約を受け続けている
2023年2月27日、イギリス政府と欧州委員会は、北アイルランド議定書、すなわち“ウィンザー枠組み”と呼ばれる新たな手配の代わりに、原則的な政治的合意を達成することを発表した。この新しい枠組みは北アイルランド議定書下の現行制度を根本的に変え,特にイギリスの医療製品の規制において,MHRAはイギリス市場(すなわち大ブリテンおよび北アイルランド連合王国)に輸送されたすべての医療製品の承認を担当するが,EMAは北アイルランドへの医療製品の承認に何の役割も果たすことはないであろう。MHRAは、イギリスで販売されているすべての医薬製品に全イギリス範囲のMAを付与し、製品がイギリス全体で単一パッケージおよび単一ライセンスで販売できるようにする。ウィンザー枠組みは2023年3月24日にEU·英国合同委員会の承認を得たため、英国政府とEUは立法措置を制定し、法律にする。2023年6月9日,MHRAはウィンザーフレームワークの薬品について2025年1月1日から適用すると発表した。“2012年ヒト医薬品条例”(SI 2012/1916)(改正された)、またはHMR、主要な法的文書 連合王国の薬品を管理するために,HMRは国内法に連合王国が欧州連合から離脱する前から存在していた医薬製品に関するEU法律文書を取り入れている。
二次立法によって連合王国法律に転換された欧州法は引き続き“保留EU法”として適用されている。しかし、CTRのような新しい立法はイギリスに適用されないだろう。イギリスの薬品監督管理の枠組みの中で薬品の品質、安全性と有効性、臨床試験、MA、薬品の商業販売と流通をカバーする大部分はEU指令と法規に由来するため、イギリスの離脱はイギリスの候補製品の開発、製造、輸入、承認と商業化の方面の監督管理制度に実質的な影響を与える可能性がある。例えば、イギリスはEMAからEU範囲内のMAを取得する集中プログラムによってカバーされなくなり、もし私たちの任意の候補製品が承認された場合、イギリスでマーケティングするためには単独のMAが必要となる。新たな国際認可枠組みは2024年1月1日から発効し,この枠組みにより,MHRAは新たなイギリスMAの申請を決定する際に,EMAと何らかの他の規制機関によるMAの承認決定を考慮する
知的財産権
私たちは、当社のADCプラットフォーム、物質の固有成分、ADC製品の候補製品および使用および製造方法をカバーする特許の出願、および当社のビジネス発展に商業的重要性を有すると考えられる他の任意の発明および改善を含む、私たちのビジネスに重要と考えられるノウハウを保護することを積極的に求めています。私たちはまた、ビジネス秘密と機密情報、技術ノウハウ、持続的な技術革新に依存して、私たちの独自と知的財産権の地位を発展させ、維持しています。
私たちのビジネス成功は、私たちが私たちの業務に重要だと思う技術、発明、および改善のために特許および他の固有の保護を獲得し、維持する能力があるかどうか、私たちの特許を保護すること、私たちの商業秘密および他のノウハウの機密性を保護すること、および第三者の特許および独自の権利を侵害することなく運営する能力があるかどうかに大きく依存するだろう。私たちの政策は、私たちのノウハウ、発明、改善に関連する米国、国際(特許協力条約またはPCTによる)の提出、および外国特許出願を含む、私たちの独自および知的財産権の地位の保護を求めることであり、これらの出願は、私たちの業務の発展と実施に非常に重要であると考えられます。私たちはまた、私たちの非特許商業秘密とノウハウを保護し、私たちの技術革新を続けて、私たちの業務を発展させ、私たちの競争地位を維持すると信じています。
個別特許の期限は、特許を取得した国の法律用語に依存する。米国を含む多くの国では,特許期間は非臨時特許出願が最初に提出された日から20年である。米国では、特許期間の延長は、特許期限調整によって延長することができ、これは、特許権者が米国特許商標局による特許審査および承認時の行政遅延を補償することができ、または、1つの特許が以前に提出された特許によって最終的に放棄された場合、短縮することができる。法定と規制要件を満たしていれば、薬物や生物製品をカバーする特許期限はFDA承認時にも延長する資格がある。将来,我々の候補薬剤がFDAまたは外国規制機関の承認を得た場合,各薬剤の臨床試験時間や他の要因に応じて,可能な場合には,これらの薬剤をカバーする発行済み特許の特許期間の延長を申請する予定である。私たちのいかなる係属中の特許出願が発行されるか、または私たちは任意の特許期間の延長または私たちの任意の特許条項の有利な調整から利益を得ることが保証されない。
他のバイオテクノロジーや製薬会社と同様に、私たちが私たちの候補薬物や技術のために私たちの特許や知的財産権の地位を維持し、強化する能力があるかどうかは、有効な特許主張を成功させ、承認された後にこれらの主張を実行するかどうかにかかっている。しかし、私たちが係属中の特許出願、および将来第三者から提出または許可される可能性のある任意の特許出願は、特許の発行につながらないかもしれません。私たちはまた私たちの特許で許可または実行される可能性のある特許請求の範囲の広さを予測することができない。私たちが現在所有しているか許可されているか、または将来得られる可能性のある任意の発行された特許は、挑戦、無効、回避、またはその権利要件の範囲を縮小する可能性がある。例えば、私たちは審理中の第三者特許出願によってカバーされている発明の優先権を決定することができない。もし第三者が米国で準備して提出した特許出願も私たちが獲得する権利のある技術や療法を持っていると主張すれば、私たちは発明の優先権を決定するために米国特許商標局の介入手続きに参加しなければならないかもしれないが、これは最終的な結果が私たちに有利であっても、非常に予測不可能である。さらに、我々が開発する可能性のある候補薬剤の臨床開発および規制審査には長い時間がかかるため、任意の候補薬剤が商業化される前に、任意の関連特許は商業化後短期的に失効または有効に維持される可能性があり、それにより、特許が対応する製品に提供される保護および特許が提供可能な任意の競争優位性を制限することができる。私たちの知的財産権に関連するリスクに関するより多くの情報は、“リスク要因-私たちの知的財産権に関するリスク”を参照してください
私たちはアメリカと外国の特許出願を提出し、起訴し、維持することによって、私たちの研究と開発で生まれたいくつかの発明を保護することを求めています。任意の特定の特許出願の地域出願範囲は、我々が研究および開発している新規プラットフォームおよび候補製品へのこの発明の予期される適用性を含むが、これらに限定されない多くの要因に依存するであろう。私たちの新型Dolasyntenプラットフォームと関連製品候補および私たちの新型免疫合成プラットフォームと関連製品候補に関連する発明については、私たちはアメリカ、ヨーロッパ、オーストラリア、カナダ、中国、日本、韓国などの国で特許保護を求め、他の外国司法管轄区で保護を求める可能性もある。我々の特許の組み合わせが、我々のDolasyntenプラットフォームおよび免疫合成プラットフォームおよび候補製品に関連する態様は、以下でより詳細に説明される。Synaffixから非排他的内部許可を取得し,その特定サイト共役技術に関するいくつかの特許や特許出願を取得した.これらのライセンスされたSynaffix特許および特許出願は、8つの認可された米国特許、3つの出願されている非一時的な米国特許、13件の許可された外国特許、および14件の出願中の外国特許を含み、中国、ヨーロッパ、インド、日本、オランダを含むがこれらに限定されない複数の外国司法管轄区域に関連する。ライセンス特許は、追加の特許期間調整または特許期間延長を含まず、2031年から2040年までに満了する予定である。今まで、私たちは特許保護を提供する可能性のあるすべての国と地域の司法管轄区域で特許保護を申請していません。しかも、私たちは承認前に国と地域の特許出願を放棄することに決定するかもしれない。最後に、各国または地域特許の付与手続きは、いくつかの法域において、出願が関連登録当局によって拒否され、他の法域によって承認される可能性がある独立した手続きである可能性がある。同様に、国によっては、同じ候補製品または技術に異なる範囲の特許保護が付与される可能性がある。
以下に、我々のDolasynThenおよび免疫合成ADCプラットフォームに関連する知的財産権の組み合わせおよび私たちの候補製品、構成要素および使用について概説する。その中のいくつかのポートフォリオはまだ非常に早い段階にあり、いくつかの未解決の特許出願はまだ起訴され始めていない。起訴は長い過程であり、その間、最初にUSPTOによって提出された審査請求の範囲は、それらが本当に発行された場合に縮小される可能性がある(場合によっては顕著である)。私たちは次に言及される保留特許出願もそうだと予想する。
Dolasyntenプラットフォーム
我々の新規Dolasyntenプラットフォームの知的財産権の組み合わせは、新規金フラビンペイロードのための物質組成物、新規ステント及びそのADC、並びにこれらの新規物質組成物の使用及び製造方法を対象としている。私たちのDolasyntenプラットフォームの知的財産権については、2023年12月31日現在、発行された5つの米国特許(2032年満期(物質成分)、2037年(物質成分)、2041年満期(物質成分および使用および製造方法)、追加の特許期限調整または特許期限延長を含まず)、29件の発行された外国特許(オーストラリア、カナダ、中国、欧州、日本、韓国で発行された特許を含む)、2032年満期(物質成分)および2037年(物質成分および使用成分)、特許期限調整または特許期間延長の追加期限は含まれていない)、米国と4つの特許家族内のいくつかの外国司法管轄区域での審査待ちの出願。新規Dolasyntenプラットフォームに関連する係属中の出願によって発行される任意の米国または外国特許は、追加の特許期限調整または特許期間延長を含まず、2032年から2041年の間に満了すると予想される。
XMT-1660
XMT-1660は私たちの現場で特定のB 7-H 4 ADC製品候補品であり、その知的財産権の組み合わせは、私たちの新しいB 7-H 4抗体と私たちのDolasyntenプラットフォームに基づく新しいADCに関連する物質組成、およびこれらの新しい結合体の使用、製造と管理方法を対象としている。XMT−1660に関連する知的財産権については、2023年12月31日現在、米国特許出願(2042年に満了する予定である(物質および使用方法の成分)、追加の特許期限調整または特許期限延長を含まない)、および米国および複数の外国司法管轄区域における3つの特許系列の係属中の特許協力条約(PCT)に基づく許可を有している。未決定出願で発行されたXMT−1660物質組成物およびその使用、管理および/または製造方法に関連する任意の米国または外国特許は、追加の特許期限調整または特許期限延長を含まず、2042年から2044年の間に満了すると予想される。
免疫合成プラットフォーム
我々の新規免疫合成プラットフォームの知的財産権の組み合わせは、新規刺激剤及びそのADCの物質組成、及びこれらの新規ペイロード及びADCの使用方法及び製造方法である。私たちの免疫合成プラットフォームに関連する知的財産権については、2023年12月31日現在、発行された米国特許(2040年に満了(物質成分)、追加の特許期限調整または特許期限延長を含まない)と、米国での裁判待ち出願と、2つの特許家族内の複数の外国司法管轄区域とを有している。新規STINGアゴニストおよび免疫合成ADCプラットフォームに関連する係属中の出願によって発行される任意の米国または外国特許は、追加の特許期限調整または特許期間延長を含まず、2040年から2041年の間に満了すると予想される。
XMT-2056
我々の新規免疫合成ADC XMT-2056に対する我々の知的財産権の組み合わせは、我々の新規HER 2抗体および免疫合成プラットフォームに基づく新規ADCの物質組成、ならびにXMT-2056を含むこれらの新規結合体を使用、製造、および管理する方法である。XMT-2056に関連する知的財産権については、2023年12月31日現在、発行された3つの米国特許(2035年満期(物質および使用方法の組成物)、追加の特許期限調整または特許期限延長を含まない)、発行された34件の外国特許(2035年満期(物質成分、製造方法および使用組成物)、追加の特許期限調整または特許期限延長を含まない)、PCTに従って米国および各外国司法管轄区で待っている4つの特許シリーズの出願を有している 新規HER 2標的免疫合成ADC XMT-2056に関連する係属中の出願によって発行される任意の米国または外国特許は、追加の特許期限調整または特許期間延長を含まず、2035年から2044年の間に満了すると予想される。
特許に加えて、私たちは、非特許の商業秘密および機密技術、発明、独自情報、および持続的な技術革新に依存して、私たちの競争地位を発展させ、維持している。私たちは、私たちの専門情報の機密性の保護と維持を求めており、一部は、私たちの従業員やコンサルタントと発明秘密および譲渡プロトコルを実行することによって、レベルおよび役割に応じた適切なスポーツ禁止および非募集プロトコル、ならびに私たちの協力者および科学コンサルタントとの秘密協定を含むことができます。私たちはまた、選定された科学コンサルタントと協力者と発明の譲渡を要求する協定に署名した。私たちが締結した機密協定は、私たちの固有の情報を保護することを目的としており、私たちに発明を譲渡することを要求する協定または条項は、対応する取引相手との関係によって開発された技術の所有権を付与することを目的としています。しかし、私たちは、私たちがすべての適用可能な従業員や請負業者とこのような合意に署名したことを保証することができません。またはこれらの合意は、私たちの知的財産権および独自の情報権利に十分な保護を提供することになります。商業秘密とノウハウを保護することは難しいかもしれない。特に,独自の情報を保護するための措置をとっているにもかかわらず,時間の経過とともに,我々の技術プラットフォーム,ビジネス秘密,ノウハウに関する情報が,独立した開発や記述方法の公開プレゼンテーションによって業界内に伝播する可能性が予想される.私たちのビジネス秘密に関連するリスクに関するより多くの情報は、“リスク要因-当社の知的財産権に関連するリスク-従業員および第三者と締結された秘密保護協定は、不正な商業秘密および他の固有情報の漏洩を阻止できない可能性があります”を参照してください
競争
生物技術と生物製薬業界及び腫瘍子業界の特徴は技術進歩が迅速で、競争が激しく、知的財産権の保護が力強いことである。私たちが開発と商業化に成功したどの候補製品も、既存の治療法や将来出現する可能性のある新しい療法と競争しなければならない。独自のADCプラットフォームや科学的専門知識が競争優位を提供してくれると信じているが,大手バイオ製薬会社,専門バイオテクノロジー会社,学術研究部門,公共·民間研究機関を含む様々な機関が潜在的な競争力のある製品や技術を積極的に開発している。これらの競争相手は一般に以下のように分類される
癌新治療法の研究開発者:多くの世界の製薬会社と中小生物技術会社は新しい癌治療法を求めており、小分子、生物製剤、ADCを問わない。これらの治療法のいずれも我々の製品より臨床的に優れていることが証明されている可能性がある。
細胞毒性ADCプラットフォームを持つ会社です多くの会社は、新しいペイロードクラス、新しい共役方法、新しい目標部分を含むADC分野の革新に投資している。これらの取り組みのいずれも、我々が開発または開発しているプラットフォームよりも優れた特性を有するプラットフォームをもたらす可能性がある。ADC技術を持つ複数の会社が、エバービー社、Ambrx Biophma,Inc.(ジョンソンに買収されることが予想されている)、第一三共株式会社、ジリッド科学会社、ファイザーを含む当社のプラットフォームを競争する可能性があることも知られています。これらの会社または彼らのパートナーや協力者は、アステラス製薬会社、アスリコン、ロー氏グループのメンバー遺伝子テーク、武田を含み、現在および未来の候補製品と同じ競争優位を持つ候補製品を開発する可能性があります。複数の会社もBolt BioTreateutics,Inc.;Sutro Biophma,Inc.およびTakedaを含む我々の免疫合成プラットフォームとその候補製品と競合できるADCを開発しており,異なる免疫刺激法を採用しているにもかかわらず。我々の革新技術に基づいて、他の候補製品と比較して、我々のADCの有効性、安全性、耐性に基づいて競争したい。しかし、もし私たちの開発センターがこのような点で明らかな利点がなければ、私たちは効果的に競争できないかもしれない。
B 7-H 4ターゲット製品を持つ会社:複数の研究されているB 7−H 4 ADCが初のヒト臨床試験を行っており,SGN−B 7 H 4 V(ファイザー,レーガン社から買収),HS−20089/GSK 5733584(グラクソ史克社,ハンソン製薬有限会社から許可を得た),AZD 8205(アスリーカン),他のB 7−H 4 ADCが臨床前開発段階にある。また,CLN−418(クリナム腫瘍社からのB 7−H 4 x 4−1 BB BsAb),GEN 1047(Genmab A/SからのB 7−H 4 x CD 3 BsAb),ABL 103(ABL Bio,Inc.由来のB 7−H 4 x 4−1 BB BsAb)を含む他の非ADCモデルに基づくB 7−H 4標的薬の臨床試験が行われている。すべての上述の計画は現在1期或いは1/2期の臨床試験にあり、広範な固形腫瘍に集中しているが、多くの計画は既知の高B 7-H 4発現の固形腫瘍を優先し、乳癌、子宮内膜癌と卵巣癌を含む。著者らが他のB 7-H 4方案と有効に競争する能力は、標的腫瘍タイプ、ペイロード、治療効果と耐性によってXMT-1660を他の治療法と区別する能力に依存する。XMT-1660を他のB 7-H 4に対する候補製品と効率的に区別できないことは,我々の競争能力に悪影響を与える
詐欺誘導プラットフォームと候補製品を持つ会社:現在臨床試験が行われている免疫刺激ADCで使用されているペイロードは,STINGアゴニストとToll様受容体あるいはTLRアゴニストの2つに分類される。他の臨床前および発見計画には、CD 73およびPD-(L)1のような腫瘍関連抗原に対する計画が含まれている。現在臨床的に活発な免疫刺激ADCには、ボルト治療会社が含まれている。S社のBDC−1001(HER 2指向TLR 7/8アゴニスト)と武田のTAK−500(CCR 2誘導刺激剤)は,いずれも多くの固形腫瘍型で検討されている。臨床的にもCDNと非CDN STINGアゴニストを含む様々な系で投与されるSTINGアゴニストレジメンがある。このような製品と効果的に競争する能力は、標的および/またはバイオマーカーの選択、有効性、安全性および耐性に応じて、XMT−2056および他の潜在的な免疫合成ADCをこれらの他の療法と区別する能力に依存する。
HER 2ターゲットADCを持つ会社:承認されたHER 2標的ADCは2つありますロー氏のKadcylaは®乳がん治療に承認されたAdo−trastuzumab emtansine,アスリコン社のSと第一三共株式会社のENHERTU®乳癌、非小細胞肺癌および胃癌の治療のために承認された(fam-trastuzumab deruxtecan-nxki)。この2つの承認されたHER 2標的ADCは,それぞれの適応の治療パターン,特に乳癌において変化した。現在、全世界でも40個以上のHER 2に対するADCが臨床開発を行っており、そのペイロード、連結子と腫瘍標的はすべて異なる。我々とこれらのエージェントとの有効な競合能力は,XMT-2056のSとこれらの他のエージェントとの区別や組合せ可能性に依存する
私たちの多くの競争相手は、単独でも戦略的パートナーとも、私たちよりも多くの財力、技術、人的資源を持っている。したがって、私たちの競争相手は私たちよりも治療の承認を得ることに成功し、広範な市場受け入れを得ることができ、それによって私たちの治療が時代遅れになったり、競争力がないようにすることができる。バイオテクノロジーとバイオ製薬業界で加速されたM&A活動は、より多くの資源を私たちの少ない競争相手に集中させる可能性がある。これらの会社はまた合格した科学と管理人員を募集と維持し、臨床試験場を創立し、臨床試験のために患者を募集し、著者らの計画と相補的或いは必要な技術を獲得する方面で著者らと競争を展開している。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。もし私たちの競争相手が私たちの同類製品よりも効果的で、安全で、より便利で、あるいはもっと安い製品を開発し、商業化すれば、私たちのビジネス機会は大きく制限されるかもしれない。私たちのビジネス成功に重要な地域では、競争相手も私たちの前に規制部門の承認を得て、私たちの競争相手が私たちの製品が入る前に強力な市場地位を築くことができるかもしれません。私たちは私たちの計画の成功を決定する要素が私たちの候補製品の有効性、安全性、耐性になると信じている。
従業員と人的資本
2023年12月31日現在、私たちは123人の常勤従業員がいて、そのうち83人が医学博士や他の高度な学位を持っています。これらの従業員のうち,84人が研究·開発に従事し,39人が一般·行政活動に従事している。私たちの職員たちは労働組合代表もなく、集団交渉協定のカバー範囲もない。私たちは私たちが従業員と仲がいいと思う。
私たちの未来の成功は、私たちが高技能従業員を引きつけ、維持し続ける能力に大きくかかっていると信じている。私たちの人的資本目標は、適用された場合、私たちの既存と新設された従業員を識別、募集、維持、激励、統合し、従業員の福祉と職場の安全に集中することを含む。私たちは従業員に競争力のある給料とボーナス、株式機会、持続的な学習と成長を支援する発展計画、及び医療保健、退職計画、有給休暇を含む彼らの生活のあらゆる面の健康を促進する穏健な雇用プランを提供する
多様性,公平性,包摂性,帰属感を促進することが癌患者に治療法を発見,開発,もたらす重要な要因であると考えられる。2023年12月31日まで、私たちの労働力の56%は女性だ。私たちは、私たちがサービスするコミュニティと患者を代表する労働力チームを構築し、包容的な文化を育成するために努力しており、このような文化の中で、すべての声が歓迎され、耳を傾け、尊重されている。
施設
私たちの会社の本社はマサチューセッツ州ケンブリッジ市にあります。私たちは会社本部のあるマルチテナントビルで約四十五,000平方フィートのオフィスと実験室空間を借りました。この空間の大部分の賃貸契約は2026年3月に満期になる。その後、私たちはレンタル期間をさらに5年間延長することを選択することができる。私たちは、このオフィスと実験室空間は私たちの現在の需要を満たすのに十分であり、必要な時に適切な追加空間を提供すると信じている。
企業情報
私たちは2001年にデラウェア州の会社として設立しました私たちの主な実行事務室はマサチューセッツ州カンブリッジ記念通り840号にあります。郵便番号:02139、電話番号は6174980020です。私たちのサイトはwww.mersana.comです。我々は、定期的に、我々の年間報告(Form 10-K)、四半期報告(Form 10-Q)、現在の報告(Form 8-K)、および1934年の証券取引法第13(A)または15(D)節に提出または提出された報告の改訂版を含む重要な情報を無料で提供し、これらの報告は、米国証券取引委員会または美銀美林に電子的に提出された後、合理的で実行可能な範囲でできるだけ早く提出または提供される。私たちのサイトは、公衆投資家に接触する主要な配信ルートであり、“米国証券取引委員会FD規則”に基づいて負う開示義務を遵守するための重大な非公開情報を開示する手段でもあることを認識している。我々のサイト上の情報は,本Form 10-K年次報告に含まれていると見なすべきではなく,Form 10-K年次報告の一部と見なすべきではなく,どのサイトの引用もアクティブなハイパーリンクで行うつもりはない.
項目1 A.リスク要因は含まれていない
我々の業務及び財務結果は、以下に述べるリスク及び不確定要因を含む様々なリスク及び不確実性の影響を受ける。我々の普通株への投資を決定する前に、以下のようなリスクおよび不確実性に関する情報と、本年度報告書の他の10-K表の他の情報は、我々の合併財務諸表および関連付記を含むことをよく考慮しなければならない。以下に説明するリスクと不確実性は私たちが直面している唯一の危険と不確実性ではない。私たちは現在知られていないか、または現在重要ではないと考えている他のリスクや不確実性もまた、私たちの業務に悪影響を及ぼす可能性があり、あるいは私たちの実際の結果は、本報告書で行われた前向きな陳述と、私たちが時々行う可能性のある展望的な陳述に含まれる結果とは大きく異なる。以下のいずれかのリスクが発生すれば、私たちの業務、財務状況、経営業績、将来の成長見通しは重大な悪影響を受ける可能性がある。私たちは次のように議論されたどんな事件も起こらないという保証がない。
我々のADC候補品の開発と承認に関連するリスク
私たちは現在臨床試験で限られた数のADC候補製品を評価している。もし私たちのすべての候補製品が臨床開発に失敗した場合、私たちの業務に悪影響を与え、同じ技術に基づく他のADC候補製品の開発を停止することが要求されるかもしれない。
XMT−1660およびXMT−2056は,我々が現在臨床試験評価を行っている唯一の候補品である。われわれは2023年7月に,われわれの単腕登録試験において,従来の主要候補製品であるupifitamab rilsotinやupRiのデータがその主要な終点に達していないことを評価し,Uplit−Ritと呼び,その主要な終点に達しておらず,UpRiに関する開発活動を終了し,UpRiとカルボプラチンを併用する第1段階の併用試験を中止し,Carplatinは白金感受性卵巣癌治療に広く用いられる標準白金系化学療法薬であり,グレードアップ−Aと呼ぶことを発表した。われわれの第3段階臨床試験はUpRiを再発性白金感受性卵巣癌の二重白金併用治療後の維持治療として行った。これをUP-NEXTと呼んでいますまた,われわれのXMT−2056臨床試験は2023年3月から2023年10月までの間に米国食品·薬物管理局(FDA)に臨床的に放置され,現在も回復していない。他にもいくつかの臨床前プロジェクトが開発されているが,これらのプロジェクトが臨床開発段階に達するためには,追加の投資と時間,規制部門の承認が必要である。さらに、我々が現在開発している他の候補製品は、XMT−1660およびXMT−2056と同じプラットフォームに基づいている。候補製品が我々のプラットフォームの任意の潜在的な問題によって開発に失敗した場合、同じ技術に基づく候補製品の開発を停止することが要求される可能性がある。もし私たちがXMT-1660またはXMT-2056または任意の他の現在または未来の候補製品の開発を停止することを要求された場合、またはXMT-1660またはXMT-2056または任意の他の現在または未来の候補製品が規制の承認を得られなかった場合、または十分な市場受容度を得ることができなかった場合、私たちは利益を達成することを阻止または大幅に延期される可能性がある。
発見計画や候補製品の失敗は臨床前あるいは臨床開発の任意の段階で発生する可能性があり,また,我々と我々の協力者の発見計画や製品候補は臨床前あるいは臨床開発の早期段階にあるため,失敗のリスクが高い。私たちまたは私たちの協力者は決して規制部門の承認を得ることに成功し、そのような発見計画または候補製品から収入を得ることができないかもしれない。
私たちは私たちの臨床開発努力の初期段階にあり、私たちの主な候補製品です。我々はXMT−1660とXMT−2056の第1段階臨床試験を行っているが,これら2つの候補製品の臨床試験は完了していない。私たちが製品収入を作る能力は、私たちの候補製品の成功開発、マーケティング承認、最終商業化に大きく依存します。これは永遠に起こらないかもしれません。製品収入は何年も起こらないと予想しています。我々はXMT-1660およびXMT-2056の臨床前研究の結果、および任意の他の現在または未来の候補製品の臨床前研究または早期臨床試験の結果について、必ずしも私たちが行っている或いは未来の発見計画、臨床前研究或いは臨床試験の結果を予測するとは限らない。候補薬物の臨床前研究と早期鼓舞的な臨床結果は後期臨床前研究或いは臨床試験期間中の人類における類似結果を予測できないかもしれない。製薬と生物技術業界の多くの会社は臨床開発の早期段階で積極的な成果を得た後、臨床試験で重大な挫折を経験し、私たちはすでに再び類似の挫折に直面する可能性がある。例えば,2023年7月,われわれのUpRiのUplift第二段階臨床試験はその主要な治療効果の終点に達していないことを発表し,われわれのUpRi 1 b期臨床試験の奏効率データは有望であるにもかかわらず。他社の挫折は,主に臨床試験施行期間中の臨床前発見,あるいは臨床前あるいは臨床試験における安全性や有効性イベント,以前に報告されていない有害事象を含むものである。同様に,われわれの臨床試験の進展に伴い,われわれも新たな安全信号を決定し,例えば,UpRi治療を受けた患者に重篤な出血事件が発生したようであり,その発生率がバックグラウンドよりも高いことを示し,この評価により,2023年6月にまとめたデータ安全報告書をFDAに提出した。
同様に,臨床試験の設計はその結果が製品の承認を支持するかどうかを決定することができるが,臨床試験設計における欠陥は臨床試験の進展が良好になるまで明らかにならない可能性がある。2023年3月、FDAは、XMT-2056第1段階試験の一時停止を発表したことを発表し、これまで、XMT-2056に関連すると考えられるレベル5(致命的な)深刻な有害事象(SAE)のため、自発的に試験を一時停止したことをFDAに通知した。SAEは第一段階の臨床試験用量増加部分に初期用量レベルで登録された第二名の患者に発生した。2023年10月31日、FDAは臨床制限を廃止し、私たちの第1段階用量漸増設計において開始用量を低下させたと発表した。2023年10月に臨床試験を廃止して以来、著者らはまだ私たちのXMT-2056第一段階臨床試験に参加する患者を募集していない。
私たちが行う可能性のあるどんな臨床試験も、規制部門の許可を得て私たちの候補製品を市場に出すために必要な有効性と安全性を証明しないかもしれない。また,我々の候補製品や類似技術を用いた競争相手製品の臨床試験結果は,我々が開発している他の候補製品の安全性や有効性への懸念を引き起こす可能性がある。もし私たちが行っているあるいは未来の臨床試験結果が私たちの候補製品に対する治療効果が確定していない結果であれば、もし私たちが統計学的に意味のある臨床終点に達していなければ、あるいは私たちの候補製品に安全問題や有害事象が存在すれば、私たちの候補製品の発売承認を得ることを阻止または延期されるかもしれない。例えば,2023年6月にわれわれがすべてのUpRi臨床試験のまとめた安全性分析報告をFDAに提出したところ,重篤な出血事象の発生頻度が背景よりも高いようであることが評価され,FDAはUpgrade−AとUp−Next臨床試験を部分的に休止し,2023年7月,われわれのUplit臨床試験が主要な終点に達しなかった後,Up−NextとUpgrade−A臨床試験を含めてUpRiの将来開発を中止することにした。また,われわれのXMT−2056第1段階臨床試験では,1人の患者が5級SAEを有しており,FDAは2023年3月から2023年10月までこの試験を休止した。我々が行っているXMT−1660とXMT−2056臨床試験および将来の臨床試験では,われわれの候補製品が臨床開発過程で進展するにつれて,ある患者は死亡を招く可能性のある事象を含む不良事象を経験することが予想される。
多くの要素のため、同一候補製品の異なる臨床試験間の安全性或いは有効性結果は有意差が存在する可能性があり、これらの要素は方案中に規定された試験プログラムの変化、患者群の大きさとタイプの差異、投与方案とその他の臨床試験方案の変化と遵守状況及び臨床試験参加者の退学率を含む。そのほか、臨床前と臨床データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品が臨床前研究と臨床試験で満足できると考えているが、依然としてFDAの承認を得られなかった。私たちまたは私たちの協力者が私たちの候補製品の臨床試験結果が発売承認に値すると思っていても、FDAまたは類似の外国規制機関は同意しない可能性があり、私たちの候補製品の発売を承認しないかもしれない。
代替的に、私たちが規制部門の承認を得ても、この承認は、予期されたまたは期待されているような広範な適応や患者集団に適用されないか、または重大な使用または流通制限または安全警告を含むラベルが必要とされる可能性がある。私たちはまた、承認を得るために追加的または予期しない臨床試験を行うことを要求されるか、または規制部門の承認を維持するために追加の上場後試験要求を受ける可能性がある。さらに、規制当局は、リスク評価および緩和戦略またはREMS計画の形態のような製品の承認を撤回するか、またはその流通に制限を加えることができる。監督管理機関の候補製品に対する承認を適時に得ることができず、いかなる製品マーケティング制限或いは製品撤回も私たちの業務、運営結果と財務状況に負の影響を与える。
著者らが時々発表或いは公表した臨床試験の初歩、中期と主要なデータは更に多くの患者データの獲得に従って変化する可能性があり、そして監査と検証プログラムの影響を受け、これは最終データの重大な変化を招く可能性がある。
私たちは時々私たちの臨床試験の初歩的、中期的、あるいは主要なデータを公表したり公表したりするかもしれない。肯定的な初歩的なデータはこのような実験の後続や全体的な結果を予測できないかもしれない。著者らが完成する可能性のある臨床試験の中期データは必ずしも最終結果を予測するとは限らず、しかも患者登録の継続とより多くの患者データの獲得に伴い、1つ以上の臨床結果は実質的に変化するリスクが発生する可能性がある。予備データやトップラインデータも監査と確認手続きを受ける必要があり、これは最終データが私たちが公表する可能性のある予備データやトップラインデータと大きく異なる可能性がある。我々は2024年に我々のXMT−1660第1段階臨床試験の初期データを開示する予定であるが,これらのデータは試験中の最終データと実質的に異なる可能性がある。したがって、最終データを取得する前に、予備データ、中間データ、および主要データを慎重に見るべきである。初期または中期データと最終データとの間の不利な違いは、私たちのビジネスの将来性を深刻に損なう可能性があります。
私たちの候補製品の臨床試験の開始、登録または完了を遅延または阻止する可能性があるイベントは、私たちのコスト増加を招く可能性があり、規制の承認を得ることができないか、あるいは臨床試験を一時停止または中止させることを招き、私たちの候補製品をタイムリーに商業化することを阻止し、甚だしきに至っては実現できないかもしれない。
私たちが行っているおよび将来の任意のXMT-1660、XMT-2056、または任意の他の現在または未来の候補製品の臨床試験を含む臨床試験は、計画通りに行われるか、または計画的に完了することを保証することはできない(もしあれば)。1つまたは複数の臨床試験の失敗は、試験の任意の段階で発生する可能性があり、他のイベントは、臨床試験を一時的または永久的に停止させる可能性がある。臨床開発の成功またはタイムリーな開始、登録または完了を妨げる可能性があるイベントは、以下のことを含む
•遅延と規制機関は試験設計について合意した
•遅延または予期される臨床研究組織またはCRO、サイト管理組織またはSMOおよび臨床試験サイトと受け入れ可能な条項と合意することができなかった
•各臨床試験地点で必要な機関審査委員会(IRB)または倫理委員会(EC)の承認を得ることは困難である
•適切な患者を募集し、臨床試験方案の標準に符合する臨床試験に参加する上で直面している挑戦
•監督機関、IRBs、またはECsは任意の理由で、安全考慮のため、あるいは臨床手術または試験場所を検査した後、臨床保留を実施することを含む
•私たちまたは私たちの任意のサプライヤーまたはサプライヤーが契約した第三者による必要なスクリーニングの遅延
•CRO、SMO、他の第三者、または私たちは臨床試験の要求を遵守できなかった
•FDAの良好な臨床実践またはGCPまたは他の国/地域に適用される規制ガイドラインに従って動作することができなかった
•候補製品または臨床試験を行うために必要な他の材料の数または品質不足、例えば、遅延試験、検証、製造、または臨床現場への候補製品の送達を含む;
•試験を完了していない、または治療後にフォローアップしていない患者;
•臨床試験における任意の候補製品に関連するSAEの発生を含む予期されるまたは予期されない安全問題であって、これらの候補製品は、候補製品の潜在的利点、または癌療法のような他の臨床前または臨床試験によって生じる可能性のある報告を超えているとみなされ、これらの試験は、私たちの候補製品の安全性または有効性の懸念を引き起こす
•新しい臨床プログラムを修正または提出するか、または追加のデータを提出する必要がある法規要件またはガイドラインの変化;
•1つ以上の臨床試験を継続するのに十分な資金が足りない;または
•ロシアとウクライナの間の持続的な衝突、およびイスラエルとガザ地区を支配するパレスチナ組織ハマスとの戦争を含む地政学的または他の事件は、意外にも、私たちの臨床試験場所、CRO、SMO、または関連する開発活動を展開するための他の行動に適した地域または世界的な業務を妨害、延期または普遍的に妨害している。
遅延は、上述の要素による遅延を含み、コストが高い可能性があり、著者らが現在と予想される臨床試験を開始、登録或いは完成する能力に負の影響を与える可能性がある。2023年6月、著者らはUp-NextとUpgrade-A臨床試験がFDAの一部の臨床で保留されたことを発表し、これまでFDAは著者らのすべてのUpRi臨床試験の総合安全性分析をFDAに提出し、著者らは深刻な出血事件の発生率が背景より高いように評価したことを報告した。2023年7月,UpRiのUplit臨床試験におけるデータが主な終点に達していないことを発表し,UpRiに関する開発活動を段階的に終了する予定であり,UpRiのアップグレード−AとUp−Next臨床試験を終了した。また,2023年3月にわれわれのXMT−2056の第1段階臨床試験はレベル5 SAEに達した後にFDAによって臨床的に棚上げされたことを発表した。FDAは2023年10月にこの臨床休止を廃止し,この臨床試験の登録回復に努めているが,現時点では患者登録はない。もし私たちや私たちの協力者が臨床試験に成功できなければ、私たちまたは彼らは規制部門の承認を得ることができず、私たちの技術に基づいて私たちの候補製品や私たちの協力者の候補製品を商業化することもできないだろう。
私たちの臨床試験で十分な数の患者を募集できないことは、私たちの候補製品コストの増加と開発周期の延長を招く可能性がある。
臨床試験には十分な患者登録が必要であり、これは多くの要素の関数である
•患者群の大きさと性質;
•調査中の病気の重症度は
•試験の資格基準を含む試験案の性質と複雑さ
•実験の設計
•臨床試験地点の数と患者のこれらの地点への接近度
•病気の看護基準を調査しました
•臨床研究者は条件に適合する患者の能力と約束を識別する
•研究中の薬物の他の既存療法に対する潜在的な優位性およびリスクに対する臨床医および患者の見方は、我々が調査している適応のための任意の新薬が承認される可能性がある;および
•臨床試験に参加した患者は,試験完了前に試験を終了するリスク,あるいは進行癌患者であるため,臨床試験の全期間を過ごすことができない。
また、私たちの臨床試験は他の臨床試験と私たちの現在と未来の候補製品と同じ治療分野の製品を奪い合う。この競争は、私たちの試験に参加することを選択する可能性のある患者の一部が、私たちの競争相手による試験に参加することを選択する可能性があるので、私たちが利用可能な患者の数およびタイプを減少させる。合格した臨床研究者の数が限られているため、著者らはいくつかの競争相手が使用した同じ臨床試験場所で著者らのいくつかの臨床試験を行い、これは著者らの臨床試験に応用できる患者数を減少させることが予想される。さらに、我々の現在および将来のいくつかの候補製品は、インターフェロン遺伝子免疫合成刺激または刺激剤プラットフォームに基づく製品を含むので、より一般的な癌治療方法(他の承認されたADC薬を含む)よりも革新的であり、潜在的患者およびその医師は、我々が行っているまたは任意の将来の臨床試験に参加する患者を募集するのではなく、従来の腫瘍療法または他の承認されたADC薬を使用する傾向があるかもしれない。
プロトコル基準に適合する臨床試験への適切な患者の募集および募集における挑戦は、コストを増加させる可能性があり、XMT−1660、XMT−2056、または任意の他の現在または将来の候補製品の開発計画のための現在の遅延をもたらす可能性がある。
私たちの候補製品は不良や予期せぬ深刻な副作用を引き起こす可能性があり、その規制部門の承認を延期または阻止し、承認されたラベルの商業イメージを制限したり、上場承認後に重大な負の結果を招く可能性がある(あれば)。
私たちの候補製品による不良または予期しない深刻な副作用は、私たちの臨床前研究を中断、延期、または停止させる可能性があり、または私たちまたは規制機関の臨床試験の中断、延期、または停止を招く可能性があり、より厳しいラベル、またはFDAまたは同様の外国の規制機関が規制承認を遅延または拒否する可能性がある。私たちが候補製品を開発している多くの重篤な疾患の治療法のように、私たちの候補製品を使用することは、死を含む治療に関連する重篤な有害事象またはTRAEを含む副作用を生じる可能性が高い。私たちの実験結果は、これらまたは他の副作用の重症度と流行度を明らかにする可能性があり、これは受け入れられない。この場合、私たちの実験は一時停止または終了される可能性があり、FDAまたは同様の外国規制機関は、私たちの任意またはすべての目標適応を承認する候補製品の開発を停止または拒否するように命令することができます。TRAEはまた,患者の募集や患者の試験完了能力に影響したり,潜在的な製品責任クレームを招いたりする可能性がある。このようなどんな状況でも、私たちの業務、財政状況、そして見通しに大きな被害を及ぼす可能性がある。
例えば,われわれは2023年にわれわれのDolaflexinプラットフォームを用いて開発したUpRiの臨床試験を中止し,この試験に参加した患者は死亡,出血,AST上昇,嘔気,血小板減少(血小板減少を含む),疲労,貧血,発熱,ALT上昇,血ALP/LDH上昇,蛋白尿,嘔吐,虚弱,下痢,頭痛,末梢神経病変,好中球減少,肺炎を含む重篤なTRAEを経験した。また,われわれのXMT−1592臨床試験でも重篤な貧血と肺炎TRAEを経験し,われわれは2022年5月にXMT−1592の開発を中止し,われわれのDolasyntenプラットフォームを用いて開発を行った。また,われわれのXMT−2056第一段階臨床試験はわれわれの免疫合成プラットフォームを用いて開発され,5級重篤な有害事象(SAE)が発生した後,FDAは2023年3月から2023年10月まで臨床試験を一時停止した
我々のDolasyntenプラットフォームを用いて開発したXMT−1660の第1段階臨床試験も行っている。我々の候補製品はいくつかを共有するが、すべてのプラットフォーム技術、ペイロード、および目標ではないので、任意の候補製品報告のセキュリティイベントがこれらの共有属性に関連しているかどうかを予測または評価することは困難である可能性がある。我々の候補製品(XMT−1660およびXMT−2056を含む)が開発された任意の段階では、死亡を引き起こす可能性のある副作用、または他のSAEまたは潜在的な安全問題を含む深刻なTRAEを含む非臨床研究または臨床試験において不良副作用が観察される可能性がある。任意のこのような深刻なTRAE、SAE、または他の潜在的なセキュリティ問題は、UpRi、XMT−1592または任意の他の候補製品の臨床試験において我々が以前に観察した他の深刻なTRAE、SAEまたは他の安全問題、またはこれらの問題を除いても類似している可能性がある。
また,われわれとわれわれの臨床試験調査者は現在,重篤な不良や副作用が薬物に関与しているかどうかを確認している。FDAまたは同様の規制機関は、私たちまたは私たちの臨床試験調査者の臨床試験データの解釈、および私たちまたは私たちの臨床試験調査者によって得られたSAEまたは副作用が薬物とは無関係であるという結論に同意しないかもしれない。FDAまたは同様の規制機関は、承認を支援する追加の臨床前または臨床データを含む、私たちの候補製品の安全性に関するより多くの情報を提供することを要求する可能性があり、これは、私たちに追加費用を発生させ、私たちのある候補製品の承認を延期または阻止し、および/または商業化計画を延期または変更させるか、または候補製品の開発を完全に放棄することを決定する可能性がある
また,設計により,臨床試験は潜在患者集団のサンプルに依存している。患者数と接触時間が限られているため、私たちの候補製品は希で深刻な副作用は、より多くの患者が候補製品に接触した時にのみ発見される可能性がある。もし私たちの候補製品が発売許可を得て、私たちあるいは他の人が承認を得た後にこのような候補製品による不良副作用を発見すれば、多くの潜在的な重大なマイナス結果を招く可能性がある:
•規制当局は、“ブラックボックス”警告や禁忌症のようなラベル説明書の追加を要求するかもしれない
•患者に配布するためにこのような副作用のリスクを概説するための薬物ガイドラインの作成を要求されるかもしれません
•規制当局は、医薬品ガイドライン、医師のコミュニケーション計画、または制限された分配方法、患者登録、および他のリスク最小化ツールのような安全な使用を確保する要素を含む可能性があるリスクを低減するためのREMS計画の策定を要求することができる
•私たちはこれらの候補製品の分配または管理方法を変更し、追加の臨床試験を行うか、または候補製品のラベルを変更することを要求されるかもしれない
•私たちは規制調査と政府の法執行行動の影響を受けるかもしれない
•規制部門はこのような製品候補製品の承認を撤回または制限することができる
•私たちはこれらの候補品を市場から除去することに決定するかもしれない
•私たちは起訴され、私たちの候補製品に接触したり服用したりする個人による傷害に責任を負うかもしれない
•私たちの名声は損なわれるかもしれない。
これらの事件のいずれも、特定の候補製品に対する市場の受け入れ度を達成または維持することを阻止することができ、承認されれば、私たちの業務、運営結果、および将来性を深刻に損なう可能性がある
同様に、我々の協力者または競合他社によって開発または商業化されたADCの不良または深刻な副作用は、FDAまたは同様の規制機関に行動をもたらす可能性があり、我々の候補製品の臨床試験または上場承認があれば、これらの候補製品を商業化する能力に実質的かつ悪影響を及ぼす可能性がある。
私たちは、潜在的な候補製品を開発しないことを選択することができ、1つまたは複数の発見または臨床前計画または候補製品を一時停止または終了することができる。
いつでも、任意の理由で、私たちは、私たちの1つまたは複数の発見計画、臨床前計画、または候補製品が、その計画または候補製品にリソースを割り当てることを保証するのに十分な潜在力がないことを決定するかもしれない。また,我々の財力と人的資源が限られているため,XMT-1660とXMT-2056,UpRiとXMT-1592を含む歴史的な製品の開発に重点を置いている.したがって、私たちは候補製品を開発しないことを選択することができ、私たちの1つまたは複数の発見または臨床前計画を一時停止または終了することを選択することもできる。もし私たちが大量の資源を投入した計画または候補製品を一時停止または終了すれば、私たちはすべての投資収益を提供できない計画または製品候補製品に資源を費やすだろう。例えば,2023年7月,われわれの第二段階Uplift臨床試験はその主要終点に達しなかったため,UpRiのさらなる開発中止を発表した。また,2022年5月にXMT−1592の開発中止が決定し,NaPi 2 bバイオマーカーの非小細胞肺癌(NSCLC)における罹患率の低さと,このような適応の競争が激しくなっていることが原因である。私たちはまた特定の適応のための候補製品の開発を止める可能性がある。例えば,2021年11月にUpRiを非小細胞肺癌患者への単一薬剤としての使用を中止することにし,卵巣癌患者に重点を置くことにした。したがって、我々は、既存または将来の計画または候補製品を含む、UpRiおよびXMT−1592を開発するために最初に使用されたリソースを潜在的により生産的な用途に割り当てる予期された機会を逃す可能性がある。もし私たちが特定の未来の候補製品の商業潜在力や目標市場を正確に評価しなければ、私たちは協力、許可、または他の印税手配を通じて未来の候補製品に対する貴重な権利を放棄するかもしれない。
私たちまたは私たちの協力者は他の潜在的な候補製品を発見して開発することができないかもしれない。
私たちと私たちの協力者が新製品候補製品を決定する研究計画は大量の技術、財政と人的資源を必要とし、私たちあるいは私たちの協力者は新製品候補製品を決定することに成功できないかもしれません。もし私たちまたは私たちの協力者が臨床前と臨床開発のために適切な他の候補製品を決定することができなければ、私たちまたは彼らが候補製品を開発する能力と、私たちの製品を商業化することによって収入を得ること、あるいは私たちの協力者が将来その製品を販売することから印税を得る能力が影響を受ける可能性があり、これは私たちの財務状況に重大な損害を与え、私たちの株価に悪影響を及ぼす可能性がある。
私たちの財務状況と追加資本需要に関連するリスク
私たちは私たちの目標を達成するために大量の追加資金を必要とし、必要な時に必要な資本を得ることができなければ、私たちの製品開発や将来の商業化努力を延期、制限、減少、または終了させることができるかもしれない。
2023年12月31日まで、私たちの現金、現金等価物、有価証券は2.091億ドルです。設立以来、私たちはすでに大量の現金を使用しており、予測可能な未来には、XMT-1660、XMT-2056、および任意の他の現在または未来の候補製品を開発するために多くの資源を費やし続けていくと予想される。これらの支出には、研究開発、臨床前研究および臨床試験、規制承認および製品の製造、および販売が許可された製品(あれば)および新しい技術を得る可能性のあるコストが含まれる可能性がある。さらに、他の予期しない費用も発生する可能性がある。著者らの計画と予想される臨床試験の結果は高度に不確定であるため、著者らは候補製品の開発と商業化に成功するために必要な実際の数量を合理的に見積もることができない。もし私たちが現在または未来の候補製品の臨床試験で任意の遅延に遭遇すれば、患者登録の遅延を含めて、私たちのコストは増加するだろう。将来的に上場企業として運営し、追加人員を募集し、私たちの施設を拡大する上で関連コストが生じる可能性もあります。
私たちの将来の資本需要は多くの要素に依存しています
•XMT-1660、XMT-2056、および任意の現在または未来の候補製品の研究と開発、ならびに臨床前研究と臨床試験を行う範囲、進捗、結果とコスト;
•承認および商業化のための規制準備のためのXMT−1660、XMT−2056、および臨床試験のための任意の他の現在または将来の候補製品のコスト;
•前臨床研究および臨床試験が成功した場合、XMT-1660、XMT-2056および任意の他の現在または将来の候補製品の規制承認を得る時間および関連するコスト;
•XMT−1660、XMT−2056、および任意の現在または将来の候補製品の商業化活動コストは、任意の候補製品が販売を許可された場合、製造、マーケティング、販売および流通コストを含む
•私たちは、戦略的協力、許可または他の手配、およびそのような合意の財務条項の能力を確立し、維持する
•請求項の準備、提出、起訴、保守、弁護、および執行に関連する費用は、訴訟費用および任意のそのような訴訟の結果を含む
•私たちの未来の製品または私たちの協力者が開発した製品の時間、収入、販売金額、または印税
•競争する癌療法や他の不利な市場の発展;
•任意のセット診断および/または補充診断の要件またはコストを開発する。
私たちは、私たちの現在の現金、現金等価物、および有価証券が、2026年まで私たちの現在の運営計画に資金を提供するのに十分な資金を提供すると信じている。しかし、私たちのこれらの推定は間違っていることが証明される可能性があるという仮定に基づいており、私たちの運営計画は現在私たちが知らない多くの要素によって変わる可能性があり、私たちは計画よりも早い追加資金が必要かもしれない。私たちが追加的な資金が必要な時、私たちは私たちが受け入れられる条項でこのような資金を得ることができないかもしれない。もし私たちが十分な資金をタイムリーに得ることができない場合、私たちは、私たちの1つまたは複数の候補製品の臨床前研究、臨床試験または他の開発活動の遅延、制限、減少、または終了を要求されるかもしれないし、私たちの将来の販売およびマーケティング能力の確立、または他のは、私たちの候補製品を商業化するために必要な活動である可能性がある。現在または将来の運営計画のために十分な資金があると考えても、有利な市場条件や戦略的考慮のため、追加的な資本を求めることができるかもしれない。
私たちは設立以来純損失が発生しており、商業販売のための製品は何も承認されていません。少なくとも今後数年以内に、大きな運営損失を受け続けると予想しています。私たちは絶対に利益を達成したり維持したりしないかもしれない。
設立以来、私たちは純損失を被った。2023年、2022年、2021年12月31日までの年度の純損失はそれぞれ171.7ドル、2.042億ドル、170.1ドルだった。2023年12月31日現在、私たちの累計赤字は8.264億ドルです。私たちの損失は主に私たちの発見と開発活動によるコストによるものです。私たちの純損失は四半期ごとと毎年大きく変動するかもしれません。私たちは今まで何の製品も商業化していませんし、販売製品から何の収入も得ていません。予測可能な未来には、私たちはいかなる製品収入も発生しないと予想されています。もし十分な製品販売収入がなければ、私たちは未来に永遠に利益を達成しないかもしれない
私たちは大部分の財務資源を研究開発に投入して、私たちの臨床と臨床前開発活動を含む。これまで、私たちの運営資金は、主に私たちの戦略協力収益、私たちの優先株の私募と私たちの普通株の公開、私たちの初公募株、2019年と2020年の後続公募株、そして私たちの市場での株式発行計画から来ています。私たちの未来の純損失額は私たちの未来の支出速度にある程度依存するだろう。著者らはまだ候補製品の重要な臨床試験を完成しておらず、現在或いは計画されている臨床試験中の候補製品の数は限られている。もしあれば、私たちは商業化できる候補製品を持つためにあと数年かかる。私たちが規制部門の承認を得て候補製品をマーケティングしても、私たちの将来の収入は、私たちの候補製品が承認された1つ以上の市場の規模と、私たちが十分な市場受容度、第三者支払者の清算、そしてこれらの市場での私たちの候補製品の十分な市場シェアを得る能力に依存するだろう。
私たちは今後数年間巨額の費用と運営損失が発生すると予想している。私たちは持続的な活動に関連した費用が増えるかもしれません
•XMT−1660およびXMT−2056の臨床開発および製造活動を継続する
•XMT−2068およびXMT−2175を含む他の候補製品の発見、検証、および開発活動を継続する
•私たちの協力の下で研究開発活動を展開し
•臨床試験を完成させるための現在と未来の候補製品は市場の承認を得ている
•第三者とのビジネス上実行可能な供給および製造関係の確立および維持を含む、私たちの候補製品のための持続可能かつ拡張可能な製造プロセスを開発すること
•競争的な技術や市場の発展に対応しています
•私たちの知的財産権の組み合わせを維持し、拡大し、保護する
•より多くの研究、開発、そして一般的で行政的な人たちを雇う。
FDAまたは任意の同等の外国規制機関が、現在行われていることに基づいて臨床試験または前臨床試験を行うことを要求する場合、またはXMT−1660、XMT−2056、または任意の他の現在または将来の候補製品の臨床試験を完了する上で何らかの遅延が生じた場合、私たちの費用は増加する可能性がある。
利益を維持するためには、規制部門の承認を得、規制部門の承認を得る可能性のある製品を製造、マーケティング、販売するために、私たちの候補製品の開発に成功しなければならない。私たちはこれらの活動で成功しないかもしれません。私たちは製品販売や戦略的協力から利益を達成するのに十分な収入を得ることはできないかもしれません。私たちが未来に利益を達成しても、私たちはその後の時期に利益を維持できないかもしれない。もし私たちが利益を上げたり、利益を維持できなければ、私たちの市場価値を下げ、資金を調達し、業務を拡大し、他の候補製品を発見したり、開発したり、運営を継続する能力を弱める可能性がある。
追加資本の調達は、私たちの既存の株主を希釈し、私たちの運営を制限するか、あるいは私たちの技術またはADC製品の候補製品に対する権利を放棄することを要求するかもしれません。
これまで、相当な製品収入を生み出すことができれば、私募と公募による株式発行、債務融資、協力、戦略連合、許可手配を含む様々な方法で資本需要に融資する予定です。私たちが株式または転換可能な債務証券を売却することによって追加資本を調達する場合、私たちの普通株主の所有権権益は希釈され、このような株式または転換可能な債務証券の条項は、清算または他の優先的、または他の方法で私たちの普通株主の権利に悪影響を及ぼす特典を含む可能性がある。追加の債務融資は、実行可能であれば、将来の債務の発生、資本支出の発表、配当の発表、または将来の債務を保証するために、私たちのいくつかの行動をとる能力を制限または制限する契約を含み、いずれも、私たちが業務を展開し、運営計画を実行する能力に悪影響を及ぼす可能性がある。もし私たちが第三者との戦略的協力を通じてより多くの資金を調達すれば、私たちは私たちのプラットフォームや候補製品を含む、私たちの技術に対する貴重な権利を放棄し、あるいは私たちに不利な条項でライセンスを付与しなければならないかもしれない。もし私たちが必要な時に株式または債務融資を通じてより多くの資金を調達できない場合、私たちはXMT-1660、XMT-2056、または任意の他の現在または未来の候補製品に対する製品開発または将来の商業化努力を延期、制限、減少、または終了する必要があるかもしれません。または第三者開発およびマーケティングは、私たちがより開発し、マーケティングしたい候補製品の権利を与えています。
私たちは私たちにいくつかの肯定的で否定的な条約を遵守し、私たちの経営と財政的柔軟性に制限を加えることを要求する信用計画を持っている。
2021年10月、私たちは新しい信用手配、オックスフォード金融有限責任会社を担保代理とし、貸手であるシリコンバレー銀行を貸手として、First-Citizens Bank&Trust Companyの子会社を貸手として、他の貸手または共同融資者として融資と安全協定を達成した。改訂された新しい信用手配によると、私たちはすでに2,500万ドルを借りていますが、改訂された新しい信用手配は追加の借入金額がありません。新しい信用配置は、知的財産(ただし、知的財産から支払いおよび収益を得る権利を含む)、および知的財産の負の質権を含まない、私たちが所有または後に獲得したほとんどの個人財産を担保とする。
新しい信用手配はまた慣例陳述と保証、肯定と否定契約、及び慣例違約事件を含む。いくつかの慣用的な負の条約は、私たちが未来の債務を発生させ、留置権を付与し、投資を行うこと、買収を行うこと、配当金を分配すること、特定の制限的な支払いおよび資産を売却する能力を制限するが、それぞれの場合にはいくつかの例外がある。私たちがこのような条約を遵守しないことは、融資と保証協定の下での違約事件を招き、新しい信用計画によって不足している債務の加速を招く可能性がある。
私たちは、より利益や成功の可能性が高い可能性の高い候補製品を利用することなく、特定の候補製品を追求するために資源を使うかもしれない。
私たちの財務と管理資源が限られているので、私たちは特定の候補製品に集中している。したがって、私たちは他の候補製品を探す機会を放棄したり延期したりするかもしれないが、これらの製品は後により大きな商業潜在力を持っていることが証明された。私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。潜在的な候補製品を正確に評価できなかったことは、市場潜在力の低い候補製品に重点を置いてしまう可能性があり、これは私たちの業務や財務状況を損なうことになる。現在および将来の研究開発計画および特定の適応の候補製品への支出は、いかなる商業的に実行可能な候補製品も生じない可能性がある。もし私たちが特定の候補製品の商業的潜在力や目標市場を正確に評価しなければ、私たちは協力、許可、または他の印税手配によって候補製品に価値のある権利を放棄するかもしれないが、この場合、候補製品の独占開発権と商業化権利を維持することは私たちに有利だ。
私たちの第三者への依存に関するリスクは
私たちは第三者メーカーとサプライヤーに依存しているため、私たちの研究開発、臨床前と臨床開発材料の供給は限られたり中断したりする可能性があり、あるいは数量や品質は満足できないかもしれない。
私たちは第三者契約メーカーに依存して、私たちの臨床前と臨床試験製品の供給を生産し、私たちの現在の協力によって私たちの製造義務を支持しますが、私たちはいかなる臨床または商業規模の候補製品を製造するための内部資源と能力が不足しています。私たちの契約製造業者が活性医薬成分および最終医薬製品を生産するための施設は、FDAおよび他の同様の外国規制機関が受け入れることができなければならず、検査は、適用される規制機関にマーケティング申請または関連する外国規制書類を提出した後に行われる。著者らの臨床前と臨床開発製品の供給が十分で、途切れない或いは品質が満足できることを保証することができず、或いは引き続き受け入れ可能な価格で供給する。もし私たちの契約メーカーが私たちの規格やFDAまたは適用される外国規制機関の厳しい規制要件に適合した材料を生産することに成功しなければ、彼らはその製造施設の規制承認を確保または維持することができないだろう。また、地政学的事件が私たちのコントロール範囲や私たちの契約メーカーの制御範囲を超えて業績障害をもたらし、彼らが私たちに製造品を生産したり、私たちに製品を渡す能力を阻害したら、十分な臨床前と臨床開発製品の在庫を確保できないかもしれません。私たちのメーカーのどの交換にも大量の努力と専門知識が必要です。合格した交換数は限られているかもしれません。
候補製品の製造過程はFDAと外国規制機関の審査を受けなければならない。サプライヤーとメーカーは適用された製造要求を満たし、監督機関の要求された厳格な施設と技術検証テストを受けなければならず、現在の良好な製造実践のような監督基準に適合する。私たちの契約製造業者が十分な品質管理、品質保証、合格者の能力を維持することを直接制御することはできません。もし私たちのどのメーカーも規制要件を遵守していない場合、または品質、時間、または他の側面での私たちの義務を履行していない場合、または私たちの部品や他の材料の供給が他の理由で限られたり、中断されたりした場合、私たちは自分で材料を製造することを余儀なくされるかもしれませんが、私たちは現在能力や資源を持っていない、あるいは他の第三者と合意している場合、私たちは合理的な条項でそうすることができないかもしれません。場合によっては、我々の候補製品を製造するために必要な技術的スキルまたは技術は、元の製造業者固有または独自である可能性があり、そのようなスキルまたは技術を他の第三者に譲渡することは困難である可能性があり、実行可能な代替案が存在しない可能性がある。これらの要素は、私たちのこれらの製造業者への依存を増加させ、あるいは他の第三者が私たちの候補製品を生産するために、これらのメーカーからライセンスを取得することを要求するだろう。もし私たちが何らかの理由でメーカーの交換を要求された場合、新しいメーカーの施設とプログラムが品質基準とすべての適用された法規とガイドラインに適合しているかどうかを確認するように要求されます。新メーカーの検証に関する遅延は、タイムリーまたは予算内で候補製品を開発する能力に悪影響を及ぼす可能性があります。私たちの契約製造業者への依存はまた、彼らまたはその施設にアクセスする権利のある第三者が、私たちの商業秘密や他の固有の情報を取得し、占領する可能性に直面させる。
もし私たちが規制部門の任意の候補製品の承認を得たら、私たちは引き続き第三者メーカーに依存すると予想される。私たちが第三者とすでにまたは将来製造手配を達成した範囲内で、私たちはこれらの第三者に依存して、品質管理と保証に関する要求を含む契約と監督管理の要求に適合する方法でその義務を適時に履行する。もし私たちが候補製品の第三者製造を獲得したり維持したり、商業的に合理的な条項でそうすることができなければ、私たちは候補製品の開発と商業化に成功できないかもしれない。私たちまたは第三者が私たちの製造要件を実行し、現在の良好な製造実践またはcGMPを遵守できなかった場合、様々な態様で私たちのビジネスに悪影響を及ぼす可能性があります
•開発中の候補製品の臨床試験を遅延または開始または継続できない;
•規制申請の提出を遅延したり、規制部門の候補製品の承認を遅延させたりすることができなかった
•既存または未来の戦略的パートナーの協力を失う;
•第三者製造施設や私たちの製造施設に対して規制部門の追加検査を行う
•私たちの候補製品ロットの流通を停止したり、リコールしたりすることを要求する
•製品候補製品の発売と商業化が許可された場合、私たちの製品のビジネスニーズを満たすことができない
•罰金、否定的な宣伝、そして民事と刑事法執行と制裁。
私たちまたは私たちの第三者製造業者は、十分な品質と数量で私たちのADC候補製品の生産規模を拡大することに成功できないかもしれません。これは、私たちが私たちのADC候補製品を開発し、承認された製品を商業化することを延期または阻止することになります。
私たちの候補製品の臨床試験を行い、任意の承認された候補製品を商業化するためには、私たちまたは私たちの第三者メーカーはこれらの製品を大量に生産する必要があるだろう。私たちまたは私たちの第三者製造業者は、私たちの任意の候補製品の製造能力をタイムリーに、または費用効果的に向上させることに成功できないかもしれません。また,拡張活動中に品質の問題が生じる可能性がある.もし私たちまたは任意の第三者製造業者が十分な品質と数量で私たちの候補製品の生産規模を成功的に拡大することができなければ、その候補製品の開発、テスト、および臨床試験は延期または不可能になる可能性があり、いかなる最終製品の規制承認や商業発表は延期または獲得できない可能性があり、これは私たちの業務を深刻に損なう可能性がある。もし私たちが第三者が私たちの候補製品を商業的に供給するために製造したり、商業的に合理的な条項でそうすることができなければ、私たちの候補製品の開発と商業化に成功できないかもしれない。
私たちは、サードパーティがXMT-1660、XMT-2056、および私たちの他の候補製品のための臨床前研究および臨床試験を行うことに依存しており、これらの第三者が我々の義務を正確かつタイムリーかつ成功的に履行していなければ、XMT-1660、XMT-2056、または任意の他の現在または未来のADC候補製品の規制承認を得ることができないかもしれない。
われわれは進行中のXMT−1660とXMT−2056の臨床試験を設計し,XMT−1592の試験は2022年に終了し,われわれのUpRiの向上,アップグレード−AとUp−Next臨床試験は,2023年にこれらの試験の開発を中止し,任意の未来の候補製品のために任意の未来の臨床試験を設計する予定であり,臨床前研究が成功すれば,戦略パートナーがこのような試験設計を担当していない。しかしながら、我々は、管理、監視、および他の方法での多くのこのような試験を実行するために、CRO、SMO、臨床サイト、研究者、および他の第三者に依存している。したがって,われわれ自身に完全に依存しているスタッフに比べて,これらの臨床試験の進行,スケジュールと完成および臨床試験により開発されたデータの管理の直接制御は少ない。これらのCRO、SMO、調査員、他の第三者は私たちの従業員ではなく、私たちのプロジェクトに使用する時間と資源の制御は限られています。私たちは他の多くの会社とこのような第三者の資源を争っている。これらの第三者は他のエンティティと契約関係にある可能性があり、その中のいくつかは私たちの競争相手である可能性があり、これは私たちの計画から時間と資源を消費するかもしれない。著者らが契約した第三者は著者らの臨床前研究或いは臨床試験を行う時に勤勉、慎重或いは適時でない可能性があり、或いは現在の良好な実験室実践或いは現在の良好な臨床実践(状況によります)を守らず、それによって臨床前研究或いは臨床試験の遅延或いは不成功を招く。
私たちが通常依存している第三者はいつでも彼らの契約を終了することができ、代替手配を達成しなければならないことは、候補製品の開発と商業化を延期します。外部の当事者とのコミュニケーションも挑戦的である可能性があり、ミスや協調活動の困難を招く可能性がある。外部の当事者は
•人員配置が困難である
•契約義務を履行しない者
•規制コンプライアンスの問題に直面しています
•優先順位が変化したり、財務的苦境に陥ったりする
•他の実体との関係を構築し、その中のいくつかは私たちの競争相手かもしれない。
FDAと類似の外国監督機関は、データと結果が信頼性と正確であることを保証し、試験参加者の権利、完全性と機密性を保護するために、GCPを含む、臨床試験結果を設計、進行、監視、記録、分析と報告する際に法規と標準を遵守することを要求する。私たちは第三者に依存して引き続き私たちの臨床試験を行うつもりであるが、彼らは私たちの従業員ではなく、私たちはすべての臨床試験がその全体的な研究計画、方案とその他の要求に従って行われることを保証する責任がある。私たちのこれらの第三者の研究開発活動への依存は、これらの活動に対する私たちの統制を減少させるだろうが、私たちの責任を軽減することはない。私たちの臨床試験中の任意の法律または法規違反行為について、私たちは、最高刑事起訴までの民事罰を含む可能性のある無見出しの警告状または法執行行動を受ける可能性があります。
もしこれらの第三者がそのプロトコル下の職責を成功裏に履行できなかった場合、もし彼らが獲得したデータの品質または正確性が彼らが臨床試験規程または規制要求を遵守できなかったことによって影響を受けた場合、あるいは彼らが臨床試験規程を遵守できなかった場合、あるいは予想された期限内に完成できなかった場合、私たちの候補製品の臨床試験は規制要求に適合しない可能性がある。FDAは臨床試験スポンサー,主要研究者,試験地点の定期検査によりGCP規定を実行している。もし私たちまたは私たちのCROが適用されたGCPまたは他の法規の要求を遵守できなかった場合、私たちの臨床試験で発生した臨床データは信頼できないと思われる可能性があり、第三者を交換する必要があるかもしれず、私たちはマイナスの宣伝、罰金と民事或いは刑事制裁を受ける可能性があり、臨床前開発活動或いは臨床試験は延長、延期、一時停止或いは終了する可能性がある。上記のいずれかの事件が発生した場合、私たちは規制部門の候補製品に対する承認をタイムリーにあるいは根本的に得ることができないかもしれない。
私たちは他社とのいくつかの戦略関係に依存して、私たちのADCプラットフォームとADC候補製品の研究、開発、商業化を支援します。もし私たちの既存の重要なパートナーのパフォーマンスが期待されていない場合、これは、ADC候補製品を商業化したり、技術許可によって収入を得る能力に悪影響を与えたり、私たちの業務に負の影響を与える可能性があります。
私たちはすでに戦略協力を構築し、第三者と戦略的協力や他の関係を構築し続けて、私たちのプラットフォームと既存と未来の候補製品を研究、開発し、商業化するつもりです。2022年12月、私たちはメルクKGaA、ドイツのダムシュタットまたはメルクKGaAの付属会社Ares Trading、S.A.と協力と許可協定を締結し、私たちの免疫合成プラットフォームを用いて候補ADC製品の研究、開発と商業化を行い、2022年2月にヤンソンバイオテクノロジー会社またはジョンソンと協力協定を締結し、私たちのDolasynThenプラットフォームを用いて候補ADC製品の研究、開発、商業化を行った。また、2022年8月には、グラクソ·スミスクライン知的財産権(第4号)有限公司とオプション、協力、ライセンス契約を締結し、この合意に基づき、グラクソ·スミスクラインに独占的な選択権を付与し、独占的な許可を得て、XMT-2056を含む製品を共同開発して商業化した。これらの計画によると、私たちは私たちの協力者に依存して彼らの臨床試験を設計し、行います。したがって、私たちの協力者がこれらの計画を実行することを制御したり監督したりすることはできません。これらの計画は成功しないかもしれません。これは私たちの業務運営にマイナスの影響を与える可能性があります。さらに、これらの協力者のいずれかが、これらの計画または提案された製品の支援を撤回するか、または他の方法で彼らの開発を損なうか、または負の結果に遭遇した場合、私たちの業務および私たちの候補製品は負の影響を受ける可能性がある。
場合によっては、私たちの協力者は理由で私たちとの合意を終了するかもしれないし、場合によっては私たちとの合意を勝手に終了し、私たちの技術の使用を停止するかもしれない。さらに、私たちの協力者が私たちの技術を使用したり、採用したりする製品に投入される可能性のある資源の数量と時間を制御することはできません。また,我々と協力者との関係は,我々の科学者や管理チームの多大な時間と労力を分散させ,我々の資源を複数の内部や連携プロジェクトに効率的に割り当てる必要がある可能性がある.私たちの協力者は協力協定で規定された義務を履行できないかもしれないし、直ちに義務を履行できないかもしれない。もし私たちの協力者と私たちの間に衝突が発生すれば、相手は私たちに不利な方法で行動し、私たちの戦略を実施する能力を制限するかもしれない。もし私たちの任意の重要なパートナーが私たちと彼らとの合意を終了したり、違反したり、他の方法で彼らの義務をタイムリーに達成できなかったり、またはGSKがその共同開発および商業化XMT-2056のライセンス選択権を行使しないことを最終的に決定した場合、技術アクセスおよび許可料、マイルストーンおよび特許権使用料、開発コストの精算を減少または除去することによって、より多くの努力を投入し、製品候補製品の内部開発に関連するコストを発生させることが要求され、財務状況に悪影響を及ぼす可能性があります。また、私たちの協力者が私たちの候補製品や候補協力製品に関する計画を優先的に順位付けして十分な資源を投入しなければ、私たちまたは私たちの協力者はこれらの候補製品を商業化できない可能性があり、これは私たちの収入と利益を創出する能力を制限するだろう。
我々の協力者は,競合製品,治療法または技術を単独で開発し,我々または我々の協力者が対象とする疾患の治療法を開発することができる。競合製品は、私たちの協力者によって開発されたものであっても、私たちの協力者が使用する権利があるものであっても、協力者が私たちの候補製品の支持を撤回することにつながる可能性があります。我々の協力者が戦略関係に貢献し続けても、彼らはいかなる結果製品を積極的に開発しないか、商業化することを決定する可能性がある。さらに、もし私たちの協力者が私たちのプラットフォームまたは技術に基づいて彼らの候補製品に対して異なる臨床的または監督管理戦略をとる場合、彼らの候補製品の有害事象は、類似技術を使用する候補製品に負の影響を与える可能性がある。このような進展のいずれも私たちの製品開発努力を損なう可能性がある。
今まで、私たちの収入の大部分は少数の協力者に依存してきた。このような協力者たちのいずれかを失うことは、私たちが予想された収入支払いを達成できないことを招くかもしれない。
私たちは限られた数の会社と戦略的協力に到達した。これまで、私たちの収入の大部分は、私たちの戦略パートナーといくつかの合意に基づいて支払われたお金から来ており、私たちの収入の一部は戦略的協力から続くと予想されています。私たちの任意の協力者を失ったり、私たちの協力者が私たちとの合意に基づいて負う義務を履行できなかった場合、許可や技術費用の支払い、マイルストーンの支払い、印税または精算を含めて、私たちの財務業績に重大な悪影響を及ぼす可能性があります。私たちの既存と未来の戦略協力下での支払いも時間と金額に大きな変動の影響を受け、これは私たちの収入が証券アナリストや投資家の予想を下回って、私たちの株価を下落させる可能性がある。
私たちはより多くの戦略的協力を求めるかもしれませんが、ビジネス的に合理的な条件でそれらを構築したり維持したりできなければ、私たちの開発と商業化計画を変えなければならないかもしれません。
私たちは引き続き戦略的に私たちの協力を評価し、適切な状況で、将来的には主要なバイオテクノロジーやバイオ製薬会社との協力を含むより多くの戦略的協力を行うことが予想される。私たちは私たちの製品候補とプラットフォームのために適切なパートナーを探す面で激しい競争に直面しており、交渉過程は時間がかかって複雑である。私たちが私たちのプラットフォームを利用したり、私たちの候補製品を向上させるために、第三者との協力を成功させるために、潜在的な協力者は、私たちが求めている条項と他の会社が許可できるプラットフォームと製品だから、これらのプラットフォームと候補製品を、彼らが魅力的だと思う市場で経済的価値のある製品とみなさなければならない。たとえ私たちが戦略協力の構築に成功したとしても、私たちが合意した条項は私たちに不利になる可能性があり、例えば、候補製品の開発や承認が延期または承認された製品販売に失望した場合、私たちはこのような戦略的協力を維持できないかもしれない。我々の候補製品またはプラットフォームに関連する戦略協力協定の任意の遅延は、既存または将来の候補製品の開発および商業化を遅延させ、市場に進出してもそれらの競争力を低下させる可能性がある。もし私たちの期待や業界アナリストの予想に基づいて私たちの戦略的協力の下で収入を生み出すことができなければ、この失敗は私たちの業務を損ない、直ちに私たちの普通株の取引価格に悪影響を及ぼす可能性がある。
もし私たちが私たちの候補製品に関する他の戦略的協力を確立し、維持することができず、私たちはまだ戦略的協力を達成していない場合、私たちはそのような候補製品の開発に関連するすべてのリスクとコストを負担し、追加の融資を求め、追加の従業員を雇用したり、他の方法で予算のない追加の専門知識を発展させる必要があるかもしれない。もし私たちが追加資金を求めたり、より多くの従業員を雇用したり、より多くの専門知識を開発することができなければ、必要であれば、私たちの現金消費率が増加するか、あるいは私たちの候補製品開発速度を下げる措置が必要になるだろう。これは私たちが現在パートナーを持っていない候補製品の開発に否定的な影響を及ぼすかもしれない。
ADC候補製品の商業化に関するリスク
私たちの未来の商業成功は私たちのADC製品の候補製品が承認されれば、医者、患者と医療支払者の中で著しい市場受容度を得ることができるかどうかにかかっている。
私たちが将来開発または買収する可能性のある他の現在または将来の候補製品に対する規制部門の承認を得ても、この候補製品は医師、医療支払者、患者、およびより広範な医療コミュニティの市場受け入れを得ることができないかもしれない。すべての承認された製品に対する市場の受け入れ度は、多くの要素に依存する
•臨床試験で証明されたこの製品の有効性と安全性
•製品が承認された適応および規制部門は、ラベル上で要求される可能性のある任意の警告を含む製品と共に使用されるラベルを承認する
•医師と患者は安全で効果的な治療法としてこの製品を受け入れる
•代替療法と比較して、治療のコスト、安全性、有効性
•第三者支払者と政府当局が十分な補償と定価を持っているかどうか
•相手が便利で管理しやすい
•副作用の発生率と重症度
•私たちの販売とマーケティング努力の有効性。
どの製品に対する見方も競争相手製品に対する見方の影響を受けます。したがって、私たちの競争相手製品に対する大衆の否定的な見方は私たちの候補製品の市場受容度にマイナス影響を与えるかもしれない。市場受容度は私たちが相当な収入と収益性を作るために必須的だ。任意の候補治療薬は,承認され商業化されれば,限られた能力でしか受け入れられないか,あるいはまったく受け入れられない可能性がある。もしどんな承認された製品も市場に受け入れられなければ、私たちは相当な収入を生むことができないかもしれません。私たちの業務は影響を受けるでしょう。
著者らの候補薬物の目標患者群の発病率と流行率はまだ正確に確定されていない。もし私たちの候補薬物の市場機会が私たちが推定したものよりも小さい場合、または私たちが得た任意の承認が患者集団のより狭い定義に基づいている場合、私たちの収入と利益を達成する能力は不利な影響を受け、実質的な可能性がある。
B 7-H 4を発現する癌とヒト表皮増殖因子受容体2-或いはHER 2-を発現する癌の正確な発病率と流行率はまだ確定されていない。これらの疾患に罹患している人数および候補製品治療から利益を得る可能性のある人数の推定値は推定に基づいている。XMT−1660、XMT−2056、または我々の現在または将来の任意の他の候補製品の総潜在的市場機会は、最終的には、そのような各候補製品の最終ラベルに含まれる診断基準(私たちの候補製品がこれらの適応のための販売が許可された場合)、医学界の受容度、ならびに患者参入、薬品定価および精算に依存するであろう。XMT-1660、XMT-2056、または私たちの現在または未来の任意の他の候補製品が治療可能な患者の数は予想を下回る可能性があり、患者は他の方法で私たちの薬物治療を受けることができないかもしれない、または私たちは新しい患者を識別または接触する上でますます大きな困難に直面する可能性があり、これらのすべては私たちの手術結果および業務に悪影響を及ぼすであろう。
もし私たちが販売、マーケティング、流通能力を確立できなければ、私たちの候補製品が承認されれば、私たちはそれを商業化することに成功できないかもしれない。
私たちは販売やマーケティングインフラもなく、販売、マーケティング、流通製品の経験もありません。上場承認された任意の製品をビジネスに成功させるためには、販売·マーケティング組織を構築したり、そのような販売·マーケティングの協力手配を求めたりする必要がある。
将来的には、米国およびいくつかの外国司法管轄区域でXMT-1660および任意の他の現在または将来の候補製品を販売するための重要な販売およびマーケティングインフラを構築したいと考えており、もしそれらが承認されれば、XMT-2056にそうするかもしれない。私たち自身の販売、マーケティング、そして流通能力を確立することはリスクに関するものだ。例えば、販売チームの採用と訓練は高価で時間がかかり、どんな製品の発表も延期される可能性がある。販売チームを募集し、マーケティング能力を確立する候補製品の商業発表が何らかの理由で延期または発生していない場合、これらの商業化費用を早期または不必要に発生させる。これは費用が高いかもしれません。もし私たちが私たちの販売とマーケティング担当者を維持したり再配置できなければ、私たちの投資は損失します。
私たち自身が製品を商業化するのを阻害するかもしれません
•十分な数の効果的な販売とマーケティング担当者を募集し、訓練し、維持することはできません
•販売員は医者に触れることができない
•未来の製品を処方するのに十分な数の医師が足りません
•販売者が提供するセット製品の不足は、より広い製品ラインを持つ会社と比較して競争劣勢になる可能性がある
•独立した販売およびマーケティング組織の作成に関連する予測不可能なコストおよび費用。
もし私たちが自分の販売、マーケティング、流通能力を確立し、第三者と合意してこれらのサービスを提供することができなければ、私たちの製品収入と私たちの収益力(もしあれば)は、私たちが自分で開発したどの製品も私たちのマーケティング、販売、流通よりも低くなるかもしれない。
さらに、私たちは第三者との合意に成功しないかもしれません。アメリカ国外で私たちのいくつかの候補製品を販売、マーケティング、流通するか、または私たちに有利な条項でそうすることができないかもしれません。私たちはこれらの第三者に対して限られた支配権を持っているかもしれません。彼らのいずれも必要な資源と注意力を投入して、私たちの製品を効果的に販売し、マーケティングすることができないかもしれません。もし私たちが販売、マーケティング、流通能力を成功的に確立できなければ、私たち自身も第三者と協力しても、私たちの候補製品を商業化することに成功しないだろう。
私たちのADC候補製品のいくつかの細分化市場での精算は限られているか、使えないかもしれません。これは私たちの製品を利益的に販売することを難しくするかもしれません。
国内外市場では、私たちの任意の候補製品の販売が承認されれば、政府医療計画、商業保険、管理医療機関のような第三者支払者がどの程度支払うかにある程度依存する。これらの第三者支払人はどの薬品が保険範囲に入るかを決定し、これらの薬品の精算レベルを確立する。医療費の抑制は国内外の政府および個人第三者支払者の優先順位となっている。薬品価格はこの努力の重点だった。政府や個人第三者支払者は、特定の薬物のカバー範囲や精算額を制限することでコストを抑制しようとしており、候補製品を販売する収益性に影響を与える可能性がある。コスト制御措置は、製品のために制定される可能性のある価格を低下させる可能性があり、これは製品収入が予想を下回る可能性があります。
第三者支払人の精算は、第三者支払人の製品の使用が以下の条件を満たすかどうかの決定を含む多くの要素に依存する可能性がある
•健康計画の下で保障された福祉
•安全で効果的で医学的に必要なものです
•特定の患者に適しています
•費用対効果があります
•実験的でも調査的でもない。
不利な価格設定制限は、これらの候補製品が市場承認を得たとしても、XMT-1660、XMT-2056、または任意の他の現在または将来の候補製品への投資を回収する能力を阻害する可能性がある。
政府または他の第三者支払人から製品の保証と精算承認を得ることは時間がかかり、高価なプロセスであり、支払人に私たちの製品の使用を支援する科学的、臨床的、費用効果的なデータを提供する必要があるかもしれない。また,第三者支払者のカバー範囲や新規承認薬品の精算に関する不確実性が大きい。私たちは保証と補償の承認を得るのに十分なデータを提供できないかもしれない。私たちは私たちのどんな候補製品も保険や十分な精算を受けることができることを確実にすることができない。また、精算金額が私たちの製品への需要を減らしたり、私たちの製品の価格を下げたりしないことは確認できません。もし精算が得られない場合や限定精算だけでは、私たちは特定の製品を商業化できないかもしれない。また,米国では,第三者決済者が新薬のカバー範囲や精算レベルを制限することで医療コストを抑制しようとするようになってきている。したがって,第三者支払者が患者に新たに承認された薬物を多少補償するかどうかには大きな不確実性があり,逆に薬品定価に圧力を与える。製造業者はまた、販売または有利なカバレッジを達成するために、価格割引を提供することを要求される可能性がある。
米国や海外市場では価格規制が実施される可能性があり、これは私たちの将来の収益力に悪影響を及ぼす可能性がある。
アメリカでは、薬品価格はますます審査と立法行動の影響を受け、政府のこのような製品コストに対する監督管理を実施する。また、一部の外国国では、欧州連合加盟国を含め、処方薬の価格設定は政府によってコントロールされている。他の国は処方薬の価格設定に対して似たような接近をするかもしれない。これらの国では、製品の発売許可を受けた後、政府当局との定価交渉にかなりの時間がかかる可能性がある。また、費用抑制措置の一部として、各国政府や他の利害関係者は価格や補償レベルにかなりの圧力をかける可能性がある。政治、経済、規制面の事態は定価交渉をさらに複雑化させる可能性があり、補償を受けた後、定価交渉が継続される可能性がある。EU加盟国が使用する参考価格と平行分配、あるいは低価格と高価な会員国の間の裁定は、価格をさらに下げる可能性がある。いくつかの国では、私たちの候補製品の費用効果を他の既存療法と比較して、精算または価格設定の承認を得るか維持するために、臨床試験または他の試験を行う必要があるかもしれない。私たちはそのような価格と精算が私たちや私たちの戦略パートナーに受け入れられるかどうかを確信できない。第三者支払者や主管当局が割引を公表することは、公布国や他の国の価格や補償レベルにさらなる圧力を与える可能性がある。価格設定が満足できないレベルに設定されている場合、または私たちの製品が精算できない場合、または範囲または金額に制限された場合、私たちまたは私たちの戦略パートナーの販売収入およびこれらの国における候補製品の潜在的な収益力は否定的な影響を受けるだろう。
私たちは激しい競争に直面しています。もし私たちの競争相手が私たちの現在または未来の任意の候補製品よりも効果的で、より安全で、あるいはより安い製品を開発·販売すれば、私たちのビジネス機会は負の影響を受けるでしょう。
生物技術と生物製薬業界の特徴は技術が迅速に進歩し、競争が激しく、独自製品を高度に重視していることである。多くの第三者が癌治療の様々な方法の開発に競争している。それらは製薬会社、バイオテクノロジー会社、学術機関、そして他の研究機関を含む。私たちの競争相手が開発したどんな治療法も候補品より優れているかもしれません。これらの競争相手は、私たちのプラットフォームや候補製品よりも効率的な技術の開発に成功したり、私たちのプラットフォームを時代遅れにしたり、競争力に欠けたり、経済的ではないかもしれない。より多くの会社が私たちの市場に参入し,他の癌療法をめぐる科学開発が加速し続けるにつれて,将来的にはますます激しい競争に直面することが予想される。
ADC技術を持つ複数の会社が私たちのプラットフォームと競争する可能性があり、これらの会社または彼らのパートナーおよび協力者が、私たちの現在および未来の候補製品と同じ指標を持つ候補製品を開発する可能性があることも知っている。複数の会社でも,異なる免疫刺激方法を採用しているにもかかわらず,我々の目標の同じバイオマーカーに対するADC,あるいは異なる免疫合成候補製品と競合する可能性のあるADCを開発している。私たちは私たちの革新技術と私たちのADCが他の候補製品と比較した有効性、安全性、耐性に基づいて競争したいが、もし私たちのADCがこれらの点で明らかな優位性がなければ、私たちは効果的に競争できないかもしれない。私たちが将来開発する可能性のある製品も他の製品や療法からの競争に直面する可能性があり、その中のいくつかはまだ意識されていないかもしれない。
私たちと比べ、私たちの多くの競争相手は研究開発、製造、臨床前研究、臨床試験を行い、監督管理の許可とマーケティングの面でもっと多くの財力と専門知識を持っている。また、多くの競争相手は特許保護や許可手配を積極的に求め、彼らが開発した技術を使って使用料を徴収することを期待している。特に,大手製薬会社は臨床試験,市場承認,臨床試験地点の構築,患者募集や薬品製造において豊富な経験を有しており,我々の分野の製品の発見,開発,商業化に成功する可能性がある。規模が小さいかスタートアップ段階にある会社も重要な競争相手となる可能性があり、特に大手や成熟会社との戦略関係を通じて。これらの第三者は、合格した科学·管理職の採用と維持、私たちのプロジェクトと相補的な技術の獲得で競争しています。
さらに、私たちの候補製品が承認されて商業化されれば、私たちは生物模倣薬からの競争に直面するかもしれない。2010年3月の“医療改革法案”の成立に伴い,生体模倣薬の市場進出経路が確立された。2009年の生物製品価格競争と革新法案、またはBPCIAは、FDAが後続の生物製品を承認するための方法を確立し、参考製品に12年間のデータ排他性を提供した。BPCIAは複雑であり、FDAによって解釈され、実施され続けている。また、政府の提案は12年間の参考製品専門期間を短縮しようとしている。また,“BPCIA”は“全体医療改革法案”の一部として公布されているため,現在のこの法案に対する訴訟挑戦は“BPCIA”の有効性に影響する可能性があり,以下ではより詳細な検討を行う。そのため、“BPCIA”の最終的な影響、実行と規制解釈についてはまだ重大な不確実性が存在する。
ヨーロッパでは、ヨーロッパ薬品管理局(European Medicines Agency、EMAと略称する)は簡略化経路を通じて製品を承認するガイドラインを発表し、生物模倣薬もすでにヨーロッパで承認された。もし私たちの潜在製品の生物類似バージョンがアメリカやヨーロッパで承認されれば、潜在製品の販売と毛利益および私たちの財務状況に負の影響を与えるかもしれない。
私たちの現在と潜在的な未来の候補製品について、私たちは、低コストで製造され、成功的にマーケティングできる製品を効果的に競争し、開発することができるかどうかは、私たちの能力にかかっていると信じています
•私たちの技術プラットフォームを向上させ
•私たちの技術と製品の知的財産権保護を獲得して維持します
•必要な政府と他の公共と個人の承認をタイムリーに得る
•重要な人材を誘致し、維持する
•効率的にビジネス化しています
•承認された適応に基づいて私たちの製品の精算を受けました
•私たちの製品の商業化に関連する適用法律、法規、規制要件および制限を遵守し、任意の変更または増加した規制制限を含む
•私たちの候補製品の開発と商業化を進めるために、より多くの戦略的協力を加える。
私たちの知的財産権に関するリスクは
もし私たちが私たちの技術やADC候補製品に関連する知的財産権を獲得したり保護したりできない場合、あるいは私たちの知的財産権が不足している場合、効果的に競争できないかもしれません。
私たちの成功は、私たちの知的財産権とノウハウの保護を獲得し、維持する能力に大きく依存する。我々は、XMT-1660、XMT-2056、XMT-2068、およびXMT-2175を含む、特許、商業秘密、および機密技術保護および秘密協定の組み合わせによって、我々のプラットフォームおよび私たちの候補製品に関連する知的財産権を保護する。生物製薬会社の特許地位は通常不確定であり、複雑な法律や事実を考慮しているため、近年多くの訴訟の対象となっている。したがって,我々の特許権の発行,範囲,有効性,実行可能性,商業的価値は高い不確実性を持っている.米国特許商標局(USPTO)と外国特許庁が特許を付与する際に採用する基準は,常に統一的または予測可能であるわけではない。例えば、特許出願または特許において許容される特許請求の範囲については、世界的に統一された政策はない。また,米国や他の国の特許法や特許法解釈の変化は,我々の特許の価値を低下させたり,我々の特許保護の範囲を縮小したりする可能性がある。特許訴訟過程は高価で、複雑で、時間がかかり、私たちは必要または望ましいすべての特許および特許出願を合理的なコストまたはタイムリーに提出、起訴、維持、強制執行または許可することができないかもしれない。我々は,特許保護を得るのが遅くなる前に,我々の研究開発成果で特許を申請できることを決定できなかった可能性もある.私たちの特許および特許出願に関連するすべての潜在的に関連する以前の技術が発見されたことは保証されない。私たちは、既存技術が発行された特許を無効にするために使用されるか、または私たちの係属中の特許出願を特許発行として阻止するために使用されることができることを知らないかもしれない。
私たちが所有しているまたは許可されている特許出願は、発行された特許を生成できない可能性があり、たとえそれらが特許として発行されていても、このような特許は、米国または他の国/地域における私たちのプラットフォームおよび候補製品をカバーしていない可能性がある。特許の発行は,その発明性,範囲,有効性または実行可能性に関する決定的な要素ではなく,我々の特許は米国や海外の裁判所または特許庁で挑戦される可能性がある。このような挑戦は、排他的な喪失または特許主張の縮小、無効、または実行不能をもたらす可能性があり、これは、他人が類似または同じ技術および製品を使用することを阻止するか、またはそれを商業化する能力を制限するか、または我々の技術および候補製品の特許保護期間を制限する可能性がある。例えば、たとえ私たちが許可または所有している特許出願が確実に特許として発行されたとしても、そのような特許が私たちのプラットフォームおよび候補製品をカバーしていても、第三者はその有効性、実行可能性、または範囲に疑問を提起する可能性があり、これは、そのような特許が縮小または無効になる可能性がある。さらに、それらが挑戦されていなくても、私たちの特許および特許出願は、私たちのADCプラットフォームまたは候補製品に十分な保護または排他性を提供することができず、他の人が私たちの主張を中心に設計されることを阻止したり、他の方法で競争優位性を提供したりすることができないかもしれない。これらの結果のいずれも、第三者の競争を阻止する能力を弱める可能性があり、第三者競争は私たちの業務に悪影響を及ぼす可能性がある。
もし私たちが私たちのプラットフォームまたは私たちの候補製品に関連する特許出願を所有または許可して特許として発行できなかった場合、それらの保護の広さや強度が脅かされたり不足したりした場合、またはそれらが意味のある排他性を提供できなかった場合、それは会社が私たちと協力することを阻止するかもしれない。私たちは、どのような特許が発行されるか(あれば)、任意のそのような特許の広さ、または発行された任意の特許が無効であることが発見されるか、強制的に実行できないか、または第三者の脅威にさらされるかどうかを保証することはできない。関連するライセンス特許を得ることができない、またはこれらの特許に成功したり、私たちが私たちに所有したり、許可したりすることができない任意の他の特許は、任意の候補製品の開発に成功し、それを商業化するために必要な権利を奪う可能性がある。米国や他のほとんどの国の特許出願は出願後しばらくは秘密であるため,一部の特許出願は発表前には依然として秘密であり,候補品に関連する特許出願を初めて提出した会社であることは確認できない.さらに、少なくとも私たちのいくつかの特許および特許出願について、第三者がそのような特許出願を提出した場合、米国特許商標局または第三者は、誰が最初に私たちが出願した特許特許請求の範囲によってカバーされた任意の主題の人であるかを決定するために、米国で介入手続きを開始することができる。しかも、特許の寿命は限られている。アメリカでは、特許の自然失効期間は一般的に申請後20年だ。様々な延期があるかもしれない;しかし、特許の有効期限とその提供される保護は限られている。新製品候補製品の開発、テスト、規制審査に要する時間を考慮すると、これらの候補製品を保護する私たちが所有または許可している特許は、他社が私たちの候補製品と競合する製品の商業化を効果的に阻止できる前に満期になる可能性があります。もし私たちが規制承認を得るのに遅延があれば、私たちは特許保護の下で薬を販売する時間がさらに短縮されるかもしれない。私たちの候補製品をカバーする特許を取得しても、製品の特許有効期限が満了すると、類似または模倣薬からの競争に直面する可能性がある。特に私たちの製品の模造バージョンの発売は、私たちの製品に対する需要が直ちに大幅に減少する可能性があり、これは私たちの業務、財務状況、運営結果、見通しに実質的な悪影響を及ぼす可能性があります。
2011年9月16日、“ライシー·スミス米国発明法”または“ライシー·スミス法案”が法律に署名された。“ライシー·スミス法案”は米国特許法を多くの重大な改正を行った。これらの条項には,特許出願起訴方式に影響を与える条項が含まれており,既存技術を再定義し,特許訴訟に影響を与える可能性があり,米国特許制度を“先発明”制度から“先発明者申請”制度に転換する可能性がある。第1の発明者から出願制の下で、特許可能性の他の要件が満たされていると仮定すると、第1の特許出願を提出した発明者は、通常、他の発明者がその前に発明を行ったか否かにかかわらず、1つの発明の特許を得る権利があるであろう。これらの規定はまた、第三者が特許起訴中に米国特許商標局に既存技術を提出することを可能にし、米国特許商標局によって管理される許可後のプログラムが特許有効性を攻撃する追加のプログラムを規定する。米国特許商標局は、“ライシー·スミス法案”の管理を管理する追加法規及びプログラムを制定し、“ライシー·スミス法案”に関連する特許法の多くの実質的な改正、特に最初に出願を提出した発明者条項が、2013年3月16日に発効した。したがって,Leahy-Smith法案が我々の業務運営にどのような影響を与えるかは不明である(もしあれば).Leahy-Smith法案とその実施は、私たちの特許出願をめぐる起訴と、私たちが発行した特許の実行または保護の不確実性とコストを増加させる可能性があり、これらのすべては、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。米国または他の国/地域の知的財産権管理法がさらに変化する可能性がある場合、または米国で“ライシー·スミス法案”の条項を解釈·実施し続けることは、私たちの知的財産権を取得、維持、実行する能力に不確実性をもたらす可能性があり、私たちのプラットフォームおよび候補製品を保護する方法でこれを行う能力に悪影響を及ぼす可能性がある。
どんな特許保護の喪失も私たちの業務に実質的な悪影響を及ぼす可能性がある。私たちは競争相手が私たちの候補製品と似ているか同じ製品で市場に入るのを防ぐことができないかもしれない。
XMT−1660、XMT−2056、および任意の他の現在または将来のADC候補製品をカバーする発行された特許は、法廷または米国特許商標局または同様の外国当局で疑問が提起された場合、無効または実行不可能である、競合製品によって侵害されることはない。
場合によっては、第三者が私たちの知的財産権を侵害していることを発見することは困難かもしれませんが、検出されても、侵害行為を証明することは困難かもしれません。XMT-1660、XMT-2056、または現在または未来の候補製品をカバーする任意の他の特許を強制的に実行するために、私たちまたは私たちの許可パートナーのうちの1つが第三者に対して法的訴訟を提起した場合、被告は、その製品が主張する特許を侵害していない、または私たちの候補製品をカバーする特許を無効または強制的に実行できないと反訴することができる。米国の特許訴訟では、被告が特許の無効または強制執行不可能と主張する反訴はありふれており、第三者は様々な理由に基づいて特許が無効または強制執行できないと断言することができる。有効性を疑問視する理由は、他にも、新規性の欠如、明らかな書面記述の欠如、または実施できないことを含む、いくつかの法定要件のいずれかを満たしていないと言われているからかもしれない。主張を実行できない理由は,他にも,特許起訴に関連する者が起訴中に米国特許商標局に関連情報を隠蔽したり,誤った陳述をしたりすることである可能性がある.第三者も米国や海外の行政機関に類似したクレームを出すことができ、訴訟範囲外でも同様である。このようなメカニズムには、再審査、当事者間の審査、付与後審査、介入手続き、派生手続き、および外国法ドメインにおける同等の手続き(例えば、反対手続き)が含まれる。このような訴訟は、私たちの候補製品をカバーして保護しないように、私たちの特許を撤回、キャンセル、または修正することをもたらす可能性がある。法的に権利侵害、無効、そして実行不可能と断言した後、結果は予測できない。権利侵害に関しては、裁判所は、第三者製品がこれらのクレームを侵害しないことを決定する方法でクレームを解釈することができるか、または第三者製品がクレームの各要素を体現または実践していることを決定することができず、クレームを侵害する可能性がある。例えば、私たちの特許の有効性については、私たち、私たちの許可者、私たちの特許弁護士、特許審査員が起訴中に知らなかった無効な以前の技術を確認することはできません。もし第三者が法的に無効または強制執行できないと主張した場合、私たちは少なくとも部分的、さらにはすべて、私たちの1つまたは複数の候補製品の特許保護を失うだろう。このような特許保護の喪失または第三者の競争製品が私たちの特許を侵害していないことが発見された場合、私たちの業務、財務状況、運営結果、および将来性に重大な悪影響を及ぼす可能性がある。
もし私たちがいかなる許可、戦略協力、または他の合意の下での義務を履行できなかった場合、私たちは損害賠償金の支払いを要求され、ADC候補製品の開発と保護に必要な知的財産権を失う可能性があります。
私たちは許可、協力、そして他の合意にある程度依存している。私たちは私たちの研究を進めたり、私たちの候補製品の商業化を許可するために他の人から追加の許可を得る必要があるかもしれませんし、私たちは合理的なコストや合理的な条項で追加の許可を得ることができないかもしれません(もしあれば)。第三者知的財産権の許可や買収は競争分野であり、より多くの老舗企業が魅力的であると考えられる第三者知的財産権許可または買収戦略をとる可能性がある。これらの老舗会社はその規模、資本資源及び更に強い臨床開発と商業化能力のため、私たちより競争優位を持っているかもしれない。しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。私たちはまた私たちの投資が適切な見返りを得るための条項の許可や第三者知的財産権を得ることができないかもしれない。
さらに、私たちの既存の許可および協力協定は、メルクKGaAと免疫合成プラットフォームに関連する知的財産権の許可、グラクソ·スミスクラインとXMT-2056の知的財産権の潜在的な許可を含む;私たちとジョンソンは、DolasynThenプラットフォームの知的財産権の許可、Synaffix B.V.またはSynaffixとの許可、DolasynThenプラットフォームに含まれるコンポーネントの許可、および私たちが将来締結した任意の許可、協力、または他の合意は、様々な開発、商業化、資金、マイルストーン、許可使用料、勤勉、再許可、保険、訴訟、挑戦、および強制執行またはその他の義務を課す可能性がある。私たちがこれらの義務に違反したり、許可されていない方法で私たちに許可された知的財産権を使用した場合、私たちは損害賠償の支払いを要求される可能性があり、許可者は、メルクKGaAとの合意の場合、免疫合成プラットフォームをカバーする権利の許可を含む許可を終了する権利がある可能性があり、グラクソとの合意の場合、XMT-2056をカバーする権利の潜在的許可を含み、DolasynThenプラットフォーム内のコンポーネントの権利の許可を含む。上記のいずれも、許可された知的財産権によって保護された製品を開発、製造、販売することができない、または競争相手が許可技術を得ることができないようにする可能性がある。許可、協力、または他の合意によると、知的財産権に関する論争が生じる可能性がある:
•ライセンス契約に従って付与された権利範囲および他の解釈に関する問題;
•私たちの技術およびプロセスは、ライセンス契約に拘束されていないライセンス側の知的財産権をどの程度侵害しているか
•私たちの協力開発関係に基づいて、特許と他の権利を再許可する
•私たちのライセンス契約の下での義務と、どのような活動がこれらの職務義務を満たしていますか
•私たちの許可者、私たちと私たちの協力者が共同で知的財産権を創造または使用することによって生成された発明および知識の発明および所有権;
•特許技術発明の優先権。
さらに、我々が現在、第三者または第三者から知的財産権または技術を許可するプロトコルは複雑であり、そのようなプロトコルのいくつかの条項は、様々な解釈の影響を受ける可能性がある。可能性のある契約解釈の相違の解決は、関連する知的財産権または技術に対する私たちの権利と考えられる範囲を縮小するか、または関連協定の下での私たちの財務または他の義務を増加させる可能性があり、いずれも、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。また、我々が許可している知的財産権をめぐる紛争が、商業的に許容可能な条項で現在の許可手配の能力を維持していることを阻害したり、損害を与えたりすれば、影響を受けた候補製品の開発に成功し、商業化することができない可能性がある。
場合によっては、私たちは、特許出願の準備、提出、起訴を制御する権利がないか、または第三者から許可された技術をカバーする特許を維持する権利がないかもしれません。したがって、私たちはこれらの特許と出願が私たちの業務の最適な利益に合った方法で起訴され、維持され、実行されるかどうかを確認することができない。私たちのライセンシーがそのような知的財産権を獲得または維持できなかった場合、またはそのような知的財産権の権利を失った場合、私たちが許可を得た権利および私たちの独占経営権は、そのような許可権利に拘束された任意の製品の権利を開発または商業化することに悪影響を受ける可能性がある。
さらに、ライセンス内特許および特許出願に対する我々の権利は、そのようなライセンス内特許と特許出願の共同所有者との間の機関間または他の運営プロトコルにある程度依存する。1つまたは複数の共通の所有者がそのような機関間または運営プロトコルに違反している場合、私たちは、このようなライセンス内の特許および特許出願の権利に悪影響を受ける可能性がある。また、将来の製品の販売にどのくらいの印税義務が支払われるかは確定できませんが、金額は大きいかもしれません。私たちの将来の印税義務の金額は、開発や商業化に成功した製品で使用される技術や知的財産権にかかっています(あれば)。したがって、私たちが製品を開発して商業化することに成功しても、私たちは利益を達成したり維持することができないかもしれない。上記のいずれも、我々の競争地位、業務、財務状況、経営結果、および見通しに重大な悪影響を及ぼす可能性がある。
もし私たちが必要な第三者知的財産権を得ることに成功したり、私たちの既存の知的財産権を維持することができなければ、関連計画や候補製品の開発を放棄しなければならないかもしれません。私たちの業務、財務状況、運営結果、見通しは影響を受ける可能性があります。
私たちは私たちの知的財産権を保護したり強制したりする訴訟に巻き込まれたり、知的財産権クレームを弁護したりする可能性があり、これは高価で時間がかかり、成功しない可能性がある。
競争相手や他の第三者は、私たちの特許を侵害したり、私たちが持っているおよび権限内の知的財産権を流用したり、または他の方法で侵害する可能性がある。侵害または無許可の使用に対抗するためには、私たちが所有しているおよび許可されていない知的財産権を強制または擁護し、私たちの機密情報および商業秘密を保護するため、または私たち自身の知的財産権または他の人の独自の権利の有効性および範囲を決定するために、訴訟または他の知的財産権訴訟手続きが必要となる可能性がある。このような訴訟または訴訟は高価で時間がかかる可能性があり、どのようなクレームも、私たちが彼らの特許または他の知的財産権を侵害したことを告発することを含む、被告に私たちに反クレームを提起させる可能性がある。私たちはそのような訴訟や法的手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちは現在、潜在的な多くの競争相手と私たちよりも多くの資源を投入して知的財産権を起訴し、より成熟かつより発達した知的財産権の組み合わせを持っている。したがって、私たちは努力したにもかかわらず、私たちは第三者が私たちの知的財産権を侵害したり流用したりすることを防ぐことができないかもしれない。解決策が私たちに有利であっても、訴訟や他の知的財産権訴訟は、巨額のコストや経営陣の注意力や資源の移転を招く可能性があり、これは私たちの業務や財務業績を損なう可能性があります。
さらに、訴訟または他の手続きでは、裁判所または行政裁判官は、私たちが私たちに与えることができるかもしれない特許を無効または強制的に実行できないと判断することができ、または裁判所は、私たちの特許が関連技術をカバーしていないことを理由に、他方の関連技術の使用を阻止することを拒否することができる。任意の訴訟または他の手続きにおける不利な結果は、私たちの1つまたは複数の特許を無効が宣言されるか、強制的に実行できないか、または狭い解釈される危険性に直面させるかもしれない。さらに、知的財産権訴訟や他の訴訟手続きが大量に開示される必要があるため、このような訴訟の間に、私たちのいくつかの機密情報が開示によって漏洩される可能性がある。任意の特許または他の知的財産権訴訟または他の訴訟において、公聴会の結果、動議および他の一時的手続きまたは発展の裁決が公開される可能性があり、証券アナリストまたは投資家がこれらの発表が負であると考える場合、私たちの候補製品、計画または知的財産権の知覚価値は低下する可能性がある。したがって、私たちの普通株の市場価格は下がるかもしれない。上記のいずれも、当社の業務、財務状況、経営結果、見通しに重大な悪影響を及ぼす可能性があります。
第三者による知的財産権侵害や流用のクレームは、我々の開発と商業化努力を阻害または延期する可能性がある。
私たちのビジネス成功は、私たちと私たちの戦略的パートナーと候補製品を開発、製造、マーケティング、および販売し、第三者特許および独自の権利を侵害、流用、または他の方法で侵害することなく、私たちの独自技術を使用する能力にある程度依存する。アメリカ国内外では、生物製薬業界の特許とその他の知的財産権に関連する訴訟が大量にあり、特許侵害訴訟、介入、異議、再審、当事者間審査、派生と許可後の審査手続きを含み、これらの訴訟はアメリカ特許商標局と対応する外国特許庁に関連する。我々が開発している分野には,米国や外国から発行された特許や第三者が所有する未解決特許出願が多く存在し,我々の候補製品を開発することが可能である.バイオ製薬業界の拡張や特許の発行に伴い、我々の候補製品が第三者特許権侵害のクレームを受けるリスクが増加する可能性がある。
第三者は、私たち、私たちの顧客、許可された当事者、または私たちによって賠償された当事者が彼らの独自技術を不正に使用すること、またはそれらの是非曲直にかかわらず、彼らの知的財産権または他の権利を侵害、流用、または他の方法で侵害すると断言するかもしれない。例えば、私たちは、第三者の特許、商標または著作権を侵害した、または私たちの従業員が彼らの前の雇用主の商業秘密または機密情報を流用したり、漏洩したりした疑いを受ける可能性がある。我々の候補製品の使用または製造に関連する第三者特許または特許出願が存在する可能性があるが、材料、処方、製造方法または治療方法に対するこれらの特許または特許出願の権利を識別することはできない。例えば、2000年11月29日までに提出された出願と、その日以降に提出されたいくつかの出願とは、特許として発行される前に、米国以外に提出されることはなく、依然として秘密である。いくつかの例外を除いて、前の例外を含めて、米国および他の地方の特許出願は、通常、最初に提出されてから約18ヶ月の待機期間後に公表され、時々全く公表されない場合がある。したがって、私たちのプラットフォームまたは私たちの候補製品に関する特許出願は、私たちが知らずに他の人によって提出されたかもしれない。さらに、いくつかの制限の下で、公表された係属中の特許出願は、私たちのプラットフォーム、私たちの候補製品、または私たちの候補製品の使用または製造をカバーするために、後で修正することができる。
第3の方針が私たちのクレームに法的根拠がないと考えても、管轄権のある裁判所は、第三者の特許が有効で強制的に実行可能であり、材料、処方、製造方法、分析方法、または治療方法を含む候補製品の様々な側面をカバーすることができ、この場合、第三者は、特許が満了するまで、または許可を得ない限り、このような第三者の金銭損害賠償を要求される可能性があり、これは巨額である可能性がある。このようなライセンスは、もしあれば、受け入れ可能な条項で提供されないかもしれない。たとえ私たちが許可を得ることができても、これらの権利は非排他的である可能性があり、これは私たちの競争相手が同じ知的財産権を獲得することを招き、大量の許可と印税を支払うことを要求するかもしれない。最終的に、実際または脅威の特許侵害クレームにより、許容可能な条項で許可を得ることができない場合、製品の商業化を阻止されたり、何らかの態様の業務運営を停止させられたりする可能性がある。
私たちにクレームを出した当事者たちも禁止または他の公平な救済を受ける可能性があり、これは、私たちの技術または1つ以上の候補製品をさらに開発して商業化する能力を効果的に阻止することができるかもしれない。特許侵害、商業秘密の流用、あるいは他の知的財産権侵害の告発を弁護することは、結果がどうであっても、費用が高く、時間がかかる可能性がある。したがって、私たちが最終的に勝訴したり、早い段階で和解が成立したりしても、このような訴訟は私たちに予期せぬ費用をもたらす可能性がある。また,訴訟や脅威訴訟は,我々の管理チームの時間や注意力に大きな要求を与え,会社の他の業務への追求を分散させる可能性がある.もし私たちに対する侵害クレームが成功した場合、可能な禁止救済に加えて、故意の侵害の3倍の損害賠償と弁護士費の支払い、特許使用料の支払い、私たちの侵害製品の再設計、または第三者から1つ以上の許可を得ることを含む実質的な損害賠償を支払わなければならないかもしれないし、あるいは大量の時間とお金の支出が必要かもしれない。
第三者が第三者がその第三者の機密情報や商業秘密を適切に取得して使用していないと考えた場合、公金を流用した疑いに直面する可能性がある。もし私たちが第三者の機密情報や商業秘密を盗用したことが発見された場合、私たちはこのような機密情報や商業機密をさらに使用することを阻止され、製品候補製品を開発する能力を制限することができ、商業的に合理的な条項では得られないかもしれないし、独占的ではないかもしれないし、損害賠償金の支払いを要求される可能性があります。これは巨額かもしれません。上記のいずれも、当社の業務、財務状況、経営結果、見通しに重大な悪影響を及ぼす可能性があります。
私たちは世界各地で私たちの知的財産権と固有の権利を保護できないかもしれない。
私たちの製品に巨大な市場があると予想される世界のすべての国では、候補製品の特許出願、起訴、弁護の費用は目を引くほど高いかもしれないし、外国の法律はアメリカの法律のように私たちの権利を保護できないかもしれない。しかも、私たちの知的財産権許可協定は常に世界的な権利を含むわけではないかもしれない。したがって、私たちは第三者がアメリカ以外のすべての国で私たちの発明を実施することを防ぐことができないかもしれない。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの技術を使用して自分の製品を開発することができ、また、私たちに特許保護や許可証を持っているが、法執行力がアメリカの地域に比べて他の侵害製品を輸出することもできる。これらの製品は私たちの製品と競争するかもしれませんが、私たちの特許や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。
また、一部の外国の法律は知的財産権の保護程度が米国の法律に及ばず、多くの会社は外国の管轄区域でこのような権利を保護し、守る際に重大な問題に直面している。特定の国の法制度、特に特定の発展途上国の法律制度は、特許および他の知的財産権保護、特にバイオテクノロジーに関連する特許の強制執行に賛成せず、これは、私たちの許可および所有特許の侵害、または全体的に私たちの知的財産権および独自の権利を侵害する競争製品のマーケティングを阻止することを困難にするかもしれない。外国の管轄区域で私たちの知的財産権と独自の権利を強制的に執行する訴訟手続きは巨額のコストを招く可能性があり、私たちの努力と注意を私たちの業務の他の側面から移すことは、私たちの特許を無効または狭義に解釈されるリスクに直面させる可能性があり、私たちの特許出願を特許として発表しないリスクに直面させ、第三者が私たちにクレームを提起する可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。したがって、私たちの知的財産権および独自の権利を世界各地で強制的に実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネス的優位性を得るのに十分ではないかもしれません。
多くの国に強制許可法があり,これらの法律により特許権者は第三者に強制的に許可を付与される可能性がある。また、多くの国は政府機関や政府請負業者に対する特許の実行可能性を制限している。これらの国では,特許権者の救済措置は限られている可能性があり,このような特許の価値を大幅に低下させる可能性がある。もし私たちまたは私たちの任意の許可者が第三者に私たちの業務に関連する任意の特許の許可を付与することを余儀なくされた場合、私たちの競争的地位が損なわれる可能性があり、私たちの業務、財務状況、運営結果、および見通しは不利な影響を受ける可能性があります。
従業員および第三者との秘密保持協定は、商業秘密や他の固有情報の不正流出を防ぐことができない可能性がある。
特許提供の保護に加えて、私たちは、商業秘密保護および秘密協定によって、申請不可能な特許を保護すること、または特許を出願しない独自技術を選択すること、強制実行が困難なプロセス、および私たちのプラットフォーム技術の任意の他の要素、ならびに特許が含まれていないノウハウ、情報または技術に関する発見および開発プロセスを含む、当社のセキュリティ技術の保護に依存する。しかし、商業秘密を含む機密技術を保護することは難しいかもしれない。私たちは、従業員、コンサルタント、外部科学コンサルタント、請負業者、協力者と秘密保護協定を締結することで、私たちのノウハウやプロセスを保護することを求めています。私たちは私たちが可能であるか、または私たちの商業秘密またはノウハウとプロセスに接触したすべての当事者とこのような合意に到達したことを保証することができない。さらに、私たちの秘密保護協定および他の契約保護は、不正な開示、第三者侵害、または流用から私たちの知的財産権を保護するのに十分ではないかもしれません。このような合意に違反した場合、私たちは十分な救済措置を持っていない可能性があり、私たちの商業秘密および他の固有情報は、私たちの競争相手に開示される可能性があり、または他の人は、実質的に同等またはより高度な独自情報および技術を独立して開発するか、または他の方法で私たちのビジネス秘密を取得するか、またはそのような技術を開示することができる。
第三者に我々の任意の機密技術または商業秘密を不正に入手して使用することを強制することは高価で時間がかかり、結果は予測できない。また、米国国内外のいくつかの裁判所も商業機密を保護することをあまり望まないこともある。私たちの機密技術や商業秘密を流用または不正に開示することは、私たちの競争的地位を損なう可能性があり、私たちの業務に実質的な悪影響を及ぼす可能性があります。
私たちは、私たちの許可者、従業員、コンサルタント、コンサルタント、または私たちが彼らの知的財産権を流用したと主張したり、私たち自身の知的財産権の所有権を要求したりする第三者のクレームを受けるかもしれない。
私たちと私たちの許可側の多くの従業員は、私たちの上級管理職、コンサルタント、コンサルタント、現在、あるいは以前、私たちの競争相手や潜在的な競争相手を含む大学や他のバイオテクノロジーや製薬会社に雇われています。その中の一部の従業員は、私たちの上級管理職のメンバーを含み、以前の雇用に関連する所有権、秘密、およびスポーツ禁止協定または同様の協定に署名した。私たちは、私たちの従業員、コンサルタント、およびコンサルタントが、私たちのために働いているときに他人の固有情報またはノウハウを使用しないことを保証するために努力しているが、私たちまたはこれらの個人は、商業秘密または他の固有情報を含む任意のそのような個人の現職または前任雇用主の知的財産権を使用または開示している疑いを受ける可能性がある。このようなクレームを弁護するために訴訟を提起する必要があるかもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭的損害賠償を支払う以外に、私たちは貴重な知的財産権や人員を失ったり、損害を受けたりする可能性がある。このような知的財産権は第三者に付与することができ、私たちの技術や製品を商業化するためには第三者から許可を得る必要があるかもしれない。そのような許可は商業的に合理的な条項や根本的に存在しないかもしれない。このようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣の注意を分散させる可能性がある。上記のいずれも、当社の業務、財務状況、経営結果、見通しに重大な悪影響を及ぼす可能性があります。
また、私たちの政策は、知的財産権の概念や開発に参加する可能性のある私たちの従業員と請負業者が合意に署名し、このような知的財産権を私たちに譲渡することを要求しているにもかかわらず、実際に私たちが自分の知的財産権と見なしているすべての当事者とこのような合意を実行することができないかもしれない。知的財産権の譲渡は自動的に実行されない可能性があり、あるいは譲渡協定が違反される可能性があり、私たちは第三者にクレームをつけさせられたり、私たちが私たちの知的財産権の所有権だと思うことを決定するために、私たちが提起する可能性のあるクレームを弁護したりすることができます。
もし私たちが開発する可能性のある候補製品のために特許期間の延長とデータ独占権を得なければ、私たちの業務は実質的に損害を受ける可能性がある。
FDAが私たちが開発する可能性のある任意の候補製品の上場承認の時間、期限、および詳細に基づいて、私たちが所有または許可している1つまたは複数の米国特許は、例えば、米国で1984年に“医薬品価格競争および特許期限回復法”またはハッジ-ワックスマン修正案に基づいて限られた特許期間を延長する資格がある可能性がある。ハッジ·ワックスマン改正案は、FDA規制審査中に失われた特許期間の補償として、特許期間を最大5年間延長することを許可している。1つの特許期間の延長は、製品が承認された日から14年を超えてはならず、1つの特許を延長することしかできず、承認された薬物、その使用方法又はその製造方法に関する請求項を延長することしかできない。しかし,テスト段階や規制審査中に職務調査を行うことができなかったこと,適用の最終期限内に出願できなかったこと,関連特許の満了前に出願を提出できなかったこと,適用要件を満たしていなかったことなどにより延期が得られなかった可能性がある.しかも、特許保護の適用期間や範囲は私たちが要求しているものよりも短いかもしれない。もし私たちが特許期間の延長を得ることができない場合、あるいはこのような延長の期限が私たちが要求したものよりも短い場合、私たちの競争相手は私たちの特許が満期になった後に競争製品の承認を受ける可能性があり、私たちの業務、財務状況、運営結果、見通しは実質的に損なわれる可能性がある。
特許や他の知的財産権保護に加えて、FDAの生物製品ライセンス申請やBLAプログラムに基づいて、私たちの生物候補製品のために市場とデータの独占経営権を求める可能性があり、現在アメリカでは12年、ヨーロッパでは10年、他の国/地域では他の利用可能期間となっている。私たちの候補製品をカバーする特許の有効期限はデータと市場排他性を超えてはいけない。米国や現在このような保護されている他の国の立法行動により、このようなデータと市場排他性が短縮される可能性があり、生物類似競争が予想よりも早く市場に参入するリスクをもたらす可能性がある。また、どのような生物類似の競争製品も、承認されると、どの程度私たちの関連参考製品を代替できるかは不明であり、多くの不確定な市場と規制要素に依存する
私たちの特許保護の獲得と維持は、政府特許機関によって提出された様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
私たちが所有または許可している特許および特許出願の有効期間内に、定期維持費、継続費、年会費、および様々な他の政府特許および特許出願費用は、米国特許商標局および米国以外の様々な政府特許機関に支払われる。場合によっては、私たちは私たちの許可パートナーに依存して、これらの費用をアメリカと非アメリカの特許機関に支払う。米国特許商標局および様々な非米国政府機関は、特許出願過程において、いくつかのプログラム、文書、費用支払い、および他の同様の条項を遵守することを要求する。私たちはまた、私たちが許可した知的財産権に関するこれらの要求を遵守するために、私たちの許可側が必要な行動を取ることに依存している。場合によっては、不注意は、滞納金を支払うことによって、または規則を適用する他の方法によって救済することができる。しかしながら、場合によっては、規定を遵守しないことは、特許または特許出願が放棄または失効される可能性があり、それにより、関連法ドメインの特許権の一部または全部が失われる可能性がある。この場合、潜在的な競争相手は、同様または同じ製品または技術で市場に参入する可能性があり、これは、私たちの業務、財務状況、運営結果、および将来性に重大な悪影響を及ぼす可能性がある。
知的財産権はすべての潜在的な脅威を解決できるとは限らない。
私たちの知的財産権が提供する未来の保護の程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できないか、あるいは競争優位性を維持することができるかもしれない。例えば:
•他の人は、私たちが開発または利用する可能性のある同様のADC関連技術の任意の候補製品と同様のADC製品を製造することができるかもしれないが、これらの製品は、私たちが許可または将来所有する可能性のある特許の請求項の範囲内ではない
•私たち、または私たちのライセンスパートナーまたは現在または未来の戦略パートナーは、私たちが許可または将来所有する可能性のある発行された特許または係属中の特許出願がカバーする最初の発明をした会社ではないかもしれない
•私たち、または私たちの許可パートナー、または現在または未来の戦略的パートナーは、私たちまたは彼らのいくつかの発明に関する最初の特許出願ではないかもしれない
•他の会社は、私たちが持っているまたは許可された知的財産権を侵害することなく、類似または代替技術を開発したり、私たちのいかなる技術を複製したりすることができる
•私たちが未解決の許可された特許出願または将来所有する可能性のある特許出願は、発行された特許をもたらすことができないかもしれない
•私たちが権利を持っている発行された特許は、私たちの競争相手による法的挑戦を含む無効または強制執行不可能と認定される可能性があります
•私たちの競争相手は特許権のない国で研究や開発活動を行い、これらの活動から学んだ情報を利用して競争力のある製品を開発し、私たちの主要な商業市場で販売するかもしれません
•私たちは他の特許を申請できる独自技術を開発しないかもしれない
•他人の特許は私たちの業務を損なうかもしれません
•いくつかの商業秘密または秘密技術を保護するために、私たちは特許を提出しないことを選択することができ、第三者はその後、そのような知的財産権をカバーする特許を提出する可能性がある。
これらの事件が発生すると、それらは私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
規制承認およびその他の法律コンプライアンス事項に関するリスク
たとえ著者らが必要な臨床前研究と臨床試験を完成しても、監督管理の審査過程は高価で、時間と不確定であり、私たちが一部或いはすべての候補製品の商業化の許可を得ることを阻止するかもしれない。そのため、いつ、どの地域にあるか、どの地域であるかを予測することはできず、候補製品を商業化するマーケティング承認を得ることになる。
製品の研究、テスト、製造、ラベル、承認、販売、マーケティング、普及と流通はすべてFDAと類似外国の監督管理機関の広範な監督管理を受けている。FDAのBLA承認や米国以外の関連規制機関のマーケティング承認を得るまで、米国や他の国/地域で候補製品を販売することは許可されていません。我々の候補製品は異なる開発段階にあり,開発過程に固有の失敗リスクの影響を受けている.私たちはまだアメリカや他の任意の司法管轄区で私たちの任意の候補製品の申請を提出したり、マーケティングの許可を得ていません。また、企業としては、マーケティング承認を得るために必要な申請の提出や支援に経験がなく、その過程で第三者CROに依存して支援することが予想される。
米国でも海外でも、上場承認を得る過程は長く、高価で不確実だ。最終的に承認される場合、これは数年を要する可能性があり、関連する候補製品のタイプ、複雑性、および新規性を含む様々な要因によって大きく異なる可能性がある。上場承認を得るためには、候補製品の安全性と有効性を決定するために、広範な臨床前と臨床データ及び支持情報を監督機関に提出する必要がある。FDAまたは他の規制機関は、私たちの候補製品が安全で有効ではなく、適度に有効であるか、あるいは不良または意外な副作用、毒性または他の特徴を持っていると認定し、上場承認を得ることができないようにしたり、商業用途を阻止したり制限したりする可能性がある。
さらに、小児科研究公平法またはPREAによれば、いくつかの生物学的製品のBLAまたは補足BLAは、すべての関連する小児科亜集団における生物製品の安全性および有効性を評価し、スポンサーがFDAの延期または免除を受けない限り、製品の安全に有効な各小児科亜集団の用量および投与をサポートしなければならない。延期は、小児科試験が完了する前に、製品または候補治療薬が成人で使用を許可する準備ができていることを発見するか、または小児科試験が開始される前に追加の安全性または有効性データを収集する必要があることを含むいくつかの理由があるかもしれない。欧州連合の適用立法は,スポンサーがEMA小児科委員会が承認した小児科調査計画に基づいて小児科群で臨床試験を行うか,その委員会によるこれらの研究の免除や延期を得ることも求められている。私たちがアメリカやEUの規制承認を求めている任意の候補製品については、私たちが免除されたり、代替的に必要な研究や他の要求をタイムリーに達成できる保証はありません。これは関連する名声の損害を招き、法執行行動の影響を受ける可能性があります。
さらに、開発中の市場承認政策の変化、追加法規、法規またはガイドラインの制定または公布の変化、または各提出された製品申請に対する規制審査の変化は、申請の承認または拒否の遅延を招く可能性がある。例えば、2022年12月、FDORAの採択に伴い、国会は、各新薬または生物製品の3期臨床試験または任意の他の“重要な研究”のための多様な行動計画を策定し、提出することをスポンサーに要求する。これらの計画はより多くの異なる患者群がFDA監督製品の後期臨床試験に参加することを奨励することを目的としている。また,2022年1月には,従来の臨床試験指令2001/20/ECの代わりに,新たな臨床試験条例(EU)第536/2014号がEUで施行された。この規定はEUの臨床試験の許可、進行と透明性を単純化して簡素化することを目的としている。臨床試験承認の調整手続きによると、1つ以上のEU加盟国で行われる臨床試験のスポンサーは、承認申請を提出するだけでよい。提出された材料は臨床試験情報システムを通じて提出され、これはEMAが監督する臨床試験ポータルサイトであり、臨床試験スポンサー、EU加盟国の主管当局と公衆に使用することができる。
監督管理機関は審査過程中にかなりの自由裁量権を持っており、臨床前と臨床試験で得られたデータに対する異なる解釈は候補製品の上場承認を延期、制限或いは阻止する可能性がある。私たちが最終的に得たどのマーケティング承認も限られているかもしれないし、制限されたり、承認された後の約束は、承認された製品が商業的に実行できないようにすることができる。
外国の管轄区域でマーケティングの許可を得られなければ、私たちの候補製品は海外で販売できないだろう。私たちの製品候補製品がアメリカで得られたいかなる承認も、私たちの製品候補製品が外国司法管轄区で承認されることを保証することはできません。私たちのどの候補製品も外国司法管轄区で発売される可能性があれば、外国業務に関連するリスクに直面します。
承認されれば、私たちは現在の候補製品XMT-1660およびXMT-2056を直接または協力によって国際市場で販売するつもりだ。EUや他の外国司法管轄地域で我々の製品をマーケティング·販売するためには、単独のマーケティング承認を得、多くの異なる規制要件を遵守しなければならない。承認手続きは国によって異なり、追加的なテストが含まれるかもしれない。承認を得るのに要する時間は、FDA承認を得る時間とは大きく異なる可能性がある。米国以外の上場承認プロセスには、通常、FDA承認の取得に関するすべてのリスクが含まれている。もしあれば、私たちはアメリカ以外の規制機関から直ちに承認されないかもしれない。FDAの承認は、他国又は管轄区域の規制機関の承認を確保するものではなく、米国以外の1つの規制機関の承認も、他の国又は司法管区の規制機関又はFDAの承認を確保することができない。マーケティング承認を申請するかもしれませんが、どの市場でも私たちの製品を商業化するために必要な承認は得られません。
米国以外の多くの国でも、候補製品は精算承認を得なければ、その国で販売することができない。場合によっては、私たちは私たちの製品のための価格を取るつもりで、承認されれば、承認される必要があります。非米国規制の承認を得て非米国規制要求を遵守することは、私たちに重大な遅延、困難、コストをもたらす可能性があり、私たちの候補製品がある国/地域で発売されることを延期または阻止する可能性がある。また、もし私たちが米国国外で私たちの候補製品を販売するために必要な非アメリカの承認を得られなかった場合、あるいは適用された非アメリカの規制要求を遵守できなかった場合、私たちの目標市場は減少し、私たちの候補製品のすべての市場潜在力を達成する能力は損なわれ、私たちの業務、財務状況、運営結果、見通しは不利な影響を受ける可能性がある。
また、イギリスのEU離脱により、私たちはイギリスでマーケティング許可を得るより高いリスクに直面する可能性があり、一般にイギリスの離脱と呼ばれる。イギリスはこれ以上ヨーロッパ単一市場とEU関税同盟の一部ではない。2021年1月1日から、薬品と保健品規制機関(MHRA)はイギリスの薬品と医療機器の監督を担当し始め、国内法によると、イギリスはイングランド、スコットランド、ウェールズを含み、北アイルランド議定書の条項によると、北アイルランドは現在EU規則の制約を受けている。しかし、連合王国と欧州連合は、連合王国の医薬製品の規制を含む“北アイルランド議定書”下の現行制度を根本的に変更した“ウィンザー枠組み”に同意した。実施されると、ウィンザーフレームワークによる変化はMHRAがイギリス市場(すなわち大ブリテンと北アイルランド連合王国)に輸送されたすべての医薬製品を承認することを担当させ、EMAは北アイルランドへの医薬製品の輸送を許可する上でいかなる役割を果たすことができなくなる。イギリスの離脱やその他の理由により、いかなるマーケティング許可を得る上でのいかなる遅延も、いかなるマーケティング許可も得ることができず、イギリスで私たちの候補製品のための規制承認を求める努力を制限または延期させる可能性があり、これは私たちの業務に重大で実質的な損害を与える可能性がある。
また、外国の規制機関はその承認政策を変更し、新たな規定を制定する可能性がある。例えば、欧州委員会が2020年11月に開始した“欧州薬物戦略”イニシアティブを背景に、欧州連合の薬物立法は現在全面的な検討が行われている。欧州委員会は、医薬製品に関するいくつかの立法文書の改正に関する提案(規制データ保護の期限を短縮する可能性があり、迅速通路の資格を改訂するなど)。2023年4月26日に出版された。提案された改正は欧州議会と欧州理事会の合意と採択が待たれるため,提案は採択前に重大な修正が行われる可能性があり,2026年初めまではないと予想される。しかし、長期的には、これらの改正は製薬業と私たちの業務に大きな影響を与える可能性がある。
私たちは、私たちがアメリカ国外でマーケティング承認を得た任意の候補製品を商業化する際に、関税、貿易障壁、規制要件を含む追加のリスクに直面すると予想しています。インフレや特に外国経済や市場の政治的不安定を含む経済的疲弊、居住または海外旅行の従業員が税収、雇用、移民、労働法を遵守している場合、外国為替変動は、運営費用の増加と収入の減少、他の国での業務を展開する他の義務、および労働騒動が米国よりも一般的な国の労働力不確実性を招く可能性があります。
私たちはアメリカ以外の場所で臨床試験を行う予定です。FDAはこれらの地点で行われた試験のデータを受け入れない可能性があり,米国以外で試験を行うことで余分な遅延や費用に直面する可能性がある。
私たちはアメリカ以外の1つまたは複数の試験地点で1つまたは複数の臨床試験を行う予定だ。FDAや他の規制機関がその管轄外で行われた臨床試験を受けた研究データは,何らかの条件によって制限される可能性があり,まったく受け入れられない可能性もある。外国の臨床試験データが米国の上場承認の唯一の根拠となることを目的としている場合、FDAは通常、(I)データが米国の人口および米国の医療実践に適用されない限り、(I)データが米国の人口および米国の医療実践に適用されない限り、(Ii)試験はGCP規定に基づいて公認能力を有する臨床研究者によって行われ、(Iii)データはFDAによる現場検査を必要とせず、あるいはFDAがこのような検査を行う必要があると考えられ、FDAは現場検査や他の適切な方法でデータを検証することができる
また,海外の研究データが承認の唯一の根拠としようとしなくても,FDAはこれらのデータを上場承認申請の支援として受け入れず,研究設計が良好でGCP要求に適合しない限り,FDAは必要と考えた場合に現場検査により研究データを検証することができる。多くの外国の規制機関もまた似たような承認要求を持っている。また、このような外国裁判は、裁判を行う外国司法管轄区域に適用される現地法によって管轄される。FDAや同様の外国規制機関が米国や適用司法管轄区域以外で行われた試験データを受け入れることは保証されない。FDAまたは同様の外国の規制機関がこれらのデータを受け入れない場合、追加の試験が必要になり、これは高価で時間がかかる可能性があり、私たちが開発する可能性のある現在または未来の候補製品が適用される司法管轄区域で商業的な承認を得ることができない可能性がある
アメリカ以外で臨床試験を行うことで、以下のようなリスクを含む多くのリスクに直面することになります
•他の外国の規制要件;
•外国為替変動
•海外の製造、税関、輸送、倉庫の要求を守る
•医療実践と臨床研究における文化的違い
•一部の国では知的財産権の保護力が弱まっている
•私たちの裁判は戦争やテロなどの地政学的事件によって中断されたり遅延されたりする。
どの規制機関も私たちの製品の販売を許可することは指標によって制限されるだろう。FDA規制に違反して私たちの製品を未承認の用途に使用する規定を遵守できなかったり、発見されたりした場合、刑事罰、巨額の罰金、または他の制裁、損害賠償を受ける可能性があります。
未承認用途の製品の普及に関する法規は複雑であり,FDA,EMA,MHRA,他の政府機関から実質的に解釈されている。FDAは2021年9月、医薬品の予期される用途を決定する際に機関が考慮すべき証拠タイプを記載した最終法規を公表した。それにもかかわらず,医師は承認されたラベルと一致しない方法で,ラベル外で患者に我々の製品を処方する可能性がある。私たちは私たちの販売とマーケティング実践が適用された法規に適合することを確実にするために、コンプライアンスと訓練計画を実施するつもりだ。これらの計画にもかかわらず、FDAや他の政府機関は、私たちのやり方が未承認の用途への私たちの製品の普及を禁止するようになっていると主張したり発見したりするかもしれない。私たちの従業員が会社の政策や適用された法規を遵守して、承認されていない用途のための製品を普及させるかどうかも確認できません。
ラベル外販売促進に規制制限があるにもかかわらず、FDAと他の監督管理機関は会社がある場合にその製品について真実、非誤解性と非販売促進の科学的な交流を行うことを許可する。例えば,FDAは2023年10月にガイドラインを発表し,未承認用途に関する科学的情報を医療保健提供者に配布する拘束力のない政策を管理することについて概説した。本ガイドライン草案は、このような通信が真実で、誤解性がなく、事実と偏らないことを要求し、医療保健提供者が許可されていない使用に関する情報の利点および欠点、ならびに有効性および実用性に必要なすべての情報を説明することを含む。また,FDAの最近の指導や,2023年の総合支出法案の一部として法律に署名した“承認前情報交換法”によると,会社は処方情報と一致する情報を普及させ,支払人の処方委員会メンバーと未承認薬物や承認済み薬物の未承認用途のデータについて能動的に会話することも可能である。私たちはこれらの議論に参加し、すべての適用された法律、法規指導、業界ベスト実践を遵守し、ヘルスケア提供者、支払人、他の顧客とコミュニケーションを取ることができるかもしれない。FDAの様々な法規、ガイドライン、政策、そして最近公布された立法をよく考慮して、我々の製品普及に関する制限を遵守することを確保する必要がある
近年、大量の製薬と生物技術会社は様々な連邦と州監督管理、調査、検察と行政実体調査と調査の目標となり、これらの実体は司法省と各種アメリカ検事事務室、衛生·公衆サービス部監察長事務室、FDA、連邦貿易委員会または連邦貿易委員会、および各州総検察長事務室を含む未承認用途および他の販売行為のための製品の使用を促進することに関する。これらの調査容疑は、独占禁止法違反、FDCA違反、虚偽申告法、処方薬営業法および逆控除法の違反、および他の承認されていない用途の製品の販売促進、定価および連邦医療保険および/または医療補助精算に関連する行為に違反する疑いがあるなど、様々な連邦および州の法律および法規に違反している。これらの調査の多くは“りっぱな担い手“”虚偽申告法“による行動。虚偽請求法によれば、どの個人も政府を代表してクレームを提出することができ、個人または実体が虚偽クレームを提出したこと、または虚偽クレームが政府に支払いを要求することになると主張することができる。この人は1部持ってきたりっぱな担い手訴訟はどんな回復や和解から一杯のスプーンを分ける権利があるりっぱな担い手訴訟は、通常“密告者訴訟”とも呼ばれ、通常は現職または前任社員によって提起される。1つはりっぱな担い手訴訟を提起する時、政府はこの事件に介入して起訴するかどうかを決定しなければならない。もしそれが拒否すれば、個人は単独で起訴することができる。
FDAや他の政府機関が私たちに法を執行しているならりっぱな担い手許可されていない用途への製品の普及に関する禁止令に違反すれば、法令や会社の誠実な合意に同意するような巨額の民事または刑事罰金や損害賠償、その他の制裁に直面する可能性があり、これらの制裁に基づいて、私たちの活動は、適用される法律や法規の遵守を確保するために継続的な審査と監督を受けることになる。このような罰金、報酬、または他の制裁は、私たちの収入、業務、財務の見通し、そして名声に悪影響を及ぼすだろう。
私たちが将来発売許可を得たどの製品も発売後に市場から制限されたり撤退したりする可能性があります。もし私たちが規制要求を守っていない場合、あるいは承認された後にこのような製品の意外な問題に遭遇した場合、私たちは重大な処罰を受けるかもしれません。
私たちが発売許可を得た任意の製品、およびその製品の製造プロセス、承認後の研究と措置、ラベル、広告、販売促進活動などは、FDAと他の監督管理機関の持続的な要求と審査を受ける。これらの要求には、安全およびその他の発売後の情報と報告の提出、登録と上場要求、製造、品質管理、品質保証と記録と書類の対応する維持に関する要求、医師へのサンプルの配布と記録の保存に関する要求が含まれる。製品が上場承認されても、承認は、再生可能エネルギー管理システムを実施する要求を含む、その製品が発売可能な指定用途の制限または承認条件に制限される可能性がある。
FDAはまた、製品の安全性或いは有効性を監視するために、高価な発売後の研究或いは臨床試験とモニタリングを要求する可能性がある。FDA及び司法省を含む他の機関は、製品の製造、販売及び流通が承認の適応にのみ適用され、承認されたラベルの規定に適合することを確実にするために、製品の承認後のマーケティング及び販売促進活動を密接に規制し、監督する。FDAは、ラベル外使用に関するメーカーのコミュニケーションに厳格な制限を加えており、もし私たちが許可されていない製品を販売すれば、ラベル外マーケティングの警告や法執行行動を受ける可能性がある。“虚偽請求法”を含むFDCAおよび他の処方薬販売促進および広告に関する法規に違反することは、連邦および州医療詐欺および法律乱用および州消費者保護法違反の調査または告発を招く可能性がある。
さらに、市場承認を得る可能性のある任意の製品およびその製造業者または製造プロセスの以前に未知の有害事象または他の問題、または法規要件を遵守できなかったことが後に発見された場合、様々な結果が生じる可能性がある
•このような製品、製造業者、または製造プロセスの制限;
•製品のラベルやマーケティングの制限
•製品の流通や使用の制限;
•発売後の研究や臨床試験が求められている
•警告状や見出しのない手紙
•製品が市場から撤退した
•私たちが提出した保留申請または承認された申請を承認する補充申請を拒否する;
•製品をリコールする
•第三者支払者の保証の制限
•罰金、利益または収入の返還、
•上場承認の一時停止または撤回
•この製品の輸出入許可を拒否した
•製品を検収する
•民事または刑事処罰を禁令または適用する。
最後に,新薬製品を開発·マーケティングする能力は,行われている挑戦FDAによるミフェプリストン承認訴訟の影響を受ける可能性がある。具体的には、2023年4月7日、アメリカテキサス州北区地区裁判所はFDAのミフェプリストンに対する許可を保留し、ミフェプリストンは1種の薬物製品であり、最初は2000年に承認され、その流通はREMS下で採用された各種条件の制約を受けた。この決定を下した際、地域裁判所は、米国の薬品の開発、承認、流通に悪影響を及ぼす可能性のある調査結果を下した。他の裁決では、地域裁判所は、原告が彼らの告発に勝つ可能性が高い、すなわちFDAがミフェプリストンを承認する際に独断的で気まぐれな行為を行った可能性が高く、そのラベルで決定された条件下での薬剤の使用が安全であるかどうかに関する証拠を十分に考慮していない。また,地域裁判所は連邦裁判所訴訟の長期的な要求を,原告がそのNDAを承認する決定についてFDAを提訴することを許可するか,あるいはREMSに基づいて要求を確立し,原告またはそのメンバーが被害を受ける程度,すなわちFDAの薬品承認決定が実際に原告に与えられた薬物による不良事件を受けた患者に看護を提供させることに基づいている
2023年4月12日、米国第五巡回控訴裁判所は地域裁判所の裁決をある程度棚上げした。その後、2023年4月21日、米国最高裁判所は地域裁判所のすべての決定を棚上げにし、第5巡回控訴裁判所が地域裁判所の裁決の控訴を処理し、任意のまたは最高裁判所に提出した移審令申請を処理するのを待った。第五巡回控訴裁判所は2023年5月17日にこの事件に対して口頭討論を行い、2023年8月16日に裁決を下した。裁判所はミフェプリストンの市場からの除去を拒否し,FDAが2000年に最初に承認した挑戦は訴訟時効で禁止されていることを発見した。しかし控訴裁判所は、原告は彼らの訴訟で勝つ可能性が高く、FDAが2016年と2021年にミフェプリストンの使用範囲を拡大することを許可した変化は独断的で気まぐれだと考えている。2023年9月8日、司法省とミフェプリストンのメーカーは、米国最高裁に控訴裁判所の裁決の再審査を要求する移審令の請願書を提出した。2023年12月13日,最高裁は控訴裁判所の裁決に関するこれらの移審令の請願書を承認した。
似たような制限はEUでの私たちの製品の承認にも適用される。販売許可の保持者は、医薬製品の製造、マーケティング、普及と販売に適用される一連の要求を守らなければならない。これらの問題は、欧州連合の厳格な薬物警戒または安全報告規則を遵守すること、これらの規則は認可後の研究および追加の監視義務を強制することができること、許可された薬品を製造することは、それに対して単独のメーカー許可証を持たなければならないこと、および許可薬品のマーケティングと普及を含むことであり、これらの薬品は欧州連合で厳格に規制され、EU加盟国の法律によっても制約されている。これらと他の連合の要求を守らないことはまた重大な処罰と制裁につながる可能性がある。
したがって、私たちまたは私たちの協力者が1つまたは複数の候補製品のマーケティング承認を得たと仮定すると、私たち、私たちの協力者、および私たちと彼らの契約製造業者は、製造、生産、製品監視、および品質管理を含むすべてのコンプライアンス分野で時間、お金、エネルギーをかけ続けるだろう。もし私たちと私たちの協力者が承認後の法規要件を遵守できなければ、私たちまたは私たちの協力者が未来の製品をマーケティングする能力が制限される可能性があり、これは私たちが利益を達成したり維持したりする能力に悪影響を及ぼす可能性があります。また、承認後の法規を遵守するコストは、私たちの経営業績や財務状況に悪影響を及ぼす可能性があります。
私たちは私たちの候補製品のためにいくつかの指定を求めるかもしれませんが、画期的な療法、アメリカの迅速なチャネル、優先審査指定、EUの優先薬品やPrime指定を含むが、私たちはこのような指定を受けないかもしれません。私たちが受け取っても、そのような指定はより速い開発や規制審査や承認過程をもたらすことはないかもしれません。
私たちは過去にFDAの審査と承認を加速するために、私たちの1つまたは複数の候補製品のためのいくつかの指定を求め、将来的に私たちの1つまたは複数の候補品のために特定の指定を求め、将来的に私たちの1つまたは複数の候補製品のために特定の指定を求めることができた。画期的な治療製品は、重篤な疾患の治療のために単独でまたは1つまたは複数の他の製品と組み合わせて使用することを意図した製品として定義され、初期臨床証拠は、製品が1つまたは複数の臨床的重要終点において、臨床開発早期に観察された実質的な治療効果のような既存の療法よりも実質的に改善された効果を示す可能性があることを示す。画期的な治療法として指定された製品に対して,FDAと試験スポンサーとの相互作用やコミュニケーションは,臨床開発の最も有効な方法を決定するのに役立つとともに,無効なコントロールレジメン中の患者数を最小限に抑えることができる。
FDAはまた、深刻なまたは生命に危険な疾患または状態を治療するために1つまたは複数の他の製品と単独でまたは組み合わせて使用することが意図されている場合、迅速なチャネル審査のための製品を指定することができ、そのような疾患または状態が満たされていない医療需要を解決する可能性を示す。Fast Track製品については,スポンサーがFDAとより多くのインタラクションを行う可能性があり,FDAは申請完了前にFast Track製品申請部分の審査を開始する可能性がある。FDAがスポンサーから提出された臨床データを初歩的に評価した後にFast Track製品が有効である可能性があると判断すれば,スクロール審査が可能である。FDAはすでに末期或いは転移性三陰性乳癌の成人患者の治療にXMT-1660を許可した。
私たちはまた私たちの1つ以上の候補製品のための優先検討指定を求めることができる。FDAが候補製品が治療において大きな進展を遂げたと判断した場合、または適切な治療方法が存在しない治療方法を提供する場合、FDAは候補製品を優先的に検討するように指定する可能性がある。優先審査指定は、FDA審査申請の目標が6ヶ月であり、標準的な10ヶ月の審査期間ではないことを意味する。
このような指定はFDAの裁量権だ。したがって,我々の候補製品の1つがこれらの指定の基準を満たしていると考えても,FDAは同意せず,このような指定を行わないことにする可能性がある.また、指定を受けても、FDAの従来の手順に従って承認を考慮した製品と比較して、候補製品の指定を受けることは、より速い開発や規制審査や承認過程につながらない可能性があり、FDAの最終承認を確保することはできない。さらに、我々の1つまたは複数の候補製品がこれらの認証を取得する資格があっても、FDAは、これらの候補製品がもはや資格条件を満たしていないと後で決定することができ、またはFDAの審査または承認を決定する期間が短縮されないことができる。
連合で、私たちは未来に私たちの候補製品のために良質な称号を求めるかもしれない。PRIMEはEMAの役割を強化し、科学と監督管理支持を強化し、開発を最適化し、未満足の医療需要を解決する潜在力を有する重大な公衆衛生利益の新薬の評価を加速することを目的とした自発的計画である。この計画の重点は,EUに満足できる治療法がない疾患に対する薬物,あるいはそのような方法が存在しても,既存の治療法よりも大きな治療優位性を提供することである。Primeは開発中でEUで許可を得ていない薬品に限られており,出願人は集中手順で予備上場許可申請を申請しようとしている。Primeとして受け入れられるためには、候補製品はその主要な公共健康利益と治療革新方面の資格基準に適合しなければならず、この基準は声明を実証できる情報に基づいている。
良質な認証の利点は、医薬品委員会またはCHMP調査委員を任命し、上場許可申請の前に継続的な支援を提供し、知識の確立を支援すること、重要な開発マイルストーンで早期対話および科学的提案を行うこと、および製品の加速的な審査を行うことが可能であることを含み、これは、申請中に承認意見をより早期に発表するために審査時間を減少させることを意味する。PRIMEは申請者が同時にEMA科学提案と衛生技術評価提案を要求することができ、適時な市場進出を促進することができる。いずれの候補製品の良質な認証を得ても,従来のEMAプログラムと比較して,この認証は実質的に速い開発過程,審査または承認をもたらさない可能性がある。さらに、Prime称号を取得することは、EMAがマーケティング許可を付与する可能性を保証または増加させることを保証しない。
XMT−2056の孤児薬物指定を受けているが,他の候補製品の孤児薬物排他性は得られない可能性があり,我々が獲得しても,この排他性はFDAやEMAが他の競合製品を承認することを阻止しない可能性がある。
孤児医薬品法によれば、1つの製品がまれな疾患または疾患を治療するための医薬または生物学的製剤である場合、FDAはその製品を孤児薬として指定することができる。EU EMAは孤児製品の承認に対しても似たような規制制度を持っている。一般に、孤児薬物指定を有する候補製品がその後、その指定された適応を有する最初の発売許可を得た場合、製品は、一定期間内に市場排他期間を得る権利があり、これにより、FDAまたはEMAは、同じ製品の同じ治療適応の別のマーケティング申請をその期間内に承認することができない。適用期間はアメリカでは7年、EUでは10年だ。1つの製品が指定された孤児薬の基準をもはや満たしていない場合、特にその製品の利益が十分に高く、市場排他性がもはや合理的でない場合、EUの排他的期限は6年に短縮されることができる。
FDAが私たちの製品が孤児薬物の独占経営権を獲得することを承認するために、この機関は、米国の年間患者数が20万人未満の疾患または疾患の治療に指定されていることを発見しなければならない。FDAは孤児薬物の排他的な条件や疾患がこの基準を満たしていないと結論するかもしれない。私たちが製品の孤児薬物排他性を獲得しても、この排他性は、異なる製品が同じ条件で承認されることができるので、その製品を競争から効果的に保護することができない可能性がある。特に,遺伝子療法を背景に,孤児薬物排他性については,“同一薬物”を構成する概念は何が変化しているのか,FDAは最終指導意見を発表し,与えられたベクタークラス内の遺伝子組換えやベクターのわずかな違いだけで両遺伝薬物製品を異なる薬剤と見なすことはないことを示した。さらに、孤児薬が承認された後であっても、FDAが、より安全で、より効果的であることが証明されているので、後の製品が臨床的に優れていると結論した場合、FDAはその後、同じ疾患に対して同じ製品を承認することができる。FDAまたはEMAが、指定された要求に重大な欠陥があると判断した場合、または製造業者が稀な疾患または疾患を有する患者の需要を満たすのに十分な数の製品を保証できない場合、孤児薬の排他性を失う可能性もある。FDAは2022年5月、胃癌患者を治療するためのXMT-2056の孤児薬物指定を許可したが、将来的には任意の他の候補製品の孤児薬物独占経営権を得ることができない可能性がある。
2017年、国会はFDA 2017年再認可法案、FDARAと略称した。その他の事項以外に、FDARAはFDAが以前に存在した監督管理解釈を編纂し、薬品スポンサーに1種の孤児薬物の臨床優位性を証明することを要求し、この薬物は他の方面で以前許可された同じ稀な疾病を治療する薬物と同じであって、孤児薬物の排他性を獲得することができる。トランプ総裁が2020年12月27日に署名した総合的な立法によると、製品の臨床的優位性を示す要求は、2017年FDARA公布前に孤児薬物指定を受けたがFDAの許可や許可を得ていない薬物と生物製品に適用される。
FDAと議会は“孤児薬物法案”とその法規と政策をさらに再評価するかもしれない。第11巡回控訴裁判所の2021年9月の判決が特にそうである可能性があることを考慮すると、この判決は、排他的範囲を決定するために、用語“同じ疾患または状態”は、指定された“まれな疾患または状態”を意味し、FDAは、それを“適応または使用”を意味すると解釈することはできないと考えている。裁判所は,孤児薬の排他性は“適応や使用”ではなく,指定された疾患や状況全体に適用されると結論した。この決定を覆すための立法提案があったが、それらはまだ法律になっていない。FDAは2023年1月23日、裁判所の命令範囲を超える事項において、FDAは、孤児薬の独占性と孤児薬の使用または適応のために許可された使用または適応とを束ねて、その既存の法規を適用し続けると発表した。FDAや国会が将来、いつ、あるいはどのように孤児の薬物法規や政策を変更するかどうかもわかりませんし、どのような変化が私たちの業務に影響を与える可能性があるかどうかもわかりません。FDAがその孤児の薬物規制および政策を変更する可能性があることによると、私たちはXMT-2056のために得られた孤児薬物指定の任意の予想される利点を失う可能性があり、私たちの業務は悪影響を受ける可能性がある。
米国食品医薬品局、米国証券取引委員会および他の政府機関の資金不足は、政府の閉店やこれらの機関の運営の他の中断を含め、重要な指導部や他の人員の能力を雇用·保留し、新製品やサービスのタイムリーな開発や商業化を阻止するか、あるいはこれらの機関が私たちの業務運営に依存する可能性のある正常な業務機能を履行することを阻止することを阻害する可能性があり、これは私たちの業務に負の影響を与える可能性がある。
FDAの新製品の審査と承認能力は、政府予算と資金レベル、肝心な人員の雇用と維持及びユーザー費用の支払いを受ける能力、及び法定、監督と政策の変化を含む様々な要素の影響を受ける可能性がある。したがって、その機関の平均検討時間は近年変動している。FDAや他の機関の中断も、新製品候補製品が必要な政府機関によって審査および/または承認されるのに要する時間を遅らせる可能性があり、これは私たちの業務に悪影響を及ぼすだろう。また、政府が米国証券取引委員会や我々の業務に依存する可能性のある他の政府機関に提供する資金は、研究開発活動に資金を提供する機関を含め、政治プロセスの影響を受けており、政治プロセス自体が不安定で予測不可能である。
FDA、EMA、および他の機関の中断も、必要な政府機関の新薬の審査および/または承認に要する時間を遅らせる可能性があり、これは私たちの業務に悪影響を及ぼすだろう。例えば、近年、2018年や2019年を含め、米国政府は何度も閉店しており、米国食品·医薬品局や米国証券取引委員会などのある規制機関は、キー従業員を休暇させ、キー活動を停止せざるを得ない
また,新冠肺炎の大流行のような事件は中断する可能性がある。新冠肺炎の大流行期間中、一部の会社は完全な返信を受けたことを発表した。原因はアメリカ食品と薬物管理局がその応用に対する必要な検査を完成できないからである。将来的に類似した公衆衛生緊急事態が発生すれば、FDAは現在のペースを継続できない可能性があり、審査スケジュールが延長される可能性がある。米国以外で類似した状況に直面している規制当局は、類似した公衆衛生緊急事態に対応するために、同様の制限や他の政策措置をとる可能性があり、規制活動において遅延に遭遇する可能性がある
したがって、政府が長期的に停止したり、他の中断が発生したりすると、FDAが私たちが提出した規制文書を適時に審査して処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。今後の停止や他の中断は、米国証券取引委員会のような他の政府機関にも影響を与える可能性があり、これは、私たちの公開届出文書の審査(必要があれば)を延期し、私たちが公開市場に入る能力によって、私たちの業務に影響を与える可能性もあります。
FDAの加速承認は、現在または将来の任意の候補製品を承認しても、より速い開発や規制審査や承認過程をもたらすことはなく、私たちの候補製品が上場承認される可能性を増加させることはありません。
私たちはFDAの加速された承認手続きを使用して、私たちの現在と未来の任意の候補製品の承認を求めることができる。1つの製品が深刻または生命に危険な疾患を治療する場合、通常、既存の治療方法よりも有意な利点を提供し、臨床的利益を合理的に予測する可能性のある代替終点に影響を与える場合、製品は加速承認を得る資格がある可能性がある。FDA或いは他の適用される監督管理機関は代替終点が長期臨床利益を合理的に予測する可能性があるかどうかについて決定を下す。
加速承認を求める前に、FDAからのフィードバックを求め、このような加速承認を求める能力を他の方法で評価します。承認の一つの条件として,FDAは承認を加速させた製品のスポンサーに十分かつ良好に制御された発売後検証性臨床試験を行うことを求めている。これらの実証試験は職務調査で達成されなければならず、私たちはこれらの上場後の実証試験で異なるまたは追加的な終点を評価する必要があるかもしれない。これらの検証試験は、現在予想されているよりも多くの患者を募集する必要がある可能性があり、追加のコストをもたらす可能性があり、これは、現在予想されている推定コストよりも高い可能性がある。また、FDAは現在、承認を加速させる条件として販売促進材料を事前に承認することを要求しており、製品の商業発売時期に悪影響を及ぼす可能性がある。
FDAが提案されたエージェント端末に同意する保証はありませんし、加速承認または任意の他の形態の加速開発、審査、または承認を得るために、現在または未来の任意の候補製品のためにBLAを求めたり、提出したりすることを決定する保証もありません。同様に、FDAフィードバック後、私たちが最初にそうすることを決定しても、承認または任意の他の形態の加速開発、審査または承認を求めたり申請したりすることは保証されない。さらに、加速承認または他の迅速な規制指定された申請を提出することを決定した場合、そのような提出または申請が受け入れられるか、または任意の迅速な審査または承認がタイムリーに承認されるか、または全く保証されない保証はない。
例えば、私たちの候補製品の予期される臨床的利益を検証するために必要な試験が、そのような利点を検証できなかった場合、または薬剤に関連するリスクが合理的であることを証明するのに十分な臨床的利益を示さなかった場合、FDAは、加速承認経路に従って承認された候補製品の承認を撤回することができる。もし他の証拠が私たちの候補製品が使用条件下で安全でないか有効でないことを示している場合、私たちは私たちの候補製品に対して必要な承認後試験を行っていない、あるいは私たちの候補製品に関連する虚偽や誤った宣伝材料を散布しても、FDAは承認を撤回することができる。私たちの候補製品のための加速承認または任意の他の形態の加速開発、審査または承認を得ることができなかったか、または候補製品を撤回することは、候補製品の商業化の時間をより長くもたらし、候補製品の開発コストを増加させ、市場での競争的地位を損なう可能性がある。
2022年12月に可決された“食品·薬物総合改革法案”(FDORA)に伴い、国会は薬品や生物製品の承認を加速するためのいくつかの条項を改正した。具体的には、新しい立法認可FDAは、スポンサーが加速承認を得る前に検証的臨床試験を行うことを要求し、加速された承認された製品のスポンサーが6ヶ月ごとに承認後の研究の進展報告をFDAに提出することを要求し、検証性試験が製品の臨床的利益を検証できなかった後、加速手順を使用して新薬申請またはBLAの加速承認を撤回することを要求する。また,FDORAは,承認加速後にこのような研究を要求しないことを決定した場合には,そのサイト上で“なぜ承認後研究に適していないのか,あるいは不要な理由”を公表することを求めている。
最近,FDAは2023年3月にガイドライン案を発表し,現在承認を加速させている考え方と方法について概説した。FDAによると、癌の深刻性と生命に危害を及ぼす性質のため、加速承認経路は通常腫瘍薬物の審査に用いられる。単一アーム試験は通常加速承認を支持するために使用されるが、無作為対照試験はより信頼性の高い治療効果と安全性評価を提供し、利用可能な治療との直接比較を可能にするため、第一選択方法である。そのため、FDAは実験データの設計、進行と分析の考慮要素を概説し、これらの試験は腫瘍治療薬物の加速承認を支持することを目的としている。このガイドラインは現在草案形式であり,最終的に決定されても法的拘束力はないが,我々がどの製品の承認を加速するかを求める場合には,FDAのガイドラインを慎重に考慮する必要がある。したがって、私たちが加速的な承認を得たとしても、私たちはより速い開発や規制審査や承認過程を経験することができず、加速された承認を得ることは、最終的にFDAの完全な承認を得ることが保証されないかもしれない。
EUでは、必要なすべてのセキュリティおよび有効性データが取得されていない場合には、“条件付き”上場許可が付与される可能性がある。条件付きマーケティング許可は、欠落データを生成すること、またはセキュリティ対策を増加させるために必要な条件を保証することに依存する。条件付きマーケティング許可の有効期間は1年であり、すべての関連条件が満たされるまで年に1回更新しなければならない。適用可能な未定研究が提供されると、条件付きマーケティング許可は“標準”マーケティング許可となることができる。しかしながら、EMAによって設定された時間範囲内で条件が満たされていない場合、マーケティング許可は更新を停止する
FDA、EMA、または同様の規制機関が、私たちの任意の候補製品または治療製品のセットの承認に関連するセット診断テストの承認または承認を得ることを要求する場合、私たちは診断テストの承認または承認を得るために遅延に直面しており、候補製品を商業化することができない可能性があり、私たちの収益能力は実質的な損害を受ける可能性がある。
FDA、EMA、または同様の規制機関が、私たちの任意の候補製品を承認する際にセット診断テストの承認または承認を得ることを要求する場合、そのようなセット診断テストは、私たちのより高度な臨床試験および私たちの候補製品の商業化に使用される。これらのセットの診断技術を結合して候補製品を開発し、それを商業化することに成功するために、著者ら或いは著者らの協力者はいくつかの科学、技術、監督管理と後方勤務方面の挑戦を解決する必要がある。FDAの指導によれば、FDAがキット診断装置を決定することが、新しい治療製品または新しい適応を安全かつ有効に使用することを確保するために不可欠であり、セット診断が同時に承認または承認されない場合、FDAは通常、その治療製品または新しい治療製品の適応を承認しない。しかしながら、場合によっては(例えば、治療製品が深刻なまたは生命に危険を及ぼす疾患の治療を意図しており、満足できる利用可能な治療方法がない場合、または承認された製品のラベルが深刻なセキュリティ問題を解決するために修正される必要がある場合)、FDAは、診断の事前または同時マーケティング許可を伴うことなく治療製品を承認することができる。この場合、承認に伴う診断は発売後の要求や約束である可能性がある。
セットの診断と治療製品を共同開発することは精確な医学を推進するキーポイントである。共同開発は、開発開始時においても後の時点においても、治療製品および関連診断に伴う同期マーケティング許可の獲得に役立つ方法で行われるべきである。どの患者がその製品の治療から最も利益を得る可能性が最も高いかを決定するためにセット診断が必要である場合、どの患者が特定の治療製品を使用する治療によってより大きな深刻な有害事象リスクに直面しているか、またはより良い安全性または有効性を達成するために治療を調整するために特定の治療製品の治療反応を監視する必要がある場合、FDAは、癌治療のために治療製品を安全かつ有効に使用するために重要なすべてのセット診断テストが市場の承認を得ることを要求する。複数の外国の規制機関も体外随伴診断を医療機器として規制しており,これらの規制の枠組みの下では,我々が開発可能な任意の将来の診断の安全性と有効性を証明するための臨床試験が要求される可能性があり,これらの国が商業化される前に,単独の規制承認や承認が必要となると予想される。
診断に伴う治療製品ラベルの一部として承認され、治療製品の使用を、診断開発に伴う検出のための特定のゲノム変化または変異変化を伴う発現患者に制限する。FDA、EMA、または同様の規制機関が私たちの任意の候補製品の同時診断の承認または承認を要求する場合、製品候補の承認前、同時に、承認後であっても、私たちおよび/または将来の協力者は、これらの随伴診断の承認または承認を開発および取得する際に困難に直面する可能性がある。このような診断を受けたり作成したりする過程は時間も費用もかかる。FDAは以前、治療候補薬物を承認しながら発売前に承認或いはPMAを得るために、候補製品に反応する患者を選択するために体外随伴診断を要求してきた。臨床前および臨床データの収集およびFDAの提出および審査を含むPMAプロセスは、数年またはそれ以上を要する可能性がある。これは、厳格な上場前審査を含み、その間、スポンサーは、デバイスの安全性および有効性の合理的な保証を準備してFDAに提供し、デバイス設計、製造、およびタグを含むデバイスおよびそのコンポーネントに関する情報を提供しなければならない。設備が市場に投入された後、それは依然として重要な監督管理要求を受けて、開発、テスト、製造、流通、マーケティング、販売促進、ラベル、輸入、輸出、記録保存と不良事件報告の方面の要求を含む。
私たちまたは第三者パートナーは、規制機関の承認または承認を得たり、診断手順のいかなる遅延または失敗を承認したりしても、関連する候補製品の承認または営業継続を延期または阻止する可能性がある。さらに、2020年4月、FDAは、個別治療製品ではなく、より広範なラベル声明をサポートする提案を含む、特定の腫瘍治療製品の開発およびラベル随伴診断に関する新しいガイドラインを発表した。私たちはこのガイドラインが私たちのセット診断開発と戦略に与える影響を評価し続けるつもりだ。この指導およびFDA、EMAおよび他の規制機関の将来の発表は、私たちの候補製品のためのキット診断プログラムの開発に影響を与える可能性があり、規制承認または承認の遅延を招く可能性があり、あるいは私たちの候補製品にまだセット診断手続きが必要かどうかの決定を変更する可能性があります。私たちは、サブセットの人々のためのより広い主張またはより狭い主張を支持するために、追加の研究を行う必要があるかもしれない。また、他の承認された診断製品がそのラベル宣言を拡大し、将来承認される任意の候補製品がカバーする適応を含めることができれば、私たちの診断セット開発計画を継続する必要がなくなるかもしれないし、これらの診断セット開発戦略を変更する必要があるかもしれません。これは、販売セット診断テストから収入を得る能力に悪影響を及ぼすかもしれません。
また、私たちは第三者が私たちの候補製品の設計、開発、製造のためのセット診断テストに依存するかもしれません。もし私たちがこのような協力合意に到達すれば、私たちはこれらの診断に伴う許可や承認の開発と獲得における私たちの将来の協力と努力に依存するだろう。開発と監督管理承認或いは承認過程において、診断に伴う選択性/特異性、分析検証、再現性或いは臨床検証などの問題を解決する必要があるかもしれない。また,前臨床研究や早期臨床試験からのデータが候補製品開発に伴う診断を支持しているようであっても,その後の臨床試験で発生したデータは診断に伴う分析や臨床検証を支援できない可能性がある。私たちと将来のパートナーは、規制許可または承認を得て、私たちが直面している候補製品自体と類似した随伴診断方法を開発、取得すること、規制許可または承認を得ること、商業規模と適切な品質基準で十分な数の製品を生産すること、市場の承認を得ることなど、困難に直面する可能性があります。
もし私たちが候補製品のための診断プログラムを開発することに成功しなかった場合、あるいは開発を遅延させることができなければ、私たちの候補製品の開発は不利な影響を受ける可能性があり、私たちの候補製品はマーケティング承認を得ることができない可能性があり、マーケティング承認を得た候補製品のすべての商業潜在力を実現できないかもしれない。したがって、私たちの業務、運営結果、そして財務状況は実質的な被害を受ける可能性がある。また、当社と契約した診断会社は、候補製品の開発や商業化に期待されるセット診断テストの販売中止や製造を決定したり、その診断会社との関係が終了したりする可能性があります。私たちは、代替診断テストの供給を得るために、私たちの候補製品の開発および商業化のために、または商業的に合理的な条項でそうすることができない可能性があり、これは、私たちのセット診断および治療製品候補の共同開発または商業化に悪影響および/または遅延をもたらす可能性がある。
承認されれば、生物製品として許可され、規制された候補製品は、規制経路を簡略化することで承認された生体模倣薬の競争に直面する可能性がある。
BPCIAは“患者保護と平価医療法案”の一部として公布され、この法案は“衛生保健と教育負担能力調整法”によって改正され、あるいは総称して“ACA”と呼ばれ、生物類似と交換可能な生物製品を承認するための簡略化された方法を確立することを目的としている。規制経路はFDAのために生物類似生物製品を審査および承認する法律的権威を確立し、生物類似体と承認された生物製品との類似性に基づいて、それを“交換可能な”生物製剤として指定することを含む
BPCIAによると、基準生物製品は、初めて許可を得た日から12年以内にデータ独占権が付与され、FDAは、参照生物製品に基づく生物類似または交換可能製品の申請を受け入れず、基準製品が初めて許可された日から4年後になる。また、生物類似製品の許可は、基準製品が初めて許可された日から12年後にFDAによって発効する可能性がある。この12年間の独占期間内に、別の会社は、BLAが参照製品、スポンサーのデータを返信しない限り、または生物学的に類似した申請の形態で出願を提出する限り、競合生物の許可を開発して得ることができる
BLAによりバイオ製品として開発されたどの候補製品も12年の専門期間を得る資格があるべきであると考えられる。しかしながら、国会の行動または他の理由により、このような排他性は短縮される可能性があり、またはFDAは、我々の製品候補製品を競合製品の参考製品とみなさないかもしれず、これは、予想よりも早く生物類似競争のための機会を創出する可能性がある。さらに、承認されると、生物学的類似体が、どの程度、非生物製品に類似した伝統的な模倣薬代替の方法で任意の参照製品を置換するかは、まだ発展中のいくつかの市場および規制要因に依存するであろう。それにもかかわらず、競争の激化と価格設定圧力の増大により、私たちの候補製品と類似した生物の承認は、私たちの業務に実質的な悪影響を及ぼすだろう。
私たちの活動は、ヘルスケア提供者、第三者支払者、患者、政府関係者との交流を含み、医療、反腐敗、データプライバシーと安全、および消費者保護法律に関する広範な規制を受け続けている。適用法を遵守しないことは、巨額の罰金、契約損害、名声損害、収入減少、および私たちの業務の削減または再編を招く可能性があります。
私たちの活動は現在または将来的に医療保健、反腐敗、データプライバシー、および安全消費者保護に関連する様々な連邦や州法律に直接または間接的に支配される可能性がある。もし私たちの候補製品がFDAの承認を得て、アメリカでこれらの製品を商業化し始めたら、私たちのこのような法律の下での潜在的なリスクは著しく増加し、私たちはこのような法律を遵守することに関連するコストも増加する可能性がある。これらの法律には限定されません
•連邦虚偽声明、虚偽声明、および民事罰金法律は、他の者に加えて、政府資金を支払うために虚偽声明を故意に提出または提出させることを禁止するか、または虚偽クレームを得るために虚偽声明を作成または誘導することを禁止する
•連邦逆控除法は、他の人に加えて、個人の商品またはサービスの紹介または購入または注文を誘導するために、直接または間接的に提供、請求、受け入れ、または任意の報酬を提供することを禁止し、これらの報酬は、連邦医療保険および医療補助などの連邦保健プランに従って支払うことができる
•2018年に公布された連邦リベート禁止は、患者の特定の提供者(リハビリテーションの家、臨床治療施設、および実験室)への患者の転任に関するいくつかの支払いを禁止し、個人医療計画および政府医療計画によって精算されるサービスに適用されるリハビリテーションにおけるリベート禁止法案と呼ばれている
•連邦法は、1996年の健康保険携帯性および責任法、またはHIPAAと呼ばれ、医療提供者および他のエンティティのプライバシーを保護することに加えて、任意の医療福祉計画(個人医療計画を含む可能性がある)を詐欺する計画または医療保健事項に関する虚偽陳述を行うことを禁止する
•“食品、薬品および化粧品法”は、薬品マーケティングを厳格に管理し、メーカーがラベル外用途のためにこのような製品を販売することを禁止し、サンプルの配布を監督する
•連邦法律は、製薬業者に、特定の計算された製品価格を政府に報告するか、または政府当局または個人実体に、通常、政府医療計画下の精算条件として何らかの割引またはリベートを提供することを要求している
•いわゆる“連邦陽光”法は、製薬および医療機器会社に、教育病院、医師、およびいくつかの非医師従事者との間のいくつかの財務的相互作用を監視し、連邦政府に報告して、再公開することを要求する
•“HIPAA”のプライバシー、セキュリティ、および違反条項は、特定の“保険エンティティ”(ヘルスケア提供者、健康計画および医療情報交換所)およびその特定の“業務パートナー”請負者が、個人が識別可能な健康情報のプライバシー、安全、および送信を保護する義務があることを規定する
•州安全違反通知法、州健康情報プライバシー法、連邦と州消費者保護法を含む連邦と州法律法規は、健康に関連する個人情報および他の個人情報の収集、使用、開示および保護を管理する。
•市場活動を広く規制し、消費者の活動を損なう可能性がある連邦消費者保護法と不正競争法
•“海外腐敗防止法”、あるいは“海外腐敗防止法”と呼ばれる米国の法律であり、外国政府関係者(例えば、特定の医療専門家を含む可能性がある)との間のいくつかの財務関係を規定している
•州法は、個人医療計画、州プライバシー法、州消費者保護法、および製薬業者と医療提供者との間の相互作用を規制する州法を含む、任意の第三者支払人精算の項目またはサービスに適用可能な反リベートおよび虚偽請求法律のような、上記の各連邦法律と同様であり、このような財務的相互作用の開示を要求するか、または特定のコンプライアンス基準を強制的に採用することを要求し、多くの基準は重大な点で互いに異なり、通常は連邦法によって先制されず、コンプライアンス作業を複雑化させる。
また,我々の任意の候補製品の米国以外での規制承認や商業化も,上記の医療保健法や他の外国法の外国等価物の制約を受ける可能性がある。我々の業務手配が適用される医療保健法に適合することを確保する努力は巨額のコストに及ぶ可能性がある。政府と法執行当局は、私たちの商業行為は、解釈が適用される詐欺や乱用、または他の医療保健法律および法規の現在または未来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。私たちの運営が上記の任意の法律または私たちに適用される任意の他の政府法規に違反していることが発見された場合、私たちは、民事、刑事および行政処罰、損害賠償、罰金、返還、連邦医療保険、医療補助および他の連邦医療保健計画から除外される可能性があり、契約損害、名声損害、利益減少、および将来の収益減少、ならびに私たちの業務の削減または再編を含む罰を受ける可能性があります。
現在と未来の立法は候補製品のための私たちの精算の難しさとコストを増加させるかもしれない。
米国や一部の外国司法管轄地域では、医療システムの立法や規制の変化、提案された変化について多くの変化が続いており、これらの変化は、候補製品の上場承認を阻止または延期し、承認後の活動を制限または規制し、マーケティング承認を得た任意の製品を収益的に販売する能力に影響を与える可能性がある。現在の法律、および将来取られる可能性のある他の医療改革措置は、より厳しいカバー基準をもたらす可能性があり、私たちが受け取る可能性のある任意の承認製品の価格に追加の下振れ圧力をもたらす可能性があると予想しています。もし私たちの製品が清算や範囲が限られていなければ、私たちの業務は実質的な損害を受けるかもしれません。
2010年3月、オバマ総裁は“医療·教育負担能力調整法案”によって改正された“患者保護·平価医療法案”に署名し、総称して“平価医療法案”と呼ばれた。さらに、ACAが公布されて以来、他の立法改正も提案され、採択された。2011年8月、“2011年予算制御法案”(Budget Control Act Of 2011)などの法案は国会のための支出削減措置を制定した。赤字削減合同特別委員会の任務は、2013年から2021年までの間に少なくとも1.2兆ドルの赤字削減を提案することであるが、同委員会は必要な目標を達成できず、立法をいくつかの政府プロジェクトに自動的に削減することを触発した。これらの変化には,コロナウイルス援助,救済,経済安全法案やCARE法案により,プロバイダに支払う医療保険総金額が前期ごとに2%削減され,2013年4月に発効し,2031年まで有効となる
現在の立法により、医療保険支出の実際の減少幅は4%に達する可能性がある。総合支出法案は2022年12月に総裁·バイデンが署名して法律となり、医療保険計画の自動減額にいくつかの改正がなされた。総合支出法案第1001条は、2010年の4%の法定現金支払法またはPAYGO自動減額を2024年末まで2年延期する。“2021年米国救援計画法案”の公布により,医療保険計画を4%削減する計画が2023年1月に発効する。総合支出法案の医療補償タイトルには、2011年の連邦医療保険自動減額の2%予算制御法案を6ヶ月~2032年度に延長し、2030年度と2031年度の支払減少率を低減する第4163条が含まれている。
2012年の“米国納税者救済法”は,いくつかの医療サービス提供者に支払う連邦医療保険を減少させ,政府が提供者に多額の金を取り戻す訴訟時効を3年から5年に延長した。これらの法律は、連邦医療保険および他の医療資金のさらなる減少をもたらす可能性があり、他の方法で、規制によって承認される可能性のある任意の製品または候補製品の価格、またはそのような任意の製品の処方または使用頻度に影響を与える可能性がある。
ACAが公布されて以来、この法律の条項を廃止し、代替するために、多くの法的挑戦と国会行動が続くだろう。例えば、2017年に公布された“雇用減税法案”(Tax Act)に伴い、国会は“個人強制令”を廃止した。この条項は、ほとんどのアメリカ人が最低レベルの医療保険を購入することを要求する条項が2019年に施行される。2021年6月17日、米国最高裁はいくつかの州のPPACAに対する最新の挑戦を却下したが、PACAの合憲性を具体的に裁くことはなかった。ACAに関する訴訟と立法は継続される可能性があり、結果は予測不可能で不確実である。
トランプ政権はまた、ACAの実施を破壊または延期するための行政行動をとっており、ACAに従って権力および責任を有する連邦機関がACAの実施を放棄、延期、免除、または延期するように指示する任意の条項を含み、これらの条項は、各州、個人、医療保健提供者、医療保険会社または医薬品または医療機器製造業者に財政的または規制的負担をもたらす。しかし、2021年1月28日に、バイデン総裁はこれらの命令を撤回し、連邦機関に米国人の医療保険取得を制限するルールや他の政策を再検討するよう指示し、この獲得を保護し強化するための行動を検討するように指示した。この命令によれば、連邦機関は、新しい冠肺炎に関連する合併症を含む以前の疾患を有する人の保護の政策を弱めること、医療補助およびACAによるデモおよび免除によって、作業要求を含むカバー範囲または破壊計画を減少させることが可能な政策、医療保険市場または他の医療保険市場を破壊する政策、連邦医療補助およびACAに参加しにくくする政策、および扶養者の負担能力を含む保険または経済援助の負担可能性を低減する政策を再検討するように指示されるであろう。
2021年12月13日,EUは衛生技術評価又はHTAに関する第2021/2282号条例を採択し,第2011/24/EU指令を改正した。この規定は2022年1月に施行されたが、2025年1月から適用され、その間に実施に関する準備と手順が取られるだけである。適用されると、それは関連製品に従って段階的に施行されるだろう。この条例は、新しい医療製品およびいくつかの高リスクの医療機器を含むEU加盟国の衛生技術評価における協力を促進し、これらの分野で共同臨床評価を行うEUレベルの協力に基礎を提供することを目的としている。これは、EU加盟国がEU範囲内で汎用的なHTAツール、方法、およびプログラムを使用することを可能にし、患者に最大の潜在的影響を有する革新的な衛生技術の共同臨床評価、共同科学協議、開発者がHTA当局にアドバイスを求め、新興衛生技術および早期発見の将来性のある技術を決定し、他の分野で自発的な協力を継続することを含む4つの主要分野で協力することを可能にする。個別EU加盟国は、衛生技術の非臨床(例えば、経済、社会、倫理)の評価を引き続き担当し、定価と精算について決定する。
我々は,これらの医療改革,および将来とりうる他の医療改革措置は,連邦医療保険や他の医療資金のさらなる減少,より厳しいカバー基準,新たな支払い方法,および任意の承認された製品のために得られる価格および/または医師が市場に進出する可能性のある任意の承認された製品を管理することによって得られる補償レベルの追加的な下振れ圧力をもたらす可能性があると予想している。精算レベルの低下は私たちが受け取った価格や私たちの製品の処方や管理頻度に悪影響を及ぼす可能性があります。連邦医療保険や他の政府が計画している精算のいかなる減少も、個人支払者の支払いの同様の減少を招く可能性がある。したがって、これらの改革が発効すれば、市場で承認された候補製品の期待収入の開発に成功する可能性があり、我々の全体的な財務状況および候補製品の開発または商業化の能力に影響を与える可能性がある。
処方薬の米国や外国の司法管轄区域における価格はかなりの立法や行政行動の影響を受けており、許可を得ると、我々の製品の価格に影響を与える可能性がある。
アメリカでも処方薬の価格はずっと話題になっています。アメリカ議会は最近数回の調査を行い、州と連邦立法を提案し、公布し、薬品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、連邦医療保険と医療補助下の薬品コストを下げることを目的とした。2020年には、総裁·トランプ氏が処方薬のコスト削減を目的としたいくつかの行政命令を発表し、これらの命令のいくつかが条例に盛り込まれている。これらの規定には、価格最恵国モデルを実施し、連邦医療保険B部分のある医師が管理する薬品に対する支払いを他の経済先進国が支払う最低価格とリンクさせ、2021年1月1日から発効する臨時最終規則が含まれている。しかし,この規定は全国的な予備禁止の制約を受け,2021年12月29日,医療保険·医療補助サービスセンター(Center for Medicare&Medicaid Services,略称CMS)が最終規則を発表し,この規定を廃止した。CMSは、この規則の発表に伴い、それは価値をMedicare B部分の薬品支払いのすべての選択に組み入れ、そして受益者が根拠に基づく看護を獲得する機会を改善することを探索する。
また、2020年10月、HHSとFDAは、各州と他のエンティティが804条の輸入計画、すなわちSIPを制定し、いくつかの処方薬をカナダから米国に輸入することを許可する最終規則を発表した。この規定はPhRMAの訴訟で挑戦されたが,裁判所でPhRMAがHHSを起訴する資格がないことが発見された後,2023年2月に連邦地方裁判所に却下された。いくつかの州はカナダからの麻薬の輸入を許可する法律を採択した。いくつかの州は第804条輸入計画提案を提出しており、FDAの承認を待っている。FDAは2023年1月5日にフロリダ州のカナダ薬物輸入計画を承認した。この規定はまた、販売時点での値下げを反映するための新しい安全港を創出し、薬局福祉マネージャーと製造業者との間のいくつかの固定費用スケジュールのための新しい安全港を創出し、この計画の実施は、インフレ低減法案(IRA)によって2032年1月1日に延期された。
2021年7月9日、総裁·バイ登は14063号行政命令に署名し、その中で薬品価格などの問題に重点を置いた。この命令は衛生と公衆サービス部(HHS)に45日以内に1つの計画を制定し、処方薬の過度な定価を打撃し、国内の薬品サプライチェーンを強化し、連邦政府がこのような薬品に支払う価格を下げ、そして繰り返し出現した価格詐欺問題を解決するように指示した。2021年9月9日、HHSは薬品値下げ計画を発表した。この計画の主な特徴は,(A)メーカーとの薬品価格交渉を支援することにより,薬品価格がすべての消費者や医療システム全体により負担と公平になること,(B)サプライチェーンの強化を支援し,生体模倣薬や後発薬を促進し,透明性を増加させる市場改革により,処方薬業界全体の競争を改善·促進すること,(C)公共·民間研究を支援し,市場インセンティブを確保することで価値と入手可能な新しい療法の発見を促進し,科学的革新を促進し,より良い医療保健と健康改善を促進することである。
最近、2022年8月16日に“アイルランド共和軍”が総裁·バイデンによって法律に署名された。新しい立法は連邦医療保険D部分に影響を与え、D部分は計画であり、連邦医療保険A部分または連邦医療保険B部分に加入する個人は毎月外来処方薬保険料を支払うことを選択することができる。他の事項を除いて、アイルランド共和軍はある薬品のメーカーに連邦医療保険との価格交渉(2026年から)を要求し、価格は交渉できるが上限があり、連邦医療保険B部分と連邦医療保険D部分に基づいてリベートを実施し、インフレを超える価格上昇を処罰し(2023年に初めて満了)、D部分のカバーギャップ割引計画の代わりに新しい割引計画を用いる(2025年から)。アイルランド共和軍は衛生·公衆サービス部長官が最初の数年に規制ではなく指導によってその多くの規定を実施することを許可した。
具体的には、価格交渉では、国会はMedicareが、競争相手の模倣薬や生体模倣薬を持たず、Medicare B部分とD部分によって精算されることを許可した。CMSは、2026年からMedicare D部分によって支払われた10種類の高コスト薬物の価格を交渉することができ、その後、2027年に15種類のMedicare D部分薬剤を増加させ、2028年に15種類のMedicare B部分またはD部分薬剤を増加させ、2029年以降、毎年20種類のMedicare B部分またはD部分薬剤の価格を増加させることができる。この規定は、少なくとも9年間承認された医薬品および許可13年の生物製品に適用されるが、単一のまれな疾患または疾患のための許可された医薬および生物製品には適用されない。それにもかかわらず、CMSは価格交渉でこれらの製品の最高価格を決定する可能性があるため、もし私たちの製品がMedicare価格交渉の対象であれば、私たちは完全に政府行動のリスクに直面するだろう。また、存在する可能性のあるリスクを考慮すると、IRAのこれらの条項は、私たちの任意の候補製品が承認された場合、予想されるリターンを達成できない、またはこのような承認された医薬製品の特許を保護して、どのような承認された製品が発売されてから9年後に価格を制定するか、またはそのすべての価値を達成することができないというリスクをさらに増加させる可能性がある
また、この立法は、製薬業者が提供する価格が法律で規定された交渉で達成された“最高公平価格”以下でない場合、または値上げ幅がインフレを超える場合、製薬業者は民事罰金と潜在的な消費税を受けることになると規定している。この立法はまた、メーカーに連邦医療保険D部分の価格上昇幅がインフレを超えた薬品にリベートを支払うことを要求している。新法律では,2024年の連邦医療保険の自己払い薬品コスト上限は年間4000ドルと規定されており,その後2025年から年間2000ドルが上限となっている。さらに、Medicare Part D処方薬計画に参加した個人については、彼らが計画の高いハードルや“悲劇的な時期”に達する前に要求された保証範囲がその初期の年間保証限度額を超えていれば、アイルランド共和軍は個人に関する法的リスクを増加させる可能性がある。最初の年間保証限度額を超え、悲劇的な時期を下回るサービスを必要とする個人は、壊滅的な時期に達するまで100%の処方費用を支払わなければならない。他にも、アイルランド共和軍には、共同保険と共同支払いコストの削減、低所得補助金計画の資格の拡大、年間自己負担費用に価格上限を設定することで、個人の財務負担を軽減するための多くの条項が含まれており、各規定は定価と報告書に潜在的な影響を与える可能性がある。したがって,IRAがどのように実施されるかは不明であるが,いかなる連邦や州医療改革が我々にどのような影響を与えるかは確定できないが,このような変化は我々の活動に新たなあるいはより厳しい規制要求を加えたり,製品の精算を減少させたりする可能性があり,いずれも我々の業務,運営結果,財務状況に悪影響を及ぼす可能性がある。
2023年6月6日、メルク社はHHSとCMSを提訴し、アイルランド共和軍の連邦医療保険に対する薬品価格交渉計画が憲法第5改正案に違反した無償徴収を構成したと主張した。その後、米国商会、百時美施貴宝会社、PhRMA、アステラス、ノとノッド、楊森製薬、ノワワ、アスリコン、ブリンガー-インゲルハイムを含むいくつかの他の当事者も異なる裁判所に訴訟を提起し、HHSとCMSに対して類似した憲法クレームを提出した。私たちはIreland共和軍のこのような条項と他の条項に関連した訴訟が継続され、結果的に予測可能で不確実だと予想する。したがって,IRAがどのように実施されるかは不明であるが,いかなる連邦や州医療改革が我々にどのような影響を与えるかは確定できないが,これらの変化は我々の活動に新たなあるいはより厳しい規制要求を加えるか,あるいは我々の製品の精算を減少させる可能性があり,承認されれば,これらはいずれも我々の業務,運営結果,財務状況に悪影響を及ぼす可能性がある。
州レベルでは、各州はますます積極的に、価格または患者の精算制限、割引、ある製品への参入の制限、マーケティングコストの開示および透明性措置を含む薬品および生物製品の価格設定を制御するための法規を立法と実施し、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。さらに,地域医療機関や個別病院では,どの薬品やサプライヤーが処方薬や他の医療計画に含まれるかを決定するために入札プログラムが使用されるようになってきている。これらの措置が承認されると、私たちの製品に対する最終的な需要を下げたり、私たちの製品の価格設定に圧力をかけるかもしれません。私たちは将来、より多くの州と連邦医療改革措置を取ることが予想され、そのいずれも連邦と州政府が医療製品やサービスのために支払う金額を制限する可能性があり、これは私たちの候補製品に対する需要の減少または追加の価格設定圧力を招く可能性がある。
EUでは、承認されれば、同様の政治、経済、規制発展が候補製品を利益的に商業化する能力に影響を与える可能性がある。米国やEU以外の市場では,精算や医療保健支払い制度は国によって異なり,多くの国で特定製品や療法に価格上限が設定されている。欧州連合を含む多くの国では、処方薬の定価は政府の統制と参入を受けている。これらの国では、製品の発売許可を受けた後、政府当局との定価交渉にかなりの時間がかかる可能性がある。いくつかの国で精算或いは定価の承認を得るために、私たち或いは私たちの協力者は臨床試験を行い、私たちの製品の費用効果を他の利用可能な治療法と比較する必要があるかもしれない。もし私たちの製品が精算を受けられない場合、あるいは範囲や金額が制限されている場合、あるいは定価レベルが満足できなければ、私たちの業務は実質的な損害を受ける可能性があります。
私たちは、データプライバシーやセキュリティに関する厳しいプライバシー法、情報セキュリティ法律、法規、政策、契約義務の制約を受けており、これらの要求を守らなければ、巨額の罰金と処罰を受ける可能性があり、これは私たちの業務、財務状況、または運営結果に大きな悪影響を及ぼす可能性があります。
私たちは、個人情報の収集、送信、記憶、および使用に適したデータプライバシーおよび法律法規の保護の制約を受けており、その中には、米国、EU、英国の包括的な規制制度を含む、個人情報のプライバシー、セキュリティ、および伝送に何らかの要求を加えることが含まれている。プライバシーとデータ保護の立法と規制構造は世界各地の司法管轄区域で発展し続けており、プライバシーやデータ保護問題にますます注目されており、これは私たちの業務に影響を与える可能性がある。これらの法律および法規を遵守しないことは、罰金、監禁会社の役人および大衆の非難、影響を受けた個人の損害クレーム、私たちの名声被害、および名誉損失を含む、私たちに対する法執行行動を引き起こす可能性があり、これらはいずれも、私たちの業務、財務状況、運営結果、または将来性に重大な悪影響を及ぼす可能性がある。
アメリカには多くの連邦と州の法律法規が個人情報のプライバシーと安全に関連している。特に、HIPAAが公布した条例に基づいてプライバシーおよびセキュリティ基準が規定され、個別に識別可能な健康情報または保護された健康情報の使用および開示が制限され、保護された健康情報のプライバシーを保護し、保護された電子健康情報の機密性、完全性および可用性を確保するための行政、物質および技術保障措置の実施が要求される。保護された健康情報が適用されたプライバシー基準や我々の契約義務に従って処理されているかどうかを決定することは複雑である可能性があり,変化する解釈の影響を受ける可能性がある.このような義務は私たちの現在または未来の業務活動の一部または全部に適用されるかもしれない。
もし私たちが保護された健康情報のプライバシーと安全を適切に保護できなければ、私たちは私たちの契約に違反していることが発見されるかもしれない。また、適用されるHIPAAプライバシーやセキュリティ基準を含めて適用されるプライバシー法に従わなければ、民事や刑事罰に直面する可能性があります。HHS法執行活動は財務的責任と名声の損害を招く可能性があり、このような法執行活動に対する応答は大量の内部資源を消費する可能性がある。また、州総検察長は民事訴訟を起こし、禁止令や損害賠償を求め、州住民のプライバシーを脅かす侵害行為に応える権利がある。私たちはこのような規定が私たちの業務にどのように解釈、実行されるかを決定することができない。法執行活動や潜在的な契約責任に関連するリスクに加えて、私たちは連邦や州レベルで変化する法律と法規を遵守するために努力している可能性が高く、私たちの政策、手続き、システムを絶えず修正する必要があります。
HHSの潜在的な法執行に加えて、連邦貿易委員会(FTC)のプライバシー法執行を受ける可能性がある。連邦貿易委員会は、その最近の法執行行動を通じて、特に健康および遺伝子データの不正処理に注目し、連邦貿易委員会法案第5条に基づいて“不公平”と解釈されるプライバシー侵害のタイプと、健康違反通知ルールをトリガできると考えられる活動タイプを拡大している(連邦貿易委員会もこのルールを実行する権利がある)。その機関はまた、私たちの業務に影響を及ぼす可能性のある商業監視とデータセキュリティに関する規則を制定している。私たちは、私たちが取る可能性のある法執行行動のリスクを低減するために、連邦貿易委員会が進化していく規則と適切なプライバシーとデータセキュリティ慣行の指導を考慮する必要があるだろう。もし私たちが潜在的なFTC法執行行動の影響を受けたら、私たちは非常に具体的なプライバシーと(告発された違反の性質に依存する)遵守を要求する和解命令の制約を受けるかもしれない。もし私たちが連邦貿易委員会と合意したすべての同意命令に違反したら、私たちは追加的な罰金とコンプライアンス要求を受けるかもしれない。
各国では個人情報処理に関する具体的なルールも積極的に制定されている。2018年、カリフォルニア州は“カリフォルニア消費者プライバシー法”(California Consumer Privacy Act)を採択し、CCPAと略称し、2020年1月1日に施行され、カリフォルニア住民の個人情報を処理する企業に多くの要求を提出した。CCPAの多くの要件は、企業に、収集された彼らに関する情報およびそのような情報の使用および共有方法に関する通知をデータ主体に提供することを要求し、データ主体にそのような個人情報へのアクセスを要求する権利を付与し、場合によっては、そのような個人情報の削除を要求することを含む、以下にさらに説明する“一般データ保護条例”(GDPR)における要件と同様である。CCPAはまた,カリフォルニア住民にその個人情報を販売しないことを選択する権利を与えている。CCPAにはその要求に違反した会社に対する重大な処罰が含まれている。2020年11月、カリフォルニア州有権者は、2023年1月1日に施行されるカリフォルニアプライバシー権法案(CPRA)を採択し、CCPAを大幅に拡大し、カリフォルニア州住民に個人情報の使用、保持、共有を要求することが合理的に必要であり、収集または処理の目的に応じて、敏感な個人情報の追加保護を提供し、住民への保留情報の通知に関するより多くの開示を要求することを含むGDPRのような追加条項を採択した。CPRAはまた、CPRAおよびカリフォルニアの他のプライバシー法を実行することで、コンプライアンスリスクをさらに増加させる新しい実行機関であるカリフォルニアプライバシー保護局を作成した。CPRAの規定は私たちのいくつかの商業活動に適用されるかもしれない。
カリフォルニア州を除いて、少なくとも11州がCCPAとCPRAのような包括的プライバシー法を採択した。このような法律は施行されたか、2026年末までに施行されるだろう。これらの法律は,CCPAやCPRAと同様に,個人情報の処理に関する義務と,“敏感”データ(場合によっては健康データを含む)を処理する特殊な義務を規定している。このような法律のいくつかの規定は私たちの商業活動に適用されるかもしれない。またいくつかの州は、2024年以降に施行される2023年の立法会議中に包括的なプライバシー法を採択しているか、またはニューハンプシャー州およびニュージャージー州を含む。他の州は今後このような法律を考慮するだろうし、国会も連邦プライバシー法が採択されるかどうかを議論してきた。私たちの業務の健康情報に影響を及ぼすかもしれないいくつかの州の専門的な規制もある。例えば、ワシントン州は最近、健康情報の収集と共有を規範化する健康プライバシー法を採択し、この法律はまた個人的な訴権を有し、関連するコンプライアンスリスクをさらに増加させる。コネチカット州とネバダ州も同様の法律を採択して消費者の健康データを規制している。これらの法律は、私たちの研究対象の決定、ビジネスパートナーとの関係、そして最終的に私たちの製品のマーケティングと流通を含む、私たちのビジネス活動に影響を与えるかもしれません。
米国の法律と同様に、ヨーロッパや他の国にも重要なプライバシーやデータセキュリティ法が適用されている。欧州経済地域または欧州経済区に位置する個人の個人データ(個人健康データを含む)の収集、使用、開示、移転またはその他の処理、および欧州経済地域で行われる個人データ処理については、2018年5月に施行された一般データ保護条例(GDPR)によって規制されており、この条例は、当業界で運営する会社が個人データや国境を越えてこのようなデータを処理することに義務を負っている。GDPRは重い問責義務を規定し、データ管理者と処理者にそのデータ処理と政策の記録を保存することを要求した。私たちまたは私たちのパートナーまたはサービスプロバイダのプライバシーまたはデータセキュリティ対策がGDPR要件を遵守できない場合、私たちは、個人データを使用する方法および/または2000万ユーロまたは前期の世界年商4%までの罰金、および影響を受けた個人の賠償要求、負の宣伝、名声損害、および潜在的な商業および営業権損失を変更することを要求する訴訟、監督管理調査、法執行通知に直面する可能性があります。
GDPRは、個人データをEUから欧州委員会に国境を越えて移行させ、十分なデータ保護立法を提供していないと考えている国(例えば米国)に制限を加えている。会社が個人データをEUから他国に移す能力について、人々は懸念してきた。2020年7月、欧州連合裁判所(Court of the European Union、略称CJEU)は、個人データを欧州経済地域から米国に移転することを合法化する仕組みの一つである欧州連合-米国プライバシー保護法案(EU-U.S.Privacy Shield)の無効を発表した。CJEUの決定は、個人データを欧州経済圏から米国に移転する標準契約条項である別のデータ伝送手段の長期的な可能性も疑問視される。我々はプライバシー保護による自己認証を行っていないが,CJEUのこの決定は,EEAから米国へのデータ転送をより厳密に審査し,データプライバシー法規を遵守するコストや,サプライヤやパートナーと適切なプライバシーやセキュリティ協定を交渉するコストを増加させる可能性がある.
2022年10月、EU·米国プライバシーの盾の代替品として、EU-米国のデータプライバシーの枠組みを実施する行政命令に署名した。欧州委員会は2023年7月10日に充足性決定を採択した。十分な決定は、EU-米国データプライバシーフレームワークを自ら認証する米国社が、EUから米国にデータを送信するための有効なデータ転送機構とすることを可能にする。しかし、いくつかのプライバシー提唱団体は、EU-米国のデータプライバシーの枠組みに挑戦すると表明している。これらの挑戦が成功すれば、EU-米国のデータプライバシーの枠組みに影響を与えるだけでなく、標準契約条項や他のデータ伝送機構の生存能力もさらに制限される可能性がある。この問題をめぐる不確実性は私たちの業務に影響を及ぼす可能性がある。
連合王国のEU離脱後,2018年にイギリスデータ保護法はイギリスで行われた個人データ処理に適用され,GDPRが規定している義務と平行した義務が含まれている。英国の2018年の“データ保護法”は“施行”と“GDPR”の補完であり,2018年5月23日に王立の承認を得てイギリスで発効した。GDPRによると,欧州経済圏からイギリスへの個人データの移行は現在合法であり,欧州委員会が2021年6月に十分な決定を下したためである。しかし、この決定は法廷で挑戦されるかもしれない。連合王国は、連合王国からEU/欧州経済区へのデータが影響を受けないことを確実にするために、すべてのEU 27カ国および欧州経済圏加盟国がデータ保護において十分であると判断した
GDPRに加えて,世界ではプライバシーやデータセキュリティ法が制定されている国が増えている.多くの法律はGDPRに倣って手本としているが、他の法律は異なるまたは互いに衝突する条項を含む。これらの法律は、コンプライアンスコスト、契約に関連するコスト、および潜在的な法執行行動を増加させることによって、承認されれば、私たちの臨床試験および商業製品の販売および流通を含む業務活動を展開する能力に影響を与えるだろう。
最近のデータプライバシー規制の変化の影響を扱い続けているが,新法規の発効と継続的な法的挑戦に伴い,データプライバシーは国内レベルでも国際的にも変化しており,変化するデータ保護ルールを遵守する努力は成功しない可能性がある.これらの法律の解釈と適用は私たちのやり方と一致しない可能性があり、これは私たちの業務に悪影響を及ぼすかもしれない。私たちはこの変化する状況を理解して順応するために多くの資源を投入しなければならない。個人情報のプライバシーや安全に関する連邦、州、国際法律を守らないと、このような法律で定められた罰金や処罰に直面する可能性があります。
私たちが将来所有する可能性のある任意の国際業務を管理する法律法規は、米国以外で特定の製品を開発、製造、販売することを阻止し、コストの高いコンプライアンス計画の制定と実施を要求するかもしれない。
私たちの業務をアメリカ以外に拡張すれば、国際業務に関する米国の法律と、私たちが運営·計画している各司法管轄区域の法律を遵守するために、より多くの資源を投入する必要があるだろう。“海外腐敗防止法”は、いかなる米国の個人または企業が、いかなる外国人官僚、政党または候補者に直接的または間接的に支払い、支払いを提供または許可するか、または任意の価値のあるものを提供することを禁止し、その個人または企業が業務を獲得または保持することを支援するために、外国の実体の任意の行為または決定に影響を与えることを目的としている。“海外腐敗防止法”はまた、米国に上場する証券会社にある会計条項を遵守することを要求し、これらの条項は、会社(国際子会社を含む)のすべての取引の帳簿と記録を正確かつ公平に反映し、国際業務のために適切な内部会計制御システムを設計し、維持することを要求する。
“反海外腐敗法”を遵守することは高価で困難であり、特に腐敗は公認問題である国である。また、海外腐敗防止法は製薬業に特別な挑戦をもたらしており、多くの国では病院が政府によって運営されているため、医師や他の病院従業員は外国人官僚とされている。臨床試験やその他の仕事に関連して病院に支払われた何らかの金は、政府関係者に支払われた不正金と考えられ、“海外腐敗防止法”の法執行行動につながった。
様々な法律、法規、および行政命令はまた、国家安全目的のために秘密にされた情報、および特定の製品およびこれらの製品に関連する技術データ、またはいくつかの非米国国民との共有を米国国外で使用および伝播することを制限する。さらに、欧州連合は、医師の処方、推薦、裏書き、購入、供給、注文、または医療製品の使用を誘導または奨励するために、医師に福祉または利益を提供することを禁止する。医師への福祉や利点の提供も、英国の“2010年反賄賂法”のようなEU加盟国の反賄賂法の管轄を受けている。このような法律に違反することは巨額の罰金と監禁につながるかもしれない。いくつかのEU加盟国では、医師に支払われる費用は公開されなければならない。また、医師との合意は、通常、医師の雇用主、医師の主管専門組織、および/またはEU加盟国の監督当局に事前に通知し、承認しなければならない。これらの要件は、EU加盟国に適用される国家法律、業界規則、または専門行為規則に規定されている。このような要求を守らないことは、名声のリスク、公開非難、行政処罰、罰金、または監禁につながる可能性がある。
もし私たちがアメリカ以外での業務を拡大すれば、これらの法律を遵守するためにもっと多くの資源を投入する必要があります。これらの法律は、私たちがアメリカ以外で特定の製品や候補製品を開発、製造、販売することを阻止するかもしれません。これは、私たちの成長潜在力を制限し、私たちの開発コストを増加させるかもしれません。国際ビジネス慣行に関する法律を遵守しなければ、重大な民事·刑事罰を受け、政府契約の資格を一時停止または廃止する可能性がある。米国証券取引委員会は、発行者が海外腐敗防止法の会計規定に違反したため、発行者の米国取引所での証券取引を一時停止または禁止する可能性もある。
私たちと私たちの第三者契約製造業者は環境、健康、安全法律法規を守らなければなりません。これらの法律法規を守らなければ、私たちは大きなコストや責任に直面するかもしれません。
私たちと私たちの第三者製造業者は、研究室プログラムおよび危険材料と廃棄物の使用、生成、製造、流通、貯蔵、処理、処理、救済、処分を管理する法律法規を含む多くの環境、健康、安全法律法規の制約を受けている。可燃性および生物学的材料を含む危険化学品は、当社の業務のいくつかの態様に関連して、使用、発生、製造、流通、貯蔵、運搬、処理、または危険材料および廃棄物の処理による傷害または汚染リスクを除去することができません。汚染や傷害が発生したり、環境、健康、および安全法律法規を遵守できなかった場合、私たちはそれによって生じる任意の損害に責任を負う可能性があり、任意のこのような責任は私たちの資産および資源を超える可能性がある。民事や刑事罰金やこのような法律や法規を遵守しない罰に関する巨額の費用を招く可能性もある。危険材料の使用による従業員の負傷により生じる可能性のあるコストや支出を支払うために労働者補償保険を維持しているが、潜在的な責任を支払うのに十分ではない可能性がある。私たちは私たちが生物、危険または放射性物質を貯蔵したり処分したりすることによって、私たちが提起した環境責任や有毒侵害に対して保険を維持することはできません。
環境、健康、そして安全の法律法規はますます厳しくなっている。現在または未来の環境、健康、安全法律法規を遵守するために、私たちは巨額のコストを発生させるかもしれない。このような現行または未来の法律法規は私たちの研究、開発、または生産努力を損なうかもしれない。私たちがこのような法律を守らないことはまた巨額の罰金、処罰、または他の制裁につながるかもしれない。
また、私たち第三者契約製造業者の運営については、適用される環境、健康および安全法律法規に準拠していない場合、または私たちの製品に関連する廃棄物を適切に処理することができなければ、それによって生じるいかなる損害、名声被害、または私たちの候補製品または製品の製造および供給中断に責任を負わなければならないかもしれません。
私たちの従業員は規制基準と要求を遵守しないこと、インサイダー取引を含む不当な行為や他の不正活動に従事する可能性がある。
私たちは従業員詐欺や他の不適切な行為の危険に直面している。従業員の不正行為には、FDAの規定を故意に遵守しないこと、FDAに正確な情報を提供しないこと、私たちが確立した製造基準を遵守しないこと、連邦および州医療詐欺および法律法規の乱用を遵守しないこと、州および連邦証券法を遵守しないこと、財務情報やデータを正確に報告しないこと、または不正な活動を開示することが含まれるかもしれない。特に、医療業界の販売、マーケティング、商業配置は、詐欺、リベート、自己取引、その他の乱用を防止するための広範な法律法規によって制約されている。これらの法律法規は、広範な価格設定、割引、マーケティングおよび販売促進、販売手数料、顧客インセンティブ計画、および他のビジネス計画を制限または禁止する可能性があります。従業員の不当行為はまた臨床試験過程で得られた情報を不当に使用する可能性があり、これは規制制裁と著者らの名声に深刻な損害を与える可能性がある。従業員の不正行為を常に識別し、阻止することができるわけではなく、このような行為を発見し、防止するための予防措置は、未知または未管理のリスクや損失を効果的に制御することができないか、またはそのような法律や法規を遵守しないことによる政府の調査または他の行動または訴訟から私たちを保護することができない可能性がある。また、私たちは、起きていなくても、このような詐欺や他の不正行為を告発する可能性があるというリスクに直面している。もし私たちにこのような訴訟を提起した場合、私たちは私たちの権利を弁護または維持することに成功しませんでした。これらの訴訟は、民事、刑事および行政処罰、損害賠償、罰金、返還、連邦医療保険、医療補助および他の連邦医療保健計画から除外される可能性があり、契約損害、名声損害、利益減少および将来の収益減少、および私たちの業務の削減または再編を含む、私たちの業務運営能力および運営結果に悪影響を及ぼす可能性があります。
米国と国際貿易政策の変化、特に中国に関する政策変化は、我々の業務や経営業績に悪影響を及ぼす可能性がある
米国政府が最近発表した声明と取られたいくつかの行動は、中国製のいくつかの製品に対する数回の関税と輸出規制制限を含む米国と国際貿易政策の潜在的な変化を招く可能性がある。2018年3月、トランプ政権は米国に進出した鉄鋼とアルミニウムに関税を課すことを発表し、2018年6月、トランプ政権は中国から輸入された商品に対するさらなる関税の徴収を発表した。最近、中国と米国はそれぞれ関税を徴収しており、米国商務省が大量の中国実体をその“未確認リスト”に追加することを含む、より多くの貿易障壁が出現する可能性があることを示しており、米国の輸出業者にこのような実体に商品を輸出する前により多くの手続きを経ることが求められている。新たな関税、輸出規制、または他の新しい法律や法規が取られるかどうか、またはそのような行動が私たちまたは私たちの産業にどのような影響を与えるかは不明であり、バイデン政府がこれらの措置を逆転させたり、同様の政策措置を取るために努力するかどうかも不明である。最近では2024年2月、米議会議員が中国バイオテクノロジー会社の薬明カントと薬明生物(あるいは総称して無錫)の調査を呼びかけ、中国軍との関連がある疑いがあることを理由に経済制裁を実施する可能性がある。輸出規制、資本規制または関税のような不利な政府の国際貿易政策は、私たちの候補製品およびプラットフォーム材料の製造コストを増加させ、私たちの薬品に対する需要に影響を与え(承認されれば)、私たちの候補製品の競争地位に影響を与え、私たちと私たちの協力者が臨床前研究および臨床試験で使用した原材料と候補完成品の輸出入、特に私たちが中国から輸入した任意の候補製品および材料について、私たちと無錫による製造サービスの手配を含む。任意の新しい関税、輸出規制、立法および/または法規が実施される場合、または既存の貿易協定が再交渉される場合、または特に米国または中国政府が最近の貿易緊張のために報復貿易行動をとる場合、これらの変化は私たちの業務、財務状況、および運営結果に悪影響を及ぼす可能性がある
私たちのビジネスや産業に関するリスクは
もし私たちが高級管理者と肝心な科学者を引き付けることができなければ、私たちは私たちのADC候補製品の開発に成功し、私たちの臨床試験を行い、そして私たちのADC候補製品を商業化することができないかもしれない。
私たちが競争の激しい生物技術と生物製薬業界の中で競争できるかどうかは、私たちが高い素質の管理、科学と医療人員を誘致、激励と維持できるかどうかにかかっている。私たちはマーティン·フーベル医学博士、私たちの総裁、最高経営責任者など、私たちの上級管理職のメンバーに強く依存しています。彼らは2023年9月にアンナ·プロットパースの後を継いでいます。私たちはまた、首席医療官と首席人事官が2023年9月に退職することを発表しました。他の上級管理職のメンバーを失ったサービスは、研究、開発、商業化の目標の実現を阻害する可能性があります。しかも、この人たちのみんなはいつでもわが社での彼らの雇用関係を終わらせることができる。私たちは私たちの役員や他の職員たちに“キーパーソン”保険を提供しない。
合格した科学、臨床、販売とマーケティング人員を募集し、維持することも私たちの成功の鍵になるだろう。私たちはマサチューセッツ州ケンブリッジ市の工場で業務を展開しており、この地域は多くの他の生物製薬会社と多くの学術·研究機関の本部所在地である。技術人材に対する競争は非常に激しく、流出率は非常に高いかもしれないが、これは私たちが受け入れ可能な条件で高い素質の人員を採用と維持する能力を制限し、甚だしきに至っては全くできないかもしれない。多くの製薬とバイオテクノロジー会社の間の類似者に対する競争を受けて、私たちは受け入れ可能な条件でこれらの人員を吸引し、維持することができないかもしれない。また,2023年7月にわれわれのUplit臨床試験が主要終点に達していないことを発表した後,当時の従業員基盤を約50%削減するすなわち再編を発表し,2023年12月31日現在で再編がほぼ完了している。再編成は未来に合格者を維持して採用することをもっと難しくするかもしれない。また、私たちは、科学と臨床コンサルタントを含むコンサルタントとコンサルタントに依存して、私たちの研究開発と商業化戦略の制定を助けてくれます。私たちのコンサルタントやコンサルタントは採用されたり、他のエンティティと相談や相談契約を締結したりする可能性があり、これは彼らに対する私たちの利用可能性を制限するかもしれません。
システム障害、セキュリティホール、またはネットワーク攻撃が発生した場合、私たちの業務と運営は影響を受けます。
当社のCROおよび他の請負業者、コンサルタント、および法律および会計会社を含む、私たちと協力し、私たちが依存または将来依存する可能性のある様々な第三者のコンピュータシステムは、ネットワーク攻撃、コンピュータウイルス、恐喝ソフトウェア、マルウェア、不正アクセス、自然災害、テロ、戦争、および電気通信および電気故障を含むサービス中断またはセキュリティ破壊の影響を受けやすい。 私たちは、私たちの第三者プロバイダに有効なセキュリティ措置を実施し、そのような任意の故障、欠陥、または違反を識別して修正することに依存します。世界各地からの未遂攻撃と侵入の数、強度と複雑性の増加に伴い、セキュリティホール或いは破壊のリスクは普遍的に増加し、特にコンピュータハッカー、外国政府、民族国家行為者とネットワークテロリストを含むネットワーク攻撃或いはネットワーク侵入を含む。特に、深刻な恐喝ソフトウェア攻撃が一般的になってきており、当社の運営を深刻に中断させる可能性があり、当社の製品やサービスを提供する能力、敏感なデータや収入の損失、名声被害、資金移転を招く可能性がある。恐喝支払いは恐喝ソフトウェア攻撃の負の影響の持続時間を短縮する可能性があるが、例えば、適用された法律または法規によってそのような支払いを禁止するため、そのような支払いを望まないか、または支払うことができない可能性がある。しかしながら、何らかの故障、事故、またはセキュリティホールが発生し、私たちの運営または私たちと契約した第三者の運営中断を招く場合、私たちの計画および業務運営の実質的な中断を招く可能性があります。
私たちのほとんどの従業員たちは混合方式で働いていて、私たちはまた遠隔勤務の従業員たちを持っている。 このような配置は私たちの情報技術システムとデータのリスクを増加させます。私たちはより多くの従業員が私たちのオフィスやネットワークの外でネットワーク接続、コンピュータ、設備を使用して、家で、途中、公共の場所で働くことを含むからです
私たちは過去に未遂のネット釣り攻撃を経験したが、私たちの運営に実質的な影響を与えなかった;しかし、将来私たちは重大なシステム故障やセキュリティホールに遭遇する可能性があり、これは私たちの運営を中断させたり、私たちの開発計画を実質的に破壊したりする可能性がある。私たちは私たちの商業秘密や他の固有の情報を得ることができないかもしれないし、他の中断を経験することは、修復するために多くの資源がかかるかもしれない。例えば、私たちの候補製品の臨床試験データの紛失は、私たちの規制承認作業を遅延させ、データを回復または複製するコストを著しく増加させる可能性があります。
また、情報システムおよびネットワークにおいて維持されている情報(従業員または他の人の個人情報を含む)が盗用、誤用、漏洩、改ざん、または故意または意外な漏洩または損失によって引き起こされるリスクに直面する可能性がある。外部側は、私たちのシステムまたは私たちと契約した第三者のシステムに侵入しようと試みたり、詐欺的な手段で私たちの従業員またはそのような第三者の従業員に敏感な情報を開示させて、私たちのデータにアクセスするように強要したりすることができます。時間が経つにつれて、このような脅威の数と複雑さは増加している。これらの事件の発生を防ぐためのシステムや制御を開発·維持しているにもかかわらず、脅威を識別·緩和する流れもありますが、このようなリスクは解消できません。また、私たちまたは私たちと契約した第三者が、もしあれば、このような破壊やセキュリティホールを迅速に発見する保証はありません。また、これらのシステム、制御、プロセスの開発と維持費用が高く、技術の変化と安全対策の克服に伴う努力がより複雑になり、監視と更新を続ける必要がある。任意の中断またはセキュリティホールが、私たちの技術または候補製品に関連するデータやアプリケーションまたは他のデータまたはアプリケーションの損失または破損、または機密または独自の情報を適切に開示しない場合、私たちは責任を招く可能性があり、私たちの競争地位や市場の私たちのセキュリティ対策の有効性に対する見方が損なわれる可能性があり、私たちの信頼が損なわれる可能性があり、私たちの候補製品のさらなる開発が延期される可能性がある。
ソーシャルメディアおよび人工知能ベースのプラットフォームをますます使用することは、責任、データセキュリティおよびプライバシー法違反、または名声被害をもたらす可能性がある。
私たちと私たちの職員たちは内部と外部のコミュニケーションの手段としてソーシャルメディアツールをますます利用している。変化するソーシャルメディア伝播ガイドを監視し、適用規則を遵守しようと努力しているにもかかわらず、私たちまたは私たちの従業員がソーシャルメディアを使用して私たちの製品やビジネスを伝播することは、適用要件に違反していることが発見される可能性があります。さらに、私たちの従業員は、わざとまたは意図的に、私たちのソーシャルメディア政策または他の法律または契約要件に適合しない方法でソーシャルメディアを使用することができ、これは、商業秘密または他の知的財産権の損失をもたらす責任が生じる可能性があり、または私たち従業員、臨床試験参加者、および他の人の個人データ公開をもたらす可能性がある。さらに、ソーシャルメディア上の私たちまたは私たちの候補製品に関する否定的な投稿やコメントは、私たちの名声、ブランドイメージ、および名声を深刻に損なう可能性があります。また,人工知能やAIに基づく解決策は,生成性AIを含めて,我々を含むバイオテクノロジーやバイオ製薬業界でますます利用されている.私たちの従業員または私たちが依存する第三者が人工知能解決策を使用することは、引き続き増加する可能性があり、私たちの内部政策、データ保護法、他の適用可能な法律または契約要件に違反する機密情報(個人データおよび固有情報を含む)の開示をもたらす可能性がある。人工知能解決策の濫用は責任を生じ、商業秘密或いは他の知的財産権の損失を招き、名声損害を招くか、或いは意外な偏見或いは他の結果の結果を招く可能性がある。人工知能ソリューションの乱用はまた、私たちの従業員、臨床試験参加者、協力者、または他の第三者の個人データへの不正アクセスおよび使用をもたらす可能性があります。これらの事件のいずれも、私たちの業務、見通し、経営結果、財務状況に重大な悪影響を及ぼす可能性があり、私たちの普通株の価格に悪影響を及ぼす可能性があります。
私たちは未来の成長と事業の成功的な拡大を管理する上で困難に直面するかもしれない。
2023年にUpRiの開発を中止した後に再編を実施したにもかかわらず,臨床試験と商業化による現在の候補製品の推進を求めるにつれ,我々の開発,規制,製造,マーケティング,販売能力を拡大し,あるいは第三者と契約を締結し,これらの能力を提供する必要がある。私たちの業務は過去に拡大しているので、私たちの業務が今後再び拡大すれば、様々な戦略パートナー、サプライヤー、他の第三者とのより多くの関係を管理し続ける必要があると予想されます。未来の成長は経営陣の会員たちにもっと多くの重大な責任を負わせるだろう。私たちの将来の財務業績と私たちが候補製品を商業化し、効果的に競争する能力は、私たちが未来の成長を効果的に管理する能力にある程度依存するだろう。そのためには、私たちの開発作業や臨床試験を効率的に管理し、より多くの管理、行政を採用、訓練、統合することができなければなりません。必要であれば、販売やマーケティング担当者も含めなければなりません。私たちの限られた財務資源と、このような予想成長に関連する物流と運営の変化を管理するために、私たちはこれらの任務を達成できないかもしれません。もし私たちがそのいずれかの任務を達成できなければ、私たちが会社の発展に成功したり、私たちの運営を中断したりするのを阻害するかもしれません。
もし私たちに製品責任訴訟や他のクレームを提起すれば、私たちは大量の責任を負い、私たちのADC候補製品の商業化を制限することを要求されるかもしれません。
私たちの候補製品の臨床テストのため、私たちは固有の製品責任リスクに直面しています。もし私たちがどんな製品を商業化すれば、私たちはもっと大きなリスクに直面します。例えば、私たちが開発した任意の製品が臨床試験、製造、マーケティング、または販売中に引き起こされ、または傷害をもたらすと考えられ、または他の態様では適切でない場合、私たちは起訴されるか、または他のクレームを提起される可能性がある。このような製品責任クレームは、製造欠陥、設計欠陥、製品固有の危険について警告、不注意、厳格な責任と保証違反の告発を含む可能性がある。クレームはまた国や外国消費者保護法や同様の計画に基づいて提起されることができる。もし私たちが製品責任クレームで自分自身を弁護することに成功できなければ、私たちは重大な責任を招いたり、私たちの候補製品の商業化を制限することを要求されるかもしれません。成功的な防御であっても、多くの財政的で管理的な資源が必要だ。事件がどうであっても最終的な結果がどうであろうと、賠償責任は次のようになるかもしれない
•私たちの名声を損なう
•私たちの候補製品や私たちが開発する可能性のある製品の需要が減少した
•臨床試験参加者の脱退
•関連訴訟の弁護費用
•経営陣の時間と資源を移転する
•臨床試験参加者や患者に大量の金銭的報酬を与える
•製品のリコール、撤回またはラベル付け、マーケティング、または販売促進制限;
•収入損失
•候補品を商業化することはできません
•私たちの株価は下落しています。
潜在的な製品責任クレームを防止するために、許容可能なコストで十分な製品責任保険を得ることができず、我々が開発した製品の商業化を阻止または阻害する可能性がある。私たちは現在私たちの臨床試験に製品責任保険を提供して、総金額は1000万ドルです。私たちはこのような保険を受けますが、私たちに対するいかなるクレームも、裁判所の判決や和解の金額が私たちの保険の全部または一部の保険範囲内ではないか、あるいは私たちの保険範囲を超えてしまう可能性があります。私たちの保険証書にも様々な例外があります。私たちは製品責任クレームの影響を受けるかもしれません。私たちは保険範囲を持っていません。この場合、私たちは、裁判所によって裁決されたり、和解協定で協議された、私たちの保険範囲を超えたり、私たちの保険カバー範囲内にない金額を支払わなければならないかもしれません。私たちは、これらの金額を支払うために十分な資本を得るか、または得ることができません。許容可能なコストで十分な保険範囲を獲得または維持することができない場合、または潜在的な製品責任クレームに対して他の方法で保護を提供することができない場合、候補製品の開発および商業生産および販売を阻止または抑制する可能性があり、これは、私たちの業務、財務状況、運営結果、および見通しに悪影響を及ぼす可能性がある。
私たちは将来資産を買収したり戦略同盟を構成したりするかもしれないが、私たちはこの買収のメリットを意識しないかもしれない。
私たちはより多くの技術や資産を獲得し、第三者と戦略同盟を結んだり、合弁企業を設立したりする可能性があり、これらは私たちの既存の業務を補完または拡大すると信じています。将来性のある市場や技術の資産を買収すれば、既存の技術と組み合わせることに成功しなければ、これらの資産を買収するメリットを実現できないかもしれません。私たちは戦略連合や買収によって生じた任意の新製品を開発、製造、マーケティングする時に多くの困難に直面するかもしれません。これらの新製品は私たちが期待した利益を達成したり、私たちの業務を強化したりすることを延期したり阻止したりすることができます。このような買収の後、取引が合理的であることを証明するために、予想される相乗効果を達成することは保証できません
私たちの普通株に関するリスクは
もし私たちの株価が変動すれば、私たちの株主は大きな損失を受けるかもしれない。
私たちの株価はずっと変動していて、変動し続けるかもしれない。2021年2月23日から2024年2月23日までの間、私たちの普通株の終値は1株19.78ドルの高位から1株1.06ドルの低位まで様々である。我々普通株の市場価格は、本“リスク要因”部分に列挙された多くのリスク要因および他の制御できない要因によって、広範な変動の影響を受ける可能性がある
•XMT-1660およびXMT-2056を含む、現在または未来の候補製品の臨床前研究および臨床試験の結果および時間;
•競争相手製品の臨床試験の結果
•私たちのビジネス秘密を十分に守ることができませんでした
•私たちが追加資本を調達する条件や資金を集める能力
•戦略的協力や許可の設定を開始または終了します
•規制の動きは、私たちの製品や競争相手の製品に対する行動を含む
•財務状況と経営業績の実際または予想変動
•証券アナリストは私たちまたは私たちの競争相手や私たちの業界に関する研究報告を発表した
•私たちまたは私たちの競争相手が私たちまたは私たちの競争相手が市場のアナリストに与える予測や導きに到達できなかった
•キーパーソンの増減
•買収、剥離、剥離、合弁企業、戦略投資、または業務戦略の変化など、私たちの競争相手の戦略決定
•私たちやこの業界の立法や他の規制発展に影響を与えることで;
•医療支払い制度の構造を変え
•投資家は会社の評価の変動は私たちと互角だと思っています
•私たち(私たちのATM機の発売計画を含む)、私たちの内部者、あるいは他の株主は私たちの普通株を売却します
•ジャーナリズムや投資界の投機行為
•追加的な資金調達努力が発表されるか、または予想される
•バイオ製薬株の市場状況の変化
•一般的な市場と経済条件の変化、ロシアとウクライナの間の持続的な衝突、イスラエルとハマスの間の持続的な戦争を含む地政学的紛争など、金利とインフレが高止まりしている。
また,株式市場は従来から大きな変動,特に製薬,バイオテクノロジー,その他の生命科学会社の株を経験してきた。製薬、バイオテクノロジー、その他の生命科学会社株の変動性は、その株に代表される会社の経営業績とは無関係であることが多い。このような変動により、株主は彼らが株式を購入したかそれ以上の価格で普通株を売ることができない可能性がある。私たちは単一の業界を経営しているので、私たちは特にこれらの要素の影響を受けやすいです。これらの要素は私たちの業界や私たちの製品に影響を与え、あるいは私たちの市場に小さい程度影響を与えます。また、このような変動により、私たちはナスダック株式市場の上場要求を守ることができないかもしれない。過去、証券集団訴訟は会社の株価が変動した後に提起されることが多かった。このようなタイプの訴訟は、巨額の費用を招き、私たちの経営陣の関心や資源を分散させる可能性があり、判決を履行したり、訴訟を終わらせたりするために多額のお金を支払う必要があるかもしれない。
私たちは予測可能な未来に何の現金配当金も支払わないと予想している。
私たちは予測可能な未来に、私たちは普通株式保有者に現金配当金を支払わないと予想している。代わりに、私たちは私たちの業務を維持して拡大するためにすべての収益を維持する予定だ。さらに、私たちの新しい信用計画には条項が含まれており、任意の将来の債務融資計画は、私たちの普通株が発表または支払い可能な配当金の追加条項を禁止または制限する追加条項を含む可能性があります。そのため、投資家は、投資リターンを実現する唯一の方法として、価格上昇後に普通株を売ることに依存しなければならない。
当社の会社登録証明書の改訂と再記述、私たちの第二次改正と再記述の定款、デラウェア州法律の条項は逆買収効果がある可能性があり、買収が株主に有利になるとしても、他の会社の買収を阻止する可能性があり、私たちの株主が現在の経営陣を交換または更迭しようとすることを阻止する可能性があります。
当社の改正及び再記載された会社登録証明書は、改正、二回目の改正及び再記載された会社定款及びデラウェア州法律に含まれる条項が、株主が有利と考える可能性のある私たちの支配権の変更、又は私たちの経営陣の変更を阻止、延期、又は阻止する可能性があり、株主がその株式から割増取引を得る可能性があることを含む。当社の改訂および再記載された会社登録証明書、および改訂および再記載された第2の改正および再記載の付例は、以下の条文を含む
•“空白小切手”優先株を許可し、株主の承認なしに私たちの取締役会から発行することができ、投票権、清算、配当、その他の私たちの普通株より優れた権利が含まれている可能性がある
•メンバーが3年間交互に勤務している分類された取締役会を作成した
•株主特別会議は当社の取締役会でしか開催できないことを明確に規定している
•株主が書面の同意の下で行動することを禁止する
•株主の年次株主総会での承認のための事前通知プログラムを作成し、指名された取締役会メンバーの人選を提案することを含む
•私たちの取締役会の空きは、当時在任していた取締役の大多数が埋めることしかできないことになっていた
•私たちの役員は理由がある場合にのみ免職されることになっています
•いかなる株主も取締役選挙で投票権を累積してはならないことを明確に規定している
•当社の取締役会に当社の第二次改正と再記載の定款を修正、変更または廃止する権利があることを明確に許可する
•私たちが改正して再記載した会社登録証明書の特定の条項と、第二次改正と再記載の定款を改正するためには、私たちの普通株式保有者の絶対多数票が必要です。
また、私たちはデラウェア州に登録して設立されたので、私たちはデラウェア州会社法第203条の規定によって管轄されています。この条項は、規定された方法で合併または合併が承認されない限り、私たちが発行した議決権のある株を15%以上保有している人が取引日後3年以内に私たちと合併または合併することを禁止しています。
私たちが改訂し、再記載した会社登録証明書は、改正、第2回改正と再記載された定款またはデラウェア州法律の遅延または抑止権変更の条項は、私たちの株主が彼らが持っている普通株から割増の機会を得ることを制限する可能性があり、また一部の投資家が私たちの普通株に支払う価格に影響を与える可能性がある。
私たちが純営業損失とある税収控除を使う能力は一定の制限を受ける可能性があります。
2023年,2022年,2021年12月31日までの年度では,これらのプロジェクトから収益を実現する不確実性のため,毎年発生する純営業損失やNOL記録所得税割引はない。私たちは最初からNOLを招いた。2023年12月31日現在,連邦NOLは約4.79億ドル,州NOLは約4.148億ドルである。4.79億ドルの連邦NOLでは、3410万ドルが2037年前の異なる日に満期になる。残りの4.448億ドルの連邦NOLは満期にならないだろう。この州のNOLは2043年まで異なる日に満了されるだろう。2022年12月31日まで、私たちは約2320万ドルと680万ドルの連邦と州研究開発税の繰越免除があります。これらの控除は2043年までの異なる日に満期になります。税法によると、2018年と今後数年に発生する連邦NOLは無期限に繰り越すことができるが、このような連邦NOLの控除は限られている。各州がどの程度税法を遵守するかはまだ確定されていない。また、国税法第382条及び州法律の対応規定によると、1社が3年間に価値でその持分所有権を計算する50%を超える“所有権変更”を経験した場合、当該会社は変更前のNOL及び他の変更前の税収属性を用いて変更後の収入又は税収を相殺する能力が制限される可能性がある。私たちは会社が設立されてから、所有権が変化し、いくつかのNOLと研究開発税収の繰越が制限されることを確定した。私たちの株式所有権の未来の変化、その中のいくつかは私たちがコントロールできなくて、本規則第382節の所有権の変化を招き、更に私たちのNOLと研究開発税収を利用して繰越を免除する能力を制限するかもしれません。州法によると、私たちのNOLもまた被害を受ける可能性がある。したがって、私たちは私たちのNOLと研究開発税収の繰越の大きな部分を利用できないかもしれない。また、NOLと研究開発税収控除の能力を利用することは、利益を実現し、米国連邦課税収入を生み出すことができるかどうかにかかっている。私たちが設立されて以来、私たちは純損失が発生しており、少なくとも今後数年以内に、私たちは引き続き重大な損失を受けると予想されている;したがって、私たちはいつ私たちのNOLを使用するために必要なアメリカ連邦課税収入を生むことができるのか分からない。これらの資産の将来収益の不確実性を最終的に実現するため、我々のNOLや他の繰延税金資産に関する全額推定準備金を記録した。
税法またはその実施または解釈の変化は、私たちの業務および財務状況に悪影響を及ぼす可能性がある。
税法の変化は私たちの業務や財務状況に悪影響を及ぼすかもしれない。“税法”は“CARE法”の改正により、“税法”の重大な改正を行った。その他の事項以外にも、税法には、会社税率を35%の最高限界税率から21%の統一税率に引き下げ、NOLの控除額を2017年12月31日以降に納税年度に発生した損失の今年度課税収入の80%に制限することが含まれており、いずれのNOLも無期限に繰り越すことができるにもかかわらず、法人税への大きな変化が含まれている。また、税法は2022年から現在の研究開発費を差し引く選択肢を廃止し、外国研究の支出に起因できる場合には、5年または15年以内に資本化·償却することを企業に求めている。
CARE法案に加えて,国会の新冠肺炎対策の一部として,2020年と2021年に税収条項を含む経済救済立法が公布された。2022年8月に法律に署名したアイルランド共和軍は、上場企業のある株の買い戻しに1%の消費税を課すなど、新たな税収条項も導入した。1%の消費税は、一般に、金銭または他の財産(会社自体の株式を除く)と交換するために、上場企業(またはそのいくつかの付属会社)が会社株主から株を買収するのに適用されるが、極めて最低限の例外がある。したがって、消費税は非伝統的な株の買い戻しのいくつかの取引に適用される可能性がある。
税法、アイルランド共和軍、そして他の立法下の規制指導は継続されており、これらの指導は最終的に私たちの業務と財務状況に及ぼすそれらの影響を増加または減少させる可能性がある。また,各州が税法,アイルランド共和軍,その他の税法をどの程度遵守するかは不明である。
私たちが改訂して再記述した会社登録証明書は、改訂された後、デラウェア州内の州または連邦裁判所が私たちの株主のために開始する可能性のあるいくつかのタイプの訴訟および訴訟の独占フォーラムを指定し、これは、私たちまたは私たちの取締役、上級管理者、または従業員との紛争において有利な司法フォーラムを得るための私たちの株主の能力を制限することができる。
当社の改正及び再記載された会社登録証明書の規定は、限られた例外状況を除いて、デラウェア州衡平裁判所は、以下の事項の専属裁判所となる:(1)当社を代表して提起された任意の派生訴訟又は法律手続き、(2)当社の任意の取締役、高級社員又は他の従業員が当社又は当社の株主に対して負う信頼責任を主張するいかなる訴訟、(3)DGCL、改訂及び再記載された会社登録証明書又は当社の第2の改訂及び重述付例のいずれかの条文に基づいて生じる当社の任意の訴訟、(4)任意の解釈、適用、適用、私たちの改正および再記載された会社登録証明書(改正または第二次改正および再記載)の有効性を執行または決定するか、または(3)(1)施行または決定(2)改正および再記載された会社定款または(5)内部事務原則によって管轄される私たちのクレームに対する任意の他の主張の訴訟は、各事件において、被告に指定された不可欠な当事者が当事者管轄権を有する大裁判官によって管轄される。いかなる者又は実体が自社株の株式を購入又はその他の方法で取得するいかなる権益も、当社の改訂及び再記載された上記会社の登録証明書の規定に了承され、同意されたものとみなされる。このような裁判所条項の選択は、株主が司法裁判所において、私たちまたは私たちの役員、上級管理者、または他の従業員と紛争することに有利であると考えるクレームを提出する能力を制限する可能性があり、これは、私たちと私たちの役員、上級管理者、および従業員に対するこのような訴訟を阻止する可能性がある。あるいは、裁判所が、私たちが改訂および再記載した会社登録証明書のこれらの条項が、1つまたは複数の指定されたタイプの訴訟または法的手続きに適用されないことを発見した場合、またはそれを実行できない場合、私たちは、他の管轄区域でそのような問題の解決に関連する追加費用を発生させる可能性があり、これは、私たちの業務および財務状況に悪影響を及ぼす可能性がある。
この排他的裁判所条項は,1934年に改正された“証券取引法”(Securities Exchange Act)を実行するために生じた義務や責任のための訴訟には適用されず,連邦裁判所の排他的管轄権を規定している。しかし、専属裁判所条項に列挙された1つまたは複数のカテゴリの訴訟に適用することができ、改正された1933年の証券法または証券法に基づいて提出されたクレームを主張することができ、証券法第22条が連邦裁判所および州裁判所が提起したすべての訴訟に対して同時に管轄権を有する限り、証券法またはその規則および法規の下の任意の義務または責任を実行することができ、証券法の下でのクレームについては、私たちの株主は連邦証券法およびその規則および法規の遵守を放棄したとみなされない。
もし証券アナリストが私たちの業務に関する研究や報告を発表しなければ、あるいは彼らが私たちの株に対するマイナス評価を発表すれば、私たちの株価と取引量は低下する可能性があります。
私たちの普通株の取引市場は、業界または金融アナリストが発表した私たちまたは私たちの業務に関する研究と報告にある程度依存している。もし私たちの業務を追跡している一人以上のアナリストが私たちの株に対する彼らの評価を引き下げたら、私たちの株価は下落するかもしれない。もしこのようなアナリストの一人以上が私たちの株を追跡しなくなったり、私たちに関する報告書を定期的に発表できなかったら、私たちの株の市場での可視度を失うかもしれません。これは逆に私たちの株価を下落させる可能性があります。
私たちの総流通株の一部は近い将来市場に売却される可能性があり、これは私たちの業務が良好であっても、私たちの普通株の市場価格を大幅に低下させる可能性がある。
公開市場で私たちの大量の普通株を売ることはいつでも起こる可能性がある。これらの売却、あるいは市場で大量の普通株を持っていると思っている人が株を売却しようとしているという見方は、私たちの普通株の市場価格を下げる可能性があります。
私たちは基本的に私たちの株式補償計画によって発行される可能性のあるすべての普通株を登録した。これらの株は発行時に公開市場で自由に販売することができ、付与されると、関連会社に適用される数量制限を受ける。これらの追加株のいずれかが公開市場で販売されている場合、またはそれらが売却されると考えられている場合、私たちの普通株の市場価格は低下する可能性がある。
一般リスク因子
改正された1933年証券法によると、私たちは“小さな報告会社”であり、より小さい報告会社に適した様々な報告要求のいくつかの免除を利用することを決定すれば、私たちの普通株の投資家に対する吸引力が低下する可能性がある
私たちが“小さな報告会社”になる資格がある限り、定期報告書や依頼書における役員報酬に関する開示義務の削減、および今後何らかの新たな開示義務を遵守する発効日を含む、他の“より小さい報告会社”ではない他の上場企業に適用される様々な報告や他の要求に適した何らかの免除を選択することができます。また、私たちが大型加速申告機関とも加速申告機関ともみなされない限り、私たちは免除を使用し続け、2002年に改正されたサバンズ-オキシリー法案404条またはサバンズ-オクスリ法案の監査人認証要件を遵守しない。(I)非関連会社が本会計年度第2四半期最終営業日に保有する公開流通株が2.5億ドル未満、または(Ii)最近終了した会計年度の年収が1億ドル未満であれば、本年度第2四半期最終営業日までの公開流通株が7億ドル未満であれば、依然として規模の小さい報告会社となる。
もし私たちが資格を持っていて、規模の小さい報告会社が獲得できる免除に依存していれば、投資家が私たちがこれらの免除に依存している可能性があるかどうかによって、私たちの普通株の吸引力が低下するかどうかを予測することはできません。したがって、一部の投資家が私たちの普通株の吸引力が低下していることを発見すれば、私たちの普通株はそれほど活発ではない取引市場がある可能性があり、私たちの株価はもっと変動するかもしれない。
不利なグローバル経済または地政学的条件は、私たちの業務、財務状況、または経営結果に悪影響を及ぼす可能性がある。
我々の経営業績は、インフレ、金利、全体の経済状況の変化および不確実性を含む世界経済全体の状況、地政学的考慮と世界金融市場状況の不利な影響を受ける可能性がある。例えば、世界金融危機は資本と信用市場の極端な変動と混乱をもたらした。私たちは、世界的な信用と金融市場の悪化が、私たちの株価、私たちの現在の現金等価物、またはポートフォリオ、または融資目標を達成する能力に悪影響を与えないことを株主に保証することはできない。現在の株式と信用市場が悪化すれば、任意の必要な債務や株式融資をより困難にし、コストがより高く、希釈程度をより高くする可能性がある。適時に有利な条件で任意の必要な融資を得ることができなければ、私たちの成長戦略、財務業績、株価に実質的な悪影響を与える可能性があり、臨床開発計画の延期または放棄を要求する可能性がある。経済が疲弊したり低下したりして、私たちの臨床開発活動に関連するサプライヤーとサプライヤーに圧力を与える可能性もある
また、2022年2月に開始されたロシアとウクライナの間で持続的な衝突と世界的な反応は、米国と他の国の制裁や、イスラエルとハマスの間の戦争を含む、我々の業務が直面しているリスクをもたらしたり悪化させたりする可能性がある。私たちは私たちの運営、サプライヤー契約、臨床試験手配を評価しました。現在、これらの衝突は私たちの財務状況や運営結果に実質的な悪影響を与えないと予想されています。しかし、もしこのような敵対行動が持続的に、エスカレートしたり、拡大したりすれば、私たちが本報告書で決定した他のリスクは悪化するかもしれない。例えば、私たちの供給スケジュールや臨床場所が制裁の拡大や私たちの業務や関係のある国の参加によって中断された場合、私たちの業務は実質的な妨害を受ける可能性があります。また、国が支援するネットワーク攻撃の使用が拡大する可能性があり、衝突の一部として、ネットワークセキュリティやデータ保護対策の能力を維持または強化する能力に悪影響を及ぼす可能性がある。上記のいずれも私たちの業務を損なう可能性があり、現在の経済、地政学的気候、金融市場の状況が私たちの業務に悪影響を及ぼす可能性のあるすべての方法を予測することはできません。
財務報告や開示制御や手続きに対して有効な内部統制を維持できなければ、当社の業務を損なう可能性があり、投資家のわが社への信頼と我々普通株の価値に悪影響を及ぼす可能性がある。
財務報告に対する効果的な内部統制は、信頼できる財務報告を提供するために必要であり、適切な開示制御や手順とともに詐欺を防止することを目的としている。必要な新しい制御措置や改善された制御措置を実施できなかったり、実行中に遭遇した困難は、私たちの報告義務を履行できない可能性があります。さらに、私たちがサバンズ-オキシリー法第404条に基づいて行った任意のテスト、または私たちの独立公認会計士事務所がその後に行った任意のテストは、財務報告の内部統制における私たちの欠陥を明らかにするかもしれません。これらの欠陥は、実質的な弱点と考えられているか、または私たちの財務諸表を前向きまたは追跡的に変更する必要があるかもしれません。または、さらなる関心または改善が必要な他の分野が発見される可能性があります。悪い内部統制はまた、投資家が私たちが報告した財務情報に自信を失う可能性があり、これは私たちの株式の取引価格にマイナス影響を与える可能性がある。
私たちは四半期ごとに内部統制と手続きの変化を開示することを要求され、私たちの経営陣は毎年これらの統制の有効性を評価することを要求されている。しかし、私たちが小さい報告会社である限り、私たちの独立公認会計士事務所は、サバンズ·オクスリー法案第404条に基づいて、財務報告書の内部統制に対する私たちの有効性を証明する必要はありません。
制御過程を維持したり改善したりする努力が最終的に成功する保証はなく、将来起こりうる重大な弱点を回避することもできない。 我々は2023年に再編を実施したが,場合によっては内部統制活動を行う従業員は,従来これらの活動に従事していた従業員とは異なる。変化する経営環境は,我々の内部制御システムが効率的な設計や内部制御活動が設計通りに行われないリスクを増加させる. 会計又は財務機能部門の従業員又はコンサルタント、又はキー内部統制を監督する他の業務分野の個人の再編及び任意のさらなる離職は、将来の内部統制欠陥の可能性を増加させる可能性がある。 将来の財務報告の内部統制におけるいかなる重大な弱点の救済にも成功できなければ、あるいは他の重大な弱点が発見された場合、私たちの財務報告の正確性とタイミングは不利な影響を受ける可能性があり、定期報告のタイムリーな提出に関する証券法の要求、および適用される証券取引所の上場要求を守り続けることができない可能性があり、投資家は私たちの財務報告に自信を失う可能性があり、私たちの株価はしたがって下落する可能性がある。私たちはまた、ナスダック、米国証券取引委員会、または他の規制機関の調査の対象になる可能性があり、これは、私たちの名声や財務状況を損なうか、あるいは私たちの核心業務の財務·管理資源を移転する可能性がある。
私たちまたは私たちが依存している第三者は深刻な災難の悪影響を受けるかもしれない。
洪水、火災、爆発、地震、極端な天気状況、医療疫病、電力不足、通信故障、または他の自然または人為的事故または事件のような計画外の事件は、私たちの施設や私たちが契約した第三者の施設を十分に利用できず、私たちの業務運営能力に重大かつ不利な影響を与える可能性があり、私たちの財務や運営状況に大きなマイナス影響を与える可能性がある。これらの施設や運営へのアクセスを失うことは、コスト増加、現在または将来の候補製品の開発遅延、または私たちの業務運営中断をかなり長く招く可能性があります。
私たちは深刻な災害や同様の事件が発生した時、私たちが維持している保険金額がどんな損害や損失を補うのに十分な保証がないだろう。もし私たちの工場や私たちの第三者契約メーカーの製造施設が事故や事件やその他の理由で稼働できない場合、たとえ短い時間であっても、私たちのいかなる研究開発計画や商業化努力も損害を受ける可能性があります。
プロジェクト1 B.--未解決スタッフの意見。
ない。
第1 C項目はネットワークセキュリティに関するものだ。
ネットワークセキュリティリスク管理と戦略
著者らは、私たちの全体企業リスク管理計画に統合し、他の関連機能と統合し、例えば情報技術、IT、システムアーキテクチャとサプライヤー管理などのネットワークセキュリティリスク管理計画を設計し、維持した;この計画は最適な実践と標準を利用して、ネットワークセキュリティとその他の情報セキュリティ脅威からのリスクを評価、識別、管理することを目的としている。 この計画の一部として、私たちはネットワークセキュリティ脅威によるリスクを定期的に評価し、これは私たちのより広範なリスク管理活動の一部であり、私たちの内部制御システムの構成要素でもある。 評価過程では,我々の内部で管理されている情報技術やIT,システムおよび重要な業務機能,および我々が協力する第三者サービスプロバイダ,プロバイダ,協力者によって運営または管理される可能性のある敏感なデータに関連するリスクを考慮した.
我々は,ガイドラインとして国家標準と技術研究所ネットワークセキュリティフレームワークやNIST CSFを用いて,我々の業務に関連するネットワークセキュリティリスクの識別,評価,管理を支援している.NIST CSFに基づいて計画を設計し評価しましたこれは私たちが特定の技術基準、規範、または要求を満たすことを意味するのではない。
私たちのネットワーク安全リスク管理計画は私たちの全体企業リスク管理計画に統合され、他の法律、コンプライアンス、戦略、運営と金融リスク分野に適用される汎用的な方法、報告ルートと管理プロセスを共有する。私たちの全体的なリスク緩和戦略の一部として、私たちはまたネットワーク保険の保証範囲を維持しているが、このような保険のタイプまたは金額は、セキュリティホール、サイバー攻撃、および他の関連違反に対する私たちのクレームをカバーするのに十分ではないかもしれない。
私たちのネットワークセキュリティリスク管理計画の主な側面は
•リスク評価は、私たちの重要なシステムと情報が直面している重大なネットワークセキュリティリスクを識別することを目的としています
•専任者は主に私たちのネットワークセキュリティリスク評価の流れ、私たちのセキュリティコントロール、サイバーセキュリティ事件に対する私たちの反応を管理する
•潜在的な脆弱性の発見を支援するために、私たち自身のシステムに対して定期的な演習とテストを行います
•適切な状況では、私たちのITチームの監督の下で、外部サービスプロバイダを使って私たちのシステムの評価に協力してくれます 脅威情報を利用してシステムの脆弱性を能動的に認識することと、ネットワーク攻撃に対する我々の防御を能動的に識別し、ネットワークセキュリティ脅威警報をタイムリーに提供することを含むネットワークセキュリティ脅威を監視する
•従業員、イベント応答者、および高度管理者に新入社員、年間、および一時的なネットワークセキュリティ意識トレーニングを提供します
•ネットワークセキュリティイベントに対応するプログラムを含むネットワークセキュリティイベント応答計画または応答計画と、
•セキュリティアンケートを完了し、状況に応じて重要なデータまたは情報を維持する第三者のリスク評価を行い、第三者情報セキュリティ能力の評価および検証を支援する職務調査プロセスを含む、当社のITチームによって監督される主要サービスプロバイダ、サプライヤー、および協力者に対する第三者リスク管理プロセス。
私たちの応答計画は、ネットワークセキュリティ脅威とネットワークセキュリティイベントおよびイベントに対する私たちの応答プロトコルを列挙し、少なくとも年に1回応答計画を検討する当社のネットワークセキュリティイベント応答チーム(CSIRT)によって維持される。CSIRTは,我々の首席運営官兼首席財務官上級副総裁に仕事を報告する情報技術部副総裁と,我々の実行チームメンバーや他の上級管理者を含むIT部門リーダーから構成されている.私たちの応答計画は、データセキュリティホールが発生したときにどのように識別、評価、報告、応答、および回復し、これらの機能を担当する人員を指定するための枠組みを提供することを目的としている。我々のITチームは、外部プロバイダとソフトウェア製品のサポートを利用して、様々なソースから受信したセキュリティ警報を評価し、CSIRTは、ネットワークセキュリティイベントと判断された任意の警報または脅威を直ちに評価し、さらなる評価のために応答計画に基づいて報告する。サイバーセキュリティ事件が発生したことを確認した後
CSIRTは、我々の内部部門の代表と、内部または外部の法律顧問または他の外部ネットワークセキュリティコンサルタントまたはサービスプロバイダとを含むことができるイベント応答チームを確立する。CSIRTはリスク制御、通知プロセス、システム回復、事件記録と評価、データ保存と法医学分析を含む協調対応戦略を制定することを目的としている
我々の対応計画は、潜在的な重要性をさらに評価し、適切な場合には、上級管理チームの他のメンバー、取締役会監査委員会議長または監査委員会、および取締役会メンバー全員(必要に応じて)に通知し、必要に応じて開示するために、私たちの業務戦略、財務状況、および運営結果に重大な影響を与える可能性の高いネットワークセキュリティ事件を迅速に関連幹部に報告することを目的としている。
これまで、ネットワークセキュリティ脅威は、まだ私たちの業務戦略、運営結果、または財務状況に実質的な影響を与えていませんが、私たち、私たちの協力者、および私たちの第三者サプライヤーとサービスプロバイダは将来的にネットワークセキュリティ脅威の目標となる可能性があり、どのネットワークセキュリティ脅威も私たちの業務に実質的な悪影響を及ぼす可能性があります。 我々が直面しているネットワークセキュリティリスクと我々の潜在的な影響に関する説明については,本年度報告表格10−K第I部第1 A項の“リスク要因”を参照されたい。
サイバーセキュリティの管理と監督
我々の取締役会はネットワークセキュリティリスクをそのリスク監督機能の一部と見なし、監査委員会にネットワークセキュリティやその他の情報技術リスクの監督を依頼した。私たちの監査委員会は、経営陣が私たちのネットワークセキュリティリスク管理計画に関連した持続的な活動を監督している。
私たちの監査委員会は私たちのネットワークセキュリティリスクに関する管理層の定期的な報告を受けてフィードバックを提供する。また、経営陣は、必要に応じて重大なネットワークセキュリティの脅威や事件の最新状況を監査委員会に通報する。
私たちの監査委員会は、ネットワークセキュリティに関する活動を含む全体取締役会にその活動を報告した。取締役会のメンバー全員が定期的に私たちの実行チームのプレゼンテーションを聞き、私たちの情報技術副総裁が私たちのネットワークセキュリティリスク管理計画を通報しました。
我々の実行チームには、最高運営官兼首席財務官上級副総裁と首席法務官上級副総裁が含まれており、我々のネットワークセキュリティ脅威による重大なリスクの評価と管理を担当している。実行チームは私たちの全体的なネットワークセキュリティリスク管理計画に主な責任を持っている。私たちの首席運営官兼首席財務官の上級副総裁は私たちの情報技術副総裁を指導して、彼は私たちの全社のネットワークセキュリティ戦略、政策、標準とプロセスの運営監督を指導し、そして関連部門の間で仕事を展開して、私たちの会社と私たちの内部ネットワークセキュリティ人員の準備を評価して助け、そして外部のネットワークセキュリティ顧問を招いてネットワークセキュリティリスクに対応します。 我々の情報技術副総裁は、端末セキュリティ、ネットワークセキュリティ、イベント応答計画、エンドユーザトレーニングプロジェクトを20年間実施した経験を含む25年以上のITとネットワークセキュリティプロジェクト管理経験を持っている。我々の内部ネットワークセキュリティ担当者は,製薬/バイオテクノロジー,技術,電気通信や金融サービスなどの複雑かつ国際業務の垂直分野でネットワークセキュリティ,情報セキュリティ,データ保護,プライバシー,規制コンプライアンス,リスク管理に関する経験を持ち,情報システム管理やセキュリティに関する第三者認証を複数持っている.
我々の実行チームは、内部セキュリティ担当者のプレゼンテーション、政府、公共またはプライベートソース(私たちが招聘した外部コンサルタントを含む)から得られた脅威情報および他の情報、ならびにIT環境に配備されたセキュリティツールによって生成された警報および報告を含む様々な方法で、キーネットワークセキュリティリスクおよびイベントの予防、検出、評価、緩和および修復を理解し、監視する。
ネットワーク脅威を阻止し、発見するために、私たちは毎年すべての従業員(アルバイトと臨時従業員を含む)にデータ保護、ネットワークセキュリティとイベント応答と予防訓練とコンプライアンス計画を提供し、社会工学、ネットワーク釣り、暗号保護、機密データ保護、資産使用と移動安全を含むタイムリーかつ関連するテーマをカバーし、従業員にすべての事件の重要性を直ちに報告するように教育している。我々はまた,技術に基づくツールを用いてネットワークセキュリティリスクを低減し,従業員ベースのネットワークセキュリティ計画を支援している
第2項:財産を含む.
私たちの会社の本社はマサチューセッツ州ケンブリッジ市にあります。私たちは会社本部のあるマルチテナントビルで約四十五,000平方フィートのオフィスと実験室空間を借りました。この空間の大部分の賃貸契約は2026年3月に満期になる。その後、私たちはレンタル期間をさらに5年間延長することを選択することができる。私たちは、このオフィスと実験室空間は私たちの現在の需要を満たすのに十分であり、必要な時に適切な追加空間を提供すると信じている。
三番目の項目:法的手続きを継続する。
私たちは時々様々な法的手続きやクレームの影響を受けるかもしれません。これらの訴訟とクレームは私たちの正常な商業活動の過程で発生します。結果にかかわらず、弁護と和解コスト、管理資源の移転、その他の要素により、訴訟は私たちの業務、財務状況、運営結果、将来性に悪影響を及ぼす可能性がある。私たちは現在、実質的な法的手続きに参加していない
四番目の項目:炭鉱の安全情報開示。
適用されません。
第II部
第五項:登録者普通株、関連株主事項及び発行者が株式証券を購入する市場
普通株取引に関するいくつかの情報は
我々の普通株のナスダック世界精選市場での取引コードは“MRSN”である。2024年2月23日現在、14名の普通株式保有者が登録されている。株主の実際の数は受益者である株主を含む記録保持者の数を超えているが、その株は仲介人や他の被著名人が街頭名義で保有している。
配当政策
私たちは普通株の現金配当金を発表したり支払ったりしたことがない。私たちは現在、私たちの未来のすべての収益を維持するつもりで、もしあれば、私たちの業務の成長と発展に資金を提供します。予測可能な未来に、私たちは私たちの普通株に現金配当金を支払うつもりはない。しかも、私たちの現在の信用スケジュールには、いくつかの例外的な場合に普通株の配当金を支払うことを禁止する制限的な契約が含まれている。将来的に任意の現金配当金を派遣する決定は、私たちの取締役会が適宜決定し、関連する制限や他の要素を考慮する可能性があることにかかっている。投資家は現金配当金を得ることを期待して私たちの普通株を購入してはいけない。
株式表現グラフ
1934年証券取引法(改正)または取引法(18)節の場合、以下の業績グラフおよび関連情報は、“募集材料”または米国証券取引委員会(または米国証券取引委員会)“保存”とみなされてはならないし、このような情報を参照することによって、1933年の証券取引法または改正された証券法または証券法に基づいて提出された任意の将来の文書に組み込まれてはならない。
次の図は2018年12月31日から2023年12月31日まで、つまり今年の最後の取引日を比較し、私たちの普通株とナスダック総合指数とナスダックバイオテクノロジー指数の表現を比較した。この比較は,2018年12月31日の終値後,我々の普通株と上記各指数に100ドルが投資されたと仮定し,配当再投資(あれば)を仮定している.この図に含まれる株価表現は,必ずしも未来の株価表現を示唆しているとは限らない.
最近売られている未登録証券
ない。
発行者および共同会社の購入者が株式証券を購入する
2023年第4四半期に、私たちまたは任意の関連バイヤーまたは私たちを代表する誰も、または関連バイヤーは、私たちの普通株式のどの株式も購入しませんでした
第6項。以下の内容:[保留されている]
プロジェクト七、会社経営陣の財務状況と経営成果の検討と分析
以下、当社の財務状況及び経営結果の検討及び分析は、本年度報告10-K表の他の部分の監査財務諸表及び関連付記とともに読まなければならない
いくつかのイベントに対する我々の実際の結果および時間は、任意の前向き陳述において議論、予測、予想、または指摘された結果と大きく異なる可能性がある。展望的陳述は将来の業績の保証ではなく、私たちの実際の運営結果、財務状況と流動性、および私たちが経営している業界の発展は、本年度報告に含まれる前向き陳述と実質的に異なる可能性があることを想起させます。また,我々の経営業績,財務状況,流動性,および我々が経営している業界の発展が本年度報告に含まれる前向き陳述と一致していても,将来の結果や発展を予測できない可能性がある。
以下の情報および任意の前向き陳述は、第2の部分1 Aの下で決定されたリスクを含む、本年度報告書10-K表の他の部分的に議論された要因に関連して考慮されなければならない。リスク要因です
我々は読者に,我々が行ったいかなる前向き陳述にも過度に依存しないように注意し,これらの陳述はそれらの発表日の状況のみを反映している.私たちは、法律および米国証券取引委員会(米国証券取引委員会)の規則が、私たちの予想またはそのような声明に基づく可能性のあるイベント、条件、または状況の任意の変化を反映するために、または実際の結果および前向き声明に記載されている内容の可能性に影響を与える可能性があるために、任意のそのような声明を公開的に更新または修正することを明確に要求しない限り、いかなる義務も負わない。
2022年12月31日までの年度と2021年12月31日までの年度の検討と分析については,2023年2月28日に米国証券取引委員会に提出された2022年12月31日現在の10−K表年次報告の項目7.経営陣の財務状況と経営成果の検討と分析を参照されたい。
概要
著者らは臨床段階の生物製薬会社であり、抗体-薬物結合体或いはADCの開発に専念し、重大な需要を満たしていない癌患者に臨床上意義のあるメリットを提供する。著者らは数十年の業界経験を利用して、DolasynThenと免疫合成の2つの独自と差別化のADCプラットフォームを開発した。DolasynThenは我々の細胞毒性ADCプラットフォームであり、サイトに特定の同質ADCを生成することを目的としている。次に、Dolasynnは、特定の標的に対して薬物対抗体比(DAR)を最適化することを可能にし、用量制限性の深刻な好中球減少症、末梢神経病変、および眼毒性を回避するために、臨床的に証明された独自の黄金色ペイロードを利用する。免疫合成は著者らの独自のSTIN(インターフェロン遺伝子刺激物)アゴニストプラットフォームであり、系統的に管理するADCを産生し、抗原を発現する腫瘍細胞と腫瘍常駐免疫細胞の中でSTINGシグナルを局部的に活性化し、先天性免疫刺激の抗腫瘍潜在力を放出することを目的としている。私たちはこれらのプラットフォームを利用して私たちの会社とパートナーのためにADC候補製品を生成しています。これらの製品は現在の看護基準を改善する潜在力があると信じています
私たちの2つの臨床段階候補製品はXMT-1660とXMT-2056である。XMT-1660はB 7-H 4を目標としたDolasynten ADCであり、そのDARは正確、目標最適化され、6であり、著者らは一期臨床試験を行っており、現在乳癌、子宮内膜癌、卵巣癌を含む様々な腫瘍患者を募集している。XMT-2056はシステム投与された免疫融合ADCであり、新型のヒト表皮増殖因子受容体2(HER 2)エピトープに対して、DARは8であり、著者らはHER 2発現の末期或いは再発固形腫瘍患者に対して第一段階の臨床試験を行っており、乳癌、胃癌、結腸直腸癌と非小細胞肺癌を含む
私たちは2つの初期段階の臨床前候補薬があり、私たちはXMT-2068とXMT-2175と呼ばれ、それらは私たちの免疫合成プラットフォームを利用している。
2023年7月、著者らはUplit登録試験XMT-1536、upifitimab rilsodotin、あるいはUpRiとも呼ばれ、その主要な終点に到達できなかったことを発表し、著者らはUpRiの開発を停止することを決定し、そしていくつかのUpRiの臨床試験、及び著者らの監督と商業準備を含むUpRi関連開発活動を段階的に終了する。同時に、私たちの取締役会は、当時の従業員基数を約50%削減したり、再編したりすることを含むいくつかの費用削減措置を承認したと発表しました。2023年12月31日現在,UpRi関連活動の清盤と再編がほぼ完了している。また,2022年5月には,卵巣癌と非小細胞肺癌(NSCLC)患者の第1段階用量探索試験であるDolasynten ADCの開発を中止し,2022年9月に完成したDolasynten ADCの開発を中止することにした。
我々はすでにグローバル協力を達成し、グラクソ·スミスクライン知的財産権(第4号)有限会社またはGSKにXMT-2056の共同開発と商業化の独占的選択権を提供している。また、Janssen Biotech、Inc.またはジョンソン、およびドイツのダムシュタットの完全子会社Ares Trading、S.A.またはその各エンティティ(例えば、適用可能)と戦略的研究開発協力関係を構築し、私たちの独自のプラットフォームを利用して、私たちの協力者が選択した限られた数の目標を利用して他のADC候補製品を開発および商業化する。私たちのADC候補製品とプラットフォームの潜在力、私たちの科学と技術専門知識及び私たちの知的財産権戦略はすべて私たちの独立と協力の努力を支持し、癌患者に抵抗するために生活を変えるADCを発見と開発すると信じている。
設立以来、私たちの業務はずっと私たちのプラットフォームを構築し、潜在的な候補製品を確定し、臨床前研究のための薬物物質と薬物製品材料を生産し、臨床前と毒理学研究を行い、臨床試験材料の製造と臨床試験を行い、私たちの知的財産権を創立と保護し、私たちの会社に人員を提供し、資金を集めてきた。私たちはどんな製品も販売を許可されていないし、製品販売から何の収入も得ていない。私たちは主に私たちの戦略協力、私たちの転換可能な優先株と私たちの普通株の公開を通じて、私たちの市場やATMによる株式発行計画を含む私たちの業務に資金を提供します。
設立以来、私たちは累積的に巨額の運営損失を発生させた。2023年、2022年、2021年12月31日までの年度の純損失はそれぞれ171.7、2.02億、1.701億ドルだった。2023年12月31日現在、私たちの累計赤字は826.4ドルです。私たちは今後数年間巨額の費用と運営損失が生じると予想しています
•XMT−1660およびXMT−2056の臨床開発および製造活動を継続する
•XMT−2068およびXMT−2175を含む他の候補製品の発見、検証、および開発活動を継続する
•ジョンソン、メルク、グラクソ·スミスクラインと協力して研究開発活動を展開した
•臨床試験を完成させるための現在と未来の候補製品は市場の承認を得ている
•第三者とのビジネス上実行可能な供給および製造関係の確立および維持を含む、私たちの候補製品のための持続可能かつ拡張可能な製造プロセスを開発すること
•競争的な技術や市場の発展に対応しています
•私たちの知的財産権の組み合わせを維持し、拡大し、保護する
•より多くの研究、開発、そして一般的で行政的な人たちを雇う。
財務運営の概要
収入.収入
今まで、私たちは販売製品から何の収入も得ていない。私たちのすべての収入は戦略的協力から来ている。
2022年12月、ドイツのダムシュタット·メルクKGaAの完全子会社Ares Trading S.A.と、2022年メルクKGaA協定である協力·商業許可協定を締結した。“2022年メルクKGaAプロトコル”は、我々の最大2つの標的抗原の免疫合成プラットフォームを利用してADC候補製品を開発および商業化することを規定している。マクKGaAは標的抗原に対する抗体の産生を担当し、私たちは生物結合活動を実行してADCを作成し、いくつかの化学、製造と制御開発と早期製造活動を担当し、費用はメルクKGaAが負担する。メルクKGaAは、これらのADC候補製品のさらなる開発および商業化を担当する独占的権利を有し、担当する。2023年12月31日までの年間で,2022年のメルクKGaA協定に関する1070万ドルを確認した。2022年12月31日までの1年間、2022年のメルクKGaA協定に関する収入は確認されていません
2022年8月、GSKに独占的なオプションを提供し、XMT-2056を含む製品を共同開発し、製品を商業化することができるかもしれないGSKに独占的なオプションを提供するために、グラクソ·スミスクラインと協力、オプションおよびライセンス契約、またはGSKプロトコルを締結した。グラクソ·スミスクラインがその選択肢を行使する前に、我々は我々のXMT−2056計画に関する製造、研究、早期臨床開発を担当している。グラクソ·スミスクラインがその選択権を行使すれば、グラクソ·スミスクラインはさらに共同開発と商業化ライセンス製品を所有し、責任を負うことになる。GSKプロトコルに関する340万ドルと200万ドルの連携収入を,2023年と2022年12月31日までの年間でそれぞれ確認した。
2022年2月、著者らは楊森生物技術会社或いはジョンソンと研究協力と許可協定、即ちジョンソン協定を締結し、著者らのDolasyntenプラットフォームを利用してADC候補製品を開発と商業化し、最大3種類の標的抗原を生産することができる。我々は、2023年7月14日と2023年9月25日に改正されたこのような協定を“ジョンソン協定”と呼ぶ。ジョンソンは標的抗原に対する抗体の産生を担当し,我々は生物結合活動を行ってADCを作成し,ある化学,製造,制御開発や早期製造活動を担当し,費用はジョンソンが負担する。ジョンソンはこのようなADC候補製品のさらなる開発と商業化を持って責任を負う。2023年,2023年,2022年12月31日までの年間で,ジョンソン合意下の業績に関する連携収入1,660万ドルと2,420万ドルをそれぞれ確認し,発展マイルストーンの実現を含む
2014年6月、著者らはドイツDarmstadtのMerck KGaAと協力と商業許可協定、あるいは2014年Merck KGaA協定を締結し、著者らのDolaflexinプラットフォームを利用してADC候補製品を開発と商業化し、最大6種類の標的抗原に応用できる。2018年5月、私たちはドイツのダムシュタットのメルクKGaAと、2018年のメルクKGaA供給協定を締結し、研究性新薬またはIND研究と臨床試験に使用可能な材料を供給した。2023年12月15日、私たちはメルクKGaAと共同で、2014年のメルクKGaA協定と2018年のメルク供給協定を終了することに同意しました。2023年,2023年,2022年12月31日までの年間で,2014年メルクKGaA協定と2018年メルクKGaA供給協定に関する370万ドルと極めて低い金額の収入をそれぞれ確認した。
2023年12月31日,2023年12月31日,2022年12月31日までの年間で,発展マイルストーンの実現に関する収入250万ドルと,アザナ生物科学有限責任会社やアザナ生物科学社との協力協定に関するサービス収入30万ドルをそれぞれ確認した
予測可能な未来に、私たちは私たちのほとんどの収入がグラクソ·スミスクライン、ジョンソン、そしてメルクKGaAと行われている協力協定から来ると予想している。臨床開発の不確定な性質と時間を考慮して、私たちはいつ、あるいはこれらの協力の下でさらなる記念碑的支払いまたはいかなる印税支払いを受けるかどうかを予測できない。
費用.費用
研究開発費
研究と開発費用には、私たちの薬物発見努力、製造、および私たちの候補製品の開発が含まれています
•従業員に関する費用には、賃金、福祉、株式ベースの報酬費用が含まれる
•我々が研究、臨床前活動、製造、臨床試験を行う第三者に代表される研究と開発に資金を提供するコスト
•研究室用品
•賃貸料、減価償却、修理費用を含む施設費用;
•私たちの第三者許可協定に基づいて前払いとマイルストーン支払いが行われます。
研究·開発コストは発生時に費用を計上する。製造、臨床前研究、臨床試験のようないくつかの活動のコストは、通常、特定の任務を達成する進捗の評価によって確認される。いくつかの開発活動のコストは、臨床試験のように、特定のタスクを達成する進捗を評価した上で確認され、これらの評価に使用されるデータは、患者登録、臨床サイト活性化、および私たちと契約した第三者が提供してくれる情報を含む。
研究開発活動は私たちのビジネスモデルの核心だ。臨床開発後期段階にある候補製品は通常,臨床開発早期段階の候補製品よりも高い開発コストを有しており,これは主に後期臨床試験の規模と持続時間の増加および製造コストによるものである。我々の将来の研究·開発コストは現在のレベルで増加し続けており,これはわれわれの臨床開発計画の進展にかかっていると予想される。私たちの任意の候補製品の開発成功と商業化に関連する要素はたくさんあり、未来の試験設計と各種法規要求を含み、その中の多くの要素は私たちの現在の開発段階では正確に確定できない。また、私たちがコントロールできない将来のビジネスや規制要因は、私たちの臨床開発計画や計画に影響を与えるかもしれない。
私たちの従業員や他の資源は複数の開発されているプロジェクトに配置されているため、私たちは歴史的に計画通りにすべての内部研究開発費を割り当てていません。内部研究と開発費用を総額として列記する。我々の内部研究開発コストは,主に人員に関するコスト,在庫による報酬コスト,減価償却や実験室消耗品を含む施設コストである
著者らは著者らの候補製品とプラットフォームを生産し、著者らを代表して臨床試験を行う臨床研究機関に巨大な外部コストを発生させた。臨床開発における候補製品ごとのこれらの外部費用を収集した。著者らの臨床開発において関連候補製品を持つプラットフォームのコストは通常このプラットフォームに基づいて臨床上最も先進的な候補製品に割り当てられる。2022年第2四半期に卵巣癌および非小細胞肺癌患者の第1段階用量探索試験のDolasynThen ADC XMT-1592のさらなる臨床開発を停止することを決定したことを考慮して、この決定を下した後、私たちのDolasyntenプラットフォームに関連するすべてのコストはXMT-1660に再分配される可能性があり、それは現在私たちの主なDolasynThenに基づく候補製品である。臨床開発において我々の候補製品に属さないすべての外部研究と開発費用は臨床前と発見コストに計上されている。これらのコストは、我々の候補製品XMT−2068およびXMT−2175および追加の早期発見段階計画およびいくつかの割り当てられていないコストと関連している。次の表は、2023年12月31日、2022年12月31日、2021年12月31日までの年間ごとの外部研究開発費をまとめたもので、上記の計画通りに示しています。
| | | | | | | | | | | | | | | | | |
| 現在までの年度
十二月三十一日 |
| (単位:千) | 2023 | | 2022 | | 2021 |
| UpRi外部コスト | $ | 48,902 | | | $ | 66,119 | | | $ | 45,511 | |
| XMT-1660外部コスト | 14,098 | | | 15,032 | | | — | |
| XMT-2056外部コスト | 5,812 | | | 4,981 | | | — | |
| XMT-1592外部コスト | 434 | | | 3,802 | | | 9,126 | |
| 臨床前と発見コスト | 4,441 | | | 14,991 | | | 28,464 | |
| 内部研究開発コスト | 74,582 | | | 68,460 | | | 48,912 | |
| 研究開発総コスト | $ | 148,269 | | | $ | 173,385 | | | $ | 132,013 | |
私たちの候補製品が開発に成功するかどうかは大きな不確実性を持っている。したがって、候補製品の余剰開発を完了するために必要な作業の性質、時間、および推定コストを合理的に推定または知ることはできない。私たちはまた、いつ(もしあれば)私たちの規制承認された候補製品から商業化されて販売される候補から収入を得ることを予測できない。これは,薬物開発に関連する多くのリスクと不確実性に起因しており,以下のような不確実性を含む
•臨床前研究とINDを支持する研究に成功した
•臨床試験の登録に成功しました
•関連規制部門の上場承認を受けた
•商業製造能力を確立するか、または第三者製造業者との手配を行う
•私たちの候補製品のために特許と商業秘密保護および規制排他性を獲得し、維持する
•単独でまたは他人と協力して、承認された後に候補製品を商業化すること;および
•承認された後、このような薬物の安全性は引き続き受け入れられる。
我々の任意の候補製品の開発、製造、または商業化に関連するこれらの変数のいずれかの結果は変化し、候補製品開発に関連するコスト、タイミング、および生存能力を著しく変化させるであろう。
例えば,2023年7月27日,UpRiの臨床開発中止が決定したと発表した。したがって、私たちは以前この計画に特化した資源を私たちの次世代ADCとプラットフォーム、DolasynThenと免疫合成に割り当てた。著者らは引き続きXMT-1660とXMT-2056の臨床開発と製造を行い、著者らの臨床前パイプラインを推進し、著者らのADC技術の改善に投資し、今後数年で巨額の研究開発費用が発生すると予想している。
一般と行政費用
一般および行政費用は、主に、株式ベースの給与を含む行政、財務、会計、業務発展、法律業務、情報技術および人的資源機能者の賃金、および従業員に関連する他の費用を含む。その他の重大なコストには、研究開発費に含まれていない施設コスト、特許や会社事務に関する法律費用、会計や他のコンサルティングサービスの費用が含まれている。
今後数年間、外部コンサルタント費用や特許費用に関連するコスト増加、その他の費用を含む持続的な研究開発活動を支援するために、多くの一般的かつ行政的費用が発生することが予想される。
再編成費用
再構成費用には主に解散費と福祉、賃金通知、再配置サービス、契約終了費用が含まれる。2023年12月31日までの1年間に、870万ドルのこのような費用を確認しました。2023年12月31日現在、再編はほぼ完了している。
その他の収入(費用)
その他の収入(支出)には、主に私たちの信用手配下の借金に関する利息支出、繰延融資コストに関する償却と債務割引の増加が含まれる。利息収入には現金等価物と有価証券が稼いだ利息が含まれている。
経営成果
2023年12月31日までと2022年12月31日までの年次比較
以下の表は、2023年12月31日、2023年12月31日、2022年12月31日までの3年度の業務結果、およびこれらの項目の変化をまとめたものである | | | | | | | | | | | | | | | | | |
| 現在までの年度
十二月三十一日 | | ドルの小銭 |
| (単位:千) | 2023 | | 2022 | |
| 協力収入 | $ | 36,855 | | | $ | 26,581 | | | $ | 10,274 | |
| 運営費用: | | | | | |
| 研究開発 | 148,269 | | | 173,385 | | | (25,116) | |
| 一般と行政 | 59,543 | | | 56,963 | | | 2,580 | |
| 再編成費用 | 8,713 | | | — | | | 8,713 | |
| 総運営費 | 216,525 | | | 230,348 | | | (13,823) | |
| その他の収入(支出): | | | | | |
| 利子収入 | 12,073 | | | 2,883 | | | 9,190 | |
| 利子支出 | (4,073) | | | (3,328) | | | (745) | |
| その他の収入を合計して純額 | 8,000 | | | (445) | | | 8,445 | |
| 純損失 | $ | (171,670) | | | $ | (204,212) | | | $ | 32,542 | |
協力収入
2023年12月31日までの年間で,協力収入が2022年12月31日までの年度より1,030万ドル増加したのは,主に2022年メルクKGaAプロトコルと2014年メルクKGaAプロトコルにより確認された連携収入が1,430万ドル増加したことと,ジョンソン合意による早期開発マイルストーンに関する320万ドル増加であったが,ジョンソン合意により確認された連携収入の1,080万ドル減少分によって相殺されたためである。
研究開発費
研究開発支出は2022年12月31日現在の1兆734億ドルから2023年12月31日現在の1億483億ドルに減少し、2510万ドル減少した。
研究開発費が減少した要因は
•再編成によりUpRiの製造と臨床開発活動は1730万ドル減少した
•550万ドルの削減は、主にXMT-1660およびDolasyntenプラットフォームの製造活動と関連している
·XMT-2056の製造と臨床開発活動に関連する320万ドルの削減が予想される;
·第三者ライセンス契約により、払い戻し不可能なライセンス支払いに関する費用が200万ドル削減されました。
XMT−1660の臨床開発活動に関連する280万ドルの増加分は,これらの減少した費用を相殺した。
一般と行政費用
一般·行政費は260万ドル増加し、2022年12月31日現在の5700万ドルから2023年12月31日現在の5950万ドルに増加した。一般的かつ行政費用が増加した要因は、再編前の従業員数の増加により、従業員報酬(株ベースの報酬を含まない)が290万ドル増加したが、コンサルティングや専門サービスに関する30万ドルの減少がこの増加を部分的に相殺したためである。
その他の収入を合計して純額
その他の収入(支出)総額は840万ドル純増加し、2022年12月31日までの年度の40万ドルから2023年12月31日までの年度の800万ドルに増加した。純残高増加の主な原因は現金等価物と有価証券の利子収入増加である。
流動性と資本資源
流動資金源
これまで、私たちは主に戦略協力、私たちの転換可能な優先株と私たちの普通株の公開を通じて、私たちの初公募株、2019年と2020年の後続公募株、私たちのATM株式発行計画を含む資金を提供してきました
2020年5月には,ATM持分発行計画である2020 ATMを構築し,この計画により,Cowen and Company,LLCまたはCowenを販売代理として,1.00億ドルまでの普通株を現行市場価格で随時公衆に提供·販売することができる。2021年12月31日までの年間で,2020年にATMにより約400万株の普通株を売却し,毛収入と純収益はそれぞれ4410万ドルと4310万ドルであった。2022年12月31日までの年間で、2020年にATMで約1170万株の普通株を売却し、毛収入と純収益はそれぞれ5590万ドルと5480万ドルだった。2022年12月31日現在、2020年までにATMで未販売や販売可能な金額はありません。
2022年2月、私たちはコーエンと新しい販売契約、すなわち2022年2月のATMを締結し、この協定によると、私たちは時々当時の市場価格でコーエンを介して100.0ドルまでの普通株を提供し、販売することができる。2022年12月31日までの年間で,2022年2月のATMにより約1880万株の普通株を売却し,毛収入と純収益はそれぞれ9840万ドルと9640万ドルであった。2023年12月31日までの年間で,2022年2月のATMにより約30万株の普通株を売却し,160万ドルの毛収入と純収益を生み出した。2023年12月31日現在、2022年2月現在、ATMでは未販売と販売可能な金額があります。
2022年11月、私たちはコーエンと販売代理店として追加の販売契約、すなわち2022年11月のATMを締結し、この協定によると、私たちは時々当時の市場価格でコーエンを介して150.0ドルまでの普通株を一般に提供し、販売することができる。2023年12月31日までの年間で,2022年11月のATMにより約1420万株の普通株を売却し,毛収入と純収益はそれぞれ9410万ドルと9220万ドルであった。2023年12月31日現在,約5590万ドルが販売されておらず,2022年11月のATMによる販売が可能である。
2019年5月8日、私たちはシリコンバレー銀行または前SVBと融資と保証契約、すなわち優先信用手配を締結し、その後、2019年6月29日、2020年8月28日、2021年8月27日に改訂を行った。2021年10月29日、私たちは新しい信用手配、オックスフォード金融有限責任会社が担保代理と貸金人として、前SVBは貸手として、他の貸手は時々当事者として、または共同で貸手として、融資と保証協定を締結した。2023年3月,前SVBの利息相続人として,シリコンバレーブリッジ銀行(Silicon Valley Bridge Bank,N.A.)が前SVBに代わって貸手となり,その後第一公民銀行と信託会社(First−Citizens Bank&Trust Company,略称SVB)傘下のシリコンバレー銀行(Silicon Valley Bank,SVB)がSVBBに代わって貸手となった。2023年12月31日現在、私たちは2022年2月17日、2022年10月17日、2022年12月27日、2023年3月23日に改訂された新しい信用手配に基づいて2500万ドルを借り入れましたが、これまで改訂された新しい信用手配によると、追加の借入金額はありません。2024年11月1日までに利息のみを支払い、2026年10月1日満期日までに月等額で元金と適用利息を支払う義務があります。新しい信用配置は、知的財産(ただし、支払いおよび知的財産収益を得る権利を含む)を含まず、私たちが所有または後に取得したほとんどの個人財産を担保とし、知的財産権に関する負の質権は、清算が発生したときに、融資者が償還を得る権利が私たちの普通株式所有者の権利よりも優先されることを保証する。新しい信用手配を締結した後、吾らは終了前にSVBが以前の信用手配の下で更なる信用のすべての約束を提供し、そして吾らが以前の信用手配に従って前SVBが提供したすべての保証と担保権益を提供した。
2023年12月31日まで、私たちは2.091億ドルの現金と現金等価物と有価証券を持っています。私たちの既存の現金、現金等価物、有価証券を除いて、私たちがグラクソ·スミスクライン、ジョンソン、メルクKGaAと行っている協力協定によると、私たちはマイルストーン支払いと他の支払いを得る資格がある。私たちが記念碑的な支払いを稼ぐ能力と、これらの金額を稼ぐ時間は、私たちの発展、規制、ビジネス活動のタイミングと結果にかかっているため、まだ確定していない。
キャッシュフロー
次の表は、2023年12月31日、2022年、2021年12月31日までの3年間のキャッシュフロー情報を提供しています
| | | | | | | | | | | | | | | | | |
| 現在までの年度
十二月三十一日 |
| (単位:千) | 2023 | | 2022 | | 2021 |
| 経営活動のための現金純額 | $ | (168,882) | | | $ | (49,363) | | | $ | (139,988) | |
| 投資活動提供の現金純額 | 119,883 | | | (152,716) | | | (648) | |
| 融資活動が提供する現金純額 | 94,675 | | | 153,017 | | | 63,646 | |
| 現金、現金等価物、および制限現金の増加(減少) | $ | 45,676 | | | $ | (49,062) | | | $ | (76,990) | |
経営活動に使われている現金純額
2023年12月31日現在、経営活動で使用されている純現金は1兆689億ドルで、主に私たちの純運営資本の変化によって調整された1.717億ドルの純損失、私たちの協力協定に関連する繰延収入、および2110万ドルの株式報酬と460万ドルの有価証券プレミアムおよび割引償却を含む他の非現金プロジェクトを含む。2022年12月31日現在、運営活動で使用されている現金純額は4,940万ドルで、主に経営運資本の純額変動調整の純損失2.042億ドル、協力協定に関する繰延収入およびその他の非現金項目を含み、2,150万ドルの株式給与を含む
投資活動による現金純額
2023年12月31日までの年間で、投資活動が提供する現金純額は119.9億ドルだったが、2022年12月31日までの年間で、投資活動に使用された現金純額は152.7億ドルだった。2023年12月31日までの年度内に、投資活動が提供する現金純額は主に有価証券の満期日を含み、一部は有価証券の購入によって相殺される。2022年12月31日までの年度、投資活動で使用される現金純額は主に有価証券の購入を含み、一部は有価証券の満期日によって相殺される
融資活動が提供する現金純額
2023年12月31日までの1年間で、融資活動が提供した純現金は9,470万ドルだったが、2022年12月31日までの年間は153.0ドルだった。融資活動が提供する現金純額は、2022年12月31日までの年間で、主に2022年2月と2022年11月の現金自動支払機での普通株の売却益9350万ドルを含む。12月31日までの1年間、融資活動が提供する現金純額には、2020年の現金自動支払機と2022年2月のATMの使用収益1兆509億ドルが主に含まれている。
資金需要
私たちは、特に研究開発と製造を続け、臨床試験を開始し、候補製品のマーケティング承認を求める時に、私たちが行っている活動に関連した現金支出が増加すると予想している。さらに、私たちの候補製品が市場の承認を得た場合、このような販売、マーケティング、および流通は潜在的なパートナーの責任ではないので、薬品販売、マーケティング、製造、流通に関連した巨額の商業化費用が発生すると予想される
2023年12月31日まで、現金、現金等価物、有価証券2.091億ドルを持っています。私たちは、私たちの現在の利用可能な資金が、2026年まで私たちの現在の運営計画に資金を提供するのに十分だと信じている。私たちの財政資源がどのくらいの間私たちの業務を支持するのに十分な予測は前向きな陳述であり、リスクと不確定要素に関連し、実際の結果は様々な要素によって異なるかもしれない。私たちは間違っていることが証明される可能性があるという仮定に基づいてこの推定をして、私たちは現在予想されているよりも早く私たちが利用できる資本資源を利用することができる。私たちの将来の資本需要は多くの要素に依存します
•著者らの候補製品の薬物発見、臨床前開発、実験室テストと臨床試験の範囲、進捗、結果とコスト
•私たちの研究開発プロジェクトの範囲、優先順位、数量
•私たちの候補製品に対する規制審査のコスト、時間、結果
•私たちが有利な条件で協力を確立し維持する能力があれば
•私たちが獲得した任意の協力協定に基づいて、マイルストーンまたは支払いをトリガする他の事態が発生した
•将来の協力協定によると、私たちは臨床試験費用の補償程度を返済するか、または受ける権利がある
•特許出願を準備し、提出し、起訴し、私たちの知的財産権を維持し、実行し、知的財産権に関するクレームを弁護するコスト;
•他の候補品や技術をどの程度得ることができるか
•臨床と商業生産の製造手配のコストを確保すること
•もし私たちが規制機関の承認を得て私たちの候補製品をマーケティングする場合、販売とマーケティング能力の契約を確立または締結するコスト。
潜在的な候補製品を確定し、臨床前テストと臨床試験を行うことは時間がかかり、高価かつ不確定な過程であり、長年を必要とし、しかも著者らは永遠に発売許可を獲得し、薬物販売を実現するために必要なデータ或いは結果を生成できないかもしれない。しかも、私たちの候補製品が承認されれば、商業的成功を得られないかもしれない。私たちの商業収入は、もしあれば、薬物の販売から来ます。これらの薬物は何年も商業的に得られないと予想されます。したがって、私たちは追加的な資金調達に依存して私たちの業務目標を達成し続ける必要があるだろう。私たちは受け入れ可能な条項で十分な追加融資を受けることができないかもしれないし、根本的にできないかもしれない。
これまで、相当な製品収入を生み出すことができれば、戦略協力、許可手配、株式発行、債務融資を通じて私たちの現金需要を満たす予定です。グラクソ·スミスクライン、ジョンソン、メルクKGaAとの協力で研究開発活動が成功すれば、GSK、ジョンソン、メルクKGaAとの合意から現金マイルストーン支払いを稼ぐことが可能になる。もし私たちが追加の戦略協力や第三者との許可手配を通じて資金を調達すれば、私たちは私たちの技術、将来の収入源、研究計画、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になる可能性のある条項で許可を与えなければならないかもしれない。もし私たちが必要な時に株式や債務融資を通じてより多くの資金を調達できない場合、私たちは私たちの薬物開発や将来の商業化努力を延期、制限、減少、または中止する必要があるかもしれないし、私たちが自分で開発し、マーケティングすることをより望んでいた候補製品の権利を与える必要があるかもしれない。
もし私たちが株式または転換可能な債務証券を売却することで追加資本を調達する場合、私たちの普通株主の所有権権益は希釈され、これらの証券の条項は清算または他の私たちの普通株主の権利に悪影響を及ぼす特典を含む可能性がある。将来の追加債務融資は、可能であれば、追加債務を招く、資本支出を行う、または配当を宣言するなど、私たちが具体的な行動をとる能力を制限または制限する契約を含むいくつかの合意に関連する可能性がある。
契約義務
2023年12月31日まで、私たちは契約義務が知られている重大な現金需要から、主に運営と融資リース負債と、私たちの新しい信用手配による元金と利息支払いを含みます。私たちの将来の融資とレンタルの最低賃貸支払い総額は付記12に含まれています賃貸借契約連結財務諸表に付記する。2023年12月31日現在、運営と融資リースを含む将来の未割引最低賃貸支払い総額は970万ドル。私たちの新しい信用計画によると、私たちの将来の最低元金支払い総額は付記8に含まれています借金を抱え連結財務諸表に付記する。2023年12月31日まで、私たちの新しい信用手配での今後の最低元本支払い総額は2500万ドルです。
私たちは正常な業務過程で第三者と協定を締結して、臨床前、臨床、製造、その他の運営目的のための製品とサービスを提供することを支援します。これらのプロトコルは、通常、我々が合理的な通知後にいつでもキャンセルすることができ、いくつかのプロトコルは停止権を含むが、終了料または清算コストを支払う必要がある。このような債務の正確な金額は終了の時間と合意に関する正確な条項に依存し、合理的に見積もることができない。
我々とライセンス契約を締結した第三者への将来の支払いを含む、いくつかのマイルストーンの実現時に期限が満了し、支払われるべき第三者に未来のお金を支払う義務があります。私たちはこのようなマイルストーンの達成と時間が固定的で確定的ではないので、このような約束を私たちの貸借対照表に含まなかった。
2019年1月、私たちはSynaffix B.V.またはSynaffixと商業許可協定を締結し、私たちは2021年11月と2022年2月にこの協定を修正して再説明し、Synaffixとの関係を拡大した。修正して再記述したプロトコルをSynaffixライセンスと呼ぶ.Synaffixライセンスによれば,Synaffix独自サイト固有の共役技術を用いたターゲットに対するADCを開発,製造,商業化する権利がある.私たちは私たちの開発計画と協力に関する5つの目標を許可し、私たちは最大6つの追加的な目標を許可する権利がある。Synaffixライセンスに関連する680万ドルの費用を支払いました。その中には、400万ドルの予約料と許可料、180万ドルのマイルストーン支払い、100万ドルが将来の予約費および許可料、および将来の潜在開発マイルストーンの一部に使用される可能性があります。発行、開発、規制、ビジネスマイルストーンに4800万~1.32億ドルを支払う義務があります。ライセンスターゲットに対するADC製品が商業販売を開始する場合、もしあれば、Synaffixにそれぞれの製品の純売上高の1桁パーセント以下の階層印税を支払う必要がある。Synaffixライセンスは、製品のSynaffixライセンスの下で許可された特許のうちの最後の有効な権利要件が満了するまで、各国/地域および個々のライセンス製品に基づいて有効に維持される。各国/地域の各許可製品のSynaffixライセンスが満了した後、私たちがその国/地域で私たちに付与したこのような製品のライセンスは、全額支払いおよび永久的になります。私たちは製品を全部または一つずつ許可するSynaffix許可をいつでも終了することができる。どちらも,他方が合意の重要な条項に違反していることや,他方が破産に関するイベントが発生したことが原因で,所定の通知や救済期間内にSynaffixライセンスを終了することができる.
重要な会計政策と重大な判断と見積もり
私たちの経営陣は私たちの財務状況と経営結果の討論と分析は私たちの財務諸表に基づいています。これらの報告書はアメリカ公認会計原則に基づいて作成されています。これらの財務諸表を作成するには、私たちの財務諸表に報告されている資産、負債、収入および費用、または資産および負債の開示に影響を与える判断および推定を行う必要があります。私たちの推定は、歴史的経験、既知の傾向、および事件、およびこのような場合に合理的と考えられる様々な他の要素に基づいている。異なる仮定または条件では、実際の結果は、これらの推定値とは異なる可能性がある。持続的な基礎の上で、私たちは状況、事実、経験の変化に基づいて私たちの判断と推定を評価する。推定に重大な改訂が行われた影響は,推定が変化した日から財務諸表に前向きに反映される。
我々の最も重要な会計政策は,収入確認と計算すべき研究開発費に関する政策であり,これらの政策は本年度報告Form 10−Kにおける総合財務諸表付記で検討されていると考えられる。
収入確認
“会計基準更新2014-09”の範囲で協力協定を締結しました取引先と契約した収入またはASC 606は、この合意に基づいて、私たちの技術およびいくつかの候補製品の権利を許可し、第三者に研究開発サービスを提供する。これらの手配の条項は、一般に、払い戻し不可能な前払い費用、研究開発コストの精算、開発、規制、および商業マイルストーン支払い、製品の純売上を許可する費用および印税のうちの1つまたは複数の費用を支払うことを含む。
ASC 606によれば、顧客が約束された商品またはサービスの制御権を取得した場合、1つのエンティティは、そのエンティティがこれらの商品またはサービス交換から取得することを望む対価格を反映する収入を確認する。ASC 606の範囲内で決定された予定が確認すべき適切な収入額を決定するために、(1)顧客との契約を決定するステップ(S)、(2)約束された商品またはサービスが履行義務であるかどうかを決定するステップ、(3)可変価格の制限を含む取引価格の計量、(4)取引価格を履行義務に割り当てるステップ、の5つのステップを実行する。(V)各履行義務を履行する場合(または義務を履行する場合)に収入を確認します。私たちは、エンティティが顧客に譲渡された商品やサービスと引き換えに獲得する権利のある対価格を受け取る可能性がある場合にのみ、5段階モードを契約に適用します。
私たちの手配では、約束された商品やサービスには、通常、私たちの知的財産権と研究開発サービスの許可権が含まれています。また、契約にはオプションの追加項目があり、これらの項目はマーケティング特典とみなされ、お客様がそのオプションを選択した場合は、お客様との個別契約として計算されます。選択権が契約を締結する前に提供されない実質的な権利を提供しない限り。履行義務は、契約において約束されたユニークな商品またはサービスを顧客の商品またはサービスに譲渡することである。約束された商品またはサービスは、以下の場合にユニークとみなされる:(I)顧客は、単独でまたは他の既製の資源と共に商品またはサービスから利益を得ることができる、または(Ii)約束された商品またはサービスは、契約中の他の約束とは別に識別することができる。約束された商品またはサービスがユニークであるかどうかを評価する際には、潜在的知的財産権の発展段階、顧客が自ら知的財産権を開発する能力、あるいは必要な専門知識をいつでも得ることができるかどうか。
私たちは、譲渡契約で約束された貨物またはサービスが受信する予定されている金額から取引価格を推定します。対価には、固定対価格と可変対価格が含まれている可能性があります。可変対価格を含む各手配の開始時と、各報告期間において、潜在的な支払い金額と支払いを受ける可能性を評価します。可能性の高い金額法または予想金額法を用いて、予想される受信金額を推定し、どの方法に基づいて予想される受信金額をよりよく予測しますか。i重大な収入逆転が発生しない可能性が高い場合、可変対価格は取引価格に含まれます。
私たちの契約は一般的に開発と規制マイルストーン支払いを含む。契約開始時および各報告期間において、マイルストーンが達成可能であると考えられているかどうかを評価し、最も可能な金額法を用いて取引価格に含まれる金額を推定する。重大な収入逆転が発生しない可能性が高い場合、関連するマイルストーン値は取引価格に含まれる。規制承認など、規制承認のような制御または許可側がコントロールしているマイルストーンではない。*その後の各報告期間の終了時に、このような開発マイルストーンを実現する可能性や関連制限を再評価し、必要に応じて全体の取引価格の推定を調整する。
販売ベースの特許使用料を含む手配については、販売レベルに基づくマイルストーン支払いを含み、許可が特許使用料に関連する主要項目とみなされる場合には、(I)関連販売が発生した場合、または(Ii)特許使用料の一部または全部が割り当てられた履行義務が履行された(または部分的に履行されている)ときに収入を確認する。
私たちは基本履行義務の推定独立販売価格に基づいて取引価格を分配するか、またはいくつかの可変対価格の場合に1つ以上の履行義務に割り当てます。私たちは契約で決定された各履行義務の独立販売価格を決定するために判断が必要な仮説を立てなければなりません。私たちは重要な仮定を利用して、他の比較可能な取引を含む可能性がある独立販売価格を決定します。取引を交渉する際に考慮される定価と、それぞれの履行義務を達成する推定コスト。可変対価格の条項が履行義務の履行状況に関連している場合、ある可変対価格は、契約のうちの1つまたは複数の履行義務に特化して割り当てられ、各履行義務に割り当てられた結果金額は、各履行義務から得られることが予想される金額と一致する。
ライセンスやその他の承諾からなる履行義務については、合併履行義務の性質を判断して評価し、合併履行義務が時間の経過とともに満たされているか、時間が経過するにつれて払戻不可能な前払い費用からの収入を確認するために、進展を測定する適切な方法を決定する。各報告期間で進捗の測定基準を評価し、必要に応じて業績および関連収入確認の測定基準を調整する。知的財産権許可が手配で決定された他の履行義務と異なると判定された場合、払戻不可能な収入からの収入を確認し、ライセンスが顧客に譲渡され、顧客がライセンスを使用して利益を得ることができる場合、ライセンスに割り当てられた前払い費用。
協力手配
我々は,ASC 808により連携運営活動を代表する連携プロトコルの要素を記録した協力手配それは.そのため,双方を代表する積極的な参加者の活動と,双方とも活動のビジネス成功に依存した重大なリスクとリターンに直面する協力合意の要素が,協力手配として記録されている.我々と我々の協力者との間の取引および第三者との間の取引を決定する際に、ASC 606における指導を考慮する。一般に,協調スケジューリングによる取引分類は,手配の性質と契約条項および参加者の業務的性質に基づいて決定される.提携による収入については,顧客との取引で依頼者とみなされれば,純売上高における我々のシェアを毛数で確認し,顧客との取引におけるエージェントとみなされれば,純額で確認することは,ASC 606の指導と一致する.
研究と開発費用を計算すべきである
財務諸表作成プロセスの一部として、各貸借対照表の日付までの計上費用を見積もる必要がある。このプロセスは、未締結契約および調達注文を審査し、私たちの担当者とコミュニケーションして、私たちに代わって実行されるサービスを決定し、インボイスを受け取っていない場合、または他の方法で実際のコストを通知している場合に、実行されたサービスレベルおよびサービスによって生じる関連コストを推定することを含む。私たちのほとんどのサービスプロバイダはサービスを履行したり契約のマイルストーンに達した時に毎月私たちに借金の領収書を発行してくれます。私たちは、私たちが当時知っていた事実と状況に基づいて、各貸借対照表の日付までの課税費用を推定した。私たちは定期的にサービスプロバイダと私たちの推定の正確性を確認し、必要に応じて調整します。私たちの計算すべき研究開発費用の大きな見積もりには、私たちのサプライヤーが提供する研究開発活動に関連するサービスによるコストが含まれていますが、領収書は受け取っていません。
我々は、我々を代表して研究·開発を行っているサプライヤーとの見積もりや契約に基づいて、受信したサービスや費用の見積もりに基づいて、研究·開発活動に関連する費用を記録する。これらの合意の財務条項は交渉が必要であり、契約によって異なり、支払いの不均衡を招く可能性がある。場合によっては、私たちのサプライヤーに支払われるお金は、提供されたサービスレベルを超え、研究開発費の前払いにつながる可能性があります。サービス料を計算する際には、各期間内にサービスを提供する時間帯、被験者の登録人数、アクティブなサイト数、かかる努力度を推定します。サービスの実行時間や努力度が私たちの推定と異なる場合、それに応じて計算または前払い費用残高を調整します。実際に発生した金額と実質的に差はないと予想されていますが、提供されたサービスの状態や時間の推定が実際に行われているサービスの状態や時間と異なる場合には、任意の特定の時期に高すぎたり低すぎる金額を報告することができます。私たちの見積もりは実際に発生した金額と実質的な差はない。上記の推定を行う際には、重大な判断に関連する。
最近の会計声明
付記2を参照重要な会計政策の概要当社の業務に適用される最新の会計声明の説明を理解するために、総合財務諸表付記を参照してください。
第7 A項には、市場リスクに関する定量的かつ定性的な開示が含まれる
金利リスク
私たちは金利の変化と関連した市場リスクに直面している。2023年12月31日まで、現金、現金等価物、有価証券2.091億ドルを持っています。私たちの市場リスクに対する主な開口は金利敏感性であり、金利敏感性はアメリカ金利全体レベルの変化の影響を受け、特に私たちの投資は現金等価物と有価証券を含み、アメリカ国債、商業手形、社債、アメリカ政府機関証券に投資するからである。しかし、私たちのポートフォリオの短期的な持続期間と私たちの投資の低リスク状況により、最低金利が直ちに100ベーシスポイント変化しても、私たちポートフォリオの公平な市場価値に実質的な影響を与えないと信じている。
私たちの新しい信用ツールの金利は金利の変化に敏感だ。信用手配による借入利息は利息を計算しなければならず、浮利は(I)8.50%及び(Ii)の最優遇金利プラス5.25%の両者の中で大きい者に等しい。私たちは現在金利変化に対するヘッジキャンペーンをしていない。2023年12月31日現在、新信用手配による未返済資金は2,500万ドルであり、関連金利の潜在的な変化は私たちの業務結果にとって重要ではない。
外貨為替リスク
2023年12月31日現在、外貨為替レートの変化に関する市場リスクにさらされていませんが、アジアやヨーロッパにあるサプライヤーと契約を結ぶ可能性があり、外貨為替レートの変動の影響を受ける可能性があります。
第8項:財務諸表及び補足データ
Mersana治療会社
連結財務諸表索引 | | | | | |
| ページ |
独立公認会計士事務所報告(PCAOB ID:42) | 121 |
合併貸借対照表 | 123 |
合併経営報告書と全面赤字 | 124 |
株主権益合併報告書 | 125 |
統合現金フロー表 | 126 |
連結財務諸表付記 | 127 |
独立公認会計士事務所報告
Mersana治療会社の株主と取締役会に。
財務諸表のいくつかの見方
添付されているMersana治療会社(当社)が2023年12月31日までと2022年12月31日までの総合貸借対照表、2023年12月31日までの年間ごとの関連総合経営と全面赤字報告書、総合株主権益表と総合現金フロー表および関連付記(総称して“総合財務諸表”と呼ぶ)を監査した。総合財務諸表は、すべての重要な点で、2023年12月31日、2023年12月31日、2022年12月31日までの会社の財務状況、および2023年12月31日までの3年度の経営結果とキャッシュフローを公平に反映しており、米国公認会計原則に適合していると考えられる。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査することを求められておらず、私たちも招聘されて監査を行っていません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性に対する意見を表明するためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
以下に述べる重要な監査事項とは、(1)財務諸表に対して重大な意味を有する勘定又は開示に関し、(2)特に挑戦的、主観的、又は複雑な判断に係る財務諸表を監査して生じた事項である。重要監査事項の伝達は、総合財務諸表に対する私たちの全体的な意見をいかなる方法でも変えることはなく、以下の重要な監査事項を伝達することによって、重要な監査事項またはそれに関連する勘定または開示について個別の意見を提供することはない。
| | | | | | | | |
| | 課税と前払いの臨床費用 |
| | |
| 関係事項の記述 | | 総合財務諸表付記7に記載されているように、2023年12月31日現在、同社の臨床費用は総額510万ドルである。また,同社の前払い費用と他の流動資産は合計500万ドルであり,2023年12月31日までに臨床試験に基づいてあらかじめ支払われたサービス金額が含まれている。総合財務諸表付記2に記載されているように、当社は、サプライヤーが提供したいくつかの資料に基づいて、発生した実際のコストまたは払った努力レベルを含めて、発生した臨床コストおよび関連する計算すべきまたは残りの前払い費用を推定しなければならない。このような活動の支払いは個別に手配された条項によって、発生した費用パターンとは異なる可能性がある
監査会社の計算すべきおよび前払い臨床費用は、これらの金額は、第三者サプライヤーの様々な推定、および管理層メンバー推定の他の入力、例えば、発生したが請求書が発行されていない実際のコスト、推定されたプロジェクトスケジュール、およびこれらのサービスに関連するコストに基づくので、複雑かつ判断される。また,会社の研究·開発活動の継続時間や第三者から伝票を受信した時間のため,実際に発生した金額は通常財務諸表発行日には不明である
|
私たちが監査でどのようにこの問題を解決したのか | | 計算すべき臨床費用および前払いの臨床費用をテストするために、私たちの監査プログラムは、記録された金額を推定するための基礎データの正確性および完全性をテストすることを含む。私たちは会社が研究開発プロジェクトを監督する研究開発者との討論を通じて、研究開発活動の進展を実証した。また、会社と第三者との契約や未解決の変更書をチェックして、記録金額への影響を評価しました。さらに、当社は、第三者がこれまでに生成したコストの推定値を含む、会社が特定のサイトおよび他の第三者から直接受信した情報を独立して確認および/または検討した。著者らはまた、サプライヤー、研究或いはその他の重要な伝票対応計と前払い臨床費用の変動に基づいて分析を行い、第三者から受信した後続領収書を検査し、対応計と前払い残高の影響を評価した。 |
/s/ 安永法律事務所
2013年以来、当社の監査役を務めてきました。
ボストン、マサチューセッツ州
2024年2月28日
Mersana治療会社です。
合併貸借対照表
(千単位で1株当たりおよび1株当たりのデータは含まれていない) | | | | | | | | | | | |
| 十二月三十一日
2023 | | 十二月三十一日
2022 |
| 資産 | | | |
| 流動資産: | | | |
| 現金と現金等価物 | $ | 174,561 | | | $ | 128,885 | |
| 短期有価証券 | 34,523 | | | 151,827 | |
| 売掛金 | — | | | 30,000 | |
| 前払い費用と他の流動資産 | 4,973 | | | 8,507 | |
| 流動資産総額 | 214,057 | | | 319,219 | |
| 財産と設備、純額 | 3,831 | | | 3,985 | |
| 経営的リース使用権資産 | 7,694 | | | 10,475 | |
| 他の非流動資産 | 478 | | | 661 | |
| 総資産 | $ | 226,060 | | | $ | 334,340 | |
| 負債と株主権益 | | | |
| 流動負債: | | | |
| 売掛金 | $ | 7,319 | | | $ | 13,951 | |
| 費用を計算する | 21,898 | | | 43,184 | |
| 収入を繰り越す | 28,147 | | | 30,610 | |
| リース負債を経営する | 3,252 | | | 2,798 | |
| 短期債務 | 2,083 | | | — | |
| その他流動負債 | 938 | | | 990 | |
| 流動負債総額 | 63,637 | | | 91,533 | |
| 非流動経営賃貸負債 | 5,149 | | | 8,575 | |
| 長期債務、純額 | 23,148 | | | 24,929 | |
| 繰延収入、非流動収入 | 97,167 | | | 117,043 | |
| 他の非流動負債 | 55 | | | 203 | |
| 総負債 | 189,156 | | | 242,283 | |
| 約束(注15) | | | |
| 株主権益 | | | |
優先株、$0.0001額面価値25,000,000ライセンス株;0それぞれ2023年12月31日と2022年12月31日に発行された株式 | — | | | — | |
普通株、$0.0001額面価値350,000,000ライセンス株;120,711,745そして105,144,864それぞれ2023年12月31日と2022年12月31日に発行された株式 | 12 | | | 11 | |
| 追加実収資本 | 863,242 | | | 746,889 | |
| その他の総合収益を累計する | 11 | | | (152) | |
| 赤字を累計する | (826,361) | | | (654,691) | |
| 株主権益総額 | 36,904 | | | 92,057 | |
| 総負債と株主権益 | $ | 226,060 | | | $ | 334,340 | |
付記はこれらの連結財務諸表の構成要素である。
Mersana治療会社です。
合併経営報告書と全面赤字
(千単位で1株当たりおよび1株当たりのデータは含まれていない) | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | 2021 |
| 協力収入 | $ | 36,855 | | | $ | 26,581 | | | $ | 43 | |
| 運営費用: | | | | | |
| 研究開発 | 148,269 | | | 173,385 | | | 132,013 | |
| 一般と行政 | 59,543 | | | 56,963 | | | 36,888 | |
| 再編成費用 | 8,713 | | | — | | | — | |
| 総運営費 | 216,525 | | | 230,348 | | | 168,901 | |
| その他の収入(支出): | | | | | |
| 利子収入 | 12,073 | | | 2,883 | | | 65 | |
| 利子支出 | (4,073) | | | (3,328) | | | (1,267) | |
| その他の収入を合計して純額 | 8,000 | | | (445) | | | (1,202) | |
| 純損失 | $ | (171,670) | | | $ | (204,212) | | | $ | (170,060) | |
| その他の全面的な損失: | | | | | |
| 有価証券の未実現収益 | 163 | | | (152) | | | — | |
| 総合損失 | $ | (171,507) | | | $ | (204,364) | | | $ | (170,060) | |
| 普通株主は純損失を占めるべきである−基本損失と希薄損失 | $ | (171,670) | | | $ | (204,212) | | | $ | (170,060) | |
| 普通株主1株当たり純損失−基本損失と希釈損失− | $ | (1.48) | | | $ | (2.18) | | | $ | (2.41) | |
| 加重−普通株主が1株当たり純損失の平均普通株数を占めるべき−基本と償却− | 116,112,891 | | | 93,654,243 | | | 70,580,949 | |
付記はこれらの連結財務諸表の構成要素である。
Mersana治療会社です。
株主権益合併報告書
(千単位で1株当たりおよび1株当たりのデータは含まれていない) | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 普通株 | | その他の内容
支払い済み
資本 | | その他の総合収益を累計する | | 積算
赤字.赤字 | | 株主権益 |
| 株 | | 金額 | | | | |
| 2020年12月31日残高 | 68,841,288 | | | $ | 7 | | | $ | 508,499 | | | $ | — | | | $ | (280,419) | | | $ | 228,087 | |
市場取引から普通株を発行し、発行コスト#ドルを差し引く988 | 3,961,074 | | | — | | | 43,087 | | | — | | | — | | | 43,087 | |
| 株式オプションの行使 | 421,381 | | | — | | | 1,837 | | | — | | | — | | | 1,837 | |
| 帰属制限株式単位は、従業員の納税義務を差し引いた純額 | 407,060 | | | — | | | (259) | | | — | | | — | | | (259) | |
| ESPPにより普通株を購入する | 78,253 | | | — | | | 640 | | | — | | | — | | | 640 | |
| 株に基づく報酬費用 | — | | | — | | | 18,409 | | | — | | | — | | | 18,409 | |
| 純損失 | — | | | — | | | — | | | — | | | (170,060) | | | (170,060) | |
| 2021年12月31日の残高 | 73,709,056 | | | $ | 7 | | | $ | 572,213 | | | $ | — | | | $ | (450,479) | | | $ | 121,741 | |
市場取引から普通株を発行し、発行コスト#ドルを差し引く3,476 | 30,497,875 | | | 4 | | | 150,758 | | | — | | | — | | | 150,762 | |
| 株式オプションの行使 | 414,914 | | | — | | | 1,331 | | | — | | | — | | | 1,331 | |
| 普通株式承認証の行使 | 16,654 | | | — | | | — | | | — | | | — | | | — | |
| 株式単位の帰属を制限する | 235,591 | | | — | | | — | | | — | | | — | | | — | |
| ESPPにより普通株を購入する | 270,774 | | | — | | | 1,065 | | | — | | | — | | | 1,065 | |
| 株に基づく報酬費用 | — | | | — | | | 21,522 | | | — | | | — | | | 21,522 | |
| その他総合損失 | — | | | — | | | — | | | (152) | | | — | | | (152) | |
| 純損失 | — | | | — | | | — | | | — | | | (204,212) | | | (204,212) | |
| 2022年12月31日の残高 | 105,144,864 | | | $ | 11 | | | $ | 746,889 | | | $ | (152) | | | $ | (654,691) | | | $ | 92,057 | |
市場取引から普通株を発行し、発行コスト#ドルを差し引く2,082 | 14,464,531 | | | 1 | | | 93,669 | | | — | | | — | | | 93,670 | |
| 株式オプションの行使 | 102,596 | | | — | | | 427 | | | — | | | — | | | 427 | |
| 制限株式単位及びその他の株式奨励の帰属 | 618,246 | | | — | | | — | | | — | | | — | | | — | |
| ESPPにより普通株を購入する | 381,508 | | | — | | | 1,121 | | | — | | | — | | | 1,121 | |
| 株に基づく報酬費用 | — | | | — | | | 21,136 | | | — | | | — | | | 21,136 | |
| その他総合収益 | — | | | — | | | — | | | 163 | | | — | | | 163 | |
| 純損失 | — | | | — | | | — | | | — | | | (171,670) | | | (171,670) | |
| 2023年12月31日の残高 | 120,711,745 | | | $ | 12 | | | $ | 863,242 | | | $ | 11 | | | $ | (826,361) | | | $ | 36,904 | |
付記はこれらの連結財務諸表の構成要素である。
Mersana治療会社です。
統合現金フロー表
(単位:千) | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | 2021 |
| 経営活動のキャッシュフロー | | | | | |
| 純損失 | $ | (171,670) | | | $ | (204,212) | | | $ | (170,060) | |
| 純損失と経営活動で使用される現金純額の調整: | | | | | |
| 減価償却 | 1,517 | | | 927 | | | 855 | |
| | | | | |
| | | | | |
| 有価証券の割増と割引の純償却 | (4,569) | | | (1,462) | | | — | |
| 株に基づく報酬 | 21,136 | | | 21,522 | | | 18,409 | |
| 非現金でレンタル料金を扱っております | 2,780 | | | 2,777 | | | 1,829 | |
| 他の非現金プロジェクト | 687 | | | 763 | | | 723 | |
| 経営性資産と負債変動状況: | | | | | |
| 売掛金 | 30,000 | | | (30,000) | | | — | |
| 前払い費用と他の流動資産 | 3,534 | | | 3,863 | | | (2,734) | |
| その他の資産 | — | | | — | | | (718) | |
| 売掛金 | (6,080) | | | 947 | | | 483 | |
| 費用を計算する | (20,935) | | | 13,594 | | | 12,570 | |
| | | | | |
| リース負債を経営する | (3,143) | | | (2,539) | | | (1,827) | |
| 収入を繰り越す | (22,139) | | | 143,709 | | | (43) | |
| その他負債 | — | | | 748 | | | 525 | |
| 経営活動のための現金純額 | (168,882) | | | (49,363) | | | (139,988) | |
| | | | | |
| 投資活動によるキャッシュフロー | | | | | |
| 有価証券の満期日 | 277,970 | | | 97,000 | | | — | |
| 有価証券を購入する | (155,919) | | | (247,519) | | | — | |
| 財産と設備を購入する | (2,168) | | | (2,197) | | | (648) | |
| 投資活動提供の現金純額 | 119,883 | | | (152,716) | | | (648) | |
| | | | | |
| 融資活動によるキャッシュフロー | | | | | |
| | | | | |
| 市場施設の純収益 | 93,539 | | | 150,893 | | | 43,087 | |
| | | | | |
| 株式オプションを行使して得られる収益 | 427 | | | 1,331 | | | 1,837 | |
| ESPPによる普通株購入収益 | 1,121 | | | 1,065 | | | 640 | |
| 帰属制限株式単位に関する従業員の納税義務を支払う | — | | | — | | | (259) | |
| 債券を発行して得られる収益は,発行コストを差し引く | (150) | | | — | | | 24,042 | |
| 債務を返済する | — | | | — | | | (5,486) | |
| 融資リース義務下の支払い | (262) | | | (272) | | | (215) | |
| 融資活動が提供する現金純額 | 94,675 | | | 153,017 | | | 63,646 | |
| | | | | |
| 現金、現金等価物、および制限現金の増加(減少) | 45,676 | | | (49,062) | | | (76,990) | |
| 期初現金、現金等価物、および限定現金 | 129,363 | | | 178,425 | | | 255,415 | |
| 現金、現金等価物、制限された現金、期末 | $ | 175,039 | | | $ | 129,363 | | | $ | 178,425 | |
| | | | | |
| 非現金活動の追加開示: | | | | | |
| | | | | |
| | | | | |
| 売掛金と売掛金のうち財産·設備購入 | $ | 132 | | | $ | 753 | | | $ | — | |
| 計上すべき費用のうち債務融資コスト | $ | — | | | $ | 150 | | | $ | — | |
| 売掛金と売掛金のうち普通株式発行コスト | $ | — | | | $ | 131 | | | $ | — | |
| 利子を支払う現金 | $ | 3,380 | | | $ | 2,463 | | | $ | 429 | |
| 経営性リース負債と引き換えに使用権資産 | $ | — | | | $ | 298 | | | $ | 3,783 | |
| 融資リース負債と引き換えに使用権資産 | $ | — | | | $ | — | | | $ | 609 | |
| | | | | |
付記はこれらの連結財務諸表の構成要素である。
カタログ表
Mersana治療会社です。
連結財務諸表付記
1. 業務の性質と列報根拠
Mersana治療会社は臨床段階の生物製薬会社であり、抗体-薬物結合体(“ADC”)の開発に集中し、重大な需要を満たしていない癌患者に臨床上意義のあるメリットを提供する。同社の次世代ADCプラットフォームは、独自の黄金色薬物ペイロードを提供するDolasynThenと、固有のインターフェロン遺伝子(“STINT”)アゴニストペイロードを提供する免疫合成とを含む
同社はXMT-1660、B 7-H 4ガイドのDolasynten ADCを研究しており、一期の臨床試験で固形腫瘍患者を募集しており、乳癌、子宮内膜癌、卵巣癌患者を含む。同社は2023年1月にXMT-2056を研究する第一段階の臨床試験を開始し、XMT-2056は1種の免疫合成刺激剤ADCであり、ヒト表皮増殖因子受容体2(“HER 2”)の新しいエピトープに対して、以前治療を受けたHER 2を発現する末期/再発固形腫瘍患者、乳癌、胃癌、結腸直腸癌と非小細胞肺癌を含むことを目的とした。2023年3月、会社が自発的にこの臨床試験を一時停止した後、この臨床試験はアメリカ食品と薬物管理局(FDA)に臨床的に保留され、FDAは2023年10月にこの臨床保留を解除した。同社はまだ持っている二つ同社の免疫合成プラットフォームを利用した他の早期臨床前候補薬物XMT-2068とXMT-2175。
2023年7月、同社は、その主要な終点に達していないupifitamab rilsodotin(“upRi”)第2段階Uplifitamab Rilsodotin臨床試験の主要なデータを発表した。この声明について、同社は2023年7月27日、その将来の主な重点は、その次世代ADCプラットフォーム、DolasynThenと免疫合成推進候補製品と協力を利用することであると発表した。そこで,同社はUpRiに関する開発活動および規制と商業準備を終了し,UpRiのアップグレード−A期と第3段階Up−Next臨床試験を終了し,FDAは2023年6月に一部の臨床休止を行った
同社は生物技術業界会社によく見られるリスクに直面しており、追加資本の需要、臨床前研究と臨床試験失敗のリスク、それが確定と開発可能な任意の候補薬物は上場許可と精算を得る必要がある、その候補製品は商業化に成功し、市場認可を得る必要があり、肝心な者への依存、ノウハウの保護、政府法規の遵守、競争相手開発技術革新、第三者メーカーへの依存及び試験生産から大規模生産への移行能力を含む。
会社は設立以来累計純損失を出しています。会社の純損失は#ドルです171.7百万、$204.2百万ドルとドル170.12023年、2022年、2021年12月31日までの年度はそれぞれ100万ドル。少なくとも今後数年以内に、会社は経営赤字を続けることが予想される。2023年12月31日現在、会社の累積損失は$となっている826.4百万ドルです。他の要因を除いて、同社の将来の成功は、その候補製品を識別·開発する能力に依存し、最終的には収益運営を実現する能力に依存する。同社は,そのほとんどの財源と努力を研究·開発および一般·行政費用に投入し,このような研究·開発を支援している。純損失とマイナス運営キャッシュフローはすでに、会社の株主権益や運営資本に悪影響を与え続けている。
会社は,本年度報告がForm 10−K形式で発表されてから,少なくとも今後12カ月以内に,その既存資金は会社の運営に資金を提供すると信じている。経営陣が業務に資金を提供する能力に対する信念は、リスクや不確定要因の影響を受ける推定に基づいている。実際の結果が経営陣の見積もりと異なれば、会社は追加の資金を求める必要があるかもしれない。
添付されている総合財務諸表は、米国公認会計原則(“米国公認会計原則”)及び米国証券取引委員会(“米国証券取引委員会”)の規則及び規定に基づいて作成される。本付記内の適用指針に対するいかなる言及も、財務会計基準委員会(“FASB”)の会計基準編纂(“ASC”)及び会計基準更新(“ASU”)に掲載されている権威あるアメリカ公認会計原則を指す。
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
2. 重要会計政策の概要
合併原則
添付されている連結財務諸表には、当社とその完全子会社Mersana Securities Corp.の連結財務諸表が含まれています。すべての会社間残高と取引は無効になりました。
予算の使用
米国公認会計原則に基づいて会社の総合財務諸表を作成する際には、管理層は、財務諸表日の資産、負債、権益、収入、費用及び又は有資産と負債の関連開示及び報告期間内の収入及び支出の報告金額に影響を与える推定と仮定を行う必要がある。会社経営層は、その収入スケジュール内の業績義務の決定と、これらの業績義務の独立販売価格、計算すべき臨床前、製造および臨床費用、株式に基づく報酬の推定値、所得税の判断を含むが、これらに限定されない推定値を評価し続けている。実際の結果はこれらの推定とは異なる可能性がある。
市場情報を細分化する
運営部門は企業の構成要素として定義され、その独立した離散情報は、首席運営意思決定者または意思決定グループが資源をどのように割り当てるかを決定し、業績を評価する際に評価することができる。同社はその運営·管理業務を単一の運営部門、すなわちADCの発見·開発業務と見なしている。
研究と開発
研究と開発コストは、発生時に費用を計上します
•従業員に関する費用には、賃金、ボーナス、福祉、出張、株式報酬費用が含まれる
•契約研究組織、研究場所と他の実体と臨床前研究、臨床試験と関連サービスについて合意したことによる費用と支出
•ADC候補製品、臨床試験材料、および他の研究開発材料の取得、開発および製造のコスト
•規制の準備と活動に関する費用と費用
•協力協定に関連するコストおよびライセンス契約に関連する許可料およびマイルストーン支払い;
•NaPi 2 bのバイオマーカー診断の開発に伴うコスト
•賃貸料、光熱費、施設修理、保険およびその他の用品の直接および分配費用を含む施設、減価償却およびその他の費用
•臨床,臨床前,発見,その他の研究活動に関する他の費用。
臨床前研究、臨床試験、製造開発活動のようないくつかの開発活動のコストは、特定の任務を達成する進捗を評価した上で確認され、使用されるデータは、患者登録、臨床現場活性化、およびサプライヤーが会社に提供するその実際のコストまたは支出レベルに関する情報を含む。これらの活動の支払いは、発生したコストモデルとは異なる個別手配の条項に基づいており、合併貸借対照表に前払いまたは計算すべき臨床前、製造、および臨床費用として反映される可能性がある。
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
収入確認
当社はASC 606の範囲内の協力協定を締結した取引先と契約した収入これらの取り決めの条項は、一般に、払い戻し不可能な前払い費用、研究開発費用の精算、開発、規制、および商業マイルストーン支払い、ライセンス製品の純売上高の費用および使用料のうちの1つまたは複数の費用を支払うことを含む。
ASC 606によれば、顧客が承諾した商品またはサービスの制御権を取得した場合、1つのエンティティは、そのエンティティがこれらの商品またはサービス交換から取得することを望む額を反映する収入を確認する。ASC 606の範囲内で決定された手配が確認すべき適切な収入額を決定するために、会社は、(1)顧客との契約を決定するステップ(S)、(2)約束された商品またはサービスが履行義務であるかどうかを決定するステップ、(3)可変対価格の制限を含む取引価格の計量、(4)取引価格を履行義務に割り当てる、の5つのステップを実行する。(V)会社が各履行義務を履行する際に収入を確認する。会社は、エンティティが顧客に譲渡された商品またはサービスと交換する権利のある対価格を受け取る可能性が高い場合にのみ、5ステップモードを契約に適用する。
会社の手配で約束された商品やサービスには、通常、会社の知的財産権と研究開発サービスの許可権が含まれています。会社はまた、契約にオプションの追加項目があり、これらの項目はマーケティング要約とみなされ、顧客がそのオプションを選択した場合は、顧客との単独契約として計算されます。オプションが契約を締結する前に提供されない実質的な権利を提供しない限り。履行義務とは、契約において約束されたユニークな貨物またはサービスを顧客に譲渡する貨物またはサービスを意味する。約束された貨物またはサービスは、以下の場合にユニークであると考えられる:(I)顧客は、単独でまたは他の既製の資源と共に貨物またはサービスから利益を得ることができ、または(Ii)約束された貨物またはサービスは、契約中の他の約束から分離して識別することができる。約束された貨物またはサービスが異なるかどうかを評価する際に、会社は潜在的な知的財産の開発段階などの要因を考慮することができる。顧客が独自に知的財産権を開発する能力と必要な専門知識の可用性。
当社は、譲渡契約で約束された貨物またはサービスが受信すると予想される金額に基づいて取引価格を推定します。この対価には、固定対価格および可変対価格が含まれている可能性があります。可変対価格を含む各手配開始時と各報告期間において、当社は潜在的な支払い金額と支払いを受ける可能性を評価します。当社は、最も可能な金額法または予想値法を用いて予想される受信金額を推定し、どの方法に基づいて、当社が獲得する権利のある対価格金額をよりよく予測することができます。大きな収入逆転が起こらない可能性が高い場合、可変対価は取引価格に含まれています。同社は、(A)約束された対価格が約束された貨物およびサービスの現金販売価格に近いので、重要な融資部分が存在するかどうかを決定するために、その各創設計画を評価し、(A)約束された対価格が約束された貨物およびサービスの現金販売価格に近いため、(B)支払いの時間が貨物およびサービスの譲渡に近く、契約期間全体にわたって相対的に短いからであると結論付けた。
その会社の契約には一般的に開発と管理マイルストーン支払いが含まれている。契約開始時および各報告期間において、当社はマイルストーンに達する可能性があるかどうかを評価し、最も可能な金額法を用いて取引価格に含まれる金額を推定する。重大な収入逆転が発生しない可能性が高い場合には、関連する記念碑値は取引価格に計上される。規制承認のような当社または許可者の制御範囲内でのマイルストーンの支払いはない。その後の各報告期間の終了時に、会社は、そのような開発マイルストーンを実現する可能性や関連制限を再評価する必要がある。全体の取引価格の推定値を調整します。どのような調整も累積追跡に基づいて記録されており、これは調整期間中の許可証、協力、および他の収入および収益に影響を与えます。
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
販売に基づく特許権使用料を含む手配については、販売レベルに基づくマイルストーン支払いを含み、許可が特許権使用料に関連する主な項目とみなされている場合、当社は、(I)関連販売が発生した場合、または(Ii)特許権使用料の一部または全部が割り当てられた履行義務が履行された(または部分的に満たされている)ときに収入を確認する。これまで、当社は、会社の任意の協力手配からのいかなる特許権使用料収入も確認していない。
当社は、基本履行義務の推定独立販売価格に基づいて取引価格を割り当てるか、またはある可変対価格の場合には1つ以上の履行義務に割り当てる。*会社は、契約で決定された各履行義務の独立販売価格を決定するために判断すべき仮定を作成しなければならない。当社は、キー仮説を用いて独立販売価格を決定し、その中に他の可比取引が含まれている可能性がある。交渉取引時に考慮される定価と、それぞれの履行義務を達成する推定コスト。可変対価格の条項が履行義務の履行状況に関連している場合、ある可変対価格は、契約のうちの1つまたは複数の履行義務に特化して割り当てられ、各履行義務に割り当てられたそれによって生じる金額は、会社が各履行義務から得られると予想される金額と一致する。
ライセンス及びその他の承諾からなる履行義務については、当社は、合併履行義務が時間の経過とともに履行されているか、またはある時点で履行されているかを決定する判断を利用して、経過とともに進行を測定する適切な方法を決定し、払戻不可能な前払い費用からの収入を確認する。企業は、各報告期間において進捗の測定基準を評価し、必要に応じて業績測定基準および関連収入確認を調整する。会社の知的財産権許可が手配で決定された他の履行義務と判定された場合、会社は払戻不可能な収入からの収入を確認する。ライセンスが顧客に譲渡され、顧客がライセンスを使用して利益を得ることができる場合、ライセンスに割り当てられた前払い費用。
その会社は各契約で決定された請求書スケジュールに基づいて顧客から支払いを受ける。このような勘定書は普通あります30-日条項。前払い金及び費用は、会社がこれらの手配の義務を履行するまで、受領又は満了時に契約負債(繰延収入)と表記する。代金に対して無条件であり、かつ支払いまでに時間が経過するだけであれば、金額を売掛金とする。価格権に対して時間経過以外の条件の制約を受けると,その額は支払権が無条件になるまで契約資産として記録される.米国会計基準第606条によれば、会社は顧客契約に応じて契約資産及び契約負債を純額で列記する。
協力手配
当社はASC 808に基づいて、その共同経営活動を代表する連携協定の要素を記録する協力手配8(“ASC:808”)。そのため,双方を代表する積極的な参加者の活動と,双方とも活動のビジネス成功に依存した重大なリスクとリターンに直面する協力合意の要素が,協力手配として記録されている.当社は、当社とその協力者との取引や当社と第三者との取引を決定する際にも、ASC 606における指導意見を類推的に考慮しています。一般に,協調スケジューリングによる取引分類は,手配の性質と契約条項および参加者の業務的性質に基づいて決定される.提携による収入については,会社が顧客との取引で依頼者とみなされれば,会社は純売上高におけるシェアを毛数で確認し,顧客との取引でエージェントとみなされれば,会社が純額で確認することはASC 606の指導と一致する.
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
公正価値計量
公正価値は、資産を売却する際に受信された価格または計量日に市場参加者間で負債を移転するために支払われる価格として定義される。ASC:820,中国公正価値計量公平な価値に応じて計量するツールのために、三級評価階層構造を構築した。この階層構造は、計量日までの資産または負債推定値の投入の透明性に基づく。この3つのクラスは以下のように定義される
第1レベル-推定方法の投入は、アクティブ市場における同じ資産または負債の見積もり(未調整)である。
第2レベル--推定方法の投入には、市場における同様の資産および負債の見積もりの活発化と、金融商品のほぼ全期間にわたって直接または間接的に観察可能な資産または負債の投入が含まれる。
第三級-推定方法への投入は観察できず、公正価値計量に重要な意義がある。
有価証券
会社の投資戦略は保本に重点を置いています。当社は当社の投資政策で概説した信用品質基準に適合するツールに投資しています。短期有価証券には債務証券への投資が含まれており、これらの債務証券の満期日は3カ月を超え、貸借対照表の日から1年も経っていない。その会社はそのすべての有価証券を販売可能なものに分類した。したがって、このような投資は公正な価値で入金される。公正価値は見積もりの市場価格に基づいて決定される。割引と割増の償却と増加は他の収入(費用)で利息収入、純額と記す。実現した損益は他の収入(費用),純額に計上される
当社は、ASC 326における売却可能な債務証券減価モデルに基づいて、債務証券を売却することができることを評価する金融商品--信用損失各報告日までに、債務証券の売却確認のために使用可能な公正価値が帳簿価値よりも低い任意の低下が信用損失の結果であるか否かを決定する。当社は総合経営報告書に信用損失と全面損失を他の収入(費用)の純額の構成要素として記録しており、純額は証券の公正価値と償却コストとの差額に限られている。現在まで、同社は売却可能な債務証券にいかなる信用損失も記録していない。
現金と現金等価物
当社はすべての原始満期日または購入時の残り満期日が三ヶ月以下の高流動性投資を現金等価物と見なしています。同社は余分な現金を主に通貨市場基金、商業手形、政府機関証券に投資し、これらの証券は非常に高い流動性と高い信用格付けを持っている。このような投資の信用と市場の危険はわずかだ。現金と現金等価物はコスト別に記載され、市場価値に近づいている。
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
株式報酬の会計計算
当社はASC 718に基づいて株式報酬を会計処理し、報酬--株式報酬(“ASC 718”)。ASC 718は、従業員、取締役、および非従業員に支払われたすべての株式を、運営報告書において、その付与日の公正価値に基づいて費用として確認することを要求する。同社はブラック·スコアーズオプション定価モデルを用いて付与されたオプションの公正価値を推定している。ブラック·スコアーズオプション定価モデルは、(A)予想株価変動、(B)予想報酬期間の計算、(C)無リスク金利、および(D)期待配当を含むいくつかの主観的仮定に基づく投入を必要とする。期待株価変動率は,期待期限の仮定に見合った一定期間に基づいて計算される.歴史的に見ると、会社の普通株は初公募株が完成するまで市場を公開していないため、特定の会社の歴史と隠れた変動率データが不足しているため、会社の予想変動率の推定は1組の上場企業の類似会社の歴史変動率に基づいている。予想変動率の計算は代表的な会社の歴史変動率に基づいており、これらの会社は製品開発段階と生命科学業界の重点を含む当社と類似した特徴を持っている。2022年の間、当社は株式市場データを持つ時間長の株価履歴と、毎回付与された期待期間内の上場企業に類似した履歴変動性を総合的に用いて変動性を推定するようになった。2023年の間、同社はその株価履歴のみを用いて変動性を推定し始めた。当社は“米国証券取引委員会従業員会計公報第107号”に規定されている簡略化方法を採用している株式支払従業員に付与されたオプションの期待期間を計算し、期待期間を推定するために合理的な基礎を提供するのに十分な履歴行使データがないためである。期待期間を有するオプション付与については、会社が十分な株価履歴を有している場合には、付与日までの予想期限の変動平均値を用いて期待株価変動率を算出する。非従業員に付与されたオプションについては、当社は、期待期限仮定の基礎としてオプション手配の契約条項を利用する。無リスク金利は、期限と株式オプション予想期限が一致する国庫ツールをベースとしている。期待配当収益率はゼロ同社は配当金を派遣したことがないため、今のところそうする計画もない。
当社は、当社の普通株による授出日の市場価格、あるいは授出日が当社の第一取引市場開設日でなければ、直前の取引日の前の取引日に応じて、制限された株式単位(“RSU”)ごとに授与日の公平な価値を定める。サービス帰属条件に適合した株式報酬については、自社は株式報酬支出が付与日の公正価値に等しいことを確認し、必要なサービス期間内に直線的に株式報酬を行う。
当社は没収記録を没収期間の累計調整としています。
財産と設備
財産と設備はコストから減価償却累計を引いて申告する.減価償却は、資産ごとの推定耐用年数内で直線法を用いて以下のように計算される
| | | | | |
| コンピュータ装置、オフィス機器、およびソフトウェア | 3年.年 |
| 実験室装置 | 5年.年 |
| 賃借権改善 | 耐用年数や賃貸年数が短い |
廃棄又は売却時には、処分資産のコスト及び関連減価償却は貸借対照表から除外され、関連損益は経営報告書及び全面赤字に反映される。2023年12月31日、2022年、2021年12月31日までの4年間、重大な資産売却は発生していない。
事件や業務環境の変化が資産の額面が完全に回収できない可能性があることを示すたびに、当社はその長期資産の減値を審査する。もし会社が減価審査を行って資産の回収可能性を評価すれば、会社は資産使用と最終処分で予想される未割引キャッシュフローの予測をその帳簿価値と比較する。資産の帳簿金額がその推定されていない未割引の将来のキャッシュフロー純額を超えた場合、当社は資産帳簿金額が資産公正価値を超えた金額の中で減価費用を確認する。“会社”ができた違います。2023年12月31日まで,2022年と2021年12月31日までの年度の減価費用は確認しない。
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
賃貸借証書
ASC 842と一致した賃貸借証書当社は、開始時に賃貸契約であるか否かを決定します。経営リースには、会社総合貸借対照表上の使用権賃貸資産(“ROU資産”)、賃貸債務の現在部分、長期賃貸債務が含まれています。融資リース制約を受けた資産は不動産や設備に計上され、関連賃貸義務は当社総合貸借対照表中の他の流動負債及びその他の長期負債に計上される。リース資産の減価テスト方式は運営に使用されている長期資産と同じである。経営的リースのリース費用は直線法でリース期間内に営業費用であることが確認されているが、融資リースの費用は実利法を用いて減価償却費用と利息費用であることが確認されている。当社は短期賃貸確認免除を選択しており、当社は元の期限が12ヶ月以下の賃貸の総合貸借対照表でリース負債とROU資産を確認しないことを許可しています。
ROU資産は会社がリース期間内に対象資産を使用する権利を代表し,リース義務は会社がリースによるリース金を支払う義務を代表する。レンタル負債およびそれに対応するROU資産は、最初に予想される残りのレンタル期間の賃貸支払い現在値に基づいて入金される。レンタル期間を決定する際には、会社は、選択権の行使を合理的に決定した場合にリース契約を延長または終了する選択権を含む。受け取った報酬などの項目については、ROU資産を何らかの調整する必要があるかもしれない。レンタル契約に隠されている金利は一般的に確定しにくい。そのため、同社はその逓増借款金利を利用して賃貸支払いを割引している。逓増借款金利は、当社が類似した経済環境下で、同じ通貨、同じ期限、担保方式で借金した固定金利を反映している
当社は賃貸契約を賃貸と非レンタル部分とに分けて計算します。
特許費用
当社は特許出願及び関連法律費用を発生済み費用とし、添付の総合経営報告書及び総合損失において当該等の費用を一般及び行政費用としている。
所得税
当社は米国会計基準第740条に基づいて所得税を計算した所得税会計(ASC 740)は、貸借対照法で繰延税項目を提出する。*当社は、財務諸表または納税申告書に登録されたイベントの予想される将来の税務結果を反映するために繰延税金項目資産および負債を確認する。当社は、財務報告と資産および負債税ベースとの差に基づいて繰延税金資産および負債を決定し、資産および負債の差額は、制定された税率および差額予想が逆転したときに発効する法律に基づいて計量される。既存の証拠の量に基づいて、繰延税金資産の一部または全部が現金化できない可能性が高い場合には、推定免税額を提供することができる。
所得税の頭寸がさらに持続する可能性がある場合にのみ、会社はこれらの頭寸の影響を確認する。確認された所得税の頭寸は最も実現可能な最大金額で計量されている。確認や計測の変化は変化が発生したと判断した期間に反映される.
総合収益(赤字)
総合収益(赤字)には純損失とその他の総合損失が含まれている。2023年12月31日、2023年12月、2022年12月31日までの年度、その他の全面収益(赤字)には、未実現収入と有価証券損失の変化が含まれている。2021年12月31日までの年度、総合損失は純損失に等しい。
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
信用リスクと表外リスク集中
会社を集中的な信用リスクに直面させる可能性のある金融商品には、主に現金等価物と有価証券が含まれる。その投資政策によると、同社は信用格付け、満期日、業界グループ、投資タイプ、発行者によってこのような証券に投資する金額を制限しており、米国政府が発行した証券は除く。当社はこのような金融商品がそれに対していかなる重大な信用リスク集中を構成しているとは考えていない。その会社は所有している違います。外国為替契約、オプション契約、または他の外国ヘッジ手配のような表外リスクを有する金融商品。
最近発表された会計公告
財務会計基準委員会または他の基準制定機関は時々新しい会計公告を発表し、会社は指定された発効日からこのような公告を採用する。以下で別途議論されない限り、当社は、最近発表された基準を採用することが、当社の総合財務諸表または開示に重大な影響を及ぼす可能性があるとは考えていない。
2023年11月、FASBは会計基準更新2023-07を発表した分部報告(テーマ280):改善可能報告分部開示主に重要部門の費用に関する情報を追加開示することによって、報告可能な部門の開示要求を改善することを目的としている。この基準は,2023年12月15日以降に開始される財政年度と,2024年12月15日以降に開始される財政年度内の移行期間に適用され,早期採用が許可されている。これらの修正は、財務諸表に記載されている以前のすべての期間にさかのぼって適用されるべきである。当社は現在、新しい基準に関連した開示要求を評価しています。
FASBは2023年12月に会計基準更新2023-09を発表した所得税開示を改善するこれは、各エンティティに、その有効税率調節に関する分類情報、及び司法管轄区域に納められた所得税に関する拡大情報を開示することを要求する。開示要求は予想に基づいて実施され、遡及適用を選択することができる。この基準は2024年12月15日以降の会計年度に施行され、早期採用が許可されている。当社は現在、新しい基準に関連した開示要求を評価しています。
3. 協力協定
グラクソ·スミスクライン
2022年8月6日、当社はグラクソ·スミスクライン知的財産権(第4号)有限会社(“GSK”)と協力、選択権及び許可協定(“GSK協定”)を締結し、これにより、当社はGSK独占選択権(“選択権”)を付与し、独占許可(“選択権”)を取得し、XMT−2056を含む製品(“許可製品”)を共同開発及び商業化し、同社がGSKにXMT−2056を交付する第1期単薬臨床試験における乳癌患者の投与量の増加及び濃縮による特定時間帯(“選択期”)内で行使することができる。グラクソ·スミスクラインの選択権行使は、1976年の“ハート-スコット-ロディノ反トラスト改良法”(Hart-Scott-Rodino反トラスト改良法)によって承認される必要があるかもしれない(“HSR承認”、GSKは任意の適用されたHSR承認後に選択権を行使する、すなわち“GSK選択権行使”)。グラクソ·スミスクライン選択権を行使する前に、同社は先頭に立ってそのXMT−2056計画に関連する製造、研究、早期臨床開発のコストを担当する。グラクソ·スミスクラインが選択権を行使した後、もしあれば、グラクソ·スミスクラインはXMT-2056の生産を選択する権利があり、グラクソ·スミスクラインは当社とXMT-2056を共同開発し、米国とEUの特許製品(S)を承認するために、グラクソ·スミスクラインは大部分の開発コストを負担する。グラクソ·スミスクラインは、米国とEU以外の承認を得るためだけのすべての開発コストを担当する
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
GSKプロトコルによると、GSK株購入権を行使した後、いくつかの例外的な場合および指定支払い義務の規定の下で、当社の合計開発コスト上限は固定金額であり、任意の超過金額はGSKが負担し、当社が任意の特許製品について指定シェア米国利益(または損失)の選択権を徴収(または負担)しない限りGSKが負担する(“利益シェア選択”)。超過した開発コストは、GSKプロトコルで規定された利息に計上され、将来の規制および販売マイルストーンまたは満期になる可能性のある当社の特許使用料が後で当社によって償還または相殺されます。会社がその利益シェア選択権を行使すれば、会社が開発コストに占めるシェアの上限は適用されなくなり、会社は当時返済されていなかった超過部分に計算すべき利息コストを支払わなければならない。また、会社がその利益分額選挙を行使すれば、米国で任意のライセンス製品を共同で普及させることを同時に選択することもできる。
グラクソ·スミスクライン協定によると、グラクソ·スミスクラインは同社に払い戻し不可能な前払い費用を支払った100.02022年8月は1.2億だった。グラクソ·スミスクラインがオプションを行使した後、もしあれば、グラクソ·スミスクラインは会社に#ドルのオプション行使金を支払う義務がある90.02000万ユーロ(“オプション支払い”)。同社は将来の開発、規制、ビジネスマイルストーンの支払いを受ける資格があり、最高約$に達する1.330億ドルで、会社がその利益シェア選択権を行使しなければ、ライセンス製品の世界売上高に応じて印税を20%程度に分類する。マイルストーンの支払い総額に含まれる金額は$30会社はGSKオプション行使前に発生する可能性のある早期臨床開発マイルストーンを満たして1000万ドルを稼ぐ資格がある。会社がその利益シェア選択権を行使すれば、会社は減少した開発、規制、商業マイルストーン支払い、米国以外の販売の特許権使用料を減少させる資格がある。グラクソ·スミスクラインは、会社が利益シェア選択権を行使するか否かにかかわらず、特定の第三者に何らかの記念碑的な支払いや特許権使用料を支払う責任を負い、現在同社はこれらの第三者とXMT-2056計画に関する合意を締結している。
グラクソ·スミスクラインがその選択権を行使しない場合、GSKプロトコルはオプション期限終了時に終了する。GSKオプションを行使する場合、GSKプロトコルは、事前に終了しない限り、すべての国/地域のすべてのライセンス製品の特許使用料期間およびすべての支払い義務が満了する日まで有効に継続される。
会計分析
同社はASC 606に基づいてGSKプロトコルを評価し、契約相手GSKが顧客であると結論した。同社は以下のことを決定した二つGSKプロトコルで規定されている物質履行義務:(I)GSKプロトコルで規定されている一括データ、情報、材料を提供するために必要な開発活動は、製造、研究、早期臨床開発活動(“開発活動”)と(Ii)共同開発と商業化許可製品のオプション(“許可オプション”)を含む
同社は,開発活動は異なる履行義務であり,基本活動は契約範囲内で区別できないため,総合開発計画への投入であり,データや情報を生成し,グラクソ·スミスクラインが選択権を行使するかどうかを決定するために価値を提供すると結論している。許可選択権は実質的な権利と考えられ,許可の価値が選択権支払いの金額を超えているため,独自の履行義務である
ASC 606によれば、当社は、GSKプロトコルの下での初期取引価格が#ドルに等しいと判断した100.0グラクソ·スミスクラインが支払った前金、払い戻し不可、および貸切不可の金を含む2000万ドル。グラクソストックオプションを行使する前に出現する可能性のある早期臨床開発マイルストーンは初期取引価格に含まれておらず,すべてのマイルストーンの金額が完全に制限されているためである。評価制限の一部として,開発段階やマイルストーンの実現に必要な開発に関する余剰リスク,実現マイルストーンが会社やGSKの制御範囲を超えているかどうかなど,多くの要因が考えられている。グラクソ·スミスクラインオプション支払いには、契約開始時の初期取引価格と、GSKオプション行使後の任意の将来の開発、規制、および商業マイルストーン支払い(特許権使用料を含む)は含まれていません
ASC 606項の分配目標に基づき、会社は$を割り当てた100.0義務履行ごとの相対独立販売価格に応じて,取引価格において開発活動と許可選択権に固定前払いを支払う.開発活動の独立販売価格はコストプラス利益の方法で計算され,見積りのための代替案前開発スケジュールである.ライセンスオプションの独立販売価格について,同社は収入に基づく方法を採用しており,オプション後の開発スケジュールとコスト,収入予測,割引率および技術と規制成功の可能性を含む重要な仮定を含んでいる
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
当社は、予備株式開発の予定期間内に開発活動実績義務に関する収入を確認し、使用する基本アクティビティの比例パフォーマンスモデルとして実行される.これは…E C会社は発生したコストと予想される総コストの割合に基づいて業績を測る
同社はライセンスオプションに関連した収入確認を延期した。許可選択権を行使し,GSKが独占許可を得た場合,会社はGSKプロトコルでの義務を履行する際に収入を確認する.オプションが行使されていなければ、会社はオプション満期時に全収入を確認する。
同社が記録した協力収入は、2023年12月31日と2022年12月31日までの年間で3.41000万ドルと300万ドルです2.0それぞれグラクソ·スミスクライン協定の下での努力と関連がある。2023年12月31日、2023年12月31日、2022年12月31日まで、会社はドルを記録しました94.61000万ドルと300万ドルです98.0GSKプロトコルでの履行義務未履行に関する繰延収入はそれぞれ1,000,000,000ドルであった.この繰延収入は、残りの業績期間中に確認され、履行義務の期待履行時間に応じて連結貸借対照表上で当期または非当期に分類される。
強生
2022年2月、当社はヤンソン生物科学技術会社と研究協力及び許可協定(“ジョンソン”及び2023年7月14日及び2023年9月25日に改訂された同等の協定、すなわち“ジョンソン協定”)を締結し、新型ADCの研究、開発及び商業化に専念した三つMersanaのADC専門知識とジョンソン独自抗体を用いたDolasyntenプラットフォームを利用することにより,腫瘍標的を実現した。ジョンソン契約に署名したとき、当社は返金できない前払い#ドルを受け取りました40.0ジョンソンから100万ドルを得ましたジョンソン合意に基づいて,当社はジョンソンを授与する二つ独占的で譲渡不可能なグローバルライセンスである研究許可と商業化許可(総称して“ジョンソン許可”と呼ぶ)。ジョンソン許可の一部として,研究許可は目標ごとに自社技術下でジョンソンの権利および共同開発による技術における当社の権益を1つずつ付与し,目標ごとに研究と化学,製造および制御計画下でのジョンソンの活動のみを行っている。ジョンソン·ライセンスの一部を構成する商業化ライセンスは、当社の技術と当社が共同開発した技術の下でターゲットごとに付与された権利金ライセンスに基づいて、目標のためのライセンスADCおよびライセンスADCを含む任意のライセンス製品を開発、製造、商業化、および他の方法で開発することである。ジョンソンは一番多く選ぶことができます三つ目標は、代替締め切りの前に各目標を1回置き換えることができる。第二世代権をジョンソンが行使した後,その第一次ビット権に費用を支払う必要はないが,後続のビット権を獲得する費用を一度に支払わなければならない.2023年12月31日までの年度内に、ジョンソンはある指標に対する第1回交代権を行使した
双方が合意した研究とCMC計画によると、当社は生物結合、生産開発、臨床前製造及びいくつかの関連研究と臨床前開発活動を行い、研究用新薬申請(“IND”)を提出してジョンソンの更なる開発、製造と商業化のために目標を実現することに同意した。ジョンソンは単独で許可を得たADCのINDイネーブル研究,IND提出,臨床開発,規制活動,商業化を担当する。会社とジョンソンは共同研究委員会と共同製造委員会で平等な代表を持ち、研究とCMC活動を監督する。同社は、目標研究計画の下での活動は2025年まで続くと予想している
ジョンソンは会社CMC活動の費用を約束の料率で支払うことを要求された。Allの開発成功と商業化を仮定する三つジョンソンの目標は、会社が最高$を得る資格があります5052000万ドルが開発と規制のマイルストーンと530600万ドルの販売マイルストーンと、ADC製品の総純売上高の中間桁から低い2桁の印税
事前に終了しない限り、ジョンソン·プロトコルは、ジョンソン·プロトコルの下の製品の最後の特許使用料の期限が満了したときに終了します。ジョンソン·プロトコルには、ジョンソン·プロトコルに違反するが救済しなければならないが、ジョンソンが便宜のために終了することと、当社が特許に挑戦する際に終了する慣用規定と、終了の効力に関する慣用規定が含まれている
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
ジョンソンは当社に単独の臨床供給プロトコルに基づいて臨床製造サービスを提供することを要求することができる。ジョンソンはまた,当社に約束したレートでバイオカップリングや製造プロセス技術の技術移転を要求することができ,費用はジョンソンが負担する。
会計分析
当社はASC 606に基づいてジョンソン契約を評価し、契約取引相手が顧客にジョンソンしていると判断した。同社は以下のことを決定した7人ジョンソン·プロトコルの下での物質履行義務:(I)独占ジョンソン許可証と研究活動三つターゲットを指定し,(2)CMCアクティビティごとに三つ標章と(3)第1標の代替権を指定する。
当社は,ジョンソンライセンスと研究活動は目標ごとの総合業績義務であり,ジョンソンライセンスはその独自性により研究活動と区別できないと結論している。CMCアクティビティは、これらのアクティビティが第三者プロバイダによって実行されることができるので、各ターゲットの異なる業績義務であると考えられる。第1の目標代替権は、オプション使用料がないため、独自の履行義務である実質的な権利であると考えられる。
ASC 606によると、当社はジョンソン·プロトコルの初期取引価格が#ドルに等しいことを決定しました40.0前払い、払い戻し不可、融資不可の支払いを含む1000万ドル。最初の取引価格にはすべてのマイルストーンの金額が完全に制限されているため、開発プロジェクトや規制マイルストーンは含まれていない。評価制限の一部として,当社は発展段階やマイルストーンの実現に必要な開発に関する余剰リスク,マイルストーンの実現が当社やジョンソンを超えているかどうかの制御など,多くの要因を考慮している。販売に基づくマイルストーンに関連するいかなる対価格(特許権使用料を含む)も関連販売発生時に確認され、このようなマイルストーンは主にジョンソンの許可付与に関連していると決定されているため、取引価格からも除外される。2023年12月31日現在、改訂されたジョンソン合意取引の総価格はドルです48.01000万ドルです。2023年の間、当社は推定取引価格を$に改訂しました6.0いくつかの発展マイルストーンの制約と、これらのマイルストーンの実現に必要な発展に関する余剰リスクの再評価により、600万ドルを得ることができる。
当社はCMC活動の対価格を可変対価と決定した。CMC活動の1つ三つ指定されたターゲットが起動されました。当社は、ASC 606項のCMCアクティビティに関連する実際の便宜的な請求書を適用する権利を選択しました。したがって、サービスが所与のターゲットに対する対応するCMC計画を超える場合、会社は、CMCアクティビティに関連する収入を確認する
ASC 606項の分配目標と一致し、当社は契約義務ごとの相対独立販売価格に基づいて、取引総価格をジョンソン許可と研究活動および優先代替権に割り当てる。ジョンソンライセンスと研究活動および最初の代替権の各独立販売価格は、収益法および代替権行使の可能性を用いて推定され、その中には、開発スケジュール、収入予測、割引率、および技術と規制が成功する確率が含まれている。ジョンソンはその第一世代権を行使するため、その義務履行に関する取引価格はジョンソン許可証と研究活動に再割り当てられている三つ指定された目標。
当社は比例業績モデルを採用し、研究サービスの推定期間内にジョンソン許可証と研究サービス業績義務に関連する収入を確認した。同社は発生したコストと予想される総コストの割合に基づいて業績を測定している
同社が記録した協力収入は、2023年12月31日と2022年12月31日までの年間で16.61000万ドルと300万ドルです24.2それぞれ“ジョンソン·プロトコル”の項での履行義務に関係している.2023年12月31日、2023年12月31日、2022年12月31日まで、会社はドルを記録しました10.41000万ドルと300万ドルです15.8ジョンソンプロトコルに関する繰延収入はそれぞれ1,000,000,000ドルであり,残りの業績期間で確認し,それぞれの業績義務を期待する時間に応じて総合貸借対照表上で流動または非流動に分類される
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
メルクKGaA
免疫合成プラットフォームプロトコル
2022年12月、当社はドイツDarmstadt Merck KGaAの完全子会社であるAres Trading S.A.(“Merck KGaA”及び/又はその付属会社、“Merck KGaA”及び同等のプロトコル、即ち“2022 Merck KGaAプロトコル”)と研究協力及び許可プロトコルを締結し、新型ADCの研究、開発及び商業化に専念し、最高で達することができる二つMersanaのADC専門知識とMerck KGaAとの独自抗体を利用した免疫合成プラットフォームにより,特定の標的抗原を実現した。2022年のメルクKGaA協定に関連して、同社は払い戻しできない前払い#ドルを受け取った30.01000万ドルです。“2022年メルクKGaA協定”によると、会社はメルクKGaAを授与します二つ独占的で譲渡不可能なグローバルライセンスである研究ライセンスと商業化ライセンス(総称して“Merck KGaAライセンス”と呼ばれる)。メルクKGaAライセンスは、マークKGaAライセンスの一部であり、この研究ライセンスは、研究およびCMC計画の下で各ターゲットについてメルクKGaAの活動を展開するためにのみ使用される会社の技術下の権利および協力によって共同開発された技術における会社の権益をターゲットごとにマークKGaAに提供する。Merck KGaAライセンスの一部を構成する商業化ライセンスは、共同開発技術によって共同開発された技術によって会社によって付与された個々のターゲットの印税許可に基づいて、ターゲットのための許可ADCおよび許可ADCを含む任意のライセンス製品を開発、製造、商業化、および他の方法で利用する。
双方が合意した研究とCMC計画によると、同社は、IND(または海外同等製品)を提出することによって目標を推進し、メルクKGaAによるさらなる開発、製造および商業化のために、生物結合、生産開発、臨床前製造、およびいくつかの関連研究および臨床前開発活動を行うことに同意した。メルクKGaAは独自にINDイネーブル研究、IND提出、臨床開発、監督活動、許可を得たADCの商業化を担当する。同社とメルクKGaAは、共同研究委員会と共同製造委員会で平等な代表を持ち、研究とCMC活動を監督する。同社は、目標の研究計画に基づいた活動が2026年まで続くと予想している
会社のCMC活動はメルクKGaAによって合意されたレートで補償される。開発と商業化に成功したと仮定して二つマークKGaAの目標で、同社は最高$を得る資格があります2002000万ドルが開発と規制のマイルストーンと600販売マイルストーンおよびADC製品の総純売上高の段階的な桁数から低い二桁版税。
事前に終了しない限り、2022年メルクKGaAプロトコルは、2022年メルクKGaAプロトコルの下で製品の最後の特許使用料の期限が満了する。“2022年メルクKGaA協定”には、“2022年メルクKGaA協定”に違反した場合を含むいずれか一方が終了する習慣条項が含まれているが、Merck KGaAが便宜上、会社がライセンス特許に挑戦した際に終了し、影響を終了する習慣条項を修復しなければならない
メルクKGaAは当社に単独の臨床供給プロトコルに基づいて臨床製造サービスを提供することを要求する可能性がある。メルクKGaAはまた,会社に合意したレートで生物結合技術の技術移転を要求することができ,費用はメルクKGaAが負担する。
会計分析
当社はASC 606に基づいて2022年のメルクKGaAプロトコルを評価し、契約相手側Merck KGaAが顧客であると結論した。同社は以下のことを決定した四つ“2022年メルクKGaA協定”に規定されている材料性能義務:(I)メルクKGaA独占ライセンスと研究活動二つ目標の指定と(2)各コミュニティ管理委員会の活動二つ指定された目標。
同社の結論は、メルクKGaAライセンスと研究活動は各目標の総合的な業績義務であり、メルクKGaAライセンスの独自の性質を考慮して、それらは研究活動と区別できないからである。CMCアクティビティは、これらのアクティビティが第三者プロバイダによって実行されることができるので、各ターゲットの異なる業績義務であると考えられる
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
ASC 606によれば、会社は2022年のメルクKGaAプロトコルでの初期取引価格が#ドルに等しいと決定した32.02000万ドルが含まれています30.0100万ドルの前金、払い戻し不可、および計上不可能な費用、およびいくつかの最近の発見マイルストーン。これは1ドルです30.0会社は2022年12月31日現在、前払い費用100万ドルを受け取っておらず、2022年12月31日現在の年度は売掛金と相応の繰延収入負債として記録されている。その会社はその後、2023年2月にこのお金を受け取った。取引価格に含まれていない開発と規制マイルストーンは制限されている。規制の評価の一部として,同社は開発段階やマイルストーンの実現に必要な開発に関する余剰リスク,実現マイルストーンが会社やメルクKGaAの制御範囲を超えているかどうかなど,多くの要因を考慮している。販売ベースのマイルストーンに関連する任意の対価格(特許使用料を含む)は、このようなマイルストーンが主にメルクKGaAに付与されるライセンスに関連すると決定されたため、取引価格から除外される関連販売が発生したときに確認されるであろう。
当社はCMC活動の対価格を可変対価と決定した。当社は、ASC 606項のCMCアクティビティに関連する実際の便宜的な請求書を適用する権利を選択しました。したがって、サービスが所与のターゲットに対する対応するCMC計画を超える場合、会社は、CMCアクティビティに関連する収入を確認する。ターゲットのCMC活動はまだ開始されていない.
ASC 606項の分配目標に基づき、会社は$を割り当てた32.0契約義務ごとの相対独立販売価格によると、メルクKGaAライセンスと研究活動の推定取引価格は100万ドルである。メルクKGaAライセンスおよび研究活動の各独立販売価格は、比較可能な取引に基づいて確立された調整された市場評価方法を用いて推定される。
同社は比例業績モデルを使用して、研究サービスの推定期間中にメルクKGaA許可証と研究サービス業績義務に関連する収入を確認した。同社は発生したコストと予想される総コストの割合に基づいて業績を測定している
2023年12月31日までの年間で、同社が記録した協力収入は10.72000万ドルは2022年のメルクKGaA協定の下でのその努力と関連がある。“会社”ができた注釈2022年12月31日までの1年間に、2022年のメルクKGaA協定に関する協力収入が記録的に記録された。2023年12月31日、2023年12月31日、2022年12月31日まで、会社はドルを記録しました20.21000万ドルと300万ドルです30.0それぞれ2022年のメルクKGaAプロトコルで履行されていない履行義務に関する繰延収入である。この繰延収入は、残りの業績期間中に確認され、それぞれの業績義務を履行する予想時間に基づいて連結貸借対照表上で当期または非当期に分類される。
Dolaflexinプラットフォームプロトコル
当社は2014年6月、ドイツ·ダムシュタットのMerck KGaAと協力·商業許可協定(時々改訂された“2014 Merck KGaA協定”)を締結した。2014年のメルクKGaAプロトコルは、メルクKGaAに6つの独占目標のためのADCを開発する権利を提供した。2018年5月、当社はドイツのダムシュタットのメルクKGaAと“供給協定”(時々改訂された“2018メルクKGaA供給協定”)を締結しました。2018年Merck KGaA供給協定の条項に基づいて、会社はMerck KGaAに臨床前非良好生産実践ADC薬物物質と臨床良好生産実践薬物物質を提供し、2014年Merck KGaAプロトコルが指定した抗体の一つと関連する臨床試験に用いることができる。2023年第4四半期、当社はMerck KGaAと共同で2014年Merck KGaAプロトコルと2018年Merck KGaA供給プロトコルを終了することに同意した
会計分析
2014年メルクKGaA協定と2018年メルクKGaA供給協定は2023年第4四半期に終了しました。これ以上の履行義務がないため、会社は#ドルを確認した3.72014年のメルクKGaA協定の終了と2018年のメルクKGaA供給協定の終了に関する収入は、2023年12月31日までの年間で1.8億ユーロである。
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
契約が終了する前に、当社は2014年のメルクKGaA協定に基づいて、以下の履行義務を決定しました:(I)独占許可と研究サービス6人指定された目標は、(Ii)将来の技術改善の権利、および(Iii)プロジェクトチーム責任者の参加および共同研究委員会サービスを提供する。
当社の結論は、マークKGaAが関連研究サービスなしにライセンスのメリットを得ることができないため、指定されたターゲットの各ライセンスは、指定されたターゲットに関連する研究サービスと区別されない。指定されたターゲットの各ライセンスおよび関連サービス履行義務は、各研究計画が他の指定されたターゲットに対する他のすべての研究計画とは独立して行われるので、指定された目標に関連する研究サービスと区別される。
2023年12月31日までの年間で,2014年メルクKGaA協定と2018年メルクKGaA供給協定に関する確認連携収入は$である3.71000万ドルです。2022年12月31日と2021年12月31日までの年間で確認された2014年メルクKGaA協定と2018年メルクKGaA供給協定に関する連携収入は重要ではない。
2023年12月31日現在、同社は注釈2014年のメルクKGaA協定と2018年のメルクKGaA供給協定に関連する収入を延期した。2022年12月31日までに会社はドルを記録しました3.92014年メルクKGaA協定と2018年メルクKGaA供給協定に関する繰延収入は合計2.5億ドル。
契約資産と負債集計表
以下の表に会社の契約負債残高の変化を示す
| | | | | | | | | | | | | | | | | | | | | | | |
| 残高は 初めから 周期の | | 足し算 | | 控除額 | | 残高は 期末 |
| 2023年12月31日までの年度 | | | | | | | |
| | | | | | | |
契約責任: | | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| 繰延収入総額 | $ | 147,653 | | | $ | 5,517 | | | $ | 27,856 | | | $ | 125,314 | |
| | | | | | | |
2022年12月31日までの年度 | | | | | | | |
契約責任: | | | | | | | |
| | | | | | | |
| 繰延収入総額 | $ | 3,944 | | | $ | 170,000 | | | $ | 26,291 | | | $ | 147,653 | |
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
“会社”ができた注釈2023年12月31日までと2022年12月31日までにその連携協定に関連する任意の契約資産を記録する。
2023年12月31日、2023年12月31日、2022年12月31日までの年間で、それぞれの期間の契約負債残高の変化により、会社は以下の収入を確認した
| | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 |
| 当期確認の収入は: | | | |
| | | |
| | | |
| | | |
| | | |
| 期初めに契約負債の額を計上する | $ | 27,870 | | | $ | 43 | |
| | | |
| | | |
| | | |
| | | |
| | | |
その他の収入
二零一二年の研究、開発および許可協定(“アザナ生物科学協定”)によると、当社は協力者のアザナ生物科学有限会社(“アサナ生物科学”)に限られたサービスを提供している。2022年12月31日までの年間で、会社が確認した収入は0.3100万ドルはこれらのサービスに関連しています注釈2023年12月31日と2021年12月31日までの年度内のこれらのサービスに関する収入を確認した。2023年12月31日までの年間で、会社が確認した収入は2.51000万ドルはアザナ生物科学協定の次の発展マイルストーンの実現と関連がある。
4. 公正価値計量
以下の表は、当社の公正価値によって日常的に計量された資産に関する資料を提供し、この等価値を特定するための公正価値評価技術の公正価値階層内のレベルを示す
| | | | | | | | | | | | | | | | | | | | | | | |
| 2023年12月31日 |
| (単位:千) | 合計する | | 見積もり:
非活発状態で
市場
(一級) | | 意味が重大である
他にも
観察できるのは
入力量
(二級) | | 意味が重大である
見えない
入力量
(第3級) |
| 現金等価物 | | | | | | | |
| 貨幣市場基金 | $ | 90,649 | | | $ | 90,649 | | | $ | — | | | $ | — | |
| アメリカ国債 | 24,889 | | | 24,889 | | | — | | | — | |
| $ | 115,538 | | | $ | 115,538 | | | $ | — | | | $ | — | |
| 有価証券 | | | | | | | |
| アメリカ国債 | $ | 29,548 | | | $ | 29,548 | | | $ | — | | | $ | — | |
| アメリカ政府機関証券 | 4,975 | | | — | | | 4,975 | | | — | |
| $ | 34,523 | | | $ | 29,548 | | | $ | 4,975 | | | $ | — | |
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
| | | | | | | | | | | | | | | | | | | | | | | |
| 2022年12月31日 |
| (単位:千) | 合計する | | 見積もり:
非活発状態で
市場
(一級) | | 意味が重大である
他にも
観察できるのは
入力量
(二級) | | 意味が重大である
見えない
入力量
(第3級) |
| 現金等価物 | | | | | | | |
| 貨幣市場基金 | $ | 50,471 | | | $ | 50,471 | | | $ | — | | | $ | — | |
| アメリカ政府機関証券 | 9,993 | | | — | | | 9,993 | | | — | |
| $ | 60,464 | | | $ | 50,471 | | | $ | 9,993 | | | $ | — | |
| 有価証券 | | | | | | | |
| アメリカ国債 | $ | 107,810 | | | $ | 107,810 | | | $ | — | | | $ | — | |
| アメリカ政府機関証券 | 44,017 | | | — | | | 44,017 | | | — | |
| $ | 151,827 | | | $ | 107,810 | | | $ | 44,017 | | | $ | — | |
2023年12月31日および2022年12月31日までの年間推定技術や公正価値計量レベルの間に変動はなかった。
評価レベルで1レベルに分類される投資には、一般に米国国債と通貨市場基金が含まれており、公正価値は同じ証券の日常的な活発な市場によって決定されやすいからである。評価レベルで第2レベルに分類される投資には、一般に、公正価値が類似した証券および他の観察可能な投入の活発な毎日市場に基づいて決定されやすいため、米国政府機関証券が含まれる。当社は第三者価格源から得られた推定値を考慮して投資の公正価値を推定しています
総合貸借対照表に反映される前払い支出及びその他の流動資産、売掛金、売掛金及び売掛金の帳簿価額は短期的な性質に属するため、その公正価値に近い。
2023年12月31日、2023年12月31日及び2022年12月31日まで、当社は新信用手配(付記8参照)の項で借金を返済していない帳簿価値が公正価値(第2級公正価値計量)に近く、当社が現在獲得可能な金利を反映している。新しい信用計画は付記8でより詳細に議論されています債務.
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
5. 現金、現金等価物、短期有価証券
現金と現金等価物
次の表は、会社が2023年12月31日まで、2023年12月31日と2022年12月31日までの現金、現金等価物と制限現金をまとめたものです。
| | | | | | | | | | | | | | | | | | | | | | | |
| (単位:千) | 現在までの年度
2023年12月31日 | | 現在までの年度 2022年12月31日 |
| 初めから
周期の | | 端部
周期の | | 初めから
周期の | | 端部
周期の |
| 現金と現金等価物 | $ | 128,885 | | | $ | 174,561 | | | $ | 177,947 | | | $ | 128,885 | |
| 他の資産に含まれる制限された現金、非流動 | 478 | | | 478 | | | 478 | | | 478 | |
| 現金フロー表の現金総額、現金等価物、および制限現金 | $ | 129,363 | | | $ | 175,039 | | | $ | 178,425 | | | $ | 129,363 | |
有価証券
下記表は、会社が2023年12月31日、2023年12月31日、2022年12月31日に保有する有価証券をまとめたものである。
| | | | | | | | | | | | | | | | | | | | | | | |
| 2023年12月31日 |
| (単位:千) | 償却する
コスト | | 毛収入
実現していない
収益.収益 | | 毛収入
実現していない
損 | | 公平である
価値がある |
| | | | | | | |
| | | | | | | |
| 有価証券 | | | | | | | |
| | | | | | | |
| | | | | | | |
| アメリカ国債 | $ | 29,535 | | | $ | 13 | | | $ | — | | | $ | 29,548 | |
| アメリカ政府機関証券 | 4,977 | | | — | | | (2) | | | 4,975 | |
| $ | 34,512 | | | $ | 13 | | | $ | (2) | | | $ | 34,523 | |
| | | | | | | | | | | | | | | | | | | | | | | |
| 2022年12月31日 |
| (単位:千) | 償却する
コスト | | 毛収入
実現していない
収益.収益 | | 毛収入
実現していない
損 | | 公平である
価値がある |
| 有価証券 | | | | | | | |
| アメリカ国債 | $ | 107,964 | | | $ | 7 | | | $ | (161) | | | $ | 107,810 | |
| アメリカ政府機関証券 | 44,016 | | | 24 | | | (23) | | | 44,017 | |
| 合計する | $ | 151,980 | | | $ | 31 | | | $ | (184) | | | $ | 151,827 | |
その会社のすべての有価証券は1年以下で満期にならなければならない。当社は、2023年、2023年および2022年12月31日までに、有価証券の売却による損益は生じていませんので、当社は累計全面損失から何の金額も再分類していません。
2023年12月31日現在、会社の債務証券組合は1つは未実現損失状態で公正価値合計#ドルの証券5.01000万ドルです。いくつありますか違います。2023年12月31日現在、損失が12カ月を超える証券は実現していない。会社の有価証券の未実現損失は市場金利の上昇によるものです。当社は取り戻すまでこのような証券を保有する意欲と能力があります。“会社”ができた注釈2023年12月31日と2022年12月31日までの年度内に、その有価証券の任意の信用関連減価費用を記録する。
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
6. 財産と設備
財産および設備は、2023年12月31日、2023年12月31日、2022年12月31日まで
| | | | | | | | | | | |
| (単位:千) | 十二月三十一日
2023 | | 十二月三十一日
2022 |
| 実験室装置 | $ | 8,246 | | | $ | 7,960 | |
| 賃借権改善 | 2,206 | | | 1,943 | |
| コンピュータ、ソフトウェア、オフィス機器 | 2,449 | | | 1,824 | |
| | | |
| コストで計算した財産と設備総額 | 12,901 | | | 11,727 | |
| 減算:減価償却累計 | (9,070) | | | (7,742) | |
| $ | 3,831 | | | $ | 3,985 | |
2023年12月31日現在、2022年と2021年12月31日までの4年度の減価償却費用は1.51000万、$0.91000万ドルと300万ドルです0.92億5千万ドルと2億5千万ドルです
7. 費用を計算する
2023年12月31日、2023年、2022年までに、支出には以下が含まれている
| | | | | | | | | | | |
| (単位:千) | 十二月三十一日
2023 | | 十二月三十一日
2022 |
| 給料と関連費用を計算しなければならない | $ | 8,807 | | | $ | 11,558 | |
| 臨床費用を計算すべきである | 5,063 | | | 14,822 | |
| 研究と非臨床費用を計算すべきである | 3,090 | | | 2,767 | |
| 製造費用を計算する | 2,566 | | | 11,536 | |
| 再編成費用を計上すべきである | 1,047 | | | — | |
| 専門費用を計算する | 936 | | | 1,865 | |
| その他の措置を講じる | 389 | | | 636 | |
| $ | 21,898 | | | $ | 43,184 | |
8. 債務
2019年5月8日、当社はシリコンバレー銀行(“前SVB”)と融資及び担保協定(“優先信用手配”)を締結し、これにより、当社は$を借入する5.01000万ドルです。優先信用手配は変動年利率に基づいて利子を引き出し、金利は(I)の中の大きい者に等しい4.0%および(Ii)1.50最割引金利より1%低いです。以前の信用計画は2020年8月31日まで利息しかありません。
2020年8月28日、当社は先の信用手配を第2回改訂(“第2回改訂”)した。第二修正案によると、同社は$を抽出した5.2第二改正案を作成する際には,当社は2百万ドルを支払い,得られた金は自社の先行信用メカニズム下での既存残高の償還に用いられ,前SVBへの義務を果たす。“第二修正案”によって修正された優先信用手配は、利息は変動年利率が(I)の中の大きい者に等しいことを計算しなければならない4.25%および(Ii)1.00最割引金利より%高いです
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
二零二一年十月二十九日、当社は、前SVBおよびOxford Finance LLC(“オックスフォード”と、前SVBおよびその他の時々の融資者(“融資者”)と共に融資および保証協定(“新しい信用手配”)を締結した。2023年3月には,シリコンバレーブリッジ銀行(“シリコンバレーブリッジ銀行”)が前SVBに代わって貸手となり,前SVBの利息相続人となり,その後シリコンバレー銀行(First−Citizens Bank&Trust Company(“SVB”)傘下のシリコンバレー銀行がSVBBのすべての預金と融資を負担し,SVBBの代わりに貸手となった。2022年2月17日、2022年10月17日、2022年12月27日、2023年3月23日に改訂された新しい信用手配は、会社が所有または後に獲得したほとんどの個人財産を担保とし、知的財産権(ただし、知的財産権から支払いおよび収益を得る権利を含む)および知的財産権の負の質権を含まない。会社は$を引いた25.0新しい信用計画を実行する際には1000万ドルです5.5得られた金のうち1百万ドルは先行信用メカニズム下の既存残高の返済に使用され、SVBに対する債務を履行する。新しい信用手配を締結した後、当社はSVBが以前の信用手配に基づいて更に信用を発行するすべての約束を終了し、そして当社が以前の信用手配に基づいてSVBに付与したすべての保証と担保権益を確立した。2023年12月31日現在、改訂された新しい信用手配によると、当社には追加借款金額はありません。
新しい信用ツールの利息は変動年利率であり、(I)の大きい者に等しい8.50%および(Ii)5.25最割引金利より%高いです。利息は月ごとに支払い、毎月の初日に延滞する。同社は2024年11月1日までに利息のみを支払い、2026年10月1日(“満期日”)までに元金と適用利息を月等額で支払う義務がある。何らかの発展の節目に達した場合、利息のみの期限は2025年11月1日まで延長される。
会社は貸手に支払うことを要求されています4.25当時当社に支給されていた定期ローン元金の%です。この最終支払いは各定期ローンの有効期限内に有効利息法で累積されています。定期融資は会社のほとんどの資産を担保としているが、負の質権やいくつかの他の慣行によって排除された知的財産権は除外されている。
会社の選択により、会社はすべて可能ですが、定期ローンの未返済元金残高を部分的に前払いすることができますが、前払い費用は1.0事前返済が当該定期融資の融資日の2周年後に発生したが、満期日までに、当社に発行されている定期融資の割合。新しい信用手配には当社に適用される慣用的な肯定と制限契約が含まれています。その中には、会社がその会社の存在と政府の承認、特定の財務報告の提出、保険カバー範囲の維持、預金口座に関するいくつかの要求を満たすことを要求する条約が含まれている。制限的契約は当社が担保を譲渡し、追加債務を招く、合併或いは買収を行い、配当金を支払う或いは他の分配を行う、投資を行う、留置権の設立、資産の売却及び同意制御権の変更などの能力に関する要求を含み、すべての状況はある常習例外状況の規定によって制限される。
特定の違約事件が発生した場合、会社の新しい信用手配下での支払い義務は加速する可能性があり、これらの違約事件は、会社の業務、運営または財務、またはその他の条件が重大な不利な変化を含むが、これらに限定されない。違約事件が発生した時に返済されなかった金額は貸金人の要求に応じて支払わなければならず、以下の追加金利で利息を計算しなければなりません5.0未払い金の年利率。当社は2023年12月31日まで、新しいクレジット手配のすべてのチェーノを遵守しています。このため、2023年12月31日現在、貸借対照表に記載されている融資残高の分類は、決定された将来の支払義務の時期に基づいて決定される。
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
以下は、2023年12月31日までの新クレジット手配の債務の概要です
| | | | | |
| (単位:千) | 十二月三十一日
2023 |
| 債務総額 | $ | 25,000 | |
| 差し引く:長期債務の現在部分 | (2,083) | |
| 総債務,当期分を差し引く | 22,917 | |
| 債務融資コストは,増額後の純額を差し引く | (237) | |
| 最終支払いに関する累積 | 468 | |
| 長期債務、純額 | $ | 23,148 | |
2023年12月31日現在、将来の満期予想元金は以下の通りです
| | | | | |
| (単位:千) | |
| 2024 | $ | 2,083 | |
| 2025 | 12,500 | |
| 2026 | 10,417 | |
| |
| 債務総額 | $ | 25,000 | |
2023年12月31日まで及び2022年12月31日までの年度新信用手配に関する利息支出は3.91000万ドルと300万ドルです3.22億5千万ドルと2億5千万ドルです当社は2021年12月31日までの年度中に新クレジット手配に関する利息支出は何も確認していません。2021年12月31日までの年度先のクレジット手配に関する利息支出は$0.81000万ドルです。“会社”ができた違います。2023年12月31日と2022年12月31日までの年間では、先の信用手配に関連したいかなる利息支出も確認されていない。
9. 株主権益
優先株
2023年12月31日までに会社は25,000,000優先株の株式を授権する違います。優先株は既に発行された.
場内(“ATM”)株式発行計画
2020年5月、同社はATM株式発行計画(“2020年ATM”)を設立し、同計画によると、同社は最高$まで発行·販売できるようになった100.0その普通株1.8億株を当時の市場価格で時々売却した。当社は2021年12月31日までに販売しております3,961,074普通株、純収益は#ドルです43.11000万ドルです。当社は2022年12月31日までに販売します11,740,2102020年のATMでの普通株、純収益は$54.81000万ドルです。2022年12月31日現在、2020年のATMはすべて使用が完了している。
2022年2月、同社は新たなATM株式発行計画(“2022年2月ATM”)を設立し、同計画によると、同社は最高$まで発行·販売できるようになった100.0その普通株1.8億株を当時の市場価格で時々売却した。当社は2022年12月31日までに販売します18,757,6652022年2月のATMでの普通株、純収益は$96.41000万ドルです。当社は2023年12月31日までに販売します256,3862022年2月のATMでの普通株、純収益は$1.61000万ドルです。2023年12月31日現在、2022年2月にATMはすべて使用が完了している。
2022年11月、会社は追加のATM株式発売計画(“2022年11月ATM”)を設立し、この計画により、会社は最高$まで発行·販売することができる150.0その普通株1.8億株を当時の市場価格で時々売却した。当社は2023年12月31日までに販売します14,208,1452022年11月のATMでの普通株、純収益は$92.21000万ドルです。2023年12月31日までの約55.92022年11月のATMによると、まだ100万台が販売されておらず、販売できる。
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
株式承認証
2013年のA-1シリーズ優先株発行では、会社はある投資家に普通株を購入する引受権証を付与した。その条項によると、このような期限が切れていないすべての引受権証は2023年9月27日に満期になった。株式承認証には1ドルがあります0.051株当たりの行使価格および契約期間は10何年もです。これらの株式承認証の公正価値は発行時に権益構成部分として入金される。2023年12月31日までに違います。発行された普通株の引受権証を購入する。当社は2022年12月31日までに年度中に発送します16,654株式証純行使時の普通株。
普通株
2022年6月9日の会社2022年株主総会において、会社株主は、普通株式の法定株式数を増加させるために、会社の第5回改訂後の会社登録証明書の改正案を承認した0.00011株当たりの額面は175,000,000至れり尽くせり350,000,000それは.この増加は2022年6月9日にデラウェア州国務長官に修正案証明書を提出して発効した
普通株保有者には権利がある1つは保有する株式1株に投票する。普通株主は、会社の取締役会(“取締役会”)によって発表されない限り、配当金を得る権利がない。
2023年12月31日と2022年12月31日までに14,736,953そして11,944,664それぞれ,発行済み株式オプションを行使するために予約された普通株,制限株式単位(“RSU”)と引受権証である.
| | | | | | | | | | | |
| 十二月三十一日
2023 | | 十二月三十一日
2022 |
| 株式オプション | 10,902,845 | | | 10,051,283 | |
| 制限株式単位 | 3,834,108 | | | 1,870,791 | |
| 株式承認証 | — | | | 22,590 | |
| 14,736,953 | | | 11,944,664 | |
10. 株に基づく報酬
株式激励計画
初公開前に、会社は会社の2007年株式激励計画(“2007年計画”)に基づいて株式オプションを付与した。2007年は2017年6月に満期になる予定だ。2007年計画に基づいて付与されたオプションの任意のキャンセルまたは没収は、以下に述べるように、会社の2017年株式インセンティブ計画(“2017計画”)下の利用可能なオプションを増加させる。
2017年6月、会社株主は2017年計画を承認しました。2017年計画によると、普通株オプション、制限株式単位(RSU)または他の株式ベースの奨励の形で、会社の従業員、高級管理者、取締役、コンサルタント、コンサルタントに授与されます。2017年計画により発行可能な普通株式数は毎年1月1日に累計増加する(A)4(B)取締役会が指定した当該等の他の金額。奨励条項は取締役会によって決定されるが、2017年計画の規定を守らなければならない。2017年6月に満了した2007計画に基づいて付与されたオプションのいかなるキャンセルや没収も、2017年計画で付与可能な株式数を増加させます。2023年1月1日、株式数 2017年計画により普通株式を発行できる割合が増加した4,205,794株式です。当社は2023年12月31日までに合算して授与します7,211,153RSUは、普通株のオプションを購入し、2017年の計画に基づいて従業員と非従業員取締役に普通株を提供する。2023年12月31日までに2,043,3282017年計画によると、将来発行可能な株。
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
2017年計画によると、奨励的株式オプションであっても非適格株式オプションであっても、1株当たりの権利価格は付与日普通株の公正時価を下回らず、従業員に付与されるオプションの帰属期限は一般に4年それは.当社が時々実施している非従業員役員の報酬政策によると、非従業員取締役に付与された現金採用費を稼ぐための購入権は授出時にすべて帰属し、初めて取締役会に入った時に非従業員取締役に付与された株式購入権は3年そして,株主総会の終了日に非従業員取締役のオプションを付与する1年それは.2017年計画によると付与されたオプションは10授与の日から数年。2007年計画項の下の株式購入権は取締役会(或いはその許可委員会)によって決定された行使価格によって授与され、この価格は授出日関連普通株の公平な市価を下回らず、そして取締役会(或いはその許可委員会)によって決定された帰属条項によって規定されなければならない。会社合併、合併、解散または清算の場合、取締役会は、付与オプションの条項の付与を加速するか、または他の方法で調整することができる。
誘因賞
取締役会又はその認可委員会の承認を経て、当社は時々ナスダック上場規則第5635(C)(4)条に基づいて、その従業員に普通株及び/又はRSU株式を雇用誘因として購入する選択権を付与する。2022年2月までには、普通株式を購入するオプションのみがインセンティブ報酬として付与され、既存の持分インセンティブ計画外で付与される。このようなオプションの条項は2017年計画とほぼ同じだ
2022年2月、取締役会は不正定期権、株式付加価値権、制限株、RSU、その他の株式ベースの奨励を付与することを規定する会社2022年インセンティブ株式インセンティブ計画(“インセンティブ計画”)を採択し、合計に関連した2,000,000会社普通株式(インセンティブ計画の規定により調整)。当社は2023年12月31日までに合算して授与します877,575インセンティブ計画により新たに雇用された従業員に普通株のRSUとオプションを購入する。2023年12月31日までに1,170,752インセンティブ計画によると、将来発行される株を提供することができる
2023年12月31日まで、オプションが購入できます457,500インセンティブ計画が制定される前にインセンティブ奨励として付与された発行された普通株。
株式オプション活動
株式オプション活動の概要は以下のとおりである | | | | | | | | | | | | | | | | | | | | | | | |
| 番号をつける の株 | | 重み付けの- 平均値 行権価格 | | 加重平均 残り 契約条項 | | 骨材 内在的価値 |
| 2023年1月1日現在返済されていない | 10,051,283 | | | $ | 9.84 | | | 7.2 | | $ | 8,197 | |
| 授与する | 4,017,763 | | | $ | 4.76 | | | | | |
| 鍛えられた | (102,596) | | | $ | 4.25 | | | | | |
| 取消·没収 | (3,063,605) | | | $ | 12.00 | | | | | |
| 2023年12月31日現在の未返済債務 | 10,902,845 | | | $ | 7.42 | | | 6.8 | | $ | 1,786 | |
| | | | | | | |
| 2023年12月31日に行使できます | 6,499,540 | | | $ | 8.23 | | | 5.5 | | $ | 711 | |
2023年,2023年,2022年,2021年12月31日までの年次授期権の加重平均授受日の公正価値は#ドルである3.82, $3.97そして$11.71それぞれ1株です。2023年,2023年,2022年と2021年12月31日までの年度内に行使されたオプションの内的価値総額は$である0.3百万、$1.5百万ドルと$4.3それぞれ100万ドルです総内的価値とは,オプション所有者が期内で株式オプションを行使する際に受け取った行権価格と販売入札との差額である.
株式オプションを行使して受け取った現金は#ドルです0.4百万、$1.3百万ドルとドル1.82023年12月31日まで、2022年、2021年12月31日までの3年間はそれぞれ100万ドル。
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
制限株式単位およびその他の株式奨励
会社は定期的にある高級管理者や他の従業員にサービス条件付きRSUを配布しています1年そして4年授与の日から。その非従業員役員報酬政策によると、時々発効すれば、会社は毎年非従業員取締役にサービス条件付きRSUを発行し、通常授与される1年与えられた日から、当社もいくつかの非従業員取締役に普通株を発行し、稼いだ現金予備招聘費の代わりに、当該株式は付与時に全数帰属する。
以下にRSU活動の概要を示す | | | | | | | | | | | | | | | | | | | | | | | |
| 番号をつける
の株 | | 加重平均
残り
契約条項 | | 骨材
内在的価値 | | 加重平均
授与日
公正価値 |
| 2023年1月1日に帰属していない | 1,870,791 | | | 1.5 | | $ | 10,963 | | | $ | 8.55 | |
| 授与する | 4,025,544 | | | — | | | | | $ | 4.42 | |
| 既得 | (572,825) | | | — | | | | | $ | 8.83 | |
| 没収される | (1,489,402) | | | — | | | | | $ | 6.25 | |
| 2023年12月31日に帰属していません | 3,834,108 | | | 1.4 | | $ | 8,895 | | | $ | 5.01 | |
2023年12月31日,2022年,2021年12月31日までの年度内に帰属するRSUの公正価値総額は$である3.1百万、$1.5百万ドルとドル5.8それぞれ2億5千万ドルです
従業員株購入計画
2017年12月31日までに、取締役会通過および当社株主が2017年度従業員による株購入計画(“2017 ESPP”)を採択しました。その会社は最初に保留した225,0002017年にESPPが発行した普通株によると、毎年1月1日から増加する年間増額に加え、(I)の最小値に相当する450,000普通株株1つは前年12月31日終値までに発行された普通株式数の割合、及び(3)取締役会がその日又はその日までに決定した当該年度普通株式数は、最高で超えてはならない4,725,000普通株の合計。2017年ESPP発行普通株式数の増加450,0002023年1月1日。当社は、2023年12月31日および2022年12月31日までの年間で発行します381,508そして270,774それぞれ2017年のESPP下の株です。2023年12月31日までに364,2832017年にESPPが発行できる株によると。
株に基づく報酬費用
会社はASC 718の規定を使用して従業員と非従業員のすべての株式奨励を計算する。
株式による補償費用は必要なサービス期間内に確認され,このサービス期間は通常帰属期間であり,直線法が採用されている。
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
以下の表は、企業合併経営報告書と総合損失のうち、株式ベースの補償費用を奨励タイプ別に示しています
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| (単位:千) | 2023 | | 2022 | | 2021 |
株式オプション | $ | 14,171 | | | $ | 15,814 | | | $ | 14,528 | |
制限株式単位およびその他の株式奨励 | 6,184 | | | 5,175 | | | 3,522 | |
| 従業員株購入計画 | 781 | | | 533 | | | 359 | |
| 総運営費に計上された株式報酬費 | $ | 21,136 | | | $ | 21,522 | | | $ | 18,409 | |
以下の表に、会社合併経営報告書と全面赤字に反映された株式ベースの補償費用を示す
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| (単位:千) | 2023 | | 2022 | | 2021 |
| 研究開発 | $ | 11,043 | | | $ | 11,386 | | | $ | 9,984 | |
| 一般と行政 | 10,093 | | | 10,136 | | | 8,425 | |
| 総運営費に計上された株式報酬費 | $ | 21,136 | | | $ | 21,522 | | | $ | 18,409 | |
2023年12月31日までに1ドルあります14.8百万ドルとドル11.8未確認株式ベースの報酬支出は,それぞれ帰属していない株式オプションと帰属していないRSUに関連しており,加重平均中に確認される予定である1.6年和2.4それぞれ数年です。
各オプション報酬の公正価値は、付与された日にブラック·スコアーズオプション定価モデルを用いて推定され、その加重平均は以下のように仮定される | | | | | | | | | | | | | | | | | |
| 十二月三十一日 |
| 2023 | | 2022 | | 2021 |
無リスク金利 | 3.8 | % | | 2.1 | % | | 0.9 | % |
期待配当収益率 | — | % | | — | % | | — | % |
所期期間(年) | 6.05 | | 5.99 | | 6.06 |
株価の変動を予想する | 103 | % | | 88 | % | | 82 | % |
同社の普通株の予想変動率は現在、その歴史変動率に基づいて決定されている。注2を参照重要会計政策の概要もっと多くの情報を知ります。無リスク金利は、オプション予想期限と一致した米国債収益率に基づく違います。配当率は仮定されており、同社は歴史的になく、その普通株の配当金を支払うことも望まないからである。付与されたオプションの予想期限は、ホーム日と契約期限終了との間の中間点として推定される簡略化された方法の使用に基づく。
RSUの公正価値は、付与された日の会社普通株の終値によって決定される。
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
11. 1株当たり純損失
普通株1株当たりの基本純損失の計算方法は、普通株株主が純損失をその期間に発行された普通株の加重平均株式数で割るべきであり、希釈可能な証券はさらに考慮しない。1株当たり純損失の計算方法は、普通株株主が純損失を在庫株方法を用いて決定した期間の普通株と潜在的希薄化証券の加重平均で割るべきである。
1株当たりの純損失を計算する場合、株式オプション、RSUに帰属していないおよび普通株を購入する引受権証は潜在的な希薄化証券とされているが、その影響は逆薄となるため、すべての届出期間の基本および当株当たりの純損失は同じであるため、1株当たりの純損失の計算範囲は計上されない
次の表は、それらを含めることが反ダンピング(普通株等値株)になるので、1株当たりの純損失計算から除外された発行された潜在的希薄化証券を示している
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 | | 2021 |
| 株式オプション | 10,902,845 | | | 10,051,283 | | | 8,342,429 | |
| 未帰属限定株式単位 | 3,834,108 | | | 1,870,791 | | | 817,609 | |
| 株式承認証 | — | | | 22,590 | | | 39,474 | |
| 14,736,953 | | | 11,944,664 | | | 9,199,512 | |
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
12. 賃貸借証書
同社はマサチューセッツ州ケンブリッジ市にあるオフィスと実験室空間に対して運営賃貸契約を持ち、ある設備に対して運営と融資レンタルを行った。当社は2020年3月にオフィスビル及び実験室賃貸借契約(“オフィスビル賃貸借契約”)第7改正案を締結し、レンタル期間を2026年3月に延長する。会社はオフィスビルのレンタル期間を延長して、増加する権利があります5年.
2021年4月5日、会社はオフィスビルレンタルの第8項改正案を締結し、会社の既存ビルに追加のオフィススペースを付与した5年2021年7月1日から、会社がレンタル料を支払うことを約束しました5.0その間(“拡張テナント”)。オフィスビル賃貸項目の選択権とは独立して、当社は拡張賃貸期間を追加延長する権利があります5年それは.当社は2023年12月31日現在、オフィスビル賃貸延長·拡張賃貸条項の選択権を行使しており、合理的に確定されているとはみなされていない。
拡張協定は、オフィス賃貸の範囲を拡大し、追加の賃貸支払いが市場賃貸料に見合っているため、単独の契約入金として賃貸修正である。当社は署名日に拡張賃貸契約の賃貸種別を評価し,拡張賃貸を経営賃貸入金とすることを決定した。使用権資産および対応する経営リース負債は、レンタル期間内の賃貸支払いの現在値に基づいて計算されています。当社は、信用と財務状況が類似している会社の未償還債務の分析に基づいて推定される総合信用格付けを用いることにより、割引率として適切な増量借款金利を決定する。経営リースは純賃貸であるため、非レンタル部分(すなわち公共エリアメンテナンス)は、実際に発生したコストに応じてレンタル料とは別に支払われるため、これらの非レンタル部分はROU資産や負債には含まれず、発生した期間に料金として反映される。
同社は家主の利益のための予備信用状協定を持っており、金額は#ドルである0.52023年12月31日、2023年12月31日、2022年12月31日までのオフィスビル賃貸·拡張賃貸に関する費用は100万ドル。
当社の残りの融資リース条項は1年至れり尽くせり5年いくつかのデバイスの場合、いくつかは、公正な価値で購入されたオプションを含む
経営と総合損失表に研究と開発および一般と行政費用を含めたリース費用構成は以下のとおりである | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| (単位:千) | 2023 | | 2022 | | 2021 |
| リースコストを経営する | $ | 3,838 | | | $ | 3,793 | | | $ | 3,502 | |
| | | | | |
| 融資リースコスト: | | | | | |
| 使用権資産の償却 | $ | 185 | | | $ | 194 | | | $ | 169 | |
| 賃貸負債利息 | 16 | | | 28 | | | 28 | |
| $ | 201 | | | $ | 222 | | | $ | 197 | |
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
リースに関する補足貸借対照表情報は以下のとおりである | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2023 | | 2022 |
| 経営リース: | | | |
| 経営的リース使用権資産 | $ | 7,694 | | | $ | 10,475 | |
| 賃貸負債を経営し、流動 | $ | 3,252 | | | $ | 2,798 | |
| リース負債を経営する | $ | 5,149 | | | $ | 8,575 | |
| | | |
| 融資リース: | | | |
| 財産と設備、毛額 | $ | 1,038 | | | $ | 1,038 | |
| 財産と設備、減価償却累計 | $ | (723) | | | $ | (539) | |
| その他流動負債 | $ | 144 | | | $ | 240 | |
| その他負債 | $ | 54 | | | $ | 203 | |
| | | |
| 加重平均残余レンタル期間: | | | |
| 賃貸借契約を経営する | 2.3年.年 | | 3.3年.年 |
| 融資リース | 1.1年.年 | | 2.0年.年 |
| | | |
| 加重平均割引率: | | | |
| 賃貸借契約を経営する | 10.8 | % | | 10.8 | % |
| 融資リース | 4.4 | % | | 5.4 | % |
レンタルに関する補足キャッシュフロー情報は以下のとおりである | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| (単位:千) | 2023 | | 2022 |
| 賃貸負債に含まれる金額を計量するために支払う現金: | | | |
| レンタル経営からの経営キャッシュフロー | $ | 4,029 | | | $ | 3,865 | |
| 融資リースの運営キャッシュフロー | $ | 16 | | | $ | 28 | |
| 融資リースによるキャッシュフロー | $ | 262 | | | $ | 272 | |
2023年12月31日まで、レンタルをキャンセルできない将来のレンタル支払いは以下の通りです | | | | | | | | | | | |
| (単位:千) | 賃貸借契約を経営する | | 融資リース |
| 2024 | $ | 3,975 | | | $ | 141 | |
| 2025 | 4,187 | | | 48 | |
| 2026 | 1,310 | | | 8 | |
2027年とその後 | — | | | — | |
| | | |
| 賃貸支払総額 | 9,472 | | | 197 | |
| 現在価値調整 | (1,069) | | | — | |
| 賃貸負債現在価値 | $ | 8,403 | | | $ | 197 | |
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
13. 所得税
当社は、2023年、2023年、2022年および2021年12月31日までの年度を記録しています違います。会社の営業損失と繰延税金資産の全額推定手当により、毎年発生する純営業損失(“NOL”)の所得税優遇。
2023年、2023年、2022年12月31日終了年度の実際の税率は以下の通り
| | | | | | | | | | | | | |
| 2023 | | 2022 | | |
| アメリカの法定連邦金利 | 21.0 | % | | 21.0 | % | | |
| 連邦福祉を差し引いた州税 | 5.5 | % | | 5.7 | % | | |
| 恒久的差異 | (0.1) | % | | (0.1) | % | | |
| | | | | |
| 一般商業信用 | 4.0 | % | | 4.4 | % | | |
| 株の報酬 | (1.9) | % | | (1.4) | % | | |
| | | | | |
| 評価免除額を変更する | (28.5) | % | | (29.6) | % | | |
| — | % | | — | % | | |
繰延所得税は、財務報告目的のための資産および負債の帳簿金額と所得税目的のための金額との間の一時的な差異の純税影響を反映する当社の2023年12月31日まで、2023年12月31日と2022年12月31日までの繰延税項目純資産の重要な構成要素は以下の通りです
| | | | | | | | | | | |
| (単位:千) | 2023 | | 2022 |
| 繰延税金資産: | | | |
| 純営業損失 | $ | 126,798 | | | $ | 113,981 | |
| 研究開発資本化 | 67,107 | | | 40,380 | |
| 税金の繰り越しを免除する | 28,409 | | | 21,429 | |
| 株に基づく報酬 | 6,899 | | | 6,522 | |
| 費用を計算する | 3,142 | | | 3,340 | |
| 賃貸負債 | 2,274 | | | 3,096 | |
| 大文字許可証 | 2,725 | | | 3,077 | |
| 収入を繰り越す | 3,688 | | | 1,062 | |
| 減価償却 | 353 | | | 437 | |
| | | |
| 他にも | 156 | | | 116 | |
| 繰延税項目の総資産総額 | 241,551 | | | 193,440 | |
| 推定免税額 | (239,468) | | | (190,588) | |
| 繰延税項純資産から推定免税額を差し引く | 2,083 | | | 2,852 | |
| 繰延税金負債 | | | |
| 使用権資産 | (2,083) | | | (2,852) | |
| 繰延税金負債総額 | (2,083) | | | (2,852) | |
| 繰延税金純額 | $ | — | | | $ | — | |
当社は設立以来NOLを生み出しています。2023年12月31日現在,会社の連邦と州NOL繰り越し額は約$である479.0百万ドルとドル414.8それぞれ100万ドルですドルの中で479.0連邦NOLは100万ドルを繰り越しました34.12037年までに100万人の人々が違う日付で満期になった。残りの$444.8数百万の連邦NOL繰り越しは期限が切れないだろう。国NOL繰越は2043年までに異なる日付で満期になります。2023年12月31日まで、会社は連邦と州の研究開発税の繰越免除$を持っています23.2百万ドルとドル6.8それぞれ100万ドルで、2043年までの異なる日に満期になります。
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
米国会計基準第740条の要求に基づき、会社管理層は、その繰延税金資産の信頼性に関する証拠を評価している。既存の証拠の重要性によると、積極的でも消極的であっても、経営陣が確定しており、会社はこれらすべての資産のメリットを実現できない可能性が高い。そのため、同社は#ドルの推定手当を記録した239.5百万ドルとドル190.62023年12月31日と2022年12月31日はそれぞれ100万円。推定免税額は#ドル増加した48.92023年12月31日までの年度内に、主に当社の今年度の純損失と信用によるものである。
NOL及び研究開発税収控除繰越の使用は、第382節に規定する重大年度制限を受ける可能性があり、その理由は、第382節及び類似の国の規定により、以前に発生した又は将来発生する可能性のある所有権変更制限を受けているからである。これらの所有権の変化は、将来の課税収入と税収を相殺するために毎年使用できるNOLと研究開発税収の繰越免除の金額を制限する可能性がある。当社が設立されてからのいつでも第382節で定義された支配権変更が発生した場合、そのNOLまたは研究開発税収控除の使用は、まず所有権変更時に会社株の価値に適用される長期免税率を乗じて決定され、必要に応じて追加的に調整される第382節に規定される年次制限を受ける。いかなる制限も一部のNOLや研究開発税の使用前の繰越期限を招く可能性がある。当社は過去に所有権変更が発生したことを確定しており、あるNOLと研究開発税収の繰越が制限される。
会社は、ASC 740における所得税不確実性会計処理に関する会計基準を採用している。当社の税収関連準備金は、当社がその税務申告または立場で得た税収割引がどれだけあるか、税収割引に関連する任意の潜在的または有事項を解決した後に実現される可能性があるかどうかの決定に基づいています。2023年、2023年、2022年12月31日まで、会社は違います。未確認の税金割引。
会社は2023年に長年の研究開発信用研究を依頼し、その結果、2023年12月31日までの会社累計繰延税金資産を調整した。繰延税金資産の変動は全額推定準備金で相殺される。
税務状況の不確定に関連する利息および罰金は、添付されている経営報告書および全面損失報告書の中で所得税費用に分類される。2023年、2023年、2022年12月31日まで、会社は違います。不確定な税務状況に関連するものは利息または罰金を計算しなければならない。
会社はアメリカ連邦税務管区に所得税申告書を提出し8人州司法管区。当社は2023年、2023年、2022年および2021年12月31日までの4年間海外業務を行っていません。国税局と州税務機関が評価した訴訟時効は2019年までの納税年度で閉鎖されており、2019納税年度までに発生した繰越属性にもかかわらず、今後しばらく審査状況に応じて調整することができる。いくつありますか違います。現在行われている連邦や州監査。
14. 従業員福祉計画
当社には、規則401(K)節(“401(K)計画”)に基づいて設立された固定供出計画があり、基本的に全従業員をカバーしています。満18歳の従業員21仕事をしています1,000工数は401(K)退職計画に参加する資格がある。従業員は最高で95税引前ベースで連邦年度限度額の合格報酬の%に達する。2023年12月、2022年、2021年12月までの年間で、会社は相当の貢献をしました100従業員の出資比率は、最高限度額は4条件を満たした報酬の%です。当社は2023年、2023年、2022年および2021年12月31日までに支出$を記録しました1.3百万、$1.1百万ドルとドル0.8それぞれ401(K)プランへの貢献に関係している.
15. 支払いを引き受ける
許可協定
2023年12月31日までの年間で、当社は注釈払い戻し不可能なライセンス支払いに関する研究開発費を記録する。当社は、2022年12月31日および2021年12月31日までの年間で、払戻不可能なライセンス支払いに関する研究開発費$を記録しました1.5百万ドルとドル3.1それぞれ100万ドルです
カタログ表
Mersana治療会社です。
連結財務諸表付記
(続)
2023年12月31日までの年間で、当社は注釈開発マイルストーンに関する研究開発費を記録した。当社は、2022年12月31日および2021年12月31日までの年間で、発展マイルストーンに関する研究および発展費$を記録しました0.7百万ドルとドル2.1それぞれ100万ドルです2022年と2021年の発展マイルストーンはそれぞれXMT-1660とUpRiと関係がある。
付記12を参照賃貸借契約2023年12月31日現在、会社はレンタルに関する将来義務を負っている。
16. 再編成する
2023年7月27日,同社が白金耐性卵巣癌患者に対するUpRi第2期臨床試験のトップデータを評価した後,同社は重点領域の優先順位を再決定し,UpRiの臨床開発を中止することを発表した。これらの決定について,会社取締役会は2023年7月26日に削減約を含む何らかの費用削減措置を承認した50会社の当時の総従業員基数の割合(“再編成”)を占める。影響を受けた従業員は、リストラに関する解散費や福祉、賃金通知、再就職サービスを受ける資格がある。
2023年12月31日現在、再編はほぼ完了している。発生した再編コストは合併経営報告書の再編費用と全面赤字に計上される
次の表は、再構成に関する費用をまとめています
| | | | | | | | | | | | | | | | | | | | | | |
(単位:千) | | 解散費と従業員関連費用 | | | | 契約終了その他の費用 | | 総コスト |
| | | | | | | | |
| これまでの累積コスト | | $ | 6,924 | | | | | $ | 1,789 | | | $ | 8,713 | |
2023年12月31日までの年間発生コスト | | $ | 6,924 | | | | | $ | 1,789 | | | $ | 8,713 | |
次の表は再編成に関する費用をまとめたものです研究と開発活動や一般的で行政活動:
| | | | | | | | | | |
(単位:千) | | 2023年12月31日までの年度 | | |
研究や開発に関わっています | | $ | 5,393 | | | |
一般事務と行政事務 | | $ | 3,320 | | | |
連結貸借対照表に計上すべき費用の計上すべき再編成費用は以下のとおりである
| | | | | | | | | | | | | | | | | | | | | | |
| (単位:千) | | 解散費と従業員関連費用 | | | | 契約終了費用 | | 総コスト |
| 2022年12月31日の残高 | | $ | — | | | | | $ | — | | | $ | — | |
追加料金 | | 6,924 | | | | | 372 | | | 7,296 | |
現金払い | | (5,917) | | | | | — | | | (5,917) | |
その他の調整 | | (252) | | | | | (80) | | | (332) | |
2023年12月31日の残高 | | $ | 755 | | | | | $ | 292 | | | $ | 1,047 | |
項目9.報告会計·財務開示面の変化と会計担当者との相違
ない。
項目9 A:管理制御とプログラム
情報開示制御とプログラムの評価
我々は、取引法に基づいて提出または提出された報告書において開示を要求する情報を確保するために、1934年の証券取引法(改正)または“取引法”の下で第13 a-15(E)および15 d-15(E)規則で定義された“開示制御および手順”を維持し、(1)証券取引委員会の規則および表が指定された期間内に記録、処理、まとめ、および報告すること、(2)必要な開示に関する決定をタイムリーに行うために、我々の主要幹部および首席財務官を含めて蓄積し、適切に伝達することを目的とする。私たちの経営陣は、どんな制御やプログラムも、どんなに設計や操作が良くても、その目標を達成するために合理的な保証を提供するしかないことを認識しており、私たちの経営陣は、可能な制御とプログラムの費用対効果関係を評価する際にその判断を適用しなければならない。私たちの開示制御と手続きはその制御目標を達成するための合理的な保証を提供することを目的としている。
我々の経営陣は、CEOと財務責任者の参加の下、2023年12月31日現在、すなわち本年度報告Form 10−Kがカバーする期間が終了した時点で、制御及びプログラムの有効性を開示することを評価した。上記の評価に基づき、我々の最高経営責任者及び最高財務官は、我々の開示制御及び手続がその日の合理的な保証レベルで有効であると結論した。
財務報告の内部統制
財務報告の内部統制に関する経営陣の報告
私たちの経営陣は私たちの財務報告書に対する十分な内部統制を確立して維持する責任がある。“取引法”に基づいて公布された規則13 a-15(F)および15 d-15(F)における財務報告の内部制御は、私たちの主要幹部と主要財務官が設計またはその監督の下で、米国公認会計原則またはGAAPに基づいて外部目的財務報告の信頼性と財務諸表の作成に合理的な保証を提供するために、私たちの取締役会、管理層および他の人員によって実施されるプログラムと定義されている。私たちの財務報告書に対する内部統制は以下の政策と手続きを含む
•私たちの資産に対する私たちの取引と処置を合理的で詳細かつ正確に反映した記録を保存することと関連がある
•アメリカ公認会計原則に基づいて財務諸表を作成するために取引が必要と記録されていることを保証し、私たちの収支は私たちの経営陣と取締役の許可のみに基づいて行われることを保証します
•当社の財務諸表に重大な影響を及ぼす可能性のある不正な取得、使用、または処理を防止またはタイムリーに発見することができる合理的な保証を提供します。
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。将来的にどのような有効性評価を行うかの予測には,条件の変化により制御が不十分になる可能性がある,あるいは政策やプログラムを遵守する程度が悪化する可能性があるというリスクがある.
我々の経営陣は、2023年12月31日までの財務報告内部統制の有効性を評価した。この評価を行う際には,管理層は,トレデビル委員会後援組織委員会が2013年の内部統制−総合枠組みで提案した基準を用いた。我々の評価によると、我々の経営陣は、2023年12月31日現在、財務報告に対する内部統制がこれらの基準に基づいて有効であると結論している。
財務報告の内部統制の変化
2023年12月31日までの3ヶ月間、財務報告の内部統制(取引法第13 a-15(F)および15 d-15(F)条の定義による)に大きな影響を与えなかったか、または合理的に財務報告内部統制に大きな影響を与える可能性が高い。
プロジェクト9 B:他の資料の提供
役員と上級社員の取引手配
我々役員及び上級管理者の報酬(改正された証券取引法又は証券取引法第16 a-1(F)条を参照)の大部分は、株式報酬の形態であり、取締役及び上級管理者は、株式報酬の帰属又は行使時に源泉徴収義務を履行すること、及び多様化又はその他の個人的な理由から、そのような株式報酬に基づいて得られた証券又は他の会社証券について公開市場取引を行うことができる。取締役や上級管理者による我々証券の取引は、適用される米国連邦証券法に適合することを要求する我々のインサイダー取引政策に適合しなければならず、重大な非公開情報を有する場合の取引を禁止する法律である。取引法の下の10 b 5-1規則は、重大な非公開情報を持った場合に取引を開始する懸念を回避するために、取締役や上級管理者が私たちの証券の取引を事前に手配できるように肯定的な抗弁を提供している。
2023年第4四半期に2023年11月7日, マーティン·フーベル, 医学博士社長兼CEOと取締役会のメンバーは, 通過するルール10 b 5-1は、取引法下のルール10 b 5-1(C)(1)(Ii)(D)(3)に記載されているように、“合格売りから購入取引まで”の資格に適合することが意図されている。このような売却からカバーへの配置は、帰属が時間の推移および/または業績目標の達成に基づくにかかわらず、時々彼に付与される制限的な株式単位、またはRSUに適用される(その条項に従って米国が帰属および決済に関連する源泉徴収義務のために株を抑留するRSUは含まれない)。この手配では、私たちの普通株は自動的に販売され、そうでなければ、RSUを保証する各決済日に適用予定義務を履行するために必要な金額が発行され、売却されたお金は適用される予定義務を満たすために私たちに渡されます。このスケジュールによれば、売却される株式数は、帰属条件が満たされる程度、決済時に我々の普通株の市場価格、およびこのスケジュールによって制限される将来的に追加のRSUが付与される可能性があるので、現在決定されていない。
以上を除いて,ほかに役員や役人はいない通過するあるいは…終了しましたルール10 b 5-1取引スケジュールまたは非10 b 5-1取引スケジュール (定義はS-K条例第408(C)項参照)2023年第4四半期に。
プロジェクト9 C.検査妨害の開示を禁止する外国司法管区
適用されません。
第III部
プロジェクト10.管理役員、役員、および企業管理
以下に述べる以外に、本プロジェクトに要求される情報は、2023年12月31日までの財政年度終了後120日以内に米国証券取引委員会に提出された最終依頼書又は2024年依頼書の“第1号提案−取締役選挙”、“執行者”、“取締役会及び会社管理に関する情報”及び“延滞第16条(A)報告”部分の情報を参考に統合したものである。
取締役、上級管理職、従業員、最高経営責任者、最高財務官、首席会計官または主計長、または同様の機能を実行する者を含む、会社のウェブサイトhttps://mersana.com/“投資家とメディア”部分(https://ir.mersana.com)の“コーポレート”部分の“コーポレートガバナンス”部分に適用されます。私たちは、表格8-K第5.05項の開示要件に基づいて開示される必要がある“商業行為および道徳基準”の任意の改正または免除を、私たちのウェブサイトで開示する予定です。
第11項:役員報酬の増加
本条項に要求される情報(S-K法規第402(V)条に要求される情報を除く)は、2024年の委託書に“役員報酬”と“取締役会と会社のガバナンス−報酬委員会連動及び内部人参加に関する”と題する章の情報を参考にして格納されている。
第十二項:特定の実益所有者及び経営陣の保証所有権及び関連株主事項の決定
本プロジェクトによって要求される情報は、2024年の委託書に“いくつかの実益所有者および経営陣の保証所有権”と題する情報および“持分補償計画に従って発行された証券”の節の情報を参照して組み込まれる。
第十三項:特定の関係及び関連取引の確立、並びに取締役の独立性
本プロジェクトが提供を要求する情報は,2024年の依頼書の“何らかの関係と関連者取引”と“取締役会とコーポレートガバナンスである取締役独立性”の部分の情報を参考にした.
14項目目:総会計士費用とサービス料
本プロジェクトに必要な資料は、当社の2024年依頼書“主要会計士費用及びサービス”及び“監査委員会事前承認政策及びプログラム”の節に記載されている資料を参考にします。
第IV部
プロジェクト15.展示および財務諸表の添付表
(A)(1)財務諸表
当社の総合財務諸表は、本年度報告表格10-K第II部第8項の“総合財務諸表索引”に記載されている。
(A)(2)財務諸表付表
必要または適用されないので、必要な資料が連結財務諸表またはその付記に含まれるので、すべての付表は省略される。
(A)(3)展示品
| | | | | | |
展示品
番号をつける | 展示品の紹介: | |
| 3.1 | 2023年6月8日現在、第5回改訂·再改訂された会社登録証明書(2023年6月9日を参照して米国証券取引委員会に提出された会社の現在の8−Kレポートの添付ファイル3.1が組み込まれている)。 | |
| | |
| 3.2 | 2回目の改正及び再改訂の定款(当社が2023年3月31日に米国証券取引委員会に提出した現在の8−K表報告書の添付ファイル3.1を引用して編入)。 | |
| | |
4.1 | 証券説明(2023年2月28日に米国証券取引委員会に提出された10-K年報(文書番号001-38129)添付ファイル4.3編入参照)。 | |
| | |
10.1† | 賠償協議表(2017年6月16日に米国証券取引委員会に届出されたS−1号登録説明書修正案第1号添付ファイル10.1(フレット番号333−218412)合併参照)。 | |
| | |
| 10.2 | 商業リースは,日付は2009年2月24日であり,Mersana Treateutics,Inc.とRivertech Associates II,LLC(2017年6月1日に米国証券取引委員会に提出されたS-1表登録声明(文書番号333-218412)の添付ファイル10.2参照により統合されている). | |
| | |
| 10.3 | Rivertech Associates II LLCとMersana Treateutics,Inc.間のリースの第7回延長·修正協定については,2020年3月10日にMersana Treateutics,Inc.とRivertech Associates II LLC間のリース契約(2020年5月8日に米国証券取引委員会に提出されたForm 10-Q四半期報告(ファイル番号001-38129)の添付ファイル10.1を参照して組み込む)である。 | |
| | |
| 10.4 | Rivertech Associates II LLCとMersana Treateutics,Inc.の間でMersana Treateutics,Inc.とRivertech Associates II LLCの間で締結されたリースの8件目のリース修正協定については,2021年4月5日に発効する(合併内容は2021年5月10日参照により米国証券取引委員会の10−Q四半期報告(文書番号001−38129)の添付ファイル10.1)に提出される。 | |
| | |
10.5+ | Synaffix B.V.とMersana Treateutics,Inc.の間で2021年11月23日に署名された商業許可およびオプション協定が改訂および再署名された(2022年2月28日に米国証券取引委員会に提出された10-K表年次報告書(文書番号001−38129)の添付ファイル10.15を参照して組み込まれる)。 | |
| | |
10.6+ | Mersana Treateutics,Inc.およびSynaffix B.V.は、2022年2月2日に改正され、再署名された商業許可およびオプション協定の第1号修正案(2022年5月9日に米国証券取引委員会に提出された10-Q表四半期報告書(文書番号001-38129)の10.3添付ファイルを参照して編入される)。 | |
| | |
10.7+ | マーサナ治療会社とヤンソンバイオテクノロジー社が2022年2月2日に署名した研究協力および許可協定(2022年5月9日に米国証券取引委員会に提出された10-Q表四半期報告書(ファイル番号001~38129)の添付ファイル10.2を参照して組み込まれる)。 | |
10.8+ | Mersana Treateutics,Inc.とJanssen Biotech,Inc.の間で2023年7月14日に署名された研究協力·許可協定(期日は2022年2月2日発効)の修正案第1号(2023年11月7日に米国証券取引委員会に提出された10-Q表四半期報告書(ファイル番号001-38129)の添付ファイル10.1を参照して組み込まれる)。 | |
| | | | | | |
10.9+ | マーサナ治療会社とヤンソンバイオテクノロジー会社との間で2023年9月25日に署名された研究協力·許可協定(2022年2月2日発効)の修正案第2号(2023年11月7日に米国証券取引委員会に提出された10-Q表四半期報告書(文書番号001−38129)の10.2号添付ファイルを参照して編入される)。 | |
| | |
10.10+ | マーサナ治療会社とグラクソ·スミスクライン知的財産権(第4号)有限会社が2022年8月6日に署名した協力、選択権、および許可協定(2022年11月7日に米国証券取引委員会に提出された10-Q表四半期報告書(文書番号001-38129)の添付ファイル10.1を参照して編入される)。 | |
| | |
10.11+ | Mersana Treateutics,Inc.およびAres Trading S.A.が2022年12月22日に署名した協力および商業許可協定(2023年2月28日に米国証券取引委員会に提出された10-K表年次報告書(文書番号001-38129)の添付ファイル10.18を参照して編入される)。 | |
| | |
10.12+ | 融資および担保協定は、2021年10月29日に、オックスフォード金融有限責任会社、シリコンバレー銀行、メルサナ治療会社によって締結される(2022年2月28日に米国証券取引委員会に提出された10-K表年次報告書(文書番号001-38129)の添付ファイル10.14を参照して編入される)。 | |
| | |
10.13+ | 融資·担保協定第1修正案は、2022年2月17日に、オックスフォード金融有限責任会社、シリコンバレー銀行を含む融資者とMersana Treateutics,Inc.(2022年5月9日に米国証券取引委員会に提出された10-Q表四半期報告(文書番号001-38129)の添付ファイル10.4を参照して統合された)である。 | |
| | |
10.14+ | “融資·担保協定第2修正案”は、2022年10月17日に、オックスフォード金融有限責任会社、文書に記載されているシリコンバレー銀行を含む貸手とメルサナ治療会社によって締結される(2023年2月28日に米国証券取引委員会に提出されたForm 10-K年次報告書(文書番号001-38129)の添付ファイル10.21を参照して合併)。 | |
| | |
| 10.15 | “融資·担保協定第3修正案”は、2022年12月27日に、オックスフォード金融有限責任会社、文書に記載されているシリコンバレー銀行を含む貸手とメルサナ治療会社とが締結されている(2023年2月28日に米国証券取引委員会に提出されたForm 10-K年次報告書(文書番号001-38129)の添付ファイル10.22を参照して合併)。 | |
| 10.16 | オックスフォード金融有限責任会社が2023年3月23日に署名した融資および保証協定第4修正案では、融資者は、シリコンバレーブリッジ銀行およびMersana Treateutics,Inc.(合併内容は、2023年5月9日に米国証券取引委員会に提出された10-Q表四半期報告書(文書番号001~38129)の添付ファイル10.1を参照することによって提出される)。 | |
| | |
| 10.17 | マーサナ治療会社とコーエン有限責任会社が2022年11月7日に署名した販売協定(2022年11月7日に米国証券取引委員会に提出された現在の8-K表報告書の添付ファイル1.1(ファイル番号001-38129)を参照して合併)。 | |
| | |
10.18† | Mersana Treateutics,Inc.とAnna Protopapasの間で2017年3月17日に発行された招待状が改訂され再発行された(2017年6月1日に米国証券取引委員会に提出されたS-1表登録声明(文書番号333-218412)の添付ファイル10.16を参照して編入された)。 | |
10.19† | Mersana治療会社とTimothy B.Lowingerとの間で2017年3月8日に発行された招待状(2017年6月1日に米国証券取引委員会に提出されたS−1表登録声明(文書番号333−218412)の添付ファイル10.18を参照して編入)が改訂·再発行された。 | |
| | |
10.20† | Mersana Treateutics,Inc.およびBrian DeSchuytnerによって2019年6月10日に発行された招待状(2020年5月8日に米国証券取引委員会に提出されたForm 10-Q四半期報告書(ファイル番号001-38129)の添付ファイル10.3を参照して組み込まれる)。 | |
| | |
| | |
10.21† | Mersana Treateutics,Inc.およびArvin Yangによって2020年11月5日に発行された招待状(2021年5月10日に米国証券取引委員会に提出されたForm 10-Q四半期報告(ファイル番号001-38129)の添付ファイル10.2を参照して組み込まれる)。 | |
10.22† | Mersana Treateutics,Inc.とAlejandra Carvajalの間の招待状は,2021年3月5日(2022年5月9日参照により米国証券取引委員会に提出されたForm 10-Q四半期報告書(文書番号001−38129)の添付ファイル10.5への編入)である。 | |
| | |
| | | | | | |
10.23† | Mersana Treateutics,Inc.およびTushar Misraが2021年6月15日に発行した招待状(2022年5月9日に米国証券取引委員会に提出されたForm 10-Q四半期報告(ファイル番号001-38129)の添付ファイル10.6を参照して組み込まれる)。 | |
10.24*† | Mersana治療会社とMohan Balaの間の招待状は,2021年7月20日である。 | |
10.25† | 退職および別居協定は、2023年9月5日に、Mersana Treateutics,Inc.とAnna Protopapasによって締結される(2023年11月7日に米国証券取引委員会に提出されたForm 10-Q四半期報告書(ファイル番号001-38129)の添付ファイル10.3を参照して編入される)。 | |
10.26† | Mersana Treateutics,Inc.とMartin Huberの間の招待状は、2023年9月5日(2023年11月7日に米国証券取引委員会に提出されたForm 10-Q四半期報告書(ファイル番号001-38129)の添付ファイル10.4を参照して組み込まれる)である。 | |
| | |
10.27† | Mersana Treateutics,Inc.とArvin Yangの間で2023年9月6日に署名された書簡協定(2023年11月7日に米国証券取引委員会に提出されたForm 10-Q四半期報告書(ファイル番号001−38129)の添付ファイル10.5を参照して組み込まれる)。 | |
10.28† | 改訂された2007年株式インセンティブ計画(2017年6月1日に米国証券取引委員会に提出されたS-1表登録説明書(文書番号333-218412)添付ファイル10.19を参照)。 | |
| | |
10.29† | 2007年株式インセンティブ計画下の株式オプションインセンティブプロトコル表(2017年6月1日に米国証券取引委員会に提出されたS−1レジストリ(ファイル番号333−218412)添付ファイル10.20参照)。 | |
| | |
10.30† | 2007年株式インセンティブ計画下の不適格株式オプション表(2017年6月1日に米国証券取引委員会に提出されたS-1表(文書番号333-218412)添付ファイル10.21参照)。 | |
| | |
10.31† | 2017年株式インセンティブ計画(2017年6月16日に米国証券取引委員会に届出されたS-1表登録説明書修正案第1号添付ファイル10.22(フレット番号:333-218412)を参照して組み入れられます)。 | |
| | |
10.32† | 2017年株式インセンティブ計画下のインセンティブ株式オプション協定表(2017年6月16日に米国証券取引委員会に届出されたS-1表登録説明書修正案第1号(第333-218412号文書)第10.23号添付ファイルを参照)。 | |
| | |
10.33† | 2017年株式インセンティブ計画下の非法定株式オプション協定表(2017年6月16日に米国証券取引委員会に提出されたS-1表登録説明書修正案第1号(333-218412号文書)第10.24号添付ファイル参照)。 | |
| | |
10.34† | 2017年株式インセンティブ計画下の限定株式単位表は、2023年4月までに付与された奨励に適用される(2021年8月6日に米国証券取引委員会に提出された10-Q四半期報告(文書番号333-38129)の添付ファイル10.1を参照して組み込まれる)。 | |
10.35† | メルサナ治療会社2017年株式インセンティブ計画下の従業員制限株式単位プロトコル表は、2023年4月以降に付与された奨励に適用されます(添付ファイル10.2を参照して2023年8月8日に米国証券取引委員会に提出された10-Q四半期報告書(文書番号001-23819)に組み込まれています)。 | |
10.36† | Mersana Treateutics,Inc.2017年株式インセンティブ計画下の非従業員取締役限定株式単位プロトコル表は、2023年4月以降に付与された奨励に適用される(添付ファイル10.3を参照して2023年8月8日に米国証券取引委員会に提出された10-Q四半期報告書(文書番号001-23819)に組み込まれる)。 | |
| | |
10.37† | 2022年インセンティブ株式インセンティブ計画(2022年2月28日に米国証券取引委員会に提出された10-K年報(ファイル番号001-38129)添付ファイル10.28を参照)。 | |
| | |
10.38† | 2022年インセンティブ株式インセンティブ計画下の非法定株式オプション協定表(2022年2月28日に米国証券取引委員会に提出された10-K年報(ファイル番号001-38129)添付ファイル10.30を参照)。 | |
10.39† | 2022年インセンティブ株式インセンティブ計画下のインセンティブ制限株式単位テーブルは、2023年4月までに付与された奨励に適用される(添付ファイル10.29を参照して2022年2月28日に米国証券取引委員会に提出された10-Kフォーム年次報告(ファイル番号001-38129)に組み込まれる)。 | |
10.40† | Mersana Treateutics,Inc.2022年誘導株式インセンティブ計画下の制限株式単位プロトコルテーブル(添付ファイル10.1を参照して2023年8月8日に米国証券取引委員会に提出されたForm 10-Q四半期報告書(ファイル番号001-23819))を組み込む。 | |
| | |
10.41† | 改訂された2017年従業員株購入計画(2023年2月28日に米国証券取引委員会に提出された10-K年報(文書番号001-23819)添付ファイル10.40を参照)。 | |
| | |
| | | | | | |
10.42† | 2017年現金配当計画(2017年6月16日に米国証券取引委員会に届出されたS-1表登録説明書修正案1号添付ファイル10.26(文書番号333-218412)合併参照)。 | |
| | |
10.43*† | 非従業員役員報酬政策は、2023年12月15日に改正された。 | |
| | |
| 21.1 | メルサナ治療会社の子会社(2023年2月28日に米国証券取引委員会に提出された10-Kフォーム年次報告書(ファイル番号001-38129)の添付ファイル21.1合併を引用することにより)。 | |
| | |
23.1* | 独立公認会計士事務所安永会計士事務所の同意を得ました。 | |
| | |
31.1* | 2002年のサバンズ·オキシリー法第302節で可決された1934年の証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて発行された首席執行幹事証明書。 | |
| | |
31.2* | 2002年サバンズ−オキシリー法第302節で可決された1934年証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて首席財務幹事が認証された | |
| | |
32.1** | 2002年の“サバンズ-オックススリー法案”906節で採択された“米国法典”第18編1350条による最高経営責任者と最高財務官の認証。 | |
| | |
| 97*† | 改正·再決定された還付政策 | |
| | |
| 101.INS* | XBRLインスタンスドキュメントを連結する | |
| | |
| 101.Sch* | イントラネットXBRL分類拡張アーキテクチャ文書 | |
| | |
| 101.カール* | インラインXBRL分類拡張計算リンクライブラリ文書 | |
| | |
| 101.def | インラインXBRL分類拡張Linkbase文書を定義する | |
| | |
| 101.実験所* | XBRL分類拡張ラベルLinkbase文書を連結する | |
| | |
| 101.前期* | インラインXBRL分類拡張プレゼンテーションLinkbaseドキュメント | |
| | |
| 104* | 表紙対話データファイル−表紙XBRLタグは、内蔵されたXBRL文書(添付ファイル101に含まれる)に埋め込まれている。 | |
*ここで提出されたファイルです。
**本10-K表年次報告書に添付されている添付ファイル32.1の証明によれば、米国証券取引委員会に届出されたものとはみなされず、参照によって1933年“証券法”(改訂本)または1934年“証券取引法”(改訂本)に従って提出された任意の文書に組み込むことはできない。このような文書に含まれる任意の一般登録言語にかかわらず、本表の10-K年度報告日の前または後に行われてはならない。
イは管理契約又は補償計画を示す。
S-K法規第601(B)(10)(Iv)項によれば、本展示品の以下の部分は漏れている。
第16項:表格10-K要約
ない。
サイン
1934年“証券取引法”第13節又は第15節(D)節の要求に基づき、当社は、正式に許可された次の署名者が代表して本報告に署名することを正式に手配した。
| | | | | |
| Mersana治療会社 |
| |
| 日付:2024年2月28日 | /投稿S/マーティン·フーベル |
| マーティン·フーベル医学博士です 社長と最高経営責任者 (首席行政長官及び権限を受けた署名者) |
本報告は、1934年の証券取引法の要求に基づき、以下の者によって登録者として指定された日に署名された。 | | | | | | | | | | | | | | |
サイン | | タイトル | | 日取り |
| | | | |
/寄稿S/寄稿マーティン·フーベル | | 取締役CEO社長(最高経営責任者) | | 2024年2月28日 |
マーティン·フーベル医学博士です | | |
| | | | |
寄稿S/寄稿Brian DeSchuytner | | 首席運営官兼首席財務官(首席財務官)上級副総裁 | | 2024年2月28日 |
ブライアン·ドシュイターナー | | |
| | | | |
/S/エヒシュ·マンダリア | | 総裁副、首席会計官(首席会計官) | | 2024年2月28日 |
アシュシュ·マンダリア | | |
| | | | |
寄稿S/査読者David·モント | | 取締役会議長 | | 2024年2月28日 |
デヴィッド·モント | | |
| | | | |
/S/ローレンス·M·アレバ | | 役員.取締役 | | 2024年2月28日 |
ローレンス·M·アレバ | | |
| | | | |
/著者S/ウィラード·H·ディール | | 役員.取締役 | | 2024年2月28日 |
ウィラード·H·ディール医学博士 | | |
| | | | |
/S/エリン·M·ディアス | | 役員.取締役 | | 2024年2月28日 |
| アリーン·M·ディアス | | |
| | | | |
/S/アンドリュー·A·F·ハック | | 役員.取締役 | | 2024年2月28日 |
アンドリュー·A·F·ハック医学博士 | | |
| | | | |
寄稿S/寄稿クリスチャン·ヘイグ | | 役員.取締役 | | 2024年2月28日 |
クリスチャン·ヘイグ医学博士 | | |
| | | | |
/S/アンナ·プロトーパース | | 役員.取締役 | | 2024年2月28日 |
アンナ·プロトーパース | | |
| | | | |