
アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
|
1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
本財政年度末まで
あるいは…。
|
1934年証券取引法第13条又は15(D)条に基づいて提出された |
手数料書類番号
(登録者の正確な氏名はその定款に記載)
(明またはその他の司法管轄権 会社や組織) |
(税務署の雇用主 |
(主にオフィスアドレスを実行) |
(郵便番号) |
登録者の電話番号は市外局番を含んでいます(
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
|
取引コード |
|
登録された各取引所の名称 |
|
|
同法第12条(G)に基づいて登録された証券:ありません
登録者が証券法第405条に規定する有名な経験豊富な発行者である場合は、再選択マークで表示してください
登録者がこの法第13又は15(D)条に従って報告書を提出する必要がないか否かを、再選択マークで示す。はい。☐
登録者が(1)過去12ヶ月以内(または登録者がそのような報告を提出する必要があるより短い期間)に1934年の証券取引法第13条または15(D)条に提出されたすべての報告書を再選択マークで示すかどうか、および(2)このような提出要求を過去90日以内に遵守してきた
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
|
☒ |
|
ファイルマネージャを加速する |
|
☐ |
|
|
|
|
|
|||
非加速ファイルサーバ |
|
☐ |
|
規模の小さい報告会社 |
|
|
|
|
|
|
|
|
|
新興成長型会社 |
|
|
|
|
|
|
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に第240.10 D−1(B)条に従って受信されたインセンティブベースの補償に従って回復分析を行う必要があるかどうかを再選択マークで示す☐
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)。はい、そうです
登録者普通株の総時価Sトック$
2024年2月16日現在、登録者が発行する普通株式数は
引用で編入された書類
登録者はその2024年年度株主総会に提出された委託書の一部であり,登録者はこれよりも遅れずに米国証券取引委員会に提出しようとしている 登録者は,2023年12月31日までの財政年度後120日を超えて,本年度報告の表格10−Kの第III部を引用して組み込む
カタログ表
|
|
ページ |
第1部 |
|
|
第1項。 |
業務.業務 |
3 |
第1 A項。 |
リスク要因 |
59 |
項目1 B。 |
未解決従業員意見 |
108 |
プロジェクト1 C。 |
ネットワーク·セキュリティ |
109 |
第二項です。 |
属性 |
109 |
第三項です。 |
法律訴訟 |
109 |
第四項です。 |
炭鉱安全情報開示 |
109 |
|
|
|
第II部 |
|
|
五番目です。 |
登録者普通株市場、関連株主事項及び発行者による株式証券の購入 |
110 |
第六項です。 |
保留されている |
111 |
第七項。 |
経営陣の財務状況と経営成果の検討と分析 |
112 |
第七A項。 |
市場リスクの定量的·定性的開示について |
125 |
第八項です。 |
財務諸表と補足データ |
125 |
第九項です。 |
会計と財務情報開示の変更と相違 |
132 |
第9条。 |
制御とプログラム |
132 |
プロジェクト9 B。 |
その他の情報 |
134 |
プロジェクト9 Cです。 |
検査妨害の外国司法管轄権を開示する |
134 |
|
|
|
第三部 |
|
|
第10項。 |
役員·幹部と会社の管理 |
135 |
第十一項。 |
役員報酬 |
135 |
第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
135 |
十三項。 |
特定の関係や関連取引、取締役の独立性 |
135 |
14項です。 |
最高料金とサービス |
135 |
|
|
|
第4部 |
|
|
第十五項。 |
展示·財務諸表明細書 |
136 |
プロジェクト16 |
表格10-Kの概要 |
138 |
i
我々の業務に関連する重大なリスクとその他のリスクの概要
II
前向き陳述に関する特別説明
このForm 10−K年次報告または年次報告書には、改正された1933年証券法第27 A節および改正された1934年証券取引法第21 E節の安全港条項に基づいて行われた前向きな陳述が含まれている。本年度報告では歴史的事実に関する陳述を除き,他のすべての陳述は前向き陳述である。場合によっては、前向き陳述は、“可能”、“将”、“すべき”、“予想”、“意図”、“計画”、“予想”、“信じ”、“推定”、“予測”、“潜在”、“継続”などの用語、またはこれらの用語または他の同様の用語の否定語によって識別されることができる。このような陳述は未来の結果や業績の保証ではなく、重大なリスクと不確実性に関するものだ。本年度報告における展望的陳述は、以下の明示的または暗示的な陳述を含むが、これらに限定されない
1
本年度報告中の任意の展望性陳述は私たちの未来の事件と私たちの未来の財務表現に対する現在の見方を反映し、既知と未知のリスク、不確定性とその他の要素に関連し、私たちの実際の結果、業績或いは成果はこれらの展望性陳述と明示或いは暗示する任意の未来の結果、業績或いは成果とは大きく異なるかもしれない。実際の結果が現在の予想と大きく異なる可能性がある要因には,第I部第1 A項“リスク要因”と本年度報告の他の部分に記載されている要因がある。このような不確実性を考慮して、あなたはこのような前向きな陳述に過度に依存してはいけない。法的要求がない限り、私たちは未来に新しい情報があっても、これらの前向きな陳述を任意の理由で更新または修正する義務がない。
私たちのすべての展望的な陳述は本年度報告書までの日に限られている。いずれの場合も、実際の結果は、これらの前向き情報とは大きく異なる可能性がある。私たちはそのような期待や展望的な陳述が正しいことが証明されることを保証できない。本年度報告書に言及されているか、または我々の他の開示または他の定期報告または他の文書に言及されているか、または米国証券取引委員会または米国証券取引委員会に提出された他の文書または文書に記載されている1つまたは複数のリスク要因またはリスクおよび不確定要因の発生または任意の重大な悪影響は、私たちの業務、見通し、財務状態、および運営結果に重大な悪影響を及ぼす可能性がある。法律に別の規定があることを除いて、私たちは、本年度報告日後に発生した実際の結果、計画、仮説、推定または予測の変化、またはそのような展望的表現に影響を与える他の状況を反映するために、そのような前向き表現の更新または修正を承諾または計画しない。本年度報告の後になされた任意の開示声明または開示は、本年度報告に含まれる任意の前向き陳述に修正または影響を与える場合、本年度報告に含まれるそのような陳述を修正または置換するものとみなされる。
私たちは時々、これらの市場の潜在規模および特定の疾患の推定発病率および流行率の推定を含む、私たちの業界、一般的な商業環境、および特定の疾患に関する市場の推定、予測、および他の情報を提供するかもしれない。推定、予測、予測、市場研究或いは類似方法に基づく情報自体は不確定要素の影響を受け、実際の事件、状況或いは数字は、実際の疾病罹患率と市場規模を含み、本年度報告に反映された情報とは大きく異なる可能性がある。他に明確な説明がない限り、我々は、市場研究会社および他の第三者、業界、医療および一般出版物、政府データおよび同様のソースによって準備された報告、研究調査、研究および類似データから、当業界、商業情報、市場データ、流行率情報および他のデータを取得し、場合によっては、私たち自身の仮説および分析を適用し、これらの仮説および分析は、将来的に不正確であることが証明される可能性がある。
2
第1部
プロジェクト1.ビジネス
私たちは生物製薬会社で、新しい小分子療法の発見と開発に専念し、人体自身の天然タンパク質分解システムを利用することで病原タンパク質を選択的に分解する。我々の独自の標的蛋白質分解或いはTPDプラットフォームはPegasusと呼ばれ、高度な選択性の小分子蛋白質分解物を発見でき、全身病原蛋白質に対して活性を有する。我々の小分子タンパク質分解器は,既存の療法に比べて独自の利点があり,従来のモデルでは解決が困難であったヒトゲノムの大部分を解決できると信じている。我々はすでに臨床的に検証された生物経路に焦点を当てているが,重要な生物ノード/タンパク質は投与されていないか,あるいは十分に投与されていない。我々はこれまで,我々の飛馬プラットフォームを用いて免疫学−炎症と腫瘍学分野に焦点を当てた新しいタンパク質分解器を設計し,われわれのプラットフォームの能力を他の治療領域に応用し続けてきた。我々の使命は,TPDを用いてヒト細胞中のすべての標的類の薬物治療を行うことである。
著者らの現在の臨床分期計画はIRAK 4、STAT 3とMDM 2であり、各計画は生物検証経路中の高影響標的に対して、広範な免疫性炎症性疾患、血液系悪性腫瘍と/或いは固形腫瘍の治療に機会を提供した。著者らの計画は高い影響目標の解決に対する関心を体現しており、これらの目標は伝統的なモデルにとってつかみにくいものであり、重大な医療需要を満たしていない多種の深刻な疾病の発病機序を推進した。われわれが開示した臨床前計画はSTAT 6とTYK 2に対して十分に検証された経路にあり,競合他社の療法に比べてわれわれの分解器技術が独自の優位性を提供する可能性があると信じている。この二つのプロジェクトは現在INDを支持する研究にある。
われわれのIRAK 4計画については,サイノフェイ社と協力し,腫瘍学や免疫腫瘍学分野以外のIRAK 4に対する候補薬を開発している。我々は,化膿性汗膜炎(HS),炎症性皮膚疾患およびアトピー性皮膚炎(AD),および潜在的な他の適応を含むインターロイキン1受容体/Toll様受容体またはIL−1 R/TLR駆動の免疫学的炎症条件および高度に満たされていない医療ニーズを治療するためのIRAK 4分解剤KT−474を開発している。健康ボランティアおよびHSおよびAD患者の列を含むKT−474の第1段階試験を完了した。サイノフェイによるKT−474の第二段階臨床試験は,HSとADにおける潜在力を初歩的に調査した。この2つの適応の臨床試験が開始され,患者用量が行われている。
われわれの臨床腫瘍学的計画については,侵襲性リンパ腫を含む再発/難治性液体と固体腫瘍患者に対する第一段階臨床試験においてSTAT 3分解剤KT−333を評価している。試験の1 a段階では患者登録と投与量が行われており,2024年にはより多くの臨床データが提供される予定である。2023年9月、FDAは、以前に孤児薬指定を得た適応である再発/難治性末梢T細胞リンパ腫の治療のためのKT-333高速チャネル指定を承認することを発表した。われわれのMDM 2分解剤KT−253の第一段階臨床試験は2023年3月に開始された。この研究は再発或いは難治性高度髄系悪性腫瘍、急性リンパ球性白血病或いはALL、リンパ腫と固形腫瘍の成人患者におけるKT-253増量用量の安全性、耐性、薬物動態/薬効学と臨床活性を評価している。試験の1 a段階で、患者登録と投与量は進行中であり、著者らは2023年11月に初歩的な安全性、メカニズム検証と概念検証データを提供した。私たちは2024年にもっと多くの臨床データを提供する予定だ。KT-253は2023年6月、FDAによって急性骨髄性白血病を治療する孤児薬として承認された。2023年11月、予想される分解レベルに達し、用量制限毒性が不足しているにもかかわらず、増加する免疫学的導管を集中的に支援するために、我々のKT-413(IRAKIMiD)計画の開発を中止することを決定した。
私たちの戦略
我々の使命は,重篤な疾患患者の生活を改善するために,新たな変革的な療法を発見,開発,商業化することである。我々は1種の独特な標的選択策略があり、標的蛋白分解(TPD)に焦点を当て、唯一或いは最適なロック解除薬物モードの未投与/不十分な投与の標的である。我々の随一のプロジェクトは,強力な遺伝学と臨床経路の有効性を有するタンパク質を対象としており,巨大な臨床とビジネス機会を有する分野にサービスしている。TPDは1種の疾病知らない技術であり、著者らは現在いくつかの疾病領域でこの方法を推進しており、主に免疫学と選定された腫瘍学の肝心な治療標的に集中している。我々の目標は,我々のTPD分野でのリーディング能力を利用して,完全に集積したバイオ製薬会社となり,一連の新しい分解剤薬を持つことである。
3
私たちは以下の戦略目標を達成することでこの目標を達成するつもりだ
標的タンパク質分解の背景
タンパク質は組織と器官の構造、機能、調節を担当する。人体内の細胞は絶えずタンパク質を合成と分解し、蛋白質動態バランスと呼ばれるバランスを維持している。多くの疾患は、タンパク質自体の活性化、変異または下方制御、または特定のタンパク質の転写および翻訳を担当する遺伝子によって駆動されるタンパク質挙動異常の結果である。各種の疾病分子に対する認識の深まりと人類全ゲノム特徴の確定に伴い、研究努力はますます薬物の開発に集中し、腫瘍、自己免疫、心臓代謝、神経退行性疾患と稀な遺伝病を招く故障蛋白を解決する。
“薬”のゲノム挑戦
近年,タンパク質活性異常の問題を解決するためのいくつかの治療法が開発されてきた。その中には、タンパク質機能の小分子阻害剤、治療用抗体、RNA干渉療法、アンチセンスオリゴヌクレオチドまたはASO、および他の遺伝療法などのオリゴヌクレオチドベースの療法が含まれる。
その中のいくつかのモードは疾病の治療と患者の生活の質に巨大な影響を与え、他のいくつかのモードは早期段階にあるが、潜在力を提供した。しかし、これらの伝統療法は特定の挑戦に直面し、それらの治療効果とカバー範囲を制限している。既存のモデルのいくつかの限界は、以下の点を含む
4
これらの制限から,これまでに完全ヒトゲノムの20%しか有効に投与されていないと考えられる。その中のいくつかの挑戦を克服できる新しい治療法は,投与されたプロテオーム/ゲノムの拡大や,それを必要とする患者に新たな有効な薬剤を提供するために必要である。TPDはこのようなモデルであると考えられる.
蛋白質標的分解
ユビキチン-プロテアソーム系(UPS)は細胞が新しいタンパク質の合成と分解と損傷および/または誤ったフォールディングタンパク質の間のバランスを制御するための方法の一つである。ユビキチンを介した蛋白質分解の発見は細胞分裂とDNA修復などの特定過程に重要な知見を提供し、そして各種の細胞経路におけるUPSの重要な作用を発見し、細胞周期、シグナル経路、遺伝子発現制御と酸化ストレスに対する反応を含む。UPSの発見はまた,この細胞過程を利用して疾患を治療する新しい方法を示している。
以下の図1に示すように,UPSは一連の丁寧に編成された酵素配列からなり,最終的にタンパク質の多ユビキチン化と細胞中のプロテアソームの分解を招く。タンパク質ユビキチン化はユビキチン活性化酵素(E 1)、ユビキチン結合酵素(E 2)とユビキチン蛋白リガーゼ(E 3)からなる下落酵素が関与する細胞過程である。ヒトには,2種類のユビキチン活性化E 1酵素,30種類以上のE 2酵素と約600種類のE 3リガーゼがある。
E 3/E 2/ユビキチンリガーゼ複合体(青色で示す)は基質タンパク質(オレンジ色で示す)に結合し、ユビキチンの転移を媒介し、それによって標的タンパク質のプロテアソームによる分解を引き起こす。
標的タンパク質分解はこの固有の細胞過程を利用した新しい方法である。TPDモードのコアは1つの小分子から構成されており、私たちは異二機能分解物と呼ばれる。この異二機能分解物分子の作用は病原蛋白とE 3リガーゼの間に1つの三元複合体を形成することによって、1種の“新しい”相互作用を調節することである。E 3は一連のユビキチンを連結することによって分解すべきタンパク質標的を標識し、プロテアソームは標識されたタンパク質を認識し、それを小ペプチドに分解する.
有効な三元錯体を形成することは,次の図ステップ2に示すように,TPDの重要なステップであり,その形成,機能,細胞と体内系への影響は分解の成功及び疾患への影響に重要である。また,分解物分子は様々な異なる細胞タイプや環境で分解でき,患者の治療に正確な薬物特性を持つ必要がある。
図ステップ3とステップ4に示すように,分解物が目的タンパク質のユビキチン化を促進した後,タンパク質がプロテアソームに分解されるにつれて,分子がタンパク質から分離され,別の三元複合体を形成して再び分解過程を行うことができる。この反復機構は触媒的であり,低い濃度でも効力を向上させることができ,これは小分子阻害剤や治療用抗体などの他の方法とのもう一つの重要な違いである。
5
図1
TPDの独特な利点により,この変革的な方式は従来小分子に投与されていないタンパク質を標的にすることができる。具体的には、TPDはステント蛋白や転写因子などの触媒機能のないタンパク質を標的とすることができ、小分子様の薬物特性を有し、潜在的に経口と系統分布が可能であり、RNAiなどのオリゴマーに基づく治療法ではない。TPD分子はまた既存の小分子製造原理に従っており、これらの原理は他の治療方法よりもコストが低い。分解過程の触媒的性質から,この方法は潜在的な治療効果があり,従来療法に比べて薬物物質の量が少なく,投与量が少ないと信じられている。
この20年間,小分子を用いてタンパク質の動態バランスに影響を与えることが多くの薬物によって臨床的かつ商業的に検証されてきた。Bortezomibやfulvestrantなどの薬物はそれぞれプロテアソームと標的エストロゲン受容体を抑制し,プロテアソーム依存の分解を実現することが知られている。最近、免疫調節イミン系薬物、例えばレナドアミンとポマ度アミンは、UPSを通じて一連の転写因子の分解を指導すると理解されている。
これらの免疫調節薬はUPSを用いてタンパク質を分解する概念を検証し,疾患環境中で薬理と治療効果を生じた。しかし、この領域の早期方法とは異なり、TPDはさらにこの概念を検証し、異二機能分解物を合理的に設計することによって、標的蛋白とE 3リガーゼの慎重な結合を調整することによって、必要な蛋白質分解を駆動し、前向きにより広範な蛋白質に対して分解を行う。
分解物効率の重要な要素はE 3標的化リガーゼに対する特異性と親和性である。異なるE 3リガーゼは異なる分布と細胞局在特徴を有しており,これはどのE 3リガーゼを特定の疾患蛋白標的に用いるかを考慮する際の重要な因子である。自然界には約600種類のE 3リガーゼがあるが,これまで少数のE 3リガーゼのみが治療目的に評価されており,標的に利用できるゲノムの大部分が残されている。
6
私たちの飛び馬座TMホーム.ホーム
我々の特許薬物発見プラットフォーム名はPegasusであり、細胞中のすべての目標クラスに対して薬物治療を行う潜在力のある標的タンパク質分解剤を合理的に設計することができる。著者らの方法はE 3ユビキチンリガーゼと標的蛋白との関係の理解に根ざし、これは私たちが標的をリガンドと分解可能な属性を決定することができ、そして多種の要素がどのように効力、選択性、薬物動態学(PK)と薬効学(PD)に影響するかを決定することができる。著者らはすでに広範な能力と知識を確立し、著者らの臨床前と臨床チャンネルの発展に貢献した。
著者らの独自の化学専門知識はE 3リガーゼと標的蛋白結合剤の設計と最適化に人工知能(AI)の洞察力を持たせ、それによって私たちに最適な薬用性能を有する標的蛋白分解器を設計する機会を与えた。また,我々のE 3リガーゼ全身プロファイルを用いて約600個のユニークなE 3リガーゼの発現プロファイルを含む。この画像を用いて,目的タンパク質を発現,分布,細胞内定位,生物学的に適切なE 3ユビキチンリガーゼにマッチングさせることができ,この過程は我々の機械学習に基づくアルゴリズムによって実現されている。体外および体内および健康および罹患組織および細胞タイプのPK/PDの理解に基づいて,目的蛋白レベルに影響を与える様々なパラメータを測定·予測する我々の定量系薬理学モデルを用い続けた。我々はまた,新たな組織制限性や選択性E 3リガーゼの同定に焦点を当て,従来のCereblon/IMiD相互作用を超えて,分子の設計が小分子相互作用により未薬物や未リガンドのタンパク質を標的とすることができるようにした。
私たちの治療管は
我々が準備しているプロジェクトには,臨床と臨床前開発の異なる段階の免疫学と腫瘍学プロジェクトがある。われわれの免疫学的項目はIRAK 4,STAT 6,TYK 2である。われわれの腫瘍パイプラインにはSTAT 3とMDM 2がある。我々はまだ開発の早い段階で複数の項目があるが,ここでは述べておらず,腫瘍学や免疫学以外の治療分野の目標を探索している。
以下の表は著者らが公開開示した臨床と最近の臨床段階パイプラインをまとめた。

図2
1 KT-474(SAR 444656)はセノフィと協力し、Kymeraは米国での開発と商業化に参加する権利があり、利益は半分である。2桁の等級別印税を連続して徴収する
2 現在の適応:HSとAD。IL-1 R/TLR経路が発症機序に関与しており、追加の潜在的な機会であることが示された他の疾患
3 STAT 3 I/I機会の評価を行っている。
7
われわれの免疫学的項目:IRAK 4,STAT 6,TYK 2
IRAK 4
IL−1 R/TLR駆動の免疫学的−炎症条件および高度に満たされていない医療ニーズを治療するための、IL−1 R/TLR駆動の免疫学的−炎症条件および高度に満たされていない医療ニーズを治療するためのKT−474、高活性かつ選択的な経口生物用IRAK 4分解剤を開発している。求められている初歩的な適応はHSとADを含む。我々がIRAK 4の分解を選択したのは,IL−1 R/TLR経路の免疫学的および炎症における作用がよく検証され,他の方法と比較して,多くの異なる炎症メディエーターの単一ノードに対する薬物治療が潜在的な優位性を有し,これらの方法はIRAK 4ノードを刺激する多くのサイトカインの1つに集中しているからである。IRAK 4はIL-1 R/TLRシグナル経路中の重要なノードであり、IRAK 4のSキナーゼ活性と足場機能に依存する。われわれは体外および体内研究により,KT−474がIRAK 4の分解を誘導し,キナーゼや足場機能に影響することを観察し,IL−1 R/TLRを介した炎症を有効かつ選択的に遮断することができ,IRAK 4キナーゼ阻害剤よりも優れていると考えられた。したがって,KT−474は現在の治療案や現在開発されている他の薬剤の結果を改善する可能性があると信じている。われわれはサイノフェイと協力し,腫瘍学や免疫腫瘍学分野以外のIRAK 4に対する候補薬を開発している。サイノフェイは最近KT−474をHSとAD患者に対する2つの第2段階臨床試験に進め,第1群の患者は2023年第4四半期の2つの試験で用量を得た。より多くの情報については、本年度報告の他の部分タイトルが“業務-連携-サイノフィとの連携プロトコル”となっている部分を参照されたい。
統計データ6
我々はSTAT 6に対する分解物を開発しており,STAT 6はIL−4/IL−13シグナル経路に特定される重要な転写因子であり,アレルギー性疾患における2型炎症の中心駆動因子でもある。STAT 6は遺伝子検証の標的であり、著者らはこの経路はすでに承認されたIL-4/IL-13標的生物製品を通じて臨床検証を行い、例えばdupilumabなどを信じている。臨床前研究において、著者らの一流の経口STAT 6分解剤KT-621はすべての関連するヒト細胞環境においてIL-4/IL-13経路に対する完全な抑制を示し、その強力な皮モル効力はdupilumabなどの経路生物製剤と類似或いは優れている。KT-621は多くの臨床前治療効果研究においても強い活性を示した。また,低経口投与ではKT−621は体内でSTAT 6をほぼ完全に分解し,多くの臨床前毒性研究で耐性が良好であった。KT−621は1日1回の経口小分子分解剤として開発されており,アトピー性皮膚炎,喘息,慢性閉塞性肺疾患,好酸性食道炎,慢性副鼻腔炎合併鼻ポリープなどを含む様々な疾患に対して広範な活性を有する可能性が信じられている。我々は2024年下半期に一期臨床試験を開始する予定である。
TYK 2
TYK 2に対する分解物を開発しており,TYK 2はI型インターフェロンやインターフェロン,IL−12やIL−12やIL−23シグナルに必要なJanus KinaseやJAKファミリーのメンバーである。TYK 2は遺伝子と臨床的に検証された自己免疫性と炎症性疾患の標的である。TYK 2は成熟した足場機能を有し,サイトカイン受容体表面の発現や活性化に重要な役割を果たしている。臨床前研究では,われわれ一流の経口TYK 2分解剤KT−294は,すべての関連するヒト細胞環境におけるピコ分子からナノモルへの潜在力を示しており,唯一のTYK 2標的であり,ヒト機能喪失生物学的潜在力,すなわちI型インターフェロン,IL−12,IL−23をほぼ完全に抑制する経路を有し,IL−10やIL−10も保持していると考えられる。TYK 2の分解は小分子阻害剤の挑戦を克服する可能性があり,小分子阻害剤は選択性が乏しく,標的結合が限られていること,および/またはI型インターフェロンに対する有効活性が乏しいため限界がある。KT−294は1日1回の経口小分子分解剤として開発されており,潜在的な生物製剤様活性特徴を有し,炎症性腸疾患,乾癬,乾癬関節炎,狼瘡などの疾患の治療が可能であると考えられている。私たちは2025年上半期に一期臨床試験を開始する予定だ。
私たちの腫瘍学プロジェクト:STAT 3とMDM 2
統計データ3
われわれは血液系悪性腫瘍や固形腫瘍の治療に選択的STAT 3分解剤を開発している。STAT 3が自己免疫疾患で分解する可能性も探索している。STAT 3は転写因子であり,複数の異なるサイトカインや増殖因子受容体を介してJAK,および癌遺伝子融合蛋白やSTAT 3自体の変異により活性化される。STAT 3の腫瘍生物学における異なる機能,腫瘍細胞の免疫監視からの逃避,炎症や線維化は様々な高需要疾患を解決する機会を提供していると信じている
8
適応は単一の遺伝子を標的とし臨床的に検証する方法である。JAK−STAT経路はいくつかの臨床的に成功したJAK標的薬で部分的に解決されているが,すべての関連細胞型のSTAT 3に広く影響を与える薬剤はないと考えられる。SH 2ドメインに対する小分子STAT 3ダイマー化阻害剤は開発されてきたが,第一に,すべてのSTATファミリーメンバーのSH 2ドメインの相同性がSTAT 3特異的な能力を得る能力に影響し,第二に,STAT 3の二量化独立転写活動を阻止できないという大きな挑戦がある。これらの理由から,STAT 3分解剤は標的と選択性薬物の開発に変革的な解決策を提供し,多様なSTAT 3依存の病理に対応する可能性が考えられる。われわれは現在,侵襲性リンパ腫を含む再発/難治性液体および固体腫瘍患者のためのSTAT 3分解剤KT−333を評価している。試験の1 a段階用量アップ部分では,患者登録と投与量が進行中であり,2024年により多くの臨床データを提供する予定である。
MDM 2
われわれはMDM 2を標的とした分解剤を開発しており,固形腫瘍や血液系悪性腫瘍の治療に用いられている。MDM 2は最もよく見られる腫瘍抑制遺伝子P 53の重要な調節因子であり、P 53は50%近くの癌において完全(或いは野生型)を維持する。小分子阻害剤と異なり、著者らのMDM 2分解剤KT−253は、MDM 2フィードバック回路を克服し、迅速にアポトーシスを誘導する能力があることが臨床前に証明されており、一時的な暴露下でも同様である。著者らは2023年5月にKT-253の一期臨床試験を開始し、成年液体と固体腫瘍患者におけるKT-253の安全性、耐性、PK/PDと臨床活性を評価することを目的とした。試験の1 a段階用量アップ部分では,患者登録と投与量が進行中であり,2024年により多くの臨床データを提供する予定である。
私たちの目標選択方法は
TPDの将来性を実現するために、著者らは1種の独特かつ差別化された方法を採用して標的を選択し、これにはいくつかの重要な原則があり、図3に示すように、著者らの研究と開発を指導した。著者らは未使用或いは十分に投与されていない標的、例えば転写因子と骨格蛋白に集中し、明確な臨床検証とヒト遺伝学/因果生物学による検証の経路内にある。著者らはTPDが最適或いは唯一の解決策の標的であることを確定し、強力な分解剤の理由と視線を持って、著者らの方法がこれらの経路における既存の薬物に対する優位性を示す。しかも、私たちは重大な患者ニーズと巨大なビジネス機会のある分野に集中している。

図3
9
臨床免疫学:IRAK 4
要約.要約
IL-1 R/TLR駆動の免疫炎症条件およびHS、ADおよび他の疾患を含む、IL-1 R/TLR駆動の免疫炎症条件および医療ニーズを高度に満たしていない疾患の治療のための高活性かつ選択的な経口生物用IRAK 4分解剤を開発している。我々がIRAK 4の分解を選択したのは,IL−1 R/TLR経路の免疫学的および炎症における作用がよく検証され,他の方法と比較して,多くの異なる炎症メディエーターの単一ノードに対する薬物治療が潜在的な優位性を有し,これらの方法はIRAK 4ノードを刺激する多くのサイトカインの1つに集中しているからである。IRAK 4はIL-1 R/TLRシグナル経路中の重要なノードであり、IRAK 4のSキナーゼ活性と足場機能に依存する。インビトロおよび体内研究では,KT−474がIRAK 4の分解を誘導し,キナーゼや足場機能に関与することが観察され,IL−1 R/TLRを介した炎症を選択的に遮断することができ,IRAK 4キナーゼ阻害剤よりも優れていると考えられた。したがって,KT−474は現在の治療案や現在開発されている他の薬剤の結果を改善する可能性があると信じている。KT-474の第一段階試験は健康ボランティア及びHSとAD患者を含み、2022年10月に完成した。われわれはサイノフェイと協力し,腫瘍学や免疫腫瘍学分野以外のIRAK 4に対する候補薬を開発している。サイノフェイはKT−474をHSとAD患者の第2段階臨床試験に進めており,いずれも2023年第4四半期に開始された。より多くの情報については、本年度報告の他の部分タイトルが“業務-連携-サイノフィとの連携プロトコル”となっている部分を参照されたい。
生物学と作用機序
IRAK 4はMyddosomeの重要な構成部分であり、myddosomeは先天性免疫に参与する多蛋白複合体であり、TLRsとIL-1 Rsを介してシグナル伝達を媒介する。IRAK 4蛋白は皮膚、リンパ組織、骨髄、胃腸と肺を含む多種の異なる組織タイプに広く発現している。
IRAK 4の機能はそのキナーゼ活性と足場機能に依存し,TLRやIL−1 R関与とMYD 88活性化後にMyddosome複合体を組み立てるために必要である。キナーゼ機能は主にIRAK 4-JNK軸のリン酸化イベントを担当するが、ステント機能は主に核因子-KBの活性化といくつかの重要な炎症性サイトカインとケモカインの下流遺伝子牽引を担当する。
IRAK 4の分解はIRAK 4キナーゼの抑制よりも優れていると考えられ,われわれの臨床前データは,IRAK 4蛋白のキナーゼ活性や足場機能を遮断することが重要であることが示唆されているため,抑制だけでなくこの蛋白を除去する必要がある。IL-1ファミリーサイトカインは、IL-1 a、IL-1?、IL-18、IL-36とIL-33を含み、多種の異なる免疫炎症状態と疾患と関連している。TLRsとIL-1 RsはすべてこれらのIL-1ファミリーサイトカインの産生と応答に参与しているため、単一の小分子分解物を標的とするIRAK 4は多種の異なるサイトカインとケモカインに作用することができ、それによってIL-1 R/TLR駆動の疾病の治療に変革性の方法を提供した。

図4
10
発展のチャンス
米国,ヨーロッパ,日本では1.5億人以上がTH 1による疾患を有していると推定されている。多くの皮膚、リウマチと胃腸免疫炎症疾患の適応があり、その発病機序はIL-1ファミリーサイトカインとTLR刺激に関連する。効率的かつ選択的なIRAK 4分解剤は現在承認されている治療方案と臨床開発方案よりも顕著な優位性を提供すると信じている。われわれは最初にHSとADを優先したが,これは自己免疫性皮膚疾患であり,臨床的にはIL−1 R/TLR経路の影響を受けるサイトカインを標的にすることができるという概念があるが,これらの疾患に対する需要は依然として高い。多くの他の疾患において、IL-1 R/TLR経路は発病機序に関連し、呼吸器系、胃腸疾患、リューマチに対する追加の潜在的な機会になる可能性がある。
化膿性汗腺炎
HSは慢性、破壊性、痛みと虚弱な炎症性皮膚病であり、アメリカと全世界の人口の1%に影響している。HS患者は多くの痛み、ドレナージの結節と膿瘍があり、通常皮膚のしわ内にあり、炎症と細菌定植を特徴とする。現在HSは対症療法を行い、コルチコステロイド、抗生物質と手術を使用している。FDAによって承認されたHSを治療する唯一の薬剤は、抗腫瘍壊死因子抗体アダモモノクロナル抗体であり、約50%の中~重度疾患の患者にいくつかの利点を提供するが、治癒するものではない。したがって,HS治療に対するより良い療法の需要は依然として高く,満足されていない。
TLRsに対する細菌の活性化,および角質形成細胞や炎症細胞はIL−1 a,IL−1?とIL−36を産生し,高レベルの腫瘍壊死因子−a,IL−6とIL−17を特徴とする炎症を招き,HSの発症機序に中心的な役割を果たしている。IL-1 a(Bermekimab)、IL-1 a/?受容体(Anakinra)とIL-17(secukinumabとbimekizumab)などの単一サイトカインに対する単一抗体は、すでにHSにおいて初歩的な臨床活性を示し、HSにおけるIL-1 R/TLR経路を標的とするために臨床検証を提供した。したがって,多くのサイトカインやTLRに作用するIRAK 4分解剤は,現在開発されている単一サイトカイン標的化試薬よりも大きな利点を提供する可能性がある。
アトピー性皮膚炎
ADは慢性掻痒性炎症性皮膚病であり、児童に最もよく発生するが、成人にも影響を与える。全世界の主要な市場において、ADの確定診断罹患率は6000万を超え、その中の約40%、即ち2400万人は中から重度の分類に属する。ADは数ヶ月から数年の慢性再発過程を持続し、皮膚の乾燥と深刻な掻痒を主な症状とし、慢性掻爬や裂傷による皮膚肥厚を伴うこともある。ADの対症治療は局所治療を採用し、皮膚形成剤、コルチコステロイドとホスホジエステラーゼ阻害剤を含む。FDAが承認した主要な系治療薬はIL−4 Ra標的抗体DUPILUMAであり,その3期試験では約40%の中から重度疾患患者のみが主要な終点に達しているにもかかわらず,かなりの割合を残した患者は現在十分なサービスを得られていない。
また,IL−18とIL−1はいずれもADや他の自己免疫性や炎症性疾患の炎症の産生に関与しており,好酸喘息や慢性副鼻腔炎を含む証拠がある。IL-18(GSK 1070806)とIL-1 a(Bermekimab)に対するモノクロナル抗体はすでにADにおいて初歩的な臨床活性を示したが、抗IL-1 a/bのモノクロナル抗体(Lutikizumab)はHSにおいて初歩的な臨床活性を示した。したがって,IRAK 4分解物はTLRシグナルを完全に遮断することによりIL−18やIL−1の産生に関与する能力と,IL−1 Rシグナル経路を完全に遮断することによる両サイトカインの細胞反応が,ADやHSおよび他の自己免疫性や炎症性疾患の進展に納得できる機序基盤を提供していると考えられる。
臨床研究とデータ
2021年12月、私たちは健康ボランティアにおいて、KT-474段階1試験の単一上昇用量またはSADおよび複数の上昇用量またはMAD部分の用量増加を達成した。この試験は105名の健康ボランティアの安全性、耐性と薬物動態学を評価した。SAD部分は25~1600 mgの単回用量で構成されている。MAD部分は50 mgから200 mgまで14日間連続投与する漸増用量を含む。試験した健康ボランティア部分のハイライトは、IRAK 4の単回と複数回の毎日用量での強力(>95%)と持続分解(図5)、および体外TLRを介したサイトカイン誘導の広範な抑制を含む(図6)。KT−474はすべての用量群において全体的な耐性が良好であった。
11

図5

図6
試験された健康ボランティア部分の後、HSおよびAD患者を含むKT-474段階1試験の患者列またはC部分の用量を決定するための単回用量食品効果行列を完成させ、この試験は2022年10月に完了した。HSとAD患者列はHSとAD患者21名で行われたオープンタグ研究である。これらの患者は毎日75 mgを食物とともに服用しており,われわれのSAD/MAD試験における投与量の一つである100 mg絶食に相当すると推定される。患者列投与量は75 mgで28日間連続服用した。
KT−474血漿PKは1日1回75 mg,または患者のqd用量(給餌状態)は第1段階臨床試験MAD部分の健常ボランティアに相当し,後者は絶食状態で1日1回100 mgを受け,MAD 3と呼ぶ。また,C部分は定常状態の平均Cmax(6時間用量後濃度)とC(用量前濃度)レベルは14日目のMAD 3レベルと一致し,44時間の平均半減期はMAD(34−59時間)の観察範囲内であった。さらに、血漿中のKT−474濃度は、同様のレベルのIRAK 4をもたらす
12
健康ボランティアとHS/AD患者の分解性。具体的には,濃度が3 ng/mlより高い場合,両個体群とも通常80%以上の分解率であった。また,評価試料の患者のPBMCでは,IRAK 4レベルは28日目に定量下限に近づいていた。
KT−474は,評価可能なHSとAD患者の皮膚に高皮膚曝露を示し,以下の図7に示す。

図7
そのほか、評価可能なHSとAD患者の皮膚損傷中のベースラインIRAK 4レベルは健康ボランティアの約2倍である。投与28日目までに,ADとHS患者の皮膚損傷中の平均IRAK 4レベルは健常被験者とほぼ同じレベルまで低下し,図8に示すようになった。

図8
KT−474は一般的に耐性が良好であった。重篤な有害事象は認められず,薬物関連感染もなく,中止や中止をきたす有害事象も認められなかった。患者群でも健常ボランティア研究のMAD部分で観察された7日目と一致した温和で非不利なQTc値の延長が認められたが,28日間の投与期間で服薬を継続したところ,QTc値は自発的にベースラインレベルに回復した。
13
KT-474がHSとAD患者に対して全身性抗炎症作用を有するかどうかを確定するため、それぞれ治療前、治療期間と治療後の異なる時間に血漿IL-6、CRP、SAAとIL-1 bレベルを測定した。ベースラインレベルが正常上限より高い患者では,評価可能な患者はすべて4種類の分析物の抑制を示し,42日目の平均最大低下幅は41%から63%と様々であった。患者列はまた、血液と皮膚中の全身性IRAK 4の分解がAD或いはHSに関連することが知られている炎症促進遺伝子の発現にどのように影響するかを評価した。ADで影響を受ける遺伝子には,Th 2サイトカインIL−5,炎症体NLRP 3,CXCL 1とIL−2 Rbがある。HSに影響を与える遺伝子はIL-1ファミリーサイトカインIL-1とIL-36 A、Th 1炎症メディエーター、例えばインターフェロン-gとGZMB、Th 17サイトカインIL-17 A、および天然免疫駆動因子IL-8とCSF 3を含む。この2つの疾患では,多くの遺伝子が90%以上抑制されており,引き下げ幅が大きい。

図9
倍変化:−1=50%減少,−2=75%減少,−3=87.5%減少。
C部にはHSとADのための探索的臨床終点がある。これらの終点を選択してKT-474治療が皮膚病負担及びHSとAD患者の生活の質に影響する疼痛と掻痒などの症状への影響を評価した。AD患者の中で、図10に示すように、湿疹面積と重症度指数(EASI)採点を用いて測定し、皮膚皮膚損傷は平均37%減少し、個別患者の減少は76%に達した。最大下げ幅は28日目に現れ、42日目に維持された。

[図10]
14
図11に示すように,AD患者の過去1週間または過去24時間の平均最大掻痒回数はそれぞれ52%と63%減少し,最大減少は42日目に発生した。ピーク掻痒反応は,過去1週間または過去24時間以内にピーク掻痒の4単位減少と定義され,それぞれ57%と71%のAD患者に出現し,28日目以降まで反応が持続した。

図11
AD患者7名のうち,2名の患者の重症度の有効調査者は全世界評価(VIG)が改善し,残りの患者は42日目まで安定していた。
HS患者では,すべての患者が非常に重篤な疾患を有する2名の患者を含む治療効果分析を行った。また,中から重度疾患のみのHS患者の奏効率分析も行われており,これらの患者は本研究の目標群である。全HS患者においてANカウントは平均46%減少し,中から重度患者では平均51%,個別患者では100%減少し,最大減少は42日目に発生した。すべてのHS患者において,28日目に0,1または2カウントに達した患者の割合は42%,中から重度疾患患者では50%であった。HiSCR 50反応は、計数が50%以上減少し、膿瘍またはドレナージ性瘻管が増加しないと定義される。図12に示すように,42日目のHiSCR 50応答者の割合は全HS患者で42%,中から重度疾患患者で50%であった。

[図12]
15
HiSCR 75反応は,計数が75%以上減少し,全HS患者で25%,中から重度疾患の患者で30%と定義されている。
痛みやかゆみの症状も測定された。図13に示すように,すべてのHS患者と中重度疾患を有するHS患者における疼痛デジタル分級尺度(NRS)の平均低下幅はそれぞれ49%から55%であり,最大低下は28日目から42日目の間に発生した。疼痛NRS 30反応は、疼痛NRSがベースラインから少なくとも30%および少なくとも1単位減少すると定義される。図13に示すように,すべてのHS患者において疼痛NRS応答率は50%であり,それらの中から重度疾患までのHS患者では疼痛NRS応答率は60%と28日目まで持続している。

[図13]
また,全HS患者のかゆみピークは平均62%,中重度疾患を有するHS患者は68%減少し,全HS患者の最大掻痒減少期間は42日目,中等度から重度疾患を有するHS患者の最大減少期間は28日目であった。また,HS患者5名のうち,医師の疾患重症度のグローバル評価は改善し,そのうち1名の中等度疾患患者の病態はベースラインレベルで除去されたが,他の評価可能な患者では42日目まで安定していた。
臨床発展計画
2023年第4四半期、第1回目の患者はHSおよびADのKT-474第2段階臨床試験で服用した。
HSの第二段階臨床試験、或いはZENは、二重盲検、プラセボ対照の両腕ランダム試験であり、KT-474経口錠剤或いはプラセボから構成され、毎日一回である。主な結果の測定指標は、総膿瘍と炎症性結節(AN)計数のベースラインよりのパーセンテージ変化である。選択された副次的結果指標は、HISCR 50に達した患者の割合、2;HIS 4とベースラインの絶対的な変化;Hurley分期が改善した患者の割合AN 50;報告された毎日の最も深刻な痛みHS-皮膚疼痛-NRSとベースラインの変化;およびHS-皮膚疼痛-NRSを使用する参加者の毎日の最も深刻な痛みは少なくとも30%および少なくとも1単位の割合を減少させた。
ADの第二段階臨床試験、或いはAdvantaと呼ばれ、二重盲検、プラセボ対照の三群のランダム試験であり、KT-474用量1経口錠剤、KT-474用量2経口錠剤或いはプラセボから構成され、毎日一回である。主な結果測定基準はEASIにおけるベースラインの百分率変化である.選択された副次的結果は、VIGA−ADが0または1であり、ベースラインよりも2点低下した参加者の割合、−50 EASI−75−90に達した参加者の割合、基線より毎週1日平均PP−NRSが4点減少した参加者の割合、1日あたりPP−NRSの週平均とベースラインとの百分率変化、および1日あたりPP−NRSの週平均とベースラインとの絶対的な変化を含む。
行われている2つの試験の背線データは2025年上半期に発表される予定だ。また,KymeraとSanofiはHSやAD以外に適応を拡張する機会を評価している。
16
臨床前免疫学:STAT 6とTYK 2
統計データ6
要約.要約
我々はSTAT 6に対する分解物を開発しており,STAT 6はIL−4/IL−13シグナル経路に特定される重要な転写因子であり,アレルギー性疾患における2型炎症の中心駆動因子でもある。STAT 6は遺伝子検証の標的であり、この経路はすでに承認されたIL-4/IL-13標的生物製品を通じてdupilumabを含む臨床検証を行っている。
生物学と作用機序
STAT 6はIL−4と13サイトカインシグナル伝達に必要な特異的転写因子である。IL−4受容体は2つのタイプがあり、I型はIL−4受容体αおよびγCからなり、II型はIL−4受容体αおよびIL−13受容体α1からなる。IL−4はI型受容体およびII型受容体の両方を介しており、IL−13シグナルはII型のみを通過する。IL−4あるいはIL−13が受容体に結合すると,下流で活性化したJAKキナーゼがリン酸化しSTAT 6を活性化し,アレルギー性TH 2炎症を引き起こす。

図14
IL−4とIL−13シグナルに対する生物製剤の臨床効果は,STAT 6調節サイトカインがアレルギー性疾患の臨床検証標的であることを証明している。JAK阻害剤とは異なり,STAT 6はIL−4と13種類のサイトカインによって特異的に活性化され,IL−4と13シグナル伝達を選択的に遮断する手段としてSTAT 6を支持している。STAT 6の発病作用もヒト遺伝学的支持を得ており、STAT 6機能突然変異の獲得は深刻な早発性アレルギー反応性疾患を引き起こすことが示唆された。また,マウスではSTAT 6ノックアウトは多様なアレルギー性疾患モデルにおいて保護作用を有し,これらのマウスは発育が正常であり,活力と繁殖力を有している。
発展のチャンス
米国,ヨーロッパ,日本では1.5億人以上がTH 2による疾患を有していると推定されている。既存の経路療法では,アトピー性皮膚炎,結節性痒疹,喘息,慢性閉塞性肺疾患,慢性副鼻腔炎合併鼻ポリープ,好酸性食道炎など,多くの疾患の有効性が確認されている。
STAT 6の分解は生物製品に相当する包括的な経路抑制を示す可能性があるが,利点は簡単で毎日の口腔画像であると考えられる。多くの患者の経口レジメンに対する選好は、現在注射可能な生物製剤よりも多くの全世界の患者に接触することができ、TH 2疾患の治療モデルを潜在的に変化させることができるかもしれない。
17
臨床前研究とデータ
我々の主要STAT 6分解剤KT−621は非常に有効なSTAT 6分解剤である。図15に示すように、KT-621は研究されたすべての疾患に関連するヒト初代細胞タイプにおいて皮モル分解の潜在力を示し、KT-621をKymeraで設計とテストした最も有効な異二機能分解剤の一つにした。STAT 6の分解はすべてのTH 2疾患に関連する造血細胞,皮膚と呼吸適応に関与する上皮細胞,角質形成細胞と肺上皮細胞を含む,肺と食道由来の平滑筋細胞,呼吸器や胃腸の適応,および血管内皮細胞がすべてのTH 2疾患の炎症細胞浸透に関与していることが分かった。

[図15]
図16に造血細胞と組織細胞の用量依存性分解を示した。

図16
18
STAT 6を含むすべてのプログラムの分解選択性曲線を質量分析計を用いて測定した。下の図はKT-621分解選択性を描いた火山図である。図に示すように,KT−621 DC 90の100倍の濃度でSTAT 6はKT−621が質量分析計により検出された約10,000種類のタンパク質の中で唯一分解されたタンパク質である。具体的には,他のSTAT蛋白は分解されず,KT−621は非常に高い分解選択性を有することが示唆された。

図17
サイトカイン分析では,以下の表とグラフに示すように,KT−621の他のすべてのSTAT蛋白に対する機能選択性を測定した。観察されたプロテオミクス選択性と一致して,KT−621はSTAT 6の機能のみを抑制し,他のSTATタンパク質に影響を与えず,平坦な用量反応曲線に示すように,KT−621の高機能選択性をさらに証明した。

図18
TH 2機能検出においてもKT−621を評価し、IL−4および13シグナルへの影響を評価した。特に,IL−4と13誘導ヒトPBMCにおけるTARCの放出,IL−4と13誘導ヒトCD 19 B細胞のCD 23発現(B細胞活性化マーカーであり,IgE変換に関与している),IL−13誘導ヒト気管支および食道平滑筋細胞のPeriostin放出の解析を検出した。我々は,いずれもTH 2疾患の臨床的に成熟したバイオマーカーであるため,PDのバイオマーカーとしてTARC,IgE,Periostinを選択した。KT-621を比較しました
19
デュピロマブで通路を遮断する。以下の図19に示すように,われわれの臨床前試験では,KT−621はヒトTH 2機能検出でIL−4/IL−13経路を完全に遮断し,IC 50‘Sはデュピマブより低かった。

[図19]
また,KT−621の体内での分解レベルを評価することによりKT−621を評価した。われわれの研究では,KT−621はマウス,ラット,イヌ,非ヒト霊長類を含む多くの臨床前種でSTAT 6を強く分解している。具体的には、KT−621は、低経口用量でSTAT 6の用量依存性深さ分解を達成することができる。図20では,犬体内でのKT−621‘Sの分解を示し,用量依存的な分解を示している。約1~3 MPKの用量が枯渇時のSTAT 6の最大分解をもたらす0.2~12.8 MPKの範囲を測定した。また,われわれの研究では,KT−621は迅速な分解開始を示し,特に単回経口投与後数時間以内であった。これらの結果、および私たちが完成した他のPK/PD研究は、KT-621がヒトにおいて低用量および開発可能な有効用量の潜在力を有することを示している。

図20
20
非ヒト霊長類動物のキー疾患関連組織におけるSTAT 6の分解も評価した。下図21に示すように,KT−621は10 MPK用量で14日間連続し,血液,脾,皮膚,肺を含む重要疾患関連組織でSTAT 6を分解した。

図21
上記の分解は、私たちの用量範囲内の最低用量レベルの用量発見安全性研究を表す。その研究では,300 MPKまでの用量を14日間連続して服用し,有効濃度の40倍以上の濃度であった。著者らが研究したすべての用量レベルで、最高用量レベルを含み、KT-621耐性は良好であり、有害事象或いは関連発見はなかった。
皮膚と肺の前臨床モデルでは体内にあるKT−621の活性。まず,低カルシウムビタミンD 3類似体MC 903を局所応用して誘導した2週間のアトピー性皮膚炎モデルにおけるKT−621の役割を評価した。このモデルはDupiumabに対する反応を許容し,臨床前の活性を比較できるようにIL−4/IL−4 Rαヒト化マウスを用いた。KT−621は2,8と32 MPKで経口投与し,1日1回,11日間連続した。これらの投与量はそれぞれ約70%、80%と90%の脾臓分解を招き、図22に示す。この臨床前研究では,DUPILUMA皮下注射は週2回,毎回25 MPKであり,このレベルではIL−4 Rαの飽和を確保し,IL−4/13を完全に遮断することが予想される。人間ではこの投与量は1週間ごとに300ミリグラムに相当すると推定されていますこれは人間が承認した最高用量プログラムです

[図22]
21
図23に示すように,PO賦形剤とSubQ IgG 4対照群の血清総IgEはNO MC 903群と比較して上昇し,著明なTH 2炎症を示した。逆に,この臨床前研究では,KT−621は用量依存的にIgEの上昇を強力に抑制し,dupiumabに相当するレベルにした。STAT 6分解の90%をもたらすKT−621用量は,この臨床前モデルにおいてIL−4 Rα飽和用量に類似したドゥピロマブ活性を産生し,KT−621がIL−4とIL−13を完全に遮断していることが示唆されたことに注意されたい体内にある.

図23
*PO Vehicle(MC 903)に対する意味;#SC IGG 4 25 MPK BIWへの意味
TH 2炎症に最も関与する臨床前モデルと考えられるKT−621の1カ月の肺炎症モデルにおける役割を評価した。卵白蛋白とは異なり,卵白蛋白は通常マウスモデルに用いられているが,ヒト体内で気道炎症を引き起こすことはないが,ダニは現実世界のアレルゲンであり,ヒトで喘息を引き起こすことができる。鼻腔HDMモデルはTH 2炎症を主とし,DUPILUMAの臨床前開発に用いられている。前述の皮膚モデルと同様にIL 4/IL 4 Rαヒト化マウスを用いた。KT−621は同じ3用量レベル,すなわち2,8と32 MPKで1日1回経口投与し,31日間連続し,脾の分解率はそれぞれ72%,85%と91%であり,従来の皮膚モデルと一致した。DUPILUMAは25 MPKの速度で9回皮下注射し、週2回、IL-4 Rαの飽和を確保し、IL-4/IL-13を完全に遮断することを目的とし、ヒトが1週間ごとに許可した最高用量300 mgの用量と類似している。
図24に示すように,主なTH 2炎症は血清中のIgEの著明な上昇を示し,気管支肺胞洗浄液中のTARCとPeriostinの放出,および肺内への好酸球の集積を示している。これらはいずれもTH 2バイオマーカーとして公認されている。KT-621はすべての3つの用量レベルで強い抑制作用を示した。この前臨床研究ではKT−621はTH 2炎症を抑制した体内にあるこれらすべてのTH 2指標について,鼻腔内HDM喘息モデルでは,IL−4 RTH 2飽和用量のデュピロマブはIL−4 Rαと同等かそれ以上であった。
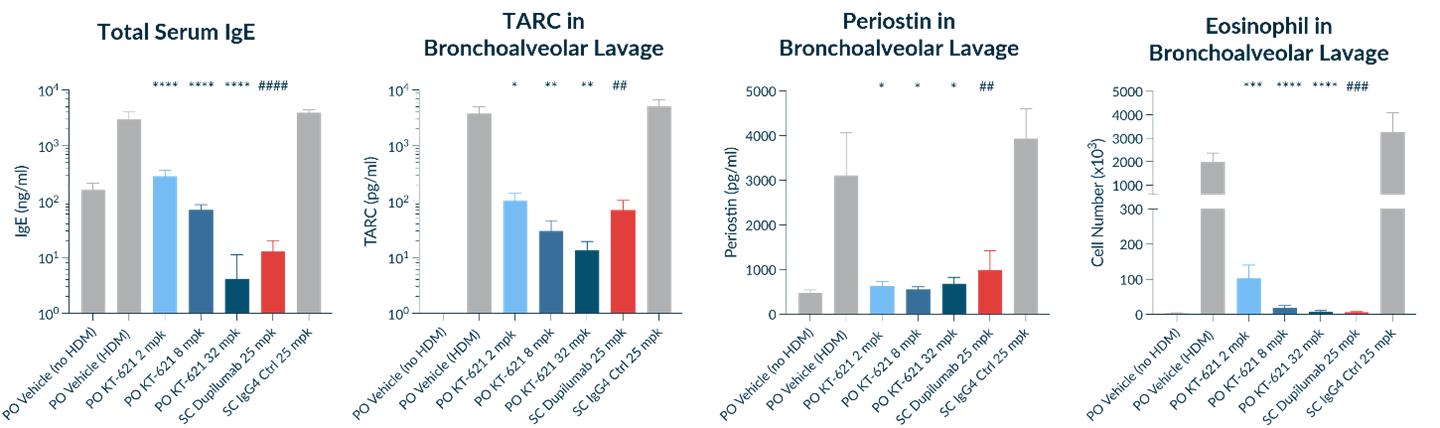
[図24]
*PO Vehicle(HDM)に対する意味;#SC IGG 4 Ctrl 25 MPKへの意味
22
臨床発展計画
我々の主要STAT 6分解剤KT−621は現在IND作動研究を行っている。2024年下半期に一期臨床試験を開始し,2025年に一期結果を報告する予定である。
TYK 2
要約.要約
我々はTYK 2の効率的かつ選択的低下物を開発しており,TYK 2はI型インターフェロンに必要なJAKファミリーのメンバー,あるいはインターフェロン,IL−12やIL−23シグナル伝達であり,自己免疫や炎症性疾患において遺伝的かつ臨床的に検証されている。
生物学と作用機序
図25に示すように,TYK 2はJAKファミリーの一員であり,IL−12,IL−23およびI型インターフェロン受容体に結合し,シグナル伝達や転写活性化(STAT)転写因子を再募集しリン酸化する。また,TYK 2は成熟した足場機能を有し,サイトカイン受容体表面の発現や活性化に重要な役割を果たしている。機能欠損変異体は自己免疫疾患において保護作用を有し、TYK 2のアロステリック阻害剤(Deucravitnib)およびIL-12、IL-23およびインターフェロン-αに対する複数の生物学的製剤は、複数の自己免疫疾患の治療に承認され、TYK 2を高度に有効な標的とする。

[図25]
TYK 2の分解は,タンパク質を完全に除去することでヒトノックアウト生物学を完全に要約することができ,選択性の欠如,限られた標的結合,および/またはI型インターフェロンに対する有効活性の欠如により限界がある小分子阻害剤の挑戦を克服する可能性がある。したがって,TYK 2分解者はI型インターフェロン,IL−12,IL−23の全経路抑制を実現し,潜在的生物活性を有する経口錠剤中のIL−10を1日1回保持している可能性がある。
発展のチャンス
米国,ヨーロッパ,日本では2000万人以上がI型インターフェロンとIL−12/IL−23を介した疾患を有していると推定されている。皮膚科,胃腸科,リウマチ科,中枢神経系など多くの免疫治療領域で適応の機会が多い。経路生物製品やTYK 2小分子阻害剤は,TYK 2が複数の適応にまたがる有効な潜在力を支持している。TYK 2の分解は抑制とは異なり,生物製品に相当する網羅的な経路抑制を示す可能性が考えられるが,毎日の経口プロファイルのおかげであると考えられる。
23
臨床前研究とデータ
我々は,効率的かつ選択的なTYK 2分解剤KT−294を開発した。図26において,我々のプロテオミクスデータは,KT−294は高選択性マイクログラムTYK 2分解物であり,TYK 2ヒト欠乏の生物学的特性を要約し,IL−10/IL−22を含まずI型インターフェロンおよびIL−12/23シグナルを完全に抑制していることを示している。特に,KT−294はJAKファミリーメンバーに対して極めて高い選択性を示した。

図26
図27では,ヒトPBMCと角質形成細胞の皮モル分解を実現し,一連の細胞型でわれわれの機能解析を低NM抑制した。著者らはPBMC中のIL-23とIL-12シグナル経路に対して非常に強い臨床前治療効果があり、PBMC中のI型インターフェロン経路に対して非常に強い抑制作用があり、特異性B細胞測定においても非常に強い抑制作用がある。IL−10とIL−22経路は完全に回避されていることが証明された。

図27
24
IL−10は腸管動態バランスにおいて重要な役割を果たしており,上皮修復や粘膜癒合など,炎症性腸疾患やIBDなどの疾患において非常に重要である。事実,IL−10機能変異の喪失はヒト早発性難治性大腸炎を引き起こす。図28に,細胞にTYK 2が存在しない場合にIL−10でpSTAT 3を誘導できることを示すウエスタンブロットを示し,TYK 2がIL−10シグナルに必要でないことを示した。TYK 2小分子阻害剤であるdeucravisitinibは,TYK 2を介するのではなくJAK 1を抑制するため,この経路のシグナル伝達を完全に遮断することが示唆された。

図28
図29において,IL−10を用いて単球にリン酸化STAT 3を誘導する実験があり,この場合,IL−10経路を抑制しないため,我々の分解物はpSTAT 3に影響を与えなかった。しかし、それとは逆に、デクラビチニブは、オレンジ色のデータ点の減少が示すように、IL-10信号を効果的に抑制する。

図29
同様に、図30において、IL-10を使用してエンドトキシン誘導性腫瘍壊死因子-αを阻害する。KT−294はIL−10がエンドトキシン誘導性腫瘍壊死因子−αの放出を抑制することには影響しないが,ノルビチニブはIL−10‘がエンドトキシン誘導性腫瘍壊死因子−αの放出を抑制し,腫瘍壊死因子−αを上昇させることを抑制し,オレンジデータ点でこの作用を示した。
25
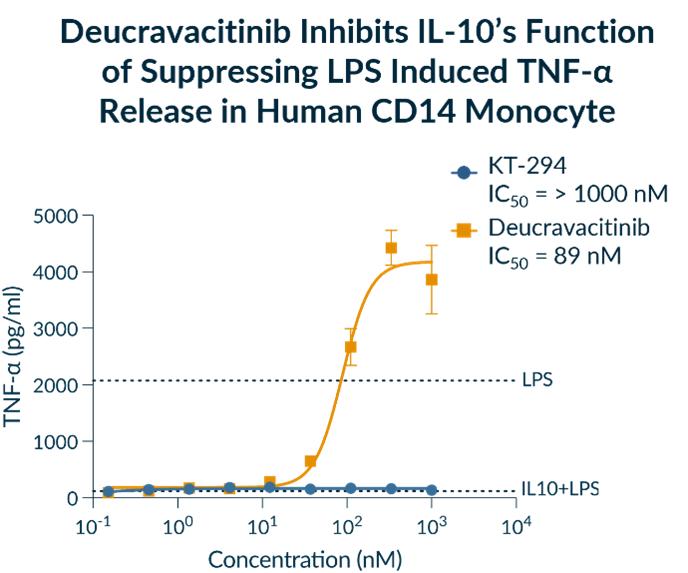
図30
また,KT−294をTAK−279と比較したところ,TAK−279は研究中のTYK 2阻害剤であり,JAKよりも選択的であったため,IL−10を省略した。我々の実験では,TYK 2を完全に占有する予定のTAK−279濃度とTYK 2を完全に分解する予定のKT−294濃度を用いて,両化合物間に生物学的差があるかどうかを評価した。これらの濃度はインターフェロンαpSTAT 2で検出されたIC 95値から決定され,この検出は最も抑制が困難なTYK 2関連サイトカイン検出である。RNAseqデータはKT-294がインターフェロン経路署名遺伝子に対してもっと良い抑制作用を有することを示し、図31中の21個の遺伝子署名点数に示すように。図32に示すように,先天性免疫経路の違いが観察された。これはKT−294がタンパク質,およびすべての可能な足場機能を完全に除去し,表現型複製ヒト遺伝子ノックアウトのためと考えられる。臨床結果によると,TAK−279は繰り返し投与量35 mgの場合,臨床曝露は77 nm以下であり,TAK−279段階3,30 mg用量に近い。臨床関連曝露では,TAK−279は図31のKT−294に示す経路抑制レベルに達しない可能性が高いと考えられる。

図31
26

[図32]
KT-294は、インビボでのTYK 2の用量依存性深分解を低用量で達成することができる。具体的には,非ヒト霊長類動物やNHPでは,1日低用量経口投与を繰り返し,KT−294は用量応答的にTYK 2を分解し,TYK 2の完全分解を達成し,図33に示すように薬理標的への経路を提供する。
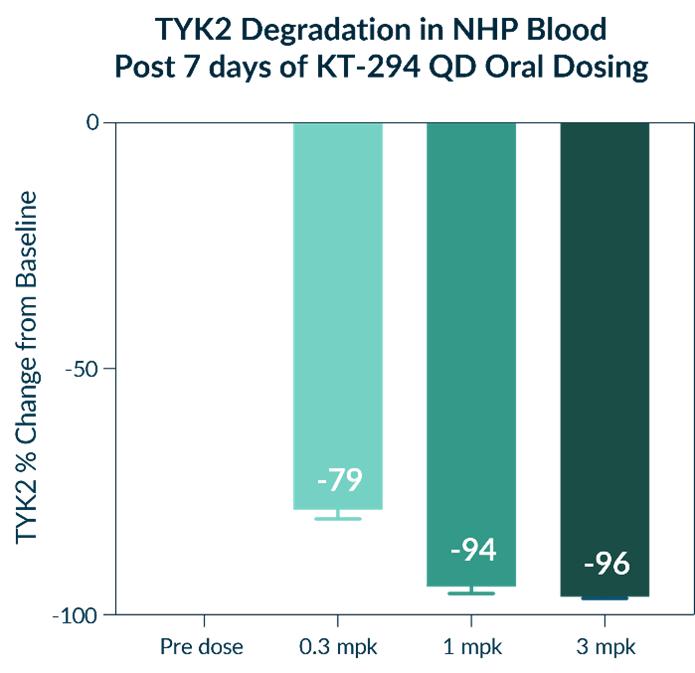
[図33]
臨床発展計画
2025年上半期にTYK 2分解剤KT−294の第1段階臨床試験を開始し,2025年にデータを報告する予定である。
27
臨床腫瘍学:STAT 3とMDM 2
統計データ3
要約.要約
われわれは血液系悪性腫瘍や固形腫瘍の治療に選択的STAT 3分解剤を開発している。STAT 3が自己免疫疾患で分解する可能性も探索している。STAT 3は転写因子であり、JAKsによって多種の異なるサイトカインと増殖因子受容体を活性化し、癌遺伝子融合蛋白とSTAT 3自体の突然変異によって活性化される。STAT 3の腫瘍生物学的,腫瘍細胞からの免疫監視および炎症と線維化の異なる機能は,単一の遺伝子や臨床検証の経路を標的とすることにより,様々な需要の高い疾患適応を解決する機会を提供していると信じている。JAK−STAT経路はいくつかの臨床的に成功したJAK標的薬で部分的に解決されているが,すべての関連細胞型のSTAT 3に広く影響を与える薬剤はないと考えられる。SH 2ドメインに対する小分子STAT 3ダイマー阻害剤はすでに開発されているが,大きな課題がある。すべてのSTATファミリーメンバーのSH 2ドメインの相同性がSTAT 3特異性を得る能力と,STAT 3の二量化独立転写活動を阻止できないことに影響している。これらの理由から,STAT 3分解剤は標的と選択性薬物の開発に変革的な解決策を提供し,多様なSTAT 3依存の病理に対応する可能性が考えられる。われわれは現在,侵襲性リンパ腫を含む再発/難治性液体および固体腫瘍患者のためのSTAT 3分解剤KT−333を評価している。試験の1 a段階では患者登録と投与量が行われており,2024年にはより多くの臨床データが提供される予定である。
生物学と作用機序
STAT 3は転写因子であり,STAT蛋白ファミリーの一員である。サイトカインや増殖因子によりSTAT 3は受容体に関連するセリン/トレオニンキナーゼでリン酸化され,STAT 3やp−STAT 3をリン酸化し,二量体を形成し,核内に転位し,DNAに結合し,腫瘍発生,炎症,線維化に関与する各種遺伝子の転写を調節する。STAT 3は多くの癌の中でよく突然変異と活性化が発生し、臨床では高度に医療需要を満たしていない侵襲性血液系悪性腫瘍を含む。機序的には,STAT 3の異常活性化は癌細胞の生存,増殖,転移の促進に直接関与している。また,STAT 3は腫瘍,間質と免疫細胞間のクロストークを調節し,免疫抑制の腫瘍微小環境を促進する。IL−6と形質転換増殖因子βによって活性化されたSTAT 3も自己免疫と線維化の発症機序に関与している。STAT 3の疾病発病機序における異なる作用はそれを癌、自己免疫性疾患と繊維化疾病の薬物開発を治療する魅力的な標的にさせた。

[図34]
JAKファミリーキナーゼに対する小分子阻害剤、例えばJAK 1、JAK 2、JAK 3およびTYK 2は、関節リウマチ、乾癬性関節炎および潰瘍性大腸炎などの自己免疫疾患の治療に許可されており、JAK 2/STAT 5経路に対して使用されている。腫瘍学的には、JAK阻害剤は、原発性骨髄線維化および真性赤血球増加症、および急性移植片を含むJAK 2/STAT 5経路活性化を引き起こす血液系悪性腫瘍の変異のために許可されている
28
宿主疾患に対抗する。JAK阻害剤は一連のサイトカインや増殖因子のシグナル伝達を遮断し,STAT 3の活性化だけでなく,STAT 1とSTAT 5の活性化も減少した。抗腫瘍効果の調節に対して、このような広範な活動は互いに矛盾した結果を生じる可能性がある。特に,STAT 1活性の抑制は細胞溶解T細胞と抗原提示細胞の抗腫瘍免疫反応を抑制し,STAT 3を抑制するだけで実現できる生産性免疫反応を相殺した。したがって,JAK阻害剤は骨髄増殖性腫瘍以外の癌では臨床活性を示さなかった。JAK阻害剤の広範な活性は特定のカテゴリーの副作用にも関与している。STAT 3を選択的に標的とすることにより,JAK抑制によりSTAT 1とSTAT 5をより広く抑制することに関する免疫抑制や安全リスクを回避することができるとともに,JAK依存やSTAT 3の独立活性化の問題を効率的に解決することができる。IL-6またはその受容体IL-6 Rなどの炎症性サイトカインに対するモノクロナル抗体も、一部の自己免疫疾患への使用が許可されている。しかし、自己免疫性と線維性疾患およびある癌は通常多種のサイトカインによって調節される。そのため、標的STAT 3はIL-6のシグナル伝達に参与するだけでなく、形質転換成長因子とサイトカイン、例えばIL-12、IL-2とIL-15のシグナル伝達にも参与するため、もっと有効である可能性がある。したがって,STAT 3を直接標的とすることはSTAT 3に収束する複数のシグナル経路を阻止し,腫瘍が微小環境を許容する病理過程を逆転することに寄与する可能性がある。
発展のチャンス
STAT 3分解物の腫瘍発生、腫瘍細胞のチロシンキナーゼ阻害剤と化学療法に対する耐性及び免疫監視の逃避などにおける多重作用は血液系悪性腫瘍と固形腫瘍に多種の発展機会を提供した。
悪性血液病
発ガン性STAT 3突然変異と/或いはSTAT 3経路活性化末梢T細胞リンパ腫(PTCL)と皮膚T細胞リンパ腫(CTCL)に非常によく見られる。全世界の主要市場では,毎年約8000名の患者がPTCLと診断され,約6000名の患者がCTCLと診断されている。STAT 3変異と経路活性化およびPTCL亜群とCTCLの免疫チェックポイント阻害剤に対する反応性は,これらの適応にSTAT 3依存性が示唆され,単一療法としてSTAT 3分解剤を開発する機会があった。PTCLの第一線治療の標準看護はCD 30誘導抗体-薬物結合物ブツキシマブと化学療法を結合することである。大多数のPTCL患者はALK-ALCL、PTCL-非特指、AITLとNK/Tリンパ腫亜型を含み、最終的に進展と死亡する。難治性/再発性疾患の患者では,現在の治療選択は限られており,承認されたプラトリシンやロミジキシンの治療効果は限られている。これらの高度に需要を満たしていない難治性/再発性PTCL亜群では,STAT 3変異(約13−38%)とSTAT 3経路活性化(90%と高い)の高い罹患率が認められた。STAT 3低下がプログラム死リガンド1やPD−L 1レベルに及ぼす影響は文献に記載されていることから,われわれのSTAT 3分解剤はこれらの患者に二重の影響を及ぼすと予想される。晩期疾患とSTAT 3活性化レベルが最も高いCTCL患者では,根治的治療法もなく,標準的な看護もなかった。抗体-薬物結合体、HDAC阻害剤と免疫検査点阻害剤は一定の活性を有し、早期或いは難治/再発患者に応用できるが、腫瘍内在作用と免疫調節作用を有する有効な治療に対してまだ非常に高い需要がある。
STAT 3経路の活性化もほとんどのT細胞とNK細胞大顆粒リンパ球性白血病患者に存在し,70%までの患者が発癌のSTAT 3変異を有していた。これらの発現はSTAT 3への依存を高く示唆しており,さらにこれらの患者におけるJAK阻害剤の初歩的な臨床活性が支持されている。STAT 3の活性化はAMLやDLBCLにもよく見られるが,STAT 3変異はあまり見られないにもかかわらず。DLBCLにおけるPD-L 1の過剰発現はもっと悪い疾病の結果と関係があり、すでにこれらの患者のPD-1/PD-L 1薬物に対する反応が報告されている。STAT 3がPD−1/PD−L 1に下流に影響していることから,STAT 3分解剤の単一療法および他の活性薬との併用は深い臨床効果を実現する可能性があると信じられている。
固形腫瘍
PD−1/PD−L 1免疫チェックポイント阻害剤(ICIS)とチロシンキナーゼ阻害剤(TKI)に反応する癌は,非小細胞肺癌(NSCLC),頭頸部扁平上皮癌(HNSCC),乳癌と結腸直腸癌を含み,STAT 3は固形腫瘍のICISやTKIに対する耐性に確立された役割を果たしているため,注目されている発展機会である。具体的には,STAT 3分解者はこれらの方法に合わせてあらかじめ反応を改善したり,二線付加療法として獲得性耐性を克服したりする可能性がある。
29
臨床研究とデータ
2022年、我々は、再発および/または難治性リンパ腫、白血病および固形腫瘍の成人患者の治療のために、KT-333の28日目、8および15日目の毎週投与の安全性、耐性、PK/PDおよび臨床活性を評価するためのKT-333の第1段階臨床試験を開始した。以下の図35に実験の詳細を示す.

図35
2023年12月,我々は2023年米国血液学会(ASH)年次総会と博覧会のポスター会議でKT−333第1段階臨床試験の臨床データを示した。このポスターは臨時更新を提供し、データ締め切りは2023年10月18日です。その日まで、29名の患者、中位年齢65歳、すでに5つの用量レベル(DL 1-5)の平均8回の治療を受け、5例の皮膚T細胞リンパ腫、2例の大顆粒リンパ球性白血病、或いはLGL-L、末梢T細胞リンパ腫、B細胞とホジキンリンパ腫各1例、及び19例の各種固形腫瘍悪性腫瘍を含む。全体的には,抗腫瘍活性の早期徴候を示し,用量は通常耐性が良好であり,血液や腫瘍で著明なSTAT 3ノックアウトに関与している。
次の表は,2023年10月18日までのデータ締め切り,5種類の用量レベル治療を受けた29名の患者の人口統計データを重点的に紹介した。

図36
⌂=間変性T細胞リンパ腫;データ締め切り:2023年10月18日。
KT-333は一般的に耐性が良好であり、主に便秘、疲労、吐き気と貧血を含む1級と2級の副作用である。KT−333に関連するレベル3以上の唯一の有害事象は、口腔炎、関節痛、および体重減少である。LGL-L患者はDL 5に口腔炎と関節痛の2種類の用量制限毒性(DLTS)が出現し、固形腫瘍/リンパ腫患者にDLTSは観察されなかった。これらの発見に基づいて、研究方案は固形腫瘍とリンパ腫患者と白血病患者(LGL-LとT細胞前リンパ球性白血病、或いはT-PLL患者を含む)に改訂され、それぞれ継続投与量が増加した。
30
機構を証明するために,循環1とサイクル2では図37に示す標的質量分析計を用いてSTAT 3の末梢血液中での分解を評価した。KT−333注入後のPBMCにおけるSTAT 3の平均最大分解率は,用量レベル1と5の間でそれぞれ70%から84%に増加した。STAT 3のパーセンテージ変化は、ベースラインより2つのSTAT 3ポリペプチドの平均パーセント変化を表す。投与量4レベルで、患者の末梢血単球中のSTAT 3蛋白の最大抑制率は96%に達し、異なる用量間のSTAT 3蛋白の回復。

図37
図38に示すように,多重免疫蛍光データの半定量分析により,KT−333投与後~24時間のCTCL生検ではSTAT 3陽性細胞が69%,リン酸化STAT 3陽性細胞が87%減少した。


図38
31
著者らは5例のCTLC患者中3例の臨床活動状況を観察し、その中の2例は部分的に緩和し、1例は病状が安定した。典型的なホジキンリンパ腫(CHL)の部分反応も1例認められた。まとめると,これらの結果は液体腫瘍における単一薬物活性を証明しており,臨床前データの支持を得ている。固形腫瘍では,臨床前に強い単剤活性は認められず,計4名の患者に頭頸部腫瘍でより持続時間の長い安定疾患やSDのパターンが認められた。以下の表では,2023年10月18日までのデータ遮断日の応答データを重点的に紹介した。

図39
*耳下腺粘液上皮様癌(C 7+)、副鼻腔腺癌(C 5)、胆管細胞癌(C 3)、腎細胞癌(C 3+)。

図40
*Sezary症候群に起因する症状を治療するためにC 1の最初の週にステロイド治療を受けた;袁はd/t AE(Dr.を停止した。2皮膚扁平上皮癌);d/t PI離散度の中止(中止時の病状は安定);HNC 1=耳下腺粘液表皮様癌;HNC 2=副鼻腔腺癌
KT−333はCTCL患者の腫瘍におけるSTAT 3,PSTAT 3とSOCS 3の発現を著しく低下させ,同時にインターフェロンγ刺激の遺伝子を誘導し,腫瘍微小環境中の陽性免疫調節反応を示唆し,臨床と臨床前に抗PD−1薬物の活性を増強することが証明され,KT−333と抗PD−1薬物の組み合わせに拡張することを支持した。第一段階試験では,皮膚T細胞リンパ腫(CTCL)患者の腫瘍生検においてPD−1に対する治療感受性を予測するインターフェロンガンマシグナルが誘導され,KT−333とPD−1抗体治療が相乗的に作用することが示唆された。
32

[図41]

[図42]
臨床発展計画
試験の1 a段階用量増加部分は進行中である。私たちは2024年にもっと多くの臨床データを提供する予定だ。
33
MDM 2
要約.要約
われわれはMDM 2を標的とした分解剤を開発しており,固形腫瘍や血液系悪性腫瘍の治療に用いられている。KT-253はMDM 2を標的とし、MDM 2は最もよく見られる腫瘍抑制因子p 53の重要な調節因子である。P 53は、癌細胞の成長を調節する能力を保持していることを意味する、癌の50%近くで完全に維持されている(野生型)。小分子阻害剤(SMI)はすでにP 53の発現を安定と上昇させるために開発されたが、それらは1つのフィードバック循環を誘導し、MDM 2蛋白レベルを増加させ、それによってP 53を抑制し、その治療効果を制限できることを発見した。臨床前研究において、KT-253はすでにMDM 2フィードバック回路を克服し、短い暴露下で迅速に癌細胞死亡を誘導する能力を示し、治療効果と安全性を高めるために機会を提供した。2023年5月、我々はKT-253の一期臨床試験を開始した。本研究は成人液体と固体腫瘍患者におけるKT-253の安全性、耐性、PK/PDと臨床活性を評価することを目的とした。KT-253はすでに1期試験で臨床機序の検証を実現し、液体と固体腫瘍タイプにおいて抗腫瘍活性の兆候を示した。試験の1 a段階では患者登録と投与量が行われており,2024年にはより多くの臨床データが提供される予定である。
生物学と作用機序
マウス二分2(MDM 2)癌蛋白は主要なE 3リガーゼであり、腫瘍抑制遺伝子P 53を制御する。P 53は1種の転写因子であり、細胞のストレスに対する反応を調節し、そして細胞運命の決定を指導し、例えば細胞周期の停滞、DNA修復、老化と細胞アポトーシスであり、50%近くの液体と固体癌において機能を有し、多くのP 53機能細胞系はMDM 2の過剰発現に依存してP 53と生存を抑制する。MDM 2を分解除去することによってP 53を安定と上昇させることは、細胞死及び/或いは細胞周期の停滞を招くことができる。MDM 2小分子阻害剤は様々な腫瘍タイプで臨床活性を示したが,MDM 2抑制によりフィードバック回路が制限され,その活性は図43に示すように制限された。このフィードバック循環はMDM 2蛋白発現を上昇させ、これは逆に駆動を占有する小分子をMDM 2を抑制しにくい。そのため、小分子阻害剤のP 53に対する上昇作用は比較的に小さく、通常細胞アポトーシスではなく細胞周期の停滞を招き、MDM 2/P 53小分子阻害剤の治療効果を制限した。このようなフィードバック循環もより多くの長期接触薬物を必要とし、腫瘍中の適度なMDM 2抑制を維持し、これは正常細胞に対する毒性効果を招き、それによってこれらの阻害剤の安全性と耐性を制限するかもしれない。分解者は触媒によりタンパク質を完全に除去することでMDM 2フィードバック回路を克服することが可能である。これは高効率薬物の開発を可能にし、これらの薬物は腫瘍細胞中で強力なp 53上昇と不可逆的な急性アポトーシス反応を誘導することができ、一時的な暴露だけで、それによって治療効果を最大化し、そして正常細胞の回復時間を許可することによって安全性を改善する。

[図43]
34
発展のチャンス
MDM 2に依存する大量のp 53 wt細胞系は,図44に示すように,腫瘍学においてこの経路に有効かつ耐性の良好な薬物の潜在的機会を提供する高レベルビューを提供した。これらの腫瘍細胞タイプはMDM 2遺伝子増幅と過剰発現の癌を含むが、これらに限定されない。MDM 2のP 53に対する脱安定作用は細胞周期の停滞とアポトーシスを遮断することによって細胞を生存させることができる。機会は非常に多様であるが,われわれの開発努力は,分解物による急性アポトーシス反応の影響を最も受けやすい腫瘍に集中する予定であり,これらの腫瘍の中で,最大の治療指数と治療効果が実現できると信じている。著者らが最初に興味を持った疾患領域は血液系悪性腫瘍と固形腫瘍の適応であり、臨床前にMDM 2分解による急性アポトーシス反応が見られ、間欠投与の臨床活動を予測した。

[図44]
血液系悪性腫瘍に対して、KT-253は急性骨髄性白血病(AML)、骨髄繊維化、骨髄異形成症候群(MDS)、急性リンパ球性白血病(ALL)とTP 53 WTリンパ腫において潜在的な単一治療と連合治療機会を有する。固形腫瘍に対して、KT-253は成人と児童腫瘍のサブセットに単一の治療機会があり、新しく出現した腫瘍の不知性発展の道を有する潜在的な遺伝子署名を通じて情報を得た。著者らは、1 a期後の次の段階の患者のためのバイオマーカーの選択を開発するために、KT-253間欠投与による多種の異なる実体および液体腫瘍タイプの体内反応に影響する因子を検査する包括的な臨床前および臨床データセットを集約している。
臨床前研究とデータ
KT-253‘SはP 53を有効に安定させ、そして一時的に暴露し、癌細胞のアポトーシスを促進する。図45において,KT−253は小分子阻害剤と比較して体外細胞増殖抑制,アポトーシスともに200倍以上の改善を示している。

[図45]
35
KT−253は小分子阻害剤と異なりタンパク質を除去することができ,p 53に依存したフィードバック回路を克服し,後者はMDM 2の産生を上昇させる。図46に示すように,小分子阻害物(フィードバックループ)はMDM 2レベルを増加させ,P 53の安定を損なう。

図46
臨床前モデルでは、KT−253の4時間標的被覆は、図47に示すように、アポトーシスを誘導するのに十分である。これらのデータはKT−253の間欠投与計画が治療指数を向上させながら治療効果を向上させることを支持している。
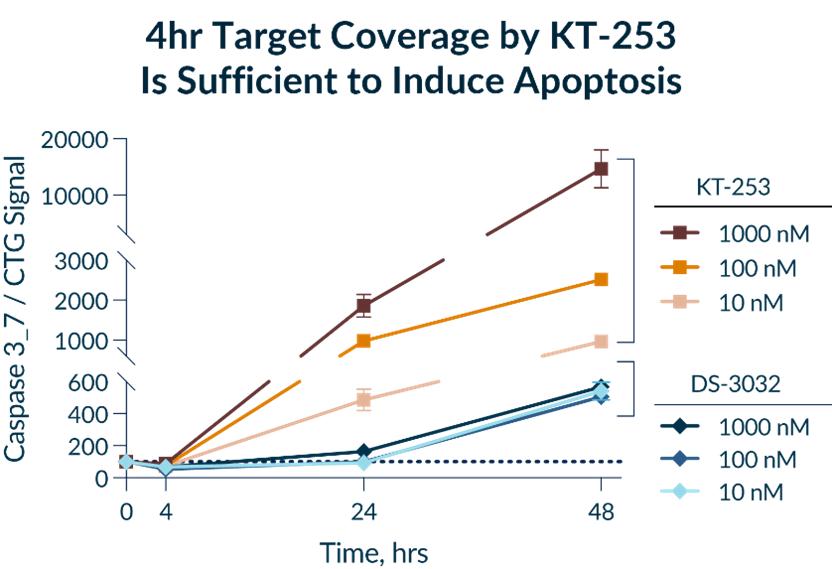
[図47]
臨床前モデルにおいて、KT-253は有効にMDM 2を分解し、経路影響と抗腫瘍活性は小分子阻害剤より優れている。図48に示すように、RS 4;11(ALL)腫瘍の標的プロテオミクス解析は、投与1時間後にMDM 2および関連経路活性化バイオマーカーがp 53およびGDF 15を含む強力な分解を示す。
36

図48
図49にMV 4;11(AML)CDXモデルの単回3 mg/kg KT−253用量後の持続腫瘍消退を示す。また,KT−253はMCCモデルで強い抗腫瘍活性を示した。小分子阻害剤(DS-3032)の臨床関連用量方案は治療効果が観察されなかった。

図49
37
臨床研究とデータ
第一段階試験は2023年5月に患者投与を開始し、AML、ALL、リンパ腫と固形腫瘍を含む再発或いは難治性高レベル髄系悪性腫瘍患者におけるKT-253の安全性、耐性、PK/PDと臨床活性を評価している。第一段階用量漸増研究の患者は、3週間に1回の静注用量のKT-253を受ける。この開放ラベル研究は、推奨される第2段階用量を決定することを目的としており、2つのアームからなり、各アームの増加用量はKT-253である。第一グループはリンパ腫と末期固形腫瘍患者からなり、第二グループは高度悪性髄系腫瘍とALL患者から構成されている。

図50
2023年10月20日までのデータ遮断日まで、9例の固形腫瘍患者はA群の1~3個の投与量レベル、平均2.3周期、1~6周期に入選した。進行中の1 a段階試験のARM Aからのデータは、KT-253は患者の前の2つの用量レベルにおいて臨床機序証明と臨床活動の初歩的な兆候を有することを示した。用量レベル1のすべての患者の臨床反応結果は利用可能である。曝露により臨床的には機能しないと予想されるが,用量レベル1で治療した固形腫瘍患者3名のうち,6周期後に治療を継続した4周期後に1つの確認された部分反応があり,4周期後に1つ確認された安定した疾患が認められ,6周期後に無反応で検討を中止し,1名の患者が1周期後に経過したものを図51に示す。部分的に応答した患者はメルケル細胞癌(MCC)であり、腹部リンパ節と皮膚に転移し、これまで化学療法と多種の異なる免疫検査点阻害剤の治療を受けたことがある。

[図51]
用量レベル1−3の間に用量制限毒性はなかった。データ遮断日まで、最もよく見られる薬物関連不良事件。1/2級吐き気およびレベル1下痢を含む、2つまたは2つ以上の患者に発生する。1用量レベル1の患者が第4周期で重篤な有害事象,すなわちレベル3低血圧が出現したのは,研究薬に関連すると考えられる経口摂取量の減少によるものであった。治療は静脈輸液を含み、患者はまだ研究中であり、投与量の減少或いは低血圧再発はなかった。データ締め切りまで,6周期治療を受けた患者でも,好中球減少や血小板減少の副作用は認められなかった。
38
臨床発展計画
KT-253 1 a期試験は開放ラベルの投与量の増加研究であり、再発或いは難治性高度髄系悪性腫瘍、ALL、リンパ腫と固形腫瘍の成年患者は3週間に1回の静脈投与量のKT-253治療を受ける。この研究は安全性、耐性、PK/PDと初期臨床活動性を評価し、推薦された第二段階用量を決定することを目的としている。2つの腕で構成されており、各腕には漸増用量のKT-253がある。ARM Aは末期固形腫瘍およびリンパ腫患者に用いられ、ARM Bは再発または難治性高度悪性髄系腫瘍(AMLとALLを含む)患者に応用される。第1の患者は2023年5月に用量治療を受け、A群の最初の2つの用量レベルは完全に組み込まれ、登録作業が行われている。ARM Aの前2用量レベルで目標薬理学を示した後,ARM Bの登録も開始された。同社は2024年により多くの臨床データを共有する予定である。
セノフィ(前身はGenzyme Corporation)との協力協定
2020年7月7日,セノフィの子会社Genzyme Corporationと,最初のセノフィ合意で,2つの生物学的標的に対する候補薬を共同開発することで合意した。最初のセノフィ協定は2020年第3四半期に施行された。
2022年11月15日、我々は、元のセノフィ協定に規定されているいくつかの研究条項および責任を修正するために、元のセノフィ協定を修正する改訂された協力および許可協定、または修正されたセノフィ協定をセノフィと締結した。修正されたセノフィ協定は,協力条項に要求される第2段階試験の時間と数も詳細に説明した。改訂されたセノフィ協定は2022年12月5日に発効する。改訂されたセノフィプロトコル改訂後の元セノフィプロトコルを本稿ではセノフィプロトコルと呼ぶ.
サイノフェイ協定によれば、Kymeraは、協力中に生成されたIRAK 4および他の開示されていない用途分野の不開示目標のための特定の先導化合物を開発、製造および商業化するために、サイノフィにグローバル独占ライセンスを付与する。このような許可は、指定されたマイルストーンの後にのみ、連携目標ごとに行使することができる。IRAK 4に対する化合物の使用領域は、腫瘍学および免疫腫瘍学を含まず、診断、治療、治癒、緩和または予防のいずれかの疾患、障害または状態を含む。
セノフェイプロトコルによると,この2つの目標については,発見および臨床前研究を担当し,IRAK 4に対する少なくとも1つの分解剤および最大3つの予備分解剤について第1段階臨床試験を行い,場合によっては費用は私たちが負担する。この2つの目標に対して,セノフィは協力候補製品ごとに特定の開発マイルストーンが出現した後,候補製品の開発,製造,商業化を担当している。
また、セノフィ協定によれば、セノフィは独占選択権または選択加入権を付与し、(I)適用分野におけるこのような目標に対する協力製品の米国での開発コストの50%の資金を提供する権利と、(Ii)米国の適用分野におけるこのような目標に対して協調製品を商業化する純利益と純損失とを平均的に分担する権利があることを、我々の単独判断に基づいて行使することができる。また、もし私たちが選択権を行使すれば、セノフィは各協力目標に適用される独占的な選択権を与えてくれます。一度行使すれば、アメリカである分野での共同普及活動を許可します。
セノフィがセノフィ協定に基づいてセノフィに独占許可を与えたことを考慮すると、セノフィは私たちに1億5千万ドルを前払いした。前金を除いて、合意に基づいて、いくつかの開発または規制活動が完了した後、合計14.8億ドルに達するいくつかの開発マイルストーン支払いを得る資格があり、その中で10億ドル以上がIRAK 4計画に関連している。また、合計7.00億ドルにのぼるいくつかのビジネスマイルストーン支払いを受ける資格があり、そのうち4.0億ドルはIRAK 4計画に関連しており、ある純売上高のハードルに達したときに支払う。計画ごとの純売上高のために等級別印税を取得する資格もあり、上位から高青少年まで範囲があり、場合によっては1桁に上方調整される可能性がある。
セノフィ協定が早期に終了しない限り、この協定は、セノフィ協定の下で製品に関連するすべての支払い義務が満了した日から製品ごとに満了する。私たちまたはセノフィは、相手の重大な違約または資金で借金をしないか、またはいくつかの特許挑戦のために合意を終了することができます。また、セノフィは、事前に書面で通知した場合、便宜や重大なセキュリティ事件の目的で合意を終了することができ、セノフィが開発責任を負う場合には、任意の協力候補との合意を終了することができる
39
特定のターゲットの協調候補オブジェクトの商業化または製造のために、サイノフェイは、特定の期間内に、そのターゲットに対する任意の協調候補オブジェクトの利用を停止する。
また,2022年12月,セノフィは第二段階臨床試験にKT−474を使用する意向を示した書面通知を行った。2023年第4四半期に、同社はそれぞれ4000万ドルと1500万ドルの2つのマイルストーンを実現し、第1および第2の適応に関連する第2段階の臨床試験における第1人の患者の投与量を決定した。2023年12月31日現在、会社は4000万ドルのマイルストーンを受け取り、連結貸借対照表の売掛金には1500万ドルが含まれている。
2023年9月、当社とセノフィは共に、開示されていない目標に関連する活動を停止することに同意し、第2の目標に関連するマイルストーンおよび特許権使用料の支払いを得る資格がなくなりました。
製造業/サプライチェーン
私たちは私たちの候補薬物を生産するための製造施設を持ったり運営したりしていませんが、現在は私たち自身の臨床や商業規模の製造能力を構築する計画もありません。我々は現在,第三者契約製造組織やCMOと協力して,臨床前研究のための候補薬を生産しており,今後も継続していく予定である。私たちは依存して第三者メーカーが薬品と完成品を生産することに依存し続けると予想される。私たちは第三者メーカーを招いて私たちの候補薬物に薬物物質を提供し、第三者メーカーを招いて臨床試験で使用した完成薬を開発し、製造した。私たちは現在、これらのメーカーから注文を購入する方式で供給を受けており、長期的な供給スケジュールを立てていない。もしこれらのメーカーのいずれかがどんな理由でも私たちに提供できなければ、私たちはこのような代替製品を決定して同定する時にいくつかの遅延があるかもしれないが、多くの潜在的な代替製品があると信じている。
我々のすべての候補薬物は低分子有機化合物であり,通常は小分子と呼ばれるが,従来の小分子療法よりも大きい。我々がこれらの化合物を選択したのは,潜在的な治療効果と安全性だけでなく,合成が容易であり,商品コストも高いと予想されるからである。私たちはすでに臨床試験のための薬物物質と薬物製品を生産し、私たちの生産プロセスを改善し続けている。薬物物質や薬物製品プロセスは拡大しやすく、製造過程で特殊な設備を必要としない。後期臨床と商業製造に対する需要を十分に満たすために、私たちのサプライヤーは彼らの生産規模を拡大する必要があります。そうでなければ、代替サプライヤーを探す必要があります。
競争
バイオテクノロジー産業は新製品を開発する競争で異常に競争が激しい。標的タンパク質分解、臨床開発専門知識と知的財産権の地位における著者らの長年の専門知識によって、著者らは著しい競争優位を持っていると信じているが、現在、標的タンパク質分解或いは標的タンパク質分解開発プラットフォームを使用する会社と、より伝統的な治療方式(例えば小分子や抗体)に専念する会社からの開発計画の競争に直面している。競争はより大きな製薬会社、生物技術会社と学術界を含む多種の源から来る可能性がある。
患者のための小分子タンパク質分解剤療法を開発する会社には、Arvinas,Inc.,C 4 Treateutics,Inc.,Nurix Treateutics,Inc.およびFoghorn Treateutics,Inc.があるが、これらに限定されない。また、いくつかの大手製薬会社がこの分野への臨床前投資を開示している。我々の競争相手には,他の標的化されたタンパク質分解法や小分子,抗体あるいは遺伝子療法を開発している会社も含まれており,これらの方法の適応は我々の目標と同じである。我々が小分子タンパク質分解器の開発に直面しているライバルのほかに,我々のIRAK 4,STAT 6,TYK 2,STAT 3,MDM 2プロジェクトで競争に直面する.これらの適応の多くは、より伝統的な治療法を含む可能性がある承認された看護基準を有する。これらの既存療法と効率的に競合するためには,われわれのタンパク質分解剤療法が既存療法に有利であることを証明する必要がある。
知的財産権
私たちの成功は、私たちの候補製品と未来の製品のために知的財産権保護を確保する能力、および私たちのプラットフォームタンパク質分解技術および任意の他の私たちの業務に商業的重要性があると考えられる関連発明と改善にある程度依存する。私たちの成功は私たちの擁護と実行にかかっています
40
知的財産権は、第三者の効果的かつ強制的に実行可能な特許および独自の権利を侵害、流用、または他の方法で侵害することなく、我々の固有情報の機密性を保護する。
他のバイオテクノロジーや製薬会社と同様に、私たちの候補製品、将来の製品、および他の独自技術の知的財産権保護の能力を保護し、維持することは、有効な特許カバー範囲を得ることができるかどうかに依存し、許可された場合にこれらの特許を実行するだろう。しかし、私たちは、私たちが係属中の特許出願および将来提出される可能性のある任意の特許出願が特許の発行をもたらすか、または私たちが獲得する可能性のある任意の発行された特許が、競争相手から保護されるのに十分な特許保護を提供することを保証することはできない。私たちが獲得したいかなる発行された特許も第三者の挑戦、無効、または回避を受けるかもしれない。
特許以外にも、私たちは商業秘密、技術ノウハウ、持続的な技術革新に依存して、私たちの競争地位を発展させ、維持しています。従業員、コンサルタント、科学コンサルタント、請負業者、潜在的協力者と守秘協定や発明譲渡協定を締結することで、私たちのノウハウをある程度保護することを求めています。
特許組合
私たちの知的財産権は、物質組成、薬物組成、使用方法、治療方法、および他の関連化合物および方法の請求項を含む、私たちのプラットフォームE 3リガーゼリガンド技術および私たちの新しい二機能分解剤候補製品をカバーする一連の完全所有特許シリーズを含む。私たちの知的財産の組み合わせはまだ非常に早い段階にあり、2023年12月31日現在、私たちの知的財産権の組み合わせは、19件の許可された米国特許、約100件の米国特許出願、約25件の国際特許出願、5つの許可された外国特許、および約449件の外国特許出願を含む。我々の特許の組み合わせは、一般に、(1)プラットフォームE 3リガーゼ配位子特許ファミリーと(2)様々な標的固有の分解物特許ファミリーを含むタンパク質分解物特許ファミリーとに分類される。
Platform E 3リガーゼリガンド特許ファミリー
我々のプラットフォームE 3リガーゼ特許ファミリーは、Cereblon E 3ユビキチンリガーゼの新しいリガンドに対する4つの特許ファミリー、および治療方法および他の関連方法を含む完全所有である。2023年12月31日現在、当社のプラットフォームE 3リガーゼリガンド特許シリーズは、3つの許可された米国特許、5つの米国特許出願、および3つの欧州特許出願を含む。これらの出願によって生成された任意の米国または外国特許は、承認され、すべての適切な維持費が支払われる場合、特許期間の調整または延長を行うことなく、2038年から2044年の間に満了することが予想される。
タンパク質分解剤特許ファミリー
我々のタンパク質分解剤特許ファミリーは完全所有であり,目的タンパク質のユビキチン化に寄与する新規二機能分解剤化合物,および治療法や他の関連方法を対象としている。2023年12月31日現在、私たちのタンパク質分解器特許シリーズは、2つの許可された米国特許、5つの米国特許出願、およびオーストラリア、カナダ、ヨーロッパ、イスラエル、日本、メキシコ、ニュージーランド、ロシア連邦のような他の司法管轄区で提出された約19件の外国特許出願を含む。これらの出願によって生成された任意の米国または外国特許は、承認され、すべての適切な維持費が支払われる場合、特許期間の調整または延長を行うことなく、2038年から2043年の間に満了することが予想される。
ターゲット特定分解器特許ファミリー
我々の目標とする特定の分解物特許ファミリーは完全所有であり、特定のタンパク質を分解することを目的とした分解物化合物、および治療方法および他の関連方法を重点的に保護する。これらの標的は、例えば、IRAK(インターロイキン1受容体関連キナーゼ)およびSTAT(シグナル伝達および転写活性化剤)を含む。2023年12月31日現在、我々の目標特定分解剤特許シリーズには、14件の米国特許出願、約87件の米国特許出願、約22件の国際特許出願、3つの外国特許出願、およびオーストラリア、ブラジル、カナダ、中国、ユーラシア大陸、ヨーロッパ、イスラエル、インド、日本、メキシコ、ニュージーランド、シンガポール、南アフリカ、台湾などの他の司法管轄区で提出された約423件の特許出願が含まれている。私たちの目標特定の分解器特許シリーズによって生成された任意の米国または外国特許は、承認され、すべての適切な維持費が支払われる場合、2038年から2044年の間に満了し、特許期限の調整または延長は行われないと予想される。
IRAK専用特許家
我々のIRAK専用特許シリーズは,IRAK分解に特化した分解剤化合物の特許シリーズと,新規IRAKリガンドをカバーする特許シリーズを含む全資所有である。2023年12月31日現在、私たちのイラク特許シリーズは、11件の許可された米国特許、約34件の米国特許出願、約8つの国際特許出願、3つの許可された外国特許、および約293件の外国司法管轄区域で提出された特許出願を含む
41
オーストラリア、アルゼンチン、ブラジル、カナダ、中国、ヨーロッパ、ユーラシア大陸、湾岸協力委員会、イスラエル、インド、日本、メキシコ、ニュージーランド、シンガポール、南アフリカ、台湾など。私たちのイラク特許家族によって取得された任意の米国または外国特許は、許可され、すべての適切な維持費が支払われれば、2038年から2044年の間に満期になり、いかなる特許期限の調整や延長も行われないと予想される。
KT−474候補品については、2023年12月31日現在、オーストラリア、ブラジル、カナダ、中国、欧州、イスラエル、インド、日本、韓国、メキシコ、ニュージーランド、シンガポール、南アフリカ、台湾を含む3つのライセンスされた米国特許、8つの出願されている国際特許、1つの許可された外国特許、および他の管轄区で提出された約128の特許出願を有しており、各出願はKT−474の物質組成および/またはKT−474の製造または使用方法に関連している。これらの特許ファミリーによって取得された任意の米国または外国特許によれば、承認され、すべての適切な維持費が支払われる場合、特許期間の調整または延長を行うことなく、2039年から2044年の間に満了することが予想される。
統計データ専用特許シリーズ
我々のSTAT専用特許ファミリーは完全所有であり、シグナル伝達と転写活性化剤(STAT)の分解に特化することを目的とした分解剤化合物に集中している。2023年12月31日現在、私たちの統計特許シリーズは、2つの許可された米国特許、約18件の米国特許出願、3つの国際特許出願、およびオーストラリア、カナダ、中国、ユーラシア大陸、ヨーロッパ、インド、イスラエル、日本、韓国、メキシコ、台湾などの他の司法管轄区で提出された約49件の特許出願を含む。私たちの統計データ特定特許シリーズによって生成された任意の米国または外国特許は、承認され、すべての適切な維持費が支払われれば、2040年から2044年の間に満了し、いかなる特許期限の調整や延長も行われないと予想される。
その他のターゲット特定特許シリーズ
2023年12月31日現在、我々は、許可された米国特許、約31件の米国特許出願、10件の国際特許出願、およびオーストラリア、アルゼンチン、ブラジル、カナダ、中国、欧州、湾岸協力委員会、イスラエル、インド、日本、メキシコ、ニュージーランド、シンガポール、南アフリカ、台湾で出願された約81件の特許出願を有しており、これらの特許は、他のタンパク質に特化した分解性化合物に特化している。これらの特許ファミリーによって取得された任意の米国または外国特許によれば、許可され、すべての適切な維持費が支払われる場合、特許期間の調整または延長を行うことなく、2040年から2044年の間に満了することが予想される。
個別特許の期限は、これらの特許を取得する国によって異なる可能性がある。一般に,米国で提出された出願から発行される特許は,最も早く発効した非仮出願日から20年以内に有効である。場合によっては、FDA規制審査中に実際に失われた期間の一部を再取得するために、特許期限を延長することができる。ハッジ·ワックスマン法は、特許期間が特許満了後最大5年間延長されることを許可しているが、任意の延長を含む総特許期間はFDA承認後14年を超えてはならない。1つの特許が1回しか延期できない、すなわち、1つの特許が複数の製品に適用される場合、1つの製品に基づいて延期するしかない。
米国以外の特許の期限は適用される現地法の規定によって異なるが,通常は最も早く発効した国の出願日から20年である。
ヨーロッパおよび他の外国司法管轄区域にも同様の特許期限延長条項があり、承認された薬物をカバーする特許期間を延長する。可能な場合には、私たちの候補製品及びその使用方法をカバーする特許出願特許期間を延長することが望ましい。
商標
私たちはすでに米国を含む異なる司法管轄区域に私たちの候補製品や他の技術に関する商標登録申請を提出する予定です。
私たちはアメリカ、ヨーロッパ、カナダにKYMERAマーカーとKYMERA治療マーカーの登録を申請しました。腫瘍学,自己免疫,免疫腫瘍学,その他関連疾患を治療するための薬物や医療製剤および診断試薬のために,同一管轄区でIRAKIMID商標を申請した。また,薬物開発と薬物開発·発見サービスに関するE 3ヒトATLASとE 3リガーゼ全身ATLASの申請を提出した。イギリスの離脱により、2020年12月31日までに存在するすべてのEU商標が自動的にイギリスにクローン登録されました
42
最近、私たちはアメリカで私たちのK&Designマークの申請を提出しました。私たちは適切な時にこの申請中のアメリカの優先日に基づいてEU、イギリス、カナダの申請を提出する予定です。
政府の監督管理
FDAと連邦、州と地方の各級及び外国のその他の監督管理機関は、その他の以外に、薬品の研究、開発、テスト、製造、品質管理、輸出入、安全、有効性、ラベル、包装、貯蔵、流通、記録保存、承認、広告、販売促進、マーケティング、承認後の監視と承認後の報告などの方面に対して広範な監督管理を行った。私たちのサプライヤー、契約研究組織、契約メーカーとは、私たちの候補製品を承認したい国家規制機関の様々な臨床前、臨床、製造、商業承認要件を満たすことを要求されます。薬品規制の承認を得て、その後適切な連邦、州、地方と外国の法規と条例を遵守する過程を確保するには大量の時間と財政資源が必要である。
米国では,FDAは改正された連邦食品,薬物,化粧品法案(FD&C Act),その実施条例,その他の法律に基づいて薬品を規制している。もし私たちが適用されるFDAまたは他の製品開発、臨床試験、承認に関する要件、または製品製造、加工、運搬、貯蔵、品質管理、安全、マーケティング、広告、販売促進、包装、ラベル、輸出、輸入、流通、または販売に関連する任意の他の法律要件を遵守できない場合、私たちは行政または司法制裁または他の法的結果を受ける可能性がある。これらの制裁または結果は、FDAが承認待ちの申請を拒否すること、進行中の研究のための臨床封印、承認された申請の一時停止または撤回、警告または無見出しの手紙の発行、製品の撤回またはリコール、製品の差し押さえ、再標識または再包装、製造または流通の完全または部分的な一時停止、禁止、罰金、民事処罰または刑事起訴を含むことができる。
FDAが我々の候補製品が治療適応薬として承認され、米国で発売される前に必要なプログラムには、一般に以下のような態様が含まれる
薬物の前臨床研究と臨床試験
人体でいかなる薬物をテストする前に、候補製品は厳格な臨床前テストを経なければならない。臨床前研究は薬物化学、調合と安定性の実験室評価、及び体外と動物研究を含み、安全性を評価し、ある場合に治療使用の理論基礎を確立する。臨床前研究の進行は連邦政府の制約を受けている
43
安全/毒理学研究のGLP要件を含む国家法規と要求。臨床前研究の結果および生産情報と分析データはINDの一部としてFDAに提出されなければならない。INDはFDAがヒトに研究製品の使用を許可する要求であり,臨床試験開始前に発効しなければならない。IND提出後,いくつかの長期臨床前試験が継続される可能性がある。INDはFDAが受け取った30日後に自動的に発効し、FDAが30日以内に臨床試験の進行に対して懸念或いは問題を提出しない限り、人類研究患者が不合理な健康リスクに直面することを心配し、そしてすべて或いは一部の臨床一時停止を強制的に実施することを含む。FDAは一時停止の理由をスポンサーに通知しなければならず,臨床試験が開始される前に決定された欠陥を解決しなければならない。INDの提出は、FDAが臨床試験の開始を許可しないか、または臨床試験をINDに最初に指定された条項に従って開始することを許可しない可能性がある。
開発された臨床段階は、合格した研究者の監督の下で、GCP要求に応じて、すべての研究患者に任意の臨床試験へのインフォームドコンセントを提供することを含む、試験スポンサーによって雇用または制御されない医師である健康ボランティアまたは患者に候補製品を提供することに関する。臨床試験は,臨床試験の目標,用量プログラム,被験者の選択と排除基準,および安全性と有効性をモニタリングするためのパラメータと基準を詳細に説明するレジメンで行われた。INDの一部として、すべての議定書とその後の議定書のいかなる修正もFDAに提出されなければならない。また、各臨床試験は臨床試験を行う各機関の内部審査委員会によって審査と承認され、臨床試験に参加する個人が直面するリスクが最小限に減少し、期待される利益と合理的に関連することを確保しなければならない。IRBはまた、各臨床試験対象またはその法律代表に提供されなければならないインフォームドコンセントを承認し、完成まで臨床試験を監視しなければならない。FDA、IRBまたはスポンサーは、患者が受け入れられない健康リスクに直面していることを発見することを含む、様々な理由で臨床試験を一時停止または終了することができる。実施中の臨床試験や完成した臨床試験を公的登録機関に報告することに関する要求もある。適用される臨床試験に関する情報は,臨床試験結果を含め,www.Clinicaltrials.govサイト上で発表するために特定の時間枠で提出しなければならない。
米国国外で臨床試験を行うスポンサーはFDAの認可を得ることができるが,INDによる臨床試験を希望している。外国の臨床試験がINDで行われていなければ,研究がGCP要求に基づいて行われていれば,FDAはNDAを支持する研究結果を受け入れ,必要であればFDAは現場検査でデータを検証することができる。
治療適応を評価してNDAの発売承認を支持する臨床試験は通常3つの連続段階に分けて行われ、重なる可能性がある。
2022年3月、FDAは“拡張行列:最初のヒト臨床試験のために腫瘍薬物と生物製品の開発を加速するための”と題する最終ガイドラインを発表し、その中で、薬物開発者がどのように腫瘍薬物開発の初期段階(すなわち最初の人体臨床試験)で通常シームレス試験設計と呼ばれる適応試験設計を利用して、伝統的な3段階の試験を拡張行列試験と呼ばれる連続試験に圧縮するかについて概説した。個人拡張キュー設計をサポートする情報はINDアプリケーションに含まれ,FDAによって評価される.コホート試験を拡大することは薬物開発の効率を向上させ,開発コストや時間を削減する可能性がある。
承認後試験は,4期臨床試験や上場後研究と呼ばれることがあり,最初の上場承認後に行われる可能性がある。これらの試験は、通常、臨床環境において製品を使用するための追加の安全データを生成するために、予期される治療適応下で患者の治療から追加の経験を得るために使用される。場合によっては,FDAはNDAを承認する条件として4期臨床試験を強制的に実行することができる。
44
その他の情報に加えて,臨床試験結果を詳細に説明する進捗報告は少なくとも毎年FDAに提出されなければならない。IND安全性書面報告は試験スポンサーがこの情報に深刻かつ意外な疑わしい不良事件、他の研究或いは動物試験或いは体外試験の結果を報告する資格があることを確定しなければならないことを表明し、人類ボランティアに重大なリスクがあること、及び方案或いは研究者マニュアルに列挙された深刻な疑わしい副作用の発生率といかなる臨床重要な増加があった後15日後にFDAと調査人員に提出しなければならない。スポンサーはまた、FDAに任意の意外、致命的、あるいは生命に危害を及ぼす疑いのある副作用をできるだけ早く通知しなければならないが、いずれの場合も、スポンサーが初めて情報を受け取った後の7つの日数に遅れてはならない。
臨床試験と同時に、会社は通常追加の動物研究を完成し、候補製品の化学的および物理的特性に関する追加情報を開発し、cGMPの要求に基づいて最終的に商業的に薬物製品を量産するプロセスを決定しなければならない。製造過程は一貫して高品質の候補製品ロットを生産できる必要があり、メーカーは最終薬物製品の特性、強度、品質と純度をテストする方法などを開発しなければならない。また,適切な包装を選択·テストし,候補製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
アメリカは薬品の発売を許可した
必要な臨床試験,臨床前研究や臨床試験の結果,製品の化学,製造,制御,提案されたラベルなどに関する詳細な情報を想定し,NDAの一部としてFDAに提出し,1つまたは複数の適応の市場への承認を要請した。NDAは1つまたは複数の適応を指定する新薬発売に対する承認要求であり,要求される適応におけるその薬物の安全性と有効性の証拠を含まなければならない。マーケティング申請は臨床前研究と臨床試験の否定と曖昧な結果、そして積極的な発見を含まなければならない。データは、製品使用の安全性および有効性を試験するために、または研究者によって開始された研究を含む多くの代替源からの臨床試験からのものである可能性がある。上場承認を支持するためには、提出されたデータは研究製品の安全性と有効性を確定し、FDAを満足させるために、品質と数量で十分でなければならない。薬物がアメリカで発売される前に、NDAに対するFDAの承認を受けなければならない。
FDAは、提出されたすべてのNDA出願を受け入れる前にそれを検討し、NDA出願を受け入れるのではなく、より多くの情報の提供を要求する可能性がある。FDAはNDAを受信してから60日以内にNDA届出を受け入れる決定をしなければならず,このような決定にはFDAが届出を拒否することが含まれる可能性がある。提出された申請が受け入れられると,FDAはNDAの深い実質的な審査を開始する。FDAは、薬物が求められた適応に対して安全かつ有効であるかどうか、およびその製造、加工、包装、または保有施設が、製品の持続的な安全、品質および純度を確保するための基準に適合しているかどうかを決定するために、セキュリティプロトコルを検討する。FDAが“処方薬使用料法案”(PDUFA)により達成した目標と政策によると,FDAは提出日から10カ月以内に新分子実体NDAの予備審査を完了して出願人に応答することと,新分子実体NDA提出日から6カ月以内に優先審査を行うことを目標としている。FDAは、そのPDUFA規格または優先NDAの目標日を常に満たすわけではなく、審査プロセスは、FDAがより多くの情報を提供または明確にすることを要求することによって延長されることが多い。
また,改訂されたPDUFAにより,秘密プロトコルごとに使用料が付加されなければならない.FDAは毎年PDUFAユーザ料金を調整する。場合によっては、小企業が初めて出願した出願料を免除することを含む、費用を免除または減免することができる。また,孤児薬として指定された製品については,NDAに対して使用料を評価せず,非孤児適応も含まれていない限りである。
FDAがリスク評価および緩和策が薬物の利益をそのリスクよりも大きく確保するために必要であると考えている場合、リスク評価および緩和策またはREMS計画の提出も要求される可能性がある。REMS計画は、投与ガイドライン、医師コミュニケーション計画、評価計画、および/または分配方法、患者登録、または他のリスク最小化ツールのような安全な使用を保証する要素のようなリスク評価および緩和戦略を使用することを含むことができる。
FDAは新薬の申請を諮問委員会に提出するかもしれない。諮問委員会は,臨床医や他の科学専門家を含む独立した専門家からなるグループであり,審査,評価を担当し,申請を承認すべきかどうか,どのような条件で提案すべきかについて提案している。FDAは諮問委員会の提案に制限されていないが、それは決定を下す時にこれらの提案を慎重に考慮するだろう。
秘密協定を承認する前に、FDAは通常、製品を生産する1つまたは複数の施設を検査する。FDAは、製造プロセスおよび施設がcGMP要件に適合していることを決定し、要求された仕様の下で製品が一貫して生産されることを保証するのに十分でない限り、申請を承認しないであろう。さらに、NDAを承認する前に、FDAは、GCPおよび他の要件に適合することを保証し、FDAに提出された臨床データの完全性を保証するために、1つまたは複数の臨床試験場所を検査することができる。
45
NDAとすべての関連情報を評価した後,諮問委員会のアドバイス(あれば)や製造施設や臨床試験地点に関する検査報告を含めて,FDAは承認状を発行する可能性があり,場合によっては完全な返信が発行される可能性がある。完全な返信状は、一般に、NDAの最終承認を保証するために満たされなければならない特定の条件の宣言を含み、FDAが出願を再検討するために追加の臨床または臨床前試験が必要とされる可能性がある。この補足情報を提出しても、FDAは最終的にその申請が承認された規制基準を満たしていないと決定する可能性がある。もしこのような条件がFDAの満足を得たら、FDAは通常承認書を発行するだろう。この薬物の商業マーケティングを許可し、特定の適応の具体的な処方情報を提供する。
FDAが解決すべき特定のリスク(S)に基づいて製品を承認しても、製品の承認適応を制限する可能性があり、製品ラベルに禁忌症、警告または予防措置を含むことが要求され、承認後に薬剤の安全性をさらに評価するための第4段階の臨床試験を含む承認後の研究が要求され、発売後に製品を監視するための試験および監視計画が要求されるか、または販売および使用制限またはREMS下の他のリスク管理メカニズムを含む他の条件が適用され、これらの条件は、製品の潜在的な市場および利益に大きな影響を与える可能性がある。FDAは発売後の研究或いはモニタリング計画の結果に基づいて、製品の更なるマーケティングを阻止或いは制限することができる。承認後、新たな適応の追加、製造変更、および追加のラベル宣言など、承認された製品のいくつかのタイプの変更は、さらなるテスト要件およびFDAの審査および承認を受けることになる。
孤児薬の指定と排他性
1983年の“孤児医薬品法”によれば、FDAは、米国で20万人未満の影響を与える珍しい疾患または状態を治療するための薬剤を孤児の称号を与えることができ、または米国で20万人を超える影響を与える場合、米国でこのような疾患または状態を治療する製品を開発および提供するコストを製品の販売から回収することができる合理的な期待がない。秘密保護協定を提出する前に、孤児としての指定を要請しなければならない。孤児製品を開発する会社は、合格した臨床試験に税金免除を提供し、申請料を免除することを含むいくつかのインセンティブを得る資格があるにもかかわらず、孤児製品の指定は、監督審査および承認過程においていかなる利点を伝達したり、監督審査および承認過程の持続時間を短縮したりすることはない。
孤児として指定された製品がその後、このような指定された疾患または状況を有するFDAの最初の承認を得た場合、製品は、7年間の市場排他期を有する権利があり、その間、FDAは、限られた場合、例えば、後続の製品が孤児排他性を有する製品よりも優れた臨床的利点を示すか、または元の申請者が十分な数の製品を生産することができない限り、同じ適応の同じ治療薬を販売するために、任意の他の出願を許可することができない。しかし,競争相手は孤児製品が排他的な適応を有する異なる治療薬の承認,あるいは孤児製品が排他的な適応を有するのではなく,同一の治療剤の異なる適応の承認を得る可能性がある。もし競争相手が私たちの前に同じ適応の同じ治療剤の承認を得たら、孤立した製品排他性は私たちの製品が臨床的に優れていることを証明できない限り、7年以内に私たちの製品が承認されることを阻止するかもしれない。指定された孤児製品が発売承認された場合,その適応範囲は指定された範囲よりも広く,孤児排他性を得る資格がない可能性がある。さらに、FDAが後に指定された要求に重大な欠陥があると判断した場合、または製品を承認する製造業者が、このようなまれな疾患または疾患患者の需要を満たすのに十分な数の製品が保証されない場合、米国における孤児薬の独占営業権を失う可能性がある。
KymeraのSTAT 3分解剤KT−333は2022年9月に開発されており,再発および/または難治性リンパ腫や固形腫瘍の治療に用いられ,2022年6月に末梢T細胞リンパ腫(PTCL)治療の孤児薬に指定されたのに続き,KT−333は米国食品医薬品局により皮膚T細胞リンパ腫(CTCL)を治療する2番目の孤児薬として承認されている。2023年6月、Kymeraが開発しているMDM 2分解剤KT-253は、FDAによって急性骨髄性白血病の孤児薬として承認された。これらの指定は,まれな疾患を治療する薬剤の開発を奨励するインセンティブを提供する。
薬品開発と審査計画を加速する
FDAは新薬開発と審査を促進と加速するためのいくつかの計画を維持し、深刻或いは生命に危害を及ぼす疾病或いは条件の治療において満たされていない医療需要を満たす。これらの計画には、迅速チャネル指定、突破的治療指定、優先審査、加速承認、およびプラットフォーム技術指定が含まれており、これらの計画の目的は、FDA規格の開発および審査手順よりも早く患者に提供するために、重要な新薬の開発または審査を加速することである。
新薬が重篤または生命に危険な疾患または状態を治療することを意図し、そのような疾患または状態を解決する満たされていない医療需要の潜在力を示す場合、迅速なチャネル指定を受ける資格がある。迅速チャネルはスポンサーに指定され、臨床前と臨床開発中にFDAと相互作用することで、より多くの機会を提供します
46
マーケティング申請が提出されると、スクロール審査が行われる可能性があり、これは、機関がスポンサーが完全な出願を提出する前に、マーケティング申請の内容の一部を審査することができ、以下に説明する優先審査を行うことができることを意味する。
さらに、1つの新薬が、深刻または生命に危険な疾患または状態を治療することを意図し、初歩的な臨床証拠が、1つまたは複数の臨床的重要な終点において、例えば、臨床開発早期に観察された重大な治療効果のような既存の治療法よりも顕著な改善を示す可能性がある場合、画期的な治療指定を受ける資格がある可能性があることを示す。画期的な治療指定は迅速チャネル指定のすべての機能を提供し,また,第1段階では効率的な薬物開発計画の密な指導や,適切な場合に高度管理者や経験豊富な審査者を学際的な審査に参加させることを含むFDAの開発加速に対する組織承諾が開始された。
迅速なチャネルまたは突破療法の称号を有する製品を含むFDA承認を提出する製品は、優先的な審査指定および承認の加速を含むFDAの審査および承認プロセスを加速させるための追加の計画を得る資格がある可能性もある。製品が重篤な疾患または状態の治療、診断または予防において顕著な安全性または有効性を提供する可能性がある場合、優先審査を受ける資格がある。優先審査によると、FDAは6ヶ月以内に申請を審査しなければなりませんが、標準審査は10ヶ月です。
さらに、代替終点に対して臨床的利益を合理的に予測する可能性のある製品の影響が証明されるか、または臨床終点への影響が、不可逆的発症率または死亡率の影響よりも早く測定されることができ、病状の重症度、希少性または流行率、および代替治療が利用可能または不足していることを考慮すると、加速承認を得る資格がある。
加速承認は、通常、スポンサーが追加的な承認を行うことに同意した後の研究に依存して、製品の臨床的利益を検証および説明するために、2022年の食品·薬物総合改革法案(FDORA)によれば、FDAは、必要に応じて、承認前または承認が加速された製品の承認日後の特定の期間内に、そのような試験を要求することが許可されている。FDORAによれば、FDAは、例えば、検証試験が製品の予期される臨床的利益を検証することができない場合、承認の加速下で承認された薬物または適応の承認を撤回することができるように、手続きを加速させる権限を増加させる。さらに、承認を加速させることが検討されている製品については、FDAが別の通知がない限り、FDAは、通常、上場承認後120日以内に伝播または発行しようとしているすべての広告および販売促進材料を、承認前審査期間内に機関に提出し、上場承認後120日後に、すべての広告および販売促進材料を少なくとも最初の伝播または発行の所定の時間前に30日前に提出しなければならない。
医薬品および生物製品条例によれば、以下の場合、医薬品または生物製品またはそれによって使用されるプラットフォーム技術が、指定されたプラットフォーム技術として指定される資格がある:(1)プラットフォーム技術が機密協定に従って承認された医薬品の使用に組み込まれているか、または(2)許可または許可された薬品の発起人によって提出された予備証拠、または薬物出願において提出されたデータに対する参照権が付与された発信者によって提出された予備証拠は、プラットフォーム技術が品質、製造または安全に悪影響を与えることなく、1つ以上の薬物使用に組み込まれる可能性があることを示す。(3)適用者が提出したデータまたは情報は、プラットフォーム技術の導入または利用が薬物開発または製造過程および審査過程に顕著な効率をもたらす可能性があることを示している。スポンサーは、IND出願を提出した間または後の任意の時間に、IND出願を要求対象とするプラットフォーム技術を組み込むか、または使用する指定されたプラットフォーム技術としてFDAに指定することを要求することができる。指定された場合、FDAは、プラットフォーム技術の使用または組み込まれた薬物の任意の後続の元のNDAの開発および検討を加速することができる。
1つの製品が1つまたは複数のこれらの計画の条件に適合していても、FDAは、製品がもはや資格条件を満たしていないと後で決定することができ、またはFDAが審査または承認する期間が短縮されない可能性がある。また、迅速チャネル指定、突破的治療指定、優先審査、加速承認は承認の科学的あるいは医学的標準を変えることはなく、承認を支持するために必要な証拠の質を変えることもないが、開発や審査過程を加速する可能性がある。
小児科情報と小児科排他性
改訂された“小児科研究平等法”によれば、いくつかのNDAおよびいくつかのNDA補充剤は、すべての関連する小児科亜群において主張される適応の安全性および有効性を評価し、安全かつ有効な各小児科亜群に対する製品の投与および投与をサポートするためのデータを含まなければならない。FDAは小児科データの提出を延期することを許可するか、またはすべてまたは部分的な免除を与える可能性がある。FD&C法案は、新しい活性成分、新しい適応、新しい剤形、新しい用量レジメンまたは新しい投与経路を含む薬物がマーケティング申請を提出することを計画しているスポンサーは、第2段階会議終了後60日以内に予備小児科研究計画を提出しなければならない、またはそのような会議がない場合には、第3段階または第2/3段階試験が開始される前に予備小児科研究計画またはPSPを早期に提出しなければならない。
47
最初のPSPは、研究目標および設計、年齢群、関連する終点および統計方法、またはそのような詳細な情報を含まない理由、ならびに小児科研究データおよび支援情報の提供を延期または完全または部分的に免除することを要求する任意の要件を含む、スポンサー計画によって行われる1つまたは複数の小児科研究の概要を含まなければならない。FDAとスポンサーはPSPについて合意しなければならない。臨床前研究,早期臨床試験および/または他の臨床開発計画から収集したデータに基づいて小児科計画の変化を考慮する必要があれば,スポンサーは合意した初期PSPに対する修正案を随時提出することができる。
1つの薬物はまた、米国で小児科市場の独占経営権を獲得することができる。小児科独占経営権を獲得した場合、すべての製剤、剤形、活性部分の適応、および特許条項の既存の独占経営期間は6ヶ月増加する。この6ケ月の専営権は他の独占保護終了時から始まり、自発的に1つの小児科試験或いは複数の小児科試験を完成させた上で授与することができ、条件は小児科専営権を付与する時に、9ヶ月以上の残りの期限があることである。
アメリカの薬品承認後の要求
FDAによって生産または流通を許可された医薬品は、記録保存、定期報告、製品サンプルおよび流通、追跡および追跡、製品不良反応の報告、販売促進および広告要件の遵守に関する要求を含むFDAの普遍的かつ持続的な規制を受けなければならず、その中には、未承認の用途または患者集団に限定された製品(“ラベル外使用”と呼ばれる)、および業界支援の科学的および教育活動の制限が含まれる。処方薬製品のメーカーと他の薬品サプライチェーンに参加する各当事者はまた、製品追跡と追跡要求を遵守し、偽、移転、窃盗、故意に混入した製品または本来米国で流通するのに適していない製品をFDAに通報しなければならない。医師はラベル外の用途のために合法的な製品を開く可能性があるが、製造業者および製造業者を代表する個人は、そのような用途をマーケティングまたは普及させてはならない。FDAや他の機関はラベル外用途の普及を禁止する法律や法規を積極的に実行しており、ラベル外用途を不当に普及させていないことが発見された会社は、連邦や州当局の調査を含む重大な責任に直面している可能性がある。処方薬宣伝材料は初回使用または最初の発表時にFDAに提出されなければならない。さらに、薬物が適応、ラベルまたは製造プロセスまたは施設の変化を含む任意の修正がある場合、出願人は、新しいNDAまたはNDAサプリメントの承認を得るために提出および提出を要求される可能性があり、これは、追加のデータまたは臨床前研究および臨床試験を開発する必要があるかもしれない。
FDAはNDAを承認するための条件として、いくつかの承認後の要求を加えるかもしれない。例えば、FDAは、製品の商業化後の安全性および有効性をさらに評価および監視するために、第4段階の臨床試験およびモニタリングを含む上場後試験を要求する可能性がある。
さらに、医薬品の生産および流通承認に参加する医薬品メーカーおよびその下請け業者、および製品、成分およびコンポーネントを提供する下請け業者は、FDAおよびいくつかの州機関にその機関を登録し、我々と私たちの契約製造業者に特定の手続きおよび文書要求を適用したcGMPを含む現在行われている規制要件を遵守することを確実にするために、FDAおよびいくつかの州機関の定期的な抜き打ち検査を受けなければならない。法律および法規の要求を遵守しない製造業者は、警告状、生産停止、製品差し押さえ、禁止、民事処罰、または刑事起訴のような法律または法規行動に直面する可能性がある。また、持続的で年に1回の処方薬製品計画使用料もある。
その後、製品には、予期されない深刻度または頻度の不良事象、または生産プロセス、または規制要件を遵守できなかったことを含む、以前に未知の問題が存在し、新しいセキュリティ情報を追加するために承認されたラベルの改訂を招く可能性があり、新しい安全リスクを評価するために発売後の研究または臨床試験を要求するか、またはREMSに従って流通または他の制限を実施することが可能であることが分かった。他の他の潜在的な結果には
48
仲間診断学の規制
診断に伴い、特定の治療製品から利益を得る可能性が最も高い患者を決定すること、特定の治療製品を使用することによって深刻な副作用のリスクを増加させる可能性がある患者を決定すること、またはより高い安全性または有効性を達成するために治療を調整することを目的とする特定の治療製品治療の反応を監視すること。診断に伴いFDAは医療機器として規制されている。アメリカでは、医療機器の設計と開発、臨床前と臨床試験、発売前の承認或いは承認、登録と上場、製造、ラベル、貯蔵、広告と販売促進、販売と流通、輸出と輸入及び発売後の監督などの方面は、すべて“食品と薬物管理局法案”及びその実施条例とその他の連邦と州の法律法規の管轄を受けている。免除またはFDAが法執行自由裁量権を行使することを得ない限り、診断テストは、一般に、商業化前に市場許可またはFDAの承認を得る必要がある。医療機器に適用されるFDAマーケティング許可の2つの主要なタイプは、上場承認前通知または510(K)、および上場前承認申請またはPMAを承認することである。
医療デバイスの510(K)許可を得るか、または510(K)の許可を得たデバイスをいくつか修正するためには、製造業者は、提案されたデバイスが以前に承認された510(K)デバイスと実質的に等しいか、または1976年5月28日以前に商業販売中の改訂前デバイス、またはFDAがPMAの述語デバイスの提出を要求していないことを証明する発売前通知を提出しなければならない。装置が用語装置と実質的に等しいと判断した場合、FDAは、提案された装置を言語装置と比較し、予期される用途、技術、設計、および安全性および有効性に影響を与える可能性のある他の特徴において、主語装置が述語装置に相当するかどうかを評価する。FDAが、対象装置が述語装置と実質的に等しいと判断した場合、対象装置の上場を許可することができる。510(K)の発売前通知経路は、通常、申請完了日から3~12ヶ月を必要とするが、より長い時間を要する場合がある。
PMAは有効な科学的証拠支持が必要であり、これは通常大量のデータを必要とし、技術、臨床前、臨床と製造データを含み、この設備の安全性と有効性を証明し、FDAを満足させる。診断テストの場合、PMAは通常、分析および臨床検証研究に関するデータを含む。PMA審査の一部として、FDAは、製造業者が設計、テスト、制御、文書、および他の品質保証手順に従うことを要求する品質システム法規(QSR)に適合することを保証するために、1つまたは複数の製造施設を承認前に検査する。法規によると、FDAの初期PMAの審査には6ヶ月を要するが、この過程は通常より長い時間を必要とするが、完成には数年かかる可能性がある。FDAがPMAおよび製造施設の評価に有利である場合、FDAは、PMAの最終承認を保証するために、一般に満たされなければならないいくつかの条件を含む承認状または承認状を発行するであろう。PMAまたは製造施設に対するFDAの評価が有利でない場合、FDAは、PMAの承認を拒否するか、または承認できない手紙を発行する。承認できない手紙は、申請の不足点を概説し、可能な場合には、PMAを承認するために必要な条件を決定する。承認されると、承認後の要求、承認条件、または他の規制基準が遵守されていない場合、または最初のマーケティング後に問題が発見された場合、FDAはPMAの承認を撤回する可能性がある。
FDAは2014年7月31日、“体外随伴診断装置”の開発と承認手順を述べた最終指導文書を発表した。ガイドラインによれば、診断テストに依存して使用される新しい治療製品のために、診断装置が対応する治療製品を安全かつ有効に使用するために不可欠である可能性がある場合、FDAは同時開発が不可能である可能性があることを認識しているにもかかわらず、治療の同時開発および承認または承認の前に適用されなければならない。しかしながら、1つの薬剤がセット診断なしに安全または有効に使用できない場合、FDAのガイドラインは、診断装置の承認または許可がない場合、通常はその薬剤を承認しないことを示している。FDAは2016年7月にガイドライン草案を発表し、体外随伴診断設備と治療製品の共同開発の原則を述べた。ガイドライン草案は治療製品及びそれに対応する体外随伴診断の開発と同時マーケティング許可を指導する原則を記述した。
承認或いは承認を得ると、セット診断設備はFDAのQSR要求、不良事件報告、リコールと是正及び製品マーケティング要求と制限を含む上場後の要求を守らなければならない。医薬品メーカーと同様に,キット診断メーカーはいつでもFDAから抜き打ち検査を受け,その間,FDAは製品(S)と我々の施設を審査し,その当局の規定に適合することを確保する。
49
その他の規制事項
候補製品の製造、販売、普及、および製品の承認または商業化後の他の活動は、適用される場合にも、米国FDA以外の多くの規制機関によって規制されなければならず、これらの規制機関は、連邦医療保険および医療補助サービスセンター(CMS)、衛生·公衆サービス部の他の部門、法務省、薬品監督管理局、消費財安全委員会、連邦貿易委員会、職業安全·健康管理局、環境保護局、および州と地方政府と政府機関を含む可能性がある。
他の医療保険法
医療提供者、医師、第三者支払者は、私たちが市場で承認された任意の製品の推薦と処方において主な役割を果たすだろう。私たちの業務運営および現在または将来の第三者支払者、医療提供者、医師との任意の手配は、私たちが上場許可を得た任意の薬物の業務または財務的配置および関係を制限する可能性があり、広範に適用される詐欺や乱用、および他の医療法律および法規に直面する可能性があります。米国では、これらの法律には、州と連邦反リベート、虚偽声明、医師透明性、薬品価格報告、および患者データプライバシーおよび安全法律法規が含まれているが、これらに限定されない。私たちの業務がこのような法律または任意の他の適用可能な政府法規に違反していることが発見された場合、行政、民事および刑事罰、損害賠償、罰金、返還、削減または再構成業務、誠実な監督と報告義務、連邦および州医療計画から除外され、責任個人が監禁される可能性があるが、処罰を受ける可能性がある。
これらの法律の範囲も執行も不確定であり,現在の医療改革環境下では,特に適用の前例や法規が乏しい場合には,急速な変化が生じる可能性がある。連邦と州法執行機関は最近、医療保険会社と医療保健提供者の間の相互作用の審査を強化し、医療保健業界の一連の調査、起訴、有罪判決と和解を招いた。政府当局は、私たちの業務やり方が現在または未来に適合していないと結論するかもしれないが、詐欺および乱用または他の医療保健法律および法規の適用に関する現行または未来の法規、法規または判例法に関連している。もし私たちの運営がこれらの法律または任意の他の私たちに適用される可能性のある政府法規に違反していることが発見された場合、私たちは重大な民事、刑事と行政処罰、損害賠償、罰金、監禁、返還、政府援助の医療計画から除外された(例えばMedicareおよびMedicaid)、名声損害、追加の監督、報告義務に直面する可能性があり、もし私たちが会社の誠実な合意または同様の和解の制約を受けて、これらの法律違反に関する疑惑を解決し、私たちの業務を削減または再編する。もし私たちがそれと業務往来があることを期待している任意の医師や他の医療提供者や実体が適用法を遵守していないことが発見された場合、彼らは類似した行動、処罰、そして制裁を受けるかもしれない。ビジネス手配が適用される医療保険法に適合していることを確保し,政府当局が行う可能性のある調査に対応することは,時間や資源がかかる可能性があり,会社の業務への関心を分散させる可能性がある。
保険カバー範囲と精算
米国や他国の市場では,自分の病状に応じて処方治療を受けている患者や処方サービスを提供する提供者は,通常第三者支払者に依存してすべてまたは一部の関連医療費を精算している。したがって,候補製品が承認されても,その製品の販売は,米国の連邦医療保険や医療補助などの政府健康計画,商業健康保険会社,管理型医療機関を含む第三者支払者にある程度依存し,その製品に保険を提供し,十分な精算レベルを確立する程度である。米国では,新薬精算に関する主な決定は通常医療保険と医療補助サービスセンター(CMS)によって行われており,これは米国衛生·公衆サービス部の機関である。CMSは新薬がどの程度連邦医療保険の下でカバーと精算されるかを決定し、個人支払者はよくCMSに大きく従う。第三者支払者の間には統一された薬品保険と精算政策がない。そのため、薬品の保証範囲と精算範囲は支払人によって異なる。第三者支払者が製品に保険を提供するか否かを判定する過程は、価格又は販売率を設定する過程と分離することができ、保険が承認されると、支払者は製品に料金を支払うことができる。第三者支払者は、徴収された価格に挑戦し、医療の必要性を審査し、医療製品およびサービスの費用対効果を審査し、コストを管理するために制御を実施することが増えている。第三者支払者は、保証範囲を承認リスト上の特定の製品に制限することができ、特定の適応のすべての承認製品を含まない可能性がある配合表とも呼ばれる。
販売が許可される可能性のある任意の製品の保険および精算を確保するために、会社は、製品の医療必要性および費用効果を証明するために高価な薬物経済学的研究を行う必要があるかもしれないが、これは、FDAまたは他の同様の規制承認を得るのに必要なコスト以外の追加支出が必要となるであろう。また、会社は購入者、個人健康計画、または政府医療計画に割引を提供する必要があるかもしれない。それにもかかわらず、候補製品は医学的に必要または費用効果があると考えられないかもしれない。
50
製品が承認されると、第三者支払者が製品を保証しない決定は医師の使用率を減少させ、販売、私たちの運営、財務状況に実質的な悪影響を及ぼす可能性がある。また、第三者支払者が製品に保険を提供することを決定することは、十分な販売率を承認することを意味するわけではない。また、1人の支払人が1つの製品に保険を提供することを決定することは、他の支払人もその製品に保険や精算を提供することを保証することはできず、また、支払者によって保険や精算レベルが大きく異なる可能性がある。
医療コストの抑制は連邦、州と外国政府の優先順位となっており、製品価格はこの努力の重点である。各国政府はコスト制御計画の実施に大きな興味を示し、価格制御、精算制限と代替後発薬の要求を含む。価格制御及びコスト制御措置を講じ、既存の制御及び措置を有する司法管区においてより限定的な政策をとることにより、任意の承認された製品の販売から得られる収入をさらに制限することができる。保証政策と第三者支払人の販売率はいつでも変化する可能性があります。1社またはその協力者が規制承認を受けた1つまたは複数の製品が有利な引受·精算状態を獲得しても、将来的にはあまり有利ではない保証政策や精算料率が実施される可能性がある。
現在と未来の医療改革立法
米国や一部の外国司法管轄地域では、医療システムに関する複数の立法·規制改革および提案された改革が継続して存在する可能性があり、医療の獲得性を拡大し、医療の質を向上させ、医療のコストをコントロールまたは低減することを目的としている。例えば、2010年3月、米国議会は、政府医療計画下の製品のカバー範囲の変更や支払い方法を含む“平価医療法案”を公布した。“平価医療法案”には、私たちの潜在的な候補製品に非常に重要な条項が含まれている
公布以来、“平価医療法案”のいくつかの面は多くの司法、行政、行政、立法方面の挑戦を受けており、今後も“平価医療法案”に対してより多くの挑戦と修正案を提出することが予想される。2021年6月17日、米国最高裁はいくつかの州がACAに対して提出した最新の司法挑戦を却下したが、ACAの合憲性を具体的に裁くことはなかった。バイデン総裁はすでに複数の行政命令を発表し、処方薬のコスト問題を解決しようとしている。バイデン政府の他の医療改革措置や他の挑戦,ACAの廃止または代替の努力(あれば)が我々の業務にどのように影響するかは不明である。
“平価医療法案”が公布されて以来、米国は他の立法改正も提出し、可決した。他の事項を除いて、2011年の“予算制御法”には、医療保険提供者に支払われる医療保険支払いを前期当たり合計2%削減することが含まれており、2013年4月に施行され、その後、国会が追加行動をとらない限り2032年まで有効となる立法改正が行われている。2012年の“米国納税者救済法”の署名が法律となり、病院、画像センター、がん治療センターを含むいくつかの医療サービス提供者に支払う医療保険をさらに減少させ、政府が医療サービス提供者に多額の支払いを取り戻す訴訟時効を3年から5年に延長した。
51
また、支払い方法は医療立法と規制措置の影響を受ける可能性がある。例えば、CMSは、バンドル支払いモードを含む新しい支払いおよび配信モードを開発することができる。また、最近政府はメーカーがその商業製品の価格設定の方式に対してより厳格な審査を行い、国会が数回の調査を行い、州と連邦立法を提出し、製品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、政府の薬品に対する計画補償方法を改革することを提出し、公布した。その中のいくつかの措置および他の措置は発効するために追加の許可を必要とする可能性があるが、国会と現政府は薬品コストを制御するために新しい立法および/または行政措置を求め続けると表明している。米国の個別州も、価格または患者の精算制限、割引、ある製品への参入の制限、マーケティングコストの開示および透明性措置を含む立法および実施により、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。
2022年の“インフレ率低減法案”(IRA)中のいくつかの条項は異なる程度で私たちの業務に影響を与える可能性があり、その中には2024年から連邦医療保険D部分の受益者の自己支出上限を7,050ドルから2,000ドルに下げ、それによってカバー不足を解消する;連邦医療保険D部分精算の多くの薬物に対してメーカーの新しい財務責任を規定する;アメリカ政府はある高コストの薬物と生物製品の連邦医療保険B部分とD部分の価格設定について交渉することを許可し、模造薬や生物類似競争が存在しない;及び会社に薬品価格の増加がインフレより速い速度で連邦医療保険にリベートを支払うことを要求する。また,IRAによると,孤児薬は連邦医療保険薬品価格交渉計画の影響を受けないが,孤児名があることを前提としており,唯一承認された適応はこの疾患や状況に対するものである。1つの製品が複数の孤児の称号または複数の承認の適応を得た場合、それは孤児薬物免除を受ける資格がない可能性がある。アイルランド共和軍の実施は現在行われている訴訟の影響を受けており,これらの訴訟はアイルランド共和軍の医療保険薬品価格交渉計画の合憲性を疑問視している。Ireland共和軍が私たちの業務と医療産業全体に及ぼす影響はまだ明確ではない。
米国以外では、製品のカバー範囲の確保と十分な支払いも課題に直面している。多くの国で、処方薬の価格設定は政府によって規制されている。政府当局との価格交渉は製品が監督管理の許可を得る範囲をはるかに超えている可能性があり、臨床試験を行い、製品のコスト効果を他の利用可能な治療法と比較する必要があるかもしれない。このような臨床試験を行うことはコストが高く、商業化の遅延を招く可能性がある。
連合では、様々な国の価格設定と補償プログラムの差が大きい。一部の国では、補償価格を合意した後にのみ、製品を販売することができると規定されている。いくつかの国は、特定の候補製品のコスト効果を現在利用可能な治療法またはいわゆる衛生技術評価と比較して、精算または価格設定の承認を得るために、追加の研究を達成することを要求するかもしれない。例えば、欧州連合は、その国の健康保険制度が補償を提供する製品範囲を制限し、人が使用する医療製品の価格を制御するために、その加盟国に様々な選択を提供している。EU加盟国は製品の具体的な価格を承認することができ、製品を市場に投入する会社の収益力を直接または間接的に制御する制度を採用することもできる。他の会員国は会社が自分の製品価格を固定することを許可しているが、処方数を監視し、処方を制限するために医師に指導意見を発表した。最近、EUの多くの国が薬品割引要求を高め、各国が医療支出を管理しようとしていることに伴い、これらの努力は継続される可能性があり、特にEUの多くの国が深刻な財政危機と債務危機を経験した場合である。全体的には,医療コスト,特に処方薬の下り圧力が大きくなっている。そのため、新製品の参入にはますます高い壁が設けられている。政治、経済、規制面の事態は定価交渉をさらに複雑化させる可能性があり、補償を受けた後、定価交渉が継続される可能性がある。欧州連合の各加盟国が使用する参考定価、および平行貿易、すなわち低価格と高価な加盟国間の裁定は、価格をさらに下げることができる。薬品に対して価格規制や精算制限を実施することが保証されていない国は、これらの国で承認されれば、任意の製品に対して有利な精算と定価手配を許可する。
52
他の連邦や州の法律または要求に適合する
もし私たちが開発可能な任意の製品が総務庁連邦供給スケジュールの許可ユーザに提供される場合、他の法律と要求が適用される。製品はアメリカの“毒物防止包装法”に適用される児童保護包装要求に適合しなければならない。製造、ラベル、包装、流通、販売、販売促進、および他の活動は、連邦および州消費者保護および不正競争法によって制限される可能性があり、私たちが受ける可能性のある他の要求を受ける可能性がある。
医薬製品の流通は他の規定と条例を遵守しなければならず、広範な記録保存、許可、貯蔵と安全要求を含み、許可されていない医薬製品の販売を防止する。
これらの法律または規制要件のいずれかを守らなければ、会社は法律または規制行動に直面する可能性がある。場合によっては、適用される規制要件を満たさないことは、刑事起訴、罰金またはその他の処罰、禁止、連邦医療計画から除外されること、リコールの要求、製品の差し押さえ、生産の完全または部分的な一時停止、製品の承認の拒否または撤回、再表示または再包装、または政府契約を含む会社の供給契約の締結を拒否する可能性がある。私たちがこれらの法律に違反する請求や行動は、たとえ私たちが弁護に成功しても、巨額の法的費用を招き、私たちの経営陣の業務運営への注意をそらす可能性があります。私たちの販売を禁止したり、販売したり、撤回したりする将来の製品は、不利な方法で私たちの業務に大きな影響を与える可能性があります。
規制、法規、または既存の規制の解釈の変化は、(I)私たちの製造スケジュールを変更すること、(Ii)製品ラベルまたはパッケージを追加または修正すること、(Iii)私たちの製品をリコールまたは停止すること、または(Iv)追加の記録保存要件を要求することなど、私たちの将来の業務に影響を与える可能性がある。このような変化を強制的に実施すれば、私たちの業務運営に悪影響を及ぼす可能性がある。
他のアメリカの環境、健康、安全の法律法規
私たちは多くの環境、健康と安全法律と法規によって制約される可能性があり、それらの管理実験室手続きと危険材料と廃棄物の処理、使用、貯蔵、処理と処理の法律と法規を含むかもしれない。時々、将来、私たちの業務は、化学品および生物学的材料を含む危険かつ可燃性材料の使用に関連する可能性があり、危険な廃棄物製品を生成する可能性もある。これらの材料や廃品を処分する契約を第三者と締結しても、これらの材料による汚染や傷害のリスクを完全に解消することはできません。私たちの危険な材料を使用または処分して汚染または損傷をもたらす場合、私たちはそれによるいかなる損害に責任を負う可能性があり、いかなる責任も私たちの資源範囲を超える可能性がある。民事や刑事罰金やこのような法律や法規を遵守しない罰に関する巨額の費用を招く可能性もある。
私たちは従業員の負傷によって生じる可能性のある費用と費用を支払うために労働者補償保険を維持していますが、この保険は潜在的な責任に対応するのに十分ではないかもしれません。しかし、私たちは私たちが提起した環境責任や有毒侵害請求に保険を提供することはできない。
また、現在または未来の環境、健康、安全法律法規を遵守するためには、巨額のコストが生じる可能性がある。現在または未来の環境法律法規は私たちの研究、開発、または生産努力を損なうかもしれない。しかも、このような法律法規を遵守しないことは巨額の罰金、処罰、または他の制裁につながる可能性がある。
アメリカ国外の薬品に対する政府の監督管理
米国国外で任意の製品をマーケティングするためには、他の国/地域の安全性と有効性に関する多くの異なる監督管理要求、及び臨床試験の管理、マーケティング許可或いは代替規制経路の決定、わが製品の製造、商業販売と流通などの要求を遵守する必要がある。例えば、連合王国および欧州経済地域、または欧州経済区(EU加盟国にアイスランド、リヒテンシュタイン、およびノルウェーを加えて構成される)では、医薬製品は中央プログラムまたは国家プログラムを介して販売許可を得なければならない。
53
現在連合王国(大ブリテンおよび北アイルランド連合王国を含む)はEUを離脱しており、イギリスは集中マーケティング許可のカバーを受けなくなっている(北アイルランド議定書によると、現在北アイルランドでは集中マーケティング許可が認められ続けている)。2024年1月1日、イギリスの医薬品·保健製品規制機関(MHRA)は新しい国際認可枠組みを構築し、この枠組みによると、MHRAはイギリスのマーケティング許可を付与するかどうかを考慮する際に、EMAおよびいくつかの他の規制機関がマーケティング許可を承認することに関する決定を考慮する可能性がある。MHRAはまた、EU加盟国が分散または相互承認手続きによって承認されたマーケティング許可を考慮して、イギリスまたはイギリスでマーケティング許可をより迅速に承認することを期待する権利がある。
EUでは、発売が許可されている治療適応革新薬(すなわち参考製品)は、8年間のデータ独占と追加2年間の市場独占販売許可を得る資格がある。データ排他期は、模倣薬または生物類似薬申請者がEUで模倣薬または生物類似薬の発売許可を申請することを禁止する時、参照製品が初めてEUで許可を得た日から8年以内に、製品ファイルに含まれる革新者の臨床前および臨床試験データを参照する。市場排他期は、参考製品がEUで最初の許可を得た10年後まで、成功した模倣薬または生物類似申請者がその製品をEUで商業化することを禁止している。10年の最初の8年間に、マーケティング許可保持者が1つまたは複数の新しい治療適応の許可を得た場合、許可前の科学的評価期間中に、これらの適応は既存の療法と比較して有意な臨床的利益をもたらすことができると考えられる場合、10年間の市場専門期間は最大11年に延長することができる。1つの製品がEMAによって革新的な医薬製品とみなされることは保証されず、しかも製品はデータ独占性を得る資格がない可能性がある。1つの製品が革新的な医薬製品とみなされても、革新者が所定のデータ独占期間を得るために、別の会社は、同社が薬物試験、臨床前試験、および臨床試験の完全かつ独立したデータパケットを有するMAAに従ってマーケティング許可を得ることを前提として、その製品の別のバージョンを販売することができる。
EUでは、“孤児医療製品”を指定する基準は原則として米国と類似している。EUでは、1つの医療製品が孤児として指定されることができ、条件は、(1)生命または慢性衰弱に危険な疾患の診断、予防または治療を目的としていること、(2)申請されたとき、そのような疾患がEUで10,000人中5人以下に影響を与えること、または(B)孤児の身分によるメリットがなければ、その開発に必要な投資が合理的であることを証明するためにEUで十分な見返りをもたらす可能性が低いことである。および(3)このような疾患を満足できる診断、予防または治療する方法がEUに市販されていないか、または、そのような方法があれば、製品はその疾患の影響を受けている人に大きな利益を与えるであろう。孤児医薬製品は減料や無料などの財政的奨励を受ける資格があり、マーケティングの許可を得た後、
54
承認された治療適応は10年間の市場独占経営権を得る権利がある。この10年間の孤児市場の専門期間内に、MAAを受け入れてはならず、同じ治療適応の類似薬品に対して発売許可を与えてはならない。類似医薬製品“の定義は、承認された孤児医薬製品に含まれる1つ以上の類似活性物質を含む医薬製品であり、同じ治療適応のためのものである。孤児製品はEUで追加2年間の市場排他性を得ることもでき,EUでは小児科研究の取り決め小児科調査計画が遵守されている。いかなる補充保護証明書やSPCの延期も孤児適応に対する小児科研究に基づくことはできない。5年目の終了時に、製品が指定された孤児の基準を満たしていないと判断された場合、例えば、製品の利益が十分に高く、市場排他性を維持するのが合理的であることを証明するのに十分でない場合、10年間の市場排他性は6年に減少することができる。さらに、以下の場合、いつでも同じ治療適応の類似製品に上場許可を付与することができる:(I)第2の出願人は、その製品が類似しているが、より安全で、より効果的で、または臨床的により良いことを証明することができる;(Ii)孤児製品を許可するマーケティング許可保持者は、2回目の孤児医薬品の申請に同意することができる、または(Iii)孤児製品を許可するマーケティング許可保持者は、十分な孤児薬を供給することができない。
EUのマーケティング許可を得る前に、出願人は、EMAが特定の製品の免除、カテゴリ免除、またはPIPに含まれる1つまたは複数の措置を許可しない限り、EMAによって承認された小児科調査計画(PIP)に含まれるすべての措置を証明しなければならない。すべての販売許可手続のそれぞれの要求は(EC)第1901/2006号条例、いわゆる小児科条例に規定されている。会社が認可された薬物のために新たな適応,薬物形式あるいは投与経路を増加させようとした場合にも,この要求は適用される。EMAの小児科委員会(PDCO)は、ある薬物の開発を延期することを許可する可能性があり、会社が成人に対する有効性と安全性を証明する十分な情報があるまで、会社が児童薬物の開発を延期することを許可する。小児薬の開発を必要としない場合、または適切でない場合、PDCOは、高齢者人口のみに影響を与える疾患のような免除を与えることもできる。MAAを提出するか、または既存のマーケティング許可を修正する前に、EMAは、会社が各関連PIPに列挙された合意された研究および措置を実際に遵守していると判断する。出願人がEUのすべての加盟国の上場許可を取得した場合、または欧州委員会が集中手続で付与されたマーケティング許可を取得し、小児科群に対する研究結果が製品情報に含まれている場合、否定的な場合であっても、最高特許保護期間を延長することによって6ヶ月の合格特許保護を追加的に取得する資格があり、最高技術特許出願を提出すると同時に、または最高特許保護期限の2年前のいつでも延長出願を提出することを前提としている。孤児薬品の場合、孤児市場の独占権は2年間延長することができる。この小児科奨励は特定の条件の制約を受け,PIPに適合したデータを開発·提出する際に自動的に獲得されない。
アメリカと類似して、EUの非臨床と臨床研究の各段階は厳格な監督管理によってコントロールされている。
2014年4月、EUは“臨床試験条例”(EU)第536/2014号(“臨床試験条例”)を採択し、2022年1月31日に“臨床試験指令2001/20/EC”に代わった。臨床試験条例はすべてのEU加盟国に直接適用され、これは各EU加盟国で国家立法を制定する必要がないことを意味する。“臨床試験条例”の一時的な条項は、2025年1月31日までに、すべての臨床試験はこの条例に移行しなければならないと規定しており、“臨床試験条例”はEUの臨床試験の承認を簡略化と簡素化することを目的としている。
この条例の主な特徴は,単一入口点による申請プログラムの簡略化,すなわち“臨床試験情報システム”,申請のための準備と提出のための単一文書,および簡略化された臨床試験スポンサー報告プログラム,および統一的な臨床試験申請評価プログラムであり,2つに分けられる。第1部はEUのすべての加盟国の主管当局によって調整評価され、加盟国の審査を参考にした後、臨床試験許可申請(関係加盟国)が提出された。二番目の部分は関連する各会員国によって個別的に評価される。臨床試験申請の評価には厳しい期限が設定されている。評価手続きにおける道徳管理委員会の役割は、EU加盟国に関する国家法律によって引き続き管轄されるだろう。しかし、全体的に関連するスケジュールは臨床試験条例によって定義されるだろう。
上記のEU規則は一般に欧州経済地域(EEA)に適用される。
EU規制枠組みの改革
欧州委員会は2023年4月に立法提案を提出し,実施されれば,EUの現在のすべての薬品(まれな疾患や児童を治療する薬品を含む)の規制枠組みに代わる。欧州委員会はその審査と承認のために欧州議会と欧州理事会に立法提案を提供した。2023年10月、欧州議会は立法提案を修正した報告書を発表した
55
これは欧州議会によって討論されるだろう。欧州委員会の立法提案が承認されると(修正または修正されない)、それらはEU法律として採択されるだろう。
アメリカの海外データ収集に対する政府の規制
もし私たちがEUで臨床試験を行ったら、私たちは追加的なプライバシー制限を受けるだろう。欧州経済圏の個人健康データの収集と使用は、2018年5月25日に施行された“一般データ保護条例”(GDPR)の規制を受けている。GDPRは、欧州経済地域内のデータ主体に商品またはサービスを提供するか、または欧州経済地域内のデータ主体の行動を監視することに関する個人データを処理する限り、欧州経済区に設立された任意の会社および欧州経済区以外に設立された会社の個人データの処理に適用される。GDPRは,データ当事者の同意に関する厳しい要求,個人データの使用に関するより多くの開示,“高リスク”処理に対するプライバシー影響評価,保持個人データの制限,強制的なデータ漏洩通知,“プライバシー設計”要求を含む個人データ制御者のデータ保護義務を強化し,処理者であるサービスプロバイダに直接義務を規定している.GDPRはまた,個人データをヨーロッパ経済圏以外に移転する国に対して厳しいルールを実施しており,これらの国では米国のような十分な保護レベルを確保できていない。GDPRや欧州経済圏加盟国の関連国データ保護法の要求を守らないと,GDPRからややずれる可能性があり,前会計年度の世界収入の4%または20,000,000ユーロの罰金を招き,金額の大きい者を基準とする可能性がある。また,GDPRはデータ主体にGDPR侵害による物質や非物質被害の権利を付与する。データ保護義務の変化の広さと深さを考慮して、GDPRの遵守を維持するには大量の時間、資源、費用が必要であり、新たなデータ保護ルールの遵守を確保するために追加の制御およびプログラムを作成する必要があるかもしれない。これまでGDPRの実行状況は限られており,特にバイオ製薬開発において,将来の任意の試験の新たな要求に対する確実な解釈の不確実性に直面しており,データ保護当局や裁判所が新しい法律を解釈する際に要求されるすべての措置を実施することに成功していない可能性がある。また、連合王国が2020年1月31日にEUを離脱したのに続き、GDPRは2020年12月31日の移行期間終了時に連合王国への適用を停止した。しかし、2021年1月1日現在、イギリスの2018年“EU(離脱)法案”は、GDPR(2020年12月31日に存在するものと同様だが、イギリスの何らかの具体的な改正が必要)をイギリスの法律に組み入れ、イギリスGDPRと呼ばれている。英国GDPRと2018年の英国データ保護法は,EUのデータ保護制度から独立した連合王国のデータ保護制度を規定しているが,EUのデータ保護制度と一致している。イギリスのGDPR違反は、金額が高い者を基準に、1750万GBまたは世界収入4%の罰金を招く可能性があります。英国はEU GDPR下の第3の国とされているが、欧州委員会は現在、英国がEU GDPRで十分な保護を提供していることを認める決定を発表しているため、EU由来の個人データの英国への移行は依然として制限されていない。EU GDPRと同様に,イギリスGDPRは英国以外の国への個人データの移行を制限しており,これらの国はイギリスから十分な保護を提供しているとはみなされていない。イギリス政府は、イギリスからヨーロッパ経済圏への個人データ転送が依然として自由に流動していることを確認した。
データ保護当局はGDPRをどのように強制的に遵守するかを求めており,大きな不確実性がある。たとえば,欧州経済圏で業務を行っている会社を当局がランダムに監査しているかどうか,あるいは当局がその権利が侵害されていると主張する個人の訴えを待っているかどうかは不明である.法執行の不確実性とGDPRコンプライアンスの確保に関連するコストは重く、私たちの業務、財務状況、運営結果、将来性に悪影響を及ぼす可能性があります。
もし私たちが第三者流通業者を使用すれば、これらの外国政府法規を遵守することは通常、これらの流通業者の責任であり、彼らは私たちが限られた統制された独立請負業者である可能性がある。
イギリスの離脱とイギリスの規制枠組み
イギリスは2020年1月31日にEUから正式に離脱した。EU製薬法は2020年12月31日に満了する英国に適用され続ける過渡期がある。しかし、EUとイギリスは、2021年1月1日から暫定的に適用され、2021年5月1日から正式に適用される貿易·協力協定すなわちTCAに合意した。TCAは、相互承認GMP、医薬製品の製造施設の検査、発表されたGMPファイルを含む薬品に関する具体的な条項を含むが、イギリスとEUの薬品法規の卸売相互承認は提供されない。現在、イギリスは“2012年人類薬品条例”(改正された)(北アイルランド議定書に基づき、EU規制枠組みは現在引き続き北アイルランドに適用されている)を採択し、医薬製品のマーケティング、普及、販売に関するEUの立法を実施している。したがって,イギリスの規制制度はEUの現行の規制制度と大きく一致しているが,イギリスの規制制度はEUから独立しており,TCAはイギリスとEUの薬品立法を相互に認めることを規定していないため,これらの制度は将来的に異なる可能性がある。しかし、TCAによると、イギリスの新しい国際承認手続きの下で、EUの薬品立法の卸売承認は行われていない
56
上述したように、MHRAは、イギリスのマーケティング許可の申請を考慮する際に、EMEA(およびいくつかの他の規制機関)がマーケティング許可を承認する決定を考慮する可能性がある。
2023年2月27日、イギリス政府と欧州委員会は、北アイルランド議定書、すなわち“ウィンザー枠組み”と呼ばれる新たな手配の代わりに、原則的な政治的合意を発表した。この新しい枠組みは、イギリスの医療製品の規制を含む北アイルランド議定書下の現行制度を根本的に変えた。原則として、MHRAはイギリス市場(すなわち大ブリテンおよび北アイルランド連合王国)に輸送されたすべての医療製品を承認する責任があり、EMAは北アイルランドへの医療製品の承認に何の役割も果たすことはないだろう。MHRAは、イギリスで販売されているすべての医薬製品のイギリス範囲での単一マーケティング許可を付与し、製品がイギリス各地で単一パッケージおよび単一ライセンスで販売できるようにする。2023年3月24日、EU·イギリス合同委員会はウィンザー枠組みを承認したため、イギリス政府とEUは立法措置を制定し、法律にする。2023年6月9日,MHRAはウィンザーフレームワークの薬品について2025年1月1日から適用すると発表した。
従業員と人的資本
2023年12月31日現在、私たちには187人の常勤従業員がいて、そのうち97人が医学や博士号を持っています。私たちの従業員チームのうち、143人の従業員が研究開発に従事し、44人の従業員が業務開発、金融、法律および一般管理と行政に従事している。私たちは従業員の知的資本が私たちの業務の重要な推進力であり、私たちの未来の見通しの鍵でもあると考えている。私たちは絶えず業務の需要と機会を評価し、内部の専門知識と能力とアウトソーシングの専門知識と能力の間でバランスを取る。現在、著者らはほとんどの臨床試験仕事を臨床研究機関にアウトソーシングし、そしてある薬物生産を契約メーカーにアウトソーシングする。薬物開発は複雑な仕事であり、広範な学科を越えた深い専門知識と経験が必要である。製薬会社は、大きさにかかわらず、限られた数の合格申請者を争って専門職を埋めている。私たちは私たちの報酬計画を密接に監視し、すべての従業員に非常に競争力があると思う報酬と保険福祉の組み合わせを提供し、私たちの株式計画に参加する。合格した応募者を誘致するために、会社は各従業員に基本賃金と現金目標ボーナス、医療とその他の福利厚生、株式給与を含む全面的な福利厚生を提供する。総報酬に占めるボーナス機会と株式報酬の割合は責任レベルに応じて増加する。実際のボーナス支出は業績に基づいている。
私たちのどの従業員も集団交渉協定の制約を受けず、労働組合や労働組合の代表にも拘束されない。私たちは私たちが従業員と仲がいいと思う。
私たちは個性化された発展計画、指導、コーチ、グループ訓練、会議出席、授業料精算を含む財務支援を通じて従業員の更なる発展を支援している。
施設
私たちの会社の本社はマサチューセッツ州のウォータータウンにあります。そこで借りて、約三十四、522平方フィートのオフィスと実験室空間を占有しました。私たちのウォータータウン賃貸契約の現在の期限は2030年3月31日に期限が切れます。私たちはレンタル期間を5年延長して、12ヶ月前に通知することができます。レンタル料は合意した市場価格で計算します。2021年12月、マサチューセッツ州ウォトタウンで100,624平方フィートのオフィスと実験室空間のレンタル契約を締結し、2024年2月に使用を開始します。この賃貸契約の初期レンタル期間は134ヶ月で、私たちは2つの連続した選択があり、それぞれ当時の市場価格でレンタル期間を5年間延長します。私たちは私たちが私たちの新しい施設に移転した後、私たちの現在の空間を第三者に又貸しするつもりだ。
私たちの新しい施設は私たちの現在の需要を満たすのに十分だと予想される。私たちの将来のビジネスニーズを満たすために、私たちは追加的または代替的な空間をレンタルするかもしれません。私たちは将来的に商業的に合理的な条項で適切な追加または代替空間を提供すると信じています。
私たちの会社情報は
私たちは2015年9月にデラウェア州法律に基づいて設立され、名称はProject HSC,Inc.です。私たちはKymera Treateutics,LLCの権益相続人であり、Kymera Treateutics,LLCはデラウェア州法律に基づいて2017年5月25日に設立された有限責任会社であり、私たちが発行したすべての普通株の前所有者でもあります。私たちの主な実行事務室はマサチューセッツ州ウォータータウン230号アーセナル広場200号にあります。郵便番号:02472、電話番号は(857285300)。ウェブサイトの住所はwww.kymeratx.comです。本サイトへの引用は非アクティブなテキスト参照のみであり,本サイトのコンテンツは引用によって本Form 10-K年次報告に組み込まれていると見なすべきではない.
57
利用可能な情報
我々のForm 10-K年次報告、Form 10-Q四半期報告、Form 8-K現在の報告、および1934年“証券取引法”第13(A)または15(D)節に提出または提供されたこれらの報告の任意の修正は、米国証券取引委員会または米国証券取引委員会に提出された後、合理的で実行可能な状況下でできるだけ早く私たちのサイトで無料で取得することができ、URLはwww.kymeratx.comである。
米国証券取引委員会は、米国証券取引委員会に電子的に提出された、我々および他の発行者に関する報告書、依頼書および情報声明、およびその他の情報を含む相互接続サイトを維持する。アメリカ証券取引委員会の相互接続サイトの住所は
当社のコーポレートガバナンス基準、商業行為と道徳基準、ならびに監査委員会、報酬委員会、指名およびコーポレートガバナンス委員会の規約のコピーは、私たちのウェブサイトwww.kymeratx.comの“Investors”の下に掲示されています。
58
第1 A項。RISK因子です。
私たちの業務は高い危険と関連がある。投資決定を下す前に、以下の説明およびまとめられた重大および他のリスクおよび不確実性、ならびに本年度報告書の他の情報、ならびに我々の連結財務諸表および関連説明、ならびに“経営陣の財務状況および経営結果に対する議論と分析”および“前向きな陳述に関する特別な説明”と題する部分を慎重に考慮しなければならない。我々の実際の結果は,本年度報告がForm 10−K形式で以下および他の場所でこれらの要因を説明したため,前向き陳述で予想された結果と大きく異なる可能性がある。以下に説明する危険は私たちが直面している唯一の危険ではない。次のいずれの事件や事態が発生しても、私たちの業務、財務状況、経営結果、将来性を損なう可能性があります。したがって、私たちの普通株の市場価格は下がるかもしれません。あなたは私たちの普通株へのすべてまたは一部の投資を失うかもしれません。新しいリスクは時々出現する可能性があり、いかなる要素や要素の組み合わせが私たちの業務、将来性、財務状況と運営結果に与える影響を予測できない。
私たちの財務状況と追加資本需要に関連するリスク
私たちは生物製薬会社で、運営の歴史は限られており、これまで薬品販売から何の収入も生じておらず、永遠に利益を上げないかもしれない。
生物製薬薬物開発は投機性の強い仕事であり、大きなリスクに関連している。私たちが2015年に設立し、2016年に初期資金を獲得して以来、これまで、私たちの運営は主に私たちの会社、業務計画、資金調達、研究と開発、私たちのパイプラインの開発、私たちの知的財産権の組み合わせの構築、臨床前研究の展開及び私たちの候補製品に対して第一段階の臨床試験を行うことに限られている。私たちは薬品販売から何の収入も得たことがない。私たちの現在のどの候補製品も規制部門の承認を受けていない。通常,発見から患者治療に利用可能な新薬の開発には数年を要する。したがって、私たちの未来の成功や生存能力に対するどんな予測も、私たちがより長い運営歴史を持っている時のように正確ではないかもしれない。また、経営歴史が限られている企業として、私たちは予測できない費用、困難、複雑な状況、遅延と他の既知と未知の要素、例えば流行病あるいはマクロ経済状況に関連する事態発展に直面する可能性がある。研究開発に専念している会社から、後期開発やビジネスを支援する能力のある会社に移行する必要があります。そのような移行で、私たちは成功しないかもしれない。
設立以来、重大な運営損失が発生しており、予測可能な将来も赤字が続くと予想されている。
設立以来、私たちのほとんどの努力と財政資源は、私たちの独自の標的タンパク質分解薬発見プラットフォーム、すなわちPegasusの開発に集中していますTMプラットフォーム、初期製品候補、そして私たちの協力とパートナー関係を支援します。これまで、私たちは主に私募株式融資における外部投資家と協力者への転換可能な優先株の発行と販売、私たちの協力と私たちの初公開株(IPO)、後続発行、パイプライン発行、市場販売計画前払い金によって、私たちの運営に資金を提供してきました。私たちが設立してから2023年12月31日まで、このような取引と協力を通じて合計約10.7億ドルの毛収入を集めました。2024年1月、引受割引と手数料、推定発行費用を差し引く前に、私たちの普通株と事前出資の引受証から約3.162億ドルを取得し、私たちの普通株を購入するために使用しました。2024年2月、Cowen and Company,LLCとの販売契約に基づき、普通株販売から販売手数料を差し引く前に5000万ドルを受け取りました。2023年12月31日現在、私たちの現金と現金等価物と投資は4.363億ドルです。設立以来、私たちは毎年純損失を出しており、2023年12月31日までの累計損失は5.308億ドルだった。2023年,2022年,2021年12月31日までの年度において,我々が報告した純損失はそれぞれ1.47億ドル,1.548億ドル,1.02億ドルであった。我々のほとんどの運営損失は,我々の研究開発計画に関するコストと我々の運営に関する一般的かつ行政的コストによるものである.今後数年と予測可能な未来には、巨額の費用と増加していく運営損失が引き続き発生すると予想される。私たちのこれまでの損失は、予想された将来の赤字に加え、株主赤字や運営資本に悪影響を与え続けるだろう。私たちは持続的な活動に関連する費用が大幅に増加すると予想しています
59
また、私たちが現在または未来の候補製品が市場の承認を得たら、販売、マーケティング、製品製造、流通に関連した巨額の費用が発生する。薬物開発に関連する多くのリスクや不確実性により,将来の損失の程度やいつ利益が達成されるかは予測できない(あれば)。私たちが本当に利益を達成したとしても、私たちは四半期や年度の収益性を維持したり向上させることができないかもしれない。
将来の財務状況に関するリスク
私たちは多くの追加資金を集める必要があるだろう。もし私たちが必要な時や魅力的な条項で資金を調達できなければ、私たちは私たちのいくつかの候補製品開発計画や将来の商業化努力を延期、削減または停止させることを余儀なくされるだろう。
医薬業界の発展は資本集約型である。我々は様々なプロジェクトの臨床開発活動に従事しており,現在もいくつかの潜在適応の臨床前開発により複数の開発候補を進めている。私たちが行っている活動に関連する費用は大幅に増加し、特に私たちが現在または未来の候補製品の臨床前と臨床活動を引き続き研究·開発し、推進し、マーケティング承認を求める場合には、大幅に増加することが予想される。さらに、私たちが現在または未来の任意の候補製品がマーケティング承認を受けた場合、このような販売、マーケティング、製品製造、流通は私たちの協力者の責任ではないので、販売、マーケティング、製品製造、流通に関連する巨額の商業化費用が発生すると予想される。もし私たちが現在または未来の候補製品のためにより多くの兆候および/または地理的位置を求めることを選択した場合、または他の方法で私たちが現在予想しているよりも早く拡張することを選択すれば、私たちはまたより早く追加資金を調達する必要があるかもしれない。また、上場企業の運営に関連した追加コストが引き続き発生すると予想される。したがって、私たちは私たちの持続的な業務と関連した多くの追加資金を得る必要があるだろう。もし私たちが必要な時や魅力的な条件下で資金を調達できない場合、私たちは私たちの1つ以上の候補製品の開発と商業化を延期、削減、または停止させられ、必要に応じて私たちの業務を拡大したり、他の方法で私たちのビジネスチャンスを利用することができない可能性があり、これは私たちの業務、財務状況、運営結果に大きな影響を与えるかもしれない。
2023年12月31日現在、我々は約4億363億ドルの現金および現金等価物と投資を持っている。2020年8月、私たちは普通株の初公募株を完成し、引受業者が彼らの選択権を全面的に行使することを含む9,987,520株の普通株を発行し、1株20.00ドルの公開発行価格で最大1,302,720株の普通株を購入した。引受割引と手数料、私たちが支払うべき他の推定発売費用を差し引く前に、今回の発行から得られた総収益は約1兆998億ドルでした。IPOと同時に、676,354株の普通株を1株当たり公開発行価格でVertexに売却することを発表した。同時に私募が私たちにもたらす総収益は約1,350万ドルだ。同時に行われた私募も2020年8月に完成した。2021年7月には、普通株の後続発行とVertexとの追加私募取引を完了し、合計約2.431億ドルの純収益を生み出した。2022年8月、私たちは普通株と事前融資権証のパイプ発行を完了し、1.5億ドルの総収益を得た。2024年1月、私たちは普通株式と事前融資権証の後続発行を完了し、引受割引と手数料、推定発売費用を差し引く前に、総収益は3.162億ドルだった。2024年2月、Cowen and Company,LLCとの販売契約に基づき、普通株販売から販売手数料を差し引く前に5000万ドルを受け取りました。2023年12月31日までの約4億363億ドルの現金と現金等価物と投資に、2024年第1四半期に発行された純収益と、2024年1月にセノフィとの協力協定に基づいて受け取った1500万ドルのマイルストーン支払いにより、2027年上半期までの運営に十分な資金を提供することが予想されます。私たちは間違っていることが証明される可能性があるという仮定に基づいてこの推定をして、私たちは現在予想されているよりも早く私たちの資本資源を使用することができる。この推定はまだ何も得られていないと仮定しています
60
協力や他の戦略連合を通じて追加的な資金を提供する。私たちの将来の資本需要は多くの要素に依存し、それによって大幅に増加するかもしれない
潜在的な候補製品の確定及び臨床前研究と臨床試験を行うことは時間がかかり、高価と不確定な過程であり、数年の時間をかけて完成する必要があり、しかも著者らは永遠に発売許可を獲得し、薬物販売を実現するために必要なデータ或いは結果を生成できないかもしれない。さらに、私たちの現在または未来の候補製品は、承認されれば、商業的に成功しないかもしれない。私たちの商業収入は、もしあれば、薬物の販売から来ます。これらの薬物は何年も商業的に得られないと予想されます。したがって、私たちは私たちの業務目標を達成するために追加的な資金に依存し続ける必要があるだろう。
いかなる追加的な拠出努力も、現在または将来の候補製品を開発し、それを商業化する能力に悪影響を及ぼす可能性がある。全体的に、金融市場の中断は株式や債務融資をより困難にする可能性があり、資金調達需要を満たす能力に重大な悪影響を及ぼす可能性がある。私たちは未来の資金調達が十分な金額や私たちに有利な条項で提供されることを保証できない。さらに、任意の融資条項は、私たちの株主の持株や権利に悪影響を及ぼす可能性があり、私たちが追加証券(株式や債務を問わず)、またはそのような証券を発行する可能性は、私たちの株式の市場価格を下落させる可能性があります。追加的な株式または転換可能な証券の売却は私たちのすべての株主を希釈するだろう。債務の発生は固定支払義務の増加を招き、私たちは追加債務を発生させる能力の制限、私たちが知的財産権を得ることができるかもしれない能力の制限、私たちの業務を展開する能力に悪影響を及ぼす可能性のある他の運営制限など、いくつかの限定的な条約に同意する必要があるかもしれない。私たちはまた、協力者との手配または他の方法によって、他の状況よりも望ましいより早い段階で資金を求めることを要求される可能性があり、私たちのいくつかの技術または現在または未来の製品候補製品の権利を放棄すること、または他の方法で私たちに不利な条項に同意することが要求される可能性があり、いずれも、私たちの業務、運営結果、および見通しに重大な悪影響を及ぼす可能性がある。
上場企業として、私たちの運営コストは引き続き増加し、私たちの経営陣は新しいコンプライアンスを実施するために多くの時間を投入する必要があるだろう。
上場企業として、私たちは個人会社として発生していない大量の法律、会計、その他の費用を引き続き発生させます。私たちは、改正された1934年証券取引法または取引法の報告要件を遵守しなければなりません。その中で、私たちは、私たちの業務および財務状況に関する年間、四半期、および現在の報告書をアメリカ証券取引委員会またはアメリカ証券取引委員会に提出することを要求します。また、2002年に改正されたサバンズ-オキシリー法案と、米国証券取引委員会とナスダックグローバル市場が後にサバンズ-オクスリー法案の条項を実施するために採択された規則は、効率的な情報開示と財務制御の確立と維持を要求し、コーポレートガバナンスのやり方を変更することを含む上場企業に多くの要求を出している。また、2010年7月には“ドッド·フランクウォール街改革·消費者保護法案”、あるいは“ドッド·フランク法案”が公布された。
61
テレス·フランク法案には、米国証券取引委員会がこれらの分野で“報酬発言権”や代理アクセスのような追加的な規制をとることを要求する重要な会社管理と役員報酬に関する条項がある。私たちは2022年からこれらの要求を実施することを要求され、実施過程で予期しない費用が発生した。株主急進主義、現在の政治環境、および現在の高度な政府介入と規制改革は、大量の新しい法規と開示義務を招く可能性があり、これは追加のコンプライアンスコストを招き、現在予見できない方法で業務を運営する方法に影響を与える可能性がある。
上場企業に適用される規則と条例は、私たちの法律と財務コンプライアンスコストを大幅に増加させ、いくつかの活動をより時間と費用を増加させる。これらの要求が私たちの経営陣と従業員の注意を他の業務から移すと、それらは私たちの業務、財務状況、運営結果に実質的な悪影響を及ぼす可能性がある。増加したコストは私たちの純収益を減少させ、あるいは私たちの純損失を増加させ、他の業務分野でコストを下げることを要求するかもしれない。例えば、これらの規則や条例は、取締役や上級管理者責任保険をより難しく、より高価にすることが予想され、同じまたは同様の保険範囲を維持するために多くのコストを発生させることが要求される可能性がある。私たちは、これらの要求に応答して生成される可能性のある追加コストの金額や時間を予測または推定することができない。これらの要求の影響は、私たちの取締役会、取締役会委員会、または役員に参加することをより難しくし、合格した人を引き付け、維持することを可能にするかもしれない。
金融サービス業の不利な事態の発展に影響を与え、例えば流動性、違約又は金融機関又は取引相手側が義務を履行しない実際の事件又は懸念は、我々の現在及び予想されている業務運営及び我々の財務状況及び運営結果に悪影響を及ぼす可能性がある。
流動性が限られている、契約違反、業績が悪い、または金融サービス業または金融サービス業の他の不利な発展に影響を与える実際の事件、または任意のこのような事件または他の類似のリスクに対する懸念または噂は、過去および未来に市場全体の流動性問題を引き起こす可能性がある。例えば、2023年3月10日、シリコンバレー銀行(SVB)はカリフォルニア州金融保護·革新部によって閉鎖され、後者は連邦預金保険会社(FDIC)を担当者に任命した。それ以来、SVBはそれらが第一市民銀行に買収され、ほぼ正常な運営に戻ったことを発表した。同様に,2023年3月12日,Signature BankとSilvergate Capital Corp.はそれぞれ破産管理プログラムに巻き込まれた.それ以来、より多くの金融機関も同様の破産を経験し、破産管理手続きに置かれている。さらに、私たちの任意の顧客、サプライヤー、または私たちと業務往来のある他の当事者が、そのようなツールやそのような金融機関との融資スケジュールに基づいて資金を得ることができない場合、これらの当事者が私たちに債務を支払ってくれたり、追加金を支払うことを要求する新しいビジネス計画を達成する能力が悪影響を受ける可能性があります。この点で、SVB信用協定や手配された取引相手、および信用証受益者(など)のような第三者は、SVB閉鎖の直接的な影響を受ける可能性があり、より広範な金融サービス業の流動性懸念の不確実性を受ける可能性がある。同様の影響は、例えば2008-2010年の金融危機の間に過去にも発生したことがある。
インフレと金利の急速な上昇は、以前に発行された金利が現在の市場金利よりも低い国債の取引価値を低下させる。米国財務省、連邦預金保険会社、連邦準備委員会は、このようなツールの売却による潜在的損失のリスクを低減するために、金融機関が保有するいくつかのこのような政府証券を保証する金融機関に250億ドルまでの融資を提供する計画を発表しているが、金融機関の顧客引き出しの広範な需要や金融機関の即時流動性の他の需要は、このような計画の能力を超える可能性がある。また、米国財務省、連邦預金保険会社、連邦準備委員会が将来他の銀行や金融機関が閉鎖された場合に未保険資金のルートを提供すること、あるいはタイムリーにそうすることは保証されない。
私たちの銀行と顧客の関係を必要または適切に評価する必要があると思いますが、私たちが獲得した資金源や他の信用手配の金額は、私たちの現在および予想される将来の業務運営に資金または資本化を提供するのに十分であり、私たちと直接信用協定や手配を持っている金融機関または金融サービス業全体または全体の経済に影響を与える深刻な被害を受ける可能性があります。他にも、これらの要因には、流動性の緊張または失敗、様々な金融、信用または流動資金協定または手配された義務を履行する能力、金融サービス業または金融市場の中断または不安定、または金融サービス業会社の将来性に対する懸念または否定的な予想が含まれる可能性がある。これらの要因は、我々と金融や業務関係にある金融機関や金融サービス業会社に関連する可能性があるが、金融市場や一般金融サービス業に関連する要因も含まれている可能性がある。
1つまたは複数のこれらの要因に関連するイベントまたは懸念の結果には、現在および予想されているビジネス運営、ならびに私たちの財務状況および運営結果に生じる様々な重大かつ悪影響が含まれている可能性がある。これらは、以下を含むことができるが、これらに限定されない
62
さらに、米国または国際金融システムに対する投資家の懸念は、より高い金利またはコスト、より厳しい財務および運営契約、または信用および流動性源を得るための体系的な制限を含む、あまり有利ではない商業融資条項を招く可能性があり、それによって、私たちは融資を受けにくく、さらには融資を受けることができない。他のリスクに加えて、利用可能な資金または現金および流動資金源の減少は、運営費用、財務義務、または他の義務を履行する能力に悪影響を及ぼす可能性があり、私たちの財務および/または契約義務に違反したり、連邦または州賃金および労働時間法違反を招いたりする可能性がある。上記のいずれかの影響、または上記の要因または他の関連または同様の要因に起因する任意の他の影響は、我々の流動資金、私たちの現在および/または予想される業務運営、ならびに財務状態および運営結果に重大な悪影響を及ぼす可能性がある。
さらに、マクロ経済または金融サービス業のいずれのさらなる悪化も、私たちの顧客またはサプライヤーの損失または違約を招く可能性があり、さらに、私たちの現在および/または予想される業務運営および運営結果および財務状況に大きな悪影響を及ぼす可能性がある。例えば、供給者たちはこれ以上顧客として私たちと付き合っていないことを決定するかもしれない。さらに、供給者は、上述した任意の流動性または他のリスクの悪影響を受ける可能性があり、これらのリスクは、未加入預金を取得する機会を遅延させたり、または苦境に陥ったり、倒産した金融機関の既存の信用手配を利用する能力を失ったりすることを含むが、これらに限定されない重大な悪影響を与える可能性がある。いかなるサプライヤーの破産または資本が債務を相殺しないか、またはサプライヤーのいかなる違約または違約、または任意の重大なサプライヤー関係の喪失は、私たちの重大な損失を招き、私たちの業務に実質的な悪影響を及ぼす可能性がある。
薬品開発と規制承認に関するリスク
臨床前と臨床開発に関するリスク
我々の開発は非常に早期の段階にあり,われわれのIRAK 4,STAT 3,MDM 2プロジェクトは早期臨床開発段階にある。もし私たちが安全または効果的な理由でこれらの製品を診療所で普及させることができない場合、あるいは私たちの候補製品を商業化することができない場合、あるいはそうする過程で重大な遅延に遭遇すれば、私たちの業務は実質的な損害を受けるだろう。
私たちの収益性は私たちが収入を作る能力にかかっている。これまで、私たちは協力収入を生み出してきましたが、私たちはまだ候補製品から何の収入も発生していません。近い将来にも薬品販売から何の収入も生じないと予想しています。1つ以上の候補製品の開発を完了し、市場の承認を得て販売を開始しない限り、製品販売から収入を得ることはないと予想される。薬物開発に関連する多くのリスクや不確実性が、以下の不確実性を含むため、いつ(あれば)これらの候補製品から収入を得ることができるかも予測できない
63
現在または将来の候補製品を商業化しようとすると、大きな販売やマーケティングコストが生じることが予想される。現在または将来の候補製品のキーまたは登録臨床試験を開始し、成功し、現在または将来の候補製品が商業販売の承認を得ても、これらのコストがかかるにもかかわらず、私たちの現在または未来の候補製品は商業的に成功しない可能性がある。薬品販売が発生した後、私たちはすぐに利益を達成しないかもしれません。もしあれば。もし私たちが収入を作ることができなければ、私たちは利益を上げないだろうし、持続的な資金がなければ、私たちは運営を続けることができないかもしれない。
私たちは飛馬プラットフォームに基づいて候補製品を発見し、開発する方法が斬新で検証されておらず、いかなる製品を開発する時間、開発コスト、開発成功の可能性を予測することは困難である。
私たちの飛び馬座TMPlatformは,標的タンパク質分解(TPD)と呼ばれる方法を用いて候補製品を発見·開発した。私たちの未来の成功はこの新しい治療法の成功にかかっている。アメリカ或いはヨーロッパはまだ異二機能分解剤の候補製品の使用を許可しておらず、このような治療製品の開発実行可能性の基礎データは初歩的であり、限られている。また、私たちはまだではなく、私たちのどの候補製品も臨床試験中または後に発売承認された有効性と安全性を証明することに成功しないかもしれない。特に,TPDの実現に成功し治療効果を得る能力は,合理的な薬物開発の流れを持つ非二機能分子の開発に成功し,タンパク質標的とE 3リガーゼの正確な組み合わせを有する分子を開発する必要がある。これは複雑な過程であり、期待された効果を達成するために、多くの構成部分あるいは生物機序の協同作業が必要である。正確な標的を理想的なE 3リガーゼと正しいリンカーと適時あるいは根本的に一致させることで分解物を発見できるかどうかを決定することはできない。私たちのすべての候補製品は臨床前または早期臨床開発段階にある。したがって、私たちの現在または未来の任意の候補製品の治療は、私たちが現在予測できない悪影響を及ぼすかもしれない。
これらの要因の影響により,製品候補開発の時間やコストを予測することは困難であり,我々の飛馬の応用が予測できるかどうかも予測できないTMプラットフォーム、または任意の類似または競合プラットフォームは、任意の製品の開発およびマーケティング承認をもたらす。私たちが未来に遭遇した飛行馬座に関する発展問題はTMプラットフォームまたは私たちの任意の研究計画は、重大な遅延または意外なコストをもたらす可能性があり、または商業的に実行可能な製品の開発を阻害する可能性がある。これらの要素のいずれも、私たちが臨床前研究および臨床試験を完成させることを阻止するか、または私たちが適時または利益的に開発する可能性のある任意の候補製品を商業化することができる。
64
私たちは他の候補製品を成功的に識別または発見することができないかもしれないし、または特定の候補製品または適応を追求するために限られた資源を使用する可能性があり、より有利または成功する可能性の高い製品候補または適応を利用することができないかもしれない。
私たちの戦略の重要な要素は私たちの飛馬座を応用することですTM幅広い目標と新しい治療分野を解決するためのプラットフォームと製品パイプライン。われわれが行っている治療発現活動は,腫瘍学,炎症,免疫学的あるいは遺伝病の治療に有用な候補品を決定することに成功しない可能性がある。私たちの研究計画は、潜在的な候補製品の識別に成功できない可能性があり、あるいは私たちの潜在的な候補製品が有害な副作用を有することが証明される可能性があるか、または製品を販売できないか、または発売許可を得ることができない他の特徴を有する可能性がある。
私たちの財務と管理資源が限られているので、私たちは限られた数の研究プロジェクトと候補製品に集中している。我々は現在,IRAK 4,STAT 6,TYK 2計画を含む我々の免疫学的製品の組み合わせに焦点を当てており,多様な炎症性や自己免疫疾患に関連する重要なシグナル経路,多様な癌に対するSTAT 3やMDM 2計画を含むわれわれの腫瘍学的製品の組み合わせを目指している。場合によっては、私たちはプロジェクトへの投資を止めることを決定するかもしれない。例えば、2023年11月に、我々は、増加する免疫学的導管を集中的に支援するために、我々のKT−413(IRAKIMID)計画の開発を停止することを決定したと発表した。したがって、私たちは他の現在または未来の候補製品を探すことを放棄または延期するか、または後により大きな商業潜在力を有することが証明された他の兆候を探すことができるかもしれない。私たちの資源分配決定は私たちが実行可能な商業薬や利益のある市場機会を利用できないかもしれない。現在および将来の研究開発計画および特定の適応の現在または将来の候補製品への支出は、いかなる商業的に実行可能な薬剤も生じないかもしれない。もし私たちが特定の候補製品の商業的潜在力や目標市場を正確に評価しなければ、私たちは将来の協力、許可、または他の印税手配を通じて候補製品に価値のある権利を放棄するかもしれないが、この場合、候補製品の独占開発権と商業化権利を維持することは私たちに有利だ。
私たちは私たちの先行プロジェクトの成功的な開発に大きく依存している。私たちは私たちが規制部門の承認を得ることができるか、私たちの現在または未来の任意の候補製品を商業化することに成功すると確信できない。
私たちは現在販売されている候補製品を承認しておらず、販売に適した候補製品を開発できないかもしれません。私たちの業務は私たちの現在または未来の候補製品の成功した開発、規制承認、商業化に大きく依存している。私たちの現在または未来の候補製品の臨床前研究および臨床試験、ならびに私たちの現在または未来の候補製品の製造およびマーケティングは、米国および他の国/地域の多くの政府機関の広範かつ厳格な審査および規制を受けるであろう。これらの国および地域では、現在または未来の任意の候補製品をテストまたは販売する予定である。現在または将来の任意の候補製品の商業販売が規制部門の承認を得る前に、各候補製品が安全かつ有効であり、各目標適応に使用できることを、臨床前研究および臨床試験によって証明しなければならない。薬物開発は長く、高価で不確定な過程であり、著者らの臨床前研究と臨床試験のどの段階でも遅延或いは失敗が出現する可能性がある。この過程は長年の時間を要する可能性があり、上場後の研究とモニタリングを含むかもしれないが、これには大量の資源がかかるだろう。米国で大量に開発されている薬物のうち,一部のみがFDAの規制承認手続きに成功して商業化されており,EUが開発している薬物は欧州医薬品局(EMA)の科学的評価を経てEU委員会の規制承認を得る成功率も同様に低い。したがって、私たちが必要な資金を得ることができても、私たちの開発、臨床前研究、臨床試験に資金を提供し続けることができても、現在または未来の任意の候補製品が開発または商業化に成功することを保証することはできません。例えば、2020年12月に、健康ボランティアおよびHSまたはAD患者において行われた最初のヒト無作為、二重盲検、プラセボ対照臨床試験を開始するために、KT−474のIND申請を提出した。この計画は最初に一部の臨床で保留され、第一段階試験の複数回の増分用量(MAD)部分に関連し、FDAが試験SAD部分の健康ボランティアの中期データの審査を待つ。2021年6月,FDAは中期SAD結果を審査した後,KT−474第1段階試験MAD部分の一部の臨床制限を解除し,それ以降,第1段階試験は完了した。
FDA、EU、またはEUから新薬申請またはNDAの承認を得るまで、私たちはEMAで科学的評価を行った後に欧州委員会からマーケティング許可申請やMAAの承認を得るまで、またはこれらの国の必要な承認を得るまで、米国で現在または未来の候補製品を販売することは許可されていません。NDAまたはMAAの承認を得ることは、様々な理由で、私たちの現在または将来の任意の候補製品の承認を延期、制限、または拒否する可能性がある複雑で、長く、高価で、不確実なプロセスである
65
これらの要素のいずれも、その多くは私たちがコントロールできないものであり、現在または未来の候補製品のための規制承認を得てマーケティングに成功する能力を危険にさらす可能性がある。私たちが規制承認を求める上でのどのような挫折も、私たちの業務と見通しに実質的な悪影響を及ぼすだろう。
もし私たちが患者を開始または募集する時に臨床試験に参加する時に遅延や困難に遭遇した場合、必要な規制承認を受けることは延期または阻止される可能性がある。
試験開始が遅れる可能性があり、FDAや米国国外の同様の規制機関の要求に応じて、これらの試験に参加するのに十分な数の合格者を見つけることができない可能性があります。また、私たちの競争相手のいくつかは、現在または未来の候補製品に対する臨床試験を行っており、これらの候補製品は、現在または未来の候補製品治療の患者群と同じであり、本来私たちの臨床試験に参加する資格がある患者は、競争相手の現在または未来の候補製品の臨床試験に参加する可能性があります。
患者の入選は他の要素の影響を受ける可能性がある
66
著者らが時々発表或いは公表した臨床試験の一時、“背線”と初歩的なデータはより多くの患者データの獲得に従って変化する可能性があり、そして監査と検証手続きの制限を受け、これは最終データの重大な変化を招く可能性がある。
著者らは時々著者らの臨床前研究と臨床試験の初歩的或いは主要なデータを公開開示する可能性があり、これらのデータは当時使用可能なデータの初歩的な分析に基づいて、特定の研究或いは試験に関連するデータに対してより全面的な審査を行った後、結果及び関連する発見と結論は変化する可能性がある。例えば,2023年12月にKT−333第1段階試験の積極中期結果を発表し,2023年11月にKT−253 1 a段階試験の積極中期結果を発表した。しかしながら、2つの試験からの最終的な背線データがそのような結果と一致するか、または陽性とみなされる保証はない。私たちはまた、私たちのデータ分析の一部として、すべてのデータを全面的かつ詳細に評価する機会がないか、または受け取る機会がないかもしれないという仮説、推定、計算、および結論を出した。したがって、私たちの報告の裏線または予備結果は、同じ研究の将来の結果と異なる可能性があり、またはより多くのデータを受信して十分に評価されると、異なる結論または考慮要因がこれらの結果を合格させる可能性がある。バックラインデータはまだ監査と確認手続きを受ける必要があり、これは最終データが私たちが以前に発表した予備データと大きく異なる可能性がある。したがって、最終データが利用可能になる前に、バックラインデータは慎重に表示されなければならない。私たちはまた臨床試験の中間データを時々開示するかもしれない。私たちが完成する可能性のある臨床試験の中期データは、私たちの臨床試験からのデータを含み、患者登録の継続およびより多くの患者データの獲得に伴い、あるいは私たちの臨床試験の患者が彼らの疾患の他の治療を継続するにつれて、1つまたは複数の臨床結果が実質的に変化する可能性があるリスクに直面する可能性がある。初期または中期データと最終データとの間の不利な違いは、私たちのビジネスの将来性を深刻に損なう可能性があります。しかも、私たちまたは私たちの競争相手が中間データを開示することは私たちの普通株の価格変動を招くかもしれない。
さらに、規制機関を含む他の人は、私たちの仮定、推定、計算、結論または分析を受け入れないか、またはデータの重要性を異なる方法で解釈またはトレードオフする可能性があり、これは、特定の計画の価値、特定の候補製品または製品の承認または商業化、およびわが社の全体的な状況に影響を与える可能性がある。さらに、私たちが開示する特定の研究または臨床試験に関する開示を選択する情報は、一般に広範な情報に基づいており、あなたまたは他の人は、私たちが決定した重大または他の適切な情報が私たちの開示に含まれることに同意しない可能性がある。
もし私たちが報告した中期、バックライン、または予備データが実際の結果と異なる場合、または規制機関を含む他の人が結論に同意しない場合、私たちが承認を得て私たちの候補製品を商業化する能力が損なわれる可能性があり、これは私たちの業務、運営結果、将来性、または財務状況を損なう可能性がある。
著者らの現在或いは未来の候補製品の早期臨床前研究と臨床試験の積極的な結果は必ずしも著者らの現在或いは未来の候補製品の後続の臨床前研究と臨床試験の結果を予測するとは限らない。もし私たちが未来の臨床試験で私たちの現在または未来の候補製品の臨床前研究の積極的な結果をコピーすることができなければ、私たちは開発に成功し、監督部門の許可を得て、私たちの現在または未来の候補製品を商業化することができないかもしれない。
著者らは現在或いは未来の候補製品の臨床前研究に対して得られた積極的な結果、及び現在或いは未来の候補製品の早期臨床試験から得られる可能性のある任意の積極的な結果、進行中のKT-474、KT-333とKT-253の臨床試験を含み、必ずしも必要な後続の臨床前研究と臨床試験の結果を予測できるとは限らないかもしれない。現在または未来の製品の計画で臨床前研究や臨床試験を完成させることができても
67
現在の開発スケジュールによれば、KT−474、KT−621、KT−294、KT−333およびKT−253を含む現在または将来の候補製品は、後続の臨床前研究または臨床試験において複製されない可能性がある。特に,KT−621とKT−294について何らかの臨床前研究が行われているが,これら2つの候補製品がこれまでの臨床前研究のようにわれわれが計画した臨床試験に現れるかどうかは不明である。例えば、臨床前研究において、(I)KT-621は、すべての関連するヒト細胞環境においてIL-4/IL-13経路の完全な阻害を示し、その皮モル効力はdupiumabよりも優れ、活性はdupiumabに相当するか、またはそれよりも優れており、(Ii)KT-294は、評価されたすべての関連ヒト細胞タイプにおける皮モル~ナノモルの効力を示す。しかし,これらの臨床前結果が臨床試験で複製される保証はない。製薬やバイオテクノロジー業界の多くの会社が早期開発に積極的な成果をあげた後,後期臨床試験で大きな挫折を経験し,類似した挫折に直面しないことは確認できない。その他を除いて,これらの挫折は,以前に報告されていない有害事象を含む臨床試験進行期間中の臨床前発見や臨床前研究や臨床試験で行われた安全性や有効性観察によるものである。そのほか、臨床前と臨床データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品が臨床前研究と臨床試験で満足できると思っているが、FDA或いは類似の外国の監督管理機関の許可を得られなかった。もし私たちが現在または未来の候補製品の任意の計画の臨床前研究または臨床試験で積極的な結果を得ることができなければ、私たちの現在または未来の候補製品の開発スケジュール、規制承認と商業化の見通し、およびそれに応じた私たちの業務および財務の見通しは、重大な悪影響を受けるだろう。
さらに、我々の計画または将来の臨床試験は、我々のKT−474、KT−333、およびKT−253の第1段階臨床試験の開放ラベル患者部分のような“開放ラベル”試験設計を採用することができる。オープンタグ“臨床試験”とは、患者および研究者の両方が、患者が研究製品候補を受け入れているかどうかを知っているかどうか、または既存の承認薬またはプラセボを意味する。最も典型的には,オープンラベル臨床試験は候補の研究製品のみをテストし,異なる用量レベルで試験を行う可能性がある。開放ラベル臨床試験は様々な制限を受けており,これらの制限は任意の治療効果を誇張する可能性があり,開放ラベル臨床試験中の患者が治療を受ける際に知られているからである。オープンラベル臨床試験は“患者偏見”の影響を受ける可能性があり,すなわち患者が症状が改善したと考えているのは,実験的治療を受けていることを意識しているだけである。また,オープンラベル臨床試験は,“調査者偏見”の影響を受ける可能性があり,すなわち臨床試験の生理結果を評価·審査する人は,どの患者が治療を受けているかを知り,その知識を知っている場合に治療群の情報をより有利に解釈することが可能である。オープンラベル試験の結果は,われわれのKT−474,KT−333およびKT−253の第1段階試験を含み,いずれの候補製品の将来の臨床試験結果も予測できない可能性があり,制御された環境でプラセボまたは能動対照を用いて研究を行う場合には,開放ラベル臨床試験を含む。
私たちの候補製品の目標患者群の発病率と流行率はまだ正確に確定されていない。もし私たちの候補製品の市場機会が私たちが推定したものよりも小さい場合、または私たちが得た任意の承認が患者集団のより狭い定義に基づいている場合、私たちの収入と利益を達成する能力は悪影響を受け、実質的な可能性がある。
我々の現在と未来の候補製品が求めている適応の確実な発生率と流行率は不明である。これらの疾患に罹患している人の数および候補製品治療から利益を得る可能性があるこれらの疾患患者のサブセットの予測は、推定に基づいている。HS,炎症性皮膚疾患,AD,関節リウマチなどの広範な免疫学的炎症疾患の治療に用いられるKT−474を開発している。私たちの候補製品のすべての潜在的な市場機会は、最終的にそれらが証明された安全性と有効性、各製品の最終ラベルに含まれる診断基準、私たちの候補製品がこれらの適応、医学界および患者参入の受容度、製品価格と精算のために承認されたかどうかに依存する。私たちの製品のアメリカおよび他の場所での候補患者数は予想を下回る可能性があり、患者は私たちの製品治療を受け入れられないかもしれないし、新しい患者はますます識別しにくくなったり、接触したりする可能性があり、これらはすべて私たちの運営結果や業務に悪影響を及ぼすだろう。
大流行、流行病、あるいは伝染病の爆発は私たちの業務や財務業績に実質的な悪影響を及ぼす可能性があり、私たちの候補製品の開発中断を招く可能性があります。
大流行や流行のような公衆衛生危機は私たちの業務に悪影響を及ぼすかもしれない。例えば、2019年12月、SARS-CoV-2(重症急性呼吸症候群コロナウイルス2)またはコロナウイルスと呼ばれる新型ウイルス株が中国武漢で浮上し、それ以来、私たちの主要なオフィスと実験室があるマサチューセッツ州東部を含む全世界に伝播してきた。この大流行や各国政府がこの大流行に対応するために実施した政策や条例は,その大部分が解除されている--企業や商業に直接的かつ間接的な大きな影響を与えている。コロナウイルスの大流行はかなりの変化を遂げており
68
政府が強制的に実施した隔離、旅行制限、その他現在キャンセルされている公衆衛生安全措置を含む様々な対応が実施されている。将来のいかなる大流行が私たちの業務あるいは私たちの第三者パートナーの業務に与えることも含めて、私たちの臨床前研究あるいは臨床試験業務の影響の程度も、未来の事態の発展に依存し、これらの事態の発展は高度に不確定であり、疫病の持続時間、出現する疾病の重症度に関する新しい情報、疾病の制御或いはその影響を治療する行動などを把握できない。例えば,他の生物製薬会社と同様に,臨床試験で被験者の募集を遅らせる可能性がある。感染症は影響を受けた地域に位置する第三者CROの従業員にも影響する可能性があり,これらの地域に依存して臨床試験を行っている。また,我々のLeadや他のコア候補製品が対象としている患者群は,特に感染症やその変種に感染しやすい可能性があり,われわれの臨床試験に参加できる患者を識別することが困難になり,登録された患者がどのような試験を完了する能力に影響する可能性がある。将来のいかなる感染症伝播が患者登録や治療にどんな負の影響を与えるか、あるいは私たちの候補製品の実行は臨床試験活動のコストの高い遅延を招く可能性があり、これは私たちの監督部門の許可を得て、私たちの候補製品を商業化する能力に悪影響を与え、私たちの運営費用を増加させ、私たちの財務業績に重大な悪影響を与える可能性がある。
そのほか、臨床試験の適時登録は臨床試験地点に依存し、臨床試験地点は全世界の衛生問題の不利な影響を受け、例えば流行病である。私たちの候補製品の臨床試験および私たちの業務に悪影響を及ぼす可能性のある公衆衛生危機のいくつかの要因は、延期または他の方法で行われる可能性がある
米国証券取引委員会や食品·医薬品局の閉鎖または中断の範囲や重症度など、他の計画や潜在的な企業や政府機関を予測することはできない。これらの要因、およびいかなる予見不可能なこのような中断に関連する他の要因も、私たちの業務および私たちの運営結果および財務状況に実質的な悪影響を及ぼす可能性がある。また、これらや関連問題をめぐる不確実性は、米国や他の経済体の経済に悪影響を及ぼす可能性があり、必要な資本を調達して候補製品を開発·商業化する能力に影響を与える可能性がある。他の世界的な健康問題はまた、私たちまたは私たちと接触している第三者のいる国の社会、経済、労働力の不安定を招く可能性がある。
69
私たちの現在または未来の候補製品は不良または他の不良副作用を招く可能性があり、その規制承認を延期または阻止し、承認されたラベルの商業イメージを制限したり、上場承認後に重大な負の結果を招く可能性がある(あれば)。
私たちのすべての候補製品は臨床前あるいは早期臨床開発段階にあり、私たちの現在あるいは未来の任意の候補製品の治療は現在予測できない悪影響を与えるかもしれない。私たちの現在または未来の候補製品によって引き起こされる副作用は、臨床前研究を中断、延期、または停止させる可能性があり、または私たちまたは規制機関の臨床試験の中断、延期、または停止を招く可能性があり、より厳しいラベルを引き起こす可能性があり、またはFDAまたは他の規制機関が規制承認を遅延または拒否する可能性がある。炎症性や自己免疫疾患,癌あるいは他の疾患を治療する多くの方法のように,我々の候補品を使用すると副作用が生じる可能性がある。さらに、任意のタンパク質分解製品における潜在的なリスクの1つは、健康なタンパク質または分解を標的としないタンパク質が分解されるか、または目的タンパク質自体の分解が有害事象、副作用または意外な特徴をもたらす可能性があることである。我々の現在または将来のどの臨床研究においても、健康なタンパク質または分解されていないタンパク質を分解するために、私たちの分解物分子を使用することが可能である。現在または未来の任意の候補製品を用いて治療を行った後、不良事象を遅延させる潜在的なリスクも存在する。
これらの副作用は,非標的活動,試験対象のアレルギー反応,または体内で望ましくない標的効果による可能性がある。われわれの臨床試験結果は,これらや他の副作用の重症度や流行度を明らかにする可能性があり,受け入れられない。この場合、私たちの実験は一時停止または終了される可能性があり、FDAまたは同様の外国規制機関は、現在または将来の候補製品の任意のまたはすべての目標適応のさらなる開発を停止または拒否するように命令することができる。薬物に関連する副作用は、患者の募集または患者の試験完了能力に影響を与える可能性があり、あるいは潜在的な製品責任クレームを招く可能性がある。このようなどんな状況でも、私たちの業務、財政状況、そして見通しに大きな被害を及ぼす可能性がある。
また,現在あるいは将来の候補製品は標的毒性に関連する臨床試験で副作用を引き起こす可能性がある。標的毒性が観察された場合、または現在または将来の候補製品が予期しない特徴を有する場合、それらの開発を放棄する必要があるかもしれないし、より狭い用途またはサブ集団に開発を制限する必要があるかもしれないが、これらの用途またはサブ集団では、リスク-収益の観点から、副作用または他の特徴がそれほど一般的ではなく、それほど深刻ではない、またはより容易に受け入れられるであろう。多くの化合物は最初は癌や他の疾患を治療する早期テストで希望を示したが,その後副作用が認められ,この化合物のさらなる発展を阻止した。
また,臨床試験の本質は潜在的な患者集団のサンプルを利用することである。患者数と接触時間が限られているため、私たちの現在または未来の候補製品の稀かつ深刻な副作用は、候補製品に接触する患者の数が明らかに増加した場合にのみ発見される可能性がある。もし私たちが現在または未来の候補製品が市場の承認を得て、私たちまたは他の人が承認を得た後、このような現在または未来の候補製品による不良副作用を発見すれば、多くの潜在的な重大な負の結果を招く可能性がある
これらの事件は、影響を受けた候補製品に対する市場の受容度を達成または維持することを阻止し、現在または未来の候補製品の商業化コストを大幅に増加させる可能性があると考えられる
70
承認され、現在または将来の候補製品を商業化し、収入を創出する能力に大きな影響を与えることに成功した。
私たちの現在または未来の候補製品を作ることは複雑で、私たちは生産で困難に直面するかもしれない。このような困難に遭遇した場合、臨床前研究および臨床試験、または商業目的のために現在または将来の候補製品を提供する能力が延期または停止される可能性がある。
私たちの現在または未来の候補製品を製造する過程は複雑で、厳格に規制されている。私たちは自分の製造施設や人員を持っておらず、現在または未来の候補製品を第三者に依存して生産し続ける予定です。これらの第三者契約製造組織またはCMOは、私たちの需要を満たすのに十分な資源または能力を提供することができず、彼ら自身の独自プロセスを私たちの候補製品製造プロセスに統合することができるかもしれない。第三者独自のプロセスの制御および監視は限られており、第三者は、私たちの同意または知ることなく、そのプロセスを修正することを選択することができる。これらの修正は、製品配合の修正に影響を与えるように、製品の損失や故障を引き起こすことを含む、私たちの製造に負の影響を与える可能性があり、これには追加の生産運転または製造業者の交換が必要であり、両方の場合は、私たちの現在または将来の候補製品のコストを著しく増加させ、生産を著しく遅らせる可能性がある。メーカーの変化は通常、製造プロセスとプロセスの変化に関連しており、これは私たちが臨床試験で使用した以前の臨床供給と任意の新しいメーカーの供給との間の過渡的な研究を要求するかもしれない。臨床用品の比較可能性の証明には成功しない可能性があり,追加の臨床試験が必要かもしれない。
立法提案は通過を待っており,可決されれば,米国がいくつかの外国相手と関係があるか,あるいは国家安全に脅威となるバイオテクノロジー提供者の資金に悪影響を与える可能性がある。任意の影響を受けたバイオテクノロジー供給者と商業関係のみの実体の潜在的下流への悪影響は不明であるが、サプライチェーン中断または遅延が含まれている可能性がある。
我々と契約を締結したCMOのいずれかがその義務を履行できなければ,別のCMOとの合意を余儀なくされる可能性があり,合意がまったくなければ合理的な条項ではできない可能性がある。これは私たちが代替供給源を確立したので、私たちの臨床試験供給を大幅に延期するかもしれない。場合によっては、我々の候補製品または製品を製造するために必要な技術的スキル(承認された場合)は、元のCMO固有または独自のものである可能性があり、困難に遭遇する可能性があり、またはそのようなスキルを予備または代替サプライヤーに譲渡することを禁止する契約制限が存在する可能性があり、またはそのようなスキルを全く譲渡できない可能性がある。また,我々が何らかの理由でCMOの交換を要求された場合,新たなCMOが品質基準とすべての適用法規に適合する施設やプログラムを保持していることを確認するように要求される.我々はまた、例えば比較可能な研究を製造することによって、任意の新しい製造プロセスが、以前にFDAまたは他の規制機関に提出された仕様に基づいて、私たちの候補製品を生産することを検証する必要がある。新しいCMO検証に関連する遅延は、私たちがタイムリーにまたは予算内で候補製品を開発したり、製品を商業化する能力に悪影響を及ぼす可能性があります。また,CMOはこのCMOが独立して持つ我々の候補製品の製造に関する技術を持つことができる.これは、私たちのこれらのCMOへの依存を増加させるか、または別のCMOが私たちの候補製品を生産するために、これらのCMOからライセンスを取得することを要求するだろう。また、私たちの現在または未来の候補製品が臨床前研究と臨床試験を通じて潜在的な承認と商業化の方向に発展することに伴い、製造過程の様々な方面は変化し、過程と結果の最適化に努力することが予想される。このような変更は、規制申請を修正する必要がある可能性があり、これは、現在または将来の任意の候補製品が改善された製造プロセスを使用することができる時間的枠組みをさらに遅らせることができ、私たちの臨床試験で使用された以前の臨床供給と任意の新しいメーカーの臨床供給との間の追加的な接続研究または試験を行う必要があるかもしれない。臨床用品の比較可能性の証明には成功しない可能性があり,追加の臨床試験が必要かもしれない。このような遅延は、私たちの業務、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
規制承認に関するリスク
現在または将来の候補製品のために必要な規制承認を得ることができない場合や、必要な規制承認を遅延させることができなければ、現在または将来の候補製品を商業化することができない、または商業化を遅延させることができず、収入を創出する能力が深刻に損なわれる。
私たちの現在または未来の候補製品とその開発と商業化に関連する活動は、設計、テスト、製造、安全、効果、記録保存、ラベル、貯蔵、承認、広告、販売促進、販売、流通、輸入と輸出を含み、すべて米国FDAと他の監督機関、その他の国と地域の類似機関の全面的な監督管理を受けている。現在および将来のいずれかの候補製品を商業化することができる前に、関連する管轄区域規制機関のマーケティング承認を得なければならない。私たちは現在の候補製品の販売許可をどの管轄区域の規制機関からも得ていません
71
私たちの現在の候補製品や私たちが将来開発を求める可能性のあるどの候補製品も規制部門の承認を得られないかもしれません。会社として、規制承認を得るために必要な申請の提出と支援に関する経験は限られており、この過程で第三者CROおよび/または監督コンサルタントに依存して支援することが予想される。監督部門の承認を得るためには、各監督機関に広範な臨床前と臨床データ及び支持情報を提出し、候補製品の安全性と有効性を確定する必要がある。監督管理機関の承認を得るためには,薬品製造過程に関する情報を関連監督機関に提出し,生産施設を検査する必要があり,通常は臨床場所である。私たちの現在または未来の候補製品は効果がないかもしれないし、適度な効果しかないかもしれないし、不良または意外な副作用、毒性、または他の特徴があることが証明される可能性があり、上場許可を得ることを阻止したり、商業使用を阻止したり制限したりする可能性がある。
アメリカと国外では、規制承認を得る過程はすべて高価であり、追加の臨床試験が必要であれば、数年かかるかもしれないが、本当に承認された場合、関連する候補製品のタイプ、複雑性、新規性を含む様々な要素によって大きく異なる可能性がある。開発期間中の市場承認政策の変化、追加法規または法規の変化、あるいは米国国外で提出された各機密協定または同等の申請タイプの監督審査の変化は、申請承認または拒否の遅延を招く可能性がある。FDAと他国の類似機関は承認過程においてかなりの自由裁量権を有しており、いかなる申請も拒否することができ、私たちのデータが承認を得るのに十分ではなく、追加の臨床前、臨床、あるいは他の研究を行う必要があると決定することもできる。私たちの現在または未来の候補製品は、以下の理由を含む、多くの理由で規制部門の承認を得ることができないか、または得られないかもしれない
私たちが承認されても、規制機関は、私たちが要求しているものよりも少ないか制限されているか、私たちの薬物のために徴収しようとしている価格を承認しないかもしれない、高価な発売後の臨床試験の表現によって承認されるかもしれない、またはその候補製品の商業化に必要または必要なラベル宣言を含まないことを承認する可能性がある現在または未来の任意の候補製品を承認することができる。上記のいずれの状況も、私たちの現在または未来の候補製品のビジネス見通しに実質的な損害を与える可能性がある。
もし私たちが承認を得る上で遅延に遭遇した場合、あるいは私たちが現在または未来の候補製品の承認を得ることができなければ、私たちの現在または未来の候補製品の商業的見通しが損なわれる可能性があり、私たちが収入を作る能力は深刻な損害を受けるだろう。
72
私たちは、現在または未来の任意の候補製品のための画期的な治療認証および/または迅速チャネル認証を求めることができるかもしれない。FDAがこれらの指定を承認しても、より速い開発、規制審査、または承認過程をもたらすことはなく、このような指定は、私たちの候補製品が米国で上場承認される可能性を増加させないだろう。
私たちは私たちの現在または未来の1つまたは複数の候補製品のための画期的な治療法の称号を求めるかもしれない。画期的な治療法は、1つまたは複数の他の薬剤と単独でまたは1つまたは複数の他の薬剤と組み合わせて重篤または生命を脅かす疾患または状態を治療することを意図した薬剤として定義され、初歩的な臨床証拠は、1つまたは複数の臨床的に重要な終点において、臨床開発早期に観察される実質的な治療効果のような既存の療法よりも有意な改善を示す可能性があることを示す。画期的な治療法として指定された薬物に対して,FDAと試験スポンサーとの相互作用やコミュニケーションは,臨床開発の最も有効な経路を決定するのに役立つとともに,無効対照案中の患者数を最小限にすることができる。FDAで画期的な治療法に指定されている薬物も優先審査や加速承認を受ける資格がある可能性がある。画期的療法に指定されたのはFDAの裁量権である。したがって,現在または将来の候補品の1つが画期的療法として指定された基準に適合していると考えても,FDAは同意せず,このような指定を行わないことにした可能性がある。いずれの場合も、現在または将来の候補製品に対する突破療法指定を受けることは、FDAの従来の手順に従って承認を考慮する療法と比較して、より速い開発プロセス、審査または承認をもたらすことはなく、FDAの最終承認を保証することもできない。さらに、現在または将来の1つまたは複数の候補製品が画期的な治療法となる資格があっても、FDAは、これらの候補製品がもはや資格条件に適合していないと決定するか、またはFDAの審査または承認を決定する期間が短縮されない可能性がある。
私たちは私たちの現在または未来の1つまたは複数の候補製品のための高速チャネル認証を求めるかもしれない。2023年第3四半期、米国食品医薬品局は、再発/難治性CTCLおよび再発/難治性PTCLの治療のためのKT-333高速チャネルを承認した。重症または生命に危険な疾患の治療に使用される薬剤が使用され、そのような疾患が満たされていない医療ニーズを解決する可能性を示す場合、薬物スポンサーは、迅速なチャネル指定を申請することができる。FDAは幅広い裁量権を持ち,この称号を付与するかどうかを決定するため,特定の現在または将来の候補製品がこの称号を獲得する資格があると考えても,FDAがそれを付与することを保証することはできない。現在または将来の候補製品のいくつかの高速チャネル認証を実際に取得したとしても、従来のFDAプログラムと比較して、より速い開発プロセス、審査、または承認を経験しない可能性がある。FDAが我々の臨床開発プロジェクトのデータが高速チャネルの指定をサポートしなくなったと考える場合,その指定を撤回する可能性がある。高速チャネル指定だけではFDA優先審査プログラムに適合する資格は保証されない。
FDAの加速承認手続きに基づいて、KT−474、KT−621、KT−294、KT−333、KT−253、または任意の他の適用可能な将来の候補製品の承認を求めることができる。このアプローチは、より速い開発や規制審査や承認過程をもたらすことはないかもしれませんし、私たちの候補製品が上場承認される可能性も増加しません。
KT−474、KT−621、KT−294、KT−333、KT−253、または将来の候補製品の承認を加速することを求めることができる。製品が重篤または生命に危険な疾患を治療し、一般に既存の療法よりも有意な利点を提供する場合、加速承認を得る資格がある可能性がある。さらに、それは、臨床的利益を合理的に予測する可能性のある代替終点への影響、または不可逆的な発病率または死亡率またはIMMよりも早く測定することができる臨床終点への影響を証明しなければならず、IMMまたは他の臨床的利益の効果を合理的に予測することができる。承認を加速させる条件として,FDAは十分かつ良好に制御された上場後臨床試験を要求する可能性があり,FDORAによると,FDAは現在,必要に応じてこのような試験を承認前または承認が加速された製品の承認日後の特定の期間で行うことが許可されている。FDORAはまた、スポンサーがこのような研究をタイムリーに行うことができなければ、必要な更新をFDAに送信するか、またはそのような承認後の研究が薬剤の期待される臨床的利益を検証できない場合、FDAは、加速的な承認を得た薬物または生物学的製剤の承認を迅速に撤回することができる。FDORAによれば、FDAは、職務調査を行っていない会社に対していかなる承認後の検証的研究を行うか、または速やかに当該機関に進捗報告を提出した会社に罰金を科すように行動する権利がある。さらに、FDAが別途通知しない限り、FDAは現在、加速された承認された製品の販売促進材料の事前承認を要求しており、製品の商業発売時間に悪影響を及ぼす可能性がある。したがって,加速的な承認経路の利用を求めても,加速的な承認を得ることができない可能性があり,加速された承認を得ても,その製品のより速い開発,規制審査,承認過程を体験できない可能性がある.また,加速承認を得ることは,製品の加速承認が最終的に従来の承認に変換される保証はない.
73
私たちのいくつかの候補製品は孤児薬物の称号を得た。私たちはまた、私たちの現在または未来のいくつかの候補製品のために孤児薬物指定を求めることができ、私たちは潜在的な市場排他性を含む孤児薬物指定に関連する利点を維持できないかもしれない。
FDAは、末梢T細胞リンパ腫および皮膚T細胞リンパ腫の治療のためのKT-333および急性骨髄性白血病の治療のためのKT-253の孤児薬物名を許可している。私たちの業務戦略の一部として、現在または未来の他の候補製品のいくつかの適応のために孤児薬の指定を求めることも可能ですが、私たちは成功しないかもしれません。米国やヨーロッパを含むいくつかの管轄区の規制機関は、患者数が比較的少ない薬物を孤児薬に指定する可能性がある。孤児医薬品法によると、薬物がまれな疾患や疾患を治療するための薬である場合、FDAは孤児薬として指定することができる。米国では、孤児薬は通常、年間患者数が20万人未満、または米国では患者数が20万人を超えていると定義されているが、米国では、薬物開発コストは米国の販売から回収できないという合理的な期待がある。米国では、孤児薬物指定側は臨床試験費用、税金優遇、ユーザー費用減免のための贈与資金を提供する機会を提供するなどの財政的インセンティブを得る権利がある。
同様に、欧州連合では、欧州医薬品局孤児医薬製品委員会の提案によると、欧州委員会は、生命または慢性衰弱にかかわる疾患の診断、予防または治療のための製品に孤児の称号を付与することができ、これらの製品は、EUの10,000人当たり5人以下に影響を与え、満足できる診断、予防または治療方法を行うことは許可されていない(またはこの製品は、適用される疾患の影響を受ける人に大きな利点を有するであろう)。さらに、生命に危害を及ぼす、深刻な虚弱または深刻かつ慢性疾患の診断、予防または治療のための製品については、インセンティブ措置がなければ、EUでの製品の販売が、製品を開発するための必要な投資が合理的であることを証明するのに十分でない可能性が高い場合には、製品名を付与することができる。欧州連合では、孤児を指定することで一方が費用を減らしたり費用を免除したりするなど、経済的奨励を受ける権利がある。
一般に、孤児薬物指定を有する製品がその後、その指定された適応を有する最初の発売許可を得た場合、製品は、限定された場合を除いて、限定された場合を除いて、FDAまたはEMAがその期間内に同じ製品および適応を承認する別のマーケティング申請を阻止する権利がある。適用期間は米国では7年、EUでは10年。5年目の終了時に、製品が市場排他性がもはや合理的でないことを証明することを含む、指定された孤児の基準に適合しない製品が決定された場合、EUの排他的期間は6年に短縮されることができる。FDAまたはEMAが、指定された要求に重大な欠陥があると判断した場合、または製造業者が稀な疾患または疾患を有する患者の需要を満たすのに十分な数の薬剤を保証できない場合、孤児薬の排他性を失う可能性がある。
我々が製品の孤児薬物排他性を獲得しても,この排他性は異なる活性成分を含む競合薬が同じ条件で許可されるため,競合から製品を効率的に保護することができない可能性がある。さらに、孤児薬が承認された後であっても、FDAが後の薬物が臨床的に優れていると結論した場合、より安全で、より有効であることが証明され、または患者ケアに大きな貢献を果たしているため、FDAはその後、同じ薬物による同じ疾患の治療を承認することができる。
2017年8月3日、米議会はFDARAと略称するFDA 2017年再認可法案を可決した。他の事項以外に、FDARAはFDAの以前に存在した監督管理解釈を薬物スポンサーに1種の孤児薬物の臨床優位性を証明することを要求し、この薬物は他の方面で以前許可された同じ稀な疾病を治療する薬物と同じであって、孤児薬物の排他性を得ることができる。この新しい立法は以前の前例を覆し、即ち“孤児薬品法”はFDAに孤児排他期を認めることを明確に要求し、その臨床優勢にかかわらず。また、2021年の“総合支出法案”では、FDARA編纂の解釈がFDARA発行前に孤児指定が発行されたが、製品承認がFDARA公布後に適用されることが明らかになったため、国会はこの解釈をさらに変更しなかった。FDAは孤児薬物法とその規制と政策をさらに再評価するかもしれない。FDAがいつ、どのように孤児薬の法規や政策を変えるかどうかもわかりませんし、どのような変化が私たちの業務に影響を与える可能性があるかどうかもわかりません。FDAがその孤児の薬物法規や政策に変化する可能性があることによって、私たちの業務は悪影響を受ける可能性がある。
74
私たちが現在または未来の任意の候補製品が規制部門の承認を得ても、私たちは持続的な義務と持続的な規制審査の制約を受けることになり、これは多くの追加費用を招く可能性がある。また,現在または将来の候補製品が承認されれば,ラベルや他の制限や市場撤退の制限を受ける可能性があり,規制要求を遵守できなかったり,任意の製品候補が承認された場合に予期しない問題に遭遇したりすると,罰を受ける可能性がある.
FDAまたは同様の外国規制機関が、現在または将来の任意の候補製品を承認する場合、薬物の製造プロセス、ラベル、包装、流通、追跡および追跡、有害事象報告、貯蔵、広告、販売促進および記録は、広範かつ持続的な規制要件によって制限されるであろう。これらの要求には,安全と他の上場後の情報と報告,登録要求の提出,および我々が承認後に行った任意の臨床試験がcGMPと良好な臨床実践(GCP)を継続して遵守しているかどうかが含まれている。いくつかの商業処方薬製品の場合、製造業者およびサプライチェーンに参加する他の当事者はまた、流通チェーン要求を満たし、製品を追跡および追跡するための電子的、相互運用可能なシステムを確立し、偽、移転、盗難、および意図的に混合された他の製品、または米国で流通するのに適していない他の製品をFDAに通報しなければならない。現在または将来の候補製品のために得られる任意の規制承認は、医薬が発売される可能性のある承認表示用途の制限または承認条件によって制限される可能性があり、または第4段階臨床試験を含む可能性の高い発売後試験要件を含む、薬物の安全性および有効性を監視する可能性がある。さらに、FDORAによれば、承認された薬物および生物製品のスポンサーは、市場状態が任意に変化したときに、薬物の撤回などの6ヶ月の通知をFDAに提供しなければならず、そうしなければ、FDAは製品を生産停止製品リストに登録する可能性があり、これは製品の発売能力をキャンセルするであろう。FDAは、このような製品が承認された適応のみを確保し、承認されたラベルの規定による販売を確実にするために、医薬品および生物製品の承認後のマーケティングおよび販売促進を密接に規制する。その後、予期しない重症度または頻度の有害事象、または私たちの第三者製造業者または製造プロセスに関連する問題、または規制要求を遵守できなかったことを含む、以前に未知の薬物問題が発見され、他を除いて、以下のようになる可能性がある
FDAの政策は変わる可能性があり、私たちの現在または未来の候補製品に対する規制承認を阻止、制限、または延期するための追加の政府法規が公布されるかもしれない。もし私たちが既存の要求の変化に適応できない場合、あるいは新しい要求や政策を採用することができない場合、または私たちがコンプライアンスを維持できない場合、私たちは私たちが得る可能性のあるマーケティング承認を失う可能性があり、これは私たちの業務、将来性、利益を達成または維持する能力に悪影響を及ぼすだろう。
米国食品医薬品局、米国証券取引委員会および他の政府機関の資金不足は、政府の閉店やこれらの機関の運営の他の中断を含め、重要な指導部や他の人員の能力を雇用·保留し、新製品やサービスのタイムリーな開発や商業化を阻止するか、あるいはこれらの機関が私たちの業務運営に依存する可能性のある正常な業務機能を履行することを阻止することを阻害する可能性があり、これは私たちの業務に負の影響を与える可能性がある。
FDAの新製品の審査と承認能力は、政府予算と資金レベル、肝心な人員の雇用と維持及びユーザー費用の支払いを受ける能力、及び法定、監督と政策の変化を含む様々な要素の影響を受ける可能性がある。したがって、その機関の平均検討時間は近年変動している。また、政府が米国証券取引委員会や我々の業務に依存する可能性のある他の政府機関に提供する資金は、研究開発活動に資金を提供する機関を含め、政治プロセスの影響を受けており、政治プロセス自体が不安定で予測不可能である。
75
FDAや他の機関の中断も、新製品候補製品が必要な政府機関によって審査および/または承認されるのに要する時間を遅らせる可能性があり、これは私たちの業務に悪影響を及ぼすだろう。政府が長期的に停止すれば、FDAが私たちが提出した規制文書を適時に審査して処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。また、将来的に政府の閉店は、適切な資本化と事業継続のために、私たちが公開市場に進出し、必要な資本を得る能力に影響を与える可能性がある。
外国規制承認と外国市場に関するリスク
私たちが現在または未来の候補製品がアメリカでマーケティング承認を得ても、私たちは決して規制部門の承認を得ず、アメリカ以外の場所で私たちの現在または未来の候補製品をマーケティングすることができるかもしれない。
私たちは、現在または未来の候補製品のために、米国国外の規制承認を求めることを計画している。しかし、米国国外で任意の製品をマーケティングするためには、他の国/地域が多く、それぞれ異なる安全性、有効性、および他の規制要件を確立し、遵守しなければならない。承認手続きは国によって異なり、追加の製品候補テストと追加の行政審査期限が含まれる可能性があります。他国で承認を得るのに要する時間は、FDA承認を得るのに要する時間とは大きく異なる可能性がある。他の国/地域の上場承認の流れは、通常、上述したFDAの米国での承認に関するすべてのリスクおよびその他のリスクに関連する。特に、米国以外の多くの国では、製品が商業化される前に定価と精算承認を受けなければならない。この承認を得ることは、このような国で製品が市場に出す時間を大幅に遅らせることになるかもしれない。一方の国の上場承認は、他の国の上場承認を確保することはできないが、一方の国の上場承認の失敗や遅延は、他国の規制プロセスに悪影響を及ぼす可能性がある。他の国で上場承認を得ることができなかった場合、または承認を得る上でいかなる遅延や他の挫折が生じても、これらの海外市場における現在または将来の候補製品のマーケティング能力を弱めることになる。このような減価はいずれも私たちの潜在市場の規模を縮小し、私たちの業務、運営業績、将来性に大きな悪影響を及ぼす可能性がある。
私たちの将来の成長は私たちの外国市場に進出する能力にかかっているかもしれませんが、そこでは追加の規制負担や他のリスクや不確定要素の影響を受け、これらは私たちの業務に実質的な悪影響を及ぼすかもしれません。
私たちが外国市場関連規制機関の規制承認を得るまで、私たちは現在または未来のいかなる候補製品もマーケティングや普及は許可されておらず、私たちは現在または未来の候補製品の規制承認を決して得られないかもしれない。多くの他の国で単独の規制承認を得るためには、これらの国の安全性と有効性に関する多くのおよび異なる規制要求、および私たちの現在または未来の候補製品の臨床試験や商業販売、定価、流通などに対する規制要求を守らなければならず、これらの管轄区域で成功するかどうかを予測することはできない。もし私たちが現在または未来の候補製品が承認され、最終的に私たちの現在または未来の候補製品を海外市場で商業化すれば、私たちは追加のリスクと不確実性に直面するだろう
76
私たちの現在または将来の候補製品の海外販売は、政府の規制、政治的、経済的不安定、貿易制限、関税変化の悪影響を受ける可能性もある。
我々は将来的には米国以外の地域で現在または将来の候補製品の臨床試験を行う可能性があり,FDAや類似の外国規制機関はこのような試験のデータを受け入れない可能性がある。
私たちは将来、ヨーロッパを含めてアメリカ以外で1つ以上の臨床試験を行うことを選択するかもしれない。FDAや同様の外国規制機関が米国や他の管轄地域以外で行われている臨床試験を受けた研究データは,何らかの条件によって制限される可能性があり,まったく受け入れられない可能性もある。外国の臨床試験のデータを米国の上場承認の基礎として利用しようとする場合、FDAは通常、(I)データが米国の人口と米国の医療実践に適用されない限り、(I)データが米国の人口および米国の医療実践に適用されない限り、(Ii)試験は公認能力のある臨床研究者が行うことができ、(Iii)データは有効と見なすことができ、FDAによる現場検査を必要としない、あるいはFDAがこのような検査を行う必要があると考えた場合、FDAは現場検査や他の適切な方法でデータを検証することができる。そのほか、十分な患者群と統計能力を含むFDAの臨床試験要求を満たさなければならない。多くの外国の規制機関もまた似たような承認要求を持っている。また、このような外国裁判は、裁判を行う外国司法管轄区域に適用される現地法によって管轄される。FDAや同様の外国規制機関が米国や適用司法管轄区域以外で行われた試験データを受け入れることは保証されない。FDAまたは同様の外国の規制機関がこれらのデータを受け入れない場合、追加の試験が必要になり、これは高価で時間がかかる可能性があり、私たちが開発する可能性のある現在または未来の候補製品が適用される司法管轄区域で商業的な承認を得ることができない可能性がある。
私たちはアメリカと外国のいくつかの反腐敗、反マネーロンダリング、輸出規制、制裁、その他の貿易法律と法規の制約を受けている。私たちは違反によって深刻な結果に直面するかもしれない。
その他の事項を除いて、米国および外国の反腐敗法(“反海外腐敗法”(FCPA)、反マネーロンダリング、輸出規制、制裁およびその他の貿易法律法規を含み、私たちは総称して貿易法と呼ぶ)は、会社およびその従業員、代理人、臨床研究組織、法律顧問、会計士、コンサルタント、請負業者および他のパートナーの許可、承諾、提供、提供、誘致、または直接または間接的に公共または民間部門の受取人の腐敗または不当な支払いまたは任意の他の価値のあるものを受け入れることを禁止する。近年、反腐敗と反賄賂法は積極的に施行され、広く解釈されている。貿易法違反は、大量の刑事罰金と民事処罰、監禁、貿易特権の喪失、資格取り消し、税収の再評価、契約違反と詐欺訴訟、名声損害、その他の結果を招く可能性がある。米国以外での活動の増加に伴い、政府機関や国有または付属実体の役人や従業員との相互作用を増加させることが含まれる可能性があり、これらの法律の下でのリスクが増加する可能性がある。このような法律を遵守しないことは、私たちを調査、制裁、和解、起訴、他の法執行行動、利益の返還、巨額の罰金、損害賠償、他の民事と刑事罰または禁止、不利なメディア報道、および他の結果に直面させるかもしれない。どんな調査、行動、または制裁も、私たちの業務、運営結果、そして財政状況を損なう可能性がある。私たちは政府機関や政府付属病院、大学、その他の組織の役人や従業員と直接または間接的な相互作用を持っている。
77
米国以外の政府は厳しい価格制御を実施する傾向があり、これがあれば私たちの収入に悪影響を及ぼす可能性がある。
いくつかの国、特にEUでは、処方薬の価格設定は政府によって統制されている。これらの国では、製品の発売許可を受けた後、政府当局との定価交渉にかなりの時間がかかる可能性がある。いくつかの国で保険と精算或いは定価の承認を得るために、私たちは臨床試験を行う必要があるかもしれません。私たちの候補製品の費用効果を他の利用可能な療法と比較します。また、米国以外の多くの国は限られた政府支援計画を持っており、私たちの候補製品などの製品の精算を提供し、個人支払者に商業製品を獲得させることに重点を置いている。もし私たちの候補製品が精算を受けることができない場合、あるいは範囲や金額が制限されている場合、あるいは定価レベルが満足できなければ、私たちの業務は実質的な損害を受ける可能性があります。
医療やその他の法規遵守に関するリスク
現在または将来の候補製品を商業化することができても、これらの薬剤は、不利な価格設定法規や第三者保険·精算政策の制約を受ける可能性があり、これは私たちの業務を損なうことになる。
私たちが規制承認を受ける可能性のあるどの製品のカバー範囲や精算状態にも、大きな不確実性がある。米国や他の国では、規制機関のマーケティング承認を得て商業販売を行う可能性のある任意の製品の販売は、保証範囲と第三者支払者の精算にある程度依存することになる。第三者支払者には、政府医療計画(例えば、MedicareおよびMedicaid)、管理型ヘルスケア提供者、個人健康保険会社、健康維持組織、および他の組織が含まれる。この第三者決済者たちは彼らがどのような薬を支払うかを決定し、清算水準を確立する。政府や他の第三者支払者が提供する保険範囲や精算範囲は,多くの患者が標的タンパク質分解療法などの治療を負担できるために重要である。
米国や他の国の市場では,患者は通常第三者支払者によって治療に関連する費用の全部または一部を精算する。政府医療保健計画(例えば連邦医療保険や医療補助)や商業支払者の十分なカバーと精算は新製品の受容度に重要である。私たちが私たちの候補製品を商業化することに成功できるかどうかは、政府衛生行政部門、個人健康保険会社、その他の組織がこれらの製品と関連治療に提供する保険範囲と十分な補償にある程度依存する。保険を提供しても、承認された精算金額が十分に高くない可能性があり、十分な投資リターンを実現するために十分な価格設定を確立したり維持したりするのに十分ではないかもしれません。政府当局と第三者支払者、例えば個人健康保険会社や健康維持組織は、どのような薬剤を支払うかを決定し、精算レベルを確立する。
新たに承認された製品の保険カバー範囲や精算に関する不確実性も大きく,カバー範囲はFDAや同様の外国規制機関が薬物を承認する目的よりも限られている可能性がある。米国では,新薬精算に関する主な決定は通常医療保険と医療補助サービスセンター(CMS)によって行われており,米国衛生·公衆サービス部(HHS)の一機関である。CMSは私たちの製品がどの程度連邦医療保険の下でカバーされ、精算されるかを決定する。個人支払者はCMSに大きく従う傾向がある。支払人が精算を決定する際に考慮する要素は、製品があるかどうかに基づいている
私たちが現在または未来の候補製品を商業化することに成功した能力はまた、政府当局、個人健康保険会社、および他の組織がこれらの現在または未来の候補製品および関連治療に保険および補償を提供する程度にある程度依存するであろう。また、支払者が製品に保険を提供することを決定することは、十分な返済率を承認することを意味するものではない。私たちは私たちが商業化されたどんな候補製品もカバー範囲を持っていることを確実にすることができない。保険を提供すれば、精算は限られたレベルに限られており、マーケティングの承認を得た任意の候補製品を商業化することに成功できないかもしれません。
78
米国では、第三者支払者は製品の保証と精算に対して統一的な政策を持っていない。したがって,異なる支払先が提供する補償範囲や金額の決定が大きく異なる可能性がある.支払者が製品に保険を提供するかどうかを決定するプロセスは、支払者が製品のために支払う支払率を設定するプロセスと分離することができる。第三者支払者は,ある特定の製品やサービスを保証することを決定し,他の支払者もその医療製品やサービスに保証を提供することを確保することはできない.第三者支払者は、承認されたリストまたは処方内の特定の製品に保証範囲を制限することができ、特定の適応のすべてのFDAによって承認された製品を含まない可能性がある。さらに、コストの低い模倣薬または他の代替品が利用可能である場合、第三者支払者は、その処方に特定のブランド製品を含むことを拒否するか、または他の方法で患者がブランド薬を得ることを制限する可能性がある。
医薬品の正味価格は、政府医療計画または個人支払者が要求する強制的な割引またはリベート、および将来、米国価格よりも低い価格で販売される可能性のある国から医薬品を輸入することを制限する法律の緩和によって低下する可能性がある。ますます多くの第三者支払人は製薬会社に価格に基づいて所定の割引を提供し、医療製品の定価に挑戦することを要求している。私たちは私たちが商業化されたどの候補製品も精算できることを確実にすることはできない。もし精算できるなら、精算のレベルもそうだ。また、多くの製薬業者は平均販売価格(ASP)と最適価格のようないくつかの価格報告指標を計算し、政府に報告しなければならない。場合によっては、これらの指標が正確かつタイムリーに提出されていない場合には、処罰が適用される可能性がある。また,これらの薬品の価格は,政府医療計画が要求する強制的な割引やリベートによって低下する可能性がある。販売が承認される可能性のある製品の保証範囲や精算を確保するためには、我々の製品の医療必要性と費用効果を証明し、FDAや同様の規制承認を得るのに必要なコストを証明するために、高価な薬物経済学的研究を行う必要があるかもしれない。さらに、購入者、個人健康計画、または政府医療計画に割引を提供する必要があるかもしれない。私たちが最善を尽くしたにもかかわらず、私たちの候補製品は医学的に必要あるいは費用効果があると思われないかもしれない。第三者支払者が1つの製品が他の利用可能な療法と比較して費用効果がないと思う場合、彼らは承認された製品をその計画下の福祉としてカバーしないかもしれない、または、もし彼らがそう思う場合、支払いレベルは利益的な方法で私たちの製品を販売するのに十分ではないかもしれない。製品が承認されると、第三者支払者が製品を保証しない決定は医師の使用率を減少させ、販売、私たちの運営、財務状況に実質的な悪影響を及ぼす可能性がある。
最後に、いくつかの外国の国では、候補製品の提案価格は合法的に発売されるために承認されなければならない。製品の価格設定に対する各国の要求の差が大きい。例えば、EUでは、薬品の定価と精算は国家レベルで個別のEU加盟国の社会保障制度に基づいて規制されている。一部の外国諸国は、その国の健康保険制度が補償を提供する医療製品の範囲を制限し、人が使用する医療製品の価格を制御することができる様々な選択を提供している。精算または定価の承認を得るために、その中のいくつかの国は臨床試験の完成を要求する可能性があり、特定の候補製品のコスト効果を現在利用可能な治療法と比較する。一国は医薬製品の具体的な価格を承認することができ、医薬製品を市場に投入する会社の収益力に対して直接或いは間接的な制御制度をとることもできる。製品の価格コントロールや精算制限がある国/地域が、私たちのどの候補製品に対しても有利な精算と定価手配を許可することは保証されません。清算が許可されても、歴史的に見て、いくつかの外国で発売された候補製品、例えばEUのいくつかの国は、アメリカの価格構造に従わず、しかも価格は通常はるかに低い。
現在と将来の医療立法改革措置は、私たちの業務や運営結果に実質的な悪影響を及ぼす可能性がある。
規制、法規、または既存の規制の解釈の変化は、例えば、(I)私たちの製造スケジュールの変更、(Ii)製品ラベルの追加または修正、(Iii)私たちの製品のリコールまたは停止、または(Iv)追加の記録保存要件のような私たちの業務に影響を与える可能性があります。このような変化を強制的に実施すれば、私たちの業務運営に悪影響を及ぼす可能性がある。
米国や一部の外国司法管轄地域では、医療を得る機会を拡大し、医療の質を向上させ、医療コストをコントロールまたは低減することを目的としたいくつかの立法·規制改革および提案された改革を継続することが可能である。例えば,2010年3月,“医療·教育協調法案”(ACAと略す)で改正された“患者保護·平価医療法案”(Patient Protection and Affordable Care Act,ACAと略す)が可決され,政府や民間保険会社が医療保険に融資する方式を大きく変え,米国製薬業に大きな影響を与えた。その他の事項以外に、ACAは生物製品をより低コストの生物模倣薬の潜在競争を受けさせ、340億薬品割引計画に符合する実体タイプを拡大し、医療補助薬物返却計画に基づいてメーカーが医療補助薬物返却計画の下で借りたリベートを計算し、メーカーの医療補助薬物返却計画の下でのリベートを増加し、リベート計画を医療補助管理の看護組織に登録した個人に拡大し、あるブランドの処方薬メーカーに対する年費と税収を計算し、そして新しい連邦医療保険D部分カバーギャップ割引計画を作成した
79
メーカーは保証空白期間内に条件を満たす受益者に50%の販売時点割引(2018年の両党予算法により70%に向上し、2019年1月から発効)を提供することに同意しなければならず、メーカーの外来薬物として連邦医療保険D部分カバーの条件を組み入れ、連邦政府の比較有効性研究の項目を増加させるために激励を提供しなければならない。
公布以来、ACAのいくつかの側面は司法、行政、行政、国会立法の挑戦に直面しており、将来的にACAに対してより多くの挑戦と修正案を提出することが予想される。2021年6月17日、米国最高裁はいくつかの州がACAに対して提出した最新の司法挑戦を却下したが、ACAの合憲性を具体的に裁くことはなかった。また,バイデン総裁は複数の行政命令を発表し,処方薬コスト問題を解決しようとしている。バイデン政府の他の医療改革措置や他の挑戦,ACAの廃止または代替の努力(あれば)が我々の業務にどのように影響するかは不明である。
また、ACAが公布されて以来、米国は他の立法改正を提案し、採択した。例えば
また、支払い方法は医療立法と規制措置の影響を受ける可能性がある。例えば、CMSは、バンドル支払いモードのような新しい支払いおよび配信モードを開発することができる。最近、政府はメーカーが製品を販売するための価格設定方式の審査を強化した。このような審査は米国議会の最近の数回の調査を招き、さらに連邦と州立法の提出と通過を招き、これらの立法は薬品定価の透明性を高め、連邦医療保険下の処方薬のコストを下げ、定価とメーカー患者計画との関係を審査し、政府計画の薬品精算方法を改革することを目的としている。
また、米国の薬品価格決定方法に対する立法と法執行への関心もますます大きくなっている。具体的には,政府はメーカーが販売する製品に価格を設定する方式をより厳しく審査し,米国議会のいくつかの調査を招き,連邦を提案し公布した
80
そして州立法は、薬品定価の透明性を高め、連邦医療保険下の処方薬のコストを下げ、定価とメーカー患者計画との関係を審査することを目的としている。2020年12月2日、HHSは薬品メーカーがD部分でスポンサーの値下げを計画している安全港の保護を取り消し、法律が値下げを要求しない限り、直接あるいは薬局福祉マネージャーを通過する規定を発表した。この規定はまた、販売時点での値下げを反映するための新しい避難港を作成し、薬局福祉マネージャーと製造業者との間のいくつかの固定料金手配のための避難港を作成する。裁判所の命令により,上記安全港の除去と増加が延期され,アイルランド共和軍はさらにこの規則の実施を2032年1月1日に延期した。いくつかの措置および他の提案された措置は発効するために追加の立法によって許可される必要があるかもしれないが、バイデン政府はこれらの措置を撤回または他の方法で変更する可能性があるが、バイデン政府と国会は薬品コストを制御するための新しい立法措置を求め続けると表明している。
州レベルでは、各州はますます積極的に、価格または患者の精算制限、割引、ある製品への参入の制限、マーケティングコストの開示および透明性措置を含む薬品および生物製品の価格設定を制御するための法規を立法と実施し、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。法律で規定されている第三者支払者の支払金額の価格制御またはその他の制限は、私たちの業務、財務状況、運営結果、将来性を損なう可能性があります。また、地域衛生保健当局や個別病院は、どの薬品やサプライヤーが彼らの処方薬や他の医療計画に含まれるかを決定するために入札プログラムを使用することが増えている。これらの措置が承認されると、私たちの製品に対する最終的な需要を減らしたり、私たちの製品の価格設定に圧力を与えたりする可能性があり、これは私たちの業務、財務状況、運営結果、見通しにマイナスの影響を与える可能性があります。
私たちは将来的により多くの州と連邦医療改革措置を取ることが予想され、そのいずれも連邦と州政府が医療製品およびサービスのために支払う金額を制限する可能性があり、これは私たちの現在または未来の候補製品に対する需要の減少または追加の価格設定圧力を招く可能性がある。米国の将来の立法や行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することはできない。もし私たちまたは私たちが接触する可能性のある任意の第三者が既存の要求の変化に緩やかに適応できない場合、または新しい要求や政策を採用することができない場合、または私たちまたはそのような第三者が規制適合性を維持できない場合、私たちの候補製品は得られた可能性のあるいかなる規制承認も失う可能性があり、私たちは利益を達成したり維持することができないかもしれない。
顧客、医療提供者、医師、第三者支払者との関係は、適用されるリベート、詐欺、乱用、および他の医療法律法規の制約を受けることになり、これは、私たちを刑事制裁、民事処罰、政府医療計画から除外され、契約損害、名声損害、および将来の利益と収益の減少に直面させる可能性がある。
医療サービス提供者、医師、および第三者支払者は、市場の承認を得た任意の現在または将来の候補製品の推薦および処方において主な役割を果たすだろう。私たちの業務運営や現在または将来の第三者支払者や顧客との任意の手配は、詐欺や乱用に関連する広範に適用される連邦および州法律、および他の医療に関する法律や法規に直面する可能性があります。製薬会社は、連邦政府およびそれらが業務を展開している州と外国の司法管轄区域当局の追加的な医療監督管理と法執行を受けており、これは、私たちが研究、販売、マーケティング、流通し、マーケティングの許可を得た任意の製品の財務的配置と関係を制限するかもしれない。このような法律には、州および連邦反リベート、詐欺および乱用、虚偽声明、ならびに薬品の価格設定および支払い、ならびに医師および他の医療提供者への他の価値移転に関連する透明性のある法律および法規が含まれているが、これらに限定されない。私たちの業務がこのような法律または任意の他の適用可能な政府法規に違反していることが発見された場合、行政、民事および刑事罰、損害賠償、罰金、返還、削減または再構成業務、誠実な監督と報告義務、連邦および州医療計画から除外され、責任個人が監禁される可能性があるが、処罰を受ける可能性がある。他の事項に加えて、これらの法律は、私たちがマーケティング承認を得た任意の現在または将来の候補製品のビジネスまたは財務的配置および関係に影響を与える可能性があります。より多くの情報については、本報告の“政府規制−その他の規制事項−他の医療保健法”というタイトルの部分を参照されたい。
これらの法律の範囲も執行も不確定であり,現在の医療改革環境下では,特に適用の前例や法規が乏しい場合には,急速な変化が生じる可能性がある。連邦と州法執行機関は最近、医療保険会社と医療保健提供者の間の相互作用の審査を強化し、医療保健業界の一連の調査、起訴、有罪判決と和解を招いた。我々の内部運営と将来の第三者の業務配置が適用される医療法律や法規に適合することを確保することは、多くのコストに及ぶ。政府当局は、私たちの業務やり方が現在または未来に適合していないと結論するかもしれないが、詐欺および乱用または他の医療保健法律および法規の適用に関する現行または未来の法規、法規または判例法に関連している。もし私たちの操作がこれらの法律または他の可能性に違反していることが発見されたら
81
もし私たちが私たちに適用されれば、私たちは重大な民事、刑事と行政処罰、損害賠償、罰金、返還、契約損害、名声損害、利益減少と将来の収益、監禁、政府援助の医療計画(例えばMedicareおよびMedicaid)から除外され、私たちの業務を削減または再編し、そして私たちがこれらの法律を遵守しない疑いを解決するために会社の誠実な合意または同様の和解の制約を受けた場合、追加の報告義務と監督に直面する可能性がある。しかも、このような行動を防御するのは高価で時間がかかる可能性があり、大量の財政と人的資源が必要となる可能性がある。したがって、私たちが私たちに提起される可能性のあるいかなる訴訟も防ぐことに成功しても、私たちの業務は損害を受ける可能性がある。もし私たちがそれと業務を展開することを期待している任意の医師または他の提供者または実体が適用されない法律を遵守していないことが発見された場合、彼らは政府の援助された医療計画および監禁から除外されることを含む刑事、民事または行政制裁を受ける可能性がある。上記のいずれかの状況が発生した場合、私たちの業務運営能力と当社の運営結果は悪影響を受ける可能性があります。もし私たちがそれと業務を展開することを期待している任意の医師や他の医療提供者や実体が適用法を遵守していないことが発見された場合、彼らは同様の行動、処罰、および制裁を受ける可能性がある。
EUはまた、医師の処方、推薦、裏書き、購入、供給、注文、または医療製品の使用を誘導または奨励するために、医師に福祉または利点を提供することを禁止している。医師への福祉や利点の提供は、イギリスの“2010年反賄賂法”のようなEU加盟国の反賄賂法の管轄を受けている。このような法律に違反することは巨額の罰金と監禁につながるかもしれない。いくつかの連合会員国で医者に支払われた費用は公開されなければならない。また、医師との合意は、通常、医師の雇用主、医師の主管専門組織、および/またはEU加盟国の監督当局に事前に通知し、承認しなければならない。これらの要件は、EU加盟国に適用される国家法律、業界規則、または専門行為規則に規定されている。このような要求を守らないことは、名声のリスク、公開非難、行政処罰、罰金、または監禁につながる可能性がある。
もし私たちが環境、健康、安全の法律法規を守らなければ、私たちは罰金や罰金を科されたり、コストを発生したりする可能性があり、これは私たちの業務の成功に実質的な悪影響を及ぼすかもしれない。
私たちは多くの環境、健康と安全法律と法規の制約を受けて、それらの研究室の手続きと危険材料と廃棄物の処理、使用、貯蔵、処理と処理を管理する法律と法規を含む。私たちの行動は化学物質、生物学的、そして放射性物質を含む危険で燃えやすい材料の使用に関するものだ。私たちの業務はまた危険な廃棄物製品を発生させるだろう。私たちは一般的に第三者と契約を結び、このような材料と廃棄物を処理する。私たちはこのような材料が汚染や傷害をもたらす危険を除去することができない。もし私たちが危険な材料を使用して汚染や損傷をもたらしたら、私たちはそれによるいかなる損害に責任を負い、いかなる責任も私たちの資源の範囲を超える可能性がある。私たちはまた民事や刑事罰金と処罰に関連した巨額の費用を発生させるかもしれない。
危険材料の使用による従業員の負傷により生じる可能性のあるコストや支出を支払うために労働者補償保険を維持しているが、潜在的な責任を支払うのに十分ではない可能性がある。私たちは私たちが生物、危険または放射性物質を貯蔵したり処分したりすることによって、私たちが提起した環境責任や有毒侵害に対して保険を維持することはできません。また、現在または未来の環境、健康、安全法律法規を遵守するためには、巨額のコストが生じる可能性がある。現在または未来の環境法律法規は、私たちの研究、開発、生産努力を損なう可能性があり、これは私たちの業務、将来性、財務状況、または運営結果を損なう可能性があります。
私たちの従業員、首席調査員、CRO、コンサルタントは、規制基準と要求、およびインサイダー取引法律を遵守しないことを含む、不適切な行為や他の不適切な活動に従事する可能性があります。
私たちは従業員、首席調査員、CRO、そしてコンサルタントが詐欺や他の不法活動に従事する可能性があるというリスクに直面している。これらの当事者の不正行為は、意図的、無謀、および/または不注意な行為、またはFDAおよび他の規制機関に規定された不正活動を開示することを含むことができ、これらの機関に真実、完全かつ正確な情報を報告することを要求する法律、米国および海外の医療詐欺および法律法規の乱用、または財務情報またはデータの正確な報告を要求する法律を含むことができる。特に、医療業界の販売、マーケティング、患者支援および業務配置は、詐欺、不正行為、リベート、自己取引、その他の乱用行為を防止するための広範な法律法規によって制約されている。これらの法律法規は、広範な価格設定、割引、マーケティングおよび販売促進、販売手数料、顧客インセンティブ計画、および他のビジネス計画を制限または禁止する可能性があります。これらの法的制約を受けた他の活動は、臨床試験中に得られた情報の不適切な使用、あるいは私たちの臨床前研究または臨床試験で虚偽のデータを作成することを含み、これは規制制裁を招き、私たちの名声に深刻な損害を与える可能性がある。私たちはすべての従業員に適用される行動規則を採択しましたが、従業員や他の第三者の不正行為を常に識別して阻止できるわけではありません。私たちがこのような活動を発見し、防止するための予防措置は、未知または未管理のリスクや損失を効果的にコントロールできない可能性があり、あるいは政府の調査や他の行動や訴訟から私たちを保護することができないかもしれません
82
これらの法律や法規。また、私たちは、起きていなくても、このような詐欺や他の不正行為を告発する可能性があるというリスクに直面している。もし私たちにこのような訴訟を提起した場合、私たちは私たちの権利を弁護したり、維持することに成功しませんでした。これらの訴訟は、民事、刑事および行政処罰、損害賠償、罰金、連邦医療保険、医療補助および他の連邦医療保健計画から除外される可能性があり、契約損害、名声損害、利益および将来の収益の減少、および私たちの業務縮小を含むかもしれません。これらはいずれも、私たちの業務運営能力および私たちの運営結果に悪影響を及ぼす可能性があります。
私たちが第三者の業務配置と適用される医療法律や法規に適合するように努力することは、多くのコストに関連することを確実にします。これらの法律の範囲が広く、法定例外状況と選択可能な避難港が限られていることから、私たちのいくつかの商業活動は1つ以上のこのような法律の挑戦を受けるかもしれない。政府当局は、私たちの業務実践が現在または未来に適用される詐欺および乱用または他の医療保健法律および法規に関連する現行または未来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。私たちの運営がこれらの法律または他の私たちに適用される可能性のある任意の政府法規に違反していることが発見された場合、私たちは、罰金、損害賠償、罰金、返還、個人監禁、名誉被害、MedicareおよびMedicaidなどの政府援助に関与する医療計画から除外され、追加の報告要件および監督を受ける可能性があります(これらの法律違反に関する告発および私たちの業務の削減または再構成に関する)これらのいずれも、私たちの業務運営能力および私たちの運営結果に悪影響を及ぼす可能性があります。
私たちはこれらの法律に違反するリスクが増加していることが発見された。その多くの法律は規制部門や裁判所の十分な説明を受けておらず、それらの条項は様々な解釈ができるからだ。これらの法律に違反して私たちにとった行動は、たとえ私たちが弁護に成功しても、巨額の法的費用を招き、私たちの経営陣の業務運営への注意をそらす可能性があります。持続的に変化するコンプライアンス環境、および異なるコンプライアンスおよび/または報告要件を有する複数の司法管轄区域の必要性に適合するために、強力かつ拡張可能なシステムを確立し、維持することは、医療会社が1つまたは複数の要件と衝突する可能性を増加させる。
商業化に関連するリスク
私たちが現在または未来の候補製品がマーケティング承認を得ても、私たちの現在または未来の候補製品は幅広い市場で受け入れられない可能性があり、これは彼らの販売から得られる収入を制限するだろう。
FDAまたは他の適用可能な規制機関が承認された場合、私たちの現在または未来の候補製品の商業的成功は、私たちの現在または未来の候補製品に対する医学界(医師、患者、および医療支払者を含む)の認知および受容の程度に依存する。市場が現在または未来の候補製品を受け入れるかどうかは、承認されれば、多くの要素に依存する
83
もし私たちが現在または未来の候補製品が承認されたが、患者、医者、および支払人が十分に受け入れられる程度に達していない場合、私たちは現在または未来の候補製品から十分な収入を生成して利益を達成または維持することができないかもしれない。精算を承認する前に、医療支払者は、現在あるいは未来の候補製品がこれらの目標適応の治療に加え、患者に増加的な健康利益を提供することを証明することを要求するかもしれない。私たちの教育医療界、患者組織、第三者支払人は、私たちの現在または未来の候補製品のメリットを理解する努力には大量の資源が必要かもしれないし、決して成功しないかもしれない。
私たちは激しい競争に直面しており、これは他の人たちが私たちよりも早く、あるいはより成功的に薬物を発見、開発、商業化することにつながるかもしれない。
新薬の開発と商業化競争は激しい。我々は、タンパク質分解、抗体治療、阻害性核酸、遺伝子編集または遺伝子治療開発プラットフォームを使用した第三者からの競争に直面し、さらに、小分子阻害剤のようなより伝統的な治療方式に専念する会社に直面する。潜在的な競争相手はまた学術機構、政府機構とその他の公共と個人研究組織を含み、これらの組織は研究を行い、特許保護を求め、そして新薬の研究、開発、製造と商業化のための協力手配を確立する。
われわれは患者のための小分子タンパク質分解剤療法の開発に努力している競争相手にはArvinas,Inc.,C 4 Treateutics,Inc.,Foghorn Treateutics Inc.およびNurix Treateutics,Inc.が含まれているが,その中のいくつかは臨床開発に入っている。また、いくつかの大手製薬会社はすでにこの分野の臨床前と臨床投資を開示している。我々の競争相手には,他の標的化されたタンパク質分解法や小分子,抗体あるいは遺伝子療法を開発している会社も含まれており,これらの方法の適応は我々の目標と同じである。我々が小分子タンパク質分解剤の開発に直面しているライバルのほかに,我々のIRAK 4,STAT 6,TYK 2,STAT 3,MDM 2プロジェクトで競争に直面する。これらの適応の多くは、より伝統的な治療法を含む可能性がある承認された看護基準を有している。これらの既存療法と効率的に競合するためには,われわれのタンパク質分解剤療法が既存療法に有利であることを証明する必要がある。
私たちの現在あるいは未来の多くの競争相手は研究開発、製造、臨床前テスト、臨床試験を行い、監督管理の承認、精算とマーケティング許可を得る薬物の面で私たちより多くの財務資源と専門知識を持っている。製薬、バイオテクノロジー、診断業界の合併と買収は、より多くの資源を私たちの数の少ない競争相手に集中させる可能性がある。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。これらの競争相手はまた、合格した科学、販売、マーケティングと管理者を募集し、維持し、臨床試験場と臨床試験の患者登録を確立し、そして私たちの計画と相補的或いは必要な技術を獲得する上で私たちと競争している。
84
もし私たちの競争相手が私たちまたは私たちの協力者が開発する可能性のある任意の薬物よりも安全で、より効果的で、副作用が少ない、より便利で、より安い薬物を開発し、商業化すれば、私たちのビジネス機会は減少または消失するかもしれない。私たちの競争相手はまた私たちよりも早くFDAや他の規制機関のその薬品の承認を得ることができ、これは私たちの競争相手が私たちまたは私たちの協力者が市場に入る前に強力な市場地位を確立することをもたらすかもしれない。私たちのすべての現在或いは未来の候補製品の成功に影響する重要な競争要素は、承認されれば、それらの治療効果、安全性、利便性、価格、模造薬の競争レベル及び政府と他の第三者支払い者が精算できるかどうかであるかもしれない。
私たちに対する製品責任訴訟は、私たちが重大な責任を負うことを招き、私たちが開発する可能性のある現在または未来の候補製品の商業化を制限する可能性があります。
私たちは人体臨床試験で現在または未来の候補製品をテストすることに関連する固有の製品責任リスクに直面しており、私たちが開発する可能性のある現在または未来の候補製品を商業販売すれば、私たちはより大きなリスクに直面する。もし私たちが私たちの現在または未来の候補製品に被害を与えるクレームを自己弁護することに成功できなければ、私たちは重大な責任を招くかもしれない。是非曲直や最終結果にかかわらず、賠償責任は
私たちはまだ製品責任保険を維持していません。臨床試験を開始する時、任意の候補製品を商業化することに成功すれば、保険範囲を増やす必要があると予想されます。保険範囲はますます高くなっています。私たちは可能などんな責任も満たすために、合理的な費用や十分な金額で製品責任保険を維持することができないかもしれない。
将来的に、私たちが現在または未来の候補製品を販売およびマーケティングするために、販売およびマーケティングおよび患者支援能力を確立することができない場合、もし私たちが現在または未来の候補製品が承認された場合、私たちはそれを商業化することに成功できないかもしれず、私たちは何の収入も生まれないかもしれない。
私たちは現在販売やマーケティングインフラがなく、販売、マーケティング、患者支援あるいは薬品流通の経験もありません。販売およびマーケティングの責務を保持する任意の承認された候補製品をビジネスに成功させるためには、私たちの販売、マーケティング、患者支援、管理、および他の非技術的能力を確立するか、または第三者とこれらのサービスを実行することを手配しなければならない。将来、私たちが現在または未来の候補製品が承認されれば、私たちは、現在または未来の候補製品を販売するために、集中的な販売およびマーケティングインフラを構築すること、または私たちの協力者と共に販売活動に参加することを選択するかもしれない。
私たち自身の販売とマーケティング、および患者支援能力の確立、および第三者とこれらのサービスを実行する計画を達成することはリスクに関連する。例えば、販売チームの採用と訓練は高価で時間がかかり、どんな薬の発売も延期される可能性がある。販売チームを募集し、マーケティング能力を確立する候補製品の商業発表が何らかの理由で延期または発生していない場合、これらの商業化費用を早期または不必要に発生させる。これは費用が高いかもしれません。もし私たちが私たちの販売とマーケティング担当者を維持したり再配置できなければ、私たちの投資は損失します。
現在または未来の候補製品の商業化を阻害する可能性がある要因は
85
もし私たちが第三者と販売、マーケティング、患者支援、流通サービスの手配を達成すれば、私たちの薬品収入またはこれらの薬品収入が私たちにもたらす収益力は、私たち自身が開発した任意の現在または未来の候補製品をマーケティングし、販売することよりも低いかもしれない。さらに、私たちは、私たちの現在または未来の候補製品の配置を第三者との販売およびマーケティングに成功させることができないかもしれないし、私たちに有利な条項でそうすることができないかもしれない。私たちはこれらの第三者に対して支配権がほとんどないかもしれません。彼らのいずれも必要な資源と注意を投入して、私たちの現在または未来の候補製品を効果的に販売し、マーケティングすることができないかもしれません。もし私たちが販売とマーケティング能力をうまく確立できなければ、私たち自身も第三者と協力しても、私たちは私たちの現在または未来の候補製品を商業化することに成功しないだろう。しかも、私たちの業務、経営業績、財務状況、そして見通しは重大な悪影響を受けるだろう。
私たちの第三者への依存に関するリスク
我々は現在と未来の候補製品のために進行中と計画中の臨床前研究と臨床試験に依存し、引き続き第三者に依存することが予想される。これらの第三者がその契約義務を成功的に履行し、法規要件を遵守し、または予想される最終期限までに完了できない場合、私たちは現在および潜在的な将来の候補製品のマーケティング承認を得ることができないか、または商業化することができず、私たちの業務は深刻な損害を受ける可能性がある。
我々は,医療機関,CRO,契約製造組織,戦略パートナーなどの独立した研究者や協力者に利用して依存し,臨床前研究を支援している。私たちは独立して臨床試験を行うことができない。私たちは医療機関、臨床研究者、契約実験室、協力パートナーを含む他の第三者に依存して、現在の候補製品の臨床試験や他の方法での支援を提供しており、将来の候補製品もこれらの第三者に依存することが予想される。私たちはこれらの方が私たちの候補製品のために臨床試験を実行し、その活動のいくつかの方面だけを制御することに深刻に依存している。しかし、私たちは私たちのすべての臨床試験が適用された方案、法律と法規の要求、および科学的な基準に従って行われていることを確実にする責任があり、私たちのCROへの依存は私たちの規制責任を解除しない。私たちが臨床前研究または臨床試験を行っている間の任意の法律および法規に違反する行為に対して、私たちはタイトルのない警告状または法執行行動を受ける可能性があり、その中には民事処罰が含まれている可能性があり、最高で刑事起訴に達することができる。
私たちと契約したいかなる第三者も、データと結果が科学的に信頼性と正確であることを保証し、試験患者が臨床試験に参加する潜在的なリスクを十分に理解し、彼らの権利を保護するために、GCPを含む臨床試験結果を行う、モニタリング、記録、報告する法規と要求を遵守しなければならない。これらの規定はFDA、欧州経済圏加盟国の主管当局と類似した外国の監督管理機関によって臨床開発中の任意の薬物に対して実行される。FDAは臨床試験スポンサー,主要研究者,試験地点の定期検査によりGCP要求を実行している。私たちが契約した第三者が適用されたGCPを遵守できなかった場合、私たちの臨床試験で生成された臨床データは信頼できないと考えられる可能性があり、FDAまたは同様の外国の規制機関は、私たちのマーケティング申請を承認する前に追加の臨床試験を行うことを要求するかもしれません。検査後、FDAはどの臨床試験もGCPに適合しているかどうかを確認することは保証できません。また,われわれの臨床試験はcGMP法規により生産された現在または将来の候補製品と行わなければならない。私たちまたは私たちが契約した第三者がこれらの規定を遵守できなかった可能性があり、特定または臨床試験全体のいくつかの側面を繰り返す必要があるかもしれません。これは、上場承認過程を遅延させ、法執行行動の影響を受ける可能性があります。私たちはまた、政府が支援しているデータベースClinicalTrials.govに特定の時間範囲でいくつかの行われている臨床試験を登録し、試験計画に関連する情報を含むいくつかの情報を提供することを要求されている。そうしないと罰金、否定的な宣伝、そして民事と刑事制裁につながるかもしれない。
KT−474,KT−333,KT−253の第1段階試験を設計し,現在または将来の候補製品のために他の臨床試験を設計したり,他のスポンサー試験時に設計に参加したりする予定であるが,第三者がすべての臨床試験を行う予定である。そのため、著者らの臨床開発の多くの重要な方面は、それらの行為とタイミングを含め、著者らの直接制御下にない。われわれ自身に完全に依存しているスタッフと比較して,第三者による臨床試験への依存も臨床試験により開発されたデータ管理の直接制御が少ない。外部の当事者とのコミュニケーションも挑戦的である可能性があり、ミスや協調活動の困難を招く可能性がある。外部の当事者は
86
これらの因子は第三者の臨床試験の意思や能力に重大な悪影響を与える可能性があり、著者らのコントロール範囲を超える意外なコスト増加に直面する可能性がある。もし私たちのCROが満足できる方法で臨床試験を行わなかった場合、彼らの私たちの義務に違反したり、法規の要求を遵守できなかった場合、私たちの現在または未来の候補製品の開発、マーケティング承認、商業化が遅れる可能性があり、私たちはマーケティング承認を得ることができず、私たちの現在または未来の候補製品を商業化することができないかもしれません。あるいは私たちの開発計画は実質的で不可逆的な損害を受ける可能性があります。CROが収集した臨床データに依存できなければ,われわれが行った臨床試験の規模を繰り返し,延長あるいは増加させることが要求される可能性があり,商業化を著しく遅らせる可能性があり,支出を著しく増加させる必要がある。
もし私たちがこれらの第三者CROとのいかなる関係も終了すれば、私たちはCROの代わりに商業的に合理的な条項で合意できないか、あるいは全く合意できないかもしれない。もし私たちのCROがその契約の義務または義務を成功裏に履行できなかった場合、または予期された最終期限内に完了した場合、彼らが交換する必要がある場合、または彼らが得た臨床データの品質または正確性が、私たちの臨床規程、規制要件、または他の理由を遵守できなかった場合、そのようなCROに関連するいかなる臨床試験も延長、延期または終了される可能性があり、私たちは現在または未来の候補製品のマーケティング承認または成功のために商業化することができないかもしれない。したがって、私たちの財務業績と私たちの現在または未来の候補製品のビジネス見通しが損なわれ、私たちのコストが増加する可能性があり、私たちの収益能力が延期される可能性があると思います。
私たちが依存する第三者供給原料薬、薬品、そして私たちの候補製品で使用される出発材料の数は限られており、これらのサプライヤーのいずれかを失っても、私たちの業務を深刻に損なう可能性があります。
私たちの候補製品の中の薬物物質と薬物製品は少数の供給者から提供されており、場合によっては唯一の供給源である。著者らは現在或いは未来の候補製品の開発に成功できるかどうか、そして最終的に十分な数量の商業薬物を提供して市場の需要を満たすことができ、ある程度私たちが法規の要求に従ってこれらの薬物の薬物製品と薬剤を獲得する能力があるかどうかに依存し、そして商業化と臨床試験に十分な数量がある。私たちは現在、私たちのいかなる薬品と薬品供給者がいかなる原因で運営を停止した場合、すべての薬品或いは薬品の余分或いは第二の供給源に供給することを手配していません。私たちの薬物物質、薬物製品、または原料送達のいかなる遅延も悪影響を及ぼす可能性があり、私たちの業務を損なう可能性があります。例えば、2020年2月、私たち武漢の原料薬原料サプライヤーの一つである中国は新冠肺炎の疫病で数週間生産を停止し、別の原料薬メーカーに原料薬出発材料を渡す時間が少し遅延した。
立法提案は通過を待っており,可決されれば,米国がいくつかの外国相手と関係があるか,あるいは国家安全に脅威となるバイオテクノロジー提供者の資金に悪影響を与える可能性がある。任意の影響を受けたバイオテクノロジー供給者と商業関係のみの実体の潜在的下流への悪影響は不明であるが、サプライチェーン中断または遅延が含まれている可能性がある。
我々の現在または将来のすべての候補製品について、我々は、NDAをFDAおよび/またはMAAに提出する前に、そのような原料薬、医薬品、および医薬物質を識別および資格のより多くの製造業者に提供する予定である。しかし、私たちは、私たちの単一ソースおよびデュアルソースサプライヤーが私たちの製品に対する需要を満たすことができるかどうか、あるいは私たちがこれらのサプライヤーと合意した性質のためか、私たちとこれらのサプライヤーとの限られた経験のせいか、あるいは私たちの顧客としてのこれらのサプライヤーに対する相対的な重要性のためである。過去の表現によると、私たちは彼らが未来に私たちの需要を満たす能力を評価することが難しいかもしれない。私たちのサプライヤーは過去に彼らの製品に対する私たちの需要を適時に満たすことができますが、彼らは将来私たちの需要を彼らの他の顧客に従属するかもしれません。
必要であれば、私たちの現在または将来の候補製品で使用される医薬品および薬剤のための追加または代替の供給者を確立することは、すぐには完了しないかもしれない。もし私たちが代替サプライヤーを見つけることができれば、その代替サプライヤーは合格を必要とし、追加の規制承認が必要になる可能性があり、これはさらなる遅延を招く可能性がある。現在または将来の候補製品で使用されている薬品および薬物の十分な在庫を維持することを求めているが、コンポーネントまたは材料供給の中断または遅延、または代替源から許容可能な価格でそのような原料薬、医薬製品、および薬物を得ることができず、私たちの開発努力を阻害、遅延、制限、または阻止する可能性があり、これは、私たちの業務、運営結果、財務状況、および将来性を損なう可能性がある。
87
私たちの成功は、当社の実行管理チームが会社の成熟とともに業務発展、戦略的パートナーシップ、投資機会を求めることに成功した能力に依存しています。私たちは将来的に戦略同盟や買収を形成したり、追加の協力と許可手配を達成したりする可能性もありますが、私たちはこのような協力、連合、買収、または許可手配の利点を達成できないかもしれません。
私たちはセノフィと協力と許可手配を達成し、将来的に戦略同盟や買収を形成したり、合弁企業を作成したり、第三者と追加の協力と許可手配を達成したりする可能性があり、現在の候補製品と私たちが開発する可能性のある任意の未来の製品の開発と商業化努力を補完または強化すると信じています。これらの関係のいずれも、非日常的な費用と他の費用を発生させ、私たちの短期的かつ長期的な支出を増加させ、私たちの既存の株主を希釈したり、私たちの管理と業務を混乱させたりする証券を発行することを要求することができるかもしれない。
また、適切な戦略的パートナーを探す上で激しい競争に直面しており、交渉過程は時間がかかり複雑である。さらに、私たちは、協力努力の開発段階が早すぎると考えられるかもしれないので、現在または未来の候補製品のための戦略的パートナーシップまたは買収または他の代替配置を確立することに成功できないかもしれないので、第三者は、私たちの現在または未来の候補製品が、安全性、有効性、純度、および有効性を示し、マーケティング承認を得るための必要な潜在力を持っているとは思わないかもしれない。
さらに、我々の技術または現在または将来の候補製品に関する協働は、以下のリスクを含む可能性がある多くのリスクに直面する可能性がある
したがって,我々が既存の運営や会社文化と組み合わせることに成功しなければ,既存の連携や許可手配の利点を実現できない可能性があり,将来達成可能な任意の戦略的パートナー関係や買収,連携,許可手配の利点を実現することができず,スケジュールを延期したり,他の方法で我々の業務に悪影響を与える可能性がある.戦略的取引、ライセンス、協力、または他の業務がパートナーシップを発展させた後に、そのような取引の合理的な収入または特定の純収入を証明することが達成されるかどうかも確認できません。私たちの現在または未来の候補製品に関連する新しい協力または戦略的パートナーシップ協定のいかなる遅延も可能です
88
私たちの現在または未来のいくつかの地域または特定の適応の候補製品の開発と商業化を延期することは、私たちの業務の将来性、財務状況、および運営結果を損なうだろう。
このForm 10−K年次報告書の他の部分で議論されているように,我々のパートナーであるサイノフェイは,HSとADにおけるKT−474の第2段階ランダム第2段階試験を評価するランダム第2段階臨床試験を行っている。セノフィは,これらの臨床試験を推進するための努力と資源を決定する上で大きな裁量権を持つ。上記の要因のため、私たちは、私たちの既存の協力および許可スケジュールの利点を達成することができないかもしれません、または私たちが達成する可能性のある任意の未来の戦略的パートナーシップまたは買収、協力、または許可スケジュールは、私たちのスケジュールを延期したり、他の方法で私たちの業務に悪影響を与える可能性があります。
私たちの製造プロセスはこのようなプロセスの品質と信頼性に関するFDAの規定を守る必要があります。関連法規を遵守しないいかなる行為も、私たちの臨床前および臨床計画の遅延または終了、およびいかなる規制承認の一時停止または撤回をもたらす可能性がある。
私たち自身の工場や第三者の工場で私たちの製品を商業的に生産するためには、FDAのcGMP法規とガイドラインを守る必要があります。私たちは品質管理と品質保証を実現する上で困難に直面する可能性があり、人材不足が発生する可能性がある。FDAと同様の外国規制機関の検査を受け、適用される規制要件に適合しているかどうかを確認する。CGMPまたは他の規制要件を遵守しない場合、または私たちの施設または第三者施設または運営が規制要件を遵守できなかった場合、または任意の規制機関の検査によって引き起こされた私たちの候補製品の製造、充填、包装または貯蔵中に発生した遅延、中断、または他の問題は、現在または将来の候補製品を開発し、それを商業化する能力を著しく弱める可能性があり、私たちの臨床試験候補製品の供給の重大な遅延、または臨床試験の終了または一時停止、または現在または未来の候補製品の上場申請の提出または承認を遅延または阻止する可能性がある。深刻な不遵守はまた、警告または無タイトル手紙、罰金、禁止、民事処罰、規制機関が、現在または未来の候補製品にマーケティング承認、遅延、一時停止または承認撤回、許可証の取り消し、製品の差し押さえまたはリコール、運営制限、刑事起訴を与えることができなかったことを含む制裁の適用を招く可能性があり、これらはいずれも私たちの名声と業務を損なう可能性がある。
もし私たちの第三者メーカーが損害を与えたり、適用法に違反したりする方法で危険と生体材料を使用すれば、損害賠償責任を負う可能性があります。
我々の研究開発活動は,我々の第三者メーカーによる潜在的危険物質(化学材料を含む)の制御使用に関するものである。私たちのメーカーはアメリカ連邦、州と地方の法律法規の制約を受けて、医療と危険材料の使用、製造、貯蔵、運搬と処分を管理している。メーカーがこれらの材料を使用,処理,貯蔵,処分する手順は法律で規定されている基準に適合していると信じているが,医療や危険材料による汚染や傷害のリスクを完全に除去することはできない。このような汚染や傷害のため、私たちは責任を負うかもしれないし、地方、都市、州、または連邦当局はこれらの材料の使用を制限し、私たちの業務運営を中断するかもしれない。事故が発生すると、私たちは損害賠償責任や罰金を要求されるかもしれません。責任は私たちの資源範囲を超えるかもしれません。私たちは医療や危険材料による責任に保険をかけない。適用される環境法律法規の遵守はコストが高く、現在または未来の環境法規は私たちの研究、開発、生産努力を損なう可能性があり、それによって私たちの業務、将来性、財務状況、または運営結果を損なう可能性がある。
知的財産権に関するリスク
もし私たちの候補技術や製品のために特許や他の知的財産権保護を獲得して維持することができない場合、あるいは獲得された知的財産権保護範囲が十分でなければ、私たちの競争相手は私たちと同じ技術や薬物を開発し、商業化する可能性があり、私たちの技術や薬物を商業化することに成功する能力は損なわれる可能性があり、私たちは私たちの市場で効果的に競争できないかもしれない。
私たちのビジネス成功は、現在または未来の候補製品および私たちのコア技術、米国および他の国で特許または他の知的財産権保護を取得し、維持する能力を含む、私たちの独自の飛馬を含む、私たちの現在または未来の候補製品および私たちのコア技術にある程度依存していますTMプラットフォームは,我々の最初のIRAK 4,STAT 3,MDM 2プログラムであり,これは我々の最先端の開発プログラム,および我々独自の合成ライブラリや他の技術ノウハウである.私たちは、私たちのノウハウ、発明、改善に関連する特許出願を米国および海外に提出することを含む、私たちの独自および知的財産権の地位の保護を求めており、これらは私たちの業務の発展と実施に非常に重要です。私たちはまた、ビジネス秘密、技術ノウハウ、持続的な技術革新によって、私たちの独自と知的財産権の地位を発展させ、維持しています。
89
我々は、物質組成、薬物組成、使用方法、治療方法、および他の関連方法を含む、我々のプラットフォームE 3リガーゼリガンド技術および我々の新規二機能性分解物に関連する特許出願および特許を有している。
2023年12月31日現在、私たちは、101個の特許シリーズを含む新規化合物およびその製造および使用方法をカバーする特許組み合わせを含む。特許期限調整、補充保護証明書出願または特許期限の延長は、異なる国/地域の満期日の延期を招く可能性があり、端末免責声明は、米国の満期日を早める可能性がある。
バイオテクノロジーと製薬会社の特許地位は通常高度に不確定であり、複雑な法律と事実問題に関連しており、近年多くの訴訟のテーマとなってきた。
私たちの現在または未来の候補製品が商業化に成功するために必要な特許保護の程度は、場合によっては入手できないか、または深刻に制限される可能性があり、私たちの権利を十分に保護することができないか、またはいかなる競争優位性を獲得または維持することを可能にするかもしれない。私たちはいかなる保証も提供することができません。私たちのいかなる未解決の特許出願も、発行された特許に成熟して、私たちの飛馬を保護するのに十分な範囲が含まれますTMプラットフォームと私たちの現在または未来の候補製品。さらに、私たちの特許出願または私たちが所有または許可する可能性のある任意の特許によって提供される保護の広さまたは強度が脅かされている場合、会社が私たちと協力し、現在または将来の候補製品を許可、開発、または商業化することを阻止するかもしれない。
また、外国の法律は、米国の法律のように私たちの権利を保護しない可能性がある。例えば、米国以外の司法管轄区域では、すべての知的財産権所有者が同意または許可に同意しない限り、許可は強制的に施行できない可能性がある。したがって、我々特許権の任意の実際または主張する共通所有者は、このような共通所有のためにこれらの特許を使用する補償または回避を支払うことを要求する金銭的または平衡法的救済を求めることができる。しかも、特許の寿命は限られている。米国、そして私たちが特許出願に従事している他のほとんどの管轄区域では、すべての維持費が支払われていると仮定し、特許の自然失効時間は通常提出後20年である。各管轄区域に基づいて、様々な延期を得ることができるが、特許の有効期間は限られているので、それが提供する保護は限られている。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちが所有する可能性のある特許は、他の人が私たちの現在または未来の候補製品と同様または同じ薬物を商業化することを阻止するのに十分かつ持続的な特許保護を提供できないかもしれない。他の当事者は、特許出願を提出したか、または特許を受信している可能性があり、要求された発明が、私たち自身の特許出願または発行された特許で主張されている発明と重複または衝突する可能性があり、同じ化合物、方法、処方または他の主題に関連する可能性があり、いずれの場合も、これらの発明によって市場における私たちの特許地位を主導することができる、私たち自身に関連するまたは競争力を有する技術を開発している。科学文献で発見された発表は、実際の発見に遅れがちであり、米国および他の司法管轄区の特許出願は、通常、特許出願の最初の優先権日の後に少なくとも18ヶ月後に発行されるか、または場合によっては全く発行されない。したがって、私たちは、私たちが所有する可能性のある特許、許可内特許、または係属中の特許出願に要求される発明を最初に提出した人であるか、そのような発明のために特許保護を申請した最初の人であるかを正確に知ることができない。したがって、私たちの特許権の発行、範囲、有効性、実行可能性、商業的価値は何の確定的な予測もできません。
さらに、特許起訴過程は高価で時間がかかり、私たちはすべての必要または望ましい特許出願を合理的なコストまたはタイムリーに提出して起訴することができないかもしれない。さらに、私たちの現在または未来の候補製品または技術に関するいくつかの係属中の特許出願は、訴訟が始まっていない。特許訴訟は長い過程であり、その間、最初に特許庁(S)によって提出された審査に関する権利請求の範囲は、もし彼らが本当にそうすれば、発表時に大幅に縮小される可能性がある。我々は,特許保護を得るのが遅くなるまで,我々の研究開発成果で特許を申請できることを確認できない可能性もある.さらに、場合によっては、私たちは、第三者に許可された技術を含む特許出願の準備、提出および起訴を制御する権利がないかもしれない。したがって、このような特許および出願は、私たちの業務の最適な利益に合った方法で起訴され、強制されてはならない。
競争優位性を確立および/または維持することを望む特許保護を取得したとしても、第三者は、その有効性、実行可能性、または範囲に疑問を提起する可能性があり、これは、このような特許が縮小され、無効にされ、または実行できない可能性がある。特許の発行は、その発明性、範囲、有効性、または実行可能性に対して決定的ではなく、我々の特許は、米国および海外の裁判所または特許庁で挑戦される可能性がある。我々は、米国特許商標局、米国特許商標局、欧州特許庁または欧州特許庁または他の国/地域において、反対、派生、再審査、当事者間の審査または付与後の審査手続きに巻き込まれ、私たちの特許権または将来、そのような権利許可を得る可能性のある他の人の特許権に挑戦する可能性がある。さらに私たちは
90
第三者は、米国特許商標局、欧州特許庁、または他の機関に提出され、これは、我々が係属中の特許出願に付与された権利要件を縮小または排除する可能性がある。競争相手は私たちの前に私たちが発行した特許に挑戦したり、特許出願を提出したりするかもしれない。競争相手はまた私たちが彼らの特許を侵害したと主張するかもしれないので、私たちは私たちの特許や特許出願で主張されている私たちの技術を実践することができない。競争相手はまた、発明が特許資格に適合していないこと、新規でないこと、明らかに、および/または創造性が不足していることを行政特許機関または裁判官に証明することができ、および/または特許出願が説明、基礎、有効化および/または支援に関連する関連要件を満たしていないことを証明することによって、私たちの特許に異議を提起することができ、訴訟では、競合他社は、我々の特許が無効であるか、または様々な理由で強制的に実行できないと断言することができる。もし裁判所や行政当局が同意すれば、私たちは疑問視されている特許の保護を失うだろう。
このような任意の提出または手続きにおいて不利な裁決を下すことは、排他的または経営の自由を失うことをもたらす可能性があり、または特許主張の全部または部分的な縮小、無効または実行不能をもたらす可能性があり、これは、他人が私たちに支払うことなく、同様の技術および薬物を使用または商業化することを阻止する能力を制限するか、または我々の技術および現在または将来の候補製品の特許保護期間を制限する可能性がある。このような挑戦はまた、第三者特許権を侵害することなく、現在または将来の候補製品を製造または商業化することができない可能性がある。さらに、私たちの特許および特許出願によって提供される保護の広さまたは強度が脅かされている場合、会社が私たちと協力し、現在または将来の候補製品を許可、開発、または商業化することを阻止するかもしれない。
さらに、私たちは将来、私たちの特許または特許出願の所有権を主張する元従業員やコンサルタントのクレームを受けるかもしれません。これは、彼らが私たちを代表して仕事をしている結果です。私たちは通常、私たちのすべての従業員、コンサルタント、コンサルタント、および私たちのノウハウ、情報または技術にアクセスできる他の任意の第三者に、その発明のような権利を譲渡または付与することを要求していますが、私たちは私たちの知的財産権に貢献する可能性のあるすべての当事者とこのような合意に署名したかどうかを決定することはできませんし、これらの当事者との合意が潜在的な挑戦に直面したときに維持されるかどうか、またはこれらの合意に違反するための十分な救済措置がないかもしれません。
挑戦を受けていなくても、私たちが発行した特許および未解決の特許出願(発行された場合)は、任意の意味のある保護を提供することができない場合があり、または、類似または代替技術または薬物を非侵害的に開発することによって、私たちが所有する可能性のあるまたは許可内の特許を回避するために、競合他社が我々の特許声明をめぐる設計を阻止することができる。例えば、第三者は、現在または将来の1つまたは複数の候補製品と同様の利益を提供する競争力のある薬剤を開発することができるが、その成分は、私たちの特許保護範囲内ではない。現在または将来の候補製品が保有または出願している特許および特許出願について提供される特許保護が十分に広くなく、そのような競争を阻害するのに不十分であれば、現在または将来の候補製品を商業化することに成功する能力は負の影響を受ける可能性があり、これは私たちの業務を損なうであろう。さらに、1つ以上の管轄区域で価値のある範囲の特許を発行することができても、すべての関連する管轄区域または十分な数の管轄区でそのようなクレームを得ることができず、競争を効果的に低減することができる可能性がある。私たちがこのような特許主張を獲得、維持、または実行できない任意の管轄区域では、私たちの競争相手は、私たちと同じ製品を含む彼らの製品を開発し、商業化することができるかもしれない。
世界のすべての管轄区域で現在または未来のすべての候補製品のために特許や他の知的財産権保護を受けることはありません。私たちが保護を求めている司法管轄区でも、私たちの知的財産権を十分に実行できない可能性があります。
私たちは現在または未来の候補製品Pegasusを特許カバーすることができないかもしれないTMプラットフォーム、あるいはすべての国の他の技術。現在または未来の候補製品の特許を出願、起訴、擁護する、飛馬TMプラットフォームや他の技術は世界のすべての国で尻込みするほど高く、アメリカ以外のいくつかの国の知的財産権はアメリカほど広くないかもしれません。また、一部の外国の国の法律は知的財産権の保護の程度はアメリカの連邦や州法律よりも低いです。そのため、私たちは第三者がアメリカ以外のすべての国で私たちの発明を侵害したり、アメリカや他の司法管轄区域で私たちの発明を使用して製造した製品を販売したり、輸入したりすることができないかもしれません。競争相手は、私たちが特許保護を受けていない司法管轄区で私たちの技術を使用して彼ら自身の製品を開発することができ、他の侵害製品を私たちが特許保護を持っているが、法執行力がアメリカに及ばない地域に輸出することもできる。これらの製品は私たちの現在または未来の候補製品と競争する可能性があり、私たちが特許を発行した司法管轄区域では、私たちの特許出願や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれない。私たちのほとんどの特許の組み合わせは非常に早い段階にある。私たちは適用の締め切りまでにどのような管轄区域で私たちのポートフォリオの様々な発明の保護を求めるかを決定する必要があります。多くの会社は外国の管轄区域の知的財産権の保護と保護に重大な問題に直面している。ある国の法律制度、特にある発展途上国の法律制度は、特許、商業秘密、その他の知的財産権保護、特に薬品に関する保護の強制執行に賛成しない
91
これは、私たちが持っている可能性のある任意の特許の侵害、または競争製品の許可内またはマーケティングを阻止することを困難にし、一般的に私たちの独占権を侵害する可能性がある。私たちが特許出願中の任意の権利または私たちが外国司法管轄区域で所有または許可する可能性のある任意の特許を強制的に執行する訴訟手続きは、巨額のコストをもたらす可能性があり、私たちの努力と注意を私たちの業務の他の側面から移すことは、私たちが所有または許可する可能性のある任意の特許が無効または狭義に解釈されるリスクに直面する可能性があり、私たちの特許出願が発行できない可能性があるリスクに直面し、第三者が私たちにクレームを請求する可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。したがって、私たちが世界各地で私たちの知的財産権を実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネスメリットを得るのに十分ではないかもしれません。
多くの国に強制許可法があり,これらの法律により特許権者は第三者に強制的に許可を付与される可能性がある。また、多くの国は政府機関や政府請負業者に対する特許の実行可能性を制限している。これらの国では,特許権者の救済措置は限られている可能性があり,このような特許の価値を大幅に低下させる可能性がある。もし私たちが私たちが所有または許可する可能性のある私たちの業務に関連する任意の特許について第三者に許可を与えることを余儀なくされた場合、私たちの競争的地位は損なわれる可能性があり、私たちの業務、財務状況、運営結果、および見通しは不利な影響を受ける可能性がある。
私たちは第三者と同等または十分な特典の条項ですべての市場で知的財産権の許可または再許可を取得したり、付与したりしないかもしれない。
第三者の特許やノウハウを用いて私たちの製品を商業化する必要があるかもしれませんが、この場合、これらの第三者から許可を得ることが求められます。第三者知的財産権の許可は競争分野であり、より多くの老舗企業は、魅力的または必要と考えられる第三者知的財産権許可または買収戦略をとる可能性がある。より成熟した会社は私たちより競争優位にあるかもしれません。それらの規模、資本資源及びより強い臨床開発と商業化能力のためです。しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。私たちはまた私たちの投資を適切なリターンを得るための条項で許可したり、第三者の知的財産権を取得することができないかもしれないし、全くできないかもしれない。もし私たちがこのような技術を許可できない場合、あるいは私たちが不利な条項でそのような技術を許可することを余儀なくされた場合、私たちの業務は実質的な損害を受ける可能性がある。必要な許可を得ることができなければ、影響を受けた現在または将来の候補製品を開発または商業化することができない可能性があり、これは私たちの業務に実質的な損害を与える可能性があり、このような知的財産権を持つ第三者は、私たちの販売禁止を求めることができ、あるいは私たちの販売については、印税や他の形態の賠償を支払う義務がある。我々が許可を得ることができても,非排他的である可能性があり,我々の競争相手が我々に許可された同じ技術にアクセスできるようにする.上記のいずれも、私たちの競争地位、業務、財務状況、経営結果、将来性を損なう可能性があります。
もし私たちが現在または任意の未来の合意の義務を履行できなければ、これらの合意に基づいて、第三者から知的財産権の許可を得ることができたり、許可者との業務関係が妨害されたりする可能性があり、私たちは私たちの業務に非常に重要な許可権を失う可能性があります。
私たちは特許、ノウハウ、そして独自の技術に依存しており、私たち自身のものもあれば、セノフィと他のパートナーが許可している。私たちのビジネス成功は、私たちの現在または未来の候補製品を開発、製造、マーケティング、販売し、第三者の固有の権利を侵害することなく、私たちおよび私たちのライセンシーの独自技術を使用する能力があることに依存します。もし私たちが許可協定の下で任意の義務を深刻に違反または違約した場合、セノフィおよび他のパートナーは、それぞれの許可協定を全面的に終了する権利があるかもしれない。これらの許可の任意の終了、または基礎特許が予期される排他性を提供できない場合、重大な権利の損失を招き、現在または将来の候補製品Pegasusの商業化能力を損なう可能性があるTMプラットフォーム、または他の技術、競争相手または他の第三者は、規制機関の承認を自由に求め、私たちと同じ製品を市場に販売し、現在または将来のいくつかの候補製品の開発および商業化を停止することを要求されるかもしれない。上記のいずれも、我々の競争地位、業務、財務状況、経営結果、および見通しに重大な悪影響を及ぼす可能性がある。
ライセンス契約によると、私たちと現在または未来のライセンシーとの間でも知的財産権に関する紛争が発生する可能性があります
92
さらに、第三者の知的財産権または技術を許可するプロトコルは複雑である可能性があり、このようなプロトコルのいくつかの条項は、様々な解釈の影響を受ける可能性がある。可能性のある契約解釈の相違の解決は、関連する知的財産権または技術に対する私たちの権利と考えられる範囲を縮小するか、または関連協定の下での私たちの財務または他の義務を増加させる可能性があり、いずれも、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。さらに、我々が許可する可能性のある知的財産権紛争が、許容可能な条項で現在または将来の許可スケジュールを維持する能力を阻害または弱める場合、影響を受ける現在または将来の候補製品または技術の開発および商業化に成功することができない可能性があり、これは、私たちの業務、財務状況、運営結果、および見通しに大きな悪影響を及ぼす可能性がある。
知的財産権は現在または未来の候補製品や他の商業活動が商業成功を得ることを保証できない。多くの要素が私たちの知的財産権によって提供される任意の潜在的な競争優位性を制限するかもしれない。
私たちの知的財産権は、所有していても許可されていても、将来提供される保護の程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できない可能性があり、私たちの競争相手や潜在的な競争相手に入る障害を提供したり、私たちの競争優位性を維持することができます。また、第三者が私たちの技術実践をカバーする知的財産権を持っていれば、私たちの知的財産権を十分に行使したり、私たちの知的財産権から価値を抽出することができないかもしれません。以下の例は例示的である
93
このような事件が発生した場合、私たちの業務、財務状況、運営結果、将来性を深刻に損なう可能性があります。
特許保護に関するリスク
私たちの特許保護は、政府特許機関によって適用された様々なプログラム、書類提出、締め切り、費用支払い、および他の要求に応じて、特許期限を含む特許保護を取得して維持し、これらの発明に対する特許保護出願の締め切りを逃した場合、またはこれらの要件を遵守していない場合、私たちの特許保護は減少またはキャンセルされる可能性がある。
米国特許商標局および外国政府特許機関は、特許出願中および任意の特許発行後に、いくつかのプログラム、文書、費用支払い、および他の同様の規定を遵守することを要求する。また、定期維持費、継続費、年会費および/または様々な他の政府費用は定期的に支払う必要がある。場合によっては、適用規則に従って滞納金を支払うことによって、または他の方法でミスを訂正することができるが、場合によっては、規定を遵守しないことは、特許または特許出願が放棄または失効される可能性があり、それにより、関連法ドメインの特許権の一部または全部の喪失をもたらす可能性がある。特許の放棄または失効をもたらす可能性のある不正事件には、規定された期限内に公式行動に対応できなかったこと、費用が支払われていなかったこと、および特許を適切に合法化することができず、正式な文書が提出されなかったことが含まれるが、これらに限定されない。この場合、我々の競争相手は同様または同じ製品またはプラットフォームで市場に参入する可能性があり、これは私たちの業務の見通しや財務状況に重大な悪影響を及ぼす可能性がある。
FDAが現在または将来の候補製品の上場承認の時間、期限および詳細に基づいて、私たちが所有または許可している1つまたは複数の米国特許は、1984年の“薬品価格競争および特許期限回復法”(Hatch-Waxman修正案と略称する)に従って限られた特許期間を回復する資格がある可能性がある。ハッジ·ワックスマン改正案は、製品開発およびFDA規制審査中に失われた特許期間の補償として、特許回復期間を最長5年とすることを許可している。ヨーロッパや他の管轄地域では、異なる法律が承認された薬品の特許延期を管理している。しかしながら、私たちは、テスト段階または規制審査中に職務調査を行うことができなかったこと、適用された最終期限内に出願を提出できなかったこと、関連する特許が満了する前に出願を提出できなかったこと、または適用された要求を満たすことができなかったことなどの理由で、特許延期を得ることができない可能性がある。例えば、私たちが承認された製品をカバーするすべての特許が、これらの特許がカバーしている製品がNDAの承認を受けた日から14年以上であれば、私たちは米国で延期されないかもしれない。しかも、特許保護の適用期間や範囲は私たちが要求しているものよりも短いかもしれない。もし私たちが特許期間の延長または回復を得ることができない場合、あるいはどのような延長の期限が私たちが要求したよりも短い場合、私たちの競争相手は私たちの特許が満了した後に競争製品の承認を受ける可能性があり、私たちの創造能力は実質的な悪影響を受ける可能性がある。
特許法の変更は、特許の全体的な価値を低下させ、現在または将来の候補製品を保護する能力を弱める可能性がある。
他の生物製薬会社と同様に、私たちの成功は知的財産権、特に特許に大きく依存している。生物製薬業界で特許を獲得と実施することは技術上の複雑性と法律上の複雑性にも関連するため、コストが高く、時間がかかり、しかも内在的な不確定性を持っている。
近年、米国最高裁判所はいくつかの特許事件に対して裁決を下し、場合によっては利用可能な特許保護範囲を縮小するか、場合によっては特許所有者の権利を弱めるかを決定している。また,最近では米国や他の国の特許法をより多く改正することが提案されており,採択されれば,我々のノウハウのために特許保護を受ける能力,あるいは我々のノウハウを実行する能力に影響を与える可能性がある。米国議会、米国裁判所、米国特許商標局、および他の国の関連立法機関の将来の行動によれば、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、それによって、私たちが新しい特許を獲得したり、私たちの既存の特許と将来獲得可能な特許を実行する能力を弱める可能性がある。
94
私たちの商標、商号、ビジネス秘密に関するリスク
もし私たちの製品または会社名の商標および商号が私たちが製品をマーケティングしようとしている1つまたは複数の国/地域で十分に保護されていない場合、私たちは製品ブランド名の発売を延期し、異なる国/地域で異なるまたはあまり有効ではない商標や商号を使用するか、あるいは私たちの製品ブランド認知度を確立する他の潜在的な不利な結果に直面する可能性がある。
私たちの商標または商号は、挑戦、侵害、希釈、回避、汎用商標として発表されるか、または他の商標を侵害していると判断される可能性がある。この場合、私たちは、関心のある市場における潜在的なパートナーと顧客の名前の承認を得るために、これらの製品名を必要とするので、これらの商標に対する私たちの権利を保護することができないかもしれません。
また、商標登録中に、米国特許商標局や外国司法管轄区同様の機関が私たちの商標登録を拒否した訴訟を受ける可能性があります。例えば、米国特許商標局は、それぞれ2021年4月と2022年10月に、E 3リガーゼ全身ATLASおよびE 3ヒトATLASおよび私たちのK&Designフラグの出願に対して予備的なオフィス行動拒否を発行した。E 3 Ligase全身ATLASおよびE 3人体ATLASが発表され、許可通知が発表されたが、米国特許商標局が私たちのマーク申請に後続のオフィス行動をとるかどうかは不明であり、私たちはこれに応答しなければならないが、最終的にはこの申請を克服できないかもしれないし、この申請も承認されるだろうかどうか。
米国特許商標局および多くの外国司法管轄区の同様の機関においても、第三者は、係属中の商標出願に反対する機会があり、および/または登録商標の抹消を求める機会がある。例えば、2019年11月、ノワ製薬は米国とEU商標局で訴訟を起こし、KYMRIAH商標の権利を理由に、KYMERAとKYMERA Treateuticsの薬品と薬物開発サービスの登録申請を却下した。この紛争は2020年10月に友好的に解決され、KYMERAおよびKYMERA治療申請に関連して現在米国で登録または許可され、EUに登録されている。
もし私たちが私たちの商業秘密を十分に保護して実行できなければ、私たちの商業と競争の地位は損なわれるだろう。
私たちが所有する可能性のあるまたは許可中の特許によって提供される保護に加えて、特許を取得できない可能性のあるノウハウ、特許を実施することが困難なプロセス、および私たちの製品発見および開発中に特許が含まれない可能性のあるノウハウ、情報、または技術に関連する任意の他の要素を商業秘密保護、秘密保護プロトコル、およびライセンスプロトコルに依存して保護することを求めている。私たちは、すべての従業員、コンサルタント、コンサルタント、および私たちのノウハウ、情報、または技術にアクセスできる任意の第三者にセキュリティ協定を締結することを要求していますが、ビジネス秘密は保護が困難かもしれませんし、私たちの協力者およびサプライヤーが使用するビジネス秘密の保護制御は限られています。私たちは私たちがすべての状況でこのような合意を取得したか、または私たちの商業秘密や独自の情報に接触した可能性があるか、または接触したことがあるすべての当事者とそのような協定を締結したことを保証することはできない。
さらに、どちらか一方が合意に違反し、私たちの商業秘密情報を意図的にまたは意図的に開示する可能性があり、私たちはこのような違反について十分な救済措置を得ることができないかもしれない。また,競争相手は我々のビジネス秘密を他の方法で取得したり,基本的に同じ情報や技術を独立して開発したりする可能性がある.強制執行側が商業秘密を不正に開示したり流用したりする主張は困難であり、高価で時間がかかり、結果は予測できない。もし私たちが第三者が私たちのどんな商業秘密を使用するのを阻止するために法廷に訴えることを選択すれば、私たちは巨額の費用を生むかもしれない。私たちが勝訴しても、このような訴訟は私たちの時間と他の資源を消費するかもしれない。また、一部の外国の法律は、専有権や商業秘密の保護の程度や方法が米国の法律に及ばない。そのため、私たちは私たちの知的財産権を保護し、守る上で米国や海外で重大な問題に直面する可能性がある。もし私たちが第三者に私たちの知的財産権を不正に開示することを阻止できなければ、私たちは私たちの市場で競争優位性を確立したり、維持することができなくなり、これは私たちの業務、財務状況、運営結果、および将来の見通しに大きな悪影響を及ぼすかもしれない。
従業員の場合、私たちが締結した合意は、個人的に構想された、私たちの現在または計画中の業務または研究開発に関連するもの、または通常の勤務時間内、私たちのオフィス内で行われる、または私たちの設備または独自の情報を使用するすべての発明が、私たちの固有財産であることを規定している。私たちのすべての従業員が彼らの発明を私たちに譲渡することを要求しているにもかかわらず、私たちは実際に私たちが自分の知的財産権と見なしているすべての当事者とこのような合意を構想したり開発したりすることに成功しないかもしれない。知的財産権の譲渡は自動的に実行されない可能性があり、あるいは譲渡協定が違反される可能性があり、私たちは第三者にクレームをつけさせられたり、私たちが私たちの知的財産権の所有権だと思うことを決定するために、私たちが提起する可能性のあるクレームを弁護したりすることができます
95
財産です。このようなクレームは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。
知的財産権訴訟と侵害クレームに関連するリスク
私たちは、費用がかかり、時間がかかり、成功しないかもしれない、被告になるか、または他の方法で私たちの知的財産権を保護または実行する訴訟の当事者になるかもしれない。
競争相手は、私たちの特許や商標を含む、私たちが持つ可能性のあるまたは許可内の任意の知的財産権を侵害するかもしれない。さらに、我々が所有する可能性のあるまたは許可内の任意の知的財産権も、発明権、優先権、有効性、または実行不可能な紛争を含む紛争のテーマとなる可能性がある。権利侵害や不正使用に対抗するために、私たちは費用が高く時間がかかるかもしれない侵害請求を要求されるかもしれない。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。また、侵害訴訟では、裁判所は、米国法第35編271(E)(1)節の規定に基づいて、私たちが所有する可能性のあるまたは許可内の任意の知的財産権を無効または強制執行できない、または他方が特許を取得する可能性のある技術を使用して特許侵害の安全港に属すると判断することができる。もう1つのリスクは、これらの特許の有効性が支持されていても、裁判所は、私たちが所有する可能性のある任意の特許が、関連技術またはそのような第三者の活動が私たちの特許出願を侵害していないか、または可能性のある任意の特許を有している可能性があることを理由に、他方の論争のある技術の使用を阻止することを拒否する可能性があるということである。任意の訴訟または弁護手続における不利な結果は、私たちが所有または許可する可能性のある任意の特許または他の知的財産権のうちの1つまたは複数を、無効、強制実行または狭義の解釈ができないリスクに直面させ、私たち自身のアプリケーションを発表できないリスクに直面させる可能性がある。このような訴訟または訴訟は、私たちの運営損失を大幅に増加させ、開発活動または任意の将来の販売、マーケティング、患者支援、または流通活動に使用することができるリソースを減少させる可能性がある。私たちはそのような訴訟や訴訟手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らがより多くの財政資源とより成熟して発展した知的財産権の組み合わせを持っているので、このような訴訟や法的手続きの費用を私たちよりも効率的に負担するかもしれない。特許訴訟または他の訴訟の開始と継続によって生じる不確実性は、市場での競争能力に重大な悪影響を及ぼす可能性がある。
第三者によって開始されるか、または米国特許商標局によって提起されたライセンス後訴訟手続は、我々の特許出願または我々が所有する可能性または許可中の任意の特許に関する発明の有効性または優先権を決定するために必要である可能性がある。これらの訴訟は費用が高く、不利な結果は私たちの既存特許権の損失を招く可能性があり、関連技術の使用を停止したり、勝利者から許可権を得ようとしたりすることを要求することができるかもしれない。もし勝利者が商業的に合理的な条件で私たちにライセンスを提供しなければ、私たちの業務は損害を受ける可能性があります。可能なUSPTOライセンス後訴訟に加えて、欧州特許庁の特許異議訴訟の一方、又は我々の特許が挑戦を受ける可能性のある他の外国特許庁又は裁判所の類似訴訟の一方となる可能性がある。これらの訴訟の費用は巨大である可能性があり、いくつかの特許請求の範囲の喪失または特許全体の損失をもたらす可能性がある。付与後の挑戦手順における不利な結果は、私たちの業務に実質的な悪影響を及ぼす可能性がある関連国または司法管轄区域で私たちの1つまたは複数の発明を実施する他者を排除する権利を失う可能性があります。特許庁内の訴訟や認可後の手続きは、私たちの利益に不利な決定を招く可能性があり、たとえ私たちが成功しても、巨額のコストを招き、私たちの経営陣や他の従業員の注意を分散させる可能性があります。私たちは私たちの商業秘密や機密情報が盗用されることを防ぐことができないかもしれません。特に法律ではアメリカのようにこれらの権利を十分に保護できないかもしれません。
さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。さらに、公聴会、動議、または他の一時的な手続き、または事態の発展の結果が発表される可能性がある。もし証券アナリストや投資家がこれらの結果がマイナスだと思っていれば、私たちの普通株の価格に大きな悪影響を及ぼす可能性がある。
私たちは私たちが所有したり許可したりする可能性のある知的財産権に対する侵害を検出できないかもしれない。第三者が私たちが所有する可能性のあるまたは許可中の任意の知的財産権を侵害していることを発見しても、第三者に訴訟を提起しないか、和解を達成することを選択することができます。もし私たちが後に第三者の権利侵害を起訴すれば、第三者がいくつかの法律を弁護することができるかもしれないが、侵害行為が初めて発見されて訴訟を提起するまでの間に遅延がない限り、ないだろう。このような法的防御は、私たちが所有または許可内の任意の特許または他の知的財産権を第三者に対して強制的に実行することができないかもしれない。
1つ以上の国の知的財産権訴訟や行政事務室の挑戦は、私たちに大量の資源を費やし、私たちの人々の正常な責務に対する関心を分散させる可能性がある。解決策が私たちに有利であっても、知的財産権クレームに関連する訴訟や他の法的手続きは、巨額の費用を発生させ、私たちの技術や管理者の正常な責任を分散させる可能性があります。2021年8月私たちはアメリカ特許庁を始めました
96
承認された後の審査を続けて、私たちの現在の候補品とは関係のない第三者特許に挑戦します。これに応じて、疑問視された第三者特許の所有者は、その特許を完全に放棄した。私たちは未来にもっと多くの第三者特許に挑戦するかもしれない。また、公聴会、動議、または他の一時的な手続きまたは事態の発展の結果が公表される可能性があり、証券アナリストや投資家がこれらの結果がマイナスだと思う場合、私たちの普通株の価格に大きな悪影響を及ぼす可能性がある。このような訴訟または訴訟は、私たちの運営損失を大幅に増加させ、開発活動または任意の将来の販売、マーケティング、患者支援、または流通活動に使用することができるリソースを減少させる可能性がある。私たちはそのような訴訟や訴訟手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。上述したように、私たちのいくつかの競争相手は、彼らがより多くの財政資源を持っているので、私たちよりもこのような訴訟や訴訟の費用を効率的に負担することができるかもしれない。特許訴訟または他の訴訟の開始および継続によって生じる不確実性は、我々の臨床前研究および将来の臨床試験を継続するために必要な資金を調達すること、我々の研究計画を継続すること、第三者から必要な技術を許可すること、または現在または将来の候補製品を商業化することを支援する能力を支援するために、市場での競争能力を損なう可能性がある。上記のいずれの事件も、私たちの業務、財務状況、運営結果、そして将来性を損なうだろう。
私たちまたは私たちの従業員が第三者の知的財産権を侵害したり、私たちの合意に違反したため、損害賠償や和解費用の影響を受ける可能性があります。私たちは、現職または元従業員、請負業者、またはコンサルタントを含む私たちまたは他方の当事者が第三者の機密情報を誤って開示していることを告発、告発されるか、または他の方法で訴訟または紛争の一方になる可能性がある。注意およびリソースを移動させることに加えて、このような紛争は、私たちのビジネス的名声および/または私たちのノウハウの保護に悪影響を及ぼす可能性がある。
私たちの候補製品や計画に関連する知的財産権環境は混雑しており、第三者は法的訴訟を提起する可能性があり、私たちが彼らの知的財産権を侵害、流用、または他の方法で侵害することを告発する可能性があり、その結果は不確実であり、私たちの業務の成功に実質的な悪影響を及ぼす可能性がある。私たちのビジネス成功は、私たちが現在および未来の候補製品を開発、製造、マーケティング、販売し、第三者の知的財産権を侵害、流用、または他の方法で侵害することなく、当社の独自技術を使用する能力にかかっています。バイオテクノロジーおよび製薬業には、派生、干渉、再審、当事者間の審査、米国特許商標局で行われる認可後審査手続き、または外国司法管轄区域における異議および他の同様の手続きを含む特許および他の知的財産権に関する訴訟、および特許に挑戦する行政訴訟が多くある。私たちまたは私たちの現在または未来の任意のライセンシーまたは戦略パートナーは、私たちの現在または未来の候補製品および/または独自技術の侵害、流用、または他の方法で彼らの知的財産権を侵害すると主張する、特許または他の知的財産権を所有する第三者の将来の対抗訴訟または訴訟に参加し、直面したり、脅したりする可能性がある。現在あるいは未来の候補製品が馬に乗ることを保証することはできませんTMプラットフォームおよび私たちが開発している、開発されている、または将来開発される可能性のある他の技術は、第三者が所有する既存または将来の特許または他の知的財産権を侵害しない、または侵害しない、流用するか、または他の方法で侵害するであろう。例えば、私たちの多くの従業員たちは以前他のバイオテクノロジーや製薬会社に雇われていた。私たちは、私たちの従業員、コンサルタント、およびコンサルタントが、私たちのために働いているときに他人の独自の情報またはノウハウを使用しないことを確実にするために努力しているが、私たちまたはこれらの個人は、商業秘密または他の固有情報を含む、そのような任意の個人の元雇用主の知的財産権の使用または開示を告発される可能性がある。私たちはまた、従業員、コンサルタント、およびコンサルタントの発明を保護するために提出された特許および出願、さらには、私たちの現在または未来の1つまたは複数の候補製品飛馬に関連する特許および出願の告発を受ける可能性があるTMプラットフォームや他の技術は、その前の雇用主またはアルバイト雇用主によって合法的に所有されている。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。
米国法第35編271(E)(1)節の規定によると、現在または将来の候補製品の開発、臨床前および臨床試験に関連するいくつかの活動は、特許侵害回避港によって制限される可能性があるが、FDAのこのような候補製品の承認を得ると、私たちまたは将来の任意の許可者または戦略パートナーは、直ちに将来の対抗訴訟の当事者になるか、または特許または他の知的財産権を有する第三者が、これらの候補製品の侵害、流用、または他の方法でその知的財産権を侵害する訴訟に直面したり、脅したりする可能性がある。我々が現在または将来の候補製品を開発している分野には,第三者が所有する米国や外国から発行される特許や係属中の特許出願が多く存在する.バイオテクノロジーや製薬業界の拡張や特許の発行に伴い、現在または将来の候補製品は、他人の特許権侵害のクレームを引き起こすリスクが増加する可能性がある。さらに、私たちを含む業界参加者は、どの特許が様々なタイプの薬剤、製品、またはそれらの使用または製造方法をカバーしているかを常に明確にしているわけではない。したがって,我々の分野では大量の特許が発行され,特許出願が提出されているため,第三者が我々の現在または将来の候補製品,技術または方法を含む特許権を所有していると主張する可能性がある.
もし第三者が私たちが知的財産権を侵害、流用、または他の方法で侵害すると主張すれば、私たちは多くの問題に直面するかもしれませんが、これらに限定されません
97
私たちのいくつかの競争相手たちは彼らがより多くの資源を持っているので、私たちよりも効率的に訴訟費用を受けるかもしれない。さらに、いかなる訴訟の開始および継続に生じるいかなる不確実性も、運営を継続するために必要な資金を調達する能力に重大な悪影響を及ぼす可能性があり、または私たちの業務、運営結果、財務状況、および見通しに重大な悪影響を及ぼす可能性がある。上記のいずれの状況の発生も、我々の業務、財務状況、経営結果、または将来性に重大な悪影響を及ぼす可能性がある。
第三者は私たちが許可されていない状況で彼らのノウハウを使用していると主張するかもしれない。米国で法に基づいて発行された特許の有効性推定は、証拠が“明瞭で納得できる”場合にのみ、米国裁判所でこの推定を覆すことができる、より高い証明基準である。第三者特許が付与されている可能性があるが、これらの特許が、現在または将来の候補製品の使用または製造に関連する成分、配合、製造方法、または治療方法の請求項を知らない。特許出願は発表するのに何年もかかるかもしれない。さらに、米国のいくつかの特許出願は特許発行前に秘密にされている可能性があるため、米国および多くの外国司法管轄区の特許出願は、通常、その最初の優先権出願日の18ヶ月後に公表され、科学文献の出版物は実際の発見よりも遅れていることが多いため、他社が現在または未来の候補製品または技術をカバーする特許出願を提出していないとは判断できない。もしそのような特許出願が特許として発行され、そのような特許が私たちの特許出願または私たちが所有または許可する可能性のある特許よりも優先的である場合、私たちは、第三者が所有している、商業的に合理的な条項では得られない、または全く得られない可能性のある権利、または非独占的基礎のみで取得できない権利を取得することを要求される可能性がある。現在処理中の第三者特許出願が存在する可能性があり、これにより、現在または将来の候補製品が発行された特許を侵害する可能性がある。私たちは第三者が所有する特許を知っているが、これらの特許は、私たちの現在または未来の候補製品や他の技術とは関係なく、私たちの現在または未来の候補製品や他の技術によって侵害される可能性もあると考えられる。さらに、第三者は将来的に特許を取得し、私たちの技術を使ってこれらの特許を侵害したと主張するかもしれない。さらに、私たちは関連特許を識別できないかもしれないし、特許が無効で、強制的に実行できない、使い切るか、または私たちの活動によって侵害されていないという結論を誤って導出することができないかもしれない。管轄権のある裁判所が、現在または将来の候補製品の製造プロセス、製造過程で使用または形成された分子または任意の最終製品自体をカバーするために任意の第三者特許を保有している場合、そのような特許の所有者は、適用特許によって許可されたか、またはそのような特許が満了するまで、無効または実行不可能であると判断されない限り、候補製品を商業化する能力を阻止することができるかもしれない。同様に、管轄権のある裁判所が任意の第三者特許を有し、共同療法または患者が方法を選択することを含む、私たちの処方、製造プロセス、または使用方法の様々な態様をカバーする場合、任意の特許の所有者は、ライセンスを取得しない限り、または特許が満了するまで、または最終的に無効または実行不可能と判定されるまで、候補製品を開発し、それを商業化する能力を阻止することができるかもしれない。いずれの場合も、そのような許可は商業的に合理的な条項や全く存在しないかもしれない。もし私たちが商業的に合理的な条項で第三者特許の必要な許可を得ることができなければ、あるいは私たちの現在または未来の候補製品や飛馬を商業化する能力を全く得ることができないならばTMプラットフォームは損傷したり遅延したりする可能性があり、私たちの業務を深刻に損なう可能性がある。我々が許可を得ても,我々の競争相手が我々に許可された同じ技術にアクセスできるように非排他的である可能性がある.
98
私たちにクレームを出した当事者は、禁止または他の公平な救済を求めて獲得するかもしれません。これは、私たちの現在または未来の候補製品のさらなる開発と商業化を効果的に阻止することができます。これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、私たちの業務における従業員資源を大量に移転する可能性がある。もし私たちの侵害、流用、または他の侵害行為に対するクレームが成功した場合、故意に侵害した3倍の損害賠償金と弁護士費を含む大量の損害賠償金を支払う必要があるかもしれません。第三者から1つ以上の許可証を取得し、印税を支払うか、私たちの侵害製品を再設計することは不可能かもしれません。あるいは大量の時間とお金の支出が必要かもしれません。私たちはそのようなライセンスがあるかどうか、あるいは商業的に合理的な条項で提供されるかどうかを予測できない。さらに、訴訟がない場合であっても、我々の研究を進めたり、現在または将来の候補製品の商業化を可能にするために、第三者からライセンスを取得することを必要または選択することが可能である。私たちはもしあれば、合理的な費用または合理的な条項でこのような許可証のいずれかを得ることができないかもしれない。この場合、私たちは現在または未来の候補製品や技術をさらに開発して商業化することができず、これは私たちの業務を深刻に損なう可能性があります。
私たちは、関連する第三者特許を識別しないか、または第三者特許の関連性、範囲、または失効を誤って解釈する可能性があり、これは、私たちが権利侵害請求を受けるか、または現在または将来の候補製品を開発およびマーケティングする能力に悪影響を及ぼす可能性がある。
私たちは、関連特許の識別、特許請求の範囲、または関連特許の満了を含む、私たちまたは私たちの許可者の任意の特許検索または分析を保証することはできず、私たちは、任意の管轄区における現在または将来の候補製品の商業化に関連する、または必要な、米国および海外のすべての第三者特許および係属特許出願を識別したことを確実にすることはできない。例えば、2000年11月29日までに出願された米国特許出願と、その日後に提出されたいくつかの米国特許出願とは、特許発行前に米国国外では提出されず、これらの出願は秘密にされている。上述したように、米国および他の地方の特許出願は、優先権を要求する最初の出願の約18ヶ月後に発行され、このような最も早い出願日は、一般に優先権日と呼ばれる。したがって、私たちの現在または未来の候補製品をカバーする特許出願は、私たちが知らずに第三者によって提出されたかもしれない。さらに、いくつかの制限された場合、公表された未定特許出願は、現在または未来の候補製品をカバーするために、または私たちの現在または未来の候補製品を使用するために、後で修正することができる。特許請求の範囲は、法律の解釈、特許における書面開示、および特許の起訴履歴に依存する。特許または係属出願の関連性または範囲の説明は正しくない可能性があり、これは、現在または将来の候補製品をマーケティングする能力に悪影響を及ぼす可能性がある。私たちは、私たちの現在または未来の候補製品が第三者特許のカバー範囲内にないか、または第三者の保留出願が関連範囲のクレームを提起するかどうかを誤って予測する可能性がある。私たちが関連する任意の米国または海外特許の満期日の決定は正しくない可能性があり、これは、現在または未来の候補製品を開発し、マーケティングする能力に悪影響を及ぼす可能性がある。私たちは関連特許を識別して正確に解釈することができず、私たちが現在または未来の候補製品を開発し、マーケティングする能力に悪影響を及ぼすかもしれない。
もし私たちが関連特許を識別して正確に解釈できなければ、私たちは侵害請求を受けるかもしれない。私たちは私たちがこのような侵害請求を成功的に解決したり、他の方法で解決できるという保証はない。もし私たちがこのような紛争で失敗した場合、損害賠償金(これは重大かもしれない)の支払いを余儀なくされることを除いて、私たちは現在または未来の任意の候補品や侵害と思われる技術を商業化することを一時的または永久的に禁止されるかもしれない。可能であれば、私たちはまた、第三者の知的財産権を侵害しないように、現在または未来の候補製品を再設計することを余儀なくされるかもしれない。これらのすべての事件は、私たちが最終的に勝っても、私たちが大量の財務と管理資源を移転する必要があるかもしれません。そうでなければ、私たちは私たちの業務に投入することができ、私たちの業務、財務状況、運営結果、見通しに悪影響を及ぼすかもしれません。
従業員事務に関するリスク、管理成長、および私たちの業務に関するその他のリスク
従業員の事務と管理成長に関するリスク
私たちの未来の成功は私たちが肝心な幹部を維持する能力、及び合格した人材を吸引、維持、激励する能力にかかっている。
私たちはNello Mainolfi博士、私たちの総裁と最高経営責任者Jared Gollob医学博士、私たちの首席医療官ブルース·ジェイコブス、私たちの最高財務官Ellen Chiniaraと私たちの最高経営責任者Jeremy Chadwickと私たちの管理、科学と臨床チームの他の主要なメンバーの研究開発、臨床と業務発展の専門知識に高度に依存している。私たちは私たちの幹部と招聘契約を締結したが、彼らの誰もが私たちとの雇用関係をいつでも終わらせることができる。私たちは私たちの役員や他の職員たちに“キーパーソン”保険を提供しない。また、私たちは私たちの研究と開発と商業化を支援するために、科学と臨床コンサルタントを含むコンサルタントとコンサルタントに依存しています
99
策略。私たちのコンサルタントやコンサルタントは、私たち以外の雇用主に雇われる可能性があり、他のエンティティと締結された相談または相談契約に基づいて約束することができ、これは、私たちが彼らを得る機会を制限するかもしれない。もし私たちが引き続き高い素質の人材を誘致し、維持することができなければ、私たちが成長戦略を推進する能力は制限されるだろう。
合格した科学、臨床、製造及び販売とマーケティング人員を募集と維持することも著者らの成功の鍵となる。幹部や他の重要な従業員を失ったサービスは、私たちの研究開発と商業化目標の実現を阻害し、業務戦略を成功させる能力を深刻に損なう可能性がある。また、幹部やキーパーソンを交換することは困難かもしれませんが、私たちの業界では開発成功、規制承認、商業化に必要なスキルや経験を持っている個人の数が限られているので、長い時間がかかるかもしれません。この限られた人材バンクから募集する競争は非常に激しく、多くの製薬と生物技術会社の間の類似人員に対する競争を考慮して、私たちは受け入れ可能な条件でこれらの肝心な人員を採用、訓練、維持或いは激励することができないかもしれない。私たちはまた、大学や研究機関から科学や臨床人を募集する競争に直面している。臨床試験で成功できなかったことは、合格した科学者の採用と維持をもっと挑戦的になる可能性がある。
私たちは私たちの会社を引き続き発展させ、拡大する必要があり、私たちはこのような発展と拡張を管理する上で困難に直面するかもしれません。これは私たちの運営を混乱させるかもしれません。
2023年12月31日現在、私たちは187人のフルタイム従業員を持っており、従業員数と業務範囲を増やす予定です。私たちが予想していた発展と拡張を管理するためには、私たちの管理、運営、財務制度を継続して実施し、改善し、私たちの施設を拡大し、より多くの合格者を募集し、訓練し続けなければならない。私たちの経営陣は、日常活動から過剰な注意を移し、これらの開発活動を管理するために多くの時間を使用する必要があるかもしれない。私たちの資源が限られているため、私たちは私たちの業務の拡張を効果的に管理できないかもしれませんし、より多くの合格者を募集して訓練することもできません。これは、私たちのインフラが弱く、操作上のミスを招き、ビジネス機会を失い、従業員を失い、残りの従業員の生産性を低下させる可能性がある。私たちのビジネスの拡大は大きなコストを招く可能性があり、現在または将来の候補製品を開発するなど、他のプロジェクトから財務資源を移転する可能性があります。私たちの経営陣が私たちの予想した発展と拡張を効果的に管理できなければ、私たちの費用は予想以上に増加する可能性があり、私たちの収入を創出したり増加させる能力が低下する可能性があり、私たちは私たちの業務戦略を実施できないかもしれない。私たちの将来の財務業績と、現在または将来の候補製品を商業化し、効果的に競争する能力は、当社の将来の発展と拡張を効果的に管理する能力にある程度依存するだろう。
私たちまたは私たちが依存している第三者は、予見できない世界的な事件や自然災害の悪影響を受けるかもしれませんが、私たちの業務の連続性や災害復旧計画は、深刻な災害から私たちを十分に保護できないかもしれません。
マクロ経済状況、暴力爆発、あるいは地政学的不安定など、予見できない世界的な事件は、私たちの業務に悪影響を及ぼす可能性がある。このような紛争は、制裁、禁輸、供給不足、地域不安定、地政学的転換、サイバー攻撃、その他の報復行動、およびマクロ経済状況、通貨レートおよび金融市場への悪影響をもたらす可能性があり、これは、私たちの運営および財務業績、および私たちと業務往来のある第三者の業務および財務業績に悪影響を及ぼす可能性がある。
さらに、洪水、火災、爆発、地震、極端な天気条件、医療流行病または流行病、電力不足、通信故障または他の自然または人為的事故や事件などの計画外事件は、私たちの施設や私たちの第三者契約製造業者の製造施設を十分に利用できず、私たちの業務運営能力に重大かつ不利な影響を与え、私たちの財務や運営状況に大きなマイナス影響を与える可能性がある。これらの施設を使用できないとコスト増加、候補製品の開発遅延、私たちの業務運営が中断する可能性があります。自然災害や流行病は、私たちの運営をさらに混乱させ、私たちの業務、財務状況、運営結果、見通しに実質的で不利な影響を及ぼす可能性がある。自然災害、停電、または他の事件が発生した場合、本社の全部または大部分を使用することができなくなり、私たちの研究施設や私たちの第三者契約メーカーの製造施設のような重要なインフラを破損したり、他の方法で運営を中断したりすることは難しいかもしれません。場合によっては、かなり長い間私たちの業務を継続することはできません。深刻な災害や同様の事件が発生した場合、我々の既存の災害復旧および業務連続計画は十分ではないことが証明される可能性がある。私たちの災害復旧と業務連続計画の限られた性質のため、私たちは大量の費用を発生する可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性があります。私たちのリスク管理政策の一部として、私たちは私たちの業務に適していると思うレベルに保険範囲を維持します。しかし、これらの施設で事故や事故が発生すると、保険金額がいかなる損害や損失を補うのに十分であるかを投資家に保証することはできません。もし私たちの工場あるいは私たちの第三者契約メーカーの製造工場が事故や事件あるいはその他の原因で運転できなければ、短い時間でも
100
しばらくの間、私たちのどんな研究開発プロジェクトも被害を受ける可能性がある。どの業務中断も、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。
データやプライバシーに関するリスク
我々の内部コンピュータシステム、または我々の第三者CROまたは他の請負業者またはコンサルタントのシステムは、故障したり、セキュリティホールに遭遇したりする可能性があり、これにより、現在または将来の候補製品の開発計画が実質的に破壊される可能性がある。
セキュリティ対策が実施されているにもかかわらず、我々の内部コンピュータシステムおよび我々の第三者CROおよび他の請負者およびコンサルタントのコンピュータシステムは、コンピュータウイルス、許可されていないアクセス、自然災害、テロ、戦争、および電気通信および電気故障の破壊を受けやすい。これまで,このようなシステムの故障,事故,セキュリティホールは経験していないが,このような事件が発生して我々の運営が中断されると,我々の計画が深刻に中断される可能性がある.例えば、私たちの現在または未来の候補製品の臨床前研究または臨床試験データの損失は、私たちの監督管理の承認作業を遅延させ、データを回復または複製するコストを著しく増加させる可能性がある。任意の中断またはセキュリティホールが、私たちのデータまたはアプリケーション、私たちの技術または現在または将来の候補製品に関連する他のデータまたはアプリケーションが失われたり、破損したり、または機密または固有の情報を適切に開示しない場合、私たちは責任を招く可能性があり、私たちの現在または未来の候補製品のさらなる開発は延期されるかもしれません。
私たちは私たちの情報システムをネットワーク攻撃から十分に保護できない可能性があり、サイバー攻撃は個人データを含む機密や独自の情報が漏洩し、私たちの名声を損なう可能性があり、私たちを重大な財務と法的リスクに直面させる可能性がある。
私たちの日常的な運営では、私たちは、私たちまたは私たちの第三者プロバイダが運営する情報技術システムに依存して、電子情報を処理、送信、保存します。私たちの製品発見作業では、名前、郵送住所、電子メールアドレス、電話番号、臨床試験情報など、様々な個人データを収集して使用することができます。成功したネットワーク攻撃は、知的財産権、データ、または他の流用資産の盗難または破壊、または他の方法で私たちの機密または独自の情報を危険にさらし、私たちの運営を混乱させる可能性があります。ネットワーク攻撃の頻度,複雑さ,強度が増加しており,発見されにくくなってきている.サイバー攻撃には、敵対する外国政府の不法行為、工業スパイ活動、電気通信詐欺および他の形態のネットワーク詐欺、有害なマルウェアの配備、サービス拒否、社会工学詐欺、または他のデータセキュリティ、機密性、完全性、および可用性を脅かす手段が含まれる可能性がある。成功したサイバー攻撃は、運営中断を含むが、運営中断に限定されず、金融情報、商業秘密、財務損失、会社戦略計画の開示を含む機密商業情報が流用されることを含む深刻な負の結果をもたらす可能性がある。私たちは私たちの情報システムを保護するために資源を投入しているにもかかわらず、私たちはネットワーク攻撃が脅威であることを認識しており、私たちの努力が情報セキュリティホールを防止することを保証することはできません。これらの抜け穴は、私たちの業務、法律、財務、または名声を損なう、あるいは私たちの運営結果と財務状況に実質的な悪影響を及ぼすことになります。安全違反または不適切なアクセスを防止または軽減することができず、私たちの臨床データまたは患者個人データを使用または開示することができないいかなる行為も、国(例えば、州違反通知法)、連邦(例えば、HITECHによって改正されたHIPAA)、および国際法(例えば、EU一般データ保護条例、またはGDPR)によって規定される重大な責任を招き、私たちの名声に重大な悪影響を及ぼす可能性があり、収集されたデータを使用して、新しい研究を行う能力に影響を与え、私たちの業務を混乱させる可能性がある。
私たちは、私たちの第三者プロバイダに有効なセキュリティ措置を実施し、そのような任意の故障、欠陥、または違反を識別して修正することに依存します。私たちはまた、私たちの従業員やコンサルタントに依存して、彼らの安全証明書を保護し、私たちの敏感な情報を含む可能性のあるコンピュータや他のデバイスの使用とアクセスに関する私たちの政策とプログラムを遵守します。私たちまたは私たちの第三者プロバイダが、私たちの情報技術システムおよびデータ完全性を効果的に維持または保護できなかった場合、または私たちの情報技術システムの重大な中断を予見、計画または管理できなかった場合、私たちまたは第三者プロバイダは、このようなネットワーク攻撃を予防、検出および制御することが困難である可能性があり、そのような攻撃は、上記の損失、および医師、患者および私たちのパートナーとの紛争、規制制裁または処罰、運営費用の増加、支出または収入損失、または他の不利な結果をもたらす可能性があり、いずれも、私たちの業務、運営結果、財務状況、将来性、およびキャッシュフローに実質的な悪影響を及ぼす可能性がある。このような第三者がセキュリティホールを防止または軽減できなかったか、またはそのような情報を取得または開示することができなかったことは、私たちに同様の不利な結果をもたらす可能性がある。このようなセキュリティやデータプライバシー漏洩の影響を防止または軽減できなければ、訴訟や政府調査に直面する可能性があり、業務の潜在的な中断を招く可能性があります。
私たちはマサチューセッツ州ウォトタウンの既存の空間を分けて、私たちが新しい工場に引っ越した後に入居するつもりです。もし私たちが優遇条件で私たちの空間を借りることができなければ、あるいは私たちのテナントが彼らのいかなる転貸の義務も履行できなければ、私たちは意外なコストを負担するかもしれません。これは私たちの財務業績に影響を与えるかもしれません。
101
私たちは現在マサチューセッツ州ウォータータウンで34,522平方フィートの研究開発とオフィス空間をレンタルし、レンタル契約は2030年3月31日に満期になります。2021年12月、マサチューセッツ州ウォトタウンで100,624平方フィートのオフィスと実験室空間のレンタル契約を締結し、2024年2月にレンタルを開始しました。私たちは私たちが私たちの新しい施設に移転した後、私たちの現在の空間を第三者に又貸しするつもりだ。もし私たちが割引条項で私たちの余分な空間を借りることができなければ、あるいは私たちの空間を転貸することができますが、私たちのテナントは私たちにレンタル料を支払うことができなかったり、私たちの義務を滞納したりすることができなければ、私たちは思わぬ支払い義務が生じるかもしれません。
私たちの普通株に関するリスクは
私たちの普通株の価格はずっと大幅に変動し続ける可能性があり、投資家は彼らのすべてまたは一部の投資を損失するかもしれない。
私たちの株価はずっと不安定で、様々な要因で大幅に変動し続ける可能性があります。株式市場全体、特に生物製薬会社の市場は極端な変動を経験し、この変動は往々にしてある会社の経営業績と関係がなく、世界の異なる地域の衝突、上昇するインフレ率と金利の変化と関係があり、これらの変化は多くの会社の株価下落を招き、それらの基本的な業務モデルや見通しは根本的に変わっていないにもかかわらず。この変動のせいで、あなたは投資の全部または一部を失うかもしれない。私たちの普通株の市場価格は多くの要素の影響を受けるかもしれません
これらおよび他の市場および業界要素は、私たちの普通株の市場価格と需要を大幅に変動させる可能性があり、私たちの実際の経営業績にかかわらず、投資家が株式支払い価格以上で彼らの株を売却することを制限または阻止する可能性があり、そうでなければ、私たちの普通株の流動性にマイナス影響を与える可能性がある。本節で説明したリスクを含む、上記の任意のリスクまたは任意の広範な他のリスクを達成することは、私たちの普通株の市場価格に重大かつ実質的な悪影響を及ぼす可能性がある。私たちの普通株の価格は比例しない影響を受ける可能性があります。市場が不確定で不安定な時期に、投資家は伝統的な利益業界と会社を好むかもしれません。
102
不安定な市場や経済状況は、私たちの業務、財務状況、株価に深刻な悪影響を及ぼす可能性がある。
広く報道されているように、世界の信用と金融市場は過去数年間に極端な変動と破壊を経験し、流動性と信用供給の深刻な減少、消費者自信の低下、経済成長の低下、失業率の上昇と経済安定の不確定性を含む。信用と金融市場のさらなる悪化と経済状況への自信が起こらない保証はない。私たちの全体的な業務戦略は、このような経済低迷、不安定なビジネス環境、または持続不可能で不安定な市場状況のいずれかの悪影響を受ける可能性がある。現在の株式や信用市場が悪化したり改善されていない場合、任意の必要な債務や株式融資をより困難にし、コストがより高く、希釈度が高くなる可能性がある。また、私たちの株価は下落する可能性があり、一部の原因は株式市場の変動と全体的な経済低迷である。
適切かつ有利な条件で必要な融資を得ることができなければ、私たちの成長戦略、財務業績、株価に実質的な悪影響を及ぼす可能性があり、私たちの1つまたは複数の候補製品の開発と商業化を延期、削減、または停止すること、または潜在的な許可内または買収を求めることを延期することを要求するかもしれない。さらに、私たちの現在の1つまたは複数のサービスプロバイダ、製造業者、および他のパートナーは、経済的に困難な時期を乗り切ることができない可能性があり、これは、私たちが時間通りに、予算で運営目標を達成する能力に直接影響を与える可能性がある。
追加資本の調達は、私たちの株主に希釈し、私たちの運営を制限するか、または私たちの技術または現在または未来の候補製品の権利を放棄することを要求するかもしれません。
これまで、私たちが相当な製品収入を生み出すことができれば、私たちは私募と公募株式発行、債務融資、協力、戦略連合、マーケティング、流通、または許可手配を通じて、私たちの現金需要に資金を提供する予定です。私たちは現在約束された外部資金源を持っていない。私たちが普通株式または転換可能な証券または普通株に交換可能な証券を売却することによって追加資本を調達する場合、あなたの所有権資本は希釈され、これらの証券の条項は、清算または普通株株主としての権利に重大な悪影響を与える他の特典を含む可能性がある。債務融資が可能であれば、私たちの固定支払義務を増加させ、追加債務を招く、資本支出を行う、または配当を宣言するなど、特定の行動をとる能力を制限または制限する契約を含む可能性がある。
もし私たちが第三者との他の協力、戦略連合またはマーケティング、流通または許可手配によって資金を調達する場合、私たちは、私たちの知的財産権、将来の収入フロー、研究計画、または現在または未来の候補製品に対する貴重な権利を放棄しなければならないか、または私たちに不利になる可能性のある条項で許可を付与しなければならないかもしれない。もし私たちが必要な時に株式や債務融資を通じてより多くの資金を調達できない場合、私たちは私たちの1つ以上の候補製品の開発と商業化を延期、削減または停止することを要求されるかもしれません。私たちの潜在的なライセンス内や買収を延期するか、あるいは私たちが自分で開発し、マーケティングすることをより望んでいた現在または未来の候補製品の権利を与えることができます。
もし証券アナリストが私たちの業務に関する研究や報告を発表しなければ、あるいは彼らが私たちの株に対するマイナス評価を発表したら、私たちの株価は下落するかもしれません。
私たちの普通株の取引市場は、業界や金融アナリストが発表した私たちまたは私たちの業務に関する研究と報告にある程度依存している。私たちはこのようなアナリストを統制できない。私たちはあるアナリストから研究報告を得たにもかかわらず、アナリストが私たちを報道し続けたり、有利な報道を提供したりする保証はない。もし私たちの業務を追跡している一人以上のアナリストが私たちの株に対する彼らの評価を引き下げたら、私たちの株価は下落するかもしれない。さらに、もしこのようなアナリストの一人以上が私たちの株を追跡しなければ、私たちは市場での私たちの株の可視性を失うかもしれません。これは逆に私たちの株価を下落させるかもしれません。
私どもの役員、役員、主要株主及びその関連会社はわが社に大きな影響力を持っており、これはあなたが会社の事務に影響を与える能力を制限し、会社の統制権の変更を延期または阻止する可能性があります。
2023年12月31日現在、我々の役員、取締役、主要株主及びその関連会社の既存保有量の合計は、私たちが発行した普通株の約31%を占めています。したがって、これらの株主が一緒に行動すれば、彼らは、私たちの管理と事務、および私たちの株主に承認された事項の結果に影響を与えることができ、選挙役員と任意の合併、合併、または私たちのほとんどの資産を売却することができます。これは、私たちの株主の一つとして、あなたの最適な利益に合致すると思うかもしれませんので、私たちの普通株に対する能動的な買収提案や要約を阻止または阻止することができます。これらの個人またはエンティティのうちのいくつかは、あなたの興味とは異なる可能性があります。例えばこれらの株主の多くは
103
もし私たちの株が現在の取引価格で売却され、私たちの株を持っている時間が長くなれば、彼らは他の投資家よりも私たちの会社を買収側に売却することに興味を持っているかもしれません。あるいは彼らは私たちが他の株主の利益から離れる戦略をとることを望んでいるかもしれません。さらに、私たちの任意の非関連株主は、時々私たちの普通株式で大量の頭金を蓄積したり、獲得したりすることができ、また、私たちの業務に影響を与えたり、株主に承認された事項にも影響を与えることができます。
投票権がこれらの株主に集中することは、私たちの支配権の変更を延期、延期、または阻止するため、私たちの普通株価格に悪影響を及ぼす可能性があります。私たちの合併、合併、買収、または他の業務合併を阻害するか、または潜在的な買収者が買収要約を提出することを阻止するか、または他の方法で私たちの支配権を獲得しようと努力しているからです。
私たちはすでに私たちの株式計画から大量の株式承認証と株式奨励を発行して、私たちの普通株の株式として行使することができて、これは私たちの既存株主の所有権権益を大幅に希釈する可能性があります。
2023年12月31日まで、予備融資承認株式証の行使時に発行するために、約3,000,000株の普通株を確保しました。さらに、私たちは、発行済み株式オプションおよび既存の制限株式単位を行使する際に発行するために、8,706,304株の普通株式を予約した。これらの証券の行使や転換は、流通株数の大幅な増加をもたらし、我々の既存株主の所有権利益を大幅に希釈する。私たちの株式計画からの持分奨励の基礎株式はS-8表レジストリに登録されています。したがって、一旦帰属すると、これらの株式は発行時に自由に行使し、公開市場で販売することができるが、関連会社に適用される数量に制限される。オプションの行使とその後の標的普通株の売却は私たちの株価を下落させる可能性がある。
将来的に私たちの株主の所有率をさらに希釈し、私たちの株式インセンティブ計画に基づいて、私たちの株主の所有率をさらに希釈し、株価を下落させる可能性があることを含む、私たちの普通株を売却して発行する権利、または普通株を購入する権利。
未来には私たちが計画している業務を継続するための追加的な資金が必要になるだろう。もし私たちが追加の持分証券を発行して資本を調達したり、私たちの持分インセンティブ計画や他の契約義務に基づいて、私たちの株主は深刻な希釈を経験するかもしれない。私たちは1回または複数回の取引で、私たちが時々決定した価格および方法で普通株、転換可能証券、または他の株式証券を販売することができる。もし私たちが1回以上の取引で普通株、転換可能証券、または他の株式証券を売却または発行する場合、投資家はその後の売却によって深刻な希釈を受ける可能性がある。これらの売却はまた、私たちの既存株主の実質的な希釈をもたらす可能性があり、新しい投資家は私たちの既存株主よりも優れた権利を得る可能性がある。
さらに、私たちの発行された普通株の相当数の株はいつでも公開市場で販売される可能性がある。これらの売却、あるいは市場で大量の普通株を持っていると思っている人が株を売却しようとしているという見方は、私たちの普通株の市場価格を下げる可能性があります。私たちが初めて公募する前に私たちの株主だった人は私たちの普通株の大量の株式を保有し続けており、その多くは現在公開市場で販売できるようになっています。これらの株式の大部分は比較的少ない株主が保有しており、これらの株主のうち誰も株式売却能力を制限する合意に達していない。我々の株主は、そのそれぞれの有限パートナーおよび他の株主に大量の株式を売却するか、保有する株式を分配するか、またはそのような売却または分配を予想するか、またはそのような売却が発生する可能性があることを予想し、我々の普通株の市場価格を大幅に低下させる可能性がある。
さらに、私たちは将来の活動に資金を提供するために追加の資本を調達する必要があると予想されているので、私たちは将来的に大量の普通株または普通株または普通株に交換可能な証券を販売することができるかもしれない。将来的に普通株式または普通株関連証券を発行し、発行された株式オプションまたは株式承認証の行使、発行された制限株式単位の帰属および決済、および我々の持分インセンティブ計画に基づいて付与された新規株式奨励(ある場合)に加えて、さらなる償却を招く可能性がある。Cowen and Company、LLCまたはCowenと締結された販売契約や販売契約によると、総額2.5億ドルの普通株を“市場”形式で発売·販売することができるが、その制限を受ける必要がある。2023年12月31日現在、販売契約に基づいていかなる普通株式も売却していない。これまでに販売契約により約5000万ドルの普通株が販売されている。販売契約に基づいて将来的に私たちの普通株の追加株式を売却すれば、私たちの普通株を購入した投資家はさらに希釈するかもしれません。
104
公開市場で私たちの大量の普通株を売ることは私たちの株価を下落させるかもしれない。
公開市場で私たちの大量の普通株を売ることはいつでも起こる可能性がある。これらの売却や市場で大量の普通株を持っていると思っている人が株を売却しようとしているという見方は、私たちの普通株の市場価格を下げる可能性があります。
我々の株式インセンティブ計画又はこれらの計画に基づいて付与された将来の奨励に基づいて、発行された株式オプション又は決済制限株式単位を行使する際に発行される株式は、適用されるホームスケジュール、任意の適用される市場行き詰まり及びロック合意の規定、並びに証券法第144条及び第701条の許容される範囲内で、公開市場で販売される。
条件によると、私たちの普通株式のいくつかの所有者は、彼らの株式に関する登録声明を提出することを要求する権利があり、または彼らの株式を私たち自身または他の株主のために提出する可能性のある登録声明に含める権利がある。私たちはまたS-8表の登録声明を提出して、私たちの株式補償計画の下で発行されたか、あるいは未来に発行する普通株の発行を登録します。本登録宣言に基づいてS-8表で登録された株式は、発行時に公開市場で自由に販売することができ、一旦帰属すると、関連会社に適用される数量に制限される。これらの追加株のいずれかが公開市場で販売されている場合、またはそれらが売却されると考えられている場合、私たちの普通株の市場価格は低下する可能性がある。
私たちの普通株をコーエン社に売却するか、コーエン社を通じて普通株を発行することは深刻な希釈を招く可能性があり、コーエン社が買収した普通株を売却したり、このような売却が発生する可能性があると考えられ、私たちの普通株の価格下落を招く可能性がある。
2021年10月1日、Cowenと販売契約を締結し、この協定によれば、販売契約期間内のいつでも、販売契約の特定の制限及び適用法律を遵守した場合に、私たちの普通株を提供して販売することができる。コーエンが配給通知を出した後に販売される株式数は、販売期間中の普通株の市場価格と当社とコーエンが設定した制限によって変動します。販売株あたりの1株あたりの価格は販売期間内に我々の普通株の市場価格によって変動するため、現段階では最終的に発行される株式数を予測することはできない。私たちがCowenに売却するか、Cowenを通じて売ることは、私たちの普通株の他の保有者の利益を大幅に希釈する可能性がある。また、私たちの普通株の大量の株式を売却したり、そのような株を売却することを期待したりすることは、将来的には株式や株式関連の証券を売却することを難しくする可能性があり、売却の時間および価格は、私たちが本来販売を実現したいと思っていたものである可能性がある。2021年10月1日から2023年12月31日まで、コーエン販売協定により普通株は販売されていません。2024年2月7日、私たちはコーエン販売協定を通じて1,519,453株の普通株を売却した。
私たちは予測可能な未来に私たちの株にいかなる現金配当金も支払わないと予想されるので、資本付加価値(あれば)は投資家の唯一の収益源になるだろう。
私たちは私たちの株の現金配当金を発表したり支払ったりしたことがない。私たちは現在、私たちの未来のすべての収益を維持するつもりで、もしあれば、私たちの業務の成長と発展に資金を提供します。したがって、私たち普通株の資本付加価値(あれば)は投資家の予測可能な未来における唯一の収益源になるだろう。
私たちはもっと大きな証券集団訴訟の危険に直面しているかもしれない。
歴史的に見ると、証券集団訴訟は通常、ある会社の証券市場価格が下落した後に提起される。このリスクは,バイオテクノロジーや製薬会社が近年大幅な株価変動を経験しているため,我々と特に関連している。もし私たちが起訴されれば、巨額のコストを招き、経営陣の注意と資源を移す可能性があり、これは私たちの業務を損なう可能性がある。また、私たちは一方としてのいかなる訴訟も、正当な理由の有無にかかわらず、不利な判決を招く可能性がある。私たちはまた不利な条件で訴訟を解決することに決定するかもしれない。このような負の結果は、巨額の損害賠償または罰金の支払い、私たちの名声を損なう、または私たちのビジネス慣行に悪影響を及ぼす可能性があります。また、訴訟中には、公聴会、動議、または他の一時的な手続きまたは事態の発展の結果を否定的に公開発表する可能性があり、これは私たちの普通株の市場価格に負の影響を与える可能性がある。
105
税金関連リスク
税法の変化は私たちや私たちの投資家に悪影響を及ぼすかもしれない。
米国連邦、州、地方所得税に関する規則は立法過程に参加する人員および米国国税局(IRS)と米国財務省の審査を受け続けている。税法の変化(これらの変化は追跡力を持つ可能性がある)は、私たちまたは私たちの普通株の所有者に悪影響を及ぼすかもしれない。これらの変化は、私たちを追加的な収入ベースの税金および非所得税(例えば、賃金税、売上税、使用税、付加価値税、純価値税、財産税、および商品およびサービス税)に直面させる可能性があり、これは逆に私たちの財務状況および運営結果に実質的な影響を与える可能性がある。例えば、守則第174条によれば、2021年12月31日以降の納税年度には、米国で発生した研究開発費が資本化·償却され、我々のキャッシュフローに悪影響を及ぼす可能性がある。さらに、新しい、変更された、修正された、または新しい解釈または適用された税法は、私たちの顧客と私たちのコンプライアンス、運営、その他のコスト、および私たちの製品のコストを増加させる可能性があります。近年、このような変化は多く発生しており、未来も変化し続けるかもしれない。
私たちの業務活動規模の拡大に伴い、アメリカと非アメリカはこのような活動に対する課税のいかなる変化も私たちの有効税率を増加させ、私たちの業務、財務状況と経営業績を損なう可能性があります。税法の潜在的な変化が私たちの普通株投資に与える影響について税務コンサルタントにお問い合わせすることを促します。
私たちは私たちの純営業損失の繰越といくつかの他の税務属性を利用して未来の課税収入を相殺する能力はいくつかの制限を受ける可能性があるため、未来の納税義務を減らすことができません
2023年12月31日現在、連邦と州の純営業損失の繰越はそれぞれ1兆605億ドルと1.613億ドルであり、2036年から異なる金額で満期になる(2017年12月31日以降の納税年度による連邦純営業赤字の繰越は含まれておらず、これらの繰越は満期の影響を受けないが、2020年12月31日以降の納税年度では、このような連邦NOLの控除額は私たちの課税収入の年間80%以内に制限される可能性がある)。2023年12月31日まで、連邦と州の研究開発税収の繰越免除もあり、それぞれ1950万ドルと810万ドルで、2036年に満期になる。これらの純営業損失と税収控除は満期時には使用されていない可能性があり、将来の所得税負債を相殺するためには使えない。また、一般に、改正後の1986年の国税法第382条及び383条又は同法によると、会社が“所有権変更”を行う際には、変更前の純営業損失又は税収控除、又は純額又は控除を利用して将来の課税収入又は税金を相殺する能力が制限される。これらの目的に関して、所有権変更は、通常、1つまたは複数の株主または会社の株式の少なくとも5%を保有する株主グループの所定の試験期間内の総株式保有量が、これらの株主または株主グループの最低所有権パーセントよりも50ポイント以上増加する場合に生じる。私たちの既存のNOLや信用は、以前の所有権変更によって制限される可能性があります。また、将来的に私たちの株式所有権の変化は、その多くが私たちの制御範囲内ではなく、規則382および383条による所有権の変化を招き、NOLまたは信用を利用する能力を制限する可能性がある。州法によると、私たちのNOLや信用もまた損なわれる可能性がある。したがって、私たちは私たちのNOLや信用の実質的な部分を使用できないかもしれない。さらに、私たちがNOLや相殺能力を利用することは、私たちの収益力とアメリカ連邦と州の課税収入にかかっています。上述したように、“リスク要因-私たちの財務状況と追加資本需要に関連するリスク”の節では、私たちは設立以来、重大な純損失が発生しており、予測可能な未来に引き続き重大な損失を受けることが予想される;したがって、私たちは、規則382および383節に制限されたNOLまたはクレジット決済を利用するために、必要な米国連邦または州課税収入がいつ発生するかどうか分からない。
私たちの統制と報告要求に関連するリスク
もし私たちが財務報告に対して適切かつ効果的な内部統制を維持できなければ、私たちの経営業績と私たちの経営業務の能力が損なわれる可能性がある。
私たちが正確な財務諸表をタイムリーに作成できるように十分な内部財務と会計制御と手続きを持っていることを確保して、これは高価で時間のかかる仕事であり、常に再評価する必要がある。財務報告の内部統制は、財務報告の信頼性を合理的に保証することを目的としたプログラムであり、公認された会計原則に基づいて財務諸表を作成する。2002年のサバンズ·オキシリー法第404条または第404条によると、我々の経営陣が提出した財務報告書の内部統制に関する報告書を、我々の独立公認会計士事務所が発行した財務報告内部統制に関する証明報告書を含めて提出しなければならない。
106
第404条の規定に適合するために、私たちはコストが高く、挑戦的である財務報告書に対する私たちの内部統制の過程を記録し、評価する過程に参加した。この点では、外部コンサルタントを招聘することが可能な内部資源を継続的に提供する必要があり、詳細な作業計画により、財務報告内部制御の十分性を評価して記録し、適宜ステップ改善制御プログラムを採用し、制御措置がファイルのように機能しているかどうかをテストすることにより、財務報告内部制御の継続報告及び改善手順を実施する。我々は努力したにもかかわらず,我々も我々の独立公認会計士事務所も,規定された時間枠内で結論を出すことが可能であり,財務報告の内部統制に有効であり,第404条の要件に適合していることを証明している。これは、私たちの財務諸表の信頼性に自信を失ったため、金融市場に不利な反応をもたらす可能性がある。また、投資家は、私たちの内部統制が不足していると思っていたり、正確な財務諸表をタイムリーに作成することができないと考えており、これは私たちの株価を損なう可能性があり、現在または将来規制機関の承認を得る可能性のある任意の候補製品を効率的にマーケティングして販売することを困難にしています。
私たちの開示統制と手続きはすべてのミスや詐欺を阻止したり検出できないかもしれない。
私たちは取引法の特定の報告書の要求事項を守らなければならない。我々の開示制御および手続きは、取引所法案に基づいて提出または提出された報告書において、開示を要求する情報が蓄積され、米国証券取引委員会規則および表で指定された期間内に管理層、記録、処理、まとめおよび報告に伝達されることを合理的に確保することを目的としている。任意の開示制御およびプログラムまたは内部制御およびプログラムは、発想および動作がどのように完全であっても、絶対的な保証ではなく、合理的な保証を提供することしかできず、制御システムの目標が達成されることを確保することしかできないと信じている。これらの固有の限界は,意思決定過程における判断が誤りである可能性があり,簡単な誤りや誤りによって故障が発生する可能性があるという現実を含む.さらに、ある人の個人的な行動、2人以上の結託、または無許可超越制御は、制御を回避することができる。したがって、我々の制御システムの固有の制限により、エラーや詐欺によるエラー陳述や開示不足が発見されることなく発生する可能性がある。
私たちの憲章と付例に関連するリスク
私たちの定款文書とデラウェア州法律における反買収条項は、制御権の変更を延期または阻止する可能性があり、これは私たちの普通株の市場価格を制限し、私たちの株主の現在の経営陣の変更を阻止または罷免する試みを阻止または挫折させる可能性がある。
私たちの4回目の改訂と再記載された会社登録証明書および私たちの第2の改正と再記述の定款には、わが社の支配権変更を延期または阻止することができ、または私たちの株主が有利と思う取締役会変更を延期または阻止する可能性がある条項が含まれています。いくつかの規定には
また、私たちはデラウェア州で登録されているので、私たちはデラウェア州会社法第203条の規定によって管轄されています。この条項は、株主が議決権を持って発行された株の15%以上のいくつかの業務統合を禁止する可能性があります。これらの逆買収条項と私たちの第四回改正と再記載の会社登録証明書、並びに改訂と再記載された会社定款の他の条項は、株主または潜在的な買収者をより難しくする可能性があります
107
わが取締役会の支配権を獲得したり、当時の取締役会が反対する行動を起こしたりして、わが社の合併、買収要約、代理権争いを延期または阻害する可能性もあります。これらの規定はまた、代理権競争を阻止し、あなたと他の株主があなたが選択した取締役を選ぶことを難しくしたり、あなたが希望する他の会社の行動を取らせたりすることができます。制御権の変更、取引、取締役会の変動を遅延または阻止することは、私たちの普通株の市場価格の下落を招く可能性がある。
私たちの改正と再記述の定款は、私たちの株主によって開始される可能性のあるいくつかの訴訟の独占フォーラムとして特定の裁判所を指定し、これは、私たちの株主が有利な司法フォーラムを得ることが私たちとの紛争を処理する能力を制限することができる。
吾等の改正及び再記載された会社細則によれば、吾等が書面で別の裁判所を選択することに同意しない限り、デラウェア州衡平裁判所は、州法律が以下の事項について提起したクレームの唯一及び独占裁判所である:(1)吾等を代表して提出された任意の派生訴訟又は法律手続き、(2)吾等に違反する任意の取締役、上級職員又は他の従業員が吾等又は吾等の株主に対する信認責任に基づいて提起した任意の訴訟、(3)DGCL、吾等の改正及び再記載された会社登録証明書又は吾等の改正及び再記載された付例のいずれかの条文に基づいて提出された訴訟;または(4)内部事務原則またはデラウェアフォーラム条項によって管轄されるクレームを主張する任意の訴訟。デラウェア州フォーラム条項は証券法や取引法によって発生したいかなる訴訟根拠にも適用されないだろう。私たちが代替裁判所を選択することに書面で同意しない限り、米国マサチューセッツ州地域裁判所は、証券法または連邦裁判所条項に基づいて提起された任意の訴えを解決する唯一のおよび独占的なフォーラムであるべきであると、私たちが改正し、再記述する付例はさらに規定されている。また、私たちは、私たちの株式株式の権益を購入または他の方法で獲得した個人または実体は、デラウェアフォーラム条項と連邦フォーラム条項に注目して同意したとみなされ、再記載された規定である;しかし、株主ができなければ、連邦証券法およびその規則および法規の遵守を放棄したともみなされない。
私たちは、私たちの定款におけるデラウェアフォーラム条項および連邦フォーラム条項を修正し、再記述することが、株主にこのようなクレームを求める際に追加の訴訟費用をもたらす可能性があり、特に株主がデラウェア州またはマサチューセッツ州連邦に居住している場合、または適用される場所または近くに住んでいる場合には、追加の訴訟費用をもたらす可能性があることを認識している。また、私たちの改正と再記述された定款におけるフォーラム選択条項は、私たちの株主が司法フォーラムで、私たちまたは私たちの役員、役員、または従業員とのトラブルに有利だと思うクレームを提出する能力を制限する可能性があり、これは、訴訟が成功しても、私たちの役員、従業員に対する訴訟を阻止することができ、私たちの株主に利益をもたらす可能性があります。また、デラウェア州最高裁判所は、他州裁判所は連邦裁判所選択条項の有効性を維持し、この条項は証券法によるクレームを要求すると主張しているが、他の裁判所が私たちの連邦フォーラム条項を実行するかどうかには不確実性があると判断した。もし連邦フォーラムの条項が実行できないことが発見されたら、私たちはこのような問題の解決に関連した追加費用を発生させるかもしれない。連邦フォーラムの条項はまた、その条項が実行不可能または無効だと主張する株主に追加的な訴訟費用を適用するかもしれない。デラウェア州衡平裁判所および米国マサチューセッツ州地域裁判所もまた、訴訟の株主が存在するか、または訴訟を提起する裁判所を選択する可能性があり、このような判決は、私たちの株主に多かれ少なかれ有利である可能性があることを含む、他の裁判所とは異なる判決または結果を下す可能性がある。
項目1 B。取消解析Dスタッフがコメントした。
ない。
108
プロジェクト1 C。ネットワーク·セキュリティ安全性
ネットワークリスク管理と戦略
Kymeraは、私たちの研究計画、臨床試験、従業員、サプライヤーに関するデータを使用し、保存し、処理します。我々は、我々のデータおよびシステムへの脅威を含むネットワークセキュリティ脅威からのリスクを評価し、識別し、管理するための情報セキュリティ計画を開発し、維持した。私たちは、私たちのリスク管理プロセスの一部として、内部と外部のセキュリティテストを含む定期的に私たちの資産を評価し、適用されるセキュリティ制御の有効性を評価します。このような評価は産業基準に基づいて行われる。私たちのネットワークセキュリティリスク管理プロセスは私たちの全体的なリスク管理計画の一部だ。ネットワークセキュリティに対する人々の認識を向上させるための訓練を含む従業員教育プログラムもある。我々は、我々の情報および情報システムに構成される重大なネットワークリスクを識別して管理する法律およびガバナンス手順、および適用されるようなネットワークイベントを評価して対応するための枠組みについて概説したイベント応答ポリシーを採択した。
ネットワークセキュリティリスクに関するガバナンス
最高経営責任者と私たちの実行管理チームの最終指導の下で、ネットワークセキュリティ監督委員会(CSSC)は主に財務、法律、運営、人的資源、情報技術からの代表を含むネットワークセキュリティリスクの管理を監督する責任がある。CSSCは,必要な場合に会議を開催し,イベント応答プログラムを審査·更新し,ネットワークセキュリティイベント応答グループのイベント応答活動を監視する.
情報技術担当者とCSSCは主に我々のネットワークセキュリティ計画の評価と管理を担当する.情報技術担当者は最高経営責任者に報告し,情報技術やセキュリティチームの構築と指導において25年以上の経験を持っている.
取締役会は、我々のリスク管理計画に対して最終監督権を有し、ネットワークセキュリティの監督を含む同計画の監督を取締役会の監査委員会に委託している。情報技術副総裁は定期的に監査委員会にネットワークセキュリティ問題に関する報告書を提出する。首席財務官および首席法律幹事は、いかなる重大なネットワークセキュリティ事件およびそのような事件によって生じる可能性のある開示義務が発生した場合に監査委員会に通知することを担当する。
プロジェクト2.専門家ペルテスです。
2030年3月に期限が切れる賃貸契約によると、私たちは現在マサチューセッツ州ウォータータウンに約34,522平方フィートのレンタルオフィスと実験室空間を持っています。私たちはレンタル期間をさらに5年間延長することを選択することができる。また、2035年3月に満期になった賃貸契約によると、私たちは2023年2月にマサチューセッツ州ウォトタウンで100,624平方フィートのオフィスと実験室空間を占有し始めました。私たちは2つのレンタル期間を延長する選択があります。それぞれ5年間延長します。私たちのオフィスと実験室空間は私たちの現在の需要を満たすのに十分であり、必要な時に適切な追加空間を提供すると信じている。
項目3.法律法律手続き。
時々、私たちは訴訟や他の法的手続きに巻き込まれるかもしれない。私たちは現在、私たちの経営陣が私たちの業務に重大な悪影響を及ぼす可能性があると考えている訴訟や法的手続きに参加していません。結果にかかわらず、弁護と和解コスト、管理資源の移転、その他の要素により、訴訟は私たちの業務、財務状況、運営結果、将来性に悪影響を及ぼす可能性がある。
プロジェクト4.地雷安全安全に開示する。
適用されません。
109
第II部
項目5.登録者普通株式市場、関連株持株者は重要で発行者が株式証券を購入する。
普通株取引に関するいくつかの情報は
我々の普通株はナスダック世界精選市場での取引コードは“KYMR”であり、2020年8月21日から公開取引されている。その前に、私たちの普通株は市場を公開しなかった。
株式表現グラフ
次の図は,2020年8月21日,すなわち我々の普通株の第1回公開取引日から2023年12月31日までに,現金が我々の普通株,ナスダック総合指数,ナスダックバイオテクノロジー指数に投資されたと仮定した累積総リターンを示している。このような見返りは歴史的結果に基づいており,未来を示唆するための表現ではない.ナスダック総合指数とナスダックバイオテクノロジー指数のデータ仮説配当は再投資.
41ヶ月の累積総リターンの比較*
ケメラ治療会社ではナスダック総合指数とナスダックバイオテクノロジー指数は

*2020年8月21日に配当金の再投資を含む株式および指数の100ドルに投資します。
12月31日までの会計年度。
本項目5の業績グラフは、1934年の証券取引法(改正)第18条の目的に基づいて米国証券取引委員会に提出された“募集資料”または“アーカイブ”とはみなされず、また、この条項に規定された責任の制約を受けず、引用によって明確にこのような文書に統合されない限り、1933年の証券法または1934年の“証券取引法”に基づいて提出された任意の文書に引用されているとみなされてはならない。
110
私たち普通株保有者
2024年2月16日現在、約21人の普通株保有者が登録されている。この数字には、“著名人”や“街”の名で株を持っている株主は含まれていない。
株式補償計画に基づいて発行された証券
表格10-K第5項に要求される株式報酬計画に関する資料は、本年度報告第3部第12項に組み込まれる。
最近売られている未登録証券
ない。
プロジェクト6.Re料理が出ました。
適用されません。
111
項目7.経営陣の以下の問題の議論と分析財務状況と経営実績。
以下、当社の財務状況及び経営結果の検討及び分析は、本年度報告10-K表の他の部分の監査財務諸表及び関連付記とともに読まなければならない。本議論および分析に含まれるいくつかの情報および本Form 10-K年次報告における他の情報は、リスクおよび不確定要因に関する前向きな陳述を含む、我々の業務計画および戦略に関する情報を含む。実際の結果が以下の議論および分析における前向きな陳述に記載されているか、または示唆された結果と大きく異なる可能性のある重要な要素の議論については、本年度報告(Form 10−K)の第I部分1 A項の“リスク要因”と題する部分を読まなければならない。
概要
私たちは生物製薬会社で、新しい小分子療法の発見と開発に専念し、人体自身の天然タンパク質分解システムを利用することで病原タンパク質を選択的に分解する。我々の独自の標的蛋白質分解或いはTPDプラットフォームはPegasusと呼ばれ、高度な選択性の小分子蛋白質分解物を発見でき、全身病原蛋白質に対して活性を有する。我々の小分子タンパク質分解器は,既存の療法に比べて独自の利点があり,従来のモデルでは解決が困難であったヒトゲノムの大部分を解決できると信じている。我々はすでに臨床的に検証された生物経路に焦点を当てているが,重要な生物ノード/タンパク質は投与されていないか,あるいは十分に投与されていない。我々はこれまで,我々の飛馬プラットフォームを用いて免疫学−炎症と腫瘍学分野に焦点を当てた新しいタンパク質分解器を設計し,われわれのプラットフォームの能力を他の治療領域に応用し続けてきた。我々の使命は,TPDを用いてヒト細胞中のすべての標的類の薬物治療を行うことである。
著者らの現在の臨床分期計画はIRAK 4、STAT 3とMDM 2であり、各計画は生物検証経路中の高影響標的に対して、広範な免疫性炎症性疾患、血液系悪性腫瘍と/或いは固形腫瘍の治療に機会を提供した。著者らの計画は高い影響目標の解決に対する関心を体現しており、これらの目標は伝統的なモデルにとってつかみにくいものであり、重大な医療需要を満たしていない多種の深刻な疾病の発病機序を推進した。われわれが開示した臨床前計画はSTAT 6とTYK 2に対して十分に検証された経路にあり,競合他社の療法に比べてわれわれの分解器技術が独自の優位性を提供する可能性があると信じている。この二つのプロジェクトは現在INDを支持する研究にある。
われわれのIRAK 4計画については,サイノフェイ社と協力し,腫瘍学や免疫腫瘍学分野以外のIRAK 4に対する候補薬を開発している。我々は,化膿性汗膜炎(HS),炎症性皮膚疾患およびアトピー性皮膚炎(AD),および潜在的な他の適応を含むインターロイキン1受容体/Toll様受容体またはIL−1 R/TLR駆動の免疫学的炎症条件および高度に満たされていない医療ニーズを治療するためのIRAK 4分解剤KT−474を開発している。健康ボランティアおよびHSおよびAD患者の列を含むKT−474の第1段階試験を完了した。サイノフェイによるKT−474の第二段階臨床試験は,HSとADにおける潜在力を初歩的に調査した。この2つの適応の臨床試験が開始され,患者用量が行われている。
われわれの臨床腫瘍学的計画については,侵襲性リンパ腫を含む再発/難治性液体と固体腫瘍患者に対する第一段階臨床試験においてSTAT 3分解剤KT−333を評価している。試験の1 a段階では患者登録と投与量が行われており,2024年にはより多くの臨床データが提供される予定である。2023年9月、FDAは、以前に孤児薬指定を得た適応である再発/難治性末梢T細胞リンパ腫の治療のためのKT-333高速チャネル指定を承認することを発表した。われわれのMDM 2分解剤KT−253の第一段階臨床試験は2023年3月に開始された。この研究は再発或いは難治性高度髄系悪性腫瘍、急性リンパ球性白血病或いはALL、リンパ腫と固形腫瘍の成人患者におけるKT-253増量用量の安全性、耐性、薬物動態/薬効学と臨床活性を評価している。試験の1 a段階で、患者登録と投与量は進行中であり、著者らは2023年11月に初歩的な安全性、メカニズム検証と概念検証データを提供した。私たちは2024年にもっと多くの臨床データを提供する予定だ。KT-253は2023年6月、FDAによって急性骨髄性白血病を治療する孤児薬として承認された。2023年11月、予想される分解レベルに達し、用量制限毒性が不足しているにもかかわらず、増加する免疫学的導管を集中的に支援するために、我々のKT-413(IRAKIMiD)計画の開発を中止することを決定した。
2015年の設立以来、私たちはほとんどの努力を投入して、私たちの会社、研究開発活動、業務計画、資金調達、私たちの知的財産権の組み合わせを構築し、これらの業務に一般的かつ行政的な支援を提供しています。これまで,転換可能優先株の売却,普通株の売却(2020年8月の初公募株(IPO)を含む)および同時に行った私募,2021年7月の後続発行,2022年8月の同時私募により14.5億ドルの総収益を得てきた
112
私募株式投資、またはPIPE、発行、2024年1月に後続発行、Cowen and Company、LLCの販売協定、およびわが社と協力します。
設立以来、私たちは重大な運営損失が発生した。私たちが利益を達成するのに十分な製品収入を生み出すことができるかどうかは、私たちの現在の候補製品または任意の未来の候補製品の成功的な開発と最終的な商業化に大きく依存するだろう。2023年、2022年、2021年12月31日までの年度の純損失はそれぞれ1.47億ドル、1.548億ドル、1.02億ドルだった。また、2023年12月31日と2022年12月31日までの累計赤字はそれぞれ5.308億ドルと3.838億ドルだった。私たちは私たちが行っている活動によって、私たちの費用と資本需要が大幅に増加すると予想しています。特に私たちは
また、私たちのすべての主要候補製品が市場承認されれば、製品製造、マーケティング、販売、流通に関連した巨額の商業化費用が発生すると予想される。したがって、私たちは私たちの持続的な運営を支援し、私たちの成長戦略を実施するために多くの追加資金を必要とするだろう。製品販売から相当な収入を得ることができる前に、他社との協力や他の戦略取引が含まれている可能性がある株式、債務融資、または他の資本源を売却することで、当社の運営に資金を提供する予定です。私たちは必要に応じて追加資金を優遇条項で調達したり、そのような他の合意や手配を達成することができないかもしれません。もし私たちが必要な時に資金を調達したり、このような合意に到達できなかったら、私たちは私たちの1つ以上の候補製品の開発と商業化を大幅に延期、減少、またはキャンセルしなければならないかもしれない。
私たちが臨床開発に成功し、候補薬物の発売承認を得ない限り、製品販売から収入を得ることはありません。新薬の発売承認を確保するための長い過程には多くの資源が必要だ。どのような遅延や規制部門の承認を得られなかった場合も、私たちの候補製品開発作業と私たちの全体業務に重大な悪影響を及ぼすだろう。製品開発に関連する多くのリスクや不確実性のため、費用が増加する時間や金額を予測することもできず、いつ実現したり、利益を維持したりすることができるかどうかも予測できない。たとえ私たちが製品販売を作ることができても、私たちは利益を上げることができないかもしれない。もし私たちが利益を上げることができない場合、または持続的に利益を上げることができない場合、私たちは計画通りに運営を継続できず、私たちの運営を減少または終了させることができないかもしれない。
2023年12月31日現在、私たちは4.363億ドルの現金、現金等価物、有価証券を持っています。私たちは、既存の現金、現金等価物、有価証券に、2024年第1四半期の純収益と、私たちとセノフィとの協力協定によって2024年1月に受け取った1500万ドルのマイルストーン支払いに基づいて、2027年上半期まで私たちの業務に資金を提供するのに十分だと信じている。これにより,KT−474の第2段階データ,KT−253とKT−333を超えた追加概念検証データ,および我々のSTAT 6とTYK 2計画のいくつかの臨床変曲点が予想される。私たちは間違っていることが証明される可能性があるという仮定に基づいてこの推定をして、私たちは予想よりも早く利用可能な資本資源を枯渇させるかもしれない。“流動性と資本資源”を見てください
113
私たちの運営結果の構成要素は
収入.収入
これまで、私たちは製品販売から何の収入も得ておらず、予測可能な未来にも製品販売から何の収入も得られないだろう。私たちの唯一の収入はVertex製薬会社とセノフィとの研究協力協定から来ている。私たちは、今後数年間、私たちの収入は主に私たちの現在の協力協定と私たちが未来に達成する可能性のある任意の追加的な協力から来ると予想している。今まで、私たちはどんな協力協定の下でどんな印税も受け取っていない。
頂点連携プロトコル
2019年5月9日、我々はVertexと、最小分子タンパク質の分解を推進し、最大6つの目標を達成するVertex合意に達した。Vertexプロトコルによると,Vertexには独占選択権が付与され,連携により開発された候補製品が許可され,Vertexは開発と商業化を制御する.Vertexプロトコルによると,目標の発見と臨床前研究を担当し,Vertexはその許可選択権を行使した後に候補製品の開発,製造,商業化を担当している。Vertexは私たちに5,000万ドルの払戻不可能プリペイドを提供し、単独で同時に署名した株式購入契約に基づいて、B-1シリーズの転換可能な優先株の3,059,695株を1株6.54ドルで購入した。
Vertexプロトコルは2023年5月9日の初期研究期間終了時に満了します.
セノフィ協定
2020年7月7日,我々はセノフィと,2つの生物学的標的に対する候補薬物を共同開発する協力協定を達成した。セノフィ協定によると、私たちは、IRAK 4および他の非開示用途分野の不開示目標のための協力中に生成された特定の先導化合物の開発、製造および商業化を可能にするグローバル独占許可をセノフィに付与した。このような許可は、指定されたマイルストーンの後にのみ、連携目標ごとに行使することができる。IRAK 4に対する化合物の使用領域は、腫瘍学および免疫腫瘍学を含まず、診断、治療、治癒、緩和または予防のいずれかの疾患、障害または状態を含む。著者らは発見と臨床前研究を担当し、少なくとも1種のIRAK 4に対する分解剤と最大3種類のバックアップ分解剤に対して第1段階臨床試験を行った。この2つの目標に対して,セノフィは協力候補製品ごとに特定の開発マイルストーンが出現した後,候補製品の開発,製造,商業化を担当している。
独自選択権または選択権を有し、(I)使用分野に適用されるこのような目標に対する連携製品の米国開発コストに対して50%の資金を提供する権利と、(Ii)米国の使用分野に適用されるこのような目標に対する協力製品の商業化の純利益と純損失を平均的に分担する権利とを含む連携目標を1つずつ行使することができる。また、もし私たちが選択権を行使すれば、セノフィは各協力目標に適用される独占的な選択権を与えてくれます。一度行使すれば、アメリカである分野での共同普及活動を許可します。
セノフィ協定が早期に終了しない限り、この協定は、セノフィ協定の下で製品に関連するすべての支払い義務が満了した日から製品ごとに満了する。私たちまたはセノフィは、相手の重大な違約または資金で借金をしないか、またはいくつかの特許挑戦のために合意を終了することができます。さらに、サイノフェイは、事前の書面通知の下で、便宜上プロトコルを終了するか、または重大なセキュリティイベントのためにプロトコルを終了することができ、セノフィが特定の目標に対する協力候補の開発、商業化、または製造責任を負う場合、サイノフィが特定の期間内にその目標のための任意の協調候補の使用を停止する場合、任意の協力候補との合意を終了することができる。
114
セノフィがセノフィ協定に基づいてセノフィの独占ライセンスを付与したことを考慮すると、セノフィは1.5億ドルを前払いした。前金以外にも、いくつかの開発や規制活動が完了した後、合計14.8億ドルにのぼるいくつかの発展マイルストーン支払いを得る資格があり、そのうち10億ドル以上がIRAK 4計画と関係がある。私たちは合計7.00億ドルに達するいくつかの商業マイルストーン支払いを得る資格があり、そのうち4.0億ドルはIRAK 4計画に関連し、ある純売上高のハードルに達したときに支払う。計画ごとの純売上高のために等級別印税を取得する資格があり、上位から高青少年まで範囲があり、場合によっては1桁に上方調整される可能性がある。2023年12月31日現在,セノフィ協定により,我々はこれまでいくつかのIRAK 4臨床マイルストーンに関連した5500万ドルのマイルストーンを実現してきた。
2022年11月15日、我々は、元のセノフィ協定に規定されているいくつかの研究条項および責任を修正するために、元のセノフィ協定を修正する改訂された協力および許可協定、または修正されたセノフィ協定をセノフィと締結した。修正されたセノフィ協定は,協力条項に要求される第2段階試験の時間と数も詳細に説明した。改訂されたセノフィ協定は2022年12月5日に発効する。
また,セノフェイについては,セノフィは2022年12月2日にセノフィ社に書面で通知し,協力目標1候補薬KT−474を第2段階臨床試験に進める意向を示し,改訂されたセノフィ協定でさらに規定された記念碑的支払いを得ることになった。KT-474の第二段階臨床試験はHSとADにおける潜在力を初歩的に調査しており、この2種類の適応の臨床試験はすでに開始され、2023年に用量を開始した。
2023年9月、同社とセノフィは共同で、連携目標2に関する活動を停止することに同意した。
運営費
設立以来、私たちの運営費用は研究開発費と一般と行政費用だけが含まれています。
研究開発費
研究·開発費には,主に標的タンパク質分解療法の発見·開発に関する費用が含まれている。これらの研究作業とコストには,外部研究コスト,人員コスト,用品,許可費,施設関連費用が含まれている。私たちは発生した費用に応じて研究と開発費用を支払う。これらの費用には
私たちが将来受け取る研究·開発活動のための商品やサービスのための前金は前払い費用として記録されています。これらの金額は、貨物が関連サービスを提供するときに費用として確認されるか、または貨物がサービスを提供することが予期されなくなるまで確認される。
臨床開発後期段階にある候補製品は通常,臨床開発早期段階の候補品よりも高い開発コストを有しており,これは主に後期臨床試験の規模と持続時間が増加しているためである。われわれが計画している臨床開発活動により,われわれの研究と開発費用は大幅に増加すると予想される
115
近いうちと未来に。現在、私たちは任意の未来の候補製品の臨床開発を完成するために必要な努力の性質、タイミングとコストを正確に推定或いは知ることができない。
以下の要因により,我々の将来の臨床開発コストは大きく異なる可能性がある
候補製品の開発成功と商業化は大きな不確実性を持っている。これは、製品開発や商業化に関連する多くのリスクや不確定要因があるためである
116
我々の候補製品開発に関連するこれらの変数のいずれの結果の変化も、候補製品開発に関連するコストおよび時間を著しく変化させる可能性がある。私たちのどんな候補製品も規制部門の承認を得ることができないかもしれない。
一般と行政費用
一般と行政費用は主に行政、財務、法律、会社と業務発展及び行政機能者の賃金と関連費用を含む。一般および行政費用には、特許および会社事務に関連する法律費用、会計、監査、税務および行政相談サービスの専門費用、保険料、施設費用、行政出張費用、マーケティング費用、およびその他の運営コストも含まれる。
私たちは、製品候補製品の開発と持続的な研究活動を支援するために従業員を増やすことに伴い、将来的に私たちの一般的かつ行政費用が増加すると予想しています。私たちはまた、上場企業としての持続的な成長により、より多くの会計、監査、法律、規制、コンプライアンスと役員および役員および役員保険コスト、および私たちの上場企業の持続的な成長に関する法律、投資家、広報費用が発生すると予想しています。
その他の収入(費用)
利子その他収入と費用純額
利息と他の収入と支出には、私たちの投資現金残高から稼いだ利息と私たちの融資リースの利息が含まれています。
経営成果
2023年12月31日までと2022年12月31日までの年次比較
次の表は、2023年12月31日までと2022年12月31日までの年間経営結果をまとめています
|
|
十二月三十一日までの年度 |
|
|
変わる |
|
||||||
|
|
2023 |
|
|
2022 |
|
|
|
|
|||
|
|
(単位:千) |
|
|||||||||
協力収入 |
|
$ |
78,592 |
|
|
$ |
46,826 |
|
|
$ |
31,766 |
|
運営費用: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
|
189,081 |
|
|
|
164,248 |
|
|
|
24,833 |
|
一般と行政 |
|
|
55,041 |
|
|
|
43,834 |
|
|
|
11,207 |
|
総運営費 |
|
|
244,122 |
|
|
|
208,082 |
|
|
|
36,040 |
|
運営損失 |
|
|
(165,530 |
) |
|
|
(161,256 |
) |
|
|
(4,274 |
) |
その他の収入、純額 |
|
|
18,568 |
|
|
|
6,448 |
|
|
|
12,120 |
|
純損失 |
|
$ |
(146,962 |
) |
|
$ |
(154,808 |
) |
|
$ |
7,846 |
|
協力収入
我々は,それぞれが決定した履行義務に関する業績モデルに基づいて個々の連携プロトコルでの収入を確認し,これは各プロトコルで研究サービスを提供する期間である.
2023年12月31日までの年間協力収入は7860万ドルで、そのうち840万ドルと7020万ドルはそれぞれVertexとセノフィとの協力合意によるものです。2022年12月31日までの1年間の協力収入は4,680万ドルであり,そのうち1,080万ドルと3,600万ドルはそれぞれVertexとセノフィとの連携協定によるものである。収入の増加は主に2023年第4四半期にセノフィ協定により5500万ドルのマイルストーンを実現したことによる。
117
研究開発費
次の表は、提出段階ごとの研究開発費(正式開発候補者が指名されるまで計画費用を開示しない)をまとめています
|
|
十二月三十一日までの年度 |
|
|
変わる |
|
||||||
|
|
2023 |
|
|
2022 |
|
|
|
|
|||
|
|
(単位:千) |
|
|||||||||
外部研究開発コスト: |
|
|
|
|
|
|
|
|
|
|||
IRAK 4 |
|
$ |
13,762 |
|
|
$ |
17,850 |
|
|
$ |
(4,088 |
) |
Irakimid |
|
|
4,641 |
|
|
|
4,914 |
|
|
|
(273 |
) |
統計データ3 |
|
|
11,490 |
|
|
|
8,332 |
|
|
|
3,158 |
|
MDM 2 |
|
|
8,170 |
|
|
|
11,823 |
|
|
|
(3,653 |
) |
他にも |
|
|
56,637 |
|
|
|
46,474 |
|
|
|
10,163 |
|
内部研究開発コスト |
|
|
94,381 |
|
|
|
74,855 |
|
|
|
19,526 |
|
研究開発費総額 |
|
$ |
189,081 |
|
|
$ |
164,248 |
|
|
$ |
24,833 |
|
2023年12月31日までの1年間の研究開発費は1兆891億ドルだったが、2022年12月31日までの年間の研究開発費は1億642億ドルだった。2,480万ドルの増加は主に研究開発機能部門の従業員数の増加による人事、株の報酬と占有コストの1,950万ドルの増加であり、主に臨床前計画の仕事の増加に関する他の研究開発費の1,020万ドルの増加と、関連する第1段階の臨床試験の持続的な進展により、我々のSTAT 3計画の支出は320万ドル増加した。IRAK 4,IRAKIMiD,MDM 2計画の発展段階の変化により,我々の活動に関する直接費用が800万ドル減少し,これは主にこれらの計画の増加によって相殺されている.
一般と行政費用
2023年12月31日までの年間の一般·行政費は5500万ドルであるのに対し、2022年12月31日までの年間は4380万ドルである。1,120万ドルの増加は、主に一般および行政機能の従業員数の増加により人事、株式ベースの報酬、占有コストが880万ドル増加し、上場企業としての増加を支援する法律および専門サービス費用が240万ドル増加したためである。
その他の収入、純額
2023年12月31日までの1年間の他の収入純額は1860万ドルだったが、2022年12月31日までの年間は640万ドルだった。1,220万ドル増加したのは,主に期間に関する現行金利によるものである。
経営成果
2022年12月31日までと2021年12月31日までの年次比較
次の表は、2022年12月31日と2021年12月31日までの年間経営実績をまとめたものです
|
|
十二月三十一日までの年度 |
|
|
変わる |
|
||||||
|
|
2022 |
|
|
2021 |
|
|
|
|
|||
|
|
(単位:千) |
|
|||||||||
協力収入 |
|
$ |
46,826 |
|
|
$ |
72,832 |
|
|
$ |
(26,006 |
) |
運営費用: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
|
164,248 |
|
|
|
137,017 |
|
|
|
27,231 |
|
一般と行政 |
|
|
43,834 |
|
|
|
36,345 |
|
|
|
7,489 |
|
総運営費 |
|
|
208,082 |
|
|
|
173,362 |
|
|
|
34,720 |
|
運営損失 |
|
|
(161,256 |
) |
|
|
(100,530 |
) |
|
|
(60,726 |
) |
その他の収入、純額 |
|
|
6,448 |
|
|
|
313 |
|
|
|
6,135 |
|
純損失 |
|
$ |
(154,808 |
) |
|
$ |
(100,217 |
) |
|
$ |
(54,591 |
) |
118
協力収入
我々は,それぞれが決定した履行義務に関する業績モデルに基づいて個々の連携プロトコルでの収入を確認し,これは各プロトコルで研究サービスを提供する期間である.
2022年12月31日までの1年間の協力収入は4,680万ドルであり,そのうち1,080万ドルと3,600万ドルはそれぞれVertexとセノフィとの連携協定によるものである。2021年12月31日までの年間協力収入は7,280万ドルであり,そのうち1,850万ドルと5,430万ドルはそれぞれVertexとセノフィとの連携協定によるものである。
研究開発費
次の表は、提出段階ごとの研究開発費(正式開発候補者が指名されるまで計画費用を開示しない)をまとめています
|
|
十二月三十一日までの年度 |
|
|
変わる |
|
||||||
|
|
2022 |
|
|
2021 |
|
|
|
|
|||
|
|
(単位:千) |
|
|||||||||
外部研究開発コスト: |
|
|
|
|
|
|
|
|
|
|||
IRAK 4 |
|
$ |
17,850 |
|
|
$ |
27,368 |
|
|
$ |
(9,518 |
) |
Irakimid |
|
|
4,914 |
|
|
|
10,847 |
|
|
|
(5,933 |
) |
統計データ3 |
|
|
8,332 |
|
|
|
10,081 |
|
|
|
(1,749 |
) |
MDM 2 |
|
|
11,823 |
|
|
|
— |
|
|
|
11,823 |
|
他にも |
|
|
46,474 |
|
|
|
35,909 |
|
|
|
10,565 |
|
内部研究開発コスト |
|
|
74,855 |
|
|
|
52,812 |
|
|
|
22,043 |
|
研究開発費総額 |
|
$ |
164,248 |
|
|
$ |
137,017 |
|
|
$ |
27,231 |
|
2022年12月31日までの1年間の研究開発費は1兆642億ドルだったが、2021年12月31日までの年間の研究開発費は1.37億ドルだった。2,720万ドルの増加は、主に、我々のMDM 2計画のIND支援研究に関する2,240万ドルの増加と、私たちの他のパイプライン計画およびプラットフォームへの投資の増加、および研究開発機能部門の従業員数の増加により、人員、株ベースの報酬、占有コストが2,200万ドル増加したためです。IRAK 4,IRAKIMiDとSTAT 3計画発展段階の変化およびIRAKIMiDの停止により,我々のIRAK 4,IRAKIMiDとSTAT 3計画活動に関する直接費用は1720万ドル減少し,これは主にこれらの増加によって相殺されている.
一般と行政費用
2022年12月31日までの年間の一般·行政費は4380万ドルであるのに対し、2021年12月31日までの年間は3630万ドルである。750万ドルの増加は、主に従業員数の増加によりG&A社員の報酬が650万ドル増加したことと、上場企業としての増加を支援する法律や専門サービス費が100万ドル増加したことである。
その他の収入、純額
2022年12月31日までの1年間、その他の純収入は640万ドルだったが、2021年12月31日までの年間は30万ドルだった。610万ドル増加したのは、主に期間に関する現行金利のせいだ。
流動資金と資本資源
私たちはまだどの製品販売からも何の収入も得ていません。設立以来、重大な運営損失が発生しています。私たちはまだ何の製品も商業化していません。もしあれば、数年以内に製品販売から収入を得ないと予想しています。これまでに,転換可能優先株の売却,普通株の売却(2020年8月の初公募株と同時私募を含む),2021年7月の後続発行と同時私募,2022年8月のパイプライン発行,2024年1月の後続発行,コーエン社との販売協定,および我々の企業協力から14.5億ドルの総収益を得てきた。2023年12月31日現在、私たちは4.363億ドルの現金と現金等価物と有価証券を持っています。
119
2021年10月、Cowen and Company、LLCまたはCowenと販売契約を締結し、この合意により、Cowenを私たちの販売代理として、総収益が2.5億ドルに達する普通株式を時々“市場”発行で発売することができます。私たちはコーエンに手数料を支払うことに同意し、最高はコーエンが販売契約に従って売却した任意の株の総収益の3.0%である。2023年12月31日現在、私たちの普通株は販売契約に基づいて売却されていません。2024年2月7日,我々は販売契約により1,519,453株の普通株の販売を完了し,総収益は約5,000万ドルであった。
キャッシュフロー
次の表は、列挙された各期間の現金源と用途をまとめています
|
|
現在までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2020 |
|
|||
|
|
(単位:千) |
|
|||||||||
経営活動用の現金 |
|
$ |
(102,826 |
) |
|
$ |
(153,085 |
) |
|
$ |
(128,946 |
) |
投資活動によって提供される現金 |
|
|
139,886 |
|
|
|
20,519 |
|
|
|
(99,835 |
) |
融資活動で提供された現金 |
|
|
4,192 |
|
|
|
152,999 |
|
|
|
250,280 |
|
現金、現金等価物、および限定的な現金の純増加 |
|
$ |
41,252 |
|
|
$ |
20,433 |
|
|
$ |
21,499 |
|
経営活動に使われている現金流量
2023年12月31日までの年間で,運営活動には1.028億ドルの現金が使用されており,その間の純損失1.47億ドルと,我々の協力合意での繰延収入が860万ドル減少したことが主な原因である。これは、他の営業資産および負債の純減少1,120万ドルによって相殺され、これは、主に売掛金、営業賃貸負債の変化、および4150万ドルの非現金項目調整によるものである(主に株に基づく報酬、減価償却および償却、および利用可能な販売証券の割増および割引を含む)。
2022年12月31日までの1年間に,運営活動に1億531億ドルの現金が使用されたのは,その間の純損失1.548億ドルと,我々の協力合意での繰延収入が3780万ドル減少したためである。これは、非現金プロジェクトの調整3930万ドル(主に株ベースの補償、減価償却および償却、および利用可能な販売証券の割増および割引を含む)および20万ドルの経営資産および負債の純増加によって相殺され、これは、主に売掛金および売掛金の変化によって、前払い費用および他の経営資産および負債の変化によって部分的に相殺される。
2021年12月31日までの1年間に,経営活動に1兆289億ドルの現金が使用されたのは,主にこの間の純損失1.02億ドルと,我々のセノフィとVertex協力協定での繰延収入が6940万ドル減少したためである。これは、非現金プロジェクトの調整3320万ドル(主に株ベースの補償、減価償却および償却、および利用可能な販売証券の割増および割引を含む)および750万ドルの経営資産および負債の純増加によって相殺され、これは、主に売掛金および売掛金の変化、一部が前払い費用および他の経営資産および負債の変化によって相殺されるためである。
投資活動によって提供されるキャッシュフロー
2023年12月31日までの年間で、投資活動が提供する現金は1兆399億ドルで、3.635億ドルの有価証券満期日を含み、一部は1.892億ドルの有価証券の購入と3440万ドルの物件と設備の購入によって相殺された。
2022年12月31日までの年間で,投資活動が提供する現金は2,050万ドルであり,その中には4.693億ドルの有価証券満期日が含まれており,4.46億ドルの有価証券の購入と280万ドルの物件や設備の購入によって相殺されている。
2021年12月31日までの年間で、投資活動のための現金は9980万ドルで、4.564億ドルの有価証券の購入と160万ドルの物件や設備の購入を含み、3.582億ドルの有価証券の満期日によって相殺される。
120
融資活動によるキャッシュフロー
2023年12月31日までの1年間に、融資活動が提供する現金純額は420万ドルで、主に従業員の株式オプションを行使する290万ドルの収益と、従業員の株式による発行株の140万ドルの収益を含み、一部は10万ドルの融資リース支払いによって相殺された。
2022年12月31日までの年間で、融資活動が提供する現金純額は1.53億ドルで、主に2022年8月にパイプラインで発売された1億498億ドルの純収益、従業員の株式オプションを行使した320万ドルの収益、従業員の株式による発行株の110万ドルの収益を含み、一部は110万ドルの融資リース支払いによって相殺された。
2021年12月31日までの年間で、融資活動が提供する現金純額は2.503億ドルで、主に2021年7月に後続発行で受け取った2億408億ドルの純収益、2021年7月に同時に私募で得られた230万ドル、従業員の株式オプションを行使する収益760万ドル、従業員の株式購入計画に基づいて株式を発行する収益80万ドルを含み、2020年8月のIPOに関する40万ドルの発行コストと80万ドルの融資リース支払いによって相殺される。
将来の資金需要
我々が行っている活動に関連する費用は大幅に増加することが予想され,特に候補製品の後期臨床開発を進めた場合である。また、上場企業として、成長に関連した追加コストが発生することが予想される。
私たちの候補製品や計画の開発に関連する多くのリスクと不確実性、および私たちが第三者とどの程度協力して私たちの候補製品を開発する可能性があるため、私たちの候補製品の研究や開発の完了に関連する資本支出や運営費用の増加の時間と金額を見積もることはできません。私たちの運営費の時間と金額は主に見られるだろう
2023年12月31日までの既存の現金、現金等価物、有価証券は4.363億ドルで、2024年第1四半期に発行された純収益と2024年に受け取った1500万ドルのマイルストーン支払いを加えたと信じている
121
私たちとセノフィとの協力協定によると、2024年1月には2027年上半期までの資金を提供するのに十分な資金が提供される。私たちは間違っていることが証明される可能性があるという仮定に基づいてこの推定をして、私たちは予想よりも早く利用可能な資本資源を枯渇させるかもしれない。私たちは、私たちの臨床プロジェクトの臨床開発を継続するために追加の資金が必要になると予想し、規制部門の承認を得たら、私たちの候補製品を商業化し、許可内や他の候補製品の買収を求める。もし私たちの候補製品が規制部門の承認を得たら、私たちは製品製造、販売、マーケティング、流通に関連した巨額の商業化費用が発生することが予想され、これは私たちの候補製品をどこで商業化するかにかかっている。
潜在的な候補製品を決定し、臨床前研究と臨床試験を行うことは時間がかかり、高価かつ不確定な過程であり、完成するのに数年かかり、しかも私たちは永遠に市場の許可を得て製品販売を実現するために必要なデータ或いは結果を生成できないかもしれない。しかも、私たちの候補製品が承認されれば、商業的成功を得られないかもしれない。私たちの商業収入は、もしあれば、数年以内に商業用途がないと予想される製品の販売から来ます。したがって、私たちは私たちの業務目標を達成するために多くの追加資金を得る必要があるだろう。
これまで、相当な製品収入を生み出すことができれば、株式発行、債務融資、協力、戦略連合、および第三者のマーケティング、流通、または許可手配との組み合わせで、私たちの現金需要を満たす予定です。しかし、私たちは必要に応じて優遇条件で、または追加資金を調達できないか、またはそのような他の計画を達成することができないかもしれない。マクロ経済要因による市場変動は、必要に応じて資本を得る能力にも悪影響を及ぼす可能性がある。私たちが株式または転換可能な債務証券を売却することで追加資本を調達する場合、私たちの既存株主の所有権権益は大幅に希釈される可能性があり、このような証券の条項には、清算または他の私たちの普通株主の権利に悪影響を及ぼす特典が含まれる可能性がある。債務融資および優先株融資に関与する可能性のある協定には、追加債務を招く、資本支出を行う、または配当を宣言するなど、特定の行動をとる能力を制限する制限的な契約が含まれる可能性がある。もし私たちが第三者との協力、戦略連合またはマーケティング、流通または許可手配によって資金を調達する場合、私たちは、私たちの技術、将来の収入源、研究計画、または候補製品に対する貴重な権利を放棄しなければならないか、または私たちに不利になる可能性のある条項でライセンスを付与しなければならないかもしれない。もし私たちが必要な時に株式や債務融資や他の手配を通じて追加資金を調達できない場合、私たちは私たちの製品開発や将来の商業化努力を延期、減少、またはキャンセルすることを要求されるかもしれません。あるいは私たちが自分で開発し、マーケティングすることをより望んでいた候補製品の権利を与えることができます。
契約義務その他の約束
私たちは契約に基づいて、私たちの未来の流動性とキャッシュフローに影響を与えるお金を支払う義務があるという合意に達した。このような計画は主に私たちのレンタル約束と関連した計画を含む。
賃貸承諾額
私たちのレンタル約束は、マサチューセッツ州ウォータータウンでの2つの実験室とオフィススペース賃貸契約の満期支払いを反映しています。この2つの合意はそれぞれ2030年3月と2035年3月に満了します。2023年12月31日まで、賃貸契約に対する約束は1兆345億ドルで、このようなレンタル期間内に支払います。私たちのレンタルと将来の支払い時間に関するより多くの情報は、本年度報告Form 10-Kに含まれる総合財務諸表の付記7、レンタルを読んでください。
その他の義務
著者らは正常な業務過程中に各種の第三者と臨床試験、臨床前研究、テスト、製造とその他の運営サービスと製品の契約を締結した。これらの契約規定は通知され次第契約を終了することができる.キャンセル時に支払われるべき金額は、通常、キャンセル日までに提供されるサービスの支払いまたは発生した費用のみを含み、当社サービスプロバイダのキャンセル不可義務が含まれています。このような支払いはこのような契約義務と他の義務開示に単独で含まれていない。
重要な会計政策と試算
私たちの経営陣は私たちの財務状況と経営結果の討論と分析は私たちの総合財務諸表に基づいています。これらの報告書はアメリカ公認会計原則あるいはGAAPに基づいて作成されました。我々の連結財務諸表および関連開示を作成する際には、総合財務諸表における資産、負債、収入、コストおよび費用に影響を及ぼす報告金額、または資産および負債の開示に影響を及ぼす推定および判断を行う必要がある。私たちの推定は、歴史的経験、既知の傾向、および事件、およびこのような状況で合理的だと思う様々な他の要素に基づいている
122
これらの結果は資産や負債の帳簿価値の判断の基礎を構成しているが,これらの資産や負債の帳簿価値は他のソースでは明らかではない。私たちは持続的な基礎の上で私たちの推定と仮定を評価する。異なる仮定や条件の下で、私たちの実際の結果はこれらの推定とは異なる可能性がある。
我々の主な会計政策は、本年度報告書末の総合財務諸表付記2により詳細に記載されているが、以下の会計政策は、総合財務諸表の作成に使用される判断と推定に最も重要であると信じている。
収入確認
ASC 606によれば、エンティティは、そのクライアントが約束された商品またはサービスの制御権を取得したときに収入を確認し、その金額は、エンティティがこれらの商品またはサービスと交換するために予期される対価格を反映する。
ASC 606の範囲内の手配で確認すべき適切な収入金額を決定するために、(I)顧客との契約を決定するステップ(S)、(Ii)契約で約束された商品またはサービスを決定し、約束された商品またはサービスが履行義務であるかどうかを決定するステップ、(Iii)取引価格を測定するステップ、(Iv)取引価格を履行義務に割り当てるステップ、および(V)(または)各履行義務を履行するときに収入を確認するステップの5つのステップを実行する。エンティティが顧客に譲渡された商品またはサービスと交換するために獲得する権利のある対価格を受け取る可能性がある場合にのみ、5段階モデルを契約に適用する。
オプションの商品またはサービスが提供される場合、オプションが顧客物質の権利を付与するかどうかを決定するためにオプションを評価します。この確定オプションを含む定価は、顧客が契約を締結していない場合に受信しない金額であるか否かを決定する。もし私たちが結論を出したら、オプションは実質的な権利を伝達し、それは単独の履行義務として入金されるだろう。契約の履行義務を決定する時、私たちはそれらの違う約束を決定する。顧客が単独でまたは既製の資源と共に貨物またはサービスから利益を得ることができ、貨物またはサービスが契約内の他の約束とは別に識別できる場合、承諾された貨物またはサービスは異なると考えられる。約束が明確でなければ、合併後の約束のセットが区別できるまで、契約中の他の約束と統合されるだろう。
私たちは譲渡契約で約束された貨物またはサービスが予想される対価格金額に基づいて取引価格を推定します。対価は固定対価格と変動対価を含むことができます。可変価格を含む各手配の開始時に、潜在的な支払いの金額と支払いを受ける可能性を評価します。大きな収入逆転が起こらない可能性が高い場合、可変対価は取引価格に含まれる。特許化合物を含む販売の特許権使用料に基づく契約については、関連販売が発生した日に収入を確認する。最後に、約束商品およびサービスの独立販売価格に対するコミットメント対価格およびコミットメント商品およびサービス譲渡に対する支払い時間を分析することによって、契約が重要な融資構成要素を含むかどうかを決定する。報告日ごとに,吾らは取引価格および責任履行の実現確率および取引価格に関する制限を再評価した。必要であれば、取引価格を調整し、これまで制限されていた金額の進捗記録に基づいて進捗を累積します。
義務を履行する制御権が顧客に移行する場合には、収入が確認される。合併の履行義務に承諾のライセンス及び関連サービス又は他の承諾が含まれている場合には、管理層は、収入確認の適切な時間を決定するために判断する必要がある。そうするとき、私たちは、収入がある時点であるか、またはある時間に確認されるかを決定するために、契約内の1つまたは複数の主な約束を決定しなければならない。もし時間が経つにつれて、私たちは適切な進展測定基準を決定しなければならない。許可が義務履行における主要な約束であると考えられる場合、時点またはタイムアウト収入確認に最適な結論を得るために、機能的知的財産権でも象徴的知的財産権でも許可の性質を決定しなければならない。機能的または象徴的な知的財産権を決定するためには、顧客がその現在の状態でライセンスを利用して利益を得ることができるかどうか、またはライセンスの効用が私たちが行っている活動に依存するか、または私たちが行っている活動の影響を受けるか、または私たちに関連しているかどうかを評価する必要がある。
各報告日において、一定期間内に移行する履行義務の進捗状況を算出する。計算には,これまでに発生した費用の推定総費用に対する投入計量に基づいて,履行義務の移行を達成するのが一般的である。そして、得られた総収入は、任意の累積追跡調整を含めて、進捗の測定を用いて計算される。
研究と開発契約コストと対策プロジェクト
連結財務諸表作成過程の一部として、我々が計上すべき研究·開発費用を見積もる必要がある。このプロセスには、未締結契約および調達注文の審査、当社の適用者とコミュニケーションして、私たちを代表して実行されるサービスを決定すること、および実行されたサービスレベルを評価することが含まれます
123
また、請求書を受信していない場合や、実際のコストを他の方法で通知している場合には、サービスに関連するコストを発生させる。私たちのほとんどのサービスプロバイダは予定のスケジュールに従って、あるいは契約マイルストーンに達した時に私たちに借金の領収書を発行しますが、前金が必要なものもあります。私たちは、当時私たちが知っている事実と状況に基づいて、連結財務諸表において、各貸借対照表の日付までの課税費用を推定します。我々は定期的にサービスプロバイダとこれらの推定の正確性を確認し,必要に応じて調整する.計算すべき研究および開発費用の推定例には、以下の者に支払われる費用が含まれる
我々は,我々を代表する非臨床研究を提供·実施·管理する複数のCMOとCROとのオファーや契約に基づいて,受信したサービスや費用の努力を推定し,外部研究や開発に関連する費用を記録した。これらの合意の財務条項は交渉が必要であり、契約によって異なり、支払いの不均衡を招く可能性がある。場合によっては、私たちのサプライヤーに支払われるお金は、提供されたサービスレベルを超え、事前支払い費用につながる可能性があります。サービス料を計算する際に、サービスを提供する時間帯と各時間の努力度を見積もります。サービスの実行時間や努力の程度が推定値と異なる場合には,それに応じて計算すべき費用や前払い費用の金額を調整する.実際に発生した金額と実質的に異なることはないと予想されるが、提供されたサービスの実際の状態および時間に対する提供サービスの状態および時間の理解が異なる可能性があり、報告された金額が任意の特定の時期に高すぎたり、過小になったりする可能性がある。これまで,我々が計算した研究や開発費の先行推定には何の大きな調整もなかった.
株式ベースの報酬費用
我々は、従業員、取締役、および非従業員に付与された公平な価値に基づいて、従業員、取締役および非従業員に付与された持分報酬を測定し、必要なサービス期間(通常は対応する報酬の帰属期間)内でそのような報酬の補償費用を確認する。株式ベースの支払いには、帰属された普通株式を含む株式オプションおよび普通株式の付与が含まれる。持分奨励の計量日を付与日とし、持分に基づく報酬コストは直線原則で必要なサービス期間(すなわち帰属期間)の支出であることを確認する。各株式オプション付与の公正価値は、付与された日にBlack-Scholesオプション定価モデルを使用して推定され、このモデルは、予想される株価変動、オプションの予想期限、オプションの予想期限に近い期間の無リスク金利、および私たちの期待配当収益率を含むいくつかの主観的仮定に基づいて入力することを要求する。予想変動率は代表的な上場企業の報告変動率データから計算され、これらの会社は歴史情報を持っている。私たちは私たちと似たような特徴を持ち、歴史的株価情報と株式ベースの奨励の期待期限が近似している会社を選択した。我々は,選定会社株の我々が計算した株式オプション期待期限と近似した同じ時期の毎日終値を用いて履歴変動性データを計算した.私たちは株価変動に関する十分な数の履歴情報が利用できるまで、この方法を適用し続けるつもりだ。無リスク金利は、付与時に有効な米国債収益率曲線に基づいており、期待期限の仮定に見合っている。我々は,期待期限が帰属日と契約期限終了との間の中点として推定される簡略化された方法を用いる.歴史的な訓練データが不足しているため、私たちはこの方法を採用した。期待配当収益率はゼロと仮定しています。私たちは現在普通株の配当金を支払う計画がないからです。
124
第七A項。定量と合格市場リスクの開示について。
私たちは金利の変化と関連した市場リスクに直面している。私たちの市場リスクに対する主な開口は金利感受性であり、金利敏感性はアメリカの金利全体レベルの変化の影響を受け、特に私たちの投資は現金等価物を含み、通貨市場基金と有価証券の形で存在し、アメリカ国債または政府債券と会社証券に投資するからである。しかし、私たちポートフォリオの持続期間の短期性と私たちの投資の低リスクのため、市場金利の即時10%の変化は、私たちのポートフォリオの公平な市場価値や私たちの財務状況や経営業績に実質的な影響を与えないと考えられます。
私たちはまた外貨為替レートの変化と関連した市場リスクに直面している。私たちはアジアとヨーロッパのサプライヤーと契約を結び、いくつかの領収書は外貨で価格を計算します。私たちはこれらの手配に関連した外貨為替レートの変動の影響を受けるだろう。私たちは現在私たちの外貨為替レートに対するリスクを持っていない。2023年12月31日現在、私たちは外貨建ての重大な負債を持っていません。
インフレは一般的に私たちの労働コスト、第三者サプライヤー、そして臨床試験コストを増加させることによって私たちに影響を及ぼす。世界的なマクロ経済環境はすでにあり、非常に厳しい挑戦を経験し続けるだろう。このようなマクロ経済要素がもたらし、私たちは引き続きコスト増加と他の懸念される問題をもたらすと予想される。これらのインフレ圧力がどのくらい続くか、あるいはそれらが時間の経過とともにどのように変化するかは予測できませんが、世界経済、私たちの業界、わが社への持続的な影響が見られると予想されています。インフレ圧力が持続的に存在すれば、引き続き私たちの総合的な財務状況、経営業績および/またはキャッシュフローに悪影響を及ぼす可能性がある。しかし、インフレ環境により金利が増加し、利息収入が以前に達成された水準よりも高くなった。
プロジェクト8.財務諸表Sと補足データ。
125
連結財務諸表索引
独立公認会計士事務所報告(上場企業会計監督委員会ID: |
F-2 |
|
|
2023年12月31日と2022年12月31日までの連結貸借対照表 |
F-4 |
|
|
2023年,2022年と2021年12月31日までの総合経営報告書と全面赤字報告書 |
F-5 |
|
|
年度までの株主権益総合報告書 2023年12月31日、2022年、2021年 |
F-6 |
|
|
2023年,2022年,2022年12月31日終了年度までの統合キャッシュフロー表1 |
F-7 |
|
|
連結財務諸表付記 |
F-9 |
F-1
独立登録公共会計基金報告書情報資源管理
Kymera治療会社の株主と取締役会に。
財務諸表のいくつかの見方
Kymera治療会社(当社)の2023年12月31日と2022年12月31日までの連結貸借対照表、2023年12月31日までの3年間の毎年の関連総合経営報告書と全面赤字、株主権益と現金流量および関連付記(総称して“総合財務諸表”と呼ぶ)を監査しました。総合財務諸表は、すべての重要な点で、2023年12月31日、2023年12月31日、2022年12月31日までの会社の財務状況、および2023年12月31日までの3年度の経営結果とキャッシュフローを公平に反映しており、米国公認会計原則に適合していると考えられる。
また、米国上場企業会計監督委員会(PCAOB)の基準に基づき、テレデビル委員会協賛組織委員会が発表した“内部統制-総合枠組み(2013枠組み)”で確立された基準に基づいて、2023年12月31日までの財務報告内部統制を監査し、2024年2月22日に発表された報告書について保留意見を発表した。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
F-2
重要な監査事項
以下に述べる重要な監査事項とは、監査委員会に伝達または要求が監査委員会に伝達された当期財務諸表監査によって生じる事項である:(1)財務諸表に対して重大な意義を有する勘定または開示に関するものであり、(2)特に挑戦的で主観的または複雑な判断に関するものである。重要監査事項の伝達は、総合財務諸表に対する私たちの全体的な意見をいかなる方法でも変えることはなく、以下の重要な監査事項を伝達することによって、重要な監査事項またはそれに関連する勘定または開示について個別の意見を提供することはない。
|
|
協力手配 |
説明: この事. |
|
総合財務諸表付記5で述べたように、当社は個々の業績義務に関する収入を確認しており、研究開発サービスは個々の計画ごとの研究開発活動に関するコストと、将来個別業績義務を履行するために生じるコストに基づいて、入力法で提供されるためである。収入が確認されていない受信金額は、会社総合貸借対照表で契約負債として繰延される。同社は2023年12月31日までの1年間で7860万ドルの協力収入を確認した。
監査会社が協力手配から得た収入の会計は複雑であり、主に評価入力法の下で時間経過とともに確認された収入の期待総コスト推定数を決定する必要がある。監査完了協力協定の進展は、業績義務履行に必要な残りの研究開発コストを見積もる際に使用される主観管理仮定に関連するため、特に挑戦的である。余剰推定研究開発コスト総額の計算には、内部従業員の仕事、材料コスト、第三者契約コストに関する予測コストが含まれる。連携スケジュールにより収入が管理職によるこれらの判断や見積りの影響を受けることを確認し,これらの仮定の変化に敏感である.
|
私たちが監査でどのようにこの問題を解決したのか |
|
経営陣の推定を支援するデータの完全性と正確性を評価する制御を含む、企業策定評価過程における内部統制の設計および運用有効性を評価·テストした。
企業の連携収入をテストするために、連携プロトコルを読むことと、上記の推定および重大な判断を評価するための基礎データの正確性および完全性をテストするテストとを含む監査プログラムを実行した。会社の重大な見積もりと判断の合理性を評価するために,コスト見積りを従来の類似活動に発生していたコストと比較し,サービス完了に必要な余剰見積り努力レベルを評価し,実際の発生コストの証拠をチェックした。また、会社の研究·開発者と重要な仮説の基礎を議論し、彼らは協力手配の完成を監督している。 |
/s/
2018年以来、当社の監査役を務めてきました。
2024年2月22日
F-3
KYMERA治療会社
強固な基礎スプレー銃のシーツ
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
|
十二月三十一日 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
有価証券(付記4) |
|
|
|
|
|
|
||
売掛金 |
|
|
|
|
|
|
||
契約資産 |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
流動資産総額 |
|
$ |
|
|
$ |
|
||
非流通有価証券(付記4) |
|
|
|
|
|
|
||
財産と設備、純額(付記6) |
|
|
|
|
|
|
||
資産を使用し、レンタルを経営する |
|
|
|
|
|
|
||
他の非流動資産 |
|
|
|
|
|
|
||
制限現金 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
負債と株主権益 |
|
|
|
|
|
|
||
流動負債: |
|
|
|
|
|
|
||
売掛金 |
|
$ |
|
|
$ |
|
||
課税費用(付記8) |
|
|
|
|
|
|
||
収入を繰り越す |
|
|
|
|
|
|
||
リース負債を経営する |
|
|
|
|
|
|
||
融資リース負債 |
|
|
|
|
|
|
||
その他流動負債 |
|
|
|
|
|
|
||
流動負債総額 |
|
$ |
|
|
$ |
|
||
非流動負債 |
|
|
|
|
|
|
||
繰延収入,当期分を差し引く |
|
|
|
|
|
|
||
賃貸負債を経営し,当期分を差し引く |
|
|
|
|
|
|
||
融資リース負債、当期分を差し引く |
|
|
|
|
|
|
||
他の非流動負債 |
|
|
|
|
|
|
||
総負債 |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|||
株主権益: |
|
|
|
|
|
|
||
普通株、$ |
|
|
|
|
|
|
||
追加実収資本 |
|
|
|
|
|
|
||
赤字を累計する |
|
|
( |
) |
|
|
( |
) |
その他の総合損失を累計する |
|
|
( |
) |
|
|
( |
) |
株主権益総額 |
|
|
|
|
|
|
||
総負債と株主権益 |
|
$ |
|
|
$ |
|
||
付記はこれらの連結財務諸表の構成要素である。
F-4
KYMERA治療会社
合併状態運営と総合損失のNTS
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
運営報告書データ: |
|
|
|
|
|
|
|
|
|
|||
*収入 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
運営費用: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
一般と行政 |
|
|
|
|
|
|
|
|
|
|||
総運営費 |
|
|
|
|
|
|
|
|
|
|||
運営損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
その他の収入(支出): |
|
|
|
|
|
|
|
|
|
|||
利子とその他の収入 |
|
|
|
|
|
|
|
|
|
|||
利息とその他の費用 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
その他収入合計 |
|
|
|
|
|
|
|
|
|
|||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
その他の全面的な損失: |
|
|
|
|
|
|
|
|
|
|||
有価証券の未実現収益 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
全面損失総額 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
普通株による純損失と純損失の照合 |
|
|
|
|
|
|
|
|
|
|||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
普通株主は純損失を占めなければならない |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
普通株主は1株当たり純損失を占めなければならない,基本的に |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
加重平均発行済み普通株式、基本普通株式、および |
|
|
|
|
|
|
|
|
|
|||
付記はこれらの連結財務諸表の構成要素である。
F-5
KYMERA治療会社
株主権益合併報告書
2023年まで,2022年および2021年12月31日まで年度
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
普通株 |
|
その他の内容 |
|
積算 |
|
積算 |
|
合計する |
|
||||||||
|
株 |
|
価値がある |
|
資本 |
|
赤字.赤字 |
|
得/(失) |
|
権益 |
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
2020年12月31日残高 |
|
|
$ |
|
$ |
|
$ |
( |
) |
$ |
( |
) |
|
|
||||
株式オプションの行使 |
|
|
|
|
|
|
|
— |
|
|
— |
|
|
|
||||
帰属制限株 |
|
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
ESPPにより株を発行する |
|
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
|||
コンサルタントに既得制限株を発行する |
|
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
株に基づく報酬費用 |
|
— |
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
||
有価証券は赤字を実現していない |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
( |
) |
|
( |
) |
公開発売に関連する株を発行する |
|
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
|||
同時私募で株式を発行する |
|
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
|||
純損失 |
|
— |
|
|
— |
|
|
— |
|
|
( |
) |
|
— |
|
|
( |
) |
2021年12月31日の残高 |
|
|
$ |
|
$ |
|
$ |
( |
) |
$ |
( |
) |
$ |
|
||||
普通株式の発行と予備資金付き |
|
|
|
|
|
|
|
— |
|
|
— |
|
|
|
||||
株式オプションの行使 |
|
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
|||
帰属制限株 |
|
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
— |
|
||
ESPPにより株を発行する |
|
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
|||
株に基づく報酬費用 |
|
— |
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
||
有価証券は赤字を実現していない |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
( |
) |
|
( |
) |
純損失 |
|
— |
|
|
— |
|
|
— |
|
|
( |
) |
|
— |
|
|
( |
) |
2022年12月31日の残高 |
|
|
$ |
|
$ |
|
$ |
( |
) |
$ |
( |
) |
$ |
|
||||
株式オプションの行使 |
|
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
|||
帰属制限株 |
|
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
ESPPにより株を発行する |
|
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
|||
株に基づく報酬費用 |
|
— |
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
||
有価証券の未実現収益 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
|
|
|
||
純損失 |
|
— |
|
|
— |
|
|
— |
|
|
( |
) |
|
— |
|
|
( |
) |
2023年12月31日の残高 |
|
|
$ |
|
$ |
|
$ |
( |
) |
$ |
( |
) |
$ |
|
||||
付記はこれらの連結財務諸表の構成要素である。
F-6
KYMERA治療会社
合併状態キャッシュフロープロジェクト
(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
経営活動 |
|
|
|
|
|
|
|
|
|
|||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
純損失と経営活動で使用される現金純額の調整: |
|
|
|
|
|
|
|
|
|
|||
株に基づく報酬費用 |
|
|
|
|
|
|
|
|
|
|||
減価償却および償却 |
|
|
|
|
|
|
|
|
|
|||
販売可能な有価証券の割増と割引 |
|
|
( |
) |
|
|
|
|
|
|
||
財産と設備処分損失 |
|
|
|
|
|
|
|
|
|
|||
経営性資産と負債変動状況: |
|
|
|
|
|
|
|
|
|
|||
前払い費用と他の資産 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
売掛金 |
|
|
( |
) |
|
|
|
|
|
|
||
契約資産 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
売掛金 |
|
|
|
|
|
|
|
|
|
|||
費用とその他の流動負債を計算しなければならない |
|
|
|
|
|
|
|
|
|
|||
収入を繰り越す |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
経営的リース使用権資産 |
|
|
|
|
|
|
|
|
|
|||
リース負債を経営する |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
他の非流動資産 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
他の非流動負債 |
|
|
( |
) |
|
|
|
|
|
|
||
経営活動のための現金純額 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
投資活動 |
|
|
|
|
|
|
|
|
|
|||
財産と設備を購入し,純額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
有価証券を購入する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
有価証券の満期日 |
|
|
|
|
|
|
|
|
|
|||
投資活動提供の現金純額 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
||
融資活動 |
|
|
|
|
|
|
|
|
|
|||
普通株発行および付随収益 |
|
|
|
|
|
|
|
|
|
|||
株式オプションを行使して得られる収益 |
|
|
|
|
|
|
|
|
|
|||
従業員の株購入計画の収益 |
|
|
|
|
|
|
|
|
|
|||
初公募に関する発売費用をお支払いいたします |
|
|
|
|
|
|
|
|
( |
) |
||
融資リースの支払い |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
その後公開された収益は、引受割引と |
|
|
|
|
|
|
|
|
|
|||
増発の収益を同時に指向しています |
|
|
|
|
|
|
|
|
|
|||
融資活動が提供する現金純額 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
現金、現金等価物、および限定的な現金の純増加 |
|
|
|
|
|
|
|
|
|
|||
期初現金、現金等価物、および限定現金 |
|
|
|
|
|
|
|
|
|
|||
期末現金、現金等価物、および制限現金 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
キャッシュフロー活動の補足開示 |
|
|
|
|
|
|
|
|
|
|||
新しい経営リース負債と引き換えに使用権資産 |
|
$ |
|
|
|
|
|
|
|
|||
利子を支払う現金 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
非現金投資·融資活動の追加開示 |
|
|
|
|
|
|
|
|
|
|||
融資·リース負債による財産·設備の購入 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
財産と設備調達は売掛金と |
|
$ |
|
|
$ |
|
|
$ |
|
|||
付記はこれらの連結財務諸表の構成要素である。
F-7
以下の表は、上記各期間の現金、現金等価物、および制限された現金残高の入金状況を示す
|
|
十二月三十一日 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
現金と現金等価物 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
制限現金 |
|
|
|
|
|
|
|
|
|
|||
現金総額、現金等価物、制限された現金 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
F-8
KYMERA治療会社
総合備考財務諸表
付記1.業務説明及び主要会計政策の概要
Kymera治療会社とその子会社であるKymera証券会社は合併に基づいて“会社”と呼ばれている。同社は生物製薬会社であり,小分子療法の発見と開発に専念し,ヒト自身の自然細胞過程を利用して原因タンパク質を選択的に分解することを標的タンパク質分解と呼ばれている。会社は設立以来、主に研究と開発に取り組んできた。当社はまだいかなる製品の開発、申請、あるいはいかなる製品の監督管理許可を得ておらず、市場のそのような製品に対する受け入れと需要も確認されていない。そのため、同社はバイオテクノロジー業界の新興会社が共同で直面しているいくつかのリスクに直面している。これらのリスクは主に製品発見と開発過程の不確定性、肝心な個人への依存、会社の競争相手開発と同じ或いは類似の技術革新、ノウハウの保護、政府法規と承認要求の遵守、会社の資本獲得能力及び市場の製品受容度の不確定性を含む。
当社はすでに予測可能な未来に引き続き損失を被り、累積赤字は#ドルとなる見通しだ
2023年12月31日現在、会社は現金、現金等価物、有価証券を持っているIESは$
同社は、既存の現金、現金等価物および有価証券を用いて、または戦略的融資機会によって、その製品の組み合わせの将来の研究開発コストに資金を提供することが予想されており、これらの機会は、将来提供される株式、協力協定、または債務を含むことができるが、これらに限定されない。しかしながら、これらの戦略または融資機会のいずれも有利な条件で実行または達成されることは保証されず、もし本当にあれば、その中のいくつかは既存の株主の権益を希釈する可能性がある。もし同社が追加の未来資本を得ることができなければ、それはその計画の臨床前研究と臨床試験を完成できないかもしれない。
2021年新株増発
2021年7月6日、会社は普通株の後続発行を完了し、発行と販売を行った
公募株式“パイプライン”発行における民間投資の応用
2022年8月18日、当社はいくつかの認可投資家と証券購入協定を締結し、この合意に基づき、当社は私募方式で当該等投資家に(I)を売却·発行することに合意した
事前出資株式証明書は会社普通株とリンクしている(その他の面で権益分類の要求に適合している)ため、会社は事前出資株式承認証で受け取った対価格を発行して追加実収資本として会社の総合貸借対照表に記録している。あらかじめ出資した引受権証は随時行使することができる.もし事前資金権証の所有者とその関連会社が実益を超えている場合
F-9
体を鍛える事前融資権証の保有者はそれを超えないように増やすか減らすことができる
2023年12月31日までの12ヶ月間で
付記2.主要会計政策の概要
添付されている連結財務諸表は、本付記および添付の連結財務諸表および付記に記載されているいくつかの重要な会計政策の適用状況を反映する。
合併原則
添付されている総合財務諸表は、同社およびその完全子会社Kymera Securities Corporationの勘定を含む。すべての会社間取引と残高は合併で販売された。
陳述の基礎
添付されている連結財務諸表は、アメリカ合衆国公認会計原則または公認会計原則に従って作成されている。本説明における適用ガイドラインへの任意の言及は、財務会計基準委員会(FASB)の会計基準編纂またはASCおよび会計基準更新(ASU)において見つけることができる権威の米国公認会計原則を意味する。
予算の使用
米国公認会計原則に基づいて財務諸表を作成し、財務諸表日の資産及び負債報告金額、又は事項の開示及び報告期間内に報告された費用金額に影響を与えるために、管理層に推定及び判断を要求する。経営陣の見積もりと判断は、既存の情報、歴史的経験、様々な他の当時の状況が合理的とされているという仮定に基づいて得られ、評価され続けている。使用見積り数は財務報告過程に固有であるため,実際の結果はこれらの見積り数とは異なる可能性がある.業務運営により発生した取引や残高を記録する際に、管理層は、推定を行う際に得られる最適な情報に基づいて推定を行う。これらの財務諸表を作成する際に依存する大きな見積もりには、セノフィやVertexとの協力協定で確認された収入、研究開発費の計上費用、株式ベースの報酬費用が含まれています。より良い情報が利用可能または実際の金額が決定可能になるにつれて、記録された推定数が修正されるであろう。したがって、以前の推定数の改訂は経営業績に影響を及ぼす可能性がある。
市場と地理情報を細分化する
経営部門は企業の構成要素として定義され、その独立した離散情報は、首席経営意思決定者あるいは意思決定グループが資源をどのように分配するかを決定し、業績を評価する際に評価することができる。会社の最高経営決定者は最高経営責任者です。会社は以下の点でその運営·管理業務を確認します
現金と現金等価物
現金等価物は高流動性の投資であり、購入時に元の満期日が3ヶ月以下の現金に変換しやすい。これらの資産には、通貨市場基金、米国国債、米国政府機関証券、会社証券(商業手形を含む)への投資が含まれる。その会社は主要金融機関でその銀行口座を維持している。
制限現金
限定現金とは、会社の施設賃貸に関する信用状を得るために持っている現金のことです。
有価証券
その会社は残り期限を大きくする
F-10
シーツです残り満期日が大きい
各報告日において、会社は、未実現の損失が信用損失によるものであるか否かを決定するために減価評価を行う。損害は個人の安全レベルで評価される。損失が信用損失或いはその他の要素に由来するかどうかを決定する時、考慮する要素は会社が投資を持ってその余剰コスト基礎が回収されるまでの意図と能力、公正価値が余剰コスト基礎より低い程度、公正価値がコスト基礎より低い時間長と程度、発行者の財務状況、発行者は従来から予定の利息或いは元金を支払うことができなかった、格付け機関の証券格付けに対するいかなる変化、発行者或いは発行者業界に影響する不利な法律或いは監督管理事件、及び経済状況の任意の重大な悪化を含む。
公正価値計量
公認会計原則に基づき、ある資産と負債は公正価値に基づいて帳簿に記入される。公正価値は、計量日市場参加者間の秩序ある取引において、資産または負債が元金または最も有利な市場で負債を移転するために徴収または支払いされる交換価格として定義される。公正価値を計量するための推定技術は,観察可能な投入を最大限に利用し,観察不可能な投入を最大限に減少させなければならない。公正価値別に列挙された金融資産および負債は、公正価値レベルの以下の3つのレベルのうちの1つで分類および開示されなければならず、そのうちの最初の2つのレベルは可視とみなされ、最後のレベルは見えないとみなされる
レベル1-同じ資産の活発な市場でのオファー
レベル2-観察可能な投入(第1レベルオファーを除く)、例えば、同様の資産または負債アクティブ市場のオファー、同じまたは同様の資産の非アクティブ市場のオファー、または観察可能な市場データによって確認されることができる他の投入。
レベル3-市場活動が非常に少ない或いは市場活動支持のない観察できない投入は、資産の公正価値を確定することに重要な意義があり、定価モデル、現金フロー方法と類似技術を含む。
短期的な性質のため、会社の現金等価物、前払い費用、売掛金とある計算すべき項目の帳簿価値はその公正価値に近い。
賃貸借証書
一つの手配の開始時に、当社は当時の独特の事実及び状況に基づいて、この手配が賃貸借契約であるか否か又は賃貸借契約を含むか否かを決定する。経済的に購入資産と類似したリースは通常、融資リースに分類され、そうでなければ、そのレンタルは経営的賃貸に分類される。その会社は原始期限を認めないことを選択した
財産と設備
属性設備は減価償却累計を差し引いたコストで入金されます。重大な増築·改築は資本化に計上する;それぞれの資産寿命を改善または延長することができないメンテナンス·メンテナンスは運営費に計上される
F-11
AS招いた
|
|
使用寿命を見込む (単位:年) |
実験室装置 |
|
|
家具と固定装置 |
|
|
事務設備 |
|
|
コンピュータ装置 |
|
|
賃借権改善 |
|
廃棄または販売時には、資産を処分するコストおよび関連する減価償却は勘定から抹消され、それによって生じる収益または損失は運営損失に計上される。
建設中の工事はコストで帳簿に記載されており、関連資産の設立または建設の直接コストが含まれている。関連資産が完成して使用された後、建設プロジェクトに減価償却費用を計上する。
長期資産減価準備
保有および使用すべき長期資産(使用権資産を含む)が、イベントやビジネス環境の変化が発生して資産の帳簿価値が完全に回収できない可能性がある場合には、回収可能かどうかをテストする。当社がいつ減値審査を行うかを決定する際に考慮する要素は、予想に関連する業務パフォーマンスの顕著な不良、業界或いは経済傾向に重大なマイナス影響が出現すること、及び資産用途の重大な変化或いは計画変更を含む。長期資産の回収可能性を評価するために減値審査を行うと、当社は長期資産の使用と最終処分による未割引キャッシュフローの予測をその帳簿価値と比較する。1つの資産の使用が予想される推定未割引将来の現金流量がその帳簿金額よりも少ない場合には、減価損失が確認される。減価損失は、減価資産の帳簿価値がその公正価値を超え、割引現金流量に基づいて決定される“会社”ができた
株式承認証
当社は、まず権利証がASC 480−10の負債分類に適合しているか否かを評価することにより、発行された権証の会計分類が負債または権益であることを決定する負債性と利益性を兼ね備えたいくつかの金融商品会計そしてASC 815-40によると自社株にリンクして決済可能なデリバティブ金融商品の会計処理それは.ASC 480−10によれば、株式承認証が強制償還可能である場合、発行者は、現金または他の資産を支払うことによって権利証または関連株式を決済する義務があるか、または可変数量の株式を発行することによって決済されなければならない場合、株式承認証は負債分類とみなされる。
株式証明書がASC 480-10での負債分類に適合していない場合、当社はASC 815-40での要求を評価し、この要求は、発行者に現金決済契約の契約が公正価値で記録された負債であることを要求する可能性があり、純現金決済特徴をトリガする取引が発生する可能性にかかわらず、どのようにするかを規定する。株式証明書がASC 815-40に基づいて負債分類を行う必要がない場合、株式分類を達成するために、会社は株式証明書がその普通株にリンクしているかどうかを評価し、株式承認証がASC 815-40または他の適用される米国公認会計原則に基づいて株式に分類されるかどうかを評価する。すべての関連評価を行った後、会社は権利証を負債または権益に分類する。負債分類株式権証は発行当日及びその後の会計期間終了日に公正価値で入金し、発行日後に価値を公正に許可するすべての変動は経営報告書の中で損益とする必要がある。株式分類は株式証を発行日に公正価値で入金し、発行後に確認された公正価値に変動はない。
研究開発コスト
研究と開発コストは主に会社の最先端の臨床段階計画のコストを含む的確なタンパク質分解療法の発見と開発に関連するコストを含む。これらの研究作業やコストも会社のPegasusの開発と強化を支援しているTM目的タンパク質分解プラットフォームは、外部研究コスト、人員コスト、用品、許可費、施設関連費用を含む。当社は発生した研究と開発費用を負担します。
F-12
特許費用
特許出願の提出と起訴に関するすべての特許に関する費用は,支出が回収できるかどうかの不確実性による費用で計上される。発生した金額は一般費用と行政費用に分類される。
融資コスト
発行権益単位と株式に関するコスト計上収益を権益帳簿価値に減額する。同社は、このような融資が完了するまで、進行中の融資に直接関連するいくつかの法律、専門会計、その他の第三者費用を繰延発売コストとしている。融資完了後、これらのコストは融資で得られた収益の減少額として入金される。計画された融資が放棄された場合、繰延発売コストは直ちに連結経営報告書と全面赤字に営業費用を計上するそれは.あったことがある
収入確認
ASC 606では、主題606、顧客との契約収入、またはASC 606顧客が承諾した商品やサービスに対するコントロール権を獲得した場合、当社は収入を確認し、その金額は、実体がそのような商品やサービスと交換するために得られると予想される対価格を反映している。ASC 606の範囲内の手配によって確認されるべき適切な収入額を決定するために、会社は、(I)顧客との契約を決定するステップ(S)、(Ii)契約で約束された貨物またはサービスを決定し、約束された貨物またはサービスが履行義務であるかどうかを決定するステップ、(Iii)取引価格の計量、(Iv)取引価格を履行義務に割り当てるステップ、および(V)会社が各履行義務を履行したときに収入を確認するステップの5つのステップを実行する。エンティティが顧客に譲渡された商品またはサービスと交換するために獲得する権利のある対価格を受け取る可能性がある場合にのみ、会社は5ステップモードを契約に適用する。契約開始時に、契約がASC 606の範囲内にあると決定されると、会社は、各契約において約束された商品またはサービスを評価し、どれが義務を履行しているかを決定し、各約束された商品またはサービスが異なるかどうかを評価する。そして、会社は、履行義務を履行する際にそれぞれの履行義務に割り当てられた取引価格の金額が収入であることを確認する。
このような手配の会計手配の一部として、当社は、a)上記(Ii)ステップの決定によって決定された履行義務の数と、その等履行義務が契約における他の履行義務と異なるか否か、b)上記(Iii)ステップでの取引価格、およびc)上記(Iv)ステップで取引価格を割り当てる契約で決定された各履行義務の独立販売価格を決定するために、重大な判断を用いなければならない。当社は、以下に述べるように、特許使用料を除くマイルストーンまたは他の可変対価格が取引価格に含まれるべきかどうかを決定するために判断を使用します。取引価格は相対独立販売価格で契約履行義務ごとに割り当てられ、会社は契約項目の履行義務を履行する際に収入を確認する。企業独自技術または顧客選択権が提供する物質的権利のライセンスの独立販売価格を決定する際には、市場機会、推定された開発コスト、成功の可能性、およびライセンスに基づいて候補製品を商業化するのに要する時間に関する仮定を含む市場条件および特定のエンティティの要因、および交渉合意時に考慮される要因および内部開発の推定を含む。独立販売価格を推定することを確認する際に、当社は、独立販売価格を推定するための主な仮定の変動が、責任履行間の手配が価格配分に大きな影響を与えるかどうかを評価する。
当社が譲渡契約で約束した貨物またはサービスにより予想される対価格金額に基づいて、取引価格を推定します。対価は固定対価格と変動対価を含むことができます。可変価格を含む各手配の開始時に、会社は潜在的な支払いの金額と支払いを受ける可能性を評価する。当社は最高額法または期待値法を用いて取引価格を推定し、どの方法によって予想される対価格額を予測することができる。大きな収入逆転が起こらない可能性が高い場合、可変対価は取引価格に含まれる。
履行義務は、契約において約束された独自の商品又はサービスを顧客に移転する義務である。約束された貨物またはサービスは、(1)顧客が単独でまたは他の既製の資源と共に貨物またはサービスから利益を得ることができ、(2)約束された貨物またはサービスを契約内の他の約束とは別に識別することができる場合とは異なると考えられる。承諾した商品やサービスが異なるかどうかを評価する際に、会社は以下のような要素を考慮します
F-13
基礎知的財産権の開発段階では、顧客が自ら知的財産権を開発する能力、及び必要な専門知識をいつでも得ることができるかどうか。
ライセンスと他の承諾からなる履行義務については,会社は合併履行義務の性質を判断を用いて評価し,合併履行義務が時間の経過とともに履行されるか,ある時点で履行されるかを決定する。その会社は各契約で決定された請求書スケジュールに基づいて顧客から支払いを受ける。前払い金及び費用は、当社が当該等の手配の義務を履行するまで、受領又は満了時に繰延収入と記す。貸借対照表の後日12カ月以内に収入と確認された金額は,付随する総合貸借対照表で繰延収入の当期部分に分類される予定である。貸借対照表後12カ月以内に収入が確認されなかった金額は繰延収入に分類され、当期分を差し引く。会社の対価格権利が無条件である場合、金額は売掛金として記録される。収入が確認されていますが領収書を受け取っていないか発行されていない金額は通常契約資産として確認されます。
許可を独占する·手配において付与された許可が、手配において決定された他の約束または義務とは異なると判定された場合、一般に、研究開発サービスが含まれ、許可が顧客に譲渡され、顧客が許可を使用して利益を得ることができる場合、会社は、許可に割り当てられた払戻不可能な前払い費用の収入を確認する。許可が他の約束と異なるかどうかを評価する際に、会社は、パートナーの研究開発能力および関連する専門知識の一般市場での利用可能性を含む、各手配に関する事実および状況を考慮する。さらに、会社は、パートナーが残りのコミットメントを受信せずにライセンスからその予期される目的から利益を得ることができるかどうか、ライセンスの価値が実行されていない約束に依存するかどうか、他のプロバイダが残りのコミットメントを提供することができるかどうか、および残りのコミットメントから分離して識別することができるかどうかを考慮するだろう。他の承諾と組み合わせたライセンスについて、会社は、合併履行義務が時間の経過とともに履行されるか、ある時点で履行されるかを決定するために、判断を利用して、合併履行義務の性質を評価し、時間が経過するにつれて、収入を確認するための進展を測定するための適切な方法を決定する。当社は報告期間ごとに進捗指標を評価し、必要に応じて推定された余剰研究開発コストに基づいて業績指標と関連収入を調整して確認します。余剰推定研究開発コスト総額の計算には、内部従業員の仕事、材料コスト、および第三者契約コストに関する予測コスト、およびこれらの活動の仮定時間および持続時間が含まれる。連携スケジュールにより収入が管理職によるこれらの判断や見積りの影響を受けることを確認し,これらの仮定の変化に敏感である.したがって、進捗の測定、及び収入を確認すべき期間は、経営陣の推定に依存し、手配の過程で変化する可能性がある。
研究と開発サービス−会社の協力およびライセンス契約における約束は、一般に、パートナーを代表して会社によって実行される研究および開発サービスを含む。研究·開発サービスを含む業績義務については,会社は通常,このような業績義務に割り当てられた収入を適切な進捗評価基準に基づいて確認している。当社は判断を用いて収入を確認するための進捗を測る適切な方法を決定しており、これは通常、発生したコストのような投入措置である。当社は上記独占ライセンスの規定に基づき、報告期間ごとに進捗状況を評価する。パートナーからの精算は、共同開発活動のような顧客関係ではなく、パートナーとの協力関係によるものであり、研究や開発費用の減少として記録されることが多い。
顧客オプション−当社の手配は、例えば、手配開始時または所定のオプション期間内に目標を許可する権利など、いくつかのオプション購入の権利を協力者に提供することができる。これらの合意によれば、費用は、手配開始時に前払い費用または支払いとして当社に支払われるか、または許可証の選択権を行使する際に支払われることができる。1つのスケジュールが、顧客が追加の商品またはサービスを取得することを可能にする顧客選択権を含むと判定された場合、顧客選択権の基礎となる商品およびサービスは、手配開始時に契約義務とみなされない。同社は、顧客の物質的権利選択を評価するか、または追加商品またはサービスの選択を無料または割引して得る。顧客選択権が実質的な権利を表すと決定された場合、実質的な権利は、手配の開始時に別個の履行義務として確認される。当社は、決定された割引と顧客がオプションを行使する可能性に基づいて決定された相対独立販売価格に基づいて材料権利に取引価格を割り当てる。重大な権利に割り当てられた金額は,オプション行使または満期まで収入として確認されない.
一里塚払い-いくつかのイベントに基づくマイルストーン支払いを含む各スケジュールの開始時に、会社は、マイルストーンが実現可能であると考えられるかどうかを評価し、金額を推定する
F-14
最も可能な金額法を用いた取引価格に含まれる。大きな収入逆転が起こらない可能性が高い場合、関連するマイルストーン価値は取引価格に含まれるだろう。規制承認のような会社または被許可者の統制範囲内にない記念碑的支払いは、これらの承認を受ける前に実現可能であるとは考えられない。同社の評価の要因には、この評価における特定のマイルストーンを達成するために、科学、臨床、規制、商業、および他の克服しなければならないリスクが含まれている。重大な収入逆転が発生しない可能性があるかどうかを決定する際には,かなりの判断が必要である。その後の各報告期間が終了した時点で、当社はすべての制限されたマイルストーンの実現確率を再評価し、必要に応じて全体の取引価格の推定を調整する。いずれの調整も累積追跡をもとに記録されており,調整期間中の収入や収益に影響を与える.一里塚やその他の可変対価格が特に会社が単一の業績義務を履行する努力や業績義務を履行する特定の結果に関連すれば、重大な収入逆転が発生しない可能性が高い場合、会社は通常、マイルストーン金額をすべてその業績義務に分配する。
印税販売ベースの特許使用料を含む予定については、販売レベルに基づくマイルストーン支払いを含み、ライセンスは、特許使用料に関連する主要項目とみなされ、会社は、(I)関連販売が発生した場合、または(Ii)特許使用料の一部または全部が割り当てられた履行義務が履行された(または部分的に履行された)ときに収入を確認する。現在まで、同社はそのいかなる許可手配によって生じたいかなる特許権使用料収入も確認していない。
協力収入-当社はASC 808の範囲内にあるかどうかを評価するために協力スケジュールを分析した協力的な計画やASC 808は、このようなスケジュールが、そのような活動の商業的成功に依存する活動の積極的な参加者であり、重大なリスクおよびリターンに直面する締約国による共同経営活動に関連するかどうかを決定するために使用される。この評価は手配された全ライフサイクル内で手配当事者の責任の変化に基づいて行われる。複数の要素を含むASC 808の範囲内の協調配置の場合、会社は、最初に、協働するどの要素がASC 808の範囲内にあるとみなされ、どの要素がプロバイダ−顧客関係をより反映することができ、したがって、主題606の範囲内にあると判断する。ASC 808に従って示される協調スケジュールの要素の場合、適切な識別方法は、一般に、トピック606をクラス比することによって一貫して決定され、適用される。特別テーマ606に基づいて説明された手配要素については、当社は上記5ステップモデルを適用する。
許可や連携手配に関するコスト
協力手配の一部として取得した技術ライセンスに関するコストは発生時に費用を計上し,通常は総合経営報告書における研究·開発費用に計上される。
売掛金
会社は顧客の財務状況の評価に基づいて顧客に信用を発行する。標準条項によると、金額が満期になった場合、会社はすべての請求書の売掛金を記録します。売掛金は控除適用の即時賃金割引とその他の契約調整及び不良債権準備後の未収金額に記載されています。当社は、売掛金の超過時間の長さ、顧客の債務返済能力、および全体の経済と業界全体の状況を考慮して、融資準備が必要かどうかを評価するために、複数の要素を考慮します。会社が売掛金が回収できないと判断した場合、会社は再発行する。全体的に言えば、同社と顧客の間に重大な入金問題はありません.
株に基づく報酬
当社は、従業員、取締役、非従業員に付与された公正な価値に基づいて、従業員、取締役、非従業員に付与されたすべての株式報酬を会計処理し、必要なサービス期間内にこれらの報酬の補償費用を確認し、これは通常、対応する報酬の帰属期間である。株式ベースの支払いには、帰属された普通株を含む株式オプションと普通株の贈与が含まれる。株式報酬の計量日は付与日であり、株式の報酬コストに基づいて直線原則で必要なサービス期間(すなわち帰属期間)の費用として確認される。当社は業績帰属条件付き株式オプションと制限株を発行しており、業績条件に達する可能性があると結論した場合にはこれらの奨励金を記録している。株式に基づく補償は、添付の総合業務報告書および総合損失報告書において、関連サービスを提供する機能に基づいて分類される。同社は一部の奨励金が付与された株式ベースの報酬支出を確認した。没収は発生状況に応じて計算されます。
F-15
各株式オプション付与の公正価値は、付与された日にブラック·スコアーズオプション定価モデルを使用して推定され、このモデルは、予想される株価変動、オプションの予想期限、オプションの予想期限に近い期間の無リスク金利、および会社の期待配当収益率を含むいくつかの主観的仮定に基づく投入を必要とする。初公募まで、当社の普通株は活発な市場がなかったため、当社は当時の事実と状況から付与日普通株の公正価値を推定した。上場企業になると、関連普通株の公正価値は会社株の付与日の終値に等しい。当社は2020年に初めて公募したため、当社は十分な期間の特定会社の歴史と隠れた変動率資料が不足している。そこで、上場企業グループの履歴変動率に基づいて予想される株価変動率を推定し、自身の取引株価変動性に関する十分な履歴データを有するまで継続する予定である。当社の株式オプションの期待期限は、“簡略化”方法で決定され、“通常”オプション資格に適合する株を奨励するために使用されます。非従業員に付与された株式オプションの期待期限は、オプション付与の契約期間に等しい。無リスク金利は、奨励付与時に発効した米国債収益率曲線を参考にして決定され、期間は奨励の予想期限にほぼ等しい。予想配当収益率は、当社が現金配当金を支払ったことがないことに基づいており、予測可能な未来には現金配当金は一切支払われないと予想される。各制限された普通株奨励の公正価値は、付与された日に会社普通株の同一日の公正価値に基づいて推定される。
従業員株式購入計画下での割引購入の補償費用は,Black-Scholesモデルを用いて回顧準備金と購入割引の公正価値を計算し,要約期間の補償費用であることを確認した.
所得税
当社はFASB会計基準に基づいて740のテーマを符号化して所得税を記録した所得税、または繰延税金は、貸借対照法を使用して規定されるASC 740。この方法によれば、繰延所得税資産および負債は、既存資産および負債の財務諸表の帳簿金額とそれぞれの所得税ベースとの間の差によって生じる将来の所得税結果に基づいて確認される。繰延所得税資産と負債は,制定された所得税税率計量を用いており,これらの仮差額の回収または決済が予想される年度の課税所得額に適用される予定である。所得税税率変動が繰延所得税資産や負債に及ぼす影響は、将来実現不可能な任意の所得税割引の推定準備期間中に収入または費用であることが確認される。
当社は不確定な税務状況に関連して各税務機関に支払うことが可能な税金のための準備金を保留しています。記録された税収割引は、当社がその税務申告または立場で得た税収割引が、税収割引に関連する任意の不確実性が解決された後に“より可能性がある”かどうかの決定に基づいており、関連問題が税務機関によって提起されると仮定する。
表外リスクと信用リスク集中
当社には外国為替契約、オプション契約、その他の海外ヘッジ手配などの重大な表外リスクはありません。会社を集中的な信用リスクに直面させる可能性のある金融商品には、主に現金、現金等価物、および限定的な現金が含まれる。会社の現金、現金等価物、および限定現金は大型金融機関の口座に保管されている。当社は、現金、現金等価物、制限された現金を持つ預金機関の財務力により、当社は重大な信用リスクに直面しないと信じている。同社は通貨市場基金に現金等価物の一部を保有しており、これらの基金は米国債と米国機関債務に投資している。現金等価物は、個人米国債、米国政府機関証券、商業手形を含む会社証券にも投資されている。会社の有価証券には主に社債、米国機関債券、米国債が含まれており、会社を集中的な信用リスクに直面させる可能性がある。会社はどの投資タイプにも投資できる金額を制限する投資政策をとっている。当社はいかなる信用損失も経験しておらず、そのような基金に重大な信用リスクがあるとも信じていない。
総合損失
総合損失には,純損失,有価証券の未実現収益と損失,株主との取引や経済事件以外の取引や経済事件による他の株主権益変動(損失)がある。
F-16
1株当たり純損失
会社が参加証券の定義に合った株を発行する場合、会社は2段階法を用いて普通株株主が1株当たりの基本と希薄純収益(損失)を計算すべきである。2級法は、発表または累積された配当金および未分配収益の参加権に基づいて、各種類の普通株および参株証券の1株当たり純収益(損失)を決定する。二段階法は、普通株式株主がその期間に獲得可能な収益(損失)が、その期間のすべての収益(損失)が分配されたように、彼らがそれぞれ収益を共有する権利に基づいて普通株と参株証券との間に分配することを要求する。会社の転換可能な優先株参加会社が発表した任意の配当金であるため、参加証券とみなされている。参加した証券は当社の損失を負担する必要がないため、赤字期間中は、2段階法による分配を行う必要はない。
普通株株主が1株当たりの基本純収益(損失)を占めるべき計算方法は、普通株株主は純収益(損失)を当期に発行された普通株の加重平均株式数で割るべきであり、その中に所与の名義行権価格の事前出資承認株式証を含むそれは.普通株株主は希釈純収益(損失)を占めるべきであり、普通株株主の1株当たり純収益(損失)を調整して、希釈証券の潜在的な影響に基づいて未分配収益を再分配することによって計算される。普通株株主が希釈純収益(損失)を占めるべき計算方法は:普通株株主は希釈純収益(損失)を当期発行普通株の加重平均で割るべきであり、潜在的な希釈性普通株を含む。この計算では、普通株を購入した未償還オプション、未付与制限株式奨励、転換可能優先株の株式は潜在的な希釈性普通株とみなされる。当社は記載されているすべての期間に純損失を記録しているため、普通株株主が占めるべき1株当たりの基本的および償却純損失は、潜在的な希薄化証券に組み入れられた1株当たりの損失と同じであり、赤字である。
付記3.公正価値計量
以下の表は、同社が公正価値で常時計測している金融資産の情報を示し、2023年12月31日および2022年までの公正価値を決定するための公正価値レベル(千単位)を示している
|
|
公正価値に応じて計量する |
|
|||||||||||||
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
|
合計する |
|
||||
資産: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
現金等価物 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
アメリカ国債 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
商業手形 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
流通有価証券 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
アメリカ政府機関は |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
社債 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
非流通有価証券 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
アメリカ政府機関は |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
社債 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
制限現金 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
合計する |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|||
F-17
|
|
公正価値に応じて計量する |
|
|||||||||||||
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
|
合計する |
|
||||
資産: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
現金等価物 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
流通有価証券 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
アメリカ政府機関は |
|
|
— |
|
|
|
|
|
|
|
|
|
|
|||
社債 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
非流通有価証券 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
アメリカ政府機関は |
|
|
— |
|
|
|
|
|
|
|
|
|
|
|||
社債 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
制限現金 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
合計する |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|||
付記4.有価証券
次の表は、2023年12月31日と2022年12月31日に保有する売却可能な債務証券(単位:千)をまとめている
説明する |
|
償却する |
|
|
実現していない |
|
|
実現していない |
|
|
公平である |
|
||||
2023年12月31日 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
アメリカ政府機関証券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
会社証券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
合計する |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
説明する |
|
償却する |
|
|
実現していない |
|
|
実現していない |
|
|
公平である |
|
||||
2022年12月31日 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
アメリカ政府機関証券 |
|
|
|
|
|
|
|
|
( |
) |
|
$ |
|
|||
会社証券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
合計する |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
2023年12月31日現在、会社は
2023年12月31日までに会社は
当社は売却可能な証券の公正価値が償却コストより低い基礎まで低下したことが信用関連要素によるものであるかどうかを確定しなければならない。各報告日において、会社は、未実現の損失が信用損失によるものであるか否かを決定するために減価評価を行う。損害は個人の安全レベルで評価される。信用損失或いはその他の要素による損失の有無を決定する時、考慮する要素は会社が投資を持ってその余剰コスト基礎を回収するまでの意図と能力、公正価値が余剰コスト基礎より低い程度、公正価値がコスト基礎より低い時間の長さと程度、財務状況を含む
F-18
発行者の責任、発行者がこれまで計画通りに利息或いは元金を支払うことができなかったこと、格付け機関の証券格付けに対するいかなる変動、発行者又は発行者の属する業界に影響を与える不利な法律或いは監督管理事件、及び任意の経済状況の重大な悪化。
前表に示した売却可能証券の未実現損失は簡明総合経営報告書で確認されていないが、これらの証券は高信用品質の投資級証券であるため、会社は売却しようとせず、予想回復前に売却することも要求されず、公正価値の低下は信用損失以外の要因に起因することができる。その評価によると、その会社はそれを確定した
注5.連携
セノフィが協力して
協議条項
2020年7月7日、同社はセノフィと、2つの生物標的に対する候補薬物を共同開発する協力協定を達成した。セノフィ協定によると、同社は、IRAK 4またはCollaboration Target 1の協力のために生成されたいくつかの先導化合物の開発、製造、商業化を許可し、未開示の使用分野またはCollaboration Target 2のもう1つの不開示の目標をセノフィに付与した。指定されたマイルストーンに達した後にのみ、Collaboration Targetごとにこのような許可を行使することができる。IRAK 4に対する化合物の使用領域は、腫瘍学および免疫腫瘍学を含まず、診断、治療、治癒、緩和または予防のいずれかの疾患、障害または状態を含む。
セノフェイプロトコルによると、同社は発見と臨床前研究を担当し、IRAK 4および最大3つの予備分解剤に対して少なくとも1つの分解剤の第1段階臨床試験を行った。この2つの目標に対して,セノフィは協力候補製品ごとに特定の開発マイルストーンが出現した後,候補製品の開発,製造,商業化を担当している。
また、セノフィ協定によれば、セノフィは、(I)適用分野におけるこのような目標に対する協力製品の米国での開発コストに対して50%の資金を提供する権利(I)と、米国の使用分野におけるこのような目標に適用される協力製品の商業化のための純利益と純損失とを二分する権利を含む独占選択権または選択権を当社に付与する。また、当社が選択権を行使すれば、セノフィは当社に協力目標ごとに適用される独占選択権を付与し、一度行使すれば、当社が米国で何らかの共同普及活動を行うことを許可します。
セノフィ協定が早期に終了しない限り、この協定は、セノフィ協定の下で製品に関連するすべての支払い義務が満了した日から製品ごとに満了する。当社またはセノフィは、他方で重大な違約または資金を相殺しないか、またはいくつかの特許挑戦のために合意を終了することができます。さらに、セノフィは、便宜上、セノフィプロトコルを終了すること、または重大なセキュリティイベントのためにセノフィプロトコルを終了することを書面で通知することができ、サイノフェイが特定の目標に対する協力候補の開発、商業化、または製造責任を負う場合、セノフィが特定の期間内にその目標のための任意の協力候補の使用を停止する場合、会社は任意の協力候補とのセノフィプロトコルを終了することができる。
セノフィ協定によりセノフィ独占ライセンスが付与された代償として、セノフィは同社に#ドルを前払いした
F-19
2022年11月15日、我々は、元のセノフィ協定に規定されているいくつかの研究条項および責任を修正するために、元のセノフィ協定を修正する改訂された協力および許可協定、または修正されたセノフィ協定をセノフィと締結した。修正されたセノフィ協定は,協力条項に要求される第2段階試験の時間と数も詳細に説明した。改訂されたセノフィ協定は2022年12月5日に発効する。
また,セノフィについては,セノフェイは2022年12月2日に同社に書面通知を行い,連携目標1候補薬KT−474を第二段階臨床試験に進める意向を示した。2023年第4四半期に会社は2つのマイルストーンを実現しました
2023年9月、同社とセノフィは共同で、連携目標2に関する活動を停止することに同意した。
会計処理
同社は発見と臨床前研究活動およびセノフィプロトコルでの独占許可付与を分析し,この手配はサプライヤーと顧客の関係を示し,ASC 606に計上すると結論した。
同社は,合意により,(1)連携目標1の研究サービス,(2)連携目標1の研究ライセンス,(3)連携目標1の独占ライセンス,(4)連携目標2の研究サービス,(5)連携目標2の研究ライセンス,(6)連携目標2の独占ライセンス,(7)研究期間延長の選択肢,および(8)開発期間のオプション研究サービスを決定した。
同社は連携目標1と2が互いに異なることを決定した。目標ごとに分解剤に関する研究は異なる段階にあり,開発活動が成功すれば許可分野も異なる。したがって、各目標に関連するすべての約束は、他の目標に関連する約束とは異なると考えられる。
各連携目標の研究開発サービスは、研究許可証や独占ライセンスと区別なく決定され、連携目標ごとの単一業績義務に統合されている。すなわち,Collaboration Target 1の総合研究サービス,研究許可と独占許可,Collaboration Target 2の総合研究サービス,研究許可,独占許可の2つの義務が決定された.個々の目標の独占許可はセノフィプロトコルでの臨床前と臨床研究開発サービスと変わらず,主に研究の高度な専門性とタンパク質分解物の開発に関わる新技術である臨床前活動と研究および第1段階臨床試験が他方で必要な方法で行うことができないためである。
研究期間延長の選択と発展期間のオプション研究サービスを実質的な権利として評価した。各オプションに関連する費用は独立販売価格以上です。したがって,標的オプションは履行義務ではなく,入札オプションが行使されるまで,個々のオプションに関する費用は取引価格に含まれない.
同社が確定した取引の総価格は$
会社が相対的に独立した販売価格に基づいて前払いを各履行義務に割り当てることは以下の通りです
F-20
会社はドルの配分を決めた
同社は個々の業績義務に関する収入を確認しており,研究·開発サービスは入力法を用いて提供されているため,個々のプロジェクトの研究·開発活動に関するコストと,将来その単独業績義務を履行するために予想されるコストに基づいて確認されている。統制権の移譲はこの時期に発生し、経営陣から見れば、各履行義務の履行に進展した最良の測定基準である。受け取った収入が確認されていない金額は、会社総合貸借対照表上の契約負債として繰延され、履行義務が履行されるまで、残りの研究開発期間中に確認される。取引価格に加えられたマイルストーンと精算対価格は収入として確認され,制限されない場合には累積追跡が行われる.2023年12月31日現在、連携目標1に関する業績義務は完全に履行されていない。2023年12月31日現在、連携目標2に関する業績義務は完全に履行されている当社は2023年12月31日までに年度内に確認します
業績マイルストーンに関する任意の追加対価格は、逆転可能なリスク解消時に確認され、会社はそれに応じて合意で決定された取引価格に調整し、累積追跡に基づいて収入を確認し、改訂された手配対価格を契約義務に再分配しなければならない。販売マイルストーン支払いおよび特許権使用料に関連する任意の対価格は、関連マイルストーン事件または販売発生時に確認されるので、関連販売発生または関連履行義務が履行されたときに確認される。規制マイルストーンの評価の一部として,研究や開発マイルストーンの実現は基本研究や開発活動の結果に依存することを含む多くの要因が考えられているため,会社の制御範囲内ではない.2023年第4四半期、会社は2つの発展マイルストーンを実現したKT−474第2段階臨床試験における第1および第2の適応における第1の患者の用量に関連する。これらのマイルストーンに関連して会社は制限なく$を投入しています
頂点合意
2019年5月9日(“発効日”)に,当社はVertexと連携プロトコル(“Vertexプロトコル”)を締結し,小分子タンパク質分解剤の最大6つの目標を達成することを推進した。Vertexプロトコルによると,Vertexはターゲット開発を指定するための候補製品の独自選択権を持ち,Vertexは開発と商業化を制御する.Vertexプロトコルによると,会社は目標の発見と臨床前研究のみを担当しており,Vertexはその許可選択権を行使した後に候補製品の開発,製造,商業化を担当している。協力の初期研究期間は4年(
その会社は最高$を取得する資格がある
F-21
“頂点合意”の期限は発効日から2023年5月9日に初期研究期限が終了した時点で満了する。
会計処理
同社はVertexプロトコルに要求される共同研究活動を分析し,サプライヤーと顧客の関係を示し,ASC 606項目に計上すると結論した。
同社は、(1)非独占的で印税免除の研究ライセンス、(2)最大6つの目標で行われる研究および開発サービス、および(3)各目標の開発、製造、および商業化努力のために各目標を許可することができるという重大な約束を決定した。研究·開発サービスは,研究·開発ライセンスと区別なく,単一の履行義務に統合されていることが決定された。当社は、将来のライセンス対象のオプションは割引価格で定価されているのではなく、各標的のオプション執行費は現段階で開発段階で検討されている独立販売価格以上であることを確認している。そのため、オプションと入札ライセンスは履行義務から除外され、入札オプションが行使される前に、オプション行権料は取引価格から除外される。
研究開発制限マイルストーンの評価の一部として、当社は研究開発マイルストーンの実現が基本的な研究開発活動の結果に依存することを含む多くの要因を考慮しているため、当社の制御範囲内ではない。
手配開始時には、2つの会計単位、すなわち
同社は,個々のプロジェクトの研究·開発活動に関するコストと,将来業績義務を履行するために予想されるコストに基づいて,業績義務に関する収入を確認し,研究·開発サービスは入力法を用いて提供されているためである。統制権の移譲はこの時期に発生し、経営陣から見れば、履行義務の履行に進展する最良の測定基準である。Vertex連携プロトコルは2023年5月の初期研究期間終了後に満了する.そこで,会社はその履行義務を完全に履行し,2023年5月にVertex連携に関するすべての余剰繰延収入を確認した。D.D当社は、2023年、2022年および2021年12月31日までの年度内に確認します
次の表に2023年12月31日までの年間売掛金,契約資産,負債の変化(単位:千)を示す
|
|
残高は |
|
|
足し算 |
|
|
控除額 |
|
|
残高は |
|
||||
売掛金と契約資産: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
売掛金-セノフィ |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
未開売掛金-セノフィ |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
売掛金と契約資産総額 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
契約責任: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
繰延収入-頂点 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
繰延収入-セノフィ |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
契約総負債 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
F-22
付記6.財産と設備
財産·設備には、2023年12月31日と2022年12月31日まで、以下の内容が含まれる(単位:千)
|
|
2023年12月31日 |
|
|
2022年12月31日 |
|
||
財務使用権資産項目の下の実験室及び事務装置 |
|
$ |
|
|
$ |
|
||
実験室装置 |
|
|
|
|
|
|
||
コンピュータ装置 |
|
|
|
|
|
|
||
家具と固定装置 |
|
|
|
|
|
|
||
賃借権改善 |
|
|
|
|
|
|
||
まだ使用していない資産 |
|
|
|
|
|
|
||
総資産と設備 |
|
|
|
|
|
|
||
減価償却累計を差し引く |
|
|
( |
) |
|
|
( |
) |
財産と設備、純額 |
|
$ |
|
|
$ |
|
||
2023年12月31日現在、2022年と2021年12月31日までの減価償却支出は$
不動産と設備には融資リース項目の下の実験室と事務設備使用権資産が含まれており、コストベースは#ドルである
使用権資産に関する償却費用s $
注7.賃貸借契約
2019年10月、当社は撤回不可施設賃貸協定(“2019年レンタル”)を締結しました
2000年12月に当社は賃貸借契約を取り消すことができないことを締結した
2021年の賃貸借契約は、所有者が会社の不動産を建設する前に基地ビルを建設することを要求する。当社は、会計開始日が所有者が基地建築の拡張を完了し、当社に制御権を移管した場合、その日付は2023年1月初めに発生したと考えている。当社は会計開始日に2021年の賃貸借契約の分類を評価し,その賃貸借を運営賃貸として入金すべきであると考えている。同社は#ドルの経営リース負債を記録した
F-23
会社の結論は,大家が支払うテナント改善手当に関する改善はテナント資産であるため,#ドルを記録した
当社の融資リース義務には、資本リースによる融資のある財産と設備が含まれています。
2023年、2023年、2022年12月31日終了年度のレンタル費用構成要素(単位:千):
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
リースコストを経営する |
|
$ |
|
|
$ |
|
||
融資リースコスト: |
|
|
|
|
|
|
||
使用権資産の償却·融資 |
|
|
|
|
|
|
||
融資リース負債利息支出 |
|
|
|
|
|
|
||
可変リースコスト |
|
|
|
|
|
|
||
総賃貸コスト |
|
$ |
|
|
$ |
|
||
同社の2023年12月31日までと2022年12月31日までの賃貸契約に関する補足キャッシュフロー情報は以下の通り(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
計量に含まれる金額のための現金 |
|
|
|
|
|
|
||
経営的リース使用の経営的キャッシュフロー |
|
$ |
|
|
$ |
|
||
融資リースのための経営的キャッシュフロー |
|
$ |
|
|
$ |
|
||
融資リースのための融資キャッシュフロー |
|
$ |
|
|
$ |
|
||
2023年12月31日と2022年12月31日までの加重平均残存賃貸期間と割引率は以下の通り
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
残りのレンタル期間: |
|
|
|
|
|
|
||
レンタルを経営する |
|
|
|
|
||||
融資リース |
|
|
|
|
||||
割引率: |
|
|
|
|
|
|
||
レンタルを経営する |
|
|
% |
|
|
% |
||
融資リース |
|
|
% |
|
|
% |
||
F-24
2023年12月31日現在、賃貸と融資リースを経営する未割引未来賃貸支払いは以下の通り(千計)
財政年度 |
|
運営中です |
|
|
融資する |
|
||
2024 |
|
|
|
|
|
|
||
2025 |
|
|
|
|
|
|
||
2026 |
|
|
|
|
|
|
||
2027 |
|
|
|
|
|
|
||
2028 |
|
|
|
|
|
|
||
その後… |
|
|
|
|
|
|
||
最低賃貸支払総額 |
|
|
|
|
|
|
||
利子または推定利息に相当する額を差し引く |
|
|
( |
) |
|
|
( |
) |
賃貸負債現在価値 |
|
$ |
|
|
$ |
|
||
2024年の未割引の将来のレンタル支払いには約#ドルが含まれています
付記8.課税料金
料金には、2023年12月31日と2022年12月31日まで、以下の内容が含まれています
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
研究開発費 |
|
$ |
|
|
$ |
|
||
給与明細と給与明細との関連 |
|
|
|
|
|
|
||
専門費 |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
費用を計算する |
|
$ |
|
|
$ |
|
||
注9.その他の引受金及びその他の事項
法律訴訟
正常業務過程において、会社が経営する業界は特許法律要求の影響を受けやすいため、会社は法的訴訟、クレーム、訴訟の影響を受ける可能性がある。法律訴訟やクレームの推定損失が可能かつ推定可能な場合、当社はこのような損失を会計処理します。これらの事項に関連する法的費用は発生時に費用を計上する。当社は現在いかなる法的手続きの当事者でもありません。
弁済手配
デラウェア州の法律で許可されている場合、当社は協定を締結し、賠償を受けている側がその要求に応じてこのような身分でサービスを提供している場合、ある事件或いは事件についてその投資家、従業員、高級管理者と取締役(総称して“賠償を受ける側”)に賠償を行う。賠償期限は被賠償当事者の終身である。同社はこれらの賠償協定の推定公正価値はわずかだと考えている。当社は正常業務中に標準賠償協定を締結しました。これらの合意に基づいて、当社は、損害を受けないように、損害を受けた当事者(通常は当社の業務パートナーまたは顧客)に対して、任意の米国特許または任意の第三者が自社製品に対して提出した任意の著作権または他の知的財産権侵害クレームによって損害または発生した損失を賠償し、損害を受けないように賠償することに同意する。これらの賠償協定によると、会社が将来支払うことを要求される可能性のある最大潜在金額は無限だ。その会社は賠償手配のクレームがあることを知りませんでした。そしてそれはすでに
F-25
注10.株式ベースの報酬
2018年株式オプションと付与計画
2018年11月、取締役会または取締役会のいずれかのグループ委員会は、取締役会または取締役会の任意のグループ委員会が、会社の従業員、高級管理者、取締役および独立請負者に株式オプションおよびその他の株式ベースの奨励を適宜付与することができる2018年株式オプションおよび付与計画(“2018計画”)を採択した。2018年の計画によると、これ以上いかなる贈与も提供されません。しかし、2018年には、この計画に基づいて付与された未償還持分奨励金の管理を継続する予定だ。2018年計画に基づいて付与された未行使株式購入権が取り消され、没収され、または他の方法で行使されず、他の方法で2018年計画下の株式備蓄に返却される限り、当該等の奨励関連株式の数は、将来的に2020年の株式購入及び奨励計画に基づいて付与されることができる。
2020年株式オプションとインセンティブ計画
2020年8月、会社とその株主は“2020年株式オプションとインセンティブ計画”(略称“2020年計画”)を承認し、2020年8月20日から発効した。2020計画は2018計画に取って代わった。会社の取締役会が会社の初公募終了後に2018計画に基づいて追加奨励を行わないことを決定したからだ。2020年には、会社がその高級管理者、従業員、役員、コンサルタントに対して株式と現金に基づく奨励を行うことを許可する計画だ。その会社はすでに初歩的に保留した
2020年従業員株購入計画
2020年8月、会社とその株主は“2020年従業員株購入計画”を承認し、2020年8月20日から発効した。2020年にESPPの予備保留と承認発行は最大合計
株式オプション
2023年12月31日までの1年間において、2020計画下の株式オプション活動の概要は以下の通りである(株と1株当たりのデータを除いて、千計)
|
|
量 |
|
|
重みをつける |
|
|
重みをつける |
|
|
骨材 |
|
||||
2022年12月31日に返済されていません |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
授与する |
|
|
|
|
|
|
|
|
|
|
|
|
||||
鍛えられた |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
没収される |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
2023年12月31日現在の未返済債務 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2023年12月31日に行使できます |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2023年,2022年及び2021年12月31日までに年度内に行使される株式オプションの内在的価値s $
F-26
2023年,2022年および2021年12月31日までの年度授権の加重平均公正価値は$
2023年12月31日現在、株式オプションに帰属していない未確認株式報酬支出総額は$
次の表は,本年度までの株式オプションの持分報酬支出について概説した2023年12月31日、2022年、2021年:
|
|
十二月三十一日までの年度 |
|||||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|
|||
研究開発 |
|
$ |
|
|
$ |
|
|
$ |
|
|
|||
一般と行政 |
|
|
|
|
|
|
|
|
|
|
|||
株式に基づく報酬総額 |
|
$ |
|
|
$ |
|
|
$ |
|
|
|||
会社がBlack-Scholesオプション定価モデルで使用している加重平均仮定は、従業員および非従業員の年間までの株式オプションの付与日公正価値を決定するために使用される2023年12月31日、2022年、2021年:
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|||
予想期限(年単位) |
|
|
|
|
|
|
|
|
||||
波動率 |
|
|
% |
|
|
% |
|
|
% |
|||
無リスク金利 |
|
|
% |
|
|
% |
|
|
% |
|||
配当率 |
|
|
% |
|
|
% |
|
|
% |
|||
限定株単位
当社はサービスベースと業績に基づく帰属条件に制限株式単位の株式を付与しています
|
|
量 |
|
|
授与日 |
|
||
2022年12月31日に帰属していない |
|
|
|
|
$ |
|
||
授与する |
|
|
|
|
|
|
||
既得 |
|
|
( |
) |
|
|
|
|
没収される |
|
|
( |
) |
|
|
|
|
2023年12月31日に帰属していません |
|
|
|
|
$ |
|
||
その会社は授与した
2023年12月31日現在、未帰属制限株の未確認株式補償費用総額は$
2023年まで,2022年および2021年12月31日まで年度会社は限定株のために株式ベースの報酬費用#ドルを記録した
F-27
株式ベースの報酬費用
この年度までの年度内に,研究と発展及び従業員,役員及び非従業員の一般及び行政支出の権益報酬支出総額を計上する2023年12月31日、2022年12月31日、2021年12月31日の状況は以下の通り(単位:千)
|
|
12月31日までの年度 |
|||||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|
|||
研究開発 |
|
$ |
|
|
$ |
|
|
$ |
|
|
|||
一般と行政 |
|
|
|
|
|
|
|
|
|
|
|||
株式に基づく報酬総額 |
|
$ |
|
|
$ |
|
|
$ |
|
|
|||
付記11.関連者取引
付記5に記載の協力を除いて、当社は添付されている総合財務諸表に記載されている期間に関連先取引はないが、総合財務諸表付記ではこのような取引を他の方法で検討していない。
注12.所得税
同社は米国の運営子会社が実現した利益に関する所得税支出を記録している。2023年、2022年及び2021年12月31日までに、グループ純営業損失(“NOL”)及び全額推定額が計上されたため、無形所得税支出が入金された。
2023年12月31日、2022年、2021年12月31日までの年度、米国の法定所得税税率と会社の実際の税率の調整状況は以下の通り
|
|
十二月三十一日 |
||||||
|
|
2023 |
|
|
2022 |
|
2021 |
|
法定税率下の税収効力 |
|
% |
|
% |
% |
|||
州税 |
|
|
|
|
|
|||
株の報酬 |
|
( |
|
|
|
|
||
恒久的差異 |
|
|
|
|
|
|||
連邦研究開発信用 |
|
|
|
|
|
|||
他にも |
|
( |
|
|
( |
|
( |
|
評価免除額を変更する |
|
( |
|
|
( |
|
( |
|
合計する |
|
% |
|
% |
% |
|||
繰延所得税は、財務報告目的のための資産および負債の帳簿金額と所得税目的のための金額との間の一時的な差異の純税影響を反映する
|
|
十二月三十一日 |
|||||||
|
|
2023 |
|
|
2022 |
|
|
||
繰延税金資産: |
|
|
|
|
|
|
|
||
連邦純営業損失繰越 |
|
$ |
|
|
$ |
|
|
||
国有純営業損失を繰り越す |
|
|
|
|
|
|
|
||
研究開発信用繰り越し |
|
|
|
|
|
|
|
||
賃貸負債 |
|
|
|
|
|
|
|
||
収入を繰り越す |
|
|
|
|
|
|
|
||
課税項目及び準備金、株式その他 |
|
|
|
|
|
|
|
||
資本化研究と開発 |
|
|
|
|
|
|
|
||
繰延税金資産総額 |
|
$ |
|
|
$ |
|
|
||
推定免税額 |
|
$ |
( |
) |
|
$ |
( |
) |
|
繰延税金資産 |
|
$ |
|
|
$ |
|
|
||
固定資産と無形資産 |
|
|
( |
) |
|
|
( |
) |
|
使用権資産 |
|
|
( |
) |
|
|
( |
) |
|
繰延税項目純資産 |
|
$ |
|
|
$ |
|
|
||
F-28
会社は設立以来営業赤字による所得税支出はありません。証拠の成分に基づいて、繰延税金資産の一部または全部が現金化されない可能性が高い場合、ASC 740は、報告された繰延税金資産を低減するために推定値を計上する必要がある。当社はすでにその繰延税金資産の現金化に影響するプラスと負の証拠を評価した。これに基づき、当社は繰延税項純資産の全額計上について試算しており、繰延税金資産が現金化される可能性は低いためだ。2023年の間、推定手当は#ドル増加した
2022年から減税·雇用法案(TCJA)が第174条を改正し,現在米国と非米国での研究·実験(R&E)支出をそれぞれ5年または15年以内に資本化·償却することが求められており,これらの支出は2021年12月31日以降の納税年度に支払われている。TCJA改正案の前に、第174条は、納税者が直ちに支払いまたは発生した年間にR&E支出を差し引くことを許可する。同社は2022年から会計方法をこの必要な変更を行っており、計算方法は将来の米国国税局の指導の下で調整される可能性がある。
2023年12月31日現在、同社は約
2023年12月31日現在、同社は連邦と州の研究開発信用繰越および孤児薬物信用繰越約$を持っている
同社は、財務諸表において不確定な税収状況の税収利益をどのように確認、計量、記録するか、不確定税収事項の特定の開示を要求すること、不確定税収状況を規定する準備金を貸借対照表上でどのように分類すべきか、過渡期および中期指導、および他の規定を提供することを規定するASC 740-10“所得税における不確実性会計”の規定に従う。当社は、2023年12月31日と2022年12月31日現在、未確認の税収割引に関する税収準備金を記録していません。同社の政策は、その損益表において、いかなる不確定な税収状況から生じる利息及び罰金を所得税費用の構成要素(あれば)として確認することである。2023年12月31日と2022年12月31日まで、会社は
12月31日までの連邦およびマサチューセッツ州の所得税申告表
注13.1株当たり純損失
1株当たり純損失
1株当たり基本損失と希釈損失の算出方法は、純損失を当期に発行された加重平均普通株で割ったものであり、その名義行使価格を与えた事前出資の引受権証(千で計算するが、株式と1株当たりデータを除く)を含む
|
|
十二月三十一日 |
|||||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|
|||
分子: |
|
|
|
|
|
|
|
|
|
|
|||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
分母: |
|
|
|
|
|
|
|
|
|
|
|||
加重平均は普通株式を発行し、基本株は |
|
|
|
|
|
|
|
|
|
|
|||
1株当たり基本と希釈して純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
F-29
当社の潜在的希薄化証券は、限定株と株式オプションを含み、1株当たり純損失の計算から除外されており、1株当たり純損失を減少させるためである。したがって、普通株株主が基本純損失と希釈後の1株当たり純損失を占めるべき加重平均既発行普通株数は同じである
|
|
十二月三十一日 |
|||||||||||
|
|
2023 |
|
|
2022 |
|
|
2021 |
|
|
|||
未帰属限定株 |
|
|
|
|
|
|
|
|
|
|
|||
普通株購入オプション |
|
|
|
|
|
|
|
|
|
|
|||
合計する |
|
|
|
|
|
|
|
|
|
|
|||
注14.後続イベント
2024年1月9日、同社は普通株式の後続発行を完了し、一部の投資家に普通株式の代わりにその普通株の株式を購入する予備融資承認株式証を提供した。会社が発行·販売する
2024年2月7日、Cowen and Company,LLCとの販売契約により、同社は完成した
F-30
項目9.Accoとの変更と分岐“会計と財務開示”誌。
ない。
第9条。制御するプログラムがあります
情報開示制御とプログラムの管理職の評価
我々は、“1934年証券取引法”(改正)又は“取引法”の下で第13 a-15(E)及び15 d-15(E)条の規則又は“取引法”で定義された“開示制御及び手続”を維持し、取引法に基づいて提出又は提出された報告書において開示を要求する情報を確保することを目的としている:(1)米国証券取引委員会規則及び表で指定された期間内に記録、処理、まとめ、報告される、(2)我々の主要幹部及び主要財務官を含めて、我々の主要幹部及び主要財務官を適宜伝達して、必要な開示に関する決定を直ちに行う。私たちの経営陣は、どんな制御やプログラムも、どんなに設計や操作が良くても、その目標を達成するために合理的な保証を提供するしかないことを認識しており、私たちの経営陣は、可能な制御とプログラムの費用対効果関係を評価する際にその判断を適用しなければならない。私たちの開示制御と手続きはその制御目標を達成するための合理的な保証を提供することを目的としている。
我々の経営陣は、CEO(CEOを務める)と財務責任者(CEOを務める)の参加の下、2023年12月31日現在、すなわち本年度報告Form 10-Kがカバーする期間が終了した場合の制御やプログラムの有効性を評価した。この評価に基づき、我々のCEOおよび最高財務官は、我々の開示制御および手続きがその日の合理的な保証レベルで有効であると結論した。
財務報告の内部統制
財務報告の内部統制に関する経営陣の報告
私たちの経営陣は財務報告書の十分な内部統制を確立して維持する責任がある。財務報告の内部統制は、取引法第13 a-15(F)及び15 d-15(F)条において、会社の主要執行者及び主要財務官又は同様の機能を果たす者が設計又はその監督の下で行われ、公認会計原則に基づいて財務報告の信頼性及び外部目的の財務諸表を作成するために合理的な保証を提供するために、会社の取締役会、管理層及びその他の人員によって実施されるプログラムとして定義されている
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。将来的にどのような有効性評価を行うかの予測には,条件の変化により制御が不十分になる可能性がある,あるいは政策やプログラムを遵守する程度が悪化する可能性があるというリスクがある.
最高経営責任者と最高財務責任者の監督と参加の下、我々の経営陣は、テレデビル委員会後援組織委員会が内部統制-総合枠組み(2013年の枠組み)で提出した基準に基づいて、2023年12月31日までの財務報告書の内部統制に対する有効性を評価した。この評価に基づき、経営陣は、財務報告書に対する内部統制は2023年12月31日から有効であると結論した。
私たちの独立公認会計士事務所は私たちの財務報告書内部統制の認証報告書を発行しました。この報告を以下に示す。
132
の報告独立公認会計士事務所
Kymera治療会社の株主と取締役会に。
財務報告の内部統制については
我々は、トレデビル委員会後援組織委員会が発表した“内部統制-総合枠組み”(2013年枠組み)(COSO基準)で確立された基準に基づいて、Kymera治療会社の2023年12月31日までの財務報告を内部統制監査を行った。Kymera治療会社(当社)はCOSO基準に基づき,2023年12月31日からすべての重要な面で財務報告に対する有効な内部統制を維持していると考えられる。
我々はまた、米国上場会社会計監督委員会(PCAOB)の基準に従って、当社の2023年12月31日まで、2023年12月31日と2022年12月31日までの総合貸借対照表を監査し、2023年12月31日までの3年度の関連総合経営報告書と全面赤字、株主権益と現金流量、および関連付記と2024年2月22日に発表した報告書について保留意見を発表した。
意見の基礎
当社経営陣は、効果的な財務報告内部統制を維持し、添付されている“経営陣財務報告内部統制報告”に掲載されている財務報告内部統制の有効性を評価する責任がある。私たちの責任は私たちの監査に基づいて、会社の財務報告書の内部統制に意見を述べることです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務報告の有効な内部統制がすべての重要な面で維持されているかどうかを決定するために、合理的な保証を得るために監査を計画し、実行することを要求する。
我々の監査には,財務報告の内部統制を理解すること,重大な弱点があるリスクを評価すること,評価されたリスクテストに基づいて内部統制の設計·運用有効性を評価すること,および状況下で必要と考えられる他のプロセスを実行することが含まれる。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
財務報告の内部統制の定義と限界
会社の財務報告に対する内部統制は、公認された会計原則に基づいて、財務報告の信頼性と外部目的の財務諸表の作成に合理的な保証を提供することを目的としたプログラムである。会社の財務報告に対する内部統制には、(1)合理的で詳細かつ正確かつ公平に会社の資産を反映した取引および処分に関する記録の保存、(2)公認された会計原則に従って財務諸表を作成するために必要な取引が記録されている合理的な保証を提供し、会社の収入および支出は会社の経営陣と取締役の許可のみに基づいて行われる。(三)財務諸表に重大な影響を及ぼす可能性のある不正買収、使用、または処分会社の資産を防止またはタイムリーに発見する合理的な保証を提供する。
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来的にどのような有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムの遵守度が悪化する可能性がある.
/S/安永法律事務所
ボストン、マサチューセッツ州
2024年2月22日
133
財務報告の内部統制の変化
2023年12月31日までの12ヶ月間、財務報告の内部統制(“取引法”第13 a-15(F)および15 d-15(F)条参照)に大きな影響を与えなかったか、または合理的に財務報告の内部統制に大きな影響を与える可能性のある変化はなかった。
プロジェクト9 B。他にも情報です。
次の表は、2023年12月31日までの3ヶ月以内に、任意の上級管理者(1934年証券取引法下の第16 a-1(F)条を参照)または取締役が2023年12月31日までの3ヶ月以内にルール10 b 5-1の取引スケジュールまたは非ルール10 b 5-1の取引スケジュールを締結、修正または終了することを開示する
名前と肩書き |
取引手配の種別 |
取る行動(行動日) |
継続時間または終了日 |
販売待ち証券総数 |
取引スケジュールについての説明 |
証券取引法の正面抗弁条件を満たすための取引計画 |
端末.端末 ( |
適用されない |
適用されない |
先に採用した取引計画を中止する |
|
証券取引法の正面抗弁条件を満たすための取引計画 |
端末.端末 ( |
適用されない |
適用されない |
先に採用した取引計画を中止する |
プロジェクト9 Cです。検査を妨害する外国司法管轄区域を開示する。
適用されません。
134
第三部
プロジェクト10.役員·役員職ICERSと会社管理。
第10条に要求される情報は、2024年年次総会に関する我々が米国証券取引委員会に提出した最終委託書に含まれ、引用により本明細書に組み込まれる。
プロジェクト11.行政官イー補償します。
第11項に要求される情報(“報酬および業績”のタイトル下の情報は含まれていない)は、2024年年次総会に関する米国証券取引委員会に提出された最終依頼書に含まれ、参照によって本明細書に組み込まれる。
プロジェクト12.特定の受益者の保証所有権従業員と経営陣と関連した株主問題。
第12条要求された情報は、2024年年次総会に関する我々が米国証券取引委員会に提出した最終委託書に含まれ、参照により本明細書に組み込まれる。
第13条に要求される情報は、2024年年次総会に関する我々が米国証券取引委員会に提出した最終委託書に含まれ、引用により本明細書に組み込まれる。
第14項.元金口座TING料金とサービスです。
第14条要求された情報は、2024年年次総会に関する我々が米国証券取引委員会に提出した最終委託書に含まれ、引用により本明細書に組み込まれる。
135
第4部
プロジェクト15.物証、資金請求書の明細書です。
本報告に記載されている連結財務諸表一覧については、本年度報告F−1ページの表格10−Kにおける連結財務諸表インデックスを参照して本項目に組み込む。
展示品 番号をつける |
|
説明する |
|
|
|
3.1 |
|
Kymera治療会社の第4回改訂及び再登録証明書(参照登録者によって2020年8月25日に証券取引委員会に提出された8−K表現在報告書の添付ファイル3.1(第001−39460号ファイル)に編入)。 |
|
|
|
3.2 |
|
第2の改正および再改訂は、Kymera治療会社の定款を改正した(登録者を引用して2020年8月25日に証券取引委員会に提出された8−K表の現在報告書の添付ファイル3.2(書類番号001−39460))。 |
|
|
|
4.1 |
|
普通株式証明書サンプル(2020年8月17日に米国証券取引委員会に提出された登録者登録説明書S−1/A表(文書番号333−240264)添付ファイル4.1登録成立参照)。 |
|
|
|
4.2 |
|
第2回改正·再改訂は、登録者とその一部株主との間の投資家権利協定を改正し、2020年3月11日から発効する(登録者が2020年7月31日に米国証券取引委員会に提出したS-1表登録説明書(第333-240264号文書)添付ファイル4.2を会社として設立)。 |
|
|
|
4.3 |
|
証券説明(登録者が2021年3月11日に米国証券取引委員会に提出した10−K表年次報告添付ファイル4.3(第001−39460号文書)を参照して法団として成立)。 |
|
|
|
4.4 |
|
事前出資株式証表(登録者が2022年8月19日に証券取引委員会に提出した8−K表に基づいて現在報告されている添付ファイル4.1(書類番号001−39460)を統合)。 |
|
|
|
4.5 |
|
事前出資株式証表(登録者が2024年1月5日に証券取引委員会に提出した8-K表現在報告の添付ファイル4.1を参照)。 |
|
|
|
10.1# |
|
2018年株式オプション及び付与計画及びその付与協定のフォーマット(2020年7月31日に米国証券取引委員会に提出された登録者登録説明書S−1表(文書番号333−240264)添付ファイル10.1を参照して編入)。 |
|
|
|
10.2# |
|
2020年株式オプション及びインセンティブ計画及びその奨励協定のフォーマット(2020年8月17日に米国証券取引委員会に提出された登録者登録説明書S−1/A表(文書番号333−240264)添付ファイル10.2を参照)。 |
|
|
|
10.3# |
|
2022年1月17日の非従業員役員報酬政策を改正し、再調整した。 |
|
|
|
10.4# |
|
上級管理者現金奨励金計画(2020年8月13日に米国証券取引委員会の登録者登録説明書S-1/A表(書類番号333-240264)添付ファイル10.4)に提出されます。 |
|
|
|
10.5# |
|
2020年従業員株購入計画を改訂·改訂する(登録者が2020年11月5日に米国証券取引委員会に提出した10-Q四半期報告添付ファイル10.1(ファイル番号001-39460)合併参照)。 |
|
|
|
10.6 |
|
登録者とその役員及び上級管理者それぞれとの間の賠償協議表(登録者が2020年8月17日に米国証券取引委員会に提出した登録者登録説明書S−1/A表(書類番号333−240264)添付ファイル10.6が法団として成立する)。 |
|
|
|
10.7 |
|
登録者とアーセナル造船所持株有限公司との間のリースは,2019年8月15日(登録者が2020年7月31日に米国証券取引委員会に提出したS-1表登録声明(書類番号333-240264)添付ファイル10.8が会社として設立された)。 |
136
|
|
|
10.8# |
|
雇用契約表の改訂及び再発注(2020年8月13日に証券取引委員会の登録者登録声明S−1/A表(アーカイブ番号333−240264)添付ファイル10.9を法団に提出)。 |
|
|
|
10.9 |
|
登録者とVertex PharmPharmticals Inc.との間の主協力協定は,2019年5月9日(登録者が2020年7月31日に米国証券取引委員会に提出されたS−1表登録説明書(文書番号333−240264)添付ファイル10.11)である。 |
|
|
|
10.10 |
|
登録者とGenzyme社との協力·許可協定が改訂·再署名されたのは、2022年11月15日である。 |
|
|
|
10.11 |
|
登録者とVertex PharmPharmticals Inc.との間の主協力協定の第1修正案は,2020年8月27日(登録者が2020年11月5日に米国証券取引委員会に提出された10−Q表四半期報告の添付ファイル10.3が会社として設立された)。 |
|
|
|
10.12 |
|
登録者とVertex PharmPharmticals Inc.との間の主協力協定の第2の修正案は,2021年10月21日である。 |
|
|
|
10.13 |
|
登録者とARE-MA地域間のリース,番号75,LLC,日付は2021年12月20日である。 |
|
|
|
10.14 |
|
証券購入協定は,登録者とその当事者との間で締結され,期日は2022年8月18日である(登録者により2022年8月19日に証券取引委員会に提出された8−K表現在報告の添付ファイル10.1により法団として設立される)。 |
|
|
|
10.15 |
|
登録者と当事者との間で2022年8月18日に締結された“登録権協定”(登録者が2022年8月19日に米国証券取引委員会に提出した8−K表現行報告添付ファイル10.2(第001−39460号文書)に基づいて会社として設立された)。 |
|
|
|
10.16 |
|
登録者とGenzyme社との間の手紙は,期日は2023年11月14日である。 |
|
|
|
21.1 |
|
登録者子会社リスト。 |
|
|
|
23.1 |
|
独立公認会計士事務所安永会計士事務所の同意を得ました。 |
|
|
|
24.1 |
|
授権書(本年報の署名ページに表格10-K形式で掲載されている)。 |
|
|
|
31.1 |
|
2002年のサバンズ·オキシリー法第302節で可決された1934年の証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて発行された首席執行幹事証明書。 |
|
|
|
31.2 |
|
2002年サバンズ−オキシリー法第302節で可決された1934年証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて首席財務幹事が認証された。 |
|
|
|
32.1+ |
|
2002年にサバンズ·オクスリ法案第906条で可決された“米国法典”第18編1350条に規定されている最高経営責任者証明書。 |
|
|
|
32.2+ |
|
2002年の“サバンズ-オックススリー法案”906節で採択された“米国法典”第18編1350条による首席財務官の認証。 |
|
|
|
97# |
|
賠償追討政策 |
|
|
|
101.INS |
|
XBRLインスタンスドキュメントを連結する |
|
|
|
101.書院 |
|
イントラネットXBRL分類拡張アーキテクチャ文書 |
|
|
|
101.カール |
|
インラインXBRL分類拡張計算リンクライブラリ文書 |
|
|
|
101.def |
|
インラインXBRL分類拡張Linkbase文書を定義する |
|
|
|
101.介護会 |
|
XBRL分類拡張ラベルLinkbase文書を連結する |
|
|
|
101.Pre |
|
インラインXBRL分類拡張プレゼンテーションLinkbaseドキュメント |
|
|
|
104 |
|
カバーインタラクションデータファイル(フォーマットは、添付ファイル101に含まれる適用分類拡張情報を含むイントラネットXBRLである)。 |
+ 改正された1934年の証券取引法第18条の規定によると、本証明書は提出されたものとみなされず、当該条項の責任にも拘束されない。このような証明は、引用によってそのような出願が明示的に組み込まれない限り、改正された1933年の証券法に基づいて提出されたいかなる出願にも組み込まれているとはみなされない。
#は、管理契約または任意の補償計画、契約、またはスケジュールを表します。
米国証券取引委員会の規定によると、本展示品の一部の内容(星番号で示す)は省略されている。
137
要求された情報は適用されないか、または連結財務諸表に記載されているため、連結財務諸表のすべての付表は省略される。
項目16.表10-Kの概要
適用されません。
138
サイン
改正された1934年証券取引法第13又は15(D)節の要求に基づいて、登録者は、正式に許可された次の署名者がその代表を代表して本報告書に署名することを正式に手配した.
|
|
Kymera治療会社は |
||
|
|
|
|
|
日付:2024年2月22日 |
|
差出人: |
|
/S/Nello Mainolfi,博士 |
|
|
|
|
Nello Mainolfi博士. |
|
|
|
|
社長と最高経営責任者 |
各個人署名は以下のような者が現在Nello Mainolfi,Ph.D.とBruce Jacobs,CFA,MBA,およびそのすべての人を構成し,その真と合法的な事実権利者と代理人として完全な代替と再代替の権力を持ち,他の人なしに行動し,彼または彼女の名義,場所,代理で行動し,各人の名義と以下に述べる各署名を立て,本年次報告の任意およびすべての修正をテーブル10-Kの形で提出し,すべての証拠物とそれに関連する他の文書とともに提出する.米国証券取引委員会に、上記の事実弁護士および代理人、ならびに彼ら一人一人が各行為および事柄を行うすべての権力および権力を付与し、上述したすべての事実弁護士および代理人または彼らのいずれかまたは彼らまたはそれらの代替者が、行われたすべての行為および事柄を合法的に行うことができるか、または結果として生じるすべての行為およびことを承認および確認することができる。
本報告書は、改正された1934年の証券取引法の要求に基づき、以下の登録者によって登録者として指定日に署名された。
名前.名前 |
|
タイトル |
|
日取り |
|
|
|
|
|
/S/Nello Mainolfi,博士 |
|
社長と最高経営責任者 |
|
2024年2月22日 |
Nello Mainolfi博士 |
|
行政主任(首席行政主任) |
|
|
|
|
|
|
|
/S/ブルース·ジェイコブス、CFA、商工管理修士 |
|
首席財務官 |
|
2024年2月22日 |
ブルース·ジェイコブスCFA MBA |
|
首席財務会計官(首席財務会計官) |
|
|
|
|
|
|
|
/S/ブルース·ブス、ディフィル |
|
取締役会議長 |
|
2024年2月22日 |
ブルース·ブスディフィル |
|
|
|
|
|
|
|
|
|
/S/ジェフアルバス、工商管理修士 |
|
役員.取締役 |
|
2024年2月22日 |
ジェフ·アルバス商工管理修士 |
|
|
|
|
|
|
|
|
|
/S/パメラ·エスポシト博士 |
|
役員.取締役 |
|
2024年2月22日 |
パメラ·エスポシト博士 |
|
|
|
|
|
|
|
|
|
/S/ジョアンナ·ホロビン,MB,CHB |
|
役員.取締役 |
|
2024年2月22日 |
ジョアンナ·ホロビンMB CHB |
|
|
|
|
|
|
|
|
|
/S/Gorjan Hrustanovic、博士 |
|
役員.取締役 |
|
2024年2月22日 |
Gorjan Hrustanovic博士 |
|
|
|
|
|
|
|
|
|
/S/ジョン·M·マラガノール博士 |
|
役員.取締役 |
|
2024年2月22日 |
ジョン·M·マラガノ博士 |
|
|
|
|
|
|
|
|
|
/S/リー·モーガン |
|
役員.取締役 |
|
2024年2月22日 |
リー·モーガン |
|
|
|
|
|
|
|
|
|
/S/エレナ、リオデロフ、CFA |
|
役員.取締役 |
|
2024年2月22日 |
エレナ·リオデロフCFA |
|
|
|
|
|
|
|
|
|
/投稿S/ビクター·サンドール |
|
役員.取締役 |
|
2024年2月22日 |
ビクター·サンドール |
|
|
|
|
139