
ルール424(B)(4)に従って提出する
登録番号:333-276502
| 目論見書 |
1,375,000株普通株式
3,750,000シリーズ株式承認証最大3,750,000株普通株購入
1,875,000株Bシリーズ株式承認証最大1,875,000株普通株購入
2,375,000株事前資金(Br)株式承認証最大2,375,000株普通株購入
800万株の普通株を対象としたAシリーズ権証、B系権証、予備資本権証
150,000部の配給代理株式承認証最大150,000株普通株を購入
15万株の普通株を対象とした配給代理承認株式証
Cingate Inc.
私たちは1,375,000株の普通株を発行し、1株当たり0.0001ドルの価値があり、Aシリーズの株式承認証と一緒に、最大3,750,000株の普通株を購入し、私たちはこれを“Aシリーズ株式承認証”、及びBシリーズ株式承認証と呼び、最大1,875,000株の普通株を購入し、私たちはこれを“Bシリーズ株式承認証”と呼び、公開発行価格は1株2ドルであり、株式証を付帯する。Aシリーズ株式承認証とBシリーズ株式承認証は以下では“株式承認証”と呼ばれる。私たちの普通株は1株当たりAシリーズ株式承認証とBシリーズ株式承認証と一緒に販売し、Aシリーズ株式承認証は1株普通株を購入し、Bシリーズ株式承認証は半分普通株を購入する。Aシリーズ株式証明書の発行権価格は1株当たり2.00ドルであり、すぐに発行することができる。Aシリーズ株式承認証は発行日から5年以内に満期になります。Bシリーズ株式承認証の発行価格は1株2.00ドルで、すぐに行使することができる。Bシリーズ株式承認証は発行日から2年以内に満期になる。本募集説明書は、Aシリーズ及びBシリーズ株式承認証を発行して発行可能な普通株式とも関係している
今回の発売で私たちの普通株の株式を購入した投資家にも機会を提供し、その投資家 がその関連会社とある関連側と共に今回の発売完了後に私たちが発行した普通株の4.99%(または投資家選択時9.99%) を所有し、普通株 を購入する機会があり、そうでなければ、投資家の実益所有権が4.99%(または投資家選択時に9.99%)を超えることになります。資本金権証は、1部当たり0.0001ドルの使用価格で私たちの普通株の1株を購入し、私たちは“予備金権証”と呼ぶ。あらかじめ出資した引受権証は発行時に行使可能であり,全行使時に満期となる。各事前資本権証 はAシリーズ株式承認証とBシリーズ株式承認証と一緒に販売され、前者は普通株を購入し、後者は半分の 普通株を購入する。1部当たりの事前資本権証と付随持分証の公開発行価格は、今回の発行中に公衆に販売した普通株と付随持分証の1株価格から0.0001ドルを引いたものに等しい。本募集説明書はまた、事前資本承認株式証を行使する際に発行可能な普通株の発売に関するものである。
このbr製品は、その日までに製品を終了することを決定しない限り、2024年2月14日に終了します(いつでも適宜終了することができます) 今回の発行で購入したすべての証券を一度に成約します。今回の発行期間中、1株当たりのbr株(または予融資権証)とセット引受権証の総合公開発行価格は変わらない。
普通株式及び/又は事前資本権証の株式及び付随する引受権証は、今回の発売でのみ一括して購入することができるが、 を分けて発行し、発行後すぐに分離することができる。
我々はH.C.Wainwright&Co.,LLCまたは配給エージェントを今回の発売に関する独占配給エージェントとして招聘している.配給代理は、その合理的な努力を尽くして、本募集説明書が提供する証券の売却を手配することに同意した。配給エージェントは、私たちが提供する任意の証券を購入または販売することもなく、特定の数または金額の証券を購入または販売するように手配する必要もない。私たちは、本募集説明書で提供されるすべての証券を売却したと仮定して、次の表に記載された配給代理費を配給エージェントに支払うことに同意しました。brは、投資家資金を受け取った後に、今回の発行で発行される証券を交付するため、信託、信託、または同様の手配で資金を受け取る予定はありません。今回の発売の成約条件として最低発売要求はありません。 今回の発売には最低発売金額の要求がないため、ここで発売されたすべての証券よりも少ない可能性があり、受け取る収益を著しく減少させる可能性があり、募集説明書に記載されている業務目標を達成するのに十分でなければ、今回発売された投資家は返金を受けることはありません。また、信託口座や最低発売金額がないため、投資家は当社に投資している可能性があります。しかし、今回の発行に興味がないため、私たちはすべての期待された目標を達成することができない。また,我々がこれらの資金を用いて我々の業務計画を効率的に実施できるかどうかは不明であるが,我々が証券を売却して得られたどの収益も即座に利用できる.より多くの 情報については、“リスク要因”の部分を参照してください。私たちは今回の発行に関連したすべての費用を負担するつもりだ。これらの手配に関するより多くの情報は、本募集説明書137ページの“流通計画” を参照してください。
我々は2023年11月30日に20株1株の逆株式分割を実施し,これにより,20株当たり発行済み株式と発行済み普通株 が1株普通株に再分類された。逆株式分割は私たちの普通株の額面または私たちの普通株の法定株式数に影響を与えない。別の説明がない限り、本入札明細書のすべての株式および各株式情報は、逆株式分割を反映するように調整されている
私たちのbr}普通株はナスダックに看板を掲げて取引して、コードは“CING”です。私たちの普通株は2024年2月2日のナスダックでの終値は1株1.33ドルです。事前融資権証、Aシリーズ権証、あるいはBシリーズ権証はまだ成熟していない公開取引市場であり、このような市場は発展しないと予想される。私たちは、いかなる証券取引所または他の国で認可された取引システムに上場する事前融資権証、Aシリーズ権証、またはBシリーズ権証を申請するつもりはありません。活発な取引市場がなければ、事前融資権証、Aシリーズ権証、Bシリーズ権証の流動性が制限される。
米国証券取引委員会が適用する規則によると、我々は“新興成長型会社”であり、低下した上場企業の報告要求を遵守する。
私たちの証券に投資することは高い投機的で高い危険を持っている。我々の証券に投資する際に考慮すべき情報に関する議論は、本募集説明書の7ページ目からの“リスク要因”を参照してください。
| 一株および附随持分証 | 前払金の引受証と付随する引受権証 | 合計する | ||||||||||
| 公開発行価格 | $ | 2.00 | 1.9999 | $ | 7,499,762.50 | |||||||
| 配置代理費(1) | $ | 0.1560 | 0.1560 | $ | 585,000.00 | |||||||
| 費用を差し引く前の収益は私たちにくれます(2) | $ | 1.8440 | 1.8439 | $ | 6,914,762.50 | |||||||
| (1) | 我々は,今回の発行所の総収益の7.8%に相当する現金料金を配給エージェントに支払うことに同意した.私たちはまた、最高50,000ドルの非責任費用精算、最高100,000ドルの法的費用、および15,950ドルの決済費用を含む、その発売に関連するいくつかの費用を配給エージェントに返済することに同意します。また、吾らは、配給代理またはその指定者承認株式証の発行に同意し、今回発売中に売却された普通株式の4.0%に相当する普通株(前払い資本証を行使して発行可能な普通株株式を含む)を1株2.50ドルで購入し、行使価格は1株2.50ドルであり、公開発売価格および付随持分証の125%に相当する。配置エージェントが受信する補償の説明については、より多くの情報を知るために“割当て計画 ”を参照してください。 |
| (2) | 今回の発行には最低証券数や募集金額が発行完了条件とされていないため,実際の公開発売金額,配給代行費,我々に提供される募集資金(あれば)を決定することはできず,上記の総最高発売金額を大きく下回る可能性がある.詳細は“割当計画”を参照されたい |
米国証券取引委員会またはどの州証券委員会もこれらの証券を承認または承認しておらず、本募集説明書の正確性または十分性のために を通過していない。どんな反対の陳述も刑事犯罪だ。
通常の成約条件を満たすことを前提に、今回発売された証券は2024年2月6日頃に受け渡しされる予定だ。
H.C.ウェインライト&Co.
本募集説明書の日付は2024年2月2日です。
カタログ表
| ページ | |
| 募集説明書の概要 | 1 |
| 供物 | 5 |
| リスク要因 | 7 |
| 前向き陳述に関する警告的説明 | 58 |
| 市場価格と配当情報 | 59 |
| 商売人 | 60 |
| 経営陣の財務状況と経営成果の検討と分析 | 100 |
| 所有権と利益を保証するすべての人と管理 | 115 |
| 関係者と取引しています | 117 |
| 管理する | 119 |
| 役員と役員の報酬 | 121 |
| 収益の使用 | 128 |
| 薄めにする | 129 |
| 株本説明 | 130 |
| 私たちが提供する証券説明書は | 133 |
| 配送計画 | 137 |
| 法律事務 | 140 |
| 専門家 | 140 |
| そこでもっと多くの情報を見つけることができます | 141 |
| 財務諸表索引 | F-1 |
あなたはこの目論見書に含まれている情報だけに依存しなければなりません。本入札明細書に含まれる情報 とは異なる情報を提供することは誰にも許可されていません。本募集説明書の日付は、本募集説明書の表紙に記載されている日付である。本入札明細書に含まれる情報が、その日付以外のどの日付でも正確であると仮定するべきではありません。
米国以外の投資家について:私たちは米国以外のどの司法管轄区域でも本募集説明書を発行または発行することを可能にする措置を取っていません。あなたはあなた自身に教えて、今回の発行と配布本の説明書に関連するいかなる制限も守らなければなりません。
| i |
募集説明書 概要
この 要約は、本入札明細書の他の部分に含まれる情報を重点的に紹介しており、投資決定を行う前に考慮すべきすべての情報は含まれていません。私たちの証券に投資する前に、あなたは株式募集説明書の“リスク要因”の部分を含む株式募集説明書全体をよく読まなければならない。もしどんなリスクが現実になれば、私たちの業務、財務状況、経営業績、見通しは実質的な悪影響を受ける可能性がある。この場合、私たちの証券価格は下落する可能性があり、あなたのbr}は投資の一部または全部を損失する可能性があります。私たちが別に説明や文意が別に言及されていない限り、用語“私たち”、“私たち”、“私たちの業務”、“当社”および“cingate”は、(1)再編合併(以下、定義)が完了した時または後に、当社の初公開募集を含めて、cingate Inc.およびその合併付属会社に、cingate Treateutics LLC、またはCTX;および(2)再編合併(我々の初公開発売を含む)が完了する前に、CTXおよびその合併付属会社に提供することを意味する。
概要
私たちはバイオ製薬会社で、私たちの独自のPrecisionを使って定期的にリリースしますTM(“PTRTM)薬物brは、重い日常投与レジメンおよび二次治療を特徴とするよく見られる診断疾患を有する患者の生活を改善するために、次世代薬物製品導管を構築および推進するためのプラットフォーム技術を提供する。私たちの最初のポイントは注意欠陥/多動性障害(ADHD)の治療であり、私たちは他の治療領域を決定して評価しており、これらの分野では、私たちのPTR技術は不安障害 のような将来の候補製品の開発に使用される可能性がある。我々のPTRプラットフォームは、特定の所定の時間間隔で薬物物質を放出し、それによって1日1回、多用量錠剤を放出する可能性を可能にするために、専用の侵食バリア層(“EBL”)を採用している。
我々の目標はADHD治療市場であり,2023年11月現在,米国市場規模は230億ドルを超えると推定されており,そのうち186億ドルは覚せい剤brによるものである。覚せい剤はADHDを治療する最もよく見られる処方類薬物であり、アメリカのすべてのADHD処方の約88%を占める。これに対し,非覚せい剤は通常二線あるいは補助治療環境でしか使用されておらず,すべてのADHD薬処方の約12%を占めている。徐放或いは長時間作用刺激性薬物剤形は最もよくADHDの第一線の治療薬物として使用され、ADHD市場の総支出の約160億ドルを占め、すべての覚せい剤処方の54%を占める。これらの徐放剤の多くは朝1回の投与が許可されており,日中の再投与の必要性を解消することを目的としている。しかしながら、現在の“1日1回”徐放剤形を使用して、多くの患者は、有効な日カバーを達成するために、当日遅い時期(通常は午後早い時期)に第2剤または“促進剤”brの用量を受け、したがって、様々な副作用brを受ける。現在の治療モデルには,重大で満たされていない需要,すなわち真の1日1回のADHD刺激薬が存在し,その持続時間は全有効日カバー範囲を提供し,副作用を改善し,患者の多くの満たされていない需要をよりよく満たすことができると考えられる。
我々は、CTX-1301(酢酸ジメチル)とCTX-1302(右旋フェニルプロピルアミン)、 の3つの主要な患者集団のADHDの治療に使用している:子供(6~12歳)、青少年(13~17歳)、成人(18歳以上)。CTX-1301およびCTX-1302は、現在承認されている覚せい剤療法の主な欠点を以下の方法で解決することを目的としている:即時効果(30分以内)を提供し、“平日全体”の持続時間を提供する。“促進剤/回復” 用量の短時間効果の覚せい剤薬物の需要を除去した;早期薬物の“摩耗”に関連する反発/崩壊症状を最大限に減少または除去した;br}そして良好な耐性を提供し、同時に薬物の血液レベルの低下をコントロールした。さらに、私たちは、ADHD患者の60%までの主要な薬物と一緒に使用される“ブースター”の投与量を廃止することによって、私たちの候補製品は、短時間効果の覚せい剤に関連する乱用と転移を減少させることができると信じている;医師が2つの薬物ではなく1つの薬物を処方することを許可する;患者が2つの薬物ではなく1つの薬物の支払いを許可すること;支払人が2つの薬物ではなく1つの薬物を清算することを可能にする
| 1 |
最近の発展
債務転換
2022年8月には,Werth Family Investment Associates LLC(“WFIA”)を受益者とした500万ドル(“原始元本”)元票(“原始手形”)を発行し,2023年5月に改訂·再発行された本票(“WFIA手形”) を発行し,オリジナル手形での元金金額を300万ドルから800万ドル増加させた。2023年9月8日、Cingate Inc.およびCTX はWFIAと手形変換プロトコルを締結し、これにより、WFIAは、WFIA手形項の元の元本金額をそのすべての課税利息、または5,812,500ドルと合わせて、予備金権証(“9月WFIA事前資本権証”) に変換し、資本金権証1部あたりの転換価格17.00ドルで341,912株の普通株を購入することに同意する。
2024年1月25日に、吾ら はWFIAと手形転換協定(“2024年1月手形転換協定”)を締結し、残りの300万ドル元金にすべての課税利息を加えたり、3,287,500ドルを前資金権証(“2024年1月WFIA資本権証”) に変換し、2024年1月WFIA予資権証あたりの転換価格4.785ドルで687,043株普通株を購入した(“2024年1月債務転換”)。私たちの普通株の2024年1月24日の終値は4.35ドルです。1月のWFIA事前承認株式証は満期日がなく,ただちに1株0.0001ドルの行使価格で行使可能であり,取引法13(D)条の規定により,WFIAとその関連会社は実益が我々普通株発行株式の19.99% を超えないことを条件としている。剰余元金金額にそのすべての課税利息を加えて前払い資金引受権証を発行した後,当社はWFIA手形を全額支払いしており,当社は WFIA手形により他の責任はない。
首席財務官を任命する
2024年1月25日、私たちはジェニファー·L·カラハンを上級副総裁兼首席財務官に任命した。Callahanさんは私たちの首席財務官と首席会計官を担当するだろう。Callahanさんの任命について、CTXは2024年1月25日にCallahanさんと雇用協定を締結した。雇用協定は年間35万ドルの基本給を規定する。しかし、2023年12月15日に開示されたコスト抑制措置を受けて、Callahanさんの年間基本給は40%減少し、21万ドルに低下した。brはこれに関連して、CTX-1301の新薬申請が連邦医薬品局に提出されてから3ヶ月後の日にCallahanさんに金額を支払うことになり、減給で彼女に支払われなかったドルの総額に減給額の20%を加えることに相当する。また、カラハンには年間ボーナスを得る資格があり、目標額はカラハン年度基本給の25%(25%)だった。各ボーナスの実際の金額は私たちの報酬委員会が自ら決定し、私たちの業績とCEOが提案したCallahanさんの個人的な業績に基づいて決定されるだろう。
訓練とATMの販売を許可します
2023年10月1日から本募集説明書の発行日までの間、投資家は、私たちが発行した前払い資金承認株式証をいくつか行使し、260,261株の私たちの普通株を購入しました(当該等の行使、すなわち“株式承認証行使”)
2023年10月1日から本募集説明書期日までの間に,吾らはH.C.Wainwright&Co.,LLCと締結した市場発売プロトコル(“ATMプロトコル”)により,283,800株の普通株を販売し,純収益は3,115,303ドル(“ATM販売”)であった。
株を逆分割する
我々は2023年11月30日に20株1株の逆株式分割を実施し,これにより,20株当たり発行済み普通株と発行済み普通株が1株普通株に再分類された。逆株式分割は私たちの普通株の額面や私たちの普通株の許可数に影響を与えない。別の説明がない限り、本入札明細書のすべての株式および各株式情報は、逆方向株式分割を反映するように調整されるであろう。
| 2 |
注目を行っている
今回の発売は最大の努力で行われており、我々が販売している証券はここで発売されたすべての証券よりも少ない可能性があり、今回の発売から得られる純収益も大幅に減少する可能性がある。私たちの現在の業務計画によると、今回発行された純収益に私たちの手元の現金を加えて、2024年第2四半期末までの資本需要を満たすと信じています。今回の発行後、私たちはより多くの資金を集めて私たちの運営を支援し、私たちが計画している開発と商業化活動を支援していく必要があります。
ナスダック
私たちの普通株式と権利証は現在ナスダックで看板取引されている。2023年5月16日、ナスダック上場資格従業員(以下、“従業員”と略す)から通知を受け、“ナスダック証券市場上場規則”第5550(B)(1)条(“最低株主権益規則”)に規定されている250万ドルの最低株主権益要求に適合しなくなり、上場を継続できないことが通知された。私たちは2023年6月30日にナスダックにコンプライアンス計画を提出した。2023年7月28日、ナスダック証券市場は、追加資本を調達する計画を含む、これまでにナスダック証券市場に提出されたコンプライアンス計画に含まれるいくつかのマイルストーンを実現するために、最低株主権益規則を再遵守するための2023年11月13日までの延長を許可することを通知した。2023年11月14日,吾らはナスダックの書簡を受け取り,吾等が最低株主権益規則に違反し続けていることに基づいて,職員は吾らの要求に基づいてナスダック通信グループ(以下“委員会”と呼ぶ)で尋問を行うことにし,吾らの証券をナスダックから退市することにした。
私たちは専門家グループとナスダックが私たちの要請を承認する前に公聴会を行うことを直ちに要求します。2024年2月に予定されている専門家チームのヒアリングでは、ナスダックに上場し続けるすべての適用要件brに対するコンプライアンスと、最低株主権益ルールと独立 取締役ルールの遵守を含む長期コンプライアンスを維持する能力を再遵守する計画を提出します(以下のように定義します)。従業員の除名行動は延期され、聴聞とグループが聴聞後に承認する任意の追加の延長期間の満了を待つ。私たちは引き続き最終ステップを取って、 が最低株主権益規則、独立取締役規則、その他のナスダック上場要求を遵守したことを証明するつもりです;しかし、陪審員が私たちの上場継続の要求を承認することを保証することはできません。また、陪審員が承認する可能性のあるいかなる延期の期間内に が最低株主権益規則、独立取締役規則と他のナスダック上場要求を遵守できるかを保証することもできません。我々が最低株主権益規則を再遵守し,上場を継続するためにも,最低株主権益規則 を長期的に遵守する能力があることを証明する必要がある。
2023年7月28日、私たちはナスダック株式市場から通知を受け、私たちは1株当たりの最低購入価格を1.00ドルに維持して、ナスダック上場を継続できるという要求を守らなかったことを示した。ナスダック上場規則第5810(C)(3)(A)条によると、我々は180個のカレンダー 日のコンプライアンス期間を取得し、通知の日から2024年1月24日まで、最低成約見積要求を再遵守する。2023年11月30日、私たちは私たちの普通株を逆分割しました。 2023年12月15日、私たちはナスダック株式市場から通知を受けて、私たちは最低終値の入札価格要求を再遵守しました。最低終値要求brや他のナスダック上場要求を守り続ける保証はありません。
2023年12月26日、私たちは3人の取締役会のメンバーがそれぞれ2023年12月12日と2023年12月13日に辞任したことに基づいて、取締役上場規則第5605条(“独立取締役規則”)に記載されている独立ナスダック上場委員会、監査委員会、報酬委員会およびナスダックの取締役指名に対する独立監督規定を守らないという社員手紙を受け取った。私たちの取締役会は現在二人の役員で構成されており、彼らは独立していない。
| 3 |
我々 は潜在的な新しい取締役を探しており,グループヒアリングの前に独立取締役ルール遵守への進展 を示す予定である.私たちが“役員独立規則”を再遵守する保証はありません。
我々 は、最低株主権益ルール と独立取締役ルール を含むナスダックの持続的な上場要求を満たさなければならず、そうでなければ、私たちの業務に重大な悪影響を与える可能性のあるリスクが退市する。もし私たちの普通株式と引受権証がナスダックから撤退すれば、私たちの普通株と引受権証の流動性を大幅に減少させ、ナスダックに関連する市場効率の喪失と連邦証券法の優先購入権を失ったため、私たちの普通株と引受権証の価格がそれに応じて大幅に低下する可能性がある。また、退市は、私たちが受け入れられる条項や全く受け入れられない条項で融資源を代替することで資金を調達する能力を損なう可能性があり、投資家、サプライヤー、顧客、従業員の潜在的な自信を失い、業務発展の機会を減少させる可能性がある。もし私たちの普通株式と引受権証が取得されたら、私たちの普通株と引受権証はもっと売買しにくいかもしれないし、正確な見積もりを得ることができないかもしれません。私たちの普通株と引受権証の価格は大幅に下落する可能性があります。退市は、もしあれば、受け入れ可能な条件で資金を調達する能力を弱める可能性もある。
私たちのbr組織構造は
Cingate Inc.はデラウェア州の会社であり、設立時は持株会社であった。私たちの初公募株について、私たちはいくつかの組織取引を完了した。2021年9月29日、Cingateは、Cingateの完全買収子会社をCTXと合併(“再編合併”)することにより、Cingate Treateutics LLCまたはCTXを買収した。再編合併の結果,CTXはcingateの完全子会社となる。本明細書のすべての情報は、他の説明または文脈に別の規定があることを除いて、再構成統合の完了を反映する。
企業情報
私たちの主な執行事務室は西47号1901にありますこれは…。Pace、カンザスシティ、カンザス州66205、私たちの電話番号は(913)9422300です。私たちのサイトの住所はWwwc.cingulate.comそれは.我々のサイト に含まれている,あるいはそれを介してアクセス可能な情報は,本募集説明書の一部ではなく,本募集説明書の一部として,あるいは我々の証券を購入するかどうかを決定する際に考慮すべきではない.
Cingate、PTR、Cingate Treeutics、Enfoqis、Enfoqus、Trodesca、Ivoqus、Taylerza、Tymprezi、Finish、Master、および私たちのロゴは、本入札明細書で使用されているいくつかの商標です。本募集説明書には、他のbr組織財産に属する商標、商号、およびサービスマークも含まれる。便宜上、本入札明細書で言及されている私たちの商標および商標名は、記号およびbr記号を持たない可能性があるが、これらの参照は、適用法に従って、これらの商標および商標名に対する私たちまたは適用許可者の権利を最大限に主張しないことを意味するわけではない。
新興成長型会社としての影響
私たちが最近終了した会計年度収入が12.35億ドルを下回る会社としては、“2012年に私たちの企業創業法案を迅速に開始する”や“JOBS法案”で定義されている“新興成長型企業”の資格に適合している。新興成長型会社は、何らかの報告要求や他の一般的に上場企業に適用される負担を免除する利点を利用する可能性がある。これらの規定 は:
| ● | 本募集明細書に2年間のみ記載された監査財務諸表および2年のみの選択された財務データを含む財務データに関する債務を削減する | |
| ● | 改正された“2002年サバンズ-オキシリー法案”第404条または“サバンズ-オキシリー法案”の監査認証要件の例外を遵守する | |
| ● | 定期報告書、依頼書、および登録説明書における役員報酬スケジュールの開示を減少させること | |
| ● | 免除されたbrは、役員報酬または黄金パラシュート手配について拘束力のない諮問投票の要求を行う。 |
私たちは5年に及ぶ免除またはそれ以上の時間を利用して、私たちがもう新しい成長型会社ではないようにするかもしれない。したがって、ここに含まれる情報は、あなたが株を持っている他の上場企業から受け取った情報とは異なる可能性があります。 私たちは、以下の最初に出現した場合、新興成長型会社になることを停止します:(1)私たちの年間毛収入が12.35億ドルを超える会計年度の最終日、(2)2026年12月31日、(3)1934年証券取引法(改正)や取引法で定義されている“大型加速申告会社”の日付と考えられます。(4)私たちはこれまでの3年間に10億ドルを超える転換不可能債務証券を発行した日。
雇用法案はまた、新興成長型企業として、上場企業に適用される新しい会計基準や改正された会計基準を遵守するために、延長された過渡期を利用することを可能にし、これらの基準が非上場企業に適用されるまで、これらの基準の採用を延期することを可能にする。私たちはこの免除を取り消すことができないので、他の非新興成長型企業の上場企業と同じ新しいまたは改訂された会計基準を遵守することはできません。
| 4 |
製品
次の 要約は,その製品に関する基本情報を含む.本願明細書に含まれるより詳細な情報 が完全に限定される。私たちの証券に投資決定を下す前に、“リスク要因”、“経営陣の財務状況および経営結果の議論および分析”の項目の情報、および本募集説明書の他の部分に含まれる財務諸表を含む株式募集説明書全体をよく読みなさい。
| 普通株式発行 | 1375,000株です。 | |
| あらかじめ出資した株式承認証を提供する | 我々はまた、投資家に事前融資承認株式証を提供して、最大2,375,000株の普通株を購入するが、これらの投資家が今回の発行で私たちの普通株の株式を購入することは、その投資家とその関連会社およびある関連側 が今回の発行が完了した後、私たちが発行した普通株の4.99%以上(または投資家が選択した場合、9.99%)を所有し、そうでなければこの超過した普通株を招くことになる。各事前出資の権利証はAシリーズ株式承認証とBシリーズ株式承認証と一緒に販売され、前者は普通株を購入し、後者は半分の普通株を購入する。1部当たりの事前資本権証と付属引受権証の総合公開発行価格は、今回発行で販売された普通株と付属引受権証の1株当たりの総合公開発行価格から0.0001ドルを引いたものであり、事前出資株式証1部あたりの使用価格は1株当たり0.0001ドルとなる。本募集説明書 はまた、事前資本承認株式証を行使する際に普通株を発行可能な発行に関するものである。より多くの情報については、私たちが提供する証券説明 を参照してください。 | |
| 株式承認証 提供 | 各普通株または事前資本権証は、Aシリーズ株式承認証とBシリーズ株式承認証と一緒に発売され、前者は普通株の購入に使用され、後者は半分の普通株を購入するために使用される。Aシリーズ株式証の行使価格は1株2.00ドルであり、直ちに行使することができる。Aシリーズ株式承認証は発行日から5年以内に満期になります。Bシリーズ株式承認証の発行価格は1株2.00ドルで、すぐに行使することができる。Bシリーズ株式承認証は発行日から2年以内に満期になる。より多くの情報については、“私たちが提供する証券説明書”を参照してください。 | |
| 普通株 今回の発行前の未償還株(1) | 1,450,171株。 | |
| 今回発行後の普通株式 (1) | 5,200,171株は、すべての事前資金の引受権証が行使され、株式承認証が行使されていないと仮定する。 | |
| 収益を使用する | 配給代理費と推定発売費を差し引いた後,引受権証を行使しないと仮定すると,初回発売の純収益は約650万ドルと予想される。今回の発行から得られたすべての純収益をCTX−1301の持続的な研究開発と商業化活動,運営資本,資本支出,一般企業用途に利用する予定であり,さらなる研究開発への投資を含む。私たちの現在の業務計画によると、今回発行された純収益に私たちの手元の現金を加えて、2024年第2四半期末までの資本需要を満たすと信じています。今回の発行後、私たちの運営を支援するためにより多くの資金を集め、私たちが計画している開発と商業化活動を支援していく必要があります。“収益の使用”を参照してください |
| 5 |
| ナスダック資本市場記号 | 私たちのbr}普通株はナスダックに看板を掲げて取引して、コードは“CING”です。私たちは、いかなる証券取引所または他の国で認可された取引システムに上場する事前融資の権利証、Aシリーズ権証、またはBシリーズ権証を申請するつもりはありません。活発な取引市場がなければ、事前融資権証、Aシリーズ権証、Bシリーズ権証の流動性が制限される。 | |
| ロックする | いくつかの例外を除いて、私たちのすべての役員と幹部は、今回の発売終了後90日以内に、私たちの任意の普通株を直接または間接的に売却、譲渡または処分したり、私たちの普通株に転換したり、交換することができる証券に交換したりすることができません。より多くの情報については、“割当計画”を参照してください。 | |
| 配給代理権証 | 我々は,今回発売した普通株式総数の4.0% (事前資本証を行使して発行可能な普通株式を含む)を購入するために,配給エージェントまたはその指定者,株式承認証または配給エージェントに株式証を発行することに同意し,行使価格は,今回発売中に販売された1株当たり公開発行価格の125%および付随する株式承認証 に相当する.配給代理株式証は発行時に行使可能となり、今回の発売開始日から5年以内に満了する。より多くの情報については、“割当計画”を参照してください。 | |
| リスク要因 | 私たちの証券に投資することは高いリスクがあり、あなたのすべての投資損失を招く可能性があります。本募集説明書の7ページ目からの“リスク要因” を参照してください。私たちの証券への投資を決定する前によく考慮すべき要素 を理解してください。 |
(1) 今回発行された直後に発行される普通株数は、2024年1月25日までに発行された普通株の1,450,171株をベースとしており、この日までには、以下は含まれていない
| ● | 私たちの2021年株式激励計画(“2021年計画”)によると、私たちの2021年株式激励計画(“2021年計画”)によると、73,647株の普通株を発行することができ、加重平均価格は1株59.72ドルである | |
| ● | 私たちの普通株式174,479株は、2021年計画に従って将来発行することができます | |
| ● | 786,710株は、権利行使時に発行可能な普通株であり、加重平均行権価格は1株46.40ドルである | |
| ● | 1,028,955株の普通株は、事前融資権証を行使した後に を発行することができ、加重平均行権価格は1株当たり0.0007314ドルである。 |
本募集説明書内のすべての資料は(I)吾などが事前融資権証 及び(Ii)吾などを発行していないと仮定して、本募集説明書が提供する引受権証を行使していない。
| 6 |
リスク要因
私たちの証券に投資することはリスクと関連がある。本募集説明書に含まれる他の情報に加えて、以下のリスク要因をよく考慮しなければなりません。“前向きな陳述に関する戒め”というタイトルの節に記載されている事項を含めて、以下のリスク要因をよく考慮しなければなりません。これらのリスク要因は網羅的ではなく、投資家が私たちの業務、財務状況、将来性について自分の調査を行うことを奨励します。私たちは他のリスクと不確実性に直面する可能性があり、これらのリスクと不確実性は私たちが現在知らないこと、あるいは私たちが現在どうでもいいと思っていることであり、これはまた私たちの業務や財務状況を損なう可能性がある。もしこれらのリスクのいずれかが実際に発生すれば、私たちの業務、財務状況、運営結果は実質的な悪影響を受ける可能性があり、私たちは私たちの目標を達成できないかもしれません。私たちの証券の価値は下落する可能性があり、あなたの一部または全部の投資は損失する可能性があります。私たちが今知らない他のリスクや私たちは現在どうでもいいリスクは私たちの業務運営を深刻に損なう可能性があると思っています。これらのリスクのいずれかが発生した場合、私たちの業務、運営結果、または財務状況、および見通しが損なわれる可能性があります。この場合、私たちの普通株の市場価格と権利証のbr価値は下落する可能性があり、あなたはすべてまたは一部の投資を損失する可能性があります。以下の議論は、財務諸表および財務諸表の付記とともに読まなければならない。
リスク要約
以下 は我々に重大な悪影響を及ぼす可能性のある主なリスクと不確実性をまとめたものである。この要約は、以下に含まれる各リスク要因のより詳細な説明と共に読まれなければなりません。
| ● | 私たちbrは生物製薬会社で、運営歴史が限られており、私たちの候補製品を推進して商業化することを含む追加の資金が必要です。 | |
| ● | 我々はすでに運営赤字の歴史を受けており,予見可能な将来に巨額のコストが発生し続けることが予想される。私たちは現在利益を上げていないし、私たちは絶対に利益を達成したり維持したりしないかもしれない。 | |
| ● | 今回の発行後、我々は、CTX-1301、CTX-1302、および/またはCTX-2103の開発および商業化を完了することを含む、運営を継続するために多くの追加資金を調達する必要がある。もし私たちがもっと多くの資本を集めることができなければ、私たちは破産保護や他の代替案を求めるように要求されるかもしれません。これは私たちの証券保有者が私たちへの投資の一部または全部を失う可能性があります。 | |
| ● | もし私たちがナスダックの持続的な上場要求を再遵守できなかった場合、私たちの普通株および/または株式承認証は取得される可能性があり、私たちの普通株および/または株式承認証の価格、および私たちの資本市場に入る能力はマイナスの影響を受ける可能性がある。 | |
| ● | 私たちの は主に私たちの候補製品CTX-1301とCTX-1302の成功した開発と商業化に依存します。CTX-1301とCTX-1302はADHDの治療に使用され、CTX-2103は不安を治療するために使用され、これらの製品は製品/早期臨床開発 (CTX-2103)、臨床開発(CTX-1301)または将来の製品開発(CTX-1302)を計画しており、まだ が承認されていません。私たちはこのような候補製品や他の候補品が規制部門の承認を受けることを保証できない。これはそれらが商業化できる前に必要だ。 | |
| ● | CTX-1301、CTX-1302、およびCTX-2103の規制承認を得ても、この承認は限られている可能性があり、私たちは厳格で持続的な政府規制を受けるだろう。もし私たちの候補製品が承認されれば、その商業成功は医者、患者、第三者支払人と医学界の市場認可を得るかどうかにある程度依存する。 | |
| ● | オピオイドと覚せい剤乱用をめぐる社会問題は、法執行部門の分流に対する懸念及び乱用に打撃を与える監督管理努力を含み、私たちの候補製品の潜在市場を低下させる可能性がある。 |
| ● | 私たちの業務は幅広い規制要求を受けており、承認された候補製品は持続的なbrと持続的な規制審査を受けることになり、これは巨額の費用を招き、このような製品を商業化する能力を制限する可能性がある。 | |
| ● | 我々の CTX−1301およびCTX−1302の供給源は限られており、これらは所定の製品であるため、サプライチェーンの任意の中断は、CTX−1301およびCTX−1302の生産および販売に影響を与え、候補製品の開発および商業化の遅延をもたらす可能性がある。 | |
| ● | 私たちは第三者に依存して私たちの候補製品を製造して包装し、私たちの候補製品の臨床試験と監督申請を提出しますが、これらの第三者の表現は、私たちの候補製品の最終期限 ,このような試験の完了および/または規制提出を含む満足できないかもしれません。 |
| 7 |
| ● | 私たち は第三者に依存して私たちの候補製品を商業化し、私たちは第三者に依存して私たちの商業化された任意の製品に多くの基本サービス を提供する可能性があります。流通、顧客サービス、売掛金管理、現金入金 と不良イベント報告を含む。これらの第三者が予想通りに動作していない場合、または法律および法規の要求を遵守していない場合、我々はCTX-1301、CTX-1302、および/またはCTX-2103を商業化する能力が深刻な影響を受けるであろう。私たちは規制部門のbr制裁を受ける可能性がある。 | |
| ● | 私たちは将来、私たちの組織の規模と複雑さをさらに増加させる必要があり、私たちは成長戦略を実行し、どんな成長を管理するかについて困難に直面する可能性がある。 | |
| ● | 私たちの研究と開発は候補製品の発見と開発に集中しており、これは市場に出すことができないかもしれない。 | |
| ● | 私たち はますます情報技術に依存しており、私たちのシステムとインフラはネットワークセキュリティ とデータ漏洩リスクを含む一定のリスクに直面している。 | |
| ● | もし私たちの製品や候補製品に関連する知的財産権が不足していれば、私たちは私たちの市場で効果的に競争できないかもしれません。 | |
| ● | 私たちの証券の活発な取引市場は持続できないかもしれない。 |
私たちの財務状況と資金需要に関連するリスク
私たちbrは経営の歴史が限られている生物製薬会社です。
私たちbrは生物製薬会社で、運営履歴が限られていますので、これによって私たちの業務と将来性を評価することができます。私たちはbrの臨床研究を完成し、監督部門の許可を得て、それから製品の商業販売を始めることができます。私たちの業務計画が成功する可能性は、早期業務の発展と拡大、そして私たちが置かれている監督と競争環境の面でよく遭遇する問題、巨額の費用、困難、合併症、遅延を考慮しなければならない。医薬製品開発は高度な投機的な仕事であり、大きなリスクに関連し、資本集約型業務である。
したがって、私たちの将来性を考慮して、会社が開発の初期段階、特に私たちのような早期製薬会社がよく遭遇するコスト、不確実性、遅延、困難を考慮すべきです。潜在投資家は経営歴史が限られている会社が直面するリスクと不確実性を慎重に考慮すべきだ。特に、潜在的な投資家は、私たちができることを保証することはできません
| ● | 私たちの業務計画を成功的に実施または実行し、私たちの業務計画が健全であることを保証することはできません | |
| ● | CTX−1301、CTX−1302および/またはCTX−2103の製品開発/調製および臨床試験、ならびに任意またはすべての製品のマーケティングに成功した | |
| ● | 以前の契約製造機関(“CMO”)がCTX-1301臨床供給において経験した生産遅延に基づいて、brの製造または臨床製品の製造に成功し、商業薬品供給を確立した | |
| ● | 私たちのCTX-1301第三段階臨床計画を完成させることを含む、資本市場または他の態様で十分な資金を集めて、私たちの業務計画を実施します | |
| ● | 私たちの製品に十分な知的財産権保護を提供します |
| 8 |
| ● | 幹部と他の従業員が2023年12月に退職することを考慮して、経験豊富な管理とコンサルティングチームを誘致し、保留する | |
| ● | 私たちの候補薬が医学界や第三者支払者や消費者に受け入れられることを確実にします | |
| ● | 単独販売でも他人と協力しても候補薬の商業販売を開始します | |
| ● | 上場後の規制要件を守ること | |
| ● | 私たちの既存および/または将来調達された資金を利用して、私たちの業務戦略を効率的に実行します。 |
もし私たちが上記のいずれかを成功的に実行できなければ、私たちの業務、財務状況、経営業績、未来の成長見通しは重大な悪影響を受けるだろう。
我々はすでに運営赤字の歴史を受けており,予見可能な将来に巨額のコストが発生し続けることが予想される。私たちは現在利益を上げていないし、私たちは絶対に利益を達成したり維持したりしないかもしれない。
我々 は運営から収入が生じたことがなく,数年以内に収入が生じる可能性はあまりなく,現在運営は赤字状態 であり,配合/製造開発,候補薬物の臨床試験,上場企業運営に関するコストが発生するため,我々の運営コストは大幅に増加することが予想される。私たちは、規制部門の承認を得て、私たちの候補製品CTX-1301、CTX-1302、およびCTX-2103を商業化することに成功するまで、相応の収入なしに費用が発生することが予想されます。私たちは、私たちの候補薬をアメリカや国際的にどんな適応でマーケティングしても規制部門の承認を得ることができないかもしれません。CTX−1301、CTX−1302、および/またはCTX−2103の規制承認を得ても、任意の将来の資産の開発費用は増加し続ける。私たちが連邦薬品管理局(“FDA”)の承認を得るためにCTX-1301第三段階の臨床試験を行う時、私たちは追加の臨床開発費用を発生する。
私たちbrは設立以来経常赤字が発生しており、2023年9月30日までの累計損失は約8,600万ドルです。これらのことは、持続的な経営企業としての私たちの能力を大きく疑っており、予測可能な未来に運営を継続したり、正常な運営過程で資産と負債を返済することができない可能性があることを意味しています。もし私たちがさらなる資金を得ることができなければ、私たちは運営を続けることができないかもしれない。私たちはこれらの計画を継続して実行しているにもかかわらず、私たちが受け入れられる条項で十分な資金を得ることに成功し、持続的な運営に資金を提供することに成功する保証はない。
我々 は,我々の候補製品の臨床開発 および我々が選択可能な任意の他の適応や候補製品の開発のために,予見可能な未来に大量の現金資源を投入し続ける。これらの支出には、臨床試験、製造運営および候補製品供給のような製造および臨床開発に関連するコスト、および販売が許可された任意の製品のマーケティングおよび販売が含まれる。特に、私たちのアメリカでの第3段階試験は達成するために多くの資金が必要だ。どの臨床試験の進行と結果も高度に不確定であるため、著者らは現在と未来の候補製品の開発と商業化に必要な実際の金額を合理的に推定することができない。
私たち はいつ、または利益を達成することができるかどうか、または利益を維持することができるかどうか分からない。もし私たちが未来に利益を達成すれば、私たちは後続期間に利益を持続することができないかもしれない。実現して利益を維持できなかったことは、私たちが運営を維持する能力を弱化させ、私たちの証券の価格と私たちの資金調達能力に悪影響を及ぼす。
| 9 |
今回の発行後、運営を継続するために大量の追加資本を調達する必要があります。もし私たちが資金を集めることができなければ、私たちは破産保護や他の代替案を求めることを要求されるかもしれないが、これは私たちの証券保有者が私たちへの投資の一部または全部を失うことになるかもしれない。
今回の発行後、私たちは運営を継続するために多くの追加資金を調達し、私たちの計画の開発と商業化活動を支持し続ける必要があります。私たちの現在の業務計画によると、今回発行された純収益に手元の現金を加えることで、2024年第2四半期末までの資本需要を満たすと信じています。私たちの将来の資金需要の額と時間は多くの要素に依存するだろう
| ● | 私たちの現在とbrの開発、許可または買収の任意の候補製品の臨床試験および他の製造/製品開発活動の時間、進捗、およびコスト | |
| ● | 私たちの候補製品はアメリカや他の国での臨床試験結果です | |
| ● | 私たちの候補製品はFDA承認と任意の外国規制機関の承認のスケジュールと関連するコストを獲得した | |
| ● | 私たちが開発または獲得した任意の他の未来の候補製品の数量と特徴 | |
| ● | 戦略的協力、許可、共同推進または他の手配の能力、およびそのような計画の条項および時間 ; | |
| ● | もし私たちが現在または任意の未来の候補製品が販売を許可された場合、製造、マーケティング、販売、および流通コストを含む商業化活動のコスト | |
| ● | 承認された製品の市場受容度と受容率 | |
| ● | 我々の第三者製造および供給スケジュールによれば、現在および未来の任意の候補製品および私たちのbr商業化された任意の製品に支払われるコスト; | |
| ● | コスト および私たちが確立する可能性のある任意の他のアウトソーシング商業製造または供給スケジュールの完了時間; | |
| ● | 私たちの第三者サービスプロバイダは、私たちの限られた流動性と、サービスのプリペイドを必要としないサービスプロバイダがサービスを提供し続ける意志に基づいています | |
| ● | 私たちの候補製品に関連する任意の特許主張および他の知的財産権の費用を準備、提出、起訴、維持、弁護、および実行する; | |
| ● | 起訴または弁護私たちが巻き込まれているまたは巻き込まれる可能性のある任意の訴訟に関連する費用と、このような訴訟のために支払うべき任意の損害賠償 | |
| ● | 発生可能な任意の製品リコールに関連するコスト ; | |
| ● | 上場企業の運営コスト ; | |
| ● | 代替性および競争的製品または治療の出現、承認、獲得可能性、知覚的利点、相対コスト、相対安全性、および相対的有効性; | |
| ● | 製品および候補製品、技術またはビジネスの任意の買収に関連するコスト、および | |
| ● | 人員、施設、設備の要求。 |
| 10 |
私たち は受け入れ可能な条項で追加資金を提供するかどうか、あるいは根本的にできないかどうかを確定できない。さらに、私たちが入る可能性のある将来の債務融資は、私たちに留置権または追加債務の発生、配当金の支払い、株式の償還、特定の投資、特定の合併、合併、または資産売却取引に従事する能力の制限を含む、私たちの業務を制限する契約を適用するかもしれません。
2023年末に、私たちはいくつかのコスト削減措置を開始した。このような措置は従業員の基本賃金を減少させることと基本供給者たちを検討することを含む。また、取締役会の非経営者メンバーは、現金取締役会の費用を今後のある日に延期することに同意した。もし私たちが必要な時に受け入れ可能な条項 で追加資金を調達できない場合、またはCTX-1301、 CTX-1302および/またはCTX-2103と戦略的協力を達成できない場合、私たちの運営をさらに制限する必要があるかもしれないし、または魅力的でないbr条項協定を締結することで資金を得る必要があり、これは、少なくとも追加資金を得る前に、私たちの業務、株価、私たちのビジネス関係の第三者との関係に実質的な悪影響を及ぼすかもしれない。
追加資本の調達は、私たちの株主を希釈し、私たちの運営を制限したり、製品の権利を放棄することを要求したりする可能性があります。
私たちが相当な製品収入を生成することができる前に、私たちは、公共または私募株式または債務融資、第三者融資、マーケティングおよび流通手配、ならびに他の協力、戦略連合および許可スケジュール、またはこれらの方法の任意の組み合わせによって私たちの現金需要を満たす予定だ。私たちは約束された外部資金源を持っていない。もし私たちが株式または転換可能な債務証券を売却することによって追加資本を調達する場合、あなたのわが社での所有権権益は希釈される可能性があります。これらの証券の条項は、清算または他の特典を含む可能性があり、株主としての権利に悪影響を及ぼす可能性があります。br}債務および持分融資(利用可能であれば)は、特定のbr行動をとる能力を制限または制限するプロトコル、例えば、私たちの株を償還すること、投資を行うこと、追加債務を発生させること、資本支出を行うこと、配当金を発表すること、または私たちが取得、販売できるかもしれない知的財産を制限する能力を含むいくつかの合意に関連する可能性があります。また,WFIAはあらかじめ出資した引受権証を持っており,我々の普通株1,028,955株を購入することができる。このような事前出資の引受権証を行使すると、わが社での所有権権益が希釈されます
もし私たちが将来の協力、戦略連合、または第三者許可手配を通じてより多くの資本を調達する場合、私たちは私たちの知的財産権、未来の収入流、研究計画または候補製品に対する貴重な権利を放棄しなければならないかもしれない、または私たちに不利になる可能性のある条項でライセンスを付与しなければならないかもしれない。もし私たちが必要な時にもっと多くの資金を集めることができなければ、私たちは私たちの候補製品の開発または将来の商業化努力を延期、制限、減少または終了することを要求されるかもしれません。あるいは私たちが自ら開発し、マーケティングする候補製品の権利を与えることができます。
| 11 |
税法の変化は私たちの業務、財務状況、経営結果、キャッシュフローに重大な悪影響を及ぼす可能性があります。
私たちは私たちが業務を展開している司法管轄区の税収法律、法規と政策の制約を受けて、これらの管轄区はアメリカの連邦、州と地方政府及び外国の管轄区の税務当局を含む可能性があります。税法の変化、その他の要素は、私たちの納税義務に変動をもたらし、他の方法で私たちの納税状況および/または納税義務に悪影響を及ぼす可能性があります。税務機関と他の政府機関は、私たちの管轄区の所得税規則を検討し続けています。税法の変化(これらの変化は追跡力を持つ可能性がある)は、私たちまたは私たちの株主に悪影響を及ぼす可能性がある。私たちは将来どのような税金提案が提出されるか、あるいはこれらの変化が私たちの業務にどのような影響を与えるかを予測できませんが、これらの の変化は、それらが税務法律、法規、政策または実践に組み込まれていれば、私たちの財務状況と私たちの司法管轄区域の未来の全体的な有効税率に影響を与え、税務コンプライアンスの複雑さ、負担、およびコストを増加させる可能性があります。
私たちは純営業損失を利用して将来の課税収入を相殺する能力が制限されるかもしれません。
米国税法の制限により、私たちのbr純営業損失繰越(“NOL”)といくつかの他の税務属性は未来のbr所得税負債を相殺するために使用できない可能性がある。減税·雇用法案(TCJA)によると、2017年12月31日までの納税年度に発生する連邦NOL は無期限に繰り越すことができる。繰越はその後の年間純収入の80%を超えてはならない。
さらに、“規則”第382節および第383節には、“所有権変更”(通常は、3年以内に会社の株式所有権が50%を超える任意の変更を意味する)を経験した会社が、変更前のNOLおよび税収控除繰越を利用して将来の課税所得額を相殺する能力を制限する規則も含まれている。これらのルールは,通常,会社の5%以上の株を直接または間接的に所有する株主の所有権変更や,会社の新規発行株による所有権変更 に重点を置いている.一般に,所有権変更が発生した場合,NOLと税収控除繰越および何らかの固有損失を用いた年間課税所得額制限は,適用される長期免税税率 と所有権変更直前の会社株価値の積に等しい.したがって、このような所有権変更後、私たちはこのような損失と相殺期限が切れる前に損失で私たちの課税収入を相殺することができないか、あるいは私たちの納税義務を相殺することができないかもしれません。この場合、私たちは私たちが所有権変更を経験していない場合よりも大きな連邦と州所得税負債 が生じるかもしれません。
我々の独立公認会計士事務所は,2022年12月31日現在と2021年12月31日までの会計年度報告 に解釈的なbr段落を含み,継続的に経営していく企業としての能力があるかどうかを示している。
私たちの独立公認会計士事務所は、私たちが2022年12月31日と2021年12月31日までの財務諸表に関する報告 には、私たちが継続的に経営している企業として経営を続ける能力に大きな疑問があることを示しています。設立以来,経常的な運営損失と負のキャッシュフローを経験しており,将来的には運営損失が発生し,大量の現金資源が消費されることが予想される。追加融資がなければ、これらの条件 は、持続的な経営企業としての私たちの能力を大きく疑わせ、予測可能な未来に運営を継続できない可能性があり、正常な運営過程で資産や債務返済を実現できない可能性があることを意味する。もし私たちが資金を得ることができなければ、私たちは運営を続けることができないかもしれない。私たちはこれらの計画を継続して実行しているにもかかわらず、私たちが受け入れられる条項で十分な資金を得ることに成功し、持続的な運営に資金を提供することに成功する保証はない。
開発,臨床試験,製造と規制承認に関するリスク
私たちは主に私たちの候補製品CTX-1301およびCTX-1302の成功した開発と商業化に依存し、CTX-1301およびCTX-1302はADHDの治療のために、CTX-2103は不安を治療するために使用され、これらの製品は製品/早期臨床開発(CTX-2103)、臨床開発 (CTX-1301)または将来の製品開発(CTX-1302)を計画しており、まだ承認されていない。私たちがこのような候補製品または他の候補製品に対する規制機関の承認を得ることは保証できません。これはそれらが商業化できる前に必要です。
私たち はまだ候補製品の開発および/または規制部門の承認を得ていません。開発は大量の財政資源、広範な候補製品開発と臨床試験を投入する必要がある。この過程は長年の努力を必要とするが、最終的な成功の保証はない。
| 12 |
私たちが候補製品から収入を得る能力は、それらの成功した開発、規制承認、最終的な商業化に大きく依存し、数年以内には起こらないと予想される。私たちの候補製品の成功は多くの要素にかかっていますが、これらに限定されません
| ● | 製品開発と必要な臨床試験に成功しました | |
| ● | 私たちの臨床試験で成功しゴールを達成しました | |
| ● | 私たちの候補製品が関連するリスクが収益よりも大きいことを証明する | |
| ● | 第三者製造業者との締結および保守手配を含む、我々の候補製品のための製造プロセスの開発に成功した | |
| ● | 我々の候補製品を生産するための施設に対するFDAの承認前検査の完了と、臨床試験点の選択に成功した |
| ● | 米国薬品監督管理局(DEA)がFDAの提案を考慮して、候補製品の制御物質スケジュールを決定することを含む、適用監督機関からの適時な上場許可を受けた | |
| ● | 私たちの候補製品のために特許、商標および商業秘密保護および規制排他性を獲得し、維持し、他の側面で知的財産権の組み合わせにおける私たちの権利を保護する; | |
| ● | 現在の良好な製造仕様またはcGMPを含む法規要件の遵守を維持する; | |
| ● | 承認された後、単独または他社と協力して、候補製品の商業販売を開始する | |
| ● | 患者、医学界、第三者支払人が私たちの候補薬を承認した場合、brを受け入れる | |
| ● | 他の療法と効果的に競争しています | |
| ● | 医療保険や適切な精算を受けて維持します | |
| ● | 承認後に薬物製品の持続的に許容可能な安全性と有効性を維持する。 |
上記の1つまたは複数の要因(その多くは制御できない)をタイムリーにまたは根本的に実現できなければ、私たちは重大な遅延やコスト増加に遭遇したり、規制部門の承認を得たり、私たちの候補製品を商業化することができない可能性があります。規制部門の承認を得ても、私たちの任意の候補製品を商業化することに成功することはできません。したがって、私たちはあなたに保証することはできません。私たちは私たちの候補製品または任意の未来の候補製品を販売することによって、運営を継続するために十分な収入を生成することができます。
CTX−1301、CTX−1302、および/またはCTX−2103に対する製品開発は、様々な理由で失敗する可能性があるが、これらに限定されない
| ● | 候補製品は臨床研究で失敗しました | |
| ● | 候補製品に対する患者の副作用または他の安全問題への適応; | |
| ● | 臨床試験データが不足しており、候補製品の有効性或いは優位性を支持できない | |
| ● | 開発または商業化活動に使用可能な十分な数の製品をタイムリーかつ経済的に効率的に生産することができない | |
| ● | 価格設定と精算を含む規制環境の変化は、新しい適応に適応するために新製品あるいは既存製品を開発することが魅力的ではなくなった。 |
| 13 |
FDAまたは他の規制機関が私たちの候補製品の販売前審査を行うことは長くて不確実なプロセスであり、承認が延期される可能性があり、brが制限され、または拒否される可能性があり、いずれも私たちが運営収入をもたらす能力に悪影響を及ぼす。
新薬申請(“NDA”)の承認を得るまで,我々の候補薬物製品を米国で販売することは許されていない。FDA承認を得るのに必要な時間(あれば)は予測不可能であるが、臨床試験開始後には通常数年を必要とし、これは、規制機関の重大な裁量権および関連する候補製品のタイプ、複雑性、および新規性を含む多くの要因に依存する。我々は、FDAまたは任意の司法管轄区域の任意の他の規制機関に、brなどのマーケティング申請または任意の同様の出願を提出していない。
FDAは薬品承認プロセスにおいてかなりの自由裁量権を有しており、様々な理由で承認候補製品 を延期、制限、または拒否することができる。例えばFDA:
| ● | CTX−1301、CTX−1302、CTX−2103、または我々が決定し開発する可能性のある任意の他の候補製品の505(B)(2)規制承認経路に依存できないかどうかを決定することができるかどうか | |
| ● | INDまたはNDAの一部として提供される情報が不十分であることを決定できるかどうか、臨床的欠陥または他の側面を含むことは、任意の適応に対する任意の候補製品の安全性および有効性を証明することができない | |
| ● | 生物学的同等性研究および/または臨床試験のデータは、セキュリティプロトコルの提出をサポートするのに十分であるとは考えられないかもしれないが、任意の発見安全リスクが私たちの候補製品の臨床および他の利点を超えることを含む、セキュリティプロトコルの提出をサポートするのに十分である | |
| ● | 私たちの製品の安全性、有効性、薬物動態学または他の特性 候補製品が承認される前に、またはそのような研究を承認の条件とすることを要求するために、追加の研究を要求することができるかもしれない | |
| ● | Brは、私たちの臨床試験設計または製品開発製造データ、生物学的同等性研究および/または臨床試験からのデータの解釈に同意しないか、または私たちの試験設計を審査およびレビューした後であっても、承認要求を変更する可能性がある | |
| ● | 我々の505(B)(2)セキュリティプロトコルは、ある市販薬またはいくつかの薬剤に適切に依存しないか、または我々のCTX−1301、CTX−1302、CTX−2103、または任意の他の候補製品の出願が、上場薬物の特許または非特許排他性によって承認されることが阻止されることを決定することができる | |
| ● | 候補製品で使用される活性医薬成分(“原料薬”)を供給するために、私たちが契約を締結した第三者製造業者の製造プロセスまたは施設の欠陥を見つけることができるかどうか | |
| ● | 私たち自身の製造プロセスにおける不足点を見つけることができ、あるいは私たちの候補製品を生産するために提案した製造プロセスや施設を拡大する提案を見つけることができる | |
| ● | 私たちの候補製品の適応を承認することは、私たちが要求するよりも少ないか、または高価な承認後の臨床試験の表現に従って承認されるかもしれない | |
| ● | 承認政策を変更したり、新しい法規を採用したりすることができる | |
| ● | おそらく は私たちの候補製品の商業化に成功するために必要または望ましいと思うラベル宣言を承認しないかもしれません。 |
承認プロセスの時間と費用、および将来の臨床試験結果の予測不可能性および他の促進要因は、米国または他の司法管轄区域で規制部門の承認を得ることができなくなる可能性があり、米国または他の司法管轄区では、CTX-1301、CTX-1302、CTX-2103、br}または私たちが開発しているまたは将来的に開発を求める可能性のある任意の他の候補薬剤を招く可能性があり、これは、私たちの業務、運営結果、および将来の見通しを大きく損なうことになる。この場合、新たな臨床試験を行うリソースがない可能性もあり、および/または、任意のこのような候補薬剤のさらなる臨床開発が不合理であることを決定し、そのような任意の計画を停止する可能性がある。
| 14 |
臨床試験費用が高く、設計と実施が困難であり、完成まで数年かかる可能性があり、しかも結果は確定していない。私たちは、候補製品の商業化を完了または最終的に達成できない間に、追加の コストを生成したり、遅延に遭遇する可能性がある。
Br}は、私たちの任意の候補製品がいつ、または人体で有効または安全を証明するかどうかを予測することができず、規制部門の許可を得るだろう。規制部門から任意の候補製品を販売する市場承認を得る前に、製品/製造 開発を完了し、その後、私たちの候補製品の人体における安全性と有効性を証明するための臨床試験を行わなければならない。臨床試験は高価で設計や実施が困難であり,完成まで数年かかる可能性があり,結果は不確定である。1つまたは複数の臨床試験の失敗は、開発の任意の段階で発生する可能性がある。早期臨床試験の結果は後続の臨床試験の成功を予測できない可能性があり、臨床試験の中期結果も最終結果を予測できるとは限らない。早期研究結果の解釈には慎重が必要であり,これらの研究は通常規模が小さく,ある被験者が積極的な傾向が現れていることを示している。より多くの被験者を募集した臨床試験後期の結果 は,期待される安全性や有効性結果を示すことができないか,あるいは同じ候補製品の早期試験結果と一致しない可能性がある。後期臨床試験結果は早期臨床試験 を複製できない可能性があり、原因は非常に多く、試験設計が異なり、試験終点が異なる或いは研究中に試験終点が不足し、被験者 群、被験者数、被験者選択標準、試験持続時間、薬物投与量と調合及び早期研究は統計能力が不足していることを含む。そのほか、臨床データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品が早期と後期の臨床試験で満足できると考えているが、しかし依然としてその製品に対するbr}の発売許可を得られなかった。
私たちのbrは、臨床試験中に、または臨床試験の結果によって、多くの予見不可能なイベントに遭遇する可能性があり、これらのイベントは、市場の承認を得ることを遅延または阻止するか、または私たちの候補製品を商業化することを阻止する可能性があるが、これらに限定されない
| ● | 臨床試験の開始または継続を支持するために、満足できる臨床前、毒理学または他の体内または体外データを生成することができない; | |
| ● | 監督機関あるいは機関審査委員会は私たちまたは私たちの調査者が必要に応じて臨床試験を開始し、予想される試験地点で臨床試験を行うことを許可したり、臨床試験方案を修正したりすることを許可してはならない | |
| ● | 私たちのbrは、予想される試験場所と契約研究組織またはCROと受け入れ可能な臨床試験契約または臨床試験方案 との合意に遅れたり、合意に達しない可能性がある | |
| ● | 失敗、遅延、または十分な数の試験サイトを決定および維持することができず、その多くは他の臨床項目 に参加している可能性がある; | |
| ● | 我々の候補製品の臨床試験は否定的あるいは不確定な結果が生じる可能性があり、必要な場合には統計的意義を証明できないことを含めて、私たちは決定あるいは監督機関が追加の臨床試験を要求したり、薬物開発計画を放棄したりする可能性がある | |
| ● | 私たちの候補製品の臨床試験に必要な被験者の数は私たちが予想していたより多いかもしれません。これらの臨床試験の登録人数は私たちが予想していたよりも遅いかもしれません。あるいは参加者がこれらの臨床試験から退出する速度は私たちが予想していたよりも高いかもしれません | |
| ● | Br患者は試験或いは治療後のフォローアップを完成できなかった | |
| ● | 治療期間や治療後に患者を十分に監視することはできない | |
| ● | 臨床サイトと調査者は試験方案から外れ、法規の要求に従って試験または退出試験を行っていない | |
| ● | 私たちの第三者請負業者は、法規の要求や試用合意を遵守できないかもしれないし、私たちに対する契約義務をタイムリーに履行できなかったり、全く遵守していないかもしれない |
| 15 |
| ● | 規制機関または機関審査委員会は、規制要件に適合していないこと、または参加者が受け入れられない健康リスクに直面していることを発見することを含む、様々な理由で臨床研究を一時停止または終了することを私たちまたは私たちの調査者に要求する可能性がある | |
| ● | 我々の候補製品の臨床試験コストは、505(B)(2) 秘密協定を通じて私たちの候補製品を承認できない場合を含む、私たちが予想していたよりも高いかもしれない | |
| ● | 観察された安全発見または他の原因により、FDAまたは同様の規制機関が実施した臨床保留は、臨床試験を開始または延期することができなかった、または完成できなかった | |
| ● | 規制部門は私たちの試験設計や実施に同意しないかもしれない | |
| ● | 臨床試験のために十分な量の品質で許容できる候補薬剤を生産することができない; |
| ● | 私たちのbr候補製品は副作用または他の意外な特徴を有する可能性があり、私たちまたは私たちの調査者、監督機関 または機関審査委員会が試験を一時停止または終了させる可能性がある。 |
もし私たちの候補製品に対して、現在予想されている以上の追加の臨床試験または他の試験を行うことが要求された場合、もし私たちが候補製品の臨床試験または他の試験を成功させることができなければ、これらの試験または試験の結果が陽性でないか、または軽度陽性である場合、または安全問題がある場合、私たちは:
| ● | 私たちの候補製品の市場承認を得るために遅延された | |
| ● | 発売許可を得ていない; | |
| ● | Brの承認を得た適応または患者群は、期待または期待ほど広くない | |
| ● | Brの承認を得たが、候補製品を商業化するために必要な宣言に成功しなかった | |
| ● | 重大な使用または配布制限またはセキュリティ警告を含むラベルの承認を得ること; | |
| ● | 追加の上場後のテスト、監督またはその他の要求を受ける;または | |
| ● | 市場承認を得た後、この製品 を下積みします。 |
もし私たちがテスト、臨床試験、製造、あるいは上場承認を得るのに遅延があれば、私たちの開発コストも増加する可能性があります。例えば、CTX-1301の開発コストが増加しているのは、第3段階の固定用量研究の生産がこの研究に必要な最終用量強度を遅延させているからである。われわれのいずれの臨床試験が計画通りに開始されるかどうかは分からないが, は再構成や計画どおりに完成する必要があるかどうか,あるいは全く知られていない。2023年末,我々はFDAのフィードバックにより,CTX−1301の臨床開発計画の変更を発表し,それに応じてCTX−1301の2つの3期試験の登録を停止した。私たちは、規制部門のCTX-1301の承認を得るために、これらの試験を再起動し、および/または新しい試験を開始する必要があるかもしれない。このような臨床試験を再開したり開始したりすることは難しいか不可能であることが分かるかもしれない。重大な製品製造や臨床試験遅延は、候補製品を商業化する独占的な権利を持っているか、または競争相手が製品を市場に出すことを可能にする任意の期限を短縮し、候補製品を商業化することに成功する能力を弱める可能性がある。
小児および青少年のためのCTX-1301およびCTX-1302臨床試験の規制承認を得るには、小児科群の規制承認要求がより厳しいので、追加の研究および/またはより長い研究持続時間が必要となる可能性がある。
小児科薬物の開発には安全投与量と長期安全性を確定するための追加の研究が必要かもしれない。これらの追加的な研究は、規制機関が成人の薬品使用を許可するために必要な資源ではなく、大量の追加資源を投入する必要があるかもしれない。我々はCTX-1301の重要な3期固定用量小児科と青少年の安全性と有効性研究、ならびにCTX-1301の3期小児科用量最適化開始と持続時間研究を停止したが、許可を得るためには、これらの試験および他の試験を再開し、完成しなければならないかもしれない。これらの追加的な要件のために、CTX−1301およびCTX−1302の承認が延期される可能性があり、これは、CTX−1301およびCTX−1302のビジネス見通しに悪影響を及ぼす可能性があり、製品収入を創出する能力を深刻に遅らせる可能性がある。また,新冠肺炎(あるいは他の潜在的流行病)により,臨床試験に参加する児童数が減少する可能性がある。私たちのbrは私たちが監督部門の許可を得て、私たちの候補製品を小児科人群あるいは成人群で商業化することを保証することはできません。
| 16 |
候補製品の製造または処方を変更する方法 は、追加のコストまたは遅延をもたらす可能性がある。
候補製品は非臨床試験および早期および後期臨床試験によって開発されるため、製造方法や配合など、開発計画の様々な面で変更され、プロセスおよび結果の最適化に努める可能性がある。このような変化はこのような期待された目標を達成できないかもしれない。これらの変更のいずれも、私たちの候補製品のパフォーマンスが異なることをもたらし、計画中の臨床試験または変更材料を使用した他の将来の臨床試験の結果に影響を与える可能性があり、または候補製品の安全性またはリスク状態を変化させる可能性があり、これはFDAまたは他の規制機関のさらなる質問に関連する可能性がある。このような変化はまた、追加のテスト、FDA通知、またはFDA承認を必要とする可能性がある。これはbr臨床試験の完成を遅らせる可能性があり、移行臨床試験或いは1つ或いは複数の臨床試験を繰り返し、臨床 試験コストを増加させ、著者らの候補製品の承認を延期し、そして著者らの製品販売と収入を創造する能力を脅かす必要がある。
私たちの候補製品CTX-1301およびCTX-1302は制御された物質を含み、その製造、使用、販売、輸入、輸出、処方、および流通はDEAによって規制されている。
私たちの候補製品を商業化できる前に、DEAはFDAの提案を考慮して制御物質スケジュールを決定する必要があるだろう。これは長い過程かもしれませんし、候補製品のマーケティングを延期するかもしれませんし、私たちが資格を得る可能性のある任意の規制排他期間を短縮するかもしれません。我々のCTX-1301およびCTX-1302候補製品が承認されれば、 は1970年の“制御対象物法”(以下、“CSA”) およびDEA実施条例で定義された“制御物質”として規制され、これらの条例はDEAによって管理されている登録、安全、記録保存、報告、貯蔵、流通、輸入、輸出、在庫、割当、その他の要求を確立する。これらの要求は私たち、私たちのbr契約製造業者とディーラー、そして私たちの候補製品の処方と調剤業者に適用されます。DEAは規制対象物質の処理を閉じた流通チェーンで管理している。この規制は、製造·包装に使用される設備や原材料に延長され、不正な商業ルートへの損失や移転を防止する。いくつかの州と外国でもこれらの薬物を制御物質として独立して管理している。
DEAは制御物質を付表I,II,III,IVまたはV類と規定した。乱用の可能性および身体的または心理的依存によると、承認された薬品は、付表2、付表3、付表4または付表5とすることができ、その中で付表2物質は乱用リスクが最も高い物質と考えられ、付表5物質はこのような物質の中で相対的に乱用リスクが最も低い物質である。付表二薬brは以下の特徴に適合する薬物である
| ● | この薬には高い乱用の可能性があります | |
| ● | この薬剤が現在米国治療で受け入れられている医療用途、または厳格な制限の下で現在受け入れられている医療用途; および | |
| ● | 薬物乱用は深刻な心理的あるいは身体的依存を招く可能性がある。 |
CTX-1301およびCTX-1302中の活性医薬成分(ジメチルアセテートおよび右旋フェニルプロピルアミン)は現在、付属表二製品として登録されている。我々の将来のいくつかの候補品もDEAがCSA下の付表II制御物質 とされる可能性が予想される。したがって、承認されれば、製品の製造、輸送、貯蔵、販売、および使用は高度に規制されるだろう。付表二薬品は登録、安全、記録保存と報告の面で最も厳格な要求を受け、これらの薬品の配布、処方と配布は厳格な監督管理を受けている。
規制された物質を製造、流通、配布、輸入または輸出するいかなる施設も年次登録を行う必要がある。登録は特定の場所、活動、そして制御された物質スケジュールに対して行われる。
また、DEA割当制度は制御物質の供給と生産を制御し、制限し、私たちの製品はDEA生産と調達割当計画のbrによって制約される可能性がある。DEAはDEAの合法的な科学と医薬需要を満たすために必要な数量の推定に基づいて、アメリカが全部でどれだけの規制された物質 を生産できるかのために総割当量を確立した。制御された物質の生産者たちは年間割当量を申請しなければならない。もし私たちまたは私たちの契約の製造業者またはサプライヤーがDEAから十分な割当量を得ていない場合、もし私たちの候補製品が発売を許可された場合、私たちは私たちの臨床試験を達成したり、商業需要を満たすのに十分な数のこれらの制御された物質を得ることができないかもしれない。
Brの制限により,これらの法律法規は,我々の候補製品が制御されたbr物質を含む商業化を制限する可能性がある。これらの法律と法規を遵守しないことはまた、私たちのDEA登録が撤回され、製造と流通活動が中断され、法令に同意し、刑事と民事処罰、国家行動などの結果を招く可能性がある。
| 17 |
私たちが臨床試験の被験者登録中に遅延や困難に遭遇した場合、必要な規制承認を受けることは延期または阻止される可能性がある。
FDAや米国以外の同様の規制機関の要求に応じてこれらの試験に参加するのに十分な数の合格者を見つけることができない場合、私たちの候補製品の臨床試験 を開始または継続できない可能性がある。将来の臨床試験で被験者を募集する成功度を予測することはできませんもし著者らが1つの臨床試験でbr名の被験者の募集に成功しなかった場合、私たちがいつ次の臨床試験を開始できるかに影響する可能性があり、これは私たちの監督管理機関が私たちの候補製品を承認し、それを商業化する努力に重大な遅延が出現することを招く可能性がある。また,われわれのいくつかのライバルであるbrは,我々の候補品と同様の適応を治療するための臨床試験を行っているが,本来われわれの臨床試験に参加する資格のある被験者は,競争相手の臨床試験に移行する可能性がある。科目募集は他の要素の影響を受け、 を含むが、これらに限定されない
| ● | 試験プログラムに規定されている被験者群の規模と性質 | |
| ● | 研究に関する資格基準 | |
| ● | 研究を受けた候補製品のbr感知のリスクと収益; | |
| ● | 候補品が制御物質である可能性があります | |
| ● | 被験者が臨床試験で経験した深刻または予期しない薬物関連有害事象; | |
| ● | 研究中の疾患または状態を治療するために承認された薬物の供給状況; | |
| ● | 臨床試験の適時登録を促進するための努力程度 | |
| ● | 医師の患者の回診方法 |
| ● | 被験者のインフォームドコンセントを獲得し維持する能力は | |
| ● | 臨床試験において被験者および後続治療に戻る能力を保持している | |
| ● | 必要なテスト、プログラム、後続行動を含む臨床試験設計 | |
| ● | 治療中および治療後に被験者の能力を十分に監視すること | |
| ● | 新たな研究者と臨床サイト の追加を遅延させる; | |
| ● | 臨床試験サイトを臨床試験から撤回した | |
| ● | 同じ適応のための臨床開発における他の候補薬の存在;および | |
| ● | 潜在被験者の臨床試験地点の近似性と有用性。 |
臨床試験に十分な数の被験者を募集することはできず、深刻な遅延を招き、1つ以上の臨床試験を完全に放棄することが要求される可能性がある。これらの臨床試験の登録遅延は、候補製品の開発コストを増加させる可能性があり、これは私たちの価値を低下させ、追加融資を受ける能力を制限する可能性がある。
私たちの臨床試験は、私たちの候補製品の安全性と有効性を証明できないかもしれないし、あるいは私たちの候補製品の開発過程で深刻な不良または受け入れられない副作用 を発見する可能性があり、これは規制部門の承認および商業化を阻止または延期する可能性があり、私たちのコストを増加させるか、または私たちの一部またはすべての候補製品の開発を放棄または制限する必要がある。
私たちの候補製品の商業販売に対する規制部門の承認を得る前に、私たちの候補製品は各目標適応において安全かつ有効であり、開発のどの段階でも故障する可能性があることを証明しなければならない。臨床試験では目標適応に対して検討されている候補製品の安全性や有効性が証明されないことが多い。
| 18 |
多くの医薬製品があるため、著者らの候補製品の治療は副作用或いは副作用或いはbr事件を引き起こす可能性がある。私たちの候補製品には承認された活性医薬成分が含まれているにもかかわらず、これは、私たちの候補製品において活性医薬成分または薬剤クラスを使用することによって生じる副作用がよく知られていることを意味するが、私たちの候補製品は依然として副作用を引き起こす可能性がある。
私たちの候補製品が臨床試験において深刻な副作用または予期しない特徴を有する場合、私たちは、副作用または他の特徴があまり一般的ではない、あまり深刻ではない、またはリスク収益の観点からより受け入れやすいより狭い用途またはサブ集団に開発を制限する必要があるかもしれない。FDAまたは機関審査委員会はまた、登録対象への潜在的な深刻な傷害を制限するために、セキュリティ情報に基づいて、臨床試験を一時停止、中断、または制限することを要求する可能性がある。このような調査結果 は、規制機関が私たちの候補製品にマーケティング許可を提供できないことをさらに招く可能性がある。
我々のbr候補製品は、悪影響をもたらすか、または他の特性を有する可能性があり、その規制承認を遅延または阻止するか、または任意の承認のラベルまたは市場受容度の範囲を制限するか、または上場承認後に重大な負の結果をもたらす可能性がある(ある場合)。
もし私たちのすべての製品が市場の承認を得た後に深刻または予期しない副作用を招く場合、いくつかの潜在的な重大な負の結果を引き起こす可能性があるが、これらに限定されない
| ● | FDAは、製品の安全性および有効性を監視するために、追加の臨床試験または臨床試験、または高価な上場後試験および監視を必要とする可能性がある | |
| ● | 規制部門は、製品の承認を撤回したり、その流通に制限を加えたりすることができる | |
| ● | 私たちは、患者に配布するために、このような副作用のリスクを概説するための薬物ガイドラインを作成する必要があるかもしれないし、または、製品の利益がリスク よりも大きいことを確実にするために、リスク評価および緩和戦略(“REMS”)を実施する必要があるかもしれない | |
| ● | 規制当局は警告や禁忌症のようなラベル宣言の追加を要求するかもしれない | |
| ● | 私たち は製品の配布または管理方法の変更を要求される可能性があります | |
| ● | 私たちは自発的に製品をリコールする必要があるかもしれない | |
| ● | 私たちは起訴され、私たちの製品候補製品に接触または服用した個人による傷害に責任を負う可能性があります | |
| ● | 私たちの名声は影響を受けるかもしれない。 |
これらの事件のいずれも、影響を受けた製品または候補製品に対する市場の受容度を達成または維持することを阻止することができ、私たちの製品および候補製品の商業化コストを大幅に増加させる可能性がある。
FDAが我々の候補製品と承認されたbr参照薬が十分な生物学的同等性または類似の生物利用度を有すると結論しなければ、あるいはFDAが予想される505(B)(2)NDA経路での承認を許可しなければ、候補製品の承認経路は予想よりもはるかに長くなり、コストもはるかに高くなり、もたらす合併症やリスクも予想よりもはるかに大きくなり、FDAは最終的に候補製品を承認しない可能性がある。
連邦食品、医薬品及び化粧品法案(“FDCA”)第505条(B)(2)条は、少なくともいくつかの承認に必要な情報が、出願人又は出願人のための調査からではなく、調査を行う者から参考又は使用の権利を得ていない場合に機密協定を提出することを可能にする。FDAのFDCA第505(B)(2)節の解釈は,NDAを承認するために,出願人が発表された文献やFDA以前の承認製品の安全性と有効性の調査結果に部分的に依存することを許可することである。FDAはまた、以前に承認された製品との任意の偏差をサポートするために、出願人に追加の臨床試験または測定を要求する可能性がある。次に、FDAは、参照製品のすべてまたは一部が承認されたラベル適応および第505条(B)(2)の出願人が求めた任意の新しい適応について新製品候補を承認することができる。FDAは、ブラックボックス警告を含むすべてまたは部分的な制限、禁忌症、警告または注意事項を申請者の製品ラベルに含むことを要求することができ、またはラベルに制限、禁忌症、警告または注意事項を追加することを要求することができる。我々の戦略の重要な要素の1つは、505(B)(2)NDA経路を介して、現在の候補製品CTX−1301、CTX−1302、およびCTX−2103に対するFDAの承認を求めることである。FDAが、我々の候補製品が505(B)(2)節の要件を満たしていないと判断した場合、または候補製品が承認された製品と生物学的同等性または同様のバイオアベイラビリティを有することが証明できない場合、他のデータおよび情報を提供し、505(B)(1)節に従って提出された従来のNDAの規制承認に適した追加基準を満たすために、追加の臨床試験を行う必要があるかもしれない。さらに、FDAが505(B)(2)NDA経路に従うことを可能にしても、これは候補製品に依存して、製品の安全性または有効性を評価する臨床試験を含む追加の臨床試験を行う必要があるかもしれない。このような状況が発生すれば,我々の候補製品のFDA承認に要する時間と財政資源,および我々の候補製品に関連する合併症やリスクが大幅に増加する可能性がある。
| 19 |
さらに、505(B)(2)セキュリティプロトコル経路に従わない場合、新しい競合製品が私たちの候補製品よりも早く市場に投入される可能性があり、これは、私たちの競争地位およびビジネスの将来性を損なう可能性があります。505(B)(2)セキュリティプロトコル 経路の採用が許可されても,候補製品が必要な商業化承認(あれば)をタイムリーに得ることは保証されない.他の会社は私たちの前に類似製品の製品承認を受けるかもしれません。これは私たちが製品brの承認を得る能力を延期し、私たちをより激しい競争に直面させることになります。
さらに、FDAは過去数年間505(B)(2)に従って多くの製品を承認しているにもかかわらず、一部の製薬会社および他の会社はFDAによるFDCA 505(B)(2)の解釈に反対し、FDAの以前の安全性および有効性調査結果に依存することを可能にする。FDAが505(B)(2)条の解釈を変更する場合、または505(B)(2)に対するFDAの解釈が法廷で成功的に挑戦された場合、FDAが将来提出する任意の505(B)(2)NDA を承認することを遅延または阻止する可能性がある。さらに、FDAは3年間の排他的条項の解釈を採択し、この解釈によれば、505(B)(2)の申請は、以前に承認された排他的な薬物(またはその薬剤に関する任意の安全性または有効性情報)に依存しなくても排他的に阻止されることができる。FDAの説明によると、私たちの1つまたは複数の候補製品の承認は、私たちの候補製品といくつかの革新的な特徴を有する以前に承認された医薬製品との排他的なbrによって阻止される可能性があり、505(B)(2)申請が以前に承認された医薬製品を市販薬として識別していなくても、またはその任意の安全性または治療効果データに依存する。もし私たちの候補製品が規制部門の承認を得られなければ、私たちの収入創出能力 を大きく制限し、適切なすべての適応とラベル宣言がこのような承認を得られなければ、私たちの潜在収入 を減少させる可能性があると考えられる。
我々の候補製品が505(B)(2)法規経路に従って承認された場合であっても、承認は、我々が要求するよりも限られた被験者を含む製品が発売される可能性のある指定された用途によって制限される可能性があり、ブラックボックス警告を含む製品ラベルに禁忌症、br警告または予防措置を含むことが要求される可能性があり、他の承認条件によって制限される可能性があり、 または製品の安全性または有効性、またはREMSのような他の発売後の要求を監視するために、高価な発売後の臨床試験、試験および監督の要求が含まれる可能性がある。FDAはまた、ラベルが候補製品の商業化に成功するために必要または必要なラベルを含むことを許可しないかもしれない。
1つの管轄区域で私たちの候補製品に対する規制承認を獲得し、維持することは、私たちの候補製品の規制承認を他の管轄区域で成功的に得ることを意味するわけではない。
私たちが1つの管轄区域で私たちの候補製品の監督管理承認を獲得し、維持しても、このような承認は私たちの が他の管轄区で規制承認を得ることができるか、または維持できる保証はありませんが、1つの管轄区域で規制承認を得ることができないか、または遅延することは、他の管轄区の監督管理の承認プロセスに悪影響を及ぼす可能性があります。例えば、FDAが米国内での候補製品の販売を許可しても、外国司法管轄区の比較可能な規制機関は、候補製品のこれらの国/地域での製造、マーケティング、普及を承認しなければならない。承認手続きは司法管轄区域によって異なり、1つの管轄区で行われる調査は他の管轄区の監督機関によって受け入れられない可能性があるため、米国とは異なる要求および行政審査期限に関連する可能性がある。
外国の監督管理の許可を得て、外国の監督管理要求を遵守することは私たちに重大な遅延、困難とコストをもたらす可能性があり、私たちの製品がある国/地域で発売されることを延期または阻止する可能性がある。もし私たちが国際市場の規制要求を遵守できず、および/または相応のマーケティング承認を得ることができなければ、私たちの目標市場は減少し、候補製品のすべての市場潜在力を達成する能力は損なわれるだろう。
また,FDAや適用される外国規制機関が米国や他の管轄地域以外で行われている臨床試験を受けた研究データは,何らかの条件によって制限される可能性がある。外国の臨床試験のデータがアメリカでの上場承認の根拠として使用されることを目的としている場合、FDAは通常、(I)データがアメリカの人口とアメリカの医療実践に適用されない限り、外国のデータのみに基づいて申請を承認し、(Ii) 試験は公認能力を有する臨床研究者によって行われ、GCP規定に符合しない。また、十分な患者群と統計能力を含むFDAの臨床試験要求を満たさなければならない。多くの外国の規制機関は似たような承認要求を持っている。また、いかなる外国裁判も、裁判を行う外国司法管轄区域に適用される現地法律によって管轄される。FDAや任意の適用された外国規制機関が米国や適用司法管轄区以外で行われた試験データを受け入れることは保証されていない。
| 20 |
私たちのbrは、CTX-1301の第3段階臨床試験または任意の他の候補製品の将来の臨床試験を成功させることができないかもしれない。
3期臨床試験を行うことは複雑な過程である。われわれの管理チームメンバーは過去に他社勤務時に3期臨床試験を行ったことがあるにもかかわらず,会社としてはこれまで3期臨床試験を行ったことがないため,我々が予想していたよりも多くの時間とより大きなコストを必要とする可能性がある。私たちの第三段階の臨床試験に正確な治療方案、完成或いは遅延を含めることができず、CTX-1301の未来の臨床試験を開始することを阻止或いは延期する可能性があり、それによって監督部門の私たちの候補製品に対する承認を得て商業化することは、私たちの財務業績に不利な影響を与える。また,我々の競争相手の一部は現在CTX−1301と同じ適応を治療する候補製品に対して臨床試験を行っており,そうでなければわれわれの臨床試験に参加する資格のある患者が競争相手の候補製品の臨床試験に応募する可能性がある。
患者 登録は他の要素の影響を受け、以下を含む
| ● | 調査中の病気の重症度は | |
| ● | 研究に関する資格基準 | |
| ● | 研究を受けた候補製品のbr感知のリスクと収益; | |
| ● | 臨床試験をタイムリーに登録する努力を促進する | |
| ● | 医師の患者の回診方法 | |
| ● | 治療中および治療後に患者の能力を十分に監視する |
| ● | 潜在的患者のための臨床試験場所の距離および利用可能性を提供する | |
| ● | 私たちは、対象、主要な研究者、または作業者、または臨床サイトの利用可能性の潜在的な大流行を制限する可能性があるなど、制御できない可能性のある要因 である。 |
我々 は最近FDAのフィードバックによりCTX−301の臨床開発計画の変更を発表し,それに応じてCTX−1301の2段階3試験の登録 を停止した。私たちは、規制部門のCTX-1301の承認を得るために、これらの試験を再起動し、および/または新しい試験を開始する必要があるかもしれない。そのような臨床試験を再起動したり起動したりすることは難しいか不可能であることが分かるかもしれない。
CTX-1301、CTX-1302、および/またはCTX-2103の規制承認を得ても、このような承認は限られている可能性があり、私たちは厳格で持続的な政府規制を受けるだろう .
規制部門がCTX−1301、CTX−1302、および/またはCTX−2103の商業化を承認した場合であっても、FDAが承認する可能性のある適応またはラベル宣言範囲は、私たちが求めているか、または他の方法でその使用または販売を特別に警告または他の制限を要求する範囲よりも小さい。規制当局は、私たちまたは他の会社がCTX-1301、CTX-1302、br}および/またはCTX-2103を販売するターゲットグループを制限するか、または私たちの他の候補製品のターゲットグループを制限するかもしれない。CTX−1301、CTX−1302および/またはCTX−2103の利点は、FDAまたは他の規制機関の同意を得ない可能性があり、またはこれらの機関は、製品ラベルまたは広告に関連する 宣言を含むことに反対する可能性があり、したがって、CTX−1301、CTX−1302および/またはCTX−2103は、他の同様の製品と比較して、我々の予想される競争優位性を有さない可能性がある。特に、FDAは我々の製品の 有効期限に基づいてラベル宣言を制限する可能性があります。さらに、医薬品安全問題を解決するいかなる新しい立法も、製品開発や商業化の遅延を招くか、コンプライアンスを確保するコストを増加させる可能性がある。
| 21 |
もし私たちが監督部門の任意の候補製品に対する承認を得たら、製品の製造プロセス、ラベル、包装、流通、不良事件報告、貯蔵、広告、販売促進と記録保存などの活動は広範なbrと持続的な規制要求の制約を受けるだろう。これらの要件には、安全性および他の上場後情報および報告の提出、 登録、およびcGMPの継続を含む。 FDAまたは同様の規制機関は、製品の安全性または有効性を監視するために、高価な上場後非臨床研究または臨床試験 (一般に“第4段階試験”と呼ばれる)および発売後監視を要求する可能性もある。もし私たちまたは監督機関が製品に以前に未知の問題、例えば意外な深刻性または頻度の不良事件、 生産問題または製品生産または加工施設の問題、例えば製品汚染または深刻な不適合cGMPが存在することを発見した場合、監督管理機関はその製品、製造施設、または私たちに制限を加えるかもしれない。したがって、私たちと私たちのCMOは、cGMPのコンプライアンスを評価し、FDAに提出された任意のセキュリティプロトコルまたは任意の他のタイプの国内または海外マーケティング申請で行われた約束を遵守するために、持続的な審査および検査を受ける。もし私たちまたは私たちの第三者プロバイダが、私たちのCMOが適用された法規を完全に遵守できなかった場合、私たちは私たちのbr製品のリコールまたは撤回を要求されるかもしれません。
さらに、予想されていない重症度または頻度の有害事象を含む製品に以前に未知の問題が存在することが後に発見された場合、または当社の第三者製造業者または製造プロセスに関連する問題、または規制要件を遵守できなかった場合、以下のことが含まれる可能性がある
| ● | 製品の製造、承認された製造業者または製造プロセスの制限; | |
| ● | 製品ラベルやマーケティングに対する制限 | |
| ● | 製品流通または使用制限 ; | |
| ● | 発売後の研究や臨床試験が求められている | |
| ● | この製品を市場から撤回する; | |
| ● | 製品 リコール; | |
| ● | 警告またはFDAの無タイトルレターまたは外国規制機関の同様の違反通知; | |
| ● | FDAまたは他の適用可能な規制機関が処理すべき出願または承認された出願の追加を承認することを拒否する; | |
| ● | 罰金、利益または収入の返還、 | |
| ● | 上場承認の一時停止または撤回 | |
| ● | 私たちが行っているどんな臨床試験も一時停止します | |
| ● | 製品brは、輸出入製品の差し押さえ、差し押さえ、または拒否を拒否する | |
| ● | 法令、禁止または民事または刑事罰の適用に同意する。 |
さらに、FDAまたは他の規制機関の政策は変化する可能性があり、追加の政府法規を公布して、私たちの候補薬物の規制承認を阻止、制限、または延期する可能性がある。もし私たちが既存の 要求の変化や新しい要求や政策の採用に適応できない場合、または他の方法でコンプライアンスを維持できない場合、私たちは私たちが得る可能性のある任意のマーケティング承認を失う可能性があり、これは私たちの業務、見通し、達成、またはbr}の持続的な利益の能力に悪影響を及ぼすだろう。
FDAの政策は変わる可能性があり、追加の政府法規を公布して、私たちの候補製品の上場承認を阻止、制限、または延期する可能性があります。もし私たちが既存の要求の変化や新しい要求や政策の採用に緩やかに適応できない場合、または私たちがコンプライアンスを維持できない場合、私たちは私たちが得る可能性のあるマーケティング承認を失う可能性があり、これは私たちの業務、将来性、利益を達成または維持する能力に悪影響を及ぼすだろう。
| 22 |
我々の従業員、独立請負業者、主要な調査者、コンサルタント、サプライヤー、CRO、CMO、および私たちが協力する可能性のある任意のパートナー は、規制基準および要件を遵守しないことを含む、不適切な行為または他の不適切な活動に従事する可能性がある。
私たちbrは、従業員、独立請負業者、主要な調査者、コンサルタント、サプライヤー、CRO、CMO、および私たちが協力する可能性のある任意のパートナーが詐欺または他の不正活動に従事する可能性のあるリスクに直面しています。このような人員の不正行為は、FDAまたは他の規制機関に真、完全かつ正確な情報を報告することを要求する法律、製造基準、連邦、br州および外国の医療詐欺および乱用法律、データプライバシー法律法規、または財務情報またはデータを真に、完全かつ正確に報告することを要求する法律を含む、故意、無謀または不注意な行為、または法律または法規に違反する不正な活動を含む可能性がある。特に、医療業界の販売、マーケティング、その他の業務配置は、詐欺、リベート、自己取引、その他の乱用を防止するための広範な法的制約を受けている。これらの法律は、研究、製造、流通、定価、割引、マーケティングおよびbr}販売手数料、販売手数料、顧客インセンティブ計画、および他の商業計画を含む広範な商業活動を制限または禁止する可能性がある。これらの法的制約を受けた活動はまた、臨床試験中に得られた情報の不適切な使用または虚偽陳述、または薬品の不法流用に関するものであり、このような法律または法規を遵守しなければ、規制処罰または他の行動または訴訟を招き、私たちの名声に深刻な損害を与える可能性がある。また、連邦調達法は政府契約に関連する不当行為に対して実質的な処罰を行い、ある請負業者に商業道徳と行為準則を遵守することを要求した。また、個人や政府は、発生しなくても、このような詐欺や他の不正行為を告発する可能性があるというリスクに直面している。もし私たちにこのようなbr訴訟を提起し、私たちが自分のために弁護したり、私たちの権利を維持することに成功しなかった場合、これらの訴訟は、民事、刑事と行政処罰、損害賠償、金銭罰金、返還、監禁、FDAのマーケティング許可を得る資格を失ったbr、Medicare、Medicaidおよび他の連邦医療計画から除外される可能性があり、契約損害、br名声損害、利益減少と将来収益を含む、私たちの業務、財務状況、運営結果、および見通しに実質的かつ不利な影響を与える可能性がある。これらの法律を遵守しない疑惑を解決し、当社の業務を削減または再構築するために、会社の誠実な合意または他の合意を遵守する場合、当社の業務運営能力および私たちの経営業績に悪影響を及ぼす可能性のある追加の報告要件を任意に提供します。
政府の調査や他の法執行活動のため、私たちのビジネス慣行の修正、罰金、巨額の費用の支払い、または他の損失を受けることが要求される可能性があります。
私たち はアメリカおよび/または外国の司法管轄区域で業務を展開するために訴訟や政府調査を受ける可能性があります。私たちの業界の多くの会社のように、私たちは時々政府当局の問い合わせや伝票、他のbrタイプの情報要請を受けるかもしれません。私たちは私たちの業務活動に関するクレームや他の訴訟の影響を受けるかもしれません。
調査や法的手続きの最終結果の予測は困難であるが,これらの問題の不利な解決や和解 は:
| ● | 重大な損害賠償、罰金、罰金または他の支払い、および政府プロジェクトの資格を排除および/または廃止するなどの行政救済措置、または何らかの方法で業務を経営することを禁止する他の裁決; | |
| ● | このような訴訟や調査に関連するリスクを回避するために、私たちの業務運営を変更します | |
| ● | 製品 リコール; | |
| ● | 評判が悪く私たちの製品に対する需要が減少しました | |
| ● | 大量の時間と資源がかかり、そうでなければ、私たちの業務を運営するために使用することができます。 |
私たちはいくつかのリスクに保険を提供していますが、私たちの保険金額はすべての不利な 解決策とクレームと債務決済の総金額をカバーするのに十分ではないかもしれません。すべての潜在的な危険と責任に対する保険を受けることもできない。
| 23 |
私たちの現在と未来のパートナーは、私たちの現在と未来のパートナーが製品リコールの影響を受ける可能性があり、これは私たちのブランドと名声を損なう可能性があり、 は私たちの業務に負の影響を与えるかもしれない。
もし私たちの候補製品が発売を許可され、規格に適合していない場合、または怪我や病気を引き起こすと考えられている場合、または製造、ラベル、販売促進、販売または流通に関連する法規を含むbr政府法規に違反していると告発された場合、私たちbr、または私たちの現在および潜在的なパートナーは、製品のリコール、撤回、または差し押さえの影響を受ける可能性がある。将来のいかなるリコール、撤回、または差し押さえは、消費者が私たちのブランドの自信に重大なマイナス影響を与える可能性があり、私たちが許可した製品に対する需要の減少を招く可能性がある。また、私たちが承認した任意の製品のリコール、撤回、または差し押さえには、経営陣の高い関心が必要となり、巨額と予期せぬ支出を招き、私たちの業務、財務状況、運営結果を損なう可能性があります。
上場承認された候補製品名については,我々 はFDAの承認を得る必要があり,この命名承認に関連するいかなる失敗や遅延 も我々の業務に悪影響を及ぼす可能性がある.
米国特許商標局(“USPTO”)の正式商標登録を取得したか否かにかかわらず、候補製品のいずれの名称にもFDAの承認を得る必要があると考えられる。FDAは、通常、提案された製品名を評価することを含み、提案された名称が他の製品名と混同される可能性があるかどうかを評価する。FDAが我々が提出した任意の製品名が医療クレームを適切に示唆していないと考えた場合,その名称に反対する可能性がある。FDAが私たちが提案した任意の製品名に反対する場合、私たちは私たちの候補製品のための代替名を採用することを要求される可能性があり、これはbr提案名のさらなる評価を招く可能性があり、追加の遅延とコストを招く可能性がある。
FDAおよび他の政府機関の資金不足や世界的な健康問題による中断brは、彼らのbrの採用、保持、または重要な指導部および他の人員の能力の配置を阻害する可能性があり、または新しいまたは修正された製品やサービスの適時な開発、承認、商業化を阻止する可能性があり、これは私たちの業務に負の影響を与える可能性がある。
FDAが新製品を審査と承認する能力は、政府予算と資金brレベル、肝心な人員の雇用と維持及びユーザー費用の支払いを受ける能力、法律、法規と政策の変化、その他のFDAの通常の機能能力に影響を与える可能性のあるbr事件を含む様々な要素の影響を受ける可能性がある。そのため,この機関の平均審査時間は近年変動している である.また,研究開発活動を援助する他の政府機関への政府の援助は政治過程の影響を受けており,この過程は本質的に不安定で予測不可能である。
FDAおよび他の機関の中断はまた、必要な政府機関の新薬の審査および/または承認または承認に要する時間を遅くする可能性があり、これは私たちの業務に悪影響を及ぼす。例えば、ここ数年間、米国政府は何度か閉鎖されており、FDAのようないくつかの規制機関は、FDAの重要な従業員を休暇させ、重要なbr活動を停止しなければならない。
全世界の新冠肺炎疫病とアメリカ突発公共衛生事件に対応するため、アメリカ食品薬品監督管理局は2020年3月に外国製造施設と製品の大部分の検査を一時的に延期し、国内製造施設に対する定例監督検査を延期し、そして臨床試験の実施に対して指導を提供した。FDAは、格付けシステムを利用して、所与の州および地域のウイルス軌跡データおよび州および地方政府によって制定されたルールおよびガイドラインに基づいて、いつどこでこのような検査を行うことが最も安全であるかを決定することを支援する。FDAは類似したデータを用いて 優先の海外業務の回復を通知する.
もし 政府が長期的に停止したり減速したりした場合、あるいは世界的な健康問題がFDAまたは他の監督管理機関の定期検査、審査または他の規制活動を阻止する場合、FDAまたは他の監督管理機関が規制提出の能力を適時に審査し、処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に重大な悪影響を及ぼす可能性がある。
| 24 |
私たちの将来の成長は海外市場に進出する能力にある程度依存しており、そこでは追加の規制負担や他のリスクや不確実性の影響を受ける。
私たちの将来の収益性は候補品を海外市場で商業化する能力にある程度依存し、第三者との協力に依存するつもりです。もし他の候補製品を海外市場で商業化すれば、私たちは追加のリスクと不確実性に直面するだろう
| ● | 私たちのbr顧客は海外市場で私たちの候補製品のために市場参入と適切な精算を得ることができます | |
| ● | 私たちは第三者に依存しているので、ビジネス活動を直接統制することはできません | |
| ● | 複雑で変化の多い外国の監督管理、税務、会計、法律要求の負担を遵守する | |
| ● | 外国の異なる医療実践と風俗習慣は市場受容度に影響を与える | |
| ● | 輸入または輸出許可要件; | |
| ● | 売掛金入金時間が長い ; | |
| ● | 出荷の納期がもっと長い; | |
| ● | 言語障害 技術訓練; | |
| ● | 海外の一部の国の知的財産権の保護を減少させる | |
| ● | 外国通貨の為替レートの変動 | |
| ● | 契約紛争が発生した場合、外国の法律によって管轄される契約条項の解釈。 |
私たちの候補製品の海外販売は、政府規制、政治的、経済的不安定、貿易制限、関税変化の悪影響を受ける可能性もあり、いずれも私たちの運営結果に悪影響を及ぼす可能性があります。
大流行、流行病や伝染病(例えば新冠肺炎)の発生は、私たちの候補製品の開発を中断する可能性があります。
大流行や流行のような公衆衛生危機は私たちの業務に悪影響を及ぼすかもしれない。2019年12月、新冠肺炎は世界を風靡した。コロナウイルスの大流行により、政府の強制隔離、旅行制限とその他の公衆衛生安全措置を含む各種の対応措置が実施された。伝染病の大流行、流行或いは爆発が私たちの運営或いは私たちの第三者パートナーの運営(私たちの開発研究或いは臨床試験運営を含む)に対する影響の程度 は未来に発生する事件に依存し、これらの事件は高度な不確定性があり、いかなる疫病の持続時間及びその影響を制御或いは処理する行動などを含む自信を持って予測できない。我々の業務の大部分は米国で行われているにもかかわらず,感染症の世界的な伝播は米国や海外での候補製品開発や臨床試験運営に悪影響を及ぼす可能性がある。感染症が患者の登録または治療または私たちの候補製品の実行に与えるいかなる負の影響も臨床試験活動のコストの高い遅延を招く可能性があり、これは私たちの監督部門が私たちの候補製品の承認を得てそれを商業化する能力に悪影響を与え、私たちの運営費用を増加させ、私たちの財務業績に重大な悪影響を与える可能性がある。
大流行、流行病、または伝染病の爆発の場合、延期または他の方法で私たちの候補製品および私たちの業務に悪影響を及ぼす可能性のあるいくつかの要因は、:
| ● | 私たちが計画した臨床試験を開始するために、現地の監督管理機関の許可を得ることを遅延させる | |
| ● | 私たちの臨床試験で参加者を募集または維持する時に遅延または困難が発生した |
| 25 |
| ● | 臨床サイトの起動遅延または困難は、臨床サイト調査員と臨床サイトスタッフを募集する困難を含む | |
| ● | 臨床サイトは、臨床試験に必要な物資と材料の受信を遅延させ、臨床試験材料の輸送に影響を与える可能性のある全世界輸送中断を含む | |
| ● | 大流行,流行病あるいは感染症への対応の一部として,地方法規が変化し,臨床試験を行う方法を変更することが要求される可能性があり,意外なコスト,あるいは臨床試験を完全に停止する可能性がある | |
| ● | 医療資源を臨床試験の進行から移行させ,われわれの臨床試験場所である病院と臨床試験を支援してくれた病院スタッフの移転を含む | |
| ● | 連邦或いは州政府、雇用主と他の人が強要或いは提案した旅行制限、或いは臨床試験参加者の訪問と研究プログラムの中断、例えば臨床試験場所のモニタリングなどの肝心な臨床試験活動の中断により、臨床試験データの完全性に影響を与える可能性のある状況が発生した | |
| ● | われわれの臨床試験の参加者が臨床試験中に感染症に感染するリスクは、観察された有害事象の数を増加させることを含む臨床試験の結果に影響する可能性がある | |
| ● | 著者らの研究開発機構の操作が制限或いは限られているため、臨床前研究の中断を招いた | |
| ● | は、私たちの第三者製造業者の運営に潜在的な負の影響を与えます | |
| ● | 従業員資源制限または従業員が休暇を余儀なくされたため、現地の監督機関、道徳委員会と他の重要な機関と請負業者との必要な相互作用は遅延した | |
| ● | 第三者CROの従業員資源は制限され、そうでなければ、従業員またはその家族が病気になったか、または従業員が大勢との接触を避けることを望むことを含む、我々の臨床試験の進行に集中する | |
| ● | FDAまたは他の規制機関は、影響を受けた地域の臨床試験データの受け入れを拒否した | |
| ● | FDA承認前検査遅延 は,承認の前提条件である. |
商業化に関するリスク
最近公布され、将来の政策および立法は、候補製品のマーケティング承認を獲得し、それを商業化する難しさとコストを増加させる可能性があり、マーケティング承認を得た任意の候補製品の精算に影響を与える可能性がある。
政府の処方薬調達と精算計画に影響を与える立法と規制行動が相対的に頻繁である。アメリカにいます。例えば、2010年に“患者保護と平価医療法案”(PPACA)が公布され、医療保健のカバー範囲を拡大し、薬品精算に重大な変化を行った。PACAが公布されて以来、米国はすでに他の製薬業に影響を与える立法改正 を提出し、採択した。例えば、“2022年インフレ率低減法案”には、医療保険と医療補助サービスセンター(CMS)が毎年限られた数の高コスト、単一源の薬物交渉のための“最高公平価格”の条項と、価格上昇がインフレよりも速い場合に連邦医療保険にリベートを支払うことを要求する別の条項が含まれている。どんな新しい法規を遵守しても非常に時間がかかり、コストが高く、それによって私たちの業務に重大な悪影響を及ぼす可能性があります。
| 26 |
さらに、多くの州では、バイオ製薬メーカーが独自の価格情報を開示すること、または州機関が購入した薬品に対して最高価格上限を設定することを要求するなど、間接的または直接に薬品の価格を規範化するための立法を提出または公布している。例えば、2017年、カリフォルニア州知事は、指定された敷居を超えるいくつかの薬品の値上げに対する処方薬メーカーの事前通知および説明を要求する処方薬価格透明性州法案に署名した。国会や州立法機関は様々な法案を検討しており,これらの法案は薬品調達と価格交渉を改革し,管理ツールをより多く使用してMedicare Part Dの保証範囲を制限し,米国以外からの低価格薬品の輸入を促進し,模倣薬の使用を奨励することを許可している。このような計画と立法は私たちのbr製品に追加的な価格設定圧力をもたらすかもしれない。
連邦や州レベルの医療補助計画変更は我々の業務に実質的な悪影響を及ぼす可能性もある。私たちの製品の保証範囲と精算範囲に影響を与える可能性のある提案は、各州により大きな柔軟性を与えてMedicaid計画がカバーする薬品を管理することと、カナダや他の国/地域からの処方薬の再輸入を許可することを含み、私たちの製品の使用と保証範囲を制限することによって実質的な悪影響を及ぼす可能性がある。また,連邦基本医療補助税還付の増加により,州医療補助計画は我々の製品に対する追加的な補充還付を要求する可能性がある。民間保険会社または管理型医療計画が医療補助カバー範囲と支払い発展を追跡する場合、彼らはこれらの増加したリターンポイントを利用して私たちの製品に定価圧力をかけることができ、彼らがより低い支払いスケジュールを採用することは悪影響を拡大する可能性がある。
米国や海外の将来の立法や行政または行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することはできない。私たちは将来、より多くの州と連邦医療改革措置を取ることが予想され、そのいずれも連邦と州政府が医療製品やサービスに支払う金額を制限する可能性がある。また,バイデン政府は,米国人間·衛生サービス部長を含め,処方薬価格の低下が優先順位であることを示しているが,政府がどのような手順をとるか,あるいはこれらの手順が成功するかどうかは不明である。
メーカーに影響を与える他の提案された規制行動 は私たちの業務に重大な悪影響を及ぼす可能性がある。このような提案された立法や規制行動やそれによる州政府行動が米国での製品の使用や精算に及ぼす影響(あれば) を予測することは困難であるが,我々の運営結果は悪影響を受ける可能性がある。
不利な価格設定法規、第三者精算のやり方や医療改革の取り組みは、私たちの将来の業務を損なう可能性がある。
製薬会社は医療コストの削減を要求するますます大きな圧力に直面している。米国では,これらの圧力は管理型医療集団や機関や政府調達業者など様々な源から来ている。連邦医療保健計画と民間部門の受益者を代表して交渉を行う実体の購買力が増加し、将来の定価圧力を増加させる可能性がある。このような圧力 は,政府が定価計算で訴訟を起こしたり調査を行ったりするリスクを増加させる可能性もある。製薬産業は未来にもっと厳格な規制と政治的で法的行動に直面するかもしれない。
不利な価格設定制限は、私たちの将来の候補製品が規制部門の承認を得ても、1つ以上の将来の候補製品への投資を回収する能力を阻害する可能性がある。承認前の不利な価格制限はまた私たちのビジネス潜在力を低下させ、私たちに悪影響を及ぼすだろう。私たちが任意の潜在的製品を商業化することに成功する能力はまた、政府衛生行政当局、個人健康保険会社、その他の組織を含む、これらの製品と関連治療の保証範囲と精算範囲がどの程度第三者支払者から得られるかにある程度依存する。第三者支払者は、彼らがどのような薬を支払うかを決定し、br精算レベルを確立する。医薬製品の獲得保険や精算に適用される類似の挑戦は随伴診断にも適用される。
アメリカの医療産業と他の地域の重要な傾向の一つはコスト統制だ。第三者支払者は,特定の薬物の保証範囲や精算金額を制限することでコストを抑制しようとしている。ますます多くの第三者支払者が価格に基づいて事前に決定された割引を提供し、医療製品の料金に挑戦するように会社に要求している。将来商業化されたどの製品も保険と精算を受けることができることを保証することはできません。もし精算できるなら、精算レベルはいくらですか。精算は私たちが将来市場で承認された任意のbr製品の需要や価格に影響を与えるかもしれない。精算が得られない場合や限られたレベルに限られていれば、私たちが開発に成功した任意の候補製品を商業化することに成功しないかもしれません。
| 27 |
承認された製品の精算に重大な遅延が生じる可能性があり、カバー範囲はFDAや他の国/地域の規制機関がこの製品を承認する用途よりも限られている可能性がある。さらに、精算資格を得ることは、すべての場合に任意の製品のために支払うこと、または研究開発、製造、販売、および流通を含む私たちのコストをカバーするレートで支払うことを意味するわけではない。新製品の一時支払い(適用すれば)も私たちのコストを支払うのに十分ではない可能性があり、恒久的な支払いにならない可能性があります。支払率は製品の使用や臨床環境によって異なる可能性があり,精算された低コスト製品で許容される支払い に基づいて,他のサービスの既存の支払いに統合される可能性がある。製品の正味価格は、法律 が現在、製品販売価格が米国より低い国/地域からの製品の輸入を制限しているので、第三者支払者が要求する強制的な割引またはリベート、および将来の任意の法律の緩和によって低減することができる。第三者支払者 は,彼ら自身の精算政策を設定する際には通常Medicare保証政策と支払い制限に依存するが,Medicare保証範囲と精算決定のほかに,彼ら自身の方法と承認の流れがある.したがって、1つの第三者支払者がある製品に保険を提供することを決定することは、他の支払者もその製品に保険を提供することを保証することはできない。私たちは第三者支払者から承認製品の保証範囲と十分な補償を迅速に得ることができません。これは私たちの経営業績、潜在的な製品を商業化するために必要な資金を調達する能力、および私たちの全体的な財務状況に実質的な悪影響を及ぼす可能性があります。
我々 は、特定の候補製品または指示を追求するために限られたリソースを費やす可能性があり、より利益またはより成功する可能性の高い候補製品または指示 を利用することができない。
我々の財務と管理資源は限られているため,特定の 適応に対して決定された開発計画と候補製品に焦点を当てている。したがって、私たちは他の候補製品を求めるビジネスチャンスを放棄または延期したり、後により大きな商業潜在力を持つことが証明された他の兆候を求めることができるかもしれない。私たちの資源分配決定は私たちが実行可能な商業薬や利益の市場機会を利用できないかもしれない。私たちの現在と未来の研究開発計画と特定の適応候補製品への支出はいかなる商業的に実行可能な製品も発生しないかもしれない。もし私たちが特定の候補製品の商業的潜在力や目標市場を正確に評価しなければ、私たちは協力、br許可、または他の印税手配によって候補製品に貴重な権利を放棄するかもしれないが、この場合、私たちは候補製品の独占的開発とbr}商業化権利を維持した方が有利になるだろう。
私たちの候補製品の商業成功(承認されれば)は医師、患者、第三者支払人と医学界の市場認可を得るかどうかにある程度依存する。
私たちが製品収入を作る能力は、候補製品の最終マーケティング承認を成功させ、それを商業化する能力に大きく依存するだろう。
我々の任意の候補製品CTX−1301、CTX−1302、および/またはCTX−2103が規制部門の承認を得ても、医師、患者、第三者支払者、および医療コミュニティにおいて十分な市場受容度を得ることができない可能性がある。市場の承認を得ることができなかったことは私たちの収益能力を制限し、私たちの運営結果に影響を与えるだろう。CTX−1301、CTX−1302、および/またはCTX−2103の市場受容度は、多くの要因に依存するであろう
| ● | CTX−1301、CTX−1302および/またはCTX−2103の治療効果および潜在的利点、ならびに代替療法または競合製品との比較; | |
| ● | 我々の第三者協力者は、CTX−1301、CTX−1302および/またはCTX−2103に関する医師および患者の潜在的利益およびbr}利点の有効性を教育するために努力している | |
| ● | 医療コミュニティや患者が新しい技術を採用する意思は | |
| ● | 規制の承認を得て商業権を持つ地域では,我々の目標患者群の規模,このような候補薬物の市場規模に基づいている | |
| ● | 副作用の流行率や重症度は |
| 28 |
| ● | 広範な商業流通を通じて候補薬物の安全性を証明する | |
| ● | 競争力のある価格で候補品を販売することができます | |
| ● | 競合製品に対する私たちの候補製品の費用対効果 | |
| ● | 当社のすべての製品CTX−1301、CTX−1302、およびCTX−2103を生産することができます | |
| ● | 競合製品または治療法と比較した医師、患者、および医療コミュニティ(第三者支払者を含む)の、CTX−1301、CTX−1302および/またはCTX−2103の安全性、有効性、および潜在的利益に対する見方 | |
| ● | 他の製品承認に関連する任意のこのような上場承認の時間; | |
| ● | 他の薬の併用にはどんな制限もありません | |
| ● | 患者権益提唱団体の支援 | |
| ● | 代替療法よりも比較的便利で投与が容易である | |
| ● | 政府医療計画と第三者支払者は、他の競合製品や療法に対する価格設定に十分な保険と精算を提供する。 |
もし私たちの候補薬が承認されたが、重要な市場参加者の十分な受け入れが得られなかったら、私たちは相当な収入を得ることができなくなり、私たちは利益を上げることができないかもしれないし、利益を維持することができないかもしれないし、追加の融資を求める必要があるかもしれない。
私たちの候補製品の交渉、第三者のカバーと精算を確保し、維持する能力は、アメリカと他の司法管轄区域の政治、経済、規制発展の影響を受ける可能性があります。政府は引き続きコスト制御措置を実施し、 第三者支払人は安全性と有効性以外に、ますます薬品価格に疑問を提起し、その費用効果を審査するようになっている。これらの他と類似した発展は、将来市場で承認された私たちの任意の候補製品の市場受け入れ度を大きく制限するかもしれない。
私たちbrは他の製薬会社からの激しい競争に直面する可能性があり、効果的な競争ができなければ、私たちの経営業績は影響を受けるだろう。
製薬業界は競争が激しく、迅速で重大な技術変革の影響を受けている。もし私たちが技術変革の最前線に立つことができなければ、私たちは効果的に競争できないかもしれない。私たちの競争相手が開発した技術の進歩や製品 は私たちの技術や候補製品を時代遅れにし、競争力に欠けたり、経済的ではないかもしれません。
私たちbrは、大手多国籍製薬会社を含む米国と国際的に競争相手がいると予想されています。例えば,アンフェタミンXRは現在ShireがAdderall XRブランドで米国で販売されているのに対し,メチルフェニデートはJanssenがConcertaブランドで米国で販売されており,ノワールがFocalin XRとRitalin LAブランドで販売されている。また、ブランド薬物のメーカーも彼ら自身の処方を改善し、私たちのこれらの薬物の改善と競争することができる。私たちの多くの競争相手は、より多くの研究開発者およびより経験的なマーケティングおよび製造組織のような、より多くの財務、技術、および他の資源を持っている。バイオテクノロジーと製薬産業の合併と買収は、私たちの競争相手により多くの資源を集中させる可能性がある。したがって、これらの会社が規制承認を受ける速度は私たちよりも速いかもしれませんし、その製品を販売し、マーケティングする上でも効果的かもしれません。より小さいまたは早い段階にある会社も重要な競争相手である可能性があり、特に大手成熟会社との協力によって手配される可能性がある。技術の商業適用性の進歩とこれらの業界の投資資金のより多くの獲得可能性により、競争はさらに激化する可能性がある。私たちの競争相手は、独占的な基礎の上で開発、買収に成功するかもしれない、私たちのPTRプラットフォームよりも効果的またはコストの低い薬物br製品または薬物送達技術、または私たちの現在開発されているか、または開発可能な任意の候補製品であるかもしれない。さらに、我々の競争相手は、FDAに市民請願書を提出し、FDAを説得しようとするかもしれないが、私たちの製品またはその承認を支持する臨床試験に欠陥があるか、または候補製品または候補製品の薬物カテゴリに対応するために新たな規制要求を提出することができる。我々の競争相手のこのような行動は、FDAが505(B)(2)条に従って提出した任意の秘密協定を承認することを遅延または阻止する可能性がある。
| 29 |
たとえ私たちが競争相手の前に候補製品を商業化する規制承認を得ることに成功しても、私たちの未来の製薬製品は後発薬や他の後続薬物製品からの直接競争に直面する可能性がある。私たちが将来規制部門の承認を得る可能性のある候補製品は、米国の処方薬市場でのこのような承認された製品のパフォーマンスに応じて、予想よりも早く、またはより積極的に後発薬からの競争に直面する可能性がある。我々の競争能力は、多くの場合、非特許製品の使用を奨励しようとしている保険会社または他の第三者支払者の影響を受ける可能性がある。後発医薬品は今後数年以内に発売される予定だ。私たちの候補製品が市場で承認されても、その時までに承認されていれば、競争相手の非特許製品よりもはるかに高い価格になる可能性がある。
505(B)(2)NDA経路を作成することに加えて、FDCAに対するHatch-Waxman修正案は、認可された簡略化新薬申請(ANDA)に基づいて、以前に法規NDA条項に従って発売された薬剤と同じ模倣薬を承認することをFDAに許可する。ANDAは先に承認された参考市販薬(“RLD”)に対する臨床前と臨床試験に依存し,RLDと“生物学的同等性”を有することをFDAに証明しなければならない。いくつかのマーケティングまたはデータ排他性保護がRLDに適用される場合、法規は、FDAがANDAを承認することを禁止する。このような競合他社または第三者が、私たちの特許を侵害することなく生物学的同等性を証明することができる場合、競合他社または第三者は、その後、競合他社の模倣薬を市場に押し出すことができるかもしれない。
私たちは、私たちが競争する能力は、これらに限定されないと信じている
| ● | 第三者と開発されている上場製品および候補製品の有効性および安全性を含む、我々の製品および候補製品の有効性および安全性 ; | |
| ● | 私たちの候補製品の臨床開発と発売承認に要する時間 | |
| ● | 監督機関と良好な関係を維持する能力 | |
| ● | 私たちが規制部門の承認を得た任意の候補製品を商業化およびマーケティングする能力 | |
| ● | 私たちの製品と規制の承認を受けた候補製品の価格は、ブランドや模倣薬の競争相手との比較を含む | |
| ● | 個人と政府の医療保険計画(医療保険を含む)がカバー範囲と適切な精算レベルを提供するかどうか | |
| ● | 私たちの製品や候補製品に関連する知的財産権を保護する能力; | |
| ● | 費用対効果に基づいて私たちの製品を製造して販売し、規制の承認を受けた候補製品の商業数量を製造し販売する能力; | |
| ● | 私たちのすべての製品と医師と他の医療提供者の監督管理の許可を得た候補製品を受け入れます。 |
もし私たちの競争相手が私たちの製品よりも効果的で、安全で、安い製品(あれば)、あるいは私たちの製品よりも早く発売された製品(あれば)を販売すれば、私たちは周期的に市場に入るのが遅すぎて、商業成功が得られないかもしれません。あるいは価格を下げなければならないかもしれません。これは私たちの収益と収益力に影響を与えます。
さらに、成功した商業化は、特許および他の知的財産権所有者によって提起された任意のクレームに十分に保護し、効果的に対応できるかどうかにかかっており、私たちの製品が彼らの権利を侵害しており、私たちの製品が予期せぬ悪影響または負の宣伝を生じているかどうか、および競争相手として出現する新製品または既存製品に、より臨床的効果および費用効果があることが証明されている可能性がある。もし私たちがこれらの任務を成功させることができなければ、私たちはすぐに商業化できない、あるいは を維持し、私たちの業務を発展させるのに十分な収入を生み出すことができないかもしれない。
| 30 |
我々 は潜在的な後続競争相手の興味を予測することができず,他のライバルがどれだけ速い速度で競合製品を市場に出すことを求めているのか予測できず, がANDAの直接競争相手として承認されたのか,505(B)(2)秘密協定として我々の将来の薬物製品の1つを参考にしているのか。FDAが将来私たちの候補薬物の模倣薬のバージョンを承認し、もしそれらが商業マーケティングのために承認された場合、このような競争製品は、私たちの候補製品が承認された可能性のある各適応で直ちに私たちと競争する可能性があり、これは、私たちの将来の収入、収益性、およびキャッシュフローに負の影響を与え、これらの候補製品への投資からリターンを得る能力を大きく制限するかもしれない。
オピオイドと覚せい剤乱用をめぐる社会問題は、法執行部門の分流に対する懸念及び乱用に打撃を与える監督管理努力を含み、私たちの候補製品の潜在市場を低下させる可能性がある。
処方薬の乱用やオピオイド、覚せい剤や他の制御物質の転移に関するメディアの報道は珍しくない。br法執行や規制機関は、オピオイドや覚せい剤の供給を制限しようとする政策をとる可能性がある。このような努力は私たちが候補製品を商業化する能力を抑制するかもしれない。オピオイドの過激な法執行と不利な宣伝については、処方薬の乱用に対する規制、処方薬の乱用に対する大衆の問い合わせと調査、訴訟や規制活動、私たちの製品の販売、マーケティング、流通、あるいは貯蔵は私たちの名声を損なう可能性がある。承認されれば、このような負の宣伝は、私たちの候補製品の潜在的な市場規模を縮小し、それらの販売から得られる収入を減らすことができるかもしれない。
また、国会、州立法機関、FDA、その他の規制機関が現在と未来にオピオイドや覚せい剤乱用に打撃を与える努力は私たちの候補製品市場にマイナス影響を与える可能性がある。立法者またはFDAは、いつでも新しい立法または規制措置を発表する可能性があり、これは、私たちの製品候補製品の規制負担を増加させたり、ビジネス機会を減少させたりする可能性がある。
我々の第三者依存に関するリスク
もし私たちが要求通りに私たちの製品や候補製品を量産できなかった場合、あるいは製薬メーカーに適用される厳格な規定 を遵守できなかった場合、私たちは候補製品の開発と商業化の過程で規制処罰と遅延 に直面する可能性がある。
私たちは現在第三者サプライヤーに依存して私たちの候補製品に原料薬と補助剤を提供している。原材料供給のどのような不足も生産や他の遅延を招き、私たちに悪影響を及ぼす可能性がある。また、監督管理部門は通常医薬製品の原材料源を承認しなければならないため、原材料サプライヤーの変更により生産遅延や原材料コストが上昇する可能性がある。どのような遅延も処罰を引き起こす可能性があり、これは私たちの業務に否定的な影響を与えるだろう もし私たちの原材料メーカーが困難に直面したり、私たちへの義務を履行できなかった場合、私たちはFDAの承認を得て私たちの製品や候補製品を販売する能力が脅かされるだろう。さらに、臨床試験供給のいかなる遅延または中断も、私たちの生物学的同等性および/または臨床 試験の完了を遅延または禁止し、私たちの生物学的同等性および/または臨床試験を行うことに関連するコストを増加させ、遅延された期間に応じて、新しい試験を開始し、巨額の追加費用を支払うか、または試験を終了することを要求する。
製薬製品の製造には、先進的な製造技術と技術制御の開発を含む大量の専門知識と資本投資が必要である。製薬会社は生産規模を拡大する上で困難に直面するかもしれない。これらの問題 は生産コストと生産量に関連する製造困難、品質管理(製品の安定性と品質保証テストを含む)、合格者不足及び連邦、州と外国法規の遵守を含む。私たちはまた、他のデバイスを購入する必要があるかもしれませんが、いくつかのデバイスは、調達、設定、および検証のために数ヶ月以上かかり、増加する需要を満たすために、私たちのソフトウェアおよび計算能力を増加させる必要があるかもしれません。このような成長或いは移行を管理できなければ、回転時間遅延、製品コストの上昇、製品品質の低下或いは競争挑戦に対する応答速度が遅くなる可能性がある。これらの分野のいずれの分野の失敗も、私たちの製品に対する市場の期待を満たすことを困難にし、私たちの名声と業務の将来性を損なう可能性がある。
| 31 |
医薬品メーカー は、FDAがその施設検査計画で実行したcGMP要求を遵守する必要がある。 cGMP要求には、品質管理、品質保証、記録、文書の維持 および法規要求に合わないいかなる場合も調査·是正する義務がある。これらの要求を遵守できなかったことは、罰金や民事処罰、生産停止、製品承認の一時停止または遅延、製品の差し押さえまたは自発的なリコール、または製品承認の撤回を招く可能性がある。もし私たちの任意の製品または候補製品の安全が適用法または他の理由に従わなかったことによって損害を受けた場合、私たちはbr候補製品の規制承認を得ることができないか、またはそのような製品または候補製品を商業化することに成功することができず、それによるいかなるダメージに責任を負う必要があるかもしれない。これらの要素のいずれも、私たちの製品または候補製品の臨床開発、規制提出、承認または商業化の遅延を招く可能性があり、コスト上昇を招き、または私たちの候補製品を効率的に商業化することができません。
我々の CTX−1301および/またはCTX−1302の供給源は限られており、これらは所定の製品であるため、サプライチェーンの任意の中断は、CTX−1301および/またはCTX−1302の生産および販売に影響を与え、候補製品の開発および商業化の遅延をもたらす可能性がある
私たちが CTX−1301および/またはCTX−1302のために提出する予定のNDAは、各候補製品に提案されている製造プロセスを含む。私たちの製造プロセス、施設、またはサプライヤーへの任意の変更は、セキュリティプロトコルを修正する必要があるかもしれません。私たちの製造プロセス、施設、またはサプライヤーへの任意の変更は、セキュリティプロトコルを修正する必要があるかもしれません。また,候補製品を生産するための独自プロセスであるため,薬品の生産活動を直ちに代替サプライヤーに移すことはできず,生産施設の交換に時間がかかり,コストの高い作業である可能性がある。例えば、2022年10月に、私たちは新しいCMOを発表した。CTX-1301固定用量研究はbrを延期され、同時に新しいCMOを使用する製造技術を確立し、固定用量研究所に必要な最終用量強度 を生産する。将来の生産施設の潜在的な変化は、生産場所の変化を含めて、私たちの機密協定ファイルを補完することも求められています。私たちの候補製品または製品のために任意の1つまたは複数の成分br物質の適切な合格代替供給源を決定することは非常に時間がかかる可能性があり、私たちは候補製品の開発と商業化の過程で大きな遅延を招くことなく、これをすることができないかもしれない。任意の代替供給者はまた、NDAサプリメントで資格を得る必要があり、サプリメントを承認するためにFDAの検査を受ける必要がある可能性があり、これは、他の臨床試験に関連する遅延を含むさらなる遅延をもたらす可能性がある。
これらの要素は私たちの候補製品の臨床試験、監督提出、必要な承認或いは商業化の遅延を招く可能性があり、 は私たちにより高いコストを発生させ、私たちがその商業化に成功することを阻止する。さらに、私たちのサプライヤーが必要な商業数量のコンポーネントおよび原料薬を商業的に合理的な価格でタイムリーに渡すことができない場合、私たちのサプライヤー が特定の所定のコンポーネントを供給するのに十分なDEA割当量を得られない場合、私たちは実質的に同じコストで生産でき、私たちの候補製品の商業化と将来の潜在的候補製品の臨床試験の代替 サプライヤーを得ることができない場合、遅延する可能性があり、または潜在的収入を損失する可能性があり、私たちの業務、財務状況、運営結果、名声は不利な影響を受ける可能性がある。
私たちのbrは依存し、引き続き第三者に完全に依存して、私たちの臨床前、臨床試験と商業薬品供給を制定し、生産することが予想される。私たちの任意の候補薬物の開発および商業化は停止、延期、または利益が低下する可能性があり、もしこれらの第三者がそのような薬物の十分な数の供給を提供できなかった場合、または許容可能な品質レベルでそのような薬剤を提供できなかった場合、適用される法規要件または契約義務を含むことを含み、したがって、私たちの運営は損なわれる可能性がある。
私たちは今のところありませんし、私たち自身の製造施設のようなインフラや能力を内部で得るつもりもありません。私たちの臨床試験および臨床前研究のための私たちの臨床前および臨床薬の供給、または商業数のbr}規制部門の承認を得る可能性のある任意の候補薬を生産するつもりはありません。私たちは唯一の源、第三者サプライヤーから原料薬を調達し、brはCMOとその施設で私たちの候補薬物を生産する契約を締結しており、予測可能な未来にこのようにし続ける予定である。したがって、私たちは私たち自身の候補薬を調製または生産するための資源と専門知識が不足しており、私たちの第三者への依存は、許容可能なコストで十分な量の原料薬または私たちの候補製品を得ることができないリスクを増加させ、これは、私たちの臨床試験または私たちの他の開発または商業化努力の能力を遅延、阻止または損害する可能性がある。例えば,我々の従来のCMOの運営資源の問題により,CTX−1301固定用量研究の臨床用品の製造と交付に遅延が遭遇した。我々の新しいCMOがCTX−1301の製造プロセスを確立している間,臨床用品の生産はさらに遅れた。
| 32 |
私たちはCMOと合意して、このCMOに私たちの候補薬物CTX-1301のすべての臨床、登録、および商業ロットを生産させるつもりで、私たちは1つ以上のメーカーと合意して、私たちの未来の臨床試験および/または商業販売のために薬品の製造、供給、貯蔵、および流通を供給する予定だ。我々は候補薬物の供給のためにbrという関係を確立または継続する予定であるが,商業的に合理的な条項の下でこれらの関係を維持できる保証はない(あれば)。これらの関係を維持できなければ,新たなCMOを探し同定する際に開発作業の遅延 に遭遇する可能性がある.もし私たちの現在の任意の候補薬または私たちが将来開発または獲得可能な任意の候補薬が規制部門の承認を得たら、私たちは1つ以上のCMOに依存してそのような薬物の商業的供給を生産する。
たとえ が既存の第三者関係を維持したり、他の第三者製造業者とこのようなプロトコルを確立することができれば、 が第三者製造業者に依存することは、限定されないが、これらに限定されない追加のリスクをもたらす
| ● | 第三者に依存してFDAとDEAのコンプライアンスと品質保証を確保する | |
| ● | 私たちの個人情報は、私たちのビジネス秘密とノウハウを含めて盗用される可能性がある | |
| ● | 追加の規制申告を含む、中断 および供給者交換に関連するコスト; | |
| ● | サード·パーティは、コストが高いか、または私たちに不便をもたらす時間に、プロトコルを違反、終了、または更新しない可能性がある | |
| ● | 十分な製造能力を遅延または調達できないか、または拡大することができない | |
| ● | ビジネス上合理的な条件で第三者と製造協定を交渉することはできない | |
| ● | 第3者との製造契約を終了または更新しない方法で、私たちに代価または損害を与える方法で、 | |
| ● | 製品コンポーネントは限られた数の供給源に依存し、場合によっては単一のソースに依存するので、これらの製品コンポーネントの十分な供給を確保できない場合、私たちはタイムリーなbr方式、十分な数量、または許容可能な条件で候補製品を製造して販売することができないだろう。 |
これらのリスクの各々は、私たちの臨床試験、私たちの候補薬剤の承認(あれば)、または私たちの候補薬剤の商業化を延期する可能性があり、brは、より高いコストをもたらすか、または私たちの潜在的な製品収入を奪う可能性がある。いくつかの事件は、禁止、リコール、差し押さえ、または生産の完全な一時停止、または部分的な一時停止を含むFDAの行動の基礎となる可能性がある。
私たちは私たちの候補製品の製造に最終的な責任がありますが、私たちは自分で私たちの製品を製造するのではなく、私たちは私たちのCMOに依存してCGMPを守ります。我々がCMOと締結したプロトコルは,品質管理,品質保証,合格者に関する要求など,いくつかのcGMP要求に従って を実行することを要求しているが,これらの基準を実行して維持するためにCMOの行動を制御することはできない.もし私たちのCMOが私たちの規格やFDAや他の規制機関の厳格な規制要求に適合する材料を生産することに成功しなければ、私たちはこのような要求に適合できる代替CMOを招聘しない限り、私たちの候補薬物の規制承認 を得ることを阻止され、私たちはそれができないかもしれない。私たちのCMOのどのような失敗も、承認されれば、私たちの開発、マーケティング承認、または私たちの候補製品をマーケティングする能力に深刻な影響を及ぼすだろう。
また、もし私たちの候補製品が承認されたら、私たちのサプライヤーは製造、テスト、品質管理、私たちの候補製品に関連する記録保存を含む監督管理要求を受け、規制機関の持続的な検査を受ける。私たちの任意のサプライヤーが適用された法規を遵守できないことは、私たちの製造能力の長時間の遅延と中断を招く可能性があります。私たちは、すべての法規の要求に適合する別のサプライヤーと、任意の必要な リコールまたは他の是正措置に関連する市場中断を求めています。
| 33 |
第三者 メーカーはcGMP法規や米国以外の類似した法規要件を遵守できない可能性がある。私たちが適用された法規または第三者メーカーが関連法規を遵守できなかったことは、警告状、臨床一時停止または臨床試験の中止、罰金、禁止、返還、返還、民事処罰、br}遅延、一時停止または承認またはその他の許可の撤回、FDAの承認保留申請の拒否、製品抑留、FDA 法令が生産および流通業務に重大な制限または一時停止、禁止、輸入または輸出の拒否、製品抑留、不良宣伝、親愛なる医療保健提供者レターまたはその他の警告を含む、関連法規を遵守できなかったことを引き起こす可能性がある。ライセンスの取り消し、候補製品の差し押さえまたはリコール、操作制限、政府契約または既存の契約下の未来の注文の拒否、および虚偽のクレーム法案の責任、連邦医療保健計画から除外されたこと、およびbr社の誠実な合意などの結果を含むbr}民事と刑事責任、これらの結果はすべて私たちの製品供給に重大な不利な影響を与える可能性がある。
もし私たちの第三者契約製造業者が制御物質に関連するDEA法規を遵守できなかった場合、彼らの許可証がキャンセルされる可能性があり、私たちの製品と候補製品の生産が中断または停止する可能性がある。これは、私たちが開発し、 が市場の承認を得たり、私たちの候補製品をマーケティングしたりする能力に影響を与えるだろう。
私たちのbr候補製品と私たちが開発する可能性のある任意の薬品は他の候補製品や薬品と製造施設の使用権を争うかもしれませんが、私たちは優遇条項でこれらの施設の使用権を得ることができないかもしれません。
CGMP法規の下で運営され、DEAライセンスを持って規制された物質を調達、保有、使用するメーカーの数は限られている。私たちの既存または未来の製造業者の任意の性能障害は、臨床開発またはマーケティング承認を延期する可能性があります。私たちは現在余分な供給や二番目の契約製造業者を手配していない。もし私たちの現在の契約 製造業者が約束通りに履行できない場合、私たちはこのような製造業者を交換する必要があるかもしれません。 および任意のそのような交換の資格を決定する際に、追加のコストと遅延が生じる可能性があります。例えば,我々の従来のCMOの運営資源の問題により,CTX−1301固定用量研究の臨床用品の生産と納入に遅延があった。臨床用品の生産は更に延期されたが、著者らの新しいCMOはCTX-1301の製造技術を確立した。
私たちのbr}は、締め切り前にこのような試験および/または規制提出を完了できなかったことを含む、第三者に依存して臨床試験および/または規制提出を行うことに依存することが予想される。
私たちは私たちが計画した臨床試験と私たちの候補製品の規制のためにCROと協力したいです。我々はCRO及びその他の第三者(例えば臨床データ管理組織、監督対策士、医療機関と臨床研究者)に頼って著者らの計画した臨床試験を行い、私たちの候補製品のために適切な監督管理提出書類を用意し、適用される監督管理要求の遵守を確保することに協力することを望んでいる。このような第三者とのプロトコルは、第三者が履行されていないことを含む様々な理由で終了する可能性がある。もし私たちが代替計画を達成する必要があれば、私たちの薬物開発活動は延期されるだろう。
これらの第三者への臨床開発活動への依存は,これらの活動の制御を減少させる可能性があるが,我々の責任を軽減することはない。例えば,我々のすべての臨床試験が試験の全体的な研究計画や案に沿って行われることを確保していきたい。さらに、FDAは、データと報告の結果が信頼性と正確であることを保証し、試験参加者の権利、完全性と機密性 を保護するために、臨床試験結果を行い、記録し、報告する監督基準、すなわち一般的に良好な臨床実践またはGCPと呼ばれることを要求する。規制機関は,試験スポンサー,主要調査者,試験場を定期的に検査することでこれらのGCPを実行する。指定された臨床試験を指定された時間範囲で登録し,政府が援助したbr}データベースClinicalTrials.gov上で完了した臨床試験結果を公表することも求められている。また,cGMP要求に応じて生産された製品を用いて臨床試験を行わなければならない。これらの規定を遵守しなければ,臨床試験を繰り返す必要がある可能性があり,規制部門の承認プロセスを遅らせることになる。著者ら、私たちのCRO或いは臨床試験サイトに関連する適用要求を遵守できなかったことはまた、臨床一時停止と臨床試験の中止、資格取り消し、FDAの臨床データに基づく申請の承認拒否、警告状、製品が承認された場合、上場承認の撤回、罰金とその他の金銭的処罰、br}遅延、不利な宣伝及び民事と刑事制裁などの結果を招く可能性がある。
| 34 |
さらに, これらの第三者は他のエンティティと関係がある可能性があり,その中のいくつかは我々の競争相手である可能性がある.もしこれらの第三者 が法規の要求や私たちが規定した規程に従ってその契約の職責を履行し、期待された期限内に私たちの臨床試験を完成または実行しなければ、私たちの候補製品のマーケティング承認を得ることができないか、遅延する可能性があり、私たちの候補製品を商業化することに成功できない、あるいは私たちの努力を遅らせることができるかもしれない。
さらに、私たちの臨床試験の首席研究者は時々私たちの科学顧問や顧問を務めるかもしれないし、brはこのようなサービスによって現金または株式補償を受けるかもしれない。これらの関係と任意の関連する賠償が知覚または実際の利益の衝突をもたらし、またはFDAが財務関係が研究の解釈に影響を及ぼす可能性があると結論した場合、適用される臨床試験場所で生成されたデータの完全性が問われる可能性があり、臨床試験自体の効用が脅かされる可能性があり、これは、FDAが提出した任意の秘密協定が延期または拒否される可能性がある。このような遅延または拒否は、私たちが候補製品を商業化することを阻止することができる。また,主な調査者との手配も,連邦反リベート法規のような他の医療規制法の審査を受けている。
私たちはまた、他の第三者に依存して、私たちの臨床試験の製品供給を保存し、配布したい。私たちの流通業者がどんな性能故障や適用された法規要件(FDAまたはDEAの要求を含む)に従わない場合、私たちの候補製品の臨床開発やマーケティング承認または製品の商業化が遅延し、追加の損失をもたらし、私たちの潜在的な製品収入を奪う可能性があります。
もし私たちと契約した第三者がその契約の義務または義務を成功裏に履行できなかった場合、または予想された期限内に を達成できなかった場合、または彼らが得た臨床データの品質または正確性が私たちの臨床方案や法規の要求または他の理由に従わなかった場合、私たちの臨床試験は延長、延期または終了される可能性があり、追加の 試験が必要であり、私たちは規制部門の承認を得ることができないかもしれないし、私たちの候補製品を商業化することに成功するかもしれない。したがって、私たちの候補製品の商業見通しは損なわれ、私たちのコストは増加するかもしれません。私たちの創造能力は遅延するかもしれません。もし私たちが将来第三者サービスプロバイダの業績を識別して管理することができなければ、私たちの業務は不利な影響を受ける可能性がある。
我々 は第三者との協力に依存して我々の候補製品を開発して商業化している.これらの協力 が成功しなければ、私たちはこれらの候補製品の市場潜在力を利用できないかもしれない。
2023年3月7日, 我々はIndegene,Inc.(“Indegene”)と共同商業化プロトコル(“商業化プロトコル”) を締結した.商業化プロトコルによれば、Indegeneは、(A)医療事務および薬物警戒、(B)定価、精算および市場アクセス、(C)商業運営、および(D)マーケティングに関連するサービスを含むCTX-1301の商業化サービスを提供する。
私たちはまた、私たちの候補製品の開発および商業化のために、米国でのマーケティングが許可されている任意の候補製品の商業化を含む、より多くの第三者パートナーを探すことも可能である。潜在的なパートナー には、共同商業化パートナーおよび地域、国、国際大中型製薬会社が含まれる。
私たち は、私たちの現在および未来の任意の協力者が、私たちの候補製品の開発またはそれを商業化するためのリソースの数および時間を限られた制御に特化している可能性があります。私たちがこれらのスケジュールから収入を創出する能力は、私たちの協力者がこれらのスケジュールの中で彼らに割り当てられた機能を成功的に履行する能力に依存する。商業化協定によると、私たちはIndegeneと作業宣言を締結し、Indegeneが提供するサービス、そのようなサービスの配信可能な内容、および私たちが支払う費用を列挙する。私たちはIndegeneが提供するサービスや私たちが支払うべき費用を含む、私たちが受け入れられる条項に従って作業説明書の条項を協議することができないかもしれない。もし私たちがそれができなければ、私たちはCTX-1301の商業化のために他の協力を求めなければならず、これは商業化を延期するかもしれない。
私たちのbrは現在の協力がリスクを構成しており、将来的には私たちの候補製品に関するいかなる協力も以下のリスクをもたらすだろうが、これらに限定されない
| ● | 私たちは、私たちの知的財産権、将来の収入源、研究計画、または候補製品に対する貴重な権利を放棄しなければならないかもしれません | |
| ● | 協力者 は、彼らがこれらの協力の仕事および資源に適用されることを決定する上で大きな裁量権を有する |
| 35 |
| ● | 協力者 は予想通りに義務を履行していない可能性がある | |
| ● | 協力者 は監督部門の許可を得た任意の候補製品の開発と商業化を行ってはならず、臨床試験結果、協力者の戦略重点或いは利用可能な資金の変化或いは外部要素(例えば買収)に基づいて、引き続き を継続しないか、開発或いは商業化計画を更新しないことを選択してはならない | |
| ● | 協力者は臨床試験を延期することができ、臨床試験計画に資金不足を提供し、臨床試験を停止或いは候補製品を放棄し、新しい臨床試験を繰り返し或いは行うことができ、或いは新しい候補製品の調合に臨床試験を要求することができる | |
| ● | 私たちと協力して発見された候補製品 は、私たちの協力者によって彼ら自身の候補製品 と競争するとみなされるかもしれません。これは、協力者が資源を投入して私たちの候補製品を商業化することを停止する可能性があります | |
| ● | 規制の承認を得て、私たちの1つまたは複数の候補製品に対してマーケティングおよび流通権限を持つbr}協力者は、そのような製品のマーケティングおよび流通のために十分なリソースを投入していない可能性がある | |
| ● | 特許権、契約解釈、または第一選択開発プロセスにおける分岐を含む協力者との分岐 は、候補製品の研究、開発または商業化の遅延または終了をもたらす可能性があり、候補製品に対して追加の責任を負うことになる可能性があり、または訴訟または仲裁を引き起こす可能性があり、いずれも時間がかかり、コストが高い | |
| ● | 協力者は、私たちまたは彼らの知的財産権を正確に維持したり守ったりすることができないかもしれないし、br方式で私たちまたは彼らの固有の情報を使用して、訴訟を招き、そのような知的財産権または独自の情報を危険にさらしたり、無効にしたり、または潜在的な訴訟に直面させたりする可能性がある | |
| ● | 協力者は第三者の知的財産権を侵害する可能性があり、これは私たちを訴訟と潜在的な責任に直面させるかもしれない | |
| ● | もし私たちが協力を中止したら、私たちは停止費を支払う必要があるかもしれない | |
| ● | 協力者の便宜のために、協力 を終了する可能性があり、終了すれば、適用可能な候補製品をさらに開発するか、または商業化するために追加資金を調達する必要があるかもしれない。 |
商業化プロトコルおよび任意の将来の協調プロトコルは、最も効率的な方法で、または候補製品の開発または商業化を引き起こさない可能性がある。もし私たちの現在または未来のパートナーが業務合併に参加すれば、薬物開発または商業化計画の持続的な追求と重視は延期、減少、または終了される可能性がある。
もし私たちが協力を維持したり構築できなければ、私たちは私たちの開発と商業化計画を変更しなければならないかもしれない。
商業化プロトコルによれば、Indegeneは、(A)医療事務および薬物警戒に関連するサービス、(B)定価、精算および市場アクセス、(C)商業運営、および(D)マーケティングを含むCTX-1301の商業化サービスを提供する。
私たちの候補製品および臨床プロジェクトの開発および潜在的な商業化には大量の追加資金が必要になります。 私たちのいくつかの候補製品については、これらの候補製品を開発および/または商業化するために、パートナーまたは製薬会社と維持し、さらに協力する必要があるかもしれません。
| 36 |
私たちは適切な協力者を探すことで激しい競争に直面している。我々が連携について他の最終合意 を達成するかどうかは,協力者の資源や専門知識の評価,提案した連携の条項や条件,提案した協力者の複数の要因の評価に依存する.これらの要因には、臨床試験の設計または結果、FDAまたは米国以外の同様の規制機関が承認する可能性、候補製品の潜在的な市場、候補製品の製造と患者への配送のコストと複雑さ、競争製品の潜在性、我々の技術所有権に関する不確実性の存在、このような所有権に挑戦する場合、挑戦の是非を考慮することなく、これらの要因が含まれる可能性がある。そして、一般的な業界および市場状況を考慮することができます。br}パートナーは、協力可能な同様の指示を得るために、候補製品または技術を代替することを考慮することができ、そのような連携が、私たちの候補製品との連携よりも魅力的であるかどうかを考慮することができます。
商業化協定によると、私たちはIndegeneと作業宣言を締結し、Indegeneが提供するサービス、そのようなサービスの配信可能な内容、および私たちが支払う費用を列挙する。私たちは、Indegeneが提供するサービスや私たちが支払うべき費用を含む、私たちが受け入れられる条項に従って作業説明書の条項 を協議することができないかもしれません。もし私たちがそれができなければ、私たちはCTX-1301の商業化のために他の協力を求めなければならず、これは商業化を延期するかもしれない。
私たち はタイムリーで、受け入れ可能な条項によって、あるいは全く協力を協議できないかもしれません。もし私たちがそれができない場合、私たちは候補製品の開発を減らし、私たちの1つ以上の開発計画を減少または延期し、潜在的な商業化を延期したり、販売やマーケティング活動の範囲を縮小したり、私たちの支出を増加させ、自費で開発または商業化活動を展開する必要があるかもしれない。もし私たちが私たちの支出を増やして自分の開発や商業化活動に資金を提供することを選択すれば、私たちは追加の資本を得る必要があるかもしれません。これらの資本は受け入れ可能な条項で私たちに提供できないか、あるいは全く得られないかもしれません。もし私たちが十分な資金を持っていなければ、私たちは私たちの候補製品をさらに開発したり、それを市場に出して製品収入を生成することができないかもしれない。
私たちは第三者に依存して、流通、顧客サービス、売掛金管理、現金催促、不良事件報告など、私たちの商業化された任意の製品に多くの基本サービスを提供します。これらの第三者が予想通りに実行されていない場合、または が法律および法規の要求を遵守できない場合、私たちはCTX-1301、CTX-1302および/またはCTX-2103を商業化する能力が深刻な影響を受けることになり、私たちは規制制裁を受ける可能性がある。
商業化プロトコルによれば、Indegeneは、(A)医療事務および薬物警戒に関連するサービス、(B)定価、精算および市場アクセス、(C)商業運営、および(D)マーケティングを含むCTX-1301の商業化サービスを提供する。
私たちの は、CTX-1301、CTX-1302、およびCTX-2103を含む、我々の任意またはすべての製品の販売および流通に関連する様々な機能を実行するために追加の第三者サービスプロバイダを保持することができ、承認された場合、これらの製品の重要な側面は、私たちの直接制御範囲内ではありません。 これらのサービスプロバイダは、流通、顧客サービス、売掛金管理、および現金入金に関連するキーサービスを提供する可能性がある。
私たちはIndegeneに大きく依存し、未来の第三者プロバイダに大きく依存してサービスを提供してくれるだろう。これらの第三者サービスプロバイダが適用される法律法規を遵守できない場合、予想される期限内に作業を完了できなかった場合、またはその契約義務を履行していない場合、ビジネスニーズを満たすために製品を納入する能力は深刻な影響を受ける可能性がある。また,第三者を招いて有害事象報告,セキュリティデータベース管理,我々の候補製品に関する医療情報要求や関連サービスを満たす様々な他のサービスを提供してくれることも可能である。もしこれらのサービスプロバイダが維持しているデータの品質や正確性が不足している場合、あるいは彼らが様々な要求を満たしていない場合、私たちは規制部門の制裁を受ける可能性がある。
もし私たちが私たちの製品や候補製品のために十分な保証と精算レベルを維持することができなければ、承認されれば、彼らの商業成功は深刻に阻害される可能性がある。
私たちの製品と規制部門の承認を得た候補製品の成功販売は、第三者支払者が十分な保険とbr精算を提供するかどうかにかかっている。処方薬を服用してその病状を治療する患者は、通常、その処方薬に関連する費用の全部または一部を精算するために第三者支払者に依存する。政府医療保健計画(例えばMedicareとMedicaid)と商業支払者の十分なカバーと精算は新製品の受け入れ度に重要である。カバー範囲 は臨床と経済標準に依存する可能性があり、より成熟或いはコストの低い治療代替製品がすでに利用可能であるか、あるいはその後利用可能な場合、これらの標準は新薬製品に不利である。特定の製品の保証範囲を獲得したと仮定すると,発生する精算料率が十分に高くない可能性があり,あるいは患者が受け入れられないと考えられる高い共済額が必要となる可能性がある。患者が私たちのbr製品を使用する可能性はあまりありません。保険を提供し、私たちの製品の大部分のコストを支払うのに十分でなければなりません。
| 37 |
さらに、CTX−1301、CTX−1302、およびCTX−2103の市場は、第三者支払者の薬物処方brまたは第三者支払者が保険および精算の薬剤リストを提供するかどうかに大きく依存するであろう。業界競争はこのような 配合に組み入れられ、よく製薬会社に下行定価圧力をもたらす。さらに、コストの低い模倣薬または他の代替品がある場合、第三者支払者は、その処方に特定のbrブランド薬を含むことを拒否するか、または処方によって制御されるか、または患者がブランド薬に接触することを制限することができる。
第三者支払者は、海外でも国内でも、政府のものでも商業的でも、ますます複雑な方法を開発して医療コストを抑えています。 また、米国では、第三者支払者の間で薬品の保証と精算に統一された政策要求がない。そのため、薬品の保証範囲と精算範囲は支払人によって異なる。そのため、保証範囲の確定過程は通常時間がかかり、高価な過程であり、それぞれの支払人に著者らの製品を使用する科学と臨床支持を提供する必要があるが、保証範囲を一貫的に応用し、十分な精算を保証するか、あるいは最初に獲得することは保証されない。
また,将来の保険や精算は米国や国際市場でより多く制限される可能性があると考えられる。私たちが規制部門の承認を得る可能性のある候補製品の第三者保証範囲と精算は、アメリカや国際市場で入手できないか足りないかもしれません。これは、私たちの業務、運営結果、財務状況、見通しに大きな悪影響を及ぼす可能性があります。
第三者支払者は消費者が私たちの製品を購入する費用を十分にカバーしたり補償したりできないかもしれません。
我々の将来の収入や運営から正のキャッシュフローを生み出す能力は,政府や第三者支払者が様々な手段で医療コストを抑えたり低減したりする継続的な努力の影響を受ける可能性がある。特定の海外市場では、処方薬の価格設定は政府によって規制されている。米国では、多くの連邦や州政府が同様の政府統制措置の実施を提案し続けていると予想されている。私たちはどのような立法提案を採用するか、あるいは連邦、州、あるいは個人医療製品とサービスの支払者がいかなる薬品定価改革提案や立法に対してどのような行動を取るかもしれないことを決定することができない。このような改革は製品の開発やテストを困難にする可能性があるため、販売から収入を得て利益を実現する能力を制限する可能性があります。また、このような改革が私たちの業務やパートナーに影響を与える可能性があれば、製品を商業化する能力が損なわれる可能性があります。
米国や他の地域では,処方薬製品の販売は,消費者が第三者支払者(例えば政府や個人保険計画)から精算できるかどうかに大きく依存している。第三者支払者は医療製品の料金にますます多くの挑戦をしている。CTX−1301、CTX−1302、および/またはCTX−2103の市場は、第三者支払者が保険および精算を提供するか否かに大きく依存するであろう。業界が精算資格を得るための競争は、薬品の価格下圧力 を招くことが多い。さらに、価格の低い後発薬や他の代替品が利用可能な場合、第三者支払者は、特定のブランドの薬物や製品の精算を拒否する可能性がある。米国では,第三者支払者の間に統一された薬品製品保証と精算政策はない。各第三者支払人はそれぞれ保証範囲と精算レベルを許可するため、保証範囲と十分な精算を獲得することは時間がかかり、高価な過程である。私たちは私たちの製品を使用してそれぞれの第三者支払者に科学的と臨床的支援を提供することを要求されますが、承認される保証はありません。この過程は市場の私たちの製品に対する受け入れ度を遅らせる可能性があり、私たちの未来の収入と経営業績に負の影響を与える可能性がある。たとえ私たち がCTX−1301、CTX−1302、および/またはCTX−2103を市場に投入することに成功したとしても、費用対効果があるかどうか、または が患者に保険および十分な補償を提供できるかどうかを決定することはできない。患者は、保険が提供されない限り、CTX−1301、CTX−1302、および/またはCTX−2103を使用する可能性が高く、その費用の大部分を支払うのに十分である。
また、多くの外国、特にEU内の国では、処方薬の定価が政府によって規制されている。アメリカ以外のいくつかの司法管轄区では、薬物の提案定価は必ず承認されなければ、合法的に発売されることができない。各国の薬品定価に対する要求は大きく異なる。例えば、EUは、その加盟国にオプションを提供し、その国の医療保険システムが精算を提供する医療製品の範囲を制限し、人が使用する医療製品の価格を制御する。加盟国は医薬製品の具体的な価格を承認することができ、 も医薬製品を市場に投入する会社の収益力に対して直接或いは間接的な制御制度をとることができる。また、製品の発売承認を受けた後、これらの国·地域の政府当局との定価交渉にはかなりの時間がかかる可能性がある。米国以外の国で精算または定価の承認を得るためには、追加の臨床試験を行う必要があるかもしれませんが、特に私たちの製品の費用対効果 CTX-1301、CTX-1302および/またはCTX-2103と他の利用可能な治療法を比較する必要があります。私たちは海外の低価格製品のCTX-1301および/またはCTX-1302に対する競争に直面する可能性があり、これらの製品は薬品に対して価格規制を行う。さらに、輸入された外国製品がCTX−1301、CTX−1302、および/またはCTX−2103と競合する可能性があり、これは、私たちの収益性に悪影響を及ぼす可能性がある。
| 38 |
私たち はCTX-1301、CTX-1302とCTX-2103の価格は現在の治療法と競争力を持つ必要があり、アメリカと国際市場で全額精算を受ける資格があると考えている。もし、第三者支払者からCTX−1301、CTX−1302、br}および/またはCTX−2103の保証範囲および十分な支払いレベルを得ることができない場合、医師は彼らがどのくらいの薬を処方するか、またはどのような場合にこのような薬を処方するかを制限する可能性があり、患者のbr}は購入を拒否するかもしれない。これは逆に私たちが任意またはすべての製品を成功的に商業化する能力に影響を与え、私たちの業務を損なう可能性がある。
もし私たちがCTX-1301、CTX-1302、および/またはCTX-2103、および任意の未来の候補製品の需要をサポートできない場合、私たちは増加する需要を満たすのに十分な能力を持っているか、または私たちの薬物送達技術の発展を成功的に管理できない場合、私たちの業務は影響を受ける可能性がある。
私たちの数が増加するにつれて、私たちは、予想される回転時間内により大規模な製品生産をサポートするために、私たちのプラットフォームを拡張する必要があるだろう。私たちは、より多くのCTX-1301、 CTX-1302および/またはCTX-2103を処理するために、より多くの認証された実験室科学者および技術および製造者が必要かもしれない。私たちはまた他のデバイスを購入する必要があるかもしれませんが、いくつかのデバイスは購入、設定、および検証のために数ヶ月以上かかるかもしれません。これらの規模の増加,人員,設備の拡張やプロセス改善が成功することは保証されず,我々の施設に必要なbr}拡張を収容するのに十分な空間があるかどうかは保証されない.
顧客と第三者支払者との関係は、刑事制裁、民事処罰、契約損害、名声損害、利益および将来の収益の減少に直面する可能性がある反リベート、詐欺、乱用、および他の医療法律および法規の制約を受けています。
私たちの製品および規制の承認を得てアメリカで販売されている任意の候補製品について(もしあれば)、私たちと第三者支払者と顧客の手配は、私たちが広く適用される詐欺と乱用、および他の医療法律と法規に直面する可能性があり、私たちのマーケティング、販売、流通を制限するかもしれません。私たちがマーケティングの許可を得た任意の製品の業務または財務的配置と関係を制限するかもしれません。また、私たちはアメリカの健康情報プライバシーとセキュリティ法規の制約を受ける可能性があります。 連邦と州政府と私たちが業務を展開している外国司法管轄区域。私たちの運営能力に影響を与える可能性のある法律には
| ● | 連邦反減税条例は、他の事項に加えて、個人を推薦するために、直接または間接的に故意に誘致、受け入れ、提供または支払いを禁止することを禁止し、個人を推薦したり、連邦医療保健計画(例えば連邦医療保険と医療補助計画)に従って費用を支払うことができる項目やサービスを購入または推薦する;個人または実体は、この法規または具体的な意図を実際に理解する必要がなく、違反行為を実施することができる | |
| ● | 連邦政府への資金支払い義務を回避、減少または隠蔽するために、連邦政府への資金支払い義務を減少または隠蔽するための連邦医療保険および医療補助計画を含む連邦民事·刑事虚偽申告法および民事金銭罰法を含む連邦民事·刑事虚偽申告法および民事金銭罰法。訴訟は、政府または通報者によって提起されることができ、連邦反ダンピング法令違反による連邦医療保健計画の物品およびサービスに対する支払いクレームは、虚偽クレームまたは詐欺的クレームを構成すると断言することを含むことができる |
| ● | 1996年“連邦医療保険移行性·責任法案”(HIPAA)は、詐欺の任意の医療福祉計画を実施する計画に対して、または重大な事実を故意に偽造、隠蔽または隠蔽し、または医療福祉、プロジェクトまたはサービスの交付または支払いについて任意の重大な虚偽陳述を行い、刑事および民事責任を適用する。アメリカ連邦反バックル法規と類似しており、個人或いは実体はこの法規或いはbr違反の具体的な意図を実際に理解する必要がなく、違反を実施することができる。HIPAAは、2009年の“健康情報技術と臨床健康法”(“HITECH”)及びそのそれぞれの実施条例改正により、個人が識別できる健康情報のプライバシー、安全と伝送を保護する上で、医療保健提供者、健康計画及び医療情報交換所及びその業務パートナーに強制契約条項を含む何らかの義務を課した | |
| ● | PACAの一部として公布された“医師支払い日光法案”は、Medicare、Medicaidまたは児童健康保険計画に従って精算可能な薬品、設備、生物製品および医療用品のいくつかのメーカーが、医師および教育病院への支払いおよび他の方法での価値移転に関する情報、ならびに医師およびその直系親族が所有する所有権および投資権益をCMSに毎年報告することを要求する“医師支払い陽光法案”の一部として発行される | |
| ● | 同様の州および外国の法律、例えば州反リベートおよび虚偽クレーム法律は、非政府第三者支払者(民間保険会社を含む)によって精算される医療項目やサービスの手配およびクレーム、および場合によっては健康情報のプライバシーや安全を管理する州および外国法律に適用可能であり、多くの法律は重大な点で互いに異なり、HIPAAによって先制されず、コンプライアンス作業を複雑化させることが多い。 |
| 39 |
我々と第三者の業務配置が適用される医療法令に適合することを確保する努力 は巨額の コストに及ぶ可能性がある。政府当局は、私たちの業務実践が現在または未来の法規、br法規、または詐欺および乱用または他の医療保健法律法規の適用に関する判例法に適合していない可能性があると結論するかもしれない。もし私たちの業務がこれらの法律または任意の他の私たちに適用される可能性のあるいかなる政府法規に違反していることが発見された場合、私たちは損害賠償、罰金、監禁、MedicareおよびMedicaidのような政府援助の医療計画から除外され、私たちの業務を削減または再編することを含む重大な民事、刑事、および行政処罰を受ける可能性がある。もし私たちがそれと業務を展開する任意の医師または他の医療提供者または実体が適用法に適合していないことが発見された場合、彼らは、政府の援助に参加する医療計画から除外されることを含む刑事、民事または行政処罰を受ける可能性がある。
製品責任訴訟は私たちの資源を移転し、大量の責任を招き、私たちの製品の商業潜在力を下げるかもしれない。
われわれは臨床試験参加者から適切なインフォームドコンセントを得たにもかかわらず,われわれの候補製品の臨床試験による固有製品責任クレームリスクに直面している。CTX−1301、CTX−1302、および/またはCTX−2103または任意の他の候補製品のマーケティング承認を取得し、商業販売を行う場合、より大きなリスクに直面するであろう。例えば、私たちが開発した任意の製品が臨床試験、製造、マーケティング、または販売中にダメージを与えたと言われたり、不適切なことが発見された場合、起訴される可能性があります。任意のこのような製品責任クレームは、製造欠陥、設計欠陥、製品固有の危険、不注意、厳格な責任、または保証違反の告発を含む可能性がある。州消費者保護法によると、クレームも主張することができる。もし私たちが製品責任クレームを自己弁護することに成功できなければ、私たちは巨額の責任を負うか、あるいは私たちの候補製品の商業化を制限することを要求されるかもしれません。事件がどうであっても最終的な結果がどうであろうと、賠償責任は次のようになるかもしれない
| ● | 私たちの経営陣が業務戦略を実行するための資源を減らした | |
| ● | 私たちの候補製品や開発可能な製品への需要を減らしました | |
| ● | 私たちの名声とメディアの大きな否定的な関心を損なう | |
| ● | 臨床試験参加者は脱退しました | |
| ● | 規制当局が調査を開始した | |
| ● | 製品 リコール、撤回またはラベル付け、マーケティングまたは販売促進制限; | |
| ● | この訴訟を正当化するための巨額の費用 | |
| ● | 試験参加者または患者に巨額の金銭的報酬を支給する | |
| ● | 収入の損失 | |
| ● | 私たちが開発する可能性のあるどんな製品も商業化できない。 |
もし私たちのすべての候補製品が商業販売を許可されれば、私たちは消費者の私たちに対する見方と私たちの製品の安全性と品質に高度に依存します。もし私たちが否定的な宣伝を受けたら、私たちは不利な影響を受けるかもしれない。もし私たちの任意の製品または他の会社によって製造および流通された任意の類似製品が患者に有害であることが証明された場合、または患者に有害であると断言された場合、私たちも悪影響を受ける可能性がある。私たちは消費者の見方に依存するので、患者が私たちの製品または他の会社が販売している任意の類似した製品を使用または誤用することによる、疾患または他の悪影響に関連する任意の負の宣伝は、私たちの財務状況または運営結果に重大な悪影響を及ぼす可能性がある。
私たちの製品責任保険カバー範囲は私たちが発生する可能性のあるすべての責任とすべての責任をカバーするのに十分ではないかもしれません。
私たちの は現在全部で1,000万ドルの製品責任保険があります。これは私たちが発生する可能性のあるすべての責任とすべての 責任をカバーするのに十分ではないかもしれません。保険範囲はますます高くなっています。私たちは合理的なbrコストまたは起こりうる任意の責任を支払うのに十分な金額で保険範囲を維持できないかもしれない。予期せぬ副作用を有する薬物に基づく集団訴訟では,多くの判決が下されている。成功した製品責任クレームまたは私たちに対する一連のクレーム、特にbr}判決が私たちの保険範囲を超えた場合、私たちの現金を減少させ、私たちの業務に悪影響を及ぼす可能性があります。さらに、許容可能な費用で十分な保険範囲を得ることができないか、または潜在的な製品責任クレームに対して保護を提供することができない可能性があり、これらのクレームは、私たちの製品の商業生産および販売を阻止または抑制する可能性がある。
| 40 |
もし私たちが環境、健康、安全法律法規を遵守できなかった場合、私たちは罰金や処罰を受けたり、brコストが発生したりする可能性があり、これは私たちの業務成功に重大な悪影響を及ぼす可能性がある。
私たちのbrは多くの環境、健康、そして安全法律法規によって制限されている。私たちの行動は化学物質と生物学的材料を含む危険で燃えやすい材料の使用に関するかもしれない。私たちの業務は危険な廃棄物製品を発生させる。私たちは第三者と契約を結んで、このような材料と廃棄物を処理することを望んでいる。私たちはこれらの材料による汚染や傷害のリスクを取り除くことができません。もし危険な材料を使用して汚染や損傷を起こした場合、私たちはそれによるいかなる損害にも責任を負う可能性があります。 どんな責任も私たちの資源の範囲を超えている可能性があります。私たちはまた民事や刑事罰金と処罰に関連した巨額の費用を発生させるかもしれない。
我々は、危険材料または他の仕事に関連する傷害の使用による従業員へのダメージによるコストおよび支出を支払うために労働者補償保険を維持しているが、この保険は、潜在的なbr責任に対して十分な保険を提供することができない可能性がある。また、現在または未来の環境、健康、安全に関する法律や法規を遵守するためには、巨額のコストが生じる可能性がある。このような現行または未来の法律法規は私たちの研究、開発、または生産努力を損なうかもしれない。このような法律法規を遵守しないことはまた巨額の罰金、処罰、または他の制裁につながる可能性がある。
私たちの成長、従業員、運営に関するリスクを管理する
私たちは将来、私たちの組織の規模と複雑さをさらに増加させる必要があり、私たちは成長戦略を実行し、どんな成長を管理するかについて困難に直面する可能性がある。
私たちの既存のbr管理、人員、システム、および施設は、私たちの業務計画と短期的な将来の成長をサポートするのに十分ではありません。私たちは、私たちの計画の研究、開発、商業化活動を支援するために、私たちの製造チーム、臨床チーム、管理、運営、財務、その他の資源をさらに拡大する必要があります。
私たちの運営、成長、様々なプロジェクトを効率的に管理するためには、
| ● | 私たちの業務、財務、管理、および規制コンプライアンス制御、および報告システムとプログラムを改善していきます | |
| ● | 十分な数の優秀な従業員を引きつけて維持します | |
| ● | マーケティング、販売、流通能力を発展させる | |
| ● | 候補製品の商業化活動を効率的かつ経済的に管理しています | |
| ● | 開発と商業化パートナーとの関係を構築し、維持し、 | |
| ● | 臨床試験を効率的に管理しています | |
| ● | 我々の第三者供給と製造運営を経済的かつ効率的に管理するとともに、現在の候補製品の生産能力をビジネスレベルに向上させる | |
| ● | 我々の開発作業を効率的に管理するとともに,パートナーや他の第三者に対する契約義務を履行する. |
また,歴史的には,製品開発や臨床試験に関するタスクを含む複数のタスクをアルバイト外部コンサルタントを用いて実行していきたいと考えてきた。私たちの成長戦略はまた、これらと他の未来の任務を実施するために、私たちのbrコンサルタントの使用を拡大する必要があるかもしれない。私たちはコンサルタントに依存して私たちの業務のいくつかの機能を履行し、これらのコンサルタントを効率的に管理して、彼らが契約義務を成功的に履行し、予想される締め切りまでに仕事を完了することを保証する必要があります。私たちが既存のコンサルタントを管理したり、他の適任な外部コンサルタントを見つけることができる保証はありません。必要に応じて、経済的に合理的な条項、あるいは全くできません。新入社員の採用やコンサルタントの利用拡大で私たちの組織 を効率的に管理し、拡大することができなければ、私たちが計画している研究開発や商業化活動を効果的に実行するために必要な任務を効率的に実行することができない可能性があり、私たちの研究開発や商業化目標を実現できないかもしれません。
| 41 |
もし私たちが販売およびマーケティング能力を確立することができない場合、または第三者と合意して私たちの候補製品 をマーケティングおよび/または販売することができない場合、私たちはいかなる収入も生成できないかもしれない。
私たち は現在、CTX−1301、CTX−1302、またはCTX−2103を販売、マーケティングまたは流通する組織を有していない。したがって、CTX−1301、CTX−1302および/ またはCTX−2103を商業化するためには、 を確立するか、または第三者とマーケティング連携を行う必要がある。私たちは米国でCTX-1301、CTX-1302、および/またはCTX-2103を販売するために自分の販売チームを設立し、発展させることは高価で時間がかかり、任意の製品の発表を延期するかもしれない。私たちがこの能力を開発することに成功したかどうかは確認できませんが、私たちがそうしても、そのような組織を設立して維持するコストはそうするメリットを超える可能性があります。
販売組織の設立と管理には重大なリスクがあり、私たちは合格したbr個人を採用、保留し、激励する能力があるかどうか、十分な販売手がかりを生成し、販売とマーケティング人員に十分な訓練を提供し、異なる地理的位置に分散した販売とマーケティングチームを効果的に管理し、ホストサービスと第三者支払者との交渉に成功した。我々の内部販売、マーケティング、および流通能力の開発のいかなる失敗や遅延も、これらの製品の商業化に悪影響を及ぼすだろう。
商業化プロトコルによれば、Indegeneは、(A)医療事務および薬物警戒に関連するサービス、(B)定価、精算および市場アクセス、(C)商業運営、および(D)マーケティングを含むCTX-1301の商業化サービスを提供する。
我々 はまた,第三者と追加の戦略的パートナーシップを構築し,我々の候補製品を商業化することも可能である.
商業化協定によると、私たちはIndegeneと作業宣言を締結し、Indegeneが提供するサービス、そのようなサービスの配信可能な内容、および私たちが支払う費用を列挙する。私たちは、Indegeneが提供するサービスや私たちが支払うべき費用を含む、私たちが受け入れられる条項に従って作業説明書の条項 を協議することができないかもしれません。もし私たちがそれができなければ、私たちはCTX-1301の商業化のために他の協力を求めなければならないだろう。これは商業化が遅れる可能性がある。私たちはまた、私たちが受け入れられる条項に従って第三者との関係を構築することが難しいかもしれないし、製品を商業化したいすべての地域では関係を築くことができないかもしれない。もし私たちが十分な販売、マーケティング、流通能力を確立できなければ、 独立しても第三者と協力しても、私たちは十分な製品収入を生み出すことができず、利益を上げることができないかもしれません。私たちは現在、広く資金の豊富なマーケティングと販売業務および/または根深い流通ルートを持つ多くの会社と競争します。マーケティングおよび販売機能を実行するための内部チームまたは第三者の支援がなければ、私たちはこれらのより成熟した会社と成功的に競争することができないかもしれない。
もし私たちが管理職や他のキーパーソンを引き付けることができない場合、私たちは私たちの候補製品の開発や商業化を成功させたり、他の方法で私たちの業務計画を実施したりすることができないかもしれません。
私たちが競争の激しい製薬業界の中で競争できるかどうかは、私たちが高い素質の管理、科学、医療、販売とマーケティング及びその他の人員を誘致し、維持できるかどうかにかかっている。私たちは私たちの管理と科学者たちに非常に依存している。これらの個人のサービスを失うことは、私たちの製品ラインの成功開発を阻害、遅延、あるいは阻止する可能性があり、私たちが計画した臨床試験の完成、私たちの候補製品の商業化は内または新しい資産を獲得することができ、そして私たちが業務計画を成功的に実施する能力にマイナスの影響を与えるかもしれない。もし私たちがこの人たちの中の誰かのサービスを失ったら、私たちは適切な代替人員をタイムリーに見つけることができないか、あるいは適切な代替者を見つけることができないかもしれません。したがって、私たちの業務は損なわれるかもしれません。2023年12月、最高財務官を含む幹部2人と臨床運営社員2人が辞任した。2024年1月25日、私たちは私たちの上級副総裁兼首席財務官にbrカラハンさんを任命した。
2023年12月、私たちの取締役会の4人の独立メンバーが辞任した。私たちの取締役会の現職2人は独立していません。私たちは現在監査委員会、報酬委員会、または指名とコーポレートガバナンス委員会を持っていません。私たちの取締役会は現在非独立取締役のみで構成されており、そのうちの一人が私たちのCEOであるため、取っている行動を独立して監督するメリットを得ることができません。また、多数を占める独立取締役会メンバーの不足は、内部統制の維持と発展を困難にし、内部統制におけるいかなる重大な弱点を補い、金融市場で資金を調達することを困難にする可能性がある。2023年12月26日、私たちは3人の取締役会メンバーがそれぞれ2023年12月12日と2023年12月13日に辞任したことに基づいて、取締役上場規則 5605(“独立取締役規則”)に掲載されている独立ナスダック上場規則、監査委員会、給与委員会及び取締役の取締役指名に対する独立監督要求を遵守しないという従業員手紙を受け取った。私たちは新しい取締役を物色していますが、新しい取締役を誘致し、独立した取締役会の多数の席を維持したり、独立した取締役ルールを再遵守できる保証はありません。
私たちはすべての重要な従業員の生命保険ではなく、特定の個人の生命保険に対して、“キーパーソン”の保険証書を持っている。わが社の価値のある従業員を引き留めるために、給料と現金奨励のほか、時間とともに付与された株式オプションを提供することも可能です。時間が経つにつれて、従業員に付与された株式オプションの価値は、私たちの株価変動の大きな影響を受け、これらの変動は私たちの制御範囲を超え、いつでも他社の見積もりを相殺するのに十分ではないかもしれません。
| 42 |
生物技術、製薬とその他の業界のbr人材に対する激しい競争のため、私たちのbrは未来に合格した管理層と他の肝心な人員を引き付けることができないかもしれない。私たちは経験者を私たちの会社に誘致することは難しいかもしれませんし、従業員を募集して維持するために大量の財務資源が必要かもしれません。私たちと合格人材を競争する多くの他の製薬会社 は私たちよりも多くの財務と他の資源、異なる リスク状況とより長い業界歴史を持っている。それらはまた職業発展にもっと多様な機会とより良い機会を提供するかもしれない。業務目標を実現するために必要な人員を引き付けることができなければ、br制限に遭遇する可能性があり、業務戦略を実施し、業務目標を実現する能力を損なうことになります。
また,科学と臨床コンサルタントを持ち,開発や臨床戦略の策定を支援している。これらのコンサルタント は私たちの従業員ではなく、他のエンティティと約束、相談、またはコンサルティング契約を締結しているかもしれません。これは、彼らの私たちに対する利用可能性を制限するかもしれません。また、私たちのコンサルタントは他社と合意し、これらの会社が私たちと競争する可能性のある製品や技術の開発に協力するかもしれません。
我々の研究開発の重点は候補製品の発見と開発であるが,これらの候補製品は市場に投入できない可能性がある。
これまで、我々の開発研究および臨床開発は、ADHDを治療するための候補製品CTX−1301、CTX−1302、および不安を治療するためのCTX−2103を生成してきた。私たちの成長戦略の一部として、私たちはより多くの候補製品を決定し、開発し、マーケティングするつもりだ。私たちは私たちのパイプラインとノウハウのために様々な治療機会を探索している。私たちは、現在または将来の任意の特定の内部候補製品の開発を完了するのに数年かかる可能性があり、任意の段階で失敗する可能性があります。私たちは、生物学的等価、安全かつ効果的、および/または許可された薬剤よりも商業的に著しく改善された薬剤を開発することができないかもしれません。私たちが資源を割り当てた候補製品は成功しないかもしれない。この戦略の成功は、将来性のある候補製品および製品を識別、選択、発見、および取得する能力にある程度依存する。
許可証の提案、交渉、および実施、または候補製品または承認された製品の取得の流れは、長くて複雑である。他の会社には、より多くの財務、マーケティング、販売資源を持つ会社が含まれており、候補製品や承認された製品の許可や買収を争っている可能性があります。私たちの資源は限られており、第三者製品、業務、技術の買収や許可を識別して実行し、現在のインフラに統合することはできません。また,我々 は未完成の潜在的買収に資源を投入する可能性があるかもしれないし,このような努力の期待収益を実現できない可能性がある.私たちは私たちが が受け入れられると思う条項や他の候補製品を全く獲得できない権利がないかもしれない。
さらに、将来の買収は、多くの運営と財務リスクをもたらす可能性がある
| ● | 未知の債務にさらされるリスクは | |
| ● | 買収と統合コストは予想以上に高い | |
| ● | 買収された企業の運営や人員を私たちの運営や人員と組み合わせることは難しい。 |
さらに、我々が買収した任意の候補製品は、商業販売の前に、広範な臨床試験およびFDAおよび他の規制機関の承認を含む追加の開発作業が必要となる可能性がある。
我々のPrecision Timed Release Platform技術に基づいて候補製品の開発に成功し,商業化することができなければ,将来的に製品収入を得ることができず,我々の財務状況に大きな被害を与え,株価に悪影響を与える可能性がある.
| 43 |
私たちのbrの経営業績は大きく変動する可能性があり、これは私たちの将来の経営業績を予測しにくくし、私たちの経営業績が予想を下回ってしまう可能性があります。
これまで,我々の運営は主に我々の候補製品の策定と開発に限られており,我々の候補製品の臨床試験を行ってきた。私たちのどんな候補製品も規制部門の承認を受けていない。したがって、もし私たちがより長い運営歴史や市場で承認された製品を持っていれば、私たちの未来の成功または生存能力に対するいかなる予測 もそれらが予測できるほど正確ではないかもしれない。また、私たちの経営業績は様々な他の要素によって変動する可能性があり、その中の多くの要素は私たちがコントロールできないもので、以下の要素を含む予測が難しいかもしれません
| ● | 私たちの候補製品の開始、登録、臨床試験の時間を延期します | |
| ● | 我々の候補製品または競合候補製品の臨床試験のタイミングおよび成功または失敗、または私たちの競争相手またはパートナー間の統合を含む業界競争構造における任意の他の変化; | |
| ● | 臨床開発における候補製品の監督審査と承認に関するいかなる遅延もない | |
| ● | 我々の候補製品に関する研究や開発活動の時間、コスト、投資レベルは、時々変化する可能性がある | |
| ● | 我々の候補製品を製造するコストは、FDAのガイドラインや要求、生産数量によって異なる可能性がある | |
| ● | 私たちは候補製品を開発するために追加資金を得ることができます | |
| ● | 私たちは、他の候補製品および技術を取得または開発するための支出 ; | |
| ● | もし私たちの候補製品が承認されれば、それらの需要レベルは大きく異なるかもしれない | |
| ● | 私たちの候補製品の潜在的な副作用は、商業化を延期または阻止し、または承認された薬物の市場からの撤退を招く可能性がある | |
| ● | 承認された場合、患者または医療提供者は、候補製品のカバー範囲または十分な補償能力を得ることができる | |
| ● | 私たちは第三者製造業者が私たちの候補製品を供給したり製造したりすることに依存しています | |
| ● | 効果的な販売、マーケティング、流通インフラをタイムリーに構築したり、アウトソーシングすることができます | |
| ● | 市場は私たちの候補製品(承認されれば)を受け入れ、これらの候補製品の需要を予測する能力 | |
| ● | 私たちはアメリカ以外で承認され、私たちの候補製品を商業化する能力 | |
| ● | 私たちは協力、許可、または他の手配の能力を確立し、維持する | |
| ● | 知的財産権を保護する能力と第三者の能力 | |
| ● | 潜在的な訴訟または他の紛争に関連する費用と結果; | |
| ● | 私たちは未来の成長を十分に支援することができます | |
| ● | 私たちは私たちの業務を効率的に管理するために、重要な人員を引き付けて維持する能力がある | |
| ● | 危険物質に関わる潜在的な責任 | |
| ● | 私たちは十分な保険証書を維持することができます | |
| ● | 未来 私たちの会計政策における会計声明や変更。 |
| 44 |
私たちの経営業績と流動性需要は市場変動や経済低迷の負の影響を受ける可能性があります。
私たちの経営業績と流動性はアメリカや世界の他の地域の経済状況の負の影響を普遍的に受けるかもしれません。非必須医療製品やプログラムの市場は,特に不利な経済条件の影響を受けやすい可能性がある。一部の患者は私たちのいくつかの候補製品は自由に支配可能であると思うかもしれないが、もしこのような製品が全額精算を得ることができなければ、これらの製品に対する需要は著者らの目標患者群の自由支配可能な支出レベルとリンクするかもしれない。国内·国際経済状況と懸念に基づいて、国内·国際株式·債務市場は高度な変動や動揺を経験し続けている可能性がある。もしこれらの経済状況や懸念が持続的または悪化し、br市場が引き続き変動すれば、私たちの経営業績と流動性は多くの面でこれらの要素の不利な影響を受ける可能性があり、私たちのいくつかの製品に対する需要が弱まって、必要な時に資金を調達することが難しくなり、私たちの株価が下落する可能性がある。また、私たちは主にアメリカで私たちの製品を販売する予定ですが、私たちのパートナーは広範なグローバル業務を持っており、これは間接的に私たちをリスクに直面させています。
もし私たちの内部コンピュータシステムが故障したら、私たちの業務と運営は影響を受けるだろう。
セキュリティ対策が実施されているにもかかわらず、私たちの内部コンピュータシステムおよび私たちの現在および未来のパートナー、請負業者およびコンサルタントのコンピュータシステムは、コンピュータウイルス、許可されていないアクセス、自然災害、テロ、戦争および電気通信、および電気故障によって破壊されやすい。我々はこれまでこのような重大なシステム故障,事故やセキュリティホールに遭遇していないが,このような事件が発生して我々の運営が中断されると,我々の製造活動,開発計画,業務運営に大きな中断を招く可能性がある.例えば、完成したbrや将来の臨床試験の生産記録や臨床試験データの損失は、私たちの規制承認作業を遅延させ、データを回復または複製するコストを著しく増加させる可能性がある。任意の中断またはセキュリティホールが私たちのデータまたはアプリケーションを紛失または破損させ、機密または独自の情報を適切に開示しない場合、私たちは責任を招く可能性があり、私たちの製品および候補製品のさらなる商業化およびbr}開発が延期される可能性がある。
私たち はますます情報技術に依存しており、私たちのシステムとインフラはネットワークセキュリティ とデータ漏洩リスクを含む一定のリスクに直面している。
我々の情報技術システムに重大な中断や情報セキュリティが破壊され,我々の業務に悪影響を与える可能性がある.通常の業務プロセスでは,大量の機密情報を収集,格納,転送するため,安全な方法でこのような機密情報の機密性と完全性を確保しなければならない.我々の情報技術(Br)システムおよび我々と契約を締結した第三者サプライヤーのシステムの規模および複雑さは、当社の従業員、パートナーまたはサプライヤーの不注意または故意の行為、悪意のある第三者の攻撃、または私たちまたは第三者が維持するシステムインフラの意図的または意外な物理的損傷を含むため、サービス中断およびセキュリティ破壊の影響を受けやすい可能性がある。これらの機密、独自、または商業秘密情報を秘密にすることは、私たちの競争業務の地位に非常に重要だ。我々は、このような情報を保護し、情報技術に投資するための措置をとっているが、我々の努力は、システム内のサービス中断またはセキュリティホールを防止すること、または私たちのトラフィック運営に悪影響を及ぼす可能性があること、またはキーまたは敏感な情報の損失、伝播、または誤用をもたらす機密情報の不正使用または意図しないエラー使用または漏洩を防止することを保証することはできない。盗難、ハッカー攻撃、詐欺、brの詐欺、または他の形態の詐欺、または任意の他の理由によって、他の人が競合製品を生産すること、私たちのノウハウまたは情報を使用すること、または私たちの業務または財務状態に悪影響を及ぼす可能性がある、私たちのセキュリティ対策または意外な損失、開示意図のない伝播、許可されていない伝播、商業秘密、固有情報、または他の機密情報の流用または乱用。さらに、このような中断、セキュリティホール、機密情報の損失または漏洩は、私たちに財務、法律、業務、および名声被害をもたらす可能性があり、私たちの業務、財務状態、運営結果、またはキャッシュフローに重大な悪影響を及ぼす可能性があります。
| 45 |
私たちの知的財産権に関するリスク
もし私たちが私たちの製品や候補製品に関連する知的財産権が不足していれば、私たちは私たちの市場で効果的に競争できないかもしれない。
私たちは、特許、商業秘密保護、および秘密保護協定の組み合わせによって、私たちの製品、候補製品、および技術に関連する知的財産権を保護します。第三者による私たちの機密または独自の情報の開示または流用は、競争相手が私たちの技術的成果をコピーしたり、超えたりして、市場での私たちの競争的地位を侵食する可能性がある。
特許性、有効性、実行可能性および請求項の範囲に関連する法律基準のため、製薬およびバイオテクノロジー発明をカバーする特許は、複雑な法律、科学的および事実的問題に関連する。我々のような複雑な薬物放出の特徴を有する医薬製品の処方brは、任意の新しい特許出願の範囲を制限する密集した研究、出版、および特許分野である。したがって、私たちが特許を取得し、維持し、実施する能力は不確定であり、任意の既存特許の下の任意の権利、または私たちが取得または許可する可能性のある任意の特許は、競合他社の製品またはプロセスに対する商業的利点を提供するために、私たちの製品および候補製品に十分な保護を提供できない可能性がある。私たちが持っている特許出願は、米国または外国で特許を発行できない可能性がある。 特許が確実に発行されても、第三者は、その特許性、有効性(例えば、以前に確定されていない 以前の技術、または発明の事前開示、使用、販売または要約販売のような特許禁止事象を発見することによって)、実行可能な または範囲に挑戦する可能性があり、これは、このような特許の縮小、無効、または強制実行を招く可能性がある。例えば、米国特許 は第三者によって採択される可能性がある各方面間米国特許商標局と欧州特許庁の審査,認可後審査,派生又は妨害手続は,欧州特許庁の反対手続を介して疑問を提起することができる。また、競争相手に対して我々の特許権を主張すれば、競争相手は、主張する特許権の有効性および/または実行可能性を疑問視する可能性がある。付与された米国特許は、法定の有効性推定を有する権利があるが、その発行は、その有効性または実行可能性に関する確実な結論 ではなく、十分な独自保護や類似した製品を有するライバルに対する競争優位性を提供することができない可能性がある。
もし私たちが持っているまたは追求している特許および特許出願が私たちの製品および候補製品に提供する保護の広さまたは強度が成功的な挑戦を受けた場合、私たちは予期しない競争に直面する可能性があり、これは私たちの業務に実質的な悪影響を及ぼすかもしれない。たとえそれらが挑戦されていなくても,我々の特許や特許出願は我々の知的財産権を十分に保護できないか,あるいは我々の主張をめぐる他の人の設計を阻止する可能性がある.例えば、第三者は、我々の製品または候補製品と同様の治療利益を提供するが、私たちの特許保護範囲に属さない十分な異なる競争力を有する製品を開発することができる。
また,特定の管轄区域の臨床試験や市場進出に遅延があれば,特許保護された特定製品を販売できる時間が短縮される。
法律が保護を提供する場合であっても、私たちの固有の権利の範囲を強制して決定するために高価で時間のかかる訴訟が必要となる可能性があり、そのような訴訟の結果は不確定であろう。もし私たちまたは私たちの未来のパートナーのうちの1つが製品または私たちの技術をカバーする特許を強制的に執行するために第三者に対して法的訴訟を提起した場合、被告は私たちの特許の無効および/または強制執行ができないと反訴することができる。米国の特許訴訟では,被告が無効および/または実行不可能と主張する反訴が一般的である.有効性を疑問視する理由は、新規性の欠如、明らかな、書面記載の欠如、有効化されていない、または特許禁止事件を含むいくつかの法定要件のいずれかを満たすことができないと言われていることが可能であり、例えば、出願提出日の1年以上前の開示、使用、または販売発明などである。主張を実行できない理由は, 例えば,特許訴訟に関連する人が起訴中に米国特許商標局に重要な情報を隠蔽したり,誤った陳述をしたりしたためと考えられる.法律が無効で強制できないと断言した後の結果は予測できない。 例えば、有効性については、無効な以前の技術がないとは判断できないが、私たちと特許審査員は起訴中に知らないか、または私たちの特許に挑戦する第三者は、特許禁止事件が発生したとは断言しない。原告または被告が私たちの1つまたは複数の特許に対する無効および/または実行不可能な法的主張で勝訴した場合、私たちは私たちの1つまたは複数の製品または候補製品の少なくとも一部またはすべての特許保護を失うだろう。このような特許保護の喪失は、私たちの業務に実質的な悪影響を及ぼす可能性がある。
| 46 |
また,第3方向USPTO提出の既存技術の発行前提出を受けたり,再審に参加したりする可能性がある各当事者間 私たちの特許権の介入手続きを検討したり、挑戦したりする。私たちが将来提出する出願に基づく特許 もまた、派生および/または認可後の審査手続きの影響を受ける可能性がある。このような提出、訴訟、または訴訟において不利な裁決を下すことは、私たちの特許権の範囲を縮小したり、無効にしたりする可能性があり、第三者が私たちの技術または製品を商業化し、私たちと直接競争することを可能にします。さらに、私たちの特許および特許出願によって提供される保護の広さまたは強度が脅かされている場合、企業が私たちと協力して、現在または未来の候補製品を許可、開発、または商業化することを阻止する可能性がある。
私たちは世界のすべての管轄区域で私たちの知的財産権を保護できないかもしれませんし、私たちが保護を求めている司法管轄区でも、私たちの知的財産権を十分に実行できないかもしれません。
申請すると、世界のすべての国と司法管轄区で候補製品特許の起訴と保護の費用は目を引くほど高くなり、私たちのアメリカ以外のいくつかの国の知的財産権はアメリカほど広くない。また、いくつかの外国の国の法律は知的財産権の保護程度がアメリカの連邦や州法律よりも低い。したがって、米国外で特許権を申請することを選択しても、私たちはbr関連の権利要件を得ることができない可能性があり、および/または第三者が米国以外のすべての国/地域で私たちの発明を実施したり、米国または他の管轄地域で私たちの発明を使用して製造された製品を販売または輸入することを阻止できないかもしれません。
競争相手のbrは、私たちが特許保護を求めて獲得しない司法管轄区域で私たちの技術を使用して彼ら自身の製品を開発することができ、また、他の侵害製品を私たちが特許保護を持っている地域に輸出する可能性があるが、法執行力はアメリカ に及ばない。これらの製品は私たちの製品と競争するかもしれませんが、私たちの特許または他の知的財産権はそれらの競争を効果的にまたは阻止するのに十分ではないかもしれません。私たちが特定の管轄区域で申請して発行された特許を取得しても、私たちの特許主張や他の知的財産権は、第三者が私たちとの競争を阻止するのに有効または十分ではない可能性があります。
一部の国の法律は知的財産権の保護の程度はアメリカの法律に及ばない。多くのbr会社はある外国司法管轄区の知的財産権保護と擁護に重大な問題に直面している。 一部の国の法律制度、特に発展途上国の法律制度は、特許と他の知的財産権保護の強制執行に賛成しない。これは私たちの特許の侵害(獲得すれば)や私たちの他の知的財産権の流用を阻止することを難しくするかもしれない。例えば、多くの外国国には強制許可法があり、この法律によると、特許所有者はライセンスを第三者に付与しなければならない。また、多くの国は、政府機関や政府請負業者を含む特許の第三者への実行可能性を制限している。このような国では、特許は限られた利点を提供するかもしれないし、利益さえないかもしれない。
最終的には、国ごとに特許保護を求めなければならない。これは高価で時間のかかるプロセスであり、結果は不確定である。したがって、私たちは、将来的に特定の国で特許保護を求めないことを選択する可能性がある。また、私たちはいくつかの市場での私たちの製品の知的財産権を保護するつもりですが、私たちが私たちの製品を販売したいかもしれないすべての司法管轄区域で行われたり、類似した努力を維持することができることを確実にすることはできません。したがって、このような国で知的財産権を保護するための私たちの努力は十分ではないかもしれない。
私たちの特許保護の獲得と維持は、政府特許機関が提出した様々なプログラム、書類提出、費用支払い、および他の要求 を遵守することに依存し、これらの要件に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
米国特許商標局および各種外国政府特許機関は、特許過程において、いくつかのプログラム、文書、費用支払い、およびその他の規定を遵守することを要求している。場合によっては、規定を遵守しないことは、特許または特許出願が放棄または失効する可能性があり、それにより、関連する管轄区域の特許権の一部または全部が失われる可能性がある。この場合、競争相手は他の場合よりも早く市場に参入するかもしれない。
| 47 |
もし私たちが第三者の知的財産権侵害で起訴されたら、これは高価で時間がかかり、この訴訟の不利な結果は私たちの業務に実質的な悪影響を及ぼすだろう。
私たちのビジネス成功は、私たちと私たちの協力者が承認した製品と私たちの候補製品を開発、製造、マーケティング、販売する能力、および第三者の独自の権利を侵害することなく私たちの独自技術を使用する能力に依存します。私たちが候補製品を開発する分野には、第三者が所有する米国と外国から発行される特許や未解決の特許出願が多く存在します。製薬業界の拡張とより多くの特許の発行に伴い,我々の製品や候補製品は他人の特許権侵害のクレームを引き起こすリスクが増加する可能性がある。例えば、私たちの製品または候補製品は、私たちが現在知らない第三者が発行した特許を侵害する可能性があり、これは私たちの製品または候補製品を商業化できないかもしれない。特許出願が発行されるまでに数年かかる可能性があるため、現在処理されている出願が存在する可能性があり、これらの出願は、我々の製品 または候補製品が発行された特許を侵害する可能性がある。
製薬産業は特許保有者と後続薬品生産者の間の特許訴訟に満ちている。FDAが競争相手の製品を30カ月間承認することを阻止する可能性 はOrange Book特許について訴訟を起こす動機を増加させているが,非Orange Book特許に関する訴訟はADHD分野でもよく見られる.すでに多くの特許訴訟がADHD治療薬のほとんどに関連している。この傾向は継続する可能性があるため,正常業務過程で発生する法的問題やクレームの当事者となる可能性がある。
私たち は、私たちの製品または候補製品が彼らの知的財産権を侵害していると主張する第三者の将来の訴訟に直面したり、脅かされたりする可能性がある。もし私たちの製品または候補製品が第三者の知的財産権を侵害していることが発見された場合、私たちまたは私たちの協力者は、特許ライセンスを取得しない限り、適用された承認された製品および候補製品を商業化することができないように裁判所に命じられ、損害賠償金を支払うことができるかもしれない。許容可能な条項によると、私たちは許可証を得ることができないかもしれない(あれば)。また、訴訟期間中、特許保有者は、予備禁止または他の公平なbr救済を受けることができ、承認された製品の製造、使用、または販売を禁止し、事件の裁判を待つことができ、これは数年以内に発生しない可能性がある。
製薬業界には、特許や他の知的財産権に関する多くの訴訟が一般的に存在する。もし第三者が私たちまたは私たちの協力者がその知的財産権を侵害していると主張すれば、私たちはいくつかの問題に直面するかもしれないが、これらに限定されない
| ● | 権利侵害と他の知的財産権クレームは、事件の状況にかかわらず、訴訟を提起するのは高価で時間がかかる可能性があり、そして私たちの管理層の核心業務に対する注意力を移転する可能性がある | |
| ● | 第三者が私たちに訴訟を起こした当事者は私たちよりも多くの資源を持っているかもしれません | |
| ● | 権利侵害の巨額の損害賠償は、もし裁判所が紛争製品の侵害または第三者の権利違反と判断した場合、私たちは支払わなければならないかもしれない。もし裁判所が侵害が故意であると認定した場合、私たちは3倍の損害賠償金と特許所有者の弁護士費の支払いを命じられる可能性がある | |
| ● | 裁判所は、第三者がその権利を私たちに許可しない限り、私たちの製品または将来承認された任意の候補製品を販売することを禁止しています | |
| ● | 第三者から許可を得る場合、私たちは私たちの知的財産権に大量の使用料、費用、または交差許可を付与する必要があるかもしれない;および | |
| ● | 私たちの任意の製品および候補製品を再設計して、それらが侵害されないようにすることは不可能かもしれません。または大量のお金の支出と時間が必要かもしれません。 |
| 48 |
我々の薬物開発戦略は505(B)(2)規制承認経路に大きく依存しており,承認された薬物の第三者特許を侵害していないことを証明することが求められている。このような認証は通常、第三者が知的財産権侵害に対してクレームを出し、弁護費用が高く時間がかかり、いかなる訴訟における不利な結果も、私たちの開発と商業化努力を阻止したり、延期したりする可能性があり、これは私たちの業務を損なうことになる。
私たちのビジネス成功は、既存のbrによって承認された医薬製品に対する第三者の特許および独自の権利の侵害を回避することに大きく依存する。505(B)(2)規制承認経路を使用して私たちの製品および候補製品を承認するので、私たちは第三者によるこれらの承認された医薬製品に関する研究の全部または一部に依存します。したがって、FDAに私たちの候補製品の承認を申請する際には、(1)FDAのオレンジブックには、我々のNDAに関する特許情報が記載されていないこと、(2)オレンジブックに記載されている特許が期限切れになっていること、(3)記載されたbr}特許が有効期限が切れていないが、特定の日に満了し、特許満了後に承認を求めること、または(4)記載された特許brが無効であるか、または提案された医薬製品の製造、使用、または販売によって侵害されないことをFDAに証明することが要求される。特許 が無効または侵害されていないこと、または第4項の認証がFDAに証明された場合も、505(B)(2)セキュリティプロトコルがFDAによって届出を受けた後、特許 所有者に第4項の認証の通知を送信しなければならない。そして、第三者は私たちが通知で決定された特許を侵害したことを告発するために私たちを提訴することができる。通知を受けてから45日以内に特許侵害訴訟を提起することは、30ヶ月以内または特許が満了した日、訴訟が和解に達し、または裁判所が侵害訴訟において私たちに有利な判決を下すまで、FDAが私たちの秘密保護協定を承認することを自動的に阻止するだろう。第三者が規定された45日間の期間内に特許侵害訴訟を提起しなかった場合、私たちの秘密保護協定は30ヶ月の猶予の制限を受けないだろう。しかしながら、第三者が45日間の期間内に訴訟を提起しなくても、30ヶ月の猶予を援用しても、FDAの承認後に私たちの製品を販売する権利に挑戦する可能性があり、したがって、45日の期限が過ぎても、いくつかの侵害訴訟のリスクがある。
我々 は,ビジネス秘密や他の固有情報の漏洩を十分に防ぐことができない可能性がある.
我々 は,特に特許保護 が不適切であると考えられたり,入手できない場合には,ビジネス秘密によって我々のノウハウや技術進歩を保護している.しかし、商業秘密は保護することが難しい。我々は,我々のbr従業員,コンサルタント,外部科学協力者,協賛研究者,他のコンサルタントと締結された秘密協定に部分的に依存して,我々のビジネス秘密や他のbr}固有の情報を保護する.これらのプロトコルは、機密情報の漏洩を効果的に阻止できない可能性があり、機密情報を不正に開示することなく適切な救済措置を提供できない可能性がある。しかも、他の人たちは私たちの取引秘密と固有の情報を独立して発見するかもしれない。私たちの固有権の範囲を実行して決定することは、高価で時間のかかる訴訟を必要とする可能性がある。ビジネス秘密保護を取得または維持できない場合、競争相手は、当社の製品と競合する製品を開発するために、当社の独自の情報 を利用したり、当社の競合ビジネスの地位に追加的な重大な悪影響を与える可能性があります。
私たちは第三者からクレームを受ける可能性があり、これらの第三者は私たちの従業員や私たちが彼らの知的財産権を流用したと主張したり、私たち自身の知的財産権を持っていることを要求したりします。
私たちの一部の従業員は以前、実際または潜在的な競争相手を含む他の会社に雇われたことがある。私たちはまた、他の組織に同時に雇われたり、他のエンティティにサービスを提供するコンサルタントやコンサルタントを雇うことができます。私たちは、私たちの従業員、コンサルタント、およびコンサルタントが、私たちのために働いているときに他人の固有情報またはノウハウを使用しないことを保証するために努力しているが、私たちは、商業機密または他の固有情報を含む、または他方との合意に違反したり、他方に対する法的義務に違反したりしているとして、私たちまたは私たちの従業員、コンサルタントまたはコンサルタントが、商業機密または他の固有情報を含む任意のそのような当事者の元雇用主の知的財産権を使用または開示していることが疑われる可能性がある。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。
また、私たちは通常、知的財産権開発に参加する可能性のある従業員、コンサルタント、コンサルタント、請負業者に、このような知的財産権を譲渡する協定に署名することを要求していますが、私たちは、私たちが自分の知的財産権を実際に開発している当事者とこのような合意を実行することに成功できないかもしれません。私たちと彼らの譲渡協定は自動的に実行されないかもしれないし、違反される可能性があります。私たちは第三者にクレームをつけさせられたり、彼らは私たちが私たちの知的財産権の所有権とみなされていることを確認するために、私たちが提起したクレームに抗弁する可能性があります。同様に、私たちは、従業員、コンサルタント、またはコンサルタントが私たちのために実行してくれる仕事が、第三者(例えば、雇用主または元雇用主)に対するその人の義務と衝突する可能性があり、したがって、第三者は、私たちが実行する仕事のために生成された知的財産権に対して所有権利益を持っている。このようなクレームに対抗するためにbr訴訟が必要かもしれない。
もし私たちがこのような任意のクレームを起訴または弁護することができなかった場合、私たちは金銭損害賠償を支払うことに加えて、貴重な知的財産権または人員を失う可能性がある。このようなクレームの起訴や抗弁に成功しても、訴訟は巨額のコストを招き、経営陣の注意を分散させる可能性がある。
505(B)(2)に従ってCTX−1301、CTX−1302および/またはCTX−2103および他の候補製品の承認を求める決定は、このような候補製品に対するFDAの承認を延期する可能性がある特許侵害訴訟を提起するリスクを増加させる可能性がある。
505(B)(2)法規経路に従って提出された任意のセキュリティプロトコルについて、特許が承認されたと主張する薬剤が候補製品に含まれ、505(B)(2)セキュリティ協定に参照されている場合、特許所有者FDAオレンジブックに承認された薬剤のために掲載された任意の特許 が無効であり、強制的に実行されないか、または私たちの薬剤を製造、使用または販売することによって侵害されることがないことをFDAに証明し、通知しなければならない。私たちの認証通知を受けてから45日以内に私たちに特許侵害訴訟を提起した場合、FDAは、最初の30ヶ月、特許満了、訴訟和解、または侵害事件において私たちに有利な裁判所判決まで、または裁判所が命じたより短いまたはより長い期間まで、505(B)(2)秘密協定の承認を自動的に阻止する。このような訴訟は一般的に特許所有者によって提起される。したがって、候補製品の開発に多くの時間と費用を投入する可能性がありますが、候補製品を商業化する前に、私たちは重大な遅延と特許訴訟を受ける可能性があります。私たちはいかなる特許侵害請求を正当化することにも成功できないかもしれません。私たちが権利侵害がないことが発見された場合、あるいは原告の特許主張が無効または強制執行できないことが発見されても、どのような侵害クレームの弁護も高価で時間がかかり、私たちの製品や私たちの他の候補製品の発表を延期し、管理職の正常な責任を分散させるだろう。
| 49 |
今回の発行、証券市場と我々の証券の所有権に関するリスク
もし私たちがナスダックの持続的な上場要求を再遵守できなかった場合、私たちの普通株および/または株式承認証は取得される可能性があり、私たちの普通株および/または株式承認証の価格、および私たちの資本市場に入る能力はマイナスの影響を受ける可能性がある。
私たちの普通株式と権利証は現在ナスダックで看板取引されている。2023年5月16日、我々は“ナスダック株式市場上場規則”第5550(B)(1)条(“最低株主権益規則”)の継続上場に関する最低株主権益要求に適合しないことを指摘したスタッフから通知を受けた。私たちは2023年6月30日にナスダックにコンプライアンス計画を提出した。2023年7月28日、ナスダックは、これまでナスダックに提出されていたコンプライアンス計画に含まれるいくつかのマイルストーンを達成したことを条件に、最低株主権益ルールを再遵守するために2023年11月13日まで延長することを許可し、追加資本を調達する計画を達成したことを通知した。吾らは2023年11月14日にナスダックから書簡を受け取り、吾らが最低株主権益ルール を遵守し続けていないことに基づき、スタッフは吾らの証券をナスダックから退市することを決定し、惟須待吾らはナスダック聴取チーム(以下“委員会”と呼ぶ)に尋問を要求した。
私たちは専門家グループとナスダックが私たちの要請を承認する前に公聴会を行うことを直ちに要求します。2024年2月に予定されている専門家チームのヒアリングでは、ナスダックに上場し続けるすべての適用されるbr要求に対するコンプライアンスと、最低株主権益ルールと独立 取締役ルールの遵守を含む長期コンプライアンスの能力を再遵守する計画を提出します(以下のように定義します)。従業員の除名行動は延期され、聴聞とグループが聴聞後に承認する任意の追加の延長期間の満了を待つ。私たちは引き続き最終ステップを取って、 が最低株主権益規則、独立取締役規則、その他のナスダック上場要求を遵守したことを証明するつもりです;しかし、陪審員が私たちの上場継続の要求を承認することを保証することはできません。また、陪審員が承認する可能性のあるいかなる延期の期間内に が最低株主権益規則、独立取締役規則と他のナスダック上場要求を遵守できるかを保証することもできません。上場を続けるためには、最低株主権益規則 を長期的に遵守する能力があることを証明する必要がある。我々は、公聴会の前に財務状況を改善する方法が限られており、最低株主権益規則に適合していることを証明することができる。私たちはあまり優遇されない条項でbr資本をさらに調達しようと試みるかもしれません。これはあなたと私たちの既存の株主の権益をさらに希釈するかもしれません。私たちはより多くの資本の調達に成功したり、他の方法で最低株主権益規則を満たすことができないかもしれない。
2023年7月28日、ナスダックから通知を受け、ナスダックでの上場継続が1株1.00ドルの最低購入価格を維持しなければならないという要求に合致しないことを指摘した。ナスダック上場規則第5810(C)(3)(A)条によると、我々は180暦の適合期間を提供し、通知が出された日から、あるいは2024年1月24日まで、最低成約見積要求を再遵守する。2023年11月30日、私たちは普通株を逆分割し、2023年12月15日、私たちはナスダックから通知を受けて、私たちは最低終値要求を再遵守しました。最低終値要求や他のナスダック上場要求を遵守し続ける保証はありません。
2023年12月26日、私たちは従業員から手紙を受け取り、3人の取締役会メンバーがそれぞれ2023年12月12日及び2023年12月13日に辞任したことに基づいて、私たちは取締役上場規則第5605条(取締役上場規則“独立取締役規則”)に掲載されている独立ナスダック上場委員会、監査委員会、報酬委員会及びナスダックの取締役指名に対する独立監督規定を守らなくなった。私たちの取締役会は現在2人の取締役で構成されていますが、この2人の取締役は独立していません。
我々 は潜在的な新しい取締役を探しており,グループヒアリングの前に独立取締役ルール遵守への進展 を示す予定である.私たちが“役員独立規則”を再遵守する保証はありません。
我々 は、最低株主権益ルール と独立取締役ルール を含むナスダックの持続的な上場要求を満たさなければならず、そうでなければ、私たちの業務に重大な悪影響を与える可能性のあるリスクが退市する。もし私たちの普通株式と引受権証がナスダックから撤退すれば、私たちの普通株と引受権証の流動性を大幅に減少させ、ナスダックに関連する市場効率の喪失と連邦証券法の優先購入権を失ったため、私たちの普通株と引受権証の価格がそれに応じて大幅に低下する可能性がある。また、退市は、私たちが受け入れられる条項や全く受け入れられない条項で融資源を代替することで資金を調達する能力を損なう可能性があり、投資家、サプライヤー、顧客、従業員の潜在的な自信を失い、業務発展の機会を減少させる可能性がある。もし私たちの普通株式と引受権証が取得されたら、私たちの普通株と引受権証はもっと売買しにくいかもしれないし、正確な見積もりを得ることができないかもしれません。私たちの普通株と引受権証の価格は大幅に下落する可能性があります。退市は、もしあれば、受け入れ可能な条件で資金を調達する能力を弱める可能性もある。
今回の発行は最善を尽くした上で行われており,我々が販売する証券はここで発行されたすべての証券よりも少ない可能性があり,今回の発行から得られる純収益は大幅に減少する可能性があり,限られた運営資金のみを提供することになる
今回の発売は最大の努力で行われており、我々が販売している証券はここで発売されたすべての証券よりも少ない可能性があり、今回の発売から得られる純収益も大幅に減少する可能性がある。私たちの現在の業務計画によると、今回発行された純収益に私たちの手元の現金を加えて、2024年第2四半期末までの資本需要を満たすと信じています。今回の発行後、私たちはより多くの資金を集めて私たちの運営を支援し、私たちが計画している開発と商業化活動を支援していく必要があります。
経営陣は今回発行された純収益の使用に対して広範な裁量権を持っているため,これらの純収益をどのように使用するかに同意しない可能性があるため,このような 収益は適用できない可能性がある.
私たちの経営陣は今回の発行で得られた資金の使用に対してかなりの裁量権を持つだろう。我々は現在、今回発行した純収益 をCTX-1301の持続的な研究開発と商業化活動に応用し、さらに研究開発に投資することを含む運営資本、資本支出、一般企業用途に使用する予定である。しかし、私たちの経営陣 は、今回発行された純収益を運用する上で幅広い裁量権を持ち、必ずしも が私たちの運営業績を改善したり、私たちの証券価値を向上させる方法、あるいはあなたが同意しない方式に収益を使うことができます。あなたはあなたの投資決定の一部として、収益が適切に使用されているかどうかを評価する機会がないように、私たちの経営陣のこれらの用途の判断に依存します。もし私たちの経営陣がこれらの資金を有効に運用できなければ、不利なリターンと私たちの見通しへの不確実性を招く可能性があり、これらは私たちの証券の価格下落を招く可能性がある。
今回の発行で証券を購入すれば、あなたの投資はすぐに希釈されます。
あなたのbrは今回の発売で直ちに重大な希釈を受けることになります。普通株および付属引受権証の1株当たり公開発売価格および1株当たりの事前資本権証および付属引受権証の公開発売価格は、今回の発売後の普通株の調整後の1株当たり有形帳簿純値より大幅に高くなる。したがって、今回の発行で証券を購入された場合、今回の発行後に支払われる普通株式1株当たり価格は、私たちの予想有形帳簿純価値 を大きく上回ることになります。普通株1株2.00ドルの公開発行価格と付属のbr引受権証、および2023年9月30日までの予定有形帳簿純価値によると、直ちに1株0.15ドルの希釈を経験することになります。これは、今回の発行発効後に調整された1株当たりの有形帳簿純値と公開発行価格との差額を表しています。
| 50 |
私たちが提供した事前融資権証や株式承認証は公開市場を持っていません。
事前融資権証や株式承認証にはまだ既定の公開取引市場がなく、このような市場は発展しないと予想される。また、私たちは、どの国の証券取引所または他の国でも認められている取引システムに事前融資権証または権利証を上場することを申請するつもりはありません。活発な取引市場がなければ、事前融資権証と引受権証の流動性が制限される。
今回の発行で購入した資本金権証と引受権証の所持者は、その予資権証又は株式承認証を行使して我々の普通株を買収するまでは、普通株株主の権利を有していない。
事前資本権証及び引受権証の所有者が行使時に我々普通株の株式を取得する前に、当該等所有者は、事前融資権証及び引受権証に関連する普通株株式の権利を有しないであろう。前払い資金株式承認証及び引受権証を行使した後、所持者は普通株主の権利を行使する権利のみを有し、関連事項の記録日 は行使日の後である。
これらの株式承認証は投機的である.
株式承認証は、投票権または配当金を受け取る権利のような所有者にいかなる普通株式所有権権も付与されていない。 は、限られた時間内に普通株式を固定価格で取得する権利を表すだけである。また,今回の発行後,株式承認証の時価(あれば)は不確実であり,引受権証の時価がその推定発行価格以上になる保証もない.株式承認証はいかなる市場や取引所に上場したり、オファー取引をしたりすることはありません。brは私たちの普通株の市場価格が永遠に引受権証の使用価格に等しいかそれを超えることを保証することができません。そのため、株式承認証が満期になる可能性がある時には価値がありません。
証券購入プロトコルにより今回の発行で我々の証券を購入した購入者 は,購入者が証券購入プロトコルなしにその証券を購入する権利を持つ可能性がある.
連邦証券や州法により今回発売されたすべての購入者に提供される権利や救済措置のほか、証券購入契約を締結した購入者は、違約請求をすることができる。違約に対して請求する能力は、(I)株式をタイムリーに交付することと、(Ii)成約後1年以内に変動金利融資を行わないことに同意するが、いくつかの例外的な場合を除くことと、(Iii)成約後90日以内にいかなる融資も行わないことに同意することと、 および(Iv)違約賠償とを含む、証券購入協定において彼らに提供される独自の契約を実行する手段をこれらの投資家に提供する。
この は最善を尽くした製品であり,証券の売却を要求する最低額はなく,我々の業務計画(我々の最近の業務計画を含む)に必要な資金を調達しない可能性がある.
配給エージェントは,今回発行中の証券を購入する要約を求めるために合理的な最善を尽くすことに同意した.配給代理は、私たちの手から任意の証券を購入する義務もなく、特定の数量や金額の証券の購入や販売を手配する義務もありません。今回の発売完了には証券を売却しなければならない最低数の要求はありません。 今回の発売完了には最低発売金額の要求がないため、私たちの実際の発売金額、配給代理費、収益は現在確定できず、上記の最高金額を大幅に下回る可能性があります。 ここで発売されたすべての証券よりも少ないかもしれませんが、これは私たちが受け取った収益額を著しく減少させる可能性があります。もし私たちが販売している証券の数が私たちのbrの持続的な運営(私たちの最近の持続的な運営を含む)をサポートするのに十分でなければ、今回発行した投資家は返金を受けないだろう。したがって、私たちは、私たちが考えている運営に必要な資本額 を短期的に調達することはできないかもしれませんし、追加の資金を集める必要があるかもしれません。これらの資金は、私たちが受け入れられる条項では得られないかもしれません。
発行には最低限の要求がないため、当社が販売している証券数が本入札明細書に概説した業務目標を達成するのに不十分であれば、今回発行した投資家は返金を受けることはありません。
私たち は最低発売金額を指定していませんし、今回の発売に関するホストアカウントを設立したりしていません。信託口座や最低発売金額がないため、投資家は私たちの会社に投資しているかもしれませんが、今回の発売に興味がないため、私たちの目標を実現することはできません。また,brの運営には信託口座もなく,最低投資額もないため,これらの資金を用いて我々の業務計画を効率的に実施できるかどうかにもかかわらず,我々が証券を売却して得られたいかなる収益も即座に使用できるようになる.投資家資金はどの場合も返金されません。上場期間中も上場後も返金されません。
| 51 |
私たちの普通株式や株式承認証の活発な取引市場は持続できないかもしれない。
私たちの普通株式や株式承認証の活発な取引市場は持続できないかもしれない。私たちの普通株式または株式承認証が活発な市場を欠いていることは、彼らが売却を希望する時間、または彼らが合理的な価格で普通株または株式証を売却する能力を弱める可能性があり、その普通株または株式承認証の公平な市場価値を低下させる可能性があり、証券売却を通じて資本を調達して運営に資金を提供し続ける能力を弱める可能性があり、証券を対価格として追加の知的財産資産を得る能力を弱める可能性がある。
私たちの証券の価格は変動するかもしれません。これは私たちを証券集団訴訟を受け、私たちの株主が大きな損失を受ける可能性があります。
私たちの普通株と株式認定証の市場価格は比較的に大きく変動する可能性があり、以下の要素の影響を受けて大幅に変動する
| ● | 当社の四半期または年度の経営業績の実際または予想変動 | |
| ● | わが社の業績の速度や競争相手に対する成長率の実際の変化や予想変化 | |
| ● | 投資界や私たちが大衆に提供している財務的推定および予測を達成できなかったか、またはそれを超えることができなかった | |
| ● | 証券アナリストの最新または最新の研究報告書または報告書を発表する | |
| ● | 普通株式または株式承認証の取引量レベルが一致しない株価と出来高変動に起因することができる | |
| ● | 重要な管理職や他の人員の増加や退職 | |
| ● | 特許、訴訟事項、および私たちの技術のための特許保護を得る能力を含む紛争brまたは特許権に関連する他の発展; | |
| ● | 追加債務または株式融資努力を行うことを宣言するか、または追加的な債務または株式融資努力を期待する |
| ● | 私たち、私たちの内部人または他の株主は私たちの普通株式を売却したり、株式証明書を発行したりします | |
| ● | 米国や他の地方の一般経済、市場または政治状況(新冠肺炎の大流行による状況を含むがこれらに限定されない)。 |
特に,我々のような臨床段階では会社の市場価格は非常に不安定であり,原因としては に限らないが制限されていない
| ● | 私たちの候補製品の臨床試験における任意の遅延または失敗、またはFDAおよび他の規制機関の承認を得た | |
| ● | 製品知的財産権における事態の発展や紛争; | |
| ● | 私たちの競争相手の技術革新 | |
| ● | 投資家は我々に相当する会社の推定値が変動していると考えている | |
| ● | 私たちまたは私たちの競争相手は、重大な契約、買収、戦略的パートナーシップ、合弁企業、資本約束、新技術または特許を発表します | |
| ● | 重要な取引を完了できなかったか、またはサプライヤーと協力して私たちの製品を生産することができなかった; | |
| ● | 医療治療の価格を制限する立法を提案する。 |
| 52 |
これらの および他の市場および業界要素は、私たちの普通株式と株式承認証の市場価格と需要を大幅に変動させる可能性があり、私たちの実際の経営業績にかかわらず、投資家がその普通株式または株式承認証の株式を売却することを制限または阻止する可能性があり、そうでなければ、私たちの普通株と株式証明書の流動性に負の影響を与える可能性がある。また、株式市場、特にナスダックと新興成長型会社は、極端な価格や出来高変動を経験しており、 はこれらの会社の経営業績に関係なく、あるいは比例しないことが多い。過去に、証券の市場価格が変動した場合、その証券の保有者は、その証券を発行した会社に対して証券集団訴訟を提起する。もし私たちのいかなる株主が私たちを提訴すれば、私たちは巨額の訴訟弁護費用を生む可能性がある。このような訴訟はまた私たちの経営陣の時間と注意力を移す可能性がある。
私たちは“新興成長型会社”であり、“新興成長型会社”に適用される開示要求の低減を利用することができ、私たちの証券の投資家への吸引力を低下させる可能性がある。
我々brは,JOBS法案で定義されている“新興成長型企業”であり,我々が“新興成長型企業”であり続ける限り,他の上場企業に適用されるが“新興成長型企業”には適用されない様々な報告要求のいくつかの免除を利用したいと考えており,これらに限定されないが,サバンズ·オクスリ法案第404条の監査役認証要求に遵守することは要求されず,定期報告や依頼書における役員報酬に関する開示義務を削減している。役員報酬に対する拘束力のない諮問投票の要求を免除し、株主がこれまで承認されていなかった金パラシュート支払いの要求を免除する。我々は“新興成長型企業”とすることができ、最長5年に達するか、または(I)我々の年間総収入が12.35億ドルを超える最初の会計年度の最終日まで、(Ii)取引法第12 b-2条で定義された“大型加速申告会社”となった日、または(Iii)前3年間に10億ドルを超える転換不能債券を発行した日である。
私たちは、上記の報告書を利用して、私たちが“新興成長型会社”ではなくなるまで免除するつもりです。 雇用法案によると、“新興成長型会社”は、これらの基準が非上場企業に適用されるまで、新たな会計基準の採用や改正された会計基準の採用を延期することもできます。私たちは、この免除を利用して新しいまたは改正された会計基準 を遵守することを撤回できないので、他の“新興成長型企業”ではない上場企業と同じ新しいまたは改正された会計基準の制約を受けることはない
私たちがこれらの免除に依存することを選択すれば、投資家は私たちの証券吸引力の低下を発見するかどうかは予測できません。もし一部の投資家が私たちの証券が将来開示する選択を減らすことでそれほど魅力的ではないことを発見すれば、私たちの証券取引市場はそれほど活発ではなくなり、私たちの普通株と引受権証の価格はもっと変動するかもしれない。
上場企業として、財務報告書の適切かつ効果的な統制を策定し、維持する義務がある。もし私たちが未来に財務報告に対する適切かつ有効な内部統制を維持できなければ、私たちが正確かつ適時に財務諸表を作成する能力は損害を受ける可能性があり、これは私たちの経営業績、投資家の私たちに対する見方を損害し、それによって私たちの証券の価値を損なう可能性がある。
サバンズ-オキシリー法404節は、財務報告の内部統制の有効性を評価し、決定し、財務報告の内部統制の管理報告書を提供することを要求する。JOBS法案で定義されている“新興成長型会社”の地位を失い、加速申告のハードルに達した場合、独立公認会計士事務所は、財務報告の内部統制に対する有効性を証明することを要求される。しかし,我々がまだ新興成長型企業である限り,これらの監査人認証要求のbrに制限されない新興成長型企業が得られる免除を利用しようとしている。管理管理層が財務報告の内部統制を評価するために達成しなければならない標準的なルールは非常に複雑で、大量の文書、テスト、可能な救済措置が必要だ。“取引法”における報告会社としての要求に適合するためには、情報技術を含む我々のシステムをアップグレードする必要があり、追加の財務および管理制御、報告システムおよびプログラムを実施し、追加の会計および財務者を雇用する必要がある。もし私たちまたは私たちの監査人が財務報告の内部統制に有効であると判断できなければ、投資家は私たちの財務報告に自信を失う可能性があり、私たちの普通株式や株式証の取引価格は低下する可能性がある。
| 53 |
私たち はあなたに保証することはできません。将来私たちの財務報告に対する内部統制に重大な欠陥や重大な欠陥 が存在しないことを保証します。財務報告を内部統制できなかったいかなる行為も、財務状況、運営結果、またはキャッシュフローを正確に報告する能力を深刻に抑制する可能性がある。もし私たちが財務報告の内部統制に有効であると結論できない場合、あるいは私たちの独立公認会計士事務所が私たちの財務報告の内部統制に重大な弱点や重大な欠陥があると判断した場合、事務所がその404条の審査を開始すると、私たちの財務報告の正確性と完全性に対する投資家の自信を失う可能性があり、私たちの普通株式または株式証の市場価格は下落する可能性があり、私たちはナスダック、米国証券取引委員会、または他の規制機関の制裁や調査を受ける可能性がある。財務報告の内部統制のいかなる重大な欠陥や重大な欠陥を是正できなかったか、あるいは上場企業に必要な他の有効な制御システム を実施或いは維持できなかったことも、私たちの将来の資本市場への参入を制限する可能性がある。
我々 はこれにより大幅にコストを増加させ,大量の管理時間を上場企業として運営する.
上場企業として、私たちは巨額の法律、会計、その他の費用を発生させるだろう。例えば、私たちは、改正された1934年の証券取引法の報告要件を遵守し、有効な開示と財務制御の確立と維持、コーポレート·ガバナンスのやり方の変更、および私たちの業務および経営業績に関する年度、四半期、現在の報告書の提出を含む、Br}サバンズ-オックススリー法案およびドッド-フランクウォール街改革および消費者保護法案の適用要件の遵守を要求される。これらの要件brは、私たちの法律と財務コンプライアンスコストを増加させ、いくつかの活動をより時間とコストを高くするだろう。また,我々のbr管理層や他の人員は,これらの上場企業要求に多くの時間を投入するために,運営や他の業務事務から注意をそらす必要がある.私たちはまた、適切な上場会社の経験と技術会計知識を持つ会計と財務者をより多く招聘し、内部監査機能を構築する必要があるかもしれない。また、上場企業として運営することで取締役や上級管理職責任保険を獲得するコストが高くなり、低減された保証範囲を受け入れることや、より高い保証コストが発生することが要求される可能性があると予想されます。これはまた、合格者を私たちの取締役会、私たちの取締役会委員会、または役員に参加することを引き付け、維持することを難しくするかもしれない。また,JOBS ACTで定義されている“新興成長型会社”の資格に適合しなくなった後,加速申請者や大型加速申請者と考えられるより厳しい報告要件を遵守するための追加的な管理時間やコストが生じることが予想され,“サバンズ-オキシリー法案”404節の監査師認証要件の遵守を含む。我々は,このような要求に応じたシステムと文書を処理するプロセスのコンパイルを開始したばかりである.私たちは私たちの評価、テスト、そして必要な修復をタイムリーに終わらせることができないかもしれない。この点で、私たちは現在内部監査機能を持っておらず、適切な上場企業の経験と技術会計知識を持つ会計や財務者を雇用または契約して増加させる必要があります。
私たち は、上場企業になることによって生じる可能性のある追加コストやそのようなコストの時間を予測したり見積もることができません。
証券や業界アナリストが我々の業務に関する研究報告書を発表したり、不正確または不利な研究報告を発表しなければ、我々の株価や取引量は低下する可能性がある。
私たちの普通株式と権利証の取引市場は、証券や業界アナリストが発表した私たちまたは私たちの業務に関する研究と報告にある程度依存するだろう。私たちは現在証券と産業アナリストに対する研究報告書が限られている。もし私たちが証券や業界アナリストの十分な報道を維持できなければ、私たちの株の取引価格はマイナスの影響を受けるかもしれない。私たちの1人以上のアナリストを追跡して私たちの株式格付けを引き下げたり、私たちの業務に関する不正確または不利な研究報告を発表したりすれば、私たちの株価は下落するかもしれない。これらのアナリストのうちの1人以上が私たちへの報告を停止したり、私たちに関する報告書を定期的に発表できなかったりすれば、私たちの株に対する需要が減少する可能性があり、これは私たちの株価や取引量を低下させる可能性がある。
| 54 |
将来、私たちの普通株、株式承認証、あるいは普通株に変換できる証券を売却することは、私たちの株価を押し下げる可能性がある。
私たちの普通株または株式承認証の価格は、私たちの普通株または株式承認証の株式または株式承認証の大量売却によって下落する可能性があり、またはこれらの売却が発生する可能性があると考えられる。これらの売却、あるいはこれらの売却が発生する可能性があり、将来的に私たちが適切だと思う価格で株式証券を販売することをより困難にする可能性がある。
さらに、将来的には、融資、買収、訴訟和解、従業員手配、または他の態様のために、追加の普通株、引受権証、または普通株に変換可能な他の株式または債務証券を発行する可能性がある。このような発行は、私たちの既存株主の持分を大幅に希釈し、私たちの普通株や株式承認証の価格を下落させる可能性があります。 WFIAは、私たちの普通株の1,028,955株を購入するために、事前出資の引受証を持っています。このような事前出資の引受権証を行使すると、当社での所有権権益が希釈されます。
逆買収わが社の証明書や定款に含まれる条項、およびデラウェア州法律の条項は、買収の試みを破壊する可能性がある。
わが社の登録証明書、定款、デラウェア州法律に含まれる条項を改正し、再記述することは、brをより困難にし、取締役会が望ましくないと考えている買収を延期または阻止する可能性がある。当社のコーポレートガバナンス文書にはbr条項が含まれています
| ● | 私たちの取締役会を3つに分類します | |
| ● | Br“空白小切手”の優先株を許可して、私たちの取締役会によって発行することができて、株主の承認を必要とせず、そして私たちの普通株より優れた他の権利を含む可能性があります | |
| ● | 役員と上級管理者の責任を制限し、賠償を提供する | |
| ● | 私たちの株主が特別会議の前に業務を招集して展開する能力を制限する | |
| ● | 我々の株主会議で株主の業務提案を事前に通知し、私たちの取締役会の候補者を指名することを要求します | |
| ● | 取締役会と株主会議の開催と手配手順を制御する; | |
| ● | 私たちの取締役会に明確な権力を提供し、以前に手配された年次会議を延期し、以前に手配された特別会議をキャンセルすることができる。 |
これらの 条項は、単独または一緒に、敵意の買収、制御権の変更、または管理層の変動を延期または阻止する可能性がある。
デラウェア州会社として、私たちはまた、デラウェア州汎用会社の法律の第203条を含むデラウェア州の法律の条項を遵守しなければなりません。この条項は、私たちが発行した普通株式の15%を超える株主を保有することを禁止しています。私たちのほとんどの発行された普通株式所有者の承認なしにいくつかの業務合併を行うことを禁止しています。
私たちが改訂し、再記述した会社登録証明書、定款或いはデラウェア州法律の遅延或いは抑止権変更効果を有するいかなる条項 はすべて私たちの株主がその普通株式或いは株式承認証からプレミアムを獲得する機会を制限する可能性があり、また一部の投資家が私たちの普通株と引受権証のために支払う価格に影響を与える可能性がある。
| 55 |
私たちは予測可能な未来に私たちの普通株に現金配当金を支払わないと予想している。
私たちは予測可能な未来に私たちの普通株に現金配当金を支払わないと予想している。私たちは現在、将来のいかなる収益も維持して、私たちの業務運営と拡張に資金を提供するつもりで、私たちは予測可能な未来にいかなる配当金も発表したり支払うことはないと予想しています。したがって、株主は価格上昇後に彼らの普通株と引受権証に依存しなければなりません。これは決して起こらないかもしれません。これは彼らの投資の将来のいかなる収益を実現する唯一の方法ですから。私たちの普通株や株式承認証の株が値上がりし、株主がその株を購入したり、株式証を承認したりする価格を維持することは保証されません。
私たちの役員、役員、主要株主は私たちに対してかなりの支配権を持っていて、会社の制御権の変更を延期または阻止する可能性があります。
2024年1月25日現在、私たちの役員、現職幹部、5%を超える普通株保有者は、彼らの関連会社とともに、私たちが発行した普通株の約27%を実益しています。したがって、これらの株主は一緒に行動し、我々株主に承認された事項を承認した結果、任意の合併、合併、または私たちの所有またはほぼすべての資産を売却することを含む能力を制御することができる。また、これらの株主が共同で行動することで、わが社の管理や事務を制御することができるだろう。したがって、このような所有権集中は、以下のように私たちの普通株式と引受権証の市場価格を損害する可能性がある
| ● | 私たちの制御権の変更を延期したり、延期したり、阻止したりする | |
| ● | 私たちの合併、合併、接収、または他の業務統合に関する阻害;または | |
| ● | 潜在的買収側が買収要約を提出することを阻止したり、他の方法で私たちを制御しようとしたりする。 |
当社の役員、取締役、および5%を超える普通株を保有する株主およびその付属会社の発行済み株式の所有権についてのより多くの情報は、以下のbr“いくつかの利益所有者および管理層の保証所有権”を参照されたい。
私たちのbr改訂および再記載された会社登録証明書は、デラウェア州衡平裁判所が私たちの株主のために開始する可能性のある特定のタイプの訴訟および訴訟の唯一かつ排他的なbrフォーラムを指定し、これは、私たちまたは私たちの取締役、上級管理者、または他の従業員との紛争において有利な司法フォーラムを得る能力を制限することができる。
私たちが書面で代替裁判所を選択することに同意しなければ、デラウェア州衡平裁判所は、法律によって許容される最大範囲で、以下の項目の唯一かつ独占的な裁判所になることを要求し、再記載された会社登録証明書の要求を再記載する
| ● | 私たちが提起した任意の派生訴訟や法的手続きを代表する |
| ● | 取締役、私たちの役員、または他の従業員が、会社または私たちの株主に負う任意の受託責任に違反してクレームを提起するいかなる行為も |
| ● | 吾等又は吾等のいずれかの役員又は吾等の高級社員に対して提出された任意の 訴訟、又は吾等又は吾等の任意の役員又は高級社員に対して当社、吾等の会社登録証明書又は付例のいずれかの条文の解釈又は適用について提出された申立書;又は | |
| ● | 内務原則に支配されているクレームを主張するいかなる訴訟でも |
しかし条件は、デラウェア州衡平裁判所が管轄権の不足で上述のいずれの訴訟を却下した場合にのみ、デラウェア州の別の州裁判所にこのような訴訟を提起することができるということである。
| 56 |
排他的裁判所条項は法的に許容される範囲に限定され、取引法または他の任意の連邦証券法に基づいて提起された独占的な連邦管轄権を規定するクレームには適用されない。
また、証券法第22条は、連邦裁判所と州裁判所は、このようなすべての“証券法”訴訟に対して同時に管轄権を有していると規定している。したがって、州裁判所と連邦裁判所はこのようなクレームを受理する管轄権を持っている。複数の司法管轄区で訴訟を提起せざるを得ないことや異なる裁判所が不一致や逆の裁決を下す脅威などの考慮要因を避けるために,我々が改訂·再記述した会社登録証明書 規定は,アメリカ合衆国連邦地域裁判所が証券法に基づいて提出した任意の訴因を解決する排他的なフォーラムとなる.デラウェア州裁判所は、このようなフォーラム条項の選択が表面的な効力を有することを決定したが、株主は、米国連邦地域裁判所以外の場所で証券法に基づいて、私たち、私たちの役員、上級管理者、または他の従業員にこのようなクレームを提起することを求めることができる。この場合、私たちは、改正されたbrと再記述された会社登録証明書の独占フォーラム条項の有効性と実行可能性を強く主張する予定である。
この条項は、適用される訴訟タイプにおいてより一致したデラウェア州法律の適用を提供するため、この条項が私たちに有利であると信じているが、この条項は、私たちまたは私たちの役員、上級管理者、または他の従業員とのトラブルの請求に有利であると考える株主の司法フォーラムでのクレームを制限または阻害する可能性があり、これは、私たちおよび私たちの取締役、上級管理職、および他の従業員に対するこのような訴訟を阻止し、投資家からのクレームのコスト増加を招く可能性がある。代替的に、裁判所 が、私たちが修正および再記載した会社登録証明書に含まれる裁判所条項の選択が訴訟において適用されないか、または実行できないことを発見した場合、私たちは、他の管轄区域でそのような訴訟を解決することに関連する追加費用を生じる可能性があり、これは、私たちの業務および財務状況に悪影響を及ぼす可能性がある。
私たちbrは、裁判所がこの条項を実行するかどうかに不確実性があり、投資家は連邦証券法とその下の規則と法規の遵守を放棄することができないことに注意した。私たちはこの条項が私たちに有利であると信じているが、それはデラウェア州法律の適用された訴訟タイプでの適用をより 一致させるためであるが、この条項は私たちの役員と幹部に対するbrの訴訟を阻止する効果を果たすかもしれない。
| 57 |
前向き陳述に関する警告的説明
この目論見書には、重大なリスクと不確実性に関するいくつかの前向きな陳述が含まれている可能性がある。場合によっては、“可能”、“予想”、“予想”、“計画”、“予想”、“可能”、“目標”、“プロジェクト”、“推定”、“信じる”、“推定”、“予測”という用語によって、 前向き陳述を識別することができる。“潜在的”または“継続”またはこれらの用語の否定 または他の同様の表現は、将来に関する陳述を識別することが意図されている。これらの表現は募集説明書の発表日までの状況のみを代表し、既知と未知のリスク、不確定性とその他の重要な要素に関連し、私たちの実際の結果、業績或いは業績と展望性表現の中で明示或いは暗示する任意の未来の結果、業績或いは業績に実質的な差がある可能性がある。私たちのこれらの展望的な陳述は主に私たちの現在の未来の事件と財務傾向の予想と予測に基づいており、これらの事件と財務傾向は私たちの業務、財務状況、運営結果に影響を与える可能性があると考えられる。これらの前向きな 陳述は、以下の態様に関する陳述を含むが、これらに限定されない
| ● | 私たちはナスダックの持続的な上場要求を再遵守する能力がある | |
| ● | 私たちは運営の歴史が不足していて、追加資本が必要です | |
| ● | 私たちのbrは候補製品の開発と商業化を計画しています | |
| ● | 我々が計画したCTX−1301、CTX−1302、およびCTX−2103臨床試験の時間; | |
| ● | CTX−1301、CTX−1302、およびCTX−2103セキュリティプロトコルの時間を提出する | |
| ● | CTX−1301、CTX−1302、CTX−2103、または任意の他の将来の候補製品の規制承認を取得し、維持する時間および能力 ; | |
| ● | 候補製品の臨床的実用性は | |
| ● | 私たちのビジネス化マーケティング能力戦略 | |
| ● | 私たちのbrは現金の使用を期待しています
| |
| ● | 私たちは競争相手や業界に関連した競争地位と予測 | |
● |
私たちは重要な人を識別し、採用し、維持する能力 | |
| ● | 法律法規の影響 | |
| ● | 私たちは雇用法案に基づいて新興成長型企業になる時期を予想している | |
| ● | 私たちのbr計画は、私たちのビジネス目標と一致する、重大なビジネス潜在力を持つ他の候補製品を決定します | |
| ● | 私たちは未来の収入と支出を推定する。 |
展望性陳述は固有にリスクと不確実性の影響を受けているため、その中のいくつかのリスクと不確実性は予測または定量化できず、いくつかは私たちが制御できないので、あなたは未来のイベントの予測としてこれらの前向き陳述に依存してはならない。我々の前向き表現に反映されるイベントや状況は実現できないか発生する可能性があり,実際の結果は前向き表現で予測された結果とは大きく異なる可能性がある.我々の実際の結果と前向き陳述に明示または示唆された結果とは大きく異なる重要な要素を招く可能性のある議論については、本募集説明書の“リスク要因”の部分を参照してください。しかも、私たちは持続的な環境で運営している。新たなリスク要因や不確定要因が時々出現する可能性があり、経営陣がすべてのリスク要因や不確定要因を予測することは不可能である。これらの要素のため、本募集明細書の展望的な陳述が正確であることが証明されることを保証することはできません。法律の適用に別の要求がない限り、私たちは、任意の新しい情報、未来のイベント、br}状況の変化、または他の理由で、本明細書に含まれる任意の前向きな陳述を公開または修正するつもりはありません。しかし、あなたは、私たちが株式募集説明書の公表日後に時々アメリカ証券取引委員会に提出した報告書に記載されている要素とリスク、その他の情報を検討すべきです。
あなたは本募集説明書を読んで、その中に含まれている財務諸表と財務諸表の付記が含まれていますが、私たちの将来の実際の結果は私たちが予想していたものと大きく異なる可能性があることを理解しなければなりません。私たちはこのような警告的な声明を通じて私たちのすべての展望的声明を限定する。
| 58 |
市場価格と配当情報
市場情報
私たちの普通株はナスダックで取引され、コードは“CING”です
記録保持者
2024年1月5日までに、私たちは206人の普通株式保有者がいます。我々普通株の実際の保有者数は,実益所有者である株主を含むこの記録保有者の数を超えているが,その株式はブローカーが街頭名義で保有しているか,あるいは他の指定された人が保有している.この数の登録所有者には,その株式が他の エンティティによって信託形式で保有可能な株主も含まれていない.
配当政策
Brを除く株式配当金は2021年9月20日に発効し、私たちの初公開(“IPO”)前の推定値に基づいて正しい発行済み株式数 を決定することを目的としており、私たちは私たちの普通株のいかなる配当も発表または支払いしたことがありません。私たちは現在、すべての利用可能な資金と任意の将来の収益(もしあれば)を維持し、私たちの業務の発展と拡張に資金を提供するつもりで、予測可能な未来にはいかなる現金配当も支払わないと予想しています。将来の任意の配当金の決定は当社の取締役会が適宜決定します。
| 59 |
商売人
概要
私たちはバイオ製薬会社で、私たちの独自のPrecisionを使って定期的にリリースしますTM(PTR)TM)次世代製薬製品パイプラインを構築および推進するための薬物送達プラットフォーム技術は、煩雑な日常投与レジメンおよび二次治療結果を特徴とする頻繁に疾患を診断する患者の生活を改善することを目的としている。最初のポイントは注意欠陥/多動性障害(ADHD)と不安の治療であり,他の治療分野を決定·評価しており,これらの分野では,我々のPTR技術が将来の候補製品の開発に利用される可能性がある。我々のPTRプラットフォームは独自の侵食バリア層(EBL)を採用し,特定のあらかじめ定義された時間間隔で薬物物質を放出し,brが1日1回,多用量錠剤を放出する可能性を可能にすることを目的としている。
ADHD治療市場を狙っており、2023年11月現在、米国市場規模は230億ドルを超えると推定されており、そのうち186億ドルは覚せい剤によるものである。br覚せい剤はADHD治療に最もよく使われる薬物種別であり、米国のすべてのADHD薬物処方の約88%を占めている。これに対し,非覚せい剤は通常二線あるいは補助治療環境でしか使用されておらず,すべてのADHD薬処方の約12%を占めている。徐放或いは長時間作用刺激性薬物剤形は最もよくADHDの第一線の治療薬物として使用され、ADHD市場の総支出の約160億ドルを占め、すべての覚せい剤処方の54%を占める。これらの徐放剤の多くは朝1回の投与が許可されており,日中の再投与の必要性を解消することを目的としている。しかしながら、現在の“1日1回”徐放剤形を使用して、多くの患者は、有効な日カバーを達成するために、当日遅い時期(通常は午後早い時期)に第2剤または“促進剤”brの用量を受け、したがって、様々な副作用brを受ける。現在の治療モデルには,重大で満たされていない需要,すなわち真の1日1回のADHD刺激薬が存在し,その持続時間は活動日全体のカバー範囲を提供し,副作用 を改善し,患者の多くの満たされていない需要をよりよく満たすことができると考えられる。
我々の2種類の独自の覚せい剤:CTX−1301(酢酸ジメチル)とCTX−1302(右旋フェニルプロピルアミン)は,すべての3つの患者群のADHD:小児(6−12歳),青少年(13−17歳),成人 (18歳以上)の治療に開発されている。CTX-1301およびCTX-1302の両方は、現在承認されている覚せい剤療法の主な欠点を解決することを目的としている: は即時効果を提供する(30分以内);完全な“有効日”持続時間を提供すること;追加の覚せい剤薬を必要としない“促進剤/回復”用量;早期薬物“疲労”に関連するリバウンド/崩壊症状 を最大限に減少または除去すること;および良好な耐性を提供し、薬物br}の血中濃度の低下を制御することを目的としている。さらに、私たちは、ADHD患者の60%までの主要な薬物と一緒に使用される“推進剤”の用量を廃止することによって、私たちの候補製品は、短時間効果の覚せい剤に関連する乱用と転移を減少させることができると信じている;医師が2つの薬剤ではなく、1つの薬剤を処方することを許可する;br}患者が2つの薬剤ではなく、1つの薬剤の支払いを可能にすること;支払人が2つの薬剤ではなく1つの薬剤を清算することを可能にする。
著者らは人類被験者に概念検証試験を完成し、著者らのPTRプラットフォームを検証し、そして2020年10月にADHD患者におけるCTX-1301の1/2期研究の積極的な結果を発表し、CTX-1301とフォカリンXRの耐性を確定し、バイオアベイラビリティと投与量の割合を比較した。CTX−1301 NDAが提出した薬理学的要求を満たすために,2022年10月に食品効果研究を完了し,CTX−1301は食物と一緒に服用可能であり,食物と併用しなくてもよいことが証明された。
成人ADHD患者におけるCTX-1301の治療効果と安全性及び効果と持続時間を評価する第三段階成人用量最適化研究は2022年12月にスタートし、2023年6月に完成した。研究結果はすでに2023年精神医学大会で公表され、2024年1月に開催された2024年アメリカADHD及び関連疾病専門学会(APSARD)で公表される。この第3段階のCTX−1301研究(NCT 05631626)は,成人21名(年齢範囲:18−55歳)の成人実験室教室におけるCTX−1301使用の治療効果と安全性,およびCTX−1301の発症と持続時間を評価した。それは主要な治療効果の終点で統計学的意義に達しなかったが、CTX-1301はプラセボと比べ、永久製品業績評価(“PERMP”)得点の改善に顕著な意義がある傾向を示した。プラセボと比較して,CTX−1301 CTX−1301を用いた臨床全体印象尺度(“cgi−S”)スコアも有意な改善を示し, はこの副次的終点に対して有意な統計学的意義を示した。この試験では,CTX−1301の治療効果が有意であった −30分から,CTX−1301がアクティブな1日を通してADHD症状 を改善できることが証明された。
そのほか、著者らは2023年第3四半期に2つの児童と青少年患者に対するCTX-1301第3段階臨床研究-固定用量研究(NCT 05286762)と実験室授業環境下の投与量最適化効果と持続時間研究(NCT 05924594)を開始した。FDAから受けたCTX−1301臨床計画に関する指導により,両研究の登録を中止した。これらの研究では登録されたすべての患者が研究を完了し,それに応じてデータを報告することができる。また,このガイドラインによれば,2025年上半期に第505(B)(2)経路に基づいてCTX−1301のセキュリティ協定を提出し,フォカリンXRを市販薬の参考とし,FDA届出の有効性と安全性データを承認の基礎とし,我々CTX−1301臨床計画の生物利用度/生物学的同等性データと有効性/安全性データを用いる予定である。CTX-1301に対するFDAの承認を得たら、私たちは4回の実験を行うかもしれない。
我々は早ければ2025年にCTX−1302(右旋フェニルプロピルアミン)の臨床計画を開始する予定であり,これはわれわれがADHDを治療する第2の研究資産であり,追加のbr資本資源を待つ。
| 60 |
私たちのPTRプラットフォームは患者と医者に差別化された薬物治療選択 を提供する潜在力があり、これは患者のコンプライアンスを高め、そして他のいくつかの治療領域で健康結果を改善すると信じている。私たちは、私たちのPTR プラットフォーム技術を利用して、他の治療 領域でより多くの資産を識別し開発することによって、私たちの臨床段階チャネルを拡張し、増強する予定であり、これらの分野では、1つまたは複数の原料薬は、特定の所定の時間間隔で毎日複数回送達され、既存の療法よりも著しく改善された方法で発表される必要がある。私たちが他の未来の候補製品を選択する基準は、年間売上高が10億ドル以上に達する潜在力、明らかに差別化された治療優勢を提供する潜在力、満たされていない医療需要を克服する潜在力を含む。
私たちは連邦食品、薬物、化粧品法案505(B)(2)節に基づいて、私たちの不安障害候補薬CTX-2103(ブチロシノン)のための臨床計画を構築しており、これはより速いNDA提出時間をもたらす可能性がある。この努力の一部として,2022年9月の年度精神医学大会でCTX−2103のヒト製剤研究結果を発表した。このような3つの精密決定時用量ブチロシノンと1つの即時放出用量を提供する三連体錠剤に対して薬物動態評価を行った。さらに、シンチグラフィは、放出位置および開始を決定するために消化管におけるこれらの錠剤の輸送を示し、その後、CTX-2103製剤の完全放出曲線を確立するために薬物動態データに関連付けられる。
CTX-2103は著者らの独自のPTR薬物送達プラットフォームを結合した新型ブチロシノン徐放錠として設計された。ブチルスピロロンはアゾド誘導体および5−HT 1 Aの部分アゴニストであり、広汎性不安障害の治療に使用される最初の非ベンゾジアゼピン系抗不安薬である。ブチルスピレノンの副作用は他の抗不安薬と比較して減少する可能性がある。ベンゾジアゼピン類やバルビツール類と異なり,ブチロシドンの使用はγ−アミノ酪酸受容体に影響を与えないため,身体依存や禁断に関するリスクは存在しない。
著者らはFDAとIND前会議を開催することを要請し、2023年第4四半期に著者らが行ったCTX-2103臨床と監督管理計画の設計を討論することができるように、人類調合研究の積極的な結果は必要な肝心な情報を提供した。著者らはFDAからCTX-2103制御経路に関する情報、及びINDの臨床研究設計を受け取った。FDAのフィードバックによれば、我々は、505(B)(2)の経路の下で、通常505(B)(1)の全NDA経路よりも少ない時間およびリソースを必要とするCTX−2103の承認を求めることができると信じている。
2020年には、52億ドルの焦慮市場において、ブチロシノンの様々な形態の米国での売上高が20億ドルを超えた。CTX-2103は を毎日1回の改良放出錠に設計し、明らかな差異性と標準治療方案と比較した顕著な優勢 は毎日何度も服用しなければ治療効果を最大限に発揮できない。我々は,最終合意や追加資本資源が達成されていない場合には,不安治療のためのCTX−2103(ブチルスピレノン)の開発が早ければ2025年に開始される可能性がある積極的な許可交渉を行っている。
| 61 |
我々のbr臨床開発ルートは
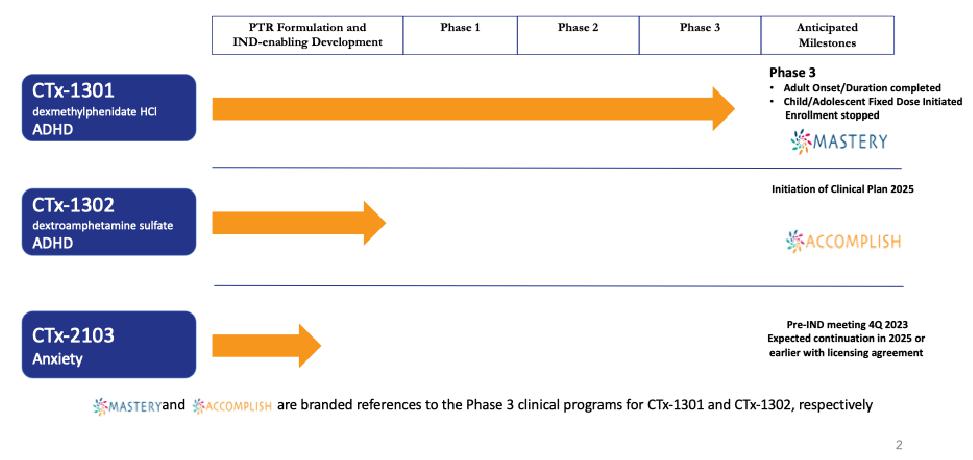
私たちの 戦略
私たちの目標は、次世代薬物製品の開発、製造、商業化に専念し、私たちのPTR薬物送達プラットフォーム技術を利用して、用量計画と薬物放出ファイルを作成して、多くのよく見られる診断疾患を有する患者の生活を改善するために、リードした革新的なバイオ製薬会社になることを目標としている。これを実現するために,我々の業務戦略の主な初期要素 は:
| ● | ADHDを治療するためのCTX−1301の開発が完了し、規制部門の承認を得た。
2020年10月,われわれはADHD患者におけるCTX−1301の1/2期研究の積極的な結果を発表し,CTX−1301とフォカリンXRの耐性,バイオアベイラビリティと用量割合を比較した。成人ADHD患者におけるCTX-1301の治療効果と安全性及び効果と持続時間を評価する第三段階成人用量最適化研究は2022年12月に開始し、2023年6月に完成した。結果は2023年精神医学大会で公表され、そのほか、著者らは2023年第3四半期に2つの児童と青少年患者に対するCTX-1301第3段階臨床研究-固定用量研究と実験室 教室環境下の用量最適化効果と持続時間研究を開始した。FDAが提供したCTX−1301(酢酸デキサメタゾン)臨床計画に関する指導意見に基づき,この2つの研究に組み込まれた。これらの研究ではすべての登録患者が研究を完了することができ,データはそれに応じて報告される。また,このガイドラインによると,2025年上半期に第505(B)(2)経路に基づいてCTX−1301のセキュリティプロトコルを提出し,フォカリンXRを参考に市販薬とし,その有効性と安全性データを我々のCTX−1301臨床計画の生物利用度/生物同等性データと有効性/安全性データとともに承認の基礎とする予定である。 | |
| ● | CTX-1301の商業化に成功.
CTX-1301に対するFDAの承認を得たら、Indegene,Inc.の助けを借りて私たちの主要候補を商業化する予定です。2023年3月、私たちはIndegeneと共同商業化協定を締結し、この合意に基づいて、Indegeneはマーケティング、販売、市場参入、流通、サービス料を含むCTX-1301の商業化サービスを提供します。 |
| 62 |
| ● | CTX−2103不安治療の臨床試験の進展2022年9月、CTX-2103のヒト製剤研究データは、トリ放出ブチロシノンを単回投与できることを示したと発表した。われわれはこれらのbr結果を用いてCTX−2103の臨床計画を設計し、1日1回の多用量錠剤として設計され、brは標準治療方案と比較して明らかな差別化と注目される優勢を有すると信じている。我々は,最終合意に達する前に,不安治療のためのCTX−2103(ブチルスピリドン)の開発が早ければ2025年に開始される可能性があり,追加の資本資源を待つ積極的な許可交渉を行っている | |
| ● | ADHDを治療するCTX-1302の研究進展我々は早ければ2025年にCTX−1302(右旋フェニルプロピルアミン)の臨床br計画を開始する予定であり,これはわれわれがADHDを治療する第2の研究資産であり,追加の資本資源を待つ。
| |
| ● | 我々PTRプラットフォームの潜在力を最大限に発揮し、重大な医療需要と数十億ドルの収入を満たす潜在力を持つ新しい適応の中でより多くの候補製品を開発した我々は,我々のPTR薬物送達プラットフォーム技術と簡略化された505(B)(2) を用いて経路を開発し,他の治療分野でより多くの治療資産を開発する予定であり,これらの分野では,1つまたは複数の活性医薬成分 を特定のあらかじめ定義された時間間隔で1日複数回投与し,既存療法を有意に改善できるように放出する必要がある。これは患者のコンプライアンスを高め,健康状態を改善すると信じている。我々が評価しようとしている他の適応としては,不眠,非オピオイド,アルツハイマー症,甲状腺機能低下,精神病,うつ病,心血管疾患,パーキンソン病,片頭痛,口腔腫瘍学,口乾(口乾),躁鬱症などがある。 |
| ● | 私たちの製品の組み合わせを補完するために、または許可された他の資産または計画を買収し、および/または私たちの技術を利用するそれは.私たちの現在の候補製品の組合せ を支援し、私たちの組織に相当な価値を提供するために、潜在的な協力機会または資産買収を継続的に評価する。早期から中期までの候補製品の開発に専念し,br}臨床データを生成し,開発の後期段階に入り,最終的に商業化に向かう可能性がある。 | |
| ● | 私たちのPTRプラットフォームを他の会社に許可し、私たちの候補製品をアメリカ以外の国に許可します。私たちは、私たちの既存の資産とPTR薬物送達プラットフォーム技術を他の会社に許可するための機会を積極的に探し、評価しており、これらの会社は、需要が満たされていない大量の患者集団にサービスを提供することを望んでおり、および/または患者の需要を満たすために毎日複数回の投与を1日1回のbrに変えることを望んでいる。私たちはまた、私たちの候補製品を米国以外で使用する機会に権限を与えることを求めて評価するための対話を行っている。 | |
| ● | さらに私たちの知的財産権の組み合わせを強化しますそれは.私たちは、私たちの多様な知的財産権の組み合わせを管理し、拡大し、私たちのビジネス秘密と技術ノウハウを維持し、私たちのPTRプラットフォーム、現在と未来の候補パイプライン、および独自のbr製造プロセスに重点を置くつもりです。私たちは、これらの活動は、私たちの治療資産および圧縮錠剤方法と競争しようとする可能性のある潜在的な競争相手から、プラットフォームおよび候補製品を保護するために重要であると信じている。 | |
| ● | 我々の既存のcGMP専門知識を利用する.我々は、リアルタイムテストと発表の潜在力を持ち、私たちの第三者製造パートナーが使用するための独自、信頼性、高生産量の専用製造設備 を開発した。我々のプロセス は,他の薬物物質を含むことができるプラットフォームを作成し,他のbr適応や治療領域への拡張を可能にすることを目的としている。これらの製造能力や設備への投資は,我々の開発スケジュールや現在と将来の資産の全体的な開発コストを大幅に短縮することが予想される.我々は現在、臨床または商業製造需要を満たすために技術移転や大規模なプロセスを必要としない商業製造装置を使用している。 2022年10月、我々は、我々の新しい契約開発 および製造組織(“CDMO”)としてSocial CDMO,Inc.(“Social”)を選択したことを発表し、この組織は、我々の主要な候補CTX-1301のすべての臨床、登録、および商業ロット を生産する。Socialは、ジョージア州ガイエンスビルに位置する工場や装備に、当社が提供する独自の設備を専用に搭載しています |
| 63 |
私たちのbrチームは
私たちの創業者と管理チームはバイオ製薬分野で長年の経験を持ち、ファイザー、ノワール国際株式会社、デュポン社、セノフィ社など、リードするバイオ製薬会社で管理職を務めています。我々のチームは薬物開発と薬品商業化の面でbrの豊富な経験と専門知識を持っており、多種の精神と神経系製品を含む。
シェイファー、私たちの共同創業者、会長兼最高経営責任者は、ファイザー、ノワール国際株式会社とセノフィ社で高級指導者を務め、薬物開発と商業化の面で25年以上の経験を持っている。マシュー·ブラームス博士は著者らの共同創業者兼首席医療官であり、彼は成人と児童精神医学領域で30年以上の臨床患者管理経験を持ち、brは多種のADHD薬物の研究、開発と評価に参与した。ラウル·R·シルバ博士は著者らの共同創業者兼首席科学官であり、勤務する児童と青少年精神病学者であり、ニューヨーク大学医学院の児童と青少年精神医学助教授と副主席を務めたことがある。私たちの首席運営官ローリー·A·マイルズは、ファイザー、ノワール国際株式会社、デュポン社、セノフィ社を含む世界的にリードしている製薬会社でリーダーを務め、薬物開発、マーケティング、商業化で25年以上の経験を持っています。
ADHD 現在の治療法の概要と欠陥
ADHDは1種の慢性神経行為と発育障害であり、数百万の児童、青少年と成人に影響を与える。米国では,4−17歳の児童·青少年の約640万人,すなわち11%がADHDと診断されている。この群では80%が治療を受け,65%が成人まで持続する臨床ADHDを示した。アメリカの成人ADHD罹患率は約1,100万人の患者で、総人口の4.4%を占め、児童と青少年人口のほぼ2倍と推定されている。現在、成人ADHD人口の約20%が治療を受けているが、ますます多くの成人患者がADHDと診断され治療を求めており、成人ADHD市場は毎年約10%増加している。米国のADHD薬の総売上高は増加を続けており,2023年11月までの12カ月間に全ADHD薬の売上高は230億ドルを超えている。
ADHD の特徴は、持続的な注意力不集中および/または多動--妨害機能および/または発育の衝動である。 はアメリカ児童と青少年精神医学学会によると、ADHDの児童と青少年におけるよく見られる表現は:
多動症: 子供たちはいつも運動しているようです。多動症の子供は動き回ったり、周りの何かに触れたり、遊んだり、会話を続けたりするかもしれません。物語や授業中、子供はくねられたり、そわそわしたり、立ち上がって部屋を歩いたりするかもしれません。足をくねらせたり指を軽く叩いたりする子供もいます。多動症の青少年や大人はイライラしているかもしれませんが、常に忙しい必要があります。
衝動: 子供たちは思わず口にしてしまうことが多い。彼らはいつも節度なく自分の気持ちを見せているかもしれない。彼らはまた彼らの行動の結果を考慮していないかもしれない。このような子供は列に並んだり交代したりするのが難しいことに気づくかもしれません。衝動的な青少年と大人は、より大きな遅延に向かって努力するのではなく、即効性のある選択をする傾向がある。
集中しない:もし活動が彼らが本当に好きなものでなければ、集中していない子供はすぐに飽きてしまうかもしれない。任務を組織して達成したり、新しいことを学ぶのは彼らにとって難しい。学生として、彼らはよく学校の宿題を書いたり、家に本を持って帰るのを忘れます。宿題を終わらせることは大きな挑戦かもしれない。どんな年齢でも、ぼんやりしている人は気が散ってしまいがちで、不注意なミスをしたり、物事を忘れたり、指示通りに行動したり、何も成し遂げていない活動から別の活動にジャンプしたりすることが多いかもしれません。
成人ADHD患者は通常騒動、衝動、時間管理困難、情緒調節困難と財務管理困難を有する。ADHDを有する成人は,心の中にそわそわしている感覚や不安を感じ,より大きなコミュニケーション困難を経験していると報告している。就職市場に入ると、多くの大人が仕事を見つけることが難しく、また何度も遅刻したり欠勤したりするため、解雇されるリスクが増加している。ADHDを有する成人の収入は,影響を受けていない同世代と比較して約30%,就業可能性は10%低い。そのほか、ADHDを有する成人は薬物乱用と飲酒、社交焦慮と抑うつを含む各種の共病を示す可能性が高い。
| 64 |
児童と成人のADHDは個人だけでなく、家族、友人、同世代にも影響を与えており、その流行は最もよく見られる診断行動障害の一つとして、社会、医療システム、経済全体に深刻な影響を与えているからである。社会レベルでは、対照群と比較してADHD患者が経験した車両事故発生率は40%以上高く、離婚率は2倍高く、意外死亡の発生率は2倍高く、前向き研究では、ADHDを有する児童と成人の監視率は対照群の約2倍であることが示されている。経済面では,米国のみでは全国で毎年増加しているADHDコストは1430億ドルから2660億ドルと様々である。
ADHDに対する単一の医学、体格または遺伝子テストがないにもかかわらず、合格した精神衛生保健専門家および医師は、ADHD症状リスト、標準化行動評価表、過去および現在の機能の詳細な病歴、および近縁または重要な他の人から得られた情報を含む様々なソースから情報を収集することができ、それによって診断評価を提供することができる。一部の事業者は,認知能力や学習成績をテストし,可能な学習障害を排除する.
覚せい剤 はADHD治療に最もよく使われる処方薬であり、すべてのADHD薬処方の約88%を占めています。 覚せい剤は付表II制御物質であり、脳中のドパミンやノルアドレナリン神経伝達物質の作用を増強することで作用していると考えられています。2023年11月までの12カ月間に約8200万枚の覚せい剤が処方された。これに対し,非覚せい剤は通常二線あるいは補助療法として使用されており,すべてのADHD薬処方の12%を占めている。現在、ADHD市場は4種類の主要な覚せい剤薬物によって主導されている:Vyvanse、AdderallXR、ConcertaとFocalinXR。 これらの製品は2000年から2007年までの間に承認され、発売され、ADHD治療モデル式の革命的変化と考えられ、最終的に1種の解決策を提供し、ADHD患者が当時必要としていた午前遅い時期の第二剤覚せい剤薬物を回避した。
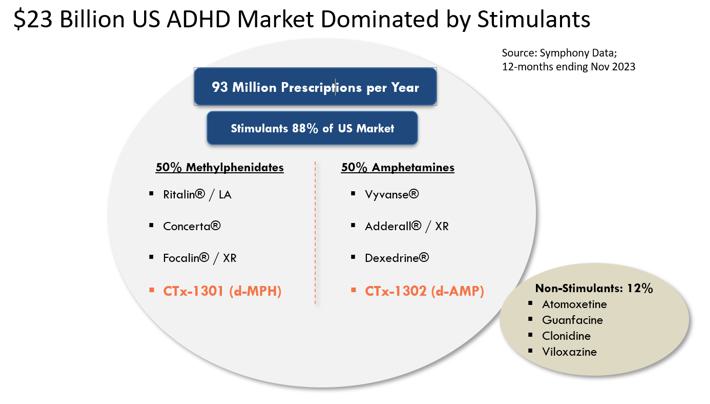
不幸にも、設計通り、すべての4種類の最もよく使われる処方覚せい剤は朝にすべての薬物物質を提供する。したがって、ほとんどの患者はまだ彼らの1日の残りの時間をカバーするための追加の薬を必要とする。現在、60%のADHD患者 は持続時間が短く、効果が遅い、午後早い時期の崩壊或いはリバウンド効果のため、午後の“増強/回復”用量 が必要である。また,それらのPK−PD放出曲線は,患者が治療中であっても衝突やリバウンド効果の深刻な影響を受ける。
| 65 |
患者や事業者は,理想的なADHD刺激薬が提供されると報告している準備万端整っている活動日全体の持続時間;直ちに行動を開始すること(30分以内)、薬物血中レベルの急速な低下に関連する崩壊/リバウンド効果を最大限に減少または回避することができ、短時間効果の覚せい剤/回復用量の必要性を除去することができるという特徴がある。
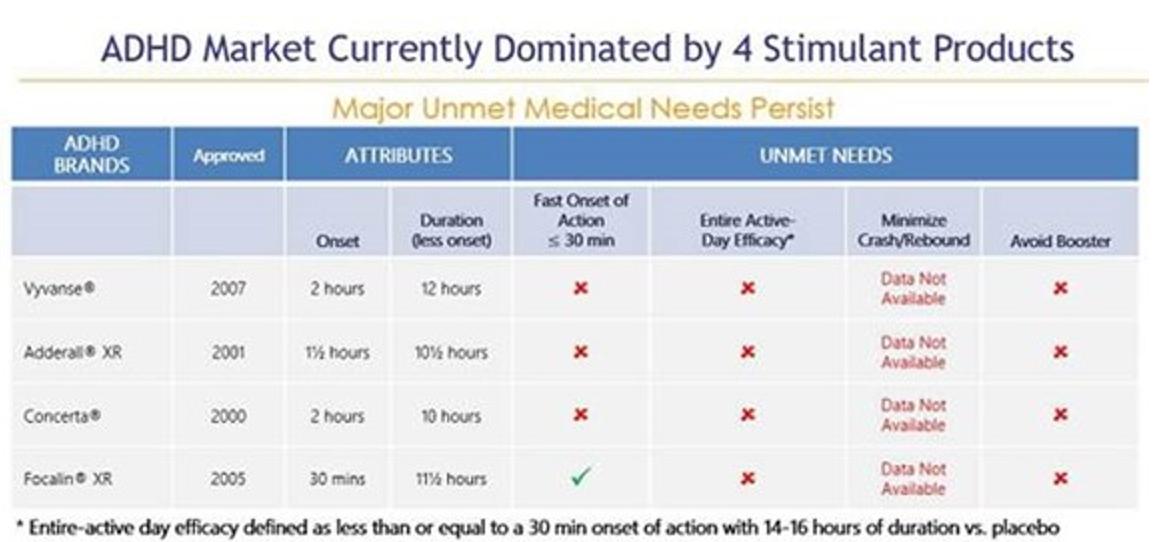
上の グラフは,承認済み製品のパッケージ挿入と承認要約ベースに基づいている.
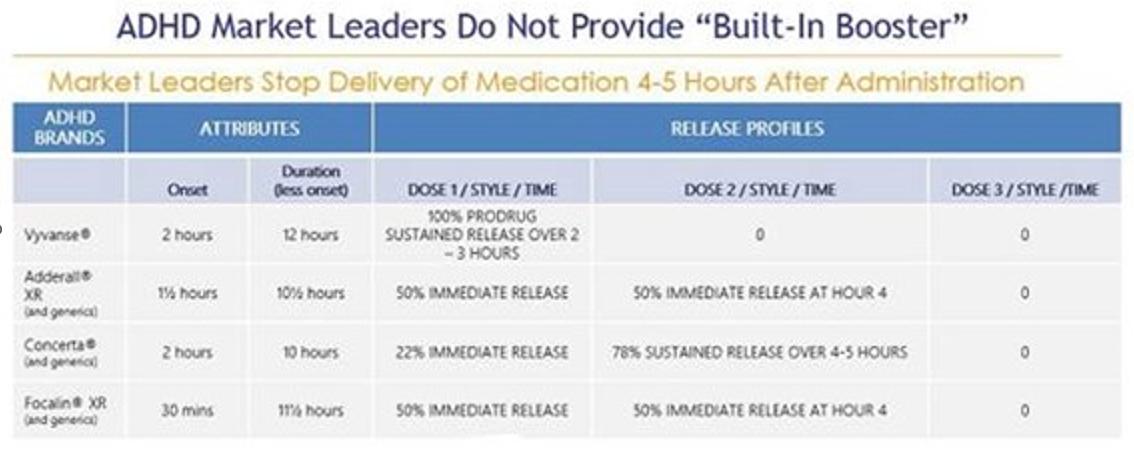
上の グラフは,承認済み製品のパッケージ挿入と承認要約ベースに基づいている.
近年、FDAは追加の覚せい剤薬物を承認し、いくつかの残りの満たされていない需要を満たすことを目的としている。咀嚼錠、液体、口腔崩壊錠がすでに発売されており、もう一つの製品は夜の投与スケジュールを持ち、早朝効果を提供することを目的としている。これらの製品はADHD患者や処方者のすべての満たされていない需要を満たすものではないため, は第一線のエージェントとしての吸引力を得ることができなかった。また,市場シェアによると,これらの最近の刺激薬は,医療従事者や彼らの患者に広く利用可能な後発薬よりも多くの利点を提供していないようである。これらはニッチ薬物であり、2020年のアメリカADHD処方総数の2.0%を占めることが証明された。したがって、迅速な効果を提供し、崩壊/反発を最大限に減少または除去し、ブースター/回復用量を除去し、最も重要なのは“有効日”全体の効果を提供する真の1日1回用量に対して、満たされていない需要が存在する。
| 66 |
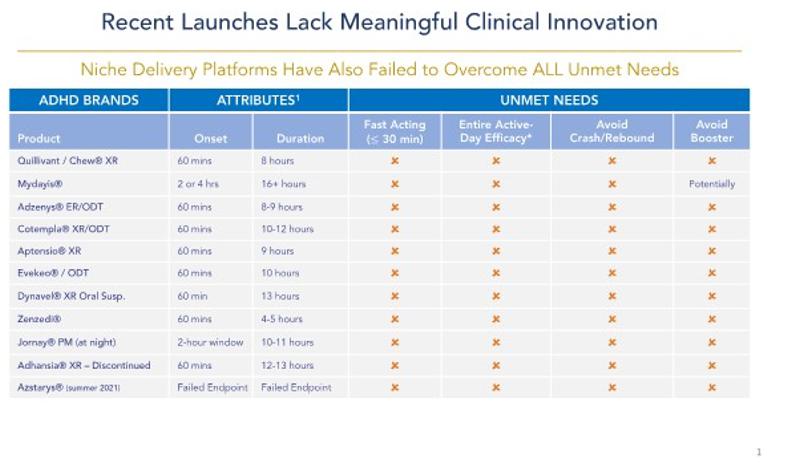
上の グラフは,承認済み製品のパッケージ挿入と承認要約ベースに基づいている.
私たちのbr解決策:私たちの独自の精密タイミング放出薬物送達プラットフォーム技術
我々は、我々の内部で開発されたPTR薬物送達プラットフォーム技術を使用して、真の1日1回の投与を可能にする薬剤を開発している。私たちのCTX-1301、CTX-1302およびCTX-2103候補薬剤は、3つの放出活性医薬成分を含み、それを小さなbr錠剤剤形に組み合わせる(多くの比較可能な単回用量ADHD製品よりも小さい)。毎回放出される原料薬は、固有のEBLによって分離され、1日に徐々に侵食され、特定の時間間隔で制御された薬物放出を提供するように設計された機能性賦形剤であり、良好な標的効果期を可能にする。
PTRプラットフォームのフィルムコーティングフィルムです
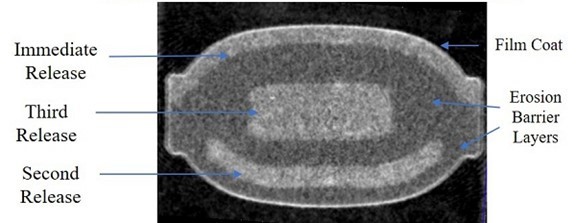
| 67 |
CTX-1301錠剤は、通常のADHDおよび他の薬剤とのサイズ比較
現在利用可能なADHD治療法と比較して、我々のPTR技術は候補薬物に以下の利点を提供すると信じている
高速 から始まります多くの現在利用可能な治療法は通常60分以上の時間を要して効果を開始するため,患者に服薬と効果の間に長い間隔を持たせる可能性がある。このような発症差をできるだけ少なくするために,患者は早く起きて薬を服用し,薬が発効するまで睡眠状態に戻ろうとするのが一般的である。我々が設計した候補薬は迅速に効果があるため,夜明け前に発症を待つのではなく,患者が1日の朝に服用することができる。
追加的な覚せい剤ブースターの要求を除去した全活動日のカバー範囲により,われわれの技術は現在処方されている治療が切れたときに午後の強化用量を服用する必要がないと信じている。私たちは、増強用量の需要を除去することによって、私たちの候補者は、特に児童および青少年患者、特に児童および青少年患者を減少させ、彼らはしばしば学校で同級生と一緒に第二剤を服用することを余儀なくされ、患者のコンプライアンスを向上させ、特にADHD集団において、br患者は彼らが必要とする追加の用量を服用して彼らの一日を過ごすことを忘れやすいからであると信じている。
乱用の潜在力を下げる承認されれば、ADHD患者に対する迅速な効果と完全な活動日解決方案は短時間効果覚せい剤の乱用と転移の発生率を低下させると信じている。短時間効果覚せい剤強化用量の需要を解消することにより,短時間作用付表II制御薬を学校や単位で午後用量のbr不正販売や娯楽使用を行う可能性が減少すると信じられている。
Br崩壊とリバウンド症状を解消した現在利用可能な治療法を使用している患者は、彼らの薬物が無効になったときに副作用またはADHD症状の爆発を報告する可能性があり、これらの影響は“崩壊”および“リバウンド”と呼ばれる。私たちの正確な投与時間、投与割合、投与方式を利用して、私たちの候補薬は血液レベルの低下をコントロールし、患者のこのような不快体験を解消できると信じています.
| 68 |
より低い コストそれは.全有効日治療効果を提供することによって、私たちの候補薬物は医者が1つ以上の薬物を処方する必要がなくなり、それによって単一の患者と医療システム全体の全体的な疾病コストを低下させた。また,覚せい剤ADHD分類における後発薬は他の分類の非制御薬よりもはるかに安いわけではない。非特許覚せい剤薬物のコストはブランド薬物コストの55%~90%である。私たちは承認されれば、私たちの候補薬は患者により費用効果的な解決策を提供すると信じている。
耐性を著しく高めた。われわれの薬物のPKとPD特徴により、患者は不眠、食欲抑制、極度の不安、イライラ不安、怒りやすさ、疲労と感情のなだらかさを含む既存の覚せい剤療法に関連する治療関連不良事件を経験すると信じている。
発売時に8種類の用量強度の可用性と単一エナンチオマー原料薬の選択を提供するそれは.我々のCTX−1301およびCTX−1302の候補製品はいずれも円形薄膜コーティングシートであり、8つの一致した用量強度を提供する予定である。事業者に適切な滴定を提供し,患者の日常用量需要を最適化する能力が重要であると考えられる。8種類の用量強度を発売することによって、業者は、患者を他の薬剤に切り替えたり、患者のためにより多くの短時間効果強化薬を補充したりする必要はない。また、8つの用量強度は、医療従事者が彼らの実践において、幼児から成人までの様々な患者 を適切に治療することを可能にすると信じている。3つの用量強度が発売された薬剤は、通常、少なくとも6~7つの用量が使用可能になるまで無視または回避される。CTX−1301およびCTX−1302の両方は、付表IIによって規制された原料薬を含む。2つの候補製品の原料薬は、複数のエナンチオマーのうちの1つのみを使用し、これは、効力、有害事象(AES)および薬物相互作用状況の改善、および増強された治療指数をもたらす可能性がある。
われわれの主要候補薬CTX−1301:ピペニルデキサメタゾン治療6年以上のADHD
著者らは著者らの最先端の候補薬物CTX-1301はADHDを治療する最初の真の毎日1回のピペラ酢デキサメタゾン錠剤になり、30分以内に効果を発揮し、そして完全な“有効日”の治療効果を有すると信じている。CTX-1301は錠剤内技術に基づく三型徐放錠であり、それは精確なbr放出回数、比率と放出方式で3種類の異なる塩酸デキサメタゾン放出を提供した。私たちのCTX-1301バージョンの概要は以下の通りです
放出#1:最初の即時放出用量、またはIRは、投与後5~6分以内に30分以内に治療効果を達成することを目的とした1日総用量の35%の提供を開始する
放出#2:投与3時間後、第1遅延徐放性(DR 1)は、90分を超える1日放出総量の45%を提供する
放出#3:投与7時間後、2回目の遅延、即時放出(DR 2、内蔵ブースター)は、約30分以内に放出される1日総用量の20%を提供する。
| 69 |
CTX−1301とフォカリンXR(市販薬参照)の放出状況の比較

私たちのbr独自の3モード解放プロファイルは、患者に症状を迅速に緩和する方法を提供し、活動日全体にわたってこの緩和を維持することを目的としている。さらに、CTX-1301は、現在利用可能な50%~50%または33%~33%の放出プロファイルではなく、このような専用の三放出設計およびユニークな35%-45%~20%放出プロファイルによって生成され、患者が同じbr}mg強度でデキサメタゾンを単独で服用すれば、より良い耐性を産生することができる、CTX-1301は、より有利な耐性を示し続けると信じている。CTX-1301は、商業的に入手可能な(ブランドまたは非特許)短時間効果および長時間効果的な処方によって複製できない解放プロファイルを提供し、ADHD患者およびプロバイダの特定の需要を満たすために正確な設計および設計を行う。
CTX-1301薄膜コーティング錠は6.25 mgから50 mgのデキサメタゾンの8種類の用量が予想される。すべての賦形剤は薬典および/または非新薬であり、経口製剤に広く使用され、薬物製品中の含有量 はFDA不活性成分データベース(IID)に列挙された最大力価よりはるかに低い。
我々のCTX-1301臨床開発計画は
CTX-1301の提案された臨床計画は2つの1/2期臨床薬理学研究と著者らの3期臨床治療効果と安全性試験を含む。
我々の1/2期生物利用度試験結果
2020年10月,絶食条件下でADHD被験者の1/2期比較バイオアベイラビリティ研究の積極的な結果を発表し,われわれのRLD Focalin XRに類似した バイオアベイラビリティを示した。調整後の主要露光パラメータの幾何平均比率(C最大値AUC0-情報 とAUC最後に高用量および低用量では、CTX−1301とフォカリンXRの間の用量はいずれも必要な80%~125%の範囲であり、brはRLDとの間のブリッジおよび用量比を示した。この研究では,意外な有害事象はなく,深刻な有害事象もなく,死亡もなく,他の安全信号も観察されなかった。
主な調査結果
フォカリンXRに橋を架ける
| ● | フォカリンXRに類似したバイオアベイラビリティを確認し,505(B)2経路を利用する能力を確認した | |
| ● | すべての個人の強度を評価する必要がないように用量比を証明しました体内にある | |
| ● | Brの任意の非臨床研究の要求やフォカリンXRタグの既存の安全性を利用する能力を解消することは,より早く市場に参入する可能性がある |
| 70 |
フォカリンXRと比較した血漿レベルを示した
| ● | CTX−1301血液レベルは発症が速く,有効日持続時間が長く,良好な耐性を示す可能性が示唆された。 | |
| ● | 設計通りに実行され、その正確な20%の内蔵ブースターの3回目の送達は、承認された場合、CTX-1301は患者の短時間効果覚せい剤に対する需要を除去し、正常な睡眠と食欲に影響を与える可能性のある非理想的な血液レベルを回避することを確認した |
フォカリンXRと比較して血漿レベルが制御可能であることを示した
| ● | 正確な20%第3次分娩は午後3時頃の血液レベルの暴落を阻止し,低下をコントロールした。 |
治療の緊急不良を著しく低下させた
| ● | 患者はPTRプラットフォームを介して正確な時間、独特の比率で25%の薬物治療を受けることが多い | |
| ● | CTX−1301患者はフォカリンXRと比較して研究薬と比較してTEAEが28.6%低下した |
図1,図2,図3にフォカリンXRとのバイオアベイラビリティデータの比較研究を示す
図1:絶食条件下での成人ADHD対象におけるCTX−1301とフォカリンXRとのバイオアベイラビリティ比較研究(低用量比較)

| 71 |
図2:絶食条件下(高用量)CTX-1301と成人ADHD被験者におけるフォカリンXRのバイオアベイラビリティの比較検討
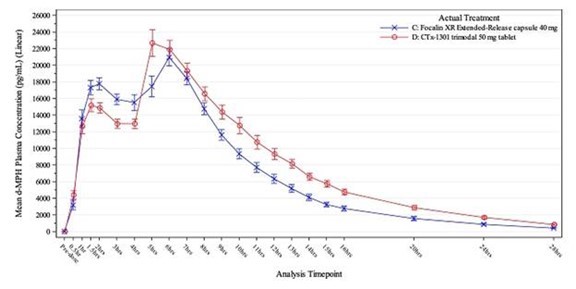
図3:絶食条件下(低用量および高用量)CTX−1301と成人ADHD対象におけるフォカリンXRのバイオアベイラビリティの比較検討
私たちの他の第一段階の研究は
我々は2022年10月に第1段階食品効果研究を完了した。主な終点は,CTX−1301は食物と一緒に服用でき,食物と一緒に服用しなくてもよいことを示している。
| FRBの研究 | |
| ● | 18~50歳の健康な成人被験者23名において、CTX-1301吸収およびバイオアベイラビリティに対する食物の影響を評価するために、第1段階の開放ラベル、ランダム、単剤、2周期、2つの治療コース(給餌および絶食)、2つの連続的な交差研究を行ったそれは.本研究の目的は,単剤CTX−1301の吸収速度と吸収程度および全体バイオアベイラビリティに対する食物の影響を評価することである。第二の目標は、絶食および給餌状態におけるCTX-1301血漿濃度の薬物動態データを提供し、単回用量CTX-1301 25 mg錠剤の安全性を評価することである。考察目標は,選定した時間間隔でさらにCTX−1301 25 mg錠剤の特性を考察することである。 |
| 72 |
私たちの第3段階はテストを把握します
成人ADHD患者におけるCTX-1301の治療効果と安全性及び効果と持続時間を評価する第三段階成人用量最適化研究は2022年12月に開始され、2023年6月に完成した。研究結果はすでに2023年精神医学大会で公表され、2024年1月に開催された2024年アメリカADHD及び関連疾病専門学会(APSARD)で公表される。この第三段階CTX-1301研究 (NCT 05631626)は成人実験室教室環境において21名の成人(年齢範囲:18-55歳)のCTX-1301の治療効果と安全性およびCTX-1301の発病と持続時間を評価した。主要治療効果の終点では統計学的有意差は得られなかったが,CTX−1301はプラセボと比較してPERMPスコアの改善に有意な改善傾向を示した。CTX−1301を用いたcgi−Sスコアもプラセボと比較して有意な改善を示し,この副次的終点に対して有意な統計的意義があった。本試験ではCTX−1301の治療効果が著明であった30分から,CTX−1301が一日中の活動でADHD症状を改善できることが証明された。
主な調査結果
PERMPはこの段階3研究で展開された技能調整数学テストである。PERMPスコアは,試みた数学問題数 に10分間会話で正解した数学問題数の和を加えたものである.得点範囲は0から800までであり,得点が高いほど の方が良いことを示している.
CGI−Sは1−7歳の精神病理学の重症度を測定する単項尺度であり,この第3段階研究で成人を測定した。
| 73 |

*RPCP:ランダム、二重盲検、プラセボコントロール期間
AISRS、またはADHD 評価表は、ADHDの基準を評価し、症状を4点制評価し、この成人研究で測定した“精神疾患診断と統計マニュアル”第5版(DSM-5)(米国精神医学協会2013)の18項目の尺度に基づいている。 は項目ごとに重症度と頻度格付けの組み合わせを用いて採点し、0(症状がないか出現しないか少ないか少ないかを反映する)から3(重症症状を反映したり非常に頻繁に反映されている)であるため、AISRS-5の総得点は0から 54までである。
これら3期CTX-1301診断ツールおよびADHD評価は、AISRS-5、CGI-SおよびPERMPを含み、一般にセキュリティプロトコルの届出を支援する研究の終点として使用される。
著者らの第三段階CTX-1301臨床安全性と有効性研究はまた2つの児童と青少年研究を含み、この2つの研究は2023年第3四半期にスタートした
| ● | A段階3、用量最適化、ランダム、二重盲検、プラセボ対照、単中心、平行群の治療効果と安全性実験室 教室研究ADHDを有する児童(6−12歳)。主な目的はPERMPを用いた実験室授業研究において、CTX-1301とプラセボによるADHD児童の治療効果を評価することである。第2の目標は、CTX−1301の臨床的治療効果の効果的時間および持続時間を決定し、プラセボと比較したCTX−1301の安全性および耐性を決定することである。 | |
| ● | A段階3、ランダム、二重盲検、プラセボ対照、多中心、固定投与量、平行群、ADHDを有する児童と青少年(6-17歳/o)に対して治療効果と安全性研究を行った。主な目標は,ADHD−RS−5を用いて固定用量のCTX−1301のプラセボと比較した治療効果を評価することである。第二の目標は、プラセボと比較した固定用量CTX−1301の治療効果の評価、CGI−Sの使用、固定用量CTX−1301の安全性および耐性、および単剤および安定状態でのPKレベルである。 |
| 74 |
| 探索的目標は、以前に患者によって報告された結果(“PRO”)と比較して、CTX-1301を使用した最適化治療の結果(“PRO”)と比較して最適化された治療の独特な利益および満足度を決定および評価するために、成人用量最適化研究において評価される。専門評価 は: |
| ● | 被験者は、有効日の全効果、疲労回避、衝突/リバウンドおよび短時間効果覚せい剤の乱用/転移を達成するために、“促進剤”用量の使用を要求した。 | |
| ● | これまでの治療の全体治療満足度をCTX−1301と比較した。 | |
| ● | 従来治療していた有害事象をCTX−1301と比較した。 | |
| ● | 1日1回のADHD真の治療の重要性を評価した。 | |
| ● | Brの乱用と/或いは転移短時間効果強化投与量の発生率を評価する。 | |
| ● | ADHD治療を必要とする患者にbrの重要な独特な点を評価するために、方法は完全な解決方案を提供し、完全な有効日の治療効果、迅速な効果、崩壊/リバウンドの回避及び除去に必要な短時間効果の覚せい剤/回復用量を有することである。 |
これらの探索的措置は臨床医に重要な情報を提供するだけでなく、支払人と市場参入チームにも重要なデータを提供する。
FDAから受けた我々のCTX−1301臨床計画に関する指導により,両研究の登録を中止した。これらの研究に参加したすべての患者が研究を完了し,それに応じてデータを報告することができる。また,このガイドラインによると,2025年上半期に第505(B)(2)経路に基づいてCTX−1301のセキュリティプロトコルを提出し,フォカリンXRを市販薬の参考とし,FDA届出の奏効率と安全性データを承認の基礎とし,我々のCTX−1301臨床計画の生物利用度/生物同等性 データと有効性/安全性データを用いる予定である。CTX-1301に対するFDAの承認を得たら、私たちは4回の実験を行うかもしれない。
CTX−1302: 右旋フェニルプロピルアミンによる6歳以上のADHD治療
我々の候補薬物CTX−1302はADHD治療の最初の真の1日1回の右旋フェニルプロピルアミン錠となり,30分以内に効果があり,活動日全体で有効であると信じている。CTX-1302は1種の三モード徐放錠であり、錠剤内錠剤技術に基づいて、正確な放出時間、比率と放出方式で3種類の放出された右旋フェニルプロピルアミンを提供することができる。我々のCTX-1302バージョン の概要は以下の通りです
| ● | 放出#1:1日の総用量の45%を提供する初期即時放出用量は、投与後5~6分以内に30分以内に治療効果を達成することを目的としたbrの開始;および | |
| ● | 放出#2:投与3時間後、DR 1は、90分を超える1日放出総量の35%を提供する | |
| ● | 放出#3:投与7時間後、内蔵ブースターは、約30分以内に放出される1日の総用量の20%を提供する。 |
CTX-1302錠剤は6.25 mgから50 mgの右旋フェニルプロピルアミンの8種類の用量強度を提供することが予想される。すべての補助剤 は薬典と/或いは非新薬であり、すでに経口製剤に広く応用されており、薬物製品中の含有量はFDA IIDに列挙された最大力価よりはるかに低い。
我々のCTX-1302臨床開発計画は
著者らが提案したCTX-1302臨床方案は1/2期の臨床薬理学研究と3期の臨床治療効果と安全性 試験を含む。著者らは2025年にADHD患者におけるCTX-1302の1/2期バイオアベイラビリティ研究を開始する予定であり、もしこの研究の結果 が成功すれば、その後2025年末或いは2026年初めに肝心な3期臨床試験、即ちブランド実現試験を開始する;すべての は資本資源に依存する。我々の1/2期試験には約100人の患者が含まれる予定であり,第3段階臨床計画には約500人の患者が含まれる。
| 75 |
私たちのbrは第一段階実験を計画しています
我々が提案した1期CTX−1302臨床薬理学的研究は,
| ● | 第1/2期比較バイオアベイラビリティ研究:成人ADHD(18+y/o)患者におけるCTX-1302とRLD、Dexedineの薬物動態学的特徴を評価と比較した。 | |
| ● | 段階 1食物効果研究:成人(18+y/o)の摂食および絶食条件下におけるCTX-1302の薬物動態学的特徴を評価する。 | |
| ● | 第1期単剤、完全反復交差研究:RLD,Dexedine Spansuleに対するCTX−1302の成人(18+y/o)における体内薬物動態曲線の被験者の内変異性を評価するために。 |
計画の第3段階で試験を完了する
| ● | A段階3、固定用量、平行設計、プラセボ対照、5週間の児童と青少年患者研究(6-17 y/o)主な治療効果の終点はADHD-RS-5である。臨床全世界改善重症度尺度(CGI-S)は副次的な終点として評価を行う。 | |
| ● | 第3段階、成人模擬職場の治療効果と安全性研究(18+):主な治療効果の終点はPERMPである。効果的な時間および効果持続時間もまた重要な副次的な終点として評価されるだろう。 | |
| ● | 長期用量最適化安全性研究は、小児科群(6~17歳/歳)の安全性を6カ月間評価する。本研究 は,研究期間中に発生した任意の有害事象を収集·モニタリングする。 |
重要な情報:シミュレーション第3段階プロトコルに含まれる探索端末は、CTX−1301第3段階計画におけるPROと同様の以前の治療に対する治療のユニークな利点および満足度を最適化するためにCTX−1302を定義し評価するであろう。
われわれ は,CTX−1302の505(B)(2)セキュリティプロトコルを参考薬としてDexedineSpansuleを用い,この薬物の有効性と安全性データをわれわれのCTX−1302臨床計画における生物利用度/生物学的同等性データと有効性/安全性データとともに承認の基礎とする予定である。
CTX−1302が完全な第3段階安全性および有効性研究パッケージを必要とする場合、同様の診断ツールおよびADHD−RS−5、CGI−SおよびPERMPを含むADHD評価がCTX−1301に組み込まれることが予想される。しかし、私たちの現在のCTX-1301臨床計画に対する理解に基づいて、私たちは再びFDAと連絡し、CTX-1302の代替、より時間的、より効果的な方法の指導を求めるかもしれない。
CTX-2103:不安関連障害を治療する候補ブチルスピレノン製品
私たちはすでに焦慮を治療するためのCTX-2103(ブチロシノン)の開発計画 を開始し、これはアメリカで最もよく見られる精神健康問題 であり、私たちはCTX-2103がブチルスピロ環ケトンの最初の毎日1回の処方になる可能性があると信じており、ブチルスピロ環ケトンは焦慮市場で最も広範な処方薬物の一つである。CTX-2103は1種の新型徐放錠であり、それは有効な薬物成分塩酸ブチロシノンを含み、1種の非ベンゾジアゼピン類薬物であり、依存或いは依存リスクが発生する証拠がない。しかし、ブチロシノンの半減期は非常に短いため、処方中に毎日何回ブスピレノンを服用して焦慮を制御することを規定しており、これは患者にとって1種の挑戦である可能性があり、そして 次の治療結果を招く可能性がある。CTX-2103は1日1回の多用量錠剤として設計され、私たちはそれが明らかな差別化 と現在利用可能な治療レジメンと比較した注目された利点を提供すると信じている。
| 76 |
我々のCTX-2103臨床開発計画は
2022年6月、私たちはCTX-2103の人体調合研究を完成した。CTX-2103の最初の人体被験者研究は単中心、開放ラベル、四腕交差研究であり、10名の健康被験者に関連する。各参加者は、異なる評価訪問中に4種類の異なる用量のブチロシノン :1つの4時間遅延したタイミングで10 mg錠剤を放出し、1つの8時間遅延したタイミングで10 mg錠剤 を放出し、1つの3パルス10 mg錠剤を0、4および8時間に薬物を放出し、および一般的なテトラスピロロン10 mg錠剤を放出した(参考製品、これは商業的処方である)。
主な目的は、遅延製剤中のブチルスピロロンの吸収と代謝産物である1-ピリミジニルピペラジン(1-PP)の血漿中の存在を評価し、点滅時間と放出データに関連付けることである。この研究の副次的な目標は、遅延ブチロシノン製品と市販製剤の薬物動態学的性能を比較することであり、また、この研究は、ブチロシノンの吸収状況および血漿中の三次放出製品の代謝産物1-PPの存在状況を評価する。
安全性 評価はCTX−2103が安全であり,耐性が良好であることを示した。この研究では,重篤,重篤あるいは臨床的意義のある治療緊急有害事象(“TEAE”)は発生していない。大多数のTEAEの重症度は比較的に軽く、ブチルスピレノンの予想イベントと一致した。TEAE、実験室検査、健康診断、心電記録とバイタルサイン(血圧と脈率)の評価により、ブチルスピレノンは安全性問題がないことを示した。
処方研究データに見られた薬物動態曲線に基づいて、CTX-2103は塩酸ブチロシノンの三次放出を実現した。この人体調合研究の陽性結果 は著者らがFDAとIND前会議を開催することを要求し、著者らのCTX-2103臨床と監督計画の設計に必要な重要な情報を提供した。IND前会議は2023年第4四半期 に開催された。著者らはFDAからCTX-2103の制御経路に関する意見、及び臨床研究の設計を受け、新薬(“IND”)を提出した。FDAのフィードバックによれば、CTX−2103は、通常505(B)(1)の全NDA経路よりも少ない時間およびリソースを必要とする505(B)(2)の経路で求められ、承認を得ることができる。
商業化する
私たちが置かれている発展段階を考慮して、私たちは現在、内部販売、マーケティング、流通インフラや能力を持っていない。CTX-1301および/またはCTX-1302が承認された場合、私たちの候補製品をアメリカで商業化することを計画しており、私たちは市場の許可を得た最初の国/地域になると予想している。私たちは彼らのbr能力から利益を得るための商業協力パートナーが必要であり、私たちにより直接的なマーケティング、販売、市場参入と流通インフラを提供し、私たちが大多数のADHD薬物の大量神経学と精神医学処方者および高処方ADHD小児科医と家庭医と交流できるようにする。
さらに、非個人戦略を含むマルチチャネル戦略を使用して、行動の推進を支援するために、適切な頻度で医師、支払人、患者、および患者介護者に接触することが予想される。個人の普及以外に、私たちは医学教育、直接マーケティング、定期刊行物広告と電子健康記録交流を通じて医者に接触するつもりである。団体,患者,介護者がADHD分野で非常に活発で歯に衣着せぬことを提唱している。患者に直接向けた戦略は、的確な教育および広告を通じて、適切なソーシャルメディアを使用して耳を傾け、参加することによって、これらの患者および介護者に通知するために、brという社会集団 に接触することができると予想される。
2023年3月、Indegeneと商業化協定 を締結しました。CTX−1301に対するFDAの承認を得た場合、Indegeneは、Indegeneによって実行されるサービス、そのようなサービスの配信内容、および支払うべき費用を含む作業説明書に従ってCTX−1301に商業化されたbr}サービスを提供する。Indegeneが提供すると予想される主要なサービスは、(A)医療事務および薬物警戒、(B)定価、精算および市場アクセス、(C)商業運営、および(D)マーケティング(現場力を含む)を含む。“材料プロトコル-Indegene,Inc.との共同商業化プロトコル”を参照。下です。Cingateはまた、戦略的製薬パートナー関係の構築を検討しており、このパートナー関係に基づいて、Cingateは米国、米国以外、または両者を兼ねてCTX-1301の許可を共同で普及または獲得する。Cingateはすべての方面で機会を探しており、私たちはすでに有名な業務発展会社のグローバル製薬パートナー(GPP)とパートナー関係を構築した。彼らは実行可能な許可パートナーを積極的に探している。
| 77 |
製造業
概要
私たち は現在製造工場を所有したり運営していません。我々はこれまで,我々のCDMOとして製薬製造研究サービス会社 を用い,臨床前研究や臨床試験のための製品の生産に用いてきた。2022年10月、私たちは、私たちの主要候補CTX-1301のすべての臨床、登録、および商業ロットを生産する新しいCDMOとして、社会CDMO,Inc.(“社会”)を保持した。Socialはジョージア州ガイエンスビルにある工場内で専用の設備を製造し、私たちが所有する独自の設備を備えている。
われわれの臨床試験で使用したどの第三者メーカー,施設およびすべてのロットの薬品や薬品もcGMPに適合しなければならない。CGMP規定には、人員、建物および施設の組織、設備、構成要素および薬品容器および閉鎖された制御、生産およびプロセス制御、包装およびラベル制御、保有および分配、実験室制御、記録および報告、ならびに返品または回収された製品に関する要件が含まれる。私たちの製品を生産する製造施設はcGMP要求とFDAの満足を満たさなければ、すべての製品を承認することができて、私たちは商業製品 を生産することができます。第三者製造業者はまた、当社の製品をテストして生産する際に使用するプログラムや操作を含むFDAや他の機関の施設の定期的な検査を受けて、私たちが適用される法規に適合しているかどうかを評価する。また,我々の薬品はII類制御物質に分類され,いずれの将来の第三者メーカーもDEAの承認と規制を受けなければならないことが求められている。
薬物br物質
我々は現在、CTX−1301(酢酸ジメチル)、CTX−1302(右旋フェニルプロピルアミン)およびCTX−2103に使用されている原料薬 および賦形剤を米国の第三者メーカーから購入している。私たちは将来、その中の多くのメーカーと商業供給協定を締結することを予想しています。br}CTX-1301とCTX-1302の原料薬はすべてアメリカ連邦の法律によって規制されています。DEAは酢酸ジメチルと右旋フェニルプロピルアミンを付表II規制物質に分類した。すべての刺激薬と同様に、乱用の可能性がある。したがって、 我々の候補製品の調達、製造、輸送、分配、貯蔵は規制され、 本入札説明書の他の部分の“DEA法規”部分はこれをより詳細に説明している。CTX−2103(ブチルスピレノン)の原料薬 は所定の制御物質ではないため,追加のDEA規定は適用されない。
知的財産権
専有 保護
私たちの商業成功は、私たちの候補薬物のために特許保護、製造と処理発見と他の技術を獲得し、維持する能力があるかどうかにある程度依存し、他人の独占権を侵害することなく運営し、他の人が私たちの独占権を侵害することを防止する。著者らはずっと私たちのADHD候補薬物に関連する知的財産権の組み合わせ、及び著者らの革新した独自のPTR薬物伝達プラットフォーム技術と私たちの技術プラットフォームを構築し、引き続き構築してきた。私たちの政策は、私たちのビジネス発展と実施に非常に重要な私たちのノウハウ、発明、改善に関連するアメリカおよびいくつかの外国特許出願を提出することによって、私たちの固有の地位を保護することを求めることです。私たちはまた、ビジネス秘密、技術的ノウハウ、持続的な技術革新、および潜在的な許可内の機会に依存して を発展させ、私たちの独自の地位を維持するつもりです。私たちは、私たちの任意の未解決特許出願または将来提出された任意の特許出願に特許が付与されるかどうかを決定することができず、また、私たちの任意の既存特許または将来私たちに付与される可能性のある任意の特許が、私たちの技術を保護する上で商業的用途を有することを保証することはできません。
| 78 |
特許 権利
我々はBDD Pharma(以下のように定義される)から許可を得ているか、米国で6つの特許および2つの特許出願を有しており、海外および地域で92件の特許および17個の特許出願を有している。米国に加えて、オーストラリア、ブラジル、カナダ、中国、エジプト、欧州(欧州特許庁に未解決の出願を提出し、欧州特許機関のいくつかの加盟国と特許検証を行った)、香港、イスラエル、インド、日本、メキシコ、ロシア、サウジアラビア、韓国 である。特許および特許出願は、候補製品の宣言、候補製品の製造方法、および候補製品を使用する治療方法を含む、我々の候補製品、我々のPTR薬物送達プラットフォーム、および我々のEBLのいくつかの特性を説明し、主張する。
私たちは、私たちの革新のための特許出願を提出し、私たちの未解決の特許出願を起訴し、私たちが発行した特許を維持し、実行することを含む、私たちの知的財産権を積極的に保護していきます。処理すべき特許出願 が特許発行をもたらすことが保証されないか、または審査過程がクレーム範囲の縮小を要求しない。また、発行された特許は、第三者によって回避される可能性があり、または裁判所または行政機関が異議を提起したときに実行不可能または無効が発見される可能性がある。したがって、第三者に対して当社の特許権を実行することに成功することができない可能性がある。他の会社が我々に付与される可能性のある任意の特許をめぐって類似または競争の技術や設計を独立して開発しない保証はない。
特許寿命の決定は、特許期限の調整及び延長など、出願の提出日及び特許法公布の他の要因に依存する。米国を含む多くの国/地域では,特許期間は一般に適用国/地域非臨時特許出願の最初の提出日から20年である。私たちが所有または許可している特許と特許出願 の有効期限は2031年から2036年までです。
私たちが持って許可された特許と特許出願の要約は以下の通りだ。
| 家庭/PCT アプリケーション | “タイトル”/(特許保護タイプ ) | 出願人/所有者 | 個の申請を処理中です | Br件の特許が発行された | 特許が満期になる | |||||
| WO 201107750 | 遅延投与“(遅延のためのプレスコーティングシート配合物、その後、活性物質の放出を延長する) | 国際薬品配達有限会社 | ドイツイギリス フランス、日本、 スイス、 アメリカ |
2031年3月 | ||||||
| WO 2011107749 | パルス薬物放出“(放出を遅延させるための錠剤製剤、次いで活性物質のパルス放出) | 国際薬品配達有限会社 | オーストリアベルギー ブルガリアチェコチェコ デンマーク、 フィンランド フランス、 ドイツ 偉大なイギリスギリシャ ハンガリーアイルランド イタリア、日本、 オランダノルウェー ポーランドポルトガル ルーマニアスロバキア スロベニアスペイン スウェーデンスイススイス トルコ、 アメリカ |
2031年3月 | ||||||
| WO 2011107755 | “即時遅延放出”(遅延放出のための錠剤配合物、次いで活性物質をパルス放出する) | 国際薬品配達有限会社 | オーストリアベルギー ブルガリアチェコチェコ デンマーク、 フィンランド フランス、 ドイツ 偉大なイギリスギリシャ ハンガリーアイルランド イタリア、日本、 オランダノルウェー ポーランドポルトガル ルーマニアスロバキア スロベニアスペイン スウェーデンスイススイス トルコ、 アメリカ |
2031年3月 | ||||||
| WO 2016075496 | “製薬br加工”(制御放出材料の製造方法) | 薬品国際配達有限会社 | ヨーロッパ.ヨーロッパ | アメリカ アメリカ | 2035年11月 |
| 79 |
| 家庭/PCT アプリケーション | “タイトル”/(特許保護タイプ ) | 出願人/所有者 | 個の申請を処理中です | Br件の特許が発行された | 特許が満期になる | |||||
| WO 2016075495 | “組成物” (有効成分の遅延放出のための錠剤) | 薬品国際配達有限会社 | エジプト |
オーストラリアオーストリア ベルギーブラジルブルガリア カナダチェコ共和国デンマーク フィンランドフランス ドイツ、イギリス、 ギリシャ、香港 ハンガリーインドアイルランド イタリア、日本 メキシコオランダ ノルウェーポーランド ポルトガルルーマニア ロシア、サウジアラビア スロバキアスロベニア 韓国スペインスウェーデン スイストルコトルコ アメリカです |
2035年11月 | |||||
| WO 2016075497 | “錠剤” (ワックス、崩壊剤および治療剤からなる徐放錠) | 薬品国際配達有限会社 | ヨーロッパ.ヨーロッパ | アメリカ アメリカ | 2035年11月 | |||||
| WO 2016138440 | “TriPulse 覚せい剤放出レシピ” | バックル治療有限責任会社 |
中国ヨーロッパ 香港インド 日本、 韓国、アメリカ |
オーストラリア、 カナダ イスラエル |
2036年2月 (リリース時) | |||||
| WO 2022/240849 | 3モード、 正確なタイミングパルス放出シート | バックル治療有限責任会社 | オーストラリア、ブラジル、カナダ、中国、ヨーロッパ、イスラエル、インド、韓国、メキシコ、アメリカ | 2042年5月(リリース時) | ||||||
| WO 2023/158694 | 三型、正確なタイミングで錠剤を放出する | バックル治療有限責任会社 | 国際パーセント | 2043年2月(リリース時) |
機密とその他の保護を取引する
特許知的財産権に加えて、私たちは特に特許保護が適切であるか、または入手可能であると信じないとき、私たちの技術を保護し、私たちの競争的地位を維持するために、商業秘密とノウハウに依存している。私たちの政策は、私たちの従業員、コンサルタント、コンサルタントが、私たちと雇用、コンサルティング、またはコンサルティング関係を構築し始める前に秘密および発明譲渡協定に署名することを要求します。合意は、個人が限られた場合でない限り、個人が私たちとの関係の過程で開発または知った任意の機密情報を他の当事者に開示してはならないことを秘密にしなければならないと規定されている。これらのプロトコルは、一般に、個人がサービスを提供する過程で発想されたすべての発明を有することを規定している。
その他 知的財産権
私たち は適切な時にアメリカで商標保護を求めている。私たちは、薬物研究開発および製品で使用されているストラップ、ストラップ治療商標、商標申請商標保護、および私たちの潜在的製品と一緒に使用される可能性のある商品名を提供しています。我々は現在米国でCingate,Cingate Treeutics,CTXとMASTHY,および我々のPTR技術の登録商標を持っている。
私たちは時々第三者知的財産権所有者から許可を得ることが必要または慎重だということを発見するかもしれない。
| 80 |
競争
私たちの業界は先進的な技術、激しい競争、そして独自製品に対する高度な重視を例示している。私たちはいくつかの短時間効果と徐放ブランド製品 が様々な配合を持っているため、製薬会社と後発薬会社からの競争に直面する可能性があり、その中のいくつかは非常に革新的であり、これらの製品の後発薬バージョンはまだ医療の需要を満たしていないからである。著者らは、候補製品の開発と商業成功に影響する重要な競争要素は: 経口投与、即時作用と全有効日持続時間を含む治療効果、安全性と耐性、br}市場参入と定価を含むと信じている。いくつかの競争相手は私たちよりも多くの財力、技術、人的資源を持っている;しかし、私たちはブランド競争のレベルが低下しており、Vyvanse独占経営権の喪失に伴い低下し続けると考えている。また、我々の潜在的な競争相手は、FDAや外国規制機関のマーケティング承認を得る上でも、より多くの経験や専門知識を持っている可能性がある。製品開発、テスト、承認、普及に加えて、製薬業界の他の競争要因には、統合、製品品質と価格、製品技術、名声、顧客サービス、技術情報の取得が含まれている。したがって、私たちの潜在的な競争相手は、競争相手またはより優れた製品を開発し、より積極的な競争を行い、私たちよりも長い間その競争優位性を維持することができるかもしれない。競争に直面して、私たちの製品は時代遅れになったり、経済効果が足りないかもしれません。
承認された場合、CTX−1301およびCTX−1302の両方は、現在市販されている、ブランドおよび非特許のピペラメチルおよびフェニルプロピルアミン製品と競合してADHDを治療するであろう。その中のいくつかの現在利用可能な製品は、JanssenのConcerta、ノワールのFocalin XR、武田のAdderall XRおよびVyvanseを含み、これらの製品は独占経営権を失っている。
近年、ADHD市場は多くの革新が出現したがニッチ市場に集中したADHD製品であり、これらの製品は以前の経口覚せい剤、特に徐放経口覚せい剤の市場シェアを占めていない。私たちはAytu、Tris、Clom、Ironshore、Rhodesを含むADHDに集中した小型バイオテクノロジー会社からの競争に直面していることに気づいた。しかし、私たちは、これらの会社の大多数が重要な競争相手であるとは思いません。それらの製品は市場全体のほんの一部を占めることができ、大量のビジネス努力を投入していないので、私たちの候補製品は、多くのADHD患者が長期的に満足していない需要を克服する潜在力を持っていると信じています。また,Cingateは潜在的な商業化や戦略的パートナー と適切な資源を利用してその資産を商業化することに成功する予定である.
FDAは最近,非特許徐放錠ピペラゾンの生物学的同等性試験に関する改訂ガイドラインを発表した。この新しいガイドラインは新しい模造薬を参考製品との生物学的同等性を証明することを困難にした。これはメチルフェニデート市場での模倣薬競争を制限すると考えられる。後発薬では,CTX−1301などの完全な有効日有効期限を有する新ブランド,ピペラゾン徐放デキサメタゾン薬との生物学的同等性を示すことは困難である可能性がある。
政府の法規
アメリカ連邦、州と地方の各級及びその他の国の政府当局は薬品の研究、開発、テスト、製造、品質管理、承認、ラベル、包装、貯蔵、記録保存、販売促進、広告、流通、承認後の監督と報告、マーケティングと輸出入などの方面に対して監督と監督を行った。通常、新薬 が発売される前に、その品質、安全性と有効性を証明するデータを大量に得なければならず、各監督管理機関に対する特定のフォーマット に組織され、審査を提出し、最終的に適用監督機関の許可を得る。
アメリカ薬品開発
米国ではFDA はFDCAとその実施条例により薬品を管理している。薬品はまた他の連邦、州、そして地方法規と規制によって制限されている。監督部門の承認を得て、適用される連邦、州と地方法規を後続的に遵守する過程を維持するには、大量の時間、人員と財力が必要だ。これらの機関及びその他の連邦、州と地方実体は薬品の研究開発活動及びテスト、製造、品質管理、ラベル、貯蔵、包装、記録保存、追跡、審査、輸入、輸出、流通、広告と販売促進を監督する。製品開発,承認過程または承認後のいつでも, が適用される米国の法規要求を遵守できない場合,出願人は行政または司法制裁を受ける可能性がある.他の行動に加えて、これらの制裁は、FDAが承認保留申請の拒否、承認撤回、臨床棚上げ、無タイトルまたは警告状、自発的製品のリコールまたは市場撤回、製品差し押さえ、生産または流通禁止の完全または部分的な一時停止、罰金、br}同意法令、政府契約の拒否、原状回復、返還、返還または民事および刑事罰を含む可能性がある。どんな機関や司法機関の法執行行動も私たちに実質的な悪影響を及ぼすかもしれない。
| 81 |
薬品 候補製品はNDAプロセスでFDAの承認を得なければなりません。その後、アメリカで合法的に販売·販売することができます。 Cingulateは505(B)(2)規制承認経路に従って私たちのNDAを提出する予定です。薬物の開発と承認は一般的に以下の内容に関連する
| ● | 良好な実験室規範(“GLP”)或いは他の適用法規に基づいて臨床前実験室テスト、動物研究と調合研究を完成した | |
| ● | ヒトに関する臨床試験が開始される前に発効しなければならないINDをFDAに提出する |
| ● | 臨床試験地点ごとに試験を開始する前に,臨床試験地点ごとの独立機関審査委員会(“IRB”)または倫理委員会の承認を得た | |
●
● |
適用されたIND法により十分かつ制御された良好なヒト臨床試験が行われている。アドバイスごとの適応に対する研究製品の安全性と有効性を評価するために、他の良好な臨床実践(“GCP”)および他の臨床試験に関する規定を評価する
編集は、製品が正確な調合、製造、保存できることを証明した |
| ● | アプリケーション使用料の支払いを含む上場承認を得るためにFDAにセキュリティプロトコルを提出すること |
| ● | 満足できるbrは、医薬品を製造するためのFDAの1つまたは複数の製造施設の承認前検査を完了し、brがcGMPに適合するかどうかを評価し、施設、方法、および薬物の特性、強度、品質、および純度を維持するのに十分な制御を保証する | |
| ● | FDAは、GCPに適合することを保証し、NDAサポートで提出された臨床データの完全性を確保するために、臨床試験場所を監査することができる | |
| ● | FDA は、適切または適用された場合に、製品が米国で任意の商業マーケティングまたは販売を行う前に、FDAコンサルタント委員会による候補製品の審査を満足的に完了することを含むNDAを審査および承認する。 |
臨床前研究
人体で任意の候補薬物をテストする前に、厳格な臨床前テストを経なければならない。臨床前開発段階は通常薬物化学、製剤と安定性の実験室評価、及び動物に対する毒性評価研究を含み、これらの研究は後続の臨床試験を支持する。スポンサーは、臨床前研究の結果を、生産情報、分析データ、任意の利用可能な臨床データまたは文献、および提案された臨床案と共にINDの一部としてFDAに提出しなければならない。INDはFDAがヒトに研究製品の使用を許可する要求であり,ヒト臨床試験が開始される前に発効しなければならない。
臨床前研究は候補製品の化学と調合の実験室評価、及び体外と動物研究を含み、不良事件の可能性を評価し、そしてある場合に治療使用の理論基礎を確立する。臨床前研究の進行は連邦法規と要求の制約を受け、GLPの安全性と毒理学研究に関する規定を含む。いくつかの長期的な臨床前テスト、例えば生殖不良事件と発ガン性の動物テストは、薬物のINDを研究してFDAに提出し、人体臨床試験を開始した後も引き続き行う可能性がある。
テストデータが505(B)(2)NDAをサポートする場合、RLDの安全性および有効性に関する文献またはFDA前の発見において、必要な臨床前データの一部または全部が参照される可能性がある。
| 82 |
臨床試験
すべての臨床試験は合格研究者の監督の下で行われなければならない。臨床試験は,研究目標を詳細に説明し,評価する安全性と有効性基準のパラメータをモニタリングするためのシナリオに基づいて行った。すべての合意はINDの一部としてFDAに提出されなければならない。対象者は臨床試験に参加する前にインフォームドコンセントに署名しなければならない。現在行われている臨床試験や臨床試験結果を公的登録機関に報告することも要求されている。それまでFDAが1つまたは複数の提案された臨床試験に懸念や問題を提起し、臨床試験を保留しない限り、FDAはFDAが受信してから30日後に自動的に発効する。この場合,INDスポンサーやFDAは臨床試験開始前に未解決の問題を解決しなければならない。したがって,INDの提出はFDAが臨床試験の開始を許可しない可能性がある。FDAは、安全考慮または要件を満たしていないために、研究前または研究中の任意の時間に臨床保留を強制的に実施することもできる。
また,臨床試験に参加する各機関を代表するIRBは,臨床試験が開始される前に任意の臨床試験の計画を審査·承認しなければならず,その後IRBは少なくとも毎年継続的な審査と再承認試験を行わなければならない。IRBは臨床試験被験者に提供する試験案やインフォームドコンセント情報などを審査·承認しなければならない。IRBの運営はFDAの規定に適合しなければならない。いくつかの臨床試験に関する情報は、プログラムの詳細および最終的な研究結果を含み、ClinicalTrials.govデータレジストリ上で公開されるために、特定の時間枠内で米国国立衛生研究院に提出されなければならない。臨床試験登録の一部として,製品,患者群,調査段階,研究場所や研究者,臨床試験の他に関する情報を公開する。スポンサーも完成後にその臨床試験の結果を開示する義務がある。場合によっては、これらの試験結果の開示は、試験完了日後2年に延期することができる。法律規定に従って登録された臨床研究や研究結果の提出が間に合わないと民事罰金を招く可能性があり、また、コンプライアンス側が連邦政府の将来の支出を獲得することを阻止する。米国国立衛生研究院(NIH)のClinicalTrials.gov登録と報告要求に関する最終規則が2017年に発効し,NIH,FDAともに,政府は要求に適合しない臨床試験スポンサーに対してこれらの要求を開始したいと述べている。
NDAを支持するための臨床試験は通常3つの連続段階で行われ,これらの段階が重なる可能性があり,合併する可能性もある。
| ● | 段階1-臨床試験は、一般に、単剤または多剤候補製品と最初に接触する比較的少ない数の健康ボランティアに関する。これらの臨床試験の主な目的は安全性、用量耐性、構造効果の関係、作用機序、吸収、代謝、分布と排泄を評価することである。候補製品または特定の患者集団に適用される汎用フォーマット変更は発生する可能性があるが、FDAの同意を事前に得られることが多い。 | |
| ● | 第2段階-臨床試験は通常、限られた患者集団で研究を行い、可能な副作用と安全リスクを決定し、特定の目標疾患に対するこの製品の治療効果を初歩的に評価し、用量耐性と最適用量を決定することに関連する | |
| ● | 第三段階-より大きな被験者集団において臨床試験を行い、拡大した患者集団における用量、臨床治療効果、および安全性をさらに評価するために、通常、地理的に分散した臨床研究場所である。これらの研究は、候補製品の全体的なリスク-収益比率を決定し、適切な場合に製品ラベルに十分な基礎を提供することを目的としている。これらの試験は、プラセボおよび/または他の対照治療との比較を含むことができる。治療期間は通常、マーケティング過程における製品の実際の使用状況をシミュレートするために延長される。これらの試験は、グローバルサイトも米国人口の代表性を有し、グローバルサイトで行われる研究がGCPに準拠するようなFDAの法規およびガイドラインに適合する限り、グローバル登録をサポートするために世界的に行うことができる。 |
| 83 |
505(B)(2)規制承認経路に従うことによって、出願人は、出願人によって行われず、出願人が参照権を得ていない調査に依存することによって、例えば、RLDに関する以前の調査のような薬物開発のいくつかの負担を軽減することができる。この場合、いくつかの臨床試験は必須ではないか、または制限される可能性があるが、一般に、候補製品のバイオアベイラビリティおよび薬物動態学的特徴を決定する第1段階試験および 505(B)(2)セキュリティプロトコルをサポートするための少なくとも1つの第3段階キー試験が必要である。
承認後 実験は,ステップ4と呼ばれる場合があり,初期マーケティング承認後に行うことができる.これらの試験は,期待される治療適応患者の治療から追加的なbr経験を得るために用いられている。場合によっては,FDAはNDAを承認する条件として4期臨床試験の性能を強制的に要求する可能性がある。
新薬製品の開発期間中、スポンサーは、INDの提出前、第2段階の終了時、およびNDAの提出前を含むFDAと会う機会がある。これらの会議はスポンサーに機会を提供し,スポンサーにこれまで収集してきたデータに関する情報を共有させ,FDAにアドバイスを提供させ,スポンサーとFDAに次の段階の開発について合意させることができる。スポンサーは通常、3期臨床試験を開始する前にこの機関と会い、彼らの重要な試験計画を提出し、新薬製品の承認を支持すると考えている。
臨床試験と同時に、会社は通常追加の動物研究を完成し、また薬物製品の物理特性に関する追加情報を開発し、cGMP要求に基づいて最終的に商業大量生産製品の技術を確定しなければならない。製造過程は一貫して高品質の候補製品ロットを生産できる必要があり、また、メーカーは最終薬物製品の特性、強度、品質、純度をテストする方法を開発しなければならない。br}また、適切な包装を選択し、テストしなければならず、候補製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
NDAとFDA審査の流れ
必要な臨床試験の完了に成功したと仮定し,臨床前研究と臨床試験の結果および製品の化学,製造と制御の詳細な説明,提案のラベルおよびその他の関連情報は,その製品の発売承認を要求するセキュリティプロトコルの一部としてFDAに提出される。秘密協定を準備して提出する費用は巨大だ。連邦法によると,多くのNDAの提出には高額な申請使用料が必要であり,現在臨床情報を含むNDAの使用料は324万ドルを超えており,承認されたNDA下のメーカーや/またはスポンサーも計画年費 を支払う必要があり,現在約394,000ドルである。これらの費用は通常毎年増加します。場合によっては費用 を減免することができる.このような費用減免は、小規模企業の申請者に適用され、これは、申請者(任意の付属会社を含む) 従業員が500人未満であり、州間商業を導入するために導入または交付された製品について承認されていないマーケティング申請 について、その最初のマーケティング申請を提出していることを意味する。
FDCA“第505(B)(1)節及び505(B)(2)節は、FDCAに従って提出可能な国家開発援助の種類に関する規定である。第505(B)(1)節は、他の新薬が同じ有効薬物成分または活性部分、すなわち薬物物質の作用を担う分子またはイオンがFDAの承認を得ていない場合の、新しい化学物質の伝統的な経路である。FDAが以前に承認された製品の新しい処方または製剤を改善する代替経路として、会社は505(B)(2)の セキュリティ協定を提出することができる。第505条(B)(2)条は、承認に必要な情報の少なくとも一部が、出願人または出願人のための研究からではなく、出願人が参照権を得ていない場合に機密協定の提出を許可する。
FDAが申請を受信すると、届出申請を受ける前に、それが実質的な審査を可能にすることが実質的に完了したかどうかを決定するために60日間の機密協定を検討し、申請を受け入れるのではなく、より多くの情報を提供することを要求することができる。提出された申請が受け入れられると、FDAは秘密保持協定の深い検討を開始する。FDAが“処方薬使用料法案”(PDUFA)VIに基づいて合意した目標と政策によると、FDAは、申請受理日から10ヶ月以内に標準審査薬物製品の申請を審査し、申請受付日から6ヶ月以内に優先審査薬の申請を審査することを求めている。br}FDAは、重篤な疾患の治療を目的とした薬物に優先審査指定を付与することができ、brが治療に重大な進展を遂げているか、または適切な治療なしに治療を提供することを決定することができる。FDAは、基準および優先NDAのPDUFA目標日 を常に達成するわけではなく、FDAは、追加の遅延提出の情報を考慮して、またはFDA審査問題の提出において提供された情報の情報 を明らかにするために、基準および優先審査の審査プロセスをさらに3ヶ月間延長することができる。
| 84 |
セキュリティプロトコルを承認する前に、FDAは、最近FDAの検査を受けない限り、候補製品の製造施設を承認前に検査して、cGMPに適合しているかどうかを決定するのが一般的である。FDAは、製造プロセスおよび施設がcGMP要件に適合していると判断しない限り、この製品 を承認せず、 製品生産が規格に適合していることを保証するのに十分である。さらに、FDAは、安全性または有効性の問題を提起する新薬製品または医薬製品の出願を諮問委員会に提出することができ、一般に、臨床医および他の専門家を含むグループであり、申請が承認されるべきかどうかを審査、評価、および提案するために、どのような条件下で承認されるべきかを審査する。FDAは、諮問委員会の提案に制限されていないが、決定を下す際にこれらの提案を慎重に考慮する。第505条(B)(2)条に基づいて提出されたNDAは、審査期間中に新たなセキュリティ情報 が開示されない限り、諮問グループ審議に通常提出されない。
FDAはセキュリティプロトコル審査過程の一部として臨床試験データを再分析する可能性があり,FDAと申請者の間で広く議論されている可能性がある。さらに、FDAは、GCPおよびINDレジメンの要件に適合することを保証し、FDAに提出された臨床データの完全性を保証するために、通常、1つまたは複数の臨床サイトを検査する。FDAによる秘密保持プロトコル の審査と評価は広く時間がかかり,元の計画よりも長い時間がかかる可能性があり,タイムリーに の承認を得ることができない可能性がある.
FDAはセキュリティプロトコルを評価した後,承認書または完全な返信状(“CRL”)を発行する.この薬剤の商業マーケティングを承認し、特定の適応の処方情報を提供する。CRLは,申請の審査周期が完了したことを示しており,現在の形で申請を承認することはない.CRLは、通常、FDAによって決定されたNDAにおける欠陥を記述し、大量の追加の臨床データ、または臨床試験、非臨床研究、または生産に関連する他の重要で時間のかかる要件を必要とする可能性がある。さらに、CRLは、申請を承認条件の下に置くために、申請者がとりうる提案操作 を含むことができる。CRLが発行された場合,申請者は秘密保持プロトコルを再提出し,手紙で指摘されたすべての欠陥を解決したり,申請を撤回したりすることができる.またはこれらの欠陥がNDAの再提出時にFDAによって満足的に解決された場合、FDAは、出願人に承認書を発行するであろう。 FDAは、そのような再提出を、2ヶ月または6ヶ月以内に含まれる情報タイプ に基づいて検討することを約束している。しかし,この補足情報を提出しても,FDAはNDAが承認された規制基準を満たしていないと認定する可能性がある。臨床試験から得られたデータは常に決定的ではなく,FDAのデータ解釈はスポンサーの同じデータに対する解釈とは異なる可能性がある。
FDAが候補製品の発売を承認する保証はなく,スポンサーが審査中に重大な困難や コストに遭遇する可能性がある。製品が発売承認されても,承認は明らかに特定の疾患や投与量に限られている可能性があり,あるいは使用適応が制限される可能性があり,製品の商業的価値を制限する可能性がある。また,FDAは製品ラベルに何らかの禁忌症,警告または予防措置を含めることを要求する可能性があり,提案ラベルの変更を条件に承認する可能性もある.FDAはまた、潜在的な効果および治療効果を監視するために、適切な生産制御および規格 を策定し、上場後の試験および監視を承諾することを許可することが可能である。例えば、FDAは、薬物の安全性および有効性をさらに評価するための4期試験を要求する可能性がある。
承認されたラベルに加えて、FDAは、薬剤の利益がそのリスクよりも大きいことを保証するために、REMS計画の形態で製品の流通、処方、または配布に制限および条件を適用することも可能である。REMSは、患者の薬品使用ガイドライン、衛生保健専門家のコミュニケーション計画、および/または安全使用を確保する要素(“ETASU”)を含むことができる。ETASUは、処方または調剤の特殊トレーニングまたは認証、調剤要件の制限、特定の場合のみの調剤、特殊な監視、および患者登録簿の使用を含むことができるが、これらに限定されない。FDAは具体的な状況に応じてREMSの要求 および具体的なREMS条項を決定する.FDAがREMS計画が必要であると結論した場合,NDAのスポンサーは提案したREMS計画を提出しなければならない。必要であれば、FDAは、承認されていないREMSなしにNDAを承認しないだろう。我々の候補製品(酢酸ジメチルおよび右旋フェニルプロピルアミン)と同じ医薬物質を含む許可された医薬品ラベルに要求される警告によれば、NDAの審査および承認プロセスの一部として、FDAは、少なくともいくつかの候補製品、特にCTX-1301およびCTX-1302を要求し、ブラックボックス警告をそのラベルの一部とすることが予想される。
| 85 |
上記の承認またはマーケティング制限のいずれも、製品の商業普及、流通、処方、または配布を制限し、商業成功を制限することが可能である。法規の要求を満たしていない場合や、初期マーケティング後に問題が発生した場合、マーケティング承認を撤回する可能性があります。
NDAの承認後、新しい適応、製造変更、およびタグ付け宣言を追加するなど、承認された製品のいくつかのタイプの変更は、さらなるテスト要件、FDA通知、およびFDA審査および承認の制約を受ける。さらに、新しいセキュリティ情報が発生した場合、追加のテスト、製品ラベル、またはFDA通知が必要となる可能性がある。
“ハッジ·ワックスマン法案”は新薬マーケティングと排他的だ
1984年の“薬品価格競争と特許期限回復法”によると、“食品薬品監督管理局のハッジ·ワックスマン改正案”とも呼ばれ、国会 はFDAが以前“薬品価格競争と特許期限回復法”法規の守秘協定条項に基づいて承認した薬品と同じ模造薬を許可し、“食品薬品監督管理局”第505(B)(2)条を公布した。後発薬の承認を得るためには,出願人は当該機関にANDA, を提出しなければならない。このような応用を支持するために、模倣薬製造業者は、以前に秘密プロトコル(RLDと呼ばれる)によって承認された医薬製品に対する臨床前および臨床試験に依存することができる。具体的には,ANDAを承認するためには,FDAは後発薬が有効成分,投与経路,剤形, と薬物強度においてRLDと同様であることを発見しなければならない。対照的に、第505条(B)(2)条は、承認が必要な情報の少なくともいくつかが、出願人によって行われていないか、または出願人のための研究から行われており、出願人が参照権を得ていないことを許可する。 第505条(B)(2)条の出願人は、以前に承認された製品に対する研究の依存を決定することができる場合には、科学的に適切であると判断することができる。生物等価型革新薬物の開発者が使用するANDA経路とは異なり、ANDA経路は、申請者がバイオアベイラビリティまたは生物学的同等性データ以外の新しい臨床データを提出することを許可しない、505(B)(2)規制経路は、後続申請者が追加のbr臨床試験または非臨床研究を行う必要がある可能性を排除しない;例えば、彼らは、以前に承認された薬物を新たな適応brまたは安全性または有効性を証明するために新たな臨床データに使用するための承認を求めることを求めている可能性がある。次に、FDAは、RLDによって承認されたラベル適応の全部または一部に対して、または 第505条(B)(2)条の出願人が求める任意の新しい適応に適用して、新製品を承認することができる。
NDAまたはその付録の承認を求める場合、NDAスポンサーは、スポンサーをカバーする製品または承認された製品使用方法の各特許をFDAにリストしなければならない。NDAの承認後、薬物出願 に記載された各特許は、FDA出版物“治療同等性評価を有する承認された医薬製品”(一般に“オレンジマニュアル”と呼ばれる)で公表される。ANDA出願人がFDAに出願する場合、出願人は、RLDオレンジブックに記載されている任意の 特許をFDAに証明する必要があるが、後続の出願人が承認を求めていない使用方法の特許は除外される。第505条(B)(2)の出願人が承認された製品の検討に依存する場合、このような出願人 はまた、オレンジマニュアルに記載されている当該承認された製品の任意の特許をFDAに証明する必要があり、その程度はANDA出願人と同程度である。
具体的には、ANDAまたは505(B)(2)NDAがオレンジブックに列挙された薬物を参照する任意の出願人は、各発表された特許について、(I)RLDの元の出願人が必要な特許情報を提出していないこと、(Ii)列挙された特許が期限切れであること、(Iii)記載された特許は期限が切れていないが、指定された日に期限が切れ、特許が満了した後に承認を求めることをFDAに証明しなければならない。又は(4)に掲げる特許は、無効、強制執行できない、又は新製品の製造、使用又は販売によって侵害されることはない。これらの認証をそれぞれ第I,II,III,IV段認証と呼ぶ.
| 86 |
第1の段落または第2の段落の認証が提出された場合、FDAは、審査が完了した直後に申請を承認することができる。第3項の証明書を提出した場合は,承認は出願中に指定された特許満期日に発効することができるが,暫定承認はその日までに発行することができる。出願が第4項の認証を含む場合、一連のイベントがトリガされ、その結果、ANDAまたは505(B)(2)出願の承認発効日が決定される。
新製品がRLDに記載されている特許またはこのような特許を侵害しない無効な認証を第(Br)段IV認証と呼ぶ。後続の出願人が第4段落の認証をFDAに提供した場合、FDAが出願人のNDA届出を受けた後、出願人はまた、第4段落の認証の通知 をNDAおよびRLDの特許所有者に送信しなければならない。そして、NDAおよび特許所有者は、第4項の認証に法的挑戦を開始することができる。第4項の認証を受けてから45日以内に特許侵害訴訟を提起すると、FDAがANDA又は505(B)(2)NDAを承認することを自動的に阻止し、第4項の通知、特許満了又は侵害事件においてANDA又は505(B)(2)出願人に有利な裁決を受けてから30ヶ月後である。代替的に、列挙された特許所有者が要求された45日間の期間内に特許侵害訴訟を提起しなかった場合、後続の出願人のANDAまたは505(B)(2)NDAは、30ヶ月の猶予期間の制限を受けない。
さらに、Hatch−Waxman修正案によれば、FDAは、参照されるRLDの任意の適用可能な非特許固有期間が満了するまで、ANDAまたは505(B)(2)NDAを承認しない可能性がある。FDCA下のこれらの市場排他性条項はまた、いくつかの申請の提出または承認を延期する可能性がある。FDCAは、新しい化学物質を含む薬物のNDA承認を得た最初の出願人に、5年間の米国内の非特許マーケティング排他性を提供している。FDA が以前に同じ活性部分を含む他の新薬を承認していなかった場合、薬物は、医薬物質の作用を担う分子またはイオンである新しい化学物質である。排他期間内に、FDAは、出願人が承認を得るために必要なすべてのデータ を得るために、出願人が合法的な参照権利を有していない場合、または薬剤の別のバージョンのために提出されたANDAまたは505(B)(2)NDAを受け入れない可能性がある。しかしながら、出願が特許無効または非侵害の証明を含む場合、4年後に提出することができる。
FDAが、出願人が行っているまたは後援する新しい臨床研究(バイオアベイラビリティ研究を除く)が申請を承認するために重要であると考えている場合、FDCAはまた、NDA、505(B)(2)NDAまたは既存のNDAの補充のために、既存の薬剤の新しい適応、用量または強度のような3年間の市場排他性を提供する。この3年間の専門権は、新しい臨床研究に関連する使用条件のみをカバーしており、FDAが原始活性成分を含む薬物の後続申請を許可することを禁止していない。5年および3年の排他性も、FDCA第505条(B)(1)に従って提出された従来のセキュリティ協定の提出または承認を延期することはない。しかしながら、従来のセキュリティプロトコルを提出する申請者は、安全性および有効性を証明するために必要なすべての臨床前研究および十分かつ制御された臨床試験を行うか、または参照権を得ることが要求される。
特許延期
国家薬品監督管理局の承認後,関連薬物特許の所有者は最長5年間の特許期間延長を申請することができる。許容される特許期間延長 は、薬物試験段階の半分−IND発効とNDA提出との間の時間− とすべての審査段階−NDA提出と承認との間の時間として計算され、最長5年である。FDAが、出願人が職務調査を経て承認を求めていないと判断した場合、時間 を短縮することができる。展示期間後の総特許期間は 14年を超えてはならない。出願段階で満了する可能性のある特許については,特許権者は臨時特許延期を請求することができる。臨時特許延期は特許期間を1年延長することができ、最大4回更新することができる。臨時特許が付与されるごとに,承認後の特許展示期間は1年減少する.特許商標局(特許商標局)の取締役は、特許出願延期された特許に含まれる薬剤が承認される可能性が高いことを決定しなければならない。秘密保持協定がまだ提出されていない薬物については、一時特許延期は利用できない。
| 87 |
小児科臨床試験と排他性
“小児科研究公平法”(“PREA”)によれば、NDAまたはいくつかのタイプのNDA補充剤は、すべての関連する小児科亜群においてこの薬剤が主張する適応の安全性および有効性を評価し、製品が安全で有効な各小児科亜群の用量および投与をサポートするためのデータを含まなければならない。FDAは小児科データの提出を延期することを許可するか、またはすべてまたは部分的な免除を与える可能性がある。
PREAの要求を満たすためには、検討すべき適切な年齢範囲が異なる可能性があり、具体的には、薬物または生物製品の薬理作用、異なる年齢群における疾患の発現、および治療反応を測定する能力に依存する可能性がある。PREAは、小児科評価を収集することを要求し、“評価が必要な各年齢群の適切な処方を使用する”(同法第505 B(A)(2)(A)節)。PREAによれば、出願人は、その小児科研究で使用される小児科処方 の承認を申請しなければならず、このような要求を提出しない場合、製品ブランドミスを引き起こす可能性がある(同法第505 B(D)節)。br}FDAは、“小児科製剤の承認を要求する”という言葉を、出願人が小児科研究を行うための任意の未承認処方(S)のための出願 または補足出願を提出しなければならないと解釈する。
2012年7月9日に法律となった“食品·薬物管理局安全·革新法案”に署名してFDCAを改正し、新活性成分、新適応、新剤形、新投与案または新投与経路のNDAのNDAのスポンサーに第2段階会議終了後60日以内に予備小児科研究計画(IPSP)を提出することを要求し、このような会議がなければ、第3段階または第2/3段階試験開始前に早期に初歩小児科研究計画を提出することを求めた。初期PSPは、目標および設計、年齢層、関連終点および統計方法、またはそのような情報を含まない理由、および小児科評価を遅らせる任意の要求 または全部または一部が小児科試験データを提供する要求および支援情報を免除することを含む、br}スポンサー計画によって行われる小児科試験(S)の概要を含まなければならない。FDAとスポンサーはPSPについて合意しなければならないが,非臨床研究,早期臨床試験,他の臨床開発計画から収集したデータから小児科計画の変化を考慮する必要があれば,スポンサーは合意した初期PSPに対する修正案を随時提出することができる。私たちは私たちのIPSPを提出して、それは受け入れられた。私たちはFDAと私たちのPREA義務について議論し続けた。
小児最適医薬品法は、FDAと達成された小児科書面請求を満たす小児科試験(S)を含むいくつかの条件が満たされている場合、NDA保有者は、その薬剤の任意の独占特許または非特許brを6ヶ月延長することができる。小児科独占試験の条件は、FDAが小児科集団における新薬の使用に関連する情報がその集団の健康に利益をもたらす可能性があることを決定することを含み、FDAは小児科臨床試験の書面請求を行い、申請者は法定時間内に要求された臨床試験を行うことに同意することを含む。NDAスポンサーから提出された小児科データがこのような データに対するFDAの書面要求に公平に応答すれば,6カ月間の排他的 を付与することができる。これらのデータは,この製品が研究されている小児科群で有効であることを証明する必要はなく,逆に臨床試験がFDAの要求に公平に応答していると考えられる場合には,追加的な保護を与える。これは特許期間の延長ではないが、FDAが別の出願を承認できない規制期間を効果的に延長している。
臨床試験情報開示
FDA規制製品の臨床試験スポンサー はウェブサイト www.Clinicaltrials.govに登録し、いくつかの臨床試験情報を開示しなければならない。そして,登録の一部として,製品,患者群,調査段階,試験地点と調査者,臨床試験の他に関する情報が公開されている。スポンサーには完成後にその臨床試験の結果を開示する義務がある。場合によっては、臨床試験結果の開示は試験完了日から2年に延期することができる。競争相手はこれらの公開された情報を用いて臨床開発計画や臨床試験設計に関する進捗状況を知ることができる。
承認後に を要求する
医薬品の承認後、製造業者および承認された医薬品は、保存活動の監視および記録、製品不良反応の報告、製品brのサンプリングおよび流通制限、許可されていない用途または患者集団へのbr薬物の普及(すなわち、“非ラベル使用”)の制限、および業界スポンサーの科学的および教育活動を制限することを含むFDAの一般的かつ持続的な規制を受けるであろう。医師はラベル外の用途のために合法的な製品を開く可能性があるが、メーカーはこのような用途を販売したり普及させたりしてはならない。FDAや他の機関はラベル外使用の普及を禁止する法律と法規を積極的に実行しており、ラベル外使用を不当に普及させていることが発見された会社は、不良宣伝、FDAの法執行行動、広告の是正、同意法令およびFDAが獲得できるすべての民事と刑事罰を含む重大な責任を負う可能性がある。処方薬宣伝材料も初回使用時にFDAに提出されなければならない。さらに、許可された医薬製品が、適応、ラベルまたは製造プロセスまたは施設の変更を含む任意の修正がある場合、申請者が新しいNDAまたはNDAサプリメントを提出して取得することを要求する可能性があり、これは、出願人が追加のデータを開発する必要があるか、または追加の臨床前研究または臨床試験を行う必要がある可能性がある。
| 88 |
FDA法規は、製品が特定の承認された施設で生産され、cGMPに適合しなければならないことを要求する。CGMP条例 は、人員、建物および施設、設備、成分および薬品容器および封口の制御、生産およびプロセス制御、包装およびラベル制御、保有および分配、実験室制御、記録および報告、および返品または回収された製品に関する要件を含む。私たちの候補製品の製造施設はcGMP要求 を満たし、FDAなどの外国規制機関の要求を満たし、その後、任意の製品を承認して私たちの商業製品 を生産することができなければならない。これらのメーカーはcGMPを遵守し,品質管理と品質保証,記録やファイルの維持を要求し,cGMPから外れた状況を調査·是正する義務がある.製造業者および承認薬品の生産および流通に参加する他のエンティティは、FDAおよびある州機関にその機関を登録し、brがcGMP要求および他の法律を遵守することを保証するために、FDAおよびいくつかの州機関の定期的な抜き打ち検査を受けなければならない。したがって,メーカーは生産や品質管理に時間,お金,労力をかけ続け,cGMPに適合したままでなければならない。CGMPに適合しないことを含む違反が発見され、 は製造活動の停止を含む法執行行動を引き起こす可能性がある。承認後に製品問題が発見されると、リコールおよび製品差し押さえを含む製品、製造業者、または承認された機密協定保持者への制限を招く可能性があります。
さらに、 生産プロセスの変更は厳しく規制されており、実施前には通常、FDA承認またはFDA通知が必要である。 薬物に適応、ラベルまたは製造プロセスまたは施設の変更を含む修正がある場合、 出願人は、追加のNDAまたは新しいNDAに対するFDAの承認を得るために提出を要求される可能性があり、これは、出願人が の追加のデータを開発するか、または追加の薬物開発/製剤研究、非臨床研究、または臨床試験を行う必要があるかもしれない。
処方薬が承認されると,法規に適合した要求や基準を保持できない場合や,製品発売後に問題が生じた場合,FDAは承認を撤回する可能性がある。後に製品に以前に未知の問題があることが発見された場合、予想されていない重症度または頻度の有害事象、または生産プロセス、または規制要件を遵守できない場合、承認されたラベルを強制的に修正して新しいセキュリティ情報を追加すること、発売後または臨床試験を実施して新しいセキュリティリスクを評価すること、またはREMS計画に従って流通または他の制限を実施することをもたらす可能性がある。他の潜在的な 結果は:
| ● | 製品の販売または製造、製品の完全な市場撤退、または製品のリコールを制限する | |
| ● | 承認された臨床試験には、罰金、警告状、または実行に関連する他の手紙または臨床保留が科される | |
| ● | FDAは、承認されるべきNDAまたは承認されたNDAの補充剤の承認を拒否するか、または製品承認を一時停止または撤回する | |
| ● | 製品の差し押さえまたは差し押さえ、または製品の輸出入を許可することを拒否する | |
| ● | 民事または刑事罰の命令または適用を禁止する | |
| ● | 法令に同意し、会社の誠実な合意に同意し、連邦医療計画の資格を取り消したり、それを除外したり | |
| ● | 宣伝材料とラベルを強制的に修正して訂正情報を配布する. |
| 89 |
そのほか、処方薬製品(サンプルを含む)の流通は“処方薬販売法”(“PDMA”)の制約を受け、この法案は連邦レベルの薬品と薬品サンプルの流通を規範化し、各州の薬品流通業者に対する登録と監督管理に最低基準を設定した。PDMAと州法はいずれも処方薬製品サンプルの分配を規制し,分配責任を確保する要求を規定している。
また、“薬品サプライチェーン安全法案”(DSCSA)は2013年に公布され、米国で流通しているいくつかの処方薬を識別し、追跡するための電子システムを構築することを目的としている。DSCSAは薬品メーカー,卸,流通業者に10年間の段階的な資源集約型義務を要求し,2023年11月に終了する予定である。時々、FDA規制を規範化する処方薬製品の承認、製造、およびマーケティングの法定条項 を著しく変更するために、新しい立法および法規が実施される可能性がある。 がさらなる立法や規制変化を公布するかどうか,あるいはFDAの法規,ガイドラインや解釈が変化するかどうか,あるいはそのような変化の影響(あれば)は予測できない。
DEA 法規
我々の現在の候補薬物中の有効成分はDEAがCSA下の制御物質とされている。CSA及びその実施条例は制御物質を処理する実体のために閉鎖的な流通チェーンを構築し、これらの実体に対して登録、記録保存と報告、安保、貯蔵、調達、製造、流通、輸入、輸出、ラベル、包装とその他の要求を提出した。DEAは,被制御物質を処理する個人や実体がこれらの要求を遵守し,制御物質の合法的な使用を確保し,被制御物質の不正な商業チャネルへの移行を防止することを要求する。
CSAは、乱用の可能性と身体または心理への依存に基づいて、制御物質を5つの付表のうちの1つ、すなわち付表1、付表2、付表3、付表4または付表5に分類する。定義によると,付表一物質は乱用の可能性が高く,現在米国では治療のための医療用途は受け入れられておらず,医療監督下での使用は公認された安全性に乏しい。米国の製薬製品でbrを販売してはならず、または患者に販売されてはならず、これらの製品は現在受け入れられている医療用途を有し、 が他の方法で発売されることが許可されている可能性は、別表II、III、IVまたはV物質とされており、その中で、付表II物質は最も高い乱用および身体または心理依存の可能性を有し、付表V物質は最も低い乱用および依存の相対的可能性を有する。付表二物質(およびいずれの付表でも麻酔品と定義されている物質)は登録,安全,記録保存,報告において最も厳しいbrが要求されており,これらの物質の分配と分配は厳しく規制されている。例えば,すべての付表2の薬物処方は医師が署名しなければならず,DEA規定に基づいて電子的に処方され,再充填できない限り,薬剤師に直接提出しなければならないことが多い。我々の候補製品(酢酸デキサメタニルと右旋フェニルプロピルアミン)中の活性成分は別表II規制物質であり,様々な制限を受けているため,製品の調達,製造,輸送,貯蔵,販売,使用が承認されれば高度なbr規制を受ける。
規制対象物質を製造,流通,輸入または輸出する施設は毎年DEAに登録しなければならない。登録は特定の場所、活動、そして制御された物質スケジュールに対するものだ。例えば、輸入および製造は個別に登録する必要があり、登録ごとにどの制御物質の付表が許可されているかを明確にする。同様に,単独の施設も単独で を登録する必要がある.
DEAは、製造業者、流通業者、輸入業者、輸出業者を検査して、安全、記録保存、報告を含むCSAとDEA法規の適合性を審査し、制御物質登録を発表する前に定期的に行う。具体的な安全要求 は,商業活動タイプおよび登録者が処理する制御物質のスケジュールや数によって異なり,その中で最も厳しい要求は付表1と付表2に適用される.必要な安全対策には,従業員の背景調査 や保険庫や在庫台帳などによる在庫の実物制御がある。メーカーや流通業者は定期的に禁毒署に別表1と付表二規制物質,別表三麻酔物質とその他の指定物質の分配状況に関する報告を提出しなければならない。すべての制御された物質を処理する記録、例えば、製造、受信、販売、交付、または他の方法で処理された各物質の完全かつ正確な記録は保存されなければならない。すべてのDEA登録者はまた、任意の制御物質の盗難または重大な損失を報告しなければならず、被制御物質の廃棄または処分の許可を得なければならない。輸入業者および/または輸出業者の登録を維持するほか、制御対象物質の輸入業者および輸出業者は、輸入または輸出ごとに表1または2に記載された物質および別表3、4および5に記載された麻酔物質のための許可証を取得しなければならない。付表3、4および5に記載されているすべての他の薬物については、輸出業者は輸出入申告書を提出しなければならない。
| 90 |
また、DEA割当システムは、付表IまたはII中の制御物質の獲得性と生産を制御し、制限している。DEA は、DEAの合法的な科学と医療需要を満たすために必要な数量の推定に基づいて、アメリカが毎年どれだけの制御物質を生産できるかの総割当量を制定した。DEAが毎年米国で生産を許可しているオピオイドや覚せい剤の有限総数は各社間で分配されており,これらの会社はDEAに個人生産と調達割当の申請を毎年提出しなければならない。私たちはDEA から年間割当量を得なければならないが,我々の製品や候補製品を製造するための付表II物質を生産または調達することができる。麻薬取締局は年内に時々総生産割当量と個別生産と調達割当量を調整することができるが、同局は調整するかどうかについてかなりの自由裁量権を持っている。任意の付表IやII制御物質を配布するには特別注文表を添付し,DEAにコピーを提供しなければならない。
が適用されるDEA要求,特に損失や転移や規制された物質の面で表現される要求を遵守できなければ,行政,民事あるいは刑事法執行行動を招く可能性がある。DEAは民事処罰を求め、必要な登録の更新を拒否し、または行政訴訟を提起してこれらの登録を撤回することができる。場合によっては、違反は刑事起訴につながるかもしれない。
各州やコロンビア特区でも制御物質を規制し,制御物質を処理する実体に対して類似した許可,記録保存,報告要求を実施している。連邦制御物質要求に加えて,実体は 中の様々な州要求を独立に守らなければならない。
薬品のカバー範囲、定価と精算
私たちが規制部門の承認を得る可能性のある任意の候補製品の保証範囲と精算状態には大きな不確実性がある。米国では、薬品の販売は、第三者支払者(例えば、MedicareおよびMedicaid計画を管理する機関、保健組織および個人保険会社を含む)が保証を提供しているかどうか、十分な精算を提供しているかどうかに大きく依存する。私たちは私たちの各候補製品に提供する保険範囲と精算金額を計画的に決定します。医療保険や医療補助計画は,個人支払者や他の政府支払人によって,彼らの薬品保険や精算政策を策定するための手本として用いられることが多い。しかし、1人の支払人が製品に保険を提供することを決定することは、他の支払者がその製品に保険を提供し、十分なbr精算を提供することを保証することはできない。各第三者支払者は,薬物に保険を提供するか否か,薬物の提供者にどの程度の金額を支払うか,薬物がその処方のどの層に置かれるかを決定する。これらの決定は,治療種別に複数の薬物製品が存在するかどうかやその計画の純コストの影響を受け,薬物メーカーがリベートした処方価格金額(あれば)を含む。一般的に、模倣薬は第一選択のレベルに置かれる。Br薬物の処方中の位置は、通常、患者がその薬物を獲得するのに必要な自己負担費用を決定し、患者や医師の薬物の採用に強く影響する可能性がある。自分の病状に応じて処方治療を受ける患者や処方サービスを行う提供者は,通常第三者支払者に依存して関連医療費の全部または一部を精算する。患者は保険を提供しなければ、私たちの製品を使用することはあまりできません。そして、私たちの製品の大部分のコストを支払うのに十分です。また,第三者支払者が薬品に保険を提供することを決定することは,適切な精算比率 を承認することを意味するものではない.また,第三者支払者は医療コストを抑えるための複雑な方法を開発している。そのため,保証範囲,精算,配置決定は非常に複雑であり,通常支払人と薬物所有者の間で広範な交渉が行われるテーマである。
私たちが戦略協力を達成し、私たちの協力者が所与の製品のためにカバー範囲と精算を求める責任がない限り、私たちは私たちの候補製品のためにカバー範囲、精算と配置決定を協議する責任があります。新製品の精算と配置決定は多くの要素に基づいており、同じ或いは類似の適応のためのすでに発売されたブランド薬物のカバー範囲、精算と配置、新製品の安全性と有効性、類似適応に使用できる模倣薬{br]、新製品に対する臨床需要及び製品のコスト効果を含む。
| 91 |
連邦医療保険計画では、CTX-1301、CTX-1302、CTX-2103は、承認されれば、自己管理の薬である可能性が高く、拡張された処方薬福祉(連邦医療保険D部分と呼ばれる)によって精算される可能性があります。この計画は、個人br計画によって管理されている自発的医療保険福祉であり、連邦政府との契約によって運営されています。これらの計画はどの製品が保証薬品に適用されるか及びどの自己支払い費用が保証薬品に適用されるかを確定する処方を制定した。これらの計画は調合と階層共同支払い構造を構築する上でかなりの自由裁量権があり、 はメーカーと交渉し、特定の製品の使用に事前許可と他の制限を加え、 CMSは差別的なやり方を検討する。これらのD部分計画は、医薬品製造業者と割引を交渉し、保険料を低減することによって、これらの割引の全部または一部を計画の各参加者に転嫁する。
もし薬品が連邦医療保険あるいは連邦医療補助によって精算される場合、定価と返却計画は適用される“連邦医療保険処方” 2003年“薬品改善と現代化法案”及び改正された“1990年総合予算調節法”(“Obra”と略称する)と1992年の“退役軍人医療法案”の医療補助税金還付要求に適合しなければならない。他の事項以外にも,Obraは薬品メーカーに州医療補助計画に処方薬のリベートを支払うことを要求し,各州に薬品br価格についてリベートを交渉することを許可しており,将来の製品の価格は本来入手可能な価格よりも低い可能性がある。製品 が総務庁連邦供給スケジュールの許可ユーザに提供される場合、他の法律および要求 が適用される。
米国政府を含む第三者支払者は、医薬製品の精算に下り圧力をかけ続けている。例えば、2022年のインフレ削減法案は、米国政府のいくつかの薬品の精算を低下させている。また,米国の医療管理傾向や健康維持組織などの組織の同時増加により薬品精算が減少する可能性がある。私たちは、これらの支払人が様々な提案や規制政策を実施するにつれて、これらの傾向が続くと予想する。現在、私たちは引き続き多くの連邦と州提案が直接または間接的に精算と価格設定の制御を実施する予定だ。
その他 ヘルスケア法律とコンプライアンス要件
私たちの候補製品を商業化しているので、FDAや同様の外国の規制機関の承認を得てマーケティングを行っていれば、追加の医療法と規制の要求、連邦政府と私たちが業務を展開している州と外国政府の執行を受けます。医療保健提供者、医師、および第三者支払者は、マーケティング承認された任意の他の候補製品を推薦し、処方する際に主な役割を果たすであろう。私たちと第三者支払者と顧客との手配は、マーケティング、販売、流通がマーケティングの承認を得た任意のbr製品の業務または財務的手配と関係を制約する幅広い適用された詐欺と乱用、および他の医療法律およびbr法規に直面させる。
適用される連邦と州医療保健法律法規の制限は:
| ● | 他の事項に加えて、“連邦反バックル条例”は、個人を推薦して商品またはサービスを購入するか、または商品またはサービスを購入または注文し、連邦医療保健計画(例えばMedicareとMedicaid計画)に基づいて支払いを行うために、誰でも直接または間接的に故意に提供、誘致、受取、または報酬を提供することを禁止する。連邦反リベート規制は違う解釈があるかもしれない。過去,政府は連邦反リベート法規を施行し,虚偽相談と医師との他の財務手配に基づいて医療会社と大規模な和解を達成した。個人または実体は、法規や具体的な意図を実際に知る必要がなく、法規に違反することができ、違反を実施することができる。また、政府は、連邦反リベート法規に違反して発生した物品やサービスを含むクレームは、民事虚偽クレーム法案については、虚偽または詐欺的クレームを構成していると断言することができる |
| 92 |
| ● | “民事虚偽請求法案”および民事罰金法律を含む連邦民事および刑事虚偽請求法は、他の事項に加えて、米国政府に虚偽、架空または詐欺的な支払い請求を提出または提出することを禁止し、虚偽、架空または詐欺的な支払いクレームの提出、または米国政府への虚偽または詐欺的クレームの作成、使用、または作成または使用を招いて、または米国政府への支払いの義務を回避、減少または隠蔽するために意図的に虚偽陳述を行うことを禁止する。これらの法律により提起された訴訟は総検事長が提起することができ、個人が政府の名義で訴訟を提起することもできる。連邦政府は、これらの法律と付随する重大な責任脅威を利用して、例えば、許可されていない用途の製品や他の不正と言われる販売やマーケティング行為の普及に関連して、全米の製薬·バイオテクノロジー会社を調査·起訴している | |
| ● | HIPAAは新しい連邦、民事および刑事法規を制定し、個人第三者支払人が医療福祉計画を知り、故意に流用または盗み取り、医療違反行為に対する刑事調査を故意に阻害し、故意に偽造、隠蔽または隠蔽または隠蔽すること、または医療福祉、プロジェクトまたはサービスの提供または支払いに関連する任意の重大な虚偽、架空、または詐欺的な陳述を含む任意の医療福祉計画を知り、故意に実行または実行しようとする計画を禁止し、または重大な事実を偽造、隠蔽または隠蔽することを含む。連邦反リベート法規と同様に、個人や実体は法規を実際に理解する必要はなく、法規違反の具体的な意図を持つ必要もなく、違反を実施することができる | |
| ● | 他の事項に加えて、PPAAは、FDAが承認した薬品、設備、生物製品および医療用品メーカーbr連邦医療保険、医療補助または児童健康保険計画brに、医師(医師、歯科医、視光師、足科医、脊椎マッサージ師、および2022年から前年に提供される支払いおよび他の価値移転brのいくつかの高級非医師医療従事者を含むと定義される)への支払いおよび他の価値移転に関する情報を毎年CMSに報告することを要求する。医師とその直系親族が持っている所有権と投資権益です | |
| ● | HITECHにより改訂されたHIPAA及びそのそれぞれの実施条例 は個人が健康情報を識別できるプライバシー、安全と伝送に対して具体的な要求を提出した。他の事項に加えて、HITECHは、特定の医療保健提供者、健康計画および医療情報交換所を含む、特定の医療保健提供者、健康計画および医療情報交換所を含む、特定の医療保健提供者、健康計画および医療情報交換所を含む、特定の医療保健提供者、健康計画および医療情報交換所を含む“事業パートナー”に直接“業務パートナー”を適用する。HITECHはまた、実体、商業パートナー、および可能な他の人に適用される民事と刑事罰を増加させ、連邦裁判所に民事訴訟を提起し、HIPAAを実行し、連邦民事訴訟に関連する弁護士費と費用を求めるために、連邦裁判所に民事訴訟を提起することができる新たな権力を与えることができる | |
| ● | 同様の国および外国の法律、例えば、国の反リベートおよび虚偽クレーム法律は、販売またはマーケティング手配、および非政府第三者支払者(民間保険会社を含む)によって精算される医療項目またはサービスに関するクレームに適用可能である | |
| ● | 州法律は、製薬会社に製薬業界の自発的コンプライアンスガイドラインおよび連邦政府が公布した関連コンプライアンスガイドラインを遵守することを要求し、医薬品メーカーに、医師および他の医療保健提供者への支払いおよび他の方法での価値またはマーケティング支出に関する情報を報告することを要求する可能性があり、これらの法律が適用する要求は、“医師支払い日光法案”および薬品販売代表登録を要求する州および地方法律よりも厳しい | |
| ● | 国の法律や外国の法律(特にヨーロッパ個人に関する個人データのEU法律)は、健康情報のプライバシーやセキュリティを管理する場合があり、その多くの法律や外国の法律は互いに大きく異なり、コンプライアンス作業を複雑化させる。 |
| 93 |
2020年11月、米国衛生·公衆サービス部(DHHS)は、医療業界により大きな柔軟性を提供し、これらの詐欺や法律乱用に関連する規制負担、特に業界参加者間の価値に基づく手配を軽減することを目的として、“反リベート法規”を実施する法規の重大な改正を完了した。
私たちが現在と未来に第三者の業務手配と適用される医療法律法規に適合することを確保することは、多くのコストに関連している。政府当局は、私たちの業務行為が、現在または未来に適用される詐欺および乱用または他の医療関連法律および法規の現行または将来の法規、法規、機関指導または判例法に適合していない可能性があると結論するかもしれない。もし私たちの運営が、これらの法律または他の任意の政府法規に違反していることが発見された場合、私たちは、金銭損失、罰金、返還、監禁、FDAの承認された資格を失い、政府契約、医療保険、または他の政府項目の清算に参加することを含む重大な民事、刑事および行政処罰を受ける可能性がある。MedicareおよびMedicaid、名声障害、利益減少、および将来の収益を含むか、または私たちがこれらの法律に違反した疑いを解決するために、当社の誠実な合意または他の合意の制約を受けている場合、および私たちの業務の縮小または再構成は、追加的なbr報告要件を必要とします。もし私たちがそれと業務を展開する任意の医師または他の医療提供者または実体が適用法に適合していないことが発見された場合、彼らは政府の援助された医療計画から除外されることを含む刑事、民事または行政制裁を受ける可能性がある。
ヘルスケア改革とヘルスケア法の潜在的変化
米国や一部の外国司法管区は、医療システムを変更するための一連の立法·規制提案を検討または公布しており、そのあり方は、将来の製品販売の収益性に影響を与える可能性がある。米国や他地域の政策立案者や支払者の中には,ヘルスケアシステムの変革を推進し,医療コストの抑制,質の向上や参入拡大を既定の目標としていることが大きな興味を持っている。米国では,製薬業はこれらの努力の重点であり,重大な立法イニシアティブの大きな影響を受けてきた。FDAと他の規制機関のbr政策は変わる可能性があり、追加の政府法規を公布して、規制部門の私たちの候補製品に対する承認を阻止、制限、または延期するかもしれません。もし私たちが既存の要求の変化や新しい要求や政策の採用に緩やかに適応できない場合、 あるいは規制適合性を維持できない場合、私たちは本来得られる可能性のあるいかなるマーケティング承認も失う可能性があり、私たち は実現したり、利益を維持することができない可能性があり、これは私たちの業務、将来性、財務状況、運営結果に悪影響を与えるだろう。 また、米国および他の地方の政策立案者および支払人は、医療保健コストの制御、品質の向上、および/または参入の拡大を既定の目標とするために、医療システムの変革を推進することに非常に興味を持っている。
例えば,PACAは2010年3月に公布され,米国の医療業界に大きな影響を与えている。PPACA は未加入者群のカバー範囲を拡大するとともに,全体の医療コストを抑えた。生物製薬製品の面で、他の事項以外に、PPACAは吸入、注入、点滴、移植或いは注射の薬物計算メーカーのMedicaid薬品返却計画下の返却点に対して、Medicaid薬品返却計画下の最低医療補助返却点を高め、そして返却計画をMedicaidホスト看護 組織に参加する個人に拡大し、あるブランドの処方薬メーカーに対する年会費を確立し、そして新しいMedicare Part D Coverage Gap割引計画を作成する新しい方法を解決した。
もう一つの例は、“2021年総合支出法案”が2020年12月27日に署名されて法律となり、広範な医療条項と既存法の改正が盛り込まれ、連邦医療保険B部分に含まれるすべての薬品とバイオ製品メーカーに2022年1月1日から国土安全保障省に製品の平均販売価格を報告し、民事による強制罰金を執行することを含む。
また,PACA以来,米国では医療支出に影響を与える他の立法変化も提案され採択されてきた。これらの変化には、2011年度の“予算制御法案”によると、医療保険提供者に支払われる医療保険総金額が2%減少し、2013年に施行され、国会がさらに行動しない限り、2030年まで有効になる。コロナウイルス援助、救済と経済安全法案(“CARE法案”)は2020年3月27日に法律に署名し、新冠肺炎の大流行の影響を受けた個人と企業にbrの財政支援と資源を提供することを目的とし、2020年5月1日から2020年12月31日まで2%の連邦医療保険自動減支 の実施を停止し、自動減支を1年から2030年まで延長し、2020年に自動減支の廃止による追加費用brを相殺する。2021年総合支出法案はその後、2020年12月27日に法律に署名し、CARE法案の暫定期間を2021年3月31日に延長する。2021年3月11日に法律となった2021年“米国救援計画法案”(American Rescue Plan Act)が署名されたのは、最新に公布された疫病緩和立法であり、重大な医療システム改革やPPAAによる保険市場の強化を目的とした計画なども含まれている。また,ACAが公布されて以来,米国では製薬業に影響を与える他の立法改正が提案され採択された。例えば、2022年の“インフレ低減法案”には、CMSが毎年限られた数の高コストであり、単一源の薬品が“最高公平価格”を交渉することを許可する条項と、価格上昇がインフレよりも速い場合に連邦医療保険にリベートを支払うことを要求する別の条項とが含まれている。
| 94 |
また、br政府はメーカーがその市場製品に価格を設定する方式に対してより厳格な審査を行い、これはいくつかの国会調査を招き、製品定価の透明性の向上、価格決定とメーカー患者計画との関係の審査及び政府の薬品精算方法を改革するための連邦と州立法を提出し、公布した。国土安全保障省はすでにいくつかの薬品価格の低減と薬品自己コストの低減のための各種措置についてフィードバックを求め、その現有の権限に基づいて他の措置を実施した。例えば,2019年5月,国土安全保障省はMedicare Advantage計画が2020年1月1日からB部分に対する階段療法の選択を許可する最終ルールを発表した。この最終規則は、2019年1月1日に施行された国土安全保障省の政策変更を編纂した。国会と行政はそれぞれ,新たな立法および/または行政措置を求めて薬品コストを制御し続けることで,この分野を持続的な不確実性に直面させていると述べている。
アメリカの個別州もますます立法を通じて、価格或いは患者の精算制限、割引、ある製品の参入とマーケティングの制限、brのコスト開示と透明性措置を含む薬品br製品の価格設定を制御するための法規を立法し、実施しており、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。2020年12月、アメリカ最高裁判所は、連邦法律は各州が薬品福祉マネージャー(“PBM”)と医療保健と薬品サプライチェーンの他のメンバーに対する監督管理能力を妨害しないと一致して判断した。この重要な決定はこの分野で各国がさらに肯定的な努力をすることをもたらすかもしれない。
FDAや他の規制機関の政策も変わる可能性があり、私たちの候補薬物に対する規制承認を阻止、制限、または延期するための追加の政府法規が公布されるかもしれない。例えば、2016年12月、“21世紀治癒法案”(“br}”治癒法案“)が法律に署名されました。その他の事項以外に、“治療法案”は薬品と設備の監督管理を現代化し、革新を奨励することを目的としているが、その最終的な実施状況はまだ確定されていない。さらに、2017年8月、FDA再認可法案は、FDAのユーザ費用計画を再許可し、“治療法案”に基づいて確立された追加の薬物および設備条項を含む法律として署名された。もし私たちが既存の要求の変化に緩やかに適応できない場合、あるいは新しい要求や政策を採用することができない場合、私たちは利益を達成したり、維持することができないかもしれません。これは、私たちの業務、見通し、財務状況、およびbr}運営結果に悪影響を与えます。
PPACAおよび将来講じられる可能性のある他の医療改革措置は、より厳しいカバー基準とより低い精算をもたらす可能性があり、承認された任意の製品の価格に追加の下振れ圧力をもたらす可能性があると予想している。連邦医療保険や他の政府援助計画を減らすいかなる精算も、個人支払者の支払いのような減少を招く可能性がある。br}が監督部門の承認を得ると、コスト制御措置や他の医療改革を実施することは、私たちが収入を創出し、利益を達成し、あるいは私たちの薬品を商業化することを阻止するかもしれない。米国や海外の将来の立法や行政または行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することはできません。私たちは将来、より多くの州と連邦医療改革措置を取ることが予想されます。そのうちのいずれも、私たちが発売許可を得た任意の未来の医薬製品を含む連邦政府と州政府が医療製品やサービスに支払う金額を制限する可能性があります。
データプライバシーと個人情報保護
私たちの業務や関連データ処理活動に適したデータプライバシー、セキュリティ、個人情報(健康情報を含む)保護の法律法規によって規制されています。プライバシーとデータ保護の立法と規制構造 は引き続き発展し、グローバルなプライバシーとデータ保護問題への関心もますます多くなってきており、これらの問題は引き続き私たちの業務に影響を与える。米国では、州データ漏洩通知法、州 が健康と個人情報のプライバシーと安全を保護する法律、および個人情報の収集、使用、開示、転送を規制する連邦と州消費者保護法の制約を受ける可能性がある。これらの法律は互いに重複して衝突する可能性があり、これらの法律のすべては裁判所と政府機関の異なる解釈を受け、複雑なコンプライアンス問題をもたらしてくれる。もし私たちが適用されたデータ保護法律と法規を遵守できなかったら、私たちは刑事罰 を含めて罰や制裁を受けるかもしれない。現在および将来の顧客および研究パートナーは、HIPAAおよび州健康情報プライバシー法を含む健康情報のプライバシーおよびセキュリティを管理する法律を遵守しなければならない。私たちが知っている限りでは、HIPAAによって保護された健康情報(“保護された健康情報”と呼ばれる)を取得し、正しい合意を遵守していない場合、業務br関連プロトコルの実行、プライバシーまたはセキュリティ対策の実施、および他の義務が含まれている可能性がある場合、私たちの顧客または研究協力者は、法執行行動の影響を受ける可能性があり、保護された健康情報の不正な受信や協力、HIPAA違反のそそのかしによって直接責任を負う可能性がある。
| 95 |
健康と個人情報を保護する州法はますます厳しくなっている。例えば、カリフォルニア州では、健康情報や他の個人識別情報の使用および開示に制限的な要求を行う“カリフォルニア医療情報秘密法”が施行されている。ワシントンは2024年3月31日に施行される“私の健康私のデータ法案”を公布し、ネバダ州の消費者健康プライバシー法は2024年3月31日に施行される。一般的な州プライバシー法の発展はさらに速い。2020年、カリフォルニアでは“2018年カリフォルニア消費者プライバシー法”(CCPA)が施行された。CCPAは欧州連合一般データ保護条例(GDPR)に含まれるいくつかの重要な概念を反映している。CCPAは、以下の方法でカバーする企業のために新しいプライバシーフレームワークを構築する:個人情報の拡張定義を作成し、カリフォルニア州の消費者のための新しいデータプライバシー権を確立し、未成年者から消費者データを収集するための特殊な規則を実施し、CCPAに違反し、合理的なセキュリティプログラムを実施できず、実践してデータ漏洩を防止する企業のために新しい、br}の深刻な法定損害賠償フレームワークを作成する。2023年1月1日、“カリフォルニアプライバシー権法案”(“CPRA”) が発効し、“カリフォルニアプライバシー権法案”が大きく改正された。これは、より多くの不確実性、コンプライアンス作業における追加的なコストおよび支出、および規則的でない追加的な潜在的な危害および責任をもたらす可能性がある。カリフォルニア州を除いて、他の12州は州データプライバシー法を公布し、そのうちの5州-コロラド州、コネチカット州、ネバダ州、ユタ州、バージニア州などの法律が2023年に施行された。米国の他州でもCCPA/CPRAのようなプライバシー法や他州プライバシー法が考えられている。
英国または欧州経済地域(すなわちEUにリヒテンシュタイン、ノルウェー、アイスランドを加えて)で業務および/または臨床試験を行う場合、GDPRおよび英国2018年EU(離脱)法案第3条および英国2018年データ保護法(DPA 2018)(“イギリスGDPR”)に基づいてイギリスの法律に保存されているGDPRも遵守しなければならない。GDPRおよびイギリスGDPRは、企業の同僚、従業員、サービスプロバイダ、試験参加者、および調査者またはCRO従業員などの他の個人に適用され、彼らはイギリスまたはヨーロッパ経済地域の住民である。イギリスのGDPRおよび/またはGDPRに違反した行為は、金額が高い者を基準として、最高2000万ユーロ/GB 1750万ユーロまたは前会計年度の世界総収入の4%の巨額の罰金を科すことができる。
アメリカの“海外腐敗防止法”
一般的に、改正された1977年の“海外腐敗防止法”または“海外腐敗防止法”は、外国人官僚に支払い、支払い、支払いを承諾し、金銭または任意の価値のあるものを支払うことを禁止し、その外国人官僚の公的な身分で行われた任意の行為または決定に影響を与え、または任意の他の不正な利益を確保して、誰かまたは誰かと業務を維持し、または業務を誰かに回すために、または業務を誰かに回すために、“いかなる外国人役人にも適用されるだけでなく、“任意の外国政党またはその役人”、“任意の外国政治職候補者”、または誰かに支払われる金も含まれているが、上記のいずれかのカテゴリの誰にも提供されることを知っているが、すべてまたは一部の報酬を与えるか、または承諾する。“海外腐敗防止法”に規定されている“外国官”には、外国政府部門、機関又は機関の役人又は従業員が含まれている。用語“ツール性” は広く、国有または国家制御のエンティティを含むことができる。重要なのは、米国当局は、公共医療および/または公共教育システムを有する国では、医療専門家の多くと外国の病院、診療所、研究機関、医学院の他の従業員が“海外腐敗防止法”の下の“外国人役人”であると考えている。私たちが海外で私たちの製品をテストして販売する時、私たちが外国の医療専門家や研究者と交流する時、私たちの任意の候補製品が将来外国の監督部門の許可を得たら、私たちと私たちを代表する代理人が私たちの製品やサービスをマーケティングすることに関連する過度または贅沢な食事、旅行、娯楽、または必要な許可と承認を得ることを防ぐために、十分な政策と手続きを制定しなければならない。“海外腐敗防止法”はまた、証券が米国に上場する会社に会計規定を遵守することを要求し、国際子会社を含む会社のすべての取引を正確かつ公平に反映した帳簿や記録を保存し、国際業務のために適切な内部会計制御制度を制定し、維持することを要求している。
| 96 |
材料 プロトコル
Indegene,Inc.と共同商業化協定を締結する.
2023年3月7日, 我々はIndegene共同商業化プロトコルを締結した
我々の要求に応じてCTX−1301に対するFDAの承認を得た場合、商業化プロトコル管轄Indegeneは、CTX−1301に商業化サービスを提供する一般的な条項を管轄する。商業化協定によると、双方は、Indegeneが提供するサービス、そのようなサービスの配信可能な内容、および私たちが支払う費用を列挙する作業宣言を締結する。“作業説明書”で明確に修正されない限り、各“作業説明書”は、“商業化合意”条項によって制限されている。 私たちは、(A)医療事務および薬物警戒、(B)定価、精算、および市場参入、(C)商業運営、(D)マーケティング(現場力を含む)を受け入れることを選択することができる。双方は,我々が決定した場合や優先順位の変化によりIndegeneが提供するサービスの任意の 変更を誠実に協議する.
商業化プロトコルは、CTX-1301送信から3年後に満了する。6ヶ月前(6)ヶ月前にIndegeneに書面で通知した場合に商業化プロトコルを終了することができ、Indegeneは12(12)ヶ月前に書面で通知したときに商業化プロトコル を終了することができる。さらに、以下の場合、いずれの場合も、商業化プロトコルの終了を30(30)日前に書面で通知することができる:(I)FDAは、我々のCTX−1301のNDAを承認しないことを通知する;(br}または(Ii)米国におけるCTX−1301の臨床開発計画を一時停止または終了する。いずれか一方は30(30)日前に“商業化協定”を終了し、重大で未治癒の違反行為に対して書面で通知するか、他方が破産した場合には直ちに終了することができる。
の間“商業化プロトコル”によると,医療情報サービス,同業者レビュー文章の開発と発表および薬物警戒サービスを除いて,Indegeneは我々以外のいずれかに戦略的医療サービスおよび/または患者マーケティング支援(または任意の他のタイプの同種サービス)を提供しないが,いかなるADHD製品についてもbr}を提供してくれない。商業化プロトコルの終了通知または特定のサービス終了の通知が発行されるまで、Indegeneの書面による承認なしに、CTX-1301の商業化については、Indegeneが提供する任意のサービスカテゴリと同じサービス を提供し、無理に抑留してはならないが、いくつかの例外は除外する。
商業化協定には、宣言、保証、守秘および賠償義務 のような合意の慣行義務が含まれている。
教師レベル は社会CDMO,Inc.と締結されたサービスプロトコルである.
2022年10月24日から社会と主サービス協定(“製造協定”)を締結した“製造協定”には、Socialまたはその付属会社が、ジョージア州ガイエンスビルにある製造工場で製造サービスを提供する一般的な条項が規定されている。このようなサービスは,合意されたワークシート によって実行される.製造契約の条項に基づいて、適用される作業説明書毎に社会履行サービスのための費用を支払うことに同意します。
| 97 |
製造協定は2027年10月に終了しますまたは 作業説明書(“初期期限”)を完了するのに必要な遅い日付は、その後、任意の一方が初期 期間または任意の継続期間の終了前少なくとも12(12)ヶ月(第1の製品のプロセス検証バッチが成功する前に) または24(24)ヶ月(第1の製品の検証バッチが成功した後)に終了しない限り、 を自動的に更新する。事前に終了しない限り、各作業説明書の期限は、その作業説明書の下でのサービスが完了したときに終了する。私たちは社会に書面通知を出してから90(90)日に製造契約または任意の作業説明書を終了することができます。社会は、いくつかの遅延または不活動(Br)またはその作業陳述の下で提供されるサービスが、適用された法規の要求に従って実行されないために、作業陳述を終了することができるが、そのような終了のいずれかの前に、社会は、商業的に合理的な努力をして、私たちと有意義な議論を行うべきであることが条件である。すべての作業説明書が終了した場合、社会は6(6)ヶ月前に書面通知で本合意を終了することができる。製造プロトコルまたは作業説明書は、重大で治癒されていない違反によって、または他方が倒産した場合に、いずれか一方によって終了することができる。
製造協定には、賠償、知的財産権保護、セキュリティ、救済措置、保証などに関連する慣行条項が含まれる。
BDD Pharma Limitedとの特許とノウハウライセンス契約
我々 は2018年8月にBDD Pharma Limited(“BDD Pharma”)と特許とノウハウライセンス契約を締結し,このプロトコルをBDD Pharmaライセンスプロトコルと呼ぶ.BDD Pharmaライセンスプロトコルによれば、我々は、BDD Pharmaが所有または制御する技術の下で独占的に許可されており、 特許およびノウハウは、制御された薬物放出バリア層に関連しており、開発、製造、販売、使用、輸入、販売、または他の方法で以下の製品を供給および商業化している:(I)3つの異なる用量の右旋フェニルプロピルアミン、ピペラメチルまたはピペラメチルエステルベースまたはフェニルプロピルアミンベースの任意の薬物を提供し、(Ii)8時間を超えるインビトロ放出または(Iii)他の方法で特許または製造されている。独自技術に基づいて開発または使用されている。私たちのbrも上場承認と臨床試験を申請して、このような製品の発売許可 を得る権利があります。我々に付与される権利は世界的であり、ヒトのどの疾患または障害の治療においても独占的であり、メチルフェニデートまたはアンフェタミンに基づく薬物またはそれらの混合物またはそれらの組み合わせで治療することができる。私たちは特定の条件に適合した場合、私たちに付与された権利を再許可する権利がある。
BDD Pharmaは、BDD Pharmaライセンス契約に署名することにより198,625ドルの支払いを得る権利がある。臨床試験および規制マイルストーンに関連する各製品のBDD Pharmaマイルストーンに合計750,000ドル(うち250,00ドルが支払われている)を支払う必要があるかもしれませんが、マイルストーン支払いの場合、同一製品の異なる用量強度は とみなされています。私たちは製品の総純売上高にBDD Pharmaが中央値に低い印税を支払うことを要求されるかもしれない。私たち はまた、製品の総純収入に応じてBDD Pharmaに中央値まで低い印税を支払うことが要求される可能性があります。これは、私たちの分許可者の売上と私たちが受け取った非印税分の許可価格に基づいています。
が早期に終了しない限り、BDD Pharmaライセンスプロトコルの期限は、私たちが取得したすべての特許の最後の満了日または私たちのすべての支払い義務の最後の満了日の遅い者まで続くであろう。私たちの特許使用料支払い義務は、製品が初めて1つの国/地域で商業販売されるか、またはその製品の1つの国/地域での製造、使用または販売をカバーする最後の満期特許brが満了してから10年以内に製品brおよび国/地域で満期になります。現在、BDD Pharmaライセンスの最後の満了特許は2035年11月に満了する。私たちの印税支払い義務が満期になった後、私たちに付与された許可は全額支払い、撤回できない、永久的になります。
私たちまたはBDD Pharmaは、他方に未治癒の重大な違約が存在する場合、または他方の破産に関連するBDD Pharma許可プロトコルを終了することができる。もし、私たち、任意の分割許可者または関係者または関連会社が、BDD Biophmaが所有している特許の有効性または強制実行を疑問提起または協力する場合、BDD Pharmaは、書面通知を出した後、直ちにBDD Pharma許可プロトコルを終了することができる。
| 98 |
人的資源 資本資源
私たちの目標を達成するためには、才能のある従業員を引きつけて維持することが必須的だ。これを促進するために、私たちは、私たちの従業員が彼らのキャリアの中で成長し、発展する機会を得るために、安全でリターンのある職場を維持し、競争力のある報酬、包括的な福祉と健康計画、そして私たち従業員の間につながりを作る計画の支持を得るために努力しています。私たちの報酬計画には、従業員を誘致、維持、激励するために株式オプションを付与することが含まれている。
2023年12月31日現在、私たちは13人の従業員を雇った。その中で,六(6)人は研究開発と製造活動に従事し,七(Br)(7)は一般と行政機能に従事している。私たちの従業員はみんなアメリカにいます。私たちは外部コンサルタントと独立請負業者を利用して私たちの常勤従業員チームを補充する。私たちの従業員の中の一つも労働機関によって代表されていないし、集団交渉の手配の下にもありません。私たちは私たちの従業員が仲がいいと思う。
企業情報
Cingate Inc.はデラウェア州会社で、2021年5月に設立され、持株会社である。CTXはデラウェア州の有限責任会社で、2012年11月に設立された。当社の初公募株の完成については、2021年9月29日、Cingate Inc.は、Cingate Inc.の完全買収子会社をCTXと合併してCTX(“合併”)に組み込むことによりCTXを買収した。 合併により、CTXはCingate Inc.の完全子会社となる。他に声明や文脈がない限り、本募集明細書のすべての情報は、再編合併およびIPOの完了を反映している。
私たちの主な執行事務室は西47号1901にありますこれは…。Pace、カンザスシティ、カンザス州66205、私たちの電話番号は(913)9422300です。私たちのサイトの住所はWwwc.cingulate.comそれは.本入札明細書には、当サイトを介してアクセス可能な情報が含まれているか、または当サイトを介してアクセス可能な情報がコスト募集説明書の一部を構成していない。私たちはこの年間報告書に私たちのウェブサイトのアドレスを含めて、ただ非アクティブなテキストとして参考にしている。
属性
私たちの会社はカンザスカンザスシティに本社を置いています。そこで約14,205平方フィートのオフィススペースを借りました。私たちのレンタル契約は2025年5月に満了し、更新を選択することができます。私たちの製造活動はジョージア州ガイエンスビルにある社会CDMOで発生しました。私たちは私たちの現在のオフィス、実験室、製造空間は私たちの需要を満たすのに十分だと信じています。私たちは新しいレンタル契約を交渉したり、運営を収容するために追加的または代替空間を評価したりすることを求めることができるかもしれない。私たちは商業的に合理的な条項で適切な代替空間を提供することが簡単だと信じている。
法律訴訟
私たちは法的手続きを待ついかなる重大な事項でもない。通常の業務過程で、私たちは時々法的手続きとクレームの影響を受けるかもしれない。
| 99 |
経営陣の財務状況と経営結果の検討と分析
以下、当社の財務状況及び経営結果の検討及び分析は、本募集説明書の他の部分に含まれる連結財務諸表及び関連注釈と共に読まなければならない。本議論および分析に含まれるいくつかの情報または本明細書の他の場所に記載された情報は、リスクおよび不確実性に関する前向きな陳述を含む、我々の業務計画および戦略に関連する情報を含む。あなたは本募集説明書の“リスク要素”の一部 を読むべきであり、討論は実際の結果が以下の討論と分析に含まれる展望性陳述に記述或いは暗示された結果と大きく異なる重要な要素を招く可能性がある。
概要
私たちはバイオ製薬会社で、私たちの独自のPrecisionを使って定期的にリリースしますTM(“PTRTM)薬物brは、重い日常投与レジメンおよび二次治療を特徴とするよく見られる診断疾患を有する患者の生活を改善するために、次世代薬物製品導管を構築および推進するためのプラットフォーム技術を提供する。私たちの最初の仕事の重点はADHDの治療です。我々のPTRプラットフォーム は、特定の所定の時間間隔で医薬物質を放出することを可能にし、それによって1日1回、多用量錠剤の可能性を放出することを可能にするための固有の侵食バリア層(“EBL”)を含む。現在の治療モードには、患者の活動日全体の需要をよりよく満たすために、真の毎日1回、持続時間が長く、副作用が突出したADHD刺激性薬物である重要な、満たされていない 需要があると考えられる。
2012年の設立以来、私たちの運営は私たちの候補製品、組織、私たちを配備した会社、業務計画、資金調達、私たちの知的財産権の組み合わせの構築と臨床試験に集中してきました。私たちは何の候補製品も販売を許可されていないし、何の収入も生じていない。私たちは公共資本と個人資本を調達することで私たちの業務に資金を提供する。これらの資金源から調達した累計資本(債務融資を含む)は2023年9月30日現在で約7900万ドルである。
私たち は設立以来大きな被害を受けた。私たちが利益を達成するのに十分な製品収入を生み出すことができるかどうかは、私たちの1つ以上の候補製品の成功した開発と商業化にかかっている。2023年9月30日と2022年9月30日までの3ヶ月間の純損失はそれぞれ600万ドルと400万ドルで、2023年9月30日と2022年9月30日までの9ヶ月間の純損失はそれぞれ1660万ドルと1310万ドルだった。我々の純損失変動の説明については、次の“経営業績”を参照されたい。2023年9月30日までの累計赤字は8600万ドルです。
私たち は短期的に巨額の費用が発生し、運営損失を増加させることを予想しています。持続的な活動に関連する費用は大幅に増加すると予想されています
| ● | CTX-1301に対する規制部門の承認を求める; |
| ● | 我々の既存および新しい候補製品(主にCTX−1301)の研究開発活動を継続する |
| ● | 著者らの臨床前研究と臨床試験のために用品を製造し、主にCTX-1301である |
| ● | CTX−1301の販売およびマーケティングをサポートするためのアウトソーシング商業インフラ; |
| ● | を上場企業として運営する. |
2023年9月30日まで、私たちは200万ドルの現金と現金同等物を持っている。私たちの現在の業務計画によると、今回発行された純収益は、私たちの手元の現金を加えて、2024年第2四半期末までの資本需要を満たすと信じています。また、FDAの潜在的承認を得るためにCTX-1301の機密協定を2025年上半期に提出するためには、約800-1,000万ドルの追加資本が必要になると考えられます。私たちはまた私たちの他の計画と商業化努力を推進するための追加的な資金が必要だ。以下の“流動資金と資本資源”を参照。
| 100 |
私たちが製品収入を作る能力は、私たちの1つ以上の候補製品の成功的な開発、規制承認、最終商業化にかかっているだろう。私たちは、製品販売から相当な収入を得ることができる前に、株式、債務融資、または他の資本源(他社との潜在的な協力や他の戦略的取引を含む)を売却することで、私たちの運営に資金を提供することが予想される。受け入れ可能な条件で、私たちは十分な資金を得ることができないかもしれないし、全く持っていないかもしれない。もし私たちが必要な時に資金を調達したり、このような合意に達しなかったら、私たちは候補製品の開発と商業化を大幅に延期、削減、または停止しなければならないかもしれない。
臨床、 製造と業務更新
CTX-1301: 成人ADHD患者におけるCTX-1301の治療効果と安全性及び効果と持続時間を評価する第三段階成人用量最適化研究は2022年12月に開始され、2023年6月に完成した。研究結果はすでに2023年精神医学大会で公表され、2024年1月に開催された2024年アメリカADHD及び関連疾病専門協会(APSARD)で公表される。この第三段階CTX-1301研究 (NCT 05631626)は成人実験室教室環境において21名の成人(年齢範囲:18-55歳)のCTX-1301の治療効果と安全性およびCTX-1301の発病と持続時間を評価した。それは主要な治療効果の終点で統計学的意義に達しなかったが、CTX-1301はプラセボと比べ、永久製品業績評価(“PERMP”)点数の改善に顕著な改善傾向があることを示した。プラセボと比較して,CTX−1301 CTX−1301を用いた臨床全体印象尺度(“cgi−S”)スコアも有意な改善を示し,この二次終点に対して有意な統計学的意義があった。この試験では,CTX−1301の治療効果が有意であった−30分から,CTX−1301が一日中の活動でADHD症状を改善できることが証明された。
そのほか、著者らは2023年第3四半期に2つの児童と青少年患者に対するCTX-1301第3段階臨床研究-固定用量研究(NCT 05286762)と実験室授業環境中の投与量最適化効果と持続時間研究(NCT 05924594)を開始した。FDAから受けたCTX−1301臨床計画に関する指導に基づき,両研究の登録を中止した。これらの研究では登録されたすべての患者が研究を完了し,それに応じてデータを報告することができる。また,このガイドラインによると, は2025年上半期に505(B)(2)節の経路によりCTX−1301のセキュリティプロトコルを提出し,フォカリンXRを参考に市販薬とし,その有効性と安全性データを我々のCTX−1301臨床計画の生物利用度/生物同等性データ と有効性/安全性データとともに承認の基礎とすることが予想される。CTX-1301に対するFDAの承認を得たら、私たちは4回の実験を行うかもしれない。
私たちは早ければ2025年にCTX-1302(右旋フェニルプロピルアミン)の臨床計画を開始する予定で、これは私たちがADHDを治療する2つ目の研究資産であり、追加の資本資源を待つ。
Socialは、複雑な調合と製造挑戦の解決に取り組んでいるCDMOであり、主に小分子治療 開発において、著者らの主要なADHD候補薬物CTX-1301のすべての臨床、登録と商業ロットを生産する。2023年4月、私たちは、ジョージア州ガイエンスビルにあるSocialの製造キットの第3段階試験で拡張可能なCTX-1301を生産し、提供した設備を備えたSocialの主要候補CTX-1301(ピペラゾンデキサメタゾン)の独自PTR製造プロセスをSocialに移管することに成功した。
2023年3月、我々は総合生命科学商業化会社Indegeneとの共同商業化合意を発表し、著者らの主要な候補薬物CTX-1301(ピペラゾンデキサメタゾン)に商業支援を提供した。この協定は独特な全方位ルートマーケティング方法 を通じて機能を跨ぐサービスをカバーし、著者らの第三段階の臨床試験期間中の商用前支持 を成功に管理し、そしてFDAが許可した後に全国範囲内で有効にCTX-1301を商業化することを目的としている。
CTX-2103: 私たちはすでにCTX-2103(ブチルスピロロン)を開発する計画を開始し、焦慮を治療するために、これはアメリカで最もよく見られる心理健康問題の一つである。処方研究を完了し、このような3つの精密決定時用量を提供するブチルスピロロンと1つの即時放出用量のトリモイルシクロケトンとの薬物動態を評価した。さらに、シンチグラフィは、放出位置および開始を確認するために消化管における錠剤の輸送を示し、その後、CTX-2103製剤の完全放出曲線を確立するために薬物動態データに関連付けられる。データに見られる薬物動態曲線から,CTX−2103はブチロシノンの三次放出を実現した。これらの結果は必要な重要な情報を提供し、私たちはbr要求がFDAとIND前会議を開催し、著者らのCTX-2103臨床と監督計画の設計を討論することができ、この計画は2023年第4四半期に開催される。著者らはFDAからCTX-2103制御経路に関する意見と臨床研究の設計を受け取り、INDを提出した。FDAのフィードバックによれば、505(B)(2) 経路の下でCTX−2103の承認を得ることができると信じており、これは、通常505(B)(1)全NDA経路よりも少ない時間およびリソースを必要とする。
| 101 |
CTX-1302: 私たちは最初に2025年にCTX-1302(右旋フェニルプロピルアミン)の臨床計画を開始する予定で、これは私たちがADHDを治療する2つ目の研究資産であり、追加の資本資源を待つ。
PTRTM プラットフォーム:私たちは、私たちのPTRプラットフォームを超えた許可と、アメリカ以外で私たちの候補製品を許可する機会を評価し続けます。また、BDD Pharma Limitedとの関係を拡大する機会を評価しています。
証券発行
ATM機 プロトコル
吾らは2023年1月にH.C.Wainwright&Co.,LLC(“HCW”) と2023年5月に改訂された市場発売協定(“ATMプロトコル”)を締結し,この合意により,吾らは時々HCWを通して普通株を発売·販売することができ,総収益は最高497万ドル(ATMプロトコルに記載されている条項や条件および制限)に達する。2023年9月30日までの3ヶ月間、ATM協定により76,943株の普通株を売却し、HCWに対する49,502ドルの補償と他の行政費用を差し引いたところ、純収益は1,595,429ドルだった。2023年9月30日までの9ヶ月間、吾らはATM協定により82,410株の普通株を販売し、当社に支払った59,898ドルの補償及びその他の行政費用を差し引いた純額は1,696,407ドルであった。2023年9月30日以降、ATM協定により283,300株の普通株 を売却し、HCWに対する97,512ドルの補償と他の管理費用を差し引いたところ、純収益は3,115,303ドル となった
株式信用限度額
2023年4月、リンカーンパーク資本基金有限責任会社(“リンカーンパーク”)と購入契約(“リンカーンパーク協定”)を締結した。リンカーン公園協定によると、リンカーン公園はリンカーン公園協定の36ヶ月以内に、リンカーン公園協定に規定されている条項と条件と制限に基づいて、時々私たちが自分で合計1200万ドルの普通株を購入することを決定することに同意した。2023年9月30日までの3ヶ月間、リンカーンパーク協定により12,000株の普通株を売却し、純収益は196,167ドルだった。2023年9月30日までの9ヶ月間、リンカーンパーク協定により25,500株の普通株を売却し、純収益は450,427ドルであった。その後、2023年9月30日まで、私たちはリンカーン公園協定に基づいてどんな普通株も売却しなかった。
個人配給
2023年8月11日、吾らはWFIAと証券購入協定(“2023年8月購入合意”) を締結し、ナスダック証券市場規則に基づき、私募方式で市場定価で91,158株自社普通株を発行し、1株当たりの購入価格は10.97ドルであり、吾等に約100万ドルの毛収入( “WFIA私募”)をもたらした。
債務(Br)換算
2022年8月、Cingate Inc.の完全子会社CTXはWFIAに元本500万ドル(“元本”)の元手形を発行し、2023年5月に改訂·再予約された承諾票 手形(“WFIA手形”)を発行し、元の手形での元本金額を300万ドルから800万ドル増加させた(“br}”2023年WFIA債務融資“)。
2023年9月8日、Cingate Inc.およびCTXとWFIAは手形変換協定を締結し、これにより、WFIAはWFIA手形項の元の元本金額をそのすべての課税利息、または5,812,500ドルと合わせて、予備金権証 (以下“9月WFIA予備資本権証”と呼ぶ)に変換し、341,912株の普通株を購入し、換算価格は1株当たり 9月のWFIA予資資権証17.00ドルであることに同意した。2023年9月8日、私たちの普通株のナスダックでの終値は1株11.55ドルです。9月のWFIA事前承認株式証は満期日がなく,ただちに1株0.002ドルで行使できるが,行使後,取引法第13(D)節の目的でWFIAとその関連会社は実益が19.99%以下の普通株式流通株を持つことになる。
2024年1月25日に、Cingate Inc.およびCTXとWFIAは手形変換プロトコル(“2024年1月手形変換プロトコル”) を締結し、これにより、WFIAは、WFIA手形項の下に残った3,000,000ドル元金を、そのすべての課税利息(または3,287,500ドル)とともに事前資本金権証(“1月WFIA予備金権証”)に変換し、687,043株普通株を購入し、1月WFIA事前資本金権証4.785ドルの転換価格で計算することに同意した。2024年1月24日、ナスダック普通株の終値は4.35ドルだった。1月のWFIA事前承認株式証は満期日がなく、直ちに1株0.0001ドルの行使価格で行使することができ、取引法13(D)条の規定により、WFIAとその関連会社は実益が我々普通株発行株式の19.99%を超えないことを条件とする。
| 102 |
WFIA手形は無担保債券であり,年利は15%である。未償還元金およびすべての未払い利息は、違約事件により加速しない限り、2025年8月8日に満期となり、支払わなければならない。WFIAは、各カレンダー四半期の最初の5営業日以内に、通知されてから120日以内にすべての未返済元本と利息を支払うことを要求する権利があります。私たち は、追加料金や罰金がないWFIAチケットの全部または一部を前払いすることが許可されています。返済された金額 の再借入は許されないことを前提としています。2023年9月30日現在、WFIA手形の元本は300万ドル、計上すべき利息は20万ドルである。 は残りの元本金額にすべての課税利息を加えて1月のWFIA事前融資権証を発行した後、WFIA手形は全額支払われており、当社はWFIA手形により何の義務も負わない。以下の“流動性と資本資源”を参照されたい。
公開サービス
2023年9月11日、吾らは1人の機関投資家と証券購入協定を締結し、この合意に基づいて、吾らは86,000株の普通株、予融資権証を発行して合計260,260株の普通株、Aシリーズ株式承認証 を購入して最大346,260株の普通株及びBシリーズ承認株式証を購入して最大173,130株の普通株を購入した({br]“2023年9月発売”)。2023年9月の株式発行は2023年9月13日に終了します。普通株及びセットAシリーズとBシリーズ株式承認証の合併購入価格は1株11.55ドルである。1株当たりの事前融資権証とセットAシリーズとBシリーズ株式承認証の合併購入価格は11.55ドルであり、普通株とセット株式証を代表する1株公開発行価格から1株事前融資承認株式証を引いた1株0.002ドルの株式価格である。事前資金調達権証は、発行日後のいつでも行使することができ、満期日がない。事前資本権証所有者がその連合会社と共に引受権証を行使した後、実益が4.99%(または所有者が選択した場合、9.99%)を超える発行された普通株式数 を有する場合、その所有者は当該等承認持分証を行使することができない。Aシリーズ株式承認証の行使価格は1株11.55ドルで、2023年11月3日に発行することができ、即ち株主は引受証を行使する時に発行できる株式の発効日に行使することができ、初期行使後5年で満期になる;Bシリーズ株式承認証の行使価格は1株11.55ドルであり、2023年11月3日、即ち株主が株式承認証の発行可能株式を行使することを許可する発効日に行使を開始し、初期行使後2年以内に満期になる。2023年9月の発売により,配給代理費やその他の発売費用を差し引く前に,約400万ドルの毛収入を受け取った。
経営成果の構成要素
収入.収入
Brが成立して以来、私たちは何の収入も生まれておらず、近い将来に製品販売から何の収入も生まれたくありません。もし私たちが候補製品の開発に成功して規制部門の承認を得たら、あるいは第三者と協力 が合意できるかもしれないなら、私たちは将来製品販売や 協力許可契約の支払いから収入を得ることができるかもしれません。
運営費用
研究と開発費
研究と開発費用には、主に、私たちの候補製品の発見と開発に生じるコストが含まれている
| ● | 我々の臨床試験および一部の臨床前活動を行うCROおよび調査地点との第三者合意に基づいて発生する費用 |
| 103 |
| ● | 原材料コストや臨床試験や他の開発試験のための材料の製造コスト |
| ● | 研究開発活動に従事している従業員の給料と福祉を含む費用 |
| ● | 製造設備コスト、減価償却、その他の分担費用; |
| ● | 契約規制サービスのために支払われる費用 と、規制機関(FDAを含む)が私たちの候補製品を承認するために支払う費用 です。 |
我々 は発生した費用に応じてすべての研究開発コストを支出しているが,研究開発に使用されている製造設備は除外されており,これらの設備は資本化されており,その使用予定寿命内に償却されている。外部開発活動のコストは,サプライヤから提供された情報を用いて特定のタスクを達成する進捗を評価することで確認される.これらの活動の支払いは、発生したコストパターンとは異なる可能性があり、前払いまたは計算コストとして我々の連結財務諸表に反映される可能性がある単一の合意の条項に基づく。
研究と開発活動は私たちの業務モデルの核心だ。予測可能な未来には,我々の候補製品の臨床開発を継続するにつれて,我々の研究開発費は増加し続けると予想される。製品が臨床開発の後期段階に入るにつれて、それらの開発コストは通常臨床開発の早期段階より高くなり、これは主に後期臨床試験の規模と持続時間が増加しているためである。歴史的に見ると、我々の研究開発コストは主にCTX-1301の開発と関係がある。私たちがCTX-1301、CTX-1302、およびCTX-2103を推進し、任意の他の潜在的な候補製品を決定するにつれて、私たちは引き続き私たちの直接外部研究開発コストをこれらの製品に分配します。私たちは、私たちの現在の現金と現金等価物、ならびに任意の将来の持分または債務融資または他の資本源から私たちの研究開発費に資金を提供する予定です。
一般料金 と管理費用
一般費用と行政費用は主に従業員の行政、行政、財務機能に関する賃金と関連コスト を含む。一般および行政費用には、法律、会計、監査、税務、コンサルティングサービスの専門費用、保険、オフィス、出張費用も含まれています。
私たちの一般と管理費用は今後増加すると予想されています。私たちは候補製品の潜在的な商業化を含めて、私たちの持続的な運営を支援するために、一般と管理者 を増やしたからです。私たちは上場企業に関連する費用増加を経験し、会計、監査、法律、規制と税務コンプライアンスサービスのコスト、 取締役と幹部保険、そして投資家と広報コストを含む。
利息 とその他の収入(費用)、純額
利子 および他の収入(支出)、純額には、関連側手形に対応する利息支出と、私たちの現金および現金等価物(通貨市場基金を含む)が稼いだ利息が含まれている。私たちの投資政策の主な目標は流動性と保証だ。
重要な会計政策と重要な判断と見積もり
我々の 連結財務諸表は米国公認会計原則(“U.S. GAAP”)に基づいて作成されている。米国公認会計原則に基づいて連結財務諸表を作成することは、報告の資産および負債額、連結財務諸表の日付までの、または有資産および負債の開示、および報告期間内の報告の費用金額に影響を与えるために、管理層に推定brおよび仮定を要求する。実際の結果 は見積りとは異なる可能性がある.
| 104 |
我々の主な会計政策は、我々の総合財務諸表付記2により詳細に記載されているが、以下の会計政策は、我々が総合財務諸表を作成するために使用される判断と推定が最も重要であると考えられる。
研究と開発コスト
研究·開発コストは,我々の候補製品開発に関するすべての直接·間接コスト を含む発生時に費用を計上する.これらの費用には、第三者に支払われる研究開発および製造サービス、人員コスト、および製造設備の減価償却が含まれる。報告期間末に、第三者サービスプロバイダに支払う金額と、研究や開発目標を達成する上での見積もり進捗を比較します。より多くの 情報が利用可能になるにつれて,このような見積りが変化する可能性がある.サービス提供者への支払い時間と、提供されたサービスによる進展を推定することによって、これらのコストに関連する前払いまたは課税費用の純額を記録することができる。
株に基づく報酬
2021年計画によると、私たちは2022年と2021年に特定の従業員と取締役に非制限株式オプションを付与した。これらのオプションは2022年に1株20ドルから31.4ドルの行使価格で付与され、2021年の行使価格は1株120.00ドルで、私たちの初公募株に関連する普通株が公衆に発行される価格だ。これらのオプションの期限は10年であり,帰属期限は即日から4年まで様々である.
2022年12月31日と2021年12月31日までの年間で,付与オプションの付与日公正価値に基づいて,それぞれ800,796ドルと43,835ドルの株式報酬支出を記録した.各オプション付与の公正価値は、付与された日にブラック·スコアーズオプション定価モデルを使用して推定される。公正価値は、報酬の必要なサービス期間(通常はホーム期間)に直線的に償却されて補償コストとなる。
ブラック·スコアーズオプション定価モデルを用いて付与された株式オプションの公正価値を推定する仮定については、我々の合併財務諸表脚注11を参照されたい。
経営成果
2023年9月30日と2022年9月30日までの3ヶ月間の比較:
次の表は、2023年9月30日と2022年9月30日までの3ヶ月間の運営結果をまとめています
| 3ヶ月まで ヶ月 | % | |||||||||||||||
| 9月30日 | 増す | 増す | ||||||||||||||
| (単位:千) | 2023 | 2022 | (減少) | (減少) | ||||||||||||
| 運営費用 : | ||||||||||||||||
| 研究開発 | $ | 3,924 | $ | 2,123 | $ | 1,801 | 84.8 | % | ||||||||
| 通常 と管理 | 1,826 | 1,845 | (19 | ) | -1.0 | % | ||||||||||
| 運営損失 | (5,750 | ) | (3,968 | ) | 1,782 | (44.9 | )% | |||||||||
| 利息 とその他の収入(費用)、純額 | (229 | ) | (59 | ) | (170 | ) | NM | |||||||||
| 純損失 | $ | (5,979 | ) | $ | (4,027 | ) | $ | 1,612 | (40.0 | )% | ||||||
| 105 |
研究開発費
次の表は、2023年9月30日と2022年9月30日までの3ヶ月間の研究開発(R&D)費用をまとめています
| 3ヶ月まで ヶ月 | % | |||||||||||||||
| 9月30日 | 増す | 増す | ||||||||||||||
| (単位:千) | 2023 | 2022 | (減少) | (減少) | ||||||||||||
| 臨床br手術 | $ | 2,346 | $ | 581 | $ | 1,765 | 303.8 | % | ||||||||
| 薬品の生産と調合 | 817 | 673 | 144 | 21.4 | % | |||||||||||
| 人件費 | 644 | 860 | (216 | ) | (25.1 | )% | ||||||||||
| 規制コスト | 117 | 9 | 108 | NM | ||||||||||||
| 研究開発費総額 | $ | 3,924 | $ | 2,123 | $ | 1,801 | 84.8 | % | ||||||||
2023年9月30日までの3カ月間の研究開発費は390万ドルで、2022年9月30日までの3カ月より180万ドル増加し、84.8%増となった。この変化は主に2022年同期と比べて臨床活動が増加した結果である。 は2023年第3四半期に、著者らはCTX-1301の2つの第3段階研究、即ち小児科用量最適化開始と持続時間研究 及び固定投与量小児科と青少年の安全性と有効性研究を開始した。また、CTX-1301の第三段階成人用量最適化研究は2023年6月に完成した。
一般料金と管理費用
次の表は、2023年9月30日と2022年9月30日までの3ヶ月の一般と行政(G&A)費用をまとめています
| 一般料金 と管理費用 | ||||||||||||||||
| 3ヶ月まで ヶ月 | % | |||||||||||||||
| 9月30日 | 増す | 増す | ||||||||||||||
| (単位:千) | 2023 | 2022 | (減少) | (減少) | ||||||||||||
| 人件費 | $ | 672 | $ | 595 | $ | 77 | 12.9 | % | ||||||||
| 弁護士費と弁護士費 | 511 | 373 | 138 | 37.0 | % | |||||||||||
| 入居率 | 144 | 109 | 35 | 32.1 | % | |||||||||||
| 保険 | 398 | 669 | (271 | ) | (40.5 | )% | ||||||||||
| 他にも | 101 | 99 | 2 | (2.0 | )% | |||||||||||
| 一般と行政費用の合計 | $ | 1,826 | $ | 1,845 | $ | (19 | ) | (1.0 | )% | |||||||
2023年9月30日までの3カ月間のG&A総支出は、2022年9月30日までの3カ月と横ばいであり、これは主に保険コストの低下によるものであり、2022年12月に更新された年間役員や上級管理職の保険料低下に関係しているが、2023年第3四半期の資金調達活動に関する法的費用増加によって相殺されている。
利息 とその他の収入(費用)、純額
次の表は、2023年9月30日と2022年9月30日までの3ヶ月の利息とその他の収入(費用)純額をまとめています
| 利息 とその他の収入(費用): | ||||||||||||||||
| 3ヶ月まで ヶ月 | % | |||||||||||||||
| 9月30日 | 増す | 増す | ||||||||||||||
| (単位:千) | 2023 | 2022 | (減少) | (減少) | ||||||||||||
| 利息 とその他の収入(費用)、純額 | $ | (229 | ) | $ | (59 | ) | $ | (171 | ) | (289.8 | )% | |||||
2023年9月30日までの3ヶ月間の利息とその他の収入(支出)総額は、主にWFIA Note項の800万ドル元金の利息と関係があり、そのうち500万ドル元金は2023年9月初めに株式に転換された。
2022年9月30日までの3カ月間の純利息とその他の収入(支出)総額は、主に2022年8月のWFIA手形項目で500万ドルの初期元金の利息と相殺される。
| 106 |
2023年9月30日までの9ヶ月と2022年9月30日の比較:
次の表は、2023年9月30日と2022年9月30日までの9ヶ月間の運営結果をまとめています
| 9ヶ月まで ヶ月 | % | |||||||||||||||
| 9月30日 | 増す | 増す | ||||||||||||||
| (単位:千) | 2023 | 2022 | (減少) | (減少) | ||||||||||||
| 運営費用 : | ||||||||||||||||
| 研究開発 | $ | 10,508 | $ | 7,064 | $ | 3,444 | 48.8 | % | ||||||||
| 通常 と管理 | 5,454 | 5,963 | (509 | ) | (8.5 | )% | ||||||||||
| 運営損失 | (15,962 | ) | (13,027 | ) | 2,935 | (22.5 | )% | |||||||||
| 利息 とその他の収入(費用)、純額 | (638 | ) | (44 | ) | 594 | NM | ||||||||||
| 純損失 | $ | (16,600 | ) | $ | (13,071 | ) | $ | 3,529 | (27.0 | )% | ||||||
研究開発費
次の表は、2023年9月30日と2022年9月30日までの9ヶ月間の研究開発費をまとめています
| 9ヶ月まで ヶ月 | % | |||||||||||||||
| 9月30日 | 増す | 増す | ||||||||||||||
| (単位:千) | 2023 | 2022 | (減少) | (減少) | ||||||||||||
| 臨床br手術 | $ | 5,071 | $ | 2,082 | $ | 2,989 | 143.6 | % | ||||||||
| 薬品の生産と調合 | 3,254 | 2,827 | 427 | 15.1 | % | |||||||||||
| 人件費 | 1,902 | 2,097 | (195 | ) | (9.3 | )% | ||||||||||
| 規制コスト | 281 | 58 | 223 | 384.5 | % | |||||||||||
| 研究開発費総額 | $ | 10,508 | $ | 7,064 | $ | 3,444 | 48.8 | % | ||||||||
2023年9月30日までの9カ月間の研究開発費は1,050万ドルで、2022年9月30日までの9カ月より340万ドル増加し、48.8%増となった。この増加は主に2022年同期と比べ、臨床活動が著しく増加したことによるものである。著者らは2022年末にCTX-1301の第三段階成人用量最適化研究を開始し、2023年6月に完成した。また、著者らは重要な第三段階児童と青少年固定投与量の安全性と有効性研究及び第三段階児童用量最適化開始と持続時間研究に関連する費用を発生し、この2つの研究はいずれも2023年第3四半期にスタートしたCTX-1301である。製造業活動は2023年にも増加しており,CTX−1301第3段階研究の臨床用品の製造が完了したからである。
一般料金と管理費用
次の表は、2023年9月30日と2022年9月30日までの9ヶ月間のG&A費用をまとめています
| 9ヶ月まで ヶ月 | % | |||||||||||||||
| 9月30日 | 増す | 増す | ||||||||||||||
| (単位:千) | 2023 | 2022 | (減少) | (減少) | ||||||||||||
| 人件費 | $ | 2,023 | $ | 1,803 | $ | 220 | 12.2 | % | ||||||||
| 弁護士費と弁護士費 | 1,495 | 1,422 | 73 | 5.1 | % | |||||||||||
| 入居率 | 396 | 353 | 43 | 12.2 | % | |||||||||||
| 保険 | 1,173 | 2,013 | (840 | ) | (41.7 | )% | ||||||||||
| 他にも | 367 | 372 | (5 | ) | (1.3 | )% | ||||||||||
| 一般と行政費用の合計 | $ | 5,454 | $ | 5,963 | $ | (509 | ) | (8.5 | )% | |||||||
| 107 |
G&A総支出は2023年9月30日までの9カ月間で540万ドルで、2022年9月30日までの9カ月間で50万ドル減少し、減少幅は8.5%だった。この変化は,主に2022年12月に更新された年次役員や上級管理者の保険料低下により保険コストが80万ドル減少したが,臨床人員の増加と年間給与の増加による人員費用の増加がこの影響を相殺したためである。
利息 とその他の収入(費用)、純額
次の表は、2023年9月30日と2022年9月30日までの9ヶ月間の利息とその他の収入(費用)純額をまとめています
| 9ヶ月まで ヶ月 | % | |||||||||||||||
| 9月30日 | 増す | 増す | ||||||||||||||
| (単位:千) | 2023 | 2022 | (減少) | (減少) | ||||||||||||
| 利息 とその他の収入(費用)、純額 | $ | (638 | ) | $ | (44 | ) | $ | (594 | ) | NM | ||||||
2023年9月30日までの9カ月間の純利息とその他の収入(支出)総額は、主に2022年8月のWFIA手形項目で最初の500万ドル元金の利息に関係し、その後2023年5月に800万ドルに増加し、2023年9月初めに株式に移行して500万ドル減少したが、投資残高が稼いだ利息はこの利息を相殺した。
2022年9月30日までの9カ月間の純利息とその他の収入(支出)総額は、主に2022年8月のWFIA手形項目で500万ドルの初期元金の利息と相殺される。
2022年12月31日までと2021年12月31日までの年度比較
次の表は、2022年12月31日現在と2021年12月31日現在の年間運営結果をまとめたものである
| 年 終わり | % | |||||||||||||||
| 十二月三十一日 | 増す | 増す | ||||||||||||||
| (単位:千) | 2022 | 2021 | (減少) | (減少) | ||||||||||||
| 運営費用 : | ||||||||||||||||
| 研究開発 | $ | 8,995 | $ | 8,410 | $ | 585 | 7.0 | % | ||||||||
| 通常 と管理 | 8,507 | 12,269 | (3,762 | ) | (30.7 | )% | ||||||||||
| 運営損失 | (17,502 | ) | (20,679 | ) | (3,177 | ) | 15.4 | % | ||||||||
| 利息 とその他の収入(費用)、純額 | (174 | ) | (31 | ) | 143 | 461.3 | % | |||||||||
| 純損失 | $ | (17,676 | ) | $ | (20,710 | ) | $ | (3,034 | ) | 14.6 | % | |||||
研究開発費
次の表は、2022年12月31日と2021年12月31日までの年間研究開発費をまとめています
| 年 終わり | % | |||||||||||||||
| 十二月三十一日 | 増す | 増す | ||||||||||||||
| (単位:千) | 2022 | 2021 | (減少) | (減少) | ||||||||||||
| 臨床br手術 | $ | 3,534 | $ | 1,086 | $ | 2,448 | 225.4 | % | ||||||||
| 薬品の生産と調合 | 2,840 | 1,429 | 1,411 | 98.7 | % | |||||||||||
| 人件費 | 2,521 | 5,874 | (3,353 | ) | (57.1 | )% | ||||||||||
| 規制コスト | 100 | 21 | 79 | 376.2 | % | |||||||||||
| 研究開発費総額 | $ | 8,995 | $ | 8,410 | $ | 585 | 7.0 | % | ||||||||
| 108 |
2022年12月31日までの年間研究開発費は900万ドルで、2021年12月31日までの年度より60万ドルか7.0%増加した。この増加はCTX-1301の臨床と製造コストの著しい増加と関係があり、著者らは2022年第4四半期に食品br効果研究を完成し、そして2022年末に第三段階の成人用量最適化研究を開始したからである。また,CTX−1301第3段階試験を生産する臨床用品に関連して,われわれの製造コストは2022年に増加した。これらの増加は, が460万ドルを2021年の研究開発費に計上することによる人員費用の減少によって相殺されているが,これはPIUSを修正した使い捨て非現金補償費用 のためである。一度の非現金費用による人員支出の減少は,2022年の年度昇給と臨床活動の増加が予想される2021年末に増加する人員によって相殺される。
一般料金と管理費用
次の表は、2022年12月31日と2021年12月31日までの年間G&A費用をまとめています
| 年 終わり | % | |||||||||||||||
| 十二月三十一日 | 増す | 増す | ||||||||||||||
| (単位:千) | 2022 | 2021 | (減少) | (減少) | ||||||||||||
| 人件費 | $ | 2,590 | $ | 9,729 | $ | (7,139 | ) | (73.4 | )% | |||||||
| 弁護士費と弁護士費 | 2,233 | 1,443 | 790 | 54.7 | % | |||||||||||
| 入居率 | 478 | 484 | (6 | ) | (0.01 | ) | ||||||||||
| 保険 | 2,611 | 325 | 2,286 | 703.4 | % | |||||||||||
| 他にも | 595 | 288 | 307 | 106.6 | % | |||||||||||
| 一般と行政費用の合計 | $ | 8,507 | $ | 12,269 | $ | (3,762 | ) | (30.7 | )% | |||||||
2022年12月31日までの年度のM&A総支出は850万ドルで、2021年12月31日までの年度より380万ドルか30.7%減少した。人事支出が710万ドルまたは73.4%減少したのは、主にG&A人員が2021年に810万ドルの使い捨て非現金補償費用を記録したためであり、この費用は利益利益単位(“PIUS”)の修正に関係している。 非現金補償費用による人員支出の減少は、2021年末に発売しようとした際に増加した人員賃金支出によって相殺される。人員費の減少は、法的費用や監査費用の増加を含む他の費用の増加によって相殺され、これは主に2022年末に将来の資本調達に備えて増加した活動によるものである。また,役員や上級管理職の保険料 は上場企業にとってはるかに高いため,保険コストは前年比で大幅に増加している。
利息 とその他の収入(費用)、純額
次の表は、2022年12月31日と2021年12月31日までの年度の利息とその他の収入(費用)純額をまとめています
| 年 終わり | % | |||||||||||||||
| 十二月三十一日 | 増す | 増す | ||||||||||||||
| (単位:千) | 2022 | 2021 | (減少) | (減少) | ||||||||||||
| 利息 とその他の収入(費用)、純額 | $ | (174 | ) | $ | (31 | ) | $ | 143 | 461.3 | % | ||||||
利息とその他の収入(支出)総額は、2022年と2021年の純額は主に未償還支払手形による利息に関連し、投資残高で稼いだ利息で相殺される。2022年の利息支出が大幅に増加したのは,2022年8月のWFIAへの500万ドル関連側手形に30万ドルの利息が生じたためである。
| 109 |
キャッシュフロー
9月30日までの9ヶ月間 | ||||||||
| 2023 | 2022 | |||||||
| 純現金経営活動 | $ | (12,472 | ) | $ | (11,676 | ) | ||
| 純額 投資活動用の現金 | (37 | ) | (10 | ) | ||||
| 純融資活動から提供された現金 | 9,139 | 4,989 | ||||||
| 現金と現金等価物純減少 | $ | (3,370 | ) | $ | (6,697 | ) | ||
経営活動のキャッシュフロー
2023年9月30日までの9カ月間、経営活動で使用された現金純額は1,250万ドル。経営活動で使用されている現金は、主に私たちの運営に資金を使って私たちの候補製品を開発し、1,660万ドルの純損失を招いたのは、株式ベースの給与支出70万ドルと減価償却支出40万ドルの2つの非現金プロジェクトの影響によるものである。経営資産と負債の変化には,雑売掛金が20万ドル減少したが,これは主に2022年12月31日現在売掛金として記録されている保険請求の回収可能額を徴収し,前払い費用や他の流動資産が140万ドル減少したためであり,これは主に我々のCMOと契約研究機関の預金を用いて,貿易帳簿や売掛金に対応する費用が150万ドル増加したためであり,これは開発活動の増加による臨床·製造費の増加,WFIA Noteの課税利息の増加によるものである。主に資金調達活動に関する法律活動に関する法的費用の増加に対応する。
2022年9月30日までの9カ月間、経営活動で使用された現金純額は1,170万ドル。経営活動で使用されている現金 は,主に我々の運営に資金を用いて候補製品を開発し,純損失1280万ドルを招いたためであり,これは2つの非現金プロジェクトの影響,すなわち株による報酬支出60万ドルと減価償却30万ドルによるものである。経営資産及び負債の変動には、2022年初めに私たちに不足している大部分の賃金及び研究開発税務相殺を受けたことによる雑債権の減少、及び臨床開発活動の前払い額に関する前払い支出及びその他の流動資産の増加がある。
投資活動のキャッシュフロー
投資活動で使用される現金純額は、2023年9月30日と2022年9月30日までの9ヶ月間、我々の研究開発を支援するために設備を購入することに関連している。
融資活動のキャッシュフロー
2023年9月30日までの9カ月間,融資活動が提供する現金純額は,主に2023年9月に発売された総収益が約400万ドル,WFIA私募発行の総収益が約100万ドル,リンカーンパーク協定とATM協定により普通株が発行された収益に関連している。しかも、私たちは2023年のWFIA債務融資から300万ドルを獲得した。
2022年9月30日までの9カ月間、融資活動が提供した現金純額は、主に2022年8月に受け取った500万ドルWFIA手形の収益に関係している。
| 年 終わり | ||||||||
| 十二月三十一日 | ||||||||
| 2022 | 2021 | |||||||
| 純現金経営活動 | $ | (15,882 | ) | $ | (10,432 | ) | ||
| 純額 投資活動用の現金 | (153 | ) | (815 | ) | ||||
| 純融資活動から提供された現金 | 4,899 | 26,542 | ||||||
| 現金と現金等価物の純増加(減少) | $ | (11,136 | ) | $ | 15,295 | |||
| 110 |
経営活動のキャッシュフロー
2022年12月31日までの年度、経営活動で使用されている現金純額は1,590万ドル。経営活動で使用されている現金は主に私たちの運営に資金を使って私たちの候補製品を開発するために1,770万ドルの純損失を出しています。これは2つの重要な非現金プロジェクトの影響を受ける前、すなわち株による報酬支出180万ドルと減価償却支出40万ドルです。経営資産と負債の変化には、2022年の研究開発と従業員の留保税免除による雑売掛金の50万ドルの減少と、CTX-1301第3段階研究の開始に関連する保証金の臨床および製造サプライヤーへの支払いと、我々の新しいCMOに製造活動保証金を支払うことによって増加した前払い費用40万ドルが含まれる。
2021年12月31日までの年度,経営活動で使用されている現金純額は1,040万ドルである。経営活動で使用されている現金は、主に私たちの運営に資金を使って私たちの候補製品を開発したため、純損失が2,070万ドルになりました。これは2つの重要な非現金プロジェクトの影響 の前で、この2つのプロジェクトは一度の非現金PIU費用1270万ドルと減価償却費用70万ドルです。営業資産と負債の変化 は、2021年12月に従業員に支払われたすべての繰延賃金金額が支払われたため、売掛金と売掛金を含めて130万ドル減少した。前払い費用も120万ドル増加しましたが、主に私たちの年間役員と上級管理職保険料の頭金があり、この保険料は私たちが初公募株を完成させた後に大幅に増加しました。
投資活動のキャッシュフロー
2022年12月31日現在と2021年12月31日までの年間投資活動で使用される現金純額は、主に設備購入に用いられ、研究開発を支援しています。
融資活動のキャッシュフロー
2022年12月31日現在、融資活動が提供する現金純額は、主にWFIAに対応した500万ドルの関連側手形による金に関係している。
2021年12月31日までの年度内に、融資活動が提供する現金純額は主に208,333株普通株および当社初公開株式に関する付帯承認株式証の発行に関連しており、株価は1株当たり120.00ドルおよび付随する株式承認証である。引受業者割引と私たちが支払った費用を差し引いた後、私たちは2500万ドルの毛収入と2040万ドルの純収益を得ました。私たちが初めて公募する前に、CTX 3,243,201個の株式単位の発行に関連した710万ドルを受け取りました。私たちは2021年12月に関連先の支払手形の返済に50万ドルを支払った。
流動性 と資本資源
流動資金源
2012年の設立以来2023年9月30日まで、私たちは収入が発生せず、運営により重大な運営損失と負のキャッシュフローが発生した。私たちの現在の業務計画によると、今回発行された純収益は、私たちの手元の現金を加えて、2024年第2四半期末までの資本需要を満たすと信じています。今回の発行後、私たちの運営を支援するために追加資金を調達し、私たちが計画している開発と商業化活動を支援していく必要があります。
2023年9月30日までの3ヶ月間、私たちはATM協定に従って76,943株の普通株を売却し、HCWに対する49,502ドルの補償と他の管理費を差し引いたところ、純収益は1,595,429ドルだった。2023年9月30日までの9ヶ月間に、ATM協定により82,410株の普通株を売却し、HCWへの補償と他の管理費59,898ドルを差し引くと、純収益は1,696,407ドルとなった。2023年9月30日以降、私たちはATM協定に基づいて何の普通株も販売していない。
| 111 |
2023年9月30日までの3ヶ月間、リンカーンパーク協定により12,000株の普通株を売却し、純収益は196,167ドルだった。2023年9月30日までの9ヶ月間、リンカーンパーク協定により25,500株の普通株を売却し、純収益は450,427ドルであった。2023年9月30日以降、ATM協定により283,800株の普通株を売却し、HCWに対する97,512ドルの補償と他の管理費を差し引いたところ、純収益は3,115,303ドルであった。
2023年8月11日,WFIA私募により,約100万ドルの毛収入を得た。
2023年9月の発売により,配給代理費とその他のbr発売費用を差し引く前に,2023年9月13日に約400万ドルの毛収入を受け取った。
管理層 はまた、他の会社との潜在的な協力または他の戦略取引を含む追加の株式発行、債券発行、または他の 資本源を含む可能性がある資金を得る他の戦略を評価している。
2025年上半期にCTX-1301の秘密保持協定を提出するためにFDA の潜在的承認については,今回の発行後,約800万~1000万ドルの追加資本が必要となると信じている。私たちはまた私たちの他の計画と商業化努力を推進するための追加資金が必要だ。しかし、FDA承認を受ける前に、候補製品への私たちの支出を予測することは難しい。また、変化する状況は、私たちが今予想していたよりも現金を使う速度が今予想されているよりずっと速いかもしれません。そして、私たちがコントロールできない状況のため、私たちは今予想しているよりも多くの現金を使う必要があるかもしれません。
私たちのbr政策は、私たちの現在の需要を超えた任意の現金を、元金残高を維持し、流動性を提供するための投資に投資し、適度な投資リターンを生成することである。したがって、私たちの現金等価物は主に通貨市場基金 に投資され、現在の金利環境では、これらの基金は現在最低リターンしか提供していない。
私たちは、少なくとも今後数年以内に、私たちが候補製品を開発し、市場承認を求め、承認された後、最終的に私たちの製品を商業化することに伴い、大量の追加運営損失を招き続けると予想しています。もし私たちの候補製品が市場の承認を得たら、私たちは大量の販売、マーケティング、アウトソーシング製造費用を発生させるだろう。また、私たちが計画している製品の商業化努力を支援する人員を含め、運営、財務、情報システム、人員を増やすための追加費用が発生すると予想されています。私たちの上場企業として適用されるコーポレート·ガバナンス、内部統制、類似の要求を守る上で巨額のコストが生じることも予想されます。
私たちの将来の現金と資本需要の使用は、多くの前向きな要素に依存するだろう
| ● | 私たちの候補製品を生産するための臨床供給のコストと時間 |
| ● | 私たちの候補製品の臨床試験の開始、進捗、時間、コスト、結果 |
| ● | 私たちは候補品ごとに臨床開発計画を立てました |
| ● | 私たちが開発または許可される可能性のある候補製品の数量と特徴 |
| ● | 私たちは実行される任意の協力または許可協定の条項を選択することができる |
| ● | FDAまたは他の同様の外国規制機関によって制定された規制要件の結果、時間、およびコストを満たす |
| ● | 私たちの特許主張と他の知的財産権の費用を提出、起訴、弁護、実行します |
| ● | 知的財産権紛争を弁護する費用は、第三者が私たちに提起した特許侵害訴訟を含む |
| ● | ビジネス規模の製造活動を実施するコストと時間; |
| 112 |
| ● | 我々が製品を商業化する地域が規制許可を得る可能性のある任意の候補製品の販売、マーケティング、および流通能力を選択することを含む、私たちの商業化作業アウトソーシングのコストと時間を含む。 |
私たちの業務を長期的に持続的に増加させるために、私たちは大量の資源を投入して研究開発を行う予定で、私たちの候補製品の臨床 試験、その他の運営と潜在的な製品買収と許可を含む。我々はすでに評価を行い,我々の内部開発チャネルを強化するために,他の製品や候補製品を買収または認可して開発する計画の一部として,一連の戦略取引を評価し続けることが予想される.私たちが求める可能性のある戦略的取引機会は、私たちの流動性と資本資源に重大な影響を与える可能性があり、追加の債務を発生させたり、株式資本を求めたり、あるいはその両方を要求することができるかもしれない。さらに、私たちは、承認されたまたは開発されている製品を新しいまたは既存の治療分野で開発、買収または許可するか、または既存の業務を拡張していく可能性があります。したがって、私たちは、私たちの業務を拡大するため、または一般会社の目的のために、他の製品、候補製品または会社を許可または買収するために、日和見主義的に追加資本を得る方法を求め続けることが予想される。 戦略取引は、1つまたは複数の公的または個人債務または持分融資によって追加資本を調達する必要があるかもしれない。または は、協力または協力手配として構築することができる。私たちは現在、買収、許可、または同様の戦略業務取引の手配、合意、または了解に達していません。2023年3月、FDAが承認した場合、マーケティング、販売、市場アクセス、および流通を含むCTX-1301の商業化サービスを提供するIndegene,Inc.と共同商業化協定を締結しました。
もし私たちが持分証券を発行することで追加資金を調達する場合、または私たちの債務が持分に変換された場合、私たちの株主は希釈に直面します。利用可能な場合、債務融資(利用可能であれば)は固定支払い義務の増加を招き、特定の行動をとる能力を制限するか、または追加債務を生成し、資本支出を行ったり、配当を宣言したりするいくつかの合意を含む可能性があります。私たちが調達した任意の債務融資または追加配当には、清算や他の特典のような私たちまたは既存の株主に不利な条項が含まれている可能性があります。もし私たちが第三者との協力および許可手配によってより多くの資金を調達する場合、 は、私たちの技術、将来の収入フロー、または候補製品の貴重な権利を放棄する必要があるか、または私たちに不利になる可能性のある条項でライセンスを付与する必要があるかもしれない。受け入れ可能な条件で、私たちは十分な資金を得ることができないかもしれないし、全く持っていないかもしれない。もし私たちが必要な時に資金を調達したり、このような合意に達しなかったら、私たちは候補製品の開発と商業化を大幅に延期、削減、または停止しなければならないかもしれない。
契約義務
以下 は我々の2023年9月30日までの契約義務をまとめており,これらの義務は我々の将来の流動性に影響を与える。
我々 は2018年8月にBDD Pharma Limitedと特許とノウハウライセンス契約を締結した。本プロトコルの説明については、本募集説明書の“業務−材料プロトコル” 部分を参照されたい。BDD Pharmaに 臨床試験や法規制マイルストーンに関する一定の金額を支払う必要がある。最初の25万ドルの記念碑的支払いは2023年2月に支払われ,当時CTX−1301の第3段階成人発症と持続時間研究で最初の患者が薬を服用していた。追加支払いは、プロトコルで定義されたいくつかのマイルストーンが完了した後に満了します。
著者らはすでにCROとCTX-1301の肝心な第三段階固定用量小児科と青少年の安全性と有効性研究について合意し、この研究は2023年7月下旬に開始し、 小児科の投与量の最適化、効果開始と持続時間の第三段階の研究は2023年8月初めに開始した。我々はすでにCDMOおよび他の第三者とCTX-1301登録ロットを生産するプロトコルを締結しており、秘密保護プロトコルを提出するにはこれらのロットが必要である。また、Indegene,Inc.と共同商業化協定を締結し、この合意によれば、Indegeneは、FDAの承認を得た後に、マーケティング、販売、市場アクセス、および流通を含むCTX-1301の商業化サービスを提供し、サービス料を徴収する。これらの契約には、事前に書面で通知すれば、キャンセルすることができる最低調達約束は含まれていません。キャンセル時に支払われるべき金額は、提供されるサービスの支払いまたは発生した費用のみを含み、 は、キャンセル日までの当社のサービスプロバイダのキャンセル不可義務を含み、場合によっては清算コストおよび回復コスト も含まれる。このような債務の正確な額は,終了時間や関連協定の条項に依存するため,不明である。
| 113 |
注目を行っている
Brが成立して以来,資金調達や研究開発活動を含む組織活動に従事してきた。私たち はまだ収入が発生しておらず、利益運営が実現されておらず、運営から正のキャッシュフローが生じたこともありません。 は利益運営が実現しても継続できる保証はありません。いずれの臨床前段階の製薬会社にも関連するリスクに直面しており,これらの会社は研究開発に多くの資金を投入している。私たちの研究開発プロジェクトが成功することは保証されず、開発された製品が必要な監督管理の承認を得ることは保証されず、いかなる承認された製品も商業的に実行可能である保証はない。また、私たちは技術が急速に変化する環境で運営しており、これは私たち従業員やコンサルタントのサービスに大きく依存しています。しかも、私たちの未来の運営は私たちが追加資本を調達する努力が成功するかどうかにかかっている。これらの不確実性は、財務諸表の発表日から一年以内に経営を続ける能力を大きく疑わせています。添付されている総合財務諸表は持続経営原則に従って作成されています。総合財務諸表には、将来的に資産の回収可能性および分類または負債の金額および分類に及ぼす可能性のある影響を反映するための調整は含まれておらず、これは、企業が持続的な経営を継続できない可能性がある企業によるものであり、企業が通常の業務中に経営、現金化資産、および負債を継続することを計画しているためである可能性がある。私たちは2023年9月30日まで、2023年9月30日と2022年9月30日までの3ヶ月と9ヶ月間に純損失が発生し、成立以来2023年9月30日までの累計損失は8600万ドルでした。現在開発中の候補製品から相当な収入を得ることができるまで、brの追加損失が予想されます。私たちの資金源には、再編合併前のCTXの各部門の私募資金、私たちの初公募株に関連する株式証券の発行、ATMプロトコル、リンカーンパーク協定、WFIA 債務融資、WFIA私募、2023年9月の発行が含まれています。私たちは私たちの運営を支援し、候補製品の開発と商業化を完成させるために追加の資金が必要になるだろう。2023年に潜在的追加資本源として達成されたこれらの合意の詳細については、上記の“流動資金および資本資源” を参照されたい。 は、必要に応じて、または許容可能な条件でこのような融資を提供することを保証することはできない。
最近発表された会計基準
2016年6月、FASBはASU 2016-13を発表した金融商品信用損失の計量これは、実体が多くの金融資産の減価を確認する方法を著しく変更し、発生時ではなく、その残存寿命内に発生すると予想される推定信用損失を直ちに確認することを要求する。2018年11月、FASBはASU 2018-19を発表した主題326“金融商品--信用損失”の編纂改善 小テーマ326-20(ASU 2016-13による作成)が改訂され、小テーマ326-20の範囲内ではないことが明確に指摘された。また、2019年4月には、FASBがASU 2019-04を発表した主題326(金融商品--信用損失)、主題815(デリバティブおよびヘッジ)、およびテーマ825(金融商品)の改善を編纂する; 2019年5月、FASBはASU 2019-05を発表しました金融商品−信用損失(特別テーマ326):方向性移行救済2019年11月、FASBはASU 2019-10を発表した金融商品-クレジット損失(トピック326)、派生ツールおよびヘッジ(トピック 815)およびレンタル(トピック842):発効日ASU 2019-11と主題326“金融商品--信用損失”の編纂改善 FASBは2020年3月にASU 2020-03を発表しました金融商品の法典化の改善ASU 2016−13のいくつかの態様については、さらなる 解明が提供される。これらの変化(改訂)は,2022年12月15日以降に開始される会計年度内の年度と中期 で発効する。当社はASU 2016−13年度の採用はその連結財務諸表に実質的な影響を与えないと予想している。
仕事法案
2012年4月5日、“雇用法案”が法律に署名された。JOBS法案に含まれる条項は,他を除いて,“新興成長型会社”に対する何らかの報告要求 を低下させている。“新興成長型企業”として、“雇用法案”の延長された過渡期を利用して新たな会計基準や改正会計基準を実施することを選択したため、新興成長型企業では、このような基準を採用する関連日に新たな会計基準または改正会計基準を遵守する必要がある。
JOBS法案に規定されているいくつかの条件に適合する場合には、“新興成長型企業”として、他の事項を除いて、第404条に基づいて我々の財務報告内部統制制度に関する監査員証明報告書を提供する必要はなく、ドッド·フランクウォールストリート改革および消費者保護法に基づいて非新興成長型上場企業に開示されるすべての報酬を提供することができる。(Iii)上場企業会計監督委員会が採択する可能性のある強制監査会社のローテーションまたは監査および財務諸表(監査人議論および分析)に関する補足情報を提供する監査人報告書の任意の要件を遵守し、(Iv)役員報酬と業績との間の相関、および最高経営責任者報酬と従業員報酬中央値との比較など、いくつかの役員報酬に関連する項目を開示する。これらの免除は、私たちのIPO完了5周年まで適用されるか、または以前に発生したものに準ずる“新興成長型企業”としての要求を満たさなくなるまで適用される。
| 114 |
セキュリティ所有権といくつかの利益所有者と管理
次の表は、2024年1月25日現在の我々の普通株式の利益所有権情報(他に説明がない限り)を示しており、具体的には以下の通りである
| ● | 私たちが知っているすべての利益は私たちの普通株の5%以上の個人または団体を持っている | |
| ● | 私たちのすべての役員は | |
| ● | 私たちが任命したすべての行政官は | |
| ● | チームとして、私たちのすべての役員と現職の幹部。 |
我々 は米国証券取引委員会の規則に基づいて実益権属を決定した。これらの規則によれば、利益所有権は、個人またはエンティティが投票権または投資権を個別にまたは共有する任意の普通株式 を含む。個人または実体実益が所有するbr株式の数およびその人の持株率を計算する際に、オプション制限された普通株式またはその人が保有する引受権証は、現在行使可能であるか、または2024年1月25日から60日以内に行使可能な普通株式または株式証明書を発行済み株式とみなされるが、他の人の持株率を計算する際に、これらの株式は発行済み株式とみなされない。
| すべての人の名前 (1) | 実益所有株式数 | パーセント クラス (2) | ||||||
| 執行幹事と役員を任命した | ||||||||
| シェン·J·シェーファー製薬会社です | 53,305 | (3) | 3.65 | % | ||||
| ローリー·A·マイルズ | 5,346 | (4) | * | |||||
| マシュー·ブラームスメリーランド州 | 33,126 | (5) | 2.28 | % | ||||
| ルイス·G·ヴァン·ホーンMBA | 10,170 | (6) | * | |||||
| クレイグ·S·ギルガロンEsq | 12,333 | (7) | * | |||||
| ピーター·J·ウォース | 327,085 | (8) | 19.99 | % | ||||
| 全役員と役員(6人)(9人) | 451,480 | (10) | 27.24 | % | ||||
| * | が1%未満であることを示す. |
| (1) | 別の説明がない限り、すべての上場株主のアドレスは1901 W.47 Place,KS 66205である。別の説明のほか,各株主は株主実益が所有する株式に対して独占投票権と投資権を持つが,適用されるコミュニティ財産法を遵守しなければならない. |
| (2) | 私たちは、1934年の証券取引法(改正された)下の規則13 d-3に基づいて、一般に証券の投票権および/または処分権によって決定される利益所有権を決定した。所有権パーセンテージは、2024年1月25日までに発行され、発行された普通株式1,450,171株に基づいており、その人が2024年1月25日まで60日以内に行使可能なオプションまたは株式証明書によって発行された任意の株式に基づく。 |
| (3) | (I)現在行使可能な引受権証を行使する際に発行可能な3537株を含む我々の普通株,(Ii)2024年1月25日から60日以内に行使可能な株式オプションは,我々の普通株7,301株と,(Iii)Fountainhead Shruged,LLCが保有する40,392株の普通株を発行することができる. Schaffer博士はFountainhead Shirded,LLCのマネージャーであり,Fountainheadが持つ証券に対して投票権と投資権を持つ.2024年1月25日から60日以内に行使できない株式オプション行使時に発行可能な普通株9,558株 は含まれていない |
| 115 |
| (4) | 2024年1月25日から60日以内に行使可能な株式オプション行使時に発行可能な2,932株普通株を含むそれは.2024年1月25日から60日以内に行使できない株式オプション行使後に発行可能な3,937株普通株は含まれていない。 |
| (5) | 2024年1月25日から60日以内に行使可能な株式オプション行使時に発行可能な普通株614株を含むそれは.2024年1月25日から60日以内に行使できない株式オプション行使後に発行可能な986株普通株は含まれていません。 |
| (6) | Louis G.Van Horn Trustが保有する普通株7,081株を含め、19年12月23日。Van HornさんはLouis G.Van Horn Trustの受託者であり、19年12月23日にLouis G.Van Horn Trustが保有する証券に対して投票権と投資権を有するVan Hornさんが当社を引退して2023年12月13日までの有効日に、株式オプションを行使することで我が普通株式2,339株を発行することができる |
| (7) | (I)現在行使可能な株式承認証を行使する際に発行可能な251株を含む我々の普通株式,(Ii)2023年12月15日現在、すなわちジガロン·さんが自社を辞任した有効日現在、株式オプション行使により発行可能な普通株式2,339株、および(3)リーマーリック集団が保有する9,265株の普通株式。ギルガロンさんは、リメルリック·グループの有限責任会社の唯一のメンバーで、リマリック·グループが保有する証券に対して投票権と投資権を持っています。 |
| (8) | 現在行使可能な引受権証を行使する際に発行可能な普通株式185,400株を含む(Ii)6752024年1月25日から60日以内に行使可能な株式オプションが発行可能な普通株と、(Iii)Werth Family Investment Associatesが保有する139,917株の普通株。WerthさんはWerth Family Investment Associates LLCのマネージャであり,Werth Family Investment Associates LLCが持つ証券に対して投票権と投資権を持っています。(X)975株が株を行使する際に発行可能な普通株 が2024年1月25日から60日以内に行使できないオプションと,(Y)843,970株の我々普通株の株式がWerth Family Investment Associates LLCが保有する予備資本金権証は含まれておらず,この権利証は所有者が行使後に発効した後に の範囲で当該等株式証を行使することを禁止している.所有者(所有者の連属会社および所有者または所有者との任意の共同経営会社が1つのグループとして行動する任意の他の者)は、実益が19.99%を超える発行済み普通株式株式を所有し、これらの株式は、当社が事前計画株式承認証を行使した後に発行可能な普通株式株式の発行が発効した後に発行される。 |
| (9) | 役員と現職幹部が含まれている |
| (10) | 2024年1月25日から60日以内に行使できない株式オプション行使時に発行可能な17,284株普通株と、2024年1月25日から60日以内に行使できない843,970株普通株式事前資本権証br}は含まれていない。 |
| 116 |
ある 関係と関連先取引
以下は、2021年1月1日以降に我々が参加する取引の説明であり、関連する金額がbrを超えるか、または(I)120,000ドルを超え、(Ii)過去2つの完了した財政年度において、我々の任意の取締役、役員、または5%を超える投票権を有する証券の所有者またはそれらの直系親族の任意のメンバーが、直接的または間接的な重大な利益(報酬スケジュールを除く)を有する最後の2つの財政年度末総資産平均値の1%を有することになる。
関連する 当事者備考
CTXは2020年2月26日、314,000ドルの約束手形(“2月手形”)を私たちの首席医療担当者Matthew Bramsに発行しました。 2020年9月30日、Bramsさんに40,000ドルの約束手形(“9月手形”を発行しました。2月手形とともにbr}“Brams手形”)を発行しました。BRAMS債券の課税利息は年利8%である。Brams博士の選択権により,BRAMSチケットの元本と受取利息は変換時のオファーの25%(25%)の割引でCTXの優先単位に変換できる.私たちは違約金や割増金を受け取ることなく、このような手形を事前に支払う権利がある。2020年12月30日と2020年7月1日にそれぞれ100,000ドルの2月期手形を返済しました。2月の手形は最初に2021年2月24日に満期になり、その後12ヶ月延長され、2022年2月24日まで延長された。9月手形は最初に2021年9月30日に満期になり、2022年2月24日に延期された。2021年7月1日、私たちは2月手形に10万ドル、2021年8月17日、2月手形に50,000ドルを支払いました。初公募が終了するまで、BRAMS手形104,000ドルと37,025ドルの元金と利息brをそれぞれ借り、2021年12月にIPOで全額支払います。
CTXは2020年7月25日、執行副総裁と首席科学官ラウル·R·シルバに100,000ドルの約束手形(“7月手形”)を発行した。7月期手形の利息は年利8%である。シルバ博士の選択によると、7月の手形の元本と応算利息はCTXの優先単位に変換でき、価格は転換時の見積もりより25%(25%)割引があります。私たちはこの手形を前払いする権利があります。違約金や保険料はかかりません。7月の手形は最初に2021年7月24日に満期になり、その後7ヶ月延長され、2022年2月24日まで延長された。IPO終了時まで、私たちはそれぞれ7月の手形100,000ドルと11,047ドルの元金と利息を借りています。この手形は2021年12月にIPOで全額支払われました。
2020年2月1日,CTXはCTX前メンバDresch,Inc.に500,000ドルの本チケット(“メンバチケット”を発行し,BRAMSチケットと7月チケットとともに“関連側チケット”と呼ぶ)を発行した.会員手形の元本および利息は,貸手から通知された後に変換時の見積単位でCTXの優先単位に変換することができる.2020年9月30日,メンバーが説明した条項によると,353,665ドルを現在の単価でCTXの246,096優先単位に変換し,146,335ドルの対応金額を残した。メンバー手形は最初に2021年2月1日に満期になり、2022年2月1日まで12カ月延期された。最初の公募終了時まで、私たちはそれぞれ会員チケット146,335ドルと39,941ドルの元金と利息を借りており、2021年12月に初公募で全額支払われた。
初公募株
パトリック·ガラゲル、取締役会メンバー、レドロー社取締役上級取締役社長、レドローリスク投資会社管理パートナー。ライドロー社(イギリス)有限公司は私たちのIPOの共同引受業者を務め、IPOは2021年12月10日に終了した。Laidlaw&Company(UK)Ltd.に支払われるIPOに関する割引および手数料は約500,000ドルである.
WFIA 備考
2022年8月9日、CTXは500万ドル(“原始元金”)の元票を発行し、WFIAを受取人(“原始 手形”)とした。私たちの取締役会のPeter WerthはWFIAのマネージャーだ。元の手形は無担保で、利息は年利15%で引き出します。2025年8月8日、またはWFIAは、2023年4月1日から始まるカレンダー四半期の前5営業日以内に書面要求を出してから120日後、未返済元金とすべての未払い利息が満期と対応しています。CTXは元のチケットの全部または一部を前払いすることを許可されており、割増または罰金を支払う必要はないが、これ以上返済された金額を借りることはできない。
| 117 |
CTXは2023年5月9日、WFIAを受益者とする改訂·再記載された元票(“WFIA手形”)に署名し、元の手形の元本を500万ドルから800万ドルに増加させた。
2023年9月8日,我々はWFIAと手形変換プロトコルを締結し,これにより,WFIAはWFIA手形項の元元本金額 をそのすべての当算利息,または5,812,500ドルと合わせて予備金権証(“9月WFIA 予資金権証”)に変換し,341,912株自社普通株を購入し,1株当たり前金金権証の転換価格を17ドルとすることに同意した。9月のWFIA事前資本金権証により,満期日なしに直ちに1株0.002ドルの取引価格 で行使できる。“証券取引法”第13(D)節では,WFIAとその関連会社は実益が当社の普通株流通株 の19.99%を超えないようにした。
2024年1月25日、我々は2024年1月の手形変換協定を締結し、WFIAに対応する手形項目の残りの3,000,000ドル元金とすべての課税利息、または3,287,500ドル、 を1月のWFIA予備資本金権証に変換し、687,043株の普通株を購入し、前資金権証1部あたりの転換価格は4.785ドルであった。私たちの普通株の2024年1月24日の終値は4.35ドルです。2024年1月の予融資権証 は満期日がなく、直ちに1株0.0001ドルの行使価格で行使することができ、取引法第13(D)条の規定により、当該等の権利証を行使した後、当社及びその関連会社は実益が19.99%以下の普通株式流通株を所有することが条件となる。
未償還元金およびすべての未払い利息は、違約事件により加速しない限り、2025年8月8日に満期になって支払わなければならない。WFIAは、2023年7月1日から、各カレンダー四半期の前5営業日以内に、CTX通知後120日以内にすべての未返済元本と利息を支払うことを要求する権利があります。CTXは割増や罰金を支払うことなく、WFIA手形の全部または一部を前払いすることが許可されている。条件は、返済された金額は再借入できないことである。 2023年9月30日現在、WFIA手形では3,000,000ドルの元金と157,339ドルの未返済利息がある。本公告日までにWFIAチケットは全額支払われており、当社はWFIAチケット項目の他の義務を負いません。
WFIA 2023年8月私募
2023年8月11日に、吾らはWFIAと証券購入協定(“2023年8月購入契約”)を締結し、私募で51,158株自社普通株を発行し、1株当たり購入価格は10.97ドルであり、吾等が対応する取引費用(“WFIA私募”)を差し引いた。 WFIA私募は2023年8月11日に終了した。
上級管理職と役員への賠償
私たちはすでに私たちの現職役員と役員一人一人と賠償協定を締結しました。これらの合意は、私たちが私たちにサービスを提供することによって生じる可能性のあるこれらの個人の責任を最大限に賠償し、彼らに対する訴訟によって発生した費用を事前に支出し、彼らが賠償を受けることができるように、デラウェア州の法律で許可された範囲内で最大限に賠償することを要求する。私たちはまた私たちの未来の役員や役員と賠償協定を締結するつもりです。
関係者取引の政策と手順
私たちbrは、私たちの役員、取締役、取締役に指名された候補者、任意のカテゴリの普通株式の5%を超える実益所有者、上記の人の任意の直系親族メンバー、パートナーまたは依頼者として雇用されているか、または類似の職にある任意の会社、会社または他のエンティティ、またはその人が5%以上の実益所有権の権益を有する任意の会社、会社または他のエンティティ、または関連側の任意の会社、会社または他のエンティティを禁止する関連側取引の政策および手順をとっている。当社の取締役会が審査委員会を介して行動していない場合やbrの場合、審査委員会の議長が事前に同意して私などと取引を締結しています。関連金額が100,000ドルを超えるか、またはそれを超える可能性があり、関連側が直接的または間接的な利益を有する場合、私たちの監査委員会に最初に提出されるか、または場合によっては私たちの監査委員会議長に提出されて検討され、承認されなければならない。このような提案を承認または拒否する場合、我々の監査委員会は、取引条項が、同じまたは同様の場合に非関連第三者が一般的に得ることができる条項 ,私たちへの利益の程度、製品またはサービスの他のソース ,および取引における関係者の利益の程度を下回ることができるかどうかを含むが、これらに限定されない取引の重要な事実を考慮するであろう。2023年12月、私たちの取締役会の4人の独立メンバーが辞任し、私たちは現在監査委員会、報酬委員会、または会社管理委員会を指名していません。
| 118 |
管理する
執行役員と役員
次の表は、2024年1月25日現在の役員および役員の年齢情報を提供しています
| 名前.名前 | 年ごろ | ポスト | ||||
| 実行官 : | ||||||
| シェン·J·シェーファー製薬会社は | 49 | CEO兼取締役会長 | ||||
| ジェニファー·L·カラハン | 53 | 上級副総裁と財務総監 | ||||
| ローリー·A·マイルズ博士 | 67 | 執行副総裁と首席運営官 | ||||
| 医学博士ラウル·R·シルバ | 66 | 執行副総裁兼首席科学官 | ||||
| マシュー·ブラームスメリーランド州 | 60 | 執行副総裁兼首席医療官 | ||||
| 取締役: | ||||||
| ピーター·J·ウォース | 85 | 役員.取締役 | ||||
シェン·J·シェーファー製薬会社です2012年にCingateを共同設立し、その後も私たちの最高経営責任者と取締役会長を務めてきた。Cingateで働く前に、Schaffer博士は2009年7月から2012年12月までSabre Science Solutions取締役社長を務めた。これまで、Schaffer博士 は2008年9月から2009年5月までPRI-Med Accessで取締役国民財務部マネージャーを務め、2004年2月から2007年12月までセノフィで高級マーケティングマネージャーを務め、2001年6月から2003年10月までノワールでマーケティングマネージャーを務めた。1999年7月から2001年6月まで,ロッグス製薬業界奨学金計画首席研究員を務め,ワーナー·ランバート/パーカー·デイビスとファイザー社で高級研究員を務めた。1997年6月から1999年7月までHoechst Marion Rousselで臨床研究アシスタントを務めた。Schaffer博士は薬物開発、商業化と生物技術商業運営において25年を超える経験を持っている。Schaffer博士はカンザス大学薬学院から薬学博士号を取得した。私たちは、Schaffer博士の製薬業界に対する広い知識、彼の広範な治療分野の臨床とビジネス背景、そして彼が私たちの最高経営責任者を務めた経験は、彼が私たちの取締役会に就く資格があると信じている。
ジェニファー·L·カラハンBrは2024年1月25日に上級副総裁兼最高財務官を務めた。彼女は2017年1月から会社で会計職を務め、2019年1月に会社の副総監総裁に任命された。当社の職務を担当する前、br}カラハンさんは2014年からカンザスシティ現地の会計士事務所子午線ビジネスサービス会社の会計会計を務めてきた。彼女はスタートアップ会社やプロセス改善が必要な会社を含む様々な業務にアウトソーシングコントローラサービスを提供している。彼女のキャリアの中で、カラハンさんは様々な業界や段階の会社にコンサルティングサービスを提供している。彼女は1992年6月から1998年12月まで徳勤でキャリアを開始し、そこで監査業務で様々な職務を担当した。Callahanさんは公認会計士の資格を持ち、クレトン大学の会計と金融学の学士号を取得した
Laurie A.Myers博士MBAです2018年4月から執行副総裁兼首席運営官を務め、2013年6月から2018年4月まで取締役会メンバーを務め、2017年11月から2018年4月まで運営総監上級副総裁を務めています。マイルズ博士はLinea System、LLC顧問委員会のメンバーも務め、2020年9月以来務めている。マイルズ博士は以前、2014年9月から2017年11月までの間にフィディア製薬米国社のマーケティング担当を務めていた。同社はフィディア製薬会社の完全子会社である。マイルズ博士は2012年1月から2014年12月までニュージャージービジネススクールの兼任教授を務め、2010年1月から2014年9月まで総裁とハレットDavid戦略グループの取締役会メンバーを務めた。マイルズ博士はロッグス医学院とロッグス薬学院から毒理学哲学博士号を取得し、聖ジョセフ大学から工商管理修士号を取得し、スクラントン大学から理学修士と学士号を取得した
ラウル·R·シルバ医学博士2013年に他人と共同でCingateを設立し、2018年1月から会社執行副総裁兼首席科学官を務めた。2009年以来、彼は個人的に働いてきた。これまで、シルバ博士は2006年から2009年までロクラン児童精神医学センターの役員を務めていた。2005年から2009年にかけて、ニューヨーク大学児童研究センターの副議長も務めた。シルバ博士は1999年から2006年までベルビユ病院センターで児童精神医学取締役副主任医師を務めた。これまで,1995年から1990年までニューヨーク市サンルーク/ルーズベルト病院で児童·青少年精神病科の取締役教授を務めていた。彼は1990年にコロンビア大学サンルーク/ルーズベルト病院センターで児童と青少年精神医学の研究を完成し、ニューヨーク大学医学センターで精神薬理研究の研究を完成した。シルバ博士は普通、児童と青少年精神医学の面で取締役会の認証を受けた。シルバ博士はロス大学で医学博士号を取得し、フェルリー·ディキンソン大学で生物学理学学士号を取得した。
マシュー·ブラームスメリーランド州2013年に共同でCingateを創設し、2018年1月から執行副総裁兼首席医療官を務め、2018年1月から2021年7月までCingateの取締役を務めた。ブラームス博士は1999年4月から2021年1月までBayou City Researchの担当を務めた。これまで,テイラーリハビリテーションセンター(2019年4月現在),レクビユ健康リハビリテーションセンター(2018年から2019年),テキサス州ヒューストンPARC(2012年から2015年),老年精神科単位メキシコ湾病院(2009年から現在)を含む複数の医療機関で医学取締役顧問や/または入院精神病学者 を務めてきた。ブラームス博士はADHD臨床分野に関与するすべての主要製薬会社の研究チームに不可欠な一員であった。ブラームス博士はベイラー医学院でそれぞれ成人精神医学と児童精神医学の実習と奨学金を完成させた。彼は成人と児童精神医学委員会証明書(1994) を持ち,米国精神医学と神経医学委員会の上級委員会審査員代理である。彼はテキサス大学科学センターから医学博士号を取得し、テキサス大学から生物学文学学士号を取得した。
| 119 |
ピーター·J·ウォース2018年6月から私たちの取締役会に勤めています。ウェスさんは、Chemwerth Inc.の創業者でCEOであり、全方位的なサービスを提供するジェネリック医薬品開発·供給会社であり、世界的に規制された市場に活性医薬成分を提供する。 ウェスさんは、1975年3月から1982年5月までの間に、ツィゲフリード社の子会社であるガンス化工の社長副社長を務めていた。br}は、1965年から1975年にかけて、Upohn製薬会社(現ファイザー)の研究開発部門で働いていた。また、Werthさんは2010年12月からVm Pharma LLC、2012年5月からVM Treateutics LLC、2012年6月からAlopexx Vaccines LLC、2014年8月からVM Oncology LLC、2015年1月からPerseus Science Group LLC、2017年8月からLikarda LLC、2017年9月からTechtona LLC、2018年6月からMedRhythms LLC、2020年9月からBastion Healthare LLCの取締役を担当している。彼はスタンフォード大学で有機化学士号を取得し、ヘスバーグ州立大学で化学と数学理学学士号を取得した。私たちは、Werthさん生命科学の業界の豊富な経験と彼はビジネスと国際市場の知識で、彼は私たちの取締役会に在任する資格を持っていると信じています。
家族関係
私たちの取締役や役員の間には家族関係はありません。
取締役会 構成
私たちの取締役会は現在2人のメンバーで構成されている。私たちの役員の任期は、彼らの後継者が選出され、資格を得るまで、または彼らが辞任または罷免される前の者まで。
著者らが改訂と重述した会社登録証明書と定款の条項によると、私たちの取締役会は の3種類に分けられ、即ちI類、II類とIII類に分けられ、各類は3年間交互に在任している。ある種の取締役の任期が満了した後、この種類の取締役は任期満了の当年の株主年会で新たな3年間の任期に当選する資格がある。私たちの取締役は以下の3つに分類されます
| ● | 1級取締役はピーター·J·ウェスで、任期は2025年に開催される株主総会(Br)で前回いっぱいになる。 |
| ● | 取締役の三番目の人物はシェン·J·シェーファーで、任期は2024年に開催される株主総会で満了する。 |
私たち は現在II類取締役は何もいません。私たちは、各レベルが可能な限り3分の1の取締役で構成されるように、取締役名の増加によって増加した任意の追加取締役ポストが3つのレベルの間で割り当てられることを予想しています。brは私たちの取締役会を3つのレベルに分け、3年間の任期を交錯させることで、私たちの経営陣の交代や制御権の変更を延期または阻止する可能性があります。
当社の改訂及び再記載された会社の登録証明書及び改訂及び重述された付例規定は、当社の取締役会が決議を通過して初めて、許可された取締役数を変更することができます。私たちの改正および再記載された会社の登録証明書および改訂および再記載された付例も、私たちの取締役は任意の理由で免職されることしかできませんが、私たちの取締役会のいかなる空きも、取締役会の拡大による空きを含めて、私たちの当時在任していた取締役が過半数投票で埋めることしかできません。
我々 は,取締役が指名されることを決定する際に多様性を考慮した正式な政策を持っていない.取締役会は取締役会全体の範囲内で各 個人を評価し、 業務の成功を最もよく維持できるグループを構築し、これらの異なる分野での異なる経験を利用して合理的な判断を行うことで株主利益を代表することを目的としている。
取締役 独立
ナスダックの規則によると、取締役会がその人が取締役責任を履行する際に独立判断を妨害する関係がないと判断した場合にのみ、同社は“独立した取締役”となる資格がある。
我々の取締役会は、現在、ナスダックモール規則5605(A)(2)で定義された“独立取締役”の資格に適合する取締役は何もいないことを確認した。華通が取締役を受取人とする約束手形のため、取締役会はウェスさんが独立したアリペイではないと認定した。また、謝恩·シェーファーは会社の最高経営責任者であるため、独立した取締役会社ではない。2023年12月6日に取締役会を辞任したScott Applebaum、2023年12月12日に取締役会を辞任したパトリックGallagher、2023年12月13日に取締役会を辞任したGregg GivensとCurt Medirosはそれぞれ独立取締役とされている。
私たちは現在監査委員会、報酬委員会もなく、指名や会社管理委員会もなく、会社は他の取締役を選んでいます。私たちの取締役会がより多くの取締役会メンバーを任命したら、私たちはナスダック規則のbr上場基準に基づいて独立性分析を行います。
2023年12月26日、3人の取締役会メンバーがそれぞれ2023年12月12日と2023年12月13日に辞任したことに基づいて、独立取締役上場規則5605(“独立取締役規則”)に記載されているナスダック上場規則brのナスダック指名に対する監督要求を遵守しなくなることを指摘した従業員からの手紙を受け取った。br}我々は潜在的な新しい取締役を探しており、グループ公聴会の前に独立取締役規則遵守への進展を示す予定である。私たちが“役員独立規則”を再遵守する保証はありません。
| 120 |
役員報酬
次の表と添付の開示は、私たちが指定した役員が2023年に稼いだ報酬情報を示しています。私たちが任命した役員には、私たちの最高経営責任者、2023年12月31日までに最高報酬を担当した2人の役員(私たちの最高経営責任者brを除く)と、2023年12月31日までに役員を務めた場合、最高報酬の2人の役員(私たちのCEOを除く)となり、以下のようになります
| ● | 最高経営責任者兼取締役会長の謝恩·シェーファー | |
| ● | ローリ·A·マイルズ執行副総裁最高経営責任者 | |
● |
マシュー·N·ブラムス常務副主任総裁首席医療官 | |
| ● | ルイス·G·ファン·ホーン元副総裁兼最高財務責任者 | |
| ● | クレイグ·S·ギルガロン、Esq.,元常務副総裁、総法律顧問兼事務総長。 |
集計表 給与表
次の表に、私たちが指定した各役員が示した年で取得、稼いだり、支払ったりする給与情報を示します。
名前 と 担当者 職 | 年.年 | 賃金.賃金 ($) | ボーナス.ボーナス ($)(1) | 在庫品 賞.賞 ($) | 選択権 賞.賞 ($)(2) | 非持分 激励する 平面図 補償する ($) | すべての その他 補償する ($) | 合計する ($) | |||||||||||||||||||||||
| シェン·J·シェーファーは | 2023 | 491,961 | - | - | 137,610 | - | - | 629,571 | |||||||||||||||||||||||
| 会長兼最高経営責任者 | 2022 | 475,000 | 29,688 | - | 78,503 | - | - | 583,191 | |||||||||||||||||||||||
| ローリー·A·マイルズ | 2023 | 415,167 | - | - | 61,160 | - | - | 476,327 | |||||||||||||||||||||||
| 執行副総裁と首席運営官 | 2022 | 400,000 | 25,000 | - | 34,890 | - | - | 459,890 | |||||||||||||||||||||||
| マシュー·N·ブラームス | 2023 | 244,792 | - | - | 22,935 | - | - | 267,727 | |||||||||||||||||||||||
| 副社長 とCMOの実行 | |||||||||||||||||||||||||||||||
| ルイス·G·ヴァン·ホーンは | 2023 | 382,205 | - | - | 76,450 | - | - | 458,655 | |||||||||||||||||||||||
| 前執行副総裁兼最高財務官(3) | 2022 | 380,000 | 23,750 | - | 34,890 | - | - | 438,640 | |||||||||||||||||||||||
| クレイグ·S·ギルガロン | 2023 | 385,250 | - | - | 61,160 | - | - | 446,410 | |||||||||||||||||||||||
| 前執行副総裁、GC、秘書(4) | 2022 | 380,000 | 23,750 | - | 34,890 | - | - | 438,640 | |||||||||||||||||||||||
| (1) | ボーナス額は、私たちが指定した役員が2022年に獲得した年間ボーナスを代表します。 |
| (2) | FASB ASCテーマ718によると, 金額は2023年2月28日,2022年2月25日,2021年12月7日の非限定株式オプション奨励の付与日公正価値を反映している.オプション報酬の公正な市場価値はブラック·スコアモデルを使用して決定された。2021年12月期株式オプション付与日公正価値を推定するための仮定については、2021年12月31日現在の年度総合財務諸表付記10を参照されたい。2022年の株式オプション付与日の公正価値を推定するための加重平均仮定については、2022年12月31日までの年度総合財務諸表付記11を参照されたい。2023年2月の株式オプション付与日の公正価値を推定するための仮定は以下のとおりである |
無リスク金利 |
0.04% | |
| 所期の 期限(年単位) | 6.08 | |
| 期待変動 | 1.13 | |
| 期待配当収益率 | 0% | |
| 日付公正価値を与える | $30.58 |
| (3) | Van Hornさんは2023年12月13日から会社を退職する |
| (4) | ギルガロンさんは、高度管理職を辞任し、Cingate Treateutics LLC(“CTX”)の採用を2023年12月15日から終了します。Gilgaronさんは、2021年9月23日にCTX社と締結された雇用契約第4節(E)によると、解雇されたことには“十分な理由がある”と主張している。同社はギガロンさんのクレームに異議を申し立てています。当社が受益者として一般的な免除を受ける場合には、GilGallonさんによる雇用契約が“十分な理由”で終了した場合、契約終了日から60日以内に、その基本給(402,800ドル)と目標ボーナス(100,500ドル)に相当する一括払いを得る権利があります。また、さんギガロンが解任日後も引き続き4(4)ヶ月間雇われていた場合、彼の行使していなかった株式オプションは解任日から行使可能となり、授任されたすべての株式オプションをその任期中に行使することができる。 |
| 121 |
従業員福祉計画
私たちbrは現在、医療、歯科、視力保険を含むすべての従業員に適用される基礎的な広範な医療と福祉を提供しています。
401(K) 計画
我々 は,条件を満たすすべての従業員に401(K)貯蓄計画(“401(K)計画”)を提供する。401(K)計画によれば、毎年必要な安全港マッチングを除いて、401(K)計画に対応するbr}貢献は提供されない。
応急手当計画
2023年12月16日から全従業員の年間基本給を下げ、従業員が当社に残るよう奨励することについて、取締役会独立メンバーは1つまたはボーナス計画を承認した。この計画によると、会社はCTX-1301新薬申請をFDAに提出した日から3ヶ月の日付(“支払日”)に従業員1人当たりの金額を支払い、支払日までに減給で従業員に支払われていない基本給ドル総額の減給額の20%に相当する。
Brと私たちが指定した幹部を採用する手配
シェン·J·シェーファー
2021年9月23日、私たちはSchaffer博士と雇用協定を締結した。シェイファー博士の雇用協定条項によると、彼は最高経営責任者を務めている。雇用協定の最初に定められた基本給は年間475,000ドルであり, は2023年1月1日から503,500ドルに増加した。コスト抑制策では,シェイファーの年間基本給は226,350ドルに低下し,2023年12月16日から発効した。また,シェイファー博士は年間ボーナスを得る資格があり,目標金額はシェイファー博士年度基本給の25%(25%)に相当する。各ボーナスの実際の金額は私たちの報酬委員会が自ら決定し、会社の業績とSchaffer博士の個人的な業績に基づく。彼の雇用契約条項によると、Schaffer博士は会社の他の幹部や高級管理者が獲得できるすべてのインセンティブ計画と繰延給与計画に参加する資格があり、私たちが採用する可能性のある任意の従業員福祉計画と株式計画に参加する資格があり、これらの計画は会社が随時適宜修正することができる。
Schaffer博士に書面で通知した後,我々 はいつでもSchaffer博士の雇用を中止することができ,Schaffer博士は正当な理由を含めていつでもその雇用関係を終了することができる(この用語はSchaffer博士の雇用合意に定義されている)。
| 122 |
Schaffer博士の雇用が会社によって無断で終了またはSchaffer博士に正当な理由で終了された場合,Schaffer博士は終了日から21(21)日以内および終了後7(7)日の満了後,Schaffer博士の基本給と年間目標ボーナスの1.5倍(1.5)倍の現金で,会社や関連個人や実体を受益者とする全面的なクレームを得る権利がある。終了日から60日以内に。また,Schaffer博士が持つすべての株式オプションと株式付加価値権は,契約終了日後に4(4)ヶ月再雇用されれば付与され,契約終了日から全任期の残り時間内に行使することができる。Schaffer博士が支配権変更後12(12)ヶ月以内に会社に無断で雇用を中止されたり,正当な理由でSchaffer博士に雇用関係を中止されたりすれば,Schaffer博士は終了日から21(21)日以内およびその後7(7)日の満了後に,Schaffer博士の基本給と年間目標ボーナスの2(2)倍に相当する現金の解散費を得る権利がある。終了した日から60日以内にしかし、任意の支払いまたは福祉が“国内税法”第280(G)節で定義された“パラシュート支払い”を構成する場合、その支払いは、国内税法第4999節に徴収された消費税および(Ii)全額支払いの金額から、“国内税法”第4999節に従って徴収された消費税を含むすべての税額を減算する。さらに、Schaffer博士が保有するすべての株式オプションおよび株式付加価値権は、終了日からその完全な任期の残りの時間内に帰属し、行使することができる。
ローリー·A·マイルズ
2021年9月23日、私たちはMyersさんと雇用協定を締結した。マイルズさんの採用協定条項によると、彼女は執行副総裁と首席運営官を務めている。雇用協定で最初に規定された基本給は年収400,000ドルで、2023年1月1日から424,000ドルに増加した。コスト抑制策では、マイルズさんの年間基本給は21.2万ドルに低下し、2023年12月16日から発効した。また、マイルズには年間ボーナスを得る資格があり、目標額はマイルズ年度基本給の25%(25%)である。各ボーナスの実際の金額は、当社の報酬委員会が自ら決定し、会社の業績と最高経営責任者が提案したMyersさんの個人業績に基づいて決定されます。雇用契約の条項によると、Myersさんには、会社の他の役員や上級管理者が獲得できるすべての報酬と繰延報酬計画に参加する資格があり、私たちが採用する可能性のある任意の従業員福祉計画や株式計画に参加する資格があり、これらの計画は会社によって適宜修正される可能性があります。
Myersさんに書面で通知した後、私たちはいつでも理由なくMyersさんの雇用を終了することができ、Myersさんは正当な理由を含めていつでもその雇用を終了することができる(Myersさんの雇用契約におけるこの用語の定義)。
会社がマイルズさんの雇用を理由なく中止したり、マイルズさんの雇用を中止する十分な理由があれば、マイルズさんは終了日から二十一(21)日以内に会社や関連個人や実体に有利な全面的なクレームに署名し、終了後七日以内にマイルズさんの基本給と年間目標ボーナスの1倍に相当する現金の解散費を受け取る権利がある。Myersさんが持っているすべての株式オプションと株式付加価値権は、彼女が契約終了日から4(4)ヶ月再雇用された場合、契約終了日から帰属し、その全任期の残りの時間内に行使することができる。マイルズさんが支配権変更後12(12)カ月以内に会社に無断で雇用を中止されたり、マイルズさんに正当な理由で雇用を中止されたりすれば、マイルズさんは終了日から21日以内と、終了後7日(7)の満了後、マイルズさんの基本給と年間目標ボーナスの1(1)倍の現金解散費を得る権利がある。終了した日から60日以内にしかし、任意の支払いまたは福祉 が“国内税法”第280(G)節で定義された“パラシュート支払い”を構成している場合、支払い金額は、(I)最大金額のうちの大きな1つとなり、これらの支払いの任意の部分が、“国内税法”第499節で徴収される消費税 および(Ii)全額支払いの金額から、“国内税法”第4999節により徴収される消費税の影響を含まないことを保証するために、すべての税金が差し引かれる。さらに、Myersさんが持っているすべての株式オプションおよび株式付加価値権は、そのすべての残り期限が終了した日から帰属し、行使することができる。
| 123 |
マシュー·N·ブラームス
2021年9月23日、我々はブラームスさんと雇用契約を締結しました。ブラームスさんの採用契約条項によると、彼は執行副社長と首席医療官を務めている。雇用協定で最初に定められた基本給は年収200,000ドルで、2023年1月1日から250,000ドルに増加した。ブラームスさんの年間基本給は、原価抑制対策で2023年12月16日から125,000ドルに引き下げられました。Bramsさんの雇用契約は2024年1月1日より、(I)Bramさんの年初来基本給を許容(A)Bramsさん401(K)計画に国税局が許容する最高額に修正し、(B)当社は法定最低金額を連邦、州、地方税の支払に差し止め、(Ii)当社は、2021年の総合持分インセンティブ計画に基づき、各カレンダー四半期の最終営業日にBramsさんに1,000株の非合格株 オプションを付与すると規定しています。
ブラームスさんはブラームスさん年の基本給の25%(25%)を目標とする年間ボーナスを得る資格があります。各ボーナスの実際の金額は、当社の報酬委員会が自ら決定し、CEOの提案に基づいて、会社の業績とBRAMSの個人業績に基づいています。雇用契約の条項によると、ブラームスさんは、当社の他の役員または上級管理職が得ることができるすべての報酬および繰延報酬計画に参加する資格があり、当社が採用する可能性のある任意の従業員福祉計画および持分計画に参加する資格を有しており、これらの計画は、企業によって随時適宜改訂されることができます。
Bramsさんに書面通知を出した後では、我々はいつでもBramsさんの雇用関係を終了することができます一方、Bramsさんは十分な理由を含めていつでもその雇用関係を終了することができます(Bramsさんの雇用契約に定義されています)。
もし当社がブラムスさんやブラームスさんの採用を無断で中止した理由があれば、ブラムスさんは終了日から21日以内およびその後7(7)日の満了後、終了日から21日(Br)日内およびその後7(7)日の満了後、終了日から60日以内に当社および関係者およびエンティティが受益者となる包括的解除債権を受け取る権利があり、ブラームスさん賃金の12(12)カ月以内にブラームスさん賃金の1倍に相当する月給を得ることができる。また、ブラームスさんが保有するすべての株式買取および株式付加価値は、それが終了契約日後に再雇用される4(4)ヶ月で、終了契约日からすべての残りの时间内に帰属及び行使する。ブラームスさんの支配権変更後十二(12)ヶ月以内に会社から無断で採用を中止したり、ブラームスさんが採用を中止する権利があるならば、ブラームスさんは終了の日から二十一日(21)日以内及び終了後七日(七)の満了後にブラームスさんの基本給の一倍に相当する現金解散料を得ることができます。ただしブラムスさんは、会社及び関連個人及び実体を受益者とするための全面的なクレームを署名しました。 終了日から60日以内;しかし、任意の支払いまたは福祉が国税法第280(G)節に定義された“パラシュート支払い” を構成する場合、支払金額は、この等支払いの任意の部分が国税法4999節で徴収された消費税および(Ii)全額支払いから国税法第4999節で徴収された消費税を含むすべての税金の影響を受けないことを保証するために、(I)最大金額のうちの大きな者を基準とする。さらに、ブラームスさんが保有するすべての株式 オプションと株式付加価値権は、終了の日から帰属し、その完全期間の残りの 期間に行使することができます。
ルイス·G·ヴァン·ホーン
2021年9月23日、我々はVan Hornさんと雇用契約を締結しました。ファン·ホーンさんの採用契約条項によると、彼は執行副総裁と最高財務責任者を務めています。雇用協定の最初に定められた基本給は年間380,000ドルであり,2023年1月1日から406,600ドルに増加した。また、ファンホーンは年間ボーナスを得る資格があり、目標金額はファンホーンの年間基本給の25%(25%)に相当する。各ボーナスの実際の金額 は、当社の報酬委員会が自ら決定し、CEOが提案した会社の業績とVan Hornさんの個人的な業績に基づいて決定されます。雇用契約の条項によると、Van Hornさんは、すべてのインセンティブおよび繰延報酬プログラムを得ることができる他の役員または上級管理職が取得する資格があり、当社が通過する任意の従業員福祉プログラムおよび持分計画に参加する資格を持っています。
Van Hornさんに書面通知を出した後、我々はVan Hornさんの雇用を随時理由なく終了する権利がある一方、Van Hornさんは正当な理由(当該用語はVan Hornさんの雇用契約に定義される)を含めていつでもその雇用関係を終了する権利がある。
| 124 |
もし会社がVan Hornさんの採用を理由なく終了した場合、またはVan Hornさんがその採用を終了する権利があるならば、Van Hornさんは終了の日から21日(21)の日以内におよび終了後7(7)の日の満了後に、Van Hornさんの基本給および年間目標賞金の1倍に相当する現金の散財料を得る権利がある。ただし、会社および関連個人および実体の受益者として包括的なクレームを受領する権利がある。終了日から60日以内に。さらに、Van Hornさんは、彼が契約終了の日から4(4)ヶ月間再雇用されていれば、契約終了日からすべての期限が満了する場合、すべての株式オプションと株価付加権を保有しています を行使します。Van Hornさんの雇用が制御権変更後12(12)ヶ月以内に会社の理由なく終了するかまたはVan Hornさんによって十分な理由で終了した場合、Van Hornさんは終了の日から21日(Br)の7日以内に満了した後7(7)日以内に終了する権利があり、終了日から21日後の7(7)日以内に会社および関連個人および実体を受益者とする債権の全面的放出を得る。契約終了日から60日以内に、ファンホーンさんの基本給と年間目標ボーナスの1倍に相当する現金を一度に支払う。ただし、任意の支払いまたは福祉が“国税法”第280(G)節で定義された“パラシュート支払い”を構成している場合、その支払いは、国税法第4999節で徴収された消費税および(Ii)国税法第4999節に徴収された消費税の大きな金額を含む全税項を減算するために、(I)最大金額となることが条件である。さらに、Van Hornさんが保有するすべての株式オプションおよび株式付加価値権は、終了の日からその全残余期間内に付与され行使可能となります。
ファン·ホーンさんは、2023年12月13日に私たちの最高財務責任者から退職します。
クレイグ·S·ギルガロンEsq
我々は2021年9月23日にキルガロンさんと雇用契約を締結した。ギガロンさんの採用契約条項に基づき、彼は常務副総裁と総法律顧問を務めている。雇用協定の最初に定められた基本給は年間380,000ドルで、2023年1月1日から402,800ドルに増加した。また、ギルガロンには年間ボーナスを得る資格があり、目標額はギルガロンの年間基本給の25%(25%)に相当する。最高経営責任者の助言によると、各ボーナスの実際の金額は、当社の報酬委員会が、企業の業績とさん·ギガロンの個人的な業績に応じて自ら決定します。彼の雇用契約の条項によると、ギルガロンさんは、他の企業の役員または上級管理職が取得することができるすべてのインセンティブおよび繰延報酬計画に参加する資格があり、他の役員退職計画および任意の 従業員福祉計画および持分計画に参加する資格があります。これらの計画は、企業によって随時適宜改訂されることができます。
われわれは理由なくギルガロンさんへの書面通知を出した後、いつでもギルガロンさんの雇用を終了する権利があるが、ギルガロンさんはその雇用を正当な理由で打ち切る権限を有している(この用語ではギルガロンさんの雇用契約には定義がある)。
なぜか、ギルガロンのさんが会社に雇用を中止したり、正当な理由で雇用を終了したりした場合、キルガロンのさんは、終了日から21日以内および終了日から7(7)日の満了後、契約終了日から60日以内に、ギルガロンさんの基本給と年間目標ボーナスの1倍の現金相当の散金を得る権利がある。また、GilGallonさんが保有するすべての株式オプションと株式付加価値権は、彼が契約終了日から4(4)ヶ月間再雇用された場合には、付与され、契約終了日からその全任期の残りの時間内に行使することができます。もしギガロンのさんが支配権変更後十二(十二)ヶ月以内に理由なく雇用を終了したり、ギガロンさんに正当な理由で雇用を打ち切られた場合は、ギガロンさんは終了の日から二十一日(二十一)日以内及びその後七(七)日の満了後に、ギガロンさんの基本給及び年度目標ボーナスの一倍に相当する現金解散料を請求する権利を得ることができます。ただし、法人及び関連個人及び実体を受益者とするクレームに署名したことを条件とする。終了した日から60日以内にしかしながら、任意の支払いまたは福祉が国税法第280(G)節に定義された“パラシュート支払い”を構成する場合、支払金額は、(I)最大金額のうちのより大きな者となり、これらの支払いの任意のbr部分が国税法第499節で徴収される消費税および(Ii)全額支払いの金額から、国税法第4999条に従って徴収される消費税を含むすべての税項を減算することを保証する。さらに、GilGallonさんが保有するすべての株式オプションと株式付加価値権は、終了の日からその完全期間の残り時間 内に帰属し、行使することができます。
ギルガロンさんは、会社の上級管理職を辞任し、Cingate Treateutics LLCの仕事を2023年12月15日に終了しました。ギルガロンさんは会社の上級管理職を辞任し、CTXでの雇用関係を2023年12月15日から終了した。Gilgaronさんは、CTXと締結した2021年9月23日の雇用契約第4条(E)によると、解雇されたことには“十分な理由がある”と主張している。会社はギルガロンさんのクレームに反駁した。当社を受益者とする一般的な免除に調印することを前提に、GilGallonさんによる雇用契約によれば、“十分理由” で契約を終了すると、契約終了日から60日以内に、基本賃金(402,800ドル) と目標ボーナス(100,500ドル)に相当する一括払いを得る権利があります。また、さんギガロンが解任された日から四(4)ヶ月間再雇用される場合には、解任の日から何の返済もされていない彼の株式オプションを付与して行使することができ、付与されたすべての株式オプションをその期限内に行使することができる。
| 125 |
未償還のbr 2023年度末の持分奨励
| 名前.名前 | 付与日 | 行使可能な未行使オプションの証券数(#)(1) | オプションの対象証券数(#)行使できない(1) | オプション 発行権価格(ドル)(1) | オプション の有効期限 | |||||||||||||
| シェン·J·シェーファー | 2-28-2023 | (2) | - | 4,500 | 35.80 | 2-28-2033 | ||||||||||||
| 2-25-2022 | (2) | 1,544 | 1,831 | 27.60 | 2-25-2032 | |||||||||||||
| 12-7-2021 | (3) | 4,492 | 4,492 | 120.00 | 12-7-2031 | |||||||||||||
| ローリー·A·マイルズ | 2-28-2023 | (2) | - | 2,000 | 35.80 | 2-28-2033 | ||||||||||||
| 2-25-2022 | (2) | 685 | 815 | 27.60 | 2-25-2032 | |||||||||||||
| 12-7-2021 | (3) | 1,685 | 1,684 | 120.00 | 12-7-2031 | |||||||||||||
| マシュー·N·ブラームス | 2-28-2023 | (2) | - | 750 | 35.80 | 2-28-2033 | ||||||||||||
| 2-25-2022 | (2) | 287 | 338 | 27.60 | 2-25-2032 | |||||||||||||
| 12-7-2021 | (3) | 113 | 112 | 120.00 | 12-7-2031 | |||||||||||||
| ルイス·G·ヴァン·ホーン(4) | 2-28-2023 | (2) | - | 2,500 | 35.80 | 2-28-2033(4) | ||||||||||||
| 2-25-2022 | (2) | 654 | 846 | 27.60 | 2-25-2032(4) | |||||||||||||
| 12-7-2021 | (3) | 1,685 | 1,684 | 120.00 | 12-7-2031(4) | |||||||||||||
| クレイグ·S·ギルガロン(4) | 2-28-2023 | (2) | - | 2,000 | 35.80 | 2-28-2033(4) | ||||||||||||
| 2-25-2022 | (2) | 654 | 846 | 27.60 | 2-25-2032(4) | |||||||||||||
| 12-7-2021 | (3) | 1,685 | 1,684 | 120.00 | 12-7-2031(4) | |||||||||||||
(1)
|
我々の普通株の株式数 対象株式オプションとオプション行権価格は、2023年11月30日に発効した我々が発行した普通株と発行済み普通株の20株1株逆株式分割を反映している。 |
| (2) | オプションの帰属は以下のとおりである:授出日の1年周年日から25%であり、残りの株式は初期帰属日後の36ヶ月間月分割 である。 |
| (3) | 付与された日から オプションは4つの均等額の年間分割払いに分けられる。 |
|
(4) |
ヴァン·ホーンさんおよびギルガロンさんは、2021年計画に基づき、それぞれの終了日から90日後までの間、既得株式オプションを行使することができます。90日の期限の満了後、Van HornさんとGilgaronさんが保有するすべてのストックオプションは、2023年2月28日に付与されたストックオプションを含めて没収されます。ギガロンさんは、会社の上級管理職を辞任し、CTXとの雇用関係を2023年12月15日から終了した。キルガロンさんは、2021年9月23日にCTXと締結した雇用契約第4条(E)に基づき、彼の離職に十分な理由があると主張している。同社はギガロンさんのクレームに異議を申し立てています。企業が受益者として包括的な解除契約を締結したことを前提に、ギルガロンさんの雇用契約に基づき、契約終了日から60日以内に、基本賃金(402,800ドル)と目標ボーナス(100,500ドル)に相当する一括払いを得る権利があると“十分な理由”で契約を終了した。さんギガロンが退職後も引き続き雇用されている4(4)ヶ月の場合、彼が保有している任意の未行使の株式オプションは、終了日の の日から帰属して行使することができ、付与されたすべての株式オプションをその全期限内に行使することができる。 |
| 126 |
役員報酬
以下の表は当社の取締役会の各非従業員メンバーが2023年に取締役として獲得、獲得或いは支払いの補償 を示したが、当社の取締役会及び委員会会議に出席する合理的な支出を精算したものを除外した。
2023年取締役補償表
| 名前.名前 | 費用.費用 勝ったのは または 支払いました 現金で ($)(1) | 選択権 賞.賞 ($)(2) | 合計する ($) | |||||||||
| スコット·アプルボム(3) | 43,341 | 12,938 | 56,279 | |||||||||
| ジェフ(Br)コンロイ(3) | 0 | 0 | 0 | |||||||||
| パトリック·ガラゲル(3) | 46,470 | 12,938 | 59,408 | |||||||||
| グレッグ·ギブンス(3) | 71,332 | 12,938 | 84,270 | |||||||||
| Curt Mediros(3) | 48,030 | 12,938 | 60,968 | |||||||||
| ピーター·ウォース | 35,000 | 12,938 | 47,938 | |||||||||
| (1) | コスト抑制策では、取締役会は2023年第2、第3、第4四半期の現金費用の支払いを延期した。 |
| (2) | 金額は,FASB ASCテーマ718により,2023年6月15日に非従業員取締役の非限定株式オプションを付与し,その日までの公正価値を反映している.オプション報酬の公正な市場価値はブラック·スコアモデルを使用して決定された。2023年6月の株式オプション付与日の公正価値を推定するための仮定は以下のとおりである
無リスク金利 0.04% 期待 期限(年)5.51 予想変動率は1.28 期待配当収益率は0% 付与日公正価値$17.25
Applebaum,Gallagher,Givens,Medirosさんは,それぞれの辞任日から90(90)日以内に既得株式オプションを行使することができる。90日の期間が満了すると、Applebaum、Gallagher、Givens、およびMedirosさんが保有するすべてのストックオプションは、2023年6月15日に付与されたストックオプションを含めて没収されます。コンロイ·さんは2023年12月31日現在、オプションを保有していない。アプルボーム·さんは1,350件のストックオプションを保有しており、うち600件が付与されている。ガラゲル、ギブンス、メイディロス、ウォースさんは1,650件のストックオプションを保有しており、うち675件が付与されている。 |
| (3) | 以下の取締役は当社取締役会から辞任します:Applebaumさん(2023年12月6日)、Conroyさん(2023年1月5日)、ガラゲルさん(2023年12月12日)、Givensさん(2023年12月13日)、Medirosさん(2023年12月13日) |
2023年役員報酬計画
我々の報酬委員会と取締役会は、2023年6月15日から発効する非従業員取締役に対する役員報酬計画を承認した。この案は、以下の年度の現金補償を規定している
| ● | 取締役予約料:35,000ドル | |
● |
Lead独立役員採用費-55,000ドル | |
| ● | 委員会議長固定器: |
| ○ | 監査 -15,000ドル | |
| ○ | 報酬 -10,000ドル | |
| ○ | Brとコーポレート·ガバナンスを指名--8000ドル |
| ● | 委員会のメンバーは採用者: |
| ○ | 監査 --7500ドル | |
| ○ | 報酬 -5,000ドル | |
| ○ | Brとコーポレート·ガバナンスを指名します4000ドル |
すべての 現金料金は四半期延滞で支払い、カレンダー 年の全部または一部の月数に比例して割り当てられます。取締役は会議費用を徴収しないが、取締役会または委員会の会議回数が1年間の通常の会議回数 を超える限り、取締役会または委員会の会議回数が1年間の通常の会議回数 を超える限り、首席独立取締役(ある場合)および報酬委員会議長 を支払うことを決定することができる。取締役会や委員会会議に参加することによる非従業員取締役の合理的な費用も精算します。非従業員取締役は、授与日1周年に一度に分割払いになる年間株式オプション奨励を受ける。2023年6月15日現在、各非従業員取締役は、この日に750件のオプションの非限定株式オプション奨励を受けている。新しく任命された非従業員取締役は、1,000件のオプションの初期非限定株 オプション報酬を得る。
私たちの最高経営責任者シェイファー博士は私たちの取締役会長を務めていますが、彼の取締役としてのサービスによって追加の報酬を得ることはありません。シェイファー博士の2023年の給与の説明については、報酬集計表を参照されたい。
| 127 |
収益を使用する
配給代理費と我々が支払うべき推定発売費用を差し引いた後,あらかじめ出資した引受権証も売却しないと仮定し,今回発行した純収益は約650万ドルと見積もられている。しかし、これは最善を尽くした発行であり、最低証券数や収益金額が成約条件としてないため、実際の発行金額、配給代行費、純収益は現在、本募集説明書の表紙に規定されている最高金額を大幅に下回る可能性があり、私たちが発行しているすべての証券やいかなる証券も販売しない可能性がある。したがって、私たちが受け取った純収益は大幅に減少するかもしれない。
我々 は,我々が今回の発行から得たすべての純収益をCTX−1301の持続的な研究開発と商業化活動,および研究·開発へのさらなる投資を含む運営資本,資本支出,一般企業用途に利用する予定である。
現在、上述したように今回発行された純収益を使用することが予想されているが、資金の再分配が必要な場合もある。私たちの実際の支出の金額と時間は、私たちの販売とマーケティング と商業化努力、私たちの製品への需要、私たちの運営コスト、および本募集説明書の“リスク要因”の項目で説明されている他の要素を含む多くの要素に依存するだろう。したがって、私たちの経営陣は今回発行された純収益を活用することができるだろう。投資家 は経済,財務,あるいは他の情報を評価する機会がなく,これらの情報に基づいて 収益をどのように使用するかを決定する.
今回発行された純収益を使用する前に、短期、投資レベル、利子計上ツール、アメリカ政府証券を含む様々な保本投資に純収益を投資する予定です。
| 128 |
薄めにする
もしあなたが今回の発行で私たちの証券に投資した場合、あなたの所有権権益は直ちに今回の発行後の1株当たりの公開発行価格と付属の引受権証と私たちの普通株の調整後の1株当たりの有形帳簿純値との差額に希釈されます。
2023年9月30日現在、私たちの有形帳簿純赤字は約10万ドル、あるいは普通株1株当たり約0.11ドルです。私たちの1株当たりの有形帳簿純赤字は、2023年9月30日までの私たちの普通株式流通株数を除いて、私たちの総有形資産から総負債を引いたものに等しい。
株式承認証とATM販売を実施した後、2023年9月30日現在、我々の予想有形帳簿純価値は約300万ドル、または普通株1株当たり約2.14ドル、1株当たり約2.25ドル増加する。
1株2.00ドルの発行価格で1,375,000株の普通株と株式承認証およびbrを伴う株式承認証と事前融資権証を実施し,最大2,375,000株の普通株と随伴株式権証を購入した後,今回発売中の1部当たりの予資権証と承認株式証に伴う発行価格は1.9999ドルであり,配給代理費と支払うべき発売費用を差し引いた後,今回発売で提出したすべての事前出資の承認証が行使されていると仮定し,br今回の発売で提供される持分証は行使されない。このような株式承認証には何の価値もないため、このような株式承認証は持分に分類され、株式に計上され、2023年9月30日まで、私たちが調整後の有形帳簿純値で計算した予備試験金額は約960万ドル、あるいは普通株式1株当たり約1.85ドルである。この額は我々の既存株主にとって、調整後の予想帳簿純価値は1株0.29ドルであり、今回の発行に参加した投資家にとっては、直ちに1株0.15ドル 減少する見通しである。今回の発行に参加した投資家が支払う1株当たりの公開発行価格と付随する引受権証から形式的な調整後の1株当たりの有形帳簿損失純額を差し引くことで、今回の発行に参加した投資家に対する1株当たりの償却を決定する。次の表 はこの希釈を説明している:
| 1株当たり価格とセット株式証明書を公開発行する | $ | 2.00 | ||||||
| 歴史 2023年9月30日までの普通株式1株当たり有形帳簿純損失 | $ | (0.11 | ) | |||||
| 株式承認証演習とATM販売の有形帳簿純赤字の増加に起因することができる | $ | 2.25 | ||||||
| 株式証明書実施とATM販売後の有形帳簿純価値を予想する | $ | 2.14 | ||||||
| 今回発行された1株当たりの有形帳簿純価値の低下によるものと考えられます | $ | (0.29 | ) | |||||
| 今回の発行後の調整後の1株当たりの有形帳簿純価として | $ | 1.85 | ||||||
| 今回の発行株を購入した新投資家の1株当たりの償却 | $ | 0.15 |
以上の表と検討は、2023年9月30日までに発行された868,940株の普通株(権証行使とATM販売発効後、発行予定の普通株は1,413,001株 )に基づいており、br}までの日付は以下を含まない
| ● | 2021年計画によると発行された既発行株式オプション行使後に発行可能な72,638株の普通株は、加重平均行権価格は1株59.80ドルである |
| ● | 2021年計画によると、将来発行可能な66,679株の私たちの普通株 |
| ● | 786,710株普通株は、発行された株式権証を行使して発行することができ、加重平均行権価格は1株46.40ドルである |
| ● | 602,173株普通株(株式承認証行使と2024年1月債務転換発効 後に形式で1,028,955株と計算)は、 未発行の事前資本権証を行使する時に発行することができ、加重平均行権価格は1株当たり0.002ドルである。 |
以上検討したbr}情報は参考までに,定価時に決定された実際の公開発行価格と今回発行した他の条項に基づいて調整する.
| 129 |
株本説明
以下の説明は、私たちの証券の中で最も重要な用語をまとめた。これはただの要約であるため,あなたに重要である可能性のあるすべての 情報は含まれていない.完全な説明については、私たちが改訂および再説明した会社登録証明書および再記載された定款を参照して、そのコピーは、登録説明書の証拠品アーカイブとして使用され、本募集説明書はその一部である。
授権 資本化
私たちの改訂と再記載された会社登録証明書によると、私たちは240,000,000株の1株当たり額面0.0001ドルの普通株と10,000,000株の1株額面0.0001ドルの優先株を含む250,000,000株の法定株式を持っています。
2024年1月25日現在、発行済み普通株は1,450,171株であり、優先株は発行されていない。
普通株 株
私たちの普通株の保有者 は、私たちの取締役会が発表したこの用途のための合法的な資金から配当する権利があります。 普通株は償還も転換もできません。普通株保有者は優先引受権や引受権 を持って私たちのどの証券も購入していません。
私たち普通株の各保有者はその名義で発行された普通株ごとに一票を投じる権利があります。普通株式保有者は役員選挙に投票する際に投票数を累計する権利がない。
私たちが清算、解散、または清算する場合、私たちの普通株式の保有者は、すべての債務と他の債務を返済した後、比例して私たちの資産の比例シェアを獲得する権利があり、これらの資産は合法的に分配することができる。私たちの普通株のすべての流通株はすでに十分に入金されていて、評価できません。
優先株
私たちのbr取締役会は、株主のさらなる行動を必要とせずに、1つまたは複数のカテゴリまたはシリーズで最大10,000,000株の優先株を発行し、その指定、権利、特典、特権、および制限を決定する権利があり、株主が にさらに投票したり、行動する必要はない。これらの権利、優先権、および特権は、配当権、転換権、投票権、償還条項、清算優先権、債務超過基金条項、およびそのようなカテゴリまたは一連の株式数を構成または指定することができ、任意または全部が普通株の権利よりも大きい可能性がある。私たちの優先株発行は普通株式保有者の投票権やその等持者が私たちの清算時に配当金や支払いを得る可能性に悪影響を及ぼす可能性があります。また、優先株の発行は、わが社の支配権の変更や他社の行動を遅延、延期、阻止する可能性があります。優先株流通株はなく、私たちは現在も何の優先株 を発行する予定もありません。
逆買収デラウェア州法律とわが社の登録証明書と付則の影響
デラウェア州の法律、私たちが改正して再記載した会社の証明書、および私たちが改正して再記載した以下の法律の条項 は、他方が私たちの統制を受けることを遅延、延期、または阻止する可能性があります。
デラウェア州一般会社法203節
私たちはデラウェア州会社法第203条の制約を受けて、この条項はデラウェア州会社が株主が利益株主になった日から3年以内にこの株主といかなる業務合併を行うことを禁止しますが、以下の場合を除きます
| ● | その日までに、会社の取締役会は、株主が利害関係者になる企業合併や取引を承認した |
| 130 |
| ● | 株主が利害関係のある株主となる取引が完了した後、利害関係のある株主は、取引開始時に会社の少なくとも85%の発行済み議決権株を持っている。 発行された議決権のある株(ただし、関連株主が所有する発行済み議決権のある株は含まれていない)を決定するためには、(I)取締役や上級管理者を務めるbr}人が所有する株式、および(Ii)従業員参加者が当該計画に従って保有する株式が入札または交換要約で入札されるか否かを秘密に決定する権利のない株式は含まれていない。あるいは… | |
| ● | またはその後、企業合併は取締役会によって承認され、株主年次会議または特別会議で承認され、書面による同意ではなく、議決権付き株を発行した賛成票の少なくとも662/3%で承認され、関心のある株主が所有していない。 |
一般に、203節の企業合併の定義には、以下が含まれる
| ● | 会社と利益関連株主に関する任意の合併または合併 | |
| ● | 会社の10%以上の資産を売却、譲渡、質権、または他の方法で処分し、利害関係に関連する株主; | |
| ● | ある例外を除いて、会社が利益関連株主に会社の任意の株の取引を発行または譲渡することを招く | |
| ● | 会社に関する任意の取引であり、その取引の効果は、その会社の株式または利益関連株主の実益が所有する当該会社の任意のカテゴリまたは一連の株式の割合を増加させることである;または | |
| ● | 利益関連株主は、会社または会社によって得られた任意の損失、立て替え、保証、質権、または他の財務利益を受け取る。 |
一般に、第203条は、“利害関係のある株主”を、その人の関連会社及び共同経営会社と共に実益が所有しているか、又は利害関係のある株主身分確定前3年以内に会社が議決権付き株を発行した15%以上の実体又は個人を確実に所有していると定義する。
会社登録証明書と別例
当社の会社登録証明書の改訂と再記載の定款規定:
| ● | 私たちの取締役会を3つに分類します | |
| ● | 許可 は“空白小切手”優先株を発行し、その条項は、その株が株主の承認なしに を発行できることを確定することができる | |
| ● | 株主の取締役罷免を制限する | |
| ● | 株主に絶対多数票で私たちの定款または会社登録証明書のいくつかの条項を修正することを要求します | |
| ● | 株主が書面の同意の下で行動することを禁止し、すべての株主の行動が私たちの株主会議で行われなければならないことを要求する | |
| ● | 株主が株主特別会議を開催する権利を取り消す; | |
| ● | 取締役会メンバーの指名又は株主総会で行動可能な事項を規定する事前通知要求; | |
| ● | デラウェア州を私たちの特定の株主訴訟に対する独占的な管轄権として確立する。 |
| 131 |
ライセンスですが未発行株の潜在的な影響
私たちが改訂して再記載した会社登録証明書によると、株主の承認を必要とせずに将来発行できる普通株と優先株があります。私たちは、将来の公開発行を含めて、追加資本を調達して、会社の買収または配当金としての支払いを促進するために、これらの増発株式を様々な会社の目的に使用することができる。
未発行および未保持の普通株および優先株の存在は、私たちの取締役会が現在の管理層に優しい人に株を発行することができ、あるいは優先株を発行することができ、その条項は、合併、要約買収、代理競争、または他の方法で私たちの支配権を獲得する第三者の試みをより困難または阻害し、私たちの経営層の連続性を保護することができるかもしれない。また、取締役会は、投票権、配当権、転換権、償還権、清算優先株を含む一連の優先株を決定する権利を決定する権利がある。デラウェア州会社法で許可されている最大範囲で、私たちの会社の登録証明書に規定されている任意の制限を受けています。取締役会が優先株を発行し、このような優先株に適用される権利と割引を決定することを許可する目的は、株主が特定の発行を採決することに関する遅延を解消することである。優先株の発行は可能な融資、買収、他社の目的に理想的な柔軟性を提供しているが、 は第三者が私たちが発行した議決権のある株の多数 を買収したり阻止したりすることを困難にする可能性がある。
フォーラム選択
私たちが書面で代替裁判所を選択することに同意しない限り、デラウェア州衡平裁判所は、(I)会社を代表して提起された任意の派生訴訟または法的手続きを提起しなければならない。(Ii)会社の任意の取締役、会社の役員または他の従業員または会社の株主が受託責任に違反する任意の訴訟を主張し、 (Iii)会社または取締役または会社の役員に対してクレームを提起する任意の唯一かつ排他的な裁判所。または当社または任意の取締役または当社の上級管理者に対するクレームは、DGCLの任意の条項の解釈または適用について、当社の登録証明書または定款、または(Iv)内部事務原則によって管轄されるクレームを主張する任意の訴訟であるが、上記の各訴訟において、デラウェア州衡平裁判所が管轄権を有していないと認定した任意のクレームは除外される。本条項は、“取引法”によって生成されたクレーム、または任意の他の規定が排他的連邦管轄権を有する連邦証券法には適用されない。しかし、独占裁判所条項は、私たちが書面で代替裁判所を選択することに同意しない限り、アメリカ合衆国連邦地域裁判所は、証券法に基づいて提起された任意の訴因を解決する独占裁判所となるだろう。したがって、この条項は、証券法第22条が連邦裁判所および州裁判所が証券法またはその下の規則および法規に規定する任意の義務または責任を執行するすべての訴訟に対して同時に管轄権を有することが規定されているので、専属裁判所条項に列挙された1つまたは複数のカテゴリに属し、証券法に基づいてクレームを主張する訴訟に適用することができる。裁判所が証券法下の債権を執行するかどうかという排他的な裁判所規定にはまだ不確実性がある。
私たちbrは、裁判所がこの条項を実行するかどうかに不確実性があり、投資家は連邦証券法とその下の規則と法規の遵守を放棄することができないことに注意した。私たちはこの条項が私たちに有利であると信じているが、それはデラウェア州法律の適用された訴訟タイプでの適用をより 一致させるためであるが、この条項は私たちの役員と幹部に対するbrの訴訟を阻止する効果を果たすかもしれない。
エージェントに接続する
我々の普通株の譲渡エージェントはノースカロライナ州のComputerShare Trust Companyである.
| 132 |
私たちが提供する証券説明
私たちは1株2.00ドルの総合公開発行価格で1,375,000株の普通株を発売し、株式証明書を添付します。私たちはまた事前融資権証を提供して、最大購入することができます2,375,000株の普通株式 が今回の発売で我々の普通株式を購入すると、買い手およびその関連会社および一部の関連者が、今回の発売完了後に4.99% (または買い手が選択した場合、9.99%)を超える実益を有することになり、超過所有権をもたらす普通株の購入者の代わりに、普通株式の購入者 を発行した。1株当たりの私たちの普通株或いは事前融資株式承認証 はAシリーズ株式承認証とBシリーズ株式承認証と一緒に販売し、前者は普通株を購入し、後者は半分 普通株を購入する。当社の普通株式及び/又は事前資本権証及び関連引受権証の株式は別途発行されます。当社も当社の普通株株式を登録しており、当社が提供する事前資本権証及び引受権証を行使した後に時々発行することができます。
普通株 株
我々普通株の主な条項と規定は、本募集説明書における“株式説明”というタイトルで説明した。
株式承認証
ここで提供されるAシリーズの権証およびBシリーズの権証のいくつかの条項および条項の要約 は完全ではなく、権利証条項の制約を受け、権利証条項の制限を受け、その形式は証拠物として 登録説明書に提出され、本入札説明書はその一部である。潜在投資家は株式証明書表の条項と条項 を慎重に検討して、株式証明書の条項と条件の完全な説明を得るべきである。
持続期間 と行権価格
Aシリーズ株式承認証の行使価格は1株2.00ドルで、すぐに行使できる。Aシリーズ株式承認証 は発行日から5年以内に満期になります。Bシリーズ株式承認証の発行権価格は1株2.00ドルで、直ちに行使することができる。Bシリーズ株式承認証は発行日から2年以内に満期になります。株式配当、株式分割、再編または類似事件が我々の普通株および行使権証価格に影響を与える場合、株式証明書の行使価格と発行可能普通株の数は適切な 調整される。株式承認証は普通株式と事前資本権証と分けて発行され、その後すぐに単独で を譲渡することができる。株式承認証は証明された形でのみ発行されるだろう。
可運動性
所有者ごとの選択により,株式証はすべてまたは部分的に行使可能となり,正式に署名された行権通知 を吾等に提出し,行権時に購入した普通株式数について全数金を支払う(以下で議論するキャッシュレス行権は除く).所有者(及びその連合会社)は、当該等所有者承認株式証の任意の部分からbrを行使してはならず、行使直後に4.99%を超える発行済み普通株式を有しているが、もし所有者がbr等に少なくとも61日間の事前通知を発行した場合、保有者は当該等承認株式証を行使した後、発行された普通株の保有量を行使直後に発行された普通株の9.99%に増加させることができ、この百分率所有権は株式証の条項に基づいて決定することができる。
| 133 |
キャッシュレストレーニング
もし, が所有者が株式承認証を行使する際に,証券法に基づいて普通株を発行または転売する登録声明 が当時発効していないか,またはそのような株式の発行に適用されていない場合,所有者は,株式承認証を行使する際に承認証に記載されている式に基づいて決定された普通株式純額を選択し,株式承認証を行使する際に吾等に支払う現金金の代わりに選択することができる.
基本取引
株式証明書に記載されている基本的な取引の場合、一般に、私たちの普通株式の任意の再編、資本再分類、またはbrの再分類、販売、譲渡、または他の方法で私たちのすべてまたはほとんどの財産 または資産を処分し、私たちは他の人と合併または合併して、私たちが発行した株式に代表される50%を超える投票権を獲得し、任意の個人または団体は、私たちが発行した株式に代表される50%を超える投票権の実益所有者となり、他のエンティティとの合併または他のエンティティに入る買収要約または交換要約、または私たちが発行した株式に代表される投票権が50%を超えるbrの承認を受けた要約または交換要約は、その後の任意の株式承認証の行使時に、所有者 は、当該基本取引が発生する直前に承認株式証を行使することによって発行可能な1株当たりの普通株について、相続人または自社の普通株式数を代替価格として受け取る権利がある。および、所有者は、イベント発生直前に引受権証を行使することができる普通株式数の取引において、またはその取引のために受信されるべき任意の追加コストを有する。上述したように、基本取引が発生した場合、株式証所有者は、基本取引が完了すると同時に、または基本取引を完了してから30日以内に、株式証未行使部分を承認するブラック-スコアーズ価値と交換するために、吾らまたは後続エンティティに引受権証を償還することを要求する(各株式承認証を参照) を要求する。
しかしながら、当社の取締役会によって承認されていない基本取引を含む我々の制御範囲内でない基本取引が発生した場合、権証所有者は、基本取引が完了した日にのみ、我々または後続エンティティから同じタイプまたは形態(かつ同じ割合)の対価格を得る権利があり、基本取引に関連する私たちの普通株式保有者に提供および支払いされた権利証の未行使部分のブラック·スコアーズ価値を有する。株式または現金と株式の任意の組み合わせ、または私たち普通株の保有者は、ファンダメンタル取引に関連する他の形態の対価格を受け入れることを選択することができるかどうか。
譲渡可能性
適用法律に適合する場合には,権証所持者は,権利証を適切な譲渡文書とともに本行に戻す際に,自ら譲渡権証を選択することができる.
断片的株
株式承認証の行使時には、普通株式の断片的な株式は発行されない。代わりに、私たちの選択によると、発行された普通株式数を次の完全なシェアに上方丸め込むか、またはその最後の“br”シェアについて現金調整を支払い、金額はそのシェアに行使価格を乗じたものに等しい。
取引 市場
権利証はまだ成熟した取引市場ではなく、私たちはそのような市場が発展しないと予想している。私たちは、いかなる証券取引所や他の国でも認められている取引システムに上場する権利証を申請するつもりはありません。活発な取引市場がなければ、権利証の流動性は非常に限られるだろう。
| 134 |
株主として権利
株式証明書に規定があるか、または所有者が当社の普通株式に対する所有権を持っていない限り、この承認持分所有者は、当該所有者が当該等の株式所有者が株式証を承認するまで、任意の投票権を含む当社の普通株式所有者の権利または特権を有していない。株式承認証は、株式証所有者が私たちの普通株の分配或いは配当に参与する権利があることを規定する。
免除と修正案
吾等の同意及び少なくともbr}の大部分の未弁済持分証所有者の同意を経て、当該等の持分証を修正又は改訂することができ、又は当該等の株式承認証の規定を放棄することができる。
前払い資金株式証明書
以下に提供される予資権証のいくつかの条項および条項の要約は完全ではなく、資本資本権証条項の制約を受け、資本資本権証条項のすべての制限を受け、その形態は登録説明書の証拠物としてアーカイブされ、本入札説明書はその一部である。潜在投資家は事前融資権証表の条項と条項 をよく読んで、事前融資権証の条項と条件の完全な説明を得るべきである。
持続期間 と行権価格
ここで発売された前払い助成権証1株あたりの普通株初期行権価格は0.0001ドルに相当する。事前融資承認株式証 は直ちに行使可能であり、すべての行使時に満期になる。行権時に普通株を発行できる行権価格と株式数は、配当、株式分割、再編または類似イベントが私たちの普通株式と行権価格に影響を与える場合には、適切な調整が行われます。
可運動性
予備資本権証は,所有者1人当たり全部または一部を選択して行使することができ,正式に署名された行使権証を吾等に通知し,権利証を行使する際に購入した普通株式数について全数金を支払う方法である(以下で議論するキャッシュレス行使は除く).保有者(およびその関連会社)は、事前計画権証の任意の部分を行使してはならない。条件は、保有者が行使直後に4.99%を超える普通株流通株を持っていることであるが、brは、保有者が少なくとも61日間の事前通知を出した後、保有者は事前資本権証を行使した後、直ちに実益所有権を私たちが発行した普通株数の9.99% に増加させることができる。したがって、所有権率は事前資本金権証の条項によって決定されます。今回の発行で予資権証の購入者は、事前資本金権証が発行される前に初期行使限度額を私たちの普通株式流通株の9.99%に設定することを選択することができます。
キャッシュレストレーニング
持株者は、前払助成権証に記載されている式に基づいて決定された普通株式純額ではなく、その等行権を行使する際に吾等に支払うことが期待される現金の代わりに、行使時に普通株式純額(全部または一部)を受け取ることを選択することができる。
断片的株
事前資本権証を行使する際には、普通株の断片的な株式は発行されない。逆に、当社の選択の下で、発行された普通株式数を次の株式全体に上方丸め込むか、あるいは当社は現金調整 を支払い、金額はその点数に行使価格を乗じたものに等しい。
譲渡可能性
適用法律に適合した場合、事前資本金権証は、所持者が事前資本金権証を適切な譲渡文書と共に返還する際に、所持者の選択に応じて譲渡することができる。
| 135 |
取引 市場
権利証はまだ成熟した取引市場ではなく、私たちはそのような市場が発展しないと予想している。私たちはいかなる証券取引所や他の国でも認められている取引システムに上場する事前融資権証を申請するつもりはありません。活発な取引市場がなければ、事前融資権証の流動性は極めて限られるだろう。
株主として権利
資本金権証に規定があり、またはその所有者による普通株式の所有権がある以外、事前資本権証の所有者は、その資本資本権証を行使する前に、当社の普通株式所有者の権利または特権を有しておらず、いかなる投票権も含む。事前融資権証は、予融資権証の所有者は私たちの普通株の分配或いは配当に参与する権利があると規定する。
基本取引
前払い権証に記載されているように、基本的な取引は、一般に、私たちの普通株式の任意の再編、資本再編または再分類、売却、譲渡、または他の方法で私たちのすべてまたはほとんどの財産のbrまたは資産を処分し、私たちは他の人と合併または合併し、私たちが発行した株式に代表される50%以上の投票権を買収し、任意の個人または団体は、私たちが発行した株式に代表される50%を超える投票権の実益所有者となり、他のエンティティとの合併又は他のエンティティに組み込まれた買収要約又は交換要約、又は我々が発行した株式に代表される投票権の50%以上の買収要約又は交換要約によって承認された場合、その後、任意の事前出資の引受権証を行使する際に、所有者は、当該基本取引が発生する直前の発行すべき普通株を行使する1株当たりの普通株について、代替価格である相続人又は買収会社又は自社の普通株式数又は買収会社(既存の会社である場合)の普通株式数を取得する権利がある。および、所有者がイベントの直前に事前計画権証を行使することができる普通株式数の取引によって受信されるべき任意の追加コスト とを含む。
配給代理権証
私たちはまた、最大150,000株の普通株 を購入するために、配給エージェント(またはその指定者)に株式承認証を発行することに同意する。配給代理権証は即時に行使可能となり,その条項は上記株式承認証とほぼ同じであり,販売代理権証の行使価格は1株2.50ドル(1株発行価格および付認株式証発行価格の125%に相当)であり,終了日は今回の発売開始から5年 年である。以下の“割当計画”を参照してください。
| 136 |
流通計画
2023年12月27日(改訂)の採用契約に基づき,H.C.Wainwright&Co.,LLCまたは配給エージェントを我々の独占配給エージェントとして招聘し,合理的な最大努力で本募集説明書に基づいて発売された証券の購入要約を求めた.契約契約は、配給エージェントに自社の証券の購入を承諾させることはありませんが、契約契約により、配給エージェントは私たちを拘束する権利がありません。配給エージェントは、本募集説明書に従って提供される任意の証券を購入または販売することもなく、特定の数または金額の証券を購入または販売するように手配する必要もない。これはベストエフォートのキャンペーンで、今回の発売終了の条件として最低発売金額要求 はありません。配給代理は私たちの証券売却を手配するために合理的な最善を尽くすことに同意した。したがって、私たちはすべての普通株、事前融資権、そして引受権を売却しないかもしれない。今回発売された条項は,市場状況と我々,配給エージェントと潜在投資家との交渉に依存する.配給エージェントはそれが予想される発行で新しい資本を調達できることを保証しない。配給エージェントは,サブエージェントや選定したトレーダーに今回の発売に協力してもらうことができる.
ここで提供される証券を購入する投資家 は、私たちと証券購入協定に署名する権利があります。連邦証券と州法律により今回発売されたすべての購入者に提供される権利とbr救済措置のほか、証券購入契約を締結した購入者は私たちに違約請求をすることができる。今回発売中の大購入者にとっては,違約クレームを要求する能力が重要であり,証券購入プロトコルに基づいて提供される以下のユニークな契約を実行する手段として,(I)発売終了後1年以内に変動金利融資を行わない契約は,いくつかの例外を除いて,(Ii)発売終了後90日以内に株式融資を行わない契約が行われているが,例外的な場合は除く.証券購入協定における陳述、保証、およびチノの性質は、以下のことを含むべきである
| ● | 組織、資格、許可、衝突なし、政府届出不要、米国証券取引委員会届出中の現行事項、無訴訟、労働その他のコンプライアンス問題、環境、知的財産権と所有権事項及び“反海外腐敗法”などの各種法律のコンプライアンスについて標準的な陳述と保証を行う | |
| ● | 株式承認証登録、他の発行と統合してはならない、株主権利計画のない、重大な非公開情報の有無、収益の使用、購入者の賠償、普通株の保留と上場及び90日以内に後続持分売却ができないなどの事項に関する契約。 |
我々 は、本募集説明書 に従って提供される証券を購入するために、投資家資金を受信した後、発行中の証券を投資家に交付する。本募集説明書に基づいて発行された証券を2024年2月6日頃に交付する予定です。 今回の発行には最低証券数や収益金額の制限はありません。
料金 と費用
今回の発行で得られた総収益の7.8%に相当する総現金費用を配給エージェントに支払うことに同意しました。 今回の発行に関する50,000ドルの非実売費用手当、最高100,000ドルの法的費用と費用、最高15,950ドルの清算費用を配給エージェントに返済します。配給代理費と費用は含まれていないと思いますが、今回発行された総費用は約216,237ドルです。
配給代理権証
また、吾らは、今回発売した普通株式総数の4.0%(任意の事前出資株式承認証の関連株式を含む)を購入するために、配給代理又はその指定者又は配給代理権証に承認株式証を発行することに同意しており、 行使価格は、今回発売された1株当たり公開発行価格の125%及び付随する引受権証に相当する。配給代理権証は発行時に行使され、今回の発売開始から5年で満了する。
| 137 |
行使時に有効な登録声明登録がない場合、またはその中に記載されている目論見書が配給代理権証所有者が株式承認証株式を転売することができない場合、配給代理権証は、このとき“キャッシュなし行使”の方法で全部または部分的に行使することができ、すなわち、所有者が販売代理権証に従って計算された数 引受権証株式を受け取る権利がある。
配給代理権証は、FINRA規則5110に基づいて、慣用的な逆希釈条項(株式配当、分割、資本再構成などに適用される) を規定している。
しっぽ
もし吾等と配給エージェントとの契約契約期間内に、配給エージェント(又は当社と発売について会議を行う)が当社に紹介された任意の投資家が、吾等と配給エージェントとの契約終了又は満了後24(24)ヶ月以内に、公開又は非公開発売又は集資取引の形で吾等に任意の資本を提供する場合、吾等は、当該等のbr投資家の総収益について提供された現金及び承認持分補償を配給エージェントに支払う。FINRAルール2010によれば,配置エージェントは,当事者が配置エージェントによって直接紹介された範囲でのみこのような費用を得る権利がある.
優先購入権
発売が完了した場合、吾らは配給代理に優先購入権を付与しており、これにより、吾らは (場内融資を含む)や私募又は任意の他の株式、株式リンク又は債務証券の資金調達方式で資金を調達することを決定し、吾等は今回の発売完了後24(24)ヶ月前の任意の時間に投資銀行又はブローカー/取引業者を唯一の帳簿管理人、引受業者又は配給代理として採用する権利がある。
ロックプロトコル
我々の上級管理者と取締役は配給エージェントと合意しており,今回の 発売終了後90日間の販売禁止期間がある.これは、適用される販売禁止期間内に、当該者が、直接または間接的に売却、契約売却、売却、流通、購入、質権、質権の付与、または当社の普通株の任意の株式を、または当社の普通株の任意の証券に変換または行使または交換することができる任意のオプション、権利または株式承認証に変換または交換してはならないことを意味する。譲渡先がこれらのロック制限に同意すれば,販売禁止期間内に何らかの限られた譲渡を許可する.今回の発行終了後90日以内に我々の証券の発行と販売に類似したロック 制限を行うことにも同意したが,例外的な場合は除外した.配給エージェントは,このようなロックプロトコルの条項を放棄することを適宜決定することができ,別途通知する必要はない.
また、いくつかの例外的な場合を除いて、吾らは、今回の発売終了後1年以内に、当社の普通株取引価格または将来の特定またはイベントに基づいて価格リセットを行う証券を発行しないか、または任意の合意を締結して、将来確定した価格で証券を発行しないことに同意している。
賠償する
私たちは、証券法下のいくつかの責任、または分配エージェントがこれらの債務について支払うことを要求される可能性がある支払いを含む、配給エージェントのいくつかの責任を賠償することに同意した。
また、当行は、ある例外を除いて、今回発売中の証券購入者が、(I)吾等が証券購入契約又は関連文書に作成した任意の陳述、保証、契約又は合意に違反したこと、又は(Ii)第三者(当該購入者に関連する第三者を除く)が証券購入協定又は関連文書及び行われる取引について購入者に対して提起した任意の訴訟によって引き起こされた、又はそれに関連する責任を賠償する
| 138 |
ルール Mを守る
配給代理は、証券法第2(A)(11)節に示される引受業者と見なすことができ、委託者を務めている間に受信した任意の費用および我々の証券を販売する際に達成された任意の利益は、証券法による引受割引または手数料とみなされる可能性がある。配給エージェントは、ルール10 b-5および取引法下の法規Mを含むが、これらに限定されない証券法および取引法の要求を遵守することが要求される。これらのルールや規定 は,配給エージェントが我々の証券を購入·販売する時間を制限する可能性がある.これらの規則および規定によれば、配給エージェントは、(I)私たちの証券に関連する任意の安定した活動に従事してはならない;および(Ii)私たちの任意の証券を入札または購入するか、または誰にも私たちの任意の証券を購入させようと試みるが、取引法によって許可されるものは除外され、彼らが流通に参加することが完了するまで。
その他 関係
我々又は我々の関連会社との通常の業務過程において、配給代理及びその関連会社は、将来的に投資銀行取引及び他の商業取引に従事することが可能となっている。配給代理店は、これらの取引の通常の費用および手数料を受信したか、または将来的に受け取る可能性がある。
また、正常な業務活動において、配給代理及びその共同経営会社は、複数の投資を行うことができ、それ自体及び顧客の口座のために債務及び株式証券(又は関連派生証券)の取引を積極的に行うことができる。当該等の投資及び証券活動は、吾等又は吾等の連属会社の証券及び/又は手形に係る可能性がある。配給代理及びその付属会社も、当該等の証券又は金融商品について投資提案及び/又は独立研究意見を発表又は発表することができ、顧客が当該等の証券及びツールの多頭及び/又は空頭倉位を保有又は提案することができる。
我々が市場で提供するサービスにより,配給エージェントが配給エージェントとして機能する.ATMプロトコルについては、配給代理は、この合意に基づいて、当社の普通株株式を1回に販売して得られた毛収入の3.0%で手数料を徴収する権利がある。ATM契約の締結について、私たちは配給代理に50,000ドルの費用を払い戻しました。また、私たちは、職務調査更新会議ごとに、最高2,500ドルの法律顧問費用および任意の付帯費用を配置エージェントに精算することに同意します
配給エージェントは2023年9月の発売で配給エージェントを担当する.2023年9月の発売については,配給エージェントは(I)280,000.01ドルの配給エージェント費,または2023年9月に発売された総収益の7.0%と,(Ii)非実売費用50,000ドル,弁護士費と支出100,000ドル,および決済費用15,950ドルを返済した.また,2023年9月発売の締め切りには,配給エージェント(またはその指定者)に配給エージェント権証を発行し,配給エージェント(またはその指定者)に合計17,316株の普通株を購入した.このような配給代理権証の使用価格は14.44ドルで、2028年9月11日に満期になる。
電子配信
配信エージェントが維持するサイト上に電子フォーマットの入札説明書を提供することができ、配給エージェントは入札説明書を電子的に配信することができる。電子形式の入札説明書を除いて、これらのサイト上の情報は本募集説明書の一部でもなく、本募集説明書の一部でもなく、私たち或いは配給代理の許可及び/又は裏書き を経ておらず、投資家は依存すべきではない。
エージェントに接続する
我々普通株の譲渡エージェントと登録機構はノースカロライナ州のComputerShare Trust Companyである.
ナスダックが発売された
私たちのbr}普通株はナスダックに看板を掲げて取引して、コードは“CING”です
| 139 |
法務
この目論見書が提供する証券の有効性は、ニューヨークLowenstein Sandler LLPによって伝達される。Ellenoff Grossman&Schole LLP,New York,New Yorkは,今回の発行に関する配置エージェントの法律顧問である.
専門家
我々は,2022年12月31日まで,2022年12月31日と2021年12月31日までの連結財務諸表,および2022年12月31日までの2年間の毎年の総合財務諸表は,本稿の他の部分に出現する独立公認会計士事務所ピマウェイ会計士事務所の報告 及び当該事務所の会計·監査専門家としての権威 に基づいて計上されている。2022年12月31日と2021年12月31日の連結財務諸表をカバーする監査報告書には解釈段落が含まれており、その中で、我々の経常的な運営損失と純資本不足は、この実体の持続経営企業としての能力に大きな疑いを抱かせていることが指摘されている。連結財務諸表 は、このような不確実性の結果に起因する可能性のあるいかなる調整も含まない。
| 140 |
ここで詳細な情報を見つけることができます
この目論見書は、我々が米国証券取引委員会に提出した登録声明の一部である。本募集説明書は、登録説明書に記載されている全ての情報および登録説明書の証拠物を含まない。当社および本募集説明書の下で提供される証券のより多くの情報については、登録声明および登録声明の一部として提出された証拠品およびスケジュールを参照してください。あなたは本募集説明書に含まれている情報だけに依存すべきです。私たちは他の誰もあなたに違う情報を提供することを許可していません。私たちは、要約を許可しない司法管轄区域でこれらの証券を要約することはありません。本募集説明書に含まれる情報は、本募集説明書の交付時間または私たちの証券の任意の販売時間にかかわらず、関連文書の日付のみが正確であると仮定しなければなりません。
我々 は“取引法”の情報要求を遵守し,“取引法”に基づいて年度,四半期,その他の報告, 依頼書,その他の情報を委員会に提出する.このような報告書、依頼書、および他の情報は、登録声明、証拠物、およびスケジュールを含み、委員会のウェブサイトwww.sec.govを介して公衆に閲覧することができる。
我々は、これらの資料を電子的に証監会に提出または他の方法で証監会に提出した後、合理的で実行可能な場合には、できるだけ早く私たちのウェブサイト上または私たちのサイトを通じて、私たちの年間報告Form 10-K、Form 10-Q四半期報告、Form 8-K現在の報告 を無料で提供し、1934年の証券取引法第13(A)または15(D)節に基づいて提出または提供された報告修正案をできるだけ早く提供する。本募集説明書に含まれる情報または本サイトを介してアクセス可能な情報は、本募集説明書の一部ではなく、本募集説明書にも含まれていない。
| 141 |
Cingate, Inc.
連結財務諸表インデックス
| 監査されたbr合併財務諸表 | |
| 独立公認会計士事務所報告 | F-4 |
| 2022年と2021年12月31日までの連結貸借対照表 | F-5 |
| 2022年と2021年12月31日までの総合業務報告書 | F-6 |
| 2022年12月31日までと2021年12月31日まで年度株主権益変動表 | F-7 |
| 2022年12月31日と2021年12月31日までの年間現金フロー表 | F-8 |
| 財務諸表付記 | F-9 |
| 監査されていない合併財務諸表 | |
| 2023年9月30日と2023年12月31日までの連結貸借対照表 | F- 25 |
| 2023年9月30日と2022年9月30日までの3ヶ月と9ヶ月の総合経営レポート | F-26 |
| 2023年9月30日と2022年9月30日までの9ヶ月間の株主権益合併報告書 | F-27 |
| 2023年9月30日と2023年9月30日までの9ヶ月間統合キャッシュフロー表 | F-28 |
| 連結財務諸表付記 | F-29 |
| F-1 |
Cingate Inc.
連結財務諸表
2022年12月31日と2021年12月31日までの年度
(独立公認会計士事務所レポート付き)
| F-2 |
Cingate Inc.
連結財務諸表
2022年までと2021年12月31日まで年度
カタログ表
| ページ | |
| 独立公認会計士事務所報告(PCAOB ID:185) | F-4 |
| 合併貸借対照表 | F-5 |
| 合併経営報告書と全面赤字 | F-6 |
| 合併株主権益変動表 | F-7 |
| 統合現金フロー表 | F-8 |
| 連結財務諸表付記 | F-9 |
| F-3 |
独立公認会計士事務所報告
株主と取締役会へ
Cingate Inc.:
連結財務諸表に関する意見
我々は、添付Cingate Inc.(当社)が2022年12月31日及び2021年12月31日までの総合貸借対照表、2022年12月31日までの2年間の各年度の関連総合経営報告書及び全面赤字、株主権益及びキャッシュフロー、及び関連付記(総称して総合財務諸表と呼ぶ)を監査している。総合財務諸表は,米国公認会計原則に従って,会社の2022年12月31日,2022年12月31日と2021年12月31日までの財務状況,および2022年12月31日までの2年間の各年度の経営成果とキャッシュフローを公平に反映していると考えられる。
注目を行っている
添付されている総合財務諸表はすでに作成時に当社は継続経営企業としていると仮定している。総合財務諸表付記1で述べたように,当社はすでに運営損失,累積損失を出しており,運営および発展のための追加資金が必要であり,これらすべてが持続経営企業としての持続経営能力を大きく疑っている。付記1は、これらの事項に関する経営陣の計画も説明しています。連結財務諸表には、このような不確実性の結果による可能性のあるいかなる調整も含まれていません。
意見を求める根拠
これらの合併財務諸表は会社経営陣が担当しています。私たちの責任は、私たちの監査に基づいてこれらの連結財務諸表に対して意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社を独立させなければならない。
私たち はPCAOBの基準に従って監査を行います。これらの基準は、連結財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても詐欺によるものであっても、監査を計画し、実行することを要求する。我々の監査には、連結財務諸表の重大な誤報のリスクを評価するためのプログラム を実行すること、エラーによるものであっても詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれる。これらの手続きは、連結財務諸表中の金額および開示に関する証拠をテストに基づいて検討することを含む。私たちの監査には、経営陣が使用する会計原則の評価と重大な推定、合併財務諸表の全体列報の評価も含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
/s/ピマウェイ法律事務所
私たちは2020年から当社の監査役を務めています。
ミズーリ州カンザス市
2023年3月10日、注19日が2024年1月12日を除く
| F-4 |
Cingate Inc.
合併貸借対照表
2022年12月31日と2021年12月31日
| 2022 | 2021 | |||||||
| 資産 | ||||||||
| 流動資産: | ||||||||
| 現金と現金等価物 | $ | 5,356,276 | $ | 16,492,745 | ||||
| 短期投資 | - | 933 | ||||||
| 雑売掛金 | 234,432 | 690,248 | ||||||
| 前払い費用と他の流動資産 | 2,278,944 | 1,698,353 | ||||||
| 流動資産総額 | 7,869,652 | 18,882,279 | ||||||
| 財産と設備、純額 | 2,904,787 | 3,145,378 | ||||||
| 経営的リース使用権資産 | 630,618 | 858,600 | ||||||
| 総資産 | 11,405,057 | 22,886,257 | ||||||
| 負債と株主権益 | ||||||||
| 流動負債: | ||||||||
| 売掛金 | 762,357 | 264,687 | ||||||
| 費用を計算する | 894,635 | 601,300 | ||||||
| 支払手形 | 5,000,000 | - | ||||||
| 融資リース負債流動 | 16,053 | 15,096 | ||||||
| 賃貸負債を経営し、流動 | 339,755 | 295,595 | ||||||
| 流動負債総額 | 7,012,800 | 1,176,678 | ||||||
| 長期負債: | ||||||||
| 融資リース負債、当期純額 | 21,487 | 37,534 | ||||||
| 賃貸負債を経営し,当期純額 | 488,748 | 828,503 | ||||||
| 長期負債総額 | 510,235 | 866,037 | ||||||
| 総負債 | 7,523,035 | 2,042,715 | ||||||
| 株主権益 | ||||||||
| 普通株、額面0.0001ドル;2022年12月31日と2021年12月31日までに、2.4億株の発行を許可し、株式565,470株を発行·発行する | 57 | 57 | ||||||
| 優先株は、額面0.0001ドル;2022年12月31日と2021年12月31日まで、認可発行株10,000,000株、発行と発行株は0株 | - | - | ||||||
| 追加実収資本 | 73,290,461 | 72,575,584 | ||||||
| その他の総合収益を累計する | - | 165 | ||||||
| 赤字を累計する | (69,408,496 | ) | (51,732,264 | ) | ||||
| 株主権益総額 | 3,882,022 | 20,843,542 | ||||||
| 総負債と株主権益 | $ | 11,405,057 | $ | 22,886,257 | ||||
合併財務諸表の付記を参照。
| F-5 |
Cingate Inc.
合併経営報告書と全面赤字
2022年と2021年12月31日までの年度
| 2022 | 2021 | |||||||
| 運営費用: | ||||||||
| 研究開発 | $ | 8,995,280 | $ | 8,410,489 | ||||
| 一般と行政 | 8,506,438 | 12,268,909 | ||||||
| 営業損失 | (17,501,718 | ) | (20,679,398 | ) | ||||
| 利息とその他の費用,純額 | (174,514 | ) | (30,593 | ) | ||||
| 所得税前損失 | (17,676,232 | ) | (20,709,991 | ) | ||||
| 所得税の割引 | - | - | ||||||
| 純損失と総合損失 | $ | (17,676,232 | ) | $ | (20,709,991 | ) | ||
| 普通株は基本的に1株当たり純損失と希釈して1株当たり純損失 | $ | (31.26 | ) | $ | (55.87 | ) | ||
| 基本普通株と希釈後の普通株1株当たり純損失の加重平均株式数の計算に用いる | 565,470 | 370,679 | ||||||
合併財務諸表の付記を参照
| F-6 |
Cingate Inc.
株主権益合併報告書
2022年と2021年12月31日までの年度
| 積算 | ||||||||||||||||||||||||||||
| 普通株 | 余分な実収- | メンバーの | 積算 | その他の総合 | 株主の | |||||||||||||||||||||||
| 株 | 金額 | 資本 | 資本 | 赤字.赤字 | 収入.収入 | 権益 | ||||||||||||||||||||||
| 残高2021年1月1日 | - | - | $ | 32,314,543 | $ | (31,022,273 | ) | $ | 165 | $ | 1,292,435 | |||||||||||||||||
| 会員の出資 | - | - | - | 7,104,957 | - | - | 7,104,957 | |||||||||||||||||||||
| 短期投資の未実現損失 | - | - | - | - | - | - | - | |||||||||||||||||||||
| IPOで発行された普通株は、発行コスト4,627,079ドルを差し引く | 208,333 | 21 | 20,374,197 | - | - | - | 20,374,218 | |||||||||||||||||||||
| 株に基づく報酬費用 | - | - | 43,835 | - | - | - | 43,835 | |||||||||||||||||||||
| 再編合併に関する有限責任会社単位を普通株に変換する | 357,137 | 36 | 39,419,464 | (39,419,500 | ) | - | - | (0 | ) | |||||||||||||||||||
| 再編合併に関する利益利益単位の変更 | - | - | 12,738,088 | - | - | - | 12,738,088 | |||||||||||||||||||||
| 純損失 | - | - | - | - | (20,709,991 | ) | - | (20,709,991 | ) | |||||||||||||||||||
| 残高2021年12月31日 | 565,470 | 57 | $ | 72,575,584 | $ | - | $ | (51,732,264 | ) | $ | 165 | $ | 20,843,542 | |||||||||||||||
| 売却可能な投資 | - | - | - | - | - | (165 | ) | (165 | ) | |||||||||||||||||||
| 株に基づく報酬費用 | - | - | 800,796 | - | - | - | 800,796 | |||||||||||||||||||||
| IPOコスト | - | - | (85,919 | ) | - | - | - | (85,919 | ) | |||||||||||||||||||
| 純損失 | - | - | - | - | (17,676,232 | ) | - | (17,676,232 | ) | |||||||||||||||||||
| 残高2022年12月31日 | 565,470 | 57 | $ | 73,290,461 | $ | - | $ | (69,408,496 | ) | $ | - | $ | 3,882,022 | |||||||||||||||
合併財務諸表の付記を参照
| F-7 |
Cingate Inc.
統合現金フロー表
2022年と2021年12月31日までの年度
| 2022 | 2021 | |||||||
| 経営活動: | ||||||||
| 純損失 | $ | (17,676,232 | ) | $ | (20,709,991 | ) | ||
| 純損失と経営活動で使用される現金純額の調整: | ||||||||
| 減価償却 | 394,011 | 708,317 | ||||||
| 利益利息単位変動に関する非現金補償費用 | - | 12,738,088 | ||||||
| 株に基づく報酬 | 800,796 | 43,835 | ||||||
| 経営性資産と負債変動状況: | ||||||||
| 雑売掛金 | 455,816 | (538,476 | ) | |||||
| 前払い費用と他の流動資産 | (580,591 | ) | (1,258,857 | ) | ||||
| 経営的リース使用権資産 | 227,982 | 100,124 | ||||||
| 貿易勘定と売掛金をまかなう | 791,005 | (1,324,686 | ) | |||||
| 賃貸負債の当期部分を経営する | 44,160 | 61,427 | ||||||
| 賃貸負債の長期部分を経営する | (339,755 | ) | (228,938 | ) | ||||
| その他負債 | - | (23,615 | ) | |||||
| 経営活動のための現金純額 | (15,882,808 | ) | (10,432,772 | ) | ||||
| 投資活動: | ||||||||
| 財産と設備を購入する | (153,420 | ) | (814,736 | ) | ||||
| 短期投資を売却して得られる収益 | 933 | - | ||||||
| 他にも | (165 | ) | - | |||||
| 投資活動のための現金純額 | (152,652 | ) | (814,736 | ) | ||||
| 融資活動: | ||||||||
| 普通株で得た金を初めて公開発行し,純額 | - | 20,374,218 | ||||||
| IPO発行コスト | (85,919) | |||||||
| 会員の出資 | - | 7,104,957 | ||||||
| 支払手形の支払い | - | (500,335 | ) | |||||
| 支払手形収益 | 5,000,000 | - | ||||||
| 融資リース債務の元金支払い | (15,090 | ) | (436,259 | ) | ||||
| 融資活動が提供する現金純額 | 4,898,991 | 26,542,581 | ||||||
| 現金と現金等価物: | ||||||||
| 現金と現金等価物の純減少 | (11,136,469 | ) | 15,295,073 | |||||
| 年初現金および現金等価物 | 16,492,745 | 1,197,672 | ||||||
| 年末現金および現金等価物 | $ | 5,356,276 | $ | 16,492,745 | ||||
| 非現金活動 | ||||||||
| 課税財産と設備は年末には支払われない | $ | - | $ | 279,730 | ||||
| 現金支払い: | ||||||||
| 支払の利子 | $ | 19,000 | $ | 114,725 | ||||
合併財務諸表の付記を参照
| F-8 |
Cingate Inc.
連結財務諸表付記
2022年12月31日と2021年12月31日までの年度
(1)業務性質と流動資金
組織する
Cingulate Inc.は生物製薬会社であり、その薬物送達プラットフォーム技術を利用して製品を開発することに集中し、brが毎日1回の多用量療法錠剤を調製と製造できるようにし、最初は注意欠陥/多動障害(ADHD)の治療に集中した。同社は2種類の独自の第一線覚せい剤薬物CTX−1301(酢酸デキサメタゾン)とCTX−1302(右旋フェニルプロピルアミン)を開発しており,ADHDの治療に用いられており,すべての患者群:児童,青少年,成人に適している。CTX-1301とCTX-1302は柔軟なコア錠剤技術と目標製品プロファイルを採用し、迅速な効果と持続的な活動br日を提供し、血漿薬物レベルは制御可能に低下し、そして良好な耐性を有することを目的とした。同社はすでにCTX-1301の第三段階臨床試験を開始し、成人用量最適化研究中の第一陣の患者は2023年初めに投与した。また、同社には3つ目の不安治療製品、CTX-2103があり、開発段階にある。
2012年11月14日、デラウェア州有限責任会社Cingate Treateutics LLC(CTX)が設立された。2021年5月10日、CTXのデラウェア州会社と完全子会社Cingate Inc.(Cingate、または当社)が設立され、ホールディングスとして当社が上場することが予想される。2021年第3四半期に発生した再編合併により,Cingate はCTXを効率的に買収し,CTXのすべての発行済単位はCingate普通株に変換された。CTXは依然として会社が業務を展開する実体 である.
CTX はCingateの前身であり,財務報告目的に用いられている。2022年12月31日と2021年12月31日までの年度の総合財務諸表と付記はCingateとその付属会社(CTXを含む)の全面的な合併を代表し、すべての言及されたbr社の内容はこの全面的な合併を代表する。
流動性
社は設立以来運営中に損失と負キャッシュフローが出現した。収益前実体として、会社は開発中の候補製品が米国食品医薬品局(FDA)の承認、生産、発売、収入を得るまで、資金を調達して運営を支援する能力に依存している。初公募株は2021年12月に完成し、会社に研究開発活動を継続させる能力がある。また、付記7で述べたように、当社は2022年8月に本票から500万ドルの収益を得ています。しかし、当社は運営や開発に追加の資金が必要となります。当社は2023年1月に販売代理(“ウェインライト”)であるH.C.ウェインwright&Co.,LLCと市場発売プロトコル(“ATM プロトコル”)を締結することにより,当社は随時Wainwrightを通して普通株を発売することができ,総収益は最高2,650,000ドルに達する。さらに、経営陣は、他の会社との潜在的な協力や他の戦略取引を含む追加株式の発行、債務の発行、または他の資本源を含む資金を得る様々な戦略を評価している。これらの計画を成功させるには会社の努力も必要であり、市場要因やFDAによる候補製品の承認など、会社のコントロール以外の要素も必要である。当社では、その計画が効果的に実施され、上記の条件やイベントが十分に緩和または緩和されることは保証されておらず、当社が財務諸表発表日から1年間経営を継続する能力が大きく疑われている。付随する総合財務諸表 は持続経営をもとに作成され、正常業務過程における資産現金化および負債返済状況 を考慮する。連結財務諸表は、このような不確実性の結果 がもたらす可能性のあるいかなる調整も反映しない。
| F-9 |
(2)重要会計政策の概要
(A)列報基礎と合併原則
添付されている総合財務諸表は、米国公認会計原則(“米国公認会計原則”)に基づいて作成されている。連結財務諸表はCingateとその完全子会社の勘定を含む。すべての企業間口座と取引は合併でキャンセルされました。
(B)予算の使用
米国公認会計原則に基づいて連結財務諸表を作成することは、報告期間中の資産及び負債の報告金額、連結財務諸表の日付又は有資産及び負債の開示及び報告期間内に報告された費用金額に影響を与えるために、管理層に推定及び仮定を要求する。実際の結果は推定とは異なる可能性がある。
および仮説は定期的に検出されると予想されるが,改訂の影響は必要と判断された 期間の総合財務諸表に反映される.
(三)信用リスク集中
会社は現金等値預金を持っており、財政年度を通して、これらの預金は連邦預金保険会社が加入している250,000ドルの限度額を超えています(入金項目は考慮していません)。経営陣はこれらの金融機関の健全性を監督しており、当社が預金中の未保険部分に関する重大な信用リスクを有しているとは考えていない。
(D)現金と現金同等物
銀行 普通預金口座と初期満期日が3ヶ月以下の短期流動投資は現金と現金等価物とみなされる。2022年12月31日現在と2021年12月31日現在の現金および現金等価物には、銀行預金および短期通貨市場基金が含まれています。現金および現金等価物は、公正な価値のコストで計算されています。
(E)雑入金
雑項目(Br)の売掛金には、会社の2021年と2020年の連邦所得税申告書(まだ受け取っていない)による賃金税控除と、この2年間に発生した賃金コストの従業員残留税控除が含まれています。同社はIAS 20を分析した政府補助金の会計計算と政府援助の開示 “は、これらの入金に対して会計処理を行う際に。当社は2022年12月31日と2021年12月31日までに、これらの売掛金に関する支出を必要としないと判断した。
(F)財産及び設備、純額
財産と設備の純額はコストから減価償却累計を引いて申告する.保修と修理は発生時に費用を計上する。物件と設備は推定余剰使用年数によって直線減価償却を採用し、あるいは賃貸改善 或いは融資賃貸項目の下で賃貸資産を場合、賃貸年期(例えば短い)に基づいて減価償却を提出する。
(G)借款書
会社は、カンザスシティ本部オフィスとニュージャージーオフィスのオフィススペースと、いくつかの家具と設備の2つの融資リースに関連する2つの取消不能な経営リースのうちの2つのテナントであり、そのうちの1つのレンタル期間は2021年12月31日に終了する。
| F-10 |
会社は契約開始時に手配がリースであるかどうかまたはテナントを含むかどうかを決定します。当社はリース開始日にROU資産と賃貸負債を確認します。経営リースについては,リース負債の最初とその後はリース開始日未払い賃貸支払いの現在値 で計測した。融資リースについては,リース負債は最初に運営リースと同様の 方式および日付で計量され,その後実際の利息法で償却コストで計測される。
会社が未払い賃貸支払いを現在値に割引するための割引率を決定するには、管理層の判断が必要だ。 ASC 842は、テナントがレンタルに隠れた金利を用いて未払い賃貸支払いを割引することを要求し、その金利が簡単に確定できない場合は、その逓増借款金利を用いて割引を行う。暗黙金利は当社の賃貸契約の1つの契約に記載されているが、他の賃貸契約については、当社はレンタル者の推定残存価値やレンタル者の繰延初期直接コスト金額を得ることができないため、暗黙金利は確定できない。そこで,会社はこれらのレンタルの割引率としてその逓増借款金利を用いている。当社の賃貸の逓増借款金利とは、似たような条項の下での賃貸支払いに等しい金額を担保に借入するのに必要な金利のことです。当社は担保に基づいて借金をすることができなかったため、信用格付け機関が実体の信用格付けを決定する際に通常分析される要素に基づいて総合信用格付けを決定した。当社は収入前実体であるため、当社はその逓増借入金利は賃貸計量日の総合CCCと低い債券利差 に基づいて特定の賃貸満期日に基づく無リスク金利を加えるべきであると決定した。
投資収益資産は、リース開始日またはリース開始日前に支払われたリース支払い調整後のリース負債初期金額に、生成された任意の初期直接コストに加えて受信した任意のリースインセンティブを減算することを含む。
経営リースについては、ROU資産はその後、レンタル期間全体にわたってリース負債の帳簿価値に 初期直接コストを加え、任意の前払い(未払い)賃貸支払いを加え、受信した累積賃貸インセンティブの未償却残高 を減算する。リース費用は、リース負債の増加とROU資産の償却を含むリース期間内に直線的に確認される。
融資リースについては,リースが対象資産の所有権を当社に譲渡するか,あるいは当社が行使対象資産の選択権を当社に譲渡することを合理的に決定しない限り,ROU資産はその後,直線的な方法を用いてリース開始日からその耐用年数終了またはリース期間終了前までの償却を行う。これらの場合、ROU資産は対象資産の使用年数内に償却される。ROU資産の償却はリース負債の支払利息 と分けて確認と列報する。
ROU 経営と融資リースの資産は減価損失を評価する。ASCのテーマ別 360-10における長期資産減価指導によると、財産と設備--全体的にそれは.ROU資産は、それが存在する資産グループと共に減少値を評価する。
経営的リースROU資産は合併貸借対照表に経営的リース使用権資産として示されている。経営リース負債の当期と長期部分 は総合貸借対照表にそれぞれ示されている。融資リースROU資産には不動産、工場、設備が含まれている。融資リース負債の当期部分と長期部分はそれぞれ総合貸借対照表 に列挙されている。
(H)長期資産減価
イベントや状況がその長期資産(物件や設備を含む)の帳簿価値が回収できない可能性があることを示す場合、会社はその等の資産の帳簿価値を評価する。これらの事件や環境変化は、経営業績の著しい悪化、業務計画の変化、あるいは将来のキャッシュフローの変化を含む可能性がある。減価指標があれば、当社は資産の帳簿価値と資産予想による未割引現金流量を比較することで回収可能性を評価する。将来のキャッシュフローの合計が帳簿金額より少ないと予想されれば、会社は減価損失を確認する。減価損失は,帳簿価値が長期資産群の公正価値を超える金額を比較することで計測される。2022年12月31日までや2021年12月31日までに減値は確認されていない。
| F-11 |
五、研究開発
研究·開発コストは,会社のbr候補製品開発に関するすべての直接·間接コストを含む発生時に費用を計上する。これらの費用には、第三者に支払う研究開発と製造サービス、人員コスト、製造設備の減価償却が含まれている。報告期間末には、同社が第三者サービス提供者に支払う金と、研究開発目標の達成に向けた見積もり進捗を比較する。このような推定値は、より多くの情報が取得されると変化する可能性がある。サービス提供者への支払い時間および会社が提供するサービスの進展を推定することにより、会社はこれらのコストに関する前払いまたは課税費用純額を記録する可能性がある。
(J)株ベースの報酬
Br社は、ブラック·スコアーズオプション定価モデルを使用して、従業員の付与日の公正価値に基づいて、すべての株式奨励の従業員と取締役株式報酬支出を測定します。サービス条件のある株式奨励については,株式報酬費用は必要なサービス期限内に直線法で確認する.没収行為は発生時に確認します。付記11の他の情報 を参照してください。
*(K)Paycheck保護計画
2020年3月27日、米連邦政府は“コロナウイルス援助、救済、経済安全法案”(略称“CARE法案”)を公布した。 CARS法案は、米国小企業管理局(SBA)によって管理され、2020年6月5日に公布されたPaycheck保護計画柔軟性法案(“PPP柔軟性法案”)によってさらに改正されるPaycheck保護計画(PPP)に関する条項を含む。2020年4月、会社は312,500ドルの購買力平価ローンを獲得した。2021年2月、同社は2番目の購買力平価ローン236,457ドルを獲得した。PPP柔軟性法案によって改正されたCARE法案の要求によると、条件に合った賃金コスト、レンタル料、担保融資利息、光熱費の支払いに収益を利用すれば、PPPローンは完全に免除される。
2021年7月、会社は通知を受け、1回目の購買力平価ローンは小企業管理局に免除され、2021年10月、会社は通知を受け、2回目の購買力平価ローンは小企業管理局に免除された。同社はIAS 20を分析した政府補助金の会計計算と政府援助の開示“、 これらのローンの会計処理。
(L)断片
運営部門は会社の構成要素であり、単独の財務情報が利用可能であり、会社の首席運営決定者が資源をどのように割り当てるかを決定し、業績を評価する際に定期的に評価を行う。同社は現在、1つの運営業務部門である薬品開発で業務を展開している。
*(M)所得税
Cingate Inc.は“国内税法”によりC類会社として課税される。Cingate Inc.は、財務報告に記録された資産および負債金額と税務法規によって計量されたこのような金額との間の一時的な差の影響を反映するために、繰延所得税を記録する。2022年12月31日まで、CTXはCingate Inc.の全額免除エンティティであり、CTXとその完全子会社Cingate Works Inc.のすべての活動は、Cingate Inc.の当期とbr繰延税金資産と負債の計算に含まれている。同社は、履歴収入レベルや将来の課税所得額などの項目によって、brの繰延税金資産を実現できない可能性があることを決定したため、brの全額推定準備を記録した。
| F-12 |
会社はまだ何の不確定な税務状況も確定していません。連結財務諸表には、不確定税務状況に関する利息や罰金は確認されていません。また、税収優遇総額が今後12ヶ月以内に合理的に大幅に増加または減少する可能性があることが確認されていない税務頭寸は存在しない。
Br社は連邦と各州の管轄区で所得税申告書を提出します。このような連邦所得税はどうでもいい。会社 は2019年前の数年以内に米国税務機関の連邦所得税審査を受けなくなった。
(N)普通株式引受権証
同社は2021年12月にそのIPOに関する引受権証を発行した。これらの権益ツールは発行時に公正な価値で評価される。注12の他の情報を参照してください。
(O)1株当たり純損失
基本的に1株当たり純損失は純損失を期間内に発行された普通株の加重平均から算出した。 は潜在的な希釈証券は考慮しない。普通株当たりの純損失の計算方法は,純損失を簡略化方法で除した期間内に発行された普通株の加重平均に期内の潜在的希薄化ツール数の総和 を加えることである。1株当たりの純損失を希釈するのは1株当たりの純損失とほぼ同じであり、潜在的な希薄化証券の影響が逆薄であるためである。
(3) 前払い料金と他の流動資産
費用を前払いする
前払い費用およびその他の流動資産は、2022年12月31日と2021年12月31日までである
前払い料金明細書
| 2022 | 2021 | |||||||
| 研究開発 | $ | 1,377,391 | $ | 643,917 | ||||
| 保険 | 472,152 | 761,594 | ||||||
| 活性薬物成分 | 209,156 | 264,361 | ||||||
| 資金調達コストを繰り下げる | 100,339 | - | ||||||
| 専門費 | 61,524 | - | ||||||
| 会費と購読料 | 37,684 | - | ||||||
| 他にも | 20,698 | 28,481 | ||||||
| 前払い費用総額 | $ | 2,278,944 | $ | 1,698,353 | ||||
| F-13 |
(4)財産と設備
2022年12月31日と2021年12月31日までの財産と設備の純資産は以下の通り
財産と設備別表
| 推定数 | ||||||||||||
| 使用寿命 | ||||||||||||
| (単位:年) | 2022 | 2021 | ||||||||||
| 装備 | 2-7 | $ | 2,565,997 | $ | 2,509,126 | |||||||
| 家具と固定装置 | 7 | 145,754 | 145,754 | |||||||||
| コンピュータ装置 | 5 | 41,898 | 41,898 | |||||||||
| 賃借権改善 | 5 | 471,505 | 471,505 | |||||||||
| 建設中の設備 | - | 1,739,699 | 1,643,150 | |||||||||
| 財産と設備、毛額 | 4,964,853 | 4,811,433 | ||||||||||
| 減算:減価償却累計 | (2,060,066 | ) | (1,666,055 | ) | ||||||||
| 財産と設備、 純額 | $ | 2,904,787 | $ | 3,145,378 | ||||||||
2022年12月31日と2021年12月31日までの年間減価償却費用はそれぞれ394,011ドルと708,317ドルである
(5)課税費用
当計 2022年12月31日現在と2021年12月31日までの料金には、
費用明細書を計算すべきだ
| 2022 | 2021 | |||||||
| 利子 | $ | 292,339 | $ | - | ||||
| 専門費 | 314,446 | 71,570 | ||||||
| 研究開発 | - | 250,000 | ||||||
| CIP-デバイス | - | 279,730 | ||||||
| 従業員ボーナス | 175,625 | - | ||||||
| 他にも | 112,225 | - | ||||||
| 計算すべき費用合計 | $ | 894,635 | $ | 601,300 | ||||
(6)または事項
Br社は、通常の業務過程やその他の面で引き起こされる法的手続きやクレームの影響を時々受ける可能性があります。私たちに対する重大な法的責任は、私たちの業務、財務状況、運営結果に悪影響を及ぼす可能性があります。
Br社は、発生または損失に関する法的コストを記録し、これらの事項に重大または損失が発生した場合に準備金を確立し、米国会計基準450に基づいて、管理層が重大または損失があることを決定し、可能性があり、合理的に評価する“意外な状況。” 損失範囲が推定され、その範囲内のある金額が 範囲内の任意の他の金額の推定値よりも良いようである場合、その金額は計算されるべきである。この範囲内のどの金額も他の金額よりも良い推定値として決定できない場合、範囲内の最小金額を累積する。これらの金額は、保険や第三者への請求によって回収可能な金額 を差し引くことはありませんが、回収可能であると考えられる場合、保険会社や他の第三者からの未割引入金は個別に計上される可能性があります。 結果の予測が困難であるため、最終的な 解決策は現在の分析とは異なる可能性があるため、経営陣が判断したり損失があったりする必要があります。当社は新しい資料に基づいて課税項目を改訂します。損失の結果を的確に予測または予測することは不可能であるが、経営陣は、財務諸表にはこのような事項に関する潜在損失計のために十分な準備金が提案されていると考えている。
| F-14 |
(7) 関係者手形の支払い
関連 支払当事者手形
2022年8月10日、会社はWerth Family Investment Associates LLC(WFIA)から5,000,000ドルの債務融資を受けた。WFIAマネージャーのPeter Werthは会社の取締役会のメンバーだ。本約束手形は無担保手形で、年率は15%です。 未返済元金とすべての課税および未払い利息は2025年8月8日に満期になりますか、または2023年4月1日からの1つのカレンダー四半期の前5営業日以内にWFIAが書面で要求してから120日以内に支払います。当社は手形の全部または一部を前払いし、割増や罰金を徴収することができますが、返済された金はこれ以上借りることはできません。2022年12月31日現在、紙幣の500万ドルはすべて返済されていない。
会社は2022年12月31日までの年間で、本手形に関する292,339ドルの利息支出を確認した。この利息支出 は2022年12月31日の総合貸借対照表の課税費用に計上される。
(8)会員資本
再編合併前に、当社は創設単位、B類、D類、E類、F類とG類優先株およびC類利益権益を含む多様な種類のメンバー資本を持っていた。B、E、F、G類優先株は、投資資本の見返りとしての現金分配に関する類似の権利を有する。クラスD優先株は創設者と他のクラス優先株のすべての権利 に加えて以下に述べるいくつかの追加権利を持つ.すべてのレベルの会員資本は投票権を持っている。同社はメンバーごとに資本br口座を維持している。2021年12月31日までの年度内に,再編合併前に3,243,201個のF類およびGクラス単位が発行された。
クラスF第一選択単位
CTX取締役会は2ロットに分けて6,984,985個のF類優先株を承認した;すべての許可されたF類株はすべて組換え 合併前に発行された。同社はFクラス単位を発行することで合計1130万ドルを調達した。新しく作成されたFレベルユニットは、CTX取締役会によって許可され、3に反映されます研究開発Fクラスユニットの作成を反映して2018年12月14日に発効する運転協定の改正と再署名が行われた。
クラスG第一選択単位
CTX取締役会は12,000,000個のG類優先株を承認した;再編合併前に2,998,184個を発行した。同社はGクラス単位の発行で670万ドルを調達した。CTX取締役会が許可した新たに設立されたG類単位は2021年2月9日に発効する 。
当社はまずB,D,E,FとG類優先株の保有者に割り当て(あれば),そのようなメンバーが返却していない出資額に比例して分配する。そして,イニシエータ単位を含めて全メンバに割り当て,メンバごとに持つ単位数の割合に比例して割り当てるとともに,メンバごとに発行する際に適用されるCクラスの利潤権益の割当て敷居を考慮して, 付記9でさらに述べたように,利益単位プロトコルごとに開示する.
単位発行に関するコスト は無関係である.再編合併の条項により,すべての単位がCingateの普通株 株に変換され,詳細は付記1を参照されたい。
(9)利益利息計画
2017年以内に、CTX取締役会はCingate Treeutics LLC持分激励計画(以下は“計画”と略称する)を制定し、採択し、 計画中に定義された会社従業員、CTXメンバー、取締役会メンバーとサービスプロバイダーにC類PIUを配布し、この計画の下でPIUの激励を受ける資格があることを提供する。PIUは当社の取締役会が適宜決定し、場合によっては当社の最高経営責任者が取締役会の許可に応じて適宜決定します。 PIUの発行敷居は、会社が発行した日の通貨前公平市場推定値に等しい。 割当敷居とは、すべてのメンバー(ある場合)に比例した現金割り当てが を超えなければならないことであり、特定のPIU所持者がその敷居を超えた割り当てに参加することができる。授標条項によると、割当ハードルは財務諸表確認の業績条件とみなされる。PIUのサービス条件における帰属期限はPIU付与プロトコルで定義され,範囲は30日から3年,すべてのPIUが付与する平均帰属期間は107日である.会社運営プロトコルの定義によると、この計画に基づいて発行されるすべてのPIUは、所定の分配のハードルを超える会社の利益を比例的に共有する権利がある。 は一般にすべてのメンバに現金分配を行うと仮定し、他のカテゴリ単位の任意の選好や優先順位に依存する。 CクラスPIUはまた、1対1で投票権を持っている。
| F-15 |
再編合併直前に、当社は8,500,000及びPIUの没収純額を承認及び発行しました。同社はFASB ASCテーマ718項目の下でこれらの賞を計算した報酬-株の報酬株式分類として奨励します。再編合併前には,将来的に敷居や目標 (業績条件)を達成して配当を実現することは不可能であるため,PIUに関する補償 費用は記録されていない。この評価は,当社の経営赤字の歴史と,必要な株を調達して運営資金を提供する上での継続的な挑戦に基づいている。組換え 合併については,付記1でさらに述べたように,850万株のPIUが57,900株のCingate普通株に交換されている。PIUと普通株の交換はPIUの条項、性質と権利の修正を生じ、業績の実現は可能であると考えられている。これにより、会社は1,270万ドル相当の非現金改装費用を確認しており、この費用 は、改訂当日控除式普通株の公正価値の評価に基づいて計算されている。この費用のうち820万ドルは一般と行政費用に計上され、450万ドルは研究開発費 に計上されている。
以下 会社計画の活動をまとめた:
株式オプション活動の概要
| 分布 | ||||||||
| 利益利息 | しきい値 | |||||||
| 職場.職場 | (百万単位で) | |||||||
| 2021年1月1日に返済されません | 8,142,461 | $25-$120 | ||||||
| 発表されました | 357,539 | $ | 160 | |||||
| 没収される | - | |||||||
| 再編合併時に普通株に転換する | (8,500,000 | ) | ||||||
| 2021年12月31日現在の未返済債務 | - | |||||||
再編合併前に、当社は、この計画の下で使用可能なすべての単位を発行し、PIUプロトコルで概説された帰属 の間にすべての単位に帰属した。
再構成統合の前(修正日でもある)には,様々な割当ての敷居でのPIUS 発行と未償還額は以下のとおりである
各種配布閾値のスケジュール
| 配布閾値$(百万): | |||||||||||||||||||||||||||||||||
| 授与された年 | $25 | $40 | $75 | $80 | $90 | $120 | $160 | 合計する | |||||||||||||||||||||||||
| 2017 | 4,753,000 | 125,200 | - | - | - | - | - | 4,878,200 | |||||||||||||||||||||||||
| 2018 | - | 661,525 | 217,725 | 22,883 | - | - | - | 902,133 | |||||||||||||||||||||||||
| 2019 | - | - | - | - | 377,524 | 458,924 | - | 836,448 | |||||||||||||||||||||||||
| 2020 | - | - | - | 1,476,126 | - | 49,554 | - | 1,525,680 | |||||||||||||||||||||||||
| 2021 | 357,539 | 357,539 | |||||||||||||||||||||||||||||||
| 合計する | 4,753,000 | 786,725 | 217,725 | 1,499,009 | 377,524 | 508,478 | 357,539 | 8,500,000 | |||||||||||||||||||||||||
(10)株主権益
当社は2022年12月31日及び2021年12月31日に額面0.0001ドル普通株240,000,000株及び額面0.0001ドル優先株10,000,000株の発行を許可し、その中で普通株565,470株を発行及び発行した。当社はまだbr優先株を発行していません。
| F-16 |
357,137株発行および発行された普通株は、再編合併に関連し、再編合併直前に発行されたCTX発行普通株式単位 に換算し、以下に説明する株式配当および逆株式分割を反映する。
208,333株普通株は1株120.00ドルで公衆に発行され、会社の初公募株に関連して、この初公募株は2021年12月に完成した。引受割引、手数料、その他の発売費用を差し引くと、会社は純収益約2,040万ドルを獲得した。
普通株式保有者は普通株を保有するごとに1票の投票権を享受することができる。当社が任意の自動または非自発的清算、解散または清算が発生した場合、当社のすべての債務および負債の清算または清算後、普通株式保有者は、当社が分配可能な残りの資産(あれば)を共有する権利がある。普通株式の保有者は取締役会が発表した時に配当金を得る権利がある。
株配当金
2021年9月30日、当社は1株当たり発行済み普通株を0.0015株普通株に交換する配当(“配当”)または合計22,402株普通株を実施した。株式配当に関連する普通株式の断片的な株式は発行されておらず、そのような断片的な権益は最も近い整数に丸められている。
2022年12月31日現在、他の配当金は発表されていない。
逆 株式分割
2021年10月12日、Cingateは発行済み普通株と発行済み普通株に対して逆株式分割を行い、比率は0.0350:1, であり、2021年11月29日に発行済み普通株と発行済み普通株に対して2回目の逆株式分割を行い、比率は :0.0321:1(“逆株式分割”)であった。いずれの逆株式分割に関連する普通株式小片株式も発行されておらず、そのような断片的権益は最も近い整数に丸められている。添付されている総合財務諸表に記載されているすべての期間のすべての株式データ、各株式データおよび関連資料は、逆株式分割の影響を反映するように をバックトラック調整し、その後、付記 1で述べたように、逆株式分割で調整した。
(11)株式ベースの報酬
2021年9月、会社取締役会および株主は、会社普通株、株式付加価値権、制限株式単位、普通株制限または非制限株、業績株、業績単位、インセンティブ配当奨励、その他の株式奨励、その他の現金奨励を付与する“2021年株式インセンティブ計画”(“2021年計画”)を採択した。2031年9月24日以降は、2021計画に従っていかなる報酬も発行することはできないが、2021計画はその後も継続され、以前に付与された報酬は完了していない。
2021年計画に基づいて付与されたオプションや他の奨励に関する発行可能な普通株の最高数は96,391株であり,2022年12月31日現在,2021年計画により発行可能な普通株は53,340株である。2021年計画によると発行可能な普通株式数は毎年1月1日に自動的に増加し、2021年計画満了まで、金額は前の年12月31日に発行された普通株式総数の5%に相当し、完全希釈ベースで計算され、取締役会がその前に当該年度の普通株備蓄が増加しないこと、またはその年度の普通株備蓄が増加する普通株式数がそれ以外の場合よりも少ないと規定されていない限り、取締役会が行動しなければならない。2021年計画によると、当社が使用価格または源泉徴収、買い戻し、または他の方法で終了するために没収、キャンセル、差し止め、または決済奨励を満たす任意の奨励は、2021計画に従って発行可能な普通株式 に再計上される。
会社が記録した株式ベースの給与支出は、2022年12月31日と2021年12月31日までの年間でそれぞれ800,796ドル、43,835ドルであり、発行済みオプションに関連している。2022年12月31日と2021年12月31日現在、2,089,509ドルと2,637,895ドルの未確認報酬コストは、2021計画に従って付与された非既得株式ベースの給与スケジュールと関連しており、今後1~4年以内に確認される予定である。
| F-17 |
2022年12月31日と2021年12月31日までの年間で、この計画下の代替案活動の概要は以下のとおりである
オプション活動の概要
| 加重平均 | 骨材 | |||||||||||||
| 加重平均 | 残り | 固有の | ||||||||||||
| 株 | 行権価格 | 契約条項 | 価値がある | |||||||||||
| 2021年1月1日に返済されません | - | |||||||||||||
| 贈与金 | 26,173 | $ | 120.00 | 9.94年 | - | |||||||||
| 鍛えられた | - | |||||||||||||
| 没収または満了 | - | |||||||||||||
| 2021年12月31日現在の未返済債務 | 26,173 | |||||||||||||
| 贈与金 | 20,882 | $ | 34.40 | 9.17年 | ||||||||||
| 鍛えられた | - | |||||||||||||
| 没収または満了 | (3,995 | ) | $ | 69.20 | ||||||||||
| 2022年12月31日に返済されていません | 43,060 | $ | 83.20 | 9.05年 | - | |||||||||
| すでに帰属しており,2022年12月31日に帰属する予定である | 43,060 | |||||||||||||
| 2022年12月31日に行使できます | 8,451 | |||||||||||||
会社が発行した株式オプションはASC 718に規定する権益会計処理資格に符合し、それに応じて授与日に公正価値で計量する。オプションの公正価値はブラック·スコアモデルを用いて推定される。会社が従業員と取締役に付与された株式オプションの付与日公正価値を推定する仮定を以下のように用いて,加重平均 をもとに示し,各年度まで:
公正価値仮定付表
| 2022 | 2021 | |||||||
| 無リスク金利 | 1.889 | % | 0.012 | % | ||||
| 予想期限(年単位) | 5.98 | 6.25 | ||||||
| 予想変動率 | 1.126 | 1.134 | ||||||
| 期待配当収益率 | 0 | % | 0 | % | ||||
無リスク金利 :当社は、付与日類似満期日までの米国債の定常満期日 に基づいて、オプション期待期間内の無リスク金利を取得する。
予想される 期限:期待期間は、付与されたオプションが未償還期間を予期し、簡略化方法(ホーム日と契約期限終了との間の中間点に基づいて)を使用して決定される
期待変動 :当社はバイオテクノロジーや製薬業界で上場企業の平均株価変動率を使用することができ、これらの会社は将来の株価傾向を代表するとされており、当社には十分な普通株取引履歴がないためである。会社は、自身の株価変動に関する十分な数の履歴情報 が利用可能になるまでこの過程を適用し続ける。
期待配当収益率 :当社はまだ支払っていませんし、近い将来に何の配当金も支払うとは期待していません。したがって, 配当収益率はゼロとなることが予想される.
2022年12月31日までの年度内に付与されたオプションの公正価値は17.20ドルから23.20ドルであり,2021年12月31日までの年度に付与されたオプションの公正価値は102.60ドルであった。
株式オプションの合計内在価値は,株式オプションの発行価格と会社普通株の公正価値との差額で計算される.執行権価格が会社の普通株公正価値を下回る株式オプションがないため、2022年12月31日と2021年12月31日まで、総内在価値はゼロである。
| F-18 |
(12) 普通株引受権証
当社初公募に関連して発行された208,333株普通株については,普通株引受権証も発行され,120.00ドルの単価が計上されている。このような権利証は直ちに分離されて取引されることができる。引受業者は15%の超過配給選択権を行使して追加の普通株を購入しなかったにもかかわらず、彼らは確かに1部の株式引受割引と手数料の価格で31,250部の追加の普通株引受権証を購入した。これら239,583株普通株引受権証の行使価格は1株120.00ドルであり、株式承認証行使時に有効な登録声明や現在の目論見書がなければ、無現金行権特徴を提供する。これらの株式承認証の有効期限は2021年12月10日から2026年12月10日までである。終了日に、これらの株式承認証は無現金行使によって自動的に行使されるだろう。
上記の普通株引受権証を除いて、当社は引受契約に基づいて引受業者に10,417部の普通株引受権証を発行し、当社が初めて公開発売する。これらの権証の発行権価格は1株あたり150.00ドルであり,キャッシュレス取引機能に を提供している.これらの株式承認証の有効期限は2022年6月7日から2026年12月10日までである。
権証の推定値はブラック·スコアモデルを用い,無リスク金利は0.0082%,期限は5.0年,変動率は1.24であった。当社の普通株の計量日における推定変動率は、生物科学技術や製薬業界内で上場企業よりも平均的な歴史株価変動率に基づいており、このような上場会社は将来の株価傾向を代表するとされている。当社の普通株には十分な取引歴史がないからである。無リスク金利は、引受権証の期待期限に基づいて、付与日までに満期日のような米国債の継続満期日を持つ。株式承認証を使用した契約期限は予想期限を推定した。
次の表は、会社の2022年12月31日と2021年12月31日までの未償還株式証明書をまとめています
未償還株式証明書要約
| 授与日 | 授与日 | |||||||||||||||
| 量 | トレーニングをする | 公正価値 | 公正価値 | |||||||||||||
| 株式承認証 | 値段 | 各授権書 | 合計する | |||||||||||||
| 残高-2021年1月1日 | ||||||||||||||||
| 引受の公開発行 | 239,583 | $ | 120.00 | $ | 95.4000993 | $ | 22,856,242 | |||||||||
| 引受権証を発行する | 10,417 | $ | 150.00 | $ | 92.7968705 | 966,665 | ||||||||||
| 残高-2021年12月31日 | 250,000 | $ | 23,822,907 | |||||||||||||
2022年にはいかなる株式承認証も発行または行使されていない。
Br社はこれらの権利証をASCサブテーマ815-40項の株式分類ツールとしているエンティティ自己資本における変換可能ツールと契約の会計処理 それらは会社の普通株式をインデックスとしているので、それら はすべての他の持分分類条件を満たしている。初公開株式取引の総収益は以下の相対公正価値方法で普通株と普通株引受権証に分配される。株式証券の公正価値 は会社の貸借対照表の追加実収資本に計上される。
株式証公正価値付表(Br)付加実収資本
| パーセント | ||||||||||||
| 公平である | 総数を占める | 金額 | ||||||||||
| 価値がある | 公正価値 | 分配された | ||||||||||
| 普通株 | $ | 25,000,000 | 51.2 | % | $ | 12,800,000 | ||||||
| 普通株引受権証 | 23,822,907 | 48.8 | % | 12,200,000 | ||||||||
| 合計する | $ | 48,822,907 | 100 | % | $ | 25,000,000 | ||||||
(13)所得税
Cingate Inc.は“国内税法”によりC類会社として課税される。Cingate Inc.は、財務報告に記録された資産および負債金額と税務法規によって計量されたこのような金額との間の一時的な差の影響を反映するために、繰延所得税を記録する。CTXはCingate Inc.の完全資本無視エンティティであり、CTXおよびその完全子会社Cingate Works Inc.のすべての活動は、Cingate Inc.の流動および繰延税金資産および負債の計算に含まれる。2022年12月31日または2021年12月31日まで、連邦または州所得税の繰延所得税の割引または費用は記録されていない。
| F-19 |
所得税税費は、米国連邦所得税税率の適用により計算される期待費用とは異なり、以下のようになる
有効所得税率明細書
| 2022年12月31日 | 2021年12月31日 | |||||||
| 法定税率の連邦所得税割引 | $ | (3,712,008 | ) | $ | (563,519 | ) | ||
| 州所得税割引 | (977,496 | ) | (148,393 | ) | ||||
| 恒久的差異 | 11,935 | 44,804 | ||||||
| 評価免除額を変更する | 4,733,324 | 713,701 | ||||||
| 他にも | (55,755 | ) | (46,593 | ) | ||||
| 所得税総支出 | $ | - | $ | - | ||||
繰延税金資産推定準備の必要性と金額を評価するには、通常、各司法管轄区のすべての利用可能な証拠に対して重大な判断と広範な分析が必要である。同等の判決は,現行税法の解釈とその適用状況の他に指針を公表することを当社に求めている。今回の評価の一部として、会社はその収益性と税務状況に関するプラスとマイナスの証拠を考慮した。既存の証拠に基づいて、繰延税金資産の全部または一部が現金化できない可能性が高い場合、推定準備が提供される。過去の収入レベルと未来の課税収入の予測によると、他の項目を除いて、当社はその繰延税金資産がさらに現金化できない可能性があると確定した。当社は2022年12月31日及び2021年12月31日にそれぞれ繰延税項目純資産計5,580,595ドル及び847,269ドルの減価準備を行い、添付の総合経営報告書及びその他の全面損失のうち所得税支出の構成要素と記す。
同社はアメリカ連邦と各州司法管轄区で所得税申告書を提出した。これらの会社は2018年までアメリカ連邦と州税務機関の所得税審査を受けなかった。
会社はFASB ASC 740の規定に従っている所得税不確実な税金状況を評価するために。本テーマでは、納税申告書において採取されたまたは予想される納税頭寸の財務諸表確認および計量に対する確認閾値および計量属性を規定する。当社は2022年12月31日現在、連結財務諸表で確認すべき重大な不確定税務状況 は何も発見していません。
| F-20 |
繰延税金貸借対照表
| 2022年12月31日 | 2021年12月31日 | |||||||
| 繰延所得税資産: | ||||||||
| 現在: | ||||||||
| 研究開発コスト | $ | 343,087 | $ | - | ||||
| 他にも | 59,018 | 4,050 | ||||||
| 現在ではない | ||||||||
| 純営業損失 | 3,381,215 | 1,201,974 | ||||||
| 研究開発コスト | 1,762,716 | - | ||||||
| 非既得性株式オプション | 204,380 | 11,835 | ||||||
| 特許 | 92,417 | 90,480 | ||||||
| 使用権資産 | 63,563 | 49,606 | ||||||
| 繰延所得税総資産 | 5,906,396 | 1,357,945 | ||||||
| 減算:推定免税額 | (5,580,595 | ) | (847,269 | ) | ||||
| 所得税純資産を繰延する | 325,801 | 510,676 | ||||||
| 繰延所得税負債: | ||||||||
| 現在: | ||||||||
| 現金を計算する | (11,228 | ) | (105,075 | ) | ||||
| 当面ではない | ||||||||
| 財産と設備 | (314,573 | ) | (405,601 | ) | ||||
| 繰延所得税総負債 | (325,801 | ) | (510,676 | ) | ||||
| 繰延税項目純資産(負債) | $ | - | $ | - | ||||
(14)賃貸証書
当社は、先に借りたカンザスシティオフィスビル空間の拡張と改修を含む2019年6月に賃貸契約を締結しました。レンタル期間は5年で、2020年5月1日から、拡張と新リフォームスペースの入居証を受け取った日です。このレンタル契約によると、レンタル料の総額は毎月30,453元から33,145元まで様々です。本レンタルは約テナントに合計201,600ドルのレンタル奨励を提供します。
付記17に記載されているように、同社は関連先からニュージャージー州の従業員のためにオフィススペースをレンタルしている。このレンタルの基本賃貸料は、2022年および2021年の月3,000ドルである。借約は2021年1月に締結され、レンタル期間は3年。
2018年に、当社は18ヶ月間の製造設備経営賃貸契約を締結しました。臨時レンタル料は設備設置期間中に発生し、レンタルは2018年6月から開始されます。これらのレンタル設備の毎月のレンタル料は六一七八六ドルです。2019年12月、本レンタルは約24ヶ月継続し、毎月のレンタル料は37,072ドルです。この日、本レンタルは会計処理として改訂され、その分類は融資型賃貸と評価され、管理層はレンタル者がレンタル資産に対して他の将来用途がないと認定したためである。リース機器に関するROU資産はリース期間内に償却される。2021年12月、レンタル期間が終了し、当社はレンタル者と購入契約を締結し、製造設備を購入します。
当社は2020年4月に、賃貸期間終了時に当社の家具所有権に譲渡する融資リースに分類された60ヶ月間のオフィス家具賃貸契約を締結した。毎月のレンタル料は1,491ドルです。レンタルした家具は直線的に償却され、7年を超えた。賃貸義務に関する推定金利は6.12%,満期日は2025年3月である。
| F-21 |
2022年12月31日までと2021年12月31日までの年間レンタル料金 は以下のように構成されています
リースコスト構成集計表
| 2022 | 2021 | |||||||
| リースコストを経営する | $ | 338,787 | $ | 338,787 | ||||
| 融資リースコスト: | ||||||||
| 使用権資産の償却 | 10,990 | 409,927 | ||||||
| 賃貸負債利息 | 2,804 | 26,504 | ||||||
| 融資リース総コスト | 13,794 | 436,431 | ||||||
| 総賃貸コスト | $ | 352,581 | $ | 775,218 | ||||
2022年12月31日と2021年12月31日現在、総合貸借対照表で報告されている金額 は以下の通りである
総合貸借対照表に報告されているリース概要
| 2022 | 2021 | |||||||
| 経営リース: | ||||||||
| 経営的リース使用権資産 | $ | 630,618 | $ | 858,600 | ||||
| その他流動負債 | 339,755 | 295,595 | ||||||
| リース負債を経営する | 488,748 | 828,503 | ||||||
| リース負債総額を経営する | 828,503 | 1,124,098 | ||||||
| 融資リース: | ||||||||
| 財産と設備 | 76,928 | 76,928 | ||||||
| 累計償却する | (44,988 | ) | (29,602 | ) | ||||
| 財産と設備、純額 | 31,940 | 47,326 | ||||||
| 融資リース下債務の今期分割払い | 16,053 | 15,096 | ||||||
| 債務の長期部分 | ||||||||
| 融資リース | 21,487 | 37,534 | ||||||
| 融資リース負債総額 | $ | 37,540 | $ | 52,630 | ||||
2022年12月31日と2021年12月31日まで、レンタルに関するその他の情報は以下の通りです
賃貸契約に関する他の資料の概要:
| 2022 | 2021 | |||||||
| キャッシュフロー情報の追加: | ||||||||
| 賃貸負債に含まれる金額を計量するために支払う現金: | ||||||||
| 営業レンタルによる営業キャッシュフロー | $ | 36,000 | $ | 433,172 | ||||
| 融資リースの営業キャッシュフロー | 3,698 | 26,504 | ||||||
| 融資リースによるキャッシュフロー | 15,090 | 436,259 | ||||||
| リース債務減少によるROU資産の減少: | ||||||||
| 賃貸借契約を経営する | 227,981 | 196,245 | ||||||
| 融資リース | 10,990 | 409,927 | ||||||
| 加重平均残余レンタル期間: | ||||||||
| 賃貸借契約を経営する | 25ヶ月 | 39ヶ月 | ||||||
| 融資リース | 27ヶ月 | 39ヶ月 | ||||||
| 加重平均割引率: | ||||||||
| 賃貸借契約を経営する | 11.62 | % | 11.57 | % | ||||
| 融資リース | 6.12 | % | 6.12 | % | ||||
| F-22 |
2022年12月31日まで、撤回不可賃貸項目におけるレンタル負債の満期日 は以下の通りです
賃貸負債満期集計表
| 運営中です | 金融 | |||||||
| 賃貸借証書 | 賃貸借証書 | |||||||
| 2023 | 414,800 | 17,900 | ||||||
| 2024 | 393,005 | 17,900 | ||||||
| 2025 | 132,580 | 4,475 | ||||||
| その後… | - | - | ||||||
| 未割引賃貸支払総額 | 940,385 | 40,275 | ||||||
| 計上された利息を差し引く | (111,882 | ) | (2,742 | ) | ||||
| リース総負債 | $ | 828,503 | $ | 37,533 | ||||
(15)1株当たり純損失
次の表に、2022年12月31日までと2021年12月31日までの1株当たり基本および償却純損失の算出方法を示す
1株基本と償却純損失付表
| 2022 | 2021 | |||||||
| 分子: | ||||||||
| 純損失 | $ | (17,676,232 | ) | $ | (20,709,991 | ) | ||
| 分母: | ||||||||
| 加重平均普通株式発行済み | 565,470 | 370,679 | ||||||
| 1株当たり基本と希釈して純損失 | $ | (31.26 | ) | $ | (55.87 | ) | ||
潜在的なbr希釈証券は、以下のように逆希釈されているので、各償却計算には含まれていない
潜在希釈証券明細書
| 2022 | 2021 | |||||||
| 2021年株式インセンティブ計画に基づいて発行された株式オプション | 43,060 | 26,164 | ||||||
| 未償還普通株引受権証 | 250,000 | 250,000 | ||||||
| 合計する | 293,060 | 276,164 | ||||||
(16)ライセンスプロトコル
CTX は、CTX−1301、CTX−1302、 およびCTX−2103を開発するための特許およびライセンス技術に関するライセンス契約を会社と締結している。CTXは、以下のマイルストーンイベントが発生した場合、以下の費用を支払います
| ● | 現場の各製品の3期臨床試験における1人目の患者の投与量に応じて250,000ドルのマイルストーン支払い を支払い,製品別に支払った。 | |
| ● | 許可された側がこの分野の製品ごとに新薬申請を提出する際には250,000ドルのマイルストーン支払いを支払い, は製品ごとに支払う。 | |
| ● | CTX-1301およびCTX-1302のマイルストーン支払いは250,000ドル、CTX-2103のマイルストーン支払いは500,000ドルであり、FDAの最初の発売承認を受けた後、製品別に支払います。 | |
| ● | ヨーロッパ医薬品局(European Medicines Agency)の最初の発売許可を受けた後、CTX-2103の250,000ドルのマイルストーン支払いを購入しました |
同社は2021年12月31日現在、CTX-1301のために250,000ドルのマイルストーンを蓄積しており、これは第3段階臨床試験における1人目の患者の投与量と関係がある。経営陣はこのマイルストーンが可能だと考えている。当社はこの2つの製品の他のマイルストーンに関する費用を記録していません。会社は2022年12月31日までにこれらの費用が発生することは不可能だと考えているからです。
| F-23 |
(17)関連者取引
当社の取締役会メンバー、取締役上級取締役、レドロー社(イギリス)有限公司。レドロー社(イギリス)有限公司は、当社初公募株の共同引受業者を務めています。2021年、Laidlaw& Company(UK)Ltd.に支払われるIPO関連の割引および手数料は約50万ドルである。
当社の総法律顧問は、当社がレンタルしたオフィス施設スペースを提供する法律事務所のパートナーである。2022年と2021年、会社が法律事務所に支払う賃貸料支出は36,000ドル。同社は2022年12月31日と2021年12月31日までに約0ドル借りている。
取締役会のPeter Werthは、付記7で述べたように、2022年に会社に500万ドルの債務融資を提供したWFIAのマネージャーである。2022年12月31日現在の年度で292,339ドルの利息支出を確認した。2022年12月31日現在、元金残高500万ドルと受取利息292,339ドルはまだ返済されていない。
(18)後続イベント
経営陣 は、2022年12月31日から2023年3月10日までの間に発生した事件を評価し、2023年3月10日は財務諸表の発表日である。
当社は2023年1月に販売代理(“ウェインライト”)であるH.C.ウェインwright& Co.,LLCと市場発売プロトコル(“ATMプロトコル”)を締結し,これにより,当社は随時Wainwrightを通して普通株 株を発売·販売することができ,総収益は最高2,650,000ドル(“株”)に達する。同社は普通株を売るたびの総収益の3%に相当する手数料をWainwrightに支払う。本出願日まで、当社はATMプロトコルに基づいて何の販売も行っていません
2023年3月7日、会社はIndegene,Inc.と共同商業化協定を締結し、この合意に基づいて、Indegeneは会社の要求に応じてCTX-1301に商業化サービスを提供する。双方はIndegeneが実行するサービス,このようなサービスの配信内容,および会社が支払う費用を示す作業説明を締結する
(19) 逆引き株
2023年11月30日、当社は1回20株1株の逆株式分割(“逆株式分割”)を完了し、逆株式分割が発効する前に発行·発行された会社普通株の数を減少させた。会社の法定普通株の数量は逆株分割の影響を受けず、会社の普通株の額面は変わらず、1株当たり0.0001ドルである。逆株式分割に関連する断片的な株式は発行されていない。開示を除いて、株式数及び1株当たり金額に関するすべての金額は、当該等の財務諸表にさかのぼって記載されている。
| F-24 |
第 部分-財務情報
項目1.財務諸表。
Cingate Inc.
合併貸借対照表(監査なし)
| 九月三十日 | 十二月三十一日 | |||||||
| 2023 | 2022 | |||||||
| 資産 | ||||||||
| 流動資産: | ||||||||
| 現金と現金等価物 | $ | 1,986,313 | $ | 5,356,276 | ||||
| 雑売掛金 | 5,836 | 234,432 | ||||||
| 前払い費用と他の流動資産 | 910,286 | 2,278,944 | ||||||
| 流動資産総額 | 2,902,435 | 7,869,652 | ||||||
| 財産と設備、純額 | 2,531,330 | 2,904,787 | ||||||
| 経営的リース使用権資産 | 436,493 | 630,618 | ||||||
| 総資産 | 5,870,258 | 11,405,057 | ||||||
| 負債と株主権益 | ||||||||
| 流動負債: | ||||||||
| 売掛金 | 1,670,436 | 762,357 | ||||||
| 費用を計算する | 692,008 | 894,635 | ||||||
| 支払手形 | 3,000,000 | 5,000,000 | ||||||
| 融資リース負債流動 | 16,805 | 16,053 | ||||||
| 賃貸負債を経営し、流動 | 353,183 | 339,755 | ||||||
| 流動負債総額 | 5,732,432 | 7,012,800 | ||||||
| 長期負債: | ||||||||
| 融資リース負債、当期純額 | 8,792 | 21,487 | ||||||
| 賃貸負債を経営し,当期純額 | 225,368 | 488,748 | ||||||
| 長期負債総額 | 234,160 | 510,235 | ||||||
| 総負債 | 5,966,592 | 7,523,035 | ||||||
| 株主権益 | ||||||||
| 普通株、額面0.0001ドル;2023年9月30日と2022年12月31日まで、発行済みと発行済み普通株はそれぞれ2.4億株と868,940株と565,470株である | 88 | 57 | ||||||
| 優先株、額面0.0001ドル;2023年9月30日と2022年12月31日まで、授権発行株10,000,000株、発行と発行株は0株 | - | - | ||||||
| 追加実収資本 | 85,912,324 | 73,290,461 | ||||||
| 赤字を累計する | (86,008,746 | ) | (69,408,496 | ) | ||||
| 株主権益総額 | (96,334 | ) | 3,882,022 | |||||
| 総負債と株主権益 | $ | 5,870,258 | $ | 11,405,057 | ||||
合併財務諸表の付記を参照。
| F-25 |
Cingate Inc.
合併営業と全面赤字報告書(監査なし)
| 9月30日までの3ヶ月間 | 9月30日までの9ヶ月間 | |||||||||||||||
| 2023 | 2022 | 2023 | 2022 | |||||||||||||
| 運営費用: | ||||||||||||||||
| 研究開発 | $ | 3,923,852 | $ | 2,123,114 | $ | 10,508,395 | $ | 7,063,626 | ||||||||
| 一般と行政 | 1,825,822 | 1,845,248 | 5,453,643 | 5,963,067 | ||||||||||||
| 営業損失 | (5,749,674 | ) | (3,968,362 | ) | (15,962,038 | ) | (13,026,693 | ) | ||||||||
| 利息とその他の収入,純額 | (229,380 | ) | (58,885 | ) | (638,212 | ) | (44,512 | ) | ||||||||
| 所得税前損失 | (5,979,054 | ) | (4,027,247 | ) | (16,600,250 | ) | (13,071,205 | ) | ||||||||
| 所得税の割引 | - | - | - | - | ||||||||||||
| 純損失 | (5,979,054 | ) | (4,027,247 | ) | (16,600,250 | ) | (13,071,205 | ) | ||||||||
| その他の全面収益(損失): | ||||||||||||||||
| 短期投資は赤字変動を実現していない | - | 3,249 | - | (166 | ) | |||||||||||
| 総合損失 | $ | (5,979,054 | ) | $ | (4,023,998 | ) | $ | (16,600,250 | ) | $ | (13,071,371 | ) | ||||
| 普通株は基本的に1株当たり純損失と希釈して1株当たり純損失 | $ | (6.05 | ) | $ | (7.12 | ) | $ | (23.24 | ) | $ | (23.12 | ) | ||||
| 基本普通株と希釈後の普通株1株当たり純損失の加重平均株式数の計算に用いる | 988,333 | 565,470 | 714,397 | 565,470 | ||||||||||||
合併財務諸表の付記を参照。
| F-26 |
Cingate Inc.
合併株主権益報告書(監査を経ていない)
| 普通株 | その他の内容 | 積算 | 積算 その他
| 株主の | ||||||||||||||||||||
| 株 | 金額 | 実収資本 | 赤字.赤字 | 収入.収入 | 権益 | |||||||||||||||||||
| 残高2022年1月1日 | 565,470 | 57 | $ | 72,575,584 | $ | (51,732,264 | ) | $ | 165 | $ | 20,843,542 | |||||||||||||
| 2022年3月31日までの3ヶ月間のイベント: | ||||||||||||||||||||||||
| 売却可能投資の未実現損失 | - | - | - | - | (2,948 | ) | (2,948 | ) | ||||||||||||||||
| 株に基づく報酬費用 | - | - | 181,518 | - | - | 181,518 | ||||||||||||||||||
| 純損失 | - | - | - | (5,003,511 | ) | - | (5,003,511 | ) | ||||||||||||||||
| 残高2022年3月31日 | 565,470 | $ | 57 | $ | 72,757,102 | $ | (56,735,775 | ) | $ | (2,783 | ) | $ | 16,018,601 | |||||||||||
| 2022年6月30日までの3ヶ月間のイベント: | ||||||||||||||||||||||||
| 売却可能投資の未実現損失 | - | - | - | - | (466 | ) | (466 | ) | ||||||||||||||||
| 株に基づく報酬費用 | - | - | 207,186 | - | - | 207,186 | ||||||||||||||||||
| 純損失 | - | - | - | (4,040,447 | ) | - | (4,040,447 | ) | ||||||||||||||||
| 残高2022年6月30日 | 565,470 | $ | 57 | $ | 72,964,288 | $ | (60,776,222 | ) | $ | (3,249 | ) | $ | 12,184,874 | |||||||||||
| 2022年9月30日までの3ヶ月間のイベント: | ||||||||||||||||||||||||
| 売却可能投資の未実現損失 | - | - | - | - | 3,249 | 3,249 | ||||||||||||||||||
| 株に基づく報酬費用 | - | - | 205,656 | - | - | 205,656 | ||||||||||||||||||
| 純損失 | - | - | - | (4,027,247 | ) | - | (4,027,247 | ) | ||||||||||||||||
| 残高2022年9月30日 | 565,470 | $ | 57 | $ | 73,169,944 | $ | (64,803,469 | ) | $ | - | $ | 8,366,532 | ||||||||||||
| 残高2023年1月1日 | 565,470 | 57 | 73,290,461 | $ | (69,408,496 | ) | $ | - | $ | 3,882,022 | ||||||||||||||
| 2023年3月31日までの3ヶ月間のイベント: | ||||||||||||||||||||||||
| 株に基づく報酬費用 | - | - | 204,479 | - | - | 204,479 | ||||||||||||||||||
| 純損失 | - | - | - | (4,004,887 | ) | - | (4,004,887 | ) | ||||||||||||||||
| 残高2023年3月31日 | 565,470 | $ | 57 | $ | 73,494,940 | $ | (73,413,383 | ) | $ | - | $ | 81,614 | ||||||||||||
| 2023年6月30日までの3ヶ月間のイベント: | ||||||||||||||||||||||||
| 市場発売と購入契約で普通株を発行し、料金を差し引いた純額 | 37,369 | 4 | 218,794 | 218,798 | ||||||||||||||||||||
| 株に基づく報酬費用 | - | - | 217,376 | - | - | 217,376 | ||||||||||||||||||
| 純損失 | - | - | - | (6,616,309 | ) | - | (6,616,309 | ) | ||||||||||||||||
| 残高2023年6月30日 | 602,839 | $ | 61 | $ | 73,931,110 | $ | (80,029,692 | ) | $ | - | $ | (6,098,521 | ) | |||||||||||
| 2023年9月30日までの3ヶ月間のイベント: | ||||||||||||||||||||||||
| 市場発売と購入契約で普通株を発行し、料金を差し引いた純額 | 88,943 | 9 | 1,621,930 | 1,621,939 | ||||||||||||||||||||
| WFIAの私募に関する普通株式発行 | 91,158 | 9 | 999,991 | 1,000,000 | ||||||||||||||||||||
| 普通株と事前資金権証を発行し,公開発行中に現金で販売し,費用を差し引く | 86,000 | 9 | 3,310,542 | 3,310,551 | ||||||||||||||||||||
| 転換関係者支払手形に関する事前融資権証の発行 | - | - | 3,949,765 | 3,949,765 | ||||||||||||||||||||
| 転換関係者支払手形に関する出資額 | 1,862,735 | 1,862,735 | ||||||||||||||||||||||
| 株に基づく報酬費用 | - | - | 236,251 | - | - | 236,251 | ||||||||||||||||||
| 純損失 | - | - | - | (5,979,054 | ) | - | (5,979,054 | ) | ||||||||||||||||
| 残高2023年9月30日 | 868,940 | $ | 88 | $ | 85,912,324 | $ | (86,008,746 | ) | $ | - | $ | (96,334 | ) | |||||||||||
合併財務諸表の付記を参照
| F-27 |
Cingate Inc.
合併キャッシュフロー表(監査なし)
| 9月30日までの9ヶ月間 | ||||||||
| 2023 | 2022 | |||||||
| 経営活動: | ||||||||
| 純損失 | $ | (16,600,250 | ) | $ | (13,071,205 | ) | ||
| 純損失と経営活動で使用される現金純額の調整: | ||||||||
| 減価償却 | 410,593 | 304,287 | ||||||
| 株に基づく報酬 | 658,106 | 594,360 | ||||||
| 経営性資産と負債変動状況: | ||||||||
| 雑売掛金 | 228,596 | 642,899 | ||||||
| 前払い費用と他の流動資産 | 1,368,658 | (293,140 | ) | |||||
| 経営的リース使用権資産 | 194,125 | 167,828 | ||||||
| 貿易勘定と売掛金をまかなう | 1,517,952 | 197,462 | ||||||
| その他流動負債 | 13,428 | 31,147 | ||||||
| リース負債を経営する | (263,380 | ) | (249,952 | ) | ||||
| 経営活動のための現金純額 | (12,472,172 | ) | (11,676,314 | ) | ||||
| 投資活動: | ||||||||
| 財産と設備を購入する | (37,136 | ) | (10,400 | ) | ||||
| 短期投資を売却して得られる収益 | - | 933 | ||||||
| 他にも | - | (165 | ) | |||||
| 投資活動のための現金純額 | (37,136 | ) | (9,632 | ) | ||||
| 融資活動: | ||||||||
| 普通株とあらかじめ出資した普通株引受権証を発行して得られた金は,費用を差し引く | 6,151,288 | - | ||||||
| 支払手形収益 | 3,000,000 | 5,000,000 | ||||||
| 融資リース債務の元金支払い | (11,943 | ) | (11,229 | ) | ||||
| 融資活動が提供する現金純額 | 9,139,345 | 4,988,771 | ||||||
| 現金と現金等価物: | ||||||||
| 現金と現金等価物の純減少 | (3,369,963 | ) | (6,697,175 | ) | ||||
| 年初現金および現金等価物 | 5,356,276 | 16,492,745 | ||||||
| 期末現金および現金等価物 | $ | 1,986,313 | $ | 9,795,570 | ||||
| 現金支払い: | ||||||||
| 支払の利子 | $ | 10,266 | $ | 10,291 | ||||
合併財務諸表の付記を参照
| F-28 |
Cingate Inc.
連結財務諸表付記(監査を経ていない)
(1) 業務の性質と流動性
組織する
Cingulate Inc.は生物製薬会社であり、その薬物送達プラットフォーム技術を利用して製品を開発することに集中し、brが毎日1回の多用量療法錠剤を調製と製造できるようにし、最初は注意欠陥/多動障害(ADHD)の治療に集中した。同社は2種類の独自の第一線覚せい剤薬物CTX−1301(酢酸デキサメタゾン)とCTX−1302(右旋フェニルプロピルアミン)を開発しており,ADHDの治療に用いられており,すべての患者群:児童,青少年,成人に適している。CTX-1301とCTX-1302は柔軟なコア錠剤技術と目標製品プロファイルを採用し、迅速な効果と持続的な活動br日を提供し、血漿薬物レベルは制御可能に低下し、そして良好な耐性を有することを目的とした。同社はCTX−1301の第1段階第3段階臨床試験を完了し,他の2つの第3段階試験が行われている。また,同社には3つ目の不安治療製品があり,CTX−2103, は配合段階にある。
2012年11月14日、デラウェア州有限責任会社Cingate Treateutics LLC(CTX)が設立された。2021年5月10日、CTXのデラウェア州会社と完全子会社Cingate Inc.(Cingate、または当社)が設立され、ホールディングスとして当社が上場することが予想される。2021年第3四半期に発生した再編合併により,Cingate はCTXを効率的に買収し,CTXのすべての発行済単位はCingate普通株に変換された。CTXは依然として会社が業務を展開する実体 である.
2023年9月30日および2022年9月30日までの総合財務諸表および付記はCingateとその付属会社(CTXを含む)の全面合併を代表し、当社に対するすべての言及はこの全面合併を代表する。
流動性
社は設立以来運営中に損失と負キャッシュフローが出現した。収益前実体として、会社は開発中の候補製品が米国食品医薬品局(FDA)の承認、生産、発売、収入を得るまで、資金を調達して運営を支援する能力に依存している。初公募株は2021年12月に完成し、約2,040万ドルの純収益を提供した。また、会社は2022年8月に1枚の約束票から500万ドルの収益を獲得し、2023年5月に本チケットを改訂·再述した際に300万ドルの収益を追加し、その一部はその後、付記7で述べたように株式に変換された。会社はH.C.Wainwrightとの市場合意と2023年のリンカーンパーク資本有限責任会社との購入契約を利用し、2023年9月30日までに合計約210万ドルの純収益を提供した。2023年第3四半期に調達された追加資本は、普通株式の発行と売却、公開発行に関連する事前出資の普通株式承認株式証、約330万ドルの純収益br、および100万ドルの非公開配給に関連する普通株の発行と販売を提供し、詳細はbr付記9を参照されたい。しかし、会社は運営と発展のために追加の資金を必要とする。経営陣は、他の会社との潜在的な協力または他の戦略取引を含む追加発行株式、債券発行、または他の資本源を含む可能性がある追加資金を得る様々な戦略を評価している。これらの計画の成功は、企業の努力にも関連しており、市場要因やFDAの候補製品の承認など、その制御できない要素にも関連している。br社は、その計画の有効な実施方式が上記の条件や事件を緩和したり、緩和したりするのに十分ではなく、財務諸表発表後1年以内に持続的な経営企業としての能力に大きな疑いを抱いている。付随する総合財務諸表 は持続経営に基づいて作成され,正常業務過程における資産現金化と負債返済を考慮した。連結財務諸表は、このような不確実性の結果がもたらす可能性のあるいかなる調整も反映しない。
| F-29 |
(2) 重要会計政策の概要
(A)列報基礎と合併原則
添付の連結財務諸表は、米国公認会計原則(米国公認会計原則)に基づいて作成されている。連結財務諸表はCingateとその完全子会社の勘定を含む。すべての会社間口座 と取引は合併でキャンセルされました。
(B)監査されていない中期財務資料
添付されている2023年9月30日までの総合貸借対照表、2023年と2022年9月30日までの3ヶ月と9ヶ月間の総合経営報告書と総合損失表、2023年と2022年9月30日までの3ヶ月と9ヶ月間の総合株主権益表、2023年及び2022年9月30日までの9ヶ月間の総合キャッシュフロー表及び関連する中期開示は監査されていません。これらの監査されていない総合財務諸表は、米国公認会計原則に基づいて中期運営および現金流量の財務状況および結果 を公平に陳述するための正常な経常的調整のみを含むすべての必要な調整を含む。中期業績は必ずしも通年またはその後のどの中期の経営業績やキャッシュフローを代表するとは限らない。添付されている総合財務諸表 は、当社の2022年12月31日までの年度の審査された総合財務諸表とその付記とともに読まなければなりません。
(三)信用リスク集中
会社は現金等値預金を持っており、財政年度を通して、これらの預金は連邦預金保険会社が加入している250,000ドルの限度額を超えています(入金項目は考慮していません)。経営陣はこれらの金融機関の健全性を監督しており、当社が預金中の未保険部分に関する重大な信用リスクを有しているとは考えていない。
(D)雑入金
2022年12月31日までの雑売掛金には、主に2020年に発生した賃金コストの従業員留保税控除、研究·開発税控除が含まれる。同社は国際会計基準20に似ている政府補助金の会計計算と政府援助の開示 はこれらの入金された会計処理中である.当社は2022年12月31日現在、これらの売掛金に関する支出を必要としないことを決定した。2023年9月30日までに、すべての従業員の留用税収控除と研究開発税収控除はすでに徴収が完了した。
(E)長期資産減価
イベントや状況がその長期資産(物件や設備を含む)や賃貸使用権(ROU)資産の帳簿価値が回収できない可能性があることを示す場合、会社はそのような資産の帳簿価値を評価する。これらのイベントや状況の変化 には、経営業績の著しい悪化、業務計画の変化、または予想される将来のキャッシュフローの変化が含まれている可能性があります。 減値指標が存在すれば、当社は資産の帳簿価値と資産予想による未割引キャッシュフローを比較することで回収可能性を評価します。将来のキャッシュフローの合計が帳簿金額より少ないと予想されれば、会社は減価損失を確認する。減価損失は,帳簿価値が長期資産群の公正価値を超える金額を比較することで計測される。2023年9月30日または2022年9月30日までの3カ月または9カ月間は減値は確認されていない。
(F)株式ベースの報酬
Br社は、ブラック·スコアーズオプション定価モデルを使用して、従業員の付与日の公正価値に基づいて、すべての株式奨励の従業員と取締役株式報酬支出を測定します。サービス条件のある株式奨励については,株式報酬費用は必要なサービス期限内に直線法で確認する.没収行為は発生時に確認します。付記11の他の情報 を参照してください。
| F-30 |
(3) 前払い費用
前払い 2023年9月30日と2022年12月31日までの料金には、
前払い料金明細書
| 九月三十日 | 十二月三十一日 | |||||||
| 2023 | 2022 | |||||||
| 研究開発 | $ | 413,431 | $ | 1,377,391 | ||||
| 保険 | 339,676 | 472,152 | ||||||
| 活性薬物成分 | 77,422 | 209,156 | ||||||
| 資金調達コストを繰り下げる | - | 100,339 | ||||||
| 専門費 | 20,775 | 61,524 | ||||||
| 会費と購読料 | 38,284 | 37,684 | ||||||
| 他にも | 20,698 | 20,698 | ||||||
| 前払い費用総額 | $ | 910,286 | $ | 2,278,944 | ||||
(4)財産と設備
2023年9月30日と2022年12月31日まで、財産と設備の純資産は以下の通り
財産と設備明細書
| 推定数 | ||||||||||||
| 使用寿命 | 九月三十日 | 十二月三十一日 | ||||||||||
| (単位:年) | 2023 | 2022 | ||||||||||
| 装備 | 2-7 | $ | 4,342,832 | $ | 2,565,997 | |||||||
| 家具と固定装置 | 7 | 145,754 | 145,754 | |||||||||
| コンピュータ装置 | 5 | 41,898 | 41,898 | |||||||||
| 賃借権改善 | 5 | 471,505 | 471,505 | |||||||||
| 建設中の設備 | - | - | 1,739,699 | |||||||||
| 財産と設備、毛額 | 5,001,989 | 4,964,853 | ||||||||||
| 減算:減価償却累計 | (2,470,659 | ) | (2,060,066 | ) | ||||||||
| 財産と設備、純額 | $ | 2,531,330 | $ | 2,904,787 | ||||||||
2023年9月30日までの9カ月分の減価償却費用は410,593ドル,2022年9月30日までの9カ月分の減価償却費用は304,287ドルであった。2023年9月30日までの3カ月分の減価償却費用は154,663ドル、2022年9月30日までの3カ月分の減価償却費用は101,429ドル。
| F-31 |
(5) 課税料金
2023年9月30日と2022年12月31日までの課税費用には、
課税費用別表
| 九月三十日 | 十二月三十一日 | |||||||
| 2023 | 2022 | |||||||
| 利子 | $ | 157,339 | $ | 292,339 | ||||
| 研究開発 | 253,524 | - | ||||||
| 専門費 | 15,000 | 314,446 | ||||||
| 従業員ボーナス | 175,625 | 175,625 | ||||||
| 活性薬物成分 | 62,393 | - | ||||||
| 他にも | 28,127 | 112,225 | ||||||
| 費用を計算する | $ | 692,008 | $ | 894,635 | ||||
(6) またはある
Br社は、通常の業務過程やその他の面で引き起こされる法的手続きやクレームの影響を時々受ける可能性があります。私たちに対する重大な法的責任は、私たちの業務、財務状況、運営結果に悪影響を及ぼす可能性があります。
Br社は、発生または損失に関する法的コストを記録し、これらの事項に重大または損失が発生した場合に準備金を確立し、米国会計基準450に基づいて、管理層が重大または損失があることを決定し、可能性があり、合理的に評価する意外な状況。 損失範囲が推定され、その範囲内のある金額が 範囲内の任意の他の金額の推定値よりも良いようである場合、その金額は計算されるべきである。この範囲内のどの金額も他の金額よりも良い推定値として決定できない場合、範囲内の最小金額を累積する。これらの金額は、保険や第三者への請求によって回収可能な金額 を差し引くことはありませんが、回収可能であると考えられる場合、保険会社や他の第三者からの未割引入金は個別に計上される可能性があります。 結果の予測が困難であるため、最終的な 解決策は現在の分析とは異なる可能性があるため、経営陣が判断したり損失があったりする必要があります。当社は新しい資料に基づいて課税項目を改訂します。損失の結果を確定的に予測したりすることはできないが、経営陣は、総合財務諸表には、このような事項に関する潜在損失計について十分な準備金が提案されていると考えている。
(7) 関連側チケットの対応
2022年8月、会社はWerth Family Investment Associates LLC(WFIA)から500万ドルの債務融資を受けた。WFIAマネージャーのPeter Werthは会社の取締役会のメンバーだ。この日付は2023年8月9日の約束手形は無担保で、年利率は15%です。2023年5月、当社はこの付記を修正し再確認することにより、元金を800万ドルに増加させ、WFIAから追加の300万ドルの債務融資を受けた。その会議の他のすべての条項は変わらないままだ。
2023年9月8日に、当社および華通証券はWFIAと手形変換プロトコル(手形変換協定)を締結し、これにより、WFIAは手形項の元元本金額500万ドルを変換することに同意し、元元金のすべての課税利息 を加えて、事前資本金権証(WFIA事前資本金権証)5,812,500ドルを発行し、341,912株自社普通株brを購入し、WFIA事前資本金権証の転換価格は17.00ドルである。当社普通株は2023年9月8日にナスダックでの市収価格である。1株11.55ドルです。WFIA事前承認株式証は満期日がなく,ただちに1株0.002ドルで行使できるが,行使後,1934年の証券取引法(改正(Exchange Act))第 13(D)節では,WFIAとその関連会社は実益が19.99%以下の会社普通株流通株 を持つことになる。
同社はASC 470-60の使用を検討しているDebtorsの問題債務再編この債務転換を計算する時に。発行された予融資権証の公正価値と取引中に決済された債務の帳簿価値との差額 は株主権益表で1,862,735ドルの出資であることが確認された。
手形の残り未償還元金300万ドルおよびすべての課税および未払い利息は、2025年8月8日またはWFIAが1カレンダー四半期の5営業日前に書面で要求した120日後に満期になり、支払いが行われます。WFIAはチケットに を支払うことを要求していない.当社は手形の全部または一部を前払いして、割増や罰金を取らないことができます。しかし、返済されたお金を借りることはできません。
| F-32 |
2023年9月30日と2022年12月31日現在、手形上の未返済元金はそれぞれ300万ドルと500万ドル。
会社は2023年9月30日までの3ヶ月間、手形に関する237,500ドルの利息支出を確認した。会社は2023年9月30日までの9カ月間、手形に関する677,500ドルの利息支出を確認した。手形に関するすべての利息支出は総合貸借対照表の計上すべき支出に計上される。
(8) 株主権益
当社は2023年9月30日及び2022年12月31日に額面0.0001ドル普通株240,000,000株及び額面0.0001ドル優先株10,000,000株 の発行を認可し、その中でそれぞれ2023年9月30日及び2022年12月31日に普通株868,940株及び565,470株を発行及び発行する。当社では優先株は何も発行していません。
普通株式保有者は普通株を保有するごとに1票の投票権を享受することができる。当社が任意の自動または非自発的清算、解散または清算が発生した場合、当社のすべての債務および負債の清算または清算後、普通株式保有者は、当社が分配可能な残りの資産(あれば)を共有する権利がある。普通株式の保有者は取締役会が発表した時に配当金を得る権利がある。
(9) 証券販売
個人配給
2023年8月11日、当社はWFIAと証券購入協定を締結し、ナスダック規則に基づいて私募で91,158株の普通株を発行し、1株当たりの購入価格は10.97ドルであり、当社に約1,000,000ドルの毛収入をもたらした。ピーター·ウォースは会社の取締役会メンバーでWFIAのマネージャーです
公開サービス
2023年9月11日、当社はある機関投資家と証券購入協定(2023年9月発売) を締結し、これにより、当社は86,000株の普通株を発行し、合計価格は1株11.55ドルであり、Aシリーズ及びBシリーズの権利証及び予資権証(予資金権証)を付随して、合計260,261株の普通株を購入し、購入価格は1部の事前資本金権証11.55ドル、及び付随するAシリーズ及びBシリーズ権証である。これは普通株の公開発行価格から1株当たり前払い資金権証を引いた1株当たり0.002ドルの発行価格を表す。2023年9月の発売 は2023年9月13日に終了します。事前資本権証は、発行日後の任意の時間に行使することができ、満期日がない。もし事前計画権証所有者およびその関連会社が権利証を行使した後、発行された普通株式数の4.99%(または所有者が選択し、9.99%を超える)を超える実益を持っている場合、当該等承認持分所有者は当該等承認持分証を行使することができない。2023年9月の発行 は会社に3,999,480ドルの毛収入をもたらし、688,929ドルの配給代理費と他の発売費用を差し引いた。 2023年9月に発行されたAシリーズとBシリーズ承認株式証の説明については、付記10を参照されたい。
関連側チケット割引
付記7で述べたように、当社は2023年9月8日にWFIA事前資本承認株式証を発行し、手形変換協定の一部として341,912株を購入することができる。この等事前資本承認株式証は満期日がなく、直ちに使用価格で1株0.002ドルで行使することができるが、取引所法令(Br)13(D)節の規定により、WFIA及びその連属会社は自社普通株既発行株式の19.99%を実益する。
| F-33 |
リンカーン公園との購入契約
当社は2023年4月にリンカーンパーク資本基金有限責任会社(リンカーンパーク)と購入契約(LP購入協定)および登録権協定(登録権協定)を締結した。有限責任会社購入契約の条項によると、リンカーン公園は当社から最大1,200万ドルの会社普通株を購入することに同意したが、有限責任会社の購入契約に記載されている条件のいくつかの制限と満たされなければならない。権利協定を登録する条項に基づいて、当社は米国証券取引委員会に登録声明を提出し、証券法に基づいて225,000株の転売が有限責任会社による合意に基づいてリンカーン公園に発行された普通株を購入するか、または可能性がある。
有限責任会社購入契約の条項によると、当社は、有限責任会社購入契約及び登録権協定に署名した際、当社がリンカーンパークに18,402株の普通株を発行し、有限責任会社購入協定により普通株を購入することを承諾した代償とする。約束株式の推定値は400,409ドルであり、普通株を発行する追加株式として記録され、有限責任会社購入協定に従って調達された資本コストの配当金の減価とみなされる。
会社は2023年9月30日までの四半期内に、有限責任会社の購入契約により12,000株の普通株を売却し、純収益は196,167ドルだった。会社は2023年9月30日までの9カ月間、有限責任会社の購入合意により25,500株の普通株を売却し、純収益は450,427ドルだった。
市場で製品
当社は2023年1月にH.C.Wainwright&Co.,LLC(HCW) と市場発売プロトコル(ATMプロトコル)を締結し,この合意により,当社は自社普通株株式を随時発行·販売することができ,総発売価格は最高497万ドルに達する。HCWは販売エージェントを担当し,ATMプロトコルにより販売ごとに3%の手数料 を得る.同社の普通株は売却時の現行市場価格で販売されるため、価格が異なる。同社は2023年9月30日までの四半期で、ATM協定により76,943株の普通株を売却し、純収益は1,595,429ドルだった。2023年9月30日までの9カ月間に、会社はATM協定により82,410株の普通株を売却し、純収益は1,696,407ドルだった。
(10) 普通株引受権証
2023年9月に発行された346,261株の普通株と事前融資権証のほか、会社はAシリーズ株式承認証とBシリーズ株式承認証を発行し、それぞれ最大346,261株の普通株と最大173,131株の普通株を購入した。AシリーズとBシリーズの株式承認証の行使価格は1株11.55ドルであり、株主が株式承認証によって発行された株式の発効日 の行使を承認することができる。A系列権証の有効期間は5年 であるのに対し,B系列権証の期限は初期行使日から2年 である.
社はASCテーマ480の規定に基づいて責任または株式分類の事前資本金権証を評価した。 負債と持分を区別するASCテーマ815派生ツールおよびヘッジそして、株式処理 が適切であることを決定する。同社は発行日3,006,008ドルの公正価値に基づいて、260,260株の普通株を購入する事前資本権証を評価した。2023年9月30日現在、事前出資の権利証はすべて行使されていない。
2023年9月の発売について、当社は最大17,313株の普通株を購入するための配給代理権証を発行しました。 配給代理権証の行使価格は1株11.55ドルです。これらの株式承認証の有効期間は5年で、2028年9月11日に終了する。
2023年9月に発行されたAシリーズ,Bシリーズ,配給代理権証はブラック·スコアモデルを用いて推定され,無リスク金利は4.5%−5.3%,期限はそれぞれ5年と2年,変動率は1.29−1.32であった。当社の普通株の計量日における推定変動率は、バイオテクノロジーや製薬業界内で上場企業よりも平均株価変動率に基づいており、これらの会社は、当社の普通株には十分な取引履歴がないため、将来の株価傾向を代表するとみなされている。無リスク金利は、引受権証の期待期限 に基づいており、ライセンス日までに満期日のような米国債の継続満期日を有する。予想期限は株式承認証の契約条項を用いて 見積もりを行った。
| F-34 |
次の表は、会社が2023年9月30日までに発行した普通株引受権証をまとめています
未弁済持分証及び権利付表
| 発行日 | 発行日 | |||||||||||||||
| 量 | トレーニングをする | 公正価値 | 公正価値 | |||||||||||||
| 株式承認証 | 値段 | 各授権書 | 合計する | |||||||||||||
| 2021年12月初公募株 | 239,584 | $ | 120.00 | $ | 95.3997 | $ | 22,856,242 | |||||||||
| 2021年12月引受業者株式証明書 | 10,417 | $ | 150.00 | $ | 92.7969 | 966,665 | ||||||||||
| 2023年9月WFIA事前計画権証 | 341,912 | $ | 0.0001 | $ | 16.9999 | 5,812,500 | ||||||||||
| 2023年9月に事前計画権証を公開発売 | 260,261 | $ | 0.0001 | $ | 11.5460 | 3,006,008 | ||||||||||
| 2023年9月にAシリーズ株式承認証を公開発行 | 346,261 | $ | 11.60 | $ | 10.7999 | 3,739,612 | ||||||||||
| 2023年9月Bシリーズ株式承認証を公開発行 | 173,131 | $ | 11.60 | $ | 8.3999 | 1,454,294 | ||||||||||
| 2023年9月の配給代理承認株式証 | 17,313 | $ | 14.40 | $ | 10.6000 | 183,518 | ||||||||||
| 残高-2023年9月30日 | 1,388,879 | $ | 38,018,839 | |||||||||||||
ASU 2020-06に基づいてこれらの権利証を株式分類ツールとして会計処理している変換可能なツールと実体自己資本における契約の会計処理それらは、会社の普通株式をインデックスとし、ASU 2020−06において株式分類のために定義された他のすべての条件 を満たすからである。2023年9月に公開発売された総収益は、以下の相対公正価値方法で普通株式/事前資本権証と普通株引受権証に割り当てられます。株式証券の公正価値は会社の貸借対照表の追加実収資本に計上される。
株式証の追加実収資本に対する公正価値を認める
| パーセント | ||||||||||||
| 公平である | 総数を占める | 金額 | ||||||||||
| 価値がある | 公正価値 | 分配された | ||||||||||
| 普通株式と事前資金権証 | $ | 3,999,480 | 42.7 | % | $ | 1,707,778 | ||||||
| Aシリーズ、Bシリーズ、配給代理承認証 | 5,377,424 | 57.3 | % | 2,291,702 | ||||||||
| 合計する | $ | 9,376,904 | 100 | % | $ | 3,999,480 | ||||||
(11) 株報酬
2021年9月、会社取締役会と株主は、会社普通株、株式付加価値権、制限株式単位、普通株制限または非制限株、業績株、業績単位、インセンティブ配当奨励、その他の株式奨励金、その他の現金奨励を付与するために、“2021年株式インセンティブ計画”(2021年計画)を採択した(2021年計画)。2031年9月24日以降は、2021計画に従っていかなる報酬も発行することはできないが、2021計画はその後も実行され、以前に付与された報酬は完了していない。
2021年計画により付与されたオプションや他の奨励に関する発行可能な普通株の最高数は139,116株であり,2023年9月30日現在,2021年計画により発行可能な普通株は66,678株である。2021年計画によると発行可能な普通株数は毎年1月1日に自動的に増加し、2021年計画満了まで、金額は前年の12月31日に発行された普通株式総数の5%に相当し、完全希釈ベースで計算すると、取締役会がその前に行動しない限り、その年度の普通株備蓄が増加しない、あるいはその年度の普通株備蓄が増加する普通株数が他の場合よりも少なくなると規定されている。2021年計画によると、当社が使用価格または源泉徴収、買い戻し、または他の方法で終了した任意の奨励を満たすために没収され、br取り消し、行使、または決済奨励時に差し止められた普通株式は、2021計画に従って発行可能な普通株式に再計上される。
| F-35 |
2023年9月30日までの9ヶ月間の会社の株式給与支出は658,105ドルであり、2022年9月30日までの9ヶ月間の会社の株式給与支出は594,363ドルであった。会社が記録した株式ベースの報酬支出は2023年9月30日までの3カ月間で236,251ドル、2022年9月30日までの3カ月間に記録された株式報酬支出は205,656ドルであり、いずれも2021年、2022年、2023年の間に発行されたオプションと関係がある。2023年9月30日と2022年12月31日現在、それぞれ2,133,614ドルと2,637,895ドルの未確認報酬コストは、2021計画で付与された非既得株式ベースの給与スケジュールと関連しており、今後1~4年以内に確認される予定である。
2023年9月30日までの3ヶ月と9ヶ月以内に、この計画下のオプション活動の概要は以下の通りです
オプション活動の概要
| 加重平均 | 骨材 | |||||||||||||||
| 加重平均 | 余剰契約 | 固有の | ||||||||||||||
| 株 | 行権価格 | 期限(年) | 価値がある | |||||||||||||
| 2023年1月1日現在返済されていない | 43,060 | |||||||||||||||
| 授与する | 19,225 | $ | 35.00 | 9.92 | ||||||||||||
| 鍛えられた | - | |||||||||||||||
| 没収または満了 | (281 | ) | ||||||||||||||
| 2023年3月31日現在の未返済債務 | 62,004 | $ | 63.20 | 9.01 | ||||||||||||
| 授与する | 6,388 | $ | 19.20 | 9.98 | ||||||||||||
| 鍛えられた | - | |||||||||||||||
| 没収または満了 | (899 | ) | ||||||||||||||
| 2023年6月30日現在の未返済債務 | 67,493 | $ | 63.20 | 9.01 | ||||||||||||
| 授与する | 5,204 | $ | 12.20 | 9.89 | ||||||||||||
| 鍛えられた | - | |||||||||||||||
| 没収または満了 | (50 | ) | ||||||||||||||
| 2023年9月30日現在の未返済債務 | 72,647 | $ | 59.80 | 8.84 | $ | 10,607 | ||||||||||
| すでに帰属しており、2023年9月30日に帰属する予定です | 72,647 | |||||||||||||||
| 2023年9月30日に行使できます | 16,816 | |||||||||||||||
会社が発行した株式オプションはASC 718に規定されている株式会計処理資格を満たしている報酬-株報酬 は,それに応じて付与日の公正価値に応じて計測される.オプションの公平価値はBlack-Scholes モデルを用いて見積もる.2023年9月30日までの9ヶ月間、会社が従業員に付与された株式オプションの付与日公正価値を推定するために使用された仮定は、以下のように加重平均で表示される
公正価値仮定付表
| 無リスク金利 | 4.073 | % | ||
| 予想期限(年単位) | 5.9 | |||
| 予想変動率 | 1.27 | |||
| 期待配当収益率 | 0 | % |
無リスク金利 :当社は、付与日類似満期日までの米国債の定常満期日 に基づいて、オプション期待期間内の無リスク金利を取得する。
予想される 期限:期待期間は、付与されたオプションが未償還期間を予期し、簡略化方法(ホーム日と契約期限終了との間の中間点に基づいて)を使用して決定される
期待変動 :当社はバイオテクノロジーや製薬業界で上場企業の平均株価変動率を使用することができ、これらの会社は将来の株価傾向を代表するとされており、当社には十分な普通株取引履歴がないためである。会社は、自身の株価変動に関する十分な数の履歴情報 が利用可能になるまでこの過程を適用し続ける。
| F-36 |
期待配当収益率 :当社はまだ支払っていませんし、近い将来に何の配当金も支払うとは期待していません。したがって, 配当収益率はゼロとなることが予想される.
2023年9月30日までの3ヶ月以内に授出した購入権の授出日の公正価値は10.20ドルから12.80ドルを介しているが、2023年9月30日までの9ヶ月以内に授受された購入権の授与日の公正価値は10.20ドルから30.60ドルを介している。
株式オプションの合計内在価値は,株式オプションの発行価格と会社普通株の公正価値との差額で計算される.我々の普通株のナスダック資本市場における終値によると、2023年9月30日まで、普通株の1株当たりの公正価値は14.20ドルで、2022年12月31日まで、1株当たりの公正価値は20.00ドルである。
(12)(br}所得税
Cingate Inc.は“国内税法”によりC類会社として課税される。Cingate Inc.は、財務報告に記録された資産および負債金額と税務法規によって計量されたこのような金額との間の一時的な差の影響を反映するために、繰延所得税を記録する。CTXはCingate Inc.の全額免除エンティティであり、CTXおよびその完全子会社Cingate Works Inc.のすべての活動は、Cingate Inc.の流動および繰延税金資産および負債の計算に含まれる。2023年9月30日および2022年9月30日までの3ヶ月間、または2023年9月30日および2022年9月30日までの9ヶ月間、繰延所得税の割引または費用は記録されていない。
所得税税費は、米国連邦所得税税率を適用することで計算される期待費用とは異なり、以下のようになる
有効所得税率調節表
| 2023年9月30日までの3ヶ月 | 3か月まで 2022年9月30日 | 9か月で終わる 2023年9月30日 | 9か月で終わる 2022年9月30日 | |||||||||||||
| 法定税率の連邦所得税割引 | $ | (1,255,601 | ) | $ | (845,722 | ) | $ | (3,486,052 | ) | $ | (2,733,779 | ) | ||||
| 州所得税割引 | (330,642 | ) | (222,707 | ) | (917,994 | ) | (719,896 | ) | ||||||||
| 恒久的差異 | 6,154 | 3,157 | 14,457 | 11,920 | ||||||||||||
| 評価免除額を変更する | 2,218,188 | 1,132,895 | 5,089,875 | 3,572,189 | ||||||||||||
| 前期を実際に調整する | (620,630 | ) | - | (620,630 | ) | - | ||||||||||
| 他にも | (17,469 | ) | (67,623 | ) | (79,656 | ) | (130,434 | ) | ||||||||
| 所得税総支出 | $ | - | $ | - | $ | - | $ | - | ||||||||
繰延税金資産推定準備の必要性と金額を評価するには、通常、各司法管轄区のすべての利用可能な証拠に対して重大な判断と広範な分析が必要である。同等の判決は,現行税法の解釈とその適用状況の他に指針を公表することを当社に求めている。今回の評価の一部として、会社はその収益性と税務状況に関するプラスとマイナスの証拠を考慮した。既存の証拠に基づいて、繰延税金資産の全部または一部が現金化できない可能性が高い場合、推定準備が提供される。過去の収入レベルと未来の課税収入の予測によると、他の項目を除いて、当社はその繰延税金資産がさらに現金化できない可能性があると確定した。当社は2023年9月30日および2022年12月31日にそれぞれ10,988,821ドルおよび5,580,595ドルの繰延税項目純資産の推定値を計上し、添付されている総合経営報告書およびその他の全面赤字のうち所得税支出の一部とした。
同社はアメリカ連邦と各州司法管轄区で所得税申告書を提出した。2018年までに、当社は米国連邦と州税務機関の所得税審査を受ける必要はありません。
会社はFASB ASC 740の規定に従っている所得税不確実な税金状況を評価するために。本テーマでは、納税申告書において採取されたまたは予想される納税頭寸の財務諸表確認および計量に対する確認閾値および計量属性を規定する。当社では、2023年9月30日または2022年12月31日現在、連結財務諸表で確認すべき重大な不確定税務状況は発見されていません。
| F-37 |
繰延税金貸借対照表
| 2023年9月30日 | 2022年12月31日 | |||||||
| 繰延所得税資産: | ||||||||
| 現在: | ||||||||
| 研究開発コスト | $ | 723,577 | $ | 343,087 | ||||
| 他にも | 59,126 | 59,018 | ||||||
| 現在ではない | ||||||||
| 純営業損失 | 5,862,906 | 3,381,215 | ||||||
| 研究開発コスト | 3,645,038 | 1,762,716 | ||||||
| 研究開発税収控除 | 756,122 | - | ||||||
| 非既得性株式オプション | 405,739 | 204,380 | ||||||
| 特許 | 99,118 | 92,417 | ||||||
| 使用権資産 | 45,265 | 63,563 | ||||||
| 繰延所得税総資産 | 11,596,891 | 5,906,396 | ||||||
| 減算:推定免税額 | (10,988,821 | ) | (5,580,595 | ) | ||||
| 所得税純資産を繰延する | 608,070 | 325,801 | ||||||
| 繰延所得税負債: | ||||||||
| 現在: | ||||||||
| 現金を計算する | - | (11,228 | ) | |||||
| 当面ではない | ||||||||
| 財産と設備 | (608,070 | ) | (314,573 | ) | ||||
| 繰延所得税総負債 | (608,070 | ) | (325,801 | ) | ||||
| 繰延税項目純資産(負債) | $ | - | $ | - | ||||
(13) 1株当たり純損失
基本的な1株当たり純損失は、普通株株主が純損失を占めるべきであることを普通株加重平均と期内に発行された事前資本権証の加重平均で割って算出される。事前資本権証は、それらの行使は株式の名義対価を交付するだけでよいので、加重平均既発行株式数 に計上される。次の表 は2023年9月30日と2022年9月30日までの3ヶ月と9ヶ月の基本と希釈後の1株当たり純損失の計算方法を示しています
1株基本と償却純損失明細書
| 2023年9月30日までの3ヶ月 | 3か月まで 2022年9月30日 | 9か月で終わる 2023年9月30日 | 9か月で終わる 2022年9月30日 | |||||||||||||
| 分子: | ||||||||||||||||
| 純損失 | $ | (5,979,054 | ) | $ | (4,027,247 | ) | $ | (16,600,250 | ) | $ | (13,071,205 | ) | ||||
| 分母: | ||||||||||||||||
| 加重平均流通株 | 988,333 | 565,470 | 714,397 | 565,470 | ||||||||||||
| 1株当たり基本と希釈して純損失 | $ | (6.05 | ) | $ | (7.12 | ) | $ | (23.24 | ) | $ | (23.12 | ) | ||||
希釈後の1株当たり計算に含まれていない潜在的希釈証券は、2023年9月30日と2022年9月30日までの3ヶ月および9ヶ月間、以下のようになる
潜在希釈証券明細書
| 2023年9月30日 | 2022年9月30日 | |||||||
| 2021年株式インセンティブ計画に基づいて発行された株式オプション | 72,647 | 44,190 | ||||||
| 未償還普通株引受権証 | 786,704 | 250,000 | ||||||
| 合計する | 859,351 | 294,190 | ||||||
| F-38 |
(14) 許可プロトコル
CTX は、CTX−1301、CTX−1302、 およびCTX−2103を開発するための特許およびライセンス技術に関するライセンス契約を会社と締結している。次のマイルストーン事件が発生したときに支払わなければなりません
| ● | 現場の各製品の3期臨床試験における1人目の患者の投与量に応じて250,000ドルのマイルストーン支払い を支払い,製品別に支払った。 | |
| ● | 許可された側がこの分野の製品ごとに新薬申請を提出する際には250,000ドルのマイルストーン支払いを支払い, は製品ごとに支払う。 | |
| ● | CTX-1301およびCTX-1302のマイルストーン支払いは250,000ドル、CTX-2103のマイルストーン支払いは500,000ドルであり、FDAの最初の発売承認を受けた後、製品別に支払います。 | |
| ● | ヨーロッパ医薬品局(European Medicines Agency)の最初の発売許可を受けた後、CTX-2103の250,000ドルのマイルストーン支払いを購入しました |
2022年12月31日までに、CTX-1301は3期臨床試験中の第1位の患者投与量に関連する250,000ドルのマイルストーンが累積 しており、管理層はこのマイルストーンが発生する可能性が高いと考えているからである。2023年初め、同社はCTX-1301第3段階臨床試験の最初の患者が用量治療を受けたため、このお金を支払った。当社は他の製品について他のマイルストーンに関するいかなる支出も記録していません。当社は2023年9月30日までにこのような支出が発生する可能性があるとは考えていないからです。
(15) 関連先取引
当社の総法律顧問は、当社がレンタルしたオフィス施設スペースを提供する法律事務所のパートナーである。当社の2023年および2022年9月30日までの9ヶ月間の賃貸料支出は27,000ドルですが、2023年9月30日および2022年9月30日までの3ヶ月間の賃貸料支出は9,000ドルと公正価値に近づいています。当社は2023年9月30日と2022年12月31日現在、本レンタル契約項での未返済金を持っていません。
取締役会のPeter WerthはWFIAのマネージャーで、同社に800万ドルの債務br融資を提供し、そのうち500万ドルは2023年9月に株式に転換した。付記7で述べたように。2023年9月30日現在、元金残高300万ドルは返済されておらず、2022年12月31日現在、初期元金残高500万ドルは返済されていない。2023年9月30日と2022年9月30日までの3ヶ月間、それぞれ利息支出237,500ドルと104,838ドルを確認した。2023年9月30日と2022年9月30日までの9ヶ月間、それぞれ677,500ドルと104,838ドルの利息支出を確認した。2023年9月30日と2022年12月31日現在、この手形に関連する課税利息のうち157,339ドルと292,339ドルが返済されていない。
2023年8月11日、当社はWFIAと証券購入協定を締結し、ナスダック規則に基づいて私募で91,158株の普通株を発行し、1株当たりの購入価格は10.97ドルであり、当社に約1,000,000ドルの毛収入をもたらした。
(16) 後続イベント
経営陣 は、中間財務諸表の発行日 である2023年9月30日から2023年11月13日までの間に発生したイベントを評価する。
(17)株式の逆分割
2023年11月30日、会社は20株1株の逆株式分割(“逆株式分割”)を完了し、逆株分割が発効する直前に発行·発行された会社普通株の数を減少させた。会社の法定普通株の数量は逆株分割の影響を受けず、会社の普通株の額面は変わらず、1株当たり0.0001ドルである。逆株式分割に関連する断片的な株式は発行されていない。開示を除いて、株式数及び1株当たり金額に関するすべての金額は、当該等の財務諸表にさかのぼって記載されている。
| F-39 |
1,375,000株普通株式
3,750,000シリーズ株式承認証最大3,750,000株普通株購入
1,875,000株Bシリーズ株式承認証最大1,875,000株普通株購入
2,375,000株事前資金(Br)株式承認証最大2,375,000株普通株購入
Aシリーズ株式承認証、Bシリーズ株式承認証と事前出資株式承認証に関する800万株普通株
150,000部の配給代理株式承認証最大150,000株普通株を購入
150,000株普通株式販売代理株式承認証

目論見書
2024年2月2日