登録番号333-276367
アメリカです
アメリカ証券取引委員会 ワシントンD.C.,20549
修正案第3号
表S-1
登録宣言 の下で1933年証券法
PANBELA治療会社は
(登録者の正確な氏名はその定款に記載)
|
デラウェア州
(明またはその他の司法管轄権 |
2834
(主な標準工業 |
88-2805017
(税務署の雇用主 |
712 Vista Blvd,キット305ミネソタ州ワコニア55387(952) 479-1196 (登録者の住所、郵便番号と電話番号を含み、市外局番を含む’S主実行オフィス)
ジェニファー·K·シンプソンCEO 712 Vista Blvd,キット305ミネソタ州ワコニア55387(952) 479-1196 (サービスエージェントの名前、住所、郵便番号と電話番号、市外局番を含む)
コピーされました
|
W.モーガン·バーンズ ジョシュア·L·コルビン W.ジェイソン·デボンFegre Drinker Bdle&Reath LLP 南七街90番地富国銀行センター2200番地 |
アル·パンジェヴァニ 普華永道現金管理有限公司 タイムズスクエア7号 ニューヨーク、ニューヨーク10036 (212) 421-4100 |
一般向けの販売開始の大まかな日付:本登録声明の発効日後に実行可能な範囲内でできるだけ早く開始することを提案します。
改正後の1933年“証券法”第415条の規定により、本表に登録されている任意の証券が遅延又は連続的に発売される場合は、次の枠を選択してください
本表が証券法第462条(B)条に発行された追加証券を登録して提出した場合は、以下のブロックを選択し、同一発売の比較的早く発効した登録書の証券法登録書番号を示してください
この表が証券法下の規則462(C)に基づいて提出された後に改正された場合、以下の枠を選択し、同じ発売された以前に発効した登録声明の証券法登録宣言番号を一覧表示してください
この表が証券法下の規則462(D)に基づいて提出された後に改正された場合、以下の枠を選択し、同じ発売された以前に発効した登録声明の証券法登録宣言番号を一覧表示してください
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
|
大型加速ファイルサーバ☐ |
加速ファイルサーバ☐ |
|
非加速ファイルサーバ☑ |
規模の小さい報告会社☑ |
|
新興成長型会社☐ |
新興成長型会社であれば、登録者が延長された移行期間を使用しないことを選択して証券法第7(A)(2)(B)節に規定する任意の新たな又は改正された財務会計基準を遵守するか否かをフックで示す
登録者は、登録者がさらなる修正案を提出するまで、本登録明細書を修正するために必要な1つまたは複数の日付を、登録者がさらなる修正案を提出するまで、その後、改正された1933年証券法第8(A)節に従って施行されるか、または登録書が証監会が第8(A)条に従って決定される日まで効力を発生させることを明確に規定する。
本募集説明書の情報は不完全であり、変更される可能性があります。アメリカ証券取引委員会に提出された登録声明が発効するまで、私たちはこれらの証券を売ることができません。本募集説明書は、証券売却の要約ではなく、いかなる要約や売却を許可しない州でもこれらの証券の購入要約を求めているわけではありません。
|
初歩募集説明書 |
完成が待たれる |
日付:2024年1月25日 |
最大8,000,000株の普通株
最大8,000,000株E類普通株購入最大2,660,000株普通株
最大8,000,000株F類普通権証購入最大14,000,000株普通株
8,000,000株までの普通株を購入する予備融資株式承認証
最大24,660,000株の普通株式関連株式承認証
これは,(A)最大8,000,000株の我々の普通株,1株当たり額面0.001ドル,(B)最大8,000,000株のE系一般権証,最大2,660,000株の我々の普通株(“E類株式承認証”),および(C)最大8,000,000株F類一般権証を購入して,最大14,000,000株の我々の普通株(“F類株式承認証”),およびE類株式権証を購入するための最大の努力公開である.仮定公開発行価格1株当たり普通株および付随株式証3.77ドル(ナスダック資本市場が2024年1月23日に発表した普通株最終販売価格に相当)で計算される。私たちの普通株は1株当たり0.25株の私たちの普通株を購入したE類株式証と1.75株の私たちの普通株を購入したF類株式証と一緒に販売します。1株当たりの一般権証の行使価格を1株当たり3.77ドル(1株当たり公開発行価格と普通権証の100%)とすると、発行時に行使でき、発行日から5年で満期となる。5営業日後、E類株式証の所有者は、現金行使時に発行可能な普通株式数の1.33倍に相当する株式総数を取得する“キャッシュレス行使の代わり”を選択するための単独の選択権を有することになる。
今回の発売で普通株を購入した買手にも機会を提供し,そうでなければ,今回の発売完了後,どの買手およびその関連会社も実益を4.99%(またはその買手が選択した場合,9.99%)我々の発行済み普通株を所有し,我々の普通株の代わりにあらかじめ出資した株式承認証を購入する機会があり,そうでなければ,その買手の実益所有権が4.99%(またはその買手が選択した場合,9.99%)我々の発行済み普通株を超えることになる.1部当たりの事前資金権証の買い取り価格は、今回発行された普通株の1株当たりの公開発行価格から1株当たりの予資権証の発行価格を引いた0.001ドルに等しい。あらかじめ出資した引受権証は発行時に行使でき、行使前に満期にはなりません。私たちが販売している事前融資の引受権証ごとに、私たちが提供する普通株式数は1対1に基づいて減少するだろう。
明確にするために、1株当たりの普通株または1株の普通株を購入する予備融資承認株式証は、1株の普通株を購入するE類株式承認証および1株の普通株を購入するF類承認株式証と共に販売される。
私たちの普通株はナスダック資本市場に発売され、コードは“PBLA”です。2024年1月23日、私たちの普通株のナスダック資本市場での最終販売価格は1株当たり3.77ドルだった。各株及び附随株式権証或いは各部分の事前融資権証及び引付株式権証の合併公開発売価格は吾らと投資家が定価時の市場状況に基づいて決定し、そして当社の普通株の現在の市価より割引する可能性がある。したがって,目論見全体で使用されている最近の市場価格とそれによる仮説価格は,実際の発行価格と大きく異なる可能性がある.一般権証も事前資本権証も国家証券取引所に上場しません。私たちはどの国の証券取引所に上場する一般権証や事前融資権証を申請するつもりはありません。活発な取引市場がなければ、一般権証と事前融資権証の流動性が制限される可能性がある。
2024年1月18日から、我々の普通株流通株に対して20株1株の逆株式分割を行った。本募集説明書に別段の規定がない限り、本募集説明書には歴史財務諸表及びその他の関連付記を除く他の株式及び1株当たりの資料は、逆株式分割の効力を有する。
私たちは、ナスダック株式市場有限責任会社(“ナスダック”)から受け取った従業員決定書を、2023年11月28日にナスダック株式市場有限責任会社(“ナスダック”)から受け取った従業員決定書に上訴することを要求し、ナスダック公聴会グループに公聴会を開催し、ナスダックの公聴会に公聴会を開催することを要求し、NSE表を米国証券取引委員会に提出することで、ナスダックの上場および登録から私たちの普通株を除去することを要求し、ナスダック公聴会グループに公聴会を開催することを要求し、退市決定を控訴させることを決定した。同社の普通株の停止と退市は放置されており、聴聞手続きの終了を待っている。したがって、同社の普通株はナスダック資本市場に上場し続ける見通しで、少なくとも公聴会後に委員会が決定する前にそうだ。この委員会がナスダック資本市場に上場し続ける同社の呼びかけを承認する保証はない。決定書によると、当社(I)はナスダック上場規則第5550(A)(2)条に規定する1株1.00ドルの最低市価(“最低購入価格要求”)を維持しておらず、及び(Ii)ナスダック上場規則第5550(B)条(“最低株主権益要求”)に規定されている継続上場の最低株主権益要求(“最低株主権益要求”)に適合していない。2024年1月18日から、普通株流通株の20株1株の逆分割を完了した。私たちが逆株式分割を実施する主な理由は、最低入札要求を満たすために、私たちの普通株の1株当たりの市場価格を潜在的に高めることである。2024年1月22日、私たちはナスダックから通知を受け、逆株式分割の発効日まで、ナスダック上場規則第5550(A)(4)条(“最低持株量要求”)に適合しなくなり、当社の普通株退市の追加的な根拠として、保有株の最小500,000株を公開することを要求した。また,より多くの株式と今回公開された純収益を発行し,最低流通株要求と最低株主権益要求を再遵守できるようにする予定である.もし私たちがすべての適用されたナスダックの継続上場要求を再遵守できなければ、私たちの普通株はナスダック資本市場から撤退する可能性が高いと思います。
私たちの証券に投資することは高度な危険と関連がある。あなたは、本募集説明書の8ページ目からの“リスク要因”のタイトルの下、および本募集説明書の任意の修正または補足文書に記載されているようなタイトルの下に記載されているリスクおよび不確定要因を詳細に検討しなければならない。
今回の発売は2024年2月15日に終了します。事前に完了しない限り、またはその日までに発売を終了することが決定されない限り(いつでも適宜終了することができます)、E類株式証、F類株式承認証、事前資本権証の基礎となる普通株は、改正された1933年証券法第415条に基づいて継続的に発売されます。私たちは、私たちが受け取った投資家の資金を受け取った後、今回の発行に関連するすべての証券を受け渡しまたは支払います。したがって、投資家の資金を受け入れるか、または信託、信託、または任意の同様の口座に入金する予定は存在しない。
我々はRoth Capital Partners,LLCを我々の独占配給エージェント(“Roth”または“配給エージェント”)として招聘し,その合理的な最大限の努力を尽くして今回の発行で我々の証券を購入するオファーを募集した.配給代理は、私たちにどんな証券を購入する義務もなく、特定の数量や金額の証券の購入や販売を手配する義務もありません。今回の募集は最低入札金額要求を募集完了の条件としていないため、実際の公募金額、配給代理費、吾などから受け取る収益(あればある)は現在決定できず、上記および本募集説明書で述べた総最高入札金額を大幅に下回る可能性がある。我々は,次の表に示す配置エージェント料金を配置エージェントに支払い,配置エージェントに何らかの他の補償を提供することに同意した.これらの手配に関するより多くの情報は、本募集説明書27ページからの“分配計画”を参照してください。
米国証券取引委員会およびどの州証券委員会もこれらの証券を承認または承認しておらず、本募集説明書の十分性または正確性に基づいて意見を発表していない。どんな反対の陳述も刑事犯罪だ。
|
1株当たりと よくある 捜査命令 |
金ごとに前払いする 手令が及ぶ ごく普通である 捜査命令 |
合計する |
||||||||||
|
公開発行価格 |
$ |
$ |
$ |
|||||||||
|
配置代理費 |
$ |
$ |
$ |
|||||||||
|
費用を差し引く前にくれた純収益(1) |
$ |
$ |
$ |
|||||||||
|
(1) |
上記の発売で得られた金額の要約は、今回の発売で発行された一般権証または事前資金権証を行使して得られたものには適用されない。 |
慣例の成約条件によると、我々の普通株と事前資本権証の株および付随する普通権証は2024年頃にある投資家に交付される予定だ。
ロス·キャピタル共同会社
本募集説明書の日付は2024年です
カタログ
|
ページ |
|
|
この目論見書について |
II |
|
前向き陳述に関する警告説明 |
三、三、 |
|
募集説明書の概要 |
1 |
|
供物 |
8 |
|
リスク要因 |
10 |
|
収益の使用 |
22 |
|
市場情報 |
23 |
|
大文字である |
23 |
|
薄めにする |
24 |
|
配当政策 |
25 |
|
経営陣の財務状況と経営成果の検討と分析 |
25 |
|
商売人 |
36 |
|
管理する |
65 |
|
役員報酬 |
69 |
|
特定の実益所有者と経営陣の保証所有権 |
71 |
|
証券説明書 |
72 |
|
配送計画 |
78 |
|
法律事務 |
83 |
|
専門家 |
83 |
|
そこでもっと多くの情報を見つけることができます |
84 |
|
財務諸表 |
F-1 |
この目論見書について
私たちは引用を通じて重要な情報を本募集説明書に統合する。“どこでより多くの情報を見つけることができますか”以下の説明に従って、参照統合による情報を無料で取得することができます。私たちの証券への投資を決定する前に、本募集説明書と“合併された情報を参照することによって”節に記載された他の情報をよく読まなければなりません。
あなたは本募集説明書に含まれている情報だけに依存すべきです。本募集説明書に記載されている資料を除いて、当社および配給代理は、誰も閣下に他の資料を提供することを許可していません。他の人があなたに提供する可能性のある他の情報の信頼性については、私たちは何の責任も負いませんし、何の保証も提供できません。この目論見書は私たちの証券の提供と販売にのみ適用されて合法的です。本募集説明書中の情報は、本募集説明書の交付時間または当社証券の任意の販売にかかわらず、本募集説明書の日付のみが正確である。それ以来、私たちの業務、財務状況、運営結果、そして見通しは変化したかもしれない。私たちはそうしません。配給代理も、これらの証券の発売が許可されていない司法管轄区でも発売されません。
アメリカ以外の投資家に対して:私たちはいません。配給代理も何もしていません。アメリカ以外のどの司法管轄区でも本募集説明書の発行、所有、配布を許可しています。米国以外の人は自分に知らせ、米国国外での証券発行や株式分配説明書の発行に関するいかなる制限も守らなければならない。
本募集説明書には、業界出版物と第三者による研究、調査と研究から得られた統計データと他の業界と市場データが含まれている。業界出版物および第三者研究、調査および研究は、一般に、それらの情報は、そのような情報の正確性または完全性を保証しないにもかかわらず、信頼できるソースから得られることを示している。これらの業界の出版物や第三者研究,調査,研究から得られたデータは信頼できると考えられる.私たちはこの目論見書のすべての開示に最終的な責任がある。
あなたはただ本募集説明書に掲載されている補充と改訂された資料に依存しなければなりません。私たちは誰もあなたに違う情報を提供することを許可していない。本募集説明書は、これらの証券を合法的に販売する場合にのみ使用することができる。本募集説明書の情報は、本募集説明書の日付のみが正確である可能性がある。
発行された任意の証券に投資するかどうかを決定する前に、目論見書をよく読んで、補充と改訂を行うことを促します。
前向き陳述に関する警告説明
本募集説明書には、改正後の1933年証券法第27 A節及び改正後の1934年証券取引法(“取引法”)第21 E節に該当する前向き陳述が含まれている。歴史的事実に関する陳述を除いて、本募集説明書に含まれるすべての陳述は、私たちの将来の経営結果と財務状況、私たちの業務戦略と計画、予想コスト及び私たちの未来の経営目標に関する陳述を含み、すべて前向きな陳述である。
場合によっては、“予想”、“仮説”、“信じ”、“継続”、“可能”、“推定”、“予想”、“可能”、“進行中”、“計画”、“潜在”、“予測”、“プロジェクト”、“すべき”、“すべき”、“将”、“将”、またはこれらの用語または他の同様の用語の否定によって、前向き陳述を識別することができる。すべての展望的声明書がこのような単語を含んでいるわけではないにもかかわらず。前向き陳述は将来の業績や結果の保証ではなく、必ずしもこのような業績や結果を実現する時間や方式を正確に説明するとは限らない。展望的陳述は、陳述を行う際に得られる情報に基づいて、既知および未知のリスク、不確定要素、および他の要素に関連し、これらのリスク、不確実性および他の要素は、我々の結果、活動レベル、業績または成果をもたらす可能性があり、本明細書における前向き陳述明示または示唆の情報とは大きく異なる。これらの要素には
|
● |
私たちは多様性とそれに応じたリスクが足りず、私たちの会社に投資しています |
|
|
● |
多元化が実現できなかったため、私たちの財務状況と業績は悪化する可能性がある |
|
|
● |
私たちは買収に成功しました |
|
|
● |
新製品候補者のために買収された会社と運営を統合することができます |
|
|
● |
私たちは受け入れ可能な条件や根本的に不要な方法で追加資本を獲得して、私たちの業務計画を実施することができます |
|
|
● |
私たちの第1段階臨床試験の最終結果は |
|
|
● |
我々のランダムII/III期臨床試験の進展と成功 |
|
|
● |
候補製品の安全性と有効性を証明することができます |
|
|
● |
私たちはアメリカ、EU、または他の国際市場で私たちの候補製品の規制を承認することができます |
|
|
● |
私たちの候補製品の市場受容度と将来の販売状況 |
|
|
● |
我々の候補製品に適用される規制変更は、製品開発コストと遅延を招く可能性がある |
|
|
● |
第三者支払者との返済計画の進展速度 |
|
|
● |
競争の技術と市場発展の影響 |
|
|
● |
特許出願の提出および起訴および特許請求の実行または弁護に関連する費用; |
|
|
● |
私たちは私たちの普通株を国家証券取引所に上場することを維持することができる |
|
|
● |
その他のリスク要因は、本募集説明書の8ページ目からの“リスク要因”というタイトルの下に含まれる。 |
“リスク要因”に記載されている事項および本明細書に記載された他の警告説明書を読まなければならず、これらの事項は、本明細書に記載されたすべての関連する前向きな陳述に適用される。私たちは、本募集明細書の展望的な陳述が正確であることが証明されることを保証することはできませんので、前向きな陳述に過度に依存しないように奨励します。あなたはこの目論見書を完全に読むべきです。法的要件を除いて、私たちの状況が未来に変化する可能性があっても、このような前向きな陳述を更新したり修正したりする義務はない。
発表の日だけに意見を発表する前向きな陳述に過度に依存しないように読者に戒め,前向き陳述が将来の結果の予測であることを認識し,予想通りには起こらない可能性がある。本明細書の“リスク要因”の項に記載されたリスクおよび不確定要因、および我々は、重要ではないと考えられるか、または現在予想されていない他の要因であるため、実際の結果は、前向き陳述で予想されるおよび歴史的結果とは大きく異なる可能性がある。私たちは展望的な陳述に反映された予想が合理的だと信じているが、私たちは私たちの予想が正しいことが証明されるかどうか分からない。私たちが展望的陳述に反映する予想は、私たちが作成する可能性のある不正確な仮定の影響を受けるか、または本明細書の“リスク要因”のタイトルに記載されたリスクおよび不確実性を含む、既知または未知のリスクおよび不確実性の影響を受ける可能性がある。本募集明細書の“リスク要因”の項に記載されているリスクおよび不確定要因は排他的ではなく、当社の財務結果や状況に重大な影響を与える可能性のある要因を含む当社および当社の業務に関するさらなる情報は、時々出現する可能性がある。実際の結果を反映して、またはそのような前向き陳述に影響を与える要素または仮説の変化を反映するために、前向き陳述を更新する義務はない。我々は、株主および投資家が、米国証券取引委員会(“米国証券取引委員会”)に提出または提出された後続の10-Kフォーム年次報告、10-Qフォーム四半期報告、および現在の8-Kフォーム報告で作成される可能性のある関連テーマの任意のさらなる開示を参照することを提案する。
募集説明書の概要
本要約では、本募集説明書の他の部分に含まれる情報を重点的に紹介し、投資意思決定を行う際に考慮すべきすべての情報は含まれていない。私たちの証券に投資する前に、私たちの財務諸表と関連する付記、見出しの次の情報を含む株式募集説明書全体をよく読まなければなりません“リスク要因”そして“管理する’財務状況と経営成果に関する検討と分析”すべての場合、本募集説明書の他の場所に含まれる。本募集説明書では、他に説明または文意が別に言及されている以外に、本募集説明書で言及されている“パンベラ”♪the the the“会社は、” “私たちは、” “私たちは、” “我々の”また類似した参考文献とはPanbela治療会社とその子会社である。
業務の概要
Panbelaは臨床段階の生物製薬会社であり,破壊的療法を開発し,緊急医療ニーズのある患者の治療に用いられている。著者らは現在、著者らが膵臓癌を治療する無作為二重盲検プラセボ対照臨床試験に参加する患者を募集しており、著者らはアメリカ国家癌研究所(NCI)が援助した第三段階臨床試験の監督と商業協力者であり、この試験は結腸癌リスクの低下と結腸腺腫治療(“CAT”)を研究することを目的としており、これは結腸直腸癌生存者或いはハイリスク結腸ポリープに対する予防的治療方法である。また、同社は家族性腺腫性ポリープ症(FAP)の第3段階登録試験のための世界的な案を設計している。FAPは稀な遺伝性疾患であり、数千個の結腸直腸腺腫(すなわち腺腫性ポリープ)の成長を招くことができ、後者は結腸癌の重要なリスク因子とされている。世界的な方案は登録経路について合意するために、連邦薬物管理局(FDA)と欧州薬品管理局(EMA)に提出される。PanbelaのFAP面と全世界登録試験の設計における豊富な経験を利用することにより,規制機関基準に適合した高品質な試験案を開発することができ,FAP治療におけるFlynpoviの潜在的安全性と有効性を示すことを目的とした。私たちはまた、(1)青少年糖尿病研究財団によって支援された早期発症1型糖尿病治療の第2段階臨床試験、(2)NCIによって援助された胃癌治療の第2段階臨床試験、(3)STK 11変異を有する非小細胞肺癌(NSCLC)のI/II段階臨床試験の治療、(4)転移性去勢抵抗前立腺癌の治療の第2段階計画、および(5)孤児疾患および癌分野で後援される前研究を含む、研究者によって開始された試験および会社が後援するいくつかの前臨床試験を支持する。
同社の主な資産はアスピリン(SBP−101),フリリンポヴィであるTMEflornithine(CPP-1 x)およびeflornithine(CPP-1 x)は、癌および自己免疫疾患のような多くのタイプの疾患に存在する障害の生物学的再配置のための多標的の方法を提供する。多くの腫瘍はそれらの成長と生存を支持するために大量の上昇したポリアミンを必要とする。これらの薬剤は相補的なリンカーであるポリアミン経路を標的としており,これらの結合が疾患で変化することが証明されている。特に、我々の鉛資産は腫瘍の成長を抑制と防止し、他の抗癌薬の抗腫瘍活性を増強し、免疫系を調節する潜在力がある。
エピピミンはポリアミン代謝抑制を誘導することを目的とした独自のポリアミン類似体である。アスピリンは膵癌患者の臨床試験で鼓舞的な抗転移疾患活性を示す。著者らが完成したI期臨床試験において、エビスタチンはゲシタビンとNAB-パクリタキセルを併用して転移性膵臓癌を治療する有効性と安全性結果は現在の無作為、二重盲検、プラセボ対照研究に支持を提供し、この研究は以前治療を受けていない転移性膵臓癌患者に伊司アスピリンとゲムシタビンとNAB-パクリタキセルの併用に支持を提供した。開発が成功すれば,伊司アスピリンは膵癌患者を有効に治療する新しい方法を代表し,この市場の主導的な製品になる可能性があると信じている。過去25年間に、アメリカ食品と薬物管理局(FDA)は3種類の第一線の治療組み合わせ、一部の患者(3%-7%)に対する単一維持治療及び1種の二線薬物のみを許可し、膵臓癌の治療に用いた。伊司アスピリンはすでに米国やヨーロッパで膵癌治療の急速チャネル状態と孤児薬物指定状態を獲得している。
Panbelaは2022年6月15日、同社の第2の主要資産であるエフロン酸を様々な形で増加させた癌予防製薬会社(CPP)を買収した。まず,研究中の新薬Flynpoviはポリアミン合成阻害剤であるエフルオロオルニチンと非ステロイド抗炎症薬であるスリン酸の組み合わせであり,次いでイプロニチンを単一薬剤とした。エフルオロオルニチンは酵素活性化の不可逆的なオルニチン脱炭酸酵素阻害剤であり、オルニチン脱炭酸酵素はポリアミン生合成における第一種の律速酵素である。シュリン酸は非ステロイド性抗炎症薬であり、ポリアミンの出力と分解代謝を促進する。Flynpoviは独特な二重作用機序を有し,新しいポリアミンの合成を抑制し,食事や微生物群におけるポリアミンの出口と分解代謝を増加させる。Flynpoviは家族性腺腫性ポリープ症(FAP)手術や結腸癌のリスク因子(例えばポリープ)を治療するために設計されているため,様々なタイプの結腸癌を予防する能力がある可能性があると信じている。FAP−310第3段階試験では,Fynpovi(エフルオロオルニチンとスリン酸)の併用による成人FAPの併用治療の有効性と安全性がいずれかの薬剤単独と比較して検討された。この研究は期待に達していないが,主な複合終点(Burkeら)である。2020年),ある専門的な分析では,併用群の患者は48カ月に及ぶ下胃腸(LGI)手術が必要となっていないのに対し,シュリン酸とエフロン群の患者はそれぞれ13.2%と15.7%(Balaguerら)であった。2022年)。このデータは,併用治療と単一治療の間で,LGI手術の必要性が100%に近いリスクを低下させていることに対応している。LGI群の統計学的意義を考慮して、新薬申請(“NDA”)をFDAに提出したが、これは発見的分析に基づく結果であるため、完全な返信(“CRL”)が発表された。CRL問題を解決するために,同社は第3段階登録実験を設計しており,現在の現金需要を増加させることなくこの計画を進めている.現在承認されていないFAPに対する薬物療法
その他の計画は、胃癌、最近発病した1型糖尿病とSTK-11変異の非小細胞肺癌の予防を含む単剤錠剤エフルオロオルニチン或いは高用量粉末エフルオロオルニチン香包のいくつかの適応を評価している。臨床前研究及び第一段階或いは第二段階の研究者が開始した試験により、エフルオロオルニチンの治療耐性は良好であり、潜在的な活性を有することを表明した。昨年12月、ケンタッキー州に本社を置く専門製薬会社US WorldMedsがFDAの承認を得て、エフロンを緩和中の神経芽細胞腫の維持療法として利用した。Panbelaは小児科神経芽細胞腫プロジェクトで何らかの資産を剥離した。この承認はエフルオロオルニチンと任意のポリアミン標的治療の癌適応における最初の承認を示している。
Flynpoviは米国でFast Track称号を獲得し,米国とヨーロッパでFAPの孤児薬物称号を獲得した。また,エフロンは米国やヨーロッパで神経芽細胞腫治療の単一薬剤として,米国で胃癌を治療する孤児薬として指定されている
臨床試験
アスピリン(SBP−101)
2015年8月、FDAは我々の研究新薬(IND)の私たちのIVOSPEMEN候補製品に関する申請を受けた。著者らはすでに過去に治療した局部末期或いは転移性膵臓癌患者に対する初歩的な臨床試験を完成した。これは第1段階の人類初の用量逓増安全性研究である。2016年1月から2017年9月まで,I期試験の用量増加段階で,29名の患者を6つのキューあるいはグループに組み入れた。いずれの用量レベルでも薬物に関連する骨髄毒性あるいは末梢神経病変は認められなかった。安全性評価を受ける以外に、29名の患者の中の23人は第1の治療周期の終了前或いは8週間の終了時に固形腫瘍反応評価標準(RECIST)を用いて初歩的な治療効果信号を評価することができ、RECISTは現在公認されている腫瘍の大きさの変化を評価する標準である。
2018年,われわれの第2の臨床試験に参加する患者を募集し始めたのはIa/Ib期研究であり,アスピリンと2種類の標準看護化学療法薬ゲムシタビンとNAB−パクリタキセルの併用の安全性,有効性,薬物動態を検討することを目的とした。投与量レベルとスケジュールを評価するために、25人の被験者が4つのキューに参加した。また25名の被験者が試験の拡張段階に参加した。中間結果は2022年1月に発表された。評価可能な被験者(キュー4とIBN=29)では,最適反応は完全緩解1例(3%),部分緩解13例(45%),安定疾患10例(34%),進行性疾患5例(17%)であった。1人の被験者はRECIST腫瘍評価のベースライン後スキャンを受けなかった。中位無進展生存(PFS)は現在6.5ケ月の最終結果であり、すでに薬物投与量中断の負の影響を受けて、潜在的な毒性を評価する可能性がある。2022年1月にデータを公表したところ,キュー4+Ib期患者の平均総生存期間は12.0カ月であり,現在最終的には14.6カ月であった。キュー2の2人の患者は、長期生存を証明している。1人は30.3ヶ月(最終データ)まで生き、1人は33.0ヶ月まで生きている。現在も7名の被験者が生存しており,そのうち1名は2番キューから,6名は4番キュープラスIbからである。
2022年1月、同社は新たな膵癌臨床試験の開始を発表した。この試験はASPIREと呼ばれ、ランダム、二重盲検、プラセボ対照試験であり、ゲムシタビンと標準膵臓癌治療方案NAB-パクリタキセルを併用し、以前転移性膵臓癌を治療したことがない患者に応用する。この実験は米国、ヨーロッパ、アジア太平洋地域の約93地点で行われる。
Aspire試験は2022年初めに開始され,米国や世界の他地域の臨床サイトの開放速度は当初の予想より遅く,一部の原因は医学界の資源疲労であるが,同社はすべての国とサイトが2024年初めに開放されると予想している。
この実験は最初にII/III期に設計され,PFSによる中期分析に必要なイベントと全体生存の主要な終点をサポートするためにサンプル量が小さい(150)。欧州やFDA規制フィードバックへの応答として,総試験サンプル量(600)と修正された設計を含むように修正され,総生存率を主な終点とし,中期分析で検査を行った。またPFSを分析し、追加の治療効果証拠を提供する。この改訂はIa/b期の一線転移性膵癌試験の最終データの支持を得ており,この試験は2020年12月に登録を完了した。この研究は600名の被験者を募集し,登録まで36カ月を要し,中期分析は2024年中に完了する予定である。独立データ安全監視委員会はすでに2回の会議を開催しており,最近では2023年11月に214名の患者の安全性を評価した。2回の会議では何の安全問題も生じず、裁判は継続され、何の修正もなかった。2024年1月25日、同社はこの試験が登録者数の50%を超えたと発表した。同社は、全面登録は2025年第1四半期に完了し、全体の生存状況に基づく中期データ分析は2024年中に提供すべきだと予想している。
FDAが推奨するすべての臨床研究を成功させることができれば,FDA,欧州医薬品局(EMA)(EU),厚生福祉部(日本),TGA(オーストラリア)の上場認可を求める予定である。アスピリンが地理的地域ごとに孤児薬に指定されている場合には,提出費用を免除することができる。
また、同社は2023年4月初め、米国癌研究協会年次総会で、卵巣癌ポリアミン代謝調節剤である伊夫スピミンの研究結果を重点的に紹介したポスタープレゼンテーションを発表した。ポスターは、アスピリンがVID 8+卵巣癌細胞を注射したC 57 BL/6マウスの治療が生存期間を著しく延長し、全体の腫瘍負担を低下させたと結論した。これらの結果は,エビスタチンが卵巣癌の臨床治療に機能している可能性を示唆しており,同社は卵巣癌の臨床前と臨床研究を継続しようとしている。
他の臨床前作業が行われており,伊夫司明(SBP−101とも呼ばれる)およびエフルオロオルニチン(CPP−1 XまたはDFMOとも呼ばれる)の多発性骨髄腫(細胞系)における役割が評価されている。2023年11月に補充発行された“血液”誌に発表されたデータは,プロポフォールとCPP−1 X抑制ポリアミンが骨髄腫細胞系の体外成長と活力に及ぼす影響を調べた。結果により、エスプミンとCPP-1 X処理は1組の多発性骨髄腫細胞系の細胞増殖を明らかに抑制し、そのアポトーシスを誘導した。アスピリンとCPP−1 Xを併用した場合,細胞成長はほぼ完全に消失した。これらの結果はエスプミンとCPP−1 Xの抗腫瘍潜在力を証明し,その臨床開発が潜在的に有望な多発性骨髄腫治療案に納得できる理由を提供した。この仕事は,同社がテキサス大学MDアンダーソン癌センターの研究者と協力し,ポリアミン代謝阻害剤療法とCAR−T細胞療法の組み合わせを臨床前モデルで評価していることを反映している。
FLYNPOVI
2009年12月、FDAは私たちの共同製品FlynpoviのIND申請を受け入れた。Flynpoviは、再発した結腸腺腫、特にハイリスク癌前ポリープを予防するために、NCIが支持する無作為·プラセボ対照のIIb/III期臨床試験で有望な結果を示した。この試験では、散発性腺腫を切除した375名の患者がエフルオロオルニチン(500 mg/d)+舒林酸(150 mg/d)の3年間の治療を受けた[N = 191])または一致するプラセボ/プラセボ(N=184)。その結果,プラセボと比較してプラセボと比較して能動併用療法は異時性腺腫発生リスク(70%),進行腺腫発生リスクは92%,多発性腺腫発生リスクは95%(Meyskensら)と有意に低下した。2008年)。この併用療法は全体的に耐性が良好であった。
散発性とFAP関連性腺腫性ポリープ症の発病機序が類似していること、及びFlynpoviが散発性腺腫とFAP患者の進行性ポリープの予防における作用機序を考慮して、FAPは第三段階計画を開始し、そして西南腫瘍グループ(SWOG)とNCIと協力して第三段階計画を開始し、結腸癌リスクの低下を研究した。
2019年に完成したFAP−310第3段階研究では,いずれかの薬剤単独と比較して,エフロンとスルリン酸の併用による家族性腺腫性ポリープ症を有する成人の治療効果と安全性を検討した(Burkeら)。2020)。患者は1:1:1の割合でランダムに分配し、エフルオロオルニチンと/またはスリン酸を毎日1回服用し、最長48ケ月間持続した。事件発生時間分析における評価の主要な終点は疾病進展であり、大手術、内視鏡下末期腺腫切除、直腸或いは直腸袋高度非典型的増殖の診断、或いは十二指腸疾患の進展と定義される。計171名の患者がランダムにグループ分けを受けた。エフロン−スルリン酸群56名中18名(32%)に疾患進展が認められ,舒林酸群58名中22名(38%),エフロン酸群57名中23名(40%)が悪化し,危険比は0.71(95%信頼区間)であった[語彙表]スリン酸と比較してエフロン−スリン酸は0.39~1.32(P=0.29)であり,エフロン酸と比較して0.66(95%CI,0.36~1.23)(Burkeら)であった。2020)。各治療群の副作用と深刻な副作用は類似している。1件後の分析では,併用群の患者は48カ月間LGI手術が必要に進展しなかったが,シュリン酸とエフロン群の患者はそれぞれ7名(13.2%)と8名(15.7%)であった(Balaguerら)。2022年)。これらのデータは,併用投与と単一投与と比較してスリン酸およびエフルオロオルニチンのHR=0.00(95%信頼区間,0.00−0.48;p=0.005)およびHR=0.00(95%信頼区間,0.00−0.44;p=0.003)の併用投与が単一投与と比較して胃腸損傷手術のリスクが100%近く低下していることを示している。LGIグループの統計学的意義を考慮して、FDAは秘密保持プロトコルを提出した。研究は主な終点に到達できなかったが,秘密保持プロトコルは探索的分析の結果に基づいているため,完全な返信を送信した.この欠陥問題を解決するためには,同社は臨床終点への影響を証明するために,1つまたは複数の十分かつ良好に制御された臨床試験の結果を提出しなければならない。
NCIとSWOGの協力のもと,結腸癌生存者としてFlynpoviが使用する治療法の利点を検討するための第3段階臨床試験が開始されている。この試験は“エフルオロオルニチンとシュリン酸による腺腫や癌予防”と命名された。PACE試験はNCIによって援助され、西南腫瘍学集団(SWOG)によって管理されている。Flynpoviは0−III期結腸癌または直腸癌患者の高危険腺腫と第二次原発結腸直腸癌再発の予防に用いられ,III期患者はエフルノニンとシュリン酸(“PACE”)を用いて結腸腺腫を予防するための二重盲検プラセボ対照試験である。この研究の目的は,Flynpovi(対応するプラセボと比較して)が1日3年間服用した後,対照ARMと比較して癌または高リスク腺腫の再発率が低下しているかどうかを評価することである。規制と商業目的のために、私たちは実験からのデータの独占的な権利を持っている。その会社は連合とアジアでのCATオプションを評価している。
2023年4月、同社は、CPPとOne-Two Treeutics Assets Limitedとの間の許可協定が2023年7月4日に発効したため、FAP患者においてFlynpoviを開発および商業化する北米権利を再取得したと発表した。
エフルオロオルニチン(CPP−1 X)とエフルオロオルニチン(CPP−1 X−S)
モノクロニチンについては,STK 11変異の非小細胞肺癌患者に対するI/II期試験と,最近発生したI型糖尿病とエフルオロオルニチンを併用したII期試験が開始され登録されている。最近、エフルオロオルニチンと大量のテストステロンとエンザルアミンの併用による転移性去勢抵抗前立腺癌を評価するII期試験が登録され始めた。最後に,エフルオロオルニチンによる胃癌予防を評価する第2段階試験は2021年に完了し,データ解析が行われている。
最新の発展動向
ナスダック従業員意見書
私たちは、ナスダック株式市場有限責任会社(“ナスダック”)から受け取った従業員決定書を、2023年11月28日にナスダック株式市場有限責任会社(“ナスダック”)から受け取った従業員決定書に上訴することを要求し、ナスダック公聴会グループに公聴会を開催し、ナスダックの公聴会に公聴会を開催することを要求し、NSE表を米国証券取引委員会に提出することで、ナスダックの上場および登録から私たちの普通株を除去することを要求し、ナスダック公聴会グループに公聴会を開催することを要求し、退市決定を控訴させることを決定した。同社の普通株の停止と退市は放置されており、聴聞手続きの終了を待っている。したがって、同社の普通株はナスダック資本市場に上場し続ける見通しで、少なくとも公聴会後に委員会が決定する前にそうだ。この委員会がナスダック資本市場に上場し続ける同社の呼びかけを承認する保証はない。決定書によると、当社(I)はナスダック上場規則第5550(A)(2)条に規定する1株1.00ドルの最低市価(“最低購入価格要求”)を維持しておらず、及び(Ii)ナスダック上場規則第5550(B)条(“最低株主権益要求”)に規定されている継続上場の最低株主権益要求(“最低株主権益要求”)に適合していない。2024年1月18日から、普通株流通株の20株1株の逆分割を完了した。私たちが逆株式分割を実施する主な理由は、最低入札要求を満たすために、私たちの普通株の1株当たりの市場価格を潜在的に高めることである。2024年1月22日、私たちはナスダックから通知を受け、逆株式分割の発効日まで、ナスダック上場規則第5550(A)(4)条(“最低持株量要求”)に適合しなくなり、当社の普通株退市の追加的な根拠として、保有株の最小500,000株を公開することを要求した。また,より多くの株式と今回公開された純収益を発行し,最低流通株要求と最低株主権益要求を再遵守できるようにする予定である.もし私たちがすべての適用されたナスダックの継続上場要求を再遵守できなければ、私たちの普通株はナスダック資本市場から撤退する可能性が高いと思います。
権利証執行権誘因とC類権証私募
2023年11月2日、私たちは既存の株式引受証のある所有者と株式承認証取引権誘因要書を締結して、私たちの普通株を購入し、それに基づいて、所有者は現金形式で既存の株主証明書を行使し、1株15.60ドルの換算権価格で私たちの普通株の合計106,500株を購入することに同意し、新しいC類普通株購入権証を発行することに同意して、合計213,000株の私たちの普通株を購入することに同意した。会社が既存の株式承認証の行使と新株式承認証の購入から得た総収益総額は約190万ドルである。新株式証の初歩的な行使価格は1株15.60ドルであり、株主が承認した日からナスダック上場規則に必要な任意の株主が承認した日から5年以内に行使することができる。我々の株主は2023年12月19日に開催された特別会議で普通株式の発行を承認した。私たちは、新権証の行使によって発行または発行可能な株式の再販売を含む登録声明を提出することに同意し、米国証券取引委員会は2023年12月20日にS-1表登録声明(文書番号333-275733)の発効を発表した。2024年1月23日現在、返済されていないC類権証は75,200件。
株を逆分割する
2024年1月18日から、20株の普通株式のうち1株の逆分割を完了した。本募集説明書に別段の規定がない限り、本募集説明書には歴史財務諸表及びその他の関連付記を除く他の株式及び1株当たりの資料は逆株式分割の効力を有する。
我々が逆株式分割を実施する主な目標は、最低入札価格要求を再遵守するために、我々の普通株の1株当たり取引価格を向上させることである。
わが社は過去2年間に逆株分割を実施し,累計比率は250株を超えているため,ナスダック市場規則第5810(C)(3)(A)条に規定されている任意のコンプライアンス治癒期を享受する資格はなく,ナスダックのスタッフは2023年11月に退市決定状を発表した。我々は引き続き我々の普通株の入札価格を積極的に監視し,利用可能オプションが最低入札価格要求に再適合することを考慮するつもりであるが,逆株分割が完了してもナスダックから撤退することは保証されない.
逆株式分割は、任意の所要日数で普通株の1株当たり入札価格を1株当たり1.00ドル以上の最低入札価格に維持し、最低入札価格要求を再遵守することを可能にすることが予想されるが、いかなる逆株式分割も将来このような効果が生じることは保証されないし、あるいは私たちの普通株が任意の特定の持続時間内に私たちの普通株をナスダック資本市場に上場することを維持することができるようになる。
どんな逆方向株式分割も期待された結果に達する保証はない。逆株分割後、私たちの普通株の1株当たりの価格は逆株式分割に伴って比例的に増加することも保証されず、会社の過去の逆株分割によって証明されたように、いかなる上昇幅もいつでも維持される保証はない。
権利証誘因とD類株式証の私募
2023年12月21日、私たちは既存の引受権証のある所有者と株式承認証取引権誘因要書を締結して、私たちの普通株を購入し、それに基づいて、所有者は現金形式で既存の引受権証を行使して、その既存の取引価格で1株15.60ドルで合計127,800株の私たちの普通株を購入することに同意し、それと引き換えに、新しいD類普通株購入権証の発行に同意し、合計255,600株の私たちの普通株を購入することに同意した。同社が既存の株式承認証の行使から得た総収益総額は約200万ドル。新株式証の初歩的な行使価格は1株19.00ドルで、ナスダック上場規則に規定されている株主の承認を得た後にのみ行使でき、株主の承認日から5年(あれば)まで行使することができる。もし会社が将来的に普通株等価物と転換可能または派生証券に基づいていくつかの希釈性普通株を発行する場合、任意の中間の逆株分割と株主の承認を受けた後、行使価格は別途低下する。2024年1月23日現在、すべての新株式証はまだ返済されていないが、行使できず、株主の承認を待っている。我々は、株主が6ヶ月以内に新株式証に関連する普通株の発行を許可し、2023年12月21日から60カレンダー日以内に新規株式証関連株の転売をカバーする登録声明を提出することを求める会議を開催することに同意した。また、当社は1年以内にいかなる変動金利取引(定義勧誘メール)も行わないことに同意していますが、6ヶ月後に発効した市場発売は除外します。
製品開発
2024年1月23日までに私たちは
|
● |
アスピリンの孤児薬名を米国食品·薬物管理局から得た |
|
● |
INDのエビスタット申請に対するFDAの承認を提出し、受信する |
|
● |
オーストラリア、フランス、イタリア、スペインでASPIRE試験の国家承認を受けた |
|
● |
転移性膵管腺癌患者のIa期単剤治療の安全性研究を完成する |
|
● |
転移性膵臓癌の治療によりFDAの“快速チャンネル”の称号を獲得した |
|
● |
私たちの2つ目の試験では、以前転移疾患で治療を受けていなかった膵管腺癌患者に対する第一線の研究であるIa/Ib期の臨床研究である中期結果を登録し、50人の被験者が参加し、そのうち25人がIa期、25人がIb期または拡張期であった |
|
● |
ジョンホプキンス医学院と2年間の研究協定を締結し、国際公認のポリアミン生物学研究員ロバート·カセロ教授が指導した |
|
● |
完成したプロセス改善措置は、商業用途に使用可能であることが予想され、このような新しいより短いアスピリン合成法に関する特許の問題通知を受けた |
|
● |
ランダム、二重盲検、プラセボ対照研究を開始し、以前転移治療を受けていなかった膵管腺癌患者の中で、ゲムシタビンとNAB-パクリタキセルを連合投与した |
|
● |
完成した臨床前評価:術前に膵臓癌を切除可能な新しい補助治療のためのエビスタチン; |
|
● |
卵巣癌腫瘍成長抑制活性の早期、臨床前適応を獲得し、ASCO-GI会議で結果を公表した |
|
● |
USANがSBP−101に対して採用した非特許名伊文司普明を受信した |
|
● |
CPPを買収·統合し、多様な形で第2のリード資産を増加させ、臨床前から登録レベルまでの臨床試験の拡張臨床開発計画 |
|
● |
欧州医薬品局(EMA)孤児薬品委員会はPanbelaが孤児に指定を申請したアスピリンとゲムシタビンとNAB−パクリタキセルを併用した転移性膵管腺癌患者の治療に積極的な意見を発表した |
|
● |
インディアナ大学によるエフルオロオルニチンによる早発性I型糖尿病治療の第2段階計画の開始を発表した |
|
● |
モフェット癌センターによるSTK 11変異を有する非小肺癌(NSCLC)のI/II期臨床試験の開始を発表した |
|
● |
テキサス大学MDアンダーソン癌センターと協賛研究協定を締結し、臨床前モデルにおいてポリアミン代謝阻害剤療法とCAR-T細胞療法の結合を評価した |
|
● |
SWOG癌研究ネットワークのPACE S 0820の第3段階試験が単一計画の無効性分析に合格し、継続することを発表した |
|
● |
米国WorldMeds NDAが児童神経芽細胞腫へのエフルオロオルニチン(DFMO)の許可を許可し、腫瘍学へのポリアミンの使用を初めて許可したことを発表した |
|
● |
ASPIRE全世界の臨床試験に参加した人数は50%を超えた。 |
わが社にかかわるリスク
当社のビジネスは、本願明細書の要約後に“リスク要因”と題する章でより全面的に説明されているように、多くの重大なリスクに直面しています。私たちの証券に投資するかどうかを決定する前に、これらのリスク、および“リスク要因”の節に記載されているリスクおよび本募集説明書の他のすべての情報を読んでよく考慮すべきであり、本募集説明書の他の部分に含まれる財務諸表および関連説明を含む。募集説明書で議論されている任意のリスクが実際に発生すれば、我々の業務、財務状況または経営業績は実質的な悪影響を受ける可能性がある。特に、私たちのリスクは以下の点を含むが、これらに限定されない
|
● |
私たちは受け入れ可能な条件や根本的に不要な方法で追加資本を獲得して、私たちの業務計画を実施することができます |
|
● |
私たちは多様性とそれに応じたリスクが足りず、私たちの会社に投資しています |
|
● |
全国的な証券取引所で上場する能力を維持しています |
|
● |
我々のランダムII/III期臨床試験の進展と成功 |
|
● |
我々の候補製品の安全性と有効性を証明することができます:プロポフォール(SBP-101)、フルオロボイル、エフルオロオルニチン(CPP-1 X); |
|
● |
私たちの候補製品SBP-101、Flynpovi、およびCPP-1 Xは、アメリカ、EU、または他の国際市場で規制承認を受ける能力; |
|
● |
私たちの候補製品SBP-101、Flynpovi、およびCPP-1 Xの市場受容度および将来の販売レベル; |
|
● |
我々の候補製品SBP-101、Flynpovi、およびCPP-1 Xに適用される規制の変化は、製品開発コストおよび遅延をもたらす可能性がある |
|
● |
第三者支払者との返済計画の進展速度 |
|
● |
競争の技術と市場発展の影響 |
|
● |
特許出願の提出及び起訴並びに特許請求の執行又は弁護に係る費用; |
|
● |
その他のリスク要因は、本募集説明書の9ページ目からの“リスク要因”というタイトルに含まれている |
小さな報告会社としての影響
我々は、取引法第12 b-2条で定義された“より小さい報告会社”であり、より小さい報告会社が入手可能ないくつかの規模の開示を利用することを選択した。
企業の歴史
Panbela治療会社の主な業務は,最初にデラウェア州の法律に基づいて“Sun BioPharma,Inc.”という名称で成立した。2011年9月。2015年、同社は当時ユタ州の法律に基づいて設立された上場会社の完全子会社との合併取引を完了し、上場企業となった。2016年には、私たちの運営子会社と合併することで、デラウェア州の法律に基づいて再登録されました。同社は“Panbela治療会社”と改称した。2020年12月2日。2022年6月15日、私たちはPanbela Treeutics,Inc.の後継者となり、取引法が公布した第12 G-3(A)規則に基づいて合併による持株会社の再編を行い、その名称を採用し、現在の構造-Panbela Research,Inc.と癌予防製薬会社の2つの完全子会社からなる。
企業情報
わが社の郵送先はミネソタ州ワコニア、ウィスタ通り712 Vista Blvd、#305、郵便番号:55387です。私たちの電話番号は(952)479-1196で、私たちのサイトはwww.panbela.comです。私たちのサイトの情報は本募集説明書の一部ではありません。我々はすでに我々のサイトアドレスを事実として参考にしており,我々のサイトへのアクティブなリンクとするつもりはない.本募集説明書に記載されている又は当社のウェブサイトに関連する資料は、参考方式で本募集説明書に組み込まれておらず、本募集説明書の一部とみなされるべきでもない。本募集明細書に登場する他社の商号、商標及びサービスマークは、それぞれ所有者の財産である。
供物
|
私たちが提供する普通株は |
予備融資権証を行使する際に発行可能な普通株と、最大16,660,000株が一般権証を行使する際に発行可能な追加普通株を含む8,000,000株の私たちの普通株。 |
|
私たちが提供した一般的な権利証は |
E類普通株式承認株式証は最大2,000,000株の普通株(あるいは2,660,000株の非現金行使普通株)を購入することができ、発行日から発行日から5年以内に行使することができ、行使価格は1株当たり普通株(1株当たり公開発行価格及び普通権証の100%)である。5営業日後、E類株式証の所有者は、現金行使時に発行可能な普通株式数の1.33倍に相当する株式総数を取得する“キャッシュレス行使の代わり”を選択するための単独の選択権を有することになる。
F類普通株式承認株式証は最大14,000,000株の普通株を購入し、発行日から発行日から5年までの期間、1株当たりの普通株行使価格$(1株当たり公開発行価格及び普通権証の100%)で行使することができる。 |
|
私たちが提供した前払い援助権証は |
また、今回の発売で私たちの普通株を購入することにより、購入者とその関連会社が今回の発売完了直後に4.99%(または購入者選択時、9.99%)を超える実益を持つことになり、事前に出資した引受権証(普通権証とともに“株式承認証”)を購入する機会があり、そうでなければ、いずれの購入者の実益所有権が4.99%を超えることになる(または購入者が選択した場合、9.99%)私たちの普通株式流通株。事前出資の株式承認証は普通株に対して行使することができるだろう。1部当たりの事前資本権証と付随する一般権証の購入価格は、今回の発行で一般株と付随する普通株を公衆に売却する価格から0.001ドルを引いたものとなり、事前資金権証1部あたりの行使価格は1株0.001ドルとなる。前払い資金株式承認証は直ちに行使可能となり、すべての行使まで随時行使することができる。私たちが販売している事前融資の引受権証ごとに、私たちが提供する普通株式数は1対1に基づいて減少するだろう。一般権証を発行し、普通株1株当たり2株普通株を購入し、今回発行中に売却された1株当たり予備資本権証1株当たり2株普通株を購入するため、今回発行中に販売される普通株数は、我々が販売する普通株と予備資本権証の株式の組み合わせによって変化することはない。 |
|
公開発行価格を仮定する |
普通株および普通権証付き1株3.77ドル、あるいは1株当たりの事前資金権証と普通権証付き3.769ドル(場合によっては)は、公開発行価格はナスダック資本市場が2024年1月23日に発表した普通株の前回売却価格3.77ドルに等しいと仮定する。 |
|
今回の発行前に発行された普通株 |
480,244株 |
|
普通株式は今回の発行後すぐに発行されます |
8,480,244株(われわれは普通株のみを売却し、事前融資権証がなく、かつ今回発行中に発行された一般権証はいずれも行使されていないと仮定する)。 |
|
収益の使用 |
配給代理費と我々が支払うべき推定発売費用を差し引いた後、普通株とセット普通権証の仮定公開発行価格は1株3.77ドルで、今回発売された純収益は約2,780万ドルに達すると予想される。今回発行された純収益を我々の候補製品であるエフアスピリンやエフルオロオルニチンの持続臨床開発,運営資金や他の一般会社用途に利用する予定であり,債務返済が含まれている可能性がある。最善を尽くした発売であり、最低金額が成約条件となっていないため、ここで発売されたすべての証券やいかなる証券も販売しない可能性がある。したがって、私たちが受け取った純収益は私たちが現在推定しているよりはるかに少ないかもしれない。21ページ“収益の使用”を参照。 |
|
リスク要因 |
あなたは、私たちの証券への投資を決定する前によく考慮すべき要素を検討するために、10ページ目から本募集説明書の“リスク要因”の部分を読むべきです。 |
|
ナスダック資本市場取引コード |
“PBLA” |
今回の発行前後で発行された普通株の数は、2024年1月23日現在発行された普通株の推定480,244株に基づいており、含まれていない
|
● |
今回の発行で販売された引受権証の行使後に発行可能なすべての株; |
|
| ● | 2024年1月18日に施行された逆株式分割に関する断片的な株式現金決済が未解決のままである | |
|
● |
本募集説明書の発行日までに、発行済み株式オプションを発行した普通株599株を加重平均発行価格で1株13,297.06ドルで行使することができる |
|
|
● |
340,952株の普通株は、加重平均発行価格で1株64.09ドルで株式引受権証発行を行使することができる。 |
別の説明がない限り、本入札明細書中のすべての情報は、行使されていないオプションまたは株式承認証を仮定しない
リスク要因
私たちの証券へのどんな投資にも高いリスクがある。投資家は、我々の証券を購入するか否かを決定する前に、以下に説明するリスク及び本入札明細書に含まれるすべての情報をよく考慮しなければならない。これらのリスクのいずれかが実際に発生した場合、私たちの業務、財務状況、または運営結果は重大な悪影響を受ける可能性がある。本入札明細書にはまた、リスクおよび不確定要因に関する前向きな陳述が含まれている。いくつかの要因の影響により、私たちの実際の結果は、以下に説明するように、本明細書の他の部分を含む、これらの前向き陳述で予想される結果と大きく異なる可能性がある。
私たちの業務や財務状況に関連するリスク
私たちは営業前の会社で、キャッシュフローを責任を持って運営していた歴史があります。
設立以来,我々の経営活動のキャッシュフローは負であったが,これは主に我々の主要候補薬物の商業化に必要な投資によるものであった。株式証券の売却と本票発行の収益により、我々の融資キャッシュフローは歴史的に正視されてきた。2022年12月31日と2021年12月31日までに,我々が経営活動に使用した現金純額はそれぞれ1,530万ドルと720万ドルであり,2022年12月31日現在,我々の運営資本はマイナス600万ドルであり,2021年12月31日現在,我々の運営資本は正960万ドルである。2023年9月30日までの四半期に、約90万ドルの現金と約700万ドルの負の運営資本があります。運営資本の定義は流動資産から流動負債を差し引くことである。
私たちの業務は新製品開発過程でよく遭遇するすべてのリスク、困難、合併症、遅延の影響を受け、私たちが競争に参加する製薬とバイオテクノロジー業界特有のリスクを受ける。投資家は新製品、サービスと技術開発市場の会社のためによく遭遇する遅延、費用、問題と不確定要素を考慮して、私たちに対して評価を行うべきである。私たちはこのような障害物を永遠に克服できないかもしれない。
私たちの現在の財務的流動性が限られているため、私たちの監査人は私たちに引き続き“経営を続ける企業。”
私たちの現在の財務流動性が限られているため、私たちの監査人が2022年の財務諸表のために作成した報告書には、私たちが“持続的な経営企業”として経営を継続する能力に関する声明が含まれている。私たちの限られた流動資金は、私たちが受け入れられる条項に従って追加融資を獲得したり、戦略関係を構築したりすることを難しくし、私たちが獲得する可能性のある任意の融資の条項と私たちの公開株価に重大で不利な影響を与えるかもしれない。
我々の“持続経営企業”としての継続経営は,運営から正のキャッシュフローを獲得し,必要に応じて外部資源を利用してキャッシュフローを増加させ,我々の現金需要を満たすことに依存している。私たちが正のキャッシュフローを達成する計画は主に証券の発行を含む。他の潜在的な資金源には、私たちの現在および潜在的な将来の候補製品の事前支払いおよびマイルストーン支払いの交渉、または私たちの製品を販売することによって規制された承認された特許権使用料、およびそのような承認された製品に関連する任意のマイルストーン支払いが含まれる。このような現金源は資金調達や他の戦略的合意によって補完されるかもしれない。しかし、私たちはこれらの目標を達成できないかもしれないし、商業的に合理的な条件で必要な資金を得ることができないかもしれないし、これらの目標を実現できないかもしれないので、経営を続けることができないかもしれません。
私たちは事業計画を実行するために必要な追加資本を得ることができないかもしれないが、これは私たちの成長能力を制限するかもしれない。
私たちの既存資本および他の既存資源は、限られた運営資本を提供するのに十分であり、私たちが予想している持続可能な機会に資金を提供するのに十分ではないだろう。2023年第3四半期末の資本および2023年9月30日以降に調達した資金は、2024年第1四半期の運営に資金を提供するのに十分である。私たちは私たちの業務を運営し続け、私たちの臨床開発計画を完成させるために追加の資金が必要になるだろう。
将来の研究開発は、臨床試験コスト、資本支出と可能な買収、そして私たちの行政要求、例えば給料、保険費用と一般管理費用、そして法律コンプライアンスコストと会計費用を含み、大量の追加資本とキャッシュフローが必要になる。私たちが商業的に合理的な条項で必要な追加資本を調達し、私たちの持続的な業務に資金を提供することができるか、あるいは根本的にできないという保証はない。
私たちは、協力手配、債務融資、株式融資、または他の方法を含めて、様々な融資取引や手配を通じて、追加資金源を求めるつもりだ。私たちは商業的に合理的な条項、必要な時間内、あるいは適切な融資取引を見つけることができないかもしれないし、他の方法で私たちに必要な資本を得ることができないかもしれない。もし私たちがもっと多くの資本を集めることに成功できなければ、私たちの資源は私たちの未来の行動が資金を提供すると思って足りないだろう。
株式を売却することで調達されたいかなる追加資本も、私たちの株主の所有権比率を希釈することができる。これはまた、私たちの資産がより大きな未償還株式プールによって所有されるので、私たちの株式証券の公平な市場価値を低下させる可能性がある。私たちが将来の資本取引で証券を発行する条項は、私たちの新しい投資家により有利になる可能性があり、割引、より高い投票権、および発行権、またはさらなる希釈効果を有する可能性のある他の派生証券を含むかもしれない。
私たちが必要な資金を得る能力は、資本市場、特に製薬と他の薬物開発業界の資本市場、私たちの活動の限られた多様性および/またはキーパーソンの流失などの要素の影響を受ける可能性がある。もし私たちが融資活動から集めた資金が私たちの資本需要を満たすのに十分でなければ、事業を減らしても、業務を停止することを要求されるかもしれません。
私たちは将来の資本融資を求める際に大量のコストが発生する可能性があり、投資銀行費、弁護士費、会課金、証券法適合費、印刷、流通費用、その他のコストを含み、これは私たちの財務状況に悪影響を及ぼす可能性がある。
私たちの候補製品の市場競争は激しく、迅速な科学的変化の影響を受けており、これは私たちの業務、運営結果、財務状況に実質的な悪影響を及ぼす可能性がある。
私たちが競争する製薬とバイオテクノロジー業界は競争が激しく、迅速で重大な技術変革を特徴としている。製薬やバイオテクノロジー会社や学術や研究機関,政府機関などからの激しい競争に直面している。その中のいくつかの組織は私たちの技術と似たような技術に基づく製品を追求している。これらの組織の他の組織は、製品を開発し、マーケティングしているか、またはこれらの競合製品が私たちの候補製品に対する疾患の治療効果において私たちの候補製品と競争力を有する製品を生産することを目的としている他の技術的方法を求めている。私たちの競争相手は、私たちが開発する可能性のある任意の製品や新技術よりも効率的で、より安全またはコストの低い製品または新技術を発見、開発、または商業化するかもしれない。私たちの競争相手はまた私たちが候補製品の承認を得るよりも早くFDAや他の規制機関の承認を得るかもしれない。
私たちの多くの競争相手は私たちよりずっと大きくて、私たちよりも多くの資本資源、研究開発者、そして施設を持っています。また、私たちの多くの競争相手は薬物の発見、開発と商業化、監督管理の承認及び薬物製造とマーケティングの方面でもっと経験がある。
私たちは、私たちの候補製品と技術との競争は、製品効率、安全性、可用性、価格を含む一連の要素に基づくと予想される。私たちが計画している将来の候補製品と競争製品の発売タイミングは製品間の競争にも影響を与えるだろう。必要であれば,我々の候補製品を開発し,必要な臨床試験を完了し,戦略的パートナーを構築し,後期試験に適切な数の候補製品の相対速度を提供することが重要な競争要因となると予想される。私たちの競争的地位はまた、私たちが現在持っていない非米国市場で特許保護を受ける能力があるかどうか、または他の方法で独自製品またはプロセスを開発し、技術構想から商業販売まで、または製薬パートナーへの許可証の発行中に十分な資本資源を有することを保証する能力があるかどうかにかかっている。もし私たちが提案された候補製品を成功的でタイムリーな方法で開発して配置できなければ、私たちは競争力がない可能性が高い。
私たちの多様性の不足はわが社への投資リスクを増加させ、私たちが多元化しなければ、私たちの財務状況と運営結果は悪化する可能性がある。
私たちの取締役会は私たちの薬物開発活動に集中しています。これらの活動は現在限られた数の候補製品に集中しています。私たちが私たちの投資を多様化できるかどうかは、私たちがより多くの資本と資金源を得ることができるかどうか、そして適切な機会を得ることができるかどうかにかかっている。
規模の大きい会社は多様な経営を通じてリスクを管理する能力がある。しかし、私たちの業務の性質と地理的範囲については、私たちは不足しており、多様性が不足し続けると予想される。したがって、私たちは、私たちの業務がより多様化し、私たちのリスク状況を向上させるために、私たちの競争に影響を与える製薬やバイオテクノロジー産業に影響を与える要素の影響を受けるかもしれない。もし私たちが私たちの業務を多様化できなければ、私たちの財務状況と運営結果は悪化するかもしれない。
もし私たちが人材を誘致して維持しなければ、私たちの業務は影響を受けるかもしれない。
私たちの成功は、私たちの経営陣や他の人たちの業務を展開する際の能力、専門知識、判断力、判断力、誠実さ、誠意に大きく依存します。私たちの管理チームは規模が小さく、キーパーソンを失ったり、適切な適格社員を引き付けることができず、私たちの業務に実質的な悪影響を及ぼす可能性があります。
私たちの成功は、適切な投資機会を見つけ、採用し、そのような投資を監視し、最終的に必要な時にそのような投資を剥離することに成功するために、私たちの経営陣、従業員、コンサルタント、戦略パートナー(できれば)が市場データを正確に解読し、対応することにかかっている。また、私たちの主要な人員が引き続き私たちに連絡したり雇用したりする保証はなく、似たような技能を持つ代替者を見つけることも保証されない。私たちは管理職とすべての重要な職員たちが適切に補償されることを確実にするために努力するつもりだが、彼らのサービスは保証されない。もし私たちが重要な人材を引きつけて維持できなければ、私たちの業務は不利な影響を受けるかもしれない。
私たちは訴訟の弁護や製品責任クレームのための損害賠償金を支払うように要求されるかもしれない。
製品責任はバイオテクノロジーと医薬製品テストとマーケティングの大きなリスクだ。人体臨床試験と監督管理許可後の製品販売において、著者らは大量の製品責任の開放に直面する可能性がある。製品責任クレームは、その是非曲直にかかわらず、保険制限を超え、経営陣の注意を移し、私たちの名声と私たちの製品の需要に悪影響を及ぼす可能性がある。このような場合でも、あなたの私たちの証券への投資は実質的な悪影響を受ける可能性があります。
新薬開発と承認に関するリスク
我々の候補製品に必要な臨床試験は高価で時間がかかり,その結果も非常に不確実である。もし私たちの任意の薬物試験が延期されたり、不利な結果が生じたりすれば、私たちは規制部門の候補製品の承認を得ることができないかもしれない。
私たちはすべての候補製品に対して広範なテストをしなければならないし、それから監督部門の承認を得て、それを市場に出して販売することができる。私たちは前臨床動物試験と人体臨床試験を行う必要がある。このような実験を行うことは長く、時間がかかり、費用がかかる過程だ。これらのテストと試験は良好な結果が得られない可能性があり、原因は非常に多く、その中には候補製品が安全性或いは有効性を証明できなかったこと、候補製品に接触することによる深刻な或いは生命に危害を及ぼす有害事象或いは副作用が発生したこと、臨床試験中に被験者を登録と維持することの困難、候補製品或いは対照薬物の供給不足、及び臨床研究者、試験監督者、請負業者、コンサルタント又は試験被験者が試験方案を遵守できなかったことを含む。測定された終点を検出するのに十分な数の患者が含まれていないため、臨床試験は失敗する可能性がある。臨床試験も失敗する可能性があり,試験に含まれる研究薬の投与量(S)が低すぎるか高すぎるため,疾患背景における薬物の最適効果を決定することはできない。多くの臨床試験は独立データモニタリング委員会(“IDMC”)の監督下で行われ、これらの独立監督機構は外部の専門家から構成され、彼らは行っている臨床試験の進展状況を審査し、現有の安全性と有効性データを含み、そして一時的、非盲目的なデータに基づいて試験の継続、修正或いは中止について提案を提出する。我々が行っているどの臨床試験も中断または修正可能であり,担当IDMCがこのような中期試験結果の審査に基づいて提案した提案に応答する。
もし私たちの候補製品のテストが良くなければ、私たちはそれらを再評価するか、高価で時間のかかる新しい試験を行うか、私たちの薬物開発計画を放棄する必要があります。われわれが前臨床試験や臨床試験から積極的な結果を得ても,将来の試験で同様の成功は得られない可能性がある。バイオ製薬業界の多くの会社は臨床試験で大きな挫折を経験し,早期の試験でも奮い立つ結果を得た。臨床試験は必要な適応の安全性と有効性を証明できず、私たちの候補製品の発展を損なう可能性があり、私たちの業務、財務状況、運営結果は実質的な損害を受ける可能性がある。
私たちは製品候補開発で重大な危険に直面している。
私たちの業務は私たちの候補製品の成功的な開発と商業化に依存する。私たちは現在、PDAを治療する初期候補製品エビ司普明の開発に集中しており、FDAまたは任意の外国司法管轄区のNDAの承認を得るまで、これらの司法管轄区の必要な承認を得るまで、米国での販売は許可されていません。新薬および/または治療製品を開発するプロセスは、本質的に複雑で予測不可能であり、時間的、高価かつ不確実である。私たちの開発計画が薬物が規制部門の承認を得て市場に受け入れられるかどうかを知るためには、長期投資を行い、大量の資源を投入しなければならない。一つは,すべての開発段階で有望に見える候補製品が出荷できない可能性があり,原因が多く,臨床計画の結果やデータからは予測できない可能性がある。候補製品は臨床試験中に無効或いは有害な副作用を招く可能性が発見される可能性があり、予想よりも長い臨床試験の進展時間を必要とする可能性があり、所定の臨床終点を実現できない可能性があり、すでに臨床利益を得ている可能性があっても、必要な監督管理の承認を得ることができない可能性があり、合理的なコストと許容可能な品質で商業量産を行うことは不可能であることが証明される可能性があり、あるいは市場の受け入れを得ることができない可能性がある。
規制部門の承認を得て私たちの候補製品を商業化するかどうかは予測できませんので、その製品や他の候補製品の将来の任意の収入時間(あれば)を予測することはできません。FDAは薬品の審査過程においてかなりの自由裁量権を持っており、多種の原因で承認候補製品を延期、制限、或いは拒否する権利があることを含む。例えばFDA:
|
● |
私たちが提供した情報が不十分で、臨床的欠陥を含むか、または私たちの任意の候補製品を証明できなかった任意の適応の安全性および有効性を決定することができる |
|
● |
臨床試験からのデータは、我々の候補製品の臨床的および他の利益がその安全リスクを超える任意の発見を含む、秘密協定の提出または米国での上場承認を支持するのに十分ではないと考えられるかもしれない |
|
● |
私たちの試験設計や臨床前研究または臨床試験データの解釈に同意しないかもしれないし、それが私たちの試験設計を審査してレビューした後であっても、承認要求を変更する可能性があります |
|
● |
私たちが候補製品を製造する契約を締結した第三者製造業者の製造プロセスまたは施設の欠陥を見つけることができる |
|
● |
私たちの候補製品が私たちが要求しているよりも少ないまたは限られた適応を承認するかもしれないし、高価な承認後の臨床試験の表現によって承認されるかもしれない |
|
● |
その承認政策を変更したり、新しい規定を採用したりすることができる |
|
● |
私たちが候補製品の商業化に成功するために必要または望ましいと思うラベル宣言を承認しないかもしれない。 |
もし私たちが開発した初期候補製品または未来の候補製品が規制部門の承認を得られなければ、私たちが収入を創出する能力を大きく制限し、適切なすべての適応とラベル宣言がこのような承認を得られなければ、私たちの潜在収入を減少させる可能性があると考えられる。
私たちの候補製品は、癌の治療のために許可されていない既存技術の新しい処方に基づいており、したがって、固有のリスクがある。私たちの候補製品の安全性と有効性に対する懸念は私たちの未来の成功を制限するかもしれない。
新技術に基づく候補製品の開発では、我々は固有の失敗リスクに直面している。これらのリスクは、私たちが作成したどの候補製品も無効になる可能性があり、私たちの現在の候補製品は安全ではない、無効であるか、必要な規制承認を得ることができないか、あるいは私たちの候補製品は大規模な生産が困難になるか、市場に割に合わないだろう。
多くの薬品は多種の潜在的な合併症と副作用を招き、すべてのものが正確に予測できるわけではなく、しかも多くの副作用は患者によって異なる可能性がある。長期的な追跡データは、私たちの候補製品に関連する追加的な合併症を明らかにするかもしれない。潜在医師や他の人の合併症に関する情報に対する反応は,市場の我々候補製品に対する受容度に実質的に影響する可能性があり,逆に我々の業務に実質的な損害を与える可能性がある。
著者らは第三者に依存して臨床試験を行うため、著者らは臨床試験の時間、進行、費用と品質を直接制御することができず、これは著者らの臨床データと結果及び関連する監督管理の承認に不利な影響を与える可能性がある。
われわれはわれわれの臨床試験活動を広く外注し,計画試験準備段階のごく一部のみを直接実行する予定である。著者らは独立した第三者CROに依存して、文書準備、場所識別、スクリーニングと準備、研究前訪問、訓練、プロジェクト管理と生物分析を含む著者らの大部分の臨床試験を実行する。CROが私たちに提供してくれた多くの重要なサービスは私たちが直接統制しているものではない。もし私たちとCROの関係に何か論争や中断があれば、私たちの臨床試験は延期されるかもしれない。また,我々が提出した規制文書では,第三者CROが行っている臨床作業の質と有効性に依存している。CROの流れ、方法、または結果が無効または不十分であると判定された場合、私たち自身の臨床データおよび結果、ならびに関連する規制承認は悪影響または無効を受ける可能性がある。
私たちは第三者サプライヤーと他の第三者に依存して私たちの候補製品を生産します。これらの第三者への依存は私たちの研究開発計画の進歩と私たちの候補製品の開発を損なう可能性があります。
我々は,臨床前研究や臨床試験に必要な原材料や薬品供給を第三者に依存して提供し続ける予定である。2021年の間,同社は我々の製造パートナーと協力し,新たなより短く,より安価な活性医薬物質の合成を確認した。しかし、第三者の生産遅延は、私たちの臨床試験を延期したり、任意の商業活動に悪影響を及ぼす可能性があります。また、私たちは第三者に依存して私たちの候補製品を製造し、調合することは、私たちが製品に製造欠陥が存在する可能性があるリスクに直面していることを意味し、私たちはこれらの欠陥を予防または制御する能力が限られている。私たちはこれらの活動を監視して、私たちの品質基準、予算、スケジュールに適合することを保証しているにもかかわらず、私たちは私たちの候補製品を生産するのではなく、私たちの候補製品の製造をもっと少なく制御し続けるつもりだ。また、私たちと付き合っている第三者は、人員配置が困難で、優先順位が変化したり、財務的苦境に陥ったりする可能性があり、これは私たちの候補製品の製造と生産に悪影響を及ぼす可能性があります。
臨床前研究と完成した臨床試験の結果が必ずしも将来の結果を予測できるとは限らず,我々の現在の候補製品は今後の研究や試験で有利な結果が得られない可能性がある。
臨床前研究と第一段階の臨床試験は主に普通の人群における候補製品の治療効果をテストするためではなく、初歩的な安全性をテストし、薬物動態学と薬効学を研究し、疾病群の中の少数の研究患者の限られた治療効果を研究し、及び候補製品の異なる用量と用量計画下の副作用を識別と理解することを試みた。臨床前研究或いは完成した臨床試験の成功は今後の研究或いは試験を確保することができず、持続的な臨床前研究と大規模な臨床試験が成功することを含み、必ずしも未来の結果を予測できるとは限らない。早期研究または試験における有利な結果は、後の研究または試験で重複しない可能性があるが、後期試験における候補製品は、早期試験に合格したにもかかわらず、許容可能な安全性および有効性を示すことができない可能性がある。
私たちの業務規制に関するリスク
連邦と州薬品マーケティングのコンプライアンスと報告要求は、私たちを州政府または他の政府当局の規制と法律行動に直面させるかもしれない。
食品と薬物管理局現代化法案(“FDMA”)は公開された臨床試験の公開登録を確立し、深刻或いは生命に危害を及ぼす疾病或いは疾病を治療することを目的とした薬物に関連し、公衆のこれらの臨床試験に対する認識を高め、これらの臨床試験の機会を獲得する。FDMAにより,医薬品メーカーや他の試験スポンサーは,これらの試験の一般的な目的,および試験の資格基準,場所,連絡情報を公表しなければならない。いかなる臨床試験の掲示要求を守らなければ、私たちはマイナスの宣伝、罰金とその他の処罰に直面する可能性があり、これらはすべて私たちの業務に実質的な損害を与える可能性がある。
近年、カリフォルニア州、バーモント州、メイン州、ミネソタ州、ニューメキシコ州、ウェストバージニア州を含むいくつかの州は立法を公布し、製薬会社にマーケティングコンプライアンス計画を確立し、販売、マーケティング、定価とその他の活動に関する報告を定期的に提出することを要求している。他の州も似たような立法を考慮している。その中の多くの要求事項は新しくて不確実であり、利用可能な指針は限られている。私たちがこのような法律を完全に遵守しない限り、私たちは法執行行動、罰金、その他の処罰に直面し、否定的な宣伝を受ける可能性があり、これらはすべて私たちの業務を損なう可能性がある。
もし私たちが開発したすべての候補製品が不利な価格設定法規、第三者精算やり方、あるいは医療改革措置の制約を受けたら、私たちの候補製品を商業化することに成功する能力は損なわれる可能性がある。
私たちの将来の収入、収益力、資本獲得の機会は、政府や個人第三者支払人が様々な手段で医療コストをコントロールしたり、低減したりする持続的な努力の影響を受けるだろう。私たちはいくつかの連邦、州、外国の提案が政府規制を通じて薬品コストを抑えることを予想している。最近の医療改革立法が私たちの業務に影響を与える可能性があるかどうかは定かではありませんし、連邦、州、外国、個人支払者が最近の改革に対応するためにどのような行動をとるかもしれません。したがって、実施されたどんな改革が私たちの業務に与える影響を予測することは難しい。私たちが成功に私たちの候補製品を商業化できるかどうかは、ある程度政府衛生行政当局(例えばアメリカのMedicareとMedicaid)、個人健康保険会社と他の組織によるこのような候補製品と関連治療費用の精算程度に依存する。新たに承認された保健製品の精算状況には大きな不確実性があり,特に現在有効な治療がないか,あるいは通常医療看護の適応が求められていない。製品研究開発への投資の適切なリターンを実現するために、十分な第三者保険がないかもしれません。もし政府と第三者支払者が私たちの候補製品を使用するために十分な保険と精算レベルを提供しなければ、私たちの候補製品は市場の承認を得られない可能性があり、私たちの運営結果は損害を受けるだろう。
医療立法改革措置は私たちの業務と運営結果に実質的な悪影響を及ぼす可能性がある。
政府の処方薬調達や精算計画に影響を与える立法や規制行動が頻繁に発生している。米国では,ACAは2010年に公布され,医療保険のカバー範囲を拡大している。その時以来、人々は腐敗防止法の全部または一部を廃止、改正、または行政制限するために多くの努力をしてきた。例えば、トランプ総裁が2017年に法律となった減税·雇用法案に署名して個人医療保険認可を廃止したことは、ACAの重要な構成要素と考えられている。2018年12月、テキサス州の連邦地域裁判所は、この判決が保留されて控訴を待っていたにもかかわらず、個人医療保険のライセンス違憲を理由にACAを覆した。ACAが直面している持続的な挑戦と新たな立法提案はACAの将来の生存能力と医療保険市場の不安定な不確実性を招いた。これによる私たちの業務への影響は不確実であり、実質的かもしれない。
処方薬の価格を統制する努力はまた私たちの業務に実質的な悪影響を及ぼす可能性がある。例えば、2018年、トランプ総裁と米国衛生·公衆サービス部長官は“米国患者優先青写真”を発表し、いくつかの部分の実施を開始した。このイニシアティブには,後発薬と生物類似薬の競争を増加させ,医療保険計画が薬品価格をより直接交渉できるようにし,薬品価格の透明性を向上させ,消費者の自腹コストを低減する方法がある。トランプ政権はまた、コストを決定する基準として“国際価格設定指数”を構築することを提案し、連邦医療保険B部での薬品の精算を制限する可能性がある。他の製薬メーカー業界に関する提案では、連邦医療保険D部分の福祉を変更し、連邦医療保険D部分でインフレに基づくリベートを実施し、福祉構造を変更し、メーカーの破滅的な段階での支払いを増加させる法案が提案されている。今国会の指導の下で,薬品定価に関する法案数が急激に増加していることから,我々の業務への影響は不確実であり,実質的である可能性がある。
また、多くの州は立法を提出或いは公布し、間接的或いは直接に薬品定価を監督することを求め、例えば生物製薬メーカーに報告独自の価格情報を公開することを要求したり、国家機関が購入した薬品に対して最高価格上限を設定することを要求したりする。例えば、2017年、カリフォルニア州知事は、指定された敷居を超えるいくつかの薬品の値上げに対する処方薬メーカーの事前通知および解釈を要求する処方薬価格透明性州法案に署名した。国会や州立法機関は様々な法案を考慮しており,これらの法案は薬品調達と価格交渉を改革し,管理ツールをより多く利用して連邦医療保険D部分のカバー範囲を制限することを許可し,米国以外からの価格の低い薬品の輸入を促進し,模造薬の使用を奨励している。国会で可決されたインフレ低減法案は、医療保険交渉処方薬の価格を許可する。このような計画と立法は私たちの製品に追加的な価格設定圧力をもたらすかもしれない。
連邦や州レベルの医療補助計画の変化も我々の業務に実質的な悪影響を及ぼす可能性がある。我々の製品のカバー範囲や精算範囲に影響を与える可能性のある提案は,各州により大きな柔軟性を与えて医療補助計画でカバーされている薬品の管理や,カナダや他の国からの処方薬の再輸入を許可することで,我々の製品の使用やカバーを制限することで実質的な悪影響を及ぼす可能性がある。また,連邦基本医療補助税還付の増加により,州医療補助計画は我々の製品に対する追加的な補充還付を要求する可能性がある。ある程度、民間保険会社あるいは管理保健計画は医療補助カバー範囲と支払い状況を追跡し、彼らはこれらの増加したリベートの公布を利用して私たちの製品に定価圧力をかけることができ、彼らはより低い支払いスケジュールを採用することは不利な影響を拡大する可能性がある。
他の提案された影響メーカーの規制行動は私たちの業務に実質的な悪影響を及ぼすかもしれない。このような提案された立法や規制行動やそれによって生じる州行動が米国での製品の使用や精算に及ぼす影響を予測することは困難であるが,我々の運営結果は悪影響を受ける可能性がある。
私たちの知的財産権に関するリスクは
もし私たちが私たちの所有権を獲得し、維持し、実行できなければ、私たちは効果的な競争や利益運営ができないかもしれない。
エビサミンについては,フロリダ大学研究財団(UFRF)とライセンス契約を結び,Flynpoviについてはアリゾナ大学取締役会とライセンス契約を結んだ。許可された知的財産権や他の生物製薬会社の特許は通常不確定であり、複雑な法律、科学、事実の問題に関連している。
私たちが薬物を開発および商業化する能力は、(I)広範で保護可能な知的財産権を取得および/または開発すること、(Ii)必要に応じて商業的に合理的な条項で他の人の固有の権利の追加的な許可を得ること、(Iii)他人の固有の権利を侵害することなく運営すること、(Iv)他人が私たちの固有の権利を侵害することを防止すること、および(V)私たちの会社のノウハウおよび商業秘密を保護することに大きく依存する。
私たちが獲得する可能性のある特許および将来発行される可能性のある特許は、挑戦、無効、または回避される可能性があり、これらの特許によって付与される権利は、独自の保護または同様の技術を有する競争相手に対する競争優位性を提供してくれない可能性がある。また,我々の競争相手は類似した技術を独立して開発したり,我々が開発した任意の技術を複製したりする可能性がある.潜在的候補製品の開発、テスト、規制審査には多くの時間がかかるため、私たちの任意の候補製品が商業化できる前に、どの関連特許も商業化後の短い期間で有効に期限が切れるか、または有効に維持され、それによってこの特許の任意の利点を弱める可能性がある。
米国および多くの外国司法管轄区の特許出願は、通常、出願後少なくとも12ヶ月後に発表されるか、または場合によっては全く発表されないため、科学文献で発見された出版物は、実際の発見よりも遅れていることが多いので、私たちも私たちの許可者も、私たちまたは私たちの許可者が、発行された特許または係属中の特許出願で主張された最初の発明を提出した人であるか、またはこれらの特許出願に規定されている発明を保護する最初の人であることを決定することはできない。
さらに、UFRFはこれまで、米国でのみライセンス技術のいくつかの要素のために保護を求めることを選択しており、国際特許保護を出願する時間が経過している。これはある市場における会社の知的財産権の地位を制限し、潜在的な企業パートナーに対する会社の全体的な価値に影響を与える可能性がある。
私たちの特許保護の獲得と維持は、政府特許機関によって提出された様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある
米国特許商標局と各種外国政府特許機関は,特許過程においていくつかのプログラム,文書,費用支払いなどの規定を遵守することを要求している。場合によっては、規定を遵守しないことは、特許または特許出願の放棄または失効をもたらす可能性があり、それにより、関連する管轄区域の特許権の一部または全部が失われる可能性がある。この場合、競争相手は他の場合よりも早く市場に参入するかもしれない。
私たちは第三者の権利侵害や流用クレームに直面する可能性があり、私たちに不利と判定されれば、重大な損害賠償金を支払うことになる可能性があります。
製薬やバイオテクノロジー産業では、特許や他の知的財産権に関する訴訟や他の訴訟が数多く発生している。私たちは時々様々な特許訴訟や他の知的財産権に関する訴訟の側になるかもしれませんが、私たちが訴訟に関連するいかなる知的財産権も使用しようとしない場合でもそうです。
特許訴訟や他の訴訟の費用は、私たちに有利な問題を解決しても、巨大である可能性がある。私たちの競争相手のいくつかは、私たちの競争相手がより多くの財政資源を持っているかもしれないので、このような訴訟や訴訟の費用を私たちよりも効果的に受けることができるかもしれない。もし私たちに対する任意の特許訴訟または他の訴訟が解決された場合、私たちまたは私たちの協力者は、相手の許可を得ずに私たちの薬物を開発、製造、販売、または輸入することを禁止され、重大な損害賠償責任を請求される可能性がある。私たちは商業的に受け入れられる条項や何の必要な許可も得られないかもしれない(S)。
特許訴訟または他の訴訟の開始と継続によって生じる不確実性は、市場での競争能力に重大な悪影響を及ぼす可能性がある。特許訴訟と他の訴訟もまた多くの管理時間を取るかもしれない。
従業員や他の人と締結された秘密保持協定は、当社のノウハウ、ビジネス秘密、その他の独自情報の漏洩を十分に防止できない可能性があり、知的財産権を十分に保護できない可能性があり、競争能力を阻害する可能性があります。
私たちは医療技術開発のハイテク分野で業務を展開しているため、私たちは私たちの独自の商業秘密と非特許技術を保護するために、商業秘密保護にある程度依存している。しかし、ビジネス秘密は保護することが難しく、他の人が同じまたは似たような技術を自ら開発しないと確信できない。私たちは、私たちのビジネス秘密および非特許技術を保護するために、私たちのすべての従業員、コンサルタント、会社のパートナーと秘密保護協定を締結することを含む措置を取っています。これらの合意は、一般に、その当事者によって開発されたもの、または私たちとの関係中に第三者に開示されたいかなる機密情報も第三者に開示しないことを相手に秘密にすることを要求する。私たちは一般に、当事者が私たちにサービスを提供する過程で構想された発明が私たちの独自の財産になることを規定するこれらの当事者からも合意を得る。しかし、このような合意は遵守されないかもしれないし、知的財産権を効果的に私たちに割り当てられないかもしれない。我々の商業秘密やノウハウを不正に取得して使用することを強制する側の告発は困難であり、高価で時間がかかり、結果は予測できない。しかも、米国以外の裁判所は商業秘密やノウハウを保護することをあまり望んでいないかもしれない。商業秘密保護を獲得または維持できなければ、私たちの競争的地位に悪影響を及ぼす可能性がある。
私たちは私たちの従業員がその前の雇用主によって言われた商業機密を間違って使用したり開示したりしたと告発されるかもしれない
バイオテクノロジー業界でよく見られるように、私たちが雇った個人は以前、私たちの競争相手や潜在的な競争相手を含む他のバイオテクノロジー会社に雇われていた。現在、私たちに対するクレームが解決されていないにもかかわらず、私たちは、これらの従業員または私たちが意図していない、または他の方法でその前の雇用主の商業秘密または他の固有情報を使用または漏洩するというクレームを受ける可能性がある。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。これらのクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣の注意を分散させる可能性がある。
今回の発行と私たちの普通株保有に関わるリスク
私たちはナスダックから撤退するかもしれないが、これは私たちの株の流動性と私たちの融資能力を深刻に損なうだろう。
2023年第1四半期に、これまでに決定した最低入札価格と最低株主権益の欠陥を是正し、ナスダックのすべての適用可能な上場基準を再遵守した。2023年4月14日、私たちはナスダック上場資産部から通知状を受け取り、私たちの普通株は30営業日連続でナスダック上場規則第5550(A)(2)条に要求された1株1.00ドルの最低終値を維持していないと指摘した。ナスダックは、ナスダック資本市場に引き続き上場する最低入札価格要件を再遵守するために、2023年10月11日まで上場期限を延長することを承認した。私たちが2023年5月25日に開催した年次株主総会で、株主たちは、私たちが発行した普通株を所定の範囲で逆株式分割するために、私たちが再登録した会社証明書に対する修正案を承認しました。この許可に基づき、我々の取締役会は30株1株の逆分割比率を承認し、わが社は2023年6月1日に逆株式分割を実施した。株式の逆分割の主な原因は、私たちの普通株の1株当たりの市場価格をナスダック資本市場の継続上場の最低購入価格要求を超えるまで向上させようとしているからだ。2023年6月15日、私たちは普通株の終値が180カレンダー日の猶予期間内に10営業日連続で1株1.00ドルに達したか、またはそれを超えたので、ナスダック上場資産部から通知状を受け取りました。
その後、私たちは公聴会を要求し、控訴を遵守する計画を紹介することを要求し、許可された。私たちは2023年11月28日にナスダックの従業員裁決状を受け取り、ナスダックに私たちの普通株取引を一時停止することを通知し、ナスダックの退市決定に上訴しない限り、ナスダックの上場と登録から私たちの普通株を除去する25-NSE表を米国証券取引委員会に提出した。同社の普通株の停止と退市は放置されており、聴聞手続きの終了を待っている。したがって、私たちの普通株は少なくとも公聴会後に委員会が決定する前に、ナスダック資本市場に上場し続ける予定だ。この委員会がナスダック資本市場に上場し続ける同社の呼びかけを承認する保証はない。センチレターは当社(I)が最低入札価格で1株1.00ドルの最低市場価格を維持することを要求していないこと、および(Ii)最低株主権益要求を満たしていないことを伝えた。2024年1月18日から、20株の普通株式のうち1株の逆分割を完了した。私たちが逆株式分割を実施する主な理由は、最低入札要求を満たすために、私たちの普通株の1株当たりの市場価格を潜在的に高めることである。2024年1月22日、私たちはナスダックから通知を受け、逆株式分割の発効日まで、ナスダック上場規則第5550(A)(4)条(“最低持株量要求”)に適合しなくなり、当社の普通株退市の追加的な根拠として、保有株の最小500,000株を公開することを要求した。また,より多くの株式と今回公開された純収益を発行し,最低流通株要求と最低株主権益要求を再遵守できるようにする予定である.もし私たちがすべての適用されたナスダックの継続上場要求を再遵守できなければ、私たちの普通株はナスダック資本市場から撤退する可能性が高いと思います。
私たちは過去にコンプライアンスを再獲得しましたが、何らかの理由で、ナスダックは私たちの証券をナスダック資本市場の取引から撤退させ、私たちは別の評判の良い国の証券取引所に上場できない場合、以下の部分または全部の減記が発生する可能性があり、そのいずれもが私たちの株主に重大な悪影響を及ぼす可能性があります
|
● |
普通株の流動性と販売可能性 |
|
● |
普通株の市場価格は |
|
● |
私たちは私たちの業務を継続するための資金を得ています |
|
● |
私たちの普通株に投資する機関と一般投資家の数を考慮する |
|
● |
私たちの普通株式のうち市営業者の数は |
|
● |
私たちの普通株式取引価格と取引量に関する情報の可用性; |
|
● |
普通株取引を行いたい自営業者の数。 |
また、ナスダック資本市場への上場を停止した場合、私たちは認知度や認知度の低い市場で取引しなければならないかもしれません。例えば場外取引市場では、私たちの株は“細価格株”として取引される可能性があり、これは私たちの株式取引をより困難かつ煩雑にし、別の市場で取引されている会社が魅力が低く、関連リスクの高い投資とみなされる可能性があり、既存または潜在的な機関投資家が私たちの普通株に投資することにあまり興味がない、あるいは投資を禁止される可能性があります。これはまた私たちの普通株の市場価格をさらに下落させる可能性がある。
逆株式分会が必要な時間内に私たちの株価を上げたり、ナスダックでの上場を維持したりすることは保証できません.
株式の逆分割は私たちの普通株市場価格の上昇を伴うが、このような逆株式分割が私たちの普通株市場価格に与える持続的な影響は確実に予測できず、他社の逆株分割の歴史もそれぞれ異なる。一部の投資家たちは最近の逆株分割に対して否定的な見方をするかもしれない。
ナスダック規則は上場企業が最低購入価格要求を維持または再遵守するために逆方向株式分割を実施する回数に具体的な制限を加えていないが、ナスダックはすでに、一連の逆方向株式分割はナスダック上場証券に対する投資家の自信を弱める可能性があり、特に逆方向株式分割が希釈取引の後に発生する可能性があると表明した。したがって、ナスダックは、私たちの上場を維持することは大衆の利益に合わないと認定するかもしれません。たとえ私たちがどんな逆株式分割でも最低入札価格要求を再遵守しても。当社は過去2年間に逆株分割を実施しており、累計比率は250株を超えていますが、普通株は最低競り要求に達していませんので、ナスダック市場ルール第5810(C)(3)(A)条に規定されているいかなるコンプライアンス治癒期も享受する資格はありませんが、上述したように、ナスダックのスタッフは最低競り要求に合わないため退市決定を出しています。
また、逆株分割は、1株当たりの価格が低価格株を取引しない投資家を引き付けることにはならない可能性がある。逆株分割は私たちの普通株のいくつかの潜在投資家に対する適切性を高める可能性があると信じていますが、逆株式分割後の私たちの普通株は投資家にもっと魅力的になることを保証することはできません。逆株分割を実施しても、逆株式分割とは無関係な要因により、将来の業績や市場全体の傾向を含めて、普通株の市場価格が低下する可能性があります。普通株の取引価格が下落すれば、絶対数字と私たちの総時価に占める割合として、逆株分割なしの下落幅よりも下落幅が大きくなる可能性がある。
最近の逆株分割は、私たちの普通株の流動性を減少させ、より高い取引コストを招く可能性がある。
私たち普通株の流動性は、このような逆株式分割が流通株数を減少させ、特に株価が逆株式分割によって増加しなければならないため、逆株式分割の負の影響を受ける可能性がある。また、逆株分割を実施すれば、100株未満の普通株を持つ“端数”株主の数を増やす。片手取引の委託手数料及びその他のコストは、通常、100株以上の普通株の取引コストよりも高い。したがって、逆株式分割は、上述したように私たちの普通株の販売可能性を増加させる期待結果に達することができない可能性がある。
逆株式分割は私たちが許可した株式の減少を伴うものではない。
逆株式分割は私たちの株主に何の希釈作用もありませんが、逆株式分割による流通株の減少は、許可発行株式に対する私たちの株主が所有する株式の割合を低下させ、取締役会が発行可能な認可株式の相対数を効果的に増加させることを適宜決定することができます。取締役会は時々、当社の普通株の発行を含む取引やその他のリスク投資を行い、当社とその株主の最適な利益に合致すると考えることができる。取締役会が逆株式分割後に追加の普通株を発行することを承認した場合、私たちの既存株主の所有権権益は、このような逆株分割を行っていない場合に発生する希釈よりも大きくなる可能性がある。
追加資本を調達することは私たちの株主を希釈したり、私たちの運営を制限するかもしれない
もし私たちが株式または転換可能な債務証券を売却することによって追加資本を調達する場合、株主の所有権権益は希釈され、条項は清算または株主としての権利に悪影響を及ぼす他の特典を含む可能性がある。債務融資が可能であれば、追加債務を招く、資本支出を行う、または配当を宣言するなど、私たちが具体的な行動をとる能力を制限または制限する契約を含むいくつかの合意が含まれる可能性がある。これらの事件のいずれも、製品開発および商業化目標を達成する能力に悪影響を与え、私たちの業務を損なう可能性がある。私たちは現在利用可能な信用配置の不足によって何の悪影響も生じないと予想する。
普通株の発行や株式購入権の行使に応じて普通株を発行することは、私たちの普通株の価格下落を招き、投資家の大部分の投資損失を招く可能性があります。
我々の株主が公開市場で我々の普通株を大量に売却したり、第144条に規定する任意の法定保有期間が満了した場合、または流通株のロック期間満了に適用された場合、または未償還オプションまたは株式承認証を行使する際に発行されるロック期間が満了した場合、通常“宙に浮いている”と呼ばれる場合があり、私たち普通株の市場価格が下落する可能性が予想される。未解決の状況が存在し、発生しているかどうかにかかわらず、将来的に合理的または適切な時間および価格で株式または株式関連証券を売却することで追加融資を調達する能力をより困難にする可能性がある。2024年1月23日現在、加重平均行権価格で1株13,297.06ドルで599株の普通株を購入した未償還オプションを有し、残り契約期間は8.7年、および加重平均行権価格で1株64.09ドルで340,952株の普通株を購入した未償還引受権証を有し、平均残存株式期間は5.24年である。
証券アナリストは私たちの普通株を自発的に報道したり、引き続き報道したりしない可能性があり、これは私たちの普通株の市場価格にマイナス影響を与える可能性がある。
普通株価格は証券アナリストが発表した会社とその業務に関する研究と報告の重大な影響を受けることが多い。私たちはこのアナリストたちに何の統制権も持っていない。証券アナリストが私たちの普通株をカバーしたり継続したりする保証はない。もし証券アナリストが私たちの普通株をカバーしなければ、研究カバーの不足は私たちの普通株の市場価格に不利な影響を与えるかもしれない。もし私たちの普通株が証券アナリストにカバーされ、私たちの株の格付けが引き下げられたら、私たちの株価は下落するかもしれない。もしこれらのアナリストのうちの1人以上が私たちを追跡しなくなったり、私たちに関する定期的な報告書を発表できなかった場合、私たちは金融市場での可視性を失う可能性があり、これは私たちの株価や取引量を低下させる可能性がある。
今回の発行で普通株を購入すれば、すぐに私たちの普通株の帳簿価値を大幅に希釈することになります。
私たちの普通株の提案公開発行価格は私たちの普通株の1株当たりの有形帳簿純価値より大幅に高いです。今回の発行で普通株を購入した投資家は、私たちの負債を差し引いた1株当たりの価格を支払い、私たちの有形資産の帳簿価値を大きく超えます。したがって、投資家が今回の発行で普通株を購入することは、1株当たり0.209ドルの即時希釈を招くことになり、これは、仮定に基づく1株3.77ドルの合併公開発行価格と付随する一般権証である(仮に公開発行価格は、ナスダック資本市場が2024年1月23日に発表した3.77ドルのような、我々の普通株の前回の売却価格に等しいと仮定する)。また、今回の発行で普通株を購入した投資家は、我々の設立以来の株主投資総額の約26%に貢献するが、このような投資により、今回発行された普通株に続く約94%を持つことになる。今回の発行で普通株を購入した投資家の株式が希釈されているため、我々が清算すれば、投資家が獲得した収益は今回の発行で支払われた購入価格を大きく下回る可能性がある。さらに、私たちは私たちが予想している運営レベルを支援するために追加資本を調達する必要があるかもしれないので、私たちは将来的に大量の普通株または普通株または普通株に交換可能な証券を販売するかもしれない。これらの将来発行される株式または株式に関連する証券は、未償還オプションおよび買収に関連する任意の追加発行株式(ある場合)を行使することに加えて、投資家の権益をさらに希釈する可能性がある。
私たちが株式証の保有者が株式証明書を行使して私たちの普通株を買収するまで、彼らは普通株主としての権利を持たないだろう。
閣下が株式承認証を行使する際に当社の普通株式を購入しない限り、閣下は引受権証を行使する際に発行可能な当社の普通株式については何の権利もありません。あなたの株式引受証を行使する際には、行使日以降の事項について普通株主の権利を行使する権利を記録する権利のみがあります
一般権証の性質は投機的である.
本募集説明書に従って提供される一般株式承認証は、投票権または配当金を受け取る権利のような所有者に任意の普通株式所有権を与えるのではなく、限られた期間内に固定価格で私たちの普通株式を買収する権利を表すだけである。具体的には、自発的な発行日から、一般権証所持者は発行日から5年前に、普通株を買収する権利を行使し、1株$の使用価格(1株当たり公開発行価格および普通権証の100%)を支払うことができ、その後いかなる未行使の一般権証も失効し、何の価値もない。5営業日後、E類株式証の所有者は、現金行使時に発行可能な普通株式数の1.33倍に相当する株式総数を取得する“キャッシュレス行使の代わり”を選択するための単独の選択権を有することになる。また、今回の発売後、一般権証の時価は確定しておらず、一般権証の時価が公開発売価格以上になる保証もない。普通株の市場価格が永遠に普通権証の執行権価格に等しいかどうかを保証することはできず、一般権証所有者がこれらの権利証を行使することが利益があるかどうかを保証することもできない。
今回発行された株式引受証には既定の公開取引市場はない。
今回発行中に発売された一般権証や事前資金権証には既定の公開取引市場はありません。私たちは今回の発行で発行された一般権証や事前資本権証をどの国の証券取引所に上場するかを申請するつもりはありませんし、一般権証や事前資金権証の場外取引市場でのオファーの資格を求めるつもりもありません。活発な取引市場がなければ、一般権証及び事前融資権証の流動性は事前に行使されることなく制限される。
これは最善を尽くした発行であり、最低額の証券の売却を要求することはなく、私たちのビジネス計画に必要な資本額を集めることはできないかもしれません。
配給代理は、今回の発行で提供された証券を購入するために、その合理的な最大限の努力を尽くして要約を求めることに同意した。配給代理は、私たちにどんな証券を購入する義務もなく、特定の数量やドルの金額の証券の購入や販売を手配する義務もありません。今回の発売は2024年2月15日に終了します。事前に完了しない限り、またはその日までに発売を終了することを決定しない限り、いつでも適宜終了することができます。今回の発行を完了する条件として,売却しなければならない証券数や収益金額の最低要求はない。今回の発売には最低発売金額が発売完了条件とされていないため、実際の発売金額、配給代行費、吾などから受け取る収益は現時点では割り切れず、前述の最高金額を大幅に下回る可能性がある。私たちが販売している証券は、私たちがここで提供しているすべての証券よりも少ないかもしれません。これは、私たちが受け取った収益金額を著しく減少させることができます。もし私たちが私たちの業務に資金を提供するのに十分な証券を販売していなければ、今回発行された投資家は、本明細書の“収益の使用”の部分で述べたように、返金を得ることはできません。したがって、私たちは、私たちが考えている運営に必要な資本額を短期的に調達することができず、追加の資金を調達する必要があるかもしれません。これらの資金は、私たちが受け入れた条項では得られないかもしれません。
私たちの定款とデラウェア州法律は株主が有利だと思う買収を阻止するかもしれない。
私たちの会社の登録証明書と定款、デラウェア州法律の適用条項は、第三者が私たちの取締役会の承認なしに私たちの支配権を得ることを難しくしたり、阻止したりするかもしれません。これらの規定には
|
● |
罷免役員に制限を設ける |
|
● |
株主特別会議を開催する人数を制限する |
|
● |
取締役会への指名または株主総会で行動可能な事項の事前通知要求を作成する |
|
● |
私たちの役員選挙での累積投票は許されません。そうでなければ、多数の株主よりも少ない取締役の選挙が許可されます |
|
● |
毎年選挙される役員の数を制限する分類取締役会を設置する |
|
● |
私たちの取締役会が株主の承認を必要とせずに条項を指定して優先株を発行する権利を持たせる。 |
さらに、デラウェア州一般会社法第203条一般的には、私たちが発行した議決権株の15%以上を保有している者、または私たちの任意の共同会社または付属会社が過去3年間に議決権を有する株式の15%以上を発行している任意の業務と合併することを制限しています。このような所有権の買収を招くことを取締役会が事前に承認していない限り、これらの規定は、我々の管理チームを強化し、現行価格よりも高い割増でその株を潜在的な買収者に売却する機会を株主に奪う可能性がある。このような制御権プレミアムが得られない可能性がある場合は、私たちの普通株の価格を下げるかもしれない。
もし私たちが優先株を発行すれば、私たちの普通株保有者の権利と普通株の価値は不利な影響を受けるかもしれない。
当社の取締役会は様々な種類や系列優先株を発行する権利があり、株主は何の行動もする必要がありません。取締役会も株主の承認なしに、投票権、配当権及び配当又は当社業務の清算、解散又は清算時の普通株に対する割引権及びその他の条項を含む任意の種類又はシリーズ優先株の条項を特定する権利がある。もし私たちが未来に優先株を発行する場合、配当金の支払いにおいて、または清算、解散または清算時に普通株より優先するか、または投票権を有する優先株を発行する場合、普通株の投票権を希釈し、普通株式保有者の権利または普通株の価値は不利な影響を受けるだろう。
もし私たちが財務報告書に対して効果的な内部統制を維持できなければ、私たちの普通株の価格は不利な影響を受けるかもしれない
私たちは財政報告書に対する適切な内部統制を確立して維持することを要求された。これらの制御が確立されていないか、またはこれらの制御が確立されると、業務、財務状態、または運営結果に関する開示に悪影響を及ぼす可能性がある。これらの統制のいかなるミスも、私たちが正確な会計記録を維持し、会計ミスと財務不正を発見することを阻止する可能性がある。経営陣の財務報告内部統制の評価は、解決すべき弱点や投資家の懸念を引き起こす可能性のある他の潜在的事項を見つける可能性がある。財務報告の内部統制または経営陣の財務報告の内部統制に対する我々の評価において解決すべき任意の実際または予想される弱点は、私たちの普通株式価格に悪影響を及ぼす可能性がある。
今回の発行が完了しても、私たちは将来、私たちの運営に資金を提供するためにより多くの資金を調達する必要があり、これらの資金は受け入れ可能な条項で提供できないか、あるいは全く得られないかもしれない。必要な時にこの必要な資金を得ることができなければ、私たちは私たちの製品開発努力や他の業務を延期、制限、または中止させられるかもしれません。
我々は経常的な運営損失があり,運営キャッシュフローは負であり,累積的な赤字がある。私たちは私たちの行動に資金を提供し続けるためにもっと多くの資金を集めなければならない。もし私たちが必要な時や受け入れ可能な条件で追加の資本を得ることができなければ、私たちは私たちの業務計画に従って私たちの業務を継続できないかもしれません。あるいは私たちは私たちの業務を完全に停止しなければならないかもしれません。株式や持分支援証券を売却することで調達された任意の追加資本は、私たちの株主の持株比率を希釈することが可能であり、私たちの株式証券の時価低下を招く可能性もある。私たちが将来の資本取引で発行する任意の証券の条項は、新しい投資家に有利になる可能性があり、割引、より高い投票権、および発行権証または他の派生証券を含む可能性があり、これは、当時返済されていなかった証券の保有者にさらなる希薄化効果をもたらす可能性がある。
もし私たちが必要な時や受け入れ可能な条件で追加資金を得ることができない場合、私たちは重大な計画支出の延期、減少、または廃止、一部または全部の業務の再編、削減または廃止、技術または資産を処分し、第三者が私たちの株主の投資損失を招く可能性のある価格で私たちの会社を買収し、破産または完全な運営停止を申請することを求められるかもしれない。これらの事件のいずれも、私たちの業務、財務状況、および運営結果に実質的な悪影響を及ぼす可能性がある。また、もし私たちがタイムリーに多くの資金を得ることができなければ、私たちは経営を続ける企業として経営を続ける能力が大きく疑われ、倒産リスクが増加し、私たちの株主の投資は最高で完全な損失に達することができる。
収益の使用
販売代行費と想定される発売費用を差し引いた後、支払うべき配給代理費と支払う予定の発売費用を差し引くと、今回発売された引受権証の行使による収益(あれば)は含まれておらず、今回の発売で売却された証券から約2,780万ドルの純収益を得ることになり、仮定に基づく1株3.77ドルの合併公開発行価格と付随する一般権証(私たちの普通株が2024年1月23日に発表した最終販売価格に基づいて、3.77ドル)となると予想される。
適用される目論見付録が別途説明されていない限り、任意の証券を販売して得られた純収益を、(I)我々の初期製品候補エヴィサプミンの持続臨床開発、(Ii)CPP Assetsのパートナーとの臨床開発のための医薬製品のコスト、および(Iii)一般会社用途に使用する予定である。一般会社の用途は、未返済債務の返済、運営資金、一般及び行政支出を含むことができる。当社は、本募集説明書の付録日までに、買収に関する計画、承諾、または合意を有していないにもかかわらず、純収益の一部を使用して、当社自身の業務または技術と相補的と考えられる業務または技術を投資または買収することもできます。私たちは上記のすべての目的に特化された純利益金額を決定していない。したがって、私たちの経営陣は、これらの証券を売る純収益を運用するためのかなりの情動権と柔軟性を持っているだろう。
私たちは保証書(“担保書”)の当事者であり,この保証書によると,期日2022年6月15日のSucampo GmbHを受益者とする元票(“手形”)担保CPPの支払義務に基づき,2023年9月30日現在,元金残高は約520万ドルであることに同意した。CPPは1月31日に4つの残りの100万ドルを支払うことを要求されて、計算すべきだが未払いの利息を加えますST2024年、2025年、2026年、残り残高は2027年1月31日に満期となる。第1期100万ドルと課税利息は2023年2月1日に支払われる。
我々の現在の計画と業務条件によると,今回の純収益の期待用途は我々の意図を表しており,これらの計画や業務条件は我々の計画や業務条件の発展に応じて変化する可能性がある.著者らの実際の支出の額と時間は多くの要素に依存し、未来に始まる可能性のある臨床研究或いは臨床研究のタイミングと成功程度、監督管理提出の時間及び監督機関のフィードバックを含む。したがって、私たちの経営陣は、今回発行された純収益を使用する上で幅広い裁量権を持つことになります。今回発行された純収益を使用する前に、純収益を一時的に投資レベルの利息証券に投資する可能性があります。
著者らは現在、この資金は膵臓癌を治療する新しいランダム、二重盲検、プラセボ対照の臨床試験(ASPIRE試験と呼ばれる)に重大な進展を得ることができると推定している。中期分析が陽性であれば,現在の試験を継続するためにFDAや他の同様の承認が必要となる。追加臨床試験のコストと時間は,われわれが求めている適応数および試験の性質と規模に大きく依存する。無作為臨床試験の完了と膵癌の治療を承認する他のステップの費用は6000万~8000万ドルである可能性が推定されている。
候補製品の開発に要するコストを予測することは困難である可能性があり,規制部門の承認を得て薬物を商業化するためには通常必要な開発作業を達成するための追加資金が必要と予想される。私たちは間違っていることが証明される可能性があるという仮定に基づいてこれらの推定をして、私たちは現在予想されているよりも早く私たちが利用できる資本資源を使用することができる。
市場情報
私たちの普通株はナスダック資本市場に発売され、コードは“PBLA”です。2024年1月23日現在、49名の普通株式保有者が登録されている。
大文字である
次の表は、2023年9月30日現在の私たちの現金および現金等価物と資本化状況をまとめています
|
● |
2023年9月30日までの実際のベースで計算します |
|
|
● |
株式承認証を行使するために発行された330,470株の普通株に基づいて調整され、これらの株式承認証の行使は2023年9月30日以降に発生し、132株は主に2023年9月30日以降に行われた権証キャッシュ交換の結果である |
|
|
● |
調整基準で8,000,000株の普通株および一般権証を発行および売却し,1株3.77ドルの仮定で価格および付随する一般権証を公開し,今回の発売で最大16,660,000株の普通株を購入し,配給代理費用や吾などが支払うべき推定発売費を差し引くと,総収益純額は最大約2,780万ドル(事前資金権証は売却しないと仮定する). |
以下、審査及び調整されていない資料は参考に供するだけであり、本行の今回の発売完了後の資本は、今回発売された実際の公開発行価格及び定価時に定められた他の条項に基づいて調整される。あなたは参考のために、“経営陣の財務状況と経営結果の討論と分析”および歴史財務諸表とその関連付記を結合して次の表を読まなければならない
|
(単位:千) |
締め切りの実際 9月 30, 2023 (未監査) |
自分から 9月 30, 2023 (その後) 反省する |
奉納する 調整、調整 |
形式的には AS 調整後の |
||||||||||||
|
現金 |
$ | 907 | $ | 5,817 | $ | 27,772 | $ | 33,589 | ||||||||
| 普通株、額面0.001ドル、許可100,000,000株;実際に発行されたと発行された株149,656株;後続事件後に発行された株480,244株;発行済み株と発行済み株8,480,244株,調整後予定 | - | 0.4 | 8.0 | 8.4 | ||||||||||||
|
追加実収資本 |
106,029 | 110,939 | 27,764 | 138,703 | ||||||||||||
|
赤字を累計する |
(109,883 |
) |
(109,883 |
) |
- | (109,883 |
) |
|||||||||
|
総合収益を累計する |
1,371 | 1,371 | - | 1,371 | ||||||||||||
|
株主権益総額 |
$ | (2,483 |
) |
$ | 2,427 | $ | 27,772 | 30,199 | ||||||||
株式募集説明書の先頭ページに記載されている普通株式と普通権証の発行株式数は一定のままであると仮定し、普通株と普通権証の発行価格が増加(減少)するごとに0.50ドル、現金、追加実収資本、株主権益総額がそれぞれ約370万ドル増加(減少)すると仮定する。同様に,公開発売価格が不変であると仮定すると,普通株と普通権証発行数が増加(減少)するごとに200,000株が現金,追加実収資本,株主資本総額約70万ドルを増加(減少)し,推定された配給代理費と推定される我々が支払うべき発売費用を差し引く.上記調整された資料の備考資料は参考になり、実際の公開発売価格と定価時に決定した今回発売された他の条項に基づいて調整される。
今回の発行前後の発行済み普通株の数は、2023年9月30日現在の発行済み普通株149,656株と、主に株式承認証の行使により発行された330,588株に基づいており、2023年9月30日以降に発生した現金株式承認証は含まれていない
|
● |
今回の発行で販売された引受権証の行使後に発行可能なすべての株; |
|
| ● | 2024年1月18日に施行された逆株式分割に関する断片的な株式現金決済が未解決のままである |
|
● |
加重平均発行権価格で1株13,297.06ドルで発行済み株式オプションを行使することができる599株普通株 |
|
● |
340,952株普通株は、発行された株式承認証を行使する際に発行することができ、2023年9月30日以降に発行された引受権証を含むが、今回の発行に関係なく、加重平均行権価格は1株64.09ドルである。 |
薄めにする
私たちの普通株の株式を購入した場合、あなたの権益はすぐに今回の発行で支払う1株当たりの発行価格と今回の発行後の私たちの普通株の調整後の1株当たりの有形帳簿純価値との差額に希釈されます。一株当たりの有形帳簿純価値は私たちの総有形資産から総負債を引いて、私たちが発行した普通株の株式数で割ることに等しい。
2023年9月30日現在、引受権証(主に現金)の行使により発行された330,588株の株式承認証を実施した後、我々の有形帳簿純価値は約240万ドル、または普通株式1株当たり5.054ドルである。
上記備考調整および吾等を実施して1株3.77ドルの仮定公開発行価格で全8,000,000株の普通株および付随する一般権証を売却し、配給代行費および吾等が支払うべき推定発売費を差し引いた後、私は2023年9月30日の調整済み有形帳簿純価値は約3,020万ドル、または1株当たり3.561ドルであった。これは、我々の既存株主の調整後、有形帳簿純価値が直ちに1株当たり約1.493ドル減少し、今回の発行株の購入者に対して1株0.701ドルを直ちに希釈することを意味する
|
1株当たり公開発行価格を仮定する |
$ | 3.770 | ||
|
2023年9月30日現在の予定1株当たり有形帳簿純価値 |
$ | 5.054 | ||
|
新投資家による1株当たり収益の増加 |
$ | (1.493) | ||
|
今回の発行後の調整後の1株当たりの有形帳簿純価として |
$ | 3.561 | ||
|
今回の発行で新投資家に1株当たりの収益を希釈する |
$ | 0.209 |
公開発行価格は1株3.77ドルおよび一般権利証が0.50ドル増加または減少し、1株当たり新規投資家の1株約0.06ドルを増加または減少させると仮定し、本募集説明書の表紙に掲載されている発売株式数は変わらないことを前提とし、推定配給代理費および推定発売費を差し引いた後である。
今回の発行前後の発行済み普通株の数は、2023年9月30日現在の発行済み普通株149,656株と、主に株式承認証の行使により発行された330,588株に基づいており、主に現金であり、2023年9月30日以降に発生している
|
● |
今回の発行で販売された引受権証の行使後に発行可能なすべての株; |
|
|
● |
2024年1月18日に施行された逆株式分割に関する断片的な株式現金決済が未解決のままである |
|
|
● |
加重平均発行権価格で1株13,297.06ドルで発行済み株式オプションを行使することができる599株普通株 |
|
|
● |
340,952株普通株は、発行された株式承認証を行使する際に発行することができ、2023年9月30日以降に発行された引受権証を含むが、今回の発行に関係なく、加重平均行権価格は1株64.09ドルである。 |
配当政策
私たちは私たちの株の現金配当金を発表したり支払ったりしたことがない。今回の発行が完了したら、私たちの将来の収益(あれば)を残して、私たちの業務のさらなる発展と拡大に資金を提供するつもりで、予測可能な未来に現金配当金を支払うことは期待していません。将来の現金配当金の支払い(あれば)は、私たちの取締役会が様々な要素を考慮して適宜決定します。これらの要素は、私たちの財務状況、経営業績、現在と予想される現金需要、未返済債務、拡張計画、融資者が適用する制限(あれば)を含みます。
経営陣によるbrの議論と分析財務状況と経営成果
当社の財務状況及び経営結果に関する以下の議論は、当社の財務諸表及び本募集明細書の他の部分に含まれる財務諸表の注釈と共に読まなければなりません。この議論には展望的な陳述が含まれており、これらの陳述は業務の将来に対する私たちの仮定に基づいている。私たちの実際の結果は展望的陳述に含まれている結果とは大きく違うかもしれない。どうぞお読みください “前向き陳述に関する注意事項” 本入札明細書で使用される前向き陳述に関するより多くの情報を取得するために、本募集説明書の他の場所に含まれる。
2023年9月30日までの3ヶ月と9ヶ月は、2022年9月30日までの3ヶ月と9ヶ月、2022年と2021年12月31日までの財政年度の経営陣との議論と分析は歴史的である。比較のため、2023年1月13日に実施された40株1株逆分割または2023年6月1日に実施された30株1株逆株式分割は、2022年9月30日までの3カ月と9カ月の株式と1株当たりの金額を再報した。2023年9月30日までの3ヶ月および9ヶ月は、2022年9月30日までの3ヶ月および9ヶ月、および2022年および2021年12月31日までの財政年度と比較し、2023年9月30日以降に発生し、経営陣の検討および分析に含まれる事件は、2024年1月18日に発効した20株1株逆株分割で重複する株または1株当たりの金額はない。
概要
Panbela治療会社(“Panbela”とその直接·間接子会社“私たち”,“私たち”,“当社”,“会社”)は臨床段階の生物製薬会社であり,破壊的療法を開発し,緊急に満たされていない医療ニーズの患者の治療に用いられている。
我々の主な候補薬はエビサミン(SBP−101)であり,フロリダ大学研究財団社とFlynpovi(エフルオロオルニチン(CPP−1 X)とシュリン酸)から独占的に許可されている。Flynpoviは口頭で提供される。同社はアリゾナ大学取締役会のFlynpoviを商業化するグローバル独占ライセンスを持っている。
Panbelaはポリアミンプラットフォームを用いた破壊的療法の開発に専念し,緊急医療ニーズのある患者の治療に専念しているため,ポリアミンの各種疾患に対する治療を単独と併用で評価するための2つのスポンサーの研究合意に参加した。現在、ジョン·ホプキンス大学医学院との協力は主に作用機序と固形腫瘍に集中しているが、MD-Anderson癌センターの協力は主に血液系悪性腫瘍に集中している。多発性骨髄腫(細胞系)におけるSBP-101およびCPP-1 X(DFMOまたはeflornithineとも呼ばれる)に関する研究要約は、2023年11月の“血液”雑誌補足刊に掲載された米国血液学会(ASH)会議サイト上のオンライン出版物によって受け入れられている。
アスピリン(SBP−101)
2015年、FDAは私たちのIVOSPEMEN候補製品に関する私たちの研究新薬(IND)の申請を受けた。2022年5月,米国ではNAMES(“USAN”)がSBP−101のUSANとして伊文アスピリンを採用しているとの通知を受けた。アンフェタミンの薬の情報 USANサイト(www.ama-assn.org/go/usan)に公開されています。
著者らはすでに過去に治療した局部末期或いは転移性膵臓癌患者に対する初歩的な臨床試験を完成した。これは第1段階の人類初の用量逓増安全性研究である。2016年1月から2017年9月まで,I期試験の用量増加段階で,29名の患者を6つのキューあるいはグループに組み入れた。いずれの用量レベルでも薬物に関連する骨髄毒性あるいは末梢神経病変は認められなかった。安全性評価を受ける以外に、29名の患者の中の23人は第1の治療周期の終了前或いは8週間の終了時に固形腫瘍反応評価標準(RECIST)を用いて初歩的な治療効果信号を評価することができ、RECISTは現在公認されている腫瘍の大きさの変化を評価する標準である。
2018年,われわれの第2の臨床試験に参加する患者を募集し始めたのはIa/Ib期研究であり,アスピリンと2種類の標準看護化学療法薬ゲムシタビンとNAB−パクリタキセルの併用の安全性,有効性,薬物動態を検討することを目的とした。投与量レベルとスケジュールを評価するために、25人の被験者が4つのキューに参加した。また25名の被験者が試験の拡張段階に参加した。中間結果は2022年1月に発表された。評価可能被験者(4列,IBN=29個)では,最適反応はCR 1(3%),PR 13(45%),SD 10(34%),PD 5(17%)であった。1人の被験者はRECIST腫瘍評価のベースライン後スキャンを受けなかった。中間PFSは、現在6.5ケ月であり、潜在的な毒性を評価するために、薬物投与量中断の負の影響を受けている可能性がある。2022年1月にデータを公表したところ,キュー4+Ib期患者の平均総生存期間は12.0カ月であり,現在最終的には14.6カ月であった。キュー2の2人の患者は、長期生存を証明している。1つは30.3カ月(最終データ)であり,もう1つは33.0カ月であり,2022年3月18日のデータベースロック時には生きている.7人の被験者はデータベースロック時に生きており、1名は2番キューから、6名は4番キューからIb期を追加した。
2022年1月、同社は新しい臨床試験を開始することを発表した。この試験はASPIREと呼ばれ、無作為、二重盲検、プラセボ対照試験であり、ゲムシタビンとNAB-パクリタキセルと併用し、これは標準的な膵臓癌治療方案であり、以前転移性膵臓癌を治療したことがない患者に適している。この実験はアメリカ、ヨーロッパ、アジア太平洋地域の約94地点で行われる。同社は2022年8月にオーストラリアで試験に参加した1人目の患者を発表した。2022年9月、同社は規制部門の承認を得て、スペイン、フランス、イタリアにサイトを開設できると発表した。2023年9月30日には81サイトが10カ国でオープンした。
米国や世界の他地域に臨床診療所を開設する速度は当初の予想より遅く,一部の原因は医学界の資源が逼迫していることであるが,同社は2023年までにすべての国や診療所が開放されると予想している。
この実験は最初に第2段階/第3段階に設計され,初期サンプル量は小さかった。欧州およびFDA規制フィードバックへの応答として,この研究は,総生存率(主要終点)を用いて中間分析で検査を行うために,総試験サンプル量(600)と修正された設計を含むように修正された。すべての国が開放されており、すべてのサイトが年末までに開放される予定だ。独立したデータ安全監視委員会(DSMB)は会議を開いて予め指定されたセキュリティ分析を行い、何の修正もせずに実験を継続することを提案した。この研究は600名の被験者の登録を完了するまで36ヶ月を要すると予想され、中期分析は2024年初めに完成する。2024年1月25日、同社はこの試験が登録者数の50%を超えたと発表した。同社は、全面登録は2025年第1四半期に完了し、全体の生存状況に基づく中期データ分析は2024年中に提供すべきだと予想している。
2023年4月初め,同社は米国癌研究協会年次総会で卵巣癌ポリアミン代謝調節剤としてのアスピリンの研究結果を重点的に紹介したポスタープレゼンテーションを発表した。ポスターの結論は,VID 8+卵巣癌細胞を投与したC 57 BL/6マウスはアスピリン化学療法を受けた後,生存期間を有意に延長し,全体の腫瘍負担を低下させたと結論した。その結果,エスピミン併用標準看護化学療法は卵巣癌の臨床治療に有用である可能性が示唆され,同社は卵巣癌の臨床前と臨床研究を継続しようとしている。
FDAや他の国の承認を得るためには追加の臨床試験が必要かもしれない。追加臨床試験のコストと時間は試験の性質と規模に大きく依存する。
フルボイル(エフルオロオルニチン(CPP−1 X)とシュリン酸)
2009年、FDAは候補製品である私たちの共同製品FlynpoviのIND申請を受けた。
1つの第3段階の研究では,エフルオロオルニチン単独あるいはスリン酸単独と比較して,エフロンとスルリン酸の併用による家族性腺腫性ポリープ症(“FAP”)の有効性と安全性が検討されている。計171名の患者がランダムにグループ分けを受けた。Flynpovi群56名中18名(32%),シュリン酸群58名中22名(38%),エフロン群57名中23名(40%)が疾患進展を認め,危険比は0.71(95%信頼区間)であった[語彙表]0.39~1.32)、シュリン酸(p=0.29)と0.66(95%CI、0.36~1.23)は、エフルオロオルニチンと比較した。一件後の分析では,Flynpovi群の患者は48カ月に及ぶ下胃腸(LGI)手術が必要に進展しなかったのに対し,シュリン酸とエフロン(CPP−1 X)群の患者はそれぞれ7名(13.2%)と8名(15.7%)であった。これらのデータは、FlynpoviおよびHR=0.00(95%CI、0.00~0.48に対応するp =0.005)、HR=0.00(95%CI、0.00-0.44;p =0.003)フルオロボイルおよびエフルオロオルニチン。LGI群の統計学的意義を考慮して,新薬申請(“NDA”)をFDAに提出した。研究は主な終点に到達できなかったが,秘密保持プロトコルは探索的分析の結果に基づいているため,完全な返信を送信した.この欠陥問題を解決するためには,同社は臨床終点への影響を証明するために,1つまたは複数の十分かつ良好に制御された臨床試験の結果を提出しなければならない。
2023年4月、同社はCPPとOne-Two Treeutics Assets Limitedとの間の許可プロトコルが終了したため、FAP患者においてFlynpoviを開発および商業化する北米権利を再取得した
0−III期結腸癌または直腸癌患者の高危険腺腫および第二原発結腸直腸癌の再発を予防するためのFlynpovi二重盲検プラセボ対照試験が行われており、III期−エフルオロオルニチンおよびスリン酸を用いた結腸腺腫(“PACE”)の予防が行われている。この研究の目的は、エフルオロオルニチンおよびスリン酸(対応するプラセボと比較して)が、高度非典型的増殖、絨毛特徴を有する腺腫、1 cm以上の腺腫、多発性腺腫、任意>/=0.3 cmの腺腫、全進行結腸直腸事象、または全ての結腸直腸事象に対して治療効果があるかどうかを評価することである。PACE試験は国家癌研究所(“NCI”)と西南腫瘍集団(“SWOG”)が協力して援助した。同社は2023年6月28日に,PACE試験があらかじめ計画された無駄な分析に合格したと発表した。
エフルオロオルニチン(CPP−1 X)/エフルオロオルニチン(CPP−1 X−S)
FDAは2009年と2018年にエフルオロオルニチンのIND申請を受けた。
STK 11変異非小細胞肺癌患者のエフルオロオルニチン香包の評価を今年から評価する試験計画がある。エフロン錠については,インディアナ大学や青少年糖尿病研究財団(“JDRF”)と協力し,2023年1月11日に早発性I型糖尿病の第二段階試験を開始した。2つのポスターデモは第1段階T 1 Dの結果を検討し,1つは内分泌学会会議,もう1つは2023年6月の糖尿病免疫学学会会議で検討した。また,あるII期試験では,エフルオロオルニチンは大量のテストステロンとベンザルアミン治療による転移性去勢抵抗前立腺癌の評価を受けている。
2023年7月17日、同社はエフルオロオルニチン児童神経芽細胞腫計画のいくつかの権利、所有権、および権益を剥離した。これらの資産には、小児腫瘍学グループ(“COG”)/米国国立癌研究所によって支持されているエフルオロオルニチン香包による再発性難治性神経芽細胞腫の治療を評価する試験が含まれている。US World Medsとの合意条項によると、同社は、これらの資産の売却と引き換えに約950万ドルの非希釈資金を得る権利がある。取引完了時に、会社は400,000ドルの初期支払いを受け取り、買収会社が臨床開発、監督許可、商業販売に関するいくつかのマイルストーンを成功させた場合、残りの支払いは受け取られる。
財務概要
2023年6月1日には、会社普通株の40株(1:30)の割合で逆株式分割を行い、2023年1月13日には、会社普通株の40株(1:40)の割合で逆株式分割を行った。私たちの普通株のすべての株と1株当たりの金額はこれらの逆株分割を反映するために遡及調整されている。
2011年以来、私たちは1.099億ドルの損失を受けた。2023年9月30日までの9ヶ月間、私たちは1880万ドルの純損失を出した。その間、私たちは経営活動から約2180万ドルのマイナスキャッシュフローも生まれた。我々が研究開発活動と商業化を継続することに伴い,引き続き重大な損失を招くことが予想され,経営活動からの負の純キャッシュフローが生じる。
2023年9月30日と2022年12月31日まで、私たちの現金はそれぞれ約90万ドルと130万ドルです。2023年9月30日までの9カ月間で現金が40万ドル減少したのは、運営からのマイナスキャッシュフロー2180万ドル分が2140万ドルの純融資活動を相殺したためだ。現在の無作為臨床試験では伊夫アスピリンの使用を除いてアブラシャインの薬剤が不足しているため,同社は臨床試験のためのこの基準の看護成分の調達を開始しており,本四半期までの最後の四半期に臨床地点に提供された場合,約310万ドルが研究·開発に用いられている。同社は調達供給のすべてのルートを模索し続けており、第3四半期には約50万ドルの前金を別途支払う必要がある。これらのプリペイドは,交付前から要求されており,前払い費用として貸借対照表に残り,業務で使用されている現金期間に反映される.純融資活動には,普通株の登録公開,あらかじめ出資した引受権証,引受権証があり,純収益は約2300万ドルである。同社はまた、市場での販売手配で普通株を売却し、純収益は約160万ドルだった。同社は同時期に160万ドルのローン返済も記録した。
私たちは、私たちの運営を継続するためにより多くの資金を調達し、2023年第3四半期以降に必要な将来の試験を完了し、米国、EU、その他の国際市場で規制承認を求めることを含む、私たちの業務計画を実行する必要がある。歴史的に見ると、私たちの運営資金は主に株式、証券、債務の売却から来ている。私たちは過去に必要な資本を獲得して私たちの運営を支援することに成功したにもかかわらず、私たちは似たような方法で追加的な融資を求めるかもしれないが、私たちが商業的に合理的な条項と条件で追加的な融資を得ることができるか、または根本的にできないという保証はない。もし私たちの臨床データが積極的でなければ、あるいは経済や市場状況が悪化すれば、このリスクは増加するだろう。添付されている簡明総合財務諸表の作成仮説は、引き続き経営を継続する企業として、正常な業務過程で資産の現金化と清算負債を考慮する。
もし私たちが必要な時に追加的な融資を得ることができない場合、私たちの運営規模を削減し、外部専門サービス提供者の使用を減らすこと、従業員や従業員の報酬を減らすこと、私たちの候補製品の開発を大幅に修正または延期すること、私たちの候補製品を商業化する権利を第三者に許可すること、または運営を停止することが含まれる可能性がある。
当社は新冠肺炎の流行で私たちの業務に重大な中断をもたらしていません。
権利証執行権誘因とC類権証私募
2023年11月2日、私たちは既存の株式引受証のある所有者と株式承認証取引権誘因要書を締結して、私たちの普通株を購入し、それに基づいて、所有者は現金形式で既存の引受権証を行使し、1株当たり0.78ドルの換算権価格で2,130,000株の私たちの普通株を購入することに同意し、それと引き換えに、新しいC類普通株購入権証の発行に同意し、合計4,260,000株の私たちの普通株を購入することに同意した。会社が既存の株式承認証の行使と新株式承認証の購入から得た総収益総額は約190万ドルである。新株式証の初歩的な行使価格は1株当たり0.78ドルであり、株主が承認した日からナスダック上場規則に必要な任意の株主が承認した日から5年以内に行使することができる。我々の株主は2023年12月19日に開催された特別会議で普通株式の発行を承認した。私たちは、新権証の行使によって発行または発行可能な株式の再販売を含む登録声明を提出することに同意し、米国証券取引委員会は2023年12月20日にS-1表登録声明(文書番号333-275733)の発効を発表した。2024年1月23日現在,未返済のC類権証は1,504,000件である。
権利証誘因とD類株式証の私募
2023年12月21日、吾らはいくつかの既存株式証明書所有者と株式承認証取引権誘因要約書簡を締結し、当社の普通株を購入することに同意し、これにより、所有者は現金方式でその既存引受権証を行使することに同意し、その既存の使用価格で1株当たり0.78ドルで合計2,556,000株自社普通株を購入し、吾らが新しいD類普通株購入承認株式証を発行することに同意し、最大5,112,000株自社普通株を購入する。同社が既存の株式承認証の行使から得た総収益総額は約200万ドル。新株式証の初歩的な行使価格は1株0.95ドルであり、ナスダック上場規則に規定されている株主の承認を得た後にのみ行使でき、株主の承認日から5年(あれば)まで行使することができる。もし会社が将来的に普通株等価物と転換可能または派生証券に基づいていくつかの希釈性普通株を発行する場合、任意の中間の逆株分割と株主の承認を受けた後、行使価格は別途低下する。2024年1月23日現在、すべての新株式証はまだ返済されていないが、行使できず、株主の承認を待っている。我々は、株主が6ヶ月以内に新株式証に関連する普通株の発行を許可し、2023年12月21日から60カレンダー日以内に新規株式証関連株の転売をカバーする登録声明を提出することを求める会議を開催することに同意した。また、当社は1年以内にいかなる変動金利取引(定義勧誘メール)も行わないことに同意していますが、6ヶ月後に発効した市場発売は除外します。
経営成果
手術成績の比較(単位:千):
| 9月30日までの3ヶ月間 | 9月30日までの9ヶ月間 | |||||||||||||||||||||||
|
2023 |
2022 |
パーセント 変わる |
2023 |
2022 |
パーセント 変わる |
|||||||||||||||||||
|
運営費 |
||||||||||||||||||||||||
|
一般と行政 |
$ | 1,107 | $ | 1,294 | -14.5 | % | $ | 4,102 | $ | 4,349 | -5.7 | % | ||||||||||||
|
研究開発 |
6,739 | 2,329 | 189.4 | % | 14,501 | 24,563 | -41.0 | % | ||||||||||||||||
|
総運営費 |
7,846 | 3,623 | 116.6 | % | 18,603 | 28,912 | -35.7 | % | ||||||||||||||||
|
その他の費用、純額 |
(4 | ) | (835 | ) | -99.5 | % | (353 | ) | (1,390 | ) | -74.6 | % | ||||||||||||
|
所得税割引 |
19 | 56 | -66.1 | % | 167 | 104 | 60.6 | % | ||||||||||||||||
|
純損失 |
$ | (7,831 | ) | $ | (4,402 | ) | 77.9 | % | $ | (18,789 | ) | $ | (30,198 | ) | -37.8 | % | ||||||||
研究と発展(“R&D”)及び一般及び行政(“G&A”)の支出は、当社が株式オプションを発行することによる非現金株式報酬支出を含む。私たちは株式補償の公正な価値に従ってその帰属中に支出する。株式に基づく奨励の条項や帰属スケジュールは、付与のタイプや引受人の就業状況によって異なる。2023年9月30日までの賞は、表現や時間条件に応じて授与されます。私たちは未来に追加的な非現金株式の給与支出を記録すると予想しているが、これはかなりのものかもしれない。
次の表は,我々の総合損失表における株による補償費用をまとめたものである
|
9月30日までの9ヶ月間 |
||||||||
|
2023 |
2022 |
|||||||
|
一般と行政 |
$ | 554 | $ | 697 | ||||
|
研究開発 |
145 | $ | 160 | |||||
|
株に基づく報酬総額 |
$ | 699 | $ | 857 | ||||
2023年9月30日と2022年9月30日までの3ヶ月
一般と行政費用
2023年第3四半期、われわれのM&A支出は14.5%低下し、110万ドルに低下し、2022年第3四半期の130万ドルを下回った。減少の主な原因は法律と他の専門サービスの減少だ。
研究開発費
我々の研究開発費は2022年第3四半期の230万ドルから2023年第3四半期の670万ドルに増加し、189.4%に増加した。この増加は,主に2023年9月30日までの3カ月間に初めて臨床サイトに供給されたASPIRE臨床試験で使用された標準看護薬Abraxaneのコストが約320万ドルまたは約6カ月の供給量であったためである。ASPIRE試験,および多くの地点への標準看護薬の供給需要は,前年よりもコストの増加を推進し続ける。
その他の費用、純額
2023年9月30日までの3カ月間、その他の費用の純額は約4000ドル。この期間の他の支出は40万ドルであり、会社間の売掛金残高からの外国為替損失と1枚の約束手形からの利息支出は、通貨市場口座売却資産収益と利息収入の40万ドルその他の収入によってほぼ相殺されている。2022年9月30日までの3カ月間、その他の費用の純額は約90万ドル。2022年9月30日までの3ヶ月間の他の費用は、会社間の売掛金残高の外貨為替損失と本チケット2枚の利息支出と関係がある。
所得税割引
2023年9月30日までの3カ月間で、所得税割引は19,000ドルに低下し、2022年9月30日までの3カ月の56,000ドルを下回った。私たちの所得税の優遇は主にオーストラリアで行われた研究開発活動に関連した返却可能な税金の免除から来ています。ASPIRE試験は世界各地のいくつかの国で行われており,2023年9月30日現在,オーストラリアを含む5つの臨床地点である。
2023年9月30日と2022年9月30日までの9ヶ月
一般と行政費用
2023年9月30日までの9カ月間で、M&A支出は5.7%減の410万ドルとなり、2022年9月30日までの9カ月間の430万ドルを下回った。減少は主に2022年9月30日までの9カ月間のCPP買収に関する専門サービスによるものである。
研究開発費
2023年9月30日までの9カ月間で、我々の研究開発費は41.0%減の1450万ドルとなり、2022年9月30日までの9カ月間より1010万ドル減少した。この減少は主に2022年第2四半期のCPP買収の知的財産権研究開発が1770万ドルを突破したためだ。この一度のログアウトを含まず,2023年までの9カ月間の研究開発は760万ドル増加し,AbraxaneのコストおよびASPire試験におけるサイトと被験者登録の増加に関与している。
その他の費用、純額
2023年9月30日までの9カ月間、その他の費用の純額は約40万ドル。この期間の他の費用は、会社間売掛金残高の外貨為替損失と本チケット2枚の利息支出と関係があるが、売却された固定資産の収益と貨幣市場口座の利息収入部分は相殺される。
2022年9月30日までの9カ月間で、その他の費用純額は約140万ドルであり、これは会社間の売掛金残高の外貨為替損失に関係している。
所得税割引
所得税優遇は2023年9月30日までの9カ月間で167,000ドルに増加し、2022年9月30日までの9カ月間の104,000ドルを上回った。我々の所得税優遇は,主にオーストラリアで行われている研究開発活動に関する払戻可能な税収控除に由来しており,ASPIRE試験により,昨年に比べて増加し始めている。
2022年と2021年12月31日終了年度業務成果比較(千計)
| 十二月三十一日までの年度 | ||||||||||||
|
2022 |
2021 |
百分率変化 |
||||||||||
|
運営費 |
||||||||||||
|
一般と行政 |
$ | 6,044 | $ | 4,587 | 31.8 | % | ||||||
|
研究開発 |
28,049 | 5,423 | 417.2 | % | ||||||||
|
総運営費 |
34,093 | 10,010 | 240.6 | % | ||||||||
|
その他の収入,純額 |
(956 | ) | (613 | ) | 56.0 | % | ||||||
|
所得税割引 |
116 | 488 | -76.2 | % | ||||||||
|
純損失 |
$ | (34,933 | ) | $ | (10,135 | ) | 244.7 | % | ||||
一般および行政(“G&A”)および研究開発(“R&D”)支出には,株式オプションの発行による非現金株報酬支出が含まれる.株式に基づく報酬の条項や授与スケジュールは、贈与タイプや被贈与者の就職状況によって異なる。2022年12月31日現在、授与された賞は時間と業績条件に基づいている。私たちは未来に追加的な非現金補償費用を記録すると予想しているが、これは相当なものかもしれない。下表は、2022年12月31日と2021年12月31日までの年度の総合経営報告書と全面赤字における株式ベースの補償費用(千単位)をまとめたものである
|
十二月三十一日までの年度 |
||||||||
|
2022 |
2021 |
|||||||
| 一般と行政 | $ | 889 | $ | 1,083 | ||||
|
研究開発 |
199 | 204 | ||||||
|
株に基づく報酬総額 |
$ | 1,088 | $ | 1,287 | ||||
一般と行政費用
2022年のM&A支出は31.8%増の600万ドルに達し、2021年の460万ドルを上回った。M&A費用の増加は主に法律と財務顧問費用の増加の結果だ。
研究と製品開発費用
我々の研究開発費は2021年の540万ドルから2022年の2800万ドルに増加し、417.2%に増加した。約1,770万ドルの非現金によるIPR&Dのログアウトを考慮したところ,残りの増加は主にわれわれのイビサミン(SBP−101)無作為試験に関する臨床試験コストの増加によるものであった。われわれの臨床研究の拡大に伴い,研究開発費は引き続き増加することが予想される。知的財産権の研究と開発のログアウトは使い捨てだ。
その他の収入,純額
2022年12月31日と2021年12月31日までの年度、その他の費用純額はそれぞれ100万ドルと60万ドルで、主に外貨取引損失からなる。
所得税割引
所得税優遇は2021年の48.8万ドルから2022年の11.6万ドルに引き下げられた。我々の所得税優遇は,主にオーストラリアで行われている研究開発活動に関する払戻可能な税収割引に由来しており,Ia/Ib段階試験の完了や,ASPIRE試験がオーストラリアにすべての計画地点を開設していないことに伴い,2022年に大幅に減少している。
流動性と資本資源
次の表は,2023年9月30日と2022年12月31日までの流動性と資本資源,および2023年9月30日と2022年9月30日までの9カ月間のキャッシュフローデータをまとめたものである。これは、以下のより詳細な議論(千計)を補足することを目的としている
|
流動性と資本資源 |
||||||||
|
2023年9月30日 |
2022年12月31日 |
|||||||
|
現金 |
$ | 907 | $ | 1,285 | ||||
|
運営資本(赤字) |
$ | (7,031 | ) | $ | (6,056 | ) | ||
|
キャッシュフローデータ |
9月30日までの9ヶ月間 |
|||||||
|
2023 |
2022 |
|||||||
|
現金提供側(使用): |
||||||||
|
経営活動 |
$ | (22,169 | ) | $ | (10,273 | ) | ||
|
投資活動 |
400 | (656 | ) | |||||
|
融資活動 |
21,393 | 5 | ||||||
|
為替レート変動が現金に与える影響 |
(2 | ) | (2 | ) | ||||
|
現金純増(マイナス) |
$ | (378 | ) | $ | (10,926 | ) | ||
運営資金
2023年9月30日と2022年12月31日まで、私たちの現金と現金等価物の総額はそれぞれ90万ドルと130万ドルです。2023年9月30日現在、890万ドルの流動負債と700万ドルの営業資本赤字がありますが、2022年12月31日現在の流動負債と運営資本赤字は780万ドルです。運営資本の定義は流動資産から流動負債を差し引くことである。
キャッシュフロー
経営活動に使われている現金純額
2023年9月30日までの9カ月間で、経営活動に使われた純現金は約2180万ドルだったが、2022年9月30日までの9カ月で約1030万ドルだった。これらの期間に使用された現金純額は主にこれらの期間の純損失を反映しており、経営資産や負債変化の影響部分によって相殺されている。2023年9月30日までの9ヶ月間、経営活動で使用された現金には、ランダム試験を指導したCROが保有する長期預金を支援するための550万ドルが含まれていたが、CROへのこれらの預金の支払い速度が鈍化したため、売掛金の増加分はこの部分を相殺した。業務に使用される現金増加は,Aspire試験を支援するためにこれらの地点に標準看護薬を供給するための約320万ドルと,標準看護薬供給を前払いするための50万ドルを反映している。
投資活動が提供する現金純額
投資活動が提供する現金には、2023年9月30日までの9カ月間の知的財産権販売収益が含まれている。2022年9月30日までの9ヶ月間に発生した現金は、CPPから買収が行われている研究開発の銀行や法的コストと関係がある。
融資活動が提供する現金純額
融資活動が提供する純現金は2023年9月30日までの9カ月間で約2,140万ドル、2022年9月30日までの9カ月間、融資活動が提供した純現金は約5,000ドルだった。2023年9月30日までの9ヶ月間に提供された現金は、普通株売却、事前出資の引受権証、引受権証の収益ですが、一部は約束手形支払いで相殺されます。2022年9月30日までの9ヶ月間に提供された現金は、株式引受権証を行使する収益である。
資本要求
私たちが引き続き業務を展開し、私たちの業務計画を実行することに伴い、私たちは膵臓癌を治療する最初の候補製品エビ司普明の臨床開発計画を完成することと、アメリカ、EUとその他の国際市場の監督管理の承認を求めて、私たちは引き続き大量と増加する損失を招くことを予想して、これは引き続き経営活動から負の純現金流を産生する。
私たちの未来の資本使用と需要は多くの現在と未来の要素にかかっている。これらの要素にはこれらに限定されない
|
● |
2022年1月に開始されたグローバルランダム第2/第3段階試験の完了を含む、我々の規制承認申請を支援するために必要な臨床試験の進展状況 |
|
● |
私たちは重要なサプライヤーと支払い条件を協議する能力; |
|
● |
卵巣癌におけるアスピリンの開発とCPP買収により得られた資産の開発作業のコスト拡大 |
|
● |
私たちの候補製品Flynpoviを開発するコストは |
|
● |
現在行われている早期臨床試験を行い,第三者協力により資金を獲得すれば,様々な適応のエフルオロオルニチンを開発するコストが成功する |
|
● |
私たちは候補製品の安全性と有効性を証明することができます |
|
● |
私たちはアメリカ、EU、または他の国際市場で私たちの候補製品の規制承認を得ることができる |
|
● |
私たちの候補製品に適用される規制規制の変化は、製品開発のコストと遅延を招く可能性がある |
|
● |
私たちの候補製品の市場受容度と未来の販売レベル |
|
● |
第三者支払者との返済計画の進展速度 |
|
● |
競争の技術と市場発展の影響 |
|
● |
特許出願の提出及び起訴、並びに特許請求の執行又は弁護に係る費用。 |
2023年9月30日まで、私たちは資金を借りることができる既存の信用計画を持っていない。歴史的に見ると、私たちの運営資金は主に株式、証券、債務の売却から来ている。私たちは過去に必要な資本を獲得して私たちの運営を支援することに成功したにもかかわらず、私たちは似たような方法で追加的な融資を求めるかもしれないが、私たちが商業的に合理的な条項と条件で追加的な融資を得ることができるか、または根本的にできないという保証はない。
負債.負債
CPPは2022年6月15日にSucampo GmbH(“貸金人”)に改訂及び再予約された引受票(“本票”)を発行し、元金金額は約620万ドル(“元本”)である。この手形はいずれの未返済元金を単利とし、年利率は5%である。すべての未払い元金は当時のいかなる未払いおよび応算利息とともに以下のように支払わなければならない:(I)100万元、別途2024年1月31日、2025年1月31日および2026年1月31日またはそれまでの未払い利息を加算し、および(Ii)すべての残り元金を2027年1月31日またはそれまでの未払い利息に加算する。同社は計画通り2023年1月31日に100万ドルと利息を支払った。2023年9月30日現在の未償還元金残高は約520万ドル。2023年9月30日現在,課税と未払い利息総額は約172,000ドルである。
Panbelaは貸主に発行された手形の全金額に貸主を受益者とする支払い保証を提供している。
2022年と2021年の普通株式と引受権証の発行
2022年10月4日、同社は登録公開を完了し、合計177,175株の普通株、予備融資権証を発行し、1株0.001ドルの使用価格で合計325,325株の普通株、および引受権証を購入し、合計753,749株の普通株を購入した。2023年1月30日に希釈公開を終了した後、これらの権利証の行使価格は1株1.51ドルに再定価された。これらの証券の発行価格は普通株と1.5権証1株当たり12ドル、または1株当たり事前融資の権証と1.5権証11.999ドルである。今回発行された純収益総額は約530万ドル。2022年12月31日現在、すべての事前出資の引受権証が行使されている。これらの証券は,S-1表中の有効登録宣言に基づいて発行される.
2022年7月19日、Panbela治療会社(“会社”)は、Roth Capital Partners,LLC(“代理店”)と販売契約を締結し、総販売総価格が8,400,000ドルに達する会社普通株を時々“市場”株式発行計画(“ATM計画”)で売却する。2022年12月31日までの1年の最後の月に、同社はATM機で28,343株の普通株を発売販売し、約93,000ドルの毛収入を生み出した。会社は約44,000ドルの融資コストを発生させ、これらのコストは2022年12月に会社がATMに基づいて株を発売する際に追加の実収資本を計上し始めた。ATM計画によると、会社が代理店に支払う手数料は、ATM計画に基づいて普通株を売却する総毛収入の3.0%に相当する。2022年12月31日までの1年間の純収益は約4.6万ドル。
2021年7月2日、同社は83,333株の普通株の貸切公開を完了し、購入価格は1株120.00ドルであった。今回発行された総収益は約1000万ドル。引受業者の割引や他の発行コストを差し引いた純収益は約910万ドル。
将来の資本需要
私たちは、必要な将来の試験を完了し、アメリカ、EU、その他の国際市場で規制承認を求めることを含む、私たちの業務を継続し、私たちの業務計画を実行するためにより多くの資金が必要です。歴史的に見ると、私たちの運営資金は主に株式、証券、債務の売却から来ている。私たちは過去に必要な資本を獲得して私たちの運営を支援することに成功したにもかかわらず、私たちは似たような方法で追加的な融資を求めるかもしれないが、私たちが商業的に合理的な条項と条件で追加的な融資を得ることができるか、または根本的にできないという保証はない。私たちの既存の現金と2023年1月に公募株で集めた現金は2023年第3四半期までの運営費用を支払うのに十分だと信じています。
もし私たちが必要な時に追加的な融資を得ることができない場合、私たちは私たちの業務を削減し、行動する必要があります。その中には、外部専門サービス提供者の使用を減らすこと、従業員や従業員の報酬を減らすこと、私たちの候補製品開発を大幅に修正または延期し、私たちの候補製品を商業化する権利を第三者に許可することが含まれている可能性があります。そうでなければ、他のアプリケーションを求めたり、運営を一時停止したりします。
もし私たちが株式または転換可能な債務証券を売却することで追加資本を調達すれば、私たちの既存株主の利益は希釈され、条項は清算または他の既存株主の権利に悪影響を及ぼす特典を含む可能性がある。もし私たちが優先株を発行すれば、私たちの株主の権利に影響を与えたり、私たちの普通株の価値を下げたりするかもしれない。特に、将来の優先株保有者に付与される具体的な権利には、投票権、配当金および清算の優先権、転換および償還権、債務返済基金条項、および第三者との合併または第三者への資産売却能力の制限が含まれる可能性がある。債務融資が可能であれば、追加債務を招く、資本支出を行う、または配当を宣言するなど、私たちが具体的な行動をとる能力を制限または制限する契約を含むいくつかの合意が含まれる可能性がある。これらのすべての事件は、規制承認と商業化目標を達成する能力に悪影響を与え、私たちの業務を損なう可能性がある。
私たちの将来の成功は、追加資金、第2/第3段階臨床試験、および必要な将来の試験を得る能力、および私たちの候補製品のために米国、EU、および他の国際市場でマーケティング承認を得る能力にかかっている。もし私たちが必要な時に追加の資金を得ることができなければ、もし私たちの第2段階/第3段階の臨床試験が成功しなければ、もし私たちが未来の試験に必要な規制の承認を得なければ、あるいはこれらの研究が終了すると、私たちのSBP-101候補製品がマーケティング承認を得られなければ、私たちは経営を続けることができず、運営を停止させることになるだろう。本募集説明書に含まれる財務諸表の作成仮説は、資産の回収可能性や分類に関するいかなる調整も含まれておらず、これらの不確実性の結果に起因する可能性のある負債額も含まれていない継続的な経営企業であると仮定する。
許可協定
UFRFとの独占ライセンス契約によると、前回の改訂は2019年10月4日であり、ライセンス技術開発のライセンス製品の純売上高の2.5%から5%の特許権使用料の支払いが要求され、期間は短い:ライセンス製品の初商業販売から10(10)年、または各国ベースの市場独占期間。最新の修正案は未来のすべてのマイルストーン支払いをキャンセルした。その会社はまだ年間10,000ドルのライセンス維持費を支払うことを約束した。
最近の会計公告
最近の会計声明の検討については、以下のF-16ページから始まる2021年と2022年12月31日終了財政年度連結財務諸表付記4を参照。
商売人
第1部
|
第1項。 |
業務.業務 |
Panbela治療会社とその完全子会社であるPanbela Research,Inc.,癌予防製薬有限会社(アイルランド)と癌予防製薬会社(総称して“私たち”,“私たち”,“Panbela”,“会社”)が存在する主な目的は,破壊的療法を開発し,緊急医療ニーズのある患者の治療に用いることである。Panbela Treateutics Pty LtdはPanbela Research,Inc.の完全子会社です。癌予防製薬有限責任会社と癌予防製薬有限会社(イギリスとウェールズ)は癌予防製薬会社の完全子会社です。わが社の前身は2011年にデラウェア州法律に基づいて設立された商業実体です。“普通株”という用語は私たちの普通株を意味し、1株当たり0.001ドルの価値がある。
がん予防製薬会社(“CPP”)買収
2022年6月15日、Panbelaは個人臨床段階会社であり、癌およびまれな疾患のリスクおよび再発を低減するための治療方法を開発し、合併の対価には、(A)164,689株の普通株、(B)18,298株が制限されている(合併協議参照)普通株を購入し、(C)加重平均発行価格で1株当たり14.00ドルで最大39,918株の普通株を購入する代替オプション、および(D)加重平均執行価格で1株当たり165.80ドルで最大8,451株の代替株式を購入する代替株式証明である。決済後や支払いがあれば、最高6000万ドルに達しますが、マイルストーンの要求を満たす必要があります。
持株会社再編
2022年6月15日からPanbelaはPanbela Research,Inc.(前身はPanbela Treateutics,Inc.,“前身”)の後継者となり,ホールディングス再編により前身はPanbelaの直接完全子会社となった。Panbelaは改正された1934年の証券取引法(“取引法”)により公布された第12 G-3(A)条規則により前身の後継者となった。
業務の概要
Panbelaは臨床段階の生物製薬会社であり,破壊的療法を開発し,緊急医療ニーズのある患者の治療に用いられている。著者らは現在、著者らが膵臓癌を治療する無作為二重盲検プラセボ対照臨床試験に参加する患者を募集しており、これはアメリカ国家癌研究所(NCI)が援助した第三段階の臨床試験であり、結腸癌リスクの低下と結腸腺腫治療(“CAT”)を研究するために用いられ、これは結腸直腸癌生存者或いはハイリスク結腸ポリープに対する予防的治療方法である。そのほか、著者らは家族性腺腫性ポリープ症(FAP)のために第三段階登録試験を設計しており、FAPは稀な遺伝性疾患であり、数千例の結腸直腸腺腫(即ち腺腫性ポリープ)の成長を招くことができ、後者は結腸癌の重要なリスク要素と考えられている。我々はまた、研究者による試験および会社が後援するいくつかの前臨床試験を支持し、(1)青少年糖尿病研究財団によって援助された早期発症1型糖尿病を治療するI期およびII期臨床試験、(2)NCIによって援助された胃癌治療のII期臨床試験、(3)STK 11変異を有する非小細胞肺癌(NSCLC)のI/II期臨床試験の治療、および(4)孤児疾患および癌領域で支援される前臨床研究を含む。
同社の主な資産はアスピリン(SBP−101),フリリンポヴィであるTMEflornithine(CPP-1 x)およびeflornithine(CPP-1 x)は、癌および自己免疫のような多くのタイプの疾患に存在する障害の生物学をリセットするための多標的の方法を提供する。多くの腫瘍はそれらの成長と生存を支持するために大量の上昇したポリアミンを必要とする。これらの薬剤は相補的なリンカーであるポリアミン経路を標的としており,これらの結合が疾患で変化することが証明されている。特に、我々の鉛資産は腫瘍の成長を抑制と防止し、他の抗癌薬の抗腫瘍活性を増強し、免疫系を調節する潜在力がある。
エピピミンはポリアミン代謝抑制を誘導することを目的とした独自のポリアミン類似体である。アスピリンは膵癌患者の臨床試験で鼓舞的な抗転移疾患活性を示す。著者らが完成したI期臨床試験において、エビスタチンはゲシタビンとNAB-パクリタキセルを併用して転移性膵臓癌を治療する有効性と安全性結果は現在の無作為、二重盲検、プラセボ対照研究に支持を提供し、この研究は以前治療を受けていない転移性膵臓癌患者に伊司アスピリンとゲムシタビンとNAB-パクリタキセルの併用に支持を提供した。開発が成功すれば,伊司アスピリンは膵癌患者を有効に治療する新しい方法を代表し,この市場の主導的な製品になる可能性があると信じている。過去25年間に、アメリカ食品と薬物管理局(FDA)は3種類の第一線の治療組み合わせ、一部の患者(3%-7%)に対する単一維持治療及び1種の二線薬物のみを許可し、膵臓癌の治療に用いた。アスピリンは米国で膵癌治療の急速チャネル状態と孤児薬物指定状態を獲得し,ヨーロッパでも孤児薬物指定を受けている。
私たちは2022年6月にCPPを買収し、様々な形で会社の第二の主要資産であるエフルオロオルニチンを増加させた。まず研究中の新薬Flynpoviであり,ポリアミン合成阻害剤であるエフルオロオルニチンと非ステロイド抗炎症薬であるスリン酸の組み合わせであり,次いで単一薬物であるエフルオロオルニチンである。エフルオロオルニチンは酵素活性化の不可逆的オルニチン脱炭酸酵素(ODC)阻害剤であり、ODCはポリアミン生合成における第一種の律速酵素である。スリン酸は非ステロイド性抗炎症薬(NSAID)の一種であり、ポリアミンの出口と分解代謝を促進する。Flynpoviは独特な二重作用機序を有し,新しいポリアミンの合成を抑制し,食事や微生物群におけるポリアミンの出口と分解代謝を増加させる。FlynpoviはFAP手術や大腸癌につながる可能性のあるリスク因子(例えばポリープ)を治療するために設計されているため,様々なタイプの結腸癌を予防する能力がある可能性があると信じている。FAP−310第3段階試験では,成人FAP患者におけるFlynpovi(エフルオロオルニチン(CPP−1 X)とスルリン酸)の治療効果と安全性を行い,いずれかの薬剤単独使用と比較した。この研究は期待に達していないが,主な複合終点(Burkeら)である。2020年),ある専門的な分析では,併用群の患者は48カ月に及ぶ下胃腸(LGI)手術が必要となっていないのに対し,シュリン酸とエフロン群の患者はそれぞれ13.2%と15.7%(Balaguerら)であった。2022年)。これらのデータは,併用療法と単一治療の間でLGI手術が必要なリスクが100%近く低下していることに対応している。LGI群の統計学的意義を考慮して、新薬申請(“NDA”)をFDAに提出したが、これは発見的分析に基づく結果であるため、完全な返信(“CRL”)が発表された。この欠陥問題を解決するためには,同社は臨床終点への影響を証明するために,1つまたは複数の十分かつ良好に制御された臨床試験の結果を提出しなければならない。FAPに対する薬物療法は現在のところ承認されていない。
その他の計画は単剤錠剤エフルオロオルニチン或いは高用量粉末エフルオロオルニチンカプセルのいくつかの適応を評価しており、胃癌、最近発病した1型糖尿病、転移性去勢抵抗前立腺癌とSTK-11突然変異の非小細胞肺癌を含む。臨床前研究及び第一段階或いは第二段階の研究者が開始した試験により、エフルオロオルニチンの治療耐性は良好であり、潜在的な活性を有することを表明した。
Flynpoviは米国でFast Track称号を獲得し,米国とヨーロッパでFAPの孤児薬物称号を獲得した。また,エフロンは米国やヨーロッパで神経芽細胞腫治療の単一薬剤として,米国で胃癌を治療する孤児薬として指定されている。
臨床試験
アスピリン(SBP−101)
FDAは2015年8月,我々のIVOSPEMEN(SBP−101)候補製品の研究新薬(IND)の申請を受けた。著者らはすでに過去に治療した局部末期或いは転移性膵臓癌患者に対する初歩的な臨床試験を完成した。これは第1段階の人類初の用量逓増安全性研究である。2016年1月から2017年9月まで,I期試験の用量増加段階で,29名の患者を6つのキューあるいはグループに組み入れた。いずれの用量レベルでも薬物に関連する骨髄毒性あるいは末梢神経病変は認められなかった。安全性評価を受ける以外に、29名の患者の中の23人は第1の治療周期の終了前或いは8週間の終了時に固形腫瘍反応評価標準(RECIST)を用いて初歩的な治療効果信号を評価することができ、RECISTは現在公認されている腫瘍の大きさの変化を評価する標準である。この完成した臨床試験の安全性と初歩的な治療効果信号の要約は後でIに含まれるボルスミン(SBP-101)の臨床研究進展–膵臓癌、第一段階臨床試験設計と完成(伊維司明単一療法)。
2018年,われわれの第2の臨床試験に参加する患者を募集し始めたのはIa/Ib期研究であり,アスピリンと2種類の標準看護化学療法薬ゲムシタビンとNAB−パクリタキセルの併用の安全性,有効性,薬物動態を検討することを目的とした。計25名の被験者が4つのキューに参加し,用量レベルとスケジュールを評価した。また25名の被験者が試験の拡張段階に参加した。中間結果は2022年1月に発表された。評価可能な被験者(キュー4とIBN=29)では,最適反応は完全緩解(CR)1例(3%),部分緩解(PR)13例(45%),安定期(SD)10例(34%),進行性疾患(PD)5例(17%)であった。1人の被験者はRECIST腫瘍評価のベースライン後スキャンを受けなかった。中位無進展生存期間(PFS)は現在最終的に6.5ケ月であり、すでに薬物投与量中断の負の影響を受けた可能性があり、潜在的な毒性を評価する。2022年1月にデータを公表したところ,キュー4+Ib期患者の平均総生存期間は12.0カ月であり,現在最終的には14.6カ月であった。列2の2人の患者は、1人の患者が30.3カ月(最終データ)であり、もう1人の患者が33.0カ月であり、2022年3月18日まで生存していることを証明している。2022年3月18日までのデータ遮断日には7名の被験者が生存しており,そのうち1名は第2群,6名は第4群プラスIbからであった。研究設計、安全性、仮治療効果信号の詳細については、以下の“ビジネス”の節を参照されたいプロポフォール(SBP−101)の臨床応用進展–膵癌、Ia/Ib期臨床試験中期結果(第一線併用治療).
著者らのIa/b期研究が示した安全性結果と腫瘍成長の抑制は2022年1月に開始したSBP-101ランダム研究に支持を提供した。この試験はASPIRE試験と呼ばれ、ランダム、二重盲検、プラセボ対照試験であり、ゲムシタビンとNAB-パクリタキセルを併用し、以前治療を受けていなかった転移性膵臓癌患者に応用されている。この実験は世界的にアメリカ、ヨーロッパ、アジア太平洋地域の約95地点で行われている。ASPIRE試験は2022年に開始され、アメリカと世界の他の地域の臨床サイトの開放速度は最初の予想より遅く、一部の原因は医学界の資源が疲弊しているが、同社はすべての国とサイトが2023年に開放されると予想している。
この実験は最初にII/III期に設計され,PFSによる中期分析に必要なイベントと全体生存の主要な終点をサポートするためにサンプル量が小さい(150)。この研究は,ヨーロッパやFDA規制フィードバックへの応答として,総試験サンプル量(600)と修正された設計を含むように修正され,総生存率を主な終点とし,中期分析で検査を行っている。またPFSを分析し、追加の治療効果証拠を提供する。この改訂はIa/Ib期の一線転移性膵癌試験の最終データの支持を得ており,2020年12月に登録を完了した。この研究は600名の被験者を募集し,登録まで36カ月を要し,中期分析は2024年中に完了する予定である。独立データ安全監視委員会はすでに2回の会議を開催しており,最近では2023年11月に214名の患者の安全性を評価した。2回の会議では何の安全問題も生じず、裁判は継続され、何の修正もなかった。研究設計と期待時間に関するより詳細な情報は本“ビジネス”の節以降であるプロポフォール(SBP−101)の臨床応用進展–膵臓癌、無作為臨床試験設計と期待時間(ASPIRE試験)。
FDAが推奨するすべての臨床研究を成功させることができれば,FDA,欧州医薬品局(EMA)(EU),TGA(オーストラリア)の上場許可を求める予定である。アスピリンが地理的地域ごとに孤児薬に指定されている場合には,提出費用を免除することができる。
同社は2022年4月初め、米国癌研究協会年次総会で、卵巣癌ポリアミン代謝調節剤としてのアスピリン(SBP-101とも呼ばれる)の研究結果を重点的に紹介したポスタープレゼンテーションを発表し、その後、2022年6月に“国際分子科学誌”(Holbertら)に発表した。2022年)。ポスターや出版物では,VDID 8+卵巣癌細胞を投与したC 57 BL/6マウスはアスピリン治療を受け,生存期間を有意に延長し,全体の腫瘍負担を低下させたと結論している。これらの結果は,エビスタチンが卵巣癌の臨床治療に機能している可能性を示唆しており,同社は卵巣癌の臨床前と臨床研究を継続しようとしている。2023年4月、同社は卵巣癌の追加臨床前作業を重点的に紹介するポスタープレゼンテーションを発表した。このポスターはSBP-101と標準看護化学療法薬物の併用による白金耐性卵巣癌の治療効果を強調した。ゲムシタビン、トポテカン、アドリアマイシンの治療は著しく増加したことが証明されています体外培養シスプラチンに対するSBP-101の感受性と薬剤耐性卵巣癌細胞の毒性。パクリタキセルとドセタキセルは追加的なメリットがないことが証明されています体外培養SBP-101だけですポスターの結論は,VID 8を含むC 57 BL/6マウスに対する治療である+SBP-101とアドリアマイシンを併用した卵巣癌患者は生存期間を明らかに延長し、総腫瘍負担を低下させた。
他の臨床前作業は進行中であり,多発性骨髄腫(細胞系)におけるエフロキサシンとエフルオロオルニチン(CPP−1 xあるいはDFMOとも呼ばれる)の役割を評価している。11月の“血液”誌増刊に発表されたデータは,エビアスピリンとCPP−1 Xが骨髄腫細胞系の体外成長と活力に及ぼす影響を調べた。結果により、エスプミンとCPP-1 X処理は1組の多発性骨髄腫細胞系の細胞増殖を明らかに抑制し、そのアポトーシスを誘導した。アスピリンとCPP−1 Xを併用した場合,細胞成長はほぼ完全に消失した。これらの結果はエスプミンとCPP−1 Xの抗腫瘍潜在力を証明し,その臨床開発が潜在的に有望な多発性骨髄腫治療案に納得できる理由を提供した。この仕事は,同社がテキサス大学MD Anderson癌センターの研究者と協力し,ポリアミン代謝阻害剤療法とCAR−T細胞療法の組み合わせを臨床前モデルで評価していることを反映している。
フリリンポヴィ
2009年12月、FDAは私たちの共同製品FlynpoviのIND申請を受け入れた。Flynpoviは、再発した結腸腺腫、特にハイリスク癌前ポリープを予防するために、NCIが支持する無作為·プラセボ対照のIIb/III期臨床試験で有望な結果を示した。この試験では、散発性腺腫を切除した375名の患者がエフルオロオルニチン(500 mg/d)+舒林酸(150 mg/d)の3年間の治療を受けた[N = 191])または一致するプラセボ/プラセボ(N=184)。その結果,プラセボと比較してプラセボと比較して能動併用療法は異時性腺腫発生リスク(70%),進行腺腫発生リスクは92%,多発性腺腫発生リスクは95%(Meyskensら)と有意に低下した。2008年)。この併用療法は全体的に耐性が良好であった。
散発性とFAP関連性腺腫性ポリープ症の発病機序が類似していること、及びFlynpoviが散発性腺腫とFAP患者の進行性ポリープの予防における作用機序を考慮して、FAPは第三段階計画を開始し、そして西南腫瘍学グループ(“SWOG”)とアメリカ国家癌研究所と協力して第三段階計画を開始し、結腸癌リスクの低減を研究した。
2019年に完成したFAP−310第3段階研究では,いずれかの薬剤単独と比較して,エフロンとスルリン酸の併用による家族性腺腫性ポリープ症を有する成人の治療効果と安全性を検討した(Burkeら)。2020)。患者は1:1:1の割合でランダムに分配し、エフルオロオルニチンと/またはスリン酸を毎日1回服用し、最長48ケ月間持続した。事件発生時間分析における評価の主要な終点は疾病進展であり、大手術、内視鏡下末期腺腫切除、直腸或いは直腸袋高度非典型的増殖の診断、或いは十二指腸疾患の進展と定義される。計171名の患者がランダムにグループ分けを受けた。エフロン−スルリン酸群56名中18名(32%)に疾患進展が認められ,舒林酸群58名中22名(38%),エフロン酸群57名中23名(40%)が悪化し,危険比は0.71(95%信頼区間)であった[語彙表]スリン酸と比較してエフロン−スリン酸は0.39~1.32(P=0.29)であり,エフロン酸と比較して0.66(95%CI,0.36~1.23)(Burkeら)であった。2020)。各治療群の副作用と深刻な副作用は類似している。一件後の分析では,併用群では48カ月以内に下胃腸(“LGI”)手術が必要に進展しなかったが,シュリン酸とエフロン群はそれぞれ7名(13.2%)と8名(15.7%)の患者(Balaguerら)であった。2022年)。これらのデータは,併用投与と単一投与と比較してスリン酸およびエフルオロオルニチンのHR=0.00(95%信頼区間,0.00−0.48;p=0.005)およびHR=0.00(95%信頼区間,0.00−0.44;p=0.003)の併用投与が単一投与と比較して胃腸損傷手術のリスクが100%近く低下していることを示している。LGIグループの統計学的意義を考慮して、FDAは秘密保持プロトコルを提出した。研究は主な終点に到達できなかったが,秘密保持プロトコルは探索的分析の結果に基づいているため,完全な返信を送信した.この欠陥問題を解決するためには,同社は臨床終点への影響を証明するために,1つまたは複数の十分かつ良好に制御された臨床試験の結果を提出しなければならない。
NCIとSWOGの協力のもと,結腸癌生存者としてFlynpoviが使用する治療法の利点を検討するための第3段階臨床試験が開始されている。この試験は“エフルオロオルニチンとシュリン酸による腺腫や癌予防”と命名された。PACE試験はNCIが援助し,SWOGが管理している。0−III期結腸癌または直腸癌患者の高リスク腺腫および第2原発結腸直腸癌の再発を予防するための第3段階二重盲検プラセボ対照試験が行われている。この研究の目的は,Flynpovi(対応するプラセボと比較して)が1日3年間服用した後,対照ARMと比較して癌または高リスク腺腫の再発率が低下しているかどうかを評価することである。規制と商業目的のために、私たちは実験からのデータの独占的な権利を持っている。同社はEUとアジアにおける結腸直腸腺腫療法(CAT)の選択を評価している。
2023年4月、同社は、CPPとOne-Two Treateutics Assets Limitedとの間の許可協定が2023年7月4日から終了したため、FAP患者においてFlynpoviを開発および商業化する北米権利を再取得したと発表した。
エフルオロオルニチン(CPP−1 X)とエフルオロオルニチン(CPP−1 X−S)
上には モノクロニチンは,STK 11変異非小細胞肺癌患者におけるI/II期試験と,最近発症したI型糖尿病患者におけるEflornithineを用いたII期試験が開始されたエフルオロオルニチンカプセル(CPP−1 X−S)を評価している試験を行っている。最後に,エフルオロオルニチンによる胃癌予防を評価する第2段階試験は2021年に完了し,データ解析が行われている。
2024年1月23日までに私たちは
|
● |
アスピリンの孤児薬名を米国食品·薬物管理局から得た |
|
● |
INDのエビスタット申請に対するFDAの承認を提出し、受信する |
|
● |
オーストラリア、フランス、イタリア、スペインでAspire試験を受けた国で承認された |
|
● |
転移性膵管腺癌患者のIa期単剤治療の安全性研究を完成する |
|
● |
転移性膵臓癌の治療によりFDAの“快速チャンネル”の称号を獲得した |
|
● |
私たちの2つ目の試験では、以前転移疾患で治療を受けていなかった膵管腺癌患者に対する第一線の研究であるIa/Ib期の臨床研究である中期結果を登録し、50人の被験者が参加し、そのうち25人がIa期、25人がIb期または拡張期であった |
|
● |
ジョンホプキンス医学院と2年間の研究協定を締結し、国際公認のポリアミン生物学研究員ロバート·カセロ教授が指導した |
|
● |
完成したプロセス改善措置は、商業用途に使用可能であることが予想され、このような新しいより短いアスピリン合成法に関する特許の問題通知を受けた |
|
● |
ランダム、二重盲検、プラセボ対照研究を開始し、以前転移治療を受けていなかった膵管腺癌患者の中で、ゲムシタビンとNAB-パクリタキセルを連合投与した |
|
● |
完成した臨床前評価:術前に膵臓癌を切除可能な新しい補助治療のためのエビスタチン; |
|
● |
卵巣癌腫瘍成長抑制活性の早期、臨床前適応を獲得し、ASCO-GI会議で結果を公表した |
|
● |
USANがSBP−101に対して採用した非特許名伊文司普明を受信した |
|
● |
CPPを買収·統合し、多様な形で第2のリード資産を増加させ、臨床前から登録レベルまでの臨床試験の拡張臨床開発計画 |
|
● |
欧州医薬品局(“EMA”)孤児薬品委員会はPanbelaが孤児に指定を申請したアスピリンとゲムシタビンとNAB−パクリタキセルを併用した転移性膵管腺癌患者の治療に積極的な意見を発表した |
|
● |
インディアナ大学によるエフルオロオルニチンによる早発性I型糖尿病治療の第2段階計画の開始を発表した |
|
● |
モフェット癌センターによるSTK 11変異を有する非小細胞肺癌(NSCLC)のI/II期臨床試験の開始を発表した |
|
● |
テキサス大学MDアンダーソン癌センターと協賛研究協定を締結し、臨床前モデルにおいてポリアミン代謝阻害剤療法とCAR-T細胞療法の結合を評価した |
|
● |
SWOG癌研究ネットワークのPACE S 0820の第3段階試験が単一計画の無効性分析に合格し、継続することを発表した |
|
● |
米国WorldMeds NDAが、腫瘍学におけるポリアミンの最初の承認であるエフルオロオルニチン(DFMO)の小児神経芽細胞腫の承認を許可することを発表した |
|
● |
ASPIRE全世界の臨床試験に参加した人数は50%を超えた。 |
すい臓がん
膵癌はヨーロッパ約151,000人(“ヨーロッパ疫学とハイリスク群スクリーニング提案”,Partykaら,2023年7月),米国では年間約64,000人(米国癌協会)を悩ませている。癌の事実と2023年のデータですジョージア州アトランタ:米国癌協会;2023年と膵臓癌の概要)および世界293,000人-欧州および米国(GLOBOCAN 2020)は含まれていない。ヨーロッパ第4位の癌死亡原因(GLOBOCAN 2020)と米国第3位の癌死亡原因(SEER癌統計概況2021)として決定されている。平均的には,膵管腺癌(“PDA”)は所与の例年診断されたすべての膵癌の約95%を占めている。2つの最もよく用いられる治療案を考慮すると,以前治療を受けておらず状態が良好であった患者の総生存期間の中央値は8.5カ月(Von Hoff 2013)から11.1カ月(Conroy 2011)の間であり,有効なPDA治療は依然として主要な未満足医療需要である。
膵臓癌は通常早期診断ができず、最初の臨床バイタルサインと症状は曖昧で非特異的であるため。最もよく見られる症状は体重減少、上腹部(腹部中央上部)および/または背部痛と黄疸を含む。背部痛は上腹痛に比べて通常鈍感で持続的,内臓起源であり,背部に放射され,上腹痛はぼやけて間欠的である。あまり一般的でない症状は、吐き気、嘔吐、下痢、拒食および新たな糖尿病(これは早期シグナルである可能性がある)またはグルコース不耐性(Hidalgo 2010)を含む。
手術は,約20%の患者のみが診断時に手術切除に適しているにもかかわらず,唯一の治療意図を有する治療選択である。根治性手術を受けた患者の生存率は依然として限られており,平均23カ月(Macarulla T,et al Clin Transl Onol 2017)であった。
切除可能な疾患を有する少数の患者にとって、手術は治療の選択である。腫瘍の位置によって、手術過程は頭部膵臓十二指腸切除術に関連する可能性があり、“ホイップル手術”と呼ばれ、即ち遠位膵臓切除術或いは全膵臓切除術である。膵臓酵素欠乏症と糖尿病はこの病気とこれらの外科手術のよく見られる合併症である。70%に達する膵臓癌患者は胆道閉塞が存在し、経皮或いは内視鏡ステント留置術によって緩和できる。しかし,腫瘍が完全に切除されても膵癌患者の結果は失望的であった(Hidalgo 2010,Seufferlein 2012)。3つの大型無作為臨床試験(Hidalgo 2010)では,術後化学療法は無進展と総生存率を改善したが,すべての3試験で治療を受けた患者の術後生存率中央値は20−22カ月と類似していた。術前(新補助)化学療法はますます人々の注目を集め、その目的は手術切除率と長期治療効果を高めることである。
切除不能、局部末期或いは転移性疾患を有する患者では、これらの患者は大多数のPDA患者の割合を占め、治療選択範囲は単純化学療法から放射線治療と化学療法を結合する治療形式までである。しかし,併用療法の毒性が増加し,このような併用レジメンの無作為試験の登録者数が低いため,化学療法に放射線治療を増加させるいかなる利点についても確実な結論は得られない(Hidalgo 2010)。
ゲムシタビンは現代管理時代に初めてPDA患者の治療に許可された化学療法薬であり、5.65ケ月の中位生存期間(Burris 1997)を提供した。ゲムシタビン単一療法はずっと転移性膵臓癌患者の標準治療方案であり、ゲムシタビンとエルロチニブ(Tarceva)の併用は中位生存期間を2週間延長できることが証明されるまでである。この適度な利点は顕著な副作用と高いコストで緩和され,標準治療案としての採用が制限されている。その後、多剤化学療法とFOLFIRINOXの併用はゲムシタビン単独投与より4.3ケ月(OS=11.1ケ月)の中位生存期間のメリットを提供することが証明されたが、その顕著な副作用はこの方案の選択が良好な患者を制限し、常に白血球増殖因子治療が必要である。NAB−パクリタキセル(Abraxane)は,ゲムシタビンとの併用による市販認可(FDA承認,2013年)を得ており,ゲムシタビン単独使用と比較して全体的な生存期間が7週間増加していることを示している(Von Hoff,2013年)。したがって,次表にまとめたゲムシタビンと比較して,併用療法は適度な生存益を示した(Thota 2014)。
現在の第一線の治療方法:三つの主要な陽性臨床試験の生存と毒性概況
|
吉西他浜と ゲムシタビン/エルロチニ 第3段階試験 |
ACCORD 11実験 |
転移性膵癌 腺癌の臨床研究 試用版(MPACT) |
||||||||||||||||||||||
|
ゲムシタビン |
ゲムシタビン/ エロチニブ |
ゲムシタビン |
FOLFIRINOX |
ゲムシタビン |
ゲムシタビン/ NAB−パクリタキセル |
|||||||||||||||||||
|
1年の生存 |
17% | 23% | 20.6% | 48.4% | 22% | 35% | ||||||||||||||||||
|
全体生存中央値(月) |
5.91 | 6.24 | 6.8 | 11.1 | 6.7 | 8.5 | ||||||||||||||||||
|
中央値無進展生存期間(月) |
3.55 | 3.75 | 3.3 | 6.4 | 3.7 | 5.5 | ||||||||||||||||||
|
全体応答率 |
8% | 8.6% | 9.4% | 31.6% | 7% | 23% | ||||||||||||||||||
|
毒性.毒性 |
||||||||||||||||||||||||
|
好中球減少症 |
– | – | 21% | 45.7% | 27% | 38% | ||||||||||||||||||
|
発熱性好中球減少症 |
– | – | 1.2% | 5.4% | 1% | 3% | ||||||||||||||||||
|
血小板減少症 |
– | – | 3.6% | 9.1% | 9% | 13% | ||||||||||||||||||
|
腹をくだす |
2% | 6% | 1.8% | 12.7% | 1% | 6% | ||||||||||||||||||
|
感覚神経障害 |
– | – | 0% | 9% | 1% | 17% | ||||||||||||||||||
|
疲れている |
15% | 15% | 17.8% | 23.6% | 7% | 17% | ||||||||||||||||||
|
皮疹 |
6% | 1% | – | – | – | – | ||||||||||||||||||
|
口内炎 |
|
0% | – | – | – | – | ||||||||||||||||||
|
感染する |
17% | 16% | – | – | – | – | ||||||||||||||||||
出典Thota Rら、腫瘍学、2014;1月28日(1):70–74
他の薬物は現在調査を受けているが、Abraxane、Lynparzaが許可して以来、PDAを治療する第一線の薬物として発売許可を得た薬物は一つもない®(Olaparib)2019年12月に、一線白金および化学療法レジメンが少なくとも16週間以内に進行しない悪性または有害胚系BRCA変異(GBR CAm)転移性膵臓癌患者の維持治療のための使用が承認された。
家族性腺腫性ポリープ症
家族性腺腫性ポリープ症(FAP)は稀で生命に危害を及ぼす可能性のある遺伝疾患であり、アメリカでは約10,000人に1人が発生する。FAPは主に結腸腺腫性ポリープ症(装甲運搬車)腫瘍抑制遺伝子装甲運搬車突然変異は通常常染色体優性遺伝性状として遺伝するが,25%ものFAP患者は同じ種系変異を有し,家族歴はない。10,000人に1人だけがFAPにかかっています米国の年間流行率は約30,000,ヨーロッパでは約50,000と推定されている。治療しなければ,患者は結腸と直腸全体に数百から数千個のポリープを形成する。FAPは通常青少年早期に発生し、治療しなければ、40歳になると、ほぼ100%の生涯リスクが結腸直腸癌に罹患する。市場で承認されていないFAP薬。
多くの患者は腺腫が大きくなり、多くなるまで症状がなく、直腸出血、甚だしきに至っては貧血、あるいは癌が進展するまで症状がない。一般的に、癌はポリープが出現して10年後に発展し始める。非特異的症状は便秘或いは下痢、腹痛、触知可能な腹部腫瘍と体重減少を含む可能性がある。
癌の予防と良好な生活の質を維持することはFAP患者管理の主要な目標である。10代や20代前半には結腸直腸癌の予防的手術が提唱されている。予防性手術は通常全腹結腸切除回腸-直腸吻合術(“IRA”)とその後の頻繁な内視鏡モニタリングが必要であり、そして必要に応じてポリープ切除と焼灼/レーザーアブレーションを行う。広範な直腸病変の患者は全直腸切除回腸袋肛門管再建術を受けなければならない。それにもかかわらず,全直腸切除回腸袋肛門再建術を受けた患者の約50%が新直腸(回腸パウチ)に腺腫性ポリープを発症する。十二指腸癌と硬線維腫は全結腸切除術後に死亡する2つの主要な原因であり、早期発見と治療が必要である。高位内視鏡検査が必要であり,壷腹と十二指腸癌のリスクを減少させる。進行期腫瘍と切除不能疾患の患者は,細胞毒性化学療法と手術(可能であれば手術)の組み合わせで緩和あるいは安定することができる。FAP患者は100%CRCに罹患するリスクがある;しかし、患者がスクリーニング治療計画に入る時、このリスクは明らかに低下する。
FAP患者の治療において、まだ満たされていない主要な需要は1種の治療手段であり、それは重大な手術干与の需要、特に結腸切除術とIRA或いは直腸切除術と回腸外科袋(IPAA)を延期或いは回避することができる。このような干与措置はよく一時或いは永久性回腸造口術が必要であり、そしてそれに伴い長期或いは永久性の生活の質の欠陥、例えば頻繁な排便(1日平均6回)、夜間便失禁、及び女性患者の生殖潜在力が低下する。重要なのは非手術代替方案を発見し、重複した内視鏡と外科手術を遅延或いは回避して患者の生活の質を維持することである。それらの結腸の特に完全な患者に対して、薬物治療は有意義にポリープの進展を制御或いは遅延する機会を提供し、そして彼らにもっと多くの選択を提供し、いつ或いは予防性結腸切除術/直腸切除術を受けるかどうかを選択し、生活の質を最適化する。
この潜在的な利点は、FAPの長期経過がほとんどの患者に最終的な結腸切除術を受けることを基本的に要求するので、実際には最も強力な潜在的利点である可能性がある。このような急進的な手術を安全に数年遅らせることは若い患者の価値をどのように強調しても過言ではない。
現在、FAP患者の治療は承認され、市販されていない。1999年、セレキシブは、FAP患者において行われた無作為二重盲検プラセボ対照研究において観察されたポリープ数の減少に基づいて、FAPの治療のためのFDAによって条件的に許可されたが、保持者ファイザー社が追加のデータを提供することを市場に許可する必要がある。2011年、FDAは、臨床的利益を検証するための発売後の研究が完了しておらず、H支部によって要求された承認条件として、Celebrex(セレキシブ)カプセルのFAP適応を自発的に市場から撤回することを要求した。2011年の手紙で、ファイザーはFDAにCelebrex(セレキシブ)カプセルのFAP適応を市場から撤回することを要求した。2012年からCelebrex(セレキシブ)カプセルのFAP適応の承認が撤回された。2003年10月にヨーロッパ薬品管理局が科学審査を行った後、セレキシブは欧州委員会に“特殊な状況”でFAP集中治療を許可された。ライセンスは、製品ライフサイクルにおける特定の義務に基づいて付与され、主に、その有効性および安全性に関するさらなるデータを提供するためのものであるが、出願人/ライセンス保持者は、この中央ライセンス後の義務を履行することができない。公開提供された情報によると、ライセンス後の研究は2004年第1四半期に開始され、EU集中マーケティング許可は所有者が必要なデータを提供できないため撤回された。
卵巣癌
世界的に卵巣癌の年間発症率は約314,000人,年間死亡者数は約207,000人(Globocan 2020)である。米国では,卵巣癌は全新発癌症例の約1%を占め,約22,000例(米国癌協会)である。癌の事実と数字2021。ジョージア州アトランタ:米国癌協会;2021年),転移性疾患の5年生存率は約29%(SEER状況説明書卵巣2022)であった。米国癌協会のデータによると、卵巣癌は女性の癌死亡の5番目の原因であり、死亡者数は女性生殖系の他の任意の癌を上回っている。
早期疾患のスクリーニング法の検出に失敗したため,70%近くの患者が末期(Giornelli 2016;Partridgeら)と診断された。2009年Bastら。2007年;Gohaganら。2000年;Chudecka-G≡az 2015年)。したがって,多くの患者は確定診断後の最初の2年以内に再発し,最適な一次細胞減少術や標準的なカルボプラチン/パクリタキセル補助化学療法6周期を行った後も同様である。
二線化学療法は主に無病間隔(“DFI”)(一線化学療法の完成と臨床再発の間の時間)或いは無進展間隔(“PFI”)(最後の化学療法が再発と進行の間の時間)に依存する。白金耐性/薬剤耐性,白金治療期間中の再発(難治),あるいは最後の白金ベースの化学療法後DFI/PFIが12カ月,あるいは白金部分に敏感であり,最後の白金ベースの化学療法後6~12カ月の無病生存(“DFS”)/PFSの3つの分類がある。
Pignataらによると。2017年,白金感受性患者では,単一薬物や非白金組み合わせと比較して,白金ベースの組み合わせ治療がPFS優位に関与していた。一部の敏感な再発患者(PFIは6ケ月から12ケ月の間)に対して、2つの選択がある:白金系薬物或いは非白金系薬物治療(単一薬物或いは連合治療)。最後に,薬剤耐性や難治性再発患者(PFI
結腸直腸癌
米国癌学会が発表した米国癌統計データによると,2022年に米国では結腸直腸癌が男性や女性に最もよく見られる第3の癌となり,癌に関連する死亡の第3の要因となると推定されている。高危険腺腫性ポリープは結腸直腸癌の重要な危険因子と考えられている。2015年に米国では5.2万人がこの病気で死亡すると推定されている。EUの発病率は更に高く、Globocan 2020概況によると、そこでは毎年約25.5万人がCRCで死亡している。
全世界で年間約1,931,000例の新診断例がある(2020年北米では約180,000例予定)。提示速度はアジアでも重要になっている(中国と日本)。結腸直腸腺腫(あるいは“ポリープ”)は結腸直腸癌の重要な危険因子と考えられている。医学界と科学界の一般的な共通認識は、これらのポリープは90%以上の結腸直腸癌の前兆であることである。
結腸癌はアメリカのすべての結腸直腸癌の4分の3近くを占めている。手術(補助化学療法を含むか、または含まない)によって結腸癌を治愈する可能性があるが、局部と局部末期結腸癌患者は依然としてかなり大きな結腸腺腫、遠隔再発、続発性結腸癌形成と結腸直腸癌関連死亡のリスクに直面している。ポリープ切除術は大腸癌死亡率を低下させる有効な方法のようであるが,コストや患者受容性に制限されている(Newcombら)。1992年;Selbyら。(1992年)。あるタイプの大腸ポリープは結腸直腸癌に進展するリスクを増加させる。ハイリスクポリープ(絨毛組織学的ポリープ,大きさ1 cm,高度非典型的増生,あるいは3または3以上と定義された多発性腺腫)が結腸直腸腫瘍発生研究の重点となっており,これらの病変の悪性の可能性が高い(Lotfiら)。1986年、スペンサーら。1984年Winawerら。1993年Martinezら。2009年)。現在、切除した結腸癌患者の看護標準は臨床検査、実験室分析と結腸鏡評価によるモニタリングである。しかし、データにより、結腸鏡検査は結腸全体の範囲内で結腸直腸癌の死亡を一致的に予測することはできない--事実、右側結腸直腸癌は結腸鏡検査からいかなる死亡益も得られなかった(Baxterら)。2009年)。結腸鏡検査の他の潜在的な問題は(非常に少ない)穿孔、感染、出血と現在の提案を守らないことを含む。そのため、安全と有効な化学予防干与は現在の結腸癌モニタリング例を補充と改善するために巨大な潜在力を提供した。 CATを治療するための他の治療法と異なり、Flynpoviは非手術と非侵襲的選択であり、患者の生活の質を高め、更に高い医療保健システムの費用負担を減少する可能性がある。
独自の技術
ポリアミンの機能と特性
ポリアミンは人体細胞内の代謝の異なる実体であり、DNA複製、RNA転写と加工及び蛋白質(例えば膵臓酵素)の合成を結合し、促進する。ヒト細胞は3種類の必要な天然に存在するポリアミンであるフミン,イミノアミン,スペルミンを含む。ポリアミンは多くの細胞増殖、アポトーシスと蛋白質合成に必要な機能を有する。細胞内ポリアミンの臨界平衡はいくつかの酵素、例えばオルニチン脱炭酸酵素(ODC)とイミノアミン/スペルミンN 1アセチルトランスフェラーゼ(SSAT)によって維持される。これらのすべての動態酵素は、天然ポリアミンライブラリーを緊密かつ持続的に調節するために、一時的で迅速に誘導可能な細胞内タンパク質である。これらの酵素は細胞内でポリアミンを非常に狭い濃度範囲に維持し続けている。
ポリアミン代謝と癌
ポリアミンは細胞増殖に必要である。多くの癌,特に癌遺伝子が駆動する癌は,ポリアミン代謝の妨害に敏感である可能性が考えられている。天然ポリアミン腐アミン、イミノアミンとスペルミンは成長に関連する過程、傷口癒合と癌の発展と密接に関連している。通常,ポリアミンライブラリーは合成,分解代謝,輸送機構による調節が厳しく制御されている(GernerとMeyskens 2004)。このような厳密な制御を失うことはポリアミンの過剰蓄積を招き,細胞の悪性形質転換に有利である可能性がある。したがって,癌細胞の成長制御の喪失に伴い,形質転換した細胞は正常細胞のポリアミン枯渇よりも敏感である可能性がある。したがって、ポリアミン代謝経路は治療介入の合理的な目標である(Casero 2018)。
免疫系は多種の可溶性と細胞成分が必要であり、ポリアミンを含めて、正常な免疫機能を発揮することができる。したがって,ポリアミンは免疫反応の重要な調節剤であり,特に腫瘍微小環境では濃度が高い。自己免疫疾患では,腫瘍細胞と自己反応性B細胞とT細胞に高いレベルのポリアミンが存在する。ポリアミンの失調は腫瘍免疫の脱出、細胞圧力の上昇と自己免疫力の増強を招くことができる。治療干与によるポリアミン経路のリセットにより、正常な免疫機能を回復する可能性がある。
ポリアミン経路をリセットする薬物治療方法
同社の主要資産はエビ司普明とフリンポヴィであり、この2種類の薬物は癌と自己免疫などの多種の疾病に存在する失調生物学をリセットするための多標的の方法を提供した。例えば、多くの腫瘍はそれらの成長と生存を支持するために極めて高いレベルのポリアミンを必要とする。これらの薬物は相補的接合部のポリアミン経路を標的としており,これらの結合は疾患の変化であることが証明されている。特に、これらの薬物は腫瘍の成長を抑制と防止し、他の抗癌薬の抗腫瘍活性を増強し、免疫系を調節する潜在力がある。
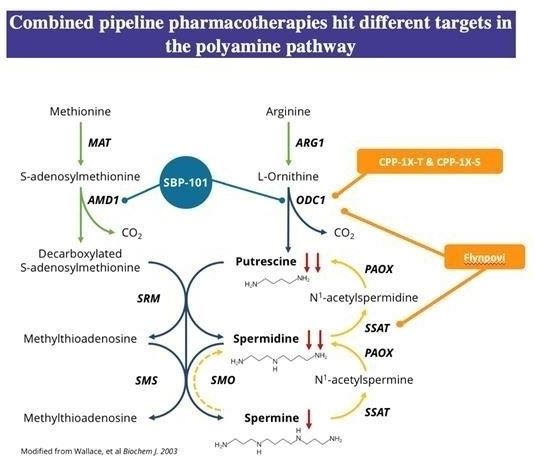
ポリアミン類似体−イビアスピリン(SBP−101)
多くの腫瘍は膵癌を含み,ポリアミンの摂取率が増加している。ポリアミン類似体,例えばアスピリンは,自然に産生されるポリアミンに構造的に類似しており,細胞のポリアミン摂取系によって認識され,これらの化合物が随時細胞に入ることができる。膵腺房細胞は,その非凡なタンパク質合成能により,ポリアミンやポリアミン類似体の増強摂取を示すと考えられる。膵臓腺房細胞のこのような優先摂取のため、ポリアミン類似物、例えばアスピリンは細胞のポリアミン平衡と生合成ネットワークを破壊し、そしてcaspase 3活性化とポリADPリボポリメラーゼ(PARP)切断などの過程を通じて細胞プログラム性死亡或いはアポトーシスを誘導する。概念検証は複数のヒト膵癌モデルで確認されており,両者は体内にあるそして体外培養膵管腺癌はアスピリンに敏感である。
エピピミンは特許のポリアミン類似体であり,その独特の化学構造により膵外分泌腺房細胞に蓄積していると考えられる。フロリダ大学薬学部のRaymond J.Bergeron教授はアスピリンを発見し,広く研究している。
実験室研究所が示したように,アスピリンの主要な作用機序には,この化合物の膵外分泌の摂取促進が含まれていることが証明されており,膵癌はこの化合物の最初の発展の論理的な原因である。動物モデルでは,十分に高い用量で対応する天然ポリアミンレベルの低下,caspase 3活性化,PARP開裂,外分泌腺胞とカテーテル細胞のアポトーシス破壊(プログラム細胞死)を招き,炎症反応がない。重要なことは,インスリンを分泌する膵島細胞は構造や機能的に腺房細胞と異なり,プロポフォールの影響を受けないことである。2つの独立した実験室の動物モデルでは,エビスタチンは転移性膵癌の成長を含めて移植されたヒト膵癌細胞の有意な抑制を示した。
伊司アスピリンは膵外分泌,肝と腎および膵管腺癌細胞の天然親和性を利用しており,インスリンを産生する膵島細胞を損なうことはないと考えられている。現在の多くの癌治療方法は、化学療法、放射線治療と手術を含み、すべて深刻な副作用を伴い、更に患者の生活の質を低下させた。しかし,これまでの臨床研究で評価されたデータから,骨髄抑制や末梢神経病変をきたす場合,アスピリンの副作用は典型的な化学療法レジメンの副作用と重なったり進行したりしないと考えられる。以下に述べるように,われわれの第1段階研究の第5列で観察された用量制限毒性は低用量では観察されず,標準化学療法に通常関連する骨髄抑制や末梢神経病変の有害事象と重複しないことが予想される。最近完成したIa/Ib期拡張段階で評価された用量と投与計画は最大耐容量(MTD)より低く,この用量レベルではヒト膵の外分泌も内分泌もアスピリンの影響を受けないことが予想されるため,治療は膵酵素やインスリンレベルに影響を与えない。新しいASPIRE試験における用量レベルと用量計画はIa/Ib研究拡張段階で評価された用量レベルと用量計画と同じである。
オルニチン脱炭酸酵素阻害剤エフルオロオルニチン(CPP−1 X)
オルニチン脱炭酸酵素はポリアミン生合成における最初の律速酵素であり、それはオルニチンのフミンへの変換を触媒し、哺乳動物と多くの真核細胞中のポリアミンの生合成を調節する。エフルオロオルニチンは、α-ジフルオロメチルオルニチンとも呼ばれ、オルニチン類似体である。エフルオロオルニチンは不可逆的にODC 1に結合し、天然ODC 1基質オルニチンの酵素活性部位へのアクセスを阻止した(MeyskensとGerner 1999)。エフルオロオルニチンの投与はODC活性とポリアミン濃度を低下させる。APC遺伝子変異を有する遺伝子マウスモデルでは,エフルオロオルニチン投与は腸管癌化を減少させ,輸送や分解代謝によりポリアミン濃度を低下させ,腫瘍進展を抑制する(Babbarら)。2003年)。
エフルオロオルニチンによる動物の治療はODC活性を抑制し,特に細胞分裂の迅速な組織や器官である。ポリアミン生合成は真核細胞の成長と分化に重要であることが証明されており,ポリアミン生合成の抑制は細胞分化を刺激あるいは抑制することができ,具体的には研究したモデルに依存する(GernerとMeyskens 2004)。そのため,エフルオロオルニチンは様々なモデルにおいて細胞分化を促進あるいは抑制する。
ポリアミンの生物合成も化学発癌、細胞転化と腫瘍細胞増殖の重要なステップであり、ますます多くの証拠により、エフルオロオルニチンの細胞増殖と腫瘍発生に対する抑制作用は癌遺伝子、ポリアミン代謝とODC活性の間の複雑な相互関係に関与する可能性があることを表明した。MYCは正常細胞増殖に必要な転写因子をコードする癌遺伝子であるが,過剰発現すると細胞異常成長を引き起こす(GernerとMeyskens 2004)。また,c−MycはODC遺伝子の転写活性子(Penaら)である。1993年)(ベロ·フェルナンデス、パーカム、クリーブランド1993年)。また,エフルオロオルニチンは神経芽細胞腫細胞中のN−Myc mRNAやヒト結腸癌細胞におけるc−Myc mRNA(Celanoら)を減少させることが証明されている。イミノアミンはc-Mycの転写と発現を優先的に刺激するが、c-Fosの転写と発現を刺激しない(TabibとBachrach 1999)。以上より,これらの結果は,ポリアミンが転写レベルである癌遺伝子の発現にフィードバック作用を果たしていることを示している。
大腸腺腫性ポリープ症突然変異を持つマウス(APC)腫瘍阻害遺伝子発生腸管腫瘍の数は、FAP患者において発見された数と類似している。遺伝子の変異APC遺伝子はODC活性を増加させ,腸管ポリアミンレベルの増加を招く。FAP動物モデルにおける研究では,エフルオロオルニチンのみで腸管腫瘍数を有効に減少させることが示唆されている(Erdmanら)。1999年b)と結腸腫瘍負担(Yerushalmiら)。2006)。エフルオロオルニチンは大腸粘膜や皮膚細胞におけるポリアミンレベルを低下させる(GernerとMeyskens 2004)。
エフルオロオルニチンの有益な主要な臨床証拠は展望性、ランダム、プラセボ対照の臨床研究から来ており、これらの研究はエフルオロオルニチン単薬によるあるタイプの癌(前立腺癌と基底細胞皮膚癌)を治療するリスクが比較的に高いことを表明した。無作為,プラセボ対照の臨床研究では,結腸ポリープ切除歴のある被験者では,エフロンが直腸粘膜組織中のポリアミンを減少させた。このマーカー研究はFAP患者に特に関連しており,FAP患者では標的組織には腸管や結腸粘膜(Meyskensら)がある。1998年)。
エフルオロオルニチンはすでに監督部門の許可を得ており、大量の静脈投与として、アフリカ昏睡病の治療、および局所薬物として多毛症を治療する(身体部位の毛髪が過度に成長し、毛髪は通常成長していない或いは極めて少ない)。いかなる経口剤形のエフルオロオルニチンもいかなる適応においても規制部門の承認を得たことはない。
イミノアミン/アルミンN-アセチルトランスフェラーゼ活性化剤(“SSAT 1”) –シュリン酸
ポリアミンの輸送はペルオキシソーム増殖剤活性化受容体−g(“PPARg”)によって維持される。この受容体はSSAT転写を順方向に調節し,ポリアミンアセチル化とポリアミンの細胞外への輸送を促進する。正常条件下では、K-RAS分子はPPARgに対して活性がなかった。しかし、K-RAS遺伝子の突然変異は1種の産物を産生し、PPARgのSSAT翻訳への影響を抑制し、それによってポリアミンバンクの増加と腫瘍発生(Babbarら)を招く。2003年)。スリン酸のような非ステロイド性抗炎症薬は、PPARgによってSSATの転写を増強し、それによってポリアミンの分解代謝と出力を増加させる。
シュリン酸は非ステロイド性抗炎症薬中のアリールアルキル酸類薬物の一員であり、プロスタグランジン合成過程におけるシクロオキシゲナーゼの非選択性阻害剤である。舒林酸の潜在的な作用機序を理解するために、著者らは舒林酸スルホン(シクロオキシゲナーゼ阻害活性に乏しい舒林酸代謝物)がヒト結腸腫瘍由来細胞を処理した後の遺伝子発現パターン(Babbarら)を測定した。2003年)。スルリン酸スルホン酸は細胞成長を抑制し,アポトーシスとイミノアミン/アルミンN−アセチルトランスフェラーゼ(SSAT 1)の発現を誘導し,ポリアミン出口に関連するポリアミン分解代謝酵素である(Xie,GillieとGerner 1997)。スリン酸スルホン酸は,シクロオキシゲナーゼに依存しない転写活性化SAT 1遺伝子中の特定のPPARγ反応エレメント(PPRE)によりSAT 1の発現を誘導する。スルリム酸で処理した細胞はSAT 1発現を誘導し,ポリアミン出力を刺激することができる。
ヒト細胞とマウスモデルにおける実験結果は、シュリン酸と他の非ステロイド性抗炎症薬がポリアミン分解代謝を活性化することを示した(GernerとMeyskens,2009)。したがって、非ステロイド性抗炎症薬は、組織中のポリアミンレベルを低下させるために、エフルオロオルニチンなどのポリアミン合成の阻害剤を補充することができる。細胞培養では,シュリン酸代謝物は用量依存的に体外細胞生存を減少させ,24時間曝露時間が150ミクロンを超えた場合(Lawsonら)。2000)。
マウス,ラット大腸癌モデルにおける実験はいずれもスリン酸の予防作用(Babbarら)を証明している。2003年)。シュリン酸は多発性腸管腫瘍(Min)マウスの腫瘍形成を阻止し,MinはFAPのAPC変異関連腸管癌化を模倣したマウスモデルである。Minマウスでは,腫瘍予防用量のシュリン酸は組織中のプロスタグランジンE 2やシクロオキシゲナーゼ−2のレベルを抑制した(Boolbolら)。1996年)。他の非臨床研究において、舒林酸はラットとマウスモデル中の膀胱、肺と前胃腫瘍の形成を抑制する作用がある(Kelloff、Booneなど。1994年Kelloff Crowellらです(1994年)。
二重目標であるFlynpovi
二重作用機序を通じてポリアミンバンクの能力を減少させ、即ち合成を抑制し、分解代謝と出力を増強し、Flynpoviが腫瘍発展を予防する方面で相互に補充し、ポリアミンバンクの増加による腫瘍形成増強を招く患者群の中で仮説を招く。エフルオロオルニチンはODCの不可逆阻害剤であり,ポリアミンの一から合成を担当しているが,スリン酸はSSATを調節し,SSATはポリアミンの出力と分解代謝に作用する。したがって,このFlynpoviという組み合わせは新たなポリアミンの産生を抑制し,食事や微生物群から得られるポリアミンも除去した。
Flynpoviは臨床前,臨床環境ともに胃腸中のポリアミンを減少させる能力がある。Igantenkoらの研究では,Eflornithine単独および非ステロイド抗炎症薬であるスリン酸またはセレキシブとの併用による腸管腫瘍の数,分級およびポリアミン含有量への影響が評価されているAPCMin/+マウス(Ignatenkoら)2008年)。エフルオロオルニチンとスリン酸の併用投与は単独投与より明らかに優れた(PApcMin/+マウス。また,本研究では,0.5%エフルオロオルニチン治療群を除いて,すべての治療群で有意な進展(PApcMin/+マウス)が認められ,高レベル腺腫のパーセンテージを低下させなかった。エフルオロオルニチンとスリン酸の併用は顕著であった(P
エフロン酸とシュリン酸治療群が高レベル腺腫を抑制する能力は重要な発見であり、このモデル中の高レベル腺腫はFAP患者に見られる高レベル腺腫と関連しているため、臨床上切除と手術イベントの指標である。これらのデータはエフルオロオルニチンとシュリン酸の併用によるFAP患者治療の理論基礎を支持し、腸管ポリアミン含有量と高レベル腸腺腫の発生率を減少させる。
さらに重要なことに,Flynpoviとの併用治療は,以前散発性腺腫を有する患者の異時性結腸腺腫の発生率(Meyskensら)を有意に低下させた。2008年)。Meyskensと同僚はIIb/III期,散発性結腸腺腫の二重盲検薬物予防研究(PSCA研究)を行い,散発性腺腫を切除した375名の被験者が3年間のエフルオロオルニチン(1日500 mg)+スリン酸(1日150 mg)の治療を受けた[N = 191])または一致するプラセボ/プラセボ(N=184)。その結果,プラセボと比較してプラセボと比較して能動併用療法は異時性腺腫のリスク(70%),進行腺腫のリスク92%,多発性腺腫のリスク95%を有意に低下させた。この併用療法は全体的に耐性が良好であった。
散発性とFAP関連腺腫性ポリープ症の発病機序、及び散発性腺腫とFAP患者の進行性ポリープの予防におけるエフロンと非ステロイド性抗炎症薬の作用機序は、FAP関連患者におけるFAP-310試験の発展を招いた装甲運搬車生殖系突然変異。
FAP−310第三段階研究は,いずれかの薬物単独と比較して,エフルオロオルニチンと舒林酸の併用による家族性腺腫性ポリープ症の治療効果と安全性を評価した(Burkeら)。2020)。患者は1:1:1の割合でランダムに分配し、エフルオロオルニチンと/またはスリン酸を毎日1回服用し、最長48ケ月間持続した。ある事件後分析では,併用群の患者は48カ月に及ぶ下胃腸(LGI)手術が必要に進展しなかったのに対し,シュリン酸とエフルニチン群の患者はそれぞれ7名(13.2%)と8名(15.7%)であった(Balaguerら)。2022年)。これらのデータは,併用投与と単一投与と比較してスリン酸のHR=0.00(95%CI,0.00−0.48;P=0.005)および併用投与のHR=0.00(95%CI,0.00−0.44;P=0.003)が,いずれも100%に近い胃腸損傷手術のリスクを低下させることを示している。
エピミン(SBP-101)の発展計画
アスピリンの膵癌適応への開発には,臨床前段階と臨床段階がある。臨床前段階は2015年に基本的に完成し、4つの主要な構成部分から構成されている:化学、製造と制御(CMC)、臨床前(実験室と動物)薬理学研究、臨床前毒理学研究及びオーストラリアとアメリカの監督管理提出。
膵臓癌の治療を準備するエスピミンIndは著者らの製造、臨床前毒理学、薬物動態学と代謝専門家、著者らの監督管理事務プロジェクト管理と著者らの内部の臨床専門知識の協力が必要である。2015年8月、FDAは私たちの申請を受け入れた。
オーストラリアでは,ヒト研究倫理委員会(“HREC”)の申請がその後の臨床試験通知(“CTN”)とともに治療薬物管理局(“TGA”)に提出されている。
著者らが以前治療した局部末期或いは転移性膵臓癌患者で行った初歩的な臨床試験はオーストラリアとアメリカの臨床地点で行われたI期臨床試験であり、これは初めての人類用量増加安全性研究である。オーストラリアメルボルンとアデレードの主要癌治療センターおよびアリゾナ州スコッツデールのMayo Clinic ScotsdaleとHonorHealthで膵癌を治療する専門臨床医を招聘した。これらの重要なオピニオンリーダーは,膵癌研究で良好であり,研究者としてわれわれの第一段階ヒト第一研究に参加することに同意した。
この実験の結果はプロポフォール(SBP−101)の臨床応用進展–膵臓癌は I期臨床試験設計と完成(イヴィ司明単一療法)下です。
我々は2020年12月に第2回臨床試験の患者募集を完了した。この第二項の臨床試験はIa/Ib期研究であり、エスプミンと2種類の標準看護化学療法薬物ゲムシタビンとNAB-パクリタキセルの併用の安全性、有効性と薬物動態学を研究した。2020年12月までに25名の被験者がIa段階の4つの列に入り,他の25名の被験者が拡張Ib段階に入った。この実験の安全性と中期的な治療効果はアスピリン(SBP−101) 臨床発展-膵癌、Ia/Ib期臨床試験中期結果(第一線併用治療)下です。
この新しい試験は無作為、二重盲検、プラセボ対照の研究であり、エスプミンと2種類の標準看護化学療法薬物ゲムシタビンとネブ-パクリタキセルの併用の安全性と有効性を研究した。試験設計と予想時間は中で検討する臨床発展-膵臓癌、無作為臨床試験設計と期待時間(ASPIRE試験)。
また,適切な膵癌患者に対する新たな補助治療が模索されている。アスピリンは膵癌以外の癌に対して潜在的な治療用途を有している可能性が示唆されている臨床前データもある。2021年2月、私たちはジョンホプキンス大学医学部と研究合意に達した。協力の重点はPanbelaの研究試薬エビスタットのさらなる開発であり,膵癌以外の細胞系の活性,診断に情報を提供するバイオマーカー,検査点阻害剤との潜在的な結合である。同社は2021年12月,卵巣癌細胞系におけるイビサプミンの活性を支持する積極的な臨床前データを発表し,2022年に提出し発表した(Holbertら)。2022年)。
エピミン(SBP−101)の臨床研究進展–すい臓がん
これまでの膵癌の臨床的進展には
|
● |
2017年に完成した第1段階SBP-101単一療法研究 |
|
● |
A段階Ia/Ib SBP-101の第一線の連合治療研究は、すでに2020年末に登録を完成した |
|
● |
ランダム、二重盲検、プラセボ対照の第一線の連合治療研究は2022年1月にスタートした。 |
これらの計画の詳細は以下のとおりである.
I期臨床試験設計と完成(イヴィ司明単一療法)
著者らはすでに過去に治療した局部末期或いは転移性膵臓癌患者に対する初歩的な臨床試験を完成した。これは第1段階の人類初の用量逓増安全性研究である。2016年1月から2017年9月まで,I期試験の用量増加段階で,29名の患者を6つのキューあるいはグループに組み入れた。いずれの用量レベルでも薬物に関連する骨髄毒性あるいは末梢神経病変は認められなかった。安全性評価を受ける以外に、29名の患者の中の23人は第1の治療周期の終了前或いは8週間の終了時に固形腫瘍反応評価標準(RECIST)を用いて初歩的な治療効果信号を評価することができ、RECISTは現在公認されている腫瘍の大きさの変化を評価する標準である。
従来の化学療法薬の使用中によく見られる有害事象と重複する可能性のない有害事象の存在は、ゲムシタビン、NAB-パクリタキセル、さらにはFOLFIRINOXのようなイスプラミンと従来の化学療法薬の併用を支持する。
Ia/Ib期臨床試験中期結果(第一線併用治療)
2018年,われわれの第2の臨床試験に参加する患者を募集し始めたのはIa/Ib期研究であり,アスピリンと2種類の標準看護化学療法薬ゲムシタビンとNAB−パクリタキセルの併用の安全性,有効性,薬物動態を検討することを目的とした。計25名の被験者が4つのキューに参加し,用量レベルとスケジュールを評価した。また25名の被験者が試験の拡張段階に参加した。中間結果は2022年1月に発表された。評価可能被験者(4列,IBN=29個)では,最適反応はCR 1(3%),PR 13(45%),SD 10(34%),PD 5(17%)であった。1人の被験者はRECIST腫瘍評価のベースライン後スキャンを受けなかった。中間PFSは、現在6.5ケ月であり、潜在的な毒性を評価するために、薬物投与量中断の負の影響を受けている可能性がある。2022年1月にデータを公表したところ,キュー4+Ib期患者の平均総生存期間は12.0カ月であり,現在最終的には14.6カ月であった。列2の2人の患者は、1人の患者が30.3カ月(最終データ)であり、もう1人の患者が33.0カ月で生きていることを証明している。
図4. SBP 101期Ib一線併用治療安全性試験評価- 最適な全体的な応答

ソース:Singhal,N.,ポスタープレゼンテーション,ASCO GI 2022
無作為臨床試験設計と期待時間(ASPIRE試験)
2022年1月、同社は新しい臨床試験を開始することを発表した。この試験はASPIREと呼ばれ、無作為、二重盲検、プラセボ対照試験であり、ゲムシタビンとNAB-パクリタキセルと併用し、これは標準的な膵臓癌治療方案であり、以前転移性膵臓癌を治療したことがない患者に適している。この実験はアメリカ、ヨーロッパ、アジア太平洋地域の約95地点で行われる。
米国や世界の他地域に臨床診療所を開設する速度は当初の予想より遅く,一部に医療界の資源が逼迫していることが原因であるが,同社は2024年初めまでにすべての国や診療所が開放されると予想している。
この試験は最初にII/III期試験として設計され,PFSと全生存(OS)の主要終点に基づく中期分析に必要なイベントを支援するためのサンプル量が小さい(150)。この研究は,ヨーロッパやFDA規制フィードバックへの応答として,総試験サンプル量(600)と修正された設計を含むように修正され,総生存率を主な終点とし,中期分析で検査を行っている。またPFSを分析し、追加の治療効果証拠を提供する。この改訂はIa/Ib期の一線転移性膵癌試験の最終データの支持を得ており,2020年12月に登録を完了した。この研究では600名の被験者を募集し,登録まで36カ月を要すると予想される。
2024年1月25日、同社はこの試験が登録者数の50%を超えたと発表した。同社は、全面登録は2025年第1四半期に完了し、全体の生存状況に基づく中期データ分析は2024年中に提供すべきだと予想している。
FDAが推奨するすべての臨床研究を成功させることができれば,FDA,EMA(EU),厚生労働省(日本),TGA(オーストラリア)の上場認可を求める予定である。エビサミン(SBP-101)が各地理的領域の孤児薬物として指定されている場合、提出費用は、“孤児薬物状態”に記載されているように免除されることができる
フルオロボイルとエフルオロオルニチンの発展計画(CPP−1 X)
FDAは2009年12月、連合製品Flynpoviに関するCPPのIND申請を受け、Flynpoviは候補製品であり、2009年11月および2018年8月にINDのエフルオロオルニチンに対する申請を受けた。
FlynpoviのCPPが実行するFAPと結腸癌予防の開発計画には,臨床前/非臨床と臨床段階がある。非臨床段階は四つの主要な部分から構成されている:CMC、臨床前(実験室と動物)の薬理学研究、臨床前毒理学研究及びアメリカとヨーロッパの監督管理提出。同様に,いくつかの異なる適応のエフロンとエフルオロオルニチン香包の開発計画には,米国規制機関が提出したほとんどの同じ主成分が含まれている。
臨床発展–フリリンポヴィ
これまでFlynpoviの臨床開発には
|
● |
FAP−310第3段階 |
|
● |
PACE第3段階試験 |
FAP−310第3段階試験
FAP-310の第三段階研究において、Flynpovi(ES併用薬)の成人FAPに対する治療効果と安全性について、エフルオロオルニチン或いはシュリン酸単独投与と比較して研究を行った。計171名の患者がランダムにグループ分けを受けた。Flynpovi群では18例(32%),舒林酸群58例中22例(38%),エフルオロオルニチン群57例中23例(40%)に疾患進展があり,Flynpovi群のリスク比はシュリン酸とEflornithine群と比較して0.71(95%CI,0.39~1.32),Flynpovi群のリスク比は0.66(95%CI,0.36~1.23)であった。1つの事後分析では,併用群の患者は48カ月以内にLGI手術が必要に進展しなかったのに対し,シュリン酸とエフロン群ではそれぞれ7名(13.2%)と8名(15.7%)の患者がLGI手術を必要とした。これらのデータは、HR=0.00(95%CI、0.00-0.48;Pに対応する =0.005)スリン酸およびHR=0.00(95%CI、0.00~0.44;P エフルオロオルニチンと組み合わせて使用するための0.003)。
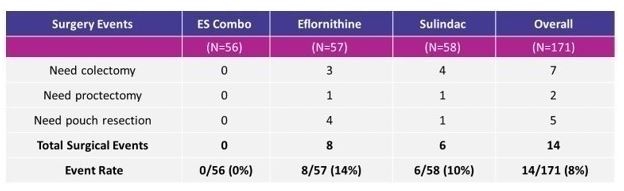
LGIグループの統計学的意義を考慮して、FDAは秘密保持プロトコルを提出した。研究は主な終点に到達できなかったが,秘密保持プロトコルは探索的分析の結果に基づいているため,完全な返信を送信した.この欠陥問題を解決するためには,同社は臨床終点への影響を証明するために,1つまたは複数の十分かつ良好に制御された臨床試験の結果を提出しなければならない。
結腸癌生存者のIII期臨床試験
NCIとSWOGの協力のもと,結腸癌生存者としてFlynpoviが使用する治療法の利点を検討するための第3段階臨床試験が開始されている。この試験は“エフルオロオルニチンとシュリン酸による腺腫や癌予防”と命名された。PACE試験はNCIが援助し,SWOGが管理している。0−III期結腸癌または直腸癌患者の高危険腺腫および第2原発結腸直腸癌の再発を予防するためのFlynpoviの二重盲検プラセボ対照試験である。この研究の目的は,フルボイル,エプタフルオロオルニチンとスリン酸の組み合わせ,(対応するプラセボと比較して)毎日投与3年後に,対照ARMと比較して癌または高リスク腺腫の再発率が低下しているかどうかを評価することである。規制と商業目的のために、私たちは実験からのデータの独占的な権利を持っている。
臨床発展–エフルオロオルニチン(CPP)– 1X)
これまでのエフロン酸の臨床開発には
|
● |
第二段階胃癌予防試験 |
|
● |
最近発症した1型糖尿病のI期とII期試験 |
|
● |
I/II期STK−11変異型非小細胞肺癌試験 |
第二段階胃癌予防試験
ヘリコバクター·ピロリ人類で最もよく見られる細菌感染であり、すべての人が胃炎を引き起こす。胃炎は下落胃域に沿って進展し,胃炎から癌前段階にかけて萎縮性胃炎(特殊胃上皮喪失)と腸化生(IM),胃腺癌に至る(コレア1992)。対.の応答としてヘリコバクター·ピロリ感染宿主は強力な先天性と獲得性免疫反応を引き起こし、粘膜炎症を招くが、生物を根絶することはできない。いくつかの研究により、免疫反応の失敗はL-アルギニン代謝とポリアミンの失調と関係がある可能性があり、マクロファージがオルニチンデカルボキシラーゼ(Chaturvediら)を上方制御することを含む。2010年Chaturvedi、de Sablet、Coburnら。2012年)(Chaturvedi、de Sablet、Peekら。2012年(Chaturvediら)2011)(Xu et al.2004年)(Chaturvediら。2014年(Chaturvediら)。2004年)。体内のポリアミンレベルが増加するピロリ菌-マウス胃炎の誘導とDFMO内服治療は胃ポリアミンレベルと重症度を低下させることができるヘリコバクター·ピロリ定住と胃炎(Chaturvediら)2010)である。スナネズミのモデルではヘリコバクター·ピロリポリアミンレベルは胃炎、DNA損傷と異型増殖/癌に発展する程度と関係がある。このモデルでは,エフルオロオルニチンはポリアミンレベルとDNA損傷を低下させ,異型増殖や癌の発生率を50%以上低下させた(Chaturvediら)。2014年)。
ファンデルビルト大学の研究者とNCIの援助の下で、この研究者による第二段階試験はホンジュラスとプエルトリコで行われ、ランダム、二重盲検研究であり、毎日一回のエフロン酸とプラセボの胃癌前病変患者の18ケ月に及ぶ治療期を比較した。この実験は完了し、データ分析が行われている。同社はすでに米国でエフルオロオルニチンを用いて胃癌を治療する孤児薬の称号を得ている。
I期とII期に新しく発症した1型糖尿病(T 1 D)試験
T 1 Dは臓器特異的自己免疫疾患であり、慢性免疫媒介膵臓β細胞破壊であり、部分的またはほとんどの場合絶対インスリン欠乏をもたらすことを特徴とする。多くの症例は自己免疫を介した膵β細胞破壊によるものであり,この破壊は異なる速度で発生している。膵β細胞の約90%が破壊された場合,臨床症状が出現する。したがって,β細胞を保護する機能は有望な治療の目標である(Couperら)。2014年)。早期糖尿病腎症ではODCの活性が上昇し,腎臓肥大や高ろ過(Pedersenら)をきたす。1992年、トウら。2003年)生体内最近発病したT 1 Dの実験モデルにおいてエフルオロオルニチンを評価する研究により、エフルオロオルニチンは腎臓肥大と増殖の発展を抑制し、糖尿病の発生率を低下させ、β細胞集団の生存と再生を増強し、ランゲルハンス島炎を減少させ、及びT細胞亜群の免疫寛容バランスを維持する上で作用があることを表明した。
同社はインディアナ大学の研究者と協力して、最近発病した1型糖尿病患者におけるエフルオロオルニチンの投与量を増加させる安全性と有効性を評価するために、JDRFによって援助された第一段階の研究を行った。完成した第一段階試験により、エフルオロオルニチン内服3ケ月の治療コースは最近発病したT 1 D児童と成人において良好な副作用特徴があることを表明した。尿ポリアミンデータは,エフルオロオルニチン治療がオルニチンデカルボキシラーゼ活性を有効に抑制し,用量の増加とともに尿フミン値が低下することを反映していることを示している。また,代謝効率は検出できなかったにもかかわらず,750 mg/mの治療を受けた被験者は2/日および1000 mg/m2エプタフルオロオルニチンはプラセボと比較して治療6カ月後により高いCペプチドAUCを示し,β細胞機能の改善が示唆された。これらのデータは、エフルオロオルニチンが、単独で、または免疫治療を含む1型糖尿病の治療または予防の組み合わせレジメンと共にβ細胞の機能を改善することができることを示している。より大きな第2段階研究が計画されており,エフルオロオルニチン治療がCペプチドの維持に及ぼす影響を検出するために,2023年初めに開始することを目標としている。
I/II期STK−11変異型非小細胞肺癌試験
STK 11は肺腺癌の中で4番目によく見られる変異遺伝子であり,30%と高い症例で機能喪失(Laderianら)が発生している。2020)。LKB 1欠損患者の細胞毒性T細胞の浸透は減少し、PDL-1状態にかかわらず、PD 1或いは抗PDL-1治療に対する反応は比較的に悪い。CHECKMate-057試験肺腫瘍はKRASとSTK 11に共通突然変異が存在し、PD-1軸阻害剤に対する反応は比較的に悪かった(Skoulidisら。2018年)。これらの結果は,KRAS状態にかかわらず,STK 11変異の腫瘍が寒冷な免疫微小環境を有することを示唆している。
2つの注釈の良好な肺腺癌データセットを用いたバイオインフォマティクス解析では,オルニチン脱炭酸酵素(エフルオロオルニチンの標的)が上昇した。また、LKB 1欠損腫瘍はいくつかの溶質輸送体の顕著な上昇を示した(SLC 7 A 2, SLC 14 A 2そして、そしてSLC 16 A 4). SLC 7 A 2カチオン性アミノ酸アルギニン,リジン,オルニチンの膜輸送を担うことが知られている。また,LKB 1はアルギニンがオルニチンと尿素(アルギナーゼを介して)とオルニチンからフミン(ODC 1を介して)に変換するアルギニン経路を失った。以上より,これらの結果はODC 1がLKB 1欠損肺癌の重要な代謝駆動因子である可能性を示唆している。
他のモデルシステムにおいて、エフルオロオルニチン治療は腫瘍微小環境を調節できることが証明された。先行研究のキューは、ODC 1が免疫抑制(Chamaillardら)に寄与する可能性があることを示している。1997年)。エフルオロオルニチンはODc 1阻害剤であることから,eflornithineによる代謝酵素odc 1の抑制はLKB 1欠損腫瘍中の腫瘍浸潤性リンパ球(TIL)の数を増加させ,PD(L)−1を回復してこれらの患者への利益を遮断すると推察される。
同社は現在,STK−11変異非小細胞肺癌患者のエフルオロオルニチンを評価するための研究者によるI/II段階試験を計画しており,2023年初めの開始を目指している。
神経芽細胞腫試験
神経芽細胞腫は未成熟神経細胞から起源する稀な癌であり、児童癌死亡の15%近くを占める。[1]Panbela治療会社の子会社癌予防製薬会社は、神経芽細胞腫髄芽細胞腫翻訳研究連盟(NMTRC)(現在児童癌を破った)、神経芽細胞腫治療の新たな進展(NANT)、児童腫瘍学グループ(COG)と国家癌研究所(NCI)などの有力な神経芽細胞腫研究グループと広く協力し、神経芽細胞腫を治療する臨床薬物としてエフロン酸を開発した
2023年7月、同社はそのエフロン小児神経芽細胞腫プロジェクトのいくつかの資産をUS WorldMedsに剥離したことを発表した®1ケンタッキー州に本社を置く専門製薬会社。合意条項によると、Panbelaはエフルオロオルニチンの小児科神経芽細胞腫計画のためのいくつかの資産を売却するために、約950万ドルまでの非希釈資金を得る権利がある。PanbelaはUSWがエフロンの臨床開発、規制承認、商業販売に関連するマイルストーンを成功させた後に支払いを受ける。
2023年12月,同社はUSFMがNDAに対するFDAの承認を得て,緩解期神経芽細胞腫の維持療法としてエフロンを用いることを発表した
総開発コスト
アスピリンの開発には臨床前と臨床開発の2段階がある。著者らはすでに膵臓癌に対する初歩的な臨床前開発と2つのI期臨床試験を完成した。第2段階/第3段階試験は始まったばかりである。もし我々のIVOSPEMEN候補製品の第一線の臨床試験の結果が開発を継続することが合理的であることが証明された場合、外国の司法管轄区でFDAまたは他の承認された追加の臨床試験を行う必要があるかもしれない。追加臨床試験のコストと時間は試験の性質と規模に大きく依存する。
Flynpoviの開発はFAPや結腸癌予防の臨床前と臨床開発にも関与している。同社はFDAと欧州医薬品局(“EMA”)が世界的な登録試験について合意に達することを確保しようとしている。
孤児薬状況
孤児薬品法は、まれな疾患を安全かつ有効に治療、診断または予防することを目的とした薬物は特殊な地位を有し、これらの疾患はアメリカでは20万人未満、あるいは20万人を超える影響を与えると規定されているが、メーカーはこのような薬物の開発と販売のコストを回収すべきではない。孤児薬物を指定する利点は,(I)FDAの承認手続きの簡略化,(Ii)薬物開発に関連する費用の税収減免,(Iii)孤児薬物メーカーのFDAの援助の許可,孤児薬物の承認に必要な臨床試験の助成,(Iv)薬物開発の促進である。さらに重要なことに,FDAは承認後7年間の米国独占営業権を付与し,この孤児薬物メーカーが薬物開発投資を回収する能力を大幅に向上させた。そのため、候補製品を孤児薬物に指定することは、そのスポンサーがより速く、より安価な方法でその製品を商業化する機会を与える可能性がある。
2014年,米国のアスピリン孤児薬の地位を獲得した。2022年12月14日,EMA孤児薬物製品委員会はPanbela申請エトプミン(SBP−101)とゲムシタビンとNABパクリタキセルを併用して転移性膵管腺癌患者の孤児指定に積極的な意見を発表した。
私たちはすでに米国(それぞれ2013年と2011年)とヨーロッパ(それぞれ2013年と2011年)でFAPのFlynpoviとEflornithineの孤児薬物指定資格を取得した。また,米国(2010年)とヨーロッパ(2011年)で神経芽細胞腫単一薬物の孤児薬物指定資格を取得し,米国で胃癌治療(2015年)の孤児薬物指定資格を取得した。
快速通路
2020年6月,ゲムシタビンとNAB−パクリタキセルとの併用時に転移性PDAの第一線患者の治療に用いられるエビスタチンの開発に用いられたFDAの迅速チャネル指定を得た。
また,2017年9月にFDAの高速チャネル指定を取得し,FAP治療のためのFlynpoviを開発した。
高速チャネル指定があれば、私たちまたは北米パートナーはFDAとより頻繁に相互作用する可能性があり、FDAは申請が完了する前に新薬申請(“NDA”)の部分を審査するかもしれない。残りの情報を提出するスケジュールを提供し、申請者が適用された使用料を支払っていれば、スクロール審査を行うことができる。しかしながら、FDA審査申請の期間目標は、NDAの最後の部分が提出されてから開始される。また,FDAが高速チャネルの指定が臨床試験中に出現したデータの支持を得なくなったと考えた場合,FDAはその指定を撤回する可能性がある。
知的財産権
我々の契約メーカーSyngene International Ltdが合成プロセスの改善に努めているため,特許(US 11,098,005 B 2)“生産(65,155)−3,8,13,18−TETRAAAAICOSANE−6,15−diolの方法”が2021年8月24日にPanbelaに発行された新たな短い合成プロセスが開発された。特許請求項は、エフズミンを製造する新しい方法をカバーし、合成ステップを19ステップから6ステップに減少させる。
Flynpoviでは,エフラニチンとスリン酸の固定用量の組み合わせである物質特許の成分が広く国有化され,2037年までの潜在的保護を提供している。また,家族性腺腫性ポリープ症,神経芽細胞腫,および最近発症した1型糖尿病の治療のためのFlynpoviおよび/またはEflornithineのいくつかの使用方法特許を有している。
私たちは追加的な知的財産権を提供する他の機会を評価している。
人的資本管理
2024年1月23日現在、私たちは8人の従業員がいて、そのうち7人はフルタイム社員です。私たちの職員たちは労働組合代表もなく、集団交渉協定のカバー範囲もない。私たちは私たちが職員たちとの関係が良いと信じている。
私たちの人的資本目標には、私たちの既存と新しい従業員、コンサルタント、コンサルタントを識別、採用、維持、激励、統合が含まれている。私たちの株式と現金インセンティブ計画の主な目的は、株と現金に基づく報酬奨励を付与することによって、これらの人員を誘致、維持、奨励し、これらの人員を激励して、私たちの目標を実現し、会社の成功を招き、私たちの株主のために価値を増加させることである。
私たちは労働力の背景と視点の多様性を重視し、私たちの政策は、人種、宗教、信仰、肌の色、民族の血統、血統、身体障害、精神障害、医療条件、遺伝情報、結婚状況、性別表現、年齢、軍人と退役軍人身分、性指向または連邦、州あるいは地方法律が確立された他の保護された特徴に基づいて差別を行わないことである。
私たちは運営責任が私たちの既存の従業員、独立コンサルタント、そして私たちのグローバルCROによって処理できると信じている。我々は従来,独立コンサルタントや請負業者のサービスを用いて様々な専門サービスを提供してきた.私たちは、第三者サービス提供者のこのような使用が、一般的で管理費用を最小限に抑える能力を強化したと信じている。私たちは、私たちの人員や人材ニーズを定期的に評価するつもりで、これがより適切な資源選択になれば、従業員が増えることが予想されます。
競争
癌治療の新製品の開発と商業化競争は激しく、迅速かつ重大な技術変化の影響を受けている。私たちの知識、経験、科学資源は私たちに競争優位を提供してくれると信じていますが、私たちは世界各地の主要な製薬会社、専門製薬会社、バイオテクノロジー会社からの激しい競争に直面しています。私たちの多くの競争相手は明らかにもっと多くの財力、技術、そして人的資源を持っている。規模の小さいスタートアップ企業も重要なライバルとなる可能性があり、特に大手や成熟会社との連携で手配されている。したがって、私たちの競争相手は私たちよりも早く、または成功的に製品を発見、開発、許可、または商業化することができるかもしれない。
私たちは現在の候補製品の面で競争に直面し、未来の候補製品の面でも競争に直面し、これらの競争は製薬、生物技術と他の関連市場の細分化市場から来ており、これらの市場は癌に関連する分子変化とシグナル経路に対する方法を求めている。私たちの競争相手は、私たちよりも早く規制機関のその製品の承認を得ることができ、あるいは特許保護や他の知的財産権を得る可能性があり、これは私たちの候補製品を開発または商業化する能力を制限する。私たちの競争相手はまた、私たちの製品よりも効果的で、便利で、より安価で、あるいはより安全性の高い薬物を開発することができます。これらの競争相手は、彼らの製品を製造し、マーケティングする上で、私たちよりも成功するかもしれません。
また、診断会社と協力して私たちの候補製品を開発する必要があるかもしれませんが、これらの協力を構築する際には、他社からの競争に直面します。私たちの競争相手はまた合格した科学、管理と商業人員を募集と維持し、臨床試験場と臨床試験の患者登録を確立し、著者らの計画を補充し、あるいは必要な技術を提供する方面で私たちと競争を展開する。
また、コスト効果と精算可能な癌治療を得るために、より広範な市場競争に直面している。癌患者を治療する最も一般的な方法は、化学療法、免疫治療、ホルモン治療および標的薬物治療またはこれらの方法の組み合わせを含む手術、放射線および薬物治療である。市販されている癌薬物療法は多種多様である。多くの場合、これらの薬物は治療効果を向上させるために併用される。我々の候補製品(承認されれば)は,これらの既存の薬物や他の療法と競合する可能性があるが,それらが最終的にこれらの療法と組み合わせて使用されるか,またはこれらの療法の添付ファイルとして使用される限り,我々の候補製品は,現在の療法と競合するのではなく,随伴療法として承認される可能性がある。その中のいくつかの薬物はブランド薬物であり、特許保護され、他の薬物は模倣薬である。保険会社および他の第三者支払者は、非特許製品または特定のブランド製品の使用を奨励することも可能である。もし私たちの候補製品が承認されれば、それらの価格はブランド模造薬を含む競争相手の模造薬より高くなると予想しています。したがって、私たちが市場に進出することに成功したどの候補製品も市場の承認を得て、かなりの市場シェアを得ることが課題となる。また,多くの会社が新たな治療法を開発しており,我々の候補製品の臨床開発における進歩に伴い,看護基準が何になるかは予測できない。
商業化する
私たちはまだ販売、マーケティング、あるいは製品流通インフラを構築しておらず、大量の管理資源を投入してこのようなインフラを計画していません。私たちの主要な候補製品はまだ早期臨床開発段階にあるからです。私たちは現在、これらの機能を果たす専門知識と能力を持つより大きな製薬機関と協力することを予想している。
承認されれば、Flynpoviは北米で1:2の速度で商業化されるだろう。
製造業とサプライヤー
私たちは所有したり経営したりしないし、現在は製造施設を設立する計画もない。私たちは現在、臨床前および臨床試験、および商業化される可能性のある任意の製品の商業生産のために、第三者に依存して、私たちの候補製品を生産し続けると予想されている。必要であれば、私たちは供給協定を締結することによって、あるいは他の手配を通じて、第三者製造業者と交渉し、追加のSBP-101臨床供給を提供するつもりだ。FDAにセキュリティプロトコルを提出する前に、私たちは、資格のある製造業者が、私たちの初期候補製品に活性医薬成分および充填および完了サービスを提供することを決定し、将来の任意の候補製品においてこの方法を継続して使用したいと考えている。
Flynpoviの生産がさらなる臨床研究や北米の商業化に使用されることを確保し,この製品が承認されればOne−Twoが担当し,同社は北米生産のための非独占ライセンスを取得した。
材料協定
我々がUFRFと2011年12月22日に締結した“標準独占ライセンス協定”(“ライセンス契約”)は、発行された米国特許番号US 5,962,533(2016年2月満了)およびUS 6,160,022(2020年7月満了)に含まれるノウハウの独占ライセンス、およびライセンス契約によって定義されたノウハウを付与し、UFRFは学術または政府用途のために保持している。この合意に基づいて、私たちはUFRFに様々な特許使用料、費用、そしてマイルストーン支払いを支払うことに同意する。ライセンス協定は2016年12月に改正(“第1改正案”)され、2019年10月に再改正される(“第2改正案”)。第二改正案によると、ライセンス契約で定義されているすべての最低特許権使用料とマイルストーン支払いは廃止される。また、特許権使用料の支払期限は、(I)第1回商業販売日から10(10)年又は(Ii)各国市場排他期間のうち短いものに変更される。UFRFはまた、重大な合意違反、破産、特許権使用料および他の習慣的条件の支払いができなかったなど、標準および同様の理由で本ライセンスを終了することができる。この協定は、2025年12月31日までに最初の商業販売が行われていなければ、UFRFは終了することを可能にする。
政府の監督管理
FDA承認プロセス
アメリカでは、医薬品はFDAによって広く規制されている。その他の事項以外に、“連邦食品、薬品と化粧品法”及びその他の連邦と州法規と条例は薬品の研究、開発、テスト、製造、貯蔵、記録保存、承認、ラベル、販売促進とマーケティング、流通、承認後のモニタリングと報告、サンプリング及び輸出入に対して管理を行う。適用された米国の要求を遵守しないことは、FDAが未解決のNDAの承認を拒否し、警告または無見出し手紙、製品リコール、製品差し押さえ、生産または流通の完全または部分的な一時停止、禁止、罰金、民事罰、および刑事起訴のような様々な行政または司法制裁を受ける可能性がある。
米国では、新製品または承認製品のいくつかの変更に対する医薬製品の開発は、通常、臨床前実験室および動物試験に関連し、臨床試験が開始される前に発効しなければならないINDと、FDA承認を求める各適応に対する薬物の安全性および有効性を決定するために、十分かつ制御された臨床試験とをFDAに提出する。FDA上場前の審査要求を満たすには通常長年の時間を要し、実際の所要時間は製品或いは疾病のタイプ、複雑性と新規性によって大きく異なる可能性がある。
臨床前試験は製品の化学、調合と毒性に対する実験室評価、及び製品特性と潜在安全性と有効性を評価する動物試験を含む。臨床前試験の進行は必ず良好な実験室実践を含む連邦法規と要求に符合しなければならない。臨床前試験の結果はINDの一部として他の情報とともにFDAに提出され,研究者のマニュアル,製品化学,製造と制御に関する情報,潜在的な副作用とリスク,提案された臨床試験案が含まれている。IND提出後,生殖毒性や発ガン性の動物試験など,長期的な臨床前試験を継続する可能性がある。
ヒト臨床試験を開始する前に,各INDの提出後30日間の待機期間が求められている。FDAがこの30日間INDにコメントもINDにも疑問を提起しなければ,INDで提案された臨床試験が開始される可能性がある。
臨床試験は合格した研究者の監督の下で、健康ボランティア或いは患者に研究用新薬を提供することに関連する。臨床試験は:(I)連邦法規に適合する;(Ii)良好な臨床実践(GCP)に適合し、これは、患者の権利と健康を保護し、臨床試験発起人、管理者および監督者の役割を定義するための国際基準であり、(Iii)試験目標を詳細に説明し、安全性を監視するためのパラメータおよび評価すべき有効性基準の方案である。米国患者のテストに関する各々のプログラムおよび後続のプログラム修正案は、INDの一部としてFDAに提出されなければならない。
FDAが臨床試験がFDAの要求に沿って行われていないと考えている場合,あるいは臨床試験患者に受け入れられないリスクとなっている場合,FDAはいつでも臨床試験の一時的または永久的な停止を命じたり,他の制裁を加えたりすることができる。臨床試験における患者の研究案やインフォームドコンセント情報も機関審査委員会(“IRB”)に提出して承認しなければならない。IRBはまた、IRBの要求を遵守できなかったために、現場の臨床試験を一時的または永久的に停止することを要求することができ、または他の条件を適用することができる。
ニューノミンの発売承認を支持する臨床試験は通常3つの連続段階で行われるが、これらの段階は重なる可能性がある。第1段階、すなわち最初に健康なヒト対象/患者に薬剤を導入し、新陳代謝、薬物動態、薬理作用、用量増加に関連する副作用、および可能であれば有効性を評価するための早期証拠を評価するために試験を行った。第2段階は、一般に、特定の適応、用量耐性、および最適用量での薬剤の有効性を決定し、よく見られる副作用および安全リスクを決定するために、限られた患者集団で試験を行うことに関連する。化合物が第2段階評価において有効性および許容可能な安全性を証明する場合、重要または第3段階試験は、多くの患者の臨床治療効果および安全性に関する追加の情報を得るために行われ、一般に地理的に分散された臨床試験場所であり、FDAが薬物の全体的な利益-リスク関係を評価し、薬物のラベルに十分な情報を提供することを可能にする。多くの場合、FDAはこの薬物の治療効果を証明するために、2つの十分かつ良好なコントロールの第三段階臨床試験を必要とする。研究が大規模な多中心試験であり,内部一致性が証明され,統計学的に非常に説得力があり,死亡率,不可逆的な発症率や疾患の予防に臨床的意義のある影響が発見され,潜在的な重篤な結果があり,第2回試験で結果を確認することは実際あるいは倫理的に不可能であれば,他の確認性の証拠を持つ単一第3段階試験で十分である可能性がある。NDAが承認された後、長期安全性、生活の質、またはコスト効果を評価するために、第4段階試験が行われる可能性がある。
必要な臨床試験が完了した後,NDAを用意してFDAに提出する。この製品が米国で発売される前に、FDAがNDAを承認する必要がある。NDAは、すべての臨床前、臨床および他の試験の結果、ならびに製品の薬理、化学、毒理学、製造、制御、および任意の提案されたラベルに関連するデータアセンブリを含まなければならない。機密協定の準備と提出のコストは高く、費用は通常毎年増加する。
FDAはNDAを受信した日から60日の時間があり,当該機関の敷居に基づいて申請が届出を受けているかどうかを決定し,申請が十分完全であると考え,実質的な審査を行うことができる。提出された申請が受け入れられると、FDAは深い検討を始めた。FDAはNDAを審査する際にいくつかの業績目標を設定し、即時性を奨励することに同意した。多くの標準審査薬品申請は提出後12カ月以内に審査を行い,多くの優先審査薬申請は提出後8カ月以内に審査を行う。優先審査は、FDAが治療において大きな進展を得るか、または適切な治療方法がない場合に治療を提供する薬剤を決定するのに適用することができる。優先審査を受ける場合、FDAの目標は6ヶ月以内に申請に行動することだ。FDAは、いくつかの遅延された情報を考慮するために、または提出中に提供された情報の情報を明確にするために、標準審査および優先審査の審査手続きをさらに3ヶ月延長することができる。FDAはまた、新薬製品の申請、または安全性または有効性の問題を有する医薬製品の申請を外部諮問委員会に提出することができる−通常、臨床医および他の専門家を含むグループである−審査、評価を行い、申請を承認すべきかどうかについて提案することができる。FDAは諮問委員会の提案によって制限されていないが、それは一般的にそのような提案に従っている。
NDAを承認する前に、FDAは、通常、GCPに適合することを確実にするために、1つまたは複数の臨床場所を検査する。さらに、FDAは薬物を製造する1つ以上の施設を検査するだろう。FDAは、現在の良好な生産実践(“cGMP”)に適合しない限り、製品を承認しないであろう(“cGMP”)、これは、標準生産の品質システムであり、NDAに含まれるデータは、研究された適応において安全かつ有効であることを証明する大量の証拠を提供する。
FDAがNDAと製造施設を評価した後、それは承認状または完全な返信を発行するだろう。完全な応答文は、一般に、提出中の不足点を概説し、FDAが出願を再検討するために、大量の追加のテストまたは情報を必要とする可能性がある。NDAが再提出されたとき、またはいつ、これらの欠陥がFDAによって満足的に解決された場合、FDAは承認書を発行するであろう。FDAは、含まれる情報タイプに応じて、そのような再提出された出願を2ヶ月または6ヶ月以内に検討することを約束している。
この薬物の商業マーケティングを許可し、特定の適応の具体的な処方情報を提供する。NDA承認の条件として、FDAは、薬物の利点が潜在的なリスクよりも大きいことを確実にするために、リスク評価および緩和戦略(“REMS”)を要求する可能性がある。REMSは,薬物ガイドライン,医療専門家のコミュニケーション計画,安全使用を確保する要素(“ETASU”)を含むことができる。ETASUは、処方または調剤のための特別なトレーニングまたは認証、場合によっては調剤、特殊な監視、および患者登録簿の使用を含むことができるが、これらに限定されない。REMSに対する要求はこの薬物の潜在的な市場と収益力に重大な影響を与える可能性がある。さらに、製品承認には、薬物の安全性または有効性を監視するために、大量の承認後の試験および監視が必要となる可能性がある。承認されると、規制基準が守られていない場合、または最初のマーケティング後に問題が発見された場合、製品承認は撤回される可能性がある。
承認された出願において確立されたいくつかの条件の変更は、適応、ラベルまたは生産プロセスまたは施設の変更を含み、変更を実施するためには、新しいNDAまたはNDA付録を提出し、FDAの承認を得る必要がある。新適応のNDAサプリメントは通常,オリジナル申請と類似した臨床データが必要であり,FDAがNDAサプリメントを審査する際に使用するプログラムや行動は,NDAを審査する際に使用するプログラムや行動と同じである。
迅速な承認指定と承認の加速
FDAは、以下の薬剤の開発に利便性を提供し、審査を加速させることが求められている:(1)深刻または生命に危険な疾患を治療するための薬剤、または(2)有効な治療方法がなく、このような疾患が満たされていない医療需要を満たす可能性があることを証明する薬剤。高速チャネル計画によれば,新候補製品のスポンサーは,候補製品のIND届出と同時にまたはその後,特定の適応の候補製品を迅速チャネル薬として指定することをFDAに要求することができる。FDAは、スポンサーの申請を受けてから60日以内に、候補製品が高速チャネル認証を受ける資格があるかどうかを決定しなければならない。
迅速チャネル計画およびFDAの加速承認規定によれば、FDAは、患者に既存の治療よりも意義のある治療利益を提供する深刻または生命に危険な疾患に対する薬物を承認することができ、その基礎は、臨床利益の代替終点を合理的に予測することができるか、または不可逆的な発病率または死亡率よりも早く測定することができる臨床終点であり、この臨床終点は、不可逆的な発病率または死亡率または他の臨床的利益への影響を合理的に予測することができ、同時に、病状の深刻性、希少性または流行率、および代替療法の利用可能性または不足を考慮することである。
臨床試験において、代替終点は1種の疾病或いは状況の実験室或いは臨床症状の測定であり、それは患者の感覚、機能或いは生存方式の直接測定の代わりになる。代替終点は通常、臨床終点よりも容易または迅速に測定される。この基礎の上で承認された候補製品は厳格な発売後のコンプライアンス要求を遵守しなければならず、IV期の完成或いは承認後の臨床試験を含めて、臨床終点への影響を確認する。必要な承認後研究を行わない場合や,発売後の研究期間中に臨床的利益が確認できなければ,FDAが市場からのリコールを加速させることが可能である。加速規制によって承認された候補製品のすべての宣伝材料はFDAの優先審査を経なければならない。
提出された申請が高速チャネル指定を受けた場合、スポンサーはFDAとより頻繁に相互作用することができ、FDAは申請が完了する前にNDAの内容の一部を審査する可能性がある。申請者が余剰情報を提出するスケジュールを提供し、申請者が適用された使用料を支払うことができれば、スクロール審査を行うことができる。しかしながら、FDA審査申請の期間目標は、NDAの最後の部分が提出されてから開始される。また,FDAが高速チャネルの指定が臨床試験中に出現したデータの支持を得なくなったと考えた場合,FDAはその指定を撤回する可能性がある。
突破的治療指定
FDAはまた、重症または生命に危険な疾患または状態を治療するための薬物承認申請の開発および検討を加速することを要求されており、初歩的な臨床証拠が示されている場合、薬剤は1つまたは複数の臨床的に重要な終点で既存の治療法よりも実質的に改善されている可能性がある。画期的な治療法計画により,新製品候補のスポンサーは,特定の適応の候補品を画期的な治療法として指定することをFDAに求めることができる。FDAはスポンサーの申請を受けてから60日以内に候補品が突破療法指定を受ける資格があるかどうかを決定しなければならない。
孤児薬の指定と排他性
孤児医薬品法は、まれな疾患または疾患の治療のための製品の開発にインセンティブを提供する。孤児医薬品法によれば、FDAは、米国では通常20万人未満に影響を与え、米国では20万人を超える影響を与え、米国ではこのような疾患または疾患を治療する薬剤の開発および生産のコストが製品の販売から回収されることが合理的に予想されていない稀な疾患または疾患を治療するための薬剤を孤児の称号を与えることができる。スポンサーが稀な疾患または疾患の治療を目的としていることをスポンサーが証明した場合、FDAは、同じ薬剤がスポンサーが孤児指定の適応を求めていることを前提として、当該製品を承認した孤児を孤児疾患適応として指定する。同じ薬物がスポンサーが孤児指定の適応を求めるために承認されていれば,スポンサーは合理的に見える臨床的優位仮説を提出しなければならず,孤児指定を得ることができる。秘密保護協定を提出する前に、孤児としての指定を要請しなければならない。FDAが孤児の称号を承認した後、FDAは治療剤のアイデンティティおよびその潜在的な孤児の使用を開示する。
孤児指定は、研究支出、税収控除、“処方薬使用料法案”(PDUFA)が費用減免を申請し、孤児薬物専門権を取得する資格など、製造業者に福祉を提供することができる。孤児として指定された製品が、その後、このような指定された疾患または疾患の活性部分を有するFDAの最初の承認を得た場合、孤児薬物排他性を得る権利があり、これは、限られた場合でなければ、FDAが同じ活性成分を含む別の製品を同じ適応のために承認することを7年以内に禁止する。場合によっては、孤児薬物独占性は、同じ適応を有する同じ活性成分を有する後続製品が、より良い治療効果または安全性に基づいて臨床的に承認された製品よりも優れていることが証明された場合、または患者ケアに重大な貢献をする場合、または孤児薬独占性を有する会社が市場ニーズを満たすことができない場合を含む別の製品の承認を阻止しない。また、FDAは、これらの製品が異なる有効成分を含む限り、1つ以上の製品を同一の孤児適応または疾患に使用することを許可することができる。また,競争相手は孤児薬に対して排他的な適応を持つ異なる製品の承認を得たり,同一製品に対して排他的であるが孤児薬物に対して排他的な異なる適応を承認したりする可能性がある。
欧州連合では,孤児薬物指定はまた,締約国に費用の削減や費用の免除などの財政的奨励を受け,薬物や生物製品の承認後10年間の市場排他性を付与する権利がある。孤児薬物指定基準が満たされなくなり、製品の利益が十分に高く、市場排他性を維持するのが合理的であることを証明するのに不十分であれば、この期間は6年に短縮される。
上場承認申請を提出する前に、孤児薬の指定を申請しなければならない。孤児薬物の指定は、監督審査と承認過程においていかなる利点も伝達されず、規制審査と承認過程の持続時間を短縮することもない。
承認後に要求する
機密協定が承認されると、製品はいくつかの承認後に要求される制約を受けるだろう。例えば、FDAは薬品の承認後のマーケティングと販売促進を密接に監督し、消費者向けの広告、ラベル外販売促進、業界賛助の科学と教育活動及びインターネットに関連する販売促進活動の基準と法規を含む。薬品は承認の適応と承認されたラベルの規定でしか販売できない。
FDAがNDAを承認した後,有害事象報告と定期報告を提出する必要がある。FDAはまた、上場後試験、いわゆる第4段階試験、リスク評価および緩和策、またはREMS、ならびに承認製品の効果を監視することを要求することができ、またはFDAは、承認時に条件を追加して、製品の流通または使用を制限する可能性がある。また、品質管理、薬品製造、包装とラベルプログラムは承認された後、現在の良好な生産実践或いはcGMPに引き続き適合しなければならない。医薬品製造業者と彼らのいくつかの下請け業者はFDAと特定の州機関に彼らの工場を登録することを要求された。FDAの登録要求エンティティはFDAの定期抜き打ち検査を受け,その間,FDAは製造施設を検査し,cGMPの遵守状況を評価する。そのため,メーカーはcGMPの遵守を維持するために,生産や品質管理の分野で時間,お金,精力をかけ続けなければならない。ある企業が規制基準を遵守できなかった場合、初期マーケティング後に問題に遭遇した場合、または後に以前に意識されていなかった問題が発見された場合、監督管理機関は製品の承認を撤回したり、製品のリコールを要求したりすることができる。
規則および環境事項を付加する
米国では、FDAに加えて、私たちの活動は、連邦医療保険や医療補助サービスセンター(CMS)、米国衛生·公衆サービス部の他の部門(例えば、監察長事務室)、米司法省(DoJ)、司法省内の個別連邦検事室、州や地方政府を含む様々な連邦、州、地方当局によって規制される可能性がある。例えば、販売、マーケティング、科学/教育支援プロジェクトは、“社会保障法”、“虚偽申告法”における反詐欺および反乱用条項を遵守しなければならず、私たちの活動は、“健康保険携行性と責任法案”(HIPAA)や同様の州法におけるプライバシー条項に関連しており、各法律が改正されている可能性がある。
他の事項に加えて、連邦反リベート法規は、任意の個人またはエンティティが、購入、レンタル、注文または購入の手配、レンタルまたは購入の手配、レンタルまたは注文として、Medicare、Medicaid、または他の連邦医療計画に従って精算可能な任意の物品またはサービスの見返りとして、故意に、または故意に現金または実物で直接または間接的に、公開または隠蔽的に提供、支払い、請求または任意の報酬を受けることを禁止する。報酬という単語は価値のあるものを含むと広く解釈されている。逆リベート法規は、一方では薬品メーカーであり、他方では処方者、購入者、処方マネージャーの間の手配に適用されると解釈される。法的な例外と規制された避難所があり、いくつかの一般的な活動を起訴されないように保護する。例外や安全港の範囲は狭く,処方,購入または推奨の報酬を誘導するために告発される可能性があるやり方に関連しており,例外や安全港の資格を満たさなければ審査される可能性がある。特定の適用された法定例外や安全港を規制するすべての要求を満たしていないことは、このような行為自体が“反リベート条例”に規定された不法行為であることを意味するわけではない。代わりに、そのすべての事実と状況の累積審査に基づいて、この手配の合法性を逐案的に評価する。私たちのやり方が“反リベート規制”に適合していると信じる理由があるが、私たちのやり方はすべての場合、法定例外や安全港保護を規制するすべての基準に適合していないかもしれない。
また、“平価医療法案”は“反リベート法規”下の意図標準を改正し、更に厳格な標準にし、個人或いは実体がこの法規或いはこの法規に違反する具体的な意図を実際に理解する必要がなく、違反を実施することができる。また,ACAは連邦“虚偽申告法”(以下さらに議論)に基づき,連邦“反リベート法令”違反による物品やサービスのクレームを含めて虚偽や詐欺的クレームを構成する判例法を編纂している。
民事金融処罰条例は、任意の個人またはエンティティに厳しい経済的処罰を科すことを許可しており、他にも、その個人またはエンティティは、連邦医療計画へのクレームを提起したか、またはそのクレームがクレームに従って提供されていないプロジェクトまたはサービス、または虚偽または詐欺的であることを知っているか、または認識すべきである。
他の事項に加えて、連邦虚偽申告法は、連邦政府の支払いまたは承認を要求するために、または虚偽または詐欺的クレームに関連する虚偽記録または陳述の作成、使用、または作成または使用を引き起こすために、任意の個人または実体が連邦政府に意図的に提出するか、または虚偽請求の提出を引き起こすことを禁止する。2009年の“詐欺法執行·回収法案”の改正により、クレームには、米国政府に提出された金銭又は財産に対する“任意の請求又は要求”が含まれている。最近,いくつかの製薬や他のヘルスケア会社がこれらの法律により起訴されており,顧客に無料製品を提供し,顧客がその製品の連邦計画に課金することを期待しているといわれている。他社も起訴され、これらの会社は未承認の用途に製品を販売し、虚偽の声明を提出したため、精算できなかった。
HIPAAは、計画を故意かつ故意に実行または実行しようとすることを禁止し、虚偽または詐欺的な言い訳、陳述または承諾の方法で任意の医療福祉計画(個人第三者支払者を含む)が所有または制御または保管している任意の金銭または財産を詐欺または取得し、悪巧み、計画または装置によって重大な事実を偽造、隠蔽または隠蔽すること、または医療福祉、プロジェクトまたはサービスの提供または支払いに関連する任意の重大な虚偽、架空または詐欺的陳述を行うことを知りながら、新しい連邦刑法を制定する。
また、多くの州で同様の詐欺や法律や法規の乱用があり、医療補助や他の州が計画して精算するプロジェクトやサービスに適用されたり、いくつかの州では支払者にかかわらず適用されている。
私たちは連邦政府と私たちが業務を展開している州のデータプライバシーと安全規制によって制限されるかもしれない。HIPAAは“経済と臨床健康を促進する健康情報技術法”(“HITECH”)及びその実施条例の改正により、個人が健康情報を識別できるプライバシー、安全と伝送に対して要求を提出した。他の事項に加えて、HITECHは、保護された健康情報を受信または取得するために、カバーエンティティに代わってサービスを提供する商業パートナー、独立請負業者、またはエージェントに、HIPAAのプライバシーおよびセキュリティ基準を直接適用させる。HITECHはまた4つの新しい民事罰金等級を作成し、HIPAAを改訂し、民事と刑事処罰を商業パートナーに直接適用し、州総検察長に新しい権力を与え、連邦裁判所に民事訴訟を提起し、損害賠償または禁止令を要求して連邦HIPAA法を執行し、連邦民事訴訟に関連する弁護士費と費用を求めることができる。また,州法は特定の場合に健康情報のプライバシーやセキュリティを管理しており,その多くの法律は互いに大きく異なり,同様の効果が生じず,コンプライアンス作業を複雑にする可能性がある。
さらに、ACAの範囲内の連邦医師は、Medicare、Medicaidまたは児童健康保険計画(いくつかの例外を除く)に支払うことができるいくつかの医薬品、器具、生物および医療用品の製造業者に、医師、他の指定された保健専門家および教育病院または医師、他の指定された保健専門家および教育病院への要求、またはその指定された実体または個人を代表して行われる特定の支払いまたは他の価値移転に関する情報を報告し、医師および他の指定された保健専門家およびその直系親族によって所有されるいくつかの所有権および投資権益を毎年報告する。一部の州でも同様の法律があり、製造業者にカバーされた個人および実体への移転のいくつかの価値を報告することを要求している。製品を商業的に流通させるためには、州の法律を遵守しなければならない。これらのメーカーまたは流通業者が州に営業場所がなくても、ある州で製品をその州に搬送するメーカーおよび流通業者を含む州に薬品および生物製品の製造業者および卸売業者を登録することを要求しなければならない。一部の州はまた、メーカーと流通業者が流通チェーン中で製品の系統を確立することを要求しており、いくつかの州はメーカーと他の州に流通チェーン中の製品の流れを追跡し追跡できる新しい技術を採用することを要求している。いくつかの州はすでに立法を公布し、製薬と生物技術会社にマーケティングコンプライアンス計画を確立し、州政府に定期報告を提出し、販売、マーケティング、定価、臨床試験およびその他の活動を定期的に公開し、および/またはその販売代表を登録し、薬局および他の医療保健実体が製薬および生物技術会社にいくつかの医師処方データを販売およびマーケティングのために提供することを禁止し、いくつかの他の販売およびマーケティング行為を禁止する。私たちのすべての活動は連邦と州消費者保護と不正競争法によって制限されるかもしれない。
もし私たちの運営が上記のいずれかの連邦および州医療保健法律または私たちに適用される任意の他の政府法規に違反していることが発見された場合、私たちは、MedicareとMedicaid、禁止、個人通報者が政府の名義で提起した個人訴訟、または政府契約、契約損害、名声損害、名声損害、行政負担、利益減少と将来の収入減少、および私たちの業務の削減と再編を含む、民事、刑事および/または行政処罰、損害賠償、罰金、返還、MedicareおよびMedicaid、禁止、個人通報者が政府名義で提起された個人訴訟を含むが、処罰を受ける可能性がある。いずれも私たちの業務運営能力と運営結果に悪影響を及ぼす可能性があります。
保証と精算を請け負う
私たちが規制部門の承認を得た任意の候補製品のカバー範囲と精算状態には、重大な不確実性がある。米国や他の国·地域の市場では、規制部門の承認を得て商業販売を行う任意の製品の販売は、第三者支払者がそのような製品に保険を提供する程度に依存し、十分な補償レベルを確立することになる。米国では,第三者支払者には連邦や州医療計画,個人管理のヘルスケア提供者,医療保険会社,その他の組織が含まれている。第三者支払者が製品に保険を提供するかどうかを決定するプロセスは、製品価格を決定するプロセス、または第三者支払者が製品のために支払うべき支払率を決定するプロセスから分離することができる。第三者支払者は、承認リスト上の特定の製品に保証範囲を制限することができ、処方表とも呼ばれ、FDAによって承認された特定の適応のすべての製品を含まない可能性がある。第三者決済者は、価格に挑戦し、医療の必要性を審査し、医療製品、療法、サービスの費用対効果を審査するとともに、それらの安全性と有効性を疑問視するようになっている。私たちは、私たちの製品の医療の必要性と費用効果、FDAの承認を得るのに必要なコストを証明するために、高価な薬物経済学的研究を行う必要があるかもしれない。私たちの候補製品は医学的に必要で費用効果があると思われないかもしれない。支払者が製品に保険を提供することを決定することは、十分な返済率を承認することを意味するものではない。また、支払者が1つの商品に保険を提供することを決定し、他の支払者もその商品に保険を提供することを保証することはできない。連邦医療保険精算も同様であり、異なるサプライヤーが支払いを処理するため、1つのサプライヤーの保険は他のすべてのサプライヤーが保険を提供することを保証することはできない。製品開発への投資の適切な見返りを実現するために、十分な価格レベルを維持することができる十分な第三者精算がないかもしれない。また、アメリカ連邦政府の薬品定価関連問題における立場は変化しており、不確定であり、いかなる変化もアメリカの薬品定価に実質的な影響を与える可能性があり、承認されれば、私たちの候補製品に実質的な影響を与える可能性もある。
他の国もまた違う価格設定と精算プログラムを持っている。EUでは,各国政府はその定価と精算規則や国家医療システムの制御により薬品の価格に影響を与え,これらのシステムは消費者にこれらの製品の大部分のコストを支払っている。いくつかの法域はプラスリストとネガティブリスト制度を実行し、補償価格を合意した後にのみ、製品を販売することができる。精算または定価の承認を得るために、その中のいくつかの国は臨床試験の完成を要求する可能性があり、特定の候補製品のコスト効果を現在利用可能な治療法と比較する。他の会員国たちは会社が自分の薬品価格を固定することを許可するが、会社の利益を監視する。英国の国家健康·介護卓越研究所(“NICE”)も費用便益分析を考慮する必要がある。医療コストの下振れ圧力は非常に大きくなっている。そのため、新製品の参入にはますます高い壁が設けられている。また、一部の国では、低価格市場からの国境を越えた輸入が一国国内の定価に商業圧力をかけている。
政府や第三者支払者が十分な保険や補償を提供できない場合、規制部門の承認を得て商業販売を行う任意の候補製品の適正性が影響を受ける可能性がある。また,米国では管理型医療への重視度が増加しており,医療定価の圧力が増加し続けることが予想される。保証政策と第三者精算料率は随時変化する可能性がある。規制部門の承認を得た1つまたは複数の製品が有利な引受·精算状態を獲得しても、将来的にはあまり有利ではない引受政策や精算料率が実施される可能性がある。
利用可能な情報
ウェブサイトはwww.Panbela.comにあります。本募集説明書に含まれている又は当社のサイトに関する情報は、本募集説明書の一部ではありません。我々はすでに我々のサイトアドレスを事実として参考にしており,我々のサイトへのアクティブなリンクとするつもりはない.
我々は、我々の10-K表年次報告、10-Q表四半期報告、現在の8-K表報告、およびこれらの報告の修正を含む、取引法第13(A)または15(D)節に従って米国証券取引委員会に提出または提供された資料を、当社のウェブサイトを通じて無料で提供します。これらの材料は、私たちがアメリカ証券取引委員会に電子的に保存または提供した後、合理的で実行可能な状況でできるだけ早く私たちのサイトに公開されます。
公衆はアメリカ証券取引委員会の公共資料室で私たちが提出した任意の資料を読むことができます。公共資料室はワシントンD.C.20549、第五街450号にあります。公共資料室の運営状況については、米国証券取引委員会1-800-米国証券取引委員会-0330に請求することができる。米国証券取引委員会には、我々や他の発行者に関する報告書、依頼書、情報声明、その他の情報が含まれているサイトが設置されており、これらの情報はwww.sec.govに電子的に提出することができる。
管理する
私たちの執行官に関する情報は
ジェニファー·K·シンプソン、博士、MSN、CRNP、現在55歳で、2020年7月以来総裁兼CEOと取締役を務めてきた。当社に入社する前に、シンプソン博士は2015年から2020年6月までナスダックシステム会社(デルカス:CTTH)の総裁兼最高経営責任者および取締役会メンバーを務めていた。2012年以来、彼女はDelcathで様々な他の指導者を務めてきた。2011年から2012年まで、Simpson博士はImClone Systems,Inc.(礼来社の完全子会社)でグローバルマーケティング、腫瘍学ブランド主管総裁副総裁を務め、すべての製品の商業化活動を担当し、その中の1つの後期資産の発売に準備した。2009年から2011年まで、シンプソン博士は製品チャンピオン総裁副主任を務め、2008年から2009年まで、ImClone社製品Ramucirumab副主任総裁製品チャンピオンを務めた。2006年から2008年まで、シンプソン博士はペンシルバニア州に本部を置く生物技術会社Ortho Biotech(現在ヤンソン生物技術会社)で製品取締役腫瘍学治療マーケティング専門員を務め、免疫学、腫瘍学と腎臓病領域の革新的な解決方案に専念した。彼女のキャリアの初期には,シンプソン博士は10年以上血液学/腫瘍学看護師従事者と教育家を務めていた。シンプソン博士は2019年8月からずっとEagle PharmPharmticals,Inc.の取締役会、指名と会社管理委員会のメンバーを務め、2021年7月以来CytRx Corporationの取締役会メンバーを務めている。シンプソン博士はピッツバーグ大学の疫学博士号,ロチェスター大学の看護学修士号,ニューヨーク州立大学バッファロー校の看護学学士号を有している。
現在64歳のスーザン·ホヴァトは2018年4月以来、副総裁兼首席財務官を務めてきた。ホーガスは製薬、医療、消費者組織で財務と運営職を務めていた。Horvathさんは同社のポストのほか、取締役会のメンバーを務め、Photonic Pharma,LLCに財務コンサルティングサービスを提供している。Photonic Pharma,LLCは個人持株会社であり、早期薬物発見の効率向上に集中している。Panbelaチームに加入する前に、Horvathさんは2016年から2018年1月までEnobobs,LLCの首席財務官を務め、視力矯正に専念した個人会社であり、2014年から2016年までプライベート持株を担当した安全製品会社Tensiious Holdings,Inc.(d/b/a Ergodyne)の首席財務官兼最高財務官、2011年から2014年まで、HealthSense,Inc.の首席財務官兼副社長を務め、HealthSense,Inc.は新世代技術(SaaS)と遠隔監視会社であり、高齢者と脆弱な成人のための安全と生活の質の改善を提供し、生活コストを削減し、医療コストを全体的に低下させる。彼は2008年から2010年まで首席財務官を務め、運営と人力資源部の副総裁を担当し、2008年から2010年まで商業化段階にある早期医療機器会社血球会社の運営と人力資源部副総監を務め、2004年から2007年まで強力な消費ブランドの開発とマーケティングに専念する上場消費保健品会社CNS,Inc.の副総裁とチーム責任者を務めた。Horvathさんはイリノイ大学シャンパン校の会計学学士号を持ち、公認会計士と公認会計士で、活躍していない。
私たちの取締役会に関する情報
我々の業務は、できるだけ多くの3つのレベルに分けられた取締役会が監督し、取締役は通常指定されたレベルに選ばれ、任期は3年である。以下は取締役会の現在のメンバーに関するいくつかの情報です
第II類取締役-任期は2024年に満了する
マイケル·T·カレン、M.D.,M.B.A.、現在77歳で、2021年5月に当社従業員として退職して以来、当社の取締役会議長と非従業員取締役を務めてきた。クーロン博士は2011年11月の共同創業以来、会社の執行議長と取締役を務めてきた。クーロン博士は開発段階の会社と協力して候補薬物の臨床前から臨床開発までの専門知識を計画、設計、推進することを含む33年間の製薬経験をわが社にもたらした。クーロン博士は2018年10月から2020年7月までの間に私たちの総裁兼最高経営責任者を務めました。彼は2011年11月から2015年6月まで私たちの首席医療官と総裁を務めた。カレン博士は2009年から2011年まで製薬業界に職務調査相談を提供し、これまで衛材製薬で1年間の移行相談サービスを提供してきた。2000年から2008年まで、彼はMGI Pharma Inc.で首席医療官を務め、以前SunPharm社でG.D.Searle社の首席医療官を務め、そして世界第5位の契約研究機構IBAH Inc.で臨床コンサルティング副主任総裁を務め、そこで彼は商業戦略コンサルティングサービスを提供し、製薬業界のいくつかの発展段階にある会社のために発展計画、監督事項と臨床試験を設計した。クーロン博士はIDD医療会社の共同創業者とCEOでもあり、製薬スタートアップ企業である。カレン博士は1988年に3 M製薬会社に入社し、心血管、肺、リューマチと免疫反応調節薬の開発に取り組んだ。彼のキャリアの中で、クーロン博士は2004年以来の3種類のALOXI、Dacogen、Lusedraを含む10種類の薬物の承認を得た。クーロン博士は内科委員会認証を持ち,1977年から1988年までミネソタ州オワトナ診療所で勤務し,そこで総裁を務めた。カレン博士はミネソタ大学で医学博士と理学学士号を取得し、聖トーマス大学で工商管理修士号を取得し、ノースカロライナ大学教会山校とノースカロライナ大学ウィルミントン校で内科入院医師と取締役会認証を完了した。
D.ロバート·スペールは現在68歳で、2015年9月以来取締役の一員を務めている。スチュアートさんは2012年3月以来、太陽生物製薬研究会社の取締役を務めてきた。スペールさんは、農業部門で39年以上の経験を持っています。1973年から2005年まで、スペールはミネソタ州カンディヨシー県に農場を所有し、5000エーカーを経営し、トウモロコシ、大豆、テンサイを生産した。スペールさんは取締役会に勤めている間に豊富な経験を持っています。1992年から1996年までValAdCoの取締役会長を務め,1996年から2003年までフェニックスバイオ複合材料会社の取締役会長を務めた。
第3種役員-任期は2025年に満了する
アーサー·J·フラタミコは58歳で、2019年12月からわが社の取締役を務めている。登録薬剤師であり,製薬業界で30年以上の経験を持ち,Radiant BioTreateuticsの最高経営責任者を務めており,Radiant BioTreateuticsは2021年5月から多抗体開発に焦点を当てた新しい抗体プラットフォームを進めており,多価と多重特異性抗体である。Radiantに加入する前に、Fratamicoさんは、2017年1月からGalera Treateutics,Inc.の首席ビジネス官を務めてきました。同社は、癌放射線治療の発見と開発に取り組む新しいジスムターゼ模倣薬の発見と開発に取り組んでいます。Galeraに加入する前に、Fratamicoさんは、2016年12月までナスダックに上場されている臨床段階バイオテクノロジー企業であるVitae PharmPharmticals,Inc.のチーフビジネス担当を2014年5月からエルビエに販売しています。Vitae製薬会社に入社する前、複数のバイオテクノロジー会社で同様の行政職を務め、GMinin X製薬会社やMGI製薬会社の販売促進を含む業務開発を指導していた。大量のライセンス取引や買収を担当したほか、外部会社の戦略や管理のコミュニケーションを指導していた。彼はまたいくつかの高級マーケティング、製品計画、新製品開発職を務めた。フラタミコさんは、フィラデルフィア薬学科学アカデミーの薬学学士号、ドレイクセル大学の工商管理修士号を取得しています。
ジェフリー·S·マティソンは、63歳で、2015年9月からわが社の取締役を務めてきた。マティソンは取締役や上場医療機器会社NeuroOne Medical Technologies Corporationの監査委員会議長も務めている。Mathiesenさんは2021年6月以来、上場医療機器会社Helius Medical Technologies,Inc.(ナスダック株式コード:HSDT)の財務責任者、財務担当者兼秘書を務めており、神経健康に焦点を当てた非侵襲的プラットフォーム技術の開発に取り組んでおり、2022年5月から取締役を務め、2020年6月から2021年6月まで取締役兼監査委員会議長を務めている。また、Mathiesenさんは、2022年3月~2022年12月に、ヘルスケア·ライフサイエンス·データ変換プラットフォーム·ソリューションの上場プロバイダHealthcare Triangle,Inc.(ナスダック·コード:HCTI)の取締役·監査委員長を務め、2018年7月~2020年2月にeNeura、Inc.の取締役·監査委員会議長を務め、eNeuraは個人所有の医療技術企業であり、片頭痛の急性治療·予防治療を提供する。Mathiesenさんは、民間のバイオ製薬会社Teewinot Life SciencesのCEOとして2019年10月から2019年12月までの間に、純粋な医薬品レベルの大麻化合物の生合成生産に専念し、2019年3月から2019年10月までの間にCEOを務めます。2020年8月、ティビノ生命科学社は米国破産法第11章に基づいて自発的な要望書を提出した。これまで、2015年9月から2018年9月までの間に上場生物製薬会社Gphire治療会社の首席財務官を務めていた。2015年8月から2015年9月まで、彼はゲフィルの顧問だった。2011年3月から2015年1月まで、上場医療機器会社サンシャイン心臓会社の首席財務官を務めた。マティソンは1993年以来、副会長総裁と首席財務長を含む上場企業の役員職を務めてきた。さん·マティソンはナンダコタ州大学の会計学の学士号を持ち、公認会計士でもあります。
第I類取締役-任期は2026年に満了する
Daniel·J·ドノワン,59歳,2022年6月以来取締役を務めている。2011年から2022年6月までパンベラがCPPの買収を完了するまで、CPPの取締役と首席商務官(非従業員職)を務めてきた。2014年の創業以来、民間会社RreLife Solutions,Inc.のCEOを務めてきた。彼は2023年1月以来強度治療会社の取締役会メンバーを務めており、監査委員会のメンバーでもある。Rare Lifeに入社する前、ドノヴァンは2001年にオンコウェイ医薬会社を設立し、2011年まで取締役と社長の社長を務めていた。EnVision Pharmaは2008年に連合生物資源会社に買収され、ドノワンさんは連合生物源会社で高級副社長戦略と市場開発部のメンバーを務め、指導チームのメンバーになりました。ドノヴァンはファイザーでキャリアを開始し、様々な責任がますます大きくなってきた職を務め、販売から米国や国際市場の市場研究やマーケティングまで、最終的に取締役やヨーロッパチームの責任者を務めた。ファイザー在任中、彼は製薬業のいくつかの最も成功した製品発表の商業化過程において重要な役割を果たした。
ジェフリー·E·ジェイコブは現在62歳で、2022年6月以来取締役の一員を務めている。彼は2009年からCPPの最高経営責任者を務め、2022年6月までPanbelaがCPPの買収を完了した。アリゾナ州バイオ製薬コンサルティング·投資会社Tucson Pharma Ventures LLCの担当者でもあり,2004年からこの職務を担当してきた。2004年、ジェイコブはシステム生物学、予測性薬物ゲノム学、臨床試験設計革新を抗癌新薬の開発に応用したスタートアップ会社であるSystems Medicine Inc.を設立した。ジェイコブは2007年まで同社の最高経営責任者を務め、2008年末まで部門最高経営責任者を務めた。1987年から2004年にかけて、ジェイコブはResearch Corporation Technologiesに雇われ、最近の職務は上級副社長。その間、彼はResearch Corporation Technologiesを率いて特許開発と許可組織から初期段階の技術孵化とリスク展開会社に転換した。彼は研究会社の技術会社の取締役会のメンバーを務め、現在取締役会の議長を務めている。彼は創設取締役会のメンバーでもあり、これまでクリティカルパス研究所の首席プロジェクト官を務めていた。ジェイコブさんは、マサチューセッツ工科大学で工学の修士号、技術および政策の修士号、アリゾナ大学で工学の学士号を取得しています。
ジェニファー·K·シンプソン博士、マイクロソフト新聞網、CRNPは、2020年7月以来、わが社の総裁兼最高経営責任者と取締役を務めてきた。参照してください“私たちの執行官に関する情報は“以上はシンプソン博士の背景と経験に関するさらなる情報です。
役員は自主独立している
“ナスダック株式市場継続上場規則”(以下、“ナスダック規則”と略す)の規定によると、取締役会の多くのメンバーは“ナスダック”で定義されている“独立取締役”でなければならない。我々の取締役会は、それぞれの非従業員、すなわちドノヴァン·さん、フラタミコ·さん、マティソン·さん、スペシャルティ·さんが“独立取締役”であることを決定しました
役員報酬
次の表には、最近終了した財政年度に非従業員役員を務めた者の報酬に関するいくつかの情報が示されている。過去に30株1株と40株1株の逆分割株金額が再申告された。
|
名前.名前 |
稼いだ費用 あるいは…。 |
選択権 賞.賞 ($) |
合計する |
|||||||||
|
マイケル·T·カレン(A) |
72,500 | 3,642 | 76,142 | |||||||||
|
アーサー·J·フラタミコ(B) |
49,000 | 3,642 | 52,642 | |||||||||
|
ジェフリー·S·マティソン(C) |
81,500 | 3,642 | 85,142 | |||||||||
|
D.ロバート·スペール |
57,500 | 3,642 | 61,142 | |||||||||
| Daniel·J·ドノワン(E) | 52,500 | 3,642 | 56,142 | |||||||||
|
ジェフリー·ジェイコブ(F) |
40,000 | 3,642 | 43,642 | |||||||||
|
(a) |
カレン博士は2023年12月31日までに計27株を購入するオプションを持っている。 |
|
(b) |
フラタミコは2023年12月31日までに計14株を購入するオプションを持っている。 |
|
(c) |
マティソンは2023年12月31日までに計14株を購入するオプションを持っている。 |
|
(d) |
スペールは2023年12月31日現在、合計14株を購入するオプションを保有している。 |
|
(e) |
ドノヴァンは2023年12月31日現在、合計14株を購入するオプションを持っている。 |
|
(f) |
ジェイコブは2023年12月31日までに計24株を購入するオプションを持っている。 |
Panbelaは2023年の間、取締役会とその委員会会議に参加したことによる自己負担費用を非従業員役員に精算した。
同時に私たちの従業員やサービスプロバイダの役員は、私たちの取締役会に勤めていることで追加の現金補償を受けることはありません。シンプソン博士は2022年度全体で総裁とCEOを務めた。
2022年2月、給与委員会は非従業員役員の現金給与の更新を承認した。以下に述べる年間額は、委員会が変更しない限り有効である2022年1月1日から発効する。
|
年間前払い金 (すべての金額はドル単位) |
一般情報 |
監査委員会 |
ノミネート&指名 統治する 委員会 |
補償する 委員会 |
||||||||||||
|
非従業員取締役 |
40,000 | |||||||||||||||
|
議長(A) |
32,500 | |||||||||||||||
|
リード独立役員(Sequoia Capital)(A) |
22,500 | |||||||||||||||
|
委員会議長 |
15,000 | 7,500 | 10,000 | |||||||||||||
|
委員.委員 |
7,500 | 4,000 | 5,000 | |||||||||||||
|
(a) |
非従業員役員招聘金に支払う。 |
役員報酬
指名された行政員の報酬
次の開示の要点は私たちが任命された幹部たちだ。2023年度には、私たちの“指定幹部”には、私たちだけの二人の幹部、シンプソン博士、ホヴァスさんが含まれています。
私たちが任命した各幹部の基本給は、最初は適用幹部との公平な交渉に基づいて決定された。私たちの取締役会の報酬委員会は毎年私たちの役員の給与を検討するだろう。基本給を交渉または審査する際には、報酬委員会は、そのメンバーの経験、役員の将来の成功への期待貢献、および他の役員の相対的な報酬および責任に基づいて市場競争力を考慮する。
報酬総額表
以下の表は、指定された実行幹事(総称して“実行者”と呼ぶ)が列挙されている間に稼いだ報酬に関する情報を提供する
|
氏名と主要ポスト |
年.年 |
賃金.賃金 |
選択権 賞(A) |
在庫品 賞 ($) |
不公平 激励計画 補償する (b) |
合計する |
|||||||||||||
|
ジェニファー·K·シンプソン |
2023 |
527,000 | 44,921 |
‐ |
‐ |
571,921 | |||||||||||||
|
社長と最高経営責任者 |
2022 |
506,000 |
‐ |
‐ |
188,324 | 694,324 | |||||||||||||
|
スーザン·ホヴァト |
2023 |
333,000 | 15,535 |
‐ |
‐ |
348,535 | |||||||||||||
|
首席財務官兼副総裁 “金融”の総裁 |
2022 |
320,200 |
‐ |
‐ |
103,459 | 423,459 | |||||||||||||
|
(a) |
この表のオプション奨励の価値は、財務会計基準委員会会計基準編纂特別テーマ718に基づいて計算された財政年度内に付与されたこのような奨励の公正価値を代表する。報酬推定値を決定するための仮定は、総合財務諸表付記9において議論される。2022年度にはいかなる持分奨励も発行されていません。 |
|
(b) |
代表がPanbelaの2022年現金インセンティブ計画に基づいて2023年に支払う金は、以下の通り。給与委員会は、2023年の現金奨励案で稼いだ現金奨励金の支払いを決定するために実績を確認していない。 |
現金奨励的報酬
2023年、給与委員会は臨床発展と財務マイルストーンに基づいて各幹部のための業績目標を策定した。各実行者の目標達成後の潜在的報酬は、以下にさらに説明する実行者雇用プロトコルに規定された目標に等しい。2023年の奨励は、あれば2024年第1四半期に支払われる予定であり、これは報酬委員会の最終決定にかかっている。
2023年12月31日現在の未償還持分奨励(調整後20株1株、30株1株、40株逆向株1株)
|
オプション大賞 |
|||||||||||||||
|
名前.名前 |
グラント 日取り |
量 証券 潜在的な 体を鍛えていない オプション(#) 練習可能である |
量 証券 潜在的な 体を鍛えていない オプション(#)キャンセル- 練習可能である |
選択権 トレーニングをする 値段 |
選択権 満期になる 日取り |
||||||||||
|
ジェニファー·K·シンプソン |
7/17/2020 |
8 | – | 239,760 |
7/17/2030 |
||||||||||
|
3/30/2021 |
4 |
2(a) |
98,160 |
3/30/2031 |
|||||||||||
|
3/27/2023 |
– | 165(b) | 300 |
3/27/2033 |
|||||||||||
|
スーザン·ホヴァト |
4/17/2018 |
1 | – | 138,000 |
4/17/2028 |
||||||||||
|
5/21/2019 |
2 | – | 70,800 |
5/21/2029 |
|||||||||||
|
9/24/2019 |
1 | – | 120,000 |
9/24/2029 |
|||||||||||
|
6/24/2020 |
1 | – | 119,520 |
6/24/2030 |
|||||||||||
|
3/30/2021 |
1 |
– |
98,160 |
3/30/2031 |
|||||||||||
|
3/27/2023 |
– | 57(b) | 300 |
3/27/2033 |
|||||||||||
|
(a) |
2024年3月30日にすべての残りの株式を帰属する予定だ。 |
|
(b) |
3月27日に3回に分けて2024年、2025年、2026年にほぼ均等に分割する予定だ。 |
雇用協定
私たちはすべての幹部たちと雇用協定を締結した。以下に概説する具体的な条項を除いて、各役員は、私たちの他の役員(あれば)を含む、私たちの従業員が一般的に得ることができる他の報酬や福祉計画に参加する資格があります。このような雇用協定には、慣例的なeスポーツ禁止契約とeスポーツ禁止契約も含まれており、知的財産権の秘密保持と譲渡に関する補充協定に署名するよう実行機関に要求している。
雇用協定によると、私たちの取締役会の給与委員会は毎年各幹部の基本給を審査しています。雇用協定によると、委員会は適用年度の昇給を承認することができるが、役員の同意を得ない限り、役員の基本給を当時の水準以下にしてはならない。
社長と最高経営責任者
彼女の雇用契約によると、シンプソンは年間業績現金ボーナスを得る資格があり、目標金額は彼女の基本給の50%を下回らない。ボーナス額の支払いは、適用される現金ボーナス期限が終わるまで、私たちの取締役会が策定する指標の実現状況と、彼女がPanbelaに雇われ続けるかどうかにかかっています。
財務副総裁兼首席財務官総裁
彼女の雇用協定によると、ホガスは年間業績現金ボーナスを得る資格があり、目標金額は彼女の基本給の40%を下回らない。ボーナス額の支払いは、適用される現金ボーナス期限が終わるまで、私たちの取締役会が策定する指標の実現状況と、彼女がPanbelaに雇われ続けるかどうかにかかっています。
終了または制御権変更時の潜在的支払い
私たちそれぞれの雇用合意によると、シンプソン博士またはHorvathさんの雇用が“理由”(雇用協定の定義が適用されているような)または彼女の“十分な理由”(雇用協定の定義が適用されている)以外の任意の理由で終了した場合、Simpson博士またはHorvathさんは、終了日または以前に蓄積された他の金額を加えた比例配分された金額を得る資格があるだろう。この金額は、彼らのそれぞれの年収に加えて、終了日または前に蓄積された他の金額に相当する。“支配権変更”の前6ヶ月以内または“支配権変更”後2年以内(適用される雇用契約の定義により)このような解雇が発生した場合、シンプソン博士またはホガスさんは、彼女のそれぞれの年収に相当する金額を獲得し、発生当時の全額現金ボーナス目標と同じ金額を終了する。
特定の実益所有者と経営陣の保証所有権
次の表は、2024年1月23日現在の発行された普通株式の実益所有権に関するいくつかの情報を示しており、これらの情報は、(I)次の表に記載されている私たちの各指名された役員、(Ii)私たちの各取締役、(Iii)私たちのすべての役員、取締役、および取締役が全体として指名された人、および(Iv)私たちが発行した普通株の5%以上の他のすべての実益所有者を保有している。所有権パーセンテージは、同じ日の取引終了時までに推定された480,244株の発行された普通株に基づく。米国証券取引委員会規則に基づいて実益権属を決定する。私たちが知っている限り、別の説明があり、適用されるコミュニティ財産法の制約を受けない限り、以下に掲げる株の保有者は、所有する株式に対して唯一の投票権および投資権を有する。次の表には、2024年1月23日から60日以内に行使可能な普通株買収権利の株式数が含まれている。以下に別途説明されない限り、表に記載されている各役員または役員の住所は、C/o Panbela Treateutics,Inc.,712 Vista Blvd#305,Waconia,Minneota 55387である。
|
名前.名前 |
金額と 自然界 有益な |
パーセント 株* |
||||||
|
行政員および役員 |
||||||||
|
ジェニファー·K·シンプソン |
33 |
(a) |
* | |||||
|
スーザン·ホヴァト |
27 |
(b) |
* | |||||
|
マイケル·T·クーロン |
60 |
(c) |
* | |||||
|
ダニエル·J·ドノヴァン |
25 |
(d) |
* | |||||
|
アーサー·J·フラタミコ |
14 |
(e) |
* | |||||
|
ジェフリー·E·ジェイコブ |
27 |
(f) |
* | |||||
|
ジェフリー·S·マティソン |
14 |
(g) |
* | |||||
|
D.ロバート·スペール |
48 |
(h) |
* | |||||
|
全役員と現執行幹事(8人) |
248 |
(i) |
* | |||||
|
* |
1.0%を下回った。 |
|
(a) |
12株の株式オプション制約を受けた株と、14株の株式証制約を受けた株とを含む。 |
|
(b) |
株式オプションに拘束されている株6株と株式証拘束された14株を含む。 |
|
(c) |
株式オプションに拘束されている株27株と、株式証制約を受けた株1株を含む。147株と10株も含まれ,それぞれカレン生活信託が保有する引受権証である。 |
|
(d) |
14株の株式オプションに制約された株が含まれている。Westport Boys,LLC(“Westport”)が持つ3株,GDB Investments,LLP(“GDB”)が持つ6株も含まれている。ドノヴァンさんは、Westportの管理メンバーとGDBの指定されたメンバーです。ドノヴァンさんは、WestportとGDBが所有する証券の実益所有権を放棄しますが、彼がその中で金銭的利益を除外しています。 |
|
(e) |
14株の株式オプションに制約された株が含まれている。 |
|
(f) |
24株のオプション制約を受けた株、配偶者と共同所有している引受権証に拘束されている株を2株含む。 |
|
(g) |
14株のオプション制約を受けた株が含まれている。 |
|
(h) |
14株の株式オプションに拘束されている株式、配偶者が保有する引受権証に拘束されている株式19株、配偶者株式証に拘束されている株式12株を含む |
|
(i) |
株式オプション制約を受けた株式125株および株式証制約を受けた53株を含む。 |
証券説明書
以下に記載するPanbela普通株(1株当たり額面0.001ドル(“普通株”)の一般条項及び条文要約は完全ではなく、当社の改訂された再予約会社登録証明書(“証明書”)及び改訂された会社再予約付例(“定款”)の規定の制限及び保留を受けなければならない。より多くの情報を知るためには、証明書、定款、デラウェア州会社法(“DGCL”)の適用条項を読んでください。
授権株
当社は最大1.1億株の株式発行を許可しており、その中の1億株は普通株であり、1000万株は優先株であり、1株当たり額面0.001ドル(“優先株”)である。
普通株
普通株式流通株は、いかなる他の株式よりも優先する権利を有しておらず、各株式は、様々な点で任意の他の株式と等しい。普通株保有者は、毎回の株主総会で、登録されている普通株を1株保有するごとに、1票を投じる権利がある。普通株式所有者はいかなる優先購入権、引受権、転換権、償還権或いは債務超過基金権を享受する権利がない。普通株を増発すれば、優先購入権がなければ、株主の利益が希釈される可能性がある。
当時発行された任意の優先株の任意の優先株の任意の優先権に基づいて、普通株式所有者は、取締役会の発表時に現金、財産または会社の株式の形態で配当金を得る権利があり、十分な純利益または黒字がこの目的に合法的に使用できることを前提としている。資本資産の任意の分配において、清算のように、自発的であっても非自発的であっても、普通株式の保有者は、債権者が全額弁済後に残った資産を比例して取得する権利がある。すべての普通株の発行された株式と流通株は評価できない。
反買収条項
憲章文書やDGCLには何らかの条項が含まれており、自社の自主的な買収を阻止したり、自社を自主的に買収することをより困難にしたりする可能性がある。以下は、同社に適用されるより重要な反買収条項である
デラウェア州反買収法
一般に、DGCL第203条は、当該株主が利害関係のある株主となってから3年以内に利害関係のある株主(以下この定義を参照)と業務合併を行うことを禁止し、又は2,000名以上の株主により登録されたデラウェア州会社が、当該業務合併が所定の方法で承認されない限り、業務合併を行うことを禁止する。“企業合併”は、利益関連株主に経済的利益をもたらすために、合併、資産または株式売却または他の取引を含む。“利害関係のある株主”とは、利害関係のある株主の身分が確定するまでの3年以内に、関連会社や共同経営会社と共に会社の15%以上の投票権を有する株を所有または確実に所有する者をいう。第二百三十条によれば、会社と利害関係のある株主との間の企業合併は、次の条件のうちの一つを満たさない限り、三年以内に禁止される
|
● |
株主が利益株主になる前に、取締役会は株主を利益株主にする企業合併や取引を承認した |
|
● |
株主が利害関係のある株主となる取引が完了した後、利害関係のある株主は、取引開始時に少なくとも会社が発行した議決権付き株の85%を有するが、発行された議決権付き株、取締役および上級管理者が所有する株式および従業員株計画を決定するためのものは含まれていない場合がある |
|
● |
株主が利害関係のある株主になったとき又はその後、企業合併は、会社取締役会によって承認され、株主年度又は特別会議において、議決権を有する株式(利害関係のない株主所有)の少なくとも3分の2の賛成票で承認される。 |
株主の指名と提案の要求をあらかじめ通知する
この付例は、株主会議が提出した株主提案及び指名取締役候補について事前通知手続を設けているが、取締役会又は取締役会の指示の下で行われた指名は除外している。
株主特別会議
証明書と付例では、株主特別会議は会社の取締役会、取締役会議長、または最高経営責任者によってしか開催できないと規定されている。
分類取締役会
この証明書では、役員は3種類に分類され、交互の任期選挙で選出されることが規定されている。毎回の年次会議では、約3分の1の役員が選出され、任期は3年になる。任期を交錯させた取締役は、理由がある場合にのみ免職され、当時取締役選挙で投票する権利があった75%以上の流通株保有者の賛成票でのみ免職することができる。
取締役会の権威
取締役会は、株主の承認を求めることなく、1つまたは複数の優先株シリーズを設立する権利があり、そのカテゴリまたはシリーズの権力、優先株、権利および制限を決定する権利を含む会社の任意または全部の株式を発行する権利がある。取締役会は付例を通過·変更する権利があるが、当時のすべての発行済み株式の少なくとも66.67%の投票権の所有者が取締役選挙において附例を通過、改正または廃止する権利があることに制限されなければならない。
優先株
私たちの取締役会は、事前に株主の承認を得ずに1つ以上の一連の優先株を設立し、決定する権利があります
|
● |
このシリーズの株の数 |
|
● |
投票権、配当権、転換権、償還権、および清算優先権を含む名称、優先権、および相対権 |
|
● |
どんな制限、制限、制限もあります。 |
私たちの取締役会は、将来可能な融資や買収、出現可能な他社のニーズを満たすための柔軟性を提供するために、1つまたは複数のシリーズの優先株を発行することができると信じています。優先株の認可株式および普通株式の授権および未発行株式は発行可能であり、普通株式所有者は、法律または任意の証券取引所または我々証券がその上に上場または取引する可能性のある自動見積システムの規則が適用されない限り、そのような行動を要求する必要はない。
我々の取締役会は、株主の承認なしに投票権及び転換権を有する優先株の発行を許可することができ、これは普通株式保有者の投票権及び他の権利に悪影響を及ぼす可能性がある。我々の取締役会は現在このようにする予定はありませんが、一連の優先株を発行する可能性があり、この一連の条項によると、これらの優先株はわが社の合併、要約買収、または他の買収の試みを阻害する可能性があります。我々の取締役会はまた優先株を発行することができ、その条項は買収の試みを阻止する可能性があり、これらの買収の試みを通じて、買収側は買収要約や他の取引を含む当社の取締役会の構成を変えることができる可能性があり、その中の一部または大多数の株主は彼らの最適な利益に適合していると考えているかもしれない、あるいは株主は当時の市場価格よりも高い株価プレミアムを得る可能性がある。したがって、どんな優先株の発行も私たちの普通株の市場価格を下げるかもしれない。
当社取締役会は、当社と株主の最適利益の判断に基づいて、このような株を発行するか否かを決定します。私たちは現在優先株を発行する計画を持っていない。
オプション
2016年には当初、付与された奨励認可に基づいて最大116株の私たちの普通株を発行する予定で、その年間常青特徴に応じて164,246株増加した。2024年1月23日現在、2016年計画により、553株の普通株を購入するオプションが発行されており、加重平均価格は1株13,297.06ドルである。同じ日まで、2016年計画によると、約163,757株の普通株が将来的に付与できる。
2024年1月23日現在、2011年計画によると、普通株5株を購入したオプションはまだ返済されておらず、加重平均価格は1株76,200.00ドルである。株主が2016年計画を承認した後、私たちは2011年計画の下での奨励を停止した。
2024年1月23日現在、CPP 2010年株式激励計画によると、我々の普通株41株を購入したオプションはまだ返済されておらず、加重平均価格は6,743.41ドルであり、これらはすべてCPPを買収する際に仮定している。
未弁済持分証
2024年1月23日現在、私たちは340,952株の普通株の引受権証を発行·発行しており、発行された優先株の株式を承認していない。同じ日まで、未弁済株式証の加重平均行権価格は1株64.09ドルであり、平均残り行の権利期間は5.24年であった。発売が下記のいずれかの株式承認証を下回る行使価格で普通株を売却するとみなされた場合、当該等株式証1部当たりの行使価格は、それぞれの条項に基づいて調整される。いずれの場合も(長期株分割を除く)、どの発行外の引受権証も、より多くの数の普通株を行使することはできない。
2022年10月株式承認証
2022年10月、私たちは最大1,256株の私たちの普通株(“2022年10月株式承認証”)を購入するために普通株引受権証を発行し、私たちの普通株と引受権証の公開に関連しています。2022年10月の権利証の最初の行権価格は、1株当たり108,96.00ドル(その後の逆株式分割調整後)であり、2027年10月4日まで発行時に行使することができる。会社が将来的に普通株のいくつかの希釈株を発行する場合、普通株等価物および変換可能または派生証券を含む場合、行使価格は別途低下する。2024年1月23日までに、285株の2022年10月の既発行株式権証があり、調整後の発行権価格は1株当たり12.39ドルである。
2023年1月株式承認証
当社は2023年1月に普通株引受権証を発行し、合計3,091株自社普通株(“当社2023年1月株式承認証”)を購入し、自社普通株及び株式承認証を公開発売した。1月の権利証の初期行権価格は、1株当たり1,650.00ドル(その後の逆株式分割調整後)であり、2028年1月30日まで発行時に行使することができる。会社が将来的に普通株のいくつかの希釈株を発行する場合、普通株等価物および変換可能または派生証券を含む場合、行使価格は別途低下する。株式承認証発行30日当日または後に、株式取得者は、(X)現金行使で発行可能な普通株式総数と(Y)0.75の積に相当する株式総数を得る“選択可能な無現金行使方式”を選択することもできる。2024年1月23日までに、128株2023年1月の既発行株式権証があり、調整後の現金行権価格は1株12.39ドルである。
A類とB類株式証
2023年6月に、著者らはA類普通株購入承認株式証(“A類株式承認証”)を発行し、最大113,500株普通株(“A類株式承認証”)及びB類普通株購入承認株式証を購入し、最大113,500株自社普通株(“B類株式承認証”及びA類株式承認証、“六月承認株式証”)を購入し、自社普通株及び株式承認証の公開発売と関係がある。6月の権証の初期行権価格は1株15.60ドル(その後の逆株式分割調整後)であり、2028年6月16日まで発行時に行使することができる。会社が将来的に普通株のいくつかの希釈株を発行する場合、普通株等価物および変換可能または派生証券を含む場合、行使価格は別途低下する。A類株式証についてのみ、一般権証所有者は、発行30日後または後に通知を出して“キャッシュレス行使の代わり”を選択することができ、これにより、(X)現金行使により発行可能な普通株式総数と(Y)0.24の積に相当する株式総数を得ることができる。2024年1月23日まで、A類はすでに株式承認証の残り2,595株を発行し、B類はすでに株式承認証の残り7,000株を発行し、調整後の現金行権価格は1株当たり12.39ドルであった。
C類株式承認証
私たちは2023年11月2日に、既存の株式承認証の行使を誘導するために、最大213,000株の私たちの普通株(“C類株式承認証”)を購入するためのC類普通株引受権証を発行した。C類株式証の初期行権価格は1株15.60ドルであり(その後の逆株式分割を調整した後)、株主が承認した日からナスダック上場規則に必要な任意の株主が承認した日から5年以内に行使することができる。我々の株主は2023年12月19日に開催された特別会議で普通株式の発行を承認した。会社が将来的に普通株のいくつかの希釈株を発行する場合、普通株等価物および変換可能または派生証券を含む場合、行使価格は別途低下する。2024年1月23日までに、まだ75,200株のC類はすでに株式権証明書を発行し、1株当たりの現金発行権価格は15.60ドルである。
我々は,C類権証の行使により発行または発行可能なC類権証関連株式転売に関する登録声明を提出することに同意し,米国証券取引委員会は2023年12月20日にS-1表登録声明(文書番号333-275733)の発効を発表した.
D類株式証明書
著者らは2023年12月21日にD類普通株引受権証を発行し、合計255,600株自社普通株(“D類株式承認証”)を購入し、現有のC類株式承認証の行使を奨励する。D類株式証の初期行権価格は1株19.00ドルであり(その後の逆株式分割を調整した後)、ナスダック上場規則に規定されている株主の承認を得た後にのみ行使することができ、株主の承認日から5年間行使することができる(あれば)。もし当社が将来的に普通株等価物と転換可能または派生証券に基づいていくつかの希釈性普通株を発行すれば、任意の介入の逆株式分割と株主の承認を受けた後、行使価格は別途低下する。2024年1月23日まで、すべてのD類株式証はまだ返済されておらず、行使できず、株主の承認を待っている。
私たちは2023年12月21日以降60(60)のカレンダー日にD類株式証関連株の転売を含む登録声明を提出することに同意した。また、当社は1年以内にいかなる変動金利取引(定義勧誘メール)も行わないことに同意していますが、6ヶ月後に発効した市場発売は除外します。
E類とF類株式証
今回の発行提案により発行されたE類とF類承認株式証のいくつかの条項の検討については、以下の“一般権証”というタイトルの下の説明を参照されたい。
あらかじめ出資して株式証明書を発行する
ここで提供される予資権証のいくつかの条項及び条項の以下の要約は不完全であり、資本資本権証の条項の制約を受け、資本資本権証の条項の制限を完全に受け、その形態は、本募集説明書の一部として登録声明の証拠物として提出される。準投資家は事前融資権証表の条項と条文を慎重に審査し、事前融資権証の条項と条件を全面的に説明しなければならない。
持続期間と行権価格。ここで発売されたあらかじめ出資した引受権証1部あたりの初期行権価格は1株0.001ドルである。前払い資金株式承認証は直ちに行使することができ、いつでも行使することができ、前払い資金株式証がすべて行使されるまで行使することができる。株式配当、株式分割、再編、または同様のイベントが我々の普通株式および行権価格に影響を与える場合、行権時に発行可能な普通株の行権価格および株式数は適切に調整される。事前資金権証は、添付されている一般権証とは別に発行され、その後すぐに単独で譲渡することができる。
運動性があります前払い資金株式承認証は、所有者毎に全部または一部を選択して行使することができ、正式に署名された行使権通知を吾等に提出し、権利証を行使する際に購入した普通株式数について全数金を支払うことができる(以下で議論するキャッシュレス行使を除く)。今回の発行における予資権証の購入者は、発行時に直ちにその予資権証を行使し、今回の発行終了時に予資権証に関連する普通株を取得するために、発行定価の後と、予資権証の発行終了前に行使通知を提出することを選択することができる。保有者(およびその連合会社)は、事前計画権証の任意の部分を行使してはならず、保有者が行使直後に4.99%を超える発行済み普通株を所有していることを条件としているが、保有者が吾等に少なくとも61日間の事前通知を出した後、保有者は事前計画権証を行使した後、発行された普通株式の保有量を行使直後の発行された普通株式数の9.99%に増加させることができる。今回の発売で事前融資権証を購入した買い手も、事前融資権証を発行する前に、初期行使限度額を発行した普通株の9.99%に設定することができる。あらかじめ出資した引受権証を行使することで普通株の断片的な株式を発行することはない。私たちは断片的な株式ではなく、次の完全なシェアに切り込むつもりだ。
キャッシュレス運動。保有者がその事前資本権証を行使する際に、証券法に基づいて事前計画権証を発行する普通株式の登録声明が当時発効していないか、または提供されていないことを登録した場合、保有者は、事前資本権証を行使する際に、事前資本権証に記載されている式に基づいて決定された普通株式純額を、事前資本権証の行使時に吾等に支払うことを期待する現金の代わりに受け取ることを選択することができる。
譲渡可能性。法律の適用の規定の下で、事前資本金権証は、所持者が事前資本金権証を適切な譲渡文書と一緒に吾に渡すなどの時に、所持者が自ら譲渡を選択することができる。
取引所が上場する。いずれの証券取引所又は国が認可した取引システムにおいても、事前資金権証のための取引市場を提供していない。私たちはどんな証券取引所や国家が認可した取引システムにも事前融資権証を上場するつもりはありません。
株主としての権利それは.事前出資株式証明書に別途規定があるか、またはその所有者による当社の普通株式の所有権がない限り、事前出資承認持分証所有者は、その事前出資承認権証を行使する前に、任意の投票権を含む当社の普通株式所有者の権利または特権を有していない。
基本的な取引。事前融資権証に記載されているような基本的な取引は、一般に、私たち普通株の任意の再編、資本再編または再分類、売却、譲渡、または他の方法で私たちのすべてまたは実質的にすべての財産または資産を処分し、私たちは他の人と合併または合併し、私たちが発行した普通株の50%以上を買収し、または任意の個人または団体が私たちが発行した普通株に代表される50%の投票権の実益所有者となり、予備資金権証の所有者は、予融資権証を行使する際に同じ種類と金額の証券を得る権利がある。もし所有者がこのような基本取引の前に直ちに事前融資権証を行使した場合、彼らは現金または他の財産を得るだろう。
普通権証
ここで提供される一般権証のいくつかの条項および条項の以下の要約は不完全であり、一般権証の条項の制約を受け、一般権証の条項によって完全に制限され、その形式は、本募集説明書の一部として登録声明の証拠物として提出される。潜在投資家は一般権証の形式の条項と規定を慎重に検討し、一般権証の条項と条件を完全に記述しなければならない。
持続期間と行権価格。ここで発売される1株当たりの一般権証の初期行権価格は$(1株当たり公開発行価格と普通権証の100%)に等しい。一般権証は直ちに行使可能であり、元の発行日の5周年に期限が切れる。株式配当、株式分割、再編、または同様のイベントが我々の普通株式および行権価格に影響を与える場合、行権時に発行可能な普通株の行権価格および株式数は適切に調整される。(I)吾などの将来的に普通株を発行するいくつかの償却行為が発生したように、普通株等価物および変換可能証券または派生証券、および(Ii)任意の逆株式分割によって、行使価格は別途引き下げられる。普通権証は普通株と分離して発行され、その後すぐに単独で保有される。0.25株を購入した私たちの普通株のE類株式証と1.75株の私たちの普通株を購入したF類株式証は、普通株1株当たりまたは今回の発行で購入した予備融資承認株式証の価格で発行されます。
可運動性それは.一般権利証は,所有者ごとに全部または一部の行使を選択することができ,正式に署名された行使通知を吾らに提出し,行使時に購入した普通株式数について全数金を支払う方法である(以下で議論するキャッシュレス行使は除く).所有者(およびその連合会社)は、所有者が行使直後に4.99%を超える発行済み普通株式を所有していない限り、一般権証の任意の部分を行使してはならないが、所持者が吾等に少なくとも61日間の事前通知を出した後、所有者は、一般権証の条項に基づいて決定されているので、所有者は、行使直後に発行された普通株式数の9.99%まで発行された普通株式の保有量を増加させることができる。普通権利証の行使によって普通株式の断片的な株式を発行することはない。私たちは断片的な株式ではなく、次の完全なシェアに切り込むつもりだ。
キャッシュレス運動それは.所有者が一般権証を行使する際に、証券法に基づいて普通株式発行を登録する登録声明が当時有効でなかったか、または登録声明を提供しておらず、証券法による免除登録が当該等の株式の発行に適用されなかった場合、所持者は、当該等株式証を行使する際に吾等に支払うことが期待される現金支払いの代わりに、通常権証を行使する際に(全部又は一部)通常権証に記載されている式に基づいて定められた普通株式純額を徴収することを選択することができる。
別のキャッシュレストレーニングそれは.権証発行後の第5営業日または後に、E類株式証所有者も通知を出して“キャッシュレス行使の代わり”を選択することができ、これにより、(X)現金行使により発行可能な普通株式総数と(Y)1.33の積に相当する株式総数を得ることができる
譲渡可能性それは.適用法律に適合する場合には、帳簿帳簿形式の通常権利証は、信託会社の施設を介して譲渡されることを所有者が選択することができ、実物形式の一般権証は、通常権証が適切な譲渡文書と共に株式所有者代理人に返却されたときに譲渡することができる。吾らとVIStock Transferとの間の引受権証エージェントプロトコルによると、株式認証エージェントとして、通常の権利証は、最初に簿記形式で発行され、預託信託会社(“DTC”)に格納された1つ以上のグローバル証明書によって代表され、DTCの代理名人CEDE&Co.の名義で登録されるか、またはDTCの他の指示に従って登録される。
取引所が上場するそれは.一般的な権利証は既定された公開取引市場がなく、私たちは市場が発展しないと予想する。また、私たちは、どの証券取引所や国家が認可した取引システムにも一般権証を上場するつもりはありません。活発な取引市場がなければ、一般的な権利証の流動性は制限されるだろう。
株主としての権利それは.一般権証が別途規定されているか、または当該所有者によって当社の普通株式株式の所有権を有していない限り、一般権証所有者は、その普通株式承認権証を行使する前に、任意の投票権を含む当社の普通株式所有者の権利または特権を有していない。
基本的な取引。一般的な権利証形式の基本取引が発生した場合、一般に、我々普通株を含む任意の再編、資本再編または再分類、売却、譲渡、または他の方法で私たちのすべてまたは実質的にすべての財産または資産を処分し、他の人との合併または合併、私たちが発行した普通株式の50%以上を買収するか、または任意の個人または団体が普通株式に代表される50%の投票権を発行した実益所有者となり、一般権証所有者は、一般権証を行使する際に同じ種類および金額の証券を得る権利がある。もし所有者がこのような基本的な取引の直前に一般権証を行使した場合、彼らは現金または他の財産を得るだろう。
譲渡エージェント、登録者、許可エージェント
我々普通株の譲渡エージェントと登録者,2022年10月権証,2022年1月権証およびA類とB類権証の権証エージェント,および提案するE類とF類権証の権証エージェントはVStock Transferであり,同社はVIStock Transfer,電話:18 Lafayette Place,Woodmel,New York,郵便番号:11598,電子メール:INFO@VSTORT.COM,または+1(212)8288436に連絡することができる.
将来売却する資格のある株
将来的には、発行されたオプションや引受権証を行使して発行された株を含む、私たちの大量の普通株を公開市場で販売したり、これらの売却を予想したりすることは、時々現行の市場価格に悪影響を与え、将来的に株式を調達する能力を弱める可能性がある。
今回の発行終了後、今回発行中に販売された一般権証を現金で全額行使すれば、2024年1月23日までに発行された普通株480,244株により、合計8,480,244株の普通株と24,480,244株の私たちの普通株の流通株があります。これらの流通株のうち,今回の発行で販売された株はすべて自由に取引可能となるが,我々の関連会社が今回の発行で購入したどの株も,証券法第144条規則の定義により,以下の制限を満たした場合にしか販売できない.
規則第百四十四条
一般に、現行の第144条規則によれば、吾等は上場企業申告規定を少なくとも90日遵守し、売却前90日以内の任意の時間に、証券法において、いかなる者も吾等の連合会社の一つとみなされず、売却を提案した株式の少なくとも6ヶ月(吾等の連営会社以外のいずれかの先行所有者の保有期間を含む)を実益している場合は、当該株式等を売却する権利があり、第144条の売却方法、出来高制限又は通知規定を遵守する必要はないが、第144条の公開資料要求を遵守しなければならない。当該者が売却予定の株式を少なくとも1年間所有しており、我々関連会社以外のいずれかの以前の所有者の保有期間を含む場合、その者は、第144条のいかなる要求も遵守することなく、以下に述べるロック契約の満了時にこれらの株式を売却する権利がある。
一般的に、現在有効な第144条の規則によれば、われわれの連合会社又はわが連合会社を代表して株式を売却する者は、以下のロック契約の満了後のいずれか3ヶ月以内に、以下のような大きな者を超えない株式を売却する権利がある
|
● |
当時発行された普通株数の1%は、今回の発行に続く約95,024株に相当する |
|
● |
もし吾らの普通株がナスダック資本市場に上場していれば、今回の売却に関する表144の通知を提出するまでの4つのカレンダー週間内に、吾らの普通株はその市場での週平均取引量を指す。 |
私たちの連合会社または私たちの連合会社を代表して株式を売却する人は、規則144に従って株式を売却し、いくつかの形態の販売条項と通知要求を遵守し、私たちに関する最新の公開情報を得ることができるかどうかを確認する必要があります。
規則第701条
ルール701は、一般に、書面補償計画または契約に従って我々普通株株の株主を購入することを許可し、直前90日以内に当社の関連会社とみなされない場合は、ルール144に依存してこれらの株を売却することができるが、ルール144の公開情報や保有期間規定を遵守する必要はない。ルール701はまた、ルール144の保有期間要求を遵守せずに、ルール144に従ってそのルール701株を売却することをわが社の関連会社に許可する。しかしながら、この規則によれば、すべてのルール701の株式の所有者は、本入札説明書の日付の90日後まで待たなければならないが、ルール701に従ってこれらの株式を販売することができるが、以下に説明する市場硬直化プロトコルおよびロックプロトコルを遵守しなければならない。
株式オプション及び株式承認証
我々が普通株式を購入した発行済株式証とオプション株式承認証の検討については,上の証券説明を参照されたい
配送計画
2024年の配給代理契約に基づいて、我々はすでにRoth Capital Partners、LLCを私たちの主要な配給エージェントとして招聘し、合理的な最大の努力で本募集説明書を購入して提供する証券の見積もりを求めた。配給代理は何の証券を売買することもなく、特定の数や金額の証券を売買する必要もないが、その“合理的な最大の努力”を尽くして当行の証券の売却を手配しなければならない。したがって、私たちは発行されたすべての証券を売却しないかもしれないし、全く売らないかもしれない。配給エージェントは、1つまたは複数のサブエージェントまたは選択されたトレーダーを招いて今回の発行に参加することができる。
我々は,投資家の選択の下で,機関投資家と直接証券購入契約を締結し,機関投資家が今回の発行で我々の証券を購入することを希望する.証券購入契約を締結していない投資家は、本募集説明書のみに基づいて今回発行された我々の証券を購入しなければならない。
配給エージェントプロトコルは,配給エージェントの義務が配給エージェントプロトコルにロードされる条件によって制約されることを規定している.
今回の発売は2024年2月15日に終了します。事前に完了しない限り、またはその日までに発売を終了することが決定されない限り(いつでも適宜終了することができます)、E類株式証、F類株式承認証、事前計画権証の基礎となる普通株は、それぞれすべて行使または満期になるまで発売され続けます。我々は、投資者が本募集説明書に基づいて提供された証券を購入する資金を受け取った後、発行された証券を投資家に渡す。したがって、投資家の資金を受け入れるか、または信託、信託、または任意の同様の口座に入金する予定は存在しない。本募集説明書に基づいて提供される証券は2024年頃に受け渡しされる予定です。最低証券数や収益額がないことが今回の発行終了の条件です。
配給代理費、手数料、支出
今回の発売完了時に、吾らは今回発売された証券の総収益の7.0%に相当する現金取引費を配給代理に支払う。また、代理弁護士を配置する費用と支出を含めて、今回の発売に関する自己負担費用を精算し、最高100,000ドルに達する。
次の表は,我々が発行したすべての証券を購入したと仮定して,我々の公開発行価格,配給エージェント費,控除費用前の収益を示している.
|
1株当たり よくある 捜査命令 |
金ごとに前払いする 手令が及ぶ よくある 捜査命令 |
|||||||
|
公開発行価格 |
$ |
$ |
||||||
|
配置代理費 |
$ |
$ |
||||||
|
費用を差し引く前の収益は私たちにくれます |
$ |
$ |
||||||
次発行の総支出であり,登録費,届出および上場費用,印刷費および法律および会計費用(配給代理費用を除く)を含めて約315,000元であり,すべて当社が支払うと予想される。この数字には,配給エージェントの実売費用が含まれており,配給代理法律顧問の法的費用は含まれているが,発売終了時に最高100,000ドルの総費用を支払うことに同意している.
販売禁止協定
吾等及び吾等の執行者及び取締役は、今回の発売開始前に代表とロック合意を締結することを期待し、この合意に基づいて、本募集説明書がその一部を構成する登録声明発効日から90日以内に、これらの者又は実体が代表者の事前同意を得ない場合には、(A)売却、売却契約、質権、質権、又は他の方法で処分すること(又は合理的に意図又は合理的に予想される処分を行うことを目的とする取引)を提出、締結しないことに同意する(実際の処置又は現金決済又はその他の方法での有効な経済処分にかかわらず)。取引法第16条に示される上昇等値を確立または増加させるか、または下落等値を確立または増加させるか、すなわち、普通株式として行使可能な任意の普通株または証券を変換または減少させることができる。(B)自己等株式株式を発売するか、又は吾等株式株式に発行又は行使可能又は自己等株式株式に交換可能な任意の証券に関する登録声明を提出又は手配するか、又は(B)前述のいずれかの株を発行又は発行するか、又は(C)任意のスワップ又は他の手配を締結し、(A)項に記載されたいずれかのこれらの取引にかかわらず、わが等株式所有権の任意の経済的結果を他の者に全部又は部分的に移転させるための任意の合意を提出又は手配する。上記(B)又は(C)項は、自社株式又は当該その他の他の証券を現金又はその他の方法で交付する。
賠償する
私たちは、証券法下の責任を含めて、配給エージェントのいくつかの責任を賠償し、配給エージェントがこれらの債務のために支払うことを要求される可能性があるお金を支払うことに同意する。
発行価格と株式証券取引権価格の確定
我々が発売している証券の実際の公開発行価格と、我々が発売している一般権証と予融資権証の行使価格は、私たち、配給代理と発売中の投資家の間で、私たちの普通株の発売前の取引価格などに基づいて交渉します。我々が発売した証券の公開発行価格および我々が発売した一般権証および予備資本権証の行使価格を決定する際には、ナスダック最低価格を含む、我々の歴史と将来性、わが業務の発展段階、将来の業務計画およびこれらの計画の実施程度、我々の経営陣に対する評価、発売時の証券市場の一般的な状況、その他関連すると考えられる要因を考慮する(ナスダック上場規則第5635(D)条参照)。
その他の補償
もし吾等が配給エージェントとの契約を終了又は終了してから6(6)ヶ月以内に、吾等が配給エージェントに吾等に紹介した任意の投資家又は配給エージェントが吾等を代表して議論した任意の投資家に株式又は株式にリンクした証券を売却することを完了した場合、配給代理は引受業者又は配給代理としてではなく(任意の個人又は実体が任意のオプション、株式証又は他の転換可能な証券を行使する場合を除く)、特定の例外を除いて、吾等は分配代理に本条に記載の手数料を支払わなければならず、それぞれの場合において、当該等の投資家から受け取った当該等の融資部分のみから手数料を支払わなければならない。
規則M
配給代理人は、証券法第2(A)(11)条に示される引受業者と見なすことができ、それが受領した任意の手数料、および依頼者を務めている間にその売却された証券を転売することによって達成された任意の利益は、証券法による引受割引または手数料と見なすことができる。引受業者として、配給エージェントは、ルール10 b-5および取引法でのルールMを含むが、ルール10 b-5および取引法でのルールMを含むが、これらに限定されない証券法および取引法の要求を遵守することが要求される。これらのルールや規定は,依頼者の配給エージェントとして我々の証券を購入·販売する時間を制限する可能性がある.これらの規則および規定によれば、配給エージェント(I)は、私たちの証券に関連する任意の安定した活動に従事してはならず、(Ii)私たちの任意の証券を競ったり購入したりしてはならず、取引法が許可されなければ、それが流通に参加するまで、誰にも私たちの任意の証券を購入させようとしてはならない。
電子化流通
電子フォーマットの入札説明書は、配給代理メンテナンスのサイト上で提供することができる。発行に関連して、配給エージェントまたは選定された取引業者は、入札説明書を電子的に配信することができる。AdobePDFとして印刷可能な目論見書を除いて、どの形式の電子入札説明書も今回の発行には使用されない。
電子形式の株式募集規約以外に、配給代理サイト上の資料及び配給代理が維持している任意の他のサイトに掲載されているいかなる資料も、募集定款或いは登録説明書の一部ではなく、吾等或いは配給代理が配給代理として承認及び/又は裏書きを得ておらず、投資家は依存してはならない。
何らかの関係がある
配給代理とその共同経営会社は将来、通常の業務中に時々投資銀行と金融コンサルティングサービスを提供することができ、通常の費用と手数料を受け取ることができる。
“ナスダック”資本市場の上場
私たちの普通株はナスダック資本市場に発売され、コードは“PBLA”です
アメリカ国外で制限を提供します
ヨーロッパ経済区
欧州経済圏の各加盟国については、募集説明書指示の下で以下の免除を除いて、発行対象となる証券は、その加盟国ではまだ、または公衆にいかなる要約も発行されていない
|
(a) |
株式募集説明書指令に定義されている適格投資家に対する任意の法律実体 |
|
(b) |
150人未満の自然人または法人(募集説明書命令で定義された適格投資家を除く)に販売するが、このような任意の要約に対する代表の同意を事前に得なければならない |
|
(c) |
募集定款指令第3条第2項の範囲内のいずれか他の場合は、 |
提供上記(A)~(C)項で述べた証券要約は、当社又は配給代理が目論見指令第3条に基づいて募集定款を掲載するか、又は目論見指令第16条に基づいて募集定款を補充しなければならない。
加盟国に位置するすべての証券要約又は証券要約に関する通信を受けた者、又は最初に我々の証券の任意の株式を買収した者は、配給代理及び会社に陳述、保証、確認及び同意したとみなされるであろう(1)目論見書指示を実施する第2(1)(E)条の当該加盟国法律でいう“適格投資家”であり、(2)目論見命令第3条(2)で使用されるこの用語は、金融仲介として買収された我々の証券の任意の株式である。その要約で買収された証券は、募集定款指令で定義されている適格投資家以外のどの加盟国の人に買収するためのものでもなく、適格投資家以外のどの加盟国の人に買収するためでもなく、又は配給代理が事前に要約又は転売に同意した場合、又は我々の証券が任意の加盟国を代表する適格投資家以外の者が買収した場合には、目論見書指令に基づいて、それに提出された当該等の証券要約は当該等の者になされたものとみなされない
当社、配給代理及びそのそれぞれの共同経営会社は、上記の陳述、確認、合意の真実性と正確性に依存する。
本募集説明書の作成根拠は、目論見書指令の免除により、どの加盟国でもわれわれ証券のどの要約に対しても提出され、目論見書の発行要求の制限を受けないことである。したがって、当該加盟国で本募集説明書に記載されている発売予定の証券について要約を作成又は提出しようとする者は、当社又は配給代理が募集規約指令第3条に基づいて当該要約について募集規約を掲載する義務がない場合にのみ要約を行うことができる。当社または配給代理に入札説明書の掲載が義務付けられている場合、当社または配給代理はいずれも許可されておらず、いかなる証券要約も許可されていません。
この条項について、“私たちの証券を公衆に公開する”という言葉は、任意の加盟国における私たちの任意の証券に関連しており、投資家が証券の購入または承認を決定することができるように、任意の形態および任意の手段で証券の購入または承認を決定することができるようにすることを意味する。この加盟国で目論見書指示を実施する任意の措置がこれらの条項を変更する可能性があるからである。“目論見書指示”とは、2003/71/EC(改正された)であり、各加盟国の任意の関連実行措置を含む命令を意味する。上記販売制限は、以下のいずれかの他の販売制限以外の追加制限である。
イギリスの潜在的投資家は
また、連合王国では、本文書は、(I)2005年の“金融サービス·市場法”(金融促進)令第19(5)条の範囲内の投資に関する事項について専門的な経験を有する者のみに配布され、その後提出される任意の要約についてのみ対象となる。(Ii)命令第49条(2)(A)から(D)条に示す高純価値会社(または他の方法でその命令を合法的に伝達することができる者)に属する(すべての者を合わせて“関係者”と呼ぶ)。連合王国では、非関係者は本文書に基づいたり、本文書に依存したりしてはならない。連合王国では、本文書に関連するいかなる投資や投資活動も関係者に提供し、それと行うことしかできません。
スイスから潜在投資家への通知
証券はスイスで公開発売されない可能性があり、スイス証券取引所(Six)やスイスの他のいかなる証券取引所や規制された取引機関にも上場しない可能性がある。本文書は,ART項で発行される目論見書の開示基準は作成時に考慮していない.652 aかArt“スイス義務法典”の1156条又は上場目論見書の開示基準による。27歳からです。六上場規則又はスイスの任意の他の証券取引所又は規制された取引機関の上場規則。本文書および当社の証券または今回の発売に関連する任意の他の発売またはマーケティング材料は、スイスで公開配布または公開提供することはできません。
本書類または今回の発行、当社または私たちの証券に関連する任意の他の発売またはマーケティング材料は、スイスの規制機関に提出されるか、またはスイスの規制機関の承認を得るであろう。特に、本文書はスイス金融市場規制機関FINMA(FINMA)に提出されることはなく、私たちの証券の要約もスイス金融市場監督局(FINMA)の規制を受けることはなく、私たちの証券の要約はスイス連邦集団投資計画法案(CISA)によって承認されることもありません。中鋼協が集団投資計画で権益を取得した買収者に提供する投資家保障によると、私たちの証券の購入者には伸びていない。
ドバイ国際金融センター潜在投資家心得
本募集説明書は、ドバイ金融サービス管理局(“DFSA”)による要約証券規則による免除要約に関するものである。本募集説明書は、DFSA“発行済み証券ルール”で指定されたタイプの方にのみ配布することを目的としています。それは他の誰にも渡すこともできないし、他の誰にも依存することはできない。DFSAは、免除オファーに関連するいかなるファイルも審査または確認する責任がありません。DFSAはこの目論見書を承認せず、ここに記載されている情報を確認する措置も取らず、目論見書に対して何の責任も負わない。本募集説明書に関連する証券は、流動性が不足している可能性があり、および/または転売制限されている可能性がある。発売された証券を購入しようとする者は自ら証券に関する職務調査を行うべきである。もしあなたが本募集説明書の内容を理解していない場合、あなたは許可された財務コンサルタントに相談しなければならない。
オーストラリアの潜在投資家の注意事項
オーストラリア証券及び投資委員会(“ASIC”)には、初回発売に関する配給書類、目論見書、製品開示声明又はその他の開示文書は提出されていない。本募集説明書は、“2001年会社法”(以下、“会社法”という。)下の目論見書、製品開示声明又は他の開示文書を構成するものではなく、目論見書、製品開示声明又は会社法が規定する他の開示文書に必要な情報も含まれていない。
会社法第708条に記載されている1つ以上の免除によれば、オーストラリアでは、“ベテラン投資家”(会社法第708(8)条に示す)、“専門投資家”(会社法第708(11)条に示される)または他の態様に属する者(“免除投資家”)にのみ提出され、会社法第6 D章に従って投資家に開示されずに証券を提供することが合法である。
免除されたオーストラリア投資家が申請した証券は、発売発行日から12ヶ月以内にオーストラリアで発売されてはならない。会社法第708条の免除又はその他の規定に基づいて、会社法第6 D章に基づいて投資家に開示する必要がない、又は要約は、会社法第6 D章に該当する開示書類に基づいて行われる。私たちの証券を買収する人は誰でもオーストラリアのこのような転売制限を守らなければならない。
本募集説明書には一般資料のみが含まれており、特定の人々の投資目標、財務状況、または特別な需要は考慮されていない。それはどんな証券推薦や金融商品提案も含まれていない。投資決定を下す前に、投資家は本募集定款内の資料が彼らの需要、目標と状況に適しているかどうかを考慮する必要があり、もし需要があれば、このようなことについて専門家の意見を聞く必要がある。
香港の潜在的投資家の心得
当該等の証券は要約や売却はなく、香港で任意の文書形式で要約や販売することもないが、(A)は“証券及び先物条例”(第章)で定義された“専門投資家”を除外する。又は(B)その他の場合、この文書は、“会社条例”(第571章)で定義された“株式募集規約”ではない。32)または、この条例によって示される公衆への要約を構成しない。香港または他の場所では、そのような証券に関連する広告、招待または文書が発行されているか、または発行されていないか、またはそれに関連する広告、招待または文書が管理されていないか、または発行されていないか、またはその内容が香港の公衆に閲覧または読まれる可能性がある)であるが、香港以外の人にのみ販売または販売されることが意図されている人または“証券および先物条例”およびこの条例によって締結された任意の規則によって定義された“専門投資家”のみに販売される証券に関連する者は例外である。
日本の潜在投資家の心得
この等の証券は、日本の“金融商品及び取引法”(1948年第25号法律改正本)に基づいて登録されていないため、直接又は間接的に日本で発売または販売されることはなく、または任意の日本人の利益のために、または他の人に直接または間接的に日本国内または任意の日本人に再発売または転売されることはなく、日本の関連政府または規制当局が関連時間に公布したすべての適用法律、法規及び部級の指針に適合しない限り、本項において、“日本人”とは、日本の法律に基づいて設立された任意の会社又は他の実体を含む日本に住む誰かを意味する。
シンガポールの潜在投資家の心得
この目論見書はシンガポール金融管理局にはまだ目論見書として登録されていません。したがって、本募集説明書及び証券要約又は販売、又は引受又は購入招待に関する任意の他の書類又は材料は、直接又は間接的にシンガポール国内の人々に配布又は配布してはならず、シンガポール国内の人々に直接又は間接的に引受又は購入の招待を行ってはならないが、以下を除く:(I)“証券及び先物法”第289章“証券及び先物法”第274条に基づいて機関投資家に配布し、(Ii)第275(1)条に基づいて関係者に、又は第275(1 A)条に基づいて任意の者に提供する。そして、“SFA”第275条に規定する条件に適合するか、または(Iii)他の方法で“SFA”の任意の他の適用条項の条件を満たし、適合する。
証券がSFA第275条に基づいて関係者によって引受または購入された場合、すなわち:
|
(a) |
その唯一の業務は投資を保有することであり、そのすべての株式は1人以上の個人が所有しており、各個人は認可投資家である |
|
(b) |
信託(受託者は認可投資家ではない)は、唯一の目的は投資を保有することであり、その信託の受益者はどの受益者も投資家を認める個人である |
当該会社の証券(証券取引条例第239条(1)に規定されているように)又は受益者の当該信託における権利及び利益(いずれにしても記載)は、当該会社又は当該信託が“証券取引条例”第275条に基づいて提出した要約買収後6ヶ月以内に譲渡してはならないが、以下の場合を除く
|
(a) |
機関投資家又はSFA第275(2)条に規定された関係者,又はSFA第275(1 A)条又は第276条(4)(I)(B)条に示される要約により生じた者 |
|
(b) |
考慮していない場合、あるいは移転を考慮するだろうか |
|
(c) |
すべての譲渡は法律の施行によって行われる |
|
(d) |
第二百七十六条第七項に規定するように、又は |
|
(e) |
シンガポール“2005年証券及び先物(投資要約)(株式及び債券)規程”第32条に述べたように。 |
カナダの潜在投資家の注意事項
証券は、国家文書45-106に定義されている認可投資家である購入者または購入元金とみなされる購入者にのみ販売される株式募集規約の免除又はこの条例第七百三十三条第一項証券法(オンタリオ州)、国家機器31-103に定義されている許可顧客である登録要件、免除、継続的な登録義務それは.証券のいかなる転売も、証券法が適用される目論見書の要求の免除または目論見書の要求を受けない取引に適合しなければならない。
本入札明細書(本募集説明書の任意の改正を含む)に不実陳述が含まれている場合、カナダのある省または地域の証券法は、購入者が購入者のいる省または地域の証券法に規定されている期限内に撤回または損害賠償を行使することを前提として、購入者に撤回または損害賠償を提供することができる。買い手は、これらの権利の詳細を理解するために、または法律顧問に相談するために、買い手の所在する省または地域の証券法の任意の適用条項を参照しなければならない。
“国家文書33−105”第3 A.3節(又は非カナダ司法管区の政府により発行又は担保された証券については、第3 A.4節)保証紛争(NI 33-105)、配給エージェントは、今回の発行に関連する利益相反に関する引受業者の開示要求を遵守する必要がない
法律事務
本募集説明書が提供する証券の有効性は,Faegre Drinker Bdle&Reath LLPによって伝達される.Pryor Cashman LLPは配給エージェントの法律顧問を務め,今回の発行に関する何らかの法務を処理する.
専門家
Panbelaは、2022年12月31日現在と2021年12月31日現在の財務諸表および2022年12月31日までの2年度の財務諸表を独立公認会計士事務所Cherry Bekairt LLPが監査し、この報告書を引用して本募集説明書に記入している。このような財務諸表は,同社が会計·監査の専門家の権威として提供した報告書に基づいて組み込まれている。
そこでもっと多くの情報を見つけることができます
我々は、証券法に基づいてS-1表の形式で米国証券取引委員会に登録声明を提出し、本募集説明書の下で提供される証券の流通を登録した。登録声明は、添付された証拠物およびスケジュールを含み、私たちおよび証券に関する他の関連情報を含む。米国証券取引委員会の規則及び規定によれば、本募集説明書には、登録説明書に含まれるいくつかの情報を省略することができる。
我々は証券取引法の情報要求に制約され、年度、四半期、現在の報告、依頼書、その他の情報を米国証券取引委員会に提出することを要求されている。我々が米国証券取引委員会に提出した任意の情報は、本入札説明書に組み込まれた文書を参照することにより、米国証券取引委員会のウェブサイトwww.sec.govでも取得することができる。私たちはまた、アメリカ証券取引委員会にこれらの書類を提出した後、合理的で実行可能な場合に、できるだけ早く私たちのウェブサイトwww.panbela.comでこれらの書類を無料で提供します。本入札説明書には、本入札明細書に含まれる情報、または本サイトを介して取得可能な情報は含まれていない。
2023年9月30日までの3ヶ月と9ヶ月は、2022年9月30日までの3ヶ月と9ヶ月、および2022年12月31日と2021年12月31日までの財政年度の財務諸表とは歴史的である。比較のため、2023年1月13日に発効した40株1株逆分割または2023年6月1日に発効した30株1株逆株式分割の株と1株当たり金額を改めて述べた。2023年9月30日までの3ヶ月と9ヶ月は、2022年9月30日までの3ヶ月と9ヶ月、および2022年12月31日と2021年12月31日までの財政年度と比較して、2024年1月18日に発効した20株1株を逆分割して再申告する株や1株当たりの金額はない。
項目1.財務諸表
Panbela治療会社簡明総合貸借対照表
(単位は千で、シェアは含まれていない)
|
2023年9月30日 |
2022年12月31日 |
|||||||
|
(未監査) |
||||||||
|
資産 |
|
|||||||
|
流動資産: |
||||||||
|
現金と現金等価物 |
$ | 907 | $ | 1,285 | ||||
|
前払い費用と他の流動資産 |
824 | 443 | ||||||
|
課税所得税 |
155 | 49 | ||||||
|
流動資産総額 |
1,886 | 1,777 | ||||||
|
他の非流動資産 |
8,742 | 3,201 | ||||||
|
総資産 |
$ | 10,628 | $ | 4,978 | ||||
|
負債と株主赤字 |
||||||||
|
流動負債: |
||||||||
|
売掛金 |
$ | 6,612 | $ | 2,865 | ||||
|
費用を計算する |
1,133 | 2,993 | ||||||
|
支払利息 |
172 | 325 | ||||||
|
支払手形 |
- | 650 | ||||||
|
債務、流動部分 |
1,000 | 1,000 | ||||||
|
流動負債総額 |
8,917 | 7,833 | ||||||
|
債務,現在分の純額を差し引く |
4,194 | 5,194 | ||||||
|
非流動負債総額 |
4,194 | 5,194 | ||||||
|
総負債 |
13,111 | 13,027 | ||||||
| 株主赤字: | ||||||||
|
優先株、$0.001 額面価値10,000,000許可された違います。2023年9月30日と2022年12月31日までに発行または発行された株式 |
- | - | ||||||
|
普通株、$0.001額面価値100,000,000許可された2,996,753そして34,761発行された株式と2,996,334そして34,7612023年9月30日と2022年12月31日までの未返済債務 |
3 | - | ||||||
|
原価計算の在庫株419そして02023年9月30日と2022年12月31日までの株 |
- | - | ||||||
|
追加実収資本 |
106,026 | 82,286 | ||||||
|
赤字を累計する |
(109,883 | ) | (91,094 | ) | ||||
|
総合収益を累計する |
1,371 | 759 | ||||||
|
株主総損失額 |
(2,483 | ) | (8,049 | ) | ||||
|
総負債と株主赤字 |
$ | 10,628 | $ | 4,978 | ||||
全期間の株式と1株データは、2023年6月1日に発効した30株1株逆株式分割と、2023年1月13日に発効した40株1株逆株分割を反映するように調整されている。
簡明な総合財務諸表付記はこれらの報告書の構成要素である。
パンバラ治療会社は
簡明合併経営報告書と全面赤字(千元、1株および1株当たりの金額は含まれていない)
(未監査)
|
9月30日までの3ヶ月間 |
9月30日までの9ヶ月間 |
|||||||||||||||
|
2023 |
2022 |
2023 |
2022 |
|||||||||||||
|
運営費用: |
||||||||||||||||
|
一般と行政 |
$ | 1,107 | $ | 1,294 | $ | 4,102 | $ | 4,349 | ||||||||
|
研究開発 |
6,739 | 2,329 | 14,501 | 24,563 | ||||||||||||
|
営業損失 |
(7,846 | ) | (3,623 | ) | (18,603 | ) | (28,912 | ) | ||||||||
|
その他の収入(支出): |
||||||||||||||||
|
利子収入 |
49 | 6 | 114 | 10 | ||||||||||||
|
知的財産権を売却する収益 |
400 | - | 400 | - | ||||||||||||
|
利子支出 |
(71 | ) | (87 | ) | (245 | ) | (107 | ) | ||||||||
|
その他の費用 |
(382 | ) | (754 | ) | (622 | ) | (1,293 | ) | ||||||||
|
その他費用合計 |
(4 | ) | (835 | ) | (353 | ) | (1,390 | ) | ||||||||
|
所得税割引前損失 |
(7,850 | ) | (4,458 | ) | (18,956 | ) | (30,302 | ) | ||||||||
|
所得税割引 |
19 | 56 | 167 | 104 | ||||||||||||
|
純損失 |
(7,831 | ) | (4,402 | ) | (18,789 | ) | (30,198 | ) | ||||||||
|
外貨換算調整 |
381 | 727 | 612 | 1,240 | ||||||||||||
|
総合損失 |
$ | (7,450 | ) | $ | (3,675 | ) | $ | (18,177 | ) | $ | (28,958 | ) | ||||
|
1株当たりの基本と償却純損失 |
$ | (2.69 | ) | $ | (257.36 | ) | $ | (14.35 | ) | $ | (2,255.96 | ) | ||||
|
加重平均流通株−基本と希釈 |
2,914,600 | 17,107 | 1,309,137 | 13,386 | ||||||||||||
全期間の株式と1株データは、2023年6月1日に発効した30株1株逆株式分割と、2023年1月13日に発効した40株1株逆株分割を反映するように調整されている。
簡明な総合財務諸表付記はこれらの報告書の構成要素である。
パンバラ治療会社は
株主(損失)権益簡明連結報告書
(単位は千で、シェアは含まれていない)
(未監査)
|
2023年9月30日までの9ヶ月間 |
||||||||||||||||||||||||||||||||
| 普通株 | 在庫株 |
余分な実収 |
積算 |
その他を累計する |
株主合計 |
|||||||||||||||||||||||||||
|
株 |
金額 |
株 |
金額 |
資本 |
赤字.赤字 |
総合収益 |
(赤字)権益 |
|||||||||||||||||||||||||
|
2023年1月1日現在の残高 |
34,761 | $ | - | - | - | $ | 82,286 | $ | (91,094 | ) | $ | 759 | $ | (8,049 | ) | |||||||||||||||||
|
普通株を売却して得た金 |
237,191 | - | - | - | 15,359 | - | - | 15,359 | ||||||||||||||||||||||||
|
こまごました株に支払われた現金 |
- | - | - | - | (4 | ) | - | - | (4 | ) | ||||||||||||||||||||||
|
権証を現金に換えます |
264,124 | 1 | - | - | (1 | ) | - | - | - | |||||||||||||||||||||||
|
株に基づく報酬 |
- | - | - | - | 180 | - | - | 180 | ||||||||||||||||||||||||
|
純損失 |
- | - | - | - | - | (5,125 | ) | - | (5,125 | ) | ||||||||||||||||||||||
|
外貨換算調整 |
- | - | - | - | - | - | 164 | 164 | ||||||||||||||||||||||||
|
2023年3月31日現在の残高 |
536,076 | $ | 1 | - | $ | - | $ | 97,820 | $ | (96,219 | ) | $ | 923 | $ | 2,525 | |||||||||||||||||
|
普通株を売却して得た金 |
2,008,881 | $ | 2 | - | - | $ | 7,711 | - | - | $ | 7,713 | |||||||||||||||||||||
|
こまごました株に支払われた現金 |
- | - | - | - | (5 | ) | - | - | (5 | ) | ||||||||||||||||||||||
|
権証を現金に換えます |
67,694 | - | - | - | - | - | - | - | ||||||||||||||||||||||||
|
断片的な株式の調整 |
(613 | ) | - | - | - | - | - | - | - | |||||||||||||||||||||||
|
株に基づく報酬 |
- | - | - | - | 329 | - | - | 329 | ||||||||||||||||||||||||
|
純損失 |
- | - | - | - | - | (5,833 | ) | - | (5,833 | ) | ||||||||||||||||||||||
|
外貨換算調整 |
- | - | - | - | - | - | 67 | 67 | ||||||||||||||||||||||||
|
2023年6月30日までの残高 |
2,612,038 | $ | 3 | - | $ | - | $ | 105,855 | $ | (102,052 | ) | $ | 990 | $ | 4,796 | |||||||||||||||||
|
普通株を売却して得た金 |
3,017 | $ | - | - | - | $ | 3 | 3 | ||||||||||||||||||||||||
|
逓増製品コスト |
- | - | - | - | (22 | ) | - | - | (22 | ) | ||||||||||||||||||||||
|
前払い資金持分証を行使する |
165,000 | - | - | - | - | - | - | - | ||||||||||||||||||||||||
|
前払い資金権証で無現金に両替します |
95,954 | - | - | - | - | - | - | - | ||||||||||||||||||||||||
|
権証を現金に換えます |
120,744 | - | - | - | - | - | - | - | ||||||||||||||||||||||||
|
在庫株 |
(419 | ) | - | 419 | - | - | - | - | - | |||||||||||||||||||||||
|
株に基づく報酬 |
- | - | - | - | 190 | - | - | 190 | ||||||||||||||||||||||||
|
純損失 |
- | - | - | - | - | (7,831 | ) | - | (7,831 | ) | ||||||||||||||||||||||
|
外貨換算調整 |
- | - | - | - | - | - | 381 | 381 | ||||||||||||||||||||||||
|
2023年9月30日までの残高 |
2,996,334 | $ | 3 | 419 | $ | - | $ | 106,026 | $ | (109,883 | ) | $ | 1,371 | $ | (2,483 | ) | ||||||||||||||||
全期間の株式と1株データは、2023年6月1日に発効した30株1株逆株式分割と、2023年1月13日に発効した40株1株逆株分割を反映するように調整されている。
簡明な総合財務諸表付記はこれらの報告書の構成要素である。
パンバラ治療会社は
株主(損失)権益簡明連結報告書
(単位は千で、シェアは含まれていない)
(未監査)
|
2022年9月30日までの9ヶ月間 |
||||||||||||||||||||||||||||||||
| 普通株 | 在庫株 |
余分な実収 |
積算 |
その他を累計する |
株主合計 |
|||||||||||||||||||||||||||
| 株 |
金額 |
株 |
金額 |
資本 |
赤字.赤字 |
総合収益 |
権益(赤字) |
|||||||||||||||||||||||||
|
2022年1月1日現在の残高 |
10,995 | $ | - | - | - | $ | 66,240 | $ | (56,160 | ) | $ | 133 | $ | 10,213 | ||||||||||||||||||
|
制限株の帰属 |
4 | - | - | - | - | - | - | - | ||||||||||||||||||||||||
|
株に基づく報酬 |
- | - | - | - | 334 | - | - | 334 | ||||||||||||||||||||||||
|
純損失 |
- | - | - | - | - | (3,666 | ) | - | (3,666 | ) | ||||||||||||||||||||||
|
外貨換算調整 |
- | - | - | - | - | - | (299 | ) | (299 | ) | ||||||||||||||||||||||
|
2022年3月31日現在の残高 |
10,999 | $ | - | - | $ | - | $ | 66,574 | $ | (59,826 | ) | $ | (166 | ) | $ | 6,582 | ||||||||||||||||
|
普通株式発行-CPP |
6,099 | $ | - | - | - | $ | 9,605 | - | - | $ | 9,605 | |||||||||||||||||||||
|
制限株の帰属 |
4 | - | - | - | - | - | - | - | ||||||||||||||||||||||||
|
株に基づく報酬 |
- | - | - | - | 293 | - | - | 293 | ||||||||||||||||||||||||
|
純損失 |
- | - | - | - | - | (22,130 | ) | - | (22,130 | ) | ||||||||||||||||||||||
|
外貨換算調整 |
- | - | - | - | - | - | 812 | 812 | ||||||||||||||||||||||||
|
2022年6月30日までの残高 |
17,102 | $ | - | - | $ | - | $ | 76,472 | $ | (81,956 | ) | $ | 646 | $ | (4,838 | ) | ||||||||||||||||
|
オプションを行使して現金を持たない |
12 | $ | - | - | - | $ | - | - | - | $ | - | |||||||||||||||||||||
|
現金持分証を行使する |
- | - | - | - | 5 | - | - | 5 | ||||||||||||||||||||||||
|
株に基づく報酬 |
- | - | - | - | 230 | - | - | 230 | ||||||||||||||||||||||||
|
純損失 |
- | - | - | - | - | (4,402 | ) | - | (4,402 | ) | ||||||||||||||||||||||
|
外貨換算調整 |
- | - | - | - | - | - | 727 | 727 | ||||||||||||||||||||||||
|
2022年9月30日までの残高 |
17,114 | $ | - | - | $ | - | $ | 76,707 | $ | (86,358 | ) | $ | 1,373 | $ | (8,278 | ) | ||||||||||||||||
全期間の株式と1株データは、2023年6月1日に発効した30株1株逆株式分割と、2023年1月13日に発効した40株1株逆株分割を反映するように調整されている。
簡明な総合財務諸表付記はこれらの報告書の構成要素である。
パンバラ治療会社は
キャッシュフロー簡明併合表 (単位:千)
(未監査)
|
9月30日までの9ヶ月間 |
||||||||
|
2023 |
2022 |
|||||||
|
経営活動のキャッシュフロー: |
||||||||
|
純損失 |
$ | (18,789 | ) | $ | (30,198 | ) | ||
|
純損失と経営活動で使用される現金純額の調整: |
||||||||
|
核販売が行っている研究と開発(IPR&D) |
- | 17,737 | ||||||
|
株に基づく報酬 |
699 | 857 | ||||||
|
非現金利子支出 |
172 | 97 | ||||||
|
知的財産権を売却する収益 |
(400 | ) | - | |||||
|
経営性資産と負債変動状況: |
||||||||
|
課税所得税 |
(112 | ) | 302 | |||||
|
前払い費用と他の流動資産 |
(381 | ) | (451 | ) | ||||
|
他の非流動資産 |
(5,541 | ) | (2,561 | ) | ||||
|
売掛金 |
4,370 | 5,392 | ||||||
|
負債を計算すべきである |
(2,187 | ) | (1,448 | ) | ||||
|
経営活動のための現金純額 |
(22,169 | ) | (10,273 | ) | ||||
|
投資活動によるキャッシュフロー: |
||||||||
|
知的財産権研究開発への投資 |
- | (660 | ) | |||||
|
知的財産権を売却して得た収益 |
400 | - | ||||||
|
合併で得た現金 |
- | 4 | ||||||
|
投資活動での純現金の提供 |
400 | (656 | ) | |||||
|
資金調達活動のキャッシュフロー: |
||||||||
|
普通株と引受権証を売却して得られた金は,純額は#ドルである2.1何百万ドルもの製品の発売コストは |
23,052.00 | - | ||||||
|
こまごました株に支払われた現金 |
(9.00 | ) | - | |||||
|
株式証明書を行使して得た金 |
- | 5 | ||||||
|
手形の元金払い |
(1,650 | ) | - | |||||
|
融資活動が提供する現金純額 |
21,393 | 5 | ||||||
|
為替レート変動が現金に与える影響 |
(2 | ) | (2 | ) | ||||
|
現金純変動額 |
(378 | ) | (10,926 | ) | ||||
|
期初現金及び現金等価物 |
1,285 | 11,867 | ||||||
|
期末現金および現金等価物 |
$ | 907 | $ | 941 | ||||
|
キャッシュフロー情報の追加開示: |
||||||||
|
利子計算期間中に支払った現金 |
$ | 398 | $ | 9 | ||||
|
非現金取引の追加開示: |
||||||||
|
資産買い入れ対価として発行される普通株·株式オプションと引受権証の公正価値 |
$ | - | $ | 9,605 | ||||
全期間の株式と1株データは、2023年6月1日に発効した30株1株逆株式分割と、2023年1月13日に発効した40株1株逆株分割を反映するように調整されている。
簡明な総合財務諸表付記はこれらの報告書の構成要素である。
Panbela治療会社簡明合併財務諸表付記
1.ビジネス
Panbela治療会社(“Panbela”)とその直接子会社Panbela Research,Inc.(“Panbela Research”)癌予防製薬会社(“CPP”)と癌予防製薬(アイルランド)有限会社が存在する主な目的は,破壊的療法の開発であり,緊急医療ニーズがあるが満足されていない患者の治療に用いられる。Panbela Treateutics Pty LtdはPanbela Researchの完全子会社で、オーストラリアの法律に基づいて設立された。癌予防は二つ全資保有の休眠子会社:イギリス実体癌予防製薬有限会社とアリゾナ州有限責任会社癌予防製薬有限会社。Panbela治療会社とその直接·間接子会社を総称して“私たち”,“会社”と呼ぶ
われわれのパイプラインの主な目標は,薬物療法を利用し,薬物療法を補充することで増加した疾患関連ポリアミンを減少あるいは正常化することである。われわれの主な候補薬はエフロンアスピリン(SBP−101)であり,フロリダ大学研究財団社から独占的に許可されており,Flynpoviはエフルオロオルニチン(cpp−1 x)とスリン酸とeflornithine(cpp−1 x)の単独錠剤あるいは香嚢形態の組み合わせである。私たちはすでにアリゾナ大学のアリゾナ州取締役会からFlynpoviを商業化する独占的な許可権を取得しましたが、北米でのFlynpoviの開発と商業化の再許可協定は4月に許可者によって終了しました4, 2023.
逆株分割
6月から発効する1, 2023,パンベラは1対1の30普通株の流通株を逆分割する。1月施行13, 2023,パンベラは1対1の40普通株の流通株を逆分割する。本文に別段の規定がない限り、当社の普通株のすべての株式と1株当たりの金額は逆株式分割を反映するように遡及調整されている。備考をご参照ください7より多くの情報を得るために。
2.リスクと不確実性
その会社は高度な規制と競争の環境で運営されている。薬品の開発,製造とマーケティングは米国食品·薬物管理局(FDA),オーストラリア治療用品管理局,EUの欧州薬品管理局および他の国の類似機関の承認を得,これらの機関の持続的な監督を受ける必要がある。新しい薬品の承認を得ることは決して確定的ではなく、何年もかかるかもしれないし、通常は大量の支出が予想される。
1ドルの損失を受けました109.9 百万私たちがいてから2011それは.上には9人9月末までの月30, 2023,私たちは純損失#ドルが発生した18.8 百万それは.私たちはまた経営活動から約#ドルのマイナスキャッシュフローを生み出した22.2 百万その間に。私たちが引き続き発展活動に従事し、商業化を求めることに伴い、大きな損失を受けることが予想され、経営活動から負の純キャッシュフローが生じる可能性がある。約$を生み出しました21.4 百万普通株の売却,あらかじめ出資した引受権証と引受権証の収益に関する融資活動による正現金流量は,以下の項目の支払いによって一部相殺される二つこの切符です。9月までに30, 2023,約一ドルの現金があります0.9 百万運営資本の赤字は$7.0 百万(運営資本の定義は、流動資産から流動負債を差し引く)と株主赤字#ドルである2.5 百万それは.同社の主要な現金源には従来から転換可能な債券の発行と株式証券の発行が含まれていた。
添付されている簡明総合財務諸表の作成仮説は、引き続き経営を継続する企業として、正常な業務過程で資産の現金化と清算負債を考慮する。簡明な総合財務諸表は、資産の回収可能性または分類、またはこれらの不確実性の結果に起因する可能性のある負債額に関連するいかなる調整も含まない。私たちの現在の独立公認会計士事務所は、彼らの監査報告書の中で、このような持続的な経営の不確実性を強調しています20223月の財務諸表16, 2023.私たちが通常の業務過程で経営を継続し、資産の帳簿価値を実現し、債務を返済する能力は、私たちが追加融資を得る能力、私たちの開発仕事の成功、私たちの候補製品のアメリカ、オーストラリア、EUまたは他の市場でマーケティング承認を得る能力、そして最終的に私たちの候補製品をマーケティングして販売する能力を含む多くの要素に依存します。他の要因を除いて、これらの要因は、私たちが継続的に経営している会社として運営を続ける能力を大きく疑わせている。備考をご参照ください4“流動性とビジネス計画”と題して
年1月2022同社は、米国、欧州、アジア太平洋地域(APAC)で行われている世界ランダム第2/第3段階臨床試験の開始を発表した。同社はこの新冠肺炎に関連する新臨床試験の進行は何の妨害も受けないと予想している。この試験はゲムシタビンとアブラシャ(NaB−パクリタキセル)の十分な供給に依存しており,供給不足が生じる可能性がある。
3.根拠を述べる
吾らはすでにアメリカ公認会計原則(“アメリカ公認会計原則”)及びアメリカ証券取引委員会(“アメリカ証券取引委員会”)10-Q表及びS-X規定に基づいて中期簡明総合財務諸表を作成した。したがって、それらは、米国公認会計基準によって要求される完全な財務諸表のすべての情報および脚注を含まない。これらの中期簡明総合財務諸表は、正常経常的な計上項目からなるすべての調整を反映しており、経営陣は、これらの調整項目は、我々の総合財務状況、総合経営業績、列挙日までの総合現金流量を公平に報告するために必要であると考えている。私たちの財政年度は12月に終わります31それは.12月現在の簡明総合貸借対照表31, 2022,監査された総合財務諸表に由来するが、米国公認会計基準要件のすべての開示は含まれていない。これらの中期簡明総合財務諸表は、私たちが最近提出した10-K表年次報告書と、その後米国証券取引委員会に提出された文書に含まれる年次総合財務諸表とその付記と一緒に読まなければなりません。私たちの業務の性質は、どんな過渡期の結果も年間の予想結果を代表できないかもしれないということだ。
4.流動資金と業務計画
6月に21, 2023,同社は普通株,事前融資権証,引受権証の登録公開を完了し,純収益は約$である7.7 百万.
1月に30, 2023,同社は普通株,事前融資権証,引受権証の登録公開を完了し,純収益は約$である13.8 百万それは.さらにここでは1つ目は1/42023会社は純収益の約$を受け取りました1.6 百万会社のATMで普通株の売却を計画している(付記参照7)それは.年内に、ATM機は何の販売も発生しない予定です二番目1/42023.
私たちは私たちの現在の事業計画を支援するためにもっと多くの資金を集めなければならない。株式や債務融資、あるいは戦略的協力や許可協定など、様々なルートでより多くの資金を調達することを求めることができる。私たちは私たちの運営を支援するために追加の資金源を得ることができるという保証はないし、もし私たちがそのような資金を持っている場合、私たちはこれらの追加資金が私たちの需要を満たすのに十分であるか、私たちが受け入れられる条件で十分になるという保証はない。私たちの臨床データが積極的でなければ、あるいは経済や市場状況が悪化すれば、このリスクは増加するだろう。
私たちの将来の成功は私たちが追加資金を得る能力、私たちの開発仕事の成功、私たちの候補製品のための伊司アスピリン、フラボヴィッド、エフルオロオルニチンがアメリカあるいは他の市場でマーケティング許可を得る能力、そして最終的に私たちが候補製品をマーケティングし、販売する能力にかかっている。もし私たちが必要な時に追加の融資を得ることができなければ、もし私たちの臨床試験が成功しなければ、あるいは上場承認を得ることができなければ、私たちは経営を続けることができなくなり、私たちの会社の運営を停止し、清算することを余儀なくされるだろう。
私たちは私たちが商業的に合理的な条件で追加的な融資を受けることができるか、あるいは根本的に保証できないという保証はない。追加の転換可能な債券や持分証券の売却は、私たちの既存株主の持分を希釈する可能性がある。
5.主な会計政策の概要
合併原則
添付されている簡明な総合財務諸表には、会社の資産、負債、費用が含まれている。すべての重大な会社間取引と残高は合併で販売された。
予算の使用
簡明な総合財務諸表を作成するには、管理層が、財務諸表の簡明な統合財務諸表の日付における資産および負債の報告金額、または有資産および負債の開示および報告期間内に報告された費用金額に影響を与えるために、推定および仮定を必要とする。実際の結果はこれらの推定とは異なる可能性があり,特に行われている大流行や制御対応による重大な社会や経済混乱や不確実性を考慮している。
研究開発コスト
研究と開発コストには臨床試験による費用が含まれています第3者サービスプロバイダは、様々なテストを実行し、私たちの臨床前研究に関連するデータを蓄積する;スポンサーの研究プロトコル;私たちの臨床前研究および人体臨床試験に必要な製造プロセスのために十分な数の薬品を開発および拡張生産するために必要な製造プロセス;私たちの候補製品開発計画の実行に関連する専門知識のリソースの問い合わせ、給与、福祉および株式ベースの報酬、およびライセンス知的財産権の許可および維持のコストを含む人員コスト。
臨床試験費用を含めて研究開発費を徴収し、発生時に費用を計上します。われわれのヒト臨床試験は現在も将来も臨床試験現場で行われ,契約研究組織(CRO)の協力の下で共同管理される。臨床試験地点を確立する費用は研究協定調印後に計算しなければならない。臨床試験業績と関連する費用は通常契約金額と合意マイルストーンの実現によって累計し、例えば患者登録、患者フォローアップなどである。著者らは臨床試験地点とCROとのコミュニケーションを通じて、患者登録とその他の活動の程度を含む各重要な契約下の業績レベルを監視し、そして必要に応じて四半期ごとに見積もり数を調整し、臨床費用に各臨床試験地点と各CROがかかった実際の努力を反映させる。
ある製品のコストを確保する第3者臨床試験のための医薬製品は通常交付前に支払われ,受信して臨床地点に搬送可能な場合には研究開発費が計上される。
研究と開発コスト2022知的財産権の研究と開発を含む。この資産はCPPの証券保有者から購入され、資産購入後すぐにログアウトして研究開発に使用されている。
すべての重要なCRO契約は私たちの書面通知によって終了することができます。私たちは通常、CROの実際にかかった努力と、いかなる場合にも発生したいくつかのキャンセルできない費用に責任を負うことができます点ターミネーター。
ライセンス制約された知的財産権に将来他の用途がないと判断された場合、特許技術許可の取得に関連するコストを支払う。
株に基づく報酬
株式ベースの報酬を計上する際には、付与日のこれらの奨励の公正価値に基づいて、持分ツールの報酬と交換するために受信された従業員および非従業員サービスのコストを計量·確認する。株式に基づく報酬費用の計算には、高度な主観的な仮定を入力する必要があり、これらの仮定は私たちの最適な推定を代表し、内在的な不確実性と経営陣判断の応用に関連している。補償コストは帰属期間内に直線分配法を用いて比例的に確認され,帰属期間は必要なサービス期間と考えられる。業績に基づく株式オプション報酬の報酬費用は、“業績”が発生した場合、または発生する可能性がある場合に確認される。
株式奨励の公正価値は付与された日にブラック·スコルスオプション定価モデルを用いて推定される。株式報酬の公正価値の決定は、私たちの株価といくつかの複雑かつ主観変数に関する仮定の影響を受ける。無リスク金利は米国債金利をもとに、個々の奨励の期待期限に適用される。予想変動率は会社の株価の歴史的変動性に基づく。配当収益率をゼロなぜなら、私たちは予測可能な未来に何の配当も発表しないと予想しているからだ。付与されたオプションの予想期限は、“簡略化”方法を使用して決定される。この方法では,期待期限は中間点平均帰属日と契約期間終了の間です。
外貨換算調整
Panbela Treateutics Pty Ltdの機能通貨はオーストラリアドルです。したがって,Panbela Treateutics Australia Pty Ltdの資産と負債および株式取引は期末レートでドルに換算される.収入と支出はその期間の有効な平均為替レートに換算する。これにより発生した換算損益は株主権益内列報の累計全面損失構成部分に計上される。.の間に9か月9月までの期間30, 2023そして2022蓄積された他の総合損失から業務の任意の再調整までは無関係である。
総合損失
総合損失には私たちの純損失と外貨換算の影響が含まれています。
1株当たり純損失
1株あたりの基本純損失の計算方法は,当期発行普通株の加重平均を純損失で割ることである。1株当たりの純損失は、期内に発行された普通株の加重平均に在庫株方法で計算した薄化潜在普通株の加重平均を加えて計算する。効果が逆希釈または1株当たり純損失を減少させる場合、このような潜在的な希薄化株は除外される。当社の潜在的希薄化株式は、発行された普通株式オプションと引受権証を含み、すべての期間の減額1株当たり純損失には計上されておらず、反ダンピングの結果となるからである。
次の表は、その影響が示された日に逆希釈されるため、希釈後の1株当たり純損失計算に計上されていない普通株潜在株式を示している
|
九月三十日 |
||||||||
|
2023 |
2022 |
|||||||
|
従業員と非従業員株式オプション |
13,455 | 3,352 | ||||||
|
普通株引受権証により発行可能な普通株 |
4,068,826 | 4,538 | ||||||
| 4,082,281 | 7,890 | |||||||
6.支払手形
スキャンブ本券
9月までに30, 2023,CPPの未清算残高は約#ドルである5.4 百万改正及び重述された引受票(“Sucampo手形”)の項下の元金及び利息。この手形の初期元本金額は約#ドルである6.2 百万Sucampo GmbHを受益者とし、日付は6月です15, 2022.9月までの元本残高30, 2023約$とする5.2 百万Sucampo手形によると、単利金利は5年利率です。すべての未払い元金は、当時のいかなる未払いと応算利息と合わせて、以下のように支払わなければなりません:(I)$1.0 百万毎年1月またはそれまでに累算したがまだ支払われていないすべての利息を加える31, 2024,1月31, 2025,1月と31,2026年;及び。(Ii)すべての残り元金に1月又はそれまでの未払い利息を加える31, 2027.2月に1, 2023,Panbelaは支払うべき残高#ドルを支払った1.0 百万約#ドルの応算と未払い利息を加える295,000それは.9月までに30, 2023,当社がSucampo手形によって支払うすべての金は当期金であり、その手形の未払い利息は約#ドルである172,000それは.Panbelaは日付が6月の保証に基づいて、Sucampo手形の下でのCPPの支払い義務に同意しました15, 2022.
Tillotts約束手形
12月まで31, 2022,CPPの未清算残高は約#ドルである0.7 百万改訂された元本項のもとの元金と利息を代表し、その初期元本金額は約#ドルである650,000Tillotts Pharma AGをサポートします。元金残高及び未払い利息は1月全数で支払いました31, 2023.
7.株主権益
普通株式と引受権証を公開発売2023年6月
6月に21, 2023,当社は登録を終えて公開発売し,あわせて発行した586,000普通株式、事前融資株式権証の株式は、最大で購入できます1,684,000普通株、行使価格は$0.0011株および株式承認証は最大購入可能である4,540,000その普通株の行使価格は$である3.75一株ずつです。これらの証券の総発行価格は1ドルです3.751株当たり普通株および引受権証二つ株、または$3.749あらかじめ出資した引受権証と引受権証に基づいて購入する二つ株式です。今回発行された純収益総額は約1ドルである7.7 百万それは.9月までに30, 2023,事前に出資したすべての引受権証は行使された。これらの証券は,S-1表中の有効登録宣言に基づいて発行される.
余剰権証のうち,購入権証2,270,000普通株式は,選択可能な無現金行使条項があり,この条項によれば,所持者は,(X)現金行使時に発行可能な普通株式総数と(Y)の積に相当する株式総数を通知して得ることができる0.24それは.この機能は6月に発売された28, 20236月まで30, 2023 違います。株式はこのオプションのキャッシュレス操作で発行されています。9月までに30, 2023 120,744普通株式はすでに発行されて株式証を承認して購入する503,100普通株です。
同じ日まで、株式証明書は最大で購入できます4,036,900普通株式はまだ発行されておらず、行権価格は#ドルである3.751株当たり、その中の引受権証は購入できます1,766,900別の種類のキャッシュレストレーニングを行う資格がある。
普通株式および株式承認証を公開発売2023年1月
1月に30, 2023,当社は登録を終えて公開発売し,あわせて発行した161,407普通株式、事前融資株式権証の株式は、最大で購入できます61,090普通株、行使価格は$0.0011株および株式承認証は最大購入可能である444.999調整された取引価格で計算される普通株式は、$2.239一株ずつです。これらの証券の総発行価格は1ドルです67.501株当たり普通株および引受権証二つ株、または$67.47あらかじめ出資した引受権証と引受権証に基づいて購入する二つ株式です。今回発行された純収益総額は約1ドルである13.8 百万それは.これらの証券は,S-1表中の有効登録宣言に基づいて発行される.
すべての前払い資金株式承認証はすでに2月までに行使された3, 2023.残りの株式証明書には別のキャッシュレス行使条項があり,この条項によれば,所有者は(X)現金行使時に発行可能な普通株式総数と(Y)の積に相当する株式総数を通知して受け取ることができる0.75それは.この機能は3月に発売された1, 20239月までに30, 2023, 331,824普通株式はすでに発行されて株式証を承認して購入する442,434株式です。株式購入承認証2,566普通株はまだ発行されておらず、調整後の発行権価格は#ドルである2.239一株ずつです。
市場で計画する
私たちは7月の販売契約を締結しました29, 2022,この条項によると,Roth Capital Partners,LLC(“代理人”)は会社普通株の株式を売却することができ,総販売価格は最高$に達する8.4 百万時々、“市場”株式発行計画(“現金自動支払機計画”)を通過する。販売契約によるとロスは3.0ATM機はどんな普通株販売総毛収入のパーセンテージを計画しています。
1月に2023会社は売りました14,694ATM機計画下の普通株の価格は約$です1.6 百万毛収入の中で。
9月に2023会社は売りました3,017ATM機計画下の普通株の価格は約$です3,900毛収入の中で。
満期後に発生した権利証取引については、当社は少なくとも5月までにATM機の計画に基づいて販売することはできません2, 2024(付記を参照)10).
逆株分割
五月五日25, 2023,会社は株主特別会議を開催し,株主は改訂された会社登録証明書の改訂を提案し,以下の割合で株式逆分割を実施した5回1セット (1:5)の最高割合は1対1 100人 (1:100)である。6月に1, 2023,会社の取締役会が会社の普通株式の逆分割の実施を許可した割合は30人に1人 (1:30)である。逆株式分割の結果として30歳 (30)発効直前に発行され、発行された会社の普通株式は、それぞれの所有者が何の行動も取らずに、自動的に合併して変換する1つは (1)会社の普通株のシェア違います。逆株式分割に関連した断片的な株を発行した。そうでなければ、逆株式分割に関連する断片的な株式を取得する権利を有する株主は、株式ではなく現金支払いを得る資格があり、この合計は実質的ではない。6月に1, 2023,同社はデラウェア州国務長官にその会社の登録証明書の改訂証明書を提出し、改訂された証明書が発効した30人に1人 (1:30)会社が発行した株式と発行済み普通株の株式を逆株式分割し、6月から発効する1, 2023.
十一月に29, 2022,会社は株主特別会議を開催し,株主は改訂された会社の登録証明書を改訂する提案を採択し,逆株式分割を実施し,割合は40人のうち1人 (1:40)である。1月に13, 2023,会社の取締役会は会社の普通株式の逆分割の実施を許可した。逆株式分割の結果として40歳 (40)発効直前に発行され、発行された会社の普通株式は、それぞれの所有者が何の行動も取らずに、自動的に合併して変換する1つは (1)会社の普通株のシェア違います。逆株式分割に関連した断片的な株を発行した。そうでなければ、逆株式分割に関連する断片的な株式を取得する権利を有する株主は、株式ではなく現金支払いを得る資格があり、この合計は実質的ではない。1月に13, 2023,同社はデラウェア州国務長官にその会社の登録証明書の改訂証明書を提出し、改訂された証明書が発効した40人のうち1人 (1:40)会社が発行した株式と発行済み普通株の株式を逆株式分割し、1月から発効する13, 2023.
保留株
指定された日まで、以下の普通株式は将来の発行のために予約されます
|
2023年9月30日 |
||||
|
未償還株式オプション |
13,455 | |||
|
持分インセンティブ計画の下で付与可能な株 |
- | |||
|
未弁済持分証 |
4,068,826 | |||
| 4,082,281 | ||||
8.株式報酬
2016総合インセンティブ計画
パンバラ治療会社は2016当社取締役会は3月に“総合インセンティブ計画”(“2016計画”を採択しました20165月に株主の承認を得ました2016それは.♪the the the2016条件を満たす従業員、取締役、コンサルタントに奨励性と非法定株式オプション、制限株、株式付加価値権、業績単位、業績株、その他の株式奨励を付与することを計画している。われわれは以下の条項に基づいて普通株を購入するオプションを付与する2016付与された日標普通株を下回らない公正な市場価値で計画する。以下の条項により付与されたオプション2016計画の最長期限は10個何年もです。9月までに30, 2023,購入するオプション12,054普通株流通株2016計画と違います。株はまだ未来の奨励に使用することができる。加重平均行の重みは$1,105.88平均残存寿命は9.0何年もです。
2011年株式オプション計画
私たちの取締役会はこれ以上Panbela Treateutics,Inc.によって賞を授与しない2011株式オプション計画(“2011年計画”)株主承認の原本を受け取った後20165月の計画2016それは.“世界保健機関は2011その計画の条項によると、その計画はまだ完成されていない。9月までに30, 2023,購入するオプション171普通株の流通株はまだ発行されていない2011計画してみます。加重平均行の重みは$3,721.58平均残存寿命は1.3何年もです。
CPP’S 2010持分インセンティブ計画
当社はCPPに関するすべての余剰権利と義務を担っています2010株式インセンティブ計画(“CPP計画”)は置換オプションを発行することで行われる。9月までに30, 2023,購入するオプション1,230CPP計画によると、普通株はまだ発行されておらず、加重平均行権価格は#ドルである361.561株当たりの平均残存契約期間は6.8何年もです。
株に基づく報酬費用
一般と行政(“G&A”)研究開発(“R&D”)支出には、私たちが株式オプションを発行することによる非現金株式報酬支出が含まれている。株式に基づく報酬の条項や授与スケジュールは、贈与タイプや被贈与者の就職状況によって異なる。9月までに授与された賞30, 2023時間と性能条件に基づくベスト。約一ドルです0.2 百万9月現在、従業員、役員、コンサルタントに付与されたオプションに関する未償却株式報酬支出30, 2023.未償却費用は来年度に確認されます12何ヶ月になりますか。
各期間の株式ベースの報酬支出は以下のとおりである(千単位)
|
9月30日までの9ヶ月間 |
||||||||
|
2023 |
2022 |
|||||||
|
一般と行政 |
$ | 554 | $ | 697 | ||||
|
研究と開発 |
145 | 160 | ||||||
| $ | 699 | $ | 857 | |||||
期間中に付与、行使、取消または没収されたオプションの詳細情報9人9月末までの月30, 2023以下は以下のとおりである
|
関連株 オプション |
重みをつける 平均値 行権価格 1株当たり |
骨材 内在的価値 |
||||||||||
|
2023年1月1日の残高 |
3,287 | $ | 4,369 | $ | - | |||||||
|
授与する |
10,240 | 15 | ||||||||||
|
鍛えられた |
- | - | ||||||||||
|
キャンセルします |
(72 | ) | 1,424 | |||||||||
|
没収または満了 |
- | - | ||||||||||
|
2023年9月30日の残高 |
13,455 | $ | 1,071 | $ | - | |||||||
9月までに発行された、帰属され、帰属された株式オプションの情報30, 2023以下に示す
| 未償還、既帰属、所期帰属 | 帰属し行使可能なオプション | ||||||||||||||||||||||
|
1株当たりの権益 |
株 |
加重平均 残り 契約期限 (年) |
重みをつける 平均値 行権価格 |
オプション 練習可能である |
加重平均 残り 契約期限 (年) |
||||||||||||||||||
| $15 | 10,240 | 9.50 | $ | 15 | 1,662 | 9.50 | |||||||||||||||||
| $264 | 1,150 | 7.26 | $ | 264 | 1,150 | 7.26 | |||||||||||||||||
| $1,764 | - | $3,540 | 582 | 5.01 | $ | 3,215 | 569 | 4.94 | |||||||||||||||
| $3,810 | - | $4,908 | 498 | 5.57 | $ | 4,493 | 398 | 5.08 | |||||||||||||||
| $5,004 | - | $7,320 | 428 | 6.14 | $ | 6,102 | 428 | 6.14 | |||||||||||||||
| $9,720 | - | $18,120 | 557 | 4.81 | $ | 12,987 | 557 | 4.81 | |||||||||||||||
|
合計する |
13,455 | 8.66 | $ | 1,071 | 4,764 | 7.19 | |||||||||||||||||
中で付与されたオプションの公平市場価値の仮定9人9月末までの月期間30, 2023含まれています
|
2023 |
||||||||||
|
普通株主公正価値 |
$15.00 | |||||||||
|
無リスク金利 |
3.59% | - | 3.60% | |||||||
|
期待配当収益率 |
||||||||||
|
期待オプション寿命 |
5.08 | - | 5.50 | |||||||
|
株価の変動を予想する |
131.0% | - | 138.0% | |||||||
9.知的財産権の売却収益
七月に17, 2023,同社はエフルオロオルニチン児童神経芽細胞腫計画のある権利、所有権と利益を剥離した。契約条項によると、同社は最高約$を得る権利がある9.5 百万非希釈資金でこのような資産の売却と交換する。初期支払いは#ドルです400,000もし買収会社が取引を完成する時にすでにこのような金を受け取った場合、買収会社が臨床開発、監督管理許可及び商業販売に関連するあるマイルストーンを成功に完成すれば、残りの金を受け取ることができる。取引が完了したとき、これらのマイルストーンを成功させることは不可能であり、会社は売却の日にこれらの将来の支払いを確認しなかった。それらは達成されたか、または現金化可能な価値がないからである。この契約は顧客と締結されたものではなく、売却企業を代表するものでもないため、初期支払いは知的財産権を販売する収益として確認され、年内の他の収入に反映されることが確認された三つ9月末までの月30, 2023.
10.後続の活動
十一月に2, 2023,当社は、いくつかの既存株式証保有者(“所有者”)と株式承認証行使誘因申立書簡(“誘因関数”)を締結して普通株式株式(“既存株式承認証”)を購入することにより、所有者は現金購入と引き換えにその既存株式承認証を行使することに同意する2,130,000会社の普通株の合計,減保有権価格は$とする0.781株につき,当社が下記の既存の株式承認証とほぼ同じ条項で新株式承認証(“誘導権証”)を発行することに同意し,最も多く購入することに同意した4,260,000会社普通株式(“誘導権証株式”)および現金支払い#0.125既存株式承認証を行使する際に全額支払われた既存株式1株当たりの引受権証株式。同社が受け取った総収益は約#ドルだった1.9 百万所有者は既存の引受権証及び売却誘導権証を行使する。その会社は$を生み出した115,659取引に関連する投資銀行手数料では、特定の費用を精算することを除いている。
当社は、誘導権証の行使により発行又は発行可能な誘導権証株式の転売を含むS-3表の登録声明を提出することに同意した(“転売登録声明”)20歳 (20)入社通知書の日付のカレンダー日。株式募集書では、当社は、株主の承認を得るまで、いかなる普通株式または普通株等価物を発行しないか、または米国証券取引委員会に任意の他の登録声明(場合によっては、いくつかの例外を除く)を提出することに同意する。当社は、以下の日までにいかなる変動金利取引も行わないことに同意しました(株式募集書で定義されているように)1つは募集書が発行された日からの1年間ですが、市場で発売されているものを除き、その日以降に発効することができます6人入社通知書が発行された日から数ヶ月。
独立公認会計士事務所報告
取締役会と株主へ
パンバラ治療会社は
ミネソタ州ワコニア
財務諸表のいくつかの見方
Panbela Treateutics,Inc.およびその子会社(“当社”)の2022年12月31日と2021年12月31日までの連結貸借対照表と、この期間までの年度ごとの総合運営と全面赤字、株主(損失)権益とキャッシュフロー表、および関連付記(総称して“財務諸表”と呼ぶ)を監査しました。財務諸表は,当社の2022年12月31日と2021年12月31日までの財務状況,および同年度までの各年度の経営結果とキャッシュフローをすべての重要な面で公平に反映しており,米国公認の会計原則に適合していると考えられる。
会社の継続経営企業としての継続経営能力に大きな疑いがある
添付財務諸表の作成は、同社が引き続き経営を継続する企業であると仮定している。財務諸表付記2に記載されているように、同社には経常赤字と業務からのマイナスキャッシュフローがあり、継続経営企業としての能力が大きく疑われている。付記3は、これらの事項に関する管理職の評価および管理職の計画を説明します。財務諸表には、このような不確実性に起因する可能性のある調整は含まれていません。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査することを求められておらず、私たちも招聘されて監査を行っていません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性について意見を述べるためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項-資産購入
以下に述べる重要な監査事項は、当社監査委員会が伝達または要求を伝達する当期財務諸表監査によって生じる事項を指すものである:(I)財務諸表に対して大きな意味を有する勘定または開示に関するものであり、(Ii)特に挑戦的、主観的または複雑な判断に関するものである。重要監査事項の伝達は、財務諸表に対する私たちの全体的な意見をいかなる方法でも変えることはなく、次の重要な監査事項を伝達することによって、重要な監査事項又はそれに関連する勘定又は開示について個別の意見を提供することもない。
重要な監査事項の説明
財務諸表付記6で述べたように、2022年6月15日、会社は癌予防製薬会社(“CPP”)の買収を完了し、取引は資産購入(“取引”)と表記されている。買収した純資産は,CPP候補製品に関する買収の進行中の研究·開発(“IPR&D”)を含む公正価値で入金され,会社の総合経営報告書に研究·開発費用として入金される。
監査会社は、取引が業務合併又は資産買収の基準に適合しているか否かを評価する際に係る判断及び他の会計考慮要因のため、この取引に対する会計が複雑である。主観的考慮は、買収された総資産の公正価値の実質的にすべてが単一の識別可能資産または同様の識別可能資産のセットに集中しているかどうかであり、その中で最も重要なのは知的財産権研究開発資産である。
監査で重要な監査事項をどのように処理するか
この取引に対する会社の会計処理をテストするために、以下の監査手続きを実行しました
|
● |
ASCトピック805に定義されている関連会計基準への会社の適用を評価した企業合併. |
|
● |
私たちはどんな隠れた営業権の評価も含めて、純資産の完全性と正確性を評価した。 |
|
● |
私たちは取引で支払われた非現金対価格の公正価値を再計算し、普通株式オプションを含む。 |
|
● |
私たちはまた連結財務諸表に関連して開示された適切性を評価した。 |
2014年以来、当社の監査役を務めてきました。
/S/さくらんぼ·ベカルテ
フロリダ州タンパ市
2023年3月16日
Panbela治療会社合併貸借対照表
(単位は千で、シェアは含まれていない)
|
2022年12月31日 |
2021年12月31日 |
|||||||
|
資産 |
||||||||
|
流動資産: |
||||||||
|
現金と現金等価物 |
$ | 1,285 | $ | 11,867 | ||||
|
前払い費用と他の流動資産 |
443 | 91 | ||||||
|
課税所得税 |
49 | 321 | ||||||
|
流動資産総額 |
1,777 | 12,279 | ||||||
|
他の非流動資産 |
3,201 | 593 | ||||||
|
総資産 |
$ | 4,978 | $ | 12,872 | ||||
|
負債と株主権益 |
||||||||
|
流動負債: |
||||||||
|
売掛金 |
$ | 2,865 | $ | 640 | ||||
|
費用を計算する |
2,993 | 2,020 | ||||||
|
支払利息 |
325 | - | ||||||
|
支払手形 |
650 | - | ||||||
|
債務、流動部分 |
1,000 | - | ||||||
|
流動負債総額 |
7,833 | 2,660 | ||||||
|
債務,現在分の純額を差し引く |
5,194 | - | ||||||
|
非流動負債総額 |
5,194 | - | ||||||
|
総負債 |
13,027 | 2,660 | ||||||
|
株主(損失)権益: |
||||||||
|
優先株、額面0.001ドル;許可1000,000,000株;2022年12月31日と2021年12月31日まで、いかなる株も発行されていない |
- | - | ||||||
|
普通株、額面0.001ドル;許可100,000,000株;発行済み株と発行済み株はそれぞれ1,049,644株と335,961株であり、それぞれ2022年12月31日と2021年12月31日までである |
1 | - | ||||||
|
追加実収資本 |
82,285 | 66,240 | ||||||
|
赤字を累計する |
(91,094 | ) | (56,161 | ) | ||||
|
総合収益を累計する |
759 | 133 | ||||||
|
株主権益総額 |
(8,049 | ) | 10,212 | |||||
|
総負債と株主権益 |
$ | 4,978 | $ | 12,872 | ||||
全期間の株式と1株当たりのデータは、2023年1月13日に発効した40株の逆株分割を反映するように調整されている。
連結財務諸表付記はこれらの報告書の構成要素である。
パンバラ治療会社は
合併経営報告書と全面損失br}(千元、1株および1株当たり金額は含まれていません)
|
十二月三十一日までの年度 |
||||||||
|
2022 |
2021 |
|||||||
|
運営費用: |
||||||||
|
一般と行政 |
$ | 6,044 | $ | 4,587 | ||||
|
研究開発 |
28,049 | 5,423 | ||||||
|
営業損失 |
(34,093 | ) | (10,010 | ) | ||||
|
その他(費用)収入: |
||||||||
|
利子収入 |
14 | 1 | ||||||
|
利子支出 |
(288 | ) | (12 | ) | ||||
|
その他の費用 |
(682 | ) | (602 | ) | ||||
|
その他費用合計 |
(956 | ) | (613 | ) | ||||
|
所得税割引前損失 |
(35,049 | ) | (10,623 | ) | ||||
|
所得税割引 |
116 | 488 | ||||||
|
純損失 |
(34,933 | ) | (10,135 | ) | ||||
|
外貨換算調整 |
626 | 517 | ||||||
|
総合損失 |
$ | (34,307 | ) | $ | (9,618 | ) | ||
|
1株当たりの基本と償却純損失 |
$ | (67.91 | ) | $ | (34.64 | ) | ||
|
加重平均流通株−基本と希釈 |
514,369 | 292,607 | ||||||
全期間の株式と1株当たりのデータは、2023年1月13日に発効した40株の逆株分割を反映するように調整されている。
連結財務諸表付記はこれらの報告書の構成要素である。
パンバラ治療会社は
合併株主権益報告書(損失)
(単位は千で、シェアは含まれていない)
| 2022年12月31日まで及び2021年12月31日まで年度 | ||||||||||||||||||||||||
|
|
その他の内容 |
|
積算 |
合計する |
||||||||||||||||||||
| 普通株 | すでに納めた | 積算 | 全面的に | 株主の | ||||||||||||||||||||
|
株 |
金額 |
資本 |
赤字.赤字 |
損 |
権益(赤字) |
|||||||||||||||||||
|
2021年1月1日の残高 |
241,495 | $ | - | $ | 54,857 | $ | (46,026 | ) | $ | (384 | ) | $ | 8,447 | |||||||||||
|
公募--普通株式の発行 |
83,319 | - | 9,054 | - | - | 9,054 | ||||||||||||||||||
|
現金持分証を行使する |
5,723 | - | 1,042 | - | - | 1,042 | ||||||||||||||||||
|
株式証明書の行使は現金なし |
4,763 | - | - | - | - | - | ||||||||||||||||||
|
制限株の帰属 |
557 | - | - | - | - | - | ||||||||||||||||||
|
オプションを行使して現金を持たない |
104 | - | - | - | - | - | ||||||||||||||||||
|
株に基づく報酬 |
- | - | 1,287 | - | - | 1,287 | ||||||||||||||||||
|
純損失 |
- | - | - | (10,135 | ) | - | (10,135 | ) | ||||||||||||||||
|
外貨換算調整 |
- | - | - | - | 517 | 517 | ||||||||||||||||||
|
2021年12月31日の残高 |
335,961 | - | 66,240 | (56,161 | ) | 133 | 10,212 | |||||||||||||||||
| 普通株式·オプション·引受権証の発行-CPP買収 | 182,988 | - | 9,605 | - | - | 9,605 | ||||||||||||||||||
|
公開発行-普通株式と引受権証の発行 |
502,500 | 1 | 5,302 | - | - | 5,303 | ||||||||||||||||||
|
普通株を売る |
28,345 | - | 46 | - | - | 46 | ||||||||||||||||||
|
制限株の帰属 |
269 | - | - | - | - | - | ||||||||||||||||||
|
オプションを行使して現金を持たない |
372 | - | - | - | - | - | ||||||||||||||||||
|
現金持分証を行使する |
25 | - | 5 | - | - | 5 | ||||||||||||||||||
|
シェアに基づく報酬 |
- | - | 1,088 | - | - | 1,088 | ||||||||||||||||||
|
純損失 |
- | - | - | (34,933 | ) | - | (34,933 | ) | ||||||||||||||||
|
未発行の断片的な株式調整、株式分割 |
(816 | ) | - | (1 | ) | - | - | (1 | ) | |||||||||||||||
|
外貨換算調整 |
- | - | - | - | 626 | 626 | ||||||||||||||||||
|
2022年12月31日の残高 |
1,049,644 | $ | 1 | $ | 82,285 | $ | (91,094 | ) | $ | 759 | $ | (8,049 | ) | |||||||||||
全期間の株式と1株当たりのデータは、2023年1月13日に発効した40株の逆株分割を反映するように調整されている。
連結財務諸表付記はこれらの報告書の構成要素である。
パンバラ治療会社は
キャッシュフロー表統合レポート (千)
|
十二月三十一日までの年度 |
||||||||
|
2022 |
2021 |
|||||||
|
経営活動のキャッシュフロー: |
||||||||
|
純損失 |
$ | (34,933 | ) | $ | (10,135 | ) | ||
|
純損失と経営活動で使用される現金純額の調整: |
||||||||
|
核販売が行っている研究と開発(IPR&D) |
17,737 | - | ||||||
|
株に基づく報酬 |
1,088 | 1,287 | ||||||
|
非現金利子支出 |
273 | - | ||||||
|
経営性資産と負債変動状況: |
||||||||
|
課税所得税 |
212 | 51 | ||||||
|
前払い費用と他の流動資産 |
(127 | ) | 247 | |||||
|
臨床試験費用の保証金 |
(2,561 | ) | (540 | ) | ||||
|
売掛金 |
2,249 | 631 | ||||||
|
負債を計算すべきである |
786 | 1,214 | ||||||
|
経営活動のための現金純額 |
(15,276 | ) | (7,245 | ) | ||||
|
投資活動によるキャッシュフロー: |
||||||||
|
知的財産権研究開発への投資 |
(660 | ) | - | |||||
|
合併で得た現金 |
4 | - | ||||||
|
投資活動のための現金純額 |
(656 | ) | - | |||||
|
資金調達活動のキャッシュフロー: |
||||||||
| 普通株を売却して得られたお金は、手数料と発売コストを差し引いて44ドルです | 46 | - | ||||||
|
普通株式と引受権証を公開発行する収益は,引受業者の割引と発行コストを差し引いて727ドルである |
5,303 | - | ||||||
|
普通株式の公開発行による収益は引受業者の割引と発行コストを差し引いて946ドルです |
- | 9,054 | ||||||
|
株式承認証を行使して得られた収益 |
5 | 1,042 | ||||||
|
融資活動が提供する現金純額 |
5,354 | 10,096 | ||||||
|
為替レート変動が現金に与える影響 |
(4 | ) | (6 | ) | ||||
|
現金純変動額 |
(10,582 | ) | 2,845 | |||||
|
年初現金および現金等価物 |
11,867 | 9,022 | ||||||
|
年末現金および現金等価物 |
$ | 1,285 | $ | 11,867 | ||||
|
キャッシュフロー情報の追加開示: |
||||||||
|
利子計算期間中に支払った現金 |
$ | 15 | $ | 12 | ||||
|
非現金取引の追加開示: |
||||||||
|
資産買い入れ対価として発行される普通株·株式オプションと引受権証の公正価値 |
$ | 9,605 | $ | - | ||||
連結財務諸表付記はこれらの報告書の構成要素である。
Panbela治療会社連結財務諸表付記
|
1. |
業務.業務 |
Panbela治療会社(“Panbela”)とその直接完全子会社:癌予防製薬会社(“CPP”)とPanbela Research,Inc.(“Panbela Research”)とそのそれぞれの子会社は,いずれも全額所有であり,破壊的療法を開発し,緊急に満たされていない医療ニーズの患者の治療に用いることが主な目的である。Panbela Treateutics Pty LtdはPanbela Researchの完全子会社で、オーストラリアの法律に基づいて設立された。CPPは2つの完全子会社を持っている:イギリス実体癌予防製薬有限責任会社とアリゾナ州有限責任会社癌予防製薬有限責任会社。Panbelaとその直接的で間接的な子会社は“私たち”、“私たち”、“私たちの”、“会社”と呼ばれている
われわれのパイプラインの主な目標は,薬物療法を利用し,薬物療法を補充することで増加した疾患関連ポリアミンを減少あるいは正常化することである。私たちの主要な候補薬はエフルオロオルニチン(CPP-1 X)とシュリン酸の混合物Flynpoviであり、私たちはすでにフロリダ大学研究基金社からこの薬の世界的な独占許可を得た。私たちはアリゾナ大学アリゾナ州取締役会からFlynpoviを商業化する独占許可権を取得し、これらの権利は北米でのFlynpoviの開発と商業化の再許可協定に支配されている。
CPPを買収する
2022年6月15日に、吾らは2022年2月21日にPanbela、CPP、Panbela Research、Canary合併付属会社I,Inc.(“合併付属会社I”)およびCanary Merge付属会社II,Inc.(“合併付属会社”)が締結した合併合意および計画(“合併合意”)に基づき、先に公表されたCPPに対する戦略的業務再編および買収(“合併合意”)を完成させた。合併合意の条項によると、(I)Sub I(当時Panbelaの全資付属会社、Panbela Research自体がPanbela Researchの完全子会社)とPanbela Researchが合併してPanbela Research(“第1回合併”)に合併し、Panbela Researchは1回目の合併を保留し、(Ii)Merge Sub II(当時Panbelaの全資付属会社)はCPPと合併してCPP(“第2次合併”、1回目の合併とともに“合併”)を保留し、CPPは2回目の合併を保留した。合併の結果,Panbela ResearchとCPPはそれぞれPanbelaの完全子会社となった。また、合併の完了と関連があれば、“Panbela治療会社”だ。“Panbela Research,Inc.”と改名しましたそして“カナリア合併持株会社”です。“パンバラ治療会社”と改名しましたその他の情報については、付記6、“資産購入”を参照されたい。
株を逆分割する
Panbelaは2023年1月13日からその普通株流通株に対して40株1株の逆株分割を行った。本文に別段の規定がない限り、当社の普通株のすべての株式と1株当たりの金額は逆株式分割を反映するように遡及調整されている。詳細については、注釈9を参照されたい。
|
2. |
リスクと不確実性 |
その会社は高度な規制と競争の環境で運営されている。薬品の開発,製造とマーケティングは米国食品·薬物管理局(FDA),オーストラリア治療用品管理局,EUの欧州薬品管理局および他の国の類似機関の承認を得,これらの機関の持続的な監督を受ける必要がある。新しい薬品の承認を得ることは決して確定的ではなく、何年もかかるかもしれないし、通常は大量の支出が予想される。
2011年の設立以来、私たちは9110万ドルの損失を受けた。2022年12月31日までの1年間に3490万ドルの純損失が発生した。2022年12月31日までの年度純損失のうち、1,770万ドルの進行中の研究開発(IPR&D)がCPP買収により研究開発(R&D)費用として抹消された。この間、私たちの経営活動は約1530万ドルのマイナスキャッシュフローを生み出した。我々が開発活動を継続し,我々の鉛資産の商業化を図るに伴い,重大な損失が予想され,経営活動から負の純キャッシュフローが生じる可能性がある。2022年12月31日現在,我々の現金は130万ドル,負運営資本は600万ドル(流動資産から流動負債を差し引いた),株主赤字は800万ドルである。同社の主要な現金源には従来、株式証券の発行と転換可能な債券が含まれていた。CPPの主要な現金源は従来から株式証券の発行、転換可能な債務と発展パートナーも含まれている。
付随する総合財務諸表を作成する際には、継続的に経営している企業として、正常な業務過程で資産や清算負債を現金化することを考慮し、資産の回収可能性や分類に関するいかなる調整も含まれておらず、これらの不確実性の結果による負債額も含まれていないと仮定する。私たちが通常の業務過程で継続的に経営し、資産の帳簿価値と債務を返済する能力は、私たちが追加融資を得る能力、私たちの開発仕事の成功、アメリカ、オーストラリア、EUまたは他の市場での候補製品であるエフルオロオルニチン(CPP-1 X)とエフルオロオルニチン香包(CPP-1 X-S)が上場承認を得る能力、そして最終的に私たちの候補製品をマーケティングし、販売する能力を含む複数の要素に依存する。他の要因を除いて、これらの要因は、私たちが継続的に経営している会社として運営を続ける能力を大きく疑わせている。“流動性と商業計画”というタイトルの付記3を参照
2023年12月は世界保健機関が新型コロナウイルス株(“新冠肺炎”)が全世界大流行33ケ月に伝播することを発表した後の日である。状況はまだいくつかの不確定性があるが、著者らは依然として新冠肺炎が会社の業務、財務状況、経営業績とキャッシュフローに与える最終的な影響を予測できない。同社は2021年春にインドで製造された活性製品物質の製造遅延も経験した。米国で完成した最終製造手順にも遅延が生じており,これは新冠肺炎とある程度関係している。これまで,われわれの臨床あるいは前臨床試験の供給は中断されていない。2022年1月、同社は世界ランダム第2/第3段階臨床試験を開始することを発表し、米国、ヨーロッパ、オーストラリアで行われる予定だ。規制部門の承認やサイト開放の速度は予想より遅いが、同社は登録やデータに大きな遅延はないと予想している。設立以来、同社の行政業務は分散してきたため、同社は大流行や関連制限によって行政中断や追加コストを受けていない。
|
3. |
流動資金と管理計画 |
私たちは私たちの現在の事業計画を支援するために追加的な資金源を求める必要があるだろう。株式や債務融資など、様々な資金源を通じてより多くの資金を調達したり、戦略的協力や許可協定を通じてより多くの資金を調達したりすることができる。私たちは私たちの運営を支援するために追加の資金源を得ることができるという保証はないし、もし私たちがそのような資金を持っている場合、私たちはこれらの追加資金が私たちの需要を満たすのに十分であるか、私たちが受け入れられる条件で十分になるという保証はない。私たちの臨床データが積極的でなければ、あるいは経済や市場状況が悪化すれば、このリスクは増加するだろう。
もし私たちが必要な時に追加の融資を得ることができない場合、私たちは私たちの業務を削減する必要があり、外部の専門サービス提供者への使用を減らすこと、従業員や従業員の報酬を減らすこと、私たちの候補製品の開発を大幅に修正または延期すること、第三者が私たちの候補製品を商業化することを許可すること、癌や自己免疫疾患の治療、または私たちが本来追求または運営を停止しようとしていた応用のための行動が含まれるかもしれない。
2022年12月31日以降、同社は普通株と引受権証の登録公開を完了し、普通株株を購入し、総収益は約1500万ドルとなった。今回発行されたより多くの情報については、付記13を参照されたい。
同社は一般株を市場(ATM)施設でも販売している。2022年12月31日までの四半期に28,343株が売却され、総収益は約93,000ドルだった。2022年12月31日以降、同社はATMで普通株の追加販売を完了し、総収益は約160万ドル。今回発行されたより多くの情報については、付記13を参照されたい。
2022年10月4日、同社は登録公開発行を完了し、合計177,175株の普通株、予備融資権証を発行し、1株0.001ドルの使用価格で最大325,325株の普通株を購入し、1株12ドルの使用価格で最大753,749株の普通株を購入する権利証を発行した。これらの証券の発行価格は普通株と1.5権証1株当たり12ドル、または1株当たり事前融資の権証と1.5権証11.999ドルである。今回発行された純収益総額は約530万ドル。2022年12月31日までに,あらかじめ出資したすべての引受権証が行使され,13,000ドルの追加収益を得た。
CPP買収で買収された資産について行われている多くの開発作業は,ライセンスパートナーから資金を提供しており,付記8,“Flynpoviの開発と商業化ライセンス契約”,あるいは国立癌研究所(“NCI”)や青少年糖尿病研究基金(“JDRF”)を含む外部組織との協力を参照されたい。
2021年7月2日,同社は1株120.00ドルで83,333株の普通株の公開発行を完了し,純収益は約910万ドルであった。2021年第1四半期、会社は普通株式承認証の行使から約100万ドルの純収益を獲得した;計5598株の普通株を発行した
私たちの現金は2023年第3四半期まで続くと予想される。
私たちの将来の成功は私たちがより多くの資金を得る能力にかかっていて、私たちの開発仕事の成功、私たちはアメリカまたは他の市場のエフルスタチン(SBP-101)、エフルオロオルニチン(CPP-1 X)とエフルオロオルニチン香包(CPP-1 X-S)の上場承認を得る能力、そして私たちの候補製品を最終的にマーケティングし、販売する能力を獲得します。もし私たちが必要な時に追加の融資を得ることができなければ、もし私たちの臨床試験が成功しなければ、あるいは上場承認を得ることができなければ、私たちは経営を続けることができなくなり、私たちの会社の運営を停止し、清算することを余儀なくされるだろう。
私たちは私たちが商業的に合理的な条件で追加的な融資を受けることができるか、あるいは根本的に保証できないという保証はない。追加の持分証券や転換可能な債券を売却することは、私たちの既存株主の持分を希釈する可能性がある。
|
4. |
重要会計政策の概要 |
陳述の基礎
当社はすでに米国公認会計原則(“公認会計原則”)に従って付随する総合財務諸表を作成している。私たちの財政年度は12月31日に終わるだろう。
合併原則
添付されている連結財務諸表には、Panbela治療会社とその直接および間接子会社の資産、負債、費用が含まれている。すべての重大な会社間取引と残高は合併で販売された。
予算の使用
連結財務諸表の作成は、連結財務諸表の日に報告された資産及び負債額、又は有資産及び負債の開示及び報告期間内に報告された収入及び支出に影響を与えるために、管理層に推定及び仮定を要求する。実際の結果はこれらの推定とは異なる可能性がある。
企業合併と資産買収
私たちは買収会計方法を用いて買収された企業に対して会計計算を行い、この方法の要求は、買収が企業合併の定義に符合すれば、買収した資産と負担した負債は買収の日にそれぞれの公正価値で入金しなければならない。買収が企業合併の定義に適合していない場合は、資産買収に計上し、購入対価格を買収した資産に割り当てる。
ASC 805は、企業合併、買収が企業合併を代表するか否かを判定するモデルを提供する。一企業になるためには、買収された実体の一連の活動は投入と実質的な過程が必要であり、これらの投入と実質的な過程は共に創造産出を大きく促進する能力を有する。買収されたエンティティはまた、買収された総資産の公正価値に基づいて“実質的にすべて”が単一資産または1組の類似資産に集中しているかどうかに基づいて、買収が実質資産買収を代表するかどうかを決定することに関連する“スクリーニングテスト”に合格しなければならない。この評価は、現金、繰延税項目、および繰延税項目に関連する営業権のようないくつかの取得された資産を含まないが、確認された資産以外の任意の移転対価格を含む他のすべての総資産を含む。
信用リスクが集中する
会社を深刻な集中信用リスクに直面させる可能性のある金融商品は主に現金です。現金は商業銀行の普通口座に入れます。時々、このような預金は保険限度額を超える可能性がある。2022年12月31日現在、会社の現金のうち100万ドルが保険限度額を超えている。同社の現金預金には何の損失もありません。
現金と現金等価物
現金等価物には、期限が3ヶ月を超えない短期·高流動性投資が含まれる。
他の非流動資産
その他の非流動資産は主に契約研究機関(“CRO”)の長期預金からなる。これらの金額は試験終了時に運営費や研究開発費であることが確認された。
研究開発コスト
研究開発コストには、人体臨床試験を行う際に生じる費用が含まれており、第三者サービスプロバイダが様々な試験を実行し、当社の臨床前研究に関連するデータを蓄積する費用は、当社の臨床前研究および人体臨床試験に使用される十分な数のSBP-101、FlynpoviおよびCPP-1 X化合物を生産するのに十分な数のSBP-101、FlynpoviおよびCPP-1 X化合物を生産するのに必要な費用を提供するための研究プロトコルであり、私たちの候補製品開発計画の実行に関連する専門知識のコンサルティングリソーススタッフコストを有しており、給与、福祉および株式ベースの給与および私たちの許可された知的財産権の許可および維持のコストを含む。
臨床試験費用を含めて研究開発費を徴収し、発生時に費用を計上します。われわれのヒト臨床試験は臨床試験現場で行われ,われわれがCROの協力の下で共同管理した。臨床試験地点を確立する費用は研究協定調印後に計算しなければならない。臨床試験業績に関連する費用は通常契約金額と合意マイルストーンの実現によって累積し、例えばサイト開放、患者登録、患者フォローアップなどである。著者らは臨床試験サイトとCROとのコミュニケーションを通じて、患者登録とその他の活動の程度を含む各重要な契約下の業績レベルを監視し、そして必要に応じて四半期ごとに推定数を調整し、臨床費用に各臨床試験サイトと各CROの実際の仕事支出を反映させる。
研究と発展コストも知的財産権の研究と発展を含む。この資産はCPPの証券所有者から購入し、資産を買収した後すぐに研究と発展のためにログアウトする。
ライセンス制約された知的財産権に将来他の用途がないと判断された場合、特許技術許可の取得に関連するコストを支払う。
臨床試験は費用を計算しなければならない
臨床前研究と臨床試験活動のコストはサプライヤーが特定の任務を達成する上での進展を評価した上で確認したものであり、使用するデータは臨床サイトの活性化、患者登録或いはサプライヤーが会社に提供するその実際のコストに関する情報を含む。これらの活動の支払いは個別契約の条項に基づいており,支払い時間はサービスを提供する期間と大きく異なる可能性がある.当社は、進捗又は完了状態又は完了したサービスに関する適用者及び外部サービスプロバイダの報告及び検討により、計算すべき推定を決定する。各貸借対照表日までの課税費用の推定は、当時知られていた事実と状況に基づいている。
株に基づく報酬
株式インセンティブ奨励の会計計算において、私たちは付与日とその等の奨励の公正価値に基づいて、従業員と非従業員サービスのコストを計量し、確認し、株式ツールの奨励と交換する。補償コストは帰属期間内に直線分配法を用いて比例的に確認され,帰属期間は必要なサービス期間と考えられる。私たちは発生した時間帯に記録を没収します。業績に基づく株式オプション奨励の補償費用は、“業績”が発生した場合または発生する可能性がある場合に確認する。
株式奨励の公正価値は付与された日にブラック·スコルスオプション定価モデルを用いて推定される。株式報酬の公正価値の決定は、私たちの株価といくつかの複雑かつ主観変数に関する仮定の影響を受ける。無リスク金利は米国債金利をもとに、個々の奨励の期待期限に適用される。予想変動率は主に会社普通株の変動率に基づいている。予想される未来には何の配当も発表されないと予想されるので、配当収益率をゼロとする。付与されたオプションの予想期限は、“簡略化”方法を使用して決定される。この方法によれば、予想期限は、平均帰属日と契約期限終了との間の中間点として推定される。
制限株式単位の公正価値は、付与された日に標的普通株の公正価値に基づいて計算される。
所得税
所得税は貸借対照法で計算される。繰延税項資産及び負債は、総合財務諸表中の既存資産及び負債の帳簿価額とそのそれぞれの課税基礎との間の差額及び営業損失及び税項相殺繰越間の差額に基づいて確認された将来の税項結果を推定する。繰延税金資産と負債は、当社が経営している管轄区ごとの法定税率で計量され、これらの仮差額の回収または決済が予想される年度の課税所得額に適用される予定です。税率変動が繰延税金資産や負債に及ぼす影響は、公布日を含む期間の経営で確認されている。繰延税金資産を最も顕在化する可能性のある額に減らすために、必要に応じて推定準備を設ける。当社は2022年12月31日および2021年12月31日までの繰延税項目総資産について全額推定を提供する準備をしています。当社の政策は、所得税に関する利息と罰金を総合経営報告書と全面赤字報告書の所得税費用に分類することです。
外貨換算
Panbera Treateutics Pty Ltdの機能通貨はオーストラリアドル(“AUD”)である.したがって,Panbela Treateutics Pty Ltdの資産と負債および株式取引は期末レートでドルに換算される.料金はその期間有効な平均為替レートに換算します。これにより発生した換算損益は,総合経営報告書と全面赤字報告書に累積総合損益の構成要素として入金される。2022年12月31日と2021年12月31日までの年間では、累積された他の総合収益から業務のどの再分類調整までも無関係である。
当社は当日有効為替レートで外貨建ての取引を記録しています。取引日と決算日の間の変動は取引損益であることが確認された。
1株当たり純損失
1株当たり純損失を計算する方法は,当期に発行された普通株の加重平均(分母)を純損失(分子)で割ることである。この間に発行された株式とその間に再買収された株式(あれば)は,その流通期間の部分に応じて重み付けを行う.希釈1株当たり収益(EPS)の計算は基本1株当たり収益の計算と類似しており、希釈性潜在普通株が発行された場合、発行される追加普通株の数を含むように分母が増加しているだけである。私たちの希釈1株当たり収益は基本的に1株当たり収益と同じである。普通株の同値株は計算に含まれていないので、それらの影響は逆希釈されているからである。
以下に発行された潜在的普通株は、それらの影響が希釈されていないので、希釈後の1株当たり純損失の計算には含まれない
|
十二月三十一日 |
||||||||
|
2022 |
2021 |
|||||||
|
従業員と非従業員株式オプション |
100,556 | 61,582 | ||||||
|
制限株式単位 |
- | 269 | ||||||
|
普通株引受権証により発行可能な普通株 |
889,910 | 127,710 | ||||||
| 990,466 | 189,561 | |||||||
最近採用された会計公告
2020年8月、アメリカ財務会計基準委員会はアメリカ会計基準委員会第2020-06号“債務と転換及びその他のオプション”(サブ題470-20)と“実体自己権益派生ツールと対沖契約”(サブ題815-40):“実体自己権益中の転換可能ツールと契約の会計処理”を発表し、現行のアメリカ公認会計基準に要求されている主要な分離モードを廃止することによって、転換可能ツールの会計処理を簡略化した。ASUは株式契約を取り消し、派生商品範囲の例外に必要なある決済条件を満たし、ある領域で1株当たりの収益を希釈する計算を簡略化した。本会計基準は、2021年12月15日以降に開始される年次報告期間に適用され、これらの会計年度内の移行期間を含む。当社は2022年12月31日までの年度にASUを採用しています。この更新は、修正された遡及または完全に追跡された移行方法を使用することを可能にする。当社は、今回のASUの影響がその連結財務諸表に実質的な影響を与えないことを決定しました。
|
5. |
費用を計算する |
計算すべき費用には、以下の項目が含まれる
|
十二月三十一日 |
||||||||
|
2022 |
2021 |
|||||||
|
臨床試験および関連費用 |
$ | 1,760 | $ | 992 | ||||
|
奨励的報酬 |
550 | 434 | ||||||
|
解散費と関連賃金税 |
448 | - | ||||||
|
専門サービス |
147 | 235 | ||||||
|
他にも |
88 | 359 | ||||||
|
負債総額を計算すべきである |
$ | 2,993 | $ | 2,020 | ||||
|
6. |
資産買い入れ |
2022年6月15日、会社は合併により、これまで発表されていたCPPの戦略的業務再編と買収を完了した。合併合意の条項によると、合併直前にCPPが発行した持株の保有者は、合併完了時にPanbelaの普通株式を取得する。Panbela Researchの株主は合併後のホールディングスPanbelaの大部分の流通株を保持している。CPP株主は、マイルストーン社から合計6000万ドルの支払いまたは支払い、および主要資産エフロンの潜在的承認および商業化に関連する特許権使用料支払いを受ける資格があるだろう。
私たちは、合併で買収された総資産の公正価値が、単一の識別可能資産または同様の識別可能資産のセットに実質的にすべて集中しているかどうかを決定するために“スクリーニングテスト”を行った。CPPの主要資産は、Flynpovi(CPP-1 X)とスリン酸)、エフルオロオルニチン(CPP-1 X)およびエフルオロオルニチン香包(CPP-1 X-S)を含む3つの形態のエフルオロオルニチンであり、知的財産権および開発からなる単一識別可能資産として確認されている。
資産を買収する契約の対価格にはいくつかの対価格が含まれており、これらの対価格は購入日に発生する可能性もなく、合理的に見積もることもできない。したがって,このまたは対価のある価値は以下の購入価格の割当てから除外されている.買収に関連する取引コストはすでに知的財産権研究開発への追加投資として入金されている。
以下は、CPP資産買収に関する購入対価格とその購入対価格の割り当て概要である
|
株 |
価値(千) |
|||||||
|
CPP株主に普通株式を発行する |
182,978 | $ | 7,839 | |||||
|
普通株式入札オプション継続 |
39,904 | 1,637 | ||||||
|
普通株関連株式承認証が置換されている |
8,452 | 129 | ||||||
|
現金ではありません |
$ | 9,605 | ||||||
|
発生する取引コスト |
$ | 658 | ||||||
|
総掛け値 |
$ | 10,263 | ||||||
|
プロセス研究と開発で* |
$ | 17,737 | ||
|
現金 |
4 | |||
|
その他流動資産 |
230 | |||
|
売掛金と売掛金 |
(811 | ) | ||
|
利子と支払手形 |
(6,897 | ) | ||
| $ | 10,263 |
*FASB ASCトピック730によれば、この資産は、統合完了直後に支出されます。
|
7. |
支払手形 |
スキャンブ本券
CPPは2022年12月31日現在,2022年6月15日の改訂·重記された元票(“Sucampo手形”)の項での未償還元金と利息は約640万ドル,初期元金金額は約620万ドル,受取人はSucampo GmbHである。Sucampo手形の場合の未償還元本残高は年利5%で利息となります。すべての未払い元金は、当時のいかなる未払いおよび累算利息とともに、以下のように支払わなければならない:(I)100万元、別途2023年1月31日、2024年1月31日、2025年1月31日および2026年1月31日またはそれまでの累算だが利息を払わない;および(Ii)すべての残り元金は2027年1月31日または以前に累算すべきだが未払いの利息を加算する。CPPまたはその親会社Panbelaが2023年1月31日までに任意の債券または株式の発行または発売から現金収益を得る場合、CPPは、そのような現金収益から同時に前金を強制することを要求され、金額は、(I)100万ドルと支払日までのSucampo手形のすべての未払い利息と、(Ii)当該現金収益の10%に相当する。CPPが2023年1月31日に支払うべき金額は、ドル対ドルに基づいて、このような前払い金額を差し引く。同社は2022年10月4日に株式募集を完了し、その後、2022年11月4日までに満期となる600万ドルの総収益の10%または60万ドルを支払う義務が生じた。当社は2022年12月31日現在この金を支払っていないため、支払日に違約し、2022年11月4日に5%の追加懲罰利息を発生させた。2023年2月1日、Panbelaは100万ドルの満期残高を支払い、約29.5万ドルの応算と未払い利息を支払った。この支払いの結果として、同社は現在、すべての満期支払いの最新の支払いであり、契約を違約しなくなった。2022年12月31日現在、この手形の課税利息と未払い利息は約243,000ドルである。Panbelaは2022年6月15日の日付の保証に基づいて、Sucampo手形でのCPPの支払い義務を保証することに同意した。
普通寝台の切符
CPPは2022年12月31日現在、Tillotts Pharma AGを受益者とする改訂された元票(“Tillotts手形”)の項目での未返済残高は約70万ドル、初期元本は約65万ドルである。Tillotts手形未返済の元金残高により年利4%で単利を計上する。2022年12月31日までの未払い利息は約82,000ドルである。改訂されたTillotts手形下のすべての未返済金額は、2023年1月31日に満期になり、全額支払いされます。
|
8. |
Flynpoviの開発と商業化許可プロトコル |
CPPはOne-Two Treateutics Assets Limited(“One-Two”)と締結された許可プロトコルの一方であり,合意日は2021年7月16日である.合意によると、One-TwoはCPP社が北米でFlynpoviを開発し商業化することを許可した。この協定はまた、CPPが規制部門がFlynpoviを承認した後に記念碑的な支払いと、Flynpoviの許可地域での純売上の特許権使用料を取得することを要求する。マイルストーン支払いおよび純販売特許権使用料は、1-2がFDA承認を確保するために必要な任意の開発活動に関連する直接従業員、臨床、および規制コストのために、ドル対ドルに基づいて1-2の金額を減少させなければならない。同社は北米でのFlynpoviの開発や規制承認に関するいかなる費用も負担していない。
|
9. |
引受金とその他の事項 |
フロリダ大学研究財団と許可合意に達しました
2011年12月22日、フロリダ大学研究財団(UFRF)と独占ライセンス契約を締結した。本ライセンス協定は、2016年12月12日に改正(“第1修正案”)され、2019年10月3日に再改正される(“第2改正案”)。ライセンス契約は,ライセンス技術開発のライセンス製品純売上高の2.5%から5%までの特許権使用料をUFRFに支払うことを求めている。“第2修正案”は、すべての最低年間特許権使用料を廃止し、特許権使用料の支払期限を、(1)特許製品の初商業販売日から10年、または(2)各国規制排出期間が満了したときのうちの1つに修正する。元の合意で想定されていたすべての未来のマイルストーン支払いは第2の修正案でキャンセルされた。
修正された許可協定は相変わらず慣行と慣用的な終了条項によって制限されている。その会社はまた年間10,000ドルのライセンス維持費を支払わなければならない。
アリゾナ大学とのライセンス契約は
CPPはアリゾナ大学(“大学”)アリゾナ州取締役会と署名したライセンス契約の一方である。機関間合意によると、カリフォルニア大学取締役会は、カリフォルニア大学オーウェン校を代表して、いくつかの特許、臨時特許、臨床試験データ、および化学的癌予防、ポリープ予防および他の技術に関連する他の知的財産権をCPPに許可することに同意した。同大学は,同大学がカリフォルニア大学オーウェン校と共同で保有する特許権を管理する権利がある。このライセンス契約は、CPPに知的財産権ベースの製品を独占的に商業化する権利を与える。知的財産権の交換として,CPPは大学に一定の費用と特許費用の補償を支払い,CPP株式を買収する引受権証を大学に付与した。合併の結果、株式承認証は株式承認証に代わり、1株11.20ドルで2,772株のPanbela普通株を購入した。
CPPはまた、いくつかの研究、開発、および規制のマイルストーンを達成する際に、合計90,000ドルまでの追加マイルストーン支払いを大学に支払うことに同意した。未来のマイルストーン支払いは掛け値があるかどうかと考えられ、支払う可能性がある時に計算されるだろう。2022年12月31日現在、記念碑的な支払いは何も可能ではない。
|
10. |
株主権益 |
普通株式および株式承認証を公開発売する
2022年10月4日、同社は登録公開発行を完了し、合計177,175株の普通株、予備融資権証を発行し、1株0.001ドルの使用価格で最大325,325株の普通株を購入し、1株12ドルの使用価格で最大753,749株の普通株を購入する権利証を発行した。これらの証券の発行価格は普通株と1.5権証1株当たり12ドル、または1株当たり事前融資の権証と1.5権証11.999ドルである。今回発行された純収益総額は約530万ドル。2022年12月31日現在、すべての事前出資の引受権証が行使されている。これらの証券は,S-1表中の有効登録宣言に基づいて発行される.
市場で計画する
2022年7月19日、Panbela治療会社(“会社”)は、Roth Capital Partners,LLC(“代理店”)と販売契約を締結し、総販売総価格が8,400,000ドルに達する会社普通株を時々“市場”株式発行計画(“ATM計画”)で売却する。
2022年12月31日までの1年の最後の月に、同社はATM機で28,343株の普通株を発売販売し、約93,000ドルの毛収入を生み出した。会社は約44,000ドルの融資コストを発生させ、これらのコストは2022年12月に会社がATMに基づいて株を発売する際に追加実収資本を計上し始めた。ATM計画によると、同社がロスに支払う手数料は、ATM計画に基づいて普通株を売却する総毛収入の3.0%に相当する。2022年12月31日までの1年間の純収益は約4.6万ドル。
普通株を公開発行する
2021年7月2日、同社は83,333株の普通株の貸切公開を完了し、購入価格は1株120.00ドルであった。今回発行された総収益は約1000万ドル。引受業者の割引や他の発行コストを差し引いた純収益は約910万ドル。これらの証券は,S-3表中の有効登録声明に基づいて発行される.
株式承認証を行使して普通株を購入する
同社は2021年12月31日までの1年間に25株を発行し、1株181.60株の価格で引受権証を行使し、合計約4,500ドルの収益を得た。
同社は2021年12月31日までの1年間に5723株の普通株を発行し、総収益は約100万ドルであり、発行された株式権証を行使した結果となった。大部分はすでに株式承認証を行使し、計5,598部で、1株当たり181.60ドルで行使され、普通株式はS-1表の有効登録によって発行される。
同時期に同社が4,762株の普通株を増発したのは,発行された株式証を発行した純無現金承認株証を行使して13,403株を購入したためである。2021年12月31日までの年度内に、14,333株の普通株の引受権証が満期になり、すべての引受権証の行使価格は1株5.00ドルとなった。また、2021年12月31日までの1年間に、2714株の普通株を15.00ドルで購入した株式引受証が満期になった。
株を逆分割する
2022年11月29日、当社は株主特別総会を開催し、株主は自社の登録証明書を改訂し、40:1(1:40)の割合で逆株式分割を実施する提案を採択した。2023年1月13日、会社取締役会は会社の普通株式の逆分割の実施を許可した。逆株式分割の結果、発効直前に発行·発行された会社普通株のうち、40(40)株毎に自動的に合併されて1(1)株会社普通株に変換され、それぞれの持株者がいかなる行動も必要としない。逆株式分割に関連する断片的な株式は発行されていない。そうでなければ、逆株式分割に関連する断片的な株式を取得する権利を有する株主は、株式ではなく現金支払いを得る資格があり、この合計は実質的ではない。2023年1月13日、会社はデラウェア州国務長官に改正された会社登録証明書修正書を提出し、2023年1月13日から会社が発行した普通株と発行された普通株のうちの1つ(1:40)の逆株式分割を行った。2023年1月13日金曜日の寄り付き時、同社の普通株は逆分割調整に基づいて取引を開始した。
保留株
|
2022年12月31日現在、将来の発行に保留されている普通株は以下の通り |
||||
|
未償還株式オプション |
100,556 | |||
|
持分インセンティブ計画の下で付与可能な株 |
50,511 | |||
|
発行済み普通株引受権証により発行可能な普通株 |
889,910 | |||
| 1,040,977 | ||||
|
11. |
株に基づく報酬 |
2016総合インセンティブ計画
2020年4月9日に施行された2016年総合インセンティブ計画(“2016計画”)は、我々の取締役会の承認を得て、株主の承認を得ています。2016年計画では、条件を満たす従業員、取締役、コンサルタントに奨励性および非法定株式オプション、制限株式、株式付加価値権、業績単位、業績株、およびその他の株式奨励を付与することが可能になります。我々は2016年計画に基づいて普通株を購入するオプションを付与し、行使価格は付与された日に関する普通株の公平な市場価値を下回らない。2016年計画に基づいて付与されたオプションの最長期限は10年。2016年の計画によると、106,825株の普通株が発行に保留されている。2022年12月31日現在、2016年計画によると、55,495株の普通株を購入したオプションを発行しており、加重平均行権価格は1株241.65ドル、平均残存契約期間は約6.5年である。同じ日に、まだ50,511株の株が未来の奨励を受けることができる。
2011年株式オプション計画
2016年計画が承認される前に、2011年株式オプション計画(“2011年計画”)に基づいて株式奨励が付与された。株主が2016年計画を承認すると同時に、取締役会は2011年計画の条項に基づいて、2011年計画の下で支払われていない賠償金は支払われていないにもかかわらず、2011年計画を終了した。2011年計画により付与されたオプションの最長期限は10年であり、従業員のオプションは一般にゼロから2年以上である。2022年12月31日現在、2011年計画によると、5,600株の普通株を購入したオプションはまだ返済されておらず、加重平均行権価格は1株118.92ドル、平均残存契約期間は約2.0年である。
CPP’S 2010持分インセンティブ計画
合併の結果,当社は代替オプションの発行によりCPPの二零一零年持分インセンティブ計画(“CPP計画”)に関するすべての余剰権利と義務を担った。2022年12月31日まで、CPP計画によると、39,461株の普通株を購入したオプションはまだ返済されておらず、加重平均行権価格は1株14.013ドル、平均残存契約期間は7.2年である。
私たちはブラック·スコアーズオプション推定モデルによって推定された各報酬の公正価値に基づいて株に基づく報酬を確認した。結局、帰属中に確認された実際の費用は、実際に帰属した株のみに向けられるだろう。
オプション活動の概要は以下のとおりである
|
関連株 オプション |
重みをつける 平均値 行権価格 1株当たり |
骨材 内在的価値 |
||||||||||
|
2021年1月1日の残高 |
53,438 | $ | 270.92 | $ | 388,461 | |||||||
|
授与する |
13,851 | 154.55 | ||||||||||
|
鍛えられた |
(197 | ) | 35.20 | |||||||||
|
キャンセルします |
- | - | ||||||||||
|
没収する |
(5,510 | ) | 410.75 | |||||||||
|
2021年12月31日の残高 |
61,582 | $ | 233.00 | $ | 8,821 | |||||||
|
CPP計画のオプション継続 |
39,904 | 14.00 | ||||||||||
|
鍛えられた |
(443 | ) | 8.80 | |||||||||
|
キャンセルします |
- | - | ||||||||||
|
没収する |
(487 | ) | 562.97 | |||||||||
|
2022年12月31日の残高 |
100,556 | $ | 145.50 | $ | - | |||||||
各期間の株式ベースの報酬支出は以下のとおりである(千単位)
| 十二月三十一日までの年度 | ||||||||
|
2022 |
2021 |
|||||||
| 一般と行政 | $ | 889 | $ | 1,083 | ||||
|
研究開発 |
199 | 204 | ||||||
|
株に基づく報酬総額 |
$ | 1,088 | $ | 1,287 | ||||
2022年12月31日までの2年間、当社の未帰属株式状況の概要は以下の通りです
|
以下の株 選択権 |
加重平均 付与日公正価値 |
|||||||
|
2021年1月1日に帰属していない |
12,653 | $ | 166.80 | |||||
|
授与する |
13,851 | 118.93 | ||||||
|
既得 |
(7,153 | ) | 144.41 | |||||
|
没収する |
(1,250 | ) | 119 | |||||
|
2021年12月31日に帰属していません |
18,101 | $ | 137.41 | |||||
|
授与する |
- | - | ||||||
|
既得 |
(8,184 | ) | 135.03 | |||||
|
没収する |
(487 | ) | 562.93 | |||||
|
2022年12月31日に帰属していない |
9,430 | $ | 139.04 | |||||
2022年12月31日現在、発行され、帰属され、帰属されると予想される株式オプション情報は以下の通りである
|
未償還、既帰属、所期帰属 |
帰属し行使可能なオプション |
||||||||||||||||||||||
|
1株当たりの権益 |
株 |
重みをつける 平均値 残り 契約書 寿命(年) |
重みをつける 平均値 行権価格 |
オプション 練習可能である |
重みをつける 平均値 残り 契約期限 (年) |
||||||||||||||||||
| $8.80 |
- |
$58.80 | 39,810 | 7.12 | $ | 14.31 | 39,810 | 7.12 | |||||||||||||||
| $90.40 |
- |
$91 | 1,578 | 8.02 | $ | 90.46 | 625 | 6.99 | |||||||||||||||
| $100 | - | 166.80 | 30,595 | 6.38 | $ | 135.94 | 24,383 | 5.90 | |||||||||||||||
| $180 |
- |
$244 | 11,676 | 5.94 | $ | 208.29 | 10,738 | 5.81 | |||||||||||||||
| $324 |
- |
$404 | 12,051 | 6.19 | $ | 365.14 | 10,724 | 6.02 | |||||||||||||||
| $604 | 4,846 | 3.95 | $ | 604.00 | 4,846 | 3.95 | |||||||||||||||||
|
合計する |
100,556 | 6.62 | $ | 145.50 | 91,126 | 6.34 | |||||||||||||||||
2022年12月31日現在、未帰属従業員の株式オプションに関する報酬支出総額は80万ドルで、1.1年の加重平均期間に支出に割り当てられる見通しだ。
2022年12月31日までの年間では、新たなオプションは付与されていない。2022年6月15日、同社は、CPPを買収する購入コストの一部として、1株当たり14.00ドルの加重平均行権価格で最大39,904株の普通株を完全に既得の代替オプションを提供した。
非従業員株報酬
会計基準更新(“ASU”)2019-07“報酬-株式報酬(テーマ718)”に基づいて、非従業員に付与された株式オプションを計算します。非従業員に付与された株式オプションについて、私たちは2022年と2021年にそれぞれ281,000ドルと222,000ドルの非従業員株報酬を記録した。
制限株式単位
最近完成した財政年度の制限性非既存普通株の数量と加重平均付与日の公正価値は以下の通りである
|
量 販売制限株 |
加重平均 贈与日交易会 価値がある |
|||||||
|
2021年1月1日制限非帰属 |
287 | $ | 138.00 | |||||
|
2021年に承認されました |
539 | 166.80 | ||||||
|
2021年に帰属する |
(557 | ) | 152.00 | |||||
|
2021年12月31日までの制限された非帰属 |
269 | $ | 138.00 | |||||
|
2022年に承認されました |
- | - | ||||||
|
2022年に帰属する |
(269 | ) | 67.60 | |||||
|
2022年12月31日までの制限された非帰属 |
- | $ | - | |||||
2022年12月31日現在、未帰属制限株式単位に関する補償費用は確認されていない。
株式報酬支出には、2022年12月31日と2021年12月31日までの年間限定株式単位に関する支出が約43,000ドル、106,000ドルが含まれている。
|
12. |
所得税 |
設立以来、私たちは純運営損失が発生した。私たちは添付の財務諸表に純営業損失の利益を反映しておらず、私たちの繰延税金資産について全額推定値を設定しています。
2022年12月31日と2021年12月31日に、会社の課税所得税はそれぞれ約49,000ドルおよび321,000ドルであり、その中には、我々の子会社Panbela Treeutics Pty Ltd.の研究および開発活動に関連する払戻可能な税金インセンティブが含まれている。
繰延所得税は、財務報告目的のための資産および負債の帳簿価値と所得税目的のための金額および営業損失と税収控除の繰越との一時的な違いによる純税収の影響を反映している。
私たちの繰延税金資産と負債の重要な構成要素は以下の通りです(千で計算)
|
2022 |
||||
|
税金: |
||||
|
現在のところ |
(116 | ) | ||
|
延期する |
- | |||
|
非電流 |
- | |||
| (116 | ) | |||
| 十二月三十一日 | ||||||||
|
繰延税金資産(負債) |
2022 |
2021 |
||||||
|
純営業損失が繰り越す |
$ | 17,564 | $ | 9,986 | ||||
|
研究単位繰り越し |
338 | 235 | ||||||
|
株に基づく報酬 |
1,928 | 1,734 | ||||||
|
第174節償却 |
1,921 | - | ||||||
|
他にも |
68 | 66 | ||||||
|
繰延税金資産 |
21,819 | 12,021 | ||||||
|
推定免税額 |
(21,819 | ) | (12,021 | ) | ||||
|
繰延税項目純資産 |
$ | - | $ | - | ||||
将来の税金優遇の実現は、私たちが繰越期間内に十分な課税収入を生み出す能力があるかどうかにかかっている。私たちの経営赤字の歴史のため、管理層は上述した未来の税金優遇による繰延税金資産は現在実現不可能であると考えているため、私たちはすでに全額評価を提供しています。
法定税率と実際の税率の入金は以下の通り
|
十二月三十一日までの年度 |
||||||||
|
2022 |
2021 |
|||||||
|
法定料率 |
21.0 | % | 21.0 | % | ||||
|
恒久的差異 |
(10.8 | ) | 2.1 | |||||
|
実際の税率変動 |
0.0 | (0.4 | ) | |||||
|
州政府 |
0.1 | - | ||||||
|
推定免税額 |
(13.0 | ) | (24.3 | ) | ||||
|
海外研究激励措置 |
0.1 | 3.1 | ||||||
|
他にも |
2.9 | 1.6 | ||||||
|
有効率 |
0.3 | % | 3.1 | % | ||||
2022年12月31日までの純営業損失と税収控除は以下の通り
|
(単位:千) |
金額 |
期限が切れる年限 |
|||
|
純営業損失-連邦 |
23,068 |
2031年から満期になる |
|||
|
2018年から2022年までの純営業損失-連邦 |
23,977 |
いつまでも期限が切れない |
|||
|
税金控除-連邦 |
338 |
2041年から |
|||
IRCが提供する所有権変更制限や類似した国の規定により,繰越や貸記に対する純営業損失の利用はかなりの年次制限を受ける可能性がある。私たちはまだIRC第382条の下で所有権変更が発生したかどうかを決定するために詳細な分析を行っていない。所有権変更の影響は,変更前期間に起因する純営業損失繰越の使用に年度制限を加えることである。
その会社はアメリカとオーストラリアで税金を払わなければなりません。2017年12月31日までの年度及び以降の納税申告書は連邦と州税務機関の審査を受けなければならない。Panbela Treateutics Pty Ltdは2015年12月31日以降の年度の納税申告書はオーストラリア税務当局の審査を経なければならない。
|
13. |
後続事件 |
2023年1月の間、会社はATM計画に従って440,830株の普通株を売却した。純収益は約160万ドル。
2023年1月30日、同社は登録公開を完了し、合計4,842,224株の普通株を発行し、1株0.001ドルの使用価格で最大1,832,776株の普通株を購入する予備融資権証と、1株2.75ドルの使用価格で最大13,350,000株の普通株を購入する承認証を発行した。これらの証券の発行価格は、普通株式と2つの株式承認証1株当たり2.25ドル、または各事前融資の引受証と2つの株式承認証2.249ドルである。今回発行された純収益総額は約1370万ドル。2023年2月2日現在、すべての事前出資の引受権証が行使されている。これらの証券は,S-1表中の有効登録宣言に基づいて発行される.
最大800万株普通株 最大8,000,000株E類普通株式承認株式証購入最大2,660,000株普通株 最大8,000,000株F類普通株式承認証購入最大14,000,000株普通株 最大800万株の普通株を購入する事前資金承認証 最大24,660,000株の普通株式関連株式承認証
パンバラ治療会社は
目論見書
ロス·キャピタル共同会社
, 2024
第II部
目論見書不要の資料
|
十三項。 |
発行、発行の他の費用。 |
Panbela治療会社(“当社”)が登録されている普通株の発行·販売に関する対応コストと支出を表に示す。証券取引委員会(“委員会”)登録料を除くすべての表示金額は見積数である。
|
アメリカ証券取引委員会登録料 |
$ |
8,856 | ||
|
FINRA届出費用 |
$ |
25,000 | ||
|
会計費用と費用 |
$ |
40,000 |
||
|
弁護士費と支出 |
$ |
200,000 |
||
|
譲渡代理費、登録費、授権証代理費 |
$ |
20,000 |
||
|
印刷費 |
$ |
10,000 |
||
|
雑類 |
$ |
11,144 |
||
|
合計する |
$ |
315,000 |
|
14項です。 |
役員と上級者への賠償です。 |
デラウェア州一般会社法第145条によると、任意の法律団は、その訴訟によって実際的かつ合理的に招いた支出(弁護士費を含む)、判決、罰金、和解のために支払われた当該人の支出に対抗するために、当該人が実際および合理的に招いた支出(弁護士費を含む)、判決、罰金、和解のために支払われたお金に対抗するために、または法律団の要求を提供すべきであると規定されているが、その人は誠実に行動し、その団体の最適な利益に適合しているか、または反対しない方法で行動しなければならない。その行為が違法であると信じる合理的な理由はないが、その人がその法律団によって提起されたか、またはその法団に基づいて提起された訴訟であれば、その人がその法団に法的責任があると判決されたいかなる申立についても弁済することはできない。
会社の会社登録証明書および改正と再記述の定款はデラウェア州法律で許可されている最大の程度でその役員の責任を制限している。デラウェア州の法律では、会社役員はその役員の受託責任に違反した金銭損害に対して個人責任を負わないと規定されているが、以下の責任を除く
|
● |
会社や株主への忠誠義務に違反する |
|
● |
善意でない行為やしないこと、または故意の不正行為に関連しているか、または違法であることを知っている |
|
● |
デラウェア州会社法第174条の規定により、配当金を不正に支払ったり株式を償還したりすること |
|
● |
取締役はそこから不当な個人的利益を得る取引をしています。 |
これらの責任制限は連邦証券法による責任には適用されず、禁止救済や撤回など、獲得可能な衡平法の救済措置にも影響を与えない。会社の改正と再記述の定款規定は、会社は法律で許可された最大限にその役員と役員を賠償し、他の高級管理者、従業員、その他の代理人に対して賠償を行うことができる。
デラウェア州会社法の許可の下で、当社はすでに当社の各取締役及び行政人員と合意を締結し、同社などの人々が実際及び合理的に招いた支出、判決、罰金、罰金、和解及びその他の金額を賠償することを要求し、誘導訴訟の支出を含むが、このような支出、判決、罰金、罰金、和解及びその他の実際或いは行う可能性のある法律手続きに関連する費用は、例えば当社のいかなる取締役或いは行政人員がかつて当社の取締役であったか、或いはかつて当社の取締役であったために一方とされる可能性があり、当社はこのような人々に賠償を行わなければならない。取締役が善意に基づいて行動し、会社の最良の利益に適合または反対しないことを彼または彼女が合理的に信じて行動する場合にのみ、会社はこれらのお金を支払う義務がある。いかなる刑事訴訟に対しても、取締役が彼や彼女の行為が不法であると信じる合理的な理由がない場合にのみ、会社はその金額を支払う義務がある。賠償協定はまた、賠償要求を提出した場合に適用されるいくつかの手続きを規定している。
デラウェア州会社法第145条(G)条は、会社が誰を代表して保険の購入及び維持を許可するか、その改正及び再記載された定款が賠償を許可するか否かにかかわらず、これらの者が会社の役員、上級管理者、従業員又は代理人であっても、これらの者が会社に提供されたサービスにより生じた行為である。当社は、現在またはかつて役員または高級社員であった任意の人のために保険を購入し、そのような身分でのクレームによるいかなる損失も防止するために保険を購入する予定であるが、いくつかの例外は除外する。
|
第十五項。 |
最近販売されている未登録証券。 |
2021年6月30日までの6カ月間、発行された株式承認証を行使したため、会社は162株の普通株を発行した。発行された普通株のうち、158株は株式承認証の純額、無現金行使によって発行され、5446株を購入し、残りの5株は25,000ドルの現金で発行された。我々は、証券法第4(A)(2)節に規定されている登録免除に依存しており、米国証券取引委員会が証券法で公布された規則D規則501に基づいて定義されているように、販売待ち証券をマーケティングまたは他の方法で提供する一般的な勧誘や広告は一切使用されていない。
2022年6月15日、期日が2022年2月21日の合併協議及び計画(“合併合意”)に基づき、当社はCPP証券保有者に以下の証券を売却·発行した:(A)5,489株の普通株、(B)609株が依然として保留されている(合併協議参照)普通株、(Iv)加重平均買付価格で1株420.00ドルで最大1,330株の普通株の置換オプションを購入し、及び(V)株式承認証をリセットし、加重平均買付価格で1株4974.00ドルで最大2,810株の普通株を購入した。2023年6月15日から、差し止めされた信託のすべての株が売り手に解放される。我々は、証券法第4(A)(2)節に規定されている登録免除に依存しており、米国証券取引委員会が証券法で公布された規則D規則501に基づいて定義されているように、販売待ち証券をマーケティングまたは他の方法で提供する一般的な勧誘や広告は一切使用されていない。
2023年11月2日、当社はその既存の引受権証のいくつかの所有者と株式承認証を締結し、誘因なく普通株式を購入し、これにより、所有者は現金で既存の引受権証を行使し、1株当たり0.78ドルの換算権価格で合計2,130,000株の普通株を購入することに同意し、これと引き換えに、当社が既存の株式承認証とほぼ同じ条項で新しいC類株式証を発行することに同意した。最大4,260,000株の普通株(“C類株式承認証”)を購入し、既存の引受証株式1株当たり0.125ドルを現金で支払い、既存の引受証を行使する際に全額支払う。C類株式証の行使条項は、上記の“証券--未償還引受権証の記述--C類株式承認証”のタイトルの下でさらに説明される。同社が保有者が既存の株式承認証とC類株式承認証の売却から得た総収益総額は約190万ドル。当社は、その公布された規則例D第4(A)(2)条及び第506条(B)に公布された証券法免除登録規定に基づいてC類株式証を発行し、C類株式証株式は同じ免除又は証券法第3(A)(9)条に規定する免除により発行されている。
2023年12月21日、当社はその既存のC類株式証所有者と持分証取引権誘因要約書簡を締結し、これにより、所有者は現金方式で既存のC類株式承認証を行使することに同意し、既存の取引価格で1株当たり0.78ドルで合計2,556,000株の普通株を購入し、当社がC類株式証とほぼ同じ条項で新しいD類株式証を発行することに同意し、最大5,112,000株普通株(“D類株式承認証”)を購入することに同意した。D類株式証の行使条項は、上記の“証券--未償還引受権証の記述--D類株式証”というタイトルの下でさらに説明されている。同社が保有者がC類株式証を行使したことから得られた総収益は約200万ドル。当社は、その公布された証券法規第4(A)(2)条及び第506条(B)条に公布された証券法登録規定の免除に基づいてC類株式証株式及びD類株式証を発行し、D類株式証株式はすでに又は同じ免除又は証券法第3(A)(9)条に規定する免除により発行される。
|
第十六項。 |
展示品と財務諸表明細書。 |
(a) 陳列品
|
展示品 違います。 |
説明する |
|
|
3.1 |
再記載された会社登録証明書(2023年1月17日に提出されたタブ8-K現在の報告書の添付ファイル3.1を参照して編入) |
|
|
3.2 |
証明書の改訂証明書の再登録(2023年5月31日に提出されたタブ8−Kの現在の報告書の添付ファイル3.1を参照して編入) |
|
|
3.3 |
別例の改訂および再改訂(2023年4月18日に提出されたタブ8−K現在の報告書の添付ファイル3.1を参照して編入) |
|
| 3.4 | 再登録証明書の改訂証明書(2024年1月19日に提出されたタブ8−K現在の報告書の添付ファイル3.1を参照して編入) | |
|
4.1 |
証券説明(2020年12月31日までの財政年度のForm 10−K年報に添付ファイル4.1を引用) |
|
展示品 違います。 |
説明する | |
|
4.2 |
2018年12月と2019年1月に発行された普通株式証券表(添付ファイル10.3を参照して2018年12月28日に提出された現在の8-K表報告書に組み込む) |
|
|
4.3 |
2019年8月から10月までに発行された普通株式証券表(添付ファイル10.2を参照して2019年8月29日に提出された現在の8-K表報告書に組み込む) |
|
|
4.4 |
2020年5月22日、6月5日、6月15日、6月22日に発行された引受権証表(添付ファイル10.2を参照して2020年6月11日に提出された現在の8-K表報告書に組み込む) |
|
|
4.5 |
Stock Transferとの権利証明代理プロトコルは、2020年9月1日の有限責任会社(添付ファイル4.1を参照して2020年9月1日に提出された8-Kフォームの現在のレポートに組み込まれています) |
|
|
4.6 |
普通株引受権証表(添付ファイル4.6に添付) |
|
|
4.7 |
VIStock Transferと署名された引受権証代理契約は、2022年10月4日の有限責任会社(2022年10月4日に提出された8-K表の現在報告書の添付ファイル4.1を参照して編入) |
|
|
4.8 |
普通株式引受権証表(2022年10月4日提出の8-K表現在報告書の添付ファイル4.3を参照して編入) |
|
|
4.9 |
VIStock Transferとの権利証明代理プロトコルは、2023年1月30日の有限責任会社(添付ファイル4.1を参照して1月31日に提出された8−Kフォームの現在の報告書に組み込まれる。2023年) |
|
|
4.10 |
Stock Transferとの権利証明代理プロトコルは、2023年6月21日の有限責任会社(添付ファイル4.1を参照して2023年6月21日に提出された8-Kフォームの現在の報告書に組み込まれています) |
|
|
4.11 |
A類普通株引受権証表(2023年6月21日に提出された8-K表現在報告の添付ファイル4.3を組み合わせて) |
|
|
4.12 |
クラスB普通株式引受権証表(2023年6月21日に提出された8-K表に添付ファイル4.4を参照して現在の報告に組み込む) |
|
|
4.13 |
C類普通株引受権証表(2023年11月3日に提出された8-K表の現在報告書の添付ファイル4.1) |
|
|
4.14 |
クラスD普通株式引受権証表(2023年12月21日に提出された8-K表の現在報告書の添付ファイル4.1を参照して編入) |
|
|
4.15+ |
Vock Transfer,LLCと締結された引受権証エージェントプロトコルフォーマット |
|
|
4.16+ |
あらかじめ出資して株式証の書式を承認する |
|
|
4.17+ |
E類普通株引受権証のフォーマット |
|
|
4.18+ |
F類普通株引受権証のフォーマット |
|
|
5.1+ |
フェグレル·デリンク·ビデルとReath LLPの観点は |
|
|
10.1* |
2015年1月1日現在改訂された2011年株式オプション計画(添付ファイル10.1を参照して2015年9月11日に提出された8-Kフォームの現在の報告書に組み込む) |
|
|
10.2* |
2011年計画報酬のインセンティブ株式オプション協定表(添付ファイル10.2を参照して2015年9月11日に提出されたタブ8-Kの現在の報告書に組み込む) |
|
|
10.3* |
2011年に奨励を計画した非限定株式オプション協定表(添付ファイル10.3を参照して2015年9月11日に提出されたタブ8-Kの現在の報告書に組み込まれる) |
|
|
10.4* |
2020年4月9日までに改訂·再記述された2016年総合インセンティブ計画(添付ファイル99.1を参照して2020年5月26日に提出された8−K表の現在の報告書に組み込む) |
|
|
10.5* |
2016年度計画奨励株式オプションプロトコルフォーマット(添付ファイル10.4を参照して2016年6月30日までの第10-Q四半期報告書) |
|
|
10.6* |
2016年総合インセンティブ計画で奨励された非制限株式オプションプロトコル表(添付ファイル10.5を参照して2016年6月30日までの四半期報告Form 10-Q) |
|
|
10.7* |
2016年度総合インセンティブ計画奨励の業績株式オプションプロトコル表(添付ファイル10.7を参照して2016年12月31日現在の財政年度Form 10-K年報) |
|
|
10.8** |
フロリダ大学研究財団社との標準独占許可協定は、2011年12月22日(添付ファイル10.5を参照して2015年9月11日に提出されたForm 8-K現在の報告書に組み込まれている) |
|
|
10.9 |
2016年12月12日にフロリダ大学研究財団と署名したライセンス契約第1修正案表(添付ファイル10.10を参照して2019年12月31日までの財政年度Form 10-K年度報告書に組み込む) |
|
|
10.10 |
フロリダ大学研究財団社とのライセンス契約第2修正案は、2019年10月3日(添付ファイル10.1を参照して2019年10月9日に提出されたForm 8-K現在の報告書の添付ファイル10.1に組み込む) |
|
展示品 違います。 |
説明する | |
|
10.11* |
Susan Horvathとの雇用契約は、2018年4月17日(添付ファイル10.4を参照して2018年3月31日までの四半期Form 10-Q四半期報告に組み込まれています) |
|
|
10.12* |
ジェニファー·K·シンプソンとの雇用協定は、2020年7月15日(添付ファイル10.1を参照して2020年7月16日に提出された現在の8-K表報告書に組み込まれる) |
|
|
10.13 |
SEED資本加速器融資協定とSEED資本融資手形の第1修正案は、2017年10月13日(添付ファイル10.1参照2019年3月31日現在のForm 10-Q四半期報告を参照) |
|
|
10.14 |
SEED資本加速器融資協定とSEED資本融資手形の2回目の改正は、2019年4月5日(添付ファイル10.2参照)2019年3月31日現在のForm 10-Q四半期報告に組み込まれています |
|
|
10.15 |
日付は2019年12月31日の種子資本加速器融資協定と種子資本融資第3改正案(添付ファイル10.25を参照して2019年12月31日までの財政年度Form 10-K年度報告書に組み込む)と明記されています |
|
|
10.16 |
証券購入契約表は、2018年12月21日と31日、2019年1月14日、25日、31日(2018年12月28日に提出された現在の8-K表報告書の添付ファイル10.1を参照して編入されます) |
|
|
10.17* |
改正癌予防製薬会社2010年株式インセンティブ計画(添付ファイル10.1を参照して2022年6月16日に提出された8−K表の現在の報告書に組み込む) |
|
|
10.18* |
株式オプション負担通知表(添付ファイル10.2を参照して2022年6月16日に提出された8-Kフォーム現在報告に組み込む) |
|
|
10.19 |
株式承認証表の交換(添付ファイル10.3を参照して2022年6月16日に提出された現在の8-K表報告書に組み込む) |
|
|
10.20 |
Sucampo GmbH(f/k/a Sucampo AG)を受益者とする転換可能な本票は、2017年9月6日、2022年4月7日に修正された(添付ファイル10.4を参照して2022年6月16日に提出された現在の8−K表報告書に組み込まれる) |
|
|
10.21 |
Sucampo GmbH(f/k/a Sucampo AG)を受益者とする保証は、日付は2022年6月15日(添付ファイル10.5を参照して2022年6月16日に提出された8−K表の現在の報告書に組み込まれる) |
|
|
10.22 |
ジェフリー·E·ジェイコブスと締結された別居·解放協定は、2022年6月15日(添付ファイル10.6を参照して2022年6月16日に提出された現在の8-K表報告書に組み込まれている) |
|
|
10.23 |
証券購入契約表(2022年10月4日に提出された8-K表の現在報告書の添付ファイル10.2) |
|
|
10.24 |
Roth Capital Partnersとの配給代理プロトコルは、2022年9月29日まで(2022年10月4日に提出された8-Kフォーム現在報告の添付ファイル10.1を参照して組み込む) |
|
|
10.25 |
Roth Capital Partners,LLCとの配信エージェントプロトコルは,2023年6月16日(2023年6月21日に提出された8−Kフォーム現在報告の添付ファイル10.1を参照して組み込まれる) |
|
|
10.26 |
Panbela Treateutics,Inc.と指名された購入者との間の証券購入プロトコルテーブル(2023年6月21日に提出された8-Kフォームの現在の報告書の添付ファイル10.2を参照して組み込まれる) |
|
|
10.27 |
2023年11月2日の求人用紙フォーム(添付ファイル10.1を参照することにより、2023年11月3日に提出された現在の8-Kフォーム報告書に組み込まれる)。 |
|
|
10.28 |
日付は2023年12月21日の求人用紙表(添付ファイル10.1を参照して2023年12月21日に提出された現在の8-K表報告書に組み込む) |
|
|
10.29+ |
Roth Capital Partners,LLCと締結した配信エージェントプロトコルフォーマット |
|
|
10.30+ |
証券購入契約フォーマット |
|
|
21.1+ |
付属会社名簿 |
|
| 23.1+ |
独立公認会計士事務所の同意 |
|
|
23.2+ |
Faegre Drinker Bdle&Reath LLPの同意書(添付ファイル5.1参照) |
|
|
24.1+ |
授権書 |
|
|
107+ |
届出費用表 |
|
+ |
前に提出しました。 |
|
++ |
本局に提出します。 |
|
* |
経営陣補償計画または手配は、本目論見書の証拠品として提出しなければならない。 |
|
** |
証券取引委員会が発表した機密待遇を与える命令により漏れた一部の証拠品。 |
(b) 財務諸表明細書。
必要な資料は適用されないか、または財務諸表または関連付記に記載されているので、すべての付表は省略される。
別表2.評価及び合資格勘定
要求された資料は適用されないか、またはこれらの資料は財務諸表または関連付記に記載されているため、他のすべての添付表は省略される。
|
17項です。 |
約束する。 |
登録者はこう約束します
|
(1) |
要約または売却の任意の期間に、本登録声明の発効後修正案を提出する |
|
(i) |
証券法第10(A)(3)節に要求される任意の目論見書を含む |
|
(Ii) |
登録説明書の発効日(または登録説明書の発効後の最近の改訂)の後に発生した、個別的に、または全体的に、登録説明書に記載されている情報が根本的に変化することを表す任意の事実またはイベントが、目論見説明書に反映される。上記の規定にもかかわらず、発行された証券の数の増加または減少(発行された証券の総ドル価値が登録されたものを超えない場合)、および最高発行範囲を推定するローエンドまたはハイエンドからのいかなる逸脱も、規則424(B)に従って委員会に提出された目論見形式に反映されてもよく、総数量および価格の変化が有効登録書の“登録料計算”表に規定されている最高発行総価格の20%を超えないことを前提としている |
|
(Iii) |
本登録明細書には、以前に登録説明書に開示されていなかった割当計画に関連する任意の重大な情報またはそのような情報の任意の重大な変更が含まれる |
しかし前提は(A)(A)(1)(1)(I)、(Ii)及び(Iii)段落が発効後修正案に含まれることを要求する情報は、登録者が取引法第13節又は第15(D)節に従って委員会に提出又は提出した報告書に含まれている場合、第(A)(1)(I)、(Ii)及び(Iii)段落は適用されず、これらの報告は引用により登録声明に組み込まれる。
|
(2) |
証券法下のいかなる責任を確定するかについては、当該等が発効するたびの改正は、本稿で提供する証券に関する新たな登録声明とみなされるべきであり、当該等の証券の発売は、その初の好意的な発売とみなされるべきである。 |
|
(3) |
施行後の改正により、発行終了時にまだ販売されていない登録中の証券は、登録から削除される。 |
|
(4) |
証券法に規定されている買い手に対する責任を決定するために、ルール424(B)に従って提出された各入札説明書は、発行に関連する登録声明の一部として、ルール430 Bによって提出された登録声明またはルール430 Aに従って提出された目論見書を除いて、登録声明の一部とみなされ、発効後初めて使用された日に含まれるべきであるしかし前提は登録声明の一部に属する登録声明または募集規約内で行われた任意の陳述、または引用によって本明細書に組み込まれたか、または登録声明または募集規約内に組み込まれた文書内で行われた任意の陳述は、最初の使用前に販売契約を締結した購入者にとって、最初の使用日の直前に登録声明または募集規約内で行われた任意の陳述を代替または修正することはない。 |
|
(5) |
証券法に基づいて任意の買い手に対する責任を決定するために、登録者は、証券法424(B)(1)または(4)または497(H)条に基づいて提出された目論見フォーマットから漏れた情報を、本登録説明書の発効時の一部とみなさなければならない。証券法の下の任意の責任を決定するために、各項目に目論見書形式を含む改正後の改正は、その中で提供される証券に関する新たな登録声明とみなされなければならず、当時、当該証券の発売は初期とみなされなければならない善意のその供え物です。 |
|
(6) |
上記の規定により、登録者の役員、上級管理者及び統制者は、“証券法”に規定された責任に基づいて賠償を受けることができるが、登録者は、米国証券取引委員会は、このような賠償は“証券法”に規定された公共政策に違反しているため、強制的に執行することができないと判断している。登録者が登録中の証券について賠償要求を提出した場合(登録者が取締役、登録者の上級者を支払うか、又は人為的にいかなる訴訟、訴訟又は法律手続きに成功して招いたか又は支払うことに成功した費用を除く)であれば、登録者の弁護士がこのことが前例をコントロールすることによって解決されたと考えない限り、登録者は、適切な司法管轄権を有する裁判所に、その賠償が証券法で表現された公共政策に違反しているか否か、及びその発行された最終裁決によって管轄されるか否かの問題を提出する。 |
サイン
改正された1933年の証券法の要求によると、登録者は2024年1月25日にミネソタ州ミネアポリス市で以下の署名者が登録者を代表して本登録声明に署名することを正式に許可した。
|
PANBELA治療会社は |
||
|
差出人: |
/S/ジェニファー·K·シンプソン |
|
|
ジェニファー·K·シンプソン 社長と最高経営責任者 |
||
授権書
1933年の証券法の要求に基づき、本登録声明は、次の者によって指定された身分及び日付で署名された。
|
サイン |
タイトル |
日取り |
||
|
/S/ジェニファー·K·シンプソン |
社長と最高経営責任者 |
2024年1月25日 | ||
|
ジェニファー·K·シンプソン |
(元金) 執行者 役員)と役員 |
|||
|
/投稿S/スーザン·ホヴァス |
総裁副財務長財務総監財務担当者 |
2024年1月25日 | ||
|
スーザン·ホヴァト |
秘書(首席) 金融 そして 会計計算 (税関職員) |
|||
| * |
取締役会長兼取締役会長 |
2024年1月25日 | ||
|
マイケル·T·クーロン |
||||
| * |
役員.取締役 |
2024年1月25日 | ||
|
ダニエル·J·ドノヴァン |
||||
| * |
役員.取締役 |
2024年1月25日 | ||
|
アーサー·J·フラタミコ |
||||
| * |
役員.取締役 |
2024年1月25日 | ||
|
ジェフリー·E·ジェイコブ |
||||
| * |
役員.取締役 |
2024年1月25日 | ||
|
ジェフリー·S·マティソン |
||||
| * |
役員.取締役 |
2024年1月25日 | ||
|
D.ロバート·スペール |
*ジェニファー·K·シンプソンはここに署名し、上記登録者の各取締役を代表して、上記各董事タールが署名するために署名した授権書に基づいて本文書に署名する。
|
差出人: |
/S/ジェニファー·K·シンプソン |
| ジェニファー·K·シンプソン | |
|
事実弁護士 |