zntl-202309300001725160偽Q312/312023045700017251602023-01-012023-09-3000017251602023-11-02エクセルリ:シェア00017251602023-09-30ISO 4217: 米ドル00017251602022-12-31ISO 4217: 米ドルエクセルリ:シェア00017251602023-07-012023-09-3000017251602022-07-012022-09-3000017251602022-01-012022-09-3000017251602021-12-3100017251602022-09-300001725160米国会計基準:普通株式会員2023-06-300001725160米国会計基準:追加払込資本構成員2023-06-300001725160米国会計基準:その他の包括利益の累計メンバー2023-06-300001725160米国会計基準:利益剰余金メンバー2023-06-300001725160米国会計基準:非支配持分メンバー2023-06-3000017251602023-06-300001725160米国会計基準:追加払込資本構成員2023-07-012023-09-300001725160米国会計基準:その他の包括利益の累計メンバー2023-07-012023-09-300001725160米国会計基準:普通株式会員2023-07-012023-09-300001725160米国会計基準:非支配持分メンバー2023-07-012023-09-300001725160米国会計基準:利益剰余金メンバー2023-07-012023-09-300001725160米国会計基準:普通株式会員2023-09-300001725160米国会計基準:追加払込資本構成員2023-09-300001725160米国会計基準:その他の包括利益の累計メンバー2023-09-300001725160米国会計基準:利益剰余金メンバー2023-09-300001725160米国会計基準:非支配持分メンバー2023-09-300001725160米国会計基準:普通株式会員2022-12-310001725160米国会計基準:追加払込資本構成員2022-12-310001725160米国会計基準:その他の包括利益の累計メンバー2022-12-310001725160米国会計基準:利益剰余金メンバー2022-12-310001725160米国会計基準:非支配持分メンバー2022-12-310001725160米国会計基準:追加払込資本構成員2023-01-012023-09-300001725160米国会計基準:その他の包括利益の累計メンバー2023-01-012023-09-300001725160米国会計基準:普通株式会員2023-01-012023-09-300001725160米国会計基準:非支配持分メンバー2023-01-012023-09-300001725160米国会計基準:利益剰余金メンバー2023-01-012023-09-300001725160米国会計基準:普通株式会員2022-06-300001725160米国会計基準:追加払込資本構成員2022-06-300001725160米国会計基準:その他の包括利益の累計メンバー2022-06-300001725160米国会計基準:利益剰余金メンバー2022-06-300001725160米国会計基準:非支配持分メンバー2022-06-3000017251602022-06-300001725160米国会計基準:追加払込資本構成員2022-07-012022-09-300001725160米国会計基準:その他の包括利益の累計メンバー2022-07-012022-09-300001725160米国会計基準:普通株式会員2022-07-012022-09-300001725160米国会計基準:非支配持分メンバー2022-07-012022-09-300001725160米国会計基準:利益剰余金メンバー2022-07-012022-09-300001725160米国会計基準:普通株式会員2022-09-300001725160米国会計基準:追加払込資本構成員2022-09-300001725160米国会計基準:その他の包括利益の累計メンバー2022-09-300001725160米国会計基準:利益剰余金メンバー2022-09-300001725160米国会計基準:非支配持分メンバー2022-09-300001725160米国会計基準:普通株式会員2021-12-310001725160米国会計基準:追加払込資本構成員2021-12-310001725160米国会計基準:その他の包括利益の累計メンバー2021-12-310001725160米国会計基準:利益剰余金メンバー2021-12-310001725160米国会計基準:非支配持分メンバー2021-12-310001725160米国会計基準:追加払込資本構成員2022-01-012022-09-300001725160米国会計基準:その他の包括利益の累計メンバー2022-01-012022-09-300001725160米国会計基準:普通株式会員2022-01-012022-09-300001725160米国会計基準:非支配持分メンバー2022-01-012022-09-300001725160米国会計基準:利益剰余金メンバー2022-01-012022-09-300001725160ZNTL: ゼンテラ治療薬メンバー2023-06-15エクセルリ:ピュア0001725160ZNTL: ゼンテラ治療薬メンバー2023-06-152023-06-1500017251602023-04-012023-06-300001725160SRT: 最低メンバー数ZNTL: 資産取得、偶発的対策、負債変数、管理マイルストーン、支払メンバー、変更の検討ZNTL: ゼンテラ治療薬メンバー2023-06-150001725160ZNTL: 資産取得、偶発的対策、負債変数、管理マイルストーン、支払メンバー、変更の検討SRT: 最大メンバー数ZNTL: ゼンテラ治療薬メンバー2023-06-150001725160ZNTL: 資産取得、偶発的対価/負債、マイルストーン、支払いメンバーZNTL: ゼンテラ治療薬メンバーUS-GAAP: 評価手法割引キャッシュフローメンバー2023-06-150001725160ZNTL: ゼンテラ治療薬メンバー2023-01-012023-09-300001725160ZNTL: カリラファーマシューティカルズ社のメンバー2017-12-212017-12-210001725160ZNTL: カリラファーマシューティカルズ社のメンバー2017-12-210001725160US-GAAP: 変動利害関係法人の主要受益者2022-12-310001725160US-GAAP: 変動利害関係法人の主要受益者2021-12-310001725160US-GAAP: 変動利害関係法人の主要受益者2023-01-012023-09-300001725160US-GAAP: 変動利害関係法人の主要受益者2022-01-012022-12-310001725160US-GAAP: 変動利害関係法人の主要受益者2023-09-300001725160米国会計基準:コマーシャル・ペーパー・メンバー2023-09-300001725160米国会計基準:企業債務証券メンバー2023-09-300001725160米国会計基準:米国政府機関債務証券メンバー2023-09-300001725160米国会計基準:米国財務省証券会員2023-09-300001725160米国会計基準:コマーシャル・ペーパー・メンバー2022-12-310001725160米国会計基準:企業債務証券メンバー2022-12-310001725160米国会計基準:米国政府機関債務証券メンバー2022-12-310001725160米国会計基準:米国財務省証券会員2022-12-31zntl: セキュリティ0001725160US-GAAP: 当事者間協働協定メンバーとの共同協定取引ZNTL: ゼンテラ治療薬メンバー2023-09-30zntl: 潜在的なマイルストーン支払い0001725160US-GAAP: マネー・マーケット・ファンド・メンバー米国会計基準:フェアバリューインプットレベル1メンバー2023-09-300001725160US-GAAP: マネー・マーケット・ファンド・メンバー米国会計基準:公正価値インプットレベル2メンバー2023-09-300001725160US-GAAP: マネー・マーケット・ファンド・メンバー米国会計基準:フェアバリューインプットレベル3メンバー2023-09-300001725160US-GAAP: マネー・マーケット・ファンド・メンバー2023-09-300001725160米国会計基準:コマーシャル・ペーパー・メンバー米国会計基準:フェアバリューインプットレベル1メンバー2023-09-300001725160米国会計基準:コマーシャル・ペーパー・メンバー米国会計基準:公正価値インプットレベル2メンバー2023-09-300001725160米国会計基準:コマーシャル・ペーパー・メンバー米国会計基準:フェアバリューインプットレベル3メンバー2023-09-300001725160米国会計基準:コマーシャル・ペーパー・メンバー2023-09-300001725160米国会計基準:米国政府機関債務証券メンバー米国会計基準:フェアバリューインプットレベル1メンバー2023-09-300001725160米国会計基準:公正価値インプットレベル2メンバー米国会計基準:米国政府機関債務証券メンバー2023-09-300001725160米国会計基準:米国政府機関債務証券メンバー米国会計基準:フェアバリューインプットレベル3メンバー2023-09-300001725160米国会計基準:米国政府機関債務証券メンバー2023-09-300001725160米国会計基準:フェアバリューインプットレベル1メンバー2023-09-300001725160米国会計基準:公正価値インプットレベル2メンバー2023-09-300001725160米国会計基準:フェアバリューインプットレベル3メンバー2023-09-300001725160米国会計基準:コマーシャル・ペーパー・メンバー米国会計基準:フェアバリューインプットレベル1メンバー2023-09-300001725160米国会計基準:コマーシャル・ペーパー・メンバー米国会計基準:公正価値インプットレベル2メンバー2023-09-300001725160米国会計基準:コマーシャル・ペーパー・メンバー米国会計基準:フェアバリューインプットレベル3メンバー2023-09-300001725160米国会計基準:フェアバリューインプットレベル1メンバー米国会計基準:企業債務証券メンバー2023-09-300001725160米国会計基準:公正価値インプットレベル2メンバー米国会計基準:企業債務証券メンバー2023-09-300001725160米国会計基準:フェアバリューインプットレベル3メンバー米国会計基準:企業債務証券メンバー2023-09-300001725160米国会計基準:フェアバリューインプットレベル1メンバー米国会計基準:米国政府機関債務証券メンバー2023-09-300001725160米国会計基準:公正価値インプットレベル2メンバー米国会計基準:米国政府機関債務証券メンバー2023-09-300001725160米国会計基準:フェアバリューインプットレベル3メンバー米国会計基準:米国政府機関債務証券メンバー2023-09-300001725160米国会計基準:米国財務省証券会員米国会計基準:フェアバリューインプットレベル1メンバー2023-09-300001725160米国会計基準:米国財務省証券会員米国会計基準:公正価値インプットレベル2メンバー2023-09-300001725160米国会計基準:米国財務省証券会員米国会計基準:フェアバリューインプットレベル3メンバー2023-09-300001725160US-GAAP: マネー・マーケット・ファンド・メンバー米国会計基準:フェアバリューインプットレベル1メンバー2022-12-310001725160US-GAAP: マネー・マーケット・ファンド・メンバー米国会計基準:公正価値インプットレベル2メンバー2022-12-310001725160US-GAAP: マネー・マーケット・ファンド・メンバー米国会計基準:フェアバリューインプットレベル3メンバー2022-12-310001725160US-GAAP: マネー・マーケット・ファンド・メンバー2022-12-310001725160米国会計基準:コマーシャル・ペーパー・メンバー米国会計基準:フェアバリューインプットレベル1メンバー2022-12-310001725160米国会計基準:コマーシャル・ペーパー・メンバー米国会計基準:公正価値インプットレベル2メンバー2022-12-310001725160米国会計基準:コマーシャル・ペーパー・メンバー米国会計基準:フェアバリューインプットレベル3メンバー2022-12-310001725160米国会計基準:コマーシャル・ペーパー・メンバー2022-12-310001725160米国会計基準:フェアバリューインプットレベル1メンバー2022-12-310001725160米国会計基準:公正価値インプットレベル2メンバー2022-12-310001725160米国会計基準:フェアバリューインプットレベル3メンバー2022-12-310001725160米国会計基準:コマーシャル・ペーパー・メンバー米国会計基準:フェアバリューインプットレベル1メンバー2022-12-310001725160米国会計基準:コマーシャル・ペーパー・メンバー米国会計基準:公正価値インプットレベル2メンバー2022-12-310001725160米国会計基準:コマーシャル・ペーパー・メンバー米国会計基準:フェアバリューインプットレベル3メンバー2022-12-310001725160米国会計基準:フェアバリューインプットレベル1メンバー米国会計基準:企業債務証券メンバー2022-12-310001725160米国会計基準:公正価値インプットレベル2メンバー米国会計基準:企業債務証券メンバー2022-12-310001725160米国会計基準:フェアバリューインプットレベル3メンバー米国会計基準:企業債務証券メンバー2022-12-310001725160米国会計基準:フェアバリューインプットレベル1メンバー米国会計基準:米国政府機関債務証券メンバー2022-12-310001725160米国会計基準:公正価値インプットレベル2メンバー米国会計基準:米国政府機関債務証券メンバー2022-12-310001725160米国会計基準:フェアバリューインプットレベル3メンバー米国会計基準:米国政府機関債務証券メンバー2022-12-310001725160米国会計基準:米国財務省証券会員米国会計基準:フェアバリューインプットレベル1メンバー2022-12-310001725160米国会計基準:米国財務省証券会員米国会計基準:公正価値インプットレベル2メンバー2022-12-310001725160米国会計基準:米国財務省証券会員米国会計基準:フェアバリューインプットレベル3メンバー2022-12-310001725160ZNTL: 資産取得、偶発的対価/負債、マイルストーン、支払いメンバー米国会計基準:フェアバリューインプットレベル3メンバーUS-GAAP: 評価手法割引キャッシュフローメンバー2023-09-300001725160SRT: 最低メンバー数ZNTL: 発生メンバーの可能性のある測定入力米国会計基準:フェアバリューインプットレベル3メンバーUS-GAAP: 評価手法割引キャッシュフローメンバー2023-09-300001725160ZNTL: 発生メンバーの可能性のある測定入力SRT: 最大メンバー数米国会計基準:フェアバリューインプットレベル3メンバーUS-GAAP: 評価手法割引キャッシュフローメンバー2023-09-300001725160ZNTL: 資産取得、偶発的対価/負債、マイルストーン、支払いメンバー米国会計基準:フェアバリューインプットレベル3メンバー米国会計基準:測定入力割引率メンバー2023-09-300001725160ZNTL: 偶発的対価負債メンバー2022-12-310001725160ZNTL: 偶発的対価負債メンバー2023-01-012023-09-300001725160ZNTL: 偶発的対価負債メンバー2023-09-300001725160ZNTL: 実験装置メンバー2023-09-300001725160ZNTL: 実験装置メンバー2022-12-310001725160米国会計基準:リースホールド改善メンバー2023-09-300001725160米国会計基準:リースホールド改善メンバー2022-12-310001725160ZNTL: オフィス機器と家具NC2023-09-300001725160ZNTL: オフィス機器と家具NC2022-12-310001725160米国会計基準:コンピュータ機器メンバー2023-09-300001725160米国会計基準:コンピュータ機器メンバー2022-12-310001725160米国会計基準:建設中メンバー2023-09-300001725160米国会計基準:建設中メンバー2022-12-310001725160ZNTL: フォローオンオファリングメンバー米国会計基準:普通株式会員2023-06-152023-06-150001725160ZNTL: フォローオンオファリングメンバー米国会計基準:普通株式会員2023-06-150001725160ZNTL: フォローオンオファリングメンバー2023-06-152023-06-150001725160米国会計基準:普通株式会員ZNTL: 2020年のインセンティブ・アワード・プランのメンバー2021-01-310001725160米国会計基準:一般クラス B メンバーSRT: 最大メンバー数ZNTL: 2020年のインセンティブ・アワード・プランのメンバー2021-01-310001725160米国会計基準:普通株式会員ZNTL: 2020年のインセンティブ・アワード・プランのメンバー2021-01-012021-01-310001725160ZNTL: 2020年のインセンティブ・アワード・プランのメンバー2023-09-300001725160ZNTL:2022年の雇用誘致インセンティブアワードプランメンバー2022-07-310001725160ZNTL:2022年の雇用誘致インセンティブアワードプランメンバー2023-09-300001725160米国会計基準:研究開発費メンバー2023-07-012023-09-300001725160米国会計基準:研究開発費メンバー2022-07-012022-09-300001725160米国会計基準:研究開発費メンバー2023-01-012023-09-300001725160米国会計基準:研究開発費メンバー2022-01-012022-09-300001725160米国会計基準:一般管理費メンバー2023-07-012023-09-300001725160米国会計基準:一般管理費メンバー2022-07-012022-09-300001725160米国会計基準:一般管理費メンバー2023-01-012023-09-300001725160米国会計基準:一般管理費メンバー2022-01-012022-09-300001725160米国会計基準:従業員ストックオプションメンバー2023-07-012023-09-300001725160米国会計基準:従業員ストックオプションメンバー2022-07-012022-09-300001725160米国会計基準:従業員ストックオプションメンバー2023-01-012023-09-300001725160米国会計基準:従業員ストックオプションメンバー2022-01-012022-09-300001725160米国会計基準:従業員株式会員2023-07-012023-09-300001725160米国会計基準:従業員株式会員2022-07-012022-09-300001725160米国会計基準:従業員株式会員2023-01-012023-09-300001725160米国会計基準:従業員株式会員2022-01-012022-09-300001725160ZNTL: 制限付株式特典と制限付株式ユニット会員2023-07-012023-09-300001725160ZNTL: 制限付株式特典と制限付株式ユニット会員2022-07-012022-09-300001725160ZNTL: 制限付株式特典と制限付株式ユニット会員2023-01-012023-09-300001725160ZNTL: 制限付株式特典と制限付株式ユニット会員2022-01-012022-09-300001725160SRT: 最低メンバー数米国会計基準:従業員ストックオプションメンバー2023-01-012023-09-300001725160SRT: 最大メンバー数米国会計基準:従業員ストックオプションメンバー2023-01-012023-09-300001725160SRT: 最低メンバー数米国会計基準:従業員ストックオプションメンバー2022-01-012022-09-300001725160SRT: 最大メンバー数米国会計基準:従業員ストックオプションメンバー2022-01-012022-09-300001725160米国会計基準:従業員株式会員2023-09-300001725160米国会計基準:従業員ストックオプションメンバー2023-09-300001725160米国会計基準:制限付株式会員2023-09-300001725160米国会計基準:制限付株式会員2023-01-012023-09-300001725160米国会計基準:RSU 加盟国の制限付株式単位2023-09-300001725160米国会計基準:RSU 加盟国の制限付株式単位2023-01-012023-09-300001725160米国会計基準:従業員ストックオプションメンバー2023-01-012023-09-300001725160米国会計基準:従業員ストックオプションメンバー2022-01-012022-09-300001725160米国会計基準:制限付株式会員2023-01-012023-09-300001725160米国会計基準:制限付株式会員2022-01-012022-09-300001725160米国会計基準:RSU 加盟国の制限付株式単位2023-01-012023-09-300001725160米国会計基準:RSU 加盟国の制限付株式単位2022-01-012022-09-300001725160米国会計基準:関連当事者メンバーZNTL: マスターサービス契約メンバー2023-06-300001725160米国会計基準:関連当事者メンバーZNTL: ゼンテラ治療薬メンバー2023-06-150001725160米国会計基準:関連当事者メンバーZNTL: ゼンテラ治療薬メンバー2023-01-012023-06-300001725160米国会計基準:関連当事者メンバーZNTL: ゼンテラ治療薬メンバー2022-01-012022-09-300001725160米国会計基準:関連当事者メンバーZNTL: マスターサービス契約メンバー2022-09-300001725160ZNTL: メリッサ・エパーリーのメンバー2023-01-012023-09-300001725160ZNTL: メリッサ・エパーリーのメンバー2023-07-012023-09-300001725160ZNTL: メリッサ・エパーリーのメンバー2023-09-30
米国
証券取引委員会
ワシントンDC 20549
フォーム 10-Q
(マークワン)
☒1934年の証券取引法のセクション13または15(d)に基づく四半期報告書
四半期終了時 2023年9月30日
または
☐1934年の証券取引法第13条または第15 (d) 条に基づく移行報告書
___________________から_______________への移行期間について
コミッションファイル番号: 001-39263
ゼンタリス製薬株式会社
(憲章に明記されている登録者の正確な名前)
| | | | | |
| デラウェア州 | 82-3607803 |
(州またはその他の管轄区域) 法人または組織) | (IRS) 雇用主
識別番号) |
| |
1359 ブロードウェイ, スイート 801 | |
| |
| ニューヨーク、 | |
ニューヨーク | 10018 |
| (主要執行機関の住所) | (郵便番号) |
(212) 433-3791
(登録者の電話番号、市外局番を含む)
N/A
(前回の報告以降に変更された場合、以前の名前、以前の住所、および以前の会計年度)
同法第12条 (b) に従って登録された証券:
| | | | | | | | |
| 各クラスのタイトル | トレーディングシンボル | 登録された各取引所の名前 |
普通株式、 一株あたり額面0.001ドル | ZNTL | ナスダック・ストック・マーケットLLC(ナスダック・グローバル・マーケット) |
登録者が、(1)1934年の証券取引法第13条または第15条(d)で提出が義務付けられているすべての報告書を過去12か月間(または登録者がそのような報告を提出する必要があったよりも短い期間)に提出したか、(2)過去90日間にそのような提出要件の対象であったかどうかをチェックマークで示してください。はい☒ いいえ ☐
登録者が、過去12か月間(または登録者がそのようなファイルの提出を求められたより短い期間)に、規則S-Tの規則405(本章の§232.405)に従って提出が義務付けられているすべてのインタラクティブデータファイルを電子的に提出したかどうかをチェックマークで示してください。はい☒ いいえ ☐
登録者が大規模な加速申告者、加速申告者、非加速申告者、小規模な報告会社、または新興成長企業のいずれであるかをチェックマークで示してください。取引法規則12b-2の「大規模加速申告者」、「加速申告者」、「小規模報告会社」、および「新興成長企業」の定義を参照してください。
| | | | | | | | | | | | | | |
| 大型加速フィルター | ☒ | | アクセラレーテッド・ファイラー | ☐ |
| 非加速ファイラー | ☐ | | 小規模な報告会社 | ☐ |
| | | 新興成長企業 | ☐ |
新興成長企業の場合は、登録者が取引法第13条 (a) に従って規定された新規または改訂された財務会計基準を遵守するために延長された移行期間を使用しないことを選択したかどうかをチェックマークで示してください。☐
登録者がシェル会社(取引法の規則12b-2で定義されている)であるかどうかをチェックマークで示してください。はい☐いいえ ☒
2023年11月2日の時点で、登録者は 70,765,771普通株式、1株あたり額面0.001ドル、発行済です。
目次
| | | | | | | | |
| | ページ |
| 第一部。 | 財務情報 | 3 |
| アイテム 1. | 財務諸表 (未監査) | 4 |
| 要約連結貸借対照表 | 4 |
| 要約連結営業報告書 | 5 |
| 要約連結包括損失計算書 | 6 |
| 要約連結キャッシュフロー計算書 | 7 |
| 要約連結株主資本計算書 | 9 |
| 要約連結財務諸表の注記 | 11 |
| アイテム 2. | 経営陣による財政状態と経営成績に関する議論と分析 | 26 |
| アイテム 3. | 市場リスクに関する定量的・質的開示 | 41 |
| アイテム 4. | 統制と手続き | 41 |
| 第二部 | その他の情報 | 43 |
| アイテム 1. | 法的手続き | 43 |
| アイテム 1A. | リスク要因 | 43 |
| アイテム 2. | 株式の未登録売却、収益の使用、および発行者による株式の購入 | 99 |
| アイテム 3. | シニア証券のデフォルト | 99 |
| アイテム 4. | 鉱山の安全に関する開示 | 99 |
| アイテム 5. | その他の情報 | 99 |
| アイテム 6. | 展示品 | 100 |
| 署名 | 102 |
将来の見通しに関する記述に関する注意事項
フォーム10-Qのこの四半期報告書、または四半期報告書には、改正された1933年の証券法、または証券法のセクション27A、および改正された1934年の証券取引法、または取引法のセクション21Eに含まれる将来の見通しに関する記述のセーフハーバー条項の意味における将来の見通しに関する記述が含まれています。この四半期報告書に含まれる歴史的事実の記述以外のすべての記述は、将来の見通しに関する記述です。場合によっては、「可能性がある」、「する」、「すべき」、「期待する」、「計画する」、「予測する」、「できる」、「意図する」、「目標とする」、「計画する」、「熟考する」、「信じる」、「見積もる」、「予測」、「可能性」、「設計」、「目的」、「サポート」、「継続」などの用語で将来の見通しに関する記述を識別できますまたは、これらの用語やその他の類似の表現の否定性。ただし、すべての将来の見通しに関する記述にこれらの言葉が含まれているわけではありません。この四半期報告書に含まれる将来の見通しに関する記述には、以下に関する記述が含まれますが、これらに限定されません。
•私たちの競争上の地位と業界。
•資本要件、追加資本の必要性、将来の現金ニーズ、コスト、費用、収益、資本資源、キャッシュフロー、財務実績、収益性、納税義務、流動性、成長、契約上の義務、当社の現金資源が現在の事業計画に充てられる期間、財務報告に対する内部統制、開示管理と手続きに関する当社の期待、予測、見積もり。
•製品候補の安全性と有効性を実証する臨床試験の能力、およびその他の肯定的な結果。
•世界のマクロ経済環境とインフレ率と金利の上昇。
•現在進行中および将来の前臨床研究と臨床試験のタイミングと焦点(それらの研究と試験からのデータの報告、そのタイミング、臨床試験への登録開始のタイミングを含む)。
•私たちの臨床試験に登録する患者数の見積もり。
•当社の製品候補の有益な特性、安全性、有効性、治療効果。
•製品候補の開発、製造、供給、承認、商品化に関する当社および協力者の戦略、計画、期待、およびその時期。
•私たちの研究のデザイン、私たちの研究から期待される情報やデータの種類、そしてそれによって期待される利益。
•マーケティング承認を取得して維持する当社の能力、およびそれに関する市販後の要件を満たす能力。
•協力者やライセンシーからの、またはそれらへの支払いのタイミングと金額、およびマイルストーンやロイヤルティに関するものを含め、コラボレーション契約やライセンス契約に基づいて予想される取り決めと利点。
•当社のパイプライン(その可能性を含む)、および関連する研究開発活動
•サイクリンE1陽性腫瘍や相同組換え欠損腫瘍など、ゲノム不安定性の高い腫瘍を対象とするバイオマーカー濃縮戦略に関する当社の計画。
•プログラムのスケジュール、登録までの潜在的な経路、および当社が追求する可能性のある追加の適応症など、製品候補のさらなる開発に関する当社の計画。
•承認されれば、候補製品について第三者の支払者と適時に、またはまったく、適切な価格設定、補償、および償還の条件とプロセスを交渉、確保、維持する当社の能力。
•あらゆる診断ツールの開発に関する当社の計画(その費用を含む)。
•パイプラインの価値を最大化するために、追加の戦略的機会を評価する計画です。
•タンパク質分解プログラムに関する進行中の研究を進める計画です。
•他の治療法と組み合わせて製品候補を開発する計画です。
•当社の既存の協力関係、および当社の製品候補を開発、製造、または商品化するために必要または望ましい協力、ライセンス、またはその他の取り決めを取得し、有利な条件を交渉する能力。
•承認された場合、重点分野や販売戦略を含む、製品候補の商品化に関する当社の計画。
•私たちの研究、開発、商品化の取り組みのタイミングと成功の可能性。
•期待されるマイルストーンのタイミングとその発表。
•当社の製品候補の市場機会の規模。
•当社の製品候補が第一、第二、二次治療ライン、または他の薬剤との併用として承認および使用されることに関する当社の期待。
•規制当局への提出と承認のタイミングまたは可能性(アゼノセルチブの最初の新薬申請提出の目標タイミングを含む)。
•製品候補の規制当局の承認を取得し、維持する当社の能力。
•米国、欧州連合、その他の管轄区域における既存の規制と規制の進展。
•特許の取得と維持を含む当社の知的財産上の立場、および当社の特許およびその他の所有権および知的財産権に関連する行政、規制、法律、その他の手続きの時期、結果、影響、およびその時期と解決。
•私たちの施設、リース契約、および将来の施設の空き状況。
•会計基準と見積もり、その影響、および完了予定時期
•サイバーセキュリティと情報セキュリティ;
•当社製品候補の開発、製造、供給、商品化を含め、第三者への継続的な依存が予想されます。
•保険の適用範囲;
•主要契約の推定履行期間。
•追加の人材を雇う必要性、人材を引き付けて維持する当社の能力、競争力のある報酬と福利厚生を提供する能力。
•最高科学責任者の後継者計画を実行するという私たちの計画。そして
•COVID-19が私たちの事業に与える影響。
この四半期報告書の将来の見通しに関する記述は予測に過ぎず、主に当社の事業、財政状態、経営成績に影響を与える可能性があると当社が考える将来の出来事や財務動向に関する現在の期待と予測に基づいています。これらの将来の見通しに関する記述は、この四半期報告書の日付時点でのみ述べられており、以下の「要約リスク要因」、この四半期報告書の「リスク要因」、「リスク要因」、「経営陣による財政状態と経営成績に関する議論と分析」というタイトルのセクション、およびこの四半期報告書の他の部分に記載されているものを含む、多くの既知および未知のリスク、不確実性、仮定、およびその他の重要な要因の影響を受けます。
将来の見通しに関する記述は本質的にリスクと不確実性の影響を受けやすく、その中には予測も定量化もできないものもあれば、当社の制御が及ばないものもあります。これらの将来の見通しに関する記述を将来の出来事の予測として当てにするべきではありません。当社の将来の見通しに関する記述に反映されている出来事や状況は達成されないか、発生する可能性があり、実際の結果、財政状態、業績、または成果は、将来の見通しに関する記述で予測されたものと大きく異なる可能性があります。さらに、私たちは進化する環境で事業を行っています。新しいリスク要因と不確実性が時々現れる可能性があり、経営陣がすべてのリスク要因と不確実性を予測することはできません。適用法で義務付けられている場合を除き、新しい情報、将来の出来事、状況の変化などの結果として、ここに含まれる将来の見通しに関する記述を公に更新または改訂する予定はありません。
ゼンタリス®およびそれに関連するロゴはZentalisの商標です。この四半期報告書に記載されているその他すべての商標、商号、サービスマークは、それぞれの所有者の財産です。この四半期報告書に記載されているすべてのウェブサイトアドレスは情報提供のみを目的としており、アクティブなリンクやウェブサイトの情報をこの文書に組み込むことを意図したものではありません。
リスク要因の概要
私たちの事業は、フォーム10-Qのこの四半期報告書のパートII、項目1A、「リスク要因」に記載されているものを含め、多くのリスクと不確実性にさらされています。私たちの普通株に投資するときは、これらのリスクと不確実性を慎重に考慮する必要があります。当社の事業に影響を与える主なリスクと不確実性には、次のものがあります。
•当社の営業履歴は限られており、商業販売が承認された製品もありません。そのため、現在の事業を評価し、将来の成功と存続可能性を予測することは難しいかもしれません。
•私たちは創業以来多額の純損失を被っており、当面は引き続き多額の純損失を被ると予想しています。
•事業資金を調達するには、多額の追加資本が必要になります。必要なときに、または許容できる条件でそのような資金を調達できない場合、研究および医薬品開発プログラムの1つ以上、または将来の商業化活動を延期、削減、および/または廃止せざるを得ない可能性があります。
•私たちは、現在臨床試験中の主力製品候補であるアゼノセルチブ(Zn-C3)やZn-D5の成功に大きく依存しています。これらの製品候補の開発を完了し、承認を得て、適時に商品化できなければ、私たちのビジネスは損なわれます。
•当社の製品候補の臨床試験では、米国食品医薬品局、FDA、またはその他の同等の米国以外の規制当局が満足できる安全性と有効性が実証されない場合や、肯定的な結果が得られない場合があります。
•患者の選択を可能にするバイオマーカーの診断ツールの開発が成功しなかったり、開発が大幅に遅れたりした場合、製品候補の商業的可能性を最大限に発揮できない可能性があります。
•私たちは、他の治療法と組み合わせて製品候補を開発していますが、それにはさらなるリスクがあります。
•FDAやその他の同等の米国以外の規制当局の規制承認プロセスは時間がかかり、本質的に予測不可能です。最終的に候補製品について規制当局の承認を得ることができなければ、製品の収益を上げることができず、事業に大きな打撃を与えることになります。
•私たちは激しい競争に直面しており、競合他社が私たちよりも早く、または私たちが開発する製品候補よりも効果的、安全、または安価な技術や製品を開発して販売した場合、私たちの商業機会は悪影響を受けます。
•私たちの成功は、知的財産と独自のプラットフォームを保護する能力にかかっています。知的財産と独自のプラットフォームを適切に保護できない場合、または製品候補を保護するのに十分な発行済み特許を取得して維持できない場合、他の企業が当社とより直接的に競争する可能性があり、それが当社の事業に悪影響を及ぼします。
•私たちの既存のコラボレーションは私たちのビジネスにとって重要であり、将来のライセンスも私たちにとって重要になる可能性があります。これらのコラボレーションを維持できない場合、またはこれらの取り決めが成功しなかった場合、私たちのビジネスに悪影響が及ぶ可能性があります。
•私たちは、前臨床試験や臨床試験の特定の側面を実施するにあたり、独立した臨床研究者やCROを含む第三者に依存しており、今後も依存し続けることを期待しています。これらの第三者が契約上の義務を首尾よく遂行しなかったり、適用される規制要件を遵守しなかったり、予定された期限を守らなかったりすると、候補製品について規制当局の承認を得たり、商品化したりすることができず、事業に大きな打撃を与える可能性があります。
•私たちの商業的成功は、第三者の特許やその他の所有権を侵害することなく事業を行う能力に大きく依存しています。私たちが彼らの所有権を侵害しているという第三者からの主張は、損害賠償責任につながったり、私たちの開発や商品化の取り組みを妨げたり遅らせたりする可能性があります。
•私たちの業界では、有能な人材をめぐる競争が特に激しいです。主要な人材を維持または雇用できない場合、ビジネスを維持または成長させることができない可能性があります。
•米国、世界、政治、経済の不利な状況は、当社の事業、財政状態、または経営成績に悪影響を及ぼす可能性があります。
•事業の中断は、当社の事業に悪影響を及ぼす可能性があります。
パートI—財務情報
アイテム 1.財務諸表。
ゼンタリス製薬株式会社
要約連結貸借対照表
(未監査)
(千単位、株式金額と額面価格を除く)
| | | | | | | | | | | |
| 9月30日 | | 12月31日 |
| | 2023 | | 2022 |
| 資産 | | | |
| 流動資産 | | | |
| 現金および現金同等物 | $ | 110,751 | | | $ | 43,069 | |
| 有価証券、販売可能 | 405,886 | | | 394,302 | |
| | | |
| 前払費用およびその他の流動資産 | 11,627 | | | 14,562 | |
| | | |
| 流動資産合計 | 528,264 | | | 451,933 | |
| 資産および設備、純額 | 6,393 | | | 7,705 | |
| オペレーティングリースの使用権資産 | 36,184 | | | 42,373 | |
| 前払費用およびその他の資産 | 8,511 | | | 9,723 | |
| グッドウィル | 3,736 | | | 3,736 | |
| | | |
| ゼンテラ・セラピューティクスへの投資 | — | | | 21,213 | |
| 制限付き現金 | 2,627 | | | 2,627 | |
| 総資産 | $ | 585,715 | | | $ | 539,310 | |
| 負債と株主資本 | | | |
| 現在の負債 | | | |
| 買掛金 | $ | 14,264 | | | $ | 11,247 | |
| 未払費用 | 44,654 | | | 45,400 | |
| 流動負債合計 | 58,918 | | | 56,647 | |
| 繰延税金負債 | — | | | 853 | |
| 長期リース負債 | 43,266 | | | 45,166 | |
| その他の長期負債 | 1,634 | | | 2,620 | |
| 負債総額 | 103,818 | | | 105,286 | |
| コミットメントと不測の事態 | | | |
| | | |
| 公平 | | | |
| | | |
| | | |
優先株式、$0.001額面価格; 10,000,000承認された株式。 いいえ2023年9月30日および2022年12月31日に発行された株式 | — | | | — | |
普通株式、$0.001額面価格; 250,000,000承認された株式。 70,640,684そして 59,280,2472023年9月30日と2022年12月31日にそれぞれ発行された株式と発行された株式 | 70 | | | 59 | |
追加払込資本 | 1,309,696 | | | 1,031,462 | |
| その他の包括損失の累計 | (359) | | | (1,353) | |
| 累積赤字 | (827,639) | | | (596,365) | |
| 株主資本の総額 | 481,768 | | | 433,803 | |
| 非支配持分 | 129 | | | 221 | |
| 総資本 | 481,897 | | | 434,024 | |
| 負債総額と株主資本 | $ | 585,715 | | | $ | 539,310 | |
要約連結財務諸表の注記を参照してください。
ゼンタリス製薬株式会社
要約連結営業報告書
(未監査)
(千単位、1株あたりの金額を除く)
| | | | | | | | | | | | | | | | | | | | | | | |
| | 3 か月が終了
9月30日 | | 9 か月が終了
9月30日 |
| | 2023 | | 2022 | | 2023 | | 2022 |
| 営業経費 | | | | | | | |
| 研究開発 | $ | 46,765 | | | $ | 42,181 | | | $ | 138,033 | | | $ | 132,118 | |
| ゼンテラの進行中の研究開発 | — | | | — | | | 45,568 | | | — | |
| 一般と管理 | 15,953 | | | 12,012 | | | 47,986 | | | 43,415 | |
| 営業費用の合計 | 62,718 | | | 54,193 | | | 231,587 | | | 175,533 | |
| 営業損失 | (62,718) | | | (54,193) | | | (231,587) | | | (175,533) | |
| その他の収入 (費用) | | | | | | | |
| 投資およびその他の収入、純額 | 7,209 | | | 1,905 | | | 15,769 | | | 2,755 | |
| | | | | | | |
| | | | | | | |
| 税引前純損失 | (55,509) | | | (52,288) | | | (215,818) | | | (172,778) | |
所得税費用(給付) | 31 | | | (159) | | | (466) | | | (109) | |
| 持分法投資による損失 | — | | | 2,371 | | | 16,014 | | | 9,460 | |
| 純損失 | (55,540) | | | (54,500) | | | (231,366) | | | (182,129) | |
| 非支配株主に帰属する純損失 | (12) | | | (99) | | | (92) | | | (294) | |
| ゼンタリスに帰属する純損失 | $ | (55,528) | | | $ | (54,401) | | | $ | (231,274) | | | $ | (181,835) | |
| 発行済普通株式1株あたりの純損失、基本および希薄化後 | $ | (0.79) | | | $ | (0.96) | | | $ | (3.64) | | | $ | (3.56) | |
| | | | | | | |
| 1株あたりの純損失の計算に使用される普通株式、基本株および希薄化後 | 70,612 | | | 56,807 | | | 63,601 | | | 51,098 | |
要約連結財務諸表の注記を参照してください。
ゼンタリス製薬株式会社
要約連結包括損失計算書
(未監査)
(千単位)
| | | | | | | | | | | | | | |
| 3 か月が終了
9月30日 | 9 か月が終了
9月30日 |
| 2023 | 2022 | 2023 | 2022 |
| 純損失 | $ | (55,540) | | $ | (54,500) | | $ | (231,366) | | $ | (182,129) | |
| その他の包括利益 (損失): | | | | |
| | | | |
| 有価証券の含み損益 (損失) | (21) | | (491) | | 994 | | (2,145) | |
| 包括損失合計 | (55,561) | | (54,991) | | (230,372) | | (184,274) | |
| 非支配株主に帰属する包括損失 | (12) | | (99) | | (92) | | (294) | |
| ゼンタリスに帰属する包括的損失 | $ | (55,549) | | $ | (54,892) | | $ | (230,280) | | $ | (183,980) | |
要約連結財務諸表の注記を参照してください。
ゼンタリス製薬株式会社
要約連結キャッシュフロー計算書
(未監査)
(千単位)
| | | | | | | | | | | |
| | 9 か月が終了
9月30日 |
| | 2023 | | 2022 |
| 営業活動: | | | |
| 連結純損失 | $ | (231,366) | | | $ | (182,129) | |
| 純損失を営業活動に使用された純現金と調整するための調整: | | | |
| 減価償却と償却 | 1,052 | | | 1,067 | |
| オペレーティングリースの使用権資産と固定資産の減損 | 4,953 | | | — | |
| | | |
| | | |
| | | |
| Zenteraの進行中の研究開発の非現金対価部分 | 15,045 | | | — | |
| 持分法投資による損失 | 16,014 | | | 9,460 | |
| 株式ベースの報酬 | 40,942 | | | 37,228 | |
機器の廃棄による損失 | (4) | | | (68) | |
| (割引の増加)/有価証券の保険料の償却、純額 | (8,602) | | | (1,249) | |
| | | |
| 繰延所得税 | (853) | | | — | |
| 営業資産および負債の変動: | | | |
| | | |
| 前払費用およびその他の資産 | (5,199) | | | (4,716) | |
| 買掛金と未払負債 | 522 | | | 10,477 | |
| オペレーティングリースの使用権資産と負債、純額 | 273 | | | 3,508 | |
| 営業活動に使用された純現金 | (167,223) | | | (126,422) | |
| 投資活動: | | | |
| 有価証券の購入 | (453,195) | | | (407,177) | |
| 有価証券の満期による収入 | 451,207 | | | 307,279 | |
| | | |
| 資産および設備の購入 | (410) | | | (2,388) | |
投資活動に使用された純現金 | (2,398) | | | (102,286) | |
| 資金調達活動: | | | |
| | | |
| 普通株式の発行による収入、純額 | 235,680 | | | 209,297 | |
| 株式インセンティブプランに基づく普通株式の発行による収入 | 1,623 | | | 2,005 | |
| | | |
| | | |
| | | |
| | | |
| 財務活動による純現金 | 237,303 | | | 211,302 | |
| 現金、現金同等物および制限付現金の純増額(減少) | 67,682 | | | (17,406) | |
| 現金、現金同等物および期首制限付現金 | 45,696 | | | 62,584 | |
| 期末の現金、現金同等物、制限付現金 | $ | 113,378 | | | $ | 45,178 | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
次の表は、連結キャッシュフロー計算書で報告された現金、現金同等物、および制限付現金の調整を示したものです。
| | | | | | | | | | | |
| 9月30日 |
| 2023 | | 2022 |
| 現金および現金同等物 | $ | 110,751 | | | $ | 42,551 | |
| 制限付き現金 | 2,627 | | | 2,627 | |
| 要約連結キャッシュフロー計算書に報告された現金、現金同等物、および制限付現金の合計 | $ | 113,378 | | | $ | 45,178 | |
要約連結財務諸表の注記を参照してください。
ゼンタリス製薬株式会社
要約連結株主資本計算書
(千単位)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 2023年9月30日に終了した3か月間 |
| | | | | | | | | | | | | |
| 共通 | | [追加]
支払い済み
資本 | | その他の包括利益の累計 | | 累積
赤字 | | 非制御 興味 | | 合計
エクイティ |
| 株式 | | 金額 | | | | | |
| 2023年6月30日の残高 | 70,604 | | | $ | 70 | | | $ | 1,295,520 | | | $ | (338) | | | $ | (772,111) | | | $ | 141 | | | $ | 523,282 | |
| 株式ベースの報酬費用 | — | | | — | | | 13,867 | | | — | | | — | | | — | | | 13,867 | |
| その他の包括損失 | — | | | — | | | — | | | (21) | | | — | | | — | | | (21) | |
| | | | | | | | | | | | | |
| 制限付株式単位の権利確定に関連する普通株式の発行 | 17 | | | — | | | — | | | — | | | — | | | — | | | — | |
| | | | | | | | | | | | | |
| オプションの行使による普通株式の発行、純額 | 3 | | | — | | | 54 | | | — | | | — | | | — | | | 54 | |
| 従業員株式購入制度に基づいて発行された株式 | 17 | | | — | | | 255 | | | — | | | — | | | — | | | 255 | |
| | | | | | | | | | | | | |
| 非支配株主に帰属する純損失 | — | | | — | | | — | | | — | | | — | | | (12) | | | (12) | |
| ゼンタリスに帰属する純損失 | — | | | — | | | — | | | — | | | (55,528) | | | — | | | (55,528) | |
2023年9月30日の残高 | 70,641 | | | $ | 70 | | | $ | 1,309,696 | | | $ | (359) | | | $ | (827,639) | | | $ | 129 | | | $ | 481,897 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 2023年9月30日に終了した9か月間 |
| | | | | | | | | | | | | |
| 共通 | | [追加]
支払い済み
資本 | | その他の包括利益の累計 | | 累積
赤字 | | 非制御 興味 | | 合計
エクイティ |
| 株式 | | 金額 | | | | | |
| 2022年12月31日現在の残高 | 59,280 | | | $ | 59 | | | $ | 1,031,462 | | | $ | (1,353) | | | $ | (596,365) | | | $ | 221 | | | $ | 434,024 | |
| 株式ベースの報酬費用 | — | | | — | | | 40,942 | | | — | | | — | | | — | | | 40,942 | |
| その他の包括損失 | — | | | — | | | — | | | 994 | | | — | | | — | | | 994 | |
| 引受割引、手数料、募集費用を差し引いた株式募集に関連する普通株式の発行 | 11,033 | | | 11 | | | 235,669 | | | — | | | — | | | — | | | 235,680 | |
| 制限付株式単位の権利確定に関連する普通株式の発行 | 235 | | | — | | | — | | | — | | | — | | | — | | | — | |
| | | | | | | | | | | | | |
| オプションの行使による普通株式の発行、純額 | 54 | | | — | | | 995 | | | — | | | — | | | — | | | 995 | |
| 従業員株式購入制度に基づいて発行された株式 | 42 | | | — | | | 628 | | | — | | | — | | | — | | | 628 | |
| 譲渡制限付株式報奨の取り消し | (3) | | | — | | | — | | | — | | | — | | | — | | | — | |
| 非支配株主に帰属する純損失 | — | | | — | | | — | | | — | | | — | | | (92) | | | (92) | |
| ゼンタリスに帰属する純損失 | — | | | — | | | — | | | — | | | (231,274) | | | — | | | (231,274) | |
2023年9月30日の残高 | 70,641 | | | $ | 70 | | | $ | 1,309,696 | | | $ | (359) | | | $ | (827,639) | | | $ | 129 | | | $ | 481,897 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 2022年9月30日に終了した3か月間 |
| | | | | | | | | | | | | |
| | 共通 | | [追加]
支払い済み
資本 | | その他の包括利益の累計 | | 累積
赤字 | | 非制御 興味 | | 合計
エクイティ |
| | 株式 | | 金額 | | | | | |
2022年6月30日時点の残高 | 56,967 | | | $ | 57 | | | $ | 961,205 | | | $ | (1,779) | | | $ | (486,993) | | | $ | 333 | | | $ | 472,823 | |
| 株式ベースの報酬費用 | — | | | — | | | 10,317 | | | — | | | — | | | — | | | 10,317 | |
| その他の包括損失 | — | | | — | | | — | | | (491) | | | — | | | — | | | (491) | |
| | | | | | | | | | | | | |
| 制限付株式単位の権利確定に関連する普通株式の発行 | 5 | | | — | | | — | | | — | | | — | | | — | | | — | |
| | | | | | | | | | | | | |
| オプションの行使による普通株式の発行、純額 | 18 | | | — | | | 327 | | | — | | | — | | | — | | | 327 | |
| 従業員株式購入制度に基づいて発行された株式 | 14 | | | — | | | 262 | | | — | | | — | | | — | | | 262 | |
| 譲渡制限付株式報奨の取り消し | (2) | | | — | | | — | | | — | | | — | | | — | | | — | |
| 非支配株主に帰属する純損失 | — | | | — | | | — | | | — | | | — | | | (99) | | | (99) | |
| ゼンタリスに帰属する純損失 | — | | | — | | | — | | | — | | | (54,401) | | | — | | | (54,401) | |
2022年9月30日時点の残高 | 57,002 | | | $ | 57 | | | $ | 972,111 | | | $ | (2,270) | | | $ | (541,394) | | | $ | 234 | | | $ | 428,738 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 2022年9月30日に終了した9か月間 |
| | | | | | | | | | | | | |
| | 共通 | | [追加]
支払い済み
資本 | | その他の包括利益の累計 | | 累積
赤字 | | 非制御 興味 | | 合計
エクイティ |
| | 株式 | | 金額 | | | | | |
| 2021年12月31日時点の残高 | 45,491 | | | $ | 45 | | | $ | 723,593 | | | $ | (125) | | | $ | (359,559) | | | $ | 528 | | | $ | 364,482 | |
| 株式ベースの報酬費用 | — | | | — | | | 37,228 | | | — | | | — | | | — | | | 37,228 | |
| その他の包括損失 | — | | | — | | | — | | | (2,145) | | | — | | | — | | | (2,145) | |
| 引受割引、手数料、募集費用を差し引いた株式募集に関連する普通株式の発行 | 11,284 | | | 11 | | | 209,286 | | | — | | | — | | | — | | | 209,297 | |
| 制限付株式単位の権利確定に関連する普通株式の発行 | 154 | | | 1 | | | — | | | — | | | — | | | — | | | 1 | |
| | | | | | | | | | | | | |
| オプションの行使による普通株式の発行、純額 | 60 | | | — | | | 1,130 | | | — | | | — | | | — | | | 1,130 | |
| 従業員株式購入制度に基づいて発行された株式 | 30 | | | — | | | 874 | | | — | | | — | | | — | | | 874 | |
| 譲渡制限付株式報奨の取り消し | (17) | | | — | | | — | | | — | | | — | | | — | | | — | |
| 非支配株主に帰属する純損失 | — | | | — | | | — | | | — | | | — | | | (294) | | | (294) | |
| ゼンタリスに帰属する純損失 | — | | | — | | | — | | | — | | | (181,835) | | | — | | | (181,835) | |
2022年9月30日時点の残高 | 57,002 | | | $ | 57 | | | $ | 972,111 | | | $ | (2,270) | | | $ | (541,394) | | | $ | 234 | | | $ | 428,738 | |
要約連結財務諸表の注記を参照してください。
要約連結財務諸表の注記
1. 組織とビジネス
組織
Zentalis Pharmaceuticals, Inc.(「Zentalis」、「当社」)は、がんの基本的な生物学的経路を標的とする小分子治療薬の発見と開発を行っている臨床段階のバイオ医薬品企業です。同社の主力製品候補であるアゼノセルチブ(Zn-C3)は、進行した固形腫瘍および血液悪性腫瘍に対する業界初かつ最高のWEE1阻害剤となる可能性があります。同社はまた、アゼノセルチブと組み合わせたBCL-2阻害剤Zn-D5を開発しています。当社は、業績の評価と経営上の意思決定を目的として、事業を単一のセグメントとして管理しています。会社の有形資産はすべて米国に保管されています。
流動性
関連する条件や事象を総合的に考えると、財務諸表の発行日から1年以内に期限が来るため、企業が義務を履行できなくなる可能性が高い場合、企業が継続企業として存続する能力について実質的な疑念が存在します。当社は、2023年9月30日に終了した四半期の中間未監査要約連結財務諸表が発行された日から1年以内に、継続企業として存続できるかどうかについて実質的な疑念を引き起こす状況や事象はないと判断しました。
2. 中間未監査財務諸表
プレゼンテーションの基礎
添付の中間未監査要約連結財務諸表は、米国の一般に認められた会計原則(「米国会計基準」)およびフォーム10-Qの四半期報告書に関連する米国証券取引委員会(「SEC」)の規則および規制に従って作成されています。期末の要約連結貸借対照表データは、当社の監査済み財務諸表から導き出されたものですが、米国会計基準で義務付けられているすべての開示は含まれていません。これらの暫定未監査要約連結財務諸表およびその注記は、2023年3月1日にSECに提出された2022年12月31日に終了した年度のフォーム10-Kの年次報告書に含まれる当社の監査済み財務諸表およびその注記と併せて読む必要があります。ここに記載されている中間期間の未監査の財務情報は、経営陣の見解では、提示された期間の財政状態と経営成績を公正に提示するために必要なすべての調整を反映しており、そのような調整は通常の定期的な調整のみで構成されています。当期の表示に合わせて、前期の要約連結貸借対照表に特定の再分類が行われました。
添付の中間未監査要約連結財務諸表には、当社の完全子会社と、当社が主な受益者である変動持分法人(「VIE」)が含まれます。連結により、会社間取引と残高はすべてなくなりました。
当社は、完全所有ではない事業体における当社の所有権、契約上およびその他の利益を評価して、これらの事業体がVIEであるかどうか、もしそうであれば、当社がVIEの主な受益者であるかどうかを判断します。私たちがVIEの主な受益者であり、したがってVIEの統合が必要かどうかを判断する際には、(1)VIEの経済パフォーマンスに最も大きな影響を与えるVIEの活動を指示する権限と、(2)そのVIEにとって重要となる可能性のあるVIEの損失を吸収する義務、またはVIEから利益を受ける権利の両方があるかどうかを判断する定性的なアプローチを適用します。
既存の関係や将来の取引が変更されると、そのようなVIEが統合または統合解除される可能性があるため、私たちは私たちがVIEの主な受益者であるかどうかを継続的に評価します。提示された期間中、私たちは契約上提供する必要のないその他の重要な財政的支援やその他の支援をVIEに提供していません。
持分法は、投資先に対して当社が大きな影響力を行使することはできても、コントロールできない投資の会計処理に使用されます。そのような投資は貸借対照表に記録され、株式投資の純利益または損失の割合は、投資およびその他の収益(費用)の純額の4分の1の遅れで計上されます。
見積もりの使用
米国会計基準に準拠した連結財務諸表を作成する場合、経営陣は、当社の連結財務諸表および添付注記で報告される金額に影響する見積もりと仮定を行う必要があります。私たちは、過去および予想される結果と傾向、および経営陣がその状況下で合理的であると考えるその他のさまざまな仮定に基づいて、見積もりや判断を継続的に評価しています。その性質上、見積もりには本質的に不確実性が伴うため、実際の結果は経営陣の見積もりと異なる場合があります。
重要な会計方針
2023年9月30日に終了した9か月間に、2022年12月31日に終了した会計年度のフォーム10-Kの年次報告書に記載されているように、会社の重要な会計方針ではこれまで報告されていなかった以下の重要な会計方針を採用しました。
買収と偶発的検討
当社は、資産の取得やその他の類似取引を評価して、その取引を企業結合または資産買収として計上すべきかどうかを評価します。まず、スクリーンテストを適用して、取得した総資産の公正価値の実質的にすべてが単一の識別可能な資産または類似の識別可能な資産のグループに集中しているかどうかを判断します。画面が合えば、その取引は資産取得として計上されます。条件を満たさない場合は、企業の要件を満たすアウトプットを生み出す能力を備えたインプットやプロセスを会社が取得したかどうかをさらに判断する必要があります。
企業結合であると判断された場合、当社は、会計基準更新(ASU)2017-01、企業結合(トピック805)に示されている会計方法に基づいて取引を会計処理します。事業の定義の明確化。これにより、企業結合の買収企業は、買収した事業体のすべての資産、引き受けた負債、および非支配持分の公正価値を認識し、買収日を公正価値の測定値として設定しますポイント。したがって、当社は、買収日現在の公正価値の見積もりに基づいて、買収した事業体の偶発的資産と負債、および買収した事業体の非支配持分を含む、企業結合によって取得された資産と引き受けた負債を認識します。会計基準体系化(ASC)805「企業結合」に従い、当社は、取得日時点でののれんを、取得した特定純資産の公正価値に対して支払われた対価の公正価値を超えるものとして認識し、測定します。
会社の事業買収の対価には、特定の1つまたは複数のイベントの発生を条件とする将来の支払いが含まれる場合があります。このような偶発的対価支払いの義務は、取得日に公正価値で計上されます。その後、条件付対価義務は報告期間ごとに評価されます。支払いによる変動以外の偶発的対価の公正価値の変動は、利益または損失として認識され、連結包括損失計算書の繰延および偶発的対価負債の公正価値の変動に記録されます。偶発的対価負債は次のように予想されます
貸借対照表日が流動負債に表示されてから12か月以内に決済されます。貸借対照表日から12か月後に決済される予定の条件付対価負債は、長期負債として表示されます。
資産取得であると判断された場合、当社はASC 805-50に基づいて取引を会計処理します。これにより、買収する事業体は、買収する事業体への費用に基づいて取得した資産と引き受けた負債を、与えられた対価に加えて取引費用を含む相対的な公正価値ベースで認識する必要があります。対価として与えられた非現金資産の公正価値が、買収主体の帳簿上の資産の帳簿上の帳簿価額と異なる場合を除き、買収日時点で損益は認識されません。現金以外の譲渡対価は、費用(提供された対価の公正価値に基づいて測定されるものとします)か、取得した資産と引き受けた負債の公正価値、どちらか確実に測定可能な方に基づいて測定されます。のれんは資産の取得では認識されず、取得した純資産の公正価値を超えて譲渡された超過対価は、相対的な公正価値に基づいて識別可能な資産に割り当てられます。進行中の研究開発(「IPR&D」)に関するライセンス契約(「IPR&D」)が事業の定義を満たさず、資産が技術的実現可能性に達しておらず、したがって将来の代替用途がない場合、当社は、連結包括損失計算書で、取得した知的財産権研究開発費用などのライセンス契約に基づいて行われた支払いを行います。資産取得における条件付対価の支払いは、不測の事態が解決され、対価が支払われるか、支払可能になったときに認識されます(偶発的対価がデリバティブの定義を満たしている場合、その金額は取得した資産の基準の一部になります)。偶発的対価の支払いが確認されると、その金額は取得した資産または資産グループの費用に含まれます。
3. 重要な取引
ゼンテラ・セラピューティクス
2023年6月15日、当社と一部の完全子会社が、上海を拠点とするがん治療薬(「ゼンテラ」)の開発に焦点を当てた臨床段階のバイオ医薬品企業であるZentera Therapeuticsとのコラボレーションおよびライセンス契約(「終了契約」)を終了する契約を締結したことを発表しました。これにより、そのような完全子会社が承認しました。Zenteraは、当社の製品候補であるアゼノセルチブ、Zn-D5、Zn-C5(「Zenteraコラボレーション製品」)の国民における特定の開発権および商品化権中華民国、マカオ、香港、台湾(総称して「中華圏」)。これらの契約が終了した結果、私たちは中華圏でZenteraからアゼノセルチブ、Zn-D5、Zn-C5の権利を取り戻し、現在、これらの資産の世界的な開発権と商品化権を保有しています。 コラボレーション契約およびライセンス契約を終了する契約と同時に、私たちはZenteraと株式購入契約(「株式購入契約」)を締結し、当社の利益を還元しました 40.3最低限の対価として、ゼンテラの株式の%。
解雇契約と株式購入契約を一緒に評価した結果、獲得した労働力、インプット、またはアウトプットの生産能力に貢献できる実質的なプロセスなしに、ライセンスされた知的財産を再取得する取引は、会計上の目的での資産取得であると判断しました。
送金された対価の合計は45.6ミリオンは次の要素で構成されていました。$の固定対価30百万、これは前払いに相当します。$の許しの考慮事項を修正しました9.4コラボレーション契約およびライセンス契約に基づく何百万もの未払いの売掛金。私たちの返品に関する考慮事項を修正しました 40.3最低限の現金対価としてZentelaの株式の割合。コストアプローチで調整後の貸借対照表法を使用すると、取引時の持分法投資の帳簿価額と、知的財産の返還後の持分法投資の公正価値との差はドルでした13.7百万。これは、2023年第2四半期の営業報告書の持分法投資項目の損失として認識されました。偶発的対価としての統制変更のマイルストーン支払いの変動対価は、次のいずれかです ゼロまたは $15.0百万。約$の偶発的対価の価値0.5百万は、将来の割引キャッシュフローの見積もり、およびマイルストーン達成確率や割引率の見積もりなどのその他の重要な見積もりを使用して計算されました。このマイルストーンの偶発的対価の価値は、各報告期間において公正価値で再測定されます。必要に応じて、損益は営業報告書に報告されます。また、ドルも発生しました0.5何百万もの買収関連
取得した資産の対価総額に含まれていた費用。Zenteraに支払うべきその他の対価には、中華圏におけるアゼノセルチブ、Zn-D5、Zn-C5の純売上高に対する一桁低いロイヤルティが含まれます。これらの追加支払いは、規制当局の承認と中華圏での商業販売の後にのみ支払われ、取引価格からは除外されます。
取得した進行中の研究開発資産の公正価値は、重要な見積もりを含む市場アプローチに基づいていました。これらの見積もりには、比較対象企業のキャリブレーション調整、コスト見積もり、管理および市場性の割引、成功確率の見積もり、適用される割引率が含まれていました。受け取ったZenteraの進行中の研究開発と引き換えに与えられた対価の公正価値の超過分は、契約終了費用として計上されました。会社は$の全額を認めることにしました45.62023年9月30日に終了した9か月間の要約連結営業報告書と包括損失。
4. ビジネスコンビネーション
カリラ製薬株式会社
2017年12月21日、私たちはドルを買収しました4.5カリラ・ファーマシューティカルズ社(「カリラ」)のシリーズB優先株は数百万株にのぼります 25鎮痛薬治療研究分野への参入を目的としたKalyraの持分の%。買収価格はすべて現金で支払われました。
権威あるガイダンスに従って、Kalyraはインプット、従業員、知的財産、およびアウトプットを生み出すことができるプロセスからなるビジネスであると結論付けました。さらに、KalyraはVIEであり、私たちが主な受益者であり、共通の経営陣と取締役会の代表を通じて、Kalyraの経済パフォーマンスに最も大きな影響を与える活動を指揮する権限を持っているという結論に達しました。2017年12月21日より前に、当社とKalyraは研究開発サービスとサポートの提供について取引を行いました。Kalyraの財政状態と経営成績は、初期投資日から当社の連結財務諸表に含まれています。
信頼できるガイダンスに従い、識別可能な資産、負債、および非支配持分を、最初の連結時に公正価値でVIEに記録しました。特定されたのれんは、労働力と、事業体の統合によって期待される相乗効果で構成されています. T2023年9月30日および2022年12月31日現在のカリラの総資産と負債は重要ではありません。Kalyraを統合した結果として計上された負債は、当社の一般資産に対する追加請求ではありません。権威あるガイダンスによると、Zentalisが所有していないKalyraの持分は、要約連結貸借対照表の非支配持分として報告されます。
以下は、非支配持分に帰属する資本(純資産)の調整です(千単位)。
| | | | | | | | | | | |
| 9月30日 | | 12月31日 |
| | 2023 | | 2022 |
| 期首における非支配持分 | $ | 221 | | | $ | 528 | |
| 非支配株主に帰属する純損失 | (92) | | | (307) | |
| | | |
| | | |
| | | |
| 期末の非支配持分 | $ | 129 | | | $ | 221 | |
| | | |
| | | |
5. 公正価値測定
売却可能な有価証券は、次のもので構成されていました(千単位)。
| | | | | | | | | | | | | | | | | | | | | | | |
| 2023年9月30日 |
| 償却コスト | | 未実現総利益 | | 未実現損失総額 | | 推定公正価値 |
コマーシャル・ペーパー | $ | 87,492 | | | $ | 1 | | | $ | (69) | | | $ | 87,424 | |
企業債務証券 | 149,539 | | | 10 | | | (82) | | | 149,467 | |
米国政府機関 | 105,258 | | | 7 | | | (110) | | | 105,155 | |
米国財務省証券 | 63,956 | | | — | | | (116) | | | 63,840 | |
| $ | 406,245 | | | $ | 18 | | | $ | (377) | | | $ | 405,886 | |
| | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | |
| 2022年12月31日 |
| 償却コスト | | 未実現総利益 | | 未実現損失総額 | | 推定公正価値 |
コマーシャル・ペーパー | $ | 296,309 | | | $ | 71 | | | $ | (587) | | | $ | 295,793 | |
企業債務証券 | 7,472 | | | — | | | (26) | | | 7,446 | |
米国政府機関 | 23,970 | | | — | | | (182) | | | 23,788 | |
米国財務省証券 | 67,904 | | | — | | | (629) | | | 67,275 | |
| $ | 395,655 | | | $ | 71 | | | $ | (1,424) | | | $ | 394,302 | |
| | | | | | | |
2023年9月30日の時点で、 五十八公正市場価値が$の売却可能な負債証券の347.6百万人が総含み損のポジションにありました0.4百万。それらのうち、 五十八総含み損のポジションが$になっています0.41年未満で百万と ゼロ1年以上にわたって総含み損の状態が続いています。 投資の減損を評価する際には、減損の深刻さ、基礎となる信用格付けの変化、予想される回復率、売却の意図、予想される市場価値の回復前に投資を売却する必要がある可能性、予定されている現金支払いが引き続き行われる確率などの要素を検討します。これらの有価証券を検討した結果、2023年9月30日現在の未実現損失はいずれも信用損失によるものではないと考えています。なぜなら、これらの証券を売却するつもりはなく、償却原価ベースの回復前にこれらの証券を売却する必要もありそうもないからです。
売却可能な負債証券の契約満期は次のとおりです(千単位)。
| | | | | | | | | | | |
| | | |
| 2023年9月30日 | | 2022年12月31日 |
| 推定公正価値 |
| 期限 1 年以内 | $ | 238,972 | | | $ | 394,302 | |
| 1年後から5年以内 | 166,914 | | | — | |
| $ | 405,886 | | | $ | 394,302 | |
会社は$を持っていました0.52023年9月30日現在、Zenteraとのコラボレーション契約およびライセンス契約を終了する契約に関連する100万ドルの偶発対価負債。条件付対価残高は以下のものに限られています 一公正価値で測定される潜在的なマイルストーン支払い(参照注3-重要な取引追加情報用)。条件付対価の公正価値は、マイルストーンの達成確率を割り引いたマイルストーンの金銭的価値と、マイルストーンが達成されると予想されるタイミングに基づく現在価値要因に基づいて推定されます。条件付対価残高の価値は、市場では観察できない重要なインプットに基づいています。これは、公正価値階層におけるレベル3の測定です。この責任は2022年12月31日時点では存在しませんでした。
次の表は、2023年9月30日および2022年12月31日現在の公正価値(千単位)で定期的に測定される金融資産と負債をまとめたものです。
| | | | | | | | | | | | | | | | | | | | | | | |
| 2023年9月30日 |
| レベル 1 | | レベル 2 | | レベル 3 | | 推定公正価値の合計 |
| 金融資産: | | | | | | | |
| 現金同等物: | | | | | | | |
マネー・マーケット・ファンド | $ | 76,333 | | | $ | — | | | $ | — | | | $ | 76,333 | |
コマーシャル・ペーパー | 10,483 | | | — | | | — | | | 10,483 | |
米国政府機関 | 14,978 | | | — | | | | | 14,978 | |
| 現金同等物の総額: | 101,794 | | | — | | | — | | | 101,794 | |
| | | | | | | |
| 販売可能な有価証券: | | | | | | | |
コマーシャル・ペーパー | — | | | 87,424 | | | — | | | 87,424 | |
企業債務証券 | — | | | 149,467 | | | — | | | 149,467 | |
米国政府機関 | — | | | 105,155 | | | — | | | 105,155 | |
米国財務省証券 | 63,840 | | | — | | | — | | | 63,840 | |
| 売却可能な有価証券の合計: | 63,840 | | | 342,046 | | | — | | | 405,886 | |
| | | | | | | |
公正価値で測定された総資産
| $ | 165,634 | | | $ | 342,046 | | | $ | — | | | $ | 507,680 | |
金融負債: | | | | | | | |
| 不測の事態への対価 | $ | — | | | $ | — | | | $ | 500 | | | $ | 500 | |
| 金融負債合計 | $ | — | | | $ | — | | | $ | 500 | | | $ | 500 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | 2022年12月31日 |
| | レベル 1 | | レベル 2 | | レベル 3 | | 推定公正価値の合計 |
| 金融資産: | | | | | | | | |
| 現金同等物: | | | | | | | | |
マネー・マーケット・ファンド | | $ | 26,811 | | | $ | — | | | $ | — | | | $ | 26,811 | |
| コマーシャル・ペーパー | | 1,998 | | | — | | | — | | | 1,998 | |
| 現金同等物の総額: | | 28,809 | | | — | | | — | | | 28,809 | |
| | | | | | | | |
| 販売可能な有価証券: | | | | | | | | |
| コマーシャル・ペーパー | | — | | | 295,793 | | | — | | | 295,793 | |
企業債務証券 | | — | | | 7,446 | | | — | | | 7,446 | |
米国政府機関 | | — | | | 23,788 | | | — | | | 23,788 | |
米国財務省証券 | | 67,275 | | | — | | | — | | | 67,275 | |
| 売却可能な有価証券の合計: | | 67,275 | | | 327,027 | | | — | | | 394,302 | |
| | | | | | | | |
公正価値で測定された総資産
| | $ | 96,084 | | | $ | 327,027 | | | $ | — | | | $ | 423,111 | |
金融負債: | | | | | | | | |
| 不測の事態への対価 | | $ | — | | | $ | — | | | $ | — | | | $ | — | |
| 金融負債合計 | | $ | — | | | $ | — | | | $ | — | | | $ | — | |
2023年9月30日の解約契約に基づいてZenteraに支払われる偶発的対価の評価には、観察不可能な以下の重要なインプットが使用されました。
| | | | | | | | | | | | | | |
| 偶発対価責任 | 公正価値 現在 2023年9月30日 | 評価手法 | 観察不能な入力 | 範囲 |
| (千単位) | | | |
| マイルストーン支払い | $ | 500 | | 割引キャッシュフロー | 発生の可能性 | 1.0% - 2.4% |
| | | 割引率 | 40% |
| | | 予定期間 | 永続性 |
次の表は、レベル3のインプット(千単位)を使用して公正価値で測定した、会社の偶発的対価の対象となる活動を反映しています。
| | | | | |
2022年12月31日時点での条件付き検討 | $ | — | |
| Zenteraへの偶発的対価の発行 | 500 |
| 偶発的対価の公正価値の変化 | — | |
2023年9月30日時点での条件付き検討 | $ | 500 | |
2023年9月30日に終了した9か月間、公正価値階層のレベル1とレベル2の間の移転はありませんでした。2023年9月30日の時点でレベル3に分類されている機器が1つありました。2022年12月31日現在、レベル3に分類された機器はありません。2023年9月30日と2022年12月31日の時点で、非金融資産と負債について重要な公正価値調整は必要ありませんでした。
6. 前払費用とその他の資産
前払い費用およびその他の資産は、次のもので構成されていました(千単位)。
| | | | | | | | | | | |
| 9月30日 | | 12月31日 |
| | 2023 | | 2022 |
| プリペイド保険 | $ | 1,011 | | | $ | 1,018 | |
| プリペイドソフトウェアライセンスとメンテナンス | 435 | | | 958 | |
| 外国の研究開発クレジットの払い戻し | 1,024 | | | 659 | |
| 前払いの研究開発費 | 14,188 | | | 15,002 | |
| 受取利息 | 1,931 | | | 508 | |
| サブリース資産 | 1,265 | | | — | |
| 売掛金ゼンテラ | — | | | 5,874 | |
| その他の前払い費用 | 284 | | | 266 | |
| 前払い費用とその他の資産の合計 | 20,138 | | | 24,285 | |
| 長期的負担が少ない | 8,511 | | | 9,723 | |
| 前払い費用およびその他の資産の合計、現在 | $ | 11,627 | | | $ | 14,562 | |
7. 資産および設備、純額
資産および設備、純額は以下のとおりです(千単位)。
| | | | | | | | | | | |
| 9月30日 | | 12月31日 |
| | 2023 | | 2022 |
| | | |
| ラボ機器 | $ | 3,069 | | | $ | 2,622 | |
| 借地権の改善 | 4,432 | | | 4,891 | |
| オフィス機器と家具 | 1,854 | | | 2,065 | |
| コンピューター機器 | 150 | | | 150 | |
| 建設中 | — | | | 37 | |
| 小計 | 9,505 | | | 9,765 | |
| 減価償却累計額と償却額 | (3,112) | | | (2,060) | |
| 資産および設備、純額 | $ | 6,393 | | | $ | 7,705 | |
2023年9月30日に終了した3か月間の減価償却費と 2022およそ $348千ドルと375それぞれ千です。2023年9月30日に終了した9か月間の減価償却費と 2022年はドルでした1.1百万と $1.1それぞれ百万。
8. 未払費用
未払費用は以下のとおりです(千単位)。
| | | | | | | | | | | |
| 9月30日 | | 12月31日 |
| 2023 | | 2022 |
| 未払研究開発費 | $ | 29,882 | | | $ | 32,310 | |
| 未払従業員経費 | 11,159 | | | 11,246 | |
| 未払訴訟費用 | 840 | | | 1,256 | |
| 未払一般管理費 | 828 | | | 662 | |
| リース責任 | 2,425 | | | 2,162 | |
| 不測の事態への対価 | 500 | | | — | |
| 支払うべき税金 | 654 | | | 384 | |
| 未払経費の合計 | 46,288 | | | 48,020 | |
| | | |
| 長期的負担が少ない | 1,634 | | | 2,620 | |
| 未払費用の合計 | $ | 44,654 | | | $ | 45,400 | |
9. 株主資本
普通株式の続発表
2023年6月15日、私たちは後続の募集を完了しました。その中で発行と売却を行いました。 11,032,656$の公募価格での普通株式22.66一株当たり。オファリングの総収入は約$でした250.0百万、ドルの募集費用を差し引く前14.3100万は当社が支払います。
株式ベースの報酬
2020年4月より、当社の取締役会は、2021年1月に改正された2020年インセンティブアワードプラン(「2020年プラン」)を採択し、会社の株主は承認しました。これにより、選ばれた従業員、コンサルタント、および取締役会の非従業員メンバーに助成金が支給されます。現在、2020年プランに基づき、ストックオプションと制限付株式ユニット(「RSU」)を付与しています。2020年プランでは、(1) の合計額を上限としてアワードが授与される場合があります 5,600,000普通株式。さらに(2)権利が確定していないクラスB普通単位の転換時に発行された当社の普通株式の譲渡制限付株式から没収された株式(最大) 1,250,000株式); プラス (3) 2021年12月31日に終了する会計年度から始まり、2030年12月31日に終了する会計年度まで続く各会計年度の初日の年間増加額は、(a) のうち小さい方に等しい 5直前の暦年の最終日に発行された普通株式の割合、および(b)取締役会が決定した非常に少ない株式数。
2023年9月30日の時点で、 10,155,201株式は2020年プランに基づく未払いの報奨の対象となり、 1,021,420株式は、2020年プランに基づく将来の株式ベースの報奨の付与に利用できました。
2022年7月、当社の取締役会は、ゼンタリスファーマシューティカルズ社の2022年雇用誘致インセンティブ報奨制度(「2022年優遇制度」)を承認しました。これは、従業員が会社に就職する際の誘因として、新入社員に株式報奨を与えるためにのみ使用されます。取締役会は留保しました 2,275,0002022年の誘致計画に基づいて付与された報奨に従って発行される当社の普通株式です。
2023年9月30日の時点で、 1,580,825株式は2022年の誘致計画に基づく未払いの報奨の対象となり、 694,175株式は、2022年の誘致計画に基づく将来の株式ベースの報奨の付与に利用できました。
株式ベースの報奨に関連する株式ベースの報酬費用の総額は、次のとおりです(千単位)。
| | | | | | | | | | | | | | | | | | | | | | | |
| | 3 か月が終了
9月30日 | | 9 か月が終了
9月30日 |
| 2023 | | 2022 | | 2023 | | 2022 |
| 研究開発経費 | $ | 5,854 | | | $ | 4,526 | | | $ | 17,712 | | | $ | 14,723 | |
| 一般管理費 | 8,013 | | | 5,791 | | | 23,230 | | | 22,505 | |
| 株式ベースの報酬費用の総額 | $ | 13,867 | | | $ | 10,317 | | | $ | 40,942 | | | $ | 37,228 | |
株式報酬の種類別の株式ベースの報酬費用(千単位):
| | | | | | | | | | | | | | | | | | | | | | | |
| 3 か月が終了
9月30日 | | 9 か月が終了
9月30日 |
| 2023 | | 2022 | | 2023 | | 2022 |
| | | | | | | |
| ストックオプション | $ | 10,408 | | | $ | 8,999 | | | $ | 31,222 | | | $ | 29,301 | |
| 従業員株式購入制度 | 87 | | | 93 | | | 303 | | | 305 | |
| RSAとRSU | 3,372 | | | 1,225 | | | 9,417 | | | 7,622 | |
| $ | 13,867 | | | $ | 10,317 | | | $ | 40,942 | | | $ | 37,228 | |
ストックオプションと譲渡制限付株式ユニット
付与されたストックオプションの行使価格は、付与日の会社の普通株式の終値と同じです。各オプション特典の公正価値は、Black-Scholesモデルを使用して付与日に推定されます。会社の営業履歴は限られており、企業固有の過去および暗示的なボラティリティデータがないため、当社は、上場している類似企業グループの過去のボラティリティに基づいて予想されるボラティリティを推定します。過去のボラティリティデータは、株式ベースの報奨の計算された予想期間と同等の期間における選択した企業の株式の日次終値を使用して計算されました。当社は、従業員オプションの予想期間を見積もるために「簡易法」を使用しています。つまり、期待期間は、権利確定期間とオプションの当初の契約期間の算術平均に等しくなります(一般的に 10年)。リスクフリー金利は、付与時に有効であったオプションの予想期間と一致する期間の米国財務省の利回りに基づいています。当社は配当を行っておらず、オプションの有効期間中に配当を行う予定もありません。その結果、当社は配当利回りを次のように見積もっています ゼロ. 2023年9月30日と2022年9月30日に終了した9か月間に付与されたストックオプションの公正価値は、次の仮定に基づいて決定されました。
| | | | | | | | | | | |
| 2023年9月30日 | | 2022年9月30日 |
| 予想されるボラティリティ | 77.5% - 80.8% | | 73.6% - 80.5% |
| 予想される平均期間(年単位) | 5.5 - 6.1 | | 6.0 - 6.5 |
| リスクフリー金利 | 3.4% - 4.2% | | 1.5% - 3.3% |
| 予想配当利回り | — | % | | — | % |
従業員株式購入制度
2020年4月より、当社の取締役会はゼンタリスファーマシューティカルズ社の2020年従業員株式購入計画(「ESPP」)を採択し、株主は承認しました。この計画はその後修正され、2021年3月15日に発効しました。2023年9月30日の時点で、ESPPに基づいて発行可能な当社の普通株式の最大総数は 1,913,160。ESPPの条件の下で、会社の従業員は最大で次のことを選択できます 20彼らの報酬の%、最大金額は$25,000暦年当たり、会社の普通株式を次の購入価格で購入するために源泉徴収されます 85(i)の初取引日の会社の普通株式の1株あたりの公正市場価値(終値)のうち低い方の% 6 か月提供期間、または(ii)該当する購入日、最終取引日として定義されます 6 か月提供期間。 終了した期間中のESPPに基づく株式購入権の公正価値を見積もるために使用された加重平均仮定は次のとおりです。
| | | | | | | | | | | | | |
| | | 2023年9月30日に終了しました |
| | | | | |
| ESP | | | | | |
| ボラティリティ | | | 94.2 | % | | |
| 期待期間 (年) | | | 0.5 | | |
| リスクフリーレート | | | 4.9 | % | | |
| 予想配当利回り | | | — | | | |
報酬費用の概要
報奨の種類別の未認識推定報酬費用の合計と、そのような費用が認識されると予想される加重平均サービス期間(特に明記されていない限り、千単位):
| | | | | | | | | | | |
| 2023年9月30日 |
| 認識されません
経費 | | 残り
加重平均認識期間
(年) |
| ストック・オプション | $ | 108,520 | | | 2.87 |
| RSA | 16 | | | 0.25 |
| RSU | 27,849 | | | 2.69 |
| ESP | $ | — | | | 0 |
2023年9月30日に終了した9か月間に、発行しました 54.3ストックオプションの行使に関連する普通株式の千株。2023年9月30日に終了した9か月間、 116.9特定の制限付株式報酬(「RSA」)に関連して発行された1,000株の普通株式、権利が確定しています。未払いのストックオプション、権利確定していないRSA、権利確定していないRSU の合計は約 10.3百万株、 4.4千株と 1.52023年9月30日の時点で、当社の普通株式はそれぞれ100万株が発行されました。
10. コミットメントと不測の事態
法的不測の事態
私たちは、知的財産、雇用、契約上の問題など、通常の業務過程で生じる訴訟や請求など、さまざまな紛争に巻き込まれる可能性があります。これらの請求はいずれも、高額な訴訟費用の対象となる可能性があります。損失がわかっている、または損失が発生する可能性が高いと考えられ、金額を合理的に見積もることができる場合、会社はこれらの事項について連結財務諸表に負債を記録します。当社は、追加情報が見つかり次第、会計期間ごとにこれらの見積もりを見直し、必要に応じて損失引当金を調整します。問題が負債につながる可能性が高く、損失額を合理的に見積もることができる場合、当社は、連結財務諸表が誤解を招かないようにするために必要な範囲で、発生する可能性のある損失または損失の範囲を見積もり、開示します。損失が見込めない、または合理的に見積もることができない場合、負債は連結財務諸表に記録されません。私たちは一般的に、さまざまな種類の負債をカバーする十分な保険に加入していると考えていますが、保険会社が補償を拒否したり、保険限度額が損害賠償や和解を完全に満たすのに不十分だったりする場合があります。この場合、そのような報奨金の支払いは、当社の連結業績と財政状態に重大な悪影響を及ぼす可能性があります。さらに、そのような主張は、成功したかどうかにかかわらず、私たちの評判とビジネスを損なう可能性があります。私たちは現在、損失責任の記録を必要とする法的手続きの当事者ではありません。
リース
私たちのコミットメントには、オペレーティングリースに関連する支払いが含まれます。 2023年9月30日現在の将来の最低オペレーティングリース支払額のおおよその年間は次のとおりです(千単位)。
| | | | | | | | |
| 年 | | オペレーティングリース |
| | |
| 2023年 (残りの) | | $ | 1,548 | |
2024 | | 6,486 | |
2025 | | 6,799 | |
2026 | | 7,278 | |
2027 | | 7,451 | |
その後 | | 38,778 | |
| 最低リース料の合計: | | 68,340 | |
少ない:帰属利息 | | (22,649) | |
オペレーティングリース負債総額 | | 45,691 | |
少ない:現在の部分 | | (2,425) | |
リース負債、当期分を差し引いたもの | | $ | 43,266 | |
| | |
オペレーティングリースの加重平均残存リース期間は約 9.0年。
2023年3月6日、私たちはサブリース契約を締結しました。この契約に基づき、ニューヨーク州ニューヨークのブロードウェイ1359、スイート1710、1800にあるオフィススペースをサブテナントに転貸しました。特定のトリガーイベントの結果、長期資産グループの帳簿価額を、割引キャッシュフローモデルを使用した収益アプローチに基づいて決定された推定公正価値と比較することにより、暫定減損テストを実施しました。モデルで使用された見積もりと仮定には、予測キャッシュフローと割引率が含まれていました。その結果、私たちは$の減損費用を計上しました5.02023年9月30日に終了した9か月間のオペレーティングリースの使用権資産およびニューヨークリースに関連する固定資産に対する当社の営業費用(百万単位)。2023年9月30日に終了した9か月間、私たちは$のリース収入を記録しました0.6このサブリースに関連する100万ドル。
11. 普通株式1株あたりの純損失
普通株式1株あたりの基本および希薄化後の純損失は、次のように計算されました(1株あたりの金額を除いて千単位)。
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 3 か月が終了
9月30日 | | | | | 9 か月が終了
9月30日 |
| 2023 | | 2022 | | | | | | | 2023 | | 2022 |
| 分子: | | | | | | | | | | | | |
| ゼンタリスに帰属する純損失 | $ | (55,528) | | | $ | (54,401) | | | | | | | | $ | (231,274) | | | $ | (181,835) | |
| 分母: | | | | | | | | | | | | |
| 基本および希薄化後の発行済普通株式の加重平均数 | 70,612 | | | 56,807 | | | | | | | | 63,601 | | | 51,098 | |
| 普通株式1株当たりの純損失 | $ | (0.79) | | | $ | (0.96) | | | | | | | | $ | (3.64) | | | $ | (3.56) | |
未払いのストックオプション、権利確定されていないRSA、権利確定されていないRSUを含む当社の潜在的かつ希薄化可能な有価証券は、希薄化防止効果が期待されるため、普通株式1株あたりの希薄化後の純損失の計算から除外されています。
次の普通株式同等物は、希薄化防止効果(千単位)になるため、普通株式1株あたりの希薄化後の純損失の計算から除外されています。
| | | | | | | | | | | | |
| | 9月30日 | |
| | 2023 | | 2022 | |
| 優れたストックオプション | 10,260 | | | 7,671 | | |
| 権利が確定していないRSA | 4 | | | 172 | | |
| 投資したRSU | 1,476 | | | 468 | | |
| 11,740 | | | 8,311 | | |
12. 関連当事者の開示
テンパス
当社の最高経営責任者で取締役会のメンバーであるキンバリー・ブラックウェル医学博士は、以前はテンパス研究所株式会社(「テンパス」)に雇用され、2023年6月までテンパスの顧問を務めていました。その時点でテンパスはゼンタリスの関連会社ではなくなりました。当社は、2020年12月にTempusとデータライセンスおよび調査サービスを提供するためのマスターサービス契約を締結しました。ドルがありました0.52023年にTempusが関連当事者であった期間と2022年9月30日に終了した9か月間にTempusが実施したサービスに対して発生した100万ドルおよび重要でない手数料は、それぞれ百万ドルです。
ゼンテラ
当社の最高科学責任者であるケビン・D・バンカー博士は、2023年6月にバンカー博士がゼンテラとのコラボレーションおよびライセンス契約の終了および当社の売却に関連して辞任するまで、ゼンテラの取締役会のメンバーを務めていました。 40.3ゼンテラの株式の%をゼンテラに返還します。追加情報については、注3を参照してください。したがって、当社は、解約日までにZenteraを関連当事者として特定していました。
2020年5月、当社の完全子会社であるゼノアルファ株式会社、Kグループアルファ株式会社、Kグループベータ株式会社はそれぞれ、コラボレーションおよびライセンス契約を締結しました。これに基づいて、開発とコミュニケーションに関してZenteraと協力しました。中華圏でのZenteraコラボレーション製品の製品化。Zenteraとのコラボレーションおよびライセンス契約の条件に基づき、Zenteraは中華圏でのZenteraコラボレーション製品の開発費用を負担し、Zenteraは中華圏外でのZenteraコラボレーション製品の開発費用を負担しました。ただし、Zenteraは、グローバルデータ管理、ファーマコビジランス、安全性データベース管理、化学、製造、管理にかかる費用の一部を当社に払い戻す義務がありました。各Zenteraコラボレーション製品に関する活動。Zenteraが2023年に関連当事者であった期間と、2022年9月30日に終了した9か月間、この取り決めに基づいて発生した金額は合計$でした3.5百万と $8.3それぞれ百万で、連結損益計算書では研究開発費として記載されています。上記で開示したように、これらのコラボレーションおよびライセンス契約は2023年6月に終了し、その時点でZenteraはZentalisの関連会社ではなくなりました。
アイテム 2.経営陣による財政状態と経営成績に関する議論と分析
以下の財政状態と経営成績に関する議論と分析は、当社の中間未監査要約連結財務諸表、フォーム10-Qのこの四半期報告書の他の場所に記載されている関連注記およびその他の財務情報、ならびに2022年12月31日に終了した年度のフォーム10-Kの年次報告書に含まれる監査済み連結財務諸表および関連注記と一緒に読む必要があります。この議論と分析に含まれる情報、またはフォーム10-Qのこの四半期報告書の他の場所に記載されている情報には、重大なリスクと不確実性を伴う将来の見通しに関する記述が含まれています。フォーム10-Qのこの四半期報告書の「リスク要因」セクションに記載されているものなど、多くの重要な要素の結果として、当社の実際の業績は、これらの将来の見通しに関する記述で予想されるものと大きく異なる可能性があります。見やすいように、以下のテキストでは数字の一部を四捨五入しています。
[概要]
私たちは、がんの基本的な生物学的経路を標的とする小分子治療薬の発見と開発に焦点を当てた臨床段階のバイオ医薬品企業です。当社の主力製品候補であるアゼノセルチブ(Zn-C3)は、進行した固形腫瘍や血液悪性腫瘍に対するクラス初かつ最高のWEE1阻害剤となる可能性があります。アゼノセルチブは、進行中の複数の臨床試験にわたって、単剤療法および併用療法として評価されています。臨床試験では、アゼノセルチブは耐容性が高く、複数の腫瘍タイプにわたって単剤として、またいくつかの化学療法の骨格と組み合わせて抗腫瘍活性を示しています。アゼノセルチブ臨床開発プログラムの一環として、サイクリンE1陽性腫瘍や相同組換え欠損腫瘍など、ゲノム不安定性の高い腫瘍を標的とする濃縮戦略を模索しています。また、アゼノセルチブと組み合わせてBCL-2阻害剤であるZn-D5を開発しており、WEE1阻害剤とBCL-2阻害剤の両方を臨床開発しているのは当社だけだと考えています。私たちは現在、アゼノセルチブとZn-d5の独占的なライセンス供与権または世界規模での開発および商品化権を独占的に所有しています。
また、がん生物学と医薬品化学における豊富な創薬経験と能力(統合発見エンジンと呼んでいます)を引き続き活用して、非公開の標的のタンパク質分解物に関する継続的な研究を進めています。私たちの製品候補は、同様の経路をターゲットとする現在のプログラムとは差別化されており、承認されれば、がん患者の臨床成績に大きな影響を与える可能性があると考えています。
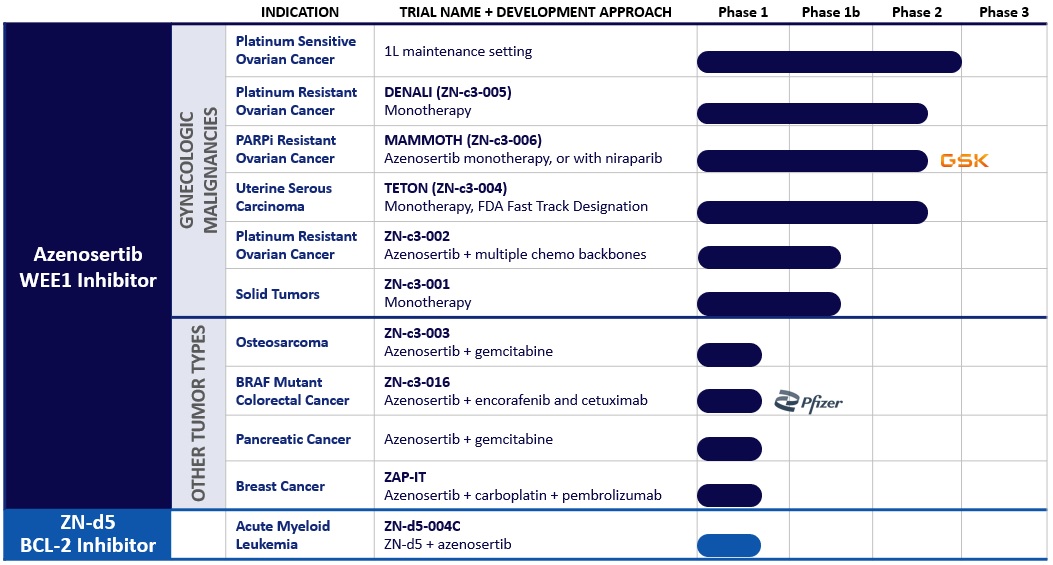
私たちのパイプライン
次の表は、当社の製品候補パイプラインをまとめたものです。
私たちの開発プログラム
アゼノセルチブ(WEE1阻害剤)
アゼノセルチブは、潜在的にクラス最高かつ業界初の経口小分子WEE1阻害剤です。DNA損傷応答キナーゼであるWEE1の阻害は、損傷したDNAを修復できずにがん細胞を有糸分裂に駆り立てます。その結果、細胞が死滅し、それによって腫瘍の増殖が妨げられ、腫瘍の退縮を引き起こす可能性があります。現在、米国食品医薬品局(FDA)によって承認されたWEE1阻害剤はありません。私たちは、優れた選択性と薬物動態(PK)特性など、WEE1を標的とする他の治験療法よりも優れているようにアゼノセルチブを設計しました。アゼノセルチブは現在、単剤療法、従来の化学療法や他のDNA損傷剤との併用、分子標的薬との併用の単剤療法として、進行した固形腫瘍や血液悪性腫瘍について臨床で評価されています。私たちは、2026年に婦人科悪性腫瘍におけるアゼノセルチブの最初の新薬申請(NDA)を提出することを目標としています。
次の臨床試験はアゼノセルチブ臨床開発プログラムの一部です:
•プラチナ感受性卵巣癌(PSOC)におけるアゼノセルチブの臨床試験。PSOC患者を対象に、一次維持療法でアゼノセルチブを評価する臨床試験を開始する予定です。2024年の後半にこの試験に関する追加の詳細を開示し、2025年にこの試験を開始する予定です。
•単剤療法-サイクリンE1による高悪性度漿液性卵巣がん、卵管、または原発性腹膜がん(HGSOC)(DENALI-Zn-C3-005)の第2相臨床試験。 サイクリンE1陽性のプラチナ耐性HGSOC患者を対象とした第2相臨床試験で、単剤療法としてアゼノセルチブを評価しています。私たちのサイクリンE1陽性濃縮戦略は、サイクリンE1タンパク質の高発現により癌細胞がアゼノセルチブの抗腫瘍効果に感作されることを示す前臨床データと、サイクリンE1タンパク質レベルが臨床と関連している可能性があるという予備的なレトロスペクティブ臨床データによって裏付けられています。
WEE1阻害の恩恵を受けます。さらに、2023年4月、2023年米国がん研究協会(AACR)年次総会で、アゼノセルチブがサイクリンE1高腫瘍細胞のがん細胞死を促進することを示す前臨床データを発表しました。 インビトロそして、患者由来のサイクリンE1-HIGHの増殖を実質的に阻害しました インビボ腫瘍モデル。この試験のトップラインデータは、2025年の前半に開示される予定です。
•単剤療法/併用-プラチナ耐性卵巣がん(PROC)(MAMMOTH-Zn-C3-006)における単剤療法およびPARP阻害剤(PARPi)との併用としてのアゼノセルチブの第1/2相臨床試験。 私たちは、アゼノセルチブを単剤療法として、またGSKのPARP阻害剤であるニラパリブ(ZEJULA)との併用で評価しています。®)、GSKとの臨床協力の一環として、ParPi治療に失敗したPROC患者を対象とした第1/2相臨床試験で。この研究では現在、3つのコホートを評価しています。1つは2つの薬剤の同時投与、もう1つは2つの薬剤の交互投与、3つ目は400 mgのアゼノセルチブの単剤療法、5日間連続休止。この臨床研究は、アゼノセルチブとニラパリブの組み合わせが卵巣癌の相乗的な細胞死をもたらすことを示した前臨床データによって裏付けられています インビボモデル。この試験のトップラインデータは、2024年の後半に開示される予定です。
•単剤療法-再発性または持続性の子宮漿液性がん(USC)の第2相臨床試験(TETON-Zn-C3-004)。 アゼノセルチブは現在、USC患者を対象とした第2相臨床試験で単剤療法として評価されています。2022年9月14日のデータカットオフの時点で、合計43人の患者が登録され、投与されました。アゼノセルチブの耐容性は良好でした。最も一般的な治療関連の有害事象(AE)は、吐き気(全グレード60.5%/グレード3以上9.3%)、疲労(全グレード46.5%/グレード3以上9.3%)、下痢(全グレード37.2%/グレード3以上7.0%)、嘔吐(全グレード32.6%/グレード3以上7.0%)でした。FDAは2021年11月、進行性または転移性疾患の管理のためにプラチナベースの化学療法を少なくとも1回受けた進行性または転移性USC患者のアゼノセルチブにファストトラック指定を付与しました。この患者集団を対象とした研究デザインは、米国での登録に役立つ可能性があると考えています。この試験のトップラインデータは、2025年の後半に開示される予定です。
•組み合わせ-PROCでのアゼノセルチブと化学療法の第1b相臨床試験(Zn-C3-002)。 アゼノセルチブは現在、PROC患者を対象とした第1b相臨床試験で、4つの別々のコホートでパクリタキセル、カルボプラチン、PLD、およびゲムシタビンのそれぞれと組み合わせて評価されています。2023年5月25日、この第1b相臨床試験の陽性データを発表しました。アゼノセルチブは、複数の種類の化学療法との併用で耐容性が高く、すべての患者で客観的奏効率(ORR)と無増悪生存期間中央値(MPF)という有望な臨床活性を示しましたが、特にサイクリンE1陽性腫瘍の患者は、化学療法による治療後に予後が不良で転帰が比較的悪いことが認められているサブグループです。すべての化学療法併用群で合計115人の患者が研究に登録されました。2023年4月10日の時点で、94件が回答評価可能でした。すべての投与スケジュールで、アゼノセルチブとパクリタキセルのORRが50.0%(MPFは7.4m、mDoRは5.6m)と最も高く、続いてアゼノセルチブ+ゲムシタビンのORRが38.5%(MPFは8.3m、MDoRは6.2m)でした。アゼノセルチブとカルボプラチンのORRは35.7%(MPFは10.4m、MDoRは11.4m)で、アゼノセルチブとPLDのORRは19.4%(MPFは6.3m、MDoRは8.3m)でした。免疫組織化学(IHC)に使用できる組織を持っていた患者のうち、87%がサイクリンE1+(Hスコア> 50)でした。サイクリンE1+の状態は、IHCデータ(ORRが40.0%対8.3%、MPFが9.86か月対3.25か月、HR = 0.37、P = 0.0078)を使用して、反応評価可能な患者集団全体でORRが優れており、MPFが長いことと関連していました。この患者集団では、WEE1阻害と化学療法の潜在的な相乗効果を示しています。すべてのアゼノセルチブ間欠投与群で頻繁にみられたグレード3以上の治療関連有害事象(TRAE)(%)は、血小板減少症(27.5%)、好中球減少症(25.5%)、貧血(15.7%)、および疲労(9.8%)でした。
•単剤療法-固形腫瘍(Zn-C3-001)を対象とした第1b相用量決定臨床試験。現在、固形腫瘍の治療のための第1b相用量決定臨床試験で、単剤療法としてアゼノセルチブを評価しています。2023年6月6日、この臨床試験の陽性データを発表しました。2023年4月24日現在、高度に前治療された進行性固形腫瘍の患者が登録され、単剤療法を受けました。
毎日の連続投与または断続的な週次投与スケジュールのいずれかで300 mg以上の用量のアゼノセルチブ。すべての腫瘍タイプで、74人の患者が連続投与スケジュールでアゼノセルチブを投与され、53人の患者が断続的な投与スケジュールでアゼノセルチブを投与されました。臨床的に意味のある同等の用量レベルで連続投与と断続的を比較した場合、データは次のとおりです。断続的投与は、連続投与と比較して、アゼノセルチブの安全性が維持され、耐容性が向上しました。胃腸、疲労、血液学のグレード3と4のTRAEは、連続投与と同等か良好でした。断続的なコホートでは、TRAEによる中止は見られませんでした。AUCで測定した定常状態の暴露0-24、連続投与で1日300 mgで観察されたAUCと比較して、400 mgの断続的投与で5日間オン、2日間オフで2倍以上になり、断続的な投与は連続投与と比較してより高い最大濃度レベルを達成しました。2023年6月2日の時点で、断続的投与で治療された患者で確認されたORRは36.8%(19年7月)でしたが、連続投与を受けた患者では19.2%(5/26)でした。アゼノセルチブを間欠投与された反応評価可能な患者では、確認されたORRはUSCで50%、卵巣癌で30.8%でした。断続的な投与スケジュールを受けた卵巣がんおよびUSC患者の89%は、ベースラインスキャンから標的病変が減少しました。断続的な投薬スケジュールを受けたこのサブグループの患者のフォローアップの中央値は4.4か月で、2023年6月2日の時点で63%(19年12月)の患者が治療を続けていました。2023年11月6日、この試験の更新データを発表しました。2023年6月2日に奏効が評価可能だった同じ患者集団(卵巣がんとUSC患者)を対象とした2023年10月25日のデータによると、これらの患者では引き続き36.8%(19年7月)のORRが続いていました。2023年6月2日のデータと比較すると、このサブグループの患者のフォローアップ期間の中央値は9.2か月に増加し、MPFは6.5か月に増加しました。2023年9月27日現在、アゼノセルチブは、安全性が評価可能な患者を追加し、フォローアップを長期化することで、良好な安全性と忍容性を示し続けています。この試験の最終結果は、2024年の後半に開示される予定です。
•併用-再発または難治性骨肉腫(Zn-C3-003)におけるアゼノセルチブと化学療法の第1相臨床試験。 R/R骨肉腫の成人および小児患者を対象に、ゲムシタビンと併用したアゼノセルチブの第1相臨床試験の用量漸増部分を完了しました。この患者集団において、ゲムシタビンと組み合わせたアゼノセルチブの第2相用量の推奨案を特定し、臨床的に有意義な活性を示しました。研究者主導の試験では、アゼノセルチブとゲムシタビンの併用が引き続き骨肉腫で評価されることを期待しています。骨肉腫のアゼノセルチブについて、FDAから希少疾病用医薬品指定と希少小児疾患指定を受けました。この試験の最終結果は、2024年の前半に開示される予定です。
•組み合わせ-BRAF V600E変異転移性大腸がん(mCRC)(Zn-C3-016)におけるアゼノセルチブとエンコラフェニブおよびセツキシマブ(BEACONレジメン)の第1/2相臨床試験。 私たちはファイザーと協力して、第1/2相臨床試験でBRAF V600E変異mCRCの患者を対象に、アゼノセルチブをエンコラフェニブおよびセツキシマブ(BEACONレジメンとして知られるFDA承認の標準治療)と組み合わせて評価しています。前臨床試験では、WEE1阻害は突然変異による癌の多くの標的薬との相乗効果を示しており、BEACONレジメンにアゼノセルチブを追加すると、細胞株由来の異種移植モデルにおける抗腫瘍活性が増強されました。私たちは2023年の第1四半期にこの臨床試験への登録を開始し、2024年の後半にこの試験の初期データを開示する予定です。
•組み合わせ-膵臓がんにおけるアゼノセルチブと化学療法の第1/2相臨床試験。 また、ダナ・ファーバーが後援する、プラチナ耐性膵がん患者を対象としたアゼノセルチブと化学療法(ゲムシタビン)を評価する第1/2相臨床試験を支援することにも同意しました。
•組み合わせ — トリプルネガティブ乳がん(TNBC)を対象とした、アゼノセルチブ、化学療法、ペムブロリズマブの第1/2相臨床試験。また、ダナ・ファーバーが後援する、TNBC患者を対象としたアゼノセルチブ、化学療法(カルボプラチン)、ペムブロリズマブを評価する第1/2相臨床試験を支援することにも同意しました。
Zn-D5(BCL-2 阻害剤)
Zn-D5は、BCL-2のクラス最高の選択的経口小分子阻害剤である可能性があります。BCL-2は、アポトーシスと呼ばれる細胞死の調節に重要な役割を果たすタンパク質です。BCL-2の過剰発現は、多くの種類の癌で頻繁に検出され、癌細胞のアポトーシスを防ぎます。医薬品化学の専門知識を活用して、クラス最高の効能、選択性、PK特性を持つZn-d5を設計しました。Zn-D5は現在、アゼノセルチブと併用したR/R AML患者を対象に診療所で評価されています。
次の試験はZn-d5臨床開発プログラムの一部です:
•単剤療法-再発または難治性軽鎖アミロイドーシス(R/R ALアミロイドーシス)の第1相臨床試験(Zn-D5-003)。 R/R ALアミロイドーシスの単剤療法としてのZn-d5の第1相臨床試験の用量漸増部分を完了しました。R/R ALアミロイドーシス患者では、1日400 mg以上のZn-D5で治療された患者の血液学的反応率が40%で、予備的な有効性シグナルが観察されました。Zn-D5はRAEがほとんどなく、耐容性が良好でした。提案された単剤療法の用量は、1日800 mgと確認されています。アゼノセルチブ+Zn-d5の組み合わせなど、アゼノセルチブのフランチャイズの機会にリソースを集中させるために、この適応症向けにZn-d5をさらに開発する予定はありません。
•単剤療法-非ホジキンリンパ腫(NHL)の第1相臨床試験(Zn-D5-001)。 私たちは、NHL患者の単剤療法としてZn-d5を評価する第1相用量漸増試験の主要安全性とPKエンドポイントを満たし、現在この試験を終了しています。アゼノセルチブ+Zn-d5の組み合わせなど、アゼノセルチブのフランチャイズの機会にリソースを集中させるために、この適応症向けにZn-d5をさらに開発する予定はありません。
•併用-再発または難治性急性骨髄性白血病(R/R AML)におけるZn-D5とアゼノセルチブの第1/2相臨床試験(Zn-D5-004c)。 Zn-D5は、R/R AML患者を対象とした第1/2相用量漸増臨床試験で、アゼノセルチブと併用して評価されています。この試験の第1相部分では、両方の薬剤の用量を増やして組み合わせの用量を特定します。これは、第2相拡大コホートで評価されます。この研究には、最大約100人の患者が登録される予定です。この臨床試験は、Zn-d5とアゼノセルチブの組み合わせが、これらの製品候補のいずれかを単剤として示した活性と比較して、R/R AMLを含むいくつかの適応症の活性が大幅に向上することを示した前臨床モデルによって裏付けられています。前臨床モデルでは、Zn-D5とアゼノセルチブの併用がマウスで良好な耐容性を持つことも示されました。私たちは、WEE1阻害剤とBCL-2阻害剤の両方を臨床開発している唯一の企業だと考えています。この試験の初期データは、2024年の後半に開示される予定です。
流動性の概要
創業以来、私たちの事業は、会社の組織と人員配置、事業計画、資金調達、知的財産ポートフォリオの確立、製品パイプラインの研究開発に限定されてきました。商業販売が承認された製品はありません。また、製品の販売による収益も得られていません。臨床開発が成功し、1つ以上の製品候補について規制当局の承認を得て商品化しない限り、製品の販売から収益を上げることはありません。継続的な事業を支援し、成長戦略を追求するために、多額の追加資本を調達する必要があります。
創業以来、私たちは多額の営業損失を被っています。2022年12月31日に終了した年度の純損失は2億3,710万ドル、2023年9月30日と2022年9月30日に終了した9か月間の純損失はそれぞれ2億3,140万ドルと1億8,210万ドルでした。2023年9月30日の時点で、累積赤字は8億2,760万ドルでした。当面の間、多額の費用と営業損失が続くと予想しています。2023年9月30日現在、現金、現金同等物、有価証券は5億1,660万ドルでした。2023年9月30日現在の現金、現金同等物、および有価証券は、2026年までの営業費用と資本支出要件を賄うのに十分であると考えています。これらの見積もりは、不正確であることが判明する可能性のある仮定に基づいており、利用可能な資本資源を予想よりも早く活用することができました。
ライセンス契約と戦略的コラボレーション
レクリウムIPホールディングス、合同会社ライセンス契約
2014年12月、当社の完全子会社であるゼノファーマシューティカルズ社は、Recurium IP Holdings、LLC(「Recurium IP」)とライセンス契約(「リキュリウム契約」)を締結しました。この契約は、その後修正され、ゼノファーマシューティカルズ社は、治療用医薬品の開発と商品化を目的として、Recurium IPが所有または管理する特定の知的財産権に対する独占的な世界的ライセンスを付与されました。または痛みを和らげる以外の病気の予防。2022年12月31日に終了した年度のフォーム10-Kの年次報告書で開示された特定の企業再編の後、当社の完全子会社であるゼノ・マネジメント株式会社(「ZMI」)は、リキュリウム契約のゼンタリス契約当事者になりました。Recurium契約に基づいてZMIが独占的にライセンスしている知的財産権には、アゼノセルチブとZn-D5を対象とする特定の知的財産が含まれます。ZMIは、特定の条件のもと、Recurium契約に基づく権利をサブライセンスする権利を有します。ZMIは、10種類の特定の生物学的標的のうちの1つを調節する化合物を含む、または含む少なくとも1つの製品を開発して商品化し、特定の開発活動を実行するために、商業的に合理的な努力を払う必要があります。
リキュリウム契約の条件に基づき、ZMIは、アゼノセルチブやZn-D5など、10種類の特定の生物学的標的のいずれかを調節する化合物を含む、または含む製品に関して、開発および規制上のマイルストーン支払い、純売上高に対するロイヤルティの支払い、および特定のサブライセンス支払いを行う義務があります。ZMIは、そのようなライセンス製品ごとに最大4,450万ドルの開発および規制上のマイルストーン支払いを行う義務があります。さらに、ZMIは、動物に使用される特定のライセンス製品に対して、最大15万ドルのマイルストーン支払いを行う義務があります。ZMIはまた、そのようなライセンス製品の販売に対して、一桁台半ばから高い割合でロイヤルティを支払う義務があります。さらに、ZMIがRecurium契約に基づいて独占的にライセンスされている特定の特許に基づく権利をサブライセンスまたは第三者に譲渡することを選択した場合、ZMIはそのような取引に関連して受け取った特定のサブライセンス収入の20%をRecurium IPに支払う必要があります。
Recurium契約は、2032年12月21日の遅くに失効します。また、国別では、その国でライセンスされているすべての製品の最終期限が切れるロイヤリティ期間の満了日に失効します。ただし、いずれかの当事者が原因または破産事由により早期に解約した場合を除きます。
ファイザー開発契約
2022年4月、私たちはファイザーと開発契約を締結し、協力してアゼノセルチブの臨床開発を進めました。私たちは、アゼノセルチブやその他のパイプラインの経済的所有権や管理権をファイザーに付与しませんでした。2022年10月、私たちはファイザーとの最初の臨床開発協力を発表しました。これは、BRAF V600E変異mCRC患者を対象に、エンコラフェニブとセツキシマブ(ビーコンレジメンとして知られるFDA承認の標準治療)と組み合わせたアゼノセルチブの第1/2相投与漸増試験を開始することです。
グラクソ・スミスクラインの臨床試験協力および供給契約
2021年4月、私たちはGSKと臨床試験協力および供給契約を締結しました。この契約に基づき、プラチナ耐性卵巣がん患者を対象に、アゼノセルチブとGSKのポリ(ADPリボース)ポリメラーゼ(PARP)阻害剤であるニラパリブの組み合わせを評価しています。この契約に基づき、当社は、四半期ごとに開催される当社の代表者とGSKの代表者で構成される共同開発委員会の監督下で、関連する研究の実施と費用を負担します。GSKは、共同研究で使用するニラパリブを無料で提供しています。研究終了時に、臨床データやその他のレポートをGSKに提供する必要があります。
この契約は、将来の臨床試験に参加するための先行交渉権を与えるものではありません。また、いずれの当事者も、単剤療法として、または他の製品または化合物と組み合わせて、あらゆる治療分野における他の臨床試験でそれぞれの化合物を評価する追加の権利または能力を相手方に付与していません。
GSKとの契約は、それに基づく両当事者のすべての義務の完了時、またはいずれかの当事者による終了時に失効します。私たちとGSKはそれぞれ、相手方による重大な違反があった場合、契約を終了する権利があります。さらに、安全上の理由から、またはどちらかの当事者が医療、科学、法律、その他の理由で独自の化合物の開発を中止することを決定した場合、または規制当局がその当事者が研究用の化合物を供給することを妨げる措置を講じた場合、または相手方が特定の破産、破産、または同様の状況にさらされた場合、契約はどちらかの当事者によって終了される可能性があります。また、GSKは、ニラパリブが安全でない方法で使用されていると合理的かつ誠実に信じており、そのような問題に対処するための変更を取り入れず、適切な当事者に昇格しても問題を解決できないことを書面で通知した場合、本契約を終了する権利を有します。
ゼンテラ・セラピューティクス
2023年6月15日、当社と一部の完全子会社が、Zentera Therapeutics(Zentera)とのコラボレーションおよびライセンス契約を終了する契約を締結したことを発表しました。これにより、そのような完全子会社は、製品候補であるアゼノセルチブ、Zn-D5、Zn-C5に対する特定の開発権および商品化権をZenteraに付与しました。中華民国、マカオ、香港、台湾、または中華圏。これらの契約が終了した結果、私たちは中華圏でZenteraからアゼノセルチブ、Zn-D5、Zn-C5の権利を取り戻し、現在、これらの資産の世界的な開発権と商品化権を保有しています。
上記に関連して、私たちはZenteraに一定の対価を支払うことに同意しました。これには、3,000万ドルの前払い、マイルストーン支払い、中華圏におけるアゼノセルチブ、Zn-D5、Zn-C5の純売上高に対する一桁台前半のロイヤルティが含まれます。さらに、最低限の対価として、ゼンテラの40.3%の株式をゼンテラに売却しました。.
当社の経営成績の構成要素
収益
これまでのところ、私たちは収益を上げていません。また、近い将来、製品の販売から収益を生み出す見込みはありません。私たちは、コラボレーション契約に基づいて受け取った支払いから収益を生み出しており、将来的には収益を生み出す可能性があります。これには、前払い金、ライセンス料、マイルストーンベースの支払い、研究開発費の払い戻しが含まれます。
研究開発費
研究開発費は、主に、発見活動や製品候補の開発などの研究活動にかかる費用で構成され、以下が含まれます。
•研究開発業務に従事する人員の給与、福利厚生、その他の関連費用(株式ベースの報酬費用を含む)。
•委託研究機関(CRO)、および当社に代わって研究、前臨床活動、臨床試験を実施するその他の第三者、ならびに当社の前臨床試験および臨床試験に使用する医薬品を製造する受託製造機関(CMO)を含む第三者との契約に基づいて発生した費用。
•外部コンサルタントの費用(手数料、株式ベースの報酬、関連する旅費を含む)
•実験用品および前臨床試験および臨床試験材料の取得、開発、製造の費用。
•研究開発活動に使用される知的財産に対して行われたライセンス支払い。そして
•施設の賃貸と維持費、およびその他の運営費に割り当てられた費用。
研究開発費は、発生したとおりに費用をかけます。特定の協力協定の下で払い戻された研究開発費は、研究開発費の減額として計上され、関連費用が発生した期間に計上されます。
外部開発費を製品候補または開発プログラムごとに追跡していますが、人件費、ライセンス契約に基づいて行われる一般的なライセンス支払い、またはその他の内部費用を特定の開発プログラムまたは製品候補に割り当てることはありません。これらの費用は、以下の表の未配分の研究開発費に含まれています。
次の表は、製品候補または開発プログラム別の研究開発費をまとめたものです。
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | 9月30日に終了した3か月間 | | 9月30日に終了した9か月間 | |
| | 2023 | 2022 | | 2023 | 2022 | | | |
| アゼノセルチブ | $ | 18,941 | | $ | 12,333 | | $ | 45,728 | | $ | 36,480 | | | | | | | |
| 亜鉛-d5 | | 3,150 | | | 5,694 | | | 13,648 | | | 7,145 | | | | | | | |
| 未配分の研究開発費 | | 24,674 | | | 24,154 | | | 78,657 | | | 88,493 | | | | | | | |
| 研究開発費の合計 | $ | 46,765 | | $ | 42,181 | | $ | 138,033 | $ | 132,118 | | | | | | | |
研究開発活動は私たちのビジネスモデルの中心です。臨床開発の後期段階にある製品候補は、一般的に臨床開発の初期段階の製品よりも開発コストが高くなります。これは主に、後期段階の臨床試験の規模と期間が増加するためです。当面の間、研究開発費は大幅に増加し続け、進行中の臨床試験を完了し、新しい臨床試験を開始し、追加の製品候補の発見と開発を継続し、臨床開発を無事に完了した製品候補の規制当局への提出書類の準備を進めるにつれて、総経費の大部分を占めると予想しています。
私たちの製品候補の開発が成功するかどうかは非常に不確実です。現時点では、製品候補または当社が開発する可能性のあるその他の製品候補の既存および将来の臨床試験の期間と費用、また、販売承認を得た製品候補の商品化と販売から収益を生み出すかどうか、いつ、どの程度収益を生み出すかを確実に判断することはできません。どの製品候補についても、販売承認を得ることは決してできないかもしれません。当社の製品候補および将来開発する可能性のあるその他の製品候補の臨床試験と開発の期間、費用、時期は、次のようなさまざまな要因によって異なります。
•患者1人あたりの試験費用。
•各試験に登録した患者の数。
•承認に必要な試験の数。
•試験に参加したサイトの数。
•試験が実施されている国。
•対象となる患者を登録するのに必要な期間。
•患者の中退率または中止率
•世界的なマクロ経済環境の結果などによる臨床試験の遅れ。
•規制当局から要請された追加の安全監視の可能性
•治験とフォローアップへの患者の参加期間。
•製品候補の開発段階。
•製品候補の有効性と安全性プロファイル。
•臨床試験デザインと患者登録率の不確実性。
•安全性と有効性、初期の臨床データ、競争、製造能力、商業的実行可能性を含む、当社の製品候補の実際の成功確率。
•重要かつ変化する政府の規制と規制ガイダンス。
•マーケティング承認のタイミングと受領。
•特許請求およびその他の知的財産権の申請、起訴、弁護、執行にかかる費用。そして
•熟練した人材を引き付けて維持する私たちの能力。
製品候補の開発に関してこれらの変数のいずれかの結果が変化すると、その製品候補の開発に関連するコストとタイミングが大幅に変化する可能性があります。たとえば、FDAや他の規制当局が、製品候補の臨床開発の完了に必要になると予想される以上の臨床試験の実施を要求した場合、または患者の登録やその他の理由で臨床試験が大幅に遅れた場合、臨床開発の完了に多額の追加財源と時間を費やす必要があります。
一般管理費
一般管理費は、主に、役員、財務、事業開発、管理職の人員に対する給与およびその他の関連費用(株式ベースの報酬を含む)で構成されています。一般管理費には、知的財産および企業問題に関連する弁護士費用、会計、監査、税務、コンサルティングサービスの専門家費用、保険費用、旅費、施設関連の費用(直接減価償却費、施設の賃貸と維持のための割り当て費用、およびその他の運営費を含む)も含まれます。
今後、臨床段階のプログラムや、開発する可能性のあるその他の製品候補に関連する研究開発活動を支援するために人員を増やすにつれて、一般管理費が増加すると予想しています。また、特に新興成長企業ではなくなった今、上場企業であることに伴う費用の増加も予想されます。これには、ナスダックとSECの要件への準拠を維持するための会計、監査、法律、規制、税務関連サービスの費用、取締役および役員の保険費用、投資家および広報費用が含まれます。
利息収入
利息収入は、現金、現金同等物、および売却可能な有価証券で得られる利息で構成されます。
所得税
創業以来、当社と当社の子会社は、それぞれの繰越期間内にこれらの税属性を利用することの不確実性のため、純税制上の優遇措置を計上していない特定の法域で、連邦、州、および外国の累積純営業損失を計上してきました。
業務結果
2023年9月30日に終了した3か月と2022年9月30日に終了した3か月との比較
次の表は、示された期間の当社の経営成績と、それらの項目の変化をドルでまとめたものです。
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | 9月30日に終了した3か月間 | | 増加
(減少) |
| | 2023 | | 2022 |
| | (千単位) |
| 営業経費 | | | | | | | | |
| 研究開発 | $ | 46,765 | | | $ | 42,181 | | | $ | 4,584 | |
| | | | | | | | |
| 一般と管理 | | 15,953 | | | | 12,012 | | | | 3,941 | |
| 営業費用の合計 | | 62,718 | | | | 54,193 | | | | 8,525 | |
| 事業による損失 | | (62,718) | | | | (54,193) | | | | (8,525) | |
| | | | | | | | |
| 投資およびその他の収入、純額 | | 7,209 | | | | 1,905 | | | | 5,304 | |
| 税引前純損失 | | (55,509) | | | | (52,288) | | | | (3,221) | |
| 所得税(給付)費用 | | 31 | | | | (159) | | | | 190 | |
| 持分法投資による損失 | | — | | | | 2,371 | | | | (2,371) | |
| 純損失 | | (55,540) | | | | (54,500) | | | | (1,040) | |
| 非支配株主に帰属する純損失 | | (12) | | | | (99) | | | | 87 | |
| ゼンタリスに帰属する純損失 | $ | (55,528) | | | $ | (54,401) | | | $ | (1,127) | |
研究開発費
2023年9月30日に終了した3か月間の研究開発(研究開発)費用は、2022年9月30日に終了した3か月間の4,220万ドルに対し、4,680万ドルでした。460万ドルの増加は主に、前期にZenteraと共有した320万ドルの費用、人件費に関連する260万ドルの増加、そのうち140万ドルは非現金の株式ベースの報酬費用、80万ドルはコンサルティング費用によるものです。これらの増加は、施設費と臨床費がそれぞれ130万ドルと70万ドルの減少によって一部相殺されました。
一般管理費
2023年9月30日に終了した3か月間の一般管理費は1,600万ドルでしたが、2022年9月30日に終了した3か月間は1,200万ドルでした。この400万ドルの増加は、主に人件費の290万ドルの増加によるもので、そのうち220万ドルは非現金の株式ベースの報酬費用に関連し、110万ドルは施設と外部サービスに関連していました。
投資およびその他の収入、純額
2023年9月30日に終了した3か月間の投資およびその他の収益は、2022年9月30日に終了した3か月間の純額190万ドルに対し、720万ドルでした。530万ドルの増加は、主に次の要因によるものです
投資された現金と有価証券の増加と、ニューヨーク事務所の転貸による40万ドルの収入による収益です。
持分法投資損失
2022年9月30日に終了した3か月間の持分法投資の損失は、2022年9月30日に終了した3か月間の240万ドルに対し、2023年9月30日に終了した3か月間は記録しませんでした。2023年6月30日までの3か月間に、私たちは持分法投資を売却しました。
2023年9月30日に終了した9か月と2022年9月30日に終了した9か月間の比較
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | 9月30日に終了した9か月間 | | 増加
(減少) |
| | 2023 | | 2022 |
| | (千単位) |
| 営業経費 | | | | | | | | |
| 研究開発 | $ | 138,033 | | | $ | 132,118 | | | $ | 5,915 | |
| ゼンテラの進行中の研究開発 | | 45,568 | | | | — | | | | 45,568 | |
| 一般と管理 | | 47,986 | | | | 43,415 | | | | 4,571 | |
| 営業費用の合計 | | 231,587 | | | | 175,533 | | | | 56,054 | |
| 事業による損失 | | (231,587) | | | | (175,533) | | | | (56,054) | |
| その他の収入 (費用) | | | | | | | | |
| 投資およびその他の収入、純額 | | 15,769 | | | | 2,755 | | | | 13,014 | |
| | | | | | | | |
| | | | | | | | |
| 税引前純損失 | | (215,818) | | | | (172,778) | | | | (43,040) | |
所得税給付 | | (466) | | | | (109) | | | | (357) | |
| 持分法投資による損失 | | 16,014 | | | | 9,460 | | | | 6,554 | |
| 純損失 | | (231,366) | | | | (182,129) | | | | (49,237) | |
| 非支配株主に帰属する純損失 | | (92) | | | | (294) | | | | 202 | |
| ゼンタリスに帰属する純損失 | $ | (231,274) | | | $ | (181,835) | | | $ | (49,439) | |
研究開発費
2023年9月30日に終了した9か月間の研究開発費は1億3800万ドルでしたが、2022年9月30日に終了した9か月間の研究開発費は1億3,210万ドルでした。590万ドルの増加は主に、2023年6月に終了したZenteraとの費用分担関係に関連する費用の480万ドルの増加、非現金株式ベースの報酬の310万ドルの増加、施設費と諸経費の220万ドルの増加によるものです。
そして、コンサルティング費用の160万ドルの増加。これらの増加は、臨床試験費用と共同研究費がそれぞれ320万ドルと260万ドルの減少によって一部相殺されました。
ゼンテラの進行中の研究開発費
2023年9月30日に終了した9か月間のZenteraの進行中の研究開発費は4,560万ドルでしたが、2022年9月30日に終了した9か月間はゼロでした。この増加は、2023年9月30日に終了した9か月間に、Zenteraとの協力の終了に関連して、進行中の研究開発のためにZenteraに送金された現金および非現金対価の合計4,560万ドルによるものです。
一般管理費
2023年9月30日に終了した9か月間の一般管理費は4,800万ドルでしたが、2022年9月30日に終了した9か月間は4,340万ドルでした。この460万ドルの増加は、主にオペレーティングリースの減損費用500万ドルと人件費に関連する80万ドルによるものです。これは、設備費と諸経費の120万ドルの削減によって部分的に相殺されました。
投資およびその他の収入、純額
2023年9月30日に終了した9か月間の投資およびその他の収益は、2022年9月30日に終了した9か月間の純額280万ドルに対し、1,580万ドルでした。1,300万ドルの増加は、主に投資された現金と有価証券の増加による収益と、ニューヨークオフィスの転貸による60万ドルの収益によるものです。
所得税給付
2023年9月30日と2022年9月30日に終了した9か月間で、それぞれ50万ドルと10万ドルの所得税控除を記録しました。Zenteraとの関係の終了により、持分法投資に関連する繰延税金負債を償却し、その結果、2023年9月30日に終了した9か月間の純税制上の優遇措置が得られました。
持分法投資損失
2023年9月30日に終了した9か月間の持分法投資の損失は、2022年9月30日に終了した9か月間の950万ドルに対し、1,600万ドルでした。650万ドルの増加は、Zenteraとの提携の終了に関連して、Zentalisに知的財産が返還された後に評価されたZenteraに返還された株式の公正価値を反映して記録された1回限りの請求によるものです。
流動性と資本資源
創業以来、私たちの事業は、会社の組織と人員配置、事業計画、資金調達、知的財産ポートフォリオの確立、製品パイプラインの研究開発に限定されてきました。商業販売が承認された製品はなく、製品の販売による収益も得られておらず、重大な営業損失を被りました。研究プログラムと製品候補の前臨床開発と臨床開発を進める中で、当面の間、多額の費用と営業損失が発生すると予想しています。現在および将来の研究プログラムと製品候補に関する追加の前臨床研究と臨床試験の実施、前臨床研究と臨床試験を支援するためのCMOとの契約、知的財産ポートフォリオの拡大、および事業に対する一般管理サポートの提供に関連して、研究開発および一般管理費が増加すると予想しています。
その結果、継続的な事業を支援し、成長戦略を追求するために、多額の追加資本を調達する必要があります。製品の販売から大きな収益を生み出すことができるようになるまでは、株式の売却、負債融資、またはその他の資本源を通じて事業資金を調達する予定です。これには、他社との協力やその他の戦略的取引が含まれる場合があります。私たちがそうなるという保証はありません
特に世界的なマクロ経済環境とインフレ率と金利の上昇を考慮して、私たちに受け入れられる条件で、またはまったく受け入れられる条件で、事業資金を調達するために必要な水準の資金を調達することに成功しました。必要に応じて十分な追加資金を確保できない場合、1つ以上の製品候補の開発と商品化を大幅に遅らせたり、縮小したり、中止したり、潜在的なライセンスや買収の追求を遅らせたりする必要があります。
治療薬の開発と商品化には数多くのリスクと不確実性が伴うため、費用の増加のタイミングや金額、いつまたは収益性を達成または維持できるかを予測することはできません。製品の販売で収益を上げることができたとしても、黒字にならない可能性があります。収益を上げられなかったり、継続的に収益性を維持できなくなったりすると、計画したレベルで事業を継続できず、事業の縮小または中止を余儀なくされる可能性があります。
現在、承認された製品はなく、製品の販売から収益を得たこともありません。これまで、私たちは主に株式の売却を通じて事業資金を調達してきました。設立から2023年9月30日まで、私たちは普通株式とシリーズA、B、Cの転換優先ユニットの売却から総収入として合計12億ドルを調達しました。これには、2023年6月の追加募集による総収入2億5,000万ドルが含まれます。2023年9月30日の時点で、現金、現金同等物、有価証券は5億1,660万ドル、累積赤字は8億2,760万ドルでした。私たちは、現金および現金同等物の大部分を主要な金融機関の口座に保管しており、これらの機関での預金は保険限度を超えています。市場の状況は、これらの機関の存続可能性に影響を与える可能性があります。現金および現金同等物を保有している金融機関のいずれかに障害が発生した場合、無保険の資金にタイムリーに、またはまったくアクセスできないという保証はありません。これらの資金にアクセスできない、またはアクセスが遅れると、当社の事業と財政状態に悪影響を及ぼす可能性があります。2023年9月30日の時点で、私たちは借金を抱えていません。
ATMプログラム
2021年5月、私たちはLeerink Partners LLCを販売代理店として売買契約または売買契約を締結しました。この契約に基づき、米国証券取引所に提出されたフォームS-3(ファイル番号333-255769)の登録届出書に基づいて、総額最大2億ドルの「市場」オファリング、またはATMで普通株を発行および売却することができます。欧州委員会、またはSEC、または2021年5月4日。普通株式の売却は、売買契約に基づき、改正された1933年の証券法または証券法の規則415(a)で定義されている「市場での募集」と見なされる販売で行うことができます。これには、ナスダックグローバルマーケットまたはその他の既存の普通株式取引市場を通じて直接行われた販売が含まれます。2023年9月30日に終了した四半期中、売買契約に基づいて普通株式を売却しませんでした。2023年9月30日の時点で、売買契約に基づいて売却可能な普通株式は1億4030万ドル残っていました。
キャッシュフロー
次の表は、提示された期間における当社の現金の出所と用途をまとめたものです。
| | | | | | | | | | | | | | | | | |
| | 9月30日に終了した9か月間 |
| | 2023 | | 2022 |
| | (千単位) |
| 営業活動に使用された純現金 | $ | (167,223) | | | $ | (126,422) | |
投資活動に使用された純現金 | | (2,398) | | | | (102,286) | |
| 財務活動による純現金 | | 237,303 | | | | 211,302 | |
| 現金および現金同等物の純増額(減少) | $ | 67,682 | | | $ | (17,406) | |
営業活動
創業以来、私たちは損失を被っています。2023年9月30日に終了した9か月間の営業活動に使用された純現金は1億6,720万ドルでした。これは主に、製品候補の研究開発活動に関連する費用と一般管理費が発生した2億3,140万ドルの純損失と、営業資産と負債の変動440万ドルで、6,860万ドルの非現金調整によって一部相殺されました。
2022年9月30日に終了した9か月間の営業活動に使用された純現金は1億2,640万ドルで、主に製品候補の研究開発活動に関連する費用と一般管理費による純損失1億8,210万ドルで、4,640万ドルの非現金調整と930万ドルの営業資産および負債の変動によって相殺されました。
投資活動
2023年9月30日に終了した9か月間の投資活動に使用された純現金は240万ドルでしたが、4億5,120万ドルの有価証券の満期からの収益が、4億5,320万ドルの超過現金の純投資と40万ドルの不動産および設備の購入によって相殺されました。
2022年9月30日に終了した9か月間の投資活動に使用された純現金は1億230万ドルでしたが、有価証券の満期からの収益が3億730万ドルで、4億720万ドルの超過現金の純投資と240万ドルの不動産および設備の購入によって相殺されました。
資金調達活動
2023年9月30日に終了した9か月間の資金調達活動によって提供された純現金は2億3,730万ドルで、主に2023年6月の追加募集に関するもので、純現金は2億3,570万ドルでした。2023年9月30日に終了した9か月間に、株式インセンティブプランに基づく普通株式の発行により、さらに160万ドルが提供されました。
2022年9月30日に終了した9か月間の資金調達活動によって提供された純現金は2億1,130万ドルで、主に1億8,880万ドルの純現金を提供した2022年5月の追加募集と、2,050万ドルの純現金を提供したファイザーへの2022年4月の直接募集に関するものです。ファイザーから受け取った総収入2,500万ドルのうち、420万ドルの収益は、投資日の当社の普通株式の公正価値を超えるプレミアムでした。この金額は、未監査の要約連結貸借対照表に未払研究開発費として計上されており、コラボレーション期間中の研究開発費の削減として認識されます。2022年9月30日に終了した9か月間に、株式インセンティブプランに基づく普通株式の発行により、さらに200万ドルが提供されました。
資金要件
当社の営業費用は2022年に増加し、継続的な活動に関連して将来も大幅に増加すると予想されています。
具体的には、次の場合に経費が増加します。
•腫瘍適応症の治療のためのアゼノセルチブとZn-D5の臨床開発を進めます。
•現在および将来の他の研究プログラムや製品候補、および該当する場合は、当社の製品候補と将来の製品候補に関連するバイオマーカーの診断ツールの前臨床および臨床開発を追求します。
•他の製品、製品候補、または技術のライセンス供与または権利の取得。
•当社の知的財産ポートフォリオの維持、拡大、保護
•研究、製造、規制、臨床開発などの追加の人員、および管理担当者を雇います。
•すべての製品候補について規制当局の承認を求め、必要に応じて、臨床開発を無事に完了できるような製品候補に関連するバイオマーカーの診断ツールを求めます。そして
•運営、財務、管理システムを拡大し、公開企業としての運営をサポートする人員を含む人員を増やします。
2023年9月30日現在の現金、現金同等物、および有価証券は、2026年までの営業費用と資本支出要件を賄うのに十分であると考えています。これらの見積もりは、不正確であることが判明する可能性のある仮定に基づいており、利用可能な資本資源を予想よりも早く活用することができました。
医薬品の研究、開発、商品化には多くのリスクと不確実性が伴うため、必要な運転資本額を確実に見積もることは困難です。私たちの将来の資金要件は、次のような多くの要因に左右されます。
•アゼノセルチブとZn-d5プログラムの臨床試験の進捗状況、費用、結果。
•今後開始する他の研究プログラムにおける追加研究と前臨床研究、および必要に応じて、当社の製品候補と将来の製品候補に関連するバイオマーカーの診断ツールの進捗状況、費用、結果。
•製品候補やその他のプログラムを前臨床開発や臨床開発を進める際の、プロセス開発や製造のスケールアップ活動の費用とタイミング。
•戦略的協力、ライセンスまたはその他の契約を確立および維持する当社の能力、およびそのような契約の金銭的条件。
•他の製品、製品候補、または技術のライセンス供与または権利を取得する範囲
•特許出願の準備、申請、審理、知的財産権の維持と保護、知的財産関連の請求に対する弁護にかかる費用と時期。そして
•熟練した人材を引き付けて維持する私たちの能力。
さらに、当社の業績は将来変化する可能性があり、そのような事業計画に関連する運営上のニーズと資本要件を満たすために追加の資金が必要になる場合があります。
製品の販売から大きな収益を生み出すことができるようになるまでは、株式の売却、負債融資、またはその他の資本源を通じて事業資金を調達する予定です。これには、他社との協力やその他の戦略的取引が含まれる場合があります。
現在、クレジットファシリティや確約された資金源はありません。株式または転換社債の売却を通じて追加資本を調達する限り、お客様の所有権は希薄化され、これらの証券の条件には、普通株主としての権利に悪影響を及ぼす清算またはその他の優遇措置が含まれる場合があります。デット・ファイナンスと優先株式融資(可能な場合)には、追加の負債の発生、資本調達など、特定の行動を取る能力を制限または制限する契約が含まれる場合があります
支出または配当の申告。他の第三者の資金調達、協力契約、戦略的提携、ライセンス契約、またはマーケティングと流通の取り決めを通じて追加の資金を調達した場合、技術、将来の収益源、研究プログラム、または製品候補に対する貴重な権利を放棄するか、私たちにとって不利な条件でライセンスを付与しなければならない場合があります。必要に応じてエクイティファイナンスまたはデットファイナンスで追加の資金を調達できない場合、製品開発または将来の商品化努力を延期、制限、削減、または終了するか、そうでなければ自社で開発して販売したい製品や製品候補を開発および販売する権利を付与する必要があります。
重要な会計上の見積もり
以下は、2022年12月31日に終了した会計年度のフォーム10-Kの年次報告書の「経営陣による財政状態と経営成績に関する議論と分析」というタイトルのセクションの「重要な会計上の見積もり」で報告された、当社の重要な会計上の見積もりの更新を示しています。
| | | | | | | | | | | | | | |
| | | | |
| 資産買収 | | |
| 方法論 | | 判断と不確実性 | | 実際の結果が仮定と異なる場合の効果 |
| 私たちは、企業結合とは見なされない資産の取得を、その資産または資産グループの取得費用(取引費用を含む)に基づいて測定し、認識します。資産の取得では、将来の代替手段がない状態で進行中の研究開発を取得するために割り当てられた費用は、取得日の費用として認識されます。 | | 進行中の研究開発のために交換される対価には、公正価値を簡単に決定できない資産が含まれる場合があります。私たちは、受け入れられているさまざまな評価モデルと手法を使用して、簡単に決定できる公正価値なしに対価の公正価値を見積もります。 | | 私たちは、受け入れられた評価モデルで裏付け可能な見積もりを使用して評価を行います。見積もりが正確でない場合、買収した資産価値を過小評価または誇張する可能性があります。
|
オフバランスシートアレンジメント
貸借対照表外の取り決めは締結していません。
アイテム 3.市場リスクに関する定量的・質的開示
該当しません。
アイテム 4.統制と手順。
統制と手続きの有効性に関する固有の制限
当社の開示管理と手続きを設計、評価するにあたり、経営陣は、どんなにうまく設計され運用されても、望ましい管理目標の達成を合理的に保証することしかできないと認識しています。さらに、情報開示の管理と手続きの設計には、リソースに制約があり、経営陣は可能な管理や手続きのメリットをその費用に対して評価する際に判断を下す必要があるという事実を反映していなければなりません。
開示管理と手続きの評価
当社の経営陣は、当社の最高執行責任者と最高財務責任者の参加を得て、フォーム10-Qのこの四半期報告書の対象期間の終わりに、当社の開示管理と手続き(取引法の規則13a-15(e)および15d-15(e)で定義されている)の有効性を評価しました。その評価に基づいて、当社の最高執行責任者と最高財務責任者は、2023年9月30日現在、当社の開示管理と手続きは妥当な保証レベルで有効であると結論付けました。
財務報告に関する内部統制の変更
2023年9月30日に終了した四半期中に発生した取引法の規則13a-15(d)または15d-15(d)に基づく経営陣の評価で特定された、財務報告に対する内部統制(取引法の規則13a-15(f)および15d-15(f)で定義されているとおり)に、財務報告に対する内部統制に重大な影響を及ぼした、または重大な影響を与える可能性が合理的に高い変更はありませんでした。
パート II-その他の情報
アイテム 1.法的手続き。
私たちは重要な法的手続きの対象にはなりません。
アイテム 1A.リスク要因。
当社の普通株式に投資するかどうかを決定する前に、以下に記載されているリスクと不確実性、およびフォーム10-Qのこの四半期報告書の他の情報を慎重に検討する必要があります。これには、フォーム10-Qのこの四半期報告書および「経営陣による財政状態と経営成績に関する議論と分析」というタイトルのセクションの他の部分に記載されている当社の中間未監査の要約連結財務諸表および関連注記が含まれます。これらのリスクのいずれかが発生した場合、当社の事業、財政状態、経営成績、または見通しに重大かつ悪影響が及ぶ可能性があり、その結果、当社の普通株式の市場価格が下落し、投資の全部または一部を失う可能性があります。フォーム10-Qのこの四半期報告書には、リスクと不確実性を伴う将来の見通しに関する記述も含まれています。「将来の見通しに関する記述に関する注意事項」を参照してください。現時点で知られていない、または私たちが現在重要ではないと判断している追加のリスクや不確実性も、当社の事業運営に支障をきたす可能性があります。当社の実際の業績は、以下に示すものを含む特定の重要な要因の結果として、これらの将来の見通しに関する記述で予想されるものと大きく異なる可能性があります。
当社の財政状態と追加資本の必要性に関連するリスク
当社の営業履歴は限られており、商業販売が承認された製品もありません。そのため、現在の事業を評価し、将来の成功と存続可能性を予測することは難しいかもしれません。
私たちは、営業履歴が限られている臨床段階のバイオ医薬品企業であり、当社の事業と展望を評価していただけます。商業販売が承認された製品はありません。また、製品の販売による収益も得られていません。これまで、私たちは、会社の組織と人員配置、事業計画、パートナーシップの実行、資金調達、潜在的な製品候補の発見、特定、開発、関連する知的財産権の確保、進行中のアゼノセルチブとZn-D5の臨床試験を含む製品候補の前臨床研究と臨床試験の実施に、実質的にすべてのリソースと労力を費やしてきました。私たちは、販売承認を得たり、商業規模で製品を製造したり、私たちに代わって第三者に手配したり、製品の商品化を成功させるために必要な販売やマーケティング活動を行う能力をまだ実証していません。その結果、営業履歴が長い場合よりも、将来の成功や存続可能性を正確に予測することが難しいかもしれません。
さらに、急速に進化する分野の臨床段階のバイオ医薬品企業が頻繁に経験する、予期せぬ費用、困難、合併症、遅延、その他の既知および未知の要因やリスクに遭遇する可能性があります。また、研究開発に重点を置いている会社から、商業活動をサポートできる会社に移行する必要があるかもしれません。これらのリスクや困難に適切に対処したり、そのような移行をうまく行わなかったりすると、私たちの事業は打撃を受けるでしょう。
私たちは創業以来多額の純損失を被っており、当面は引き続き多額の純損失を被ると予想しています。
当社は、設立以来、各報告期間に純損失を被っており、これまでのところ製品販売による収益はありません。また、主に民間資金調達、新規株式公開(IPO)、および普通株式のその後の公募を通じて事業資金を調達してきました。2022年12月31日に終了した年度には2億3,710万ドル、2023年9月30日と2022年9月30日に終了した9か月間でそれぞれ2億3,140万ドルと1億8,210万ドルの純損失が発生しました。2023年9月30日の時点で、累積赤字は8億2,760万ドルでした。私たちの損失は主に、製品候補の研究開発にかかった費用と、事業インフラを構築する際に発生した管理費やその他の費用によるものです。製品が商品化され、製品の販売から収益が得られるまでには、あるとしても数年かかると予想しています。1つまたは複数の製品候補の販売承認を得て商品化できたとしても、追加の潜在的な製品を発見、開発、販売するにつれて、多額の研究開発およびその他の費用が発生し続けると予想されます。
研究開発努力を続け、規制当局の承認取得と製品候補の商品化を目指しているため、当面の間、多額の費用と営業損失の増加が続くと予想しています。私たちが被る純損失は四半期ごとに大きく変動する可能性があるため、経営成績の期間ごとの比較は、将来の業績の良い指標にはならないかもしれません。将来の純損失の規模は、経費の将来の増加率と収益創出能力に一部依存します。当社の過去の損失と将来の予想される損失は、当社の運転資本と収益性の達成と維持能力に悪影響を及ぼしており、今後もそうなるでしょう。
収益を生み出し、収益性を達成できるかどうかは、いくつかの目標を達成できるかどうかに大きく依存します。
私たちのビジネスは、製品候補の発見、開発、商品化が成功するかどうかに完全に依存しています。現在、どの製品の販売からも収益を上げていません。商業販売が承認された製品はありません。また、今後数年間は、製品の販売から収益が得られる見込みはありません。私たちが収益を生み出し、収益性を達成できるかどうかは、私たちまたは将来の協力者が、次のような多くの目標を達成する能力に大きく依存します。
•アゼノセルチブ、Zn-D5、その他の将来の製品候補を含む当社の製品候補の前臨床および臨床開発、および関連費用(公衆衛生上の緊急事態、インフレ率や金利の上昇などの米国および世界の経済問題、または進行中の軍事紛争による前臨床研究または臨床試験の遅延の結果として発生した、または引き続き発生する可能性のある予期しない費用を含む)を無事かつタイムリーに完了しました。他の原因の中でも、
•該当する場合、当社の製品候補またはその他の将来の製品候補に関連するバイオマーカーの診断ツールの入手可能性または開発の成功。
•アゼノセルチブやZn-D5を含む当社の製品候補、およびその他の将来の製品候補の臨床開発のために、CROや臨床現場との関係を確立し、維持します。
•当社が臨床開発を無事に完了した製品候補について、該当する規制当局からの販売承認を適時に受領すること。
•製造販売承認の維持。これには、該当する規制当局に対して必要な製造販売後の承認の約束を行うことも含まれます。
•候補製品向けに効率的でスケーラブルな製造プロセスを開発します。これには、販売用に適切に梱包された最終製品の入手が含まれます。
•臨床開発を支援し、承認されれば当社が開発する製品候補に対する市場の需要を満たすために、量と質の両面で適切な製品とサービスを提供できる第三者との商業的に実行可能な供給および製造関係の確立と維持。
•社内または1人以上の協力者による商業インフラの開発を含む、マーケティング上の承認を得た後、商業的立ち上げが成功する。
•当社の製品候補が販売承認された後も、引き続き許容できる安全性プロファイル。
•患者、医学界、第三者支払者による当社製品候補の商業的承認。
•新製品候補の特定、評価、開発。
•特許、企業秘密、ノウハウ、規制上の独占権を含む当社の知的財産権を、米国および国際的に取得、維持、拡大します。
•知的財産ポートフォリオにおける私たちの権利の保護。
•第三者の干渉または侵害の申し立てがある場合、それに対する防御。
•当社の製品候補を開発、製造、または商品化するために必要または望ましい協力、ライセンス、またはその他の取り決めにおける有利な条件の交渉。
•当社が開発する製品候補について、病院、政府、第三者の支払者による適切な価格設定、補償、および償還を受けること。
•競合する治療法、技術開発、市場開発への取り組み。そして
•特に現在の労働市場において、有能な人材の誘致、雇用、維持。
私たちは目標の達成に決して成功しないかもしれませんし、たとえ成功したとしても、収益性を達成するのに十分な大きさの収益を生み出すことは決してないかもしれません。収益性を達成しても、四半期または年次ベースで収益性を維持または増加させることができない場合があります。私たちが利益を上げることができず、それを維持できないと、会社の価値が低下し、研究開発努力を維持または促進し、追加の必要な資本を調達し、事業を成長させ、事業を継続する能力が損なわれる可能性があります。
事業資金を調達するには、多額の追加資本が必要になります。必要なときに、または許容できる条件でそのような資金を調達できない場合、研究および医薬品開発プログラムの1つ以上、または将来の商業化活動を延期、削減、および/または廃止せざるを得ない可能性があります。
前臨床試験や臨床試験の実施を含む医薬品の開発は、非常に時間がかかり、費用がかかり、不確実なプロセスであり、完了するまでに何年もかかります。私たちの事業は創業以来多額の現金を消費しており、継続的な活動に関連して経費が増加すると予想しています。特に、アゼノセルチブ、Zn-D5、その他の製品候補の臨床試験を開始して実施し、市販承認を求めている場合はそうです。私たちが開発した製品候補の1つ以上が商業販売が承認されたとしても、承認された製品候補の商品化に関連して多額の費用が発生すると予想しています。FDA、欧州医薬品庁、EMA、その他の規制機関から、現在予定されているものに加えて臨床試験や前臨床試験を実施するよう要求された場合、経費は予想以上に増加する可能性があります。また、当社の製品候補および将来の製品候補に関連するバイオマーカーの診断ツールの開発、製造、供給のために、特定の診断会社と協力することに関連する費用が発生する可能性があります。その他の予期しない費用が発生する可能性もあります。さらに、アゼノセルチブやZn-d5など、当社の製品候補のいずれかの販売承認を得た場合、医薬品の販売、マーケティング、製造、流通に関連して多額の商品化費用が発生すると予想されます。計画および予定されている臨床試験の設計と結果は非常に不確実であるため、開発する製品候補の開発と商品化を成功させるために必要な実際の量を合理的に見積もることはできません。また、特に新興成長企業ではなくなった今、公開企業としての運営に関連する追加費用も発生しており、今後も発生すると予想されます。したがって、事業を継続するためには、多額の追加資金を調達する必要があります。
2023年9月30日現在、当社の現金および現金同等物と有価証券は5億1,660万ドルです。現在の事業計画に基づくと、2023年9月30日現在の現金、現金同等物、および有価証券は、2026年までの営業費用と資本支出要件を賄うには十分ですが、製品候補の開発を完了するために必要なすべての活動に資金を提供するには十分ではないと考えています。この見積もりは間違っているかもしれない仮定に基づいており、現在の予想よりも早く利用可能な資本資源を使うことができます。状況の変化により、中には私たちの手に負えないものもあり、現在の予想よりもはるかに早く資本を消費する可能性があります。また、計画よりも早く追加の資金を調達する必要があるかもしれません。
当社は、現金および現金同等物の大部分を米国の主要金融機関および多国籍金融機関の口座に保管しており、これらの機関の預金は保険限度を超えています。市場の状況は、これらの機関の存続可能性に影響を与える可能性があります。現金および現金同等物を保有している金融機関のいずれかに障害が発生した場合、無保険資金にタイムリーに、またはまったくアクセスできないという保証はありません。これらの資金にアクセスできない、またはアクセスが遅れると、当社の事業と財政状態に悪影響を及ぼす可能性があります。
私たちは、パブリックエクイティまたはプライベートエクイティの募集、債務融資、コラボレーション、ライセンス契約、またはその他の資金源を通じてさらなる資金を調達する必要があります。これにより、株主が希薄化したり、事業活動が制限されたりする可能性があります。当社には、確約された外部資金源はありません。適切な追加資金が、受け入れ可能な条件では提供されない場合もあれば、まったく提供されない場合もあります。COVID-19、米国および世界経済の問題、世界的なサプライチェーンの混乱、国際的な政情不安、インフレ率と金利の上昇、その他の要因による公衆衛生上の緊急事態、その他の要因による市場の変動も、必要に応じて資本にアクセスする能力に悪影響を与える可能性があります。必要なときに、または許容できる条件で資本を調達できない場合、当社の財政状態と事業戦略の遂行能力に悪影響を及ぼし、研究段階のプログラム、臨床試験、または将来の商業化の取り組みの1つ以上を延期、範囲の縮小、中断、または廃止しなければならない場合があります。
製品候補の発見、開発、商品化に関連するリスク
私たちは、現在臨床試験中の主力製品候補であるアゼノセルチブやZn-D5の成功に大きく依存しています。これらの製品候補の開発を完了し、承認を得て、適時に商品化できなければ、私たちのビジネスは損なわれます。
私たちの将来の成功は、臨床試験をタイムリーに完了し、市場承認を得て、主力製品候補の商品化を成功させることができるかどうかにかかっています。私たちは、製品候補の研究開発に多大な努力と財源を投資しています。そのため、製品の販売から収益を上げるには、追加の臨床開発、臨床、前臨床、製造活動の評価、政府規制当局からの販売承認、多額の投資、多額のマーケティング活動が必要になります。FDAおよび/または同等の米国外の規制当局から販売承認を受ける前に、他の製品候補を販売または宣伝することは許可されていません。また、そのような販売承認を受けることは決してないかもしれません。
当社の主力製品候補の成功は、次のようないくつかの要因によって決まります。
•進行中および計画中の臨床試験の無事かつタイムリーな完了。
•米国内および海外の製品候補の臨床開発のために、CROおよび臨床現場との関係を維持および確立します。
•臨床試験で観察されたAEの頻度と重症度。
•FDAおよび/または市販承認に関する同等の旧米国規制当局が満足できる有効性、安全性、および忍容性のプロファイル。
•該当する規制当局からの販売承認の適時の受領。
•該当する規制当局への市販後の承認義務の範囲。
•該当する場合、当社の製品候補またはその他の将来の製品候補に関連するバイオマーカーの診断ツールの入手可能性または開発の成功。
•当社の製品候補の臨床開発を目的とした、第三者の原薬および医薬品のサプライヤーおよび製造業者との既存の供給契約の維持または新規の供給契約の確立。
•承認された場合、候補製品との商業販売に適した完成品を入手するために、既存の製品を維持するか、サードパーティメーカーとの新しい大規模生産契約を確立すること。これには、候補製品と組み合わせて試験している医薬品の供給も含まれます。
•特許、企業秘密、ノウハウを含む当社の知的財産権、および規制上の独占権を、米国および国際的に取得し、維持します。
•知的財産ポートフォリオにおける私たちの権利の保護。
•マーケティングの承認を得た後、商業販売が成功裏に開始されたこと。
•販売承認後も引き続き許容できる安全性プロファイル。
•患者、医学界、第三者支払者による商業的承認。そして
•他の治療法と競合する私たちの能力。
私たちは、臨床開発や規制当局への提出プロセスの特定の側面、知的財産権に対する潜在的な脅威、将来の協力者の製造、マーケティング、流通、販売の取り組みなど、これらの要因の多くを完全に制御することはできません。これらの要因の1つ以上についてタイムリーに、またはまったく成功しなかった場合、候補製品の商品化が大幅に遅れたり、成功しなかったりして、事業に重大な損害を与える可能性があります。候補製品について販売承認を受けなければ、事業を継続できない可能性があります。
私たちは、開発する可能性のある特定の製品候補の研究、開発、商品化のために第三者と協力しており、将来的には提携する可能性があります。これらのコラボレーションのいずれかが成功しない場合、それらの製品候補の市場ポテンシャルを活用できない可能性があります。
私たちは、1つまたは複数の製品候補の研究、開発、商品化のために第三者の協力者を探しており、将来的には探す可能性があります。たとえば、私たちはファイザー、GSK、ダナ・ファーバーと協力してアゼノセルチブの開発を行っています。将来締結する可能性のあるコラボレーション契約における協力者には、大中規模の製薬会社やバイオテクノロジー企業が含まれます。第三者と協力契約を締結した場合、それらの契約により、協力者が共同開発を試みる可能性のある製品候補の開発と商品化に費やすリソースの量と時期に対する当社の管理が制限される可能性があります。私たちが参加した、または参加する可能性のあるコラボレーションの成功を予測することはできません。これらの取り決めから収益を生み出すことができるかどうかは、協力者の能力と、これらの取り決めで割り当てられた機能を首尾よく遂行するための努力にかかっています。
私たちの研究プログラム、私たちの製品候補、そして私たちが開発する可能性のある将来の研究プログラムや製品候補を含むコラボレーションは、私たちに次のようなリスクをもたらします。
•共同作業者は、これらの共同作業に費やす努力とリソースを決定する上で、大きな裁量権を持っています。
•共同事業者は、当社が開発する可能性のある製品候補の開発および商品化を追求することはできません。また、臨床試験の結果、共同研究者の戦略的焦点または市場上の考慮事項の変更(事業単位または開発機能の売却または処分、または利用可能な資金またはリソースを転用したり、競合する優先事項を生み出す買収や企業結合などの外部要因によるものを含む)に基づいて、開発または商業化プログラムを継続または更新しないことを選択したりすることもできます。この場合、該当する製品候補のさらなる開発または商品化を進めるために、追加の資本が必要になる場合があります。
•共同研究者は、臨床試験を延期したり、臨床試験プログラムに十分な資金を提供しなかったり、臨床試験を中止したり、製品候補を放棄したり、当社の製品候補を危険な方法で臨床試験に使用したり、新しい臨床試験を繰り返したり実施したり、臨床試験のために製品候補の新しい製剤を必要としたりする可能性があります。
•競合製品の方が開発が成功する可能性が高い、または当社よりも経済的に魅力的な条件で商品化できると信じている場合、協力者は当社の製品または製品候補と直接的または間接的に競合する製品を独自に開発するか、第三者と開発することができます。
•特定の注意義務を条件として、1つまたは複数の製品のマーケティングおよび販売権を持つ協力者は、そのような1つまたは複数の製品のマーケティングと流通に十分なリソースを投入できない場合があります。
•協力者は、当社の知的財産または所有権を適切に取得、維持、行使、または擁護しない場合や、当社の専有情報を危険にさらしたり無効にしたり、訴訟にさらされる可能性のある方法で専有情報を使用したりする可能性があります。
•協力者は、私たちが協力した結果として生じる当社の製品に関する知的財産を所有または共同所有する場合があります。その場合、当社にはコラボレーションの知的財産を商業化する独占権はありません。
•当社の協力者と当社の間で紛争が発生し、その結果、当社の製品または製品候補の研究、開発、または商品化が遅れたり終了したり、費用のかかる訴訟や仲裁が発生して経営者の注意とリソースをそらす可能性があります。
•私たちは、支配権の変更を受けた場合など、コラボレーションで特定された状況下で特定の権利を失う可能性があります。
•コラボレーションは終了する可能性があり、終了した場合、該当する製品候補のさらなる開発または商品化を追求するために追加の資本が必要になる場合があります。
•コラボレーション契約は、製品候補の最も効率的な方法での開発または商品化につながらない場合もあれば、まったく得られない場合もあります。私たちの現在または将来の協力者が企業結合に関与した場合、そのような協力の下での製品開発または商品化プログラムの継続的な追求と重点は、遅れたり、減少したり、中止されたりする可能性があります。
•協力者は、適正検査基準(GLP)、優良臨床基準(GCP)を含むグッドプラクティスの品質ガイドラインや規制、現在の適正製造基準(cGMP)など、適用される法律、規制、ガイダンスを遵守できない場合があります。また、FDAまたは同等の米国以外の規制当局から臨床開発計画の承認を得ることができない場合もあります。
•私たちは、特定の規制、臨床、製造、財務、およびその他の情報を協力者に要求する場合があります。これらの情報が、適時またはまったく提供されない場合、事業目標を達成したり、適用される法律、規制、ガイダンスを遵守したりする能力に影響を与える可能性があります。
これらの契約に基づいて期待される資金が得られない場合、製品候補の開発が遅れ、製品候補を開発するために追加のリソースが必要になる場合があります。さらに、協力者の1人が私たちとの契約を終了した場合、適切な代替協力者を見つけたり、新しい協力者を引き付けることがより困難になり、私たちの開発プログラムが遅れたり、ビジネス界や金融界における私たちの認識に悪影響が及ぶ可能性があります。このレポートに記載されている製品開発、販売承認、商品化に関連するすべてのリスクは、協力者の活動にも当てはまります。
将来的には、私たちが開発する可能性のある製品候補の開発と潜在的な商品化について、製薬会社やバイオテクノロジー企業と協力することを決定するかもしれません。これらおよびその他の同様の関係により、非経常費用やその他の費用を負担したり、短期および長期の支出を増やしたり、既存の株主を希薄化させる証券を発行したり、経営や事業を混乱させたりすることが必要になる場合があります。さらに、適切な協力者を探す際に激しい競争に直面する可能性があり、交渉プロセスは時間と複雑です。最終的な協力合意に達するかどうかは、とりわけ、協力者のリソースと専門知識の評価、提案された協力の条件、および提案された協力者によるいくつかの要因の評価にかかっています。私たちや協力者が開発する可能性のある製品候補に権利をライセンスしても、既存の事業や企業文化とうまく統合できなければ、それらの取引のメリットを実感できない可能性があります。
私たちの長期的な展望は、開発に失敗したり、商業的実行可能性に悪影響を及ぼす可能性のある追加の製品候補を発見、開発、商品化することに一部依存しています。
私たちの将来の業績は、現在の臨床開発段階を超える製品候補の発見、開発、規制当局の承認の取得、商品化を成功させる能力にかかっています。製品候補は、前臨床および臨床開発のどの段階でも予期せず失敗する可能性があります。安全性、有効性、臨床実施、医療基準の変化、その他の予測不可能な変数に関連するリスクにより、製品候補の過去の故障率は高くなっています。製品候補の前臨床試験または初期の臨床試験の結果は、製品候補の後の段階の臨床試験で得られる結果を予測するものではない場合があります。
私たちが開発する可能性のある他の製品候補の成功は、次のような多くの要因に左右されます。
•臨床試験の開始または継続を支援するのに十分なデータを生成すること。
•臨床試験を開始するための規制当局の許可の取得;
•臨床試験を実施するために必要な当事者との契約。
•タイムリーに患者を臨床試験に登録し、臨床試験を完了すること。
•臨床試験に使用するのに十分な量の製品候補をタイムリーに製造すること。そして
•臨床試験におけるAE
他の製品候補を臨床開発にうまく進めたとしても、その成功は、この「リスク要因」セクションの他の箇所で説明されているすべての臨床的、規制的、商業的リスクの影響を受けます。したがって、他の製品候補の発見、開発、規制当局の承認の取得、商品化、または大きな収益の創出が可能であることを保証することはできません。
FDAやその他の同等の米国以外の規制当局の規制承認プロセスは時間がかかり、本質的に予測不可能です。最終的に候補製品について規制当局の承認を得ることができなければ、製品の収益を上げることができず、事業に大きな打撃を与えることになります。
FDAの販売承認を得ずに、候補製品を米国で商品化、マーケティング、宣伝、販売することは許可されていません。元米国規制当局も同様の要件を課しています。FDAやその他の同等の米国以外の規制当局による承認を得るのに必要な時間は予測できません。通常、臨床試験の開始から何年もかかり、関連する製品候補の種類、複雑さ、新規性など、さまざまな要因に左右されます。さらに、承認方針、規制、または承認を得るために必要な臨床データの種類と量は、製品候補の臨床開発の過程で変更され、管轄区域によって異なる場合があります。そのため、承認が遅れたり、申請を承認しないという決定が遅れたりする可能性があります。規制当局は承認プロセスにおいてかなりの裁量権を持っており、申請の受理を拒否したり、当社のデータが承認には不十分で、追加の前臨床データ、臨床データ、またはその他のデータが必要であると判断したりする場合があります。最終的に臨床試験を完了し、製品候補の規制当局への申請の承認を得たとしても、FDAおよびその他の同等の米国外の規制当局は、当初の要求よりも限られた適応症またはより狭い患者集団の製品候補を承認する場合があります。どの製品候補についても、規制当局の承認を申請または取得していません。また、既存の製品候補や将来開発を試みる可能性のある製品候補のいずれも、規制当局の承認を得ることができない可能性があります。
当社の製品候補の申請は、次のようなさまざまな理由で規制当局の承認を得られない可能性があります。
•FDAまたは他の同等の米国以外の規制当局が、当社の臨床試験の設計、実施、または結果に同意しない場合があります。
•FDAまたは他の同等の米国外の規制当局が、当社の製品候補は安全で有効ではない、中程度の効果しかない、または望ましくない、または意図しない副作用、毒性、または販売承認の取得を妨げたり、商業的使用を防止または制限するその他の特徴があると判断する場合があります。
•臨床試験で研究された集団は、承認を求めている全集団における有効性と安全性を保証するのに十分な範囲または代表的でない可能性があります。
•FDAまたは他の同等の米国以外の規制当局が、前臨床試験または臨床試験からのデータに対する当社の解釈に同意しない場合があります。
•当社の製品候補の臨床試験から収集されたデータは、NDAやその他の提出物の提出を裏付けるには不十分であり、米国やその他の国で規制当局の承認を得るのにも不十分な場合があります。
•提案された適応症に対する製品候補のリスクと便益の比率が許容範囲内であることを、FDAまたは他の同等の米国以外の規制当局に証明できない場合があります。
•FDAまたはその他の同等の米国以外の規制当局が、臨床用品および商業用品の契約を結んでいる第三者メーカーの製造プロセス、試験手順、仕様または施設を承認しない場合があります。
•FDAまたは同等の旧米国規制当局が特定の製品候補のコンパニオン診断の承認または許可を要求していて、FDAまたは同等の規制当局がそのような承認または許可を提供しない場合、その製品候補はマーケティングが承認されない可能性があります。および/または
•FDAまたは他の同等の米国外の規制当局の承認方針または規制が大幅に変更され、臨床データが承認に不十分になる場合があります。
さらに、臨床試験に関するFDAおよびその他の同等の米国以外の規制当局の方針や慣行が変更され、追加の政府規制が制定される可能性があります。たとえば、近年、FDAはガイダンス草案を発行し、腫瘍薬の開発中に臨床試験スポンサーが使用する用量最適化手順の改革と近代化を目的としたプログラムを開始しました。これらの取り組みにより、まだFDAの規制や方針に正式な変更はありませんが、用量の選択と最適化に関するFDAの考え方の変化により、計画中または進行中の臨床試験の設計を変更したり、現在の予想を超える追加の前臨床、臨床、製造研究を実施したりする必要があり、コストが増加したり、製品候補の開発が遅れたりする可能性があります。FDAは、臨床試験の多様性に関するガイダンス草案も発行しました。このガイダンスの目的は、米国で過小評価されている人種や民族の集団から代表的な数の参加者を登録するための人種・民族多様性計画を策定するためのアプローチについて、医療製品を開発しているスポンサーに推奨事項を提供することです。実施されれば、FDAは人種・民族多様性計画をスポンサーの開発プログラムの重要な部分として評価します。そのためには、計画している臨床試験の登録方法を変更する必要があり、コストが増加したり、製品候補の開発が遅れたりする可能性があります。
さらに、欧州連合(EU)での臨床試験に関連する規制環境は最近変化しました。2014年4月に採択され、EU臨床試験指令を廃止するEU臨床試験規則(CTR)は、2022年1月31日に適用されました。臨床試験指令では、各加盟国で管轄の国民保健機関と独立した倫理委員会の両方に個別の臨床試験申請書(CTA)を提出する必要がありましたが、CTRは一元化されたプロセスを導入し、関係するすべての加盟国に1つの申請書を提出するだけで済みます。CTRにより、スポンサーは両方に1回の提出を行うことができます
各加盟国に所管官庁と倫理委員会があり、加盟国ごとに1つの決定が下されます。CTAの評価手順も統一されています。関係するすべての加盟国による共同評価や、倫理規則など、自国の領土に関連する特定の要件に関する各加盟国による個別の評価が含まれます。各加盟国の決定は、一元化されたEUポータルを介してスポンサーに伝えられます。CTAが承認されれば、臨床研究の開発を進めることができます。CTRは3年間の移行期間を想定しています。進行中の臨床試験と新しい臨床試験がCTRによってどの程度管理されるかはさまざまです。(i)臨床試験指令に基づいて2022年1月31日より前に申請が提出された臨床試験、または(ii)2022年1月31日から2023年1月31日までの間に申請書が提出され、スポンサーが臨床試験指令の適用を選択した臨床試験は、2025年1月31日まで同指令に準拠します。この日以降、進行中のものも含め、すべての臨床試験がCTRの規定の対象となります。当社、協力者、およびCROなどのサードパーティのサービスプロバイダーによるCTR要件の遵守は、当社の開発計画に影響を与える可能性があります。
この承認プロセスに時間がかかり、臨床試験の結果が予測できないため、候補製品を販売するための規制当局の承認を得られない可能性があり、その結果、当社の事業、経営成績、および見通しに重大な悪影響が及ぶ可能性があります。
さらに、製品候補の承認を得たとしても、規制当局は、私たちが要求するよりも少ないまたはより限定的な適応症で当社の製品候補を承認する場合があり、狭い適応症、警告、またはリスク評価および軽減戦略(REMS)、または同様のリスク管理措置の形で重大な制限を課す場合があります。規制当局は、当社が開発する可能性のある製品に請求する予定の価格を承認しない場合や、費用のかかる市販後の臨床試験の実施を条件として承認する場合や、製品候補の商品化を成功させるために必要または望ましい表示内容が含まれていないラベルの付いた製品候補を承認する場合があります。前述のシナリオはいずれも、当社の事業に重大な損害を与える可能性があります。
当社の製品候補の臨床試験では、FDAまたは他の同等の米国以外の規制当局が満足できる安全性と有効性が実証されない場合や、肯定的な結果が得られない場合があります。
当社の製品候補の販売について、FDAまたは他の同等の米国以外の規制当局から市販承認を得る前に、製品候補の安全性と有効性を実証するための前臨床開発と広範な臨床試験を完了する必要があります。臨床検査は費用がかかり、設計と実施が難しく、完了するまでに何年もかかることがあり、最終的な結果は不明です。プロセスのどの段階でも、1つ以上の臨床試験が失敗する可能性があります。前臨床試験と初期段階の臨床試験の結果は、後の臨床試験の成功を予測するものではないかもしれません。たとえば、潜在的なバイオマーカーは、前臨床で検証されたとしても、臨床試験では機能的に検証されない場合があります。さらに、前臨床データと臨床データは、さまざまな解釈や分析の影響を受けやすく、製品候補が前臨床試験や臨床試験で満足のいく結果を出したと信じている多くの企業は、それでも医薬品の市販承認を得ることができませんでした。FDAまたは同等の米国外の規制当局が試験結果を私たちと同じように解釈することを保証することはできません。また、製品候補の承認を求める申請書を提出する前に、さらに試験が必要になる可能性があります。その場合、私たちが利用できない多額のリソースを費やしたり、計画されたスケジュールに遅れが生じたりする可能性があります。臨床試験を開始するほとんどの製品候補は、規制当局によって商業化が承認されることはありません。
さらに、製品候補の規制当局への提出について、CRO、協力者、その他の第三者によって生成された前臨床データ、臨床データ、品質データに一部依存する場合があります。これらの第三者との関係を規定する契約を締結している、または締結する予定ですが、実際の業績に対する影響は限定的です。これらの第三者が当社にデータを提供しない場合、または該当する場合は、いずれの場合も当社との契約に従って規制当局への提出を行う場合、当社の開発プログラムは大幅に遅れる可能性があり、追加の調査を実施したり、独自に追加のデータを収集したりする必要が生じる可能性があります。いずれにしても、開発費は増加するでしょう。
将来の臨床試験が予定通りに開始されるのか、予定通りに患者を登録するのか、あるいは進行中および/または将来の臨床試験が予定通りに完了するのか、それともまったく完了するのかはわかりません。臨床試験は、次のような遅延など、さまざまな理由で遅れる可能性があります。
•当社の臨床試験の設計または実施に関して意見が分かれている、FDAまたは同等の元米国規制当局。
•試験を開始するための規制当局の許可の取得、または試験デザインについて規制当局との合意に達すること。
•CROおよび臨床試験サイトとの合意に達しなかったり遅れたりした場合、その条件は広範な交渉の対象となる可能性があり、CROや試験サイトによって大きく異なる可能性があります。
•1つ以上の機関審査委員会、IRB、または倫理委員会から承認を得ること。
•IRBまたは倫理委員会が、治験実施地での試験の承認、一時停止、終了の拒否、追加の被験者の登録の禁止、または試験への承認の撤回。
•臨床試験プロトコルの変更。
•臨床施設が試験プロトコルから逸脱している、または試験を中止している。
•十分な量の製品候補を製造すること、または臨床試験で使用するのに十分な量の併用療法を入手すること。
•被験者が期待した速度で治験に登録しなかったり、継続できなかったり、治療後のフォローアップのために戻ってこなかったりします。
•被験者は、私たちが製品候補を開発している適応症に代わる治療法を選択したり、競合する臨床試験に参加したりしています。
•臨床試験を継続するための十分な資金がない。
•重度または予期しない薬物関連のAEを経験している被験者。
•他社が実施した同じクラスの薬剤の試験での重篤なAEの発生。
•長期間の臨床観察または結果データの分析を必要とする臨床エンドポイントの選択。
•cGMP規制または同様の米国外の要件またはその他の適用要件への違反、または製造プロセスにおける製品候補の感染または相互汚染により、FDAまたは同等の米国外の規制当局から一時的または恒久的な閉鎖を命じられた当社の製品候補またはそのコンポーネントを製造する施設。
•必要または望ましいと思われる製造プロセスの変更。
•第三者の臨床研究者が当社の臨床試験を実施するために必要な免許または許可を失い、予定されたスケジュールどおりに臨床試験を実施しなかったり、臨床試験プロトコル、GCP、またはその他の規制要件に従って臨床試験を実施しなかったりしました。
•第三者の請負業者が適時または正確な方法でデータ収集または分析を行わない場合。
•第三者の請負業者が、規制要件の違反を理由に、FDA、その他の政府、規制当局から禁止されたり、停止されたり、その他の罰則が科されたりします。
代替の請負業者を探す必要があり、そのような請負業者が作成したデータの一部または全部をマーケティングアプリケーションのサポートに使用できない場合があります。または
•私たちが臨床試験で第三者と協力している場合、私たちの共同研究者は私たちの臨床試験に十分なリソースを投入していないか、私たちの臨床試験に優先順位を付けていない可能性があります。
さらに、COVID-19による混乱により、計画中および進行中の臨床試験の開始、登録、実施、または完了が困難または遅延し、引き続き発生する可能性があります。また、当社、そのような試験を実施している機関のIRB、そのような試験のデータ安全監視委員会、またはFDAまたは同等の米国以外の規制当局によって臨床試験が中断または終了された場合、遅延が発生する可能性があります。そのような当局は、規制要件または当社の臨床プロトコルに従って臨床試験を実施しなかったこと、FDAまたは同等の米国外の規制当局による臨床試験運営または試験サイトの検査の結果、臨床保留が課されること、予期しない安全性の問題または副作用、薬物使用による利益の証明の失敗、政府規制または行政措置の変更、または欠如など、さまざまな要因により、そのような停止または終了を課すことがあります。に十分な資金を臨床試験を続けてください。さらに、規制要件や方針が変更される可能性があり、これらの変更に対応するために臨床試験プロトコルを修正する必要があるかもしれません。改正により、再審査のために臨床試験プロトコルをIRBまたは倫理委員会に再提出する必要が生じる場合があります。これは、臨床試験の費用、時期、または無事完了に影響を与える可能性があります。
さらに、製品候補に対して行うように、米国以外の国で臨床試験を実施すると、臨床試験の完了を遅らせる可能性のある追加のリスクが伴います。これらのリスクには、医療サービスや文化的慣習の違いにより、米国以外の国で登録された患者が臨床プロトコルを順守できないこと、米国の規制制度に関連する追加の管理上の負担を管理すること、およびそのような米国外の国に関連する政治的および経済的リスクが含まれます。
さらに、私たちの臨床試験の主任研究者は、随時私たちの科学顧問またはコンサルタントを務め、そのようなサービスに関連して報酬を受け取ることがあります。特定の状況下では、これらの関係の一部をFDAまたは同等の米国以外の規制当局に報告する必要がある場合があります。FDAまたは同等の元米国規制当局は、当社と主任研究員との金銭的関係が利益相反を引き起こした、またはその他の方法で研究の解釈に影響を与えたと結論付ける場合があります。したがって、FDAまたは同等の元米国規制当局は、該当する臨床試験サイトで生成されたデータの完全性に疑問を呈する可能性があり、臨床試験自体の有用性が危うくなる可能性があります。その結果、場合によっては、FDAまたは同等の米国以外の規制当局によるマーケティング申請の承認が遅れたり、拒否されたりする可能性があり、最終的には1つ以上の製品候補の販売承認が拒否される可能性があります。
製品候補の臨床試験の完了または終了が遅れると、製品候補の商業的見通しが損なわれ、これらの製品候補のいずれかから製品収益を生み出す能力も遅れます。さらに、臨床試験の完了が遅れると、コストが増加し、製品候補の開発と承認プロセスが遅くなり、製品の販売を開始して収益を上げることができなくなります。
さらに、臨床試験の終了または停止、または開始または完了の遅延を引き起こす、または引き起こす要因の多くは、最終的には製品候補の規制当局の承認の拒否につながる可能性もあります。その結果、臨床試験が遅れると、私たちが製品候補を商品化する独占権を有し、競合他社が私たちより先に製品を市場に出すことができる期間が短くなる可能性があり、製品候補の商業的存続可能性が大幅に低下する可能性があります。これらの出来事はいずれも、当社の事業、財政状態、および見通しに重大な損害を与える可能性があります。
患者の選択を可能にするバイオマーカーの診断ツールの開発が成功しなかったり、開発が大幅に遅れたりした場合、製品候補の商業的可能性を最大限に発揮できない可能性があります。
私たちの戦略の構成要素には、患者さんが当社の製品候補を選択する際の指針となる診断ツールの使用が含まれる場合があります。場合によっては、腫瘍プロファイリングパネルなどの診断ツールが市販されていることがあります。まだ市販されていない場合は、製品候補に関連するバイオマーカーの診断薬を開発するために、診断会社との協力を求める必要があるかもしれません。このような開発関係の確立や維持は難しいかもしれませんし、そのような協力関係を築く上で他社との競争に直面するでしょう。
バイオマーカーの同定と検証には、いくつかのリスクもあります。私たちは、他の診断パートナーと協力して、1つまたは複数のプログラムの予測バイオマーカーを特定できない場合があります。潜在的なバイオマーカー(特定のゲノム変異など)やそれらの機能的関連性を前臨床的に検証できない場合があります インビトロまたは インビボモデル。バイオマーカーとターゲットの関係の一部を特定または検証するために私たちが頼りにしているデータ分析とデータベースからの情報は、潜在的な患者集団を正確に反映していない場合や、誤った方法論に基づいている場合があります。潜在的なバイオマーカーは、たとえ前臨床で検証されたとしても、機能的に有効ではないか、人間の臨床試験で検証されていない可能性があります。
これらの関係者と協力して、製品候補の診断ツールをうまく開発できなかったり、開発が遅れたりした場合、製品候補の開発に悪影響が及ぶ可能性があります。コンパニオン診断など、特定の診断ツールの開発には多額の運転資金が必要で、将来の収入にはならない可能性があります。そのためには、追加の資金を調達する必要があり、現在の投資家が希薄になったり、将来の事業継続能力に影響を与えたりする可能性があります。
市販の診断薬にはリスクもあります。たとえば、そのような診断薬の信頼できる供給を受けることができない場合や、規制当局の承認を得ずにその使用に対する払い戻しを得ることができない場合などです。
私たちが随時発表または公開する臨床試験の中間、初期、「トップライン」、および暫定データは、より多くの患者データが利用可能になり、最終データに重大な変更をもたらす可能性のある監査および検証手続きの対象となるにつれて変更される可能性があります。
当社は、前臨床試験および臨床試験の初期データ、予備データ、またはトップラインデータを随時公開することがあります。これらのデータは、その時点で入手可能なデータの予備分析に基づいており、特定の研究または試験に関連するデータをより包括的に検討した結果、結果および関連する調査結果と結論が変更される場合があります。また、データ分析の一環として、仮定、推定、計算、結論を行いますが、すべてのデータを受け取っていないか、すべてのデータを完全かつ慎重に評価する機会がなかった可能性があります。その結果、追加のデータを受け取って完全に評価した後に、私たちが報告する初期、トップライン、または暫定的な結果が、同じ研究の将来の結果と異なる場合や、異なる結論や考慮事項がそのような結果に適格となる場合があります。これらのデータの一部は、依然として監査および検証手続きの対象となっているため、最終データが以前に公開した暫定データと大きく異なる可能性があります。そのため、最終データが入手可能になるまで、初期、トップライン、暫定データを注意して閲覧する必要があります。
また、前臨床試験や臨床試験の中間データを開示することもあります。私たちが完成する可能性のある臨床試験の中間データには、患者の登録が継続してより多くの患者データが入手可能になるにつれて、または私たちの臨床試験の患者が自分の病気に対して他の治療を続けるにつれて、1つまたは複数の臨床転帰が大きく変わるリスクがあります。中間データと最終データの不利な違いは、当社の事業見通しを著しく損なう可能性があります。さらに、当社または競合他社が中間データを開示すると、当社の普通株式の価格が変動する可能性があります。
さらに、規制機関を含む他の機関は、当社の仮定、見積もり、計算、結論、または分析を受け入れたり同意しなかったり、データの重要性を異なって解釈または評価したりする場合があります。これは、特定のプログラムの価値、特定の製品候補または製品、および当社全般の承認または商品化に影響を与える可能性があります。さらに、特定の研究または臨床試験に関して私たちが公開することを選択した情報は、一般的に広範な情報に基づいており、あなたや他の人は、私たちが開示に含めるべき重要またはその他の適切な情報であると私たちが判断した内容に同意しない場合があります。
私たちが報告する初期、中間データ、トップライン、または暫定データが実際の結果と異なる場合、または規制当局を含む他の人が、到達した結論、当社の製品候補の承認を得て商品化する能力に同意しない場合、当社の事業、業績、見通し、または財政状態に害を及ぼす可能性があります。
たとえ承認されたとしても、私たちの製品候補は、医師、患者、医療費支払者、および商業的成功に必要な医療界の他の人々の間で十分な市場受け入れを得られない可能性があります。
当社の製品候補が規制当局の承認を得たとしても、医師、患者、医療費支払者、および医学界の他の人々の間で十分な市場で受け入れられない可能性があります。当社が承認した製品候補が市場で受け入れられる度合いは、次のような多くの要因によって決まります。
•代替治療と比較して臨床試験で実証された有効性と安全性プロファイル。
•候補製品および競合製品の市場導入のタイミング。
•製品候補が承認されている臨床適応症、
•該当する場合、当社の製品候補またはその他の将来の製品候補に関連するバイオマーカーの診断ツールの入手可能性
•代替治療や競合製品には必要ない可能性のある、箱入りの警告、表示の禁忌、REMS、または同様のリスク管理措置など、当社の製品候補の使用に関する制限。
•代替治療に対する製品候補の潜在的利点と認識されている利点
•代替治療と比較した治療費;
•政府機関を含む第三者支払者による補償の有無、適切な払い戻し、および価格設定。
•併用療法として使用するための承認された製品候補の入手可能性。
•比較的便利で管理しやすい。
•対象となる患者集団が新しい治療法を試す意欲、および医師がこれらの治療法を処方する意欲。
•販売およびマーケティング活動の有効性。
•当社の製品、製品候補、または類似の承認済み製品または第三者による開発中の製品候補に関する不利な宣伝。そして
•同じ適応症に対する他の新しい治療法の承認。
当社の製品候補のいずれかが承認されたが、医師、病院、医療費支払者、患者に十分な受け入れが得られない場合、その製品候補から十分な収益を生み出せない、または得ることができず、財務結果に悪影響が及ぶ可能性があります。
臨床試験への患者の登録や維持が遅れたり困難になったりした場合、私たちの臨床開発活動が遅れたり、悪影響が出たりする可能性があります。
患者登録は臨床試験のタイミングを決定する重要な要素であり、臨床試験のタイミングは、試験に参加する患者を募集するスピードと、必要なフォローアップ期間の完了に一部依存します。FDAまたは同等の米国外の規制当局が要求する試験の結論に達するまで、これらの試験に参加するのに十分な数の適格な患者を見つけて登録できない場合、製品候補の臨床試験を開始または継続できない場合があります。さらに、将来の製品候補のための特定の臨床試験は、患者集団が比較的少ない適応症に焦点を当てている可能性があります。これにより、適格な患者の登録がさらに制限されたり、登録が予想よりも遅くなったりする可能性があります。私たちの臨床試験の適格基準は、いったん確立されると、利用可能な試験参加者のプールをさらに制限する可能性があります。
競合他社が当社の製品候補と同じ適応症で開発中の製品候補の臨床試験を進行中であり、そうでなければ当社の臨床試験の対象となる患者が、代わりに競合他社の製品候補の臨床試験に登録する場合も、患者登録が影響を受ける可能性があります。当社の臨床試験への患者登録は、次のような他の要因の影響を受ける可能性があります。
•患者集団の規模と性質。
•調査中の疾患の重症度。
•調査中の病気に対する承認された薬の入手可能性と有効性。
•プロトコルで定義されている当該試験の患者適格基準。
•検討中の製品候補について認識されているリスクとメリット。
•研究中の製品候補が他の利用可能な治療法(調査中の適応症で承認される可能性のある新製品を含む)と比較して検討中の製品候補の潜在的な利点に関する臨床医と患者の認識。
•臨床試験へのタイムリーな登録を促進するための取り組み。
•医師の患者紹介慣行;
•治療中および治療後に患者を適切に監視する能力。
•将来の患者のための臨床試験サイトの近接性と可用性;
•臨床試験サイトによる見込み患者の継続的な登録。そして
•臨床試験に登録された患者が、完了前に試験から脱落したり、後期のがん患者である可能性があるため、臨床試験の全期間にわたって生存できなくなったりするリスク。
臨床試験に十分な数の患者を登録できないと、大幅な遅延が発生するか、1つまたは複数の臨床試験を完全に中止しなければならない場合があります。臨床試験への登録が遅れると、製品候補の開発コストが増加し、製品候補の販売について販売承認を得ることができなくなる可能性があります。さらに、たとえ十分な数の患者を臨床試験に登録できたとしても、そのような患者の臨床試験への登録を維持することは難しいかもしれません。
私たちは、他の治療法と組み合わせて製品候補を開発していますが、それにはさらなるリスクがあります。
私たちは、がんやその他の病気を治療するために、アゼノセルチブとZn-D5を他の承認済みまたは未承認の治療法の1つ以上と組み合わせて開発しています。将来、他の承認または未承認の治療法と組み合わせて、追加の製品候補を開発する可能性があります。承認または未承認の治療法の供給が予期せず途絶えた場合、遅延、中断、停止、終了が発生する可能性があります。
保留中または進行中の臨床試験を再開または繰り返す必要があります。私たちが開発する製品候補が市販承認を受けたり、他の既存の治療法と組み合わせて使用するために商品化されたとしても、FDAまたは米国外の同等の元米国規制当局が、当社の製品と組み合わせて使用される治療法の承認を取り消したり、それらの既存の治療法のいずれかで安全性、有効性、製造、または供給の問題が生じる可能性があるというリスクにさらされ続けます。製品候補と組み合わせて使用する治療法が、製品候補のいずれかに対して選択した適応症の標準治療として置き換えられた場合、FDAまたは同等の米国以外の規制当局から、追加の臨床試験の実施を要求される場合があります。これらのリスクのいずれかが発生すると、承認された場合、自社製品が市場から排除されたり、商業的にあまり成功しなかったりする可能性があります。
また、FDAまたは同等の米国外の規制当局による販売がまだ承認されていない1つ以上のがん治療薬と組み合わせて、製品候補を評価することもできます。未承認のがん療法と併用適応症との組み合わせで開発した製品候補を、その未承認の治療法が最終的に単独で、または当社の製品と組み合わせて市販承認を得られない場合、その製品候補を市場に出して販売することはできません。さらに、未承認のがん治療薬は、重篤な副作用の可能性、臨床試験の遅延、規制当局の承認の欠如など、現在開発中および臨床試験中の製品候補に関して説明されているのと同じリスクに直面します。
FDAまたは同等の元米国規制当局がこれらの他の医薬品を承認しなかったり、承認を取り消したり、開発した製品候補と組み合わせて評価することを選択した医薬品に安全性、有効性、品質、製造、供給の問題が生じた場合、そのような併用療法の承認を得たり、販売したりできない場合があります。
私たちや戦略的パートナーが開発する製品候補の市場機会が、私たちが思っているよりも小さい場合、私たちの収益は悪影響を受け、私たちの事業は打撃を受ける可能性があります。
当社の製品候補による治療の恩恵を受ける可能性のある対象患者集団の予測は、私たちの推定に基づいています。科学文献、診療所の調査、患者基礎、市場調査など、さまざまな情報源から導き出されたこれらの見積もりは、正しくない場合があります。さらに、新しい研究により、これらのがんの推定発生率または有病率が変わる可能性があります。さらに、当社の製品候補にとって潜在的に対応可能な患者集団は、最終的には当社の製品候補による治療に適さない場合があります。私たちの市場機会は、市場に参入する将来の競合治療法によっても制限される可能性があります。私たちの見積もりのいずれかが不正確であることが判明した場合、私たちまたは当社の戦略的パートナーが開発する製品候補の市場機会は大幅に減少し、当社の事業に重大な悪影響を及ぼす可能性があります。
私たちは激しい競争に直面しています。競合他社が私たちよりも早く、または私たちが開発する製品候補よりも効果的、安全、または安価な技術や製品を開発して販売した場合、私たちの商業機会は悪影響を受けます。
バイオテクノロジーと製薬業界は、技術の急速な進歩、激しい競争、独自の新製品や製品候補への強い重点が置かれているという特徴があります。競合他社は、当社の製品候補と競合する製品、製品候補、プロセスを開発しているか、開発しているか、開発している可能性があります。私たちが開発して商品化することに成功した製品候補は、将来利用可能になる可能性のある既存の治療法や新しい治療法と競合することになります。私たちは、製品候補の開発を試みる可能性のある症状の治療のために、かなりの数の製品が現在開発中であり、将来的に市販される可能性があると考えています。さらに、当社の製品は、承認を求めている適応症の治療に医師が使用する適応外医薬品と競合する必要があるかもしれません。これにより、既存の治療法を当社の製品に置き換えることが難しくなる可能性があります。
特に、私たちが追求しているオンコロジーの分野では激しい競争があります。私たちは、主要な多国籍製薬会社、確立されたバイオテクノロジー企業、専門製薬会社、新興企業や新興企業、大学、その他の研究機関など、米国内および国際的に競合他社がいます。また、経営陣、科学者、臨床開発担当者を採用するためにこれらの組織と競争しています。これは、当社の専門知識レベルや事業遂行能力に悪影響を及ぼす可能性があります。
計画。また、臨床試験サイトの設立、臨床試験への被験者の登録、新製品候補の特定とライセンス供与においても競争に直面しています。
私たちは当初、十分に検証された生化学的ターゲットに取り組むことを選択しました。したがって、当社の製品候補ごとに、既存の製品や開発中の製品との競争に直面することが予想されます。多くの大手製薬会社やバイオテクノロジー企業を含め、がんの治療法を開発または販売している企業は数多くあります。これらの現在および潜在的な競合他社の多くは、私たちよりもはるかに優れた財務、製造、マーケティング、医薬品開発、技術的および人的資源、および商業上の専門知識を持っています。特に大手製薬会社やバイオテクノロジー企業は、臨床試験、規制当局の承認の取得、患者の募集、バイオテクノロジー製品の製造において豊富な経験を持っています。これらの企業はまた、私たちよりもはるかに優れた研究およびマーケティング能力を持っており、承認された製品や開発の後期段階にある製品や、ターゲット市場で大手企業や研究機関との共同契約を結んでいる場合もあります。既存の製薬会社やバイオテクノロジー企業も、新しい化合物の発見と開発を加速したり、私たちが開発する製品候補を時代遅れにする可能性のある新しい化合物のライセンス供与に多額の投資をする可能性があります。中小企業や初期段階の企業も、特に大規模で確立された企業との協力的な取り決めや、私たちのプログラムを補完する、またはプログラムに必要な技術の獲得を通じて、重要な競争相手になる可能性があります。これらすべての要因の結果として、競合他社はFDAまたは他の同等の米国以外の規制当局から承認を得たり、私たちよりも先に私たちの分野で製品を発見、開発、商品化したりする可能性があります。
競合他社が、私たちが開発するどの製品よりも安全で、より効果的で、より深刻な影響が少なく、より便利で、より広いラベルを持ち、より効果的に販売され、払い戻され、またはより安価な製品を開発して商品化した場合、私たちの商業的機会は減少または排除される可能性があります。また、競合他社は、当社が承認するよりも早く、FDAまたは他の同等の米国以外の規制当局から自社製品の販売承認を取得する可能性があります。その結果、当社が市場に参入する前に、競合他社が強力な市場での地位を確立する可能性があります。私たちが開発する製品候補が販売承認を得たとしても、それまでに承認された競合製品よりも大幅に割高な価格設定になり、競争力が低下する可能性があります。技術の進歩や競合他社が開発した製品により、当社の技術や製品候補は時代遅れになったり、競争力が低下したり、経済的でなくなったりする可能性があります。私たちが効果的に競争できなければ、私たちが開発する可能性のある製品の販売から収益を生み出す機会が、承認されれば、悪影響を受ける可能性があります。
限られたリソースを特定の製品候補または適応症を追求するために費やしても、より収益性が高い、または成功する可能性が高い製品候補または適応症を利用できない場合があります。
私たちの財源と管理資源は限られているため、特定の適応症のために特定した研究プログラム、治療プラットフォーム、製品候補に焦点を当てています。その結果、他の治療プラットフォームや製品候補、または後でより大きな商業的可能性や成功の可能性が高いことが証明されたその他の適応症についての機会の追求を見送ったり、遅らせたりする可能性があります。資源配分の決定により、実行可能な商品や収益性の高い市場機会を活用できなくなる可能性があります。現在および将来の研究開発プログラム、治療プラットフォーム、特定の適応症向けの製品候補への支出は、商業的に実行可能な製品を生み出さない可能性があります。特定の製品候補の商業的可能性またはターゲット市場を正確に評価しない場合、開発権と商品化権を単独で保持する方が有利な場合には、コラボレーション、ライセンス、またはその他のロイヤルティ契約を通じて、その製品候補に対する貴重な権利を放棄することがあります。
製品候補の製造方法や配合方法を変更すると、追加費用や遅延が発生する可能性があります。
製品候補が前臨床試験や臨床試験を経て市販承認や商品化に進むにつれて、収量と製造バッチサイズを最適化し、コストを最小限に抑え、一貫した品質と結果を達成するために、製造方法や配合など、開発プログラムのさまざまな側面が変更されることがよくあります。このような変更には、意図した目的を達成できないというリスクが伴います。これらの変更のいずれかにより、当社の製品候補の性能が異なり、計画された臨床の結果に影響を与える可能性があります
変更された材料を用いて実施される試験またはその他の将来の臨床試験。これにより、臨床試験の完了が遅れたり、橋渡し臨床試験の実施や1つ以上の臨床試験の繰り返しが必要になったり、臨床試験費用が増加したり、製品候補の承認が遅れたり、承認された場合に製品候補を商品化して収益を生み出す能力が危うくなる可能性があります。
私たちの事業には製造物責任の重大なリスクが伴い、十分な保険に加入できない場合、そのようなことが当社の事業および財政状態に悪影響を及ぼす可能性があります。
私たちの事業は、治療法の開発、試験、製造、マーケティングに内在する重大な製造物責任リスクにさらされています。製造物責任の請求により、開発プログラムの完了が遅れたり、妨げられたりする可能性があります。製品のマーケティングに成功した場合、そのような主張により、FDAまたはその他の規制当局が、当社の製品、製造プロセスと設備、またはマーケティングプログラムの安全性と有効性を調査する可能性があります。FDAやその他の規制当局の調査により、当社製品のリコールや、より深刻な執行措置、それらが使用される可能性のある承認された適応症の制限、承認の一時停止または撤回につながる可能性があります。メリットや最終的な結果にかかわらず、賠償請求は、当社製品の需要の減少、評判の低下、関連する訴訟の弁護費用、経営陣の時間とリソースの流用、試験参加者または患者への多額の金銭的賞与につながる可能性もあります。私たちは現在、開発段階に適していると思われる製造物賠償責任保険に加入しており、承認された場合、製品候補のいずれかを販売する前に、より高いレベルの保険に加入する必要があるかもしれません。私たちが加入している、または加入している保険は、潜在的な負債に対して十分な補償を提供しない場合があります。さらに、臨床試験と製造物責任保険はますます高価になっています。その結果、当社の事業や財政状態に悪影響を及ぼす可能性のある製造物責任請求による損失から私たちを守るのに、妥当な費用で十分な保険に加入できない可能性があります。医薬品や生物製剤に適用される補償や償還の取得に関する同様の課題は、私たちや私たちの共同研究者が開発する可能性のあるコンパニオン診断などの診断ツールにも当てはまるかもしれません。
私たちが開発する製品候補はすべて、価格規制だけでなく、第三者による不利な補償や償還慣行の対象となる可能性があります。
ほとんどの患者が高価な治療を受けることができるためには、政府の保健管理当局、民間の健康保険会社、マネージドケア組織、その他の第三者支払者を含む第三者支払者による補償の有無と範囲、および適切な払い戻しが不可欠です。販売承認を受けた製品候補の売上は、米国でも海外でも、製品候補の費用が第三者の支払者によってどの程度カバーされ、払い戻されるかに大きく依存します。払い戻しが利用できない場合、または限られたレベルでのみ利用できる場合、候補製品をうまく商品化できない可能性があります。補償が提供されたとしても、承認された償還額は、十分な投資収益率を実現するのに十分な価格を設定または維持できるほど高くない場合があります。補償範囲と償還は、当社が販売承認を得ている製品候補の需要または価格に影響を与える可能性があります。補償範囲と補てんが利用できない場合、または償還が限られたレベルでのみ利用できる場合、販売承認を得た製品候補をうまく商品化できない可能性があります。
第三者支払者の補償と新たに承認された製品の払い戻しに関しては、大きな不確実性があります。たとえば米国では、新製品の償還に関する主な決定は、通常、米国保健社会福祉省(HHS)内の機関であるメディケア&メディケイドサービスセンター(CMS)が行います。CMSは、新製品がメディケアの対象となるかどうか、またどの程度補償されるかを決定します。民間の第三者支払者は、補償範囲と償還に関するCMSの決定にかなりの程度従うことがよくあります。ただし、ある第三者支払者が製品候補に補償を提供することを決定したからといって、他の支払者も製品候補に対して補償を提供することが保証されるわけではありません。その結果、カバレッジの決定プロセスには時間とコストがかかることがよくあります。このプロセスでは、当社製品の使用に関する科学的および臨床的サポートを各第三者の支払者に個別に提供する必要があります。補償と適切な払い戻しが一貫して適用される、または最初に得られるという保証はありません。
第三者支払者が、製薬会社に定価からの所定の割引を提供するよう要求し、医療製品に請求される価格に異議を唱えることが増えています。さらに、そのような支払者はますます増えています
価格に異議を唱え、医療の必要性を検討し、医療製品候補の費用対効果を検討します。新たに承認された医薬品の補償と償還の取得には、特に大幅な遅延が生じる可能性があります。第三者の支払者は、フォーミュラリーと呼ばれる承認済みリストにある特定の製品候補に補償範囲を制限する場合があります。フォーミュラリーには、特定の適応症に対してFDAが承認したすべての医薬品が含まれているとは限りません。当社製品の医学的必要性と費用対効果を実証するために、費用のかかる薬物経済学的研究を実施する必要があるかもしれません。とはいえ、私たちの製品候補は、医学的に必要でも費用対効果が高いとも考えられていないかもしれません。私たちが商品化しているどの製品についても、補償や補てんが受けられるかどうか、また補てんが可能であれば、補てんのレベルはどのくらいになるのか、確信が持てません。
2022年8月、2022年のインフレ削減法(IRA)が法制化されました。とりわけ、IRAは、特定の医薬品の製造業者に、上限を条件として交渉できる価格で、メディケアと価格交渉を行うことを義務付けています(2026年から)。インフレを上回る価格上昇を罰するために、メディケアパートBとメディケアパートDに基づいてリベートを課し(2023年に最初に予定されている)、パートDの補償ギャップ割引プログラムを新しい割引プログラムに置き換えます(2025年から)。州レベルのIRAと価格規制の詳細については、「規制当局の承認およびその他の法的遵守事項に関連するリスク」を参照してください。 — 現在の規制や将来の法律の変更により、困難に直面する可能性があります。」下記をご覧ください。
米国以外では、国際事業は一般的に政府の広範な価格統制やその他の市場規制の対象となっています。ヨーロッパ、カナダ、その他の国でのコスト抑制イニシアチブへの関心の高まりは、当社の製品候補などの治療薬の価格設定と使用に圧力をかけており、今後もそうなると考えています。多くの国、特にEU加盟国では、医療製品の価格は、国民医療制度の一環としてさまざまな価格管理メカニズムの対象となっています。これらの国では、製品が販売承認を受けた後、政府機関との価格交渉にかなりの時間がかかる場合があります。一部の国で償還または価格承認を得るために、製品候補の費用対効果を他の利用可能な治療法と比較する臨床試験を実施する必要がある場合があります。一般的に、このようなシステムでの製品価格は、米国よりも大幅に低くなっています。他の国では、企業が製品の価格を自分で固定することを許可していますが、企業の利益を監視および管理しています。米国以外の価格統制の追加やその他の価格規制の変更により、候補製品に対して請求できる金額が制限される可能性があります。したがって、米国以外の市場では、当社製品の償還額が米国に比べて減少し、商業的に妥当な収益と利益を生み出すには不十分な場合があります。
将来の製品候補に対する第三者支払者からの補償範囲と適切な償還を確立または維持できない場合、それらの製品の採用と売上に悪影響が及び、ひいてはそれらの製品候補が承認された場合のマーケティングまたは販売能力に悪影響を及ぼす可能性があります。補償範囲ポリシーと第三者支払者の償還率はいつでも変更される可能性があります。規制当局の承認を受けた1つ以上の製品について有利な補償範囲と償還状況が達成されたとしても、将来的にはより不利な補償ポリシーと償還率が実施される可能性があります。
さらに、私たちまたは私たちの協力者は、当社の製品候補に使用するために、コンパニオン診断テストを含む診断テストを開発する場合があります。コンパニオン診断テストでは、コンパニオン医薬品または生物学的製剤の補償と払い戻しとは別に、補償と払い戻しが必要です。医薬品に当てはまる補償と償還の取得に関する同様の課題は、コンパニオン診断にも当てはまります。補償範囲と適切な払い戻しが利用できない場合、または利用できるレベルが限られている場合、私たちが開発した製品候補をうまく商品化できない可能性があります。
規制当局の承認やその他の法令遵守事項に関連するリスク
米国または米国外の規制当局の承認を得ることができず、その結果、当社の製品候補を商品化できない可能性があります。
当社の製品候補は、とりわけ、研究、試験、開発、製造、安全性、有効性、承認、記録管理、報告、ラベリング、保管、包装、広告とプロモーション、価格設定、マーケティング、流通に関する広範な政府規制の対象となります。米国では、厳格な前臨床試験と臨床試験、および広範な規制当局の承認プロセスを無事に完了する必要があります
新薬が市販される前に、多くの米国以外の管轄区域で。これらおよびその他の規制要件を満たすには、費用がかかり、時間がかかり、不確実であり、予期せぬ遅延が発生する可能性があります。私たちが開発する可能性のある製品候補が、必要な臨床試験を経て、販売を開始するために必要な規制当局の承認を得るという保証はありません。
私たちは、大規模または極めて重要な臨床試験を実施、管理、完了したことはありません。また、FDAやその他の規制当局による規制当局の承認プロセスを管理したこともありません。FDAやその他の規制当局から承認を得るのに必要な時間は予測不可能で、製品候補の種類、複雑さ、新規性にもよりますが、通常は何年もかかる広範な臨床試験を成功裏に完了する必要があります。FDAと米国の旧機関が臨床試験データを評価する際に使用する基準は、医薬品開発中に変更される可能性があり、しばしば変更されるため、それらがどのように適用されるかを確実に予測することは困難です。さらに、FDAと米国の旧カウンターパートは、特定の製品候補のコンパニオン診断の承認または許可を要求する場合があり、そのような規制当局がコンパニオン診断を承認または承認しない場合、製品候補のマーケティングを承認しない場合があります。また、医薬品開発、臨床試験、FDAまたは元米国規制当局の規制審査期間中の将来の法律や行政措置を含む新しい政府規制、またはFDAまたは元米国規制当局の方針の変更により、予期しない遅延やコストの増加に遭遇する可能性があります。
必要な承認を求めたり取得したりするのが遅れたりしなかったりすると、私たちが開発して承認を求めている特定の製品候補から収益を生み出す能力に重大な悪影響を及ぼします。さらに、医薬品を販売するための規制当局の承認には、その医薬品の販売対象となる承認された用途または適応症、表示、またはその他の制限に重大な制限が課される場合があります。さらに、FDAには、NDAの承認の一環として、または承認後にREMSを要求する権限があります。これにより、承認された医薬品の流通または使用にさらなる要件または制限が課される場合があります。米国以外の管轄区域でも同様の要件が存在する可能性があります。これらの要件または制限には、専門的な訓練を受けた特定の医師または医療センターへの処方を制限すること、特定の安全な使用基準を満たす患者に治療を制限すること、治療を受けた患者に登録を要求することが含まれる場合があります。これらの制限や制限は、医薬品の市場規模を大幅に制限し、第三者支払者による償還に影響を与える可能性があります。
また、臨床試験の実施、製造および販売の承認、価格設定、第三者への償還など、米国外の多数の規制要件の対象となっています。米国以外の規制当局の承認プロセスは国によって異なり、通常、上記のFDA承認に関連するすべてのリスクと、米国以外の管轄区域における現地の規制の履行に起因するリスクが含まれます。さらに、米国以外の管轄区域で承認を得るのに必要な時間は、FDAの承認を得るのに必要な時間とは異なる場合があります。
当社の現在または将来の製品候補は、単独で、または他の承認された製品や治験中の新薬と組み合わせて使用すると、重大なAE、毒性、またはその他の望ましくない副作用を引き起こす可能性があり、その結果、規制当局の承認を阻害したり、市場での受け入れを妨げたり、商業的可能性を制限したり、商業的可能性を制限したり、重大な悪影響をもたらす可能性があります。
一般的な医薬品と同様に、製品候補の使用に関連する副作用やAEが発生する可能性があります。私たちの臨床試験の結果から、許容できないほど高い重症度や、副作用の有病率や予期しない特徴が明らかになる可能性があります。当社の製品候補によって引き起こされる望ましくない副作用により、当社または規制当局が臨床試験を中断、延期、または中止する可能性があり、その結果、ラベルがより制限されたり、FDAまたは同等の米国外の規制当局による規制当局の承認が遅れたり拒否されたりする可能性があります。薬物関連の副作用は、患者の募集や登録患者の試験完了能力に影響を与えたり、製造物責任の請求につながる可能性があります。これらの出来事はいずれも、当社の事業、財政状態、および見通しに重大な損害を与える可能性があります。
当社の製品候補が、単独で、または他の承認された製品や治験中の新薬と組み合わせて使用したときに、前臨床試験や臨床試験において望ましくない副作用を伴ったり、予期しない特性を持ったりする場合、開発を中断、遅延、中止するか、望ましくない副作用やその他の特性が少ないより狭い用途や亜集団に開発を制限する必要があるかもしれません。
リスクと利益の観点から、それほど厳しくない、または受け入れやすいです。治療関連の副作用は、患者の募集や登録被験者の試験完了能力に影響を与えたり、製造物責任の請求につながる可能性もあります。このような事態が発生すると、影響を受ける製品候補の市場での受け入れの達成または維持が妨げられ、当社の事業、財政状態、および見通しが著しく損なわれる可能性があります。
現在進行中および計画中の臨床試験に参加している患者は、将来、前臨床試験や以前の臨床試験では観察されなかった重大なAEやその他の副作用に苦しむ可能性があります。当社の製品候補の中には、慢性療法として使用されたり、規制当局によって安全上の懸念が特に精査される小児集団で使用されるものもあります。さらに、当社の製品候補を他の治療法と併用すると、製品候補が治療法に関連するAEを悪化させる可能性があります。当社の製品候補で治療された患者は、手術、放射線、化学療法の治療を受けている可能性もあります。これらの治療は、製品候補とは無関係の副作用やAEを引き起こす可能性がありますが、それでも臨床試験の成功に影響を与える可能性があります。重症患者を臨床試験に含めると、その患者が使用している可能性のある他の治療法や薬が原因で、または患者の病気の重症化により、死亡やその他の医学的有害事象が発生する可能性があります。
現在または将来の臨床試験のいずれかで重大なAEまたはその他の副作用が観察された場合、臨床試験への患者の募集が困難になったり、患者が試験から脱落したり、その製品候補の試験または開発努力を完全に中止する必要がある場合があります。私たち、FDA、その他の同等の規制当局、またはIRBは、さまざまな理由で、製品候補の臨床試験をいつでも中断することがあります。たとえば、そのような試験の被験者が容認できない健康上のリスクや有害な副作用にさらされているという信念などです。バイオテクノロジー業界で開発されたいくつかの潜在的な治療法は、初期段階の試験で治療上の可能性を示していましたが、後に副作用を引き起こし、さらなる開発を妨げることが判明しました。副作用によって製品候補が市販承認の取得または維持を妨げない場合でも、望ましくない副作用が他の治療法と比較して耐容性が高いため、市場での受け入れが妨げられる可能性があります。これらの進展はいずれも、当社の事業、財政状態、および見通しに重大な損害を与える可能性があります。
さらに、当社の製品候補のいずれかが販売承認を得た場合、そのような製品候補に関連する毒性が承認後に発生し、追加の臨床安全性試験を実施する必要が生じたり、禁忌、警告、注意事項が医薬品ラベルに追加されたり、製品の使用が著しく制限されたり、製品が市場から撤退したりする可能性があります。私たちの製品候補が、前臨床試験や初期段階の臨床試験に基づく規制当局の承認の取り消しを妨げたり、取り消したりするような毒性を人間に引き起こすかどうかは予測できません。
FDAおよびその他の同等の米国以外の規制当局は、管轄外の場所で実施された試験のデータを受け入れない場合があります。
将来、国際的な臨床試験を実施することを選択するかもしれません。FDAまたは他の同等の米国規制当局が、それぞれの管轄外で行われた臨床試験の研究データを承認するには、特定の条件が適用される場合があります。FDAまたは同等の米国外の規制当局による米国または他の管轄区域外で実施された臨床試験の研究データの承認には、特定の条件が適用される場合もあれば、まったく受け入れられない場合もあります。米国外の臨床試験のデータが米国での販売承認の唯一の基礎となることが意図されている場合、FDAは通常、米国以外のデータのみに基づいて申請を承認しません。ただし、i)データが米国の人口と米国の医療現場に適用可能であり、ii)試験が能力が認められた臨床研究者によって現在のGCP要件に従って実施され、iii)FDAがデータを検証できる場合を除きます。立入検査またはその他の適切な手段を通じて。さらに、調査対象の患者集団の妥当性や統計的検出力など、FDAの臨床試験要件を満たす必要があります。さらに、米国以外の研究データが唯一の承認の基礎となることを意図していない場合でも、FDAは、研究がGCP要件に従って適切に設計され、適切に実施され、必要に応じて現場での検査を通じて試験のデータを検証できる場合を除いて、そのデータを市販承認申請の裏付けとして受け入れません。 多くの旧米国規制当局には同様の承認要件があります。さらに、このような米国外の裁判には、裁判が行われた旧米国法域の該当する現地の法律が適用されます。FDAまたは同等の米国以外の規制当局が、該当する管轄外で行われた試験のデータを受け入れるという保証はありません。FDAまたは同等の米国以外の規制当局がそのようなデータを受け入れない場合、追加の試験が必要になります。
これは費用と時間がかかり、事業計画の段階が遅れることになり、その結果、製品候補が該当する法域での商品化の承認を受けられない可能性があります。
ある法域で製品候補の規制当局の承認を得て維持しても、他の法域で製品候補の規制当局の承認をうまく取得できるわけではありません。
ある法域で製品候補の規制当局の承認を取得して維持しても、他の法域で規制当局の承認を取得または維持できることを保証するものではありません。たとえば、FDAが製品候補の販売承認を与えたとしても、米国以外の管轄区域の同等の規制当局も、それらの国での製品候補の製造、マーケティング、販売促進、および償還を承認する必要があります。ただし、ある法域で規制当局の承認を得られなかったり遅れたりすると、他の管轄区域の規制当局の承認プロセスに悪影響を及ぼす可能性があります。承認手続きは管轄区域によって異なり、ある法域で実施された臨床試験が他の法域の規制当局に受け入れられない場合があるため、追加の前臨床試験や臨床試験など、米国とは異なる要件や管理審査期間が含まれる場合があります。米国以外の多くの法域では、候補製品はその法域での販売が承認される前に償還の承認を受ける必要があります。場合によっては、製品に請求する予定の価格も承認が必要です。
米国外の規制当局の承認を取得し、米国以前の規制要件の遵守を確立して維持することは、当社にとって重大な遅延、困難、およびコストにつながり、特定の国での当社製品の導入を遅らせたり妨げたりする可能性があります。私たちや将来の協力者が国際市場の規制要件を遵守しなかったり、該当するマーケティング承認を受けなかったりすると、ターゲット市場は縮小し、候補製品の市場ポテンシャルを最大限に引き出す能力が損なわれます。
当社の製品候補が規制当局の承認を受けたとしても、それらは製造販売後の重要な規制要件と監督の対象となります。
候補製品について受け取る可能性のある規制当局の承認には、規制当局への報告書の提出と、製品候補の安全性と有効性を監視するためのサーベイランスが必要です。特定の年齢層の使用制限、警告、注意事項、禁忌に関連する重大な制限が含まれている場合があり、承認後の面倒な研究やリスク管理要件が含まれる場合があります。たとえば、FDAは、当社の製品候補を承認するためにREMSを要求する場合があります。これには、投薬ガイド、医師のトレーニング、コミュニケーション計画、または安全な使用を確保するための追加要素(制限された配布方法、患者登録、その他のリスク最小化ツールなど)が必要になる場合があります。さらに、FDAまたは元米国規制当局が当社の製品候補を承認した場合、製品候補の製造プロセス、ラベリング、包装、流通、AE報告、保管、広告、宣伝、輸入、輸出、記録管理には、広範かつ継続的な規制要件が適用されます。これらの要件には、安全性およびその他の市販後の情報およびレポートの提出、登録、ならびに承認後に実施する臨床試験におけるcGMPまたは同様の米国外の要件とGCPの継続的な遵守が含まれます。さらに、CMOとその施設は、cGMP規制または同様の米国外の要件と基準に準拠しているかどうか、FDAおよびその他の規制当局による継続的な審査と定期的な予告なしの検査の対象となります。当社または規制機関が、重症度や頻度が予想外のAEなど、製品に関するこれまで知られていなかった問題、または製品が製造されている施設の問題などを発見した場合、規制機関は、製品のリコールや市場からの撤回、製造の中止を要求するなど、その製品、製造施設、または当社に制限を課すことがあります。さらに、FDAおよびその他の同等の米国以外の規制要件に従わない場合、当社は次のような行政上または司法上の制裁の対象となる可能性があります。
•製品承認の遅延または拒否。
•進行中または計画中の試験の全部または一部の臨床保留を含む、臨床試験を実施する当社の能力に対する制限。
•製品、メーカー、または製造プロセスの制限。
•警告または無題の手紙。
•民事および刑事上の罰則。
•差し止め命令;
•規制当局の承認の一時停止または撤回。
•製品の押収、拘禁または輸入禁止。
•自発的または必須の製品リコールと広報要件。
•生産の全部または一部の停止、および
•費用のかかる新しい製造要件など、業務に制限を課すこと。
上記のイベントやペナルティが発生すると、製品候補を商品化して収益を生み出すことができなくなり、それに対応するために多大な時間とリソースを費やす必要が生じ、評判が悪くなる可能性があります。
FDAやその他の規制当局の方針が変更されたり、当社の製品候補の規制当局の承認を防いだり、制限したり、遅らせたりする可能性のある追加の政府規制が制定されることがあります。 また、米国または海外の将来の法律や行政措置によって生じる可能性のある政府規制の可能性、性質、範囲を予測することもできません。
FDAやその他の規制機関は、適応外使用の促進を禁止する法律や規制を積極的に施行しています。
当社の製品候補のいずれかが承認され、それらの製品の適応外使用を不適切に宣伝したことが判明した場合、当社は重大な責任の対象となる可能性があります。FDAやその他の規制機関は、当社の製品候補などの処方薬について行われる可能性のあるプロモーションの主張が承認されれば、厳しく規制しています。特に、FDAやその他の規制機関によって承認されていない用途では、製品の承認されたラベルに反映されていない用途で製品を宣伝することはできません。製品候補の市販承認を受けた場合でも、医師は承認されたラベルと矛盾する方法で患者にそれを処方することがあります。そのような適応外使用を促進したことが判明した場合、重大な責任の対象となる可能性があります。米国連邦政府は、適応外使用の不適切な宣伝の疑いで企業に多額の民事上および刑事上の罰金を科し、いくつかの企業が適応外販売を行うことを禁止しています。FDAはまた、特定の販売促進行為を変更または制限する同意判決または恒久的差止命令を企業に締結するよう求めています。製品候補のプロモーションをうまく管理できない場合、承認された場合、重大な責任の対象となり、当社の事業と財政状態に重大な悪影響を及ぼす可能性があります。
資金不足や世界的な健康上の懸念によってFDA、SEC、その他の政府機関が混乱した場合、これらの機関は当社の事業運営の基盤となる通常の事業機能を実行できなくなり、当社の事業に悪影響を及ぼす可能性があります。
FDAやその他の規制当局が新製品を審査して承認する能力は、政府の予算や資金水準、主要人員の雇用と維持の能力、ユーザー料金の支払いを受け入れる能力、法律、規制、ポリシーの変更、FDAや米国外の規制当局の日常業務遂行能力に影響を与える可能性のあるその他の事象など、さまざまな要因によって影響を受ける可能性があります。その結果、FDAと元米国規制当局での平均審査時間は、近年変動しています。さらに、研究開発活動に資金を提供する機関を含め、SECや私たちの事業が依存する可能性のある他の政府機関への政府資金は、本質的に流動的で予測不可能な政治的プロセスの対象となります。
アムステルダムへの移転とそれに伴うスタッフの交代により、FDAやEMAなどの他の機関が混乱すると、必要な政府機関による新薬の審査や承認に必要な時間が遅くなり、当社の事業に悪影響を及ぼす可能性があります。たとえば、2018年と2019年を含む近年、米国政府は何度か閉鎖し、FDAやSECなどの特定の規制機関は、重要な従業員を解雇し、重要な活動を停止する必要がありました。さらに、公開企業としての事業において、将来の政府の閉鎖や遅延により、適切な資本投入と事業継続に必要な資本を獲得する能力が損なわれる可能性があります。
これとは別に、COVID-19に対応して、FDAは国内および米国外の製造施設のほとんどの検査をさまざまな時点で延期しました。その後、FDAは可能な限り国内施設の標準検査業務を再開しましたが、FDAは、従業員と規制対象団体の安全を確保するために、検査活動の監視と変更を続けています。ウイルスの再発や新しい亜種の出現により、検査がさらに遅れる可能性があります。 米国外の規制当局が、COVID-19に対応して同様の制限やその他の政策措置を採用した可能性があります。
FDAまたは同等の米国外の規制当局から迅速な承認またはその他の迅速な開発またはレビューを得ることができない場合、私たちが検討しているものを超える追加の前臨床試験または臨床試験を実施する必要があります。これにより、必要な市販承認の取得にかかる費用が増加し、受領が遅れる可能性があります。FDAから早急な承認を受けたとしても、確認試験で臨床的有益性が検証されない場合、または厳格な市販後の要件に準拠していない場合、FDAは加速承認の撤回を求めることがあります。
将来的には、1つ以上の製品候補について、迅速な承認または別の形態の迅速な開発またはレビューを求める可能性があります。加速承認プログラムでは、FDAは、利用可能な治療法よりも有意義な治療効果をもたらす重篤なまたは生命を脅かす状態を治療するために設計された製品候補を、その製品候補が臨床的利益を予測する可能性がかなり高い代理エンドポイントまたは中間臨床エンドポイントに効果があると判断された場合に、加速承認を与える場合があります。FDAは、臨床的利益とは、不可逆的な罹患率や死亡率など、特定の疾患に関連して臨床的に意味のあるポジティブな治療効果であると考えています。承認を早めるために、代理エンドポイントとは、臨床的利益を予測すると考えられているが、それ自体は臨床的利益の尺度ではないマーカーです。検査室での測定、X線画像、身体的徴候、またはその他の測定値などです。中間臨床エンドポイントとは、不可逆的な罹患率または死亡率への影響、またはその他の臨床的利益への影響を予測する可能性がかなり高い、不可逆的な罹患率または死亡率への影響よりも早く測定できる臨床エンドポイントです。早期承認経路は、利用可能な治療法に対する新薬の利点が直接的な治療上の利点ではないが、患者と公衆衛生の観点から臨床的に重要な改善である場合に使用できます。承認された場合、承認が早まるには、通常、薬の臨床的利益を検証して説明するための追加の確認研究を熱心に実施するというスポンサーの同意が条件となります。そのような確認研究で薬の臨床的利点が確認されない場合、FDAは速やかにその薬の承認を取り消すことがあります。さらに、2022年12月、大統領は2023会計年度までの米国政府への資金提供を目的としたオムニバス歳出法案に署名しました。オムニバス法案には、2022年の食品医薬品オムニバス改革法が含まれています。この法律は、とりわけ、確認試験の実施に対するFDAの監視を強化するなど、承認が加速された製品を規制するFDAの能力を拡大することを目的とした改革を導入しました。ただし、これらの改革の最終的な影響は不明のままです。
EUでは、中央集権的な手続きの下で、EMAのヒト用医薬品委員会が販売承認申請の迅速な審査を行うことがあります。迅速な評価手続きを要求する申請者は、特に治療法の革新の観点から、製品候補が公衆衛生上の大きな関心事になると期待されていることを正当化する必要があります。
製品候補の迅速な承認または別の形態の迅速な開発またはレビューを求める前に、FDAまたは米国の元規制当局にフィードバックを求めるつもりです。それ以外の場合は、迅速な承認または別の形態の迅速な開発またはレビューを求め、受ける能力を評価します。フィードバックやその他の要素を評価した上で、迅速な承認やその他の形態の開発、レビュー、承認のためにNDAを追求または提出することを決定するという保証はありません。さらに、もし私たちが
当社の製品候補について、迅速な承認または別の形式の迅速な開発、レビュー、または承認の申請書を提出することを決定した場合、そのような提出または申請が受理されること、またはそのような迅速な開発、レビュー、または承認が適時に、またはまったく許可されるという保証はありません。FDAまたは他の同等の米国以外の規制当局も、申請を検討したり、あらゆる種類の承認を与える前に、さらなる調査を実施するよう要求する可能性があります。製品候補の迅速な承認またはその他の迅速な開発、レビュー、または承認を得られないと、そのような製品候補の商品化までの期間が長くなり、そのような製品候補の開発コストが増加し、市場における当社の競争力が損なわれる可能性があります。
現在の規制や将来の法律の変更により、困難に直面する可能性があります。
既存の規制方針が変更されたり、当社の製品候補の規制当局による承認を防いだり、制限したり、遅らせたりする可能性のある追加の政府規制が制定される可能性があります。米国内外を問わず、将来の法律や行政措置によって生じる可能性のある政府規制の可能性、性質、範囲を予測することはできません。既存の要件の変化や新しい要件やポリシーの採用に遅くなったり、適応できなかったり、規制遵守を維持できなかったりすると、取得した販売承認を失い、収益性を達成または維持できなくなる可能性があります。
たとえば、2010年3月、2010年の医療と教育の和解法、またはまとめてACAによって改正された2010年の患者保護および手ごろな価格の医療法が可決されました。これにより、政府と民間保険会社の両方による医療の資金調達方法が大幅に変わり、米国の製薬業界に大きな影響を与えます。ACAの制定以来、ACAの特定の側面について司法、行政、議会による異議申し立てがありました。 2021年6月17日、米国最高裁判所は、ACAの合憲性について特に判決を下すことなく、ACAに対する最新の司法上の異議申し立てを却下しました。最高裁判所の判決に先立ち、バイデン大統領は、ACAマーケットプレイスを通じて健康保険に加入する目的で、2021年2月15日から2021年8月15日までの特別加入期間を開始する大統領命令を出しました。大統領令はまた、特定の政府機関に対し、医療へのアクセスを制限する既存の方針や規則を見直し、再検討するよう指示しました。これには、就労要件を含むメディケイドの実証プロジェクトや免除プログラム、メディケイドやACAを通じて健康保険に加入するうえで不必要な障壁となる政策の再検討などが含まれます。他の医療制度改革措置が当社の事業にどのように影響するかは不明です。私たちは、将来の事業に影響を与える可能性のあるACAの変更を引き続き監視しています。
さらに、ACAが制定されて以来、米国では他の法改正が提案され採択されています。これらの変更には、2021年の米国レスキュープラン法が含まれます。これにより、2024年1月1日から、現在医薬品の平均メーカー価格の100%に設定されている法定のメディケイド医薬品リベートの上限が廃止されます。
さらに、最近、製薬会社が市販製品の価格を設定する方法について政府の監視が厳しくなっています。その結果、米国議会でいくつかの調査が行われ、とりわけ、製品価格の透明性を高め、価格設定と製造業者の患者プログラムの関係を見直し、医薬品に対する政府プログラムの償還方法を改革することを目的とした連邦法および州法が提案および制定されました。ごく最近、2022年8月16日、IRAが法制化されました。とりわけ、IRAは、特定の医薬品の製造業者に、上限を条件として交渉できる価格で、メディケアと価格交渉を行うことを義務付けています(2026年から)。インフレを上回る価格上昇を罰するために、メディケアパートBとメディケアパートDに基づいてリベートを課し(2023年に最初に予定されている)、パートDの補償ギャップ割引プログラムを新しい割引プログラムに置き換えます(2025年から)。IRAは、国土安全保障長官が最初の数年間、規制ではなくガイダンスを通じてこれらの規定の多くを実施することを許可しています。HHSは、これらのプログラムが実施されるたびにガイダンスを発行し、更新し続けます。2023年8月29日、HHSは、価格交渉の対象となる最初の10種類の医薬品のリストを発表しました。ただし、メディケアの医薬品価格交渉プログラムには現在法的問題があります。さらに、バイデン政権の2022年10月の大統領令に応えて、2023年2月14日、HHSは、メディケアおよびメディケイドイノベーションセンターによる3つの新しい検査モデルの概要を示すレポートを発表しました。これらのモデルは、医薬品コストの削減、アクセシビリティの促進、および医療の質の向上を実現する能力について評価されます。このモデルが将来、何らかの健康改革策に活用されるかどうかは不明です。
州レベルでは、価格や患者への償還の制約、割引、特定の製品アクセスやマーケティング費用の開示制限、透明性対策など、医薬品や生物製品の価格を管理するための法律を議会が可決し、規制を実施することが増えています。また、場合によっては、他国からの輸入や大量購入を促進することを目的としたものもあります。
将来採用される可能性のある他の医療改革措置により、補償基準がより厳しくなり、承認された製品に対して受け取る価格がさらに下がる可能性があると予想しています。メディケアやその他の政府プログラムからの償還額が減額されると、民間の支払者からの支払いも同様に減少する可能性があります。コスト抑制策やその他の医療改革の実施は、収益の創出、収益性の達成、または製品候補の商品化を妨げる可能性があります。
承認後の要件を拡大し、バイオテクノロジー製品の販売および販促活動を制限するための立法上および規制上の提案がなされました。追加の法改正が制定されるのか、FDAや米国外の規制、ガイダンス、解釈が変更されるのか、そのような変更が製品候補の販売承認に与える影響があるとしても、どうなるかはわかりません。さらに、FDAの承認プロセスに対する議会による監視が強化されると、販売承認が大幅に遅れたり妨げられたりする可能性があります。また、より厳しい製品ラベル貼付、市販後テスト、その他の要件の対象となる可能性があります。
現在および将来の事業活動に関連する医療従事者、臨床研究者、CRO、および第三者支払者との関係は、詐欺および虐待に関する法律およびその他の医療法および規制の対象となる可能性があります。
医療提供者と第三者の支払者は、当社が販売承認を得た製品候補の推奨と処方において主要な役割を果たします。医療従事者、臨床研究者、CRO、第三者の支払者および顧客との現在および将来の取り決めにより、広く適用される詐欺や虐待、およびその他の医療法規制にさらされる可能性があります。これらの法律は、当社がマーケティング承認を得た製品のマーケティング、販売、流通における事業上または財務上の取り決めおよび関係を制約する可能性があります。適用される連邦、州、および米国外の医療法および規制に基づく制限には、次のものがあります。
•連邦反キックバック法は、とりわけ、個人や団体が、連邦医療プログラムに基づいて支払いが行われる可能性のある商品やサービスを誘発したり、報奨したり、その見返りとして、直接的または間接的に、現金または現物で報酬を故意かつ故意に求め、提供したり、受け取ったり、提供したりすることを禁じています。メディケアやメディケイドなど。個人または団体は、連邦反キックバック法について実際に知っている必要はなく、違反する特定の意図を持っている必要もありません。
•民事上の虚偽請求法を含む連邦の虚偽請求法(民事上の虚偽請求法を含む)は、個人または団体が、とりわけ、虚偽または詐欺的な支払い請求を故意に連邦政府に提示したり、提示させたり、金銭の支払い義務を回避、軽減、または隠蔽するために虚偽の陳述をしたりすることを禁止しています。連邦政府。さらに、政府は、米国連邦反キックバック法の違反に起因する品目またはサービスを含む請求は、民事虚偽請求法の目的上、虚偽または詐欺的な請求を構成すると主張する場合があります。
•1996年の連邦医療保険の相互運用性と説明責任に関する法律(HIPAA)は、とりわけ、医療給付プログラムを詐欺するスキームを実行または実行しようとすること、または医療問題に関して虚偽の陳述を行うことを禁じています。連邦反キックバック法と同様に、個人または団体は、違反を犯すために、法律に関する実際の知識や違反する特定の意図を持っている必要はありません。
•連邦医師支払いサンシャイン法(オープンペイメント法に改称)は、対象となる医薬品、デバイス、生物製剤、および医療用品の該当するメーカーを義務付けています。
支払いは、特定の例外を除いて、メディケア、メディケイド、または児童健康保険プログラムの下で利用できます。法律で定義されている医師、助手やナースプラクティショナーを含む特定の非医師開業医、教育病院への支払いやその他の価値移転に関する情報、および医師とその近親者が保有する所有権と投資権に関する情報を毎年CMSに報告します。報告された情報は、検索可能なWebサイトで公開されており、毎年開示する必要があります。そして
•民間保険会社を含む非政府の第三者支払者によって払い戻される医療品またはサービスに関する販売またはマーケティングの取り決めや請求には、州のキックバック防止法や虚偽請求法など、類似の州法および米国以前の法律および規制が適用される場合があります。
一部の州法では、バイオテクノロジー企業がバイオテクノロジー業界の自主コンプライアンスガイドラインと連邦政府によって公布された関連するコンプライアンスガイダンスを遵守することを義務付けており、製薬会社に対し、支払いやその他の価値の移転に関する情報を医師やその他の医療提供者またはマーケティング支出に報告するよう要求する場合があります。一部の州法では、バイオテクノロジー企業が特定の医薬品の価格に関する情報を報告することを義務付けています。
適用されるデータ保護、プライバシーおよびセキュリティに関する法律、規制、基準、その他の要件を実際に遵守していない、または違反していると認識されると、当社の事業、経営成績、および財政状態に悪影響を及ぼす可能性があります。
世界のデータ保護環境は急速に進化しており、臨床試験に関連して収集する可能性のある情報などの個人情報の収集、使用、開示、保持、およびセキュリティに関する多くの州、連邦、および米国外の法律、要件、規制の対象となっているか、対象となる可能性があります。実施基準と執行慣行は、当面の間、不確実なままである可能性が高く、将来の法律、規制、基準、またはそれらの要件に対する認識が当社の事業に与える影響はまだ判断できません。この進化は、当社の事業に不確実性をもたらし、特定の法域での事業遂行、または個人情報の収集、保管、転送、使用、共有の能力に影響を与え、契約におけるより厄介な義務の受け入れを必要としたり、責任が生じたり、追加費用を課したりする可能性があります。これらの法律、規制、標準を遵守するためのコストは高く、将来的には増加する可能性があります。当社が連邦、州、または米国以前の法律または規制、当社の内部方針および手続き、または個人情報の処理に適用される契約に従わなかった、または遵守していないと認識された場合、否定的な宣伝、政府の調査および執行措置、第三者による請求、および当社の評判の低下につながる可能性があり、そのいずれも当社の事業、経営成績、および財政状態に重大な悪影響を及ぼす可能性があります。
米国では、HIPAAは、とりわけ、個人を特定できる健康情報のプライバシー、セキュリティ、送信、侵害報告に関する特定の基準を課しています。私たちは現在、HIPAAの対象事業体または事業提携先として行動しているとは考えていないため、その要件や罰則を直接受けることはありませんが、HIPAAに基づくプライバシーおよびセキュリティ要件の対象となる第三者(臨床試験データを入手する研究機関を含む)から健康情報を入手する場合があります。事実と状況によっては、HIPAAに違反した場合、重大な罰則が科せられる可能性があります。一部の州では、個人情報のプライバシー、処理、保護について、同等のプライバシーおよびセキュリティに関する法律や規制を採用しています。たとえば、カリフォルニア州はカリフォルニア州消費者プライバシー法(CCPA)を制定し、2020年1月1日に施行されました。CCPAは、カリフォルニア州の消費者に個人のプライバシー権を与え、特定の個人情報を扱う団体のプライバシーとセキュリティに関する義務を強化します。CCPAは、違反に対する民事罰のほか、データ侵害訴訟の可能性とそれに伴うリスクを高めたデータ侵害に対する私的訴訟権を規定しています。さらに、カリフォルニア州プライバシー権法(CPRA)は、通常2023年1月1日に施行され、CCPAを大幅に改正します。CPRAは、追加の消費者権利プロセス、データ使用の制限、リスクの高いデータに対する新しい監査要件、機密データの特定の使用のオプトアウトなど、対象となる企業に追加のデータ保護義務を課しています。また、実質的な規制の発行を許可された新しいカリフォルニアのデータ保護機関が設立され、プライバシーと情報セキュリティの強化につながる可能性があります。追加のコンプライアンス投資や潜在的なビジネスプロセスの変更が必要になる場合があります。他の州でも同様の法律が可決され、現在も続いています
米国におけるプライバシー法がより厳しくなる傾向を反映して、州および連邦レベルで提案されます。このような法律が制定されると、要件が相反する可能性があり、コンプライアンスが困難になる可能性があります。私たちがHIPAA、CCPA、CPRA、またはその他の国内のプライバシーおよびデータ保護法の対象または影響を受ける場合、これらの法律の要件に従わなかったことによる責任は、当社の財政状態に悪影響を及ぼす可能性があります。
海外での事業は、データ保護当局からの監視や注意の強化の対象となる可能性もあります。たとえば、EU一般データ保護規則(GDPR)は2018年5月に発効し、欧州経済領域(EEA)内、またはEEAでの活動に関連して、個人の個人データを処理するための厳しい要件を課しています。さらに、臨床試験参加者に関して当社が処理する個人データの一部は、GDPRに基づく特別なカテゴリーまたは機密の個人データであり、追加のコンプライアンス義務および現地法の特例の対象となります。GDPRを遵守しなければならない企業は、データ保護要件のより強固な規制施行、行政上の罰則、違反した場合の罰金、または違反した企業の世界年間収益の4%、いずれか大きい方の金額の罰金など、コンプライアンス義務とリスクの増大に直面します。罰金に加えて、GDPR違反は、規制調査、評判の低下、データ処理活動の中止/変更の命令、執行通知、査定通知(強制監査用)、および/または民事訴訟(集団訴訟を含む)につながる可能性があります。他の要件の中でも、GDPRは、GDPRの対象となる個人データを、米国など、そのような個人データに対して適切な保護を提供していない第三国に転送することを規制しています。欧州経済領域(EEA)と米国間の現在の移転メカニズムの有効性と存続性は依然として不明です。欧州連合司法裁判所(CJEU)の判例法では、標準契約条項(適切な個人データ転送メカニズムとして欧州委員会によって承認された標準形式の契約)に依存するだけでは必ずしもすべての状況で十分ではなく、転送はケースバイケースで評価する必要があると定められています。2022年10月7日、バイデン大統領は「米国の情報活動に対する保護措置の強化」に関する大統領令に署名しました。この命令は、EEAから米国へのデータ転送に関してCJEUが提起した懸念に対処するための新しい救済メカニズムと拘束力のある保障措置を導入し、2022年12月13日に発表された新しいEU-米国データプライバシーフレームワーク(DPF)の基礎を形成しました。DPFはまた、EUと英国の市民を対象とした新しい救済メカニズムを導入しました。これは、以前のCJEUの判決における主要な懸念事項に対処するものであり、標準契約条項に基づく移転が将来異議を申し立てられる可能性が低くなることを意味する可能性があります。欧州委員会は2023年7月10日にDPFに関する妥当性決定を採択し、DPFはDPFの下で自己認証された米国企業へのGDPR移転メカニズムとして有効になりました。現在、グループ内および第三者移転の両方に関して、個人データをEEA外および米国を含む英国外に転送する際には、EU標準契約条項、EU標準契約条項の英国補遺、および関連する英国国際データ転送協定に依存しています。また、特定の状況では、個人の同意に基づいて個人データを転送する場合もあります。国際的な個人データの移転、特に米国への移転に関する法的な複雑さと不確実性の時代を経て、米国などへの移転に関して、規制上のガイダンスと執行の状況は引き続き発展すると予想されます。国際的な個人データ移転に関する既存の法的複雑さと不確実性は今後も続くと予想しています。特に、DPF妥当性決定には異議が唱えられ、米国およびその他の法域への国際移転は、引き続き規制当局による厳しい監視の対象となると予想されます。その結果、特定の運用上の変更を行う必要があり、既存のデータ転送に関する改訂された標準契約条項やその他の関連文書を必要な期間内に実施する必要があります。
さらに、2021年1月1日から、GDPRと英国のGDPRの両方を遵守する必要がありました。英国のGDPRは、2018年の改正英国データ保護法とともに、英国の国内法でGDPRを維持しています。英国のGDPRは、GDPRに基づく罰金、つまり1750万ポンドまたは世界の売上高の4%のいずれか大きい方の罰金を反映しています。2023年10月12日、DPFの英国拡張の下で自己認証された米国法人への英国GDPRデータ転送メカニズムとして、英国DPFの延長が(英国政府の承認を得て)発効しました。他の国や管轄区域への事業拡大を続けるにつれて、ビジネスの遂行方法に影響を与える可能性のある追加の法律や規制の対象となる可能性があります。
当社の従業員、独立請負業者、コンサルタント、商業協力者、主任研究者、CRO、CMO、サプライヤー、ベンダーは、規制基準や要件への違反など、不正行為またはその他の不適切な活動に従事する可能性があります。
私たちは、従業員、独立請負業者、コンサルタント、商業協力者、主任研究者、CRO、CMO、サプライヤー、ベンダーが不正行為やその他の不適切な活動に従事するリスクにさらされています。これらの当事者による不正行為には、FDAやその他の旧米国当局の規制に従わなかったり、FDAまたは米国外の規制当局に正確な情報を提供しなかったり、連邦、州、および米国外の医療詐欺や乱用に関する法律や規制を遵守しなかったり、財務情報やデータを正確に報告したり、不正行為を当社に開示したりすることが含まれる可能性があります。特に、ヘルスケア業界における販売、マーケティング、ビジネスの取り決めには、詐欺、不正行為、キックバック、自己取引、その他の不正行為を防ぐことを目的とした広範な法律や規制が適用されます。これらの法律や規制は、価格設定、割引、マーケティングとプロモーション、販売手数料、顧客インセンティブプログラム、その他のビジネス上の取り決めを幅広く制限または禁止している場合があります。これらの関係者による不正行為には、臨床試験の過程で得られた情報の不適切な使用が含まれる可能性があり、規制上の制裁措置や当社の評判への重大な損害につながる可能性があります。これらの当事者による不正行為を特定して阻止することが常に可能というわけではなく、この活動を検出して防止するために講じる予防措置は、未知または管理されていないリスクや損失を管理したり、これらの法律や規制に従わなかったことに起因する政府の調査やその他の措置や訴訟から私たちを保護したりするのに効果的ではない場合があります。私たちに対してそのような訴訟が提起され、私たちが自分自身を守ることや権利を主張することに成功しなかった場合、それらの行動は、民事、刑事、行政上の罰則を含む重大な罰則、損害賠償、罰金、解雇、個人投獄、メディケアやメディケイドなどの政府資金による医療プログラムへの参加の除外、完全性監視と報告義務、契約など、当社の事業に重大な影響を与える可能性があります損害、評判の低下、利益の減少、そして将来収益と事業の縮小または再編。
環境、健康、安全に関する法律や規制を遵守しないと、罰金や罰則の対象になったり、事業に重大な悪影響を及ぼす可能性のある費用が発生したりする可能性があります。
私たちは、実験室での手順や、有害物質や廃棄物の取り扱い、使用、保管、処理、廃棄に関するものを含め、環境、健康、安全に関する多数の法律や規制の対象となっています。私たちの事業には、化学物質や生物学的物質を含む危険で可燃性の物質の使用が含まれます。私たちの事業では有害廃棄物も生産しています。私たちは通常、これらの材料や廃棄物の処分について第三者と契約しています。これらの材料による汚染や怪我のリスクを排除することはできません。危険物の使用により汚染やけがが発生した場合、その結果生じた損害について当社が責任を負う可能性があり、いかなる責任も当社の資源を超える可能性があります。また、民事または刑事上の罰金や罰則に関連して多額の費用が発生する可能性もあります。
私たちは、危険物の使用による従業員の傷害によって発生する可能性のある費用や費用をカバーするために労働者災害補償保険に加入していますが、この保険は潜在的な負債に対して十分な補償を提供しない場合があります。私たちは、化学物質や生物物質を含む危険で可燃性の物質の保管または廃棄に関連して当社に対して請求される可能性のある環境責任または有毒な不法行為の請求に対する保険に加入していません。
さらに、現在または将来の環境、健康、安全に関する法律や規制を遵守するために多額の費用が発生する可能性があります。これらの現在または将来の法律や規制は、私たちの研究、開発、または商品化の取り組みを損なう可能性があります。これらの法律や規制に従わないと、多額の罰金、罰則、その他の制裁措置が科せられることもあります。
動物実験が制限される可能性があるため、私たちの研究開発活動が影響を受けたり、遅れたりする可能性があります。
特定の法律や規制では、ヒトを対象とした臨床試験を開始する前に、製品候補を動物で試験することが義務付けられています。動物実験活動は論争と不利な宣伝の対象となっています。動物の権利団体やその他の組織や個人は、次のことを迫って動物実験活動を止めようとしています
これらの分野における法律や規制、そして抗議活動やその他の手段を通じてこれらの活動を妨害することによる。これらのグループの活動が成功する限り、私たちの研究開発活動は中断されたり、遅れたり、より高価になったりする可能性があります。
当社の事業活動は、米国海外腐敗行為防止法(FCPA)、および当社が事業を展開する他の国の同様の贈収賄防止法および腐敗防止法、ならびに米国および特定の米国以外の輸出規制、貿易制裁、輸入法および規制の対象となる場合があります。これらの法的要件を遵守すると、米国以外の市場での競争力が制限され、違反した場合は責任を問われる可能性があります。
米国外に事業をさらに拡大する場合、事業を計画している各管轄区域の多数の法律や規制を遵守するために、追加のリソースを投入する必要があります。当社の事業活動は、FCPAおよび当社が事業を展開する他の国の贈収賄防止または腐敗防止に関する法律、規制、または規則の対象となる場合があります。FCPAは通常、企業とその従業員、および第三者の仲介業者が、公的な行動に影響を与えたり、事業を獲得または維持したりするために、直接的または間接的に、米国政府以外の役人に価値のあるものを提供、約束、譲渡、または許可することを禁じています。FCPAはまた、上場企業に対し、企業の取引を正確かつ公正に反映した帳簿と記録を作成して保管し、適切な内部会計管理システムを考案して維持することを義務付けています。私たちの事業は厳しく規制されているため、米国以外の政府の職員を含む公務員との重要なやり取りが必要です。さらに、他の多くの国では、政府が所有・運営する病院、医師、その他の病院従業員は、FCPAでは元米国公務員とみなされます。最近、SECと米国司法省は、バイオテクノロジーと製薬会社に対するFCPAの執行活動を強化しました。当社の従業員、代理人、請負業者、または当社の関連会社全員が、特にこれらの法律が非常に複雑であることを考えると、すべての適用法および規制を遵守するかどうかは定かではありません。これらの法律や規制に違反すると、罰金、当社、当社の役員または従業員に対する刑事制裁、剥奪処分、その他の制裁や是正措置、および当社の事業遂行の禁止につながる可能性があります。このような違反には、1つ以上の国で製品を提供する能力の禁止が含まれ、当社の評判、ブランド、国際活動、従業員を引き付けて維持する能力、事業、見通し、経営成績、財政状態に重大な損害を与える可能性があります。
さらに、当社の製品および活動は、米国および米国以外の輸出規制、貿易制裁、輸入法および規制の対象となる場合があります。当社製品の輸出入に関する政府の規制、または該当する場合、当社製品に必要な輸入または輸出許可を取得していないと、当社の海外販売に悪影響を及ぼし、収益に悪影響を及ぼす可能性があります。当社製品の輸出に関して適用される規制要件を遵守すると、国際市場での当社製品の導入が遅れたり、場合によっては、一部の国への当社製品の輸出が完全に妨げられたりする可能性があります。さらに、米国の輸出管理法と経済制裁により、米国の制裁対象国、政府、および個人への特定の製品やサービスの出荷が禁止されています。輸出入規制やそのような経済制裁を遵守しなかった場合、罰金や特定の輸出特権の拒否などの罰則が科せられる可能性があります。さらに、新しい輸出入制限、新しい法律、既存の規制の施行または範囲、またはそのような規制の対象となる国、個人、または製品におけるアプローチの変化により、当社製品の使用が減少するか、国際事業を展開する既存または潜在的な顧客に製品を輸出する能力が低下する可能性があります。当社製品の使用が減少したり、当社製品へのアクセスを輸出または販売する能力が制限されたりすると、当社の事業に悪影響を及ぼす可能性があります。
従業員に関するリスク、当社の成長管理、および当社の事業に関連するその他のリスク
私たちの成功は、高度に熟練した執行役員と従業員を引き付けて維持する能力に大きく依存しています。
成功するには、資格のある臨床、科学、技術、管理の人材を募集、維持、管理、意欲を高める必要があり、経験豊富な人材をめぐって激しい競争に直面しています。私たちは、経営陣の主要メンバー、科学・医療スタッフに大きく依存しています。特に管理レベルで、有能な人材の誘致と維持に成功しなければ、事業計画を実行する能力に悪影響を及ぼし、経営成績に悪影響を及ぼす可能性があります。特に、1人以上の執行役員の喪失は
適切な交代要員を適時に採用できなければ、私たちにとって有害です。バイオテクノロジー分野での有能な人材をめぐる競争は激しく、その結果、将来の事業の成功に必要な有能な人材を引き付けて維持し続けることができない可能性があります。将来、経験豊富な人材を当社に引き付けることが困難になり、従業員の採用と維持の取り組みに多額の財源を費やす必要があるかもしれません。
私たちが有能な人材を求めて競争している他のバイオテクノロジー企業の多くは、私たちよりも多くの財源やその他のリソース、さまざまなリスクプロファイル、業界での長い歴史を持っています。また、キャリアアップのためのより多様な機会とより良い展望を提供してくれるかもしれません。これらの特徴の中には、質の高い候補者にとって、私たちが提供しているものよりも魅力的なものもあります。質の高い人材を引き付けて維持し続けることができなければ、製品候補を発見、開発、商品化できる速度と成功が制限され、ビジネスを成功裏に成長させる可能性が損なわれます。
効果的な販売またはマーケティング能力を確立できない、または第三者と製品候補を販売またはマーケティングする契約を締結できない場合、規制当局の承認を得た製品候補の販売またはマーケティングを成功させることができない場合があります。
製品候補を商品化したことはありません。当社が商品化権を保持している製品候補が承認された場合、商品化するには、製品候補の販売またはマーケティングが承認されている地域ごとに、マーケティング、販売、流通、管理、およびその他の非技術的能力を構築するか、第三者と取り決めをしてこれらのサービスを実施する必要があります。これらの必須のタスクをうまく達成できないかもしれません。また、当社が商品化権を保持していない製品候補については、承認された製品候補を商品化するために協力者の支援を頼りにします。
技術的な専門知識を持ち、製品候補を商品化するための流通能力を備えた営業またはマーケティングチームを社内に設立することは、費用と時間がかかり、経営幹部の多大な注意を必要とします。製品候補を自社で商品化する能力に影響を与える可能性のある要因には、十分な数の有能な営業およびマーケティング担当者の採用と維持、製品候補を処方するために十分な数の医師にアクセスまたは説得すること、および独立した販売およびマーケティング組織の設立に関連するその他の予期しない費用が含まれます。社内の販売、マーケティング、流通能力の開発に失敗したり遅れたりすると、特に私たちに代わってそのようなサービスを提供するための第三者と契約を結んでいない場合、市場への承認を得た製品候補の商品化に悪影響を与える可能性があります。あるいは、自社の営業部隊と流通システムを強化するため、または自社の営業部隊と流通システムの代わりに、直接販売部隊と確立された流通システムを持つ第三者とグローバルに、または地域ごとに協力することを選択した場合、提案された協力に関連してそのような第三者と交渉し、取り決めを締結する必要があります。必要なときに、許容できる条件で、またはまったくそのような取り決めを締結できない場合、規制当局の承認を受けた製品候補を正常に商品化できないか、そのような商品化に遅延または制限が生じる可能性があります。承認された製品候補を、自社で、または1つ以上の第三者との協力を通じて正常に商品化できない場合、将来の製品収益が損なわれ、さらに大きな損失を被る可能性があります。
私たちの計画と戦略を成功裏に実施するためには、組織の規模を拡大する必要があり、この成長を管理するのが難しいかもしれません。
開発と商品化の計画と戦略を成功裏に実施し、公開企業として事業を継続するためには、管理、運営、販売、マーケティング、財務、法務、コンプライアンス、その他の人員を追加する必要があると予想されます。将来の成長により、経営陣には次のような重大な責任が追加されます。
•追加の従業員の特定、採用、統合、維持、モチベーションの向上
•当社が負う可能性のある請負業者やその他の第三者に対する契約上の義務を遵守しながら、臨床検査、FDA、その他の米国以外の規制機関による製品候補の審査プロセスを含め、社内の開発努力を効果的に管理します。そして
•運用、財務、管理の統制、報告システムおよび手順を改善します。
将来の財務実績と、製品候補の開発を成功させ、承認されれば商品化する能力は、将来の成長を効果的に管理する能力に一部依存します。また、経営陣は、これらの成長活動の管理にかなりの時間を費やすために、日常の活動から不釣り合いな注意をそらさなければならない場合もあります。さらに、経営陣のメンバーを含む一部の従業員は、会社間契約に従ってKalyra Pharmaceuticals, Inc. に代わってサービスを行っています。その結果、そのような個人はすべての時間とリソースを当社や他の子会社に割り当てていません。成長活動を管理する必要性と相まって、当社の事業の日々の活動に十分な注意を向ける能力がさらに制限される可能性があります。
私たちは現在、臨床開発と製造の重要な側面を含む特定のサービスの提供を、特定の独立組織、アドバイザー、コンサルタントに大きく依存しており、近い将来も引き続き依存していきます。独立した組織、アドバイザー、コンサルタントのサービスが、必要なときに適時に引き続き利用できること、または適格な代替機関を見つけることができることを保証することはできません。さらに、外部委託された活動を効果的に管理できない場合、または第三者のサービスプロバイダーが提供するサービスの質や正確性が何らかの理由で損なわれた場合、当社の臨床試験は延長、延期、終了する可能性があり、製品候補の販売承認を得たり、事業を進めたりできない場合があります。既存の第三者サービスプロバイダーを管理したり、経済的に合理的な条件で他の有能な外部請負業者やコンサルタントを見つけたりできることを保証することはできません。
新入社員を雇用したり、サードパーティのサービスプロバイダーを追加したりして組織を効果的に拡大できない場合、製品候補のさらなる開発と商品化に必要なタスクをうまく実行できず、その結果、研究、開発、商品化の目標を達成できない可能性があります。
情報技術システムの障害、サイバー攻撃、またはサイバーセキュリティの欠陥が発生した場合、当社の事業と運営が損なわれる可能性があります。
セキュリティ対策が実施されているにもかかわらず、当社の情報システム、および現在および将来のCRO、CMO、その他の請負業者、コンサルタント、協力者、サードパーティのサービスプロバイダーの情報システムは、コンピューターウイルスやマルウェア(ランサムウェアなど)、悪意のあるコード、自然災害、テロ、戦争、電気通信および電気障害、ハッキング、サイバー攻撃、フィッシング攻撃、その他のソーシャルエンジニアリングスキーム、従業員の盗難による攻撃、損害、中断に対して脆弱です。または誤用、ヒューマンエラー、詐欺、サービス拒否またはサービス低下攻撃、洗練された国民国家および国民国家が支援する行為者、または組織内の人物、または組織内のシステムにアクセスできる者による不正アクセスまたは使用。情報技術システムへの攻撃は、その頻度、持続性、高度さ、激しさも増しており、さまざまな動機と専門知識を持つ高度で組織化されたグループや個人によって行われています。COVID-19と継続的なハイブリッドな職場環境の結果として、インターネット技術への依存とリモートで働く従業員の数により、サイバーセキュリティリスクの増大に直面する可能性もあります。これにより、サイバー犯罪者が脆弱性を悪用する機会が増える可能性があります。さらに、システムへの不正アクセスやシステム妨害に使用される手法は頻繁に変更され、ターゲットに対して導入されるまで認識されないことが多いため、これらの手法を予測したり、適切な防止策を講じたりできない場合があります。また、長期間検出されないままになるセキュリティ違反が発生することもあります。たとえ特定されたとしても、攻撃者が統制を回避し、検出を回避し、法医学的証拠を削除または難読化するように設計されたツールや手法をますます使用するようになっているため、インシデントや違反を適切に調査または修正できない場合があります。
私たちと一部のサービスプロバイダーは、サイバー攻撃やセキュリティインシデントの対象になることがあります。このような事象が発生して当社の業務が中断されたり、当社の企業秘密、個人情報、その他の専有情報や機密情報が不正に取得またはアクセスされたりした場合、当社の創薬および開発プログラムが重大な混乱を招く可能性があります。連邦、州、および旧米国政府の要件には、特定の個人を特定できる情報に関するセキュリティ違反を個人に通知する企業の義務が含まれます。これは、当社、または当社のベンダー、請負業者、または当社が戦略的関係を築いている組織が経験した侵害に起因する可能性があります。セキュリティ違反に関する通知とフォローアップ措置は、当社の評判に影響を与え、訴訟費用や是正費用などの多額の費用を負担する可能性があります。たとえば、完了した臨床試験または将来の臨床試験から臨床試験データが失われると、規制当局の承認作業が遅れ、失われたデータを回復または再現するためのコストが大幅に増加する可能性があります。また、候補製品の製造を第三者に頼っています。その会社のコンピューターシステムに関連する同様の出来事も、当社の事業に重大な悪影響を及ぼす可能性があります。何らかの混乱またはセキュリティ違反により、データの損失または損傷、または機密情報や専有情報の不適切な開示につながる場合、訴訟や政府の調査にさらされる可能性があり、製品候補のさらなる開発と商品化が遅れる可能性があり、特定の州、連邦、および/または国際的なプライバシーおよびセキュリティ法に違反した場合、多額の罰金または罰則の対象となる可能性があります。
当社の保険契約は、そのような中断、故障、またはセキュリティ違反によって生じる潜在的な損失を補償するには不十分な場合があります。また、そのような保険は、今後、経済的に合理的な条件では利用できなくなったり、まったく利用できなくなったりする可能性があります。さらに、私たちの保険は、私たちに対してなされたすべての請求をカバーしているわけではなく、いかなる場合でも高い控除額になる可能性があります。訴訟の弁護は、そのメリットにかかわらず、費用がかかり、経営陣の注意をそらす可能性があります。
EUの価格設定、医薬品のマーケティング、および償還規制は、欧州加盟国で当社製品を販売し、補償を受ける能力に重大な影響を与える可能性があります。
私たちは、米国および米国以外の特定の管轄区域の両方で、候補製品を販売するための承認を求めるつもりです。候補製品について1つ以上の米国以外の管轄区域で承認を得た場合、それらの管轄区域の規則や規制の対象となります。一部の米国以外の国、特にEU諸国では、医薬品の価格設定が政府の管理やその他の市場規制の対象となっており、候補製品の価格設定と使用に圧力がかかる可能性があります。これらの国では、候補製品の販売承認を得た後、政府機関との価格交渉にかなりの時間がかかる場合があります。さらに、当社製品候補の市場での受け入れと販売は、当社の製品候補に対する適切な補償と第三者支払者からの償還の有無に大きく依存し、現在および将来の医療改革措置の影響を受ける可能性があります。
米国の連邦反キックバック法禁止法と同様に、医薬品の処方、推奨、承認、購入、供給、注文、または使用を誘発または奨励するために医師に利益または利益を提供することもEUでは禁止されています。医師への給付または給付の提供は、2010年の英国贈収賄法など、EU加盟国の国内法に準拠しています。これらの法律に違反すると、多額の罰金や懲役が科せられる可能性があります。
特定のEU加盟国の医師への支払いは、公に開示する必要があります。さらに、医師との契約は、多くの場合、医師の雇用主、その管轄の専門機関、および/または個々のEU加盟国の規制当局による事前の通知および/または承認の対象でなければなりません。これらの要件は、EU加盟国で適用される国内法、業界規範、または職業行動規範に規定されています。これらの要件に従わないと、評判上のリスク、公の叱責、行政処罰、罰金、または懲役につながる可能性があります。
薬の価格設定と償還に関する要件は、国によって大きく異なります。たとえば、EUは加盟国に対し、国民健康保険制度が払い戻しを提供する医薬品の範囲を制限したり、人間が使用する医薬品の価格を管理したりするオプションを提供しています。さまざまなEU加盟国で使用されている参考価格設定と、並行配布、つまり低価格加盟国と高価格加盟国の間の裁定取引により、価格をさらに引き下げることができます。加盟国が医薬品の特定の価格を承認したり、
代わりに、医薬品を市場に出す会社の収益性を直接的または間接的に管理するシステムを採用する場合があります。一部の国では、償還または価格承認を取得または維持するために、当社の製品候補の費用対効果を他の利用可能な治療法と比較する臨床研究またはその他の研究を実施する必要がある場合があります。バイオ医薬品に価格統制や償還制限がある国が、当社製品のいずれに対しても有利な償還および価格設定を許可しているという保証はありません。歴史的に、EUで発売された製品は米国の価格構造に従わず、一般的に価格は大幅に低くなる傾向があります。第三者の支払者または当局による割引の公表は、公表国およびその他の国の価格または償還水準にさらなる圧力をもたらす可能性があります。価格が不十分なレベルに設定されている場合、または当社製品の償還が利用できない、または範囲または金額が制限されている場合、販売による収益と、それらの国における当社の製品候補の潜在的な収益性に悪影響が及びます。
米国、世界、政治、経済の不利な状況は、当社の事業、財政状態、または経営成績に悪影響を及ぼす可能性があります。
当社の業績は、米国および世界経済、米国および世界の金融市場の一般的な状況によって悪影響を受ける可能性があります。たとえば、最近の世界経済の低迷はインフレ率と金利の上昇を引き起こし、資本市場と信用市場には極端なボラティリティと混乱をもたらしました。景気後退や景気後退の悪化や長期化は、私たちの事業にさまざまなリスクをもたらす可能性があります。その中には、必要に応じて許容できる条件で追加の資本を調達できることも含まれます。信用市場や金融市場のさらなる悪化や経済状況への信頼が起こらないという保証はありません。景気の低迷や衰退は、サプライヤーに負担をかけ、供給の中断を招き、供給品の価格が上昇したり、お客様が当社のサービスに対する支払いを遅らせたりする可能性もあります。さらに、現在の軍事紛争は、当社および当社が依存する第三者の事業を混乱させたり、その他の悪影響を与える可能性があります。関連する制裁、輸出規制、またはその他の措置が、米国、EU、ロシアなどの国によって開始され、将来開始される可能性があり(たとえば、潜在的なサイバー攻撃、エネルギーの流れの中断など)、当社の事業および/またはサプライチェーン、CRO、CMO、および当社が取引を行うその他の第三者に悪影響を及ぼす可能性があります。上記のいずれかが当社の事業に害を及ぼす可能性があり、現在の経済情勢と金融市場の状況が当社の事業に悪影響を与える可能性があるすべての方法を予測することはできません。
事業の中断は、当社の事業に悪影響を及ぼす可能性があります。
私たちの事業は、火災、悪天候、停電、電気通信障害、テロ活動、公衆衛生上の危機、COVID-19などのパンデミック疾患、および当社の制御が及ばないその他の自然災害や人為的災害や出来事による中断に対して脆弱です。たとえば、COVID-19の結果、前臨床試験や臨床試験など、事業に悪影響を与える可能性のある混乱を経験したことがあり、今後も経験する可能性があります。私たちの施設は、時々悪天候に見舞われる地域にあります。私たちは、大規模な竜巻、洪水、火災、地震、停電、テロ活動、公衆衛生上の危機、パンデミック病、その他の災害が事業や財務結果に及ぼす潜在的な影響について体系的な分析を行っておらず、そのような災害に対する復旧計画もありません。また、事業の中断による実際の損失を補償する十分な保険に加入しておらず、当社が被った損失や損害は事業に悪影響を及ぼす可能性があります。これらの事業中断が発生すると、当社の事業と財政状態に深刻な悪影響を及ぼし、コストと費用を増大させる可能性があります。
純営業損失の繰越やその他の特定の税属性を活用する能力は限られている場合があります。
2022年12月31日現在、連邦および州の純営業損失繰越(NOL)は、それぞれ約3億9,030万ドルと1億9,240万ドルでした。3億6,940万ドルの連邦NOLは、2017年12月31日以降に開始する課税年度に発生し、無期限に繰り越すことができますが、将来の期間の課税所得の最大80%を相殺するためにのみ使用できます。この制限により、過去数年間に連邦NOLが発生していたとしても、今後は米国連邦所得税を支払う必要があるかもしれません。2018年1月1日より前に開始する課税年度に発生した当社の連邦NOLにはこの制限はありませんが、適用される米国連邦税法に基づいて20課税年度のみ繰り越すことが許可されており、使用されない場合は2033年に失効します。私たちの州のNOLは2033年に期限切れになります。
さらに、改正された1986年の内国歳入法、または同法第382条および第383条に基づき、企業が「所有権の変更」(通常、1人以上の「5パーセント株主」による所有権の累積的な変更であり、3年間で50パーセントポイントを超える累積的な変化として定義されます)を受けた場合、法人は所有権変更前の連邦NOL、およびその他の特定の変更前の税属性を相殺することができます。変更後の課税所得と所得税の負債は制限される場合があります。州の税法にも同様の規則が適用される場合があります。私たちは過去にそのような所有権の変更を経験したかもしれませんし、将来的には私たちの制御の及ばない株式所有権の移転の結果として所有権の変更を経験するかもしれません。そのような所有権の変更が発生したかどうか、またはそのような所有権の変更によって生じる可能性のある年間制限(ある場合)を特定するための調査は実施していません。NOLやその他の特定の税属性を利用する能力は、上記の所有権の変更によって制限される可能性があり、その結果、NOLやその他の特定の税属性の大部分を利用できなくなり、キャッシュフローや経営成績に重大な悪影響を及ぼす可能性があります。
当社の製品候補を国際的にマーケティングすることに関連するさまざまなリスクは、当社の事業に重大な悪影響を及ぼす可能性があります。
候補製品については、米国外で規制当局の承認を求める予定です。したがって、必要な承認を得た場合、米国以外の国での事業に関連して、次のような追加のリスクにさらされると予想されます。
•米国以外の国におけるさまざまな規制要件と償還制度。
•関税、貿易障壁、価格および為替管理、その他の規制要件の予期しない変更。
•インフレを含む経済的弱さ、または特に米国経済と市場を除く政治的不安定。
•海外に居住または旅行する従業員の税法、雇用法、移民法、労働法の遵守。
•給与税の源泉徴収を含む米国以外の税金。
•米国以外の通貨の変動。これにより、営業費用の増加や収益の減少、および他国で事業を行う際に付随するその他の義務が生じる可能性があります。
•米国外事業の人員配置と管理の難しさ。
•労働不安が米国よりも多い国における労働力の不確実性
•FCPAまたは同等の米国以外の規制に基づく潜在的な責任
•特に米国と同程度に知的財産権を尊重および保護していない米国以外の国では、契約上および知的財産権の行使が困難になります。
•海外での原材料供給または製造能力に影響を及ぼすあらゆる事象による生産不足、および
•戦争やテロなどの地政学的行動による事業の中断。
当社の海外事業に関連するこれらおよびその他のリスクは、収益性の高い事業を達成または維持する能力に重大な悪影響を及ぼす可能性があります。
将来買収や戦略的パートナーシップを行う場合、資本要件が増加し、株主が希薄化し、負債や偶発的負債を引き受け、その他のリスクにさらされる可能性があります。
当社は、補完的な製品、知的財産権、技術、事業のライセンス供与や買収など、さまざまな買収機会や戦略的パートナーシップを随時評価することがあります。買収や戦略的パートナーシップの可能性には、次のような多くのリスクが伴う可能性があります。
•営業費用と現金要件の増加。
•追加債務または偶発債務の引き受け。
•私たちの持分証券の発行。
•買収した会社の業務、知的財産、製品の同化(新しい人材の統合に伴う困難を含む)
•このような戦略的合併や買収を追求する上で、既存のプログラムやイニシアチブから経営陣の注意をそらすこと。
•主要な従業員の定着、主要な人員の喪失、および主要なビジネス関係を維持する能力の不確実性。
•そのような取引の相手方に関連するリスクと不確実性。これには、その当事者とその既存の製品または製品候補の見通し、および販売承認が含まれます。
•買収した技術や製品から、買収を行う際の目標を達成したり、関連する買収や保守の費用を相殺したりするのに十分な収益を上げることができません。
さらに、将来買収を行ったり、パートナーシップを追求したりする場合、希薄化可能な有価証券を発行したり、債務を引き受けたり負担したり、多額の一時的な費用を負担したり、無形資産を取得したりして、将来多額の償却費用が発生する可能性があります。さらに、適切な買収機会を見つけることができない場合があり、そのようにできないと、事業の発展に重要な技術や製品を成長させたり、アクセスしたりする能力が損なわれる可能性があります。
上場企業であるという要件は、私たちのリソースに負担をかけ、訴訟が増え、経営者の注意をそらす可能性があります。
上場企業として、当社は取引法、サーベンス・オクスリー法、ドッド・フランク・ウォール街改革・消費者保護法、またはドッド・フランク法の報告要件、ナスダックの上場要件、およびその他の該当する証券規則および規制の対象となっています。これらの規則や規制を遵守することは増加しており、法律上および財務上のコンプライアンスコストが増加し、一部の活動がより困難になり、時間とコストがかかり、システムとリソースに対する需要が高まるでしょう。取引法では、とりわけ、事業と経営成績に関する年次、四半期、および最新の報告書を提出することが義務付けられています。サーベンス・オクスリー法では、とりわけ、財務報告に対する効果的な開示管理と手続き、内部統制を維持することが義務付けられています。財務報告に関する内部統制の変更を四半期ごとに開示する必要があります。財務報告に関する開示管理と手続き、および内部統制をこの基準を満たすように維持し、必要に応じて改善するには、多大なリソースと管理監督が必要になる場合があります。その結果、経営陣の注意が他の事業上の懸念からそらされ、当社の事業と経営成績に悪影響を及ぼす可能性があります。また、これらの要件を満たすために、追加の従業員を雇ったり、外部のコンサルタントを雇ったりする必要があるかもしれません。これにより、コストと経費が増加します。
さらに、サイバーセキュリティリスクとガバナンスに関する新しい開示要件を含む、コーポレートガバナンスと公開に関する法律、規制、基準の変更は、上場企業に不確実性をもたらし、法的および財務上のコンプライアンスコストを増大させ、一部の活動により時間がかかるようになっています。これらの法律、規制、基準は、多くの場合、具体性に欠けるため、さまざまな解釈の対象となります。その結果、規制機関や統治機関によって新しいガイダンスが提供されるにつれて、実際の適用は時間の経過とともに進化する可能性があります。その結果、コンプライアンス問題に関する不確実性が続き、開示とガバナンスの慣行の継続的な見直しによってコストが高くなる可能性があります。私たちは、進化する法律、規制、基準に準拠するためのリソースに投資してきましたが、今後も投資を続けるつもりです。この投資により、一般管理費が増加し、経営陣の時間と注意が収益創出活動からコンプライアンス活動に転用される可能性があります。新しい法律、規制、基準を遵守するための当社の取り組みが、その適用や慣行に関する曖昧さのために規制機関や統治機関が意図した活動と異なる場合、規制当局は当社に対して法的手続きを開始し、当社の事業に悪影響を及ぼす可能性があります。
これらの新しい規則や規制により、取締役および役員の賠償責任保険に加入する費用が高くなる可能性があります。また、将来的には、補償範囲の軽減を受け入れる必要があったり、補償を受けるために大幅に高額な費用が発生したりする可能性があります。これらの要因により、特に監査委員会と報酬委員会に参加する資格のある取締役会のメンバー、および資格のある執行役員を引き付けて維持することがより困難になる可能性もあります。公開会社として私たちに要求される書類に情報を開示することで、私たちの事業と財政状態は引き続き目に見えるようになり、競合他社やその他の第三者による脅迫や実際の訴訟につながる可能性があると私たちは考えています。これらの主張が成功した場合、私たちのビジネスは深刻な被害を受ける可能性があります。クレームが訴訟に至らなかったり、私たちに有利に解決されたとしても、解決に必要な時間とリソースは、経営陣のリソースをそらし、ビジネスに深刻な害を及ぼす可能性があります。
当社の主力製品候補の製造の一部は、第三者メーカーを通じて、中国を含む米国以外の国で行われています。これらの製造業者の運営における重大な混乱、貿易戦争、または中国を含む米国以外の国での政情不安は、当社の事業、財政状態、および経営成績に重大な悪影響を及ぼす可能性があります。
私たちは現在、製造業務を第三者に委託しており、当社の主力製品候補の臨床量は、中国を含む米国外の特定の第三者によって製造されています。そのような製品候補については、引き続きそのような第三者メーカーを利用する予定です。自然災害やその他の原因によるかどうかにかかわらず、中国を含む米国以外の国で生産が中断されたり、メーカーが当社のニーズを満たす十分な量を生産できなくなったりすると、日常的に事業を運営し、製品候補の開発を継続する能力が損なわれる可能性があります。さらに、これらのメーカーは米国外にあるため、米国または旧米国政府の政策が変更されたり、中国を含む米国以外の国で政情不安や不安定な経済状況が発生した場合に、製品の供給が途絶えたり、コストが増加したりする可能性があります。たとえば、貿易戦争は、私たちが使用する中国で製造された化学中間体に関税を課す可能性があります。これらの事項はいずれも、当社の事業と経営成績に重大かつ悪影響を及ぼす可能性があります。さらに、これらのメーカーのいずれかが製造を中断したり、規制要件を遵守しなかったりすると、潜在的な製品の臨床開発が大幅に遅れ、提案された試験に対する第三者または臨床研究者の関心と支持が低下する可能性があります。これらの中断や障害は、製品候補の商品化を妨げ、競争力を損なう可能性もあります。さらに、米国以外の国では、現地通貨の価値の変動にさらされる可能性があります。将来現地通貨が高くなると、コストが増加する可能性があります。さらに、熟練労働者の需要の増加と中国を含む米国以外の国での熟練労働者の確保の減少により賃金率が上昇するにつれて、人件費は上昇し続ける可能性があります。
当社の知的財産に関連するリスク
私たちの成功は、知的財産と独自のプラットフォームを保護する能力にかかっています。
私たちの商業的成功は、私たちの製品候補、専有技術とその使用に関する特許保護と企業秘密保護を取得して維持する能力、私たちと私たちのライセンサーが他者の所有権を侵害することなく事業を行う能力、そして私たちと私たちのライセンサーが首尾よく防御する能力に一部依存しています。
第三者の異議申し立てに対する当社の特許(当社がライセンスしているものを含む)。私たちまたは私たちのライセンサーが私たちの知的財産権を保護できない場合、または私たちの知的財産権が私たちの技術や製品候補にとって不十分である場合、私たちの競争力が損なわれる可能性があります。当社とライセンサーは通常、当社の事業にとって重要な製品候補、専有技術、およびそれらの使用に関連して、米国および米国外で特許出願を行うことにより、当社の所有権を保護しようとしています。私たちの特許出願は、そのような出願から特許が発行されない限り、そして発行された請求がその技術を対象とする範囲でのみ、そのような出願で主張されている技術を実践している第三者に対して強制することはできません。私たちの特許出願によって特許が発行されるという保証も、発行された特許が同様の技術を持つ競合他社に対して十分な保護を提供するという保証もありません。また、特許が発行された場合、侵害されたり、第三者によって設計されないという保証もありません。発行された特許でさえ、後で無効または執行不能であることが判明したり、さまざまな特許庁や裁判所で第三者が提起した手続きで変更または取り消されたりする可能性があります。私たちと私たちのライセンサーの所有権が将来どの程度保護されるかは不明です。利用できる保護は限られており、当社または当社のライセンサーの権利を適切に保護したり、当社または当社のライセンサーが競争上の優位性を獲得または維持したりできない場合があります。製品候補に関する知的財産権を適切に保護する当社および当社のライセンサーの能力におけるこれらの不確実性および/または制限は、当社の財政状態および経営成績に重大な悪影響を及ぼす可能性があります。
私たちは米国および米国以外の国で発行された特許のライセンスを取得していますが、他の米国出願中の特許出願、対応する国際特許出願、および特定の米国以外の国での特許出願における請求が、米国特許商標庁(USPTO)、米国の裁判所、または米国以外の国の特許庁や裁判所によって特許可能と見なされるかどうかは定かではありません。また、当社が発行した特許の請求が次のものにならないことも確信できません。異議を申し立てられた場合、無効または執行不能であることが判明しました。
特許出願プロセスには多くのリスクと不確実性が伴い、私たち、私たちのライセンサー、または将来の協力者が、特許を取得して擁護することによって製品候補を保護することに成功するという保証はありません。これらのリスクと不確実性には、次のものが含まれます。
•USPTOおよびさまざまな外国政府の特許機関は、特許手続き中に多くの手続き、書類、料金支払い、およびその他の規定の遵守を要求します。これに従わないと、特許または特許出願が放棄または失効し、関連する法域における特許権の一部または全部が失効する可能性があります。
•特許出願によって特許が発行されるわけではありません。
•特許は、異議申し立て、無効化、変更、取り消し、回避されたり、執行不能であることが判明したり、その他の方法で競争上の優位性をもたらさない可能性があります。
•競合他社の多くは、当社やライセンサーよりも大幅に大きなリソースを持っており、その多くが競合技術に多額の投資を行っており、製品候補の製造、使用、販売の能力を制限、妨害、または阻止する特許を求めたり、申請したり、すでに取得している場合があります。
•世界中の健康問題に関する公共政策の問題として、成功したことが証明された病気の治療について、米国内外で特許保護の範囲を制限するよう米国および旧米国政府および国際政府機関に大きな圧力がかかる可能性があります。そして
•米国以外の国では、米国の裁判所が支持する特許法よりも特許権者にとって不利な特許法がある場合があります。これにより、米国の元競合他社が競合製品を製造、開発、販売する機会が増えます。
特許出願手続きも費用と時間がかかり、当社または当社のライセンサーは、必要または望ましいすべての特許出願を、妥当な費用で、適時に、または保護が商業的に有利なすべての法域で提出および審査することができない場合があります。また、私たちやライセンサーがそうする可能性もあります
特許保護を受けるには手遅れになる前に、私たちの研究開発成果の特許性のある側面を特定しないでください。さらに、状況によっては、私たちは、ライセンサーや第三者からのものを含め、私たちがライセンスしている技術に関する特許出願の作成、出願、審査を管理したり、特許を維持したりする権利がありません。また、ライセンスされた特許権を行使するために、ライセンサーの協力を必要とする場合がありますが、そのような協力は提供されない場合があります。したがって、これらの特許や出願は、私たちのビジネスの最善の利益と一致する方法で訴追および執行されない場合があります。ライセンサーによる特許出願および維持活動が、適用法および規制に従って行われている、または実施される予定であるかどうかは定かではありません。これにより、そのような特許またはそのような出願から発行される可能性のある特許の有効性と執行可能性に影響を与える可能性があります。そうしないと、当社がライセンスしている該当する知的財産の権利を失う可能性があり、その結果、製品や製品候補を開発および商品化する能力に悪影響が及び、競合他社が競合製品を製造、使用、販売することを阻止できなくなる可能性があります。
さらに、従業員、外部の科学協力者、CRO、CMO、コンサルタント、アドバイザー、ライセンサー、その他の第三者など、研究開発成果の特許性のある側面にアクセスできる当事者と秘密保持契約を締結していますが、これらの当事者のいずれかが特許出願前にそのような契約に違反し、そのような成果を開示して、特許保護を求める当社の能力を危険にさらす可能性があります。
ライセンサーや第三者から知的財産権をライセンスする契約における義務を遵守しなかったり、ライセンサーとのビジネス関係に混乱が生じたりした場合、ビジネスにとって重要なライセンス権を失う可能性があります。
私たちは、当社の事業にとって重要な知的財産権が付与されるライセンス契約の当事者であり、将来的には追加のライセンス契約を締結する可能性があります。たとえば、当社の完全子会社であるZMIは、Recurium IPとのライセンス契約の当事者です。この契約に基づき、当社は、アゼノセルチブとZn-D5製品候補を対象とする特定の知的財産を含む特定の知的財産権の独占ライセンスを取得しています。
これと他の既存のライセンス契約は私たちに課されており、私たちが知的財産をライセンスする将来のライセンス契約により、さまざまな開発、規制、商業的調査義務、マイルストーンの支払い、ロイヤルティの支払い、その他の義務が課されると予想されます。私たちがこれらの契約に基づく義務を遵守しなかった場合、または破産関連の手続きの対象となる場合、ライセンサーはライセンスを終了する権利を有する可能性があり、その場合、ライセンスの対象となる製品を販売することができなくなります。
研究を進めたり、製品候補の商品化を許可したりするために、第三者からライセンスを取得する必要がある場合があります。また、そのようなライセンスがない場合に製品候補に対して強制される可能性のある第三者の特許が存在しないという保証はありません。たとえあったとしても、商業的に合理的な条件でこれらのライセンスを取得できない可能性があります。たとえ私たちがライセンスを取得できたとしても、それは非独占的であり、それによって競合他社は私たちにライセンスされているのと同じ技術にアクセスすることができます。その場合、代替技術の開発またはライセンス供与に多大な時間とリソースを費やす必要があるかもしれません。それができない場合、影響を受ける製品候補を開発または商品化できず、当社の事業に重大な損害を与える可能性があり、そのような知的財産権を所有する第三者は、当社の販売を禁止する差し止め命令を求めるか、販売に関して、ロイヤルティやその他の形態の報酬を支払う義務を当社側に求めることができます。知的財産のライセンス供与は私たちのビジネスにとって非常に重要であり、複雑な法的、ビジネス的、科学的な問題が関係しています。ライセンス契約の対象となる知的財産に関して、当社とライセンサーの間で、次のような紛争が発生する可能性があります。
•ライセンス契約に基づいて付与された権利の範囲とその他の解釈関連の問題。
•当社の技術とプロセスが、ライセンス契約の対象ではないライセンサーの知的財産を侵害しているかどうか、またどの程度侵害しているか。
•特許やその他の権利を第三者にサブライセンスする私たちの権利。
•製品候補の開発と商品化に関連するライセンス技術の使用に関する当社の注意義務、およびそれらの調査義務を満たす活動は何か
•ライセンスを譲渡または譲渡する当社の権利。そして
•私たちのライセンサーとその関連会社とサブライセンシー、そして私たちと私たちのパートナーとサブライセンシーによる知的財産の共同作成または使用から生じる発明とノウハウの所有権。
当社がライセンスした知的財産をめぐる紛争により、現在のライセンス契約を容認できる条件で維持することが妨げられたり、損なわれたりした場合、影響を受ける製品候補の開発と商品化を成功させることができず、事業に重大な悪影響を及ぼす可能性があります。
さらに、私たちの契約の中には、特定の取引を完了する能力を制限または遅延させたり、それらの取引の価値に影響を与えたり、特定の活動を遂行する能力を制限したりするものがあります。たとえば、Recurium契約に基づいて独占的にライセンスされている特定の特許権を第三者にサブライセンスまたは譲渡する場合、そのような取引に関連して受け取る特定のサブライセンス収入の特定の割合をRecuriumに支払う必要がある場合があります。
ライセンサーが取得する特許保護の範囲が十分に広くない場合、またはライセンサーが私たちがライセンスする特許保護のいずれかを失った場合、競合他社が類似または同一の製品候補を商品化するのを防ぐ当社の能力に悪影響が及びます。
バイオ医薬品企業の特許ポジションは一般的に非常に不確実で、複雑な法的および事実上の問題が伴い、近年多くの訴訟の対象となっています。その結果、私たちの特許権の存在、発行、範囲、有効性、執行可能性、および商業的価値は非常に不確実です。私たちの保留中および将来の特許出願の結果として、当社の製品候補を保護する特許や、他社が競合製品候補を商業化することを効果的に妨げる特許が発行されない場合があります。
さらに、特許出願における請求の範囲は、特許問題における請求よりも大幅に縮小することができ、請求範囲は発行後に再解釈することができます。現在または将来ライセンス供与する特許出願が特許として発行される場合でも、有意義な保護を提供したり、競合他社やその他の第三者が当社と競争するのを防いだり、その他の方法で競争上の優位性をもたらしたりするような形で発行することはできません。私たちがライセンスする特許は、第三者によって異議を申し立てられたり回避されたり、第三者による異議申し立ての結果として狭められたり無効になったりする可能性があります。したがって、当社の製品候補が保護されるのか、それとも有効で法的強制力のある特許によって保護され続けるのかはわかりません。競合他社やその他の第三者は、類似または代替の技術または製品を非侵害的な方法で開発することで、当社の特許を回避できる場合があります。これは、当社の事業、財政状態、経営成績、および見通しに重大な悪影響を及ぼす可能性があります。
特許の発行は、その発明者、範囲、有効性、執行可能性に関して決定的なものではありません。また、当社のライセンス特許は、当社の製品候補を対象としない場合や、米国および海外の裁判所や特許庁で異議を申し立てられる場合があります。当社は、先行技術の発行前に第三者からUSPTOに提出されたり、異議申立、派生、取り消し、再審査、付与後審査、PGR、当事者間審査、IPR、または当社の特許権に異議を唱えるその他の同様の手続きに関与したりする可能性があります。無効性と法的強制力がないという主張に続く結果は予測できません。たとえば、私たちの特許の有効性に関しては、私たち、私たちのライセンサー、特許審査官が審査中に気づかなかった先行技術を無効にするものがないかどうかは定かではありません。私たちの特許、特許出願、または私たちのライセンサーの特許に関連する可能性のある先行技術がすべて見つかったという保証はありません。また、当社またはライセンサーが認識していた、または認識しているが、当社の特許および特許出願における請求の有効性または執行可能性に影響を与えるとは考えていない先行技術がないという保証もありません。それでも、最終的には請求の有効性または執行可能性に影響を与える可能性があります。そのような提出、手続き、または訴訟における不利な決定は、当社の特許権の範囲を狭めたり、無効にしたり、執行不能になったり、第三者が当社の製品候補を商品化して、支払いなしで直接当社と競争することを可能にする可能性があります。
私たち。このようなライセンスされた特許権の喪失、独占権の喪失、または特許請求が絞り込まれたり、無効になったり、執行不能であるとされたりすると、他者が類似または同一の技術や製品を使用または商品化することを阻止したり、製品候補の特許保護期間を制限したりする可能性があります。そのような手続きには、たとえ最終的に私たちにとって有利な結果になったとしても、多額の費用がかかり、科学者や経営陣に多大な時間を要する可能性があります。さらに、私たちの特許と特許出願によって提供される保護の幅広さや強さが脅かされた場合、結果がどうであれ、企業が現在または将来の製品候補のライセンス、開発、または商品化のために当社と協力することを思いとどまらせる可能性があります。
一部の製品候補の特許保護と特許出願は、ライセンサーと第三者に依存している場合があります。
私たちまたは私たちのライセンサーは、開発および商品化活動の過程で行われた発明の特許性のある側面を、特許保護を取得するには手遅れになる前に特定できない場合があります。したがって、特許権を強化する潜在的な機会を逃す可能性があります。たとえば、適切な優先権請求、発明権、書面による説明、請求範囲、または特許期間の調整の要求、特許期間の延長、またはそれらの外国同等物に関して、当社の特許または特許出願の準備または出願における形式上の欠陥が存在するか、将来発生する可能性があります。当社または当社のライセンサーが、現在または将来を問わず、そのような特許やその他の知的財産権を確立、維持、保護できない場合、そのような権利は減少または廃止される可能性があります。私たちのライセンサーが特許権の訴訟、維持、または行使に関して完全に協力的でなかったり、私たちと意見が合わなかったりすると、そのような特許権が侵害される可能性があります。ライセンスされている特許または特許出願の形式、準備、訴訟、または執行に重大な欠陥がある場合、そのような特許は無効および/または執行不能であり、そのような出願は決して有効で執行可能な特許をもたらさない可能性があります。これらの結果のいずれかが第三者との競争を防ぐ能力を損ない、当社の事業に悪影響を及ぼす可能性があります。
ライセンシーとして、私たちは特許出願や訴訟、特許の維持、その他のライセンス契約に基づくライセンスされた知的財産の保護を第三者に依頼することがあります。一部の特許、特許出願、その他の知的財産権について、これらの活動を当社が主に管理できない場合があります。第三者によるそのような活動が適用される法律や規制に従って行われている、または今後行われているかどうか、あるいは有効で法的強制力のある特許やその他の知的財産権がもたらされるかどうかは定かではありません。一部のライセンサーとのライセンス契約の条件に従い、ライセンサーは、当社のライセンス特許の執行またはこれらの特許の無効性を主張する請求に対する抗弁を管理する権利を有する場合があります。また、そのような執行または弁護を行うことが許可されている場合でも、ライセンサーの協力が必要です。私たちのライセンサーが、ライセンスされた特許に対する私たちの利益を守るために、十分なリソースを割り当てたり、そのような特許の執行やそのような請求の弁護を優先したりするかどうかは定かではありません。たとえ私たちがこれらの法的措置の当事者でなくても、不利な結果が生じると、事業運営に必要な知的財産のライセンス供与を継続できなくなる可能性があるため、事業に悪影響を及ぼす可能性があります。当社のライセンサー、将来のライセンサー、または将来の協力者が、当社の製品候補のいずれかを対象とする特許を適切に審査し、特許保護を維持しなかった場合、それらの製品候補を開発および商品化する当社の能力に悪影響が及ぶ可能性があり、競合他社が競合製品を製造、使用、販売することを阻止できない場合があります。
さらに、私たちが第三者から取得またはライセンスした特許および特許出願の特許出願を管理する権利を私たちが持っている場合でも、私たちが特許出願の管理を引き継ぐ前に行われたライセンサーとその弁護士の行動または不作為によって、私たちは依然として悪影響や偏見を受ける可能性があります。
ライセンサーを含むさまざまな第三者から取得またはライセンスされた当社の技術は、留保権の対象となる場合があります。当社のライセンサーは、多くの場合、当社との契約に基づき、基盤となる技術を当社にライセンスされた分野以外の分野で使用したり、非営利の学術・研究用途に使用したり、技術に関連する研究から得られた一般的な科学的発見を発表したり、技術に関連する情報を科学的および学術的に慣習的に開示したりする権利など、特定の権利を保持しています。ライセンサーがテクノロジーの使用をこれらの用途に限定しているかどうかを監視することは難しく、誤用があった場合、ライセンスされたテクノロジーに対する権利を行使するために多額の費用が発生する可能性があります。
取得またはライセンスされた技術を利用する能力が制限された場合、または重要なライセンス技術に対する権利を失った場合、製品の開発、ライセンス供与、マーケティング、販売を成功させることができなくなる可能性があります。
新製品の導入を防止または延期します。私たちの事業戦略は、ライセンスされ取得した技術を商用製品にうまく開発できるかどうかにかかっています。したがって、これらの技術を利用する能力に制限があると、候補製品を開発、ライセンス供与、または市場投入、販売する能力が損なわれる可能性があります。
当社の知的財産の一部は、「マーチイン」権、特定の報告要件、および当社の知的財産が政府資金によるプログラムを通じて発見されたものであると判断された場合、米国を拠点とする企業に対する優先権などの連邦規制の対象となる場合があります。このような規制を遵守すると、当社の独占権が制限され、米国以外の製造業者との契約が制限される可能性があります。
当社が取得またはライセンスした、または将来取得またはライセンス供与する可能性のある知的財産権の一部は、米国政府の資金を使用して生成された可能性があるため、特定の連邦規制の対象となる可能性があります。これらの米国政府の権利には、あらゆる政府目的で発明を使用するための非独占的、譲渡不能、取消不能な世界規模のライセンスが含まれます。さらに、米国政府は、特定の限られた状況下で、(i)発明を商品化するための適切な措置が講じられていない、(ii)公衆衛生または安全上のニーズを満たすために政府の措置が必要である、または(iii)連邦規制に基づく公共利用の要件を満たすために政府の措置が必要であると判断した場合、これらの発明の独占的、部分的に排他的、または非独占的なライセンスを第三者に付与するよう要求する権利を有します。「マーチイン権」として)。助成金受領者が政府に発明を開示しなかった場合、または指定された期限内に知的財産の登録申請を提出しなかった場合、米国政府はこれらの発明の所有権を取得する権利もあります。政府資金によるプログラムの下で生み出される知的財産にも一定の報告要件があり、それを遵守するには多大なリソースを費やす必要があるかもしれません。さらに、米国政府は、これらの発明のいずれかを具体化する製品、またはこれらの発明のいずれかを使用して製造される製品は、実質的に米国で製造されることを義務付けています。知的財産の所有者または譲受人が、米国で実質的に製造する可能性が高い潜在的なライセンシーに同様の条件でライセンスを付与するための合理的ではあるが失敗した努力がなされたこと、または状況下では国内製造が商業的に実現不可能であることを示すことができれば、資金を提供した連邦政府機関は米国産業に対するこの優先権を放棄することができます。このような米国産業の選好により、そのような知的財産に関連する製品について、米国以外の製品メーカーと契約することが制限される可能性があります。将来の知的財産が米国政府の資金を利用して生み出される場合は、ベイドール法の規定が同様に適用される可能性があります。
知的財産権は、必ずしも当社の競争上の優位性に対するすべての潜在的な脅威に対処するわけではありません。
知的財産権には限界があり、当社の事業を十分に保護したり、競争上の優位性を維持したりできない可能性があるため、当社の知的財産権によって提供される将来の保護の程度は不明です。例えば:
•他の人は、当社の製品候補に類似しているが、当社が所有またはライセンスしている特許の請求の対象とならない製品を開発できる場合があります。
•私たちまたは私たちのライセンサーは、私たちが所有またはライセンスしている発行済み特許または特許出願の対象となる発明を最初に行っ?$#@$ではないかもしれません。
•私たちや私たちのライセンサーは、私たちの発明の一部を対象とする特許出願を最初に提出したわけではないかもしれません。
•他者は、当社の知的財産権を侵害することなく、類似または代替技術を独自に開発したり、当社の技術を複製したりすることができます。
•私たちのライセンサーの保留中の特許出願が、特許の発行につながらない可能性があります。
•競合他社による法的異議申し立ての結果、当社が所有またはライセンスしている発行済み特許が無効または執行不能と判断される可能性があります。
•競合他社は、当社が特許権を持たない国で研究開発活動を行い、そのような活動から得た情報を使用して、主要な商業市場で販売する競争力のある製品を開発する可能性があります。
•特許性のある独自の技術を追加開発することはできません。そして
•他者の特許は当社の事業に悪影響を及ぼす可能性があります。
•これらの事象のいずれかが発生した場合、当社の事業、経営成績、および見通しに重大な損害を与える可能性があります。
私たちの商業的成功は、第三者の特許やその他の所有権を侵害することなく事業を行う能力に大きく依存しています。私たちが彼らの所有権を侵害しているという第三者からの主張は、損害賠償責任につながったり、私たちの開発や商品化の取り組みを妨げたり遅らせたりする可能性があります。
私たちの商業的成功は、第三者の特許と所有権の侵害を回避することに一部依存しています。ただし、当社の研究、開発、および商品化活動は、第三者が所有または管理する特許またはその他の知的財産権を侵害または侵害しているという申し立ての対象となる場合があります。他の団体が特許や所有権を持っているか、取得している場合があります。これにより、当社の製品候補や将来承認される可能性のある製品の製造、使用、販売、販売、販売、販売、販売、販売、販売、または輸入が制限されたり、妨げられたり、当社の競争力が損なわれたりする可能性があります。米国内外で、バイオ医薬品業界における特許およびその他の知的財産権に関する訴訟および行政手続きが数多く行われています。これには、USPTO、旧米国特許庁、および/または法廷での特許無効および侵害訴訟、異議申し立て、再審査、知的財産権手続およびPGR手続きが含まれます。私たちが製品候補を開発している分野には、米国および米国以外の第三者が発行した特許と出願中の特許が多数あります。当社の製品候補の使用または製造に関連する材料、製剤、製造方法、または処理方法に関するクレームを伴う第三者の特許または特許出願があるかもしれません。
バイオ医薬品業界が拡大し、より多くの特許が発行されるにつれて、当社の製品候補が第三者の特許権侵害の請求の対象となるリスクが高まります。特許出願は一定期間秘密として保持されるため、関連する出願が公開されるまで、製品候補の商品化によって侵害される可能性のある第三者の特許に気付かない場合があります。また、製品候補または技術に関連する特許を最初に申請したのが当社であったかどうかは定かではありません。さらに、特許出願は発行までに何年もかかることがあるため、現在保留中の特許出願があり、その結果、当社の製品候補が侵害する可能性のある特許が後で発行される可能性があります。また、特許間の用語の違い、データベースの不完全、特許請求の意味の評価が難しいために特許検索が不完全であるため、当社の技術に関連する可能性のある第三者の特許権を特定することは困難です。さらに、第三者が将来特許を取得し、当社の技術の使用がこれらの特許を侵害していると主張する可能性があります。第三者による特許侵害の申し立てには時間がかかり、次のような可能性があります。
•その結果、否定的な宣伝につながる可能性のある費用のかかる訴訟につながります。
•当社の技術担当者と経営陣の時間と注意をそらすこと。
•開発の遅れの原因となります。
•主張されている特許の有効期限が切れるまで、または最終的に無効または法的強制力がないと判断されるまで、または法廷で侵害されていないと判断されるまで、当社の製品候補のいずれかを商品化できないようにします。
•非侵害技術の開発を私たちに要求しますが、費用対効果の高い方法では不可能かもしれません。
•第三者に対して重大な責任を負わせる、または
•商業的に合理的な条件では利用できない、またはまったく利用できない、または非独占的である可能性のあるロイヤルティまたはライセンス契約の締結を私たちに要求します。その結果、競合他社が同じ技術にアクセスできるようになる可能性があります。
この定期報告の日付の時点で、第三者が当社に対して特許侵害を主張したことはありませんが、他の第三者が当社の製品候補の販売を妨げる所有権を持っている可能性があります。損害賠償を請求し、当社の製品候補またはプロセスに関連する活動を禁止しようとする当社に対する特許関連の訴訟は、当社が故意に侵害していると判断された場合の3倍の損害賠償を含む損害賠償責任を負う可能性があり、製品候補を製造または開発するためのライセンスの取得を要求する可能性があります。これらの請求の弁護は、そのメリットにかかわらず、多額の訴訟費用を必要とし、経営陣と従業員のリソースを当社の事業から大幅に流用することになります。そのような訴訟で私たちが勝てるかどうか、またはこれらの特許のいずれかで要求されるライセンスが、たとえあったとしても、商業的に受け入れられる条件で利用可能になるかどうかは予測できません。さらに、私たちや将来の戦略的パートナーがライセンスを取得できたとしても、その権利は非独占的である可能性があり、その結果、競合他社が同じ知的財産にアクセスする可能性があります。また、必要に応じて、権利侵害を避けるために製品候補やプロセスを再設計できるかどうか確信が持てません。したがって、司法上または行政上の手続きにおいて不利な決定が下されたり、必要なライセンスを取得できなかったりすると、候補製品の開発と商品化が妨げられ、事業、財政状態、経営成績に悪影響を及ぼす可能性があります。
当社に対して請求を行う当事者は、はるかに多くのリソースを持っているため、複雑な特許訴訟の費用を私たちよりも効果的に負担できる可能性があります。さらに、知的財産訴訟や行政手続きに関連してかなりの量の証拠開示が必要なため、私たちの機密情報の一部が開示によって危険にさらされるリスクがあります。さらに、訴訟の開始と継続に起因する不確実性は、当社の追加資金調達能力に重大な悪影響を及ぼしたり、その他の方法で当社の事業、経営成績、財政状態および見通しに重大な悪影響を及ぼす可能性があります。
私たちは、当社の特許またはライセンサーの特許を保護または執行するために訴訟に巻き込まれる可能性があり、費用と時間がかかり、成功しない可能性があります。さらに、私たちが発行した特許は、法廷で異議を申し立てられた場合、無効または執行不能とされる可能性があります。
競合他社は、当社またはライセンサーの知的財産権を侵害する可能性があります。侵害や不正使用を防ぐために、当社および/または当社のライセンサーは、費用と時間がかかる権利侵害の申し立てを要求される場合があります。さらに、私たちのライセンサーは侵害の申し立てをする必要があるかもしれませんが、そのような申し立てをしないこともできます。さらに、特許侵害訴訟では、裁判所は、当社が所有またはライセンスしている特許は無効、執行不能、および/または侵害されていないと判断することがあります。当社、当社のライセンサー、または将来の協力者となる可能性のある人が、当社の製品候補の1つを対象とした特許を行使するために第三者に対して法的手続きを開始した場合、被告は、当社の特許は無効であり、侵害されていない、および/または全部または一部が執行不能であると主張する可能性があります。特許訴訟では、主張されている特許の無効性および/または執行不能性に関する被告人の主張が一般的です。有効性異議申し立ての根拠には、特許の対象とならない主題、実用性の欠如、新規性の欠如、明白性または書面による説明の欠如、明白性または非有効性など、いくつかの法的要件のいずれかを満たしていないという申し立てが含まれます。法的強制力のない主張の根拠には、特許の出願に関係する誰かが、USPTOまたは元米国特許庁からの重要な情報を意図的に差し控えた、または訴追中に誤解を招くような発言をしたという申し立てが含まれます。
被告が無効および/または執行不能の法的主張で勝訴した場合、そのような製品候補の特許保護の少なくとも一部、場合によってはすべてを失うことになります。さらに、当社の特許、特許出願、またはライセンサーの特許出願によって提供される保護の幅広さまたは強さが脅かされた場合、企業が現在または将来の製品候補のライセンス、開発、または商品化のために当社と協力することを思いとどまらせる可能性があります。このような特許保護の喪失は、当社の事業に重大な悪影響を及ぼします。
たとえ私たちに有利に解決されたとしても、私たちの知的財産権に関連する訴訟やその他の法的手続きにより、多額の費用がかかり、技術および管理担当者の通常の責任から注意をそらす可能性があります。このような訴訟や手続きにより、当社の営業損失が大幅に増加し、収益が減少する可能性があります
開発活動または将来の販売、マーケティング、流通活動に利用できるリソース。そのような訴訟や手続きを適切に行うのに十分な財源やその他のリソースがない可能性があります。競合他社の中には、財源が大きいため、そのような訴訟や手続きの費用を私たちよりも効果的に負担できるものもあります。特許訴訟やその他の手続きの開始と継続に起因する不確実性により、市場での競争力が損なわれる可能性があります。
さらに、知的財産訴訟または当社の知的財産権に関連するその他の法的手続きに関連してかなりの量の証拠開示が必要なため、この種の訴訟またはその他の手続き中に開示することにより、当社の機密情報の一部が危険にさらされるリスクがあります。
知的財産訴訟は、当社の評判を傷つけ、普通株式の市場価格を下落させる不利な宣伝につながる可能性があります。
知的財産訴訟の過程で、訴訟の開始や、公聴会、申立てに関する判決、その他の訴訟の中間手続きの結果が公表される可能性があります。証券アナリストや投資家がこれらの発表を否定的と見なした場合、既存の製品、プログラム、または知的財産の知覚価値が低下する可能性があります。したがって、当社の普通株式の市場価格が下落する可能性があります。このような発表は、当社の評判や将来の製品市場を傷つける可能性があり、それが当社の事業に重大な悪影響を及ぼす可能性があります。
発明の優先順位を決定するために、派生または干渉手続きが必要な場合があり、不利な結果になると、関連技術の使用を中止するか、勝訴した当事者に権利をライセンスすることを試みる必要が生じる場合があります。
当社または当社のライセンサーの特許または特許出願に関する発明の優先順位を決定するには、第三者によって誘発された、当社または当社のライセンサーによって提起された、またはUSPTOによって宣言された派生または干渉手続き、または米国以外の特許庁での同様の手続きが必要になる場合があります。不利な結果になると、関連技術の使用を中止するか、勝訴した当事者にその権利をライセンスしようとする可能性があります。勝訴した当事者が商業的に合理的な条件でライセンスを提供しない場合、当社の事業が損なわれる可能性があります。そのような手続きに対する当社または当社のライセンサーによる弁護は失敗する可能性があり、たとえ成功したとしても、多額の費用がかかり、当社の経営陣や他の従業員の注意をそらす可能性があります。さらに、そのような手続きに関連する不確実性は、臨床試験の継続、研究プログラムの継続、必要な技術の第三者からのライセンス供与、または製品候補の市場投入に役立つ開発または製造パートナーシップの締結に必要な資金を調達する能力に重大な悪影響を及ぼす可能性があります。
特許改革法は、特許出願の審査と発行済み特許の執行または防御を取り巻く不確実性とコストを増大させる可能性があります。
2011年9月、リーヒー・スミス・アメリカ発明法、またはリーヒー・スミス法が法制化されました。リーヒー・スミス法には、米国特許法に対するいくつかの重要な変更が含まれています。これらには、特許出願の審査方法に影響を及ぼす条項や、特許訴訟にも影響する条項が含まれます。特に、リーヒー・スミス法に基づき、米国は2013年3月に「最初に出願する発明者」制度に移行しました。この制度では、特許性に関する他の要件が満たされていれば、第三者が請求された発明を最初に発明したかどうかにかかわらず、最初に特許出願を行った発明者が特許を取得する権利があります。したがって、2013年3月以降にUSPTOに特許出願を行った第三者は、たとえそのような第三者によって発明される前に私たちが発明したとしても、私たちの発明を対象とする特許を授与される可能性があります。そのためには、発明から特許出願までの時間を把握しておく必要があります。さらに、有効で法的強制力のある特許を取得して維持できるかどうかは、当社の技術と先行技術の違いにより、当社の技術が先行技術よりも特許可能になるかどうかにかかっています。米国およびその他のほとんどの国での特許出願は、出願後または発行までの一定期間秘密にされます。そのため、当社が(1)製品候補に関連する特許出願を最初に提出したか、(2)当社の特許または特許出願で主張されている発明のいずれかを発明したかどうかは定かではありません。
リーヒー・スミス法には、特許出願の審査方法に影響を与え、特許訴訟にも影響を与える多くの重要な変更も含まれています。これには、特許審査中に第三者が先行技術をUSPTOに提出することを許可することや、PGR、IPR、および派生手続を含むUSPTOが管理する付与後の手続による特許の有効性を攻撃する追加手続きが含まれます。そのような提出または手続きにおける不利な決定は、当社の特許権の範囲または執行可能性を狭めたり、無効にしたりする可能性があり、それが当社の競争力に悪影響を及ぼす可能性があります。
USPTOの手続きにおける証拠基準は、特許請求を無効にするために必要な米国連邦裁判所の証拠基準と比較して低いため、地方裁判所の訴訟で最初に提示された場合、同じ証拠では請求を無効にするには同じ証拠では不十分であっても、第三者がUSPTOの手続きにおいてUSPTOの手続きにおいて十分な証拠を提供する可能性があります。したがって、第三者がUSPTOの手続きを利用して、地方裁判所の訴訟で被告として最初に異議を申し立てても無効にならなかったであろう特許請求を無効にしようとする可能性があります。したがって、リーヒー・スミス法とその施行は、当社または当社のライセンサーの特許出願の審査と、当社が発行した特許の執行または弁護を取り巻く不確実性とコストを増大させる可能性があり、これらはすべて、当社の事業、財政状態、経営成績、および見通しに重大な悪影響を及ぼす可能性があります。
米国特許法、または他の国の法律の変更は、一般的に特許の価値を低下させ、それによって製品候補を保護する当社の能力が損なわれる可能性があります。
他のバイオ医薬品企業と同様に、私たちの成功は知的財産、特に特許に大きく依存しています。バイオ医薬品業界における特許の取得と執行には、非常に複雑な技術的および法的複雑さが伴います。したがって、バイオ医薬品特許の取得と執行には費用と時間がかかり、本質的に不確実です。米国およびその他の国の特許法または特許法の解釈のいずれかの変更は、当社の知的財産の価値を低下させ、特許出願の審査および発行済み特許の執行または防御を取り巻く不確実性とコストを増大させる可能性があります。私たちの特許や第三者の特許で許可または執行される可能性のあるクレームの幅を予測することはできません。さらに、議会やその他の元米国立法機関は、私たちにとって不利な特許改革法を可決する可能性があります。
たとえば、米国最高裁判所は近年、特定の状況で利用できる特許保護の範囲を狭めるか、特定の状況で特許所有者の権利を弱めるかのどちらかで、いくつかの特許訴訟について判決を下しました。当社または当社のライセンサーが将来特許を取得する能力に関する不確実性が高まることに加えて、このような事象の組み合わせにより、一度取得された特許の価値に関しても不確実性が生じています。米国議会、米国連邦裁判所、USPTO、または米国以外の管轄区域にある同様の当局の決定によっては、特許に関する法律や規制が予測できない形で変更され、当社または当社のライセンサーが新しい特許を取得したり、既存の特許や将来取得する可能性のある特許を行使したりする能力が弱まる可能性があります。
当社または当社のライセンサーは、当社の特許およびその他の知的財産の発明者または所有権に異議を申し立てる請求の対象となる場合があります。
当社または当社のライセンサーは、元従業員またはその他の第三者が当社の特許またはその他の知的財産の所有権を持っているという主張の対象となる場合があります。発明や所有権に異議を唱えるこれらの主張やその他の主張から身を守るには、訴訟が必要な場合があります。私たちまたは私たちのライセンサーがそのような請求を弁護しなかった場合、金銭的損害賠償の支払いに加えて、私たちは貴重な知的財産権を失う可能性があります。このような結果は、当社の事業に重大な悪影響を及ぼす可能性があります。私たちや私たちのライセンサーがそのような請求に対する弁護に成功したとしても、訴訟は多額の費用をもたらし、経営陣や他の従業員の注意散漫になる可能性があります。
特許条件は、製品候補に対する当社の競争力を十分な期間守るには不十分かもしれません。
特許の寿命は限られています。米国では、すべての維持費が適時に支払われた場合、特許の自然有効期限は通常、最も早い米国非仮出願日から20年です。さまざまな延長が可能ですが、特許の存続期間とそれによって提供される保護には限りがあります。たとえ特許が当社の製品候補に向けられたとしても
が買収され、特許期間が終了すると、競合製品との競争にさらされる可能性があります。製品候補の開発、試験、規制審査に必要な時間を考えると、当社の製品候補を対象とした特許は、そのような候補が商品化される前または直後に失効する可能性があります。その結果、当社の特許ポートフォリオには、当社と同様または同一の製品の商品化から他者を排除する十分な権利が与えられていない可能性があります。
私たちまたは私たちのライセンサーが製品候補の特許期間の延長を取得しない場合、私たちの事業は重大な打撃を受ける可能性があります。
当社の製品候補に対するFDAの市販承認の時期、期間、および詳細によっては、1984年の薬価競争および特許期間回復法またはハッチ・ワックスマン改正に基づく限定特許期間回復の対象となる米国特許の1つ以上が、限定的な特許期間回復の対象となる場合があります。ハッチ・ワックスマンの改正により、製品開発およびFDAの規制審査プロセス中に失われた特許期間の補償として、最長5年の特許回復期間が認められています。FDAの規制審査プロセス中に失われた特許期間の補償として、FDAが承認した製品ごとに最大1つの特許を延長できます。特許期間の延長は、特許の残存期間を製品承認日から合計14年を超えて延長することはできません。延長できるのは、そのような承認された医薬品、FDAが承認した適応症に使用する方法、またはその製造方法に関する請求のみです。特許期間の延長またはそれに相当するものは、当社の製品候補の規制当局の承認を得て、米国以外の特定の国でも利用できる場合があります。ただし、当社または当社のライセンサーは、たとえば、該当する期限内に申請しなかった、関連特許の有効期限が切れる前に申請しなかった、またはその他の方法で該当する要件を満たさなかったなどの理由で、延長を許可されない場合があります。さらに、適用される期間や与えられる特許保護の範囲は、私たちが要求するよりも短くなる可能性があります。私たちまたは私たちのライセンサーが特許期間の延長または回復を取得できない場合、またはそのような延長の期間が私たちが要求するよりも短い場合、競合他社は特許の有効期限が切れた後に競合製品の承認を取得する可能性があり、私たちの収益は、おそらく大幅に減少する可能性があります。さらに、これが発生した場合、競合他社は、当社の臨床および前臨床データを参照することで開発と試験への投資を活用し、他の場合よりも早く製品を発売する可能性があります。
私たちは世界中で知的財産権を保護できないかもしれません。
私たちは米国およびその他の特定の国で特許を発行し、出願中の特許出願を行ってきましたが、世界中のすべての国での特許の出願、訴訟、および保護には法外に費用がかかり、米国以外の一部の国における当社の知的財産権は、米国の知的財産権よりも範囲が小さい場合があります。さらに、一部の旧米国国の法律は、米国の連邦法および州法と同程度に知的財産権を保護していません。したがって、第三者が米国以外のすべての国で私たちの発明を実践したり、私たちの発明を使用して作られた製品を米国または他の法域で販売または輸入したりすることを防ぐことができない場合があります。競合他社は、当社が特許保護を取得していない法域で自社製品を開発するために当社の技術を使用する可能性があり、さらに、当社または当社のライセンサーが特許保護を受けているが執行が米国ほど強力ではない地域に侵害製品を輸出する可能性があります。これらの製品は当社の製品候補と競合する可能性があり、当社または当社のライセンサーの特許またはその他の知的財産権は、それらの競合を防ぐのに有効または十分ではない可能性があります。
多くの企業が、米国以外の管轄区域における知的財産権の保護と防御において重大な問題に直面しています。米国以外の多くの国の法制度は、特許やその他の知的財産保護の執行を支持していません。そのため、当社または当社のライセンサーの特許の侵害や、当社の所有権を侵害する競合製品のマーケティングを阻止することが難しい場合があります。米国外の法域で当社または当社のライセンサーの特許権を行使する手続きは、多額の費用を要し、当社の努力と注意を当社の事業の他の側面からそらす可能性があり、当社または当社のライセンサーの特許が無効または狭義に解釈されるリスクにさらされ、当社または当社のライセンサーの特許出願が発行されないリスクにさらされ、第三者が当社に対して請求を主張するきっかけとなる可能性があります。私たちまたは私たちのライセンサーは、私たちまたは私たちのライセンサーが提起した訴訟で勝訴することはできません。また、裁定された損害賠償やその他の救済措置は、もしあれば、商業的に意味がないかもしれません。したがって、私たちまたは私たちのライセンサーが世界中で私たちの知的財産権を行使しようとしても、私たちが開発またはライセンスしている知的財産から大きな商業的利益を得るには不十分かもしれません。
多くの国には、特許所有者が第三者にライセンスを付与することを強制される強制ライセンス法があります。さらに、多くの国では、政府機関や政府請負業者に対する特許の執行可能性を制限しています。これらの国では、特許権者が利用できる救済手段が限られている場合があり、それによって特許の価値が大幅に低下する可能性があります。当社または当社のライセンサーが、当社の事業に関連する特許に関して第三者にライセンスを付与することを余儀なくされた場合、当社の競争力が損なわれ、当社の事業、財政状態、経営成績、および見通しに悪影響が及ぶ可能性があります。
当社の特許保護の取得と維持は、規制や政府の特許機関によって課されるさまざまな手続き、書類、料金の支払い、その他の要件の遵守にかかっています。これらの要件に違反した場合、当社の特許保護は軽減または廃止される可能性があります。
特許および/または出願に関する定期的な維持費、更新料、年金手数料、およびその他のさまざまな政府手数料は、特許および/または出願の有効期間のさまざまな時点で、USPTOおよびさまざまな元米国特許庁に支払われる必要があります。私たちはこれらの料金を支払うように通知するシステムを導入しており、期日までにこれらの料金を支払うのは第三者に頼っています。さらに、USPTOおよびさまざまな旧米国特許庁は、特許出願手続き中に、手続き、書類、手数料の支払い、およびその他の同様の規定の遵守を求めています。私たちは、評判の良い法律事務所やその他の専門家を雇って遵守しています。多くの場合、不注意による不注意による不注意は、延滞料の支払いや、特定の法域に適用される規則に従ったその他の手段によって解決できます。ただし、違反した結果、特許または特許出願が放棄または失効し、関連する法域における特許権の一部または全部が失われる場合があります。このような事態が発生した場合、当社の事業に重大な悪影響を及ぼす可能性があります。
企業秘密を守ることができなければ、ビジネスと競争上の地位が損なわれるでしょう。
さらに、私たちは、競争力を維持するために、特許を取得していないノウハウ、技術、その他の専有情報を含む企業秘密の保護に依存しています。私たちは、第三者との機密保持契約、従業員、コンサルタント、ライセンサー、アドバイザーとの機密情報および発明契約の締結など、企業秘密と特許を取得していないノウハウを保護するための措置を講じていますが、そのような契約がすべて適切に締結されたことを保証することはできません。また、これらの当事者のいずれかが契約に違反し、企業秘密を含む当社の専有情報を開示する可能性があり、当社ではできない場合があります。そのような違反に対して適切な救済策を講じてください。当事者が企業秘密を違法に開示または流用したという申し立てを執行することは難しく、費用と時間がかかり、結果は予測できません。さらに、米国内外の一部の裁判所は、企業秘密を保護する意思がないか、したくない場合があります。
さらに、第三者は依然としてこの情報を入手したり、この情報や類似の情報を独自に出くわしたりする可能性があり、当社には、第三者がその技術や情報を使用して当社と競争することを妨げる権利はありません。これらの事象が発生したり、企業秘密の保護が失われたりすると、この情報の価値が大幅に低下し、競争力が損なわれる可能性があります。私たちまたは私たちのライセンサーが公開前に特許保護を申請しない場合、または独自の技術やその他の機密情報の機密性を維持できない場合、特許保護を取得したり、企業秘密情報を保護したりする能力が危険にさらされる可能性があります。
私たちは、競合他社から従業員を不当に雇用した、または当社または当社の従業員が以前の雇用主の機密情報または企業秘密を不当に使用または開示したという申し立ての対象となる場合があります。
バイオ医薬品業界ではよくあることですが、私たちは従業員に加えて、コンサルタントのサービスを利用して製品候補の開発を支援しています。これらのコンサルタントの多く、および当社の従業員の多くは、競合他社や潜在的な競合他社を含む他のバイオ医薬品会社で以前に雇用されていたか、以前にコンサルティングサービスを提供したか、現在提供している可能性があります。私たち、当社の従業員、またはコンサルタントが、以前の雇用主、以前または現在のクライアントの企業秘密またはその他の情報を不注意で使用または開示したという申し立ての対象となる可能性があります。これらの主張に対する弁護には訴訟が必要な場合があります。そのような請求に対する弁護に失敗した場合、金銭的損害賠償の支払いに加えて、貴重な知的財産権や人員を失い、事業に悪影響を及ぼす可能性があります。
これらの請求に対する弁護に成功したとしても、訴訟は多額の費用をもたらし、経営陣や他の従業員の気を散らす可能性があります。
第三者への依存に関連するリスク
私たちは、前臨床試験や臨床試験の特定の側面を実施するにあたり、独立した臨床研究者やCROを含む第三者に依存しており、今後も依存し続けることを期待しています。これらの第三者が契約上の義務を首尾よく遂行しなかったり、適用される規制要件を遵守しなかったり、予定された期限を守らなかったりすると、候補製品について規制当局の承認を得たり、商品化したりすることができず、事業に大きな打撃を与える可能性があります。
私たちは、前臨床研究と臨床試験の特定の側面を実施し、進行中の前臨床および臨床プログラムのデータを監視および管理するために、独立した臨床研究者や第三者のCROを含む第三者に依存してきましたが、今後も信頼していく予定です。私たちは、前臨床研究と臨床試験の実施をこれらの関係者に依存しており、その活動の特定の側面のみを管理しています。とはいえ、私たちは、それぞれの研究や試験が適用されるプロトコル、法律、規制、科学の基準に従って実施されるようにする責任があり、これらの第三者に依存しているからといって、規制上の責任が免除されるわけではありません。当社とCROを含む第三者請負業者は、GCP要件を遵守する必要があります。GCP要件は、臨床開発中のすべての製品候補について、FDAおよび同等の米国外の規制当局によって施行される規制とガイドラインです。規制当局は、治験依頼者、主任研究者、治験実施施設を定期的に検査することで、これらのGCPを実施しています。当社、これらの第三者または当社のCROが該当するGCPを遵守しない場合、当社の臨床試験で生成された臨床データは信頼できないと見なされ、FDAまたは同等の米国外の規制当局が、当社のマーケティング申請を承認する前に、追加の臨床試験を実施するよう要求することがあります。特定の規制当局による検査の結果、そのような規制当局が当社の臨床試験のいずれかがGCP規制に準拠していると判断することを保証することはできません。さらに、当社の臨床試験は、cGMP規制および同様の米国外の要件に基づいて製造された製品を使用して実施する必要があります。これらの規制に従わないと、臨床試験を繰り返す必要があり、規制当局の承認プロセスが遅れる可能性があります。さらに、これらの第三者が連邦、州、または米国外の詐欺や虐待、虚偽請求に関する法律や規制、または医療のプライバシーとセキュリティに関する法律に違反した場合、当社の事業に悪影響が及ぶ可能性があります。
さらに、これらの研究者とCROは当社の従業員ではないため、契約による以外に、彼らが当社の製品候補や臨床試験に費やすリソース量(時間を含む)を管理することはできません。これらの第三者は、競合他社を含む他の営利団体とも関係があり、そのために臨床試験やその他の製品開発活動を行っている可能性があり、それが当社に代わって彼らの業績に影響を与える可能性があります。独立治験責任医師またはCROが製品候補の開発に十分なリソースを投入しなかった場合、またはCROが契約上の義務または義務を首尾よく果たせなかったり、予定された期限を守れなかったり、交代が必要になったり、当社の臨床プロトコルや規制要件に従わなかったり、その他の理由で入手した臨床データの質や正確性が損なわれたりした場合、当社の臨床試験は延長、延期、終了することがあります。規制当局の承認を得られないか、または正常に得られない可能性があります私たちの製品候補を商品化してください。その結果、当社の業績と製品候補の商業的見通しが損なわれ、コストが増加し、収益を生み出す能力が遅れたり、完全に妨げられたりする可能性があります。
当社のCROは、未解決の重大な違反が発生した場合、当社との契約を終了する権利を有します。さらに、私たちのCROの中には、私たちの臨床試験に参加している被験者の安全がそのような終了を正当化することが合理的に証明できる場合、私たちが債権者の利益のために一般的な譲渡を行った場合、または私たちが清算された場合、私たちとのそれぞれの契約を終了することができます。
COVID-19とそれに対応して講じられた政府の措置もCROに大きな影響を及ぼしました。CROはさらなる混乱に直面し、前臨床試験と臨床試験の開始と完了に影響を及ぼす可能性があります。
これらの第三者CROとの関係のいずれかが終了した場合、代替のCROと契約を締結したり、商業的に合理的な条件で契約を結ぶことができなくなる可能性があります。CROの切り替えまたは追加には、
追加費用と管理時間と集中力が必要です。また、新しいCROが仕事を始めるという自然な移行期間があります。その結果、遅延が発生し、希望する臨床開発スケジュールを満たす能力に重大な影響を与える可能性があります。さらに、CROは、より高い作業負荷を吸収する能力が不足していたり、私たちのニーズをサポートするために追加の容量を引き受けたりする可能性があります。私たちはCROとの関係を慎重に管理していますが、将来同様の課題や遅延に遭遇しないという保証や、これらの遅延や課題が当社の事業、財政状態、見通しに重大な悪影響を及ぼさないという保証はありません。
私たちは、前臨床研究や進行中の臨床試験のための製品候補の製造について第三者と契約していますが、追加の臨床試験、そして最終的には商品化のために、今後も契約を結ぶ予定です。このように第三者に依存していると、十分な量の製品候補や医薬品、またはそのような量を許容できる費用で手に入れることができないリスクが高まり、開発や商品化の取り組みが遅れたり、妨げられたり、損なわれる可能性があります。
現在、当社には、開発や商品化に使用する製品候補品を製造するためのインフラストラクチャや内部能力がありません。私たちは、組織のメンバーの指導のもと、前臨床研究や臨床試験用の製品候補の製造を第三者メーカーに依存しており、今後も依存し続けることを期待しています。私たちは長期供給契約を結んでおらず、必要な供給品を発注書ベースで購入しています。さらに、候補製品の原材料は、場合によっては単一のサプライヤーから調達されます。現在、在庫管理を通じて、アゼノセルチブとZn-d5(もしあれば)の潜在的な供給リスクを軽減しています。製造、供給、保管上の問題などの結果として、何らかの理由で、当社の製品候補または将来の製品候補の供給が予期せず失われた場合、保留中または進行中の臨床試験の遅延、中断、中断、中断、終了、または再開または再開または繰り返しが必要になる場合があります。
私たちは、販売承認を得た製品候補の商業供給について、引き続きサードパーティメーカーに頼っていきたいと考えています。サードパーティの製造業者との必要な契約を維持または確立できない場合や、受け入れ可能な条件で契約を結ぶことができない場合があります。たとえサードパーティの製造業者と契約を結ぶことができたとしても、サードパーティの製造業者に依存することには、次のような追加のリスクが伴います。
•第三者製造業者が当社のスケジュールに従って、またはまったく当社の製品候補を製造しなかったこと。これには、当社の第三者製造業者が当社の製品候補よりも他の製品の供給を優先する場合や、当社と第三者との間の契約条件に従って満足のいく性能を発揮しない場合が含まれます。
•サプライヤーによる生産または納品の削減または終了、または価格の引き上げや条件の再交渉。
•当社にとって費用がかかる、または不都合な時期に、第三者の製造業者による取り決めまたは契約を終了または更新しないこと。
•第三者製造業者による当社との契約の違反。
•サードパーティメーカーが適用される規制要件を遵守していないこと。
•第三者メーカーが当社の仕様に従って当社の製品候補を製造しなかったこと。
•臨床用品の誤ったラベル付け。その結果、間違った投与量が供給されたり、有効成分やプラセボが適切に特定されなかったりする可能性があります。
•臨床用品が予定通りに臨床現場に届けられないため、臨床試験が中断されたり、医薬品が商業ベンダーにタイムリーに配布されなかったりして、売上の損失につながります。そして
•企業秘密やノウハウを含む当社の専有情報の不正流用。
私たちは、活性薬物と最終医薬品の両方を製造するためのcGMP規制または同様の米国外の要件の遵守について、製造プロセスのすべての側面を完全に管理しているわけではなく、サードパーティの契約製造パートナーに依存しています。サードパーティメーカーは、米国外ではcGMP規制または同様の規制要件に準拠できない場合があります。サードパーティの委託製造業者が、当社の仕様やFDAなどの厳しい規制要件に適合する材料をうまく製造できない場合、製造施設の販売承認を確保したり、維持したりすることはできません。さらに、委託製造業者が適切な品質管理、品質保証、有能な人材を維持する能力を管理することはできません。FDAまたは同等の旧米国規制当局が、これらの施設を当社の製品候補品の製造に承認しない場合、または将来そのような承認を取り下げた場合、代替製造施設を探す必要が生じる可能性があります。これは、承認された場合、製品候補の開発、販売承認の取得、または販売の能力に大きな影響を与えます。当社または第三者メーカーが適用される規制を遵守しなかった場合、罰金、差し止め命令、民事罰則、遅延、承認の一時停止または撤回、ライセンスの取り消し、製品候補または医薬品の差し押さえまたはリコール、営業制限、刑事訴追などの制裁措置が課される可能性があります。これらはすべて、当社の製品候補または医薬品の供給に重大かつ悪影響を及ぼし、当社の事業と業績に害を及ぼす可能性があります。オペレーション。製品候補または医薬品の製造を現在および予想される将来の依存は、将来の利益率と、適時かつ競争力のある方法で市販承認を受けた製品候補を商品化する能力に悪影響を与える可能性があります。
医薬品の製造は複雑で、サードパーティのメーカーは製造が困難になることがあります。サードパーティメーカーのいずれかがこのような問題に直面した場合、承認されれば、臨床試験用の製品候補または患者向け製品の適切な供給が遅れたり、妨げられたりする可能性があります。
医薬品の製造、特に大量製造は複雑で、革新的な技術の使用が必要になる場合があります。承認された医薬品の各ロットは、同一性、強度、品質、純度、効力について徹底的なテストを受ける必要があります。医薬品の製造には、この目的のために特別に設計され検証された施設が必要であり、高度な品質保証と品質管理手順が必要です。充填、ラベリング、梱包、保管、出荷、品質管理とテストなど、製造プロセスのどこかでわずかな偏差があると、ロットの不具合、在庫の回収、腐敗につながる可能性があります。製造ロットの在庫回収や、臨床試験に使用された当社の製品候補に関する同様の措置により、試験が遅れたり、試験データの完全性や将来の規制当局への申請での使用の可能性が損なわれたりする可能性があります。製造プロセスに変更が加えられた場合、変更前と変更後の製品の同等の同一性、強度、品質、純度、または効力を示す前臨床データおよび臨床データの提供を求められる場合があります。製造元の施設で微生物、ウイルス、またはその他の汚染が発見された場合、汚染の調査と改善のためにそのような施設を長期間閉鎖する必要がある場合があります。これにより、臨床試験が遅れ、当社の事業に悪影響を及ぼす可能性があります。生物由来の成分を使用すると、感染症やアレルギー反応などの危害の申し立て、または汚染の可能性による製品施設の閉鎖につながる可能性もあります。これらの課題の結果として、メーカーが臨床試験や商品化に十分な量を生産できない場合、またはその他の方法では、開発と商品化の取り組みが損なわれ、事業、財政状態、経営成績、成長見通しに悪影響を及ぼします。
将来的にコラボレーションを確立することを決めたが、商業的に合理的な条件でそれらのコラボレーションを確立できない場合、開発および商品化の計画を変更しなければならない可能性があります。
私たちの医薬品開発プログラムと製品候補の潜在的な商品化には、経費を賄うために多額の追加現金が必要になります。私たちは、能力を拡大し、研究開発活動を加速させ、第三者による商業化活動を提供するために、選択的に協力関係を築くことを引き続き模索する可能性があります。これらの関係のいずれにおいても、非経常費用やその他の費用を負担したり、短期および長期の支出を増やしたり、既存の株主を希薄化する証券を発行したり、経営や事業を混乱させたりする可能性があります。
適切な協力者を探す上で激しい競争に直面することになり、交渉プロセスは時間と複雑です。共同作業について最終的な合意に達するかどうかは、とりわけ、協力者のリソースと専門知識の評価、提案された協力の条件、および提案された協力者によるいくつかの要因の評価に左右されます。これらの要因には、臨床試験の設計または結果、FDAまたは同等の米国外の規制当局による承認の可能性、対象製品候補の潜在的な市場、そのような製品候補の製造と患者への提供のコストと複雑さ、競合医薬品の可能性、知的財産と業界の所有権に関する不確実性の存在、および一般的な市場状況が含まれます。潜在的な協力者は、共同研究が可能な、類似の適応症のための代替製品候補または技術を検討し、そのようなコラボレーションが、私たちの製品候補にとって私たちとのコラボレーションよりも魅力的かどうかを検討することもあります。さらに、将来の製品候補のための協力やその他の代替案を確立するための取り組みが成功しない可能性があります。なぜなら、それらは共同作業には開発の初期段階であると見なされ、第三者はそれらを安全性と有効性を実証するために必要な可能性を秘めていると見なさない可能性があるからです。
さらに、最近、大手製薬会社の間でかなりの数の企業結合が行われ、その結果、将来の潜在的な協力者の数が減少しています。コラボレーションの締結に成功したとしても、そのコラボレーションの条件により、潜在的なコラボレーターと特定の条件で将来の契約を締結することが制限される場合があります。
コラボレーションを締結しようとしても、タイムリーに、受け入れ可能な条件で、あるいはまったくコラボレーションについて交渉できない場合があります。それができない場合、製品候補の開発を縮小したり、開発プログラムやその他の開発プログラムの1つ以上を削減または延期したり、潜在的な商品化を遅らせたり、販売またはマーケティング活動の範囲を縮小したり、支出を増やして自費で開発または商品化活動を実施したりする必要があります。開発や商業化活動に自力で資金を提供するために支出を増やすことを選択した場合、追加の資本を獲得する必要があるかもしれませんが、許容できる条件では入手できないか、まったく利用できない場合があります。十分な資金がないと、製品候補をさらに開発したり、市場に投入して製品の収益を生み出すことができない場合があります。
普通株式の保有に関するリスク
当社の株価は変動しやすく、投資の全部または一部を失う可能性があります。
当社の普通株式の取引価格は、変動が激しく、さまざまな要因に応じて大きく変動する可能性がありますが、その中には制御できないものもあります。株式市場全般、特に製薬会社やバイオテクノロジー企業は、多くの場合、これらの企業の業績とは無関係だったり、不均衡だったりする極端な価格や量の変動を経験しています。
実際の業績にかかわらず、幅広い市場および業界要因が当社の普通株式の市場価格に悪影響を及ぼす可能性があります。この「リスク要因」セクションで説明した要因に加えて、これらの要因には次のものがあります。
•当社または競合他社の製品候補の前臨床試験と臨床試験のタイミングと結果。
•競合製品の成功、または潜在的な競合他社による製品開発努力の発表。
•当社製品または競合他社の製品に関する規制措置
•競合他社に対する当社の成長率の実際の変化または予想される変化
•米国およびその他の国における規制または法的発展。
•特許出願、発行済み特許、またはその他の所有権に関する進展または紛争
•主要人員の採用または出発。
•当社または競合他社による、重要な買収、戦略的協力、合弁事業、協力、または資本拠出に関する発表。
•財務結果、開発スケジュール、または証券アナリストの推奨に関する見積もりの実際の変化または予想される変化。
•投資家が当社に匹敵すると考える企業の評価額の変動
•製薬およびバイオテクノロジー部門の市況。
•医療費支払いシステムの構造の変化。
•当社の普通株式の投機的取引と空売り、および「ショートスクイーズ」などの取引現象。
•当社株式の取引高水準に一貫性がないことに起因する株価および出来高の変動
•追加の資金調達努力の発表または期待
•当社、内部関係者、またはその他の株主による当社の普通株式の売却。
•市場スタンドオフまたはロックアップ契約の満了。そして
•一般的な経済、産業、市場の状況。
さらに、COVID-19、米国および世界の経済状況の結果として、他のバイオ医薬品会社の普通株式の取引価格は非常に変動しやすくなっています。これらの出来事が当社の事業、前臨床試験、臨床試験にどの程度影響するかは、将来の進展にかかっています。将来の展開は非常に不確実で、自信を持って予測することはできません。
上記のリスクのいずれか、またはこの「リスク要因」セクションに記載されているものを含め、その他のさまざまなリスクのいずれかが実現すると、当社の普通株式の市場価格に劇的かつ悪影響を及ぼす可能性があります。
当社の四半期経営成績は、大きく変動したり、投資家や証券アナリストの予想を下回ったりする可能性があり、そのたびに当社の株価が変動または下落する可能性があります。
当社の業績は四半期ごとに変動すると予想しています。当社の純損失やその他の経営成績は、次のようなさまざまな要因の影響を受けます。
•当社の製品候補または将来の開発プログラムの継続的な開発に関連する費用の水準の変動。
•臨床試験の結果、または臨床試験の追加または終了、または当社または将来の潜在的なパートナーによる資金支援。
•当社が行うコラボレーション、ライセンス、または類似の取り決めの実施、および将来の取り決めに基づいて当社が行うまたは受け取る可能性のある支払いの時期、またはそのような将来の取り決めの終了または変更。
•当社が関与する可能性のある知的財産の侵害、不正流用、違反、訴訟、異議申し立て、干渉、取り消しの手続き。
•主要人員の増員および離職。
•買収、売却、スピンオフ、合弁事業、戦略的投資、事業戦略の変更など、当社または競合他社による戦略的決定。
•当社の製品候補のいずれかが規制当局の承認を受けた場合、その承認条件、およびそのような製品候補に対する市場での受け入れと需要は、
•当社の製品候補または競合他社の製品に影響を与える規制の進展、および
•一般的な市場および経済状況の変化。
四半期ごとの業績が投資家や証券アナリストの期待を下回った場合、当社の普通株式の価格は大幅に下落する可能性があります。さらに、四半期ごとの経営成績の変動により、株価が大幅に変動する可能性があります。四半期ごとの財務結果の比較は必ずしも意味があるわけではなく、将来の業績の指標として当てにするべきではないと考えています。
当社の主要株主と経営陣は、当社の株式のかなりの割合を所有しており、株主の承認を条件とする事項に大きな影響力を行使することができます。
2023年6月30日現在、当社の執行役員および取締役は、当社の普通株式の5%以上を所有する株主と、それぞれの関連会社を合わせると、当社の発行済み普通株式のかなりの割合を所有しています。その結果、これらの株主が一緒に行動することを選択した場合、株主に承認を求めるために提出されるすべての事項、および当社の経営や業務に関連する事項に大きな影響を与えることができるでしょう。たとえば、これらの株主は、取締役の選出、組織文書の修正、合併、資産の売却、その他の主要な企業取引の承認を管理できる場合があります。これにより、お客様が当社の株主の一人として最善の利益になると思われるような、一方的な買収提案や当社の普通株式のオファーを防止または阻止することができます。この株主グループの利益は、必ずしもあなたの利益や他の株主の利益と一致するとは限らず、普通株式のプレミアム価値を求めるなど、必ずしも他の株主の利益ではなく、自分たちの最善の利益を促進するような行動をとることもあり、私たちの普通株式の実勢市場価格に影響を与える可能性があります。
公開市場でかなりの数の普通株式を売却すると、株価が下落する可能性があります。
公開市場でのかなりの数の普通株式の売却、または多数の普通株式の保有者が株式を売却するつもりであるという市場の認識は、当社の普通株式の市場価格を下げる可能性があります。当社の普通株式の発行済み株式は、証券法の規則144および701で許可されている範囲で、またはそのような株式がすでに証券法に基づいて登録されており、当社の非関連会社が保有している範囲で、いつでも公開市場で自由に売却することができます。また、株式報酬プランに基づいて発行できる、または発行済みオプションの行使時に発行可能な普通株式をすべて登録します。これらの株式は、発行時に公開市場で自由に売却でき、権利が確定すると、関連会社に適用される数量制限が適用されます。これらの追加株式のいずれかが公開市場で売却された場合、または売却されると認識された場合、当社の普通株式の市場価格は下落する可能性があります。
追加の資本を調達すると、既存の株主が希薄化したり、事業が制限されたり、当社にとって不利な条件で製品候補の権利を放棄したりする可能性があります。
私たちは、パブリックエクイティやプライベートエクイティ、債務融資、または戦略的協力による前払いやマイルストーン支払いを含むその他の資金源など、さまざまな方法で追加資本を求めることがあります。たとえば、2020年8月、2021年7月、2022年5月、2023年6月に、私たちは当社の引受公募を完了しました
普通株式。2022年4月に普通株式の直接募集を完了しました。株式、転換社債、株式の売却を通じて追加資本を調達する場合、お客様の所有権は希薄化され、条件には株主としての権利に悪影響を及ぼす清算またはその他の優遇措置が含まれる場合があります。このような資金調達は、株主への希薄化、債務契約の賦課、固定支払い義務の増加、または当社の事業に影響を与える可能性のあるその他の制限につながる可能性があります。第三者との戦略的協力に基づいて前払いまたはマイルストーン支払いを通じて追加の資金を調達した場合、製品候補に対する貴重な権利を放棄するか、私たちにとって不利な条件でライセンスを付与しなければならない場合があります。さらに、現在または将来の事業計画に十分な資金があると考えられる場合でも、有利な市況または戦略的考慮事項により、追加の資本を求める場合があります。
証券または業界のアナリストが、当社、当社の事業、または市場に関する調査やレポートを公開しない場合、または不利または誤解を招くような調査またはレポートを発表した場合、当社の株価と取引量は下落する可能性があります。
私たちの普通株式の取引市場は、証券または業界のアナリストが当社、私たちの事業、または私たちの市場について発表する調査やレポートの影響を受けます。当社を担当するアナリストの誰かが、当社、当社のビジネスモデル、知的財産、株式実績、または市場に関して不利または誤解を招くような調査または報告を行った場合、または当社の業績がアナリストの期待に応えられない場合、当社の株価は下落する可能性があります。これらのアナリストの1人以上が当社の報道をやめたり、定期的にレポートを公開しなかったりすると、金融市場での可視性が失われ、ひいては株価や取引量が下落する可能性があります。
当社の設立証明書、付随定款、およびデラウェア州法の規定は、当社の支配権の変更または経営陣の変更を阻止、遅延、または妨げて、その結果、当社の普通株式の市場価格を押し下げる可能性があります。
当社の設立証明書と細則には、当社の株主が有利と見なす可能性のある当社の支配権の変更または経営陣の変更を阻止、遅延、または阻止することにより、当社の普通株式の市場価格を押し下げる可能性のある条項が含まれています。これらの規定、とりわけ:
•取締役会のメンバー全員が一度に選出されないように、機密扱いの取締役会を設立してください。
•取締役の人数を設定し、取締役会の欠員を埋めることができるのは取締役会だけです。
•ただし、取締役は「正当な理由がある場合」、株主の3分の2の承認がある場合にのみ解任できることを条件とします。
•取締役会が株主権利計画(「ポイズンピル」とも呼ばれます)を実施するために使用できる「空白の小切手」優先株の発行を承認します。
•株主が特別株主総会を招集できないようにします。
•書面による同意による株主の行動を禁止します。これにより、株主の行動はすべて株主総会で行う必要があります。
•累積投票を禁止します。
•当社の取締役会に細則の改正を許可します。
•取締役会への選任のための指名、または年次株主総会で株主が行動できる事項を提案するための事前通知要件を定めます。そして
•上記のいくつかの条項を修正するには、株主の圧倒的多数の投票が必要です。
さらに、デラウェア州の一般会社法(DGCL)の第203条は、デラウェア州の上場企業が利害関係のある株主、一般的にはその関連会社とともに議決権の15%を所有している、または過去3年以内に議決権のある株式の15%を所有していた人物と企業結合を行うことを禁じています。その人は、その人が利害関係のある株式になった取引日から3年間保有者。ただし、企業結合が所定の方法で承認された場合を除きます。
支配権の変更を遅らせたり妨げたりする効果のある当社の設立証明書、細則、またはデラウェア州法の規定は、株主が当社の資本株式のプレミアムを受け取る機会を制限する可能性があり、一部の投資家が当社の普通株式に支払う意思のある価格にも影響を与える可能性があります。
当社の法人設立証明書には、デラウェア州司法裁判所が当社と株主との間の実質的にすべての紛争の専属管轄裁判所であると記載されています。そのため、当社の株主は、当社または当社の取締役、役員、従業員との紛争について有利な司法裁判所を取得することが制限される可能性があります。
当社の法人設立証明書には、デラウェア州司法裁判所が以下の専属管轄裁判所であることが規定されています。
•当社に代わって提起された派生訴訟または手続き
•受託者責任違反の申し立てを主張するあらゆる訴訟。
•DGCL、当社の設立証明書、または当社の細則に基づいて生じた、当社に対する請求を主張するあらゆる訴訟。そして
•内務原則に基づく当社に対する請求を主張するあらゆる行為。
この独占フォーラム条項は、株主が当社または当社の取締役、役員、その他の従業員との紛争に有利であると判断した司法フォーラムに請求を行う能力を制限する可能性があり、その結果、当社および当社の取締役、役員、その他の従業員に対する訴訟を思いとどまらせる可能性があります。当社の有価証券を購入したり、その他の方法で持分を取得したりする個人または団体は、この条項について通知し、同意したものとみなされます。裁判所が、当社の法人設立証明書にあるこの独占フォーラム条項が訴訟に適用されない、または法的強制力がないと判断した場合、他の法域での紛争解決に関連する追加費用が発生し、当社の事業に重大な損害を与える可能性があります。当社の設立証明書には、証券法または取引法に基づいて請求を主張する株主が、適用法に従って州または連邦裁判所にそのような請求を提起することを妨げるものはありません。
現在、当社の普通株式に配当を支払う予定はありません。したがって、投資収益率を達成できるかどうかは、当社の普通株式の価値の上昇にかかっています。
私たちは、持分証券の現金配当を申告したり支払ったりしたことはありません。私たちは現在、事業の発展、運営、拡大のために将来の収益を留保すると予想しており、当面の間、現金配当の申告や支払いは予定していません。したがって、株主への利益は、当社の普通株式の価値の上昇に限定されますが、これは確実ではありません。
財務報告に対する効果的な内部統制システムを維持しないと、財務結果を正確に報告できないか、詐欺を防止できない可能性があります。その結果、株主は当社の財務報告やその他の公開報告に対する信頼を失い、当社の事業と普通株式の取引価格に悪影響を及ぼす可能性があります。
信頼できる財務報告を提供するためには、財務報告に対する効果的な内部統制が必要であり、適切な開示管理と手続きとともに、詐欺を防止するように設計されています。必要な新しい統制や改善された統制を実施できなかったり、実施中に問題が発生したりすると、報告義務を履行できなくなる可能性があります。さらに、第404条に関連して当社が実施したテスト、またはそれに続く独立登録公認会計事務所によるテストにより、財務報告に対する内部統制の欠陥が明らかになり、重大な弱点と見なされたり、将来的または遡及的な変更が必要になったりする可能性があります。
財務諸表に追加するか、さらに注意または改善が必要な他の分野を特定してください。内部統制が不十分だと、投資家は私たちが報告した財務情報に対する信頼を失い、私たちの株式の取引価格に悪影響を及ぼす可能性があります。
内部統制と手続きに加えられた変更を四半期ごとに開示する必要があり、経営陣はこれらの統制の有効性を毎年評価する必要があります。私たちはもはや新興成長企業ではなくなったので、独立登録公認会計士事務所は、第404条に基づく財務報告に対する内部統制の有効性を証明する必要があります。財務報告に対する内部統制の有効性を独自に評価することで、経営陣の評価では検出できないかもしれない問題を発見することができます。財務報告に対する内部統制の重大な弱点が発見されないと、財務諸表が再表示され、是正費用を負担する必要が生じる可能性があります。
私たちは証券訴訟の対象となる可能性があり、これは費用がかかり、経営陣の注意をそらす可能性があります。
私たちの普通株式の市場価格は変動する可能性があり、過去に、株式の市場価格の変動を経験した企業は、証券集団訴訟の対象となっていました。将来、私たちはこの種の訴訟の対象になるかもしれません。当社に対する証券訴訟は多額の費用をもたらし、経営陣の注意を他の事業上の懸念からそらし、当社の事業に重大な損害を与える可能性があります。
アイテム 2.株式の未登録売却、収益の使用、および発行者による株式の購入。
該当しません。
アイテム 3.シニア証券のデフォルト.
該当しません。
アイテム 4.鉱山の安全に関する開示。
該当しません。
アイテム 5.その他の情報
(a)最高科学責任者の承継
2023年11月1日、ケビン・バンカー博士は、2023年12月31日(「辞任発効日」)をもって会社の最高科学責任者を辞任することを会社に通知しました。バンカー博士は、辞任発効日後も引き続き会社の顧問を務めます。
2023年11月6日、当社は、同社の最高翻訳責任者でバイオマーカー戦略の責任者であるマーク・ラックナー博士が、辞任発効日をもって、バンカー博士の後任として会社の最高科学責任者に就任すると発表しました。
また、2023年11月1日、バンカー博士は会社と移行およびリリース契約を締結しました。移行および解放契約に基づき、バンカー博士は辞任発効日まで、同じ報酬で最高科学責任者の地位を維持します。バンカー博士の雇用終了時に、当社は、12か月の基本給に2023年の目標ボーナスを加えたものの一括払い、および会社の費用によるCOBRA保険料の12か月間の継続的な支払いを含む、一般的な請求の解除と引き換えに特定の退職金を提供します。さらに、バンカー博士は未払いのオプションと制限付株式ユニットの権利確定を12か月間受けます。バンカー博士は、2028年12月31日まで既得のストックオプションを行使することができます。
バンカー博士と当社はまた、2024年3月31日までの辞任発効日以降、バンカー博士が会社に助言サービスを提供するというコンサルティング契約を締結しました。
上記の情報は、フォーム8-Kの「項目5.02 — 取締役または特定の役員の辞任、取締役の選出、特定の役員の任命、特定の役員の報酬的取り決め」で要求される開示を提供する目的で含まれています。
(b) なし。
(c)取締役兼役員 取引の手配
オン 2023年9月22日, メリッサ・エパーリー、会社の 最高財務責任者, 採用されたa 規則10b5-1 (c) の肯定的な抗弁を満たすことを目的とした規則10b5-1取引契約 22,0652024年12月22日までの当社の普通株式。
アイテム 6.展示品。
| | | | | | | | | | | | | | | | | | | | | | | |
示す 番号 | | 説明 | 参考により組み込み | |
| フォーム | ファイル番号 | 示す | ファイリング
日付 | ここにファイル/家具付き |
| | | | | | | |
| 2.1 | | ゼンタリスファーマシューティカルズLLC(デラウェア州の有限責任会社)をゼンタリスファーマシューティカルズ社(デラウェア州の法人)に転換する計画です。 | 10-Q | 001-39263 | 2.1 | 05/15/2020 | |
| 2.2 | | Zentalis Pharmaceuticals, LLC(デラウェア州の有限責任会社)をゼンタリスファーマシューティカルズ社(デラウェア州の法人)に転換する転換証明書 | 10-Q | 001-39263 | 2.2 | 05/15/2020 | |
| 3.1 | | ゼンタリス製薬株式会社の設立証明書 | S-8 | 333-237593 | 4.1 | 04/07/2020 | |
| 3.2 | | 2023年6月16日付けのゼンタリス製薬株式会社の設立証明書の修正証明書 | 8-K | 001-39263 | 3.1 | 06/16/2023 | |
| 3.3 | | ゼンタリス製薬株式会社の細則 | 8-K | 001-39263 | 3.1 | 03/19/2021 | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
31.1 | | 取引法規則13a-14(a)に基づく最高経営責任者の認定。 | | | | | * |
31.2 | | 取引法規則13a-14 (a) に基づく最高財務責任者の認定。 | | | | | * |
32.1 | | 18 U.S.C. セクション1350に基づく最高経営責任者の認定。 | | | | | ** |
32.2 | | 18 U.S.C. セクション1350に基づく最高財務責任者の認定。 | | | | | ** |
| 101.インチ | | インライン XBRL インスタンスドキュメント | | | | | * |
| 101.SCH | | インライン XBRL タクソノミー拡張スキーマドキュメント | | | | | * |
| 101.CAL | | インライン XBRL タクソノミー拡張計算リンクベースドキュメント | | | | | * |
| 101.DEF | | インライン XBRL タクソノミー拡張定義リンクベースドキュメント | | | | | * |
| 101.LAB | | インライン XBRL タクソノミー拡張ラベルリンクベースドキュメント | | | | | * |
| | | | | | | | | | | | | | | | | | | | | | | |
示す 番号 | | 説明 | 参考により組み込み | |
| フォーム | ファイル番号 | 示す | ファイリング
日付 | ここにファイル/家具付き |
| | | | | | | |
| 101.PRE | | インライン XBRL タクソノミー拡張プレゼンテーションリンクベースドキュメント | | | | | * |
| 104 | | 表紙インタラクティブデータファイル(インラインXBRL形式で、別紙101に含まれています) | | | | | * |
* ここに提出。
**付属しています。
署名
1934年の証券取引法の要件に従い、登録者は署名者に代わってこの報告書に正式に署名させ、正式に権限を与えられました。
| | | | | | | | |
| ゼンタリス製薬株式会社 |
| | |
日付:2023年11月6日 | 作成者: | /s/ キンバリー・ブラックウェル、M.D。 |
| | キンバリー・ブラックウェル、M.D。 |
| | 最高経営責任者 |
| | (最高執行役員) |
日付:2023年11月6日 | 作成者: | /s/ メリッサ・B・エパーリー |
| | メリッサ・B・エパーリー |
| | 最高財務責任者 |
| | (最高財務会計責任者) |