米国
証券取引委員会
ワシントンDC 20549
フォーム
(マークワン)
1934年の証券取引法のセクション13または15 (d) に基づく四半期報告書 |
四半期終了時
または
1934年の証券取引法第13条または第15条 (d) に基づく移行報告書 |
___________から___________への移行期間について
コミッションファイル番号:
(憲章に明記されている登録者の正確な名前)
(州またはその他の管轄区域) 法人または組織) |
(IRS) 雇用主 |
(主要執行機関の住所) |
(郵便番号) |
登録者の電話番号 (市外局番を含む): (
同法第12条 (b) に従って登録された証券:
各クラスのタイトル |
|
取引 シンボル (複数可) |
|
登録された各取引所の名前 |
|
|
登録者が (1) 1934年の証券取引法第13条または第15条 (d) 条により提出が義務付けられているすべての報告書を過去12か月間(または登録者がそのような報告を提出する必要があったほど短い期間)に提出したかどうか、および(2)過去90日間にそのような申告要件の対象であったかどうかをチェックマークで示してください。
登録者が過去 12 か月間(または、登録者がそのようなファイルの提出を求められたほど短い期間)に、規則 S-T の規則 405(本章の §232.405)に従って提出する必要のあるすべてのインタラクティブデータファイルを電子的に提出したかどうかをチェックマークで示してください。
登録者が大規模な加速申告者、加速申告者、非加速申告者、小規模な報告会社、または新興成長企業のいずれであるかをチェックマークで示してください。取引法規則12b-2の「大規模加速申告者」、「加速申告者」、「小規模報告会社」、および「新興成長企業」の定義を参照してください。
大型加速フィルター |
|
☐ |
|
アクセラレーテッド・ファイラー |
|
☐ |
|
|
|
|
|||
|
☒ |
|
小規模な報告会社 |
|
||
|
|
|
|
|
|
|
新興成長企業 |
|
|
|
|
|
|
新興成長企業の場合は、登録者が取引法第13条 (a) に従って規定された新規または改訂された財務会計基準を遵守するために延長された移行期間を使用しないことを選択したかどうかをチェックマークで示してください。
登録者がシェル会社(取引法の規則12b-2で定義されている)であるかどうかをチェックマークで示してください。はい ☐いいえ
2023年8月14日の時点で、登録者は
目次
|
|
ページ |
第一部。 |
財務情報 |
|
アイテム 1. |
要約連結財務諸表(未監査) |
1 |
|
貸借対照表 |
1 |
|
運用ステートメント |
2 |
|
株主(赤字)資本(赤字)に関する声明 |
3 |
|
キャッシュフロー計算書 |
5 |
|
財務諸表に関する注記 |
6 |
アイテム 2. |
経営陣による財政状態と経営成績に関する議論と分析 |
24 |
アイテム 3. |
市場リスクに関する定量的・質的開示 |
62 |
アイテム 4. |
統制と手続き |
62 |
第二部 |
その他の情報 |
|
アイテム 1. |
法的手続き |
63 |
アイテム 1A. |
リスク要因 |
63 |
アイテム 2. |
持分証券の未登録売却および収益の使用 |
64 |
アイテム 3. |
シニア証券のデフォルト |
65 |
アイテム 4. |
鉱山の安全に関する開示 |
65 |
アイテム 5. |
その他の情報 |
65 |
アイテム 6. |
展示品 |
66 |
署名 |
67 |
|
i
パートI — 財務すべての情報
アイテム 1.財務すべての声明。
タイシャ・ジーン・セラピーズ株式会社
コンデンスコンソリダテッド貸借対照表
(千単位、1株あたりのデータを除く)
(未監査)
|
|
|
|
|
|
|
||
|
|
6月30日 |
|
|
12月31日 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
現金および現金同等物 |
|
$ |
|
|
$ |
|
||
前払費用およびその他の流動資産 |
|
|
|
|
|
|
||
流動資産合計 |
|
|
|
|
|
|
||
制限付き現金 |
|
|
|
|
|
|
||
不動産、プラント、設備、純額 |
|
|
|
|
|
|
||
オペレーティングリースの使用権資産 |
|
|
|
|
|
|
||
その他の非流動資産 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
負債と株主(赤字)資本 |
|
|
|
|
|
|
||
現在の負債 |
|
|
|
|
|
|
||
買掛金 |
|
$ |
|
|
$ |
|
||
未払費用およびその他の流動負債 |
|
|
|
|
|
|
||
繰延収益 |
|
|
|
|
|
|
||
流動負債合計 |
|
|
|
|
|
|
||
繰延収益、当期分を差し引いたもの |
|
|
|
|
|
— |
|
|
タームローン、純額 |
|
|
|
|
|
|
||
オペレーティング・リースの負債、当期分を差し引いたもの |
|
|
|
|
|
|
||
その他の非流動負債 |
|
|
|
|
|
|
||
負債総額 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
株主(赤字)資本 |
|
|
|
|
|
|
||
優先株式、$ |
|
|
|
|
|
|
||
普通株式、$ |
|
|
|
|
|
|
||
追加払込資本 |
|
|
|
|
|
|
||
累積赤字 |
|
|
( |
) |
|
|
( |
) |
株主資本(赤字)総資本 |
|
|
( |
) |
|
|
|
|
負債総額と株主(赤字)資本 |
|
$ |
|
|
$ |
|
||
添付の注記は、これらの未監査の要約連結財務諸表の不可欠な部分です。
1
タイシャ・ジーン・セラピーズ株式会社
コンデンスコンソリデーテッドセット運用明書
(千単位、1株あたりのデータを除く)
(未監査)
|
|
3 か月間 |
|
|
6 か月間 |
|
||||||||||
|
|
2023 |
|
|
2022 |
|
|
2023 |
|
|
2022 |
|
||||
収益 |
|
$ |
|
|
$ |
— |
|
|
$ |
|
|
$ |
— |
|
||
営業経費: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
研究開発 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
一般管理と管理 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
営業費用の合計 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
事業による損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
その他の収入 (費用): |
|
|
|
|
|
|
|
|
|
|
|
|
||||
利息収入 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
支払利息 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
その他の収入 (費用) |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
その他の収益(費用)の合計、純額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
普通株式1株あたりの純損失(基本および希薄化後) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
加重平均発行済普通株式、基本株式および希薄化後普通株式 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
添付の注記は、これらの未監査の要約連結財務諸表の不可欠な部分です。
2
タイシャ・ジーン・セラピーズ株式会社
要約連結計算書株主(赤字)資本のセントです
(千単位、共有データを除く)
(未監査)
2023年6月30日に終了した3か月間
|
|
|
|
|
|
|
|
[追加] |
|
|
|
|
|
合計 |
|
|||||
|
|
普通株式 |
|
|
支払い済み |
|
|
累積 |
|
|
株主の |
|
||||||||
|
|
株式 |
|
|
金額 |
|
|
資本 |
|
|
赤字 |
|
|
赤字 |
|
|||||
2023年3月31日現在の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|||
株式報酬制度 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
私募による普通株式の発行、募集費用$を差し引いたもの |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
権利確定と制限付株式単位の決済に伴う普通株式の発行 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2023年6月30日現在の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|||
2022年6月30日に終了した3か月間
|
|
|
|
|
|
|
|
[追加] |
|
|
|
|
|
合計 |
|
|||||
|
|
普通株式 |
|
|
支払い済み |
|
|
累積 |
|
|
株主の |
|
||||||||
|
|
株式 |
|
|
金額 |
|
|
資本 |
|
|
赤字 |
|
|
エクイティ |
|
|||||
2022年3月31日現在の残高 |
|
|
|
|
$ |
— |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
株式報酬制度 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
普通株式の発行、販売手数料およびその他の募集費用を差し引いたもの |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
権利確定と制限付株式単位の決済に伴う普通株式の発行 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2022年6月30日現在の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||
添付の注記は、これらの未監査の要約連結財務諸表の不可欠な部分です。
3
タイシャ・ジーン・セラピーズ株式会社
株主(赤字)資本の要約連結計算書
(千単位、共有データを除く)
(未監査)
2023年6月30日に終了した6か月間
|
|
|
|
|
|
|
|
[追加] |
|
|
|
|
|
合計 |
|
|||||
|
|
普通株式 |
|
|
支払い済み |
|
|
累積 |
|
|
株主の |
|
||||||||
|
|
株式 |
|
|
金額 |
|
|
資本 |
|
|
赤字 |
|
|
資本 (赤字) |
|
|||||
2022年12月31日現在の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||
株式報酬制度 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
私募による普通株式の発行、募集費用$を差し引いたもの |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
権利確定および制限付株式単位の決済時の普通株式の発行、純額 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
ESPPに基づく普通株式の発行 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2023年6月30日現在の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|||
2022年6月30日に終了した6か月間
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
[追加] |
|
|
|
|
|
合計 |
|
|||||
|
|
普通株式 |
|
|
支払い済み |
|
|
累積 |
|
|
株主の |
|
||||||||
|
|
株式 |
|
|
金額 |
|
|
資本 |
|
|
赤字 |
|
|
資本 (赤字) |
|
|||||
2021年12月31日現在の残高 |
|
|
|
|
$ |
— |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
当初の累積赤字への調整 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
||
株式報酬制度 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
権利確定と制限付株式単位の決済に伴う普通株式の発行 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
普通株式の発行、販売手数料およびその他の募集費用を差し引いたもの |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2022年6月30日現在の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||
添付の注記は、これらの未監査の要約連結財務諸表の不可欠な部分です。
4
タイシャ・ジーン・セラピーズ株式会社
コンデンスコンソリデーテッドセットキャッシュフロー計算書
(千単位)
(未監査)
|
|
6 か月間 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
営業活動によるキャッシュフロー |
|
|
|
|
|
|
||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
純損失を営業活動に使用された純現金と調整するための調整: |
|
|
|
|
|
|
||
減価償却費 |
|
|
|
|
|
|
||
研究開発ライセンス費用 |
|
|
|
|
|
|
||
株式報酬制度 |
|
|
|
|
|
|
||
非現金リース費用 |
|
|
|
|
|
|
||
その他 |
|
|
|
|
|
|
||
営業資産および負債の変動: |
|
|
|
|
|
|
||
前払費用およびその他の資産 |
|
|
|
|
|
|
||
買掛金 |
|
|
|
|
|
|
||
未払費用およびその他の負債 |
|
|
( |
) |
|
|
( |
) |
繰延収益 |
|
|
( |
) |
|
|
— |
|
営業活動に使用された純現金 |
|
|
( |
) |
|
|
( |
) |
投資活動によるキャッシュフロー |
|
|
|
|
|
|
||
研究開発ライセンスの購入 |
|
|
— |
|
|
|
( |
) |
不動産、プラント、設備の購入 |
|
|
( |
) |
|
|
( |
) |
投資活動に使用された純現金 |
|
|
( |
) |
|
|
( |
) |
財務活動によるキャッシュフロー |
|
|
|
|
|
|
||
普通株式の発行による収入、販売手数料を差し引いたもの |
|
|
— |
|
|
|
|
|
私募による普通株式の発行による収入 |
|
|
|
|
|
— |
|
|
棚登録費用の支払い |
|
|
( |
) |
|
|
( |
) |
ESPPに基づく普通株式発行による収入 |
|
|
|
|
|
|
||
その他 |
|
|
( |
) |
|
|
( |
) |
財務活動による純現金 |
|
|
|
|
|
|
||
現金、現金同等物および制限付現金の純減少 |
|
|
( |
) |
|
|
( |
) |
期首における現金、現金同等物および制限付現金 |
|
|
|
|
|
|
||
期末の現金、現金同等物および制限付現金 |
|
$ |
|
|
$ |
|
||
現金および現金同等物 |
|
|
|
|
|
|
||
制限付き現金 |
|
|
|
|
|
|
||
期末の現金、現金同等物および制限付現金 |
|
$ |
|
|
$ |
|
||
キャッシュフロー情報の補足開示: |
|
|
|
|
|
|
||
利息として支払われた現金 |
|
$ |
|
|
$ |
|
||
非現金投資および財務活動の補足開示: |
|
|
|
|
|
|
||
買掛金および未払費用の有形固定資産、プラント、設備 |
|
|
— |
|
|
|
|
|
リース負債と引き換えに取得した使用権資産 |
|
|
— |
|
|
|
|
|
提供費用はまだ支払われていません |
|
|
— |
|
|
|
|
|
私募に関連するワラントの発行 |
|
|
|
|
|
— |
|
|
研究開発ライセンスの購入はまだ支払われていません |
|
|
|
|
|
|
||
添付の注記は、これらの未監査の要約連結財務諸表の不可欠な部分です。
5
注1—構成と説明事業運営のオプション
Taysha Gene Therapies, Inc.(以下「当社」または「Taysha」)は、もともと2019年9月20日(「インセプション」)にテキサス州の法律(「インセプション」)に基づいて設立されました。Tayshaはデラウェア州の法人に転向しました
Tayshaは患者中心の遺伝子治療会社で、まれな患者集団と大規模な患者集団の両方における中枢神経系の単一遺伝子疾患の治療のためのAAVベースの遺伝子治療の開発と商品化に焦点を当てています。
販売契約
2021年10月5日、当社はSVB Securities LLC(f/k/a SVB Leerink LLC)およびウェルズ・ファーゴ証券LLC(総称して「販売代理店」)と売買契約(「販売契約」)を締結しました。これに基づき、当社は独自の裁量により、総額が最大$の普通株式を随時発行および売却することができます。
流動性と継続性
添付の要約連結財務諸表は、継続企業に適用される一般に認められた会計原則に従って作成されています。この原則は、通常の事業過程における資産の実現と負債の履行を考慮しています。
ASC 205に従い、 財務諸表の提示、当社は、要約された連結財務諸表が発行された日から1年以内に継続企業として存続する能力について大きな疑念を生じさせる状況または事象があるかどうか、年次および中間期間ごとに評価することを義務付けられています。
将来の義務を果たす会社の能力の評価は、本質的に判断的で主観的であり、変更される可能性があります。会社の現在の予測に基づくと、経営陣は、2023年8月に完了する予定の私募による予想純収入(注記15を参照)を含め、これらの要約連結財務諸表の発行後12か月間、会社が計画していた事業を維持するのに十分な現金があると考えています。しかし、予測に内在する不確実性を踏まえ、当社は、これらの要約連結財務諸表が発行された日時点で既知の、または合理的に分かっている量的要因と質的要因の両方を考慮し、全体として、当社が継続企業として存続する能力について大きな疑念を抱く状況が存在すると結論付けました。
同社は創業以来営業損失を被っており、当面は引き続き大きな営業損失を被り、利益を上げることはないと予想しています。2023年6月30日の時点で、同社の累積赤字は $
6
注2—重要な会計方針の要約
プレゼンテーションの基礎
未監査の要約連結財務諸表は、財務会計基準審議会(「FASB」)の会計基準体系化(「ASC」)によって決定された、米国で一般に認められている会計原則(「GAAP」)、および暫定財務情報のフォーム10-Qと規則S-Xの第10条に従って作成されており、すべての重要な点で当社の年次報告書に記載されているものと一致しています。2023年3月28日に証券取引委員会に提出された、2022年12月31日に終了した年度(以下「2022年」)の10-K年次報告書」)。経営陣の見解では、未監査の要約連結財務諸表にはすべての調整が反映されています。これには、提示された期間の残高と結果を公正に計算するために必要な、通常の定期的な調整のみが含まれます。2022年12月31日現在の連結貸借対照表は監査済み財務諸表から導き出されていますが、GAAPが財務諸表全体に必要なすべての情報と脚注が含まれているわけではありません。これらの未監査の要約連結財務諸表は、当社の2022年次報告書の連結財務諸表および関連する注記と併せて読む必要があります。
統合の原則
添付の中間要約連結財務諸表には、Tayshaとその完全子会社の勘定が含まれています。連結により、会社間の取引と残高はすべて削除されました。
見積もりの使用
GAAPに準拠した財務諸表を作成するには、経営陣は、財務諸表の日付における報告された資産と負債の金額、偶発資産と負債の開示、および報告期間中の報告された費用額に影響する見積もりと仮定を行う必要があります。会社の財務諸表で最も重要な見積もりと仮定は、新規株式公開(「IPO」)前の普通株式の公正価値の決定(株式ベースの報酬へのインプット)、製造発生額と未払いの研究開発費の見積もり、長期資産の減損の測定、およびアステラス製薬との取引に関連して受け取った対価の配分に関するものです。これらの見積もりと仮定は、現在の事実、過去の経験、および状況下で妥当と考えられるその他のさまざまな要因に基づいており、その結果は、資産と負債の帳簿価額に関する判断や、他の情報源からはすぐには明らかにならない費用の記録の基礎となります。実際の結果は、これらの見積もりと大きく異なる場合があります。見積もりと実際の結果に重大な違いがある限り、会社の将来の経営成績に影響します。
重要な会計方針
2022年の年次報告書に含まれる監査済み連結財務諸表の注記2に開示されているように、当社の重要な会計方針には、以下に記載されている場合を除き、変更はありません。
ワラント
当社は、ワラントの具体的な条件とFASB ASC 480の該当する権威あるガイダンスの評価に基づいて、ワラントを株式分類商品または負債分類商品のいずれかとして会計処理しています。 負債と資本の区別(「ASC 480」) と ASC 815、デリバティブとヘッジング(「815を尋ねる」)。評価では、ワラントがASC 480に基づく独立型金融商品であるかどうか、ASC 480に基づく負債の定義を満たしているかどうか、およびワラントがASC 815に基づく株式分類のすべての要件を満たしているかどうか(ワラントが会社の普通株式に連動しているかどうか、ワラント保有者が会社の制御が及ばない状況で「純現金決済」を要求する可能性があるかどうかなど)が考慮されます。、株式分類の他の条件の中でも。この査定は専門家の判断が必要ですが、令状の発行時とその後の各報告期間中に、令状が未払いの期間中に行われます。
株式分類のすべての基準を満たす発行または変更されたワラントについては、発行時に追加払込資本の構成要素としてワラントを記録する必要があります。株式分類のすべての基準を満たしていない発行または変更されたワラントについては、ワラントは発行日の初期公正価値で記録され、その後は各貸借対照表日に記録される必要があります。会社はワラントを公正価値では負債として分類し、各報告期間でワラントを公正価値に調整します。この負債は、新株予約権の行使または期限が切れるまで、各貸借対照表日に再測定される可能性があります。公正価値の変動は、会社の要約連結営業報告書に反映されます。
包括的損失
包括損失は、添付の要約連結損益計算書に示されている純損失と同じです。
7
最近採択された会計上の宣言
2016年2月、FASBは、リースの会計処理と開示に関するガイダンスとともに、修正後のASU番号2016-02、リース(トピック842)を発行しました。この更新により、借手は貸借対照表に12か月を超える期間を持つすべてのリース(オペレーティングリースを含む)に関連する負債を認識する必要があります。この更新では、借手と貸手がリース取引に関する重要な情報を開示することも義務付けられています。
オン
当社は以下の実務上の手段を選択しました。これらはパッケージとして選択され、移行日にすべてのリース(法人が借手または貸主であるものを含む)に一貫して適用されなければなりません。i)期限切れまたは既存の契約にリースが含まれているかどうかを再評価しませんでした。ii)期限切れまたは既存のリース(つまり、既存のリース)のリース分類を再評価しませんでした。ASC 840に従ってオペレーティングリースとして分類されたものはオペレーティングリースとして分類され、既存のものはすべて分類されますASC 840に従ってキャピタルリースとして分類されたリースは、(3) ファイナンスリース (iii) 既存のリースの初期直接費用を再評価しませんでした。ASC 842の最初の適用日より前に存在していたリース(以前はオペレーティングリースとして分類されていました)の場合、借手は、ASC 840に基づくリース開始時に測定された合計リース期間、またはASC 842の初回適用日現在の残りのリース期間のいずれかを使用して、増分借入金利を測定する期間を決定することができます。ASC 842への移行にあたり、同社はリースの残りのリース期間を利用して、適切な増額借入金利を決定しました。
この基準が採用された結果、オペレーティングリースの使用権資産と$のオペレーティングリース負債が認識されました
8
次の表は、ASC 842の採用が2022年6月30日までの6か月間の要約連結営業報告書とキャッシュフロー計算書に与えた影響(千単位)をまとめたものです。
|
|
ASC 842以前の6ヶ月が終了しました |
|
|
ASC 842の調整 |
|
|
ASC 842の6ヶ月が終了した後 |
|
|||
統合運用明細書 |
|
|
|
|
|
|
|
|
|
|||
営業経費: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
その他の収入 (費用): |
|
|
|
|
|
|
|
|
|
|||
支払利息 |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
純損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|||
要約連結キャッシュフロー計算書 |
|
|
|
|
|
|
|
|
|
|||
純損失を営業活動に使用された純現金と調整するための調整: |
|
|
|
|
|
|
|
|
|
|||
減価償却費 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
非現金リース費用 |
|
|
— |
|
|
|
|
|
|
|
||
営業資産および負債の変動: |
|
|
|
|
|
|
|
|
|
|||
未払費用およびその他の流動負債 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
財務活動によるキャッシュフロー |
|
|
|
|
|
|
|
|
|
|||
その他 |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
非現金投資および財務活動の補足開示: |
|
|
|
|
|
|
|
|
|
|||
リース負債と引き換えに取得した使用権資産 |
|
|
— |
|
|
|
|
|
|
|
||
次の表は、2022年6月30日までの3か月間の要約連結営業報告書に対するASC 842の採用による影響(千単位)をまとめたものです。
|
|
ASC 842より前の3か月が終了しました |
|
|
ASC 842の調整 |
|
|
ASC 842の3か月が終了した後 |
|
|||
統合運用明細書 |
|
|
|
|
|
|
|
|
|
|||
営業経費: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
その他の収入 (費用): |
|
|
|
|
|
|
|
|
|
|||
支払利息 |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
純損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
注3—貸借対照表の構成要素
前払費用およびその他の流動資産は、以下のとおりです(千単位)。
|
|
6月30日 |
|
|
12月31日 |
|
||
前払いの研究開発 |
|
$ |
|
|
$ |
|
||
前払いの臨床試験 |
|
|
|
|
|
|
||
繰延オファリング費用 |
|
|
|
|
|
|
||
プリペイド保険 |
|
|
|
|
|
|
||
前払いボーナス |
|
|
|
|
|
|
||
その他 |
|
|
|
|
|
|
||
前払費用とその他の流動資産の合計 |
|
$ |
|
|
$ |
|
||
9
不動産、プラント、設備、純額は次のとおりです(千単位)。
|
|
6月30日 |
|
|
12月31日 |
|
||
借地権の改善 |
|
$ |
|
|
$ |
|
||
実験室用機器 |
|
|
|
|
|
|
||
コンピューター機器 |
|
|
|
|
|
|
||
家具と備品 |
|
|
|
|
|
|
||
建設中 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
減価償却累計額 |
|
|
( |
) |
|
|
( |
) |
不動産、プラント、設備、純額 |
|
$ |
|
|
$ |
|
||
2022年11月、当社は現金以外の減損費用を認めました
減価償却費は $
未払費用およびその他の流動負債は、以下のとおりです(千単位)。
|
|
6月30日 |
|
|
12月31日 |
|
||
これまでに蓄積された研究開発 |
|
$ |
|
|
$ |
|
||
未払ライセンス料 |
|
|
|
|
|
— |
|
|
これまでに行われた臨床試験 |
|
|
|
|
|
|
||
未払報酬 |
|
|
|
|
|
|
||
未払退職金 |
|
|
|
|
|
|
||
リース負債、流動部分 |
|
|
|
|
|
|
||
未払いの専門家費用とコンサルティング料 |
|
|
|
|
|
|
||
保証責任 |
|
|
|
|
|
— |
|
|
未払資産、プラント、設備 |
|
|
— |
|
|
|
|
|
その他 |
|
|
|
|
|
|
||
未払費用およびその他の流動負債の合計 |
|
$ |
|
|
$ |
|
||
注4— リース
同社は特定のオフィス、研究所、製造スペースをリースしています。
ダラスリース
2021年1月11日、当社はデラウェア州の有限責任会社であるPegasus Park, LLC(以下「ダラス・ランドロード」)とリース契約(「ダラスリース」)を締結しました。この契約に基づき、当社はおおよその賃貸借契約を結びます
ダラスのリースは始まりました
ダラスの家主は、会社が適時に家賃を支払わなかった場合や、会社に関して特定の破産事由が発生した場合など、特定の債務不履行事由が発生した場合に、ダラスリース、またはダラスリースを終了せずにオフィススペースを所有する会社の権利を終了する権利を有します。
10
ダラスのリース拡大
2021年12月14日、当社はダラスの家主とのダラス・リース(「ダラス・リースの改正」)を修正しました。これに基づき、当社はおおよその賃貸借契約を結ぶ予定です。
ダラスのリース改正は2022年7月1日に開始され、期間は約
会社は拡張施設に適用される運営費と光熱費を支払う義務があります。当初のダラスリース修正条項に基づく将来の最低リース支払い額の合計です
同社は、15日に特定のオフィススペースを追加することに関して、最初に拒否する権利があります番目のダラスの家主がそのようなスペースのオファーを受け入れる前に、テキサス州ダラスのペガサスパークドライブ3000番地75247番地にあります。
ダーラムリース
2020年12月17日、当社はデラウェア州の有限責任会社であるパトリオット・パーク・パートナーズII、LLC(以下「ダーラム・ランドロード」)とリース契約(「ダーラム・リース」)を締結しました。この契約に基づき、両社はおよそ賃貸することに合意しました。
同社は、ダーラムリースへの参入に関連して保証金を支払う必要はありませんでした。会社は施設内の内装改善を行う責任があります。会社は$を配置する必要がありました
ASC 842で認められたすべてのリース費用の概要
次の表は、2023年6月30日および2022年6月30日までの3か月および6か月間のASC 842で計上されたリース費用および当社のオペレーティングリースに関連するその他の情報(千単位)をまとめたものです。
|
6月30日に終了した3か月間 |
|
|
6月30日までの6か月間、 |
|
|||||||||
|
2023 |
|
2022 |
|
|
2023 |
|
|
2022 |
|
||||
オペレーティングリース費用 |
$ |
|
$ |
|
|
$ |
|
|
$ |
|
||||
変動リース費用 |
|
|
|
|
|
|
|
|
|
|
||||
リース費用合計 |
$ |
|
$ |
|
|
$ |
|
|
$ |
|
||||
残りのリース期間と割引率に関する補足情報は次のとおりです。
|
|
2023年6月30日 |
|
2022年12月31日 |
|
||
加重平均残存リース期間(年単位)-ファイナンスリース |
|
|
|
|
|
||
加重平均残存リース期間(年)-オペレーティングリース |
|
|
|
|
|
||
|
|
|
|
|
|
||
加重平均割引率-ファイナンスリース |
|
|
% |
|
% |
||
加重平均割引率-オペレーティングリース |
|
|
% |
|
% |
||
11
会社のオペレーティングリースに関連する補足的なキャッシュフロー情報は次のとおりです(千単位)。
|
6月30日までの6か月間、 |
|
|||||
|
2023 |
|
|
2022 |
|
||
オペレーティングリースの営業キャッシュフロー |
$ |
|
|
$ |
|
||
2023年6月30日現在、当社のオペレーティングリースおよびファイナンスリースに基づくASC 842に基づく将来の最低契約額は次のとおりです(千単位)。
12月31日に終了する年度 |
オペレーティング |
|
金融 |
|
||
2023 |
$ |
|
$ |
|
||
2024 |
|
|
|
|
||
2025 |
|
|
|
|
||
2026 |
|
|
|
|
||
2027 |
|
|
|
— |
|
|
その後 |
|
|
|
— |
|
|
リース料総額 |
|
|
|
|
||
控える:帰属 |
|
( |
) |
|
( |
) |
リース負債総額 |
$ |
|
$ |
|
||
|
|
|
|
|||
|
|
|
|
|||
$ |
|
$ |
|
|||
注5—アステラス契約
2022年10月21日(「発効日」)に、当社はアステラス製薬とオプション契約を締結しました。これにより、当社はアステラス製薬に対し、研究、開発、製造、製造、製造、使用、販売、販売の申し出、販売、輸入、輸出、販売、輸入、輸出、またはこれらを総称して利用するための独占的権利とライセンス(A)を取得する独占的オプションを付与しました。それは、発効日時点でTSHA-120(「120 GAN製品」)として知られている製品、およびGANの治療に使用するためのそれに関するバックアップ製品、またはTayshaまたはその関連会社が管理する、または当社またはその関連会社が管理するGANの治療に使用されるその他の遺伝子治療製品、またはGAN製品、および(B)そのような搾取に関してTayshaまたはその関連会社が管理する知的財産権(「GANオプション」)に基づいています。一定の延長を条件として、GANオプションは、発効日から指定の期間まで行使できます。これは、アステラス製薬が、120 GAN製品について2022年9月19日にFDAに送付した会議出席依頼(「タイプB フェーズ2終了会議」)に対するアステラス製薬とFDA間のタイプBフェーズ2会議の正式な議事録、(ii)すべての書面によるフィードバックタイプBのフェーズ2終了会議に関するFDA、および(iii)タイプBの終了に関してTayshaがFDAに送付したすべての説明文書フェーズ2の会議。
オプション契約に基づき、当社はまた、アステラスに対し、(A)レット製品(以下に定義)を利用するための独占的かつ世界規模のロイヤリティおよびマイルストーンとなる権利とライセンス、および(B)そのような利用に関連してテイシャまたはその関連会社が管理する知的財産権(「レットオプション」)を取得する独占的オプション(「レットオプション」)を付与し、GANオプションとともにそれぞれ「オプション」を付与しました。」)。一定の延長を条件として、Rettオプションは、発効日からアステラスが (i) 女性小児科試験の特定の臨床データと (ii) TSHA-102に関する特定のデータ、その期間、(i) 発効日時点でTSHA-102として知られている製品に関連するRettオプション期間、およびそれに関連するバックアップ製品を受け取った後の指定期間まで行使できます。レット症候群の治療に使用するため、および(ii)レット症候群の治療に使用されるその他の遺伝子治療製品で、以下によって制御されます。Tayshaまたはその関連会社、または当社またはその関連会社がそれらの利用に関する知的財産権(「Rett製品」)を管理しているもの。
両当事者は、アステラスがオプションを行使する場合、両当事者がオプション契約に定められた条件に基づいてライセンス契約を誠実に交渉することに合意しています。これには、アステラスによる未定の前払金、未定のマイルストーン支払い、および該当する場合はGAN製品および/またはレット製品の純売上高に対する今後決定されるロイヤルティが含まれます。
12
レットオプション期間中、当社は、(A)支配権の変更(オプション契約で定義されているとおり)に関する問い合わせ、提案、提案または提案、または支配権の変更につながると合理的に予想されるもの、または(B)潜在的な支配権変更の手続きを開始することに同意しました。いずれの場合も、最初にアステラスに通知し、アステラスに提案または提案を提出する機会を提供する必要はありません。支配権の変更につながる取引の会社。アステラス製薬がそのような通知を受け取ってから指定された期間内にそのようなオファーを提出しなかったり拒否したりした場合、会社は支配権変更取引のために第三者に入札を求めることができます。アステラスが支配権の変更につながる取引の申し出を行った場合、当社とアステラス製薬は、特定の期間、支配権の変更につながる可能性のある取引条件について、誠意を持って交渉するよう努めます。その期間は、相互の合意により短縮または延長される場合があります。
オプション契約に基づいてアステラスに付与された権利の一部の対価として、アステラスは会社に前払いを支払いました。これは$です
証券購入契約
2022年10月21日、当社はアステラス製薬と有価証券購入契約を締結し、これに基づいてアステラス製薬に私募による株式の発行(以下「私募」)を行うことに合意しました。
会計処理
2022年10月、私募の終了と移転時に
同社は、オプション契約はASC 606の範囲内にあると判断しました。 顧客との契約による収入レット症候群の治療のためのTSHA-102とGANの治療のためのTSHA-120の開発は、会社では通常の活動と見なされています。ASC 606に従って、当社はオプション契約を評価し、(1)GANへのライセンス権を取得するオプション、(2)Rettへのライセンス権を取得するオプション、(3)Rett開発計画における研究開発活動の遂行という3つの異なる履行義務を特定しました。取引価格は $に決定されます
当社が重要な権利であると判断したRettオプションとGANオプションの独立販売価格(「SSP」)を決定するために、当社は確率加重期待収益(PWERM)法を利用しました。PWERM法は、オプション行使の確率とタイミングを考慮します。契約開始時に、同社は行使の確率は
13
次の表は、契約開始時の3つの履行義務に対する取引価格の配分(千単位)をまとめたものです。
|
|
取引価格の配分 |
|
|
Rettのライセンスを取得するオプション |
|
$ |
|
|
GANのライセンスを取得するオプション |
|
|
|
|
Rettの研究開発活動 |
|
|
|
|
合計 |
|
$ |
|
|
重要な権利に割り当てられた収益は、各オプション期間が終了した時点、またはアステラス製薬が各オプションを行使するか行使しないかを決定した時点で計上されます。Rettの研究開発活動による収益は、履行義務を履行するために発生すると予想される総費用に関連して発生した費用に応じて、インプットメソッドを使用して活動が行われたときに認識されます。支配権の移転はこの期間に行われ、履行義務の履行に向けた進捗状況を示す信頼できる尺度です。
2023年6月30日までの6か月間に、当社は、Rettの研究開発活動に関連する履行義務を履行するために発生する推定費用の合計が、2022年12月31日に終了した年度および2023年3月31日に終了した3か月間に使用された費用見積もりから増加したと判断しました。この変更の累積的な影響により、$が発生することになります
会社は$の収益を計上しました
注6 — シリコンバレー銀行への融資
2021年8月12日(「締切日」)に、当社は、随時その当事者である当社(以下「貸し手」)と、貸し手の管理代理人および担保代理人であるシリコンバレー銀行(「代理人」)との間で、貸付および担保契約(「タームローン契約」)を締結しました。タームローン契約では、(i) 締日に、$が規定されています
タームローンに適用される金利は、(a) WSJプライムレートにプラスした金額の大きい方です
タームローンは、2023年8月12日までに全額前払いできます。
タームローン契約に基づく義務は、タームローン契約の条件に従って、知的財産およびその他の慣習的に除外されている特定の財産を除く会社のすべての資産に対する完全な担保権によって担保されます。金銭的な契約はなく、
14
タームローン契約には、慣習的な表明と保証のほか、支払い不履行、契約違反、支配権の変更、重大な悪影響など、慣習的な債務不履行事由も含まれます。同社は、2023年6月30日現在、タームローン契約に基づくすべての規約を遵守していました。債務不履行事由が発生した場合、追加のデフォルト金利
2023年6月30日までの3か月と6か月の間に、当社は$のタームローンに関連する支払利息を認識しました
2023年6月30日時点で支払われるローンの将来の元本債務支払い額は次のとおりです(千単位)。
12月31日に終了する年度 |
|
|
|
|
2023 |
|
$ |
— |
|
2024 |
|
|
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
元本支払い総額 |
|
|
|
|
未償却債務割引 |
|
|
( |
) |
タームローン、純額 |
|
$ |
|
|
2023年3月10日、カリフォルニア州サンタクララに本拠を置くシリコンバレー銀行は、カリフォルニア州金融保護イノベーション局によって閉鎖され、連邦預金保険公社(「FDIC」)が受領者に任命されました。2023年3月27日、ファースト・シチズンズ・バンクはシリコンバレー銀行の残りの資産、預金、ローンを購入しました。その結果、シリコンバレー銀行が以前保有していた当社のタームローンの一部は、現在はシリコンバレー銀行がファーストシチズンズバンクアンドトラストカンパニーの一部門として保有しています。会社のタームローンの残りの部分は、現在破産手続き中のSVBキャピタルが保有しています。
注7—研究、コラボレーション、ライセンス契約
UTサウスウェスタン合意
2019年11月19日、当社はテキサス大学サウスウェスタンメディカルセンター(「UTサウスウェスタン」)に代わって、テキサス大学システムの理事会と研究、協力、ライセンス契約(「UTサウスウェスタン契約」)を締結しました。UTサウスウェスタン協定に基づき、UTサウスウェスタン大学は、特定の適応症(治験中の新薬申請を可能にする研究まで)で使用されるライセンス製品に関する前臨床開発活動を主に担当し、会社はライセンス製品に関するその後のすべての臨床開発および商品化活動に責任を負います。UTサウスウェスタン大学は、このような前臨床活動を行います
UTサウスウェスタン大学との契約に関連して、当社はUTサウスウェスタン大学の特定の特許権に基づく独占的、世界的、ロイヤリティフリーのライセンスと、UTサウスウェスタン大学の特定のノウハウに基づく非独占的、世界的、ロイヤリティフリーのライセンスを取得しました。いずれの場合も、特定の用途で使用するライセンス製品の製造、製造、使用、販売、販売、販売、提供、および輸入が可能です。さらに、当社は、UT Southwesternの特定の特許とノウハウに基づいて、すべての人間が使用するための非独占的で世界規模のロイヤリティフリーのライセンスを取得しました。また、特定の特許権に基づく独占ライセンスの取得を最初に拒否する権利と、そのような特許権以外の特許権に基づく独占的ライセンスを交渉するオプションも付いています。会社は、少なくとも開発、規制当局の承認の取得、および商品化のために、商業的に合理的な努力を払う必要があります
2020年4月2日、当社はUTサウスウェスタン協定を改正し、別のライセンス製品と特定の適応症の追加、および特定の患者投与特許に対する当社への先行拒否権を含めました。この修正に関連して、追加の対価はありませんでした。2022年3月、当社とUTサウスウェスタン大学は、UTサウスウェスタン協定に基づく現在のスポンサー研究契約の支払いスケジュールと現在の業績予想を修正し、支払いを15か月延期することに相互に合意しました。
UTサウスウェスタン協定は、国ごと、ライセンス製品ごとに、その国でライセンスされた特許の最後の有効な請求が満了した時点で失効します。最初の研究期間の終了後、当社は、適応症およびライセンス製品ごとに、いつでも契約を終了することができます
15
UTサウスウェスタン大学への特定の書面による通知が必要です。いずれの当事者も、未解決の重大な契約違反または相手方の破産があった場合、契約を終了することができます。
2019年11月、UTサウスウェスタン協定に基づいて付与されたライセンス権の一部として、当社は
アベオナ CLN1契約
2020年8月、当社はAbeona Therapeutics Inc.(「Abeona」)と、セロイドリポフスチョン1型疾患(小児性バッテン病としても知られる)に対するAAVベースの遺伝子治療薬であるABO-202の研究、開発、製造に関連する特定の知的財産権およびノウハウに対する世界的な独占権に関するライセンスおよび在庫購入契約(総称して「アベオナ契約」)を締結しました。Abeona契約の条件に基づき、当社はAbeonaに最初に$の現金支払いを行いました
2021年12月、この契約に関連して規制上のマイルストーンが開始されたため、会社は$を記録しました
アベオナ・レット契約
2020年10月29日、当社はAbeonaとライセンス契約(「Abeona Rett契約」)を締結しました。この契約に基づき、当社は、ノースカロライナ大学チャペルヒル校、エジンバラ大学、およびAbeonaが研究、開発、製造、製造するために最初に開発した特定の特許、ノウハウ、および材料に基づいてサブライセンスを付与する権利を含む、世界規模の独占的なロイヤリティを伴うライセンスを取得しました。レット症候群の遺伝子治療および関連する導入遺伝子の使用のためのライセンス製品を使用し、商品化します。
アベオナの特定の義務に従い、当社は商業的に合理的な努力を払って、少なくとも1つのライセンス製品を米国で開発し、少なくとも1つのライセンス製品を商品化する必要があります。
Abeona Rett契約に関連して、当社はAbeonaに1回限りの前払いライセンス料を支払いました
Abeona Rett契約は、ライセンス製品の最後のロイヤリティ期間の満了時に、国ごと、ライセンス製品ごとに失効します。いずれの当事者も、未解決の重大な契約違反または相手方の破産があった場合、契約を終了することができます。会社は、便宜上、事前にAbeonaに書面で通知した上で、契約を終了することができます。
2022年3月、当社のレット症候群治療のためのTSHA-102の臨床試験申請(「CTA」)申請がカナダ保健省によって承認され、アベオナ・レット契約に関連する規制上のマイルストーン支払いのきっかけとなりました。会社は$を記録しました
16
の 2022年6月30日までの6か月間の運用。$
GANの治療のためのTSHA-120の世界的権利の取得
2021年3月、当社は巨大軸索ニューロパチー(「GAN」)の治療を目的とした、臨床段階のAAV9遺伝子治療プログラム(現在はTSHA-120として知られています)の世界的な独占権を取得しました。TSHA-120は髄腔内投与のAAV9遺伝子療法で、現在GANの治療に関する臨床試験で評価されています。この試験は、国立衛生研究所(「NIH」)が、GANの治療法と治療法の発見に焦点を当てた主要な患者擁護団体と緊密に協力して実施しています。TSHA-120は、GANの治療薬として米国食品医薬品局から希少小児疾患と希少疾病用医薬品の指定を受けています。全世界の権利は、2021年3月29日に発効したハンナのホープ・ファンド・フォー・ジャイアント・アクソナル・ニューロパシー(「HHF」)と当社との間のライセンス契約(以下「GAN契約」)によって取得されました。
GAN契約の条件に基づき、TSHA-120の全世界での独占権を会社に付与することと引き換えに、HHFは$の前払い金を受け取りました
CLN7のライセンス契約
2022年3月、当社はUTサウスウェスタン大学とライセンス契約(「CLN7契約」)を締結しました。この契約に基づき、当社は、バッテン病の一種であるセロイドリポフスチノーシス7の遺伝子治療のライセンス製品を開発、製造、使用、および商品化するためのサブライセンスを付与する権利を含む、独占的でロイヤリティのあるライセンスを取得しました。CLN7契約に関連して、会社は$の1回限りの前払いライセンス料を支払いました
注記8—株式ベースの報酬
2020年7月1日、当社の取締役会は、従業員、取締役、役員、コンサルタントにインセンティブストックオプション、非法定ストックオプション、株式評価権、制限付株式報酬(「RSA」)、制限付株式ユニット(「RSU」)、およびその他の株式ベースの報奨を付与することを許可する2020年株式インセンティブプラン(「既存プラン」)を承認しました。新計画(以下に定義)の承認日である2020年9月16日以降、既存の計画に基づく追加の賞は付与されません。既存プランの条件は、新プランの承認前に付与された未払いの株式報奨の条件に引き続き適用されます。
2020年9月16日、当社の株主は2020年株式インセンティブプラン(「新プラン」)を承認しました。このプランは、IPOに関連する引受契約の締結時に発効しました。
さらに、2020年9月16日、当社の株主は従業員株式購入計画(「ESPP」)を承認しました。この計画は、IPOに関連する引受契約の締結時に発効しました。ESPPに基づいて発行できる普通株式の最大数は超えません
17
インクレESPPに基づいて発行のために留保されている普通株式の数を足しました
会社のインセンティブプランに基づいて付与できる株式数は次のとおりです。
|
|
既存 |
|
|
新規 |
|
|
|
|
|||
|
|
プラン |
|
|
プラン |
|
|
合計 |
|
|||
助成対象です-2022年12月31日 |
|
|
— |
|
|
|
|
|
|
|
||
計画の調整と修正 |
|
|
( |
) |
|
|
|
|
|
|
||
助成金 |
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
没収 |
|
|
|
|
|
|
|
|
|
|||
助成を受けることができます-2023年6月30日 |
|
|
— |
|
|
|
|
|
|
|
||
ストックオプション
2023年6月30日に終了した3か月間、
次の加重平均仮定を使用して、2023年6月30日と2022年6月30日に終了した3か月と6か月の間に付与された時間ベースの権利確定ストックオプションの公正価値を見積もりました。
|
|
6月30日に終了した3か月間 |
|
|
6月30日に終了した6か月間 |
|
||||||||||
|
|
2023 |
|
|
2022 |
|
|
2023 |
|
|
2022 |
|
||||
リスクフリー金利 |
|
|
% |
|
|
% |
|
|
% |
|
|
% |
||||
予想配当利回り |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
期待期間 (年単位) |
|
|
|
|
|
|
|
|
|
|
|
|
||||
予想されるボラティリティ |
|
|
% |
|
|
% |
|
|
% |
|
|
% |
||||
次の表は、2023年6月30日までの6か月間の期間ベースのストックオプション権利確定活動をまとめたものです。
|
|
|
|
|
|
|
|
加重 |
|
|
|
|
||||
|
|
|
|
|
加重 |
|
|
平均 |
|
|
集計 |
|
||||
|
|
|
|
|
平均 |
|
|
残り |
|
|
固有の |
|
||||
|
|
株式 |
|
|
エクササイズ |
|
|
契約上 |
|
|
価値 |
|
||||
|
|
[オプション] |
|
|
価格 |
|
|
寿命 (年単位) |
|
|
(千単位) |
|
||||
2022年12月31日時点で未処理です |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
オプションが付与されました |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
||
オプションの取り消しまたは没収 |
|
|
( |
) |
|
|
|
|
|
— |
|
|
|
— |
|
|
オプションは期限切れです |
|
|
( |
) |
|
|
|
|
|
— |
|
|
|
— |
|
|
2023年6月30日に素晴らしいです |
|
|
|
|
$ |
|
|
|
|
|
$ |
— |
|
|||
オプションは2023年6月30日に行使可能 |
|
|
|
|
$ |
|
|
|
|
|
$ |
— |
|
|||
上の表の総本質的価値は、それぞれの報告日における会社の普通株式の公正価値とストックオプションの行使価格の差として計算されます。2023年6月30日の時点で、付与された権利が確定していない時間ベースの権利確定ストックオプション報奨に関連する認識されない報酬の合計は$でした
パフォーマンス・ストック・オプション
2023年2月、同社は購入オプションを発行しました
18
2023年5月、同社は購入オプションを発行しました
|
|
終了した3か月間 |
|
|
終了した6か月間 |
|
||
|
|
2023年6月30日 |
|
|||||
リスクフリー金利 |
|
|
% |
|
|
% |
||
予想配当利回り |
|
|
— |
|
|
|
— |
|
期待期間 (年単位) |
|
|
|
|
|
|
||
予想されるボラティリティ |
|
|
% |
|
|
% |
||
市場ベースのストックオプション
2023年2月、同社は購入オプションを発行しました
制限付株式ユニット
2023年2月、同社は発行しました
2023年以前に付与されたRSUに対する当社のデフォルトの源泉徴収方法は、源泉徴収義務と同等の市場価値を持つ株式が、源泉徴収義務をカバーするための権利確定と決済の際にRSUの保有者に代わって売却され、そのような売却による現金収入は会社から税務当局に送金されます。2023年に付与されたRSUの場合、RSU保有者は、源泉徴収義務のために会社に現金を支払うか、源泉徴収義務と同等の市場を持つ株式を源泉徴収し、純株式がRSU保有者に発行される純源泉徴収方法を選択するかを選択できます。
2023年3月、同社は発行しました
2023年6月30日までの6か月間の同社のRSU活動は次のとおりです。
|
|
|
|
|
加重 |
|
||
|
|
|
|
|
平均 |
|
||
|
|
|
|
|
付与日 |
|
||
|
|
番号 |
|
|
公正価値 |
|
||
|
|
株式の |
|
|
一株当たり |
|
||
2022年12月31日時点では権利が確定していません |
|
|
|
|
$ |
|
||
制限付きユニットが付与されました |
|
|
|
|
|
|
||
既得 |
|
|
( |
) |
|
|
|
|
キャンセルまたは没収 |
|
|
( |
) |
|
|
|
|
2023年6月30日に権利が確定していません |
|
|
|
|
$ |
|
||
2023年6月30日の時点で、付与された未確定RSUに関連する認識されない報酬の合計は$でした
19
パフォーマンスと市場ベースの制限付株式ユニット
2023年2月、同社は発行しました
譲渡制限付株式報酬
同社の元社長兼最高経営責任者が受賞しました
2023年6月30日までの6か月間の同社のRSA活動は次のとおりです。
|
|
|
|
|
加重 |
|
||
|
|
|
|
|
平均 |
|
||
|
|
|
|
|
付与日 |
|
||
|
|
番号 |
|
|
公正価値 |
|
||
|
|
株式の |
|
|
一株当たり |
|
||
2022年12月31日時点では権利が確定していません |
|
|
|
|
$ |
|
||
譲渡制限付株式付与 |
|
|
— |
|
|
|
— |
|
既得 |
|
|
( |
) |
|
|
|
|
キャンセルまたは没収 |
|
|
( |
) |
|
|
|
|
2023年6月30日に権利が確定していません |
|
|
— |
|
|
$ |
— |
|
従業員株式購入制度
2022年2月、当社の取締役会はESPPに基づく初回募集を承認しました。ESPPでは、対象となる従業員は、給与控除によりTayshaの普通株式を以下の価格で購入できます。
株式報酬費用
次の表は、2023年6月30日および2022年6月30日までの3か月および6か月間の要約連結営業報告書に記録されたストックオプション、ESPP、RSA、およびRSUの株式ベースの報酬費用の総額(千単位)をまとめたものです。
|
|
6月30日に終了した3か月間、 |
|
|
6月30日までの6か月間、 |
|
||||||||||
|
|
2023 |
|
|
2022 |
|
|
2023 |
|
|
2022 |
|
||||
研究開発経費 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
一般管理費 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
合計 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
注9—保証
2023年4月、当社はSSI Strategy Holdings LLC(「SSI」)の2つの関連会社(「SSI投資家」)と証券購入契約(「SSI証券購入契約」)を締結しました。これに基づいて、当社はSSI投資家に私募で発行および売却(「SSI私募事業」)することに合意しました。
20
同社は、SSIワラントはASC 815のガイダンスに基づく株式分類の基準を満たしていないと結論付けました。これは、特定の基本取引が発生した場合に保有者が可変数の株式を受け取ることを許可する決済条項と、保有者が配当に参加することを許可する条項によるものです。SSIワラントは株式分類の基準を満たしていないため、会社はワラントを公正価値での負債として記録しました。この負債は、新株予約権の行使または期限が切れるまで、各貸借対照表日に再測定される可能性があります。公正価値の変動は、会社の要約連結営業報告書に反映されます。
同社は、発行時のSSIワラントの公正価値を$と判断しました
リスクフリー金利 |
|
|
% |
|
予想配当利回り |
|
— |
|
|
期待期間 (年単位) |
|
|
|
|
予想されるボラティリティ |
|
|
% |
|
2023年6月30日時点の公正価値調整は重要ではありませんでした。2023年6月30日の時点で、
注10—普通株式1株あたりの純損失
普通株式1株あたりの基本純損失は、普通株主に帰属する純損失を、その期間の発行済み普通株式の加重平均数で割ることによって計算されます。表示されているすべての期間で会社の純損失があったため、普通株式1株あたりの基本純損失と希薄化後の純損失は同じです。
次の表は、普通株式1株あたりの基本純損失と希薄化後の純損失(千単位、1株あたりのデータを除く)の計算を示しています。
|
|
6月30日に終了した3か月間、 |
|
|
6月30日までの6か月間、 |
|
||||||||||
|
|
2023 |
|
|
2022 |
|
|
2023 |
|
|
2022 |
|
||||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
発行済普通株式の加重平均株式数を使用して、基本株式および希薄化後の普通株式1株あたりの純損失を計算します |
|
|
|
|
|
|
|
|
|
|
|
|
||||
普通株式1株あたりの純損失(基本および希薄化後) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
2023年6月30日および2022年6月30日の時点で発行されている以下の普通株式同等物は、表示されている期間の普通株主に帰属する希薄化後の1株当たり純損失の計算から除外されました。なぜなら、それらを含めると希薄化防止効果があったからです。
|
|
6月30日 |
|
|
6月30日 |
|
||
権利が確定していない RSU |
|
|
|
|
|
|
||
権利が確定していないRSA |
|
|
— |
|
|
|
|
|
ストック・オプション |
|
|
|
|
|
|
||
ワラント |
|
|
|
|
|
— |
|
|
合計 |
|
|
|
|
|
|
||
注記11—所得税
繰延税金資産および負債は、資産および負債の財務諸表と課税基準との差異に基づいて、その差が逆転すると予想される年に制定された税率を使用して決定されます。入手可能な証拠の重みに基づいて、繰延税金資産の一部または全部が実現されない可能性が高い場合は、評価引当金が支給されます。同社は、繰延税金資産の実現可能性に関係するプラスとマイナスのエビデンスを評価しました。あります
2023年6月30日の時点で、2022年12月31日に終了した年度に以前に決定された不確実な課税ポジションの性質や金額に重大な変更はありませんでした。
21
注12—コミットメントと不測の事態
訴訟
当社はいかなる重要な法的手続きの当事者でもありません。また、係属中または差し迫った請求についても認識していません。時々、会社は通常の事業活動の過程で生じるさまざまな法的手続きや請求の対象となることがあります。
コミットメント
通常の業務では、会社は従業員、ライセンサー、サプライヤー、サービスプロバイダーとさまざまな補償を含む契約を締結します。これらの取り決めに基づく同社の最大被ばく量は、2023年6月30日時点では不明です。当社は、これらの取り決めに関連する重大な損失を認識するとは考えていません。
注13 — 戦略的優先順位付け
2022年3月、同社はGANとレット症候群の特定の臨床段階のプログラムに集中できるように、会社の組織構造を変更し、より広範な運用コスト削減計画を実施しました。業務効率を上げるため、実質的に他のすべての研究開発活動は一時停止されています。
プログラムの優先順位付けに関連して、同社は人員を約削減しました
同社は、これらの費用の支払いが2024年3月31日までに完了することを期待しています。2023年6月30日の時点で記録されている未払退職金の額は次のとおりです(千単位)。
|
|
2023年6月30日現在 |
|
|
2022年12月31日現在の未払退職金残高 |
|
$ |
|
|
退職金の記録 |
|
|
|
|
退職金支給 |
|
|
( |
) |
2023年6月30日現在の未払退職金残高 |
|
$ |
|
|
注14 — 退職金制度
2021年7月、当社は全正社員に退職給付を提供する401 (k) 退職貯蓄制度を採用しました。対象となる従業員は、内国歳入庁の制限に従い、年間報酬の一定割合を拠出することができます。会社は貢献しました $
注15— その後の出来事
私募制度
2023年8月14日、当社は特定の機関投資家およびその他の認定投資家(「購入者」)と証券購入契約(「購入契約」)を締結し、これに基づいて当社は私募取引(「私募取引」)で購入者に売却および発行することに合意しました(i)
事前資金付き新株予約権には、1株あたりの行使価格が$です
22
可能性があります 行使直後に保有者が受益所有する普通株式の総数が、特定の受益所有権制限を超える場合は行使しないでください。ただし、保有者は、以下の方法で受益所有権の制限を増減できます。
私募のクロージングは、慣習的なクロージング条件に従い、2023年8月16日またはそれ以前に行われる予定です(以下「クロージング」)。クロージング時の会社への総収入は、約$になると予想されます
購入契約の条件に基づき、当社は、購入契約に基づいて発行された普通株式と、購入契約に基づいて発行された事前資金調達型ワラント(以下「ワラント株式」)の転換時に発行される普通株式(以下「ワラント株式」)の転換時に発行される普通株式(以下「ワラント株式」)の転売のために登録するための1つ以上の登録届出書を締結後(「提出期限」)に証券取引委員会(「SEC」)に提出することに同意しました(まとめて、「登録可能な証券」)、および該当する登録届出書は提出期限後の指定期間内(「有効期限」)に有効になります。また、当社は、(i)購入契約に定義されている発効日の3周年、または(ii)保有者が保有または発行可能なすべての株式およびワラント株式(キャッシュレス行使を想定)が規則144に基づいて売却できる日のいずれか早い方まで、そのような登録届出書の有効性を維持するために最善の努力を払うことに同意しました。
また、当社は、とりわけ、購入契約で定義されているとおり、すべての登録費用を支払うことに同意しています。これには、購入契約で定義されている保有者の弁護士費用は含まれません。登録有価証券の売却に適用されるすべての販売手数料、および購入契約で定義されている保有者の弁護士費用はすべて、当該保有者が負担するものとします。
登録届出書が提出期限までに提出されなかった場合、または購入契約で定義されている有効期限までにSECによって有効と宣言されなかった場合、特定の限定的な例外を除いて、会社は各購入者に対し、清算損害として次の金額を比例配分して支払うことに同意しています。
購入契約には、購入契約の当事者の利益のみを目的としてなされた慣習的な表明、保証、および契約が含まれています。このような表明、保証、および契約は、(i)事実の記述ではなく、購入契約の当事者間でリスクを配分する方法を目的としています。(ii)会社の株主や他の投資家が重要と見なすものとは異なる方法で重要性基準を適用する場合があります。したがって、この申告書には、投資家に取引条件に関する情報を提供するためだけに購入契約が含まれており、会社に関するその他の事実情報を投資家に提供するものではありません。投資家は、会社またはその子会社や関連会社の実際の事実や状況を特徴づける表明、保証、契約、またはそれらの説明に頼るべきではありません。さらに、表明および保証の対象に関する情報は、購入契約の日付以降に変更される場合があり、その後の情報は公開情報に完全に反映される場合と反映されない場合があります。
当社は、登録届出書に関連して購入者に慣習的な補償権を付与しています。購入者はまた、登録届出書に関連して会社に慣習的な補償権を付与しています。
23
アイテム 2.経営陣の議論と分析 財政状態と経営成績。
当社の財政状態と経営成績に関する以下の議論と分析は、フォーム10-Qのこの四半期報告書に含まれる未監査の要約連結財務諸表および関連注記、および2022年12月31日に終了した年度の監査済み財務諸表とその注記、および12月31日に終了した年度のフォーム10-Kの年次報告書に含まれる関連する経営陣による財政状態と経営成績に関する議論と分析と併せて読む必要があります。、2022、または年次報告書(で提出)証券取引委員会、またはSEC、2023年3月28日。文脈上特に必要な場合を除き、フォーム10-Qのこの四半期報告書の「私たち」、「私たち」、および「私たち」とは、Taysha Gene Therapies, Inc. とその連結子会社を指します。
将来の見通しに関する記述
この議論の情報には、将来の見通しに関する記述や、改正された1933年の証券法のセクション27A、または改正された1934年の証券取引法のセクション21E、または証券取引法の意味における情報が含まれています。これらはこれらの条項によって作成された「セーフハーバー」の対象となります。これらの将来の見通しに関する記述には、当社の戦略、将来の事業、将来の財政状態、将来の収益、予測コスト、見通しと経営計画および目的に関する記述が含まれますが、これらに限定されません。「予測する」、「信じる」、「見積もり」、「期待する」、「意図」、「かもしれない」、「計画」、「プロジェクト」、「意志」、「だろう」などの表現は、将来の見通しに関する記述を識別するためのものですが、すべての将来の見通しに関する記述にこれらの識別語が含まれているわけではありません。将来の見通しに関する記述で開示されている計画、意図、または期待を実際に達成できない場合があります。また、当社の将来の見通しに関する記述に過度に依存しないでください。実際の結果や出来事は、当社が行う将来の見通しに関する記述で開示されている計画、意図、期待とは大きく異なる可能性があります。これらの将来の見通しに関する記述にはリスクと不確実性が伴い、当社の実際の結果が将来の見通しに関する記述と大きく異なる可能性があります。これには、以下に記載されているリスクが含まれますが、これらに限定されません。 フォーム10-Qのこの四半期報告書のパートII、項目1A、「リスク要因」と、年次報告書のパートII、項目1A、「リスク要因」。将来の見通しに関する記述は、作成された日付時点でのみ適用され、当社は将来の見通しに関する記述を更新する義務を一切負いません。
商標に関する注意事項
このレポートに記載されているすべてのブランド名または商標は、それぞれの所有者に帰属します。文脈上特に必要な場合を除き、このレポートでの「会社」、「私たち」、「私たち」、および「私たち」とは、Taysha Gene Therapies, Inc.を指します。
[概要]
私たちは、中枢神経系(CNS)の単一遺伝子疾患の治療のためのAAVベースの遺伝子治療法の開発と商品化に焦点を当てた、患者中心の遺伝子治療会社です。私たちは、トランスフォーマティブ遺伝子治療法の開発と商品化を目的として、テキサス大学サウスウェスタンメディカルセンター(UTサウスウェスタン大学)と提携して設立されました。UTサウスウェスタン大学と共同で、私たちは遺伝子治療薬候補のポートフォリオを保有しており、いくつかの追加開発プログラムを無料で取得するための独占的なオプションを備えています。遺伝子治療薬の開発と商品化における経営陣の確かな経験と、UT Southwesternの世界クラスの遺伝子治療研究能力を組み合わせることで、私たちは患者の生活を劇的に改善する革新的な治療法を開発するための強力なエンジンを作り上げたと信じています。2022年3月、私たちは巨大軸索ニューロパチー(GAN)とレット症候群に焦点を当てた戦略的パイプラインの優先順位付けイニシアチブを発表しました。その後、業務効率を高めるために、他のすべての研究開発活動をさらに大幅に一時停止しました。
2021年4月、私たちはGANの治療のための臨床段階の髄腔内投与によるAAV9遺伝子治療プログラムであるTSHA-120の世界的な独占権を取得しました。TSHA-120の第1/2相臨床試験は、承認された治験薬申請(IND)に基づいて、国立衛生研究所(NIH)によって実施されています。神経筋疾患向けに開発された運動機能測定の検証済み32項目尺度である臨床安全性と機能性MFM32を報告しました。この試験のデータは、3.5x10の最高用量コホートを対象としています14 ベクターゲノムの総数、またはvg、(ドットブロット)と1.0x1014 2022年1月の総vg(ddPCRによる)。低用量コホートで達成されたのと同様に、臨床的に有意義な疾患の進行の遅延が続いており、これは疾患の修正の裏付けと見なされました。私たちは最近、TSHA-120の商業的に代表的な適正製造基準(GMP)バッチを完成させました。これにより、商用グレードの材料の重要なロットは、一般的に元の臨床試験材料と分析的に同等であることが示されました。このバッチのリリーステストは、2022年の第4四半期に完了しました。2022年9月、私たちは米国食品医薬品局(FDA)に会議出席依頼を提出し、2022年12月13日の電話会議でタイプBのフェーズ2終了会議を許可されました。2023年1月、正式な議事録の受領後、タイプBのフェーズ2終了会議で得たフィードバックをFDAに報告しました。FDAはTSHA-120についてさらに明確にしました。MFM32は許容可能なエンドポイントとして認められ、生物製剤ライセンス申請(BLA)をサポートするために、二重盲検プラセボ対照設計で患者を追加投与することを推奨しました。FDAは、計画されている化学、製造、製造の見直しが行われるまで、商業用資材の製造に対する当社の全体的なアプローチが適切であることを認めました
24
TSHA-120のコントロール、またはCMCデータパッケージ。その後、正式な議事録への回答としてフォローアップの質問を提出しました。FDAは、無作為化二重盲検プラセボ対照試験の設定における関連する主要評価項目としてMFM32を明らかにし、GANは非常にまれな性質のため、そのような研究の設計におけるTayshaの課題を認めました。FDAは、十分な数の患者を登録することが難しく、対照試験の環境では規制が柔軟なため、不確実性が増しても受け入れていました。さらに、FDAは、自明で臨床的に意味のある比較的大きな治療効果を実証するために、客観的な測定値を利用した代替研究デザインを検討する用意があることを示しました。FDAは、安全性データベースの規模は審査上の問題であり、治療を受けた患者からの既存の安全性データが受け入れられるかどうかは、製品の比較可能性の証明にかかっていることを認めました。薬物比較可能性データを詳述したCMCモジュール3の修正案の提出を完了し、2023年7月にフィードバックを受け取りました。FDAは、比較可能性研究(初期の臨床ロットとピボットロットの比較)と計画された臨床試験で使用するための重要なロットリリースを裏付けるには、分析データで十分であると結論付けました。
私たちは、REVEAL第1/2相臨床試験でTSHA-102を評価しています。これは、レット症候群の成人女性患者におけるTSHA-102の安全性と有効性を調べる、オープンラベル、用量漸増、ランダム化、多施設共同研究です。2023年の前半に、最初の成人患者にレット症候群の投薬を行いました。最初の患者からの最初の安全性データを検討するための独立データ監視委員会(IDMC)は、2023年の第3四半期の初めに開催されました。その時点で、IDMCは次の患者に投与する許可を与えました。治療後6週間の評価時点では、治療に伴う重篤な有害事象は発生していません。成人試験で入手可能な臨床データについて、四半期ごとに最新情報を報告していきます。2023年の第3四半期の初めに、レット症候群の小児患者を対象とした臨床試験申請(CTA)を英国の医薬品・ヘルスケア製品規制庁(MHRA)に提出し、レット症候群の小児患者を対象としたIND申請書をTSHA-102のFDAに提出しました。2023年8月、私たちはレット症候群の小児患者を対象としたTSHA-102のINDに関する許可をFDAから受けました。
私たちの営業履歴は限られています。創業以来、私たちの事業は、製品候補の前臨床研究開発活動を実施するための会社の組織化と人員配置、事業計画、資金調達、および協力契約の締結に重点を置いてきました。私たちの主力製品候補は両方ともまだ臨床段階です。販売が承認された製品候補はなく、製品の販売による収益も得ていません。私たちは主に、(i)株式の売却、つまり当社の売買契約(以下に定義)および2022年10月の追加募集に基づく新規株式公開またはIPO、普通株式の売却による総収入4億3,900万ドルの調達、(ii)転換優先株式のIPO前の私募およびその他の私募による総収入4億3,900万ドルの調達、(iii)任期ローン契約(以下に定義)、および(iv)アステラス製薬の取引。
2021年8月12日、または締切日に、私たちは随時貸し手当事者と、または貸し手の管理代理人および担保代理人である貸し手とシリコンバレー銀行、またはその代理人と、貸付および担保契約、またはタームローン契約を締結しました。タームローン契約は、(i) 2021年12月31日までに利用可能なタームローンの元本総額4,000万ドル、(ii) 2022年1月1日から2022年9月30日まで、抽選時に代理人の裁量で決定された3つの明確でアクティブな臨床段階プログラムを実施した場合に、会社のオプションでさらに2,000万ドルのタームローンファシリティを利用できるようにすることを規定しています。(iii) 2022年10月1日から2022年10月1日まで 2023年3月31日、当社のオプションでさらに2,000万ドルのタームローンファシリティが利用可能になりました実施中の臨床段階のプログラムは、抽選時に代理人の裁量により決定され、(iv) 2023年4月1日から2023年12月31日までの間に、代理人と貸し手の承認を得て、さらに2,000万ドルのタームローンファシリティ、または総称してタームローンが利用可能になります。締切日に3,000万ドルのタームローンを引き出し、2021年12月29日にさらに1,000万ドルのタームローンを引き出しました。2022年9月30日と2023年3月31日の有効期限が切れる前に、追加の2,000万ドルのトランシェを引き出していませんでした。ローン返済スケジュールでは、2024年8月31日までの利息のみの支払いと、その後に元本と利息を毎月連続して支払うことが定められています。各タームローンに関する未払いの元本、未払利息、未払利息はすべて、2026年8月1日に全額支払う必要があります。
創業以来、私たちは大きな営業損失を被っています。当社の純損失は、2023年6月30日までの6か月間で4,220万ドル、2022年6月30日までの6か月間で8,440万ドルでした。2023年6月30日の時点で、私たちの累積赤字は4億4,370万ドルでした。当面の間、引き続き多額の費用と営業損失が発生すると予想されます。私たちの継続的な活動に関連して、次のような経費が大幅に増加すると予想しています。
25
私たちのパイプライン
私たちは、まれな患者集団と大規模な患者集団の両方を対象に、中枢神経系の単一遺伝子疾患の遺伝子治療薬候補のポートフォリオを保有しています。また、いくつかの追加開発プログラムを無料で取得できる独占的なオプションもあります。私たちの遺伝子治療候補のポートフォリオは、神経変性疾患、神経発達障害、遺伝性てんかんという3つの異なる治療カテゴリーにわたる幅広い神経学的適応を対象としています。当社の現在のパイプラインは、各製品候補の開発段階も含めて、以下の表に示されています。
レット症候群の場合はTSHA-102
TSHA-102は、神経発達障害であり、重度の知的障害の最も一般的な遺伝的原因の1つであるレット症候群の臨床評価において、自己補完的な髄腔内投与型AAV9遺伝子導入療法製品候補です。急速な発達退行を特徴とし、多くの場合、脳の神経機能およびシナプス機能に不可欠な遺伝子であるMECP2のヘテロ接合性機能喪失性変異によって引き起こされます。TSHA-102は、マイクロRNAまたはmiRNA標的結合部位をウイルスゲノムの3'未翻訳領域に挿入することにより、遺伝子の過剰発現に関連する毒性を防ぐように設計されています。このMECP2の過剰発現は、MECP2重複症候群と呼ばれる状態の患者に臨床的に見られます。MECP2のレベルが上昇すると、症状と重症度の両方の点でレット症候群に似た臨床表現型になります。TSHA-102は、神経細胞特異的プロモーターであるmep426に、MECP2の短縮版であるminiCP2導入遺伝子と、miRNA応答性自動調節エレメント(MiraRE)、自己相補的なAAV9にパッケージされた当社の新しいmiRNAターゲットパネルであるmiRAREと結合して構築されます。これにより、内因性と外因性の両方のMECP2発現を細胞で調節できます。レット症候群リサーチトラストによると、レット症候群は世界中で35万人以上の患者に影響を及ぼしています。病原性/病原性の可能性の高いMECP2の突然変異によって引き起こされる典型的なレット症候群の対処可能な患者数は、米国、欧州連合、英国で15,000人から20,000人の患者と推定されています。
2021年5月、TSHA-102の前臨床データがオンラインで公開されました 脳、高く評価されている神経科学の査読付きジャーナルです。この前臨床試験は、サラ・シネット博士のUTサウスウェスタン医療センター研究所によって実施され、生後4〜5週齢の思春期のマウスに、IT投与による調節されたミニCP2遺伝子導入、TSHA-102(AAV9/Mini-CP2-MIRARE)の安全性と有効性が評価されました。TSHA-102は、規制されていない全長MECP2(AAV9/MECP2)および規制されていないMiniMCP2(AAV9/MiniCP2)と比較されました。
26
TSHA-102は、ITデリバリーによってKO(MECP2-/y)マウスの生存率を 56% 延長しました。対照的に、規制されていないminiCP2遺伝子導入は、試験したすべての用量でKOの生存率を大幅に延長できませんでした。さらに、規制されていない全長MECP2コンストラクトは生存期間の有意な延長を示さず、野生型マウスでは許容できない毒性プロファイルを示しました。
生存率に加えて、行動上の副作用も調査されました。マウスに表現型スコアリングと、歩行、後肢の留め具合、振戦などを含む一連のテストを行って、行動スコアを集計しました。MIRAREは、野生型の集約表現型重症度スコアの最小CP2を介した悪化を軽減しました。マウスは、確立されたプロトコルを使用して総合的な重症度尺度で採点されました。AAV9/MECP2およびAAV9/MINIMECP2で治療された野生型マウスは、生理食塩水で治療されたマウスで観察されたものと比較して、平均行動重症度スコアが有意に高かった(p
ミラーレを介した遺伝子型依存性遺伝子調節は、髄腔内投与されたAAV9ベクターで治療した野生型およびKOマウスの組織切片を分析することによって実証されました。以下に示すように、miRare要素の有無にかかわらずMini-MeCP2導入遺伝子を発現するベクターをKOマウスに注射したところ、Mirareは野生型マウスの無制御のMini-CP2導入遺伝子の発現を全体的に低下させました。
左のグラフは、調節されたベクターと調節されていないベクターで治療されたKOマウスが同等のベクターDNA生体内分布を示し、力価と注射精度が一致していることを確認したことを示しています(n = 1バーあたり3〜8匹のマウス)。
右のグラフは、制御されていないベクターコンストラクトによってもたらされるものと比較して、Mirareが導入遺伝子の発現を大幅に減少させたことを示しています(n = 1バーあたり6〜8匹のマウス、P 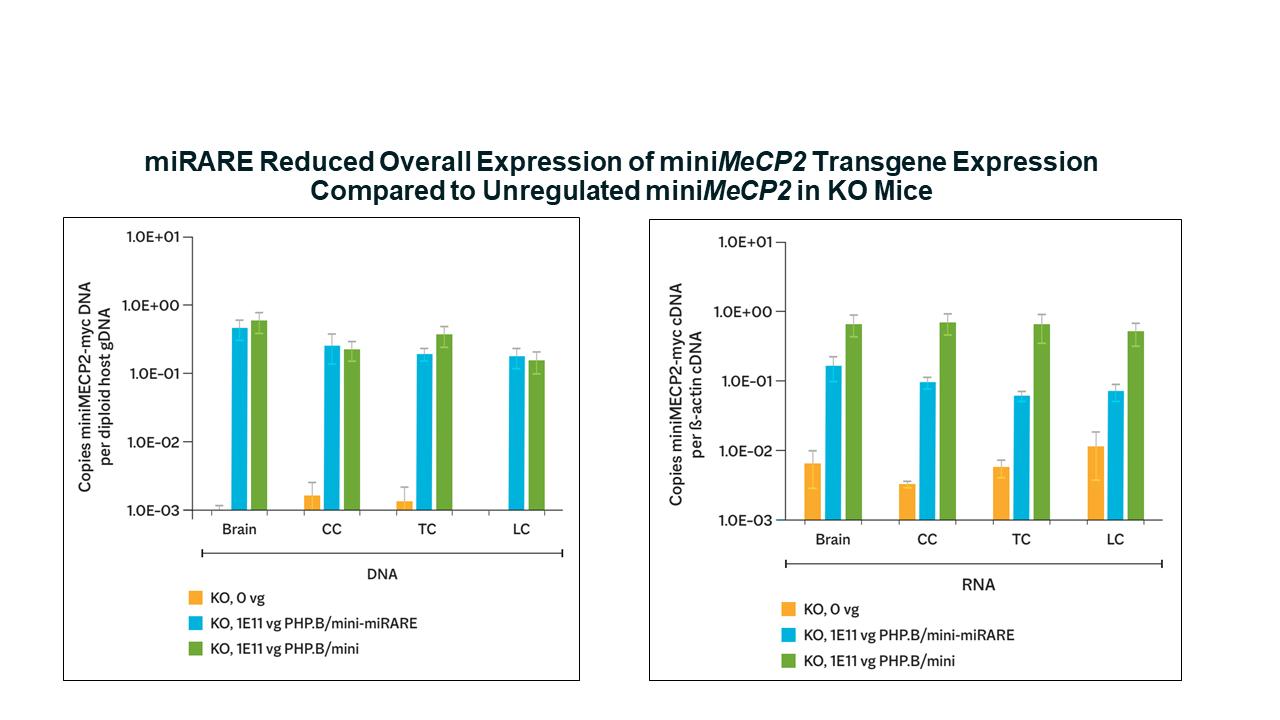
出典:シネットら2021年の補足図6BとCの変種
27
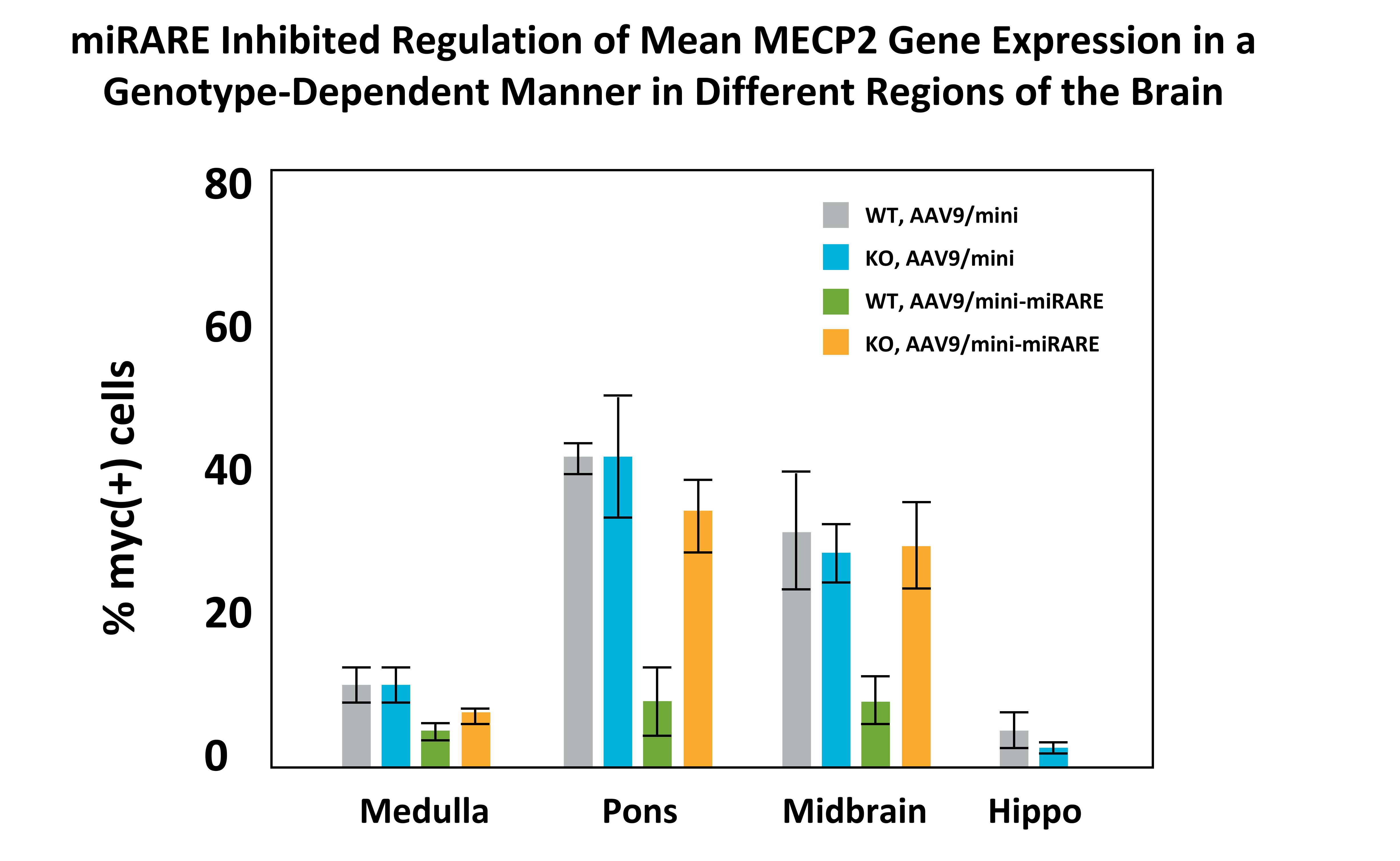
TSHA-102は、脳のさまざまな領域で調節された発現を示しました。下のグラフと写真に示すように、頭と中脳では、miRareは遺伝子型依存的に平均MECP2遺伝子発現を抑制しました。これは、KOマウスと比較して野生型マウスで観察されたmyc(+)細胞が有意に少ないことからもわかります(p
出典:シネットSE、ボイルE、ライオンズC、グレイSJの図6Bの変種。設計されたマイクロRNAベースの調節要素により、Rettマウスの安全な高用量Mini-CP2遺伝子治療が可能になります。脳。2021; 144 (10): 3005-3019
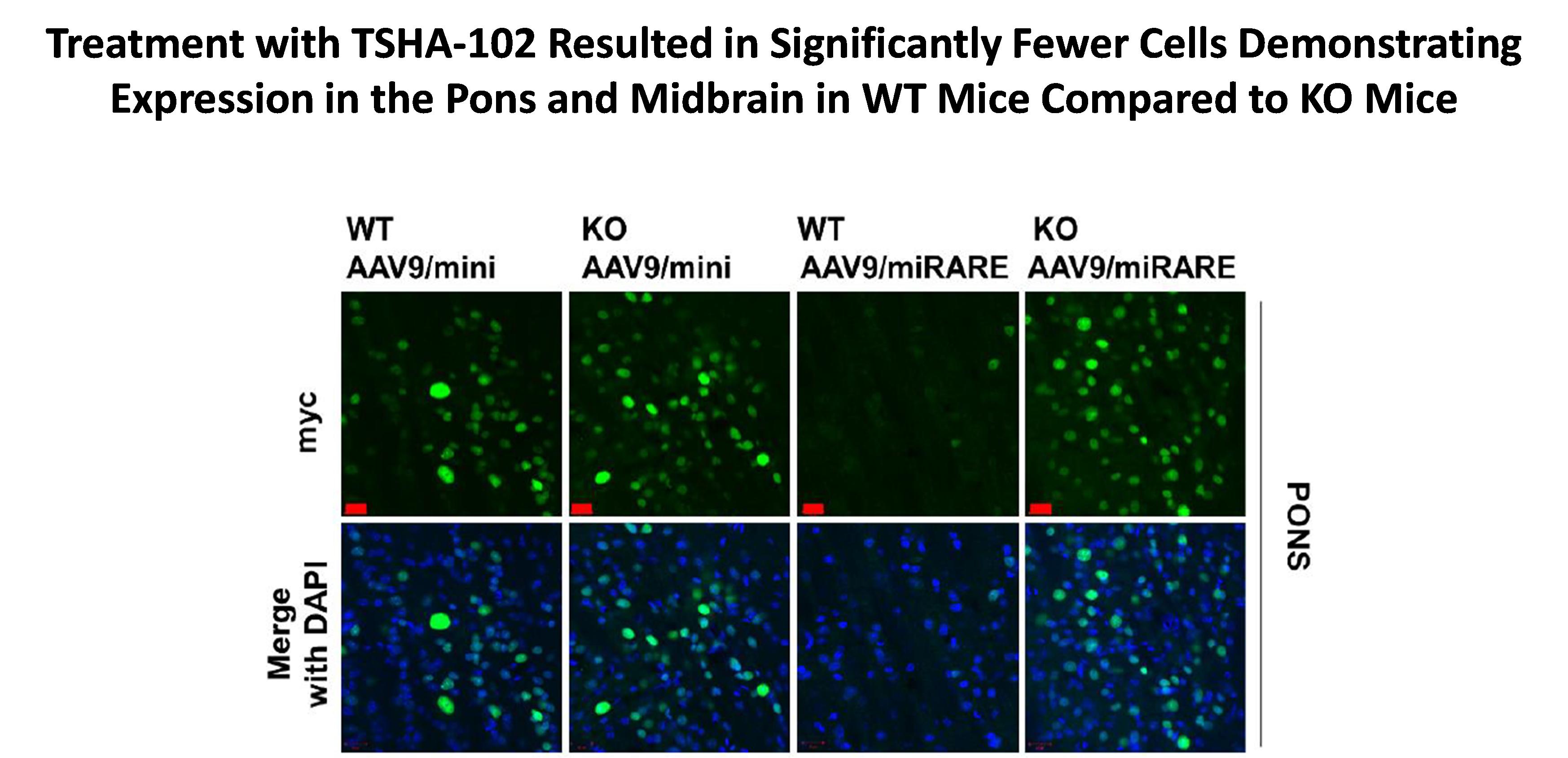
出典:シネットSE、ボイルE、ライオンズC、グレイSJの図6C。設計されたマイクロRNAベースの調節要素により、Rettマウスの安全な高用量Mini-CP2遺伝子治療が可能になります。 脳. 2021; 144(10): 3005-3019.
前臨床動物モデルでは、髄腔内mycタグ付きTSHA-102は早期死亡とは関係がなく、野生型マウスでは有害な行動副作用を引き起こさなかったため、無調節のMECP2遺伝子治療コンストラクトと比較して、最小CP2タンパク質の発現が適切にダウンレギュレーションされました。さらに、前臨床データは、Mirareが全体的に減少したことを示しました
28
miniCP2導入遺伝子の発現と、細胞ごとに異なる脳領域にわたる遺伝子型依存性のmycタグ付きminiCP2の発現が調節され、幼若マウスでの有効性を損なうことなくTSHA-102の安全性が向上しました。IT投与後のTSHA-102の薬理活性は、レット症候群のMECP2 KOマウスモデルで、3つの用量レベルと3つの年齢層(n=252)にわたって評価されました。TSHA-102を1回IT注射すると、すべての用量レベルで生存率が大幅に向上しました。中用量から高用量では、車両治療を受けた対照と比較して、すべての年齢層の生存率が向上しました。TSHA-102による治療は、MECP2 KOマウスの体重、運動機能、呼吸評価を大幅に改善しました。新生児マウス(n=45)を対象とした追加の研究が完了し、生存期間の延長がデータで示されました。最後に、IND/CTAを有効にする6か月のGLP毒性学研究では、最大2.0x10の3つの用量レベルでNHPとラットに髄腔内投与した場合のTSHA-102の生体内分布、毒物学的影響、および作用機序を調べました15vg/animal、どちらのWT種でも耐容性が良好でした。以下に示すように、DNAコピー数に反映される生体内分布は、脳の複数の領域、脊髄の一部、DRGで観察されました。
NHP毒性学研究-脳と脊髄への幅広い生体内分布
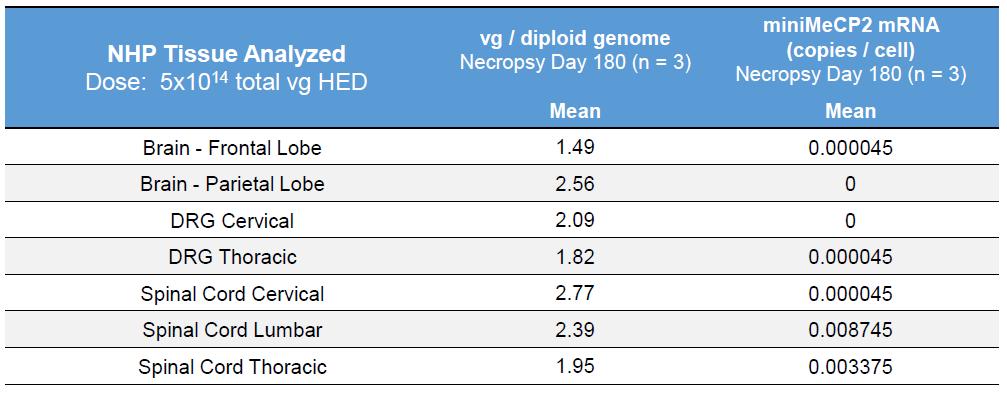
ソース:会社データ
重要なのは、複数の組織のmRNAレベルが低かったことです。これは、高レベルのDNAが送達されたにもかかわらず、miRAREの調節により、内因性MECP2の存在下でのコンストラクトからの導入遺伝子発現が予想どおり最小限に抑えられていることを示しています。導入遺伝子の過剰発現による毒性は観察されず、機能的および組織病理学的評価によって確認され、神経行動学的評価に有害な変化はなく、剖検でも有害な組織所見も見られませんでした。
新生児のKOレットマウスの有効性研究では、TSHA-102による治療により、以下に示すように生存率がほぼ正常化しました。8.8x10の用量でTSHA-102を服用してください10vg/mouse、これは2.86x10のヒト等価用量に相当します14 vg/Participentは、生後日マウスに脳室内(ICV)注射で投与した場合、生存率を大幅に改善しました。最も寿命の長い車両処理が施されていますが Mecp2‐/Y 男性は13.3週までしか生存しませんでした、治療を受けた人の47% Mecp2‐/Y 男性は36週齢まで生存しました。これが研究の結論であり、その後、すべての被験者が犠牲になりました。TSHA-102で治療したKOマウスでは、6つの異なる表現型を組み合わせた尺度であるバードスコアが大幅に改善されました。TSHA‐102は、重度のクラスピングの平均発症年齢を約7週から21週に、重度の歩行異常を約8週から20週間に大幅に遅らせました。8.8x10でのTSHA‐102のトリートメント10vg/mouseは新生児のMecp2の生存期間を大幅に延長しました‐/Y男性は、四肢の留め具と歩行に重度の異常があったため、体重が改善し、全体的な表現型スコアが改善し、発症年齢を遅らせました。
29
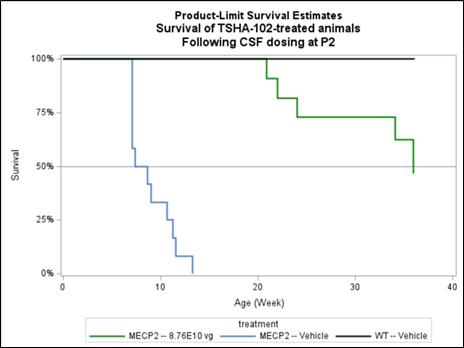
ソース:会社データ
さらに、新生児のKOレットマウスは、6つの異なる表現型能力を組み合わせた尺度であるBird Scoreで評価されたTSHA-102による治療後の行動の正常化を示しました。KO動物は当初、生後4週間で平均鳥スコアが4で評価されました。研究の過程で、TSHA-102はTSHA-102で治療したマウスの行動(鳥の総スコアで評価)を以下のように改善しました。
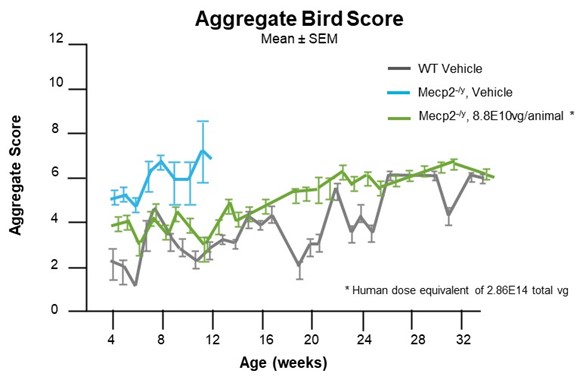
出典:LystMJ、Bird A. Rett症候群:単純な原因を持つ複雑な障害。Nat Rev Genet。2015 5月; 16 (5): 261-75. doi: 10.1038/nrg3897。エパブ 2015年3月3日。PMID: 25732612
下の表に示すように、TSHA-102は脳と脊髄への広範な生体内分布を示し、NHPの組織にはmRNAが少ないことが示されました。脳、脊髄、DRGで検出されたベクターゲノムのコピー数は、当然1.0vg/二倍体ゲノムを超えていました。脳、脊髄、DRGで測定されたminiCP2 mRNAはゼロで、miRNAコピーのダウンレギュレーションを示しています。
30
要約すると、最小有効用量を確認するためのマウス薬理学研究、2つの毒性学研究(野生型ラットと野生型NHP)、マウスの分布と遺伝子発現に関する研究、および最近の新生児マウスデータを含む、これまでに生成された前臨床データの合計が、以下に示すようにレット症候群におけるTSHA-102の臨床的進歩を裏付ける最も強力なパッケージであると考えています。
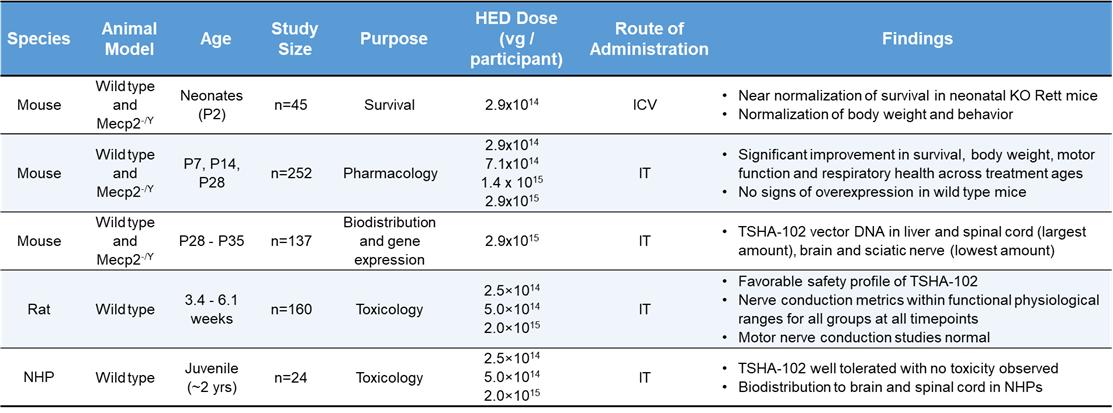
NHPの安全性と生体内分布の評価は、2022年5月に国際レット症候群財団(IRSF)の会議で、成人期のレット症候群に関する介護者の視点とともに発表されました。ASCEND全国サミットでは、「患者を中心に」という口頭発表がありました。最後に、マウスの薬理学、ラット、NHP毒性学のデータは、米国遺伝子細胞療法学会(ASGCT)の第25回年次総会と欧州遺伝子細胞療法学会(ESGCT)の会議で発表されました。
TSHA-102の治療の可能性を最大限に説明するために、新生児マウスにおける有効性を評価する追加データが生成されました。TSHA-102は新生児KOに関する薬理学研究で評価されました(Mecp2)-/Y) ネズミ。TSHA-102は、脳室内(ICV)経路で生後2日目(P2)の老齢マウスの子犬に投与されました。評価された有効性パラメータには、体重(BW)、生存率、表現型スコア(鳥スコア)が含まれていました。8.8x10の用量でP2マウスにTSHA-102をKOさせてください10vg/マウス [ヒト等価線量、(ヘッド)2.86x1014vg/参加者]生存率、体幅、行動が大幅に改善されました。車両治療を受けたKOマウスの生存期間の中央値は8.1週間でした。最も寿命の長い車両治療KOマウスは13.3週間しか生存しませんでしたが、TSHA-120で治療したKOマウスの半数(47%)は、モニタリングが終了した年齢である36週まで生存しました。TSHA‐102は、車両治療したKOマウスと比較して、KOマウスの体重を大幅に改善しました。TSHA-102で治療したKOマウスでは、6種類の表現型を組み合わせた指標である鳥のスコアが大幅に改善されました。TSHA‐102は、重度のクラスピングの平均発症年齢を約7週から21週に、重度の歩行異常を約8週から20週間に大幅に遅らせました。全体的に、8.8x102でのTSHA‐102のトリートメント10vg/mouseは新生児KOの生存期間を大幅に延長しました(Mecp2)‐/Y) 男性では、四肢の留め具と歩行に重度の異常があったため、体重が増え、全体的な表現型スコアが向上し、発症年齢が遅くなりました。
フェーズ 1/2 REVEAL臨床試験
2021年11月にTSHA-102のCTAを提出し、2022年3月にカナダ保健省によって承認されたCTAに基づく臨床開発の開始を発表しました。私たちは、REVEAL第1/2相臨床試験でTSHA-102を進めています。これは、レット症候群の成人女性患者18人を対象にTSHA-102の安全性と有効性を調べる、オープンラベルの用量漸増と用量拡張、ランダム化多施設共同研究です。参加者はTSHA-102の腰部髄腔内注射を1回受けます。用量漸増は、TSHA-102の2つの用量レベルを順番に評価します。コホート1では、最初の患者に5.7x10の用量が投与されました14合計vg、そしてコホート1の残りの患者は同じ用量を受け、2番目のコホートには1x10の用量が与えられます15合計vg。次に、設定された最大許容用量または最大投与量が、用量拡大中に投与されます。キー
31
評価には、以下に示すように、RETT固有の評価とグローバルな評価、生活の質、バイオマーカー、神経生理学および画像評価が含まれます。
カナダのケベック州モントリオールにあるサント・ジャスティン母子大学ホスピタルセンターは、神経科学および小児科の助教授で主任研究者であるエルザ・ロシニョール博士の指導の下、最初の臨床試験施設として選ばれました。2023年の前半に、最初の成人患者にレット症候群の投薬を行いました。最初の患者からの最初の臨床データを検討するためのIDMC会議は、2023年の第3四半期の初めに開催されました。その時点で、IDMCは次の患者に投与する許可を与えました。次の患者への投薬は、2023年の第3四半期に、成人患者への投与は2023年後半も継続する予定です。TSHA-102は、治療後6週間の評価時点で、治療に伴う重篤な有害事象がなく、耐容性が高い安全性プロファイルを示しました。2023年の第3四半期の初めに、レット症候群の小児患者を対象としたMHRAにCTAを提出し、レット症候群の小児患者を対象としたTSHA-102のIND申請書をFDAに提出しました。2023年8月、私たちはレット症候群の小児患者を対象としたTSHA-102のINDに関する許可をFDAから受けました。
レット症候群の治療を目的としたTSHA-102について、FDAから希少疾病用医薬品の指定と希少小児疾患の指定、欧州委員会から希少疾病用医薬品の指定を受けています。
TSHA-102は臨床試験の安全性と有効性のエンドポイントを明らかにします
主な有効性評価項目は、臨床グローバルインプレッション尺度(改善)(CGI-I)、レット症候群手機能尺度、および改訂運動行動評価(R-MBA)を使用した臨床医による患者評価です。副次評価項目には、臨床グローバルインプレッション尺度(重症度)(CGI-S)、レット症候群行動アンケート(RSBQ)、およびその他の臨床評価尺度を使用した臨床医や介護者による患者の評価が含まれます。
最初に投与された成人患者では、TSHA-102は、治療後6週間の評価の時点で、治療に伴う重篤な有害事象がなく、耐容性が高い安全性プロファイルを示しました。さらに、主任研究者(PI)は、自律神経機能(睡眠と呼吸)、発声、総運動能力(10年以上ぶりに補助なしで3分間座れるようになった)、細かい運動能力(物を保持する能力の習得)など、複数の領域で著しい臨床的改善を観察しました。これは、初期の臨床データとビデオによる証拠によって裏付けられています。
32
2023年8月、私たちはREVEAL試験で最初に投与された患者からの最初の臨床観察を報告しました。REVEAL試験で最初に投与された患者は、下のグラフに示すように、TSHA-102投与の4週間後に主要な有効性エンドポイントの改善を示しました。
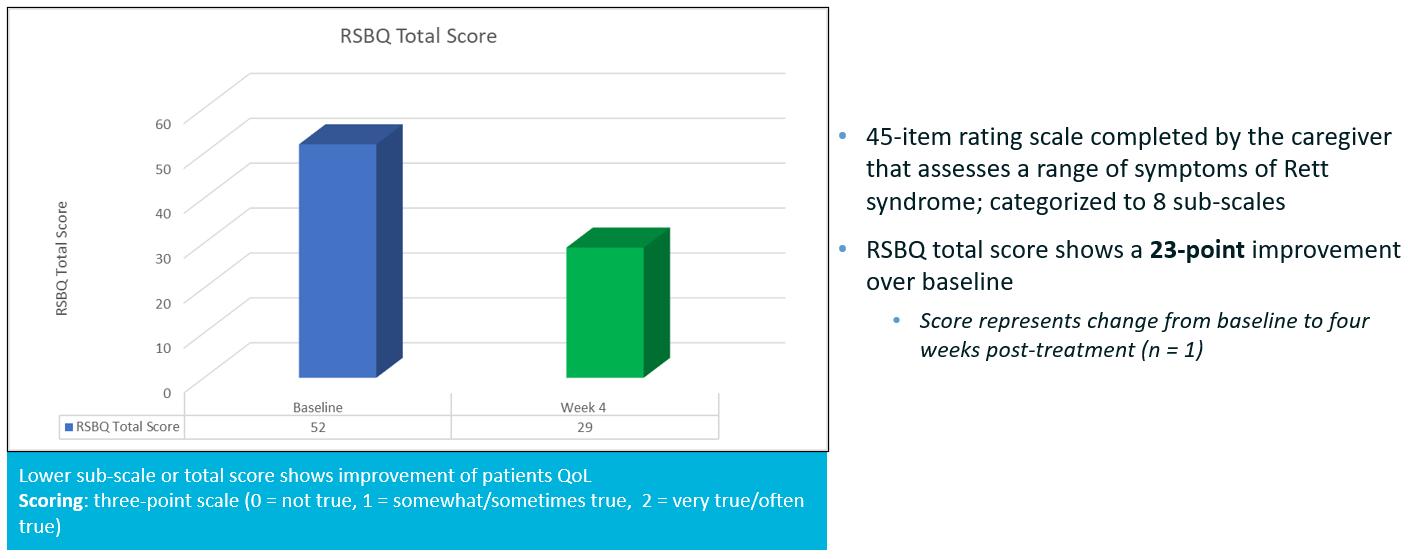
下のグラフに示すように、患者はTSHA-102投与の4週間後にRSBQトータルスコアが臨床的に有意に改善しました。
33
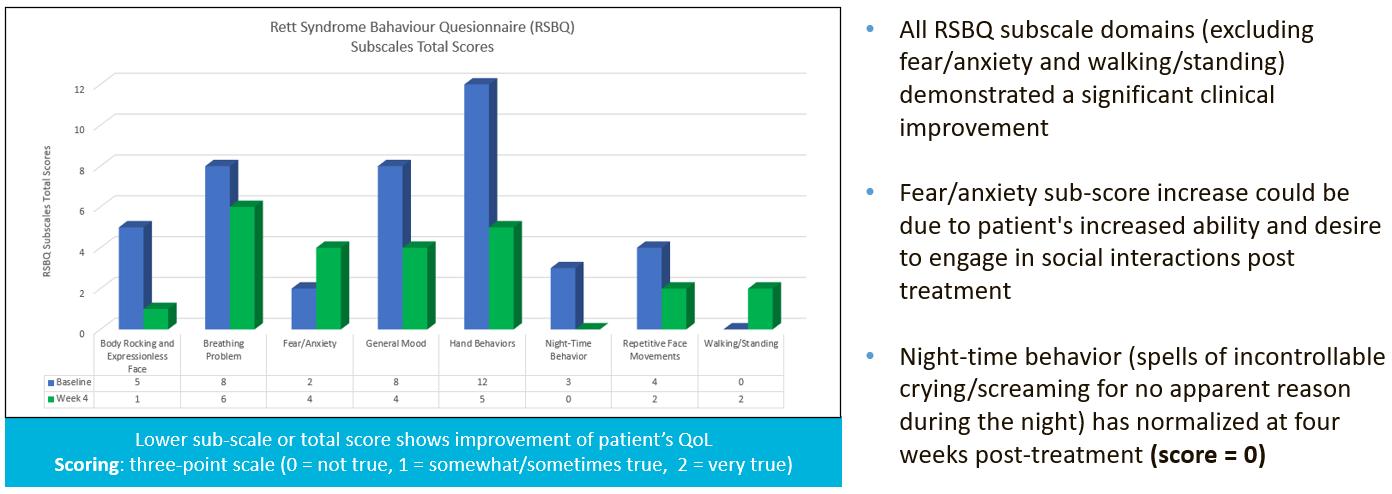
下のグラフに示すように、TSHA-102投与の4週間後に評価されたRSBQサブスケールドメインのほとんどで、患者は有意な臨床的改善を示しました。
下のグラフに示すように、TSHA-102投与後4週間のR-MBAデータに著しい変化は見られませんでした。
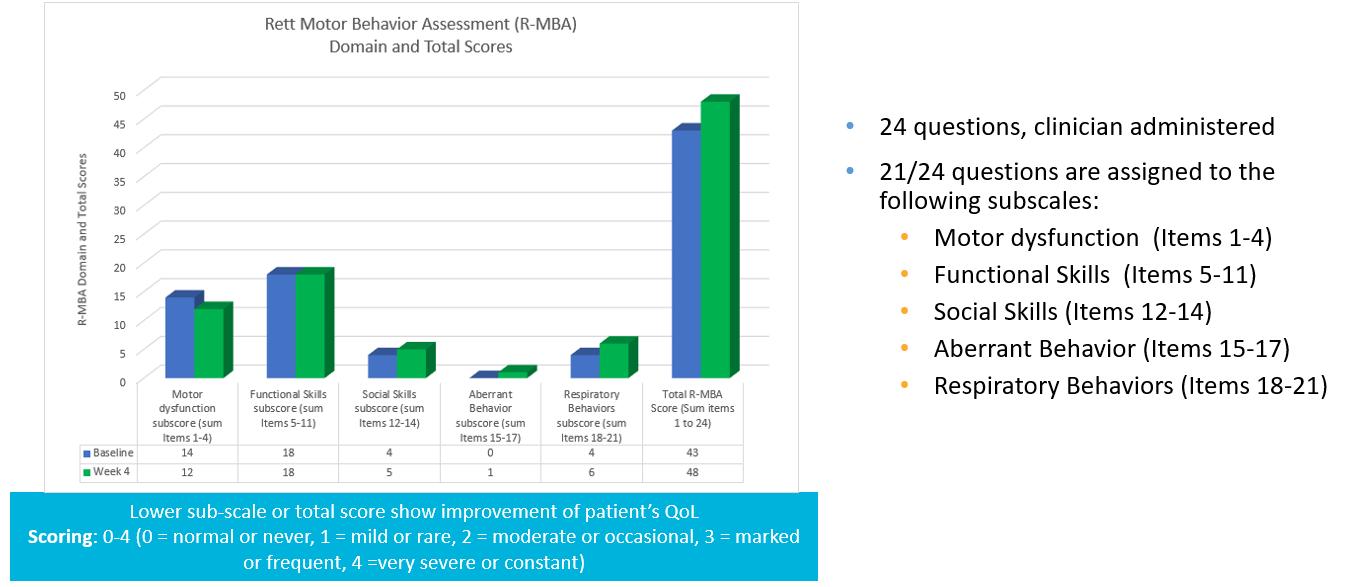
34
下のグラフに示すように、TSHA-102投与後の35日目まで、定量化できる発作イベントはありませんでした。
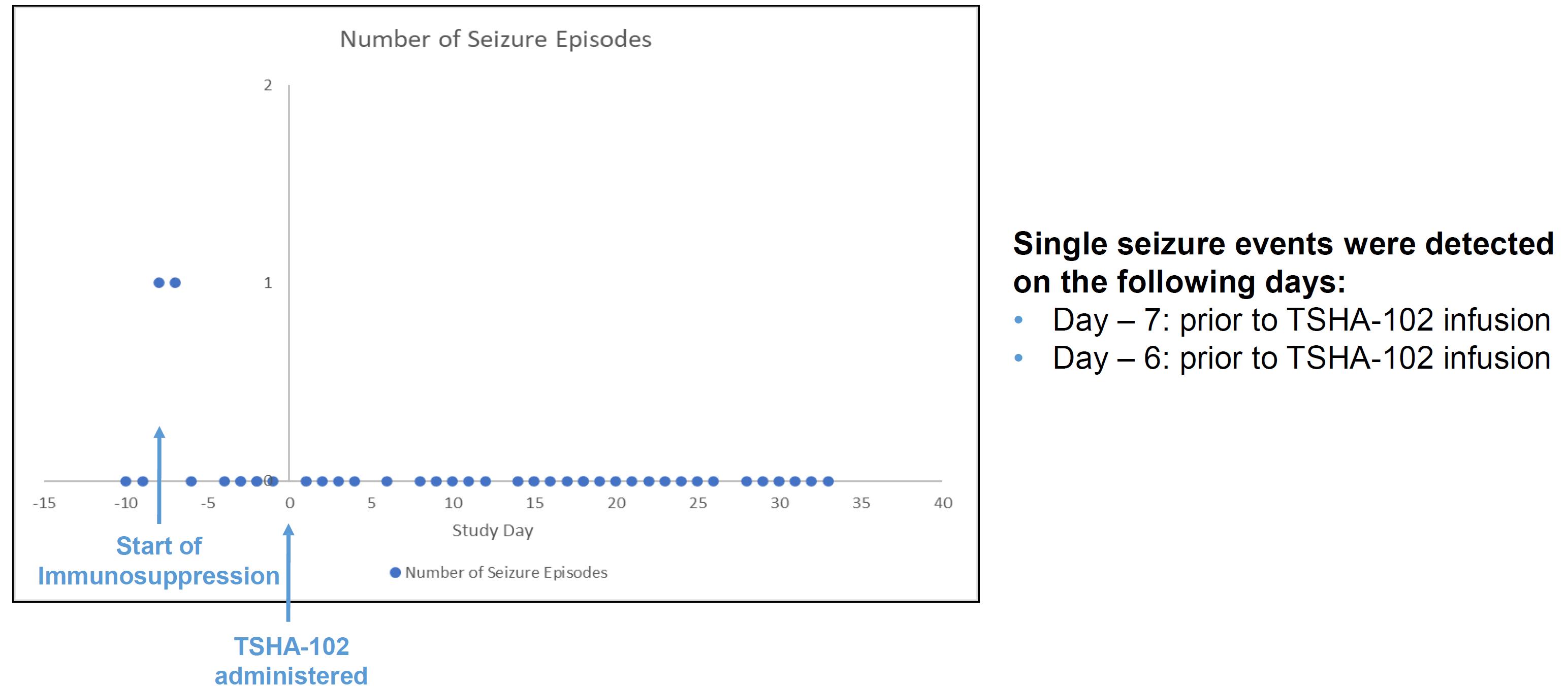
TSHA-102注入の6日前と7日目の発作は、免疫抑制の結果である可能性が高く、バックグラウンドの抗けいれん薬のレベルに影響を与えます。REVEAL試験では、第3四半期に次の患者に投与する予定です。Rett患者に関する最新の臨床情報を四半期ごとに提供します。
巨大軸索ニューロパチー(GAN)のTSHA-120
2021年3月、ハンナのホープ・ファンド・フォー・ジャイアント・アクソナル・ニューロパシー(HHF)とのライセンス契約に基づき、GANの治療を目的とした臨床段階の髄腔内投与によるAAV9遺伝子治療プログラム(現在はTSHA-120として知られています)の世界的な独占権を取得しました。契約条件に基づき、HHFは550万ドルの前払い金を受け取り、TSHA-120の商品化時に、臨床、規制、商業上のマイルストーンとして合計1,930万ドルのほか、純売上高に対して一桁の低額のロイヤリティを受け取る資格があります。
GANは、ギガキソニンの欠乏または完全な機能喪失、および中間フィラメントの蓄積によって引き起こされる、中枢神経系、末梢神経系、自律神経系の非常にまれな常染色体劣性進行性神経変性疾患です。疫学研究によると、米国、欧州連合、英国には治療可能なGAN患者が1,000人から1,500人います。
この疾患に関連する表現型には、早期(古典的)と遅発性(非古典的)の表現型があり、中間フィラメントの蓄積により病態生理学は共通しています。古典的GANの子供の症状と特徴は通常、軸索感覚運動ニューロパチーによる遠位筋力低下と感覚喪失を伴う5歳前に発症し、両側性の足下降と細かい運動協調障害として現れます。中枢神経系と小脳の病変による異常で広範囲にわたる不安定な歩行も、一般的な初期臨床症状です。古典的表現型の子供は通常、くすんだ、きつくカールした、粗い髪(「縮れた」髪)、神経生検では中間フィラメントの蓄積による「巨大な」軸索が病態性で、MRI画像では、最初は小脳歯状核周辺に進行性脊髄萎縮症と白質異常が見られます。症状が進行し、子供が大きくなるにつれて、進行性の近位筋が衰弱し、腕を上げたり、床や椅子から立ったりするのが困難になり、脊柱側弯症、遠位拘縮、進行性歩行と四肢運動失調が起こり、20年後には歩行ができなくなります。病気の初期に見られる進行性視神経萎縮症は、後の段階で視力の低下を増加させます。最近になって報告されています。確かに、自然史研究に登録された3〜21歳のGAN患者の約半数で、ベースライン時に視力低下が見られました [脳。2021年11月29日; 144(10):3239-3250]。重度の脊柱側弯症による呼吸筋力低下の増加と制限性呼吸不全のため、青少年には補助換気が必要です。GAN患者は、10代後半または20代前半に、通常は呼吸不全が原因で死亡することがよくあります。
35
遅発性または非古典的表現型は、シャルコー・マリー・トゥース2型、またはCMT2に分類されることがよくあります。これは、典型的な縮毛や古典的表現型のような中枢神経系への浸潤がなく、進行が比較的遅い典型的な早期発症の軸索感覚運動ニューロパチーであるためです。この表現型は、すべてのCMT2診断の最大6%を占める可能性があります。発症が遅い集団では、患者の生活の質が低く、日常生活活動が著しく損なわれています。この病気は生命を制限しますが、従来のGANほど深刻ではありません。従来のGANでは、対症療法は身体の発達を最大化し、悪化の速度を最小限に抑えようとします。現在、承認されている疾患修正療法はなく、緩和療法のみです。
TSHA-120は、全長のヒトギガキソニンタンパク質をコードする遺伝子組み換え遺伝子を持つAAV9自己相補性ウイルスベクターです。このコンストラクトはスティーブン・グレイ博士によって発明され、至る所での発現を促進するJetプロモーターの制御下で、最適な向性と迅速な発現を伴うGAN遺伝子のコドン最適化機能コピーを送達した最初のAAV9遺伝子治療候補です。
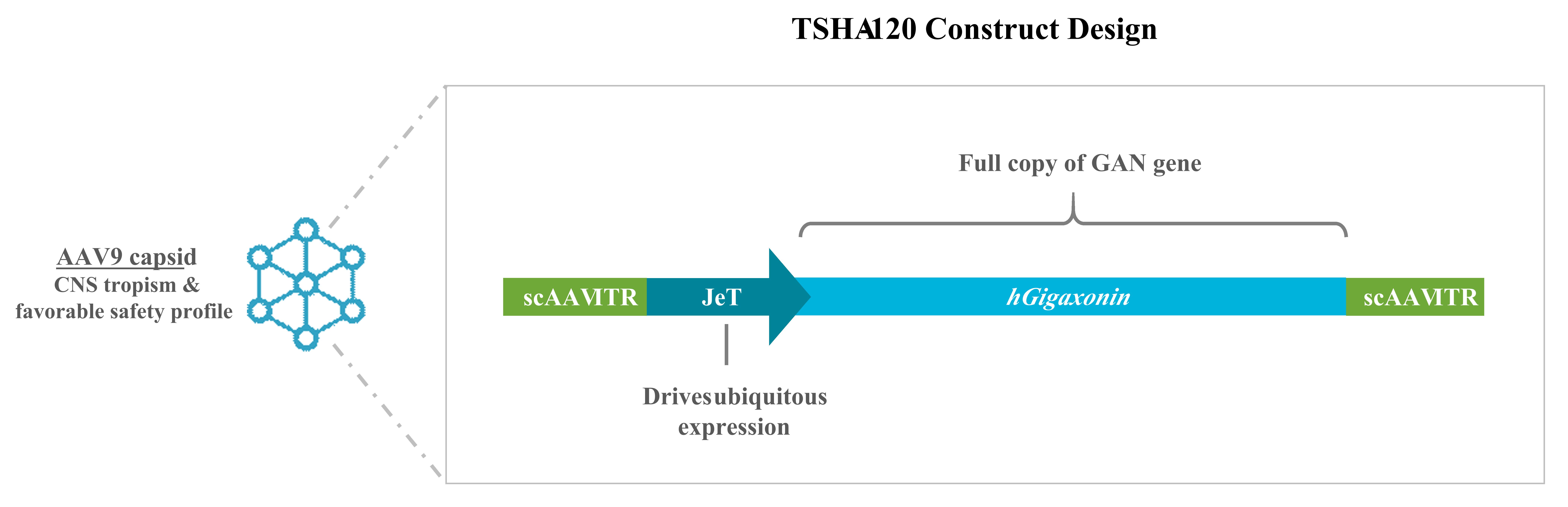
GANの治療のためのTSHA-120について、FDAから希少疾病用医薬品の指定と希少小児疾患の指定を受けています。2022年4月、私たちは欧州委員会からGANの治療薬としてTSHA-120の希少疾病用医薬品の指定を受けました。
36
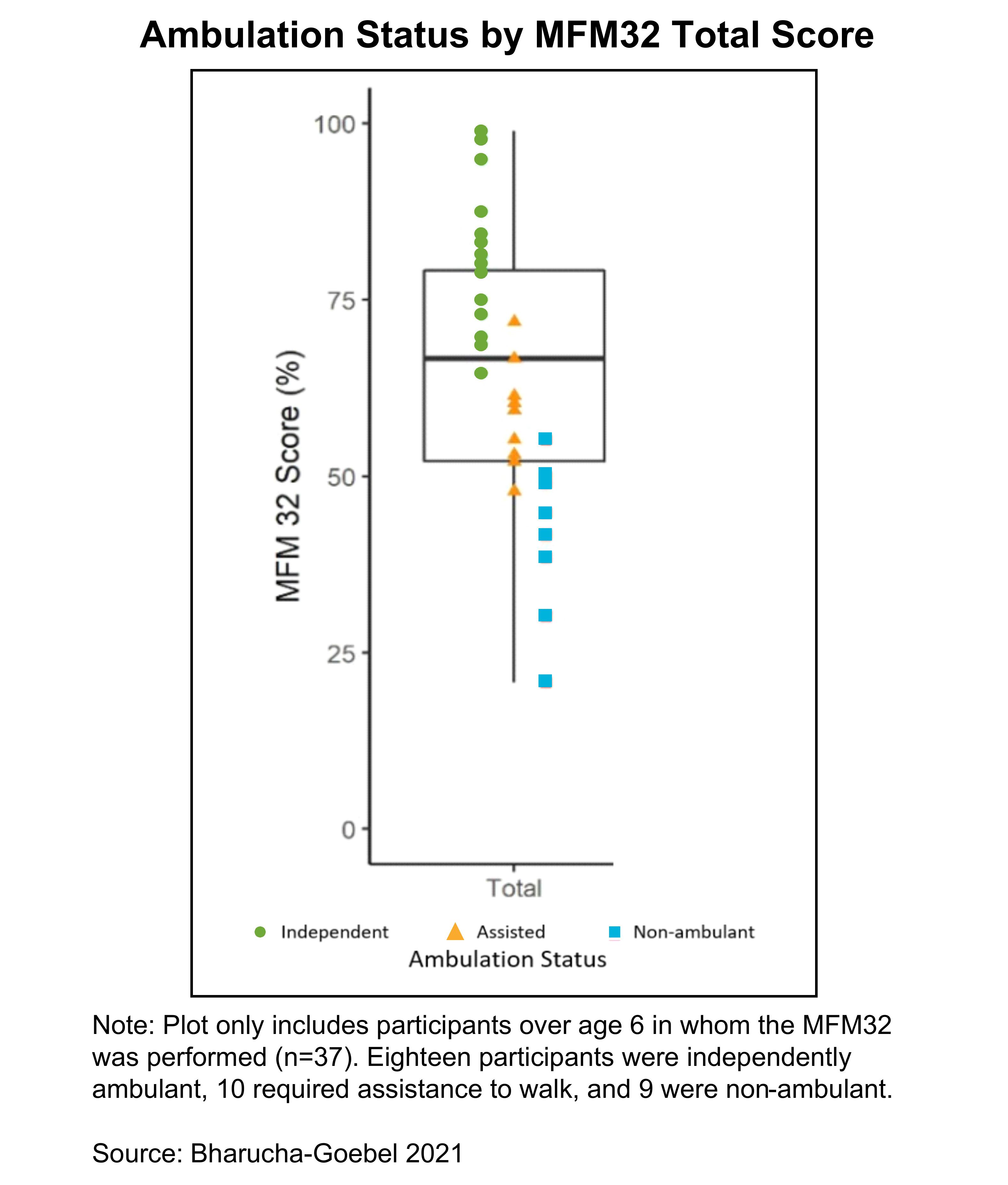
NIHが主導する縦断的前向き自然史研究が進行中です。この研究では、すでに多くの臨床評価によって特徴付けられる疾患の進行を伴う多くのGAN患者を特定し、追跡しています。これはバスケット型の自然史研究で、GANは希少疾患の1つでした。3歳から21歳までの最初の45人のGAN患者のベースライン特性が公開されています。この研究の画像データは、小脳の歯状核を取り巻く小脳白質内でT2信号異常が著しく増加していることを示しています。これは、GAN患者で最も初期の脳画像検査所見の1つです。これらの知見は、進行した疾患に伴う脳室周囲および深部白質の信号異常、より広範囲にわたる異常よりも先行しています。さらに、皮質と脊髄の萎縮は、疾患の重症度が高く年齢が高いことに対応するようです。GAN患者の肺機能障害も観察され、強制肺活量はMFM32などのいくつかの機能的転帰とよく相関していました。夜間の低換気と睡眠時無呼吸は時間とともに進行し、歩行機能が低下するにつれて睡眠時無呼吸が悪化しました。MFM32の合計スコアは、歩行状態とも相関していました。下のグラフに示すように、独立して外来した人の方が非外来群よりも成績が良く、MFM32スコアも高かったです。
37
患者はまた、COMPASS 31の自己評価アンケートに基づいて重大な自律神経機能障害を報告しました。さらに、神経伝導機能は年齢とともに進行性の感覚運動性ポリニウロパチーを示しました。神経変性疾患で予想されるように、若い患者の方がベースラインのMFM32スコアが高くなります。神経障害の重症度を評価する他の複合スコア、神経障害スコア(NIS)、運動失調、フリードライヒ運動失調評価尺度(FARS)は、MFM32と同様に、古典的なGAN患者の年齢と非常に有意な相関関係を示しました。3つの複合スコアはすべて外来状態とよく一致していました。したがって、これら3つの臨床的に関連する複合スコアは、古典的なGAN表現型の機能マーカーです。
縦断的フォローアップからの予備データおよび分析では、10人の患者が異なる時間間隔で少なくとも2回目のタイムポイントを経験しています。MFM32スコアの低下率は、すべての年齢の患者にある程度の一貫性を示しました。ほとんどの患者では、下の自然史プロットに示すように、年齢やベースラインのMFM32スコアに関係なく、計算された年平均8ポイントの低下が示されました。
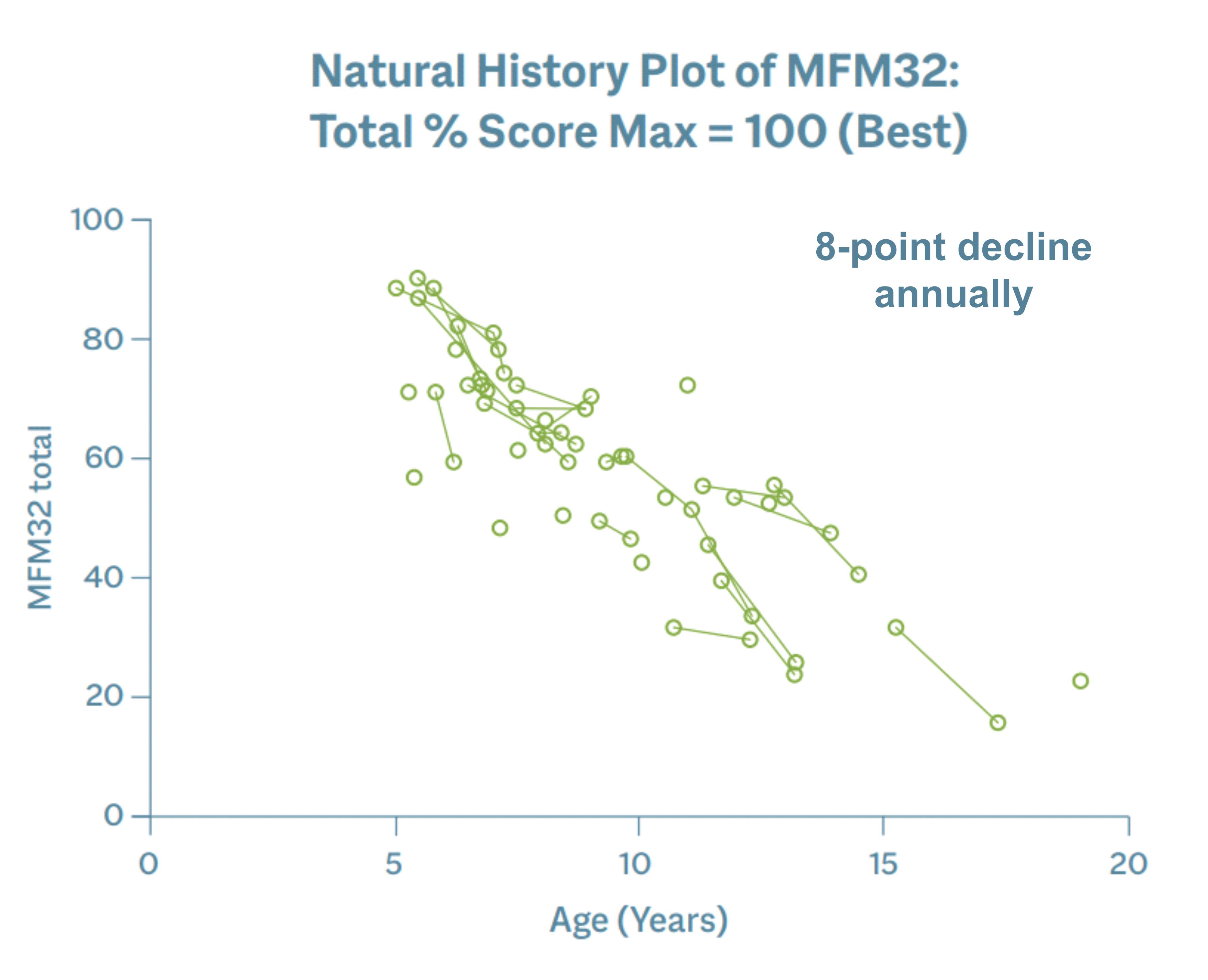
MFM32の4点スコアの変化は、脊髄性筋萎縮症などの他の小児神経筋障害において臨床的に意味があると考えられており、GANの患者は毎年重要な機能を失うことを示唆しています。現在までに、この調査から最長8年間分の確固たるデータがあります。
前臨床データ
TSHA-120は、in vitro研究とin vivo研究で良好に機能し、いくつかの動物モデルで運動機能と神経病変の改善、および長期的な安全性を示しました。注目すべきことに、TSHA-120で治療したGANノックアウト(KO)マウスでは、後根神経節(DRG)の病状の改善が実証されました。これらの前臨床結果は、多くの査読付きジャーナルに掲載されています。
38
AAV9を介したGAN遺伝子治療を受けたGAN KOげっ歯類モデルからの追加の前臨床データから、16か月で治療されたGANげっ歯類は、生後18か月の未治療のGANげっ歯類よりも有意に優れており、対照と同等であることが示されました。これらのげっ歯類は、げっ歯類の持久力、バランス、握力、運動協調性を評価するために設計されたロータロッド性能試験を使用して評価されました。潜伏時間と呼ばれるロータロッドから落ちるまでの時間も評価され、以下のデータは、治療を受けたGANげっ歯類と未処理のGANげっ歯類で潜伏時間に明らかな違いがあることを示しました。
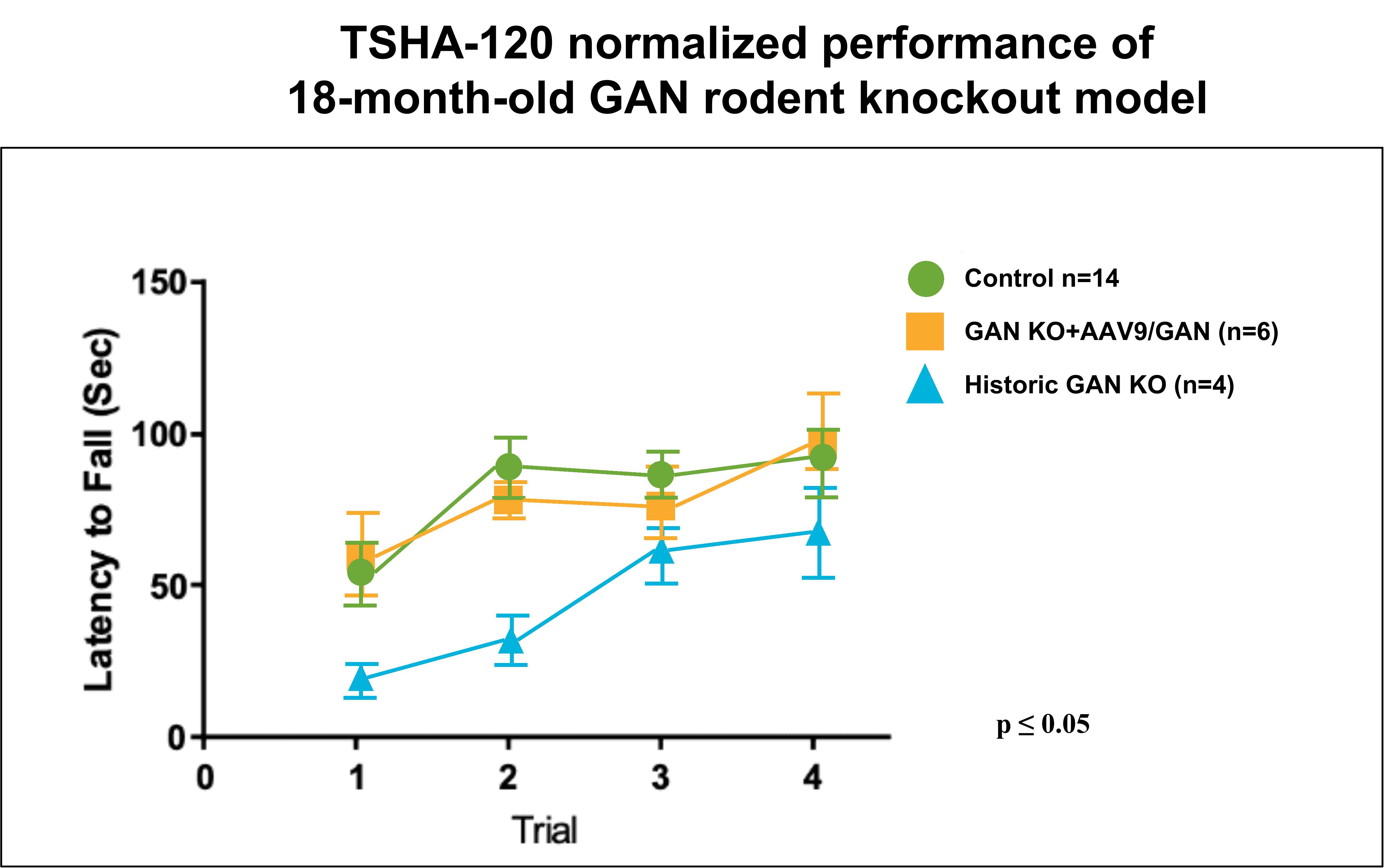
出典:スティーブン・グレイ博士によるGAN(?$#@$接合型A49E変異体)のラットモデルのロタロッド性能データ(未公開)
治療の有効性ではなく、偶然に結果が出る確率が十分に低い場合、結果は統計的に有意であると見なされます。結果の統計的有意性を判断する従来の方法は「p値」と呼ばれ、ランダムな偶然が結果をもたらした確率を表します(たとえば、p値= 0.01は、対照群と治療群の差が純粋に偶然によるものであるという確率が 1% あることを意味します)。一般的に、0.05未満のp値は統計的に有意であると見なされます。
遺伝子治療の分野で非常に興味深いトピックであるDRGの炎症に関して、DRGの組織学的外観と機能は基礎疾患の病態生理学の結果として著しく異常です。TSHA-120による治療により、GAN KOマウスのDRGの病理学的外観が大幅に改善されました。以下に示すのは、黄色の矢印で示されているように、DRGに損傷したニューロフィラメントの凝集体を含む異常なニューロン介在物が多数存在するGAN KOマウスモデルの組織です。画像Cでは、TSHA-120の髄腔内(IT)注射で治療したGAN KOマウスの組織で、DRG中のこれらの神経包有物の減少に顕著な改善が見られました。
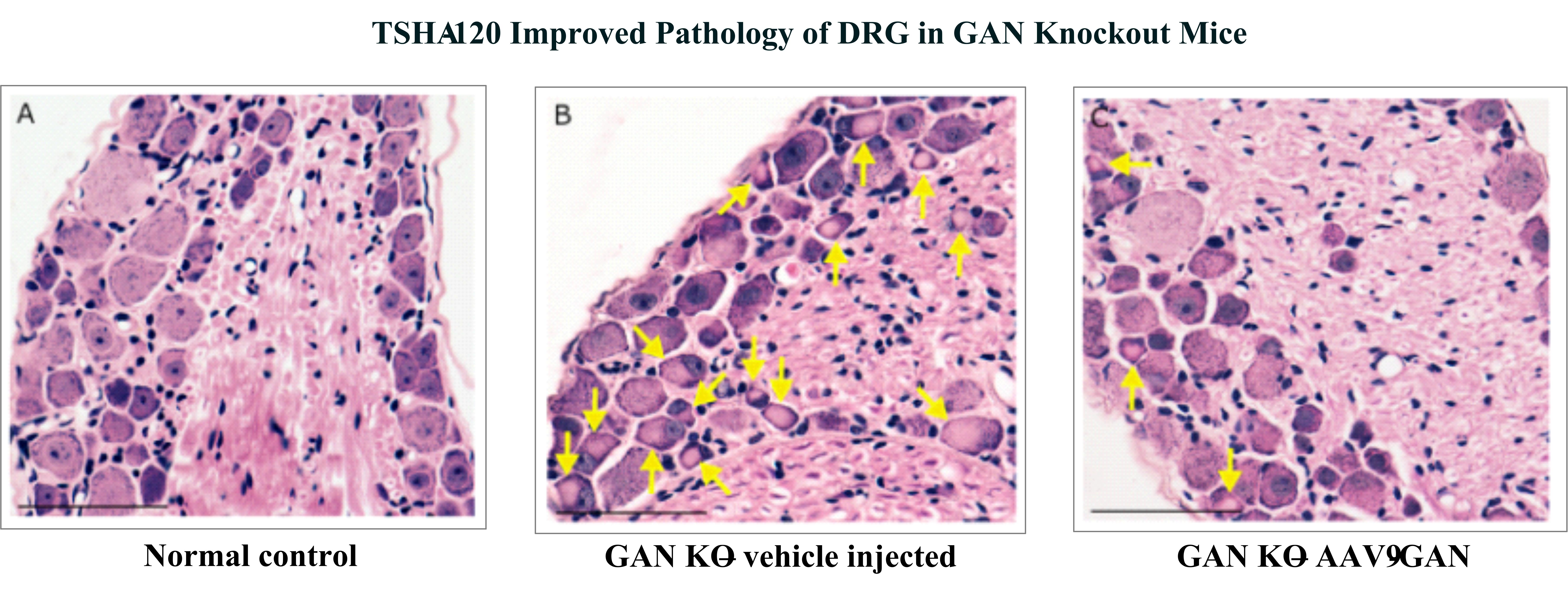
出典:ベイリーRM、アルマオD、カルブルギSN、グレイSJ(2018)の図6A〜Cの変種 巨大軸索ニューロパチーに対する髄腔内Aav9遺伝子療法の開発。 分子療法-方法および臨床開発 9160-171。
39
DRGへの介在物を減らすための定量的アプローチを適用したところ、TSHA-120で治療したマウスは、下図のようにビークルを投与されたGAN KOマウスと比較して、ニューロンの平均介在物数が統計的に有意に減少したことが観察されました。
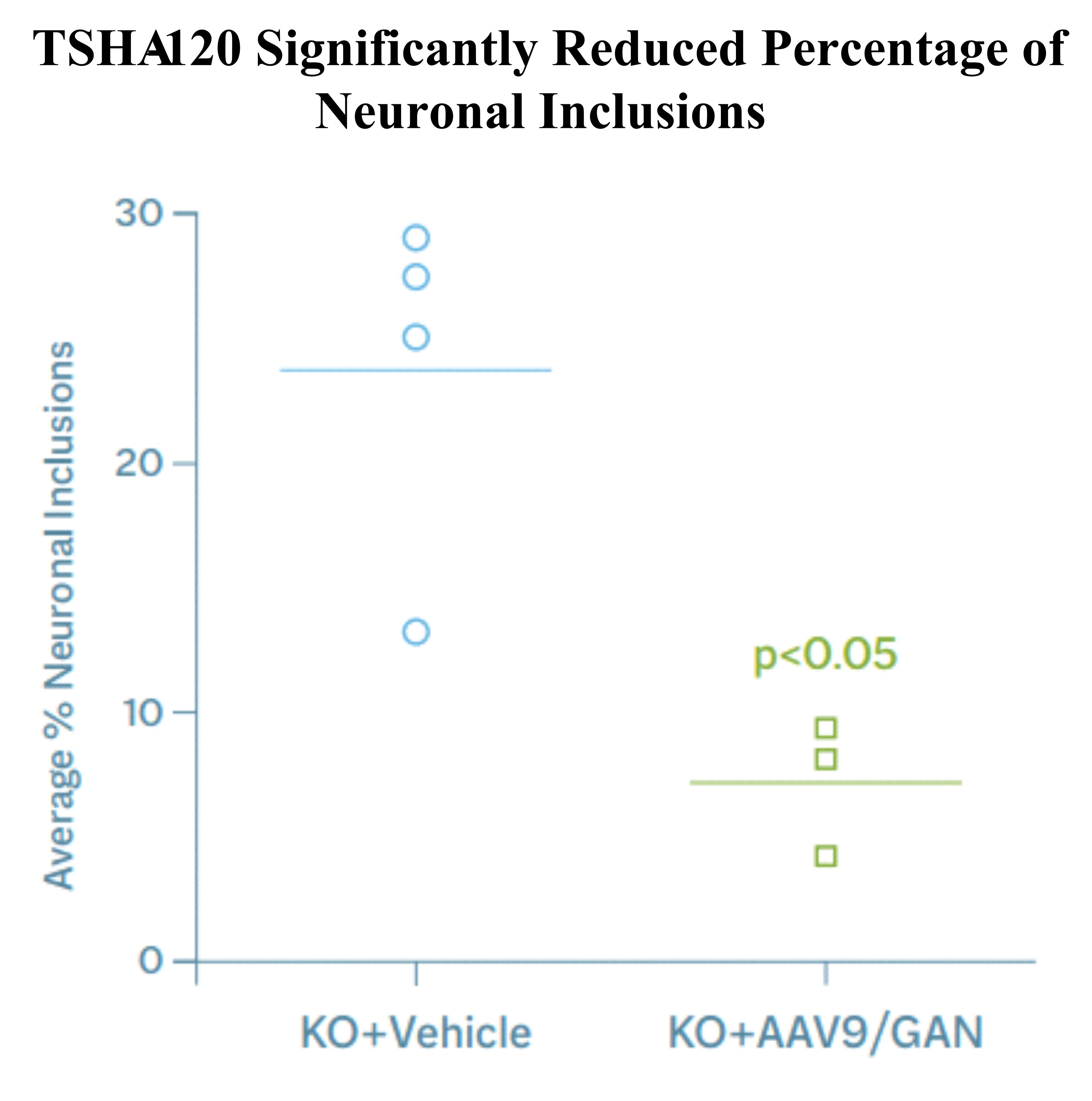
出典:ベイリーRM、アルマオD、カルブルギSN、グレイSJ(2018)の図6 Dの変異体。巨人軸索ニューロパチーに対する髄腔内Aav9遺伝子療法の開発。分子療法-方法および臨床開発 9160-171。
40
さらに、TSHA-120は、以下に示すように、GAN KOマウスの坐骨神経の病状が改善されたことを示しました。
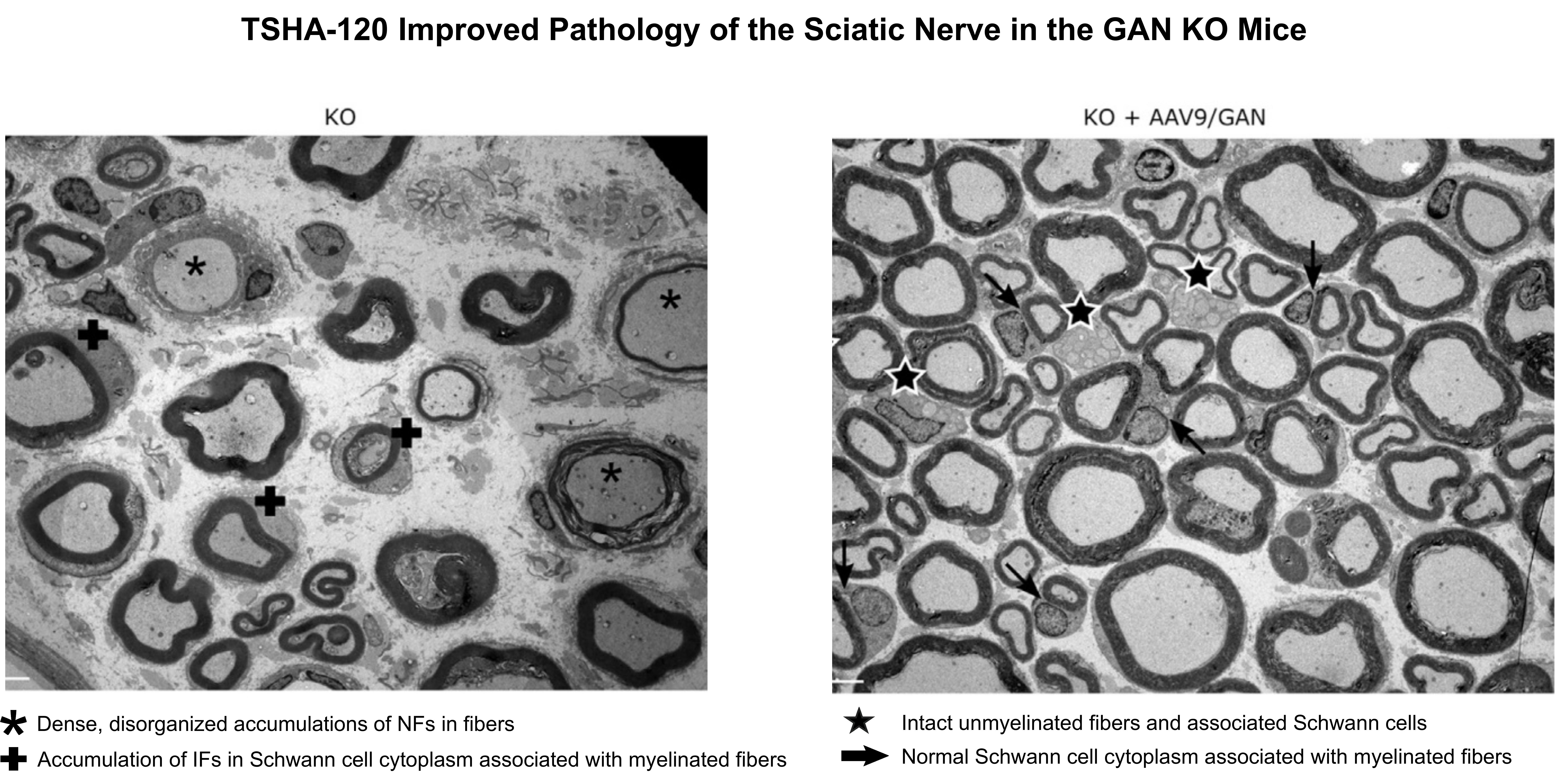
出典:ベイリーRM、アルマオD、カルブルギSN、グレイSJ(2018)の図7AとBの変種 巨大軸索ニューロパチーに対する髄腔内Aav9遺伝子療法の開発。分子療法-方法および臨床開発 9160-171。
進行中の第1/2相臨床試験の結果
TSHA-120の第1/2相臨床試験は、NIHによって承認されたINDの下で実施されています。進行中の試験は、単一施設のオープンラベル非ランダム化用量漸増試験で、患者にTSHA-120 — 3.5x10の4つの用量レベルのいずれかを髄腔内投与します。13合計サイズ、1.2x1014 合計サイズ、1.8x1014合計総重量または3.5x1014合計g(ドットブロット)。主要評価項目は安全性の評価であり、副次評価項目は病理学的、生理学的、機能的、および臨床的マーカーを使用して有効性を測定することです。現在までに、14人の患者が髄腔内投与され、12人の患者が最長3年分の長期フォローアップデータを入手しています。これらの患者の治療前の訪問間隔は3.7か月から31.5か月の範囲で、患者は治療を受ける前に少なくとも2回の訪問がありました。このデータに基づいて計算された年間の自然史減少は、すべての治療後の分析の比較として役立ちました。
遺伝子導入後1年で、最高用量の3.5x10でTSHA-120を使用すると、臨床的に有意で統計的に有意な疾患の進行の遅延または停止が見られました14合計g(n=3)。MFM32スコアの低下率の変化は、3.5x10で5ポイント改善しました14自然史における8ポイントの減少と比較したVGコホートの総数。
MFM32の一次ベイズ有効性分析は、投与後1年目の間隔で、プロトコルごとの集団、用量グループごとに実施されました。反復測定の階層モデルを使用して、試験参加者の治療前の低下と比較したMFM32パーセントスコアの勾配の変化の事後分布を推定しました。MFM32パーセントスコアの合計におけるベースラインからの変化の頻度主義的分析も、線形混合モデルを用いて用量群ごとに実施されました。
41
すべての治療用量を合わせたMFM32スコアの低下率の変化(n = 12)は、MFM32スコアの低下率の変化が5.20%ポイント(p = 0.0022)低下したことを示しており、遺伝子導入前の年間減少率は7.73%ポイントでした。
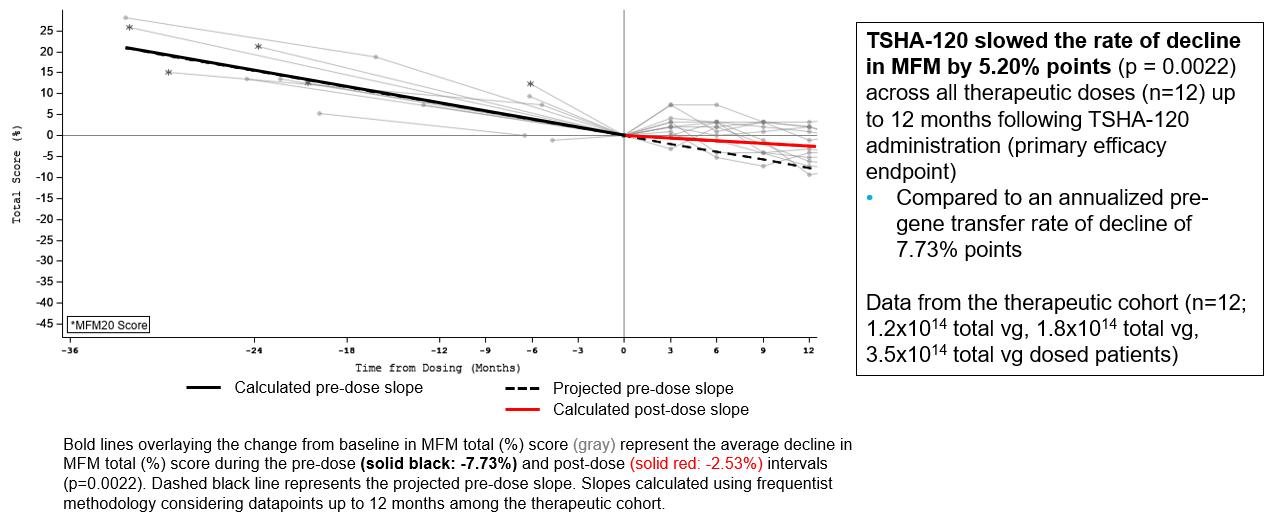
出典:2022年7月現在のデータカットによる予備分析
1.2x10でベイジアン分析が行われました14 合計サイズ、1.8x1014合計値と3.5x1014自然史と比較して、臨床的に有意な意味で疾患の進行を遅らせる可能性を評価するために、1年目のVG用量コホートの総投与量コホートです。この種の統計分析は、直接的な確率計算を可能にし、希少疾患や少数の患者集団を対象とした介入研究において有用であり、規制当局にも受け入れられています。下の表に示すように、自然史データと比較して、TSHA-120による治療後、すべての治療用量コホートで、疾患の進行が遅くなる確率はほぼ 100%、臨床的に有意な低下の確率は79.4%で、50%以上でした。
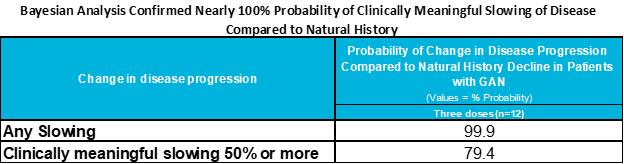
出典:2022年10月にFDAに提出され、2022年12月に開催されたタイプBのフェーズ2終了会議パッケージの付録3の表12の変種。
42
治療用量コホートの患者のMFM32スコアのベースラインからの平均変化に対するTSHA-120の効果は、治療前の期間における数年にわたる推定減少と比較して、経時的に一貫して改善し続けました。長期のMFM合計(%)スコアの分析では、下のグラフに示すように、MFM%の合計スコアに対して病気の進行が2.49%(p = 0.0065)ポイント遅れたことがわかりました。
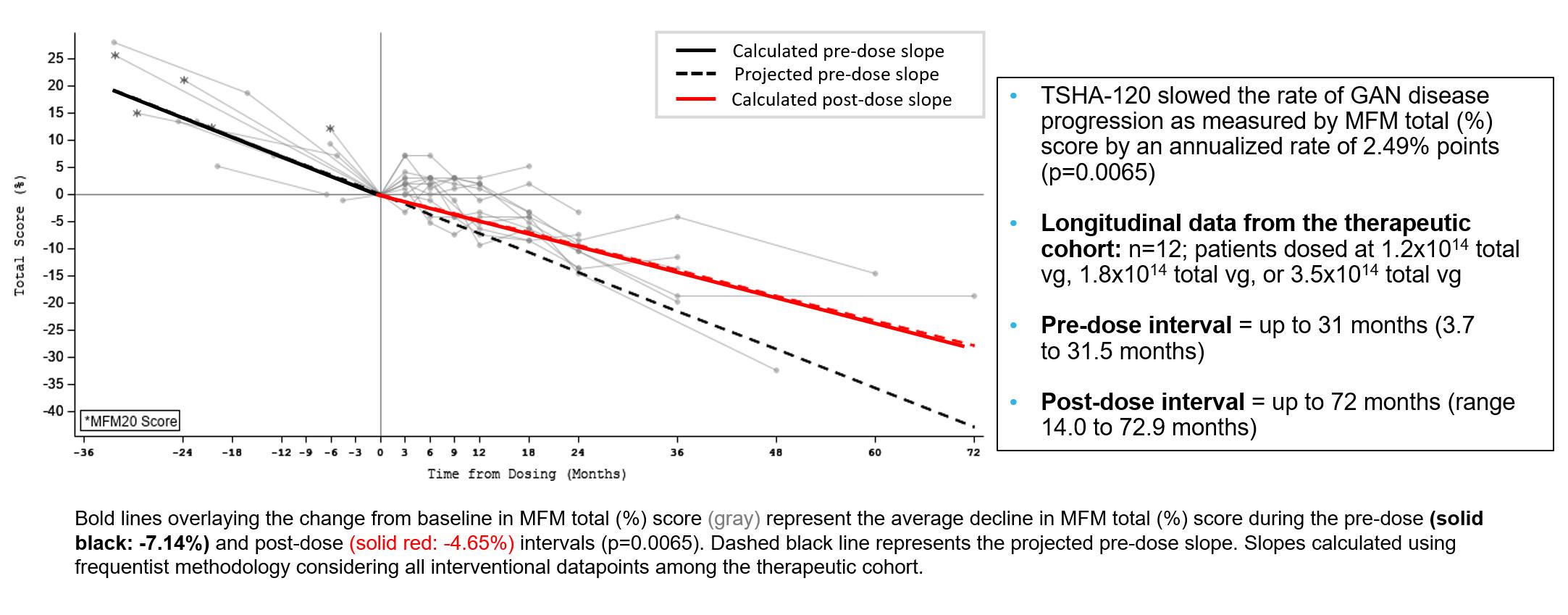
出典:2022年7月現在のデータカットによる予備分析
新しい包括的なデータ分析
NIHからのデータ転送が完了しました。タイプBの第2相終了時にFDAと会って以来、GANの疾患進行の不均一性や主要評価項目としてのMFM32の努力依存性についてのFDAのフィードバックに対応するために、自然史と介入研究データの両方から得られるデータの合計を広範囲に分析してきました。その際、盲検化されていない研究デザインを検討しました。
自然史研究データの分析によると、GANは急速に進行する重度のCNSと末梢神経系(PNS)の変性を特徴とし、すべての患者に衰弱と早期運動失調をもたらすことがわかりました。この迅速で予測可能な進行は、MFM32の一次分析で確認されたように、GAN患者とその介護者の生活の質に大きな影響を与えます(Bharucha-Goebel、Norato et al。、2021)。私たちは、超希少疾患における他の疾患モデルと同様に、疾患の進行を定量的に特徴付けるために、疾患進行モデル(DPM)を開発し、すべての自然史の古典的GAN患者の断面的および縦断的データに適用しました(Garbade、Zielonka et al. 2021、Barrett、Nicholas et al. 2022)。DPMは、関連するさまざまなCNSおよびPNSエンドポイントで進行性かつ単調な低下に続く同種性疾患として古典的GANを確立しました。そのため、このDPMは、外部対照試験に関するFDAガイダンス草案(医薬品および生物製剤に関する外部対照試験の設計と実施に関する考慮事項、2023年;(食品医薬品局、2023年))と一致して、TSHA-120の治療効果を評価するための外部コントロールとして自然史研究患者を活用するための適切な方法論を提供すると考えています。
この柔軟なモデルは直線性を前提とせず、患者の加齢に伴う減少率の変化を考慮しています。これは、多くのエンドポイントで下限効果と上限効果が観察されているGANのような進行性疾患において非常に有用です。DPMの使用は、オープンラベルの非ランダム化介入研究の設定において、一部の臨床エンドポイントが労力に依存する可能性があるというFDAのフィードバックにも対応し始めます。未治療の患者におけるDPMの予測される疾患進行と比較すると、TSHA-120で治療されたGAN患者の複数の臨床成績と客観的な生物学的エンドポイントにわたって観察された陽性治療反応は、偶然に起こる可能性はほとんどありません。
その他のエンドポイント
この臨床試験では、治療前と治療後の多くの追加エンドポイントが収集され、長期のフォローアップデータが収集されています。MFM32、MFAR(運動失調スケール)、LogMar(視力)などの機能的エンドポイント、感覚神経活動電位(SNAP)、複合筋活動電位(CMAP)などの電気生理学的エンドポイント、および皮膚生検における神経線維密度などの生物学的エンドポイントを特定しました。TSHA-120で治療を受けた個人を、DPMから予想される疾患進行率と比較したところ、複数のエンドポイントで疾患経過の軌跡に好ましい偏差が見られました。これらの分析は、事前に指定されたプログラム諮問監督委員会による分析を裏付けるものです
43
主要評価項目と二次評価項目。グループレベルでは、集計データとして見ると、測定された有効性エンドポイントの大部分は薬効を示しており、病気の進行を遅らせる可能性が高いです。
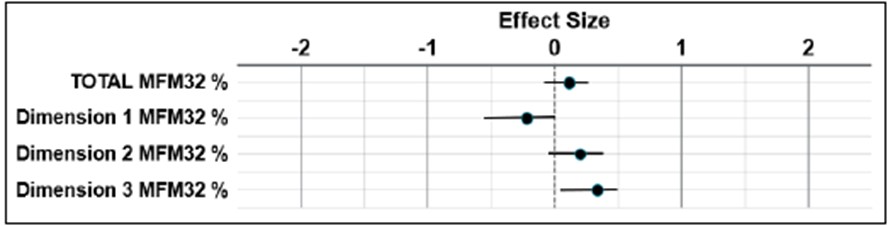
DPMに対する分析では、MFM32の推定年間減少率合計では、治療効果の大きさは7%、病気が遅くなる確率は81%でした。治療効果は主に、軸方向および近位の運動機能(13%、95%CI = -5%~30%)を捉えたドメイン3と遠位運動機能(28%、95%CI = 7~47%)を捉えたドメイン3で観察されました。99%の確率で疾患の進行を遅らせる治療効果が得られ、推定治療効果は28%でした。ベースラインの外来機能に障害がある患者集団で予想されたように、立位、転換、運動の自立性を評価するドメイン1の項目で推定された治療効果は最小限(-27%、95%CI = -54%~-3%)で、自信を持って疾患が遅くなることを予測していませんでした(1%)。病気の期間に基づくと、GAN患者は不可逆的な下肢の筋線維症にかかりやすく、神経再支配によって運動機能が回復することは期待できません。
MFM32の合計スコアとドメインスコアのDPM推定値
出典:2023年5月現在のデータカットによる予備分析
FARSとmFARSを利用した追加の分析では、TSHA-120の有意義な治療効果が示され、運動失調症の予想される毎年の悪化を遅らせました。mFARのスコア範囲は0から93で、スコアが高いほど障害が大きいことを示します。DPMは治療効果の大きさを31%(MFAR、95%CI = 7%から52%)と29%(FARS、95%CI = 8%から47%)と推定しており、99%の確率で運動失調症の予想される機能低下を遅らせます。これらの結果は、MFARの年間相対変化率は約-1.28%、FARSスコアは-2.25です。
臨床的には、これらのデータは患者にとって重要な機能の観点から解釈する必要があります。特に、手と腕の機能は患者の日常生活活動に不可欠です。したがって、手と腕の機能の低下を遅らせることは直接的かつ臨床的に重要であり、GAN患者の日常機能を改善することが期待されます(Johnson-Kerner、Roth et al.2014、Bailey、Armao et al.2018)。2021年にDuongらによって説明された方法論(Duong、Staunton et al.2021)を使用して、臨床アウトカムスコアと実際の日常生活活動(ADL)との関係を評価しました。ADLは9つの機能カテゴリー(発話、嚥下、食事の切断、取扱い道具、着替え、個人衛生、転倒、歩行、座位の質、膀胱機能)にまたがり、5段階のリンカート尺度で採点されます。スコアが高いほど介護者への依存度が高いことを示します。ほとんどの参加者は、介入研究中に治療を受けるまでに、すでに運動能力が大幅に低下しているか、完全に運動できなくなっています。機能的影響が最も小さかったのは、歩行に関連する活動(歩行など)でした。個人の自立とセルフケアに関連するADLのカテゴリーでは、自己栄養、個人衛生作業(髪や歯のブラッシング)の完了、テクノロジーとの視覚的および身体的相互作用、対人コミュニケーション、その他多くの方法での環境との関わりなど、中程度から強い治療効果が観察されました。これらの活動は、患者の自立と介護者の負担軽減に不可欠であり、特に上肢の使用を維持することに依存しています。
44
DPM:日常生活活動の推定値
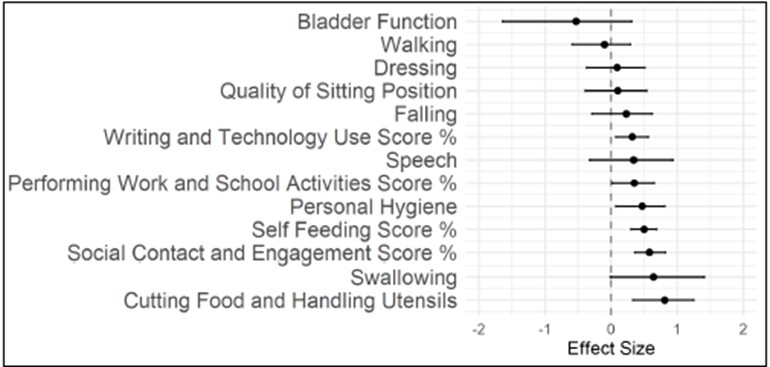
ソース: 2023年5月現在のデータカットによる予備分析。ADLは、MFM32とFARSの評価から報告または決定されました。治療効果の大きさ:0 = 疾患の自然史経過、1 = 疾患の停止、>1 = 疾患の絶対的改善。負の値は、予測される自然史疾患の経過に対する加速を示します。
GAN患者は、病気の進行中に視力が徐々に低下し、それに応じて参加者の視力が上昇します(LogMarスコアで測定)。GANの自然史研究では、ログマーは0.0から1.2の範囲でした(平均±SD:0.4±0.3)(Zein、Bharucha-Goebelら2016年)。介入研究では、治療前の最大36か月の間に患者の視力が低下しました。これは、予想される自然史の低下と一致していました。DPMは、治療により、51%(左目、95%CI = 33%から68%)および70%(右目、95%CI = 54%から84%)の治療効果を推定し、疾患の進行を遅らせる上で治療が陽性になる確率はおよそ100%です。これらの結果は、左眼で-0.06、右眼で-0.09のおよその相対的な年率変化に相当します。
自然史研究と介入研究の両方から得られた臨床機能エンドポイントの拡張分析に加えて、客観的な電気生理学的および生物学的エンドポイントをさらに検討しました。DPMに対する分析では、尺骨SNAPと中央SNAP振幅で神経反応に測定可能な改善が見られる確率が100%と推定され、治療効果はそれぞれ189%と152%と推定されました。同様に、ベースライン時に異常なCMAP反応が記録され、GAN患者の広範囲にわたる運動単位接合機能障害が示されました。DPMに対する分析では、尺骨神経のCMAP振幅に測定可能な改善が見られる確率が94%と推定され、治療効果は 29% と推定されています。
SNAPが安定または回復した5人の患者のうち4人は、神経生検で再生クラスターが増加しました。再生クラスタースコアの増加は、新しい神経線維損傷または再生プロセスの両方に関連しており、再生クラスターの一般的な評価は、3回連続の測定(電気生理学と生検)に依存します。複数の治療を受けた患者における軸索の完全性に依存するSNAP活動の回復を考慮すると、神経線維損傷に対する活発な軸索再生反応の解釈に有利な妥当な自信を持つことができます。介入研究の参加者から、遺伝子導入の1年後にベースラインと対側部位で採取した大腿近位部と脚遠位部の皮膚生検標本を、組織学的検査の対象としました。12か月目に、下肢の近位または遠位部の少なくとも1か所で皮膚の神経線維密度の安定化または増加が見られました。これには、高用量患者3人のうち3人と中高用量患者の1人が含まれます。表皮神経線維を調べたところ、遺伝子導入後のレスキューが用量依存的に起こったという客観的な証拠が明らかになり、これらの結果はTSHA-120投与との特定の関係を示唆しています。
TSHA-120による治療は、電気生理学的反応と神経組織の再生の改善につながるという結果が出ました。これはGANの自然史では見られない発見です。末梢神経の改善を組み合わせることで、MFAR、MFM32ドメイン3、ADLスコアに見られるように、手の筋力と機能を維持することができます。特に下肢の機能は病気の初期段階で失われることが多いため、腕の機能に関する知見はGAN患者の日常機能を維持するために重要です(Bharucha-Goebel、Norato et al. 2021)。これらの努力に依存しない機能的および生物学的データからの証拠は、MFARやMFM32など、臨床医が報告した治療成績で観察された改善を裏付けたと考えています。
45
個人の自立やセルフケア、特に上肢の使用を伴う活動に関連するカテゴリーで見られる治療効果。
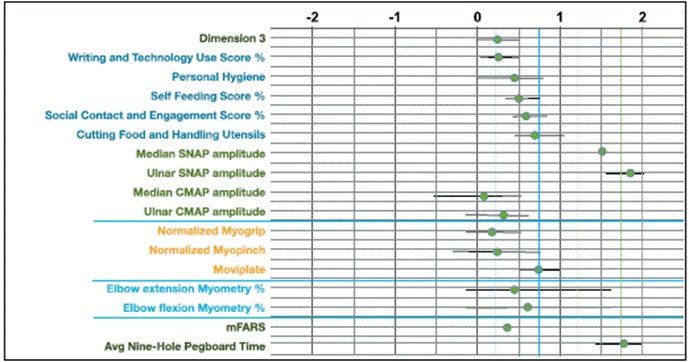
ソース:2023年5月現在のデータカットによる予備分析
TSHA-120の治療後、GAN患者では幅広い薬効が観察され、ADL、機能的臨床医が報告したアウトカム、パフォーマンスアウトカム、電気生理学的アウトカム、客観的な生物学的データなどにおいて顕著な安定化が見られました。個別に、またまとめると、TSHA-120は複数の機能領域にわたって有効性を示す傾向を示しており、GAN患者にとって非常に有意義な方法で疾患の進行速度を遅らせることができます。まとめると、これらのデータは、GAN患者におけるTSHA-120の幅広い有効性を合理的に確認できる確固たるデータセットを提供すると考えています。私たちはFDAとの正式な会合のリクエストを提出しました。2023年の第3四半期に会議を開き、この新しいデータ分析について話し合い、GANに関する規制経路における次のステップについてお知らせします。
安全性と耐容性
TSHA-120は複数回の投与で十分な耐容性を示しており、重大な急性または亜急性の炎症の徴候はなく、突然の感覚変化もなく、薬物関連または持続性のトランスアミノ炎もありません。免疫抑制や研究手順に関連する有害事象は、他の遺伝子治療で見られたものと同様で、本質は一過性でした。用量を増やしても有害事象の発生率は増加しませんでした。重要なことに、TSHA-120は、投与経路、投与、免疫抑制療法を組み合わせた結果、中和抗体の存在下で安全に投与されました。
46
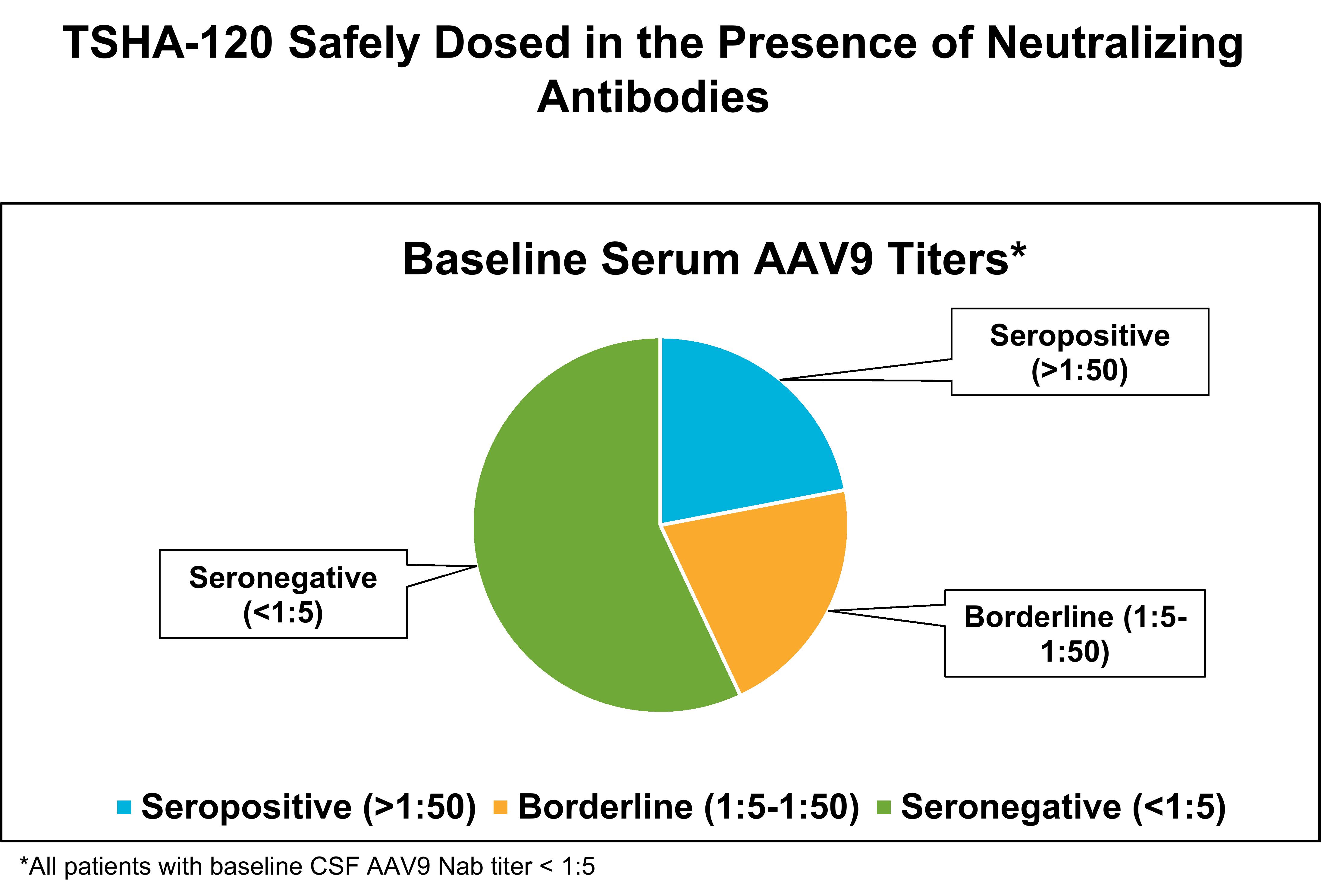
ソース:NIH
現在、進行中の臨床研究から、個々のGAN患者に関する7年以上にわたる縦断的データがあります。TSHA-120による治療は忍容性が高く、重大な安全上の問題はありませんでした。用量の増加、用量制限毒性、急性または亜急性の炎症の徴候、突然の感覚変化、およびトランスアミナーゼの薬物関連または持続的な上昇を伴わないことによる有害事象の発生率の明らかな増加はありませんでした。免疫抑制や研究手順に関連する有害事象は、他の遺伝子治療で見られたものと同様で、本質は一過性でした。TSHA-120に関連する可能性がある、またはおそらく関連すると考えられる最も一般的な重篤でない有害事象には、脳脊髄液(CSF)、髄球増加症、白血球増加、CSF Ig増加、血小板増加症、頭痛、および心臓障害(心電図異常)が含まれていました。すべての重篤な有害事象は、高用量で治療された参加者に発生した、TSHA-120に関連している可能性があると考えられる発熱の1つの重篤な有害事象を除いて、TSHA-120とは考えにくい、または無関係と評価されました。
安全性サーベイランスは継続されていますが、これまでに報告されたほとんどの事象は投与後3か月以内に始まり、治療に伴う有害事象(TEAE)の総数と割合の発生パターンに基づいて、少なくともTSHA-120に関連していると評価されたが、TSHA-120の投与量の増加に伴う事象の増加はなかったようです。
関連治療-緊急有害事象の報告率(患者1人あたり、用量別)— 3年目まで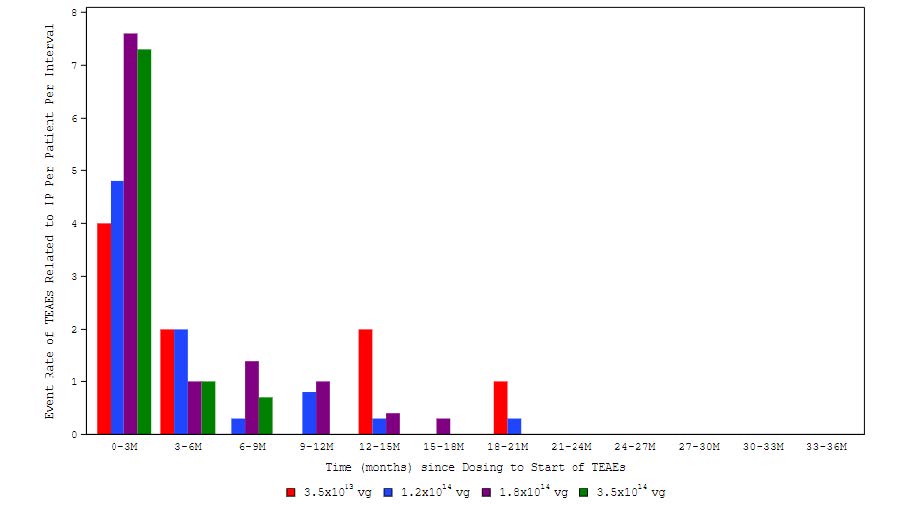
M = 月; TEAE = 治療に伴う緊急有害事象
ソース:2023年5月現在のデータカットによる予備分析。
47
ライセンス交渉に役立つ堅牢なCMCデータパッケージを提供するために、受託開発製造組織(CDMO)パートナーとの間でTSHA-120の開発ロットとGMPロットを6回無事完了しました。また、現在および以前の臨床ロットの包括的な生化学的および生物物理学的分析を完了しました。当社のCDMOは、比較リスクを減らすことを目的として、オリジナルのGANロットの製造に使用したのと同じPro10TM製造プラットフォームを利用しています。2Lから250Lスケールまでの5つの開発ロットと1つのフルスケール500L GMPロットが、現在のTSHA-120臨床ロットと並行して、製品とプロセスの残留物、空/全比率、遺伝的完全性、効力、強度などの重要な属性のアッセイを含む現在の規制要件を満たすことが期待される包括的な分析パネルを使用して分析されました。
ddPCRにより最高用量コホートである1.0x1014gで50回以上の患者用量のTSHA-120が得られたTSHA-120ピボットロットは、2022年11月にリリースされました。この資料は、将来のBLA対応活動と商業生産に向けて私たちを導きます。これらのロットは、重要な保存期間データを提供するために安定性も置かれています。
2021年9月、私たちはTSHA-120の科学諮問会議のリクエストをMHRAに提出し、2022年1月の会議を許可されました。MHRAは、効能アッセイを含む当社の商業製造およびリリースアッセイ試験戦略に合意しました。最後に、MHRAは、主要な臨床エンドポイントとしてGANのMFM32の検証作業を行い、このプロセスの一環として患者と家族を対象にMFM32項目を調査するという私たちの提案を支持してくれました。2022年9月、私たちはFDAに会議出席依頼を提出し、2022年12月13日に電話会議によるタイプBのフェーズ2終了会議を許可されました。2023年1月、正式な議事録の受領後、タイプBのフェーズ2終了会議で得たフィードバックをFDAに報告しました。FDAはTSHA-120についてさらに明確化しました。MFM32は許容可能なエンドポイントとして認められ、BLAを支援するために二重盲検プラセボ対照設計で患者に追加の投与を推奨しました。FDAは、TSHA-120用に計画されているCMCデータパッケージの審査が終わるまで、商用原料の製造に対する当社の全体的なアプローチが適切であると判断したことを認めました。その後、正式な議事録への回答としてフォローアップの質問を提出しました。FDAは、対照試験を設定する際に関連する主要評価項目としてMFM32を明確にし、GANは非常にまれな性質のため、そのような研究の設計における私たちの課題を認識しました。FDAは、十分な数の患者を登録することが難しく、対照試験の環境では規制が柔軟なため、不確実性が増しても受け入れていました。さらに、FDAは、自明で臨床的に意味のある比較的大きな治療効果を実証するために、客観的な測定値を利用した代替研究デザインを検討する用意があることを示しました。FDAは、安全性データベースの規模は審査上の問題であり、治療を受けた患者からの既存の安全性データが受け入れられるかどうかは、製品の比較可能性の証明にかかっていることを認めました。医薬品比較可能性データを詳述したCMCモジュール3の修正案の提出を完了し、2023年の第3四半期にフィードバックを受け取りました。FDAは、比較可能性研究(初期の臨床ロットとピボットロットの比較)と計画された臨床試験で使用するための重要なロットリリースを裏付けるには、分析データで十分であると結論付けました。
他のプログラム
現時点では、SLC13A5の前臨床製品候補であるTSHA-105、セロイドリポフスチノーシス1のTSHA-118、セロイドリポフスチノーシス7のTSHA-121の評価の優先順位を下げました。現在、TSHA-105、TSHA-118、TSHA-121の可能性を評価していませんが、将来、パイプライン拡張計画の一環としての製品候補としてこれらのいずれかを再評価したり、これらのプログラムを進めるためのパートナーシップを追求したりする可能性があります。
セロイドリポフスチノーシス1型疾患にはTSHA-118です
リソソーム蓄積障害であるセロイドリポフスチノーシス1型疾患(バッテン病の一種)は、幼児期に発症する進行性で致命的な神経変性疾患で、世界の出生数は約138,000人に1人と推定されています。セロイドリポフスチノーシス1型疾患の推定有病率は、米国と欧州連合で1,000人の患者です。セロイドリポフスチノーシス1は、特定の脂質修飾タンパク質の分解に関与する小さな糖タンパク質であるパルミトイルタンパク質チオエステラーゼ-1という酵素をコードするCLN1遺伝子の機能喪失性変異によって引き起こされます。CLN1遺伝子の機能喪失変異は、これらの脂質修飾タンパク質を細胞に蓄積させ、最終的には凝集、神経細胞の機能障害、そして最終的には神経細胞死につながります。
乳児発症のセロイドリポフスチノーシス1型では、臨床症状は6~24か月の間に現れ、言語機能や運動機能の急激な低下、難治性てんかん、運動失調、視覚障害などがあります。乳児発症のセロイドリポフスチノーシス1の患者は、通常、5歳までには反応が悪くなり、通常は7歳までに死亡するまでコミュニケーションが取れなくなります。乳児遅発性セロイドリポフスチノーシス1型は、2歳から4歳の間に始まり、最初の視覚および認知機能の低下を伴い、運動失調症とミオクローヌス、または急激な不随意筋痙攣の発症が続きます。若年性セロイドリポフスチノーシス1型疾患の患者は、5歳から10歳の間に発症し、最初の症状が視力喪失で、その後に認知機能低下、発作、運動機能低下が続きます。米国でセロイドリポフスチノーシス1型と診断された子供の約60%は早期発症の乳児型を呈し、残りの40%は遅発性の小児型です。
48
セロイドリポフスチノーシス1型の患者さんに対して現在利用可能な治療法はすべて症状の治療を目的としており、疾患を改善する治療法は承認されていません。遺伝子治療は、可溶性酵素の突然変異を伴う神経性セロイドリポフスチン症の形態を矯正する上で有望であることが示されています。その理由の1つは、隣接する非形質導入細胞の交差矯正です。
私たちは、機能性の導入を信じています CLN1中枢神経系に髄腔内送達されるAAV9ベクターを用いた遺伝子は、この疾患に対する疾患改変治療アプローチの可能性を秘めています。TSHA-118は、ニワトリβ-アクチンハイブリッドプロモーターの制御下でヒトコドンに最適化されたCLN1相補性デオキシリボ核酸を発現する自己相補性AAV9ウイルスベクターです。私たちは、2020年8月にアビオナ・セラピューティクス)とのライセンス契約に基づき、TSHA-118(旧ABO-202)の研究、開発、製造に関連する特定の知的財産権とノウハウに対する独占的な世界的権利を取得しました。
TSHA-118は、セロイドリポフスチノーシス1型疾患の治療を目的として、FDAから希少小児疾患の指定、ファストトラック指定、および欧州医薬品庁から希少疾病用医薬品の指定を受けています。
現在、CLN1プログラムには公開されているINDがあります。TSHA-118のCTA申請書を提出し、2021年にカナダ保健省によって承認されました。臨床試験材料が製造され、公開され、臨床試験の場で使用できる状態になりました。
SLC13A5の欠損症に対するTSHA-105
私たちは、生後数日以内に発作が始まることを特徴とするまれな常染色体劣性てんかん性脳症であるSLC13A5欠乏症の治療のためにTSHA-105を開発しています。SLC13A5欠損症は、主に脳と肝臓で発現するナトリウム依存性クエン酸トランスポーター(NaCT)をコードするSLC13A5遺伝子の双対立性機能喪失変異によって引き起こされます。現在までに、テストされたすべての突然変異では、細胞内のクエン酸塩の量がまったくないか、大幅に減少しています。nACT機能が低下すると、脳のエネルギー、代謝、機能に重要なクエン酸塩やコハク酸などの他の代謝産物の神経への取り込みが失われます。影響を受けた子供は、細かい運動能力と受容的な発話が比較的保たれている状態で、総運動機能と言語生成に障害があります。現在、SLC13A5欠乏症の治療法は承認されておらず、治療は主に症状の改善を目的としています。SLC13A5欠乏症の推定有病率は、米国と欧州連合で1,900人の患者です。
SLC13A5欠損症の遺伝子補充療法としてTSHA-105を開発しています。TSHA-105は、自己相補的なAAV9キャプシドにパッケージされたコドン最適化ヒトSLC13A5遺伝子から構築されています。
SLC13A5欠乏症によるてんかんの治療を目的としたTSHA-105について、FDAから希少疾病用医薬品の指定と希少小児疾患の指定、欧州委員会から希少疾病用医薬品の指定を受けています。臨床試験材料が製造され、公開され、臨床試験の場で使用できる状態になりました。
タウオパシーのためのTSHA-113
タウオパシーの治療のためにTSHA-113を開発しています。タウオパシーは、微小管関連タンパク質タウ(MAPT)タンパク質が人間の脳の神経原線維または神経原線維のもつれに凝集する神経変性疾患の大部分を占めます。これらには、MAPT関連前頭側頭型認知症(FTD)、進行性核上性麻痺(PSP)、皮質基底核変性(CD)、アルツハイマー病が含まれます。米国とヨーロッパでは、MAPTを介したFTDの影響を受ける患者は推定11,000人で、MAPTを介したPSPとCDの影響を受ける患者は2,000〜2,500人と推定されています。,アルツハイマー病は、推定620万人のアメリカ人と780万人のヨーロッパ人が罹患しています。
第1相試験でBiogen/IonisがタウmRNAを標的としたアンチセンスオリゴヌクレオチド(ASO)の髄腔内送達により、アルツハイマー病患者の脳脊髄液中のタウタンパク質とホスホタウが、持続的で強固に、時間と用量に依存して低下することが示されました。これらの結果に支えられて、バイオジェンは2022年8月に、アルツハイマー病による軽度の認知障害または軽度の認知症の人を対象とした第2相試験を開始しました。このASOターゲットの検証は、タウオパチーを治療するための細胞間タウmRNA(タウタンパク質の産生を減らす)を標的とする他のアプローチへの道を開きました。
生涯にわたる反復投与を必要とするASO治療とは異なり、タウオパチーの1回限りの治療法を開発しています。TSHA-113はタウ特異的miRNAをパッケージしたAAV9キャプシドで、タウオパシーの治療のために脳脊髄液に送られます。このmiRNAはタウmRNAの6つのアイソフォームすべてをターゲットにします。
タウオパシーの検証済みマウスモデルであるPS19マウスでTSHA-113の有効性をテストしました。これらのマウスはヒトMAPTを発現し、生後9〜12か月頃に著しいタウ病変、神経変性、体重減少、進行性後肢麻痺を示します。生後3か月、6か月、9か月のPS19マウスにTSHA-113を嚢内マグナ注射で投与することで、治療の有効性をテストしました。タウのmRNAとタンパク質レベルが大幅に低下していることがわかりました
49
TSHA-113トリートメント。一貫して、タウシードアッセイでは、TSHA-113で治療されたPS19マウスの脳内の病的タウレベルの低下が示されました。さらに、TSHA-113治療は、生後3か月、6か月、9か月で治療した場合、PS19マウスの生存率、体重減少、後肢のクラスピング表現型を回復することができました。まとめると、これらの結果は、タウ特異的miRNAを1回だけベクター化して送達することが、タウオパシーの治療に有望なアプローチであることを示しています。現在進行中および今後の研究は、INDを可能にする研究のための最適な用量の決定に焦点を当てています。
エンジェルマン症候群の場合はTSHA-106
私たちは、母体のUBE3A遺伝子の欠乏によって引き起こされる神経発達障害であるエンジェルマン症候群の治療のためにTSHA-106を開発しています。アンジェルマン症候群は、重度の発達遅延、運動失調と歩行障害、睡眠障害、発作、不安の高まり、攻撃性、重度の言語障害を特徴としています。アンジェルマン症候群は、世界中の患者12,000〜20,000人に約1人が罹患しています。
アンジェルマン症候群は、母方の遺伝子が欠損していて、父方のUBE3Aのコピーは損なわれていないが、長鎖ノンコーディングRNA、UBE3Aアンチセンス転写物、またはUBE3A-ATSによって沈黙している刷り込み障害です。UBE3A-ATSを標的としたASOの投与は、トランスジェニックマウスモデルでエンジェルマン症候群の症状を改善するという有望な結果を示しました。
ノースカロライナ大学から新しい遺伝子補充療法のライセンスを取得しました。この新しいコンストラクトは、同じコドン最適化導入遺伝子カセットからUBE3A mRNAの2つのアイソフォームを発現するように設計されており、この病気の1回限りの治療になる可能性があります。独自の設計上の特徴により、Hube3aアイソフォームをほぼ内因性の3:1(短い/長い)の比率で短くて長いHube3aアイソフォームを発現できます。この機能は、最適な治療結果をサポートするのに役立つ可能性があります。さらに、このコンストラクトはヒトシナプシン1プロモーターを使用して、主にエンジェルマン症候群の治療の主要な治療標的であるニューロンでのUBE3Aの発現を制限します。
公表された研究では、この二重アイソフォーム発現カセットをPHP.Bキャプシドにパッケージ化し、新生児マウスモデルに脳室内注射によって投与しました。この治療により、エンジェルマン症候群マウスの運動学習と先天的行動が大幅に改善されました(PMID:34676830)。これにより、エンジェルマン症候群のマウスは、てんかん発生および関連する海馬神経病変に対して回復力を持つようになりました。これらの結果は、ASの治療における二重アイソフォームHube3a遺伝子導入の実現可能性、忍容性、および治療の可能性を示しました。
これらの発見を翻訳可能な介入へと進めるために、私たちの共同研究者は二重アイソフォーム発現カセットをAAV9キャプシドにパッケージ化し、動物の概念実証研究を実施しました。全体として、これらの結果は、同様の用量のphp.b/hube3Aoptベクター(PMID:34676830)の新生児ICV送達に関する公開データと非常に一致しており、継続的な開発を裏付けています。現在進行中および今後の研究は、IND対応研究のための最適な用量と投与経路の決定に焦点を当てています。
米国とヨーロッパには、エンジェルマン症候群の患者が推定55,000人います。
フラジャイルX症候群のTSHA-114
私たちは、自閉症と認知障害の最も一般的な単一遺伝子の原因であるFragile X症候群の治療のためにTSHA-114を開発しています。Fragile X症候群は、世界中の約6,000人に1人が罹患しています。フラジャイルX症候群は3歳頃に診断され、不安、攻撃性、多動性、注意障害、睡眠障害、コミュニケーション障害を特徴とします。
フラジャイルX症候群は、FMR1遺伝子の5'非翻訳領域におけるCGGトリプレットリピートの病理学的拡大によって引き起こされます。トリプレットが通常の5〜55回以上で200回以上に拡大すると、遺伝子プロモーターが高メチル化され、コードされたタンパク質、壊れやすいX精神遅滞タンパク質、またはFMRPの転写と翻訳が停止します。リピートが拡大すると、RNA(エピジェネティックな遺伝子サイレンシングを誘導するDNAヘテロデュプレックス)の形成も誘導します。フラジャイルX症候群のほとんどの患者はFMRPを発現しませんが、完全変異を起こした人の中には、タンパク質の産生量が少ない(正常レベルの10%未満)人もいます。影響を受けていない人のFMRPの発現は人によって大きく異なります。フラジャイルX症候群の現在の薬物療法治療は、症状の緩和のみを目的としています。
TSHA-114を使用して、フラジャイルX(Fmr1 KO)の動物モデルで概念実証研究を実施しました。野生型マウスにTSHA-114を髄腔内投与してから12か月後まで、行動、血清、または病理組織学的マーカーに重大な副作用は観察されませんでした。TSHA-114で処理したFMRKOは、投与後、脳全体で広範囲にわたるFMRPの発現が観察されたことを示しました。TSHA-114で処理したFMRKOマウスは、聴覚性発作の強力な抑制と恐怖条件付け行動の正常化を示しました。さらに、概日運動活動の評価により、多動性と睡眠の回復が明らかになりました。個々のマウスの導入遺伝子発現と行動反応の評価では、FMRPの発現レベルと薬効との間に相関関係があることが示されました。
50
この研究の結果は、継続的な開発を強く裏付けています。現在進行中および今後の研究は、IND対応研究のための最適な用量と投与経路の決定に焦点を当てています。
米国とヨーロッパには、フラジャイルX症候群の患者が推定75,000人います。
ライセンス契約
テキサス大学サウスウェスタンメディカルセンターとの研究、協力、ライセンス契約
2019年11月、私たちはUTサウスウェスタン大学に代わってテキサス大学システムの理事会と、2020年4月に改正された研究、協力、ライセンス契約、つまりUTサウスウェスタン協定を締結しました。
UTサウスウェスタン大学との契約に関連して、UTサウスウェスタン大学の特定の特許権に基づく独占的で世界規模のロイヤリティフリーのライセンスと、UTサウスウェスタン大学の特定のノウハウに基づく非独占的、世界的、ロイヤリティフリーのライセンスを取得しました。いずれの場合も、特定の用途で使用するためのライセンス製品の製造、製造、使用、販売、販売、販売、販売、提供、および輸入が可能です。さらに、UTサウスウェスタン大学の特定の特許とノウハウに基づき、人間があらゆる用途に使用するための非独占的、世界的、ロイヤリティフリーのライセンスを取得しました。特定の特許権に基づく独占的ライセンスの取得を最初に拒否する権利と、そのような特許権以外の特許権に基づく独占的ライセンスを交渉するオプションもあります。私たちは、少なくとも1つのライセンス製品を開発、規制当局の承認を得て、商品化するために、商業的に合理的な努力を払う必要があります。
UTサウスウェスタン協定に関連して、UTサウスウェスタン大学に2,179,000株の普通株式を発行しました。特許の維持に関連する費用以外に、UTサウスウェスタン大学との契約に基づく将来のマイルストーン義務やロイヤルティ義務はありません。
UTサウスウェスタン協定は、国ごと、ライセンス製品ごとに、その国でライセンスされた特許の最後の有効な請求が満了した時点で失効します。最初の研究期間の終了後、UTサウスウェスタン大学への特定の書面による通知により、適応症別およびライセンス製品ごとのライセンス製品ベースで、いつでも契約を終了することができます。いずれの当事者も、未解決の重大な契約違反または相手方の破産があった場合、契約を終了することができます。
アベオナ(セロイドリポフスチノーシス1)とのライセンス契約
2020年8月、私たちはアベオナ・セラピューティクス社(アベオナ)とライセンス契約、つまりアベオナ CLN1契約を締結しました。Abeona CLN1契約に関連して、当社は、ノースカロライナ大学チャペルヒル校とアベオナが独自に開発した特定の特許、ノウハウ、および材料に基づいて、セロイシス1型疾患の予防、治療、または診断のための遺伝子治療のライセンス製品を研究、開発、製造、使用、および商品化するためにサブライセンスを付与する権利を含む、独占的かつ世界規模のロイヤリティを伴うライセンスを取得しました(1 ヒトのバッテン病(の一種)の
ライセンス付与に関連して、2020会計年度中にAbeonaに300万ドルの1回限りの前払いライセンス料を支払いました。私たちは、ライセンス製品1件あたり最大2,600万ドル、販売関連のマイルストーンとして最大3,000万ドル、およびライセンス製品の純売上高に対して1桁の高額なロイヤルティをAbeonaに支払う義務があります。ロイヤルティは、ライセンス製品が販売されている国で当該ライセンス製品を対象とする最後のライセンス特許の有効期限、取り消し、または完全に却下されるまで、製品が販売されている国で市場独占権が失われるまで、またはライセンス製品がそのような国に存在せず、その国に市場独占権が存在しない場合、最初から10年後まで、ライセンス製品ごと、国ごとに支払われます。そのような国でのそのようなライセンス製品の商業販売。さらに、Abeona CLN1契約と同時に、当社はAbeonaと購入および償還契約を締結しました。この契約に基づき、当社はAbeonaから特定の在庫を購入し、以前に発生した特定の研究開発費をAbeonaに払い戻し、2020会計年度に支払われた総対価は400万ドルでした。
2021年12月、セロイドリポフスチノーシス1型の治療を目的としたTSHA-118のCTA申請がカナダ保健省によって承認され、アベオナセロイドリポフスチノーシス契約に関連する規制上のマイルストーン支払いのきっかけとなりました。2021年12月31日に終了した年度の連結営業報告書に300万ドルの研究開発費を記録しました。マイルストーン手数料は2022年1月に支払われ、2022年6月30日までの6か月間の要約連結キャッシュフロー計算書では投資流出として分類されています。2023年6月30日までの6か月間、追加のマイルストーン支払いは行われたり開始されたりしませんでした。
51
Abeona CLN1契約は、ライセンス製品の最後のロイヤリティ期間の満了時に、国ごと、ライセンス製品ごとに失効します。いずれの当事者も、未解決の重大な契約違反または相手方の破産があった場合、契約を終了することができます。当社は、便宜上、事前にAbeonaに書面で通知した上で、契約を終了することがあります。
アベオナ(レット症候群)とのライセンス契約
2020年10月、私たちはAbeonaとライセンス契約、つまりAbeona Rett契約を締結しました。この契約に基づいて、ノースカロライナ大学チャペルヒル校、エジンバラ大学、アベオナが研究、開発、製造、使用、および商品化のために最初に開発した特定の特許、ノウハウ、および材料に基づいてサブライセンスを付与する権利を含む、独占的で世界規模のロイヤリティを伴うライセンスを取得しました。レット症候群の遺伝子治療および関連導入遺伝子の使用に関するライセンス製品。
アベオナの特定の義務に従い、当社は商業的に合理的な努力を払って、少なくとも1つのライセンス製品を米国で開発し、少なくとも1つのライセンス製品を商品化する必要があります。
アベオナレット契約に関連して、2020会計年度中に300万ドルの前払いライセンス料をアベオナに支払いました。私たちは、ライセンス製品1件あたり最大2,650万ドル、販売関連のマイルストーンとして最大3,000万ドルの規制関連のマイルストーンをAbeonaに支払う義務があります。また、ライセンス製品の純売上高に対して1桁の高額なロイヤルティを支払う義務があります。ロイヤルティは、ライセンス製品が販売されている国で当該ライセンス製品を対象とする最後のライセンス特許の有効期限、取り消し、または完全に却下されるまで、製品が販売されている国で市場独占権が失われるまで、またはライセンス製品がそのような国に存在せず、その国に市場独占権が存在しない場合、最初から10年後まで、ライセンス製品ごと、国ごとに支払われます。そのような国でのそのようなライセンス製品の商業販売。
2022年3月、レット症候群の治療を目的としたTSHA-102のCTA申請がカナダ保健省によって承認され、レット契約に関連する規制上のマイルストーン支払いのきっかけとなりました。2022年6月30日までの6か月間の研究開発費として要約連結損益計算書に100万ドルを記録しました。このマイルストーン料金は2022年7月に支払われました。2023年5月、私たちはレット症候群の成人患者におけるTSHA-102の安全性と予備的有効性を評価する第1/2相REVEAL試験で、最初の患者にTSHA-102を投与しました。その結果、この契約に関連するマイルストーン支払いのきっかけとなりました。2023年6月30日までの6か月間の研究開発費を要約連結営業報告書に350万ドル計上しました。このマイルストーン手数料は、2023年6月30日の時点で支払われておらず、2023年6月30日現在の要約連結貸借対照表の未払費用およびその他の流動負債に計上されています。2023年6月30日までの6か月間、本契約に関連して追加のマイルストーン支払いが行われたり開始されたりすることはありませんでした。
Abeona Rett契約は、ライセンス製品の最後のロイヤリティ期間の満了時に、国ごと、ライセンス製品ごとに失効します。いずれの当事者も、未解決の重大な契約違反または相手方の破産があった場合、契約を終了することができます。便宜上、契約を終了することがあります。
アステラス製薬とのオプション契約
発効日である2022年10月21日に、オーデンテス・セラピューティクス株式会社(d/b/a Astelas Gene Therapy)、またはアステラス製薬とオプション契約、つまりオプション契約を締結しました。
TSHA-120巨大軸索ニューロパチー
オプション契約に基づき、アステラス製薬に対し、発効日時点でTSHA-120または120として知られている製品を研究、開発、製造、使用、販売、販売、販売、販売、その他の開発、または総称して搾取または搾取するための独占的オプションを取得する独占的オプションをアステラスに付与しました。GAN製品、およびGANの治療に使用するためのバックアップ製品、または当社が管理するGANの治療に使用されるその他の遺伝子治療製品または当社の関連会社、または当社または当社の関連会社が管理する知的財産権、および(B)そのような利用またはGANオプションに関して当社または当社の関連会社が管理する知的財産権、および(B)当社または当社の関連会社が管理する知的財産権、またはGANオプション。一定の延長を条件として、GANオプションは、発効日から指定の期間まで行使できます。(i) アステラス製薬が、120 GAN製品について2022年9月19日にFDAに送付した会議出席依頼に対する当社とFDAとの間のタイプBフェーズ2終了会議の正式な議事録、(ii) タイプBのフェーズ2終了会議に関するFDAからのすべての書面によるフィードバック、および (iii) タイプBのフェーズ2終了会議に関連して当社がFDAに送付したすべての説明文書。
52
TSHA-102レット症候群
オプション契約に基づき、アステラス製薬に対し、(A) Rett製品(以下に定義)を利用するための独占的かつ世界規模のロイヤリティおよびマイルストーンを持つ権利とライセンスを取得する独占的オプション、および(B)当該利用に関連して当社または当社の関連会社が管理する知的財産権、またはRettオプション、およびGANオプションと合わせてそれぞれオプションを付与しました。一定の延長を条件として、レットオプションは、アステラス製薬が (1) 女性小児科試験の特定の臨床データと (2) TSHA-102に関する特定のデータ、または (i) 発効日時点でTSHA-102として知られている製品に関連するレットオプション期間と、それに関連するバックアップ製品を受領した後の指定期間まで行使できます。レット症候群の治療、および(ii)当社またはいずれかが管理しているレット症候群の治療に使用されるその他の遺伝子治療製品当社の関連会社、または当社または当社の関連会社が、その利用を対象とする知的財産権、またはRett製品を管理しています。
両当事者は、アステラスがオプションを行使する場合、両当事者がオプション契約に定められた条件に基づいてライセンス契約を誠実に交渉することに合意しています。これには、アステラスによる未定の前払金、未定のマイルストーン支払い、および該当するGAN製品および/またはRett製品の純売上高に対する未決定ロイヤルティが含まれます。
ゴーイング・コンサー
創業以来、私たちは営業損失を被っており、当面は引き続き大きな営業損失を被ると予想されており、決して利益を上げることはないかもしれません。研究開発活動に投資し続けるにつれて、損失は続くと予想されます。将来の義務を果たす能力の評価は、本質的に判断に基づくものであり、主観的であり、変更される可能性があります。現在の予測に基づくと、2023年8月に完了すると予想される私募による純収入(フォーム10-Qのこの四半期報告書に含まれる財務諸表の注記15と「—流動性と資本資源」を参照)を含め、これらの要約連結財務諸表の発行後12か月間、計画された事業を維持するのに十分な現金があると考えています。ただし、予測には内在する不確実性を考慮して、これらの要約連結財務諸表が発行された日時点で既知または合理的に知られている量的要因と質的要因の両方を考慮した結果、全体として継続企業として存続する能力について大きな疑念を抱く状況が存在すると結論付けました。
最近の動向
NASDAQ通知
2023年4月25日、ナスダック株式市場(ナスダック)から、手紙の日付より前の30営業日連続で、当社の普通株式の終値が1.00ドルを下回っていたことを知らせる手紙を受け取りました。ナスダック上場規則5810(c)(3)(A)に従い、ナスダックの入札価格要件の順守を取り戻すために、当初180暦日、つまり2023年10月23日までの期間が与えられました。2023年10月23日より前に、当社の普通株式の入札価格が最低10営業日連続で1.00ドル以上で取引を終えた場合、ナスダックのスタッフがナスダックの規則に従ってこの10日間の期間を延長する裁量を行使しない限り、当社は入札価格要件の遵守を取り戻します。2023年10月23日までに最低入札価格要件の遵守が回復しなかった場合、ナスダック・キャピタル・マーケットへの移転を選択して、その市場で提供されている追加のコンプライアンス期間を利用することを選択した場合、さらに180日間のコンプライアンス期間の対象となる可能性があります。資格を得るには、公開株式の市場価値に関する継続的な上場要件、およびナスダック・キャピタル・マーケットのその他すべての新規上場基準(入札価格要件を除く)を満たす必要があります。また、第2コンプライアンス期間中に入札価格の不足を是正する意向を書面で通知する必要があります。この期間中にコンプライアンスを取り戻せなかった場合、上場廃止になる可能性があります。2023年6月30日までの6か月間に、当社の取締役会および株主は、当社の普通株式の逆株式分割を実施するために、修正および改訂された定款に対する一連の代替改正を承認しました。この場合、取締役会は、株式併合比率を5対5から1対20の範囲から選択する裁量権を有します。このような株式併合および株式併合比率は、2024年の年次株主総会の前であればいつでも、取締役会の独自の裁量に委ねられます。
2023年8月3日、当社はナスダック上場規則5450 (b) (2) (A) またはMVLS要件に定められている、ナスダック・グローバル・セレクト市場への継続的な上場に必要な上場有価証券(MVLS)の最低市場価値(MVLS))を遵守していないことを示す書面による通知、つまりMVLS通知を新たに受け取りました。ナスダック上場規則5810(c)(3)(C)に従い、MVLS要件の遵守を取り戻すには、180暦日、つまり2024年1月30日、つまりMVLSコンプライアンス日までの期間を設けています。コンプライアンスを取り戻すには、MVLSコンプライアンス日の最低10営業日前に、MVLSのMVLSを50,000,000ドル以上で成約する必要があります。コンプライアンス日より前にMVLS要件の遵守が回復しない場合、ナスダックは当社の有価証券が上場廃止の対象であることを通知します。その際、当社は上場廃止の決定をナスダックの聴聞会に上訴することができます。
53
経営成績の構成要素
収益
2023年6月30日までの6か月間の収益は、アステラス製薬との取引から得られました。収益は、Rettプログラムに関連する研究開発活動が行われたことと認識しています。また、RettおよびGANオプションに関連する収益は、オプションが行使された時点またはオプション期間が終了した時点で計上します。
これまでのところ、製品の販売による収益は計上されていません。また、承認されても、近い将来、製品の販売による収益は見込めません。製品候補の開発努力が成功し、規制当局の承認、または第三者とのライセンス契約に至った場合、将来的には製品の販売から収益を上げる可能性があります。しかし、そのような収益がいつ生み出されるかについては、たとえあったとしても、保証はありません。
営業経費
研究開発費用
研究開発費は主に、前臨床試験の実施、製造開発、臨床試験の準備、および製品候補の規制申請に関連する活動を含む、製品候補の前臨床開発と発見作業で構成されます。研究開発費は発生したものとして認識され、研究開発に使用される商品やサービスの受領前に行われた支払いは、商品またはサービスが受領されるまで資産計上されます。資産の取得を通じて技術ライセンスを取得する際に発生した費用は、ライセンスされた技術が技術的実現可能性に達しておらず、将来の代替用途がない場合、研究開発費に計上されます。研究開発費には以下が含まれる、または含まれる可能性があります。
研究開発活動は私たちのビジネスモデルの中心です。一般的に、臨床開発の後期段階の製品候補は、臨床開発の初期段階の製品候補よりも開発コストが高くなります。これは主に、後期段階の臨床試験の規模と期間が長いためです。2021年から2022年にかけて、研究開発費と一般管理費を削減しましたが、近い将来、製品候補と製造プロセスの開発を継続し、前臨床プログラムの発見と研究活動を行う中で、特にRett臨床試験に関する研究開発費を増やす予定です。前臨床開発および臨床開発は本質的に予測不可能な性質があるため、現在または将来の前臨床試験および製品候補の臨床試験の開始時期、期間、または完了費用を確実に決定することはできません。臨床および前臨床開発のタイムライン、成功の確率と開発コストは、予想と大きく異なる場合があります。現在進行中および将来の前臨床研究と臨床試験、規制の進展、および各製品候補の商業的可能性に関する継続的な評価の結果に応じて、どの製品候補を追求し、各製品候補にどれだけの資金を投入するかを継続的に決定することを期待しています。将来的にはかなりの追加資本を調達する必要があります。臨床試験を開始すると、臨床開発コストは大幅に増加すると予想されます。当社の将来の費用は、次のような要因に基づいて期間ごとに大きく異なる場合があります。
54
一般管理費
一般管理費は、主に、株式ベースの報酬、退職金、旅費、採用費用など、幹部および管理職員の給与および関連費用で構成されます。その他の一般管理費には、法律、コンサルティング、会計、監査、税務関連サービスの専門家費用と保険費用が含まれます。
2022年と2023年に人員を削減してインフラストラクチャをサポートし、RettとGANのより優先度の高いプログラムに重点を置くことで、一般管理費の一部が将来減少すると予想しています。また、会計、監査、法務、コンサルティングサービスの支払いに伴う一般管理費のほか、ナスダックの上場規則とSEC要件の遵守維持に関連する費用、取締役および役員の賠償責任保険、投資家および広報活動、および上場企業としての運営に関連するその他の費用は、近い将来は一定ですが、時間の経過とともに増加する可能性があると予想しています。
長期資産の減損
長期資産の減損は、資産グループの帳簿価額が公正価値を上回った結果です。2022年11月、私たちはノースカロライナ州の製造施設の建設を続けないことに決めました。進行中の建設および製造施設の使用権リース資産に関連する、現金以外、非経常的な減損費用を記録しました。
55
業務結果
2023年6月30日および2022年6月30日に終了した3か月間の経営成績
次の表は、2023年6月30日および2022年6月30日に終了した3か月間の経営成績(千単位)をまとめたものです。
|
|
6月30日に終了した3か月間、 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
収益 |
|
$ |
2,395 |
|
|
$ |
— |
|
営業経費: |
|
|
|
|
|
|
||
研究開発 |
|
|
19,791 |
|
|
|
23,506 |
|
一般管理と管理 |
|
|
5,988 |
|
|
|
9,867 |
|
営業費用の合計 |
|
|
25,779 |
|
|
|
33,373 |
|
事業による損失 |
|
|
(23,384 |
) |
|
|
(33,373 |
) |
その他の収入 (費用): |
|
|
|
|
|
|
||
利息収入 |
|
|
223 |
|
|
|
27 |
|
支払利息 |
|
|
(1,440 |
) |
|
|
(743 |
) |
その他の収入 (費用) |
|
|
3 |
|
|
|
(3 |
) |
その他の収益(費用)の合計、純額 |
|
|
(1,214 |
) |
|
|
(719 |
) |
純損失 |
|
$ |
(24,598 |
) |
|
$ |
(34,092 |
) |
収益
アステラス製薬取引に関連する収益は、2022年11月に実行された2023年6月30日までの3か月間で240万ドルでした。記録された収益は、2023年の第2四半期に行われたRettの研究開発活動の結果です。
研究開発費用
研究開発費は、2022年6月30日までの3か月間の2,350万ドルに対し、2023年6月30日までの3か月間の研究開発費は1,980万ドルでした。370万ドルの減少は、人員削減と製造バッチやその他の原材料の購入の減少による報酬費用の減少によるものです。
一般管理費
2023年6月30日までの3か月間の一般管理費は600万ドルでしたが、2022年6月30日までの3か月間は990万ドルでした。390万ドルの減少は、人件費、コンサルティング費用、専門家費用の削減による報酬の減少によるものです。
その他の収入 (費用)
支払利息
2022年6月30日までの3か月間の支払利息は70万ドルでしたが、2023年6月30日までの3か月間の支払利息は140万ドルでした。約70万ドルの増加は主に、2023年6月30日までの3か月間のタームローンの金利が前年の比較期間と比較して高かったため、タームローン契約に基づいて発生した支払利息が増加したことによるものです。
56
2023年6月30日および2022年6月30日に終了した6か月間の経営成績
次の表は、2023年6月30日および2022年6月30日までの6か月間の当社の業績(千単位)をまとめたものです。
|
|
6 か月間 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
収益 |
|
$ |
7,101 |
|
|
$ |
— |
|
営業経費: |
|
|
|
|
|
|
||
研究開発 |
|
|
32,305 |
|
|
|
61,688 |
|
一般管理と管理 |
|
|
14,739 |
|
|
|
21,336 |
|
営業費用の合計 |
|
|
47,044 |
|
|
|
83,024 |
|
事業による損失 |
|
|
(39,943 |
) |
|
|
(83,024 |
) |
その他の収入 (費用): |
|
|
|
|
|
|
||
利息収入 |
|
|
542 |
|
|
|
41 |
|
支払利息 |
|
|
(2,814 |
) |
|
|
(1,415 |
) |
その他の収入 (費用) |
|
|
(5 |
) |
|
|
(11 |
) |
その他の収益(費用)の合計、純額 |
|
|
(2,277 |
) |
|
|
(1,385 |
) |
純損失 |
|
$ |
(42,220 |
) |
|
$ |
(84,409 |
) |
収益
アステラス製薬取引に関連する収益は、2022年11月に実行された2023年6月30日までの6か月間で710万ドルでした。記録された収益は、2023年前半に行われたRettの研究開発活動の結果です。
研究開発費用
研究開発費は、2022年6月30日までの6か月間の6,170万ドルに対し、2023年6月30日までの6か月間は3,230万ドルでした。2,940万ドルの減少は、2022年3月に行われた戦略的優先順位変更の取り組みの影響によるものです。その結果、研究開発、製造、その他の原材料の購入が920万ドル削減され、人員削減の結果として報酬費用が900万ドル減少しました。また、主に前臨床試験とINDを可能にする毒性学研究に関連する第三者研究開発コンサルティング費用の費用も870万ドル削減されました。これは、Rett REVEAL研究で進行中の臨床試験の取り組みに関する活動の増加によって部分的に相殺されました。
一般管理費
一般管理費は、2022年6月30日までの6か月間は2,130万ドルだったのに対し、2023年6月30日までの6か月間は1,470万ドルでした。約660万ドルの減少は、主にコンサルティング費用の削減と、一般管理報酬費用の削減によるものです。
その他の収入 (費用)
支払利息
2022年6月30日までの6か月間の支払利息は140万ドルでしたが、2023年6月30日までの6か月間の支払利息は280万ドルでした。約140万ドルの増加は主に、2023年6月30日までの6か月間のタームローンの金利が前年の比較期間と比較して高かったため、タームローン契約に基づいて発生した支払利息の増加によるものです。
流動性と資本資源
[概要]
創業以来、製品の販売による収益はなく、大きな営業損失を被っています。2023年6月30日の時点で、当社の現金および現金同等物は4,510万ドルでした。私たちは主にエクイティ・ファイナンシングを通じて事業資金を調達し、エクイティ・ファイナンスから総収入4億3,900万ドルを調達してきました。これには、IPO、売買契約に基づく普通株式の売却、2022年10月の追加募集、普通株式および転換優先株の私募増資、シリコンバレー銀行との融資契約、アステラス取引などが含まれます。具体的には、2020年3月から7月にかけて、シリーズA転換優先株式の合計1,000万株を総収入3,000万ドルで売却しました。2020年7月と8月に、シリーズBの合計5,647,048株の売却を完了しました
57
総収入9,600万ドルの転換優先株です。2020年9月、私たちは新規株式公開で総収入1億8,100万ドルを調達しました。
2021年8月12日、または締切日に、私たちは随時貸し手当事者と、または貸し手の管理代理人および担保代理人である貸し手とシリコンバレー銀行、またはその代理人と、貸付および担保契約、またはタームローン契約を締結しました。タームローン契約は、(i) 2021年12月31日までに利用可能なタームローンの元本総額4,000万ドル、(ii) 2022年1月1日から2022年9月30日まで、抽選時に代理人の裁量で決定された3つの明確でアクティブな臨床段階プログラムを実施した場合に、会社のオプションでさらに2,000万ドルのタームローンファシリティを利用できるようにすることを規定しています。(iii) 2022年10月1日から2022年10月1日まで 2023年3月31日、当社のオプションでさらに2,000万ドルのタームローンファシリティが利用可能になりました実施中の臨床段階のプログラムは、抽選時に代理人の裁量により決定され、(iv) 2023年4月1日から2023年12月31日までの間に、代理人と貸し手の承認を得て、さらに2,000万ドルのタームローンファシリティ、または総称してタームローンが利用可能になります。締切日に3,000万ドルのタームローンを引き出し、2021年12月29日にさらに1,000万ドルのタームローンを引き出しました。2022年9月30日と2023年3月31日の有効期限が切れる前に、追加の2,000万ドルのトランシェを引き出していませんでした。ローン返済スケジュールでは、2024年8月31日までの利息のみの支払いと、その後に元本と利息を毎月連続して支払うことが定められています。各タームローンに関する未払いの元本、未払利息、未払利息はすべて、2026年8月1日に全額支払う必要があります。
2021年10月5日、当社は、普通株式、優先株式、負債証券、新株予約権およびユニット、またはそれらの任意の組み合わせの募集価格の総額が3億5,000万ドルを上限とする登録に関する登録届出書をフォームS-3でSECに提出しました。また、同時に、SVB Leerink LLCおよびWells Fargo Securities、LLC、または販売代理店と販売契約または販売契約を締結しました。これに基づいて、当社の裁量により、販売代理店を通じて総額が最大1億5,000万ドルの当社の普通株式を随時発行および売却することができます。2022年3月、私たちは、とりわけゴールドマン・サックス・アンド・カンパニーを含むように販売契約を修正しました。追加の販売代理店としてのLLC。当社の普通株式はすべて、フォームS-3(ファイル番号333-260069)の棚上げ登録届出書、またはSECが2021年10月14日に発効を宣言した本棚登録届出書に従って発行されます。ただし、当社による本棚登録届出書の使用は、当社が本棚登録届出書に基づいて売却できる金額を制限するフォームS-3の一般指示I.B.6に従う限り制限されます。販売契約。2022年4月、私たちは売却契約に基づいて200万株の普通株式を売却し、1,160万ドルの純収益を受け取りました。2023年6月30日現在、売買契約に基づいて他の普通株式が発行および売却されていません。
2022年10月21日、アステラス製薬とオプション契約を締結し、TSHA-120およびTSHA-102に関連する独占的、全世界的、ロイヤリティおよびマイルストーンを含む権利とライセンスを取得する独占オプションをアステラスに付与しました。オプション契約に基づいてアステラスに付与された権利の一部対価として、アステラス製薬は、2022年11月に2,000万ドルの一括払い、つまり前払いを支払いました。
また、2022年10月21日に、アステラス製薬と有価証券購入契約を締結し、アステラス製薬に対し、当社の普通株式7,266,342株または私募株式を総額約3,000万ドルで私募または私募で発行して売却することに合意しました。私募は2022年10月24日に終了しました。有価証券購入契約に基づき、私募に関連して、アステラス製薬は、議決権のないオブザーバーとして取締役会の全会議に出席する個人を1人指名する権利を有します。また、私募株式に関する特定の登録権をアステラスに付与しました。
2022年10月26日、当社は、フォームS-3の有効な登録届出書および関連する目論見書および目論見書の補足に基づき、引受公募により14,000,000株の普通株式(額面価格1株あたり0.00001ドル)を発行および売却する引受契約を締結しました。一般への公開価格は1株あたり2.00ドルで、引受人は引受契約に従って1株あたり1.88ドルの価格で当社から株式を購入しました。さらに、引受会社に30日間、当社の普通株式を最大210万株まで追加購入するオプションを付与しました。フォローオンオファリングは2022年10月31日に終了し、引受割引、手数料、およびオファリング費用を差し引いた純収益2,600万ドルを受け取りました。2022年11月10日、引受人は当社の普通株式765,226株を追加購入するオプションを行使し、引受割引と手数料を差し引いた純収入は140万ドルでした。
2023年4月、私たちはSSI Strategy Holdings LLC(SSI)(その中に名前が付けられているSSI)の2つの関連会社またはSSI投資家と証券購入契約(SSI証券購入契約)を締結しました。これに基づいて、SSI投資家に私募またはSSI私募で当社の普通株式705,218株、またはSSI株とワラントを発行して売却することに合意しました。またはSSIワラント、つまり当社の普通株式を合計525,000株購入するワラント、またはワラント株式。SSIは会社に特定のコンサルティングサービスを提供しています。各SSIワラントの行使価格は、ワラント1株あたり0.7090ドルです。これは、2023年4月4日のナスダックグローバルマーケットにおける当社の普通株式の終値です。SSI私募で発行されるSSIワラントは、SSIワラントの保有者は、SSIワラントの達成まで、SSIワラントのいかなる部分も行使する権利を有しないと規定しています
58
当社の臨床プログラムに関連する特定の臨床上および規制上のマイルストーン。SSI私募は2023年4月5日に終了しました。SSIの私募の総収入は50万ドルでした。
2023年8月、私たちは特定の機関投資家やその他の認定投資家、または購入者と証券購入契約、または2023年8月の証券購入契約を締結し、これに基づいて私募取引、または2023年8月の私募で、(i)122,412,376株の普通株式または(ii)特定の購入に関して(ii)特定の購入に関して ASER、44,250,978株の普通株式を購入するための事前資金付ワラント、または株式の代わりに事前資金付きワラント。購入価格は1株あたり0.90ドル、または購入価格と事前資金付きワラントの購入価格は、購入価格から事前資金付きワラントあたり0.001ドルを引いたものです。私募のクロージングは、慣習的なクロージング条件に従い、2023年8月16日またはそれ以前に行われる予定です。総収入は、当社が支払うプレースメントエージェントの手数料と募集費用を差し引いた後、約1億5000万ドルになると予想されます。
資金要件
これまでのところ、承認された医薬品の商業販売による収益は得られておらず、少なくとも今後数年間は大きな収益を生み出すとは考えていません。製品候補の開発をタイムリーに完了できなかったり、規制当局の承認を得られなかったりすると、将来の収益を生み出す能力が損なわれます。製品候補からいつ、または収益を生み出すかどうかはわかりませんし、製品候補の規制当局の承認を得て商品化しない限り、大きな収益を生み出すことは期待できません。プログラムの優先順位付けと人員削減の結果、2021年から2022年にかけて経費が減少しました。RettとGANの開発に焦点を当てた戦略的パイプラインの優先順位付けイニシアチブにより、2023年には2022年のレベルと比較して支出がさらに削減されると予想しています。製品候補の承認を得た場合、販売、マーケティング、製造、流通に関連する多額の商品化費用が発生すると予想されます。継続的な事業に関連して、多額の追加資金が必要になると予想しています。必要なときに、または魅力的な条件で資金を調達できない場合、研究開発プログラムや将来の商業化の取り組みを延期、削減、または廃止せざるを得ない可能性があります。
2023年6月30日の時点で、当社のキャンセル不可の機器、実験室スペース、オフィススペースのリース支払い総額は3,370万ドルでした。これらのリースは、「パートI — 財務情報、項目1」にある未監査の要約連結財務諸表の注記4にさらに詳しく説明されています。フォーム10-Qのこの四半期報告書の「財務諸表」。私たちの最も重要な購入契約は、CROやその他の臨床試験ベンダーに対する約1,560万ドルの取り消し可能な購入義務です。
2023年8月の私募による予想純収入を含む、既存の現金および現金同等物により、2025年第3四半期までの営業費用と資本要件の資金を調達できると考えています。製品候補の研究開発、製造活動、プログラムの商業化前の活動、運転資金、一般的な企業目的のために、追加の資本が必要になります。将来の義務を果たす能力の評価は、本質的に判断に基づくものであり、主観的であり、変更される可能性があります。現在の予測に基づくと、これらの要約連結財務諸表の発行後、今後12か月間、計画された事業を維持するのに十分な現金があると考えています。ただし、予測には内在する不確実性を考慮して、これらの要約連結財務諸表が発行された日時点で既知の、または合理的にわかっている量的要因と質的要因の両方を考慮した結果、全体として継続企業として存続する能力について大きな疑念を抱く状況が存在すると結論付けました。
生物学的製剤の研究、開発、商業化には数多くのリスクと不確実性が伴うため、必要な運転資金の正確な金額を見積もることはできません。私たちの将来の資金調達要件は、以下を含むがこれらに限定されない多くの要因に左右されます。
59
潜在的な製品候補を特定し、前臨床試験や臨床試験を実施することは、時間と費用がかかり、不確実なプロセスであり、完了するまでに何年もかかります。また、販売承認を得て製品販売を達成するために必要なデータや結果が得られない可能性があります。さらに、当社の製品候補は、承認されても、商業的に成功しない可能性があります。私たちの商業収益は、もしあれば、短期的には市販されないと予想される製品候補の販売から得られます。したがって、事業目標を達成するためには、引き続き追加の資金調達に頼る必要があります。十分な追加融資が、受け入れ可能な条件では提供されない場合もあれば、まったく提供されない場合もあります。株式または転換社債証券の売却を通じて追加の資本を調達する場合、これらの株式またはこの負債の条件により、当社の事業能力が制限される可能性があります。タームローン契約には、負債、先取特権投資、合併、処分、その他の債務の前払い、配当、その他の分配に関する制限など、否定的な契約が含まれています。将来的に追加の債務融資やエクイティファイナンス(可能な場合)には、追加債務の発生、設備投資、利益分配やその他の取り決めの締結、配当の申告など、特定の措置を講じる当社の能力を制限および制限する規約を含む契約が含まれる場合があります。第三者とのコラボレーション、戦略的提携、マーケティング、流通、ライセンス契約を通じて追加の資金を調達した場合、当社の技術、将来の収益源、研究プログラム、または製品候補に対する貴重な権利を放棄したり、当社にとって有利ではない条件でライセンスを付与したりする必要がある場合があります。
キャッシュフロー
次の表は、2023年6月30日および2022年6月30日に終了した6か月間のキャッシュフロー(千単位)の概要を示しています。
|
|
6 か月間 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
営業活動に使用された純現金 |
|
$ |
(38,952 |
) |
|
$ |
(74,292 |
) |
投資活動に使用された純現金 |
|
|
(3,852 |
) |
|
|
(19,540 |
) |
財務活動による純現金 |
|
|
7 |
|
|
|
10,968 |
|
現金、現金同等物および制限付現金の純変動額 |
|
$ |
(42,797 |
) |
|
$ |
(82,864 |
) |
営業活動
2023年6月30日までの6か月間、営業活動に使用された3,900万ドルの純現金は、主に研究開発費への支出に起因する4,220万ドルの純損失でした。4,220万ドルの純損失は、主に株式ベースの報酬費用390万ドルと、TSHA-102第1/2相REVEAL試験における最初の成人患者の投薬に関連するAbeonaへの350万ドルのマイルストーンライセンス料など、現金以外の項目の900万ドルの調整によって相殺されました。営業活動に使用された710万ドルの追加現金は、繰延収益の減少によるものです。
2022年6月30日までの6か月間、営業活動に使用された7,430万ドルの純現金は、主に研究開発費への支出に起因する8,440万ドルの純損失でした。8,440万ドルの純損失は、現金以外の項目、主に株式ベースの報酬と1,000万ドルの減価償却費の調整によって一部相殺されました。
投資活動
2023年6月30日までの6か月間、投資活動には390万ドルの現金が使用されました。これは主に、社内製造施設プロジェクトの完了に関連する資本支出によるものです。2022年6月30日までの6か月間、投資活動には1,950万ドルの現金が使用されました。これは主に、CLN1契約に基づいてAbeonaに支払われた300万ドルの規制上のマイルストーン支払いと、当社の社内製造施設に関連する1,630万ドルの資本支出によるものです。
60
資金調達活動
2023年6月30日までの6か月間に、資金調達活動によって提供された現金は10万ドル未満でしたが、これは主にSSI戦略の私募による収益によるものですが、棚登録費用の支払いやその他の資金調達取引によって一部相殺されました。2022年6月30日までの6か月間に、資金調達活動により1,100万ドルの現金が提供されました。これは主に、売却契約に基づく200万株の普通株式の売却による1,160万ドルの純収入と、30万ドルのESPP拠出によるものです。収益は、棚登録費用の支払いやその他の資金調達取引によって一部相殺されました。
オフバランスシートアレンジメント
提示された期間には、SECの規則および規制で定義されているように、貸借対照表外の取り決めはありませんでした。また、現在もありません。
重要な会計方針と重要な判断と見積もり
2023年3月28日にSECに提出された2022年12月31日に終了した年度の監査済み連結財務諸表に開示されている当社の重要な会計方針に重大な変更はありませんでした。
最近の会計上の宣言
「パートI — 財務情報」、項目1にある未監査の要約連結財務諸表の注記2を参照してください。フォーム10-Qのこの四半期報告書の「財務諸表」には、当社の要約連結財務諸表に適用される最近の会計発表の説明が記載されています。
新興成長企業と小規模報告会社の状況
2012年4月、2012年のジャンプスタート・アワ・ビジネス・スタートアップ法、またはJOBS法が制定されました。JOBS法の第107条では、「新興成長企業」は、改正された1933年の証券法のセクション7(a)(2)(B)に規定されている延長された移行期間を利用して、新規または改訂された会計基準を遵守できると規定しています。したがって、新興成長企業は、特定の会計基準が民間企業に適用されるまで、その基準の採用を延期することができます。新しい会計基準または改訂された会計基準の遵守を理由に、移行期間の延長を選択しました。そのため、これらの会計基準の採用は、民間企業に適用されるまで延期されます。
さらに、新興成長企業として、公開企業に一般的に適用される特定の縮小開示要件やその他の要件を利用する場合があります。これらの規定には以下が含まれます:
新興成長企業としての資格がなくなるまで、これらの規定を利用することがあります。当社は、新興成長企業としての資格を失います。(i) 2025年12月31日、(ii) 当社の年間総収益が12億3500万ドルを超える会計年度の最終日、(iii) SECの規則に基づき、当社が「大規模加速申告者」とみなされる日付、つまり当社の共通の市場価値のことをいいます。非関連会社が保有する株式は、6月30日現在、または(iv)過去3年間に当社が10億ドルを超える非転換社債を発行した日付を超えていますピリオド。これらの報告負担の軽減のすべてではありませんが、一部を活用することもできます。このForm 10-Qの四半期報告書や、SECへのその他の提出書類では、特定の軽減された報告要件を利用しています。したがって、ここに含まれる情報は、お客様が株式を保有する他の公開会社から入手できる情報とは異なる場合があります。
61
また、私たちは「小規模な報告会社」でもあります。つまり、非関連会社が保有する株式の市場価値が7億ドル未満で、直近に完了した会計年度における当社の年間収益は1億ドル未満でした。(i) 非関連会社が保有する当社株式の市場価値が2億5000万ドル未満、または (ii) 直近に完了した会計年度における当社の年間収益が1億ドル未満で、かつ非関連会社が保有する当社の株式の市場価値が7億ドル未満の場合、当社は引き続き小規模な報告会社であり続ける可能性があります。当社が新興成長企業でなくなった時点で小規模な報告会社であれば、小規模な報告会社が利用できる特定の開示要件の免除を引き続き頼りにする可能性があります。具体的には、小規模な報告会社として、Form 10-Kの年次報告書に監査済み財務諸表の最新2会計年度のみを表示することを選択できます。また、新興成長企業と同様に、小規模な報告会社は役員報酬に関する開示義務が軽減されます。
アイテム 3.定量的・質的e 市場リスクに関する開示
当社は、取引法の規則12b-2で定義されている小規模な報告会社であり、この項目で要求される情報を提供する必要はありません。
アイテム 4.コントロール と手順。
開示管理と手続きの評価
当社の経営陣は、最高経営責任者および最高財務責任者の参加を得て、このフォーム10-Qの対象期間の終了時点で、当社の開示管理および手続き(取引法の規則13a-15(e)および15d-15(e)で定義されている)の有効性を評価しました。このような評価に基づいて、当社の最高経営責任者および最高財務責任者は、2023年6月30日現在、当社の開示管理および手続きは、このフォーム10-Qで開示する必要のある情報が(a)報告されたことを合理的に保証するために有効であったと結論付けました SECの規則や規則で定められた期間内に、また (b) 必要な開示に関してタイムリーな決定ができるよう、最高経営責任者や最高財務責任者を含む当社の経営陣に伝えます。
財務報告に関する内部統制の変更
フォーム10-Qのこの四半期報告書の対象期間中に、取引法の規則13a-15(d)または15d-15(d)に基づく経営陣の評価で特定された財務報告に対する内部統制に、財務報告に対する当社の内部統制に重大な影響を及ぼした、または重大な影響を与える可能性がかなり高い変更はありませんでした。
内部統制の有効性に対する固有の限界
開示の管理と手順を設計および評価するにあたり、経営陣は、どのような統制と手順も、どれほど適切に設計および運用されていても、望ましい管理目標の達成について絶対的な保証ではなく、合理的な保証しか提供できないことを認識しています。さらに、開示管理と手続きの設計には、リソースの制約があり、経営陣は可能な統制と手続きの利点をコストと比較して評価する際に判断を下す必要があるという事実を反映する必要があります。最高経営責任者や最高財務責任者を含む当社の経営陣は、当社の開示管理と手続き、および財務報告に関する内部統制は、目標の達成を合理的に保証するように設計されており、合理的な保証レベルで有効であると考えています。しかし、当社の経営陣は、当社の開示管理と手続き、または財務報告に対する内部統制がすべての誤りや詐欺を防止することを期待していません。
62
パートII—その他情報
アイテム 1.リーガル 議事録。
私たちは重要な法的手続きの対象にはなりません。時々、私たちは業務から生じる請求に関連するさまざまな請求や法的手続きに関与することがあります。私たちは現在、経営陣の意見では、当社の事業に重大な悪影響を及ぼす可能性のある法的手続きの当事者ではありません。結果にかかわらず、訴訟は、弁護および和解費用、経営資源の転用、その他の要因により、当社に悪影響を及ぼす可能性があります。
アイテム 1A.リスクk ファクター。
当社の事業は、リスクや事象の影響を受けやすく、それらが発生した場合、当社の財政状態、経営成績、および有価証券の取引価格に悪影響を及ぼす可能性があります。フォーム10-Qのこの四半期報告書に記載されているその他の情報に加えて、パートIの項目1Aに記載されている要素を慎重に検討する必要があります。2023年3月28日に証券取引委員会に提出された2022年12月31日に終了した会計年度のフォーム10-Kの年次報告書の「リスク要因」。以下に説明されている以外に、そのレポートに記載されているリスク要因に重大な変更はありません。
製品候補の開発に関するリスク
当社が随時発表または公開する臨床試験の中間的な「最上位」および暫定的な結果は、より多くの患者データが入手可能になるにつれて変更される可能性があり、監査および検証手続きの対象となるため、最終データに重大な変更が生じる可能性があります。
時々、臨床試験の中間的な結果や暫定的な結果を公表することがあります。私たちが実施する可能性のある臨床試験の中間結果は、患者登録が継続し、より多くの患者データが利用可能になるにつれて、1つ以上の臨床転帰が大きく変わるリスクがあります。暫定的な結果や最終結果も、引き続き監査および検証手続きの対象となるため、最終データは、以前に公開した暫定データと大きく異なる可能性があります。そのため、最終データが入手できるまでは、中間データや暫定データを注意して確認する必要があります。たとえば、2023年8月、TSHA-102の第1/2相REVEAL試験で治療を受けた最初の成人患者からの最初の臨床観察を発表しました。ただし、これらの観察結果は、2回目の投与を受けた患者や、TSHA-102の高用量の投与を受けた患者を含め、年齢や疾患の重症度に関係なく、持続しないか、繰り返されない場合があります。最初の臨床観察も、52週目までのREVEAL試験の主要エンドポイントでの成功につながらない場合があります。暫定または中間データと最終データの違いは、当社の事業見通しを著しく損なう可能性があり、当社の普通株式の取引価格を大幅に変動させる可能性があります。
当社の普通株式の所有権に関連するリスク
現在および将来の第三者との知的財産ライセンスにおける義務を順守しないと、事業にとって重要な権利を失う可能性があります。
私たちは、製品候補の開発において、特定の特許権と専有技術のライセンス、特にUTサウスウェスタン協定やアベオナとのライセンス契約に大きく依存しています。これらのライセンス契約は、デリジェンス、開発、商品化のタイムライン、マイルストーン支払い、ロイヤリティ、保険、その他の義務を私たちに課します。当社が義務を履行しない場合、当社のライセンサーは当社のライセンスを終了する権利を有することがあります。その場合、当社は、当該ライセンサーからライセンスを受けた知的財産の対象となる製品の開発、製造、販売ができなくなり、その他の罰則が科せられる可能性があります。このような事態は、当社の事業見通しに重大な悪影響を及ぼします。たとえば、2023年7月、ハンナズ・ホープ財団から、GANの治療に関するTSHA-120に関するライセンス契約に違反しているという通知を受け取りました。私たちはそのような契約に違反していないと信じており、この紛争を解決するためにハンナズ・ホープ財団とあらゆる手段を講じるつもりです。しかし、そのような取り組みが成功する保証はありません。
当社の開発プログラムに必要となる可能性のあるその他の第三者の技術や材料のライセンスは、将来利用できなくなったり、商業的に合理的な条件で入手できなくなったり、まったく利用できなくなったりする可能性があり、当社の事業や財政状態に重大な悪影響を及ぼす可能性があります。当社は、製品候補に関連するライセンスおよびサブライセンスされた知的財産の訴訟、維持、および執行を管理していますが、当社のライセンサーや上流のライセンサーの協力を要求する場合があり、その場合はそうならない場合があります。したがって、これらの特許権の訴訟、維持、執行が当社の事業の最善の利益と一致する方法で行われるかどうかは定かではありません。私たちまたは私たちのライセンサーがそのような特許を維持しなかった場合、または私たちまたは私たちのライセンサーがそれらの特許または特許出願に対する権利を失った場合、私たちがライセンスしている権利は減少または排除され、そのようなライセンスされた権利の対象となる当社の製品候補を開発および商品化する私たちの権利に悪影響が及ぶ可能性があります。上記に加えて、当社が第三者からライセンス供与する特許権に関連するリスクは、将来当社が所有する可能性のある特許権にも当てはまります。さらに、ライセンス契約に基づく開発義務を順守しない場合、地域ごとに当該契約に関する特許権を失う可能性があり、これは世界中の当社の特許権に影響を及ぼします。
63
現在または将来のライセンス契約を終了すると、これらの契約に基づく当社の権利が減少または排除され、その結果、有利でない条件で新規または復活した契約を交渉しなければならなくなったり、重要な知的財産や技術に対する権利を含め、これらの契約に基づく当社の権利が失われる可能性があります。上記のいずれかが原因で、他の製品候補を商品化できなくなり、当社の業績や全体的な財政状態に重大な悪影響を及ぼす可能性があります。
さらに、将来私たちがライセンスする知的財産権は、第三者が所有する知的財産に基づくサブライセンスであり、場合によっては複数の階層を通じて行われることもあります。したがって、たとえ当社がライセンス契約に基づく義務をすべて遵守していても、当社のライセンサーの行動は、サブライセンスされた知的財産を使用する当社の権利に影響を与える可能性があります。当社のライセンサーまたは上流のライセンサーが、当社にサブライセンスされた権利を取得するための契約に基づく義務を遵守しなかった場合、またはそのような契約が終了または修正された場合、当社の製品候補を開発および商品化する能力が著しく損なわれる可能性があります。
当社がナスダックに適用される要件をすべて満たさず、ナスダックが当社の普通株式を上場廃止することを決定した場合、上場廃止は当社の普通株式の市場流動性に悪影響を及ぼし、当社の普通株式の市場価格が下落する可能性があります。
2023年4月25日、ナスダックから、手紙の日付より前の30営業日連続で、当社の普通株式の終値入札価格が1.00ドルを下回っていたことを知らせる手紙を受け取りました。ナスダック上場規則5810(c)(3)(A)に従い、ナスダックの入札価格要件の順守を回復するために、180暦日、つまり2023年10月23日までの初期期間が与えられています。2023年10月23日より前に、当社の普通株式の入札価格が最低10営業日連続で1.00ドル以上で取引を終えた場合、ナスダックのスタッフがナスダックの規則に従ってこの10日間の期間を延長する裁量を行使しない限り、当社は入札価格要件の遵守を取り戻します。2023年6月30日までの6か月間に、当社の取締役会および株主は、当社の普通株式の逆株式分割を実施するために、修正および改訂された定款に対する一連の代替改正を承認しました。この場合、取締役会は、株式併合比率を5対5から1対20の範囲から選択する裁量権を有します。このような株式併合および株式併合比率は、2024年の年次株主総会の前であればいつでも、取締役会の独自の裁量に委ねられます。株式併合による継続的な影響は、仮に実施されたとしても、確実には予測できません。株式併合後の当社の普通株式の1株あたりの価格は、株式併合による発行済株式数の減少に比例して上昇せず、実質的に時価総額が減少する可能性があります。また、逆分割後の株式1株あたりの市場価格が、一定期間にわたって最低入札価格1.00ドルを超えるか、超え続けるという保証はありません。
2023年8月3日、当社はナスダック上場規則5450 (b) (2) (A) またはMVLS要件に定められている、ナスダック・グローバル・セレクト市場への継続的な上場に必要な上場有価証券(MVLS)の最低市場価値(MVLS))を遵守していないことを示す書面による通知、つまりMVLS通知を新たに受け取りました。ナスダック上場規則5810(c)(3)(C)に従い、MVLS要件の遵守を取り戻すには、180暦日、つまり2024年1月30日、つまりMVLSコンプライアンス日までの期間を設けています。コンプライアンスを取り戻すには、MVLSコンプライアンス日の最低10営業日前に、MVLSのMVLSを50,000,000ドル以上で成約する必要があります。コンプライアンス日より前にMVLS要件の遵守が回復しない場合、ナスダックは当社の有価証券が上場廃止の対象であることを通知します。その際、当社は上場廃止の決定をナスダックの聴聞会に上訴することができます。
ナスダックの継続上場の基準を満たせない場合、当社の普通株式は上場廃止の対象となります。当社の普通株式の上場廃止は、とりわけ、当社の普通株式の流動性と市場価格の低下、当社の普通株式を保有または取得する意思のある投資家の数の減少、株式資金調達の能力に悪影響を及ぼす可能性がある、当社のニュースやアナリストの報道量を減らす、将来的に追加の証券を発行したり追加の資金調達を行う能力を制限するなど、当社に悪影響を与える可能性があります。さらに、ナスダックからの上場廃止は、当社の評判、ひいては当社の事業に悪影響を及ぼす可能性があります。
アイテム 2.株式の未登録売却y 有価証券および収益の使用。
(a) 最近の未登録株式の売却
2023年4月5日、当社はSSI投資家とSSI証券購入契約を締結しました。この契約に基づき、SSI投資家に当社の普通株式705,218株、またはSSI株式、ワラント、またはSSIワラントを発行して売却し、当社の普通株式を合計525,000株またはワラント株式を購入することに合意しました。SSIは特定のコンサルティングサービスを提供しています。各SSIワラントの行使価格は、ワラント1株あたり0.7090ドルです。これは、2023年4月4日のナスダックグローバルマーケットにおける当社の普通株式の終値です。SSI私募で発行されるSSIワラントは、SSIワラントの保有者は、会社の臨床プログラムに関連する特定の臨床上および規制上のマイルストーンが達成されるまで、SSIワラントのいかなる部分も行使する権利を有しないと規定しています。SSI私募は2023年4月5日に終了しました。SSIの私募の総収入は50万ドルでした。証券に基づいて当社が発行した普通株式
64
購入契約は当初、証券法に基づいて登録されていませんでした。私たちは、証券法のセクション4(a)(2)に基づく登録要件の免除に頼りました。
(b) 収益の使用
[なし]。
(c) 発行者による株式の購入
[なし]。
アイテム 3.デフォルトはn シニア証券。
該当しません。
アイテム 4.マインセーフ情報開示を試してください。
該当しません。
アイテム 5.その他 情報。
[なし]。
65
アイテム 6.E展示品。
展示物索引に記載されている展示品は、このレポートとともに提出または提出されるか、参照によりここに組み込まれます。
示す 番号 |
|
説明 |
3.1 |
|
修正および改訂された法人設立証明書(2020年9月29日に証券取引委員会に提出されたフォーム8-K(ファイル番号001-39536)の最新報告書の別紙3.1を参照して組み込まれています)。 |
3.2 |
|
修正および改訂された細則(2020年9月29日に証券取引委員会に提出されたフォーム8-K(ファイル番号001-39536)の最新報告書の別紙3.4を参照して組み込まれています)。 |
10.1*+ |
|
非従業員取締役の報酬ポリシーを修正および改訂しました. |
31.1* |
|
2002年のサーベンス・オクスリー法第302条に従って採択された、1934年の証券取引法に基づく規則13a-14 (a) および15d-14 (a) に基づく最高経営責任者の認定 |
31.2* |
|
2002年のサーベンス・オクスリー法第302条に従って採択された、1934年の証券取引法に基づく規則13a-14 (a) および15d-14 (a) に基づく最高財務責任者の認定 |
32.1# |
|
2002年のサーベンス・オクスリー法第906条に基づいて採択された、米国法第18条第1350条に基づく最高経営責任者の認定 |
32.2# |
|
2002年のサーベンス・オクスリー法第906条に基づいて採択された、米国法第18条第1350条に基づく最高財務責任者の認定 |
101.インチ |
|
XBRL インスタンスドキュメント-インスタンスドキュメントは XBRL タグがインライン XBRL ドキュメントに埋め込まれているため、インタラクティブデータファイルには表示されません。 |
101.SCH |
|
インライン XBRL タクソノミー拡張スキーマドキュメント |
101.CAL |
|
インライン XBRL タクソノミー拡張計算リンクベースドキュメント |
101.DEF |
|
インライン XBRL タクソノミー拡張定義リンクベースドキュメント |
101.LAB |
|
インライン XBRL タクソノミー拡張ラベルリンクベースドキュメント |
101.PRE |
|
インライン XBRL タクソノミー拡張プレゼンテーションリンクベースドキュメント |
104 |
|
表紙インタラクティブデータファイル(インラインXBRLとしてフォーマットされ、別紙101に含まれています)。 |
* ここに提出。
# これらの証明書は、18 U.S.C. セクション1350に従ってこの四半期報告書に添付するためにのみ提供されており、改正された1934年の証券取引法第18条の目的のために提出されたものではなく、本書の日付より前または後に作成されたかどうかにかかわらず、そのような出願における一般的な法人化文にかかわらず、参照によって登録者の提出書類に組み込むことはできません。
+ 管理契約または補償計画を示します。
66
信号トゥーレス
1934年の証券取引法の要件に従い、登録者は署名者に代わってこの報告書に正式に署名させ、正式に権限を与えられました。
|
|
タイシャ・ジーン・セラピーズ株式会社 |
|
|
|
|
|
日付:2023年8月14日 |
|
作成者: |
/s/ ショーン・ノーラン |
|
|
|
ショーン・ノーラン |
|
|
|
最高経営責任者 (最高執行役員) |
|
|
|
|
日付:2023年8月14日 |
|
作成者: |
/s/ カムラン・アラム |
|
|
|
カムランアラム |
|
|
|
最高財務責任者 (最高財務会計責任者) |
67