アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
1934年証券取引法第13条又は15(D)条に規定する四半期報告 |
本四半期末まで
あるいは…。
1934年証券取引法第13条又は15条に基づいて提出された移行報告 |
移行期になります 至れり尽くせり .
依頼書類番号:
(登録者の正確な氏名はその定款に記載)
(明またはその他の司法管轄権 会社や組織) |
(税務署の雇用主 識別番号) |
(主な行政事務室住所)(郵便番号)
登録者の電話番号、市外局番を含む:(
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
取引コード |
登録された各取引所の名称 |
登録者が(1)過去12ヶ月以内(または登録者がそのような報告の提出を要求されたより短い期間)に、1934年の証券取引法第13または15(D)節に提出されたすべての報告を提出したかどうか、および(2)このような提出要求を過去90日以内に遵守してきたかどうかを、再選択マークで示す
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
☒ |
|
ファイルマネージャを加速する |
☐ |
|
|
|
|
|
|
非加速ファイルサーバ |
☐ |
|
規模の小さい報告会社 |
|
新興成長型会社 |
|
|
|
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する☐
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)。はい、そうです
2023年8月1日までに
前向き陳述に関する注意事項
このForm 10-Q四半期報告書または四半期報告書には、リスクおよび不確定要因に関する前向きな陳述が含まれている。我々は1995年の個人証券訴訟改革法と他の連邦証券法における安全港条項に基づいてこのような前向きな声明を行った。本四半期報告では歴史的事実に関する陳述を除いて、他のすべての陳述は前向き陳述である。場合によっては、前向き陳述は、“可能”、“将”、“すべき”、“予想”、“意図”、“計画”、“予想”、“信じ”、“推定”、“予測”、“潜在”、“継続”などの用語、またはこれらの用語または他の同様の用語の否定語によって識別されることができる。これらの前向きな陳述は、以下の態様に関する陳述を含むが、これらに限定されない
2
本四半期報告中の任意の展望性陳述は私たちの未来の事件及び私たちの業務と未来の財務表現に対する現在の見方を反映し、既知と未知のリスク、不確定性とその他の要素に関連し、私たちの実際の結果、業績或いは成果はこれらの展望性陳述と明示或いは暗示する任意の未来の結果、業績或いは業績とは大きく異なるかもしれない。実際の結果が現在の予想と大きく異なることをもたらす可能性のある要素は、本四半期報告第2部分1 A項目のリスク要因および他の部分に記載された要因を含む。このような不確実性を考慮して、あなたはこのような前向きな陳述に過度に依存してはいけない。法的要求がない限り、私たちは未来に新しい情報があっても、これらの前向きな陳述を任意の理由で更新または修正する義務がない。
私たちは時々、これらの市場の潜在規模および特定の疾患の推定発病率および流行率の推定を含む、私たちの業界、一般的な商業環境、および特定の疾患に関する市場の推定、予測、および他の情報を提供するかもしれない。推定、予測、予測、市場研究或いは類似方法に基づく情報自体は不確定要素の影響を受け、実際の事件、状況或いは数字は、実際の疾病罹患率と市場規模を含み、著者らが本四半期報告で提供した情報とは大きく異なるかもしれない。他に明確な説明がない限り、私たちは報告、研究調査、研究、および市場研究会社および他の第三者によって準備された類似データ;業界、医療および一般出版物;政府データおよび同様のソースから当業界および商業情報、市場データ、流行情報および他のデータを取得し、場合によっては私たち自身の仮説および分析を適用し、これらの仮説および分析は将来的に不正確であることが証明される可能性がある。
3
私たちの業務に関するリスクの概要
私たちの業務、見通し、財務状況、および経営結果は、多くのリスクと不確実性の影響を受けており、あなたは投資決定を下す前に、本四半期報告第II部第1 A項目のリスク要因および他の部分でより全面的に説明されているように、これらのリスクおよび不確実性を認識すべきである。これらのリスクは、以下のリスクを含むことができるが、これらに限定されない
4
5
Sage治療会社
索引.索引
|
|
|
|
ページ |
第1部-財務情報 |
|
|
||
|
|
|
|
|
第1項。 |
|
財務諸表(監査なし) |
|
7 |
|
|
2023年6月30日と2022年12月31日までの簡明総合貸借対照表 |
|
7 |
|
|
2023年6月30日と2022年6月30日までの3ヶ月と6ヶ月の簡明総合経営報告書と全面赤字 |
|
8 |
|
|
2023年6月30日と2022年6月30日までの6ヶ月間簡明合併現金フロー表 |
|
9 |
|
|
2023年6月30日と2022年6月30日までの3ヶ月と6ヶ月間の株主権益変動表 |
|
10 |
|
|
簡明合併財務諸表付記 |
|
11 |
第二項です。 |
|
経営陣の財務状況と経営成果の検討と分析 |
|
32 |
第三項です。 |
|
市場リスクの定量的·定性的開示について |
|
52 |
第四項です。 |
|
制御とプログラム |
|
52 |
|
|
|
||
第2部-その他の資料 |
|
|
||
|
|
|
|
|
第1項。 |
|
法律訴訟 |
|
53 |
第1 A項。 |
|
リスク要因 |
|
53 |
五番目です。 |
|
その他の情報 |
|
95 |
第六項です。 |
|
陳列品 |
|
96 |
|
|
サイン |
|
98 |
6
最初の部分は資金調達ですAL情報
プロジェクト1.融資ALレポート
Sage治療会社とその子会社
濃縮Consolidaテッド貸借対照表
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
(未監査)
|
|
六月三十日 |
|
|
十二月三十一日 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
有価証券 |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
協同すべき関係者 |
|
|
|
|
|
|
||
流動資産総額 |
|
|
|
|
|
|
||
財産と設備、純額 |
|
|
|
|
|
|
||
制限現金 |
|
|
|
|
|
|
||
経営性資産使用権 |
|
|
|
|
|
|
||
その他長期資産 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
負債と株主権益 |
|
|
|
|
|
|
||
流動負債: |
|
|
|
|
|
|
||
売掛金 |
|
$ |
|
|
$ |
|
||
費用を計算する |
|
|
|
|
|
|
||
賃貸負債を経営し、今期の部分 |
|
|
|
|
|
|
||
流動負債総額 |
|
|
|
|
|
|
||
賃貸負債を経営し,当期分を差し引く |
|
|
|
|
|
|
||
その他負債 |
|
|
|
|
|
|
||
総負債 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
株主権益: |
|
|
|
|
|
|
||
優先株、$ |
|
|
|
|
|
|
||
普通株、$ |
|
|
|
|
|
|
||
国庫株は、原価で計算する |
|
|
( |
) |
|
|
( |
) |
追加実収資本 |
|
|
|
|
|
|
||
赤字を累計する |
|
|
( |
) |
|
|
( |
) |
その他の総合損失を累計する |
|
|
( |
) |
|
|
( |
) |
株主権益総額 |
|
|
|
|
|
|
||
総負債と株主権益 |
|
$ |
|
|
$ |
|
||
付記はこのような簡明な総合財務諸表の構成要素である。
7
Sage治療会社とその子会社
業務部簡明連結報告書損失と全面的損失
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
(未監査)
|
|
6月30日までの3ヶ月間 |
|
|
6月30日までの6ヶ月間 |
|
||||||||||
|
|
2023 |
|
|
2022 |
|
|
2023 |
|
|
2022 |
|
||||
製品収入、純額 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
協力収入 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
総収入 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
運営コストと支出: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
販売原価 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
研究開発 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
販売、一般、行政 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
総運営コストと費用 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
運営損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
利子収入,純額 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
その他の収入,純額 |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
|
|
||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
1株当たり純損失--基本損失と赤字 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
普通株式加重平均 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
総合的な損失: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
その他の総合プロジェクト: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
有価証券が収益を実現していない |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
その他総合収益(損失)合計 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
全面損失総額 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
付記はこのような簡明な総合財務諸表の構成要素である。
8
Sage治療会社とその子会社
集約状態を簡素化するキャッシュフロープロジェクト
(単位:千)
(未監査)
|
|
6月30日までの6ヶ月間 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
経営活動のキャッシュフロー |
|
|
|
|
|
|
||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
純損失を純現金に調整する |
|
|
|
|
|
|
||
株に基づく報酬費用 |
|
|
|
|
|
|
||
有価証券割増 |
|
|
( |
) |
|
|
( |
) |
有価証券の割増償却 |
|
|
( |
) |
|
|
|
|
減価償却費用 |
|
|
|
|
|
|
||
経営性資産と負債変動状況: |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
( |
) |
|
協同すべき関係者 |
|
|
( |
) |
|
|
( |
) |
その他長期資産 |
|
|
( |
) |
|
|
( |
) |
経営性資産使用権 |
|
|
|
|
|
|
||
賃貸負債を経営し、流動 |
|
|
|
|
|
|
||
非流動経営賃貸負債 |
|
|
( |
) |
|
|
( |
) |
売掛金 |
|
|
( |
) |
|
|
( |
) |
費用とその他の負債を計算すべきである |
|
|
|
|
|
|
||
経営活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
投資活動によるキャッシュフロー |
|
|
|
|
|
|
||
有価証券の販売収益と満期日 |
|
|
|
|
|
|
||
有価証券を購入する |
|
|
( |
) |
|
|
( |
) |
財産と設備を購入する |
|
|
( |
) |
|
|
( |
) |
投資活動が提供する現金純額 |
|
|
|
|
|
|
||
融資活動によるキャッシュフロー |
|
|
|
|
|
|
||
株式オプションの行使と従業員株購入の収益 |
|
|
|
|
|
|
||
制限された株の帰属に関する従業員税の支払い義務 |
|
|
( |
) |
|
|
( |
) |
融資活動が提供する現金純額 |
|
|
|
|
|
|
||
現金、現金等価物、および限定的な現金純減少 |
|
|
( |
) |
|
|
( |
) |
期初現金、現金等価物、および限定現金 |
|
|
|
|
|
|
||
期末現金、現金等価物、および制限現金 |
|
$ |
|
|
$ |
|
||
非現金経営活動を補充開示する |
|
|
|
|
|
|
||
売掛金に掲げる財産と設備の購入 |
|
$ |
|
|
$ |
|
||
付記はこのような簡明な総合財務諸表の構成要素である。
9
Sage治療会社とその子会社
中国の簡明合併報告書株主権益のNES
(単位:千、共有データを除く)
(未監査)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
積算 |
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
その他の内容 |
|
|
他にも |
|
|
|
|
|
合計する |
|
||||||||
|
|
普通株 |
|
|
在庫株 |
|
|
支払い済み |
|
|
全面的に |
|
|
積算 |
|
|
株主の |
|
||||||||||||||
|
|
株 |
|
|
金額 |
|
|
株 |
|
|
金額 |
|
|
資本 |
|
|
損 |
|
|
赤字.赤字 |
|
|
権益 |
|
||||||||
2021年12月31日の残高 |
|
|
|
|
$ |
|
|
|
|
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
|||||
株式オプションを行使して普通株を発行する |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
従業員株購入計画による普通株の発行 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
売却可能証券は赤字変動を実現していない |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2022年3月31日の残高 |
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
|
|
|||||
株式オプションを行使して普通株を発行する |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
売却可能証券は赤字変動を実現していない |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
帰属制限株式単位は、従業員の納税義務を差し引いた純額 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2022年6月30日の残高 |
|
|
|
|
$ |
|
|
|
|
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
2022年12月31日の残高 |
|
|
|
|
$ |
|
|
|
|
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
|||||
株式オプションを行使して普通株を発行する |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
従業員株購入計画による普通株の発行 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
売却可能証券は赤字変動を実現していない |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
帰属制限株式単位は、従業員の納税義務を差し引いた純額 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2023年3月31日の残高 |
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
|
|
|||||
株式オプションを行使して普通株を発行する |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
売却可能証券は赤字変動を実現していない |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
帰属制限株式単位は、従業員の納税義務を差し引いた純額 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2023年6月30日の残高 |
|
|
|
|
$ |
|
|
|
|
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
|||||
付記はこのような簡明な総合財務諸表の構成要素である。
10
Sage治療会社そして付属会社
簡明総合についての注記財務諸表
(未監査)
Sage治療会社(“Sage”や“会社”)は、誰もがすくすくと成長できるように、生活を変える脳健康薬を提供することを使命とするバイオ製薬会社である。
同社の第二製品ZURZUVAE(ズラノロン)は2023年8月4日にアメリカ食品医薬品局(FDA)の許可を得て、成人産後うつ病(PPD)の治療に用いられている。そのほか、FDAは深刻な抑うつ障害(MDD)を治療するズランノドン新薬申請(NDA)に関連する完全な返信状を発表した。CRLは、NDAがズランノドンのMDD治療の承認を支持するための実質的な有効性証拠を提供しておらず、1つまたは複数の追加の臨床試験を行う必要があることを指摘している。同社はFDAのフィードバック意見を検討し、次の行動を評価している。
同社の最初の製品ZULRESSO(イブサノドン)CIV注射剤は米国で承認され,成人産後うつの治療に用いられている。同社は2019年6月に米国でZULRESSOを商業化した。同社には一連の他の候補製品があり、現在の重点は2つの重要な中枢神経系(“CNS”)受容体系、即ちGABAとNMDAを調節することである。GABA受容体ファミリーは中枢神経系における主要な抑制性神経伝達物質と考えられ,GABAを活性化することにより下流の神経や身体機能を調節するA受容器です。グルタミン酸受容体系のNMDA型受容体は中枢神経系の主要な興奮性受容体系である。これらの系統の機能障害は広範な中枢神経系疾患と関係がある。同社は現在、脳疾患と乱れを目指し、うつ病、神経学、神経精神医学の3つの分野に重点を置いている。
当社は2010年4月16日にデラウェア州法律登録により設立され、2011年1月19日に運営を開始し、名称はSerogen Biophma,Inc.である。2011年9月13日、会社はSage Treeutics,Inc.と改称された。
リスクと不確実性
同社は生物製薬業界会社に普遍的に存在するリスクと不確定要素の影響を受け、これらのリスクおよび不確定要素は、非臨床および臨床開発の各段階で候補製品を開発する関連リスク、これらの候補製品の規制機関の承認を得るための挑戦、医薬製品のマーケティングと販売に関連するリスク、第三者開発が同社の製品や候補製品と競争する可能性のある新技術革新の潜在力、キーパーソンへの依存、ノウハウを保護する挑戦、政府法規を遵守する必要性、薬物開発の高いコスト、必要に応じて運営の不確実性を追加的に得ることができるが、これらに限定されない。マクロ経済環境と地政学的事件がその発展活動、業務と財務状況に与える直接或いは間接的な影響。
同社が開発した候補製品は商業販売の前にFDAや外国規制機関の承認を得る必要がある。会社の現在と将来の候補製品が必要な承認を得ることは保証されず、会社の現在の製品ZULRESSOとZURZUVAEが必要な承認を維持する保証もない。会社が臨床開発に成功し、規制部門の承認を申請するのに十分な結果を生成できなかった場合、またはその任意の候補製品(MDDを治療するズランノケトンを含む)の承認が拒否または延期された場合、このようなイベントは、会社の業務およびその財務状況に重大な悪影響を及ぼす可能性がある。
♪the the the会社はまた、マクロ経済環境や地政学的事件の変化による追加リスクと不確実性の影響を受けている。インフレ上昇、景気後退リスク、ロシアとウクライナ間の持続的な衝突などのマクロ経済と地政学的事件により、米国と世界の金融市場は変動と中断を経験した。また、過去と潜在的な未来を含む株や信用市場が悪化すれば
11
銀行.銀行失敗すれば、将来の任意の債務や株式融資をより有利な条件で得ることが困難になり、既存の株主により大きな希釈をもたらす可能性がある。同社は現在、それらの協力者、従業員、サプライヤー、契約製造業者および/またはサプライヤーがこれらのイベントの負の影響をどの程度受ける可能性があるかを予測することができない。
経営を続ける企業
会計基準更新(“ASU”)2014-15号によると、財務諸表の列報−継続経営−(小主題205-40)、当社は、財務諸表の発行日から1年以内に満了するので、条件および/またはイベントが将来の財務義務を履行する能力を有するかどうかを評価する責任があります。会社設立以来、純収益#ドルを除いて、毎年経営赤字とマイナスキャッシュフローが発生している
会社は,その現在の運営計画に基づき,会社の既存の現金,現金等価物,有価証券が現在計画している運営に資金を提供するのに十分であると予想しているこの等の審査されていない中期簡明総合財務諸表(“簡明総合財務諸表”)が提出された日から少なくとも12ヶ月以内であるその後のある時点で、同社は将来の運営に資金を提供するための追加融資が必要になると予想している。会社が現在または将来の運営計画に十分な資金を持っていると信じていても、市場条件が有利または他の戦略的考慮を考慮すれば、企業は追加資本の調達を求める可能性がある。
以下は、これらの簡明な連結財務諸表を作成する際に従う主な会計政策の概要である。
陳述の基礎
本稿に含まれる会社の簡明総合財務諸表は、米国証券取引委員会の規則と規定に基づいて作成されている。米国公認会計原則(“GAAP”)に従って作成された財務諸表に一般的に含まれるいくつかの情報および脚注開示は、このような規則および法規によって許可される場合には、本報告で濃縮または省略されている。したがって、これらの簡明な総合財務諸表は、会社の2022年12月31日現在の年次報告Form 10-Kに含まれる2022年12月31日現在、2022年12月31日現在の年度監査された総合財務諸表と共に読まれなければならない。
簡明総合財務諸表は、審査された総合財務諸表と同じ基準で作成される。当社経営陣は、添付されている簡明総合財務諸表には、2023年6月30日までの財務状況、2023年6月30日と2022年6月30日までの3ヶ月と6ヶ月の経営業績と全面赤字、2023年6月30日と2022年6月30日までの6ヶ月の現金流量および2023年6月30日と2022年6月30日までの現金流量変動表を公平に反映するための正常経常的調整のみが含まれているとしている
12
2023年6月30日と2022年6月30日までの3ヶ月と6ヶ月の株主権益。2022年12月31日現在の総合貸借対照表は、監査された財務諸表から来ているが、GAAP要求のすべての開示は含まれていない。2023年6月30日までの3カ月と6カ月の業績必ずしも2023年12月31日までの年度または今後のどの時期の業績を代表するとは限らない。
合併原則
簡明な連結財務諸表には、付記2に開示された当社及びその完全子会社の勘定が含まれている重要会計政策の概要2022年12月31日までの年次報告Form 10−Kに添付されている“連結財務諸表付記”には,それは.会社間口座と取引はキャンセルされた。
予算の使用
公認会計基準に基づいて簡明な連結財務諸表を作成し、管理層に、連結財務諸表の日付における資産及び負債額、又は有資産及び負債の開示、並びに報告期間内の収入及び費用の報告金額に影響するように推定及び仮定を行うことを要求する.
研究と開発コストと対策プロジェクト
研究開発費には、賃金と福祉、間接費用、減価償却、契約サービス、その他の関連コストが含まれる研究開発活動を行うことによるコストが含まれる。研究·開発コストは関連債務が発生した場合に運営費用を計上する。
当社はすでに米国国内外の研究機関や他社と様々な研究·開発契約を締結しています。これらの協定は通常廃止でき、関連コストは発生時に研究や開発費用に計上されています。同社は想定されている持続研究開発コストの計上項目を記録している。これらの契約の請求書条項が作業完了時間と一致しない場合、会社は、報告期間の終了までにこれらの第三者に対する未済債務を推定しなければならない。どの計算すべき推定も一連の要素に基づいており、会社の研究と開発活動の進展状況に対する理解、契約項の下でこれまでの領収書発行状況、研究機関或いはその他の会社がこの期間に発生した領収書を発行していないいかなる実際のコストについてのコミュニケーション、及び契約に含まれるコストを含む。いずれの報告期間終了時の当計残高を決定する際には,重大な判断と見積もりを行う。実際の結果は同社の見積もりとは異なる可能性がある。当社が作成した歴史計算制見積もりと実際のコストに大きな差はありません。
収入確認
当社はZULRESSOの売却および会社提携と供給契約から収入を得ており、ZULRESSOは2019年3月にFDAの承認を得ており、当社はその後2019年6月に販売を開始します。これまで,協力協定の収入は,会社の協力者に割り当てられた知的財産権ライセンスの初期前払いからであり,供給プロトコルに基づいて臨床試験に材料を提供してきた。
会計基準編纂(ASC)主題606では、取引先と契約した収入(“特別テーマ606”)、約束された貨物またはサービスの制御権を顧客に譲渡することによって義務を履行する場合、または義務を履行する際に、エンティティは、エンティティがこれらの貨物またはサービス交換から得られると予想される対価格を反映する収入を確認する。エンティティが主題606の範囲内のスケジュールを決定する収入確認を決定するために、エンティティは、(I)クライアントとの契約を識別するステップ(S)、(Ii)契約内の履行義務を識別するステップ、(Iii)可変対価格(ある場合)を含む取引価格を決定するステップ、(Iv)契約内の履行義務に取引価格を割り当てるステップ、および(V)エンティティが履行義務を履行するとき(または履行義務として)収入を確認する5つのステップを実行する。顧客によって適宜行使可能な追加の商品またはサービスを含む権利の配置は、一般に代替方法と考えられる。同社はこれらの選択肢が提供されているかどうかを評価しています
13
顧客の物質的権利は、もしそうであれば、それらは義務を履行するとみなされる。会計目的のために、物質的権利の行使は、契約の修正または契約の継続と見なすことができる。
特定テーマ606の範囲に属すると判定された契約について、当社は、契約を履行する義務であるかどうかを決定するために、各契約において約束された貨物またはサービスが異なるかどうかを評価する。このような評価は、管理層が個別に約束された貨物またはサービス、およびこれらの貨物またはサービスが契約関係の他の態様とは別に判断できるかどうかを判断することを要求する主観的決定に関するものである。約束された貨物およびサービスは、(1)顧客が単独で、または顧客がいつでも利用可能な他のリソースと共に貨物またはサービスから利益を得ることができること、および(2)エンティティが貨物またはサービスを顧客に譲渡する約束を契約内の他の約束とは別に識別することができることを前提としている。
会社は、約束された貨物またはサービスと交換するために取引価格(顧客から得られた対価格金額を期待する権利があると予想される)を履行義務ごとに割り当て、各履行義務を履行する際に関連収入を確認する。各契約に対する会社の取引価格の推定には、会社が獲得する権利があると予想されるすべての可変対価格が含まれている。
協力とライセンス収入
主題606によって制約された協調または許可スケジュールの評価において約束された商品またはサービスが異なるかどうかを評価する際に、会社は、協力パートナーの研究、製造および商業化能力、および関連する専門知識の一般市場における利用可能性などの要因を考慮する。約束された貨物またはサービスが契約中の他の約束とは別に識別できるかどうかを評価する際に、会社はまた、契約の予想利益を考慮する。約束された商品またはサービスがユニークでない場合、会社は、異なる商品またはサービスを識別するまで、その商品またはサービスを他の約束された商品またはサービスと統合することを要求される。
そして、取引価格が決定され、SSP基準に対する独立販売価格(“SSP”)に従って確認された履行義務に割り当てられる。SSPは契約開始時に決定され、契約開始から義務履行までの変化を反映するように更新されない。履行義務を決定するSSPは重大な判断が必要である。履行義務のSSPを策定する際には,会社は適用される市場条件と関連する実体特定要因を考慮し,顧客と合意を交渉する際に考慮する要因と見積もりコストを含む。場合によっては、独立販売価格が高度に可変または不確定と考えられる場合、会社は、商品またはサービスのSSPを決定するために残差法を適用することができる。当社は,SSPを決定するためのキー仮説の変化が複数の履行義務間の手配が価格の割当てに大きな影響を与えるかどうかを評価することで,SSPの履行義務を確認した。
契約で約束された対価格に可変金額が含まれている場合、会社は、約束された貨物またはサービスを顧客に移転するために、獲得する権利のある対価格金額を推定する。当社は期待値法または最大可能値法を用いて可変対価金額を決定します。同社は制限されない推定可変対価金額を取引価格に計上している。取引価格に含まれる金額は,確認された累積収入が大きく逆転しない可能性の高い金額に制限される.その後報告期間ごとに終了すると,当社は取引価格に含まれる推定変動コストおよび任意の関連制限を再評価し,必要に応じて全体の取引価格の推定を調整する.いずれの調整も調整期間内に累積追跡方式で記録されている.
14
開発と規制マイルストーン支払いを含めて手配すれば、当社はマイルストーンが達成可能であると考えられているかどうかを評価し、可能な金額法を用いて取引価格に含まれる金額を推定する。大きな収入逆転が起こらない可能性が高い場合、関連するマイルストーン価値は取引価格に含まれるだろう。規制承認のような会社統制または被許可者制御の範囲内でのマイルストーン支払いは、通常、これらの承認を受ける前に実現可能とは考えられない。
取引価格を決定する際に、支払時間が当社に重大な融資利益を提供していれば、当社は貨幣時間価値の影響を対価格調整する。当社は、契約に重大な融資部分があるかどうかを評価することはなく、契約開始時の予想が、ライセンサーへの支払いからライセンス者への譲渡承諾された貨物またはサービスの譲渡までの間の時間が1年以下となる。同社はShionogiとBiogenとの手配を評価し、いずれも重大な融資部分は存在しないと結論した。知的財産権ライセンス取り決めについては、販売された使用料又は販売水準に基づくマイルストーン支払いを含み、ライセンスが使用料又はマイルストーン支払いに関連する主要項目とみなされ、会社は、(I)関連販売が発生した場合、又は(Ii)使用料又はマイルストーン支払いの履行義務が履行された場合には、使用料収入及び販売のマイルストーンに基づくものであることを確認する。
そして、各履行義務がある時点または一定期間にわたって履行された場合、会社は、当該履行義務に割り当てられた取引価格の金額を収入として確認し、時間が経過するにつれて、これが産出または投入方法に基づいているとする。同社とShionogiとの協力協定の収入は,プロトコル実行時の予備前払い考慮からであり,Shionogiの臨床試験に薬物製品を供給している。同社と生物遺伝研究会社の協力協定の収入は,生物遺伝研究会社の協力協定の実行に関する予備前払い費用から来ている。もっと知りたいのは付記6を参照してください協力協定.
製品収入、純額
同社は、その簡明な連結財務諸表で製品収入を確認し、期待値法を用いて決定されたいくつかの手当や課税項目に関連する可変対価格後の純額を控除し、制御権が顧客に移行する際に、製品が顧客の所在地に納入された場合である。取引価格に含まれる金額は,確認された累積収入が大きく逆転しない可能性の高い金額に制限される.ZULRESSOのために会社が決定した唯一の履行義務は、顧客の注文が指定された場所に製品を渡すことです。同社は顧客への製品納入に関する輸送·運搬コストを販売、一般と行政費用、簡明総合経営報告書、全面赤字に計上している。もし資産の予想償却期限が
2023年6月30日と2022年12月31日まで、会社は売掛金に対応していかなる不良債権も準備しておらず、売掛金金額は大きくない。
15
会社は契約条項に基づいて報告期間内に販売された製品に関する以下の可変対価格部分の準備金と,報告期間終了時に顧客流通ルート在庫に保持されている製品見積数を記録した。必要があれば、当社は四半期ごとに見積もり数字を更新し、確定期間中に任意の重大な調整を記録します。
記憶容量に応じて料金を計算する:当社は、当社から製品を直接購入した顧客が、契約がその顧客から受け取った価格よりも低い価格で条件に合った医療機関に製品を販売することを約束したことによる割引を想定しています。顧客が会社に支払った製品価格と条件を満たした医療機関の販売価格との差額は会社から料金を徴収する。記憶容量別使用準備金には、各報告期間終了時に条件に適合する医療機関の流通チャネル在庫に販売されると予想される機器のクレジット限度額と、顧客が申請したが会社がクレジット額を発行していない場合の記憶容量別使用課金とが含まれる。
政府の税金の払い戻し:医療補助を含む政府計画によると、同社は割引義務を負わなければならない。同社は関連製品収入を確認した同時期にリベート準備金を記録し、ZULRESSO製品収入の減少を招き、その簡明総合貸借対照表の計上費用に流動負債を計上した。同社のこれらのリベートに対する負債には、受信した前の四半期にまだ支払われていないか、または請求書を受け取っていないクレームの請求、本四半期のクレーム推定、および収入が確認されたが、各報告期間の終了時にまだ流通チャネル内にある製品の将来のクレームの推定が含まれている。
貿易割引と手当:会社は、通常、その顧客にZULRESSO販売の通常の請求書割引を提供して、タイムリーに支払い、販売注文管理、データ、および流通サービスの費用を支払います。同社は顧客がこれらの割引と費用を獲得すると推定し、会社が関連収入を確認する際にZULRESSO総収入と売掛金からこれらの割引と費用を全額差し引く。
経済援助:会社は商業保険を持つ患者に自発的な経済援助計画を提供し、これらの患者は保険範囲を持ち、経済援助を許可する州に住んでいる。同社はZULRESSOの財政援助金額を見積もり、その簡明総合貸借対照表の課税費用に任意の金額を記録している。財政援助請求費用の計算は,クレーム推定数と会社が受信予定の各クレームのコストに基づいており,登録され援助を許可された患者の人口統計データを用いている。いずれの調整も、関連収入の確認と同期間に入金され、製品収入の減少を招き、計上すべき費用の一部として簡明総合貸借対照表に計上された流動負債を構築する。
製品を返品する:業界慣行と一致して、会社は製品が会社の返品政策で規定されている製品納期付近の指定期限内にあることを前提とした破損、欠陥または期限切れ製品の製品返品権を顧客に提供する。当社は、顧客が返却可能な製品販売金額を推定し、この推定を関連製品収入確認期間の収入減少と、簡明総合貸借対照表の課税費用に計上した準備金と記録している。今まで、製品のリターンは顕著ではなく、未来も顕著ではないと予想されている。
協力手配
当社は、このような手配が双方による共同経営活動に関与しているかどうかを評価するために、その協力スケジュールを分析し、これらの当事者は活動の積極的な参加者であり、このような活動の商業成功に依存する重大なリスクとリターンに直面しているため、ASCテーマ808の範囲内で、協力手配(“主題808”)。この評価は手配された全ライフサイクル内で手配当事者の責任の変化に基づいて行われる。808の範囲内に複数の要素を含む連携スケジュールについて、会社は、連携のどの要素がそうであると考えられるかを最初に決定する
16
主題808の範囲内で、および協働するどの要素がプロバイダ−クライアント関係をより反映することができるかは、主題606の範囲内である。特別テーマ808によって計算された協力手配の要素に対して、類比権威会計文献或いは合理的かつ合理的な政策選択を適用することによって、適切な確認方法を確定し、一致して適用する。主題606に基づいて入金された手配要素については、当社は上記5ステップモデルを採用し、簡明総合経営報告書と全面損失のうちに連携収入列報として手配する。
主題808の範囲内の協調スケジュールの場合、会社は、各個々の活動の性質に基づいて、協調スケジュール内の複数の活動に関連する他の参加者の対応または不足した金額を列記するために、損益表分類を評価する。共同開発および共同商業化活動のような顧客関係の支払いまたは精算ではなく、連携関係として、一定期間協力パートナーに支払う場合、研究開発費または販売、一般および管理費用として記録されるか、または協力パートナーが一定期間内に償還される場合、これらの費用行項目の減少として記録される。
公正価値計量
公正価値とは、計量日に市場参加者間の秩序ある取引において資産を売却することによって受信された価格または負債を移転して支払う価格を意味する。公正価値台帳の金融資産と負債は、次の3種類のうちの1つに分類して開示する
レベル1 |
— |
同じ資産や負債の活発な市場見積もり。 |
|
|
|
レベル2 |
— |
第1レベル価格以外の観察可能な投入、例えば、同様の資産または負債のオファー、非アクティブな市場のオファー、または資産または負債の大部分の期限の観測可能な、または観測可能な市場データによって確認される他の投入。 |
|
|
|
レベル3 |
— |
市場活動支援のない、資産または負債の公正価値に重大な影響を与える観察不可能な投入は少ないか、または全くない。 |
当社は2023年6月30日及び2022年12月31日の現金等価物及び有価証券を公正価値階層に基づいて決定し、公正価値に基づいて帳簿を作成する公正価値計量.
短期満期日はそれぞれ2023年6月30日と2022年12月31日であるため、簡明総合貸借対照表に反映された協力売掛金、売掛金、売掛金の帳簿金額はその公正価値に近いそれぞれ,である.
最近発表された会計公告
財務会計基準委員会又は他の準則作成機関が発表又は提案した会計基準は、今後ある日までに採用する必要がなく、採用時に会社の簡明総合財務諸表に実質的な影響を与えないことが予想される。
同社の現金等価物は、公正価値レベルの第1レベルおよび第2レベルに分類される。その会社の有価証券への投資は公正価値等級の第2級に分類される。
当社の有価証券の公正価値は、独立定価源から得られた価格に基づいています。付記2で述べた公正価値階層構造と一致する重要会計政策の概要さらに、価格設定サービスからの有効なオファーを有する有価証券は、主に同様の資産の観察可能な価格設定または他の市場で観察可能な投入に基づくので、第2のレベルに反映される。これらの価格設定サービスが使用する典型的な情報には、報告された取引、基準収益率、発行者利益差、入札、要約、またはキャッシュフロー推定が含まれるが、これらに限定されない
17
早期返済利差と違約率。会社はこれらのデータの合理性を確保するために検証プログラムを実行する。当社は、独立定価サービスから受け取った価格を他のソースの価格と比較することにより、これらの価格を独自の審査を行う。検証手続きが完了した後、会社は2023年6月30日と2022年12月31日まで、価格設定サービスが提供する公正な価値計量を調整またはカバーしていない。
次の表では,以下の日までの会社の現金等価物と有価証券について概説する2023年6月30日および2022年12月31日:
|
|
2023年6月30日 |
|
|||||||||||||
|
|
合計する |
|
|
引用する |
|
|
意味が重大である |
|
|
意味が重大である |
|
||||
|
|
(単位:千) |
|
|||||||||||||
現金等価物: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
||
アメリカ商業手形 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
現金等価物合計 |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|||
有価証券: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ政府証券 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
アメリカ社債 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
国際社債 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
アメリカ商業手形 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
国際商業手形 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
アメリカ預金証書 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
アメリカ市政証券 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
有価証券総額 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
|||
|
|
2022年12月31日 |
|
|||||||||||||
|
|
合計する |
|
|
引用する |
|
|
意味が重大である |
|
|
意味が重大である |
|
||||
|
|
(単位:千) |
|
|||||||||||||
現金等価物: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
||
現金等価物合計 |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
||
有価証券: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ政府証券 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
アメリカ社債 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
国際社債 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
アメリカ商業手形 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
国際商業手形 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
アメリカ預金証書 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
アメリカ市政証券 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
有価証券総額 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
|||
2023年6月30日と2022年6月30日までの6ヶ月以内いくつありますか
18
下表は以下の日までの会社有価証券の未実現損益総額をまとめたものである2023年6月30日および2022年12月31日:
|
|
2023年6月30日 |
|
|||||||||||||||||
|
|
償却する |
|
|
未実現総額 |
|
|
未実現総額 |
|
|
信用損失 |
|
|
公正価値 |
|
|||||
|
|
(単位:千) |
|
|||||||||||||||||
資産: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
アメリカ政府証券 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
— |
|
|
$ |
|
|||
アメリカ社債 |
|
|
|
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
|
|||
国際社債 |
|
|
|
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
|
|||
アメリカ商業手形 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
|
||
国際商業手形 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
|
||
アメリカ預金証書 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||
アメリカ市政証券 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
|
||
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
— |
|
|
$ |
|
|||
|
|
2022年12月31日 |
|
|||||||||||||||||
|
|
償却する |
|
|
未実現総額 |
|
|
未実現総額 |
|
|
信用損失 |
|
|
公正価値 |
|
|||||
|
|
(単位:千) |
|
|||||||||||||||||
資産: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
アメリカ政府証券 |
|
$ |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
— |
|
|
$ |
|
||
アメリカ社債 |
|
|
|
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
|
|||
国際社債 |
|
|
|
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
|
|||
アメリカ商業手形 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
|
||
国際商業手形 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
|
||
アメリカ預金証書 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||
アメリカ市政証券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
|
|||
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
— |
|
|
$ |
|
|||
以下の表に当社の有価証券の公正価値及び現在までをまとめる2023年6月30日および2022年12月31日:
|
|
2023年6月30日 |
|
|||||||||||||||||||||
|
|
12ヶ月以下です |
|
|
12ヶ月以上 |
|
|
合計する |
|
|||||||||||||||
|
|
公平である |
|
|
実現していない |
|
|
公平である |
|
|
実現していない |
|
|
公平である |
|
|
実現していない |
|
||||||
|
|
(単位:千) |
|
|||||||||||||||||||||
アメリカ政府証券 |
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|||
アメリカ社債 |
|
|
|
|
|
( |
) |
|
|
|
|
|
( |
) |
|
|
|
|
|
( |
) |
|||
国際社債 |
|
|
|
|
|
( |
) |
|
|
|
|
|
( |
) |
|
|
|
|
|
( |
) |
|||
アメリカ商業手形 |
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
( |
) |
||
国際商業手形 |
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
( |
) |
||
アメリカ市政証券 |
|
|
|
|
|
( |
) |
|
|
|
|
|
( |
) |
|
|
|
|
|
( |
) |
|||
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|||
19
|
|
2022年12月31日 |
|
|||||||||||||||||||||
|
|
12ヶ月以下です |
|
|
12ヶ月以上 |
|
|
合計する |
|
|||||||||||||||
|
|
公平である |
|
|
実現していない |
|
|
公平である |
|
|
実現していない |
|
|
公平である |
|
|
実現していない |
|
||||||
|
|
(単位:千) |
|
|||||||||||||||||||||
アメリカ政府証券 |
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|||
アメリカ社債 |
|
|
|
|
|
( |
) |
|
|
|
|
|
( |
) |
|
|
|
|
|
( |
) |
|||
国際社債 |
|
|
|
|
|
( |
) |
|
|
|
|
|
( |
) |
|
|
|
|
|
( |
) |
|||
アメリカ商業手形 |
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
( |
) |
||
国際商業手形 |
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
( |
) |
||
アメリカ市政証券 |
|
|
|
|
|
( |
) |
|
|
|
|
|
( |
) |
|
|
|
|
|
( |
) |
|||
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|||
2023年6月30日と2022年12月31日現在、米国政府証券、米国社債、国際社債への会社の投資の未実現損失は利上げによるものである。その会社はその額面に対する割増価格でこれらの投資を購入した。現在の信用格付けはすべて当社の投資政策のガイドラインに合致しており、当社は発行者が投資の余剰コスト基準を下回る価格でいかなる証券も決済しないと予想している。当社は当該等の投資を売却するつもりはなく、その償却コスト基準を回収する前に当該等の投資を売却することを当社に要求する可能性も低い。
2023年6月30日まで会社が保有するすべての有価証券の残り契約満期日は
2022年12月31日まで会社が保有するすべての有価証券の残り契約満期日は
残りの契約満期日が1年を超える有価証券を含むすべての有価証券は、貸借対照表上で流動資産に分類され、販売可能とされているので、会社はそれらを現金に変換し、現在の業務に資金を提供することができる.
あったことがある
下表でまとめたところ2023年6月30日および2022年12月31日:
|
|
六月三十日 |
|
|
十二月三十一日 |
|
||
|
|
2023 |
|
|
2022 |
|
||
|
|
(単位:千) |
|
|||||
研究と開発コストを計算すべきである |
|
$ |
|
|
$ |
|
||
従業員と関係がある |
|
|
|
|
|
|
||
専門サービス |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
|
|
$ |
|
|
$ |
|
||
20
賃貸借契約を経営する
その会社はオフィススペースと特定の設備を借りている。簡明総合貸借対照表に記録されているすべてのレンタルは経営的リースである。同社の賃貸借契約の余剰賃貸借契約条項から
同社は2022年1月1日現在、マサチューセッツ州ケンブリッジ市の2棟のマルチテナントビルでオフィススペースをレンタルしている
許可協定
CyDexライセンスプロトコル
2015年9月、当社はLigand PharmPharmticals Inc.の完全子会社CyDex PharmPharmticals,Inc.(“CyDex”)との既存の商業許可協定を改訂し、再説明した。改訂および再記載された商業許可協定条項によれば、CyDexは、ブレサノドンおよび同社がSAGE-689と呼ばれる化合物を含む医薬製品を製造し、生成された製品をヒトまたは動物の任意の疾患または症状の治療、予防または診断のために開発および商業化するために、CyDexのCaptisol医薬製剤技術および関連知的財産権の独占的ライセンスを付与しているが、(I)ホルモンを含む製剤で任意の疾患または状態を治療する;(Ii)炎症状態を局所的に治療する;(Iii)ヒト真菌感染を治療および予防する;(Iv)網膜変性眼科治療もあります同社はブシャノロンの販売についてCyDexに特許使用料を支払うことを要求され、将来このような候補製品の開発に成功すれば、任意のSAGE-689の販売に特許権使用料を支払うことも要求される。純売上高水準により、特許権使用料は低い1桁になっている。協定の発効日から2023年6月30日まで会社はCyDexに$を支払いました
CyDexとの改訂と再記述の許可協定によると、同社は臨床開発と規制のマイルストーンを実現するために記念碑的なお金を支払うことに同意し、金額は最高で$に達する
2023年6月30日まで2022年6月30日まで6カ月その会社はできました
カリフォルニア大学許可協定
はい二零一三年十月、当社はカリフォルニア大学取締役会(“取締役”)と非独占許可協定を締結し、この合意に基づいて、当社はブシャノンに関するいくつかの臨床データ及び臨床材料の非独占許可を付与し、バイオ製薬製品の開発及び商業化に使用する
21
はい許可領域には,てんかん重積状態や出産後うつ病が含まれている。2014年5月,ライセンス契約が改正され,合意の許可使用分野,材料,マイルストーン費用条項に特発性振戦の治療内容が追加された。同社は麗晶社の臨床開発マイルストーンに$を支払った
2015年6月、当社は麗晶社と独占ライセンス契約を締結し、これにより、当社はプロゲステロンによる各種疾患の治療に関するいくつかの特許権の独占許可を取得した。この許可証と交換するために、同社は#ドルを前払いした
2023年6月30日まで2022年6月30日まで6カ月その会社はできました
ヒノキ
2018年6月、会社はShionogiと戦略的協力を結び、日本、台湾、韓国(Shionogi領土)でMDDや他の潜在的適応を治療するズランノドンを開発し、商業化した。2018年10月、当社はShionogiと当社がShionogiにズランノロン臨床材料を供給することについて供給協定を締結した。
協力協定の条項によれば、ShionogiはShionogi地域MDDおよび他の適応におけるZuranoloneのすべての臨床開発および規制文書を担当し、ZuranoloneがShionogi地域での商業化を担当し、Zuranoloneが開発に成功し、Shionogi地域内の任意の国でマーケティング許可を得た場合、Zuranoloneを担当する。Shionogiは会社に#ドルの前払いを要求されました
同社の結論は、Shionogiは、当事者がリスクとリターンを共同で分担していない地域を支援するために、Zuranolone計画に知的財産権とノウハウを提供しているため、顧客の定義に適合している。また、当社はShionogiの協力が契約入金としての要求に適合していると認定し、当社がShionogiに渡す商品やサービスと引き換えに、当社が獲得する権利のある対価格を徴収する可能性が高いことを含む。
22
当社は,Shionogi協力協定における履行義務には,スラノロンへの許可の配布と,有効な薬物成分(“原料薬”)の供給を含むいくつかの材料の臨床開発段階での供給が含まれていることを確認した。Zuranoloneライセンスに関する履行義務は,他の履行義務とは異なることが決定されているため,その制御権は署名時に移行する単独の履行義務である。開発中に使用された原料薬を含む特定の臨床材料を提供する義務は、単独の履行義務として決定された。Shionogiが最低金額または数量の商業原料薬を購入する義務がないことを考慮して、Shionogiに商業用途のための原料薬を供給することは、契約開始時の当社の履行義務ではなく、Shionogiの選択として決定され、契約を行使する際に入金される。同社はまた,予想価格が割引されていないため,商業用途原料薬供給に関する単独材料権利がないことを確認した。この事実モデルに鑑み、当社はこの合意には2つの履行義務があると結論した。
臨床供給協定によると、当社はShionogiに(I)Shionogiを製造し、(I)Shionogiが協力及び許可協定に従ってShionogi地域の許可製品を開発するために合理的に必要な臨床数量の原料薬を供給する責任があり、及び(Ii)Shionogiは協力及び許可協定に従ってShionogi地域でズランノロンの第1期臨床試験に使用する際に合理的に必要な薬物製品の数量、数量は双方が協定する。$は含まれていない臨床供給プロトコルからの協調収入
会社は契約履行義務ごとの独立販売価格の評価を完了し,許可履行義務の独立販売価格を#ドルと決定した
生物遺伝研究
2020年11月,当社は生物遺伝研究協力協定を締結し,MDD,PPDなどの疾患を治療するSAGE−217製品と特発性振戦やその他の疾患を治療するSAGE−324製品を共同開発·商業化した。また、当社もBIMAと株式購入協定(“生物遺伝株式購入協定”)を締結し、この合意に基づき、BIMAは自社普通株株式を購入します。生物遺伝研究協力協定は#年に発効した
“生物遺伝研究協力協定”の条項によると、当社は、生物遺伝研究会社に、米国でSAGE-217製品およびSAGE-324製品(各製品カテゴリおよび合計“特許製品”)の開発および商業化の独占許可、および米国およびShionogi地域以外の世界のすべての国でSAGE-217製品を開発·商業化する独占許可を付与する。そして、米国を除く世界のすべての国でSAGE-324製品を開発·商業化する独占ライセンス。当社は米国以外の地域を“生物遺伝分野”と呼び、生物遺伝協力協定により、生物遺伝は適用許可製品に関する権利を持っている。
生物遺伝研究協力協定の有効性と2020年12月にBIMAへの株式売却完了について、当社は$を受け取りました
23
その会社は$までの追加支払いを受ける資格がある
薬物開発の不確実性と薬物開発の高い歴史的失敗率、および製品の発売と商業化の挑戦により、製品が承認されれば、会社は記念碑的な支払いや生物遺伝研究協力協定に従って支払ういかなる特許権使用料も受けないかもしれない。
米国での開発·商業化活動は、会社と生物遺伝会社が合意した計画に基づいて行われ、常に双方の同数の代表で構成される共同指導委員会によって監督される。会社と生物遺伝会社は,米国での開発と商業化コスト,FDA承認·製品販売開始後の利益と損失を平均的に分担するが,会社は以下に述べることから撤退する権利がある。生物遺伝会社は、生物遺伝研究分野の任意のSAGE−217製品およびSAGE−324製品の任意の開発および商業化に関連するすべての開発活動およびコストを完全に担当するであろう。上述したように、会社は生物遺伝研究分野の任意の販売から特許使用料を得るであろう。生物遺伝会社はSAGE-217製品の世界での主要かつ記録的な販売となる。同社はSAGE-324製品の米国での主要な売上と記録的な売上であり、生物遺伝会社はSAGE-324製品の生物遺伝専門区における主要かつ記録的な売上である。
同社は生物遺伝分野に原料薬と原料薬製品を提供し、米国に原料薬、原料薬、最終薬物製品を提供し、開発と商業化活動を支援する。生物遺伝は合意期間内の任意の時間に生物遺伝区域原料薬の生産責任を負担する権利があり、発効日後の合理的な時間内に、生物遺伝区域原料薬の生産責任を負う。
事前に終了しない限り、“生物遺伝的協力協定”は、(A)生物遺伝領域内の任意の国/地域において、その国/地域内のある製品カテゴリ内のすべての許可製品の印税期限が満了するまで、個々の許可製品および個々の国/地域の許可製品に基づいて継続し、(B)米国において、製品カテゴリ内のすべての許可製品の商業化を永久的に停止することに同意する。生物遺伝会社は,あらかじめ書面で通知する権利がある場合には,便宜上,製品種別や特定地域ごとに生物遺伝研究会社連携協定の全内容を終了する権利がある。同社は撤退を選択する権利があり、米国での共同独占ライセンスを製品別に製品別の生物遺伝会社の独占ライセンスに変換する権利がある。選択脱退権を行使した後、同社は米国の利益と損失を平均的に共有することはなく、10代から20歳以下の割合の特定の特許権使用料支払いと追加販売マイルストーンを得る権利があるだろう。
当社の結論は、Biogen協力協定とBiogen株式購入協定を統合し、単一の手配として扱うべきであり、この2つの合意は同時に締結され、相互に考慮されているからである。同社は、合併後の合意の内容が特別テーマ606および特別テーマ808の範囲に属すると判断した。
発効日まで、会社は“生物遺伝研究協力協定”において、主題606の範囲内で評価する次の約束を決定した:(I)米国でSAGE-217製品の共同独占許可を提供する;(Ii)生物遺伝研究分野でSAGE-217製品の独占許可を提供する;(Iii)米国でSAGE-324製品の独占許可を提供する;(Iv)生物遺伝研究分野でSAGE-324製品の独占許可を提供する;(V)生物遺伝研究分野でSAGE-217製品のための原料薬および原料薬の臨床製造を提供する。および(Vi)生物遺伝研究分野でSAGE-324製品に原料薬および原料薬の臨床生産供給を提供した。
24
同社はまた,Biogen協力協定で概説されているいくつかの選択肢が義務履行の重大な権利を代表するかどうかを評価し,これらの選択肢はいずれもBiogenに実質的な権利を伝達していないため,Biogen協力協定における単独履行義務とはみなされていないと結論した。
同社は、上述した約束を評価し、米国におけるSAGE−217製品およびSAGE−324製品の共同独占許可がサプライヤーと顧客との関係を反映していることを決定したため、主題606の範囲内の契約履行義務を代表している。米国におけるSAGE-217製品およびSAGE-324製品の共同独占許可は、契約における他の約束とは異なる機能的知的財産とみなされる。生物遺伝研究分野のSAGE-217製品およびSAGE-324製品の独占的許可は、生物遺伝が単独でまたは他のいつでも利用可能なリソースと共にこれらの許可から恩恵を受けることができるので、生物遺伝研究協力プロトコルの背景では機能許可と考えられる。米国での共同独占許可と生物遺伝分野での独占許可は同時に交付されるため,契約開始時に義務を果たすとみなされている。生物遺伝研究協力プロトコルの範囲では,生物遺伝研究分野の原料薬とSAGE−217製品とSAGE−324製品の原料薬と原料薬製品の臨床製造供給は異なると考えられ,生物遺伝研究会社は製造サービスおよび当社が合意開始時に譲渡したライセンスから利益を得ることができるからである。したがって、それぞれは、契約開始時に主題606の範囲内で顧客との契約における個別的な履行義務を表す。
同社は、米国でのSAGE-217製品の共同開発、共同商業化、およびSAGE-324製品の共同製造に関連する協力活動は、テーマ808の範囲内の単独課金単位であると考えている。会社および生物遺伝会社はいずれも開発および商業化活動の積極的な参加者であり、重大なリスクとリターンに直面しており、これは手配中の活動の開発と商業成功に依存する。2023年6月30日と2022年6月30日までの6ヶ月以内,
共同開発,共同商業化,共同製造活動に関する生物遺伝研究会社への支払いや精算,および各当事者がこれらの活動のコストを平均的に分担することに同意し,活動の性質に応じて,研究·開発費や販売,一般·行政費用の増加または減少として入金する。
2023年6月30日までの3ヶ月と6ヶ月以内に、当社は純返済を記録しました$
2023年6月30日現在、会社記録の協同応募先$
25
以下の表は,当社で発生したBiogen連携協定に関する費用と,運営費用別に反映されたBiogenまたはBiogenへの精算をまとめたものである
|
|
6月30日までの3ヶ月間 |
|
|
6月30日までの6ヶ月間 |
|
||||||||||
|
|
2023 |
|
|
2022 |
|
|
2023 |
|
|
2022 |
|
||||
|
|
(単位:千) |
|
|||||||||||||
生物遺伝研究協力協定に関する費用 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
Biogenへの純精算は |
|
|
|
|
|
|
|
|
|
|
|
|
||||
研究開発費 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
販売、一般、行政費用 |
|
|
|
|
|
( |
) |
|
|
|
|
|
( |
) |
||
|
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
生物遺伝研究連携に関する総純費用 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
同社は生物遺伝研究協力協定の開始時に主題606項目での取引価格を$と決定した
上述したように、同社は、生物遺伝研究協力協定において、(I)米国でSAGE-217製品およびSAGE-324製品の共同独占許可を交付し、生物遺伝研究分野でSAGE-217製品およびSAGE-324製品の独占許可を交付するステップと、(Ii)生物遺伝研究分野でSAGE-217製品に原料薬および原料薬の臨床製造供給を提供するステップと、(Iii)生物遺伝研究分野でSAGE-324製品に原料薬および原料薬を提供する臨床製造供給との3つの履行義務を決定した。生物遺伝研究協力協定における履行義務ごとの販売価格は,当社のSSPによって決定され,その製品が定期的に独立して販売されていれば,どのような価格で販売されるかを決定することを目的としている。同社は、製造義務に関連する可変対価を生物遺伝研究地域の将来の臨床供給のSAGE-217製品およびSAGE 324製品に割り当て、残りの固定対価は許可義務に割り当てられる。製造義務に関連した可変対価格は実質的ではない。ドルの全ては
26
収入.収入臨床製造に対する納入義務は,ある時点,すなわち生物遺伝会社に納入された場合である。
バイオ遺伝会社株式購入プロトコルの会計処理
Biogen協力協定の締結について、当社はBIMAとBiogen株購入協定を締結した。Biogen株購入契約によると,会社はBiogen株をBIMAに売却し,価格は約$である
生物遺伝研究会社の株式購入プロトコルには、あるポーズ条項、ロック制限、および生物遺伝研究会社の株に関する投票プロトコルが含まれている。Biogen株購入契約の条項によると、BIMAはすでに同意していないし、その連合会社が当社の証券を直接或いは間接的に買収しないことを招くこともなく、当社とBiogenとの間の入札或いは交換要約或いは合併を求めたり、提案したりして、任意の事項について依頼書或いは同意書を求め、或いは他の指定行動を取らなければならず、すべての場合は指定された条件に制限されなければならない
保有期間の制限を考慮して、当社はオプション定価推定モデルを用いて発行された普通株の公正価値を決定する。会社普通株の公正価値は公正価値等級中の第二級公正価値計量とされている。モデルにおいて最も重要な仮定は,会社の株価,制限の期限,および会社株の履歴変動率と隠れ変動率に基づく株価変動性である.経営陣による公正価値調整に基づき、発行済み株式の公正価値は#ドルに決定された
株式計画
2014年7月2日、株主は2014年度の株式オプションとインセンティブ計画(“2014計画”)を承認し、同社の初公募が完了する前にすぐに発効する予定だ。2014年計画では、制限株式奨励、制限株式単位、奨励的株式オプション、非法定株式オプションを付与することが規定されている。2014年計画は、会社の2011年の株式オプションと付与計画(“2011計画”)に取って代わった。会社(The Company)
2014年計画では、毎年初日に最大4%の会社が前年最終日までの普通株式流通株を増加させることが規定されている。2023年1月1日
開ける2016年12月15日、会社取締役会(“取締役会”)は、2016年度インセンティブ株式計画(改訂·再記述、“2016計画”)を承認した。2016年計画では、以前会社役員や非従業員になっていなかった個人に株式奨励を付与し、受け入れを誘導することが規定されています
27
就職する彼らに会社の所有権を提供します2018年9月20日、取締役会は2016年計画を修正し、予約発行株式総数を増加させました
株式付与の条項は、帰属要求を含み、取締役会又は取締役会の報酬委員会によって決定されるが、適用計画の規定により制限されなければならない。当社が付与した業績ベースでない株式オプションは、授与者が付与後の特定期間内に当社への継続サービスに基づいて付与されたものとされている。これらの報酬は従業員に授与される場合、通常は比例して授与されます
2023年6月30日現在、全株式計画下の未償還奨励関連株式総数は
2022年6月16日、会社株主はこれまで取締役会が承認した改訂後の2014年従業員株式購入計画修正案を承認し、増加した
限定株単位
次の表は、時間に基づく制限株式単位と業績制限株式単位に関する活動をまとめたものである
|
|
株 |
|
加重平均付与日公正価値 |
|
||
2022年12月31日現在の未返済債務 |
|
|
|
$ |
|
||
授与する |
|
|
|
$ |
|
||
既得 |
|
|
( |
) |
$ |
|
|
没収される |
|
|
( |
) |
$ |
|
|
2023年6月30日現在の未返済債務 |
|
|
|
$ |
|
||
時間に基づく制限株式単位
2021年12月31日までの年間で、当社は授与します
2020年12月31日までの年間で、当社は
2023年3月31日までの3ヶ月以内に、当社は授与します
28
周年記念次の項目の帰属開始日
2022年6月30日までの6ヶ月以内に、当社は授与する
2023年6月30日までに
業績制限株式単位
当社は2023年及び2022年6月30日までの3ヶ月以内に授与します
当社は2023年及び2022年6月30日までの6ヶ月以内に授与します
業績制限株式単位に関する株式ベースの報酬支出の確認は、業績状況が実現可能とされた場合から、マイルストーンの将来結果に関する内在的リスクや不確実性を考慮した経営陣の最適な見積もりを使用するが、マイルストーンを持つ株主総リターン評価のマイルストーンは除く。業績制限株式単位に関する株式による報酬の確認は授与日から始まり、付与された帰属結果に関係なく、マイルストーンは株主総リターンの尺度である。
2023年6月30日と2022年6月30日現在,未完成の業績制限株式単位については,株主の総リターンを測るマイルストーンを持つ単位を除いて,まだ実現されていないマイルストーンは実現不可能と考えられているため,
2023年3月31日までの3ヶ月間で
2023年6月30日までに
29
株式オプション前転出
次の表は、時間と業績に基づく株式オプションに関する活動をまとめたものである
|
|
株 |
|
|
重みをつける |
|
|
加重平均 |
|
|
骨材 |
|
||||
2022年12月31日現在の未返済債務 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
授与する |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
鍛えられた |
|
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
没収される |
|
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
2023年6月30日現在の未返済債務 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2023年6月30日から行使可能 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2023年6月30日現在、会社には未確認株式報酬支出があり、その未償還および未付与の時間ベース株式オプション奨励に関連している$
2023年6月30日および2022年6月30日までの6ヶ月以内に行使された株式オプションの内在的価値は$
業績に基づく株式オプション
業績状況が実現可能であると考えられた場合には、業績に基づく株式オプションに関する株式ベースの報酬支出の確認を開始し、マイルストーンの将来結果に関する内在的リスクや不確実性を考慮した管理層の最適な推定を用いる。
2023年6月30日と2022年6月30日までまだ完成していない業績に基づく株式オプション付与については,まだ達成されていないマイルストーンの実現は不可能であると考えられる
2023年6月30日と2022年6月30日までの6ヶ月以内会社が承認します
2023年6月30日と2022年6月30日までの6ヶ月以内,
2023年6月30日までに
株に基づく報酬費用
下表は年度確認の株式による報酬支出をまとめたものである2023年6月30日と2022年6月30日までの3ヶ月と6ヶ月:
|
|
6月30日までの3ヶ月間 |
|
|
6月30日までの6ヶ月間 |
|
||||||||||
|
|
2023 |
|
|
2022 |
|
|
2023 |
|
|
2022 |
|
||||
|
|
(単位:千) |
|
|||||||||||||
研究開発 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
販売、一般、行政 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
30
次の表は奨励タイプ別に株式ベースの報酬支出をまとめた2023年6月30日と2022年6月30日までの3ヶ月と6ヶ月:
|
|
6月30日までの3ヶ月間 |
|
|
6月30日までの6ヶ月間 |
|
||||||||||
|
|
2023 |
|
|
2022 |
|
|
2023 |
|
|
2022 |
|
||||
|
|
(単位:千) |
|
|||||||||||||
株式オプション |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
制限株式単位 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
従業員株購入計画 |
|
|
|
|
|
( |
) |
|
|
|
|
|
|
|||
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
株式オプション奨励については、公正価値は、付与日にBlack-Scholesオプション定価モデルを用いて推定されるとともに、株式オプションを付与する条項と条件を考慮して推定される。株式オプションの公正価値は、株式オプション奨励の必要なサービス期間内に従業員、非従業員取締役、非従業員顧問に直線的に償却される。
2023年6月30日及び2022年6月30日までの6ヶ月以内に、当社の株式購入計画に基づいて付与された1株当たりの株式購入権の加重平均授受日の公正価値は$
次の表に同社の1株当たり基本と償却純損失の計算を示す2023年6月30日と2022年6月30日までの3ヶ月と6ヶ月:
|
|
6月30日までの3ヶ月間 |
|
|
6月30日までの6ヶ月間 |
|
||||||||||
|
|
2023 |
|
|
2022 |
|
|
2023 |
|
|
2022 |
|
||||
1株当たりの純損失は |
|
|
|
|
|
|
|
|
|
|
|
|
||||
分子: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
分母: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
加重平均普通株式発行済み |
|
|
|
|
|
|
|
|
|
|
|
|
||||
普通株の希薄化効果 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
加重平均普通株式発行済み |
|
|
|
|
|
|
|
|
|
|
|
|
||||
1株当たり純損失--基本損失と赤字 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
下表は希釈後の1株当たり純損失を計上していない発行済み普通株等価物をまとめたものであり,これらの等価物を計上することは逆薄となるからである2023年6月30日および2022年6月30日:
|
|
6月30日までの3ヶ月間 |
|
|
6月30日までの6ヶ月間 |
|
||||||||||
|
|
2023 |
|
|
2022 |
|
|
2023 |
|
|
2022 |
|
||||
株式オプション |
|
|
|
|
|
|
|
|
|
|
|
|
||||
制限株式単位 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
従業員株購入計画 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
未償還株式オプションと制限的株式単位は、業績条件を満たしていない業績に基づく帰属基準を含み、発行された普通株等価物の計算には計上されていない。
31
プロジェクト2.経営陣の議論と分析F財務状況と経営成果
以下の財務状況および経営結果の議論および分析、および本四半期報告書10-Q表または四半期報告書の他の場所における簡明な総合財務諸表および関連注釈、ならびに2022年12月31日までの年次報告書10-K表または年次報告書に含まれる監査財務諸表および関連注釈を読まなければなりません。歴史的情報に加えて、本議論および分析は、リスク、不確実性、および仮定に関する前向きな陳述を含む。展望的陳述は将来の業績の保証ではなく、私たちの実際の運営結果、財務状況と流動性、および私たちが経営している業務と業界の発展は、本四半期報告に含まれる展望的陳述に含まれる議論や予測の結果とは大きく異なる可能性があることを想起させます。我々は、本四半期報告の他の部分において、本四半期報告第2部1 A項“リスク要因”および“前向き陳述に関する警告”を含む、これらの潜在的差異をもたらすか、または促進する可能性があると考えられるリスクおよび他の要因について議論した。また、我々の経営結果、財務状況、流動性、および当社の業務および当社が経営している業界の発展が、本四半期報告に含まれる前向きな陳述と一致していても、将来の結果や発展を予測することができない可能性がある。このような陳述は、それらの発表日の状況のみを代表するので、読者に、私たちがしたいかなる前向きな陳述にも過度に依存しないように注意しましょう。私たちは、法律および米国証券取引委員会(米国証券取引委員会)の規則が、私たちの予想またはそのような声明に基づく可能性のあるイベント、条件、または状況の任意の変化を反映するために、または実際の結果および前向き声明に記載されている内容の可能性に影響を与える可能性があるために、任意のそのような声明を公開的に更新または修正することを明確に要求しない限り、いかなる義務も負わない。
概要
私たちは生物製薬会社で、解決策を開拓し、生活を変える脳健康薬を提供し、誰もがすくすくと成長できるようにすることを使命としています。私たちの現在の目標は脳の病気と乱れで、うつ病、神経学、神経精神医学の3つの重要な重点領域がある。会社として、私たちの重点は脳の健康であり、私たちの現在の目標は2つの重要な中枢神経系、即ち中枢神経系、即ちGABAとNMDAである。GABA受容体ファミリーは中枢神経系における主要な抑制性神経伝達物質と考えられ,GABAを活性化することにより下流の神経や身体機能を調節するA受容器です。グルタミン酸受容体系のNMDA型受容体は中枢神経系の主要な興奮性受容体系である。これらの系統の機能障害は広範な中枢神経系疾患と関係がある。
32
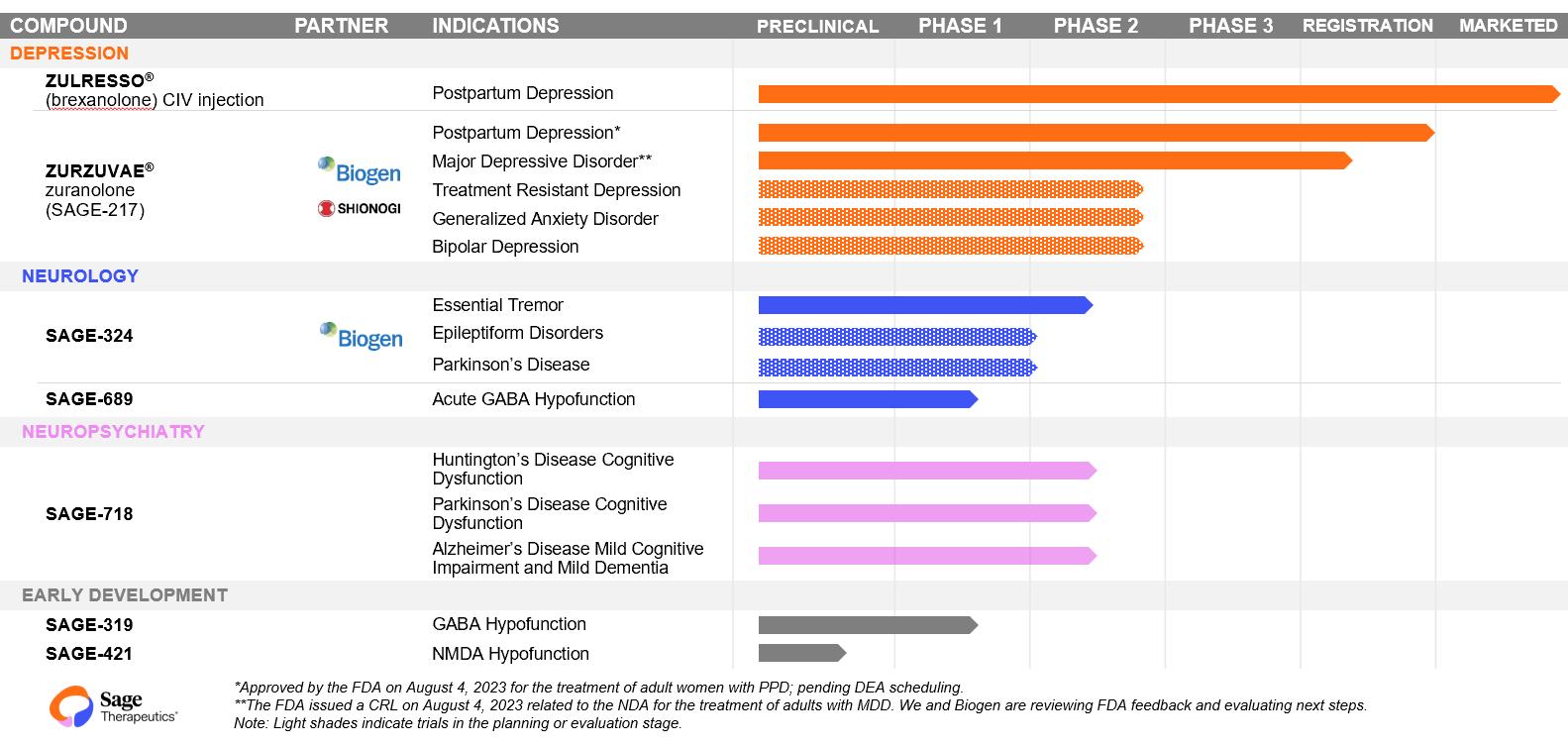
次の表は,本四半期報告提出日までの我々の製品と候補製品の組み合わせの状態をまとめたものである。
2023年8月4日、私たちの第2の製品ZURZUVAE(ズラノロン)は、成人の出産後うつ病の治療のために、米国食品医薬品局(FDA)の承認を得た。ZURZUVAEは神経活性ステロイドであり,GABAの正アロステリック調節剤であるAシナプスとシナプス外GABAに対する受容体A受容器です。ZURZUVAEは2023年第4四半期に米国で女性産後うつ治療のための薬物を商業的に発売する予定であり,これまで米国麻薬取締局(DEA)が制御物質をスケジューリングしており,FDA承認後90日以内に完了する予定である。ZURZUVAEは成年産後抑うつ患者に特化した最初の経口治療であり、毎日1回、14日間である。ZURZUVAEは、ヘルスケア提供者が患者にZURZUVAEが中枢神経系抑うつ効果により運転障害を引き起こすことを提案することを示すブロック警告を含み、ZURZUVAEを服用した人は、ZURZUVAEを服用してから少なくとも12時間後に自動車を運転することができるか、または完全な精神的警戒を必要とする他の潜在的危険活動に従事することができ、14日間の治療コースを含む。ZURZUVAEは傾眠と意識不明などの中枢神経系抑うつ作用を引き起こすことができる。医療専門家は、うつ病の悪化または緊急自殺の考えおよび行動を経験した患者においてZURZUVAEの使用を停止することを含む治療計画を変更することを考慮すべきであり、生育可能性のある女性は、ZURZUVAE治療中および最後の服薬後1週間に有効な避妊措置を使用することを推奨すべきである。ZURZUVAEの全体的な耐性は良好であり、成年女性産後抑うつ患者の2つの重要な臨床試験において一致した安全性を示した。最もよく見られる副作用は傾眠或いは傾眠、眩暈、風邪、下痢、疲労を感じる、虚弱或いは精力がない、及び尿路感染を含む。
さらに、2023年8月4日、FDAは、深刻な抑うつ障害(MDD)を治療するズランノドンの新薬申請またはNDAに関連する完全な返信、またはCRLを発表した。CRLは、NDAがズランノドンのMDD治療の承認を支持するための実質的な有効性証拠を提供しておらず、1つまたは複数の追加の臨床試験を行う必要があることを指摘している。我々は、Biogen MA Inc.またはBIMAおよびBiogen International GmbH、またはBIMAと共に、私たちの協力および許可プロトコル、または生物遺伝的協力プロトコルに基づいて、FDAのフィードバックを検討し、次の行動を評価している。
ZURZUVAEによる成人PPDの治療を許可することは2つの成人女性PPDに対する重要な臨床試験に基づく。著者らはまた4つの重要な臨床試験を完成し、成人MDDの治療に応用した。すでに完成したズランノロンによる産後抑うつ治療の重要な試験とズランノロンによるMDD治療を評価する4つの肝心な試験の中の3項目はそれらの主要な終点に達した。我々は現在MDDを治療するズランノドンの開放ラベル3期臨床試験を行っており、海岸線研究と呼ばれている
33
成人がズランノドンを繰り返し服用する安全性、耐性と必要性を評価し、最長1年に達する。海岸線研究の50 mgコホート研究の登録が完了し,研究が行われている。2023年8月,海岸線研究からの追加データ,特にサンゴ研究から海岸線研究に移行した患者行列(n=277)からのデータを公表した。このキューでは,有害事象の概要や重複治療によるデータは,先に報告された海岸線研究の他のキューのデータと類似しており,新たなセキュリティ信号は発見されなかった。SAGEは将来の医学大会で海岸線研究のさらなる分析を提出する予定である。私たちとBiogenは未来に他の感情障害のためのズランノドンを開発するかもしれない。
生物遺伝研究会社の協力協定によると、私たちは米国で生物遺伝研究会社と共同でズランノドンと私たちの別の末期化合物SAGE-324を開発しており、もし私たちの開発努力が成功すれば、私たちは許可製品と呼ばれるZURZUVAE、および許可製品324と呼ばれるSAGE-324を含む製品を、生物遺伝研究会社と共同で米国で商業化する。217個のライセンス製品と324個のライセンス製品を総称してライセンス製品と呼びます。さらに、我々は、日本、台湾、韓国またはShionogi地域以外の米国以外の地域でZuranoloneに関する特許製品を開発および商業化するBiogen独占権利を付与しており、そこでこのような権利をShionogi&Co.,Ltd.,またはShionogiに付与している。米国以外の地域を生物遺伝分野,すなわち生物遺伝研究会社が“生物遺伝研究協力協定”に基づいて適用を許可する製品が持つ権利と呼ぶ。ライセンス製品がアメリカで販売されている限り、私たちと生物遺伝会社はそのライセンス製品から発生したすべての営業利益と損失を折半します。生物遺伝研究会社の協力協議では、生物遺伝研究会社は世界規模で217種類の特許製品の販売状況を記録することが規定されている。私たちは324種類の許可製品のアメリカでの販売状況を記録し、承認された場合、生物遺伝会社は324種類の認可された製品のアメリカ以外での販売状況を記録する。
私たちはまたShionogi領土でZuranoloneを開発することについてShionogiと協力合意に達した。2021年9月,Shionogiは日本で重度MDD患者を治療するためのZuranoloneの第2段階臨床試験を完了したことを報告し,Shionogiはこの試験が主要な終点に達したと報告している。Shionogiは,単一療法や他の抗うつ薬の付加剤として重度MDD患者を治療する2つのZuranoloneの3期試験を行っていると報告し,これらの試験の結果を待つ前に,2024年第1四半期に日本医薬品·医療機器庁にNDAを提出し,ZuranoloneのMDD治療の承認を求めることを目標としていると発表した。
私たちの最初の承認された製品ZULRESSO(イブサノドン)CIV注射剤はアメリカで承認され、成人産後うつの治療に使用された。私たちは2019年6月にアメリカで産後うつを治療するZULRESSO商業薬を発売した。ZULRESSOは合格した医療監督の医療環境でしか使用できない。Brexanoloneはallopregnanoloneと化学的に同じであり,allopregnanoloneは天然に存在する神経活性ステロイドであり,zuranoloneと同様にGABAの正変異調節剤であるA受容器です。
GABAに対する他の新しい化合物も開発していますA受容体は新規GABAを含むSAGE-324A慢性経口投与のための受容体陽性アロステリック調節剤。われわれは現在,特発性振戦を有する患者をSAGE−324の2 b期用量範囲臨床試験,すなわちよく知られているKine 2研究に参加している。2022年5月、著者らはまた開放ラベルの第二段階臨床試験を開始し、特発性振戦患者におけるSAGE-324の長期安全性と耐性を評価し、緊急不良事象の発生率を主要な終点とすることを目的とした。これは長年の臨床試験であり、最初はKYNAMIC 2研究を含む他のSAGE-324臨床試験からの特発性振戦患者に開放される。SAGE-324はまた、てんかんとパーキンソン病を含む他の神経系疾患を治療する潜在力があると信じている。SAGE-324の他の開発計画は、生物遺伝会社との戦略的協力の一部として決定されるであろう。
我々が開発した第2の重点領域は,NMDA受容体を標的とした新規化合物である。我々がこの分野で選択した主要な候補製品はSAGE−718であり,これは酸素ステロイドに基づくNMDA受容体ポジ型アロステリック調節剤であり,ハンチントン病,パーキンソン病,アルツハイマー病などの疾患に関連する認知機能障害を含むNMDA受容体機能障害に関連するいくつかの認知関連障害の治療に用いられる薬剤を探索している。
FDAはSAGE-718快速チャネルがハンチントン病患者の潜在的治療法として指定されることを許可した。また,2023年2月,欧州医薬品局はSAGE−718に孤児薬名を付与した
34
ハンティントン病の潜在的治療に使われていますSAGE-718は現在ハンチントン病認知障害患者に対して3つの臨床試験を行っている:
2022年2月、用量はDIMAGE研究において開始され、ハンチントン病認知障害患者における二重盲検プラセボ対照のSAGE-718の第2段階臨床試験である。この次元研究は、3ヶ月以内のSAGE-718の1日1回の治療効果を評価することを目的としている。
2022年3月、我々は調査者研究を開始し、これはプラセボ対照の第二段階臨床試験であり、SAGE-718はハンチントン病認知障害患者に健康なボランティア成分を使用し、証拠を生成し、認知表現の効果シグナルを実世界の機能領域に関連付けることを目的としている。
2022年12月、著者らはハンチントン病認知障害患者におけるSAGE-718の長期安全性と耐性を評価するための第3段階開放ラベル研究であるPERVIEW研究を開始した。
パーキンソン病およびアルツハイマー病に関連する認知問題の治療のためのSAGE−718も評価されている。2022年3月、パーキンソン病による軽度認知障害患者におけるSAGE-718の二重盲検、プラセボ対照の第2段階臨床試験を開始し、前例研究と呼ばれた。この前例研究は42日以上のパーキンソン病による軽度認知障害患者におけるSAGE-718の安全性と有効性を評価し、その後、制御されたフォローアップ期間を行うことを目的としている。
2022年12月、著者らはLightwave研究を開始し、これは無作為プラセボ対照の第二段階臨床試験であり、SAGE-718はアルツハイマー病による軽度認知障害と軽度認知症患者に応用した。Lightwave研究は、84日以内にSAGE-718を服用する安全性と有効性を評価し、その後、制御されたフォローアップ期間であることを目的としている。
私たちは急性と慢性脳健康疾患に重点を置いた開発初期段階の他の計画もある。私たちの初期製品はSAGE-689、バランスのとれたGABAを含みますA 受容体陽性アロステリック調節剤は、第1段階臨床開発における筋肉注射、およびSAGE-319、シナプス外GABAのためのものであるA 第一段階臨床開発における社会相互作用障害を治療する口腔療法としての受容体偏愛陽性アロステリック調節剤の潜在的使用。我々はまた、SAGE-421、NMDA受容体陽性アロステリック調節剤を含むNMDA受容体調節に焦点を当てた早期化合物を有し、神経発達障害および認知回復における潜在的用途を研究する予定である。GABAアロステリック調節に関する仕事を続けたいと思いますA脳内のNMDA受容体システムですGABAANMDA受容体システムは情緒、てんかん、認知、焦慮、睡眠、痛みと運動などの障害を含む多くの精神と神経障害に影響するため広く受け入れられている。私たちの費用とパイプライン優先順位作業の結果、私たちは私たちの内部ポートフォリオから分子を開発する機会があると信じています。目標は将来このような病気を解決することであり、私たちは私たちの科学的方法を使ってGABA以外の目標を探索する機会があるかもしれないと信じていますANMDA受容体システムと、脳健康以外の満足されていない需要領域で化合物を開発する。
私たちは2019年第2四半期から製品販売から収入を得て、2019年6月に私たちの最初の製品ZULRESSOを発売しました。2020年第4四半期には、生物遺伝との戦略的協力と株式購入の収入を記録した。
我々の設立以来,毎年純損失が発生しているが,2020年12月31日までの年間純収益は6.061億ドルであり,生物遺伝研究協力協定により確認された収入を反映しており,2023年6月30日現在の累計赤字は23億ドルである。2023年6月30日までの6ヶ月間の純損失は3.072億ドルでした。これらの損失は主に研究や開発活動に関する費用によるものである
35
そして私たちの運営や商業建設に関する販売、一般、行政コスト。私たちは予測可能な未来に巨額の費用と運営損失が生じると予想している。
私たちの現在の見積もりによると、2023年6月30日までの既存の現金、現金等価物、有価証券に、私たちの持続的な協力と潜在収入の予想資金を加えることで、2025年までの運営を支援できるようになると予想されています。“流動性と資本資源”を見てください
また、最近の事態を受けて、パイプライン優先順位や労働力再編を含めて資源配分を評価しており、現金滑走路の拡大を目指しています。したがって,2024年の運営費は2023年より低下すると予想される。私たちは私たちが行っている活動に関するコストが発生し続けると予想されています
製品販売および/または協力から相当な収入を持続的に発生させることができるまで、私たちは主に収入、株式、または債務の組み合わせによって、私たちの運営に資金を提供することが予想される
36
資金調達と他の出所は、私たちのBiogenとShionogiとの協力と未来に可能な協力を含む。私たちはZULRESSO、ZURZUVAE、または任意の他の製品を商業化することに成功しないかもしれませんし、私たちの投資および目標を支援するために必要なレベルまたは時間的に有意な収入または収入を生成しないかもしれません。私たちは、現在または将来の候補製品の開発を成功させたり、必要な規制承認を成功させたり、最終的に承認された任意の製品の商業的可能性を実現したりすることは決して成功しないかもしれない。私たちは私たちの製品や候補製品のために十分な特許保護または他の排他性を得ることができないかもしれない。私たちは受け入れ可能な条項で十分な追加融資を受けることができないかもしれないし、根本的にできないかもしれない。私たちは必要な時に資金を集めることができなくて、これは私たちの財務状況と私たちの業務戦略を実施する能力にマイナス影響を与えるだろう。既存のパートナーとの合意は、私たちがいくつかの技術または候補製品の権利を放棄することを要求し、将来のどの協力も私たちに追加の権利を放棄することを要求するかもしれない。私たちは利益を達成するために相当な収入を作らなければならないだろうし、私たちは決してこれをしないかもしれない。
財務運営の概要
収入.収入
私たちは2019年第2四半期から製品販売から収入を得て、2019年6月にPPDの治療薬として最初の製品ZULRESSOを発売しました。
ZULRESSOは持続輸液として2日半を超えて使用されている。ZULRESSO輸液中の過鎮静や突然の知覚喪失は深刻な傷害をもたらすリスクがあるため,ZULRESSOはリスク評価と緩和策(REMS)計画認証を経てREMS計画の他の要求(輸液中の患者監視に関する要求を含む)に適合した医療環境でしか使用できない。医療保健機関が産後うつを患っている女性を治療するために必要な行動は複雑で時間がかかる。これらの行動は、REMS認証を取得すること、処方承認を得ること、ZULRESSOを管理するプロトコルを確立すること、および満足できる精算を確保することを含む。ビジネスカバー範囲によると、サイトは支払者ごとに補償を交渉しなければならないことが多い。このような要求は未来のZULRESSOの収入増加を制限し続けると予想される。
私たちの顧客管理現場チームと販売代表を含むZULRESSOビジネス運営は、主に既存の活発なZULRESSO治療サイトを持つ地域に集中しています。私たちはこのようなビジネス努力の方法がZULRESSOの収入機会を大きく制限し続けると予想する。
私たちはZULRESSOの収入が四半期ごとに変動する可能性があると予想する。私たちとBiogenがアメリカで女性産後うつを治療するZURZUVAEの発売に成功し、それを商業化しなければ、私たちとBiogenはZURZUVAEから収入を得ません。そして私たちまたは私たちのどの協力者も規制部門の承認を得て、私たちの現在または未来の候補製品の一つを商業化するまで、他の製品から収入を得ることはありません。もし私たちが第三者と私たちの候補製品について追加的な協力協定を締結すれば、私たちはこれらの協力から収入を得ることができるかもしれない。ライセンス料、臨床材料または製造サービス支払い、マイルストーン支払い、私たちに支払われる特許権使用料、および任意の商業化製品販売で生じる協力利益または損失における当社のシェア、および他の支払いの時間および金額により、既存または将来の協力協定によって生じる可能性のある収入(ある場合)は四半期ごとに変動すると予想されます。もしZURZUVAEが成功的に発売され、商業化され、女性産後うつを治療するために使用されれば、ZULRESSOに対するビジネス機会をさらに制限するかもしれない。
2018年6月、我々はShionogiと戦略的協力を達成し、Shionogi領土のMDDと他の潜在的適応を治療するズランノドンの臨床開発と商業化を行った。合意条項によれば、Shionogiは、MDDおよび可能な他の適応の治療のためのShionogi地域のZuranoloneのすべての臨床開発、監督管理、および商業化および製造を担当する。2018年10月、Shionogiと供給契約を締結し、この合意に基づいて、Shionogiにズランノロン臨床材料を供給した。これまでにShionogiと連携した収入は,連携プロトコルを実行する際の初期前払い許可料9000万ドルから来ており,
37
2018年12月31日までの年間協力収入と,Shionogiの臨床試験に有効な薬剤プロトコルまたは原料薬を提供した。
2020年11月、私たちはBiogenと協力協定を締結し、許可された製品を開発、製造、商業化した。生物遺伝研究協力協定の実行については,BIMAに6,241,473株の我々の普通株を売却·発行する株式購入協定も締結され,総代償は6.5億ドルであった。生物遺伝研究協力協定は2020年12月に発効し,株式購入協定での普通株の売却は2020年12月31日に完了した。BIMAが普通株を購入したため、Biogenは私たちの関係者となった。生物遺伝研究協力協定の条項によると、私たちは共同開発し、成功した場合には米国で共同商業化ライセンス製品を開発し、生物遺伝は単独で生物遺伝研究分野で開発と商業化ライセンス製品を開発する。私たちとBiogenは、FDAが許可製品を承認した後、米国内のBiogen協力協定についてのみ、活動のすべてのコストと利益と損失を二分することに同意した。Biogenは、Biogen地域内のBiogen協力協定の下で活動するすべての費用を完全に担当する。生物遺伝会社はSAGE-217製品の世界での主要かつ記録的な販売となる。私たちはSAGE-324製品のアメリカでの主要な売上と記録的な売上であり、Biogenは生物遺伝専門区におけるSAGE-324製品の主要かつ記録的な売上であるだろう。2020年12月31日までの年間で、8.75億ドルの前払と株式購入契約項目の持分投資の超過収益2.325億ドル(公正価値で計算)を含む11億ドルを記録した。“生物遺伝研究協力協定”のさらなる議論については、付記6を参照されたい協力協定や本四半期報告第1項第1項に添付されている簡明連結財務諸表に付記する。
協力手配
私たちはこのような手配が双方が行った共同経営活動に関連しているかどうかを評価するために、著者らの協力手配を分析し、これらの各方面は活動の積極的な参加者であり、重大なリスクとリターンに直面し、これはこのような活動の商業成功に依存するため、会計基準編纂或いはASCテーマ808の範囲に属する協力手配808、または主題808。この評価は手配された全ライフサイクル内で手配当事者の責任の変化に基づいて行われる。複数の要素を含むトピック808の範囲内の協調スケジュールについては、まず、協働するどの要素がトピック808の範囲内にあると考えられるか、および協働するどの要素がベンダー−クライアント関係をより反映することができ、したがって、ASCトピック606の範囲内であることを決定する取引先と契約した収入または主題606。特別テーマ808によって計算された協力手配の要素に対して、類比権威会計文献或いは合理的と理性的な政策選択を適用することによって、適切な確認方法を確定し、一致して適用する。主題606に基づいて入金された手配要素については、5段階収入確認モデルを適用し、簡明な連結経営報告書および全面損失のうち、連携収入列報として配置する。
主題808の範囲内の協調スケジュールについて、各個々の活動の性質に基づいて、協調スケジュール内の複数の活動に関連する他の参加者の対応または不足した金額を列挙するための損益テーブル分類を評価する。顧客関係の支払いや精算ではなく、連携関係として、例えば共同開発および共同商業化活動であり、一定期間協力パートナーに支払う場合には、研究開発費や販売、一般および管理費用、または協力パートナーが一定期間返済した場合には、これらの費用行項目の減少額として記録される。協同手配会計の更なる検討については、付記6を参照されたい協力協定や本四半期報告第1項第1項に添付されている簡明連結財務諸表に付記する。
私たちは、許可料、臨床材料または製造サービス支払い、マイルストーン支払い、私たちに支払われる特許権使用料、および任意の商業化製品販売で生じる協力利益または損失における私たちのシェア、および他の支払いの時間と金額によって、私たちの協力合意によって生じる可能性のある収入は四半期ごとに変動すると予想しています。私たちは生体遺伝会社から7500万ドルの記念碑的支払いを得ることができますアメリカでPPDを治療するZURZUVAEの最初の商業販売です
38
米国ではMDDの治療のためのズラノロンが初めて商業販売され、マイルストーン支払いは1.5億ドル。ShionogiとBiogenとの協力協定および協力協定収入の会計処理のさらなる検討については、付記2を参照されたい重要会計政策の概要そして注6協力協定や本四半期報告第1項第1項に添付されている簡明連結財務諸表に付記する。
販売原価
販売コストには、第三者製造コスト、パッケージサービス、送料、当社の製品純収入に応じて支払われる第三者特許権使用料、およびZULRESSOに関連する無形資産の償却を含むZULRESSO製造および流通に関連する直接および間接コストが含まれます。販売コストにはまた、いくつかの在庫製造サービスに関連する期間コスト、在庫調整費用、および製造差異が含まれる可能性がある。予測可能な未来には、製品純収入に占める私たちの商品販売コストの割合は1桁から2桁までのパーセンテージの範囲内に維持されると予想される。私たちはZULRESSOのゼロコスト在庫を長い間利用する予定です。
運営費
我々の運営費には,主に研究や開発活動や販売,一般,行政活動に関するコストが含まれている。
研究と開発費
研究·開発費用には,主に我々の製品研究や開発作業に関するコストが含まれており,発生時に費用を計上している。研究と開発費は主に
我々は、SAGE-217製品とSAGE-324製品の米国での共同開発、共同商業化、共同製造に関連する協力活動はテーマ808の範囲内の単独計算単位であると考えている。私たちとBiogenはすべて開発と商業化活動の積極的な参加者であり、重大なリスクとリターンに直面しており、これらのリスクとリターンは手配中の活動の開発と商業成功に依存する。共同開発·共同製造活動に関する生物遺伝研究会社への支払いや生物遺伝研究会社から得られた補償は,研究·開発費用の増加または減少に計上されている。2023年6月30日までの3ヶ月と6ヶ月以内に、それぞれ2,240万ドルと3,970万ドルを記録しました。2022年6月30日までの3ヶ月と6ヶ月以内に、私たちはBiogenからそれぞれ2,100万ドルと3,950万ドルを計算しました。この2つの費用はすべて私たちの研究開発費から差し引かれています。これらの費用はBiogenよりも多いからです。
39
いくつかの開発活動のコストは,我々のサプライヤーと我々の臨床サイトが提供してくれた情報やデータを用いて特定のタスクを達成する進捗を評価することによって確認された。
我々は,我々の候補製品を開発し,探索的な作業を含めて他の研究や開発計画に焦点を当て,新たな化合物を決定し,決定された化合物の目標検証を行い,先に検証した計画の最適化をリードしてきた。私たちの直接研究開発費用は計画に基づいて追跡され、主に外部コスト、例えば研究者、センター実験室、CROおよび契約製造組織に支払われる私たちの非臨床研究および臨床試験に関連する費用、私たちの候補製品に関連する第三者許可費、および私たちの計画のために働く外部コンサルタントに支払われる費用が含まれる。これらのコストは、研究開発下の複数の製品計画に配置されているため、研究開発費における未分配または株式ベースの報酬に個別に分類されるため、従業員に関連するコストや他の間接コストを特定の研究開発計画に分配しない。
研究開発活動は私たちの業務の核心だ。臨床開発後期段階にある候補製品は通常,臨床開発早期段階の候補品よりも高い開発コストを有しており,これは主に後期臨床試験の規模と持続時間が増加しているためである。予測可能な未来には、いくつかの候補製品の臨床試験と非臨床研究を継続または開始し、私たちの候補製品の臨床開発を継続するにつれて、私たちの研究と開発費用は引き続き増加すると予想される。
私たちは私たちの候補製品の現在または未来の臨床試験の持続時間とコストを決定することができない。私たちの候補製品の臨床試験と開発の持続時間、コスト、時間は様々な要素に依存します
また,新冠肺炎の流行および/または他のマクロ経済や地政学的条件の影響により,医療やサプライヤーの人員不足や米国のヘルスケアシステムが中断され始め,我々が行っており計画中の開発活動に負の影響を与え,研究開発コストを増加させる可能性もある。懸念、予防、制限、人員不足、あるいはマクロ経済環境の他の変化は、臨床サイトの決定と活性化を大幅に遅らせる可能性があり、私たちの臨床試験における登録を大幅に遅らせる可能性があり、私たちの試験の進行、監査、監視または完了を損害または延期する可能性があり、私たちのデータ収集と分析仕事の即時性と完成または私たちのデータの完全性を損害または阻害する可能性があり、あるいは私たちの試験の一時停止を招く可能性があり、すべての場合、これは私たちの予想スケジュールを満たす能力に著しく影響し、あるいは私たちの計画を変更し、私たちの研究開発コストを著しく増加させる可能性がある。例えば,いくつかの臨床試験では,我々が行っている特発性振戦患者に対するSAGE−324の運動学的2研究を含め,我々の募集速度は予想よりも遅く,我々が最初に予測した2022年末ではなく,2023年末に募集を完了する予定である。
候補製品の開発のために、これらの変数のいずれかの結果の変化は、候補製品の開発に関連するコストおよび時間の大きな変化を意味する可能性がある。例えば、FDAまたは他の規制機関が、候補製品の臨床開発を達成するために、または規制部門の承認を得るために、現在予想されている以上の臨床試験を行うことを要求する場合、または任意の臨床試験において重大な遅延に遭遇した場合、またはより多くの患者を募集する必要がある場合、臨床開発を完了するために多くの追加の財政資源と時間を要する可能性がある。
いかなる潜在的候補製品開発のいかなる段階も適時に完成できなかったことは、私たちの運営、財務状況、流動性に重大な悪影響を及ぼす可能性がある。いくつかのリスクと
40
私たちの計画を予定通りに達成できなかったか、または全く達成できなかったことに関する不確実性と、予定通りに達成できなかった潜在的な結果は、本四半期報告第II部第1 A項“リスク要因”のタイトルに記載されている。
販売、一般、行政費用
販売、一般および行政費用は、主に役員、財務、業務、商業、会社発展および他の行政機能の賃金、福祉および出張費用、株式ベースの給与費用を含む人員コストを含む。販売、一般および管理費用は、第三者によって達成されたZURZUVAEおよびZULRESSOの商業化に関連する合意に従って生成された専門費用、ZURZUVAEに関連する送信許可および発射準備活動、知的財産権保護を求める法的費用を含む広報、監査、税務および法律サービス、ならびに賃貸料、減価償却、施設維持、保険および用品を含む当社の情報技術、施設および他の関連費用の一部をさらに含む。
私たちが行っているZULRESSOに関するビジネス努力は、私たちの現場での顧客管理チームや販売代表を含め、主に既存の活発なZULRESSO治療場所を持つ地域に集中しています。私たちは、ZULRESSOに関連した持続的なビジネス活動を支援するために、賃金や関連費用を含む巨額の商業化費用を引き続き発生させる予定です。最近の事態を受けて、我々は現在、パイプライン優先順位や労働力再編を含めて資源配分を評価しており、現金滑走路の拡大を目指している。そのため、2024年の販売、一般、管理費は2023年より低下すると予想されています。ZURZUVAE計画が2023年第4四半期に米国で発売され、PPD女性患者を商業化治療するための準備を進めているため、販売、一般、管理費が引き続き発生する予定であり、DEAスケジュールに従い、FDA承認後90日以内に完了する予定である。これらの費用には、許可された投入前および投入準備活動、ZURZUVAEの雇用および訓練に参加した直販チーム、および現在または将来の候補製品のための私たちの開発作業の進展、およびこれらの製品の商業化(成功した開発および承認が成功すれば)に関するコストが含まれる。会計や法律サービスに関連する費用、役員や役人の保険料、施設や他社のインフラ費用、情報技術費などの事務関連費用を含む一般業務に関連する大量の費用が引き続き発生する見通しだ。
我々は、SAGE-217製品とSAGE-324製品の米国での共同開発、共同商業化、共同製造に関連する協力活動はテーマ808の範囲内の単独計算単位であると考えている。私たちとBiogenはすべて開発と商業化活動の積極的な参加者であり、重大なリスクとリターンに直面しており、これらのリスクとリターンは手配中の活動の開発と商業成功に依存する。共同商業化活動に関連して生物遺伝会社に支払われるお金または生物遺伝会社から得られる補償は、販売、一般および行政費用の増加または減少に計上される。2023年6月30日までの3ヶ月と6ヶ月間に、Biogenにそれぞれ750万ドルと1050万ドルの純精算を記録し、これは私たちの販売、一般、行政費用に追加された。2022年6月30日までの3ヶ月と6ヶ月以内に、販売、一般、管理費用から差し引かれたBiogenは、それぞれ280万ドルと430万ドルを純精算します。
41
経営成果
2023年6月30日までと2022年6月30日までの3ヶ月間の比較
次の表は、2023年6月30日と2022年6月30日までの3ヶ月間の運営結果をまとめています
|
|
6月30日までの3ヶ月間 |
|
|
増す |
|
||||||
|
|
2023 |
|
|
2022 |
|
|
(減少) |
|
|||
|
|
(単位:千) |
|
|||||||||
製品収入、純額 |
|
$ |
2,460 |
|
|
$ |
1,501 |
|
|
$ |
959 |
|
協力収入 |
|
|
14 |
|
|
|
— |
|
|
|
14 |
|
総収入 |
|
|
2,474 |
|
|
|
1,501 |
|
|
|
973 |
|
運営コストと支出: |
|
|
|
|
|
|
|
|
|
|||
販売原価 |
|
|
205 |
|
|
|
200 |
|
|
|
5 |
|
研究開発 |
|
|
97,161 |
|
|
|
77,297 |
|
|
|
19,864 |
|
販売、一般、行政 |
|
|
75,565 |
|
|
|
52,411 |
|
|
|
23,154 |
|
総運営コストと費用 |
|
|
172,931 |
|
|
|
129,908 |
|
|
|
43,023 |
|
運営損失 |
|
|
(170,457 |
) |
|
|
(128,407 |
) |
|
|
(42,050 |
) |
利子収入,純額 |
|
|
10,173 |
|
|
|
2,102 |
|
|
|
8,071 |
|
その他の収入,純額 |
|
|
(41 |
) |
|
|
45 |
|
|
|
(86 |
) |
純損失 |
|
$ |
(160,325 |
) |
|
$ |
(126,260 |
) |
|
$ |
(34,065 |
) |
製品収入、純額
2023年6月30日と2022年6月30日までの3ヶ月間で,ZULRESSO販売に関する250万ドルと150万ドルの製品純収入をそれぞれ確認した。販売手当と課税費用には、記憶容量による使用料金、割引、販売費、リベートと患者の経済援助が含まれており、この2つの時期はいずれも顕著ではない。
販売原価
販売コストは、2023年6月30日と2022年6月30日までの3ヶ月間、それぞれ20万ドル、ZULRESSO製造および流通に関連する直接および間接コストが含まれており、第三者製造コスト、パッケージサービス、送料、当社の製品純収入に応じて支払うべき第三者特許権使用料、およびZULRESSOに関連する無形資産の償却が含まれています。販売コストにはまた、いくつかの在庫製造サービスに関連する期間コスト、在庫調整費用、および製造差異が含まれる可能性がある。2019年3月にFDAによるZULRESSOの予備承認を得る前に、商業化時に販売するためにZULRESSO在庫を生産し、この在庫蓄積に関する890万ドルを研究開発費として記録した。このため、FDA承認前に発生したZULRESSO在庫蓄積に関する製造コストは前の時期に支出されているため、2023年6月30日と2022年6月30日までの3ヶ月間の販売商品コストには含まれていない。予測可能な未来には、製品純収入に占める私たちの商品販売コストの割合は1桁から2桁までのパーセンテージの範囲内に維持されると予想される。私たちはZULRESSOのゼロコスト在庫を長い間利用する予定です。
42
研究と開発費
次の表は、2023年6月30日と2022年6月30日までの3ヶ月間の研究開発費をまとめています
|
|
6月30日までの3ヶ月間 |
|
|
増す |
|
||||||
|
|
2023 |
|
|
2022 |
|
|
(減少) |
|
|||
|
|
(単位:千) |
|
|||||||||
ズルーヴィ(ズランノドン) |
|
$ |
30,502 |
|
|
$ |
27,827 |
|
|
$ |
2,675 |
|
SAGE-324 |
|
|
10,356 |
|
|
|
8,469 |
|
|
|
1,887 |
|
SAGE-718 |
|
|
13,132 |
|
|
|
9,015 |
|
|
|
4,117 |
|
他の研究と開発計画 |
|
|
21,691 |
|
|
|
16,201 |
|
|
|
5,490 |
|
未分配費用 |
|
|
39,402 |
|
|
|
30,310 |
|
|
|
9,092 |
|
株に基づく報酬 |
|
|
4,496 |
|
|
|
6,458 |
|
|
|
(1,962 |
) |
Biogenからの純精算 |
|
|
(22,418 |
) |
|
|
(20,983 |
) |
|
|
(1,435 |
) |
|
|
$ |
97,161 |
|
|
$ |
77,297 |
|
|
$ |
19,864 |
|
2023年6月30日までの3カ月の研究開発費は9720万ドルだったが、2022年6月30日までの3カ月の研究開発費は7730万ドルだった。1 990万ドル増加した主な理由は以下の通りです
43
販売、一般、行政費用
次の表は、2023年6月30日と2022年6月30日までの3ヶ月間の販売、一般、行政費用をまとめています
|
|
6月30日までの3ヶ月間 |
|
|
増す |
|
||||||
|
|
2023 |
|
|
2022 |
|
|
(減少) |
|
|||
|
|
(単位:千) |
|
|||||||||
関係者の |
|
$ |
33,393 |
|
|
$ |
20,338 |
|
|
$ |
13,055 |
|
株に基づく報酬 |
|
|
7,197 |
|
|
|
8,178 |
|
|
|
(981 |
) |
専門費 |
|
|
12,929 |
|
|
|
15,120 |
|
|
|
(2,191 |
) |
他にも |
|
|
14,570 |
|
|
|
11,610 |
|
|
|
2,960 |
|
生物遺伝会社への純精算 |
|
|
7,476 |
|
|
|
(2,835 |
) |
|
|
10,311 |
|
|
|
$ |
75,565 |
|
|
$ |
52,411 |
|
|
$ |
23,154 |
|
2023年6月30日までの3カ月間の販売、一般、行政費は7560万ドルだったが、2022年6月30日までの3カ月は5240万ドルだった。2320万ドル増加しました主な理由は以下の通りです
利子収入純額とその他の収入純額
2023年6月30日と2022年6月30日までの3ヶ月間の純利息収入とその他の収入の純額はそれぞれ1010万ドルと210万ドルだった。利上げの主な原因は、2022年6月30日までの3カ月から2023年6月30日までの3カ月間の金利引き上げだ。
44
2023年6月30日までと2022年6月30日までの6ヶ月間の比較
次の表は、2023年6月30日と2022年6月30日までの6ヶ月間の運営結果をまとめています
|
|
6月30日までの6ヶ月間 |
|
|
増す |
|
||||||
|
|
2023 |
|
|
2022 |
|
|
(減少) |
|
|||
|
|
(単位:千) |
|
|||||||||
製品収入、純額 |
|
$ |
5,754 |
|
|
$ |
3,082 |
|
|
$ |
2,672 |
|
協力収入 |
|
|
14 |
|
|
|
— |
|
|
|
14 |
|
総収入 |
|
|
5,768 |
|
|
|
3,082 |
|
|
|
2,686 |
|
運営コストと支出: |
|
|
|
|
|
|
|
|
|
|||
販売原価 |
|
|
435 |
|
|
|
486 |
|
|
|
(51 |
) |
研究開発 |
|
|
189,987 |
|
|
|
155,315 |
|
|
|
34,672 |
|
販売、一般、行政 |
|
|
141,273 |
|
|
|
98,888 |
|
|
|
42,385 |
|
総運営コストと費用 |
|
|
331,695 |
|
|
|
254,689 |
|
|
|
77,006 |
|
運営損失 |
|
|
(325,927 |
) |
|
|
(251,607 |
) |
|
|
(74,320 |
) |
利子収入,純額 |
|
|
19,003 |
|
|
|
3,270 |
|
|
|
15,733 |
|
その他の収入,純額 |
|
|
(229 |
) |
|
|
22 |
|
|
|
(251 |
) |
純損失 |
|
$ |
(307,153 |
) |
|
$ |
(248,315 |
) |
|
$ |
(58,838 |
) |
製品収入、純額
2023年6月30日と2022年6月30日までの6ヶ月間で、ZULRESSO販売に関する580万ドルと310万ドルの製品純収入をそれぞれ確認した。販売手当と課税費用には、記憶容量による使用料金、割引、販売費、リベートと患者の経済援助が含まれており、この2つの時期はいずれも顕著ではない。
販売原価
販売コストは、2023年6月30日と2022年6月30日までの6ヶ月間、それぞれ40万ドルと50万ドルであり、ZULRESSO製造および流通に関連する直接および間接コストが含まれており、第三者製造コスト、包装サービス、送料、当社の製品純収入に応じて支払うべき第三者特許権使用料、およびZULRESSOに関連する無形資産の償却が含まれています。販売コストにはまた、いくつかの在庫製造サービスに関連する期間コスト、在庫調整費用、および製造差異が含まれる可能性がある。2019年3月にFDAによるZULRESSOの予備承認を得る前に、商業化時に販売するためにZULRESSO在庫を生産し、この在庫蓄積に関する890万ドルを研究開発費として記録した。このため、FDA承認前に発生したZULRESSO在庫蓄積に関する製造コストは前の時期に支出されているため、2023年6月30日と2022年6月30日までの6ヶ月間の販売商品コストには含まれていない。予測可能な未来には、製品純収入に占める私たちの商品販売コストの割合は1桁から2桁までのパーセンテージの範囲内に維持されると予想される。私たちはZULRESSOのゼロコスト在庫を長い間利用する予定です。
45
研究と開発費
次の表は、2023年6月30日と2022年6月30日までの6ヶ月間の研究開発費をまとめています
|
|
6月30日までの6ヶ月間 |
|
|
増す |
|
||||||
|
|
2023 |
|
|
2022 |
|
|
(減少) |
|
|||
|
|
(単位:千) |
|
|||||||||
ズルーヴィ(ズランノドン) |
|
$ |
53,641 |
|
|
$ |
50,109 |
|
|
$ |
3,532 |
|
SAGE-324 |
|
|
16,612 |
|
|
|
18,504 |
|
|
|
(1,892 |
) |
SAGE-718 |
|
|
26,196 |
|
|
|
18,436 |
|
|
|
7,760 |
|
他の研究と開発計画 |
|
|
39,597 |
|
|
|
34,856 |
|
|
|
4,741 |
|
未分配費用 |
|
|
80,372 |
|
|
|
57,834 |
|
|
|
22,538 |
|
株に基づく報酬 |
|
|
13,269 |
|
|
|
15,073 |
|
|
|
(1,804 |
) |
Biogenからの純精算 |
|
|
(39,700 |
) |
|
|
(39,497 |
) |
|
|
(203 |
) |
|
|
$ |
189,987 |
|
|
$ |
155,315 |
|
|
$ |
34,672 |
|
2023年6月30日までの6カ月間の研究開発費は1.9億ドルだったが、2022年6月30日までの6カ月の研究開発費は1兆553億ドルだった。3470万ドル増加しました主な理由は以下の通りです
46
販売、一般、行政費用
次の表は、2023年6月30日と2022年6月30日までの6ヶ月間の販売、一般、行政費用をまとめています
|
|
6月30日までの6ヶ月間 |
|
|
増す |
|
||||||
|
|
2023 |
|
|
2022 |
|
|
(減少) |
|
|||
|
|
(単位:千) |
|
|||||||||
関係者の |
|
$ |
62,280 |
|
|
$ |
38,605 |
|
|
$ |
23,675 |
|
株に基づく報酬 |
|
|
18,462 |
|
|
|
18,116 |
|
|
|
346 |
|
専門費 |
|
|
24,490 |
|
|
|
25,629 |
|
|
|
(1,139 |
) |
他にも |
|
|
25,531 |
|
|
|
20,847 |
|
|
|
4,684 |
|
生物遺伝会社への純精算 |
|
|
10,510 |
|
|
|
(4,309 |
) |
|
|
14,819 |
|
|
|
$ |
141,273 |
|
|
$ |
98,888 |
|
|
$ |
42,385 |
|
2023年6月30日までの6カ月間の販売·一般·行政費は1兆413億ドルだったが、2022年6月30日までの6カ月間は9890万ドルだった。4240万ドル増加しました主な理由は以下の通りです
利子収入純額とその他の収入純額
2023年6月30日と2022年6月30日までの6ヶ月間の純利息収入とその他の収入の純額はそれぞれ1880万ドルと330万ドルだった。利上げの主な原因は、2022年6月30日までの6カ月から2023年6月30日までの6カ月間の金利引き上げだ。
流動性と資本資源
私たちは2019年第2四半期から製品販売から収入を得て、2019年6月に私たちの最初の製品ZULRESSOを発売しました。会社設立以来,毎年純損失が発生しているが,2020年12月31日までの年間純収益は6.061億ドルであり,生物遺伝研究協力協定により確認された収入を反映している。2023年6月30日現在、私たちの累計赤字は23億ドルです。2020年12月31日,私募でBIMAに6,241,473株の普通株を売却することを完了し,価格は1株当たり約104.14ドル,総収益は6.5億ドルであった。設立から2023年6月30日まで、初公募前に償還可能な優先株を売却し、転換可能手形を発行し、2014年7月の初公募株で普通株を売却し、後続発売および生物遺伝協力協定(バイオ遺伝株式買収と呼ぶ)に関する普通株を生物遺伝研究会社に売却し、計28億ドルの純収益を得た。BiogenとShionogiとの協力を通じて、私たちはまた10億ドルの前払いを受けた。
2023年6月30日現在、私たちの主な流動性源は現金、現金等価物、有価証券で、合計10億ドルです。私たちは現金を通貨市場基金、アメリカ政府証券、社債、商業手形、預金証券、市政証券に投資し、私たちの主な目標はリスクを著しく増加させることなく元本を保留し、流動性と最大化収入を提供することである。
47
次の表は、2023年6月30日と2022年6月30日までの6ヶ月間の主要な現金源と用途をまとめています
|
|
6月30日までの6ヶ月間 |
|
|||||
|
|
2023 |
|
|
2022 |
|
||
|
|
(単位:千) |
|
|||||
提供された現金純額(使用): |
|
|
|
|
|
|
||
経営活動 |
|
$ |
(285,200 |
) |
|
$ |
(214,239 |
) |
投資活動 |
|
|
240,460 |
|
|
|
124,704 |
|
融資活動 |
|
|
3,519 |
|
|
|
1,812 |
|
|
|
$ |
(41,221 |
) |
|
$ |
(87,723 |
) |
経営活動
2023年6月30日までの6ヶ月間、経営活動のための現金純額は主に私たちの純損失3.072億ドルから来ています。これは主に私たちの研究開発活動と私たちの販売、一般と行政費用、そして私たちの運営資産と負債の変化が280万ドルですが、2470万ドルの非現金プロジェクトによって部分的に相殺されています。
2022年6月30日までの6ヶ月間、経営活動のための現金純額は主に私たちの純損失2.483億ドルから来ています。これは主に私たちの研究開発活動と私たちの販売、一般と行政費用、そして私たちの運営資産と負債の変化が430万ドルですが、3840万ドルの非現金プロジェクトによって部分的に相殺されています。
投資活動
2023年6月30日と2022年6月30日までの6ヶ月間、投資活動が提供した純現金はそれぞれ2億405億ドルと1兆247億ドルだった。私たちは、2023年6月30日と2022年6月30日までの6ヶ月間、有価証券を購入し、私たちの現金とポートフォリオを管理する一部として、有価証券を売却し、満期にした。
融資活動
融資活動が提供する純現金は、2023年6月30日と2022年6月30日までの6カ月間でそれぞれ350万ドル、180万ドルだった。これは主に従業員が株を購入する計画に基づいて株式を購入することによるお金の増加によるものだ。
運営資本要求
予測可能な未来には、ZURZUVAEを米国PPD女性患者の治療に使用する商業化に伴い、損失が生じ続けることが予想され、FDAのフィードバックを検討し、MDDの治療に使用されるズランノロンに関連するCRLの次の措置を評価し、MDD患者において1つ以上の追加の臨床試験を行う可能性があるZuranoloneを治療するためのZuranoloneの承認を支援することを進め、現在および将来の候補製品の開発を継続し、開発に成功した候補製品の規制承認を求めることが可能である。開発と承認に成功したZULRESSOおよびZURZUVAE以外の候補製品の潜在的商業化に備え、投入前および投入準備活動に参加することを含み、承認されれば、そのような任意の製品の商業化を開始し、私たちの既存製品の組み合わせ以外で新たな候補製品を発見し、開発するために努力し続けている。私たちはまた一般業務に関連した多くの費用が発生すると予想している。さらに、ZULRESSO、ZURZUVAE、および任意の他の開発および承認された将来の製品に関連する製品販売、マーケティング、アウトソーシング製造には巨額の商業化費用が発生すると予想されています。したがって、私たちは私たちの持続的な業務と関連した多くの追加資金が必要になると予想する。
現在の見積もりによると、2023年6月30日までの既存の現金、現金等価物、有価証券に、私たちが行っている協力と潜在収入の予想資金を加えて、私たちは私たちが行っている協力と潜在収入の期待資金を加えることができると予想しています
48
2025年まで私たちの運営を支援する。また、最近の事態を受けて、パイプライン優先順位や労働力再編を含めて資源配分を評価しており、現金滑走路の拡大を目指しています。したがって,2024年の運営費は2023年より低下すると予想される。ZURZUVAE計画が2023年第4四半期に米国で発売され商業化されたPPD女性患者の治療に向けて準備しているため,DEAスケジュールに従い,FDA承認後90日以内に完了する予定であるため,運営費が発生し続ける予定である。これらのコストには、許可された投入前および投与計画活動に関与することに関連する費用、私たちの計画および進行中のSAGE−718およびSAGE−324臨床試験の推進、リソース分配およびパイプライン優先順位付け作業の継続的な評価結果に基づいて、様々な研究活動を継続すること、および我々の戦略計画を実施することが含まれる。
私たちの現在の業務計画は、私たちが展開可能な他の活動を考慮していない、あるいは私たちが現在計画しているすべての活動が同じ速度で行われるか、またはこれらのすべての活動がこの期間内に完全に起動または完了するだろう。私たちの推定は変化する可能性があるという仮定に基づいており、私たちは現在予想されているよりも早く利用可能な資本資源を使用するかもしれない。私たちはまた私たちの開発、商業化、または他の努力を変えたり増加させることを選択することができる。任意の製品または候補製品の開発および商業化に関連する多くのリスクおよび不確実性のため、現在または将来の候補製品の開発が完了したり、任意の承認された製品を商業化するために必要な増加した資本支出および運営支出の金額を推定することはできない。
私たちの将来の資本需要は多くの要素に依存します
49
私たちはバイオ遺伝会社から7500万ドルの記念碑的支払いを得ることが可能であり、この支払いは産後うつの治療のためのズランノドンの初の商業販売と関連がある。これまで、相当な製品収入および/または協力収入を生成し、持続的な利益を達成することができれば、株式発行、債務融資、協力、戦略連合、許可手配、および他の資金源によっても私たちの現金需要を満たすことができると予想されています。現在または将来の運営計画のために十分な資金を持っていると考えても、市場状況が有利または他の戦略的考慮を考慮すれば、追加的な資本を求めることができるかもしれない。もし私たちが株式または転換可能な債務証券を売却することで追加資本を調達すれば、私たちの株主の所有権権益は希釈され、これらの証券の条項は清算または他の私たちの普通株主の権利に悪影響を及ぼす特典を含む可能性がある。債務融資が可能であれば、関連する可能性のある合意は、追加債務を招く、資本支出を行う、または配当を発表するなど、私たちが具体的な行動をとる能力を制限または制限する契約を含み、株式承認証の発行を要求する可能性があり、これは私たちの株主の所有権利益を希釈する可能性がある。もし私たちが協力、戦略連合、許可手配、または第三者との他の合意を通じて追加資金を調達する場合、私たちは、私たちの技術、将来の収入源、または研究計画に対する貴重な権利を放棄しなければならないか、または私たちに不利になる可能性のある条項で許可を付与しなければならないかもしれない。資金調達は挑戦をもたらすかもしれない。マクロ経済または地政学的条件、景気後退、企業および消費者支出の減少、長期失業、または他の一般経済状況に悪影響を及ぼす可能性がある場合を含む様々な理由により、将来の市場は変動したり、撹乱されたりする可能性がある。もし私たちが必要な時に株式や債務融資や他の方法で追加資金を調達することができない場合、私たちは私たちの製品開発や将来の商業化努力を延期、制限、減少または終了することを要求されるかもしれません。あるいは私たちが自分で開発し、マーケティングすることをより望んでいた製品または候補製品の権利を与えることができます。
契約義務と約束
私たちの年間報告書に含まれる契約義務と約束に大きな変化はありません。
重要な会計政策の応用
私たちはアメリカで公認されている会計原則に従って簡明な連結財務諸表を作成します。これらの簡明な連結財務諸表を作成することは、計算、仮説、判断を行うことを要求します。これらの推定、仮説、判断は、簡明な連結財務諸表の日付報告書の資産、負債、費用、および関連して開示された金額、記録された収入と費用に影響を与えます
50
報告している間。私たちは持続的な基礎の上で私たちの推定と判断を評価する。我々は歴史的経験や当時の状況では合理的な様々な他の要因を推定していると考えられるが,これらの要因の結果は資産や負債の帳簿価値を判断する基礎となっており,そのような資産や負債の帳簿価値は他の源から容易に見られるものではない。したがって,異なる仮定や条件では,実際の結果はこれらの推定値と大きく異なる可能性がある.我々の主な会計政策は、年報の総合財務諸表付記により詳細に記載されているが、私たちの最も重要な会計政策は、収入確認、協力手配、計算すべき研究開発費、株式ベースの報酬に関する政策であると考えられる。
私たちの年間報告書の“経営陣の財務状況と経営結果の議論と分析--キー会計政策および重大な判断と推定”に記載されているものと比較して、私たちのキー会計政策には実質的な変化はありません。
51
項目3.定量と合格IVE市場リスクに関する開示
2023年6月30日まで、私たちは10億ドルの現金、現金等価物、有価証券を持っている。私たちの投資活動の主な目標は、リスクを著しく増加させることなく元本を保存し、流動性を提供し、収入を最大化することです。私たちの市場リスクに対する主な開放は金利変動と関係があり、金利変動はアメリカの金利全体の水準変化の影響を受けている。私たちの現金、現金等価物、有価証券の短期的な性質を考慮して、市場金利の突然の変化は私たちの財務状況および/または経営業績に大きな影響を与えないと予想される。私たちはどんな派生金融商品も持っていない。
私たちは海外のサプライヤーと契約を結び、ヨーロッパとカナダに子会社を設置している。そのため、私たちは私たちの海外取引に関連した外貨為替レートが不利に変化するリスクに直面している。私たちはこのような危険を開放することは重要ではないと思う。私たちはこのような為替レート変動の危険を開放しないつもりだ。
私たちは私たちの現金、現金等価物、有価証券に重大な違約リスクや流動性不足があるとは思わない。私たちの現金、現金等価物、および有価証券は過度のリスクを含まないと信じていますが、私たちの投資が将来的に市場価値の悪影響を受けないことを絶対に保証することはできません。さらに、私たちは1つ以上の金融機関で連邦保険限度額を超える大量の現金、現金等価物、および有価証券を持っている。
インフレは一般的に労働コストと臨床試験費用を増加させることで私たちに影響を及ぼす。私たちはインフレが2023年6月30日と2022年6月30日までの6ヶ月間の運営業績に実質的な影響を与えるとは思わない。
項目4.制御プログラムがあります
情報開示制御とプログラムの評価
我々は、1934年の証券取引法または1934年の証券取引法第13 a-15(E)および15 d-15(E)規則によって定義されたような開示制御および手順(例えば、1934年の証券取引法に基づいて提出または提出された報告書で開示を要求する情報を確保することを目的としている:(1)米国証券取引委員会規則および表で指定された期間内に記録、処理、まとめ、報告、(2)私たちの最高経営責任者総裁および最高経営責任者および私たちの最高財務官を含む私たちの経営陣に蓄積されて伝達される。WHOはまた、必要な開示に関する決定をタイムリーに下すために、私たちの最高財務·会計幹事でもある。
我々の経営陣は、2023年6月30日現在、最高経営責任者及び財務会計官の参加の下、開示制御及び手続の有効性を評価している。我々の経営陣は,どのような制御やプログラムも,どんなに設計や操作が良好であっても,その目標を実現するために合理的な保証を提供することしかできず,管理層は,可能な制御とプログラムのコスト-収益関係を評価する際にその判断を適用しなければならないことを認識している.我々の最高経営責任者及び最高財務会計官は、上記の評価に基づいて、2023年6月30日現在、我々の開示制御及び手続は合理的な保証レベルで有効であると結論した。
財務報告の内部統制の変化
本四半期報告に関連する期間、我々は財務報告の内部統制(1934年証券取引法第13 a-15(F)および15 d-15(F)規則の定義による)に大きな影響を与えなかったか、または合理的に我々の財務報告内部統制に大きな影響を与える可能性のある変化を生じなかった。
52
第2部-その他R情報
項目1.法律規定訴訟手続き
私たちは時々私たちの正常な業務過程で発生したクレームに関する法的手続きに巻き込まれるかもしれない 契約、雇用手配、経営活動、知的財産権またはその他の事項に関するクレームを含む。私たちは現在、私たちの財務状況、経営業績、キャッシュフロー、または他の重大な法的手続きに重大な悪影響を及ぼすと考えられる法的手続きの影響を受けていません。
第1 A項。国際ロータリーSK因子
私たちの普通株に投資することは高い危険と関連がある。投資決定を下す前に、以下に述べるリスクと、本四半期報告書のForm 10-Qまたは四半期報告書の他の情報、ならびに私たちの他の公開文書内の他の情報をよく考慮しなければなりません。私たちの業務、見通し、財務状況、または経営業績は、これらのリスクの損害を受ける可能性があり、私たちが現在知らないこと、または現在重要ではないと考えている他のリスクを受ける可能性がある。このようなリスクまたは不確定要因が実際に発生した場合、当社の業務、財務状況または経営結果は、本四半期報告における“経営層の財務状況および経営結果の議論および分析”と題する節および本報告の他の場所、ならびに私たちの他の公開文書および公開声明を含む、本四半期報告における計画、予測、および他の展望的陳述とは大きく異なる可能性がある。私たちの普通株の取引価格はこれらのリスクのいずれかによって下落する可能性があり、その結果、私たちの株主は彼らの全部または一部の投資を損失する可能性がある。
製品開発,規制承認,商業化に関するリスク
私たちの将来のビジネスの見通しは、私たちのパートナーであるBiogen MA Inc.とBiogen International GmbH、または共同で協力して、アメリカでZURZUVAE(Zuranolone)を商業化することに成功し、出産後うつ病(PPD)を治療する能力に大きく依存しています。私たちは米国で出産後うつ病を治療するZURZUVAEの商業化努力が成功することを保証することはできませんし、私たちが期待しているレベルで、あるいは私たちの目標を支持するのに必要な時間に収入を作ることができるという保証はありません。
我々の第2製品ZURZUVAEは2023年8月4日にFDAによって米国成人PPDの治療に許可された。ZURZUVAEは2023年第4四半期に米国でPPD女性治療のための商業製品を発売する予定で、これまで米国麻薬取締局(DEA)が制御物質を手配しており、FDAの承認後90日以内に完成する予定だ。ZURZUVAEは成年産後抑うつ患者に特化した最初の経口治療であり、毎日1回、14日間である。ZURZUVAEは、ヘルスケア提供者が中枢神経系抑うつ効果による運転障害を患者に推奨することを示すブロック警告を含み、ZURZUVAEを服用した人は、14日間の治療コースでZURZUVAEを少なくとも12時間服用した後に自動車を運転すべきか、または完全な精神的警戒を必要とする他の潜在的危険活動に従事すべきであり、これは処方またはZURZUVAEの使用意欲を低下させる可能性がある。このタグはまた、有害事象および他の警告および予防措置に関する情報を含む。我々の現在の業務は,米国でZURZUVAEを産後うつ治療薬として商業化できるかどうかに大きく依存している。私たちはZURZUVAEを発売したり、商業化に成功したりすることができないかもしれませんし、販売収入や利益に対する私たちの期待を達成することもできません。我々とBiogenがZURZUVAEの米国におけるPPD治療のための女性患者の商業化のために構築したインフラ、システム、プロセス、政策、関係、材料は私たちを成功させるのに十分であることは保証されない。たとえ私たちが米国で産後うつの治療のためのZURZUVAEの発売に成功したとしても、ZURZUVAEタグに含まれるブロック警告を含む、我々の予想される産後うつ女性の臨床的利益、臨床使用、または市場受容度を達成できない可能性がある。産後うつ女性の数、産後うつ女性の追加治療選択に対する満足されていない需要、およびZURZUVAEの潜在的市場は、私たちが予想していたよりはるかに少ないかもしれない、あるいは私たちは産後うつ女性のZURZUVAEの治療を商業化する際に、精算に関連するまたは他の市場に関連する問題に遭遇する可能性がある。さらに、DEAの制御物質スケジューリングは、私たちが予想していた時間内に完了できない可能性があり、これは、私たちの商業化計画を延期または阻害する可能性がある。たとえ私たちが産後うつの治療に成功したZURZUVAEを発売し、それを商業化したとしても、ZURZUVAEによる産後うつの治療収入は、私たちが監督部門の許可を得て深刻な抑うつ障害(MDD)の治療を許可した収入よりも大幅に低いと予想される。
53
私たちの将来の業務の将来性は、新薬申請(NDA)に関連する完全返信(CRL)において、MDDを治療するためのズランノドンに対するFDAのフィードバックの能力を評価し、MDDを治療するためのZuranoloneの規制承認を得る方法を見つけることに大きく依存する。FDAの立場は、MDDの承認を支持するために追加の1つ以上の祖ラノドン臨床試験が必要であり、私たちが将来控訴を求めても、FDAは彼らの立場を変えないかもしれない;このような1つ以上の試験は時間がかかり、私たちの費用を著しく増加させ、実行できない可能性がある;私たちがこのような臨床試験を行っても、それらは成功しないかもしれない。我々がMDD上でより多くの試験を行っても,我々が行った他の臨床試験の設計と結果が監督部門の承認を得るのに十分であることは保証されない。私たちはMDDを治療するためのズランノドンの規制承認を決して得られないかもしれない。MDD治療のためのZuranoloneの規制承認を得ても,MDDを治療するためのZuranoloneの商業化努力は成功しない可能性がある。
FDAは2023年8月4日、米国がMDDを治療しているズランノドンのNDAに関するCRLを発表した。CRLは、NDAがZuranoloneによるMDDの治療を許可するための多くの有効な証拠を提供しておらず、1つまたは複数の追加の臨床試験が必要であることを示している。私たちとBiogenはFDAのフィードバック意見を検討し、次の行動を評価している。私たちの将来の業務の将来性は、私たちが生物遺伝会社と協力してCRLを解決する能力に大きく依存し、アメリカの規制機関がズランノロンがMDDを治療することを許可する方法を見つけることに成功することは保証されません。ズランノロンによるMDDの治療が規制の承認を得るために成功することは保証されません。FDAの立場は,MDDの承認を支持するためには追加の臨床試験やズラノロンの臨床試験が必要であり,将来控訴を求めてもFDAは彼らの立場を変えない可能性がある。FDAのフィードバックを評価した後、MDDにおけるズランノドンの承認を支持するための1つまたは複数の追加の臨床試験を行った場合、このような試験は時間がかかり、私たちの費用が著しく増加し、成功しないかもしれない。FDAは、我々およびBiogenがMDD患者において行った任意の追加の臨床試験または非臨床研究の実施、またはFDAに提出された臨床試験または非臨床研究データを準備、収集または分析して、規制機関がMDD治療のためのズランノケトンの使用を許可することを支持するために欠陥があることを発見するかもしれない。さらに、MDDにおけるZuranoloneの承認を支援するために追加の臨床試験を行う場合、サイト起動が予想より遅いこと、登録が予想より遅いこと、必要に応じて試験または他の変化を拡大することを含む、開始、進行、登録完了、またはそのような臨床試験の完了に遅延が生じる可能性があり、これは予想スケジュールを満たす能力に影響を与え、コストを増加させる可能性がある。規制部門がMDDの治療を許可することを支援するための追加の臨床試験データを提供することができても、FDAは、それらが完全であるか、または承認を得るのに十分であると考えるデータまたは追加の情報を受け入れない可能性がある。FDAの諮問委員会が将来会議を開いてMDDを治療するズランノドンのNDAを検討する場合、諮問委員会は申請を承認しないことを提案するか、またはFDAが追加の非臨床研究または臨床試験を要求し、承認されたラベルまたは配布および使用制限を制限することを提案する可能性があり、FDAは最終的に諮問委員会の提案に同意する可能性がある。
FDAがMDDの治療のためにZuranoloneを最終的に承認したとしても、FDAは、MDD患者の特定のサブセットの治療のためにのみ承認することができ、またはブロック警告、禁忌症またはリスク評価および緩和戦略またはREMS計画要件のような承認されたラベルにおける制限または制限などの他の制限を適用する可能性がある。例えば、ZURZUVAEは、ヘルスケア提供者が患者にZURZUVAEが中枢神経系抑うつ効果によって運転障害を引き起こすことを提案することを示すブロック警告を含み、ZURZUVAEを服用する人は、ZURZUVAEを服用して少なくとも12時間後に自動車を運転すべきか、または完全な精神的警戒を必要とする他の潜在的な危険活動に従事すべきであることを示す。FDAは予想されたスケジュールに到達できない可能性があり、または彼らが私たちに提出した任意の他の情報または作業の審査時間枠を延長することを選択することができ、または審査期間を規制する任意の時点で遅延が生じる可能性があり、それにより、ズランノドンに関する私たちの計画および予想に否定的な影響を与える可能性がある。FDAまたは他の規制機関の他の決定または行動は、Zuranolone計画、私たちの計画、進展または結果、ならびにZuranoloneによるMDD治療の潜在的な製品プロファイルおよび成功の有無にも悪影響を及ぼす可能性がある。ZuranoloneがMDDの治療のために承認されても、それは、MDD発売後に臨床現場で予想されるプロファイルまたは市場受容度を有さない可能性がある;MDDの新しい治療選択に対する需要が私たちが期待しているほど大きくないかもしれないし、またはMDDのZuranoloneの商業化過程で精算または他の市場に関連する問題に遭遇する可能性がある。私たちと生物遺伝会社がMDD治療のためのズランノドンの規制を受けることができても、私たちと生物遺伝会社は決して成功できないかもしれません
54
承認された場合、ZuranoloneはMDDで商業化され、承認された場合、任意の収入が記録されるか、またはMDDでZuranoloneを販売することによって利益が得られる。
私たちの将来の事業の将来性は、私たちが単独であるいは協力を通じて開発に成功し、監督部門の承認を得て、私たちの現在と未来の候補製品をズランノロンを超える能力に大きく依存しています。計画中の臨床試験を開始し,行われている臨床試験を完成させることができるか,あるいは我々の任意の他の候補製品に関するこのような試験の結果を発表することができるかどうかは,我々が予想しているスケジュールにおいても,我々の開発計画下での臨床試験や他の活動の結果も積極的であろう。私たちまたは私たちの協力者がこれらの候補製品を追加的な実験に進めることができるかどうか、または開発に成功し、規制された承認を得ることができるかどうか、または私たちのこのような候補製品を商業化することに成功したかどうかを決定することはできない(承認されれば)。
私たちの将来の事業見通しは、ズランノロンではなく、私たち単独または協力による開発成功と規制部門の承認を得て、私たちの現在と未来の製品候補製品を承認する能力に大きく依存しています。薬物開発と製品の規制承認は長い、高価で不確定な過程に関連し、高度なリスクに関連する。
任意の候補製品の商業販売の規制承認を得る前に、非臨床研究および臨床試験は、候補製品が各目標適応において使用されることが安全かつ有効であることを証明しなければならない。私たちまたは私たちの協力者(適用すれば)は、臨床開発の各段階で、私たちの任意の他の現在の候補製品または任意の未来の候補製品の有効性および安全性を証明することができない可能性があり、または、提出を規制するために必要な任意の臨床試験または非臨床研究の他の問題に遭遇する可能性がある。非臨床研究または早期臨床試験または臨床試験中期結果の成功は、同じ化合物または他の候補製品に関連する進行中、将来的にまたは完成した研究または試験において繰り返しまたは観察されない可能性がある。著者ら或いは著者らの協力者の一部或いはすべての臨床試験はその主要或いは肝心な副次的終点を満たすことができず、安全問題を引き起こし、或いは一喜一憂の結果を生じる可能性がある。例えば,2019年12月にMDDを治療したZuranoloneの3期臨床試験−Mountain研究−がその主な終点に達していないことを発表した。より高いまたは低い患者曝露を得るために、私たちの候補製品の代替処方または用量を研究することは、予期しない有害事象を引き起こすか、または他の安全問題を引き起こす可能性があり、または負の結果をもたらす可能性があることを発見するかもしれない。例えば、我々が行っているSAGE−324の用量範囲研究、すなわち動力学2研究では、我々が以前の研究で評価した同じSAGE−324最大用量を含む複数の用量を評価している。私たちは、将来の他の研究または計画において、私たちの任意の候補製品の異なる用量、製剤、および用量持続時間を評価することを決定するかもしれない。私たちの候補製品の任意の段階の臨床試験または非臨床研究の結果は、さらなる開発を支持しないかもしれないし、あるいは私たちが予想しているまたは全くないスケジュールで規制部門の承認を申請し、得るのに十分ではないかもしれない。FDAまたは他の規制機関の他の決定や行動は、私たちの計画、進行、または結果に影響を及ぼすかもしれない。
製剤の変化は、任意の候補製品に対して行われたように、生産プロセスを改善または拡大する必要があり、開発が延期される可能性があり、または追加の臨床試験または非臨床研究を要求するか、承認後分析を行うか、または早期処方またはプロセスとは異なる結果をもたらす可能性がある。私たちまたは私たちの協力者は、私たちの期待した時間内に私たちの臨床試験を開始したり、完成させたり、臨床試験の結果を発表することができないかもしれません。私たちまたは私たちの協力者は、私たちの臨床試験において、予想よりも遅いサイト活性化または患者登録とランダム化に遭遇する可能性があり、特に入院または頻繁なサイト訪問が必要な臨床試験では、患者数が少なく、登録基準は歴史的に使用されているものよりも選択的であり、既存の治療法が存在し、他社が大型臨床試験を行っているか、あるいは関連する臨床サイトあるいは私たちのサプライヤーが医療者の不足や人員の大量流出に直面している。また、臨床サイトの開始が予想よりも遅く、データ分析が遅延または問題が発生する可能性があり、より多くの分析またはデータが必要であるか、またはより多くの患者を募集する必要があるか、または我々の任意の臨床試験における有害事象のような他の予期しない問題が生じる可能性がある。このような遅延や問題は裁判の完了と結果発表の遅延を招く可能性がある。
55
私たちが進行していて計画中の開発活動はいくつかの否定的な影響を受けるかもしれない。普遍的な医療保健とサプライヤーの人員不足及び患者と臨床サイトに対する競争は激化し、患者が著者らの臨床試験に登録及び/或いは著者らの試験のために参加する臨床サイトを確定と活性化することを困難にする可能性があり、臨床試験サイト及び/或いはサプライヤーの他の遅延を招く可能性があり、そして患者が登録後に著者らの臨床試験から退出する比率を増加させる可能性がある。容量のせいで、いくつかの臨床サイトは私たちの試験への参加を拒否または延期する可能性があります資源があります条件を制約する。これらの要素は、私たちの臨床試験における臨床サイトの識別、活性化と登録を大幅に遅らせることができ、あるいは私たちの試験を一時停止させる可能性があり、いずれの場合も、予想スケジュール、予算、または他の計画を満たす能力に著しく影響する可能性がある。
新冠肺炎の大流行中のこれらの挑戦に対応するために、著者らまたは私たちの臨床サイトは、臨床試験参加者のサイトへのアクセス回数を最小限にすることを助けるために、臨床試験プログラムおよびデータ収集アクセスを制限または修正すること、およびいくつかの臨床サイトが臨床研究機関のサイトに対する監視に関連する制限など、重要な臨床試験活動に他の制限または制限を加えることを支援するために、いくつかの措置を実施した。その中のいくつかの制限および制限は、将来可能な任意の大流行に対応することを含む未来に再実施されるかもしれない。研究プログラム、研究アクセスまたはデータ収集の制限または修正、監視または監査などの重要な臨床試験活動の制限、またはデータ分析活動に影響を与える可能性のある他の制限は、機関審査委員会が追加的な評価および評価を行う必要があるかもしれない;私たちの試験データの完全性または完全性、試験の動力性、臨床研究終点の完全性または関連性に負の影響を与える;または結果の利用可能な時間に影響を与える。
薬物開発過程は長年の時間を要する可能性があり、発売後の研究とモニタリングを含む可能性があり、これは大量の資源が必要となる。米国で大量に開発されている薬物のうち,一部のみがFDAの規制承認手続きに成功し,商業化されている。したがって、私たちが必要な時に必要な財源が引き続き私たちの開発に資金を提供しても、私たちの現在または未来の任意の候補製品がアメリカやアメリカ以外のどの国でも開発または商業化に成功することを保証することはできません。私たちまたは私たちの協力者がFDAに要求した試験やFDAと議論した試験を行っても、FDAは最終的に決定する可能性があり、設計、試験数、タイプ、研究の患者数や結果は、陽性であっても、規制部門が私たちの研究の適応のいずれかの候補製品の承認を申請または得るのに十分ではありません。または、FDAによって発行されたCRLが、MDDを治療するために使用されるズランノドンのNDAに関連する場合のような、製品の安全性または有効性または我々の予想されるプロファイルをサポートしない。
私たちまたは私たちのパートナーが私たちの現在または未来の任意の候補製品の承認を得ても、私たちとパートナーは、承認された適応でそのような新製品の商業化に成功したり、その製品の販売時間と収入または利益の面で私たちの期待に達することができないかもしれません。
私たちは私たちの投資と目標を支援するのに必要なレベルや時間でZULRESSO(ブシャノロン)CIV注射剤の販売から有意義な収入を得ることができないかもしれない。
我々の最初の製品ZULRESSOは、成人PPDの治療のために、2019年3月にFDAの承認を受け、2019年6月に発売されました。著者らはZULRESSOからの販売収入は複雑な治療要求による重大な障害の負の影響を受け、しかも歴史的に見て、新冠肺炎疫病の影響も受けた。このような要素の一部または全部が未来の収入に否定的な影響を与え続けると予想される。
ZULRESSOは持続輸液として2日半を超えて使用されている。ZULRESSO輸液中の過鎮静や突然の知覚喪失による深刻な傷害のリスクにより,ZULRESSOはREMS計画認証を経てREMS計画の他の要求(輸液中の患者モニタリングに関する要求を含む)を満たす医療監督保健環境でのみ使用されることが承認された。医療保健機関が産後うつを患っている女性を治療するために必要な行動は複雑で時間がかかる。これらの行動は、REMS認証を取得すること、処方承認を得ること、ZULRESSOを管理するプロトコルを確立すること、および満足できる精算を確保することを含む。ビジネスカバー範囲によると、サイトは支払者ごとに補償を交渉しなければならないことが多い。このような要求は産後うつを患っている女性に大きな治療障害をもたらす。これらの障害はZULRESSOの収入増加に悪影響を与え続けることが予想されるが,予想される影響の程度は知られていない。新冠肺炎疫病、アメリカ医療システムに対する関連破壊的影響及びマクロ経済環境の他の変化はこれらの障害を悪化させた。
56
私たちの顧客管理現場チームと販売代表を含むZULRESSOのためのビジネス努力は、主に既存の活発なZULRESSO治療場所を持つ地域に集中しています。このやり方はZULRESSOの収入機会を大幅に制限し続け、収入増加と収入目標の達成を困難にする可能性があると予想される。この方法に鑑み,ZULRESSOの治療地点となる新たなヘルスケア設置数も限られると予想される。ZULRESSOの管理やREMSに関する複雑な要求,関連する制限や制限,あるいは実際や予想されている満足できる精算やカバー範囲や精算上の制限が得られないため,あるいは人員不足を含む他の理由で,過去に活躍してきた治療場所のいくつかの医療環境は輸液準備状態を維持したくない可能性があることも明らかになった。活発な治療場の医療設置もZULRESSO輸液のための容量を制限することが可能である。
私たちはZULRESSOの商業化と収益の面で他の問題と挑戦に直面し続けています
私たちはまたZULRESSOのカバー範囲と返済に関する挑戦に直面し続ける予定だ。これらの制限には,ZULRESSOが精算されるPPD症例の重症度に関連する制限,ZULRESSOの前に他の治療を使用する要求,あるいはZULRESSOや輸液の精算範囲,広さ,獲得可能性または金額の他の制限が含まれている。例えば、各州の医療補助システムはZULRESSOのカバー範囲、条項と時間は引き続き州によって異なると予想され、私たちが遭遇した州はZULRESSOの精算に重大なカバー制限或いは長期遅延を加えている。同様に、ある医療機関や患者が治療の経済的負担を決定することは受け入れられないかもしれない。商業保険を持つPPD女性にZULRESSOを実施したい多くの医療機関は現在医療補助患者を治療しておらず,ZULRESSOから収入を得る能力に悪影響を与えている。
このような問題のいずれも、私たちが収入を作ったり、収入の額や時間に対する私たちの予想を満たす能力を損なう可能性がある。私たちの商業化努力に関連する問題や障害は、私たちの業務、運営結果、財務状況、見通しに実質的な悪影響を及ぼす可能性があり、私たちの努力の範囲や性質をさらに大きく変える可能性があります。私たちはZULRESSOでの私たちの商業化努力が成功することを保証することはできないし、私たちの投資と目標を支援するために必要なレベルや時間に意味のある収入や収入を生み出すことができる保証はない。
57
ZULRESSO、ZURZUVAE、私たちの現在の製品が他の適応のために承認された場合、私たちの現在または未来の候補製品、および任意の未来の製品は、開発と承認に成功すれば、副作用を引き起こす可能性があり、その商業イメージを制限する可能性がある;さらなる開発または規制承認を延期または阻止する;規制機関がボックス警告またはREMSのようなラベル声明を要求する、または他の負の結果をもたらす。
私たちは、非臨床研究において、私たちの候補製品開発の任意の段階の臨床試験において、拡張参入計画の一部として、私たちの任意の製品または候補製品の開始時、商業使用中、または任意の承認された製品の承認後の研究において、不良副作用または他の潜在的な安全問題を観察することができるかもしれない。臨床試験の本質は潜在患者集団のサンプルを利用することである。患者数および曝露時間が限られているため、ZULRESSO、ZURZUVAE、任意の他の現在または未来の候補製品、または任意の未来の製品のいくつかの副作用は、開発および承認に成功すれば、製品に接触するより多くの患者にしか露出しない可能性がある。このような副作用は深刻で、あるいは命に危険があるかもしれない。ZULRESSO、ZURZUVAE、任意の現在の製品(S)、任意の他の既存または将来の候補製品、または任意の将来の承認製品によって引き起こされる副作用が発見された場合:
これらの事件のいずれも、影響を受けた製品に対する市場の受容度を達成または維持することを阻止する可能性があり、私たちの候補製品の開発や私たちの製品を商業化するリスクとコストを大幅に増加させる可能性があり、私たちと私たちの協力者が開発に成功し、規制部門の承認を得て、私たちの現在の候補製品または未来の製品を商業化し、収入を創出する能力に大きな悪影響を及ぼす可能性があると考えられる。
58
規制機関の承認を得て私たちの任意の候補製品を市場に出すことは複雑で長く、高価で不確実な過程であり、FDAと米国以外の規制機関は様々な理由で私たちの任意の候補製品の承認を延期、制限、または拒否するかもしれない。私たちの任意の候補製品に対する規制機関の承認を得ることや、私たちが私たちの製品をマーケティングし始める能力に関するいかなる挫折や遅延も、承認されれば、私たちの業務や将来性に大きな悪影響を及ぼす可能性があります。
私たちまたは私たちの協力者がFDAまたは任意の他の国/地域の機密協定の承認を得るまで、私たちまたは私たちの協力者がこれらの国/地域から必要なマーケティング承認を得るまで、アメリカで私たちの候補製品を販売することは許されません。米国で秘密保持協定の承認を得るか、または米国以外のどの国でも上場承認を得ることは、複雑で、長く、高価で、不確実な過程である。例えば、FDAは、2023年8月4日に、MDDの治療のためのズランノドンのNDAに関連するCRLを発行する。CRLは、NDAがズランノドンのMDD治療の承認を支持するための実質的な有効性証拠を提供しておらず、1つまたは複数の追加の臨床試験を行う必要があることを指摘している。FDAおよび米国以外の規制機関は、様々な理由で、私たちの任意の候補製品の承認を延期、制限、または拒否する可能性があります
59
これらの要素のいずれも、私たちまたは私たちの協力者が規制機関の承認を得て、私たちの候補製品をマーケティングする能力を危険にさらしたり、延期したりする可能性があり、その多くの要素は私たちがコントロールできない。私たちまたは私たちの協力者が私たちの任意の候補製品のマーケティング承認を得たとしても、規制機関または他の政府機関は、指定された用途またはマーケティングの制限、または可能性の高い承認後の研究に持続的な要求を加えることを含む重大な制限を加える可能性がある。例えば、FDAはZULRESSOおよびZURZUVAEの承認に関する承認後義務を規定している。ZURZUVAEに対して、FDAは2つの発売後の研究を要求している:すでに思春期を完成した思春期女性における薬物動態学と安全性研究、及び第二種における胚胎-金属毒性研究を行う。私たちはFDAのスケジュールに従ってこのような義務を履行できないかもしれないし、根本的に履行できないかもしれない。FDAはZURZUVAEのスケジューリングを提案しており、FDA承認後90日以内に完了する予定であり、承認された場合、FDAは、現在または将来の任意の他の候補製品のスケジューリングを提案する可能性がある。この場合,ZULRESSOの場合のように,製品発売前にDEAはFDAの提案を考慮して製品の制御物質スケジュールを決定する必要がある。計画過程のスケジュールは私たちが開発と承認に成功した候補製品をマーケティングする能力を遅らせるだろう。例えば、ZURZUVAEは2023年第4四半期、すなわちDEAスケジュールの後、米国で産後うつ女性を治療する薬物を商業的に発売する予定である。ZURZUVAEのDEAスケジュールが我々の予想どおりに完了する保証はない.DEAスケジュール完了のいかなる遅延も、私たちが計画したZURZUVAE商業発射を延期する可能性があります。
他の候補製品に対する開発努力が成功すれば,将来のNDAへの優先審査をFDAに求める可能性があるが,FDAはこのような優先審査を与えない可能性がある。FDAがNDAを優先審査することを許可しても,FDAは適用される審査スケジュールを満たすことができない場合や,審査を延長する時間枠を選択することが可能である.FDAおよび他の機関の遅延、資源制限、および他の中断は、必要な政府機関の新薬の審査および/または承認に要する時間を遅らせる可能性があり、これは私たちの業務に悪影響を及ぼすだろう。例えば、米国政府は最近の歴史的に何度か閉鎖されており、FDAを含むいくつかの規制機関はキー従業員を休暇にし、キー活動を停止せざるを得ない。今後長期的な政府停止が発生すれば、FDAが提出した規制文書をタイムリーに審査して処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。
FDAの迅速なチャネルと突破療法指定或いはヨーロッパ薬品管理局(EMA)の優先薬物或いはPrime指定は必ずしもより速い開発経路或いは監督審査過程を招くとは限らず、規制承認の可能性を増加させることもない。例えば,FDAはMDDを治療するZuranoloneのNDAに関連するCRLを発表しており,これまでZuranoloneによるMDD治療の迅速かつ治療指定突破が承認されてきた。関係機関が迅速チャネルや突破的治療の指定がわれわれの臨床開発計画のデータ支持を得られなくなったと考える場合,FDAは迅速チャネルや画期的治療の指定を撤回する可能性があり,EMAはPrime指定を撤回する可能性がある。
私たちの製品と候補製品が対象とする疾患や障害患者の数は私たちが予想していたより少ないかもしれないし、あるいは私たちの製品と候補製品の潜在的な市場に対する私たちの他の仮定は正しくないかもしれません。市場は私たちが予想しているよりずっと小さいかもしれません。
どの地域においても、任意の期間において、私たちの製品が対象とする疾患および障害の実際の患者数、および私たちの製品候補対象を決定する正確な方法はない。PPDを有する女性の場合、ZULRESSOおよびZURZUVAEの場合、ZuranoloneのMDDのような規制部門が我々の製品の他の適応を承認することを求めているか、または開発している候補製品の任意の適応を開発または計画しており、疾患や障害の罹患率を推定し、推定を決定する際に適用される仮定を含め、正確ではない可能性がある。いずれの場合も、出版された文献および市場研究には、私たちの推定よりも低い範囲の推定が含まれている一連の推定がある。例えば、産後うつ罹患率の推定値は推定より高い
60
いくつかの発表された文献とクレームデータベースを分析したいくつかの研究で得られた結果で報告されている。これらの違いは,分析方法の違いによる可能性があり,スクリーニングや報告の不足や,一部の患者が臨床現場で治療を求めたくないことによるPPD診断不足である可能性が考えられる。しかし、私たちが私たちの候補製品を開発するPPDまたは任意の他の適応を選択しているか、または選択している可能性がある実際の女性の数は、私たちが考えているよりもはるかに低いかもしれない。たとえ私たちの罹患率推定が正しいとしても、私たちが開発した任意の承認された製品は、関連疾患または障害患者のサブセットにのみ適用されるか、またはそれによって使用される可能性がある。私たちは女性産後うつを治療するZURZUVAEの潜在市場、ZULRESSOの市場、及び私たちの現在と未来の他の候補製品に対する仮説と推定は正確ではないかもしれない。もし私たちが研究している疾患や障害患者の数が明らかに私たちの予想を下回っていれば、私たちまたは私たちの協力者は患者を募集して私たちの臨床試験に参加する際に困難に直面する可能性があり、これは私たちの候補製品の開発を延期したり阻害したりする可能性がある。もし私たちがどんな適応の流行率の推定や私たちの他の市場仮定が正確でなければ、これらの適応の任意の承認製品の市場は私たちが予想しているより小さいかもしれません。これは私たちの収入を制限し、利益を達成する能力を制限したり、収入または利益レベルと時間に対する私たちの期待を満たすことができます。
著者らの候補製品の非臨床研究と臨床試験の陽性結果は必ずしも著者らの候補製品の同じ適応或いは他の適応の後続の非臨床研究と臨床試験の結果を予測するとは限らない。非臨床研究と臨床試験の中期結果が完成すると、このような非臨床研究或いは臨床試験の結果を予測できない可能性がある。私たちが後の非臨床研究と臨床試験で私たちの候補製品の早期非臨床研究と臨床試験の積極的な結果を同じ適応や他の適応にコピーすることができなければ、あるいは私たちが完成した非臨床研究と臨床試験で私たちの中期結果をコピーすることができなければ、私たちは私たちの候補製品の開発に成功し、監督部門の承認を得て商業化することができないかもしれない。
私たちの候補製品の非臨床研究と臨床試験の陽性結果は必ずしも私たち或いは私たちの協力者が同じ候補製品或いは他の候補製品を使用する後続の非臨床研究或いは臨床試験から得られる結果を予測するとは限らない。例えば,MDDやPPDの早期治療に用いられたZuranolone試験とは異なり,MDD患者を評価するZuranoloneの第3段階山岳研究はその主要な終点に達していない。私たちまたは私たちの協力者は、進行中または未来に行われている祖ラノロンまたは私たちの任意の他の候補製品の臨床試験も、その主要な終点を満たすことができない可能性があることを発見するかもしれない。同様に,非臨床研究や臨床試験の中期結果は,いったん完成した非臨床研究や臨床試験の結果を予測できない可能性がある。
我々または我々の協力者はまた、用量の増加または投与頻度または持続時間の増加による、異なる患者集団の研究、または以前の研究とは異なる適応、またはそれに伴う候補製品の使用を含む、臨床試験または非臨床研究において、早期臨床研究または非臨床研究において観察または認識されていない候補製品の安全性の問題、または用量の増加または投与頻度または持続時間の増加を含むイベントの異なる比率または重症度を観察することができる。例えば、我々が行っているSAGE−324用量範囲研究では、我々が以前の研究で評価した同じ最大SAGE−324用量を含む複数の用量を評価している。これらの研究のいずれも、予期しない有害事象を引き起こす可能性があり、または他のセキュリティ問題を引き起こす可能性があり、または負の結果を生じる可能性がある。
非臨床動物モデルからの結果は臨床試験で重複しない可能性がある。多くの候補製品は、多くの中枢神経系疾患に対する候補製品を含み、非臨床方面で有望であるが、人体で類似の安全性、無毒性と有効性を証明できなかった。
製薬やバイオテクノロジー業界の多くの会社が早期開発に積極的な成果をあげた後,後期臨床試験で大きな挫折を経験し,類似した挫折に直面しないことは確認できない。多くの薬物はもっと大きい或いはもっと複雑な後期試験において治療効果と安全性結果を複製できなかった。そのほか、非臨床と臨床データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品が非臨床研究と臨床試験で満足できると考えているが、依然としてFDAの承認を得られなかった。もし私たちまたは私たちの協力者が私たちが進行して計画中の任意の候補製品の非臨床研究または臨床試験で積極的な結果を得ることができなければ、私たちの候補製品の開発スケジュール、規制承認と商業化の見通し、およびそれに応じた私たちの業務および財務の見通しは、重大な悪影響を受けるだろう。
61
現在および将来の候補製品の進行中および計画中の臨床試験の開始、登録または完了中の失敗または遅延は、予想されるスケジュールを達成できないか、または私たちのコストを増加させる可能性があり、そのような候補製品のいずれかに対する規制部門の承認および最終製品(できれば)から収入を得る能力を遅延、阻止、または制限する可能性がある。
開発された各適用段階で臨床試験を成功させることは、秘密保持協定をFDAに提出するか、または米国国外に同等の文書を提出するための前提条件であり、最終的に任意の候補製品を承認し、これらの製品の適応を開発するビジネスマーケティングに使用することである。私たちが行っているどの臨床試験が完成するかどうか、結果が発表されるかどうか、あるいは将来の試験が計画的または予想通りに始まるかどうかはわかりません。もしあれば、臨床試験の開始、登録と完成、および結果の公表は様々な原因で延期または阻止される可能性があります
さらに、臨床試験は、IRBsまたはECsが臨床試験を監督する場所で、IRBまたは道徳委員会またはECによって一時停止または終了するか、または関連する臨床試験のデータおよび安全監視委員会または他の規制機関によって終了または終了を監督することができる
62
さらに、私たちの非臨床研究と臨床試験の間、法規の要求或いはガイドラインの変化或いは意外な事件は私たち或いは私たちの協力者に非臨床研究と臨床試験方案の修正を強要する可能性があり、あるいは適用される規制機関は追加の非臨床研究と臨床試験要求を強要する可能性がある。臨床試験プログラムの改訂または変更は、FDAおよびIRBsの審査および承認を再提出する必要があり、これは、臨床試験のコスト、時間、または成功達成に悪影響を及ぼす可能性がある。もし私たちまたは私たちの協力者が遅延完了に遭遇した場合、または私たちまたは私たちの協力者が私たちの任意の非臨床研究または臨床試験を終了した場合、または私たちまたは私たちの協力者が追加の非臨床研究または臨床試験を要求された場合、私たちの候補製品の開発経路および最終的な商業見通しが損なわれる可能性があり、最終製品から製品収入を得る能力は延期されるだろう。
最後に,我々が既存の要求の変化に緩やかあるいは適応できない場合や,新たな要求や臨床試験を管理する政策を採用すれば,われわれの発展計画は影響を受ける可能性がある。例えば、2022年12月、“食品·薬物総合改革法案”の成立に伴い、国会は、各新薬や生物製品の第3段階臨床試験または任意の他の“重要な研究”のための多様な行動計画の策定と提出をスポンサーに要求している。これらの計画はより多くの異なる患者群がFDA監督製品の後期臨床試験に参加することを奨励することを目的としている。同様に,EUの臨床試験に関する規制構造も最近変化している。EU臨床試験条例,あるいはCTRと呼ばれ,2014年4月に採択され,EU臨床試験指令が廃止され,2022年1月31日に施行された。EU臨床試験指令は、各加盟国で主管する国家衛生当局および独立した倫理委員会に単独の臨床試験申請を提出することを要求しているが、CTRはすべての関連加盟国に申請を提出することのみを要求する集中的なプロセスを導入している。これらの新たな要求を満たすことができなければ,臨床試験を行う能力は延期または停止される可能性がある。
私たちまたは私たちのパートナーは、アメリカ海外で私たちの任意の製品や候補製品をマーケティングするために、またはアメリカ海外で許容可能なレベルの価格設定および精算を得るために、規制部門の承認を求めたり、得られないかもしれません.
私たちまたは私たちのパートナーは、米国以外または任意の特定の国または地域で私たちの製品または候補製品をマーケティングするために、求めることができないかもしれないが、規制部門の承認を得ることはできないかもしれない。米国国外で任意の製品を販売するためには、私たちまたは私たちの協力者は、他の国/地域が多く、それぞれ異なる安全性、有効性、および他の法規要件を確立し、遵守しなければならない。承認手続きは国によって異なり、追加の非臨床研究または臨床試験、制御の製造および分析試験に関連する追加の作業、および追加の行政審査期間に関連する可能性がある。他国で承認を得るのに要する時間は、FDA承認を得るのに要する時間とは異なる可能性がある。一方の国の上場承認は、他の国の上場承認を確保することはできないが、一方の国で上場承認を取得する失敗や遅延は、他国の規制プロセスに悪影響を及ぼす可能性がある。他国·地域での上場承認の流れには、上記で詳述したFDAの米国での承認に関するすべてのリスクやその他のリスクが関与する可能性がある。特に、米国以外の多くの国では、製品が商業化される前に定価と精算承認を受けなければならない。この承認を得るためには、より多くの研究やデータが必要となる可能性があり、これらの国における製品の市場進出の重大な遅延を招く可能性があり、ビジネス的には、市場機会や必要な投資レベルを考慮すると、このような投資は不合理である可能性がある。私たちまたは私たちの協力者が、アメリカ以外の地域や国/地域で私たちの任意の製品や候補製品に規制承認申請を提出するのに十分だと思うデータや情報を生成したとしても、関連規制機関は、私たちが承認要求に適合していないことを発見するかもしれません、あるいは私たちの申請が承認されても、重大な承認後義務がある可能性があります。
63
私たちまたは私たちの協力者が私たちの候補製品の開発に成功し、アメリカ以外の国/地域でマーケティング承認を得ることができても、私たちまたは彼らはその国/地域で許容可能なレベルの価格設定および精算承認を得ることができない可能性があり、私たちまたは彼らが得る可能性のある任意の価格設定および精算承認は、上限、リターン、または他の障害や精算制限のような厳しい制限を受ける可能性がある。価格設定に関連する煩雑な制限や制限なしに米国以外の国でマーケティングおよび定価の承認を得ることができない場合、またはそのような承認を得たときに遅延または他の挫折が生じた場合、私たちまたは私たちの協力者がこのような海外市場で成功したか、または私たちの候補製品を販売する能力を損なうことになる。どのような減価も、私たちの潜在市場の規模や収入潜在力を減少させ、これは私たちの業務、運営結果、および将来性に大きな悪影響を及ぼす可能性がある。
もし私たちの候補製品がアメリカ以外の国や地域で規制の承認を得たり、マーケティングを開始したり(承認されれば)どんな挫折や遅延もあれば、私たちまたは私たちの協力者は、ビジネス的な意味のある国や地域を継続することが、私たちの業務や将来性に実質的な悪影響を及ぼす可能性があると考えています。
私たちは完全に第三者サプライヤーによるZULRESSOおよびZURZUVAEの商業供給、および私たちの候補製品の臨床薬物供給に完全に依存しており、私たちはMDDを治療するための第三者生産の商業供給(承認されれば)、および私たちが承認した製品および候補製品の非臨床、臨床、および商業供給に依存するつもりである。
私たちは現在はありませんし、商業用途のためのZURZUVAEまたはZULRESSO供給を製造するために内部でインフラまたは能力を獲得または開発するつもりもありません。もし私たちが最終的に他の適応または任意の他の既存または未来の候補製品で規制の承認を得た場合、私たちの臨床試験および非臨床研究または将来の商業用途のために使用することを含み、私たちは第三者供給業者に活性医薬物質および完成薬を提供することに完全に依存しています。
私たちは私たちの契約メーカーにZULRESSOに関する活性医薬物質、完成薬、および包装とラベル製品の商業供給を提供することに依存しています。私たちはまた、商業的に供給される活性医薬物質、完成薬、および包装およびラベル製品を生産するのに十分な数のZURZUVAEを生産するために、契約製造業者に依存している。また、私たちの契約メーカーが進行と計画中の臨床試験と非臨床研究のために十分な数の候補製品を生産することに依存し、将来の臨床試験のために私たちの製造プロセスを拡大することに依存したいと思います。これは、私たちのパイプライン優先順位付けの結果と、私たちの開発努力が成功するかどうかに依存します。私たちは私たちの契約メーカーが私たちの製品を生産する時にcGMPを守ることを願う。私たちの契約製造業者が活性医薬成分および最終医薬製品を生産するための施設は、通常、適用される規制機関に関連するNDAまたは同等の外国規制文書を提出した後、FDAおよび他の類似した外国規制機関の承認前検査を完了し、cGMPを含む適用要件に適合するかどうかを評価しなければならない。契約メーカーはFDAの検査を受けなければならない。FDAが契約製造業者による私たちの製品または任意の候補製品の検査に関連する欠陥を発見した場合、開発および承認に成功した場合、FDAは、表483を発行してこれらの欠陥を記録し、修正行動計画を提供して遵守することを要求することができ、開発および承認に成功すれば、製品または任意の候補製品を供給する能力に影響を与える可能性がある。私たちの契約製造業者が、私たちの規格およびFDAまたは適用される外国規制機関の厳しい規制要件に適合した材料を成功裏に生産できず、私たちが予想していた時間内に、または規制検査を通過できなかった場合、彼らは、私たちの製品の製造施設に対する規制承認を確保および/または維持することができないだろう。
しかも、私たちの契約製造業者が十分な品質管理、品質保証、合格者の能力を維持することを直接制御することはできません。さらに、私たちのすべての第三者契約メーカーは、これらの会社のために材料または製品を供給および/または製造する他の会社と協力して、私たちの第三者契約メーカーがそのような材料および製品を生産する際に規制リスクに直面するようにします。したがって、これらの材料や製品を生産する規制要求を満たすことができなければ、私たちの契約メーカー工場の規制許可に影響を与える可能性がある。FDAや適用される外国規制機関が現在または将来、私たちの製品や候補製品を生産するためのこれらの施設が要求を満たしていないと判断した場合、私たちは代替製造施設を探す必要があるかもしれません。これは、私たちの任意の承認された製品の商業化努力や、私たちの候補製品開発と規制承認を得る能力に悪影響を及ぼすでしょう。私たちの契約製造業者への依存はまた彼らがそれを得る権利があるという可能性に直面させます
64
施設は、私たちの商業秘密や他の固有情報にアクセスして盗用する可能性があるだろう。さらに、自然災害が当社の第三者契約メーカーの薬物物質や医薬品の生産を中断または停止したり、ロット損失を招いたりすると、供給不足や供給再建に直面する巨額のコストに直面する可能性があります。
我々は,我々の契約メーカーとZURZUVAE薬物製品の長期供給協定を締結し,適切なときに我々の少なくとも1つの契約製造組織またはCMOとZURZUVAE薬物物質の長期供給協定を締結する予定である。私たちは私たちの契約メーカーとZULRESSO薬物物質と薬物製品に関する長期供給協定を持っている。私たちはすべての潜在的な供給リスクを緩和するのに役立つZULRESSO薬品と薬物の在庫を持っているが、この在庫が十分であることは保証できない。私たちは、SAGE-324またはSAGE-718に薬物または医薬製品を長期的に供給または余分に供給する予定はない。私たちの候補製品の各ロットの薬物物質と薬物製品は、主サービスと品質協定によって管轄された調達注文によって単独で締結された。
もし私たちの他の候補製品の既存のCMOが長期供給協定を締結したくない、あるいは私たちに薬物や薬物製品を供給することを望まない場合、もし私たちが成功して承認されれば、私たちは新しい契約メーカーと交渉する必要があるかもしれない。これらのメーカーは製造プロセスを拡大する必要があり、その後、彼らが製造した薬物製品や薬物を臨床試験または将来の商業化に使用することができる。さらに、どの契約メーカーも、FDAや他の適用可能な外国規制機関の検査を介して、規制機関によって私たちのメーカーとして承認され、その後、彼らが製造した薬品や薬物を商業目的に使用することができ、製品供給の大きな遅延や不足を招く可能性がある検証ロットを完成させる必要がある。承認されれば、私たちは契約メーカーに依存して私たちの製品の商業ロットを生産し続ける予定です。もし私たちが第三者製造の手配を維持できない場合や、商業的に合理的な条項でそうすることができない場合や、私たちの契約製造業者に関連する規制承認をタイムリーに得ることができない場合、任意の承認された製品を商業化することに成功したり、現在または未来の候補製品の開発を成功させることができないかもしれません。
ZURZUVAEまたは私たちの現在または未来の任意の他の製品または候補製品は、私たちが行っている開発努力が成功すれば、十分なレベルで広範な市場で受け入れたり精算することができない可能性があり、販売から得られる収入を制限するだろう。
ZURZUVAEは、米国で産後うつの女性患者の治療に使用されているか、またはMDD患者または私たちの現在または将来の任意の製品または候補製品の治療のための商業的成功であり、FDAまたは他の適用可能な規制機関の許可を得ることに成功した場合、医療専門家、患者、政策立案者、および医療保険支払者の認識および受け入れの程度、および十分なレベルの精算に依存する。
多くの患者が得られ,治療を負担できるようにするためには,カバー範囲や精算の十分性が重要である。処方薬を服用して疾患を治療する患者は、通常、その処方薬に関連する費用の全部または一部を精算するために第三者支払者に依存する。政府当局は、医療保険·医療補助サービスセンター(CMS)、米国衛生·公衆サービス部(HHS)内の機関、個人健康保険会社や健康維持組織などの第三者支払者を含み、どの薬剤に支払うかを決定し、これらの薬剤の精算レベルを設定している。費用統制はアメリカの医療産業と他の地域の主な懸念だ。政府当局とこれらの第三者支払人は,特定の薬物のカバー範囲や精算金額を制限することでコストを抑制しようとしている。支払者は、患者が私たちの製品を清算する前に他のコストの低い療法を試みることを要求するなど、私たちの製品(ZURZUVAEを含む)の保証範囲を制限することができ、患者は、私たちの製品の承認ラベルよりも厳しい深刻性または他の基準を満たすことを要求するか、または煩雑で時間のかかる事前許可手続きを要求するか、または精算金額を制限する可能性があります。これらの制限や制限は、承認された適応における私たちの製品の適切な使用を阻害するかもしれない。精算や遅延保険の獲得制限や制限は、支払者と支払人のタイプによって大きく異なる可能性があります。したがって、開発および承認に成功した場合、ZURZUVAEまたは私たちの任意の候補製品の第三者支払者カバー範囲および補償には大きな不確実性がある。第三者支払者が製品を使用することがその健康計画下の保証福祉であることを決定することを含む、第三者支払者の保証および補償は、多くの要素に依存する可能性がある
65
効果的で医学的に必要な;特定の患者に適している;費用効果が高い;実験的でも研究的でもない。薬品の監督管理審査、定価と精算は国によって異なる。
私たちまたは私たちの協力者は、PPD女性に対するZURZUVAE治療の保険および十分な販売率を迅速に政府援助および個人支払者から得ることができない場合、または承認された場合、MDD患者の治療のためのZURZUVAE、および私たちが開発した他の承認された製品は、私たちの経営業績、製品を商業化することに成功する能力、私たちの資金調達能力、および私たちの全体的な財務状況に実質的な悪影響を及ぼす可能性がある。保険が提供されても、私たちは十分な投資収益を達成するために十分な価格を確立したり維持することができないかもしれない。
政府や他の第三者支払人から製品の保証と精算承認を得ることは高価で時間のかかる過程である可能性があり、支払人に私たちの製品使用を支援する科学的、臨床的、費用効果的なデータを提供する必要があるかもしれない。業界競争は、第三者支払者の薬品処方、すなわち第三者支払者が保険と精算を提供する薬物リストに含まれることになり、これは薬品の定価圧力を低下させることが多い。さらに、価格の低い後発薬または他の代替品がある場合、第三者支払者は、その処方に特定のブランド薬を含むことを拒否するか、または他の方法で患者がブランド薬を得ることを制限することができる。政府の医療計画や個人支払者が要求する強制的な割引やリベート、および将来、米国より低い価格で販売される可能性のある国からの薬品の輸入を制限する法律が緩和されることで、薬品の純価格が低下する可能性がある。製薬会社が価格の予定割引を提供し、医療製品の料金に挑戦することを求める第三者支払者が増えている。また、多くの製薬業者は平均販売価格と最適価格のようないくつかの価格報告指標を計算し、政府に報告しなければならない。このような指標が正確でタイムリーに提出されなければ、処罰されるかもしれない。精算を承認する前に、支払人は私たちの候補製品が治療目標の適応に加え、患者に増量健康福祉を提供したり、医療コストを節約したりすることを証明することを要求するかもしれない。我々はZURZUVAEの支払者と価値に基づくプロトコル戦略を実施し、アメリカの産後うつ女性の治療に使用する予定である。支払者は、価値に基づくプロトコルの使用を受け入れないか、または私たちの方法に同意しない可能性があり、そのような戦略は、市場受容度またはアクセスを増加させない可能性がある。支払者が私たちの価値ベースのプロトコル方法に同意しても、彼らは、ZURZUVAEを精算する前にPPDを治療するために他の低コスト療法を試みることを患者に要求するように制限を加える可能性があり、患者は、私たちの製品によって承認されたラベルよりも厳しい深刻性または他の基準を満たすことを要求するか、または事前許可を要求することができ、これらの制限または要件は、私たちが予想しているよりも煩雑である可能性がある。もし私たちが価値に基づく合意戦略が成功しないと思うなら、私たちは私たちの接近を変えるかもしれない。ZURZUVAEや私たちまたは私たちの協力者が商業化した任意の他の候補製品が十分な保険または精算を受けるか、または保険が合理的な条項で提供されることを保証することはできません。
市場がZURZUVAEを受け入れるかどうかは、米国で女性産後うつの治療のための、または成人MDDの治療のためのズランノドンを含む任意の候補製品を開発することに成功しており、多くの要因に依存する
66
私たちは、特定の疾患の治療パターンを変更するために努力しているか、または許容される範囲内で、産後うつ女性の治療のためのZURZUVAEを含む任意の現在または将来の製品の利点を理解するように医学界および第三者支払者を教育し、将来成人MDDの治療のために許可されたズランノロンであれば、大量の資源を必要とする可能性があり、決して成功しないかもしれない。ZURZUVAEまたは私たちの現在または未来の任意の他の製品または候補製品は、FDAまたは他の適用可能な規制機関によって開発および承認に成功した場合、患者、医師、医療機関、および支払人の十分な許容度、または合理的なレベルの精算を得ることができない場合、またはそのような製品のいずれかが承認された患者の数が私たちの予想よりも少ない場合、利益を達成したり、維持したり、運営に十分な資金を提供することができないか、または予想された程度またはタイムラインに達することができない可能性がある。
たとえ一つの製品が発売承認されても、私たちは重大な発売後の義務と将来の開発と規制の困難に直面する可能性があります。
監督管理当局は発売後の研究、CMCの追加仕事と追加の小児科研究を含む任意の製品の承認に重大で、費用が高いかもしれない発売後の義務を加えるかもしれない。例えば、FDAはZULRESSOとZURZUVAEの承認に上場後の約束を適用しており、これらの上場後の承諾の実施過程で問題や遅延に遭遇する可能性があり、あるいは予期しない結果が生じる可能性がある。ZURZUVAEに対して、FDAは2つの発売後の研究を要求している:すでに思春期を完成した思春期女性における薬物動態学と安全性研究、及び第二種における胚胎-金属毒性研究を行う。
もし私たちまたは私たちの協力者が任意の適応において私たちの任意の候補製品に対して小児科研究を行うことを要求される場合、規制機関はまた、そのような小児科研究を開始する前に追加の非臨床研究または臨床試験を完了することを要求するかもしれない。
ブレシャノロンの場合のように、FDAは私たちの現在または未来の候補製品に制御された物質スケジュールを使用することを提案するかもしれない。例えば,FDAはZURZUVAEの制御物質スケジュールを推奨しており,2023年第4四半期にDEAスケジュール後の第4四半期に米国で発売され商業的に使用される予定であり,PPDを有する女性の治療に用いられ,FDA承認後90日以内に完了する予定である。DEAは制御物質スケジュールを決定するためにFDAの提案を考慮する必要があるだろう。製品が規制物質として決定された場合、製品の製造、輸送、貯蔵、販売、および使用は追加的に規制される。このような薬物の流通、処方、そして調剤も規制されている。これらの制限により、これらの法律と法規は、私たちの候補製品が制御物質を含む商業化を制限するかもしれない。これらの法律と法規を遵守しないことはまた、私たちのDEA登録が撤回され、製造と流通活動が中断され、法令に同意し、刑事と民事処罰、国家行動などの結果を招く可能性がある。DEAは制御物質を付表I,II,III,IVまたはV物質として管理している。定義によると、別表1物質は既定の医療用途がなく、米国で販売または販売されてはならない。医薬品は別表II、III、IVまたはVとすることができ、その中で付表II物質は最も乱用リスクが高いと考えられ、付表V物質はこのような物質の中で相対的に乱用リスクが最も低い物質と考えられている。Brexanoloneは現在表IVの制御物質として規制されている。その他の付表IV規制物質には,ベンゾジアゼピン類などの鎮静催眠薬が含まれている。
ZULRESSOとZURZUVAEは、将来任意の承認された製品もFDAの持続的な要求を受け、製品のラベル、包装、貯蔵と普及を管理し、安全およびその他の発売後の情報を記録保存し、提出する。FDAは重要な上場後の権力を持っています例えば
67
新しい安全情報に基づいてラベルを変更することを要求し、発売後の研究或いは臨床試験による小児科群或いは代替用量或いは投与量方案の深刻な安全リスク、安全性と有効性を評価することを要求する権利がある。
FDAは、セキュリティプロトコルまたは承認後の一部としてREMSの提出を要求する権利もある。例えば、FDAはZULRESSOのREMSを要求する。FDA要件のいずれのREMSも、REMSの遵守、および追加の承認後の規制要件および承認製品販売に対する潜在的な要件または制限を保証するためにコスト増加をもたらす可能性があり、これらすべてが販売量および収入の低下をもたらす可能性がある。さらに、ZULRESSO REMSまたは将来の製品に実施される任意のREMSに準拠できない場合、私たちは追加的な制限、制限、または重大な処罰に直面する可能性があり、いずれも私たちの業務および運営結果に実質的な悪影響を及ぼす可能性があります。
私たち、私たちの協力者、私たちの薬品と薬品の第三者メーカー、そして私たちのそれぞれの工場は、私たちの製品と候補製品(GMPを含む)を生産する際に広範な規制を受け、FDAと他の規制機関の持続的な審査と定期検査を受けて、GMPと他の法規に適合することを確実にする。もし、私たち、私たちの協力者、または規制機関が、私たちが承認した製品または候補製品に問題があることを発見した場合、例えば、生産プロセスのコントロールが悪い、あるいは私たちの製品を製造する施設、または製造過程で他の問題、汚染物質の導入、または意外な深刻または頻度の不良事件が発生した場合、規制機関は、このような製品の市場からの撤退を要求したり、生産を一時停止したりすることを含む、私たちの製品、製造業者、または私たちの協力者に制限を加えるかもしれない。もし私たち、私たちの協力者、私たちが承認した製品、私たちの候補製品、あるいは私たちの製品のメーカーまたは候補製品が適用される規制要求を遵守できなければ、規制機関は他の措置を取るかもしれません
開発および承認に成功すれば、競争療法が存在または出現する可能性があり、ZULRESSO、ZURZUVAE、または私たちの現在または未来の任意の他の候補製品の販売から生じる収入に悪影響を及ぼすことができる。
バイオ製薬業界は競争が激しい。多くの公共·民間会社、大学、政府機関、その他の研究組織が製品の研究·開発に積極的に参加しており、これらの製品は私たちの製品や候補製品と似ているか、あるいは類似した市場に向けられている可能性がある。我々の製品と類似した製品や類似適応に対する製品や療法の開発を求める会社数が増加する可能性がある。私たちの多くの潜在的な競争相手は、単独で、または彼らの戦略パートナーと共に、私たちよりも多くの財力、技術、人的資源を持っており、候補製品の発見と開発、FDAおよび他の規制機関の治療の承認、およびこれらの治療の商業化の面で著しく多くの経験を持っている。バイオテクノロジーと製薬業界の合併と買収は、私たちの数の少ない競争相手により多くの資源を集中させる可能性がある。我々が求めている適応の競争は,治療効果,安全性,利便性,可用性,価格に集中することが予想される。もし私たちの競争相手が私たちが開発する可能性のあるどの製品よりも安全で、より効果的で、副作用が少ない、より便利で、より安い製品と考えられたら、私たちのビジネス機会は減少または消失するかもしれない。
私たちの競争相手も私たちよりも早くFDAや他の規制機関のその製品の承認を得ることができ、これは私たちの競争相手が私たちが市場に入る前に強力な市場地位を確立することをもたらすかもしれない。例えば、規制部門がズランノロンのMDD治療を承認する方法を見つけることに成功したとしても、FDAは治療法としてのズランノロンの承認を支援するための追加の試験またはデータを必要とする可能性がある
68
MDDの場合、予期される審査スケジュールに到達できない可能性があり、またはその審査の時間枠を延長することを選択することができ、または審査期間を規制する任意の時点で遅延が存在する可能性があり、これらは、MDDの治療においてズランノルノンを使用する私たちの商業的機会を減少または除去するために、我々の競争相手に市場地位を確立するための時間を提供する可能性がある。
現在,ZURZUVAEやZULRESSO以外にPPD治療に特化した薬剤は承認されていない。現在、産後うつに対する治療基準は通常心理治療を含む;しかし、中度または重度産後うつの患者は通常、選択的セロトニン再取り込み阻害剤(SSRI)、セロトニンおよびノルアドレナリン再取り込み阻害剤(SNRI)などの抗うつ薬の処方を処方される。もしZURZUVAEが成功的に発売され、商業化され、女性産後うつを治療するために使用されれば、ZULRESSOに対するビジネス機会をさらに制限するかもしれない。また,ZULRESSOとZURZUVAEはLPCN 1154によるPPD治療の競争に直面する可能性もあり,LPCN 1154はLPCN 1154が505(B)(2)調節経路に従って開発されている神経活性ステロイドBrexanoloneの経口製剤である。
もし将来承認されれば、ズランノロンはまたMDD治療の競争に直面する可能性がある。MDD患者は通常SSRIs、SNRIsと非典型抗精神病薬物を含む各種の低コストの抗うつ薬物治療を使用する。Zuranoloneが米国でMDDの治療に許可された場合、AXS-05によるMDD治療の競争に直面する可能性があり、AXS-05はNMDA受容体アンタゴニスト右メサフェンとアンフェタミンの組み合わせ処方であり、アンフェタミンはFDAによって承認されたノルアドレナリンおよびドーパミンに影響を与える抗うつ薬であり、この組み合わせは2022年8月にFDAによって成人MDDの治療に許可された。Zuranoloneが米国でMDDの治療として承認された場合、それはまた、エスケタミンとカリラジンからの競争に直面する可能性がある。エスケタミンは、急性自殺願望または行動を有するMDD成人患者の難治性抑うつおよび抑うつ症状の治療のために承認され、カリラジンは最近、抗うつ薬治療を受けている患者のMDDの補助治療のために承認された。他のいくつかの会社はMDD治療のための候補製品を開発している。
神経活性ステロイド分野では,GABAの調節が主に注目されているA受容体以外にも、Marinus製薬会社やMarinusを含む多くの会社からの競争に直面している。2022年3月、MarinusはFDAがガーナ松龍、既知のGABAを承認したと発表したACDKL 5欠乏症に関連するてんかん発作の治療に用いられる陽性アロステリック調節剤神経活性ステロイドであって、まれな遺伝性てんかん。その他GABAA競争相手には,Cerevel治療会社が開発したDarigabatがあり,てんかんやパニック症の治療に用いられている。
SAGE-324、新しいGABAA受容体ポジ型アロステリック調節剤は,特発性振戦の二期開発段階にある。特発性振戦の治療のための開発に成功し、許可された場合、SAGE-324は、βアドレナリン作動性遮断薬である心得安および抗けいれん薬プロメタゾンを含む現在の第一線の治療薬からの競争に直面するであろう。他社も特発性振戦を治療する潜在薬を開発しており,Jazz製薬社が現在評価している2 b期に開発されているT型カルシウムチャネル調節剤と,Praxisが開発している2期T型カルシウムチャネル調節剤を含む。
多くの会社がNMDA受容体を調節するための製品の開発に取り組んでいる。ノワ製薬はケイド治療会社を買収した後,独自のN−メチル−D−アスパラギン酸受容体陽性アロステリック調節剤CAD−9303を開発しており,統合失調症に関連する認知障害を検討している。また、Vaccinex社はハンチントン病の認知障害を治療する薬剤としてVX 15/2503を評価している。VX 15/2503はシグナル素4 D(SEMA 4 D)蛋白に対するモノクロナル抗体である。Prilenia Treateutics,B.V.はSigma−1受容体(S 1 R)アゴニストプリドバジンも評価されており,ハンチントン病治療の3期臨床試験である。いくつかの会社はアルツハイマー病を治療する製品をすでにあるいは開発している。
69
BiogenやShionogiとの既存の協力、および将来のいかなる協力も、候補製品の開発成功や規制承認や製品の商業化を招くことはないかもしれない。私たちの協力者は、相互競争の優先順位、相互衝突のインセンティブ、または重要な決定(規制や開発戦略または適切な計画支出を含む)で私たちとは異なる観点を持っている可能性があり、これは私たちの開発と商業化努力を阻害または延期したり、私たちのコストを増加させたりする可能性がある。もし私たちが任意の協力者と深刻な相違があり、どの協力者もその義務を履行できなかった場合、または私たちの協力を全部または部分的に終了できなかった場合、または合理的なビジネス条項で私たちの業務に重要な未来の協力を作ることができなければ、私たちの業務は不利な影響を受ける可能性があり、私たちは遅延、紛争、または訴訟を受ける可能性がある。
私たちの薬物開発計画、女性産後うつを治療するZURZUVAEの商業化計画、および私たちの候補製品の任意の潜在的な商業化は支出を支払うために大量の追加の現金を必要とするだろう。私たちのいくつかの候補製品について、私たちは製薬やバイオテクノロジー会社と協力して、いくつかまたはすべての市場でこれらの候補製品を開発し、潜在的な商業化を行うことを決定するかもしれない。
私たちの既存と未来の協力は、あれば、どんな製品の開発や商業化にもつながらないかもしれない。もし私たちが単独で活動に参加すれば、私たちの協力者は候補製品の開発と商業化において同様の挑戦と障害と、協力の下での運営に関する追加的な挑戦に直面するだろう。例えば、我々は、Biogenと協力および許可協定を締結し、ZURZUVAEおよびZuranoloneおよびSAGE−324を含む他の製品を米国で共同開発および商業化し、これらの候補製品を日本、台湾および韓国以外の世界の他の地域またはShionogi領土(Zuranolone)で開発および商業化する権利をBiogenに付与している。我々はShionogi&Co.,Ltd.,Shionogiと単独で協力しており,この協力に基づいてShionogiがShionogi地域でZuranoloneを開発し商業化する権利を与えている.このような協力の下での努力は成功しないかもしれないし、私たちはBiogenまたはShionogiから任意の追加のマイルストーン支払い、利益共有収入、または特許使用料支払いを決して受けないかもしれない。例えば、ZURZUVAEは米国で成人PPDの治療に許可されているが、FDAはMDD治療のためのズランノドンCRLをNDAに発表した。BIOGenはPPD治療用のZURZUVAEの最初の商業販売から7,500万ドルの潜在的マイルストーン支払いを得ることが予想されるが、MDDを治療するための最初のZURZUVAEの商業販売に関する150.0ドルの記念碑的支払いをBIOGenから得ることは決してないかもしれない。
また,ほとんどの連携では,我々の既存の連携を含めて,意思決定のある程度の制御権が我々の協力者や我々の協力者と共有される.私たちの協力者は彼らの意思決定権を利用して、私たちの候補製品の開発と商業化の潜在力を低下させ、あるいは他の方法でそれに悪影響を及ぼす可能性があるという決定を下すかもしれない。同様に、私たちが意思決定権を共有する場合、承認されれば、意思決定で一致する必要性は、私たちの計画や製品の商業化プロセスを緩和したり阻害したりして、私たちのスケジュールを満たしたり、私たちの目標を達成できなくなったりする可能性があります。私たちの協力者は、相互競争の優先順位や異なるインセンティブを持っている可能性があり、彼らが私たちの協力から資源を移動させたり、適切な支出レベルや規制や開発戦略に合意できない可能性があります。これは、私たちの全体的な開発と商業化努力を阻害したり、私たちの全体的な支出を増加させたりする可能性があります。私たちの協力者は、競争相手と一緒に競合製品を開発したり、あるいは協力で評価された候補製品が協力者自身の製品と競争力を持っている可能性があると考えることができます。生物遺伝会社と協力した場合,両社とも特定の適応におけるある製品に対する何らかの排他的条項に同意しており,協力以外の何らかの開発機会を制限する可能性がある.また、世界的または重要な地域の主要製品開発において協力者の能力や資金に依存していれば、私たちの協力者が合意に定められた義務や協力を履行できなかった場合、私たちの業務は悪影響を受ける可能性がある。協力者と共同開発した技術や製品の所有権についても議論が生じる可能性があり、これは、影響を受ける候補製品の開発および商業化のいずれかの能力に悪影響を及ぼす可能性がある。
協力の交渉と記録は複雑で時間がかかる。また,最近では大手製薬会社間の業務合併数が大きく,将来の潜在的パートナー数の減少を招いている。私たちはタイムリーで受け入れ可能な条項でもっと多くの協力を交渉することができず、交渉することさえできないかもしれない。
70
私たちは、既存の候補製品以外の他の候補製品を成功裏に識別または発見することができないか、または新薬またはIND臨床開発申請を予想される速度で提出することができないかもしれないし、特定の候補製品または適応を追求するために限られた資源を使用する可能性があり、より利益的または成功可能性の高い可能性の高い候補製品または適応を利用することができないかもしれない。
私たちの業務の成功は、私たちと私たちのパートナーが、私たちの現在の候補製品が製品の開発に成功し、承認され、それを商業化する能力と、将来の開発のために新しい化合物を生成し、このような新しい化合物の臨床開発を行うためにINDSに提出するために必要な非臨床作業を成功させる能力に依存する。われわれの研究計画は,非臨床開発基準に適合した新しい化合物を生成できない可能性があり,また,このような化合物の産生に成功しても,INDの臨床開発応用を支援するために必要な非臨床データや他のデータを生成できない可能性があり,いずれの場合も,予想される数や速度や完全に様々な理由で生成することはできない。例えば、私たちは私たちの関心分野で十分な数の新しい目標を決定できないかもしれない。我々の研究方法は十分な数の新しい化合物の産生に成功しない可能性があり,我々が決定した目標領域での非臨床試験に適している。我々が興味のある分野で新たな化合物を生成しても,これらの化合物が非臨床開発に適していないこと,あるいは非臨床開発において臨床開発IND文書を支持しないデータを生成する可能性がある。標的を決定し、化合物を生成し、非臨床研究を行い、INDを準備するために必要な異なる段階で、著者らは十分な技術、財政と人力資源を投入して研究を行っていないかもしれない。他の潜在的な候補製品は、有害な副作用を有することが証明される可能性があり、または積極的なリスク/収益プロファイルがない可能性があり、または候補製品がさらなる開発に適合しないか、または上場承認を得ることができない可能性がある他の特徴を有する可能性がある。また、関心のある分野で新たな化合物を生成しても、承認されれば、そのような化合物の商業化の可能性に影響を与える可能性のある新しい立法を含む戦略的理由でこれらの化合物が研究される価値がないことを決定する可能性がある。
私たちの財務と管理資源が限られているため、私たちは限られた数の臨床と研究プロジェクトと候補製品に集中して、現在いくつかの脳健康障害に集中している。したがって、私たちは他の候補製品を探す機会を放棄または延期するか、または後により大きな商業潜在力を有することが証明された他の兆候を探す機会を放棄または延期するかもしれない。新製品候補製品を確定する研究プロジェクトには大量の技術、財力と人的資源が必要である。私たちは私たちの努力と資源を潜在的な計画または候補製品に集中させるかもしれません。これらの計画または製品は最終的には成功しないことが証明され、いかなる商業的に実行可能な薬物も生成されないかもしれません。私たちの資源分配決定は私たちが他の実行可能な機会を利用できないようにするかもしれない。特定の候補製品の商業的潜在力や目標市場を正確に評価していなければ、このような独占開発と商業化の権利を保持した方が有利な場合には、将来の協力、許可、または他の特許権使用料手配によって貴重な権利を放棄することができるかもしれない。もしこのような事件のいずれかが発生すれば、私たちの業務に実質的な悪影響を及ぼす可能性がある。
私たちは依存し、引き続き第三者に依存して私たちの候補製品のための任意の臨床試験を行うことが予想されます。これらの第三者がその契約義務の履行に成功し、適用基準を遵守し、予想される期限内に完了しなかった場合、規制部門の私たちの製品の承認を得ることができないか、または商業化することができない可能性があり、私たちの業務は深刻な損害を受ける可能性がある。
私たちは独立して臨床試験を行うことができない。著者らは医療機関、臨床研究者、契約実験室とその他の第三者、例えばCROに依存して、著者らの候補製品に対して臨床試験を行った。我々は第三者CROと協定を締結し,我々が行っている臨床試験にモニタリングと管理データを提供した。私たちはこれらの方が私たちの候補製品のために臨床試験を実行し、その活動のいくつかの方面だけを制御することに深刻に依存している。したがって,われわれ自身に完全に依存しているスタッフに比べて,これらの臨床試験の進行,スケジュールと完成および臨床試験により開発されたデータの管理の直接制御は少ない。外部の当事者とのコミュニケーションも挑戦的である可能性があり、ミスや協調活動の困難を招く可能性がある。外部の当事者は
71
これらの因子は第三者の臨床試験の意思や能力に重大な悪影響を与える可能性があり、著者らのコントロール範囲を超える意外なコスト増加に直面する可能性がある。しかし、私たちは私たちのすべての臨床試験が適用された方案、法律と法規の要求、そして科学的な基準に従って行われ、私たちのCROへの依存が私たちの規制責任を免除しないことを確実にする責任がある。著者ら、臨床研究者と著者らのCROは必ずGCPを含む臨床試験結果を行う、モニタリング、記録と報告する法規とガイドラインを遵守し、データと結果が科学的に信頼性と正確であることを確保し、そして試験患者が臨床試験に参与する潜在リスクを十分に理解することを確保しなければならない。これらの規定は、FDA、欧州経済地域(EEA)加盟国の主管当局、および同様の外国規制機関によって、臨床開発または臨床試験中の任意の候補製品に対して実行される。私たちまたは私たちのCROまたは契約製造業者がこれらの規定を遵守できなかった場合、または私たちの臨床レジメンまたは他の法規要件または他の理由を遵守できなかったために得られた臨床データの質または正確性が影響を受け、収集された臨床データに依存できない場合、臨床試験を繰り返したり、臨床試験の持続時間を延長したり、または私たちの臨床試験の規模を増加させることが要求される可能性がある。これは規制の承認過程を延期し、最高の民事と刑事処罰に達する法執行行動に直面させる可能性もある。第三者CROとの任意の関係が終了したり、CROを交換する必要がある場合、代替CROとの合意がタイムリーにできないか、または根本的に合意できない可能性があります。これらの問題のいずれも、規制部門が私たちの候補製品を承認することを大幅に延期または阻止する可能性があり、支出を大幅に増加させる必要がある。この場合、発売に成功して商業化すれば、私たちの財務業績が損なわれる可能性があり、私たちのコストが増加する可能性があり、ZULRESSOやZURZUVAE以外の製品から収入を得る能力が遅れる可能性があると考えられます。
私たちの未来の成功は私たちが合格した人材を引きつけ、維持し、奨励する能力にかかっている。
我々の目標を実現するためには,研究開発,臨床開発,商業化に専門知識を持つ強力な管理チームが必要である。私たちはすべての幹部と雇用協定を締結しましたが、彼らはみんな“勝手”に雇われていて、いつでも私たちとの雇用関係を終わらせることができます。私たちは私たちの役員や他の職員たちに“キーパーソン”保険を提供しない。合格した人材を採用して維持することが私たちの成功の鍵だ。多くの製薬とバイオテクノロジー会社の間の類似者に対する競争を受けて、私たちは受け入れ可能な条件でこれらの人員を吸引し、維持することができないかもしれない。私たちは大学や研究機関から科学者を募集する競争も経験した。臨床試験で成功しなかったか、あるいは監督部門の許可を得られなかったことは、合格者の採用と保留をもっと挑戦的になる可能性がある。もし私たちが引き続き高い素質の人材を誘致し、維持することができなければ、私たちの発展努力、商業化活動、業務、財務状況、経営結果と成長の見通しはすべて不利な影響を受ける可能性がある。
私たちは潜在的な製品責任のリスクに直面しています。もし誰かが私たちにクレームをつけたら、私たちは重大な責任を招くかもしれません。
ZULRESSO、ZURZUVAEを販売し、発売され商業化され、将来承認された製品と私たちの候補製品の臨床試験での使用に成功すれば、私たちは製品責任クレームのリスクに直面するだろう。患者、医療提供者、または他の使用、処方、販売、または他の方法で私たちの製品および候補製品に接触した人は、私たちに製品責任を請求するかもしれません。例えば、任意の製品または候補製品が、臨床試験、製造、マーケティング、販売、または商業使用中にダメージを与えたと言われている場合、または不適切が発見された場合、私たちは起訴される可能性がある。このような製品責任クレームは、製造欠陥、設計欠陥、製品固有の危険(アルコールまたは他の薬物との相互作用の結果を含む)、リスク知識、不注意、厳格な責任、および保証違反の告発を含むことができる。州消費者保護法によると、クレームも主張することができる。もし私たちが製品責任クレームの制約を受けて、自分を弁護することができなければ、私たちは大きな責任を招くかもしれない。是非曲直や最終結果にかかわらず、製品責任クレームは以下のようになる可能性がある
72
私たちは製品責任保険を維持して、毎年総保険限度額は二千万ドルです。しかし、私たちの保険カバー範囲は私たちが受ける可能性のあるいかなる費用や損失を補償するのに十分ではないかもしれない。さらに、将来的には、保険範囲がますます高くなると、保険範囲がますます高くなる場合を含めて、合理的なコストや十分な金額で保険範囲を維持することができない可能性がある。予期せぬ副作用を有する薬物に基づく集団訴訟では,すでに多くの判決が下されている。いかなる製品責任訴訟や他の訴訟のコストも、解決が私たちに有利であっても、巨大であり、特に私たちの業務規模や財務資源を考慮している可能性がある。私たちに提出された製品責任クレームあるいは一連のクレームは私たちの株価を下落させる可能性があります。もし私たちがこのようなクレームに対抗できなかった場合、それによって生じる判決は私たちの保険範囲を超えて、私たちの財務状況、業務と見通しは重大な悪影響を受ける可能性があります。
もし私たちが医療補助薬品リベート計画または他の政府定価計画の下での報告と支払い義務を履行できなければ、私たちは追加の精算要求、処罰、制裁、罰金を受ける可能性があり、これは私たちの業務、財務状況、運営結果、成長の見通しに実質的な悪影響を及ぼすかもしれない。
私たちが参加した医療補助薬品リベート計画と他の政府計画は連邦政府に定価データを報告することを要求して、リベートを支払って割引計画に参加することを要求します。他の計画は、特定のエンティティに請求することが許可されているZULRESSO、ZURZUVAE、または私たちが規制承認を得た任意の未来の製品の価格に制限を加えます。これらの計画およびその要求に関する法律および法規の変更または拘束力のある指導意見は、ZULRESSO、ZURZUVAE、または私たちが規制承認を得た任意の将来の製品の保証範囲および精算に負の影響を与え、私たちの運営結果に悪影響を及ぼす可能性がある。私たちはこれらの価格報告書と返金義務を守ることができず、私たちの財務業績にマイナス影響を与えるかもしれない。改正された“患者保護と平価医療法案”はここでACAと呼ばれ,この法案に基づいて公布された法規は予想できない方法で我々の義務に影響を与える可能性がある。
定価と返却計算は製品や計画によって違います。計算は複雑で、一般的に私たち、政府、あるいは規制機関、そして裁判所によって説明されるだろう。もし私たちがこれらの計画に基づいて報告した金額を再申告あるいは再計算する義務があれば、医療補助薬品リベート計画を管理する法律と法規、そして私たちの価格割引とリベートのコストが増加するかもしれません。また、医療補助薬品返却計画と他の連邦または州薬品定価計画に基づいて提出された価格設定データに関するミスに責任を負うことができるかもしれません。返却点の追跡と計画返金を含めて、虚偽の製造業者の平均価格または最適価格情報を故意に政府に提出したことが発見された場合、すべての虚偽情報に対して民事罰金を科します。必要なデータを提出できなかった場合、情報が期限を超えた毎日の民事罰金を招く可能性があり、CMSが私たちのMedicaid医薬品リベートプロトコルを終了する理由となる可能性があり、このプロトコルによると、Medicaid計画に参加しているか、または、340 B計画の要求を遵守できなかった場合、衛生資源およびサービス管理局(HRSA)は、340 B計画参加プロトコルを終了することを決定することができる。CMSが私たちのリベートプロトコルを終了するか、またはHRSAが私たちの340 B計画参加プロトコルを終了した場合、連邦医療補助または連邦医療保険B部分に基づいて、私たちが保険を受けている外来薬物にいかなる連邦金も支払わない。私たちはまた、薬品定価交渉計画およびB部分とD部分インフレリベート計画に適用される民事罰金とその他の処罰を受け、以下のように命名されたリスク要因である
73
“医療コストを削減するための医療法規は、私たちの業務や運営結果に実質的な悪影響を及ぼす可能性があります.”
私たちは他の法律と法規の制約を受けて、これは私たちを刑事制裁、民事処罰、契約損害、名声損害、利益と将来の収入の減少に直面させるかもしれない。
私たちは、連邦政府および私たちが現在または未来に業務を展開する可能性のある州と外国政府の複数の医療および他の法定および規制要件と法執行の制約を受けている。
私たちは、現在または将来的に第三者支払者、医療提供者、患者、医療機関、ZULRESSOおよびZURZUVAEの推薦、処方、精算および管理に機能する他の人との相互作用と手配を行い、将来の任意の他の候補製品に対して同様の役割を果たすことになり、開発と承認が成功すれば、広範に適用される詐欺や乱用、および他の医療保健法律および法規の制約を部分的に受け、これらの法律および法規は、私たちのマーケティング、販売および流通ZULRESSOまたは任意の将来承認された製品の業務または財務および関係を制限する可能性がある。適用される連邦と州医療法律法規の制限は
74
私たちの未来の実践と業務配置が適用された医療法律と法規に適合することを確実にして、費用が高い。政府当局は、私たちの業務実践と手配が現在または未来に適合していないと結論するかもしれないが、詐欺および乱用または他の医療保健法律および法規を適用する現行または将来の法規、法規または判例法に関連している。私たちのビジネスチームや他の従業員、コンサルタント、またはサプライヤーが行った活動を含む私たちの行動や運営が発見された場合、これらの法律または私たちに適用される可能性のある任意の他の政府法規に違反していることが発見された場合、私たちは重大な民事、刑事および行政処罰、損害賠償、罰金を受け、連邦医療保険や医療補助のような政府援助の医療計画から除外される可能性があり、これらはいずれも私たちの運営を深刻に乱し、私たちの業務や財務状況に実質的な悪影響を及ぼす可能性がある。もし政府当局が私たちの協力者の商業行為が適用された法律に適合していないと結論すれば、私たちも実質的な負の影響を受ける可能性がある。もし私たちがそれと業務を展開することを期待している任意の医師または他の提供者または実体が適用されない法律に適合していないことが発見された場合、彼らは政府の援助された医療計画から除外されることを含む刑事、民事または行政制裁を受ける可能性がある。
私たちと私たちの従業員はまた、前述したように、FDAおよび適用された非米国規制機関によって実施された法規、米国海外活動に適用される反賄賂および反腐敗法律法規、財務および他の情報またはデータをタイムリーに正確に報告するための規則、およびインサイダー取引に関連する規則を含む、私たちの業務に関連する他の法律および法規を遵守しなければならない。
行動指針を策定し、積極的なコンプライアンス計画を実施しているにもかかわらず、従業員の不正行為を常に識別し、阻止できるわけではなく、このような行為を発見し防止するための予防措置は、未知または未管理のリスクや損失を効果的に制御できない可能性があり、あるいはこれらの法律や法規に遵守できないことによる政府調査や他の訴訟から私たちを保護することができる。
データ収集は、個人情報の使用、処理、および国境を越えた伝送に関する制限された規制によって制限される. このような規定を遵守することは時間がかかって重くなるかもしれない。もし私たちが不適切な個人情報の処理を発見されたら、私たちは罰金と処罰、訴訟、名声の損害を受けるかもしれない。
私たちは健康と他の個人情報のプライバシーと安全を管理する多くの連邦、州、非アメリカの法律を守らなければならない。上述したように、HIPAAは、我々のビジネス活動に適用される範囲で、個人が健康情報を識別することができるプライバシー、セキュリティ、および送信にいくつかの要求を提示している。また,米国で臨床試験を行う際には,これらの試験に関連して収集された任意の個人情報も,当社がこれらの試験を行う際の義務を規定している連邦人間被験者保護政策(共通ルール)によって規制されている。
私たちはEU、EU、または他の国で被験者を募集して、私たちが行っているまたは未来の臨床試験に参加する予定です。私たちがそうするとき、私たちは、これらの個人に関連する個人健康データを含む、収集、使用、記憶、送信、および他の個人データの処理に関する制限を含む追加のプライバシー制限を受ける可能性がある。例えば,ヨーロッパ経済地域の臨床試験活動は,個人データ処理に関する一般データ保護条例やGDPRによって規制されている。GDPRは個人データを扱う会社に対していくつかの要求を出し,個人データを欧州経済圏(米国への移行を含む)に移行する行為に対して厳しいルールを制定し,GDPRやEU加盟国の関連国データ保護法を遵守できなかった要求に罰金と処罰を科した。GDPRはまた、データ主体と消費者協会に個人訴訟権利を与え、監督当局に苦情を提出し、司法救済を求め、場合によってはGDPR違反による損害賠償を得ることができる。GDPRの義務は重く、私たちの業務、財務に
75
経営状況、経営結果、見通し。GDPRを遵守することは厳格で時間のかかるプロセスであり、私たちの業務コストを増加させたり、私たちに業務やり方の変更を要求したりする可能性があり、私たちはこれらの努力をしたにもかかわらず、いかなるヨーロッパ活動に関連した罰金や処罰、訴訟、名声損害のリスクに直面する可能性がある。現在,個人データ転送に関する問題には大きな不確実性があり,このようなデータ転送が挑戦されれば,EU法に適合しているかどうかを合理的に決定することはできない.イギリスのEU離脱は、一般的にイギリスの離脱と呼ばれ、イギリスの未来のデータ保護規制に不確実性をもたらしている。欧州委員会はイギリスのデータ保護水準に関する十分な決定を採択した。個人データは現在、ヨーロッパ経済地域からイギリスに自由に流れることができるが、欧州委員会がイギリスが十分なレベルのデータ保護を提供しなくなったと認定した場合、十分な決定を一時停止する可能性がある。世界の多くの他の国には似たような法律があり、これらの法律(発展·拡大中)は複雑で一致しない可能性のある義務を生み出し、私たちの業務に影響を与える可能性がある。
私たちはまた、カリフォルニアの消費者のためのプライバシー権(法律で定義されているような)を創造し、消費者または家庭の個人データを処理するエンティティにより多くのプライバシーおよびセキュリティ義務を課す“カリフォルニア消費者プライバシー法”(California Consumer Privacy Act、略称CCPA)の制約を受けている。現在HIPAAや臨床試験法規に拘束されている保護された健康情報には例外があるが,CCPAは我々の業務活動に影響を与える可能性がある。カリフォルニアで最近行われた国民投票でもCCPAが改正され、2023年1月1日に発効する追加義務が追加された。2020年11月、カリフォルニア州有権者はCCPAに対して重大な改正を提出し、専門的なカリフォルニアプライバシー規制機関であるカリフォルニアプライバシー保護局(CPPA)を設立し、援助するカリフォルニアプライバシー権法案(CPRA)投票イニシアティブを承認した。CPRAによると、新しい施行条例が発表され、これは私たちが新しいまたは追加的な義務を負うことにつながるかもしれない。CCPAを遵守しないことは、重大な民事処罰や禁止救済、または法定または実際の損害賠償を招く可能性がある。しかも、カリフォルニアの住民たちは特定の種類の事件について個人的な訴訟を提起する権利がある。このようなクレームは重大な責任と損害を招くかもしれない。カリフォルニアを除いて、少なくとも11州でもCCPAに似た全面的なプライバシー法が採択された。このような法律は施行されたか、2026年末までに施行されるだろう。これらの法律は、CCPAと同様に、個人情報の処理に関する義務と、健康データが含まれる場合がある“敏感”データを処理する特別な義務とを規定している。このような法律のいくつかの規定は私たちの商業活動に適用されるかもしれない。また、2023年の立法会議中に全面的なプライバシー法を制定することを積極的に考慮している州もあり、これらの立法会議は、ニューヨーク州およびニュージャージー州を含む2024年以降に発効する。他の州も将来的には似たような法律を考慮し、国会でも連邦プライバシー法の成立について議論されてきた。私たちの業務の健康情報に影響を及ぼすかもしれないいくつかの州の専門的な規制もある。例えば、ワシントン州は最近、健康情報の収集と共有を規範化する健康プライバシー法を採択し、この法律も個人的な訴権を持っている。コネチカット州とネバダ州も同様の法律を採択して消費者の健康データを規制している。
しかも、連邦レベルでは、私たちのビジネス活動に影響を与える可能性のある国家データプライバシー法を採択するために大きな努力をしている。CCPAおよび他の州および連邦レベルの他の制定されたまたは潜在的な法律の実施および解釈をめぐる不確実性、曖昧性、複雑性、および潜在的な不一致は、私たちの業務が、個人データおよび保護された健康情報のプライバシー、セキュリティおよびセキュリティに関連して変化する規制環境の影響を受けやすいことを示している。もしこのような法律を守らなければ、私たちは罰金、処罰、または個人訴訟を受けるかもしれない。これらの法律は、私たちの研究対象の決定、ビジネスパートナーとの関係、そして最終的に私たちの製品のマーケティングと流通を含む、私たちのビジネス活動に影響を与えるかもしれません。我々はすでにCCPA遵守の流れを管理し,より多くの情報や指導を得た後,CPRAや他の連邦や州立法が我々の業務に与える影響を評価し続けている.
上記に加えて、プライバシー法またはデータセキュリティ法に違反するいかなる行為、特に流用、紛失、または他の不正使用または機密患者または消費者情報の漏洩に関連する重大なセキュリティ事件または違反行為は、私たちの業務、名声、および財務状態に重大な悪影響を及ぼす可能性がある。データ制御員として、私たちが招聘した代表者は、私たちのCROを含む個人データを処理する任意の第三者サービスプロバイダに責任を負います。我々が実施しているプライバシーやセキュリティに関する保障措置は、第三者がこのような情報を処理、記憶、送信することに関連するすべてのリスクから私たちを保護してくれる保証はありません。場合によっては、米国でも他の国でも、セキュリティホールのために、これらの脆弱性を個人および/または政府エンティティに通知する義務があるかもしれません。
76
また、2022年10月、Joeバイデン総裁はEU-米国プライバシーの盾に代わるEU-米国データプライバシーの枠組みを実施する行政命令に署名した。EUは2022年12月にEU-米国データプライバシーフレームワークによる十分性決定を開始し,欧州委員会は2023年7月10日に十分性決定を採択した.十分な決定は、EU-米国データプライバシーフレームワークを自ら認証することを可能にする米国社が、EUから米国にデータを送信するための有効なデータ転送機構とすることである。しかし、いくつかのプライバシー提唱団体は、EU-米国のデータプライバシーの枠組みに挑戦すると表明している。これらの挑戦が成功すれば、EU-米国のデータプライバシーの枠組みに影響を与えるだけでなく、標準契約条項や他のデータ伝送機構の生存能力もさらに制限される可能性がある。この枠組みがいつ最終的に決定されるか、法廷で挑戦されるかどうかはまだ分からない。この枠組みがいつ最終的に決定されるか、法廷で挑戦されるかどうかはまだ分からない。この問題をめぐる不確実性は、私たちとEU会社の活動や、将来のEUでの任意の潜在的な業務運営に影響を及ぼす可能性がある。
FDAや他の規制·法執行機関は、非ラベル使用の普及を禁止する法律法規を積極的に実行している。もし私たちが非ラベル用途を不当に普及させることが発見されたら、私たちは重大な責任を負うかもしれない。
FDAおよび他の規制および法執行機関は、処方薬に対して提示される可能性のある販売促進主張を厳格に規制し、許可されていないまたは“ラベル外”の使用を宣伝することを禁止する法律および法規を実行する。特に、製品は、製品の承認ラベルに反映されるように、FDAまたは他の規制機関によって承認されていない使用に使用されてはならない。例えば、ZURZUVAEは、米国では成人PPDの治療のみのために許可されており、MDDを含むFDAによって承認されていない使用を拡張することはできない。もし私たちがどんな製品のラベル外用途を普及させることが発見されたら、私たちは重大な責任を負うかもしれない。連邦政府は不正販売促進の疑いのある会社に巨額の民事と刑事罰金を科し、これらの会社の販売促進活動を制限する措置を講じている。製薬会社も起訴され,虚偽申告法により重大な民事,刑事と行政処罰,損害賠償,罰金を受けたが,ラベル外で薬品を販売していた疑いがあるためである。私たちまたは私たちの従業員がラベルの外でZULRESSO、ZURZUVAE、または私たちが将来承認した任意の製品のいかなる宣伝を使用しても、私たちに重大な責任を負わせる可能性があり、これは私たちの業務および財務状況に重大な悪影響を及ぼすだろう。
私たちの将来の成長は外国市場に進出する能力にある程度依存するかもしれませんが、そこでは追加の規制負担、価格コントロール、精算問題、その他のリスクと不確実性の影響を受け、私たちのアメリカ業務にマイナスの影響を与える可能性があります。
私たちの将来の収益性は、私たち自身または私たちの協力者を通じて私たちの製品と候補製品を海外市場で商業化する能力にある程度依存するかもしれません。
処方薬の海外市場での定価は外国政府によってコントロールされている。これらの国では、製品の規制承認を受けた後、政府当局との定価交渉にかなりの時間がかかる可能性がある。一部の国で精算或いは定価の承認を得るためには、私たちの候補製品のコスト効果を他の利用可能な治療法と比較する臨床試験を行う必要があるかもしれない。もし私たちの製品が精算を得られない場合、あるいは範囲や金額が制限されている場合、あるいは定価が満足できないレベルに設定されている場合、収入と収益性が損なわれる可能性があります。
いくつかの国では、EU加盟国を含めて、処方薬の価格設定は政府によって統制されている。他の国は処方薬の価格設定に対して似たような接近をするかもしれない。政府や他の利害関係者は、費用制御措置の一部として、価格や補償レベルにかなりの圧力をかける可能性がある。政治、経済、規制の発展は定価交渉をさらに複雑化させる可能性があり、保険や補償を受けた後、定価交渉は継続される可能性がある。各国で使用されている参考定価と平行配分、あるいは低価格と高価な国の間で裁定を行うことで、価格をさらに下げることができる。アメリカでは、最近の立法と行政政策と提案は、私たちがアメリカの薬品価格を下げることを望んでいることを示しています。したがって、私たちまたはアメリカ以外のパートナーは、将来、私たちがアメリカで私たちの製品に対して受け取る価格を制限するかもしれません。第三者支払者や当局が割引を発表することは、出版物の所在国や他の国/地域の価格や精算レベルにさらに圧力をかける可能性があります。もし価格が満足できないレベルに設定されている場合
77
もし私たちの製品が精算を得られない場合、あるいは範囲や金額に制限されていれば、私たちまたは私たちの協力者の販売収入と、これらの国/地域における私たちの製品の潜在的な収益力はマイナスの影響を受けるだろう。
私たちの製品と候補製品を海外市場で商業化することは、私たちをより多くのリスクと不確実性に直面させるだろう
私たちの候補製品の海外販売は、政府規制、政治的·経済的不安定、貿易制限、関税変化の悪影響を受ける可能性もある。例えば、イギリスが離脱し、ヨーロッパおよび/または世界の規制状況に悪影響を与え続ける可能性がある。英国の離脱は引き続きEUとイギリスの法的不確実性を招く可能性があり、処方薬の価格設定に関する法律を含むEUとイギリスの国家法律法規の違いを招く可能性がある。イギリスはどのEUの法律を複製または置換することを決定するので、私たちがEUとイギリスで私たちの任意の製品を商業化することを求めることを選択すれば、将来EUとイギリスで取引する能力を弱める可能性がある。
私たちの知的財産権に関するリスクは
私たちの独自技術を十分に保護できない場合、あるいは私たちの候補製品を保護するのに十分な発行された特許を得ることができない場合、他の会社は私たちとより直接的に競争する可能性があり、これは私たちの業務、運営結果、財務状況、および見通しに大きな悪影響を及ぼすだろう。
私たちは、私たちの製品および成分、それらの使用方法、および私たちの業務発展に重要である他の発明をカバーするための特許を求めることを含む、私たちの業務に重要と考えられるノウハウの保護と強化に努めています。私たちはまた私たちの業務を特許保護から保護するために商業秘密に依存するかもしれないし、特許保護に適していないと思う側面もあります。
私たちの成功は、私たちの業務に関連する重要な商業技術、発明およびノウハウの特許および他の独自保護を取得して維持する能力があるかどうか、私たちの特許を擁護し実行する能力があるかどうか(彼らが発表すれば)、私たちの商業秘密の機密性を保護する能力があるかどうか、および第三者の効果的かつ実行可能な特許および独自の権利を侵害することなく運営されるだろう。私たちはまた、独自技術、持続的な技術革新、許可内の機会に依存して、私たちの候補製品の独自の地位を強化し、維持しています。
私たちは私たちのいかなる係属中の特許出願も発行された特許に成熟することを保証できない。例えば、米国特許商標局、または米国PTOは、我々の固有GABAを主張する我々の特許出願の最終拒絶を発表しているA正アロステリック調節剤化合物は、新規性と非顕著性に乏しい。私たちは却下に控訴しており、却下を覆すことに成功しないかもしれない。
私たちは私たちの固有の化合物をカバーする特許を得ることができないかもしれない。私たちが発行した任意の特許が強制的に実行可能であること、または私たちの候補製品を保護するのに十分な範囲、または他の方法で任意の競争優位性を提供するのに十分な範囲のクレームを含むことは保証できない。例えば、ZULRESSOにカバー範囲を提供する発行された特許および特許出願は、特定のレシピのみをカバーし、これらのレシピを使用する
78
PPDやMDDなどのうつ病を治療する。したがって、このような発行された特許およびそのような特許出願から発行される可能性のある任意の特許は、第三者の競争相手が特許請求の範囲に属さないブロサノケトンの代替処方の創造、製造、および販売を阻止するか、または代替方法を実施することを阻止しない。さらに、他の当事者は、我々の方法に関連しているか、または我々の方法と競合する可能性のある技術を開発しており、特許出願が提出されているか、または提出されている可能性があり、同じ方法または処方を要求するか、要求によって私たちの特許の地位を支配する可能性のある標的を要求することによって、我々の特許出願と重複または衝突する可能性のある特許を受信している可能性がある。このような第三者特許地位は、いくつかの発明のために私たちが特許保護を受ける能力を制限することさえ失われるかもしれない。
バイオテクノロジーおよび製薬会社の特許地位は、我々の特許地位を含み、複雑な法律および事実の問題に関連しているため、私たちが得ることが可能な任意の特許請求の発行、範囲、有効性、および実行可能性は確定的に予測できない。特許が発行されると、挑戦され、実行不可能とみなされ、無効にされ、または回避される可能性がある。米国特許や特許出願もプログラムや派生プログラムに妨害される可能性がある一方的再試験、あるいは各方面間再審査手続き、支出後再審査手続き、補充審査、地方裁判所での質疑。特許は、反対、認可後の審査、または様々な外国、国、地域特許庁で提起された同様の訴訟を受ける可能性がある。これらの訴訟手続きは、特許の喪失または特許出願の拒否、または特許または特許出願の1つまたは複数の特許請求の範囲の喪失または縮小をもたらす可能性がある。しかも、そのような訴訟は費用が高いかもしれない。例えば、私たちが許可したヨーロッパ特許はブレシャノドン静脈注射をカバーしている。第三者の反対を受けて、反対手続きが進行中だ。したがって、私たちは彼らが発行すれば、競争相手に対する保護を提供しないかもしれない、または独占的に許可された任意の特許を持っているかもしれない。さらに、干渉プロセスまたは派生プロセスにおける不利な決定は、第三者が我々が求める特許権を取得することをもたらす可能性があり、これは、逆に、私たちの候補製品を開発、マーケティング、または他の方法で商業化する能力に影響を与える可能性がある。さらに、特許の発行は有効かつ強制的に実行可能と推定されているにもかかわらず、その発行はその有効性または実行可能性を決定することができず、類似した製品を有する競争相手に対抗するために十分な独自保護または競争優位を提供することができない可能性がある。特許が発行され、有効かつ強制的に実行可能であると考えられていても、競争相手は、予め存在するまたは新たに開発された技術を使用するように、我々の特許を迂回して設計することができる。他の締約国は、より効果的な技術、設計、または方法のために開発され、特許保護を得ることができる。
私たちはまた、コンサルタント、サプライヤー、元従業員、および現職の従業員が私たちの技術知識や商業機密を不正に開示したり、使用したりすることを阻止できないかもしれない。いくつかの外国の法律は私たちの専有権に対する保護の程度はアメリカの法律よりも悪く、私たちはこれらの国で私たちの固有の権利を保護することは重大な問題に直面するかもしれない。もしこれらの発展が発生すれば、私たちのすべての候補製品がこれらの国で承認されれば、それらは私たちの販売に実質的な悪影響を及ぼすかもしれない。私たちが特許権を行使する能力は私たちが侵害行為を検出する能力にかかっている。その製品で使用されているコンポーネントを宣伝しない侵害者を発見することは困難である.さらに、競争相手または潜在的な競争相手の製品侵害の証拠を得ることは困難または不可能である可能性がある。私たちの特許権を強制または擁護する訴訟は、たとえ私たちが勝訴しても、費用が高く時間がかかる可能性があり、私たちの経営陣とキーパーソンの私たちの業務運営に対する注意をそらすことになります。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、もし私たちが勝てば、得られた損害賠償や他の救済措置は商業的な意味がないかもしれない。
さらに、私たちの特許を強制的に実行または保護するプログラムは、もし発表された場合、私たちの特許が無効にされるか、実行できないか、または狭い解釈に直面する可能性がある。このような訴訟はまた、私たちの1つまたは複数の特許の一部または全部のクレームが無効であるか、または他の方法では実行できないことを含む、第三者からのクレームを引き起こす可能性がある。もし私たちの製品または候補製品の任意の特許が失効したり、強制的に実行できない場合、私たちの財務状況と経営結果は実質的で不利な影響を受ける可能性があります。また、裁判所が第三者が保有する有効で強制的に実行可能な特許が私たちの候補製品をカバーしていることが発見された場合、私たちの財務状況および運営結果も実質的な悪影響を受ける可能性がある。
未来の私たちの所有権の保護の程度は不確定で、私たちは保証できない
79
私たちは非特許のビジネス秘密に依存し、非特許の技術的ノウハウと持続的な技術革新に依存して私たちの競争地位を発展させ、維持することができ、私たちの従業員および私たちのCRO、協力者、コンサルタントと秘密保護協定を締結することで、私たちの競争地位を部分的に保護することを求めている。我々の業務に関連する技術は、このような合意締約国ではない者によって独立して開発される可能性があります。さらに、これらの合意当事者である従業員やコンサルタントがこれらの合意の条項に違反したり、違反したりした場合、そのような違反または違反を救済するための十分な救済措置がない可能性があり、このような違反または違反によって私たちのビジネス秘密が失われる可能性があります。しかも、私たちの商業秘密は私たちの競争相手に知られたり独立して発見されるかもしれない。
私たちは他人の知的財産権を侵害する可能性があり、これは私たちの製品開発を阻害または延期し、ZULRESSO、ZURZUVAE、および私たちの他の候補製品を商業化したり、商業化コストを増加させたりすることを阻止し、もし開発と承認に成功すれば。
私たちの成功は、第三者の知的財産権および独自の権利を侵害することなく運営する私たちの能力にある程度依存するだろう。私たちの業務、製品、方法が第三者の特許や他の知的財産権を侵害しないか、または侵害しないことを保証することはできません。製薬業の特徴は特許と他の知的財産権に関する広範な訴訟だ。他の当事者は、私たちの製品や候補製品、または私たちの技術を使用して、彼らが所有している特許主張または他の知的財産権を侵害するか、または私たちが彼らの独自技術を不正に使用したと主張するかもしれない。我々が現在の候補製品を開発し続け、ZULRESSO、ZURZUVAE、および任意の将来の製品を商業化するにつれて、競争相手は、私たちの技術が彼らの知的財産権を侵害していると主張するかもしれません。これは、私たちの商業化の成功を阻害するためのビジネス戦略の一部です。我々の候補製品の使用または製造に関連する第三者特許または材料、配合、製造方法または治療方法の請求項に記載の特許出願が存在する可能性がある。特許出願が発行されるまでに数年かかる可能性があるので、第三者が現在処理している特許出願を有する可能性があり、これは、私たちの製品または候補製品が発行された特許を侵害する可能性があるか、またはこれらの第三者が私たちの技術がこれらの特許を侵害していると主張する可能性がある。知的財産権訴訟の結果は不確定要因の影響を受け,これらの不確定要因は事前に十分に定量化できない。特許のカバー面は裁判所の解釈にかかっており、解釈は常に統一されているわけではない。もし私たちが特許侵害を起訴された場合、私たちは私たちの候補製品、製品、または方法が関連特許の特許主張を侵害していないか、または特許主張が無効または実行不可能であることを証明する必要があり、私たちはそれができないかもしれない。私たちがこれらの訴訟で勝訴しても、私たちは巨額のコストを招く可能性があり、私たちの経営陣と科学者の時間と注意はこれらの訴訟に移される可能性があり、これは私たちに実質的な悪影響を及ぼすかもしれない。しかも、私たちはこのような行動を円満に達成するための十分な資源がないかもしれない。
80
特許や他のタイプの知的財産権訴訟は複雑な事実と法律問題に関連する可能性があり、その結果はまだ確定していない。特許訴訟は高くて時間がかかる。知的財産権侵害に関する任意のクレームが成功した場合、他方の特許を故意に侵害することが発見された場合、損害賠償金および弁護士費の3倍を支払い、許可を受けさせられた場合には、過去に主張された知的財産権の使用および使用料および将来の他の考慮を含む実質的な損害賠償の支払いを要求する可能性がある。さらに、私たちのこのようなクレームが成功的に主張され、私たちがそのようなライセンスを得ることができない場合、私たちは開発、製造、販売、または他の方法で私たちの製品または候補製品を商業化することを停止または延期させることを余儀なくされるかもしれない。商標クレームの場合、もし私たちが権利侵害を発見された場合、私たちは第三者の知的財産権の侵害を避けるために、私たちの一部またはすべての候補製品の再設計または再命名を要求される可能性があり、可能であっても、高価で時間がかかる可能性がある。私たちがこれらの訴訟で勝訴しても、巨額の費用が発生し、経営陣がこれらの訴訟を行う際の時間と注意力を移す可能性があり、これは私たちに実質的な悪影響を及ぼす可能性がある。
これらのリスクのいずれも、私たちの業務、運営結果、財務状況、および見通しに実質的な悪影響を及ぼす可能性がある。
私たちは私たちの特許と他の知的財産権の発明権または所有権のクレームに疑問を受けるかもしれない。
私たちは私たちの従業員、コンサルタント、CRO、外部科学協力者、および他のコンサルタントと秘密保護および知的財産権分配協定を締結します。これらの合意は、一般に、当事者が私たちにサービスを提供する過程で発想された発明が私たちの固有財産となることを規定している。しかし、このような合意は遵守されないかもしれないし、知的財産権を効果的に私たちに割り当てられないかもしれない。例えば、私たちが学術コンサルタントとコンサルティング契約を締結しても、この協定によれば、この学術コンサルタントは、私たちにサービスを提供するために開発された任意の発明を私たちに譲渡しなければならない。学術コンサルタントは、彼または彼女がそのようなすべての知的財産権を彼または彼女の雇用機関または他方に譲渡する義務と衝突する可能性があるので、このような発明を私たちに譲渡する権利がないかもしれない。
私たちのほとんどの従業員は以前にも私たちの競争相手や潜在的な競争相手を含む他のバイオテクノロジーや製薬会社に雇われたことがある。私たちはまた大学や他の実体にサービスを提供するために同時に雇われたコンサルタントとコンサルタントを招いた。私たちは、従業員、コンサルタント、またはコンサルタントが私たちのために実行してくれた仕事が、雇用主のような第三者に対するこの人の義務と衝突し、したがって、第三者は私たちが実行してくれた仕事に生じる知的財産権の所有権利益を持っているというクレームの影響を受ける可能性がある。
訴訟は、これらと他の挑戦在庫または所有権のクレームに対抗するために必要かもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭損害賠償を支払うことに加えて、貴重な知的財産権、例えば貴重な知的財産権の独占所有権や使用権を失う可能性がある。このような結果は私たちの業務に実質的な悪影響を及ぼすかもしれない。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。
私たちの特許保護の獲得と維持は、政府特許機関によって提出された様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
米国特許商標局および各種外国政府特許機関は、特許過程において、いくつかのプログラム、文書、費用支払いおよびその他の手続きおよび規定を遵守することを要求する。場合によっては、規定を遵守しないことは、特許または特許出願の放棄または失効をもたらす可能性があり、それにより、関連する管轄区域の特許権の一部または全部が失われる可能性がある。この場合、競争相手は他の場合よりも早く市場に参入するかもしれない。
私たちは私たちの特許を保護または強制的に執行するかもしれない特許の訴訟に巻き込まれるかもしれないが、これは高価で時間がかかり、成功しないかもしれない。
81
たとえ私たちが所有または許可した特許出願が発行されても、競争相手はこのような特許を侵害する可能性がある。権利侵害や不正使用に対抗するために、私たちは費用が高く時間がかかるかもしれない侵害請求を要求されるかもしれない。さらに、侵害訴訟では、裁判所は、私たちまたは私たちのライセンシーの特許が無効であること、強制的に実行できないこと、および/または侵害されていないことを判断することができ、または、私たちの特許が狭く解釈され、関連する技術を含まないことを理由に、他方の関連する技術の使用を阻止することを拒否することができる。任意の訴訟または弁護手続における不利な結果は、私たちの1つまたは複数の特許を無効または狭義に解釈されるリスクに直面させ、私たちの特許出願を発行できないリスクに直面させる可能性がある。
我々の特許または特許出願または我々の許可者の特許または特許出願に関連する発明の優先度を決定するために、第三者によって引き起こされるか、または我々によって提起された干渉プログラムまたは派生プログラムが必要である可能性がある。不利な結果は、私たちが関連技術の使用を停止することを要求するか、または勝利者から許可を得ようとすることを要求するかもしれない。もし勝利者が商業的に合理的な条件で私たちにライセンスを提供しなければ、私たちの業務は損害を受ける可能性があります。訴訟や介入訴訟に対する私たちの弁護は失敗する可能性があり、成功しても巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性があります。私たちは、特に米国のようにこれらの権利を十分に保護していないかもしれない国では、私たちの知的財産権の盗用を単独でまたは許可者と一緒に防ぐことができないかもしれない。
さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。聴聞、動議、または他の一時的手続き、または事態発展の結果を公開することもできる。もし証券アナリストや投資家がこれらの結果がマイナスだと思っていれば、私たちの普通株の価格に実質的な悪影響を及ぼす可能性がある。
法廷で挑戦された場合、私たちの製品または私たちの任意の候補製品に関連する発行された特許は、無効または実行不可能と認定される可能性がある。
もし私たちまたは私たちの協力者が、私たちの製品または私たちの任意の候補製品をカバーする特許を強制的に執行するために、第三者に対して法的訴訟を提起することができる場合、被告は、私たちの製品または私たちの任意の候補製品をカバーする特許の無効および/または実行不可能を反訴することができる。米国の特許訴訟では、被告が無効および/または実行不可能と主張する反訴はありふれている。有効性を疑問視する理由には、新規性の欠如、明らかな書面記述の欠如、または実施できないことなど、いくつかの法定要件のいずれかを満たすことができなかったと言われている。主張を実行できない理由には,特許起訴に関連する者が起訴中に米国特許商標局に関連情報を隠蔽したり,誤った声明を発表したりした疑いがある.第三者も米国や海外の行政機関に類似したクレームを出すことができ、訴訟範囲外でも同様である。この仕組みには再審査、贈与後審査、一方的再試験しました各方面間審査、派生手続き、または介入、および外国法ドメインの同等の手続き、例えば反対または撤回手続き。このような訴訟は、私たちの候補製品や競争製品をカバーしないように、私たちの特許が撤回または修正される可能性がある。例えば、私たちが許可したヨーロッパ特許はブレシャノドン静脈注射をカバーしている。第三者の反対を受けて、反対手続きが進行中だ。法的に無効と実行不可能と断言された後の結果は予測できない。有効性に関しては、例えば、無効な以前の技術がないことは確認できませんが、私たちと特許審査員は起訴中にこれを知りません。もし被告が無効および/または強制執行できない法的主張に勝った場合、私たちは適用される製品または候補製品の少なくとも一部またはすべての特許保護を失うだろう。このような特許保護の喪失は私たちの業務に実質的な悪影響を及ぼすだろう。
私たちは世界中のすべての管轄区域で私たちの知的財産権を保護することを求めません。私たちが保護を求める管轄区域でも、私たちの知的財産権を十分に実行できないかもしれません。
世界のすべての国と司法管轄区で特許出願、起訴、保護候補製品特許を提出する費用は目を引くほど高く、私たちはアメリカ以外のいくつかの国の知的財産権には米国の知的財産権が広くない可能性があり、権利は米国で獲得されたと仮定しているかもしれない。また、一部の外国国の法律は知的財産権の保護度が米国の連邦や州法に及ばない。そのため、第三者が米国以外のすべての国で私たちの発明を実施することを阻止することができないかもしれない。または私たちの発明を使用して製造された製品を米国または他の管轄区域で販売または輸入する。♪the the the
82
個別外国司法管轄区で特許保護を求める法定締め切りは、私たちの各特許出願の優先日に基づいています。
競争相手は私たちが特許保護を求めない司法管轄区域で私たちの技術を使用することができる。彼らは自分の製品を開発するために自分の特許保護を申請して得ることができる。さらに、彼らは私たちが特許保護を持っている地域に他の侵害製品を輸出するかもしれないが、法執行力はアメリカほど強くない。これらの製品は私たちの製品と競争する可能性があり、私たちの特許や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれない。特定の管轄区域で出願して発行された特許を取得しても、私たちの特許主張や他の知的財産権は、第三者の競争を効果的にまたは阻止するのに十分ではない可能性があります。
一部の外国法律は知的財産権の保護程度はアメリカの法律に及ばない。ある外国司法管轄区では、多くの会社が知的財産権の保護と保護に深刻な問題に直面している。一部の国の法律制度、特に発展途上国の法律制度は、特許及び他の知的財産権保護の実行、特にバイオテクノロジー及び医薬品に関連する特許及び他の知的財産権保護を支持していない。例えば、米国貿易代表事務所の2022年の報告書は、インドや中国を含むいくつかの国が特許権調達と法執行上の挑戦を報告していると指摘している。1989年以降、インドと中国を含むいくつかの国が毎年この報告書に登録されている。これは私たちがこのような司法管轄区域で私たちの特許を侵害したり、私たちの他の知的財産権を流用することを防ぐことを難しくするかもしれない。多くの外国国には強制許可法があり,これらの法律によると,特許権者は第三者に許可を付与しなければならない。また、多くの国は、政府機関や政府請負業者を含む第三者に対する特許の実行可能性を制限している。このような国では、特許は限られた利点を提供するかもしれないし、利益さえないかもしれない。特許保護は最終的には国ごとに求めなければならないが,これは高価で時間のかかるプロセスであり,結果は不確定である。したがって、私たちは特定の国で特許保護を求めないことを選択することができ、私たちはこれらの国で特許保護の利点を享受しないだろう。
また、外国の管轄区域で私たちの特許権を強制的に執行する訴訟手続きは、大量のコストを招く可能性があり、業務の他の方面への私たちの努力と注意力を移転させることは、私たちの特許が無効または狭義に解釈されるリスクに直面する可能性があり、私たちの特許出願を発表できないリスクに直面させ、第三者が私たちにクレームを提起する可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。したがって、私たちが世界各地で私たちの知的財産権を実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネスメリットを得るのに十分ではないかもしれません。
ZULRESSOと私たちのいくつかの候補製品のために、私たちは許可された知的財産権に依存している。もし私たちが知的財産権を許可する権利を失ったら、承認されれば、私たちは私たちのいくつかの製品や候補製品を開発したり商業化したりすることができないかもしれない。もし私たちがいかなる合意に違反しても、これらの合意に基づいて、私たちは第三者から私たちの製品、候補製品または技術の使用、開発、商業化の権利を獲得しましたか、または場合によっては、特定の開発期間内に完了できなかった場合、私たちは私たちの業務に非常に重要な許可権を失うかもしれません。
私たちは多くのライセンス契約の締約国であり、これらの合意に基づいて、私たちは私たちの業務に重要な知的財産権を付与され、将来的に追加のライセンス協定を締結する必要があるかもしれません。私たちの既存のライセンス協定は、将来のライセンス協定は、私たちが様々な開発、規制、および/またはビジネスの職務調査義務、マイルストーンおよび/または特許使用料の支払いおよびその他の義務を規定することを規定すると予想されています。もし私たちがこれらの合意の下で私たちの義務を履行できなかった場合、あるいは私たちが破産に直面した場合、許可側は許可を終了する権利があるかもしれません。この場合、許可がカバーする製品を販売することができません。例えば、現在または将来のライセンスが終了した場合、許可者がライセンスの条項を遵守できなかった場合、許可された特許または他の権利が無効または強制的に実行できないことが発見された場合、または許容可能な条項で必要なライセンスを締結できない場合、私たちの業務は影響を受ける可能性がある。
私たちが以前にしたように、私たちは第三者からライセンスを取得して、私たちの研究を進めたり、私たちの候補製品の商業化を許可する必要があるかもしれませんし、私たちはいかなる第三者特許の存在も提供することができません。そのようなライセンスがなければ、私たちの現在の候補製品や未来の製品に対してこれらの特許を強制的に実行するかもしれません。私たちはもしあれば、商業的に合理的な条項でこのような許可証のいずれかを得ることができないかもしれない。たとえ許可証を取得できても
83
私たちの競争相手が私たちに許可された同じ技術にアクセスできるように非独占的かもしれない。もし私たちが許可を得ることができなければ、私たちは代替技術を開発または許可するために多くの時間と資源を必要とするかもしれない。もし私たちがそれができなければ、私たちは影響を受けた候補製品を開発したり商業化することができないかもしれません。これは私たちの業務に実質的な損害を与えるかもしれません。このような知的財産権を持つ第三者は、私たちの販売禁止を求めることができますし、私たちの販売に関しては、印税および/または他の形態の賠償を支払う義務があります。
知的財産権許可は複雑な法律、商業、そして科学的な問題に関する私たちの業務に重要だ。ライセンス契約によると、私たちとライセンシーとの間で知的財産権紛争が発生する可能性があります
私たちが許可している知的財産権をめぐる論争が許容可能な条項で現在の許可手配の能力を維持していることを妨害または損害した場合、関連製品の商業化に成功したり、影響を受けた候補製品の開発と商業化に成功することができない可能性がある。
私たちは私たちの様々な計画を支援するためにいくつかのライセンスを締結した。私たちは私たちの業務に必要または有用な第三者知的財産権の追加許可を締結するかもしれない。私たちの現在のライセンスと私たちが加入する可能性のある任意の未来のライセンスは私たちに様々な特許権使用料の支払い、マイルストーン、そして他の義務を課している。例えば、ライセンス者は、ライセンス契約に従って特許起訴およびメンテナンスの制御権を保持することができ、この場合、特許起訴に十分な影響を与えることができない場合や、保守費の支払いができないことによる予期せぬカバーミスを防止することができない可能性がある。現在または将来のライセンス契約のいずれかの義務を履行できない場合、ライセンス側は、私たちがライセンス契約に違反していると主張し、それに応じて私たちの許可を終了することを求めるかもしれません。しかも、未来の許可者たちは彼らと私たちの許可を終わらせることを自由に決定することができる。私たちの現在または未来の任意の許可を終了することは、私たちが知的財産権を使用する権利を失う可能性があり、これは、候補製品を開発したり、製品を商業化する能力に重大な悪影響を与え、競争業務の地位や業務の将来性を損なう可能性があります。
また、我々のライセンシーがライセンスの条項を遵守できない場合、ライセンス者が第三者の侵害行為を阻止できない場合、許可された特許又は他の権利が無効又は強制執行できないことが発見された場合、又は許容可能な条項で必要なライセンスを締結できない場合、私たちの業務は深刻な影響を受ける可能性がある。
私たちが許可を得たいくつかの知的財産権は、政府が援助したプロジェクトによって発見される可能性があり、したがって、“パレード”の権利、いくつかの報告要件、およびアメリカの工業に対する選好のような連邦法規の制約を受ける可能性がある。これらの規定を遵守することは、私たちの独占権を制限し、報告要求に資源を費やし、非米国メーカーと契約を締結する能力を制限しなければならないかもしれない。
私たちが許可したいくつかの知的財産権はアメリカ政府の資金を使用することによって生じる可能性があるため、いくつかの連邦法規の制約を受ける可能性がある。例えば、カリフォルニア大学理事会との許可協定によると、私たちが得たいくつかの知的財産権は、米国政府の資金を使って生まれた可能性がある。したがって、1980年の“ベハ·ドール法案”によると、アメリカ政府は、私たちの現在の製品または現在または未来の候補製品に具現化された知的財産権に対していくつかの権利を持っているか、または
84
ベハ·ドール法案です米国政府が政府援助計画の下で開発したいくつかの発明におけるこれらの権利は、任意の政府の目的のために発明を使用する非排他性、譲渡不可能、撤回不可能な世界的許可を含む。さらに、以下の場合、米国政府は、(I)発明を商業化するのに十分なステップを取っていない、(Ii)公衆衛生または安全需要を満たすために政府行動をとる必要がある、または(Iii)連邦法規の公衆使用に対する要求を満たすために政府行動をとる必要がある、または(Iii)連邦法規の公衆使用に対する要求を満たすために、第三者に上述した任意の発明の独占的、部分的独占的または非独占的許可を付与することを要求する権利がある。もし私たちが適用できなかったか、または適用できなかった許可者が政府に発明を開示できず、かつ所定の期限内に知的財産権登録申請を提出できなかった場合、米国政府もこれらの発明の所有権を得る権利がある。さらに、米国政府は、所定の期間内に特許出願を提出していないいかなる国でも、これらの発明の所有権を取得することができる。政府援助の計画の下で発生する知的財産権もいくつかの報告要求によって制約されており、これらの要求を遵守するには、私たちまたは適用可能な許可者が大量の資源を費やす必要があるかもしれない。さらに、米国政府は、主題発明を含む任意の製品、または主題発明を使用して生産された任意の製品を米国で大量に製造しなければならないことを要求している。知的財産権所有者が合理的ではあるが成功しない努力をしていることを証明することができれば、同様の条項で潜在的な被許可者に許可を付与することができ、これらの許可は米国で大量に製造される可能性が高く、またはこの場合、国内製造が商業的に不可能である場合には、製造優先権要求を免除することができる。このような米国メーカーの選好は、このような知的財産権がカバーする製品について非米国製品メーカーと契約する能力を制限する可能性がある。
もし私たちが政府援助に関する未来の計画を達成し、私たちがこのような援助のために化合物または候補製品を発見した場合、このような発見された知的財産権は“ベハ-ドール法案”の適用条項によって制限される可能性がある。
もし私たちが私たちの候補製品のために新しい化学実体または他のタイプのマーケティングおよびデータ排他性を獲得しなければ、1984年の“薬品価格競争および特許期限回復法”(一般にHatch-Waxman修正案と呼ばれる)および同様の外国立法が私たちの候補製品の特許期間を延長することによって追加的な保護を受けなければ、私たちの業務は実質的な損害を受ける可能性がある。
連邦食品、医薬品、および化粧品法案(FDCA)下のマーケティング排他的条項は、当社が販売または将来販売する可能性のある製品と同じ活性部分を有する製品のいくつかのマーケティング申請の提出または承認を延期する可能性がある。FDCAは新しい化学実体(NCE)NDAの許可を得た最初の申請者に5年間のアメリカ国内の非特許マーケティング排他性を提供した。排他期間内に、FDAは、元の革新薬と同じ適応のためのものであるか、別の適応のためのものであるかにかかわらず、出願人が承認を参照するために必要なすべてのデータを所有していない場合、または参照承認に必要なすべてのデータの合法的な権利を有する場合、同じ活性部分に基づく別の薬物の簡略化された新薬出願またはANDAまたは505(B)(2)NDAを検討するために、別の会社によって提出された別の薬剤を受け入れない可能性がある。しかしながら、出願がイノベーターNDA所有者がFDAに記載された特許のうちの1つを含む特許が無効または未侵害証明である場合、4年後に出願することができる。FDAが、出願人によるまたはスポンサー付きの新しい臨床研究が、既存の薬物の新しい適応、用量または強度のような承認申請に重要であると考えた場合、FDCAはまた、完全なNDAまたは既存のNDAの補充に3年間の市場排他性を提供する。この3年間の排他性は,この薬物が新たな臨床研究に基づいて承認された改正のみを含み,原適応や使用条件の活性成分を含む薬物のANDAをFDAが承認することは禁止されていない。我々はすでにBrexanoloneのNCE独占経営権を獲得し,現在と将来の候補製品のためにNCE独占経営権を求める予定である。私たちの候補製品がこれらの規定に基づいてマーケティングやデータ独占経営権を獲得する資格があることは保証されませんし、私たちのどの製品のこのような独占経営権だけで私たちの業務需要を満たすのに十分な保証もありません。適用される5年および3年間のNCE固有期間またはFDCA下のデータ固有期間は、完全なNDAの提出または承認を遅延させることはない。
FDAが我々の候補製品の上場承認の時間、期限、詳細に基づいて、私たちが所有または許可している1つ以上の米国特許は、将来的にハッジ·ワックスマン修正案に従って限られた特許期間を回復する資格がある可能性がある。ハッジ·ワックスマン改正案は、製品開発およびFDA規制審査中に失われた特許期間の補償として、特許回復期間を最長5年とすることを許可している。関連時間においても、私たちは私たちの製品をカバーする有効な発行特許を持っています。もし私たちが適用される要求を満たすことができなければ、延期されないかもしれません。さらに、特許保護の適用期間または範囲は、
85
私たちが要求したものより少ない。もし私たちが特許期間の延長や回復を得ることができない場合、あるいはどのような延長の期間が私たちが要求しているよりも短く、他の独占経営権を持っていなければ、私たちの競争相手は私たちの特許が満了した後に競争製品の承認を受けるかもしれません。私たちの業務、財務状況、または運営結果は不利な影響を受ける可能性があります。
米国特許法の変化は特許の全体的な価値を低下させ、製品を保護する能力を弱める可能性がある。
私たちの成功は知的財産権、特に特許に大きく依存する。生物技術業界で特許を獲得し、実施することは技術上の複雑性と法律上の複雑性にも関連するため、コストが高く、時間がかかり、内在的な不確定性を持っている。また、米国では最近、範囲の広い特許改革立法:“ライシー·スミス米国発明法”、略称“米国発明法”が公布された。米国発明法は米国特許法のいくつかの重大な修正を含む。このような条項は特許出願起訴方式に影響を与える条項を含み、特許訴訟に影響を及ぼす可能性もある。米国の発明法が我々の業務運営にどのような影響を与えるかは不明である(あれば)。しかしながら、米国発明法およびその実施は、我々の特許出願をめぐる起訴および我々の特許出願によって発行される可能性のある任意の特許の実行または保護の不確実性およびコストを増加させる可能性があり、これらすべては、私たちの業務および財務状況に実質的な悪影響を及ぼす可能性がある。
さらに、米国最高裁の裁決は、場合によっては入手可能な特許保護範囲を縮小し、場合によっては特許所有者の権利を弱める。例えば2012年3月にメオ連携サービス,DBAメオ医学実験室ら。V.プロメテウス研究所は米国最高裁は、患者サンプルから薬物代謝物レベルを測定し、それを薬物用量に関連させるいくつかのクレームは特許テーマではないとしている。この裁決は一連の一般的な仕事を通じて自然規則を応用するだけの診断特許に影響を与え、いくつかの発明が特許保護を獲得する能力に不確定性をもたらしたようである。さらに2013年6月には分子病理学協会はMyriad Genetics,Inc.を訴えている米国最高裁は,単離されたゲノムDNAは特許を申請できないと主張しているが,相補DNA分子は天然製品ではないため特許条件に適合していると主張している。2014年6月にアリス社です。Ltd.はCLS Bank Internationalらの事件を訴えた。特許請求の範囲に関連する事件は、和解のリスクを軽減する方法に関し、米国最高裁判所は、抽象概念、自然製品、および自然法則の権利要件のための特許資格が使用されるべきであると考えているプロメテウスそれは.米国特許商標局は、抽象概念、自然製品、および自然法則に対する権利要求の対象物資格を決定するプログラムをリストし、適合するガイドラインを発表したプロメテウス, 何千ものそして、そしてアリス決めます。本マニュアルでは、以下の適用は制限されていません何千ものこの決定を他の天然製品に適用することですこのような決定が私たちの業務に及ぼす全面的な影響はまだ明確ではない。
2023年5月彼は最高裁判所ですアンがセノフィらを訴えた事件では能力が乏しいため,機能定義を有するモノクロナル抗体は無効であり,主張されている抗体の製造や使用に十分な指導を提供できなかったためと考えられる。裁判所は、全体的な原則は依然として、すべてのクレームが“十分に機能しなければならない”ことであり、より広範なクレームはより多くの支援を必要とすると指摘した。
我々が将来の特許を取得する能力に関する不確実性の増加に加えて,このような事件の結合は,いったん特許を取得する価値に関する不確実性をもたらしている.米国議会、連邦裁判所、および米国特許商標局のこれらおよび他の決定によれば、特許を管理する法律および法規は、予測不可能な方法で変化する可能性があり、それにより、新しい特許を取得したり、将来発行される可能性のある任意の特許を実行する能力を弱めることができる。
Creates Actの採択に伴い、私たちは競争相手の訴訟と損害に直面するかもしれない。また,Creates Actを含む既存の法規や国会で提案された立法は,採択されて法律となると,我々の製品の特許専有権を制限したり,より早く後発薬競争に参入することを促進したりする可能性がある。
模倣薬および生物類似製品の開発を促進するための立法によれば、競争相手が提起する可能性のある訴訟および損害賠償に直面する可能性があり、彼らは、ANDAおよび505(B)(2)アプリケーションの試験を支援するために、商業的に合理的で、市場ベースの条項に基づいて十分な数の承認された製品を提供していないと主張するかもしれない。このような訴訟は私たちを追加的な訴訟コスト、損害賠償、名声損害に直面させ、これは収入の低下を招く可能性がある。ZULRESSOとの模造薬競争リスクが増加している
86
ZURZUVAEと私たちの任意の候補製品は、CREATES法案の結果を含めて承認されれば、製品収入を最大化する能力に影響を与える可能性があります。
また、国会議員は、医薬品の特許排他性を制限したり、承認薬の後発薬のより早い市場進出を促進したりするための多くの立法イニシアティブを提案している。提出された法案の例は、可決された場合、医薬製品をカバーする第1の特許以外の特許の無効推定を確立し、これらの後続特許が単独で特許を申請可能であることを証明する発明の責任を革新者に移すことを含み、可決された場合、許可連邦貿易委員会は、医薬製品をカバーする大量の特許の組み合わせが反競争行為を構成するかどうかを調査し、この場合に独占訴訟を提起することを含む。可決されればFDAが後発薬の承認を30カ月猶予することを制限し,ANDA訴訟が薬物物質の成分特許を要求する場合に限られる法案となった。このような立法が法律になると、ZULRESSO、ZURZUVAE、または任意の未来の製品に悪影響を及ぼすか、または私たちの薬物の模倣薬がより早く市場に参入することになるかもしれない。
私たちの業界に関わるリスクは
医療コストを低減するための医療法規は,我々の業務や運営結果に実質的な悪影響を及ぼす可能性がある。
米国では,連邦や州レベルでも,多くの外国司法管区においても,医療コスト低減のための立法や立法,行政,規制提案が継続されている可能性がある。コスト制御、薬品価格制御、または他の改革の実施は、MDDの治療のために最終的に承認されたズランノドンを含む、ZULRESSO、ZURZUVAE、または任意の他の成功して開発および承認された製品からの販売収入に悪影響を及ぼす可能性があり、利益を達成する能力を制限する可能性がある。
例えば,2010年3月,ACAが採択され,政府や民間保険会社が医療に融資する方式を大きく変え,米国製薬業に大きな影響を与えた。その他の事項以外に、ACAは生物製品を低コストの生物模倣薬の潜在的な競争を受けさせ、メーカーが医療補助薬物リベート計画の下で吸入、注入、点滴、移植或いは注射薬物に対するリベートを計算するための新しい方法を提供し、医療補助薬物リベート計画下のメーカーが不足している最低医療補助リベートを増加し、リベート計画を医療補助管理の看護組織に登録された個人に拡大し、あるブランドの処方薬メーカーに対する年会費と税収を構築し、そして新しい連邦医療保険D部分カバー割引計画を作成し、メーカーは70%を提供することに同意しなければならない(2018年両党予算法により、2019年から発効)販売時点割引適用ブランド薬品は、保証期間内に条件を満たす受益者に交渉価格の割引を提供し、メーカーの外来薬品として連邦医療保険D部分(その後IRAによって修正され、以下に述べる)の条件に組み入れられる。
2011年8月、“2011年予算制御法案”(Budget Control Act Of 2011)などの法案は国会のための支出削減措置を制定した。赤字削減合同特別委員会の任務は、2013年から2021年までの間に少なくとも1.2兆ドルの赤字削減を提案することであるが、同委員会は必要な目標を達成できず、立法をいくつかの政府プロジェクトに自動的に削減することを触発した。これらの変化には、各年度に提供者に支払われる連邦医療保険総額が2%に達し、2013年4月に発効し、2031年まで続くことが含まれている。“コロナウイルス援助,救済·経済安全法”とその後の立法により,これらの医療保険の自動減支削減は減少·停止され,2022年7月には2%のすべての削減が回復した。これらの法律および他の法律は、連邦医療保険および他の医療資金のさらなる減少をもたらし、他の方法で、規制によって承認される可能性のある任意の製品または候補製品の価格、またはそのような任意の製品の処方または使用頻度に影響を与える可能性がある。例えば,総裁·バイデンは2022年12月に法律となる“総合支出法案”に署名し,医療保険計画の自動減額にいくつかの改正を行った。総合支出法案1001条は、2010年の4%の法定現金現金支払法(PAYGO)の自動減額を2024年末まで2年延期する。“2021年米国救援計画法案”の公布により,医療保険計画を4%削減する計画が2023年1月に発効する。この法案の医療補償タイトルには、2011年の連邦医療保険自動減額の2%予算制御法案を6ヶ月~2032年度に延長し、2030年度と2031年度の支払減少率を低減する第4163条が含まれている。
87
反腐敗条約のいくつかの条項は司法的挑戦を受け、その解釈または実行を修正または変更する努力を受けている。例えば、2017年12月に法律に署名した2017年の米国減税·雇用法案には、2019年1月1日からACAが1年の全部または一部で合格医療保険を維持できなかった個人の税収ベースの分担責任支払いを廃止する条項が含まれており、これは一般的に“個人強制医療保険”と呼ばれている。ACA、その実施、挑戦、またはACAまたはその実施条例またはその一部規定を修正する努力、および将来取られる可能性のある他の医療改革措置は、承認されれば、私たちの業界全体および候補製品を商業化する能力に実質的な悪影響を及ぼす可能性があると予想される。
アメリカでは、薬品の価格設定に関する立法と法執行の関心が増加している。具体的には、米国議会はすでにいくつかの調査を行い、連邦と州立法を提出し、薬品定価の透明性を高め、連邦医療保険と医療補助を含む処方薬のコストを下げることを目的としており、これは商業支払者との定価と割引に関する交渉、定価とメーカー患者計画との関係の審査、政府計画の薬品精算方法の改革に影響を与える可能性がある。国会や行政は、2022年の“インフレ低減法案”(IRA)を含む薬品定価問題を解決するために多くの努力をしてきた。他の立法や公共政策を通過するかどうかは不明であり、できれば、私たちの業務にどのような影響を与える可能性がある。
アイルランド共和軍は2022年8月に総裁·バイデンによって法律に署名された。この新しい法案は連邦医療保険D部分に影響を与え、連邦医療保険A部分または連邦医療保険B部分に参加する資格のある個人に利用可能であり、ある外来処方薬のカバー範囲として毎月保険料と連邦医療保険B部分を支払うことを許可する計画である。他の事項以外に、IRAはある薬品のメーカーが連邦医療保険と価格交渉を要求し、交渉価格以上を制限し、2026年に初めて発効した;連邦医療保険B部分と連邦医療保険D部分に基づいてリベートを実施し、インフレを超える価格上昇を処罰する(第1の部分Bインフレリベート期間は2023年第1四半期である;第1部分D部分インフレリベート期間は2022年第4四半期から2023年第3四半期)であり、D部分カバーギャップ割引計画(2025年から開始)の代わりに新しいD部分割引計画を採用した。アイルランド共和軍は、これらの計画の最初の数年間、衛生·公衆サービス部長官が規制ではなく指導によってその多くの規定を実施することを許可した。交渉計画によると、メーカーは何らかの交渉やインフレ税還付条項に違反した行為で民事罰金を受け、交渉計画を守らない間に消費税を支払う可能性がある。
いくつかの製薬会社および米国商会と米国製薬研究·メーカーはHHSとCMSを提訴し,アイルランド共和軍の連邦医療保険に対する薬品価格交渉計画が米国憲法第5改正案に違反した無償徴収を構成していると主張した。私たちはIreland共和軍のこのような条項と他の条項に関連した訴訟が継続され、結果的に予測可能で不確実だと予想する。
具体的には、価格交渉では、国会は、競合していない模倣薬や生体模倣薬のいくつかの高価な単一由来薬物および生物製品の低い価格を交渉することをCMSに許可し、連邦医療保険B部分およびD部分によって精算される。CMSは、2026年から連邦医療保険D部分によって支払われる10種類の高コスト薬物の価格を交渉することができ、その後、2027年の15種類のD部分薬、2028年の15種類のB部分またはD部分薬、および2029年以降の20種類のB部分またはD部分薬剤の価格を交渉することができる。医薬品は、承認後少なくとも7年後にのみ交渉を選択することができる(このように、最初の最高交渉価格を時間通りとすると、それらは9年後に承認される)、バイオ製品は、承認後11年に交渉のために選択することができる(このように、最初に最高交渉価格を時間とした場合、最初に承認された後13年になる)。単一のまれな疾患または疾患のために許可された医薬および生物学的製品には適用されない。将来私たちのすべての製品が連邦医療保険価格交渉の対象になれば、私たちは政府が行動するリスクに直面するかもしれない。この場合,公開される連邦医療保険価格交渉の結果も,商業支払者との定価や割引の交渉に影響する可能性がある。
これらの価格設定リスクは、もし私たちのどの製品の価格が連邦医療保険価格交渉のテーマであれば、私たちは医薬製品の予想されるリターンや私たちの製品の特許のすべての価値を保護することができないというリスクをさらに増加させるかもしれない。例えば、私たちが規制部門がズラノドンのMDD治療を承認する方法を見つけることに成功したとしても、アイルランド共和軍は私たちの潜在的な未来の収入に否定的な影響を及ぼすかもしれない。したがって、これらのリスクはまた、私たちの製品と候補製品(ズランノケトンを含む)に対する私たちの開発決定に影響を及ぼす可能性がある。
88
また、アイルランド共和軍は、彼らが提供する価格が法律で規定された協議の“最高公平価格”以下であるか、または彼らの値上げ幅がインフレを超えているため、アイルランド共和軍に従わなかった製薬業者に民事罰金と潜在的な消費税を科した。アイルランド共和軍はまた,メーカーに連邦医療保険D部分で精算された薬品にリベートを支払うことを求め,薬品の価格上昇幅がインフレを超え,2025年から連邦医療保険の自己払い薬品コストを年間2,000ドルに限定し,その後インフレに応じて調整することを求めている。医薬品製造業者たちはまたこのような計画を遵守して民事罰金を受ける可能性がある。さらに、アイルランド共和軍はMedicare Part D処方薬計画に参加する個人に関連するリスクを増加させる可能性があり、もし彼らが計画の高いハードルや“悲劇的な時期”に達する前に保証がその初期の年間保証限度額を超えることを要求すれば、保証不足を経験する可能性がある。最初の年間保証限度額を超え、悲劇的な時期を下回るサービスを必要とする個人は、壊滅的な時期に達するまで100%の処方費用を支払わなければならない。他にも、アイルランド共和軍には、2025年からカバー格差の解消、共同保険と共同支払いコストの低減、低所得補助金計画の資格拡大、年間自己負担費用に価格上限を設けることで、個人の財務負担を軽減することを目的とした多くの条項が含まれており、どの規定も定価や報告に潜在的な影響を与える可能性がある。
アイルランド共和軍がどのように施行されるのかはまだ分からない。さらに、アイルランド共和軍や任意の他の連邦や州医療改革が私たちにどのような影響を与えるかを確実に予測することはできませんが、このような変化は、私たちの活動に新たなまたはより厳しい規制要求を加えたり、私たちの製品の精算を減少させたりする可能性があります。これらは、私たちの業務、運営結果、財務状況に悪影響を及ぼす可能性があります。薬品の価格設定問題を解決するためにもっと多くの国会と行政が努力するかもしれない。
州レベルでは、立法機関はますます立法を通じて、機関は価格または患者の精算制限、割引、特定の製品への参入およびマーケティングコスト開示の制限、および価格透明性措置を含む薬品および生物製品の価格設定を制御するための法規を実行し、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。
外国、連邦と州の各レベルにはずっと存在し、医療コストを抑制或いは低減或いは薬品専門期間を制限するための立法と監督管理提案が引き続き存在する可能性がある。私たちは未来に取られる可能性のある計画を予測できない。政府、保険会社、医療組織、その他の医療サービスを管理する支払人は、医療コストの抑制または低減、および/または価格規制の実施に努力し続けており、悪影響を及ぼす可能性がある
これらの措置や将来とりうる他の医療改革措置は,連邦医療保険や他の医療保健資金のさらなる削減,より厳しいカバー基準,より低い精算,および新たな支払い方法につながる可能性が予想される。これは私たちが受け取ったすべての承認された製品の価格を下げるかもしれない。連邦医療保険または他の政府援助計画の支払いを拒否または減少させるいかなる精算も、同様の民間支払者の支払い拒否または減少をもたらす可能性があり、これは、ZULRESSOおよびZURZUVAEの販売から十分な収入を得ることを阻止する可能性があり、将来承認されれば、任意の他の製品を商業化し、利益を達成することに成功できない可能性がある。
89
私たちの内部コンピュータシステムまたはネットワーク、クラウドプラットフォーム、または私たちの協力者、当社の第三者CROまたは私たちの他の請負業者、コンサルタントまたはサービスプロバイダは、障害やセキュリティホールに遭遇する可能性があり、これは、私たちの開発計画が深刻に中断し、私たちの業務に関連する個人または敏感な情報を漏洩させるか、または私たちの業務に悪影響を及ぼす可能性のある重大な責任を負う可能性があります。
私たちはますます情報技術システム、インフラ、データに依存して、私たちの業務を運営しています。私たちの内部コンピュータシステムと私たちの協力者、私たちの第三者CROと私たちの他の請負業者、コンサルタント、サービス提供者は、許可されていないアクセス、窃盗、自然災害、テロ、戦争、電気通信および電気故障、システム故障による損害、または悪意のある第三者からのネットワーク攻撃(有害なマルウェア、恐喝ソフトウェア、ウイルス、ワーム、サービス拒否攻撃、サプライチェーン攻撃、社会工学計画、および他の手段を含む)を導入し、サービスの信頼性および機密性に影響を与えます。情報の完全性と可用性).このような事件が発生し、私たちの運営が中断された場合、私たちの計画が深刻に中断されたり、お客様の個人情報の開示に責任を負うことになる可能性があります。例えば、候補製品の臨床試験データの紛失は、規制提出や承認作業の遅延を招く可能性があり、可能な場合には、データを回復または複製するコストを著しく増加させる可能性があります。任意の中断、災害、またはセキュリティホールが、私たちのデータまたはアプリケーション、または私たちの候補技術または製品に関連する他のデータまたはアプリケーションが失われたり、破損したり、または機密または独自の情報を適切に開示しない場合、私たちは責任を招く可能性があり、私たちの候補製品のさらなる開発は延期または阻止されるかもしれません。
これらの脅威や侵入に対応し、情報システム、ネットワーク、またはクラウドプラットフォームを修復または交換するために、多くの資金および他の資源が必要になるかもしれません。私たちはまた経済的損失や貴重な機密情報の損失を受ける可能性がある。さらに、個人訴訟において個人および団体が提起した規制行動および/またはクレームの影響を受ける可能性があり、これらの訴訟は、データの乱用または不当開示のクレーム、および連邦貿易委員会法案第5(A)条または連邦貿易委員会法案第5(A)条に違反する不公平または詐欺的な行為またはやり方を含むデータ収集および使用実践および他のデータプライバシー法律および法規に関連するプライバシー問題に関するものである。米国連邦貿易委員会(Federal Trade Commission,略称FTC)は、会社が持っている消費者情報の敏感性と数、業務の規模と複雑さ、および安全性の向上と脆弱性の削減に利用可能なツールのコストを考慮して、会社のデータセキュリティ対策が合理的で適切になると予想している。個人が識別可能な健康情報は敏感なデータと考えられ,より強力に保護されるべきである。FTCの消費者個人情報の適切な保護に関する指導はHIPAAセキュリティ規則が要求する指導に類似し、HIPAAセキュリティ規則はカバー実体のために個人電子個人健康情報を保護する国家標準を確立した。HIPAAセキュリティルールは、保護された電子健康情報の機密性、完全性、および安全性を確保するのを助けるために、カバーされたエンティティが適切な行政、物理的、および技術的保障措置を有することを必要とする。プライバシーの面では,連邦貿易委員会は,会社が個人に対するプライバシー約束を果たすべきである,すなわち,会社が消費者の個人情報をどのように処理すべきかという期待を設定している.プライバシー政策やウェブサイト上で行われた声明のような約束を履行できなかったいかなる行為も、連邦貿易委員会法案に違反する不公平または詐欺的な行為またはやり方を構成する可能性がある。不公平や詐欺的な行為ややり方に従事するつもりはないが、FTCはその説明約束時に約束を強制的に実行する権利があり、データ漏洩などの完全に制御できないイベントは、FTC強制実行を招く可能性がある。連邦貿易委員会が連邦貿易委員会法に基づいて執行することは、民事処罰または執行行動を招くことができる。
これらの事件の発生を防止するためのシステムや制御措置を開発·維持し、脅威を識別·緩和する流れもあるが、これらのシステム、制御、プロセスの開発と保守はコストが高く、技術の変化やセキュリティ対策の克服に伴う努力がますます複雑になり、継続的に監視·更新する必要がある。さらに、私たちまたは私たちの第三者CROまたは私たちの他の請負業者、コンサルタント、またはサービスプロバイダのセキュリティ対策は、データ損失および他のセキュリティホールを防止するのに十分であることを保証できません。私たちが努力したにもかかわらず、これらの事件が発生する可能性は完全には解消されず、長い間発見されなかった可能性のあるセキュリティホールを含む、私たちの業務に悪影響を及ぼす可能性のあるネットワーク攻撃やセキュリティホールを防ぐことができるという保証もありません。これは、この脆弱性が大きな悪影響を及ぼす可能性を大幅に増加させる可能性があります。
90
私たちの財務状況と資金需要に関連するリスク
私たちは生物製薬会社で、今まで著しい収入が出ていません。設立以来、重大な運営損失が発生しており、予測可能な将来に損失が出ることが予想される。
私たちは生物製薬会社で、1つの承認された製品しかなく、2019年第2四半期に製品販売を通じて収入が発生し始めました。バイオ製薬製品開発は投機性の強い仕事であり、大きなリスクに関連している。
これまで、私たちの運営資金は、BIMAへの株売却、初公募前に償還可能な転換可能優先株、少量の転換手形の発行など、普通株売却の収益から主に得られてきた。私たちが設立してから2023年6月30日まで、私たちはこのような取引から合計28億ドルの純収益を得た。BiogenとShionogiとの協力を通じて、私たちはまた10億ドルの前払いを受けた。2023年6月30日まで、私たちの現金、現金等価物、有価証券は10億ドルです。会社設立以来,毎年純損失が発生しているが,2020年12月31日までの年間純収益は6.061億ドルであり,生物遺伝会社との協力やライセンス契約により確認された収入を反映している。2023年6月30日までの6カ月間の純損失は3.072億ドルで、2023年6月30日までの累計赤字は23億ドルだった。
私たちのほとんどの運営損失は,我々の研究開発計画に関するコストと,我々の運営に関する販売,一般,行政コストによるものである。今後数年と予測可能な未来には、運営損失レベルが上昇していくと予想される。私たちのこれまでの損失は、予想された将来の損失に加え、私たちの株主権益や運営資本に悪影響を与え続けるだろう。最近の事態を受けて、我々は現在、パイプライン優先順位や労働力再編を含めて資源配分を評価しており、現金滑走路の拡大を目指している。したがって,2024年の運営費は2023年より低下すると予想される。運営費は引き続き発生する予定であり,特にDEAスケジュールに従って2023年第4四半期に米国でPPD女性治療のZURZUVAEを発売し,商業化する予定である。これらのコストには、許可された送信前および発射準備活動に参加することに関連する費用が含まれており、FDAのフィードバック意見を評価した後にこのような作業を推進することが決定された場合、私たちは、規制部門がMDDの治療のためのズランノドンの治療を承認することを支援するために事前に行われた任意の作業のコスト、および事前に計画されたものを含む SAGE−718およびSAGE−324の臨床試験を行っている。成人産後うつの治療のための最近のZURZUVAEの発売承認に関連する巨額の販売、マーケティングおよびアウトソーシング製造費用、およびZULRESSOおよびZURZUVAE以外の任意の既存または将来の候補製品のマーケティング承認を得た場合、このような費用が生じると予想される。上場企業として、私たちは大量の法律と会計コストを招いた。私たちは予測可能な未来に、私たちはより多くの重大な損失と運営損失を被ることを予想している。医薬品開発に関連する多くのリスクや不確実性により,将来の損失の程度やいつ利益が達成されるかは予測できない(あれば)。私たちが本当に利益を達成したとしても、私たちは四半期や年度の収益性を維持したり向上させることができないかもしれない。
私たちの収益性は、私たちが製品収入を創造し続け、および/または協力から収入を得る能力に依存する。私たちは2019年第2四半期から製品販売から収入を得て、2019年6月に私たちの最初の製品ZULRESSOを発売しました。私たちは私たちがZULRESSOに提供する収入機会が引き続き限られると予想する。ZULRESSO、ZURZUVAE、および将来承認された製品から相当な製品収入を得ることができるかどうかは、多くの要因に依存しますが、これらに限定されません
91
もし私たちが相当な製品収入および/または協力収入を持続的に生産できなければ、私たちは利益を得ることができなくなり、持続的な資金がなければ、私たちは運営を続けることができないかもしれない。
私たちは将来のある時点で追加的な資金を調達する必要があるかもしれないが、これらの資金は受け入れ可能な条件で提供できないかもしれない、あるいは全くできないかもしれない。必要な時にこの必要な資金を得ることができなければ、私たちは私たちの製品開発努力や他の業務を延期、制限、または中止させられるかもしれません。現在または将来の運営計画のために十分な資金を持っていると考えても、市場状況が有利または他の戦略的考慮を考慮すれば、追加的な資本を求めることができるかもしれない。 普通株または転換可能または普通株に交換可能な証券を売却することで追加資本を調達すれば、わが社における株主の所有権権益は希釈される。
私たちは現在ZULRESSOを商業化し、アメリカで女性産後うつを治療するためのZURZUVAEの商業製品を発売する準備をしており、SAGE-718とSAGE-324を含む非臨床と臨床開発を通じて私たちの候補製品を推進している。製品を商業化して追加的な小分子製品を開発することは高価だ。私たちは現在、パイプラインの優先順位と労働力の再構成を含めて資源配分を評価しており、私たちの現金滑走路の拡大を目指しています。したがって,2024年の運営費は2023年より低下すると予想される。運営費は引き続き発生する予定であり,特にDEAスケジュールに従って2023年第4四半期に米国でPPD女性治療のZURZUVAEを発売し,商業化する予定である。これらの予想される運営コストには、許可された投与前および投与準備活動に関与することに関連するコスト、規制部門がズランノドンによるMDD治療を承認することを支援するために進められている任意の作業のコスト(FDAのフィードバックを評価した後にこのような作業を推進することが決定された場合)、およびSAGE−718およびSAGE−324の計画および行われている臨床試験のコストが含まれる。私たちは未来に運営需要を満たすためにもっと多くの資本が必要になると予想する。もし私たちが私たちの候補製品のためにより多くの適応および/または地理的位置を求めることを選択したら、私たちがすでに求めている予想以上の試験の適応に対して追加の臨床試験を行い、新しい潜在的な機会を探したり、現在予想されているよりも速い速度で私たちの活動を拡大したりすると、私たちはより早く追加資金を集める必要があるかもしれない。
2023年6月30日まで、私たちの現金、現金等価物、有価証券は10億ドルです。私たちの現在の見積もりによると、私たちの既存の現金、現金等価物、有価証券は、私たちが行っている協力と潜在収入の予想資金に加えて、2025年までの運営を支援するのに十分になると予想されています。私たちの現在の業務計画は、私たちが展開可能な他の開発活動、または現在計画されているすべての活動が同じ速度で行われるか、またはすべての活動がこの期間内に完全に起動または完了することを考慮していません。私たちは、私たちが予想した時間内にパートナーが私たちに支払った現金に関連するマイルストーンを達成することができないかもしれないし、私たちが予想しているレベルでPPD女性の治療のためにZURZUVAEによって生成された収入を達成することはできないかもしれないし、MDDの治療のために使用されるズランノンまたは私たちが成功して開発に成功する可能性のある任意の他の製品から収入を生成することは、私たちの予想されるレベルでは達成できないかもしれない。私たちは、予期せぬ事件や計画の変化による結果を含む、私たちの現在の運営計画の予想よりも早く利用可能な資本資源を使用するかもしれない。しかも、私たちの運営計画は変化するかもしれない。私たちは、株式または債務融資、政府または他の第三者資金、マーケティングおよび流通スケジュール、ならびに他の協力、戦略連合、許可スケジュール、および他の権利に関する手配、またはこれらまたは他の方法の組み合わせによって、計画よりも早く追加資金を求めることを必要または選択することができる。いずれにしても、今後の開発努力を拡大し、規制部門の承認を得て、候補製品を商業化するための追加資金が必要と予想されます。現在または未来の経済状況が長い間資本市場に影響を与える場合、または私たちの業務の見通しが損なわれたり、資本市場が他の理由で中断された場合、私たちは許容可能な条件で追加資本を得ることができないか、または追加資本を全く得ることができないかもしれない。必要な時に資金を得ることができなければ、私たちの製品開発努力や他の運営を延期、制限、中止させることができないかもしれません。十分な資金があると信じていても
92
私たちの現在または将来の運営計画については、市場状況が有利または他の戦略的考慮を考慮すれば、追加的な資本を求めることができるかもしれない。
私たちは未来の融資が十分な金額または私たちが受け入れられる条項で提供されることを保証できない。私たちが開発や規制活動で大きな挫折を経験するたびに、FDAがNDAに発表したMDD治療のためのズランノドンCRLのように、あるいは私たちの商業化努力中、あるいは重要な臨床計画から負のデータを受け取ると、私たちの株価が下落する可能性が高く、未来の融資をより困難にし、既存の株主により大きな希釈をもたらす可能性がある。例えば、2019年12月5日にズラノロン三期山地研究の背線結果を発表した後、我々の株価は大幅に下落した。また、将来のグローバル経済の不確実性、流動性の減少、資本市場の混乱、その他のマクロ経済や地政学的条件は、より有利な条件でより多くの資金を調達することを困難にする可能性がある。しかも、どんな融資条項も私たちの株主の持株や権利に悪影響を及ぼす可能性がある。私たちは追加の証券を発行し、株式でも債務でも、このような発行の可能性は、私たちの株式の市場価格を下落させる可能性がある。債務の発生は固定支払義務の増加を招き、私たちは追加債務を発生させる能力の制限、私たちが知的財産権を得ることができるかもしれない能力の制限、私たちの業務を展開する能力に悪影響を及ぼす可能性のある他の運営制限など、いくつかの限定的な条約に同意する必要があるかもしれない。
私たちはまた、他の場合ではなく、パートナーとの手配や他の方法で資金を求めることが要求される可能性があり、私たちは、私たちのいくつかの技術または製品候補の権利を放棄するか、または他の方法で私たちに不利な条項に同意することを要求されるかもしれません。いずれも、私たちの業務、運営結果、および見通しに大きな悪影響を及ぼす可能性があります。
普通株または転換可能または普通株に交換可能な証券を売却することで追加資本を調達すれば、わが社における株主の所有権権益は希釈される。債務融資が可能であれば、私たちの固定支払義務を増加させ、追加債務を招く、資本支出を行う、または配当を宣言するなど、特定の行動をとる能力を制限または制限する契約を含む可能性がある。もし私たちが第三者との協力、戦略的パートナーシップ、許可手配を通じてより多くの資金を調達すれば、私たちは私たちの候補製品、私たちの知的財産権、未来の収入流の貴重な権利を放棄しなければならないかもしれないし、私たちに不利な条項で許可を与えなければならないかもしれない。
もし私たちがタイムリーに資金を得ることができなければ、私たちは私たちの1つ以上の研究開発計画や任意の承認された製品の商業化を大幅に削減、延期、または停止することを要求されるかもしれません。あるいは必要に応じて私たちの業務を拡大したり、他の方法で私たちのビジネスチャンスを利用することができません。これは、私たちの業務、財務状況、運営結果に大きな影響を与えるかもしれません。
私たちの普通株に関するリスクは
市場の変動は私たちの株価と投資価値の変動を招くかもしれない。
他の生物製薬会社と同じように、私たちの普通株の市場価格も不安定だ。私たちの普通株の市場価格は私たちがコントロールできない要素によって大きく変動するかもしれません
93
私たちは既存の現金と将来の潜在的な公募株収益をどのように使用するかについて広範な裁量権を持っており、これらの現金や収益を有効に使用できない可能性があり、これは私たちの運営業績に影響を与え、株価の下落を招く可能性がある。
私たちは私たちの現金の使用と将来の潜在的な後続公開の純収益を適用する上でかなりの自由裁量権を持っている。私たちは私たちの株主に著しい見返りや何の見返りも与えない目的に現金と純収益を使用することができる。さらに、使用する前に、私たちは、収入を生じたり、価値を失ったりしない方法で、潜在的な未来に後続して発行される純収益に投資する可能性がある。
私たちの定款文書やデラウェア州法律によると、反買収条項は買収をより困難にする可能性があり、私たちの株主に有利な買収であっても、私たちの株主が現在の経営陣を交換または更迭しようとすることを阻止するかもしれません。
当社の会社登録証明書の改訂及び再記載の定款の条項は、私たちの買収や私たちの経営陣の変動を延期または阻止する可能性があります。これらの規定には、我々の株主の書面による行動を禁止する秘密保護取締役会と、株主の承認なしに優先株を発行する権利がある。また、私たちはデラウェア州で登録されているので、私たちはデラウェア州会社法第203条の規定によって管轄されています。この条項は、私たちが発行した議決権株の15%以上の株主が私たちと合併または合併する能力を持つことを制限しています。これらの条項は,潜在的な買収者に我々の取締役会との交渉を要求することで,株主により大きな価値を得る機会を提供していると信じているが,我々の取締役会が拒否した要約が一部の株主から有益であると考えられていても,これらの条項は適用される.また、これらの規定は、株主が責任を持って私たちの経営陣に命じられた取締役会のメンバーを交代させることを難しくし、株主が現在の経営陣を交代または罷免しようとしていることを挫折または阻止する可能性がある。
94
未来に私たちの普通株を売却することは私たちの株価を下落させるかもしれない。
公開市場で私たちの普通株の大量の株を売却したり、これらの売却が発生する可能性があると考えたりすることは、私たちの普通株の市場価格を大幅に低下させ、追加株式証券を売却することで十分な資本を調達する能力を弱める可能性がある。例えば、BIMAが購入した6,241,473株のうちの普通株は18ヶ月のロック期間があり、ロック期間は2022年6月30日に満了し、その後、BIMAは一定の数の株式を販売することができるが、いくつかの販売および数量によって制限されるか、またはBIMAがその登録権に応じてその株の登録を要求する場合、そのような販売および数量に制限されない。BIMAは、2023年12月31日に満了した第2の18ヶ月の期限後に、制限なしに株を売却することができる。
項目5.その他の情報
私たちの役員や上級社員は
95
プロジェクト6.eXhibit
本四半期報告10-Q表の一部として提出された展示品は“展示品索引”に記載されており、ここに組み込まれて参考となる。
展示品索引
展示品 違います。 |
|
説明する |
3.1 |
|
登録者規約の改訂と再改訂(登録者が2023年6月16日に提出した現在の8−K表報告書の添付ファイル3.1(書類番号001−36544)を参照) |
|
|
|
10.1* |
|
改訂された2014年従業員株購入計画 |
|
|
|
10.2*# |
|
登録者とCyDex製薬会社との間の供給協定は、2012年12月13日で、2013年8月21日と2014年4月30日に改訂された |
|
|
|
10.3*# |
|
登録者とCyDex製薬会社との間の供給協定の第3号改正案は,日付は2015年9月25日である |
|
|
|
10.4*# |
|
登録者とCyDex製薬会社との間の商業許可協定は、2013年8月21日で、2014年4月30日に改訂された |
|
|
|
10.5*# |
|
登録者とカリフォルニア大学校董間の非排他的許可協定は,日付は2013年10月23日であり,2014年5月14日に改訂された |
|
|
|
10.6*# |
|
登録者とワシントン大学との間で締結された独占許可協定は,2013年11月11日である |
|
|
|
31.1* |
|
2002年サバンズ·オキシリー法第302条に基づいて可決された“1934年証券取引法”第13 a−14条及び第15 d−14(A)条に基づいて発行された最高経営責任者証明書 |
|
|
|
31.2* |
|
2002年サバンズ·オキシリー法第302条に基づいて成立した“1934年証券取引法”第13 a−14条及び第15 d−14(A)条に基づく首席財務官の証明 |
|
|
|
32.1+ |
|
2002年にサバンズ·オクスリ法案第906条で可決された“米国法典”第18編1350条による最高経営責任者と最高財務責任者の認証 |
|
|
|
101.INS* |
|
相互接続されたXBRLインスタンス文書(このインスタンス文書は、そのXBRLタグがイントラネットXBRL文書に埋め込まれているので、対話データファイルには現れない) |
|
|
|
101.Sch* |
|
イントラネットXBRL分類拡張アーキテクチャ文書 |
|
|
|
101.カール* |
|
インラインXBRL分類拡張計算リンクライブラリ文書 |
|
|
|
101.定義* |
|
インラインXBRL分類拡張Linkbase文書を定義する |
|
|
|
101.実験所* |
|
XBRL分類拡張ラベルLinkbase文書を連結する |
|
|
|
101.前期* |
|
インラインXBRL分類拡張プレゼンテーションLinkbaseドキュメント |
|
|
|
104* |
|
カバー相互データファイル(添付ファイル101に含まれるカテゴリ拡張情報を有するイントラネットXBRLのフォーマットである。*) |
*アーカイブをお送りします。
#本四半期報告と共に提出された10-Qテーブルは、これらの以前に提出された証拠(これらの証拠は、満期直前の秘密処理令の標的である)をS-K規則の下で米国証券取引委員会のCF開示ガイドライン第601(B)(10)(Iv)項に基づいて編集した証拠の保存規則:主題7にのみ使用される。S-K規則第601(B)(10)(Iv)項によれば、本展覧会の内容の一部は省略されている。
96
+本契約添付ファイル32.1で提供された証明は、本四半期報告の10-Kフォームと共に提供されたものとみなされ、改正された1934年証券取引法第18節の目的に基づいて提出されたとはみなされません。登録者が引用によって明確に組み込まれていない限り、このような証明は、参照によって1933年証券法(改訂本)または1934年証券法(改訂本)の下に組み込まれたいかなる文書ともみなされない。
97
登録する解決策
1934年の証券取引法の要求によると、登録者はすでに正式に本報告を正式に許可した署名者がそれを代表して署名することを促した。
|
|
Sage治療会社 |
||
|
|
|
|
|
2023年8月7日 |
|
差出人: |
|
/S/バリー·E·グリーン |
|
|
|
|
バリー·E·グリーン |
|
|
|
|
社長と取締役CEO (首席行政主任) |
|
|
|
|
|
2023年8月7日 |
|
差出人: |
|
/S/井口木美 |
|
|
|
|
井口木美 |
|
|
|
|
首席財務官 (首席財務会計官) |
98