アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表:
(マーク1)
本年度まで
あるいは…。
過渡期:_
手数料書類番号:

(登録者の正確な氏名はその定款に定められている)
| (登録設立又は組織の国又はその他の管轄区域) | (国際税務局雇用主身分証明書番号) |
| (主な行政事務室住所) | (郵便番号) |
登録者の電話番号は、
市外局番を含む:
同法第12(B)項に基づいて登録された証券:
| クラスごとのタイトル | 取引コード | 登録された各取引所の名称 | ||
| 普通株式購入の引受権証 | ATNFW | ナスダック株式市場有限責任会社(ナスダック資本市場) |
同法第12(G)項により登録された証券:
ない。
チェックマークは、登録者が証券法第405条に規定する有名な経験豊富な発行者であるか否かを示す。はい、そうです☐
登録者が当該法第13節又は第15節(D)節に基づいて報告書を提出する必要がない場合は,複選マークで示してください。はい、そうです☐
登録者
(1)が過去12ヶ月以内(または登録者がそのような報告の提出を要求された短い期間内)に1934年の“証券取引法”第13または15(D)節に提出されたすべての報告を再選択マークで示し、(2)過去90日間、登録者
はこのような提出要求を遵守してきた
登録者
が過去12ヶ月以内に(または登録者がそのような文書の提出および掲示を要求されたより短い時間内に)登録者
が過去12ヶ月以内に提出されたかどうかをチェックマークで示すことは、S-T規則405条(本章232.405節)に従って提出されたすべての対話データファイルを電子的に提出する
再選択マークで登録者 が大型加速申請者、加速申請者、非加速申請者、小さい報告会社か新興成長型会社かを示します。 参照してください“大型加速ファイルサーバは” “ファイルマネージャを加速する“と”小さな報告会社 “と”新興成長型会社“取引法第12 B-2条にある。
| 大型加速ファイルサーバ | ☐ | ファイルマネージャを加速する | ☐ |
| ☒ | 規模の小さい報告会社 | ||
| 新興成長 | |||
新興成長型会社である場合、登録者が、取引法第13(A)節に従って提供された任意の新しいまたは改正された財務会計基準を遵守するために、延長された移行期間を使用しないことを選択したかどうかをフックで示す☐
登録者
がその経営陣が“サバンズ-オキシリー法案”(“米国法典”第15編7262(B)節)第404(B)条に基づいてその財務報告を内部統制する有効性について報告書を提出したか否かを再選択マークで示し、その監査報告を作成または発表する公認会計士事務所の財務報告書の内部統制に対する有効性を証明した
証券がこの法(Br)12(B)節に基づいて登録されている場合は、届出文書に含まれる登録者の財務諸表が以前に発表された財務諸表の誤りを反映して訂正されたか否かをチェックマークで示してください☐
これらのエラー のより真ん中に再記載が必要であるかどうかをチェックマークで示すことは、登録者の任意の幹部が関連回復中に受信したインセンティブベースの報酬を§240.10 D−1(B)に従って回復分析する必要がある☐
登録者
が空殻会社であるかどうかをチェックマークで表す(“取引法”第12 b-2条で定義される)。はい、そうです☐いいえ、違います
登録者が最近完成した第2財期の最終営業日までに,登録者の非関連会社が保有する投票権と無投票権普通株の総時価を$とする
2023年3月31日までに
引用で編入された書類
登録者は,その2023年度株主総会の最終委託書の一部について(“2023年依頼書)本年度報告書の表格10-K第III部分(明記)を参照して組み込む。2023年に依頼書は、本報告に関連する会計年度終了後120日以内に米国証券取引委員会に提出される
| 監査事務所ID: | 監査役の名前: | 監査役場所:北京 |
カタログ
| 語彙表 | II | |||
| 前向き情報に関する警告 声明 | v | |||
| 第 部分I | ||||
| 第1項。 | 商売をします。 | 1 | ||
| 第1 A項。 | リスク 要因。 | 47 | ||
| 項目1 B。 | 未解決 従業員の意見。 | 106 | ||
| 第二項です。 | 属性です。 | 106 | ||
| 第三項です。 | 法的手続き . | 106 | ||
| 第四項です。 | 鉱山 安全情報. | 106 | ||
| 五番目です。 | 登録者普通株,関連株主事項及び発行者が株式証券を購入する市場 | 107 | ||
| 第六項です。 | [保留されている] | 108 | ||
| 第七項。 | 経営陣の財務状況と経営結果の検討と分析。 | 108 | ||
| 第七A項。 | 市場リスクに関する定量的で定性的な開示。 | 120 | ||
| 第八項です。 | 財務諸表と補足データ。 | 120 | ||
| 第九項です。 | 会計·財務開示面での変化と会計担当者との相違。 | 121 | ||
| 第9条。 | とプログラムを制御する. | 121 | ||
| プロジェクト9 B。 | その他 情報. | 123 | ||
| プロジェクト9 Cです。 | 検査を阻止する外国司法管轄区に関する情報を開示する。 | 123 | ||
| 第 第3部分 | ||||
| 124 | ||||
| 第10項。 | 役員、幹部、会社が管理する。 | 124 | ||
| 第十一項。 | 役員報酬。 | 124 | ||
| 第十二項 | S安全 いくつかの実益所有者の所有権と管理層および関連する株主事項。 | 124 | ||
| 十三項。 | いくつかの 関係と関連取引、および取締役独立性。 | 124 | ||
| 14項です。 | 依頼者 会計士費用とサービス。 | 124 | ||
| 第4部 | ||||
| 第十五項。 | 展示品、財務諸表、明細書 | 125 | ||
i
語彙表
以下は、本報告で使用されるいくつかの用語の縮約および定義であり、これらの用語は、一般に製薬およびバイオテクノロジー産業で使用される
“ACABrは患者保護と平価医療法案を指し、通常は平価医療法案と略称され、ニックネームはオバマ医療改革であり、これはアメリカの連邦法規であり、多くの権利と保護を提供し、医療保険をより公平で、理解しやすくし、br補助金(通過)を提供する保険料税額控除“と”コスト分担を減らす“)を負担しやすくするために。この法律は、より多くの低所得者をカバーするための医療補助計画も拡大している。
“鎮痛剤Brは痛みを緩和するために設計された薬物である。
“安達Brは、審査および承認可能な後発薬製品のためにFDAに提出されたデータを含む簡略化された新薬出願を意味する。
“抗腫瘍壊死因子Brは腫瘍壊死因子に対する生理的反応を抑制する薬剤である。
“BLABrは、FDAが販売およびマーケティングのための新しい生物を承認することを提案する米国の生物学的スポンサーが正式に提案したFDAの生物製品許可証申請を意味する。
BPCIAとは、生物製品の価格競争と革新法を意味する。
“カンナビノイド“ 中国で発見された平均化合物大麻草.は、本出願明細書で使用する場合、大麻br植物において発見されたTHCを含まない化合物を意味する。
“中央商務区大麻ビスフェノールは、大麻植物から抽出された大麻中の有効成分である。CBDはTHCの非精神活性酸化分解産物である。
“CBG“大麻フェノールは、大麻植物において発見された化合物の一種である。
“CCMO は、中央委員会Mensgenebonden Onderzoek(CCMO)、またはヒト被験者研究に関連する中央委員会であり、オランダのヒト被験者に関する医学研究の審査および管理を担当する組織を意味する。
“CHMP“br”とは,ヒト用薬品委員会であり,前身は特許薬品委員会であり,欧州薬品管理局の委員会であり,この機関の人用薬品に関するすべての問題に対する意見の述べを担当している。
“細胞質“br”とは,HHS内の連邦機関であり,医療保険計画 を管理し,州政府と協力して医療補助を管理する医療保険·医療補助サービスセンターである。
“コルチコステロイドBrは体内の炎症を低下させる薬である。
“CRO“br”とは,研究サービスを契約形式でアウトソーシングし,製薬,バイオテクノロジー,医療機器業界を支援する契約研究機関である。
“シクロスポリンA“br”は“制御物質法”、すなわちアメリカ連邦薬品政策を制定する法規を指し、この法規によると、ある物質の製造、輸入、所有、使用と流通が規制されている。
“CTABrは、特定の国での臨床試験の許可を得るために、国家監督管理機関に提出された臨床試験申請を意味する。医薬品研究に必要な情報を含むアプリケーションです。 CTAの目的は、 製品の承認を得るために、衛生当局に臨床試験に関するすべての重要な詳細を提供することです。
II
“DEA“麻薬取締庁とは、米国司法省に属する米国連邦法執行機関であり、その任務は米国内の麻薬密売と流通に打撃を与えることである。
“EMA“br”とは欧州薬品管理局を指し、これはEUが薬品評価と監督を担当する機関である。
“EU”とは EUのことです。
“FDC法案Brとは連邦食品、薬物と化粧品法案を指し、これは1938年に国会で可決された米国法であり、FDAが食品、薬品、医療機器、化粧品の安全を監督することを許可した。
“林業局Brは、米国衛生·公衆サービス部の連邦機関である米国食品医薬品局を意味する。FDAは、人間および獣薬、生物製品および医療機器の安全性、有効性および安全性を確保し、米国の食品供給、化粧品、および放射線製品の安全を確保することによって公衆の健康を保護する責任を負う。
“FS肩関節凍結とは,肩関節の硬直と痛みのことである。
“GCPBrは国際品質基準である良好な臨床実践を意味し、その後、政府はそれをヒト被験者の臨床試験に関連する法規に変換することができる。GCPは国際人用薬品登録技術要求調整委員会(ICH)に従い,臨床研究の倫理面で厳しいガイドラインを実行している。
“プロス“br”は良好な実験室操作規範を指し、非臨床健康と環境安全研究計画、実行、モニタリング、記録、アーカイブと報告の組織過程と条件に関連する品質体系である。
“GMPBrは、FDAがFDA法の許可に基づいて公布した良好な生産実践法規を意味する。これらの法的効力を有する規定は薬品、医療機器、一部の食品と血液のメーカー、加工業者と包装業者に積極的な措置を取り、その製品の安全、純粋と有効を確保することを要求している。
“HHS“米国衛生·公衆サービス部は、衛生部とも呼ばれ、米国連邦政府の内閣級部門であり、すべてのアメリカ人の健康を保護し、基本的な人間サービスを提供することを目標としている。
“HIPAA は1996年の“健康保険携帯性と責任法案”を指し、この法案の目標は、人々が健康保険を獲得しやすくし、医療情報の機密性と安全性を保護し、医療保健業界が行政コストをコントロールするのを助けることである。
“HMGB 1Brは、ヒトHMGB 1遺伝子によってコードされるタンパク質である高遷移率族タンパク質1を意味する。活性化マクロファージと単球は炎症のサイトカインメディエーターとしてHMGB 1を分泌する。
IBD“は、炎症性腸疾患を指し、慢性消化管炎症に関連する疾患を記述するための包括的な用語である。
“工業“ とは研究性新薬申請のことである。臨床試験を開始する前に、研究は承認されなければならない。研究者が人体で薬物を研究したい場合には,研究用新薬やIND申請や請求をFDAに提出しなければならない。INDアプリケーション は、例えば、FDAがこの療法が人体で安全であるかどうかを決定することができるように研究結果、薬物がどのように製造されているか、誰が製造されているか、その中に何が入っているか、その安定性はどうかなど、いくつかの情報を含まなければならない。審査計画中の臨床 研究の詳細な大綱は、人々が不必要なリスクに直面する可能性があるかどうかを決定するための研究案と呼ばれ、臨床試験を行う知識と技能を有するかどうかを決定するための臨床試験 チームの詳細な情報が含まれていなければならない。
“単独で識別可能な健康情報 HIPPAは、個人から収集された人口統計情報を含む健康情報のサブセットの情報として定義し、(1)保健提供者、健康計画、雇用主または保健情報交換によって作成または受信された情報、 および(2)個人の過去、現在または未来の身体または心理的健康または状態に関する情報、個人への健康ケア、または過去、現在または将来の個人への医療の支払い、および(A)個人識別を識別する情報;または(B)情報が個人の識別情報を決定するために使用されることができると信じる合理的な根拠がある。
“IRBBrとは、ヒト被験者に関連する生物医学研究の審査および監督を担当する正式に指定されたグループである機関審査委員会を意味する。FDAの規定によれば、IRBは、(承認を得ることを確実にするために)、または研究を承認する権利があり、修正を要求する権利がある。このグループ審査は,人間の研究対象者の権利や福祉の保護に重要な役割を果たしている。
“医療補助“brは、収入および資源の限られた人たちが医療費を支払うのを助ける米国の連邦および州医療保険計画である。Medicaidはまた、老人ホームケアおよびパーソナルケアサービスを含む、通常Medicareカバー範囲内にない福祉を提供する。
三、三、
“医療保険“br”は米国の国家医療保険計画である。主に65歳以上のアメリカ人に医療保険を提供しているが、社会保障管理局が決定した障害状態のある若者や末期腎臓疾患や筋萎縮性側索硬化症(ALSまたはLou Gehrig病)を有する人にも医療保険を提供している。
“MHRA“br”は薬品と保健製品の監督管理機関であり、イギリスの衛生と社会保健部の一つの執行機関であり、薬品と医療機器の有効かつ受け入れ可能な安全を確保することを担当している。
“資材需要計画“br”とは、1つのEU加盟国で付与され、他のEU加盟国で認められた市場許可である相互承認手続きを意味する。
“NDA“br”はFDAの新薬申請を意味し、これは米国の薬品スポンサーがFDAに新薬の販売およびマーケティングのための新薬を承認することを正式に提案するツールである。
“NIHR“br”は国家健康研究所がイギリスの政府機関であり、健康と看護方面の研究に資金を提供することを意味し、brはヨーロッパ最大の国家臨床研究資金提供者である。
“孤児薬名“ は医療疾患を治療するために開発された薬剤であり,これらの疾患は非常にまれであるため,政府の助けがなければ を生産することは利益にならない。
“ステップ1Br試験は通常、薬物を最初に健康なヒト被験者に導入し、安全性、用量耐性、吸収、代謝、分布、および除去をテストする。癌のような深刻または生命に危険な疾患の候補薬剤の場合、特に候補薬剤が生まれつき毒性が大きすぎて道徳的に健康ボランティアに使用できない可能性がある場合、最初の人体試験は通常患者で行われる。
“第二段階Br試験は通常限られた患者群で臨床試験を開始し、目的は可能な副作用と安全リスクを確定し、候補薬物の特定の目標疾患に対する治療効果を初歩的に評価し、そして用量 耐性と最適投与量を決定することである。第2段階試験はさらに:2 a段階と2 b段階試験-2 a段階に分けられ、用量要求に特化している。少数の患者は用量-反応関係、すなわち用量増加に関連する反応増加が存在するかどうかを評価するために、異なる数量の薬物を投与された。そのほか、最適な投与量の頻度を検討した;2 b期試験は専門的に厳格なテスト薬物の治療、予防或いは疾病の診断における治療効果を設計した。
“第3段階Br試験とは、拡大した患者群における投与量、臨床治療効果と安全性をさらに評価するために、地理的に分散した臨床試験地点で臨床試験を行うことである。これらの臨床試験は候補薬物の全体的なリスク-収益比を確定し、監督管理の承認と製品ラベルに十分な基礎を提供することを目的としている。
“第4段階Br試験は、異なる集団における薬剤のbr効果および長期使用に関連する任意の副作用に関するより多くの情報を収集するために、承認条件として行われる必要がある研究である。
“理学療法Brは患者の活動能力,機能,福祉を回復,維持,最大限に利用した治療である。
“POCD“ は術後認知機能障害/精神錯乱のことである。
“ラ“関節リウマチのこと。
“REMSBrは、薬物の利点がそのリスクよりも大きいことを保証するために、深刻な安全問題が存在するいくつかの薬剤の薬物安全計画を要求することができるリスク評価および緩和戦略を意味する。
“政制事務局長Brは、炎症性疾患および疼痛の治療のためのCBDのような合成医薬レベル分子、非精神活性カンナビノイド化合物の近似物または類似体である合成カンナビノイド類似体を意味する。
“スポンサー?スポンサー“brは、FSC法案および関連法規適用条項の遵守に対する責任を含む、出願人または薬品スポンサー、すなわち新薬の販売に責任を負う個人または実体を意味する。”本明細書で使用されるように、用語“保険者”は、この用語を使用する文脈に応じて、私たちのIPOの保証人、KBLIV保証人有限責任会社を指すこともできることに留意されたい。
“THCBrは、大麻の主要な精神活性成分であるテトラヒドロカンナビノールを意味する。
“腫瘍壊死因子Brとは腫瘍壊死因子であり,身体の炎症反応の一部である。
四
前向き情報に関する警告声明
本年度報告は表 10-K(以下は“報告”と略称する)を採用し、連邦証券法で規定された展望性陳述を含み、1995年“プライベート証券訴訟改革法”が指摘した前向き陳述を含む。場合によっては、 “予想”、“信じ”、“継続”、“可能”、“推定”、“予想”、“可能”、“可能”、“進行中”、“計画”、“潜在”、“予測”、“プロジェクト”、“ ”、またはこれらの用語の否定または他の同様の用語によって前向き表現を識別することができる。前向き陳述は、将来の業績または結果の保証ではなく、必ずしもその業績または結果を実現する時間または時間に対する正確な指示でもない。展望的陳述は、陳述を行う際に得られる情報に基づいて、既知および未知のリスク、不確定要素、および他の要素に関連し、これらのリスク、不確定要素、および他の要素は、本報告の第1 A項の“リスク要因”のタイトルに記載された情報を含む、本報告の前向き陳述によって明示または示唆された情報と実質的な差 をもたらす可能性がある。前向き 陳述は、以下の態様に関する陳述を含むが、これらに限定されない
| ● | 私たちの候補製品の臨床と臨床前開発、製造、規制承認と商業化への期待 |
| ● | 不確定性は同社の候補薬物の臨床開発と監督許可と関係があり、臨床試験の登録と完成の潜在的遅延、アメリカ食品と薬物管理局(FDA)とイギリスの薬品と医療製品監督管理機関(MHRA)が提出した問題を含む | |
| ● | アメリカと他の国の規制動態 ; |
| ● | 私たちは私たちの上級管理職、キーパーソン、役員を引き留めることに成功したり、彼らを募集したり、必要な変更をしたりしました |
| ● | 現在の経営キャッシュフローは負であり、私たちは業務発展を促進する潜在的な能力、および任意のさらなる融資の条項を獲得し、高度に希釈され、煩雑な条項を含む可能性がある |
| ● | Br新冠肺炎疫病は著者らの業務運営と研究開発措置に対する持続的な影響; |
| ● | 費用、将来の収入、および資本需要の推定の正確さ ; |
| ● | 同社は第三者に依存して臨床試験を行い、患者を募集し、その臨床前と臨床薬物の供給を生産する | |
| ● | このような第三者およびパートナーと双方が合意した条項を達成する能力、およびそのような合意の条項; | |
| ● | 会社が計画している製品の患者数の見積もり | |
| ● | 候補薬物の予期しない副作用または治療効果が不足し、承認および/または商業化を制限する可能性があり、またはリコールまたは製品責任クレームを引き起こす可能性がある; |
| ● | 会社はその製品開発活動に適用される多くの連邦,州や地方の法律や法規の要求,米国以外のルールや法規を完全に遵守する能力がある | |
| ● | 製品研究開発固有の挑戦と不確定性は、臨床成功と監督管理承認の不確定性を含む;br}商業成功の不確定性; | |
| ● | 会社が新薬製品計画を開発·マーケティングする能力と、これらの開発計画の時間とコスト | |
| ● | 潜在的な衰退、およびマクロ経済、地政学、衛生および工業傾向、流行病、戦争行為(持続的なウクライナ/ロシア紛争を含む)および他の大規模な危機を含む高インフレ、金利上昇、および景気後退 | |
| ● | 私たちの現在の資本資源の充足性と、未来の期待キャッシュフローを推定して、私たちの運営需要を満たす |
| ● | 私たちは普通株式と株式承認証をナスダックに上場する能力を維持します |
| ● | その他のリスクと不確実性, には,以下の“リスク要因”以下のリスクと不確実性が含まれる。 |
本報告の任意の展望的陳述 は、既知と未知のリスク、不確実性および他の要素に関連して、未来の事件または私たちの未来の財務表現に対する私たちの現在の見方を反映しており、私たちの実際の結果、業績または成果は、これらの展望的陳述に明示的または暗示されている任意の未来の結果、業績または業績とは大きく異なる可能性がある。これらの不確実性を考慮して、あなたはこれらの展望的陳述に過度に依存してはならない。本稿に含まれるすべての前向き陳述は,本報告を提出した日の のみを指す.会社または会社を代表して行動することができるすべての後続の書面および口頭前向き陳述は、上記の警告的声明によって明確に制限されている。法律には別の規定がある以外に、私たちは未来に新しい情報があってもbrを利用できるいかなる理由でもこれらの前向きな陳述を更新または修正する義務を負いません。
v
第 部分I
プロジェクト1.ビジネス
序言:序言
一般情報
本年度報告に含まれる表格10−Kに含まれる情報は、本報告の末尾に含まれる連結財務諸表と関連付記とともに読まなければならない。
参照してください“語彙表“br}は、本報告書で使用されるバイオテクノロジー産業の略称および定義されたリストを表示する。
この報告書では私たちのロゴと私たちのいくつかの商標と商号が使用されている。本報告には,他人の財産に属する商標,商号,サービスマーク も含まれる.便宜上、本報告で言及された商標、商標名、およびサービスマークは、( およびSM記号を有しない場合がある。私たちの商標、商標、およびサービスマークについて言及することは、私たちが適用法律の下で私たちの権利または適用許可者の権利(あれば)を最大限に主張しないことを意味するわけではなく、適用法律の下で他の知的財産権の所有者が彼らの権利を主張しないことを最大限主張しないということではない。私たち は、任意の他社との関係、または私たちの裏書きやスポンサー を暗示するために、他社の商標や商品名を使用または展示するつもりはありません。
本報告で使用される市場データおよびいくつかの他の統計情報は、独立した業界出版物、市場研究会社の報告、または信頼できると考えられる他の独立したソースに基づいているが、私たちは、本報告で提供されるいかなる市場または調査データも委託していない。業界出版物および第三者研究、調査および研究は、一般に、彼らのbr情報は、このような 情報の正確性または完全性を保証しないにもかかわらず、信頼できると考えられるソースから得られることを示している。私たちは本報告書に含まれるすべての開示に責任があり、私たちはこれらの業界の出版物と第三者研究、調査、および研究が信頼できると信じている。本報告ではいかなる第三者情報に関するいかなる誤った陳述も知らないが,彼らの推定,特に予測に関する推定は,多くの仮説に関連しており,リスクやbr}の不確実性の影響を受け,様々な要因によって変化する可能性がある“と題するリスク要因 “この報告書の内容。このような要素と他の要素は私たちの未来の業績が私たちの仮定と見積もりと大きく違うことをもたらすかもしれない。本稿に含まれるいくつかの市場や他のデータ,および競争相手が180生命科学会社に関するデータも,我々の好意的な推定に基づいている.
私たちの財政年度は12月31日に終わるだろう。中間業績は四半期ごとに発表され、それぞれ3月31日、6月30日、9月30日までの第1四半期、第2四半期、第3四半期であり、ここでは12月31日までの四半期を私たちの第4四半期と呼ぶ。2022年度とは2022年12月31日現在の年度を指し、2021年度は2021年12月31日現在の年度を指す。
株を逆分割する
2022年12月15日、180生命科学会社株主特別総会において、会社株主は、会社の第2回改正と再改訂された会社登録証明書の修正案を承認し、私たちの普通株の発行済み株式と発行済み株を逆方向に株式分割し、1株当たり額面0.0001ドル、4対1と20対1の間で、4対1と20対1を含み、具体的な割合は我々の取締役会またはその正式に許可された委員会が自ら決定する。改正案が承認されてから2023年12月15日までのいつでも(“株主管理局”)。2022年12月15日、会社取締役会(“取締役会”)は、株主管理局と共に、我々の普通株の逆株式分割に影響を与える割合が1:20(“逆 株式分割”)である2つ目の改正と再署名した会社登録証明書の修正案を承認した。
1
特別会議と取締役会の承認に続いて、私は2022年12月15日にデラウェア州州務卿に改訂及び再改訂された2つ目の会社登録証明書(“改訂証明書”)を提出し、株式の逆分割を実施した。修正案証明書のコピーは、添付ファイル3.1として本ファイルに添付され、参照によって本明細書に組み込まれる。
改訂証明書 によると、逆株式分割は2022年12月19日午前12:01に発効した。アメリカ東部時間(“発効時間”). 会社の普通株式或いは公共株式証株式の取引記号“ATNF”及び“ATNNW”は変更がなく、 は株式の逆分割に関係している。
改訂証明書brは、私たちの普通株の許可株式数を減少させず、私たちの普通株の額面を変更することもなく、私たちの普通株の投票権や他の条項も修正していません。
逆株式分割に関する断片的な株式は発行されておらず,登録されている株主はもともと断片的な株式を獲得する権利があり, はその断片的な株式を最も近い全体の株式に丸める権利がある.
本報告は20株1株逆分割の影響を遡及的に反映している。
定義する
文脈が に別の規定を要求しない限り、参照“会社は、” “私たちは、” “私たちは、” “私たちの、” “180人の生活”, “180 ls“と”180生命科学社特に180生命科学会社とその合併した子会社を指す。“への引用”KBL線“2020年11月6日までの会社を指す(議論と定義は以下の通り)。
また,文脈 に別の要求がある以外は,本報告の目的のみである:
| ● | “コンピュータ支援設計“ は加元; | |
| ● | ““取引所法案”“br”とは、1934年に改正された証券取引法を指す | |
| ● | “£“ または”ポンド“ポンドのこと | |
| ● | “アメリカ証券取引委員会“ または”選挙委員会“米国証券取引委員会のこと | |
| ● | “証券法 は改正された1933年の証券法のことです。 |
2
どこで他の情報を見つけますか
年度,四半期, と現在の報告,依頼書,その他の情報をSECに提出した。私たちのSECファイルはインターネット を介してSECサイトで公衆に提供することができます。サイトは:Wwwv.sec.govこのような報告がSECに報告された後、またはSECに提供された直後に無料でダウンロードすることができ、アドレスは投資家”—“アメリカ証券取引委員会の届出書類”—“すべてのSEC届出書類“私たちのサイトのホームページの住所は:Wwwww.180 life ciences.comそれは.また、私たちの秘書に口頭または書面で私たちがSECに提出した文書のコピーを無料で提供することができ、本報告の表紙に規定されている住所と電話で秘書に連絡することができます。私たちのサイトの住所はWwwww.180 life ciences.comそれは.当サイト上の情報または当サイトを介してアクセス可能な情報は、参照によって本報告に組み込まれていないので、本報告の一部とみなされてはならない。
会社の概要
我々は臨床段階バイオテクノロジー会社であり,カリフォルニア州パロアルトに本部を置き,革新的な研究と適切な併用療法を採用することにより,慢性疼痛,炎症,線維化に満足していない医療ニーズに対する治療法の開発に専念している。わが社はバイオテクノロジーと製薬分野の世界をリードする何人かの科学者によって設立された。私たちの世界的に有名な科学者マーク·フェルドマン教授、ローレンス·スタンマン教授、最近亡くなったラファエル·メフラム教授、ジョナサン·ロスバード博士、ジャグディップ·ナンチャハル教授は薬物発見に豊富な経験と重大な成功を持っている。これらの科学者はオックスフォード大学(“オックスフォード”)、スタンフォード大学(“br}”)とエルサレムヘブライ大学(“ヘブライ大学”)から来ており、管理チームは早期医療会社の援助と成長に豊富な経験を持っている。Raphael Mechoulam教授は2023年3月に亡くなったが、彼のヘブライ大学での研究は、本稿の後の“SCASプラットフォーム”の部分で述べたように、他の科学者によって継続されている。
私たちは3つの異なる製品 開発プラットフォームを持っていて、異なる疾病或いは医療条件に集中して、異なる要素、分子或いは蛋白質に対して、 は以下の通りである
| ● | 抗腫瘍壊死因子プラットフォーム:繊維化と抗腫瘍壊死因子(“抗腫瘍壊死因子”); |
| ● | SCASプラットフォーム:合成カンナビノール(CBD)または大麻フェノール(CBG)類似体(“SCA”);および |
| ● | α7 nAChRプラットフォーム:α7ニコチン型アセチルコリン受容体(“α7 nAChR”)に注目する。 |
我々の主要な候補製品 は最近イギリスで2 a期と2 b期の概念検証臨床試験を完了した。オランダ は早期Dupuyten筋拘縮症であり,手掌線維結合組織の発育に関与する疾患である。現在,われわれは,計画中の術後認知機能低下の第2段階試験,計画中の肩凍結の第2段階試験など,抗腫瘍壊死因子プラットフォーム下でのいくつかの適応の臨床試験のみを計画または行っている。同社はbr患者を募集して肩凍結可能性試験を行っており,イギリスの規制機関が緩やかな募集試験の終了を要求しているため,9名の患者で募集を終了した。実験終了の結果は,将来的にはより多くの参加者を募集するために別の実験を行う必要がある可能性があることを意味する.我々の3つの製品開発プラットフォームのうち,1つのみがSCASプラットフォームであり,カンナビノール(CBD)(大麻やテトラヒドロカンナビノール(THC)ではなく)に関連する製品に関連しており,SCASプラットフォームでの適応や製品については米国や海外での臨床試験は行われていない。著者らは現在SCAとα7 nAChRプラットフォームのための臨床前研究と開発活動を行っている。
業務戦略
我々の目標は,慢性疼痛,炎症,線維化に関する我々の研究を以下の戦略で利用することである
| ● | 我々の早期Dupuyten‘s Constraint臨床候補製品を現在の後期開発からイギリス,欧州連合(EU),米国(U.S.)に推進し,承認を得た このような候補製品については、潜在的にイギリス、EU、米国で商業化され、世界の他の市場での最適なビジネス経路が決定される |
3
| ● | 私たちの臨床前候補製品を臨床試験に移し、イギリス、EU、アメリカでこのような未来の候補製品の承認を求め、そしてアメリカ、イギリス、EUでこのような未来の候補製品を商業化する可能性がある |
| ● | 慢性疼痛、炎症、線維化を治療する新しい一流製品を発見、開発、商業化するために、当社独自のbr製品開発プラットフォームを利用した |
| ● | 慢性疼痛、炎症、繊維化研究における著者らの地位を強化する。 |
製品開発プラットフォームの概要
以下の表は,現在臨床試験段階にある製品と適応を含む,我々の3つの製品開発プラットフォームの製品と適応をまとめたものである。
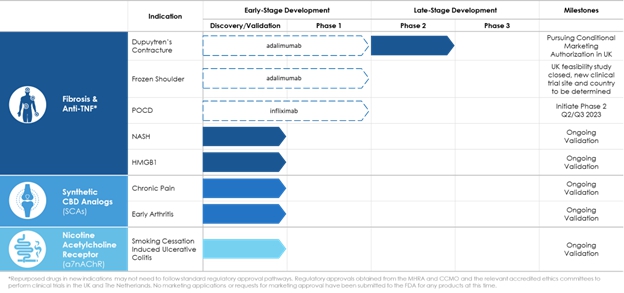
*MHRAおよびCCMOおよび関連する認可された道徳委員会から得られた、イギリスおよびオランダにおける臨床試験の規制承認。brは現在、いかなる製品の発売申請または上場承認要求もFDAに提出されていない
2021年12月1日,br社はDupuyten‘s Constraint 2 b期臨床試験の正面頂線データを発表した。
2023年2月22日,会社は凍肩実行可能性試験の患者募集を中止することを発表し,イギリスの規制機関が緩やかな募集試験の終了を要求したため,9名の患者の募集を終了した。実験終了の結果は, は将来,より多くの参加者を募集するために別の実験を行う必要がある可能性があることを意味する.
製品開発プラットフォーム は、以下でより詳細に説明する。
線維化と抗腫瘍壊死因子プラットフォーム
著者らの抗腫瘍壊死因子(TNF)プラットフォームは著者らの完全子会社180 Treeutics L.P.(“180 LP”)から始まった。このプラットフォームは,炎症性疾患や線維化の分子機序の研究や,腫瘍壊死因子を線維化のメディエーターとして発見し,他の免疫駆動疾患に焦点を当てている。この研究は1980年代にわれわれの連合議長Marc Feldmann教授によって最初に行われ,関節リウマチ(RA)患者組織の分析に基づいていた。同様の方法を活動性線維化患者のヒト疾患組織の分析に応用しており,この研究はオックスフォードのJagDeep Nanchahal教授(彼もわれわれの臨床諮問委員会議長)がリードしており,我々が開発している新たな治療標的や方法の決定につながっている。教授たち。Nanchahal とFeldmannは他の科学者と協力して,抗炎症薬開発における彼らの経験と専門知識 を利用して抗腫瘍壊死因子療法の新たな応用を探索している。われわれは,抗腫瘍壊死因子薬,例えばアダリアモノクロナル抗体が,Dupuytren筋拘縮,肩凍結や術後認知機能障害/精神錯乱(POCD)などの新たな適応に積極的に作用することを証明することを求めている。
4
著者らが臨床開発した最初の候補製品 は手の早期線維化を治療するための潜在的な薬物Dupuytren‘s Constraintであり、 は現在イギリス或いはEUではまだ承認されていない治療法である。クロストリジウム溶解菌由来のコラゲナーゼは,米国で末期Dupuytren筋拘縮症の治療に許可されている。提案された治療は,抗腫瘍壊死因子抗体アダリアモノクロナル抗体を早期疾患組織に局所投与することにより実施される。Dupuyten‘s Constraintの2 a期臨床試験結果は2018年7月に発表され、この試験は恵康信託、イギリス衛生部と同社が支持を提供した。この研究は,積極的な組織反応 が抗線維化機序を示し,指導用量の後続試験を示している。最も有効な投与量と製剤を決定した後,これらの積極的な概念検証データに基づいて,同社は恵康信託基金やイギリス衛生部とともに早期デュプイトレン筋拘縮症患者に対する2 b期試験を開始した。最初の計画は138名の患者を1:1の割合でランダムに分配し,3カ月ごとにアダマブまたはプラセボ注射を4回受け,ベースラインから合計18カ月間追跡観察を開始することであった。恵康信託の追加資金支援のもと、2 b期試験は2019年4月に174人の患者募集を完了し、2017年2月に服薬を開始した。最後の患者は2019年4月に登録された。早期デュプイトレン筋拘縮症に対する2 b期臨床試験が完了した。2021年12月1日、同社は試験の頂線データを公表し、超音波スキャン結節硬度の主要な終点と結節サイズの副次的終点は統計学的に有意差があることを示した。関連された深刻な不良事件は発生しなかった。すべての結果はすでに同業者審査の定期刊行物に提出され、発表後に発表される。この線維化と抗腫瘍壊死因子製品開発プラットフォームにより,凍結した肩,肝,肺線維化,POCDの潜在的治療法を開発するためのbr研究も行われている。
以下のグラフは抗腫瘍壊死因子プラットフォーム下で現在の提案に基づく現在と未来の臨床試験の時間スケジュールをまとめた。
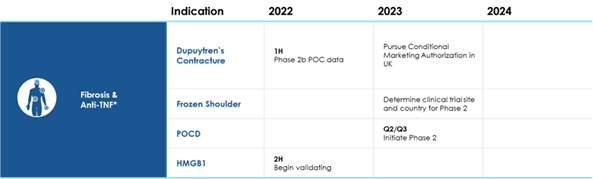
著者らはすでにイギリス薬品と保健品監督局(MHRA)とオランダ中央委員会Mensgenebonden Onderzoek(CCMO)及び関連認可道徳委員会の監督許可を得て、抗腫瘍壊死因子プラットフォーム下の適応のみに対してイギリスとオランダで臨床試験を行う。現在、抗腫瘍壊死因子プラットフォーム下のいかなる適応或いは製品に対してアメリカ食品と薬物管理局(FDA)に発売申請或いは発売許可要求を提出していない。2022年3月29日、抗腫瘍壊死因子プラットフォームによる早期Dupuyten病治療の臨床試験の臨床結果評価を検討するために、C型会議の開催要請をFDAに提出した。FDAは2022年4月26日に会議要求を承認し、会議の代わりに書面回答 を提供することに同意した。FDAは2022年6月9日、結節の硬度と大きさがこのような研究の適切な終点を構成するかどうかを疑問視する上述の書面回答を提供した。FDAは“提案された結節硬度と結節の大きさの結果指標 は患者の感覚、機能或いは生存状況を測定する臨床結果指標ではないようであり、 はあなたの未来の登録研究においてこれらの指標が治療効果の展示を支持する必要がある”と明確に表明した
2023年2月22日,会社は凍肩実行可能性試験の患者募集を中止することを発表し,イギリスの規制機関が緩やかな募集試験の終了を要求したため,9名の患者の募集を終了した。実験終了の結果は, は将来,より多くの参加者を募集するために別の実験を行う必要がある可能性があることを意味する.
HMGB 1計画
私たちの取引先HMGB 1:プロジェクト は2021年11月2日にオックスフォード大学の技術許可を得た。われわれのHMGB 1計画は線維化と抗腫瘍壊死因子プラットフォームに属する。著者らはすでにHMGB 1が1種の治療標的であり、多数の内因性成体幹細胞に作用し、現在或いは未来の損傷に対する生理的再生反応を加速することを確定した。これらの知見は幹細胞生物学や再生医学分野と広く関連しており,NASHの肝臓再生など組織修復を促進する治療法が提案されている。
5
この技術はジャグディップ·ナンチャハル教授がオックスフォード大学の実験室で許可を得る前に開発された。HMGB 1の開発はまだ初期段階であるため, 我々はマイルストーンと開発スケジュールを評価している.HMGB 1の許可には肝線維化の先行候補開発 がある。私たちのHMGB 1計画は緩やかに推進され続けている。この分子結合の分子動力学は極めて複雑であり、主要な候補を決定するために、より広範な研究が必要かもしれない。
HMGB 1プラットフォーム下のいかなる製品或いは適応について関係当局の監督管理許可を求め或いは獲得していない。
SCASプラットフォーム
私たちのSCASプラットフォームは私たちの完全子会社CannBioRex PharmPharmticals Corp.(“CBR Pharma”)から始まり、その創始者、故Mechoulam教授とFeldmann教授が共同で努力した。このプラットフォームは、炎症性疾患および疼痛の治療のためのCBDのような近距離または遠隔の非精神活性大麻類似体の開発に専念する。これらの開発はフェルドマン教授とMechoulam教授の20年間の協力の結果であり、フェルドマン教授はRAとその後の炎症性疾患の治療に使用され、抗腫瘍壊死因子療法を発見し、商業化した。これは現在世界で最も売れている薬物カテゴリーである。Mechoulam教授は世界有数の大麻化学の専門家であり、THC、CBD、その後の内因性カンナビノイドの識別に成功した。私たちはオックスフォードケネディ研究所の研究チーム(Feldmann教授、Richard Williams教授と他の人からなる)とヘブライ大学の研究チーム(Avi Domb教授、Amnon Hoffman教授と他の人からなる)と協力して、新薬を生産し、それらをテストし、それらの摂取を最適化し、疾病目標に伝達している。合成化合物に基づく新規経口活性鎮痛と消炎薬の開発を目的とし,慢性疾患の治療を目的としている。これらの合成化合物を総称して“人工合成されたCBD類似体“(”規制計画協定“)。われわれの主な開発目標は関節炎および慢性·再発性疼痛であり,われわれの副次的開発目標は糖尿病/糖尿病神経病変,線維筋痛,多発性硬化症,肥満,脂肪肝である。
現在、SCASプラットフォーム下の任意の製品或いは適応について関係当局の監督管理許可を求め或いは獲得していない。
α7 nAChRプラットフォーム
私たちの取引先α7 nAChR:プラットフォーム は私たちの完全子会社KatExcoから始まり、その創始者はそこで確定したα7 nAChR:アルツハイマー病やパーキンソン病などの疾患に関連するアミロイドの重要な受容体であるα7 nAChR:脳の神経細胞や免疫系の重要な細胞表面に発現している。ジョナサン·ロスバード博士とスタンマン教授の研究により,経口薬物の小分子はこの受容体に結合し,炎症性疾患を潜在的に減少させることが示唆された。ロスバード博士とスタンマン教授も証明したα7 nAChR:多発性硬化症や関節リウマチの動物モデルや心臓発作や脳卒中を減少させる上で重要である。私たちの取引先α7 nAChR:製品開発プラットフォームは現在開発に集中しているα7 nAChR:炎症性疾患の治療に用いられるアゴニストは,最初は禁煙後に起こる潰瘍性大腸炎であった。
今のところ関係部門に監督部門の許可を求めたり、許可を得たりしていませんα7 nAChR プラットフォーム。
候補製品
私たちは慢性疼痛、炎症と繊維化の治療のための広範かつ多様な候補製品ルートを構築することを試みている。私たちの候補製品は以下の要素選択に基づいて開発されている:満たされていない医療需要を満たす潜在力;著者らの臨床前研究と開発仕事によって確定された開発可能性;測定しやすい検証された監督端末に基づいて概念検証を迅速に実現する潜在力; 及び巨大な商業潜在力。
6
抗腫瘍壊死因子プラットフォームDupuyten筋拘縮症
概要
Dupuyten筋拘縮症は,手線維化とも呼ばれ,進行性で治癒できない疾患であり,手掌に線維索が出現することが特徴であり,通常は指環や/または小指に影響を与え,通常は複数の関節に影響を与え,けいれんや影響を受けた指を伸ばすことができない。医師の前に出現した場合,症状には手掌に結節が出現することが含まれており,これは無痛である可能性もあり,痛みの可能性もあり,通常患者を不安にさせ,収縮した指の機能を失っている可能性がある。現在のところ承認されていない治療案 は,症状が出現した早期疾患患者に用いられている。
手術はDupuytren筋拘縮症患者の標準的な治療法であるが,回復期や再発リスクの延長に関与している。
我々は,以前に承認され,いくつかの自己免疫性br条件にHumiraブランドで使用されており,早期Dupuytren筋拘縮症の治療のための抗腫瘍壊死因子治療用モノクロナル抗体の使用を変化させることにより療法 を開発している。オックスフォード大学の研究により、1種の抗腫瘍壊死因子の機序は筋芽細胞の増殖を緩和或いは防止でき、それによって手掌中の繊維結節/索状物の形成と成長を招き、そして手指痙攣を招く可能性がある。著者らはすでに2 b期の臨床試験を通じて開発計画を推進し、複数回の病巣内注射が疾病の進展と機能改善に与える影響を評価した。
末期疾患を有するDupuyten患者は主に整形外科或いは整形外科医師によって治療され、痙攣が手の機能に影響する時、彼らは侵襲性干与に依存する。現在の治療選択は開放手術(筋膜切開術或いは筋膜切開術)と侵襲性の小さい針状腱膜切開術(NA)或いはコラゲナーゼ注射を含む。侵襲性の小さい手術は,収縮した臍帯の完全性を破壊することを目的としているため,指を伸ばすことができる。不幸にも、このような選択は高い再発率と関連がある。医学界とDupuytren患者群の末期疾病結果に対する不満、及び早期/痙攣前段階での干与の選択が不足していることは、早期干与に対する医療需要がまだ満たされていないことを表明した。
Dupuyten財団のデータによると、Dupuytenの罹患率はアメリカ人口の7%に達すると推定されている。財団によると、約300万人の患者が治療すべきミオクローヌス症を患っていると推定されているが、そのうちの10%から20%の患者しか治療されていない。治療不足の原因には、使用可能な介入措置のタイプ、不良な長期結果、および精算障害が含まれている可能性がある。
2021年末に8名の整形外科/整形外科医師に対して行った初歩的なインタビューの中で、外科医師と患者は末期筋拘縮症が発展する前に非侵襲性方式でこの疾病を治療し、病状の更なる発展を制限し、機能を保留し、侵襲性手術を防止或いは延期することを強く望んでいる。この満たされていない需要を解決するための外科医の基本原理に対する外科医の反応は全体的に積極的であり,抗腫瘍壊死因子化合物の機械的概念は納得できると考えられている。調査を受けた多くの手外科医から見ると,非侵襲的で安全な製品特性はadalimumabを重要な治療選択とし,現在通常治療されているよりも広い患者に適している可能性がある。すでに発表されたデータが臨床治療効果と安全性を支持すると仮定し、アダリアモノクロナル抗体は手術、針状腱膜切開術或いはコラゲナーゼの魅力的な代替案となると信じている。また,現在治療を受けていない多くの早期患者において潜在的な用途があると信じている。
初期(これらの医師インタビューのフィードバック)と二次研究によると、Red Sky Partnersは、最初のラベルは明らかな筋拘縮を有する患者に集中し、その中でアダリマブは結節を軟化させ、進展を制限することは、現在の治療方法とは高度に異なり、アメリカでは毎年3億~3.5億ドルの収入を産生することができると結論した。より重要なのは、安全で非侵襲的な治療を提供し、機能を改善する機会を提供することは、治療可能な人々を著しく拡大する可能性があり、より多くの患者が治療を求め、より多くの医師が患者に別の選択を提供し、彼らの疾患が進展するかどうかを待つために、治療可能な人々を著しく拡大する可能性がある。これは彼らが今日できないことだ。このような製品の位置づけは最初の市場機会の2~3倍の収入機会を創出することができる。
第二段階臨床試験
著者らの完全子会社180 LPは恵康信託とイギリス衛生部と共にDupuytren‘s Constraintの2 a期臨床試験に資金を提供し、この試験は実験薬物の臨床試験設計を採用し、陽性の組織反応、及び後続試験の指導投与量と耐性を示した。このデータは2018年6月に発表された。
7
2 a期試験では28名の患者を募集し,そのうち8名が15 mg(Mg)群,12名が35 mg群,8名が40 mg群に割り当てられたACTA 2、COL 1 A 1、COL 3 A 1何度も何度もCDH 11それは.プラセボ群(1.51±0.09 ng/g)と比較して,アダマブ40 mg治療を受けた患者(1.09±0.09 ng/αg総蛋白)のμ−SMA蛋白発現レベルは有意に低下した(p=0.006)。プラセボ群(817±78 pg/μg)と比較して,40 mg群(474±84 pg/μg総蛋白)のI型前コラーゲン発現レベルも有意に低下した(p=0.019)。2つの深刻な有害事象があり、いずれも研究薬とは関係がないと考えられている。この用量範囲の検討では,アダリムマブ40 mg投与により0.4 mlで筋線維芽細胞表現型が低下し,2週間でα−SMAとI型前コラーゲンの発現が減少した。
最も有効な投与量と製剤を決定した後,これらの積極的な概念検証データに基づいて,同社は恵康信託基金やイギリス衛生部とともに,早期デュプイトレン筋拘縮症患者に対する2 b期試験を開始した。最初の計画は138名の患者を1:1の割合でランダムに分配し,3カ月ごとにアダマブまたはプラセボ注射を4回受け,ベースラインから合計18カ月間追跡観察を開始することであった。2 b期試験はWellcome Trustとイギリス衛生部が資金援助し、180 LPが出資してこの薬剤を購入し、2019年4月に174人の患者の募集を完了し、2017年2月にイギリスとオランダのグローニンゲンで服薬を開始した。
早期Dupuyten筋拘縮症に対する2 b期臨床試験が完了した。2021年12月1日、同社は試験の頂線データを公表し、このデータは、超音波スキャンの結節硬度の主要な終点と結節の大きさの副次的な終点が を満たし、統計学的意義があることを示した。関連された深刻な不良事件は発生しなかった。全研究結果は2022年4月29日に“柳葉刀リウマチ学”誌に発表された。
他の候補製品や適応
潜在的な治療法に加えて,上記Dupuyten筋拘縮症を治療する薬剤が開発されており,肩凍結などの他の線維化疾患の治療に用いられる抗腫瘍壊死因子の使用を変化させることが求められている。フェルドマン教授は20世紀80年代の研究により、抗腫瘍壊死因子は1種の有効な抗炎症薬であり、多種の可能な用途を有し、その後様々な形式の炎症性関節炎と炎症性腸疾患(IBD)及びその他の適応のために許可された。それ以来,現在世界で最も売れている薬物種別“br}抗腫瘍壊死因子療法が生じ,2022年11月3日に発表された研究報告世界報告によると,2022年のこの薬物の価値は427億ドルである。広く知られ広く使用されている治療薬であるアダリマブを用いることにより,研究·開発過程は過去20年間に数百万人の患者で広く使用されているため,既存の安全性に関する製品情報によって切断される可能性がある。
凍った肩
肩こりは,癒着性嚢炎とも呼ばれ,極めて苦痛や虚弱な疾患であり,睡眠を含めた個人の日常活動に影響を与える。アメリカ国立衛生研究院のデータによると、肩こりは40歳から60歳の人の中で最もよく見られます。brによると、2%から5%の人がある程度肩こりの影響を受け、女性の肩こりは男性よりもよく見られると推定されています。糖尿病患者では特に肩凍結が出現する可能性がある:約10%から20%の人がこの病気になるが,なぜこのようなことが発生したのかは不明である。また,肩凍結者の約20%は別の肩部で同様の問題が生じるであろう。2010年に発表された“肩肘”に発表された記事によると、30%に達する糖尿病患者は肩凍結が出現すると推定されており、この群では、症状はより持続的で頑固であることが多い。
疼痛を中心とした炎症期には,鎮痛剤,理学療法,コルチコステロイド注射治療が一般的である。持続的に硬直した患者は二次ケアに移行される可能性があり,麻酔,水拡張や外科関節鏡下で手技により被膜が放出される可能性がある。現在承認されていない標的治療は,イギリス国立健康研究所と協力し,疾患の早期疼痛を中心とした炎症段階で患者brを募集し,抗腫瘍壊死因子の局所投与の可能性を検討している。この局所注射抗腫瘍壊死因子による凍結肩治療の第二段階臨床試験は2021年6月に準備段階を開始した。NIHRはオックスフォード大学に250,000 GBの贈与を付与し,brの実行を支援しており,臨床試験場所が決定中である。その会社はこの実験を支援するために追加的な資金を提供している。2022年5月、イングランドは早期肩こりを有する男性と女性を募集し、大型無作為対照試験の実行可能性を確定し、関節内に抗腫瘍壊死因子(Adalimumab)を注射することが痛みを軽減し、痛みを主とする早期肩こり患者の機能を改善できるかどうかを評価し、この試験はanti-Freaze-F試験と呼ばれている。逆Freaze−F実験 はオックスフォード大学が行い,最初に84名の参加者の募集を求めた。新冠肺炎と該当スタッフの空きにより国家衛生条例システムが滞って承認が遅延した後、9名の参加者を募集して試験に参加し、2023年2月中旬まで続いた。その後、国家衛生研究院の研究回復とリセット計画により、著者らは新冠肺炎期間中に求人サイトを開設し、十分な参加者を募集することはかなりの挑戦に直面しているため、実験の進展は遅い。そこで,NIHRは首席調査員に裁判を終了させ,さらなる募集を要求した。これまで登録された参加者は彼らのbr注射を受け、既定のプランに従ってフォローする。同社は以前、無料延期を要請していたが、拒否された。 実験が終了した結果は、将来的により多くの 参加者を募集するために別の実験を行う必要がある可能性があることを意味する。
8
ヒト肝線維化
肝繊維化は瘢痕組織が正常肝組織に代わるため、器官に与える長期損害である。このことが最もよく見られる原因は,非アルコール性脂肪性肝疾患(NAFLD)であり,非アルコール性脂肪肝(NFL)や非アルコール性脂肪性肝炎(NASH)を含む。“自然評論”が2016年に発表した記事によると、非アルコール性脂肪肝は約30%の米国人口に影響を与えている。約2%のNFL患者と約15%から20%のNASH患者は肝硬変,肝線維化,主要な健康問題に進展している。
われわれの知る限り,NASHを治療する方法は現在のところ承認されていない。したがって、私たちは効果的な予防治療を作ることに大きな潜在的市場があると信じている。共同市場研究会社のデータによると、2018年に肝線維化を治療する市場は約130億ドルで、2022年には約200億ドルに増加し、年間複合成長率(CAGR)は11%を超えると予想されている。われわれは2020年第2四半期にヒト肝臓試料に基づくNASHの臨床前研究を開始した。
術後認知機能低下(POCD)
POCDはよく見られる神経精神症候群であり、注意力、意識と認知障害と定義され、短時間で発展し、一日で変動する。寛骨骨折患者ではPOCD発生のリスクが特に高い。イギリスの2018年国家監査データによると、すべての寛骨骨折患者の25%が精神錯乱を患っている。POCDは機能予後が悪く,生活の質の低下と入院期間の延長と関係がある。寛骨骨折患者が精神錯乱後に死亡する可能性は入院患者の2倍であり,老人ホームに設置される必要がある可能性は入院患者のほぼ4倍である。POCDも長期的な認知障害と密接に関連している
寛骨骨折は老年患者と医療保健システムが直面している主要な挑戦の一つである。2017年に発表された“柳葉刀公衆衛生”によると、米国とヨーロッパでは、寛骨骨折は平均2.7%の中高年人口の健康予想寿命損失に関連している。寛骨骨折患者の平均年齢は83歳,身体虚弱であり,3分の2は女性であった。彼らの30日間の死亡率は7%であり、パーキンソン病や多発性硬化症の診断と類似した健康関連生活の質が低下し続けている。様々な研究によると,心臓手術後13−40%の患者にPOCDが発生する。米国では年間500,000例の心内直視手術と450,000例の股関節手術があり,高齢者にとってPOCD治療の有益な療法はこれらの患者に大きなメリットを与える。著者らは2023年第2四半期または第3四半期にPOCDに対する抗腫瘍壊死因子を使用した第2段階研究を開始し、患者の募集を開始する予定である。ケネディリューマチ研究信託基金はこの潜在的な用途を保護する特許を承認した。
SCASプラットフォーム
概要
カンナビノイドは大麻植物から抽出された化合物である。大麻に含まれる2種類の主要大麻類はCBDとTHCである。草本大麻の使用に関する精神反応を引き起こすカンナビノイド系化合物が知られているが,これらのbr特性を有するカンナビノイド系化合物はない。ここ数十年来、重大な科学進歩は新しい植物由来のカンナビノイドと内因性カンナビノイドシステムの発見を招いた。ヒト内因性カンナビノイド系には、カンナビノイド受容体1(“CB 1”)とカンナビノイド受容体2(“CB 2”)の少なくとも2種類のカンナビノイド受容体がある。CB 1受容体は脳の中で最も広く発現するG蛋白共役受容体の一つと考えられ、脳の中で運動と姿勢制御、痛みと感覚知覚、記憶、認知、感情及び自主と内分泌機能に関連する領域は特に豊富である。CB 1受容体も周囲組織に認められ、末梢神経と非神経組織、例えば筋肉、肝臓組織と脂肪などを含む。CB 2受容体は主に免疫系の組織に発現し、カンナビノイドの免疫効果を媒介すると考えられている。CBDはCB 1受容体と相互作用せず,CB 2受容体の弱アゴニストのみである。CBDは人体内の他の重要な神経伝達物質と神経調節系と相互作用し、一過性受容体電位チャネル、アデノシン取り込みとセロトニン受容体を含む。大量のカンナビノイドの深遠と多様な薬理作用は大麻類薬物の多くの適応と疾病領域の開発に巨大な潜在力を提供したが、研究の複雑性も増加した。
9
SCA計画については,ヘブライ大学やオックスフォード大学とbr合意を達成しており,これらの合意に基づき,ある目標適応を治療するための新しいSCAを開発·特徴づけ,早期臨床試験を行う予定である。ヘブライ大学とオックスフォード大学と締結した研究協定により、私たちはヘブライ大学とオックスフォード大学に研究機関を設立し、ヘブライ大学で設計と合成された新しい大麻類化合物の開発と試験を促進する。ヘブライ大学の研究室はこれらの化合物を合成し、初歩的な治療効果と安全性研究を行う。
ヘブライ大学でこれらの初歩的な研究が完了すると,化合物はオックスフォード大学のリチャード·ウィリアムズ教授に送られ,そこでさらなる評価が行われ,最も潜在力のある臨床治療効果と商業開発の候補薬が決定される。その後、著者らは先導化合物の臨床開発を支持し、最終的に第二段階の臨床試験に入り、br}慢性疼痛と炎症適応の臨床実用性を確定する。
研究の重点は安全と耐性の良好な化合物を開発し、鎮痛と免疫調節活性を有し、そして現在下流の炎症過程に対する治療方法と協同作用ができることである。予備研究と開発を行った後,brが最も有望な化学誘導体を選択して1/2期臨床試験に入り,成功した毒性研究を待った。また,我々はすでに動物研究からCBDの2種類の鉛含有固体用量経口製剤を決定し,健康なヒトボランティアの薬物動態分析を促進する準備を進めている。
候補品や適応症
経口的で比較的安全な抗炎症薬,特に鎮痛特性を有する薬剤には,満たされていない需要があると考えられる。SCAはこれらの需要を満たす可能性があると信じており、関節炎、多発性硬化症、糖尿病、乾癬、肥満および脂肪肝、および様々な苦痛の状態の治療のための新しい、経口可能で特許出願可能な候補薬brの開発を開始している。我々のSCA方面の仕事は現在臨床前開発段階にある。
医療用大麻は、植物由来の化合物の複雑な混合物であるため、関心のある活性化合物の一致レベルを提供するか、または他の天然化合物のレベルを制御することは困難である。そこで,これらの医療用大麻の有害問題を解決するために,非植物由来の経口特殊用途大麻に取り組んでいる。成功すれば,これらのSCAは承認された薬物製品となり,強力な一致と安全な投与量を提供し,患者の摂取量を慎重に制御することができる。
SCAの開発と臨床研究は、SCAが医療用大麻に対していくつかの重要な利点を持つことを明らかにすると信じている
| ● | 混合物ではなく純化合物(>99.5%)を使用した |
| ● | 投与量を測定し、制御し、治療効果と副作用レベルを制御することができる |
| ● | 複製可能な製品を作ること |
| ● | 受容体を選択するために結合選好を制御するために新しい合成類似体を設計することができ、受容体結合のアゴニストまたはアンタゴニスト効果(薬物動態および動態学)を制御し、薬物の体内での半減期を変化させ、特定の組織においてのみ活性化されるプロドラッグ形態を作成し、それによって標的副作用を潜在的に減少させることができる。 |
これらの利点に加えて、科学的二重盲検臨床試験でSCAを試験することは、大麻化合物の治療的使用に対する医師の懸念を軽減するのに役立つ。この変化は,これらの薬物療法を獲得する患者数を増加させる可能性がある。臨床試験が成功すれば、SCAは多くの潜在的な市場と適応を私たちの目標とすることができ、その中には慢性と再発性疼痛、糖尿病、骨関節炎、肥満と脂肪肝を有する患者を含む。
10
α7 nAChRプラットフォーム
概要
我々の2人の首席科学者,スタンマン教授とロスバード博士は,以前にアミロイドの重要な受容体の1つであり,α7 nAChRと呼ばれ,アルツハイマー症やパーキンソン病などの疾患に関与していることが発見された。α7 nAChRは脳神経細胞と免疫系の細胞表面に発現している。ロスバード博士とスタンマン教授による研究では,経口薬である小分子がこの受容体に結合し,炎症性疾患を減少させるのに有効であることが示唆されている。Rothbard博士とSteinman教授は,この受容体は多発性硬化症や関節リウマチ動物モデルや心臓発作や脳卒中の疾患を減少させる上で重要であることを証明している。
高濃度の小分子熱ショックタンパク質の役割を理解しようと努力しています(“SHSP多発性硬化症患者の大脳病変に認められる)は,多発性硬化症,心臓と網膜虚血および脳卒中の動物モデルにおいて(I)免疫抑制と(Ii)治療作用を有することを意識させた。1つの重要な認識は,タンパク質や小ペプチドからなるアミロイド線維がsHSPと同様の生物学的反応を示すことである。線維およびsHSPはマクロファージ(“MΦ”)および制御性B細胞に特異的に結合して活性化する。架橋と沈殿実験では,両種ともnAChRに結合し,JAK 2/STAT 3シグナル伝達を介していることが示唆された。ニコチンによる実験的自己免疫性脳脊髄炎(“EAE”)の治療は,われわれの治療法と同様の免疫抑制パターンを誘導し,EAEモデルで試験時に多発性硬化症(MS)の治療に承認された多くの薬剤に匹敵する臨床前効果を示すことが認識された。つまり,これらの観察は,小分子α7 nAChRアゴニストを経口投与して炎症や自己免疫疾患を治療する戦略の開発に根拠を提供している。
α7 nAChRのα7サブユニットは内因性免疫抑制経路の構成部分であり、この経路において、迷走神経の活性化はアセチルコリンの分泌を刺激し、更にα7 nAChRをMΦsに結合し、Bリンパ細胞を調節する。MΦsの活性化は免疫抑制低下[br}を起動して炎症促進サイトカインの減少を招き、BとT細胞の活性化を抑制し、炎症を制御する。
関節リウマチのような自己免疫疾患では,重篤な炎症が関節を破壊し,多発性硬化症では脳が重要な神経回路の破壊を受け,身体の免疫系が逆に自分の組織を攻撃する。粥状動脈硬化から痛風までの他の疾患も不必要な自己免疫攻撃の所見を示した。
α7 nAChR の活性化はJAK 2とSTAT 3に関連するシグナルの低下を招き、マクロファージが免疫抑制表現型 に変化し、IL-10を産生する。IL−10は炎症性サイトカイン,特に腫瘍壊死因子,IL−1,IL−6を減少させることが知られている。そのため、α7 nAChRアゴニストは抗腫瘍壊死因子治療の補充であるべきであり、これは1種類の新しい経口薬物の開発に可能性を開拓し、これらの薬物 は抗炎症性であるが、既存の薬物、例えば非ステロイド性抗炎症薬、COX 2阻害剤、メトトレキサートとJAK阻害剤よりずっと安全である。これは、α7 nAChRアゴニストが様々な過程を遮断するために必要な重要な経路ではなく、内因性調節経路を活性化しているからである。市場機会はいくつかの大きさのバイオテクノロジー会社がα7 nAChRに対する一連の経口部分アゴニストの開発における複雑で高価な努力から来ている。これらの化合物は広範な臨床前評価を経て、2670人の被験者を含む18の研究で使用された。
11
これらの薬剤は一般的に安全であることが証明されているが、神経および精神疾患(すなわちアルツハイマー病および統合失調症)の試験では無効である。アルツハイマー病および統合失調症認知障害に対する無作為、プラセボ対照臨床試験では、化合物 はその主要な終点に到達できなかった。
我々は、これまでのこれらの研究をもとに、潜在的にこのファミリーにおいて、関節リウマチ、炎症性腸疾患、再発性および進行性多発性硬化症、アテローム性動脈硬化症、痛風および骨関節炎を含む一連の炎症および自己免疫徴候の治療のための免疫抑制薬として特許出願可能なα7 nAChR類似体を開発する予定である。我々の科学者は,マクロファージ上のα7受容体や制御性Bリンパ球がこれまで開発されてきた薬物の標的とは異なることを発見した。
候補品や適応症
我々は,製薬会社によって定義されている多くの既知のアゴニストの非特許類似体をスクリーニングすることにより,α7 nAChRの経口小分子アゴニストを同定,特徴づけ,合成し,特許を出願する予定である。この仕事をEvotec GmbHにアウトソーシングする予定であり,統合された早期発見組織であり,我々は過去に協力し,イオンチャネルやトランスポーターに特化し,目的化合物から先導化合物までの専門技術と科学専門知識を顧客に提供してきた。
安全性と有効性評価計画により,臨床前開発の候補を選択し,臨床研究を開始する可能性のある前奏として,その後FDAに新薬研究申請(IND)を提出する可能性がある。我々がそのα7 nAChR開発プラットフォームのために設計した第1の目標適応は禁煙誘導の潰瘍性大腸炎である。
アウトソーシングと製造業
著者らは現在著者らの臨床試験をアウトソーシングしており、これらの試験はイギリスエジンバラオックスフォード大学とオランダグローニンゲンで行われ、抗腫瘍壊死因子プラットフォーム下のいくつかの適応のみに関連している。われわれの臨床試験を引き続きアウトソーシングし,(1)抗腫瘍壊死因子プラットフォーム,オックスフォード大学,オランダグローニンゲンでの試験,(2)SCASプラットフォームの場合,ヘブライ大学とオックスフォード大学での臨床試験,および(3)以下の場合に臨床試験を行う予定であるα7 nAChRプラットフォーム、未定です。
私たちはまた、SCAがヘブライ語大学とα7 nAChRはEvotec GmbHによって生産され、抗腫瘍壊死因子プラットフォームは既製のadalimumabを使用する。また、私たちの製品は良好な製造仕様(GMP)レベルに達し、認可された契約研究機関(CROS)によって生産されることが予想される。
材料協定
私たちは、潜在的な候補製品を開発するために、異なる大学や各方面と材料研究·許可協定(“br}”研究協定“)を締結しました。私たちはまた、このような研究に協力するために、異なる科学者と他の材料コンサルティング·コンサルティングサービス協定(”コンサルティングプロトコル“) を締結しました。
研究協定の概要
研究協定はヘブライ大学とオックスフォード大学とのbr協定を含む。抗腫瘍壊死因子プラットフォームに対して、オックスフォード大学と締結した研究協定に基づいて、当社はbr出資出資して抗腫瘍壊死因子プラットフォームに対する研究を行うことができる。その見返りに、会社は独占的なbrオプションを獲得し、研究協定によって生成された任意の知的財産権を許可する。また、私たちはオックスフォード大学のいくつかの知的財産権を独占的に許可したライセンス契約を締結した。
SCA計画については,ヘブライ大学やオックスフォード大学とbr合意を達成しており,これらの合意に基づき,ある目標適応を治療するための新しいSCAを開発·特徴づけ,早期臨床試験を行う予定である。ヘブライ大学とオックスフォード大学と締結した研究協定により、私たちはヘブライ大学とオックスフォード大学に研究機関を設立し、ヘブライ大学で設計と合成された新しい大麻類化合物の開発と試験を促進する。
12
研究プロトコルはそれぞれ 以下のとおりである.
ヘブライ大学との研究協定は
2018年5月13日、我々の完全子会社CBR Pharmaは、Yissm Research Development 社(“Yissm”)と研究·許可協定(“2018ヘブライ語協定”)を締結し、この合意に基づいて、YissmはCBR Pharmaに特定の特許(“2018ヘブライ語許可特許”)、技術ノウハウおよび研究成果(総称して“2018ヘブライ語ライセンス技術”)を開発および商業化するために、CBR Pharmaにグローバル独占許可(“2018ヘブライ語ライセンス”)を付与した(“2018ヘブライ語ライセンス技術”)。製品を流通または販売し、すべて2018年ヘブライ語許可技術を使用して、肥満、疼痛、炎症および関節炎を含む任意およびすべての獣医およびすべてのヒト疾患を治療するために使用される(“2018領域”)。
“2018ヘブライ語 プロトコル”によると、Yissmは2018年のヘブライ語許可が付与されたにもかかわらず、ヘブライ大学を代表して(I) をヘブライ大学自身の研究および教育目的のために製造し、2018年のヘブライ語許可技術を使用および実践する権利を保持する;(Ii) 許可または2018年ヘブライ語ライセンス技術を他の学術および非営利研究組織に2018年以外の研究または商業的応用のために任意の第三者に譲渡する。
2018年ヘブライ語プロトコル は、CBR Pharmaが2018年のヘブライ語ライセンス技術に2018年の分野の開発のための1つまたは複数の再ライセンスを付与する権利があることをさらに規定している。
2018年ヘブライ語ライセンス技術の権利、所有権および権益 はYissmのみに帰属し、CBR Pharmaは2018年ヘブライ語プロトコルの条項のみに従って、 によって付与された2018年ヘブライ語ライセンスの権利を保有および使用する。
CBR Pharmaは2018年のヘブライ語許可の対価格として、Yissmに75,000ドルの許可料を支払い、毎年の許可維持費 (“br}”)を継続して支払うことに同意したライセンス維持費“)50,000ドルは、2019年5月1日から始まり、その後毎年5月1日になります。 ライセンス維持費は返金されませんが、毎年5月1日から4月30日までの製品純売上高により、毎年の印税から差し引くことができます。
Yissmはまた化合物の研究と合成に同意し,オックスフォード大学やヘブライ大学の追加研究により,CBR Pharmaはこれらの化合物を用いて経口活性鎮痛および消炎薬を開発する。化合物はヘブライ大学からオックスフォードに運ばれ、臨床前研究に使用され、痛みと炎症に対する治療効果を決定する。
2018年にヘブライ語許可技術由来化合物に関していくつかのマイルストーンに達した後、CBR PharmaはYissmに一定のbrを支払う義務があるが、以下に限定されない
| 一里塚 | 一里塚費用 | |||
| FDAに最初のINDテストを提出します | $ | 75,000 | ||
| FDAとの1/2期試験を開始します | $ | 100,000 | ||
| FDAとの3期試験を開始します | $ | 150,000 | ||
| 各製品の発売許可/許可(最大500,000ドル) | $ | 100,000 | ||
| (最大500,000ドル) | ||||
| 製品の累計売上高は売上高が10億ドルに達するまで2億5千万ドル増加した | $ | 250,000 | ||
CBR Pharmaは(I)純売上高前5億ドルの3%に相当する印税と,(Ii)純売上高が5億ドル以上に達した後の純売上高の5%をYissm に支払う。
13
CBR Pharma株主がその普通株式を売却するか、または2018年ヘブライ語プロトコルを譲渡する場合、CBR Pharmaは、そのような会社の取引に従って受信された対価格の5%の費用をYIsmに支払う義務がある。初公募株や上場活動では,CBR Pharmaは取引完了とともに,完全希釈に基づいてYIsmに発行済み普通株の5%に相当する登録普通株と発行済み普通株を発行することが義務付けられている。2020年11月6日に完成した業務合併は上場活動とされており,これにより,会社は業務合併終了前にYissmに12,028株の普通株 を発行した。研究·許可協定に基づいてYIsmに発行された株式についてのより多くの情報は、付記11-承諾およびまたは事項および付記12-株主 現在の2022年12月31日までの財務諸表の権益を参照されたい。
CBR Pharmaはまた がYissmに特許費用(最高30,000ドル)を返済することに同意した。
YissmとCBR Pharmaはまた、修正案によって延長されている2019年5月までの12ヶ月間に40万ドルの予算を提供する研究プロジェクトを構築することに同意した。
2018ヘブライ語協定は終了する:(I)2018年ヘブライ語ライセンス特許の最後の満了、 (Ii)任意の規制機関または政府機関によって付与された任意の製品の最後の独占特許権が満了する;(Iii)20年連続してどの国/地域でもいかなる製品の商業販売もない;または(Iv)2018ヘブライ語協定の条項に従って独自技術の独占ライセンスを取得することを選択した場合、その独占ライセンスは満了する。
2019年11月11日、CBR PharmaはYissmと追加の研究および許可協定(“2019ヘブライ語協定”)を締結し、この合意に基づいて、YissmはCBR Pharmaに ある特許(“2019ヘブライ語許可特許”)、独自技術および研究成果(総称して“2019ヘブライ語許可技術”、および2018ヘブライ語許可技術“ヘブライ語許可技術”)を開発および商業化するために、CBR PharmaとYissmとを締結し、 開発、製造、マーケティング、流通、修理、および製品、製品、および研究成果を提供した。これらのすべては、2019年のヘブライ語許可技術[br}の範囲内で使用される:(I)Li、ナトリウム、カリウム、カルシウム、マグネシウム、亜鉛、鉄およびアルミニウムなどの一価、二価および三価金属、例えばLi、ナトリウム、カリウム、カルシウム、マグネシウム、亜鉛、鉄およびアルミニウムなどの一価、二価および三価金属を含むマリファノール金属塩、経口および外用のための医薬製剤;およびbr(2)肥満、疼痛、炎症および関節炎を含む任意およびすべての獣医およびヒトの医療状態を含む大麻化学誘導体を管理するための医薬製剤(“2019年実地”)。
“2019年ヘブライ語協定”によると、Yissmは2019年のヘブライ語許可を付与したにもかかわらず、ヘブライ語大学を代表して、以下の権利を保持する:(I) は、ヘブライ大学独自の研究および教育目的のために、2019年のヘブライ語許可技術を作成し、使用し、実践するが、商業目的のためには ではなく、2019年のヘブライ語許可技術に含まれる任意の独自技術または未発表の特許情報の秘密brを遵守する;(Ii)2019年のヘブライ語ライセンス技術を他の学術および非営利研究機関 2019年ヘブライ語ライセンス技術を非商業研究のために許可または他の方法で譲渡し、2019年ヘブライ語ライセンス技術に含まれる任意の独自技術または発表されていない特許情報の秘密規定を遵守する;および(Iii)2019年のヘブライ語ライセンス技術を2019年以外の研究または商業用途に任意の第三者に譲渡することを許可するか、または2019年のヘブライ語ライセンス技術に含まれる任意の独自技術または未発表の特許情報を秘密にしなければならない。
2019年ヘブライ語プロトコル はさらに、CBR Pharmaは、2019年の分野での開発 のために、2019年のヘブライ語許可技術に1つまたは複数の再許可を付与する権利があると規定している。
2019年ヘブライ語ライセンス技術のすべての権利、所有権、および権益 はYissmのみに帰属し、CBR Pharmaは2019年ヘブライ語プロトコルの条項のみに従って2019年ヘブライ語許可に従って付与された権利を保有し、使用する。
14
2019年ヘブライ語ライセンス技術は、(I)2019年ヘブライ語ライセンス特許の最後の満了、(Ii)任意の規制機関または政府機関によって付与された任意の製品の最後の独占特許権の満了、(Iii)20年連続の満了に適用される特許延長期間を加え、その間、どの製品もどの国/地域でも商業販売されていない場合に終了する。または(Iv)“2019年ヘブライ語協定”の条項に基づいて独自技術の独占許可を取得することを選択した場合、その独占許可が満了したときに。
2020年1月1日、CBR Pharma とYissmはYissm で特定の分子の新しい派生商品について追加的な研究を行うことが規定されている2018年ヘブライ語協定の最初の修正案を締結した。第1改正案の条項によると、会社は2019年5月1日から毎年Yissmに200,000ドルを支払い、35%の追加大学管理費用を加え、各教授が18ヶ月以内に行う追加研究に使用する。追加的な研究は2021年4月に終了し,成功可能な薬物送達方法を研究·開発した後にさらなる臨床前作業が行われる予定であり,後期開発段階にある。
オックスフォード大学との研究協定
2013年11月1日、当社の完全子会社180 LPが契約を締結しました(最初のオックスフォード合意は)オックスフォード大学と協力し、これにより、180 LPはオックスフォード大学のDupuytren‘s Constraintの抗腫瘍壊死因子の再定位の研究と開発を支援する。
最初のオックスフォード協定によると、各お金はプロジェクトの異なるマイルストーンでISIS革新(現在オックスフォード大学革新)に支払われ、以下のように概説される
| 一里塚 | マイルストーン 費用 | |||
| 最小投資を完了しました | £ | 10,000 | ||
| ライセンス製品の第2段階試験を開始する | £ | 10,000 | ||
| ライセンス製品の第3段階試験を開始する | £ | 10,000 | ||
| ライセンス製品は登録可能なフェーズ3試用の主な終点を実現している | £ | 20,000 | ||
| 知的財産権を許可するいかなる発行された米国特許も | £ | 5,000 | ||
| FDAが新薬申請を承認した(“NDA)は、180 LPまたはその子ライセンス者のうちの1つによってライセンス製品として提出される | £ | 30,000 | ||
| EMAによって承認された180 LPまたはその子許可者のうちの1つがライセンス製品として提出されたMAA | £ | 30,000 | ||
| 180 LPまたは任意のサブ許可製品が米国で初の商業販売許可製品 | £ | 50,000 | ||
| 180 LPまたはEUのいずれかのサブライセンスが初めてビジネス形態でライセンス製品を販売 | £ | 50,000 | ||
有効なクレームが存在する任意の国/地域では、ISIS Innovationには、純売上高の0.5%に相当する印税、他の国/地域での純売上高の0.25%、および180 LPに基づいて許可技術によって付与されたすべてのサブライセンスおよび他の契約項の下またはそれに関連するすべての前払い、マイルストーンおよび他の一括払いに7.5%の費用収入印税を支払う資格がある。事前に終了しない限り、最初の“オックスフォードプロトコル”は、指定された特許出願が、発行された特許、係属中の特許出願または補足のbr保護証明書として有効である限り、またはより長いものを基準として20年の有効期間を有する。
2018年8月15日、イングランドとウェールズ法に基づいて設立された会社CannBioRex Pharma Limited(“CannU.K。)は、我々の完全子会社CBR Pharmaの完全子会社brと研究契約を締結しました(2つ目のオックスフォード合意は“オックスフォード大学と協力し、この合意に基づき、CBR Pharma(CannU.K.)オックスフォード大学がヘブライ語ライセンス技術から開発したSCAの研究と開発を賛助する。オックスフォード大学、ヘブライ大学製SCAはすでに確立された臨床前モデルで鎮痛と抗炎症効果テストを行っている。
15
2つ目のオックスフォード協定によるとオックスフォード大学は研究プロジェクトを実施しました研究プロジェクト)SCAに基づく臨床開発は、抗炎症および免疫調節特性を有することが知られている。この研究プロジェクトの目的は、ヘブライ大学で合成された化合物を開発し、特徴付けることであり、慢性疼痛、関節リウマチおよび他の慢性炎症性疾患を治療する方法を創造し、最終的に監督部門の許可を得て、2022年中下旬またはその後に早期臨床試験をできるだけ早く開始することである。第2のオックスフォード協定の初期期限は1年であり、2019年3月22日から開始されるが、事前に終了しない限り、2020年3月31日または各当事者が合意した任意の遅い日まで修正によって延長される。二番目のオックスフォード協定は2020年3月31日以降は延長されず、CannUKとオックスフォードの関係は引き続き維持され、次のようにオックスフォードと他の協定が締結された。
英国政府は研究プロジェクトのスポンサーとして、2つ目のオックスフォード協定に基づいてオックスフォード大学に以下の金を支払った
| 一里塚 | 一里塚 費用 | |||
| オックスフォード協定に調印する | £ | 166,800 | ||
| 研究プロジェクト開始後6ヶ月 | £ | 166,800 | ||
| 研究プロジェクト開始後9ヶ月 | £ | 166,800 | ||
| また別の報告書によると、中国の研究開発プロジェクトは12ヶ月後に実施される | £ | 55,600 | ||
2020年9月18日、CannU.K. はオックスフォード大学と別の研究協定(“第三牛津協定”)を締結し、この合意に基づいて、CannUKのスポンサーはオックスフォード大学のNanchahal教授が指導して繊維化のメカニズムを研究した。この協定によると、カナダ政府は最初に100,000ドルを提供し、その後、リン·ウィリアムズ博士の給料と消耗品を支援するために6ヶ月ごとに資金を提供した。
カナダ連合王国は発起国として、第3項オックスフォード協定に基づいてオックスフォードに以下の金を支払うことに同意した
| 一里塚 | 金額 満期 (含まれない) 付加価値税) | |||
| 3つ目のオックスフォード協定が調印されて30日後 | £ | 80,000 | ||
| 3つ目のオックスフォード協定が調印されて6ヶ月後 | £ | 178,867 | ||
| 3つ目のオックスフォード協定に署名して12ヶ月後 | £ | 178,867 | ||
| 3つ目のオックスフォード協定が調印されて24ヶ月後 | £ | 178,867 | ||
| 3つ目のオックスフォード協定が調印されてから36ヶ月 | £ | 178,867 | ||
2020年9月21日、CannU.K. はオックスフォード大学と別の研究協定(“第4項オックスフォード協定”)を締結し、協定によると、CannUKは3年以内にオックスフォード大学が炎症性疾患を治療するための新しい大麻素由来新化学実体(NCEs)を開発と特徴づけることに同意し、患者における早期臨床試験を開始した。
16
カナダ連合王国は発起国として、第4項オックスフォード協定に基づいてオックスフォードに以下の金を支払うことに同意した
| 一里塚 | 金額 満期 (含まれない) 付加価値税) | |||
| オックスフォード第4協定の署名から30日後 | £ | 101,778 | ||
| 4つ目のオックスフォード協定に署名してから6ヶ月後 | £ | 101,778 | ||
| 4つ目のオックスフォード協定に署名して12ヶ月後 | £ | 101,778 | ||
| オックスフォード第4協定の署名から18ヶ月 | £ | 101,778 | ||
| オックスフォード第4協定の署名から24ヶ月 | £ | 101,778 | ||
2022年3月22日、CannU.K. は第4項オックスフォード協定の修正案に署名し、研究期間を2023年12月31日まで延長し、追加費用を支払うことなく、2023年12月31日に延長した。
2021年5月24日、カナダ政府はオックスフォード大学と別の研究協定(“第五項オックスフォード協定”)を締結し、この協定に基づいて、カナダ政府はオックスフォード大学で多中心、無作為、二重盲検、平行グループの研究を展開し、抗腫瘍壊死因子注射による成人肩周囲炎疼痛主導期の治療の実行可能性研究を行う。
カナダ連合王国は発起国として、第5項オックスフォード協定に基づいてオックスフォードに以下の金を支払うことに同意した
| 一里塚 | 金額 満期 (含まれない) 付加価値税) | |||
| 5つ目のオックスフォード協定に署名した時 | £ | 70,546 | ||
| オックスフォード第5協定締結後6ヶ月 | £ | 70,546 | ||
| オックスフォード第5協定調印後12ヶ月 | £ | 70,546 | ||
| オックスフォード第5協定締結から24ヶ月 | £ | 70,546 | ||
オックスフォード許可協定
2021年11月3日、オックスフォード大学革新有限公司と独占ライセンス契約を締結しました(“オックスフォード許可協定)により、肝臓再生のためのHMGB 1分子に関するいくつかの特許を取得する権利がある。
オックスフォードライセンス協定によると、会社は以下の支払い条項に同意する
| 支払い | 支払額 | |||
| 過去の特許コスト | £ | 49,207 | ||
| 許可料 | £ | 10,000 | ||
| 年間維持費 | £ | 3,000 | ||
17
| 一里塚 | 満期金額 | |||
| INDに提出する | £ | 25,000 | ||
| 1ST第1段階研究で製品ごとに適応ごとに服用した被験者 | £ | 25,000 | ||
| 1ST第2段階研究で製品ごとに適応ごとに服用した被験者 | £ | 100,000 | ||
| 1ST第3段階研究で製品ごとに適応ごとに服用した被験者 | £ | 50,000 | ||
| 適応ごとの製品ごとに新薬申請を提出する | £ | 50,000 | ||
| 全ての特許が発行されたアメリカ特許は | £ | 5,000 | ||
| すべての適応のすべての製品はアメリカで規制されています | £ | 1,250,000 | ||
| すべての適応のすべての製品はEUやイギリスで規制されています | £ | 550,000 | ||
| それぞれの適応の各製品は日本で規制されています | £ | 150,000 | ||
| 総純売上高は50億ドルを超えた | £ | 10,000,000 | ||
| 総純売上高は100億ドルを超えた | £ | 50,000,000 | ||
| 純売上高(ドル) | 印税 レート | |||
| 1.00 | % | |||
| 2億5千万ドルから10億ドル | 2.00 | % | ||
| 10億ドルから100億ドル | 3.00 | % | ||
| > $10B | 3.50 | % | ||
スタンフォード大学許可協定
2018年5月8日、我々の完全子会社KatExcoの完全子会社Katexo PharmPharmticals は、ラン·スタンフォード初級大学(“Stanford”)取締役会とオプション協定(“Stanfordオプション”) を締結し、この合意に基づいて、スタンフォード大学はKatExco にいくつかの発明の開発および商業化の独占許可を得るオプションを付与した。スタンフォードのオプションに対する対価格として,Katexoはスタンフォードに10,000ドル(“オプション支払い”)を支払い,ライセンス発行費用プロトコルに基づいて入金することができる。
2018年7月25日(“Stanford 発効日”)、KatexoはStanfordオプションを行使し、Stanfordと独占ライセンス契約(“Stanfordライセンス契約”)を締結した。この特許によれば、Katexoは、(I)自己免疫脱髄のアルファB結晶タンパク質 および(Ii)B-1細胞およびマクロファージを活性化することができるアミロイド線維を形成し、自己免疫および神経変性疾患に対して抗炎症および治療作用を有する6つのアミノ酸のポリペプチドを形成することに関連するいくつかの米国特許の権利を取得する(“スタンフォード大学特許”)。われわれは先導化合物の臨床開発を支持し,最終的に第1期と第2期臨床試験において潰瘍性大腸炎の潜在的臨床用途を決定する。
18
スタンフォードライセンス協定によれば、スタンフォードライセンス特許によって付与された権利を除いて、スタンフォードのいかなる権利(知的財産権を含む)もKatExcoに付与されない。
KatExcoはスタンフォード許可特許を付与する価格として、オプション支払いを含めてスタンフォードに50,000ドルの初期費用を支払った。同社はスタンフォード大学に5574株の普通株を発行し、これらの株式の価値を説明する手紙を提供した。スタンフォード大学に発行された株式の一部はその後、私たちの首席科学官と連合席会長を含む5人に割り当てられた。
スタンフォード発効日の最初のbr周年からその後の各記念日から、KatExcoは最初と2回目の記念日にスタンフォード大学に年間20,000ドルのライセンス維持費を支払い、その後の各記念日にスタンフォード大学に40,000ドルを支払う。また,Katexoは,(I)第2段階試験開始時に100,000ドルを支払うこと,(Ii)FDAが初めてbr製品を承認した場合に500,000ドルを支払うことを含む支払い義務がある(“許可を得た製品)スタンフォードライセンス特許によって生成され、(Iii)その後の新規ライセンス製品当たりの費用は250,000ドルである。純売上高の2.5%で計算された特許使用料(計算方式はKatExcoまたはその再許可者、その流通業者または指定者がスタンフォードライセンス特許に基づく製品の販売、譲渡または他の処置から得られた毛収入から5%を差し引く)。さらに、Katexoは、スタンフォード大学が2018年3月3日以降に発生した任意の干渉および/または再審査を含むスタンフォード大学ライセンス特許のすべての特許費用を相殺するために、スタンフォード大学に51,385ドルを返済した。
私たちは30日間の通知を提供することでスタンフォード許可プロトコルを理由なく終了することができる。コントロール権変更の場合、スタンフォード許可プロトコルを譲渡した後、Katexoはスタンフォードに200,000ドルの制御権変更費用を支払う義務がある。スタンフォード大学ライセンス協定はまた、スタンフォード大学が、最大(I)10%または(Ii)スタンフォード大学でKatexoの所有権権益に比例する割合の非公開発行Katexo持分を維持する権利があることを規定している。スタンフォードライセンス契約に関連してStanfordに発行された株式は、Stanfordと一部の株式を取得した5名の個人がKatexo株の2.11%の総所有権を獲得し、2019年7月までに180 LP、Katexo、CBR Pharmaがそれぞれ会社再編を完了し、180 LP、Katexo、CBR Pharmaが180 LS(“同社”)の完全子会社となった再編成する“) 本プロトコルの下で“ビジネス会社法”です(ブリティッシュコロンビア州)
“Evotecプロトコル”
2018年6月7日、当社の完全子会社であるKatExcoは、いくつかの 研究サービスを実行するためにEmotecが保持されているリーディングCRO EvotecとEvotecプロトコルを締結しました。Evotec協定によると、共同プロジェクトの目標(“Evotecプロジェクト)の目的は、ヒトChrFam 7 a受容体および機能を薬理的に刺激する小分子を識別することである。Evotecプロジェクトは24カ月間に2段階で行われ,指導委員会が資源を割り当て,この委員会は四半期ごとにEvotecプロトコルの各当事者が平等に制御している。
“研華技術協定”に記載されているいくつかの免除 を除いて、Katexoは研華がそのサービス実行中に構想、発明、発見または製造するすべての知的財産権を持っているが、研華が所有または制御する知的財産権を除いて、これらの知的財産権は研華プロトコルの下で提供されるサービスで使用される既存の技術とコンポーネントに関連する。
Evotecプロトコルの最低支払金額は4,937,500ドル,最高支払い金額は5,350,250ドルである.この計画は2019年中に停止され、会社 は2023年に研華と再交渉する予定だ。2022年12月31日までに、会社は研華科技に約110万ドルを支払った。
“ペトカナ協定”
2018年8月20日、私たちはMarc Feldmann(私たちの共同執行議長)とYissm教授によって設立されたプライベート会社(“Petcannaプロトコル”)とPetcanna Pharma Corp.(“Petcanna”)と再許可契約(“Petcanna合意”)を締結した。
Petcannaプロトコルによれば、著者らは、シクロヘキセン化合物に関連し、Petcannaプロトコル(“Petcanna IP”)に列挙されたいくつかの特許brを商業用途のために商業用途に付与し、製造、販売、または獣医医療 疾患(最初は骨性関節炎)を治療するための製品にPetcanna IPを使用する製品を開発、製造、販売することができる。
19
Petcannaは再許可の対価格として、2018年第4四半期に約9,000,000株のPetcannaの普通株を発行することに同意した。本書類の提出日まで、Petcannaはまだどの株主にも株式を発行しておらず、運営も開始されていない。私たちは85%の株式 を保留し、15%の株式をYissmに譲渡するつもりだ。もしYIsmがこのような株を受け入れなければ、私たちはYissmにそのような株の当時の公平な時価の15%を支払う義務があるだろう。PetcannaはまたPetcannaにPetcanna IPを含む製品の純売上高について1%の印税を支払う。
はい、Petcanna IPの所有権と権益 は、Petcanna IPのいかなる改善も含めて、完全にわが社に属します。
Petcanna合意の当事者が別途書面の約束を持っていない限り、Petcannaプロトコルは以下の状況が発生した時に終了する:(I)最後のPetcanna知的財産権の満了日、(Ii)任意の監督管理機関または政府機関が付与した任意の製品の独占経営権が最終的に満了した日、および(Iii)連続20(20)年の間にいかなる製品の初の商業販売もない。“製品”および“最初の商業販売”という用語は、Petcannaプロトコルの定義に適用される。 Petcannaに今回の許可を付与する能力は、(I)YIsmがすべての適用当事者からそれに割り当てられたヘブライ語特許出願に必要な権利を有し、(Ii)YIsmがヘブライ語協定の条項に従って許可を付与することができ、ヘブライ語特許出願および任意の関連特許がヘブライ語ライセンスプロトコルおよびPetcannaプロトコルのそれぞれの条項の下で有効かつ良好な状態を維持することができることに依存する。
ケネディ許可協定
2019年9月27日、我々の完全子会社180 LPは、ケネディリウマチ研究信託基金(“ケネディ”)と独占許可協定(“ケネディ許可協定”)を締結し、ケネディは、再許可を付与する権利、および研究、開発、販売、または任意の医薬品(I)を製造する権利を含む180 LPに、ケネディリウマチ研究信託基金(“ケネディ許可特許”)と独占許可協定(“ケネディ許可協定”)を締結した。ケネディ許可協定による許可なしにケネディ許可特許を侵害するか、または(Ii)抗体断片または抗体由来抗体を含む抗体の製造、使用、輸入、または販売であって、ケネディ許可協定による許可なしにケネディ許可特許を侵害し、疾患の診断、予防および治療および条件を含むすべてのヒト用途のために、抗体の研究、開発、製造、使用、輸入、または販売。
ケネディ許可協定によれば、ケネディは、ケネディが許可した特許およびその付属会社、従業員、学生、および他の研究者が、このような研究および開発のための外部支援を受ける権利と、同じ目的で二次許可を付与する権利とを含む、ケネディ許可の世界的範囲内で永久的、撤回不可能、非排他的、非排他的、印税免除、再許可可能な権利を保持し、ケネディ許可のための特許およびその付属会社、従業員、学生、および他の研究者が教育および研究開発を行う目的のためにケネディ許可特許を侵害する可能性のある任意の行為を実施する。
ケネディライセンス特許を付与する代償として,180 LPはケネディに60,000 GBの前払い費用を支払い,ケネディに純売上高に相当するbrから(I)初年度純売上高10億ドルの純売上高の1%と,(Ii)純売上高が10億ドルに達した後の純売上高の2%,およびすべての再許可収入の25%をケネディに支払う。条件は,印税を構成する金額に応じて,再許可収入の割合が,このような再許可又はその付属会社が販売している製品の最初の累計純売上高の1%を下回ってはならないこと,及びこのような再許可又はその付属会社が販売している製品の累計純売上高が10億ドルを超える部分の2%を下回ってはならないことである。
ケネディに支払われる特許権使用料の有効期限は、(I)ケネディ許可特許に含まれる適用国/地域の開発された特許をカバーする製品の最後の有効な主張、(Ii)当該製品のbr}国/地域における規制の排他性が失効する、または(Iii)当該製品がその国で初めて商業販売される10年以内に満了する。
私たちは90日間の通知を提供することで、ケネディ許可協定を理由なく終わらせることができる。
20
Kinexum協定
2023年1月13日、我々は通常の業務中にKinexumと契約(“MSA”)を締結した。MSAによると,Kinexumは条件付き上場許可(CMA)と上場承認申請(MAA)に関する援助を会社に提供し,会社 はアダリアモノクロナル治療を計画している進行性早期Dupuytren病に関する申請をMHRAに提出する予定である。Kinexum契約に関連するコストを含め、同社は、MHRA申告および他の規制準備に関する活動のために、2023年9月30日までの3四半期の累計支出約900,000,000ドル~1,000,000ドルを予定している。
相談協議
問合せプロトコル は以下のとおりである.
ジャグディップ·ナンチャハル教授諮問協定
2021年2月25日、私たち(後に合意に加入したCannBioRex Pharma Limited)は、JagDeep Nanchahal教授と相談協定を締結し、2021年2月22日、2020年12月1日に施行された(改訂された相談協議“)”南チャハル教授は2014年以来ずっと当社と/あるいはその子会社にサービスを提供しており、現在当社の5%を超える株主であり、私たちの臨床諮問委員会の議長でもある。
2021年3月31日、私たちは南チャハル教授とコンサルティング協定第1修正案を締結し、2021年2月25日に南チャハル教授と締結した諮問協定を修正し、イングランドとウェールズに設立され登録される会社CannBioRex Pharma Limited(“CannBioRex Pharma Limited”)を登録したCannBioRex社)と、契約先である自社間接全額付属会社を更新し、CannBioRexが支払うべきNanchahal教授への現金支払いを規定するために、税務目的で、 CannBioRexが当該合意のいくつかの他の条項の契約者となることを規定し、当該合意の 条項に基づいて支払うべきいくつかの現金ボーナスの時間について規定する。
Nanchahal教授は外科医br科学者であり、よく見られる疾患の分子機序の確定に集中し、そして彼の発見を早期臨床試験に転化する。彼はイギリス医学研究委員会が援助した博士号を取得し、当時ロンドンの医科学生で、外科訓練期間中に外部から資金援助された実験室グループを指導していた。米国とオーストラリアで顕微外科と手外科の奨学金を修了した後、帝国工科大学の上級講師に任命された。彼の研究の重点は内因性幹細胞の標的化と繊維化の減少を通じて組織再生を促進することである。2013年、彼のチームは、一般的な手線維性疾患であるDupuytren筋拘縮症を治療する標的として、抗腫瘍壊死因子(TNF)薬を決定した。彼は現在恵康信託基金と衛生部が援助した2 b期の臨床試験を指導し、局部抗腫瘍壊死因子の早期デュプイテルン筋拘縮症患者に対する治療効果、及び早期肩周囲炎患者に対する臨床試験を評価している。彼は根拠に基づく医学の支持者であり、NICE複雑と非複雑骨折指導発展グループの中で唯一の整形外科メンバーでもある。彼は2020年に出版された“開放性骨折管理基準”を書いたグループのメンバーの一人である。これはこれらの深刻な負傷患者の看護を促進することを目的としたオープンソース出版物である。
相談br協定によると、南チャハル教授は合意期間内に当社の顧問を務めることに同意し、当社の最高経営責任者および/または取締役会が時々要求すべきサービスを提供するが、これらに限定されない:(1)Dupuytren筋拘縮症、肩凍結および術後精神錯乱/認知衰退領域での臨床試験、および(2)肝と肺線維化を含む他の線維化疾患の実験室研究(総称して“と呼ぶ)サービス.サービス”).
21
サービス提供の代償として,会社(CannBioRex Pharma Limitedにより)は,合意期間内にNanchahal教授に毎月15,000ポンド(約20,800ドル)を支払うことに同意し,23,000ポンド(約32,000ドル)に増加し,締め切り(A)Dupuyten‘s Constraint(RIDD)2 b期臨床試験データが公表された日,および(B)社が1,500万ドルを超える資本調達に成功した日である。その後、費用は毎年増加し、取締役会が許可した他の臨床試験と実験室研究の進展状況を反映する。同社はまた,同業者評議誌にbrを発表するためにDupuytren収縮臨床試験データを2021年12月に提出することに同意し,Nanchahal教授に総額100,000ポンドのボーナス (“ボーナス1”)を支払い,ボーナスは2021年12月に支払う。また、これまでに完成した仕事については、RIDD(Dupuyten‘s)試験の採用を含め、会社はNanchahal教授に434,673ポンド(約605,000ドル)を支払うことに同意した(“ボーナス2”)。Nanchahal教授が当選した時、ボーナス2はNanchahal教授の選択で少なくとも50%(50%)以上、1株当たり60.00ドル、または付与された日の株価(低い者を基準)に少なくとも50%(50%)以上支払われ、残りはポンドで支払われる。2020年12月1日(“帰属日”)の後、会社が債務または持分を売却することによって少なくとも1,500万ドルの追加資金を調達した場合、ボーナス2,000,000ドルを稼いだとみなされ、その帰属日までに計算、満期、または支払うべきではない。当社は帰属日から30暦以内に第2期ボーナスを支払わなければならない。最後に,Nanchahal教授は別の使い捨てボーナス(“ボーナス3”)を獲得し,金額は5,000ポンド(約7,000ドル)であり,第1人の患者を第2期凍結肩試験,もう1つの使い捨てボーナス(“ボーナス4”),金額5,000ポンド(約7,000ドル)を募集し,第2期精神錯乱/POCD 試験に参加するために1人目の患者を募集する。2021年3月30日、会社はNanchahal教授に217,337ポンドの代わりに5,035株の会社普通株を発行し、2021年4月15日、会社はNanchahal教授に1,886株会社の普通株を発行し、82,588ポンドの代わりにした。会社はNanchahal教授が株式を発行することで合計30万ポンドの要求を得ることに同意するために1500万ドルを調達しなければならないことも免除された。Nanchahal教授は、ボーナス2が支払うべき残り134,673ポンドが会社で少なくとも1,500万ドルの追加資金を調達して支払うことに同意した。2021年8月23日、Nanchahal教授の要求に応じて、会社は残り31%(または134,749ポンド、または184,606ドル)のボーナス2に対する対価格として、1株当たり60.00ドルの価格に基づいて、Nanchahal教授3,077株の普通株の発行に同意した。これらの株式は、株主の承認を得た会社2020年総合インセンティブ計画に基づいて発行されている。
2022年4月27日から、CannBioRexとJagDeep Nanchahal教授と“諮問協定第2修正案”(“第2 Nanchahal修正案”)を締結した。“ナンチャハル修正案”によると、Nanchahal教授は、2022年3月1日から11月1日まで、会社がデュプイトレン病2 b期の臨床試験のデータを受けた後(編集と最終承認を待たなければならない)、彼の月費は23,000 GBに増加し、増加した4,000 GBと毎月19,000 GBは会社の給料に応じて現金で支払うことが条件であることに同意した。2022年または(B)取締役会は、当社の手元にこの等課税金を支払うのに十分な現金があると判断しましたが、当社は最低15,000,000ドル(“資金確定日”)を調達してから支払うことを予想しており、すべての課税金が満期になると予想されています。
2022年12月28日、CannBioRexとNanchahal教授と諮問協定第3修正案(“第3 Nanchahal修正案”)を締結した。 第3 Nanchahal修正案は、この協定に基づいてNanchahal教授に支払う毎月現金費用を2022年12月31日までその月23,000 GB/月を維持し、相談協定有効期間(2023年1月1日から相談契約終了まで)を35,000 GB/月(総称して“費用”)に増加させることを規定している。第三条南チャハル修正案はまた、費用は会社の取締役会または報酬委員会の提案に基づいて毎年調整され、取締役会または報酬委員会は増加した金額を決定する際に、イギリスの消費者物価指数や南チャハル教授の会社の使命推進への貢献などを考慮すると規定されている。br}第三の南チャハル修正案はまた、会社がbrの原因以外のいかなる理由で相談合意を終了した場合、南チャハル教授は終了日の月費12ヶ月に相当する一次支払いを得る権利があると規定している。
上記の規定にもかかわらず、会社の取締役会または報酬委員会は、時々Nanchahal教授に現金、株式、またはオプション形式の追加ボーナスを支給することができる。
22
コンサルティング契約の初期期限は3年であり、その後3年間継続して、合意の規定に従って終了するまでです。 のいずれも12ヶ月前に相談契約の終了を書面で通知することができます(ただし、会社が合意を終了する権利は、南チャハル教授が諮問協定の規定の役割を履行できなかった場合にのみ行使されます)。(A)Nanchahal教授が効率的で勤勉にサービスを履行することを怠ったり、合意に基づいて付与された任意の同意を含む合意規定の義務に違反した場合、会社は直ちに上記の決定を下すことができる。(B)Nanchahal教授 は、任意の詐欺または不誠実な行為、またはその行為(サービスの履行または他の態様にかかわらず)が、Nanchahal教授、当社またはその任意の連属会社の名誉を損なわせる可能性があるか、または逮捕可能な罪(非監禁処罰された道路交通犯罪を除く)、または(C)Nanchahal教授を破産させるか、またはその債権者と任意の手配または和解を達成させる可能性がある。もしコンサルティング契約がbr社によっていかなる理由で中止された場合、南チャハル教授は12ヶ月の費用を得る権利があり、金額は終了日の12ヶ月である。
コンサルティング協定には、南チャハル教授の12ヶ月の競争禁止と非入札義務が含まれており、彼が会社の業務に積極的に従事しているどの国のどこでも会社のbrとの競争を禁止しているが、オックスフォード大学での研究を含む例外的な場合は除外されている。コンサルティング協定はまた、一般的な秘密および発明譲渡条項(Br)を含み、各条項は、会社が以前に複数の大学(オックスフォード大学、南チャハル教授が手、整形および再建外科教授を担当している)との既存の合意によって制限されている。
マーク·フェルドマンとのサービス契約です
2018年6月1日,CannBioRex Pharma Limited(“CannBioRex”)は我々の執行連合議長Marc Feldmann Ph.D.教授とサービス協定 (“Feldmann雇用協定”)を締結した。フェルドマン雇用協定によると、サー·フェルドマンはCannBioRexの会長、最高経営責任者兼取締役CEO、あるいはその身分に応じた他のポストに就く。サー·フェルドマンの役割には、彼が担当している役割の慣用的な役割が含まれている。サー·フェルドマンは毎年115,000ポンドの報酬を得ており、年間報酬は取締役会が審査し、適宜ボーナスを得る資格があるかどうかを取締役会が決定する。CannBioRexはまたサー·フェルドマンの出張と他のビジネス費用を精算する。
フェルドマン雇用協定によると、サー·フェルドマンが創造した、またはその雇用に関連するすべての知的財産権は、CannBioRexに属し、帰属する。
フェルドマン雇用協定は、フェルドマン卿が在任中にいかなる競争企業のために働くか、または他の企業の株式を保有することを禁止する慣例の非競争条項を含むが、彼とその家族の実益権益の合計がこの種類の証券の5%を超えない場合、彼は上場会社の証券を保有または実益することができる。
サー·フェルドマンはまた、契約終了後12ヶ月以内に(“契約終了後期限”)イギリスまたは任意の他の国/地域の競争的ビジネスまたは潜在的な合弁企業に任意の身分で参加することを禁止されている。解約期間後、彼はCannBioRexおよびその付属会社の顧客、あるいは彼が雇われている間に活動に参加している任意の会社、あるいは彼が機密情報を持っている任意の会社から業務を誘致してはならない。フェルドマン教授はさらに、契約解除後にサプライヤーを誘導または誘導しようと試みることによって、CannBioRexのビジネス関係を妨害しないことを約束した。彼はまた、退職後の間にいかなるCannBioRex従業員を誘導または誘導しようとしないかに同意した。フェルドマン雇用協定は一般的な守秘義務と守秘義務、病気休暇、そして休暇を含む。
フェルドマン雇用協定 には固定期限がない。どちらも9ヶ月前に書面通知を出して本プロトコルを終了することができます。CannBioRexも書面で通知され,フェルドマン雇用協定を随時終了し,直ちに発効することができる。もしCannBioRexが9ヶ月の書面通知を提供せずにサー·フェルドマンの採用を終了した場合、彼は9ヶ月に彼に通知する権利があると通知した基本賃金に相当する支払いを得る権利があるだろう。フェルドマン雇用協定の適用法 はイングランド法である。
取締役会は、当社の報酬委員会(および/または報酬委員会)の提案または単独の提案に基づいて、時々(株式、オプション、現金または他の形態の代価で)フェルドマン教授にボーナスを支給することを適宜決定することができる。
23
2021年11月17日、取締役会は報酬委員会の提案に基づき、サー·フェルドマンの年収を22.5万ドルに引き上げた
CannBioRexとフェルドマン卿は2022年4月27日から諮問協定改正案を締結し、この協定によると、2022年3月1日からフェルドマン卿の賃金を225,000ドル(100%)削減し、資金で日計を決定し、減少した金額を支払うことに同意した。
ローレンス·スタンマン医学博士諮問協定
2021年11月17日およびbrは2021年11月1日から当社と共同議長Lawrence Steinman,M.D.がコンサルティングプロトコル(“コンサルティングプロトコル”)を締結した。コンサルティング協定によると、Steinman博士は会社にいくつかのコンサルティングサービスを提供することに同意しているが、これらに限定されず、会社の戦略目標の定義と設定、買収と合併候補の積極的な 探し、および会社の´7 nAChRプラットフォーム (総称して“サービス”と呼ぶ)に対して主要な科学的責任を持っている。本プロトコルの期限は1年(“初期期限”)であり、双方が初期期限または任意の自動継続期間の終了前に少なくとも30日前に他方に書面通知を発行していない場合、プロトコルは初期期限後に1年間自動的に延長しなければならない(各期間は“自動継続期間” および初期期限はすべての自動継続条項(あれば))とともに、継続要求(以下に述べる )を遵守しなければならない。(I)スタンマン博士が自動継続開始日直前の会社株主年次総会で取締役会(“取締役会”)メンバーに再選されることを条件に、自動継続期間を延長することしかできない。(Ii)取締役会は、自動継続期間を適用する連合席議長(またはこの適用自動継続期間前に他の人を連合席議長に任命できなかった)および(Iii)Steinman博士が引き続きその職責を履行し、当社の´7 nAChRプラットフォームの科学発展(“継続要求”)を担当することを確認した。コンサルティング協議も以下の早い期日ですぐに満了する: (I)Steinman博士はもはや連合席議長を務めなくなり、私たちの´7 nAChRプラットフォームに主要な科学責任を負う日ではない;および(Ii)当社(取締役会の多数のメンバー(Steinman博士を含まない)が取締役会会議で投票証明した)または(2)Steinman博士(Steinman博士が取締役会に書面通知証明を出した)に要求された任意の早い日である。さらに、Steinman博士がサービスの実行を拒否することができない場合、会社は事前通知を必要とすることなく、相談プロトコルを直ちに終了することができ、他方が相談プロトコルのいかなる実質的な規定に違反している場合には、事前に通知することなく、相談プロトコルを直ちに終了することができる。
会社は合意期間内にSteinman博士に毎年225,000ドルを支払うことに同意し、2021年4月1日からの旧報酬と新報酬との差額である43,750ドルを一度に支払うことに同意した。コンサルティング協定によると、Steinman博士は、取締役会の書面による承認を得ない限り、合意期間内に会社と競合しないことに同意し、いくつかの慣用的な秘密条項および発明譲渡要件に同意する。諮問協定は終了後に12ヶ月の非入札禁止があります。
Steinman博士は2021年12月8日、1,250株の会社普通株を購入する株式オプションを付与され、期限は10年である;行使価格は日会社普通株に付与された公平な市場価値に等しく、1株79.00ドル、br}は会社の2020年総合激励計画の制約を受ける。また、2022年から、コンサルティング契約の有効期間内の毎年、会社は取締役会が将来承認した場合、Steinman博士に125,000ドルの持分補償 を支払う。将来の株式付与は48ヶ月以内に付与され、その計画に基づいて行われるだろう。将来付与される時間、株式付与の性質(例えば、RSU、PSU、限定株など)将来の株式価値の任意の変動は、会社報酬委員会および/または監査委員会によって提案され、取締役会によって承認される。
2022年4月27日から発効した当社はSteinman博士と諮問協定を改訂し、2022年3月1日からSteinman博士の賃金を56,250ドル(25%)削減し、資金確定日 に計上してこの減少した金額を支払うことに同意した。
24
知的財産権
私たちの成功は、私たちの候補製品、技術、およびノウハウの独自の要素を保護する能力があるかどうか、他人の固有の権利を侵害することなく運営され、他人の挑戦と反対を防ぎ、他の人が私たちの固有の権利を侵害することを防止する能力があるかどうかに大きく依存する。私たちはすでに米国、イギリス、ヨーロッパ、および他の国/地域で私たちのノウハウのための特許保護を求め続けている。2023年3月31日現在、私たちの知的財産権の組み合わせは、公開および/または係属中の特許請求の範囲、医薬製剤、薬物送達およびSCAの治療用途、ならびに独占的権利を有するパートナーが所有する特許を含む場合、技術的ノウハウおよび商業秘密を含む16個の特許シリーズを含む。
米国内では、私たちおよび/または私たちのパートナーは、発表された12件の特許および12件の積極的に起訴されている係属中の特許出願を許可している。米国以外では,EUが単一司法管轄区域であると仮定し,また12件の発行済み特許と23件の係争特許出願が積極的に起訴されている。私たちの政策は、業務発展に重要だと考えている技術、発明、改善のために特許保護を求めることですが、特許保護を得るコストが技術のビジネス潜在力によって合理的であると考えている場合にのみ、一般的には、私たちが大きなビジネス機会が存在すると考えている司法管轄区域に限られています。私たちはまた、商標、商業秘密、ノウハウ、持続的な革新に依存して、私たちの競争地位を発展させ、維持しています。
個別特許の期限 は,特許を取得した国/地域に依存する.我々が出願を提出したほとんどの国/地域では,特許期間は が非臨時特許出願を提出した最初の日から20年である。米国では,特許期限は特許期限を調整することで延長することができ,これは特許権者が米国特許商標局により補償するためである(“USPTO)、 が特許を付与するか、または1つの特許が別の特許よりも最終的に放棄された場合、短縮することができる。
FDAによって承認された薬物の特許期間も延長される資格があり、これは、FDA規制審査中に失われた期限の補償として回復期限を可能にする。1984年の“薬品価格競争と特許期限回復法”(“ハッジ-ワックスマン法”)は、特許満了後最大5年間の延長を許可した。特許期間の延長の長さは,薬物が規制審査を受ける時間の長さと関係がある。特許延長の残り期間は、製品承認日から14年を超えることができず、かつ、承認薬物に適用される特許を延長することしかできない。ヨーロッパおよび他の非米国司法管轄区域にも同様の条項があり、承認薬物をカバーする特許の有効期限 を延長する。
私たちが発行された特許および独自の情報を取得する権利を保護するためには、権利侵害第三者に対して訴訟を提起し、brを使用して、裁判所または公聴会に参加して、これらの特許または他の固有の権利の範囲および有効性を決定する必要があるかもしれない。
私たちはまた商業秘密に基づいて私たちの機密と固有の情報を保護する。私たちの政策は、私たちの従業員、コンサルタント、外部科学協力者、賛助された研究者、他のコンサルタントが私たちと雇用やコンサルティング関係を開始したときに秘密協定 を実行することを要求しています。
私たちの正常な運営過程で、私たちは時々知的財産権に関する訴訟や他の論争事項やクレームの側になるだろう。
180 LSの研究開発およびライセンスプロトコル
180 LSは、エルサレムヘブライ大学およびオックスフォード大学を含む研究および許可協定を当事者と締結している。これらのプロトコルの情報については、“を参照されたい”材料協定“、上の図。
25
競争
以下に我々の候補製品開発プラットフォームと潜在候補製品ごとの競争環境について説明する.
デュプイトレン筋拘縮症
われわれの治療は早期のDupuyten筋拘縮症に対するものであり,現在のところ承認されていない治療法は知られていない。現在の治療方法は主に末期のDupuyten筋拘縮、すなわち指が不可逆的に手のひらにカールすることに集中している。手術は依然として典型的な標準治療方法であるが、比較的に長い術後回復は侵襲性の小さい技術の発展を推進した。Auxiliumにより開発された薬剤XIAFLEXは,多くの患者の副作用が比較的軽いにもかかわらず,筋拘縮を生じた患者に有効であることを示している。もう一つの方法は末期臍帯を針で切断することであり、2018年に“整形外科雑誌”と“関節外科”(米国)に発表された比較臨床試験のデータによると、コラゲナーゼと経皮針刺筋膜切開術の再発率は2年以内に類似している。br}は米国国家健康研究所健康技術評価計画によって援助された臨床試験である。この研究は現在イギリスで行われており,手術治療デュプイトレン筋拘縮症とコラゲナーゼ治療の費用対効果を比較している。この研究の目的は,(I)コラゲナーゼ注射によるこの疾患の治療が手術と同様に有効かつ安全であるかどうか,および(Ii)の2つの治療法のコストを決定することである。
政制事務局長
を買収した後GW製薬会社そのEpidiolex(カンナビノール)とSativex(THCとCBD)の特許経営権は、Jazz製薬(アイルランド), Jazz Pharmaは大麻ジオール分野の重要な参加者となっている.Epidiolexは承認されたカンナビノール経口溶液であり、Draves症候群(以前は乳児期重症ミオクローヌスてんかんと呼ばれていた)、Rett症候群とLennox-Gastaut症候群を含む一連の児童てんかん疾患のてんかん発作の治療に用いられる。Jazz PharmaはEpidiolexがステッチ−ウェーバー症候群に有効であるかどうかを探索しており,この症候群は血管発育異常による出生時の脳,皮膚,眼に欠陥が出現し,より広く言えば自閉症スペクトラム障害に有効である。Jazz Pharmaが協賛した臨床試験では,多発性硬化症,潰瘍性大腸炎,クローン病などの自己免疫疾患に対するEpidiolexの有効性が試験されている。全般的に、これらの努力は最も広範な大麻ジオール臨床計画を代表する。
私たちの知る限り、複数の会社は大麻治療分野で活動しており、その候補製品の規制承認を求めている
| ● | 心臓治療会社(カナダ)同社は急性心筋炎を示す患者の心筋回復に対する彼らのCBD内服液製剤の効果を評価している。 |
| ● | Zynerba製薬会社(ペンシルバニア州)同誌は,薬物生産の経皮的大麻系薬物治療に集中しており,まれかつまれに近い神経精神疾患を治療している。Zynerbaは現在ZyGelを評価していますTM脆弱X症候群の治療のための特許保護CBD経皮ゲルであって、同社が、発育およびてんかん脳症、22 q欠失症候群および自閉症スペクトラム障害に関連するNDAをFDAに提出した、CBD経皮ゲル。 |
| ● | オコサ(ニュージャージー州)彼らのCBD,Oravexx(口腔崩壊錠)をテストして痛みや炎症をコントロールし,オピオイドへの臨床依存を減少させることが望まれている。1つの第2段階の試験では,特に適応は膝関節骨関節炎に関連する痛みである。 |
| ● | STROバイオテクノロジー(イスラエル)協賛したGVHD II期試験では,CBD(オリーブオイル合成CBD)はステロイド本来の治療効果を維持または改善するとともに,ステロイドの治療効果を増強したりステロイド用量を減少させたりすることが示唆された。その他の臨床試験には,ステロイド依存型クローン病治療のIIa期多中心試験,慢性蕁麻疹(蜂房)治療のIIa期試験,重症新冠肺炎治療のI/II期試験がある。 |
A 7 nAChR
TNFa(Humira)やTNpa受容体(Remicade)に対する抗体と核酸アプタマーは注射可能な試薬であり,その固有の性質 には臨床的限界がある。経口生物利用のTNFa阻害剤は抗体やアプタマー戦略の天然補充試薬である。brは治療試薬によって作用パターンが異なれば特に魅力的である。抗体もアプタマーもTNFaシグナルを妨害するために設計されているが,経口生物利用薬は迷走神経と大脳免疫系インターフェースを活性化することが知られているA 7ニコチン型アセチルコリン受容体アゴニスト である。
経口生物に利用可能なTNpa分泌阻害剤を開発する180 LS計画は激しい競争に直面している。最も突出しているのは,関節リウマチ,乾癬性関節炎,若年性特発性関節炎,軸性脊椎性関節炎,潰瘍性大腸炎,アトピー性皮膚炎および円形脱毛症の治療に許可されているJAK阻害剤集合(Xeljanz,Cibinqo,OLumant,Rvoq,Jyeleca)である。作用パターンはJak−Stat経路を抑制することであり,主にTNFaを含む各種炎症性サイトカインを分泌することが知られているマクロファージ系の細胞にある。これらの試薬のビジネス成功は,経口生物利用型製品の開発の重要性を実際に支援している。これらの薬物はアセチルコリン受容体経路に関与していない。Attenua Pharma、一家は とα7 nAChRアゴニストBradaniclineは、慢性咳の治療の第2段階臨床試験においてCoda Treateutics社によって得られ、計画が中止された。
26
電子会社は競争と見なすことができ、あるいは巨大な概念検証と見なすことができる。多くの点で,α7 nAChR計画 は迷走神経への化学刺激と考えられ,各適応は電気刺激から利益を得るべきであり,化学刺激に従うべきであるからである。炎症性適応が承認された製品に最も近い会社はSetPoint 医療会社であり,炎症性腸疾患や関節リウマチを治療する活性薬を有していることが知られている。彼らの装置はマイクロ刺激器brインプラントであり,頸部左側の小さな切り口を介して全身麻酔下で迷走神経を手術する。予期しない結果,短い電気パルスにより炎症性サイトカインの減少時間が約8−10時間延長する。
最後の考えは,α7 nAChRアゴニストを最初に開発した各大手製薬会社が計画を立て直すことができ,炎症適応の臨床試験に用いることである。
電子会社 は競争と見なすことができ、あるいは巨大な概念検証と見なすことができる。多くの点で、α7 nAChR計画は迷走神経への化学刺激と見なすことができるので、電気刺激から利益を得る各適応は化学刺激に従うべきである。
最後に,最初にα7 nAChRアゴニストを開発した大手製薬会社ごとに計画を立て直すことができ,臨床試験に応用した。
政府の監督管理
著者らはすでにイギリス薬品と保健品監督局(MHRA)とオランダ中央委員会Mensgenebonden Onderzoek(CCMO)及び関連認可道徳委員会の監督許可を得て、抗腫瘍壊死因子プラットフォーム下の適応のみに対してイギリスとオランダで臨床試験を行う。2 b期Dupuytenの収縮臨床試験の成功結果から,MHRAに提出された条件付きマーケティング許可申請を準備している。現在,我々はbrといかなる会議も開催しておらず,抗腫瘍壊死因子プラットフォーム下のいかなるbr適応や製品に関する申請や承認要求も米国食品医薬品局(FDA)に提出していない。
FDA承認プロセス
米国では,薬品や生物製品を含む製薬製品はFDAによって広く規制されている。アメリカ連邦食品医薬品化粧品法案(FDC法案)によると、a薬物“含むと定義する”ヒトまたは他の動物疾患の診断、治癒、緩和、治療または予防のための物品“と”物品(食品を除く)は、ヒトまたは他の動物の身体構造または任意の機能に影響を与えることが意図されている。USC 321(G)。すべての薬物と同様に、生物製品はヒト疾患の治療、予防、または治癒にも使用される。しかし、化学合成された小分子薬物とは異なり、小分子薬物は明確な構造を持ち、徹底的に表現することができるが、生物製品は通常生体材料(例えば人、動物あるいは微生物)から抽出され、構造が複雑であるため、常に十分に表現されている。米国の“公衆衛生サービス者法案”(以下、“PHS法案”)は生物製品を生物製品と定義しているウイルス、治療血清、毒素、抗毒素、ワクチン、血液、血液成分または誘導体、感作製品または類似製品…ヒトの病気や状況の予防、治療、または治癒に適している。42 USC 262(I)。FDAの法規および政策は、生物製品には、血液由来製品、ワクチン、体内診断アレルギー製品、免疫グロブリン製品、細胞または微生物を含む製品、およびほとんどのタンパク質製品が含まれることが決定されている。PHS法案によって制限されている生物製品もbrの定義に適合している麻薬問題FDC法案によると。生物製品は薬物のサブセットであるため、両者ともFDC法案条項によって規制されている。しかし、PHS法案第351条に基づいて生物製品のみが許可されているが、一部の治療用タンパク質製品はPHS法案ではなくFDC法案505条によって許可されている。)
FDC法案、PHS法案とその他の連邦と州法規は薬品と生物製品の研究、開発、テスト、製造、貯蔵、記録保存、審査、ラベル、普及とマーケティング、流通、承認後の監視と報告、サンプリングと輸入を管理する。適用される米国の要件を遵守しないことは、臨床的封印、FDAが未解決のNDAまたはFDAの承認を拒否するバイオ製品ライセンス申請(BLAS)または承認されたNDA/BLASの補充、承認撤回、警告状、製品リコール、製品差し押さえ、生産または流通の完全または部分的停止、禁止、罰金、民事処罰、および刑事起訴のような様々な行政または司法制裁を受ける可能性がある。
米国の薬物と生物開発は通常,臨床前実験室と動物試験,すなわちFDAにINDを提出し,このINDは臨床試験を開始する前に発効しなければならない。商業承認のためには、スポンサーは、提案ラベルにおいて規定、推薦、または提案された条件下での薬物/生物の使用が安全であることを証明するために、合理的に適用されるすべての方法の十分なテストを提出しなければならない。スポンサーはまた、一般に、FDAの安全性、有効性および薬物または生物学的製品の純度および効力基準に適合することを含む、提案されたラベルに規定され、推奨され、または提案された使用条件下で、FDAの安全性、有効性および薬物または生物学的製品の純度および効力基準に適合することを証明するために、十分な制御良好な臨床試験を含む実質的な証拠を提出しなければならない。FDA上場前の審査要求を満たすには通常数年の時間が必要であり、実際の所要時間は候補製品或いは疾病のタイプ、複雑性と新規性によって大きく異なる可能性がある。
27
臨床前試験は候補製品の化学成分、調合と毒性に対する実験室評価、及び候補製品の特性と潜在安全性と有効性を評価する動物試験を含む。臨床前試験の進行は必ず連邦法規と要求に符合しなければならず、FDAの良好な実験室規範(“GLP”)、良好な臨床規範(“GCP”)とbr}良好製造規範(“GMP”)法規及びアメリカ農業部が1996年の動物福祉法を実施した法規を含む。臨床前試験の結果はINDの一部として他の情報とともにFDAに提出され,候補製品化学,製造と制御に関する情報,提案された臨床試験案が含まれている。IND提出後,生殖毒性や発ガン性の動物試験など,長期的な臨床前試験を継続する可能性がある。
ヒト臨床試験が開始されるまで,各INDを提出してから30日以内に待つ必要がある。FDAがこの30日間にINDに対して臨床 を実施していない場合、またはINDを一時停止または他の方法でレビューまたは疑問視した場合、INDは発行されたとみなされ、INDで推奨されている臨床試験 は開始することができる。
臨床試験は、合格した研究者の監督の下で健康ボランティアまたは患者に研究中の新薬/生物製剤を提供することに関する。臨床試験は、(I)GCPを遵守しなければならない。これは、患者の権利および健康を保護し、臨床試験発起人、管理者および監督者の役割を決定するための国際基準および米国の法律要件であり、(Ii)他の連邦法規を遵守すること、および(Iii)試験目標、安全性を監視するためのパラメータ、および評価すべき有効性基準を詳細に説明するための方案である。米国患者の検出および後続案の修正に関連するすべてのスキームは、INDの一部としてFDAに提出されなければならない。
FDAが臨床試験がFDAの要求に沿って行われていないと考えている場合や,臨床試験患者に対して受け入れられないリスクとなっている場合,FDAはいつでも一時的,brあるいは永久的に臨床試験を停止させたり,他の制裁を加えたりすることができる。臨床試験患者の試験案とインフォームドコンセント情報は機関審査委員会にも提出しなければならない(“IRB“), は承認を待つ.IRBはまた、臨床試験の開始を阻止することができ、またはIRBの要求を遵守できないために、現場の臨床試験の一時的または永久的な停止を要求するか、または他の条件を適用する可能性がある。
上場承認を得るためにNDA/BLASを支持する臨床試験は通常3つの連続段階に分けて行われるが、特定の場合、この3つの段階は重複または異なる可能性がある。第1段階では、まず、健康なヒト対象または患者に薬物/生物学的製剤を導入し、次いで、新陳代謝、薬物動態、薬理作用、用量増加に関連する副作用を評価し、可能な場合に有効性の早期証拠を評価するために、薬物/生物学的製剤を試験する。第二段階は、一般に、特定の適応、用量耐性および最適用量に対する薬物/生物学的製剤の有効性を決定し、よく見られる副作用および安全リスクを決定するために、限られた患者集団で試験を行うことを含む。第二段階評価において、化合物が有効性および許容可能な安全性を証明した場合、より多くの患者の臨床治療効果および安全性に関する追加情報を得るために第三段階試験を行い、通常は地理的に分散した臨床試験場所である。FDAが薬物/生物の全体的な利益-リスク関係を評価し、薬物/生物のラベルに十分な情報を提供することを可能にする。多くの場合,FDAは薬物/生物の治療効果を証明するために十分かつ良好にコントロールされた2つの3期臨床試験を必要とする。しかしながら、FDAは、場合によっては、他の確証的証拠を有する単一の3期試験で十分である可能性があると判断するかもしれない。場合によっては、FDAは、様々な集団における薬剤の生物学的効果および長期使用に関連する任意の副作用に関するより多くの情報を収集するための承認条件として、上場後研究、いわゆる第4段階研究を要求することができる。薬物/生物製品によるリスクにより,他の発売後の要求brが課せられる可能性がある。
“21世紀治療法案”で提出された患者のより多くの薬物開発と評価需要を満たす具体的な要求に応えるために、FDAは“重点薬物開発ガイドライン発表計画”を発表し、この計画によると、FDAは一連のガイドラインを発表し、利害関係者がどのように患者と看護人員の患者体験データとその他の関連情報を収集し、提出し、医療製品開発と監督決定を行うために、漸進的な方法で患者と看護人員の患者体験データとその他の関連情報を収集と提出することを目的とする。これらのガイドラインは、信頼性があり、意義のある患者と看護者の意見を収集し、使用するために、brシステム方法の進歩と使用を促進し、それによって、医療製品の開発と監督決定に情報を提供することが予想される。FDAはこれまで、これらの問題について計画中の4つのガイドラインのうち3つを発表してきた。患者を中心とした薬物開発と評価問題がより優先され,臨床試験設計において がより重要な要素となり,前進していくことが予想される。
28
必要な臨床試験が完了した後,新薬申請(“NDA”)/BLAを作成しFDAに提出する。米国で候補製品の販売を開始する前に、FDAの承認を得る必要がある。NDA/BLAは、すべての臨床前、臨床および他の試験の結果、および候補製品の薬理、化学、製造、br}および対照に関連するデータアセンブリを含まなければならない。秘密プロトコル/BLAの準備と提出の費用はかなり高い.連邦法によると,NDA/BLAの提出の多くはアプリケーション使用料を支払う必要があり,2023年度の費用は約320万ドル(臨床データが必要な場合)である。
FDAはNDA/BLAを受信した日から60日の時間があり、この機関の敷居 に基づいて申請が十分に完全であるかどうかを決定し、実質的な審査を許可して、申請を受け入れるかどうかを決定する。提出された申請が受け入れられると、FDAは深い検討を始めるだろう。“処方薬使用者費用法案”によると、FDAはNDA/BLASのいくつかの業績目標を審査することに同意した。 FDAの現在の業績目標は、FDAが受信後10ヶ月以内に90%の基準(非優先)NDA/BLASの審査を完了し、優先NDA/BLASについては6ヶ月以内に審査を完了することを要求しているが、新しい分子実体/参照生物については、基準と優先NDA/BLASは2ヶ月以内の審査を追加する。1つの薬剤/生物学的薬剤が、深刻または生命に危険な疾患または状態において満たされていない医療ニーズを解決した場合、優先的に審査する資格がある。FDAは、いくつかの遅延された情報を考慮するために、または提出中に提供された情報を明らかにすることを目的とした情報を明らかにするために、標準審査および優先審査の審査プロセスをさらに3ヶ月延長することができる。このようなスケジュールはFDAに法的拘束力がない。
FDAはまた、新薬/生物製品または安全性または有効性の問題を提起する薬物/生物製品の出願を諮問委員会に提出することができ、この委員会は、一般に、申請を承認すべきかどうかを審査、評価、および提案するために、臨床医および他の専門家からなるグループである。FDAは諮問委員会の提案によって制限されていないが、それは一般にそのような提案に従っている。 NDA/BLAを承認する前に、FDAは通常、GCPに適合することを保証するために1つまたは複数の臨床サイトを検査する。
さらに,FDAは製造薬の1つまたは複数の施設を検査する。FDAは、GMPsに適合しない限り候補製品を承認しないであろうし、NDA/BLAは、この薬剤が安全かつ有効であることを証明するデータを含み、または研究された適応の安全性、純度、および効力基準に適合することを証明するデータを含むであろう。
FDAがNDA/BLAと製造施設を評価した後,FDAは承認状または完全な返信を発行する。完全な返信状(Br)は、一般に、提出中の不足点を概説し、FDAが出願を再検討するために、大量の追加のテストまたは情報を必要とする可能性がある。FDAがNDA/BLAを再提出する際にこれらの欠陥を満足的に処理した場合、FDAは承認書を発行する。FDAは、含まれる情報のタイプに依存して、2~6ヶ月以内に90%の再提出を検討することを約束した。任意の要求の追加情報が提出されているにもかかわらず、FDA は最終的に、その申請が承認の規制基準を満たしていないことを決定する可能性がある。
医薬/生物学的薬物の商業マーケティングを許可し、特定の適応の具体的な処方情報を提供する。NDA/BLA承認の条件として、FDAはリスク評価と緩和策を必要とする可能性がある(“br}FDAREMS)が、薬物/生物学的利益が潜在的リスクよりも大きいことを保証するのを助ける。REMSは,薬物ガイドライン,医療専門家のコミュニケーション計画,安全使用を確保する要素(ETASU)を含むことができる。ETASUは、処方または調剤のための特殊なトレーニングまたは認証、特定の場合にのみ調剤、特殊な監視、および患者登録簿の使用を含むことができるが、これらに限定されない。REMSに対する要求は薬物/生物の潜在市場と収益力に重大な影響を与える可能性がある。また、候補製品の承認には、薬物/生物の安全性や有効性を監視するために、大量の承認後のテストと監視が必要となる可能性がある。一旦承認されると、規制基準に適合していることが維持されていない場合や、予備発売後に問題が発見された場合、候補製品の承認は撤回される可能性がある。
29
COVID公衆衛生緊急事態申告期間中、COVID公衆衛生緊急事態FDAは、ある種類の製品に対して法執行自由裁量権を行使し、緊急使用許可(EUA)を発行することを規定し、多くの製品がFDAの正式な通常許可或いは許可なしに市場に入ることができ、またFDAの資源を非COVID関連製品から抽出することができるようにした。それ以降、米国政府は2023年5月11日から突発的な公衆衛生事件を終了することを発表したため、EUASの許可またはCOVID緊急事態に基づいてbr法執行の自由裁量権を付与した製品は、従来の承認要求に戻る可能性がある。
臨床試験情報の開示
FDA規制されたいくつかの製品(処方薬/生物製品を含む)の臨床試験のスポンサーは、米国国家衛生研究院が維持している公共ウェブサイトに特定の臨床試験情報を登録して開示しなければならない。登録の一部として,製品候補,患者br群,研究段階,研究場所や研究者,臨床試験の他に関する情報が公開されている。スポンサーもまた、完成後にこのような実験の結果を開示する義務がある。これらの試験結果を提出する締め切り スポンサー証明書が承認されていない製品を求めている場合,あるいは が1年以内に承認された製品の新適応を申請すれば,最大2年間延長することができる.競合他社は公開された情報を用いて我々の開発計画の設計や進捗に関する知識を獲得することができる.
迅速な承認指定と承認の加速
もし私たちの候補薬物/生物がFDA迅速チャネル計画の要求に適合すれば、この計画を通じて私たちの候補薬物/生物を加速させることを求める。FDAは薬物/生物製品の開発と審査を加速する計画があり、これらの薬物/生物製品は深刻或いは生命に危害を及ぼす疾病或いは病状を治療することを目的としているが、有効な治療方法がなく、そしてこの疾病が満足されていない医療需要を解決する潜在力を示した。迅速チャネル計画によれば、新薬/生物候補のスポンサーは、薬剤/生物候補薬物のIND届出と同時にまたは後に、特定の適応の候補薬剤/生物を迅速チャネル薬剤/生物として指定するようにFDAに要求することができる。FDAは、スポンサー要求を受けてから60日以内に、候補薬剤/生物が迅速チャネル指定を受ける資格があるかどうかを決定しなければならない。他の利点に加えて、FDAとより頻繁に相互作用させることができれば、FDAは、申請が完了する前に、高速チャネル薬/生物のNDA/BLA 部分の審査を開始する可能性がある。出願人が残りの情報を提出するスケジュールを提供し、FDAがスケジュールを承認し、出願人が適用された使用料を支払った場合、スクロール審査を行うことができる。しかしながら、FDA審査申請の期間目標は、NDA/BLAの最後の部分が提出されてから開始される。また,FDAが迅速チャネル指定が臨床試験中に出現したデータの支持を得なくなったと考えた場合,その指定を撤回する可能性がある。
FDAの加速承認規定によれば、FDAは、臨床利益を合理的に予測することが可能な代替終点に基づいて、または不可逆的な発病率または死亡率の前に測定可能な臨床終点に基づいて、既存の治療よりも意義のある治療のメリットを患者に提供することができる医薬/生物学的製剤を承認することができ、br}病状の重症度、希少性または流行度、および代替療法の利用可能性または不足を考慮して、不可逆的な発症率または死亡率または他の臨床的利益に合理的な影響を及ぼす。
臨床試験において、代替終点は疾病或いは状況の実験室或いは臨床バイタルサインの測定であり、患者の感覚、機能或いは生存状況に対する直接測定の代わりになる。代替終点は通常、臨床終点より簡単あるいは迅速に測定を行う。この上で承認された薬物/生物候補薬物は、臨床治療効果を確認するために、4期または承認後の臨床試験を完成することを含む厳格な発売後コンプライアンス要求を遵守しなければならない。必要な承認後研究が行われなかったか、または発売後の研究中に臨床的利益が確認されなかった場合、FDAは、薬物/生物を市場からの撤回を加速させることを可能にする。FDAが別途通知しない限り、承認が加速された製品/生物については、出願人は、発売承認後120日以内に配布または出版するために、販売促進ラベルおよび広告を含むすべての宣伝材料のコピーを承認前審査期間内にFDAに提出しなければならない。上場承認後120日後、FDAから別の通知がない限り、出願人は、少なくとも最初にラベルを発行するか、または最初に広告を発行する予定時間 の30日前に宣伝材料を提出しなければならない。
30
突破的治療指定
FDAのFAST 追跡計画と同様に,われわれの候補薬物/生物がFDAの画期的な治療指定を得る要求に適合すれば,この計画によりわれわれの薬物/生物候補を加速させることを求める。FDAの画期的な治療指定計画brは、深刻な或いは生命に危害を及ぼす疾患或いは状況の治療を加速する製品の開発と審査を加速することを目的としている。“食品および薬物管理局安全·革新法案”によれば、突破療法は、1つまたは複数の他の薬物/生物製品との併用治療のための、または生命に深刻な疾患または状態を危険にさらすことを目的とした薬物/生物製剤として定義され、初歩的な臨床証拠は、薬物/生物療法が臨床的意義を有する1つまたは複数のbr終点において、例えば臨床開発早期に観察される重大な治療効果のような既存の治療法よりも顕著な改善を示す可能性があることを示す。この指定は、高速チャネル指定のすべての機能 と、より密なFDA相互作用およびガイダンスとを含む。画期的な治療指定は,加速承認と優先審査とは異なる 状態であるが,関連する 基準を満たせば,同一の候補製品を付与することも可能である.FDAは、迅速に会議を開催し、提案を提供し、画期的な治療法の開発と審査承認の申請を加速するなど、いくつかの行動を取らなければならない。すべての画期的な治療指定の出願は、受信後60日以内に検討され、FDAは、この要求を承認または拒否するであろう。
また,“21世紀治療法案”は再生医学高度療法(RMAT)の称号を創設した。RMATは再生薬の一種として適している。ある細胞療法、治療用組織工学製品、ヒト細胞および組織製品、およびいくつかの組み合わせ製品のスポンサーがbr}が深刻または生命に危険な疾患または状態を治療するために使用することを意図しており、初歩的な臨床証拠がある場合、薬物が疾患または状態が満たされていない医療需要を解決する可能性があることを示す場合、その薬物製品のRMAT称号を得ることができる。発起人は研究用新薬申請を提出する際または後にこの要求を提出することができる。
RMATが指定したbr製品のスポンサーは,FDAとより多くかつより早いインタラクションを行う資格があり,突破的な指定 療法のスポンサーとのインタラクションのようなものである。しかも、それらは優先的な検討を受けて承認を加速させる資格があるかもしれない。RMAT指定製品のスポンサーとの会議は、長期的な臨床的利益の代替物または中間終点を合理的に に基づいて予測するのに適しているかどうか、または大量のサイトから得られたデータに依存するかどうかを検討することを含むことができる。
承認が得られると、適切なとき、FDAは、臨床証拠、臨床研究、患者登録、または電子健康記録のような他の真の証拠源を提出することによって、より大きな検証データセットを収集することによって、または承認前にすべての治療を受けた患者を承認後に監視することによって、加速承認の下で承認後の要求を満たすことを可能にすることができる。
迅速なチャネル指定、承認の加速、優先審査、画期的な治療指定は承認基準を変更することはありませんが、開発や承認の流れを加速させる可能性があります。FDAがそのうちの1つの指定を承認しても,FDAは後でこの薬物/生物製品が資格条件を満たしていないと決定する可能性がある。
みかん本発売と特許認証
1984年の“医薬品価格競争および革新法”(通称“ハッジ·ワックスマン法案”)によるFDC法案の改正によれば、秘密協定を介して薬物承認を求める場合、出願人は、出願人の候補製品または主張されている候補製品使用方法をカバーする各特許をFDAにリストしなければならない。1つの薬剤が承認された後、医薬出願に記載された各条件に適合する特許は、FDAによって承認された治療同等性評価を有する医薬製品において発行され、一般にbr}オレンジマニュアルと呼ばれる。オレンジマニュアルに記載されている薬物は、簡略化されたbr新薬申請者が特殊認証を行わなければなりません(“安達)は、薬物の模倣バージョンのために、または505(B)(2)出願と呼ばれる混合出願の出願者 によって出願される。ANDAは候補薬品のマーケティングを規定し、この候補薬品は参考に発売された革新薬物と同じ強度と剤形の有効成分を有し、そしてすでに参考発売された薬物と生物学的同等性 を有することが証明された。生物学的同等性試験の要求(無免除)を除いて、ANDA申請者は、その候補薬物の安全性または有効性を証明するために、臨床前または臨床試験または提出結果を必要としない。このようにして承認された薬物は、一般に“と呼ばれる汎用等価物“br”に列挙された薬物は,治療上は列挙された薬剤と同様と考えられ,通常は元に列挙された薬剤のために書かれた処方 によって薬剤師が代替することができる。
31
ANDA申請者は、FDAのオレンジマニュアルにおいて承認された候補製品のために記載された任意の特許をFDAに証明することを要求される。具体的には、出願人は、(I)必要な特許情報がまだ提出されていないこと、(Ii)記載された特許が期限が切れていないこと、(Iii)に記載されている特許が満了していないが、特定の日に期限が切れ、特許が満了した後に承認を求めること、または(Iv)に記載された特許が無効であるか、または新製品候補製品の侵害を受けないことを証明しなければならない。ANDA申請者も1部提出することができる“第八節声明“、その提案されたANDAタグが、記載された使用方法特許を証明するのではなく、特許使用方法に関するいかなる言語も含まない(または刻まれた)ことを証明する。
出願人が列挙された特許に挑戦していない場合、ANDA出願は、参照製品を要求するすべての特許が期限切れになったときに承認されるであろう。
新製品候補製品が承認された候補製品に列挙された特許を侵害しないか、又はそのような特許が無効又は強制的に実行できない認証を第4項認証と呼ぶ。ANDA出願人が第4項の認証をFDAに提供した場合、ANDAがFDAに届出を受けた後、出願人はまた、NDA及び特許所有者に第4項の認証の通知 を送信しなければならない。そして、NDAおよび特許所有者は、第(Br)IV段認証の通知に対して特許侵害訴訟を提起することができる。第4項の認証通知を受けてから45日以内に特許侵害訴訟を提起すると、30ヶ月前、特許満了、訴訟和解、侵害事件におけるANDA出願人に有利な裁決または裁判所の他の命令まで、FDAがANDAを承認することを自動的に阻止する。
スポンサーはまた、出願人が完全な安全性および有効性文書に基づいて医薬製品の承認を求めることを可能にする厳格な秘密協定経路である505(B)(2)条を介して医薬品の販売バージョンを求めることができ、いくつかの文書は文献からまたは他の人によって行われる可能性があり、出願人に対して参考にする権利がない。国家医薬品監督管理局“第505条(B)(2)の医薬品の出願は、新たな適応または新しい剤形のような以前に承認された医薬品を修正することができる。505(B)(2)節の申請 は、505(B)(2)の申請者によって得られた以前に承認された薬物の修正を支援する情報に加えて、以前に承認された薬物の安全性および有効性に関するFDAの調査結果に依存する可能性がある。準備505(B)(2)申請 は、新しいデータおよび情報に完全に基づいて秘密保護プロトコルを準備するコストおよび時間よりも低い可能性がある。第505条(B)(2)条出願 は、ANDAと同様の特許認証プログラムを遵守しなければならない。
新しい化学実体排他性と臨床研究排他性
NDAで新しい 化学実体(“NCE)、すなわち、FDAが任意の他のセキュリティプロトコルで承認された活性部分を含まない薬剤であって、5年間の市場排他性を得る薬剤であり、その間にFDAは、NCE薬剤バージョンの参照を承認するための薬剤の承認を求めるために、ANDAまたは505(B)(2)申請 を受け取ることができない。薬物のいくつかの変更、例えば、承認を必要とする新しい臨床研究のパッケージ挿入に新しい適応 を追加することは、その間にFDAが変更を含むANDAまたは505(B)(2)出願の3年間の排他的期間を承認することができないことに関連する。
第IV段落認証が提出された場合、ANDAまたは505(B)(2)の申請は、NCE排他性満了の1年前に提出することができる。Orange Bookに記載されている特許 がなければ,第IV段認証がない可能性があるため,排他期間満了 までANDAや505(B)(2)を提出することはできない.
植物薬の場合、FDA は、活性部分が1つ以上の主成分であるか、または全体として複雑な混合物であることを決定することができる。この決定は、任意の5年間の独占特許権の効用に影響し、任意の潜在的模倣薬競争相手は、この薬剤が原始植物薬と同じ能力であることを証明するであろう。
32
505(B)(1)の安全性および有効性を証明するために必要なすべての臨床前研究および十分かつ良好に制御された臨床試験の参照権を前提として、5年および3年の排他性は、FDAが排他的期間内に505(B)(1)バージョンの薬物出願を承認することを排除しない。
孤児指定と排他性
FDAは、米国で200,000人未満のまれな疾患または疾患を治療するための薬剤を付与することができ、または米国で200,000人を超える影響を与え、そのような疾患または疾患を治療する薬剤の開発および製造のコストが米国での販売から回収されることを合理的に期待できない場合、孤児の薬物名を付与することができる。
孤児薬物指定は、一方が臨床研究コスト、税収優遇、ユーザー費用減免に贈与資金を提供する機会などの財政的インセンティブを得る権利がある。孤児薬物指定は、監督審査と承認過程においていかなる利点も伝達されず、規制審査と承認過程の持続時間を短縮することもない。さらに、特定の薬物を取得した孤児薬物指定NDAまたはBLA申請者が最初に孤児薬物排他性を得る権利があり、これは、FDAが限られた場合でなければ、同じ適応で米国で同じ薬物 を販売する他の出願を7年以内に承認しない可能性があることを意味する。孤立薬物排他性は、FDAが同じ疾患または状態に対する異なる薬剤、または異なる疾患または状態に対する同じ薬剤を承認することを妨げるものではない。
小児科研究と排他性
NDAおよびBLASは、各安全で有効な小児科亜集団に対する薬剤の用量および投与をサポートするために、すべての関連する小児科の集団において新薬製品が主張される適応の安全性および有効性を評価するためのbrデータを含まなければならない。br}FDAは、申請者の要求に応じて、一部または全部の小児科データの提出を成人のために許可された後に延期することを許可することができ、またはいくつかの基準が満たされた場合、すべてまたは部分的な免除を承認することができる。小児科開発計画に関する議論は随時FDAと議論することができるが,通常は2期会議終了とNDAやBLAの提出との間の任意の時間に行われる。法規が別途要求されない限り、小児科データに対する要求は、孤児に指定された適応が付与されたいかなる薬剤にも適用されない。
小児科固有権は、米国における別のタイプの非特許固有権であり、FDAのいくつかの要件、例えばFDAが小児科集団における新薬の使用に関連する情報が健康利益をもたらす可能性があると判断し、出願人が特定の時間範囲内でFDA要件の研究を実行および報告することに同意する場合、このタイプの非特許固有権を付与することができる。小児科専門権は6ケ月の専有期間 を増加させ、スポンサーのこの有効部分に対するすべての既存のマーケティング専門権と特許を終了した。これは特許期間の延長ではないが、NDAまたはBLAスポンサーデータに依存した別の出願をFDAが受け入れまたは承認できない規制期間を効果的に延長する。
生物製品排他性と生体模倣薬
2010年の患者保護および平価医療法案、またはBPCI法案は、PHS法案第351(K)節に基づいてPHS法案第351(K)節に基づいてPHS法案第351(A)節によって許可されたFDA許可の参照生物製品と類似または交換可能であることが証明された生物学的許可経路として、PHS法案351(K)節に基づいて副題を含む。
参考製品が初めて許可された日から,参考生物には12年の市場独占経営権が付与され,この間,参考製品生物に類似した351(K)申請は承認されない可能性がある。参照生物はまた、4年間のいわゆるデータ排他性を付与され、その間、審査のために、参照製品の生物学的類似体の351(K)申請を提出することはできない。簡略化承認経路に従って提出された最初の生物学的製品が参照製品と交換可能であると決定されたことに基づいて、簡略化承認経路に従って提出された他の生物学的製品に対して排他的 を有し、より短い時間を基準として、(I)最初の商業マーケティングの1年後、(Ii)法的挑戦がない場合、(Ii)承認後18ヶ月、(Iii)申請が提出された場合、出願人 は、訴訟の決議から18ヶ月後、または(Iv)42ヶ月以内に訴訟が行われている場合、出願が承認されてから42ヶ月後である。
33
生物特許情報
小分子薬物とは異なり,小分子薬物では,出願人はそのNDAや何らかのサプリメントの特許情報を提出する必要があるが,生物製品の許可を求める出願人は,そのBLAやサプリメントに特許情報を提出する必要はない。また,小分子薬物とは異なり,小分子薬については,ANDAと505(B)(2)NDAの承認は,NDA医薬製品の発売特許状況を参照して影響を受け,現在第351(K)条の生体類似製品出願の承認は,特許紛争解決のための様々なプロセスと脱フックしている。生物学的に類似した申請者は、決定された特許リストを決定して訴訟するために、生物製品価格競争に参加するか否かと革新法(BPCIA)の特許訴訟条項、通称“特許の舞”と呼ばれる特許リストを選択することができる。しかし、“オレンジマニュアル”に記載されている小分子が発売薬や特許を参照しているのとは異なり、FDAの許可生物製品リスト(通称“紫書”)には特許の手続きが記載されていない。2020年12月、米議会は“生物製品特許透明法”(BPPT)を公布した(当初は“紫書連続性法案”で“小霊通法案”第351(K)(9)条を創設する形で導入された)。この部分は,生物製品参考資料brスポンサーが生物類似出願人に特許リストを提供することを要求しており,BPCIA特許訴訟プログラムの一部として現在30日以内にこれらのリストをFDAに提出しなければならず,また,FDAは2021年6月からPurple Bookデータベースにこれらのリスト(および の任意の改訂または更新)を公開しなければならない
特許期間を延長する
NDAまたはBLAの承認後,関連薬物または生物特許の所有者は,最長5年間の特許延期を申請することができる。許容される特許期間の延長期間は、製品試験段階の半分(IND提出とNDAまたはBLA提出との間の時間)と のすべての審査段階(NDA提出と承認との間の時間、最長5年)である。FDAが、出願人が職務調査を経て承認を求めていないと判断した場合、時間 を短縮することができる。展示期間後の総特許期間は14年を超えてはならない。
出願段階で満了する可能性のある特許については,特許所有者は臨時特許延期を申請することができる。臨時特許延期は特許期間を1年延長することができ、最大4回更新することができる。一時特許が付与されるたびに延期され,承認後特許延期は1年減少する.米国特許商標局(USPTO)の取締役は,特許出願延期された特許に含まれる製品 が承認される可能性が高いことを確認しなければならない。秘密保持プロトコルやBLAを提出していない薬物 は一時的な特許延期を得ることができない。
広告と販売促進
NDAまたはBLAが承認されると, 候補はいくつかの承認後に要求される制約を受ける.例えば、FDAは、直接消費者向け広告、ラベル外販売促進、業界スポンサーの科学と教育活動、およびインターネットに関連する販売促進活動の基準と法規を含む、薬品と生物製品の承認後のマーケティングと販売促進を密接に規制する。
薬品や生物製品は承認の適応と承認されたラベルの規定に基づいてしか販売できない。
承認後に変更する
適応、ラベルまたは製造プロセスまたは施設の変更を含む承認申請において決定されたいくつかの条件を変更するには、新しいNDA/BLAまたはNDA/BLA補充物の提出およびFDAの承認が必要である。新しい 適応に対するNDAサプリメントは通常,オリジナル申請と類似した臨床データが必要であり,FDAはNDAサプリメントの審査にNDAと同様のプログラムや行動 を使用している。
34
有害事象報告とGMPコンプライアンス
FDAがNDAまたはBLAを承認した後,有害事象報告を加速し,定期的な有害事象報告を提出することが要求される。FDAはまた、承認製品の効果を監視するために、いわゆる第4段階試験、REMSおよび監督を要求することができ、またはFDAは、承認時に条件を付加して、その製品の流通または使用を制限する可能性がある。また,品質管理,薬品製造,包装,ラベルプログラムは承認された後もGMPに適合し続けなければならない。医薬品製造業者と彼らのいくつかの下請け業者はFDAと特定の州機関に彼らの工場を登録しなければならない。FDAの登録要求エンティティはFDAの定期抜き打ち検査を受け,その間,FDAは製造施設を検査し,GMPに適合しているかどうかを評価する。そのため,メーカーは生産や品質管理の分野で時間,お金,労力をかけ続け, でGMPに適合したままでなければならない。もしある会社が監督管理基準を遵守できなかった場合、もし同社が最初のマーケティング過程で問題に遭遇した場合、あるいは後に以前に発見されなかった問題が発見された場合、監督管理機関は製品の審査を撤回し、警告状を出し、製品のリコールを要求し、あるいは他の法執行行動を取ることができる。
特殊プロトコル評価
スポンサーは特別合意に基づいてFDAとの合意を評価することができる(“スパ.スパ)は、治療効果宣言の主な根拠を形成するための臨床試験の必要な設計および規模に関するプロセスである。その業績目標によると、FDAは提案試験が十分であるかどうかを評価する要求を受けてから45日以内に90%の方案を評価することを約束しており、この評価は議論と要求により多くの情報を提供する可能性がある。提案した実験開始前にSPA要求 を提出しなければならず,実験開始前にすべての未解決問題を解決しなければならない.書面合意に達したら、それは記録され、行政記録の一部になるだろう。FDA法案およびFDAが法定要求を実施するガイドラインによると、SPAは通常、FDAに対して試験設計に拘束力があるが、限られた場合を除いて、例えば、FDAが研究開始後に安全性または有効性を決定するために重要な重大な科学的問題を発見した場合、案評価時に意識されていない公衆衛生問題が発生し、スポンサーとFDAが書面で変更に同意した場合、または研究スポンサーがFDAと合意した案に従わなかった。
規制対象物質
改正された“規制物質輸出法”(“シクロスポリンA)および実施条例は、規制された物質の登録、安全、記録保存および報告、貯蔵、製造、流通、配布、輸入およびその他の要件を実施し、米国麻薬取締局によって監督される(br})DEA“)”DEAは連邦機関であり、制御物質の監督管理を担当し、生産、輸入、輸出、流通、制御物質の個人或いは実体の研究或いは配布に規制要求を遵守し、制御物質の移転と濫用を防止することを要求する。
DEAは規制されたbr物質を付表I、II、III、IVまたはV物質とし、具体的にはこの物質の医療効力と乱用の潜在力に依存する。br}は現在医療用途に使用可能であることが認められ、発売が許可された薬品は別表II、III、IVまたはV物質とすることができ、その中で付表II物質の濫用の可能性が最も高く、身体や心理への依存度が最も高く、付表V物質の濫用と依存の相対的な可能性が最も低い。DEAはマリファノールを含むいくつかの薬品 を付表Vに入れた。
国家薬品監督管理局が表I制御物質を含む薬剤を承認した後,この物質は別表II,III,IVあるいはV物質に再配置しなければ発売できない。2015年11月25日に公布された“新医療療法規制透明性向上法”およびその施行された法規は、NDA承認後のDEA再配置プロセスの時間に関する不確実性を除去し、この規定によれば、メーカー は、以下の遅い日後にその製品を販売することができる:(1)DEAがFDAから科学的およびbr}医療評価および計画提案を受けた日、または(2)DEAがFDAからFDAからその薬剤の通知を受けた日。この法案はまた、7年間の孤児排他期がNDAまたはDEAスケジュールの承認から始まり、遅い時間を基準とすることを明らかにした。これは孤児の昔の状況を変えました“時計.時計FDAのNDA承認後に運転を開始し, この製品がDEA計画完了後に発売されたとしても。
35
CSAは任意の制御物質を製造,流通,分配,輸入または輸出する施設を毎年DEAに登録しなければならないことを要求している。輸入と製造活動は単独で登録する必要があり,各登録認可登録者が処理可能な制御物質の具体的なスケジュール である。DEAは、制御対象物質の登録を発行する前に、制御対象物質の安全、記録保存、報告、処理を審査するために、すべての生産施設を検査する。具体的な安全要求は,従事する商業活動のタイプや処理される制御物質のタイプ,形式,数量によって異なる。
また,各州 には,制御物質に対する許可,分配,分配,記録保存,報告要求を含む独自の制御物質法律法規がある。州薬局委員会や同様の当局は州ごとの制御物質の使用を管理している。規制対象物質の損失や移転等の適用要件を遵守しないことは、行政罰金、一時停止又は許可証の取り消し、民事及び刑事責任を招く可能性がある。
イギリス/ヨーロッパ/世界の他の地域の政府規制
米国の法規brを除いて、私たちは現在も将来も、臨床試験および任意の商業販売(定価および精算を含む)、および私たちの製品の流通(承認されれば)を含む、私たちの流通パートナーを通じて、他の司法管轄区域の様々な法規を遵守するだろう。
製品がFDAの承認を得ているか否かにかかわらず、これらの国/地域で臨床試験またはマーケティングを行う前に必要な承認を得るためには、米国以外の国/地域の規制機関でbrを開始しなければならない。
EUでは、医薬製品は発売前と発売後にEUと国家監督管理機関の広範な監督管理を受けている。国家レベルの他の規則は規制された物質の製造、輸入、輸出、貯蔵、流通、販売にも適用される。多くのEU加盟国では、医薬製品を担当する規制当局も規制物質に責任を持っている。しかし、責任 はいくつかの会員国で分離されている。通常、EUで制御物質を含む医薬製品を生産または流通する会社は、国家主管機関が発行する制御物質許可証を持ち、具体的なbr記録保存と安全義務に制限される必要がある。メンバーbr国に出入りする各貨物は単独の輸入または輸出証明が必要である。
イギリスでは、薬品は薬品と保健製品監督局の広範な監督管理を受けている(“MHRA“)、衛生·社会保健省が後援する執行機関である。MHRAは公衆の健康に有益な革新と研究開発を支持するほか、薬品、医療器械と輸血に用いる血液成分が適用の安全、品質と治療効果標準に符合することを確保することによって監督管理を行う。
臨床試験と上場承認
米国 以外のある国/地域の流れは ヒト臨床試験を開始する前に臨床試験申請を提出することが要求されており,INDと非常に類似している。例えば,EU/欧州経済圏では,EU/欧州経済区で行われているすべての臨床試験について,臨床試験申請(“CTA”)は単一入口ポータルサイト(“CTI”)を介して提出されなければならない。CTAが一国の要求に応じて承認され,ある会社が道徳委員会の良好な承認を得ると,臨床試験開発はその国で行うことができる。
2014年4月臨床試験条例(EU)は“臨床試験指令2001/20/EC ”(“前の指令”)の代わりに第536/2014号(“新条例”)を採択した。臨床試験規則がEU/欧州経済区全体で一致することを確保するため、EUはEU/欧州経済区加盟国に直接適用する“規定”として、新しい臨床試験立法を採択した。新法規は、スポンサーに、許可プロセスを簡略化したオンラインポータルサイトである、すべてのEU/欧州経済圏加盟国計画のための単一CTAを提出することを要求する。CTIS許可手続きは2つの部分から構成されている:加盟国は共同で協力して第1の部分評価を行い、第2の部分は各加盟国によって個別に評価される。これは,これまでの指令 によると,スポンサーは試験を行う各国の国当局の単独承認を求めなければならないため,大きな変化である。新法規 は2022年1月31日に施行された−同じ日からこれまでの指令が廃止された−1年間の過渡期 (2023年1月31日まで)が導入され,その間,スポンサーは 以前の指令によって管轄された旧制度か新しいCTIに基づいて新たなCTAを提出するかを選択することができる。移行期間の後,2023年1月31日からスポンサーは新法規に基づいて臨床試験を申請しなければならず,2025年1月31日には,先の指令により承認されたすべての進行中の臨床試験が新法規brに移行する必要がある。
36
イギリスでは,これまでの指令 (ヒト用薬物(臨床試験)法規2004/1031による)が適用されている。2020年1月31日、イギリスがEUを離脱し、“2018年EU(離脱)法”(略称EUWA)が正式に発効した。EUWA第1節は、EU法の英国への適用を許可してきた“1972年欧州共同体法”(ECA 1972)を廃止した。しかし、EUWA第1 A節は過渡期にECA 1972(改正された形式)の効力の大部分を保持し、第1 B節ではEUが要求した英国立法 を保持している。したがって,EUWAの実際の効果は,イギリスには適用されていない新条例 を廃止したが,先の指令を保持していることである。本節で言及されたイギリスは主にイギリスを意味する。北アイルランド議定書第182条の規定により、EU/欧州経済圏が北アイルランドで制定されたいくつかの管理基準は廃止されていないため、場合によっては適用される。現在,北アイルランド議定書の新たな交渉が行われており,2023年に改正される予定である。イギリスでは,臨床試験を計画する際には,MHRAの承認を得なければならない。“ヒト用薬品(臨床試験)条例”2004/1031とMHRAが提供した他の適用指導文書によると,CTAは提出しなければならず,研究用薬品ファイルや他の支援情報から提供されなければならない。また,担当倫理委員会がイギリスの臨床試験申請に積極的な意見を発表した後にのみ,臨床試験を開始することができる。
臨床試験、製品許可、定価、精算を管理する要求と流れは国/地域によって異なり、EUの基本法規の国が実施されているにもかかわらず、EU加盟国はすでにある程度の法律協調を持っている。すべての場合、臨床試験はGCPに関する国際協調会議のガイドラインと他の適用される法規の要求に従って行われなければならない。
規制部門の承認を得るためには,イギリスやEU/欧州経済圏の国/地域の市場に薬物を投与するためには,上場許可申請を提出しなければならない。このアプリケーション は米国のセキュリティプロトコルに類似しており,国/地域に特定された文書要求である点が異なる.すべての出願プログラムは,製品製造や品質に関する詳細な情報の提出,非臨床·臨床試験情報の提出を含む汎用技術文書形式の出願が必要である。国家認可手続き(関連するEU/欧州経済圏加盟国で有効な医薬品上場許可をもたらす)の使用に加えて、(I)集中認可手順、(Ii)相互認識手順、または(Iii)分散手順を使用してEU/欧州経済圏で使用可能な医薬品を使用することができる。イギリスは、EU/欧州経済地域で使用可能なこれら3つの認可方法を提供しない。イギリスでは、医薬品は、(I)革新的許可およびアクセス経路を使用することによって、MHRAによって(I)150日間評価経路を評価することができる。(3)“スクロール審査” 新製品およびバイオテクノロジー製品の評価経路,(4)欧州委員会(EC)決定依存手順,(V)分散および相互承認依存手順,または(Vi)北アイルランドで許可された販売許可の“制限されないアクセス”経路 。
欧州委員会は、EU全体およびアイスランド、リヒテンシュタイン、ノルウェーでの有効な販売許可を促進するために、人間の薬物を集中的に承認する手続きを制定し、これらの国はEU加盟国 とともに欧州経済区を構成した。申請者はヨーロッパ医薬品局(EMA)にマーケティング許可申請を提出し、関連する科学委員会が審査を行い、多くの場合人用薬品委員会(“CHMP“。 欧州医薬品局(”EMA)CHMP意見を欧州委員会に転送し、欧州委員会は、マーケティング許可を付与するか否かを決定するための基礎として使用する。この手続きは欧州委員会によって付与された単一のマーケティング許可がEU全体およびアイスランド、リヒテンシュタイン、ノルウェーで有効であることをもたらした。以下のヒト薬物の場合、集中手順は強制的である:(I)遺伝子工学のようなバイオテクノロジープロセスに由来し、(Ii)HIV/エイズ、癌、糖尿病、神経変性疾患、自己免疫および他の免疫機能障害およびウイルス疾患などの特定の疾患の治療のための新しい活性物質を含み、(Iii)公式に指定されている孤児薬“(ヒトまれな疾患のための薬剤) および(4)遺伝子治療、体細胞治療または組織工学薬などの先進的な治療薬。ヒト薬物(A)が2004年11月20日に承認されていない新しい活性物質を含むことに同意する場合、(B)重大な治療、科学的または技術的革新を構成するか、または(C)集中型プログラム下の許可がEU一級患者の利益に適合する場合、集中型プログラムは、申請者の自発的な要求の下で、上記のカテゴリに属さないヒト薬物にも使用することができる。イギリスではEU集中プログラムを使用することはできないが,2023年12月31日までにMHRAはEU集中プログラム下の欧州共同体で承認され,分散と相互承認のプログラム依存認可薬を利用することができる。2023年12月31日以降,MHRAは新たな国際薬物依存枠組み制度を構築しようとしている。
37
EUの中央手続き によれば、EMAがマーケティング許可申請を評価する最長期限は210日(CHMPによって提起された質問に応答するために出願人によって追加的な書面または口頭情報を提供する時間を含まない)、 の後に、欧州委員会によって実際のマーケティング許可が採用される。
特殊な場合、CHMPは加速評価を承認する可能性があり、すなわち治療革新の観点から1種の医薬製品は重大な公共健康利益を有することが予想され、これは3つの累積標準によって定義される:治療すべき疾患の深刻さ;適切な代替治療方法の不足、および極めて高い治療効果が期待される。この場合,環境保護局 はCHMP意見の評価を150日以内に完了することを確保し,その後意見を発表する。
集中的に手続きされていない医薬製品の場合、出願人は、以下の3つの手順のうちの1つを介して、国家薬品監督管理機関に上場許可申請を提出しなければならない:(I)相互認識手順(製品が少なくとも1つの他のEU/欧州経済圏加盟国で許可されている場合、その手続きを使用しなければならず、EU/欧州経済区加盟国に、公衆の健康に深刻なリスクがあることが発見されない限り、別のEU/欧州経済区加盟国に既存の許可を付与することを要求する)。(Ii) 分散手順(2つ以上のEU/欧州経済圏加盟国で同時に出願を提出する)または(Iii)国家認可手続き(単一のEU/欧州経済圏加盟国におけるマーケティング許可をもたらす)。
条件付きマーケティング許可
欧州委員会は満たされていない医療需要を満たす薬品に条件付き上場許可を与える可能性がある。このマーケティング許可は、許可を実行する際に課せられたいくつかのアクティビティ(例えば、進行中または新しい研究を完了するか、または追加の データを収集する)を前提として行われる である。これは、出願人が客観的で確認可能な理由および特定の理由に基づいて、通常の使用条件下での医薬品の有効性および安全性に関する包括的なデータを提供することができず、以下のすべての基準を満たすのに適している
| ● | 薬の利益-リスクバランスは正である | |
| ● | これは、出願人が許可後に完全なデータを提供する可能性がある | |
| ● | この薬は満たされていない医療ニーズを満たしています | |
| ● | まだもっと多くのデータが必要であるため、患者がすぐに薬物を得ることができる利点は固有のリスクよりも大きい。 |
条件付きマーケティング許可 の有効期間は1年で、毎年更新することができます。
イギリス(イングランド、br}スコットランド、ウェールズ)では、MHRAは2021年1月1日から発効する国の条件付きマーケティング許可を導入し、規範化している;北アイルランドでは、条件付きマーケティング許可の申請はEMAに提出されなければならない。イギリス計画はEU計画と同じ資格基準 を持っている。
相互認識プログラム
相互認識プログラム (‘’資材需要計画)を承認することは、EU/欧州経済圏内の個別の国の販売許可を促進する別の方法である。基本的にMRPは,強制的な集中プログラムではないすべてのヒト薬物に適用可能である。MRP は、ほとんどの従来の医薬製品に適用され、製品が1つまたは複数の加盟国で許可されている場合、その製品を使用しなければならない。
38
MRPの特徴は、他のEU/欧州経済圏加盟国のマーケティング許可を得るために、注文 において参照として使用される加盟国の既存のマーケティング許可に基づいて確立されることである。MRPでは、医薬品のマーケティング許可は、EU/欧州経済地域の1つまたは複数の加盟国に存在しており、その後、他の加盟国では、初期マーケティング許可を参照することによって、他の加盟国でマーケティング許可申請を行う。最初にマーケティング許可を得た会員国はbr参考会員国として機能するだろう。その後、販売許可を申請した加盟国は、関連加盟国として許可されなければならない。 関連加盟国は、それらが公衆健康に深刻なリスクとならない限り、参照加盟国の既存の許可を承認する許可を付与しなければならない。
MRPは、EU/欧州経済圏加盟国がそれぞれの国家マーケティング許可を相互承認する原則に基づいている。出願人は、加盟国のマーケティング許可を参照して、他の会員国でマーケティング許可を申請することができる。この場合、参照会員国は、その薬剤に関する既存の評価報告書を90日以内に更新する必要がある。評価が完了した後、報告書のコピー、および承認された製品特性要約、ラベル、および包装チラシがすべての関連加盟国に送信される。そして、関連加盟国は、加盟国の決定と製品特性、ラベル、包装チラシの要約を参照する90日間の期間がある。契約を確認してから30日以内に全国マーケティング許可を付与しなければなりません。
いずれの会員国も潜在的な深刻な公共健康リスクを理由に加盟国の販売許可を参照することを拒否した場合、問題は調整グループに提出される。60日間の期限内に、会員国は合意に達するために調整グループ内ですべての努力をしなければならない。もし失敗したら、手続きはEMA科学委員会に仲裁に提出されるだろう。そして、EMA委員会の意見を欧州委員会に転送して、意思決定過程を開始する。集中型プログラムと同様に,この過程は各欧州委員会本部長とヒト医薬製品常設委員会に諮問する必要がある。
データ独占性
イギリスおよびEUでは、後発薬のマーケティング許可申請は、臨床前および臨床試験の結果を含む必要はないが、 は、規制データ排他性期限切れの参照製品のマーケティング許可に含まれるデータを参照することができる。新しい活性物質を含む医薬製品にマーケティング許可を付与する場合、この製品はbrの8年間のデータ独占期間から利益を得ることができ、その間、監督機関はこの製品データに関連する模倣薬マーケティング許可申請を受け入れない可能性があり、他の2年間の市場独占期間を受け、その間、このような模造薬は市場に投入することができない。最初の8年以内に新しい治療適応が承認され、既存の療法と比較して有意な臨床的利益があれば、2年間の期間は3年に延長される可能性がある。
孤児医薬製品
EMAの孤児薬品委員会(“COMP)は、生命または慢性衰弱に危害を及ぼす疾患の診断、予防または治療を目的とした製品の開発を促進するために、指定孤児医薬製品を推奨することができ、EUの10,000人当たり5人以下に影響を与える。さらに、生命に危害を及ぼす、深刻な虚弱または深刻かつ慢性疾患の診断、予防または治療のための製品、およびインセンティブがない場合、製品のEUでの販売は、医療製品を開発するための必要な投資を証明するのに十分ではない可能性が高く、孤児の称号が与えられる。関連する製品が関連する適応において既存の承認された製品よりも顕著な臨床的利益を有する場合にのみ、COMPは指定孤児薬品を推薦することができる。委員会の肯定的な意見によると、欧州委員会は一般的に30日以内に孤児の身分を与える。欧州委員会の決定草案がCOMPの意見と一致しない場合、COMPは、EMAが発売許可申請を審査しながら孤児状態を再評価し、薬物が孤児基準を満たさなくなった場合(例えば、新製品が適応のために承認され、新製品が新製品に顕著な利益を有することを証明するデータがないため)、孤児状態は で撤回される可能性がある。孤児薬品に指定された側は、費用の減免または費用の減免などの財政的奨励を受ける権利があり、発売許可を得てから10年間の市場独占経営権を得る権利がある。この期間、主管部門は、同じ治療適応のための任意の類似の医薬製品を受け入れたり承認したりしてはならない。brは、その製品が顕著な臨床的利益を有していない限り、または元の孤児薬の発売許可保持者が十分な数の薬物を供給できない場合である。孤児薬品指定基準を満たさなくなった場合、この期間は、製品の利益が十分に高いことを証明することを含む6年に短縮される可能性があり、市場排他性を維持するのが合理的であることを証明するのに不十分である。
39
EU/ヨーロッパ経済地域孤児名 は北アイルランドまで延びている。そうでなければ、イギリス離脱後のイギリスでは、孤児薬品の指定申請はMHRAに提出され、MHRAはEUと同じ実質的な基準と同等の実質的な効力を採用する(ただし、EUの手続きとは異なり、早期の孤児指定を得ることは不可能である;指定申請はイギリスのマーケティング許可申請 と同時に提出されなければならない)。1つの医薬製品がEUで孤児として指定された場合、MHRAにイギリス 孤児マーケティング許可申請を提出することができ、EU孤児指定がない場合、MHRAに全イギリス(北アイルランドを含む) 孤児マーケティング許可申請を提出することができる。
小児科発展
EUやイギリスでは、新しい医薬製品を開発する会社は小児科研究計画に同意しなければならない(“ピップ)であり、例えば、関連疾患または状態が成人においてのみ発生するため、免除が適用されない限り、PIPに従って小児科臨床試験を行わなければならない。製品のマーケティング許可申請は、申請免除または延期が承認されない限り、PIPによる小児科臨床試験の結果を含まなければならず、この場合、小児科臨床試験は遅く完了しなければならない。PIPによる小児科臨床試験により発売許可を得た製品は、補充保護証明書によって6ケ月の保護期限 を延長する資格がある(もしそれがカバーする製品が承認時に条件を満たす場合)。この小児科奨励は特定の条件に制限されており,PIPに適合するデータを開発·提出する際には自動的に獲得されない。
* * * * *
もし私たちが適用された外国の監督管理要求を遵守できなかった場合、私たちは罰金、臨床試験の一時停止、brの一時停止、監督管理の承認撤回、製品のリコール、製品の差し押さえ、経営制限、刑事起訴などの処罰を受ける可能性がある。
また,多くの国は1961年麻酔薬単一条約と1971年精神薬条約の締約国であり,大麻抽出物を含む麻酔物質の国際貿易と国内規制を管理している。国/地域はその条約義務brを解釈して履行する可能性があり、これは、これらの国/地域で私たちの候補製品のマーケティング承認を得ることに法的障害をもたらす可能性がある。これらの国/地域 は、私たちの候補製品の発売を可能にするために、またはその法律法規を修正することを望まないか、または が法律法規をこのように修正するのに長い時間を要するかもしれない。この場合、私たちは近い将来、あるいはこれらの国/地域で私たちの製品を販売することができないだろう。
精算する
米国での薬品の販売は、政府医療計画、商業保険、管理医療機関のような第三者支払者が製品コストを支払う程度にある程度依存する。これらの第三者支払者は医療製品やサービスの料金にますます大きな挑戦をしている。また,医療コストの抑制は連邦と州政府の優先順位となっており,薬品価格はこの努力の重点となってきた。アメリカ政府、州立法機関と外国政府はコスト制御計画の実施に大きな興味を示し、これらの計画は価格制御、使用管理と後発薬代替要求を含む。価格制御とコスト制御措置、および既存の統制·措置を講じている司法管轄区では、より厳しい政策を採用することで、我々の純収入や業績をさらに制限する可能性がある。もしこれらの第三者決済者が私たちの未来の製品が他の利用可能な療法と比較して費用効果があると思わない場合、彼らは私たちの製品をその計画下の福祉として承認された後に私たちの製品をカバーしないかもしれない。あるいは、もし彼らがそう思うなら、支払いレベルは私たちの利益に基づいて私たちの製品を販売するのに十分ではないかもしれない。
40
2003年“医療保険処方薬品、改善と現代化法案”(“MMA)は、Medicare受益者向け処方薬の流通および定価を要求し、Medicare第D部分下の処方薬福祉を大幅に拡張することを含む。 D部分によれば、Medicare受益者は、個人実体によって提供される処方薬計画を登録することができ、これらの計画は、外来処方薬保険を提供する。D部分は独立した処方薬福祉計画と処方薬保険を通じてMedicare Advantage計画の補充として提供することができる。連邦医療保険A部やB部と異なり,D部のカバー範囲は標準化されていない。D部分処方薬計画発起人はすべての保証を受けたD部分薬物に支払う必要がなく、各薬物計画は自分の薬物処方表を開発することができ、それがどの薬物及びレベル或いはレベルをカバーするかを決定することができる。しかし、D部分処方薬処方は、カバーされたD部分薬剤の各治療カテゴリおよびカテゴリの薬剤を含む必要があるが、必ずしも各カテゴリまたはカテゴリのすべての薬剤 またはカテゴリを含むとは限らない。D部分処方薬計画で使用されるどの処方も薬局と治療委員会によって開発され、審査されなければならない。br政府の処方薬の一部のコスト支払いは、マーケティング許可を得た製品に対する需要を増加させる可能性がある。しかし、D部分処方薬計画がカバーする製品のいかなる交渉価格も、他の方法で得られた価格よりも低い可能性がある。また,MMAは連邦医療保険受益者の薬品福祉にのみ適用されているが,個人支払者は自分の支払率を設定する際には通常 連邦医療保険カバー政策と支払制限に従っている。MMAによるいかなる支払いの減少も、非政府支払者の支払いの同様の減少をもたらす可能性がある。
2009年2月17日、オバマ総裁は“2009年米国回復と再投資法案”を法律に署名した。この法律は連邦政府 に資金を提供し、同一疾患の異なる治療法の有効性を比較する。この研究は衛生·公共サービス部、医療保健研究と品質局と国家衛生研究院が監督し、研究状況と関連支出に関する定期報告を国会に提出しなければならない。比較有効性研究の結果は、公共または個人支払者に対する保険政策ではないが、このような結果をどのように回避するかは不明であり、研究が我々の候補製品の販売に何らかの影響を与える場合、そのような製品または治療のための条件が研究のテーマである場合には不明である。有効性研究を比較すると,競争相手の 製品が優位であることが示されれば,我々の候補製品の販売にも悪影響を与える可能性がある.私たちの候補製品の第三者精算減少brあるいは第三者支払者は、私たちの候補製品をカバーしないことを決定することは、医師の候補製品に対する使用を減少させ、私たちの販売、運営結果、財務状況に実質的な悪影響を与える可能性がある。
患者保護·平価医療法案は,2010年に“医療·教育負担能力調整法案”により改正された(総称してACA“ は2010年3月に公布された。ACAを公布する目的は未加入者のカバー範囲を拡大することであり、同時に全体の医療コストを制御することである。医薬製品では,他の事項に加えて,ACAは医療補助計画がカバーする薬品の業界リベート を拡大·向上させ,連邦医療保険D計画のカバー要求を変更した。まだACAの製薬会社への影響を完全に予測することはできません。多くのACA改革はまだ完成していない法定条項を実施するために詳細な法規を公布する必要があるので、医療保険·医療補助サービスセンターは公開されており、発表されたACA法規や政策を分析して、変更すべきかどうかを決定しています。さらに、米国最高裁はACAの大部分の合憲性を維持しているにもかかわらず、一部の州はACAのいくつかの条項を実施しない意向を示しており、一部の国会議員はACAの廃止に努力している。これらのチャレンジは,ACAの一部として実施される変更の不確実性 を増加させる.また,ACAに対する現在の法的挑戦や,ACA廃止への国会の努力は,ACAの一部として公布された立法変化の不確実性を増加させている。
そのほか、一部の外国の国/地区では、薬品の提案定価は必ず承認されなければ、合法的に発売されることができない。薬品の定価管理の要求は国によって異なる。例えば、いくつかのEU司法管轄区はプラスリストとネガティブリスト制度を実施しており、このような制度の下で、製品 は価格を取り決めてから販売することができる。精算または定価の承認を得るためには、その中のいくつかの国/地域の臨床試験を完成し、特定の候補製品のコスト効果を現在利用可能な治療法と比較する必要があるかもしれない。他の会員国たちは会社が自分の薬品価格を固定することを許可するが、会社の利益を監視する。各国の定価制度のこのような差 はEU加盟国間の価格差をもたらす可能性がある。薬品に対して価格制御や精算制限を行うことが保証されないいかなる国/地域でも、私たちのどの製品に対しても割引の精算と定価手配が許可されています。歴史的に見ると、EUで発売された製品は米国の価格構造に沿っていない。EUでは、全体的な医療コスト、特に処方薬の下振れ圧力が大きくなっている。そのため,新製品の参入ハードルが高くなり,政府や他の公共あるいは個人支払者から精算されていない薬品を使用することは不可能である。
41
他の医療法やコンプライアンス要件は
米国では,FDAに加えて,我々の活動は,CMS,米国衛生·公衆サービス部の他のbr部門(例えば,監察長室),米国司法省br,司法省内の個別の米国検事室,州や地方政府など,様々な連邦,州,地方当局によって規制されている可能性がある。例えば、販売、マーケティング、科学/教育補助計画は、“社会保障法案”、“虚偽クレーム法案”、“健康保険携行性と責任法案”のプライバシー条項、および同様の州法の反詐欺·乱用条項に適合しなければならない。 定価と返却計画は、1990年の“総合予算調節法”および1992年の“退役軍人医療法案”の医療補助税還付要求に適合しなければならない(“VHCA)は、各条が修正されている。将来の製品が連邦供給スケジュールの許可ユーザ に使用可能である場合、他の法律および要件が適用される。VHCAによると、製薬会社はアメリカ退役軍人事務部とアメリカ国防省、公衆衛生サービスとある個人公共衛生サービス指定実体を含む複数の連邦機関に特定のbr薬品を提供しなければならず、MedicareとMedicaidを含む他の連邦援助計画に参加することができる。さらに、米国国防総省がそのTRICARE計画に提供するいくつかの調達は、返金システムによって割引価格を提供しなければならない。VHCA参加により、定価データを提出し、複雑な法定公式に基づいて割引とリベートを計算し、連邦調達法規に管轄される政府調達契約を締結する必要がある。
製品を商業的に流通させるためには、ある州で製品を州のメーカーおよびディーラーに搬送することを含む州の法律を遵守しなければならない。この法律は、そのようなメーカーまたは卸売業者が州に営業場所がなくても、1つの州に薬品の製造業者および卸売業者を登録することを要求する。いくつかの州はすでに立法を公布し、製薬会社のマーケティングコンプライアンス計画を確立し、州政府に定期報告を提出し、定期的に販売とマーケティング活動を公開するか、あるいはその販売代表を登録することを要求した。いくつかの州はまた他の法律を公布し、いくつかの他の販売とマーケティング行為を禁止する。私たちのすべての活動は連邦と州消費者保護および不正競争法律によって制限される可能性があります。 同様に、これらの活動は、EUまたはEU加盟国の法律または私たちが製品を運営、製造または流通する他の国/地域の法律に基づいて、許可または許可要件または他の法律要件によって制限される。
環境法遵守のコスト
私たちの運営は、空気や水中への汚染物質排出の法律、危険物質と廃棄物の管理と処分、汚染された場所の整理など、様々な連邦、州、地方、外国の環境法律によって規制されている。私たちが将来環境法に違反したり、環境法に基づいて責任を負うと、整理費用、罰金、民事または刑事制裁、第三者損害や人身傷害クレームなど、巨額のコストを招く可能性があります。私たちは環境法を遵守するためのいかなるコストや影響を知らない。
気候変動に関する法規
我々の業務は医薬製品の研究·開発に集中しており,その大部分の研究·開発は我々の施設外で行われ,アウトソーシング契約研究機関や大学が行っている。したがって、私たちは気候変化に関するどんな規制も私たちの運営に影響を与えないと予想する。
しかし,より頻繁で悪天候イベントや水供給挑戦が発生する可能性があり,我々のパートナーや将来のサプライヤーの施設に影響を与える可能性がある。私たちは将来気候変動によって私たちのパートナーと未来のサプライヤーの施設やサプライチェーンに実際のリスクを与えないという保証はありません。私たちは潜在的な天気関連リスクや他の自然災害に対する私たちの脆弱性を定期的に検討し、それに応じて私たちの評価を更新する。私たちの審査によると、これらの潜在的なリスクは現在、私たちの運営に実質的な ではないと思います。
42
従業員と人的資本管理
2023年3月31日現在、私たちと私たちの子会社の全5人のフルタイム従業員がいます。従業員の1人はイギリスに、4人の従業員はアメリカにいる。
また,限られた数の臨時アルバイト従業員や科学コンサルタント,コンサルタント,サービスプロバイダを雇用し,主に学術機関や契約研究組織を介している。
私たちは一度も労働を停止したことがなく、私たちの従業員たちは集団交渉協定のカバー範囲にもいないし、労働組合代表もいない。私たちは私たちが従業員と良い関係を持っていると信じている。
私たちの人的資本資源br目標は識別、採用、維持、激励と統合既存と新入社員、コンサルタントとコンサルタントを含む。私たちの株式と現金インセンティブ計画の主な目的は、株と現金に基づく報酬奨励を付与することで、これらの人々を激励してできる限りのことをし、私たちの目標を実現することで、株主価値と会社の成功を増加させることです。
企業の歴史
形成する
私たちは2016年9月7日に設立され、デラウェア州の法律に基づいて設立された空白小切手会社です。私たちの設立の目的は、合併、資本株式交換、株式購入、資産買収などの他の同様の業務と1つまたは複数の運営企業との合併を実現することです。brは設立以来、潜在的な目標企業を決定するための努力は特定の業界に限定されないにもかかわらず、医療および関連健康業界の運営会社の買収に集中しています。
初公募株
2017年6月7日、当社初公開(“初公募株)、当社は、2017年6月23日の引受業者選挙後に引受業者に売却された1,500,000株を含む11,500,000株を1株10.00ドルの買い取り価格で売却し、その超過配給選択権を十分に行使し、毛収入115,000,000ドルを生成した。どれも“職場.職場会社普通株の20分の1 を含め、企業合併完了後に会社普通株の20分の1を得る権利(br})そうだそうだ)と、引当可能な引受権証と、会社普通株の40分の1株を購入することができるbr(“株式証を公開する“)”各公共株式承認証は所有者に1/40ドル当たり5.75ドルの使用価格で1株の普通株を購入する権利を持たせるこれは…。1株(1株当たり230.00ドル)は、調整することができます。 公募株式証を行使する際に断片的な株式を発行しません。公開株式証は初公募完了から12ヶ月で行使でき、業務合併完了後5年(2025年11月6日)に満了する。
会社は30日前に通知した後、公共株式証1部当たり0.01ドルの価格ですべて及び一部の公共株式証明書を償還することができる(“30日間両替期間 )は、普通株が償還通知日前の第3取引日に終了した30取引日以内の任意の20取引日以内の最終販売価格が1株360.00ドル以上である場合にのみ、当該等公開株式証明書に関する普通株式の有効登録声明があり、30日間にわたる償還期間内に当該普通株式に関する現行目付説明書があることが条件となる。もし会社が上述したように公共株式証の償還を要求した場合、会社管理層は公共株式証の行使を希望するすべての所有者に要求する権利があるだろう“現金ベースがありません。“すべての所有者がその公共持分証を行使することを要求するかどうかを判断する際に”現金のない上に経営陣は、会社の現金状況、発行された公開株式証の数、公開株式証を行使した後に発行された最高数の普通株が会社株主に与える希薄な影響など、他の要因を考慮する。企業合併が完了した後、各権利保持者は1/200(1/200) 普通株式を獲得する。権利交換時には,断片的な株式は発行されていない.
43
私募する
IPOが終了すると同時に、KBLIVホールディングスLLC(“スポンサー?スポンサー“),引受業者は合計450,000個の未登録 単位を購入した(”個人単位“)は、単位あたり10.00ドル、私募で4,500,000ドルの毛収入を生む。また、2017年6月23日、当社は1単位10.00ドルで追加52,500個の個人単位の販売を完了し、これらの単位は保証人と引受業者がbrを購入し、毛収入525,000ドルを生成した。このうち,保証人は377,500個のプライベートユニットを購入し,引受業者は125,000個のプライベートユニットを購入した.個人単位からの収益は、信託口座に保有するIPOの純収益に加算される信託口座“)”個人単位(その構成要素証券を含む)は、企業合併(以下の定義を参照)が完了してから30日後、プライベート単位内に含まれる引受権証(“その構成要素証券を含む)が譲渡、譲渡または売却されてはならない(”私募株式証明書“償還することはできません。保証人、引受業者、またはその譲り受けを許可された人が所有すればいいです。私募株式引受証が保証人、br引受業者又はその譲渡許可者以外の者が所有している場合、この等持分証は自社で償還することができ、この等持分者は初回公開発売先に含まれる引受権証と同じ基準で行使することができる。また、私募株式権証が引受業者又はその指定者又は共同経営会社が所有している限り、IPOに関連する登録声明の発効日から5年後に行使することはできない。そうでなければ,私募株式証の条項や条項は,IPO単位の一部として販売されている引受権証の条項と同じであり,純現金決済条項はない.
業務合併
2019年7月25日、私たちは“企業合併協定”(随時改訂)を締結しました企業合併協定“)、KBL と連結子会社(”合併子)、180生命科学会社(f/k/a 180生命科学会社)(“180“)、Katexo 製薬会社(”Katexo)、CannBioRex製薬会社(CBR製薬)、180 Treateutics(br}L.P.(‘’180 LPKatexoやCBR Pharmaと一緒に180社の子会社180生命科学と一緒に180の締約国)と180の締約国株主代表としてLawrence Pemble株主代表“)”業務統合協定に記載されている業務統合(“業務合併2020年11月6日に閉鎖され終業する)。 業務合併協議(これを含む)により、合併子会社は180社に合併し、そのうち180社は引き続き存続実体と当社の完全子会社としている(合併する“)”取引が終了する前に、180生命科学社はデラウェア州でその登録証明書の改訂証明書を提出し、その名称を180生命会社に変更し、わが社(私たちが業務合併に入る際にKBL合併会社IVと呼ばれ、私たちの会社名を180生命科学社に変更しました)。
180は2019年1月28日にデラウェア州で登録設立されました。業務合併が終了する前に、180は3つの子会社で運営されている:180 LP、デラウェア州有限組合企業で、2013年9月6日に設立された。Katexoは、2018年3月7日にカナダブリティッシュ·コロンビア州に登録設立された会社、およびCBR Pharmaは、2018年3月8日にカナダブリティッシュ·コロンビア州に登録設立された会社である。
2019年7月,180および180 LP,KatexoとCBR Pharmaはそれぞれ1つの会社再編を完了し,これにより,180 LP,Katexo,CBR Pharmaは180 LS(再編成する“)”KatexoとCBR Pharmaに関する会社再編計画は合意に基づいて完了した“ビジネス会社法”です(ブリティッシュコロンビア州)
2020年11月6日( )“締め切り)であり、当社は2020年11月5日に株主特別総会を開催して業務合併を完了し、会議では当社株主が業務合併の提案を審議し、承認する。業務合併契約(含まれる)によれば、合併付属会社は、180と合併して180に組み込まれ、180は、存続エンティティおよび当社としての完全子会社として継続される。合併は2020年11月6日に施行されます(この場合、 有効時間また,マージの終了を本稿ではと呼ぶ終業するこれまで,180社はデラウェア州で会社登録証明書改訂証明書を提出し,その名称を180生命会社に変更したが,KBL合併会社第4社は180生命科学会社と改称した。
44
発効時期には、発効時間前に発行および発行された普通株1株当たりの権利を自動的に当社の約8.41892株の普通株を受け取る権利に変換し、1株当たり額面0.0001ドル(このなどの普通株は企業合併協定により180名の普通株株主に発行することができる)合併対価株“)”これまでに180名の普通株主に合計874,737株の普通株を合併対価株式として発行しており、信託 株式を含む(以下の定義)。同様に、発効時間前に発行·発行された180株の優先株の1株当たり基礎株式は、1株会社のC類特別投票権株式または1株会社のK類特別投票権株式(当該等株式、)に変換される特別議決権株“)”特別投票権株式は、その所有者が、任意の特定の事項、命題または問題について、カナダ子会社CannBioRex Puracheco ULCおよびKatexo Puracheco ULCがそれぞれ時々発行する交換可能な株式数に相当する総投票権を獲得する権利を有するようにする。
統合の結果として, 既存の交換可能株式(総称して“と呼ぶ)交換可能株交換可能株式を管理するCannBioRex Puracheco ULCまたはKatexo Puracheco ULC(Br)の定款における株式条項に基づいて,合併の交換比率を乗じて交換可能普通株となるように調整した。交換可能株式は、株式保有者が経済的に普通株式保有者に相当する配当金及び他の権利を獲得する権利を有することを可能にし、交換可能株式保有者は会社の株主会議で投票する権利がある。交換可能な株式交換時にその保有者に発行するために、合計264株の普通株式を予約する。
業務合併協定によると、合併対価株式のうち52,500株(当該等株式、預かり取り分“)ホスト·アカウントに入金されました(”第三者預かり口座)企業合併協定下での会社の賠償権利の担保及び独占支払い源として、これらの権利は、企業合併終了後12ヶ月以内に同じ株主に発行される予定であったが、クラウス博士のこれらの株式に対するクレームはまだ行われている。
業務合併により、180の前株主が当社の持株株主となり、180は当社の完全子会社 となる。この事業合併は逆合併とみなされるため、180は会計·財務報告において買収側とみなされる。
取引が終了したため、会社は信託口座(定義は以下参照)から9,006,493ドルの資金を抽出し、40,824株の株式を償還した。
株を逆分割する
2022年12月15日、180生命科学会社株主特別総会において、会社株主は、会社の第2回改正と再改訂された会社登録証明書の修正案を承認し、私たちの普通株の発行済み株式と発行済み株を逆方向に株式分割し、1株当たり額面0.0001ドル、4対1と20対1の間で、4対1と20対1を含み、具体的な割合は我々の取締役会またはその正式に許可された委員会が自ら決定する。改正案が承認されてから2023年12月15日までのいつでも(“株主管理局”)。2022年12月15日、会社取締役会(“取締役会”)は、株主管理局と共に、我々の普通株の逆株式分割に影響を与える割合が1:20(“逆 株式分割”)である2つ目の改正と再署名した会社登録証明書の修正案を承認した。
特別会議と取締役会の承認に続いて、私は2022年12月15日にデラウェア州州務卿に改訂及び再改訂された2つ目の会社登録証明書(“改訂証明書”)を提出し、株式の逆分割を実施した。修正案証明書のコピーは、添付ファイル3.1として本ファイルに添付され、参照によって本明細書に組み込まれる。
改訂証明書 によると、逆株式分割は2022年12月19日午前12:01に発効した。アメリカ東部時間(“発効時間”). 会社の普通株式或いは公共株式証株式の取引記号“ATNF”及び“ATNNW”は変更がなく、 は株式の逆分割に関係している。
45
改訂証明書brは、私たちの普通株の許可株式数を減少させず、私たちの普通株の額面を変更することもなく、私たちの普通株の投票権や他の条項も修正していません。
逆株式分割に関する断片的な株式は発行されておらず,登録されている株主はもともと断片的な株式を獲得する権利があり, はその断片的な株式を最も近い全体の株式に丸める権利がある.
本報告は20株1株逆分割の影響を遡及的に反映している。
会社の構造
次の図は私たちの現在の組織構造を示しています
私たちについて
私たちの主な実行オフィスはカリフォルニア州パロアルト、94306、ElCamino Real、Bld.4、Suite 200にあり、電話番号は(650)507-0669です。ウェブサイトを維持していますWwwww.180 life ciences.comそれは.私たちは、私たちのウェブサイトの情報や私たちのウェブサイトを介してアクセスできる情報を引用することによって本報告書に組み込まれていません。あなたはそれを本報告の一部と見なしてはいけません
私たちの起業法を開始します
2012年4月、JumpStart 私たちの企業創業法案(“JOBS法案”)が法律として制定されました。その他の事項を除いて、“雇用法案”は、“新興成長型企業”は、5年に及ぶ期間内に特定の財務開示と管理要求を免除し、小企業に新たな融資形態を提供すると規定している。2022年12月31日、私たちはもはや“新興成長型会社”ではない。
46
第1 A項。リスク要因です
リスク要因をまとめる
私たちは私たちの業務に関連するリスクと不確実性に直面しており、その多くのリスクと不確実性は私たちがコントロールできない。特に、私たちの業務に関連するリスクは、
| ● | 私たちは臨床段階のバイオテクノロジー会社で、2022年12月31日と2021年12月31日までの年度には何の収入もなく、近い将来収入は発生しないと予想されています |
| ● | 私たちは、私たちの運営を支援するための追加の短期的および長期的な融資が必要であり、必要に応じてそのような融資を調達する能力、そのような融資の条項(利用可能であれば)、それに関連する潜在的な重大な希釈、およびそのような融資に関連する契約および制限を遵守する必要がある |
| ● | 私たちは将来の候補製品の成功に依存しています。その中のいくつかの製品は規制の承認または商業化に成功できない可能性があります。私たちの新製品の製造過程に問題があり、および/または私たちは製造法規を遵守できなかった、あるいは私たちの製造コストは意外に増加しました。私たちの製品の流通問題;そして私たちの製品を十分にマーケティングすることができませんでした |
| ● | 私たちのビジネスの成長、私たちのこのような成長を維持する能力、私たちの成長を管理し、私たちの成長戦略を実行することの困難に関連するリスク |
| ● | 以前に述べた財務諸表および無効な制御および手続きに関する負債、および現および前任上級管理者および取締役の賠償に関する費用および支出 ; |
| ● | 私たちのキーパーソンへの依存と従業員やコンサルタントを引き付ける能力を持っています |
| ● | 私たちよりも多くの資源と経験を持つ会社からの激しい競争のリスク ; |
| ● | 私たちは、企業候補薬の臨床開発と規制承認に関する不確実性を含む、私たちの候補製品の規制承認を得る能力と、これに関連するスケジュールとコスト、登録と臨床試験の完了の潜在的な遅延、FDAとMHRAによって提起された問題を含む | |
| ● | リスク:私たちの将来の候補製品 規制部門の承認を得ると、予想される市場受容度を達成できない可能性があり、新製品から収入を創出する能力を制限する |
| ● | 現在の未解決および将来のクレームおよび訴訟、将来の政府調査および他の訴訟の結果は、私たちの業務および運営結果に悪影響を及ぼす可能性がある |
47
| ● | 私たちのほとんどのライセンスプロトコルは、そのようなライセンス知的財産権を使用、所有、および/または使用する権利を許可者および/または取引相手に提供する |
| ● | 臨床前研究と比較的に早い臨床試験は必ずしも未来の結果を予測するとは限らないかもしれないし、有利な結果がないかもしれない;私たちのマーケティング経験は限られていて、私たちのいかなる候補製品が未来に承認されても、私たちが未来にそれを商業化することに成功する能力は未知である;業務中断は私たちの未来の候補製品の開発過程を遅延させる可能性があり、そして私たちの製品販売を混乱させる可能性がある |
| ● | 第三者支払者は、将来のどの製品にも保証範囲と十分な精算レベルを提供しないかもしれない |
| ● | 現在の未解決訴訟の結果、将来可能な政府調査、および私たちの業務および運営結果に悪影響を及ぼす可能性のある他の訴訟を含む訴訟責任(製品責任訴訟、株主訴訟、および規制事項を含む)、判決、損害賠償、罰金および処罰を含む |
| ● | セキュリティホール、データ損失、および他の可能性は、私たちが重要な情報にアクセスすることを阻止したり、私たちを責任や損害の中断に直面させる可能性があります |
| ● | 臨床試験に関連するリスク、これらの試験は高価で、時間がかかり、不確定であり、変更、遅延または終了が容易であり、異なるbr解釈、候補薬剤の試験、試験、申請または承認中の遅延、および/または将来性のある候補薬のためのbr承認を得る能力、およびそれに関連するコストを受ける可能性がある |
| ● | 私たちは連邦、州、外国の医療法律と法規、そしてこのような医療法律と法規の実施または変更を含む既存と未来の規則と法規を遵守することができる |
| ● | 私たちは市場で私たちの未来の候補製品または私たちのノウハウを保護する能力と、第三者が私たちの知的財産権侵害の疑いのあるクレームと責任を十分に保護しています |
| ● | 税法および制御物質法を含むが、これらに限定されないが、いかなる法律および法規も遵守されていないことを含む、国と他の管轄区域との間の法律および法規の違い、および法律または法規の変化 |
| ● | 私たちの役員、役員、コンサルタント、科学者の間の利益の衝突 |
| ● | 私たちは、いくつかの事前に約束された契約義務と制限に関する処罰を遵守できなかった |
| ● | 将来の資金調達による赤字、発行された転換可能な証券、およびこのような未来の発行/販売が私たちの証券価値に与える下振れ圧力 ; |
| ● | 新冠肺炎の流行と他の将来可能な流行病が私たちの業務に与える負の影響 |
48
| ● | 私たちの証券の非常に不安定な性質と潜在的な流動性の不足 |
| ● | 私たちの会社登録証明書は高級管理者と取締役に対する賠償を規定し、高級管理者と取締役の責任を制限し、brが株主の承認なしに優先株を許可することを許可し、いくつかの他の反買収条項と排他性 フォーラム条項を含む |
| ● | 私たちは、私たちの普通株式と引受権証がナスダックに上場する能力と、アメリカ証券取引委員会とナスダック規則と要求を遵守するコストを維持します |
| ● | ネットワークセキュリティ攻撃や他のデータセキュリティイベントを含む我々の情報技術システムに障害が発生し、私たちの業務の運営を大きく乱す可能性があります |
| ● | 私たちは他の会社を買収するかもしれません。これは私たちの経営陣の注意を分散させ、私たちの株主の追加的な希釈を招く可能性があります。そうでなければ、私たちの運営を乱し、私たちの経営業績を損なうかもしれません。もし私たちがどんな買収をすれば、彼らは私たちの業務を混乱させたり、私たちの業務にマイナスの影響を与えるかもしれません | |
| ● | 潜在的な景気後退、およびマクロ経済、地政学的、健康と業界の傾向、流行病、戦争行為(持続的なウクライナ/ロシア衝突を含む)および他の大規模な危機の影響、および米国債務上限問題における議会の行き詰まりまたは将来のアメリカ政府が予算の相違によって停止する可能性のある潜在的な影響を含む高インフレ、金利上昇および景気後退の影響 |
| ● | 私たちは最終的に私たちの経営業績を改善したり、私たちの証券価値を増加させない用途に運営資本と未来の資金を使用するかもしれません |
| ● | 私たちの成長は私たちと第三者の戦略的関係の成功にある程度かかっている。 |
あなたは私たちの普通株に投資することに大きな危険があるということを知らなければならない。私たちの普通株に投資することを決定する前に、あなたはこのような危険要素を慎重に考慮しなければならない。
以下のいずれかの リスクが発生した場合、私たちの業務、財務状況、運営結果、または他の見通しは重大な悪影響を受ける可能性があり、 のいずれのリスクの発生も私たちの成功可能性に大きな影響を与える可能性がある。このような状況が発生すれば、私たちの普通株の市場価格(あれば)が下落する可能性があり、潜在投資家は私たちの普通株へのすべてまたは一部の投資を失うことになる。
私たちの業務、財務状況、経営結果は、以下に述べるリスクと不確定要素を含む様々なリスクと不確実性の影響を受ける。本節で議論する要因 は,単独でも総合的にも,我々の実際の結果が期待や歴史的結果と大きく異なる要因をもたらす可能性がある.私たちの業務、財務状況、または運営結果はこのようなリスクのいずれかの重大な悪影響を受けるかもしれない。このすべての要素を予測したり識別することは不可能だ。したがって、以下のリスク要因の説明は、我々の業務に適用されるすべての潜在的リスクまたは不確実性について完全な議論ではない。
49
私たちの業務運営に関するリスク
私たちの現在の現金残高は、2023年第2四半期まで、私たちの計画中の業務運営に資金を提供するのに十分です。利用可能な追加資本がなければ、計画された業務運営を実行できない可能性があり、計画された業務運営の変更を余儀なくされる可能性があり、または株主に悪影響を及ぼす可能性のある他のbr行動をとる可能性がある。
私たちは臨床段階バイオテクノロジー会社で、現在収入がありません。したがって、私たちの業務は、私たちが計画している業務運営に資金を提供するために必要な現金を生成しません。 私たちは、(I)FDAおよび/またはMHRAによって承認された製品を開発し、そのような製品を商業化すること、 (Ii)私たちの候補製品に関する研究開発活動に資金を提供し、規制部門の承認を得ること、(Iii)私たちの知的財産権を保護すること、(Iv)高い人材を誘致し、維持すること、(V)競争圧力に効果的に対応すること、 および(Vi)補足業務または技術を取得することを必要とする。
私たちの将来の資本需要 は、(I)私たちの研究、開発、商業化努力に関連する範囲、持続時間および支出 ;(Ii)私たちのプロジェクトの持続的な科学的進歩、(Iii)潜在的な協力または許可取引の結果、 があれば、(Iv)競争技術の発展、(V)私たちの特許地位、および(Vi)私たちの製品の規制承認 過程を含む多くの要素に依存する。
公開または私募株式発行、債務融資または戦略連盟、許可手配を通じて大量の追加資金を調達し、計画中の業務運営に資金を提供する必要があります。私たちはもしあれば、私たちに有利な条項で追加的な資金調達を得ることができないかもしれない。一般的な市場状況、および持続的な新冠肺炎の大流行、利上げおよびインフレ、および世界的な衝突(例えば、ウクライナとロシアの間の持続的な衝突)、および米国の債務上限問題で議会が行き詰まっているか、または将来的に予算の相違によって米国政府の停止を招く可能性のある潜在的な影響は、資本市場から融資を求めることを困難にする可能性があり、どの融資条項も私たちの株主の保有量や権利に悪影響を及ぼす可能性がある。例えば、私たちが株式証券を発行することで追加資金を調達すれば、私たちの株主がさらに希釈することになり、彼らの投資価値を大きく希釈することができるかもしれない。任意の株式融資は、私たちが発行した転換可能な証券または行使可能な証券の転換または行使価格を低下させる可能性もあり、これは、大量の追加の普通株式 の発行(または潜在的発行)をもたらす可能性がある。また、私たちに追加資金を提供する条件として、将来の投資家は、既存の株主よりも高い権利を要求し、付与される可能性がある。債務融資(実行可能であれば)は、限定的な契約に関連する可能性があり、これは、将来の業務活動を展開する私たちの柔軟性を制限する可能性があり、破産が発生した場合、株式証券所有者が私たちの資産の任意の割り当てを受ける前に支払う可能性がある。私たちは、私たちの技術または候補製品の権利を放棄すること、または連合、合弁企業、または合意を通じてbrライセンスを付与することを要求されるかもしれません。条項は、追加資金を調達するために私たちに不利です。もし が十分な資金がない場合、私たちは私たちのbr業務に関連する1つまたは複数の計画活動を延期、減少またはキャンセルしなければならないか、または私たちの運営を終了しなければならないかもしれない。このような行動は私たちの普通株の市場価格を下げるかもしれない。
私たちは追加的なbr資本が必要になりますが、これは商業的に受け入れ可能な条項では得られないかもしれません。これは、持続的な経営を継続することができる企業としての私たちの能力があるかどうかに対する疑問を引き起こします。
2022年12月31日までの累計赤字は107,408,545ドル,運営資本は3,270,608ドルであり,2022年12月31日までの年度の純損失は38,726,259ドル,運営活動に使用された現金は12,127,585ドルであった。2023年3月29日現在、私たちの手元の現金は約270万ドルです。合併財務諸表を作成する際には、会社は引き続き経営を続ける企業とすると仮定しています。私たちは収入が発生していないので、私たちは債務返済と運営コストの支払いのために大量の資本を調達する必要がある。同社は最近、2022年7月(650万ドルの総収益)と2022年12月(600万ドルの総収益)に株式売却で資金を調達したが、追加の必要な資本を調達できる保証はなく、これらの資本が優遇条項で獲得される保証もない。
競争の激しい業界で、私たちは新しい企業を発展させるために固有のすべての重大なリスクに直面している。長期的な運営歴史と私たちの市場の新しい性質が不足しているため、私たちはすべての関連する収入フローを含む当社の業務戦略を成功するまで、運営損失が予想される。私たちは収益性を達成したり、相当な収入を生み出すことができないかもしれない。
私たちの現在の毎月の現金需要支出は約900,000ドルだ。全体的に、私たちは、私たちの製品の研究開発とマーケティングを支援し、拡大し、将来の臨床試験を支援し、債務を返済するために大量の追加資本資金を必要とし、追加設備と開発コスト、支払い義務、オフィス空間、業務管理システムの資本支出を提供し、私たちが計画している製品収入フローが完全に実施され、私たちの運営コストを相殺し始めるまで、他の運営コストを支払うと信じています。
50
私たちが設立して以来、私たちは株式と債務融資の収益で私たちの運営に資金を提供してきた。私たちは流動性の問題を経験したが、その理由の一つは、私たちが受け入れ可能な条件で十分な資本を調達する能力が限られているからだ。私たちは従来、普通株に転換可能な株式と債務融資を売却して私たちの運営に資金を提供し、このリスクを減らすための多くの努力を投入してきた。予測可能な未来には、私たちは株式を発行して私たちの運営に資金を提供し、私たちの運営費用に資金を提供する必要があると予想される。もし私たちが運営利益を実現できない場合、あるいは私たちが他の形態の融資を得ることに成功しなかった場合、私たちは運営費用の削減と現金節約の代替措置を評価しなければならない。
これらのことは,本稿で述べた監査人が発行した日から今後12カ月以内に経営を継続する能力があるかどうかが疑われる。付随する総合財務諸表はアメリカで一般的に受け入れられている持続経営会計原則に基づいて作成され、この原則は正常業務過程における資産現金化と負債返済を考慮したものである。したがって,総合財務諸表には資産回収性や負債分類に関するいかなる調整も含まれておらず,会社が経営を継続できなければ,これらの調整が必要となる可能性がある。本明細書に含まれる連結財務諸表には、持続的な経営脚注も含まれる。
しかも、可能性がある限り、私たちの取締役会は非現金対価格を使用して義務を履行しようと試みるだろう。多くの場合、非現金 対価格には、私たちの普通株の制限株式、優先株、または私たちの普通株式を購入する承認証が含まれると考えられます。br}私たちの取締役会は、株主の行動や投票を行わない権利がありますが、ナスダックの規則と規定を守らなければなりません(任意の取引が、私たちが当時発行した普通株の20%を超える普通株または私たちが当時発行した株式の20%以上に相当する投票権を発行する場合は、一部の例外を除く)場合は、株主の承認を得る必要があります。すべてまたは部分的に許可されているが発行されていない普通株、優先株または株式承認証を発行して、普通株 を購入する。また、私たちは、私たちの普通株の株式を売却することで資金を調達しようとすることができ、将来的には市場を下回る価格で販売されるかもしれない。これらの行動は、既存の株主の所有権利益を希釈することになり、普通株の帳簿価値をさらに希釈する可能性があり、このような希釈は実質的である可能性がある。このような発行は、株式が既存の管理層を支援するために努力している当事者またはエンティティに発行される可能性があるので、既存の管理職が私たちのコントロールを維持する能力を強化するのに役立つかもしれない。
私たちは大きな流動性需要を持っていて、増加しており、追加的な資金が必要だ。
新たな研究開発計画、臨床試験、持続的な製品商業化努力と将来の候補製品の発売により、研究開発、管理、行政費用(法律費用を含む)、運営のための現金は引き続き大幅に増加し、将来的には大幅に増加する可能性がある。私たちは、潜在的な規制部門のマーケティング申請の承認を支援し、将来の候補製品の商業化に資金を提供するために、私たちの運営を支援するために、より多くの資金を調達する必要があります。
私たちの将来の資金需要の額と時間は多くの要素に依存するだろうが、これらに限定されない
| ● | FDAおよび/またはMHRA承認の時間(ある場合)、および将来の候補製品の他の国際市場での承認(あれば) |
| ● | 私たちの製品の収入または贈与または他の出所の収入を販売する時間と金額 |
| ● | 私たちの臨床試験や他の製品開発計画の進捗とコスト |
| ● | 販売、マーケティング、および流通能力を確立またはアウトソーシングするコスト |
| ● | 私たちの将来の候補製品のアウトソーシング、成長、商業製造供給スケジュールのコストと時間 |
| ● | 私たちの将来の候補製品に関連する任意の特許主張および他の知的財産権の費用の提出、起訴、弁護、および実行; |
| ● | 競争の技術と市場発展の影響 |
| ● | 人員、施設、設備の要求; |
| ● | 私たちが確立する可能性のある他の任意の協力、許可、共同販売促進、または他のスケジュールの条項および時間。 |
51
私たちは、複数のソース(例えば、キャッシュフローの運営およびさらなる公開および/または非公開発行の収益)から私たちの将来の資本需要に資金を提供することが予想されていますが、これらの資金源のいずれも割引条項で提供されるか、または全く提供されないことを保証することはできません。また 上記のすべてのソースから資金を集めることができても、調達した金額は私たちの将来の資本需要を満たすのに十分ではないかもしれません。
私たちはbrの追加的な資本を調達する必要があるかもしれません。これらの資本は優遇的な条件で獲得できないかもしれません。もしあれば、私たちの株主の株式を希釈し、私たちの運営を制限したり、私たちの業務運営能力に悪影響を与えたりします。
私たちは深刻または長期的な経済低迷のようなマクロ経済状況による追加融資を私たちに有利な条項で得ることができないかもしれない。資本市場の中断、不確実性、または変動は、私たちの資本コストを増加させたり、事業運営に必要な資金を調達する能力を制限したりする可能性がある。中断は、FRBの政策と行動、為替レート懸念、インフレ、経済低迷または不確定性、通貨政策、金融機関の倒産、米国債務管理懸念、および政府の閉店、欧州と世界の主権債務懸念、その他の世界的または地政学的事件またはその他の要素を含む米国債務上限と予算紛争によって引き起こされる可能性がある。現在のマクロ経済状況は米国銀行業界に負の影響を与えており、例えば、最近のシリコンバレー銀行とSignature Bankの閉鎖とFDIC倒産である。私たちはこれらの銀行には何の口座もなく、これらの銀行との業務関係もありませんが、これらのような事態の発展は米国銀行システムによる他の中断が私たちにマイナスの影響を与える可能性があります。
我々の運営結果 は貨幣価値変動の悪影響を受ける可能性がある.
私たちはドル以外の通貨で を支出する。同社のレポート通貨はドルです。ある子会社のビットコインはカナダドル(“CAD”)またはポンド(“GBP”)である。これによる換算調整は,株主資本の中で他の全面的な収益を累積する構成要素として確認されている.包括収益は、所有者投資または所有者への分配以外のすべてのソースの実体権益変動 として定義され、上述した外貨換算調整 を含む。当社は2022年および2021年12月31日までに他の全面(赤字)収入 を記録し、それぞれ(3,702,963ドル)および180,554ドル(外貨換算調整)となった。
外貨収益 と外貨建て取引(会社間取引を含む)による損失を経営実績 に計上する。同社は2022年と2021年12月31日までの年度にそれぞれ(12,777ドル)と(69ドル)外貨取引(赤字)を確認した。このような金額は、添付されている総合経営報告書と全面赤字のうち、一般費用と行政費用に分類されている。
私たちが支払う費用(そして将来収入を得る)の通貨とドルの間の価値変化は、不利なbr費用が私たちの損益表に記録される可能性があります。
グローバル経済状況 は会社の業務、経営業績、財務状況、成長に重大な悪影響を及ぼす可能性がある。
インフレ、成長減速または衰退、新たな関税または増加した関税、財政·通貨政策の変化、信用の引き締め、より高い金利、高い失業率、為替変動、および米国議会が米国債務上限問題で行き詰まっているか、または将来の米国政府が予算分岐によって停止する可能性のある潜在的な影響を含む不利なマクロ経済状況は、会社の運営、費用、資本獲得の機会、および会社計画における将来の製品の市場に重大な悪影響を及ぼす可能性がある。また、消費者自信と支出は、金融市場の変動、負の金融情報、不動産と抵当ローン市場の状況、収入或いは資産価値の低下、燃料とその他のエネルギーコストの変化、労働力と医療コスト、その他の経済要素の不利な影響を受ける可能性がある。
また、世界や地域の経済状況の不確実性 や低下は、会社の資金源、サプライヤー、パートナーに大きな影響を与える可能性がある。潜在的な影響には、財務不安定;会社の将来計画された製品の運営と購入を支援するために信用を得ることができない;および債務不履行が含まれる。
経済環境の低迷は、当社の株式売却や新債発行能力が制限され、流動資金を減少させ、当社の金融商品の公正価値を低下させる可能性もある。上記及びその他の経済要因は、当社の業務、経営業績、財務状況及び成長に重大な悪影響を及ぼす可能性がある。
52
私たちの業界とより広範なアメリカ経済は2022年に予想以上のインフレ圧力を経験し、これはサプライチェーンの持続的な中断、労働力不足、地政学的不安定と関係がある。これらの状況が続けば、将来の経営業績やキャッシュフローは実質的な悪影響を受ける可能性がある。
供給制限、サプライチェーンの中断、需要の増加、アメリカの労働力の十分な雇用に関連する労働力不足、高インフレとその他の要素により、“2022カレンダー2022”のある材料、製品と輸送コストが大幅に上昇した。供給と需要brは,複数の地政学的事件(ロシアとウクライナ間の持続的な衝突を含む)により世界的なエネルギー供給が中断され,ファンダメンタルズはさらに悪化している。サービス、材料と輸送コストも相応に増加し、アメリカ各地に普遍的に存在するサプライチェーンとインフレ問題は運営コストの増加を招いた。最近のサプライチェーン制限とインフレ圧力は私たちの運営コストに悪影響を及ぼす可能性があり、私たちの将来の製品コスト、コンサルティングコスト、支出にマイナスの影響を与える可能性があり、これは私たちの業務、財務状況、運営結果、キャッシュフローに重大な悪影響を及ぼす可能性がある。
経済的不確実性 は、私たちが資本を獲得する機会に影響を与え、および/またはそのような資本のコストを増加させる可能性がある。
世界経済状況が引き続き動揺し、不確定である理由は、将来の経済状況に対する消費者の自信、景気後退と貿易戦争への懸念、エネルギー価格、金利変動、消費信用の獲得可能性とコスト、政府刺激計画の可用性とタイミング、失業率レベル、インフレ上昇、税率、およびウクライナとロシアの間で2022年2月に開始された戦争、および議会が米国債務上限問題で膠着状態に陥っているか、または未来の米国政府が予算の相違によって停止する可能性のある潜在的な影響を含む。これらの状況は依然として予測不可能であり,将来の資金調達能力に不確実性 をもたらしている。必要な資金が将来的に利用できなくなったり、コストが高くなったりすれば、私たちの業務、将来の運営結果、および財務状況に実質的な悪影響を及ぼす可能性がある。
特定の訴訟事項に関連する合併前役員および上級管理職保険証の下のいかなる金額も受け取っていないかもしれません。
2022年6月29日、KBLの合併前役員及び高級社員保険証書保証人AmTrust 国際保険者DAC(“AmTrust”)は、米国カリフォルニア州北区地域裁判所 に当社に対する宣言的救済訴訟(“宣言的救済行動”)を提起し、取締役及び上級職員保険リストの下でのAmTrustの義務の申告を要求した。救済発表訴訟では,AmTrustは合併により当社は対象保険証書下の被保険者ではなく,当社がAmTrustへの回収費用が合併前に発生した事項 に及ぶことを求めているにもかかわらず,AmTrustが主張している。2022年9月20日、当社はAmTrustに対して、AmTrustが対象取締役と上級管理者保険リストに基づいて当社に対して負担している保険責任に違反し、少なくとも200万ドルの補償性損害賠償、および適用される懲罰的賠償を要求する答弁と反訴を提起した。また,会社 はその超過保険引受会社自由専門保険会社(“自由”)に対して第三者訴えを行い,AmTrustが当社の保険責任金額を使い切った後,自由もその保険契約範囲の履行を要求される.AmTrustは2022年10月25日に会社反訴に対する回答 を提出し,自由会社は2022年10月27日に第三者苦情に対する回答を提出した。
2022年11月22日、会社はAmTrustとFreedomに対する簡易裁決動議を提出した。*動議は十分なプレゼンテーションを得て、2023年3月9日に公聴会 を行った。裁判所はこの事件を受理し、まだ裁決が下されていない。当社はAmTrustを起訴する理由が強いと信じていますが、当社がこの訴訟で勝つ保証はありません
私たちは私たちの未来の候補製品の成功に依存しており、その中のいくつかは規制部門の承認や商業化に成功しないかもしれない。
私たちの成功は、私たちの開発計画を通じて、私たちの将来の候補製品の開発と商業化に成功する能力にかかっています。デュプテロン筋拘縮症の候補製品の治療と、私たちの線維化治療および抗腫瘍壊死因子、CBD誘導体、およびα7 nAChR開発プラットフォームによって開発された他の任意の候補製品を含む。私たちはアメリカや他の場所で規制部門の承認を受けた製品を開発することができないかもしれない。FDA、MHRA、EMA、または任意の他の規制機関がこれらの 候補製品を承認することは保証されない。
著者らが未来の候補製品の商業化に成功できるかどうかは、著者らが臨床前と他の非臨床研究と臨床試験を成功的に完成できるかどうか、及びFDA、MHRA、EMAと類似の外国監督機関の監督管理許可を得ることができるかどうかに依存する。規制過程における遅延は、私たちの業務、運営結果、財務状況に実質的な悪影響を及ぼす可能性がある。
53
私たちが任意の潜在的製品から収入を得ることができるかどうかは、私たちが規制部門の承認を得て、多くの他の要求を満たすことができるかどうかにかかっており、私たちは決して収入を生み出すことに成功したり、利益を達成したりすることはできないかもしれない。
私たちが利益を達成して利益を維持する能力は、私たちが収入を作ったり、他の業務発展計画を実行したりする能力にかかっています。私たちが規制部門の承認を得て、私たちが開発または開発可能な候補製品 を商業化することに成功しない限り、私たちは相当な収入(あれば)が生じないと予想される。ある程度、成功した商業化は多くの重要なマイルストーンを実現する必要があり、臨床試験で安全性と有効性を証明し、これらの候補製品の監督管理許可、製造、マーケティングと販売を獲得し、あるいは他の合意に達して私たちが監督管理許可を得ることができる製品を商業化し、任意の発売後の要求を満たし、そして個人保険あるいは政府支払人から私たちの製品の精算を得ることができる。これらの活動に関連する不確実性とリスクのため、私たちは収入の時間と金額(あれば)、さらなる損失の程度、またはいつ利益を達成する可能性があるかを正確かつ正確に予測することができない。私たちはこのような活動で決して成功しないかもしれないし、たとえ私たちが成功しても、私たちは私たちが利益を達成するのに十分な収入を生むことができないかもしれない。たとえ私たちが利益を達成したとしても、私たちは四半期や年間収益性を維持したり向上させることができないかもしれない。
私たちは利益を達成できず、利益を維持することは私たちの普通株の市場価格を下げる可能性があり、資金を集め、業務を拡大し、私たちの製品を多様化したり、運営を継続する能力を弱める可能性があります。
我々は最近業務を拡大しており,将来的には組織の規模や複雑さを増加させる必要があり,成長戦略の管理や成長戦略の実行に困難がある可能性がある。
私たちの現在の管理、人員、そしてシステムは私たちの業務計画と未来の成長を支援するのに十分ではないかもしれない。私たちは、将来の製品に対して第1、第2、第3段階の臨床試験を行い、商業組織および商業インフラを構築するために、フルタイム従業員に相当する人数を増やす必要がある。これらの将来の活動により、私たちの業務運営の複雑さは大幅に増加すると予想される。私たちは、私たちの計画の研究、開発、製造、製造、商業化活動を支援するために、私たちの科学、製造、販売とマーケティング、管理、コンプライアンス、運営、財務、その他の資源を発展させ、拡大する必要があるだろう。
私たちは私たちの運営、成長、様々なプロジェクトを効果的に管理する必要があります
| ● | 私たちの業務、財務、管理、規制およびコンプライアンス制御および報告システムとプログラムを引き続き改善します |
| ● | 十分な数の優秀な従業員を引きつけて維持します |
| ● | 経済的に効率的な方法で私たちの商業化活動を効果的に管理する(現在、私たちの臨床試験の試験と開発は非常に費用効果がある) |
| ● | 私たちの開発作業を効率的に管理しながら、請負業者や他の第三者に対する契約義務を履行します。 |
私たちは、コンプライアンス計画、臨床試験管理、法規制事務、製剤開発、および他の薬物開発機能に関連するタスクを含む複数のタスクを当社のために複数のタスクを実行し続けています。私たちの成長戦略は、これらおよび将来の他のタスクを実施するために、コンサルタントや請負業者への使用を拡大する必要があるかもしれません。もし私たちが新入社員の募集やコンサルタントや請負業者の使用を拡大することで私たちの組織を効果的に拡大することができなければ、私たちは私たちが計画した研究開発、製造、商業化活動を効果的に実行するために必要な任務を実行できない可能性があるため、私たちの研究、開発、商業化目標を実現できない可能性がある。
54
私たちは、以前に記載された財務諸表および/またはそのような再説明をもたらす前の管理職のいくつかの行動に責任を負う。
我々は、2020年12月31日に現在のForm 8−K報告書を提出し、2021年2月3日に別の現在のForm 8−K報告書を提出し、業務統合前に発見されたKBL関連事項のため、いくつかの歴史的財務諸表が信頼できないことを発表した。そこで、このような財務諸表にエラーがあるため、2020年6月30日までの3ヶ月と6ヶ月と、2020年9月30日までの3ヶ月と9ヶ月の財務諸表を再記述した。これらのエラーは、証券取引委員会に提出されたこのような財務諸表が2020年6月30日と2020年9月30日までの元の四半期報告書で発見されたbr}である。これらの再記述はKBL経営陣の行為の結果であり、KBL経営陣の責任でもあると考えられるが(いずれも当社に雇われ続けることはない)、私たちbrはこのような重述のために株主訴訟、SEC行動、罰金と処罰、格付け引き下げ、マイナス宣伝、および重要な顧客、従業員、管理者の誘致と維持の困難を受ける可能性がある。また、我々の証券 の取引価格は、財務諸表を再報告する必要がない同様の場合の会社よりも低い可能性がある。
重大な一次取引によるプロセスを適切に調整することができず、財務諸表の誤った陳述を招く可能性がある。
私たちの年間監査過程で、私たちは記録する前に、brにエラーが発生したことを発見し、私たちの公共株式証明書の公正な価値が一部誇張された。このエラーは2022年の財務諸表提出前に訂正されました。2022年12月31日までの財務諸表の株式証明書の公正価値は正しく述べられていると信じていますが、類似したミスは私たちの財務状況と運営結果に重大な悪影響を及ぼす可能性があり、前期または未来の財務諸表を再申告する必要があるかもしれません。
今後しばらくの間、運営結果は大きく変わるかもしれない。
私たちの財務結果は予測できません。他の原因によって変動する可能性があります。その理由は、私たちの未来の候補製品の商業販売、私たちの製品開発目標とマイルストーンの実現状況、臨床試験登録と費用、研究開発費用、および契約製造と契約が支払いの時間と性質を研究することです。我々のコストの大部分は年ごとにあらかじめ定められており, の部分は我々の研究開発コストが高いためである.このため、将来の収入の小幅低下は第1四半期の財務業績に比例しない影響を与える可能性がある。
私たちは私たちの重要な人員と従業員を引きつけて維持する能力に依存している。
私たちの未来の成長と成功は、私たちが従業員を募集、維持、管理、激励する能力にかかっている。私たちは私たちの最高経営責任者ジェームズ·N·ウディ博士、私たちの共同議長マーク·フェルドマン卿博士、ローレンス·スタンマン医学博士、私たちの最高科学者ジョナサン·ロスバード博士、私たちの科学者ジャグディップ·ナンチャハルなど、私たちの現在の管理と科学者に非常に依存している。経験豊富な管理職を採用したり引き留めることができないことは、私たちの業務計画を実行する能力に悪影響を与え、私たちの経営業績を損なう可能性があります。私たちの業務は専門的な科学と管理性質を持っているため、私たちは私たちの合格した科学、技術と管理者を誘致し、維持する能力に大きく依存している。生物技術領域の合格人材に対する競争は非常に激しく、私たちは引き続き業務発展に必要な合格者を誘致し、維持することができないかもしれない、あるいは適切な後継者を募集する。
55
私たちの製造過程で発生する問題 私たちの未来の化学実体、製造法規を守らない、あるいは私たちの製造コストが意外に増加して、私たちの業務、運営結果、財務状況を損なう可能性があります。
私たちは、商業用途および臨床試験のために、将来のCBDデリバティブおよびα7 nAChR計画における候補製品の製造と供給を担当している。我々の将来の候補製品の製造はGMPと国際司法管轄区の他の法規要求 を遵守する必要がある。私たちが未来の候補製品を製造することに成功した能力は、厳格なbr制御プロセスとプログラムの下で、製品情報、改ざん防止証拠、偽造防止機能を含む完成品およびラベルと包装を製造することに含まれる。さらに、私たちは臨床試験ロットと上場ロットを含む私たちのロット間の化学的一貫性を確実にしなければならない(承認されたら)。このような一致性を証明するためには、典型的な生産制御と臨床データが必要かもしれない。私たちはまた私たちのロットが複雑なバージョン仕様に適合することを確実にしなければならない。もし私たちが規制規範に従って私たちの未来の候補製品を生産できない場合、あるいは私たちの製造プロセスが破損、br損失、その他の原因で中断されたり、私たちの製造施設の規制検査に合格できなかった場合、私たちは臨床試験のために需要を満たしたり、十分な製品を提供できない可能性があり、これはまた、将来の候補製品をタイムリーに商業化したり、コスト競争力を持つ能力を損なう可能性がある(もしあれば)。
私たちは私たちの候補製品の需要を満たすために私たちの製造能力を開発し、拡張することができないかもしれません。FDA、MHRAまたは他の外国の規制機関は、私たちまたは私たちの契約メーカーの施設が私たちの製品と候補製品 を生産するのに適していることを受け入れないかもしれません。私たちの製造過程のどんな問題も、私たちの業務、運営結果、財務状況に重大な悪影響を及ぼす可能性があります。
我々とCelltrion Healthcareとの了解覚書 は双方の最終合意に至らない可能性がある.
2021年9月、私たちは生物製薬会社Celltrion Healthcareと拘束力のない了解覚書を締結し、私たちが行っている抗腫瘍壊死因子製品の開発のための生物類似薬を供給した。これまで双方はこのような関係について最終的な合意に達しておらず,最終的には予想された条項に沿ってこのような合意に達しない可能性がある.もし私たちがCelltrion Healthcareと双方の同意の最終条項を達成できない場合、私たちは抗腫瘍壊死因子生物類似薬物の代替供給業者を見つける必要があり、私たちは代替供給者を見つけることができないかもしれない、あるいはこの代替供給業者が要求する条項は現在予想されている割引に及ばないかもしれない。上記のいずれの状況も、私たちの業務、運営結果、および財務状況に大きな悪影響を及ぼす可能性があります。
私たちは、私たちよりも多くの資源と経験を持つ会社からの激しい競争に直面することが予想され、505(B)(2)条の申請に従って私たちの製品をマーケティングしようとする競争相手の競争に直面する可能性があります。
製薬業界は競争が激しく、変化が速い。ますます多くの競争相手と潜在的な競争相手が市場に参入するにつれて、この業界は絶えず拡大し、発展している。これらの競争相手と潜在的な競争相手の多くはわが社よりも多くの財務、技術、管理、研究開発資源と経験を持っている。その中のいくつかの競争相手と潜在競争相手は薬品開発においてわが社よりも多くの経験を持っており、検証プログラムと 規制事項を含む。また、開発が成功すれば、私たちの将来の候補製品は、大手brや有名会社が提供する製品と競争し、これらの会社はわが社や私たちの協力パートナーよりも多くのマーケティングや販売経験と能力を持っています。特に,Insys Treateutics,Inc.は乳児痙攣を治療するCBDを開発している(はい。“)、および潜在的な の他の兆候。Zgenix社はすでに2つのDrave症候群を治療する小用量フェンフルラミン3期試験の陽性データを報告し、すでにこの製品によるLennox Gastaut症候群の3期試験を開始した。Biocodexは最近FDAの監督管理許可を得て、Dravet症候群を治療する薬物司チプタール(Diacomit)に応用した。わが社よりも多くの資源を持つ他の会社は将来的に似たような計画を発表するかもしれません。さらに、医療用大麻業界の会社は、FDA承認されていないCBD製剤を提供しており、これは、私たちの将来の候補製品と競争しようとするかもしれない。
56
私たちの多くの競争相手は以下の点で顕著に多くの財務と技術資源、経験、専門知識を持っている
| ● | 研究と開発 |
| ● | 臨床前試験 |
| ● | 臨床試験の設計と実施; |
| ● | 規制手続きと承認 |
| ● | 生産と製造 |
| ● | 承認された製品の販売とマーケティング。 |
私たちの業界の主な競争要因は:
| ● | 組織技術の質と広さ |
| ● | 組織の管理と組織戦略の実行 |
| ● | 従業員の技能と経験を組織し、技能と経験のある従業員を募集し、維持する能力 |
| ● | 組織の知的財産権の組み合わせ |
| ● | 能力の範囲、目標の識別と検証から薬物の発見と開発、製造とマーケティングまで |
| ● | 大量の資本資源を獲得し、発見、開発、商業化活動に資金を提供することができる。 |
さらに、競合他社 は、わずかに縮約されたbr}セキュリティプロトコルである505(B)(2)条を介して、私たちの医薬品バージョンの販売を申請することができる。国家医薬品監督管理局“第505条(B)(2)条:以前に承認された薬剤の新しい適応または新しい剤形のような修正された医薬品に代表される出願を提出することができる。505(B)(2)節−出願は、以前に承認された薬剤の安全性および有効性の調査結果 および505(B)(2)出願人によって得られた、以前に承認された薬剤修正をサポートする情報 に依存する可能性がある。準備505(B)(2)申請は、新しいデータおよび情報に完全に基づいて秘密保護プロトコルを準備するよりもコストが低く、時間が少ない可能性がある。第505条(B)(2)条:出願はANDAと同様の特許認証手続を遵守しなければならない。
もし私たちが競争に成功できなければ、私たちのビジネス機会は減少し、私たちの業務、運営結果、財務状況は実質的な損害を受ける可能性がある。
57
私たちの将来の候補製品 が承認されれば、予想される市場受容度に達しない可能性があり、新製品から収入を創出する能力を制限しています。
製品開発が成功し、規制部門の承認を得ても、私たちが十分な収入を生むことができるかどうかは、医師と患者の私たちの製品に対する受け入れの程度にかかっている。もし私たちの未来の候補製品が必要な規制承認を得たら、それらは予想される市場受容度と収入レベルに達するという保証はできません。市場のいかなる製品に対する受容度は多くの要素に依存し、適応声明、監督機関が製品ラベル上で要求する警告及び新しい 競争製品を含む。市場受容度は、商業用途における有効性と安全性の持続的な展示、医師 が製品を発行したいかどうか、第三者支払人(例えば政府医療計画と個人第三者支払人)の精算状況、製品価格、監督管理機関が規定する任意の承認後のリスク管理活動の性質、競争、 およびマーケティングと流通支援の影響を受ける可能性がある。また,米国での流通は,精算支援センターと閉鎖流通ネットワークで契約した専門薬局の適切な表現に依存する。発表時の非効率的あるいは非効率的な米国流通モデルは需要を満たすことができず、収入損失を招く可能性がある。一方の国/地域における製品の成功および受容度は、他の国/地域での活動の負の影響を受ける可能性がある。もし私たちの方法をアメリカ市場の臨床試験に適応させて、EMA、MHRAまたは他のヨーロッパ規制機関のbr需要を満たすことができない場合、あるいはヨーロッパの定価と精算交渉や決定を支持するために必要な衛生経済学と結果研究データを生成することができなければ、私たちはEMA/欧州委員会またはMHRAから私たちの製品のマーケティング許可を得ることが困難かもしれませんし、私たちのbr製品は国レベルで定価と精算承認を得ることが困難かもしれません。市場が私たちの製品を受け入れることを阻害したり制限したりする要素は、私たちの業務、運営結果、財務状況に大きな悪影響を及ぼす可能性があります。
われわれの抗腫瘍壊死因子や線維化計画におけるすべての特許は使用方法特許であり,われわれの許可を得ずに生体類似薬の使用につながる可能性がある。
著者らの最先端の薬物開発プラットフォームの成功は著者らの使用方法特許の実行可能性に依存しており、現在市場には多くの生物類似の抗腫瘍壊死因子br薬物があるからである。もし私たちが組成物特許を取得し、これらの特許を強制的に実行できなければ、私たちが抗腫瘍壊死因子プラットフォームから収入を産生する能力は大きく制限される可能性があり、競争相手は私たちの研究を利用して競争相手の薬物を市場に投入する可能性があり、これは私たちの市場シェアを減少させる。
我々のほとんどのライセンスプロトコルは、そのようなライセンス知的財産権を使用および/または使用する権利を許可者および/または取引相手に提供する。
私たちのほとんどのライセンスプロトコル は、そのようなライセンス知的財産権を使用および/または利用する権利を許可者および/または取引相手に提供し、場合によっては、そのような知的財産権、ノウハウ、および研究成果の所有権も彼らに提供する。したがって、私たちは私たちの許可協定のある当事者 と競争するかもしれません。彼らは私たちの候補製品を貨幣化、販売、または流通する独占的な権利を持っていないかもしれません。 は使用および販売地域によって制限されるかもしれません。上記のいずれかまたは全部は、私たちの運営およびキャッシュフロー結果に重大な悪影響を与え、最終的には私たちの証券の価値に影響を与える可能性があります。
更に多くの患者データの出現に伴い、著者らの臨床試験中の中期データ、TOPLINEデータと初歩データは変化する可能性があり、そして監査と検証プログラムの影響を受け、これは最終データの重大な変化を招く可能性がある。
著者らは時々私たちの臨床前研究と臨床試験の初歩的、中期或いは主要なデータを公開開示する可能性があり、これらのデータは当時使用可能なデータに対する初歩的なbr分析に基づいており、結果と関連する結果と結論は患者登録と治療の継続及び更に多くの患者データの獲得に従って変化する可能性がある。例えば、任意の候補製品に対する臨床前試験、第1段階および第2段階の臨床試験のいずれの積極的な結果も、必ずしもこのような候補製品の計画brまたは将来の臨床試験の結果を予測できるとは限らない。製薬と生物技術業界の多くの会社は臨床前と早期臨床開発で積極的な成果を得た後、臨床試験で重大な挫折に遭遇し、著者らは私たちが類似した挫折に直面しないことを確定することができない。これらの挫折は,臨床試験進行中の臨床前発見や臨床試験における安全性や有効性観察(有害事象を含む)などによるものである。前の初期データまたは中間データと将来の中期データまたは最終データとの間の不利な差は、私たちのビジネスの将来性を大きく損なう可能性があります。臨床前研究や臨床試験完了後にbr背線データを公表することも可能であり,特定の研究や試験に関するデータをより網羅的に審査すると,これらのデータが変化する可能性がある。私たちはまた、私たちのデータ分析の一部として、仮定、推定、計算、および結論を、すべてのデータを全面的かつ詳細に評価する機会がないか、または受け取る機会がないかもしれない。したがって,我々が報告した 中期,背線または予備結果は,同じ研究の将来の結果と異なる可能性があり,あるいは他のデータを受信して十分な評価を行った後,異なる結論や 考慮要因がこれらの結果を合格させる可能性がある。初期、中期、またはバックラインデータも引き続き審査および検証手順を受けることになり、これは、最終データが私たちが以前に発表したbrデータと大きく異なる可能性がある。したがって、最終データ が利用可能になる前に、予備データ、中期データ、およびバックラインデータを慎重に見るべきである。
58
さらに、規制機関を含む他の機関は、私たちの仮定、評価、計算、結論または分析を受け入れないか、またはデータの重要性を異なる方法で解釈またはトレードオフする可能性があり、これは、特定の計画の価値、特定の候補製品または製品の承認または商業化 およびわが社の全体的な状況に影響を与える可能性がある。さらに、私たちが開示する特定の研究または臨床試験に関する情報を選択することは、一般的に広範な情報に基づいており、あなたまたは他の人は、私たちが開示に含めるために重要な情報または他の適切な情報として決定することに同意しない可能性がある。そのほか、著者らの臨床データの解釈或いは著者らは臨床前の体外と体内モデルに基づいて得られた結論は不正確であることが証明される可能性があり、臨床前と臨床データは異なる解釈と分析の影響を受ける可能性があるため、しかも多くの会社は彼らの候補製品が臨床前研究と臨床試験で満足できるbrと考えているが、依然としてFDAの承認或いはヨーロッパ 委員会が授与した上場許可を得られなかった。
もし私たちが将来の臨床試験で積極的な結果を得られなかった場合、このような候補製品の開発スケジュール、規制承認と商業化の将来性、および対応する私たちの業務と財務の見通しは、実質的な悪影響を受けるだろう。
私たちのマーケティング経験は限られていて、未来の候補製品が承認されても、私たちはそれらを商業化することに成功できないかもしれません。
私たちが収入を作る能力は、最終的には、承認された製品を販売し、十分な第三者精算を得る能力にかかっている。私たちは今私たちの製品をマーケティングして販売した経験がありません。私たちの未来の製品の商業成功は、医者が私たちの未来の製品を患者に提供する意志、支払者が私たちの未来の製品に支払う意欲と能力、実現された価格設定レベル、患者の未来の製品に対する患者の反応、および私たちの未来のマーケティングパートナーが販売を創造する能力を含む、私たちの未来の製品の商業成功は多くの私たちがコントロールできない要素に依存する。将来的に私たちの未来の製品またはFDA、MHRA、または他の規制機関によって承認された任意の候補製品を商業化することに成功するために、必要な人員、システム、br手配および能力を確立または維持することができる保証はない。成功したマーケティング、販売、精算能力を確立または維持できなかった場合、あるいは第三者と成功したマーケティング計画を達成できなかった場合、私たちの製品収入は影響を受ける可能性がある。
もし私たちが将来承認した任意の製品の価格が低下した場合、あるいは政府や他の第三者支払人が保険を提供しておらず、十分な精算レベルがなければ、私たちの収入と利益の見通しは影響を受けるだろう。
病状を治療するために処方された患者は、通常、その処方薬に関する費用の全部または一部を第三者支払者によって精算される。国際市場の精算制度は国や地域によって大きく異なり、精算は通常国/地域に基づいて承認されなければならない。政府医療保健計画(例えばMedicareとMedicaid)と商業支払者のカバー範囲と十分な精算は新製品の受容度に重要である。カバー範囲の決定は、臨床的および経済的基準に依存する可能性があり、より成熟またはより低い治療代替製品が利用可能になった場合、またはその後に利用可能である場合、これらの基準は歓迎されない。将来の候補品のために保険を受けても,それによる精算料率 は,患者が受け入れられないほど受け入れられないほど高い共同支払いを必要とする可能性がある。保険を提供していない場合や患者の費用の大部分を支払うのに十分でない場合、患者は私たちの将来の候補製品を使用しないかもしれません。
また,我々の将来の候補製品の米国市場では,第三者支払者の薬物処方が得られるかどうか,あるいは第三者支払者が保証·精算の薬物リストを提供するかどうかに大きく依存する。業界競争はこのような調合 に組み入れられることは往々にして製薬会社に下振れ価格圧力をもたらす。さらに、価格の低い後発薬または他の代替品がある場合、第三者支払者は、その処方に特定のブランド薬を含むことを拒否するか、または他の方法で患者のブランド薬 の使用を制限することができる。
59
第三者支払者は、海外でも国内でも、政府のものでも商業的でも、ますます複雑な方法を開発して医療コストをコントロールしている。現在の環境は会社に圧力を与え、製品の定価が適切と思われる価格より低いことを要求している。もし私たちが最適化価格より低い価格で未来の候補製品を販売すれば、私たちの未来の収入 と全体の成功は負の影響を受けるかもしれない。また、米国では、第三者支払者の間に統一された薬品引受や精算政策はない。そのため、私たちの将来の候補製品の保証範囲とbrの精算は支払先によって異なる可能性があります。そのため、保証範囲決定過程は通常時間がかかり、高価な過程であり、私たちのbrを使用して未来の候補製品を使用するために単独で各支払人に科学と臨床支持を提供する必要があるが、保証範囲を獲得することは保証されない。もし私たちが将来の候補製品の保証範囲と十分な支払いレベルを得ることができない場合、医師は彼らがどれだけ処方されるか、あるいはどのような場合にこれらの製品を処方または管理するかを制限するかもしれず、患者は購入を拒否するかもしれない。これは私たちが候補製品を商業化することに成功する能力に影響を与え、私たちの収益性、運営結果、財務状況、将来の成功に悪影響を及ぼす可能性がある。
また、候補製品の開発と商業化について第三者と協力することを選択した場合、私たちのパートナーは、精算承認を得る可能性を高めるために、私たちの製品のbr価格を下げることを選択するかもしれません。多くの国/地域では、精算が承認される前にのみ、製品が商業的に使用されることができ、ある国/地域では、交渉過程が12ヶ月を超える可能性がある。さらに、ある国/地域の価格決定および精算決定は、他の国/地域の決定の影響を受ける可能性があり、これは、他のいくつかの国/地域の強制値下げおよび/または追加の精算制限をもたらす可能性があり、これは、販売および利益に悪影響を及ぼす可能性がある。もし、国/地域が実施する価格が、私たちまたはパートナーが利益を得るのに十分でない場合、パートナーは、これらの国/地域での製品の発売を拒否したり、製品を市場からリコールしたりすることを拒否する可能性があり、販売および収益性に悪影響を及ぼす。
業務中断は将来の候補製品の開発過程を遅延させ、私たちの製品販売を混乱させる可能性があります。
私たちの将来の製造施設、在庫、実験室施設は火災、窃盗、その他の原因で失われ、将来の候補製品の需要を満たしたり、製品開発活動を継続したり、業務を展開する能力に悪影響を及ぼす可能性があります。パートナーに商業製品を供給できないことは、パートナーが製品供給責任 を負担する権利があることを含む不良な結果を招く可能性がある。私たちがこのような業務中断を賠償するために保険を受けても、このような保険は、在庫のいかなる重大な財産や意外な損失によって私たちの業務に与える損害を完全に補償するのに十分ではないかもしれません。
もし私たちの製品に対する責任訴訟が成功すれば、私たちは大量の責任を負い、私たちの将来の候補製品の商業化を制限することを要求されるかもしれません。
私たちは私たちの製品責任に対するクレームや訴訟を受けたことがありませんが、私たちは人体臨床試験で私たちの未来の候補製品をテストすることに関連する潜在的な製品責任リスクに直面しています。そして私たちは未来に私たちのマーケティングと流通の司法管轄区でクレームリスクに直面します。私たちがアメリカと他のところで商業マーケティングと私たちの製品の流通を始めた時、私たちはもっと多くの人からのクレームに直面するかもしれません。 将来、個人は私たちの未来の候補製品の一つが被害を与えたと主張する責任クレームをするかもしれない。私たちは適切だと思う予防措置をとる予定ですが、私たちにどんな製品責任訴訟を起こしても、重大な責任は避けられないかもしれません。集団訴訟や予期せぬ副作用を有する薬物に基づく個人訴訟では多額の判決が下された。私たちは保険を購入して製品責任を負うことを計画していますが、製品責任クレームに対して会社の弁護に成功できなければ、あるいはこのような保険カバー範囲が不足していれば、私たちは大量の責任を負います。是非曲直或いは最終結果にかかわらず、責任クレームはすべて私たちの製品に対する需要の減少、名声損傷、臨床試験参加者の脱退、訴訟コスト、製品リコールコスト、金銭 賠償、責任保険コストの増加、収入損失と業務中断を招く可能性がある。
60
私たちの従業員は以前または将来、監督管理基準と法律要求の違反を含む不当な行為または他の不当な活動に従事する可能性がある。
私たちは従業員詐欺や他の不適切な行為の危険に直面している。従業員の不正行為は、FDA、米国証券取引委員会または監察長事務室の規定を故意に遵守しないこと、または任意の他の適用規制機関の規定を遵守しないこと、FDAまたは米国証券取引委員会に正確な情報を提供していないこと、米国証券取引委員会の届出文書に正確な情報を開示していないこと、適用された製造基準を遵守していないこと、他の連邦、州または外国の法律法規、情報またはデータを正確に報告すること、または許可されていない活動を開示することを含むことができる。従業員の不当行為はまた不適切な使用情報に関連する可能性があり、臨床試験過程で得られた情報、あるいは薬品の不正流用を含む可能性があり、これは政府の調査と私たちの名声を深刻に損害する可能性がある。私たちは“道徳的基準”を採択したにもかかわらず、従業員の不適切な行為がいつも識別されて阻止されるわけではない。これらの禁止された活動を検出し、防止するための予防措置は、未知または未管理のリスクまたは損失を効果的に制御することができないか、または政府の調査またはそのような法律や法規を遵守しないことによる他の行動または訴訟から私たちを保護することができない可能性がある。もし私たちの会社にこのような行動を取ったら、私たちは私たちの会社を守ることに成功したり、私たちの権利を維持することができませんでした。これらの行動は、巨額の罰金や他の制裁を加えることを含めて、私たちの業務に大きな影響を与える可能性があります。
米国の“海外腐敗防止法”やその他の反腐敗法律、輸出規制法、税関法、制裁法、その他の我々の業務を管理する法律を守らなければならない。もし私たちがこれらの法律を守らなければ、私たちは民事または刑事処罰、他の救済措置、法的費用を受ける可能性があり、これは私たちの業務、運営結果、財務状況に悪影響を及ぼすかもしれない。
我々の業務は米国の“反海外腐敗法”(“反腐敗法”)を含む反腐敗法律によって拘束されている“反海外腐敗法”)と、私たちが業務を展開している国/地域に適用される他の反腐敗法律。“海外腐敗防止法”およびこれらの他の法律は、一般に、私たちの会社、私たちの従業員および仲介機関が政府関係者または他の人に贈賄、収賄、または他の禁止されたお金を支払い、ビジネスのbrを獲得または保留し、またはいくつかの他の業務的利点を得ることを禁止している。私たちと私たちのビジネスパートナーは、“海外腐敗防止法”に潜在的に違反するリスクのある多くの司法管轄区で業務を展開しており、私たちは第三者との協力や関係に参加しており、これらの第三者の行為は、“海外腐敗防止法”や現地の反腐敗法律で規定されている責任を負わせる可能性がある。さらに、私たちは将来の規制要求の性質、範囲、または効果を予測することができず、私たちの国際業務はこれらの要求によって制約される可能性があり、既存の法律が管理または解釈される可能性がある方式を予測することもできない。
私たちはまた、アメリカ、カナダ、イスラエル、イギリス政府とEU当局が管理する法規、適用される輸出規制法規、国と人員に対する経済制裁、税関要求と通貨両替法規を含む、私たちの国際業務を管理する他のbrの法律と法規に制限されています。総称して貿易規制法律と呼ばれています。
しかし、“海外腐敗防止法”や他の法律要件、貿易統制法を含むすべての適用された反腐敗法律を遵守することを完全に効果的に確保することは保証されません。もし私たちが“海外腐敗防止法”や他の反腐敗法律や貿易規制法を遵守しなければ、私たちは刑事と民事処罰、返還と他の制裁と救済措置、そして法的費用を受ける可能性があり、これは私たちの業務、財務状況、運営結果、流動性に悪影響を及ぼす可能性がある。同様に、米国や他の当局によるいかなる潜在的な“海外腐敗防止法”違反、その他の反腐敗法律の調査も、私たちの名声、業務、財務状況、運営結果に悪影響を及ぼす可能性がある。
61
セキュリティホール、データ損失、および他の中断は、私たちの業務に関連する敏感な情報を危険にさらし、私たちが重要なbr情報にアクセスすることを阻止したり、私たちに責任を負わせたりする可能性があり、これは私たちの業務と私たちの名声に悪影響を及ぼす可能性があります。
通常の業務プロセスでは、法律で保護された患者健康情報、クレジットカード情報、私たち従業員に関する個人識別情報、知的財産権、および独自の業務情報を含む敏感なデータを収集して保存したい。これらの重要な情報のセキュリティ処理、保存、維持、転送は、私たちの運営および業務戦略に重要であり、私たちはこれらの情報を保護するために大量のリソースを投入します。不正なアクセスや漏洩から敏感な情報を保護する措置をとっているにもかかわらず、私たちの情報技術およびインフラは、ハッカーの攻撃を受けやすいか、従業員のミス、汚職、または他の中断によるウイルス、侵入または中断、またはプライバシーおよびセキュリティ規定の遵守におけるミスを受けやすい可能性がある。このようなウイルス、侵入、または中断は、私たちのネットワークに危険を及ぼす可能性があり、そこに格納されている情報は、許可されていない当事者のアクセス、開示、紛失、またはbrによって盗まれる可能性がある。我々は,このようなセキュリティイベントやプライバシーやセキュリティ規定に違反する行為を防止し,必要に応じて検出·対応するための措置を策定した.しかしながら、将来的には、そのような情報のアクセス、開示、または他の損失は、HIPAAおよびEU一般データ保護条例のような個人情報のプライバシーを保護する法律に基づいて法的クレームまたは訴訟を引き起こす可能性がある(“GDPR)、政府の法執行行動、規制処罰。許可されていないbrのアクセス、紛失、または伝播は、私たちのサンプルの処理、テスト結果の提供、セキュリティデータの共有と監視、支払人または患者への支払い、顧客支援サービスの提供、研究開発活動の提供、会社の財務情報の処理と準備、私たちの業務の様々な一般的および行政的側面を管理する能力を含む可能性があり、私たちの名声を損なう可能性があります。これらはいずれも、私たちの業務、財務状況、および運営結果に悪影響を及ぼす可能性があります。
すべてのEU加盟国に適用されるGDPRは、データ保護ルール違反に対する巨額の罰金を含み、新たで変化するデータ保護ルールの遵守を保証するための追加のメカニズム を確立することを要求する可能性がある。GDPRは複雑な法律であり、監督指導はまだ発展中であり、 はGDPRを臨床試験にどのように応用するか、あるいは私たちがそこから個人データを得ることができる他の取引に関する背景 を含む。GDPRは私たちのコンプライアンスコストを増加させ、より大きな法的リスクをもたらす。
私たちの研究開発計画と候補製品は開発中です。したがって、私たちは私たちの候補製品の開発や商業化に成功するかどうかを予測できない。
私たちの臨床段階の候補製品および私たちの他の候補薬はbrが商業化される前に大量のさらなる投資と規制承認が必要になるだろう。私たちのすべての候補製品はFDA、MHRA或いは他の類似外国監督機関の標準と要求に符合する臨床試験設計が必要であり、私たちの臨床試験のために適切な終点と患者、及びbr}追加の臨床開発、臨床前と製造活動の管理、監督管理の許可を得、製造供給を獲得し、商業組織を設立し、大量の投資と重大なマーケティング仕事を選択し、その後、私たちは製品販売から任意の収入を得ることができる。FDA、MHRA、または他の同様の外国規制機関の規制承認を得るまで、私たちは私たちの候補製品のマーケティングや普及を許可することはできませんし、私たちはいかなる候補製品の規制承認も得られないかもしれません。その時、私たちの候補製品が必要な規制承認を得る可能性があれば、私たちの候補製品に関連する疾患を治療する看護基準が変化している可能性があるので、私たちの全面的な承認計画を修正する必要があり、私たちの製品の商業的受容度は看護基準の変化によって制限される可能性があります。
私たちが必要なbr資金を獲得したり、協力を確立したりして、私たちの候補製品やパイプライン資産を後期臨床開発できるようにしても、このような臨床開発が成功するかどうか、あるいは規制部門の承認を得るかどうか、あるいは私たちの任意の候補製品を商業化して収入を生み出すことができるかどうかを決定することはできません。臨床前試験と早期臨床試験の成功は後の臨床試験が成功することを確保できず、臨床試験過程は著者らの製品 候補製品がその提案の用途に対して安全かつ有効であることを証明できない可能性がある。このようなどんな失敗も、私たちが任意の1つまたは複数の候補製品のさらなる開発を放棄し、他の候補製品の開発を遅延させる可能性がある。臨床試験後期段階の候補製品は必要な安全性と有効性特徴を示すことができないかもしれないが、すでに臨床前研究と初歩的な臨床試験を通じて進展を得たが。我々の臨床試験のいかなる遅延または終了も遅延し、FDAへの任意のNDAの提出を阻止することができ、 がMHRAにマーケティング許可申請(MAA)または条件マーケティング許可(CMA)を提出するか、または同様の許可をFDAに提出することを阻止し、最終的に私たちの候補製品を商業化し、製品収入を生成する。
私たちは以前にFDAやMAAまたはCMAに任意の候補製品のNDAを提出したことがなく、またはMHRAに同様の薬品承認文書を提出したことがあり、 であり、私たちの任意の候補製品が規制部門の承認を受けるかどうかを確認することはできない。また,我々の候補製品は臨床試験で成功しても,規制部門の承認が得られない可能性がある。もし私たちの候補製品が規制部門の承認を得られなければ、私たちは運営を続けることができないかもしれない。私たちが規制部門の承認を得て私たちの1つ以上の候補製品 を販売することに成功したとしても、私たちの収入は、必要に応じて私たちの製品候補製品と、規制部門の承認を得て商業権を持つ地域の市場規模をセットで診断するために、私たちまたは私たちの協力者と将来の協力者が規制部門の承認を得る能力があるかどうかにある程度依存する。もし私たちが狙っている患者亜群市場が私たちが予想しているほど大きくなければ、承認されれば、私たちはこのような製品の販売から相当な収入を得られないかもしれない。
62
さらに、我々が開発した任意の候補製品 が、他の既存療法と組み合わせて使用するために発売承認または商業化されても、我々 は、FDA、MHRAまたは他の同様の外国規制機関が、私たちの候補製品と組み合わせて使用される可能性のある療法の承認を取り消す可能性があるか、またはこれらの既存療法が安全性、有効性、製造、または供給問題を生じる可能性があるリスクを引き続き担う。
私たちの候補製品の商業化に成功した場合、承認されれば、市場が認められるかどうかに依存し、十分な承認 を得ることができず、相当な収入を得ることができない可能性がある。
我々の候補製品brが開発に成功し,規制部門の承認を得ても,医師,患者,医療保険支払者(例えば,個人保険会社や政府および他の助成者や医療界)の市場認可を得ることができない可能性がある。私たちのどの製品に対する市場の受け入れ度は多くの要素に依存するだろう
| ● | 私たちの製品の臨床効果と安全性を示しています |
| ● | どんな副作用の流行率や重症度も |
| ● | 製品の承認されたラベルに含まれている制限または警告 |
| ● | 費用効果と許容可能な価格設定の利用可能性 |
| ● | 競争力のある製品概要 と代替治療法および代替治療或いは治療法の利点; |
| ● | マーケティングの有効性および製品の流通方法および支援; |
| ● | もし私たちの製品が監督部門の許可を得たら、保証範囲 を引き受け、第三者支払者から十分な補償を受ける。 |
疾患指標は、疾患のより小さいサブセットとすることができ、異なる疾患サブセットが定義されるとき、それをますます小さい指標として解析することができる。このような病気のますます精密な説明は否定的な結果をもたらすかもしれない;承認された適応を作ることを含めて、それは小さすぎて、私たちは実行可能な市場がない。将来の技術が、大規模な重要な研究のための特徴とは異なる方法で疾患を記述することを可能にする場合、これらの研究を無効にするか、またはその有効性を低下させる可能性があり、 の全部または一部の研究を繰り返す必要があるかもしれない。将来の技術はより良い予後能力を提供する可能性があり,新たな治療法が必要と予想される患者の割合を減少させる可能性がある。規制部門の承認後であっても、1つの製品はその後、安全ではないか、またはその主張の効果がないことが証明され、その広範な使用を阻止したり、市場からの撤退を要求したりする可能性がある。
私たちは開発と商業化協力の構築に成功しないかもしれませんが、これは私たちが候補製品を開発する能力に悪影響を与え、製品を開発することを禁止するかもしれません。
製薬製品を開発し,臨床試験を行い,規制承認を得,製造能力やマーケティング承認を確立する製品は高価であるため,我々は将来的により多くの資源や経験を持つ会社との協力を求め,我々の候補製品を開発して商業化していくことが可能である。財務的または科学的な制限のために、私たちはまた、私たちの候補製品を研究および/または開発し、それを商業化するために、他の協力協定を締結する必要があるかもしれない。このような協力を確立して達成することは不可能かもしれないし、問題があるかもしれない。私たちが優遇条項でこのような追加的な協力を確立することができることは保証されないし、もしあれば、私たちの現在または未来の協力計画が任意の特定の候補製品または指示のために成功または維持されることを保証することもできない。もし私たちが適切なパートナーと私たちの候補製品の持続的な開発と商業化について成功したbr合意に到達できなければ、私たちは増加したコストに直面する可能性があり、私たちは商業的に開発できる候補製品の範囲と数量を制限することを余儀なくされるかもしれません。あるいはこれらの候補製品を商業化する地域 を制限し、適切なパートナーを見つけることができない製品や計画を商業化することができないかもしれません。もし私たちが成功した協力を実現できなかったら、私たちの経営業績と財務状況は実質的なbrと不利な影響を受けるだろう。
63
さらに、任意の協力協定の条項は、他の第三者との、または異なる適応、疾患または地理的位置で許可証または製品を開発する能力を制限すること、または候補製品の開発または商業化において私たちに追加的な義務を課すことを含む、他の製品に関する私たちの活動を制限するかもしれない。私たちがbrまたは協調協定に違反する任意の条項を遵守できない場合、協力者は、そのような合意 を全部または部分的に終了する権利があるか、または損害賠償を求める権利があるかもしれない。
私たちの協力および許可br協定は非常に複雑で、各当事者間でいくつかのデータ、ノウハウ、および知的財産権の所有権を共有または分割することに関連しています。したがって、私たちの協力者は、私たちまたは私たちのbrの他の協力者とは異なるいくつかの条項を説明するかもしれません。これは、協力者との意外または意図しないトラブルを引き起こす可能性があります。また,これらのプロトコルは, の他の連携,連携,あるいは合併や買収を困難にする可能性がある.
第三者に買収された協力者が、私たちの連携に悪影響を及ぼす可能性のある契約条項の変更を試みない保証はありません。 買収会社も、私たちの契約の条項や譲渡を受け入れず、合意の終了を求めることができるかもしれません。私たちのどのパートナーも、契約、制限、および/またはライセンス契約条項に違反する可能性があり、私たちは紛争に陥って、私たちは他のパートナーとの合意に違反する可能性があります。
内部でまたはパートナーを通じて販売およびマーケティング能力を確立しなければ、私たちの候補製品を商業化することに成功することはできません。
私たちは私たちのすべての候補製品のために適切な販売とマーケティング担当者、そして協力者を見つけることができないかもしれません。マーケティングと販売能力の発展には大量の支出、管理資源、時間が必要となる。このような販売チームを構築するコストは、任意の潜在的な製品収入を超える可能性があり、あるいは私たちのマーケティングと販売努力は失敗する可能性がある。もし私たちが内部マーケティングおよび販売能力をタイムリーにまたは根本的に発展させることができない場合、または受け入れ可能な条項で第三者とマーケティングおよび販売スケジュールを達成できない場合、私たちは承認された製品を効果的にマーケティングおよび販売することができない可能性があり、これは私たちが収入を創出し、利益を達成することができないだろう。また、候補製品の開発に成功し、規制部門の承認を求めることができなければ、内部マーケティングや販売能力を開発することができない可能性がある。
私たちは第三者請負業者とサービスプロバイダに依存して、私たちの開発計画のいくつかの側面を実行するつもりだ。これらの協力者が許容可能な時間範囲で品質の適切なサービスを提供できなければ,我々の開発計画の遅延や失敗を招く可能性がある.
私たちはある機能、テストとサービスを第三者、パートナー、医療機関と協力者にアウトソーシングし、生産を協力者および/または契約メーカーにアウトソーシングすることを計画しており、私たちは第三者に品質保証、臨床監視、臨床データ管理、監督管理の専門知識を提供することに依存する。特に、私たちは私たちの臨床試験を実行するために私たちのパートナーに依存している。このような個人または組織が約束または許容可能な品質基準に従って機能、テスト、薬品供給またはサービスを提供できることは保証されず、 であり、私たちの製品またはプロセスの開発に重大な遅延が生じる可能性がある。特に、いくつかの第三者サービスプロバイダ は、新冠肺炎の流行による中断によって、運営またはリストラを減少させること、または他の原因を含む契約義務を履行できない可能性があり、場合によっては、契約義務を遵守しないことがサービスプロバイダが制御できない要因によるものである場合、私たちの請求権は限られている可能性がある。
私たちの業務は私たちの候補製品の成功に高い 依存しています。もし私たちが臨床開発を成功させることができなければ、規制部門の私たちの1つ以上の候補製品の承認を得ることができない、あるいはそれを商業化することができない、あるいはもし私たちがこの点で遅延に遭遇した場合、私たちの業務は深刻な被害を受けるだろう。
私たちの将来の成功と私たちの候補製品から相当な収入を生み出す能力brは数年以内には起こらないと予想されています。これは、最近イギリスで2 b期の臨床試験に成功し、手掌線維結合組織の発育に影響を与えるbr}DupuytenのConstrature候補製品を含む、規制部門の承認を得て、私たちの1つ以上の候補製品を商業化する能力に依存しています。私たちの他のすべての候補製品は比較的に早いbr開発段階にあり、製造、臨床前テスト、臨床開発及び1つ以上の司法管轄区域の監督審査と承認の面で大量の追加投資を行う必要がある。もし私たちの任意の候補製品が安全や効果の問題、br}の開発遅延や規制の問題、あるいは他の問題に遭遇したら、私たちの開発計画と業務は実質的に損害を受けるだろう。
64
私たちは私たちの候補製品を開発し続ける財力がないかもしれない。臨床試験を完了しても、私たちは他の問題に直面する可能性があり、規制部門が私たちの候補製品を承認したり、商業化を阻止したりすることを延期したり、阻止したりする可能性があります
| ● | FDA、MHRA、または他の同様の外国の規制機関に、私たちの候補製品が安全で有効であることを証明することはできません | |
| ● | 私たちの財政と他の資源が不足していて、必要な臨床試験と臨床前研究を完成することができない |
| ● | 著者らの臨床試験、臨床前研究或いは他の候補製品の臨床試験結果は著者らと類似しており、追加の臨床試験或いは臨床前研究或いは計画の放棄を決定或いは要求する | |
| ● | 被験者が我々の臨床試験において経験した、予期しない毒性結果を含む製品に関連する有害事象、または私たちの候補製品に類似した薬物または治療用生物製剤を使用した個人; | |
| ● | 臨床試験を開始するために、研究新薬出願またはINDまたは同様の外国出願の提出を遅延させるか、または規制機関から必要なbrの承認を得ることができなかったか、または臨床試験が開始されると一時停止、終了または保留される | |
| ● | FDA、EMA、MHRA、または他の類似した外国の規制機関は、私たちの臨床試験の範囲または設計に適用された条件について | |
| ● | 私たちのbr候補製品は臨床試験期間中の効果が良くなかった | |
| ● | 対照群の表現は期待より良く、例えばプラセボ群、これは著者らの臨床試験結果が陰性或いは不確定であることを招く可能性がある | |
| ● | 被験者を臨床試験に組み込むことを遅延させた; |
65
| ● | 臨床試験の被験者の中退率は高い | |
| ● | 私たちの臨床試験に必要な候補製品や他の材料の供給や品質が不足している | |
| ● | 予想以上のbr臨床試験または製造コスト; |
| ● | FDA、EMA、MHRA、または他の同様の規制機関による臨床試験場の検査および審査 | |
| ● | 私たちの第三者請負業者または調査者は、規制要件または臨床試験案または他の方法でその契約義務をタイムリーにまたは根本的に遵守できなかった | |
| ● | FDA、EMA、MHRA、または他の同様の規制機関は、製造施設の検査および審査に不利であるか、またはこれらの施設は、FDA、EMA、または同様の規制機関が許容できるコンプライアンス状態を維持することができない | |
| ● | 一般的な臨床試験または特に私たちの療法に追加的な規制監視を適用することを含む、規制要件、政策、およびガイドラインの遅延および変更;または | |
| ● | FDA、EMA、MHRAと他の類似外国規制機関はデータの異なる解釈 を解釈している。 |
商業販売の前に、私たちの候補製品は、臨床前研究、臨床試験およびFDA、EMA、MHRAおよび/または他の適用可能な外国規制機関の承認を含む追加的かつ時間のかかる開発作業を必要とする。すべての候補製品は薬品開発固有の失敗リスク が発生しやすく,このような候補製品を含めて十分に安全かつ有効であることが証明されず,規制機関の承認を得ることができない可能性がある。さらに、承認されたこのような製品 は、経済的に生産または生産され、商業化に成功したり、市場に広く受け入れられたり、または他の商業代替製品よりも有効であることを株主に保証することはできない。
私たちの製品ラインを開発するには大量の資源が必要であり、資金を得る能力に応じて、いくつかの候補製品の開発を他の候補製品よりも優先しなければならない。さらに、私たちは、より利益があるかもしれない、またはより成功する可能性のある候補製品または指示に限られたリソースを使用することができないかもしれない。
我々は,(1)抗腫瘍壊死因子を検討する新たな臨床機会,(2)経口投与の決定,(α7ニコチン型アセチルコリン受容体アゴニストの小分子としてのbr}の3つの異なる計画があり,および(3)臨床前に開発された異なる段階にある特許可能なCBD類似体の決定,br}は最初に鎮痛剤として使用される。我々の候補製品開発には大量の資源が必要であるため,特定の疾患や疾患経路に集中し,どの候補製品を追求し推進するか,各候補製品に割り当てられる資源数を決定しなければならない。特定の候補製品または治療分野に研究、開発、協力、管理および財務資源を割り当てる決定は、実行可能な商業製品の開発を招くことがなく、より良い機会から資源を移転することが可能である。もし私たちが任意の候補製品の生存能力や市場潜在力について不正確な判断をしたり、製薬業界の傾向を曲解したりすれば、私たちの業務、財務状況、運営結果は重大な悪影響を受ける可能性がある。したがって、(I)可能な商業製品または利益の市場機会を利用できない可能性があり、(Ii)他の候補製品または他の疾患および疾患経路とのビジネスチャンスを求めることを放棄または延期することが要求される可能性があり、これらの製品または疾患または疾患経路は、私たちが選択した製品よりも大きな商業潜在力を有することが証明される可能性があり、または(Iii)私たちに有利な場合には、協力、許可、または他の印税配置によって、そのような候補製品の貴重な権利brを放棄して、独占的な開発および商業化権利 を維持することができる。
66
我々の臨床試験のスケジュールは様々な要素の影響を受ける可能性があり、どの遅延も現在の業務戦略を実行する能力に悪影響を与える可能性がある。
臨床試験は費用が高く,設計と実施が困難であり,完成まで数年かかる可能性があり,結果はまだ確定していない。私たちの候補製品の開発とテストのどの段階でも、私たちは臨床試験の遅延に遭遇する可能性がある。われわれが計画している臨床試験は時間どおりに開始されない可能性があり,有効な設計がない可能性があり,十分な数の被験者を募集することもなく,時間どおりに完成しない可能性もある。
臨床試験の遅延または不成功をもたらす可能性のあるイベントは、
| ● | 起動や実験継続に必要な資金は調達できない ; |
| ● | 規制部門の承認を得て試験を開始するのを遅延させる |
| ● | 遅延はFDA、MHRAあるいは他の外国監督機関と最終試験設計について合意した |
| ● | FDA、MHRAあるいは他の監督機関は著者らの臨床試験操作或いは試験地点を検査した後、臨床一時停止を実施した |
| ● | 遅延は予想されるCROや臨床試験地点と受け入れ可能な条項で合意した |
| ● | 各場所で必要な機関審査委員会の承認を得る上で遅延が発生した |
| ● | 被験者を完全に試験に参加させるか、または治療に戻った後のフォローを遅延させる |
| ● | 被験者が副作用やその他の理由で試験を終了したことによる遅延 |
| ● | 臨床サイトは試験からbrを除外し、登録人数を損害した |
| ● | 新たな臨床サイトの追加に要する時間 ;および |
| ● | 私たちの契約製造業者は十分な臨床試験材料の生産を遅延させて提供する。 |
もし私たちの候補製品の任意の臨床試験の開始または完了が上記の任意の原因やその他の原因で延期されれば、私たちの開発コストは増加する可能性があり、私たちの承認過程は遅延する可能性があり、商業発売後と特許保護が満期になるまでのいつでも時間が短縮される可能性があり、競争相手は私たちの前に製品を市場に出すことができるかもしれません。これらの事件のいずれも、私たちの候補製品のビジネス潜在力を損なう可能性があり、私たちの業務に重大な悪影響を及ぼす可能性があります。
67
私たちの候補製品の臨床試験は、患者が経験する可能性のあるすべての可能な副作用を明らかにすることができないかもしれない。
臨床試験は潜在患者集団の代表的なサンプルで行われ、これらのサンプルは顕著な変異性を有する可能性がある。臨床試験は限られた数の被験者と接触製品の有限持続時間に基づいて設計 を通じて確定し、潜在的な統計的意義に基づいて、任意の候補製品の計画安全性と有効性を実現できるかどうかを決定した。いずれの 統計サンプリングの結果と同様に、候補製品のすべての副作用が発見される可能性があることを保証することはできず、候補製品に長時間接触する患者数が有意に増加した場合にのみ、より完全な安全性 プロファイルを決定することができる可能性がある。また,より規模の大きい臨床試験であってもまれな重篤な副作用を決定できない可能性があり,あるいはそのような研究の持続時間は,これらのイベントが発生する可能性のある時間を決定するのに不十分である可能性がある。我々の製品を用いて治療した患者は,承認されると副作用が生じる可能性があり,FDA,MHRAまたは他の規制機関は追加のセキュリティデータを要求する可能性があり,我々の候補製品承認を得るための条件である。もし私たちのbr候補製品が発売された後に安全問題が発生したり、発見されたりすれば、私たちは私たちの製品ラベルの修正、私たちのbr製品のリコール、さらには私たちの製品に対する承認を撤回することを要求するかもしれません。
失敗は我々の薬物開発のどの段階でも起こる可能性がある 。
私たちは、規制機関による私たちの候補薬物の承認を得ることを遅延または阻止することができるかもしれないが、これらに限定されないが、これらに限定されない、テスト期間またはテスト結果において多くの予見不可能なイベントに遭遇する可能性がある
| ● | 規制機関や機関審査委員会(IRBs)は、私たちが臨床試験を開始するか、あるいは予想される試験場所で臨床試験を行うことを許可してはならない |
| ● | FDAおよび/またはMHRAは、私たちの臨床試験の範囲や設計について条件を課したり、規制環境の変化により、私たちの臨床試験案をIRBsに再提出して審査する必要があるかもしれません |
| ● | 私たちの臨床試験に必要な被験者の数はもっと多いかもしれません。患者登録にはより長い時間がかかるかもしれません。または患者は私たちの予想以上の速度で私たちの臨床試験を脱退するかもしれません |
| ● | もし私たち、規制機関、またはIRBsが参加者が不合理な健康リスクを受けていると判断した場合、私たちは私たちの1つ以上の臨床試験を一時停止または終了しなければならないかもしれない |
| ● | 私たちの第三者請負業者、臨床研究者、または契約協力者は、法規の要求を遵守できないか、または私たちに対する契約義務をタイムリーに履行できないかもしれない |
| ● | FDAは,医療基準が米国とは異なる国/地域で行われる可能性のある臨床試験の臨床データを受け入れない可能性がある |
| ● | 私たちのテストは陰性または不確実な結果をもたらす可能性があり、私たちは決定または規制機関が追加的なテストを要求するかもしれない; |
| ● | 私たちの臨床前および/または臨床試験のコストは私たちが予想していたより高いかもしれない。 |
我々は第三者 に依存してわれわれの臨床前研究と臨床研究と試験を行い,彼らが我々の義務を履行しなければ,追加適応の承認を得られない可能性がある。
われわれは現在,臨床前研究や臨床研究や試験を独立して行う能力がなく,これまで第三者契約 研究や政府組織や医療機関に依存して研究や試験を行わなければならなかった。私たちの第三者開発活動への依存はこれらの活動に対する私たちの統制を減少させた。これらの第三者は計画通りに活動を達成できない可能性があり,あるいは法規的要求やわれわれの研究設計に従ってわれわれの臨床前研究および臨床研究や試験を行うことができない可能性がある。これらの第三者がその契約責任を成功的に履行できない場合、または予想される期限までに完了すれば、悪影響を受ける可能性があり、規制部門の承認を得て適応を商業化する努力が延期される可能性がある。
68
我々が の他の当事者と一緒に研究を行うと,この実験に関するすべての決定を制御できない可能性がある.我々が研究設計,研究時間などで相手と食い違いがあれば,我々の薬物開発計画に悪影響を及ぼす可能性がある。
我々も第三者に依存して我々の研究や実験のデータを管理しているが,我々の個々の研究や実験がその全体的な調査計画と案に基づいて行われていることを確認する責任がある.そのほか、FDAと外国監督機関は、良好な実験室規範(GLP)と良好な臨床実践(GCP)、 を含む適用された法規と標準を遵守することを要求し、このような研究と試験の結果を記録し、報告し、データと結果が信頼性と の正確性を確保し、人体研究と試験参加者が十分に保護されることを確保する。我々の第三者への依存は我々のこれらの義務や要求を解除しておらず,これらの要求を満たさなければ,規制部門の他の適応に対する承認を得ることができない可能性がある。
私たちは成功するために、流通と生産能力や関係を開発し、維持しなければならない。
私たちは独立生産でも第三者製造業者とも製造スケジュール(あれば)を達成することに成功しないかもしれません。また、もし任意のメーカーが私たちとの業務往来を停止したり、遅延、供給不足、あるいは生産能力の需要が大きすぎる場合には、十分な数の製品をタイムリーにあるいは根本的に得ることができない可能性がある。製造者およびそのサプライヤーは、場合によってはFDAが実行する現行守秘協定約束および現行良好製造規範(CGMP)および要求、および他の国/地域の類似した要求を遵守しなければならない。メーカーがこれらの要求を遵守できなかったことは 製品を提供する能力に影響を与える可能性がある。私たちは第三者契約メーカーに依存するつもりですが、最終的に私たちの製品がcGMPに従って生産されることを確実にする責任があります。さらに、当社の第三者製造業者の1つまたは複数の工場の承認前検査または他の検査が行われた場合、FDAは、工場がcGMPに適合していないと判断し、工場を製造業者とするいかなるマーケティング申請も承認されない可能性があり、またはbr}工場がcGMPに適合し、FDAの成功した再検査が完了するまで承認を延期する可能性がある。
製造問題、製造施設に影響を与える自然災害や流行病や契約メーカーの損失は、私たちのbr運営を中断し、販売損失を招く可能性があります。しかも、私たちは第三者に依存して私たちの製品を生産するために必要な原材料を提供するつもりだ。サプライヤーへのいかなる依存も、重要な材料が得られない可能性があること、生産コスト、交付スケジュール、信頼性、品質の制御を減少させることを含むいくつかのリスクに関連する可能性がある。サプライヤー問題は未来の契約製造によるいかなる意外な中断 も製品の出荷が遅延する可能性があり、私たちの商品を販売するコストを増加させ、販売損失を招く可能性がある。もし私たちのサプライヤー が私たちに十分な薬物を供給できなければ、候補薬物の商業化に成功した能力に重大な悪影響を及ぼすかもしれない。
他の候補製品を発見し,開発して商業化できなければ,我々の業務を発展させることができず,我々の戦略目標を実現する能力も影響を受けるであろう.さらに、私たちは私たちの固有の治療法ではないかもしれないいくつかの治療法を商業化することを求めることもできる。
我々は現在候補製品の開発と商業化が最初の重点であるが,長期成長戦略の一部として 他の候補製品の開発を計画している。我々が計画した製品は他の用途の潜在的な適用性がある可能性があると信じているが,これらの他の用途についてはいかなる臨床試験も行われていないため,他の用途のための候補製品の開発に成功していない可能性がある。また,我々は,より多くの候補製品を発見し決定するために,基礎研究に資金と資源を投入する予定である.これらの研究計画 は最終的に任意の候補製品を決定するか否かにかかわらず,技術,財政,人的資源を必要とする。我々の研究計画 は当初,潜在的な候補製品の決定に希望を示す可能性があるが,多くの原因で臨床開発のための候補製品 が生じなかったことには,以下の理由が含まれている
| ● | 使用された研究方法 は潜在的な候補製品の決定に成功しない可能性がある | |
| ● | 競争相手は私たちの候補製品を時代遅れにする代替製品を開発するかもしれない |
69
| ● | 私たちが開発した候補製品はまだ第三者特許または他の独占的権利によって保護されているかもしれない | |
| ● | さらなる研究では、候補製品は、有害な副作用または他の特徴を有することが証明される可能性があり、それが有効である可能性が低いこと、または適用される規制基準を満たしていないことを示す | |
| ● | 候補製品は、許容可能なコストで商業的に量産できないか、または生産できない可能性がある | |
| ● | 候補製品は患者、医学界、または第三者支払人に受け入れられない可能性がある。 |
予想される時間範囲で所期の開発·商業化目標を達成できなければ、候補製品の開発·商業化が遅れる可能性があり、我々の業務·運営結果が損なわれる可能性がある。
計画目的のため、私たちは各種の科学、臨床、法規と他の製品開発目標の完成時間を推定することを求めています。 これらのマイルストーンは、科学研究と臨床試験の開始或いは完成、法規届出書類の提出、或いは商業化目標に対する期待を含むかもしれません。私たちは、進行中の臨床試験の完了、他の臨床計画の開始、br市場の承認、または製品の商業発表のようないくつかのマイルストーンの予想される時間を時々公開するかもしれない。その中の多くのマイルストーンの潜在的な実現は、私たちの制御範囲内ではないかもしれません。 これらのマイルストーンの各々は、予想通りに実現できなければ、それぞれのマイルストーンの潜在的な実現時間が私たちの推定と大きく異なることをもたらす可能性があります
| ● | 私たちの利用可能な資本資源や私たちが遭遇した資本制約; | |
| ● | 私たちの臨床試験と研究開発活動の進捗、コストと結果は、参加した臨床医と協力者のスケジュールとの衝突の程度を含む |
| ● | 臨床試験の資格基準に適合する患者の能力を識別し募集します | |
| ● | FDA、MHRA、および他の規制機関から許可されたbrとその時間 |
70
| ● | 臨床結果 | |
| ● | 規制当局が発表した他の行動、決定、または規定 | |
| ● | 私たちは候補品を生産するために十分で信頼性があり価格の安い材料を得ることができます | |
| ● | 私たちの協力者が候補製品の商業化に努力しています | |
| ● | 製品製造および販売やマーケティング活動に関する安全、コスト、時間の問題。 |
もし私たちが予想された時間範囲でいかなる発表されたマイルストーンも達成できなければ、私たちの候補製品の開発と商業化は延期される可能性があり、私たちの業務と運営結果は損害を受ける可能性があり、私たちの株価表現に負の影響を与える可能性がある。参照してください“業務.業務“ より多くの情報を知る.
もし私たちが唯一の供給源に依存して私たちの製品を生産すれば、私たちはサプライヤーの生存能力の影響を受けるかもしれない。
私たちは複数のサプライヤーから私たちの製品を調達しようと努力するつもりだ。私たちはまた、私たちの製品のどのサプライヤーとも契約を結び、彼らに契約の中で私たちの要求を満たす義務を負わせるつもりです。しかし、もし私たちが供給者に依存している場合、その供給者が私たちの要求を満たすことができないか、または満たさない場合(どのような理由でも)、私たちの業務は悪影響を受ける可能性があります。
私たちは私たちの候補薬物を生産する規模を十分に拡大できないかもしれない。
私たちは、第三者製造業者と協力しても、私たち自身であっても、タイムリーまたは費用対効果のある方法で、または全くできない、私たちの候補薬物の製造能力を十分な方法で増加させることに成功することができないかもしれない。もし契約製造業者が私たちの候補薬物の製造過程を改善した場合、私たちはこれらの改善された知的財産権を所有していないか、または共有しなければならないかもしれない。
大規模な生産には追加的な検証研究が必要となる可能性があり,これらの研究はコストが高く,FDAの審査と承認を得なければならない。さらに、候補薬剤自体の固有特性、または候補薬剤と製造および包装中に添加される他の成分との組み合わせ、または完成品または活性医薬成分の輸送および貯蔵中に添加される他の成分の固有属性により、これらの拡大活動中に品質問題が生じる可能性がある。もし私たちが私たちの任意の候補薬剤を十分な品質と数量で大規模に生産することに成功できなければ、その候補薬剤の開発および任意の最終薬物製品の規制承認や商業発売が延期される可能性があり、あるいは供給不足が発生する可能性があり、これは私たちの業務を深刻に損なう可能性がある。
我々の業務 は,持続的かつ将来的な潜在的なグローバル衝突に関するリスクの影響を受けている。
現在、ロシアとウクライナの間の紛争は続いており、軍事活動の進行と追加制裁に伴い、両国間の戦争は変化し続けている。この戦争は、経済や世界の金融市場にますます影響を与え、インフレ上昇やグローバル·サプライチェーンの中断など、持続的な経済課題を悪化させている。この衝突は現時点では我々の財務会計や報告に実質的な影響を与えないと考えられるが,我々の将来影響を受ける程度は不確定かつ予測不可能なイベントの性質や持続時間に大きく依存し,我々の業務は影響を受ける可能性がある.さらに、将来のグローバル衝突または戦争は、インフレ上昇およびグローバル·サプライチェーンのさらなる中断を含むが、これらに限定されないさらなる経済的課題をもたらす可能性がある。したがって、持続的なロシア/ウクライナ紛争および/または将来の他の世界的衝突は、運営費用の増加および/または将来の任意の収入減少をもたらす可能性があり、さらに、私たちの運営結果およびキャッシュフローに実質的な悪影響を及ぼす可能性がある
71
我々の将来の候補製品の開発と規制承認に関するリスク
臨床試験は高価で、時間がかかり、不確定であり、しかも変化、遅延或いは終了が発生しやすい。臨床試験の結果は異なる解読ができる。
抗炎症薬を製造するための3つの異なる項目がある:(1)抗腫瘍壊死因子を研究する新しい臨床機会、(2)経口投与の決定、α7ニコチン型アセチルコリン受容体アゴニストの小分子としてのbr}の決定、および(3)最初に鎮痛剤として使用されるCBDの特許類似体の決定。しかし,これらの計画は,関連する臨床試験を含めて高価で時間のかかるbrであり,設計や実施は困難である。
監督管理機関は著者らが提出した臨床試験設計を受け入れない可能性があり、著者らとは異なる方式で臨床試験結果を分析或いは解釈する可能性がある。たとえ我々の臨床試験結果が有利であっても,そのいくつかの将来の候補製品の臨床試験は数年続くと予想され,完成にはより長い時間がかかるかもしれない。また、FDA、MHRAまたは他の規制機関は、州、地方、外国当局またはIRBを含み、我々の機関の試験については、いつでもその臨床試験 を一時停止、延期、または終了することができ、追加の臨床試験を行うことを要求し、特定の臨床試験持続時間が最初に計画されたものよりも長いことを要求し、その開発計画を変更することを要求し、異なる 順序で候補製品に対して臨床試験を行うために、例えば、同じ候補製品の2つの試験を並行して実行するのではなく、段階的な方法で実施する。または、以下の理由を含む、制御された物質の調達および処理に必要な登録および割当量の割り当てを様々な理由で一時停止または終了する可能性があり、いずれも、私たちの業務、財務状態、および経営結果に実質的な悪影響を及ぼす可能性がある
| ● | 臨床試験中に候補製品の有効性が不足している |
| ● | 試験参加者が経験した深刻または予期しない毒性または副作用または他の安全問題、例えば薬物相互作用は、他の薬物レベルの混同変化をもたらす問題を含むことを発見した |
| ● | 予想より低かった被験者募集速度と臨床試験の入院率 |
| ● | すでに臨床試験を開始したが、治療副作用、治療効果不足、臨床試験過程疲労或いは任意の他の原因で随時退出する可能性のある被験者を維持することは困難である |
| ● | 規制および製造面の制限により、臨床試験のための十分な数の材料の生産または獲得に遅延が生じたり、入手できなかったりする |
| ● | 製造プロセスまたは製品配合の不足または変更 ; |
| ● | 規制部門の許可を得て試験を開始する上で遅延が発生した“臨床棚上げ“または試験が開始される前または後に、遅延は、FDAのような規制機関が試験を一時停止または終了することを必要とする |
72
| ● | DEA関連記録は、臨床サイトの安全または他の違反を報告し、DEAまたは州当局がサイトの制御物質許可証を一時停止または停止または停止させ、計画中または進行中の試験の遅延または終了をもたらす |
| ● | 適用される規制政策と法規の変化は、研究範囲、性質、または時間に対する要求の変化を含む |
| ● | 臨床試験契約またはプログラム中の許容可能な条項と予期される臨床試験場所との合意を遅延または達成できなかった |
| ● | 適切な用量の不確実性について |
| ● | 規制基準に適合した臨床試験用製品を遅延または提供できなかった |
| ● | 臨床前研究や臨床試験の不利な結果が行われています |
| ● | 我々のCROまたは他の第三者請負業者は、すべての契約要件を遵守できなかったか、またはそのサービス をタイムリーにまたは受け入れ可能に履行できなかった; |
| ● | わが社、私たちのCROまたは彼らの従業員は、適用されるFDAまたは他の臨床試験に関連するすべての法規要件を遵守できませんでした |
| ● | スケジュールは参加した臨床医や臨床機関と衝突しています |
| ● | 適切な臨床試験案を設計できなかった |
| ● | 規制機関は、非植物性、非精神活性製品にのみ適用されるが、CBD由来製品に対する一般的な懸念および乱用の可能性を懸念している |
| ● | データが不足していて、規制承認をサポートできない ; |
| ● | 医療研究者は私たちの臨床計画に従ってはいけません |
| ● | 治療期間または治療後に患者とbrの連絡を維持することは困難であり、これはデータが不完全である可能性がある。 |
我々の臨床試験に患者を参加させることは困難であることが発見されるかもしれないが,候補製品の臨床試験を延期あるいは阻止する可能性がある。
患者を確定し、それを著者らの候補製品の臨床試験に参加させることは著者らの成功に非常に重要である。私たちの臨床試験の時間は私たちが条件に合った患者を募集して私たちの候補製品のテストに参加する速度に依存します。私たちはいくつかの臨床試験で遅延に遭遇し、未来にも似たような遅延に遭遇する可能性がある。これらの遅延はコスト増加、製品開発遅延、あるいは臨床試験の完全終了を招く可能性がある。
73
十分な数の患者を識別、募集、募集することができないか、または研究において多様性を実現するために必要なまたは必要な特徴を有する患者は、予想される時間内に臨床試験を完了することができない可能性がある。患者登録は、様々な要因の影響を受ける可能性があるが、これらに限定されない
| ● | 研究プログラムに要求される設計および複雑さおよび/または参加承諾; | |
| ● | 患者集団の規模は |
| ● | 研究に関する資格基準 ; | |
| ● | 臨床用品の供給状況 |
| ● | 登録のために遅延参加サイト識別、資格、および後続のアクティブ化; | |
| ● | 類似または競合療法で観察される副作用を含む、研究を受けた候補製品の知覚リスクおよび収益 |
| ● | 潜在患者の臨床試験地点の近似性と可用性 ; | |
| ● | 競争療法と臨床試験の有用性 |
| ● | 臨床試験の適時登録を促進するために現場努力の競争 | |
| ● | サイトの動機に参加し |
| ● | 医師の患者紹介やり方 ; | |
| ● | 患者権益提唱団体の活動 ; |
| ● | 治療期間および治療後に患者を十分に監視する能力;および | |
| ● | 調査中の疾患重症度 である。 |
特に,われわれの候補製品のいずれも患者池の限られた疾患であり,臨床試験に利用できることを評価する予定である。私たちの臨床試験の資格基準は利用可能な研究参加者をさらに制限するだろう。さらに、患者の発見と診断の過程は高価であることが証明される可能性がある。われわれの臨床試験中の主治医も彼らの医学決定権を使用する可能性があり,われわれの臨床試験に参加した患者はわれわれの研究から撤退し,代替療法を試みることを提案している。また,隔離やその他の制限により,行われている新冠肺炎の大流行は,患者の臨床試験地点への能力や意思に影響を与える可能性があり,われわれの臨床試験の登録に悪影響を及ぼす可能性がある。
74
合意に基づいて必要な条件に適合する患者を募集することができなければ、FDAやEMA、MHRAまたは他の規制機関が要求する臨床試験に参加できなければ、臨床試験を開始または継続できない可能性がある。私たちが外国で臨床試験計画を開始、登録、完成することに成功する能力は、外国で業務を展開するために独自の多くのリスクの影響を受ける
| ● | CROや医師との関係を構築したり管理したりすることは困難です | |
| ● | 臨床試験を行う異なる基準; |
| ● | 地元のコンサルタントや医師やパートナーを見つけることはできません | |
| ● | 各種の外国法律、医療標準と監督管理要求の潜在的負担を遵守し、薬品と生物技術製品と治療に対する監督管理を含む |
| ● | 研究を行うために必要な臨床試験材料の調達と交付能力; | |
| ● | 対面訓練が完了できない場合には,遠隔参加現場で十分な訓練を実施することができない. |
計画通りの臨床試験を行うのに十分な数の患者を募集することが困難であれば,進行中や計画中の臨床試験を延期,制限または終了する必要がある可能性があり,いずれも臨床試験を遂行する能力や最終的な手術結果に悪影響を及ぼす。
もし私たちの会社が既存の法規を遵守できなかったら、私たちの名声と経営業績を損なうかもしれません。
私たちはアメリカ、ヨーロッパ、カナダのすべての市場、あるいは私たちの製品候補が承認中の市場で、アメリカ連邦、州、外国政府の広範な規制を受けて、私たちはこれらの市場で私たちの製品を販売することを計画しています。
FDAのGLP,GCPおよびGMP要件,薬物警戒要件,広告および販売促進制限,報告および記録保存要件,およびヨーロッパにおけるそれらの同等の要件を含むすべての法規要件を遵守しなければならない。私たちまたは私たちのサプライヤーがFDA承認前または承認後の要求を含む適用された法規を遵守できなかった場合、FDAまたは他の外国規制機関はわが社を制裁する可能性があります。医薬品がFDAまたは他の主管部門の許可を得ても、監督管理機関は製品の指示用途やマーケティングに重大な制限を加えたり、コストが高い可能性のある発売後の試験に持続的な要求を提出したりする可能性がある。私たちの米国で承認される可能性のある候補製品brは、製造、ラベル、包装、貯蔵、流通、輸入、輸出、広告、販売促進、サンプリング、記録、および連邦および州要件を含む安全および他の発売後情報の提出を含む持続的な法規要件の制約を受けるだろう。また,メーカーやメーカーの工場は,品質管理や製造プロセスがGMPに適合することを確保することを含むFDAの広範な要求を遵守しなければならない。したがって、私たちと私たちの契約製造業者は、GMPのコンプライアンスを評価するために、持続的な審査と定期的な検査を受けることになります(将来的に契約メーカーが指定された場合)。したがって、私たちと私たちと協力している他の人たちは、製造、生産、品質管理、品質保証を含むすべてのコンプライアンス分野で時間、お金、エネルギーをかけなければならないだろう。また、いくつかの副作用や生産問題をFDAに報告し、当社の製品の広告や販売促進に関する要求を遵守することが求められます。処方薬に関する販売促進情報は様々な法律や法規によって制限されており,製品承認のラベル中の情報と一致しなければならない。EUと私たちが運営する他の市場にも似たような制限と要求 がある。
75
もし監督管理機関が製品に以前に未知の問題、例えば意外な深刻性または頻度の不良イベントが存在することを発見した場合、または製品を生産する施設に問題があるか、または製品の販売促進、マーケティングまたはラベルに同意しない場合、製品または当社に制限を加えて、製品の市場からの撤退を要求することを含むことができる。もし私たちが適用される規制要件を遵守できない場合、規制機関または法執行機関は警告状を発行し、民事または刑事罰を適用し、規制の承認を一時停止し、私たちが行っている任意の臨床試験を一時停止し、承認すべき申請を拒否し、または私たちが提出した承認された申請の補充、私たちの運営に制限を加え、製品を差し押さえたり、差し押さえたり、製品のリコールを要求する可能性がある。
また、私たちの将来の製品はDEAによって規制される可能性があり、“制御物質法”や他の場所の類似法に基づいている。DEA計画 は、薬物がNDA承認日後に患者に使用可能である可能性がある場合に、このプロセスが遅延する可能性があり、このようなDEAプロセスの時間および結果 が不確定である別個のプロセスである。別に参照してください“規制された物質に関するリスク“と、以下を参照。
また,違法行為の疑いに対する政府のいかなる調査にも対応するために多くの時間と資源が必要となる可能性があり,負の宣伝が生じる可能性がある.現行の規制要件を遵守しないいかなる行為も、私たちがbrを商業化し、私たちの将来の候補製品から収入を得る能力に深刻な影響を与える可能性がある。規制制裁を実施したり、規制承認を撤回したりすれば、我々の業務価値や経営業績は悪影響を受ける可能性がある。
私たちがこれらの法律に違反する行為は、たとえ私たちが弁護に成功しても、私たちに巨額の法的費用を発生させ、私たちの経営陣の業務運営に対する注意を移し、私たちの名声を損なう可能性があります。我々はコンプライアンス作業に大量の資源 を投入する予定であり,このような支出は予測不可能である.変化する法律、法規、基準も不確実性、より高い費用、保険コスト を増加させる可能性がある。そのため,継続的に発展する基準を遵守するためにすべての合理的に必要な資源を投入する予定であり,この投資 は管理や行政費用の増加を招き,管理時間と注意を創造活動からコンプライアンス活動に移行させる可能性がある。
私たちは連邦、州、外国の医療法律法規の制約を受けて、このような医療法律法規の実施或いは変更は私たちの業務と運営結果に不利な影響を与える可能性がある。
アメリカとある外国の司法管轄区では、すでに複数の立法と規制提案が医療保健システムを変えることを目的としており、 は私たちの未来の候補製品を販売する能力に影響を与える可能性がある。もし私たちがこれらの法律のいずれかまたは任意の他の連邦、州、または外国法規に違反していることが発見された場合、私たちは行政、民事および/または刑事罰、損害賠償、罰金、個人監禁、連邦医療計画、および私たちの業務の再編を受けるかもしれない。その中のどれも私たちの業務と財務業績に実質的な悪影響を及ぼす可能性がある。これらの法律の多くは裁判所の十分な解釈を得ていないため、私たちはそのうちの1つ以上の規定に違反するリスクが増加していることが発見された。私たちがこれらの法律に違反する行為は、私たちが最終的に弁護に勝っても、巨額の法的費用が発生し、私たちの経営陣の注意を業務運営から移すことになります。また、多くの外国、特にEU諸国では、処方薬の定価が政府によって規制されている。
一部の外国の国では、薬品の提案定価は承認されなければならず、それから合法的に発売されることができる。各国の薬品定価に対する要求は千差万別である.
例えば、いくつかのEU司法管轄区 はプラスリストとネガティブリスト制度を実施し、精算価格を合意した後にのみ、製品を市場で販売することができる。Br精算或いは定価審査を得るためには、その中のいくつかの国は臨床試験を完成し、特定の候補製品のコスト効果を現在利用可能な治療法と比較する必要があるかもしれない。他の会員国は会社が自分の薬品価格を制定することを許可するが、会社の利益を監視して制御する。各国の価格設定メカニズムのこのような違いはEU加盟国間の価格差をもたらすかもしれない。薬品に対して価格制御や精算制限を実行する国/地域が私たちのいかなる製品に対しても優遇的な精算と定価手配を許可することは保証されません。歴史的に見ると、イギリスとEUで発売された製品は米国の価格構造に沿っていない。イギリスとEUでは、医療コストの全体的な下振れ圧力が大きくなり、特に処方薬が大きくなっている。そのため,新製品への参入のハードルが高くなっており,患者が政府の精算されていない薬物製品を使用する可能性は低い。私たちは海外からの低価格製品の競争に直面するかもしれません。これらの製品は薬品に対して価格規制を実施しています。また、輸入外国製品は私たちが将来販売する可能性のあるどの製品とも競争する可能性があり、これは私たちの収益性にマイナスの影響を与えるかもしれない。
76
具体的には,米国では,2010年の“平価医療法案”や将来とりうる他の医療改革措置が,より厳しい保証基準をもたらす可能性があり,承認される可能性のある任意の製品の価格に追加的な下押し圧力を与える可能性があると予想される。ACAのいくつかの態様に司法的挑戦が提起され、ACA の代わりにいくつかの立法が廃止および/または部分的に廃止および/または代替しようとしており、私たちは将来的にACAにより多くの挑戦と改正を提起することが予想される。現在,ACAが我々の将来の業務に与える全面的な影響 は不明である.米国の医療業界における政府の役割の拡大は、処方薬製品の削減、精算または規制の承認を得た他の任意の製品の価格に一般的な下振れ圧力をもたらし、製品利用率を低下させ、私たちの業務や運営結果に悪影響を及ぼす可能性がある。連邦医療保険や他の政府計画の精算を削減することは,個人支払者の支払いの類似した減少を招く可能性がある。コスト制御措置や他の医療改革を実施することは、私たちが収入を創出し、利益を達成することを阻止することができ、あるいは規制の承認を得る可能性のある任意の未来の候補製品を商業化するかもしれない。
拡大参入研究から得られた情報は、会社が後援する臨床試験における私たちの将来の候補製品の治療効果を確実に予測できず、承認を制限する可能性のある有害事象を招く可能性がある。
我々が現在支持している拡大参入研究 は制御されておらず,個別の研究者が行い,通常GCP を厳密に守らないことは,いずれもプラセボ対照試験とは異なる治療効果をもたらす可能性がある。このような研究は規制審査の有効性の逸話的証拠だけを提供する。これらの研究には参考になる対照や比較群は含まれておらず,この 患者データは研究結果としてまとめたり報告することを目的としていない。また,このような少数の患者からのデータは大きな変数を持つ可能性がある。これらの研究から得られた情報には,我々と独立した調査者がデータに適用する統計原理を選択し,システム評価会社が後援した臨床試験の治療効果によって収集されたデータや,これらの試験に適用可能な他の統計原理で評価されたデータを確実に予測できない可能性がある。これらの情報に依存して私たちの臨床試験を設計することは、治療効果を証明するために試験が十分に設計できず、私たちの将来の候補製品の承認を求める能力を遅延または阻害する可能性がある。
拡張アクセス計画 は,規制審査に支援的なセキュリティ情報を提供する.これらの研究を行っている医師は、私たちが支援している試験所の研究条件を超えた条件の子供を含む、合意と一致しない方法で、私たちの将来の候補製品 を使用するかもしれません。許可計画中の被験者が遭遇したいかなる有害事象や反応も、私たちの将来の候補製品 に起因する可能性があり、理想的なラベルを通じて規制承認を得る能力を制限することができないかもしれません。
候補薬物の臨床試験による失敗率は高かった。
一般に,臨床試験による候補薬は高い失敗率を有している。われわれの臨床試験は重大な挫折に遭遇する可能性があり,製薬やバイオテクノロジー業界の他のいくつかの会社の経験に類似しており,早期の試験でもエキサイティングな結果が得られている。また,臨床試験の結果が積極的であると考えても,FDA,MHRAや他の規制機関はデータの解釈に同意しない可能性がある。もし私たちが候補製品の臨床試験から否定的な結果を得たり、潜在的な化学、製造、制御問題、あるいは他の障害に関連する他の問題が発生した場合、私たちの未来の候補製品が承認されていない場合、私たちは私たちの運営を継続するために十分な収入や資金を得ることができない可能性があり、現在の業務計画を実行する能力は実質的に損なわれる可能性があり、業界や投資界での名声は深刻な損害を受ける可能性がある。また,われわれは臨床試験を正確に設計,起動,完成することができず,われわれの臨床試験の時間と結果,われわれの候補薬物の承認を求める能力に負の影響を与える可能性がある。
77
連邦または州の“詐欺および乱用”法律または他の管轄区域の同様の法律に違反していることが発見された場合、私たちはbrの罰金の支払いを要求され、および/または連邦または州医療計画への参加を一時停止される可能性があり、これは私たちの業務、財務状況、および運営結果に悪影響を及ぼすかもしれない。
アメリカでは、連邦および州医療保健計画における詐欺や乱用を減らすことを目的とした反バックル法、虚偽申告法、その他の法律を含む様々な連邦および州医療保健“詐欺および乱用”の法律の制約を受けている。これは、私たちの会社、特に私たちの製品がアメリカで商業化に成功したときに影響を与える可能性がある。1987年に“連邦医療保険と医療補助患者保護法”または連邦反リベート法令brは、処方薬メーカー(またはそれを代表する側)を含む誰でも、インフォームドコンセントや故意の場合に請求、受け入れ、受け入れ、規定されている。連邦医療計画(例えば、MedicareまたはMedicaid)に従って支払われる特定の薬剤の処方を含む、原資産業務を導入するための任意の報酬を提供または支払うこと。連邦法によると、安全港と呼ばれるいくつかの手配は連邦反リベート法規に違反しないと考えられている。すべての適用要件に基づいて我々の業務スケジュールを構築することを求めているが,特定の場合にどのように適用されるかを正確に決定することは困難である.したがって、私たちのやり方は連邦“反リベート法規”と“連邦虚偽申告法”の挑戦を受けるかもしれない。詐欺および法律違反の行為は、罰金および/または連邦および州医療保健計画(例えば、MedicareおよびMedicaid)の排除または一時停止、および米国政府との契約の禁止を含む刑事および/または民事制裁を受ける可能性がある。また,個人は連邦“虚偽申告法”やいくつかの州の虚偽申告法に基づいて政府を代表して訴訟を起こす権利がある。
私たちはこれらの法律を守るために私たちの業務手配を組織したと信じていますが、政府は私たちがこれらの法律に違反したことを非難したり、判定したりするかもしれません。 もし私たちがこれらの法律のうちの1つに違反していることが発見された場合、私たちは罰金の支払いを要求される可能性があり、連邦または州医療計画に参加することを一時停止または排除される可能性があり、私たちの業務、運営結果、および財務状況は不利な影響を受ける可能性がある。
EU加盟国や他の国にも反リベート法があり、権利侵害の場合に処罰を加えることができ、一部の司法管轄区では、競争相手もこの処罰を実行することができる。
深刻な有害事象(Br)または他の安全リスクは、私たちの将来の候補製品の承認を放棄し、阻止し、阻止し、または制限することを要求することができ、許可されたラベルまたは市場受容度の範囲を制限するか、またはすでに発売された製品のリコールまたは上場承認を失うことをもたらす可能性がある。
もし私たちの未来の任意の製品 の任意の候補製品が任意の商業販売承認の前または後に、深刻なまたは予期しない副作用を引き起こす場合、または誤用、乱用または転移などの他の安全リスクに関連する場合、多くの潜在的な重大な負の結果を引き起こす可能性がある:
| ● | 監督当局は臨床試験を中断、延期、または一時停止することができる |
| ● | 規制機関は、規制部門が私たちの未来の候補製品を承認することを拒否するかもしれない |
| ● | 規制当局は、警告または禁忌または使用適応制限のようないくつかのラベル宣言を要求することができ、および/または は、承認または承認後にREMS形態で配信に制限を加えることができる |
| ● | 規制機関は、その承認を撤回し、より煩雑なラベル声明を要求し、より厳しいREMSを実施するか、または承認された任意のbr製品をリコールすることを要求する可能性がある |
| ● | 私たちは製品の投与方法の変更や追加の臨床試験を要求されるかもしれません |
| ● | 私たちと協力パートナーとの関係は影響を受けるかもしれない |
78
| ● | 私たちは起訴され、患者への傷害に責任を負うことを要求するかもしれない |
| ● | 私たちの名声は影響を受けるかもしれません。私たちが小児科のbr適応のために開発している未来の候補製品よりも名声リスクが高くなります。 |
もし私たちがいつでも製品が参加者に受け入れられないリスクを構成していると思っている場合、あるいは予備データが私たちの将来の候補製品が規制部門の承認を得られないか、商業化に成功しない可能性が高いことを示している場合、私たちは自発的にbrを一時停止したり、私たちの臨床試験を中止したりすることができます。br}製品の商業販売の承認を受けた後、もし 製品の使用や接触が不良な健康結果や死亡を招く可能性があると考えた場合、私たちは自発的に市場から製品を撤回またはリコールすることができます。これまで、私たちはまだ撤回、リコール、または他の行動を取っておらず、自発的または強制的に承認された製品を市場から除去してきた。また、規制機関、IRBまたはデータ安全監視委員会が、臨床試験が適用された法規の要求に従って行われていないと判断したり、参加者に対して受け入れられないセキュリティリスクを構成していると判断した場合、規制機関、IRBまたはデータ安全監視委員会は、いつでも私たちの臨床試験を一時的または永久的に停止することを提案したり、臨床試験で研究者の使用を停止することを要求したりすることができる。br}規制機関、IRBまたはデータ安全監視委員会は、私たちが臨床試験を一時的または永久的に終了することを要求していないが、将来の候補製品の臨床試験を一時的または永久的に停止または停止させることを選択または強制された場合、この製品のビジネスの見通しは損なわれ、私たちが製品から製品の収入を得る能力は延期またはキャンセルされる可能性があります。 さらに、これらの事件のいずれも警告やタブーのようなラベル宣言を引き起こす可能性があります。さらに、このようなイベントやbrを貼り付けることは、私たちまたは私たちのパートナーが影響を受けた製品に対する市場の受容度を獲得または維持することを阻止し、私たちの将来の候補製品の商業化コストを大幅に増加させ、わが社または私たちのパートナーがこれらの製品を商業化することによって収入を生成する能力を弱める可能性がある。
将来の候補製品のためのREMSの開発は、承認プロセスの遅延を招く可能性があり、追加の規制要求 を増加させる可能性があり、これは、将来の候補製品の米国での商業化能力に影響を与え、それらの市場潜在力を低下させる可能性がある。
FDAがREMSをNDAを承認する条件として要求することなく、将来の任意の候補製品のためのNDAを承認したとしても、FDAが新しいセキュリティ情報を知り、薬物の利益が潜在的リスクよりも大きいことを保証するためにREMSを必要とする場合、FDAは承認後に任意の将来の候補製品にREMSを提供することを要求する可能性がある。REMS要素には,薬品使用ガイドライン,医療専門家のコミュニケーション計画,および安全な使用を確保する要素が含まれていることができる(“ETASU“)”ETASUは、処方または調剤のための特殊なトレーニングまたは認証、特定の場合にのみ調剤、特殊な監視、および患者登録簿の使用を含むことができるが、これらに限定されない。また、製品承認には、薬物の安全性や有効性を監視するために、大量の承認後のテストと監視が必要となる可能性がある。私たちは乱用、誤用、移転、および他の潜在的な安全問題よりも収益が大きいことを確実にするために、私たちの未来の候補製品のためにREMSを採用する必要があるかもしれない。FDAが将来の候補製品の管理可能なREMS を承認する保証はありません。これは、米国で将来の候補製品の商業化に成功する能力に大きな制限を与える可能性があります。REMS承認プロセスの遅延は、NDA承認プロセスの遅延を招く可能性があります。さらに、REMSの一部として、FDAは、製品の処方、流通、および患者使用の制限のような重大な制限を実施することを要求する可能性があり、これは、将来の候補製品を効果的に商業化する能力に深刻な影響を与え、それらの市場潜在力を著しく低下させ、それによって、私たちの業務、財務状態、および運営結果に悪影響を及ぼす可能性がある。最初のREMが厳格な制限でなくても、もし私たちの未来の候補製品が発表後に深刻な乱用/非医療 使用または不正チャネルから移転された場合、これはより厳しいREMSを含む負の規制結果を招く可能性がある。
FDAと外国機関のような規制審査の流れは冗長で、時間がかかり、本質的に予測できず、もし私たちが最終的に私たちの候補製品のために監督管理の許可を得ることができなければ、私たちの業務は深刻な損害を受けるだろう。
FDAの規制の承認なしに、私たちは米国で商業化、マーケティング、普及、または任意の候補製品を販売してはいけない。EMAやMHRAのような外国の規制機関も同様の要求を実施している。FDAや類似外国当局の承認を得るのに要する時間は本来予測不可能であるが,通常は臨床試験開始後数年が必要であり,規制機関のかなりの裁量権を含む多くの要因に依存する。また,候補製品の臨床開発過程では,承認政策,法規あるいは承認を得るために必要な臨床データのタイプや数が変化する可能性があり,管轄区域によって異なる可能性がある。私たちはこれまで、Dupuyten‘s Constraintの早期治療のための最先端の候補製品、または任意の他の候補製品 を得るために、FDAにセキュリティ協定または比較可能な外国規制機関に同様の薬品承認申請を提出していない。私たちはもっと多くの臨床前研究と臨床試験を完成して、私たちの製品の人体上の安全性と有効性を証明して、私たちはこれらの承認を得ることができます。
79
臨床試験は費用が高く,設計や実施が困難であり,完成には数年かかる可能性があり,結果自体も確定していない。我々が提出した任意の臨床試験設計がFDA,MHRAまたは他の類似した外国の規制機関によって受け入れられるか,あるいは臨床試験が計画通りに行われるか,あるいは時間どおりに完了することは保証されない(あれば)。我々の最初および潜在的な他の候補製品の臨床開発は、臨床試験中または広範な患者集団で治療効果が証明されなかったことを含む任意の開発段階固有の失敗リスクの影響を受けやすく、深刻な、医学的または商業的に許容できない有害事象が発生し、合意または適用された法規要件を遵守できなかったこと、ならびにFDA、MHRAまたは任意の他の類似した外国規制機関が候補製品が開発または承認できない可能性があると判断した。可能なことは、私たちのどの候補製品が有益な効果を持っていても、臨床評価過程でこの効果が検出されないことであり、これは、私たちの臨床試験の規模、持続時間、設計、測定、進行または分析を含む1つまたは複数の要因が原因である。 は逆に、同じ要因により、私たちの臨床試験は、その候補製品の明らかなプラス効果、すなわち が実際のプラス効果よりも大きいことを示す可能性がある(あれば)。同様に,われわれの臨床試験では,候補製品の毒性や耐性 を検出できないか,あるいは候補製品の有毒や耐性が悪いと誤って考えられている可能性があるが,事実はそうではない。深刻な有害事象またはSAEまたは他の悪影響および耐性問題は、議論された候補製品の受け入れを阻害または阻止する可能性がある。
私たちの現在と未来の製品候補製品は、以下の理由を含む様々な理由で規制部門の承認を得ることができないかもしれない
| ● | FDA、MHRA、または他の同様の外国の規制機関は、私たちの臨床試験の設計または実施に同意しないかもしれない | |
| ● | FDA、MHRA、あるいは他の類似した外国の監督管理機関を満足させ、候補製品が私たちが提案した適応に対して安全かつ有効であることを証明することはできないかもしれない | |
| ● | 臨床試験の結果はFDA、MHRA或いは他の類似した外国監督管理機関が許可した統計的意義レベル に符合しない可能性がある | |
| ● | 私たちはbr候補製品の臨床的および他の利益がその安全リスクよりも大きいことを証明できないかもしれない |
| ● | FDA、MHRA、または他の類似した外国の監督管理機関は、臨床試験または臨床前研究データの解釈に同意しないかもしれない | |
| ● | 我々の候補製品の臨床試験から収集されたデータは、セキュリティ協定のFDAへの提出または他の提出をサポートするのに十分ではない場合があり、または米国、EU、または他の場所の規制承認を得るのに十分ではないかもしれない |
| ● | FDA、MHRA、または他の同様の外国の規制機関は、私たちと臨床および商業供給契約を締結した第三者メーカーの製造プロセスに欠陥があることを発見するかもしれない | |
| ● | FDA、MHRA或いはその他の類似外国監督管理機関の承認政策或いは法規は重大な変化が発生する可能性があり、著者らの 臨床データは承認を得るのに不十分である。 |
80
この長い承認過程 や臨床試験結果の予測不可能性は,規制部門の承認を得ることができず,我々が開発した任意の候補製品 を市場に出すことができず,我々の業務,運営結果,見通しに深刻な被害を与える可能性がある。FDA、MHRA、または他の同様の外国当局は、承認過程において大きな自由裁量権を有し、私たちが開発した任意の候補製品がいつ、または規制承認を受けるかどうかを決定する。我々の候補製品の将来の臨床試験から収集されたデータが有望であると信じていても,これらのデータはFDAや任意の他の規制機関の承認を支援するのに不十分である可能性がある。
さらに、私たちが承認されても、規制機関は、私たちの要求された範囲以下またはそれ以上の適応を承認することができ、私たちが製品のために徴収しようとしている価格を承認しない可能性があり、承認は、高価な発売後の臨床試験の表現に依存するか、または候補製品のラベルを承認する可能性があるが、ラベルには、候補製品の商業化に必要または必要な声明は含まれていない。上記のいずれの場合も、私たちの候補製品のビジネス見通しに実質的な損害を与える可能性があります。
私たちの第三者への依存に関するリスク
私たちの既存の協力 計画および将来達成可能ないかなる計画も成功しない可能性があり、これは、将来の候補製品を開発し、それを商業化する能力に悪影響を及ぼす可能性がある。
私たちは、私たちの将来の候補製品を開発したり、商業化したりするために、製薬やバイオテクノロジー会社とより多くの協力計画を達成することを求めている可能性があります。我々の将来の候補製品については,米国や国際的に有力な製薬会社やバイオテクノロジー会社と各候補製品について選択的に協力することと比較して,我々自身の商業化権利を保持する利点に応じて,新たな計画を選択的に行う可能性がある。私たちが協力協定を締結することを決定した範囲では、私たちは適切なパートナーを探す上で激しい競争に直面し、 私たちが確立する可能性のある任意の協力または他の手配の条項は私たちに不利になるかもしれない。
私たちが参加するどんな既存または未来の協力 も成功しないかもしれない。我々の協調スケジュールの成功は,我々の協力者の努力や活動 に大きく依存する.協力者は通常、彼らがこれらの協力の仕事と資源に適用されることを決定する上で大きな裁量権を持っている。協力手配各方面間の開発、知的財産権、監督管理或いは商業化問題における相違は適用候補製品の開発過程或いは商業化過程の遅延 を招く可能性があり、場合によっては、協力手配を終了することもある。双方の が最終決定権を持たなければ,これらの分岐は解決が困難である可能性がある.このような終了または満了のいずれも、私たちの商業的名声を損なう可能性があり、その財務に悪影響を及ぼす可能性がある。
私たちは限られた数のサプライヤーに材料とコンポーネントを提供して、私たちの未来の候補製品を生産することを望んでいます。これらのbrサプライヤーを失ったり、彼らが適時に私たちに供給することができなくて、私たちの現在と未来の生産能力の遅延を招き、私たちの業務に悪影響を及ぼす可能性があります。
私たちは限られた数のサプライヤーに依存して、私たちの未来の候補製品を製造するために必要な材料とコンポーネントを提供する予定です。したがって、私たちは未来に十分な数の重要な材料と構成要素を得ることができないかもしれない。仕入先の遅延や中断はまた、私たちの業務、運営結果、そして財務状況を損なう可能性があります。また、新規仕入先との関係構築に必要な納期が長くなる可能性があり、新仕入先に切り替えなければならない場合、需要を満たす上で遅延が生じる可能性があります。新しいサプライヤーの資格を獲得し、場合によっては規制承認を得るためにかかる時間と労力は、追加コスト、資源移転、または製造生産量の低下を招く可能性があり、いずれも私たちの運営結果にマイナスの影響を与える。私たちの単一ソースサプライヤーへの依存は、私たちのサプライヤーが生産または納品を停止または減少する可能性があり、彼らは政府の調査と規制行動の影響を受ける可能性があり、より長い時間内に生産能力を制限または阻止し、価格を向上させたり、再交渉条項を再交渉したりする可能性がある;私たちのサプライヤーは借金を支払うことができないかもしれない;私たちは許容可能な条項や適時または根本的に適切な代替サプライヤーを見つけることができないかもしれない;供給問題による遅延は私たちの名声を損なう可能性があり、私たちの顧客を落胆させ、未来の需要を満たすために私たちの競争相手に転向させる可能性がある。
81
私たちの知的財産権に関するリスクは
私たちは私たちの未来の候補製品や私たちのノウハウを市場で十分に保護できないかもしれない。
私たちの成功は、私たちが特許を取得し、私たちのビジネス秘密を保護し、他人の独占権を侵害することなく運営する能力にある程度依存するだろう。私たちは、特許、商業秘密保護(すなわち、ノウハウ)と秘密保護協定の組み合わせによって、私たちの未来の候補製品の知的財産権を保護する。製薬分野における特許の優位性は複雑な法律や科学的問題に関連しており,不確実である可能性がある。適切な状況で、私たちは私たちの製品と技術のいくつかの側面のために特許保護を求めている。世界的に特許を申請、起訴、保護する費用は目を引くほど高いかもしれない。
私たちの政策は巨大なビジネス機会を持つ司法管轄区域で商業的潜在力のある特許技術を探すことです。しかし,特許保護は我々が開発しているいくつかの製品や技術には適用できない可能性がある.もし私たちが保護、br保護、または強制的に私たちの特許を実行するために多くの時間とお金をかけなければならない場合、他の人が持っている特許をめぐって設計することができるかもしれないし、高額な費用、特許、または他の他人が所有する独自の権利を支払う必要があるかもしれないし、私たちの業務、運営結果、および財務状況は損害を受ける可能性がある。私たちは特許を申請できる他の独自製品を開発しないかもしれない。本稿の発表日まで、私たちは多くの許可された特許と承認されている特許を含む広範な特許の組み合わせを持っています。
薬品br製品の特許地位は複雑で不確定である。私たちの未来の候補製品の特許保護範囲と程度は特に確定していません。私たちの未来の候補製品は大麻植物ではなく、薬物化学に基づいています。特許保護が求められているが, は適切な場合には,他の事項に加えて,その特定の配合の物質成分,その使用方法,製造方法についても,これら先に知られているCBD誘導体の物質成分保護 自体が得られないことはない。私たちは私たちが未来に開発した製品が私たちが発見可能な合成化合物に基づいていると予想する。私たちは、米国、ヨーロッパ、および他の国/地域で独自技術、将来の候補製品、それらの使用方法、および製造方法のために特許保護を求め続けているにもかかわらず、それらのいずれかまたは全部が有効な特許保護を受けていない可能性がある。もし私たちのどの製品も特許を発行していないために承認され販売されていれば、競争相手が私たちの商業製品の非ブランドバージョンを商業化する能力が深刻な損害を受け、淘汰される可能性がある。
わが社や他社が将来の候補製品に関する情報を発表することは、これらのbr製品や候補製品に関する特許の取得または強制実行を阻止する可能性があります。また、他の人は、類似した製品を独立して開発する可能性があり、私たちの製品を複製したり、私たちの特許権を巡って設計したりする可能性があります。さらに、私たちが発行したどんな特許も反対され、および/または無効または実行不可能だと宣言される可能性がある。もし私たちが私たちの知的財産権を十分に保護できなかったら、私たちは会社からの競争に直面するかもしれません。これらの会社は私たちの未来の候補製品と競争するために汎用製品を作成しようとしています。私たちはまた会社からの競争に直面するかもしれません。これらの会社が開発した製品は私たちの未来の候補製品とほぼ似ていますが、私たちのどの特許もその製品をカバーしていません。
多くの会社は外国司法管轄区の知的財産権を保護、保護、実行する上で重大な問題に直面している。特定の国の法律制度、特に特定の発展途上国の法律制度は、特許および他の知的財産権の強制執行、特に薬品に関連する権利を支持しておらず、これは、私たちの特許を侵害したり、私たちの独占権を侵害する方法で競争製品を販売することを阻止することを困難にする可能性がある。外国の管轄区域で私たちの特許権を強制的に執行する訴訟は巨額のコストを招く可能性があり、私たちの努力と注意を私たちの業務の他の側面から移します。
82
もし第三者がわが社が使用している知的財産権が彼らの知的財産権を侵害していると主張すれば、私たちの運営利益は悪影響を受ける可能性がある。
米国内および海外では、製薬業界の特許および他の知的財産権に関する多くの訴訟がある。私たちは時々、第三者が所有している特許、商標、著作権または他の知的財産権を侵害しているという通知を受ける可能性があり、他の会社が将来、私たち、私たちの商業パートナー、または私たちが許可しているいかなる第三者独自技術にもこのような侵害を提起しない保証はない。特許または他の知的財産権の侵害が発見された場合、または第三者から特許または他の知的財産権の許可を取得または更新できなかった場合、またはその許可技術の第三者に他の第三者の特許または他の知的財産権の侵害が発見された場合、損害賠償金を含む損害賠償金の支払いを要求される可能性があり、最高で損害賠償金の3倍まで発見または評価することができ、侵害が故意であることが発見された場合、特定の製品の生産を一時停止したり、私たちの製品を再設計したり、可能であれば、あるいは私たちは特定の新製品市場に入ることができないかもしれない。このようなクレームはいずれも費用が高くなる可能性があり、経営陣の注意と資源を弁護して移転するのに多くの時間がかかる。したがって、私たちの競争地位は影響を受ける可能性があります。 また、私たちが何らかの理由で有効なセキュリティ協定や譲渡協定を拒否または締結できなかった場合、私たちはその発明やその知的財産権を持っていない可能性があり、私たちの製品は十分な保護を受けられないかもしれません。したがって、私たちは私たちの未来の任意の候補製品やその商業化が第三者の知的財産権を侵害しないことを保証することはできない。
ビジネス秘密や他の独自の情報漏洩を十分に防ぐことができなければ、私たちの技術や製品の価値は大幅に縮む可能性がある。
私たちは、特に特許保護が不適切であるか、または入手不可能であると考えられる場合に、ビジネス秘密brに依存して私たちのノウハウを保護する。しかし、商業秘密は保護することが難しい。私たちは、現在および元従業員、コンサルタント、外部科学協力者、協賛研究者、契約メーカー、サプライヤー、および他のコンサルタントと締結された秘密協定に部分的に依存して、私たちのビジネス秘密 および他の固有情報を保護します。これらのプロトコルは、機密情報の漏洩を効果的に阻止できない可能性があり、機密情報を不正に漏洩させた場合に適切な救済措置を提供できない可能性がある。さらに、私たちは私たちが可能であるか、または私たちの商業秘密に訪問したすべての当事者とこのような合意を実行したという保証はない。私たちまたは彼らとこのような合意に署名したいずれの当事者も、その合意に違反し、私たちのビジネス秘密を含む独自の情報を漏洩する可能性があり、私たちは、そのような違反に対して十分な救済措置を得ることができないかもしれない。
一方が商業秘密を不法に開示したり流用したりする疑惑を強制することは困難で、高価で時間がかかり、結果は予測できない。また、米国国内外のいくつかの裁判所は商業秘密をあまり望んでいないか、または保護したくない。もし私たちの任意の商業秘密 が競争相手によって合法的に取得または独立して開発された場合、私たちは、彼らまたは彼らがそのような商業秘密を漏らした人がその技術または情報を使用して私たちと競争することを阻止する権利がないだろう。もし私たちのビジネス秘密がbrに漏れたり、競争相手や他の第三者によって独立して開発されたりすれば、私たちの競争地位は損なわれるだろう。
特許保護の満期やbrの喪失は、私たちの将来の収入や運営収益に悪影響を及ぼす可能性がある。
私たちは、私たちの候補製品を発見、開発、製造、販売する際に、特許、商標、商業秘密、および他の知的財産権保護に依存しています。特に、特許保護は、私たちの候補製品の開発と最終商業化の過程で非常に重要です。我々の候補製品をカバーする特許は通常市場排他性を提供しており,これは我々の候補製品 が利益を得る確率を高めるために重要である.
線維化および抗腫瘍壊死因子計画に関連する特許は2033年に満了するが、ほとんどの特許組み合わせの寿命はより長い。私たちは、これらの特許の背後にある技術を保護する可能性のある追加の特許カバー範囲を求めているが、 がこのような追加の特許保護を付与することが保証されていないか、またはこれらの特許が付与されている場合、これらの特許を侵害しないか、またはそれが強制的に実行可能であることを侵害しない。 たとえ私たちが特許を得ることに成功しても、特許の寿命は限られている。米国では,実用プログラム特許の自然失効期間は通常出願後20年である.様々な延期を使用することができる;しかし、特許の有効期間およびその提供される保護は限られている。もし私たちの候補製品が特許保護がなければ、私たちはこのような方法と組成物からの模造バージョンの競争に直面するかもしれない。
83
もし私たちが特許期間を延長することで“ハッジ·ワックスマン改正案”によって保護されなければ、私たちの業務は損なわれる可能性がある。
私たちのビジネス成功は、私たちの候補製品に対して、アメリカや他の国/地域で特許や他の知的財産権を取得し、維持する能力に大きく依存するだろう。新候補製品の開発、テスト、規制審査に要する時間を考慮すると、候補製品を保護する特許は、候補製品の商業化開始前または直後に満了する可能性がある。私たちは米国で特許期間の延長を求める予定であり,もしあれば,特許を提訴した他の国/地域でも延長を求める予定である。
私たちの候補製品の時間、持続時間、およびFDA上場承認の具体的な状況によると、私たちの1つ以上の米国特許は、1984年の“薬品価格競争および特許期限回復法”(“ハジ-ワックスマン修正案”と略称する)に従って限られた 特許期間延長またはPTEを得る資格がある可能性がある。Hatch−Waxman修正案は、開発およびFDA規制審査中に失われた特許期間の補償として、特許回復期間が特許正常満了後最大5年となることを可能にし、これは、特許がカバーする承認された適応 (および延長期間内に承認される可能性のある他の適応)に限定される。この延期は、承認された製品、製品の承認された用途、または製品の製造方法をカバーする特許に限定される。しかし、米国のFDAとUSPTO、および他の国/地域の任意の同等の規制機関を含む当局は、このような延期が利用可能かどうかの評価に同意しない可能性があり、私たちの特許延期の承認を拒否する可能性があり、または は私たちが要求したよりも限られた延期を承認するかもしれない。我々は,適用された 締め切り内に出願できなかったこと,関連特許が満期になる前に出願できなかったこと,または適用要求を満たしていなかったことなどから延期が得られなかった可能性がある.さらに、 が適用される期間または提供される特許保護範囲は、私たちの要求よりも小さい可能性がある。たとえ私たちがbr延期を得ることができても、特許期間はFDAの上場承認を得る前または直後に満期になる可能性がある。もし私たちが既存特許のbr満期日を延長できない場合、あるいはより長い満期日の新しい特許を得ることができなければ、私たちの競争相手は私たちの開発と臨床試験への投資を利用して、私たちの臨床と臨床前データを参考にして、私たちの特許が満期になった後に競争相手製品の承認を得て、他の場合よりも早く彼らの製品を発売することができるかもしれない。
規制された物質に関するリスク
制御物質 は各国の立法によって異なり,ある国/地域の立法は将来の製品を販売する能力 候補製品を制限または制限する可能性がある。
多くの国は1961年麻酔薬単一条約と1971年精神薬条約の締約国であり,この2つの条約は大麻抽出物を含む麻酔薬の国際貿易と国内規制を管理している。各国はその条約義務brを解釈して履行する可能性があり,これは我々の将来の製品がこれらの国でマーケティング承認を得ることに法的障害をもたらす可能性がある。これらの国/地域 は、私たちの将来の製品の発売を可能にするために、またはその法律法規を修正することを望まないか、または法律法規のこのような改正を達成するのに時間がかかるかもしれない。この場合、私たちは近い将来、またはこれらの国/地域で私たちの未来の候補製品 を販売することができないだろう。
我々が開発している候補製品 は米国の制御物質法律法規の制約を受ける可能性があり,これらの法律法規を遵守しない場合やこれらの法律法規を遵守するコストは,臨床 開発期間中も承認後も,我々の財務状況にも,我々の業務運営結果に悪影響を及ぼす可能性がある。
我々が開発している候補製品には,1970年の米国連邦制御物質法案やCSAで定義された制御物質が含まれている可能性がある。薬品としての制御物質はCSAの高度な規制を受けており、CSAは他の事項のほかに、DEA管理のいくつかの登録、製造割当、安全、記録、報告、輸入、輸出とその他の要求 を規定している。DEAは制御物質を5つの付表に分けた:付表I,II,III,IVまたはV物質。付表 i定義により,物質は高い濫用の可能性があり,現在はない“公認医療用途“米国では、LASK は医療監督下で使用される安全性を受けており、米国で処方、販売、販売されてはならない。米国で使用が許可されている制御物質含有薬品は、付表II、III、IVまたはVとされており、付表II物質は乱用または依存する可能性が最も高い物質とされており、付表V物質はこのような物質の中で乱用の相対的リスクが最も低い物質と考えられている。
84
大麻は別表一規制物質であるが,米国で医療用途のための大麻または大麻抽出物を含む製品は,FDAの承認が適合しているので別表2~別表5に登録しなければならない“公認医療用途“要求。もし私たちの未来の任意の候補製品がFDAの承認を受けたら、DEAは手配決定を下すだろう。FDA、DEA、または任意の外国規制機関が、私たちの将来の候補製品が乱用の可能性があると判断した場合、私たちが現在予想しているよりも多くの臨床データまたはbr}の他のデータを生成して、物質がどの程度乱用の可能性があるかどうかを決定する必要があるかもしれません。これは、コストを増加させ、および/または製品の発表を延期する可能性があります。
研究を行う施設では,これらの活動を実行するためには, 製造,配布,輸入,輸出または配布制御物質を登録(許可)しなければならず,DEAに必要な安全,制御,記録保存,報告,在庫機構を有しており,薬物 の紛失や転移を防止している。調剤施設は3年ごとに更新されなければならないほか,これらの施設は毎年更新登録されなければならない。DEAはある規制対象物質を処理する登録機関を定期的に検査している。必要な登録を得ることは、私たちの未来の製品の輸入、製造、または流通遅延を招くかもしれない。また, はCSAを遵守できず,特にCSAを遵守しないことにより損失や移行が起こり,規制行動 を招く可能性があり,これは我々の業務,財務状況,運営結果に実質的な悪影響を与える可能性がある.DEAは民事処罰を求め、必要な登録の更新を拒否したり、制限、一時停止、またはこれらの登録の手続きを開始したりすることができる。場合によっては、違反は刑事訴訟を引き起こす可能性がある。
各州でも制御物質法律法規が制定された。州制御物質法律は通常連邦法律を反映しているが,各州は単独の司法管轄区であるため,将来の候補製品を個別に手配する可能性もある。州スケジュールは、連邦規制によって承認された任意の製品の商業販売を延期する可能性があり、不利なスケジュールは、このような製品の商業的魅力に大きな悪影響を及ぼす可能性がある。私たちまたは私たちのパートナーはまた、臨床試験または商業販売のための制御された物質を取得、処理、配布するために、単独の州登録、許可または許可を取得しなければならず、適用された規制要件を満たしていない場合、DEAの要求に加えて、各州が強制的に執行し、制裁するか、または連邦法律 に基づいている可能性がある。
我々の製品は米国では制御物質である可能性があるため,米国で臨床試験を行うためには,我々の各研究サイトはDEAに研究案 を提出し,DEA研究者登録を取得して維持し,これらのサイトが我々の製品を処理·分配し,我々の輸入業者から製品を入手することを可能にしなければならない。DEAが1つまたは複数の研究サイトへの研究登録の許可を延期または拒否する場合、臨床試験は著しく遅延する可能性があり、私たちは臨床試験サイトを失う可能性がある。臨床試験の輸入業者はまた輸入業者の登録と毎回の輸入許可証を取得しなければならない。
この分野はまだ完全に統一されていないので、EU加盟国のマリファナに関する立法はそれぞれ違う。例えばドイツでは大麻は規制物質として規制されていますBit ubungsmitel)であり、その処理には特定の許可が必要である。
アメリカや海外では、医療や娯楽用大麻の合法化と使用が私たちの業務に影響を与える可能性がある。
医療や娯楽用大麻製品の使用において,米国は大きく変化している。Worldpoplulationreview.comのデータによると,FDAの承認されていない大麻製品 は連邦法で定義された付表I物質であり,連邦法律により大麻の保有と使用は許可されていないが(DEA登録により研究目的のため除外),少なくとも39州とコロンビア特区が州法を公布し,医療目的で大麻の所有と使用を許可しており,少なくとも19州とコロンビア特区が娯楽目的である。2018年に成立した米国農業法案は、大麻植物から抽出したいくつかの材料“br”のTHC含有量が極めて低いことを廃止した。私たちの業務はオンラインや薬局大麻会社とは異なるにもかかわらず、将来的に許可された販売、流通、使用、保険精算はFDAで承認されていない大麻製品の立法が私たちの業務に影響を与える可能性がある。
85
会計リスク
私たちの名誉と無形資産は過去に減価され、未来の減価リスクの影響を受けている。
以下のリスク要因で述べたように,2022年12月31日までの年間で,我々の営業権と進行中の研究開発には重大な減価費用が計上されている。
2022年12月31日現在、私たちの無形資産は約1,070万ドルで、私たちの総資産の55%を占めている。当社は、無期限無形資産と営業権の潜在的減値 を少なくとも年に1回評価し、状況の発生や変化が減値の可能性を示す証拠がある場合に評価を行う。公正な価値を評価する際に使用される多くの要素は管理職の制御範囲内ではなく、将来的に変化する可能性が高いと仮定し、推定する。これらの変化は将来の減値を招く可能性がある。 社は減値を引き起こす可能性のある重要なイベントと状況は以下の通りであると考えている
| ● | 歴史や予測された未来の経営業績と比較して、業績は著しく不良である |
| ● | 会社全体の業務戦略や資産買収使用戦略に大きな変化が生じている |
| ● | 大きな負の産業や経済的傾向 |
| ● | Br社の株価は大幅な下落を続けている |
| ● | 時価は帳簿純価値に対して低下する ; |
| ● | 予期しなかった技術的変化や競争活動 |
| ● | 消費需要の変化 |
| ● | キーパーソンの流出 |
| ● | 政府や裁判所の行動です |
無形資産の帳簿価値が上記の1つまたは複数の指標の存在により回収できない可能性があることを示す兆候がある場合、資産の帳簿価値がその公正価値を超える場合には、減価損失を確認する。営業権に減値の兆候がある場合、営業権の帳簿金額がその暗黙的公正価値を超えた場合、減値損失を確認する。
その時点または将来の他の時間に存在する状況の変化、または会社がその無期限無形資産または営業権の適切な推定値を評価する際の仮定および推定に関連する多くの変数の変化は、将来的には、このような費用が私たちのキャッシュフローに影響を与えないにもかかわらず、将来報告される運営実績および株主権益に悪影響を及ぼす会社に減価費用を記録することを要求する可能性がある。
86
私たちは過去、br、および未来に商業権や買収を含む長期資産や無形資産を損なう可能性があり、研究と開発が行われている。
状況や状況が変化して帳簿価値が回収できない可能性があることを示すたびに、当社は長期資産およびいくつかの識別可能資産(無形資産を含む)の減値を審査する。長期的に使用されているbrまたは無形資産(営業権および買収における研究開発を含む)の帳簿価値が回収できず、その推定された公正価値を超える場合、すなわち減値が存在する。営業権とは、企業合併で買収された資産と負債の購入価格と公正価値との差額を意味する。当社は毎年商業権を審査し、あるいは状況と状況が変化した時により頻繁に商業権を審査して、定性要素を初歩的に考慮して、報告単位の公正価値がその帳簿金額(営業権を含む)よりも少ない可能性があるかどうかを確定し、これを数量化分析が必要かどうかを決定する基礎とする。報告単位の公正価値がその帳簿価値よりも低い可能性が よりも大きいと判定された場合、商誉 の減少を確認するために定量化分析が行われる。
2021年12月31日現在、上場株の終値は1株78.00ドルであり、2022年の間に会社単一報告単位の時価は大幅に低下した。当社の上場株の時価は、2022年3月31日、2022年6月30日、2022年9月30日、2022年12月31日まで、それぞれ1株51.80ドル、16.96ドル、13.30ドル、3.39ドルに下落したため、当社は2022年9月30日と2022年12月31日までの売上高を定量的に分析し、減値を評価することを選択した。当社はその単一報告単位の公正時価を確定し、そしてこの価値を報告単位の帳簿価値と比較し、営業権が2つの計量日にすべて減値されたことを確定した。2022年9月30日と2022年12月31日まで、帳簿価値はそれぞれ公平市場価値より18,872,850ドルと14,674,428ドル高かった。営業権減額を確認するために、会社は第3四半期と第4四半期末にこれらの金額の損失 を記録し、2022年12月31日までの年間収益表では営業権減価損失33,547,278ドルを示した。さらなる情報については、F−1ページからの連結財務諸表の“付記5--無形資産および長期資産減価” を参照されたい。
無形資産と進行中の研究開発(“IP R&D”)資産とは、2019年7月16日に再編によって得られた技術に割り当てられた公正な価値であり、これらの技術はまだ技術可能性に達しておらず、将来的にも他の用途はない。関連する研究開発プロジェクトが完成または放棄されるまで、無期限とされている。知的財産権研究開発資産が無期限存続とみなされている間に、当社は知的財産権研究開発資産の公正価値がその帳簿額面より低い任意の事件或いは状況変化を発見した場合、毎年減値テストを行い、あるいはより頻繁に減値テストを行う。開発が完了し(通常は規制部門の承認後に完了)、企業が知的財産研究開発資産に関連する製品を商業化することができる場合、これらの資産は、使用寿命が決定されたとみなされ、その時点での推定使用寿命に基づいて償却される。もし開発を中止または放棄すれば、当社は知的財産権研究開発資産の帳簿価値がその推定公正価値を超えて計算され、知的財産権研究開発資産に関連するすべてまたは部分的な減価費用を計上することができる。
2022年12月31日現在、貸借対照表上の知的財産権研究開発資産の帳簿金額は12,405,084ドル(それぞれ自社のCBR Pharma子会社とその180 LP子会社に関連する帳簿金額1,462,084ドルと10,943,000ドルを含む)である。年末までに第三者から得られた推定値によると、当社の知的財産権研究開発資産の公正時価は9,063,000ドル(ただし、br}は当社のCBR Pharma子会社および180 LP子会社との公正価値がそれぞれ0ドルと9,063,000ドルを含む)に決定された。 は本計量日に、CBR Pharmaと180 LP子会社の資産の帳簿価値はそれぞれ公正市場価値より1,462,084ドルと1,880,000ドル高い。そこで、経営陣は総合知的財産権研究開発資産減額3,342,084ドルを確定し、減額を確認するため、当社は2022年第4四半期にこの金額に赤字を計上し、損益表では知的財産権研究開発資産減価損失を示した。2022年12月31日まで、そのCBR Pharma子会社 及び180個のLP子会社の知的財産権研究開発資産残高はそれぞれゼロと9,063,000ドルに低下した;減値後の総合知的財産権研究開発資産残高 は9,063,000ドルであった。詳しくは、F-1ページから始まる連結財務諸表の“付記5--無形資産と長期資産減価”を参照されたい。
私たちの普通株の取引価格が持続的に低迷していることは、私たちの報告単位にさらなる大きな減価を強いる可能性があり、これは私たちの資産価値に重大な影響を与え、私たちの証券価値を低下させる可能性がある。また、私たちの過去および未来は、私たちの無形資産の減値、買収を含めて行われている研究と開発が必要であり、実質的である可能性があることが確認されるかもしれない。1つの期間で確認された減少値は、私たちの普通株の将来的な価値が増加しても、その後の期間に出荷されない可能性がある。私たちは過去に、買収が行われている研究開発および営業権を含む追加の長期資産および/または無形資産減価を将来的に生成する可能性があり、これは実質的である可能性がある。
87
私たちは統制と手続き、そして財務報告書の内部統制を開示する上で私たちの大きな弱点を発見した。救済措置を講じなければ、効果的な開示制御や手続き、財務報告の内部統制を確立し、維持することができず、財務諸表に重大な誤報が発生し、私たちの報告や財務義務を履行できない可能性があり、いずれも私たちの財務状況や私たちの証券の取引価格に大きな悪影響を及ぼす可能性がある。
企業経営陣は、我々の最高財務官を含め、トレデビル委員会後援組織委員会(COSO)が“内部統制-総合枠組み”(2013)で提案した基準を用いて、2022年12月31日までの社内統制の有効性を評価した。
経営陣は、これらの基準に基づき、2022年12月31日現在、社内統制の一部は発効していないと結論した。具体的には、経営陣の結論は以下のような重大な弱点に基づいている
| ● |
当社の審査と制御プログラム は適切な精度レベルで動作しておらず、一度の逆方向株式分割に関する権証公正価値誤りと知的財産権研究開発資産の公正価値を発見することができない |
重大な欠陥は財務報告内部制御の欠陥或いは欠陥の組み合わせであるため、合理的な可能性があり、会社の年度或いは中期財務諸表の重大な誤報を適時に防止或いは発見できない可能性がある。したがって、重大な弱点は私たちが報告した財務情報に重大なミスを含む危険を増加させる。制御の設計または操作が管理層または従業員がその指定された機能を実行する正常な過程中に直ちに誤った陳述を防止または発見することを許可しない場合、制御欠陥が存在する。
財務報告に対する有効な開示 の制御プログラムと効率的な内部制御を維持することは、信頼できる財務諸表を作成するために必要であり、当社は、このような制御における重大な弱点を早急に是正することに取り組んでいる。しかし、これらの重大な弱点をいつ救済するか、または将来より多くの重大な弱点が発生しない保証はありません。重大な弱点を救済できなかった場合や、財務報告の内部統制に新たな重大な弱点が発生した場合は、私たちの財務諸表に重大なミスが発生し、私たちの報告および財務の義務をタイムリーかつ正確に履行できなくなる可能性があります。もし私たちの財務諸表が正確でなければ、投資家は私たちの運営について完全に理解していないかもしれません。あるいは私たちが報告した財務情報に自信を失ってしまうかもしれません。同様に、私たちの財務諸表がアメリカ証券取引委員会とナスダックの要求に従ってタイムリーに提出されなければ、私たちはこれらの当局の深刻な結果に直面するかもしれない。上記のいずれの場合も、それは私たちの業務、私たちの財務状況に重大な悪影響を及ぼすか、または私たちの普通株式と引受権証の取引価格に悪影響を及ぼす可能性がある。さらに、私たちがこの欠陥(または未来の任意の他の欠陥)を補うことができなかったり、私たちの開示統制と手続きおよび私たちの内部統制の十分性を維持できなかった場合、私たちは規制審査、民事または刑事罰、または株主が私たちまたは私たちの経営陣に訴訟を提起する可能性があります。
88
私たちは、発見された重大な弱点を補うために、または私たちの財務諸表が財務報告書の十分な内部統制を実施および維持することができないこと、またはこれらの制御を実施および維持することによって、未来に任意の追加的な重大な弱点または再記述が生じることを保証することができない。
また、将来的には、財務報告書の効果的な内部統制を行っていると結論できなければ、あるいは私たちの独立登録公共会計事務所が財務報告の内部統制の有効性について保留していない意見(将来必要となる程度)を提供できない場合、投資家は私たちの財務諸表の信頼性に自信を失う可能性があり、これは私たちの株価の下落を招く可能性がある。報告要件を遵守できなかったことは、米国証券取引委員会またはナスダック(状況に応じて)または他の規制機関の制裁および/または調査を受ける可能性もある。
また、我々が我々の制御プログラムやプログラムを強化することに成功しても、これらの制御プログラムは、違反 を防止または識別したり、米国証券取引委員会に提出された財務諸表や定期報告の公正な列報を促進するのに十分ではない可能性がある。これは私たちが以前の財務諸表を再説明する必要があるかもしれない。
新しい会計基準や解釈を採用するため、私たちが報告した運営結果に悪影響を及ぼす可能性があります。
新たな会計ルールや解釈を含む会計ルールの変化を実行し、遵守することは、私たちが報告した財務状況や経営業績に悪影響を与えたり、将来報告される経営業績に予期せぬ変動を招いたりする可能性があります。
私たちの普通株式と引受権証に関連するリスク
私たちの普通株の市場価格はずっと非常に不安定で、私たちがコントロールできない場合が多いため、私たちの普通株は変動し続ける可能性があります。
多くの要素のため、私たちの普通株の市場価格はずっと変動していて、引き続き大幅に変動するかもしれません。その中のいくつかの要素は私たちがコントロールできないかもしれません。これらの要因 は含まれるが、これらに限定されない
| ● | “空振りスクイズ”; |
| ● | ブログ、文章、伝言板およびソーシャルメディア、および他のメディアを含む証券アナリストまたは他の第三者のコメント |
| ● | 大株主はわが社の証券頭寸を脱退したり、“空株数”わが社証券を増減させたりした |
| ● | 財務と経営業績の実際または予想変動 ; |
| ● | 持続的な新冠肺炎の大流行に関連するリスクと不確実性 ; |
| ● | 外貨レート変動 ; |
| ● | 私たちの候補製品または競争相手の製品の計画または将来の臨床試験の開始、登録または結果 |
| ● | 競争薬や治療法の成功は |
89
| ● | アメリカと他の国の法規または法律動態 ; |
| ● | 競争力のある製品や技術の成功 |
| ● | 特許出願、発行された特許または他の固有の権利に関連する開発または論争 ; |
| ● | キーパーソンの採用や退職 ; |
| ● | 私たちの候補製品や臨床開発計画に関連する費用レベル |
| ● | 訴訟事項は、会社幹部と取締役保険政策によって回収または回収できない金額、会社の監督行動に影響するbrとその結果を含む | |
| ● | 他の候補製品かもしれない努力の結果を発見,開発,獲得した |
| ● | 財務結果、発展スケジュール、または証券アナリストの提案に関する推定実際または予想変化 ; |
| ● | 私たちはbrを得ることができないか、承認された薬物の十分な薬品供給を遅延させることができないか、または許容可能な価格で得ることができない |
| ● | 特許、訴訟事項、および私たちの技術のための特許保護を受ける能力を含む、特許権に関する紛争または他の事態の発展 |
| ● | 特許または株主訴訟を含む重大な訴訟; |
| ● | 私たちの財務業績や私たちに似ていると思われる会社の財務業績の変化 |
| ● | 任意の承認された薬物の保証範囲および十分な精算を含む医療支払いシステム構造の変化 |
| ● | 製薬とバイオテクノロジー部門の市場状況 |
| ● | 米国と海外の金融市場の全体的な経済、政治、市場状況、および全体的な変動 |
| ● | 投資家たちは私たちと私たちの業務に対する全体的な意見を持っている。 |
株式市場、特に私たちの株価は最近極端な価格や出来高変動を経験しており、これらの変動は往々にしてこれらの会社やわが社の経営業績に関係なく、あるいは比例しないことが多い。例えば、2022年の間、私たちの普通株の販売価格は、分割後調整後の高値1株80.70ドル(2022年1月5日)から安値1.18ドル(2022年12月23日)まで様々である。この間、私たちの財務状況や経営業績は何の大きな変化もなく、このような価格変動や取引量を説明することはできませんでした。しかし、私たちは既存の株主権益を希釈した株式を販売しました。これらの広範な市場変動 は我々の証券の取引価格に悪影響を及ぼす可能性がある.さらに、これらおよび他の外部要因は、私たちの普通株の市場価格や需要を大幅に変動させ続ける可能性があり、これは、私たちの株主が持っている私たちの普通株をいつでも販売することを制限または阻止する可能性があり、そうでなければ、私たちの普通株の流動性に悪影響を及ぼす可能性がある。
90
第三者によって発表されたパブリックメディア(ブログ、文章、伝言板、ソーシャルメディア、および他のメディアを含む)によって提供される情報 は、会社とは無関係な声明を含む可能性があり、信頼できないか、または不正確である可能性がある。
私たちは、第三者がブログ、伝言板、ソーシャルメディア、および他のメディアを含めて、私たちの業務に関する多くの情報を伝播していることを認識している。第三者が報告したこのような情報は正確ではない可能性があり、私たちの証券の大幅な変動を招き、最終的には私たちの普通株または他の証券価値の低下を招く可能性がある。
私たちの未償還オプションと引受権証は私たちの証券の取引価格に悪影響を及ぼす可能性があります。
2022年12月31日まで、私たちは(I)加重平均発行権価格で1株84.63ドルで合計162,956株の普通株を購入できる既発行株式オプションを持っている;(Ii)加重平均行権価格で1株33.94ドルで3,435,728株の普通株を購入できる発行済株式証を持っている。オプションおよび引受権証の有効期間内に、保有者は、所有権リスクを負うことなく、私たち普通株の市場価格上昇から利益を得る機会がある。発行済み証券の行使時に発行株も我々の既存株主の所有権利益を希釈する。
これらの株式は、公開転売可能であり、これらの株の任意の実際の転売は、私たちの普通株の取引価格に悪影響を及ぼす可能性がある。私たちは、未償還オプションや株式承認証の行使や他の証券の転換によって、将来の私たちの普通株の発行規模、または将来の私たちの普通株の株式発行と販売が私たちの普通株の市場価格に影響を及ぼす可能性がある(あれば)予測できない。私たちの普通株(買収に関連する株を含む)を大量に売却または配布したり、そのような売却が発生する可能性があると考えたりすることは、私たちの普通株の市場価格の下落を招く可能性がある。
また,発行された転換可能証券を行使/転換する際に発行可能な普通株 は過剰を表す可能性があり,我々の普通株の市場価格に悪影響を与える可能性もある.ある会社の株の市場での供給量がその株に対する需要量よりも大きい場合には、割増が生じる。このような状況が発生すると、私たちの株価は低下し、株主が市場で販売しようとしている任意の追加株式はさらに株価を下げるだけだ。もし私たちの普通株の株式数が私たちが発行した転換可能な証券所有者が売却した株式を吸収できなければ、私たちの普通株の価値は縮むかもしれません。
私たちの未償還公共株式証明書brは資金が深刻に不足している。
各公共株式承認証は所有者に1/40株当たり5.75ドルの使用価格で1/40株の普通株の1/40株を購入する権利を持たせるこれは…。1株(1株あたり230.00ドル)を調整することができる。公開株式証を行使する際には、断片的な株式は発行されない。公開株式証は初公募終了から12ヶ月から行使でき、業務合併完了後5年(2025年11月6日)に満了する。公開株式証の資金は深刻に不足しており、公開株式証を行使する際に断片的な株式を発行しないため、公開株式証は40倍の倍数でしか行使できない。したがって、公共株式証明書は何の重要な 価値もないかもしれない。さらに、少なくとも40件の公開株式証明書または普通株式断片的株式で行使可能な公開株式証を有する権利証所有者を保有しない場合には、断片的権益から価値を得るために任意の株式承認証を売却しなければならない。したがって、公共株式証の取引は有限または零細である可能性があり、このような公共株式証明書には何の重大な価値もない可能性がある。40部未満の公共株式承認証または数が40で割り切れない公共株式証明書を保有する任意の所有者は、公共株式承認証の行使時にいかなる普通株式も取得しないであろう。公共株式証を行使する際に普通株式の断片的な株式を発行することができないからである。
91
私たちは相当な数の株を売る資格があります。それらの売却や潜在的な売却は私たちの普通株の市場価格を下げるかもしれません。
私たちの普通株を公開市場で大量に売ると私たちの普通株の市場価格を損なう可能性があります。私たちの大部分の普通株は公開市場で転売することができ、(A)162,956株の普通株を購入できるオプション、加重平均行権価格は1株当たり84.63ドルである;および(B)3,435,728株の普通株を購入できる引受権証を含み、加重平均行権価格は1株当たり33.94ドルである。大量の株が売却されれば、このような売却は私たちの普通株の供給を増加させ、その価格を低下させる可能性がある。有効な登録声明および/またはルール144によれば、私たちの普通株の一部または全部が時々公開市場で発売される可能性があり、これは私たちの普通株の市場を抑制する可能性がある。ある制限を受けた場合、6ヶ月限定株を持っている人は一般に普通株を市場に売ることができます。このような株を公開販売する資格がある場合、かなりの部分を売却すると が私たちの普通株の価値を低下させる可能性があります。
私たちの証券市場には投資家がその株を売却するのに十分な流動性がないかもしれない。私たちの普通株の市場価格は引き続き変動するかもしれない.
私たちの普通株の市場価格は引き続き高度に変動するかもしれない。私たちの普通株の市場価格に大きな影響を与えるかもしれないいくつかの要素は、私たちの業界の状況や傾向、あるいは私たちの普通株の販売状況など、私たちの制御範囲を超えています。このような状況になった原因は、私たちが比較的株式アナリストに知られていない小さな会社、株式ブローカー、機関投資家、投資界で販売量に影響を与えたり、影響を与えたりする他の人が多く、そして私たちがこれらの人の注意を引いても、彼らはリスクを回避する傾向があり、私たちのような未確認の会社に追随したくなく、私たちがより成熟して実行可能になるまで、私たちの株を購入したり推薦したりしたくない。
したがって、私たちの株は、成熟した発行者と比較して、数日以上の取引活動が少ないか、全く存在しない可能性があり、成熟した発行者 は、株価に悪影響を与えることなく、多くの安定した取引量を有し、通常、持続的な販売をサポートするであろう。br}私たちの普通株のより広くまたはより活発な公開取引市場は発展または維持されない可能性があり、または取引 レベルは継続されないかもしれない。これらの要素は、私たちの表現にかかわらず、私たちの普通株の市場価格に実質的な悪影響を及ぼす可能性があります。 また、公開株式市場は極端な価格と取引量の変動を経験しています。この変動は多くの会社の証券市場価格に深刻な影響を与えており、原因は往々にして特定の会社の経営業績とは無関係である。このような広範囲な市場変動は私たちの普通株の市場価格に悪影響を及ぼすかもしれない。
もし私たちが以前に提出した登録声明がその後一時停止または終了された場合、私たちは重大なbr罰金と損害賠償に直面するだろう。
これまでのいくつかの私募証券によれば、私的に売却された株式と、その行使/転換時に発行可能ないくつかの証券の転売を登録し、一定期間内にその等の登録声明の有効性を維持するために、登録権契約を締結した。これまで、このようなすべての必要な登録声明は、米国証券取引委員会によって発効が宣言された。しかしながら、登録宣言がその後一時停止または終了された場合、または他の方法で登録権プロトコルに規定されたいくつかの要件を満たしていない場合、私たちは、私たちのキャッシュフローに悪影響を与え、私たちの証券価値を低下させる可能性がある重大なbrの罰金を支払う必要があるかもしれない。
2022年7月に付与された引受権証条項 は第三者による我々の買収を阻止する可能性がある。
私たちが2022年7月に付与した普通株式証明書のいくつかの条項は、第三者が私たちをより難しくまたは高価に買収することを可能にするかもしれない。私たちは2022年7月に付与された普通株式承認証は、私たちが“基本取引”を構成するある取引 に従事することを禁止し、まだ存在する実体が私たちが普通株式承認証によって負担する義務を負わない限り、私たちは“基本取引”を構成するいくつかの取引 に従事することを禁止する。また、2022年7月に付与された普通株式証 は、ある取引が“基本取引”を構成する場合、当該等承認持分証所有者はその選択に応じて、当該等株式証に記載された価格(当該等承認株式証に基づくBlack Scholes価値)で当該等普通株権証 を買い戻すことを要求すると規定している。普通株承認株式証のこれらの条項や他の条項は、買収が株主に有利になる可能性があっても、第三者買収を阻止または阻止する可能性がある。
92
将来的に私たちの普通株を売却して発行したり、普通株を購入する権利は、私たちの株主に追加的な希釈をもたらし、私たちの普通株の価格を下落させる可能性がある。
私たちは未来に追加的な普通株、転換可能な証券、または他の株式を発行するかもしれない。私たちはまた株式インセンティブ計画に基づいて従業員、役員、他のサービスプロバイダに普通株式を発行します。このような発行は投資家の権益を希釈し、私たちの普通株の価格を下落させる可能性がある。このような発行された新しい投資家はまた既存の株主より優先的な権利を得ることができる。
公開市場で私たちの普通株を転売することは私たちの普通株の市場価格を下落させる可能性があります。
私たちの普通株式の中で相当な数の株式がいつでも販売されるかもしれません。私たちの普通株を発行する新株は、保有株式の所有権が希釈される可能性があることを懸念して、現在の株主が私たちの普通株を転売する可能性があります。逆に、これらの転売は私たちの普通株の市場価格を下げるかもしれない。
未来に私たちの普通株を売却することは私たちの株価を下落させるかもしれない。
もし私たちの株主が公開市場で私たちの普通株を大量に売却すれば、私たちの普通株の市場価格は大幅に下がるかもしれません。公開市場は、私たちの株主が私たちの普通株の株を売却する可能性があると考えており、これは私たちの普通株の市場価格を下げる可能性もあります。私たちが1つのサイトに登録した証券の総価値は1.25億ドルに達しています棚.棚我々は2022年6月3日に米国証券取引委員会に表S-3の登録声明を提出し、2022年6月24日に発効を発表した。2023年3月28日現在、時々600万ドルを超える証券が公開市場で販売される資格がある。また、既存の株主が公開市場で私たちの普通株を大量に売却する意向を示している場合、私たちの普通株の取引価格は大幅に低下する可能性があります。私たちの普通株が公開市場で販売されている時、その市場価格は大幅に下落する可能性がある。私たち普通株株価の下落は普通株や他の株式証券の増発による資金調達能力を阻害する可能性があります。
私たちの管理書類やデラウェア州の法律に関するリスクは
私たちの会社登録証明書は、私たちが費用を負担する上級管理者と取締役の賠償を規定し、彼らの責任を制限しています。これは、当社の主要なコストを招き、当社の株主の利益を損なう可能性があります。会社資源は上級管理者やbr取締役の利益にかかる可能性があります。
私たちの会社登録証明書に規定されている賠償は以下のとおりである:“法律が適用される最大範囲内で、会社は定款条項、このような代理人または他の人との合意、株主投票または利害関係のない取締役投票または他の方法を通じて、会社のこのような代理人(およびデラウェア州法律で会社がそれに賠償を提供することを許可している任意の他の人)に賠償および立て替え費用を提供する権利があり、”デラウェア州会社法“第145条で許可された賠償および立て替え費用を超えて、デラウェア州法律(法定または非法定)の制限のみを受ける。当社、その株主及びその他の者に対する義務に違反する行為について
米証券取引委員会は,連邦証券法下で発生した責任を賠償することは“証券法”の明確な公共政策に違反しているため実行できないと言われている。もし取締役、上級職員、あるいはコントロール人が私たちの活動に関連する連邦証券法の下で発生した責任について賠償要求を提出した場合、私たちは取締役、上級職員、あるいはコントロール人がいかなる訴訟、訴訟または訴訟に成功して発生または支払いに成功した費用を支払うかを除いて、(私たちの弁護士がこの問題がコントロール前例によって解決されたと思わない限り)適切な管轄権裁判所に提出します。我々の賠償は,証券法で表現されている公共政策に違反しているかどうかは,このような問題の最終裁決 によって管轄される.もしこれが発生すれば、それに関連する法的手続きは非常に高いかもしれないし、否定的な宣伝を受ける可能性があり、この2つの要素のいずれも私たちの株の市場と価格を大幅に低下させる可能性がある。
93
当社の会社登録証明書には、当社の取締役が会社や会社の株主に与える金銭的損害に対する責任を制限する特定の条項が含まれており、場合によっては上級管理者、役員、従業員に対して賠償を請求しています。
デラウェア州法律によると、私たちの役員、高級管理者、従業員の金銭的責任が制限され、彼らに対する賠償権利が存在し、これは私たちの巨額の支出を招き、私たちの役員、高級管理者、従業員に対する訴訟を阻害する可能性がある。
私たちの会社登録証明書brは、当社の取締役が会社や会社の株主に与える金銭的損害の責任を制限する特定の条項を含んでいます。私たちが上級管理職や役員と締結した雇用·採用協定によると、私たちはまた契約賠償義務を負っています。上記の賠償義務により、当社が回収できない可能性のあるわが役員と上級管理者に対する和解や損害賠償費用を支払うための巨額の支出が生じる可能性があります。これらの条項とそれによって生じるコストも、受託責任に違反して私たちの役員や上級管理者に訴訟を提起することを阻止する可能性があり、同様に私たちの株主が私たちの役員や上級管理者にデリバティブ訴訟を提起することを阻止する可能性があり、たとえbrのような訴訟が成功すれば、私たちと私たちの株主に利益をもたらすかもしれません。
私たちの取締役は優先株と普通株の追加株式の発行を許可する権利があります。
当社の取締役は、当社の登録証明書に記載されているbrの制限および制限の範囲内で、我々の株主がさらなる行動を取らない場合、時々1つまたは複数の系列の優先株を発行する権利があり、そのような一連の株式の数および相対権利、転換権利、投票権、償還条項、清算優先権および任意の他の優遇、特殊権利および資格を決定する権利がある。どんな優先株の発行も私たちの普通株保有者の権利に悪影響を及ぼす可能性がある。もし私たちがこれから普通株を増発すれば、各投資家の私たちの株に対する所有権権益は比例して減少します。
改訂された2つ目の改訂された会社登録証明書と私たちの改正された定款における逆買収条項、およびデラウェア州法律の条項brは、わが社の統制権の変更や私たちの経営陣の変更を阻止、延期、阻止し、それによって、私たちの普通株の取引価格を下げる可能性があります。
私たちの2回目の改正と再改訂された会社登録証明書、ならびに私たちが改正して再改正した定款およびデラウェア州法律に含まれる条項brは、株主が有利と思うかもしれない合併、買収、または他の支配権変更を阻止、延期、または阻止する可能性があります。私たちの普通株式または株式承認証を持っていることによってプレミアム取引を得る可能性があります。これらの規定は、株主の交換や我々の経営陣の更迭の試みを阻止または延期する可能性もあります。当社のコーポレートガバナンス文書には、以下のbr条項が含まれています
| ● | 分類取締役会、 そのため、私たちの取締役会は2段階に分けられ、1級ごとに2年間交互に勤務している | |
| ● | 理由だけで取締役を罷免する; |
94
| ● | 我々の株主会議で行われる株主業務提案と我々の取締役会に入る候補者の指名を要求する事前通知 ; | |
| ● | 株主が書面で行動する能力を禁止する | |
| ● | 株主特別会議は取締役会議長、最高経営責任者または取締役会が取締役会の多数のメンバーが採択した決議に基づいてのみ開催されることが規定されている | |
| ● | 投票権、清算、配当金、および私たちの普通株よりも優れた他の権利を発行することができる空白小切手brを発行することができる | |
| ● | 役員と上級管理者の責任を制限し、それに賠償を提供する。 |
デラウェア州会社として、私たちはまた、デラウェア州汎用会社法第203条を含むデラウェア州の法律の条項を遵守しなければなりません。この条項は、私たちが議決権を発行した株式投票権の15%を超える株主が私たちと特定の業務を合併する能力を制限しています。私たちの二回目の改正と再登録証明書brは、私たちが改正し、再修正した定款やデラウェア州法律の遅延または制御権の変更を阻止するいかなる条項も、私たちの株主がその普通株の割増の機会を得ることを制限する可能性があり、また一部の投資家が私たちの普通株に支払いたい価格 に影響を与える可能性がある。
上記の条項と逆買収措置の存在は、投資家が将来私たちの普通株または株式承認証株式に支払うことを望む可能性のある価格を制限する可能性がある。これらはまた、潜在的な買収者がわが社を買収することを阻止することができ、買収で普通株や株式承認割増を得る可能性を下げることができる。
私たちの2回目の改正と再改訂された会社登録証明書には独占フォーラム条項が含まれており、私たちと私たちの役員や上級管理職に対する訴訟を阻止する可能性があります。
私たちの2回目の改正と再改訂された会社登録証明書は、会社が代替フォーラムを選択することに同意しない限り、デラウェア州衡平裁判所は、(I)会社を代表して提起された任意の派生訴訟または訴訟、(Ii)会社の現職または前任取締役、役員、従業員または株主が会社または会社の株主に対する受託責任に違反していると主張する任意の唯一および独占フォーラムである。(Iii)“デラウェア州会社法”、改正された第二次改正および再発行された“会社登録証明書”または定款の任意の規定に基づいてクレームを提起する任意の訴訟、または(Iv)内部事務原則によって管轄されるクレームを主張する任意の訴訟。
私たちの第二次改正と再署名された会社登録証明書の中で裁判所条項の選択は、連邦証券法及びその規則及び条例に規定されている義務を遵守することを免除しません。また、この条項は、取引法または証券法に規定されている義務または責任を執行するための訴訟には適用されない。“取引法”第27節(Br)では,連邦裁判所は“取引法”又は“規則”及びその下の規定により生じた任意の義務又は責任を執行するために提起されたすべての訴訟に対して排他的連邦管轄権を有し,証券法第22条は連邦裁判所及び州裁判所が“証券法”又はその下の規則及び条例で規定された義務又は責任を執行するために提起した訴訟に対して同時に管轄権を有することを規定する。したがって、州裁判所と連邦裁判所は証券法の下でのクレームを受理する管轄権を持っている。
これらの排他的裁判所条項は,会社株主が司法裁判所でクレームを出す能力を制限する可能性があり,これらの株主 は,会社や会社役員や役員とのトラブルに有利であると考え,このような会社や会社役員や役員に対する訴訟を阻止する可能性がある.代替的に、裁判所が、上述した排他的裁判所の1つまたは複数の規定が、上述した1つまたは複数の特定のタイプの訴訟または訴訟手続きに適用されないことを発見した場合、または強制的に実行できない場合、私たちは、他の管轄区域または裁判所がそのような問題を解決する際に追加料金を生じる可能性があり、これは、私たちの業務、財務状態、または運営結果に重大かつ不利な影響を与える可能性がある。
95
私たちの二回目の改正と再改訂された会社登録証明書には条項が含まれていますが、これらの条項によると、任意の役員または役員に提供される任意の会社の機会における任意の権益を放棄しますが、いくつかの例外は除外します。
改正された“会社登録証明書”第br節では、法律で許容される範囲内で、会社機会原則又は他の類似の原則は、会社又はその任意の上級管理者又は取締役、又はそのそれぞれの関連会社の任意の関連会社に適用されず、会社は、会社の任意の取締役又は上級管理者が、彼又は彼女が知っている可能性のある任意の会社の機会の予想を会社に提供することを放棄する。会社機会原則 が、(I)当社取締役又は上級管理者としてのみ当該者に提供される会社機会、(Ii)当社が法的に に適用され、契約に基づいて従事することが許可され、当社が他の方法で合理的に追求する会社機会、及び(Iii)いかなる法的義務にも違反することなく、取締役br又は上級管理者が当該会社への提出を許可される等の機会にのみ適用されない限り。
さらに、我々の各上級職員および取締役は、現在、他のbrエンティティに対して追加の受託責任または契約義務を有しており、これらの義務に基づいて、適用される法律に基づいて、当該上級職員または取締役は、彼または彼女の受託責任に依存して、当該エンティティに業務機会を提供することを要求される可能性がある。したがって,わが社に潜在的な業務統合機会 を提供するか否かで利益衝突が生じる可能性がある.このような葛藤は私たちに有利な方法で解決されないかもしれない。私たちが会社の機会 を放棄することは、私たちの将来の運営結果に重大な悪影響を及ぼす可能性があり、および/または利益衝突や考えられる利益衝突 を引き起こす可能性があり、これは私たちの証券の価値に大きな悪影響を及ぼす可能性がある。
私たちのbr取締役は彼らの時間を他の業務に割り当て、彼らがどのくらいの時間を私たちの事務に投入するかを決定することで利益の衝突があります。
私たちの役員は必要もなくフルタイムで私たちの事務に取り組むこともできません。私たちの一部の取締役は生命科学業界の他の会社で、brの他の取締役を含む職務を担当しています。これは、私たちの運営と彼らがサービスを提供する他の会社との間で彼らのbr時間を割り当てる際に利益の衝突を招く可能性があります。もし私たちの取締役の他のビジネスが彼らにこのような事務に投入する時間が彼らの現在の約束レベルを超えていることを要求すれば、彼らが私たちの事務に時間 を投入することを制限するかもしれません。これは私たちの運営にマイナス影響を与えるかもしれません。また,これらの人たちは,彼らの時間を様々な業務活動に割り当てる際に利益相反がある可能性がある.このような葛藤は私たちに有利な方法で解決されないかもしれない。また、我々の取締役は、上述したような会社機会免除条項 を提供するために、彼らの所属する他のbr社に会社機会を提供することを選択または要求される可能性がある。実際または予想される利益衝突は、私たちの運営結果に重大な悪影響を及ぼす可能性があり、これは私たちの証券の価値に大きな悪影響を及ぼす可能性がある。
コンプライアンス、報告書、そして上場リスク
アメリカとナスダック資本市場の報告やコーポレートガバナンス要求の遵守を確保するために、大きなコストが発生します。
我々の上場企業報告要求および適用された米国とナスダック資本市場会社管理要求は、2002年の“サバンズ-オキシリー法案”下の要求、および米国証券取引委員会とナスダック資本市場が実施した他の規則を含み、巨額のコストを発生させた。ナスダック資本市場のルールには、私たちに独立取締役を維持し、他の会社のガバナンス要求brを遵守し、年間上場と株式発行費を支払うことが求められています。これらすべてのアメリカ証券取引委員会とナスダック義務は、追加費用を含むが、これらに限定されない追加資源を約束する必要があり、私たちの上級管理職が私たちの日常運営から時間と注意を移すことを招く可能性がある。私たちはこのようなすべての適用された規則と法規が私たちの法律と財務コンプライアンスコストを著しく増加させ、特定の活動をより時間とコストを高くすると予想する。また、これらの適用される規制は、取締役や高級職員責任保険を獲得することをより困難かつ高価にする可能性があり、同じまたは同様の保証範囲を得るために、低減された保険限度額および保証範囲を受け入れることを要求される可能性があります。したがって、私たちは合格者を引き付けて維持することがもっと難しいかもしれないし、私たちの取締役会に参加したり、幹部になったりすることはもっと難しいかもしれない。
96
報告会社として、私たちはbrコストを増加させ、私たちの限られた資本資源を考慮すると、これらの追加コストは私たちの収益性に悪影響を及ぼす可能性があります。
私たちはアメリカ証券取引委員会報告会社です。“取引法”の下の規制は、報告会社に対話型データファイルを含む定期的な報告を提供することを要求しており、これは、法律、会計、監査の専門家の招聘、および拡張可能な商業報告言語(XBRL)およびサービスプロバイダの EDGAR(電子データ収集、分析、および検索)を要求しています。このようなサービスを提供するコストが高い可能性があり, 我々はより多くの損失を被る可能性があり,経営を継続する能力に悪影響を与える可能性がある.また,2002年のサバンズ−オキシリー法案および米国証券取引委員会が実施した各種関連規則はコーポレート·ガバナンスの変更を要求し,上場企業への開示要求を一般的に向上させた。例えば、報告会社として、私たちは、定期的かつ現在の報告書およびその他の情報を米国証券取引委員会に提出するように要求されており、制御およびプログラムを開示するための政策をとり、これらの制御および手続きを定期的に評価している。
報告会社として、私たちは引き続き による追加コスト(年間数十万ドルを予定)は引き続き は私たちの限られた資本資源をさらに枯渇させていく。我々は資源が限られているため,米国証券取引委員会報告会社としての義務を履行し続けるために,他の生産性br用途に資源を割り当てなければならない。また、私たちが米国証券取引委員会に提出した満期報告書や届出義務を履行し続けるのに十分な資源があるという保証はない。
私たちは過去にナスダックの持続上場基準を守っていませんでしたが、将来はナスダックの持続上場基準 を満たすことができないかもしれません。
私たちの普通株式と権利証はナスダック資本市場で取引され、コードは“ATNF“と”ATNFWは“この二つの字はそれぞれ。このような上場があるにもかかわらず、どのブローカーも私たちの証券を取引することに興味を持つ保証はない。そのため、私たちの証券を公開販売することは難しいかもしれません。 ナスダックの持続的な上場要求を永久的に満たすことで、いつでもナスダック資本市場での上場を維持できる保証はありません。私たちは現在ナスダックの持続上場基準に適合していますが、私たちは過去にこのような持続上場基準を満たしていません。私たちはこれらの要求を満たし続けることができませんでした。私たちの証券がナスダックから退市する可能性があります。
ナスダック資本市場への上場を継続するために必要な条件は、少なくとも250万ドルの株主権益の維持、3500万ドルの上場証券時価または2年前または3年間の純収入の維持、多数の独立取締役の保有、3人の監査委員会(すべての独立取締役からなる)、および入札を1株1.00ドル以上に維持することを含む。私たちの株主権益はナスダック250万ドルの最低水準を維持しないかもしれません。私たちの上場証券の時価は3500万ドル以上を維持しないかもしれません。私たちは毎年発生する純収入は50万ドルを超えないかもしれません。私たちはbrを維持できないかもしれません。あるいは株価を1.00ドル以上に維持できないかもしれません。
もし私たちがナスダックの規則と要求を守らなかったら、私たちの株は取られるかもしれない。また、私たちが上記の要求に適合していることを証明しても、私たちは引き続き他の客観的かつ主観的な上場要求を満たし続けなければならない。br}がナスダック資本市場から撤退することは、投資家が私たちの普通株式および/または株式証明を取引しにくくなる可能性があり、これは私たちの株価と流動性を低下させる可能性がある。ナスダック資本市場に上場していなければ、株主は私たちの株の売却や購入のオファーを得ることが困難になる可能性があり、私たちの株の売却や購入はより困難になる可能性があり、私たちの株の取引量や流動性は低下する可能性がある。ナスダック資本市場からの撤退はマイナス宣伝を招く可能性もあり、追加資本の調達を困難にする可能性もある。このような上場がなければ、我々の普通株式および/または株式承認証の通貨としての受容度または他の当事者が付与した価値に悪影響を及ぼす可能性がある。また、もし私たちがカードを取られたら、州青空法律に基づいて、私たちは私たちの証券の販売に関連した追加コストを発生させるだろう。これらの要求は、私たちの普通株式および/または株式承認証の市場流動性と、私たちの株主が二次市場で私たちの普通株式および/または株式証を売却する能力を深刻に制限するかもしれない。もし私たちの普通株式および/または株式承認証がナスダックによって取得された場合、私たちの普通株式および/または株式承認証はOTCQB Marketのような場外見積システムで取引する資格がある可能性があり、そこで投資家は私たちの株を売ることがより難しいことを発見したり、私たちの普通株式および/または承認株式証の市場価値に関する正確なオファーを得ることができるかもしれない。もし私たちの普通株および/または株式承認証がナスダック資本市場から撤退した場合、私たちは私たちの普通株式および/または株式承認証を別の全国証券取引所に上場したり、場外見積システムでオファーを得ることができないかもしれません
97
一般リスク因子
わが社の登録証明書とデラウェア州法律の条項は、私たちの買収を阻止するかもしれません。これは、投資家が将来私たちの普通株に支払う可能性のある価格を制限し、経営陣を強化する可能性があります。
当社の登録証明書brに含まれる条項は、株主がその最適な利益に適合していると考える可能性のある能動的な買収提案を阻止する可能性があります。 これらの条項には、交差する取締役会および取締役会が新しいシリーズの優先株を指定して発行する権利がある条項が含まれており、これは、管理層の解除の難しさを増加させる可能性があり、そうでなければ、我々の証券の現在の市場価格よりも高いプレミアムを支払う取引に関連する可能性があります。デラウェア州の法律によると、私たちはまた反買収条項の制約を受けており、これは制御権の変更を延期または阻止する可能性がある。つまり、これらの規定は経営陣を解除する難しさを増加させる可能性があり、現在の市場価格よりも高い割増を私たちの証券に支払うことに関連する取引を阻害する可能性がある。
私たちの計画における積極的な成長戦略を十分に管理できなかったことは、私たちの業務を損なったり、私たちの失敗のリスクを増加させたりする可能性があります。
予測可能な未来に、私たちは製品開発とマーケティングを増加させることで、私たちの業務を拡大するために積極的な成長戦略を実施するつもりです。私たちが私たちの業務を迅速に拡大できるかどうかは、規制された環境で働く能力、独立薬局に付加価値製品を効率的に販売する能力、サプライヤーと戦略関係を構築し、維持すること、および許容可能な条件下で十分な資本資源を得ることを含む多くの要素に依存します。私たちの拡張能力に対するいかなる制限も、私たちの業務、運営結果、財務状況に重大な悪影響を及ぼす可能性があります。したがって、私たちは私たちの販売増加目標を達成できないかもしれませんが、 私たちの運営は成功していないか、予期した運営結果を達成できないかもしれません。
また、私たちの成長は私たちの管理、行政、運営、財務資源、そして私たちのインフラに大きな圧力をもたらすかもしれない。私たちの未来の成功は私たちの上級管理職が成長を効果的に管理する能力にある程度依存するだろう。これは私たちに他の事項を除いて要求されるだろう
| ● | 追加管理 情報システムを実施する; | |
| ● | 私たちの運営、行政、法律、財務、会計システム、統制をさらに発展させる |
| ● | 人員を増任する | |
| ● | 私たちの社内ではより多くの管理階層を発展させています |
| ● | 追加のオフィススペース ; |
98
| ● | 私たちのプロジェクト、運営、法律、財務、販売とマーケティング、ならびに顧客サービスと支援組織の間で密接な協調を維持し、 | |
| ● | 私たちが拡大していく国際業務 を管理します。 |
したがって、私たちは私たちのサービスをタイムリーかつ経済的に効率的に配置するための資源が足りないかもしれない。上記のいずれかの要求を満たすことができなければ、私たちが適時にサービスを提供したり、新しい顧客を誘致したり、維持する能力に影響を与える可能性があります。
私たちの固有の情報や、私たちの顧客、サプライヤー、およびビジネスパートナーの固有の情報が失われる可能性があり、またはセキュリティホールに遭遇する可能性があります。
私たちの正常な業務プロセスでは、価値があり、商業的に敏感な知的財産権、臨床試験データ、その独自の業務情報、および私たちの将来の顧客、サプライヤーおよび業務パートナーの独自の業務情報、ならびに私たちの顧客、臨床試験対象および従業員、患者、データセンター、および私たちのネットワークにおける個人識別可能な情報を含む敏感なデータを収集し、保存したい。この情報の安全な処理、維持、そして伝達は私たちの運営に必須的だ。私たちはセキュリティ措置を取っているにもかかわらず、私たちの情報技術とインフラはハッカーの攻撃を受けやすいか、あるいは従業員のミス、汚職、その他の中断によって破られる可能性があります。どのような侵入も私たちのネットワークを危険にさらす可能性があり、そこに格納されている情報はアクセス、公開、紛失、または盗まれる可能性がある。このような任意のbrアクセス、開示、または他の情報損失は、法的クレームや訴訟、個人情報のプライバシーを保護する法的責任(Br)、規制処罰、私たちの運営を妨害し、私たちの名声を損なう可能性があり、私たちの製品と私たちの臨床試験を行う能力に自信を失うことを招き、これは私たちの業務と名声に悪影響を与え、私たちの将来の候補製品が規制部門の承認を得る時間を遅らせる可能性がある。私たちは業務中断保険を継続していますが、私たちの保険は将来のいかなるシステム違反によるすべての損失も保証しないかもしれません。
我々の情報技術システムに障害が発生し,ネットワークセキュリティ攻撃や他のデータセキュリティイベントを含めて,我々の業務の運営を大きく乱す可能性がある.
私たちの業務はますます情報技術の使用に依存しており、これは研究開発、生産、販売などのいくつかの重要な分野が私たちまたは第三者プロバイダの情報システムに大きく依存していることを意味する。我々は、データ制御およびデータ完全性に関する規制機関の要求を遵守する業務計画を実行し、当社の情報技術システムまたは第三者サービスプロバイダによって提供されるITシステムおよびITシステムの持続的かつ断続的な性能にある程度依存する。わが社、私たちの業務パートナー、サプライヤーと顧客の情報システムとソフトウェアおよび関連アプリケーションの使用がよりクラウドに基づくようになるにつれて、グローバルネットワークセキュリティホールと脅威も増加しており、br}のより複雑で的確なネットワーク関連攻撃を含み、これらの攻撃は私たちの情報システムとネットワークのセキュリティ、データと情報のセキュリティ、可用性と完全性にリスクを構成している。また、我々のITシステムは、電気通信やネットワーク障害、悪意のある人為的行動、自然災害など、様々なソースの損傷を受けやすい。さらに、ネットワークセキュリティおよびバックアップ措置がとられているにもかかわらず、私たちのいくつかのサーバは、物理的または電子的な侵入、コンピュータウイルス、および同様の破壊的な問題の攻撃を受けやすい可能性があります。br}私たちと私たちの第三者サービスプロバイダは、私たちのITシステムに影響を与える可能性のある意外な問題を防止するための予防措置を取っていますが、成功したネットワークセキュリティ攻撃または他のデータセキュリティイベントは、 機密または個人情報の盗用および/または損失を招き、システム中断をもたらし、または私たちのシステムを攻撃するマルウェアを配備する可能性があります。しばらくネットワークセキュリティ攻撃に気づかなかった可能性もある.さらに、継続的または反復的なシステム障害 や、私たちの任意のITシステムのアップグレード中に発生する問題は、私たちのデータを生成して維持する能力、特に私たちのノウハウプラットフォームを操作する能力を中断し、私たちの業務運営能力に悪影響を及ぼす可能性があります。ネットワークセキュリティ攻撃またはイベントの発生は、当社のITシステムの中断、または否定的な宣伝が、当社の株主および他の利害関係者の名声の損傷をもたらし、および/またはネットワークセキュリティイベントの予防、対応、または緩和のコストを増加させるため、業務中断を引き起こす可能性がある。さらに、敏感な個人情報または独自または機密情報を不正に伝播することは、私たちのbrまたは他の第三者を規制罰金または処罰、訴訟および潜在的責任に直面させるか、または他の方法で私たちの業務を損なう可能性があります。
99
私たちは他のbr会社を買収するかもしれません。これは私たちの経営陣の注意を分散させ、私たちの株主の株式をさらに希釈し、他の方法で私たちの運営を乱し、私たちの経営業績を損なうかもしれません。
私たちは将来、私たちの製品を補完または拡張し、私たちの技術br能力を強化したり、成長機会を提供するビジネス、製品、または技術を買収することを求めるかもしれません。潜在的な買収を追求することは、管理層の関心を分散させ、適切な買収を決定、調査、求める際に様々な費用を発生させる可能性があります。これらの買収が完了したかどうかにかかわらず、私たちがより多くの業務を買収すれば、買収の人員、運営、技術を統合することができない可能性があり、買収後に合併後の業務を効率的に管理したり、予想されるコスト節約や相乗効果を実現することができません。様々な要因により、買収された業務から期待される収益を得ることができない可能性もある
| ● | 買収に関するコスト ; |
| ● | 経営陣の関心を他の業務から移す |
| ● | 買収に関連した意外なコストや負債 |
| ● | 我々の既存業務への損害brと連携パートナーの関係を買収する |
| ● | 私たちのブランドと名声を損なう |
| ● | 重要な従業員の潜在的な流出 |
| ● | 私たちの業務の他の部分に必要な資源を使用して |
| ● | 私たちが現金で購入できる大部分のbrを使って買収を完了します。 |
将来、私たちの買収が期待されるリターンを生むことができなければ、私たちは減価評価過程で発生した経営結果に費用を計上する必要があるかもしれません。 買収は株式証券の希釈発行や債務の発生を招く可能性もあり、これは私たちの経営業績に悪影響を与える可能性があります。また、買収された業務が私たちの予想に達していない場合、私たちの業務、運営結果、財務状況は悪影響を受ける可能性があります。
もし私たちがどんな買収をすれば、それらは私たちの業務を混乱させたり、私たちの業務に負の影響を与えるかもしれません。
もし私たちが将来買収を行えば、資金が許可された場合、買収された会社の資産、人員、運営を私たち自身の資産、人員、運営と統合することは困難かもしれない。私たちは、将来可能ないかなる買収や合併も、会社の支配権の変更にはならないと予想している。しかも、買収された事業のキーパーソンは私たちのために働きたくないかもしれない。私たちは拡張が私たちの核心業務に及ぼす影響を予測できない。私たちが買収に成功したかどうかにかかわらず、交渉は私たちが行っている業務を乱し、私たちの経営陣と従業員の注意を分散させ、私たちのbr費用を増加させる可能性があります。これらのリスクに加えて、買収にはいくつかの固有のリスクが伴い、以下のリスクを含むが、これらに限定されない
| ● | 買収された製品、サービス、運営の難しさを統合する | |
| ● | 現在行われている業務の潜在的な中断と、私たちの経営陣と買収された会社の経営陣の気晴らし |
100
| ● | 統一された基準、統制、手続き、そして政策を維持することは困難だ | |
| ● | 任意の新しい管理者の統合により、従業員と顧客との関係が損なわれる可能性がある | |
| ● | 新しい顧客と既存の顧客に製品を交差マーケティングすることで、より多くの販売を実現し、私たちの顧客基盤を強化することができない場合があります | |
| ● | 買収された企業に関するいかなる政府法規の影響も |
| ● | 買収された企業または製品ラインに関連する潜在的に未知の負債、または買収された製品または事業のマーケティングおよび販売の再配置、再配置または修正、または買収された企業の私たちの買収前のbr行動に起因する任意の訴訟の弁護、および勝訴の有無にかかわらず、多くの資金がかかる必要がある | |
| ● | 司法管轄区域の労働者、環境、その他の法律によって規定された潜在的費用。 |
もし私たちが買収に関連するこれらのリスクや他の問題を解決することに成功しなければ、私たちの業務は深刻なbr損害を受ける可能性があり、その多くの問題は現在確定できない。これらのリスクや問題は、私たちが行っている業務を乱し、私たちの経営陣や従業員の注意を分散させ、私たちの費用を増加させ、私たちの運営結果に悪影響を及ぼす可能性があります。
私たちは、最終的に私たちの経営業績を改善したり、私たちの証券価値を増加させない用途に、運営資本と将来の資金を使用するかもしれません。
全体的に,我々は運営資本と将来獲得可能な任意の新しい投資資本の使用に対して完全な 決定権を持つ.我々の使用資金の数やbrの様々な要因を決定する可能性があるため,我々の最終的な資金支出(とその用途)は,現在のこのような資金に対する我々の予想運営計画とは大きく異なる可能性がある。
私たちは既存のbr運営資金と未来の資金を使って、私たちの製品とサービスの開発、私たちの卸売流通部門の製品調達、私たちのマーケティングを拡大し、あるいは私たちの運営を支援して私たちの顧客を教育するつもりです。私たちはまた資本を市場、ネットワーク拡張、買収、および一般運営資金用途に使用する。しかし、資本の使用と支出について、私たちはもっと具体的な計画を持っていない。私たちの経営陣は私たちの任意またはすべての利用可能な資本備蓄を使用することができる幅広い自由裁量権を持っている。私たちの資本は、私たちの経営業績を改善したり、他の方法で株主の投資価値を増加させない方法に使うことができます。
101
私たちは私たちの普通株についてどんな配当金を支払ったり発表したりしたことがない。
私たちは私たちの普通株や優先株のどんな配当金も支払ったり発表しなかった。同様に、私たちは近い将来、私たちの普通株に配当金や割り当て を支払わないと予想される。将来の普通株の任意の配当は、私たちの取締役会によって適宜発表され、私たちの収益、将来の運営と成長に対する私たちの財務的要求、そして私たちが適切だと思う他の事実に依存するだろう。私たちは普通株に現金配当金を支払わないことが予想されますので、あなたの投資収益(あれば)は私たちの普通株の時価の増加(あれば)に完全に依存します。
私たちが追加の普通株を発行することで融資と義務履行の努力を得ることで、株主は大幅に希釈される可能性がある。
可能な限り、私たちの取締役会 は非現金対価格を使用して義務を履行しようと試みるだろう。多くの場合、非現金対価格 は、私たちの普通株の制限株、または私たちの上級管理者、役員、適用されたコンサルタントに株式を発行する場合が含まれると考えられます。私たちの取締役会は、行動や株主投票を行う権利がありますが、ナスダックの規則と規定を遵守しなければなりません(任意の取引が20%を超える私たちが当時発行した普通株または私たちが当時発行した株式の20%以上に相当する投票権を発行する場合は、いくつかの例外を除いて、通常株主承認が必要です)。 すべてまたは部分的に許可されていますが発行されていない普通株式を発行します。また、普通株を売ることで資金を調達しようとしているかもしれませんが、市価を下回る価格であるかもしれません。これらの行動は、既存の株主の所有権権益を希釈することになり、これは普通株の帳簿価値をさらに希釈する可能性があり、この希釈は実質的である可能性がある。このような発行は、株式がbrの既存の管理層を支援するために努力している当事者またはエンティティに発行される可能性があるので、既存の 管理層が当社の制御能力を維持する能力を強化するのに役立つ可能性もある。
私たちの成長は私たちと第三者の戦略的関係の成功にある程度かかっている。
我々の業務を発展させるためには,我々の技術提供者を含む第三者との関係に依存し続ける必要があると予想される. のパートナーを決定し,彼らと交渉して関係を記録するには,膨大な時間と資源が必要である.私たちの競争相手は、第三者が彼らの製品またはサービスを偏愛するように効果的に激励するか、または私たちの製品とサービスを使用するかもしれません。また、私たちの競争相手が私たちのパートナーを買収することは、私たちの既存と潜在顧客の数を減少させるかもしれない。もし私たち が私たちと第三者との関係の構築や維持に成功しなかったら、私たちが市場で競争したり、収入を増加させる能力が損なわれる可能性があり、私たちの運営結果は影響を受ける可能性がある。私たちが成功しても、これらのbr関係が私たちの製品に対する顧客の使用を増加させたり、収入を増加させることを保証することはできません。
クレーム、訴訟、政府調査、その他の手続きは、私たちの業務と運営結果に悪影響を及ぼす可能性があります。
私たちは現在、実際および脅威クレーム、訴訟、審査、調査、および他の訴訟手続きの影響を定期的に受けていると予想されています。また、業務合併前に発見されたKBLに関する事項についていくつかの当事者に訴訟を提起しています。このような訴訟手続きは、法的費用、私たちの運営中断、br}管理資源の移転、負の宣伝、および他の要因によって私たちに悪影響を及ぼす可能性があります。我々の現在の法律手続きは、F-1ページから始まる連結財務諸表“訴訟とその他または損失がある”というタイトルの下の“付記11--支払いおよびまたは有事項”で説明している。これらの事項の結果は本質的に予測不可能であり,重大な不確実性の影響を受けている. 法的準備金やこのような事項による可能性のある損失が判断に関与していることを決定し,様々な不確実性 や予測不可能な結果を全面的に反映できない可能性がある.このような問題が最終的に解決されるまで,我々は記録金額を超える損失に直面する可能性があり, これらの金額は実質的である可能性がある.もし私たちの任意の推定と仮定が変化したり、正しくないことが証明された場合、私たちの業務、総合財務状況、運営結果、またはキャッシュフローに実質的な影響を与える可能性があります。さらに、和解の結果として、将来的に多くのお金を支払うことを要求し、特定の製品またはサービスを提供することを阻止することを要求することを含む1つまたは複数のこのような訴訟の解決策は、私たちの業務に不利な方法で私たちの業務慣行を変更し、権利侵害または他の方法で変更されない製品または技術の開発を要求し、私たちの名声を損なう、または他の方法で私たちの運営に実質的な影響を与えることを要求する可能性がある。
102
法律や法規の変更、またはいかなる法律法規にも従わないことは、私たちの業務、投資、経営結果に悪影響を及ぼす可能性があります。
私たちは国、地域、そして地方政府によって制定された法律、法規、そして規制の制約を受けている。私たちは特にアメリカ証券取引委員会、ナスダック、そしてbrの他の法律または法規の要求を遵守する必要がある。適用される法律、法規、規則を遵守して監視することは困難で、時間がかかり、コストが高いかもしれない。これらの法律、法規と規則とその解釈と応用も時々変化する可能性があり、これらの変化は私たちの業務、投資、運営結果に重大な悪影響を及ぼす可能性がある。また, が解釈や適用の適用法律,法規,規則を遵守できなければ,我々の業務や運営結果に大きな悪影響を与える可能性がある.
我々のいくつかの役員 管理者と取締役は現在,我々が行っている業務活動と類似した業務活動に従事するエンティティに所属しているため,どのエンティティに特定の業務機会 を提供すべきかを決定する際に利益衝突が存在する可能性がある.
我々の上級管理者やbr取締役は,現在または将来,我々が行っている業務活動と類似した業務活動に従事するエンティティに所属している可能性がある.私たちの上級管理職や役員は、私たちと彼らが何らかの受託責任や契約義務を持っている他のエンティティに紹介するのに適している可能性があるビジネスチャンスを認識しているかもしれません。したがって,彼らは特定の業務機会をわが社に提示すべきか他のエンティティに提示すべきかを決定する際に利益衝突 がある可能性がある.これらの紛争は私たちに有利な方法で解決されないかもしれないし、潜在的な機会は私たちに提示される前に別のエンティティに提示されるかもしれない。私たちの会社登録証明書は、私たちは、取締役または上級管理者に提供される任意の会社の機会における利益を放棄することを規定しており、この機会が取締役またはわが社の上級管理者の身分でのみその人に明示的に提供されない限り、その機会は、私たちが合法的かつ契約的に許可されている限り、私たちがこの機会を行うことは合理的である。
私たちの役員、役員、証券所有者およびその付属会社には、私たちの利益と衝突する競争的な金銭的利益が存在する可能性があります。
私たちは、私たちの取締役、役員、証券保有者、または関連会社が、私たちが買収または処分する任意の投資において、または私たちが参加または権利を有する任意の取引において、直接的または間接的な金銭的または財務的利益を有することを政策によって明確に禁止していない。実際、私たちは私たちの役員や役員に関連するターゲット企業と戦略的取引を行うかもしれません。私たちはまた、このような人たちが自分の口座のために私たちが行っているタイプの商業活動に従事することを明確に禁止する政策を明確にしていない。したがって、このような個人や実体は彼らの利益と私たちの利益の間で衝突するかもしれない。私たちの一部の幹部と役員は私たちの競争相手かもしれない会社に勤めています。また、上級管理職及び取締役の伝記を参照して、以下引用により統合してください“取締役·上級管理職·会社管理”.
私たちの業務はずっと新冠肺炎の疫病の悪影響を受けており、 そして引き続き影響を受ける可能性がある。
2019年12月、1種の新型コロナウイルス(新冠肺炎)株が武漢で浮上し、中国で発表された。2020年1月、新冠肺炎は米国やヨーロッパを含む世界の他の地域に伝播し、その伝播を抑制する努力が強化され、ある程度の成功を収めた。そのため、企業が閉鎖され、旅行や日常活動が制限されている。新冠肺炎の我々の業務への影響の程度は将来の事態の発展に依存し,これらの事態の発展は高度に不確定であり,疫病持続時間 ,米国と他の国の旅行制限や社交距離,企業閉鎖や業務中断,米国や他の国が疾患をコントロール·治療するための行動の有効性を把握できない。もし新型肺炎の流行が続いたら、私たちの計画は延期されたり中断されるかもしれない。新冠肺炎の伝播も世界経済の不確定性をもたらし、これはbrパートナー、サプライヤーと潜在顧客が彼らのコストを密接に監視し、彼らの支出予算を減少させる可能性がある。これらのことは,わが社の臨床試験,サプライチェーン,財務状況,財務業績に重大な悪影響を及ぼす可能性がある。
103
われわれの臨床試験では患者を募集し,われわれが行っている臨床試験では患者を維持し,募集した患者をフォローアップし,データを収集し,我々のいくつかの臨床試験地点で現場スタッフが制限されているか,あるいは新冠肺炎の大流行と政府の持続的な制限により一時閉鎖されている可能性がある。さらに、連邦または州政府によって課せられた旅行および物理的距離制限、または患者が大流行中に臨床試験場所に訪問することを望まないため、患者は用量またはデータ収集のために臨床試験場所にアクセスできないか、または訪問することができない可能性がある。新冠肺炎疫病によるこれらの要素は著者らの臨床試験の予想読み取りを延期或いは阻止する可能性があり、これは最終的に私たちの収入を創造する能力を遅延或いは阻止する可能性があり、そして私たちの運営結果に実質的な不利な影響を与える可能性がある。これらのことは,わが社の臨床試験,サプライチェーン,財務状況,財務業績に重大な悪影響を及ぼす可能性がある。
また,我々の“アナと雪の女王” 試用は悪影響を受けている.この試験は2022年5月末に募集を開放し、これまで新冠肺炎と相応のスタッフの空きにより国家衛生研究所のシステムが滞っており、審査が遅延した。2023年2月中旬まで9名の参加者 を募集した。新冠肺炎疫病発生後、イギリスの研究システムは支持サービスと臨床看護提供の面でかつてない挑戦に直面している。これによりNIHRは課題に直面している実験を識別·終了するために彼らの回復とリセット計画を策定した。私たちの冷凍肩試験は、求人サイトの開放と十分な参加者の募集にかなりの課題に直面しているため、このような実験の一つと考えられています。したがって、NIHRはさらに募集するために首席調査員に裁判を終わらせるように要請した。このような閉鎖または将来の閉鎖、または将来の研究で参加者を募集することに関連する困難は、私たちが研究を達成する能力、将来の薬物のスケジュール、および私たちが収入を創出し、私たちの運営を支援する能力に実質的な悪影響を及ぼすかもしれない。
私たち はこのような変化に対する気候変化または法律、規制、または市場の反応に悪影響を受ける可能性がある。
気候変化の長期的な影響は予測が難しいが、この影響は広いかもしれない。気候変動の影響は、物理的リスク(例えば、海面上昇または極端な気象条件の頻度および重症度を含む)を含む可能性があり、これは、他を除いて、私たちの運営の大部分がカリフォルニアにあり、これは悪天候の影響を受けやすい)、社会および人間の影響(例えば、人口失調または健康および福祉への損害)、コンプライアンスコストおよびbr}移行リスク(例えば、法規または技術変化)、および他の悪影響を含む可能性がある。気候変動の影響は,ある製品,大口商品,エネルギー(公共事業を含む)のコストを増加させ,さらに我々が業務運営に必要な商品やサービスを調達する能力に影響を与える可能性がある。気候変動はまた、私たちの施設が物理的な損傷や破壊、在庫損失、および気候変動に起因する可能性のある天気イベントによる業務中断が原因でコストを増加させる可能性がある。これらのイベントおよび影響は、私たちの業務運営、財務状況、または運営結果に重大な悪影響を及ぼす可能性があります。
環境、社会、そして統治問題は私たちの業務と名声に影響を及ぼすかもしれない。
政府当局、非政府組織、顧客、投資家、外部利害関係者および従業員は、多様性および包容性、気候変化、水、包装の回収可能性または回収可能性、およびプラスチックゴミのような環境、社会および管理またはESG問題にますます敏感になっている。このようなESG問題への関心は、新たな要求をもたらす可能性があり、これは、我々の製品の開発、製造、および流通に関連するコスト増加をもたらす可能性がある。私たちの競争能力はまた、より環境に優しい製品、パッケージ、またはサプライヤーの実践に対する需要が増加しているか、またはそのような顧客の期待または需要を満たすことができないなど、顧客の選好および要求変化の影響を受ける可能性がある。もし私たちが無責任に行動する場合、または重要なESG分野で責任を持って行動しないと考えられる場合、医薬品の公平な獲得、製品の品質と安全、多様性と包摂性、環境管理、地域コミュニティへの支援、会社の管理と透明性、および私たちの運営における人的資本要素を解決することを含む場合、代理コンサルティングサービス、および私たちのブランドや名声が損なわれることを含む株主の負の反応に直面する可能性がある。もし私たちが投資家、顧客、および他の利害関係者のESGに対する期待を満たすことができなければ、私たちの製品に対する需要の減少、顧客流失、br、および私たちの業務および運営結果に対する他の負の影響に直面する可能性がある。
英国のEU離脱は規制や法律の複雑さを増加させる可能性があり、これは私たちがイギリスおよび/またはヨーロッパで業務を展開することをより困難にし、私たちの製品候補製品がイギリスおよび/またはヨーロッパで規制の承認を得ることを確保する上で追加の挑戦をもたらすかもしれない。
イギリスは2020年1月31日からEUを離脱し、移行期間は2020年12月31日まで、すなわち一般的に言われている“イギリスの離脱”であり、これはイギリスとEUでの私たちの運営や候補製品に適用される規制枠組み を含む政治的·経済的不確実性をもたらし、この不確実性は数年続く可能性がある。その他の結果を除いて、イギリスの離脱はイギリスとEU間の商品、サービス、人員の自由な流動を乱す可能性があり、法律と監督管理の複雑さを増加させ、ヨーロッパで業務を展開するコスト の上昇を招く可能性がある。イギリスの離脱の結果の一つとして、EMAはイギリスからオランダに移転した。これはEMA従業員の大幅な減少を招き、これはさらにその行政手続きの深刻な中断とbr}遅延を招く可能性があり、例えば臨床試験許可またはマーケティング許可意見の承認、新薬調合中の活性物質と他の成分の輸出入 を中断し、臨床試験製品と最終認可製剤のサプライチェーンを中断する。
104
規制フレームワーク中断の累積影響は、EUおよび/またはイギリス製品のマーケティング許可および商業化の開発サイクル を大幅に増加させる可能性がある。規制の複雑さが増加する可能性があり、これは、私たちの臨床試験と規制承認の時間を乱す可能性がある。また、国と国際法律法規の変化と法的不確実性は、私たちの臨床と規制戦略に困難をもたらす可能性がある。イギリスの離脱やその他の理由で、いかなる遅延が得られても、または得られないいかなるマーケティング承認も、イギリスおよび/またはEUで私たちの候補製品を商業化することを阻止し、収入を創出し、利益を達成し、維持する能力を制限するだろう。
また、英国の離脱により、他の欧州諸国はEUに滞在し続けるかどうかについて国民投票を求める可能性がある。これらのbrの可能性と私たちが予想していないかもしれない他の可能性と、類似した前例がないことを考慮すると、イギリスのEU離脱がどのような財務的、規制、法律的影響をもたらすか、このような脱退が私たちにどのように影響し、私たちの業務が受ける可能性のある悪影響の全面的な程度に影響を与えるかは不明である。
ソーシャルメディアプラットフォームの使用は日々増加しており、私たちの業務に新たなリスクと挑戦をもたらしている。
ソーシャルメディアは,製薬会社の研究,候補製品,候補製品の交流に利用されるようになってきており,予防のための疾患が開発されている。製薬業界のソーシャルメディア実践は発展し続けており,このような使用に関する法規は常に明確ではない。この変化は不確実性と我々の業務に適用される法規を遵守しないリスクをもたらし、 は私たちに対する規制行動を招く可能性がある。例えば、被験者は、進行中の盲目的臨床試験における彼らの経験をレビューするためにソーシャルメディアチャネルを使用することができ、またはいわゆる有害事象を報告することができる。このような事件が発生した場合、私たちはbrを監視し、適用される有害事象報告義務を守ることができないかもしれないし、あるいはソーシャルメディアによって生じる政治的および市場的圧力の下で、私たちの業務または公衆の合法的なbrの利益を守ることができないかもしれない。これは、私たちの調査製品候補者に対する私たちの発言が制限されているからである。さらに、任意のソーシャルメディアまたはSNS上で敏感な情報または負のまたは不正確な 投稿またはコメントが開示されるべきではないリスクもある。GDPRのようないくつかのデータ保護法規は、ソーシャルメディアに含まれる個人データに適用される。もしこのような事件が発生したり、私たちが他の方法で適用された法規を遵守できなかった場合、私たちは責任を負い、規制行動に直面したり、私たちの業務に損害を与えたり、私たちの名声を損なうことを含むかもしれません。
私たちは将来債務を発生させるかもしれません。これは私たちの財務的柔軟性を低下させ、利息支出を増加させ、私たちの運営とコストに悪影響を及ぼすかもしれません。
私たちは未来に多くの借金を生むかもしれない。私たちの負債水準はいくつかの点で私たちの業務に影響を及ぼすかもしれません
| ● | 私たちのキャッシュフローの大部分は借金を返済する必要があります | |
| ● | 高い水準の債務は一般的に不利な経済的および産業的条件下での私たちの脆弱性を増加させる | |
| ● | 私たちの未返済債務を管理する協定に含まれる契約は、私たちの追加資金の借り入れと追加保証を提供する能力を制限し、資産を処分し、配当金を支払い、いくつかの投資を行う | |
| ● | レバレッジ率の低い競争相手に比べて、高いレベルの債務は私たちを競争劣勢にさせる可能性があるので、私たちの負債を利用して私たちが追求する機会を阻止することができるかもしれない | |
| ● | 債務契約は経済と産業の変化を計画して対応する私たちの柔軟性に影響を及ぼすかもしれない。 |
高い負債水準 は私たちが債務を滞納する可能性のある危険を増加させる。私たちは債務元金や利息を支払うのに十分なキャッシュフローを作ることができないかもしれません。将来の運営資金、借金または株式融資は、このような債務を支払うことができないかもしれません。もし私たちが十分な資金がなく、融資を手配できない場合、私たちは大量の資産を売却したり、一部の資産の担保償還権をキャンセルしなければならないかもしれません。これは、私たちの業務、財務状況、および運営結果に大きな悪影響を及ぼす可能性があります。
私たちは会計基準の変化の悪影響を受けるかもしれない。
私たちの連結財務諸表は、定期的に改訂または再解釈される米国公認会計基準の適用を受ける。私たちは時々、財務会計基準委員会を含む公認権威機関によって発表された新しいまたは修正された会計基準の採用を要求される(“FASB)“とアメリカ証券取引委員会。今後の会計基準は、連結財務諸表における会計処理方法を変更する必要がある場合があり、財務システムの重大な変更を要求する可能性があります。このような変化 は私たちの財務状況や運営結果に大きな悪影響を及ぼす可能性があります。
上記の理由と本稿で述べた他の理由から,我々証券への投資は高度なリスクに関連している。
105
項目1 B。未解決 従業員の意見。
ない。
項目2.財産
会社の本社はカリフォルニア州パロアルトにあります。当社は既存の賃貸オフィススペースがその業務に適していると信じています。
第3項.法的手続き
時々、私たちは が私たちの正常な業務過程で現れる訴訟の側になるかもしれません。
このような現在の訴訟または他の法律手続きは、本年度報告の表格10-Kの“第3項.法律訴訟”に記載されており、本年度報告書の“付記11-引受およびまたは事項”のタイトル下の“訴訟および他または損失”、“br}のF−1ページからの連結財務諸表に組み込まれている。当社は、現在の未解決事項の解決は、個別または全体的に私たちの財務状況または経営結果に重大な悪影響を及ぼすことはないと信じている。しかし、会社または裁判官、陪審員または他の事実調査者によって発見された未知の事実に基づいて、現在の訴訟または他の法律クレームの評価が変化する可能性があり、これは、このような訴訟またはクレームに対する管理層の可能な責任または結果の評価と一致しない。
また,訴訟の結果 自体も不確定である.報告期間内に当社に対する1つ以上の法的問題が解決され、金額が経営陣の予想を超えた場合、当該報告期間内の当社の財務状況及び経営業績は重大な悪影響を受ける可能性がある。
項目4.鉱山安全状況を開示する。
適用されません。
106
第II部
項目5.登録者普通株、関連株主事項及び発行者が株式証券を購入する市場
市場情報
我々の普通株と引受権証 はそれぞれナスダック資本市場に上場し,コードは“ATNF”と“ATNNW”である。
所持者
2023年3月31日までに、3,746,906株の普通株が発行·発行され、123名の記録所有者が発行済み普通株を保有し、及び3,435,728株普通株 既発行株式権証1,1500,000株を発行し、私たちの普通株の株式を購入するために使用された。各公開株式証は登録所有者に1/40株当たり5.75ドルで私たちの普通株の1/40株を購入する権利を持たせますこれは…。2020年12月6日(当社の初業務合併後30日)から2025年11月6日(当社の初期業務合併後5年)、ニューヨーク時間午後5:00以上の償還または清算時に開始された任意の時間において、1株当たり1株当たり230ドル、以下に説明するように調整する。もし1人の株式承認証所有者が40部の引受権証を持っている場合、この等株式証は1株自社普通株で を行使することができる。株式承認証を行使する際には、断片的な株式は発行されず、株式承認証は株式全体にのみ適用されなければならない。
株式補償計画に基づいて発行された証券
私たちは2020年の総合激励計画に基づいて185,907株の普通株式を予約しました(“2020年OIP)であり、このうち2,111株は将来の奨励に使用することができ、120,000株の普通株は、私たちの2022年総合インセンティブ計画に従って付与することができます(“2022年OIP2020年のOIPとともにOIP)であり、113,526株は、本報告日までの将来の報酬のために使用することができる。OIPは私たちの報酬計画と株式計画の重要な構成要素となることを目的としており、私たちはこれらの計画を使用して取締役、高級管理者、従業員、コンサルタントに株式に基づくインセンティブ奨励を付与する。我々の取締役会は、OIPによる株式奨励の付与は、当社とその子会社の主要サービスプロバイダの利益と当社のbr株主の利益を一致させることに役立ち、このような奨励を付与することは当社とその株主の最適な利益に合致すると信じている。
配当政策
私たちは普通株についてどんな現金配当金を支払うか発表したことがなく、予測可能な未来にも現金配当金を支払わないだろう。私たちは、私たちの業務運営と一般会社用途のために、すべての将来の収益を維持することを予想しています。未来に配当金を送るかどうかは私たちの取締役会が自ら決定するだろう。したがって、投資家は価格上昇後に彼らの普通株を売ることに依存しなければならないが、このような状況は決して起こらない可能性があり、これが将来の投資収益を実現する唯一の方法である。
最近売られている未登録証券
ない。
発行人が株式証券を購入する
ない。
特別議決権株
2種類の優先株を指定し,それぞれC類特別議決権株とK類特別議決権株(総称して“と呼ぶ)と命名した特殊投票権株)を有し、以下に指定される権利および最初のオプションを有する。
107
特別投票権株 の額面は1株当たり0.0001ドルである。各特別投票権株の権利と優先権は以下のものを含む
| ● | 私たちの普通株が投票権を持っているすべての場合の投票権は、普通株である |
| ● | 特別投票権株式brは、以前に発行されたCannBioRex Puracheco ULCおよび/またはKatexo Puracheco ULCのカナダ子会社180(“Katexo Puracheco ULC”に発行可能な普通株式数に相当する総投票権を所有者オデッセイ信託会社(受託者)に発行する権利を有する交換可能株”); |
| ● | 特別投票権株式保有者(および間接的交換可能株式保有者)は、普通株式保有者と同等の権利を有し、通知、報告、財務諸表、およびすべての株主総会に出席する |
| ● | 配当の権利はない |
| ● | 会社の終了、解散、または清算時に、特別投票権株式の所有者は、関連して割り当てられた任意の部分を得る権利がない |
| ● | 交換可能な株式が流出せず、CannBioRex Puracheco ULCおよびKatexo Puracheco ULCが選択権または他の約束がない場合、当社は特別な投票権株式 をキャンセルすることができ、これはCannBioRex Puracheco ULCとKatExco Puracheco ULCがより多くの交換可能な株式brを発行する必要があるかもしれない。 |
以上のように,交換可能株式の所有者 は,適用される特別投票権株式により普通株に対応する投票権と他の属性を持つ.交換可能株式は、CBR PharmaまたはKatExco証券のいくつかの元カナダ住民に、再構成に関連するカナダ所得税に関する課税資本利益の延期を得る機会を提供する。
本報告日までに,C類特別議決権株式と我々のK類特別議決権株式はそれぞれ0と264株の議決権を有している.
第六項です[保留されている]
項目7.経営陣の財務状況と経営結果の検討と分析。
以下、180生命科学社の2022年及び2021年12月31日現在及び2021年12月31日現在の経営業績及び財務状況の検討及び分析は、我々の合併財務諸表及び本年度報告に他の部分に含まれる連結財務諸表の注釈と共に読む。本経営陣の財務状況と経営業績の検討と分析には前向きな陳述が含まれている。参照してください“前向き情報に関する警告声明 “上です。実際の結果は,本年度報告の他の“リスク要因”で検討されている要因と,我々が知らない可能性のある他の要因とは大きく異なる可能性がある。
2022年12月31日までの累計赤字は107,408,545ドル,運営資本は3,270,608ドルであり,2022年12月31日までの年度の純損失は38,726,259ドル,運営活動に使用された現金は12,127,585ドルであった。添付されている総合財務諸表が作成されたことは,会社が継続的に経営する企業であると仮定している。私たちは収入が発生していないので、私たちは債務返済と運営コストの支払いのために大量の資本を集める必要がある。会社は2021年8月、2022年7月、2022年12月に資本を調達したが、より多くの必要な資本を調達できる保証はなく、これらの資本が優遇条項で獲得される保証はない
108
競争の激しい業界で、私たちは新しい企業を発展させるために固有のすべての重大なリスクに直面している。長期的な運営歴史と私たちの市場の新しい性質が不足しているため、私たちはすべての関連する収入フローを含む当社の業務戦略を成功するまで、運営損失が予想される。私たちは収益性を達成したり、相当な収入を生み出すことができないかもしれない。
私たちの現在の最低月額現金支出は約900,000ドルです。全体的に、私たちは、私たちの製品の研究開発とマーケティングを支援し、拡大し、将来の臨床試験を支援し、債務を返済し、追加設備と開発コスト、支払い義務、オフィス空間、業務管理システムの資本支出を提供し、私たちが計画した製品収入フローが完全に実施され、私たちの運営コスト を相殺し始めるまで、私たちの製品の研究開発とマーケティングを支援し、拡大するために多くの追加資金が必要になると信じている。
私たちが設立して以来、私たちは株式と債務融資の収益で私たちの運営に資金を提供してきた。私たちは流動性の問題を経験したが、その理由の一つは、私たちが受け入れ可能な条件で十分な資本を調達する能力が限られているからだ。我々は従来,株式発行と普通株に変換可能な約束票に依存して運営に資金を提供し,このリスクを減らすための多くの努力を投入してきた。私たちは、予測可能な未来に、私たちの運営に資金を提供し、私たちの未返済債務を返済するために株を発行する必要があると予想しています。もし私たちが運営利益を達成できない場合、あるいは私たちは他の形態の融資を得ることに成功できない場合、私たちは他の行動を評価して、私たちの運営費用を減らし、現金を節約しなければなりません。
添付されている総合財務諸表は、米国が公認している持続経営会計原則に基づいて作成されており、持続経営をもとに、正常な業務過程における資産の現金化と負債の返済を考慮している。したがって、総合財務諸表には、資産回収可能と負債分類に関するいかなる調整も含まれておらず、会社が持続経営企業として経営を継続できなければ、調整する必要があるかもしれない。本募集説明書に含まれる総合財務諸表 には、持続経営脚注も含まれている。
しかも、可能性がある限り、私たちの取締役会は非現金対価格を使用して義務を履行しようと試みるだろう。多くの場合、非現金 対価格には、私たちの普通株の制限株式、優先株、または私たちの普通株式を購入する承認証が含まれると考えられます。brは、私たちの取締役会は、株主投票を経ていない普通株の全部または一部を発行する権利がありますが、ナスダックの規則および規定(任意の取引が、私たちが当時発行した普通株の20%を超える発行された普通株、または当時発行された普通株の20%以上に相当する)を発行する可能性があり、取締役会は、brを発行していますが、発行されていない普通株の全部または一部を発行する権利があります。普通株の優先株または株式承認証を購入する。また、私たちは私たちの普通株の株を売ることで資金を調達して、将来的には市場を下回る価格で売ることを試みるかもしれません。これらの行動は、既存の株主の所有権権益を希釈し、普通株式の帳簿価値をさらに希釈する可能性があり、このような希釈は実質的である可能性がある。brのような発行は、既存の管理層を支援するために努力している当事者またはエンティティに発行される可能性があるので、既存の管理職が私たちの制御能力を維持する能力を強化するのにも役立つかもしれない。
MD&A組織
我々の経営陣の議論と財務状況と経営結果の分析(TheMD&A)添付されている連結財務諸表および付記に加えて、読者が私たちの運営結果、財務状況、br、およびキャッシュフローを理解するのを助けるために、他の情報が提供されています。MD&Aの組織形式は以下のとおりである
| ● | 業務概要と 最新イベント。会社の業務概要と最近発生したいくつかの重大な事件。 | |
| ● | 重要な財務諸表構成要素。-会社の重要財務諸表構成要素の概要。 |
109
| ● | 経営実績。 2022年12月31日と2021年12月31日までの12ヶ月間の財務業績を比較分析した。 |
| ● | 流動性と資本 資源。私たちの貸借対照表とキャッシュフローの変化を分析し、私たちの財務状況を検討する。 |
| ● | 重要な会計 政策と見積もりです。私たちは、私たちの報告書の財務結果と予測に含まれる仮説と判断を理解するために非常に重要な会計見積もりだと思います。 |
業務概要と最新の事件
本MD&A及び関連する2022年12月31日までの財務諸表は主に180の業務をカバーしており、これはカリフォルニア州パロアルトに本部を置く臨床段階バイオテクノロジー会社であり、慢性疼痛、炎症、繊維化と他の炎症性疾患で満足されていない医療需要の開発療法に専念し、その中で抗腫瘍壊死因子療法は革新的な研究、br}及び適切な場合の併用療法を採用することにより、患者に明らかなメリットを提供する。3つの製品開発プラットフォームがあります
| ● | 線維化と抗腫瘍壊死因子(“腫瘍壊死因子”); |
| ● | カンナビノールの誘導体の薬(“中央商務区“);及び |
| ● | α7ニコチン型アセチルコリン受容体(“α7 nAChR”). |
私たちはいくつかの未来の候補製品を開発中で、その中の一つの候補製品は最近イギリスで2 b期の臨床試験に成功し、Dupuytren‘s Constraintを治療するために使用され、これは手掌繊維結合組織の発育に影響を与える疾患である。br}180はバイオテクノロジーと製薬分野のいくつかの世界をリードする科学者によって創立された。
我々は,進行中の臨床計画を成功させ,新たな候補薬物を発見し,新たな分子を開発し,我々の既存の管路上に を構築し,未満足の臨床需要を満たすために資源brを投入する予定である。候補製品は,定義されたユニット 操作と技術からなるプラットフォームで設計される.この作業は,最も信頼できる生産条件を決定するために,プロセスの各ステップの可変性 を決定するために研究開発環境で行われている。
私たちは第三者の代行組織に依存するかもしれない(“Cmos)および他の第三者が候補製品を将来的に製造および加工する。第一陣の候補臨床製品の使用契約製造とテストに対してコスト効果があり、著者らの開発計画に基づいて臨床試験を迅速に準備できると信じている。 第三者メーカーは、期待される 臨床試験需要を満たすために、十分な数のこれらの候補製品を提供し、加工することができると予想される。
重要財務諸表構成要素
研究と開発
これまで,180の研究と開発費用は主にその3つの製品プラットフォームの発見作業および臨床前と臨床開発に関連している:(1)線維化と抗腫瘍壊死因子,(2)CBD誘導体としての薬物,および(3)α7 nAChR。研究開発費には、これら3つの製品プラットフォームに関するコストが主に含まれている
| ● | 180のパートナーと第三者との契約組織、それを代表して研究·開発活動を行う調査的臨床試験場所およびコンサルタントの合意による費用 |
| ● | 契約製造業者への費用を含む臨床材料の製造に関連するコスト; |
110
| ● | 臨床前と臨床試験の実行に関連する実験室とサプライヤー費用 ; |
| ● | 従業員に関する費用は、賃金、福祉、株式報酬を含む |
| ● | 施設やその他の費用は、施設の賃貸料と維持費用、減価償却と償却費用、その他の用品費用を含む。 |
私たちはすべての研究と開発コストが発生している間にそれを支出する。我々は,各項目の状態と外部サービスプロバイダから受信した伝票を監視することで,サービス提供によるコストを積算する.私たちは実際のコスト状況に応じて課税項目を調整します。 研究開発手配や許可協定に基づいて第三者またはマイルストーン支払いが不足している場合、マイルストーン支払い義務はマイルストーンの結果を実現する際に支出されます。
研究開発活動は私たちの業務モデルの核心だ。臨床開発後期段階にある候補製品は通常,臨床開発早期段階の候補品よりも高い開発コストを有しており,これは主に後期臨床試験の規模と持続時間が増加しているためである。臨床プロジェクトの進展や,より多くの候補製品の臨床試験の開始を求めることにより,今後数年間の研究開発費が増加することが予想される。また,より多くの候補製品を選択的に決定·開発するに伴い,研究開発費が増加することが予想される。しかしながら、現在または未来の候補製品の臨床前計画および臨床試験の持続時間および完成コストを決定することは困難である。
臨床試験と候補製品開発の持続時間、コスト、時間は様々な要素に依存するが、以下の :
| ● | 患者1人当たりの試験コストは |
| ● | 試験に参加した患者数 ; |
| ● | 実験に含まれるサイト数は ; |
| ● | 実験を行った国·地域 |
| ● | 条件に適合する患者を登録するのに要する時間長; |
| ● | 患者が受ける用量の数 |
| ● | 患者の中退率または中断率 ; |
| ● | 潜在的な追加の安全規制機関に要求される監視または他の研究; |
| ● | 新冠肺炎が私たちの実験時間に与える影響は |
| ● | 患者のフォローアップの持続時間; |
| ● | 候補製品の有効性と安全性 概要。 |
さらに、各候補製品の成功確率は、競争、製造能力、および商業生存能力を含む多くの要素に依存する。各候補製品の科学的および臨床的成功、および各候補製品のビジネス潜在力の評価に基づいて、どのような計画を実施し、援助するかを決定する。
111
候補製品 はまだ臨床および臨床前開発段階にあり、これらの努力の結果はまだ確定していないため、候補製品の開発と商業化に成功するのに必要な実際の金額を見積もることはできず、私たちがいつ利益を達成する可能性があるかどうかを推定することもできない。これらの計画は初期段階にあるため、プロジェクトごとにコストを追跡することはない。これらの計画がより先進的になるにつれて, 我々は個々の計画の外部と内部コストを追跡する予定である.
一般と行政
一般及び行政支出は主に賃金及びその他の従業員に関連するコストを含み、すでに発行された普通株の株式給与及び創業者、取締役及び行政、商業、財務、会計、法律、投資家関係、施設、業務発展及び人力資源機能の購入権を含み、そして帰属条件を含む。
その他の重大な一般および行政コストには、施設や管理費用に関するコスト、会社や特許事務に関する法的費用、br}訴訟、米国証券取引委員会届出、保険、投資家関係コスト、会計およびコンサルティングサービス費用、その他の一般および行政費用が含まれる。一般と行政コストは発生した費用に応じて計上され,提供されたサービスの状態を監視し,我々のサービスプロバイダから見積もりを受け取り,実コストに応じて我々の計上項目を調整することで,第三者が提供するサービスに上記の費用に関する金額を計上する.
今後数年間の一般費用と管理費が増加することが予想され,我々の持続的な研究開発活動,製造活動,候補製品の潜在的商業化,上場企業の運営コストの増加を支援する。これらの増加には,より多くの人員の募集,ビジネスインフラの開発に関するコスト増加,外部コンサルタント,弁護士,会計士に支払う費用の増加,上場企業に関するコスト増加,ナスダック上場規則の遵守や米国証券取引委員会が要求するサービスに関する費用,保険,投資家関係コストが含まれると予想される。
その他の収入
その他の収入は主に他社(一部の会社は関連側)が行った研究開発作業のために稼いだ費用である。
利子支出
利息支出には主に債務ツールに関連した利息支出が含まれる。
転換手形の清算収益(損失)
変換可能チケット清算収益 (損失)とは,その帳票価値に対する変換可能チケットの再買収コストの差額(超過)である.
営業権減価損失
営業権減価損失 は報告期間中の資産の帳簿価値がそれを超えて公平市価を推定した部分であり、回収できない。
知的財産権研究開発資産減価損失
知的財産権研究開発資産の減価損失 は報告期間内の資産の帳簿価値がそれを超えて公平な市価を推定した部分であり、回収できない。
IR研究開発資産の公正価値変動
知的財産権研究開発資産公正価値変動 シリーズは報告期間内にこの資産公正価値の非現金変動を指す。
112
派生負債の公正価値変動
派生ツール負債公正価値変動 は報告期間内の派生ツール負債公正価値の非現金変動を指す。 現在2022年及び2021年12月31日までの年度派生ツール負債公正価値変動による収益/損失は、期間内の株価下落/上昇によって牽引され、関連負債の公正価値が比較的に低い/比較的に高い。
株式証明書負債を計上した発売コスト
株式証明書負債に割り当てられた発売コスト変動 は配給代理費と発売支出を指し、この費用はすでに個人投資公衆株式証(“PIPE”)株式証に分配され、そしてこの等株式証が負債種別に属する時に即時に支出を計上する。
請求すべき発行権益は価値変動を公正に承諾することができる
発行可能権益の公正価値変動とは、応算権益の正式発行前の公正価値の非現金変動である。
総合経営成果
総合経営成果
2022年12月31日までの年度と2021年12月31日現在の年度との比較
| ここ数年で 12月31日、 |
||||||||
| 2022 | 2021 | |||||||
| 運営費用: | ||||||||
| 研究開発 | $ | 2,191,834 | $ | 1,000,769 | ||||
| 研究開発に関連した各方面 | 240,731 | 2,947,536 | ||||||
| 一般と行政 | 15,459,788 | 11,230,118 | ||||||
| 一般と行政関係の当事者 | 5,612 | 462,580 | ||||||
| 総運営費 | 17,897,965 | 15,641,003 | ||||||
| 運営損失 | (17,897,965 | ) | (15,641,003 | ) | ||||
| その他(費用)収入: | ||||||||
| 債務収益を返済する | - | 926,829 | ||||||
| その他の費用 | - | (146,822 | ) | |||||
| 利子支出 | (28,175 | ) | (135,953 | ) | ||||
| 利子支出関係者 | 1,508 | (50,255 | ) | |||||
| 転換支払手形支払損失純額 | - | (9,737 | ) | |||||
| 営業権減価損失 | (33,547,278 | ) | - | |||||
| 知的財産権研究開発資産減価損失 | (3,342,084 | ) | - | |||||
| 派生負債の公正価値変動 | 15,144,986 | (4,677,388 | ) | |||||
| 請求すべき発行権益は価値変動を公正に承諾することができる | - | (9,405 | ) | |||||
| 株式証明書負債に割り当てられた要約コスト | - | (604,118 | ) | |||||
| その他の費用の合計 | (21,771,043 | ) | (4,706,849 | ) | ||||
| 所得税前損失 | (39,669,008 | ) | (20,347,852 | ) | ||||
| 所得税割引 | 942,749 | 23,204 | ||||||
| 純損失 | $ | (38,726,259 | ) | $ | (20,324,648 | ) | ||
113
研究と開発
2022年12月31日までの年間で発生した研究開発費は2,191,834ドルであり,2021年12月30日までの年度の1,000,769ドルと比較して1,191,065ドルまたは119%増加した。この増加には、株式ベースの報酬支出が1,000,000ドル増加し、オックスフォード大学協定に関連する支出が43,000ドル増加し、賃金支出が29万ドル増加し、科学顧問委員会関連費用が27万ドル増加し、コンサルティング費用が120,000ドル増加し、特許および許可関連費用が100,000ドル増加することが含まれる。この活動は、YissmおよびGallly Ruthの契約に関連する費用がそれぞれ46万ドルおよび25万ドル減少し、210,000ドルの税金相殺および関連側相談費用の110,000ドル減少によって相殺される。
研究開発に関連した各方面
2022年12月31日までの年間で発生した研究開発関連費用は240,731ドルであり,2021年12月31日までの年度の2,947,536ドルに比べて2,706,805ドルまたは92%減少した。この減少は、株式ベースの報酬支出が2,300,000ドル減少したことを含み、この減少は、Dupuyten‘s Contracture(RIDD)の第2段階臨床試験(RIDD)の研究と、前年にNanchahalさんに支払われた約1,400,000ドルのNanchahalさんへの支払いを含む前年のJagDeep Nanchahalへの支払いを含む約1,400,000ドルの株式報酬支出を含む。相談費も46万ドル減少した。
一般と行政
2022年12月31日までに発生した一般および行政支出は15,459,788ドルであり,2021年12月31日までの11,230,118ドルより4,229,670ドルまたは38%増加した。この増加は、法的費用が3,700,000ドル増加し、役員および上級管理職の保険費用が88,000ドル増加し、賃金支出が550,000ドル増加したが、交換に関連する罰金が530,000ドル減少し、和解費用が360,000ドル減少し、株式補償費用が180,000ドル減少し、コンサルティング費用が40,000ドル減少したためである。
一般的に行政関連の当事者
2022年12月31日までの年間で5,612ドルの一般·行政費用関連者が発生し,2021年12月31日までの年度の462,580ドルに比べて456,968ドル減少し,減少幅は99%であった。減少の要因は,関連側相談費用が125,000ドル減少したことと,関連側売掛金に関する不良債権費用が300,000ドル減少したことである
その他の費用、純額
本グループは2022年12月31日までに他の支出純額21,771,043ドルを発生させたが,2021年12月31日までは4,706,849ドルであり,その他の支出は17,064,194ドルまたは363%増加した。支出増加の要因は,i)今年度の営業権減額33,547,278ドル,ii)今年度の知的財産権研究開発資産減額3,342,084ドル,およびiii)前年度に計上されていなかった今年度の負債926,829ドルの収益を決算し,iv)今年度のデリバティブ負債の公正価値変動19,822,374ドルおよびv)前年度の発売による604,118ドルの株式証明コストの変動に相殺されたことである。
114
流動性と資本資源
2022年12月31日と2021年12月31日までの現金残高はそれぞれ6,970,110ドルと8,224,508ドルであり,運営資本は3,270,608ドル,運営資本赤字はそれぞれ8,498,193ドルである。
2022年,2022年,2021年12月31日までの年間運営活動用現金はそれぞれ12,127,585ドルと19,371,428ドルであった。私たちが2022年12月31日までの年間運営で使用した現金は主に私たちの純損失38,726,259ドル、非現金支出調整後の総金額は23,876,048ドル、運営資産と負債レベルの変化のための現金純額は2,722,626ドルです。年内の非現金支出のかなりの一部は商業権と知的財産権研究開発資産の減価に関する非日常的支出3,690万ドル(付記5-無形資産と長期資産減価参照)に関連しているが、今年度15,144,986ドル由来負債の公正価値変動によって相殺された。私たちが2021年12月31日までの年間運営に使用した現金は、主に私たちの純損失20,324,648ドル、非現金支出調整後の総金額は9,760,161ドル、および運営資産と負債レベルの変化のための8,806,941ドルの現金純額です。今年度の運営で使用されている現金の大部分は業務合併に関する480万ドルの非日常的な費用に関連している。
12月31日まで、2022年、2021年12月31日まで、投資活動は現金を提供していません。
融資活動が提供する現金は、2022年と2021年12月31日までの年間でそれぞれ10,873,606ドル、25,411,919ドルである。2022年12月31日までの年間で、融資活動が提供する現金には、主に2022年7月の普通株売却と普通株式承認証から得られた6,499,737ドル、2022年12月の普通株売却と一般権証による5,999,851ドルとローン対応所得1,060,890ドルが含まれるが、発行コストはそれぞれ529,982ドルと484,991ドル、およびローン返済と対応関係者ローンの返済はそれぞれ1,591,035ドルと81,277ドルで部分的に相殺される。2021年12月31日までの年度中に、融資活動が提供する現金には、普通株の売却および株式承認証による金26,666,200ドルおよびローン対応金1,618,443ドルが含まれるが、転換可能債務の返済および支払ローンはそれぞれ10,000ドルおよび(807,594ドル)および発売コスト(2,055,130ドル)を支払った部分で相殺される。
私たちの候補製品は決して商業化されないかもしれません。予測可能な未来には損失が続くと予想されています。私たちの研究開発費、一般と管理費用、資本支出は引き続き増加すると予想されています。したがって、その前に、相当な製品収入を生成することができれば、潜在的な協力、ライセンス および他の同様の配置を含む株式発行、債務融資、または他の資本源の組み合わせによって、割引条項では提供できない可能性がある(全くない場合)私たちの現金需要を満たすことが予想される。追加の株式や債務証券を売却し、 が完成すれば、私たちの当時の株主を希釈するかもしれない。私たちの資本の主な用途は、私たちは引き続き 給与と関連費用、第三者臨床研究開発サービス、出現可能なライセンス支払いまたはマイルストーン義務{br)、実験室と関連用品、臨床コスト、潜在製造コスト、法律と他の規制費用、およびbr}一般管理費用であると予想される。
我々の重大な現金需要(Br)および既知の契約およびその他の義務におけるこのような需要の期間には、オックスフォード大学およびYissmの許可協定に関するマイルストーンおよび特許使用料支払い、D&O保険に関する支払い、コンサルタントへの支払い、外部コンサルティング会社(例えば、法律顧問、監査師、会計士など)に関する支払いが含まれる。これらの現金需要総額は2023年には約10,000,000ドル、2024~2027年は約27,000,000ドルである
115
また、私たちの運営計画は変わる可能性があり、臨床試験や他の研究開発活動の運営需要と資本要求を満たすために追加の資金が必要かもしれません。私たちは現在信用計画もなく、約束された資金源もない。私たちの候補製品の開発や商業化に関連する多くのリスクと不確実性のため、現在および予想されている製品開発計画に関連する資本支出や運営支出が増加したbr金額を見積もることができません。
私たちはまだ利益 を実現しておらず、運営からの現金流出が続く見通しだ。私たちの研究開発や一般的な行政費用は引き続き増加すると予想されていますので、最終的には、私たちの運営を支援するためにより多くの資金を集める必要があります。もし私たちが合理的な条項で十分な資金を得ることができない場合、私たちは大幅に削減したり、運営を停止したり、魅力的でない条項で融資協定を締結することで資金を得ることが要求される可能性がある。私たちの運営需要には運営業務の計画コストが含まれており,運営資本や資本支出に資金を提供するために必要な金額が含まれている。2022年12月31日現在、上記の条件は、財務諸表発表日から1年以内に、私たちが経営を継続している企業として経営を継続する能力が大きく疑われていることを示している。しかし,2021年8月,2022年7月,2022年12月には,それぞれ約1,390万ドル,600万ドル,550万ドルの追加資本を調達し,2023年3月29日現在,手元現金は約270万ドルであり,会社 は2023年第3四半期まで経営を継続できる企業として経営を継続できると予想されている。
我々の連結財務諸表は米国公認会計原則(“米国公認会計原則”)に従って作成され、この原則は会社の持続経営企業としての持続経営及び正常業務過程における資産現金化と負債返済を考慮したものである。連結財務諸表に記載されている資産や負債の帳簿価値は、必ずしも現金化または決済価値を代表するとは限らない。連結財務諸表には、この不確実性の結果がもたらす可能性のあるいかなる調整も含まれていない。
最近の融資と決済取引
転換可能債務 変換
2020年11月27日から2021年2月5日まで、当社の転換可能な本チケット所持者は、改訂された転換可能手形条項に基づいて、1株当たり40.00ドルから65.80ドルの換算価格で、この等変換可能手形項目で不足している合計4,782,107ドルの普通株を合計99,338株普通株に変換することができる。
2021年第3四半期に、いくつかの手形保有者は元金総額が1,234,334ドルおよび累計利息残高が105,850ドルであるいくつかの対応転換可能手形を23,357株会社の普通株に変換し、転換価格は1株49.00ドルから65.80ドルに転換することを選択した。変換可能なチケットを発行する際に発行される株式の発行時の公正価値は1,941,125ドルである.
2021年2月にサービス提供
2021年2月19日、当社は複数の機関投資家と証券購入協定を締結した購入者“) これにより、当社は合計128,200株の株式を買い手に売却することに同意しました(”株“)購入合計最大128,200株当社普通株式および引受権証(”スパ ライセンス)、総買付価格は1株91.00ドルであり、SPA承認株式証が付属している(奉納する“。 配給代理費と予定発売費用を差し引く前に、今回発売された総収益は約1,170万ドルです。 社が支払うべき発売費用です。配給代理費および当社が支払うべき発売費を差し引くと、当社の発売による純額は約1,080万元です。今回の発行は2021年2月23日に終了した。
116
Maxim Group LLC(The)代理を配置する“)当社と配給エージェントが2021年1月26日(2021年2月18日に改訂)した契約書に基づき、初発売に関する独占配給エージェントを担当しています。招聘状によると,配給エージェントは発行総毛収入の7%(7%)に相当する手数料 または816,634ドルを受け取る.
Bridge備考の変換
2021年3月8日に、当社が2019年12月27日および2020年1月3日に異なる買い手に発行した転換可能なブリッジ手形の所持者は、改訂された当該等の手形の条項に基づいて、合計432,383ドル(このような変換可能なブリッジ手形に借りられた利息66,633ドルを含む)を合計7,920株の普通株式に変換し、換算価格は1株54.60ドルとした。
早鳥資本と和解協定
当社は2021年4月23日に、2017年10月17日にEarlyBird Capital,Inc.(“EarlyBird”)と締結したある発見者合意(“発見者合意”)に基づいて支払金を清算する。当社取締役会は、提出または提出可能なすべての請求事項について和解を達成することが最も利益に合致することを決定し、和解合意(“和解合意”)を締結した。和解協定によると、当社はEarlyBirdに現金275,000ドルを支払い、EarlyBirdに11,250株の付与日価値が1,973,250ドルの会社限定普通株を発行し、1,750,000ドルの売掛金を全額返済する。当社の和解協議に関する損失223,250ドルは、添付されている簡明総合経営報告書の負債決済収益(損失)に計上されている。
アルファ資本決済 協定
2021年7月31日、当社は、Alpha Capital Anstalt(“Alpha Capital Anstalt”)と締結されたある転換手形合意によって満期になったといわれる金を弁済することで合意したAlpha)を2020年9月8日に(“Alpha音符“)”会社取締役会は、アルファ手形が提出されたか、または提出可能なすべてのクレームについて和解合意に達し、会社の最良の利益に合致すると考えている(“アルファ和解協定アルファ和解協定に基づき、当社は7,500株の普通株及び3年期株式承認証を発行し、1株141.40ドルの使用価格で1,250株の当社普通株を購入し、アルファ手形 の全面的かつ全面的な清算と交換する。
2021年8月提供
2021年8月23日、当社はいくつかの買い手と証券購入協定を締結した(“2021年8月購入者)、 これにより、当社は合計125,000株の普通株の売却に同意しました(“2021年8月株“) と株式承認証購入最大125,000株普通株(”2021年8月管道証)、合計買い取り価格は1株120.00ドル、2021年8月管材承認株式証(2021年8月提供“)”今回発行された総収益は約1,500万ドル。配給代理費用と見積会社が支払うべき発売費用を差し引いたところ、今回発売された当社への純収益は約1390万ドルだった。配給代理費と発売費用 は追加実収資本の減少額に計上される。配給代理が獲得した手数料は,今回発行された総毛収入の7% (7%),すなわち1,050,000ドルに相当する。今回の発行は2021年8月23日に終了した。
2021年8月の発売についても、当社は2021年8月23日に買い手と登録権協定を締結します(“2021年8月登録権協定“)”2021年8月の登録権利協定に基づいて、当社は、2021年8月の株式の登録及び2021年8月の管状株式承認証の行使後に発行可能な普通株の売却を登録するために、2021年9月12日又はそれまでに証券取引委員会に登録声明を提出することに同意する株式引受証)と、2021年10月22日または以前に登録声明を発効させる(または、発生した場合全面的に審査する(米国証券取引委員会、2021年11月21日)。登録声明は2021年8月31日にSECに提出され、SECは2021年9月9日に発効すると発表した。
117
係り先ローンと変換可能チケットの交換
2021年9月30日、それぞれ取締役会の共同執行議長を務めたローレンス·スタンマン博士とマーク·フェルドマン博士は会社と合意し、合意条項により、元金残高総額693,371ドル、累計利息残高157,741ドルの未返済融資項目の借金を会社普通株総額7,093株に転換し、1株120.00ドルに転換した。どの転換率が 社普通株が拘束力のある合意を締結した日の終値合併入札よりも高いですか。(詳細は付記9-対応ローンおよび付記10-対応変換可能手形 を参照
ミンツ·レヴィンは和解した
2021年9月、当社はミンツ、レヴィン、コーエン、フェリス、グロフスキー、Popeo、P.C.(“ミンツ“), これにより,会社は提供された法的サービスを支払うためにMintzに800,000ドルを支払うことに同意した.任意の利息を差し引く前に、ミンツが同社に発行した請求書の総額は1,454,240ドルである。当社は和解協定に基づいて支払いを行った後,約650,000ドルの収益 を記録した。
コント·フィッツジェラルド社は和解を訴えました
2021年10月12日、当社はCantor Fitzgerald&Co.と和解合意に達し、この合意に基づき、当社はCantorに200,000ドルを支払うことに同意し、当社に対する訴訟を却下することに同意した。会社は2021年10月13日にカントーに資金を送ります。 2021年9月30日まで、会社は合意に基づいて和解金額の課税項目を記録しました。
2021年10月21日、当社は生産停止通知を受けましたので、当社とコントール間の問題が解決されて終了しました。
2022年7月提供
2022年7月17日、当社はいくつかの買い手と1つの証券購入合意を締結し、これにより、当社は合計175,000株の普通株を売却し、最大131,604株の普通株を購入可能な予備株式承認証(“2022年7月予備融資承認株式証”)及び最大306,604株の普通株を購入できる普通株式承認株式証(“2022年7月普通株承認株式証”)、合併購入価格は1株21.20ドル及び引受株式証(“2022年7月発売”)であることに同意した。2022年7月に発売された総収益は6,499,737ドル。配給代理費と当社が支払うべき他の推定発売費用を差し引いたところ、当社の発売による純額は約600万ドルでした。配給代理費と発売費用は約530,000ドルを計上して追加実収資本の減少を図る。2022年7月の発売は2022年7月20日に終了します。彼は言いました
2022年12月提供
2022年12月20日、当社は複数の買い手と証券購入契約を締結し、これにより、当社は合計215,000株の普通株の売却、最大1,499,286株の普通株を購入する予備資本権証(“2022年12月 予資権証”)と、最大2,571,429株の普通株を購入する普通株式権証(“2022年12月 普通株式証”)に同意した。1株3.50ドルプラス株式証の総合購入価格で計算します(“2022年12月発売”)。 2022年12月発売の総収益は5,999,851ドルです。配給代理費と当社が支払うべき他の推定発売費用を差し引いたところ、当社の発売による純額は約550万ドルでした。配給代理費と発売費用約500,000ドルを追加実納資本の減少に計上した。2022年12月の株式発行は2022年12月22日に終了した。彼は言いました
118
重要な会計政策と試算
会社の連結財務諸表はアメリカ公認の会計原則に基づいて作成されています。これらの連結財務諸表を作成する際には、管理層は、その資産、負債、収入、費用の報告金額に影響を与える推定および仮定を行う必要がある。当社はいくつかの政策と推定 がその業務運営及びその過去或いは現在の運営結果に対する理解に重要であることを確定し、このような運営は(I)営業権及び (Ii)無形資産及び進行中の研究開発(“知的財産権研究開発”)と関係がある。これらの政策および推定は、会社の連結財務諸表に重大な影響を与えたり、大きな影響を与える可能性があるため、重要であると考えられ、それらは管理層に重大な判断、仮定、または推定を要求する。当社は,以下の項目を計算する際に作成した推定,判断,仮説が合理的であると信じ,これらの推定,判断,仮説を作成する際に把握した資料に基づいている。しかし、実際の結果はこれらの推定とは異なる可能性があり、これらの違いは実質的である可能性がある。
営業権/無形資産と進行中の研究と開発
当社は大量の商業権、無形資産、知的財産権研究開発資産を持っており、これらの資産は少なくとも毎年減値評価を行っている。これらの資産の減価分析は会社にとって重要であることから重要であると考えられている。業務合併或いは買収によって発生した無形資産、例えば商業権、特許と知的財産権研究開発資産は、初歩的に承認価値によって入金される。許可された特許は特許の残りのライフサイクルで償却されるだろう。知的財産権研究開発資産は無期限とされている--関連研究開発プロジェクトが完成または放棄されるまで。私たちの営業権は買収価格が買収された純資産の公正価値を超える買収から来ており、内部報告構造を再編して報告単位の構成を変えさえすれば、会社は営業権を報告単位に再分配しなければならない。その会社はその唯一の運営部門を代表する報告機関を決定した。
当社は少なくとも毎年1回の商業権/無形資産及び知的財産権研究開発資産を評価しなければならず、或いは更に頻繁に商誉/無形資産及び知的財産権研究開発資産を評価しなければならず、もし事件或いは情況が変化した場合、当社は商業誉/無形資産及び知的財産権研究開発資産 を評価しなければならず、当社の報告部門の公平価値はその帳簿価値より低い可能性が高いことを表明した。Br商誉/無形資産と知的財産権研究開発資産の減値を評価する時、当社はまず定性要素を評価して、その報告単位の公正価値がその帳簿価値より低い可能性があるかどうかを確定するかもしれない。
営業権の減価。潜在的減価を識別するための営業権資産減価テストの第1のステップは、報告単位の公正価値をその帳簿金額(営業権資産を含む)と比較する。当社は、単一報告単位の2021年12月31日までの公平時価132,760,914ドル、すなわち1株78.00ドル(市場終値)に2021年12月31日までの1,702,063株(1,701,799株普通株に264株特別投票権株を含めると、無料で普通株に変換できる)と決定した。2021年12月31日現在、報告単位の帳簿金額は39 322 695ドル(総資産6270万ドルから総負債2340万ドル)である。
2021年12月31日現在,br社の公正価値(132,760,914ドル)が会社の帳簿価値(39,322,695ドル)を超えているため,br社の帳簿価値はゼロより大きく,経営陣は報告先の営業資産に減値はないと結論した。
2021年12月31日現在、上場株の終値は1株78.00ドルであり、2022年の間に会社単一報告単位の時価は大幅に低下した。当社の上場株の時価は、2022年3月31日、2022年6月30日、2022年9月30日、2022年12月31日まで、それぞれ1株51.80ドル、16.96ドル、13.30ドル、3.39ドルに下落したため、当社は2022年9月30日と2022年12月31日までの売上高を定量的に分析し、減値を評価することを選択した。当社はその単一報告単位の公正時価を確定し、そしてこの価値を報告単位の帳簿価値と比較し、営業権が2つの計量日にすべて減値されたことを確定した。2022年9月30日と2022年12月31日まで、帳簿価値はそれぞれ公平市場価値より18,872,850ドルと14,674,428ドル高かった。営業権減額を確認するために、会社は第3四半期と第4四半期末にこれらの金額の損失 を記録し、2022年12月31日までの年間収益表では営業権減価損失33,547,278ドルを示した。詳細は、付記5-無形資産および長期資産減価を参照されたい。
119
知的財産権研究開発資産の減価
2022年12月31日現在、貸借対照表上の知的財産権研究開発資産の帳簿金額は12,405,084ドル(それぞれ自社のCBR Pharma子会社および180 LP子会社に関連する帳簿価値1,462,084ドルおよび10,943,000ドルを含む)である。年末までに第三者から得られた推定値によると、当社の知的財産権研究開発資産の公平時価は9,063,000ドルに決定されている(ただし は自社のCBR Pharma子会社と180 LP子会社に関する公平な市場価値をそれぞれ0ドルと9,063,000ドルである), 本計量日までに、CBR Pharmaと180 LP子会社の資産の帳簿価値はそれぞれ公平市場価値より1,462,084ドルと1,880,000ドル高い。そこで、経営陣は総合知的財産権研究開発資産減額3,342,084ドルを確定し、減額を確認するために、当社は2022年第4四半期にこの金額の損失を記録し、損益表では知的財産権研究開発資産減価損失を示した。2022年12月31日まで、そのCBR Pharma子会社 及び180個のLP子会社の知的財産権研究開発資産残高はそれぞれゼロと9,063,000ドルに低下した;減値後の総合知的財産権研究開発資産残高 は9,063,000ドルであった。詳細については、付記5−無形資産および長期資産減値を参照されたい。
当社は引き続き 毎年商誉/無形資産と知的財産権研究開発資産の減価テストを行ったり、その報告単位の構成が変化した場合に必要に応じてテストを行ったりする。
最近発表された会計公告
最近発表·採択された会計声明の概要については、本年度報告書に含まれる我々の連結財務諸表の主な会計政策概要 を参照されたい。
第七A項。市場リスクに関する定量的で定性的な開示。
S−K条例第305(E)項(第229.305(E)節)によれば、会社は、本プロジェクトに要求される情報を提供する必要がない小さなbr報告会社は“しかし、当社は、ルール229.10(F)(1)の定義に基づき、以下の金利リスクに関する情報を提供している。
金利リスク
私たちは正常な業務過程で市場の危険に直面している。このような危険は主に金利敏感性を含む。2022年12月31日まで、6970,110ドルの現金と現金等価物を持っています。私たちは現金を決済通貨市場口座に保管するつもりだ。私たちの市場リスクに対する主な開口 は金利感度であり、それはアメリカの金利全体のレベル変化の影響を受ける。我々の現金等価物は短期的な 満期日であるため,その投資のリスクは低く,ただちに金利100ベーシスポイントを調整することは我々の現金等価物の公平な市場価値に実質的な影響を与えない。
項目8.財務諸表および補足データ。
本プロジェクトによって要求される情報は、本報告の署名ページの後に位置し、参照によって本報告に組み込まれる本報告の“連結財務諸表インデックス”に含まれる
120
項目9.会計·財務開示面の変化と会計担当者との相違。
ない。
第9条。制御と プログラム.
情報開示制御とプログラムの評価
我々の経営陣は、最高経営責任者と最高財務官の参加の下で、我々の開示制御及び手続きの有効性を評価し、 という用語は、1934年に公布された証券取引法(改正)に基づいて公布された規則13 a-15(E)および15 d-15(E)で定義され、2022年12月31日までである(“取引所 法案”)。同等の評価によると、主要行政総裁及び主要財務総監は、当社の開示制御及びプログラムが有効であると結論した。
経営陣の財務報告に関する内部統制報告書
180生命科学会社の経営陣は、取引規制13 a~15(F)および15 d~15(F)に定義されている財務報告の十分な内部統制の確立および維持を担当する。財務報告の内部統制とは、当社の主要な行政人員と主要な財務官或いは類似の機能を実行する人員が設計或いは監督するプログラムであり、そして当社の取締役会 によって実施され、公認会計原則に基づいて財務報告と財務諸表の信頼性に対して合理的な保証 を提供する。会社の財務報告に対する内部統制は、(I)合理的で詳細かつ正確かつ公平に会社の資産を反映した取引および処置の記録を維持することに関連し、(Ii)公認された会計原則に基づいて財務諸表を作成し、会社の収入および支出 が会社の管理層および取締役の許可のみに基づいて行われるように、取引が必要として記録されている合理的な保証を提供することと、以下の政策および手順を含む。および(Iii)不正買収の防止またはタイムリーな発見、当社資産の使用または処分について合理的な が財務諸表に重大な影響を与える可能性のある保証を提供する。
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。したがって,有効と判断されたシステムであっても財務諸表の作成や列報について合理的な保証しか提供できず,将来の期間のどの有効性評価の予測も条件の変化により不十分になる可能性があり,あるいは が政策やプログラムを遵守する程度が悪化する可能性がある.
我々は,証券法の下でS-K法規第10(F)(1)項で定義されている“小さな報告会社”である.私たちが小さな報告会社であり続ける限り、私たちは他の非小報告会社の他の上場企業に適した様々な報告要件を利用して免除することができる。
180生命科学社の経営陣は、我々の首席財務官を含め、トレデビル委員会後援組織委員会(COSO)が“内部統制-総合枠組み”(2013)で提案した基準を用いて、2022年12月31日までの社内財務報告の内部統制の有効性を評価した。
経営陣は、これらの基準に基づき、2022年12月31日現在、社内統制の一部は発効していないと結論した。具体的には、経営陣の結論は以下のような重大な弱点に基づいている
| ● | 当社の審査及び制御プログラムは適切な精度レベルで動作しておらず、一度の逆方向株式分割に関連する権証公正価値誤り及び知的財産権研究開発資産の公正価値を発見した。 |
121
重大欠陥とは 制御欠陥或いは制御欠陥の組み合わせであり、年度或いは中期財務諸表の重大な誤報 が適時に予防或いは発見されない可能性は低い。会計資源が限られている会社として、経営陣は、これらの規制要件の遵守を確保するために、当社の業務から多くの時間と精力を移しています。
救済計画
経営陣は、財務報告の内部統制を発展させ、強化することを意図している
| ● | 四半期ごとに損益表の変動と差異を分析し、ドル金額と百分率変化の観点から口座残高の重大な変動を検出し、所定の閾値を超える任意の差異を検討する。 |
| ● | 米国証券取引委員会報告プロセスに対して追加的な審査を実施し、全体的な財務諸表と作成が米国証券取引委員会報告チームメンバー(米国証券取引委員会報告マネージャーではなく)の共同審査を受けることを確実にする。 |
財務報告の内部統制における重大な欠陥を補う
当社はこれまで、2021年12月31日現在、財務報告の内部統制に以下の2つの重大な欠陥が発見されたと報告している
| ● | 財務報告システム:当社は全期間にわたって完全に統合された財務合併·報告システムを保持していないため、外部報告のための財務諸表を作成するために、大量の人工分析、入金、調整を行う必要がある。 |
| ● | 株式ベースの給与及び賃金料金分類に関する期末財務開示及び報告フローの審査制御に力が入らない。 |
122
当社は、2022年12月31日までの年度内に、財務報告制度及び株式報酬プログラムの審査制御を含む、上述した2つの重大な弱点を解決するための是正措置及び/又は制御措置を実施している。我々は2022年に以下のステップを実行した:
| 1. | プロセスの連続性を確立し、財務報告および統合ツールおよびプログラムにおいて持続可能な改善および効率を実施するために、2022年のすべての報告期間内に同じ会計担当者を保持する。 | |
| 2. | 我々は以下の を実現したERPにリンクした統合ツールを実施することに成功した | |
| a. | 会計課目表を強化し、まとめを要求した | |
| b. | 将来の変化の制御を強化するために、実施前に描画および新しいプログラムを全面的に検討すること、および | |
| c. | できるだけ自動的に報告して計算します。 | |
| 3. | 財務開示および報告中の費用の完全性および適切な分類を保証するために、考慮要因を財務報告および年度計画の詳細なレビューに対応させるなど、管理レビューの一部として追加のハイレベルステップを実施する。 |
上記の是正措置 および2022年12月31日までの年次テストにより、経営陣は、上記2021年12月31日現在に存在する2つの重大な弱点が救済されたと結論した。
制御とプログラムの有効性に対する制約
開示制御およびプログラムを設計·評価する際、管理層は、任意の制御およびプログラムは、設計および動作がどんなに良くても、予想される制御目標を達成するための合理的な保証しか提供できないことを認識している。また、開示制御およびプログラムの設計は、そのコストに対する可能な制御およびプログラムの利点を評価する際にその判断を適用することが要求されるリソース制限が存在するという事実を反映しなければならない。
財務内部統制の変化 報告
2022年12月31日までの四半期内に、当社の財務報告内部統制に大きな影響を与えたり、合理的に当社の財務報告内部統制に重大な影響を与える可能性のある変化はありません。
プロジェクト9 B。他の情報。
本Form 10−K年次報告は、以下に議論する報告可能イベントの発生日から4営業日以内に提出されるため、現在のForm 8−K報告書ではなく、本年度報告書にForm 10−Kを開示することを選択した項目8.01状況によります:
8.01項目の他の活動。
先に開示されたように、全武さんは2023年1月18日に当社の最高運営·首席業務官を辞任し、同日より当社と“離職·離職契約”を締結した別居協定“)”2023年3月29日、双方は“別居協定第1修正案”(“別居協議第1修正案”)を締結し、“別居協議”の誤りを訂正した第一修正案“、 は、元の合意の日から発効し、Vuさんが退職契約に基づいて受け取ったいかなる金も、2021年のボーナスとは無関係であることを明らかにします。第一修正案は、別居協議又はその他の条項のいずれかに基づいてVuさんに支払う合計価格を変更しない。
プロジェクト9 Cです。検査を阻止する外国司法管轄区に関する情報開示。
ない。
123
第三部
第III部第10、11、12、13及び14項に要求される情報は、本年度報告では省略され、本年度報告に含まれる会計年度終了後120日以内に最終依頼書又は本年度報告書の改訂方法で提出される。
プロジェクト10.役員、役員、および会社管理。
本プロジェクトに必要な情報 はタイトルに含まれている“役員を選挙する”, “行政員”, “会社管理 ”, “道徳的規則”, “取締役会委員会のメンバー和、和債務延滞の 第16(A)節報告会社が2022年12月31日後120日以内に米国証券取引委員会(“米国証券取引委員会”)に提出する2023年依頼書のうち,会社2023年度株主総会のための依頼書の募集に関連し,引用により本明細書に組み込む。
第11項.行政職報酬
本プロジェクトに必要な情報 はタイトルに含まれている“役員と役員の報酬”, “役員報酬”, “役員報酬”, “財政年度終了時の優秀株奨励”, “報酬委員会と内部関係者の参加“と”報酬委員会報告“(必要な範囲内で)、当社は、2022年12月31日後120日以内に米国証券取引委員会の2023年依頼書に提出し、参照により本明細書に組み込む。
第12項所有権を保証するいくつかの受益者と管理層および関連株主事項。
本プロジェクトに必要な情報 はタイトルに含まれている“投票権と大株主“と”株式報酬プラン情報会社が2022年12月31日後120日以内に米国証券取引委員会に提出する2023年依頼書のうち,2022年12月31日に引用により本明細書に組み込む。
13.特定の関係 および関連取引、および取締役独立性。
本プロジェクトに必要な情報 はタイトルに含まれている“いくつかの関係や関連取引“と”会社管理 ” - “役員は自主独立している会社が2022年12月31日後120日以内に証券取引委員会に提出する2023年依頼書にbr}を提出し,引用により本明細書に組み込む。
プロジェクト14.チーフ会計士 費用とサービス
私どもの独立公共会計事務所はMarcum LLP、サンフランシスコ、カリフォルニア州、PCAOB監査師ID監査役事務所ID:688です。
本プロジェクトに必要な情報 はタイトルに含まれている“核数師の委任を承認する”-“料金を審査する“会社が2022年12月31日後120日以内に証券取引委員会に提出する2023年依頼書には、参照によって本明細書に組み込まれる。
124
第4部
項目15.物証、財務諸表、および付表
(A)本年度報告の一部として提出された書類:
以下は、10-Kテーブルに含まれる、または参照によって本テーブルに組み込まれた財務諸表、明細書、および証拠物のインデックスである。
(1)すべての財務諸表
| 連結財務諸表索引 | ||
| 独立公認会計士事務所報告 | F-2 | |
| 合併貸借対照表 | F-4 | |
| 合併経営報告書と全面赤字 | F-5 | |
| 合併株主権益変動表 | F-6 | |
| 統合現金フロー表 | F-8 | |
| 連結財務諸表付記 | F-10 |
(2)連結財務諸表の付表
上記の規定を除いて、すべての財務諸表明細書は省略されており、必要な情報が適用されていないか、または金額が明細書の提出を要求するのに十分ではないか、または必要な情報が本10-K表に含まれる連結財務諸表およびその付記に含まれているため、すべての財務諸表明細書は省略されている。
(3)展示品
展示品索引
| 提出済み/ 家具を提供します |
引用により を組み込む | |||||||||||
| 添付ファイル 番号: | 説明する | ここから声明する | 表 | ファイル 第 | 展示品 | 提出日 | ||||||
| 1.1 | 180生命科学社とAG.P./Alliance Global Partnersが2022年7月17日に締結した配給代理契約 | 8-K | 001-38105 | 1.1 | 7/19/2022 | |||||||
| 1.2 | 配給代理プロトコルは,2022年12月20日,180生命科学社とAG.P./Alliance Global Partnersとの間の合意である | 8-K | 001-38105 | 1.1 | 12/22/2022 | |||||||
| 3.1 | 会社登録証明書。 | S-1 | 333-217475 | 3.1 | 4/26/2017 | |||||||
| 3.2 | 会社登録証明書の改訂と再予約。 | 8-K | 001-38105 | 3.1 | 6/7/2017 | |||||||
| 3.3 | 改訂および再予約した会社登録証明書。 | 8-K | 001-38105 | 3.1 | 3/8/2019 | |||||||
| 3.4 | 改正された会社登録証明書第2修正案。 | 8-K | 001-38105 | 3.1 | 6/6/2019 | |||||||
| 3.5 | 改正された会社登録証明書第3修正案。 | 8-K | 001-38105 | 3.1 | 12/6/2019 | |||||||
| 3.6 | 改訂·再発行された会社登録証明書の第4回改訂 | 8-K | 001-38105 | 3.1 | 4/8/2020 | |||||||
| 3.7 | 改訂後の会社登録証明書の5回目の改訂 | 8-K | 001-38105 | 3.1 | 7/13/2020 | |||||||
| 3.8 | 2回目の改訂と再署名された会社登録証明書 | 8-K | 001-38105 | 3.1 | 11/12/2020 | |||||||
| 3.9 | 180生命科学会社第2次改訂·再登録証明書の改訂証明書は、2022年12月15日にデラウェア州国務長官に提出されました | 8-K | 001-38105 | 3.1 | 12/16/2022 | |||||||
| 3.1 | 180生命科学会社の定款を改正して再制定した。 | 8-K | 001-38105 | 3.1 | 8/10/2021 | |||||||
| 4.1 | 単位証明書サンプル。 | S-1 | 333-217475 | 4.1 | 4/26/2017 | |||||||
| 4.2 | 普通株式証明書サンプル。 | S-1 | 333-217475 | 4.2 | 4/26/2017 | |||||||
| 4.3 | 授権書の見本。 | S-1 | 333-217475 | 4.3 | 4/26/2017 | |||||||
| 4.4 | 権利証明サンプル。 | S-1 | 333-217475 | 4.5 | 5/26/2017 | |||||||
| 4.5 | 大陸株式譲渡信託会社と当社が2017年6月1日に締結した引受権証協定。 | 8-K | 001-38105 | 4.1 | 6/7/2017 | |||||||
| 4.6 | 改正された1934年証券取引法第12節に登録された登録者証券の説明 | X | ||||||||||
125
| 4.7 | 2020年6月に発行された10%プレミアム保証は、本チケット形式に変換することができます。 | 8-K | 001-38105 | 4.8 | 7/2/2020 | |||||||
| 4.8 | 10%プレミアム保証により、本チケット形式に変換できます | 8-K | 001-38105 | 4.1 | 9/14/2020 | |||||||
| 4.9 | 株式承認証表(2021年2月非公開発売) | 8-K | 001-38105 | 4.1 | 2/24/2021 | |||||||
| 4.1 | 事前融資株式証明書表(2022年7月発売) | 8-K | 001-38105 | 4.1 | 7/19/2022 | |||||||
| 4.2 | 共同株式証用紙(2022年7月発売) | 8-K | 001-38105 | 4.2 | 7/19/2022 | |||||||
| 4.3 | 事前融資株式証明書表(2022年12月発売) | 8-K | 001-38105 | 4.1 | 12/22/2022 | |||||||
| 4.4 | 共同株式証用紙(2022年12月発売) | 8-K | 001-38105 | 4.2 | 12/22/2022 | |||||||
| 4.5 | 180生命科学会社と買い手との間の授権証修正案、期日は2023年1月12日 | 8-K | 001-38105 | 4.1 | 1/12/2023 | |||||||
| 10.1 | 大陸株式譲渡信託会社と会社との間の投資管理信託口座協定。 | 8-K | 001-38105 | 10.1 | 6/7/2017 | |||||||
| 10.2 | 当社といくつかの証券保有者との間の登録権協定。 | 8-K | 001-38105 | 10.3 | 6/7/2017 | |||||||
| 10.3 | 契約書のフォーマットを完済する。 | S-1 | 333-217475 | 10.8 | 4/26/2017 | |||||||
| 10.4 | 当社が買い手と署名した証券購入協定は、日付は2020年6月12日。 | 8-K | 001-38105 | 10.1 | 7/2/2020 | |||||||
| 10.5 | 登録権協定は,日付が2020年6月12日であり,当社とその署名側が署名した。 | 8-K | 001-38105 | 10.2 | 7/2/2020 | |||||||
| 10.6 | 当社が買い手と署名した証券購入契約は、期日は2020年9月8日です | 8-K | 001-38105 | 10.1 | 9/14/2020 | |||||||
| 10.7 | 会社とサイン側との間で2020年9月8日に締結された“登録権協定” | 8-K | 001-38105 | 10.2 | 9/14/2020 | |||||||
| 10.8 | 本チケットの修正と再発行は、日付が2020年9月で、KBLIV保証人有限責任会社に発行されます | S-1 | 333-249539 | 10.24 | 10/19/2020 | |||||||
| 10.9 | 登録者、大陸株式譲渡と信託会社とLawrence Pembleとの間で2020年11月6日に署名された信託協定。 | 8-K | 001-38105 | 10.2 | 11/12/2020 | |||||||
| 10.10£ | 日付は2021年2月19日の証券購入協定で、180生命科学社と署名ページで決定された購入者が署名した。 | 8-K | 001-38105 | 10.1 | 2/24/2021 | |||||||
| 10.11 | 2021年1月26日、180生命科学会社とMaxim Group LLCの間の婚約状。 | 8-K | 001-38105 | 10.2 | 2/24/2021 | |||||||
| 10.12 | 180生命科学社とMaxim Group LLCの間で2021年2月18日に発行された招聘状の改正案。 | 8-K | 001-38105 | 10.3 | 2/24/2021 | |||||||
| 10.13 | 180生命科学会社と購入者との間で2021年2月23日に署名された登録権協定。 | 8-K | 001-38105 | 10.4 | 2/24/2021 | |||||||
10.14# |
180生命科学とジャグディップ·ナンチャハル教授の間で2021年2月22日に署名された諮問協定 | 8-K | 001-38105 | 10.1 | 3/3/2021 | |||||||
| 10.15# | 180生命科学社とジェームズ·N·ウディが2021年2月25日に改訂·再署名した雇用協定が、2020年11月6日に発効した | 8-K | 001-38105 | 10.2 | 3/3/2021 | |||||||
| 10.16# | ジェームズ·N·ウディ-株式オプション協定2021年2月26日発効(70,000株) | 8-K | 001-38105 | 10.3 | 3/3/2021 |
126
| 10.17# | 180生命科学会社とOzan Pamirの間で2021年2月24日に締結され、2020年11月6日に施行された雇用協定および2021年3月1日の改正·訂正 | 8-K | 001-38105 | 10.4 | 3/3/2021 | |||||||
| 10.18# | Ozan Pamir-株式オプション協定2021年2月26日発効(9000株) | 8-K | 001-38105 | 10.5 | 3/3/2021 | |||||||
| 10.19# | 180生命科学会社とジャグディップ·ナンチャハル教授との間で2021年3月31日に署名された諮問協定の第1修正案 | 8-K | 001-38105 | 10.2 | 4/2/2021 | |||||||
| 10.20# | 180生命科学会社、Katexo製薬会社、Ozan Pamirの間で2021年5月27日に署名され、2020年11月6日に施行される雇用協定第2改正案 | 8-K | 001-38105 | 10.2 | 5/27/2021 | |||||||
| 10.21# | 取締役被指名者招待状形式(2021年5月) | 8-K | 001-38105 | 10.1 | 5/27/2021 | |||||||
| 10.22# | KBL合併会社がジョナサン·ロスバードと2019年8月21日に締結した雇用契約表 | 10-K | 001-38105 | 10.44 | 7/9/2022 | |||||||
| 10.23£ | マーク·フェルドマン卿は2021年2月25日に施行された付記修正案の撤回を確認しました | 10-Q | 001-38105 | 10.15 | 7/19/2021 | |||||||
| 10.24 | ローレンス·スタンマン博士は2021年2月25日に施行された付記修正案の撤回を確認しました | 10-Q | 001-38105 | 10.16 | 7/19/2021 | |||||||
| 10.25 | 2021年7月31日の普通株購入承認株式証、180生命科学社の普通株1,250株を購入し、Alpha Capital Anstaltに付与する | 8-K | 001-38105 | 4.1 | 8/2/2021 | |||||||
| 10.26 | 株式オプション協定フォーマット(独立取締役は2021年8月に付与) | 10-Q | 001-38105 | 10.9 | 8/6/2021 | |||||||
| 10.27 | 買い手保証書用紙(2021年8月発売) | 8-K | 001-38105 | 4.1 | 8/24/2021 | |||||||
| 10.28£ | 180生命科学会社と署名ページで決定された購入者との間で2021年8月19日に署名された証券購入契約 | 8-K | 001-38105 | 10.1 | 8/24/2021 | |||||||
| 10.29 | 2021年8月17日180生命科学会社とMaxim Group LLCの間の婚約状 | 8-K | 001-38105 | 10.2 | 8/24/2021 | |||||||
| 10.30 | 180生命科学会社と購入者の間で2021年8月23日に署名された登録権協定 | 8-K | 001-38105 | 10.3 | 8/24/2021 |
127
| 10.31# | 180生命科学社2020総合インセンティブ計画 | S-8 | 333-259918 | 4.1 | 9/30/2021 | |||||||
| 10.32 | 株式オプション協定フォーマット180生命科学会社2020総合インセンティブ計画 | S-8 | 333-259918 | 4.2 | 9/30/2021 | |||||||
| 10.33 | 制限株式付与プロトコルフォーマット株式オプション協定180生命科学会社2020総合インセンティブ計画 | S-8 | 333-259918 | 4.3 | 9/30/2021 | |||||||
| 10.34# | 2021年10月29日、180生命科学社はアウン武と雇用協定を締結 | 8-K | 001-38105 | 10.1 | 10/29/2021 | |||||||
| 10.35# | マーク·フェルドマン卿は2021年2月25日に施行された付記修正案の撤回を確認しました | 10-Q | 001-38105 | 10.15 | 7/19/2021 | |||||||
| 10.36# | ローレンス·スタンマン博士は2021年2月25日に施行された付記修正案の撤回を確認しました | 10-Q | 001-38105 | 10.16 | 7/19/2021 | |||||||
| 10.37 | 2021年7月31日の普通株購入承認株式証、180生命科学社の普通株1,250株を購入し、Alpha Capital Anstaltに付与する | 8-K | 001-38105 | 4.1 | 8/2/2021 | |||||||
| 10.38# | 株式オプション協定フォーマット(独立取締役は2021年8月に付与) | 10-Q | 001-38105 | 10.9 | 8/16/2021 | |||||||
| 10.39# | 諮問協議日は2021年11月17日で,180生命科学社とローレンス·スタンマン医学博士が署名した。 | 8-K | 001-38105 | 10.1 | 11/18/2021 | |||||||
| 10.40# | 改正と再署名された雇用協定第1改正案は,2022年4月27日,180生命科学社とジェームズ·N·ウディ医学博士との合意である。 | 8-K | 001-38105 | 10.1 | 4/28/2022 | |||||||
| 10.41# | 2022年4月27日180生命科学会社と全オン呉との雇用協定第1改正案 | 8-K | 001-38105 | 10.2 | 4/28/2022 | |||||||
| 10.42# | 180生命科学社とジョナサン·ロスバード博士が2022年4月27日に署名した雇用協定第1修正案。 | 8-K | 001-38105 | 10.3 | 4/28/2022 | |||||||
| 10.43# | Cannbiorex製薬有限公司とマーク·フェルドマン卿が2022年4月27日に署名した雇用協定第1改正案。 | 8-K | 001-38105 | 10.4 | 4/28/2022 | |||||||
| 10.44# | 2022年4月27日に180生命科学社とローレンス·スタンマン医学博士が署名した諮問協定第1改正案。 | 8-K | 001-38105 | 10.5 | 4/28/2022 | |||||||
| 10.45# | Cannbiorex Pharma Ltd.とJagDeep Nanchahal教授が2022年4月27日に署名した諮問協定第2修正案 | 8-K | 001-38105 | 10.6 | 4/28/2022 | |||||||
| 10.46# | 180生命科学会社とジェームズ·N·ウディ医学博士との雇用協定第2改正案は2022年5月26日に発効し、2022年6月1日から発効する。 | 8-K | 001-38105 | 10.1 | 5/26/2022 | |||||||
| 10.47# | 180生命科学と全昂武が2022年5月26日に署名し、2022年6月1日に発効する雇用協定第2改正案 | 8-K | 001-38105 | 10.2 | 5/26/2022 | |||||||
| 10.48# | 180生命科学社とジョナサン·ロスバード博士が2022年5月26日に署名し、2022年6月1日に発効する雇用協定第2改正案。 | 8-K | 001-38105 | 10.3 | 5/26/2022 | |||||||
| 10.49# | 180生命科学社とLawrence Steinman,M.D.は2022年5月26日に署名し,2022年6月1日に発効した諮問協定第2修正案に署名した | 8-K | 001-38105 | 10.4 | 5/26/2022 | |||||||
| 10.50# | 180生命科学2022総合インセンティブ計画 | 8-K | 001-38105 | 10.1 | 6/14/2022 | |||||||
| 10.51£ | 180生命科学会社と買い手の間で2022年7月17日に署名された証券購入協定 | 8-K | 001-38105 | 10.1 | 7/19/2022 |
128
| 10.52£ | 180生命科学会社と買い手との間の証券購入契約、期日は2022年12月20日 | 8-K | 001-38105 | 10.1 | 12/20/2022 | |||||||
| 10.53 | 180生命科学会社と大陸株式譲渡信託会社が2022年12月22日に署名した前払い助成権証の権証代理契約 | 8-K | 001-38105 | 10.2 | 12/20/2022 | |||||||
| 10.54 | 180生命科学会社と大陸株式譲渡信託会社が2022年12月22日に署名した一般権証の権証代理協定 | 8-K | 001-38105 | 10.3 | 12/20/2022 | |||||||
| 10.55# | ロックアッププロトコルフォーマット(2022年12月発売) | 8-K | 001-38105 | 10.4 | 12/20/2022 | |||||||
| 10.56# | 2022年12月28日180生命科学社、Cannbiorex製薬有限会社とJagDeep Nanchahal教授との諮問協定第3修正案 | 8-K | 001-38105 | 10.1 | 12/29/2022 | |||||||
| 10.57# | 分離·解放協定は,2023年1月18日,180生命科学社と全武社が署名した | 8-K | 001-38105 | 10.1 | 1/20/2023 | |||||||
| 10.58 | 180生命科学会社と引受権証代理人との間で2023年1月13日に締結された引受権証代理人協定の改正案 | 8-K | 001-38105 | 10.1 | 1/18/2023 | |||||||
| 10.59*# | “別居と釈放協定第一改正案”は、2023年3月29日、180生命科学社と全武社が署名した | X | ||||||||||
| 14.1# | 商業と道徳的規則 | S-1 | 333-217475 | 14 | 4/26/2017 | |||||||
| 21.1* | 付属会社名簿 | X | ||||||||||
| 23.1* | 馬ゴム法律事務所 | X | ||||||||||
| 31.1* | サバンズ·オクスリ法第302条による主要行政官の認証 | X | ||||||||||
| 31.2* | サバンズ·オクスリ法第302条に基づく認証首席会計官 | X | ||||||||||
| 32.1** | サバンズ·オクスリ法第906条による主要行政官の認証 | X | ||||||||||
| 32.2** | サバンズ·オキシリー法第906条に基づいて首席会計官を認証する | X | ||||||||||
| 99.1 | 監査委員会の規約の形式。 | S-1 | 333-217475 | 99.1 | 4/26/2017 | |||||||
| 99.2 | 給与委員会規約の形式。 | S-1 | 333-217475 | 99.2 | 4/26/2017 | |||||||
| 99.3 | 指名及び企業管理委員会定款 | 8-K | 001-38105 | 99.8 | 11/12/2020 | |||||||
| 99.4 | リスク、安全、規制委員会規約 | 10-K | 001-38105 | 99.4 | 7/9/2021 | |||||||
| 101.INS* | 連結されたXBRLインスタンス文書-インスタンス文書は、そのXBRLタグがイントラネットXBRL文書に埋め込まれているので、対話データファイルには表示されない | X | ||||||||||
| 101.Sch* | イントラネットXBRL分類拡張アーキテクチャ | X | ||||||||||
| 101.カール* | イントラネットXBRL分類計算リンクライブラリ | X | ||||||||||
| 101.定義* | XBRLを連結してLinkbaseドキュメントを定義する | X | ||||||||||
| 101.実験所* | XBRL分類ラベルLinkbaseを連結する | X | ||||||||||
| 101.前期* | XBRLを連結してLinkbaseドキュメントを定義する | X | ||||||||||
| 104* | 本四半期報告表紙のタブ10-KのイントラネットXBRLは、添付ファイル101に内蔵されたXBRL文書セットに含まれています | X |
| * | 同封してアーカイブする。 |
| ** | 同封で提供します。 |
| # | 契約または補償計画または手配を管理します。 |
| £ | S−K条例第601(A)(5)項によれば、いくつかの付表及び証拠物は省略されている。要求されなければ、任意の漏れたスケジュールまたは証拠品のコピーは、証券および取引委員会に提供される。しかし、180生命科学会社は、改正された1934年の証券取引法第24 b-2条に基づいて、このように提供された任意の明細書または展示品の機密処理を要求することができる。 |
第 項16.テーブル10−Kまとめ
ない。
129
サイン
1934年の証券取引法第13節または第15節(D)節の要求に基づいて、登録者は、本報告を署名者が代表して署名することを正式に促し、正式な許可を得た
| 180生命科学社です | ||
| 日付:2023年3月31日 | /s/ ジェームズ·N·ウディ | |
| 差出人: | James N.Woody,CEO 幹事 (CEO) | |
| 日付:2023年3月31日 | /s/ オザン·パミール | |
| 差出人: | 臨時最高財務官オザン·パミール (首席財務·会計官) | |
1934年の証券取引法の要求によると、本報告は、以下の者代表登録者によって下で署名され、指定された日に登録者として署名された。
| サイン | タイトル | 日取り | ||
| /s/ジェームズ·N·ウディ | 取締役CEO兼最高経営責任者 | 2023年3月31日 | ||
| ジェームズ·N·ウディ | (首席行政主任) | |||
| /s/Ozan Pamir | 臨時首席財務官 | 2023年3月31日 | ||
| オザン·パミール | (首席財務会計官) | |||
| /s/マーク·フェルドマン | 共同執行議長兼取締役 | 2023年3月31日 | ||
| マーク·フェルドマン | ||||
| /s/ローレンス·スタンマン | 共同執行議長兼取締役 | 2023年3月31日 | ||
| ローレンス·スタンマン | ||||
| /s/Larry Gold | 役員.取締役 | 2023年3月31日 | ||
| ラリー·ゴルド博士 | ||||
| ドナルド·A·マゴヴィン | 役員をリードする | 2023年3月31日 | ||
| ドナルド·A·マゴヴィン | ||||
| /s/パメラ·G·マロン | 役員.取締役 | 2023年3月31日 | ||
| パメラ·G·マーロン | ||||
| /s/フランシス·ヌエテル2世 | 役員.取締役 | 2023年3月31日 | ||
| フランシス·クエテル2世 | ||||
| /s/ラッセル·T·レイ | 役員.取締役 | 2023年3月31日 | ||
| ラッセル·T·レイ | ||||
| /s/テレサ·デ·ルカ | 役員.取締役 | 2023年3月31日 | ||
| テレサ·デ·ルカ |
130
180生命科学社ですそして付属会社
2022年12月31日と2021年12月までの連結財務諸表
カタログ
| ページ | |
| 独立公認会計士事務所レポート(PCAOB ID 688) | F-2 |
| 連結財務諸表: | |
| 2022年12月31日現在と2021年12月31日現在の連結貸借対照表 | F-4 |
| 2022年と2021年12月31日までの総合経営報告書と全面赤字 | F-5 |
| 2022年と2021年12月31日までの総合株主権益変動表 | F-6 |
| 2022年12月31日と2021年12月31日までの年間現金フロー表 | F-8 |
| 連結財務諸表付記 | F-10 |
F-1
独立公認会計士事務所報告
当社の株主および取締役会へ
180生命科学作物です
財務諸表のいくつかの見方
私たちは添付された180個の生命科学作物の合併貸借対照表を監査した。(“当社”)2022年12月31日及び2021年12月31日までの3年度の関連総合経営及び全面収益(赤字)報告書、2022年12月31日までの各年度の株主権益及び現金流量変動報告書及び関連付記(総称して“財務諸表”)財務諸表は,すべての重要な面で当社の2022年12月31日までの財務状況と,2022年12月31日までの2年度の経営業績とキャッシュフローを公平に反映しており,米国公認の会計原則に適合していると考えられる。
解釈的段落--継続的な関心
添付財務諸表は、当社が引き続き経営を継続する企業として作成されると仮定して作成されています。付記2で述べたように、当社の運営資金は深刻に不足しており、重大な損失を被っており、その責任を履行し、そのbr業務を維持するために追加資金を調達する必要がある。これらのことは、同社の持続経営企業としての持続的な経営能力を大きく疑わせている。付記2は、これらの事項における経営陣の計画も説明しています。財務諸表には、このような不確実性の結果によって生じる可能性のあるいかなる調整も含まれていません。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちのbrはアメリカ上場会社会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、アメリカ連邦証券法及びアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければならない。
我々はPCAOBの 基準に従って監査を行った。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても詐欺によるものであっても、監査を計画し、実行することを要求する。当社は必要とせず、brを招聘してその財務報告の内部統制を監査していません。
私たちの監査の一部として、財務報告の内部統制を理解する必要がありますが、会社財務報告の内部統制の有効性について意見を述べるためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、財務諸表の重大な誤報のリスク(エラーによるものであっても詐欺によるものであっても)を評価するためのプログラムの実行と、これらのリスクに対応するプログラムを実行することとが含まれる。これらの手続きには、財務諸表中の金額および開示に関する証拠をテストに基づいて検討することが含まれています。私たちの監査には、使用されている会計原則および経営陣による重大な推定の評価、財務諸表の全体的なレポートの評価も含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
以下に述べる重要な監査事項とは、当期に財務諸表を監査して生じる事項であり、これらの事項は、(1)財務諸表に対して重大な意義を有する勘定又は開示に関するものであり、(2)特に挑戦性、主観性又は複雑性に係る判断を監査委員会に伝達又は要求している。重要な監査事項のコミュニケーションは、財務諸表全体に対する私たちの意見 をいかなる方法でも変えることはなく、以下の重要な監査事項を伝達することによって、重要な監査事項またはそれに関連する勘定または開示について単独の意見 を提供することもない。
F-2
中研究開発資産と減価の評価を行う
関係事項の記述
総合財務諸表付記3および付記5に記載されているように、当社は毎年(またはいくつかの場合、よりよく)定性的または定量的方法テストで中研究開発資産(IPR&D)の減値を行う。知的財産権研究開発資産の減価を評価する時、当社はまず定性要素を評価して、その知的財産権研究開発資産の公正価値 がその帳簿価値より低い可能性があるかどうかを確定する可能性がある。知的財産権研究開発資産減価テストの数量化方法の下で、 会社は各資産構成レベルの知的財産権研究開発資産の公正価値とその帳簿純値を比較する。 会社は一般的に多期超過収益法を用いて、各資産構成レベルにおける知的財産権研究開発資産の公正価値を推定する。
監査当社の量的減価テストは、経営陣が知的財産権や開発の公正価値を決定する際に重大な推定を行う必要があるため、監査人の主観的判断に関連している。重大な見積もりは、主に、市場予測、承認確率、EBITDA利益率、加重平均資本コスト、割引率、br}特許権使用料、出資費用に基づく予想 収入を含む基本仮定の敏感性によるものである。これらの仮定は会社の知的財産権研究開発資産の期待未来の収益と関係があり、 は展望性であり、経済、業界と会社の特定の品質要素に敏感であり、その影響を受ける。
私たちが監査でどのようにこの問題を解決したのか
当社の知的財産権研究開発資産の推定公正価値を評価するために、評価に使用した推定方法と、当社が分析に用いた上記で議論した重要な仮定をテストする監査プログラムを実行した。私たちの評価専門家にテスト会社が使用している重要な仮定と複雑な推定方法を協力してもらいました。また,重要な仮定を会社の歴史見積もり,実績,現在の業界,市場,経済動向と比較した。
/s/Marcum有限責任会社
馬ゴム有限責任会社
2019年以来当社の監査役を務めております
カリフォルニア州サンフランシスコ
2023年3月31日
PCAOB ID#688
F-3
180生命科学会社brとその子会社
合併貸借対照表
(ドルで表す)
| 十二月三十一日 | 十二月三十一日 | |||||||
| 2022 | 2021 | |||||||
| 資産 | ||||||||
| 流動資産: | ||||||||
| 現金 | $ | $ | ||||||
| 前払い費用と他の流動資産 | ||||||||
| 流動資産総額 | ||||||||
| 無形資産、純額 | ||||||||
| 現在行われている研究と開発 | ||||||||
| 商誉 | ||||||||
| 総資産 | $ | $ | ||||||
| 負債と株主権益 | ||||||||
| 流動負債: | ||||||||
| 売掛金 | $ | $ | ||||||
| 費用を計算する | ||||||||
| 計上すべき費用--関係者 | ||||||||
| ローンに対応しています | ||||||||
| ローン対応--関係者 | ||||||||
| 派生負債 | ||||||||
| 流動負債総額 | ||||||||
| ローンに対応しています非流動部分 | ||||||||
| 繰延税金負債 | ||||||||
| 総負債 | ||||||||
| 引受金及び又は有事項(付記11) | ||||||||
| 株主権益: | ||||||||
| 優先株、$ |
||||||||
| C類優先株; |
||||||||
| K類優先株; |
||||||||
| 普通株、$ |
||||||||
| 追加実収資本 | ||||||||
| その他の総合収益を累計する | ( |
) | ||||||
| 赤字を累計する | ( |
) | ( |
) | ||||
| 株主権益総額 | ||||||||
| 総負債と株主権益 | $ | $ | ||||||
付記はこれらの連結財務諸表の構成要素である。
F-4
180生命科学会社brとその子会社
合併経営報告書と全面損失
(ドルで表す)
| この年度までに | ||||||||
| 十二月三十一日 | ||||||||
| 2022 | 2021 | |||||||
| 運営費用: | ||||||||
| 研究開発 | $ | $ | ||||||
| 研究開発に関連した各方面 | ||||||||
| 一般と行政 | ||||||||
| 一般と行政関係の当事者 | ||||||||
| 総運営費 | ||||||||
| 運営損失 | ( |
) | ( |
) | ||||
| その他(費用)収入: | ||||||||
| 債務収益を返済する | ||||||||
| その他の費用 | ( |
) | ||||||
| 利子支出 | ( |
) | ( |
) | ||||
| 転換支払手形支払損失純額 | ( |
) | ||||||
| 営業権減価損失 | ( |
) | ||||||
| 知的財産権研究開発減価損失 | ( |
) | ||||||
| 派生負債の公正価値変動 | ( |
) | ||||||
| 請求すべき発行権益は価値変動を公正に承諾することができる | ( |
|||||||
| 株式証明書負債に割り当てられた要約コスト | ( |
|||||||
| その他の費用の合計 | ( |
) | ( |
) | ||||
| 所得税前損失 | ( |
) | ( |
) | ||||
| 所得税割引 | ||||||||
| 普通株主は純損失を占めなければならない | $ | ( |
) | $ | ( |
) | ||
| その他の全面的(赤字)収入: | ||||||||
| 外貨換算調整 | ( |
) | ||||||
| 全面赤字総額 | $ | ( |
) | $ | ( |
) | ||
| $ | ( |
) | $ | ( |
) | |||
| 発行済み普通株式加重平均: | ||||||||
付記はこれらの連結財務諸表の構成要素である。
F-5
180生命科学会社brとその子会社
合併株主権益変動表
(ドルで表す)
| 2022年12月31日までの年度 | ||||||||||||||||||||||||
| 普通株 | その他の内容 実収 |
積算 その他 全面 |
積算 | 合計する 株主の |
||||||||||||||||||||
| 株 | 金額 | 資本 | 収入.収入 | 赤字.赤字 | 権益 | |||||||||||||||||||
| 残高-2022年1月1日 | $ | $ | $ | $ | ( |
) | $ | |||||||||||||||||
| 逆株解体に関する調整 | ( |
) | ||||||||||||||||||||||
| 2022年7月の事前融資権証を発行する | - | |||||||||||||||||||||||
| 2022年7月の事前融資権行使で発行された株式 | ||||||||||||||||||||||||
| 2022年7月発行に関連する株式 | ||||||||||||||||||||||||
| 2022年12月の事前融資権証を発行する | - | |||||||||||||||||||||||
| 2022年12月の事前融資権行使で発行された株式 | ||||||||||||||||||||||||
| 2022年12月発行に関連する株式 | ||||||||||||||||||||||||
| 取締役に専門サービスを提供するために発行された株式 | ||||||||||||||||||||||||
| 株に基づく報酬 | ||||||||||||||||||||||||
| 総合的な損失: | ||||||||||||||||||||||||
| 純損失 | - | ( |
) | ( |
) | |||||||||||||||||||
| その他総合損失 | - | ( |
) | ( |
) | |||||||||||||||||||
| 残高-2022年12月31日 | $ | $ | $ | ( |
) | $ | ( |
) | $ | |||||||||||||||
付記はこれらの連結財務諸表の構成要素である。
F-6
180生命科学社ですそして付属会社
合併株主権益変動表 続
(ドルで表す)
| 2021年12月31日までの年度 | ||||||||||||||||||||||||
| 普通株 | 余分な実収 | 積算 他にも 全面的に | 積算 | 合計する 株主の | ||||||||||||||||||||
| 株 | 金額 | 資本 | 収入.収入 | 赤字.赤字 | 権益 | |||||||||||||||||||
| 残高-2021年1月1日 | $ | $ | $ | $ | ( | ) | $ | |||||||||||||||||
| KBL債務転換後発行株式(付記10) | ||||||||||||||||||||||||
| 180債務転換後に発行された株式(付記10) | ||||||||||||||||||||||||
| 融資に関する既発行株式は、融資コストを差し引く(注12) | ||||||||||||||||||||||||
| 株式証明書負債に割り当てられた要約コスト(付記12) | - | |||||||||||||||||||||||
| 非公開発行に関連して発行された権証は,派生負債に再分類される(付記8) | - | ( | ) | ( | ) | |||||||||||||||||||
| 普通株等価物交換時に発行された株(付記 12) | ( | ) | ||||||||||||||||||||||
| 売掛金を清算するために発行された株式(付記11) | ||||||||||||||||||||||||
| 2021年8月発行に関連する株は、融資コストを差し引く(注12) | ||||||||||||||||||||||||
| アルファ資本と転換可能な債務とデリバティブ債務を決済するために発行された株 (注10) | ||||||||||||||||||||||||
| 関係者ローンと転換可能手形の償還により発行された株式(付記12) | ||||||||||||||||||||||||
| 株式ベースの報酬(注12): | ||||||||||||||||||||||||
| 普通株 | ||||||||||||||||||||||||
| オプション | - | |||||||||||||||||||||||
| 解約済み株式 | ( | ) | ( | ) | ||||||||||||||||||||
| 総合収益(損失): | ||||||||||||||||||||||||
| 純損失 | - | ( | ) | ( | ) | |||||||||||||||||||
| その他総合収益 | - | |||||||||||||||||||||||
| 残高-2021年12月31日 | $ | $ | $ | $ | ( | ) | $ | |||||||||||||||||
付記はこれらの連結財務諸表の構成要素である。
F-7
180生命科学社そして付属会社
キャッシュフロー簡明統合レポート
(ドルで表す)
| ここ数年で | ||||||||
| 十二月三十一日 | ||||||||
| 2022 | 2021 | |||||||
| 経営活動のキャッシュフロー | ||||||||
| 純損失 | $ | ( |
) | $ | ( |
) | ||
| 純損失と経営活動で使用される現金純額の調整: | ||||||||
| 株に基づく報酬 | ||||||||
| サービスのために発行した株 | ||||||||
| 株式オプションと制限株式単位の償却 | ||||||||
| 営業権の減価 | ||||||||
| 知的財産権研究開発資産減価準備 | ||||||||
| 無形資産の償却 | ||||||||
| 貸倒費用の関連先 | ||||||||
| 債務収益を返済し,純額 | ( |
) | ||||||
| 転換支払手形清算損失 | ||||||||
| 繰延税金負債 | ( |
) | ( |
) | ||||
| 株式証明書負債に割り当てられた要約コスト | ||||||||
| 派生負債の公正価値変動 | ( |
) | ||||||
| 請求すべき発行権益は価値変動を公正に承諾することができる | ||||||||
| 経営性資産と負債変動状況: | ||||||||
| 前払い費用と他の流動資産 | ( |
) | ||||||
| 売掛金 | ( |
) | ||||||
| 売掛金--関係者 | ( |
) | ||||||
| 費用を計算する | ( |
) | ||||||
| 計上すべき費用--関係者 | ( |
) | ||||||
| 売掛金を発行することができる | ( |
) | ||||||
| 総額を調整する | ||||||||
| 経営活動に使われている現金純額 | ( |
) | ( |
) | ||||
| 融資活動によるキャッシュフロー | ||||||||
| 現金で発行された株は,発行コストを差し引く | ||||||||
| 2021年の株式売却および株式承認証に関する発売費用 | ( |
) | ||||||
| 2022年7月の普通株式売却及び普通株式承認証に関する発売費用 | ( |
) | ||||||
| 2022年12月の普通株式売却及び普通株式承認証に関する発売費用 | ( |
) | ||||||
| 融資収益に対処する | ||||||||
| 転換可能な債務の返済関係者 | ( |
) | ||||||
| ローンを返済し、調整後の純額を差し引く(付記9) | ( |
) | ( |
) | ||||
| ローン返済--関係者 | ( |
) | ( |
) | ||||
| 2022年7月の普通株式及び普通株式承認証を売却して得られた金 | ||||||||
| 2022年12月普通株式及び普通株式株式承認証を売却して得られた金 | ||||||||
| 2022年7月に事前出資株式証の収益を行使する | ||||||||
| 2022年12月前払い助成権証の収益を行使する | ||||||||
| 融資活動が提供する現金純額 | ||||||||
付記はこれらの連結財務諸表の構成要素である。
F-8
180生命科学社ですそして付属会社
現金流量簡明連結報告書 ,続
(ドルで表す)
| ここ数年で | ||||||||
| 十二月三十一日 | ||||||||
| 2022 | 2021 | |||||||
| 為替レート変動が現金に与える影響 | ( | ) | ||||||
| 現金が純増する | ( | ) | ||||||
| 現金--期初 | ||||||||
| 現金--期末 | $ | $ | ||||||
| キャッシュフロー情報の補足開示: | ||||||||
| その間に支払われた所得税現金 | $ | $ | ||||||
| 期日内支払利息現金 | $ | $ | ||||||
| 非現金投資と融資活動: | ||||||||
| KBL債務転換後発行された普通株 | $ | $ | ||||||
| 180債務転換後に発行される普通株式 | $ | $ | ||||||
| 関連側融資と変換可能手形の返済のために発行される普通株 | $ | $ | ||||||
| Alpha和解のために発行された株式および株式承認証 | $ | $ | ||||||
| 普通株式等価物交換普通株 | $ | $ | ||||||
| 売掛金を決算するために発行された株 | $ | $ | ||||||
| 売掛金の再分類ができる | $ | $ | ||||||
付記はこれらの連結財務諸表の構成要素である。
F-9
180生命科学会社brとその子会社
連結財務諸表付記
(ドルで計算された金額、ただし株式金額を除く)
注1-業務組織と業務性質
180生命科学会社は、前身がKBL合併会社IV(“180 LS”、またはその子会社とともに“会社”と呼ばれる)であり、デラウェア州法律に基づいて2016年9月7日に設立された空白小切手会社である。当社の設立の目的は、1つまたは複数の企業との合併、株式交換、資産買収、株式購入、再編または同様の業務合併を行うことです。
180生命会社(“180”, f/k/a 180生命科学社とCannBioRx生命科学社)当社の完全子会社であり、2019年1月28日にデラウェア州に登録設立されました。当社はアメリカにあります(“アメリカ”)炎症性疾患,線維化,慢性疼痛領域では満たされていない医療ニーズに集中した医療製薬会社であり,180の完全子会社180生物治療会社(“180 LP”),CannBioRex製薬会社(“CBR Pharma”)とKatexo製薬会社(“Katexo”)により革新的研究 を採用し,適切な場合に併用療法を採用している。180 LP、CBR Pharma、Katexoは共同で“180の子会社”を構成した。Katexoは2018年3月7日にブリティッシュコロンビア州“イギリス会社法”の規定に基づいて成立した。また、180年5月31日にカナダブリティッシュ·コロンビア州に完全子会社Katexo Callco、ULC、KatExco、ULC、CannBioRex Callco、ULCとCannBioRex Puracheco、ULCを設立し、Katexo、CBR Pharmaと180 LPの買収を促進した。2021年7月1日、カナダ社(KatexoとCBR Pharma)の資産と負債は、それぞれの子会社KatExco PharmPharmticals Corp.(“Katexo U.S.”)に移された。CannBioRex Pharma Limited(“CBR Pharma U.K.”)と。
同社は臨床段階のバイオテクノロジー会社であり,慢性疼痛,炎症,線維化,他の炎症性疾患では満たされていない医療ニーズに対する療法の開発に専念しており,これらの疾患のうち,抗腫瘍壊死因子療法は革新的な研究と適切な併用療法を用いることで患者に明らかな利点を提供する。3つの製品開発プラットフォームがあります
| ● | 肝繊維化と抗腫瘍壊死因子(“TNF”); |
| ● | カンナビノール(“CBD”)の誘導体系薬剤; |
| ● | ニコチン型アセチルコリン受容体(“α7 nAChR”)。 |
2022年の逆株式分割
2022年12月15日、会社は株主特別総会を開催し、会社株主は改訂された2つ目の“会社登録証明書”の改訂を許可し、発行された普通株と発行された普通株を逆株式分割し、額面は$とした
F-10
“証明書修正案”は普通株の認可株式数を減少させていないため、逆株式分割の効果は、発行済み株式および発行済み株式数に対して発行可能な普通株式数を増加させることである。逆株式分割は普通株の額面を変えることはなく、普通株のいかなる投票権や他の条項も修正しない。逆株式分割後に残った任意の断片的な株式は、最も近い完全株式に上方丸め込まれる。
会社の2020年総合インセンティブ計画と2022年総合インセンティブ計画について、会社報酬委員会と取締役会は、会社とその株主の最適な利益のために、(I)インセンティブ計画の下で発行可能な会社の普通株式数を20倍に引き下げた(どの断片的な株式も最も近い完全株式に切り捨てる)。(Ii)自社普通株株式を購入した未行使引受権毎に発行可能な普通株式数および他のすべての未償還報酬を20倍に減少させる(任意の断片的株式を最も近い全体株式に切り捨てる);および(Iii)奨励計画に従って以前に付与された普通株式を購入するための任意の未行使購入持分の行使価格を20倍(最も近い整数仙に丸め込む)を引き上げ、いずれの場合も逆株式分割の交換比率で公平に調整し、このような調整は逆株式分割が発効したときに自動的に発効する。財務諸表と財務諸表付記には20株反転株式分割の影響がトレーサビリティに反映されている。
リスクと不確実性
経営陣は引き続き 新冠肺炎の大流行と露烏戦争が経済と資本市場に与える影響を評価し、そして、このような事件は合理的な可能性があり、会社の財務状況にマイナス影響を与えるが、具体的な影響は本財務諸表作成日までまだ確定できないと結論した。財務諸表にはこのような不確実性の結果がもたらす可能性のあるいかなる調整も含まれていない。
現在挑戦に満ちている経済環境は、キャッシュフロー、運営資金レベル、および/または債務残高の不利な変化を招く可能性があり、これは会社の将来の経営業績や財務状況にも直接影響を与える可能性がある。 政府介入経済の最終持続時間と規模及び政府介入が会社の財務影響に与える効果はまだ不明である。この影響の程度は将来の事態発展に依存し、これらの事態発展には高度な不確実性があり、当社のコントロール下ではない。
注2-継続経営と経営陣の計画
当社は設立以来何の収入も生じておらず、重大な損失を出している。2022年12月31日までの累計赤字は
F-11
これらの総合財務諸表 は持続経営仮説の下で作成され、会社が正常な業務過程で資産と負債を清算できると仮定している。会社が運営を継続できるかどうかは が継続的に運営するために新たな融資を得るかどうかにかかっている。当社の将来の融資選択には、株式融資と融資が含まれており、当社が適時または優遇条件でこのような追加融資を得ることができない場合、当社はその発展、マーケティング、販売促進活動を削減しなければならない可能性があり、これはその業務、財務状況、経営業績に重大な悪影響を与え、最終的には運営と清算を停止させられる可能性がある。このような事項は、当社が合理的な時間内に経営を継続する能力があるかどうかに大きな疑問を抱かせ、合理的な時間は総合財務諸表発表日から1年以内と定義されている。当社の資産現金化は、当該等の総合財務諸表に記載されている帳簿額面と大きな違いがある可能性がありますが、添付されている総合財務諸表には、当社が経営を継続できないように、必要な調整は一切含まれていません。
注3-重要会計政策の概要
陳述の基礎
添付されている連結財務諸表は、米国公認会計原則(“米国公認会計原則”)に基づいて作成された。
合併原則
連結財務諸表には、当社とその完全子会社180 LP、CBR Pharma、Katexo、180 Life Corp.(“180 LC”)の勘定が含まれています。合併後の会社間のすべての残高および取引は無効になりました。連結財務諸表をドルで列記する。
予算の使用
米国公認会計原則に基づいて財務諸表を作成することは、管理層に、資産、負債、収入および支出報告金額に影響を与える推定、判断と仮定、および連結財務諸表関連付記に開示された金額を行うことを要求する。これらの財務諸表で使用される会社の重大な推定と仮定は、金融商品、株式証、オプションおよび派生負債の公正価値を含むが、研究開発税収控除および課税項目、ならびに商業権および他の無形資産減値分析に関する推定とbr}仮説を含む。会社のいくつかの見積もりは、会社独自の状況や一般経済状況を含む外部条件の影響を受ける可能性がある。これらの外部要因は、会社の見積もりに影響を与える可能性があり、実際の結果が見積もりと異なる可能性があり、合理的な可能性がある。
外貨換算
会社は通貨をドルと報告した
包括収益は、所有者投資または所有者への分配以外のすべてのソースの実体資本変動として定義され、上述した外貨換算調整を含む。当社は2022年12月31日及び2021年12月31日までに、その他の総合(赤字)収入(ドル)を記録しました
外貨建て取引(会社間取引を含む)による外貨収益とbr}損失を
経営実績に計上する。当社は確認しました($
F-12
現金と現金等価物
当社はすべての原始期限が三ヶ月以下の高流動性投資を財務諸表中の現金等価物と見なしています。2022年12月31日または2021年12月31日まで、会社
は現金等価物を持っていません。同社は2022年12月31日現在、米国とイギリスに銀行口座を所有している。その利用可能な現金残高は$
商誉
無形資産と進行中の研究と開発(“知的財産権研究開発”)
無形資産には、KatExcoが保有するライセンス特許と、再編に関連する技術的ライセンスが含まれている。許可された特許は特許の残りのライフサイクルで償却されるだろう。技術ライセンスは、あるライセンスと知識の開発と商業化のために得られたライセンスの公正な価値を表す。技術許可は関連特許の予定使用期限内に直線的に償却する。会社の研究開発活動の結果、ライセンス特許や技術ライセンスの使用期限を監視し、調整する必要がある。
金融商品の公正価値
当社は会計基準に基づいて編集(“ASC”)820“公正価値計量”(“ASC 820”)の案内に基づいて金融資産と負債の公正価値を計量し、この準則は公正価値を定義し、公正価値計量枠組みを構築し、そして公正価値計量の開示を拡大した。
F-13
ASC 820は、公正価値 を、計量日市場参加者間の秩序ある取引において、資産または負債が元金または最も有利なbr市場で負債を移動させるために課金される交換価格(退出価格)として定義する。ASC 820はまた、公正価値レベルを確立し、実体が公正価値を計量する際に観察可能な投入を最大限に使用し、観察できない投入 を最大限に減少させることを要求する。ASC 820は、公正価値を計量するために使用されることができる3つのレベルの投入を説明する
| ● | 第1レベル--アクティブ市場における同じ資産または負債のオファー ; | |
| ● | 第2レベル--アクティブ市場における資産および負債のような見積もりまたは観察可能な投入; | |
| ● | レベル3-観察できない投入(例えば、仮想キャッシュフローモデリング投入に基づく)。 |
このようなツールの短期的な性質のため,当社のいくつかの金融商品(主にローン対応からなる)の帳簿価値は,これらの 総合財務諸表に掲載されている公正価値と一致している。当社の派生負債は第三級投入を用いて推定されています(詳細は付記8-派生負債を参照)。
請求すべき権利を発行することができる
当社は、契約規定により株式の発行が義務付けられており、当該等の株式の発行に遅延が生じた場合には、計上すべき発行可能株を記録する。計算すべきは発行権益を公正価値に基づいて入金し、その公正価値は当社の総合経営報告書に変動して を確認することができる。普通株の関連株式が発行されると、発行可能株は普通株当時の公允市価によって再分類され、株式発行日から計算される。
株に基づく報酬
当社は、付与権益ツールの公正価値に基づいて、当該付与を交換するために得られたサービスコストを計量する。報酬の公正価値は、付与日に計量され、一般株の最近の現金販売価格の観察に基づいて管理層によって推定される。 次いで、報酬と引き換えにサービスを提供する必要がある期間内に公正価値金額が確認され、 は通常授権期間である。オプションまたは株式承認証を行使する際には、会社は、その許可されているが発行されていない株式から新たな普通株式を発行する。
派生負債および変換可能ツール
当社は、その債務及び株式発行を評価し、当該等契約又は当該等契約の埋め込み部分が、会社財務諸表において単独で確認することを要求するデリバティブの資格に適合しているか否かを判定する。実体は、株式証明書のような自己の株式で決済可能な契約を、実体の権益または資産または負債に分類するかどうかを考慮しなければならない。エンティティ制御範囲内にないイベントが現金純額決済を必要とする可能性がある場合、契約は権益ではなく資産または負債に分類されるべきである。
この会計処理の結果、派生ツールに埋め込まれた公正価値は、資産負債表毎に市価で計算され、負債 として記録され、公正価値変動は総合経営報告書中の他の(費用)収入純額に計上される。分岐を必要とする複数の組み込み機器がある場合、すべての分岐派生ツールは、単一の複合派生ツールとして入金される。派生ツールの分類は、そのようなツールが負債または資本として記入されるべきかどうかを含み、各報告期間の終了時に再評価される。最初に権益に分類され再分類すべき権益ツールは,ツールによる再分類日の公正価値を負債に再分類する.派生ツール負債は、貸借対照表内で流動または非流動に分類され、派生ツールが貸借対照表の日から12ヶ月以内に現金純額決済に基づくかどうかをベースとする。
F-14
埋め込み変換 オプションが分岐を必要としない場合,会社は約束日までの会社の対象株式の公正価値とツールの有効変換価格( 内価値)を比較することで有益な変換機能が存在するかどうかを評価する.
これらの 手配された債務割引は、関連債務の期限内にそれに記載された償還日に償却され、総合経営報告書で利息支出 に分類される。償還が可能になった場合にのみ、優先株割引はその償還価値に増加する。
変換可能なチケットの修正は、それが元のチケットの修正とみなされるべきかどうかを決定するために評価され、 会計を変更することなく、または条項が大きく変化した場合、元のチケットの終了および新しいチケットの発行とみなされるべきである。
当社はBlack-Scholesオプション定価モデルを用いて発行された権証とオプションの公正価値を計算した。株式承認証、転換可能手形、転換可能優先株使用の期待期限は契約期限であり、発行されたオプション使用の期待期限は付与されたオプションが未償還と予想される時間帯である。当社は“簡略化”方法を用いて“通常”オプション付与の期待期限を試算している。当社が使用している予想変動率数字は、評価ツールの期待寿命に相当する業界内の地位が類似している上場企業の一定期間の履歴変動性に基づく審査である。無リスク金利は、米国財務省のゼロ金利債券の暗黙的収益率に基づいて決定され、その残り期限は推定ツールの予想期限と一致する。
普通株1株当たり純損失
普通株1株あたりの基本純損失は,純損失を期間中に発行された普通株の加重平均で割ったものである。普通株式1株当たり純(損失) の計算方法は,純損失を発行済み普通株の加重平均数で割ったものに,普通株等価物が発行されていれば(在庫株を用いて計算し,換算法であれば )発行される余分な 普通株数を加える
以下の普通株式等価物 は、 逆希釈に計上されるので、加重平均発行された普通株式の計算には含まれない
| 31年度までに | ||||||||
| 2022 | 2021 | |||||||
| オプション | ||||||||
| 株式承認証 | ||||||||
| 潜在希釈株式総数 | ||||||||
研究と開発
研究開発費
は発生時に運営費用を計上する。当社は2022年12月31日および2021年12月31日までの年度内に発生した
所得税
当社はASCテーマ740“所得税”(“ASC 740”)の規定に従って所得税を納付している。
財務諸表または納税申告書に計上されているまたは計上されていない項目の将来の税務結果について、当社は繰延された納税資産および負債を確認します。繰延税項資産および負債は、資産および負債の課税基準と、それぞれ現行税率で計算された財務報告金額(“一時的差異”)との差額(“一時的差異”)に基づいて決定される。
当社は確認敷居と計量プログラムを用いて財務諸表を確認し,納税申告書に採取されたまたは予想されている納税状況を計測した。当社の政策は、税務関連利息の評価(あれば)を利息支出に分類し、罰金 を総合経営報告書と全面赤字における一般·行政費用に分類することである。
F-15
最近発表された会計公告
最近採用された会計公告
2020年8月、財務会計基準委員会(“FASB”)は、会計基準更新(“ASU”)第2020-06号“転換可能債務及びその他のオプション(特別テーマ470)、並びに派生ツール及びヘッジ--エンティティ自己資本契約(特別テーマ815)”を発表した。ASU 2020-06の改訂は、転換可能な債務ツール分類470-20中のいくつかの会計モードを廃止することによって、負債および権益の特徴を有するいくつかの金融商品の会計計算を簡略化することを目的としている。今回の更新における改訂によれば、変換 を埋め込む機能は、変換機能を有する変換可能ツールのホスト契約とは独立しておらず、これらの変換機能は、 が主題815-派生ツールおよび対沖下の派生商品に計上されることを要求しないか、または実収資本としての大量の割増を招くことはない である。転換可能債務ツールはその余剰コストで計量された単一負債として入金され、転換可能優先株はその歴史コストで計量された単一株式ツールとして入金され、他の特徴がない限り 分岐と派生商品として確認する必要がある。分離モードを解除することによって、変換可能な債務ツールの金利は、一般に、適用対象835(利息)における指導時に額面金利に近づく。これらのデリバティブ範囲例外の改訂 エンティティ自身の権益における契約例外は、資産または負債と確認された契約総数を変更する。 独立ツールに対して、そのツールが改訂中の派生商品範囲例外に適合する場合、エンティティはそれを権益に記録すべきである。組込み機能については、修正中の派生ツール範囲の例外に適合する場合、エンティティ は、この機能を分離して個別に説明すべきではない。当社は発表時にASU 2020-06を採用しており、当社の総合財務諸表に大きな影響はありません。
2021年5月3日、FASB はASU番号2021-04、1株当たり収益(主題260)、債務修正と補償(主題470-50)、補償-株補償(主題718)、およびエンティティ自身の株式のデリバティブとヘッジ契約(主題815-40):発行者 は独立株式分類書面コールオプションのいくつかの修正または交換に対して会計処理を行った。この基準は、brを明確にし、独立株式分類の修正または交換に対する発行者の書面償還オプション(例えば、株式証)の会計多様性を減少させ、これらのオプションは修正または交換後も持分分類を維持する。この基準は,2021年12月15日以降に開始される財政年度に適用され,この財政年度内の過渡期を含む。発行者は、新しい基準の発効日の後に発生する修正または交換に新しい基準を前向きに適用しなければならない。過渡期での採択を含めて早期採用を許可する。過渡期に新たな基準を早期に採用することが選択された発行者が選択された場合、当該過渡期を含む会計年度開始時から指導意見を適用すべきである。当社はASU 2021-04を採用し、2022年1月1日から発効しており、その採用は当社の総合財務諸表や関連開示に大きな影響を与えていません
2021年7月19日、FASB は会計基準更新(ASU)2021-05、レンタル(テーマ842):レンタル人-いくつかのレンタル料金が可変のレンタルを発表しました。賃貸実施後審査(PIR)(FASB会計基準コード(FASB ASC)842)の一部として、FASBは、レンタル者が手配しても全体的に利益を得ることが予想されるレンタル開始時に可変支払いのいくつかの販売型賃貸の損失 を確認するようにFASB ASC 842の指導に従って問題に直面していることを知った。当社はASU 2021-05を採用し、2022年1月1日から発効し、当社の総合財務諸表及び関連開示に大きな影響を与えていません。
注4-前払い料金と他の流動資産
プリペイド料金には、2022年12月31日と2021年12月31日までが含まれています
| 十二月三十一日 | ||||||||
| 2022 | 2021 | |||||||
| 保険(1) | $ | $ | ||||||
| 研究開発費の課税額控除 | ||||||||
| 専門費(1) | ||||||||
| 付加価値税売掛金 | ||||||||
| 税金.税金 | ||||||||
| 他にも | ||||||||
| $ | $ | |||||||
| (1) |
F-16
付記5−無形資産と長期資産減価
無形資産には、2022年12月31日と2021年12月31日まで、 :
| 余剰償却 期間 | 2022年12月31日まで | 2021年12月31日まで | ||||||||||||||||||||||||
| 年数単位で 12月31日、 2022 | 毛収入 資産 価値がある | 積算 償却する | ネットワークがあります 携帯する 価値がある | 毛収入 資産 価値がある | 積算 償却する | ネットワークがあります 携帯する 価値がある | ||||||||||||||||||||
| 許可を得た特許 | $ | $ | ( | ) | $ | $ | $ | ( | ) | $ | ||||||||||||||||
| 技術許可証 | ( | ) | ( | ) | ||||||||||||||||||||||
| $ | $ | ( | ) | $ | $ | $ | ( | ) | $ | |||||||||||||||||
許可された特許と技術許可の総資産価値の獲得日からの変化は,外貨為替レートの変化によるものである
会社は償却費用を記録した
無形資産に関する将来の償却状況は以下の通り
| 12月31日までの年度 | ||||
| 2023 | $ | |||
| 2024 | ||||
| 2025 | ||||
| 2026 | ||||
| 2027 | ||||
| その後… | ||||
| $ | ||||
営業権の減価
同社が公開取引した株の終値は$だった
F-17
以下は、上記に記録された営業権減価損失を含む、当社単一報告単位の2022年12月31日までの年間営業権活動概要である。
| CBR医薬 営業権 | 180 LP 営業権 | 統合された 営業権 | ||||||||||
| バランス、2021年12月31日 | $ | $ | $ | |||||||||
| 貨幣換算 | ( | ) | ( | ) | ||||||||
| バランス、2022年3月31日 | ||||||||||||
| 貨幣換算 | ( | ) | ( | ) | ||||||||
| バランス、2022年6月30日 | ||||||||||||
| 貨幣換算 | ( | ) | ( | ) | ||||||||
| 減価前残高 | ||||||||||||
| 営業権の減価 | ( | ) | ( | ) | ( | ) | ||||||
| バランス、2022年9月30日 | ||||||||||||
| 貨幣換算 | ||||||||||||
| 減価前残高 | ||||||||||||
| 営業権の減価 | ( | ) | ( | ) | ( | ) | ||||||
| バランス、2022年12月31日 | $ | $ | $ | |||||||||
知的財産権研究開発資産の減価
2022年12月31日現在、貸借対照表上の知的財産権研究開発資産の帳簿価値は$
F-18
以下は、上記知的財産権研究開発資産の記録済み損失 を含む当社の2022年12月31日までの知的財産権研究開発活動の概要である。
| CBR 医薬知的財産権 研究開発資産 | 180 LP
IP 研究開発 資産 | 統合された 知的財産権研究開発 資産 | ||||||||||
| バランス、2021年12月31日 | $ | $ | $ | |||||||||
| 貨幣換算 | ( | ) | ( | ) | ||||||||
| バランス、2022年3月31日 | ||||||||||||
| 貨幣換算 | ( | ) | ( | ) | ||||||||
| バランス、2022年6月30日 | ||||||||||||
| 貨幣換算 | ( | ) | ( | ) | ||||||||
| バランス、2022年9月30日 | ||||||||||||
| 貨幣換算 | ||||||||||||
| 減価前残高 | ||||||||||||
| 知的財産権研究開発資産減価計 | ( | ) | ( | ) | ( | ) | ||||||
| バランス、2022年12月31日 | $ | $ | $ | |||||||||
付記6--計上すべき費用
2022年12月31日と2021年12月31日まで、計算すべき費用には :
| 十二月三十一日 | ||||||||
| 2022 | 2021 | |||||||
| 相談料 | $ | $ | ||||||
| 専門費 | ||||||||
| 訴訟は費用を計算する(1) | ||||||||
| 従業員報酬と役員 | ||||||||
| 研究開発費 | ||||||||
| 利子 | ||||||||
| 他にも | ||||||||
| $ | $ | |||||||
| (1) |
2022年12月31日と2021年12月31日まで、費用関連各方面は$とします
F-19
付記7--課税株式の発行が可能
2021年12月31日までの年度内の配当金発行可能活動の概要は以下のとおりである
| 2021年1月1日の残高 | $ | |||
| 持分に再分類する | ( | ) | ||
| 2021年12月31日の残高 | $ |
当社は2020年12月31日までに、当社の普通株式と交換する固定br元でサービス契約を締結します。契約協定によると、会社は総額#ドルの債券を発行する
付記8--派生負債
以下の表は、公正価値の恒常的な計量による3級派生負債(以下定義する公共特殊目的買収会社(“SPAC”)権証を除く)の公正価値変動をまとめたものである
| 2022年12月31日までの年度 | ||||||||||||||||||||||||
| 株式承認証 | ||||||||||||||||||||||||
| 公衆 | 私 | オープンカー | ||||||||||||||||||||||
| 空間 | 空間 | パイプ.パイプ | 他にも | 備考 | 合計する | |||||||||||||||||||
| 2022年1月1日現在の残高: | $ | $ | $ | $ | $ | $ | ||||||||||||||||||
| 派生負債の公正価値変動 | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||
| 2022年12月31日現在の残高 | $ | $ | $ | $ | $ | $ | ||||||||||||||||||
| 2021年12月31日までの年度 | ||||||||||||||||||||||||
| 株式承認証 | ||||||||||||||||||||||||
| 公衆 | 私 | オープンカー | ||||||||||||||||||||||
| 空間 | 空間 | パイプ.パイプ | 他にも | 備考 | 合計する | |||||||||||||||||||
| 2021年1月1日までの残高 | $ | $ | $ | $ | $ | $ | ||||||||||||||||||
| 債務転換に関する派生債務の償還 (1) | ( | ) | ( | ) | ||||||||||||||||||||
| 融資に関する引受権証 | ||||||||||||||||||||||||
| 発行された引受権証はアルファ和解に係る(1) | ||||||||||||||||||||||||
| Alpha和解に関する派生債務弁済 (1) | ( | ) | ( | ) | ||||||||||||||||||||
| 派生負債の公正価値変動 | ( | ) | ( | ) | ||||||||||||||||||||
| 2021年12月31日現在の残高 | $ | $ | $ | $ | $ | $ | ||||||||||||||||||
| (1) | 付記10-変換可能支払手形 を参照 |
F-20
デリバティブ負債2022年12月31日現在と2021年12月31日現在の公正価値は、ブラック·スコアーズオプション定価モデルを用いて推定され、以下の仮定brを用いた
十二月三十一日 2022 | ||||
| 無リスク金利 | % | |||
| 予想期限(年単位) | ||||
| 予想変動率 | % | |||
| 配当を期待する | % | |||
SPAC株式証明書
公共SPAC捜査令状
KBL初公募の参加者は全部で
F-21
プライベートSPAC株式証明書
KBL初の私募参加者は全部で
喉頭管捜査令状
2021年2月23日、br社は購入のために5年間の引受権証(“管道権証”)を発行した
以下では を発行時にパイプ株式証を推定するために用いると仮定する:
| 2021年2月23日 | ||||
| 無リスク金利 | % | |||
| 予想期限(年単位) | ||||
| 予想変動率 | % | |||
| 配当を期待する | % | |||
他の手令
AGP許可
事業合併が2020年11月6日に完了したことを受けて、当社は以下の株式を購入する5年間の株式承認証を負担する義務があります
F-22
2021年3月12日、当社はAGP(“AGP株式承認証”)に引受権証を発行し、合算を購入した
以下では を発行時にAGP株式証券を推定するために用いると仮定する:
| 2021年3月12日 | ||||
| 無リスク金利 | % | |||
| 予想期限(年単位) | ||||
| 予想変動率 | % | |||
| 配当を期待する | % | |||
Alpha Capital Anstalt(“Alpha”) 株式証明書
2021年7月29日(2021年7月31日に署名)で合意されたAlpha和解協定(付記10-転換手形参照)について、br社は購入のための3年間の引受権証を発表した
以下では を発行時にAlpha承認株式証を推定するために用いると仮定する:
| 七月二十九日 2021 | ||||
| 無リスク金利 | % | |||
| 予想期限(年単位) | ||||
| 予想変動率 | % | |||
| 配当を期待する | % | |||
転換可能な手形
2020年に発行される変換可能チケットは、2つに分割され、派生負債として記録される埋め込み的な特徴を有する。2021年1月15日から2021年2月5日までの間に、ブラック·スコアーズオプション定価モデルを用いて、債務転換に関する派生債務弁済の公正価値(付記10-転換手形 参照)を推定し、以下の仮定を用いた
| 2021年1月15日まで | ||||
| 2021年2月5日 | ||||
| 無リスク金利 | % | |||
| 予想期限(年単位) | ||||
| 予想変動率 | % | |||
| 配当を期待する | % | |||
F-23
アルファ資本手形(付記10-対応変換可能手形参照)は、2021年第2四半期末に唯一の残高未返済の転換可能手形であり、2021年7月31日にアルファ和解協定の全金額がこの日に計上される。2021年7月31日、会社 はアルファ資本手形の清算、関連派生債務、および決済すべき残高を記録した。 のより多くの詳細は付記10-対応転換手形を参照されたい。
授権活動
以下は、2022年12月31日と2021年12月31日までの年間株式証活動の概要である(2021年8月、2022年7月、2022年12月に非公開発行の一部として付与された特定の引受権証を含み、これらの株式承認証はすべて持分種別に属する;付記12-株主権益参照)
|
番目 株式承認証 |
重み 平均行重み | 重み 平均残存寿命(年) | 固有の 値 | |||||||||||
| 未完成、 2021年1月1日 | $ | |||||||||||||
| 発表されました | ||||||||||||||
| 鍛えられた | ||||||||||||||
| キャンセルします | ||||||||||||||
| 期限が切れる | ||||||||||||||
| 未返済、2021年12月31日 | $ | |||||||||||||
| 発表されました | ||||||||||||||
| 鍛えられた | ( |
) | (1) | |||||||||||
| キャンセルします | - | |||||||||||||
| 期限が切れる | - | |||||||||||||
| 未完成、2022年12月31日 | $ | |||||||||||||
| 行使可能、2022年12月31日 | $ | |||||||||||||
| (1) | 株式承認証はすべて行使され期限がない日までに行使可能であり, であるため,今回の計算から除外されていることに注意されたい. |
2022年12月31日現在の未償還権証と行使可能権証の概要は以下の通り
| 未弁済持分証 | 行使可能な引受権証 | |||||||||||||
| 重みをつける | ||||||||||||||
| 平均値 | ||||||||||||||
| トレーニングをする | 量 | 残り | 量 | |||||||||||
| 値段 | 株 | 年単位の寿命 | 株 | |||||||||||
| $ | ||||||||||||||
| $ | ||||||||||||||
| $ | ||||||||||||||
| $ | ||||||||||||||
| $ | ||||||||||||||
| $ | - | |||||||||||||
| $ | - | |||||||||||||
2021年12月31日現在の未償還権証と行使可能権証の概要は以下の通り
| 未弁済持分証 | 行使可能な引受権証 | |||||||||||||
| 重みをつける | ||||||||||||||
| 平均値 | ||||||||||||||
| トレーニングをする | 量 | 残り | 量 | |||||||||||
| 値段 | 株 | 年単位の寿命 | 株 | |||||||||||
| $ | ||||||||||||||
| $ | ||||||||||||||
| $ | ||||||||||||||
| $ | ||||||||||||||
| $ | ||||||||||||||
F-24
備考9--ローン対応
次の表は、2022年12月31日と2021年12月31日までの年度のローン対応活動をまとめています
| 元金残高は 1月1日 2022 | 調整する | 元金 返済済みの債務 現金 | 新株発行 | 効果があります 外国.外国 取引所 レート | 元金 残高は 十二月三十一日 2022 | |||||||||||||||||||
| 賃金保障計画 | $ | $ | $ | ( | ) | $ | $ | $ | ||||||||||||||||
| 鳴り物入りの融資計画を立て直す | ( | ) | ( | ) | ||||||||||||||||||||
| 最初の保障-2021年 | )(1) | ( | ) | |||||||||||||||||||||
| 初の保障--2022年 | ||||||||||||||||||||||||
| その他の融資 | (2) | |||||||||||||||||||||||
| 融資総額に対処する | $ | $ | ( | ) | $ | $ | ( | ) | ||||||||||||||||
| 減算:ローン対応--当期分 | ||||||||||||||||||||||||
| ローン対応--非流動部分 | $ | $ | ||||||||||||||||||||||
| (1) |
| (2) |
| 元金 残高は 1月1日 2021 | 許し/許す/ 他の 収入に調整する | 元金 返済済みの債務 現金 | 新株発行 | 効果があります 外国.外国 取引所 レート | 元金 残高は 十二月三十一日 2021 | |||||||||||||||||||
| キングズブルック | $ | $ | $ | ( | ) | $ | $ | $ | ||||||||||||||||
| 賃金保障計画 | ( | ) | ( | ) | ||||||||||||||||||||
| 鳴り物入りの融資計画を立て直す | ( | ) | ( | ) | ||||||||||||||||||||
| 初保障-2020年 | ( | ) | ||||||||||||||||||||||
| その他の融資 | ||||||||||||||||||||||||
| 融資総額に対処する | $ | ( | ) | $ | ( | ) | $ | $ | ( | ) | ||||||||||||||
| 減算:ローン対応--当期分 | ||||||||||||||||||||||||
| ローンに対応しています非流動部分 | $ | $ | ||||||||||||||||||||||
F-25
ローンに対応し、今期の部分
| 単金利 | 十二月三十一日 2022 | 十二月三十一日 2021 | ||||||||||
| 2019年9月18日に発行された融資 | | % | $ | $ | ||||||||
| 2019年9月18日に発行された融資 | % | |||||||||||
| 2019年10月8日に支給される対応ローン | % | |||||||||||
| 2019年10月29日に発行された融資 | % | |||||||||||
| 2019年12月31日に発行された対応ローン | % | |||||||||||
| 2020年2月5日に支給されるローン | % | |||||||||||
| 2020年2月5日に支給されるローン | % | |||||||||||
| 2020年3月31日に支給されるローン | % | |||||||||||
| 2020年3月31日に支給されるローン | % | |||||||||||
| 2020年6月8日に支給されるローン | % | |||||||||||
| 2020年6月8日に支給されるローン | % | |||||||||||
| 2020年6月17日に支給されるローン | % | |||||||||||
| 2020年7月15日付融資** | % | |||||||||||
| 2020年7月15日に支給されるローン | % | |||||||||||
| 2020年10月8日に発行されるローン* | % | |||||||||||
| 2020年10月13日に支給されるローン | % | |||||||||||
| 2020年10月14日に支給されるローン | % | |||||||||||
| 購買力平価ローンの当期部分 (1) | % | |||||||||||
| ローンの現在分を差し戻す (1) (2) | % | |||||||||||
| 最初のbr 2021年12月10日に発行された保証資金を支払うべきです(2) | % | |||||||||||
| $ | $ | |||||||||||
| * |
| (1) |
| (2) |
F-26
ローンに対応して、非流動部分
2022年12月31日と2021年12月31日まで、会社が融資に対応する非流動部分は以下の通り
| 単金利 | 2022年12月31日 | 十二月三十一日 2021 | 成熟性 日取り | |||||||||||
| 2020年5月5日に支給されるPPP対応ローン | % | $ | ||||||||||||
| BBLS対応ローンは2020年6月10日に発行 | % | |||||||||||||
| 小計 | ||||||||||||||
| 減算:BBLS/PPPローンの現在部分(前文参照) | ( | ) | ( | ) | ||||||||||
| 非流動部分 | $ | $ | ||||||||||||
Katexoは2020年4月と5月に総額$の融資を受けた
2020年3月27日から、疫病の深刻な影響を受けている米国企業に恩恵を与えるために、“コロナウイルス援助、救済、経済安全法案”(“CARE法案”)という立法が可決された。CARE法案の条項によると、2020年にPaycheck
保護計画柔軟性法案が改正された後、会社は申請し、それぞれの
の全部または一部のPPPローンの免除を受ける資格がある。制限がある場合、このような許しは、賃金コスト(購買力平価計画に従って定義される)および担保融資利息、融資後24週間以内に発生する賃貸料または公共事業コスト(総称して“資格に適合した費用”と総称される)と、購買力平価ローンが資金を獲得した後に定義された従業員および補償レベルを維持することとを含むが、これらに限定されない、購買力平価協定に規定されているいくつかの許可の目的に基づいて融資収益を決定する。同社は、PPPローンの収益を条件に合った費用に使うとしている。猶予されていない金額はすべて利息が発生します
2021年5月19日、会社はローン減免を申請し、金額は$
2021年9月30日、会社は一部の購買力平価ローンを調整し、金額は$
2021年12月31日現在、会社が記録した受取利息は$
2020年6月10日、会社はポンドGBを獲得しました
2020年6月12日,当社
はKingsbrook Opportunities Master Fund LPと本票合意を締結し,借金元金総額
$
当社は2021年12月31日までに合計$を支払います
2021年12月10日,br社は役員および高級社員保険証券(“D&O保険”)の融資手配を締結し,
第一保険基金は$となる
F-27
ローン対応--関係者
関連側への融資(“関連側融資”)には、会社のある上級管理者、取締役、超過に対応することが含まれています
簡単だよ 金利 | 十二月三十一日 2022(1) | 2021年12月31日 | ||||||||||
| 2019年9月18日に発行された融資 | % | $ | $ | |||||||||
| 2019年10月8日に支給される対応ローン | % | |||||||||||
| 2020年2月5日に支給されるローン | % | |||||||||||
| 2020年3月31日に支給されるローン | % | |||||||||||
| 2020年6月17日に支給されるローン | % | |||||||||||
| 2020年7月15日に支給されるローン | % | |||||||||||
| 2020年10月8日に発行されるローン* | % | |||||||||||
| 2020年10月14日に支給されるローン | % | |||||||||||
| $ | $ | |||||||||||
| * |
| (1) |
発行時には,関連する 側融資が規定する満期日は,(A)業務合併完了,(B)2020年6月30日;または (C)それぞれの発行日後60日とする.2020年7月1日、当社は関連側融資の条項を改訂し、満期日を(A)適格融資終了または(B)2020年11月1日早い者に延長する。すべてのローン延期条項 を審査し、終了ではなく修正だと考えました。
2021年2月10日、同社はいくつかの融資協定の条項を修正するための改訂された融資協定を締結し、元金総額を$とした
2021年4月12日、当社はマーク·フェルドマン卿·取締役会合同執行議長のローレンス·スタンマン博士と改訂された融資協定を締結し、すべての未返済ローン契約の満期日を2021年9月30日に延長した。
当日,彼らは交換総額$の元金を選択した
F-28
融資利息支出に対処する
当社は2022年12月31日までに、未返済ローンに関する利息支出および利息収入がそれぞれ14,156ドルおよび1,490ドルであることを確認した。
2021年12月31日現在、当社が確認した利息支出および未返済ローンに関する利息支出はそれぞれ24,019ドルおよび38,874ドルである。
2022年12月31日現在、当社の利息と未返済ローンに関する計上利息はそれぞれ37,960ドルと16,770ドルです。 より多くの詳細は14-関係者に記載されている。
2021年12月31日現在、当社の計上利息と未返済ローンに関する課税利息はそれぞれ24,212ドルと812ドルです。 より多くの詳細は14-関係者に記載されている。
注10-変換可能支払手形
次の表は,2021年12月31日までの年度内の変換可能手形活動(2022年期間には活動がなく,すべての変換可能手形
の残高から詳細に説明している
| 成熟性 | ||||||||||||||||||||||||
| 日取り | ||||||||||||||||||||||||
| (改訂後、 | 01/01/21 | 影響 | 転換する | ごく普通である | 12/31/21 | |||||||||||||||||||
| 効き目がある | もし…。 | 元金 | のです。 | 普通に回す | 株 | 元金 | ||||||||||||||||||
| 日取り | )を適用する | てんびん | 火を消す | 在庫品 | 発表されました | てんびん | ||||||||||||||||||
| 自治領 | $ | $ | $ | ( | ) | $ | ||||||||||||||||||
| キングズブルック | ( | ) | ||||||||||||||||||||||
| アルファ資本 | ( | ) | ( | ) | ||||||||||||||||||||
| 運転台ノート | ( | ) | ||||||||||||||||||||||
| 合計する | $ | $ | ( | ) | $ | ( | ) | $ | ||||||||||||||||
F-29
次の表は,2021年12月31日までの年度内に変換可能な支払手形の関連先活動(2022年期間には活動がない,残高があるため)を詳細に説明している
| 2021年12月31日までの年度 | ||||||||||||||||||||||||||||
| 発効日 | 期日(適用されれば改訂される) | 01/01/21元金残高 | 発行済み債務 | 未払い利息資本が元金となる | 債務を清算する | 普通株に転換する | 12/31/21元金残高 | |||||||||||||||||||||
| 180 LP変換可能チケット | ( | ) | ||||||||||||||||||||||||||
| 180 LP変換可能チケット | ( | ) | ||||||||||||||||||||||||||
| 180 LP変換可能チケット | ( | ) | ||||||||||||||||||||||||||
| 合計する | $ | $ | $ | $ | ( | ) | $ | ( | ) | $ | ||||||||||||||||||
Dominion、Kingsbrook、Alpha変換可能チケット
業務 の統合が完了した後,Dominion(定義は後述),KingsbrookおよびAlpha(定義は後述)がチケットを変換できると仮定する.
Dominion変換可能チケット
| 自治領 | ||||||||||||
| 元金 | 債務割引 | ネットワークがあります | ||||||||||
| 2021年1月1日の残高 | $ | $ | $ | |||||||||
| 改装の影響 | ( | ) | ( | ) | ||||||||
| 2021年12月31日の残高 | $ | $ | $ | |||||||||
2020年6月12日(“Dominion br}発行日”)、
当社は2021年12月31日までの年間で利息支出$を記録した
キングズブルックは本チケットに変換できます
| キングズブルック | ||||||||||||
| 元金 | 債務割引 | ネットワークがあります | ||||||||||
| 2021年1月1日の残高 | $ | $ | $ | |||||||||
| 改装の影響 | ( | ) | ( | ) | ||||||||
| 2021年12月31日の残高 | $ | $ | $ | |||||||||
F-30
2020年6月12日(“Kingsbrook
発行日”),KBLは$を達成した
当社は2021年12月31日までの年間で利息支出$を記録した
アルファ変換可能チケット
| Alpha | ||||||||||||
| 元金 | 債務割引 | ネットワークがあります | ||||||||||
| 2021年1月1日の残高 | $ | $ | $ | |||||||||
| 消火の影響 | ( | ) | ( | ) | ||||||||
| 改装の影響 | ( | ) | ( | ) | ||||||||
| 2021年12月31日の残高 | $ | $ | $ | |||||||||
2020年9月8日(アルファ発行日)、KBLは$を達成しました
当社は2021年12月31日までの年間で利息支出$を記録した
2021年Dominion、Kingsbrook、およびAlpha変換可能チケットの変換可能債務転換/償還
保証されたbr変換可能なチケット保有者は、2021年に元本と利息を会社普通株に変換することを選択した株式は以下の通りです
| 損失は発生している | ||||||||||||||||||||||||||||
| 公正価値 | 火を消す | |||||||||||||||||||||||||||
| 元金 | 導関数 | 合計する | ごく普通である | のです。 | のです。 | |||||||||||||||||||||||
| てんびん | 利子 | 負債.負債 | 金額 | 株 | 株 | オープンカー | ||||||||||||||||||||||
| 転換された | 転換された | 転換された | 転換された | 発表されました | 発表されました | 備考 | ||||||||||||||||||||||
| Dominion変換可能チケット | $ | $ | $ | $ | $ | $ | ( | ) | ||||||||||||||||||||
| キングズブルックは本チケットに変換できます | ||||||||||||||||||||||||||||
| アルファ資本は本チケットを転換できます | ||||||||||||||||||||||||||||
| 合計する | $ | $ | $ | $ | $ | $ | ( | ) | ||||||||||||||||||||
F-31
2021年第3四半期に、ある手形所有者は、いくつかの元金残高#ドルの変換可能な手形を選択する
アルファ火を消す
2021年2月3日,Alpha Capital Anstaltが持つ変換可能手形(“Alpha”および“Alpha Capital Note”)の項目で違約事件
;
は2021年7月29日に,当社はAlphaと和解合意(“Alpha和解合意”)を達成し,Alphaによって変換可能手形に関する余剰元金および応算利息をAlphaによって変換した
艦橋ノート
2020年1月3日と2019年12月27日、会社は転換可能なブリッジ手形を発行し、総金額は$
180 LP変換可能チケット
再編について、会社は$を負担した
転換可能手形の利子
当社は2021年12月31日までの年間で利息支出$を記録した
F-32
付記11--支払引受及び又は事項
訴訟やその他の損失
当社は、クレーム、評価、訴訟、罰金、その他のソースによる損失または損失によって発生した負債 を記録しており、負債 が発生しており、損失金額が合理的に推定できる場合を記録している。2022年12月31日現在、当社には記録や損失のある負債 はありません。2022年12月31日の課税項目に関する情報は、次の潜在的な法律事項であるKBLとCantor Fitzgerald&Co. 前幹部に対する違約訴訟を参照されたい。
潜在的法的問題
KBL前行政官に対する行動
2021年9月1日、当社はデラウェア州衡平裁判所で、会社の前最高経営責任者兼取締役最高経営責任者Marlene Krauss博士(“Krauss博士”)とその2つの関連会社KBLIVスポンサー有限責任会社とKBLHealthcare Management Inc.(総称して“KBL関連会社”と呼ぶ)に対して法律訴訟を提起した。株主の償還を不当に許可しています会社の起訴状
はKrauss博士及び/又はKBL関連会社が受託責任、越権行為、不当な利益、不注意と救済を声明し、$を超える補償性損害賠償を要求することを告発した
2021年10月5日、クラウス博士およびその付属会社は、現在または会社役員および/または役員であった12人の個人、すなわちMarc Feldmann、Lawrence Steinman、James N.Woody、TeresaとDeLuca、Frank Knuettel II、Pamela Marrone、Lawrence Gold、Donald A.McGoven,Jr.,Russell T.Ray,Richard W.Barker,Shoshana Shendman,Ozan Pamir(総称してOzan Pamir)に対して、“第三者被告”)。2021年10月27日、会社とOzan Pamirはクラウス反訴に対する回答を提出し、他のすべての第三者被告は第三者苦情を却下するための動議を提出した。
2022年1月28日、クラウス博士とその付属会社は解散動議に反対するのではなく、改正の反訴と第三者訴えの許可を求め、以前に任命された6人の現職と元役員、すなわちテレサ·デ·ルカ、フランク·クエテル二世、パメラ·マロン、ラッセル·T·レイ、リチャード·W·バック、ショサナ·デルマンを罷免することを求めた。条項によって承認され、クラウス博士は2022年2月24日に改訂の回答を提出した。反訴と第三者訴え(“改訂された反訴”)。実質的に、修正された反訴疑惑:(A)会社と残りの第三者被告はクラウス博士への受託責任に違反し、米国証券取引委員会が提出した文書でクラウス博士に不実な陳述をし、取引可能なように会社の株式に登録されておらず、(B)会社とクラウス博士との間のこのような株式の登録に関する契約に違反し、クラウス博士にもこのチケットに基づいてクラウス博士の元金金額を$とすると言われている
2022年3月16日、ドナルド·A·マゴヴィン。ローレンス·ゴルドは発議を提出し、彼らの改訂後の反訴を却下し、会社と残りの第三者被告は改訂された反訴に対して回答し、否定した。-2022年4月19日、クラウス博士は小ドナルド·A·マゴヴィンに対する彼女のすべての反訴と告発を却下することを規定した。ローレンス·ゴルドは彼らの動議を提出し、彼らの改正反訴を却下した。会社と第三者被告はすべての改訂された反訴を有力に弁護し続けるつもりであるが,このようなbr改正の反訴の法的弁護に成功する保証はない。2022年4月ドナルド·A·マゴヴィンはローレンス·ゴルドは当事者として訴訟から解任されました事件中の発見 はまだ始まっていない.会社と第三者被告はすべての修正された反訴に対して積極的な抗弁を継続しようとしているが,このような改正された反訴の法的弁護に成功する保証はない
F-33
クラウス博士の会社に対する行動は
2021年8月19日、クラウス博士はデラウェア州衡平裁判所で同社を提訴した。最初の起訴状は迅速な救済を求め、(1)会社に弁護士費、クラウス博士に米国証券取引委員会に対抗する費用と米国証券取引委員会がクラウス博士に出した特定の伝票を含む前払い費用を義務化したと告発した。そして(2)会社は、クラウス博士が当社を提訴した費用の返済も要求されていると主張している。2021年9月3日頃、クラウス博士は、以下に述べるTyche Capital LLC訴訟において第三者の訴えを弁護するための弁護士費を含む会社からの立て替え費用を得る権利があるとされている別の容疑を補完している。そして、クラウス博士に対する会社自身の訴えに対する上記の弁護費用。2021年9月23日頃、会社は修正された訴えに対する回答を提出し、会社はクラウス博士のすべての告発を否定し、さらに多くの積極的な抗弁を提出した。
2021年11月15日、クラウス博士は事件のいくつかの問題について簡易判決動議を提出し、会社の反対を受けた。2021年12月7日に公聴会が行われ、2022年3月7日、裁判所はこの件について裁決を行い、簡易裁決動議を一部却下し、その動議を部分的に承認した。裁判所はその後、2022年3月29日にこの決定を実行する命令を発表した。現在、各当事者はこの実施令で規定された訴訟手続きを行っている。裁判所はクラウス博士がその動議で要求した法律費用の一部を事前に支払うことに関するクラウス博士の請求を承認し、会社はこれらの費用の一部を支払うことを要求され、それは論争費用の残りの部分に反対する。これらの法的費用は会社貸借対照表 に計上されている。
2022年10月10日、クラウス博士は、クラウス博士が要求した2022年5月から7月までの費用を全額支払うことを会社に要求し、裁判所の命令を修正する申請を行った。会社は反対意見を提出した。2023年1月18日、クラウス博士は2回目の申請を提出し、会社にクラウス博士が要求した2022年8月から10月までの全額費用を支払い、裁判所の命令を修正することを要求した。会社は反対意見を提出した。2023年3月13日、裁判所は双方の代理弁護士に電話で通知し、クラウス博士の2つの申請を承認しようとした。しかし、これまで、裁判所はこのような裁決を下していなかった。裁判所はこのような明らかな裁決を下し、裁判所は会社にクラウス博士に弁護士費を前払いすることを要求したが、この裁決はクラウス博士が最終的にこのような前払いを永久的に保留する権利があるかどうかの最終判決を構成しない。同社はその役員や上級社員保険証書からこのような金額の大部分を支払うことを求めているが,役員や高級社員保険証書がこのような金額を保証する保証はない。参照してください“宣言的 AmTrust Internationalが会社に対して行った救済行動“下です。
Tyche Capital LLCに対する訴訟
会社は2021年4月15日にニューヨーク州最高裁でTyche Capital LLC被告(“Tyche”)に対する訴訟を開始した。起訴状では、Tycheは2019年7月25日の“保証·承諾協定”と2019年4月10日の“KBLとCannBioRex業務合併の条項説明書”に規定されている会社への書面契約義務に違反していると告発している。Br社は訴状で、Tycheに主体保証項の下での義務の履行を要求したにもかかわらず、Tycheは義務を履行して履行を拒否しておらず、現在のところ会社の債務は#ドルであると主張している
2021年5月17日頃、Tycheは会社の苦情に対応し、Tycheではなく、標的保証に違反したのは会社であると主張した会社に対する答弁と反訴を行った。Tycheは、会社経営陣のメンバー3人のMarc Feldmann卿、James Woody博士、Ozan Pamir(総称して会社被告個人)を含む6人の第三者被告 に第三者訴えを行い、Tycheへの受託責任に違反したと主張した。2021年6月25日、各個別会社の被告は動議を提出し、テッチの第三者への苦情を却下した。
2021年11月23日、裁判所は会社の請求を承認し、テキが保有しているすべての信託会社株に対して差し押さえ命令を出した。このようにする過程で、裁判所は、会社訴状に呼ばれている事実に基づいて、会社が事件の事件によって勝訴の可能性を証明したと考えている。
F-34
2022年2月18日、Tyche は修正された答弁書、反訴、第三者訴えを提出した。2022年3月22日、会社と各個別会社の被告が動議を提出し、Tycheのすべてのクレームを却下した。この却下動議の公聴会は2022年8月25日に行われ、裁判所は動議を承認し、各会社の被告および会社に対する4つの反訴のうちの3つを完全に却下し、Tycheの明確な救済クレームしか残っていない。2022年9月9日、Tycheは裁判所の裁決について控訴通知を提出したが、まだ通報や裁決は行われていない。2022年8月26日、Tycheは、Tycheが信託方式で保有している会社株を解除または修正するために、会社の既存の抵当令を空けたり修正したりする動議を提出した。当社はこれに反対意見を提出し、裁判所は2023年1月3日に審理を経ずにこの動議を却下した。Tycheはその後、控訴通知の提出を拒否し、2023年1月30日に開廷ブリーフィングを提出した。当社は2023年3月2日に反対意見要約を提出したが、公聴会の期日は確定していない。
2023年1月30日、会社は簡易手続き判決動議通知を提出し、Tycheに対する平権抗弁を却下した。Tycheは最近反対意見を提出し、会社は現在回答を提出する。当社や他の個別会社の被告は、テキのすべてのクレームに積極的に抗弁しようとしているにもかかわらず、このようなクレームの法的弁護に成功する保証はない。双方はすでに書面証拠提示手続きと証言 を行った
ロナルド·バウアーとサマンサ·バウアーの訴訟は
2022年2月25日、当社及びその2つの完全子会社KatExco PharmPharmticals Corp.とCannBioRex PharmPharmticals Corp.(総称して会社原告と呼ぶ)はブリティッシュコロンビア省最高裁判所でRonald BauerとSamantha Bauer及び彼らの2社のTheseus Capital Ltd.とAstatine
Capital Ltd.(総称して“Bauer被告”と呼ぶ)に対して訴訟を提起した。会社の原告はBauer被告に資金流用と株,無許可株式販売と不当出張費用の賠償を要求し,合計金額は少なくとも$である
AmTrust Internationalが同社に対して行った宣言的救済行動
2022年6月29日、KBLの合併前役員と高級社員保険証書保証人AmTrust 国際保険者DAC(“AmTrust”)は、米国カリフォルニア州北区地域裁判所(“宣言的救済行動”)に会社に対する明確な救済訴訟を提出し、AmTrustが取締役と高級社員保険証券に基づいて負担する義務を宣言することを求めた。救済行動では、AmTrustは合併によりbr社が保険対象下の被保険者ではないと主張した。当社 はAmTrustへの回収を求めているにもかかわらず,合併前に発生した事項に触れている
2022年9月20日、会社はAmTrustに対して答弁と反訴を行い、AmTrustはAmTrustが標的役員と上級管理者保険リストに基づいて会社に対して負担する保険義務に違反し、少なくとも賠償を要求したことを告発した
2022年11月22日、会社はAmTrustとFreedomに対する簡易裁決動議を提出した。*動議は十分なプレゼンテーションを得て、2023年3月9日に公聴会 を行った。裁判所はこの事件を受理し、まだ裁決が下されていない。当社はAmTrustを起訴する理由が強いと信じていますが、当社がこの訴訟で勝つ保証はありません
Yissm研究と許可協定
2018年5月13日、CBR Pharma は、CBR Pharmaが特定の特許(“ライセンス特許”)を使用することを可能にするグローバル研究および許可協定(“Yissmプロトコル”)をエルサレムヘブライ大学Yissm研究開発会社 有限会社と締結した。ライセンスを取得した特許は,次の時間に終了しなければならない(“YISSIMプロトコル”の規定により早期に終了しない場合):(I)ライセンス技術に含まれる最後のライセンス特許がその国での満了日; (Ii)その国の規制機関又は政府機関によって付与された当該製品に対する任意の独占経営権が満了した日;又は(Iii)当該国で初めて商業販売された日から20(20)年間が終了した日。上記(Br)(I)または(Ii)項に記載の期限が、上記(Iii)項に記載の期間前の特定の国/地域で満了した場合、その国または これらの国/地域におけるライセンスは、満期後の期限内の独自技術ライセンスとみなされるべきである。
使用、開発、またはライセンス特許でカバーされた技術を採用した任意の製品の売上が500,000,000ドル(“純売上高”)に達した場合は、YIsmにbr版税を支払わなければならない。上位500,000,000ドルの年間純売上高は3%で計算され、その後は純売上高の5%で計算される。
F-35
“Yissmプロトコル”によると、 Yissmが以下のマイルストーンを実現した場合、CBR Pharmaは以下の金額を支払う義務がある
I)75,000ドル動物の成功のためのケアポイント
2)最初の試験新薬試験を提出するための75,000ドル
3)第1段階/第2段階試験を開始するための100,000ドル
4)第3段階試験を開始するために150,000ドル
5)製品1製品あたり100,000ドル 市場許可/許可(最大500,000ドル)
Vi)製品の累計売上高は1,000,000,000ドルまで250,000,000ドルであった。
脱退イベント
(“イベント”)が発生した場合、当社またはその株主が任意の対価、通貨または他の態様の任意の代価を受け取る取引または一連の取引として定義することができ、当社の株式または当社の株主を売却する代償として、
または当社の初公開(IPO)であるが、より大きな確実性のために、当社の再構成
を含まない再構成エンティティの最終持分所有者が当社と実質的に同じであれば、当社は
を発行する
CBR PharmaもYissmとのコンサルティングプロトコルの一方であり,このプロトコルによりYissmは2人の従業員をコンサルタントとして#ドルで会社に提供することに同意している
2020年1月1日、CBR Pharma
はYissm協定の第1の修正案(“第1修正案”)をYissmと締結し、CBR Pharmaが2人のYissm教授による追加
研究を後援することを許可した。第1修正案の条項によると、会社はYissmに$を支払う
追加のYissmプロトコル
CBR Pharmaは、2019年11月11日にYIssmとCBR Pharmaが許可され、関節炎や疼痛治療に関連する大麻塩研究のbr許可特許(“ライセンス特許”)の研究、開発、商業化を行う新しいグローバル研究·許可協定(“追加YIssm協定”)を締結した。YIsmは、発効日1周年終了後60日以内に、彼らの研究結果をまとめた詳細な書面報告書を会社に提出する。
ライセンス特許が“追加合意”の規定に従って事前に終了していない場合は、(I)ライセンス技術に含まれるライセンス特許が最後に満了した日、(Ii)その国の監督管理機関または政府機関によって付与された当該製品に対する任意の専有権が満了した日、(I)ライセンス技術に含まれるライセンス特許の最終期限、(Ii)その国の監督管理機関または政府機関によって付与された当該製品の任意の固有権の満了に基づいて終了しなければならない
YIsmプロトコルを付加した条項によると、CBR PharmaはYIsmに返却不可能な許可料$を支払いました
F-36
会社は以下のマイルストーンを実現するためにYIsm に以下の金額を支払うべきです
| ● | 新薬の初研究申請提出:$75,000 |
| ● | 第2段階試験で1人目の患者の投与量:100,000ドル |
| ● | 第1段階試験患者の投与量:150,000ドル |
| ● | 最初の市場許可/許可:150,000ドル |
| ● | 2回目の市場許可/許可:75,000ドル |
| ● | 製品の250,000,000.00ドルあたりの累計純売上高は,1,000,000,000,000.00ドルまで:250,000ドル |
ライセンスが商業化された後、会社はYIsmに最初の合計500,000,000ドルの年間純売上高の3%に相当する特許使用料を支払い、その後5%を支払わなければなりません。 2022年12月31日と2021年12月31日現在、会社はYIsm協定の付加に関する売掛金や売掛金残高を持っていません。2022年12月31日と2021年12月31日までの年間で、会社はそれぞれ0ドルと246,753ドルの研究開発費を記録した。
スタンフォード大学許可協定
2018年5月8日、Katexoはスタンフォード大学(“Stanford”)と6ヶ月間のオプション協定(“Stanfordオプション”)を締結し、この合意に基づいて、Stanford
は、自己免疫疾患の治療のための生体物質に関する特許(“授権済み特許”)の独占許可を得るために、会社に6ヶ月間のオプションを付与した。スタンフォードオプションの対価格として、同社はスタンフォードに#ドルを支払いました
2018年7月25日、Katexo
は6ヶ月間の選択権を行使し、スタンフォード大学と独占許可協定(“スタンフォード許可協定”)
を締結した。“スタンフォードライセンス協定”によると、発効日1周年から、当社はスタンフォードに毎年$$のライセンス維持費を前払いする
F-37
また、会社 は以下の記念碑的支払いを義務化しています
I)第2段階試験を開始して100,000ドル
Ii)米国食品医薬品局がライセンス特許から生成された製品(“ライセンス製品”)を初めて承認した場合、500,000ドルを支払う
Iii)その後の新ライセンス製品あたりの費用は250,000ドルである。
当社は30日前にスタンフォード許可協定の廃止を通知することができます。製品の純売上高の2.5%で計算された印税はスタンフォード大学に支払われます。また、協定によると、同社はスタンフォード大学の特許費用を精算する。当社はスタンフォード大学に年間ライセンス維持費20,000ドルを支払い,前払い費用に記録し,12カ月以内に直線的に支出し,2021年12月31日現在の残高はゼロである。当社は,2022年および2021年12月31日までの年度中に,スタンフォードライセンス契約に関する特許料および許可費をそれぞれ69,278ドルおよび78,245ドルとし,添付の経営報告書および全面赤字の一般および行政費用を計上した。
オックスフォード大学協定
2020年9月18日、CBR Pharmaは、合計1,085,738ドル(GB 795,468)の総代価と引き換えに、合計1,085,738ドル(GB 795,468)の総代償と引き換えに、3年間の研究開発協定(“3年オックスフォード協定”)を締結し、そのうち109,192ドル(GB 80,000)はプロジェクト開始日の30日後に支払い、残りの金額はプロジェクト開始日の6ヶ月の周年日と毎年の記念日に4回に分けて244,136ドル(GB 178,867)を支払う。本プロトコル は、いずれか一方が書面通知後に終了するか、または会社が本プロトコルの下で満期になった任意のお金を30日以上延滞した場合に終了することができる。当社は2022年および2021年12月31日までの年度までに,それぞれ322,767ドル(国標265,156)および364,673ドル(国標264,938), が3年間のオックスフォード合意に関する研究および発展費を確認した。
2020年9月21日、CBR Pharmaは、炎症性疾患を治療する大麻薬の臨床開発と交換するための2年間の研究開発協定(“2年オックスフォード協定”)をオックスフォード大学と締結し、総費用は625,124ドル(GB 458,000)であり、138,917ドル(GB 101,778)はプロジェクト開始日後30日に支払い、残りの金額は項目開始日後6ヶ月に1回支払い、4期に分けて支払い、138,917ドル(GB 101,778)は前3期に69,456ドル(50 GB,888)を最終期として支払う。本プロトコルは、いずれか一方が書面で通知した後に終了することができ、または当社が本プロトコルの下で満期になった任意のお金を30日以上延滞した場合には、いずれか一方によって終了することができる。2022年及び2021年12月31日までの年度内に、当社はそれぞれ123,891ドル(国標101,778)及び139,977ドル(国標101,778)が2年間のオックスフォード合意に関する研究及び発展支出を確認し、この等の支出は添付された総合貸借対照表の計算すべき支出に反映されている。
2022年12月31日と2021年12月31日まで、当社はオックスフォード大学に何の借金もありません
F-38
2021年5月24日、会社はオックスフォード大学と研究協定(“オックスフォード”と“オックスフォード第五協定”)を締結し、これにより、会社はオックスフォード大学を賛助して多中心、無作為、二重盲検、平行な研究グループを展開し、抗腫瘍壊死因子の注射による成人肩こり疼痛主導期の治療の実行可能性を研究する。価格として、当社はオックスフォード大学に以下の金額を支払うことに同意した
| 支払額 | ||||
| 一里塚 | (付加価値税抜き) | |||
| £ | ||||
| £ | ||||
| £ | ||||
| £ | ||||
同社は最初のbrマイルストーン$を支払いました
2021年11月2日,会社はオックスフォード大学と20年間の組織再生に関するHMGB 1分子の許可技術協定を締結し,合意に基づき,オックスフォード大学は研究,開発,使用許可のための特許を会社に許可することに同意した。同社はオックスフォード大学に過去の特許費用$を支払うことに同意した
ケネディ許可協定
2019年9月27日、180 LPは、米国、日本、イギリス、EU諸国のケネディリウマチ研究信託基金(“Kennedy Trust for Rheumatology Research”) について、再許可を付与する権利、および任意の医薬製品を研究、開発、販売または製造する権利(I)の研究、開発、製造、使用、および任意の医薬製品を研究、開発、販売または製造する権利(I)を含む許可協定(“ケネディライセンス協定”)を締結した。ケネディ許可協定によって付与された許可がない場合、ケネディ許可特許の輸入または販売は、ケネディ許可特許を侵害するか、または(Ii)抗体断片または抗体由来抗体を含み、抗体の研究、開発、製造、使用、輸入または販売は、診断、予防および治療および状態を含むすべてのヒト用途のために、ケネディ許可協定によって付与された許可されていないケネディ許可特許を侵害するであろう。
F-39
ケネディ許可特許を付与する価格として、180 LPはケネディにポンドGBの前金を支払いました
当社がケネディに支払う特許料br}の有効期限は、(I)ケネディ許可特許に含まれる対象または適用国/地域で製品を開発する特許の最後の有効主張 ;(Ii)その製品の同国での規制排他性が満了する、または(Iii)当該製品が同国で初めて商業販売された10年以内に満了する。ケネディ許可プロトコル は、90日間の通知を提供することによって、理由なく終了することができる。
Petcanna許可契約書
2018年8月20日、CBR Pharma は、その完全子会社Petcanna Pharma Corp. (“Petcanna”)と分割許可協定(“分許可協定”)を締結し、同社の元首席財務官は取締役の首席財務官である。Petcannaは個人会社で、会社と共通の依頼人がいる。
360生命科学社の合意関係者(Reform PharmPharmticals Corp.)
2020年7月1日、当社 はReform PharmPharmticals,Corp.(“Reform”)および360生命科学社(“360”)と改訂合意を締結し、これにより、360は2020年7月31日(“締め切り”)またはそれまでにReform 100%の株式を買収するという合意を締結した。 当社はReformおよび360とそれぞれ上級職員および取締役を割り当てる。締め切りは,360が180 LPに分 を支払い,総金額は300,000ドルである.契約側は360から180 LPが債務を支払い、360の融資活動で調達した1,000,000ドルあたり100,000ドル、最大300,000ドルを支払うことに同意したが、360が受信したすべての融資で得られた純額の10%以上の は、すべて返済されるまで当社への債務の返済に適用される。この取引 は2020年7月31日に完了する。
180 LPは2019年2月26日、180 LP役員および上級職員を持つ関連側Reformと1年間の合意(“薬剤協議”)を締結し、この合意に基づき、180 LP$の支払いに改革同意した
F-40
180 LPは、プロトコル期間内に“医薬品プロトコル”に関連する収入を直線的に確認する。180 LPは,2022年および2021年12月31日までに薬物合意に関する収入がないことを確認し,付随する総合経営報告書やその他の全面収入損失中の他の収入
に計上している。2021年12月31日まで、会社はbrドルを受け取ります
賃貸借契約を経営する
2016年2月、FASB はASU 2016-02を発表した賃貸借契約関連する会計基準コード化特集842は賃貸借証書(“ASC 842”). 新基準は、多くのリースが貸借対照表上で使用権(“ROU”)資産および賃貸負債であることを確認することを必要とする。 使用権資産は、最初にリース期間内に予想される支払金額の現在値で計量される。損益計算書では、これらのレンタルのコストが分解され、運営費用(使用権資産のための償却)および利息支出(賃貸支払いのうち利息に関連する部分のため)として確認される。本基準はリリース時に当社に採用されました。
ASC 842によれば、当社は、資産負債表に期限が12ヶ月以下の賃貸契約を記録せず(資産別)選択することができ、テナントが行使する購入選択権を合理的に決定することは含まれていない。当選した場合、レンタルは、以前のGAAPでの運営リースとみなされ、支払いは、レンタル期間内に直線ベースで確認される。レンタルが今回選択した資格に適合しているかどうかを決定する際には、会社は、更新オプションがレンタル期間の一部とみなされた場合にのみ、これらのオプション、すなわち、会社が行使するオプションを合理的に決定する。レンタル期間が12カ月以上に増加した場合,あるいは会社が購入選択権を行使することを決定する理由があれば,会社はこの実際の便宜策を適用することができず,ASC 842指導を適用する。
当社とその賃貸契約について(現在あります
2022年12月31日と2021年12月31日まで、当社には有効なレンタルがなく、レンタルやレンタル料もありません。
相談協議
南チャハル諮問協定
2021年2月22日、会社は関連側のジャグディップ·ナンチャハル教授(“コンサルタント”)とコンサルティング協定(改訂された“コンサルティング協定”)を締結した。その諮問協定は2020年12月1日に施行される。
コンサルティング契約によると,会社は合意期間内にコンサルタントに毎月15,000ポンド(約20,800ドル)を支払い,月23,000ポンド(約32,000ドル)に増加することに同意し,これは(A)Dupuyten‘s Constraint(RIDD)2 b期の臨床試験データ公表日と(B)会社が1,500万ドルを超える資本調達に成功した日に月23,000ポンド(約32,000ドル)に増加する。同社はまた、コンサルタントに以下のボーナスを支払うことに同意した
| ● | Dupuyten‘s Constraint臨床試験データを提出して、同業者レビューの定期刊行物に発表された金額は100,000 GB(約138,000ドル)(“ボーナス1”)である |
| ● | 2020年12月1日(“帰属日”)の後、会社は債務または株式を売却することによって少なくとも15,000,000ドルの追加資金を調達する際に、434,673ポンド(約605,000ドル)を稼ぎ、支払うべきである(“ボーナス2”)。ボーナス2は、ホーム日の後30日以内に支払われ、ホーム日の前に計算、満了、または支払われてはならない。配当2コンサルタント選択時に支払い、少なくとも50%(50%)の会社普通株式を、(I)1株当たり60.00ドルまたは(Ii)付与された日の取引価格のうちの低い者で支払い、残りはポンドで支払う |
| ● | 第2段階凍結肩甲試験(“報酬3”)に参加するための第1の患者を募集するための5,000ポンド(約7,000ドル) |
| ● | 第1の患者を2期精神錯乱/POCD試験(“報酬4”)に参加するために5,000 GB(約7,000ドル)を募集した。 |
F-41
コンサルティング契約の初期期限は3年であり、その後3年間継続し、合意の規定に従って終了するまで継続する。 のいずれも12ヶ月前にコンサルティング契約の終了を書面で通知することができる(ただし、会社が合意を終了する権利は、コンサルタントがコンサルティングプロトコルに規定された職責を履行できない場合にのみ行使される)。(A)コンサルタントが合意に規定されたサービスを効果的かつ勤勉に履行することができなかったか、または合意に従って付与された任意の同意を含むプロトコルに規定された義務に違反した場合、または以下の場合、企業は、コンサルティングプロトコルに規定された特定の条件の下で直ちに、(A)コンサルタントが合意に従って付与された任意の同意を含む)を有効かつ勤勉に履行することができない、または以下の場合。(B)コンサルタント犯は、任意の詐欺または不誠実な行為、またはその行為(サービス実行時または他の態様にかかわらず)が、コンサルタント、当社またはその任意の連属会社の名誉を損なうことができるか、または逮捕可能な罪(非監禁刑の道路交通犯罪を除く)、または(C)コンサルタントを破産させるか、またはその債権者と任意の手配または債務改質を行う可能性があることを命令または相当する可能性がある。コンサルティング契約が原因以外のいかなる理由でも会社によって終了された場合、コンサルタントは終了日から12ヶ月の費用の一括払いを受ける権利がある。
2021年3月30日から発効し、顧問を返済するために
2021年8月27日より返済不履行コンサルタントの余剰ボーナス2,会社発行
2021年12月,Dupuytenの収縮臨床試験データが同業者評議の定期刊行物発表に提出され,コンサルタントにボーナス1が支払われた。
2022年4月27日、会社は相談協定改正案を締結し、Dupuytren契約の2 b期臨床試験データの公表を受けた後、相談費はGBに増加する
2022年12月28日、br社はコンサルティング協定修正案を締結し、コンサルタント月費はGBに増加する
ラーソン相談契約
2021年4月29日、会社brは、180 LPの前最高経営責任者Glenn Larsenと、交渉代表として4つの特許の許可を得るコンサルティング契約を締結した。提供されたサービスの対価として、会社はさん#ドルのラーソン賠償に同意しました
2023年2月22日、会社はGlenn Larsenと諮問サービスを提供する第2の諮問協議を締結し;提供されたサービスに対する補償として、会社はLarsenさん#ドルの賠償を同意する
F-42
スタンマン諮問協定
当社は2021年11月17日、2021年11月1日から、当社執行合同議長のローレンス·シュタインマン(医学博士)と諮問協定を締結した相談協議“)”コンサルティング契約によると、Steinman博士は、限定される訳ではないが、会社の戦略目標の決定と制定に参加すること、買収および合併候補を積極的に探すこと、および会社に対するa´7 nAChRプラットフォーム(総称してこれを総称して呼ぶ)を含むいくつかのコンサルティングサービスを会社に提供することに同意したサービス.サービス“)”協議の期限は
同社は合意期間内に毎年スタンマン博士に22.5万ドルを支払い、#ドルを一度に支払うことに同意した
行政総裁登用協定
当社は2021年2月25日、当社の行政総裁James Woody博士(“行政総裁”)と改訂合意(“A&R合意”)を締結し、期日は2021年2月24日であり、2020年11月6日に発効し、行政総裁と当社の先の合意
に代わっている。A&R協定によると、最高経営責任者は、いずれの方向も少なくとも90日間の書面通知を提供しない限り、任期3年、その後自動的に1年間継続することができる当社の上級管理者を務めることに同意している。合意によると、CEOの年間基本給は最初に#ドルだった
最高経営責任者が契約締結に同意した追加代償として、会社は70,000株会社の普通株購入の選択権 を付与し、期限は10年、行使価格は1株88.60ドル(取締役会承認授与日(br})の終値(2021年2月26日))である。この等購入持分は当社の2020年総合激励計画によって制限され、以下の比率 で帰属する:(A)授出日に月ごとに当該等購入株権の5分の1に帰属する;及び(B)その後の 36ケ月以内に各月の最終日に月ごとに当該等購入株権の5分の4に帰属する;しかし、この等購入持分は行政総裁の死去或いはbr}障害がなく、終了或いは行政総裁が合理的な理由(定義は合意参照)で終了し、当社の制御権の変更或いは当社の売却時に直ちに帰属しなければならない。
F-43
最高経営責任者には年間ボーナスを得る資格があり、目標ボーナスは
A&Rプロトコルは、会社が正当な理由(合意に適合する救済条項)または正当な理由なし(60日前に書面でCEOに通知する)、CEOが十分な理由(合意に記載されており、合意に適合する救済条項)、br}またはCEOが十分な理由がない場合に随時終了することができる。いずれか一方が上述したような非更新通知を提供する場合、本プロトコルも、初期期限または任意の更新期間の終了時に自動的に満了する。
A&R プロトコルが会社またはCEOに十分な理由がある場合に終了した場合、会社は、18ヶ月の賃金または協議の残り期間の少ない を彼に支払い、前年の任意の課税ボーナスを支払うことに同意し、現在の“br}年間ボーナスに比例して割り当てられた部分と、解散費の同時期の医療保険料を得る(上述した)。
A&Rプロトコルは、標準および慣行の発明譲渡、賠償、秘密および非募集条項を含み、これらの条項は、合意終了後24ヶ月以内に依然として有効である。
2022年4月27日、会社は雇用協定改正案を締結し、この改正案に基づき、会社は提供する
首席財務官採用協定
2021年2月25日、会社は会社のOzan Pamir臨時財務官と2021年2月24日に発効した雇用協定(“CFO協定”)を締結した。協定によると、首席財務官は会社の臨時首席財務官(“首席財務官”)を務めることに同意し、賃上げは#ドルである
最高財務官が契約締結に同意した追加代償として、会社は9000株会社の普通株を購入するオプション を付与し、期限は10年、行使価格は1株88.60ドル(取締役会許可授与日(br})の終値br})。この等購入持分は当社の2020年総合激励計画によって制限され、以下の比率 で帰属しなければならない:(A)授出日に月ごとに当該等購入持分の5分の1を授与する;及び(B)その後各月の最終日の36ケ月以内に月ごとに当該等購入持分の5分の4に帰属する;ただし、この等購入持分は財務総監の死亡或いは障害、財務総監の無断終了或いは財務総監が良好な理由(定義合意参照)で終了し、当社の制御権変更或いは当社の販売時に直ちに帰属しなければならない。
協定によると、首席財務官は年間ボーナスを得る資格があり、目標金額は
F-44
本契約は、企業が60日前に書面で通知した場合にいつでも終了することができ、理由があるか否かにかかわらず、首席財務官がいつでも60日前に書面で通知して終了することができます。合意が何らかの理由で終了した場合もあれば、会社は6日前に合意終了を通知することもできる。当社が理由なくCFOの合意またはCFOを終了するのに十分な理由があった後、当社は3ヶ月の解散費を支払うことに同意しました。
このプロトコルは、標準 および習慣発明譲渡、賠償、秘密および非募集条項を含み、これらの条項は、合意終了後24ヶ月以内に依然として有効である。
首席運営官/首席業務官採用プロトコル
2021年10月29日、会社は全武と2021年10月27日に発効した雇用協定(“COO/CBO協定”)を締結した。契約によると、呉さんは、当社の最高経営責任者/最高経営責任者(“最高経営責任者/首席業務官”)を担当することに同意しました
,賃上げは#ドルです
最高経営責任者/最高経営責任者が協定を締結することに同意した追加の代価として、会社は彼に購入を与えた
協定によると、首席運営官/首席運営官は年間ボーナスを得る資格があり、目標金額は
本協定は、会社が30日前に書面で通知した場合にいつでも終了することができ、原因の有無にかかわらず、首席運営官/首席運営官が30日前に書面で通知した場合にはいつでも終了することができる。合意が何らかの理由で終了した場合もあれば、会社は10日前に合意終了を通知することもできる。会社が理由なく首席運営官/首席運営官の協議 や首席運営官/首席運営官を中止する十分な理由があった後、会社は彼に12ヶ月の解散費を支払うことに同意したが、役員が1周年記念日までに会社を出た場合は除外した。
このプロトコルは、標準 および習慣発明譲渡、賠償、秘密および非募集条項を含み、これらの条項は、合意終了後24ヶ月以内に依然として有効である。
2022年4月27日、会社は雇用協定改正案を締結し、この改正案に基づき、会社は提供する
F-45
付記12--株主権益
2022年の逆株式分割
2022年12月15日、180生命科学会社株主特別総会において、会社株主は、当社の普通株の発行および流通株の逆株式分割、額面$を実現するために、会社の第2回改正および再発行された会社登録証明書の改正案を承認した
“証明書修正案”は普通株の認可株式数を減少させていないため、逆株式分割の効果は、発行済み株式および発行済み株式数に対して発行可能な普通株式数を増加させることである。逆株式分割は普通株の額面を変えることはなく、普通株のいかなる投票権や他の条項も修正しない。逆株式分割後に残った任意の断片的な株式 は、最も近い完全株式に上方丸め込まれる。
会社の2020年総合インセンティブ計画と2022年総合インセンティブ計画について、会社報酬委員会と取締役会は、会社とその株主の最適な利益のために、(I)インセンティブ計画の下で発行可能な会社の普通株式数 を20倍に引き下げた(任意の断片的な株式を最も近い整数に切り捨てる)。(Ii)自社普通株株式を購入した未行使引受権毎に発行可能な普通株式数および他のすべての未償還報酬を20倍に減少させる(任意の断片的株式を最も近い全体株式に切り捨てる);および(Iii)奨励計画に従って以前に付与された普通株式を購入するための任意の未行使購入持分の行使価格を20倍(最も近い整数仙に丸め込む)を引き上げ、いずれの場合も逆株式分割の交換比率で公平に調整し、このような調整は逆株式分割が発効したときに自動的に発効する。財務諸表と財務諸表付記には20株反転株式分割の影響がトレーサビリティに反映されている。
優先株
2020年11月6日に提出された会社の第2次改正·再登録証明書によると、会社は
F-46
普通株
当社は発行を許可しています
2021年2月の非公開発行で普通株と引受権証 を売却
2021年2月19日,当社は複数の買い手(“買い手”)と証券購入合意を締結し,これにより,当社
は合算販売に同意した
管権証の執行権価格は$である
今回の発売については、当社も買い手と2021年2月23日に発効する登録権協定(“登録権協定”)を締結している。登録権協定に基づき、当社は、2021年4月24日またはそれまでに米国証券取引委員会に登録声明を提出することに同意し、パイプライン株式の登録及び配管株式承認証の行使により発行可能な普通株式(“管路株式承認証”)の再販売を行い、この登録声明を2021年6月23日又はそれまでに発効させることを宣言する(又は米国証券取引委員会が“全面審査”を行う場合は8月22日)。2021).会社は、2021年4月24日までに配管株式及び配管株式証株式の登録説明書を提出していないため、登録権協定のbr条項に違反している。しかし、この登録説明書はアーカイブに送られなければなりません。この違約により、会社は購入者に合計#ドルの損害賠償金を支払うことを要求された
橋接メモ変換
2021年第1四半期に、ある手形所持者は元金総額ドルのブリッジ手形を転換することを選択した
F-47
変換可能チケットの変換
2021年第1四半期に、ある手形所有者は、いくつかの元金残高#ドルの変換可能な手形を選択する
早鳥が集まる
当社は2021年4月23日に、EarlyBird Capital,Inc.(“EarlyBird”)と2017年10月17日に締結したある発見者合意(“発見者合意”)に基づいて支払金を清算する。当社取締役会は、提出されたまたは提出可能なすべての請求について和解を行い、和解合意(“和解合意”)を締結することが最適な利益
に合致すると考えている。和解協定によると、会社はEarlyBirdに現金で#ドルを支払った
2021年8月に普通株式と引受権証 を発売
2021年8月23日,当社は複数の買い手と証券購入協定を締結し,これにより,当社は合算して販売することに同意した
2021年8月の管権証brの発行価格は1ドルに相当する
2021年8月の発売については、当社は買い手と2021年8月23日の登録権協定(“2021年8月 登録権協定”)も締結しています。2021年8月の登録権協定に基づき、当社は、2021年8月に米国証券取引委員会が2021年8月に発売された管状株式承認証(“株式承認証”)を行使する際に販売された株式と普通株式を発行可能な株式を登録し、2021年10月22日またはそれまでに発効を宣言するために、2021年9月12日またはそれまでに米国証券取引委員会に登録声明を提出することに同意する(または米国証券取引委員会が2021年11月21日に“全面審査”を行う場合は、登録声明は2021年8月31日に提出する)。2021年9月9日、米国証券取引委員会は、2021年8月の“登録権協定”に規定された最終期限よりも早く発効すると発表した。
係り先ローンと変換可能チケットの交換
2021年9月30日、それぞれ取締役会の共同執行議長を務めたローレンス·スタンマン博士とマーク·フェルドマン博士が会社と合意し、未返済融資項目の借金を元金総額#ドルに転換した
F-48
アルファ資本決済
2021年第3四半期に会社は
2021年にサービス業に発行された普通株
2021年12月31日までに当社は共同で発行します
2022年7月提供
2022年7月17日、当社は複数の買い手と証券購入協定を締結し、これにより、当社は合算して販売することに同意した
2022年7月の事前資金の引受権証の発行価格はドルに相当する
2022年7月の一般権証brの発行価格は$に相当する
2022年12月31日まで、
はすべて
F-49
2022年12月提供
2022年12月20日,当社は複数の買い手と証券購入協定を締結し,これにより,当社は合共
の売却に同意した
2022年12月の事前資金の引受権証の発行価格は$に相当する
2022年12月の一般権証の発行価格は$に相当する
2022年12月31日まで、
はすべて
2022年にサービス業に発行された普通株
2022年12月31日までに当社は共同で発行します
F-50
2022年発行の限定株
当社は2022年12月31日までに年度中に発送します
次の表は、2022年12月31日までの年間で付与·発行された限定株式数をまとめたものである
| 既存の制限ではない | 重みをつける 平均値 グラント 日取り | |||||||
| 在庫品 | FV価格 | |||||||
| 2022年1月1日現在帰属していません | $ | |||||||
| 授与する | ||||||||
| 既得 | ||||||||
| 2022年12月31日現在帰属していません | ||||||||
| 未確認料金残高合計 | $ | |||||||
| 確認される加重平均年限 | ||||||||
特別議決権株
特別議決権株式 は、CBR PharmaとKatExcoの前株主に発行され、業務合併前の180再編に関連しています。 特別議決権株式は、保有者が会社の普通株式の株式に交換することができ、会社の普通株主と単一の カテゴリとして投票することができます。特別議決権株式はいかなる割り当て配当金も得る権利がない。
特別投票権株式に関する普通株式等価物を交換して株式を発行することは、2022年12月31日までの年度中にはない。
2021年12月31日までの年間で
次の表は、2022年12月31日と2021年12月31日までの年度の特別投票権株式活動をまとめたものである
| 残高、2021年1月1日 | ||||
| 既発行株 | ||||
| 交換株 | ( | ) | ||
| バランス、2021年12月31日 | ||||
| **新株発行済み | ||||
| *株式交換 | ||||
| バランス、2022年12月31日 |
F-51
株式オプション
2022年12月31日と2021年12月31日までの年間のオプション活動の概要 は以下のとおりである
番目 オプション | 重みをつける 平均値 トレーニングをする 値段 | 加重平均残期限( 年単位) | 固有の 価値がある | |||||||||||||
| 未返済、2021年1月1日 | ||||||||||||||||
| 授与する | ||||||||||||||||
| 鍛えられた | ||||||||||||||||
| 期限が切れる | ||||||||||||||||
| 没収される | ||||||||||||||||
| 未返済、2021年12月31日 | ||||||||||||||||
| 授与する | ||||||||||||||||
| 鍛えられた | ||||||||||||||||
| 期限が切れる | ||||||||||||||||
| 没収される | ||||||||||||||||
| 未返済、2022年12月31日 | $ | |||||||||||||||
| 行使可能、2022年12月31日 | $ | |||||||||||||||
2022年12月31日現在の未償還株式オプションと行使可能株式オプションの概要は以下の通り
| 未償還株式オプション | 行使可能株式オプション | |||||||||||||
| 重みをつける | ||||||||||||||
| 平均値 | ||||||||||||||
| トレーニングをする | 量 | 残り | 量 | |||||||||||
| 値段 | 株 | 年単位の寿命 | 株 | |||||||||||
| $ | 49.80 | |||||||||||||
| $ | 88.60 | |||||||||||||
| $ | 151.20 | |||||||||||||
| $ | 79.00 | |||||||||||||
| $ | 27.20 | |||||||||||||
2021年2月26日、
社発表-購入した年間オプション合計
F-52
2021年8月4日、会社は
を授与しました
2021年12月8日、
会社は購入を授与しました
これらのオプションは2021年に発行され,ブラック·スコアーズ推定手法で用いられている仮定は以下のとおりである
| 無リスク金利 | ||
| 所期期間(年) | ||
| 予想変動率 | ||
| 配当を期待する |
以上に示した2021年と2022年以下のこれらの仮定は,i)FRBが付与日に公表した無リスク金利, ii)期待期間は契約期間に加重平均帰属期間を加えた平均値,iii)変動率は適用四半期の他の金融商品の第三者推定報告における金利を用いたもの,iv)使用した期待配当金 金利は適用されたオプション奨励プロトコルである。
2022年5月19日、会社
が購入を授与しました
2022年5月19日、会社
は購入も授与されました
これらのオプションは2022年に発行され,ブラック·スコアーズ推定手法で用いられている仮定は以下のとおりである
| 無リスク金利 | ||
| 所期期間(年) | ||
| 予想変動率 | ||
| 配当を期待する |
会社は株ベースの報酬支出$を確認した
F-53
ナスダックコンプライアンス
2022年9月30日、吾らはナスダック上場資産部(“ナスダック”)を受け取り、書面通知(“通知状”)を出し、当社が“ナスダック”上場規則 上場規則第5550(A)(2)条のナスダック資本市場への上場継続に関する最低購入価格要求に適合しないことを通知した。ナスダック上場規則第5550(A)(2)条は上場証券 が最低購入価格を1株1.00ドルに維持することを要求しているが、上場規則5810(C)(3)(A)条は、30(30)営業日連続で最低購入価格が不足し続けていれば、最低購入価格要求に達しなかった行為があると規定している。会社普通株が2022年8月18日から2022年9月29日まで30営業日連続の終値入札価格によると、会社 は最低入札価格要求を満たさなくなった。通知状は、会社が180暦または2023年3月29日にナスダック上場規則第5550(A)(2)条を再遵守することを指摘している。コンプライアンスを再獲得するためには、会社普通株の入札価格は少なくとも10営業日連続で1株1.00ドルの終値を達成しなければならない。同社は2022年12月に逆株式分割を実施し、ナスダック基準の再遵守を支援した。2023年1月4日、ナスダックはナスダック上場規則 第5550(A)(2)条に基づき、ナスダック上場の最低入札価格を完全に遵守し直したことをナスダック社に通知した。
付記13--所得税
当社はアメリカ、カナダ、イギリスで連邦と州/省所得税を支払う必要があり、どの法人実体も非 合併方式で届出しています。再編前の180社の有限責任会社の純営業損失のメリットはその所有者に転嫁されている。
所得税前の損失は以下の国内と国際部分を含む:
| ここ数年で | ||||||||
| 十二月三十一日 | ||||||||
| 2022 | 2022 | |||||||
| 国内では | $ | ( |
) | $ | ( |
) | ||
| 国際的に | ( |
) | ( |
) | ||||
| $ | ( |
) | $ | ( |
) | |||
所得税準備金には、以下の福祉 (準備金):
| ここ数年で | ||||||||
| 十二月三十一日 | ||||||||
| 2022 | 2021 | |||||||
| 繰延税の割引: | ||||||||
| 国内: | ||||||||
| 連邦制 | $ | $ | ||||||
| 状態.状態 | ||||||||
| 国際的に | ||||||||
| 評価免除額を変更する | ( |
) | ( |
) | ||||
| 所得税純額割引 | $ | $ | ||||||
F-54
一部の繰延税金負債 はドル以外の通貨で価格され、外貨換算調整の影響を受ける。所得税条項は米国連邦法定税率と異なり,以下のようになる
| ここ数年で | ||||||||
| 十二月三十一日 | ||||||||
| 2022 | 2021 | |||||||
| アメリカ連邦法定金利 | % | % | ||||||
| 国内と海外の連邦金利の違い | ( |
)% | ( |
)% | ||||
| 連邦福祉を差し引いた州税と省税 | % | % | ||||||
| 永久的な違い: | ||||||||
| 営業権の減価 | ( |
)% | ||||||
| 株に基づく報酬 | ( |
)% | ||||||
| 派生ツール及び当計は株式の公正価値変動を発行することができる | % | ( |
)% | |||||
| 他にも | - | ( |
)% | |||||
| 評価免除額を変更する | ( |
)% | ( |
)% | ||||
| 有効所得税率 | % | % | ||||||
繰延税金資産と負債 は以下の項目からなる:
| 自分から 12月31日、 |
||||||||
| 2022 | 2021 | |||||||
| 繰延税金資産: | ||||||||
| 純営業損失が繰り越す | $ | $ | ||||||
| 償却する | ||||||||
| 計算すべき報酬は現在差し引かれない | ||||||||
| 株の報酬 | ||||||||
| 応算利息 | ||||||||
| 他にも | ( |
) | ||||||
| 繰延税金負債: | ||||||||
| 帳簿と課税基礎との差異は以下の点に関連している | ||||||||
| 技術許可証 | ( |
) | ( |
) | ||||
| 現在行われている研究と開発を買収する | ( |
) | ( |
) | ||||
| 他にも | ( |
) | ( |
) | ||||
| ( |
) | ( |
) | |||||
| 繰延税金資産と負債 | ||||||||
| 推定免税額 | ( |
) | ( |
) | ||||
| 繰延税金資産と負債、純額 | $ | ( |
) | $ | ( |
) | ||
F-55
繰延税金資産の価格準備金の変動状況は以下の通りである
| ここ数年で | ||||||||
| 十二月三十一日 | ||||||||
| 2022 | 2021 | |||||||
| 期日の初め | $ | ( |
) | $ | ( |
) | ||
| 税務規定による評価変動 | ( |
) | ( |
) | ||||
| -前年までの納税申告書 | ( |
) | ||||||
| 期末 | $ | ( |
) | $ | ( |
) | ||
2022年12月31日現在、各管轄区における会社の純営業損失(“NOL”)の繰越は、将来の課税所得額を相殺するために利用できる場合は以下の通り
| ● | 約$ |
| ● | 約$ |
| ● | 約$ |
所有権変更により、国内NOLを利用して将来の課税所得額を相殺することは、国税法第382条と類似したbr州法規の年間制限を受ける可能性がある。
当社はASC 740条項による繰延税金資産の可能性を評価した所得税(“ASC 740”)。ASC 740は、繰延税金負債の所定の打抜き、将来の課税所得額の予想、および税務計画戦略を含む、すべての利用可能な肯定的および負の証拠を考慮することを要求する。ASC 740は、“可能性が高い”繰延税金資産の全部または一部が現金化できない場合に、推定準備を確立することを要求する。2022年12月31日および2021年12月31日までの業績評価後、経営陣は繰延税金資産の将来的な変動に不確実性があると判断し、全額評価を確立した。当社が記録した評価は増加に備えている
経営陣はbrを評価し、2022年12月31日と2021年12月31日現在、会社合併財務諸表に確認すべき重大な不確定税務状況はないと結論した。当社はその未確認の税務割引は報告日から12ヶ月以内に大きな変化はないと予想しています。
2022年12月31日及び2021年12月31日までの年度内に、税務審査を展開したり、税務審査を行ったりすることはなく、その期間中に税務関連の利息或いは罰金が発生することもない。会社が設立されてからアメリカ、カナダ、イギリスで提出された納税申告書はまだbr審査を受ける必要があります。
付記14--関係者
計上すべき費用--関係者
課税費用に関する
当事者は$
ローン対応--関係者
ローン対応−関係者
は$からなる
F-56
研究開発費に関する当事者
研究と開発
費用に関する当事者$
一般的な行政費用に関する当事者
2022年12月31日と2021年12月31日までの年間で,一般·行政費用に関する当事者は$である
利子支出関係者
2022年12月31日までに、当社は$を記録しました
2021年12月31日まで、当社は$を記録しました
付記15--後続活動
財務諸表発表日から、会社は2022年12月31日以降の事件と取引を評価した。以下の事項を除いて、 は財務諸表に開示すべき後続イベントは発見されなかった。
ナスダックからのコンプライアンス通知
2023年1月4日、ナスダックは当社に通知し、“ナスダック上場規則”第5550(A)(2)条(詳細は付記12参照)に基づき、当社はナスダック上場規則の下でナスダック上場を継続する最低購入価格を完全に遵守した。
2022年12月に発売された共同株式証の改訂協定
2023年1月12日、br社は2022年12月22日付の“普通株購入承認証協定改正案”を締結し、この合意により、所有者は株式引受証を取得し、最大購入が可能となった
Kinexum協定
2023年1月13日、会社はKinexumと協定を締結し、会社がアダリムマブによる進行性早期Dupuytren病の治療を計画していることについてイギリスの薬品と保健品監督機関(MHRA)に条件付き上場許可(CMA)と上場承認(MAA)申請を提出して協力を提供することに同意した。Kinexum契約に関連するコストを含めて、同社は約$かかると予想されています
全武分離
当社は2023年1月15日から全武(当社前首席運営官/首席業務官)と180 LSとの雇用関係の終了に合意した。契約終了により、双方は退職協定を締結し、これにより、会社はVuに合意した解散費を支払うことに同意し、計上された未払い、合意された健康保険料、計算すべき有給休暇
を含み、総額は#ドルとなった
グレン·ラーソン相談契約
同社は、2023年2月22日にグレン·ラーソンとコンサルティング·サービスを提供する契約を締結し、提供されたサービスの対価として#ドルをラーソンさんに賠償することに同意しました
F-57