アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
本財政年度末まで
あるいは…。
1934年証券取引法第13条又は15(D)条に基づいて提出された |
手数料書類番号
(登録者の正確な氏名はその定款に記載)
(明またはその他の司法管轄権 会社や組織) |
(税務署の雇用主 識別番号) |
(主にオフィスアドレスを実行) |
(郵便番号) |
登録者の電話番号は市外局番を含んでいます
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
|
取引 記号 |
|
登録された各取引所の名称 |
|
|
|||
|
|
同法第12条(G)に基づいて登録された証券:ありません
登録者が証券法規則405で定義されている経験豊富な発行者である場合は、再選択マークで示してください。はい、そうです ☐
登録者が当該法第13条又は第15条(D)に従って報告書を提出する必要がないか否かを、再選択マークで示す。 はい、そうです☐
登録者が(1)過去12ヶ月以内(または登録者がそのような報告の提出を要求されたより短い期間)に、1934年の証券取引法第13条または15(D)条が提出を要求したすべての報告書を再選択マークで示すかどうか、および(2)過去90日以内にそのような提出要求を遵守してきた。
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
大型加速ファイルサーバ |
|
☐ |
|
ファイルマネージャを加速する |
|
☐ |
|
|
|
|
|||
|
☒ |
|
規模の小さい報告会社 |
|
||
|
|
|
|
|
|
|
|
|
|
|
新興成長型会社 |
|
|
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる☐
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する☐
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に第240.10 D−1(B)条に従って受信されたインセンティブベースの補償に従って回復分析を行う必要があるかどうかを再選択マークで示す☐
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)。はい、そうです☐違います
A類普通株2022年6月30日のナスダック市場での終値によると、登録者の非関連会社が保有する投票権と無投票権普通株の総時価は1ドルである
2023年3月27日現在、登録者のA類普通株流通株数は
引用で編入された書類
登録者は,2023年株主総会の最終委託書又は委託書の一部を引用して本年度報告のForm 10−K第3部に組み込む。委任状は,登録者が2022年12月31日までの財政年度の120日以内に米国証券取引委員会に提出される。
カタログ表
|
|
ページ |
第1部 |
|
|
第1項。 |
業務.業務 |
1 |
第1 A項。 |
リスク要因 |
37 |
項目1 B。 |
未解決従業員意見 |
72 |
第二項です。 |
属性 |
72 |
第三項です。 |
法律訴訟 |
72 |
第四項です。 |
炭鉱安全情報開示 |
73 |
|
|
|
第II部 |
|
|
五番目です。 |
登録者普通株市場、関連株主事項及び発行者による株式証券の購入 |
74 |
第六項です。 |
[保留されている] |
74 |
第七項。 |
経営陣の財務状況と経営成果の検討と分析 |
75 |
第七A項。 |
市場リスクの定量的·定性的開示について |
86 |
第八項です。 |
財務諸表と補足データ |
86 |
第九項です。 |
会計と財務情報開示の変更と相違 |
88 |
第9条。 |
制御とプログラム |
88 |
プロジェクト9 B。 |
その他の情報 |
89 |
プロジェクト9 Cです。 |
検査妨害の外国司法管轄権を開示する |
89 |
|
|
|
第三部 |
|
|
第10項。 |
役員·幹部と会社の管理 |
90 |
第十一項。 |
役員報酬 |
90 |
第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
90 |
十三項。 |
特定の関係や関連取引、取締役の独立性 |
90 |
14項です。 |
最高料金とサービス |
90 |
|
|
|
第4部 |
|
|
第十五項。 |
展示·財務諸表明細書 |
91 |
プロジェクト16 |
表格10-Kの概要 |
94 |
2021年7月16日、私たちは、2021年1月8日の日付のいくつかの合併プロトコルおよび再構成計画または合併合意に基づいて、以前に発表された合併を完了しました。合併プロトコルは、私たち、私たちの完全子会社、およびCelularity LLC(前身はCelularity Inc.)またはLegacy Celularityによって完了します。
合併合意の条項によると、吾らは以下のように業務合併を完了している:(A)吾らの完全合併付属会社はLegacy Celulityと合併し、Legacy Celulityは吾等の完全合併付属会社として存続している;および(B)第1回合併に続いて、同一全体取引の一部として、Legacy Celulityと第2完全合併付属会社は第2完全合併付属会社に合併し、この第2完全合併付属会社は吾などの全額直接付属会社となり、最終的にLegacy Celulityは吾などの完全直接付属会社となる。この等合併を“合併”と呼び,合併プロトコルで述べた他の取引と総称して“業務統合”と呼ぶ.締め切りには,我々の名前をGX Acquisition Corp.からCelulity Inc.に変更する.
文脈が別に説明されていない限り、本年度報告で言及した“会社”、“Celulity”、“私たち”および類似タームはいずれもCelulality Inc.(F/k/a GX Acquisition Corp.)を指すLegacy Celularityを含むその合併子会社。“GX”系とは、企業合併完了前の前身会社を指す。
本年度報告に登場するCelularityロゴ、Celularity Impact、Biovance、Biovance 3 L、Interfyl、Lifebank、CentaFlex、Celularity Inc.の他の商標またはサービスマークは、いずれもCelularity Inc.の財産である。本Form 10-K年度報告には、他社の登録マーク、商標、商号も含まれている。本明細書に記載されている他のすべての商標、登録マーク、および商号は、それぞれの所有者の財産である。
i
リスク要因をまとめる
私たちの業務は重大な危険と関連がある。以下は我々の業務が直面する重大なリスクの概要であり,これらのリスクは我々の証券への投資に投機的かつリスクを持たせている.この要約はこのようなすべての危険を扱っていない。以下、本年度報告表格10−K第I部第1 A項の“リスク要因”というタイトルの下で、これらのリスクについてより全面的に説明する。私たちの証券に投資決定を下す前に、あなたはこのような危険を慎重に考慮しなければならない。以下に述べる任意のイベントまたは開発の発生は、当社の業務、経営結果、財務状況、見通し、および株価に重大な悪影響を及ぼす可能性があります。この場合、私たちの証券の市場価格は下落する可能性があり、あなたは投資の全部または一部を失うかもしれません。また、以下に説明しない他のリスクもあり、これらのリスクは私たちが現在知らないことか、現在重要ではないと考えているかであり、これらの追加的なリスクは、私たちの業務、運営、またはAクラス普通株の市場価格にも大きな影響を与える可能性がある。
II
三、三、
前向き陳述に関する特別説明
本年度報告におけるForm 10-Kに関する部分表現は前向き表現であり、改正後の1933年“証券法”第27 A節又は“証券法”、1934年“証券取引法”第21 E節又は“取引法”の規定に適合している。展望的陳述は、予想、信念、予測、未来計画および戦略、予期されたイベントまたは傾向、および非歴史的事実に関連する同様の表現に関する。これらの陳述は私たちの未来の事件と関係があり、私たちが期待している運営、研究、開発と商業化活動、臨床試験、経営結果と財務状況を含む。これらの展望性陳述は既知と未知のリスク、不確定要素とその他の要素に関連し、私たちの実際の結果、表現或いは成果と展望性陳述の明示或いは暗示の任意の未来の結果、表現或いは成果とは大きく異なる可能性がある。前向きな陳述は、以下の態様に関する陳述を含むことができるが、これらに限定されない
場合によっては、これらの前向き陳述は、“予想”、“信じ”、“可能”、“予想”、“継続”、“可能”、“推定”、“予想”、“予測”、“意図”、“可能”、“可能”、“展望”、“計画”、“可能”、“潜在”、“予測”、“プロジェクト”、“求める”などの用語を使用することによって識別することができる。“すべき”、“努力”、“目標”、“将”、“将”およびこれらの語または他の類似語またはフレーズの否定バージョンは、これらの語がないことは、宣言が前向きでないことを意味するわけではない。これらの陳述は、私たちの現在の未来の事件に対する見方を反映しており、仮説に基づいており、リスクと不確定要素の影響を受けている。このような危険と不確実性を考慮して、あなたはこのような前向きな陳述に過度に依存してはいけない。このForm 10−K年次報告では、その多くのリスクを“リスク要因”と“経営陣の財務状況と経営結果の議論と分析”というタイトルでより詳細に議論している。展望性陳述自体はリスクと不確実性の影響を受けるため、その中のいくつかのリスクと不確定性は予測できないか定量化されており、いくつかは私たちが制御できないので、あなたは未来の事件の予測としてこれらの展望的陳述に依存してはならない。著者らの展望性陳述に反映された事件と状況は実現できない或いは発生できない可能性があり、実際の結果は展望性陳述中の予測結果と大きく異なる可能性がある。
しかも、私たちは持続的な環境で運営している。新たなリスク要因や不確定要因が時々出現する可能性があり、管理職がすべてのリスク要因や不確定要因を予測することは不可能である。あなたはこのForm 10-K年次報告書と私たちがForm 10-K年次報告書で引用した文書を完全に読んで、私たちの未来の実際の結果が私たちが予想していたものと大きく異なるかもしれないことを理解しなければなりません。私たちはこのような警告声明を通じて私たちのすべての展望的声明を限定する。法的要件が適用されない限り、私たちは、任意の新しい情報、未来のイベント、状況の変化、または他の理由による、本明細書に含まれる任意の前向きな陳述を公開または修正するつもりはありません。前向き陳述に過度に依存しないように注意し,これらの前向き陳述は本年度報告がForm 10−Kの形で発表された日にのみ発表された。
四
第1部
プロジェクト1.BU無邪気ですね。
概要
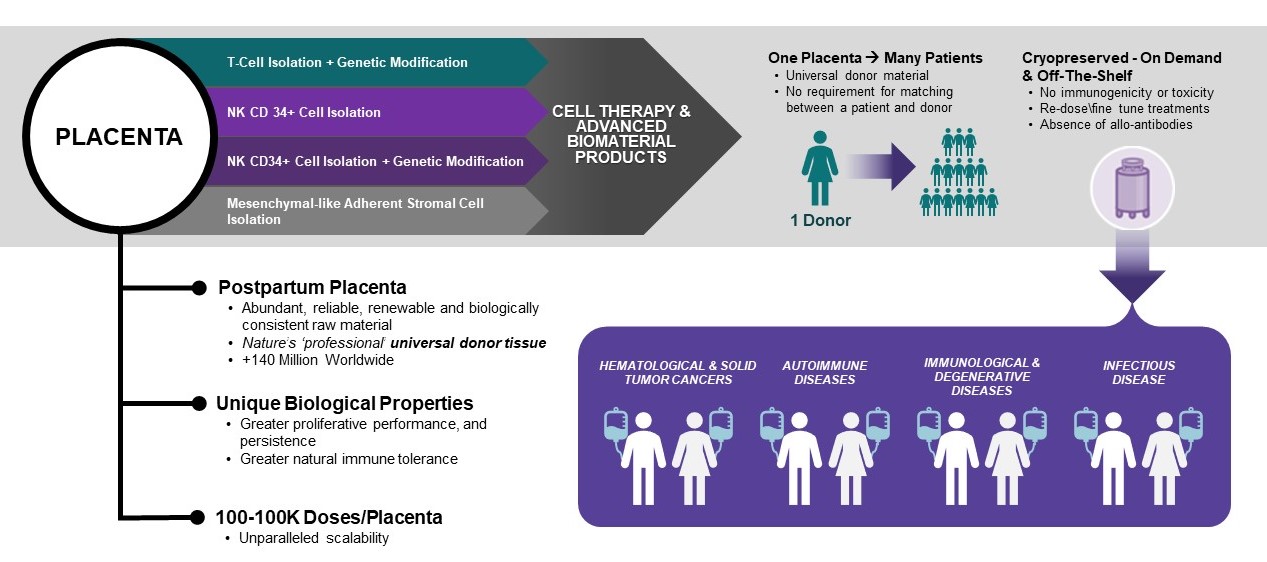
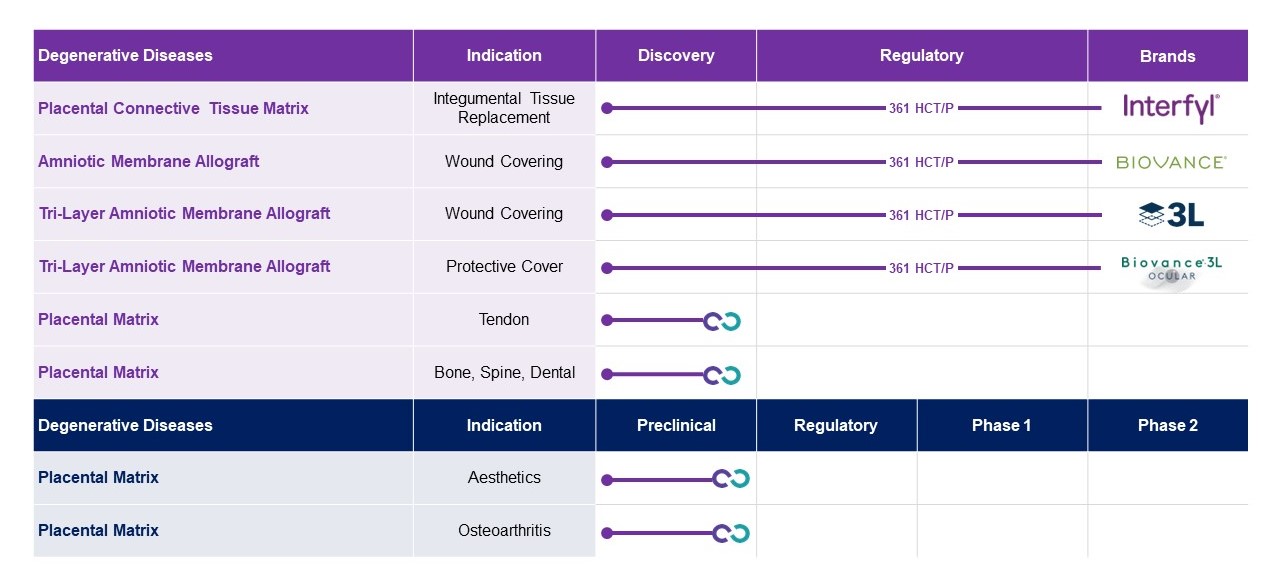
著者らは臨床段階の生物技術会社であり、既製の胎盤由来同種異体細胞療法を開発することによって癌、免疫と伝染病を治療し、細胞医学の次の発展をリードする。著者らはキメラ抗原受容体(CAR)、ナチュラルキラー細胞(NK)を持つT細胞、間葉系様接着基質細胞(MLASCs)と外体を含む一連の既製胎盤由来同種異体細胞治療候補製品を開発している。これらの候補治療薬は癌,感染症,退行性疾患に対する適応である。胎盤独特の生物学的特性と既製の獲得性を利用することによって、著者らは治療解決策を開発し、全世界の有効、獲得と負担できる治療薬物に対する重大な需要を満たすことができると信じている。胎盤から抽出した生体材料製品も積極的に開発·販売している。2023年までに、私たちは国内でこれらの製品を販売して、主に整形外科と傷口看護市場にサービスします。我々は現在,米国以外で胎盤生体材料を販売する予定であり,最初の重点は中東と北アフリカ市場である。私たちの今日の生体材料業務は主に私たちの流通ネットワークを介して私たちのBiovanceとInterfyl製品を販売することです。Biovanceは健康,臨月妊娠の胎盤から抽出した脱細胞,脱水したヒト羊膜である。それは無傷の天然細胞外基質であり、傷口の再生過程に基礎を提供し、機能組織の修復にステントを提供した。Interfylはヒト結合組織基質であり、健康、臨月妊娠の胎盤から来ている。それは各種の医学専門家によって創傷、創傷或いは手術による軟組織欠損を埋めるために使用されている。我々はBiovanceやInterfyl以外のビジネスチャネルを深化させるために新たな胎盤生体材料製品を開発している。細胞治療開発·製造における我々のコア専門知識を利用して,第三者に契約製造と開発サービスを提供することで収入を創出する予定である。この新サービスの最初のポイントは,開発段階の細胞治療会社の臨床試験のための治療候補薬の開発·製造に協力することである。2023年1月、私たちは仕事の優先順位の再調整を発表し、2023年3月現在、従業員数は約3分の1に減少した。
著者らのCelulality Impactプラットフォームは胎盤由来細胞の優勢を利用して多種の疾病に対して、そして私たちがアメリカのために建設した147,215平方フィートの施設で生物由来から凍結保存と包装を製造するまでの同種異体細胞のシームレスな統合を提供する。科学的および経済的観点から,完全に健康なインフォームドコンセントドナーからの胎盤由来の胎盤由来細胞を使用することは,潜在的な固有の利点を有すると信じられている。まず,胎盤由来細胞は成人由来細胞よりも強い乾性を示し,拡張·維持能力を有することを意味している。次に,胎盤由来細胞は免疫学的にナイーブであり,これらの細胞が特定の抗原に接触したことがないことを意味し,移植過程で毒性が低く,移植片対宿主病(GvHD)の可能性が低く,甚だしきに至ってはないことが示唆された。第三に、私たちの胎盤由来細胞は同種異体であり、これは、それらが任意の患者に使用されることを意味し、自己体細胞は単一の患者から得られ、その患者のみの使用のために使用されることを意味する。これは,既製の治療をより迅速,より信頼性,より大規模に多くの患者に提供できる重要な違いであると考えられる。
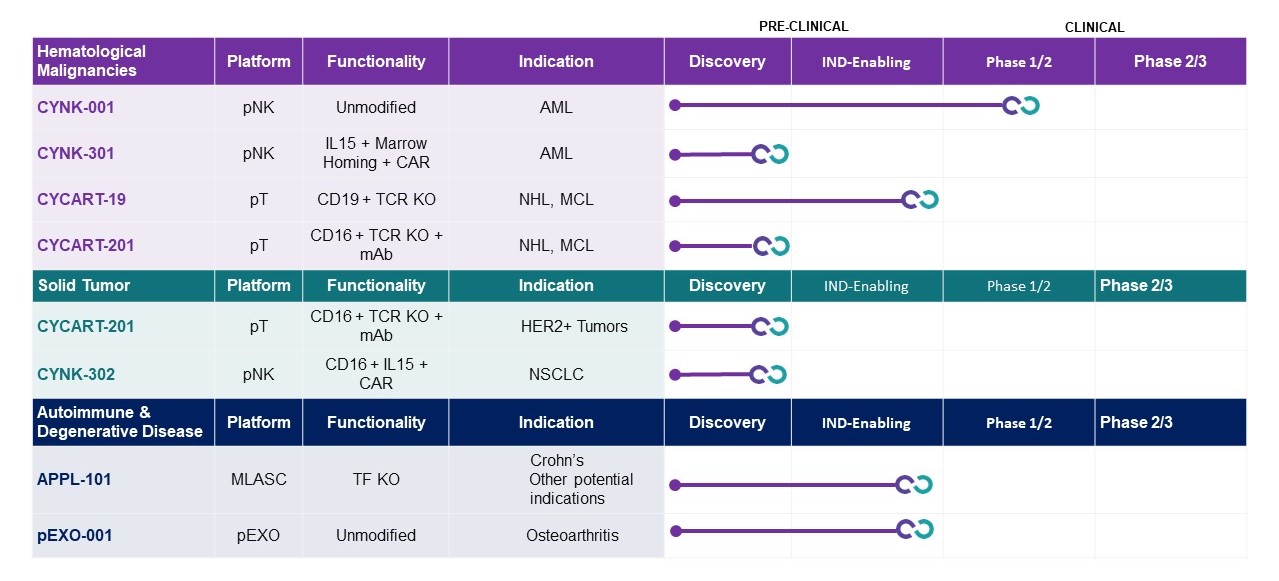
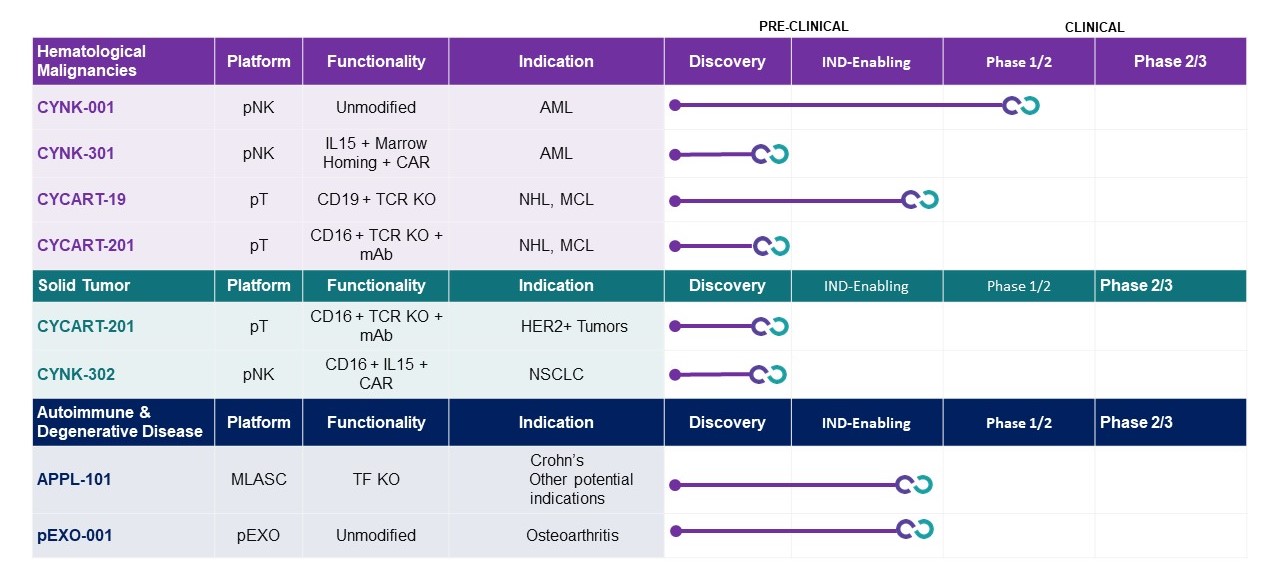
産後ヒト胎盤という単一由来の材料から、著者らは5種類の同種異体細胞或いは細胞外小胞タイプ:T細胞、未修飾NK細胞、遺伝子組換えNK細胞、MLASCsと外切体を獲得し、それらは7つの重要な細胞治療方案であるCyCART-19、CyCART-201、CYNK-001、CYNK-301、CYNK-302、APPL-001とpEXO-001に応用された。CyCART−19は胎盤由来のCAR−T細胞療法であり,B細胞悪性腫瘍の治療に開発されており,最初は分化クラスター19あるいはCD 19受容体が対象であり,その構造や関連CARSはソレントから許可されている。2022年第1四半期に、CyCART-19をB細胞悪性腫瘍の治療にINDを提出し、2022年5月下旬にFDAから正式な書面通知を受け、より多くの情報を提供し、その後、計画された1/2期臨床試験を継続することができた。私たちはFDAと協力して、その問題をできるだけ早く解決するために努力している。INDがFDAの承認を得て十分な資金が利用可能であれば,2023年下半期に試験を開始する予定である。われわれの遺伝子組換えT細胞である非ホジキンリンパ腫(NHL)や固形腫瘍においてもCyCART−201を行い,T細胞受容体(TCR)を有するCD 16を発現し,モノクローナル抗体(MAbs)に結合してノックアウトする。Cynk−001は胎盤由来の未修飾NK細胞である。2022年には、急性骨髄性白血病(AML)と多形性膠芽腫(GBM)の治療のための積極的かつ承認された臨床試験が開発されており、前者は血液癌であり、後者は固形腫瘍癌である。Cynk-001は現在AMLのアクティブな第1段階実験中である.NK療法が再発難治性AML(RrAML)を治療する際に直面するいくつかの挑戦を克服する可能性がある次世代CAR−NKとしてCYNK−301を推進する。企業資源の優先順位を決定する必要があるため,2023年1月にGBM試験における採用を停止する予定であることを発表した。しかし、私たちは私たちの固形腫瘍研究計画を引き続き推進するつもりだ。Cynk-302は、固形腫瘍において開発されている次世代CAR-NKであり、最初は非小細胞肺癌(NSCLC)に集中しており、これは持続的に高度に満たされていない需要領域である。APPL−001は胎盤由来のMLASCであり,クローン病や他の変性疾患の治療のために開発されている。PExo−001は胎盤由来の外切体であり,骨関節炎治療のために開発されている。
我々のCelulality Impact製造プロセスは、独自の処理方法CELLを使用することによって、完全に健康なインフォームドコンセントドナーから胎盤を取得する速度と拡張性を最適化することを目的としたシームレスで完全に統合されたプロセスである
1
選択的に、製品固有の化学、製造および制御、またはCMC、先進的な細胞製造および凍結保存。その結果、同種異体在庫の準備が完了し、必要に応じて胎盤由来細胞治療製品である。私たちはまた、第三者のためのいくつかの出生副産物の収集、加工と低温貯蔵を含む商業バイオバンク業務を経営と管理している。
私たちの現在の科学は私たちの経験豊富な管理チームが二十数年間蓄積してきた背景と努力の産物です。私たちは人間遺伝会社に根付いています。これは私たちの創業者で最高経営責任者のロバート·J·ハリリ医学博士が1998年にLifebankの名で設立した会社で、2002年にCelgene CorporationやCelgeneに買収されました。このチームはCelgeneで胎盤由来技術分野の専門知識を磨き続け,2017年8月までAnthrogensisを買収した。私たちは1500以上の特許と特許出願、私たちのCelularity Impactプラットフォーム、私たちの技術、技術、および現在の重要な細胞治療計画を保護する強力なグローバル知的財産権の組み合わせを持っている。このような専門知識,専門知識,知的財産権は,これらの潜在的な救命療法の迅速な開発を推進し,承認されれば,満たされていない医療ニーズを満たすために商業化されると信じている。
私たちのパイプは
我々のCelulality Impactプラットフォームを利用して、我々は4種類の胎盤由来の同種異体細胞タイプ:T細胞或いはPT、未修飾NK細胞(CYNK-001)、遺伝子組換えNK細胞(CYNK-301、CYNK-302)とMLASCsがある。胎盤由来の接着細胞外体、あるいはpEXOと呼ばれることも研究されている。
著者らは引き続き微小残留病陽性(MRD+Ve)とrrAMLでCYNK-001の第一段階試験を行ったが、著者らもrrAMLと固形腫瘍において新世代改良NKプラットフォームの進展を得た。
Cynk-301は次世代CAR-NKであり、増殖、持続性および有効性を最適化しながら、リンパ枯渇の負担を最大限に減少させることを含む、rrAMLを治療する際にNK療法が直面するいくつかの挑戦を克服する可能性がある。Cynk-301は、NK細胞の活性化、増殖および持続性を増強するために、膜結合インターロイキン15またはIL 15を結合し、それに加えて、骨髄ホーミングおよび標的CARはさらに治療効果を増強する。
Cynk-302は、固形腫瘍において開発されているCAR-NKであり、最初は非小細胞肺癌に集中しており、持続的に高度に満たされていない需要領域である。Cynk-302はCYNK-101を学習した上で構築した次世代構造である.それは、トランスジェニックによってCD 16を発現し、統合膜結合IL 15および不開示標的CARによってさらに増強される。
われわれは引き続きわれわれのCyCART−19計画を進めて臨床試験を行い,潜在的な一流あるいは最適な構造を有するわれわれのT細胞プラットフォームの開発を継続した。CyCART-201は多種の潜在的モノクロナル抗体と連合して使用するために設計され、広範な治療潜在力を持っている。CyCART−201は遺伝子改変により,TCRノックアウトを有するCD 16を発現することができた。著者らの最初の血液学的発展は非ホジキンリンパ腫と著者らの固形腫瘍で発展したヒト表皮増殖因子受容体2陽性、或いはHER 2+Ve腫瘍であり、両者は標的モノクロナル抗体と結合する。
戦略的検討の後、私たちは自己免疫と退行性疾患資産の発展に再集中した。APPL-01は遺伝子組換えMLASCであり、クローン病(CD)において初歩的な研究が行われており、私たちが以前CDで見た未修飾MLASC試験で見られた鼓舞的なシグナルを確立する。著者らは引き続き骨性関節炎の方面で進展を得て、骨性関節炎はまだ大量の満足されていない需要と市場潜在力がある。
2
TCRKO=T細胞受容体ノックアウト,TFKO=組織因子ノックアウト,MCL=マントル細胞リンパ腫
さらに、私たちは私たちの胎盤由来生体材料製品のパイプラインを拡大することを求めている。
CELULATY衝突プラットフォーム
胎盤由来細胞療法は、満足されていない医療ニーズの患者に潜在的な救命療法を提供する。著者らはすでに胎盤幹細胞の収集、加工と貯蔵の特許技術を開発し、獲得し、これらの技術は癌、伝染病と退行性疾患において潜在的な広範な治療応用を持っている。
Celulality Impactプラットフォームを使って開発しましたI私は知らない-MおいしいのはPLacenta派生A病原性のCEllTヘラピスです。胎盤独特の生物学的特性と既製の獲得性を利用することによって、著者らは治療解決策を開発し、全世界の有効、獲得と負担できる治療薬物に対する重大な需要を満たすことができると信じている。
我々のCelulality Impact製造プロセスは、独自の加工方法、細胞選択、
3
製品に固有のCMC、先進的な細胞製造および凍結保存、および同種異体在庫準備および必要に応じた胎盤由来細胞治療製品をもたらす。完全に統合されたプロセスは、私たちが専門的に建設した製造、転化研究、バイオバンク施設にある。
著者らのCelulality Impactプラットフォームは著者らの集成プロセスと胎盤由来の同種異体細胞の独特な生物学的特性を利用して、多種の疾病、癌、伝染病と退行性疾患の適応を含む。このプラットフォームは患者への治療の速度を加速し、同時に低い収入コストで高品質と純胎盤由来の細胞治療製品を製造することを確保することを目的としている。必要に応じて医師に細胞治療リストを提供し,必要な患者を治療し,再投与レジメンを許可すべきであり,他の細胞治療プラットフォームでは支持できないと考えられる。
私たちの戦略
我々の目標は,既製の同種細胞療法を提供することにより,より大きな規模とより高い品質で,魅力的な経済効果で細胞医学の次の進化をリードすることである。この目標を達成することは,胎盤由来の同種細胞療法を癌,感染症,退行性疾患の様々な適応の標準的な看護とし,世界各地のより多くの患者が潜在的な救命療法を容易に得ることができると信じている。私たちは次のようにこの使命を実現する予定です
4
私たちのチームと会社の歴史は
人類起源会社
私たちは人類の起源に根ざしています。これは私たちの創業者で最高経営責任者のロバート·J·ハリリ医学博士が1998年にLifebankの名で設立した会社です。我々のように,ヒト起源は,癌,変性疾患,感染症の治療のための胎盤由来幹細胞を用いた細胞療法の開発と提供に専念している。Celgeneは2002年12月にAnthrogenensisを株式交換合併で買収し,Celgeneの完全子会社であるCelgene Ccell TreeuticsやCCTの形でAnthrogenensisを運営した。同様に,CCTは胎盤由来幹細胞の研究と開発に専念し続けている。2016年、ハリリ博士はCelulityを設立し、私たちの今日の業務を構成する資産の買収を開始した。その中には我々の変性疾患とバイオバンク業務が含まれており,Celgeneはヒト長寿会社(HLI)に売却されており,HLIはハリリ博士と我々の取締役の一人であるディマンディス博士が共同で設立したゲノムベースの健康情報会社,および我々のコア細胞療法事業であり,我々は2017年8月にCelgeneからAnthrogenensisを買収し,株やイベント駆動あるいは価値権(CVR)と交換した。
Celgene Corporation(百時美施貴宝に買収された)
許可協定
2017年8月,Anthrogenensisの買収についてCelgeneとライセンス契約を締結した。Celgeneとの許可によれば、Celgeneには、Celgeneの許可の日から存在するAnthrogenensisの既存の知的財産権またはCelgeneが合併に関連する任意の移行サービス活動について開発された、臨床前研究目的のための、臨床前研究目的のための、および任意の自動車の製造、任意のT細胞またはNK細胞を変更して、これらの自動車に関連する製品およびサービスを表現するための開発、製造、商業化、および/または再許可可能な任意の目的のために、Celgeneとの許可に基づいて、Celgeneに世界的に範囲的な、印税免除された、十分な非独占的許可が付与されている。一方が合意に深刻に違反している場合、または他方の資金が償還されていない場合には、Celgeneライセンスを終了することができる。
あるいは価値のある権利
二零一七年八月、Anthrogenensis買収事項について、私たちはCelgeneに合併費用として私たちのXシリーズ優先株株式を発行し、Celgeneと契約または価値のある合意、またはCVR協定を締結した。CVR協定に基づき、吾らは買収中にCelgeneに発行されたXシリーズ優先株について株式1株当たりCVRを発行した。このようなCVRは、初公募または当社が売却された場合を除き、Xシリーズ優先株の株式と分離してはならない。
CVRプロトコルはCVRの保有者に各計画に基づいて、我々のいくつかの研究治療計画について合計5000万ドルの規制マイルストーンと合計1.25億ドルの商業マイルストーン支払いを得る権利があり、これらの計画は現在のCYNK-001、CYNK-101、PDA-002候補パイプライン、および伝統的なPDA-001計画を含む
5
(胎盤からの接着細胞であって、静脈注射のためのヒト発育固有のものであり、もはや発育中ではない。CVRプロトコルの項におけるこのような支払いは、PNK-007(特定のヒト遺伝子固有NK細胞を含み、ヒト遺伝子取引完了時にヒト遺伝子によって所有されるプロセスによって製造される)およびいくつかの遺伝子改変PNK-007細胞(CARを含むが、キメラ受容体を有するナチュラルキラー細胞を含まない)、および上記のいずれかの任意の誘導製品、部分、亜部分または子孫、またはヒト遺伝子取引完了時に存在する任意の関連開発計画に基づく任意の治療薬を明示的にカバーする。したがって,我々のNK細胞型特許経営権を新たな適応に拡張することに伴い,一般的な事項として,これらの支払いは開発の後期になるため,CVRプロトコルにおける具体的な条項に基づいて現在と将来の候補治療案を評価し,このような支払いの具体的な療法を決定していく予定である。また,このような計画や例年ごとに,CVR保有者は,その計画治療薬の年間純売上高の10分の1に相当する使用料を得る権利があり,この計画された治療薬が特定の国で初めて商業販売された日から最近の満了日まで,その計画治療薬をカバーする任意の有効特許主張がその国/地域での有効特許権が満了した日,このような治療薬の市場独占経営権がその国で失効した日から2027年8月(すなわちAnthrogensis買収終了10周年)までとなる。今まで、CVR協定に基づいて何のお金も支払われなかった。
“投資家権利協定”と“投資権協定”
私たちも二零一七年八月にCelgene及びいくつかの他の契約者とAnthrogenensisの買収について投資家権利協定と投資権協定を締結した。これらの合意に関するより多くの情報は、項目13を参照されたい“特定の関係や関連取引、および取締役独立許可その他の合意この年間報告書です。
同種異体胎盤由来細胞
生体材料コレクション
著者らの4種類の同種異体細胞の最初の由来材料は産後ヒト胎盤である。我々は,認可された病院と分娩センターから我々の製品を生産するためのヒト胎盤分娩材料を調達し,許可されている医療専門家が収集した。ドナーの資格はドナースクリーニング過程に依存し,ドナー計画に関する教育,ドナーのインフォームドコンセントの取得,詳細な妊産婦健康アンケートと家庭健康歴の記入が含まれている。これらの用紙はドナーが記入し,必要に応じて訓練された収集技術者の協力を得た。出産材料を提供するドナーは何の費用もかからず、再支払いもしない。
許可された医療専門家は、生体材料(臍帯血、胎盤および産婦血液サンプル)のバーコードタグおよび適切な保護チェーンファイルを含む、我々独自の収集キットを使用してドナー材料を収集する。収集されると、寄付された材料と母体血液サンプルは絶縁容器に入れられ、宅配便でニュージャージー州フロラム公園にある実験室と製造工場に運ばれる。
我々の施設に到着すると、寄付された材料は、バーコードキットのラベルの完全性および正確性を検査し、検証されたソフトウェアデータベースに電子的に符号化される。すべての品質基準に適合する場合、寄付された材料は、個別に評価され、適切な生産キットに転送されて加工および製造される。私たちの調達は迅速に拡張可能であると信じています。大量の調達関係を構築し、現在と未来の製造需要を満たすために持続可能な再生可能な供給を提供しているからです。
胎盤由来細胞のユニークな生物学
胎盤由来の細胞は独特な免疫幼稚、乾性、持続性と増殖に関連する生物学的特性を有し、これによりそれらは1種の生物選択の出発材料になり、成人骨髄或いは末梢血由来細胞と比べ、毒性がもっと低く、生物活性がもっと高い潜在力を持っている。
研究により、ヒト胎盤は1種の新しい、価値のある間葉系と造血由来の多能性幹/前駆細胞源であり、多種の治療応用を持っている。我々の同定データは、胎盤由来細胞の約1%~5%がCD 34+造血幹細胞であることを示しており、いくつかのマーカーの発現は、このような造血幹細胞がより多くの自己更新能力を有し、胎盤由来細胞の早期移植を促進する可能性があることを示している。そのほか、さらなる同定により、T細胞含有量が低く、T亜群が成熟していないことが示唆された。このことは免疫学的に幼稚であることを証明しており,さらに移植における移植片対宿主病や移植片対宿主病の可能性は低いかないことが示唆された。また、間葉系様細胞は他の特徴、能力と作用を持っている(例えば:骨形成、軟骨形成、脂肪形成分化能力と免疫調節作用)。大量の間葉系様細胞とTreg細胞は胎盤由来の細胞がGvHDの予防と宿主微環境の調節に役立つ可能性があることを表明した。つまり木の幹は
6
胎盤由来細胞の潜在増殖能力と持続性は、著者らが開発しているものを含む多くの潜在的な治療応用を支持する。
著者らはまた胎盤由来の外切体を研究し、潜在的な治療応用に応用している。細胞外体は細胞外小胞であり、細胞間のコミュニケーションチャネルとして、受容体細胞の機能変化を引き起こす。外体は特定の貨物内容物を受容体細胞に転移することによって細胞間の通信を実現し、microRNAs或いはmiRNAsを伝達することによって受容体細胞のエピジェネティクス変化を実現することができる。エキソソームはすべてのタイプの幹細胞において傍分泌効果を検出する主要な要素として決定され、遺伝物質を幹細胞から再生を必要とする組織特異的細胞に転移させる責任がある。外切除体はすでに強力な再生潜在力を有することが証明され、免疫調節特性と抗炎症特性を含む。PEXOという外体を発見しましたPEXOは増殖因子、デオキシリボ核酸或いはDNA、断片、miRNAsとメッセンジャーRNAを豊富に含み、独特なマーカーを示し、それらを他の胎盤由来ではない接着細胞の外体と区別した。精製したpEXOを医薬組成物に調製し,ヒト投与に用いて血管新生および/または血管形成を促進し,免疫活性を調節し,組織損傷を修復することを検討している。
CAR−T細胞の概要
白血球は免疫系の構成要素であり,感染性病原体や他の外来物質から身体を防御することを担当している。T細胞は白血球であり、癌細胞を含む感染または異常細胞を感知および死滅させ、免疫反応における他の細胞の活性化を調整する。
成人末梢血単核細胞或いはPBMC由来のT細胞と異なり、胎盤由来のT細胞の多くは幼稚であり、比較的に早い分化表現型を維持することができ、例えばもっと多くのナイーブ/記憶マーカーと比較的に低い発現の効果/不全マーカーを維持することができる。これらの特性はこれらの細胞により大きな増殖潜在力を持たせる離体するそれは.周知のように、胎盤由来のT細胞は更に強い免疫耐性を有し、損傷した同種異体の活性化を示し、深刻なGvHDの発生率を下げることに役立ち、これはそれらを1種の魅力的な細胞集団にし、同種異体養子細胞治療に応用する。著者らはすでに胎盤由来のT細胞を分離、形質導入と増幅し、“既製”同種異体のCAR-T細胞を産生する信頼できる方法を開発した。
同種異体ヒト胎盤T細胞は健康なドナー胎盤に由来する。凍結保存する前に,単球分離法を用いて単核細胞を分離し,胎盤T細胞を分離した。我々の同種CAR−T細胞製品は、まず、単離された胎盤T細胞を解凍および活性化し、次いで癌に対するCAR構造のウイルス形質導入、およびGvHDのリスクを最小限に低減するための追加の遺伝子改変工程を含む。形質導入およびトランスフェクトされると、CAR-T細胞は、収穫、最終調製、および凍結保存細胞治療の前に増幅され、大量のこのような細胞を産生する。
ナチュラルキラー細胞の概要-未修飾と遺伝子組換え
NK細胞は天然免疫系の有効なエフェクター細胞であり、異常とストレスの宿主細胞の識別と除去を担当する。これらはNK細胞特異的活性化受容体を備えており、細胞ストレス誘導保存抗原を認識しながら、阻害性受容体と共に調節して、誤って健康細胞に対して標的化されないようにすることができる。NK細胞はウイルス感染と抗腫瘍免疫を媒介する上で特に重要であり、抗腫瘍免疫において、正常な細胞過程を強調し、ウイルス感染と癌細胞増殖を維持する。
NK細胞療法の商業化はずっと大規模化されて臨床用量に応用された成熟NK細胞を生産する難度とコストの制限を受けている。我々のCelulality Impactプラットフォームを用いて,我々の独自の流れは,胎盤由来幹細胞を35日間で増幅しNK細胞に分化させることにより,これらの制限を緩和した。著者らは健康なドナー胎盤からHSCsを抽出し、その後、これらの細胞を増殖し、NK細胞に分化させた。この過程はすべてのドナーの胎盤に数百剤を生産することができる。胎盤HSCsの形質導入や下流で安定した遺伝子修飾CYNK細胞を産生することにより高い遺伝子修飾効率を実現する技術も開発し,これらの細胞は増強した癌傷害活性を有する。これらの細胞はその後凍結保存され,要求に応じて出荷できる。
我々の遺伝子組換えNK細胞の場合、私たちの同種修飾NK細胞製品は、単離された胎盤NK細胞の解凍および活性化から始まる。そして、レンチウイルスベクター形質導入を用いてNK細胞の効果機能を増強し、それらの腫瘍殺傷特性を維持した。我々の遺伝子組換えNK細胞は、抗体依存性細胞毒性またはADCCの潜在力を向上させるために、治療用モノクロナル抗体と組み合わせて使用することができると信じている。
MLASCの概要
胎盤由来のMLASCsは新しい、培養増幅された胎盤組織由来の間葉系様細胞群である生体内MLASCsの免疫調節特性は自己免疫を軽減し、抗炎症活性を有することを証明した。第1世代MLASCsの静脈と筋肉投与製剤が開発され、研究されている
7
クローン病,多発性硬化症,関節リウマチ,脳卒中,糖尿病足潰瘍,糖尿病末梢神経病変の臨床研究。我々は変性疾患の治療に次世代遺伝子組換えMLASCsを開発している。
同種異体ヒト胎盤MLASCsは健康ドナー胎盤に由来する。著者らの同種異体MLASC製品はまず分離した胎盤由来のMLASCsを解凍と活性化し、その後組織因子に対して遺伝子改造を行い、潜在的な毒性と副作用のリスクを減少させる。修正されると、私たちは収穫、最終処方、および細胞治療の凍結保存の前にMLASCsを大量に拡大した。
外切体研究概況
細胞外体は細胞から細胞外空間に放出される無細胞ナノレベル脂質二重層膜粒子であり,細胞と細胞,組織と組織,器官と器官の通信に重要な役割を果たしている。外切体は直径30−200 nmの晩期内切体から生成される。標的細胞と融合する時、外切体が携帯する分子貨物(蛋白質、脂質、DNA、mRNAとmicroRNAs)は細胞に挿入されて機能する。
最近,外切体は変性疾患の治療の有望な候補とされている。観察された一部の細胞治療作用は外切体を介していることが証拠されている。細胞療法と比較して,外切除療法は免疫原性が低い/免疫原性がない,保存しやすい,投与しやすいなどの利点がある。また,そのナノサイズにより,外切体は脳−血関門を通過することができ,細胞による療法よりも標的組織や臓器に広く輸送することができる。
PExo-001は1種のヒト産後胎盤由来の外切体産物であり、サイトカイン、ケモカインと成長因子から構成され、すでに再生と免疫調節活性を有することが報告されている。
同種細胞療法は既存の方法です
工学細胞療法には主に2つの方法がある:自己と同種異体。自己療法は個体患者からの工学細胞を用い,同種療法は無関係第三者健常ドナー由来細胞を用いた。著者らの人類胎盤由来同種異体プラットフォームは細胞医学の次の発展をリードしていると信じている。著者らの目標は既製の同種異体細胞療法を提供することであり、規模がもっと大きく、質が高く、経済的に魅力的で、世界各地のより多くの患者が生命を救う治療法を獲得しやすい可能性があるからである。
著者らのヒト胎盤由来の同種異体プラットフォームは現在胎盤CARR-T細胞(CyCART-19とCyCART-201)、NK細胞(CYNK-001、CYNK-301とCYNK-302)、MLASCs(APPL-001)と外体(pEXO-001)を含む。
Cycart
現在,自己CAR−T製品は白血球分離と呼ばれる過程で患者の血液からT細胞を分離することにより製造されている。特定のCARタンパク質を発現する癌標的構築物はウイルスによってT細胞に形質導入され、その後、工程T細胞は十分な数のT細胞が注入できるまで増殖される。そして,改造されたT細胞が臨床センターに運ばれ,患者に投与された。白血球分離から臨床センターまでの経過は約4週間を要した。自己方法は革命的であり、多くの患者の中で納得できる治療効果を示したが、著者らは静脈から静脈までの時間が長く、生産コストが高く、効力が可変と製造失敗の負担を受けた。
逆に、私たちの同種胎盤由来のT細胞は健康なドナーから来ており、彼らは厳格なドナースクリーニングと選択を経ている。投与は患者の細胞源と単一の薬物製品の拡張の制限を受けないため、製造された薬物製品は直ちに十分な量で患者に配置することができる。CyCART細胞は“既製品”の治療法として,必要に応じて患者に再投与する可能性も提供している。全世界に数億の健康な新生児がおり、胎盤は豊富で再生可能な健康リンパ細胞源を提供した。また、胎盤由来のT細胞は豊富な幹細胞記憶T細胞を含み、これらのT細胞は高度な増殖性と持続性を有する。胎盤T細胞は免疫特免性を有し,ドナーの宿主に対する毒性は低い(GvHD)ことが知られている。したがって、私たちは全体的により安全な細胞集団かもしれない。また,同種異体胎盤T細胞は遺伝子工学によりGvHDのリスクを最小限に抑え,患者の免疫系による破壊を回避することができる。したがって、CyCART細胞は有利な安全性を有する可能性があり、同時に患者において有効な腫瘍除去活性と持続性の持続性を提供する。
シンカー
同様に,自己NK細胞や遺伝子組換え自己NK細胞も免疫腫瘍学的研究に用いられている。NK細胞は細胞ストレスシグナルを識別することで癌細胞を直接殺傷し、移植片対宿主病のリスクは存在しない。しかし、体外末梢血由来NK細胞の増殖能力は限られており、通常白血病細胞系に基づく技術で生産を補助する必要がある。また、末梢NK細胞におけるCARベクターの伝達効率が低いため、自己CAR-NK細胞は技術的挑戦に直面していることが証明された。我々のNKプラットフォームは胎盤由来のHSCsを培養し、これらの細胞をNK細胞(CYNK)に分化させた。この過程はすべての胎盤ドナーに数百剤を生産することができる。高性能を実現する技術も開発しました
8
胎盤HSCsを形質導入することによって遺伝子修飾効率を高め、そして下流で安定した遺伝子修飾CYNK細胞を産生し、腫瘍殺傷活性を増強する。これらの細胞はその後凍結され,要求に応じて直ちに臨床管理部門に搬送されることができる。
MLASC
自己と異体骨髄或いは脂肪組織由来のMLASCsはすでにヒト臨床試験に応用されている。自己MLASC療法は、ドナー細胞に関連する有害事象がなく、細胞製品がドナー自身の細胞から由来するため、より少ない調節障害を含む利点がある。しかし,自己MLASC製品はドナーの遺伝性あるいは老化に関連する生物学的欠陥を持ち,治療価値を損なう可能性がある。また,多くの場合,患者に投与される前に自己体細胞は培養が必要であり,製造失敗のリスクがある。
逆に、同種異体MLASCsは高品質と柔軟に投与できる既製製品を提供することができる。MLASCsはその主要な組織適合性複合体I、II類蛋白の発現レベルが比較的に低いため、免疫特異体と考えられている。我々の胎盤組織由来のMLASCsは胎児由来であるため、より多くの免疫特権を有する可能性がある。また,APPL細胞はより高い増殖能を有するため,遺伝子操作により細胞を改造し,その機能を増強したり危険因子を軽減したりするために特定の機能を持たせるのに適していることが期待される。
候補治療パイプラインと発展戦略
我々は多くの胎盤由来の同種異体細胞治療候補薬を研究·開発しており,癌,感染症,退行性疾患の適応を治療している。単一由来の胎盤材料から、CAR-T細胞、未修飾NK細胞、トランスジェニックNK細胞、およびMLASCsの4つの同種細胞タイプに焦点を当てた。私たちもPEXOを研究しています。私たちの製品ラインは次の図のようになります
CYCART-19
著者らの胎盤由来のCAR-T細胞に基づく先導治療方案はCyCART-19であり、これはCD 19受容体を標的とする同種異体CAR-T細胞である。我々は重要な株主ソレントからCD 19受容体構築および関連CARSに関連する権利を獲得し、これらのCARSはCyCART-19において胎盤由来細胞および/または臍帯血由来細胞領域に使用されている。ソレントライセンス条項の説明および胎盤由来細胞および/または臍帯血由来細胞領域以外の私たちの権利については、タイトルを参照してください“ライセンス契約であるSorrento治療会社“。
現在FDAが承認しているすべてのCAR−T細胞療法,および約75%の臨床資産が開発されており,自己のものと推定されている。自己治療は末梢血由来T細胞がCARを発現する免疫細胞ベクターであり、患者を自分のドナーにすることを意味する。これらの自己CAR−T細胞療法を作製することは複雑で高価であり,静脈から静脈までの時間が長く,治療結果に影響する可能性がある。また,再発や難治性患者では,多輪リンパ球除去治療により分離細胞回復が一致しない。胎盤由来CAR-T細胞CyCART-19は分離能力の制限がなく大量生産のために設計され、必要に応じて交付される拡張可能な解決策だと信じています
9
既製の、冷凍保存された包装製品。そのほか、胎盤由来の細胞は豊富な幹細胞記憶細胞を含み、これはより大きな増殖潜在力とより長い持続性を与える体内にある.
臨床前データ
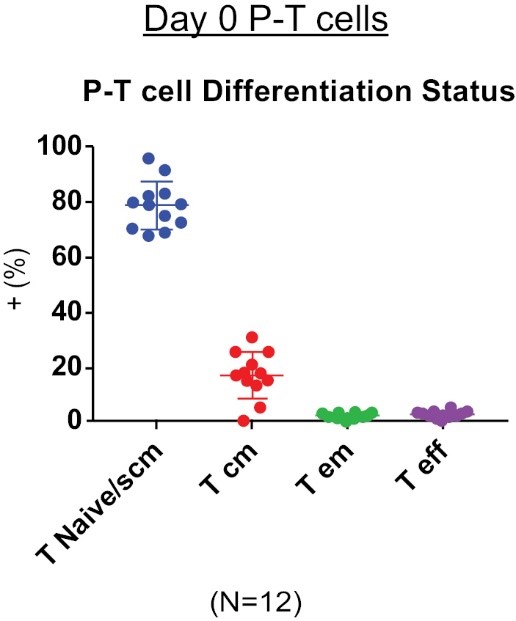
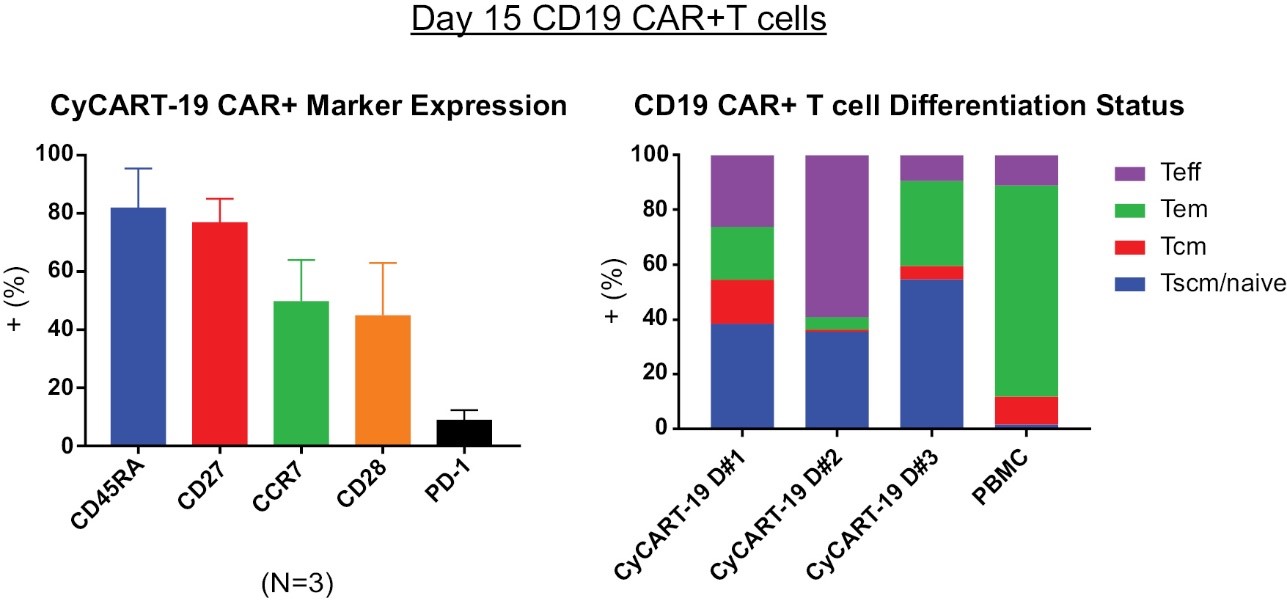
臨床前研究では,胎盤由来のT細胞は主にナイーブ/T幹細胞記憶細胞あるいはT SCMからなることが証明され,一部の中央記憶T細胞もあり,CyCART−19の出発材料を構成している。実験室で遺伝修飾と増殖/増幅を行った後、CyCART-19細胞は高レベルのナイーブ/記憶マーカーと低レベルの免疫抑制分子PD-1を発現した。また,CyCART−19細胞はPBMC由来のCD 19 CAR+T細胞に比べて高いT SCMの割合を維持しており,より大きな自己更新,増殖潜在力,リンパホーミング,より強い持続能力を意味している体内にある.
10
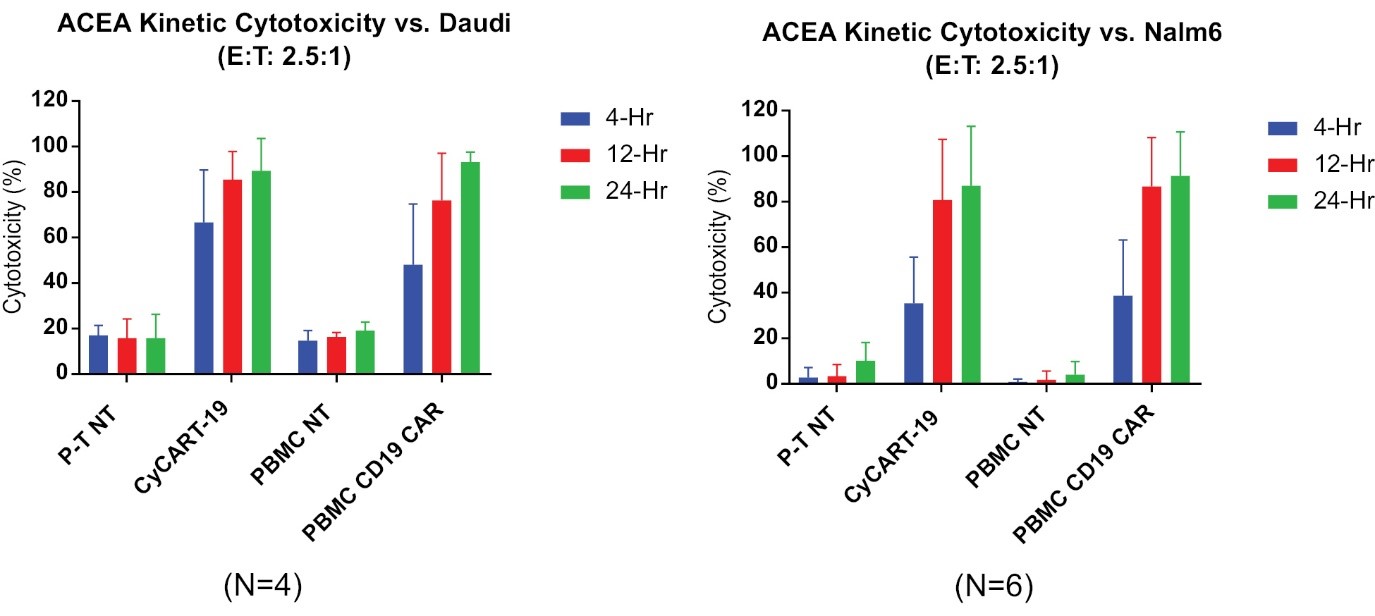
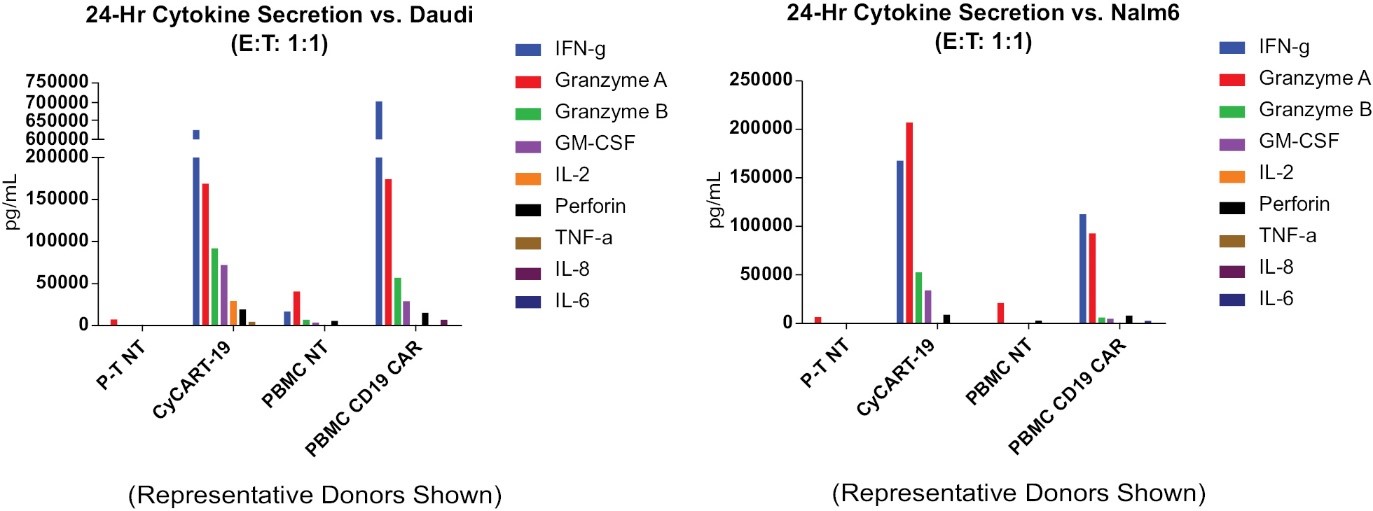
離体するCyCART-19細胞は、CD 19+標的細胞系Daudi(Burkitt‘s Lymphoma)およびNalm 6(急性リンパ芽球性白血病)を特異的に殺傷し、これらのCD 19+標的細胞に応答するために炎症性サイトカインおよびエフェクタータンパク質を分泌する。
11
12
13
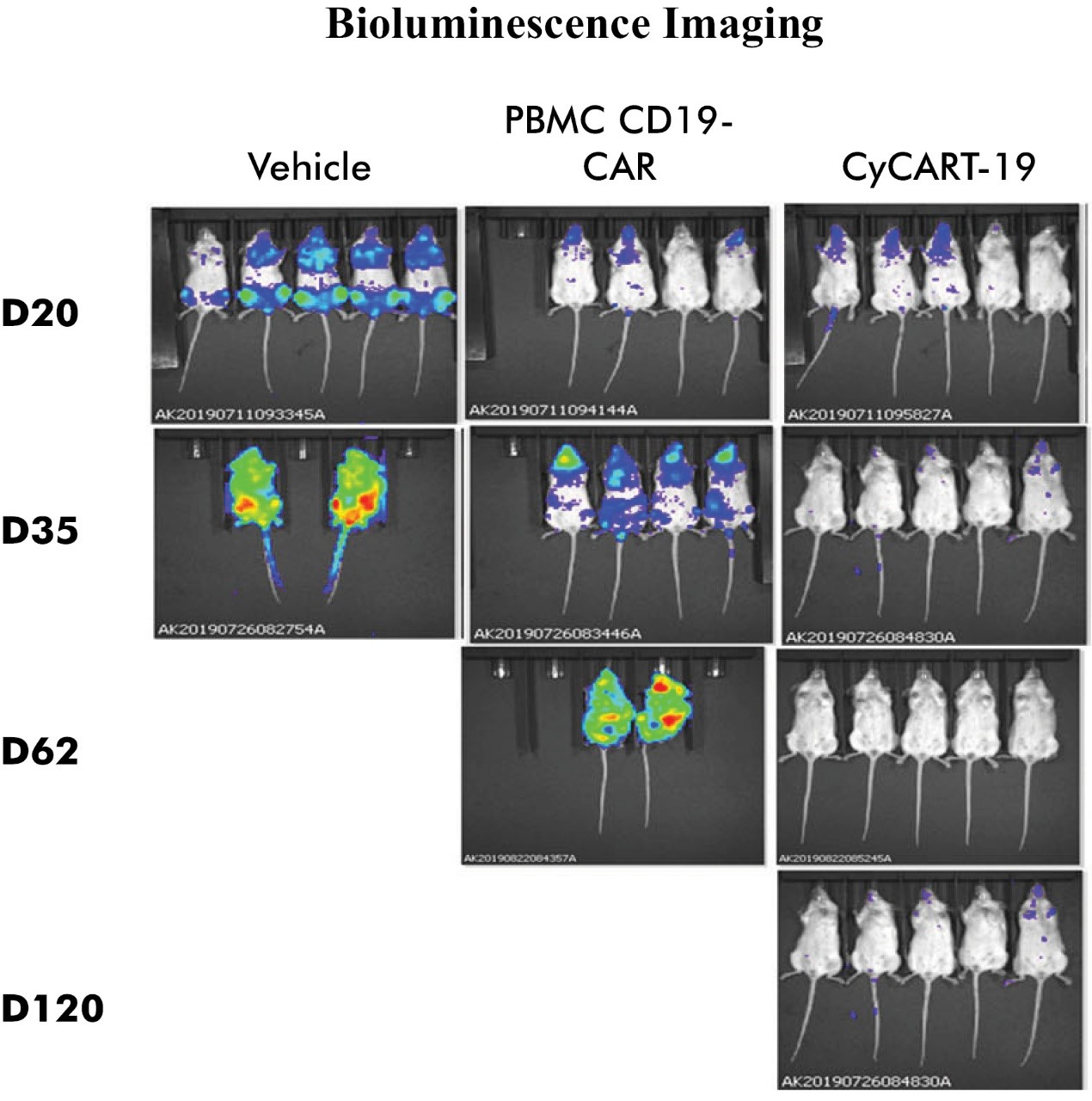
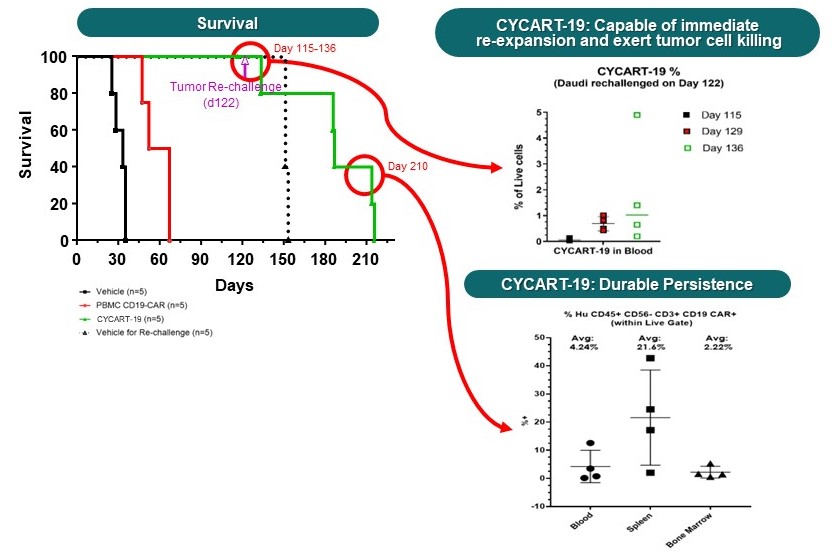
上図に示すように,マウスモデルではCyCART−19がより良好な抗リンパ腫活性と生存率を示し,成人血液由来のCD 19 CAR−T細胞よりも腫瘍再灌流において活性であり,持続性と長い免疫攻撃が示唆された。CyCART-19は腫瘍を除去し、100%が120日まで生存した。CyCART-19の“記憶”特徴は、122日目の腫瘍再挑戦後215日まで延長し、より長い持続性を示し、研究終了時により多くのリンパホーミングを脾臓に帰巣して、より長い抗腫瘍活性を誘導することである。動物研究に用いたCyCART−19細胞は,T細胞受容体αの持続ノックアウトやTRACKOに修飾されていない。
胎盤由来のT細胞は独特であり、それらは同種異体反応性反応を減少でき、そして比較的に低いGvHD発生率と重症度と関係があるからである。以下の図に示すように,増幅された胎盤由来T細胞は異種GvHDを誘発しない体内にあるマウスモデルです。治療マウスでは100%の生存率,体重減少はなく,いずれのヒトCD 3+T細胞の検出も増加しなかったことが証拠である。PBMC処理したマウスは28日目に体重が有意に減少し,全マウスが死亡し,ヒトCD 3+T細胞の検出率が増加した。


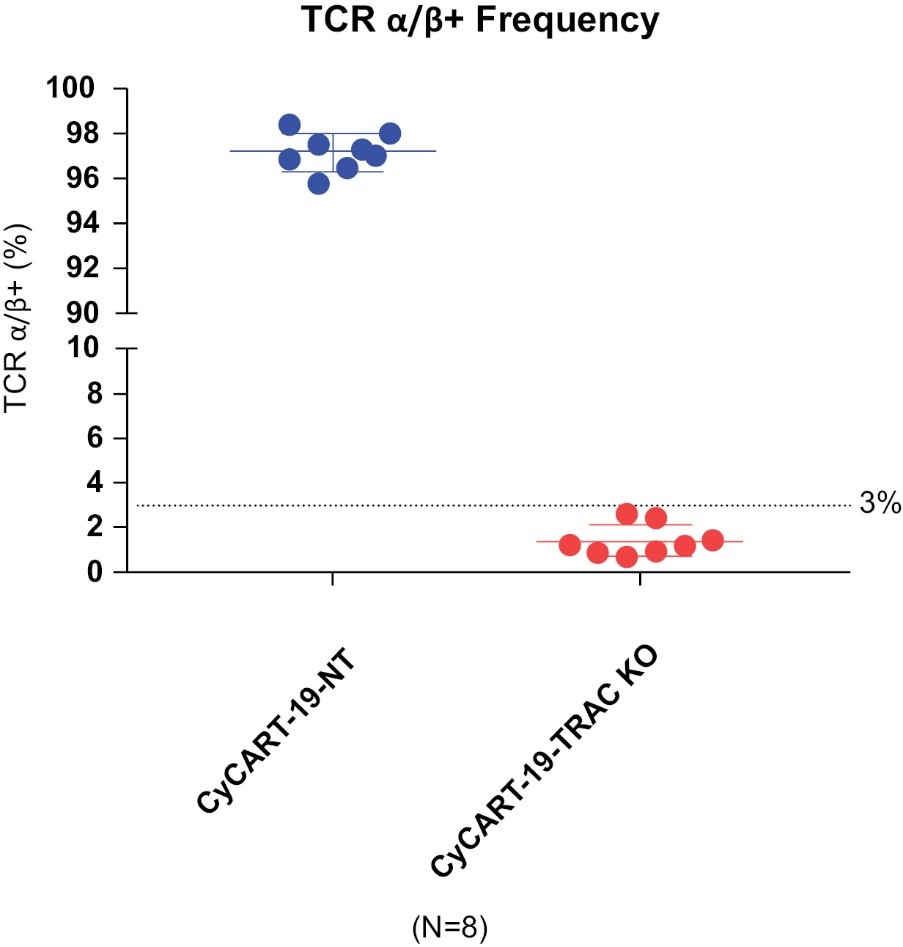
GvHDが胎盤由来のT細胞の拡張に関連しているという証拠はないが、我々の過程には、凝集した規則的な間隔の短い回文反復配列、またはCRISPR、媒介T細胞受容体α定数、またはTRAC、ノックアウト、またはKOが含まれている
14
GvHDを予防するためのさらなるリスク緩和戦略。CyCART−19トランスジェニック細胞は97−99%のTRAC KO効率を得,CD 3再刺激に対する反応(増殖)の欠如により機能性T細胞受容体を失った。
計画的1/2期臨床試験
著者らは1/2期臨床試験においてCyCART-19を評価し、B細胞悪性腫瘍(標的CD 19受容体)の治療に用いる予定である。
15
計画中の第一段階試験は安全性と投与量を評価し、そして3つの用量列(40、120と360×106形質導入の生存CAR-T細胞)を含み、3 x 3試験設計を用いて、最大18名の患者を募集する。主な終点は安全性と最大耐容量の決定である。副次的終点は総応答率(OOR)(完全応答と部分応答(CR+PR)の和),応答持続時間(DOR),無進行生存期間(PFS)と総生存期間(OS)である。CyCART-19の永続性も探る予定である.
計画中の第二段階試験はCyCART-19の治療効果を評価し、198名の患者を募集する。主な終点はORR(CR+PR)を決定することである.第2の端末は、セキュリティ、応答時間、DOR、PFS、およびOSを含む。CyCART-19の永続性も探る予定である.
2022年第1四半期に、CyCART-19をB細胞悪性腫瘍の治療にINDを提出し、2022年5月下旬にFDAから正式な書面通知を受け、より多くの情報を提供し、その後、計画された1/2期臨床試験を継続することができた。私たちはFDAと協力して、その問題をできるだけ早く解決するために努力している。INDがFDAの承認を得て十分な資金が利用可能であれば,2023年下半期に試験を開始する予定である。INDの継続が許可されることは保証されず、予期される時間枠内で継続されることが許可されるか、または研究が予期される時間枠内で開始されることが許可されるであろう。
CYCART-201
CyCART-201はCD 16を発現するトランスジェニックT細胞であり、TCR遺伝子ノックアウトを有する。CyCART-201は多種の潜在的モノクロナル抗体と連合して使用するために設計され、広範な治療潜在力を持っている。著者らの最初の血液学的発展は非ホジキンリンパ腫であり、著者らの固形腫瘍はHER 2+VE腫瘍に発展し、両者は標的モノクロナル抗体と結合する。
Cynk-001
私たちの胎盤由来の未修飾NK細胞タイプに基づく主導的な治療レジメンはCYNK-001であり、AMLを治療するために開発されている同種異体未修飾NK細胞である。
AMLは成人と児童の中で二番目によく見られる白血病タイプであり、すべての成人白血病病例の約3分の1を占める。大多数の患者は誘導化学療法に対する反応が良好で、そして完全な緩和を実現したが、3分の2の患者は第一線で治療した後に再発する。標準治療後に再発した患者(再発/難治-R/R急性骨髄性白血病)と完全に緩和したが測定可能な残留疾患(MRD+AML)を残留した患者の予後は比較的に悪く、依然として新しい治療法に対する医療需要を満たしていない。R/R AMLあるいはMRD+AMLを有するAML患者へのCYNK−001の使用を評価している。看護基準に従ったCYNK−001が、R/R AMLおよび/またはMRD+AMLのAML負担を測定可能な残留病よりもさらに低下させることができるかどうかを決定することを試みた(
臨床前データ
臨床前研究により、CYNK-001は慢性骨髄性白血病(或いはML、AMLとMMと呼ばれる)、細胞系と初代AMLサンプルに対して顕著な殺傷作用を有することが示唆された。Cynk−001の活性化はTh 1抗腫瘍反応に有利なサイトカインであり,効果標的比(E:T)が3:1の場合,CYNK−001は原発AML試料に60%と高い特異的切断を加えた高濃度のインターフェロン−gを放出した。

16

第1段階実験
我々はすでに一期用量増加試験を完了し、11名の再発/難治性AML患者を組み入れ、その中の10名の患者は単剤PNK-007で治療され、PNK-007は凍結保存されていない以前の処方のCYNK-001である。細胞療法の一般的な耐性は良好であり,用量制限毒性はなく,移植片対宿主病もなく,ヒト白血球抗原同種抗体も検出されなかった。10人の患者のうち8人が治療効果を評価することができ(2人の患者は評価のための骨髄不足のためではない)、そのうちの4人の最高用量(約7億NK細胞)治療を受けた患者のうち2人が一時的な生物学的効果を有する証拠である。
著者らは現在CYNK-001(PNK-007に相当する凍結保存NK細胞製剤)のために再発/難治性AML患者と血液学的緩和合併微小残留病(MRD)患者において1期後続試験を行っている。CYNK-001第1段階試験の一部として、血清IL-15レベルをベースラインよりも高く維持し、T調節細胞を28日間まで低レベルに維持するか、またはNK細胞の効力および持続性を潜在的に増強するために、リンパ浄化の用量を評価した。合計16名の患者がR/R AML治療を受け,10名の患者がMRD+AML治療を受けており,これまでどの用量レベルでも総用量レベル1.8億,3.6億と54億個のCYNK−001細胞を含む用量制限性毒性は認められなかった。注入0日後28日目には骨髄と末梢血中のCYNK−001細胞が持続的に存在し,最高細胞用量レベルのMRD陰性状態に達した。CYNK-001の効力と持続性を潜在的にさらに増強するために、MRDおよびR/R AMLの拡張アームは、強化リンパ除去スキーム、シクロホスファミド3600 mg分4日(前900 mg分3日)、フルダラビン120 mg分4日(前75 mg分3日前)を使用して、リンパ除去後のIL-15レベルを向上させる。管理層は,これまでの実験結果(6 Bキューを含む)を検討した後,AML研究の残りの部分の潜在的経路を評価する.2021年12月、我々はFDAからAMLを治療するためのCYNK-001の高速チャネル指定を取得した。
2021年第4四半期に、著者らは投与量、NK細胞の帰巣と持続性、安全性と生物学的効果を評価するために、静脈および腫瘍内再発に対するGBMの用量増加試験を開始した。われわれは2021年3月にFDAから再発GBM治療のためのCYNK−001の迅速チャネル指定を取得し,2021年4月にGBM治療用CYNK−001の孤児薬物指定を取得した。企業資源を優先的に配置する必要があるため,2023年1月,GBM試験における採用を中止する予定であることを発表した。
シンk-301
AMLにおける我々の経験に基づいて、我々の次世代CAR-NK-301は、リンパ枯渇の負担を最大限に減少させながら、増殖、持続性、および有効性を最適化することを含むNK療法がrrAMLを治療する際に直面するいくつかの挑戦を克服することができる。Cynk-301は膜結合IL 15を結合してNK細胞の活性化、増殖と持続性を増強し、そのほか、骨髄ホーミングと標的CARは更に治療効果を増強した。
17
シンカー-302
Cynk-302は、固形腫瘍において開発されているCAR-NKであり、最初は非小細胞肺癌に集中しており、持続的に高度に満たされていない需要領域である。Cynk-302はHER 2+VE胃癌治療においてCYNK-101を学習した上で構築した次世代構築物である。遺伝子改変によってCD 16を汎用結合タンパク質として発現し、膜結合IL 15を組み込むことによって増殖および持続性を支持し、開示されていない標的CARによって治療効果をさらに増強する最適化された構築物である。
APPL-001
現在、著者らの胎盤源性MLASCタイプの鉛治療候補薬物はAPPL-001であり、遺伝子組換えの胎盤源性MLASCである。私たちはAPPL-001によるクローン病の治療について初歩的な評価を行っている。未修飾MLASCsの臨床研究において、50名以上の患者はMLASCsを用いてクローン病を治療した。プラセボ群と比較して,治療群の臨床反応率は有意に高かった。
第1/2 a段階試験設計
計画中の1/2 a期試験は、コルチコステロイド薬に無効な重度クローン病患者へのAPPL−001の使用を評価する。主な目標は安全性と耐性を評価し、推奨される第2段階用量を決定することである。計画中の2 a段階試験の主な目標は、中重度クローン病患者の臨床緩和と臨床反応を測定することによって臨床活動を評価することである。第2の目標は,内視鏡測定や生活の質評価などの疾患改善措置を評価することである。計画の主な終点は6週間後と1年後の臨床緩解/反応である。計画の副次的な終点は,粘膜癒合の評価と,炎症性腸疾患アンケートにより測定された患者報告の生活の質の結果である。
PEXO-001
PExo-001は1種のヒト産後胎盤由来の外切体産物であり、サイトカイン、ケモカインと成長因子から構成され、すでに再生と免疫調節活性を有することが報告されている。PExo−001の最初の開発は骨関節炎に用いられる。
将来のパイプ機会
我々は,我々のCelulality Impactプラットフォームを用いて,より多くの興味のある目標を追求する予定である.これらには,現在研究中の4つの同種細胞タイプの追加適応と,将来検証可能な他の目標が含まれている。著者らの胎盤由来T細胞プラットフォームは他の受容体を標的とする潜在力を有する。
また,我々は定期的に科学や業界の構造を調査し,許可,協力あるいは技術を得る機会を探しており,これらの技術は現在あるいは新たな細胞療法を推進し,患者に利益を得るのに役立つかもしれない。
私たちがこれらの機会を追求するために十分な資本を集めることができるかどうか、あるいはその発展に一部の資金を提供する能力のある商業パートナーを見つけることができるかどうかは、科学的価値と潜在的な商業価値を持つ機会を含む未来の機会の把握に影響を与える可能性がある。共同開発または協力するプロジェクトは、内部援助プロジェクトよりも長期的な経済的利点を有する可能性があるが、これらのプロジェクトは、資本が十分であり、調査中の疾患状態で特定の専門知識を有する開発パートナーと協力し、より高い成功確率を得る可能性もある。
商業企業
私たちは引き続き新しい生体材料プロジェクトに投資して、私たちの胎盤由来生体材料製品のパイプラインを拡大します。我々は現在,腱損傷の治療と保護のための腱被覆を開発しており,この損傷では腱組織が実質的に失われていない。私たちはまた整形外科市場のための骨洞充填製品を開発している。私たちは
18
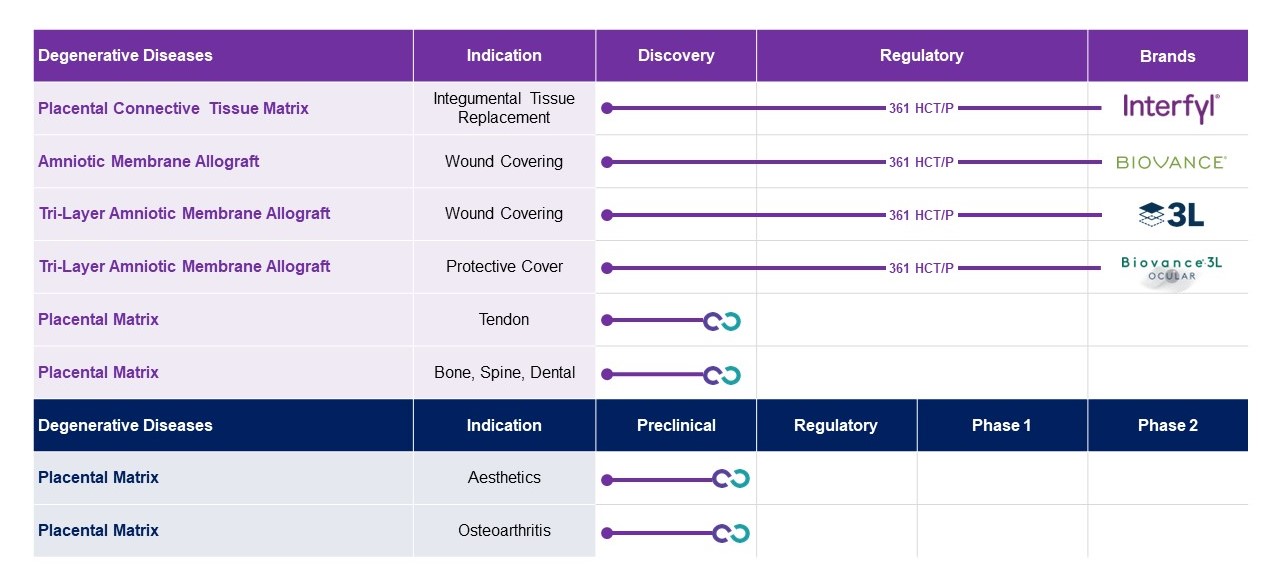
膝骨性関節炎動物モデルからの初歩的なデータは、胎盤化細胞外基質は関節痛を軽減し、損傷軟骨の軟骨生成を促進できることを表明した。私たちの製品ラインは次の図のようになります
退行性疾患
私たちは私たちが変性疾患と呼ばれる部分を含む私たちの運営結果を報告した。アメリカ国立癌研究所は退行性疾患を影響を受ける組織或いは器官の機能或いは構造が時間の経過とともに悪化する疾患と定義している。私たちの現在の変性疾患業務は主に私たちの流通ネットワークを介して私たちのBiovanceとInterfyl製品を販売することを含む。Biovanceは健康,臨月妊娠の胎盤から抽出した脱細胞,脱水したヒト羊膜である。それは無傷の天然細胞外基質であり、傷口の再生過程に基礎を提供し、機能組織の修復にステントを提供した。Interfylはヒト結合組織基質であり、健康、臨月妊娠の胎盤から来ている。それは各種の医学専門家によって創傷、創傷或いは手術による軟組織欠損を埋めるために使用されている。我々は、変性疾患の治療のためのより多くの生体材料製品、および1つ以上の生体材料を用いて細胞療法と組み合わせた変性疾患における我々固有の細胞療法および可能な組み合わせ療法の使用を検討している。BiovanceとInterfylはCelgene買収の前にAnthrogenensisで開発され,CelgeneによってHLIに販売され,2017年5月にHLIから買収され,HLIが第三者に権限を与えたマーケティングと流通権利に制限され,これらの権利は2018年5月にMistやUltraMist治療システムとともに買収された。2020年8月、我々はSanuWave Health Inc.またはSanuWaveと、(I)創傷ケア市場での流通および商業化の独占的Biovanceライセンス、および(Ii)グローバル創傷ケア市場での流通および商業化Interfylの非独占ライセンスを含む5年間のライセンス契約を締結し、あるアジア司法管轄区を除いて、この合意に基づいて、最低販売ハードルに基づいて印税を取得する。未治癒材料違反により,SanuWaveとのライセンス契約は2021年第3四半期に終了した。
私たちの退行性疾病領域におけるマーケティングと販売戦略の重点は私たちの製品のために強力な流通パートナーを発展させることであり、私たち自身の直販チームを構築することではない。2021年5月7日、私たちはArtrex,Inc.と6年間の供給および流通協定を締結し、(I)米国内の整形外科領域での流通および商業化の独占的Biovance、InterfylおよびCentaflex許可証を取得すること、および(Ii)米国内の急性慢性非癒合創傷看護領域の商業化およびInterfylおよびCentaflexの独占ライセンスを販売することを含む。2021年9月1日、私たちはEvolution Biologyx,LLCと、Interfylがオフィスまたは入院環境で使用され、Medicare Part B部分またはアメリカ衛生と公衆サービスセンター医療保険サービスセンターまたは他の政府当局によって設立された任意の後続、同等または同様のカテゴリで精算されることを含む3年間の供給および流通協定を締結したが、整形外科の医療専門科は除外され、医療専門科または整形外科または神経外科における創傷または脊柱応用は除外された。
2023年1月、私たちは米国以外の地域での中東と北アフリカへの拡大に関連する2つの新しい流通協定を発表した。我々は、国際輸出入貿易会社CH Trading Group LLCやCH Trading Groupと独占的な領土流通協定を締結した。CH貿易グループは、中東と北アフリカ100カ国以上の独自の地域で当社のハラール認証製品として販売されます。また私たちは独占を発表しました
19
タメルグループや中東ヘルスケア流通会社タメルと流通協定を結び,サウジアラビアで我々のブランド生体材料製品を流通させている。2023年3月には、アラブ首長国連邦アブダビに本部を置く世界的に有力な貿易、物流、工業促進業者アブダビ港会社(AD Ports)と独占販売協定を締結し、アラブ首長国連邦、カタール、バーレーン、オマーン、クウェート、エジプトで当社の生体材料製品を流通させることも発表しました。
私たちは私たちの成熟した商業製品BiovanceとInterfylの販売を補充するために、変性疾患分野のための新しいまたは差別化された製品を創造することに投資し続けている。私たちはBiovance 3 Lを作りましたこれは眼科と外科市場に集中した3層ヒト羊膜製品です適用状況に応じて,Biovance 3 Lはシート状と円盤の2つの形式で選択可能である.臍帯由来の脱細胞ヒト胎盤基質であるCentaFlexも作製した。CentaFlexは、損傷組織の修復を保護し、支持するために、外科的カバー、小包またはバリアとして使用することができる。我々は他にもヒト胎盤組織に基づく製品が開発中であり,これらの製品は様々な規制経路に沿って,潜在的に商業準備を実現する可能性がある。
バイオバンク
著者らは会社と契約して、臍帯血と胎盤由来細胞と組織を含む、ある生物材料を収集、加工、低温保存と貯蔵する準親に有料の生物バンクサービスを提供する。私たちは生体材料の収集、処理、低温保存の使い捨て費用と、私たちのバイオバンクで生体材料を維持する貯蔵費を受け取り、通常18年から25年の間毎年支払われている。2017年5月、我々はHLI(HLIはLifebank USA名義で運営)および退行性疾患製品BiovanceとInterfylから私たちのバイオバンク業務を買収し、2018年10月にCariCord Inc.,またはCariCord、一家家庭臍帯血バンクを買収した。
製造業
私たちはニュージャージー州フロラム公園に147,215平方フィートの専門的に建設された施設を持っています。その中にはcGMPをサポートする製造センターと、専門的な研究とオフィス空間と共有サービス空間が含まれています。我々の工場は9つのC/ISO−7と6つのD/ISO−8レベル製造キットを含み,細胞療法と先進生体材料の商業生産に特化して設計されている。私たちはニュージャージー州フロラム公園にある製造工場ですべての完成品を生産するつもりです。私たちは改善された分析方法の開発を含めて、私たちの製造プロセスを最適化するために資源を投入した。時間の経過に伴い、私たちは引き続き工芸科学、製品表現と製造に投資して、私たちの生産とサプライチェーン能力を高めることを計画しています。また,必要に応じて非排他的にCMOを使用し,将来的にはいくつかの候補治療にCMOを使用する可能性がある。例えば,CMOを用いて2022年のCYNK−001に臨床的に製造·供給し,その生産を内部化し,すべての完成品が将来的に内部生産されることが予想される。他のすべての完成品は内部で生産されている。それにもかかわらず、私たちは必要な時にCMOを招聘して、臨床と商業レベルの製品が需要に応じて持続的に供給できることを確保する。
我々の細胞治療候補案は,定義されたユニット操作と技術からなるプラットフォームによって設計·製造されている。この過程は小から大へと発展していき,cGMP条件を創出するコンプライアンスプログラムに組み込まれている.このようなプラットフォームベースのモデルがあるにもかかわらず、各治療法は唯一無二であり、各新しい候補治療法については、各工程ステップを個別にカスタマイズし、強力なプログラムを作成するための開発段階が必要であり、後でcGMP環境で実施して、臨床ロットの生産を確保することができる。この作業は、最も信頼できる生産条件を決定するために、プロセスの各ステップの可変性を評価および評価するために、研究および開発環境において行われる。
我々は,細胞治療開発と製造における我々のコア専門知識を利用して,第三者に契約製造と開発サービスを提供することで収入を創出する予定である。この新サービスの最初のポイントは,開発段階の細胞治療会社の臨床試験のための治療候補薬の開発·製造に協力することである。私たちは、現在この市場にサービスしているより大きな契約製造組織に柔軟で費用効果のある代替案を提供することができると信じている。
許可協定
私たちは正常な業務過程で許可協定を締結した。我々はすでにソレントからいくつかの技術の許可を得ており,これは我々のCyCART-19計画を研究·開発するために必要である.私たちの胎盤由来細胞治療候補薬は広範な潜在的適用性があるため、私たちはまた私たちの技術を第三者に許可して開発し、私たちが追求しようとしない他の適応あるいはいくつかの地域に使用することができる。例えば,2017年6月に肺バイオテクノロジーPBCとライセンス合意に達した。この許可協定に基づき、我々は肺生物技術会社に肺疾患と臓器移植領域における胎盤幹細胞の独占的許可を付与した。この協定は2021年3月に終了した。我々はまた,我々の変性疾患製品BiovanceとInterfylをSanuWaveに分配する権利を5年間獲得し,2020年8月に他の非コア資産の売却に関連しているが,2021年第3四半期にこの許可を終了した。
また,CelgeneからAnthrogenensisを買収する一部として,世界的に印税免除,全額支払いの非独占的許可をCelgeneに付与し,ある知的財産権を研究やビジネス目的に用い,Celgene CVRを付与した
20
これは私たちにいくつかの場合に未来のマイルストーンと特許権使用料を支払う権利を提供する。“”というタイトルの部分を参照-私たちのチームと会社の歴史-Celgene社OutライセンスプロトコルおよびCVRを含むCelgeneとの間の持続的な関係を説明する。
ソレント治療会社
2020年9月、私たちはソレントと許可および譲渡協定を締結し、ソレント特有の抗CD 19 CAR-T構築および関連CARの権利を獲得し、胎盤由来または臍帯血由来細胞のために使用した。ソレントは私たちの重要な株主です。我々は,我々の胎盤由来T細胞をソレント技術を用いて遺伝子改変してCD 19受容体を有するCAR T細胞を作製しており,これはわれわれのCyCART−19治療候補薬である。
“ソレント協定”によると、私たちは、ソレントの同意の下で、特定のCD 19自動車構造の使用によって侵害された特許権を含むソレントの特定の知的財産権に基づいて、胎盤由来細胞および/または臍帯血由来細胞の分野で、任意の疾患または疾患の治療、ならびに製造、製造、使用、販売、提供、販売、輸入、輸出および流通に関連する製品の研究、開発、輸出および流通に関連する製品を含むソレントの同意の下で、ソレントの特定の知的財産権を取得する権利がある。ソレント社独自の抗CD 19 CARR−T構造と関連CARSと胎盤由来細胞あるいは臍帯血由来細胞を組み合わせた製品の商業化と開発。前述の許可は、特定の米国仮特許出願に対して独占的であり、ソレントの他のすべての許可の知的財産権については非独占的である。
ソレントは、胎盤由来細胞および/または臍帯血由来細胞領域以外で任意の疾患または障害を治療するための権利、ならびに任意のCD 19 CAR-T構造または関連自動車を使用または結合するための非CD 19 CAR-T許可製品またはサービスを使用または結合するための、製造、製造、使用、販売、提供、販売、輸入、輸出、ならびに他の方法でCD 19 CAR-T構造または関連自動車を使用または結合するためのCD 19 CAR-T許可製品を保持する。
ソレント協定によれば、私たちはライセンス製品の開発と商業化を独占的に担当していますが、ソレントによるCD 19 CAR-T製品のいくつかの留保権利に制限されています。私たちは現在、ソレント協定に基づいて自動車構造と特許製品の持続的な供給を得るために、ソレントと供給協定を交渉している。また、私たちは商業的に合理的な努力を使って許可された製品を開発し、商業化する義務がある。
ソレント協定によれば、私たちは、私たちが生成した任意の改善されたすべての権利、所有権、および利益をソレントの背景知的財産権に譲渡することに同意しました。さらに、我々は、CD 19 CAR−T許可製品および構造(上述した)におけるSorrentoの保持権利に関連する任意の新しい発明の下で、Sorrentoプロトコルに従って生成されたCD 19 CAR−T構造に関連するか、またはCD 19 CAR−T構造をカバーするために、Sorrentoの非排他的で再許可可能で、全額支払い、印税免除のグローバル許可を付与した。ソレントは、特許製品に関連する任意の特許または特許出願の起訴および維持を含むソレント協定によって生成されたまたはそれに関連する特許および特許出願に対して主要な制御権を有し、私たちは、ソレントが放棄した任意のそのような特許および特許出願について訴訟を提起する副次的な権利を有する。
ソレント協定によると、CD 19 CAR-T許可製品に付与された任意の再許可に関連する非特許権使用料再許可収入の2桁の数百分をソレントに支払う義務がある。また,CD 19 CAR−Tライセンス製品の永久純売上高の低桁特許権使用料をSorrentoに支払う義務がある。最終的に決定された供給協定によると、自動車構造と許可製品の供給費用をソレントに支払う義務があり、この協定はコストに一定の割合を加え、保証されていない最低要求に基づいていると予想される。2022年12月31日現在、私たちはソレント協定に基づいてソレントに何の支払いもしていませんが、供給協定の交渉を継続しながら製品供給金を支払いました。
どちらか一方がソレント協定に違反した場合,ソレントプロトコルを終了することができる.また、“ソレント協定”の発効1周年後、私たちはいつでもソレントに書面で通知し、“ソレント協定”を終了する権利があります。2023年2月13日、ソレントは米国破産法第11章に基づいてテキサス州南区米国破産裁判所で自発的な訴訟手続きを開始すると発表した。現在、私たちは破産が許可協定の下で履行を続ける能力にどのような影響を与えるのか予測できない。
知的財産権
私たちのビジネス成功は、私たちのCelulality Impactプラットフォームを支持する技術、私たちのリードする細胞治療候補薬物CyCART-19、CYNK-001、CYNK-101、APPL-001、PDA-002と未来の候補治療薬の特許保護の能力、および新しい発見、製品開発技術と技術ノウハウを獲得し、維持することにある程度依存する。私たちのビジネスの成功は、他人の所有権を侵害することなく運営する能力と、他人が私たちの所有権を侵害することを防止する能力にもある程度依存しています。私たちの政策は以下のように私たちの独自の地位を発展させて維持することです
21
我々の技術、発明および改善に関連する他の方法、出願は、米国および外国の特許および出願を可能にすることができるかもしれないが、これらは、私たちの業務の発展および実施に非常に重要である。
私たちはまた、商標、商業秘密、技術ノウハウ、持続的な技術革新、秘密保持協定、および発明譲渡協定に依存して、私たちの独自の地位を発展させ、維持しています。セキュリティプロトコルは、当社の独自の情報を保護することを目的としており、発明譲渡プロトコルは、当社の従業員、コンサルタント、または他の第三者によって開発された技術の所有権を付与することを目的としています。我々は,我々のオフィスの物理セキュリティと我々の情報技術システムの物理と電子セキュリティを維持することで,我々のデータとビジネス秘密の完全性とセキュリティの保護を図る.私たちは私たちの合意と安全措置に自信があるが、その中のどれも違反される可能性があり、私たちは十分な救済措置を持っていないかもしれない。しかも、私たちのビジネス秘密は競争相手に知られたり独立して発見されるかもしれない。
許可されたおよび会社のすべての知的財産権については、私たちの任意の未解決特許出願または将来提出された任意の特許出願が特許を取得することを保証することはできず、私たちの任意の既存特許または将来私たちに付与される可能性のある任意の特許が、私たちの商業療法を保護し、その特許を使用して製造する方法において商業的用途を有することを保証することはできません。
私たちは私たちのCelularity Impactプラットフォーム、私たちの4種類の同種細胞タイプ、そして私たち自身の知的財産権と許可知的財産権に基づく治療候補案をめぐる私たちの知的財産権の組み合わせを積極的に構築している。私たちは米国と世界各地の1500以上の特許および特許出願の所有者、共同所有者、または許可者であり、私たちのCelularity Impactプラットフォーム、私たちのプロセス、技術、および現在の重要な細胞治療計画を保護する。
我々の特許の組み合わせは、CAR-T細胞、未修飾NK細胞、トランスジェニックNK細胞、MLASCsおよびエキソソーム、以下のように、我々の5つの同種胎盤由来細胞および細胞外小胞タイプに対する特許および特許出願を含む
22
より広く言えば、我々の特許組み合わせおよび出願戦略は、物質組成、製造方法、および使用方法などのための特許請求の範囲を求めることによって多層保護を提供することを意図している。私たちは、私たちの技術および関連技術およびその使用をカバーするための特許保護を求めることを含む、私たちの業務に非常に重要であると考えられるノウハウの保護と強化に努めています。
個別特許の期限は特許を取得した国の法的期限に依存する。私たちが出願を提出したほとんどの国では,特許期間は最初の優先権を要求する非仮出願が提出された日から20年である。米国では、特許期限調整によって特許期限を延長することができ、このような調整は、特許権者が米国特許商標局の特許付与時の行政遅延による損失を補償することができ、または、1つの特許が以前に提出された特許によって最終的に放棄された場合、特許期間を短縮することができる。米国では、“ハッチ·ワックスマン法案”(Hatch-Waxman Act)によると、FDAが承認した薬物をカバーする特許期間も最大5年延長する資格があり、FDA規制審査中に失われた特許期間を補うことを目的としている。特許期間延長の長さは,我々が規制審査を行う時間長に基づいて計算される。“ハッジ·ワックスマン法”によると延長された特許期間は,製品承認日から14年を超えてはならず,承認された薬物に適用される特許のみを回復することができる。また,1つの特許が1回しか回復できないため,1つの特許が複数の製品に適用されれば,1つの製品に基づいて延期するしかない.ヨーロッパおよび他のいくつかの外国司法管轄区域にも同様の規定があり、承認された薬物をカバーする特許の期限を延長する。
競争
既存の看護治療基準に加えて,我々の製品は生物製薬会社,学術研究機関,政府機関および公共·民間研究機関が開発した新しい療法と競争する。
細胞療法は臨床試験において良好な治療効果を有するため,これらの療法を開発した既存会社や新会社や同種細胞療法開発からの競争が激しくなることが予想される。
潜在的な細胞治療と生体材料の競争相手は
競争は小型バイオテクノロジー会社や大手製薬会社が求めている非細胞療法にも由来し,これらの会社には安進,アスリーカン,百時美施貴宝社,Incell社,メルク社,F·ホフマン−ラロー社がある。
私たちの多くの競争相手は、単独あるいはパートナーと協力しても、研究開発、臨床前試験、臨床試験、製造とマーケティング方面の財力と専門知識はすべて私たちよりずっと多い。将来の協力とM&Aは、より少ない数の競争相手に資源をさらに集中させる可能性がある。
もし私たちの競争相手が私たちが開発する可能性のある細胞療法よりも安全で、より効果的で、副作用が少なく、より便利で、より安価な療法を開発し、商業化すれば、私たちの商業的潜在力は減少または除去されるかもしれない。私たちの競争相手も私たちよりも早くFDAや他の規制機関の治療法の承認を得ることができ、これは私たちの競争相手が私たちが市場に入ることができる前に、あるいは開発作業をより複雑にする前に強力な市場地位を確立することをもたらすかもしれない。私たちのすべてのプロジェクトの成功に影響を与える重要な競争要素は有効性、安全性、そして利便性かもしれない。
これらの競争相手はまた臨床試験のために類似の合格科学と管理人材バンク、場所と患者集団、及び著者らの計画のために補充或いは必要な技術を奪い合う可能性がある。
23
政府規制と製品審査
私たちはアメリカで運営されているバイオ製薬会社として広く規制されている。私たちの細胞療法は生物製品として規制されるだろう。この分類があれば,われわれの細胞療法の商業生産は登録施設で行われ,生物製剤に適合したcGMPが必要となる。FDAはヒト細胞或いは組織製品を最低限操作或いは最低操作以上に分類し、最低限操作を超える製品は製品の安全性と有効性を証明するために臨床試験を行う必要があることを確定し、発売許可を得るために生物製品許可証申請(BLA)を提出した。われわれの細胞治療候補薬は最小限の操作だけではないと考えられており,市販される前に臨床試験で評価し,BLAを提出·承認する必要がある。
アメリカ(連邦、州と地方の各レベル)とその他の国の政府当局は私たちが開発している生物製薬製品の研究、開発、テスト、製造、品質管理、承認、ラベル、包装、貯蔵、記録保存、販売促進、広告、流通、承認後のモニタリングと報告、マーケティングと輸出入などの方面に対して広範な監督管理を行った。我々の候補治療薬は米国が合法的に発売される前にFDAの承認を得なければならず,外国の合法的な発売前に適切な外国規制機関の承認を得なければならない。一般的に、私たちの他の国での活動は、重要な違いがあるかもしれないにもかかわらず、米国と類似した性質と範囲の規制を受けるだろう。また、欧州規制のいくつかの重要な側面は集中的に処理されているが、具体的な国の規制は多くの点で不可欠である。規制マーケティングの承認を得る過程とその後、適切な連邦、州、地方、外国の法律法規を遵守する過程には、多くの時間と財力が必要だ。
アメリカ製品開発プロセス
米国では,FDAは連邦食品,薬物と化粧品法,公衆衛生サービス法あるいはPHSA及びその実施条例に基づいて薬品と生物製品を規制している。規制の承認を得て、その後、適切な連邦、州、地方、外国の法規と条例を遵守する過程には、多くの時間と財政資源が必要だ。製品開発過程,承認過程又は承認後のいずれかの場合,出願人が適用される米国の要求を遵守できない場合には,行政又は司法制裁を受ける可能性がある。他の行動に加えて、これらの制裁は、FDAが承認保留申請の拒否、承認の撤回、臨床封印、警告状、製品のリコールまたは市場からの撤回、製品の差し押さえ、生産または流通禁止の完全または部分的な一時停止、罰金、政府契約の拒否、原状回復、返還、または民事または刑事罰を含むことができる。どんな機関や司法法執行行動も私たちの運営と業務に実質的な悪影響を及ぼす可能性がある。米国食品医薬品局が生物製品が米国市場に参入する前に必要なプログラムには、通常、以下のような態様が含まれる
ヒトで任意の候補生物製品をテストする前に、私たちの細胞治療候補製品を含み、治療候補製品は臨床前試験段階に入る。臨床前試験は、製品の実験室評価を含む非臨床研究とも呼ばれる
24
候補製品の潜在的な安全性と活性を評価するために、化学、毒性および配合、および動物研究。臨床前試験の進行はGLPを含む連邦法規と要求に適合しなければならない。臨床試験スポンサーは臨床前試験の結果を生産情報、分析データ、任意の利用可能な臨床データ或いは文献、提案された臨床方案と共にFDAに提出し、INDの一部としなければならない。IND提出後も,いくつかの臨床前試験が継続される可能性がある。INDはFDAが提案された臨床試験に対して懸念または問題を提起し、30日以内に臨床試験を一時停止しない限り、FDAが受信後30日以内に自動的に発効する。この場合,INDスポンサーやFDAは臨床試験開始前に未解決の問題を解決しなければならない。FDAはまた、臨床試験の前または期間のいつでも、安全考慮または規定に適合しない理由で、候補生物製品に臨床的制限を加えることができる。FDAが臨床一時停止を強制した場合、試験はFDA許可なしに再開されず、その後、FDA許可の条件下でのみ再開される可能性がある。したがって,INDの提出によりFDAが臨床試験の開始を許可するか,あるいは開始すると,このような試験を一時停止または終了するという問題はないとは判断できない。
米国で臨床試験を開始する前にFDAにINDを提出するほか,組換えや核酸分子を合成するヒト臨床試験に関連する機関生物安全委員会(IBCs)の監督を受けなければならないことは,米国国立衛生研究院(NIH),組換えあるいは合成核酸分子に関する研究ガイドライン,あるいはNIHガイドラインに掲載されている。米国国立衛生研究院のガイドラインによれば、組換えおよび合成核酸は、(1)核酸分子が結合して生細胞中で複製可能な分子(すなわち、組換え核酸)、(2)化学的または他の方法で合成または増幅された核酸分子、または化学的または他の方法で修飾されたが自然に産生される核酸分子(すなわち、合成核酸)塩基対を含む分子、または(3)第(1)または(2)項に記載の分子を複製する分子を含む、と定義されている。具体的には,NIHのガイドラインによると,ヒト遺伝子転移試験の監督にはIBCによる評価と評価があり,IBCは地方機関委員会であり,組換えや合成核酸分子を用いた研究の審査·監督を担当している。IBCは、研究の安全性を評価し、公衆の健康または環境に対する任意の潜在的リスクを決定し、このような審査は、臨床試験開始前のいくつかの遅延をもたらす可能性がある。NIHガイドラインは強制的ではないが,関連研究がNIH組換えや合成核酸分子研究助成を受けた機関で行われているか,あるいはその助成によって行われていない限り,多くの会社や他のNIHガイドラインに拘束されていない機関は自発的にこれらのガイドラインに従っている。
臨床試験は合格した調査者の監督の下で患者にバイオ製品候補薬を投与することに関連し、通常は試験スポンサーに雇われたりコントロールされたりしない医師である。臨床試験は,いくつかの有害事象発生時に臨床試験が停止されることを確保する停止ルールを含む,臨床試験の目標,投与手順,被験者選択と排除基準,および被験者の安全性を監視するためのパラメータを詳細に説明するプロトコルで行われる。各スキームおよびスキームの任意の修正は、INDの一部としてFDAに提出されなければならない。臨床試験はFDAの規定に従って行わなければならず、この規定はすべての研究患者にインフォームドコンセントを提供することを含む良好な臨床実践またはGCP要求を含む。さらに、各臨床試験は、臨床試験を行う各機関の独立したIRBによって審査および承認されるか、またはサービスを提供しなければならない。IRBは試験参加者の福祉や権利の保障を担当し,臨床試験に参加する個人のリスクが最低に低下するかどうか,期待利益と比較して合理的かどうかなどの項目を考慮している。IRBはまた、各臨床試験対象またはその法律代表によって署名されなければならないインフォームドコンセントの形態および内容を承認し、完成まで臨床試験を監視しなければならない。いくつかの研究はまた、データ安全監視委員会と呼ばれる臨床研究スポンサーによって組織された独立した合格専門家グループの監視を含み、この委員会は、研究のいくつかのデータへのアクセスに基づいて、研究が指定されたチェックポイントで行うことができるかどうかを許可し、被験者が受け入れられない安全リスクまたは他の原因があると判断した場合、治療効果を示さない場合、臨床試験を停止する可能性がある。現在行われている臨床研究や臨床研究結果を公的登録機関に報告することに関する要求もある。
人体臨床試験は通常3つの連続段階に分けて行われ、これらの段階は重複或いは合併する可能性がある
承認後の臨床試験は,4期臨床試験と呼ばれることがあり,最初の上場承認後に行われる可能性がある。これらの臨床試験は,治療適応が予想される患者の治療から追加的な経験を得るためのものであり,特に長期安全なフォローアップのためである。臨床開発のすべての段階で、規制機関は広範なモニタリングを必要とする
25
すべての臨床活動、臨床データ、臨床試験調査者の監査。臨床試験結果を詳細に説明する年次進展報告はFDAに提出しなければならない。書面のIND安全報告は直ちにFDAと調査者に提出しなければならない、深刻かつ予期しない不良事件、他の研究からのいかなる発見、実験室動物試験或いは体外試験を含み、これらの発見は人類患者に対して重大なリスクがあることを表明し、或いは任意の臨床上重要な深刻な疑わしい副作用の発生率は方案或いは研究者マニュアルに列挙されたより増加することを表明した。スポンサーは15日以内にINDセキュリティ報告書を提出し,スポンサーがその情報有資格報告を確定した後でなければならない。スポンサーはまた、スポンサーが初めて情報を受け取ってから7日以内に、任意の意外、致命的、あるいは生命に危害を及ぼす疑いのある副作用をFDAに通知しなければならない。第1段階、第2段階、および第3段階の臨床試験は、もしあれば、任意の指定された時間で成功しない可能性がある。FDAまたはスポンサーまたはそのデータ安全監視委員会は、他の無関係な免疫療法試験から推定されるリスクを含む、患者が受け入れられない健康リスクに直面していることを発見することを含む、様々な理由で臨床試験を一時停止または終了することができる。同様に,臨床試験がIRBの要求に沿って行われていない場合,あるいは生物製品が患者に予期せぬ深刻な傷害を受けた場合,IRBはその機関の臨床試験の承認を一時停止または終了することができる。
ヒト細胞治療製品は新しい治療法である。これは比較的に新しく、絶えず拡大する新型治療干与措置領域であるため、試験期間の長さ、FDAが細胞治療製品の安全性、有効性、純度と効力を確定するために試験に入れる必要がある患者の数を保証することができず、これらの試験で発生したデータがFDAに受け入れられ、発売許可を支持することも保証されない。
臨床試験と同時に、会社は通常追加の研究を完成しなければならず、生物製品の物理的特徴に関する追加情報を開発し、cGMP要求に基づいて商業大量生産製品のプロセスを最終的に決定しなければならない。PHSAは,生物製品を用いた外来製剤導入のリスク低減を支援するために,属性が正確に定義できない製品の製造制御の重要性を強調した。製造過程は一貫して高品質の候補製品ロットを生産することができなければならず、他の以外に、スポンサーは最終生物製品の特性、強度、品質、効力と純度をテストする方法を開発しなければならない。また,適切な包装を選択·試験し,候補生物製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
アメリカの審査と承認の流れ
生物製品の臨床試験が完了した後,生物製品の商業販売の前に,FDAによるBLAの承認を得なければならない。提出されたBLAには、製品開発、実験室と動物研究、人体試験、製品製造と成分の情報、提案されたラベル、その他の関連情報が含まれなければならない。テストや承認過程には多大な時間と労力が必要であり,FDAがBLAの届出を受ける保証はなく,届出してもどの承認もタイムリーに承認される保証はない.
改正された“処方薬使用料法案”(PDUFA)によると、各BLAは相当な使用料を伴わなければならない。FDAは毎年PDUFAユーザ料金を調整する。PDUFAは生物製品に年間計画費も徴収している。場合によっては、小企業が初めて出願した出願料を免除することを含む、費用を免除または減免することができる。また,孤児薬として指定された製品については,この製品が孤児適応も含まれていない限り,BLASに対して使用料を評価しない。
出願提出後60日以内に、FDAは、機関が提出を受け入れる前に実質的に完了したかどうかを決定するために、提出されたBLAを審査する。FDAは、それが不完全であるか、または提出時に適切に審査できないと考えられる任意のBLAの提出を拒否することができ、より多くの情報の提供を要求することができる。この場合,BLAおよび付加情報を再提出しなければならない.再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。提出された申請が受け入れられると,FDAはBLAの深い実質的な審査を開始する。FDAは、提案された製品が安全であるかどうか、有効であるかどうか、および/またはその予期される用途に有効であるかどうか、許容可能な純度プロファイルを有するかどうか、および製品がcGMPに従って製造されているかどうかを決定して、製品の特性、安全性、強度、品質、効力および純度を確保および保存するためにBLAを審査する。FDAは、新規な生物製品または安全性または有効性の問題を提起する生物製品の申請を諮問委員会に提出することができ、一般に、申請を承認すべきかどうか、およびどのような条件下で承認すべきかを審査、評価および提案するための臨床医および他の専門家を含むグループである。FDAは諮問委員会の提案に制限されていないが、それは決定を下す時にこれらの提案を慎重に考慮するだろう。バイオ製品の承認過程において、FDAはまた、バイオ製品の安全な使用を確保するために、リスク評価および緩和戦略、またはREMSを策定する必要があるかどうかを決定する。REMSは、既知または潜在的な薬物に関連する深刻なリスクを管理し、薬物の安全な使用を管理することによって、患者がそのような薬物を継続的に得ることができるようにするための安全戦略であり、制限された分配方法、患者登録、および他のリスク最小化ツールのような薬物ガイドライン、医師のコミュニケーション計画、または安全使用を保証する要素を含むことができる。FDAがREMSが必要であると結論した場合,BLAのスポンサーは提案したREMSを提出しなければならない。必要であれば、FDAはREMSのないBLAを承認しないだろう。
26
BLAを承認する前に、FDAはこの製品を生産する施設を検査する。FDAは、製造プロセスおよび施設がcGMP要件に適合していることを決定し、要求された規格内で治療薬を一貫して生産することを確実にするのに十分でない限り、治療薬を承認しないであろう。細胞療法については,メーカーが適用範囲内でGTPSに適合していなければ,FDAもこの製品を承認しない。これらは、ヒト細胞、組織、および細胞および組織に基づく製品またはHCT/Pを製造するための方法、施設および制御を管理するFDAの法規制および指導文書であり、HCT/Pは、ヒトレシピエント内に移植、移植、注入または転移するためのヒト細胞または組織である。さらに、BLAを承認する前に、FDAは通常、IND試験要求およびGCP要求に従って臨床試験が行われることを確実にするために、1つまたは複数の臨床場所を検査する。CGMP,GTP,GCPに適合することを確保するためには,申請者は訓練,記録保存,生産,品質管理に多大な時間,お金,労力をかけなければならない。
2017年11月、FDAは“ヒト細胞、組織、細胞と組織製品の規制考慮:最小限の操作と同源使用-工業と食品薬品監督管理局職員ガイドライン”と題する指導文書を発表し、2020年7月に改訂、再発表、またはガイドラインを発表した。この文書は,21 CFR Part 1271によりバリアやカバーとして使用しようとしているシート状羊膜組織を21 CFR Part 1271により製造し,361条Hct/Psのみによる適切な規制を行うFDAの立場を確認している。GTP要求の主な目的は,細胞や組織に基づく治療薬の製造方式の確保であり,感染症の導入,伝播,伝播を防止することである。FDAの規定はまた、組織機関がFDAに彼らのHCT/Pを登録し、適用した場合にスクリーニングとテストを通じてドナーを評価することを要求する。FDAはその指導意見の中で、この機関は限られた条件下でいくつかのHCT/PのIND申請および上場前の承認に対して法執行裁量権の行使を要求すると表明しているが、この執行裁量期間は2021年5月31日までである。
関連データおよび情報が提出されたにもかかわらず、FDAは、BLAがその承認の規制基準を満たしていないことを最終的に決定し、承認を拒否する可能性がある。臨床試験から得られたデータはつねに決定的ではなく,FDAのデータ解釈は我々の同じデータに対する解釈とは異なる可能性がある。FDAが現在の形態のBLAを承認しないと決定した場合、FDAは、FDAによって決定されたBLA内のすべての特定の欠陥を記述する完全な返信を発行するであろう。決定された欠陥は微小である可能性があり、例えば、ラベル変更が必要であるか、または重大であり、例えば、追加の臨床試験が必要である。さらに、完全な返信状は、出願人がとり得る、申請を承認条件に置くための提案行動を含むことができる。完全な返信が発行された場合、出願人は、BLAを再提出し、手紙で決定されたすべての不足点を解決するか、または出願を撤回することができる。
ある治療法が規制部門の承認を得た場合,承認は特定の疾患や投与量に限られる可能性があり,あるいは使用の適応が制限される可能性があり,この療法の商業的価値を制限する可能性がある。さらに、FDAは、ラベルにいくつかの禁忌症、警告、または予防措置を含むことを要求する可能性がある。FDAは、リスク管理計画の形態で、配布、処方または調剤に制限および条件を適用することができ、または他の方法で任意の承認範囲を制限することができる。また、FDAは市販後の臨床試験を要求する可能性があり、第四段階臨床試験と呼ばれることがあり、生物製品の安全性と有効性をさらに評価することを目的とし、商業化された承認された療法の安全性を監視するためにテストと監督計画を要求する。
さらに、小児科研究公平法またはPREAによれば、BLAまたはBLA補充剤は、すべての関連する小児科亜群において主張される適応の安全性および有効性を評価し、各安全で有効な小児科亜群に対する製品の用量および投与をサポートするためのデータを含まなければならない。FDAはデータの提出を延期することを許可するか、またはすべてまたは部分的な免除を与えることができる。規制が別途要求されない限り、PREAは孤児の称号が付与されたいかなる製品にも適用されない。しかし、1つの治療適応のみが孤児の称号を有する場合、どのアプリケーションも同じ治療法を孤児適応に進めるために小児科評価が必要となる可能性がある。
孤児薬名
孤児医薬品法によれば、FDAは、米国では通常20万人未満に影響を与え、米国では20万人を超える影響を与え、かつ合理的な期待はなく、米国でそのような疾患または疾患を治療する薬剤または生物学的薬剤を開発および提供するための薬剤または生物学的薬剤の称号を米国で提供することができ、米国でそのような疾患または生物学的薬剤または生物学的薬剤の米国での販売から回収される。BLAを提出する前に,指定孤児薬を申請しなければならない。FDAが孤児薬物指定を承認した後、FDAは、治療薬の模倣薬識別情報およびその潜在的孤児の使用を開示する。孤児薬物の指定は、規制審査または承認過程においていかなる利点も伝達されず、規制審査または承認過程の持続時間を短縮することもない。
孤児薬指定を有する治療薬が、その後、このような指定された疾患に対するFDAの最初の承認を得た場合、この治療薬は、孤児製品の独占経営権を得る権利があり、これは、FDAが完全な血乳酸を含む他の出願を承認しない可能性があり、限られた場合、例えば孤児薬に対して排他的な治療薬に対する臨床的利点を示さない限り、7年以内に同じ適応の同じ生物学的薬剤を販売することを意味する。孤児薬の排他性はFDAを妨げていません
27
同じ疾患または状態のために異なる医薬または生物学的製剤を承認するか、または異なる疾患または状態のために同じ医薬または生物学的製剤を承認する。孤児薬を指定する他の利点は、いくつかの研究の税金控除およびBLA申請使用料の免除を含む。
指定された孤児薬物が孤児が指定された適応よりも広い用途で承認された場合,孤児薬物の排他性を得ることはできない。さらに、FDAが指定された要求に重大な欠陥があると後に判断した場合、または製造業者がこのような稀な疾患または疾患患者の需要を満たすのに十分な数の製品を保証できない場合、米国での独占営業権を失う可能性がある。
2021年4月、FDAは、悪性グリオーマ患者の孤児薬の治療のための、我々の非遺伝子組換え凍結保存ヒト胎盤造血幹細胞由来NK細胞療法、CYNK-001を承認した。
開発と審査計画を加速する
FDAはいくつかの計画を制定し、新薬の開発と審査を促進し、加速し、生命に深刻な或いは生命を脅かす疾病治療中に満足されていない医療需要を解決することを目的としている。これらの計画には、高速チャネル指定、画期的な治療指定、加速承認、優先審査指定が含まれています。具体的には,新たな治療法が重篤あるいは生命に危険な疾患や状況を治療し,その疾患や状況が満たされていない医療需要を解決する潜在力を示す場合には,迅速なチャネル指定を受ける資格がある。高速チャネル指定は,治療と検討されている特定の適応の組み合わせに適している。高速チャネル製品の場合、FDAは、完全な申請を提出する前にBLAを審査する部分をスクロールすることを考慮することができ、スポンサーがBLA部分を提出するスケジュールを提供した場合、FDAはBLAの部分を受け入れることに同意し、スケジュールが許容可能であると判断し、スポンサーはBLAの第1の部分を提出する際に任意の必要な使用料を支払う。
迅速なチャネル指定を有する療法を含むFDA承認を提出する任意の療法は、優先的な審査および承認の加速など、FDAの開発および審査を加速するための他のタイプの計画に参加する資格がある可能性がある。1つの療法が満足できる代替療法なしに安全かつ有効な療法を提供する可能性がある場合,あるいは市場の療法と比較して疾患の治療,診断または予防に有意な改善がある場合には,優先審査する資格がある。FDAは、優先審査として指定された新しい療法の申請を評価するために追加のリソースを使用することを試み、審査を促進するために努力する。しかも、治療薬は加速的な承認を受ける資格があるかもしれない。重篤または生命を脅かす疾患または状態の安全性および有効性の治療に関して研究された治療薬は、製品が臨床的利益を合理的に予測する可能性のある代替終点に有効であるか、または不可逆的な発症率または死亡率よりも早く測定可能な臨床終点に有効であると判定され、病状の重症度、希少性または流行率、および代替治療を利用可能または不足し、不可逆的な発症率または死亡率または他の臨床的利益への影響を合理的に予測することを考慮すると、加速承認を得ることができる。承認の条件として,FDAは,承認を加速させた薬物や生物製品のスポンサーが十分かつ制御された上場後の臨床研究を行うことを要求することができ,2022年の食品·薬物総合改革法案(FDORA)によると,FDAは現在,このような試験を承認前または承認が加速された製品の承認日後の特定の期間で行うことを適宜要求することが可能である。FDORAによれば、FDAは、例えば、検証試験が製品の予期される臨床的利益を検証することができない場合、承認の加速下で承認された薬物または適応の承認を撤回することができるように、手続きを加速させる権限を増加させる。また、承認の加速を検討している製品については、FDAは現在、機関がスポンサーに別途通知しない限り、上場承認後120日以内に伝播または発表しようとするすべての広告および販売促進材料を承認前審査期間内に機関に提出しなければならず、これは、製品の商業発売時間に悪影響を及ぼす可能性がある。さらに、画期的な治療指定は、重篤または生命に危険な疾患の治療方法の開発と審査を加速することを目的としている。FDAの指定は初歩的な臨床証拠を必要とし、候補治療薬の単独或いは他の薬物と生物製品との併用は、1つ以上の臨床的重要な終点で現在利用可能な治療方法よりも実質的な改善を示し、例えば臨床開発早期に観察された実質的な治療効果を示す。FDAが画期的な治療法を指定した場合、申請の開発および審査を加速させるために適切な行動をとることができ、これは、(I)治療開発全体にわたってスポンサーおよび審査チームとの会議を行うこと、(Ii)薬物の開発についてスポンサーにタイムリーなアドバイスを提供し、相互作用を行うことを含むことができ、承認に必要な非臨床および臨床データを収集する開発計画が可能な限り有効であることを確保すること、(Iii)高度管理者および経験豊富な審査者を適宜協力させる学際的審査を含むことができる。(Iv)開発計画の効率的な審査を促進し、審査グループとスポンサーとの間の科学的連絡人として機能するために、FDA審査グループに学際的プロジェクト担当者を割り当てること、および(V)科学的に適切な場合に代替臨床試験設計を考慮することは、より小さい試験またはより効果的な試験をもたらす可能性があり、これらの試験は、達成するためにより少ない時間を必要とし、可能性の悪い治療に曝露された患者数を最大限に減少させる可能性がある。画期的な治療指定は、高速チャネル指定のすべての利点を伴うが、これは、いくつかの条件が満たされる場合、スポンサーが、一部の申請を提出する提案スケジュールについてFDAと合意すること、およびFDAが審査を開始する前に適用される使用料を支払うことを含む、BLAの提出部分をスクロールして審査することができることを意味する。画期的な治療指定は加速承認や優先審査とは異なり,後者も関連基準を満たす場合に同一製品を付与することができる。製品が画期的な治療法として指定されている場合、FDAは製品の開発と審査を加速するだろう。
28
迅速チャネル指定、優先審査、および画期的な治療指定は、承認の基準を変更することはありませんが、開発や承認プロセスを加速させる可能性があります。
2021年3月、我々は、非遺伝子凍結保存ヒト胎盤造血幹細胞由来NK細胞治療のためのFDAの迅速チャネル認証を取得した。
承認後に要求する
FDAによって承認されたどの治療法も、記録要件、製品副作用の報告、FDAへの最新の安全性および有効性情報の提供、製品サンプルおよび流通要件、およびFDAの宣伝および広告要件の遵守を含むFDAの継続的な規制を受けなければならない。これらの要件は、消費者向けの広告基準を含むが、製品の承認されていないラベルに記載されていない製品(“非ラベル使用”と呼ばれる)のための製品の普及を制限するか、または製品の承認されていないラベルに記載されている製品(“非ラベル使用”と呼ばれる)、業界支援の科学的および教育活動の制限、およびインターネットに関連する販促活動のための製品の普及を制限する要件を含む。医師は合法的な製品を非ラベル用途のために処方することができるが、医師がその製品が彼/彼女の専門的な医学判断において適切であると思っている場合、メーカーはラベル外用途を販売または普及させてはならない。しかしながら、会社は、FDAによって承認された製品ラベルと一致する真で誤解されない情報を共有するかもしれない。その製品ラベル外使用を促進することが発見された会社は、行政、民事、刑事制裁を含む重大な責任を受ける可能性がある。
また、品質管理及び製造プロセスは、製品の長期安定性を確保するために、承認された後も適用される製造要件に適合し続けなければならない。CGMP条例では,他の事項のほかに,品質管理と品質保証およびそれに応じた記録やファイルの保守が要求され,cGMPから外れた状況を調査·是正する義務がある.承認された製品の製造および流通に参加する製造業者および他のエンティティ、ならびに製品、成分およびその構成要素を提供するエンティティは、FDAおよびいくつかの州機関にその工場を登録し、cGMPおよび他の法律に準拠することを確実にするために、FDAおよびいくつかの州機関の定期的な抜き打ち検査を受けなければならない。処方薬製品のメーカーと他の薬品サプライチェーンに参加する各当事者はまた、製品追跡と追跡要求を遵守し、偽、移転、窃盗、故意に混入した製品または本来米国で流通するのに適していない製品をFDAに通報しなければならない。そのため、メーカーはcGMPコンプライアンスを維持するために、生産と品質管理に時間、お金、労力をかけ続けなければならない。承認後に製品が発見された問題は、製品のリコールまたは市場からの撤回を含む製品、製造業者、または承認されたBLA所有者の制限をもたらす可能性がある。また,製造プロセスの変更は厳しく規制されており,変更の重要性に応じてFDAの承認を得て実施する必要がある可能性がある。新たな適応や声明を増やすなど、製品の他のタイプの変化を承認するには、FDAのさらなる審査と承認も必要である。
FDAはまた、上場後テスト、いわゆる第4段階テストを要求し、許可された製品の効果を監視するために監視を行う可能性がある。製品が以前に未知の問題を発見し、あるいは適用されたFDA要求を遵守できなかったことは、負の結果をもたらす可能性があり、負の宣伝、司法或いは行政法執行、FDAの警告状、強制要求の是正広告或いは医師とのコミュニケーション、民事或いは刑事罰などを含む。新たに発見または開発された安全性または有効性データは、新たな警告や禁忌症を増加させることを含む製品承認のラベルを変更する必要がある場合があり、他のリスク管理措置を実施する必要がある可能性もある。また、新しい立法による要求、またはFDAの政策が変わる可能性がある新しい政府要求を確立することが可能であり、規制部門が私たちが開発している療法を承認することを延期または阻止する可能性がある。
アメリカマーケティング排他性
生物製品価格競争および革新法案、またはBPCIAは、PHSAを改正し、FDAに類似のバージョンを承認することを許可する革新的な生物製品であり、一般に生物模倣薬と呼ばれる。生物類似物の承認を求める競争相手は,その分子が承認されたイノベーター生物と高度に類似していること,および他の要求を証明する申請を提出しなければならない。しかし、BPCIAはFDAが革新者生物製品が初歩的なマーケティング許可を得てから12年以内に生物類似申請を許可することを禁止した。小児科専門権を付与すれば,この12年間のデータ専有期間は6カ月,合計12.5年延長できる。小児科排他性はアメリカのもう一つの規制市場排他性だ。この6ヶ月間の排他的保護は、他の排他的保護終了時から、FDAが発表したこのような試験の“書面請求”に公平に応答した自発的な小児科試験に基づいている可能性がある。
FDAが我々の候補治療薬使用の時間、期限、および詳細を承認することによれば、その一部の米国特許が承認されれば、1984年の“薬品価格競争および特許期限回復法”(一般にHatch−Waxman法と呼ばれる)によって限られた特許期間延長を受ける資格がある可能性がある。ハッジ·ワックスマン法案は、製品開発およびFDA規制審査中に失われた特許期間の補償として、最長5年間の特許回復期限を許可する。しかし,特許期限の回復は特許の残存期間を延長することはできず,製品承認日から合計14年を超えることはできない。特許期間回復期間は、一般に、INDの発効日とBLA提出日との間の時間の半分に、BLA提出日とその出願が承認された日との間の時間を加える。承認された治療薬に適用される特許は1つのみ延期する資格があり,延期出願は特許が満了する前に提出されなければならない。アメリカ特許
29
商標局はFDAと協議し,任意の特許期限の延長または回復の出願を審査·承認する。将来、私たちは、臨床試験の期待長と関連BLAの提出に関連する他の要因に依存して、現在の期限後の特許寿命を延長するために、現在所有または許可されている特許出願のうちの1つの特許期間を回復することを意図しているかもしれない。
連邦と州の免許と登録
医療産業は広範囲な当局によって厳格に規制されている。したがって、私たちの業務は、いくつかの許可、登録、許可、許可、承認、認証、認可、その他のタイプの連邦、州、地方政府の許可を維持し、私たちが運営している各司法管轄区の様々な法規を遵守することを要求します。例えば、私たちはいくつかの州で許可証と登録を維持することを要求され、私たちの製品とサービスがこのような許可証を必要とする州で生物製品、組織バンク、血液バンク許可証、許可証、登録を取得しました。ニューヨーク州のようないくつかの州は、連邦法律下で発表された指導意見(HCT/Pに関するFDAの指導意見を含む)の解釈に基づいて、BLAのテーマとなっていない製品に対して州法的制限を実施し、これは、各州が私たちの変性疾患製品に適用される異なる、互いに衝突する可能性のある規制枠組みをもたらす可能性がある。私たちは毎年FDAで組織銀行として登録され、アメリカ血液バンク協会の国家認証を受けています。このようなライセンス要件を遵守しないことは、許可証の取り消しまたは一時停止、登録または認証、または是正、監視、民事罰金、民事強制措置、および/または刑事罰を受ける計画を含む実行行動をもたらす可能性がある。
他のアメリカの医療保険法やコンプライアンスの要求は
アメリカでは、FDAに加えて、私たちの活動は、医療保険や医療補助サービスセンター、あるいはCMS、アメリカ衛生公衆サービス部(DHS)の他の部門を含むが、これらに限定されない様々な連邦、州、地方当局によって規制される可能性があります(例えば:監察長室)、米司法省、司法省内の各連邦検事室、そして州や地方政府。例えば、私たちの商業行為は、私たちの研究および販売、マーケティングおよび科学/教育援助計画を含み、改正された“社会保障法”、“虚偽申告法”、“反リベートおよび収賄法”、“健康保険携帯および責任法案”または“HIPAA”のデータプライバシーおよび安全条項、連邦透明性要件、および同様の州法における詐欺および乱用条項を遵守する必要があるかもしれない。
他の事項に加えて、連邦反バックル法規は、個人または実体が知られている場合、直接または間接的に、現金または実物の形態で直接または間接的に提供、支払い、請求または任意の報酬(任意のリベート、賄賂またはリベートを含む)を提供し、個人の推薦または購入、レンタル、注文または手配を誘導し、購入を誘導し、レンタルまたはレンタルし、連邦医療保険、医療補助、または他の連邦医療計画に従って精算可能な任意の物品またはサービスを注文することを禁止する。報酬という単語は価値のあるものを含むと広く解釈されている。連邦反リベート法規は、薬品メーカーと処方者、購入者、および処方マネージャーの間の手配に適用されると解釈される。いくつかの法的例外と規制避難所がいくつかの一般的な活動を保護することは起訴されない。例外と安全港は狭義であり、保護を提供するためには厳格に遵守されなければならない。処方、購入、または推薦された報酬を誘発することを目的としていると告発される可能性があるやり方に関連し、例外または安全港の資格を満たしていなければ審査される可能性がある。特定の適用された法定例外や安全港を規制するすべての要求を満たしていないことは、このような行為自体が“反リベート条例”に規定された不法行為であることを意味するわけではない。代わりに、そのすべての事実と状況の累積審査に基づいて、この手配の合法性を逐案的に評価する。私たちの接近はすべての場合、法定例外や安全港保護を規制するすべての基準を満たしていないかもしれない。
また、連邦“反リベート法令”の下の意図基準は2010年に“患者保護平価医療法案”によって改正され、この法案は2010年に“保健·教育和解法案”(総称して“平価医療法案”と呼ばれる)で改正され、より厳しい基準を達成し、個人や実体が連邦の“反リベート法令”を実際に理解する必要がなくなり、あるいはその法令に違反する具体的な意図があれば違反行為を実施することができる。代わりに、報酬の“一つの目的”が推薦を誘導するためのものであれば、連邦反リベート規制は違反される。また,“平価医療法”は,連邦民事虚偽申告法(以下,議論)については,連邦“反リベート条例”違反による物品やサービスを含むクレームが虚偽や詐欺的クレームを構成する判例法を編纂している。
連邦民事罰金法規は、任意の個人またはエンティティに処罰を加え、他に、故意に、または連邦医療保健計画に虚偽または詐欺的なクレームを提出することをもたらすと判断され、その人は、クレームまたは虚偽または詐欺的に提供されていないプロジェクトまたはサービスのために提供されていることを知っているか、または知るべきである。また、反リベート法規に違反した行為は民事と刑事罰金に処せられ、違反ごとに報酬の3倍までの罰、監禁、および政府医療計画から除外される。
連邦民事および刑事虚偽請求法は、他の事項に加えて、個人または実体が知られている場合に、Medicare、Medicaidまたは他の連邦政府計画からの支払いまたは承認クレームの提出または提出を引き起こすことを禁止する連邦民事および刑事虚偽請求法を含み、これらのクレームは、虚偽または詐欺的であるか、または不正に回避、減少するために意図的に虚偽声明を作成することを禁止する
30
連邦医療計画を含めて連邦政府に資金を支払う義務を隠すか。2009年の“詐欺法執行·回収法”の改正により、クレームには、連邦政府に提出された金銭または財産に対する“任意の請求または要求”が含まれている。製薬や他のヘルスケア会社は調査を受けているか、または過去にこれらの法律に基づいて起訴されており、顧客に製品を無料で提供する疑いがあり、顧客がその製品の連邦計画に課金することを期待している。また,製薬や他のヘルスケア会社も起訴されており,これらの会社のマーケティング製品が未承認の用途に使用されており,虚偽の声明が提出されているため,精算できない。連邦虚偽申告法は、連邦虚偽申告法違反を告発し、いかなる金銭回収も共有する“密告者”としての個人代表が連邦政府を代表して訴訟を起こすことも許されている。
HIPAAは、計画を故意かつ故意に実行または実行しようとすることを禁止し、虚偽または詐欺的な言い訳、陳述または承諾の方法で任意の医療福祉計画(個人第三者支払者を含む)が所有または制御または保管している任意の金銭または財産を詐欺または取得し、悪巧み、計画または装置、重大な事実、または任意の重大な虚偽、架空または詐欺的陳述によって、医療福祉、プロジェクトまたはサービスの提供または支払いに関連する情報を故意に偽造、隠蔽、または隠蔽する追加の連邦刑法を制定する。連邦反リベート法規と同様に、個人または実体は、この法規やこの法規に違反する具体的な意図を実際に知る必要がなく、違反を実施することができる。
私たちは連邦政府と私たちが業務を展開している州のデータプライバシーと安全規制によって制限されるかもしれない。“2009年衛生情報技術促進経済と臨床衛生法案”(HITECH)及びその実施条例改正されたHIPAAは、保証実体を含むいくつかのタイプの個人と実体に対して要求を提出した(HITECH)及びその実施条例改正されたHIPAAはあるタイプの個人と実体に対して要求を提出した(即.個人が健康情報を識別することができるプライバシー、セキュリティ、および送信に関する特定のヘルスケア提供者、健康計画、および医療情報交換所)。他の事項に加えて、HITECHは、HIPAAのプライバシーおよびセキュリティ基準を、保証エンティティまたは代表の保証エンティティにサービスを提供することに関連する保護された健康情報を受信または取得する保証エンティティである独立請負者またはエージェントの商業パートナー(およびその下請け業者)に直接適用させる。HITECHはまた4つの新しい民事罰金等級を作成し、HIPAAを改訂し、民事と刑事処罰を商業パートナーに直接適用し、州総検察長に新しい権力を与え、連邦裁判所に民事訴訟を提起し、損害賠償または禁止令を要求して連邦HIPAA法を執行し、連邦民事訴訟に関連する弁護士費と費用を求めることができる。
州法はまた個人情報のプライバシーとセキュリティを管理している。多くの州の法律は大きく異なり、コンプライアンス作業を複雑にしている。例えば、カリフォルニア消費者プライバシー法(California Consumer Privacy Act、略称CCPA)は、カリフォルニアに位置する個人のためにデータプライバシー権を確立し、企業がこのような個人の個人情報をどのように収集して使用するかを要求している。カリフォルニアプライバシー権法案(CPRA)は2023年1月1日に施行され、立法がカバーする会社に追加的な義務を課し、特定の敏感な個人情報に関する消費者の権利を拡大し、CCPAを実行する権利を有する州機関を設立することを含むCCPAの重大な改正を行った。“包括的平和協定”(“包括的平和協定”改正)がどのように実行されるか、どのように解釈されるかはまだ完全には明らかではない。CCPAの絶えず変化する性質は、私たちのデータ収集や処理やり方と政策を修正し、遵守するために大量のコストと支出を発生させることを要求するかもしれない。
CCPA(CPRA改正により)は、バージニア州、コロラド州、ユタ州、コネチカット州などの他の州に同様の全面的なプライバシーとデータ保護立法を公布し、これらの立法は2023年に施行される。また,米国の他のいくつかの州でも同様のプライバシーやデータ保護立法が提案されており,いくつかの提案が採択される可能性がある.多くの既存の州プライバシー法はHIPAAが管轄する臨床試験情報や健康情報を免除しているにもかかわらず,将来のプライバシーやデータ保護法の範囲が広くなる可能性がある。また、国家プライバシー法の急増は、私たちの個人情報の収集と使用のリスクと不確実性を増加させた。これは私たちの業務に巨大なコンプライアンスコストと支出をもたらす可能性があり、規制法執行および/または訴訟における私たちの潜在的なリスクを増加させ、私たちの新しい顧客を誘致し、維持する能力にマイナスの影響を与えるかもしれない。
さらに、“平価医療法案”によって作成された連邦“医師支払陽光法案”およびその実施条例の要求に基づいて、連邦医療保険、医療補助または児童健康保険計画(いくつかの例外を除いて)に支払うことができるいくつかの薬品、器具、生物および医療用品の製造業者は、医師(医師、歯科医師、視光師、足科医師および脊椎マッサージ師を含むと定義されている)および教育病院または医師および教育病院の要求または指定された実体または個人の支払いまたは分配を表すいくつかの支払いまたは他の価値移転に関する情報を毎年協力医療センターに報告し、医師およびその直系親族が所有するいくつかの所有権および投資利益を毎年報告する。2022年1月1日から,適用されるメーカーは,前年に医師アシスタント,勤務看護師,臨床看護師専門家,登録看護師麻酔科医,麻酔科医アシスタント,登録看護師助産師への支払いと移転価値の情報を報告しなければならない。
“反海外腐敗法”(FCPA)は、個人または企業が業務を獲得または保持するのを助けるために、任意の米国人または企業が、任意の外国人官僚、政党または候補者に直接的または間接的に支払い、提供または許可支払い、または任意の価値のあるものを提供することを禁止し、個人または企業が業務を獲得または保持することを目的とする。“海外腐敗防止法”は米国に上場する証券会社にも要求を遵守することを求めている
31
国際子会社を含む会社のすべての取引の帳簿や記録を正確かつ公平に反映し、国際業務のための適切な内部会計制御システムを策定·維持する。
また、多くの州で同様の詐欺や法律や法規の乱用があり、医療補助や他の州が計画して精算するプロジェクトやサービスに適用されたり、いくつかの州では支払者にかかわらず適用されている。治療薬を商業的に流通させるためには、州の薬品および生物製品の製造業者および卸売業者の登録を要求する州の法律を遵守しなければならない。これらのメーカーまたは卸売業者が州に営業場所がなくても、いくつかの州で製品を州のメーカーおよび卸売業者に輸送することを含む。一部の州はまた、メーカーと流通業者が流通チェーン中で製品の系統を確立することを要求しており、いくつかの州はメーカーと他の州に流通チェーン中の製品の流れを追跡し追跡できる新しい技術を採用することを要求している。いくつかの州と地方司法管轄区はすでに立法を公布し、製薬と生物技術会社にマーケティングコンプライアンス計画を確立し、製薬業界の自発的コンプライアンスガイドラインと連邦政府が公布した関連コンプライアンスガイドラインを遵守し、州政府に定期報告を提出し、販売、マーケティング、定価、臨床試験およびその他の活動を定期的に公開開示し、および/またはその販売代表を登録し、薬局と他の医療実体が製薬と生物技術会社にある医師処方データを販売とマーケティングのために提供することを禁止し、そしていくつかの他の販売とマーケティング行為を禁止する。私たちのすべての活動は連邦と州消費者保護と不正競争法によって制限されるかもしれない。
私たちの業務が上記の任意の連邦および州医療保健法律または私たちに適用される任意の他の政府法規に違反していることが発見された場合、私たちは、MedicareおよびMedicaid、禁止、契約損害、名声損害、行政負担、利益および将来の収益の減少、追加報告要件および/または監督など、民事、刑事および/または行政処罰、損害賠償、罰金、返還、監禁、MedicareおよびMedicaid、禁止、契約損害、名声損害、行政負担および将来の収益の減少、追加報告要件および/または監督など、重大な処罰を受ける可能性があり、もし私たちが会社の誠実協定または同様の合意の制約を受けて、これらの法律違反に関する告発、および私たちの業務の削減または再編を解決すれば、いずれも私たちの業務運営能力と運営結果に悪影響を及ぼす可能性があります。
保証範囲·定価·精算
私たちが監督管理の許可を得た任意の候補治療方案のカバー範囲と精算状態に重大な不確定性が存在する。米国や他の国のある市場では、規制部門の許可を得て商業販売を行う任意の治療薬の販売は、第三者支払者が保険を提供する程度にある程度依存し、そのような製品のための十分な補償レベルを確立する。米国では統一された保険範囲や精算政策はなく、支払先によって保証範囲や精算が大きく異なる可能性がある。したがって、カバー範囲を決定するプロセスは、しばしば時間がかかり、高価である。米国では,第三者支払者には連邦や州医療計画,個人管理のヘルスケア提供者,医療保険会社,その他の組織が含まれている。第三者支払者が製品に保険を提供するかどうかを決定するプロセスは、製品価格を設定するプロセスから分離されてもよく、第三者支払者が製品に支払う支払率を決定するプロセスと分離されてもよい。第三者支払者は、承認リスト上の特定の製品に保証範囲を制限することができ、処方表とも呼ばれ、FDAによって承認された特定の適応のすべての製品を含まない可能性がある。第三者決済者は、価格に挑戦し、医療の必要性を審査し、医療製品、療法、サービスの費用対効果を審査するとともに、それらの安全性と有効性を疑問視するようになっている。我々の治療法の医療の必要性と費用効果,FDAの承認を得るのに必要なコストを証明するためには,高価な薬物経済学的研究が必要かもしれない。私たちの候補治療方案は医学的に必要あるいは費用効果が高いと思われないかもしれない。支払者が製品に保険を提供することを決定することは、十分な返済率を承認することを意味するものではない。私たちの治療薬の正味価格は、政府の医療計画や個人支払者が要求する強制的な割引やリベート、および将来の法律のいかなる緩和によっても低下することができ、これらの法律は現在、米国よりも販売価格が低い国からの薬品の輸入を制限している。また、1人の支払人が1つの治療に保険を提供することを決定し、他の支払人もその治療に保険を提供することを保証することはできない。治療開発への投資の適切な見返りを達成するために、十分な価格レベルを維持することができる十分な第三者精算がないかもしれない。
他の国もまた違う価格設定と精算プログラムを持っている。EUでは,各国政府はその定価と精算規則及び国家医療システムの制御により薬品の価格に影響を与え,これらのシステムは消費者に大きな薬品コストを支払っている。いくつかの法域はプラスリストとネガティブリスト制度を実行し、補償価格を合意した後にのみ、製品を販売することができる。精算または定価の承認を得るために、その中のいくつかの国は臨床試験の完成を要求する可能性があり、特定の候補療法の費用効果を現在利用可能な治療法と比較する。他の会員国たちは会社が自分の薬品価格を固定することを許可するが、会社の利益を監視する。医療コストの下振れ圧力は非常に大きくなっている。そのため、新製品の参入にはますます高い壁が設けられている。また、一部の国では、低価格市場からの国境を越えた輸入が一国国内の定価に商業圧力をかけている。そのため、米国以外の市場では、米国に比べてわが製品の精算が減少する可能性があり、商業的に合理的な収入や利益を生み出すには不十分である可能性がある。定価とリベート計画はアメリカの1990年の総合予算調節法ともっと多くの内容の中の医療補助リベートの要求に符合しなければならない
32
2010年に“保健·教育和解法案”で改正された2010年の“患者保護平価医療法案”は,総称して“平価医療法案”の最新要求と呼ばれている。総務省連邦供給スケジュールの許可されたユーザに製品を提供する場合は、他の法律および要求が適用される。
政府や第三者支払者が十分な保険や精算を提供できない場合、規制部門の承認を得て商業販売を行う任意の候補治療薬の適正性が影響を受ける可能性がある。また,米国では管理型医療への重視度が増加しており,医療定価の圧力が増加し続けることが予想される。例えば、連邦と州政府および医療計画の行動は、薬品価格と医療コストに追加の下振れ圧力をもたらす可能性があり、承認されれば、これは私たちの製品のカバー範囲と清算、私たちの収入、および私たちが他の市場製品と競争し、私たちの研究開発コストを回収する能力にマイナスの影響を与えるかもしれない。保証政策と第三者精算料率は随時変化する可能性がある。規制部門の承認を得た1つ以上の療法が有利な保証範囲と精算状態を得ても、将来的にはあまり有利ではない保証政策や精算料率が実施される可能性がある。
医療改革
米国や一部の外国司法管轄地域では、医療システムに関するいくつかの立法や規制の変化や提案された変化が継続されており、これらの変化は、候補治療薬の発売承認を阻止または延期し、承認後の活動を制限または規範化し、マーケティングの承認を得た治療候補薬を収益的に販売する能力に影響を与える可能性がある。米国や他の地方の政策立案者や支払者の中で,医療システムの改革を推進することに大きな興味があり,医療コストの抑制,質の向上および/または参入拡大を既定の目標としている。米国では、製薬業はこれらの努力の重点であり、重大な立法計画の大きな影響を受けてきた。
例えば、“平価医療法案”は、政府や民間保険会社の医療融資や提供方法を大きく変えた。上記の条項以外に、“平価医療法”の中で製薬と生物技術業界に対して重要な意義を持つ条項は以下の通りである
33
“平価医療法案”のいくつかの面は依然として行政、法律、政治面の挑戦に直面している。例えば,2019年12月,ケディラク税,医療保険提供者税,医療機器消費税を廃止したさらなる総合支出法案(H.R.1865)が法律に署名された。また,2019年1月に発効した2018年の両党予算法は,他にも“平価医療法案”が改正され,多くの連邦医療保険薬物計画におけるカバーギャップ,すなわち通常言われている“ドーナツ穴”を埋めるために改正された。2021年6月,米国最高裁はいくつかの州で“平価医療法案”に対する最新の司法挑戦を却下したが,“平価医療法案”が憲法に適合しているかどうかは具体的には判断されなかった。最高裁が裁決を下す前に,平価医療法案市場による医療保険の獲得を目的とした2021年2月15日から2021年8月15日までの特別加入期間を開始する行政命令が発表された。行政命令はまた、仕事の要求を含む医療補助モデルプロジェクトおよび免除計画の再検討、医療補助または“平価医療法案”による医療保険獲得に不必要な障害をもたらす政策を含む、特定の政府機関に、医療保健の取得を制限する既存の政策および規則の見直しを指示する。いかなる医療改革立法がアメリカ医療業界の最終内容、時間或いは影響に与えるかはまだ不明である。
これまで、2017年10月に“平価医療法案”に基づいて保険会社に補償された費用分担手当を中止する行政命令に署名してきた。前政府の結論は,“平価医療法案”(Affordable Care Act)が保険会社に支払う費用分担削減(CSR)が国会から必要な支出を得ていないことを要求し,これらの支出が完了するまで直ちに支払いを停止することを発表した。いくつかの州の検事総長は、政府の補助金の中止を阻止することを求めた訴訟を提起したが、彼らは制限令の発行を要求し、2017年10月にカリフォルニア州の連邦裁判官に却下された。2020年8月、米国連邦巡回控訴裁判所は2つの異なる事件の中で、連邦政府はこれまで数年(2017年を含む)の未払いCSRに対して全額責任を負っていると判断した。健康保険会社が2018年以降に提出した企業社会責任クレームについては,満期額(あれば)を決定するためのさらなる訴訟が必要となる。また、2018年6月、米国連邦巡回控訴裁判所は、連邦政府は第三者支払者に120億ドルを超える“平価医療法案”リスク廊下支払いを支払う必要がないと判断し、第三者支払人はこれらの支払いが彼らに不足していると弁明した。2020年4月、米国最高裁判所は米国連邦巡回控訴裁判所の裁決を覆し、事件を米国連邦クレーム裁判所に送り、政府は関連式に基づいてこれらのリスク回廊支払いを支払う義務があると結論した。
“平価医療法案”が基本的に現在の形を維持すれば、私たちが承認された任意の治療法の保証範囲と価格に追加の下り圧力を与え続け、私たちの業務を深刻に損なう可能性があると予想される。医療保険や他の政府が計画している精算のどの減少も、個人支払者の支払いのような減少を招く可能性がある。コスト抑制措置や他の医療改革を実施することは、収入の創出、利益の実現、あるいは私たちの療法を商業化することを阻止するかもしれません。これらの改革は、規制承認された候補治療薬の期待収入の開発に成功し、私たちの全体的な財務状況に影響を与える可能性があり、治療候補薬を開発する能力に影響を与える可能性がある。
さらなる立法や規制によって、私たちの業務、財務状況、そして運営結果を損なう可能性があります。“平価医療法”が公布されて以来,他の立法改正も提案され,採択された。例えば、2011年8月、“2011年予算制御法”が署名されて法律となり、それ以外にも赤字削減合同特別委員会を設置し、国会に支出削減の提案を提案した。赤字削減合同特別委員会は2013−2021年度の少なくとも1.2兆ドルの赤字削減目標を実現せず、いくつかの政府プロジェクトの自動削減を触発した。これには,各年度にプロバイダに支払われる連邦医療保険総額が最高2%減少することが含まれている。これらの削減は2013年4月に施行され、その後の法規制の立法改正により2030年まで有効となるが、2020年5月1日から2022年3月31日までの一時停止は除外される。そして、2022年4月1日から2022年6月30日までに1%の支払い減免が出現し、2022年7月1日に2%の支払い減免が再開された。また、2013年1月、病院、画像センター、がん治療センターを含むいくつかの提供者への医療保険支払いをさらに減らし、政府が提供者に多額の支払いを取り戻す訴訟時効を3年から5年に延長する“2012年米国納税者救済法”が法律に署名された。
また、2018年5月30日には、“裁判権法案”が法律に署名された。他の事項以外に、この法律はある患者に連邦フレームワークを提供し、彼らが第一段階の臨床試験を完成し、FDAの許可を得た研究用新薬製品を獲得するために調査を行っていることを許可した。場合によっては、条件を満たす患者は、臨床試験に参加することなく、FDA拡大参入計画に従ってFDA許可を得ることなく治療を求めることができる。“試用権法案”によると,製薬業者はその薬品を条件に該当する患者に提供する義務はない。
米国の特殊薬品価格設定実践における立法と法執行の興味もますます大きくなっている。連邦レベルでは、2021年7月に行政命令に署名し、(I)立法改革を支持し、医療保険と薬品価格の交渉を許可することを含む処方薬と生物製品の価格を下げる政府の政策を確認した
34
インフレ上限を実施し、低コストの後発薬と生体模倣薬の開発と市場進出を支持する;および(2)公共健康保険代替案の公布を支持する。その他の事項に加えて、行政命令は、処方薬の価格設定が高すぎ、国内薬品サプライチェーンを強化し、連邦政府が薬品支払いの価格を下げ、業界価格詐欺を解決するための行動を説明する報告書を衛生·公衆サービス部(HHS)に指示し、2003年の“連邦医療保険処方薬、改善と現代化法案”とFDA実施条例に基づいて第804条の輸入計画を制定することを提案した州とインディアン部族との協力を指示した。FDAは2020年9月にこのような実施条例を発表し、2020年11月に発効し、各州のカナダ薬品輸入計画の制定と提出に指導を提供した。また、2020年11月、CMSは最恵国モデルを実施する暫定最終規則を発表し、このモデルによると、ある薬品と生物製品の連邦医療保険B部分の販売率は1人当たりGDPに類似した経済協力と発展組織国の薬品メーカーが受け取った最低価格に基づいて計算される。2021年12月、CMSは最恵国規則を廃止した。また、カナダ当局はカナダの薬品供給を不足から保護するための規定を採択した。実施すれば、カナダからの輸入薬は私たちの任意の候補治療薬の価格に実質的かつ不利な影響を与える可能性がある。また、2020年12月、HHSは、薬品メーカーがD部分でスポンサーの値下げを計画している安全港の保護を廃止し、法律が値下げを要求しない限り、直接あるいは薬局福祉マネージャーを通じて規制を発表した。この規定はまた、販売時点での値下げを反映するための新しい避難港を作成し、薬局福祉マネージャーと製造業者との間のいくつかの固定料金手配のための避難港を作成する。2020年12月、HHSは薬品メーカーがD部分でスポンサーの値下げを計画している安全港の保護を取り消し、法律が値下げを要求しない限り、直接或いは薬局福祉マネージャーを通過する法規を発表した。この規定はまた、販売時点での値下げを反映するための新しい避難港を作成し、薬局福祉マネージャーと製造業者との間のいくつかの固定料金手配のための避難港を作成する。裁判所の命令により,上記安全港の除去と増加は延期され,最近の立法は同規則の実施を2026年1月に一時停止した。2022年のインフレ削減法案、あるいはアイルランド共和軍は、さらにこの規則の実施を2032年1月に延期する。
2022年8月、アイルランド共和軍は法律に署名した。IRAはいくつかの著者らの業務に異なる程度の影響を与える条項を含み、Medicare Part D受益者のために2,000ドルの自己負担上限を作成し、Medicare Part D中のすべての薬品に新しいメーカーの財務責任を適用し、アメリカ政府がある模造薬或いは生物類似競争のない高コストの薬物と生物製品のMedicare B部分とD部分の価格について交渉を行うことを許可し、会社にインフレより速い薬品価格のためにMedicareにリベートを支払うことを要求し、そして薬局福祉マネージャーを受益者にリベートすることを要求するリベート規則を延期する。Ireland共和軍が私たちの業務と医療産業全体に及ぼす影響はまだ明確ではない。
その中のいくつかの措置および他の提案された措置は発効するために追加の立法許可を必要とする可能性があり、行政はこれらの措置を撤回または他の方法で変更する可能性があるが、現在の政府と国会は薬品コストを制御するための新しい立法措置を求め続けると表明している。米国の個別州も、価格や患者の精算制限、割引、ある製品への参入の制限、マーケティングコストの開示と透明性措置を含む、薬品と生物製品の定価を制御するための法規を立法と実施することをますます積極的に実施しており、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。
法規を付加する
これらの規定に加えて,環境保全や有害物質に関する州や連邦法は,“職業安全と健康法”,“資源節約と回収法”,“有毒物質制御法”を含めて,我々の業務に影響を与える。これらの法律と他の法律は私たちの様々な生物、化学、放射性物質の使用、処理と処理、これらの物質は私たちの行動、そして私たちの行動によって生成された廃棄物を規範化している。もし私たちの運営が環境汚染を招いたり、個人を危険物質に曝露させたりすれば、損害賠償と政府罰金の責任を負う可能性があります。私たちは、私たちが適用される環境法律を実質的に遵守し、これらの法律を遵守し続けることは、私たちの業務に実質的な悪影響を与えないと信じている。しかし、私たちはこのような法律の変化が私たちの未来の運営にどのように影響するか予測できない。
ヨーロッパ/世界の他の地域の政府規制
米国の法規に加えて、臨床試験および私たちの治療薬の任意の商業販売と流通を含む他の管轄区域の様々な法規によって制限される。FDAによる治療薬の承認を得るか否かにかかわらず,これらの国での臨床試験やその治療薬の販売を開始する前に,外国の規制機関の必要な承認を得なければならない。米国以外のある国にも類似したプログラムがあり,ヒト臨床試験開始前に臨床試験申請を提出することが求められており,INDに似ている。例えば,EUでは,臨床試験申請は各国の国家衛生当局と独立した倫理委員会に提出されなければならず,FDAやIRBのようなものである。臨床試験申請が国の要求に応じて承認されると,臨床試験開発が可能となる。生物由来の原材料は独特の汚染リスクに直面しているため、それらの使用はいくつかの国で制限される可能性がある。
35
臨床試験、製品許可、定価と精算を指導する要求と手続きは国によって異なる。いずれの場合も,臨床試験はGCPおよび“ヘルシンキ宣言”からの適用法規要求と倫理原則に基づいて行われなければならない。
EUの規制制度の下で研究薬物や生物製品の監督管理の承認を得るためには、市場許可申請を提出しなければならない。米国で“BLA”を提出するための申請はEUでの要求と類似しているが、他の国を除いて具体的な国の文書要求は除外されている。
EU以外の他の国,例えば東欧,ラテンアメリカ,アジアの国では,臨床試験,製品許可,定価,精算を行う要求は国によって異なる。繰り返しますが,すべての場合,臨床試験はGCPおよび“ヘルシンキ宣言”からの適用法規要求と倫理原則に基づいて行われなければなりません。
もし私たちが適用された外国の規制要求を遵守できなかった場合、私たちは罰金、規制承認の一時停止または撤回、製品のリコール、治療薬の差し押さえ、操作制限、刑事起訴などを受ける可能性がある。
従業員と人的資本
2022年12月31日現在、225人のフルタイム従業員と35人の非レンタル従業員を持っています。これらの従業員のうち,41人が博士または医学博士号を持ち,35人が研究に従事し,12人が臨床開発に従事し,89人が技術操作に従事している。私たちのほとんどの従業員はニュージャージー州のフロラム公園にいます。私たちの職員たちは労働組合代表もなく、集団交渉協定のカバー範囲もない。私たちは私たちが従業員と仲がいいと思う。2023年1月、私たちは仕事の優先順位の再調整を発表し、2023年3月現在、従業員数は約3分の1に減少した。
私たちの人的資本資源目標は、私たちの既存の、より多くの従業員を識別、採用、維持、激励、統合することを含む。我々のインセンティブ計画の主な目的は、株に基づく報酬奨励と現金に基づく業績ボーナス奨励を付与することによって、選定された従業員、顧問、取締役を誘致、維持、激励することである。
利用可能な情報
私たちが電子的にアメリカ証券取引委員会に提出したり、材料を提供したりした後、私たちの年間報告書(Form 10-K)、四半期報告(Form 10-Q)、現在の報告書(Form 8-K)、および取引法第13(A)または15(D)節に提出または提出された報告書の任意の改正を、私たちの公共サイト(www.celularity.com)の投資家部分に無料で掲示することができるようにする。また、インターネットで私たちのアメリカ証券取引委員会の届出書類を読むことができます。サイトはアメリカ証券取引委員会のウェブサイトwww.sec.govです。このウェブサイトの内容はこの10-K表の年間報告書に含まれていない。また,これらのサイトのURLへの参照は非アクティブテキスト参照のみに用いた.
36
第1 A項。RISK因子です。
以下のリスク要因と、本年度報告Form 10-Kおよび私たちの他の公開ファイルの他の情報をよく考慮しなければなりません。これらのリスクの発生は、私たちの業務、財務状況、運営結果、および/または成長の見通しを損なう可能性があり、または私たちの実際の結果は、本報告書および私たちが時々行う可能性のある前向きな陳述に含まれる結果とは大きく異なる可能性がある。私たちの業務を評価する際に、あなたは私たちの公開申告書類に記載されているすべてのリスク要因を考慮しなければならない。
商工業にかかわるリスク
我々は設立以来,各時期に純損失が発生しており,細胞療法が商業販売に承認されておらず,将来的に大量の純損失が予想される。
私たちは臨床段階の生物製薬会社であり、細胞療法が商業販売のために承認されておらず、これまで細胞治療販売から何の収入も得られておらず、私たちの変性疾患やバイオバンク業務による収入は限られており、私たちの持続的な運営に関連する重大な研究開発とその他の費用が生じ続ける。生物製薬製品開発への投資は非常に高い投機性があり、それは大量の前期資本支出を必要とし、そして重大なリスクが存在するため、即ちいかなる潜在的な候補治療薬物は十分な治療効果或いは受け入れ可能な安全性を証明できず、監督管理部門の許可を得ることができず、商業上実行可能ではない。したがって、私たちは利益を上げておらず、設立以来どの時期も純損失を出している。2022年12月31日と2021年12月31日までに、1420万ドルの純利益と1.01億ドルの純損失をそれぞれ報告した。2022年12月31日現在、私たちの累計赤字は6.455億ドルです。
私たちは予測可能な未来に巨大な支出を産生することが予想され、私たちが引き続き研究し、私たちの4種類の胎盤由来の同種異遺伝子細胞タイプに基づく細胞治療候補薬物を開発し、規制承認を求めることに伴い、これらの支出は増加する:CAR-T細胞、未修飾NK細胞、トランスジェニックNK細胞およびMLASCs。私たちが1つ以上の候補治療薬を商業化することに成功しても、より多くの候補治療薬を開発し、マーケティングするために、大量の研究開発や他の支出を生み出し続けていく。また,我々が予想している生体材料製品のバージョンアップに関連したコストが発生し,米国以外での拡張を支援し,最初は中東と北アフリカ市場に重点を置く予定である。私たちは予測できない費用、困難、合併症、遅延、および私たちの業務に悪影響を及ぼす可能性のある他の未知の要素に直面するかもしれない。私たちの将来の純損失の規模は、私たちの将来の費用の成長率と候補細胞治療から収入を創出する能力にある程度依存するだろう。私たちの以前の損失と予想された未来の損失はすでに私たちの株主権益と運営資本に悪影響を与え続けるだろう。
私たちの歴史的経営実績は、私たちが経営を続けている企業として経営を続ける能力に大きな疑問があることを示しています。
設立以来,経営活動中に純損失が発生し,大量の現金を使用しており,商業販売が許可されていない細胞治療候補薬は,将来的に大量の純損失が予想される。2022年12月31日現在、私たちの累計赤字は6.455億ドル、現金と現金等価物は1400万ドルです。また、私たちがヨークビルと合意した前払い前払い合意によると、私たちは現在毎月現金の支払いを要求されており、このような義務を履行するのに十分な現金がない。私たちの株価改善やヨークビルが現金支払い義務を放棄しない限り、私たちはヨークビルへの義務を滞納する可能性があり、これは私たちの返済義務の満期を加速させる可能性があり、これは私たちの流動性に影響を与え、私たちの業務を停止または深刻に修正したり、アメリカ破産法に基づいて保護を求めたりすることを要求するかもしれない。そのため、私たちが経営を続ける能力には大きな疑問があり、これは将来の融資を得る能力に影響を与える可能性があり、私たちの業務をさらに削減する必要があるかもしれません。より詳細は項目7“経営陣の財務状況と経営成果の検討と分析”では“概要--持続経営”での議論を参照。私たちは私たちの運営を支援するために追加的な資本を調達する必要があるだろう。この追加的な資金は受け入れ可能な条項では得られないかもしれないし、全く得られないかもしれない。必要な資本を得ることができない場合、あるいは私たちの流動性需要を満たすことができなければ、私たちの業務を延期、制限、または中止させ、さらにリストラし、私たちの生体材料製品および他の臨床試験計画の商業化努力を停止し、私たちの資産の全部または一部を清算したり、他の戦略的選択を求めたり、および/またはアメリカ破産法の規定に基づいて保護を求めることができるかもしれない。
私たちは私たちの治療法を発展させ、私たちの運営計画を実施するために多くの追加資金を必要とするだろう。もし私たちが追加的な資金を得ることができなければ、私たちは私たちの候補治療薬の開発と商業化を達成できないかもしれない。
私たちは私たちの候補治療薬を開発して製造するために多くの資金がかかると予想される。私たちは私たちの治療法を発展させ、私たちの運営計画を実施するために多くの追加資金を必要とするだろう。特に,我々の治療薬を商業的に生産し,多様な細胞治療薬の登録試験を開始·完成させるために多くの追加資金が必要となる。また、承認されれば、私たちの候補治療薬を発売し、商業化するために多くの追加資金が必要になるだろう。
2022年12月31日まで、私たちは1400万ドルの現金と現金同等物を持っている。私たちは私たちの計画を実行するために追加的な資金を調達する必要があるだろう。また、変化する環境は、私たちの現在の予想よりも資本消費の速度を大きく速めるかもしれません
37
私たちがコントロールできない状況のため、今まで予想されていたよりもっと多くのお金がかかるかもしれない。もし私たちが現在の計画よりも早く拡張することを選択すれば、私たちは今の予想よりも早く大量の資本を集める必要があるかもしれない。いずれにしても、私たちの候補治療薬をさらに開発し、私たちの内部製造能力と私たちの変性疾患業務の成長に資金を提供することを含む、私たちの候補治療薬をさらに開発し、商業化するために追加の資金が必要になるだろう。
私たちは許容可能な条件で追加資金を提供するかどうか、あるいは根本的にできないかどうかを確認することができない。私たちは十分な追加資本を集めることができない場合、あるいは私たちが受け入れられる条項で追加資本を調達することができなければ、私たちは私たちの候補治療薬や他の研究開発計画の開発または商業化を大幅に延期、削減、または停止しなければならないかもしれない。もし私たちがソレントから得た許可を含めて合意の下での支払い義務を履行できなければ、私たちの許可協定も終了される可能性がある。私たちは、他の状況よりも望ましいより早い段階で、私たちの治療薬候補のためのパートナーを探すことを要求されるかもしれないし、他の方法よりも不利な条項で私たちの治療薬候補のパートナーを探したり、不利な条項で放棄したりすることができるかもしれませんが、そうでなければ、私たちは自分の開発または商業化を求めるでしょう。上記のどの事件も、私たちの業務、将来性、財務状況、経営結果を深刻に損害し、私たちの証券価格の下落を招く可能性があります。
著者らの胎盤由来細胞療法候補は癌、伝染病と退行性疾患を治療する新しい方法を代表し、これは巨大な挑戦をもたらした。
著者らは健康的、臨月的、ヒトドナー胎盤からの同種異体細胞治療候補方案を開発しており、ある場合、遺伝子改造を経ている。同種細胞は任意の患者に使用するために“既製”として設計されている。これらの新しい候補療法の推進は大きな課題をもたらしています
私たちが使用している遺伝子編集技術は比較的新しく、私たちが期待している治療候補でこの技術を使用できなければ、私たちの収入機会は実質的に制限されるだろう。
著者らは遺伝子編集技術を用いていくつかの胎盤由来の細胞タイプを修正した。私たちはこれらの技術を使用して毒性リスクを下げたり、治療効果の潜在力を高めたりする。これらの技術は比較的新しく,臨床研究では効果的に期待される効果を達成できない可能性があり,あるいはわれわれの臨床開発計画やこれらの新技術を使用している他の人の安全問題に関与している可能性がある。新しい遺伝子編集技術のいかなる問題も,我々が経験したことがなくても,我々の開発計画に悪影響を与える可能性がある.例えば、遺伝子修正はDNAに予期せぬ変化をもたらす可能性があり、例えば非標的遺伝子編集、大断片欠損或いはDNA転座、これらのいずれも望ましくない副作用を招く可能性がある。われわれの候補治療薬の遺伝子編集は,GvHDや血栓形成のリスクを制限したり,親和性を増加させたりする上でも成功しない可能性がある。
異遺伝子細胞治療領域及び更に広範な遺伝子治療領域のいくつかの競争相手はすでにFDAに臨床試験を保留された。これらの臨床試験の結果から,FDAは追加の試験を要求し,異なるタイプの試験を要求する可能性があり,さらに類似した治療経路を求める他の会社の臨床試験を評価するための方法を大幅に修正する可能性がある。われわれは競争相手の行動を制御することができず,彼らの臨床試験結果に影響を与えることはできず,FDAが別の臨床試験に出現する特定の事実パターンにどのように反応する可能性があるかも分からない。追加的なテスト、異なるタイプのテスト、または修正された規制方法は、私たちの臨床試験を延期し、試験コストを増加させるか、あるいは私たちの大量の時間、エネルギー、資源なしに、私たちの試験が継続する許可を得ることを阻止するかもしれない。例えば、2022年第1四半期に、B細胞悪性腫瘍の治療のためのCyCART-19の研究にINDを提出し、2022年5月下旬にFDAから正式な書面通知を受けた
38
計画中の1/2期臨床試験を継続できるまで,より多くの情報を提供してください。私たちはFDAと協力してその問題を解決するために努力している。
また、遺伝子編集業界は急速に発展しており、私たちの競争相手は新しい技術を導入し、候補薬物の治療に採用された技術を時代遅れにしたり、そんなに魅力的ではないようにしているかもしれない。我々の候補治療薬の開発サイクル中のどの時点でも新しい技術が出現する可能性がある。競争相手が新しい技術を使用したり開発したりするにつれて、このような技術のいかなる故障も私たちの計画に悪影響を及ぼす可能性がある。私たちはまた競争的に不利になる可能性があり、競争圧力は私たちに高いコストで新しい技術を実施させるかもしれない。また、私たちの競争相手は、より大きな財力、技術、人的資源を持って、彼らが技術的優位性を享受できるようにし、将来的に私たちの前に新しい技術を実施することを可能にするかもしれない。私たちは私たちが技術をタイムリーにまたは許容可能な費用で実施できるかどうかを確認できない。もし私たちが業界基準に合った技術的進歩を維持できなければ、私たちの運営と財務状況は不利な影響を受けるかもしれない。
私たちのCyCART-19候補治療は、ソレント治療会社のCAR-Tウイルスベクターに依存し、本ライセンスまたは任意の未来のライセンスを終了することは、私たちのビジネスを損なう重大な権利の損失をもたらす可能性がある。
私たちは特許、ノウハウ、そして独自技術に依存しており、私たち自身のものもあれば、他の人が許可しているものもある。胎盤由来のT細胞を改造して、我々のCAR-T細胞株および我々のCyCART-19候補治療レジメンを産生するために、Sorrentoから許可を得て提供されるレトロウイルス技術を使用した。私たちはソレントとの許可協定に大きく依存している。ソレントは私たちが治癒していない重大な違反によってこのライセンスを終了するかもしれない。本ライセンスの任意の終了は、重大な権利の喪失をもたらす可能性があり、CyCART-19を商業化する能力を損なう可能性があり、許可された自動車構造の任意の候補治療薬の将来の使用を損なう可能性がある。本ライセンスプロトコルでの義務を履行しなければ,ソレントライセンスプロトコルとCyCART-19の自動車構造のための我々の利点を失う可能性がある.さらに、私たちは、ソレントとの既存のプロトコルではカバーされていない標的に対する治療候補薬を研究および開発するために、ソレントから追加のライセンスを取得するか、または他の自動車製造技術を取得する必要がある。そのほか、ソレントCAR-Tレトロウイルス技術は実行可能な候補治療方案を産生できない可能性がある。CyCART−19の承認を得ていれば,ソレントがビジネス規模の生産に十分なウイルスベクターを提供できる保証はない。ソレントとの合意が終了したり、他の技術が必要な場合、私たちは合理的な条項でこのようなライセンスや技術を得ることができないか、または全く得られないかもしれません。特に市場の代替技術の数が限られていることを考慮してください。第1項を参照ビジネス-ライセンス契約-ソレント治療会社“ソレントからライセンスに関するより多くの情報を取得します。2023年2月13日、ソレントはテキサス州南区米国破産裁判所で米国破産法第11章に基づいて自発的な訴訟手続きを開始すると発表した。現在、私たちは破産が許可協定の下で履行を続ける能力にどのような影響を与えるのか予測できない。
他の遺伝子編集技術を他の細胞療法にも応用していますこれらの技術のいくつかは複数の商業サプライヤーから得ることができるが、これらのサプライヤーのいずれかが私たちへの供給を拒否した場合、これは予想される臨床利益を達成するために遺伝子改変に依存する我々の改良NK細胞およびMLASCsの開発に負の影響を与える可能性がある。さらに、現在ライセンスなしに取得可能な遺伝子編集技術のいくつかは、第三者の特許または独自技術となる可能性がある。もし私たちが必要な時に商業的に合理的な条件で許可を得ることができなければ、私たちは私たちの細胞療法の再設計および/または開発停止を余儀なくされるかもしれない。このようなどんな事件も私たちの業務の見通しに実質的な悪影響を及ぼすかもしれない。
ライセンス契約によると、私たちと現在と未来のライセンシーとの間でも、以下の点に関連する紛争を含む知的財産権に関する論争が発生する可能性があります
もし私たちが許可または将来許可を得る可能性のある知的財産権紛争が許容可能な条項で私たちの許可スケジュールを維持する能力を阻害し、弱化した場合、私たちは影響を受ける候補治療薬の開発に成功し、商業化することができないかもしれない。
39
知的財産権を保護する上で、私たちは通常、私たちが持っている知的財産権のように、私たちのライセンスと同じすべてのリスクに直面しています。もし私たちまたは私たちの現在と未来の許可者がこの知的財産権を十分に保護できなければ、私たちの製品商業化能力は影響を受ける可能性がある。
我々の候補療法は新しい技術に基づいており,候補療法開発や規制承認の時間とコストを予測することは困難である。
我々は私たちの研究、開発と製造努力を私たちの胎盤由来同種異体T細胞、NK細胞とMLASC治療候補薬物に集中し、私たちの将来の成功はこれらの治療方法の成功開発に依存する。我々は,凍結細胞を製造することによる生物源を網羅し,我々の技術の最適化と改善に投資し続けるCelulality Impactプラットフォームを開発した。我々が今後遭遇するいかなる発展問題も重大な遅延や意外なコストをもたらさないことを保証することはできず、これらの発展問題が克服できることも保証されない。私たちはまた、商業化に適した場合に生産プロセスの拡大を遅延させることが可能であり、これは、臨床研究を完成させたり、適時あるいは利益に基づいて私たちの治療法を商業化することを阻止するかもしれない。また,われわれは臨床開発の初期段階であるため,重要な試験で評価される用量,あるいは承認された場合には商業投与量に使用されることは知られていない。私たちの細胞治療候補のための適切な用量を見つけることは、私たちが予想していた臨床開発スケジュールを遅らせるかもしれない。また,治療候補薬を開発し,これらの重要な要因を知ることにより,スケーラビリティや製造コストに対する期待が大きく異なる可能性がある。
FDA、欧州薬品管理局とその他の監督機関の臨床研究要求及びこれらの監督機関が候補治療薬物の安全性と有効性を決定するための標準は、潜在的治療薬物のタイプ、複雑性、新規性及び期待用途と市場に基づいて決定された。我々のような新しい治療候補薬の規制承認プロセスは、他のより有名またはより広く研究されている薬物または他の治療候補薬と比較して、より複雑である可能性があるため、より高価で、所要時間が長い。また,NIHが発表したガイドラインによると,遺伝子治療臨床試験もIBCの審査·監督を受け,IBCは地方機関委員会であり,組換えや合成核酸分子を用いた研究の審査·監督を担当している。どの機関が臨床試験を開始する前に、機関のIRBおよびそのIBCは、研究の安全性を評価し、公衆衛生または環境に対する任意の潜在的リスクを決定する。NIHガイドラインは強制的ではないが,関連研究がNIH組換えや合成核酸分子研究助成を受けた機関で行われているか,あるいはその助成によって行われていない限り,多くの会社や他のNIHガイドラインに拘束されていない機関は自発的にこれらのガイドラインに従っている。
自家製品と比較して同種細胞治療候補薬の変異性は小さいと予想されるが,有意な臨床データは低い変異性を支持するいかなる利点もなく,健康なドナー臨月胎盤および関連スクリーニング要求を用いて,単独の変異性挑戦を生じる可能性がある。より広く言えば、任意の規制機関の承認は、任意の他の規制機関が何を承認する必要があるか、またはこれらの規制機関が何が新しい治療候補薬と関連しているかを承認する必要がある可能性があることを示していないかもしれない。また,われわれの候補療法は臨床試験では不良であるか,あるいは有害事象に関与している可能性があり,これらの有害事象は先に承認された自己療法と区別されている。例えば、同種T細胞治療候補は、自己T細胞製品ではなく、GvHDを引き起こす可能性がある。われわれはこの問題を解決しようとするために我々のCAR−T細胞候補を修正したが,CyCART−19はGvHDに関与している可能性があり,臨床試験では無効である可能性がある。有望な候補治療薬の初歩的な臨床データを収集しても,長期データは新たな有害事象や反応が持続しないことを明らかにする可能性がある。予期せぬ臨床結果は私たちの業務に大きな影響を与えるだろう。
私たちの業務は私たちの先行する候補治療薬の成功に非常に依存している。私たちの主要候補薬の承認を得られず、私たちの主要候補薬を効果的に商業化し、承認適応を有する患者の治療に使用すれば、私たちの業務は深刻な損害を受けるだろう。
私たちの業務と未来の成功は私たちが規制部門の承認を得る能力があるかどうかにかかっており、その後、CyCART-19、CYCART-201、CYNK-001、CYNK-301、CYNK-302、APPL-001、pEXO-001を含む私たちの最先端の候補薬物の商業化に成功した。これらの胎盤由来異遺伝子細胞は最初に臨床的に評価された同種異体胎盤由来細胞療法であるため、任意のこのような候補療法の失敗、或いは他の同種異遺伝子細胞療法の失敗は、著者らの候補治療薬の開発能力を阻害する可能性があり、特にGvHD或いは他の有害事象の高い或いは制御可能な比率が観察された場合、医師と監督機関の胎盤由来異遺伝子細胞療法導管全体に対する実行可能性の意見に著しく影響する。われわれの候補治療薬を服用する際に重篤な副作用が観察された場合,あるいは任意の候補治療薬が自己療法よりも安全あるいは有効であると考えられた場合,他の胎盤由来同種療法を開発する能力は深刻な被害を受ける可能性がある。
私たちの主要な候補治療薬を含むすべての候補治療薬は、より多くの臨床と非臨床開発、複数の司法管轄区域の監督審査と承認、大量投資、大規模な商業製造能力と重大なマーケティング努力を必要とし、私たちの細胞治療薬の販売から任意の収入を得ることができる。また,我々の候補療法は類似した流れ,すなわち我々のCelulality Impactプラットフォームに基づいているため,任意の先行する候補療法が安全や治療効果の問題,製造問題,開発遅延,規制問題あるいは他の問題に遭遇すれば,われわれの治療パイプラインの開発計画や業務は深刻な被害を受けるであろう。
40
著者らの候補治療薬物は不良副作用或いは他の特性を招く可能性があり、著者らの臨床開発を阻止し、著者らの監督管理の承認を阻止し、私たちの商業潜在力を制限し、或いは深刻な負の結果を招く可能性がある。
私たちの候補治療薬による不良または受け入れられない副作用は、私たちまたは規制機関の臨床試験の中断、延期、または停止を招く可能性があり、より厳しいラベル、またはFDAまたは他の類似の外国規制機関が規制承認を延期または拒否する可能性がある。われわれの臨床試験結果は副作用や予期せぬ特徴の重症度と流行度を示す可能性がある。承認され開発されている自己細胞療法はCRSと神経毒性の頻繁な発生率を示し,有害事象は患者の死亡を招いている。CyCART-19、CyCART-201、CYNK-301、CYNK-302、およびAPPL-001などのいくつかの候補治療薬が遺伝子工学を行っている。これらはすべて新技術であるため,誤りが発生する可能性があり,臨床的にヒトに使用され,有害事象を引き起こす可能性もある。我々が使用したNK細胞およびMLASCsを含む胎盤由来細胞は、固有の安全性を有し、有害事象を制限する可能性があると信じているが、これらは新しい治療法であるため、このような保証はない。
著者らの胎盤由来治療計画の発展に伴い、著者らは不良事象の結果として、いくつかの候補薬物の開発を停止或いは修正する必要があるかもしれない。例えば,APPL−001を設計する際には,Celgene細胞治療会社によるレガシー胎盤由来MLASCの第1段階臨床試験で観察された血栓形成リスクの増加による遺伝子修正を含む何らかの修正や調整が行われている。
私たちが行っているまたは計画されている任意の臨床試験では、患者は私たちの異遺伝子細胞候補治療に関連する深刻な有害事象を経験する可能性があり、その中のいくつかは死亡を引き起こす可能性がある。もし私たちの候補治療薬が開発中に受け入れられない毒性が出現した場合、私たちの試験を一時停止または終了することができ、またはFDAまたは同様の外国の規制機関は、臨床試験を停止するように命令するか、または任意またはすべての標的適応のための候補治療薬の承認を拒否することができる。データ安全モニタリング委員会はまた、他の無関係な免疫療法試験から推定されるリスクを含む、患者が受け入れられない健康リスクに直面していることを発見することを含む、様々な理由に基づいて臨床試験を随時一時停止または終了することができる。治療に関連する副作用はまた、患者募集または被験者が試験を完了する能力に影響を与えるか、または潜在的な製品責任クレームをもたらす可能性がある。また,治療医療従事者は,細胞療法による毒性が一般的な患者集団や医療従事者には出現しないため,これらの副作用を適切に認識したり処置したりしていない可能性がある。このようなどんな状況でも、私たちの業務、財政状況、そして見通しに大きな被害を及ぼす可能性がある。
私たちの臨床試験は私たちの任意の候補治療薬の安全性と有効性を証明できないかもしれないが、これは規制部門の承認と商業化を阻止または延期するだろう。
規制機関が私たちの細胞治療候補薬の商業販売を承認する前に、私たちの治療候補薬は各目標適応において安全かつ有効であることを、長い、複雑かつ高価な臨床前試験と臨床試験によって証明しなければならない。臨床試験費用は高く,完成には数年かかるかもしれないし,われわれの結果自体も確定していない。臨床試験では,いつでも失敗する可能性がある。著者らの候補治療薬物の臨床前研究と早期臨床試験の結果はいかなる承認後研究の結果を含む後期臨床試験の結果を予測できないかもしれない。
候補治療薬は臨床試験に合格できなかったため,通常極めて高い流出率が生じる。臨床前研究と初歩的な臨床試験で進展を得たが、臨床試験の後期段階では、候補治療薬物は期待した安全性と有効性を示すことができない可能性がある。生物製薬業界のいくつかの会社は高級臨床試験において重大な挫折を受け、原因は治療効果の不足、治療効果の持続性不足或いは受け入れられない安全性問題であり、早期の試験で良好な結果を得たにもかかわらず。臨床試験を開始した候補治療薬の多くは治療薬として承認されていない。
また,現在行われている試験や将来達成可能な任意の試験については,FDAや外国規制機関が我々のように結果を解釈する保証はなく,例えば,レガシーデータの再分析を含め,承認のための候補治療薬を提出する前に,より多くの試験を行う必要があるかもしれない。FDAまたは外国の規制当局がマーケティング申請を支援するために試験結果に満足していない場合、私たちの候補治療薬の承認は大幅に延期される可能性があり、または追加試験を行うためには、私たちの候補治療薬の潜在的な承認を支援するために、入手できない可能性のある大量の追加資源が必要になるかもしれない。
著者らが時々発表或いは公表した臨床試験の初期、中期と初歩データはより多くの患者データの出現に従って変化する可能性があり、そして監査と検証プログラムの制限を受け、これは最終データの重大な変化を招く可能性がある。
時々、私たちは臨床研究の初期、中期、または初歩的なデータを公表するかもしれない。著者らが完成する可能性のある臨床試験の中期データは、患者登録の継続とより多くの患者データの獲得に伴い、1つ以上の臨床結果が実質的に変化する可能性があるというリスクに直面している。初期データはまだ監査とチェック手続きを遵守しなければならず、これは最終データが以前公表された予備データと大きく異なる可能性がある。したがって、最終データを得る前に、初期、中期、および予備データを慎重に見なければならない。初期または中期データと最終データとの間の不利な違いは、私たちのビジネスの将来性を深刻に損なう可能性があります。
41
INDを提出して予想される時間内に追加的な臨床試験を開始することはできない可能性があり,たとえ我々ができてもFDAはこのような試験を許可しない可能性がある。
我々は、2023年に細胞外マトリックス生体材料製品候補のために提出される予定のINDを含む、将来的により多くの治療候補のためのINDを提出する予定である。INDまたはIND修正案の提出は、FDAが試験および臨床試験の開始を許可することをもたらすか、または開始すると、このような臨床試験を一時停止または終了させるという問題は生じないと判断することはできない。例えば,2022年にB細胞悪性腫瘍治療のためのCyCART−19のINDを提出し,FDAと連携して早急に問題を解決していきたいと考えており,このINDに基づいて臨床試験を開始する前に行わなければならないことである。同種細胞療法の製造は依然として新興で発展している分野である。したがって,製品仕様を含めて化学,製造,制御に関するテーマがIND審査の重点となり,INDの承認が遅れる可能性が予想される。また,FDAがINDや臨床試験申請に規定された臨床試験の開始を許可しても,FDAが将来われわれの要求を変えないことは保証されない。
臨床試験では大きな遅延に遭遇したり,予想されたタイムラインで試験を行うことができない可能性がある。
臨床テストは高価で時間がかかり、不確実性も存在する。もしあれば、どんな臨床研究も計画通りに行われるか、あるいは予定通りに完成することは保証できません。たとえ私たちの試験が計画通りに始まっても、問題が発生する可能性があり、著者ら或いは関連する監督管理機関はこのような臨床試験を一時停止或いは中止する可能性がある。1つ以上の臨床研究の失敗はテストの任意の段階で起こる可能性があり、私たちの将来の臨床研究は成功しないかもしれない。成功を妨げたり、臨床開発をタイムリーに完成させたりする可能性のある事件は、
42
現在行われている新冠肺炎の大流行は、新たに出現した変種或いは未来の大流行に関連する病例を含めて息を吹き返し、上述のある事件のリスクを増加させ、著者らの発展スケジュールを延期する可能性もある。例えば,2020年初めと2021年には,大流行によりAMLに対するCYNK−001第一段階臨床試験の登録に遅延があった。臨床前と臨床開発を成功させることができない場合は、私たちの追加コストを招き、あるいは私たちの収入を作る能力を弱める可能性がある。さらに、私たちの候補治療薬の生産や処方変更を行う場合、追加的な研究が要求されるかもしれませんし、修正された候補薬をより早いバージョンに接続するために追加的な研究を行うことを選択するか、または新たに発見された候補薬の追加的な研究が必要になるかもしれません。臨床研究遅延は、私たちの療法が特許保護を有する任意の期限を短縮することも可能であり、私たちの競争相手が私たちの前に細胞療法を市場に出すことを可能にすることができ、これは候補療法を商業化することに成功する能力を弱める可能性があり、私たちの業務と手術結果を損なう可能性がある。
私たちまたは私たちが依存している第三者が臨床試験地点または他の業務を集中して運営している地域では、私たちの業務は、進行中の新冠肺炎の大流行や将来の疾患爆発を含む健康流行病または流行病の影響を受ける可能性がある。
私たちの業務は持続的な新冠肺炎の大流行と未来の疾病の爆発を含む健康大流行或いは流行病の影響を受ける可能性がある。例えば,新冠肺炎の発生により,急性骨髄性白血病を治療するCYNK−001臨床試験の登録が延期されている。また、ニュージャージー州と三州地区で新冠肺炎の流行が最も深刻な時、病院資源が移転されるため、著者らは健康、完全なドナー胎盤を収集する能力は限られている。私たちのオフィスを再開し、従業員が現場に移行しているにもかかわらず、私たちの臨床試験地点間には統一的な制限と要求が不足しており、将来的には収容所や同様のタイプの制限が再実施される可能性があり、病院スタッフはドナーの同意を求めないかもしれない。私たちは今も施設爆発のリスクに直面しており、将来的には従業員の職場安全に関するクレームや意外な閉鎖や隔離の影響を受ける可能性があり、私たちの運営を乱すことになります。このような不確実性と政策と制限の変化性質は生産性に負の影響を与え、私たちの業務を混乱させ、さらに臨床計画とスケジュールを延期し、その程度は制限の長さと重症度に依存し、そして私たちが正常な過程で業務を展開する能力に対する他の制限は、私たちの業務、運営結果と財務状況に負の影響を与える可能性がある。
新冠肺炎の蔓延はすでに世界的に広範な影響を与えており、我々の経済に実質的な影響を与える可能性がある。新冠肺炎疫病がもたらす可能性のある潜在的な経済影響及びその持続時間は評価或いは予測が困難である可能性があるが、それはすでに全世界の金融市場の深刻な混乱を招いた。このような干渉が持続的または繰り返し発生すれば、私たちが資本を得ることをより難しくする可能性があり、これは未来に私たちの流動性に否定的な影響を与えるかもしれない。また,新冠肺炎の伝播による景気後退や市場コールは,我々の業務や我々A類普通株の価値に実質的な影響を与える可能性がある。
全世界の新冠肺炎の大流行は依然として変化し続けており、私たち或いは任意の類似の健康大流行或いは流行病の最終的な影響は高度に不確定である。私たちの業務、私たちが計画している臨床試験、病院と医療システム、あるいは世界経済の潜在的な遅延や影響のすべての程度はまだわかりません。これらの影響は我々の運営に実質的な影響を与える可能性があり,引き続き新冠肺炎の状況を注視していきたい。
治療候補薬を受けた患者の毒性を監視·管理することは挑戦的であり,規制の承認を得たり,治療候補薬を商業化する能力に悪影響を及ぼす可能性がある。
著者らは学術医学センターと臨床試験期間中に発生した毒性を評価と管理する経験豊富な病院と契約し、GvHD患者(CyCART-19に対する)、及び著者らの臨床試験に参与する患者の不良事件をより広くモニタリングすることを望んでいる。これらのプログラムがあっても、これらのセンターや病院は、患者や治療毒性、または任意の他の有害事象を観察することが困難である可能性があり、これは、より深刻またはより長時間の毒性、さらには患者の死亡を招く可能性がある。GvHDに何らかの深刻な問題や他の予期しない事件が発生した場合、私たちまたはFDAが私たちの1つまたは複数の臨床試験を延期、一時停止または終了させる可能性があり、これは規制機関の私たちの候補治療方案の承認を脅かす可能性がある。また,われわれの細胞療法が病院や医療センター以外で使用されている程度では,われわれの療法がビジネスベースでより広く提供されていれば,承認されると不良事象の観察や管理がより困難になる可能性がある。さらに、センターは、任意のGvHDのような、治療レジメン候補の副作用を管理するのを助けるための薬剤であり、副作用を十分に制御できない可能性があり、および/または治療効果に悪影響を及ぼす可能性がある。
臨床試験は高価で、時間がかかり、設計と実施が困難である。
人体臨床試験は費用が高く、設計と実施が困難であり、一部の原因はそれらが厳格な監督管理要求を受けているからである。著者らの同種異体胎盤由来細胞治療候補薬物は新しい技術に基づいているため、大規模に生産された既製治療薬物の在庫を確立する必要があるため、著者らはそれらは広範な研究と開発が必要であり、そして相当な製造と加工コストを持っていると予想する。さらに特定の癌患者や他のターゲットを絞った患者を治療する費用は
43
任意の潜在的副作用の治療を含む適応が重要であるかもしれない。そのため、我々の細胞治療候補薬物の臨床試験コストはより伝統的な治療技術或いは薬物製品よりはるかに高い可能性がある。
もし私たちがもっと多くの治療候補薬を開発できなければ、私たちのビジネス機会は制限されるだろう。
われわれのコア戦略の1つは,われわれの最初の7つの重要な計画であるCYCART−19,CYCART−201,CYNK−001,CYNK−301,CYNK−302,APPL−001およびpEXO−001に加えて,他の候補治療の臨床開発を継続し,最初の6つの目標適応外に拡大することである。開発、監督管理の承認を得て、より多くの細胞治療候補薬物を商業化するには大量の追加資金が必要であり、医療製品開発固有の失敗リスクが出現しやすい。これらの追加的な候補療法のいずれかを開発過程で成功させることは保証されない。
FDAの承認を得てこれらや他の候補治療薬の販売を許可しても,どのような候補治療薬も商業化に成功し,市場に広く受け入れられたり,他の商業的に利用可能な代替薬よりも有効であることは保証されない。もし私たちがより多くの候補薬の開発に成功して商業化できなければ、私たちのビジネス機会は制限されるだろう。さらに、規制部門によるより多くの治療薬候補の承認を得ることができないことは、任意の他の候補治療薬の承認過程に悪影響を与え、または任意の承認された治療候補薬の承認を失う可能性がある。
私たちの最近の組織変化と費用削減措置は成功しないかもしれない。
2022年11月と2023年1月にリストラを実施し、大部分の労働力に影響を与えた。今回のリストラの目的は,我々が臨床試験で受け取った結果と行われている臨床開発計画に基づいて,我々の労働力を再調整し,我々のニーズを満たすことである。しかし、これらの再編およびコスト削減活動は、予想以上のリストラの自然減員、私たちの残りの従業員の士気低下、および私たちがこのようなリストラ措置の期待的な利益を達成できない可能性があるリスクなど、予期しない結果とコストをもたらす可能性があり、これらはすべて私たちの運営業績や財務状況に悪影響を及ぼす可能性がある。また、役職は廃止されていますが、運営に必要ないくつかの機能が残っており、退職した従業員の役割や義務を残りの従業員に割り当てることに成功していない可能性があります。リストラやコスト削減措置は、一時停止した開発活動の再開や新たな計画の実施を困難にし、合格した代替者を雇う必要があり、追加的で予期しないコストや支出が必要になる可能性もあることが分かったかもしれません。私たちのほとんどの人員が、私たちを含む何人かの実行幹事がサービスを失ったため、私たちは私たちの業務を継続し、私たちが履行している義務を履行することができないかもしれない。このような予期しない結果は、私たちの業務、財務状況、そして運営結果に実質的な悪影響を及ぼす可能性がある。
私たちは大量の資源を必要とする独自の製造および貯蔵施設を運営している;製造または他の故障は、私たちの臨床試験および私たちの候補治療薬と私たちのバイオバンクおよび変性疾患業務の商業的可能性に悪影響を及ぼす可能性がある。
ニュージャージー州のフロラム公園に専用の工場がありますそこでは健康な完全ドナー胎盤を処理し、細胞治療と組織製品のために、私たちのバイオバンク業務を経営しています。我々の研究と既存の臨床試験需要の過程を管理した経験があるが、後期臨床試験に拡張する際の任意の候補治療薬に対する需要を満たすために、既製胎盤由来同種細胞療法を量産することができない可能性があり、あるいは承認後に商業生産に使用することができる。私たちは製造と加工方法が現在の需要を支持するのに適していると信じており、私たちは拡張可能なプロセスを持ち、ソレントを含む様々な第三者から適切な供給を得ているが、私たちの大規模化プロセスが安全で有効な同種細胞を産生することを保証することはできない。また,我々のバイオバンクや変性疾患業務を含む我々の製造·貯蔵施設は,FDAが現在ヒト細胞や組織製品を使用しているGTPを含むcGMPに適合しなければならない。そこで,適用されるGTPSや他の政府法規を含むcGMPの厳格な遵守を確保するために,FDAや他の政府機関の継続的な定期抜き打ち検査を受けている。
生物製薬製品の製造は複雑であり、先進的な製造技術と技術制御の開発を含む大量の専門知識が必要である。細胞治療製品のメーカーは生産において困難に直面することが多く,特に規模拡大や初期生産の検証や汚染のない確保が行われている。これらの問題は生産コストと生産量の困難、品質管理、製品の安定性、品質保証テスト、オペレータミス、合格者不足、及び厳格に実行されている連邦、州と外国法規を遵守することを含む。放出試験に関連するガイドラインまたはパラメータの使用のような新しい規制ガイドラインまたはパラメータの使用は、候補治療薬を製造する能力にも悪影響を及ぼす可能性がある。さらに、私たちの候補治療薬供給または製造施設で汚染物質が発見された場合、そのような供給を廃棄しなければならない可能性があり、私たちの製造施設は汚染を調査および修復するために長い時間閉鎖する必要があるかもしれない。私たちは未来に私たちの候補治療薬の製造に関連するいかなる安定性や他の問題も起こらないことを保証することはできない。
44
私たちまたは私たちの他の任意の供給者は、ドナー胎盤を含む私たちの原材料を貯蔵して輸送する物流を管理できないかもしれない。貯蔵故障、出荷遅延、および私たち、私たちのサプライヤー、または他の私たちがコントロールできない要素によって引き起こされる問題、例えば天気、衛生流行病または流行病は、治療薬の生産ができない、利用可能な治療薬を失うこと、または患者および臨床試験場所への候補治療薬の送達を阻止または遅延させる可能性がある。私たちはまた資源制限や労使紛争で製造困難に直面する可能性がある。これらの困難のいずれかに遭遇すれば,治療候補薬を患者に提供する能力が脅かされるであろう。
私たちには現在細胞療法のマーケティングチームがありません。承認されると、マーケティングおよび販売能力を確立することができない場合、または第三者と合意して私たちの候補治療薬をマーケティングおよび販売することができなければ、製品収入を生成することができないかもしれない。
私たちは現在販売、マーケティング、あるいは流通能力がありません。会社として、マーケティング細胞療法の経験もありません。私たちの現在の販売チームは私たちの変性疾患とバイオバンク業務に限られているからです。私たちは私たちの細胞治療候補のために内部専門のマーケティング組織と販売チームを作るつもりで、これらの候補が規制部門の承認を得たら、これは大量の支出、管理資源、そして時間を必要とするだろう。私たちは他の製薬とバイオテクノロジー会社と競争して、採用、採用、研修、マーケティングと販売員を維持しなければならないだろう。承認されると、細胞療法のための内部販売、マーケティング、および流通能力を確立することができないか、または決定できなければ、細胞療法の販売およびマーケティングに関する協力計画を求めるが、しかし、このような協力スケジュールを確立または維持することができることを保証することができないか、またはそれが可能であれば、彼らは有効な販売チームを持つことになる。私たちが細胞療法販売から得たどんな収入もこれらの第三者の努力にかかっているが,これは成功しないかもしれない。私たちはこれらの第三者のマーケティングと販売活動を少ないか、あるいはコントロールできないかもしれません。私たちの治療販売収入は、変性疾患製品とバイオバンク業務に対して行ったように、候補治療薬を直接商業化する場合よりも低いかもしれません。私たちは私たちの治療薬の候補を販売して販売してくれる第三者を探している時も競争に直面しています。内部販売や流通能力を発展させたり、第三者協力者と関係を構築したり、米国や他の市場で規制された承認を受けたりする任意の治療法を商業化することは保証されない。
海外での研究や臨床試験や我々の候補治療薬の国際的な売り込みに関連する様々なリスクがわれわれの業務に実質的な悪影響を及ぼす可能性がある。
私たちは世界的に私たちの候補治療薬を開発し、アメリカ以外の地域で私たちの変性疾患製品を販売する予定です。したがって、私たちは海外での経営に関する追加的なリスクに直面することが予想される
45
私たちの国際業務に関連するこれらのリスクや他のリスクは、収益性ビジネスの能力を実現または維持することに大きな悪影響を及ぼす可能性があります。
我々は複数の計画や候補治療案が開発されており,様々な標的適応が求められているため,特定の候補療法の追求に限られた資源を利用する可能性があり,より利益や成功の可能性が高い可能性の高い発展機会や候補療法を利用することはできない。
我々は,癌,感染症,退行性疾患への適応に対する細胞治療候補薬の開発に焦点を当てている。私たちの財力と人的資源が限られているため、私たちは潜在的な標的適応または候補治療薬を持つ機会を追求する機会を放棄または延期する可能性があり、これらの機会は後に私たちの現在と計画中の開発計画や候補治療薬よりも大きな商業潜在力を持っていることが証明された。私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。現在と未来の研究開発計画および他の特定の適応の将来の候補治療方案への支出は、いかなる商業的に実行可能な未来の候補治療方案を生成しないかもしれない。特定の候補治療薬の商業的潜在力や目標市場を正確に評価していなければ、協力、許可、または他の特許権使用料の手配によって、候補治療薬に対する貴重な権利を放棄する必要があるかもしれないが、この場合、将来の候補治療薬の独占開発権と商業化権利を保持することがより有利になる。
私たちは他のバイオテクノロジーや製薬会社からの激しい競争に直面しています。もし私たちが効果的に競争できなければ、私たちの経営業績は影響を受けます。
生物製薬業界の特徴は競争が激しく、革新が迅速だということだ。我々の競争相手は、類似またはより良い結果を達成することができる他の化合物、薬物または生体材料を開発することができるかもしれない。我々の細胞療法と生体材料の潜在的競争相手には,大手多国籍製薬会社,老舗バイオテクノロジー会社,専門製薬会社と大学,その他の研究機関がある。私たちの多くの競争相手はより多くの財務、技術、その他の資源、例えばより多くの研究開発者と経験豊富なマーケティングと製造組織、そして完備された販売チームを持っている。規模が小さいか早い段階にある会社も重要な競争相手になる可能性があり、特に大手老舗会社との協力で手配されている。バイオテクノロジーと製薬産業の合併と買収は、私たちの競争相手により多くの資源を集中させる可能性がある。技術の商業適用性の進歩やこれらの業界に投資する資本の増加により、競争はさらに激化する可能性がある。私たちの競争相手は、単独でも、パートナーとの協力でも、私たちの候補治療薬よりも効果的で、安全で、商業化しやすい、またはコストの低い薬物または生物学的製品の開発、買収、または独占ライセンスに成功することができ、または私たちの開発技術および製品に必要かもしれない特許技術を開発するか、または特許保護を受けることができる。
規制部門の私たちの治療薬候補の承認を得ても、競争相手の製品の供給と価格は、私たちの候補治療薬に対する需要と価格を制限するかもしれません。価格競争や医師が既存の治療法から候補治療法に切り替えたくない場合、あるいは医師が他の新薬や生物製品に変更したり、私たちの候補治療法を保留したりすることを選択した場合、私たちの業務計画を実施できないかもしれません。私たちの競争相手に関するより多くの情報は、“ビジネス-競争”の節を参照されたい
私たちは私たちのキーパーソンに高度に依存していて、私たちが高い素質の人材を誘致し、維持することに成功できなければ、私たちの業務戦略を成功的に実施することができないかもしれません。
私たちが競争の激しい生物技術と製薬業界の中で競争できるかどうかは、私たちが高い素質の管理、科学と医療人員を引き付けることができるかどうかにかかっている。私たちは私たちの創業者で最高経営責任者のロバート·ハリリ医学博士、私たちの最高経営責任者ブラッド·グローバー博士、私たちの首席医療官のロバート·キルコ医学博士を含む、私たちの管理、科学、医療者に強く依存しています。私たち幹部のサービスを失った他の重要な点は
46
従業員や他の科学や医療コンサルタント、そして適切な代替者を見つけることができず、製品開発の遅延を招き、私たちの業務を損なう可能性があります。私たちのほとんどの業務はニュージャージー州の施設で行われています。この地域は多くの他の生物製薬会社と多くの学術·研究機関の本部である。私たちの市場は技術者に対する競争が非常に激しく、私たちが受け入れられる条件で高い素質の人員を採用し、維持する能力を制限する可能性があり、甚だしきに至っては根本的にはできない。私たちは価値のある従業員を引き留めるために努力しているにもかかわらず、私たちの管理、科学、開発チームのメンバーは短時間で彼らを解雇するかもしれない。私たちは私たちの重要な従業員と雇用協定を持っていますが、これらの雇用協定は自由に雇用できることを規定しています。これは、私たちのどの従業員も通知の有無にかかわらずいつでも退職できることを意味します。私たちはこのような個人や私たちの他のどんな従業員のためにも“キーパーソン”保険証書を維持しない。私たちの成功はまた私たちが引き続き高技能の初級、中級と高級管理者及び初級、中級と高級科学と医療人員を吸引、維持、激励できるかどうかにかかっている。
私たちは業務の成長を管理する上で困難に直面するかもしれない。
2022年12月31日現在、225人のフルタイム従業員と35人の非レンタル従業員を持っています。私たちの発展と商業化計画と戦略の制定、そして私たちは業務合併後に上場企業として運営を開始し、私たちは従業員の基礎を拡大し、管理、運営、販売、研究開発、マーケティング、財務、その他の人員を増やす予定です。その後、2022年11月と2023年1月にリストラを実施し、優先順位を再決定する努力の一部として、私たちの大多数の従業員に影響を与え、戦略目標を達成しました。
現在と未来の成長は、管理職のメンバーがより多くの重大な責任を負うことを要求している
私たちの将来の財務業績と候補薬物を商業化する能力は、私たちの成長能力を効果的に管理することにある程度依存し、私たちの経営陣はまた、これらの成長活動を管理するために、不比例な注意を日常活動から移しなければならないかもしれない。
もし私たちが新入社員を雇用し、私たちのコンサルタントや請負業者チームを拡大することで、私たちの組織を効果的に拡大することができなければ、私たちの治療薬候補のさらなる開発、製造、商業化に必要な任務を遂行することができない可能性があり、したがって、私たちの研究、開発、製造、商業化目標を達成できないかもしれない。
私たちは将来的に戦略的連合を形成したり、追加的な許可計画を達成したりするかもしれないが、私たちはこのような連合や許可手配の利点を認識していないかもしれない。
私たちは戦略的同盟を結成したり、合弁企業や協力を確立したり、第三者と追加の許可手配を達成したりすることができ、これは、私たちの候補治療薬と私たちが開発する可能性のある任意の未来の候補治療薬の開発および商業化に関する私たちの努力を補完または強化すると信じています。これらの関係のいずれも、非日常的な費用や他の費用を発生させ、私たちの短期的および長期的な支出を増加させ、株主の権益を希釈したり、私たちの管理と業務を乱す証券を発行したりすることを要求する可能性がある。私たちはCelgeneにAnthrogenensis買収に関するいくつかの知的財産権を返送した。許可の広い範囲から,Celgeneは我々の知的財産権を利用して自動車分野で我々と競争する療法を開発することができる。また,CVRプロトコルによると,Celgeneには持続的な義務があり,このプロトコルによれば,CYNK−001,CYNK−301,CYNK−302を含むいくつかの療法についてCelgeneに何らかの支払いを要求される可能性がある。CVRプロトコルによると、Celgeneへの支払い義務は、もし私たちがそうすることを選択すれば、私たちがこのような資産と協力する能力を制限するかもしれない。Celgene関係に関するより多くの情報は、プロジェクト1“ビジネス-私たちのチームと会社の履歴-Celgene Corporation”を参照されたい。
また、適切な戦略的パートナーを探す上で激しい競争に直面しており、交渉過程は時間がかかり複雑である。また,我々の候補治療薬のための戦略的パートナーシップや他の代替計画を構築する努力は成功しない可能性があるが,協力努力の開発段階が早すぎると考えられる可能性があるため,第三者は我々の候補治療薬が安全性と有効性を示すために必要な潜在力を持っているとは思わないかもしれない。私たちの候補治療薬に関連する新しい戦略パートナー協定の任意の遅延は、いくつかの適応のための候補治療薬の開発および商業化を延期する可能性があり、これは、私たちの業務の将来性、財務状態、および運営結果を損なうことになる。
私たちは過去と未来に非コア分野の第三者と新しい戦略同盟、協力、許可手配を構築することを探索し続ける。このようなスケジュールは関連時間に応じて得られる情報によって達成されており,初歩的な研究や開発を行った後に長期的な協力を招くことはない可能性がある.私たちはいくつかの協定の締約国であり、将来的には、特定の分野での私たちの運営能力を禁止または他の方法で制限する新たな協定が締結される可能性がある。
47
さらに、私たちの現在または未来の戦略連合、協力または他の合意または手配の下で、私たちに知的財産権を付与すること、または私たちから知的財産権を付与すること、またはこれに関連する支払いを含む紛争が生じる可能性があり、付与された権利範囲、専有権、支払い義務、契約解釈または第一選択の研究、開発または商業化プロセスの相違を含む。このような相違のため、私たちは追加の金額を支払う必要があるかもしれません。私たちに支払われる金額は減少または遅延するかもしれません。あるいは研究、開発、商業化活動が遅延したり、手配が終了したりする可能性があり、これは私たちの業務や運営に悪影響を及ぼす可能性があります。
もし私たちが製品や業務を許可すれば、私たちがそれらを既存の運営や会社文化と組み合わせることに成功できなければ、私たちはこのような取引のメリットを実現できないかもしれません。戦略的取引または許可証の後に、このような取引の合理的な結果、収入、または特定の純収入を証明することを達成することは確認できません。
私たちは資産や他の戦略的取引を買収する利点を認識していないかもしれない。
私たちは持続的な基礎の上で様々な戦略的取引を積極的に評価する。私たちは他の事業、製品、または技術を買収し、相補的な業務で合弁や投資を求めることができる。私たちの戦略取引の成功は、私たちとソレントとのライセンス、および未来のどの戦略取引も関連するリスクと不確実性に依存しています
上記のようないかなるリスクや不確実性が発生した場合、いかなる買収や戦略取引の期待収益も実現できない可能性がある。また、外国の買収·合弁企業は、異なる文化や言語を越えた業務統合に関するリスク、為替リスク、海外業務に生じうる不利な税収結果、特定の国に関連する特定の経済、政治、規制リスクを含む他のリスクに直面している。将来の買収または処分は、私たちの株式証券の潜在的希釈発行、債務、または負債または償却費用または営業権の償却をもたらす可能性があり、これらはいずれも私たちの財務状況を損なう可能性がある。
我々の内部コンピュータシステム、または私たちのCRO、協力者、または他の請負業者またはコンサルタントが使用するシステムは、障害が発生したり、セキュリティホールに遭遇したりする可能性があります。
我々の内部コンピュータシステムおよび我々のCRO、協力者および他の請負業者またはコンサルタントのコンピュータシステムは、コンピュータウイルス、許可されていないアクセス、ネットワークセキュリティの脅威、および電気通信および電気故障の破壊を受けやすい。ネットワーク攻撃、サービス拒否攻撃、恐喝ソフトウェア攻撃、商業電子メール漏洩、コンピュータマルウェア、ウイルスおよび社会工学(ネットワーク釣りを含む)は、全体的に増加し続けている。したがって、私たちまたは私たちのサービスプロバイダのネットワークセキュリティ対策が、不正なアクセス、攻撃(複雑なネットワーク攻撃を含む可能性があります)、被害、または私たちの従業員または請負者のデータの不適切な処理を防ぐことができない場合、私たちの名声、顧客信頼、業務、運営結果、および財務状況は悪影響を受ける可能性があります。ネットワークイベントは、第三者が盗難または推定された証拠、コンピュータマルウェア、ウイルス、迷惑メール、ネットワーク釣り攻撃、恐喝ソフトウェア、盗難カードコード、および敏感なデータにアクセスするために許可されていないアクセス権限を取得しようと試みる他の意図的な攻撃を含む可能性がある、ますます複雑になっている。データを格納したり、それを介してデータを送信したりする内部コンピュータシステムを破壊または不正にアクセスするための技術はしばしば変化し、十分な予防措置を実施することができない場合や、セキュリティホールが発生したときにそれらを阻止することができない可能性がある。我々の計算機システムを浸透·破壊しようとする脅威参加者が使用する技術はしばしば変化し,ターゲットへの攻撃前には認識できない可能性があるため,これらの技術を予測できない可能性がある.これまで,このような重大なシステム障害やセキュリティホールは経験していないが,このような事件が発生して我々の運営が中断されると,我々の開発計画や業務運営に大きな中断を招く可能性がある.例えば、完成した或いは未来の臨床試験の臨床試験データの紛失は著者らの監督管理の承認作業を遅延させ、著者らのデータの回復或いは複製のコストを著しく増加させる可能性がある。私たちのシステムまたはインフラ(第三者サプライヤーが提供することを含む)内の任意の中断またはセキュリティホールが、私たちのデータやアプリケーションが失われたり、破損したり、あるいは機密または独自の情報を適切に開示しない場合、私たちは責任を招く可能性があり、私たちの候補治療薬のさらなる開発および商業化は延期される可能性がある。また,在宅勤務者への依存は,我々のネットワークセキュリティリスクを増加させ,データアクセス可能性の問題をもたらし,通信中断の影響を受けやすくなり,どの通信中断も我々の業務に悪影響を与える可能性がある.スタートアップ企業としてはありません
48
データセキュリティ保護に大きな投資を行うと、このような事件が発生しないように十分な保護が得られない可能性があり、このような努力に資源が割り当てられていない可能性もある。
FDAや他の政府機関の資金変化は、重要な指導部や他の人たちの能力を採用して保留し、新しい療法やサービスのタイムリーな開発を阻止したり、商業化したり、あるいはこれらの機関が私たちの業務運営に依存する可能性のある正常な機能を履行することを阻止することを阻害する可能性があり、これは私たちの業務にマイナス影響を与える可能性がある。
FDAが新しい療法を審査と承認する能力は、政府予算と資金レベル、肝心な人員の雇用と維持、ユーザー費用の支払いを受け入れる能力、法律、法規と政策の変化、および新冠肺炎の大流行による業務中断など、様々な要素の影響を受ける可能性がある。したがって、その機関の平均検討時間は近年変動している。また、我々の行動は、研究·開発活動に資金を提供する機関を含む政府機関の資金に依存する可能性があり、政治プロセスの影響を受け、政治プロセス自体が不安定で予測不可能である。FDAおよび他の機関の中断は、必要な政府機関によって新生物製品が審査および/または承認されるのに要する時間を遅らせる可能性もあり、これは私たちの業務に悪影響を及ぼすだろう。例えば、ここ数年間、米国政府は何度か閉鎖されており、FDAのようないくつかの規制機関は、キー従業員を休暇にし、キー活動を停止しなければならない。そのほか、2020年3月以来、新冠肺炎の疫病のため、国内外の施設に対する検査は基本的に保留され、FDAは常規のモニタリング、生物研究モニタリングと審査前検査を含む大流行前の検査活動の回復に努力してきた。FDAが承認を得るために検査を行う必要があると判断し、旅行制限のために審査期間内に検査を完了することができない場合、FDAは、遠隔相互作用評価が不十分であると考えており、機関は、場合によっては、検査が完了するまで、申請に完全な返信または行動を延期することを意図していることを示している。新冠肺炎突発公共衛生事件の期間中、アメリカ食品薬品監督管理局はその申請に対する規定検査を完成できなかったため、多くの会社は完全な回復状を受け取ることを発表した。米国以外の規制機関は、進行中の新冠肺炎の大流行に対応するために、類似した制限や他の政策措置をとる可能性があり、規制活動に遅延が生じる可能性がある。政府が長期的に停止したり中断したりすれば、FDAまたは他の規制機関が私たちが提出した規制文書を適時に審査して処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。また、将来的に政府の閉店は、適切な資本化と事業継続のために、私たちが公開市場に進出し、必要な資本を得る能力に影響を与える可能性がある。
業務中断は私たちの将来の収入と財務状況を深刻に損害し、私たちのコストと支出を増加させるかもしれない。
上述の新冠肺炎疫病による業務中断と臨床試験の遅延を除いて、私たちの業務及び私たちのCRO及びその他の請負業者と顧問の業務は電力不足、電気通信故障、水資源不足、洪水、ハリケーン、竜巻、火災、地震、極端な天気条件、医療流行病及びその他の自然災害或いは人為災害或いは業務中断を含む他の要素の影響を受ける可能性がある。このような業務中断の発生は、私たちの運営と財務状況を深刻に損害し、私たちのコストと支出を増加させる可能性がある。もし私たちまたは私たちのサプライヤーの業務が人為的または自然災害や他の業務中断の影響を受けたら、私たちの候補薬物を生産する能力は妨害される可能性があります。また、私たちの核心業務は私たちがニュージャージー州フロラム公園に専門的に建設された施設に集中しているため、この場所のいかなる中断が続けば、私たちの業務と将来性に実質的な損害を与える可能性がある。
もし私たちが現在と未来の運営に必要な連邦と州許可証と登録を取得して維持しなければ、私たちが収入を作る能力は制限されるだろう。
医療産業は広範囲な当局によって厳格に規制されている。したがって、私たちの業務は、いくつかの許可、登録、許可、許可、承認、認証、認可、その他のタイプの連邦、州、地方政府の許可を維持し、私たちが運営している各司法管轄区の様々な法規を遵守することを要求します。例えば、私たちはいくつかの州で許可証と登録を維持することを要求され、私たちの製品とサービスがこのような許可証を必要とする州で生物製品、組織バンク、血液バンク許可証、許可証、登録を取得しました。私たちは毎年FDAで組織銀行として登録され、アメリカ血液バンク協会の国家認証を受けています。このようなライセンス要件を遵守しないことは、許可証の取り消しまたは一時停止、登録または認証、または是正、監視、民事罰金、民事強制措置、および/または刑事罰を受ける計画を含む実行行動をもたらす可能性がある。私たちが現在提案している業務を考慮して、私たちは現在、私たちの製品やサービスをマーケティングするために追加のライセンスや登録を得る必要はないと信じていますが、将来追加の規制承認が必要かどうかは予測できません。必要であれば、私たちが開発しているか、または開発を試みる可能性のある幹細胞および/または他のサービスのためにも、このような承認が得られるかどうかは予測できません。私たちは必要な連邦と州許可証と登録を得ることができず、これは私たちが収入を作る能力を制限するだろう。
49
私たちと顧客、医者、第三者支払人との関係は多くの法律法規によって制限されている。もし私たちまたは私たちの従業員、独立請負業者、コンサルタント、ビジネスパートナー、サプライヤーがこれらの法律に違反すれば、私たちは巨額の処罰に直面する可能性があります。
私たちは高度に規制された業界で運営されており、私たちと顧客、医師、第三者支払人との関係は多くの法律と法規によって制限されている。“”というタイトルの部分を参照企業-政府法規と製品承認-その他アメリカの医療法律とコンプライアンス要件“と。米国や他の地域の医療提供者、医師、第三者支払者は、私たちが市場で承認された任意の候補治療薬の推薦と処方において主要な役割を果たす。私たちの現在と未来の医療提供者、第三者支払者、顧客、そして他の人たちの手配は、広く適用される詐欺と乱用、および他の医療に関する法律と法規に直面するかもしれません。これらの法律は、私たちの臨床研究と開発計画、ならびに私たちの計画と将来の細胞療法販売、マーケティングと教育計画、ならびに私たちの変性疾患製品とバイオバンク業務の販売とマーケティングに影響を与える可能性があります。特に、医療製品およびサービスの販売促進、販売およびマーケティングは、詐欺、リベート、自己取引、および他の乱用を防止するための広範な法律および法規によって制限されている。これらの法律法規は、広範な定価、割引、マーケティングおよび販売促進、販売手数料、顧客インセンティブなどの業務スケジュールを制限または禁止することができる。私たちはまた、患者情報を識別できるプライバシーと安全を管理する連邦、州、外国の法律の制約を受ける可能性がある。
これらの法律の広さ、および既存の法定例外および安全港の規制の狭さのため、私たちのいくつかの商業活動または私たちと医師の手配(一部の医師は私たちの科学顧問委員会でのサービス報酬として株式オプションを得る可能性がある)は、1つ以上のこのような法律の挑戦を受ける可能性がある。もし私たちまたは私たちの従業員、独立請負業者、コンサルタント、商業パートナー、サプライヤーがこれらの法律に違反した場合、私たちは調査、法執行行動、または重大な処罰を受ける可能性があります。私たちはすでに商業行為と道徳規範を採択したが、従業員の不当な行為や商業違反を常に識別し、阻止できるわけではなく、不正行為を発見し防止するための予防措置は、未知または未管理のリスクや損失を効果的に制御できない可能性があり、あるいは政府の調査や他の行動や訴訟から私たちを保護することができない可能性がある。これらの調査または行動または訴訟は、このような法律や法規を遵守できなかったことによるものである。私たちの業務計画が適用される医療保険法に適合することを確保する努力は巨額のコストに及ぶ可能性がある。政府および法執行当局は、私たちの業務実践が、詐欺および乱用または他の医療保健法律および法規を適用する現在または未来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。もし私たちにこのような訴訟が提起された場合、私たちは自分自身の弁護や私たちの権利の維持に成功できませんでした。これらの行動は、重大な処罰および是正措置を含む、私たちの業務に大きな影響を与える可能性があり、いずれも私たちの業務運営能力および私たちの運営結果に悪影響を及ぼす可能性があります。さらに、私たちの任意の候補治療薬または私たちの変性疾患製品のアメリカ国外での承認と商業化はまた、医療保健法および他の外国法律の外国等価物の追加的なカバーに直面する可能性がある。
これらの法律の範囲も執行も不確定であり,現在の医療改革環境下では,特に適用の前例や法規が乏しい場合には,急速な変化が生じる可能性がある。連邦と州法執行機関はよく医療会社と医療提供者との相互作用を審査し、これは医療保健業界の一連の調査、起訴、有罪判決、和解を招いた。ビジネス手配が適用される医療保険法に適合することを確保することや,政府当局が行う可能性のある調査に対応することは,時間や資源がかかる可能性があり,会社の業務への関心を分散させる可能性がある。
医薬製品の流通は広範な記録保存、許可、貯蔵と安全要求を含む追加の規定と条例を遵守し、許可されていない医薬製品の販売を防止しなければならない。
個人が識別可能な健康情報を含む個人情報の収集、使用、処理、国境を越えた伝送は、制限的な法規によって制限されている。
私たちの業務はアメリカと外国の規制機関によって広く規制されており、私たちは私たちの使用、処理、処理、維持、個人情報の保護に関するすべての適用規則と法規を遵守しなければならない。米国では、HIPAAは連邦レベルで個人が識別可能な健康情報のプライバシー、安全、伝送に要求を提出しているが、個別の州、例えばカリフォルニア州やバージニア州は、すでにプライバシー法規を通過し、個人情報の使用を制限し、そのデータを収集し、使用する上で個人に何らかの権利を提供している。第1項を参照ビジネス-他のアメリカの医療法とコンプライアンス要件米国のプライバシーとデータ保護法に関するより多くの情報。また,欧州で個人情報を収集·利用することは,EUの一般データ保護条例やイギリスでこの条例やGDPRの管理を実施している。GDPRやEU加盟国やイギリスの他の適用データ保護法の要求、あるいは他の国/地域の他の適用プライバシールールや法規の要求を遵守しないことは、巨額の罰金や他の行政処罰を招く可能性がある。私たちは、私たちの業務に適用される現在および未来のプライバシーおよびデータ保護法規を遵守するために、追加のメカニズムを確立することを要求されるかもしれない。これは、私たちの開発活動を中断または延期することができ、および/または、私たちの業務慣行を変更することを要求する可能性があり、これは、私たちの業務、財務状況、運営結果、および見通しに悪影響を及ぼす可能性があります。
50
私たちに製品責任訴訟を提起すれば、重大な責任を招く可能性があり、候補治療薬の商業化を制限することが要求される可能性がある。
私たちの候補治療薬の臨床試験のため、私たちは固有の製品責任リスクに直面しています。もし私たちがいかなる細胞療法を商業化すれば、私たちの退行性疾患製品を販売するリスクに加えて、私たちはもっと大きなリスクに直面します。例えば、私たちの候補治療薬または変性疾患製品が臨床試験、製造、マーケティング、または販売中に傷害を引き起こすか、または不適切に発見された場合、起訴される可能性がある。このような製品責任クレームは、製造欠陥、設計欠陥、治療または製品固有の危険に対する警告、不注意、厳格な責任、または保証違反の告発を含む可能性がある。州消費者保護法によると、クレームも主張することができる。もし私たちが製品責任クレームで自分自身を弁護することに成功できなければ、私たちは重大な責任を招き、あるいは私たちの候補治療薬の商業化を制限することを要求されるかもしれない。成功的な防御であっても、多くの財政的で管理的な資源が必要だ。是非曲直または最終結果にかかわらず、責任クレームは多くの悪影響をもたらす可能性があり、そのいずれも私たちの財務状況および運営結果に実質的な損害を与える可能性がある。
潜在的な製品責任クレームを防止するために、許容可能なコストで十分な製品責任保険を得ることができず、これは、私たち単独または会社パートナーと開発した治療薬の商業化を阻止または阻害したり、私たちの変性疾患業務に負の影響を与える可能性がある。私たちの保険証書にも様々な例外があるかもしれません。私たちは製品責任クレームの影響を受けるかもしれません。私たちは保険範囲を持っていません。臨床試験のために臨床試験保険を受けることを期待していますが、裁判所が裁決したり、和解協議で協議した、私たちの保険範囲の制限を超えたり、私たちの保険カバー範囲内にない金額を支払わなければならないかもしれませんし、これらの金額を支払うのに十分な資本がないか得ることができないかもしれません。私たちが未来の会社のパートナーと合意しても、私たちは損害賠償を受ける権利があります。何かクレームがあれば、この賠償は利用できないか十分かもしれません。
私たちの第三者への依存に関するリスクは
私たちは依存して第三者に依存して臨床試験を続けるつもりだ。もしこれらの第三者が彼らの契約義務を成功的に履行できなかったり、予想された最終期限までに完成しなければ、私たちは規制部門の承認を得ることができず、私たちの候補治療薬を商業化することもできないかもしれない。
私たちは大学、医療機関、CROと戦略パートナーのような独立した研究者と協力者に依存し続け、私たちの臨床前と臨床試験を行う。私たちはCROや学習サイトと予算や契約を交渉しており、これは私たちの開発スケジュールの遅延とコスト増加を招く可能性があります。私たちの臨床試験では、私たちはこれらの第三者に深刻に依存し、私たちは彼らの活動のいくつかの側面だけをコントロールするだろう。しかし、私たちは私たちのすべての研究が適用された合意、法律、法規、そして科学的基準に従って行われ、私たちの第三者への依存が私たちの規制責任を免除しないことを確実にする責任がある。我々とこれらの第三者はGCPを遵守しなければならないが,これはFDAや同様の外国規制機関が臨床開発における候補治療案に対して実行する法規やガイドラインである。規制当局は,試験スポンサー,主要調査者,試験地点を定期的に検査することでこれらのGCPを実行している。もし私たちまたはこれらの第三者のいずれかが適用されたGCP法規を遵守できなかった場合、私たちの臨床試験で生成された臨床データは信頼できないと考えられる可能性があり、FDAまたは同様の外国の規制機関は、私たちの上場申請を承認する前に追加の臨床試験を行うことを要求するかもしれない。検査後、これらの監督機関は私たちのいかなる臨床試験がGCP規定に適合しているかどうかを確認することを保証することはできません。また,われわれの臨床試験はcGMP製生物製品を用いて行わなければならず,大量の試験患者が必要である。私たちまたはこれらの第三者がこれらの規定を遵守できなかったか、または十分な数の患者を募集できなかった場合、私たちは臨床試験を繰り返す必要があるかもしれません。これは規制承認過程を延期します。さらに、その中のいずれかの第三者が連邦または州詐欺および乱用または虚偽クレーム法律法規または医療プライバシー·セキュリティ法に違反した場合、私たちの業務は巻き込まれる可能性がある。
私たちの臨床試験を行ういかなる第三者も私たちの従業員でもなく、私たちの従業員でもなく、また、私たちがこのような第三者と合意した解決策を提供する以外に、彼らが私たちが行っている臨床前、臨床、非臨床プロジェクトに十分な時間と資源を投入するかどうかを制御することができない。これらの第三者はまた、我々の競争相手を含む他の商業実体と関係がある可能性があり、彼らはまた、これらの実体のための臨床研究または他の薬物開発活動を行っている可能性があり、これは彼らの業績に影響を与える可能性がある。これらの第三者がその契約の義務または義務の履行に成功しなかった場合、または予想される期限前に完了した場合、彼らが交換する必要がある場合、または彼らが得た臨床データの品質または正確性が、私たちの臨床方案や規制要件または他の理由を遵守できなかった場合、私たちの臨床試験は延長、延期、または終了される可能性があり、私たちは候補治療薬の開発を完了し、監督部門の承認を得たり、商業化に成功したりすることができないかもしれない。したがって、私たちの財務業績と私たちの候補治療薬のビジネス見通しが損なわれ、私たちのコストが増加する可能性があり、私たちが収入を生み出す能力が遅れる可能性がある。
もし私たちが試験場所や私たちが将来使用する可能性のある任意のCROとの任意の関係で終了すれば、私たちは代替試験場所やCROと計画を達成できないか、または商業的に合理的な条項でそうすることができないかもしれない。第三者を交換或いは増加させて著者らの臨床試験を行うことは大量のコストに関連し、大量の管理時間と重点が必要である。さらに自然な移行があります
51
新しい第三者が仕事を始めた時間帯。したがって,遅延が生じ,期待される臨床開発スケジュールを満たす能力に実質的な影響を与える可能性がある。
私たちは健康な人の臨月胎盤のドナーに依存して私たちの治療候補胎盤を製造し、もし私たちが合格したドナーから十分な胎盤供給を得なければ、私たちの胎盤由来の同種細胞の発育は不利な影響を受ける可能性がある。
われわれは生物由来の健康ドナー胎盤によりわれわれの候補治療案を作製し、病院員によって必要なドナー同意を得た。健康なドナー胎盤はタイプと品質がそれぞれ異なり、このような差異は標準化候補治療を生産することを更に困難にし、そして著者らの治療候補胎盤の開発と商業化の道を更に不確定にした。我々はすでに、3種類の同種細胞タイプ(CAR-T細胞、NK細胞および間葉系様基質細胞)を産生するための胎盤由来細胞の品質と一貫性を向上させるためのプロセスを開発したが、私たちのプロセスは適切なドナーを決定できないかもしれないし、すべての問題を検出し、生産後に材料故障を発見する可能性がある。私たちはまた、新しいウイルスをスクリーニングするなど、出現する可能性のある新しいリスクに対応するために、私たちの仕様を更新しなければならないかもしれない。
我々は,規制当局が要求する仕様を含め,ドナーの材料に厳しい仕様を持ち,インフォームドコンセントに依存している。規格に適合したドナー材料を決定して得ることができない場合、規制機関と適切な仕様で合意することができず、ドナー胎盤の可変性を提供することを奨励したり、ドナー胎盤の可変性を解決することができない場合、私たちが産生する候補治療薬に不一致が存在する可能性があり、あるいは進行中の臨床試験を所望の時間で開始または継続できない可能性があり、あるいは後期臨床試験や商業化のために私たちの生産プロセスを拡大することができず、私たちの名声を損なう可能性があり、私たちの業務と将来性に悪影響を及ぼす可能性がある。
細胞による治療は特殊な原材料の利用可能性に依存し,これらの原材料は許容可能な条件で提供できないか,あるいは全く得られない可能性がある。
私たちの候補治療は、ソレントからCAR配列を輸送するウイルスベクター、および他の原材料を含む多くの特殊な原材料を必要とし、その中のいくつかは、ビジネス治療をサポートするために、または私たちの規格に適合した原材料を提供するために、資源および経験の限られた小さな会社によって製造されている。私たちは現在ソレントと供給協定を交渉していますが、私たちは通常多くのサプライヤーと専門的な供給契約を締結していません。私たちは受け入れ可能な条件で彼らと契約を結ぶことができないかもしれません。新冠肺炎の流行期間中、多くのサプライヤーは業務を削減し、私たちは原材料を調達する能力が影響を受けた。また、私たちが後期臨床試験あるいは商業化に入るにつれて、私たちのいくつかのサプライヤーは規模を拡大できないかもしれない。そのため,臨床や商業製造を支援するキー原材料の受け入れが遅れたり,完全に確保できない場合がある可能性がある。ある原材料も第三者検査が必要で、いくつかの検査サービス会社は能力がないか、あるいは私たちが要求した検査を行うことができないかもしれません。
私たちはまた他の細胞治療会社からの供給競争に直面している。このような競争は、私たちが商業的に合理的な条件で、またはタイムリーな方法で原材料を獲得したり、そのような材料をテストすることを困難にするかもしれない。
いくつかの原材料は現在単一サプライヤーまたは少数のサプライヤーから得ることができる。私たちは、これらのサプライヤーが経営を続けるかどうか、あるいは私たちの競争相手のうちの1つまたは他の私たちの予想された目的のためにこれらの材料を生産し続けることに興味のある会社によって購入されないかどうかを確認することができません。また、新仕入先との関係構築に必要な納期が長くなる可能性があり、新仕入先に切り替えなければならない場合、需要を満たす上で遅延が生じる可能性があります。新しいサプライヤーに対する資格認証にかかる時間と労力は、このような資格を満たすいかなる規制要求も含めて、追加コスト、資源移転或いは製造生産量の低下を招く可能性があり、その中のいずれも私たちの運営結果に負の影響を与える。また、私たちはビジネス的に合理的な条項で新しいサプライヤーと合意できない可能性があり、これは私たちの業務に大きな悪影響を及ぼす可能性があります。
もし私たちまたは私たちを代表して行動する第三者供給者が被害をもたらしたり、適用法に違反したりする方法で危険、非危険、生物、または他の材料を使用する場合、私たちは損害賠償責任を負わなければならないかもしれない。
我々の研究開発と製造活動は,化学や生体材料を含む潜在的危険物質の制御使用に関するものである。私たちはアメリカ連邦、州と地方の法律法規の制約を受けて、医療と危険材料の使用、製造、貯蔵、運搬と処分を管理する。私たちの手続きと私たちの第三者サプライヤーがこれらの材料を使用、処理、貯蔵、処分する手続きは法律で規定された基準に適合していると信じているが、私たちも第三者サプライヤーも医療や危険材料による汚染や傷害のリスクを完全に除去することはできない。このような汚染や傷害のため、私たちは責任を負うかもしれないし、地方、都市、州、または連邦当局はこれらの材料の使用を制限し、私たちの業務運営を中断するかもしれない。事故が発生すると、私たちは損害賠償責任や罰金を要求されるかもしれません。責任は私たちの資源範囲を超えるかもしれません。私たちは医療や危険材料による責任に保険をかけない。適用される環境法律法規の遵守はコストが高く、現在または未来の環境法規は私たちの研究、開発、生産努力を損なう可能性があり、それによって私たちの業務、将来性、財務状況、または運営結果を損なう可能性がある。
52
政府の規制に関連するリスク
FDAの監督管理許可過程は長くて時間がかかり、著者らは臨床開発と監督管理が著者らの候補治療薬物を承認する方面で重大な遅延に遭遇する可能性がある。
生物製品を含む薬品の研究、テスト、製造、ラベル、承認、販売、輸入、輸出、マーケティングと流通はすべてFDAとアメリカの他の監督管理機関の広範な監督管理を受けている。FDAからバイオ医薬品ライセンス申請やBLA承認を得るまで、アメリカではいかなるバイオ医薬品製品の販売も許可されていません。我々はこれまでFDAにBLAを提出したこともなく,類似した外国当局に類似した承認文書を提出したこともない.BLAは、各必要な適応に対する候補治療レジメンの安全性および有効性を決定するために、広範な臨床前および臨床データおよび支持情報を含む必要がある。BLAはまた,製品の化学,製造,制御に関する重要な情報を含まなければならない。
私たちは、私たちの候補治療薬の新規性が規制の承認を得る上でさらなる挑戦をもたらすと予想している。例えば,同種細胞療法の商業開発におけるFDAの経験は限られている。私たちはまた、癌のタイプや出所を考慮することなく、目標に応じて将来の治療案の候補を承認することを規制部門に要求する可能性があり、もし私たちの臨床試験が特定の由来の癌のみに関連している場合、FDAはこれを受け入れることが困難かもしれない。FDAはまた,諮問委員会と呼ばれる専門家グループを要求し,許可を支援する安全性や有効性データの十分性を審議することも可能である.諮問委員会の意見には拘束力はないが,FDAが諮問委員会の提言を常に遵守しているため,完成した臨床試験から治療薬候補ライセンスを取得する能力に大きな影響を与える可能性がある。したがって、私たちの候補治療薬の規制承認経路は不確定で、複雑で、高価で、長く、承認されないかもしれない。
安全性や有効性のプロファイルが確立された既存の治療法に関する未解決の倫理的問題を処方するのではなく,医師が患者を募集してわれわれの治療案候補に参加する臨床試験に遭遇した場合など,様々な理由で計画中の臨床試験が遅延する可能性もある。また,多くの要因により,臨床試験はわれわれ,臨床試験を行う施設のIRBs,FDA,あるいは他の規制機関によって一時停止または終了する可能性がある。我々が行っている臨床試験のデータに対するFDAの審査はまた、私たちの1つ以上の臨床試験の遅延、一時停止、または終了をもたらす可能性があり、これはまた、私たちの他の計画における臨床試験の開始を延期または阻止するだろう。もし私たちの候補治療薬の任意の臨床試験の終了或いは遅延が完了すれば、私たちの候補治療薬の商業的な将来性は損害を受け、私たちの創収能力は延期される。また、臨床試験を完成するいかなる遅延も私たちのコストを増加させ、私たちの開発と承認過程を緩和し、そして私たちの治療販売と収入を創造する能力を危険にさらす。臨床試験の開始または完了遅延を引き起こす多くの要素は、最終的に私たちの候補治療方案が規制承認を拒否される可能性がある。
もし私たちのBiovanceとInterfyl製品がPHSA第361条だけによってHCT/Pの規制資格を満たしていなければ、これらの製品が市場から上下する可能性がある。
2017年11月、FDAは“ヒト細胞、組織、細胞と組織製品の監督管理考慮:最小操作と同源使用-工業と食品薬品監督管理局スタッフガイドライン”と題する指導文書を発表し、2020年7月に改訂し、再発表した。この文書は,21 CFR Part 1271によりバリアやカバーとして使用しようとしているシート状羊膜組織を21 CFR Part 1271により製造し,361条Hct/Psのみによる適切な規制を行うFDAの立場を確認している。しかし、傷口癒合は羊膜組織の同源使用ではなく、著者らが退行性疾病業務中の2種類の製品BiovanceとInterfylに対して声明を提出しさえすれば、同源使用の範囲を超えて、著者らはFDAの強制執行を受けるかもしれない。指導意見は、FDAは2021年5月31日に期限が切れる期間内に、あるHCT/PのIND申請と上場前承認に対して、限られた条件下で法執行自由裁量権を行使することを要求すると指摘した。FDAの方法はリスクに基づいており、このガイドラインは、高リスク製品および用途が即時実行行動の影響を受ける可能性があることを明らかにしている。ニューヨーク州のガイドラインの解釈は、このガイドラインには現在例外があるにもかかわらず、そのような製品のマーケティングを制限しており、このガイドラインには現在例外があるにもかかわらず、他の州は同様の決定を下す可能性があり、これはBLAが承認されるまで、このような製品の市場を制限するであろう。
PHSA第361節によると、羊水組織は一般にHCT/Pの規定のみに適合しており、これは、議論された特定の製品およびそれに対して提案された宣言がFDA適用の最小操作および相同使用基準に適合するかどうかに依存する。これらの最小操作および対応する使用基準に適合しないHct/Pは、医薬品、医療機器、生物製品、または組み合わせ製品のようなより広範な規制を受けるであろう。これらのHCT/Pは、発売前の承認またはFDAの承認を含む、HCT/Pに対するFDAの要求および生物製品、設備または薬物に適した要求に適合しなければならない。
私たちはFDAがBLAを承認するまで、私たちの声明を修正したり、BLAで許可された適応製品しか販売できなくなるまで、BiovanceとInterfyl製品の販売を停止する必要があるかもしれない。これらの製品をマーケティングし、販売する能力を失うことは、私たちの収入、業務、財務状況、および経営結果に悪影響を及ぼすだろう。また、第351条の要件に適合するコストのため、当社製品を製造するコストが増加することが予想されます
53
現在のcGMPや現在行われている製品試験コストのような生物製品。コンプライアンスに関連したコスト増加は、当社の業務、財務状況、運営結果に悪影響を及ぼす可能性があります。
さらに、FDAは、将来のある時点で、どのような現在または未来の製品が第361条のHCT/Psの規定に適合するかという立場を修正する可能性がある。いかなる規制変更も私たちに不利な結果をもたらす可能性があり、これらの製品の上場前の承認或いは承認を要求し、追加の上場後の監督管理要求を遵守することによって、私たちの業務をもっと展開しにくくしたり、コストを高くしたりする。FDAはまた私たちのBiovanceとInterfyl製品のリコールを要求するかもしれない。
我々が開発した候補治療薬は生物製品や生物製品として規制され,予想よりも早く競争される可能性が予想される。
BPCIAは“平価医療法案”の一部として公布され、生物類似と交換可能な生物製品を承認するために短い方法を確立することを目的としている。この規制経路は、承認された生物学的製品との類似性に基づいて生物類似体を“交換可能”に指定することを含む、FDAのための生物類似生物製品を審査および承認する法的権威を確立する。BPCIAによると、バイオ類似製品の申請は、BLAによる参考製品の承認12年後にのみFDAの承認を得ることができる。この法律は複雑であり、FDAはまだ説明して施行している。したがって、その最終的な影響、実施、そして意味には不確実性がある。FDAがいつこれらのBPCIAを実施するためのプロセスを完全に採用できるかどうかは不明であるが、どのようなプロセスも私たちの生物製品の将来のビジネスの将来性に重大な悪影響を与える可能性がある。
我々が開発したBLAにより米国でバイオ製品として承認された候補治療薬はいずれも12年の排他期を得る資格があるべきであると考えられる。しかしながら、国会の行動または他の理由により、このような排他性は短縮される可能性があり、またはFDAは対象治療候補製品を競合製品の参考製品とみなさず、予想よりも早く後発薬競争の機会を創出する可能性がある。さらに、承認されると、生物学的類似体が、どの程度、非生物製品に類似した伝統的な模倣薬代替の方法で任意の参照製品を置換するかは、まだ発展中のいくつかの市場および規制要因に依存するであろう。
著者らの候補治療薬物を管理する監督管理環境はまだ確定されていない;より成熟した細胞治療製品に関連する規制規定はまだ制定中であり、監督管理要求の変化は私たちの候補治療薬物の開発遅延或いは停止、或いは監督管理の承認を得る上で意外なコストを招く可能性がある。
我々は独自の生体実体の新しい細胞治療候補薬を開発しているため,我々が受ける規制要求は完全に明確ではない。遺伝子治療製品と細胞治療製品の監督管理要求は常に変化し、将来も引き続き変化する可能性がある。また、既存の遺伝子治療製品と細胞治療製品の監督を担当する人員の間には大量の重複があり、時には調和していない。FDAは個別治療案を継続できるかどうかを決定しているにもかかわらず,他の審査機関の審査過程や決定は臨床研究の開始を阻害または延期する可能性があり,たとえFDAがこの研究を審査し,その開始を承認したとしても。対照的に、FDAは、他のエンティティが有利な審査を提供しても、IND申請を臨床的に保留することができる。さらに、各臨床試験は、臨床試験を行う各機関の独立したIRBによって審査および承認されるか、またはサービスを提供しなければならない。さらに、他の人が行った遺伝子または細胞治療製品の臨床試験の不利な発展は、FDAまたは他の規制機関が私たちの任意の候補治療薬に対する承認要求を変化させる可能性がある。他の管轄区域には複雑な規制環境が存在し、私たちの候補治療のための規制承認を求めることが考えられ、規制構造をさらに複雑化させる可能性がある。
規制審査に参加する各委員会や諮問グループ、および彼らが時々公表している新しいガイドラインや改訂されたガイドラインは、規制審査過程を延長し、追加的な研究を要求し、私たちの開発コストを増加させ、規制の立場や解釈の変化を招き、候補治療薬の承認と商業化を延期または阻止したり、重大な承認後の制限や制限を招いたりする可能性がある。著者らの胎盤由来細胞候補治療の監督管理環境は新しいため、著者らはより伝統的な薬物或いは生物製品よりもっと煩雑で複雑な監督管理に直面する可能性がある。また,我々の候補治療薬が必要な規制承認を得ても,これらの承認は後に法規の変化や適用された規制機関の規制解釈によって撤回される可能性がある。潜在的治療薬を市場に投入するために必要な規制承認を遅延または獲得できなかったか、または規制承認を得るための予期しないコストは、業務を維持するために十分な収入を生成する能力を低下させる可能性がある。
FDAは私たちの規制計画に同意しないかもしれないし、私たちは規制部門の私たちの細胞治療候補案の承認を得ることができないかもしれない。
計画の1期と1/2 a期の臨床試験を完了し,積極的なデータを得ると,潜在的な登録試験に入る予定である。FDAが新しい生物または薬物を承認する一般的な方法は、スポンサーが関連患者集団における関連生物または薬物の2つの良好な制御の第3段階臨床研究の処置データを提供することである。3期臨床研究は通常数百名の患者に関連し、コストが高く、完成するまで数年かかる。結果が十分納得できれば,FDAと関連する治療案候補のBLA提出を検討する予定である。しかし私たちは何の合意もありませんでした
54
FDAの指導意見、すなわちその規制開発計画はBLAを提出するのに十分である。例えば、FDAは、承認された自己細胞療法を含む可能性がある承認された療法を比較試験することを要求する可能性があり、これは、我々の開発スケジュールを著しく遅らせ、大量の資源を必要とする。さらに、FDAは、非常に治療が困難な患者および進行性侵襲性癌患者である失敗または自己治療資格を満たしていない患者のみを評価することを可能にすることができ、私たちの候補治療レジメンは、そのような患者の結果を改善できない可能性がある。
FDAが1/2 a期の臨床試験結果に基づいて私たちの加速承認を承認した場合、もしこのような試験が発生した場合、承認を加速する条件として、FDAは不可逆的な発病率または死亡率または他の臨床終点に対する期待効果を検証し、説明するために、上場後の研究を要求する可能性があり、この薬物または生物はFDAの薬剤撤去手順の影響を受ける可能性がある。しかし、FDAは最終的に承認前に3期の臨床試験を要求する可能性があり、特に私たちの候補治療は新しい治療法を代表するからである。また,われわれが検討している同じ適応では,新しい療法の承認に伴い看護基準が変化する可能性がある。これは、我々の候補治療薬と新たに承認された治療薬との関係を評価するために、FDAまたは他の規制機関が追加的な研究を要求することにつながる可能性がある。
私たちの臨床試験結果も承認を支持しないかもしれない。また、私たちの候補治療薬は、以下の理由を含む、多くの理由で規制部門の承認を得ることができないかもしれない
我々は,我々の一部またはすべての候補治療薬のために様々な適応の孤児薬指定を求める予定であるが,市場排他性を含めてこのような指定や孤児薬指定に関する利点を維持することができず,収入減少を招く可能性がある(あれば)。
孤児医薬品法によれば、FDAは、まれな疾患または疾患を治療するための医薬または生物の孤児の称号を付与することができる。米国では,臨床試験費用に贈与資金を提供する機会,税収割引,ユーザ費用減免など,孤児薬を指定して一方が財政的インセンティブを得る権利がある。孤児薬物指定は、規制審査および承認中にいかなる利点も伝達されないか、または持続時間を短縮することはないが、孤児薬物指定を有する製品がその後、そのような指定された疾患を有する特定の製品に対するFDAの最初の承認を得た場合、この製品は、限られた場合を除いて、同じ生物(すなわち、同じ主要分子構造特徴を有する製品)の同じ適応を販売するためにBLAを含む他の出願を承認しない可能性があることを意味する。“”というタイトルの部分を参照企業--政府の規制と製品の承認孤児薬の指定に関するより多くの情報を得る。FDAは、2021年4月に、悪性グリオーマ患者の治療のための我々の非遺伝子組換え凍結保存ヒト胎盤造血幹細胞由来NK細胞療法CYNK-001を承認し、2022年2月にFDAが胃/胃食道境界癌の治療のために研究しているナチュラルキラー細胞療法CYNK-101の孤児薬物指定を承認したにもかかわらず、FDAは、同じ主要な分子構造特徴を有さない他の生物学的製品を排出期間内に同じ適応または疾患または同じ生物の異なる適応または疾患を治療するために承認することができる。また、もし私たちが十分な治療薬を生産できない場合、あるいはその後の申請者が私たちの製品が私たちの製品よりも臨床的に優位であることを証明した場合、FDAは孤児の排他性を放棄することができる。
特定の孤児適応のうち,我々の一部またはすべての候補治療薬のために孤児薬の指定を求める予定であり,これらの適応には,これらの治療法を用いて医学的に信頼できる基礎がある。孤児薬の名前を獲得しても中国での独占販売権は
55
孤立指定適応よりも広い適応の承認を求める場合、米国は制限される可能性があり、FDAが後に適応指定を決定する要求に重大な欠陥がある場合、またはまれな疾患または疾患を有する患者の需要を満たすのに十分な数の治療薬が保証されない場合、またはその後の申請者がその臨床的優位性がわれわれの治療法を超えることを証明する場合(承認されれば)、米国は制限される可能性がある。
FDAの画期的な治療または迅速なチャネル指定を得る治療候補者の場合、私たちは、加速された開発または規制された審査および承認手続きを選択または利用できない可能性がある。
FDAと規制戦略について継続的に検討し続ける予定であり,これらの戦略は,我々の候補治療薬の加速開発経路を利用することができるかもしれないが,我々の候補治療薬がいかなる加速開発経路を得る資格があるかどうか,あるいは規制機関が関連する合格指定を付与または維持することを可能にしているかどうかを決定することができるかもしれない。私たちが追求できる潜在的な加速発展の道は突破的な治療と迅速な通路指定を含む。
画期的な治療指定は、深刻または生命を脅かす疾患を治療するための候補治療薬の設計の開発および審査を加速することを目的としており、“初歩的な臨床証拠は、薬物が1つまたは複数の臨床的に重要な終点で、例えば臨床開発早期に観察された実質的な治療効果のような既存の治療方法よりも実質的な改善を示す可能性があることを示す”としている。候補療法を画期的な治療法に指定することは,FDAとより頻繁な会議を行い,候補療法の開発計画を検討し,承認支援に必要な適切なデータの収集を確保すること,FDAは臨床試験の設計やバイオマーカーの使用を提案することなどについてより頻繁に書面通信を提供すること,最初に第1段階から効率的な薬物開発計画について密な指導を提供すること,高度な管理者が参加する組織承諾,審査と優先審査の資格を転動することを含む潜在的な利点を提供している。迅速チャネル指定は、深刻なまたは生命を脅かす疾患または状態を治療するための候補治療方案のために設計されており、これらの場合、非臨床または臨床データは、このような疾患または状態が満たされていない医療需要を解決する可能性があることを示している。
いくつかの細胞治療候補薬の迅速チャネル指定を得ているが,画期的な治療や他の候補治療薬を行わない迅速チャネル指定を選択することができ,FDAが広範な裁量権を有してこれらの指定を承認するかどうか。
したがって,特定の候補治療が画期的な治療や迅速チャネル指定を得る資格があると考えても,FDAがこのような指定を決定することは保証されない。画期的な治療指定および迅速チャネル指定は、製品承認の基準を変更することはなく、このような指定または資格が迅速審査または承認をもたらすことも保証されず、承認された適応が画期的な治療指定または迅速チャネル指定によってカバーされる適応範囲よりも狭くならないことも保証されない。したがって,画期的な治療や迅速なチャネル指定を受けても,従来のFDAプログラムよりも速い開発過程,審査,承認を経験しない可能性がある。FDAがこの製品がもはや資格基準を満たしていないと思う場合、突破的治療または迅速チャネル指定を撤回する可能性がある。もし私たちがこのような他の開発と規制を加速させる方法を利用できなければ、私たちの業務は損害を受けるかもしれない。
一つの管轄区域で私たちの候補治療薬の規制承認を獲得し、維持することは、私たちが他の管轄区域で私たちの候補治療薬の規制承認を得ることに成功することを意味するものではない。
1つの管轄区域で私たちの候補治療薬の規制承認を獲得し、維持することは、任意の他の管轄区域で規制承認を得ることができるか、または維持することができる保証はなく、1つの管轄区で監督管理承認を得ることができなかったり、遅延したりすることは、他の管轄区の規制承認過程に悪影響を及ぼす可能性がある。例えば、FDAが候補治療薬の発売を許可したとしても、外国司法管轄区の比較可能な監督管理機関はこの治療候補薬物のこれらの国での製造、マーケティングと普及を許可しなければならない。承認手続きは司法管轄区域によって異なり、1つの管轄区域で行われる臨床研究は他の管轄区の監督当局に受け入れられない可能性があるので、米国と異なるか、または米国より大きい要求と行政審査期間に関連する可能性がある。米国以外の多くの管轄区では、候補治療薬は精算承認を受けなければならず、この司法管轄区で販売が許可される。場合によっては、私たちが私たちの製品のために受け取る価格もまた承認されなければならない。
私たちはまた他の国でマーケティング申請を提出することができる。アメリカ以外の管轄区の監督機関は候補治療薬の承認に要求があり、私たちはこれらの管轄区が発売される前にこれらの要求を守らなければならない。外国の監督管理の承認を得て、外国の規制要求を遵守することは、重大な遅延、困難、コストを招く可能性があり、私たちの製品がある国/地域で発売されることを延期または阻止する可能性がある。もし私たちが国際市場の規制要求を遵守できず、および/または適用されたマーケティング承認を得ることができなければ、私たちの目標市場は減少し、私たちの治療薬候補の完全な市場潜在力を達成する能力は損なわれるだろう。
56
私たちが規制機関から私たちの治療薬候補の承認を得ても、私たちは持続的な規制義務と持続的な規制審査の制約を受けることになり、これは多くの追加費用を招く可能性があり、もし私たちが規制要件を遵守できなかったり、私たちの治療候補薬が予期しない問題に遭遇したら、私たちは処罰を受けるかもしれない。
著者らは候補治療薬が獲得したいかなる規制承認に対してもモニタリングを行い、候補治療薬の安全性と有効性を監視する必要がある。FDAはまた、私たちの候補治療方案を承認するためにREMSを必要とする可能性があり、これは、薬物ガイドライン、医師のコミュニケーション計画、または分配方法の制限、患者登録、および他のリスク最小化ツールのような安全な使用を確保する他の要素を必要とする可能性がある。さらに、FDAまたは同様の外国規制機関が私たちの候補薬物を承認すれば、私たちの候補薬物の製造プロセス、ラベル、包装、流通、有害事象報告、貯蔵、広告、宣伝、輸入、輸出、記録保存は広範で持続的な規制要求を受けるだろう。これらの要求は、安全と他の発売後の情報と報告、登録の提出、および著者らが承認後に行った任意の臨床試験が引き続きcGMPと現在のGCPに適合しているかどうか、および適用される製品追跡と追跡要求に適合しているかどうかを含む。したがって、cGMPの遵守状況、および任意のBLA、他のマーケティング申請、および以前の検査意見に対する応答で約束された遵守状況を評価するために、持続的な審査および検査を受ける。したがって、私たちと私たちと協力している他の人たちは、製造、生産、品質管理を含むすべての規制コンプライアンス分野で時間、お金、エネルギーをかけ続けなければならない。さらに、FDAは、より多くの安全性またはバイオマーカー情報を得るために、別の研究を要求するかもしれない。さらに、FDAの宣伝および広告ルールの遵守が要求されるであろう。その中には、他に加えて、消費者向け直接広告の基準、製品の用途または製品承認用途に記載されていない患者集団に対する製品の普及(“ラベル外使用”と呼ばれる)、業界支援の科学的および教育活動の制限、およびインターネットおよびソーシャルメディアに関連する販売促進活動への要求が含まれる。FDAや他の機関はラベル外用途の普及を禁止する法律法規を積極的に実行しており,ラベル外用途の普及が不適切であることが発見された会社は重大な責任を負う可能性がある。しかし、医師はその独立した医学判断に基づいて、ラベル外で使用される合法的に使用可能な製品のために処方することができる。FDAは医師が治療を選択する行為を規範化していないが,FDAはラベル外でその製品を使用するメーカーのコミュニケーションを制限している。
その後、私たちの候補治療薬には、予期せぬ深刻または頻度の有害事象、または私たちの第三者サプライヤーまたは私たちの製造プロセスを含む、以前に未知の問題が存在することが発見され、あるいは規制要件を遵守できなかった場合、承認されたラベルを修正して新しいセキュリティ情報を追加し、発売後研究または臨床研究を実施して新しいセキュリティリスクを評価するか、またはREMS計画に従って流通制限または他の制限を実施する可能性がある。他の他の潜在的な結果には
FDAおよび他の規制機関の政策は変わる可能性があり、私たちの候補治療案の規制承認を阻止、制限、または延期するための追加の政府法規が公布されるかもしれない。米国や海外の将来の立法や行政または行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することはできない。もし私たちが既存の要求の変化に緩やかに適応できない場合、あるいは新しい要求や政策を採用することができない場合、または規制適合性を維持できない場合、私たちは私たちが得る可能性のあるマーケティング承認を失う可能性があり、利益を達成したり維持することができないかもしれない。
遺伝子編集または修飾細胞に関連する遺伝子研究および治療に対する否定的な世論および規制審査の強化は、私たちの候補治療案に対する公衆の見方を損なう可能性があり、または私たちの業務を展開するか、または規制機関から私たちの候補治療方案を承認する能力に悪影響を及ぼす可能性がある。
私たちが使っている遺伝子編集技術は珍しいです公衆の認知は遺伝子編集が安全でないという説の影響を受ける可能性があるが,遺伝子編集を含む製品は公衆や医学界の受け入れを得られない可能性がある。特に,我々の成功は,我々の目標疾患を専門的に研究している医師が,既存のより熟知した治療法を代替または補完するための治療候補薬を開発することに依存し,これらの治療法はより多くの臨床データが利用可能である可能性がある。遺伝子編集に対する否定的な見方の増加は、より少ない医師が私たちの治療レジメンを開発することをもたらすかもしれないし、または患者が私たちの治療レジメンを使用したり、私たちの候補治療レジメンに参加する臨床試験に参加する意欲を低下させる可能性がある。また、遺伝子編集と細胞治療技術の新規性を考慮して、各国政府は輸入、輸出或いはその他の方面を制限して、これらの技術の制御或いはその使用を制限する可能性がある。
57
米国または国際的には、ますます多くの負の世論またはより厳しい政府法規が、私たちの業務または財務状況に悪影響を与え、候補療法の開発および商業化、またはそのような候補療法の需要を遅延または損害する可能性がある。
規制部門の候補治療案の承認を得ても,細胞療法は医師,患者,病院,癌治療センター,医学界の他の人の市場受け入れを得られない可能性がある。
工学胎盤由来の細胞を潜在的な治療法として使用することは最近の発展であり、医師、患者、病院、癌治療センターと医学界の他の人に広く受け入れられない可能性がある。多くの理由で、私たちはこの人たちに私たちの候補治療薬を使用する利点を教育できないかもしれない。例えば,我々が開発したいくつかの治療候補薬は,癌細胞および非癌細胞上に存在する可能性のある細胞表面マーカーである。私たちの候補治療薬はこれらの非癌細胞を殺す可能性があり、これは死を含む受け入れられない副作用を引き起こす可能性がある。他の要素は私たちの候補治療案が市場に受け入れられているかどうかに影響を与えるだろう
もし私たちの候補治療薬が承認されたが、医師、患者、病院、癌治療センター或いは医学界の他の人の市場認可を得られなければ、私たちは相当な収入を得ることができないだろう。私たちの細胞療法が市場に受け入れられても、私たちの療法よりも人気があり、費用効果があり、あるいは私たちの療法を時代遅れにする新製品や技術が発売されれば、時間の経過とともに市場受容度を保つことができないかもしれない。
我々の治療候補に対しては,いくつかの細分化された市場では,カバー範囲や精算範囲が限られているか,利用できない可能性があり,我々の細胞療法の販売が困難になる可能性があり,承認されれば利益になる可能性がある。
承認されれば,我々の候補療法の成功販売は,MedicareやMedicaid,管理型医療組織,商業支払者などの政府医療保健計画を含む第三者支払者が保険を提供するかどうかと十分な精算を提供するかどうかに依存する。私たちが監督管理の許可を得た任意の候補治療方案のカバー範囲と精算状態に重大な不確定性が存在する。また,我々の候補治療薬は癌,感染症,退行性疾患を治療する新しい方法を代表しているため,候補治療薬の潜在的収入を正確に見積もることはできない。保証範囲と補償要求の詳細については、タイトルを参照してください“企業-政府の規制と製品の承認-カバー範囲、定価、精算
自分の病状に医療サービスを提供する患者は通常,第三者支払者によってその治療に関連する費用の全部または一部を精算する。第三者支払者から保険と十分な補償を受けることは新製品の受容度に重要です。
第三者支払者は彼らがどのような薬と治療と費用をカバーするかを決定する。第三者支払人の精算は多くの要素に依存するかもしれないが、これらに限定されず、治療薬の使用に対する第三者支払人の決定:
58
政府あるいは他の第三者支払人から治療薬物の保証と精算を獲得することは時間がかかり、高価な過程であり、支払人に私たちの治療薬物の使用を支援する科学、臨床と費用効果データを提供する必要があるかもしれない。特定療法の保険を受けても,それによる報告率が不足していれば,病院はわれわれの療法をその施設で使用することを許可しない可能性があり,あるいは第三者支払者が受け入れられない高額の自己負担費用の支払いを要求する可能性がある。患者が我々の候補療法を使用することは不可能であり,保険を提供しない限り,候補療法を支払うのに十分な大部分の費用を精算する。治療自体は単独で精算されるかもしれないし、そうではないかもしれない。対照的に、病院または治療を実行する医師は、私たちの治療方法を使用する治療またはプログラムを提供することによってのみ補償を得ることができる。また,CMSは連邦医療保険医料金表や外来予想支払い制度を含む医療提供者への精算のための精算制度を時々改正し,医療保険支払いの減少を招く可能性がある。場合によっては、個人第三者支払者は、連邦医療保険支払いシステムの全部または一部に依存して支払率を決定する。政府医療計画を変更し,これらの計画下での支払いを減らすことは,個人第三者支払者の支払いに負の影響を与え,医師が我々の候補治療案を使用する意欲を低下させる可能性がある。
米国では、第三者支払者の間に統一された製品保証や精算政策はない。そのため、製品の保証範囲と精算範囲は支払人によって異なる。また、支払者が1つの商品に保険を提供することを決定し、他の支払者もその商品に保険を提供することを保証することはできない。製品開発への投資の適切な見返りを実現するために、十分な価格レベルを維持することができる十分な第三者精算がないかもしれない。
私たちはアメリカと選定された外国司法管轄区域で私たちの候補治療薬の販売を承認することを求めるつもりだ。もし私たちの候補治療薬が1つ以上の外国の管轄区域で承認されたら、私たちはこれらの管轄区域の規則によって制限されるだろう。一部の外国国家、特にヨーロッパ諸国では、生物製品の定価は政府によって規制されている。これらの国では,候補治療薬の発売承認を得た後,政府当局との定価交渉にかなりの時間を要する可能性がある。その中のいくつかの国は臨床試験の完成を要求し、特定の候補療法の費用効果を現在利用可能な治療法と比較する可能性がある。他のEU加盟国は会社が自分の薬品価格を固定することを許可したが、会社の利益を監視した。医療コストの下振れ圧力は非常に大きくなっている。そのため、新製品の参入にはますます高い壁が設けられている。また、一部の国では、低価格市場からの国境を越えた輸入が一国国内の定価に商業圧力をかけている。
政府や他の第三者支払者が保険や十分な補償を提供できない場合、規制部門の承認を得て商業販売を行う任意の候補治療薬の適正性が影響を受ける可能性がある。私たちは薬品価格の下振れ圧力が続くと予想している。また、保証政策と第三者精算料率は随時変化する可能性がある。規制部門の承認を得た1つ以上の療法が有利な保証範囲と精算状態を得ても、将来的にはあまり有利ではない保証政策や精算料率が実施される可能性がある。
医療改革の推進は,われわれの候補治療薬を売る能力に悪影響を及ぼす可能性があり,承認すれば利益になる。
第三者決済者は,国内でも海外でも,政府のものでも商業的でも,ますます複雑な方法を開発して医療コストを抑えている。米国や一部の外国司法管轄地域では、医療システムのいくつかの立法や規制の変化が、候補治療薬を販売する能力に影響を与える可能性があり、承認されれば利益になる可能性がある。さらなる立法や規制によって、私たちの業務、財務状況、そして運営結果を損なう可能性があります。“企業--政府の監督管理と製品審査”の一節を参照--医療改革このような法律法規に関する討論。外国、連邦と州の各レベルはすでに立法と監督管理提案を引き続き提出することが可能であり、医療保健の獲得性を拡大し、医療保健コストをコントロール或いは低減することを目的としている。コスト抑制措置や他の医療改革を実施することは、収入の創出、利益の実現、あるいは私たちの療法を商業化することを阻止するかもしれません。これらの改革は、規制承認された候補治療薬の期待収入の開発に成功し、私たちの全体的な財務状況に影響を与える可能性があり、治療候補薬を開発する能力に影響を与える可能性がある。
また、特殊薬品の価格設定実践におけるアメリカの立法と法執行の興味もますます大きくなっている。具体的には,米国議会は最近,薬品定価の透明性を増加させ,価格決定とメーカー患者援助との関係を審査するために,いくつかの調査や連邦や州立法活動を行っている
59
政府が計画した薬品精算方法を計画し、改革する。米国の個別州も、価格または患者の精算制限、割引、ある製品への参入の制限、マーケティングコストの開示および透明性措置を含む薬品の価格設定を制御するための法規を立法と実施することにより、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。
私たちは未来に取られる可能性のある計画を予測できない。さらに、政府、保険会社、管理医療組織、および他の医療サービス支払者は、医療コストの抑制または低減、および/または価格制御の実施に努力し続けており、以下の点に悪影響を及ぼす可能性がある
連邦医療保険や他の政府計画精算のいかなる減少も、個人支払者の支払いの同様の減少を招く可能性があり、これは私たちの将来の収益性に悪影響を及ぼす可能性がある。
私たちの知的財産権に関するリスクは
もし私たちが私たちの技術に関連する知的財産権の固有性を保護する努力が十分でなければ、私たちは私たちの市場で効果的に競争することができないかもしれない。
他の生物製薬会社と同様に、私たちの成功は私たちが知的財産権保護を獲得し、維持する能力に大きく依存する。私たちは特許、商業秘密保護、許可協定の組み合わせによって、私たちの技術に関連する知的財産権を保護します。私たちの機密固有情報を第三者に開示したり盗用したりすることは、競争相手が私たちの技術的成果を迅速にコピーしたり、超えたりして、市場での私たちの競争地位を侵食する可能性がある。私たちはより多くの特許出願を提出しており、今後も必要に応じて米国や他の国でより多くの特許出願を提出することが予想される。しかし予測できません
生物製薬特許の取得および実行は高価で、時間がかかり、複雑であり、私たちは、必要または望ましいすべての特許出願を合理的なコストで、またはそのような特許出願から発行される可能性のある任意の特許を維持、実行および許可することができないかもしれない。我々は,特許保護を得るのが遅くなるまで,我々の研究や開発成果の特許可能性を確認できない可能性もある.私たちは、第三者から許可された特許出願の準備、提出および起訴を制御する権利がないか、または第三者に許可された特許の権利を維持する権利がないかもしれない。したがって、このような特許および出願は、私たちの業務の最適な利益に合った方法で起訴され、強制されてはならない。
私たちが係属中の特許出願中の権利要件が、米国特許商標局または外国特許庁によって出願可能特許とみなされるか、または私たちが発行した任意の特許における権利要件が、米国または外国裁判所によって有効かつ強制的に実行可能であるとみなされることを決定することはできない。バイオテクノロジーや製薬分野の特許実力は複雑な法律や科学問題に関連しており,不確定である可能性がある。私たちが所有しているまたは許可されている特許出願は、発行された特許を生成できない可能性があり、その声明は、米国または他の国/地域における私たちの候補療法またはその使用をカバーする。特許が確実に発表されたとしても、第三者はその特許性、有効性、実行可能性または範囲に疑問を提起する可能性があり、これは、このような特許がキャンセルされ、縮小され、無効にされ、または強制的に実行できなくなる可能性がある。さらに、それらが挑戦されていなくても、私たちの特許と特許出願は、私たちの知的財産権を十分に保護したり、他の人が私たちのクレームによってカバーされないように製品を設計することを阻止することができないかもしれません。もし私たちが持っている特許や特許出願が私たちの候補療法に提供する保護の広さや強度が脅かされれば、会社が私たちと協力して私たちの候補療法を開発することを阻止し、私たちがそれを商業化する能力を脅かすかもしれない。さらに臨床試験で遅延があれば
60
私たちが特許保護の下で私たちの候補治療薬を販売できる時間は短縮されるだろう。また、米国特許法の変化は、特許の全体的な価値を低下させ、製品を保護する能力を弱める可能性がある。
米国議会、連邦裁判所、および米国特許商標局の決定によると、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、それにより、私たちが新しい特許を獲得したり、私たちの既存の特許と将来獲得可能な特許を強制的に執行する能力を弱める可能性がある。
従業員および第三者との秘密保持協定は、商業秘密や他の固有情報の不正流出を防ぐことができない可能性がある。
特許提供の保護に加えて、特許を出願できないノウハウ、特許を実施することが困難なプロセス、および私たちの製品発見および開発プロセスにおいて特許がカバーされていないノウハウ、情報または技術に関する任意の他の要素を商業秘密保護およびセキュリティプロトコルに依存して保護することが求められている。私たちは、当社の知的財産権およびノウハウを保護し、当社の従業員、コンサルタント、会社パートナー、および必要に応じて、機密協定、秘密協定、知的財産権譲渡協定を含むコンサルタントと協定を締結する措置を取っています。しかし、商業秘密は保護するのが難しいかもしれない。
許可されていない開示を規制し、許可されていない開示を発見することは困難であり、私たちはまた、私たちがこのような開示を防止するための手順が十分であるか、または十分であるかどうかを知らない。もし私たちが第三者が私たちの商業秘密を不正に取得して使用した疑いを強制するなら、これは高価で時間がかかるだろうし、結果は予測できないだろう。
私たちのすべての従業員に彼らの発明を譲渡することを要求し、私たちのノウハウ、情報、または技術にアクセスできるすべての従業員とキーコンサルタントとの秘密協定を締結することを要求していますが、私たちのビジネス秘密および他の機密固有情報が漏洩しないか、あるいは競争相手が他の方法で私たちのビジネス秘密や独立開発と実質的に同じ情報および技術を獲得しないかを決定することはできません。また、一部の国の法律の専有権に対する保護の程度や方式は米国の法律とは異なる。したがって、私たちはアメリカでも海外でも、私たちの知的財産権を保護して守ることで大きな問題に直面するかもしれない。もし私たちが許可されていない第三者に私たちの機密情報や知的財産権を開示することを阻止できなければ、私たちは私たちの市場で競争優位性を確立したり、維持することができなくなり、これは私たちの業務、経営業績、財務状況に大きな悪影響を及ぼすかもしれない。
私たちは、私たちの従業員、コンサルタント、または独立請負業者が第三者の機密情報を誤って使用または開示することに関するクレームを受けるかもしれない。
私たちは第三者から機密と固有の情報を受け取った。しかも、私たちは他のバイオテクノロジーや製薬会社に以前雇われていた個人を雇っている。私たちは、私たちの従業員、コンサルタント、コンサルタント、および独立請負業者が、私たちのために働いているときに他人の固有情報またはノウハウを使用しないことを保証するために努力しているが、私たちは、これらの第三者または従業員の前の雇用主の商業秘密または他の独自または機密情報を含む知的財産権を、私たちまたは私たちの従業員、コンサルタント、または独立請負業者が無意識にまたは他の方法で使用または開示しているというクレームを受ける可能性がある。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭損害賠償を支払う以外に、貴重な知的財産権を失い、より激しいビジネス競争に直面する可能性がある。重要な研究者の作業製品の損失は、潜在的な技術や解決策を商業化する能力を阻害または阻止する可能性があり、これは私たちの業務を損なう可能性がある。私たちがこれらのクレームを弁護することに成功しても、訴訟は巨額のコストを招き、私たちの管理チームと従業員の注意を分散させる可能性がある。
また、私たちの政策は、知的財産権の概念や開発に参加する可能性のある私たちの従業員と請負業者が合意に署名し、このような知的財産権を私たちに譲渡することを要求しているにもかかわらず、実際に私たちが自分の知的財産権と見なしているすべての当事者とこのような合意を実行することができないかもしれない。知的財産権の譲渡は自動的に実行されない可能性があり、あるいは譲渡協定が違反される可能性があり、私たちは第三者にクレームをつけさせられたり、私たちが私たちの知的財産権の所有権だと思うことを決定するために、私たちが提起する可能性のあるクレームを弁護したりすることができます。上記のいずれも、私たちの業務、財務状況、経営結果、および将来性を損なう可能性があります。
知的財産権侵害に対する第三者のクレームは、私たちの製品発見と開発を阻害または延期する可能性があり、候補治療薬を商業化する能力を持っている。
私たちの商業的成功は私たちが第三者の特許と固有の権利の侵害を避けることにある程度かかっている。バイオテクノロジーと製薬産業では、特許と他の知的財産権に関する多くの訴訟がある。我々が治療候補薬を開発している分野には,第三者が所有する米国や外国から発行された特許や係属中の特許出願が多く存在する。バイオテクノロジーや製薬業界の拡張やより多くの特許の発行に伴い,我々の候補治療薬は他人の特許権侵害のクレームを引き起こすリスクが増加する可能性がある。
61
第三者は私たちが彼らの特許を侵害したり、他の方法で彼らのノウハウを不正に使用したと主張し、訴訟を提起する可能性があるかもしれない。特許出願が発行されるまでに数年かかる可能性があるため、現在処理されている特許出願が存在する可能性があり、これらの出願は、私たちの治療薬候補が侵害されたと告発された発行された特許をもたらす可能性がある。さらに、第三者は将来的に特許を取得し、私たちの技術を使ってこれらの特許を侵害したと主張するかもしれない。管轄権のある裁判所が、私たちの候補治療薬、製造中に使用または形成された構造または分子の製造プロセス、または任意の最終治療薬自体をカバーするための任意の第三者特許を有する場合、任意のそのような特許の所有者は、適用特許に基づいてライセンスを取得するか、またはそのような特許が満了するまで、またはそれらが侵害されていない、非特許、無効、または強制的に実行されないと判断しない限り、候補治療薬を商業化する能力を阻止することができるかもしれない。同様に、管轄権のある裁判所が任意の第三者特許を有し、共同療法または患者が方法を選択することを含む、私たちの処方、製造プロセス、または使用方法の様々な態様をカバーする場合、任意の特許の所有者は、ライセンスを取得しない限り、または特許が満了するまで、または最終的に侵害されていない、非特許、無効、または強制的に実行されない限り、候補療法を開発し、それを商業化する能力を阻止することができるかもしれない。いずれの場合も、そのような許可は商業的に合理的な条項や全く存在しないかもしれない。もし私たちが商業的に合理的な条項で第三者特許の必要な許可を得ることができない場合、あるいは根本的にできなければ、候補治療薬を商業化する能力が損なわれたり、延期されたりする可能性があり、逆に私たちの業務を深刻に損なう可能性がある。
私たちにクレームを出した当事者は、禁止または他の公平な救済を求めて獲得する可能性があり、これは、私たちの候補治療薬のさらなる開発と商業化を効果的に阻止することができるかもしれない。これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、私たちの業務における従業員資源を大量に移転し、私たちの名声に影響を与える可能性がある。もし私たちに対する侵害クレームが成功した場合、私たちは故意に侵害した3倍の損害賠償金と弁護士費、第三者から1つ以上の許可証を取得し、印税を支払うか、または私たちの侵害製品を再設計することを含む大量の損害賠償を支払わなければならないかもしれないし、大量の時間とお金の支出が必要かもしれない。私たちはそのようなライセンスがあるかどうか、あるいは商業的に合理的な条項で提供されるかどうかを予測できない。また、訴訟がない場合でも、第三者からライセンスを取得して、私たちの研究を進めたり、候補治療薬の商業化を可能にしたりする必要があるかもしれません。私たちはもしあれば、合理的な費用または合理的な条項でこのような許可証のいずれかを得ることができないかもしれない。この場合、私たちの候補治療薬をさらに開発して商業化することはできません。これは私たちの業務を深刻に損なう可能性があります。
私たちは、買収およびライセンス内の許可によって、私たちの開発プロセスにおける製品コンポーネントおよびプロセスの必要な権利を取得または維持することに成功できないかもしれません。
現在,我々は第三者からの許可と我々が所有または所有する特許出願,知的財産権を有する権利により,これらの知的財産権が候補治療薬の開発を促進すると信じている。将来、私たちは、新しい技術やサービスを開発したり、それを商業化したりすることを含む、当社の業務に従事するために、取得または許可の第三者知的財産権および技術が必要である可能性があると判断することができ、私たちの業務の成長は、私たちが取得、許可、または使用する能力にある程度依存するかもしれません。
私たちは私たちが決定した第三者から第三者知的財産権を得ることができないかもしれない。私たちはこのようなライセンスのいずれかを合理的な費用または合理的な条項で得ることができないかもしれないが、これは私たちの業務を損なうだろう。たとえ我々が許可を得ることができても,我々は非排他的であり,我々の競争相手が我々に許可された同じ技術にアクセスできるようにする可能性がある.この場合、私たちは代替技術を開発または許可するために多くの時間と資源を必要とするかもしれない。このような第三者知的財産権によってカバーされる成分または方法の使用を停止する必要があるかもしれません。どのような第三者ライセンスでも私たちのライセンスを維持することができないまで。
第三者知的財産権の許可および買収は競争分野であり、私たちよりも成熟しているか、またはより多くの資源を持っている可能性のある会社も、私たちの候補治療薬を商業化するために、必要または魅力的だと思う第三者知的財産権許可または買収戦略を実施しているかもしれない。より成熟した会社は私たちより競争優位にあるかもしれません。それらの規模、現金資源、そしてより強い臨床開発と商業化能力のためです。
しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。このようなライセンスが利用可能な場合、私たちは、そのような許可者の技術の使用、一括払い、いくつかのマイルストーンに基づく支払い(例えば、売上)または売上ベースの印税と交換するために、ライセンス側に費用を支払う必要があるかもしれない。しかも、私たちの免許はまた私たちの未来のビジネス機会に制限を加えるかもしれない。
私たちが最善を尽くしたにもかかわらず、私たちの許可側は、許可協定に深刻に違反していると結論するかもしれませんので、許可協定を終了し、これらの許可協定がカバーする技術を開発し、商業化することができません。これらの許可が終了した場合、または基礎知的財産権が予想される排他性を提供できなかった場合、競争相手は、規制部門の承認を求め、我々が許可を得た技術と同じ技術を使用した製品を販売する権利がある。これは私たちの競争地位、業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼすかもしれない。さらに、これらの合意を終了するか、またはこれらのプロトコルの下での私たちの権利を減少またはキャンセルするか、または私たちの業務利益に適合した場合に、このような合意の下での私たちの権利を自由に譲渡または再許可する能力を制限します
62
したがって、私たちは、重要な知的財産権または技術に対する私たちの権利や阻害、またはそのような合意に依存する1つまたは複数の技術のさらなる開発または商業化を含む、新しい条項または回復された条項の悪い合意を交渉しなければならない、またはこれらの合意の下での権利を失わせなければならない可能性がある。
上述したリスクに加えて、現在または将来許可を得る可能性のある知的財産権は、第三者が所有する知的財産権項目の二次ライセンスを含むことができ、場合によっては多層ライセンスを通過する場合がある。したがって、私たちライセンシーの行動は、ライセンス契約下のすべての義務を遵守していても、再ライセンス知的財産権を使用する権利に影響を与える可能性があります。もし私たちのライセンシーまたは任意の上流ライセンシーがプロトコルに従って負担する義務を履行できなかった場合、これらの合意に基づいて、彼らは私たちに転任する権利を獲得した場合、またはそのようなプロトコルが終了または修正された場合、候補治療薬を開発および商業化する能力は実質的に損なわれる可能性がある。
さらに、私たちは私たちがこのような権利を持っていても、私たちが許可と再許可を得たすべての知的財産権の起訴、維持、実行を制御する権利がないかもしれないが、私たちは私たちのライセンシーと上流ライセンシーの協力を必要とするかもしれないが、これは達成されないかもしれない。私たちまたは私たちのライセンシーが許可および再許可の知的財産権を効率的に起訴、維持、実行できない場合、私たちの業務は悪影響を受ける可能性があります。
私たちのライセンス者は、第三者コンサルタントまたは協力者または第三者からの資金に依存する可能性があり、したがって、私たちのライセンス者は、ライセンス中の特許および特許出願の唯一および独占所有者ではない。もし他の第三者が私たちが許可した特許または特許出願の所有権を持っている場合、彼らはこれらの特許を私たちの競争相手に許可するかもしれません。私たちの競争相手はそれと競争する製品と技術を販売することができます。これは私たちの競争地位、業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼすかもしれない。
許容可能な条項で必要な合意に到達できない場合、または任意の必要な許可がその後終了される場合、許可の条項を遵守できない場合、または第三者の侵害を防止することができない場合、または買収または許可された特許または他の権利が無効または強制的に実行されないことが発見された場合、私たちの業務、財務状況、運営結果、および見通しは、重大で不利な影響を受ける可能性がある。しかも、私たちが代替案を開発しようとする時、私たちはサービスの導入に遅延があるかもしれない。さらに、任意の訴訟の弁護または有利な条項でこのような許可を得ることができなかった場合、製品の商業化を阻止することができ、これは、私たちの業務、財務状況、または運営、および将来性を損なう可能性があります。
私たちは私たちの特許または私たちの許可者の特許を保護または強制するために訴訟や他の法的手続きに巻き込まれるかもしれないが、これは高価で時間がかかり、成功しないかもしれない。
競争相手は、私たちの特許または私たちのライセンシーの特許を侵害したり、当社の知的財産権または私たちのライセンシーの知的財産権を流用したり、または他の方法で侵害する可能性があります。将来的には、私たちまたは私たちのライセンス者は、私たちの知的財産権または私たちのライセンシーの知的財産権を強制または保護し、私たちの商業秘密または私たちのライセンシーの商業秘密を保護し、または私たちが所有または制御する知的財産権の有効性または範囲を決定するために法的訴訟を提起することができる。
権利侵害や不正使用に対抗するために、私たちは費用が高く時間がかかるかもしれない侵害請求を要求されるかもしれない。第三者はまた、私たちまたは私たちのライセンシーに対して法的訴訟を提起することができ、私たちが所有、制御、または所有する権利のある知的財産権の有効性または範囲に挑戦することができる。侵害訴訟では、裁判所は、私たちの1つまたは複数の特許が無効または強制執行できないと判断するか、または私たちの特許が関連技術をカバーしていないことを理由に、他方の関連技術の使用を阻止することを拒否することができる。任意の訴訟または弁護手続きにおける不利な結果は、私たちの1つまたは複数の特許が無効が宣言されるか、強制的に実行できないか、または狭義に解釈されるリスクに直面する可能性があり、私たちの1つまたは複数の係属中の特許出願が発表できないリスクに直面する可能性がある。これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、当社の業務における従業員資源を大量に移転することになる。さらに、このような訴訟では、私たちの多くの相手や私たちの許可側の相手は、私たちよりも多くの資源を投入して、このような法的行動を起訴する能力があるかもしれない。もし私たちに対する侵害クレームが成功した場合、私たちは故意に侵害した3倍の損害賠償金と弁護士費、第三者から1つ以上の許可証を取得し、印税を支払うか、または私たちの侵害製品を再設計することを含む大量の損害賠償を支払わなければならないかもしれないし、大量の時間とお金の支出が必要かもしれない。
第三方向米国特許商標局は、既存技術の発行前出願、または反対、派生、撤回、再審、各方面間米国又は他の司法管区において,第三者によって引き起こされたか,又は我々又は我々のライセンス者によって提起された審査又は妨害手続,又は他の発行前又は認可後の手続又は他の特許庁手続又は訴訟は,我々又は我々のライセンス者の特許又は特許出願に関する発明の発明性,優先権,特許性又は有効性に挑戦又は必要な決定を行うことができる。不利な結果は、第三者が私たちの技術または候補治療薬を商業化し、私たちと直接競争することを可能にするために、私たちの技術または候補治療薬の特許保護を得ることができず、または第三者特許権を侵害することなく、私たちまたは私たちの許可者に関連技術の使用を停止するか、または第三者特許権を侵害することなく私たちの候補治療薬を製造または商業化することができるように、関連技術の使用を停止することを要求することができるかもしれない。
もし勝利者が商業的に合理的な条件で私たちにライセンスを提供しなければ、私たちの業務は損害を受ける可能性があります。訴訟や他の法的手続きは私たちの利益に不利な決定を招く可能性があり、たとえ私たちが勝訴しても、巨額の費用を招く可能性がある
63
私たちの経営陣と他の従業員たちの注意を分散させる。私たちは私たちの商業秘密や機密情報の盗用を単独でまたは私たちの許可者と一緒に防ぐことができないかもしれません。特に法律ではアメリカのようにこれらの権利を十分に保護していないかもしれません。
さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。さらに、公聴会、動議、または他の一時的な手続き、または事態の発展の結果が発表される可能性がある。もし証券アナリストや投資家がこれらの結果がマイナスだと思っていれば、私たちの普通株の価格に大きな悪影響を及ぼす可能性がある。もし私たちまたは私たちの許可者の特許および特許出願によって提供される保護の広さまたは強度が脅かされていれば、会社が私たちと協力し、許可、開発、または商業化した治療候補薬を阻止するかもしれない。また,訴訟に関する不確実性は,臨床試験の継続,研究計画の継続,第三者から必要な技術許可や協力に必要な資金を調達する能力に実質的な悪影響を及ぼす可能性がある。
私たちの特許保護の獲得と維持は、政府特許機関によって提出された様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
発行された特許の定期維持費は、特許有効期間内にいくつかの段階で米国特許商標局および外国特許代理機関に支払われなければならない。米国特許商標局および各種外国政府特許機関は、特許出願過程において、いくつかのプログラム、文書、費用支払い、およびその他の同様の規定を遵守することを要求する。多くの場合、適用規則に従って滞納金を支払うか、または他の方法で不注意を是正することによって失効を是正することができるが、場合によっては、規定を遵守しないことは、特許または特許出願が放棄または失効され、関連法ドメインの特許権の一部または全部の喪失をもたらす可能性がある。特許または特許出願の放棄または失効をもたらす可能性のある不正事件には、規定された期限内に公式行動に応答できなかったこと、費用が支払われていなかったこと、および適切に合法化され、正式な文書が提出されなかったことが含まれるが、これらに限定されない。この場合、私たちの競争相手が市場に参入する可能性があり、これは私たちの業務に実質的な悪影響を及ぼすだろう。
私たちの特許の生命期は私たちの製品と業務を効果的に保護するのに十分ではないかもしれない。
特許の寿命は限られている。米国では、すべての維持費が適時に支払われる場合、特許の自然失効時間は、通常、その最初の有効出願日から20年後である。様々な延期がある可能性があるにもかかわらず、特許の有効期限とその提供される保護は限られている。私たちの候補治療薬をカバーする特許を取得しても、製品の特許有効期限が切れると、生物学的類似薬や模倣薬からの競争に直面する可能性がある。さらに、米国で特許が発行された場合、USPTOによるいくつかの遅延により、特許の寿命を延長することができるが、特許出願人の特許訴訟中に生じるいくつかの遅延により、このような増加は、減少または除去することができる。もし私たちの技術が開発および/または規制審査を延長する必要があれば、私たちの技術を保護する特許は、私たちが商業化に成功する前または近いうちに満期になるかもしれません。もし私たちが私たちの製品を保護するのに十分な特許期間がなければ、私たちの業務と経営結果は悪影響を受けるだろう。
私たちまたは私たちの許可者は、私たちの特許と他の知的財産権の発明権のクレームを問われるかもしれない。
私たちまたは私たちのライセンシーは、将来、元従業員、協力者、または他の第三者が発明者または共同発明者として私たちの特許または他の知的財産権において権利を有するというクレームを受ける可能性がある。例えば,コンサルタントや他の我々の治療案の候補開発に参加している人の義務衝突により在庫紛争が生じる可能性がある。訴訟はこれらと他の挑戦在庫のクレームに対抗するために必要かもしれない。もし私たちまたは私たちのライセンシーがこのようなクレームを弁護できなかった場合、金銭損害賠償を支払うことに加えて、価値のある知的財産権の独占所有権や使用権などの貴重な知的財産権を失う可能性がある。このような結果は私たちの業務に実質的な悪影響を及ぼすかもしれない。私たちまたは私たちの許可者がこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。
私たちは世界各地で私たちの知的財産権を保護できないかもしれない。
私たちはアメリカ以外で私たちの知的財産権を保護できないかもしれない。世界のすべての国で候補治療薬の特許を申請、起訴、擁護するのは目を引くほど高価であり、私たちのアメリカ以外のいくつかの国での知的財産権はアメリカほど広くないかもしれない。また、一部の国の法律は知的財産権の保護程度は米国の連邦や州法律に及ばず、名目上このような保護があっても、このような知的財産権に対する司法や政府の法執行が不足している可能性がある。米国でも海外で出願しても,我々の特許や特許出願は挑戦される可能性があり,あるいは発行された特許を取得できない可能性がある.したがって、私たちは、第三者がアメリカ以外のすべての国で私たちの発明を実施したり、その発明を使用して製造された製品を米国または他の管轄区域で販売したり、輸入したりすることを阻止できないかもしれない。競争相手は特許保護を受けていない管轄区で私たちの技術を使って自分の製品を開発することができて、輸出することができます
64
他の侵害製品は私たちが特許保護を持っている地域に行きますが、法執行力はアメリカほど強くありません。これらの製品は私たちの製品と競争するかもしれませんが、私たちの特許や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。また,ある国には強制許可法があり,これらの法律により特許権者は他の者に許可を強制的に付与される可能性がある。また、多くの国は、政府機関や政府請負業者を含む他の当事者に対する特許の実行可能性を制限している。これらの国では、特許権者の救済措置は限られている可能性があり、これは任意の特許の価値を大幅に低下させる可能性がある。
多くの会社は外国の管轄区域の知的財産権の保護と保護に重大な問題に直面している。多くの他の国の法制度は、特許や他の知的財産権保護、特にバイオテクノロジーに関する保護の強制執行を支持しておらず、これは、これらの国で私たちの特許を侵害することを含め、私たちの知的財産権の流用や他の侵害を阻止することを困難にする可能性がある。外国の管轄区域で私たちの特許権を強制的に執行する訴訟手続きは、巨額のコストを招き、私たちの努力と注意を私たちの業務の他の側面から移転させる可能性があり、私たちの特許が無効または狭義に解釈されるリスクに直面する可能性があり、私たちの特許出願を発表できないリスクに直面させ、第三者からのクレームを引き起こす可能性がある。私たちは私たちが始めたまたは私たちのためのいかなる訴訟でも勝利しないかもしれないし、判決された損害賠償または他の救済措置(もしあれば)は商業的な意味がないかもしれない。また、米国や外国の法律や裁判所の法的裁決の変化は、私たちの技術や知的財産権の法執行のために十分に保護される能力に影響を与える可能性がある。したがって、私たちが世界各地で私たちの知的財産権を実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネスメリットを得るのに十分ではないかもしれません。
特許法の変化は,最近の特許改革立法を含めて,我々の特許出願をめぐる起訴や,我々が発表した特許の実行または保護の不確実性やコストを増加させる可能性がある.
米国又は他の国又は地域の特許法又は特許法解釈の変化は、我々の知的財産権の価値を低下させる可能性がある。私たちは、私たちの特許または第三者特許で許容または強制的に施行される可能性のある特許の広さを予測することができない。私たちは特許を申請できる他の独自技術を開発しないかもしれない。
他の特許性要件を満たしていると仮定すると,2013年3月16日までは,米国では,最初に発明して保護された発明を要求した者が特許を有しているが,米国以外では,最初に特許出願を提出した者が特許を有している。2013年3月16日またはその後、2011年9月16日に公布された“ライシー·スミス米国発明法”または“米国発明法”によれば、米国は、第1の発明者が出願制に移行し、このような制度の下で、他の特許性要件を満たすと仮定して、第1の特許出願を提出した発明者は、第三者が第1の発明によって要求された発明であるか否かにかかわらず、発明の特許を取得する権利があるであろう。したがって、2013年3月16日以降であるが、我々の前に米国特許商標局に特許出願を提出した第三者は、たとえ第三者が発明を行う前に本発明を作成したとしても、我々の発明をカバーする特許を付与することができる。これは私たちが発明から特許出願までの提出時間を認識することを要求するだろう。米国およびほとんどの他の国/地域の特許出願は、提出後または発行前の一定期間秘密であるため、私たちまたは私たちのライセンス者が、私たちの技術に関連する任意の特許出願または(Ii)発明が私たちまたは私たちのライセンシーの特許または特許出願に要求される任意の発明であることを最初に決定することはできない。
米国の発明法にはいくつかの重大な変化も含まれており、これらの変化は特許出願の起訴方法に影響を与え、特許訴訟に影響を与える可能性もある。これらの措置には、第三者が特許訴訟中に米国特許商標局に以前の技術を提出することを可能にすることと、米国特許商標局によって管理されたライセンス後のプログラム(ライセンス後審査を含む)が特許有効性を攻撃することを可能にする追加のプログラムとが含まれる各方面間審査と派生手続き。USPTO手続きの証拠基準は、米国連邦裁判所が特許請求の無効を宣言するために必要な証拠基準よりも低いため、第三者は、USPTO手続において、USPTOが権利請求を無効にするのに十分な証拠を提供する可能性があり、同じ証拠が最初に地域裁判所訴訟で提出された場合には、権利請求を無効にするのに十分ではない。したがって,第三者は米国特許商標局の手続きを用いて我々の特許主張の無効を宣言しようとする可能性があり,第三者が地域裁判所訴訟で最初に被告として疑問を提起すれば,我々の特許主張は無効ではない.したがって、米国発明法およびその実施は、私たちの所有または許可をめぐる特許出願の起訴および私たちが所有または許可した発行された特許の実行または弁護の不確実性およびコストを増加させる可能性があり、これらのすべては、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
しかも、バイオテクノロジー分野の会社の特許地位は特に不確実だ。米国最高裁判所を含む複数の裁判所による裁決は、バイオテクノロジーに関連するいくつかの発明または発見された特許範囲に影響を及ぼす。これらの裁決規定は,他の事項に加えて,抽象概念,自然現象または自然法則(例えば,特定の遺伝子変異と癌との関係)を陳述する特許請求自体が特許を出願することができない。一体何が自然法則や抽象概念を構成しているのかは不確定であり,我々の技術のいくつかの面は自然法則と考えられる可能性がある.したがって、米国および海外で進化していく判例法は、私たちおよび私たちのライセンシーが新しい特許を取得したり、既存の特許を強制的に実行する能力に悪影響を及ぼす可能性があり、第三者が所有または許可された特許に挑戦することに便利である可能性がある。
65
知的財産権は必ずしも私たちの競争優位に対するすべての潜在的な脅威を解決できるとは限らない。
私たちの知的財産権が提供する未来の保護の程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できないか、あるいはいかなる競争優位性を維持することができるかもしれない。例えば:
これらの事件が発生すると、それらは私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
A類普通株所有権に関連するリスク
私たちの証券は活発な取引市場がないかもしれませんが、これはA類普通株の売却を困難にする可能性があります。
活発な証券取引市場は発展しないかもしれないし、発展すれば、どの市場も続かないだろう。これは私たちが魅力的な価格で私たちの証券を売ることを難しくするだろう。
私たちの証券の市場価格は変動する可能性があり、これは投資価値の低下を招く可能性がある。
私たちの証券の価格は一般市場と経済状況によって大きく変動する可能性があります。活発な証券取引市場は発展しない可能性があり、発展すれば持続できない可能性もある。しかも、私たちの証券価格の変動は私たちへの投資損失の全部または一部を招くかもしれない。私たちの証券市場が発展し続けて活発になっていても、私たちの証券の取引価格は変動し、様々な要因に応じて大幅に変動する可能性があり、その中のいくつかの要因は制御できません。次のいずれの要素もあなたが私たちの証券の投資に重大な悪影響を及ぼす可能性があります。私たちの証券の取引価格はあなたが支払う価格よりはるかに低いかもしれません。この場合、私たちの証券の取引価格は回復できない可能性があり、さらに下落する可能性がある。
66
私たちの証券取引価格に影響を与える要素は
67
また、株式市場全体、特にナスダックとバイオ製薬会社は、極端な価格と出来高変動を経験しており、これらの変動は往々にしてこれらの会社の経営業績に関係なく、あるいは比例しない。広範な市場と業界要素は、その実際の経営業績にかかわらず、私たちA類普通株の市場価格にマイナス影響を与える可能性がある。従来、証券集団訴訟は、会社証券の市場価格が変動した後に会社に提起されることが多かった。このような訴訟を提起すれば、巨額の費用と経営陣の注意と資源の移転を招く可能性があり、これは私たちの業務、経営業績、あるいは財務状況を損なうことになる。
予測可能な未来に、私たちは現金配当金を支払うつもりはない。
私たちは私たちの株の現金配当金を発表したり支払ったりしたことがない。私たちは現在、将来のどんな収益も残して、私たちの業務の運営と拡張に資金を提供するつもりで、予測可能な未来に現金配当金を発表したり、支払うことはないと予想しています。したがって、もしあなたの株式取引価格が上昇すれば、あなたは私たちA種類の普通株への投資が報われるかもしれません。
もし私たちがナスダックの継続上場の基準を再遵守することに成功できなければ、私たちのA類普通株はナスダックから退市し、場外市場で取引を開始する可能性があり、これは私たちの普通株の価格と私たちの資本市場に入る能力にマイナスの影響を与えるかもしれない。
2023年3月14日、我々はナスダック上場資格部門から通知を受け、ナスダック上場規則第5450(A)(1)条に規定されているナスダック資本市場に継続して上場する最低購入価格要求を遵守しないことを通知した。A類普通株の終値は30営業日連続で1株1.00ドル以下に下落したからである。ナスダック上場規則第5810(C)(3)(A)条によると、最低入札値要求を再遵守するために、180暦または2023年9月11日までがある。コンプライアンスを再獲得するためには、我々A類普通株の終値は2023年9月11日まで10営業日連続で1株1.00ドルを達成または超えなければならない。もし私たちが2023年9月11日までにコンプライアンスを再獲得しなければ、公開保有株式の時価継続上場の要求とナスダック資本市場の他のすべての初期上場基準を満たしていれば、入札価格要求を除いて、私たちは追加の180日間の猶予を得る資格があるかもしれない。私たちは私たちA種類の普通株の終値を積極的に監視し、利用可能なオプションを評価して、最低入札要求を再遵守するつもりです。しかし、私たちは180日目のコンプライアンス期間内に最低入札要求を再遵守することを保証することはできません。私たちが2番目の180日目のコンプライアンス期間内にコンプライアンスを回復するか、または他のナスダック上場要求に対するコンプライアンスを維持することは保証されません。
もし私たちが成功しなかった場合、あるいは逆株式分割を実施しないことを選択すれば、私たちの証券は場外取引市場で取引を開始することが予想される。ナスダックから退市して場外取引市場で取引することは私たちの証券の流動性に悪影響を及ぼす可能性があります。国家証券取引所に上場する証券に比べて、場外取引市場で取引される証券取引量は通常限られており、売買見積間の価格差が大きい。したがって、緊急事態や他の理由が発生した場合、あなたはあなたの投資を清算できないかもしれません。
もしナスダックが上場基準に達しなかったため、私たちの証券をその取引所から退市したら、私たちと私たちの株主は深刻な負の結果に直面する可能性があります
68
将来的に私たちのA種類の普通株を売却し、発行する権利は、私たちの株式計画によると、私たちの株主の所有権の割合をさらに希釈し、私たちの株価を下落させる可能性があることを含む。
将来的には,臨床試験,商業化努力,研究開発活動の拡大,上場企業の運営に関するコストなど,我々の計画中の運営を継続するために大量の追加資本が必要となる可能性が予想される。資本を調達するために、私たちは1回または複数回の取引で時々決定された価格および方法で普通株、転換可能証券、または他の株式証券を販売することができる。私たちはまた、戦略連合の設立、合弁企業の設立、または第三者との追加許可手配の一部として、私たちの普通株を売却することも可能であり、これらの計画は、私たちの開発と商業化努力を補完または強化すると信じています。もし私たちが普通株、転換可能証券、または他の株式証券を販売する場合、投資家はその後の売却によって深刻に希釈される可能性がある。このような売却は既存の株主の実質的な希釈を招く可能性もあり、新しい投資家は私たちA種類の普通株式保有者よりも優先的な権利、優遇、特権を得ることができる。
私たちの定款文書とデラウェア州法律の反買収条項は、制御権の変更を延期または阻止する可能性があり、これは私たちA種類の普通株の市場価格を制限し、私たちの株主の現在の経営陣の変更を阻止または罷免する試みを阻止または挫折させる可能性がある。
第二次改正及び再記載された会社登録証明書及び企業合併を完了するためにわれわれが採択した改正及び再記載の定款には、わが社の支配権変更を遅延又は阻止する可能性がある条項又は当社の株主が有利であると考えられる取締役会変更の条項が含まれています。いくつかの規定には
また、私たちはデラウェア州会社法第203条の規定によって管轄されており、この条項は特定の企業の合併を禁止する可能性があり、株主は私たちが発行した議決権のある株の15%以上を持っている。これらの反買収条項や私たちの定款や定款の他の条項は、株主や潜在的な買収者が私たちの取締役会に対する支配権を得ることを困難にしたり、当時の取締役会が反対する行動を開始したり、わが社の合併、買収要約、代理権に関する争いを延期したり阻害したりする可能性があります。これらの規定はまた、依頼書の競争を阻止し、あなたが選択した取締役を選出しにくくしたり、ご希望の他の会社の行動を取らせたりすることができます。制御権の変更、取引または取締役会の変動を遅延または阻止することは、私たちA類普通株の市場価格の下落を招く可能性がある。
私たちの規約では、デラウェア州衡平裁判所とアメリカ合衆国連邦地域裁判所は、私たちと株主とのほとんどの紛争の独占的なフォーラムとなり、これは、私たちまたは私たちの役員、役員、または従業員との紛争を処理するために、私たちの株主が有利な司法フォーラムを得ることを制限するかもしれません。
我々の憲章は、デラウェア州衡平裁判所(または、デラウェア州衡平裁判所に主題管轄権がない場合のみ、デラウェア州域内に位置する任意の州裁判所、またはこのようなすべての州裁判所に主題管轄権がない場合にのみ、デラウェア州連邦地域裁判所)がデラウェア州成文法または一般法の下で以下のタイプの訴訟または訴訟の独占法廷となると規定している
69
この規定は、取引法で規定されている義務や責任を執行するための訴訟にも適用されず、米国連邦裁判所が排他的管轄権を有する他のクレームにも適用されない。
また、証券法第22条は、連邦裁判所と州裁判所は、このようなすべての“証券法”訴訟に対して同時に管轄権を持っていると規定している。したがって、州裁判所と連邦裁判所はこのようなクレームを受理する管轄権を持っている。複数の司法管轄区域で訴訟を提起せざるを得ないことや、異なる裁判所が不一致や逆の裁決を行わなければならない脅威、およびその他の考慮要因を避けるために、我々の憲章は、米国連邦地域裁判所が証券法に基づいて提出された任意の訴因を解決するための独占的なフォーラムとなると規定している。デラウェア州裁判所は、このような選択の裁判所条項が事実上有効であることを決定しているが、株主は依然として専属裁判所条項が指定された場所以外の場所でクレームを出すことを求めることができる。このような状況で、私たちは私たちの憲章の専属フォーラム条項の有効性と実行可能性を強力に主張することを期待する。これは、他の法ドメインでこのような訴訟を解決することに関連する多くの追加費用を必要とする可能性があり、これらの規定がこれらの他の法ドメインの裁判所によって実行されることを保証することはできない。
これらの排他的フォーラム条項は、司法フォーラムにおいて、私たちの会社または私たちの役員、役員、または他の従業員と紛争することに有利であると考える株主のクレームを提出する能力を制限する可能性があり、これは、私たちと私たちの役員、役員、および他の従業員に対する訴訟を阻止するかもしれない。もし裁判所が私たちの憲章のいずれかの専属法廷条項が訴訟で適用されないか、実行できないことを発見した場合、私たちは他の司法管轄区域で紛争解決に関連する追加費用を発生させるかもしれません。これは私たちの業務を深刻に損なうかもしれません。
一般リスク因子
不安定な市場や経済状況は、私たちの業務、財務状況、株価に深刻な悪影響を及ぼす可能性がある。
世界の信用と金融市場は過去に極端な変動と中断を経験したことがあり、最近の原因は持続的な新冠肺炎疫病、及び最近のシリコンバレー銀行の倒産を含む。これらの干渉は流動性と信用供給の深刻な減少、消費者自信の低下、経済成長の低下、失業率の上昇及び経済安定性の不確定性を招く可能性がある。信用と金融市場のさらなる悪化と経済状況への自信が起こらない保証はない。私たちの全体的な業務戦略は、このような経済低迷、不安定なビジネス環境、または持続不可能で不安定な市場状況のいずれかの悪影響を受ける可能性がある。現在の株式と信用市場が悪化すれば、任意の必要な債務や株式融資をより困難にし、コストがより高く、希釈程度をより高くする可能性がある。適時に有利な条件で任意の必要な融資を得ることができなければ、私たちの運営、成長戦略、財務業績と株価に実質的な悪影響を与える可能性があり、臨床開発計画の延期または放棄を要求する可能性がある。また、現在の1つまたは複数のサービスプロバイダは、経済低迷の中で生き残ることができない可能性があり、これは、時間通りと予算で運営目標を達成する能力に直接影響を与える可能性がある。
私たちの純営業損失の繰越といくつかの他の税務属性を利用する能力は限られているかもしれません。
1986年の国税法(改正)第382条および383条、またはこの法典と州法の対応条項によると、会社が“所有権変更”(一般に3年間のスクロール期間中にある株主の持分所有権の変化が50ポイントを超えると定義されている)を経験した場合、変更前の連邦純営業損失、繰越およびその他の変更前の税収属性を使用して変更後の収入および税金を相殺する能力が制限される可能性がある。私たちは未来に株式所有権の変化のために所有権の変化を経験するかもしれない。2022年12月31日現在、約8460万ドルの米国連邦NOL繰り越しと1820万ドルの州NOL繰り越しがあり、これらのNOL繰り越しは満期時に使用されていない可能性があり、将来の所得税債務を相殺するためにも使用できず、収益性に悪影響を及ぼす可能性がある。私たちは予測可能な未来に重大な追加純損失が発生することを予想し、私たちはこのような損失に関連するNOL繰り越し能力を利用している
70
将来の課税所得額を相殺することは、私たちの未来に発生する所有権変更に限られるかもしれない。さらに、州レベルでは、一定期間停止または他の方法でNOL繰り越しの使用を制限する可能性があり、これは州の課税税を加速または永久的に増加させる可能性がある。したがって、私たちは私たちのNOL繰り越しと他の税金属性のすべてまたは実質的な部分を使用することができないかもしれないが、これは私たちの未来のキャッシュフローに悪影響を及ぼすかもしれない。
税法の変化は私たちの業務と財政状況に悪影響を及ぼすかもしれない。
米国連邦、州、地方所得税に関する規則は立法過程に参加する人員およびアメリカ国税局とアメリカ財務省の審査を受け続けている。税法の変化(これらの変化は追跡力を持つ可能性がある)は、私たちまたは私たちの証券の所有者に悪影響を及ぼす可能性がある。近年、このような変化は多く発生しており、未来も変化し続けるかもしれない。将来の税法の変化は、私たちの業務、キャッシュフロー、財務状況、または運営結果に実質的な悪影響を及ぼす可能性があります。例えば、アイルランド共和軍は最低税と1%の株式買い戻し消費税を15%の会社を含む。私たちは投資家に税法の変化が私たちの証券投資に与える影響について彼らの法律と税務顧問に相談することを促す。
原材料、設備、労働力、輸送のコストと利用可能性の変動は、製造遅延や私たちのコストを増加させる可能性があります。
我々の製品を製造するための重要な部品の価格と可獲得性は上昇しており、大幅な変動を続ける可能性がある。また、規制やインフレ圧力により、私たち内部または第三者メーカーの労働コストが大幅に増加する可能性がある。また,物流や輸送コストは石油価格の変動によるところが大きく,供給は政治的·経済的問題によって制限される可能性がある。私たちの任意の原材料、包装、または他の調達または輸送コストのコストと利用可能性の任意の変動は、私たちの毛金利を損なう可能性があります。もし私たちがこれらの製品のコスト増加や変動の大きな部分を緩和することに成功しなければ、私たちの運営結果は損害を受けるかもしれません。
71
項目1 B。取消解析Dスタッフがコメントした。
ない。
プロジェクト2.専門家ペルテスです。
2036年に満期になった賃貸契約によると、私たちはニュージャージー州フロラム公園に約150,000平方フィートの事務、実験室、製造空間を持っていて、私たちはそれを私たちの主要な業務場所とします。私たちは私たちの既存の施設が予測可能な未来で私たちの必要性を満たすのに十分だと信じている。
項目3.法律法律手続き。
時々、私たちは訴訟や他の法的手続きに巻き込まれるかもしれない。私たちは現在、私たちの経営陣が私たちの業務に重大な悪影響を及ぼす可能性があると考えている訴訟や法的手続きに参加していません。結果にかかわらず、弁護と和解コスト、管理資源の分流、その他の要素により、訴訟は不利な影響を与える可能性がある。
2021年6月9日、元従業員John Schlechtwegは、私たちが口頭契約に違反したか、または不当に利益を得なかったことを告発するために、米コネチカット州地方裁判所に訴訟を起こした。起訴状は、私たちは特に、Sanuwaveへの特定の資産の売却に関与していることに関連した追加的な賠償をSchlechtwegさんに支払うことを拒否した。予備動議慣行によると、この事件は米国ニュージャージー州地域裁判所に審理された。その後、訴状を却下する動議を提出した後、違約クレームは却下され、唯一残った不当所得クレームがあった。これらは現在発見されているすべての当事者たちだ。訴訟で主張された残りのクレームには根拠がなく,有力な弁護をしようとしているが,訴訟の結果は保証されていないと考えられる。
2021年2月4日、一人の株主がニューヨーク州最高裁に企業合併に関する仮定集団訴訟を提起した:SPEROはGX買収会社ら、インデックス番号は650812/2021年(ニューヨーク州上級裁判所)を訴えた。2021年2月4日)。2021年2月26日、同じ株主が訴訟で修正された訴えを提出し、集団訴訟疑惑や他の告発、すなわちSpero訴えを削除した。2021年2月8日、ある株主がニューヨーク州最高裁に企業合併に関する訴えを行った:RogallaはGX Acquisition Corp.,ら、インデックス番号650877/2021年(ニューヨーク州)2021年2月8日)、またはRogallaクレーム、およびSperoクレーム、クレーム)。起訴状は、当社と当社の取締役会メンバー(企業合併前)またはGXとGX取締役会をそれぞれ被告としています。また,Rogallaの起訴状はFirst Merge Sub,Second Merge Sub,Celulityを被告とした.Rogallaの起訴状は、GX取締役会が業務合併に関する受託責任クレームに違反したことを告発し、GX取締役会がGX、First Merge Sub、Second Merge Sub、Celulalityに対する受託責任クレームに違反したことを協力して教唆した。これらの疑惑は、業務合併に関連する目論見書に重大な誤解があり、および/または業務合併に関する重大な情報が漏れている疑いに基づくものである。Speroの起訴状は、GX取締役会が業務合併に関する受託責任クレームに違反したことを告発し、GX取締役会がGXに対する受託責任クレームに違反したことを協力して教唆した。これらのクレームの根拠は、Celulalityの販売過程と推定値、および企業合併に関するS-4登録声明が重大な誤解性を有していること、および/または企業合併に関する重大な情報を見落としている疑いである。起訴側は一般的に強制令救済や撤回、指定されていない損害賠償、弁護士費や専門家費の判決などの救済措置を要求する。2021年4月29日、Spero訴訟を起こした原告は自発的にこの訴訟を停止した。2021年7月20日、ロガラ訴訟を起こした原告は自発的にこの訴訟を停止した。
GX取締役会はまた、GX仮定株主が2021年2月18日、2021年3月2日、2021年3月19日、2021年3月24日に提出した4つの要求を受け取り、GXとGX取締役会が彼らの受託責任に違反し、株式募集説明書が重大な誤解性を有すると言われているため、および/または業務合併に関する重大な情報を見落としていると言われているため、連邦証券法に違反している。これらの要求は、募集説明書の修正案または付録に修正開示を発表することを要求する。GXは、上述した疑惑には根拠がないと考えているが、GXは、2021年3月29日に、いくつかの追加情報が含まれている修正されたS−4表を米国証券取引委員会に提出し、苦情および要求および補足開示で主張された開示主張を提示する。補足開示の提出については、起訴状および訴状中の原告の弁護士は、そのクレームの不確実性を考慮して、業務合併または目論見書についてさらなる行動を取らず、その後、訴状および訴えで提起された任意およびすべてのクレームを解決するために、GXと秘密協定を達成することに同意する。
ペンシルバニア州東区連邦検事室が2022年8月14日に虚偽請求法(“米国連邦法典”第31編3729節)に基づいて提出した民事調査要求を受けた。この要件は、Interfylを含むMedicare、Medicaid、または他の連邦保険会社に提出されたサービスまたはプログラムクレームに関連する文書および情報を提供することを必要とする。私たちはこの要求に協力して進行しています
72
この要求を処理するアメリカの検事補佐官と対話している。この問題はまだ初期段階にあり、この要求がどんな責任も招くかどうかはまだ確定されていない。
プロジェクト4.地雷安全安全に開示する。
適用されません。
73
第II部
項目5.登録者普通株式市場、関連株持株者は重要で発行者が株式証券を購入する。
市場情報
私たちのA類普通株はナスダック資本市場で取引されており、株式コードは“CELU”です。私たちの株式承認証の株式コードは“CELUW”で、1株11.50ドルの発行価格でA類普通株を購入することができます。
所持者
2023年3月31日現在、我々A類普通株の登録株主は約111人、株式承認証の保有者は約6人である。
配当をする
私たちは私たちの資本や普通株のどんな配当金も発表したり支払ったりしたことがない。私たちは現在、すべての利用可能な資金と任意の将来の収益(もしあれば)を維持し、私たちの業務の発展と拡張に資金を提供するつもりで、予測可能な未来にはいかなる現金配当も支払わないと予想しています。将来配当金を派遣するかどうかは、当社の取締役会が自ら決定する。
株式補償計画に基づいて発行された証券
私たちの株式報酬計画に関する情報は、本年度報告書第3部第12項に組み込まれています
最近売られている未登録証券
ない。
発行人が株式証券を購入する
ない。
第六項です[R保存された]
74
項目7.経営陣の以下の問題の議論と分析財務状況と経営実績。
以下の議論には、証券法第27 A条及び取引法第21 E条に示される“前向き陳述”が含まれる。“前向きな陳述に関する特別な説明”を参照されたい。このような展望性陳述は私たちの意図、信念或いは現在の予想を代表し、リスク、不確定要素及びその他の要素に関連し、実際の結果とある事件の時間はこのような展望性陳述の明示或いは暗示の未来の結果とは大きく異なる可能性がある。場合によっては、前向きな陳述は、“可能性”、“予想”、“推定”、“計画”、“計画”、“予測”、“潜在”、“信じる”、“すべき”などの用語によって識別されることができる。結果をもたらすか、または促進する可能性のある異なる要因は、項目1.Bに列挙された要因を含むが、これらに限定されない。“リスク要因”と本年度報告書の他の部分。法的に別の要求がない限り、私たちは、本報告日後のイベントまたは状況を反映したり、実際の結果を反映するために、これらの前向きな陳述を更新する義務がありません。
概要
著者らは生物技術会社であり、既製の胎盤由来同種異体細胞療法を開発することによって癌、免疫と伝染病を治療し、細胞医学の次の発展をリードする。著者らはキメラ抗原受容体(CAR)、ナチュラルキラー細胞(NK)、間葉系様接着基質細胞(MLASCs)と外体(Exosome)から構築されたT細胞を含む一連の既製胎盤由来同種異体細胞治療候補製品を開発している。これらの候補治療薬は癌,感染症,退行性疾患に対する適応である。胎盤独特の生物学的特性と既製の獲得性を利用することによって、著者らは治療解決策を開発し、全世界の有効、獲得と負担できる治療薬物に対する重大な需要を満たすことができると信じている。胎盤から抽出した生体材料製品も積極的に開発·販売している。2023年までに、私たちは国内でこれらの製品を販売して、主に整形外科と傷口看護市場にサービスします。我々は現在,米国以外で胎盤生体材料を販売する予定であり,最初の重点は中東と北アフリカ市場である。私たちの今日の生体材料業務は主に私たちの流通ネットワークを介して私たちのBiovanceとInterfyl製品を販売することです。Biovanceは健康,臨月妊娠の胎盤から抽出した脱細胞,脱水したヒト羊膜である。それは無傷の天然細胞外基質であり、傷口の再生過程に基礎を提供し、機能組織の修復にステントを提供した。Interfylはヒト結合組織基質であり、健康、臨月妊娠の胎盤から来ている。それは各種の医学専門家によって創傷、創傷或いは手術による軟組織欠損を埋めるために使用されている。我々はBiovanceやInterfyl以外のビジネスチャネルを深化させるために新たな胎盤生体材料製品を開発している。細胞治療開発·製造における我々のコア専門知識を利用して,第三者に契約製造と開発サービスを提供することで収入を創出する予定である。この新サービスの最初のポイントは,開発段階の細胞治療会社の臨床試験のための治療候補薬の開発·製造に協力することである。2023年1月、私たちは仕事の優先順位の再調整を発表し、2023年3月現在、従業員数は約3分の1に減少した。
最新の発展動向
私募する
2023年3月20日、私たちは主席兼最高経営責任者のRobert Hariri博士を含む2人の認可投資家と証券購入協定を締結し、私募(I)9,381,841株A類普通株、および(Ii)セット引受権証を規定し、1株0.8343ドルと付属管承認証0.125ドルの価格で最大9,381,841株のA類普通株を購入し、総購入価格は約900万ドルであった(うちHariri博士は200万ドルを承認した)。私募は2023年3月27日に終了し、常習の終了条件を満たす必要がある。
PIPE承認株式証の行使価格は1株3.00ドルであり、直ちに行使することができ、2028年3月27日に満期(発行日から5年)になり、私たちの資本のある取引の慣例調整に影響を受ける。管状株式証所有者(その連合会社と一緒に)実益が所有するA類普通株式総数が、行使直後に株式承認証所有者が規定する指定パーセント上限(61日前に通知して調整することができる)を超える場合は、株式承認証を行使することができない。
吾らも買い手と登録権契約を締結し、これにより吾等は、A類普通株株式及びPIPE株式承認証を行使する際に発行可能なA類普通株株式の転売と、拘束力のある再許可条項説明書(後述)に基づいて支払として発行可能な株式を登録することに同意した。我々は、本年度報告書の提出後30日以内に、米国証券取引委員会または米国証券取引委員会に登録声明を準備して提出することを要求され、米国証券取引委員会が審査していない場合、45日以内に登録声明の発効を宣言し、審査を行う場合は90日以内であるが、いずれにしても2023年6月30日より遅くないように商業的に合理的な努力をしなければならない。
これらの証券は、改正された1933年“証券法”第4(A)(2)条又は当該法案及びその公布されたD条例に規定された登録免除規定に基づいて発行される。私たちは購入者たちがした陳述の一部がこの免除登録に依存している。
橋越しローンを優先的に保証します
75
2023年3月17日、私たちは主要株主の一人であるC.V.Starr&Co.Inc.と融資契約を締結し、元金総額500万ドルのローンを提供し、元の発行割引100,000ドルを差し引くと、毎年12.0%の金利で利息を計算し、1年目の利息は毎月最終日に実物で支払い、2025年3月17日に満期、融資と株式証明書は合計750,000株のA類普通株、またはStarr株式証を購入し、1株0.125ドルの買い取り価格でスタール株式承認証(または93,750ドル)を購入する。Starr株式承認証の期限は5年,執行価格は1株0.71ドルである。私たちは2023年3月17日にローンとスタール権証の売買を完了した。
融資条項によると、吾らは慣用的な負の契約を遵守し、吾等の債務の返済を制限し、株主への配当金の支払い、許可されていない他の債務の償還、任意の許可されていない資産を付与または許容する担保権益、または300万ドル未満の現金および現金等価物を保有することが連続する5営業日を超えることに同意する。ローン中の負の契約のほか、ローンには慣例違約事件も含まれている。融資条項に基づいて、私たちはスタールにそのすべての資産の優先的な保証権益を付与した。
拘束力のある再許可契約条項表
上記私募証券購入協定を締結するとともに、吾等は拘束力のある条項説明書に署名し、私募取引の認可投資家側の連属会社のいくつかの資産について協議及び再許可を締結する。拘束力のある条項説明書によると、許可側に300万ドルの現金を支払い、100万ドルのA類普通株を発行した(2023年3月17日の終値から1,694,915株)。
経営を続ける企業
会計基準更新第2014-15号によれば、実体の持続的な経営企業としての能力に関する不確実性(サブテーマ205-40)、またはASU 205-40を開示し、いくつかの条件やイベントが存在するかどうかを評価し、これらの条件とイベントを総合的に考慮して、合併財務諸表の発表日から1年以内に継続経営企業として経営を継続する能力について大きな疑いを抱いている。
新興の臨床段階生物技術会社として、著者らは企業発展に関連するいくつかの内在リスクと不確定性の影響を受けている。この点で、私たちの設立以来、経営陣のほとんどの努力は、胎盤由来同種異体細胞の基礎科学研究、私たちの現在と未来の細胞療法臨床計画を支持する臨床前研究、私たちの細胞計画の臨床開発、そして私たちの核心業務の運営を支持する施設と販売、一般および行政費用(総称して“投資”)を含む研究と開発に投資しており、これらはすべて私たちの短期的な収益力を犠牲にしている。我々は従来,バイオバンクや退行性疾患業務による有限収入と,公的·個人投資家への株式や債務証券(これらの発行を総称して“外部資本”と呼ぶ)を発行することでこれらの投資に資金を提供してきた。これらの努力をしたにもかかわらず、経営陣は私たちの研究開発と商業化が成功するか、あるいは私たちの知的財産権が十分に保護されることを保証することはできません。これらの努力が成功しても、いつになったら相当な売上を生み出すことができるか、または収益的に運営して、外部資本に依存し続ける必要はありません。私たちの株価の持続的な下落は、今後しばらくの間の営業利益や長期資産の減少を招く可能性がある。
管理層は、添付の連結財務諸表発行日(“印刷日”)まで、ASU 205-40に基づいて、以下の不利な条件およびイベントの重要性を評価した
76
これらの不確実性は、私たちが経営を続けている企業として継続する能力を大きく疑っています。添付されている総合財務諸表は、継続的な経営企業として運営していく上で作成されており、予測可能な未来に正常な業務過程で資産を現金化し、負債と約束を返済することが期待されています。したがって、添付の連結財務諸表は、これらの不確実性の結果に起因する可能性のあるいかなる調整も含まない。
新冠肺炎が大流行する
新冠肺炎疫病は疫病の急性期に失業率、大口商品と株式市場の変動を激化させる。ワクチン接種率の上昇と報告症例レベルの低下は,大流行が最も深刻な部分が経過している可能性が示唆された。新たなあるいは突然変異の変種が出現すれば、さらに措置を講じてその蔓延を抑制することは、私たちの財務状況、経営業績およびキャッシュフローの時間と数量に実質的な影響を与える可能性がある。
2021年と2022年を通して継続的に運営できるにもかかわらず,必要でない従業員とその役割を遠隔実行できる従業員に対する現地の健康アドバイスに基づき,必要に応じて“在宅勤務”政策を実施した。遠隔従業員の管理は特殊な挑戦をもたらす可能性があり、遠隔従業員の作業効率はそれほど高くない可能性がある。著者らのある操作要素(例えば胎盤組織処理、ある生物測定、転化研究と臍帯血貯蔵)は遠隔実行できないため、著者らは強制体温検査、症状評価表、漸進式洗浄とよく見られる表面の消毒を含む制御と規程を制定し、従業員のリスクを下げる。これまで、私たちはまだ何の実質的な妨害にも遭遇していないが、私たちの緩和措置が引き続き有効である保証はなく、未来の私たちの業務の重要な要素が妨害されないことを保証することはできない。
経済活動の広範な低下とある医療施設の実際の使用制限により,我々の退行性疾患業務の純収入は確かに大流行により低下している。臨床試験については,純粋に新冠肺炎のリスクで募集を中止したり延期したりしていない。しかし、CYNK-001による急性骨髄性白血病治療を評価する臨床試験の登録は、ウェブサイトが彼らの安全方案を評価し、大量の新冠肺炎患者を経験したため、2021年にいくつかの遅延を経験した。2022年の間,我々は新冠肺炎による運営への大きな影響を経験しなかった。
77
新冠肺炎あるいは任意の他の健康疫病がどの程度私たちの結果に影響する可能性があるかは未来の事態の発展に依存し、これらの事態の発展は非常に高い不確定性を持っており、出現する可能性のある新冠肺炎の重症度に関する新しい情報、新冠肺炎の抑制或いはその影響を治療する行動などを含む予測もできない。そのため、新冠肺炎は私たちの業務、運営結果、財務状況と見通しに実質的な悪影響を及ぼす可能性がある。
業務の細分化
私たちは三つの異なる業務部門を評価することで、私たちの業務を管理します:細胞治療、退行性疾患、生物バンク。報告されるべき部門は、各部門が展開する活動の異なる性質に基づいて決定される。細胞療法とは,我々が研究·開発している細胞療法であり,この療法は実証されておらず,異なる開発段階にある。すべての細胞治療項目は細胞治療部分に属する。これまで承認された細胞療法製品は得られておらず,細胞療法の販売から収入も得られていない。退行性疾病会社は外科と傷口看護市場で使用する製品、例えばBiovance、Biovance 3 L、InterfylとCentaflexを生産、販売と許可した。私たちは自分の販売チームと独立した流通業者を使ってこの細分化された市場の製品を販売します。私たちは変性疾患の分野のためにもっと多くの組織ベースの製品を開発している。バイオバンクは臍帯と胎盤から幹細胞を収集し、個人に代わってこれらの細胞の保存を将来的に使用するために提供する。私たちは主にLifebank USAブランドでバイオバンク業務を経営しています。当社の報告可能な業務部門のより多くの情報については、本年度報告に含まれる他の部分に含まれる当社の総合財務諸表の付記19“部門情報”を参照してください。
買収と資産剥離
我々の現在の業務は,我々が設立以来行ってきた戦略買収と資産剥離を反映している.以下の買収のその他の詳細については、本年報のその他の部分に掲載されている2022年12月31日現在の総合財務諸表付記1“業務性質”を参照されたい。
2017年5月にHLIからHLI細胞治療会社LLCまたはHLI CTを買収しましたHLI CTはLifebank USAを運営しており,個人臍帯血幹細胞と臍帯組織バンクであり,新生児臍帯血幹細胞と臍帯組織単位を収集,加工,凍結保存する選択肢を親に提供している。HLI CTを買収することはまた、BiovanceおよびInterfylを含む一連の生体材料資産の権利を提供してくれる。HLI CTを買収する際には,BiovanceとInterfylはAliqua Biomedical,Inc.またはAliquaの独占Dealerに制御される。2018年5月、我々はBiovanceおよびInterfylのマーケティングおよび流通権を含むAliquaのバイオ創傷ケア事業を含むAliquaからいくつかの資産を買収した。
2017年8月、私たちはCelgeneの完全子会社Anthrogenensisを買収した。今回の買収には臨床前と臨床段階の資産の組み合わせが含まれており、著者らが開発を継続している重要な細胞治療資産を含む。今回の買収により、著者らはAnthrogensisのノウハウと技術を獲得することができ、産後ヒト胎盤から抽出した大量の潜在力幹細胞と細胞治療製品を回収するために使用され、各製品はAnthrogensis製品である。人類起源買収の一部として、人類起源製品のいくつかの発明者と人類起源製品開発チームの他の重要なメンバーが私たちの仲間入りをした。
2018年10月,コロラド大学医学院のためにCariCordを買収し,コロラド大学臨床免疫実験室大学臍帯血バンクとコロラド大学取締役会が設立した家庭臍帯血バンクであり,コロラド大学は法人団体である。
許可協定
通常の業務プロセスにおいて、私たちは第三者から知的財産権及び他の権利の許可を取得し、私たちの知的財産権及び他の権利の許可も、上述したように、当社の知的財産権及び他の権利を超えている。当社のライセンス契約の詳細については、本年度報告Form 10-Kの他の部分を含む2022年12月31日までの年度監査総合財務諸表の付記16“許可及び流通協定”を参照されたい。
2020年9月、私たちはソレントと許可と譲渡協定を締結した。紀万昌博士、Legacy Celularity元取締役会メンバーで、現在ソレント社の総裁とCEOを務めている。ソレントもわが社の重要株主であり、他の重要株主とともに、業務合併やPIPE融資を終了するとともに、A類普通株を私募する。ソレントプロトコルにより,CyCART−19遺伝子改造の基礎を構成したCD 19自動車構造のグローバルライセンスを取得した。私たちは現在、ソレントから許可を得たCD 19自動車製造を製造し、供給するための供給協定を交渉している。
2017年8月,Anthrogenensisの買収についてCelgeneとライセンス契約を締結し,Celgeneはその後Bristol Meyers Squibbに買収された。Celgeneライセンスによると、Celgeneには世界的な印税免除、
78
Celgene許可日に存在する既存の知的財産権またはCelgeneによる、合併に関連する任意の移行サービス活動について開発された、合併に関連する非商業臨床前研究活動、ならびに任意の自動車の製造、商業化、およびそのような自動車に関連する製品およびサービスを発現するために任意のT細胞またはNK細胞を修正する権利を開発、製造、商業化、および/またはそのような自動車またはT細胞またはNK細胞を任意の目的のために再許可することができる。一方が合意に深刻に違反している場合、または他方の資金が償還されていない場合には、Celgeneライセンスを終了することができる。
二零一七年八月、Legacy CelularityもCelgeneにそのXシリーズ優先株を合併費用として発行し、CVR協定を締結し、この合意に基づいて、Legacy CelularityはAnthrogenensis買収事項についてCelgeneに発行したLegacy Celularity Xシリーズ優先株について1つ或いは価値のある権利或いはCVRを発行した。CVR協定は,CVRの保有者が各計画に基づいて合計5000万ドルの規制マイルストーン金額と,我々のいくつかの研究治療計画に関連した合計1.25億ドルの商業マイルストーン支払いを得る権利を持たせる。また,このような計画や例年ごとに,CVR保有者は,その計画治療薬の年間純売上高の10分の1に相当する使用料を得る権利があり,この計画された治療薬が特定の国で初めて商業販売された日から最近の満了日まで,その計画治療薬をカバーする任意の有効特許主張がその国/地域での有効特許権が満了した日,このような治療薬の市場独占経営権がその国で失効した日から2027年8月(すなわちAnthrogensis買収終了10周年)までとなる。今まで、CVR協定に基づいて何のお金も支払われなかった。私たちは四半期ごとにCVRと関連した負債を推定する。この責任の変化には,われわれの臨床計画の変更に限定されないが,これらの計画の商業的価値と金銭的時間的価値の仮定が含まれている。
経営成果の構成部分
純収入
純収入には、(I)Biovance、Biovance 3 L、Interfyl、およびCentaflexを含む生体材料製品の販売が含まれ、私たちの直接販売は、製品の販売およびレンタルを含み、私たちの流通パートナーネットワークを介した販売には、許可、特許権使用料およびその他が含まれ、(Ii)臨月妊娠後の臍帯、胎盤血液および組織の収集、加工および保存は、総称して“サービス”と呼ばれ、(Iii)SanuWaveとのライセンス契約に従って受信された許可料および特許許可料は、2021年第3四半期までのすべての費用、許可費用、特許権使用料およびその他を含む。
収入コスト
収入コストには、私たちの既存の2つの商業業務部門-バイオバンクと変性疾患-に関連する労働力、材料、および管理費用が含まれている。バイオバンクコストには,新貯蔵材料の貯蔵·輸送キットのコスト,臍帯血や他の貯蔵先のタンクや施設管理費用が含まれる。退行性疾患コストは胎盤の購入、胎盤材料の合格及び胎盤組織を適切な対路製品に加工することと関連するコストを含む。変性疾患部門のコストには,Biovance,Biovance 3 L,Interfyl,Centaflex製品ラインの生産に関する労働力と管理費用がある。直売に関する収入コストは製品販売やリースの一部であり,我々の流通パートナーネットワークを介した販売に関する収入コストは許可,印税,その他に含まれる.
研究開発費
著者らの研究と開発費用は主に胎盤由来の同種異体細胞の基礎科学研究、著者らの現在と未来の細胞医学臨床計画を支持する臨床前研究、著者らのNK細胞計画と施設の臨床開発、減価償却とその他の研究と開発活動による直接と分配された費用に用いられる。臨床試験の運用を支援する第三者CROの費用,臨床試験供給費用,研究科学者の人件費,生物研究のための専門化学品や試薬,第三者試験と検証の費用,レンタル料や施設維持費用を含めた各種管理費用が発生した。基礎研究、パートナーの研究協力及び成功を実現するための監督管理提出の研究は、私たちの現在と未来の細胞治療における成功に重要である。私たちは優先順位を再調整したので、私たちの研究開発支出は短期的に減少すると予想される。著者らの研究開発支出額は多くの要素に依存し、臨床試験の時間、臨床試験の治療効果の初歩的な証拠及び著者らが追求する適応数を含む。
販売、一般、行政費用
販売、一般、および行政費用には、主に給与、ボーナス、株式給与、私たちのコア業務の運営を支援する専門家の福祉を含む人員コストが含まれる。行政管理、財務、法律、人的資源と情報技術は販売、一般と行政費用の主要な構成部分であり、これらの費用は発生時に確認する。優先順位を再決定する努力により、短期的に私たちの販売、一般、管理コストの低下を見ることができると予想しています。私たちの販売、一般、行政コストの大きさと時間は臨床試験の進展にかかっています
79
変性疾患の組み合わせにおける新製品の発表、環境の変化の規制、または私たちの業務戦略を支援する人員の需要を含む、承認された治療法の商業化努力。
価格負債の公正な価値変動があります
CelgeneからのAnthrogenensisの買収とHLIからのHLI CTの買収が業務統合とされているため,買収会計方法に従って貸借対照表上で買収に関連しているか,または対価があることを確認した。詳細は“-買収と剥離”を参照されたい。または、価格負債の公正価値が収入推定によって導出された確率加重収入法に基づいて決定され、規制および商業マイルストーン義務および特許権使用料義務が実現される可能性が確率的に評価された。買収に関連するまたは価格のある公正価値は各報告期間に再計量され、公正価値の変化は合併経営報告書に記録されている。または価格公正価値推定の変化は、私たちまたは対価格債務の増加または減少をもたらし、経営業績に応じた費用または減少をもたらす。または価格の重要な要素は、マイルストーン支払い、販売マイルストーン支払い、特許権使用料支払いを規制することだ。規制支払いはアメリカとEUのある細胞タイプに対する規制承認に基づいて決定されなければならない。規制マイルストーン支払いは使い捨てであるが、特定の適応におけるある細胞タイプの任意の潜在的なビジネスが成功する前に支払われなければならない。特許使用料は純売上高のパーセントです。いくつかの総販売の限界に達した時、販売マイルストーンの支払いを支払わなければならない。経営陣は価格の評価または価値がある場合に実質的な判断を使用しなければならない。管理職が使用する推定には、(I)前臨床データの質に基づいて可能な臨床計画の数およびタイプ、(Ii)臨床試験に要する時間、(Iii)これらの試験において規制が成功する確率、(Iv)我々が成功する適応治療可能な潜在的患者の数、および(V)商業的地位を得る治療の価格が含まれるが、これらに限定されない。このようなすべての分野は経営陣の重大な判断に関連しており、本質的に不確実だ。
経営成果
2022年12月31日までの年度と2021年12月31日までの年度比較
|
|
十二月三十一日までの年度 |
|
|
|
|
|
パーセント |
|
|||||||
|
|
2022 |
|
|
2021 |
|
|
増す |
|
|
増す |
|
||||
純収入: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
製品販売とレンタル |
|
$ |
3,749 |
|
|
$ |
3,801 |
|
|
|
(52 |
) |
|
|
(1.4 |
)% |
サービス.サービス |
|
|
5,512 |
|
|
|
5,522 |
|
|
|
(10 |
) |
|
|
(0.2 |
)% |
ライセンス、印税、その他 |
|
|
8,714 |
|
|
|
12,012 |
|
|
|
(3,298 |
) |
|
|
(27.5 |
)% |
総収入 |
|
|
17,975 |
|
|
|
21,335 |
|
|
|
(3,360 |
) |
|
|
(15.7 |
)% |
運営費用: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
収入コスト(買収を除く償却 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
製品販売とレンタル |
|
|
2,353 |
|
|
|
3,528 |
|
|
|
(1,175 |
) |
|
|
(33.3 |
)% |
サービス.サービス |
|
|
3,536 |
|
|
|
3,649 |
|
|
|
(113 |
) |
|
|
(3.1 |
)% |
ライセンス、印税、その他 |
|
|
13,776 |
|
|
|
2,476 |
|
|
|
11,300 |
|
|
|
456.4 |
% |
研究開発 |
|
|
78,363 |
|
|
|
88,353 |
|
|
|
(9,990 |
) |
|
|
(11.3 |
)% |
販売、一般、行政 |
|
|
66,021 |
|
|
|
71,341 |
|
|
|
(5,320 |
) |
|
|
(7.5 |
)% |
価格負債の公正な価値変動があります |
|
|
(126,277 |
) |
|
|
(41,145 |
) |
|
|
(85,132 |
) |
|
|
206.9 |
% |
営業権の減価 |
|
|
3,610 |
|
|
|
— |
|
|
|
3,610 |
|
|
|
100.0 |
% |
無形資産の償却を取得した |
|
|
2,193 |
|
|
|
2,192 |
|
|
|
1 |
|
|
|
0.0 |
% |
総運営費 |
|
|
43,575 |
|
|
|
130,394 |
|
|
|
(86,819 |
) |
|
|
(66.6 |
)% |
運営損失 |
|
$ |
(25,600 |
) |
|
$ |
(109,059 |
) |
|
$ |
83,459 |
|
|
|
(76.5 |
)% |
純収入と収入コスト
2022年12月31日までの1年間の純収入は1800万ドルで、前年同期比340万ドル減少し、下げ幅は15.7%だった。この低下は,主に昨年同期,SanuWaveとの800万ドルのライセンス収入を含むためである。私たちの流通パートナーに販売した製品は470万ドル増加し、許可証収入の減少を部分的に相殺した。
2022年12月31日までの年間収入コストは1,970万ドルで、前年同期比1,000万ドル増加し、103.7%増となった。増加の主な原因は、生産量と材料差が460万ドル増加したことであり、これは、変性疾患の販売が実現されないと予想される支出の増加と、流通相手への製品販売の増加による収入コストの増加である。
80
研究と開発費
2022年12月31日までの1年間の研究開発費は7840万ドルで、前年同期に比べて1000万ドル減少し、減少幅は11.3%だった。これは主に分配コストが1,940万ドル減少したためであり,前年の間に取締役会と上級管理職に報酬を付与する株式報酬支出の分配部分が含まれており,430万ドルの人件費増加と270万ドルのPalantirプラットフォーム費用増加によって相殺されたためである.
販売、一般、行政費用
2022年12月31日までの1年間、販売、一般、行政費は6600万ドルで、前年同期に比べて530万ドル減少し、減少幅は7.5%だった。減少の要因は,前年に我々の取締役会や上級管理職の報酬に関する株式報酬支出が2590万ドル減少し,その一部が研究開発費やCthの法律和解に関する前年費用に割り当てられており,これらの費用は上場企業の運営を支援するより高い人員,専門サービス,保険コストによって部分的に相殺されていることである.
営業権の減価
2022年12月31日までの年間の営業権減額は360万ドルだったが、前年同期には減値はなかった。商誉減値は退行性疾病報告部門の期待売上高と増加が低いことによるものである。
価格負債の公正な価値変動があります
2022年12月31日までの年度または対価負債公正価値変動は1.263億ドルで、8,510万ドル減少し、減少幅は206.9%であった。減少の原因は、市場の仮定および基本的な予測に基づく変化である(対価負債公正価値の変化に関するより多くの情報は、本年度報告Form 10-Kの他の部分の連結財務諸表付記4“金融資産と負債の公正価値”を参照されたい)。
その他の収入(費用)
|
|
十二月三十一日までの年度 |
|
|
|
|
|
パーセント |
|
|||||||
|
|
2022 |
|
|
2021 |
|
|
増す |
|
|
増す |
|
||||
利子収入 |
|
$ |
365 |
|
|
$ |
332 |
|
|
$ |
33 |
|
|
|
9.9 |
% |
利子支出 |
|
|
— |
|
|
|
(3,171 |
) |
|
|
3,171 |
|
|
|
(100.0 |
)% |
株式証負債の公正価値変動を認める |
|
|
42,109 |
|
|
|
13,482 |
|
|
|
28,627 |
|
|
|
212.3 |
% |
債務公正価値変動 |
|
|
(2,522 |
) |
|
|
— |
|
|
|
(2,522 |
) |
|
|
100.0 |
% |
その他の費用、純額 |
|
|
(147 |
) |
|
|
(1,682 |
) |
|
|
1,535 |
|
|
|
(91.3 |
)% |
その他収入合計 |
|
$ |
39,805 |
|
|
$ |
8,961 |
|
|
$ |
30,844 |
|
|
|
344.2 |
% |
2022年12月31日までの1年間で、他の収入(支出)は前年同期比3080万ドル増加した。増加の主な原因は、株式証負債の公正価値が我々の普通株価格の低下によって変化することである(本年度報告10-K表の他の部分の総合財務諸表付記4“金融資産と負債の公正価値”を参照)。
流動性と資本資源
2022年12月31日現在、私たちは1400万ドルの現金と現金等価物を持っており、累計赤字は6億455億ドル。私たちの資本資源の主な用途は私たちの運営費用に資金を提供することであり、その中には主に私たちの細胞治療候補薬物の研究と開発に資金を提供し、その次は販売、一般と管理費用を含む。
発行日までに、私たちは約860万ドルの無制限現金および現金等価物を私たちのビジネスに資金を提供するために使用することができ、私たちのトラフィックを発行日から12ヶ月以内に維持するための利用可能な追加の外部資本源がありません。これらの不確実性は、私たちが経営を続けている企業として継続する能力を大きく疑っています。ご参照ください経営を続ける企業より詳細については、上記を参照されたい。
これまで,細胞療法の販売は承認されておらず,われわれの細胞療法の販売も何の収入も生じていない。生物バンクと変性疾患ビジネスから得られる収入は限られている。我々が開発に成功し,規制部門から1つ以上の候補治療薬の承認を得ない限り,細胞治療製品の販売から何の収入も得られないことが予想され,数年かかると予想される。もし私たちの候補治療薬が規制部門の承認を得たら、治療販売に関連した巨額の商業化費用が発生すると予想される
81
マーケティング、製造、流通、私たちの現在の商業化努力は私たちのバイオバンクと退行性疾病業務に限られているからです。したがって、治療および私たちの生体材料製品の販売から相当な収入を得ることができる前に、私たちはBTIG、LLC、Oppenheimer&Co.およびB.Riley Securities、Inc.またはATM計画に基づいて、2022年9月8日の市場販売協定の下での引き出しを含む、株式発行、債務融資、または他の資本源を通じて私たちの現金需要に資金を提供する予定です。私たちはまた、私たちの細胞療法の許可と協力手配、そして私たちの変性疾患業務の流通手配、CH Trade Group、Tamer、AD Portsとの流通プロトコルを含めて、海外拡張をサポートしています。しかし、私たちは必要に応じて優遇条件で、または追加資金を調達できないか、またはそのような他の計画を達成することができないかもしれない。必要なときに資金を調達できなかった場合は、私たちの財務状況や私たちの業務計画や戦略を実行する能力に悪影響を及ぼす可能性があります。必要な資本を得ることができない場合、あるいは私たちの流動性需要を満たすことができなければ、私たちの業務を延期、制限、または中止させ、さらにリストラし、私たちの生体材料製品および他の臨床試験計画の商業化努力を停止し、私たちの資産の全部または一部を清算したり、他の戦略的選択を求めたり、および/またはアメリカ破産法の規定に基づいて保護を求めることができるかもしれない。
将来的には,我々の細胞治療候補薬の開発と潜在的商業化,我々の退行性疾患業務の拡張,進行中の内部研究·開発計画に巨額の費用が生じることが予想される。現在、私たちの開発、潜在的な商業化と内部研究開発計画の性質、時間あるいは総コストを合理的に見積もることはできない。しかし、私たちの現在と未来の臨床前研究と臨床試験を完成させ、規制部門の私たちの候補治療薬の承認を得る過程を完成させ、私たちが必要と思う販売、マーケティング、流通インフラを確立するためには、承認されれば、私たちの細胞治療候補薬や生体材料製品は将来的に多くの追加資金を必要とするかもしれない。
今まで、インフレは私たちの業務に大きな影響を与えなかった。しかし、インフレと金利のいずれの大幅な上昇も経済全体に重大な影響を与える可能性があり、将来の経営業績に影響を与える可能性がある。
キャッシュフロー
次の表は、示す期間のキャッシュフローをまとめたものである
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
変わる |
|
|||
現金(用)/由 |
|
|
|
|
|
|
|
|
|
|||
経営活動 |
|
$ |
(137,876 |
) |
|
$ |
(110,096 |
) |
|
$ |
(27,780 |
) |
投資活動 |
|
|
(5,236 |
) |
|
|
(5,903 |
) |
|
|
667 |
|
融資活動 |
|
|
119,838 |
|
|
|
98,562 |
|
|
|
21,276 |
|
現金、現金等価物、および限定的な現金純変化 |
|
$ |
(23,274 |
) |
|
$ |
(17,437 |
) |
|
$ |
(5,837 |
) |
経営活動
2022年12月31日までの年間で、運営に使用されている現金純額が前年同期比2780万ドル増加したのは、主に今年度の非現金プロジェクト調整後の純収益(損失)と在庫蓄積部分が前年期間に相殺され、SanuWaveライセンス契約終了に関する繰延収入の減少を含むためである。
投資活動
2022年12月31日と2021年12月31日までの年間で、投資活動ではそれぞれ520万ドルと590万ドルの現金純額を使用しており、各期間の資本支出は前年期間の本票収入の30万ドルと相殺されている。
融資活動
2022年12月31日までの1年間に、引受権証を行使して13,281,386株の普通株を買収した現金4650万ドル、2022年5月に私募した現金収益2740万ドル、ヨークビル前払い契約の現金3920万ドル、ATMが普通株を売却する予定の現金収益600万ドルを主に含む1億198億ドルの純現金を融資活動から発生させた。2021年12月31日までの1年間に、GXと合併した1.088億ドルの現金収益、パイプ融資、およびPalantir Technologies,Inc.の投資を含む9860万ドルが発生し、上記の取引に関連する専門サービス支払い1090万ドルがこの部分を部分的に相殺した。私たちはまた500万ドルの短期ローンを受けて返済した。
82
肝心な会計見積もり
我々の重要会計政策要約は、本年度報告Form 10-Kの他の部分の総合財務諸表に含まれる付記2“重要会計政策要約”に掲載されている。
公認会計原則に基づいて我々の連結財務諸表を作成する際には、管理層は、報告期間中に報告された資産および負債の金額、連結財務諸表の日付、または有資産および負債の開示、および報告期間中の費用報告金額に影響を与えるための推定および仮定を行う必要がある。これらの総合財務諸表に反映される重大な推定および仮定は、業務合併の会計処理、営業権および無形減価評価、在庫評価、または対価格、短期債務、または株式対価格、漸増借入率の決定、研究開発費の計上、および株式オプションおよび優先株式証の推定に関する仮定を含むが、これらに限定されない。私たちの推定は、歴史的経験、既知の傾向、および当時の状況で合理的だと思う他の特定の市場要素または他の関連要素に基づいている。継続的な基礎の上で、環境、事実、経験が変化した場合、経営陣はこれらの見積もりを評価する。推定された変化は、それらが知られている期間に記録される。実際の結果はこれらの推定とは異なる可能性がある。
収入確認
製品とサービスのコントロール権が私たちの顧客に移った時、私たちは収入を確認し、金額は私たちが顧客から得たい価格を反映して、これらの製品とサービスを交換します。この過程には,顧客との契約の決定,契約中の履行義務の決定,契約価格の決定,契約価格を契約中の異なる履行義務に割り当てること,および履行義務を履行した後に収入を確認することがある.
履行義務が単独または顧客がいつでも取得でき,契約で単独で決定した他の資源とともに顧客に利益を提供する場合,履行義務は契約中の他の義務とは異なると考えられる.商品やサービスの制御権をクライアントに移すと,履行義務が履行されていると考えられ,顧客が商品やサービスの利点を利用して獲得する能力があることを意味すると考えられる.製品またはサービスの取引価格は、通常、顧客と締結された契約価格に基づいており、取引価格に可変対価格が含まれる場合には、期待値方法を用いて取引価格に含まれるべき可変対価金額を推定したり、状況に応じて取得する権利があると予想される可能な金額を推定したりする。
私たちの変性疾患部門の製品は一般的に多様な要素を含まない。私たちはこれらの製品に返品の権利があることを許可していますが、今まで返品は少なかったです。
SanuWaveとのライセンス契約により、四半期許可料と販売されている製品毎の固定印税を受け取りました。SanuWaveにBiovance特許権使用料免除,最高四半期許可料を提供した。我々はこの期間の実売上高に基づいて四半期ごとの四半期許可料を確認した(2021年第3四半期に許可を終了するまで)。
企業合併の会計処理
企業合併に対して会計処理を行うには、特に買収の日に、買収の有形および無形資産、負担された負債および買収前のまたはある事項について重大な推定と仮定を行う必要がある。私たちは、私たちの最適な推定と仮定を用いて、買収日に買収された有形無形資産、負担された負債、および買収された無形資産の使用年数に対して公正な価値を正確に分配する。我々が獲得したいくつかの無形資産や営業権を評価する際には、重要な推定の例としては、開発された技術や進行中の研究·開発が含まれているが、これらに限定されない。私たちの推定はまた私たちの繰延所得税資産と負債に影響を及ぼすかもしれない。予期しないイベントや状況が発生する可能性があり、このような仮定、推定、または実際の結果の正確性または有効性に影響を与える可能性がある。
商業権と無形資産の価値評価
当社は事業合併に関連する重大無形資産を買収し、引き続き買収し、公正価値で入金することが可能である。公正価値の確定には予測、推定と仮定を使用する必要があり、これは管理層が重大な判断を下す必要がある。これらの要素の各々は不確実性の影響を受け、無形資産の価値に大きな影響を与える可能性がある。
営業権および無期限無形資産は、毎年、または減値をもたらす可能性のあるイベントが発生した場合に減値審査を行う。減値分析は管理層が重大な判断を行う必要があり、定性要素(これらの要素に不確定性が存在する可能性があり、異なる時期に重大な変化が発生する可能性がある)を評価することもでき、数量化評価を行うこともできる。我々の量子化減価テストでは,将来の数量,収入および費用成長率,適切な割引率の選択,資産グループ化,および他の仮定および推定について大きな判断を行う必要がある将来のキャッシュフローを推定する方法を用いる.使用された推定と仮定は不確実性の影響を受ける。代替推定および仮定を使用することは、資産の推定公正価値を増加または減少させる可能性があり、我々の経営業績に影響を与える可能性がある。実際の結果は私たちの推定とは違うかもしれない。
83
値段が合うかもしれない
私たちは、私たちの買収日の総合貸借対照表に推定公正価値を計上する潜在的なマイルストーンと特許使用料債務を含む買収に関連するまたは対価格があります。私たちは各報告期間に公正価値を再計量し、総合経営報告書に変化を記録した。公正な価値の決定は管理職が重大な判断と推定をする必要がある。これらには,マイルストーンの実現とスケジュールに関する見積りと仮説,収入予測と割引率計算のための仮説がある.経営陣の仮定が不正確であることが証明された場合、対価格負債の変化を招き、我々の運営結果に実質的な影響を与える可能性がある。
株式証法的責任
負債分類権証の会計処理には、管理層が判断し、その公正価値について推定および仮説を行う必要がある(分類権証負債の重大な投入および仮定を評価するためのより多くの情報については、本年度報告Form 10-Kの他の部分の統合財務諸表付記4“金融資産と負債の公正価値”を参照)。株式証負債は最初に発行当日に公正価値で入金され、その後、報告日ごとに公正価値によって再計量され、総合経営報告書で変動を確認する。負債分類株式証の公正価値変動は、株式証が行使され、満期または権益分類資格に適合するまで、引き続き確認される。
転換可能な受取手形
私たちは2020年8月のUltraMist事業の販売から転換可能なチケットを得た。我々は信用違約モデルを用いた債券推定値を用いており,管理層は,(I)発行者普通株の公正価値と変動性,(Ii)手形変換の可能性とタイミング,および(Iii)無リスク金利についての推定と判断を用いる必要がある。もし私たちの仮定と推定が不正確であることが証明されたら、それは受取変換可能なチケットの変化を招き、私たちの運営結果に実質的な影響を与える可能性がある。
株に基づく報酬
私たちは、推定付与日の公正価値に基づいて、従業員と非従業員の株式オプションに関する補償費用を確認し、没収が発生した場合に確認します。私たちはブラック·スコアーズオプション定価モデルを使用してサービスと業績ベースの報酬を推定し、付与日の公正価値とそれによって生成された株式ベースの報酬支出を推定する。市場条件のある奨励については、モンテカルロモデルを使用して、これらの奨励の公正価値を推定する。株式に基づく報酬の付与日公正価値は、必要なサービス期間内に直線的に確認され、必要なサービス期間は、通常、各報酬の帰属期間である。ブラック·スコルスオプション定価モデルおよびモンテカルロモデルは、株式に基づく報酬の公正価値を決定するために、高い主観的仮定を使用することを必要とする。ブラック·スコアーズオプション定価モデルを適用して、2022年12月31日および2021年12月31日までの年間に付与された株式オプションの推定公正価値を決定する際に使用される特定の仮定に関する情報は、本年度報告の他の部分に含まれる監査された総合財務諸表の付記14“株式ベースの報酬”を参照されたい。このような仮定は固有の不確実性と重大な判断の適用に関するものだ。したがって、要素や予想結果が変化し、私たちが使用する仮説や推定が大きく異なる場合、私たちの株ベースの報酬は実質的に異なるかもしれない。
賃貸借証書
私たちは賃貸に隠された金利を簡単に決定することができないので、私たちは増額借入金利(IBR)を使ってレンタル負債を測定します。IBRは、類似期間や類似証券の場合、使用権やROU資産と類似した価値を有する資産を取得するために必要な資金である金利、すなわち類似経済環境下で支払わなければならない金利である。したがって、IBRは、観察可能なレートがない場合に推定する必要があるか、またはレンタル条項および条件を反映するように調整する必要がある場合に、“支払わなければならない”ことを反映している。IBRは、利用可能であれば、いくつかのエンティティおよび資産特定推定を行う必要がある観察可能な入力(例えば、市場金利)を使用して推定される。リース負債を計算する際にリース支払いの現在値を計算するための内部収益率および対応する純資産収益率は、管理層が大きな判断を使用する必要がある。
短期債務
ヨークビルと締結された前払い前払い契約に基づいて、私たちは公正価値オプションを選択して金融商品に計上した。公正価値の推定値は二項格子モデルを用いて決定した.債務の公正価値計量は市場では観察できない第三級投入と仮定を用いて決定された。公正価値によって計上された債務公允価値変動は、関連する計算すべき利息支出を含み、付随する総合経営報告書と全面収益(損失)の中で公正価値変動列に従って損益として示される。以下の項目の債務公正価値変動総額の一部に起因することができる
84
ツールに特化した信用リスクの変動は,基本市場変動を含まない割引率仮説の定期変動を具体的に計測することで決定され,付随する総合経営報告書と包括収益(損失)表に包括収益(損失)の構成要素として示されている。短期債務の実際の決済は現在の見積もりと異なる可能性があり、これはヨークビルがいつを選択するか、金額を普通株に転換する時間、満期前に返済可能な現金、および私たちの普通株価格の変動に依存する。
最近の会計公告
最近の会計声明、これらの政策の採用時期、およびこれらの政策が私たちの経営業績財務状況に及ぼす潜在的な影響の評価については、本年度報告(Form 10-K)の他の部分の付記2、“重要会計政策の概要”を参照されたい。
雇用法案会計選挙
“雇用法案”の定義によると、私たちは“新興成長型会社”です。“雇用法案”によると、新興成長型企業は、これらの基準が民間企業に適用されるまで、“雇用法案”公布後に発表された新たなまたは改正された会計基準の採用を延期することができる。
私たちは、私たち(I)がもはや新興成長型企業または(Ii)が“雇用法案”に規定されている延長移行期間から撤退することを明確かつ撤回できないまで、この延長された移行期間を使用することを選択し、新しい会計基準または改正された会計基準を遵守できるようにすることを選択した。したがって、我々の財務諸表は、上場企業の発効日までに新たなまたは改訂された会計声明を遵守している会社と比較できない可能性がある。
85
第七A項。定量と合格市場リスクの開示について。
私たちは正常な業務過程で市場リスクに直面している。このような危険は主に金利敏感性を含む。
金利リスク
2022年12月31日現在、商業銀行口座に保有されている現金と元の満期日が3ヶ月未満の通貨市場基金を主に含む1400万ドルの現金と現金等価物を持っている。2022年12月31日現在、ほとんどの現金と現金等価物は商業銀行口座や通貨市場基金に保管されている。私たちの投資活動の主な目標は資本を保存し、私たちの運営に資金を提供することだ。私たちはまた大きなリスクを負うことなく投資から最大の収益を得ることを求めている。私たちの投資は主に短期的なので、私たちの金利リスクへの開放は大きくなく、市場金利1%の変動は私たちのポートフォリオの総価値に大きな影響を与えないと思います。2022年12月31日まで、私たちは未返済の可変利息債務を持っていない。
インフレの影響
インフレは一般的に労働コストと臨床試験費用を増加させることで私たちに影響を及ぼす。私たちはインフレが本報告書で述べた間の私たちの経営結果に実質的な影響を及ぼすとは思わない。
プロジェクト8.財務諸表データを補完しています
|
ページ |
独立公認会計士事務所報告 (PCAOB ID番号 |
F-1 |
合併貸借対照表 |
F-2 |
合併経営表と全面損益表(赤字) |
F-3 |
転換可能優先株と株主権益連結報告書(損失) |
F-4 |
統合現金フロー表 |
F-5 |
連結財務諸表付記 |
F-6 |
86
独立登録者の報告公認会計士事務所
Celulity Inc.の株主と取締役会に。
財務諸表のいくつかの見方
Celularity Inc.(“当社”)2022年12月31日現在と2021年12月31日までの連結貸借対照表、2022年12月31日までの両年度の関連総合経営表と全面収益(損失)、転換可能優先株と株主権益(損失)とキャッシュフロー、および関連付記(総称して“財務諸表”と呼ぶ)を監査した。これらの財務諸表は,すべての重要な点で当社の2022年12月31日と2021年12月31日までの財務状況と,2022年12月31日までの両年度の経営結果とキャッシュフローを公平に反映しており,米国公認の会計原則に適合していると考えられる。
経営を続ける企業
添付財務諸表の作成は、同社が引き続き経営を継続する企業であると仮定している。財務諸表付記1で述べたように、当社は設立以来経営損失を受け続けており、持続経営企業としての持続経営能力に大きな疑いを抱かせている。付記1は、これらの事項における経営陣の計画も説明しています。財務諸表には、このような不確実性の結果に起因する可能性のある調整は含まれていません。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査することを求められておらず、私たちも招聘されて監査を行っていません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性について意見を述べるためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
/s/
2023年3月31日
2018年以来、当社の監査役を務めてきました。
F-1
CELULARITY Inc.
合併B割当書
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
売掛金、予備金を差し引く#ドル |
|
|
|
|
|
|
||
受取手形 |
|
|
|
|
|
|
||
在庫品 |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
流動資産総額 |
|
|
|
|
|
|
||
財産と設備、純額 |
|
|
|
|
|
|
||
商誉 |
|
|
|
|
|
|
||
無形資産、純額 |
|
|
|
|
|
|
||
使用権資産--経営リース |
|
|
|
|
|
— |
|
|
制限現金 |
|
|
|
|
|
|
||
在庫は、当期分の純額を差し引く |
|
|
|
|
|
|
||
その他長期資産 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
負債、償還可能な転換可能優先株、株主権益 |
|
|
|
|
|
|
||
流動負債: |
|
|
|
|
|
|
||
売掛金 |
|
$ |
|
|
$ |
|
||
費用とその他の流動負債を計算しなければならない |
|
|
|
|
|
|
||
融資債務の当期部分 |
|
|
— |
|
|
|
|
|
短期債務(#ドル |
|
|
|
|
|
— |
|
|
収入を繰り越す |
|
|
|
|
|
|
||
流動負債総額 |
|
|
|
|
|
|
||
繰延収入,当期分を差し引く |
|
|
|
|
|
|
||
買収に関連しているか,あるいは掛け値がある |
|
|
|
|
|
|
||
非流動賃貸負債--経営 |
|
|
|
|
|
— |
|
|
融資義務 |
|
|
— |
|
|
|
|
|
株式証負債 |
|
|
|
|
|
|
||
繰延所得税負債 |
|
|
|
|
|
|
||
その他負債 |
|
|
|
|
|
|
||
総負債 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
株主権益: |
|
|
|
|
|
|
||
優先株、$ |
|
|
|
|
|
|
||
普通株、$ |
|
|
|
|
|
|
||
追加実収資本 |
|
|
|
|
|
|
||
その他の総合収益を累計する |
|
|
|
|
|
— |
|
|
赤字を累計する |
|
|
( |
) |
|
|
( |
) |
株主権益総額 |
|
|
|
|
|
|
||
総負債、償還可能な転換可能優先株、株主権益 |
|
$ |
|
|
$ |
|
||
付記はこれらの連結財務諸表の構成要素である。
F-2
CELULARITY Inc.
合併状態経営プロジェクトと総合収益
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
純収入: |
|
|
|
|
|
|
||
製品販売とレンタル |
|
$ |
|
|
$ |
|
||
サービス.サービス |
|
|
|
|
|
|
||
ライセンス、印税、その他 |
|
|
|
|
|
|
||
総収入 |
|
|
|
|
|
|
||
運営費用: |
|
|
|
|
|
|
||
収入コスト(無形資産を獲得した償却を除く) |
|
|
|
|
|
|
||
製品販売とレンタル |
|
|
|
|
|
|
||
サービス.サービス |
|
|
|
|
|
|
||
ライセンス、印税、その他 |
|
|
|
|
|
|
||
研究開発 |
|
|
|
|
|
|
||
販売、一般、行政 |
|
|
|
|
|
|
||
価格負債の公正な価値変動があります |
|
|
( |
) |
|
|
( |
) |
営業権の減価 |
|
|
|
|
|
— |
|
|
無形資産の償却を取得した |
|
|
|
|
|
|
||
総運営費 |
|
|
|
|
|
|
||
運営損失 |
|
|
( |
) |
|
|
( |
) |
その他の収入(支出): |
|
|
|
|
|
|
||
利子収入 |
|
|
|
|
|
|
||
利子支出 |
|
|
— |
|
|
|
( |
) |
株式証負債の公正価値変動を認める |
|
|
|
|
|
|
||
債務公正価値変動 |
|
|
( |
) |
|
|
— |
|
その他の費用、純額 |
|
|
( |
) |
|
|
( |
) |
その他収入合計 |
|
|
|
|
|
|
||
所得税前純収益 |
|
|
|
|
|
( |
) |
|
所得税費用 |
|
|
|
|
|
|
||
純収益(赤字) |
|
$ |
|
|
$ |
( |
) |
|
信用リスク変動による債務公正価値変動,税引き後純額 |
|
|
|
|
|
— |
|
|
その他総合収益 |
|
|
|
|
|
— |
|
|
総合収益(赤字) |
|
$ |
|
|
$ |
( |
) |
|
情報を共有する: |
|
|
|
|
|
|
||
1株当たり純収益(損失)-基本 |
|
$ |
|
|
$ |
( |
) |
|
加重平均流通株-基本 |
|
|
|
|
|
|
||
1株当たり純収益-薄めて |
|
$ |
|
|
$ |
( |
) |
|
加重平均流通株-希釈 |
|
|
|
|
|
|
||
付記はこれらの連結財務諸表の構成要素である。
F-3
CELULARITY Inc.
転換可能な会社合併報告書株と株主権益を転任する
(単位は千で、シェアは含まれていない)
|
|
第1ラウンドは償還可能である |
|
Bシリーズは償還可能です |
|
Xシリーズ償還可能 |
|
|
普通株 |
|
在庫株 |
|
その他の内容 |
|
積算 |
|
他の総合を累計する |
|
合計する |
||||||||||
|
|
株 |
|
金額 |
|
株 |
|
金額 |
|
株 |
|
金額 |
|
|
株 |
|
金額 |
|
株 |
|
金額 |
|
資本 |
|
赤字.赤字 |
|
収入(損) |
|
権益(赤字) |
2020年12月31日の残高 |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
|
$ |
|
( |
|
$( |
|
$ |
|
$( |
|
$— |
|
$( |
||||
株式オプションの行使 |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
|
— |
|
— |
|
— |
|
|
— |
|
— |
|
|||
株に基づく報酬費用 |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
— |
|
— |
|
— |
|
— |
|
|
— |
|
— |
|
||
GX買収会社合併からの資本再編、償還、株式発行コスト、合併コストを差し引く |
|
( |
|
( |
|
( |
|
( |
|
( |
|
( |
|
|
|
|
|
|
|
— |
|
— |
|
||||||
パイプ投資家に普通株式を発行する |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
|
|
— |
|
— |
|
|
— |
|
— |
|
||||
負債分類レガシー株式証明書を持分に再分類する |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
— |
|
— |
|
— |
|
— |
|
|
— |
|
— |
|
||
Palantirに普通株式を発行する |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
|
— |
|
— |
|
— |
|
|
— |
|
— |
|
|||
Cthとの債務を終わらせるために普通株を発行する |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
|
— |
|
— |
|
— |
|
|
— |
|
— |
|
|||
純損失 |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
( |
|
— |
|
( |
2021年12月31日の残高 |
|
— |
|
$— |
|
— |
|
$— |
|
— |
|
$— |
|
|
|
$ |
|
— |
|
$— |
|
$ |
|
$( |
|
$— |
|
$ |
|
累積効果調整ASU 2016−02 |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
— |
|
— |
|
— |
|
— |
|
|
|
|
— |
|
||
以前行使した株式オプションを再分類する |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
|
— |
|
— |
|
— |
|
|
— |
|
— |
|
|||
株式オプションの行使 |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
|
— |
|
— |
|
— |
|
|
— |
|
— |
|
|||
普通株の購入と廃棄 |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
( |
|
— |
|
— |
|
— |
|
( |
|
— |
|
— |
|
( |
株式証の行使 |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
|
|
— |
|
— |
|
|
— |
|
— |
|
||||
短期債務転換による普通株 |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
|
|
— |
|
— |
|
|
— |
|
( |
|
||||
株式単位の帰属を制限する |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
制限株式単位は前払税に帰属する |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
( |
|
— |
|
— |
|
— |
|
( |
|
— |
|
— |
|
( |
株に基づく報酬費用 |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
— |
|
— |
|
— |
|
— |
|
|
— |
|
— |
|
||
パイプ投資家に普通株を発行し,発行コストを差し引く |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
|
— |
|
— |
|
— |
|
|
— |
|
— |
|
|||
ATM発行における普通株の発行は、手数料と発行費用を差し引く |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
|
— |
|
— |
|
— |
|
|
— |
|
— |
|
|||
信用リスク変動による債務公正価値変動,税引き後純額 |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
||
純収入 |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
— |
|
— |
|
— |
|
— |
|
— |
|
|
— |
|
||
2022年12月31日の残高 |
|
— |
|
$— |
|
— |
|
$— |
|
— |
|
$— |
|
|
|
$ |
|
— |
|
$— |
|
$ |
|
$( |
|
$ |
|
$ |
|
付記はこれらの連結財務諸表の構成要素である。
F-4
CELULARITY Inc.
合併状態キャッシュフロープロジェクト
(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
経営活動のキャッシュフロー: |
|
|
|
|
|
|
||
純収益(赤字) |
|
$ |
|
|
$ |
( |
) |
|
純収益(損失)と業務用現金純額の調整: |
|
|
|
|
|
|
||
減価償却および償却 |
|
|
|
|
|
|
||
非現金レンタル費用 |
|
|
( |
) |
|
|
— |
|
所得税を繰延する |
|
|
( |
) |
|
|
( |
) |
不良債権準備 |
|
|
|
|
|
|
||
株式証負債の公正価値変動を認める |
|
|
( |
) |
|
|
( |
) |
廃棄在庫備蓄 |
|
|
|
|
|
— |
|
|
営業権の減価 |
|
|
|
|
|
— |
|
|
株に基づく報酬費用 |
|
|
|
|
|
|
||
価格の公正価値変動があるかもしれない |
|
|
( |
) |
|
|
( |
) |
債務公正価値変動 |
|
|
|
|
|
— |
|
|
Cthとの債務を終わらせるために普通株を発行する |
|
|
— |
|
|
|
|
|
株式価格の公正価値変動があるかもしれない |
|
|
|
|
|
— |
|
|
その他、純額 |
|
|
( |
) |
|
|
|
|
資産と負債の変動状況: |
|
|
|
|
|
|
||
売掛金 |
|
|
( |
) |
|
|
( |
) |
在庫品 |
|
|
( |
) |
|
|
( |
) |
前払い費用と他の資産 |
|
|
|
|
|
|
||
販売純営業損失と研究開発税収控除 |
|
|
— |
|
|
|
|
|
売掛金 |
|
|
( |
) |
|
|
|
|
費用とその他の負債を計算すべきである |
|
|
|
|
|
( |
) |
|
資産と賃貸負債を使用する |
|
|
|
|
|
— |
|
|
収入を繰り越す |
|
|
|
|
|
( |
) |
|
経営活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
投資活動によるキャッシュフロー: |
|
|
|
|
|
|
||
資本支出 |
|
|
( |
) |
|
|
( |
) |
元票収益 |
|
|
— |
|
|
|
|
|
投資活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
資金調達活動のキャッシュフロー: |
|
|
|
|
|
|
||
株式承認証を行使して得られた収益 |
|
|
|
|
|
— |
|
|
短期債務収益 |
|
|
|
|
|
— |
|
|
ATM発売で普通株を販売する収益 |
|
|
|
|
|
— |
|
|
ATM機は費用と手数料の支払いを提供します |
|
|
( |
) |
|
|
— |
|
短期借入金収益−関係者 |
|
|
— |
|
|
|
|
|
短期借入金の支払い--関係者 |
|
|
— |
|
|
|
( |
) |
GX買収会社から得た資本再編現金 |
|
|
— |
|
|
|
|
|
Palantir投資の収益 |
|
|
— |
|
|
|
|
|
パイプ融資収益 |
|
|
|
|
|
|
||
株式オプションを行使して得られる収益 |
|
|
|
|
|
|
||
制限株式単位は前払税に帰属する |
|
|
( |
) |
|
|
— |
|
PIPE/SPACに関する料金をお支払いください |
|
|
( |
) |
|
|
( |
) |
融資活動が提供する現金純額 |
|
|
|
|
|
|
||
現金、現金等価物、および限定的な現金純減少 |
|
|
( |
) |
|
|
( |
) |
年初の現金、現金等価物、制限現金 |
|
|
|
|
|
|
||
年末現金、現金等価物、制限現金 |
|
$ |
|
|
$ |
|
||
キャッシュフロー情報の追加開示: |
|
|
|
|
|
|
||
利子を支払う現金 |
|
$ |
— |
|
|
$ |
|
|
所得税の現金を納める |
|
$ |
— |
|
|
$ |
— |
|
非現金投資と融資活動を補完します |
|
|
|
|
|
|
||
売掛金及び売掛金に含まれる財産及び設備 |
|
$ |
( |
) |
|
$ |
( |
) |
短期債務転換発行の普通株式 |
|
$ |
|
|
$ |
— |
|
|
GX買収合併後の資本再編 |
|
$ |
— |
|
|
$ |
|
|
在庫株を解約する |
|
$ |
— |
|
|
$ |
|
|
GX買収会社と合併して得られた非現金資産。 |
|
$ |
— |
|
|
$ |
|
|
GX買収会社と合併して負う引受権証の責任 |
|
$ |
— |
|
|
$ |
|
|
パイプ·合併関連費用の支払いとして普通株を発行する |
|
$ |
— |
|
|
$ |
|
|
株式証負債を持分に再分類する |
|
$ |
— |
|
|
$ |
|
|
前年に支払われた発行コストを再分類する |
|
$ |
— |
|
|
$ |
|
|
オプション負債を権益に再分類する |
|
$ |
|
|
$ |
— |
|
|
付記はこれらの連結財務諸表の構成要素である。
F-5
CELULARITY Inc.
合併後の注釈財務諸表
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
Celularity Inc.(“Celularity”または“会社”)は、前身はGX買収会社(“GX”)であり、デラウェア州に登録設立された空白小切手会社である
2021年7月16日(“完了日”)、当社は2021年1月8日の合併合意および再編計画(“合併合意”)に基づいて先に発表された合併を完了し、合併協定はGX、Alpha First Merge Sub,Inc.,GXのデラウェア州会社およびGXの直接完全子会社(“第一合併子会社”)、Celularity LLC(f/k/a Alpha Second Merge Sub LLC)、デラウェア州の有限責任会社およびGXの直接全額付属会社(“第二連結子会社”)およびCelularity Inc.と呼ばれるエンティティで完成した。デラウェア州法律に基づいて2016年8月29日に登録が成立した(“Legacy Celulity”)。合併取引が完了した後、GXはCelularity Inc.と改名されます。この業務合併は、米国で一般的に受け入れられている会計原則に適合する逆資本再編とみなされています(付記3参照)。
業務説明
Celularityは生物技術会社であり、既製の胎盤由来同種異体細胞療法を開発することによって癌、免疫と伝染病を治療し、細胞医学の次の発展をリードした。Celularityはキメラ抗原受容体(CAR)、ナチュラルキラー細胞(NK)、細胞、間葉系様接着基質細胞(MLASCs)と外体からなるT細胞を含む一連の既製胎盤由来同種異体細胞治療候補製品を開発している。これらの候補治療薬は癌,感染症,退行性疾患に対する適応である。サイルラティは、胎盤独特の生物学的特性と既製の獲得性を利用することによって、全世界の有効、獲得と負担が得られる治療薬物に対する重大な需要を満たすための治療解決策を開発できると信じている。Celularityはまた,胎盤から抽出した生体材料製品を積極的に開発·販売している。2023年までに、Celularityは国内でこれらの製品を販売し、主に整形外科と傷口看護市場にサービスしている。Celularityは現在,米国以外で胎盤生体材料を販売しようとしており,最初の重点は中東と北アフリカ市場である。Celularityの現在の生体材料業務は主にそのBiovanceとInterfyl製品を直接或いはその流通ネットワークを介して販売することを含む。Biovanceは健康,臨月妊娠の胎盤から抽出した脱細胞,脱水したヒト羊膜である。それは無傷の天然細胞外基質であり、傷口の再生過程に基礎を提供し、機能組織の修復にステントを提供した。Interfylはヒト結合組織基質であり、健康、臨月妊娠の胎盤から来ている。それは各種の医学専門家によって創傷、創傷或いは手術による軟組織欠損を埋めるために使用されている。サイレスはBiovanceやInterfyl以外のビジネスチャネルを深化させるために新たな胎盤生体材料製品を開発している。同社はまた,細胞治療開発や製造におけるコア専門知識を利用して,第三者に契約製造と開発サービスを提供することで収入を創出する予定である。この新サービスの最初のポイントは,開発段階の細胞治療会社の臨床試験のための治療候補薬の開発·製造に協力することである。2023年1月、同社は仕事の優先順位の再決定を発表し、約20%減少した2023年3月までの従業員総数。
Celularityの本部はニュージャージー州のフロラム公園にある。Legend Celularityは2017年8月にCelgene Corporation(“Celgene”)からAnthrogensis Corporation(“Anthrogenensis”)を買収し、Celgene Corporation(“Celgene”)は百時美施貴宝社と合併したグローバルバイオテクノロジー会社である。これまで,ヒト起源会社はCelgene細胞治療会社の名で運営されており,Celgene社はCelgeneの細胞治療部門であった。Celulality Currenタリーがいる
Celulality Impactプラットフォームは胎盤由来細胞の優勢を利用して多種の疾患に対して、生物由来からそのアメリカで専門的に建設された凍結保存と包装までの同種異体細胞までシームレスな統合を提供する
F-6
非ホジキンリンパ腫(NHL)や固形腫瘍では,CD 16とT細胞受容体(TCR)を発現するトランスジェニックT細胞がモノクローナル抗体(MAbs)に結合してノックアウトされている。Cynk−001は胎盤由来の未修飾NK細胞である。2022年、同社は急性骨髄性白血病(AML)と多形性膠芽腫(GBM)の治療のための積極的かつ承認された臨床試験を行っており、前者は血液癌であり、後者は固形腫瘍である。Cynk-001は現在AMLのアクティブな第1段階実験中である.同社はまた、NK療法が再発難治性AML(RrAML)を治療する際に直面するいくつかの挑戦を克服する可能性がある次世代CAR-NK-301を推進する。企業資源の優先順位を決定する必要があるため、同社は2023年1月にGBM試験での採用を中止する意向を発表した。しかし、同社は引き続きその固形腫瘍研究計画を推進する。Cynk-302は、固形腫瘍において開発されている次世代CAR-NKであり、最初は非小細胞肺癌(NSCLC)に集中しており、これは持続的に高度に満たされていない需要領域である。APPL−001は胎盤由来のMLASCであり,クローン病や他の変性疾患の治療のために開発されている。PExo−001は胎盤由来の外切体であり,骨関節炎治療のために開発されている。
同社は生物技術業界の早期会社によく見られるリスクと不確定要素の影響を受け、競争相手の新技術革新の開発、肝心な者への依存、ノウハウの保護、政府法規の遵守及び追加資本を獲得して運営に資金を提供する能力を含むが、これらに限定されない。現在開発されている候補薬物は,商業化前に広範な臨床前と臨床試験および規制承認を含む大量の追加承認が必要となる。このような努力は大量の追加資本、十分な人員とインフラ、そして広範囲なコンプライアンス報告能力を必要とする。同社の薬物開発努力が成功しても,同社がいつ(あれば)製品販売から相当な収入を得るかは定かではない。
買収する
Legacy Celularityは2016年の設立直後に4つの業務統合を完了した。Legend Celularityは買収まで何の重大な活動もありません。
Legacy Celularityは2017年5月31日、ヒト長寿会社(HLI)からHLI細胞治療有限責任会社(HLI CT)を買収した。HLI CTはLifebank USAを運営しており,個人臍帯血幹細胞と臍帯組織バンクであり,新生児臍帯血幹細胞と臍帯組織単位を収集,加工,凍結保存する選択肢を親に提供している。HLI CTの買収はLegacy CelulalityにBiovanceを含む一連の生体材料資産の権利を提供した®Interfylと®発達段階の胎盤幹細胞計画であるPSC−100。買収前に,HLIはAliqua Biomedical Inc.(“Aliqua”)と供給契約および許可,マーケティング,開発プロトコル(総称して“HLIプロトコル”と呼ぶ)を締結した.HLIプロトコルはAliquaにBiovanceの独占マーケティングと流通の権利を与える®Interfylと®それは.HLI CTの買収により当社のHLIプロトコルの権利、所有権および権益に移行します。全体的に、HLI CTを買収する費用は#ドルである
二零一七年八月十五日にLegacy CelularityとCelgeneが合併協定に署名し、これによりCelgeneの全額付属会社Anthrogenensis(“Anthrogensis合併協定”)を買収した。今回の買収には,免疫腫瘍学,炎症性疾患,年齢関連疾患における重要な細胞治療資産を含む一連の臨床前と臨床段階資産が含まれており,Legacy Celularityはこれらの資産の開発を継続している。Anthrogensisの買収はLegacy CelulalityがAnthrogensisの独自技術と技術を獲得でき、産後ヒト胎盤から抽出した潜在力の高い幹細胞と細胞治療製品を大量に回収する(どの製品も“Anthrogensis Products”)を回収する。ヒト遺伝子を買収する一部として、ヒト遺伝子製品のいくつかの発明者とヒト遺伝子製品開発チームの他の重要なメンバーがLegacy Celularityに参加した。全体的に言えば、人類の起源を買収する代償の公正価値は$である
Legacy Celularityは2017年8月、合併対価としてCelgeneにXシリーズ優先株株式を発行し、Celgeneと契約または価値のある権利協定(“CVRプロトコル”)に基づいて発行した
2018年5月7日、当社はAliquaと資産購入協定を完了し、Aliquaは再生医療製品を商業化する再生技術会社(以下、Aliqua APA)である。AliquaのAPAにはAliquaのBiological買収が含まれています
F-7
Biovanceのマーケティングと流通権を含む傷のケア事業®Interfylと® 二次医療機器や霧化®UltraMistと®治療システムですAliqua APAについて、会社は#ドルの現金対価格を支払いました
2018年10月5日,当社はCariCord Inc.(“CariCord”),CariCord Inc.(“CariCord”)はコロラド大学医学部(以下“コロラド大学”)およびコロラド大学医学部(以下“コロラド大学”)を代表して設立された家族臍帯血バンクであり,コロラド大学臍帯血バンク(“コロラド大学”)およびコロラド大学取締役会(法人団体)によって設立された。全体的に、CariCordを買収する費用の公正価値は#ドルです
新冠肺炎
2020年3月10日、世界保健機関は新冠肺炎疫病を大流行と発表した。ウイルスやその伝播を遅らせるための行動は、企業が経営·業務を行う地理的地域、企業のパートナーが経営·業務を展開している地域など、多くの国·地域の経済·金融市場に広範な悪影響を与え続けることが予想されている。同社は現在、現地衛生当局の提案に従い、そのチームメンバーや訪問者の曝露リスクを最小限に抑えている。しかし,この流行病の規模や範囲は不明であり,業務中断の持続時間や関連する財務影響を合理的に見積もることはできない。経営陣は新冠肺炎の潜在的影響を減らすために具体的な業務連続計画を実施しているが,会社の連続計画が必ず成功する保証はない。
疫病が始まって以来,会社は継続的に運営できるにもかかわらず,必要でない従業員やその役割を遠隔実行できる従業員に対する現地の健康アドバイスに応じて,必要に応じて在宅勤務政策を実施している。会社の業務のある環節(例えば胎盤組織処理、ある生物測定、転化研究と臍帯血貯蔵)は遠隔実行できないため、会社は強制体温検査、症状評価表、漸進式洗浄と公共表面消毒を含む制御とプロトコルを制定し、従業員のリスクを下げる。
経済活動の広範な低下とある医療施設の実際の使用制限により、同社の退行性疾患業務の純収入は確実に2021年の大流行により低下している。臨床試験については,同社は純粋に新冠肺炎のリスクにより募集を中止または延期していない。しかし、CYNK-001による急性骨髄性白血病治療を評価する臨床試験は2020年上半期にいくつかの遅延を経験し、ウェブサイトは彼らの安全方案を評価し、そして大量の新冠肺炎患者を経験したからである。AML実験の募集作業は継続しており,現在も行われている.そのため、2020年の間に、学生募集が遅れているにもかかわらず、同社の研究開発費は前年比で増加している。同社はまた新冠肺炎患者の中でCYNK-001を評価する臨床試験を開始し、これには追加の研究開発とプロジェクト管理資源が必要である。同社は、もし新冠肺炎の疫病が発生しなければ、その人力と資本資源を他の努力、例えばそのCyCART-19臨床開発計画に展開すると考えている。
2021年から2022年までの間に、新冠肺炎は腫瘍学臨床試験患者の応収率に実質的な負の影響がなかった。Celulityは2021年の間,強制体温検査と症状評価表の使用を継続し,2021年第3四半期からワクチンを接種していない従業員のための追加の安全協定を作成した。Celularityはまた連絡員を利用して従業員のためのワクチン接種の予約を手配するのを手伝った。
新冠肺炎や他の任意の健康流行病が会社の業績に与える影響の程度は将来の事態の発展に依存し,これらの事態の発展は非常に不確実性が高く,出現する可能性のある新冠肺炎の重症度に関する新たな情報や,新冠肺炎の抑制やその影響を抑制する行動なども予測できない。そのため、新冠肺炎は会社の業務、運営結果、財務状況と将来性に重大な不利な影響を与える可能性がある。
経営を続ける企業
会計基準更新(“ASU”)2014-15号によると、実体の持続的な経営能力に関する不確実性(サブ特集205-40)(“ASU 205-40”)が開示され、当社はいくつかの条件と事件が存在するかどうか(総合的に考慮)を評価しており、当社が総合財務諸表の発表日から1年以内に経営を継続する能力に大きな疑いを抱かせる。
新興の臨床段階生物技術会社として、Celulalityは企業発展に関連するある固有のリスクと不確定要素の影響を受ける。この点で,会社設立以来,経営層のほとんどの努力は,胎盤由来同種異体細胞に対する基礎科学研究,その現在と未来の細胞療法臨床計画を支援する臨床前研究,その細胞計画の臨床開発およびそのコア業務運営を支援する施設や販売,一般および行政費用(総称して“投資”)を含む研究開発に投資しており,これらはすべて会社の短期的な収益力を犠牲にしている。同社はこれまでこれらの投資に資金を提供してきた
F-8
そのバイオバンクや退行性疾患業務による有限収入と,公的·個人投資家への株式や債務証券の発行(これらの発行を総称して“外部資本”と呼ぶ)。これらの努力をしたにもかかわらず、経営陣は会社の研究開発や商業化が成功することを保証することはできず、会社の知的財産権が十分に保護される保証もない。これらの努力が成功しても、同社がいつ(あれば)相当な売上を生み出したり、企業の運営を維持したりして、外部資本に依存し続ける必要がないのかは定かではない。会社の株価の持続的な下落は、今後しばらくの間の営業権や長期資産の減少を招く可能性がある。
管理層は、添付の連結財務諸表発行日(“印刷日”)まで、ASU 205-40に基づいて、以下の不利な条件およびイベントの重要性を評価した
F-9
これらの不確実性は、同社の持続的な経営企業としての持続的な経営能力を大きく疑わせている。添付されている総合財務諸表は、当社が継続的に経営企業として経営していくことに基づいて作成され、当社は予測可能な未来に正常業務過程で資産および負債を清算および負担できることが予想される。したがって、添付の連結財務諸表は、これらの不確実性の結果に起因する可能性のあるいかなる調整も含まない。
陳述の基礎
会社の総合財務諸表は、米国公認の会計原則(“公認会計原則”)に基づいて作成されている。連結財務諸表には、会社間口座と取引後の完全子会社の口座が含まれている。本文書に掲載されている総合財務資料は、経営陣が示した期間の財務状況、経営業績及び現金流量を公平に陳述するために必要なすべての財務資料を反映している。
予算の使用
公認会計原則に基づいて会社の連結財務諸表を作成することは、経営陣に推定及び仮定を要求し、これらの推定及び仮定は、連結財務諸表の日付の資産及び負債の報告金額、又は有資産及び負債の開示及び報告期間内の収入及び費用の報告金額に影響を与える。これらの総合財務諸表に反映される重大な推定および仮定は、会社の名誉および無形減価評価、在庫評価、または対価格、短期債務、逓増借款金利の決定、研究および開発費用の計上、ならびに株式オプションおよび引受権証の推定に関する仮定を含むが、これらに限定されない。当社は歴史的経験、既知の傾向、その他その当時の状況に属すると考えられる合理的な特定の市場要素あるいはその他の関連要素から推定しています。継続的な基礎の上で、環境、事実、経験が変化した場合、管理層はその推定数を評価する。推定された変化は、それらが知られている期間に記録される。実際の結果はこれらの推定とは異なる可能性がある。
公正価値計量
当社のいくつかの資産及び負債は公認会計原則に基づいて公正価値に基づいて記帳します。公正価値は、計量日市場参加者間の秩序ある取引において、資産または負債が元金または最も有利な市場で負債を移転するために徴収または支払いされる交換価格として定義される。公正価値を計量するための推定技術は,観察可能な投入を最大限に利用し,観察不可能な投入を最大限に減少させなければならない。公正価値別に列挙された金融資産および負債は、公正価値レベルの以下の3つのレベルのうちの1つで分類および開示されなければならず、そのうちの最初の2つのレベルは可視とみなされ、最後のレベルは見えないとみなされる
現金と現金等価物
現金と現金等価物には、主に商業銀行口座に保有されている現金、通貨市場基金、元の満期日から3ヶ月未満の米国債が含まれる。当社は高い流動性と満期日の投資を持っていると考えています
F-10
買収の日から三ヶ月または三ヶ月以内に現金等価物とします。2022年12月31日と2021年12月31日にほとんどの現金と現金同等物は商業銀行口座や通貨市場基金に保管されている。
制限現金
2022年12月31日と2021年12月31日までその会社は$の信用状を維持している
在庫品
在庫はコストまたは市場(可変現純値)の中の低い者に列報し、コストは先進先出原則に従って確定する。FDAまたは他の規制機関が予備承認する前に、会社は発生した期間内に生産在庫に関連するコストを支出する。この製品が初歩的な監督管理の承認を得た後、同社はその製品に関連する在庫コストを資本化する。同社は引き続き臨床試験供給コストに関するコストを研究·開発費用としている。
会社は定期的に在庫レベルを分析し、時代遅れ、期限切れ、または過剰な在庫が存在するかどうかを決定する。いずれかの在庫(I)が販売前に満了する予定である場合、(Ii)コストベースがその可変純価値を超える場合、(Iii)内部販売予測によって決定された予想販売需要を超える場合、または(Iv)商業販売仕様に達していない場合、その在庫は、収入コストを計上することによって減記される。在庫コストが実現できるかどうかを決定するためには、経営陣は販売予測に基づいて将来の予想在庫需要を推定する必要がある。実際の市場状況が経営陣が予測しているほど有利でなければ、在庫を減記する必要があるかもしれない。当社の総合貸借対照表の当期部分の純額には、1年後に保留される予定の在庫が含まれています。
財産と設備
財産と設備はコストから減価償却累計と償却を差し引く
|
|
使用寿命を見込む |
家を建てる |
|
|
家具と固定装置 |
|
|
実験室装置 |
|
|
コンピュータ装置 |
|
|
ソフトウェア |
|
|
賃借権改善 |
|
変化が適切かどうかを決定するために、推定された使用寿命を定期的に評価する。保修と修理は発生時に費用を計上する。資産が廃棄または他の方法で処分された場合、これらの資産のコストおよび関連する減価償却または償却は、総合貸借対照表から除外され、それによって生じる任意の収益または損失は、処置中の総合経営報告書に計上される。まだ投入されていない資本資産のコストは建設中工事として資本化し、投入後に減価償却に計上する。
長期資産減価準備
長期資産には、財産、工場設備、経営的使用権資産、寿命を確定する無形資産が含まれる。保有·使用待ちの長期資産が,事件やビジネス環境の変化が発生して資産の帳簿価値が完全に回収できない可能性があることを示した場合には,回収可能テストを行う。当社がいつ減値審査を行うかを決定する際に考慮する要素は、予想に関連する業務パフォーマンスの顕著な不良、業界或いは経済傾向に重大なマイナス影響が出現すること、及び資産用途の重大な変化或いは計画変更を含む。長期資産グループの回収可能性を評価するために減値審査を行うと、当社は、長期資産グループの使用と最終処分による未割引キャッシュフローの予測をその帳簿価値と比較する。資産グループの使用予想による予想未割引将来のキャッシュフローがその帳簿金額よりも少ない場合、減価損失は運営損失で確認される。減価損失は,減価資産グループの帳票価値がその公正価値を超える部分に基づいており,これは割引キャッシュフローによって決定される.2022年第4四半期に確認された以下及び付記8に記載した退行性疾患報告単位に関連する商誉減値のため、著者らは長期資産に対して回復可能性テストを行い、このテストによって確認された追加減値はないと結論した。当社は2022年12月31日および2021年12月31日までの年間で、長期資産について何の減価損失も計上していません.
F-11
企業合併
企業合併の買収価格配分は、それぞれの公正価値に基づいて買収価格を識別可能な有形無形資産および負担する負債に分配するために、会計推定と判断を広く使用する必要がある。会計基準に従って編集された(“ASC”)805企業合併まず、当社は、買収された総資産の公正価値の実質的にすべてが識別可能な資産または同様の識別可能な資産のセットに集中しているかどうかを決定する。この敷居に達した場合、単一資産または1組の資産(例えば、適用される)は企業ではない。単一資産または類似資産のセットが閾値に達していない場合、1つのエンティティは、次に、投入と実質的なプロセスとがあるかどうかを評価しなければならない。
当社は買収会計法を用いて企業合併を計算します。この会計方法を用いて、(I)買収された確認可能資産(確認可能無形資産を含む)および仮定された負債は、一般に、買収日の公正価値に応じて計量および確認され、(Ii)買収された確認可能資産および仮説負債が識別可能な資産および仮定の公正純値を超える部分は、会計目的に応じて償却されないが、少なくとも毎年減価テストが行われることを商業権として確認すべきである。買収された知的財産権研究開発は公正な価値で確認され、最初に無期限の無形資産として記述され、買収された知的財産権研究開発に代替の将来用途があるかどうかにかかわらず。企業合併に関する取引コストは発生時に費用を計上する。
企業合併で買収された資産と負担する負債の公正価値を決定するには、経営陣が重大な判断と推定を使用する必要があり、特に無形資産については。特定の識別可能な資産を推定するための重要な推定は、推定方法の選択、将来の収入およびキャッシュフローの推定、予想される長期市場成長、将来の予想運営費用、資本コスト、および適切な割引率を含むが、これらに限定されない。経営陣の公正価値の推定は合理的と考えられる仮定に基づいているが,これらの仮定自体は不確実であるため,実際の結果は推定とは大きく異なる可能性がある.
買収日から1年遅れない計量期間内に、当社は買収資産及び負担した資産及び負債の帳簿価値に対して若干の調整を行い、営業権に応じた相殺を行うことができる。計量期間後,すべての調整が営業費用または収入として合併経営報告書に記録されている。
買収に関連するまたは対価があり、潜在的なマイルストーンと特許権使用料債務(付記12参照)を含み、買収日に買収会計方法に従って総合貸借対照表を計上し、公正価値を推定する。買収に関連するまたは価格のある公正価値は各報告期間に再計量され、公正価値の変化は合併経営報告書に記録されている。公正価値計量は市場参加者が観察できない重大な投入に基づいているため、公正価値システム中の第三級投入を代表する(付記4参照)。
資産買い入れ
当社は資産買収のコスト(取引コストを含む)に基づいて企業合併の資産買収とはみなされないことを計量し、確認した。資産買収では、知的財産権の研究開発を買収し、将来的に他の用途のないコストを買収日に研究·開発費に計上する。
現在行われている研究と開発
企業合併を通じて獲得した知的財産権研究開発の公正価値は関連研究と開発活動が完成或いは放棄する前に無期限無形資産として資本化する。関連研究·開発が完了すると、資産は確定された寿命資産に再分類され、その予想耐用年数内に償却される。
知的財産権研究開発無形資産の公正価値は通常収益法を用いて確定され、管理層はこれによって資産がその推定使用年限内に予想して発生した純現金流を予測する。純キャッシュフローは資産の完成段階、技術成功の可能性、完成の予想コスト、予想される市場競争及び資産ライフサイクルの評価を反映している。そして,キャッシュフローに関するリスク要因を反映した適切な割引率を適用することにより,純キャッシュフローを現在値に調整する.
無期限知的財産権の研究開発は償却する必要はないが、毎年減値テストを行い、減値指標があれば、より頻繁にテストを行う。同社は第4四半期に毎年その無期限知的財産権研究開発の減値状況をテストした。無限寿命知的財産権の研究開発に対して減値テストを行う時、当社はまず定性要素を評価して、事件或いは状況の存在がその公正価値がその帳簿価値よりもその帳簿価値よりも低い可能性があるかどうかを確定することができるかどうかを確定することができ、あるいは当社は数量化減値分析を行い、無限寿命知的財産権研究開発の公正価値を確定し、定性評価を行わないことができる。会社が考えている定性的要因には,重大な負の業界や経済傾向や資産用途の重大な変化や計画変化が含まれている。もし当社がまず品質要素を評価することを選択し、当社が無限寿命知的財産権研究開発の公正価値がその額面よりも少ない可能性が高いと考えた場合、当社はその後、無限寿命知的財産権研究開発の公正価値を決定する。いずれの方法でも、無限寿命知的財産権研究開発の公正価値がその帳簿金額より少ない場合、総合経営報告書で減値費用を確認する。.の間に
F-12
2022年と2021年12月31日までの年度その会社はできました
商誉
営業権とは、譲渡の価格に対する公正価値が企業合併で得られた有形と識別可能な無形資産純価の公正価値を超えることである。営業権は償却する必要はありませんが、毎年減値テストを行い、減値指標があれば、より頻繁にテストを行います。同社は通常毎年第4四半期にその営業権を減値テストを行っている。
同社は以下の項目を評価することでその運営を管理している
営業権の減価をテストする際に、当社は、まず定性的要素を評価して、イベントや状況の存在が報告単位の公正価値がその帳簿価値よりも帳簿価値よりも低い可能性があることを示すかどうかを決定することを選択することができ、あるいは当社は定性的評価を行わずに定量的減価分析を行うことができる。会社の定性的評価において考慮されるこのようなイベントまたは状況の例としては、法律またはビジネス環境の重大な不利な変化、不利な規制行動、または意外な競争が挙げられるが、これらに限定されない。当社がまず品質要因を評価することを選択した場合、当社はその報告単位の公正価値がその帳簿価値よりも少ない可能性が高いと考えている場合、当社は数量化減値テストを行う。定量化テストは、まず、報告単位の公正価値と報告単位の帳簿価値(営業権を含む)とを比較する。報告単位の公正価値が帳票金額を超えていれば,減値損失は確認されない.しかし、報告単位の公正価値がその帳簿価値よりも低い場合、当社は、帳簿価値が報告単位の公正価値を超える金額について減値費用を確認するが、報告単位に割り当てられた営業権総額を超えない。2022年12月31日までの年度内会社は営業権の減価#ドルを確認しました
法的責任を負うべきである
著者らはASC 815-40“派生ツールとヘッジ-実体自身権益契約”に記載されているガイドラインに基づいて、公開株式証とプライベート配給株式証に対して会計処理を行い、このガイドラインに基づいて、公開株式証とプライベート配給承認持分証は株式処理の基準を満たしておらず、必ず負債記録としなければならない。そのため、著者らは公開株式証と私募株式証をその公正価値によって負債に分類し、各報告期間に公開株式証と私募株式証を公正価値に調整する。この負債は、行使まで各貸借対照表の日に再計量されなければならないが、公正価値の任意の変動は、総合経営報告書において、他の(費用)収入の構成要素として確認される必要がある。私募株式証の最初とその後の推定値はBlack-Scholesオプション定価モデルを使用し、このモデルは3級公正価値測定標準と考えられている。公開株式証は各関連報告日の見積市場価格を基準に推定される。
GX業務との合併前(付記1及び3参照)において、当社は、その交換可能な優先株株式を購入する引受権証(付記13参照)を総合貸借対照表上の負債に分類し、当該等株式証は独立した金融商品であるため、当社が行使時に資産を移転する必要がある可能性がある。株式証負債はBシリーズ転換可能な優先株を購入する引受権証を含み、各株式証の発行当日に最初に公正価値で入金され、その後、報告日ごとに公正価値によって再計量される。株式証負債の公正価値変動は、連結業務報告書において他の(費用)収入の構成要素であることが確認された。優先株式証負債の公正価値は、負債が決算日に権益に再分類されるまで、総合経営報告書上で2021年7月16日決算日まで再計量される。
賃貸借証書
ASU番号2016-02によると、レンタル(テーマ842)(“ASU 2016-02”または“ASC 842”)により、当社はレンタル開始日にレンタルを分類します。手配開始時に、当社はその時の状況に基づいて、手配がリース契約であるか否か又はテナントを含むか否かを決定する。レンタル期間が1年を超える賃貸契約は、総合貸借対照表において、使用権資産(“ROU”)、賃貸負債、および長期賃貸負債(適用されるように)として確認される。当社には、当該等選択権を行使することが合理的に決定された場合に、レンタル期間内にリース契約を延長する継続選択権が含まれている。レンタル負債と対応するROUは、条項に基づいてレンタル支払いの現在価値に記録されています。レンタル契約に隠されている金利は一般的に確定しにくい。したがって、当社は、類似経済環境下で類似条項で担保方式で借金することによる金利である適切な逓増借款金利を採用しており、金額は賃貸支払いに相当する。レートや指数に依存しない可変支払いはリース負債に含まれず、発生したことが確認された。賃貸契約には残存価値保証も含まれておらず、制限や他の契約も含まれていない。支払いの初期直接コスト、受信された報酬やレンタル前金などの項目については、RUSを何らかの調整する必要がある可能性がある。もし重大な事件が起きたら
F-13
場合や他のイベントがレンタル契約年や他の資料が変更されたことを示す場合、当社は賃貸契約カテゴリを再評価し、再評価日までの改訂資料を用いてレンタル負債を再計量し、純資産収益率を調整します。
同社は移行ガイドラインが許可する“3つの一括”の実際の便宜策を選択し、リース識別、リース分類、初期直接コストに関する先の結論を再評価する要求を解消した。当社はまた、初期期限が12ヶ月以下のリースおよび当社が行使する任意の購入選択権をその総合貸借対照表に計上しない賃貸使用権資産と賃貸負債を合理的に決定する会計政策を採用している。
詳細は注11を参照されたい。
短期債務
ヨークビルと締結された前払い前払い契約に基づいて、私たちは公正価値オプションを選択して金融商品に計上した。公正価値の推定値は二項格子モデルを用いて決定した.債務の公正価値計量は市場では観察できない第三級投入と仮定を用いて決定された。公正価値によって計上された債務公正価値変動は、関連する利息支出を含み、付随する総合経営報告書の中に損益として列記し、そして公正価値変動項目の次のように全面的な損失を報告する。特定のツールの信用リスク変化に起因する債務公正価値総変動部分は、具体的な計量割引率仮定の定期的な変化(基本市場変化を含まない)によって決定され、付随する総合経営報告書と全面赤字報告書の中で全面収益(損失)の構成要素として示されている。短期債務の実際の決済は現在の見積もりと異なる可能性があり、これはヨークビルがいつを選択するか、金額を普通株に転換する時間、満期前に返済可能な現金、および私たちの普通株価格の変動に依存する。
収入確認
会社は変性疾患ビジネス(すなわちBiovance)から収入を得ています®Interfyl®)、バイオバンクサービス(すなわち、臨月妊娠後に臍帯および胎盤血液および組織を収集、処理および保存する)、ならびにライセンス、特許使用料および他の収入。
製品販売とレンタル
生物群®1種の脱細胞、脱水したヒト羊膜であり、完全に保存された天然上皮基底膜と完全な細胞外基質構造及びその生化学成分を有し、生物膜カバーとして使用し、損傷組織の修復を支持すると同時に細胞外基質を提供することを目的としている。Interfyl®1種の脱細胞の同種異体ヒト胎盤結合組織基質であり、天然の人体構造と生化学細胞外基質成分から構成され、手術要求と創傷看護に用いられ、損傷或いは不十分な胎盤結合組織の代替或いは補充とする。
製品·サービスの支配権が顧客に移転すると、同社は収入を確認し、金額は、これらの製品やサービスと交換するために、顧客から期待される対価格を反映している。この過程には,顧客との契約を決定し,契約中の履行義務を決定し,契約価格を決定し,契約価格を契約中の異なる履行義務に割り当てることと,履行義務を履行する際に収入を確認することがある.第三者を代表して徴収される販売税と他の税金は収入に含まれていない。
履行義務が単独または顧客がいつでも取得でき,契約で単独で決定した他の資源とともに顧客に利益を提供する場合,履行義務は契約中の他の義務とは異なると考えられる.会社が貨物又はサービスの制御権を顧客に譲渡すると、会社は履行義務が履行されたと考え、これは、顧客が貨物又はサービスの利益を利用して得ることができることを意味する。製品又はサービスの取引価格は、通常、顧客と締結された契約価格に基づいており、取引価格に可変対価格が含まれる場合には、当社は、期待値方法を用いて取引価格に含まれるべき可変対価格金額を推定したり、状況に応じて会社が獲得する権利があると予想される可能性のある金額を推定したりする。
同社は、数量に基づく割引、リベート、および即時報酬割引、および他の様々なインセンティブを提供し、これらは可変対価格モードで計算される。顧客が特定の数の製品を購入することで販売インセンティブを得る可能性がある場合、当社はこのようなインセンティブが実現されるかどうかを推定し、これらのインセンティブを基礎収入取引の同時期における収入の減少を確認すると確認する。当社は主に期待値手法を用いてインセンティブを推定しています。期待値方法によると、会社は同様の計画の歴史的経験を考慮し、顧客ごとに販売傾向を検討し、どのレベルのインセンティブが得られるかを推定する。
F-14
当社はその変性疾患製品に対して顧客に返品権利を提供しています。これまで同社の製品は返品が最も少なかったため、返品準備金は記録されていません。同社は製品が予想と規格に従って機能することを保証するために製品保証を提供します。お客様は単独で保証を購入することができます。これらの保証は単独の履行義務が発生します。
サービス.サービス
同社はバイオバンク収集と処理サービスおよびストレージサービスの収入を単独で確認した。加工と貯蔵サービスには,臍帯血,胎盤血,組織加工と貯蔵を提供し,個人用に提供することがある。加工と貯蔵費用の確認のための収入代表は顧客にバイオバンクを販売する。同社は加工成功時に加工費収入を確認し,一定期間契約貯蔵期間に比例して計算された貯蔵費を確認した。契約の貯蔵期間は通常
契約の取引価格を決定する際に、顧客の支払いが履行前に顕著に発生するか、または履行後に顕著に発生した場合には、調整を行い、重要な融資部分を生成する。すべての計画(年度、生涯、
考えてみると
同社は販売促進割引や他の様々な奨励を提供しており、これらは可変対価モードで計上されている。当社はこのようなインセンティブが実現するかどうかを予想し,これらのインセンティブを基本収入取引の同時期の収入の減少を確認した。当社は主に期待値手法を用いてインセンティブを推定しています。期待値方法によると、会社は同様の計画の歴史的経験を考慮し、顧客ごとに販売傾向を検討し、どのレベルのインセンティブが得られるかを推定する。
会社の処理およびストレージプロトコルには複数の履行義務が含まれているため、ASC 606顧客との契約収入から履行義務ごとの承諾サービスの推定相対独立販売価格に基づいて取引価格を割り当てることが要求される.当社はすでに調整された市場評価方法を選択して加工サービスと貯蔵サービスの独立販売価格を推定し、公表された価格表の価格はその市場の顧客がそのなどの商品或いはサービスに支払いたい価格であると判断した。会社はまた,すべての顧客が登録時に当時の価格表の価格で料金を徴収し,会社はそれぞれ加工と貯蔵の価格表の価格を説明していると考えている.
ライセンス、印税、その他
ライセンス契約に基づいて、当該会社は、関連する履行義務がある時点又は一定期間内に履行されているか否かを評価する。
SanuWave Health Inc.(“SanuWave”)とのライセンスプロトコルにより,SanuWave Health Inc.はそのMISTを構成するある資産を買収した®/UltraMist®上記業務(付記16参照)を除いて、当社は四半期許可料と販売している製品毎の固定使用料を受領しています。SanuWaveにBiovance特許権使用料免除,最高四半期許可料を提供した。同社は同期間に発生した実売上高に基づいて、四半期ごとに四半期許可料を確認している。未治癒材料違反により,SanuWaveとのライセンス契約は2021年第3四半期に終了した。
当社は、ある事件に基づくマイルストーン支払いを含む各手配の開始時に、マイルストーンが実現可能であると考えられているかどうかを評価し、可能な金額法を用いて取引価格に含まれる金額を推定する。大きな収入逆転が起こらない可能性が高い場合、関連するマイルストーン価値は取引価格に含まれるだろう。規制承認のような会社または被許可者の統制範囲内にない記念碑的支払いは、これらの承認を受ける前に実現可能であるとは考えられない。同社の評価の要因には、この評価における特定のマイルストーンを達成するために、科学、臨床、規制、商業、および他の克服しなければならないリスクが含まれている。重大な収入逆転が発生しない可能性があるかどうかを決定する際には,かなりの判断が必要である。その後の各報告期間が終了した時点で,会社は以下の条件を満たすすべてのマイルストーンの実現確率を再評価する
F-15
必要に応じて全体の取引価格の見積りを調整する.いずれの調整も累積追跡をもとに記録されており,調整期間中の収入や収益に影響を与える.一里塚やその他の可変対価格が特に会社が単一の業績義務を履行する努力や業績義務を履行する特定の結果に関連すれば、重大な収入逆転が発生しない可能性が高い場合、会社は通常、マイルストーン金額をすべてその業績義務に分配する。当社のライセンス契約のさらなる検討については、付記16を参照されたい。
変性疾患製品の直接販売は、製品販売およびレンタルに含まれているが、会社の流通パートナーネットワークを介した販売は、ライセンス、特許使用料、および他の収入に含まれている。付記16に記載されているいくつかの流通プロトコルについては、当社はASC 606-10-55-83における実際の便宜を利用して、顧客の対価格が受信した価値と直接一致すれば、エンティティは領収書を発行する権利のある金額の収入を確認することができるそのため、会社はこれらの契約のために領収書を発行する際(関連調達注文を受けた後)に収入を確認する。
収入コスト
収入コストには、同社の既存の2つの商業業務部門であるバイオバンクと変性疾患に関連する労働力、材料、管理費用が含まれている。バイオバンクコストには,新貯蔵材料の貯蔵·輸送キットのコスト,臍帯血や他の貯蔵先のタンクや施設管理費用が含まれる。退行性疾患コストは胎盤の購入、胎盤材料の合格及び胎盤組織を適切な対路製品に加工することと関連するコストを含む。変性疾患部門のコストには,Biovance,Biovance 3 L,Interfyl製品ラインの生産に関する労働力と管理費用がある。2022年12月31日までの年度では,流通パートナーとの変性疾患販売が実現できなかったため,同社は生産量向上のために大量のコストを発生させた。これらのコストは、収入コストに含まれ、ライセンス、特許使用料、および他の収入に含まれる流通パートナーの収入よりも収入コストが高くなる。
研究開発コスト
同社はすでに商業会社、研究者、大学、その他の機関と様々な研究開発やその他の協定を締結し、商品やサービスを提供している。これらのプロトコルは通常キャンセル可能であり,関連コストは発生時に研究や開発費用として記録される.研究開発費には,賃金,従業員福祉,下請け業者,施設関連費用,減価償却·償却,株式ベースの補償,第三者許可費,実験室用品,および招聘による発見,臨床前·臨床開発活動と臨床試験,臨床試験材料を製造する外部サプライヤーの外部コスト,その他のコストがある。同社は想定されている持続研究開発コストの計上項目を記録している。計算すべき負債の十分性を評価する際に、会社は、イベントの段階または完了状況、受信された請求書および契約コストを含む研究または臨床試験の進行状況を分析する。いずれの報告期間終了時の当計残高を決定する際には,重大な判断と見積もりを行う。実際の結果は会社の見積もりとは大きく違うかもしれません。将来受信した研究·開発活動のための貨物またはサービスの払戻不可能な前払いは前払い費用として記録される。このような前払い料金は、貨物が交付または関連サービスが完了したか、または貨物が交付または提供されるサービスがもはや予期されていない場合には、費用として確認される。
ライセンス契約によると、プリペイド、マイルストーン支払い、年間維持費は、これらの費用が発生する間に支出される。
広告とマーケティングコスト
広告とマーケティング費用は発生時に費用を計上する。広告とマーケティング費用は販売、一般、行政費用に含まれ、費用は#ドルです
政府補助金
その会社は時々政府の研究援助を受けるかもしれない。これらの手配により、政府機関が関連マイルストーンの実現を確認した場合、会社は付与された贈与を研究·開発費の減少に確認する。“会社”ができた
特許費用
特許出願の提出及び起訴に関するすべての特許に関する費用は、支出回収状況が不確定であることによる費用に基づいて計上される。発生した金額は販売、一般、そして行政費用に分類される。
F-16
株に基づく報酬
当社は、付与日の公正価値に基づいて従業員及び取締役のすべての株式奨励を計量付与し、必要なサービス期間内に当該等の奨励の補償費用を確認し、当該サービス期間は通常、相応の報酬の帰属期間である。同社は通常、サービスベースの帰属条件でのみ株式ベースの報酬を発行し、直線的な方法を用いてこれらの報酬の費用を記録する。
会社の取締役会はまた業績に基づく株式オプションを承認して付与することができる。業績に基づく株式オプションは業績期間中に特定の目標を達成した上で稼いだものです。当社が業績条件を達成する可能性があると考えられると、関連帰属期間中に業績に基づく報酬の費用を確認する。当社は報告期間ごとに業績奨励の帰属可能性を再評価し、累積基礎に基づいて費用を調整する。
各サービスと業績に基づく株式オプション付与の公正価値は、付与された日にブラック·スコアーズオプション定価モデルを用いて推定される。同社は歴史的に民間会社であり、その株が会社の歴史と隠れた変動率情報に特定されていない。そこで、上場同業者の履歴変動率に基づいて予想株価変動率を推定し、自身の取引株価変動性に関する十分な履歴データを持つまで継続する予定である。従業員に付与される会社株式オプションの期待期限は、“一般”オプション資格に適合する報酬を得るために“簡略化”方法で決定される。非従業員コンサルタントに付与される株式オプションの期待期間は、オプションを付与する契約期間又は関連協定に基づいて推定される期限に等しいそれは.無リスク金利は、奨励付与時に発効した米国債収益率曲線を参考にして決定され、期間は奨励の予想期限にほぼ等しい。期待配当収益率は
2021年9月、当社はその前身総裁に市場化帰属条件を有するオプションを付与した。同社はモンテカルロモデルを使用して市場の奨励に基づく公正価値を計算した。同様に2021年には、当社は制限株式単位(“RSU”)を付与し、その公正価値は付与日の株価に応じて決定される(付記14参照)。
同社がその総合経営報告書において株式による補償費用を分類する方式は,受賞者の賃金コストを分類したり,受賞者のサービス支払いを分類したりする方式と同様である。当社は没収発生時に会計処理を行うことを選択したが、サービスや履行条件を満たしていないため没収された賠償金以前に確認された補償コストは没収期間中に反転するのが一般的である。
総合収益(赤字)
総合収益(赤字)とは、米国公認会計基準に基づいて全面収益(赤字)を計上するが、純収益(赤字)に含まれない収入、費用、収益、損失であり、これらの金額は累積された他の全面収益(赤字)の調整として直接記録されているからである。当社の他の全面収益(損失)の唯一の構成要素は、公正価値オプションによって計上された特定のツールの信用リスクの変化に起因する負債公正価値の総変動部分を含む。2022年12月31日までの年度内会社が記録したツール固有の信用リスク収入は$
所得税
当社は、連結財務諸表または当社納税申告書で確認された事件の将来の税務結果を予想する繰延税金資産と負債の確認を要求する貸借対照法を用いて所得税を計算する。繰延税項資産及び負債は、財務諸表と資産及び負債の税ベースとの差額に基づいて、予想差額を用いて返送される年度の現行税率を決定する。繰延税金資産と負債の変動を所得税に計上する準備。当社は、その繰延税金資産が将来の課税収入から回収される可能性を評価し、既存の証拠の重みに基づいて、繰延税金資産のすべてまたは一部が現金化できない可能性があると考え、所得税費用を計上することで推定値を設定している。予想未来の課税オーバー額及び慎重かつ実行可能な税務計画策略を考慮して、繰延税金資産を回収する潜在力を評価する。
会社は連結財務諸表で確認された所得税の不確実性を会計処理し、2ステップ法を用いて確認すべき税収割引額を決定する。まず、税収頭寸を評価して、この頭寸の技術的価値に基づいて頭寸を維持する可能性を決定しなければならない。税務状況がより継続する可能性があると考えられる場合、税務状況は、連結財務諸表で確認された利益金額を決定するために評価される。確認可能な利益額は
F-17
沈降量税務機関と付き合う。所得税の支出には、税金優遇の影響が確認されていないこと、および関連する利息および罰金が含まれる(付記18参照)。
1株当たり純収益
普通株1株当たり基本純収益(損失)の計算方法は,純収益(損失)を期間ごとに発行された普通株の加重平均株式数で割る。普通株1株当たりの純利益(損失)には、転換可能債券、株式オプション、制限株式単位、株式承認証などの証券の潜在的な行使や転換による影響(あれば)が含まれ、これは普通株の増分株を発行することにつながる。しかし、潜在的な普通株の影響が逆希釈されている場合、それらは排除される。1株当たり純利益(損失)を希釈する場合,普通株の加重平均株数は1株当たり基本純収益(損失)と同じであるが,これは純損失が存在する場合,希釈証券は計算に含まれず,影響は逆希釈であるためである。2022年12月31日までの年度今年度中に発行された株式の償却加重平均で1株当たりの純利益を計算し、当社は純収益状況にあり、在庫株方法で計算した
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
分子: |
|
|
|
|
|
|
||
純収益(赤字) |
|
$ |
|
|
$ |
( |
) |
|
分母: |
|
|
|
|
|
|
||
加重平均流通株、基本株 |
|
|
|
|
|
|
||
加重平均希釈性株式オプション |
|
|
|
|
|
— |
|
|
加重平均制限株式単位 |
|
|
|
|
|
— |
|
|
加重平均流通株、希釈した後 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
純利益(損失)、基本 |
|
$ |
|
|
$ |
( |
) |
|
薄くして純利益を出す |
|
$ |
|
|
$ |
( |
) |
|
2段階法を使用する前に、以下の希釈可能な証券は、逆希釈されるので、発行された普通株の希釈加重平均株式の計算から除外されている
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
株式オプション |
|
|
|
|
|
|
||
制限株式単位 |
|
|
|
|
|
|
||
株式承認証 |
|
|
|
|
|
|
||
転換債 |
|
|
|
|
|
— |
|
|
|
|
|
|
|
|
|
||
市場情報を細分化する
経営部門は企業の構成要素として定義され、その独立した離散財務情報は、業績を評価する際にどのように資源を割り当てるかを決定するために、首席運営意思決定者または意思決定グループによって評価することができる。同社は以下の項目を評価することでその運営を管理している
信用リスクと重要な顧客の集中度
会社を集中的な信用リスクに直面させる可能性のある金融商品には、主に現金、現金等価物、および限定的な現金が含まれる。同社は通常、経営陣が高い信用品質を持つ金融機関の各種運営口座に残高を保持しており、金額は連邦保険の限度額を超える可能性があると考えられている。同社は現金や現金等価物や制限された現金に関するいかなる損失も経験しておらず,商業銀行関係に関連する正常信用リスク以外の異常な信用リスクの影響を受けているとも考えられない。
当社は退行性疾患製品販売やバイオバンキングサービスに関連する売掛金の信用リスクに直面している。すべての貿易売掛金はアメリカで行われた製品販売とサービスの結果です。2022年12月31日現在、会社の一顧客(顧客A)を含む
F-18
毛収入売掛金。2022年12月31日までの年間で、会社の2人の顧客が
2017年11月、FDAは指導意見を提供し、ヒト細胞と組織製品(“HCT/P”)の最新の規制枠組みを構築した。同社のInterfyl製品は適用ガイドラインで概説された最小操作と相応の使用基準に符合し、HCT/P製品としてFDAの正式な称号を獲得した。したがって、FDAが2021年5月31日にその法執行自由裁量権を終了した場合、同社はInterfyl製品の販売を停止しなかった。しかし,医療保険·医療補助サービスセンター(CMS)は顧客AからのInterfylクレームを拒否し始めたそれは.当社は,CMSはInterfyl製品とその競争相手の製品を区別していないと考えている。会社と顧客AがCMSと協力して却下されたクレームを解決し続ける間、予約$
新興成長型会社
2012年JumpStart Our Business Startups Act(“JOBS Act”)第102(B)(1)条新興成長型企業が民間企業(すなわち、改正された1933年の証券法、登録声明の発効が宣言されていない、または1934年に改正された証券取引法に基づいて登録されていない証券)が、新たなまたは改正された財務会計基準の遵守を要求されるまで、新しいまたは改正された財務会計基準を遵守することが免除される。雇用法案では、新興成長型企業は、延長された移行期間からの脱退を選択し、非新興成長型会社に適用される要求を遵守することができるが、どのような選択脱退も撤回できないと規定されている。当社は、移行期間を延長することを選択しないこと、すなわち、1つの基準が発表または改訂されることを選択しており、この基準が上場企業または民間会社に対して異なる適用日がある場合、当社は新興成長型会社として、民間会社が新しい基準または改訂基準を採用する際に新しい基準または改訂された基準を採用することができる。
これにより、当社の財務諸表を、新興成長型会社でも新興成長型会社でもない別の上場企業と比較し、後者は使用する会計基準の潜在的な違いにより、延長された過渡期を使用しないことを選択する可能性がある。
再分類する
ある前期金額は本年度に該当する列報方式で再分類された。
最近採用された会計公告
開ける
同社は2022年度第1四半期に改正遡及移行法を採用し,ASU 2016−02年度を採用しており,比較期間は述べていない。当社は、履歴リース分類の継続を可能にすることを含む、新ガイドライン内の移行指針で許容される一括実際の便宜策を選択している。ASU 2016-02を採用した会社連結財務諸表への影響のさらなる情報については、付記11を参照されたい。
採用後、同社は純収益資産と賃貸負債#ドルを記録した
2021年5月、FASBはASU 2021-04を発表した1株当たりの収益(テーマ260)、債務修正と返済(主題470-50)、報酬--株式報酬(トピック718)、およびデリバティブとヘッジ−実体自己資本の契約(小テーマ815-40):発行者による独立持分分類書面コールオプションのいくつかの修正または交換の会計処理(“ASU 2021-04”)。ASU 2021−04は、修正または交換後も持分分類が元のツールとして新しいツールを交換するように維持される発行者が、修正または条件の修正または独立持分分類書面コールオプション(すなわち、権利証)の交換をどのように考慮すべきかに関するガイドラインを提供する。発行者は測るべきだ
F-19
その前に次に、4種類の取引および各種類の取引の対応する会計処理(株式発行、債務発行、債務修正、および株式発行および債務発行または修正とは無関係な修正)を含む確認モードを採用する。会社(The Company)
2021年11月、財務会計基準委員会(“FASB”)は、政府の贈与や寄付に関する開示要求を規定したASU 2021-10、“政府援助(テーマ832)”を発表した。ASUは、取引の性質および計算取引のための関連会計政策、財務諸表上の個別項目に対する政府の援助の影響(金額を含む)および引受支払いおよびまたは事項を含む取引の重要な条項および条件の開示を要求する。このASUは2021年12月15日以降の会計年度に有効である。会社(The Company)
FASBは2020年8月、ASU 2020-06(テーマ別470-20)を発表した債務--転換やその他のオプションを持つ債務負債および権益特徴を有するいくつかの金融商品にGAAPを適用する複雑さを解決するために(“ASU 2020−06”)。ASU 2020−06は、変換可能ツールに関するガイドおよびエンティティ自己資本契約の派生範囲例外の修正を含み、サブトピック470~20のいくつかの分離モードを削除することによって、変換特性または現金変換特徴から利益を得る変換可能ツールを含む会計処理を簡略化する。さらに、ASU 2020−06は、変換可能ツールの希釈1株当たり収益を計算する際に“変換する場合”の方法を使用することをエンティティに要求する。ASU 2020-06は、2023年12月15日以降の会計年度(会社は2024年度)に有効であり、早期採用を可能にする年度内の移行期間を含む。会社は駆け出しだ
最近発表された会計公告
2016年6月、FASBはASU 2016-13を発表した金融商品--信用損失(“ASU 2016-13”)は、金融資産減価確認の会計処理を変更した。新しいガイドラインの下で、いくつかのタイプの金融商品の信用損失は予想損失に基づいて推定される。ASU 2016-13年度は、債務証券の売却と、発生以来の信用悪化による購入金融資産の減価モデルも修正された。ASU 2016−13年は2022年12月15日以降の年次期間(会社の2023年度)およびこれらの期間内の移行期間が発効し、早期採用が許可されている。会社(The Company)
二零二一年七月十六日、当社は合併協議により先に発表された合併を完了し、GX、第一合併付属会社、第二合併付属会社及びLegacy Celularityの間で完成した(付記1参照)。
合併合意の条項によると、GXとLegacy Celulity間の業務合併は、(A)第1合併子会社とLegacy Celulityを合併することにより、Legacy CelulityはGXの完全子会社として存続(“存続法団”(“初合併”)(“初合併”)および(B)1回目の合併直後に、1回目の合併と同じ全体取引の一部として第2合併付属会社と合併し、第2合併付属会社は第2合併の存続実体である。最終的にLegacy CelulityはGXの完全資本直接付属会社となる(“第二次合併”、第一次合併、“合併”、および合併合意に記載されている他の取引と共に“業務合併”と呼ぶ)。締め切り,会社名はGX Acquisition Corp.からCelularity Inc.に変更される.
合併発効時期(“発効時間”)直前に、発行および発行されたLegacy Celularity優先株(“Legacy Celularity優先株”)は、複数のLegacy Celularity普通株に自動的に変換され、額面は$
F-20
米国で一般的に受け入れられている会計原則によると、企業合併は逆資本再編とみなされている。このような会計方法によると、GXは財務報告書で“買収された”とされている会社である。この決定は,主に既存のLegacy Celularity株主(合併後の会社の投票権の相対的な多数を占める),Legacy Celularityの買収前の業務(Celularityの唯一の継続業務を含む),Legacy Celularityによって任命されたCelularity取締役会の多数の株式,Legacy Celularityの上位管理層(Celularityの上位管理職の多数を含む)に基づいている。したがって,会計目的については,合併後の実体の財務諸表はLegacy Celulality財務諸表の継続であり,業務合併はLegacy Celulityの等価物とみなされ,GXの純資産に株式を発行し,資本再編を伴う。当社はGXの純資産を歴史的コストで計上している
この取引の純収益は合計#ドルです
合併合意の条項により,Legacy Celularityの既存株主はその権益をA類普通株に交換する
業務合併を完了した後、Legacy Celularityは持分証を認めて株式分類を行う資格がある。したがって,Legacy Celularity引受権証の取引日公正価値は$となる
F-21
業務合併の直後に現れた
2021年7月パイプ融資(私募)
Palantir Technologies Inc.との配置。
Palantirは,GXが2021年5月5日にPalantirと締結した引受契約により購入した
以下の表は、同社が公正価値に応じて恒常的に計量する金融資産と負債の情報を示し、このような公正価値を決定するための公正価値レベルを示す
|
|
2022年12月31日までの公正価値計測 |
|
|||||||||||||
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
|
合計する |
|
||||
資産: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
現金等価物--通貨市場基金 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
転換可能な受取手形 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
||
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
負債: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
買収に関係しているか,あるいは対価格債務がある |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
||
株の掛け値があるか |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
||
短期債務であるヨークビルは |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
||
株式証責任の承認-2022年5月管路承認株式証 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
||
株式証明書責任-保険者が株式証を承認する |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
||
株式証明書法的責任-株式証の公開承認 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
|||
|
|
2021年12月31日までの公正価値計測 |
|
|||||||||||||
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
|
合計する |
|
||||
資産: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
現金等価物--通貨市場基金 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
転換可能な受取手形 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
||
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
負債: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
買収に関係しているか,あるいは対価格債務がある |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
||
株式証明書責任-保険者が株式証を承認する |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
||
株式証明書法的責任-株式証の公開承認 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
|||
.の間に2022年12月31日までおよび2002年12月31日までの年度
同社の現金等価物は通貨市場基金で構成されている。貨幣市場基金の評価は証券活性市場で観察できるような投入を使用し、これは公正な価値レベル中の一級評価基準を代表する。売掛金、売掛金、繰延収入及びその他の流動負債の帳簿価値は短期的な性質であるため、付随する総合財務諸表における帳簿価値と公正価値とが一致する。
F-22
価格の推定値があります
あるいは対価債務のある公正価値計量は第三級投入を採用し、確率加重収入法に基づいている。この計測は,観察できない投入に基づいているが,市場活動が少ないか,市場活動がまったくないかは,会社自身の仮定に基づいている.
次の表は、以下の日までの第3レベル投入を用いて日常的な基礎で計量された、または対価格債務のある入金を示している2022年12月31日および2021年12月31日:
|
|
締め切りの残高 |
|
|
ネットワークがあります |
|
|
購入して、 |
|
|
公正価値 |
|
|
締め切りの残高 |
|
|||||
負債: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
買収に関係しているか,あるいは対価格債務がある |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
|
||
|
|
締め切りの残高 |
|
|
ネットワークがあります |
|
|
購入して、 |
|
|
公正価値 |
|
|
締め切りの残高 |
|
|||||
負債: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
買収に関係しているか,あるいは対価格債務がある |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
|
||
当社は、報告日時部分毎に第三者推定結果に基づいて、特定のイベントを実現する可能性、割引率、支払い請求及びトリガ支払いマイルストーンの条件がそれまでの時間帯を満たすことを含む様々な仮説に基づいた各種仮説に基づく割引キャッシュフロー分析に基づいて、潜在的な将来のマイルストーンおよび金銭負債を稼ぐ公正価値を推定する。これらの具体的なイベントの実際の発生状況によると、価格の実際の決済は現在の推定数とは異なる可能性がある。
報告日ごとに、当社は価格負債を公正価値を推定することを再評価し、公正価値変動を当社の総合経営報告書の収入または支出と記す。または対価格債務の公正価値の変化は、割引期間および金利の変化、収入推定の時間および金額の変化、および様々なまたは対価格債務を実現する可能性のある確率仮定の変化に起因する可能性がある。会社は2022年12月31日と2021年12月31日までに、すべてまたは対価格を総合貸借対照表の長期負債に分類している。価格に関するより多くの情報は、付記12を参照して、“引受金及び又は有事項”を参照されたい。
権証責任の評価
2022年12月31日の権証負債は、A類普通株を購入する権利証の公正価値からなる。業務合併時に想定した私募株式権証(“保権証”)および二零二二年五月の管状株式承認証(付記13参照)は,それぞれの締め切りにBlack−Scholesオプション定価モデルにより公開価値を記録し,このモデルでは,(I)標的資産価値,(Ii)運用価格,(Iii)無リスク金利,(Iv)標的資産の変動性,(V)標的資産の配当率および(Vi)満期日を含む資料を用いた。ブラック·スコアーズオプション定価モデルが保証権証と2022年5月の管理権証の公正価値を決定する際に使用する主な観測不可能な入力はA類普通株の予想変動率である。合併前、Legacy Celularityの歴史上は個人会社であり、会社の特定の歴史と隠れた変動率情報が不足していた。そのため、それは上場同業者会社の歴史変動率に基づいてその予想される株価変動性を推定する。権利証のブラック·スコルスオプション定価モデルの投入は、公正な価値を反映するために報告期間ごとに更新される。業務合併時に負担する公開株式証(“公開株式証公開”)は締め切り日にこの等承認株式証の市場価格に基づいて公正価値を計算する。その後の各報告期間内に、公共株式証明書は期末終値によって市価に計算される。
2022年と2021年12月31日現在、株式証負債の公正価値は#ドルである
F-23
次の表は会社の株式証負債の総公正価値の前転を提供し、その公正価値は第三級投入を使用して確定された
2020年12月31日の残高 |
|
$ |
|
|
公正価値変動から確認された収益 |
|
|
( |
) |
締め切り時に負う引受権証責任(保険者株式承認証) |
|
|
|
|
成約日に負う持分証法的責任(株式公開承認証) |
|
|
|
|
伝統的なCelulital権証を株式に再分類する |
|
|
( |
) |
2021年12月31日現在の残高 |
|
$ |
|
|
|
|
|
|
|
2021年12月31日現在の残高 |
|
$ |
|
|
2022年5月管権証発行 |
|
|
|
|
公正価値変動から確認された収益 |
|
|
( |
) |
2022年12月31日現在の残高 |
|
$ |
|
|
株式承認証の公正価値は$である
保険者の株式証に対する重要な意見は以下の通りである
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
普通株価格 |
|
$ |
|
|
$ |
|
||
行権価格 |
|
$ |
|
|
$ |
|
||
配当率 |
|
|
% |
|
|
% |
||
期限(年) |
|
|
|
|
|
|
||
無リスク金利 |
|
|
% |
|
|
% |
||
波動率 |
|
|
% |
|
|
% |
||
2022年5月のパイプ株式証の重要な投入は以下の通りである
|
|
十二月三十一日 |
|
|
普通株価格 |
|
$ |
|
|
行権価格 |
|
$ |
|
|
配当率 |
|
|
% |
|
期限(年) |
|
|
|
|
無リスク金利 |
|
|
% |
|
波動率 |
|
|
% |
|
同社は発行されたLegacy Celularity重みを格子モデルを用いて評価しているが,行使価格は株価の関数であるためである。各モデルの応用において、公正価値計量に影響を与える推定と仮定は、Legacy Celularity Bシリーズ転換可能な優先株関連株の1株当たりの公正価値、無リスク金利、行使日を含み、投資家が行使を要求された時間と予想脱退日のうちの1つを考慮する。長期契約モデルの中で優先持分公証価値に影響を与える最も重要な仮定はLegacy Celularityの転換可能な優先株の各再計量日における公正価値である。同社は,関連優先株の1株当たり公正価値を決定する際に,Legacy Celularityが最近販売している転換可能優先株,第三者推定値から得られた結果,その他関連と考えられる要因を考慮した。
ドラガサックに発行された引受権証の公正価値は$
|
|
|
七月十六日、 |
|
|
普通株主公正価値 |
|
$ |
|
||
行権価格a |
|
$ |
|
|
|
期限(年) |
|
|
|
|
|
無リスク金利 |
|
|
|
% |
|
波動率 |
|
|
|
% |
|
(a)
F-24
1つはLegacy Celularity普通株式は初公募で一般向けに販売されている。2020年3月16日の改訂後、株式承認証は、(A)2025年3月16日、(B)Legacy Celularityの初公開発売完了、(C)制御権変更完了、及び(D)戦略取引が完了し、この取引に基づいて、Legacy Celularityの株主が既存のLegacy Celularity株式を全国証券取引所に上場する株式の株式と交換することができる。
Legacy CelularityシリーズB優先株に関する発行の権利証の公正価値は$
|
|
|
七月十六日、 |
|
|
普通株主公正価値 |
|
$ |
|
||
行権価格b |
|
$ |
|
|
|
用語.用語 |
|
|
|
|
|
無リスク金利 |
|
|
|
% |
|
波動率 |
|
|
|
% |
|
(b)
変換可能な受取手形の価値評価
受取変換可能チケットは,2020年にUltraMist/Mist業務を販売した際に受信した.2021年1月1日以降の任意の時間に、会社は、変換可能な受取手形項目の下での未償還金額(計算すべき利息を含む)を、所定の金利でSanuWave普通株に変換することができることを自ら決定することができる。転換可能な本チケットは2021年8月6日までに支払わなければなりませんが、2022年12月31日と2021年12月31日までは全額支払われていません。この手形の公正価値は信用違約モデルを採用した債券推定値に基づく。同社は手形違約,償還,転換の結果,確率重み付けモデルに基づく第3次投入を利用している。この計測は,観察できない投入に基づいているが,市場活動が少ないか,市場活動がまったくないかは,会社自身の仮定に基づいている.
変換可能チケット推定モデルの重要な投入は以下のとおりである
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
額面.額面 |
|
$ |
|
|
$ |
|
||
額面利率 |
|
|
|
|
||||
株価.株価 |
|
$ |
|
|
$ |
|
||
期限(年) |
|
|
|
|
||||
無リスク金利 |
|
|
% |
|
|
% |
||
波動率 |
|
適用されない |
|
|
適用されない |
|
||
株の評価があるかどうか
2022年12月31日の株式対価格負債には、2021年12月31日までの年度内に署名された和解協定(付記12参照)に基づいて、将来的にCariCord参加株主にA類普通株の公正価値を発行する可能性がある。または株式対価格債務の公正価値計量は、第3レベル投入を使用して決定され、確率に基づいて予測リターン方法(“PWERM”)を重み付けする。この測定は主に観察できない投入に基づいているが、会社自身の仮定によると、市場活動は少ないか、市場活動の支持が全くない。2021年12月31日現在、適用される調達目標は実現不可能である。
次の表は次の日までの第三級投入を用いて経常的な基礎に基づいて計量された株式対価格債務の残高を示している2022年12月31日および2021年12月31日:
|
|
締め切りの残高 |
|
|
ネットワークがあります |
|
|
購入して、 |
|
|
公正価値 |
|
|
締め切りの残高 |
|
|||||
負債: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
株の掛け値があるか |
|
$ |
- |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
||
将来のA類普通株発行の負債の公正価値は、企業が報告日ごとにPWERMを用いて各種投入と仮定に基づいて推定したものであり、会社の普通株価格、割引率、
F-25
指定された未来の作戦目標を実現する確率。または株式対価格の実際の決済は、これらの指定された目標に基づく実際の実現状況および会社の普通株価格変動の現在の推定とは異なる可能性がある。
報告日ごとに、当社は株式対価格債務を公正価値を推定し、公正価値変動を収入または費用として会社の総合経営報告書に記録する。あるいは株式対価格債務の公正価値の変化は,割引率の変化,会社普通株価格の変化,特定の運営目標を実現する可能性に関する確率仮定の変化による可能性がある.2022年12月31日現在、会社はすべてまたは株式対価格を総合貸借対照表の流動負債に分類している。株式対価格に関するより多くの情報は、付記12を参照して、“引受金及び又は有事項”を参照されたい。
短期債務推定値であるヨークビルは
当社は、2022年9月15日にヨークビルと調印された金融商品を計上するために公正価値オプションを選択しました(付記10参照)。公正価値の推定値は二項格子モデルを用いて決定した.債務の公正価値計量は市場では観察できない第三級投入と仮定を用いて決定された。公正価値によって計上された債務公允価値変動は、関連する計算すべき利息支出を含み、付随する総合経営報告書と全面収益(損失)の中で公正価値変動列に従って損益として示される。特定ツールの信用リスク変化に起因する債務公正価値総変動部分は、具体的な計量割引率仮定の定期的な変化(基本市場変化を含まない)によって決定され、付随する総合経営報告書と全面収益(損失)表に全面収益(損失)の構成要素として示されている。短期債務の実際の決済は現在の見積もりと異なる可能性があり、これはヨークビルがいつを選択するか、金額を普通株に転換する時間、会社が満期前に返済可能な現金、および会社の普通株価格の変動に依存する。
次の表はヨークビル債務の入金状況を示しており、この債務は2022年9月15日までの初期推定値を使用した日と現在のものである2022年12月31日:
負債: |
|
|
|
|
2022年9月15日現在の残高 |
|
$ |
|
|
債務を普通株に転換する |
|
|
( |
) |
公正価値調整通過 |
|
|
|
|
累計で公正価値調整を行う |
|
|
( |
) |
2022年12月31日現在の残高 |
|
$ |
|
|
ヨークビル短期債務推定モデルの重要な投入は以下の通りである
|
|
十二月三十一日 |
|
|
普通株価格 |
|
$ |
|
|
信用利回りが悪い |
|
|
% |
|
配当率 |
|
|
% |
|
期限(年) |
|
|
|
|
無リスク金利 |
|
|
% |
|
波動率 |
|
|
% |
|
割引収益率 |
|
|
% |
|
同社の主な在庫種別は以下の通り
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
原料.原料 |
|
$ |
|
|
$ |
|
||
進行中の仕事 |
|
|
|
|
|
|
||
完成品 |
|
|
|
|
|
|
||
在庫、毛数 |
|
|
|
|
|
|
||
差し引く:在庫備蓄 |
|
|
( |
) |
|
|
( |
) |
在庫、純額 |
|
$ |
|
|
$ |
|
||
貸借対照表分類: |
|
|
|
|
|
|
||
在庫品 |
|
|
|
|
|
|
||
在庫は、当期分の純額を差し引く |
|
|
|
|
|
|
||
|
|
$ |
|
|
$ |
|
||
F-26
在庫は、当期分を差し引くと、資産負債表ごとに1年後も現物があると予想される在庫が含まれています。
前払い料金と他の流動資産は、
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
臨床費用を前払いする |
|
$ |
|
|
$ |
|
||
保険料を前払いする |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
|
|
$ |
|
|
$ |
|
||
財産と設備、純額は:
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
家を建てる(1) |
|
$ |
— |
|
|
$ |
|
|
賃借権改善(2) |
|
|
|
|
|
|
||
実験室と生産設備 |
|
|
|
|
|
|
||
機械、設備及び固定装置 |
|
|
|
|
|
|
||
建設中の工事 |
|
|
|
|
|
|
||
財産と設備 |
|
|
|
|
|
|
||
減算:減価償却累計と償却(3) |
|
|
( |
) |
|
|
( |
) |
財産と設備、純額 |
|
$ |
|
|
$ |
|
||
減価償却費用は$
当社は毎年第4四半期に私たちのすべての報告単位に対して年間営業権減値テストを行い、イベントや状況が減少する可能性があることを示す場合は、より頻繁にテストを行います。2022年12月31日までの四半期内に、管理層は様々な定性と定量的な要素を決定し、これらの要素は共にトリガイベントが発生したことを示し、減値テストを実行した。減値分析の結果によると、予想売上高と増加が比較的に低いため、退行性疾病報告単位の帳簿価値はその公正価値を超え、そして2022年12月31日に確定し、退行性疾病報告単位のすべての商業権はすでに減少した。
会社の報告先に割り当てられた営業権の帳簿価値は以下のとおりである
|
|
細胞療法 |
|
|
退行性 |
|
|
バイオバンク |
|
|
合計する |
|
||||
2021年12月31日の残高 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
減損する |
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
2022年12月31日の残高 |
|
$ |
|
|
$ |
— |
|
|
$ |
|
|
$ |
|
|||
F-27
無形資産、純額
無形資産純資産額には、
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
|
役に立つと思う |
||
無形資産を償却すべきです |
|
|
|
|
|
|
|
|
||
発達した技術 |
|
$ |
|
|
$ |
|
|
|||
取引先関係 |
|
|
|
|
|
|
|
|||
商品名と商標 |
|
|
|
|
|
|
|
|||
再獲得の権利 |
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
||
差し引く:累計償却 |
|
|
|
|
|
|
|
|
||
発達した技術 |
|
|
( |
) |
|
|
( |
) |
|
|
取引先関係 |
|
|
( |
) |
|
|
( |
) |
|
|
商品名と商標 |
|
|
( |
) |
|
|
( |
) |
|
|
再獲得の権利 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
|
無形資産純資産の償却可能額 |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
||
未償却無形資産 |
|
|
|
|
|
|
|
|
||
知的財産権を獲得して製品を開発する権利 |
|
|
|
|
|
|
|
|||
|
|
$ |
|
|
$ |
|
|
|
||
無形資産の償却費用は#ドルです
以下の日までに保有する無形資産に関する5年連続の年間償却費用合計2022年12月31日は以下のように推定される
2023 |
|
$ |
|
|
2024 |
|
$ |
|
|
2025 |
|
$ |
|
|
2026 |
|
$ |
|
|
2027 |
|
$ |
|
計算すべき費用と他の流動負債には:
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
臨床試験費用を計算しなければならない |
|
$ |
|
|
$ |
|
||
専門費用を計算する |
|
|
|
|
|
|
||
ソフトウェアの開発に応じて |
|
|
|
|
|
— |
|
|
給料,ボーナス,手数料,休暇を計算しなければならない |
|
|
|
|
|
|
||
建設中の工事は計画すべきプロジェクトである |
|
|
|
|
|
— |
|
|
賃料を繰延する |
|
|
— |
|
|
|
|
|
他にも |
|
|
|
|
|
|
||
|
|
$ |
|
|
$ |
|
||
ヨークビル
2022年9月15日、会社はYA II PN,Ltd.(ヨークビル)と前払い前払い契約(PPA)を締結し、この協定によると、会社は前金を最高$まで要求することができる
F-28
至れり尽くせり購買力平価は、どうしても#ドルを下回ることはない
購買力平価協定への加入について、会社は最初に前払いした前払い#ドルを受け取った
短期借入金関係者
2021年6月8日、Legacy Celularityは$に入りました
ローン条項によると、Legacy Celularityは、現金および現金等価物の合計金額が#ドルを下回ることを許可することはできません
当社は2021年12月31日までにC.V.Starrと締結した短期借入金予定に基づいて返済していない金を返済し、この予定はキャンセルされた。C.V.スタールへの支払い総額は$
賃貸契約
付記2で述べたように,
ASU 2016−02年度の採用により、リース純資産とリース負債約#ドルが追加記録された
F-29
ROU資産は会社がリース期間内に対象資産を使用する権利を代表し,リース負債は会社を代表してリースによるリース金の支払いを義務付けている。当社のリースROU資産と負債はリース開始日にレンタル期間内にレンタル支払いの現在値で確認します。賃貸支払いの現在値を定める際には、当社は賃貸開始日の資料に基づいて、借り入れ金利を増やして複数の資産種別の適切な割引率を決定する。指数に基づく可変賃貸支払いではなく、当該賃貸負債を最初に計量した後に指数変化により生じる可変賃貸支払いは、賃貸純資産又は負債の計量に計上するのではなく、当該等の支払義務が発生している期間の収益で確認する。レンタル条項は、会社が任意のこのような選択権を行使することを合理的に決定したときに、リース契約を延長または終了する選択権を含むことができる。レンタル料金は予想レンタル期間内に直線的に確認します。レンタル料金は$
Legacy Celularityは2017年9月、ニュージャージー州ウォーレンオフィススペースの運営賃貸契約を締結した
2019年9月10日、Legacy Celularityは、ニュージャージー州シダノールスのオフィスと実験室空間の運営リースを月ごとに延長した。2019年11月1日から、Legacy Celulityは大家さんに基本的な年間レンタル料とすべての追加レンタル料を支払い始めました。レンタル料は
2019年3月13日Legacy Celularityは
その会社はレンタルスペースの合法的な所有者ではない。しかし、ASC 840項目の以前のレンタル指導によると、賃貸借証書施工期間中,当社は賃貸空間(建物ハウジングを含む)の所有者とみなされており,当社がテナントの重大な改善工事の財務·運営に直接関与することが予想されているためである。付記2で述べたように、アリゾナ州2016−02年に規定されているスーツレンタルへの移行に関するガイドラインによると、以前はスーツレンタルに建設されたと決定されていたレンタル契約がキャンセル識別されていた。
ASC 842を用いた影響は以下のとおりである
|
|
2021年12月31日現在の残高 |
|
|
ASC 842を用いた調整 |
|
|
2022年1月1日現在の残高 |
|
|||
資産 |
|
|
|
|
|
|
|
|
|
|||
財産と設備、純額 |
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||
経営的リース使用権資産 |
|
|
- |
|
|
|
|
|
|
|
||
負債.負債 |
|
|
|
|
|
|
|
|
|
|||
流動賃貸負債--経営 |
|
|
- |
|
|
|
- |
|
|
|
- |
|
融資債務の当期部分 |
|
|
|
|
|
( |
) |
|
|
- |
|
|
非流動賃貸負債--経営 |
|
|
- |
|
|
|
|
|
|
|
||
融資義務 |
|
|
|
|
|
( |
) |
|
|
- |
|
|
株主権益 |
|
|
|
|
|
|
|
|
|
|||
赤字を累計する |
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
会社の賃貸コストの構成部分はその総合経営報告書の中で以下のように分類される
F-30
|
|
現在までの年度 |
|
|
|
|
2022 |
|
|
リースコストを経営する |
|
$ |
|
|
可変リースコスト |
|
|
|
|
リース総コストを経営する |
|
$ |
|
|
短期賃貸コスト |
|
$ |
|
|
下表に年内の会社の賃貸負債に関する現金と非現金活動を示す2022年12月31日までの年度:
|
|
現在までの年度 |
|
|
|
|
2022 |
|
|
賃貸負債に関連した支払い済み現金: |
|
|
|
|
レンタル経営からの経営キャッシュフロー |
|
$ |
|
|
|
|
|
|
|
非現金リース負債活動: |
|
|
|
|
レンタル義務と引き換えに使用権資産: |
|
|
|
|
賃貸借契約を経営する |
|
$ |
- |
|
自分から2022年12月31日、会社経営賃貸負債満期日は以下の通り
2023 |
|
$ |
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 |
|
|
|
|
その後… |
|
|
|
|
賃貸支払総額 |
|
|
|
|
計上された利息を差し引く |
|
|
( |
) |
合計する |
|
$ |
|
2022年12月31日までの会社経営リースの加重平均残存レンタル期間は
企業合併に関連しているか、または掛け値がある
会社がHLI細胞治療会社、有限責任会社と人類起源会社を買収することについて、会社はある監督管理と商業マイルストーンに達した後、売り手に未来の価格を支払うことに同意した。そこで会社は$
賠償協定
通常の業務過程において、会社は、このような合意または第三者に違反する知的財産侵害クレームによる損失を含むが、これらに限定されない異なる範囲および条項の賠償をサプライヤー、レンタル者、業務パートナー、および他の当事者に提供することができる。また、当社は取締役会メンバー及び行政者と合意を締結し、取締役又は行政者としての身分又はサービスとして生じる可能性のあるいくつかの法的責任について補償を行うことを当社に要求している。これらの賠償協定によると、会社が将来支払う必要がある可能性のある最大潜在金額は多くの場合無制限である。当社はこれまで、当該等の賠償により重大なコストを発生させていません。同社は現在、どのような賠償要求があるかを知らず、2022年12月31日と2021年12月31日までの総合財務諸表に当該等の債務に関する負債を計上していない。
F-31
Palantir Technologies Inc.と合意した.
2021年5月5日、Legacy CelularityはPalantirと主購読契約を締結し、協定によると$を支払います
シリウス許可協定
当社は2021年12月にシリウス生物科技有限公司(“シリウス”)と許可協定(“シリウス許可”)を締結した。Sirionライセンスに基づき,Sirionはポロキサムに関する特許権とノウハウライセンス(“特許製品”)を当社に付与した。Sirionライセンスの一部として、同社はSirionに#ドルを支払った
法律訴訟
報告日ごとに、当社は、潜在損失金額又は潜在損失範囲が権威的な案内処理又は事項の規定によって可能かつ合理的に評価されているか否かを評価する。当社はこのような法律訴訟に関連する費用が発生した場合に支出します。
2021年3月24日、Cth Biosourcing LLC(“Cth”)はテキサス州テルアビブ県地方裁判所に要望書と開示要求を提出し、Legacy Celularityが原因を理由に組織調達協定(TPA)を終了したことを疑問視する宣言的救済を求めた。当社は2021年12月31日までに、利害関係者であるCthおよびCariCord参加株主と三者和解合意(“和解合意”)を達成し、当社は上記訴訟のすべての請求を全面的に免除すると引き換えにTPAを改訂することに同意した。また、同社は発行しました
和解協定によると、CariCordは株主に最も多くの権利を得る権利がある
当社はペンシルベニア州東区連邦検事室が2022年8月14日に“虚偽申告法”(“米国法典”第31編第3729節)に基づいて提出した民事調査要求(以下“要求”と呼ぶ)を受けた。この要件は、Interfylを含むMedicare、Medicaid、または他の連邦保険会社に提出されたサービスまたはプログラムクレームに関連する文書および情報を提供することを必要とする。同社はこの要請に協力し、この要求を処理する米国の検事補佐官と継続的な対話を行っている。この問題はまだ初期段階にあり、この要求がどんな責任も招くかどうかはまだ確定されていない。
普通株
業務合併後
2022年12月31日と2021年12月31日まで、会社の登録証明書、改訂及び再記載を経て、会社が発行することを許可する
F-32
投票権
法律に規定または任意の系列優先株のいずれかの指定証明書が別途規定されているほか、普通株式保有者は、当社取締役および他のすべての株主が行動する必要がある事項を選挙する投票権を持っている。所持者
配当をする
A類普通株の保有者は、合法的に分配可能な資金から、会社取締役会が時々発表する可能性のある配当金(あれば)を得る権利がある。いずれの場合も、当時発行された普通株が平等かつ同等に扱われていない限り、いかなる普通配当金、株式分割または株式組み合わせも普通株で発表または行われない。
清盤·解散·清盤
会社が自動的または非自発的に清算、解散、資産分配または清算を行う場合、優先株保有者の権利が満たされた後、普通株式保有者は、株主に分配可能なすべての資産の同等額の1株当たり収益を得る権利がある。
優先購入権またはその他の権利
会社の株主は優先引受権や他の引受権を持たず、普通株の債務超過基金や償還条項にも適用されていない。
役員を選挙する
当社取締役会は、第I類、第II類、第III類の3つに分類され、毎年1種類の取締役のみが選択され、各種類の任期は3年であるが、GX合併に関する特別会議の役員選挙は除外されている。第1種取締役の初期任期は1年(その後3年)、第2類取締役の初期任期は2年(その後3年)、第3種取締役の初期任期は3年(その後3年)である。役員選挙では累積投票がなく,その結果,それを上回った
業務合併前に
Legacy Celularity Legacy Celularity修正および再記述された企業登録証明書のライセンス発行2020年12月31日から
それぞれの株
2020年12月31日現在Legacy Celulityは
優先株
業務合併後
会社登録証明書許可
F-33
業務合併前に
伝統的なCelulityはAシリーズの転換可能な償還可能優先株(“Aシリーズ優先株”)、Bシリーズの転換可能な償還可能優先株(“Bシリーズ優先株”)とXシリーズの転換可能な償還可能優先株(“Xシリーズ優先株”)を発行した。A系列優先株、B系列優先株、X系列優先株を総称して“優先株”と呼ぶ。2021年7月16日に業務合併が終了する直前に、優先株の流通株がLegacy Celularity普通株に変換され、発効時期に会社のA類普通株に交換される。2020年12月31日現在、Legacy Celulity社の証明書の改訂と再記載されたLegacy Celulityの発行
優先株保有者は清算とみなされた場合に清算権を持つが,場合によってはこれらの清算はLegacy Celulityの制御範囲内に完全にあるわけではない.したがって、優先株は連結貸借対照表で株主赤字外に分類される。
二零二年三月十六日、Legacy Celularityは複数の機関投資家及び複数の個人投資家(総称して“投資家”)とBシリーズ優先株購入協定(“購入契約”)を締結した。購入契約条項に基づいて、Legacy Celularityの投資家への売却と発行の合計
ASC 480によると、従来のCelularityの分類優先株は、負債と持分を区別するそれは永久株主権益以外の証券として分類される償還可能な証券を要求するか、または持っている。したがって、Legacy Celularityは2021年12月31日にすべての株式と優先株カテゴリを付属の総合貸借対照表上の中間株式に分類した。
優先株の権利、選好、特権
特に明記した以外に、優先株保有者は以下の権利と優先権を有している
投票する.
所持者
少なくともあれば
転換する
優先株の1株は発行日後のいつでも所有者の選択に応じて切り替えることができる。また,各優先株は,当時有効な適用変換比率で自動的に普通株に変換することができる:(I)約束の公開発行終了を決定した時点で,少なくとも$を生成する
F-34
配当をする
清算する
任意の自動または非自発的清算、解散または清盤レガシー株式が発生した場合、または清算イベントとみなされる場合(以下、定義を参照)、その時点で優先株を発行していなかった各所有者は、(I)一連の優先株の元の発行価格に、宣言されているがまだ支払われていない任意の配当金を加えた金額に等しいか、または(Ii)当該保有者が、清算イベントの直前に株式交換価格でその株式を普通株に変換すべき金額に変換する権利がある。株主に分配可能な資産が優先株株主に獲得権のある全金額を支払うのに十分でない場合、割り当て可能な資産は優先株保有者の間で比例して分配され、そうでなければ当該株式についてそれぞれの金額が支払われる。
優先株保有者にすべての優先金額を支払った後、利用可能な範囲内で、Legacy Celulityの残存資産は、普通株式保有者毎に保有する株式数に応じて普通株式保有者に比例して割り当てられる。
(I)Bシリーズ優先株保有者が受信した金額から元の発行価格を減算するか、または(Ii)当時発行された優先株の大部分の所有者が1つのカテゴリとして一緒に投票し、他の方法を選択しない限り、清算されたイベントには、合併または合併(ただし、遺産Celularityの株主が投票権を介して既存または買収した会社の発行済み株式の多数を除く)または売却、リース、譲渡、独占許可またはその他のLegacy Celularityの全部またはほぼすべての資産が含まれているとみなされる。
救いを求める
改訂と再記述されたLegacy Celularity社の登録証明書は、優先株保有者に償還権を提供しない。
ATM協定
2022年9月8日、会社は販売代理および/または依頼者であるBTIG,LLC,Oppenheimer&Co.Inc.とB.Riley Securities,Inc.と市場販売協定(“ATM協定”)を締結し、この協定により、会社は時々適宜普通株株を発行·販売することができ、総発行価格は最高$に達する
市場発売中に発売された任意の株式は、S-3表における当社の有効棚上げ登録声明と関連募集説明書に基づいて補編発行されます。ATM協定によれば、販売エージェントは、1933年に改正された証券法第415(A)(4)条で定義された“市場で発行される”とみなされる法律で許可された任意の方法で普通株を販売することができる。会社は販売代理に最高可達を支払います
2022年12月31日までの年度内会社が受け取った毛収入と純収益は#ドルです
株式承認証
伝統的なCelulity許可
2018年5月7日、Legacy CelulityはDragas acに引受権証を付与し、購入合計
F-35
残り株式承認証はドラガザックと交換して購入に同意した
2020年1月9日、Legacy Celularityは引受権証を発表し、購入した
Legacy Celularityは2021年1月8日、2020年1月9日にドラガサックに発行された権証を改訂し、2020年3月16日に改訂するための権証改訂協定(“改正2号”)を締結した。第2号改正案は無現金行使条項を追加し、企業合併完了時権証の満期を規定すべき条項を廃止した。業務合併完了前にまだ行使されていない任意の株式承認証部分を株式承認証に変換して自社A類普通株株式を購入し、行権価格及び株式数は交換比率及び合併協議の条項に基づいて調整する(付記3参照)。この改正はこのような株式承認証の会計に何の変化ももたらさなかった。
2020年3月16日、Legacy Celularityは投資家と購入協定を締結した。購入契約条項に基づいて、Legacy Celularityの投資家への売却と発行の合計
2022年3月1日に、Celularityといくつかの関連側投資家は投資家それぞれのLegacy Celularity引受権証を改訂及び再記述し、(I)で1株当たりの価格を$から行使する
業務合併前に,Legacy Celularisはその総合貸借対照表上で株式証を負債に分類し,株式証は独立した金融ツールであるため,Legacy Celularisが行使時に資産を移転する必要がある可能性がある。各株式証明書に関連する負債は最初に株式承認証の発行日ごとに公正価値で入金され、その後、報告日ごとに公正価値で再計量され、最後の再計量は決算日に行われる。株式証負債の公正価値変動は、連結業務報告書における純額の他の収入(費用)の構成要素として確認されている。締め切りには、ドラガサックと投資家が保有する株式承認証が株式承認証に変換され、会社A類普通株の購入に使用される。上述の株式承認証は締め切りに持分分類資格に符合し、相応して再分類された。
従来のGX株式証明書
業務合併が完了した後、公開株式証と保証権証はまだ決済されていない
F-36
はい業務合併が完了してから90日間、所持者は証券法に規定された登録免除に基づいて、有効な登録声明があるまでの時間及び当社が有効な登録声明のいかなる期間を維持できなかったまで、キャッシュレス方式で公共株式権証を行使することができる。登録免除がなければ、所持者は現金なしで彼らの公共株式証明書を行使することができない。同社は2021年8月12日に登録声明を提出した。株式公開承認証は満期になります
会社はすべての償還と一部の株式権証の公開(保険権証を含まない)を要求することができ、価格は$である
権利証明書はGXが初めて公開発売した販売先の公開株式証と同じであり、異なる点は保権証及び保権証を行使する際に発行可能な普通株は業務合併が完了する前に譲渡、譲渡或いは販売してはならないが、いくつかの限られた例外の場合は除く。さらに、保証権証は、現金なしで行使することができ、初期購入者またはその譲受人が所有することが許可されている限り、償還することはできない。権利証明書が初期購入者又はその許可譲渡者以外の者が保有する場合、保権証は自社で償還することができ、当該等所有者が株式公開承認証と同じ基準で行使することができる。
公開株式証及び保権証を行使した後に発行可能なA類普通株の使用価格及び株式数は、株式配当、資本再編、合併又は合併を含む場合に調整される場合がある。
また、いずれの場合も、当社は現金純額決済による株式承認証の公開を要求されません。もし会社が株式証の公開承認を要求した場合、管理層は公開株式証の行使を希望するすべての所有者に株式承認契約の規定に従って、“現金なしに基づいて”行使することを要求する権利がある。公共株式証明書を行使する際に発行可能なA類普通株の行使価格と数量は、株式配当金、非常配当金または資本再編、再編、合併または合併を含む場合によって調整される可能性がある。
業務合併に関する取引コストを支払うために、GXの保証人メンバーGX保証人有限責任会社(“保険人”)は、GXと本票を締結し、運営資金を提供する。業務合併については
2022年5月パイプ
2022年5月18日、当社はある機関投資家と証券購入協定の締結を認め、私募(I)について
2022年12月31日までに会社は
|
|
量 |
|
|
トレーニングをする |
|
|
|
満期になる |
||
ドラガザック保証書 |
|
|
|
|
$ |
|
* |
|
|||
株式証を公開する |
|
|
|
|
$ |
|
|
|
|||
保証人が株式証明書を承認する |
|
|
|
|
$ |
|
|
|
|||
2022年5月配管捜査令状 |
|
|
|
|
$ |
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
||
F-37
*
2021年株式インセンティブ計画
2021年7月、会社取締役会が採択され、会社株主は“2021年株式インセンティブ計画”(“2021年計画”)を承認した。“2021年計画”は、従業員に奨励的株式オプションを付与し、従業員、取締役、コンサルタントに非法定株式オプション、株式付加権、制限株式奨励、制限株式単位奨励、業績奨励、その他の形態の株式奨励を付与することを規定している。
2021年計画によると発行に予備予約するA類普通株式数は
2021年には会社の取締役会が管理する予定です。当社取締役会又はその正式に許可された委員会は、1名以上の上級管理者(I)指定上級管理者以外の従業員が指定株式奨励を受けることを許可することができ、及び(Ii)当該等の株式奨励を受けなければならない株式数を決定することができる。2021年計画条項に該当する場合、計画管理人は、受領者、株式奨励の実行価格または実行価格(ある場合)、株式報酬当たりの株式数、株式の公平な時価、報酬に適用される帰属スケジュール、任意の帰属加速、株式奨励行使または決済時に支払われる対価格形態(ある場合)、および2021年計画に従って使用される奨励協定の条項および条件を含む奨励条項を決定する権利がある。計画管理者は2021年計画下の未完了報酬を修正する権利がある。2021年計画条項に適合することを前提として、会社の取引または資本調整に関連する場合、計画管理人は会社の株主の許可を得ず、より低い行権価格、実行価格または購入価格で再定価またはキャンセルおよび再付与してはならない、または現金、財産または他の奨励と交換するために、実行価格、実行価格または購入価格を持つ任意の奨励を取り消してはならない。
2017持分インセンティブ計画
Legacy Celularity取締役会がLegacy Celularity株主の承認を経て承認した2017年株式インセンティブ計画(“2017計画”)によって、Legacy CelularityはLegacy Celularityの従業員、取締役、コンサルタントに株式オプションを授与することができる。2021年計画の業務合併と有効性の終了については、2017年計画ではさらなる贈与は提供されなくなります。
2017年計画によると本発行可能な株式オプションの総数は
2017年は会社の取締役会が管理するか、または会社の取締役会が適宜取締役会委員会が管理する予定です。執行権価格、帰属、その他の制限はLegacy Celularity取締役会またはその委員会によって適宜決定されるが、株式オプションの1株当たりの行権価格は下回ってはならない
株式オプション推定値
サービス条件付き報酬
各オプションの公正価値は、付与日にBlack-Scholesオプション定価モデルを使用して推定され、このモデルは、例えば、行権価格、関連普通株の付与日における推定公正価値、期待期限、期待株価変動、無リスク金利、および配当率などの要因を考慮したものである。株式オプションを付与する各項目の公正価値は、以下に説明する方法および仮定を用いて当社が決定する。その中のいくつかの入力は主観的であり,通常決定するために判断が必要である.
F-38
次の表では,重み付き平均に基づいてBlack-Scholesオプション定価モデルにおいて付与日株式オプション公正価値を決定するための仮定を提案した2022年と2021年12月31日までの年度:
|
|
現在までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
無リスク金利 |
|
|
% |
|
|
% |
||
予想期限(年単位) |
|
|
|
|
|
|
||
予想変動率 |
|
|
% |
|
|
% |
||
期待配当収益率 |
|
|
— |
% |
|
|
— |
% |
2022年12月31日と2021年12月31日まで年度内に付与された1株当たりオプションの加重平均付与日公正価値はい$です
次の表は、2021年計画と2017年計画のサービス条件オプション活動をまとめています
|
|
オプション |
|
|
重みをつける |
|
|
重みをつける |
|
|
骨材 |
|
||||
2020年12月31日現在返済していません |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
授与する |
|
|
|
|
|
|
|
|
|
|
|
|
||||
鍛えられた |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
没収される |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
2021年12月31日現在の未返済債務 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
授与する |
|
|
|
|
|
|
|
|
|
|
|
|
||||
鍛えられた |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
没収される |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
2022年12月31日に返済されていません |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
帰属し、2022年12月31日に帰属する予定です |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2022年12月31日に行使できます |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
行権価格が会社A類普通株公正価値より低いオプションについては、オプションの総内在価値は、株式オプションの行権価格と会社A類普通株の公正価値との差額として計算される。
会社はサービス条件#ドルの報酬に関する株式ベースの報酬支出を記録した
2021年3月、Legacy Celularity取締役会は完全既得オプションの発行を承認し、買収
F-39
2021年第2四半期、Legacy Celularity取締役会は完全既得オプションの発行を許可し、買収
2021年9月、会社取締役会はオプションの発行を許可し、共同買収
2021年7月、会社は2つの非従業員株式オプション奨励を改訂し、業務合併時のいかなる非帰属奨励も完全に帰属させる。同社はドルの確認を速めた
業績条件のある賞
メリーランド州ロビン·L·スミスとのコンサルティング契約(付記20参照)について、同社は合計で付与されました
2021年の間、会社は業績に基づく株式オプションをいくつか持っており、これらのオプションは業績期間中に達成された特定の目標に基づいて稼いでいる。当社は2021年12月31日までに年度内に確認します
市場条件のある賞
2021年9月に当社は合共を授与しました
限定株単位
同社は従業員にRSUを支給し、通常は超過を授与する
次の表は、RSUの株による支払い奨励に関する活動をまとめたものである
F-40
|
|
株式数 |
|
|
重みをつける |
|
||
2020年12月31日現在返済していません |
|
|
— |
|
|
$ |
— |
|
授与する |
|
|
|
|
$ |
|
||
没収される |
|
|
( |
) |
|
$ |
|
|
2021年12月31日現在の未返済債務 |
|
|
|
|
$ |
|
||
授与する |
|
|
|
|
$ |
|
||
既得 |
|
|
( |
) |
|
$ |
|
|
没収される |
|
|
( |
) |
|
$ |
|
|
2022年12月31日に返済されていません |
|
|
|
|
$ |
|
||
同社が記録した株式報酬支出は#ドルだった
株に基づく報酬費用
同社は、その合併経営報告書の以下の費用種別に、株式ベースの補償費用を記録している
|
十二月三十一日までの年度 |
|
|||||
|
2022 |
|
|
2021 |
|
||
収入コスト |
$ |
|
|
$ |
|
||
研究開発 |
|
|
|
|
|
||
販売、一般、行政 |
|
|
|
|
|
||
|
$ |
|
|
$ |
|
||
次の表は、製品およびサービス別の収入情報を提供します
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
製品販売とレンタル |
|
$ |
|
|
$ |
|
||
サービス.サービス |
|
|
|
|
|
|
||
ライセンス、印税、その他 |
|
|
|
|
|
|
||
純収入 |
|
$ |
|
|
$ |
|
||
次の表は、契約負債繰延収入の変動状況を提供します
|
|
2022 |
|
|
2021 |
|
||
1月1日の残高 |
|
$ |
|
|
$ |
|
||
繰延収入* |
|
|
|
|
|
|
||
未稼ぎ収入確認** |
|
|
( |
) |
|
|
( |
) |
十二月三十一日の残高 |
|
$ |
|
|
$ |
|
||
*バイオバンクサービス貯蔵契約が履行前に受信した支払いに基づいて生成された収入を延期し、これらの契約は、契約完了時に収入として確認されます。
**会社は2021年第3四半期に、未治癒材料違反によりSanuWaveとのライセンス契約を終了しました(付記16参照))である。そこで、会社は残りの繰延収入#ドルを確認した
ソレント治療会社のライセンスと譲渡協定
2017年8月15日、Legacy Celularityは、Legacy Celularityと秋明治療会社とSorrento治療会社(総称してSorrentoと呼ぶ)と許可と譲渡協定を締結し、この合意に基づき、Legacy Celularityはある材料の独占的な許可を得た
F-41
任意の疾患または障害を治療するための製品の開発および商業化のためのソレントに関連する特許および知的財産権(“2017年ライセンス契約”)。2020年第1四半期に、双方は2017年の許可協定を終了した。
2020年8月26日、Legacy CelularityとSorrentoは拘束力のある条伝票に署名し、CD 19 CAR-T構築物の全世界独占許可を得て、胎盤由来の細胞によるいかなる疾患或いは障害の治療に用いられる(“2020 Sorrento条伝票”)。2020年ソレント条項説明書は,最終許可と供給合意を達成するために組み込まれ,さらに交渉される様々な条項について概説している。
2020年9月30日、Legacy CelularityとSorrentoは、胎盤由来細胞および/または臍帯血由来細胞による任意の疾患または疾患の治療に使用するCD 19 CAR-T構築物を独占的に許可する新しい許可および譲渡協定に署名した(“2020 Sorrento許可協定”)。ソレントが事前に書面で同意した場合、当社は合意により付与された権利を再許可する権利を保持します。ライセンスの対価格として、当社は、純売上高のより低い桁数パーセント(プロトコルで定義されているような)に相当する特許権使用料と、すべての分割可能収入に相当する低い2桁数百分比(合意中の定義のような)に相当する特許権使用料とをSorrentoに支払う義務がある。2020年には、当社またはSorrentoが90日間の書面通知の下で未治癒の重大な違反により終了したり、Sorrento協定の発効日から1周年後に、便宜上、Sorrentoに書面通知を出して6ヶ月後に終了するまで、Sorrentoライセンス契約が有効になります。
同社とソレントは、2020年のソレント許可協定に関連した新たな供給協定を積極的に交渉している。2020年ソレント条項説明書は、2020年ソレント許可協定に従って提供される材料および/または可能な製品の価格設定条項を含む、本供給プロトコルのいくつかの態様を詳細に示す。“会社”ができた
肺バイオテクノロジーPBCライセンスプロトコル
二零一七年六月三十日に、Legacy Celularityと連合治療会社の完全な付属会社肺生物科学技術有限会社(“LB”)は許可協定(“この協定”)を締結し、これに基づいて、Legacy Celularityはいくつかの知的財産権の全世界独占再許可を授与し、胸腹器官移植及び肺疾患領域の製品(“この許可IP”)を開発及び商業化することができる。LBプロトコルによると、Legacy Celularityは製品の開発と商業化のためにLBに胎盤由来の幹細胞を提供することに同意した。
2020年4月3日、Legacy Celularityとドイツ銀行は彼らの戦略協力許可協定を拡大し、新冠肺炎と急性呼吸窮迫症候群(“ARDS”)の治療をその中に入れることに同意した。改訂された協力協定によると、同社は新冠肺炎の治療のための規制部門の承認を求め、ランバーズ社は急性呼吸窮迫症候群の治療のための規制部門の承認を求める。改訂された協力プロトコルによれば、LBは新冠肺炎および急性呼吸窮迫症候群を治療するCYNK-001を商業化する世界的権利を有する。協力は開発と商業化活動を監督するために共同指導委員会によって管理されるだろう。LBはLegacy Celularityの必要性と要求に応じて財政的サポートを提供し、最高限度額は$です
2021年第1四半期に、ドイツ銀行との許可協定は2021年4月11日からすべて終了した。2020年4月3日に新冠肺炎とARDSにおけるCYNK−001の治療に関する改正案の適用を中止した。
雲頂革新私設有限会社販売協定
2018年5月4日、DraasacはLegacy Celularityに株式投資を行うと同時に、Legacy Celularityと雲頂革新プライベート有限会社(“雲頂”)は流通協定を締結し、これにより、雲頂は一部のアジア市場に当社のいくつかの製品の供給と販売権(“雲頂合意”)を授与した。雲頂協定は、当社の当時の退行性疾患製品の組み合わせに対する有限販売権を提供し、当社または当社が開発した未来の製品を自動的に所有する権利を自動的に持つことを規定している。
雲頂協定は2023年1月31日に継続し、自動的に12ヶ月連続して継続し、雲頂が継続期間の少なくとも3ヶ月前に書面通知を出して意図的に継続しないことを示しない限り、または雲頂協定はいずれか一方によって他の理由で終了される。
雲頂とドラガサックはいずれも雲頂柏ハルトの直接子会社であり、雲頂柏ハルトはマレーシアに登録して設立され登録された上場有限責任会社である。
Celgene社のライセンス契約
二零一七年八月二十日にLegacy CelularityはCelgeneとライセンス契約(“Celgeneプロトコル”)を締結し、これにより、当社はCelgeneにCelgeneがAnthrogenensisを買収した日に保有または制御したいくつかの知的財産権の2つの独立許可(“Anthrogenensis IP”)をCelgeneに付与した。Celgeneプロトコルは、Celgeneの印税免除、全額支払い、世界的な非独占的なライセンスを付与します
F-42
すべての領域の臨床前研究目的および印税免除、全額支払いの全世界的許可は、任意のCAR製造分野の製品の開発、製造、商業化および開発のためのヒト発育知的財産権を付与する権利があり、任意のTリンパ球またはNK細胞を修飾してそのようなCARを発現させるために、および/または予防、診断および/または治療用途を含む任意の目的のためにそのようなCARまたはTリンパ球またはNK細胞を使用する権利がある。セルキング協定はどんな理由でもすべての理由で終わるまで効果的に続くだろう。
SanuWaveライセンスプロトコル
2020年8月6日,UltraMist業務の販売に伴い,Legacy Celularityが進出した
2021年第2四半期に、Legacy Celulalityは、既存の許可プロトコルに従ってSanuwaveに欠陥通知を送信し、Sanuwaveは2021年7月19日までに重大な違約を修復しなければならない。Sanuwaveはこの重大な違約を是正しなかったため,Sanuwaveとの合意は2021年第3四半期に終了した。
独占供給と流通協定
2021年5月7日会社は
2021年9月1日、当社は締結しました
会社は国税法第401(K)条に基づいて固定払込貯蓄計画を確立した。この計画は、最低年齢およびサービス要件に適合するすべての従業員をカバーし、参加者が税引前に年間給与の一部の支払いを延期することを可能にする。会社の取締役会はその計画に相応の貢献をすることを適宜決定することができる。2022年12月31日まで及び2021年12月31日まで年度同社は$を出資している
F-43
当社の当期と繰延税金項目の概要は以下の通りである
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
当期所得税支出: |
|
|
|
|
|
|
||
連邦制 |
|
$ |
- |
|
|
$ |
- |
|
状態.状態 |
|
|
|
|
|
|
||
当期所得税支出総額 |
|
|
|
|
|
|
||
繰延所得税費用(福祉): |
|
|
|
|
|
|
||
連邦制 |
|
|
|
|
|
|
||
状態.状態 |
|
|
( |
) |
|
|
|
|
繰延税金総額 |
|
|
— |
|
|
|
|
|
所得税支出総額 |
|
$ |
|
|
$ |
|
||
米国連邦法定所得税率と会社の有効所得税率の入金は以下の通りである
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
連邦法定所得税率 |
|
|
% |
|
|
% |
||
連邦福祉を差し引いた州所得税 |
|
|
( |
)% |
|
|
% |
|
研究開発税収控除 |
|
|
% |
|
|
% |
||
利子割増費用 |
|
|
( |
)% |
|
|
% |
|
評価免除額を変更する |
|
|
% |
|
|
( |
)% |
|
市価で価格を計算する権利証 |
|
|
( |
)% |
|
|
% |
|
延期整頓 |
|
|
% |
|
|
% |
||
株に基づく報酬 |
|
|
( |
)% |
|
|
% |
|
減損する |
|
|
% |
|
|
— |
|
|
他の永久品 |
|
|
( |
)% |
|
|
% |
|
有効所得税率 |
|
|
( |
)% |
|
|
% |
|
繰延税金負債純額2022年12月31日および2021年12月31日には、以下が含まれる
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
繰延税金資産: |
|
|
|
|
|
|
||
純営業損失が繰り越す |
|
$ |
|
|
$ |
|
||
研究と開発税収は繰越免除 |
|
|
|
|
|
|
||
株に基づく報酬費用 |
|
|
|
|
|
|
||
起動コスト |
|
|
|
|
|
|
||
無形資産 |
|
|
|
|
|
|
||
収入を繰り越す |
|
|
|
|
|
|
||
Unicap |
|
|
|
|
|
|
||
支払いの推定利息があります |
|
|
|
|
|
|
||
法律費の資本化と償却 |
|
|
|
|
|
|
||
資本化研究と開発 |
|
|
|
|
|
— |
|
|
他にも |
|
|
|
|
|
|
||
繰延税金資産総額 |
|
|
|
|
|
|
||
繰延税金負債: |
|
|
|
|
|
|
||
現在行われている研究と開発 |
|
|
( |
) |
|
|
( |
) |
繰延税金負債総額 |
|
|
( |
) |
|
|
( |
) |
推定免税額 |
|
|
( |
) |
|
|
( |
) |
繰延税金純負債 |
|
$ |
( |
) |
|
$ |
( |
) |
F-44
未確認税収割引の期初と期末金額の入金は以下の通り
|
|
識別できない |
|
|
2020年12月31日残高 |
|
$ |
|
|
今年度の税収引当に関する増加 |
|
|
|
|
2021年12月31日の残高 |
|
$ |
|
|
今年度の税収引当に関する減少額 |
|
|
( |
) |
2022年12月31日の残高 |
|
$ |
|
|
2022年12月31日と2021年12月31日まで同社はアメリカ連邦と州で純営業損失繰越$があります
アメリカ連邦と州の営業純損失の繰越と研究開発税収の繰越免除の使用は1986年の“国税法”第382と383節及び州法律の相応条項の年間制限を受ける可能性があり、原因は以前に発生或いは未来に発生する可能性のある所有権変更である。これらの所有権変化は、将来の課税所得額または納税義務を相殺するために毎年使用可能な繰越金額を制限する可能性がある。一般に、第382条の定義によれば、所有権変更とは、3年以内にある株主の会社株における所有権を50%以上増加させる取引をいう。第382条によれば、所有権変更が発生した会社は、まず所有権変更時に会社株の価値に必要に応じて追加調整可能な適用可能な長期免税税率を乗じることにより決定される年間制限を受ける。同社は2017年8月15日に所有権変更を経験した。所有権変更の年間制限は純運営損失や研究開発信用が使用前に満期になることはないと予想される。
繰延税金資産の現金化は、会社が今後数年で課税収入を生み出す能力にかかっている。ASC 740-10所得税や繰延税金資産の一部または全部が“より可能性がある”と考えて現金化できないと考えた場合には、繰延税金資産を推定値に計上する準備が求められている。同社は、繰延税金負債の予定沖販売、将来の課税収入、税務計画戦略、および最近の財務業績を含む、利用可能なすべてのプラスおよび負の証拠を考慮している。
2022年12月31日、既存の証拠の重みによると、同社は連邦と州繰延税金資産のメリットが実現する可能性が低いと結論した。そのため、当社はその連邦と州繰延税の総資産を推定値に計上する準備をしている。
不確定所得税状況の影響は、関連税務機関監査後に“より可能性が高い”継続的な最大金額で確認されている。不確実な税金状況が続く可能性が50%未満であれば、確認されないだろう。
2022年12月31日と2021年12月31日まで同社には未確認の税収割引総額$があります
2021年の間に同社は
同社はその部門と経営陣が業績を評価し、資源を分配する方法を定期的に審査している。2020年第3四半期までにLegacy Celularityは
F-45
♪the the the業務部門の収入と収益は、他の要因に加えて、業績評価とこれらの部門との間の資源配分に用いられる。
報告されるべき部門は、各部門が展開する活動の異なる性質に基づいて決定される。細胞療法とは,同社が研究·開発している療法を指す。検討されている治療法は実証されておらず,異なる開発段階にある。退行性疾患会社は外科や創傷ケア市場で使用されている製品を生産、販売、許可している。バイオバンクは臍帯と胎盤から幹細胞を収集し、個人に代わってこれらの細胞の保存を将来的に使用するために提供する。
当社は経営部門別に管理するのではなく、全社をベースに資産を管理している。このため、首席運営決定者は、経営部門別にいかなる資産情報も定期的に審査しないため、資産情報は経営部門別に報告しない。総資産は約
部門別の財務情報は以下の通り
|
|
2022年12月31日までの年度 |
|
|||||||||||||||||
|
|
細胞.細胞 |
|
|
バイオバンク |
|
|
退行性疾患 |
|
|
他にも |
|
|
合計する |
|
|||||
純収入 |
|
$ |
— |
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|||
毛利 |
|
|
— |
|
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
|
直接費用 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
細分化市場貢献 |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
間接費用 |
|
|
|
|
|
|
|
|
|
|
|
( |
) |
(a) |
|
( |
) |
|||
運営損失 |
|
|
|
|
|
|
|
|
|
|
|
|
|
$ |
( |
) |
||||
(A)他の機器の部品 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
価格負債の公正な価値変動があります |
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
||||
株式価格の公正価値変動があるかもしれない |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
営業権の減価 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
償却する |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
その他を集計する |
|
|
|
|
|
|
|
|
|
|
$ |
( |
) |
|
|
|
||||
|
|
2021年12月31日までの年度 |
|
|||||||||||||||||
|
|
細胞.細胞 |
|
|
バイオバンク |
|
|
退行性 |
|
|
他にも |
|
|
合計する |
|
|||||
純収入 |
|
$ |
— |
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|||
毛利 |
|
|
— |
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
|||
直接費用 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
細分化市場貢献 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
間接費用 |
|
|
|
|
|
|
|
|
|
|
|
( |
) |
(b) |
|
( |
) |
|||
運営損失 |
|
|
|
|
|
|
|
|
|
|
|
|
|
$ |
( |
) |
||||
(B)他の機器の部品 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
価格負債の公正な価値変動があります |
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
||||
償却する |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
その他を集計する |
|
|
|
|
|
|
|
|
|
|
$ |
( |
) |
|
|
|
||||
以下に述べる関連先交易総合貸借対照表には、2022年及び2021年12月31日には何の関連残高もない。
アンドリュー·ペコラ博士との相談と相談契約
2017年9月1日、Legacy CelularityとLegacy Celularity取締役会のAndrew Pecora博士はコンサルティングとコンサルティングサービスの提供について科学と臨床コンサルタント協定(“SABプロトコル”)を締結した。SABプロトコルは、2019年2月1日にLegacy Celularityが署名した新しいSABプロトコルに置き換えられました。
F-46
2020年4月13日、Legacy CelularityはPecora博士とSAB協定第1修正案に署名した。第一修正案の任期は六ヶ月です。それは#ドルを支払うことを規定している
2020年10月15日、Legacy CelularityはPecora博士とSAB協定第2修正案に署名した。第二修正案に基づき、ペコラ博士は臨床開発運営と戦略についてLegacy Celulalityに戦略アドバイスを提供し、長期的な臨床開発計画の構築に協力することに同意した。この手配された補償は:(1)現金対価#ドルを含む
ロビン·L·スミス医学博士との相談合意
2022年8月16日、会社は取締役会のロビン·L·スミス医学博士とコンサルティング契約を締結し、
クラ財団
当社は、2022年12月31日及び2021年12月31日までに、以下の貢献を行います$
Cota,Inc
2020年11月,Legacy Celularity and COTA,Inc.(“COTA”)はLegacy Celularity and COTA,Inc.(“COTA”)と2018年10月29日にLegacy CelularityとCOTA間の主データ許可プロトコルの注文表(“注文表2”)を締結し,この合意によりCOTAはAML患者に関する許可データを提供する。COTA注文付表2は、本明細書に記載された最終許可データが交付されてから1年以内に終了する。サイルラティ社のアンドリュー·ペコラ元社長はCOTA社の創始者と取締役会長であり、会社の取締役会メンバーのロビン·L·スミス博士はCOTA会社の投資家の一人である。会社はCOTAに費用を支払う$
Cryoportシステム会社
同社が支払った金額の総額は#ドルだ
C.V.スタールローン
2021年6月8日、Legacy Celularityは$に入りました
F-47
履歴書スタールです。C.V.Starrと締結された新しい融資と株式証明書契約に関する情報は、付記21後続事件を参照されたい。
ソレント治療会社
2020年9月、当社はソレントと2020年のソレント協定を締結しました。紀万昌博士、Legacy Celularity取締役会のメンバーで、現在ソレント社の最高経営責任者兼総裁を務めている。ソレントも同社の重要株主であり、2021年7月のパイプライン融資に投資している。2022年12月31日及び2021年12月31日までに、当社は合計で支払います$
直系親族を雇う
アレクサンドラ·ハリリはCelularity社の会長兼最高経営責任者ロバート·J·ハリリ医学博士の娘で、現在Celularity社に招聘され、取締役会社の戦略と業務発展部の幹部を務めている。2022年、2022年および2021年12月31日までの各年度ハリリさんの基本給は#ドルです
会社は後続事件を評価しており、以下の事項以外に開示すべき事項はない
ヨークビルが事件を起こした
2023年2月22日、ヨークビルは当社に通知を出し、PPA条項の規定により、“トリガ事件”が発生した。このトリガ事件のため、同社は今#ドルを返済しなければならない
ナスダック上場通知
2023年3月14日、当社はナスダック証券市場有限責任会社(“ナスダック”)の上場資格部門を受けて当社に通知し、当社A類普通株の終値が$1を割ったため、Celulityはナスダック上場規則第5450(A)(1)条に規定されているナスダック資本市場への上場継続の最低購入価格要求に適合しなくなった
ナスダック上場規則第5810(C)(3)(A)条によれば、当社は
デラウェア州第205条手続き
2021年7月14日、当時GX買収会社(“合併前会社”)というCelularityは株主特別総会(“特別会議”)を開催し、合併前会社とCelularis Operations,Inc.(“Legacy Celularity”)業務合併のいくつかの事項を承認し、合併前会社の改訂および再記載された会社登録証明書(“合併前憲章”)の改訂証明書を通じて、合併前会社普通株の法定株式数を増加させることを提案した
デラウェア州衡平裁判所(“裁判所”)最近の裁決ガーフィールドでBOXED,Inc.を訴えた2022年WL 17959766(Del.CH2022年12月27日)は、合併前憲章の追加修正案を承認するために、テルアビブ州会社法第242(B)(2)条がCelulityにA類普通株式の多数の株式を求めて獲得することを要求するかどうかの投票を不確実性としている。したがって、任意の潜在的不確実性を解消するために、Celulalityは、2023年3月14日にDGCL第205条に基づいて裁判所に請願書(“請願書”)を提出し、改正案及びそれに基づいて発行された株式の増加を含む新たな憲章の有効性の確認及び発表を要求し、タイトルはRe Celulityでは
F-48
Inc.C.A.番号2023-0317-LWW(Del.CH.)(“205条項アクション”)。DGCL第205条は、裁判所が様々な要因を考慮した後、会社の行為及び株式を適宜承認及び確認又は確認することを許可する。
2023年3月29日、大裁判官裁判所は第205条訴訟について公聴会を開催し、請願書を口頭で承認し、同日遅く、裁判所は第205条訴訟において、午前10時から改正案と会社登録証明書を追加して発効することを確認し、発表した。2021年7月16日(米国東部サマータイム)、及び増資改正案及び会社登録証明書の効力に基づいて発行されたすべての会社株は、当該株式の最初の発行日時までである。“裁判所命令”は解決し削除されましたガフィー猫これは裁判所の裁決です。
橋越しローンを優先的に保証します
当社は2023年3月17日、当社の主要株主の一人であるスタールと融資合意を締結し、元金総額#ドルの融資(“スタールローン”)を提供する
私募&再許可契約の拘束性条項説明書
当社は2023年3月20日、2人の認可投資家(議長兼最高経営責任者Robert Hariri博士を含む)と証券購入協定を締結し、私募(I)を規定した
パイプごとの株式証の行使価格は$である
当社も2023年3月27日に買い手と登録権協定を締結し、A類普通株株式の登録及び管路株式承認証行使後に発行可能なA類普通株株式の転売と、拘束力のある再許可条項説明書に基づいて支払いとして発行された株式に同意した。私募取引が終了するとともに、当社は、上記私募取引の認可投資家側の連属会社のある資産について交渉して再許可を締結する拘束力のある条項説明書に署名した。拘束力のある条項説明書によると、会社は分譲先に#ドルを支払った
労働力の減少
2023年1月、同社は仕事の優先順位の再決定を発表し、約20%減少した2023年3月までの従業員総数。
Palantir Technologies Inc.と合意した.
2023年1月、会社はPalantir Foundryプラットフォームの使用を停止し、Palantirに論争通知を出した。理由は、このソフトウェアが約束通りに動作しておらず、PalantirがFoundryプラットフォームの実施、統合、および有効化に必要な専門サービスを会社に提供できなかったからである。そのためにはASC 420はコストを終了または処理し、2023年第1四半期に、会社は契約項目の下で発生する余剰コストと負債を約$と確認する
F-49
項目9.Accouとの変更と分岐会計と財務情報開示の専門。
ない。
第9条。制御するプログラムがあります
情報開示制御とプログラムの評価
我々の経営陣は、本年度報告書10-K表に含まれる期間の終了時に、取引法第13 a-15(E)及び15 d-15(E)条に規定されている規定に適合する当社の開示制御及びプログラムの有効性を評価している。取引法第13 a-15(E)及び15 d-15(E)条の規則の定義によれば、“開示制御及び手続”という言葉は、取引法に基づいて会社が提出又は提出した報告書において開示を要求する情報が米国証券取引委員会規則及び表に規定された期間内に記録、処理、まとめ及び報告されることを保証するための会社の制御及びその他の手続を意味する。開示制御及び手続は、取引所法案の提出又は提出された報告書に基づいて企業が開示を要求する情報が蓄積されていることを確保し、その主要幹部及び主要財務官を含む適切な管理層に伝達して、開示要求に関する決定をタイムリーに行うことを目的としているが、制御及び手続に限定されない。すべての制御系には固有の限界があるため,制御系の発想や操作がどんなに良くても,絶対的な保証ではなく合理的な保証しか提供できず,制御系を確保する目標が実現される.これらの固有の限界は,意思決定過程における判断が誤りである可能性があり,簡単な誤りや誤りによって故障が発生する可能性があるという現実を含む.さらに、ある人の個人的な行動、2人以上の結託、または制御の管理を凌駕することによって制御を回避することができる。また,制御システムの設計は,資源制約が存在し,そのコストに対する制御の利点を考慮しなければならないという事実を反映しなければならない.2022年12月31日までの開示制御·手順の評価によると、我々の最高経営責任者および最高財務官は、以下に議論する財務報告内部統制に大きな弱点があることを考慮して、この日までに、私たちの開示制御および手続きは合理的な保証レベルでは有効ではないと結論した。
財務報告の内部統制
私たちの経営陣は財務報告書の十分な内部統制を確立して維持する責任がある。私たちの内部統制システムは、財務諸表の作成と公正な列報に関する合理的な保証を私たちの経営陣と取締役会に提供することを目的としています。すべての内部制御システムには,どんなに良く設計されていても,固有の限界がある.したがって,有効と判定されたシステムであっても,財務諸表の作成や列報に合理的な保証を提供することしかできない.内部制御重大欠陥は重大欠陥或いは欠陥の集合であり、財務諸表中の重大な誤報リスクを相対的に低いレベルに下げることができず、即ち従業員は正常な仕事過程中に適時に重大な誤報を防止或いは発見することができる。内部統制の重大な欠陥、または欠陥の集合は、単に無関係ではなく、財務諸表の誤った陳述をもたらす可能性がある。財務報告管理の内部統制の評価には,テレデビル委員会後援組織委員会が“内部統制−総合枠組み(2013年)”で発表した基準を採用した。我々の経営陣は、2022年12月31日までの財務報告内部統制の有効性を評価し、財務報告内部統制には以下のような大きな弱点があるため、財務報告内部統制は合理的な保証レベルでは有効ではないと判断した
私たちは現在、このような重要な弱点を解決するために私たちの救済計画を実行している。これらの措置には
88
財務報告の内部統制の変化
上記の救済措置を継続して実施する場合を除いて、2022年12月31日までの四半期内に、財務報告の内部統制に大きな影響を与えたり、合理的に私たちの財務報告の内部統制に大きな影響を与える可能性のある変化はありません。
プロジェクト9 B。他にも情報です。
先に開示したように、2023年3月14日、私たちはデラウェア州会社法第205条に基づいて、私たちの法定株式に関連する潜在的不確実性を解決するために、デラウェア州衡平裁判所に請願書を提出した。具体的には,この請願書のタイトルはRe Celularity,Inc.,C.A.No.2023-0317-LWW(Del.当社の前身が特殊目的買収会社GX Acquisition Corp.の2件目の改正及び再記載された会社登録証明書は、A類普通株の法定株式数を110,000,000株から730,000,000株に増加させるか、又はA類増資修正案に基づいて発行された株式を含むものであり、改訂及び再記載されたGX買収会社登録証明書の2つ目の確認を求めている。
2023年3月29日、衡平裁判所はSection 205訴訟について公聴会を開催し、口頭で請願書を承認し、同日遅く、裁判所はSection 205訴訟で命令を発表し、クラスA追加修正案と私たちの2回目の改正および再記載された会社証明書が午前10時に発効したことを確認し、発表した。2021年7月16日(米国東部サマータイム)、及びA級増資改正案及び当社の第2改正及び再記載された会社証明書の有効性に基づいて発行された全ての株式は、当該株の最初の発行日及び時間までである。裁判所命令の写しは,表格10−Kを本年度報告の証拠品としてアーカイブする。
プロジェクト9 Cです。外国の開示について検査を妨げる尿毒症。
適用されません。
89
第三部
プロジェクト10.役員·役員職ICERSと会社管理。
本プロジェクトに要求される情報は、2023年株主年次総会のための私たちの最終委託書または依頼書の“提案1-取締役選挙”、“取締役会と会社管理に関する情報-商業行為と道徳基準”、“延滞16(A)条報告”、“取締役会と会社管理に関する情報-指名と会社管理委員会”および“取締役会と会社統治に関する情報-監査委員会”の情報を参考にして格納される。
プロジェクト11.実行補償します。
本プロジェクトで要求される情報は、当社の依頼書の“役員報酬”と題する章の情報を参考にして組み込まれます。
プロジェクト12.特定の受益者の保証所有権従業員と経営陣と関連した株主問題。
本プロジェクトに要求される情報は、依頼書の“特定の実益所有者および管理層の保証所有権”と“役員報酬-持分報酬計画情報”と題する部分の情報を参照して統合される。
本プロジェクトに要求される情報は,依頼書の“取締役会と会社のガバナンスに関する情報である取締役会の独立性”と“関係者との取引”と題する部分の情報を参考にして格納されている.
第14項.元金口座TING料金とサービスです。
本プロジェクトで要求される情報は、私たちの依頼書の“提案3-承認独立公認会計士事務所の選択”と題する節の情報を参考にして組み込まれます。
90
第4部
プロジェクト15.物証、資金請求書の明細書です。
以下の書類は本報告の一部として提出する
必要ではない、または要求された資料が財務諸表または付記に含まれているので、他のすべての付表は省略されている
展示品 番号をつける |
|
説明する |
|
|
|
2.1+ |
|
GX Acquisition Corp.,Alpha First Merge Sub,Inc.,Alpha Second Merge Sub,LLCとCelularity Inc.との間の統合プロトコルおよび再構成計画(添付ファイル2.1を参照して2021年1月8日に委員会に提出された8-K表報告書に組み込まれる)。 |
|
|
|
3.1 |
|
修正および再改訂された2つ目の会社登録証明書(2021年7月22日に委員会に提出されたテーブル8−Kの現在の報告書の添付ファイル3.1を参照して編入)。 |
|
|
|
3.2 |
|
付則の改訂および再改訂(2021年7月22日に委員会に提出されたタブ8−K現在の報告書の添付ファイル3.2を参照して編入)。 |
|
|
|
4.1 |
|
普通株式証明書サンプル(添付ファイル4.1を参照して2021年7月22日に委員会に提出されたテーブル8−Kの現在の報告書の添付ファイル4.1を組み込む)。 |
|
|
|
4.2* |
|
証券説明(添付ファイル4.3を参照して2022年3月31日に証監会に提出された10-K表年次報告書に組み込まれる)。 |
|
|
|
10.1 |
|
改訂および再署名された“登録権協定”(添付ファイル10.3を参照することにより、2021年7月22日に委員会に提出された現在の8−K表報告書の添付ファイル3に組み込まれる)。 |
|
|
|
10.2 |
|
2021年7月16日現在、GXスポンサー有限責任会社Celularity Inc.(F/k/a GX Acquisition Corp.)と署名ページに記載された他の各者との間で署名されたホーム合意(2021年7月22日に委員会に提出された現在の8−K表報告書の添付ファイル10.4を参照して組み込まれる)。 |
|
|
|
10.3 |
|
GX Acquisition Corp.とContinental Stock Transfer&Trust Companyが権証エージェントとして署名した、2019年5月20日の引受権証契約(2019年5月24日に委員会に提出された8-K表の現在の報告書の添付ファイル4.1を参照して組み込まれます)。 |
|
|
|
10.4 |
|
授権書サンプル(添付ファイル4.2を参照することによって、2021年7月22日に委員会に提出された現在の8−Kフォーム報告書の添付ファイル4.2に組み込まれる)。 |
|
|
|
10.5# |
|
賠償プロトコル表(表S-4登録説明(第333-252402号文書参照)添付ファイル10.9に組み込まれ、2021年6月22日に委員会に提出される)。 |
|
|
|
10.6# |
|
Celularity Inc.2017年持分インセンティブ計画(添付ファイル10.10を参照してS-4表登録声明(第333-252402号文書)に組み込まれ、2021年6月22日に委員会に提出された)が改訂され、再起動された。 |
|
|
|
10.7# |
|
2017年株式インセンティブ計画下の株式オプション付与通知、オプション合意、行使通知の表(2021年6月22日に委員会に提出された表S-4登録声明(第333-252402号文書)の添付ファイル10.11を参照して編入)。 |
91
|
|
|
10.8# |
|
Celularity Inc.2021年持分インセンティブ計画(添付ファイル99.3を参照してS-8表登録説明書(第333-260025号ファイル)に組み込まれ、2021年10月4日に委員会に提出される)。 |
|
|
|
10.9# |
|
株式オプション付与通知、オプション協定、行使通知、RSU付与通知、および2021年株式インセンティブ計画下の奨励協定(添付ファイル99.4を参照してS-8表登録声明に組み込まれる(第333-260025号文書は、2021年10月4日に委員会に提出)。 |
|
|
|
10.10# |
|
繰延賠償金贈与表(添付ファイル10.15を参照することにより、2021年7月22日に提出された委員会のタブ8−Kの現在の報告書の添付ファイル10.15に組み込まれる)。 |
|
|
|
10.11# |
|
Celularity 2021年従業員株式購入計画(添付ファイル99.5を参照してS-8フォーム登録宣言(ファイル番号333-260025)に組み込まれ、2021年10月4日に委員会に提出される。 |
|
|
|
10.12# |
|
Celularity Inc.2018年年次奨励計画(添付ファイル10.14を参照してS-4表登録声明(第333-252402号ファイル)に組み込まれ、2021年6月22日に委員会に提出されます)。 |
|
|
|
10.13# |
|
CelulityとRobert J.Haririとの間の改訂および再署名された雇用協定は、2021年1月7日(添付ファイル10.15を参照してS-4表登録声明(第333-252402号文書)に組み込まれ、2021年6月22日に委員会に提出される)。 |
|
|
|
10.14# |
|
雇用協定の修正は,2023年1月25日までにCelulity Inc.とRobert J.Haririが共同で完了した。 |
|
|
|
10.15# |
|
Celularity Inc.とDavid C.Beersとの間の雇用協定は、2022年4月1日現在、(2022年11月10日に委員会に提出されたForm 10-Q四半期報告書の添付ファイル10.7を参照して組み込まれる)。 |
|
|
|
10.16# |
|
Celularity Inc.とStephen A.Brigidoとの間の雇用協定は,2022年4月1日までである(引用により2022年11月10日に委員会に提出されたForm 10-Q四半期報告書の添付ファイル10.6)。 |
|
|
|
10.17# |
|
Celularity Inc.とKeary Dunnとの間の雇用協定は、2022年7月13日現在、(2022年11月10日に委員会に提出されたForm 10-Q四半期報告書の添付ファイル10.10を参照して組み込まれる)。 |
|
|
|
10.18# |
|
Celularity Inc.とKyle H.Fletcherとの間の雇用協定は,2022年7月13日まで(添付ファイル10.9を参照して2022年11月10日に委員会に提出されたForm 10-Q四半期報告書の添付ファイル10.9に組み込まれている)。 |
|
|
|
10.19# |
|
Celulity Inc.とBradley Gloverとの間の雇用合意は、2022年10月4日現在、Celulity Inc.とBradley Gloverとの間の雇用合意(2022年11月10日に委員会に提出されたForm 10-Q四半期報告書の添付ファイル10.11を参照して組み込まれる)。 |
|
|
|
10.20#* |
|
Celulity Inc.とJohn R.Hainesとの間の雇用合意は、2022年4月1日現在、Celulity Inc.とJohn R.Hainesとの間の雇用協定(添付ファイル10.8を参照することにより、2022年11月10日に委員会に提出されたForm 10-Q四半期報告書の添付ファイル10.8に組み込まれる)。 |
|
|
|
10.21# |
|
Celularity Inc.と篠浩Kilcoyneとの間の雇用協定は、2022年9月29日現在である(2022年11月10日に委員会に提出されたForm 10-Q四半期報告書の添付ファイル10.12を参照することにより格納される)。 |
|
|
|
10.22# |
|
2022年8月16日現在、Celulity Inc.とRobin L.Smithとの間の諮問プロトコル(添付ファイル10.1を参照することによって、2022年11月10日に委員会に提出されたForm 10-Q四半期報告書の添付ファイル10.1に組み込まれる)。 |
|
|
|
10.23# |
|
Celularity Inc.とAndrew L.Pecoraとの間の諮問プロトコルは、2022年9月21日現在、Celularity Inc.とAndrew L.Pecoraとの間の諮問プロトコル(参照によって2022年11月10日に委員会に提出されたForm 10-Q四半期報告の添付ファイル10.4に組み込まれている)。 |
|
|
|
10.24# |
|
科学·臨床コンサルタント協定は,2022年9月1日現在,Celularity Inc.とAndrew L.Pecoraによって署名されている(引用により2022年11月10日に委員会に提出されたForm 10−Q四半期報告の添付ファイル10.5に組み込まれている)。 |
|
|
|
10.25 |
|
リース契約は、2019年3月13日に、LSREF 4 Turtle,LLCとCelularity Inc.によって締結される(添付ファイル10.32を参照して2021年6月22日に委員会に提出されたS-4フォーム登録声明(第333-252402号ファイル)に組み込まれる)。 |
92
|
|
|
10.26¥ |
|
Celgene CorporationとAnthrogenensis Corp.の間で2017年8月15日に署名された許可協定(添付ファイル10.23を参照して2021年6月22日に委員会に提出されたS-4表登録声明(文書番号333-252402)に組み込まれている)。 |
|
|
|
10.27¥ |
|
“又は価値ある権利協定”は、Celularity Inc.とその中で指名された所持者との間で締結され、日付は2017年8月15日であり、2021年3月4日の“又は価値ある権利協定”改正案第1号改正(2021年6月22日に委員会に提出されたS-4表登録声明(第333-252402号文書)添付ファイル10.25を参照して格納される)。 |
|
|
|
10.28 |
|
Celularity Inc.とCelgene Corporationとの間の投資権協定は、2017年8月15日であり、2021年3月4日の投資権協定第1号改正案を経て改正された(2021年6月22日に委員会に提出されたS-4表登録声明(第333-252402号文書)添付ファイル10.26参照)。 |
|
|
|
10.29¥ |
|
許可および譲渡協定は、日付が2020年9月30日であり、Celularity Inc.とSorrento Treateutics,Inc.によって締結され、改訂された(2021年6月22日に委員会に提出された表S-4登録声明(文書番号333-252402)の添付ファイル10.27を参照して編入される)。 |
|
|
|
10.30¥ |
|
Celularity Inc.,CariCord Inc.,CC子会社とGregory L.Andrewsの間で2018年8月22日に署名された合併協定と計画は,2018年9月30日の合併協定と計画第1修正案および2020年6月24日の合併協定と計画第2改正案を経て改訂された(2021年6月22日に委員会に提出されたS-4表登録声明(第333-252402号文書)添付ファイル10.28参照)。 |
|
|
|
10.31 |
|
Celularity Inc.とDragas ac Limitedの間のB系列優先株購入権証は,2020年1月9日(2021年6月22日に委員会に提出されたS-4表登録声明(文書番号333-252402)の添付ファイル10.29を参照して組み込む)である. |
|
|
|
10.32 |
|
Celularity Inc.B系列優先株を購入する引受権証の第1号修正案は,2020年3月16日にCelularity Inc.とDragas ac Limited間の引受権証(2021年6月22日に委員会に提出されたS-4表登録声明(文書番号333-252402)の添付ファイル10.30参照により格納される). |
|
|
|
10.33 |
|
Celularity Inc.B系列優先株を購入した引受権証の第2号修正案は,2021年1月8日にCelularity Inc.とDragac Limitedとの間の引受権証(2021年6月22日に委員会に提出されたS-4表登録声明(文書番号333-252402)の添付ファイル10.31を参照して編入される)である。 |
|
|
|
10.34 |
|
Celulality Inc.A類普通株を購入するA&R承認株式証表(添付ファイル10.1を参照して現在報告されている8-K表を参照することにより、2022年3月1日に委員会に提出される)。 |
|
|
|
10.35 |
|
2021年7月16日現在、GXスポンサー有限責任会社Celularity Inc.(F/k/a GX Acquisition Corp.)と署名ページに記載された他の各者との間で署名されたホーム合意(2021年7月22日に委員会に提出された現在の8−K表報告書の添付ファイル10.4を参照して組み込まれる)。 |
|
|
|
10.36 |
|
Celulity Inc.買い手と2022年5月18日に署名された証券購入協定(添付ファイル10.1を参照することによって、2022年5月20日に証券取引委員会に提出された現在の8-K表報告書に組み込まれる)。 |
|
|
|
10.37 |
|
配管保証書の形式(2022年5月20日提出委員会の8−K表現在報告書の添付ファイル10.2を参照)。 |
|
|
|
10.38 |
|
Celularity Inc.所有者と2022年5月18日に締結された登録権協定(添付ファイル10.3を参照して2022年5月20日に委員会に提出された現在の8−K表報告書に組み込まれる)。 |
|
|
|
10.39 |
|
配給エージェントプロトコルは、2022年5月18日、Celulity Inc.と配給エージェント会社との間の合意である(添付ファイル10.4を参照することにより、2022年5月20日に委員会に提出された現在の8−Kフォーム報告に組み込まれる)。 |
|
|
|
93
10.40 |
|
Celularity Inc.,BTIG,LLC,Oppenheimer&Co.Inc.およびB.Riley Securities,Inc.が2022年9月8日に署名したマーケティング協定(2022年9月8日に委員会に提出された現在の8-K表の添付ファイル1.1を参照して組み込む)。 |
|
|
|
10.41 |
|
Celularity Inc.とYA II PN,Ltd.の間で2022年9月15日に締結された前払い契約(添付ファイル10.1を参照することによって、2022年9月15日に委員会に提出された現在の8−Kフォーム報告書の添付ファイル10.1に組み込まれる)。 |
|
|
|
16.1 |
|
Marcum LLPの手紙は、日付が2021年7月21日である(添付ファイル16.1を参照して2021年7月22日に委員会に提出された現在のタブ8-K報告書に組み込まれる)。 |
|
|
|
21.1 |
|
子会社リスト(添付ファイル3.1を参照して2021年7月22日に委員会に提出された現在の8-K表報告書に組み込まれる)。 |
|
|
|
23.1* |
|
徳勤法律事務所は同意した。 |
|
|
|
24.1* |
|
授権書(署名ページに含まれる)。 |
|
|
|
31.1* |
|
2002年のサバンズ·オキシリー法第302節で可決された1934年の証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて発行された首席執行幹事証明書。 |
|
|
|
31.2* |
|
2002年サバンズ−オキシリー法第302節で可決された1934年証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて首席財務幹事が認証された。 |
|
|
|
32.1* |
|
2002年の“サバンズ-オックススリー法案”906節で採択された“米国法典”第18編1350条による最高経営責任者と最高財務官の認証。 |
|
|
|
99.1 |
|
デラウェア州衡平裁判所令 |
|
|
|
101.INS |
|
連結されたXBRLインスタンス文書−インスタンス文書は、XBRLタグがイントラネットXBRL文書に埋め込まれているので、相互作用データファイルには表示されない。 |
101.書院 |
|
イントラネットXBRL分類拡張アーキテクチャ文書 |
101.カール |
|
インラインXBRL分類拡張計算リンクライブラリ文書 |
101.def |
|
インラインXBRL分類拡張Linkbase文書を定義する |
101.介護会 |
|
XBRL分類拡張ラベルLinkbase文書を連結する |
101.Pre |
|
インラインXBRL分類拡張プレゼンテーションLinkbaseドキュメント |
104 |
|
表紙インタラクションデータファイル(イントラネットXBRL文書に埋め込む) |
|
|
|
*アーカイブをお送りします。
# I管理契約または任意の補償計画、契約または手配を指定する.
+ S-K規則601(B)(2)項により,表と証拠物が省略されている.私たちはアメリカ証券取引委員会の任意の漏れたスケジュールや展示品のコピーの追加提供を要求しなければならないということに同意する。
元本展示品のいくつかの部分は省略されています。それらは実質的ではありませんので、登録者が個人または機密とみなすタイプです。
取引法第18条の規定によると、これらの証明書は提出されたとみなされることもなく、当該条項の責任を負うこともない。このような証明は、参照によってそのような出願の範囲が明示的に組み込まれない限り、“証券法”または“取引法”に従って提出された任意の出願に参照によって組み込まれているとみなされないであろう。
項目16.表10-Kの概要
適用されません。
94
サイン
改正された1934年証券取引法第13又は15(D)節の要求に基づいて、登録者は、正式に許可された次の署名者がその代表を代表して本報告書に署名することを正式に手配した.
|
|
Celularity Inc. |
|
|
|
|
|
日付:2023年3月31日 |
|
差出人: |
ロバート·J·ハリリ |
|
|
|
ロバート·J·ハリリ医学博士 |
|
|
|
最高経営責任者 |
このような身分ですべての者を知り,ロバート·J·ハリリ,M.D.,博士,David·ビルス,K.ハルト·フレッチャーとその本人とその真の合法的な代理弁護士と代理人を任命し,その本人の名義,場所,代理,本10-K表年次報告の任意およびすべての修正案に任意およびすべての身分で署名し,それをアーカイブし,その証拠物とそれに関連する他の文書を添付する。米国証券取引委員会に、上記の代理弁護士及び代理人及びその一人一人に十分な権力及び権力を付与し、すべての必要及び必要なものを行い、実行し、その可能性又は自ら行うことができるすべての意図及び目的を尽くし、ここで上記の事実代理人及び代理人の各々を承認して確認すること、又は彼又は彼女の1人以上の代理人が本条例により合法的に又は手配することができるすべてを和事として行うことができる
改正された1934年の証券取引法の要求に基づき、本報告は署名されたD以下は次の者が登録者として指定日に登録者を代表して提出する。
名前.名前 |
|
タイトル |
|
日取り |
|
|
|
|
|
ロバート·J·ハリリ |
|
最高経営責任者兼取締役会長(執行役員) |
|
2023年3月31日 |
ロバート·J·ハリリ医学博士 |
|
|
|
|
|
|
|
|
|
/s/David C.Beers |
|
首席財務官(首席財務·会計幹事) |
|
2023年3月31日 |
デビッド·C·ビル |
|
|
|
|
|
|
|
|
|
/s/ダイアン·パークス |
|
役員.取締役 |
|
2023年3月31日 |
ダイアン·パークス |
|
|
|
|
|
|
|
|
|
/s/ピーター·ディマティス |
|
役員.取締役 |
|
2023年3月31日 |
ピーター·ディマンディス医学博士 |
|
|
|
|
|
|
|
|
|
/s/Dean C.Kehler |
|
役員.取締役 |
|
2023年3月31日 |
ディーン·C·ケラー |
|
|
|
|
|
|
|
|
|
|
|
役員.取締役 |
|
|
林角泰 |
|
|
|
|
|
|
|
|
|
/s/Marc Mazur |
|
役員.取締役 |
|
2023年3月31日 |
マーク·マズール |
|
|
|
|
|
|
|
|
|
/s/ジョン·スカリー |
|
役員.取締役 |
|
2023年3月31日 |
ジョン·スカリー |
|
|
|
|
|
|
|
|
|
ロビン·L·スミス |
|
役員.取締役 |
|
2023年3月31日 |
ロビン·L·スミス医学博士商工管理修士 |
|
|
|
|
|
|
|
|
|
アンドリュー·C·フォン·エゼンバッハ |
|
役員.取締役 |
|
2023年3月31日 |
アンドリュー·フォン·エッセンバッハ医学博士 |
|
|
|
|
95