t
f
アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
本財政年度末まで
あるいは…。
手数料書類番号
(登録者の正確な氏名はその定款に記載)
(明またはその他の司法管轄権 会社や組織) |
(税務署の雇用主 |
|
|
(主にオフィスアドレスを実行) |
(郵便番号) |
(
(登録者の電話番号、市外局番を含む)
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
取引記号 |
所在する取引所名を登録する |
♪the the the |
同法第12条(G)に基づいて登録された証券:
ありません
登録者が証券法規則405で定義されている経験豊富な発行者である場合は、再選択マークで示してくださいはい、そうです ☐
登録者が当該法第13条又は第15条(D)に従って報告書を提出する必要がないか否かを、再選択マークで示すはい、そうです ☐
I登録者が(1)過去12ヶ月以内(または登録者がそのような報告の提出を要求された短い期間内)に、1934年の証券取引法第13または15(D)節に提出されたすべての報告書を提出したかどうか、および(2)このような提出要求を過去90日以内に遵守してきたかどうかを、再選択マークで示す。
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
大型加速ファイルサーバ |
|
☐ |
|
ファイルマネージャを加速する |
|
☐ |
|
☒ |
|
小型報告会社 |
|
||
新興成長型会社 |
|
|
|
|
|
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する☐
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する☐
これらのエラーのより真ん中に登録者の任意のエンタルピーCER幹部が相関回復期間内に§240.10 D−1(B)に基づいて受信したインセンティブベースの補償に基づいて回復分析を行う必要があるかどうかを再選択マークで示す☐
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)。はい、そうです
2022年6月30日まで,すなわち登録者が最近完成した第2四半期の最終営業日,登録者の非関連会社が保有する普通株総時価は$である
2023年3月22日までに
引用で編入された書類
以下の書類(または一部の文書)は、本Form 10-Kの次の部分を参照して組み込む:Form 10-K年次報告第III部分に要求されるいくつかの情報は、2023年の株主総会に対する登録者からの最終委託書から、登録者は、2022年12月31日までの財政年度の120日以内に第14 A条に従って米国証券取引委員会に提出される。
監査役事務所ID: |
監査役の名前: |
監査役位置: |
i
カタログ表
|
|
ページ |
第1部 |
|
|
第1項。 |
業務.業務 |
2 |
第1 A項。 |
リスク要因 |
41 |
項目1 B。 |
未解決従業員意見 |
77 |
第二項です。 |
属性 |
77 |
第三項です。 |
法律訴訟 |
77 |
第四項です。 |
炭鉱安全情報開示 |
77 |
|
|
|
第II部 |
|
|
五番目です。 |
登録者普通株市場、関連株主事項及び発行者による株式証券の購入 |
78 |
第六項です。 |
[保留されている] |
78 |
第七項。 |
経営陣の財務状況と経営成果の検討と分析 |
79 |
第七A項。 |
市場リスクの定量的·定性的開示について |
92 |
第八項です。 |
財務諸表と補足データ |
92 |
第九項です。 |
会計と財務情報開示の変更と相違 |
92 |
第9条。 |
制御とプログラム |
92 |
プロジェクト9 B。 |
その他の情報 |
93 |
プロジェクト9 Cです。 |
検査妨害に関する外国司法管区の開示 |
93 |
|
|
|
第三部 |
|
|
第10項。 |
役員·幹部と会社の管理 |
94 |
第十一項。 |
役員報酬 |
94 |
第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
94 |
十三項。 |
特定の関係や関連取引、取締役の独立性 |
94 |
14項です。 |
最高料金とサービス |
94 |
|
|
|
第4部 |
|
|
第十五項。 |
展示·財務諸表明細書 |
95 |
第十六項。 |
表格10-Kの概要 |
98 |
II
前向きに陳述する
このForm 10-K年次報告書はリスクと不確実性に関する前向きな陳述を含む。我々は1995年の個人証券訴訟改革法と他の連邦証券法における安全港条項に基づいてこのような前向きな声明を行った。本稿では,歴史的事実に関する陳述を除くすべての陳述は前向き陳述である.場合によっては、前向きな陳述は、“可能”、“予想”、“すべき”、“予想”、“意図”、“計画”、“予想”、“信じ”、“推定”、“予測”、“予測”、“潜在”、“継続”またはこれらの用語または他の同様の用語の否定的意味によって識別することができる。これらの前向きな陳述は、以下の態様に関する陳述を含むが、これらに限定されない
本年度報告中の10-K表の任意の前向き陳述は、既知と未知のリスク、不確定性およびその他の要素に関連し、未来の事件または私たちの未来の財務表現に対する私たちの現在の見方を反映しており、私たちの実際の結果、業績または業績は、これらの展望的陳述と明示的または暗示的な任意の未来の結果、業績または業績とは大きく異なる可能性がある。他に加えて、実際の結果が現在の予想と大きく異なることをもたらす可能性のある要因は、第1の部分1 Aの項に列挙された要因を含む。リスク要因“および本年度報告書10-K表の他の部分。このような不確実性を考慮して、あなたはこのような前向きな陳述に過度に依存してはいけない。法的要求がない限り、私たちは未来に新しい情報があっても、これらの前向きな陳述を任意の理由で更新または修正する義務がない。
このForm 10-K年間報告書はまた、特定の疾患の発症率および流行率に関するデータを含む、私たちの業界、私たちの業務、および特定の疾患の市場の推定、予測、および他の情報を含む。見積り,予測,予測,市場研究や類似方法に基づく情報自体は不確定要素の影響を受け,実際のイベントや状況はその情報に反映されるイベントや状況とは大きく異なる可能性がある.他に明確な説明がない限り、私たちは、報告、研究調査、研究、および市場研究会社および他の第三者によって準備された類似データ、業界、医療および一般出版物、政府データ、および同様のソースから、これらの業界、商業、市場、および他のデータを取得する。
1
パー?パーT I
プロジェクト1.BU無邪気ですね。
概要
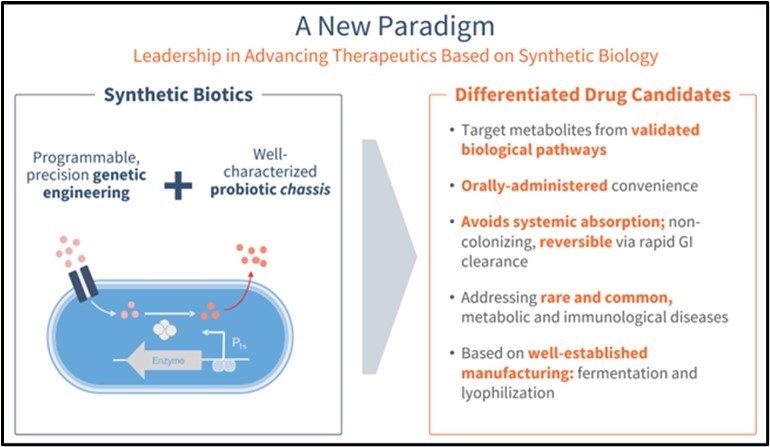
我々は後期段階にある臨床バイオテクノロジー会社であり,合成生物学に基づく生物療法の設計と開発に取り組んでいる。合成生物学は生物システム、遺伝回路と分子コンポーネント設計における工学原理の応用である。著者らは合成生物学の力を医学に応用し、精密工学と合理的な薬物開発を結合し、合成生物技術を生産する:遺伝子工学を通じて治療効果の生プロバイオティクスを獲得する。
この方法は新薬の設計,開発,商業化に優れていると考えられる。我々の候補薬物は精密工学により設計され,特定の疾患の病理生理学的に知られている有効な生物経路を目指している。我々の現在のパイプラインは、100年以上のヒト投与経験を有する大腸菌Nissle 1917のプロバイオティクスシャーシに基づくすべての経口候補薬剤を含む。われわれの候補薬物の活性は胃腸(GI)に限られており,系統的曝露や関連リスクを回避しており,これらのリスクは他の薬剤の成功を制限している。我々の候補薬物も非植民地的に設計され,GI許可により完全に可逆的である。これらの特性は,まれでよく見られる疾患に対する潜在薬物の開発を推進し,代謝や免疫学的適応に重点を置くことができる。私たちのプラットフォームは発酵と凍結乾燥を含む熟知した基礎を持つ製造技術を利用して、技術の設計と拡大を促進し、そして私たちの独特な製品のためにカスタマイズした独特と独自の革新を結合します。
2014年に設立されて以来、MIT(MIT)の技術に基づいて、私たちの合成バイオテクノロジーは、様々な段階の様々な候補薬物のパイプラインに発展してきました
2
戦略.戦略
我々の使命は,合成生物学を治療学に応用することで,新しい薬物を研究,開発,商業化し,他の方法では提供できない疾患を治療することである。私たちは合成バイオテクノロジーの方法を通じて、差別化された世界的なバイオ製薬会社になるつもりです。私たちの最近の優先順位は、SYNB 1934を私たちの重要な第3段階試験Synheny-3を通じて推進することを含む。私たちはまた私たちの早期パイプラインを推進しながら、私たちの薬物発見、開発、製造方法を最適化しています。
私たちの戦略の要素は
我々は,我々の取締役会と科学顧問委員会の支持を得ており,両委員会とも薬物発見と開発における補足経験と,上場企業の設立,管理,商業化,業務発展に関する専門知識を提供している。我々の基礎科学は,マサチューセッツ工科大学教授ジェームズ·コリンズとティモシー·Luの実験室から来ており,我々の科学顧問委員会のメンバーとして,我々の製品エンジンの開発と応用を指導することに取り組んでいる。
3
私たちのパイプライン:臨床開発における合成バイオテクノロジー
著者らの現在の臨床段階パイプラインは潜在代謝障害による重大な医療需要に対する候補薬物を含む。これらの薬剤にはSYNB 1934が含まれており,2023年上半期に北京大学の3期研究に入る予定であり,HCU,腸源性高草酸尿,痛風を治療するための候補薬剤である。我々の臨床前の仕事には,追加の代謝性疾患研究,標的探索,免疫学,特にIBDに集中する努力がある。
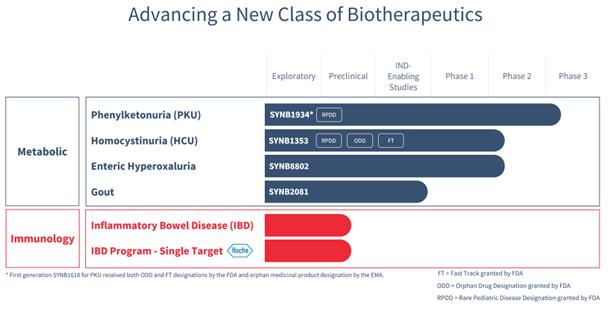
臨床管:代謝性疾患
代謝性疾患は,我々の身体が重要な代謝物や分子の欠陥や変化を分解あるいは産生することによる疾患である。これらの疾患は、PKUやHCU、あるいは臓器機能障害、例えば腸源性高草酸尿、あるいは遺伝感受性と環境要素の組み合わせ、例えば痛風などの遺伝子突然変異によって引き起こされる可能性がある。これらの疾患を有する患者では、いくつかの酵素の欠乏は、消化管および全身に代謝物の蓄積をもたらす。PKUおよびHCU患者では、これらの代謝物は有毒レベルに蓄積され、生命を脅かす医療リスクおよび/または深刻な合併症をもたらす可能性がある。
私たちの北大プロジェクトは
2022年10月,Synheny−1第2段階研究の陽性結果から,SYNB 1934がSynheny−3に入る候補薬であることが確認され,重要な第3段階研究であり,2023年上半期に開始される予定である。SYNB 1934は経口、非系統吸収の候補薬物であり、プロバイオティクスに基づく大腸菌.大腸菌Nissleは遺伝子工学によりフェニルアラニンを代謝する。これまでの研究結果はSYNB 1934を支持する潜在力が1種の有効、安全、便利な経口薬物選択になり、年齢或いはPKUの遺伝子型を問わず使用することができ、単一治療或いは補助治療にも応用できる。2023年1月、私たちはSYNB 1934がFDAによって付与された稀な小児科薬物称号(RPDD)を獲得したことを発表した。SYNB 1934が主要候補薬であることを確認する前に,第二段階研究により第一世代北京大学薬物候補薬SYNB 1618を開発した。SYNB 1618はFDAの孤児薬物と迅速通路指定、及びヨーロッパ薬品管理局(EMA)の孤児指定に対する積極的な評価を獲得した。SYNB 1618のより強力なバージョンと似ているので、SYNB 1934のこれらの称号を受け取る予定です。
北大の科学
PKUは遺伝性代謝性疾患であり、アミノ酸フェニルアラニン(Phe)の体内蓄積を引き起こす。Pheはすべての肉と乳製品、大多数の豆類、穀物とジャガイモ、およびいくつかの人工甘味料に含まれる天然タンパク質に存在する。PKU患者はフェニルアラニンヒドロキシ酵素(PAH)の遺伝欠陥を遺伝し、この酵素はフェニルアラニンを分解するために必要であり、潜在的な神経毒性レベルを招く。児童期にコントロールされていないPheレベルは正常な脳発育を妨害し、永久的な発育障害を招く可能性があり、いかなる年齢のPheレベルの深刻な上昇はすべて認知が遅くなり、注意力が集中しにくい或いは“脳霧”などの神経認知症状を招く可能性がある
4
PKU患者の合併症のリスクを減少させるためには,Pheレベルを生涯制御する必要がある。20世紀60年代に、各国は新生児のスクリーニングを開始し、高度制限性の低Phe飲食を通じて直ちにPhe制御を実施し、恒久的な深刻な知的障害を避けることを確保した。EUと米国のガイドラインによると,任意の血漿Phe濃度>400−600μMの新生児はできるだけ早く低Phe食を開始すべきである。食事には、毎日のタンパク質レベルを1日4~6 gまで低下させるための天然食品の厳格な制限が含まれており、これは、一般に、すべての動物タンパク質および乳製品、豆類およびナッツの供給源を除去し、パン、パスタ、米、およびいくつかの野菜の摂取量を制限することを含む。澱粉から作られた低蛋白パンとパスタ製品は必要なエネルギーを提供するために使用される。必要な食事レジメンは、アミノ酸ベースのフェニルアラニンを含まない配合乳または他の特定の医療食品を添加して、十分なタンパク質、ビタミン、ミネラルおよびエネルギーを提供することをさらに含む。
このような制限された食事を固守することは非常に挑戦的だ。特に挑戦は思春期の独立性が強くなる時期に現れる。高タンパク質、未修飾等価物よりも高価で栄養が悪いため、低タンパク質食品を得ることは困難をもたらす可能性があり、必要な配合粉ミルクも高価または入手が困難である可能性がある。そのため、大多数の北京大学と一緒に生活している人、特に独立して生活している成人は、Pheレベルが公認された目標よりはるかに高い。
米国、ヨーロッパ、アジアでは15万人以上が北京大学患者と診断されていると推定されている。彼らは通常新生児スクリーニングで診断されていますこれらの疾病の中で、大多数はまだ治療されておらず、これは現在の治療方案の局限性を反映している。
現在のPKU治療法の限界は
現在、2種類の薬物はすでにFDA、EMAとその他の全世界の監督管理機関によってPKUの薬物治療として許可され、血漿Pheレベルを下げる安全性と有効性に基づいている。これらの承認された薬物は臨床開発と監督手続きに前例を提供したが、著者らは異なる制限があると考えているため、以下に述べるような新しい北大医療方法が切実に必要である
5
Saproterinの応答率における限界,およびPalynziqの安全性の挑戦を考慮すると,承認された療法の限界は,ほとんどのPKU患者に経口薬物治療レジメンを必要とし,許容可能な安全性と耐性のPHE低下効果を提供すると考えられる。SYNB 1934は潜在的に安全で経口的な製品としてPheレベルを著しくかつ持続的に低下させることができ,単一療法または補助治療レジメンとしてこの需要を満たすことができると信じている。

SYNB 1934 PKUに適用されます
私たちはフェニルアラニンを合成する方法を設計しました大腸菌.大腸菌Nissleの設計はフェニルアラニンが胃腸を通過する時に代謝を行い、飲食由来のフェニルアラニンと腸管循環を介して胃腸に入るフェニルアラニンを標的とする。フェニルアラニン代謝はフェニルアラニンを消費するL−アミノ酸デアミナーゼ(LAAD)とフェニルアラニンデアミナーゼ(PAL)を添加することで達成される。これらの菌株はフェニルアラニン輸送体PhePを産生するように設計されており、フェニルアラニンを細胞に導入し、そこでPALによって代謝される。凍結乾燥粉配合の香包を含む形態で患者に提供し,水と混合し,食事とともに服用し,1日3回服用した。この製品紹介はPKU患者にとってよく知られており,フミントレキセートは香包の形で提供されているため,彼らの栄養補充製剤は通常液体と混合する必要がある。
6
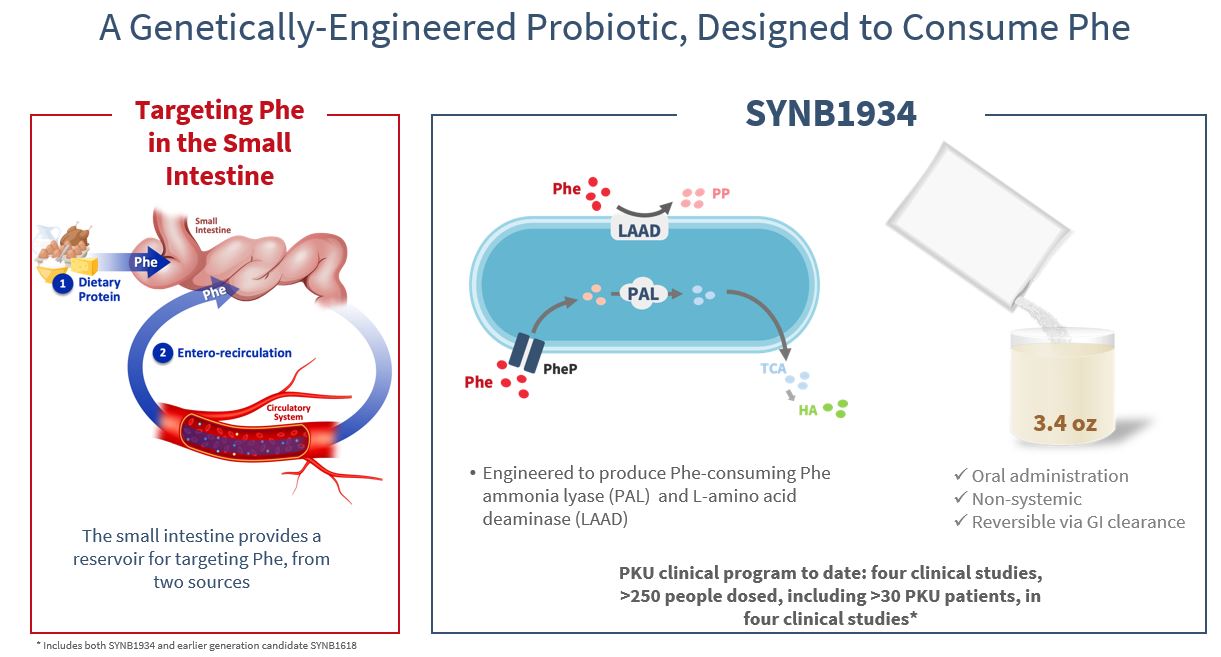
SYNB 1934は第1世代PKU候補薬物SYNB 1618の次世代バージョンであり、PAL中の5つのアミノ酸を修正することにより、PALがフェニルアラニンを代謝副産物であるトランス桂皮酸(TCA)に変換する生産性を向上させた。2018年にSYNB 1618プロジェクトの詳細な記述および疾患動物モデルと健康な非ヒト霊長類における臨床前研究のデータを発表した(ナット。バイオテクノロジーそれは.36,857-864(2018))、SYNB 1934のプロジェクトおよび早期開発概要を2021年に発表しました(NAT通信. 12, 6215 (2021)).
健常ボランティアの第1段階研究では,SYNB 1618,SYNB 1934ともに用量が2 x 10に達した場合に安全かつ耐性が良好であることが発見された12 生きている細胞に毎日3回注射して7日間連続しています高用量は軽度から中等度の胃腸症状に関与しており,主に嘔気と嘔吐である。2019年7月、私たちが発表したデータは、SYNB 1618が安全かつ耐性が良好であり、健康ボランティアとPKU患者において菌株活性の機序証明を実現したことを示している。早期液体製剤と比較して凍結乾燥SYNB 1618の耐性が向上し,患者に試験を行うために最大耐容量(MTD)を決定することができた。2021年9月、私たちは健康ボランティアにおける第1段階の結果を発表し、予測モデルはSYNB 1934がSYNB 1618よりも強い効力を有する可能性があることを示した。
2022年10月、私たちは2期Synheny-1研究からの積極的なトップライン結果を共有し、この研究はPKU患者における2種類の候補薬物の概念証明を証明し、PKU患者におけるSYNB 1934のより大きな効力を確認し、私たちはこの候補薬物が肝心な3期研究に入ることを選択した。完全な研究結果はすでに2023年3月に遺伝性代謝異常学会年会で公表された。公表された結果はSYNB 1618とSYNB 1934の主要な終点(食事挑戦後のD 5-Phe曲線下の面積変化)に成功したことを含む。この研究では、PKU患者のSYNB 1934に対する活性は、第1世代候補SYNB 1618よりも高いことが確認されている。SYNB 1934はPheを消費するための次世代合成生物である。SYNB 1934の結果はSYNB 1934の血漿Pheレベルが平均−40%低下したのに対し,考慮された応答者の血漿Pheレベルは53%(ベースラインと比較して20%以上低下した)であった。60%の患者がPhe低下>20%の基準を達成しており,臨床的に有意な反応の閾値と考えられている。この研究には,2種類の候補薬の経験も含まれており,すでにベースラインレベルでフミントレキサート(Kuvan)を服用している患者に併用したところ,補助治療の可能性が示唆された。安全性と耐性の研究結果は先の経験と一致し有利であり,北京大学計画全体に深刻な有害事象(SAE)はなく,確実に発生する有害事象は主にGIである。
7
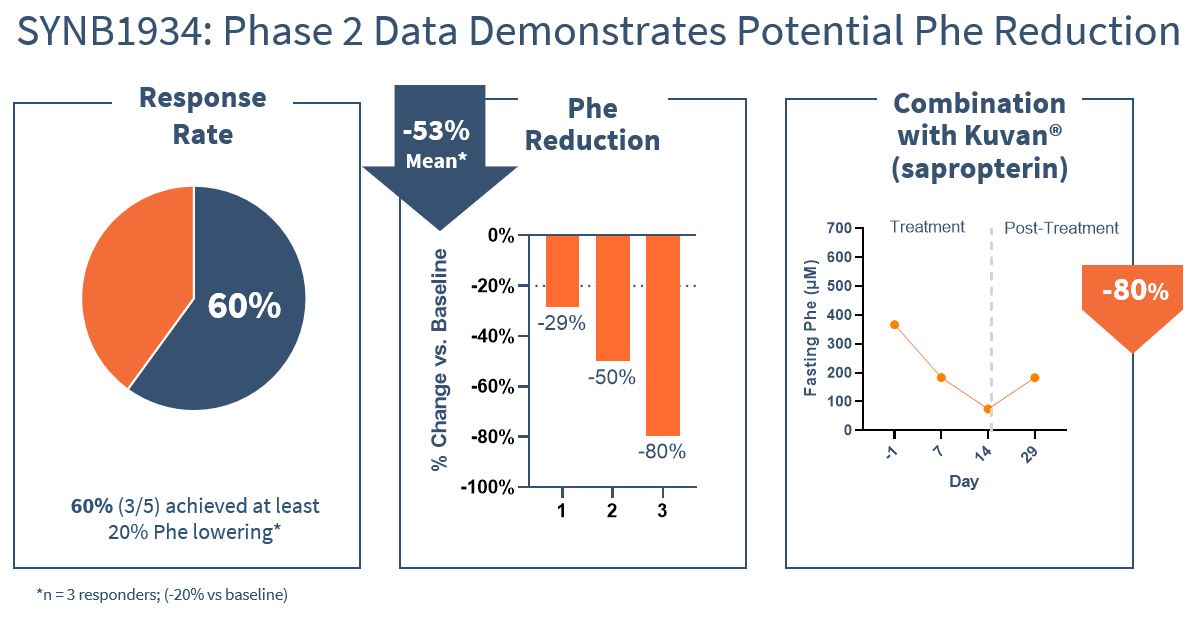
これまでに北京大学の計画には4つの臨床試験のうち240人が含まれている。PKUの潜在的治療法として,2023年上半期にSYNB 1934のキー3期研究を開始する予定である。この試験の設計は,この2つの療法が提供した成功例に基づいており,この2つの療法は規制機関によって北京大学でPHEを低下させる治療法として承認されていると予想される。現在承認されているこの2種類の薬物はいずれも単一の登録研究に基づいて承認され、フェニルアラニンの低下を主要な終点とし、彼らの第三段階試験の設計と類似した三部分構造を共有している。Synheny-3には12~18歳の成人と青少年患者が含まれる予定だ。12歳以下の患者の使用を支援するための単独の研究が必要であると予想される。
SYNB 1934のために、2041年までの物質組成物特許特許権、工学菌及びその医薬製剤に対する物質組成物、並びに治療及び投与方法及び製造方法を含む広範な米国及び前米国特許保護を求めている。計画の進展に伴い、カバー面は引き続き拡大されるだろう。
私たちのHCU計画は
2021年11月、我々は、胃腸内にホモシステイン(Hcy)の前駆体メチオニンを摂取することによって、HCU患者の血漿Hcyレベルを低下させることを目的とした新規、経口的、非系吸収候補薬剤であるSYNB 1353を指名することを発表した。私たちはまた、機械モデリングデータはSYNB 1353が血漿Hcyを58%まで低下させ、HCU患者のタンパク質摂取量を増加させる可能性があることを示した。
2022年11月,SYNB 1353はHCU食モデルを用いた健康ボランティアの第一段階研究において積極的な結果を得たことを発表し,SYNB 1353の作用機序を証明した。SYNB 1353はすでにFDAの快速チャンネル、孤児薬物と稀な小児科疾患の指定を獲得し、HCUの潜在的な治療方法とした。
8
胡適の科学
HCUは稀な遺伝性代謝障害であり、メチオニン(Met)の代謝に影響し、メチオニンはタンパク質であり、肉類、魚類と乳製品を含む多くの食物に存在する。HCUは遺伝的欠陥によるものであり,この欠陥はシスチンβ−合成酵素(CBS)という酵素の欠失を引き起こす。CBSが存在しない場合、Hcyと他の有毒化学物質とその副産物、メチオニンを含み、血液と尿中に蓄積する。HCUにおいて、上昇した総ホモシステイン(THcy)レベルは多系障害と関連し、眼の損害(異所性水晶体および/または深刻な近視)、骨格系の損害(身長が高すぎ、四肢が長すぎ、脊柱側弯、漏斗胸)、血管系(血栓塞栓症)と中枢神経系(発育遅延と知的障害)を含む。HCU治療の目標はtHcyレベルを低下と制御し、それによって急性、潜在的に生命を脅かす血栓と慢性多系統合併症のリスクを下げることである。
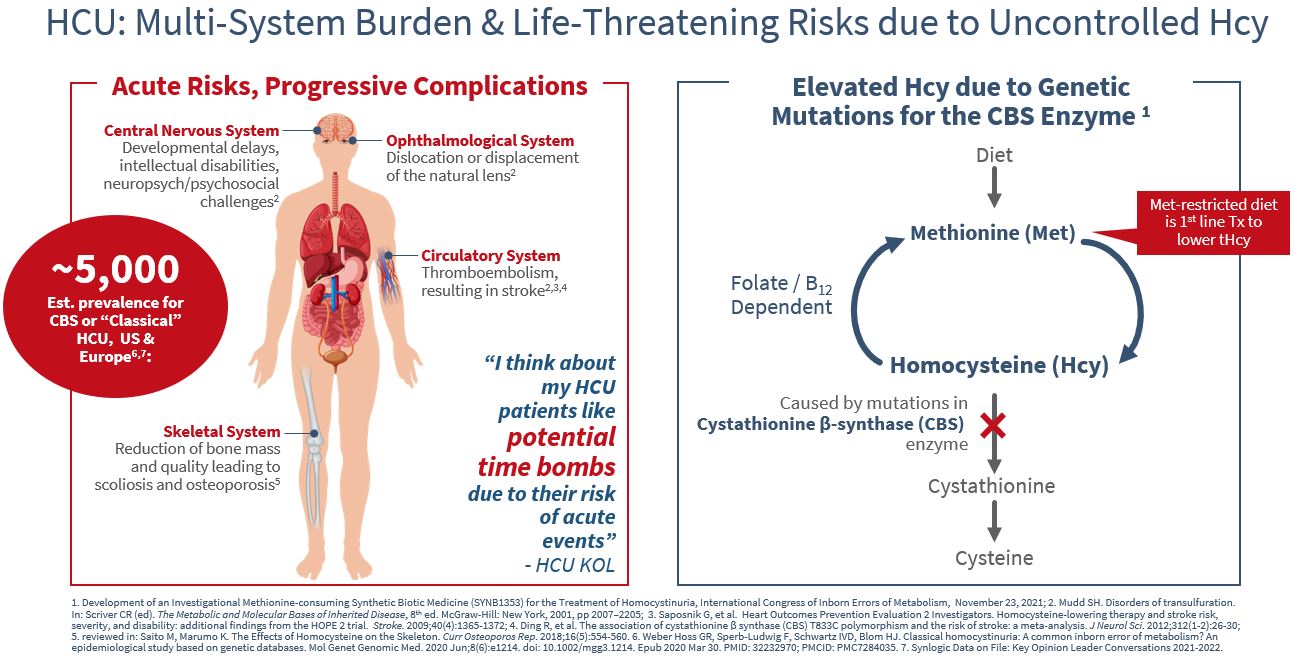
HCUを治癒する方法は現在のところなく,治療選択は限られている。患者は食事中のメチオニンの摂取を避けるために、低タンパク質の厳しい食事に従わなければならない。他の治療方法はビタミンを補充し、疾病のいくつかの副作用を最小限に下げることを含む。アメリカやヨーロッパでは約5,000人がCBSや“古典”HCUを患っています
現在のHCU治療の限界は
治療効果と耐性の原因により、承認されたHCU薬物治療の選択は限られている。表現型の軽いHCU患者は通常ビタミンB 6(ピリドキシン反応とも呼ばれる)の服用に有効であり、十分に制御されている。無反応者にはベタイン(ブランド名Cystadane)と中等度から重度メチオニンの食事制限の組み合わせが必要である。タンパク質の食事を厳しく制限することは複雑であり,継続することも挑戦的である。治療コンプライアンスはしばしば悪化し,特に思春期には,他の食事を続ける必要がある疾患のようになる。低タンパク質食の負担,アミノ酸混合物の摂取,タンパク質代替品やベタインの嗜好性への大きな挑戦を報告した。薬物療法があるにもかかわらず、tHcy治療目標を達成することは依然として挑戦である。
9
HCUに適用されるSYNB 1353
低メチオニン飲食(Hcyの前駆体)はHCU患者のtHcyレベルを低下させる標準的な治療方法である。この飲食モデルはSYNB 1353に科学的な根拠を提供し、SYNB 1353は代謝メチオニンを産生する酵素経路として設計され、それによって血漿tHcyレベルを低下させる。SYNB 1353はメチオニン脱炭酸酵素(MetDC)酵素経路を通じてMetを代謝し、MetのHcyへの転化を阻止し、それによってHCU患者のtHcyを低下させ、合併症のリスクを低下させる。SYNB 1353は経口的、非系統的に吸収された生体生物治療薬候補薬物であり、著者らの北大候補薬物と同様に、香包凍結乾燥粉の形態で提供され、1日3回の付随経口投与である。私たちはSYNB 1353の全世界開発権と商業化権利を持っており、SYNB 1353は著者らがイチョウ研究と協力した一部である。

2022年11月,SYNB 1353はHCU食モデルを用いた健康ボランティアの第一段階研究において積極的な結果を得たことを発表し,SYNB 1353の作用機序を証明した。トップラインの結果,SYNB 1353はMet食挑戦後24時間以内に曲線下面積(AUC)として測定したところ,血漿Met含量が低下した。SYNB 1353の一般的な耐性は良好であり、副作用(AEs)はすべて軽度から中度、一過性であり、しかも主に胃腸感染である。活動群と対照群の胃腸関連副作用の発生頻度と重症度は類似していた。2023年には,HCU患者におけるSYNB 1353の第2段階研究を進めていく予定である。
腸源性高草酸尿計画は
SYNB 8802は開発中の新規,経口,非系吸収の候補薬であり,腸由来高草酸尿の治療に用いられており,尿シュウ酸(Uox)レベルが高いことが特徴であり,腎結石再発の原因とされている。
腸源性高草酸尿は通常腸管に対する原発損傷、例えばクローン病、短腸症候群或いは外科手術、例えばRoux-en-Yダイエット手術によるものである。これは胃腸の吸収不良を招き、シュウ酸が胃腸を介して循環に入る吸収が増加する。シュウ酸塩結晶は腎臓を損害し、慢性腎臓疾患と末期腎臓疾患(ESRD)を招く。腸由来高草酸尿の治療法は現在のところ承認されていない。SYNB 8802は精密遺伝子工学技術を用いて設計されています大腸菌.大腸菌Nissleは3種類の異なる酵素を添加することでシュウ酸を消費し、Uoxレベルを低下させ、この3種類の酵素は一緒にシュウ酸を副産物のギ酸塩に変換する。2021年には、食事誘導高シュウ酸尿を有する健常ボランティアの尿シュウ酸レベルと糞草酸レベルの低下を証明した1 b期研究からのSYNB 8802の積極的な機序証拠を報告した。2022年12月、Roux−en−Y胃シャント手術患者のUox低下の概念に基づく証拠を発表した。
10
腸源性高草酸尿症の科学
高シュウ酸尿症は体内のシュウ酸含有量が高すぎることによる疾患である。シュウ酸は人体内に天然に存在することができ、緑葉野菜、ジャガイモ、アーモンド、コーヒーと豆類などのシュウ酸含有量の高い食物で見つけることもできる。人類はシュウ酸に対して生来の生理的需要がなく、それは通常腎臓を通じて排泄される。尿中のシュウ酸含有量が高すぎると、腎臓中のカルシウムと結合し、腎結石(腎結石形成)、腎カルシウム沈着症、慢性腎臓合併症と腎臓疾患を招く。
腸源性高草酸尿症患者において、Uox上昇と腎結石事件或いは他の腎臓不良結果の確率増加との間に直接な関係が存在する。Uoxの適度な減少でも腎結石になる確率を下げることができる。最近297名の腸源性高草酸尿症患者で行われた疫学研究により、再発性腎結石患者において、Uox 20%減少は翌年の腎結石の年間リスクを25%低下させることが示された(D‘Costa,Nephrol Dial Transpot 2020)。
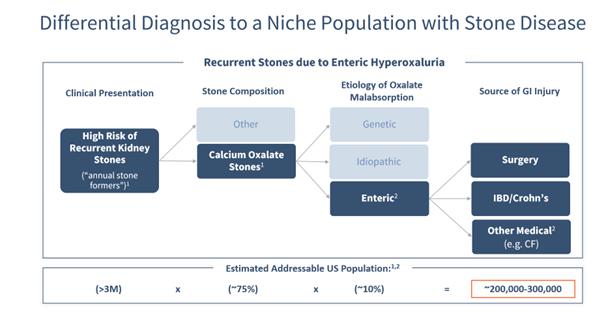
ほとんどの高シュウ酸尿症症例は特発性であり,あるいは原因は不明である。一部はまれな遺伝性疾患によるものであり,12%の高度再発腎結石形成者が明確な腸管病因によりシュウ酸塩結石を産生すると推定されている。通常腸源性高草酸尿をきたす場合には,炎症性腸疾患,あるダイエット手術,短腸症候群,嚢胞性線維化,乳糜潟がある。
原発性高シュウ酸尿症の治療法は現在ほとんど承認されておらず,肝臓に過剰なシュウ酸が産生されるまれな遺伝性疾患である。すでに承認された原発性高草酸尿症に対する治療は肝臓を標的としているため、腸源性高草酸血症患者に不利であり、その病因は胃腸吸収不良であり、肝臓の過剰生産ではない。
定性的な市場研究と実世界証拠を用いた分析によると,米国では200,000から300,000名の患者が腸由来高草酸尿症により腎結石を再発していると推定されている。現在,腸由来高草酸尿症患者の尿シュウ酸レベルを低下させる薬物療法は承認されていない。既存の治療レジメンは、一般に、尿量を1日2~3リットル以上に増加させるための高液体摂取量、低塩およびシュウ酸塩食、経口クエン酸塩および/またはカルシウムおよび/またはマグネシウムサプリメントを含む非特異的である。治療により危険な高Uoxレベルを低下させ,この患者群における腎結石再発のリスクを低下させる必要があることは明らかであると考えられる。
SYNB 8802と腸源性高草酸尿
2020年5月,腸由来高草酸尿症の臨床候補薬SYNB 8802のノミネートが発表された大腸菌.大腸菌Nissleは胃腸全体にシュウ酸塩を摂取することでUoxレベルを低下させる。
11
マウスおよび非ヒト霊長類における生体研究では,SYNB 8802は食事介入による急性高草酸尿症モデルにおけるUoxレベルを低下させた。SYNB 8802プロジェクトの詳細な記述とこれらの臨床前研究のデータは,疾患動物モデルや健康な非ヒト霊長類動物において“分子生物学”(Lubkowiczら,2022)に発表されている。
2021年には,食事誘導高シュウ酸尿を有する健常ボランティアのUoxが29%低下し,糞便シュウ酸が有意に低下した1 a段階研究からのSYNB 8802の積極的な機序証拠が報告されている。2022年12月にデータを共有し,SYNB 8802が臨床的に概念が有意に証明されたことを明らかにした−胃バイパス手術歴のある1 b期研究ではUoxが38%低下した。
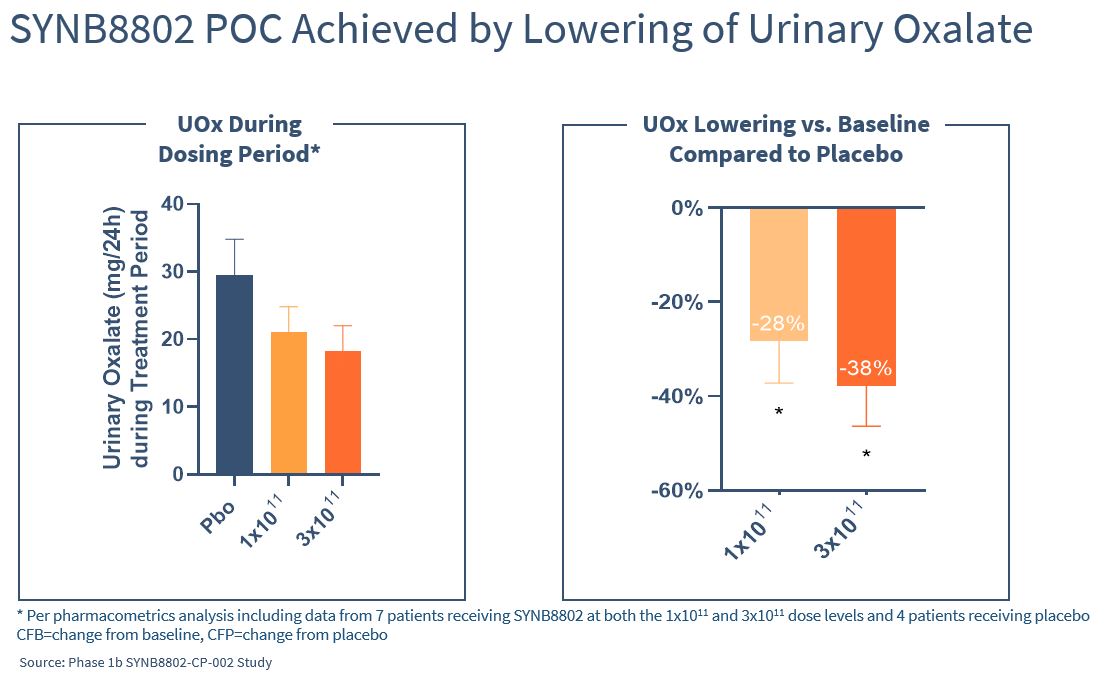
SYNB 8802は、腸由来高草酸尿を治療する最初の承認された薬剤となる可能性がある。著者らの方法は豊富で高リスクの患者群に対して、彼らは腸源性高シュウ酸尿による腎結石の高度再発の歴史による最大の満足されていない医療需要を代表する。著者らは2023年の重点は開発活動を推進し、調合と用量頻度評価に重点を置き、それから腎結石形成と/或いは事件の臨床終点を結合して未来登録の3期研究を行うことである。
私たちの痛風計画は
SYNB 2081は1種の合成生物製剤であり、胃腸中の尿酸を消費することを目的とし、目標は全身尿酸レベルを下げることであり、痛風の潜在的な治療方法として、痛風は1種の複雑な形式の炎症性関節炎である。現在の治療方案は安全性と有効性の面で局限性が存在する。SYNB 2081は著者らがリードした細胞プログラミングレベルプラットフォームイチョウの研究と協力して作成した新型、経口、非システム吸収の候補薬物を提供した。
痛風科学
痛風は体内の尿酸が関節に結晶を形成しすぎることによるものである。関節の激しい痛み,炎症,発赤,尿酸レベルの高すぎによる関節活動制限などの症状が出現する可能性がある。はい
12
また、痛風は慢性腎臓疾患で公認されている危険因子である。現在の治療方案の局限性のため、著者らは明らかに新しい治療方法が必要であると考えている。
痛風発展計画
2022年8月、私たちはSYNB 2081を私たちの潜在的な痛風治療の候補薬物として発表し、そして著者らとイチョウの協力を通じて、第二種の臨床開発に入った製品を発売した。我々の計画はSYNB 2081を我々の薬物開発パイプラインの次の段階に移行し続けることである。SYNB 2081は現在INDを支援する研究を行っている.
代謝性と免疫疾患の追加配管計画
合成生物学的方法は代謝と免疫疾患において検証された生物経路に関連する疾患標的に非常に適している。代謝性疾患については,北京大学,HCU,腸源性高草酸尿,痛風などの項目に応用した。免疫疾患では,新しい治療法に対するIBDの大きなニーズを解決する機会に焦点を当てている。
IBDは胃腸局部炎症を特徴とする疾患であり、通常T細胞、活性化マクロファージと上皮バリア機能損傷によって駆動される。IBDの発症機序は遺伝的要因にも関与しており,環境因子にも関与しており,腸管微生物と腸管免疫系との相互作用の変化による可能性がある。腸管バリア機能損傷は自己免疫性疾患の発病機序においても核心的な役割を果たしている。単層上皮細胞は腸管内容物を宿主循環系と体内の免疫細胞から分離する。上皮層を破壊することは管腔内外来抗原の病理的暴露を招き、自己免疫性疾患の感受性を増加させる。腸管微生物相と宿主間の相互作用は上皮バリアと動的平衡免疫の維持に重要な役割を果たしていると考えられている。そのため、バリア機能を増強し、胃腸炎症を減少させることは自己免疫性疾患を治療或いは予防する潜在的な治療機序である。私たちの合成生物プラットフォームは効率的にプログラミングすることができます大腸菌.大腸菌Nissleはこれらの機能を実行し,代謝が短鎖脂肪酸などの因子を産生してバリア機能を増強することや,タンパク質を分泌するなどの免疫調節サイトカインを含む。
現在IBDを治療する方法は主に免疫系の調節と炎症を抑制する療法に集中している。これらの治療法には,プレドニゾンなどのステロイドや,HUMIRA(アダリズマブ)などの腫瘍壊死因子阻害剤がある。しかし,これらの方法は感染症や癌に対するより大きな感受性を含む全身免疫抑制に関与している。米国疾病コントロール·予防センターのデータによると、2015年、米国では約300万人の成人がIBDと診断されたと報告されている。2021年6月、我々は羅氏社と研究協力協定を締結し、IBDを治療する新しい合成生物製剤を発見した。また,他の全額保有IBDの臨床前研究を進めていく。
免疫疾患では,IBDの治療に取り組んでおり,われわれの経口代謝計画から得られた知識や専門知識を利用して,腸管疾患部位に局所的に作用する活性薬剤の開発を可能にしている。我々の方法は全身投与ではなく炎症部位の局所投与に基づいているため,我々の合成バイオテクノロジーはこの治療カテゴリにおいて魅力的な安全プロファイルを提供する可能性が予想され,現在利用可能な選択と比較して安全性は製品プロファイルの中で特に理想的な属性である。
著者らは一連の合成生物技術を開発してある癌を治療し、これらの癌は腫瘍微小環境を変化させ、免疫システムを活性化し、そして腫瘍の減少を招くことを目的としている。これらの合成バイオテクノロジーは、チェックポイント阻害剤のような他の癌治療法と組み合わせて使用することができる。著者らの合成生物臨床免疫腫瘍学(IO)候補はSYNB 1891であり、これは腫瘍内投与の合成生物薬物であり、二重天然と獲得性免疫活性化剤として設計されている。2021年11月10日、著者らはSYNB 1891とPD-L 1チェックポイント阻害剤の併用による末期固形腫瘍或いはリンパ腫患者の1期試験がすでに登録を完了したことを発表した。SYNB 1891はこれ以上の研究計画を持っておらず,代謝や免疫疾患の研究·開発に取り組んでいる。
合成生物製剤の設計と開発
合成生物技術は潜在的な安全、有効、経口生物療法の独特な優勢を持っている。遺伝子工学微生物は、小分子や抗体のような伝統的な薬物治療では実現できない機能を実行するようにプログラムすることができる。遺伝子またはRNA標的治療のような他の標的方法とは異なり、合成バイオテクノロジーは、非定植として設計され、排泄によって迅速に除去されるので、可逆的である。系統的な吸収が不足しているため、それらは不良事件を発生させるリスクが低い。
13
合成生物学を利用した合成バイオテクノロジー
細菌は数百万年の進化を経て、適応、生存し、積極的に代謝し、人体内の代謝物を消費あるいは産生する。遺伝子操作の影響を受けやすいのです治療効果を与えるために,細菌の基本的な生物学的特性と合成生物学的ツールを用いて,非病原性微生物から合成生物製剤を開発し,最初は単一菌株の細菌に焦点を当てた大腸菌.大腸菌ニスラーです。
著者らの科学者はプロバイオティクス、非病原性細菌に対して遺伝子工学を行い、生物回路を作成し、回路を設計するような方法で細胞過程を指導する。私たちの目標は、特定の疾患および関心のある生物学的目標に対応するために、私たちの合成バイオテクノロジーの数、位置、および活性を正確かつ適切に制御することだ。
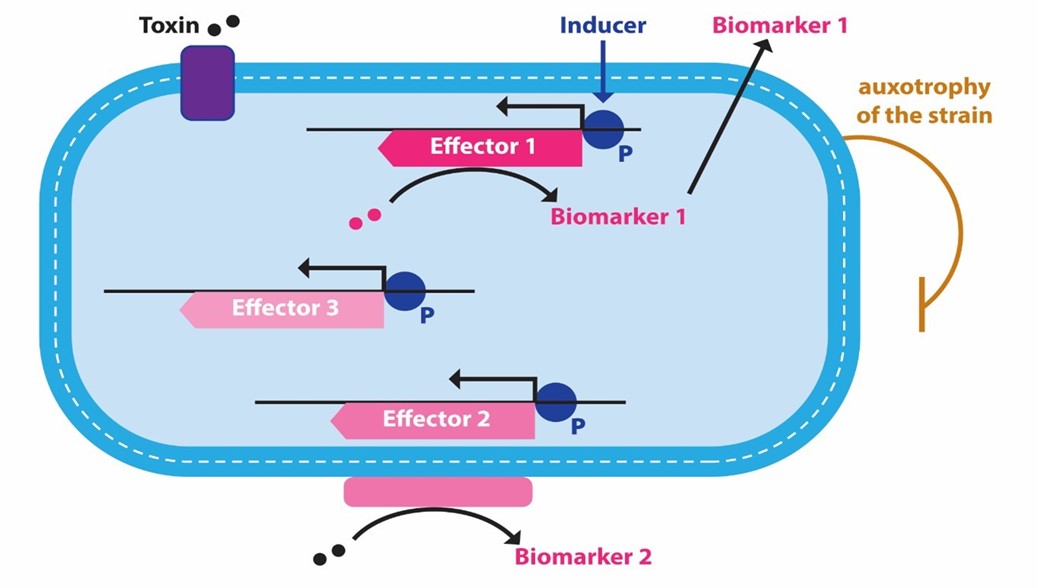
工学的合成生物医学の重要な部分は、(1)“シャーシ”、すなわち非病原性細菌、(2)治療機能を提供するコア生物活性をコードする遺伝子または経路である効果モジュール、および(3)エフェクターモジュールがどのような場合に活性化されるか、およびエフェクター自体の効力、性能、および出力を正確に決定するための調節可能スイッチを含む。
14
分子の生産や消費に必要な酵素経路は,細胞質中の位置を介して過酷なGI環境から保護される大腸菌.大腸菌Nissleは消化管全体で機能する可能性がある。著者らは独自のツール、技術ノウハウと知的財産権を利用して多種の合成生物技術を構築し、臨床前実験において治療関連の効果を産生した。これらの候補製品の進展は、高度に需要を満たしていない疾患において、可能な薬物開発経路を有する製品を優先的に考慮し、情報を得ることができる動物モデルおよびバイオマーカーの存在を含めて、有効な臨床開発を指導することに基づいている。
合成生物療法の優位性
私たちの合成生物技術会社は以下のような特徴を持つ安全で有効な治療法を提供すると信じている
有毒代謝物を分解する:他の治療方法と異なり、合成生物技術は1種の独特な機械活動を通じてプログラミングすることができる。内部代謝計画では,全経路が有毒代謝物を分解または“消費”することを目的とした合成生物製剤を設計している。後者は単一遺伝子やタンパク質欠陥に限られており,遺伝的異質性の患者集団を解決するためには,遺伝子,RNAや酵素代替療法に比べて代謝経路を用いることが有利であると考えられる。細菌伝達の全経路を通じて補償を行い、合成生物技術は単一の工学療法としてより広範な疾病群に安全、治療の解決策を提供するかもしれない。我々の臨床段階計画SYNB 1934,SYNB 8802およびSYNB 1353は合成バイオテクノロジーの例であり,胃腸中の代謝物を除去し,吸収されることを防止することを目的としている。これらの項目から学んだ知見は他の臨床前項目に応用されており,これらの項目では代謝物失調が疾患に関連している。
治療用分子の生産:また、疾患を改善する可能性のある細菌代謝産物または細菌から現地環境に分泌されるタンパク質のような有益な分子を生成するために、合成バイオテクノロジーをプログラムすることができる。この方法の例としては,短鎖脂肪酸(Reevesら)の製造が挙げられる。DDW 2022),芳香族炭化水素(AHR)アゴニスト(Shuら)。DDW 2022)やヒトIL−22蛋白の分泌(Revesら)。DDW 2022).
様々なメカニズムの組み合わせです複数のエフェクターや酵素を1つの菌株に統合することにより、合成バイオテクノロジーは多要素疾患生物学的問題をより有効に解決することが可能である。例えば、合成バイオテクノロジーは、単一エフェクターよりも毒性代謝物を効率的に消費することができる複数の酵素を用いて工学的に設計することができる。代替的に、対象の代謝産物を分解する経路は、合成生物として設計することができ、1つ以上の治療分子を生成することもできる。
現地で提供される場合,合成バイオテクノロジーは,特にシステム治療の組み合わせが必要な場合には,常にシステム治療に関連するリスクを回避する可能性があると信じている。われわれの合成生物候補薬は経口投与されており,腸管通過時に局所的に作用する。したがって、それらは血液中の有毒代謝物レベルを低下させることができ、患者に全身治療メリットを提供することができる。慢性経口投与の可能性を考慮すると,合成バイオテクノロジーは用量予測と活性可逆性の面でメリットがある可能性がある。
15
著者らの合成生物製品エンジンの特徴は高効率な薬物発見と開発に伝統的な方法よりもっと速く、更に有効に臨床候補薬物を推進する潜在力を持たせた。1年間に1つのプロジェクトを概念開発から臨床開発に移行する能力を示し,学んだ知識を各プロジェクトに応用して開発を促進した。理由はこうです
協力協定
合成バイオテクノロジーの患者開発と商業化を加速するために,他の機会を探して協力者と戦略同盟を構築し,我々の治療開発と候補製品のルートを拡大しようとしている。私たちはまた、複数の学術、研究、転化医学組織と実体と協力するより多くの機会を求めて、疾病や障害を治療する潜在力を持つ生命薬物の理解と開発を深めるつもりである。
羅氏
2021年6月、私たちはF.Hoffmann-La Roche Ltd(羅氏バーゼル)とHoffmann-La Roche Inc.(羅氏米国、および羅氏バーゼル、羅氏)とパイロット協力とオプション協定(羅氏協力とオプション協定)を締結した。羅氏の協力とオプション協定の条項によると、私たちと羅氏は炎症性腸疾患の治療の未開示の新しい標的(候補製品)を解決するために、協力研究と臨床前開発合成生物技術を求める。
16
羅氏の協力とオプション協定によると、羅氏は前払いした返却不可能な100万ドルの技術アクセス料を支払うことに同意し、この費用は2021年7月に受け取った。2022年8月、同社は研究計画第2段階の研究開発サービスを完了し、150万ドルの記念碑的支払いを実現し、この金は羅氏が2022年10月に支払った。私たちはいくつかの成功基準を達成した後、500万ドルまでの記念碑的支払いを受ける資格がある。研究期間終了後,羅氏は独占選択権(選択権)を持ち,候補製品のさらなる開発と商業化について最終的な協力と許可協定(CLA)を交渉することができる.
“羅氏協力とオプション協定”によると、この合意期間内に、双方が“羅氏協力とオプション協定”の下での義務を履行した場合にのみ、双方は、それによって制御される特定の知的財産権及びノウハウの非排他性、譲渡不可、再許可不可、印税免除の権利及び許可を他方に付与している。双方は羅氏協力とオプション合意基本研究計画の実行状況を監督·管理するための共同研究委員会(JRC)を設立した。
羅氏協力とオプション協定には各種の陳述、保証、契約、賠償とその他の習慣条項が含まれている。研究計画のいくつかの部分がいくつかの成功基準に達した場合、羅氏は書面通知を受けた後、羅氏の協力およびオプション合意を直ちに終了するか、または90日前に書面で通知することができる。いずれの一方も他方が治癒していない実質的な違約が発生した場合,羅氏協力とオプション合意を終了することができる。
私たちは引き続き羅氏協力とオプション協定に基づいて研究と開発を行う予定だ。私たちはそれぞれのマイルストーンの成功基準を達成した後、羅氏のマイルストーンの支払いを受け取る資格があります。
イチョウ生物工学会社
2019年6月、私たちは銀杏と合意に達した。この協定はSynlogicプレミアム8000万ドルに対するイチョウの株式投資を提供し、Synlogic合成生物技術パイプラインの発展を拡大と加速するための長期戦略プラットフォーム協力を達成した。著者らはイチョウの細胞プログラミングプラットフォームを用いて数千の微生物菌株を構築とテストし、早期臨床前手がかりの進展を加速し、それによって更なる臨床開発のために候補薬物を最適化する。
協議の一部として、イチョウは6,340,771株の私たちの普通株と付属の予融資権証(予融資権証)を購入して、最大2,548,117株の私たちの普通株を購入し、合併価格は1株9.00ドルと事前融資権証である。毛収入は約8000万ドルである。協定によると、イチョウに3000万ドルの前金を支払い、イチョウが提供してくれた鋳造サービスを支払うために使用され、初期期間は5年で延長できる。この初期期間および追加期間が満了した後、私たちの前払いのうち、イチョウからの購入サービスのために使用されていない任意の部分は、イチョウによって保持される。私たちは協力の一部として開発された任意の合成バイオテクノロジーの独占的な権利と、そのような製品をカバーする知的財産権を持っている。
潜在的未来協力
戦略連合は合成バイオテクノロジー目標の開発を加速させる重要な駆動力となると信じており,我々の新たな候補治療薬の援助,開発を支援し,商業化できるパートナー,特に大型代謝や免疫適応の面で引き続き探していく。著者らの合成生物技術プラットフォームの潜在的な応用は非常に広いため、著者らはまた私たちの計画に専門知識と資源を提供できる学術、研究と転化医学組織と実体を探し続け、私たちの影響をより広範な患者集団に広げることができるようにする。
知的財産権と技術ライセンス
我々は、内部開発であっても、著者らの協力者や他の第三者から許可を得ても、特許権を求め、維持し、擁護することを含む、当社の業務に重要なビジネス的意義を有するノウハウ、発明、および改善を保護し、強化するために努力している。私たちの政策は、米国および米国以外のいくつかの司法管轄区域に当社のノウハウ、発明、改善、および候補製品に関する特許出願を提出することを求めることであり、これらは私たちの業務の発展と実施に非常に重要である。
私たちはまた、私たちの独自技術と製品候補に関連する商業秘密と技術的ノウハウ、持続的な革新と許可機会に依存して、合成生物学分野における私たちの独自の地位を強化し、維持している。また、私たちはデータ独占性、市場独占性、特許期間の延長(利用可能であれば)に依存し、和を求めている
17
もし適用されれば、計画は孤児薬物指定によって提供される追加的な規制保護に依存する。私たちのビジネス成功は、私たちの技術、発明、および改善された特許および他の固有の保護を取得して維持する能力があるかどうか、私たちの商業秘密の機密性を保護すること、第三者が所有する知的財産権を使用する私たちのライセンスを維持すること、私たちの特許を含む私たちの固有の権利を擁護し実行すること、および効果的かつ実行可能な特許および第三者の他の固有の権利を侵害することなく運営される可能性がある。
私たちは知的財産権の面で有利な立場にあると信じています
私たちの知的財産権の組み合わせは私たちの合成生物プラットフォームと適用される疾患関連技術をカバーしていると信じている。2023年3月22日現在、私たちは米国および外国司法管轄区にSynlogicが所有する182件の特許および特許出願を持っており、そのうち46件が発行または承認されている。
Synlogic知的財産権
病気に関する応用
著者らの知的財産権の組み合わせにおける疾患関連応用は、高アンモニア血症、高フェニルプロピオン酸血症、高草酸尿、同型システイン尿症、高尿酸血症、ある他の遺伝性代謝疾患と状況、代謝障害、炎症状態に関連する疾患と状況、腸管炎症に関連する疾患、損傷した腸管粘膜バリア(漏れ性腸管)と各種自己免疫疾患を含むが、これらの状況と関連疾患状態に特化して設計された遺伝子回路を有する工学菌に保険を提供するいくつかの病理状況に関連する。知的財産権の組み合わせは、エンジニアリング細菌株組成物、関連処方、細菌菌株の製造方法、菌株の活性を測定する方法、および疾患を治療する方法を含む。現在、この技術に関連する知的財産権には、米国と外国の司法管轄区域で決定されている出願と、物質成分と薬物成分に対する声明のいくつかの発行された米国特許が含まれており、これらの特許は私たちの臨床候補薬物をカバーしている。我々の現在の特許と特許出願の特許期間は2035年12月から2043年12月まで様々であり,適応に応じて,いかなる特許期限の調整や延長も含まれていない。
プラットフォーム技術応用
疾患に関する技術に加えて,我々の知的財産権の組合せには,我々の内部で開発されたプラットフォーム技術への応用が含まれている.例示的なプラットフォーム技術は、例えば、遺伝子調節、細菌細胞増殖制御(その自動調節を含む)、代謝産物を輸入するためのシステム、および潜在的遺伝毒性代謝産物の産生を防止するためのシステムの改善を含む細菌シャーシ関連および遺伝回路関連技術開発を含む。これらのプラットフォーム技術及びその知的財産権のカバー範囲は著者らの治療性合成生物技術に広く適用されている。
一般的な考慮事項
個別特許の展示期間が異なり、特許出願の提出日、特許発行日及び特許を取得した国の特許の法的期限に依存する。一般に,米国で提出された出願のために発行された特許の有効期限は,最も早く発効した非仮出願日から20年である。さらに、場合によっては、米国特許商標局(USPTO)の起訴遅延および/またはFDA規制審査期間によって実際に失われた期限の一部を再取得することを示すために、特許期限を延長することができる。規制遅延については,回復期間は5年を超えてはならず,回復期間を含む総特許期間はFDA承認後14年を超えてはならない。米国以外の特許の期限は適用される現地法の規定によって異なるが,通常は最も早く発効した非仮出願日から20年である。しかしながら、特許によって提供される実際の保護は、製品によって異なり、国によって異なり、特許のタイプ、そのカバー範囲、規制に関連する延長の利用可能性、特定の国の法的救済措置の利用可能性、および特許の有効性および実行可能性を含む多くの要因に依存する。
18
私たちのような会社の特許地位は通常不確実であり、複雑な法律と事実の問題に関連している。合成生物学分野の特許で許容される特許請求の範囲については,米国ではまだ一致した政策が現れていない。アメリカ以外の特許状況はもっと不確実だ。許可されたおよび会社のすべての知的財産権については、私たちの任意の未解決特許出願または将来提出された任意の特許出願が特許を取得することを保証することはできず、私たちの任意の既存特許または将来私たちに付与される可能性のある任意の特許が、私たちの製品を保護し、これらの製品を製造するための方法において商業的用途を有することを保証することはできません。その他のリスクについては、“リスク要因−知的財産権に関するリスク”と題する節を参照されたい
商標
私たちの登録商標の組み合わせは現在、35個の登録商標、5つの許可、および4つの処理すべき出願を含んでいます。
他にも
一般的に、私たちは、従業員、請負業者、コンサルタント、協力者、コンサルタントなど、私たちの機密情報にアクセスできる人と秘密協定を締結することによって、私たちの技術や製品候補を保護することを求めています。場合によっては、私たちは商業秘密に依存して私たちの技術を保護するかもしれない。私たちは、私たちのノウハウ、ビジネス秘密、プロセスの完全性とセキュリティ、私たちの場所の実体セキュリティ、および私たちの情報技術システムの実体と電子セキュリティを維持するために努力しています。私たちはこのような個人、組織、そしてシステムに自信があるにもかかわらず、合意や安全措置が違反される可能性があり、私たちはどんな違反にも対応する十分な救済措置がないかもしれない。しかも、私たちのビジネス秘密は競争相手に知られたり独立したりするかもしれない。会社の従業員、請負業者、コンサルタント、協力者、およびコンサルタントが、私たちのために働いているときに他人が所有する知的財産権を使用する場合、関連するまたはそれによって生じるノウハウおよび発明の権利に関する紛争が生じる可能性がある。このリスクおよび当社のノウハウ、発明、改善および製品に関連するより包括的なリスクについては、“リスク要因--知的財産権に関連するリスク”というタイトルの節を参照されたい
規制事項
政府規制と製品審査
アメリカ連邦、州と地方の各級政府当局及びその他の国の政府当局は私たちが開発している製品の研究、開発、テスト、製造、品質管理、承認、ラベル、包装、貯蔵、記録保存、販売促進、広告、流通、マーケティングと輸出入などの方面に対して広範な監督管理を行った。新薬は新薬申請(NDA)プログラムを通じてFDAの許可を得なければならず、新生物は生物製品許可証申請(BLA)プログラムを通じてFDAの許可を得なければならず、このような製品はアメリカで合法的に発売されることができる。
アメリカの薬物開発プロセスは
米国ではFDAは連邦食品,薬物と化粧品法案(FDCA)に基づいて薬品や生物製品を規制し,生物製品についても公衆衛生サービス法(PHSA)や施行条例に基づいて規制を行っている。私たちの候補製品は生物製品としてFDAによって規制されるだろう。規制の承認を得て、その後適用される連邦、州、地方、外国法規を遵守する過程には多くの時間と財力が必要だ。製品開発過程,承認過程又は承認後のいずれかの場合,出願人が適用される米国の要求を遵守できない場合には,行政又は司法制裁を受ける可能性がある。これらの制裁には、FDAによる未解決申請の承認拒否、承認撤回、免許取り消し、臨床封印、警告状、製品リコール、製品差し押さえ、生産または流通の完全または部分的な一時停止、禁止、罰金、政府契約の拒否、原状回復、返還、返還、または民事または刑事罰が含まれる可能性がある。どんな機関や司法法執行行動も私たちに実質的な悪影響を及ぼすかもしれない。FDAが米国でバイオ医薬品を発売する前に必要なプログラムには、一般に以下のような態様が含まれる
19
開発する候補薬が決定されると,臨床前試験段階に入る。臨床前テストは製品の化学と調合の実験室評価、動物毒性と薬理学研究を含み、有害事象の可能性を評価し、そしてある場合に治療使用の理由を確立する。2022年12月29日に法律となった2023年総合支出法案(P.L.117−328)に署名されたFDCAと公衆衛生サービス法が改正され、薬物や生物製品の非臨床試験が含まれることが規定されているが、必要ではない体内にある動物実験です。修正された言語によると、スポンサーは様々なことを行うことができます体外培養分析(例えば、細胞ベースの分析、器官チップ、または微生理システム)シリコン片研究(すなわち、コンピュータモデリング)、人間または非ヒト生物学に基づく他のテスト(例えば、生物印刷)、または体内にある動物実験です。臨床前研究の進行はGLPの安全/毒理学研究に関する法規を含む連邦法規と要求の制約を受けている。
INDスポンサーは,臨床前試験の結果を生産情報や分析データとともにINDの一部としてFDAに提出しなければならない。2016年6月、FDAは“活性生物治療製品の早期臨床試験:化学、製造および制御情報”と題する最新の業界ガイドラインを発表し、その中には、活性生物治療製品の早期臨床試験のINDに含まれるべき化学、製造および制御情報に関するFDAの提案が含まれている。このガイドラインは、FDAまたはスポンサーに拘束力がないにもかかわらず、FDAがガイドラインを発表する際の主題に対する考え方を反映しており、私たちの合成生物候補製品INDにどのような内容が含まれるべきかに関する追加情報を提供してくれる。スポンサーはまた、臨床試験の目標、安全性を監視するためのパラメータ、および第1段階の研究が治療効果評価に役立つ場合、評価の有効性基準を詳細に説明するINDにプロトコルを含むであろう。いくつかの長期的な臨床前テスト、例えば生殖不良事件と発ガン性の動物テストは、候補製品のINDを研究してFDAに提出し、人体臨床試験を開始した後に引き続き行う可能性がある。INDはFDAが30日以内に臨床試験を保留しない限り、FDAが受領してから30日後に自動的に発効する。この場合,INDスポンサーやFDAは臨床試験開始前に未解決の問題を解決しなければならない。FDAは、進行中または提案されている臨床試験またはFDAの特定の要求に適合していない安全懸念から、FDAがスポンサーに一時停止を通知する前に、試験が開始または継続されない可能性があるため、臨床試験の前または期間の任意の時点で臨床休止を実施することもできる。
すべての臨床試験は1人或いは複数の合格研究者の監督下で行わなければならず、GCP要求に符合する。これらは,試験目標,投与手順,被験者の選択と排除基準,評価すべき安全性と有効性基準を詳細に説明するレジメンで行わなければならない。各シナリオはINDの一部としてFDAに提出されなければならず,深刻かつ予期しない有害事象については,速やかにFDAおよび調査者に安全報告書を提出しなければならない。臨床試験に参加する各機関のIRBは、その機関の臨床試験が開始される前に各案を審査および承認しなければならず、また、試験に関する情報および各試験対象またはその法律代表に提供されなければならない同意書を承認し、完成まで研究を監督しなければならず、そうでなければIRBの規則を守らなければならない。対象者はIRB承認のインフォームドコンセントに署名しなければ臨床試験に参加できない。
20
さらに、臨床試験に参加する各機関を代表するIRBは、臨床試験が開始される前に任意の臨床試験の計画を審査および承認しなければならず、IRBは少なくとも年に1回の持続的な審査および再承認試験を行わなければならない。IRBは臨床試験被験者に提供する試験案やインフォームドコンセント情報などを審査·承認しなければならない。IRBの運営はFDAの規定に適合しなければならない。
人体臨床試験は通常3つの連続段階に分けて行われ、これらの段階は重複或いは合併する可能性がある
承認後の実験は,ステップ4と呼ばれることがあり,最初の市場承認後に行われる可能性がある.これらの試験は,治療適応が予想される患者の治療から追加的な経験を得るために用いられている。場合によっては,FDAはBLAを承認する条件として4期臨床試験を強制的に実行することができる。
2023年総合支出法案では,国会はFDCAを改正し,3期臨床試験や上場認可された新薬を支援する他の“キー研究”のスポンサーにこのような臨床試験の多様な行動計画の提出を求めている。行動計画には,スポンサーの多様な学生募集目標と,目標の理由やスポンサーがこれらの目標をどのように達成するかの記述が含まれなければならない。スポンサーはスポンサーが試験案をFDA審査に提出する前にFDAに多様な行動計画を提出しなければならない。FDAは多様な行動計画の一部またはすべての要求を免除することができる。多様性行動計画が第3段階試験計画やスケジュールにどのように影響するかは不明であり,FDAがこのような計画の中でどのような具体的な情報を期待するかも不明であるが,FDAがスポンサーの多様性行動計画に反対し,スポンサーに計画の修正や他の行動を要求すれば,試験開始を遅らせる可能性がある。
FDA或いはスポンサーはいつでも様々な理由で臨床試験を一時停止或いは中止することができ、研究対象或いは患者が受け入れられない健康リスクに直面していることを発見することを含む。同様に、臨床試験が臨床レジメン、cGMPまたはIRB要求に従って行われない場合、または候補製品が患者の意外な深刻な傷害に関連している場合、IRBは、その機関の臨床試験の承認を一時停止または終了することができる。さらに、いくつかの臨床試験は、スポンサーによって組織された独立した合格専門家グループによって監督され、このグループは、データ安全監視委員会または委員会と呼ばれる。その規約によれば、当該グループは、試験のあるデータへのアクセスに基づいて、試験が指定されたチェックポイントで行うことができるか否かを決定することができる。第1段階、第2段階、および第3段階の臨床試験は、任意の指定された時間内に成功しない可能性がある(あれば)。
新しい生物の開発過程で、スポンサーはいつかFDAと会う機会がある。これらの要件は、INDを提出する前、第2段階の終了時、およびBLAを提出する前にある可能性がある。他の時間に会議を開催することを要求することができます。これらの会議は,スポンサーにこれまで収集してきたデータに関する情報を共有する機会を提供し,FDAにアドバイスを提供し,スポンサーとFDAに次の段階の開発について合意することができる。スポンサーは通常、第二段階会議終了時間を利用してFDAと彼らの第二段階臨床結果を討論し、生物研究の承認を支持すると考えられる重要な第三段階臨床試験計画を提出した。もしこのようなタイプの討論が発生すれば、スポンサーは特殊方案評価(SPA)を申請することができ、その目的はFDAと3期臨床試験方案の設計と分析の設計を達成することであり、これは治療効果声明の主要な基礎を構成する。
臨床試験と同時に,会社は通常承認が必要となる可能性のある追加の動物研究を完成させ,生化学や物理特性に関する追加情報を開発し,cGMP要求に基づいて商業量産製品のプロセスを最終的に決定しなければならない。製造過程は一貫して品質に合格した候補製品ロットを生産できる必要があり、また、メーカーは最終製品の特性、強度、品質と純度をテストする方法を開発しなければならない。さらに適切な包装は
21
選別とテストを行い,候補製品がその賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
INDが有効であり、承認される前に、少なくとも毎年FDAに進捗報告を提出しなければならず、深刻かつ意外な疑わしい副作用を含むIND安全報告書をFDAおよび調査者に提出しなければならない。他の研究からの結果は、同じまたは類似の薬剤に曝露されたヒトに重大なリスクがあることを示し、動物または体外試験からの結果は、ヒトに重大なリスクがあることを示し、プログラムまたは研究者マニュアルに記載されている場合と比較して、任意の臨床的に重要な深刻な疑わしい副作用発生率の増加を示す。年次報告書は通常、開発安全最新報告の形で提出され、IND年間報告書と同等と考えられ、EU(欧州連合)と国際調整会議(ICH)の要求にも合致する。
また,行っている臨床試験と完成した試験結果を公的登録機関に報告することが求められている。FDA規制製品の多くの臨床試験のスポンサーは、ClinicalTrials.gov上で公開して得ることができる特定の臨床試験情報を登録して開示しなければならない。そして,登録の一部として,製品,患者群,調査段階,試験地点と調査者および臨床試験の他に関する情報が公開されている。スポンサーも試験完了後に臨床試験結果を提出する義務があるが,場合によっては結果の開示が試験完了日後2年に延期される可能性がある。競争相手はこれらの公開された情報を用いて開発計画の進捗状況を知ることができる.対象の臨床研究や法律規定の研究結果を適時に登録できなかったことは民事罰金を招く可能性があり、また違反側が連邦政府の将来の支出を獲得することを阻止する。NIHのClinicalTrials.gov登録と報告要件に関する最終規則が2017年に発効し,NIHとFDAは最近,要求に適合しない臨床試験スポンサーに対してこれらの要求を実行するようになった。
アメリカの審査と承認の流れ
必要な臨床試験、非臨床研究および臨床試験の結果、ならびに製品の化学、製造および制御、安定性、品質管理および製品発表手順、提案されたラベルおよび他の関連情報と共に詳細な情報がBLAの一部としてFDAに提出されると仮定し、製品を1つまたは複数の適応の市場に使用することの承認を要求する。BLAを提出するには、相当な使用料を支払う必要があり(例えば、2023年度には、この申請料は320万ドルを超える)、いくつかの限られた場合には、生物が孤児薬として指定された場合には、そのような費用の免除を得ることができるにもかかわらず、かなりの使用料を支払う必要がある。承認されたBLAのスポンサーは計画年会費も必要であり,現在1計画あたりの費用は39万ドルを超えている。これらの費用は通常毎年増加するが、場合によっては免除および免除がある可能性がある(例えば、条件に適合する小企業が提出した最初の人間薬物申請の免除および孤児製品の免除)。
FDAは、それらが届出を受ける前に十分に完全であり、実質的な審査を行うことができることを確実にするために、すべての提出されたBLASを審査する。FDAはBLAの届出を受け入れるのではなく、より多くの情報を提供することを要求するかもしれない。この場合,BLAおよび付加情報を再提出しなければならない.再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。提出された申請が受け入れられると、FDAは深い実質的な審査を開始する。FDAが“処方薬使用料法案”(PDUFA)に基づいて原始BLASについて達成した目標と政策によると、FDAは標準出願の予備審査を完了して出願人に応答するのに10ヶ月の期間があり、優先審査の出願は6ヶ月である。FDAは常にPDUFAの目標日を達成するわけではなく、審査プロセスは、FDAがより多くの情報を提供することを要求すること、または明確にすること、およびスポンサーがそのような質問に応答するプログラムによってしばしば大幅に延長される。具体的には、FDAは、新しい情報を考慮するために、または出願人が明確な説明を提供する場合に、FDAが最初の提出後に発見された係属中の欠陥を解決するために、審査プロセスをさらに3ヶ月延長することができる。
BLAを承認する前に、FDAは通常、製造プロセスおよび施設がcGMPに適合するかどうかを決定するために、新製品の製造施設を承認前に検査する。FDAは、製造プロセスおよび施設がcGMP要件に適合していることを決定し、要求された仕様の下で製品が一貫して生産されることを保証するのに十分でない限り、この製品を承認しないであろう。これらの承認前検査は、部品製造、完成品製造、および制御試験実験室を含むBLA提出に関連するすべての施設をカバーすることができます。FDAはまた、GCP要件に適合することを保証し、FDAに提出された臨床データの完全性を保証するために、スポンサーおよび1つまたは複数の臨床試験場所を検査することができる。FDAがアプリケーション、製造プロセス、または製造施設が受け入れられないと判断した場合、それは一般に欠陥を列挙し、追加のテストまたは情報の提供を要求することが多い。これは申請に対する追加的な検討を大幅に延期するかもしれない。FDAが臨床サイトがGCPに従って臨床試験を行っていないことが発見された場合、例えば、FDAは、臨床サイトによって生成されたデータがBLAによって提供される一次治療効果分析から除外されるべきであると判断することができる。
22
FDAは、安全性または有効性の問題を提起する新しい生物学的候補の出願を含む任意のBLAを諮問委員会に提出して、申請を承認すべきかどうか、およびどのような条件下で承認すべきかを決定するために、審査、評価、および提案を行うことができる。通常,諮問委員会は臨床医や他の科学専門家を含む独立した専門家からなるグループであり,申請を審査·評価し,申請を承認すべきかどうか,承認すべきであればどのような条件で承認するかについて提案を行う。FDAは諮問委員会の提案に制限されていないが、最終承認決定を下す際にこれらの提案を考慮する。
承認過程が長く、しばしば困難であり、適用された規制基準が満たされていない場合、または追加の臨床または他のデータおよび情報が必要とされる可能性がある場合、FDAはBLAの承認を拒否する可能性がある。このようなデータや情報を提出しても,FDAは最終的にBLAが承認基準を満たしていないと決定する可能性がある.臨床試験から得られたデータはつねに決定的ではなく,FDAのデータ解釈は我々の同じデータに対する解釈とは異なる可能性がある。FDAがBLAを評価した後、承認書または完全な返信(CRL)を発行する。この薬剤の商業マーケティングを承認し、特定の適応の処方情報を提供する。CRLは,申請の審査周期が完了したことを示しており,現在の形で申請を承認することはない.CRLは、一般に、FDAによって決定されたBLAにおける特定の欠陥を記述し、追加の重要な3期試験、または臨床試験、非臨床研究、または生産に関連する他の重要で時間のかかる要件のような追加の臨床データを必要とする可能性がある。CRLを発行すると,スポンサーはBLAを再提出し,手紙で確定したすべての不足点を解決したり,申請を撤回したりしなければならない.このようなデータや情報を提出しても,FDAはBLAが承認基準を満たしていないと決定する可能性がある.FDAは、製品が安全で、純粋で、有効かつ有効であるかどうか、およびその製造、加工、包装、または保持されている施設が、製品の持続的な識別、強度、品質、効力および純度を保証するための基準に適合しているかどうかを決定するためにBLAを審査する。
1つの製品が規制部門の承認を得た場合、承認は、BLAに記載されている使用条件(例えば、患者数、適応)に限定され、これは、製品の商業的価値を制限する可能性がある。さらに、解決すべき具体的なリスクに応じて、FDAは、BLA承認後に薬剤の安全性および有効性をさらに評価することを目的とした臨床試験に関連する第4段階試験をスポンサーに行うことを要求する可能性があり、商業化された承認製品の安全性を監視するために、製品ラベルに禁忌症、警告または予防措置を含むことを要求することができる。FDAはまた、薬剤の安全な使用を確保するためにREMS(リスク評価および緩和策)を要求することを含む承認時に他の条件を設定することができる。FDAがREMSが必要であると結論した場合,BLAのスポンサーは提案したREMSを提出しなければならない。必要であれば、FDAは承認されていないREMSでBLAを承認しないだろう。REMSは、制限された分配方法、患者登録、および他のリスク最小化ツールのような薬物ガイドライン、医師のコミュニケーション計画、または安全な使用を確保する要素を含むことができる。承認またはマーケティングに関するこれらの制限は、製品の商業普及、流通、処方、または配布を制限する可能性がある。規制要件を守らない場合や最初のマーケティング後に問題が発生した場合、マーケティング承認は撤回される可能性がある。
改訂された“小児科研究公平法”(PREA)によれば、新製品の初期BLAまたはBLAのいくつかのサプリメントは、すべての関連小児科亜群において適応と主張される候補製品の安全性および有効性を評価し、安全かつ有効な各小児科亜群に対するこの製品の用量および投与をサポートするためのデータを含まなければならない。FDAは、成人のために、または小児科データ要件を完全にまたは部分的に免除するために製品が承認されるまで、申請者の要求に応じて、小児科データの提出を延期することを許可することができる。法規に別の要求がない限り、PREAは通常、孤児の称号が付与されたいかなる適応の治療製品にも適用されない。2012年に公布された“食品·薬物管理局安全·革新法案”(FDASIA)は、新活性成分、新適応、新剤形、新投与案または新投与経路を含む製品のマーケティング申請を計画するスポンサーは、第2段階会議終了後60日以内に予備小児科研究計画(PSP)を提出しなければならず、このような会議がない場合は、第3段階または第2/3段階臨床試験が開始される前に早期に提出しなければならないPREAの要求を永久的に規定している。最初のPSPは、試験目標および設計、年齢群、関連する終点および統計方法、またはそのような詳細な情報を含まない理由、ならびに小児科研究データおよび支援情報の提供を延期または完全または部分的に免除することを要求する任意の要件を含む、スポンサー計画によって実施される1つまたは複数の小児科研究の概要を含まなければならない。FDAとスポンサーはPSPについて合意しなければならない。臨床前研究,早期臨床試験あるいは他の臨床開発計画から収集したデータから小児科計画の変化を考慮する必要があれば,スポンサーは合意した初期PSPに対する修正案を随時提出することができる。
23
特許期間回復と市場排他性
FDAが私たちの薬物の時間、期限、詳細を承認することによると、私たちのいくつかのアメリカ特許は、1984年の“薬品価格競争と特許期限回復法”(Hatch Waxman修正案と略称する)によって限られた特許期間の延長を受ける資格がある可能性がある。Hatch Waxman修正案は、製品開発およびFDA規制審査中に失われた特許期間の補償として、特許回復期間を最長5年とすることを許可している。しかし,特許期限の回復は特許の残存期間を延長することはできず,製品承認日から合計14年を超えることはできない。特許期間回復期は、通常、IND発効日とBLA提出日との間の時間の半分であり、BLA提出日とその出願が承認されるまでの時間を加える。承認された薬物に適用される特許は1つのみ延期される資格があり、特許が満期になる前に延期されなければならない。米国特許商標局はFDAと協議し,任意の特許期間の延長または回復の出願を審査·承認する。将来的には,その一部が現在所有または許可されている特許出願について特許期間を回復し,特許有効期限を現在の期限を超える日まで延長する予定であり,期待される臨床試験期間および関連する“特許合意”の提出に係る他の要因に依存する。
小児科専有権は、米国で入手可能な非特許マーケティング専有権であり、承認された場合、任意の既存の規制専有権または上場特許の期間に追加的な6ヶ月の市場保護を追加することを規定する。NDA/BLAスポンサーから提出された臨床小児科データがこのようなデータに対するFDAの書面要求に公平に応答すれば,この6カ月の排他性を与えることができる。これらのデータは,この製品が研究されている小児科群で有効であることを証明する必要はなく,逆に臨床試験がFDAの要求に公平に応答していると考えられれば,追加的な保護が得られる。要求された小児科研究報告が法定期限内にFDAに提出され、FDAによって受け入れられた場合、製品の法定または規制排他性または特許保護期間にかかわらず6ヶ月間延長される。これは特許期間の延長ではないが、FDAが同一の薬物/生物の別の出願を承認できない規制期間を効果的に延長する。書面出願の発表はスポンサーに述べた臨床試験を要求しない。今まで、私たちはFDAの書面要請を受けたり要請したりしなかった。
2009年バイオ製品価格競争と革新法
“患者保護および平価医療法案”は、2010年に“医療保健と教育負担性調整法案”(総称してACAと呼ばれる)によって改正され、その中には“生物製品価格競争と革新法”(BPCIA)が含まれており、この法案はPHSAを改正し、FDA許可の参考生物製品生物と類似または交換可能な生物製品のための短い承認経路を作成した。生物類似製品は、臨床上不活性成分にわずかな差があるにもかかわらず、参考製品と高度に類似しており、しかも製品の安全性、純度と効力について、生物製品と参考製品の間に臨床的に意義のない差があると定義されている。交換可能製品は、任意の所与の患者において参照製品と同じ臨床結果を生成することが予想され、複数回投与された製品の場合、製品および参照製品は、安全リスクを増加させることなく、またはそのような代替または切り替えを行うことなく、安全リスクを増加させることなく、または参照生物製品を独占的に使用することに対して治療効果のリスクを低減することなく、以前の投与後に交互にまたは交換することができる生物学的類似製品である。FDAの承認後、基準製品の衛生保健提供者の介入を開くことなく、交換可能な生物類似体で参照製品を代替することができる。
BPCIAによれば、製造業者は、FDA許可の基準生物製品と生物学的に類似しているか、または交換可能な短い生物製品許可申請を提出することができる。このような簡略化された承認経路は、FDAが提案製品に類似した参照生物の以前の審査および承認にある程度依存することによって、生物類似体が“完全な”BLAを提出した場合よりも迅速かつ安価に発売されることを可能にすることを目的としている。
簡略化された承認経路の下で、生物類似申請者は、(1)分析研究により、生物類似製品が参照製品と高度に類似していることを示すデータに基づいて、(2)動物研究(毒性を含む)、および(3)参照製品が承認された1つまたは複数の適切な使用条件下での安全性、純度、および効力を証明するために、1つまたは複数の臨床試験に基づいて証明しなければならない。さらに、出願人は、ラベル、投与経路、用量および強度における生物学的類似製品および参照製品の使用条件が同じ作用機序を有し、生産施設が製品の安全、純度、および効力を確保するための基準に適合しなければならないことを証明しなければならない。
製品が初めて許可を得た日から、参考生物製品は12年のデータ独占期が授与され、最初に承認された交換可能な生物製品は1年に及ぶ独占期が授与され、この独占期間は最長で初の商業発売後の1年となる。FDAが書面で小児科研究を行い、受け入れなければならない場合、12年の専門期間はさらに6ケ月延長される。
24
さらに、FDAは、参照製品が最初に許可された日から4年後まで、参照生物製品に基づく生物学的類似または交換可能な製品の申請を受け入れないであろう。“初許可”とは、一般に、米国で特定の製品が許可された初期日を意味する。最初の許可日は、基準製品の同じ発起人または製造業者(おそらく人、利害関係者、または他の関連エンティティ)がその後に提案した変更(生物物品の構造の変更を含まない)は、新しい適応、投与経路、投与スケジュール、剤形、投与システム、投与装置または強度の変化をもたらすか、または生物学的製品の構造を変更して安全性、純度または効力の変化を引き起こさない後に出願される参照製品の補充物の許可日をもたらす(および新しい特定期間は適用されない)。したがって、新製品が以前の許可製品の構造の修正を含むかどうかを決定し、それにより、安全性、純度、または効力の変化をもたらし、新製品の許可がそれ自身の排他期間をトリガする最初の許可であるかどうかを評価する必要がある。その後の出願は、承認された場合には、生物製品としての“第一次許可”の排他性が保証されるか否かは、具体的な状況やスポンサーが提出したデータに依存する。
BPCIAは複雑であり、FDAはまだ説明して実施している。また、政府の最近の提案は12年間の参考製品専門期間を短縮しようとしている。BPCIAの他の面では,そのいくつかがBPCIAの排他的条項に影響を与える可能性があり,最近の訴訟のテーマでもある.したがって、協約の最終的な影響、実行、そして意味には大きな不確実性がある。
これまで、FDAはいくつかの生物模倣薬を承認し、多くの生物模倣薬はすでにヨーロッパで承認された。FDAはまたいくつかの指導文書を発表し,その生物模倣薬と交換可能な生体模倣薬を審査·承認する方法について概説した。
孤児薬名
孤児医薬品法によれば、FDAは、米国では20万人未満の疾患または疾患に影響を与えると定義されるか、または米国では20万人を超える疾患または疾患に影響を与えると一般的に定義されている稀な疾患または疾患の治療のための医薬を孤児薬として指定することができ、米国でそのような疾患または疾患を治療する薬剤を開発および提供するコストは、米国での販売から回収される。BLAを提出する前に,指定孤児薬を申請しなければならない。FDAが孤児薬物指定を承認した後、FDAは、治療薬の識別情報およびその潜在的な孤児用途を開示するであろう。この投稿は、薬物が孤児薬としてもはや指定されていないかどうかを示す。複数の候補製品は同じ適応で孤児薬物の称号を得る可能性がある。孤児薬を指定するメリットは、税収控除の開発とFDA処方薬使用料の免除を含む。しかしながら、指定孤児薬は、規制審査および承認過程においていかなる利点も伝達されず、規制審査および承認過程の持続時間を短縮することもない。
一般に、孤児指定製品が孤児適応に対するFDAの最初の承認を得た場合、この製品は、孤児薬物排他性を得る権利があり、これは、7年以内に、FDAが、以下にさらに説明する限られた場合を除いて、同じ適応の同じ薬物または生物製品を販売するために、他の任意の出願を許可することを禁止することを意味する。孤児排他性は、異なる薬物或いは生物製剤の同一の稀な疾病或いは疾病の承認を妨げることはなく、同じ薬物或いは生物製剤の異なる疾病に対する承認を阻害することもない。したがって、私たちの候補製品が孤児の独占特許を取得しても、FDAは同じ適応や疾患の治療に異なる薬物や生物製品を承認することができ、より競争力のある市場を作ることができるかもしれない。また,孤児製品に指定された薬物や生物製剤が市販承認された場合,その適応範囲は指定されたものよりも広く,孤児薬物排他性を得る資格がない可能性がある。
場合によっては、同じ薬剤または生物学的製剤を使用する別の製品は、同じ薬剤または生物学的製剤を使用して同じ疾患を治療する後続製品が、より良い治療効果または安全性または患者ケアへの重大な貢献に基づいて臨床的に承認された製品よりも臨床的に優れていることが証明された場合、または孤児の医薬品専門権を有する企業が指定された薬剤または生物学的製剤によって指定された疾患または状態患者の需要を満たすのに十分な数の薬剤または生物学的製剤を保証することを含む、同じ薬剤または生物学的製剤の使用が許可されない場合がある。したがって,競合他社がFDAで定義された同じ薬物の承認を得て,我々の薬物の臨床的優位性を示すことができない場合,あるいは候補製品が競合他社の製品に含まれていると決定され,同じ適応や疾患のために使用された場合,孤立薬物排他性も7年以内に我々の製品の承認を阻止する可能性がある。
最近の法廷事件は,FDAが孤児薬物の排他的範囲を決定する方法に疑問を提起しているが,現在,同機関は管理条例の長期的な解釈を適用し続けており,孤児薬物施行条例を変更するつもりはないことを示している。
2017年10月、FDAはSYNB 1618孤児薬物がPKUの治療に指定されることを許可し、2022年11月、FDAはSYNB 1353孤児薬物のHCU治療のための指定を許可した。
25
迅速チャネル、画期的な治療、まれな小児科疾患、優先審査指定
FDAは特定の製品を指定して加速開発或いは審査を行う権利があり、もしこれらの製品が深刻な或いは生命に危害を及ぼす疾病或いは状況の治療において満たされていない医療需要を解決することを目的としている場合。これらの計画には、高速チャネル指定、画期的な治療指定、優先審査指定が含まれる。
迅速なチャネル認証を得る資格があるために、FDAはスポンサーの要求に基づいて、臨床前研究に基づいて1種の製品を確定し、深刻な或いは生命に危害を及ぼす疾患或いは状況を治療することを目的とし、そして存在しない治療法或いは治療効果或いは安全要素に基づく可能性がある既存の治療法より優れている可能性があることを証明し、未満足の医療需要を満たすことができる。高速チャネル指定は、FDA審査チームとより頻繁な相互作用を行うための機会を提供し、製品の開発と審査を加速する。FDAはまた、完全な出願を提出する前に、スポンサーおよびFDAが出願部分のスケジュールについて合意し、スポンサーがNDAまたはBLAの第1の部分を提出する際に必要な使用料を支払うことを前提として、高速チャネル製品を審査するNDAまたはBLA部分をスクロールすることができる。さらに、迅速チャネルの指定が臨床試験中に出現するデータの支持を得なくなった場合、スポンサーが指定を撤回する可能性があり、またはFDAが指定を取り消す可能性がある。
また、2012年のFDASIAの公布に伴い、国会はINDスポンサーの要求に応じて、FDAが“画期的な療法”に指定した候補製品のための新たな規制計画を作成した。画期的な治療法は、1つまたは複数の他の薬剤または生物学的製品と単独でまたは1つまたは複数の他の薬剤または生物学的製品と組み合わせて深刻または生命に危険な疾患または状態を治療することを目的とした医薬または生物学的製剤として定義され、初歩的な臨床証拠は、1つまたは複数の臨床的に重要な終点において、臨床開発早期に観察される実質的な治療効果のような既存の治療法よりも実質的に改善された効果を示す可能性があることを示す。画期的な治療法に指定されている薬物や生物製品も,それぞれの発売申請の承認を加速する資格がある。FDAは画期的な治療法の承認申請の開発と審査を加速するために、適時に製品スポンサーと会議を行い、それに提案を提供するなど、突破的な治療法に対して何らかの行動を取らなければならない。
最後に、FDAは、重篤な疾患を治療する薬剤または生物学的製品である場合、承認されれば、安全性または有効性の面で顕著な改善を提供する優先審査製品を指定することができる。FDAがマーケティング申請を提出する際に,具体的な状況から他の既存療法と比較して,提案薬が疾患の治療,予防または診断における有意な改善を表すかどうかを決定する。顕著な改善は,ある疾患の治療の有効性の向上,治療を制限する薬物反応の除去あるいは大幅な減少,記録されている患者のコンプライアンスの向上,重篤な結果の改善,あるいは新亜群の安全性と有効性の証拠に現れる可能性がある。指定された目的を優先的に検討することは、全体的な注意およびリソースをこのような出願の評価に誘導し、FDAがマーケティング申請に行動する目標を10ヶ月から6ヶ月に短縮し、申請を提出した日から元のBLAまたは新分子実体のNDAに行動することである。
FDAは、米国で主に出生から18歳までの個人および200,000人未満の深刻かつ生命に危険な疾患に影響を与えるための稀な小児科疾患指定(RPDD)を付与している。この計画によると、スポンサーが薬物または生体薬の承認を得た後、小児科優先審査クーポン券(PPRV)を得る資格がある可能性があり、このクーポン券は、異なる製品の後続のマーケティング申請の優先審査に交換することができる。2020年12月,希少小児科疾患優先審査クーポン券は“創造希望再認可法案”で再認可され,2024年10月1日までにまれな小児科疾患製品に指定されることが許可され,2026年10月1日までに合格したNDAやBLAの承認を得た後,優先審査クーポン券を取得する資格がある。
1つの製品がこれらの計画のうちの1つまたは複数に適合していても、FDAは、製品がもはや資格条件に適合していないことを後で決定することができ、またはFDAの審査または承認を決定する期間が短縮されないことができる。また,迅速チャネル指定,画期的治療指定,優先審査は承認の基準を変更することはなく,最終的に開発や承認過程を加速させない可能性もある。
FDAは2018年4月、SYNB 1618を使用したPKU治療の高速チャネル指定を承認しました。FDAは2022年8月、SYNB 1353を使用してHCUを治療する高速チャネル指定を承認した。SYNB 1618の指定を受けており,これまで規制面でのインタラクションが存在していたため,SYNB 1934の高速チャネル指定も受ける予定である.2023年1月、SynlogicはFDAがHCUのためのSYNB 1353およびPKUのためのSYNB 1934のRPDDを承認することを発表した。
26
承認ルートを加速する
さらに、研究された製品は、深刻または生命を脅かす疾患の治療における安全性および有効性、ならびに既存の治療方法よりも意義のある治療利益を提供する製品は、FDAの加速承認を得る可能性があり、十分かつ制御された臨床試験に基づいて承認される可能性があり、これらの試験は、医薬製品が臨床的利益を合理的に予測する可能性のある代替終点に影響を及ぼすことを証明する。中間臨床終点に対する製品の影響が不可逆的な発病率または死亡率またはIMMへの影響よりも早いことができ、病状の重症度、希少性または流行率、および代替治療が利用可能または不足している場合を考慮すると、IMMまたは他の臨床的利益への影響を合理的に予測する可能性がある場合、FDAは、このような薬物または生物学的製剤の承認を加速することを許可することもできる。承認の1つの条件として、FDAは、IMMまたは他の臨床終点に対する予期される効果を検証および記述するために、承認を加速させた薬物のスポンサーに発売後の臨床試験を要求することができ、迅速な退出手順を必要とする可能性がある。加速的に承認された薬品および生物製品は、従来承認された薬物および生物製品と同じ安全と有効性法定基準に適合しなければならない。
承認を加速するために、代替終点は、例えば実験室測定、放射画像、バイタルサイン、または他の臨床的利益を予測することができると考えられるが、それ自体は臨床的利益の測定基準ではない標識である。代替終点は通常、臨床終点よりも容易または迅速に測定される。中間臨床終点は治療効果の測定であり、1種の薬物の臨床利益、例えばIMMに対する効果を合理的に予測することが可能であると考えられる。FDAは中間臨床終点に基づく加速審査の経験は限られているが、終点で測定した治療効果自体が臨床利益と伝統的な審査の基礎ではなく、もし基礎治療効果が合理的に薬物の最終長期臨床利益を予測する可能性があれば、このような終点は通常加速承認を支持することができることを表明した。
加速承認経路は病気経過が長く、薬物の期待される臨床利益を測定するために比較的に長い時間を必要とする環境に最もよく用いられ、代用或いは中間臨床終点への影響が非常に速く発生した。例えば、加速承認は、様々な癌を治療するための薬剤の開発および承認に広く使用されており、治療の目標は、通常、生存率を向上させること、または発症率を低下させることであり、典型的な病気経過の持続時間は、臨床または生存上の利点を証明するために長い、場合によっては大型の臨床試験を必要とする。
承認を加速する方法は、一般に、薬物の臨床的利益を検証および説明するために、勤勉な方法で追加的な承認後の検証的研究を行うことにスポンサーが同意することに依存する。そのため、この基礎の上で承認された候補製品は必ず厳格な発売後のコンプライアンス要求を守らなければならず、4期或いは承認後の臨床試験を完成し、臨床終点への影響を確認することを含む。必要な承認後研究を行わない場合、あるいは発売後の研究期間中にこの製品の期待される臨床的利益が確認できなければ、FDAがこの薬剤の承認を撤回することを許可する。加速承認手続きにより検討·承認されているすべての候補製品の宣伝材料はFDAの事前審査を経なければならない。
2023年の総合支出法案の一部として、国会はマーケティングを継続する前に加速的に承認された無効薬の患者への潜在的リスクを軽減するために、FDAに追加の法定権力を提供した。この法案のfdcaに対する修正案によると,fdaは加速承認を得た製品のスポンサーに承認前に検証的実験を行うことを求めることができる.スポンサーはまた、試験が完了するまで、検証性試験の進捗報告を6ヶ月ごとに提出しなければならず、これらの報告はFDAのウェブサイトに発表されている。修正案はまた、FDAがスポンサーの検証性試験が製品主張の臨床的利益を検証できなかった場合に、迅速なプログラムを使用して製品承認を撤回することを選択することを可能にする。
承認後に要求する
新製品が承認された後、製造業者および承認された製品は、保存活動の監視および記録、製品の副作用の報告、製品のサンプリングおよび流通制限、許可されていない用途または患者集団への生物製品の普及(すなわち、“ラベル外使用”)の制限、および業界支援を制限する科学および教育活動を含むFDAの一般的かつ持続的な規制を受けるであろう。医師はラベル外の用途のために合法的な製品を処方する可能性があるが、メーカーはこのような用途を販売したり普及させたりしてはならない。FDAや他の機関はラベル外用途の普及を禁止する法律法規を積極的に実行しており,ラベル外用途の普及が不適切であることが発見された会社は重大な責任を負う可能性がある。製品に適応、ラベルまたは製造プロセスまたは施設の変化を含む修正がある場合、出願人は、新しいBLAまたはBLAサプリメントの承認を得るために提出および提出を要求される可能性があり、これは、出願人に追加のデータの開発を要求するか、または追加の臨床前研究および臨床試験を行うことを要求する可能性がある。FDAはまた、製品の安全な使用を保証するためにREMSを要求することを含む、承認時に他の条件を追加することができる。REMSは、制限された分配方法、患者登録、および他のリスク最小化ツールのような薬物ガイドライン、医師のコミュニケーション計画、または安全な使用を確保する要素を含むことができる。承認またはマーケティングに関するこれらの制限は、製品の商業普及、流通、処方、または配布を制限する可能性がある。製品承認は、規制基準を満たしていないことや、初期マーケティング後に問題が発生したことで撤回される可能性があります。
27
FDAの規定は,製品は特定の承認された施設で生産され,cGMPに適合しなければならないことを要求している。CGMP条例には、人員、建物および施設、設備、部品および完成容器および閉鎖された制御、生産およびプロセス制御、包装およびラベル制御、保有および分配、実験室制御、記録および報告、ならびに返品または回収された製品に関する要件が含まれる。私たちの候補製品の製造施設はcGMP要求に適合し、FDAなどの外国規制機関の要求を満たし、その後、任意の製品と私たちの商業製品が生産できることを承認することができなければならない。私たちは依存し、第三者がcGMP法規に従って私たちの製品を生産する臨床と商業数に依存し続けることが予想される。これらのメーカーはcGMP法規、これらの法規要求、他の事項を除いて、品質管理と品質保証、記録とファイルの維持、cGMPからの逸脱を調査し、是正する義務を守らなければならない。承認された医薬品または生物製品の生産および流通に参加する製造業者および他のエンティティは、FDAおよびいくつかの州機関にその機関を登録し、cGMPおよび他の法律を遵守することを確実にするために、FDAおよび特定の州機関の定期的な事前手配または抜き打ち検査を受けなければならない。そのため、メーカーはcGMPコンプライアンスを維持するために、生産や品質管理の分野で時間、お金、労力をかけ続けなければならない。FDAや他の規制機関の将来の検査では,我々のCMO施設におけるコンプライアンス問題が発見される可能性があり,これらの問題は生産や流通を混乱させたり,是正するために大量の資源が必要となる可能性がある。さらに、cGMPに適合しないことを含むこれらのルール違反の条件が発見されると、法執行行動を引き起こす可能性があり、承認後に製品問題が発見されると、以下に説明する自発的なリコールおよび規制制裁を含む製品、製造業者、または承認されたBLA所有者への制限を引き起こす可能性がある。
承認された後、規制基準に適合したままでない場合、または製品発売後に問題が発生したり、発見されたりした場合、FDAは承認を撤回する可能性がある。後に製品に以前に未知の問題があることが発見された場合、新たなセキュリティ情報を追加するために承認されたラベルを強制的に改訂することができる;発売後または臨床試験を実施して新しい安全リスクを評価するため、またはREMS計画に従って流通または他の制限を実施する可能性がある。他の他の潜在的な結果には
そのほか、処方薬製品の流通は“処方薬販売法”(PDMA)の制約を受け、この法案は連邦一級で薬品と薬品サンプルの流通を管理し、各州の薬品流通業者に対する登録と監督管理に最低基準を設定した。PDMA,州法ともに処方薬製品サンプルの配布を制限し,配布中の責任の確保を求めている。最近、“医薬品サプライチェーン安全法案”(Drug Supply Chain Security Act、略称DSCSA)が公布され、米国で流通されているいくつかの処方薬を識別し、追跡するための電子システムを構築することを目的としている。DSCSAは薬品メーカー,卸売業者,流通業者に10年以内に段階的と資源集約型の義務を負うことを求めており,2023年11月にピークに達すると予想される。
国会は時々新しい立法を起草、提出し、採択し、これらの立法はFDA規制製品の承認、製造、マーケティングの法定条項を著しく変える可能性がある。さらなる立法変化が公布されるかどうか、FDAの法規、ガイドライン、解釈が変わるかどうか、あるいはこれらの変化の影響(あれば)が何になるかは予測できない。
28
外国監督管理
アメリカの法規に加えて、私たちは様々な外国法規の制約を受けて、これらの法規はアメリカ以外での臨床試験の表現と、私たちの製品のアメリカ以外での商業販売と流通を管理しています。私たちがFDAの候補製品の承認を得るかどうかにかかわらず、私たちはこれらの国や地域で臨床試験や製品の販売を開始するために、外国や経済地域(例えばEU)の比較可能な規制機関の承認を得なければならない。臨床試験、製品許可、定価と精算を指導する審査手続きと要求は国家と司法管轄区の間で大きく異なり、追加のテストと追加の行政審査期限に関連する可能性がある。他の国や管轄地域で承認を得るのに要する時間は、FDAの承認を得るのに要する時間とは異なり、さらに長くなる可能性がある。1つの国または管轄区域で規制承認を得ることは、他の国または管轄区域で規制承認を得ることを保証することはできないが、1つの国または管轄区域で規制承認を得ることができなかったか、または遅延して監督管理許可を得ることは、他の国または司法管轄区の規制手続きに悪影響を及ぼす可能性がある。
EUの薬品開発、審査、承認
連合では、私たちの候補製品もまた広範囲な規制要求を受ける可能性がある。アメリカと同様に、医薬製品は主管監督機関のマーケティング許可を得た後にのみ発売されることができる。アメリカと類似して、EUの臨床前と臨床研究の各段階は重要な監督管理によって制御されている。
2022年1月31日に施行された新しい“臨床試験条例”(EU)第536/2014号は、EUの臨床試験の承認を簡略化と簡素化することを目的としている。この法規の主な特徴は:単一入口点を通じて申請手続きを簡略化し、即ち“EU臨床試験門戸とデータベース”である;申請のために準備と提出する単一文書、及び簡略化された臨床試験スポンサー報告プログラム;及び統一的な臨床試験申請評価プログラムは、2つの部分に分けられる。第1の部分は、指定された報告書加盟国によって評価され、その評価報告書は、スポンサーおよび臨床試験許可申請が提出されたすべてのEU加盟国(関連加盟国)の他のすべての主管当局によって検討される。二番目の部分は関連する各会員国によって個別的に評価される。臨床試験申請の評価には厳しい期限が設定されている。評価手続きにおける道徳管理委員会の役割は、関連加盟国の国家法律によって引き続き管轄されるだろう。しかし、全体的に関連するスケジュールは臨床試験条例によって定義されるだろう。
米国と同様に,欧州連合(EudraCT)サイト:https://eudract.ema.uropa.euが臨床試験情報を公表する類似要求について述べた。
EUでの薬物の発売許可を得るために、私たちは、いわゆる集中型または国家認可プログラムに従ってマーケティング許可申請またはMAAを提出することができる。
集中手順
集中化プログラムは、欧州薬品管理局(EMA)の有利な意見に基づいて、すべてのEU加盟国およびアイスランド、リヒテンシュタインとノルウェーで有効である単一マーケティング許可を付与することを規定している。特定のバイオテクノロジーによって生産された医薬、孤児医薬製品として指定された製品、高度な治療薬(例えば、遺伝子治療、体細胞治療または組織工学薬)、およびHIV/エイズ、癌、糖尿病、神経変性疾患または自己免疫疾患、ならびに他の免疫機能障害およびウイルス疾患などの特定の疾患を治療するための新しい活性物質を含む製品については、集中的な手順が実行されなければならない。重大な治療、科学的または技術的革新を代表する製品、またはその許可が公衆の健康に有利になる製品については、集中手順がオプションである。中央プログラムによると,環境保護局がMAAを評価する最長期限は210日であり,タイマを含まず,申請者は人が薬品委員会やCHMPからの質問に答えるために追加の書面や口頭情報を提供する。CHMPは特殊な状況下で加速評価を承認することができ、特に治療革新の角度から見ると、1種の医薬製品が重大な公衆衛生利益を有することが予想される場合。加速評価プログラムによる重大な影響評価の期限は150日であり,停止クロックは含まれていない.
国家認可手続き
いくつかのEU諸国では、他の2つの可能な方法が医薬製品を許可することができ、これらの経路は集中プログラム範囲に属さない研究用医薬製品に使用することができる
29
分散したプログラム。分散プロセスを使用して、出願人は、1つ以上のEU諸国において、どのEU諸国でも許可されていない薬品を同時に許可することができ、集中手続きの強制範囲に属さない。
プログラムを相互認識する。相互認識手続きでは、EU加盟国の国家手続きに基づいて、薬物がまずその国で許可される。その後、元の国家マーケティング許可を認めることに関係国が同意する手続きにおいて、他のEU諸国にさらなるマーケティング許可を求めることができる。
上記の手順により、販売許可を承認する前に、欧州市場管理局または欧州経済区加盟国主管当局は、製品の品質、安全性、有効性に関する科学的基準に基づいて、製品のリスク-利益バランスを評価する。
条件付き承認
特定の場合、EU立法(EU第14条(7)条例(EC)第726/2004号および(EC)第507/2006号条例)は、申請者が包括的マーケティング許可を申請するために必要な包括的臨床データを取得する前に、条件付きマーケティング許可を得ることを可能にする。(1)候補製品のリスク−利益バランスが肯定的である場合,(2)出願人が必要な包括的な臨床試験データを提供できる可能性が高い,(3)満たされていない医療ニーズを満たしている場合,および(4)関連医薬製品の即時発売による公衆健康へのメリットが依然として追加データが必要であるという事実に固有のリスクを超えている場合には,候補製品(孤児医薬品として指定された薬剤を含む)に条件付き承認を与えることができる。条件付き販売許可には,現在行われているあるいは新たな研究の完了や薬物警戒データの収集義務など,販売許可保持者が履行しなければならない具体的な義務が含まれていることができる。条件付きマーケティング許可の有効期間は1年であり、リスク−収益バランスが正のままであり、条件または特定の義務を付加または修正する必要性が評価された後、毎年更新することができる。上記の集中プログラムのスケジュールは、条件付きマーケティング許可申請の審査にもCHMPに適用される。
小児科研究
EUのマーケティング許可を得る前に、申請者は、EMAが特定の製品の免除、カテゴリ免除、またはPIPに含まれる1つまたは複数の措置を許可しない限り、EMA承認に適合する小児科人口のすべてのサブクラスをカバーする小児科調査計画(PIP)に含まれるすべての措置を証明しなければならない。(EC)第1901/2006号“小児科条例”と呼ばれる、すべての販売許可手続のそれぞれの要件が規定されている。会社が認可された薬物のために新たな適応,薬物形式あるいは投与経路を増加させようとした場合にも,この要求は適用される。EMAの小児科委員会(PDCO)はある薬物の延期を承認する可能性があり、会社が成人に対する有効性と安全性を証明する十分な情報があるまで、会社が児童薬物の開発を延期することを許可する。PDCOは、小児における薬物の開発を必要としない場合、または適切でない場合にも、高齢者のみに影響を与える疾患のような免除を与えることができる。
マーケティング許可申請を提出する前に、または既存のマーケティング許可を修正する前に、EMAは、会社が各関連PIPに列挙された合意された研究および措置を実際に遵守していると判断する。
EU規制排他性
欧州連合では、許可されて発売された新製品(すなわち参考製品)は、8年間のデータ独占権と追加2年間の市場独占権を得る資格がある。データ特定期間模倣薬或いは生物類似薬申請者がEUが模倣薬或いは生物類似薬の発売許可を申請する時に参考製品ファイルに含まれる臨床前と臨床試験データに依存し、参考製品が初めてEUの許可を得た日から8年以内である。市場排他期は、参考製品がEUで最初の許可を得た10年後まで、成功した模倣薬または生物類似申請者がその製品をEUで商業化することを禁止している。10年の最初の8年間に、マーケティング許可保持者が1つまたは複数の新しい治療適応の許可を得た場合、許可前の科学的評価期間中に、これらの適応は既存の療法と比較して有意な臨床的利益をもたらすことができると考えられる場合、10年間の市場専門期間は最大11年に延長することができる。
30
EU孤児指定と排他性
EUが指定した孤児薬品の基準は原則的にアメリカの基準に似ている。(EC)条例第141/2000条によれば、以下の場合、(1)生命又は長期衰弱に危険な疾患の診断、予防又は治療を目的としている場合、(2)又は(A)申請時に、EU内の影響が10,000人中5人以下である場合、又は(B)孤児の地位のメリットがなければ、EUで十分な見返りが生じず、投資が合理的であることを証明するのに十分ではない。および(3)このような疾患を満足できる診断、予防または治療する方法がEU市場で販売されていないこと、またはそのような方法が存在する場合、製品は、(EC)847/2000条例で定義されているこのような疾患の影響を受けている人に大きな利点を有するであろう。孤児医薬製品は費用の低減や費用の免除などの財政奨励を受ける資格があり、マーケティング許可を得た後、承認された治療適応の10年間の市場排他性を得る権利がある。上場許可を申請する前に、孤児指定申請を提出しなければならない。孤児が孤児として指定された場合、出願人はマーケティング許可申請の費用減免を受けるが、マーケティング許可を提出したときにその指定が待っている場合は、そうではない。孤児指定は、規制審査および承認過程においていかなる利点も伝達されず、規制審査および承認過程の継続時間を短縮することもない。
5年目の終了時に、製品が指定された孤児の基準を満たしていないと判断された場合、例えば、製品の利益が十分に高く、市場排他性を維持するのが合理的であることを証明するのに十分でない場合、欧州連合の10年間の市場排他性は6年に減少することができる。さらに、以下の場合、同じ適応の類似製品にマーケティング許可を随時付与することができる
素数ラベル
EMAは優先薬品計画或いはPrime計画を研究薬物に授与し、そして初歩的な臨床データが満足されていない医療需要を解決する潜在力があることを確定し、そして患者に主要な治療優勢をもたらす。Primeの称号を持つ候補製品のスポンサーは多くのメリットを得ることができ、しかし限らず、早期にEMAと積極的な監督対話を行い、臨床試験設計とその他の開発計画要素を頻繁に討論し、及びファイルを提出した後にマーケティング許可申請の評価を加速する。重要なのは、ヒト医薬製品委員会(CHMP)あるいは高級治療委員会(CAT)の専門機関連絡先と調査委員はPrime計画の早期に任命され、EMA委員会レベルで製品のより多くの理解を促進することである。会議を開始してこれらの関係を開始し、EMAを含む1つの多学科専門家チームは、全体発展と監督戦略に関する指導を提供する。
マーケティング許可は原則として5年間有効であり、マーケティング許可は5年後に、EMAまたはライセンス加盟国の主管当局によるリスク-収益バランスの再評価に基づいて更新することができる。そのため、上場授権書所有者は上場授権書が失効する少なくとも6ヶ月前に、マーケティング授権書を付与してから導入されたすべての変化を含むEMA或いは主管当局に品質、安全性と有効性に関する文書の総合バージョンを提供しなければならない。一旦更新されると、上場許可の有効期限は無期限であり、欧州委員会または主管当局が薬物警戒に関する正当な理由に基づいて5年間の継続を決定しない限り、5年間の継続が決定される。医薬品は、認可が失効した後3年以内に実際にEU市場に投入されない(集中手続きの場合)、または加盟国市場のいかなる許可も許可されていない(いわゆる日没条項)。
イギリス法規
2021年1月1日から、EU法はイギリスに直接適用されなくなった。連合王国は既存の欧州連合薬品条例を採択し、それを連合王国の独立立法とし、販売許可とその他の管理規定に関する手続きとその他の要求を反映するためにいくつかの修正を行った。
イギリスで薬品を販売するために、製造業者はイギリスの許可を持っていなければならない。2021年1月1日、すべてのEUマーケティングライセンスはイギリスマーケティングライセンスに変換されましたが、メーカーは撤退を選択しなければなりません。移行期間中にイギリスは欧州委員会を通じて承認されます
31
コミュニティマーケティング許可プログラム内の新しいマーケティング許可。このような申請には,CHMPの最終的な意見を含む,関連ライセンスプログラム中に環境管理協会に提供されるすべての情報が含まれなければならない。MHRAのガイドラインは,連合王国がEU分散と相互承認プログラムによるマーケティング許可を考慮する権利があることを指摘している。さらに、MHRAのガイドラインは、新しいバイオテクノロジー製品を評価する新しい方法を含む新しい国家許可手続きに言及するために更新された。
“2021年薬品と医療機器法”によると、イギリスの薬品立法は将来的に規制が変化する可能性がある。その法案は薬品条例を採択するための新しい枠組みを作った。
北アイルランド議定書が施行された後、北アイルランドは違う規則を適用するだろう。北アイルランドでは、EU中央マーケティングアプリケーションが引き続き適用されるだろう。
貿易·協力協定には、イギリスと欧州連合との間の医薬製品の獲得性を促進し、公共健康および消費者保護を促進することを目的とした医薬製品に関する添付ファイルが掲載されている。添付ファイルは良好な製造規範(GMP)検査と証明書を相互に認めることを規定しており,これは製造施設が両市場の重複検査を必要としないことを意味する。添付ファイルは、“貿易·協力協定”の項目の下での事項を処理し、協力を促進し、技術的議論を行うための医薬製品ワーキンググループを設置する。“貿易·協力協定”のテーマに属さない規制分野について、薬物警戒を含めたさらなる二国間議論が続く見通しだ。貿易·協力協定には、ロット検出認証を認める互恵的な取り決めも含まれていない。しかし、イギリスは欧州経済地域を含む承認された国をリストしており、これにより、イギリスの輸入業者と卸売業者が特定の認証と規制基準を認めることができるようになる。欧州委員会はまだそのような承認手続きを採択していない。
連合王国に過渡的な承認手続きがあるにもかかわらず、単独の連合王国認可システムが構築されることが予想され、追加の規制費用につながる。また、ロット検査と関連する規制措置の相互承認が不足しているため、追加の規制コストが生じる。
世界の他の地域の規制
EUや米国以外の他の国,例えば東欧,ラテンアメリカ,アジアの国では,臨床試験,製品許可,定価,精算の要求は司法管轄区域によって異なる。また,臨床試験はCGCP要求および“ヘルシンキ宣言”からの適用法規要求と倫理原則に従って行わなければならない。
もし私たちが適用される外国監督管理要求を遵守できない場合、私たちは罰金、規制許可の一時停止または撤回、製品のリコール、製品の差し押さえ、経営制限、刑事起訴などに直面する可能性がある。
保証範囲·定価·精算
医薬製品の販売は第三者保険と精算があるかどうかに大きく依存する。第三者支払者には、連邦医療保険、管理医療提供者、個人健康保険会社、その他の組織などの政府医療保健計画が含まれる。私たちは第三者決済者が私たちの製品に精算を提供すると予想している。しかし、これらの第三者決済者は、価格に挑戦し、医療製品およびサービスの費用対効果を検討することが増えている。また、新たに承認された保健製品の精算状況には大きな不確実性がある。支払者が商品に保険を提供するか否かを判定するプロセスは、保険が承認された後に支払者が製品に支払う価格または販売率を設定するプロセスと分離することができる。第三者支払者は,自分の精算料率を設定する際には通常連邦医療保険カバー政策や支払い制限に依存するが,単独でカバーや精算政策を構築する独自の方法もある。したがって、保険と適切な補償を受けることは時間がかかって高価な過程になるかもしれない。第三者支払者は、承認されたリストまたは処方内の特定の製品に保証範囲を制限することができ、これは、特定の適応のすべての承認された製品を含まない可能性がある。私たちは私たちの製品の医療の必要性と費用効果を証明するために、高価な薬物経済学的研究を行う必要があるかもしれない。また,支払者が薬品に保険を提供することを決定することは,適切な返済率を承認することを意味するものではない。第三者補償は、製品開発投資の適切なリターンを達成するために、十分に高い価格レベルを維持するのに十分ではない可能性がある。私たちの候補製品は費用効果があると思われないかもしれない。私たちは第三者決済者たちに補償を求めて時間がかかって高い。精算は競争力と利益に基づいて私たちの製品を販売させることができないか足りないかもしれません。
連邦医療保険は連邦政府が管理する連邦医療保健計画であり、65歳以上の個人及びある障害者をカバーする。医薬品の性質によっては、医薬品は連邦医療保険の1つまたは複数の部分に組み込まれる可能性がある
32
薬物とそれに関連する条件と投与場所。例えば、D部分によれば、連邦医療保険受益者は、外来処方薬に保険を提供する民間エンティティによって提供される処方薬計画に参加することができる。D部分計画には,Medicare Advantage計画の補完として独立した処方薬福祉計画と処方薬カバー範囲が含まれている。連邦医療保険A部やB部と異なり,D部のカバー範囲は標準化されていない。D部分処方薬計画発起人はすべてのD部分薬物に費用を支払う必要はなく、各薬物計画は自分の薬物処方を制定することができ、それがどの薬物及びレベル或いはレベルをカバーするかを決定することができる。
連邦医療保険B部は,入院環境で使用されている注射薬の多くと,病院外来部や医師室の免許を有する医療提供者が管理する薬剤をカバーしている。連邦医療保険B部分は連邦医療保険行政請負業者が管理し,通常は保険引受決定を担当する。いくつかの支払い調整および制限に基づいて、Medicareは、通常、製造業者によって報告された平均販売価格のパーセンテージに基づいて、B部分保証薬の費用を支払い、この百分率は定期的に更新される。腫瘍内投与を予定している候補品は,連邦医療保険Bの一部規則に拘束されると考えられる。
政府の価格設定制御を実施し,処方薬コストを含む医療コストの増加を制限するために,複数の連邦や州提案が継続されると予想される。例えば,2010年3月に公布されたACAは医療業界に大きな影響を与えると予想される。ACAはほぼ採択以来米国議会の審査を受けており,ACAのいくつかの条項は十分に実施または有効に廃止されていない。したがって、その寿命はまだ確定していない。また、米国で行われている措置は増加しており、薬品価格の圧力を増加させ続けるだろう。このような措置を発表または採用することは、私たちが開発に成功する可能性のある任意の候補製品の潜在的収入に悪影響を及ぼす可能性がある。
また、一部の外国の国では、薬品の提案価格は必ず承認されなければならず、合法的に発売されることができる。各国の薬品定価に対する要求は大きく異なる。また,支払者が薬品に保険を提供することを決定することは,適切な返済率を承認することを意味するものではない。第三者補償は、製品開発投資の適切なリターンを達成するために、十分に高い価格レベルを維持するのに十分ではない可能性がある。例えば、欧州連合は、その国の健康保険制度が補償を提供する医療製品の範囲を制限し、人が使用する医療製品の価格を制御するために、その加盟国に様々な選択を提供している。加盟国は医薬製品の具体的な価格を承認することができ、直接或いは間接的に私たちが医薬製品を市場に投入する収益力を制御する制度を採用することもできる。他の加盟国は会社が自分の薬品価格を固定することを許可したが、処方量を監視し、医師に指導意見を発表し、処方を制限した。薬品に対して価格制御や精算制限を実行する国が私たちのいかなる製品にも有利な精算と定価手配を許可することは保証されません。歴史的に見ると、EUや他の国で発売された製品は米国の価格構造に従わず、通常価格ははるかに低くなることが多い。
全体的には,医療費,特に処方薬の下り圧力が大きくなっている。そのため、新製品の参入にはますます高い壁が設けられている。また、費用抑制措置の一部として、各国政府や他の利害関係者は価格や補償レベルにかなりの圧力をかける可能性がある。政治、経済、規制面の事態は定価交渉をさらに複雑化させる可能性があり、補償を受けた後、定価交渉が継続される可能性がある。EU加盟国が使用する参考価格と平行分配(低価格と高価な加盟国間の裁定)はさらに価格を下げることができる。薬品に対して価格規制や精算制限を実施する国は、有利な精算と定価手配を許可しない可能性がある。
アメリカの他の医療法律法規
もし私たちの候補製品がアメリカで承認されたら、私たちは医療詐欺や乱用に関連する様々なアメリカ連邦と州の法律、規則、法規を遵守しなければなりません。詐欺や法律違反行為は刑事·民事制裁を受けることになり、連邦医療保険や医療補助を含む連邦·州医療保健計画から除外される場合もある。これらの法律には
33
2020年11月、衛生·公衆サービス部は、医療業界により大きな柔軟性を提供し、特に業界参加者間の価値に基づく手配の面で、医療業界により大きな柔軟性を提供し、これらの詐欺や乱用に関する法律の規制負担を軽減することを目的として、“逆バックル条例”、“医師自己紹介法”(Stark法)、および受益者の誘因に関する民事罰金規則の重大な改正を完了した。
最後に、ほとんどの州にも上述した連邦法律のような法規や法規があり、その中のいくつかの法規の範囲はより広く、医療補助や他の州によって精算されるプロジェクトやサービスに適用され、あるいはいくつかの州では支払者にかかわらず適用される。いくつかの州の法律は、製薬会社に製薬業の自発的コンプライアンスガイドライン、または連邦政府が公布した関連コンプライアンスガイドラインを遵守することを要求し、また、製薬業者に医師および他の医療保健提供者への支払いまたはマーケティング支出に関する情報を報告することを要求し、これらの法律が適用される要求が“医師が日光を支払う法案”よりも厳しいことを条件とする。州法や外国法は健康情報のプライバシーやセキュリティを管理する場合もあり、多くの法律は互いに大きく異なり、HIPAAに先を越されず、コンプライアンス作業を複雑化させることが多い。
これらの法律の広汎性、およびそれらの例外および安全港の狭さのために、商業活動は1つまたは複数のこのような法律の挑戦を受けるかもしれない。これらの法律の範囲も執行も不確定であり,現在の医療改革環境下では,特に適用の前例や法規が乏しい場合には,急速な変化が生じる可能性がある。連邦と州法執行機関は最近、医療保険会社と医療保健提供者の間の相互作用の審査を強化し、医療保健業界の一連の調査、起訴、有罪判決と和解を招いた。
第三者との業務配置が適用される医療法律や法規に適合することを確保することは高価で時間がかかる。業務運営が上記の任意の法律または任意の他の適用可能な政府法規に違反していることが発見された場合、製薬業者は、民事、刑事および行政処罰、損害賠償、罰金、返還、個人監禁、政府援助の医療計画(例えば、MedicareおよびMedicaid)から除外された医療計画、契約損害、名声損害、利益および将来の収益の減少、追加の報告義務および監督(これらの法律違反に関する告発を解決するために会社の誠実な合意または他の合意によって拘束された場合)、および運営を縮小または再構成することを含む罰を受ける可能性があり、これらのいずれも、製薬業者の業務運営能力および運営結果に悪影響を及ぼす可能性がある。
34
米国の医療改革と医療保険法の潜在的変化
FDAや他の規制機関の政策は変わる可能性があり、私たちの候補製品に対する規制承認を阻止、制限、または延期するための追加の政府法規が公布されるかもしれない。例えば、議会は、FDAのユーザ費用計画を5年ごとに再認可し、これらの計画を常に変更しなければならず、また、定期的な再認可プロセスの一部として、FDAと業界利害関係者は政策や手続きについて交渉する可能性がある。国会が最近ユーザー料金計画を再認可したのは2022年9月だが、実質的な政策変化は何もない。もし私たちが既存の要求の変化に適応できない場合、あるいは新しい要求や政策を採用することができない場合、あるいは規制コンプライアンスを維持できない場合、私たちが本来得る可能性のあるマーケティング承認を失う可能性があり、私たちは利益を達成したり維持することができない可能性があり、これは私たちの業務、将来性、財務状況、運営結果に悪影響を及ぼすだろう。
前述したように,米国のヘルスケア業界や他の地域の主な傾向はコストを抑えることである。政府当局や他の第三者支払者は、特定の医療製品やサービスのカバー範囲や精算金額を制限し、医療保険や他の医療資金を削減し、新たな支払い方法を適用することでコストを抑制しようとしている。例えば、2010年3月に“平価医療法案”が公布され、他の事項を除いて、医療補助薬品還付計画の下で大多数のメーカーの最低医療補助税金還付を増加させ、メーカーが医療補助薬品還付計画の下で借りた税金還付は吸入、輸液、点滴、インプラントまたは注射の薬物に対して計算される新しい方法を導入し、医療補助薬物還付計画を医療補助管理に参加する看護計画を使用する個人処方に拡大し、ある連邦医療保険D部分の受益者に対して強制割引を導入し、メーカーが連邦医療保険D部分で外来薬をカバーする条件として;そして、アメリカ連邦医療保険と医療補助サービスセンター(CMS)に医療保険革新センターを設立し、革新的な支払いとサービス交付モードをテストし、連邦医療保険と医療補助支出を下げる。
連邦裁判所が数年間訴訟を行った後,2021年6月,米国最高裁はACAの合憲性に対する法的挑戦を却下し,ACAを支持した。ACA下のさらなる立法および規制の変化は、どのような変化または任意の法律がどのような形態をとるのか、生物製薬業界全体または私たちの将来の業務にどのように影響を与えるかどうか、または影響を与える可能性があるにもかかわらず、依然として可能である。ACA,MedicareおよびMedicaid計画の変化や増加,および他の医療改革措置による変化,特に個別州の医療参入,融資または他の立法面での変化は,米国の医療産業に実質的な悪影響を及ぼす可能性が予想される。
また,米国では“平価医療法案”以来,他の医療支出に影響を与える立法改正も提案され可決されてきた。これらの変化には、2011年の予算制御法案によると、提供者に支払われる医療保険総金額が2%減少し、2013年に始まり、2023年の総合支出法案によって延長され、追加の国会行動が取られない限り2032年まで有効になる。
また、政府はメーカーがその販売する製品に価格を設定する方式に対してより厳格な審査を行い、国会で数回の調査を行い、連邦と州立法を提出し、製品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、政府の薬品の精算方法を改革することを提出し、公布した。注目すべきは、2019年12月20日、トランプ総裁が“2019年同等のサンプル取得法案の作成と回復”(Creates Act)という“2020年更なる総合支出法案”(P.L.116-94)に署名したことである。Creates ActはFDAと他の業界人が表明した懸念、すなわちいくつかのブランドメーカーがその製品の流通を適切に制限しないことを解決し、ある製品のREMSの存在を引用して、模倣薬と生物類似製品の開発業者がブランド製品のサンプルを獲得することを阻止することを含む。模倣薬と生物類似製品開発業者はFDA要求のいくつかの比較テストを行うためにサンプルを必要とするため、一部の人は直ちにサンプルを得ることができないが、模倣薬と生物類似製品の市場進出遅延によるものである。この懸念を解消するために、Creates Actは、模倣薬または生物類似製品開発業者がブランドメーカーを起訴することを許可し、“商業的に合理的で市場に基づく条項”で必要なサンプルを提供することを強要する個人訴訟理由を確立した。模倣薬と生物類似製品の開発及びこの新しい道をどのように使用するか、及びCreates Act条項に対する任意の法律挑戦が生じる可能性のある結果は、依然として高度に不確定であり、それが私たちの未来の商業製品に対する潜在的な影響も未知である。
最近、2022年8月に総裁·バイデンが2022年インフレ削減法案、あるいはアイルランド共和軍と署名した。他の点では、アイルランド共和軍には複数の条項があり、連邦医療保険計画や米国全体に販売されている薬品の価格に影響を与える可能性がある。2023年から、薬品の価格上昇速度がインフレ率よりも速い場合、連邦医療保険BまたはD部分がカバーする薬物または生物製品のメーカーは連邦政府にリベートを支払わなければならない。この計算は薬品製品をもとに行われており、連邦政府に不足している税金還付額は、連邦医療保険B部分またはD部分が支払う薬物製品の数に直接依存する。また、CMSは2026年から毎年、模造薬や生物類似競争を含むことなく、選定された数量の単一源D部分薬物について薬品価格交渉を行う。CMSはまた,選定数のB部分薬の薬品価格を2028年から交渉する。もしCMSが薬を選んだら
35
交渉は、このような薬物による収入が減少すると予想される。アイルランド共和軍の薬品価格交渉条項を除いて、総裁·バイデンが2022年10月に発表した14087号行政命令は、アイルランド共和軍を補完し、薬品コストを低減し、革新薬物の獲得を促進する協力医療革新センターの準備とホワイトハウスに潜在的な支払いと交付モードに関する報告書を提出するよう呼びかけた。2023年2月3日現在、この報告は発表されていないが、この分野での現行政当局の優先事項や活動をさらに通報する予定だ。
州レベルでは、各州はますます積極的に、価格または患者の精算制限、割引、ある製品への参入の制限、マーケティングコストの開示および透明性措置を含む薬品および生物製品の価格設定を制御するための法規を立法と実施し、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。2020年12月、米国最高裁判所は、連邦法律は各州の監督薬局福祉マネージャー(PBM)と医療保健と薬品サプライチェーンの他のメンバーの能力を妨げないと一致して判断し、この重要な決定は各州がこの分野でさらに積極的な努力をすることを招きそうである。2022年中に、連邦貿易委員会はPBM業界のやり方に対して全面的な調査を展開し、これはこのような実体の運営、薬局ネットワーク或いは財務手配に対するより多くの連邦と州立法或いは規制提案を招く可能性がある。アメリカに現在存在しているPBM業界を変える重大な努力は薬品サプライチェーンと他の利益関係者の業務に影響を与える可能性があり、著者らのような生物製薬開発業者を含む。また,地域医療当局や個別病院では,どの薬品やサプライヤーが処方薬や他の医療計画に含まれるかを決定するために入札プログラムが使用されるようになってきている。これらの措置が承認されると、私たちの製品に対する最終的な需要を下げたり、私たちの製品の価格設定に圧力をかけるかもしれません。
これらの措置や将来とりうる他の医療改革措置は,より厳しいカバー基準をもたらす可能性があり,我々が受けた任意の承認薬の価格に追加的な下り圧力を与える可能性があり,候補製品の顧客に悪影響を及ぼす可能性が予想される。連邦医療保険や他の政府が計画している精算のいかなる減少も、個人支払者の支払いの同様の減少を招く可能性がある。
外国、連邦と州の各レベルはすでに立法と監督管理提案を引き続き提出することが可能であり、医療保健の獲得性を拡大し、医療保健コストをコントロール或いは低減することを目的としている。コスト抑制措置や他の医療改革を実施することは、私たちの収入の創出、利益の実現、あるいは私たちの製品の商業化を阻止するかもしれない。これらの改革は、私たちが開発に成功し、規制承認を得る可能性のある候補製品の予想収入に悪影響を及ぼす可能性があり、私たちの全体的な財務状況や候補製品を開発する能力に影響を及ぼす可能性がある。
その他の規制事項
私たちは多くの環境、健康と安全法律と法規の制約を受けて、それらの研究室の手続きと危険材料と廃棄物の処理、使用、貯蔵、処理と処理を管理する法律と法規を含む。このような行動は化学物質と生物学的材料を含む危険で燃えやすい材料の使用に関するかもしれない。私たちの業務はまた危険な廃棄物製品を発生させる可能性がある。私たちは第三者とこのような材料と廃棄物を処理する契約を締結した。我々の製品は遺伝子組換え生物(GMO)または遺伝子組換え微生物(GMM)として定義されており、それらの分類と制御によって規制される可能性がある。
アメリカには遺伝子組換え生物に特化した連邦立法はない。遺伝子組換え生物は伝統製品の健康、安全と環境立法を管理することによって監督管理を行う。アメリカが遺伝子組換え生物を監督する方法はこのような仮定を前提としており、即ち監督管理は製品の性質に注目すべきであり、それらの生産過程ではない。
欧州連合および他の場所では、GMMの臨床開発およびマーケティングは、各国で異なる法規および慣行を受けており、これは、環境または他の規制機関および衛生当局の承認に関連する可能性があり、製品のテストまたは許可のリスク評価の要求を規定する可能性がある。
製造業
私たちはすでに、臨床試験材料の生産を支援するための製造プロセスを確立するために、プロセス開発とcGMP生産インフラを含む、私たちの製造組織に大きな投資を続けている。製造技術は、私たちの候補製品が承認されるように、臨床規模とその後の商業規模で高品質の生命薬物を繰り返し製造できるようにすることを目的としている。マサチューセッツ州ウォルザムのcGMPクリーンルームをレンタルすることにより、内部プロセス開発グループ、品質グループ、製造能力を持つ完全に統合された開発と製造組織を構築した。この施設は私たちの臨床パイプラインの早期から中期までの研究のために臨床試験材料を生産することができる。私たちは規模拡大と商業化発展を支援するためにこの組織を建設し続ける。著者らは現在引き続き契約製造組織(CMO)と協力して末期臨床試験材料を生産することを望んでいる。
36
北京大学SYNB 1618の第一段階研究のための臨床試験材料はCMOから製造された。これらの第1回臨床試験は,薬物提示形態として凍結液体と固形製剤を用いた。その時から、私たちは私たちの臨床プロジェクトに必要な固体用量処方の生産を最適化するために、私たちの処方開発に追加の投資を行った。経口懸濁剤の粉末配合能力は、後期臨床開発と将来の商業使用のためにより友好的なプレゼンテーションを作成することができる。私たちは、私たちの合成バイオテクノロジー固体製剤(カプセルを含む)の他のプレゼンテーションの効用に投資し、研究し続けている。
高レベルの細胞やバイオマスを生産できるようにするために,スイッチを用いて合成バイオテクノロジーを設計することができる。これらのスイッチは転写因子とプロモーター対からなり,我々の合成バイオテクノロジーで生産された治療効果器の制御された発現を可能にする。細胞の代謝能力が生産過程で高レベルのバイオマスを生産するように割り当てられることを確保するために,合成生物プログラム中のエフェクター回路はこの成長段階では発現しない。製造プロセスが終了したとき、その後、回路を誘導または活性化する。このような2段階に分けた方法は,高レベルのバイオマス生産を実現し,管理に必要な活動を提供することを目的としている。私たちは工芸開発と製品の生成改善に資源を投入し続けている。
私たちの臨床開発の進歩に伴い、私たちは後期臨床試験と商業製造を支持するために、私たちの生産規模を拡大する必要がある。私たちは私たちの基準に合ったCMOを評価して、私たちの後期臨床開発と商業供給を提供しています。我々は、我々の標準に適合する1つまたは複数のCMOとの協力の利点と、内部でcGMP製造能力および能力を確立する可能性とを比較する。
競争
バイオテクノロジー産業は新製品を開発する競争で異常に競争が激しい。私たちは合成生物学と非病原性細菌代謝工学の業界のリードする専門知識、私たちの臨床開発専門知識と強力な知的財産権の地位は著しい競争優位性があると信じているが、私たちは現在、合成生物学や細胞治療開発プラットフォームを使用する会社と、より伝統的な治療方式(例えば小分子や抗体)に専念する会社からの私たちの開発計画の競争に直面している。競争はより大きな製薬会社、生物技術会社と学術界を含む複数の源から来る可能性がある。このような競争相手の多くは私たちよりも多くの資本と資源を得ることができる。これらの競争相手はまた、合格した科学と管理者を募集と維持し、臨床試験場と臨床試験の患者登録を確立し、著者らの計画を支持する技術を獲得する面で私たちと競争している。我々が最終的に商業化する可能性のある任意の製品については,任意の既存の療法や現在開発されている療法と競合するだけでなく,将来出現する可能性のある新しい療法とも競合するであろう。
競争相手には,他の合成生物学的手法を開発する他の合成生物学会社,細胞や微生物群に基づく会社,DNAやRNAに基づく会社,小分子や他の生物製品を開発する会社がある。私たちが自分の合成バイオテクノロジーを目指している兆候の場合、競争相手は含まれているが、これに限定されない
北大
HCU
腸源性高草酸尿
痛風
37
私たちのチーム:幹部創業者科学コンサルタント
我々の幹部チームは科学ビジョンを成功させた商業治療製品への転換に成功し、新しい治療法の開発の複雑な問題の解決及び監督管理を通じて新しい製品と新しい製品の開発を承認する上で良好な業績記録を持っている。私たちの科学創始者ティモシー·Lu医学博士もジェームズ·コリンズ博士も合成生物学という新興分野の専門家です。我々の管理チームや創始者に加えて,重大な満たされていない医療ニーズがある患者のための合成生物治療製品の開発に取り組んでいる研究者や臨床医と相談関係を構築し,彼らの専門知識は合成生物学,代謝工学,新陳代謝,免疫調節分野にまたがっている。私たちの科学コンサルタントには、Lu博士とコリンズ博士、ポール·ミラー博士、クリストファー·フォガット博士、カミレザー博士、医学博士、クリスタラ·パラサー博士が含まれており、合成生物学と細菌新陳代謝の専門家である。私たちの発展に伴い、私たちは私たちの顧問委員会を拡大することを望んでいる。私たちのすべての創始者とコンサルタントは私たちの株主であり、科学コンサルタントとして報酬を得ている。彼らは定期的に科学相談を行うことができるが、私たちとこれらの個人との手配は、彼らの独立した研究や他の第三者との研究から、彼らの既存または未来の任意の知的財産権を得る権利を与えてくれない。
人力資本
2023年3月22日までに72人のフルタイム従業員がいます。私たちの全従業員のうち、58人が主に研究開発活動に従事している。私たちのすべての職員たちは集団交渉協定の制約を受けない。私たちは私たちが従業員と仲がいいと信じている。
人材の獲得と引き留め
私たちは職員たちが私たちの成功に必須的だということを知っている。そのため、私たちは一流の人材を誘致し、維持することで業務成長を支援しています。私たちは空きポストに高技能の応募者を募集するために内部と外部資源を利用する。私たちは私たちの業務目標を達成するために必要な人材を引き付けて維持することができると信じている。
総奨励
私たちの総リターン理念は、競争力のある報酬と福祉プログラムを提供することで、私たちの従業員に投資してきた。私たちは従業員に基本給、年間インセンティブボーナス、長期持分インセンティブ奨励を含む報酬プランを提供します。また、生命保険、障害保険、健康保険、健康貯蓄、柔軟な支出口座、有給休暇、市場競争力のある会社が支払いをマッチングする401(K)計画および従業員株式購入計画などの全面的な従業員福祉を提供する。私たちの明確な意図は、市場競争力のある報酬と福祉プログラムを提供することで、私たちの業界の第一選択の雇用主になることです。
健康、安全、健康
私たちは投資してきて、従業員の健康、安全、そして健康に投資し続けるつもりだ。私たちは私たちの従業員たちに様々な革新的で柔軟で便利な健康と健康計画を提供する。計画福祉は、保護と安全を提供することを目的としているため、従業員は労働時間を離れる必要があるか、あるいはその財務的健康に影響を与える可能性のある事件に安心して対応できる。
新冠肺炎疫病を考慮して、著者らの投資と従業員の健康、安全と健康に対する優先考慮は特に重要な意義を持っている。私たちのチームメンバーを保護し、支持するために、私たちは個人の作業空間を最大限に利用し、勤務スケジュールを変更し、ワクチンを強制接種し、要求に応じて無症状の新冠肺炎検査を提供することを含む健康と安全対策を実施した。私たちはこの発展状況を引き続き監視し、可能な限り従業員を教育し、支援する計画を求め続けていく。
多様性公平性包括性
私たちは多様な職員たちのチームが私たちの成功に必須的だと信じている。私たちの使命は、人種、民族、宗教、国籍、性別、年齢、性的指向の違い、教育、技能、経験の違いを重視することです。私たちの重点は包括的な採用慣行、公平で公正な待遇、組織の柔軟性、そして訓練と資源だ。
38
訓練と発展
持続的な学習とリーダーシップトレーニングの機会を提供することで、従業員が生涯学習者になることを奨励すると信じている。従業員業績のリアルタイム承認を提供するために努力しているが、個人貢献に関する報酬や株式調整を決定するだけでなく、従業員が追加の訓練や発展機会から利益を得る可能性のある分野を決定する本格的な年次審査プログラムを持っている。
私たちの執行担当者と役員に関する情報
2023年3月29日現在、以下の人員は私たちの役員と役員です
名前.名前 |
|
ポスト |
行政員 |
|
|
AoifeブレナンMB、BCH、BAO、MMSc |
|
取締役CEO兼最高経営責任者 |
マイケル·延ソン |
|
首席財務官 |
アントワン·アワード |
|
首席運営官 |
役員.取締役 |
|
|
ピーター·バレット博士 |
|
アトラス·ベンチャー企業の取締役会長、パートナー |
マイケルBurgess MB CHB Ph.D |
|
Turnstone Biologics臨時首席医療官 |
リサ·ケリー-クロスウェル |
|
ボストン医療センター衛生システム上級副総裁兼首席人的資源官 |
マイケル·ヘフナン |
|
Avenge Bio,Inc.CEO |
パトリシャ·ヘット博士 |
|
リンドラ治療会社のCEO |
ニック·レシュリー |
|
277バイオ社の最高経営責任者。 |
エドワード·マーザス |
|
New Enterprise Associatesの一般パートナー |
リチャード·P·シェイ |
|
デフォス·コンサルタントのCEOにお問い合わせします |
企業情報と歴史
私たちは2007年12月にデラウェア州で設立され、名称は“Mirna Treateutics,Inc.”だった。私たちは直接私たちの子会社を通じて業務を展開します。
我々の子会社Synlogic Operating Company,Inc.は2014年3月14日にデラウェア州でTMC Treateutics,Inc.に登録されている.2014年7月15日,TMC Treateutics,Inc.はSynlogic,Inc.(統合前はプライベートSynlogic(以下定義))と改称された.2015年7月2日、Private Synlogicの普通株と優先株株主はSynlogic,LLC出資協定に調印し、この合意によると、このような普通株と優先株株主はこのような株主がPrivate Synlogicの株式に出資し、新しく設立された親会社Synlogic、LLCの普通株と優先株(2015年再編)と交換する。また,2015年の再編の一部として,IBDCoはSynlogic,LLCの子会社であり,AbbVieとライセンス,選択権,合併プロトコルを締結し,IBDの治療法の開発に用いている。2021年12月22日,IBDCoはSynlogic Operating Company,Inc.と統合され,IBDCoは独立エンティティとして存在しなくなった.
2017年5月,Private Synlogicは再構成(2017組換え)を完了し,これにより,Synlogic,LLCはPrivate Synlogicと合併してPrivate Synlogicに組み込まれ,Private Synlogicは存続した会社として継続された.2017年の再編により、Synlogic、LLCの普通株と優先株はそれぞれPrivate Synlogicの普通株と優先株に交換され、その中にはA類優先株、あるいはA類優先株とB類優先株が含まれている。また、Synlogic 2017持分激励計画(2017計画)に基づいて、Private SynlogicはSynlogic、LLC 2015持分激励計画(2015 LLC計画)の終了によってキャンセルされた励起単位の代わりに持分奨励を発表した。
2017年8月28日、Synlogic,Inc.(前身はMirna Treateutics,Inc.(ナスダック株式コード:MIRN)(MIRNA))は、2017年5月15日の日付に基づいて、Mirna、Meerkat Merger Sub,Inc.(合併子会社)とPrivate Synlogic(合併プロトコル)が署名したプロトコルおよび再編計画に従ってPrivate Synlogicとの業務統合を完了し、このプロトコルにより、Merge SubはPrivate Synlogicと統合してPrivate Synlogicに組み込まれ、Private SynlogicはMirnaの完全子会社として存在し続けている(“合併プロトコル”)。2017年8月25日、合併が完了する前に、Mirnaはその普通株を逆株式分割(逆株式分割)し、2017年8月28日、合併完了後、Mirnaは直ちに“Synlogic,Inc.”と改名した。(ナスダック:SYBX)。
39
合併プロトコルの条項により,MirnaはPrivate Synlogicの株主に普通株を発行し,逆株式分割を計上した後,使用する交換比率は,合併直前のプライベートSynlogic普通株と発行済み優先株の1株あたり交換比率(交換比率)である.さらに、Mirnaは、Synlogic 2017株式インセンティブ計画(2017計画)下のすべての未償還株式オプションを担当し、これらの株式オプションは、ここから一定数のMirna普通株を購入する権利を表し、交換比率に、以前に同オプションによって代表されていたプライベートSynlogic普通株の株式数を乗じたに等しい。MiRNAは2017年の計画も担当している。
私たちのインターネットアドレスはwww.synlogicTx.comです。我々の10-K表年次報告、10-Q表四半期報告、現在の8-K表報告、およびこれらの報告のすべての修正は、米国証券取引委員会(米国証券取引委員会)に電子的に提出するか、または米国証券取引委員会(SEC)に電子的に提出した後、合理的で実行可能な範囲内でできるだけ早く私たちのウェブサイトを介して無料で提供することができます。
40
第1 A項。RISK因子です。
私たちの普通株に投資することは高い危険と関連がある。私たちの業務、将来性、財務状況、または経営業績は、以下のリスクの重大な悪影響を受ける可能性があり、現在知られていないか、または現在重要ではないと考えている他のリスクがあります。また、これらの要素はリスクと不確定要素を代表し、実際の結果は前向き陳述が示唆した結果と大きく異なる可能性がある。そこで,我々の業務を評価する際には,本Form 10-K年次報告と我々が米国証券取引委員会に提出した他の公開文書に含まれる他の情報に加えて,以下のリスク要因の全体的な議論を考慮することを奨励する.以下のリスク要因は、私たちが将来アメリカ証券取引委員会に提出した他の報告書によって時々修正、補充、または代替されるかもしれない。
以下のリスク要因に関する議論では、“私たち”、“私たち”、“私たち”および類似用語とは、Synlogic,Inc.が2017年8月28日に統合された合併業務を意味する。
リスク要因の概要
私たちの業務は、以下の節で強調するリスクと不確実性を含む多くのリスクと不確実性の影響を受けており、これらのリスクと不確実性は、私たちの戦略を成功的に実施する上で直面している課題である。以下のリスク要因においてより詳細に説明される1つまたは複数のイベントまたは状況の発生は、単独で発生しても、他のイベントまたは状況と共に発生しても、私たちの業務、キャッシュフロー、財務状態、および経営結果に悪影響を及ぼす可能性がある。このようなリスクには限定されません
41
42
私たちの財務状況、資本要求、経営業績に関するリスク
私たちは臨床段階のバイオ製薬会社で、赤字の歴史があり、予測可能な未来に損失が続くと予想され、私たちは永遠に実現したり、利益を維持したりすることができないかもしれない。
我々は臨床段階の生物製薬会社であり,合成バイオテクノロジーの開発に専念しており,設立以来,大きな運営損失が発生している。2022年12月31日と2021年12月31日までの会計年度では、純損失はそれぞれ約6610万ドル、6060万ドルだった。2022年12月31日までの累計赤字は約3.57億ドル。今まで、私たちはまだどんな製品収入も発生していない。私たちのほとんどの損失は私たちの研究や開発計画に関する費用と私たちの業務に関する一般的かつ行政的コストによるものです。私たちはまだ製品が発売されていないので、まだ何年もかかると予想されていますが、もしあれば、製品候補が商業化されることができます。
予測可能な未来には、私たちはいかなる製品収入も発生しません。予測可能な未来には、研究開発、臨床前研究、臨床試験のコスト、候補製品の監督審査過程、および商業販売が許可された候補製品の製造とマーケティング能力の発展により、重大な運営損失が生じ続けると予想されます。私たちの未来の潜在的な損失額は不確実だ。利益を達成するためには、候補製品の開発に成功し、規制部門の承認を得て、候補製品を市場に出して商業化し、商業的に合理的な条項で任意の承認された候補製品を製造し、任意の承認された候補製品のための販売·マーケティング組織または適切な第三者代替品を構築し、私たちの業務活動を支援するのに十分な資金を調達しなければならない。私たちはこのような活動で決して成功しないかもしれないし、たとえ私たちが成功しても、利益を達成するのに十分な収入や十分な収入が生まれないかもしれない。たとえ私たちが確実に利益を達成したとしても、私たちは四半期や年度の収益性を維持したり向上させることができないかもしれない。もし私たちが達成して利益を維持できなければ、私たちの価値を下げ、資金を調達し、研究開発努力を維持し、業務を拡大し、あるいは運営を継続する能力を弱める可能性がある。私たちの価値の低下はまた私たちの株主に彼らの投資の全部または一部を損失させる可能性がある。
私たちは受け入れ可能な条件で提供できないか、または全く提供できないかもしれない大量の追加資金を必要とするだろう。
私たちは合成生物学における私たちのプロジェクトと特許薬物開発プラットフォームを発見し、開発するために大量の資金を使用し、私たちの候補製品の臨床前研究と臨床試験を行い、私たちの候補製品のための規制許可を求め、商業販売が許可されたいかなる製品を製造し、販売することを含む、大量の追加資金を必要とする。私たちの将来の資本需要と私たちは既存の資源が私たちの運営を支援する期限が私たちが予想しているのとは大きく違うかもしれないと予想しています。私たちの毎月の支出レベルは新しいものと行っている研究開発や会社の活動によって違います。候補製品の開発成功や商業化に関連する時間や活動の長さを決定することができないため、これらの製品を開発し商業化するためにどれだけの実際の資金が必要かを見積もることはできません。
私たちは予測可能な未来に製品販売や特許使用料から莫大な収入を達成しないことを予想している。私たちの収入源はまだ非常に限られています。私たちの候補製品が臨床開発を完了し、商業化承認を得て上場に成功しない限り、あるいはパートナーと第三者合意に達しなければなりません。これまで、私たちは主に証券の売却、第三者協力、合併を通じて、私たちの運営に資金を提供してきました。私たちは将来的に、協力、株式または債務融資、信用または融資手配、またはそのうちの1つまたは複数の融資源の組み合わせによって、より多くの資金を求めるつもりだ。私たちが追加資金を調達する能力は金融、経済、そして他の要素に依存し、その多くの要素は私たちがコントロールできないだろう。私たちは受け入れ可能な条項や追加的な資金を得ることができないかもしれない。もし私たちが株式または転換可能な債務証券を発行することで追加資金を調達すれば、私たちの株主は希釈され、いかなる融資条項も私たちの株主の権利に悪影響を及ぼす可能性がある。また、私たちに追加資金を提供する条件として、将来の投資家は、既存の株主よりも高い権利を要求し、付与される可能性がある。債務融資が可能であれば、将来の業務活動の柔軟性を制限する制限契約に関連する可能性があり、破産が発生した場合、債務保有者は株式証券保有者が任意の会社の資産配分を受ける前に返済される。
43
もし私たちが適時あるいは受け入れ可能な条件で資金を得ることができない場合、あるいは資金を得ることができない場合、私たちは私たちの研究開発計画と臨床前研究または臨床試験(あれば)、戦略的機会を制限したり、リストラや他の会社の再編活動を延期、制限したりしなければならないかもしれない。私たちはまた、協力者や他の人との手配を通じて資金を求めることを要求されるかもしれません。これらの手配は、私たちが自分で追求していた製品候補や技術の権利を放棄することを要求するかもしれません。
私たちの四半期と年間経営業績は将来的に変動するかもしれません。したがって、私たちは研究アナリストや投資家の期待に達することができないかもしれません。これは私たちの株価を下落させるかもしれません。
各種の要素のため、私たちの財務状況と経営業績は未来に各種の要素によって変動するかもしれません。その中の多くの要素は私たちがコントロールできません。これらの変動をもたらす可能性のある当社の業務に関連する要因には、本年度報告Form 10−Kおよび他の表に記載された他の部分に記載されている要因が含まれています
上記の様々な要因やその他の要因により、これまでのどの四半期や年度の業績にも依存して、我々の将来の経営業績の指標とすべきではない。
私たちの株価の変動は大きくて、私たちの株主は彼らが支払った価格で私たちの普通株の株式を転売できないかもしれません。
私たちの普通株の取引価格の変動は大きく、様々な要素の影響を受けて大幅に変動する可能性があり、その中のいくつかの要素は私たちがコントロールできないことであり、例えば業界アナリストの報告、投資家の見方、業界全体の傾向、マクロ経済状況、あるいは他の会社が発表した類似技術や疾病に関する負の声明である。これらの要因には、本年報10−K表の“リスク要因”の節で議論された要因、および他の要因、例えば、以下の要因も含まれる
44
また,株式市場,特に製薬,生物製薬,バイオテクノロジー株式市場は極端な変動を経験しており,これらの変動は発行者の経営業績とは無関係であることが多い。このような広範な市場変動は私たちの普通株の取引価格や流動性に悪影響を及ぼすかもしれない。従来、1株の市場価格が変動した場合、その株の保有者は発行者に対して証券集団訴訟を起こすことがあった。もし私たちのどの株主が私たちにこのような訴訟を起こしたら、私たちは巨額の訴訟弁護費用が発生する可能性があり、私たちの経営陣の関心は私たちの業務運営から移行します。
私たちの短い経営の歴史は、株主が私たちの業務のこれまでの成功度を評価することを困難にし、私たちの将来の生存能力を評価することも困難になるかもしれない。
私たちは臨床段階の生物製薬会社で、運営の歴史は限られている。私たちは2014年に積極的に運営を始めた。これまで、私たちの業務は組織会社と会社のための人員、研究開発活動、業務計画、資金調達に限られていました。研究や臨床開発に専念する会社から、ビジネス活動ができる会社に転換する必要があります。大規模な重要な臨床試験を成功させ、市場の承認を得て、商業規模の製品を製造したり、第三者代表を配置したり、成功した製品の商業化に必要な販売やマーケティング活動を行う能力があることは証明されていません。通常、新しい候補製品が発見されてから患者の治療に使用することができ、何年もかかる。私たちは予測不可能な費用、困難、複雑な状況、遅延、および他の既知および未知の要素に遭遇する可能性があり、これらの要素は、私たちが1つまたは複数の候補製品の商業化に成功することを阻害するかもしれない。また、薬物開発は資本集約型と高度な投機的な仕事であり、大きなリスクに関連している。あなたは私たちの将来性を考慮して、会社が開発と臨床試験の初期段階でよく遭遇するコスト、不確定性、遅延、困難を考慮すべきです。もし私たちがもっと長い経営歴史或いは成功して医薬製品の開発と商業化の歴史があれば、私たちの未来の将来性、計画或いは生存能力に関する展望性陳述はそんなに正確ではないかもしれない。
金融サービス業の不利な事態の発展に影響を与え、例えば流動資金、金融機関又は取引相手の違約又は不履行に係る実際の事件又は懸念は、我々が現在及び予想している業務運営及びその財務状況及び運営結果に悪影響を及ぼす可能性がある。
流動性が限られている、契約違反、業績が悪い、または金融サービス業または金融サービス業の他の不利な発展に影響を与える実際の事件、または任意のこのような事件または他の類似のリスクに対する懸念または噂は、過去および未来に市場全体の流動性問題を引き起こす可能性がある。例えば、2023年3月10日、シリコンバレー銀行(SVB)はカリフォルニア州金融保護·革新部によって閉鎖され、後者は連邦預金保険会社(FDIC)を担当者に指定した。同様に,2023年3月12日,Signature BankとSilvergate Capital Corp.はそれぞれ破産管理プログラムに巻き込まれた.米国財務省、FRB、連邦預金保険会社は声明の中で、SVBのすべての預金者は閉鎖後にただ1つの仕事後に彼らのすべての資金を抽出することができ、保険のない預金口座に保有されている資金を含むことができるが、金融サービス業全体の流動性に対する懸念には依然として不確定性があると述べた。同様の影響は、例えば2008-2010年の金融危機の間に過去にも発生したことがある。
インフレと金利の急速な上昇は、以前に発行された金利が現在の市場金利よりも低い国債の取引価値を低下させる。米国財務省、連邦預金保険会社、連邦準備委員会は、金融機関が保有するいくつかのこのような政府証券を担保とした金融機関に250億ドルまでの融資を提供し、このようなツールの売却による潜在的損失のリスクを低減する計画を発表しているが、金融機関の顧客引き出しや他の流動性需要に対する広範な需要は、この計画の能力を超える可能性がある。米国財務省、FDIC、連邦準備委員会が将来他の銀行や金融機関が倒産した場合に未保険資金を提供する保証はなく、適時にそうする保証もない。
45
私たちは適切な方法で私たちの銀行関係を検討していると思いますが、私たちが資金源や他の信用手配を得る機会は、私たちと直接手配されている金融機関や金融サービス業全体または全体経済の要素に深刻な影響を受ける可能性があります。これらの資金源および他の信用配置は、私たちの現在および予想される将来の業務運営に資金または資本を提供するのに十分です。他にも、これらの要因には、流動性の緊張または失敗、様々な金融、信用または流動資金協定または手配された義務を履行する能力、金融サービス業または金融市場の中断または不安定、または金融サービス業会社の将来性に対する懸念または否定的な予想が含まれる可能性がある。これらの要因は、我々と金融や業務関係にある金融機関や金融サービス業会社に関連する可能性があるが、金融市場や一般金融サービス業に関連する要因も含まれている可能性がある。
1つまたは複数のこれらの要因に関連するイベントまたは懸念の結果には、現在および予想されているビジネス運営、ならびに私たちの財務状況および運営結果に生じる様々な重大かつ悪影響が含まれている可能性がある。これらは以下のものを含むことができるが、これらに限定されない:
さらに、米国または国際金融システムに対する投資家の懸念は、より高い金利またはコスト、より厳しい財務および運営契約、または信用および流動性源を得るための体系的な制限を含む、あまり有利ではない商業融資条項を招く可能性があり、それによって、私たちは融資を受けにくく、さらには融資を受けることができない。他のリスクに加えて、利用可能な資金または現金および流動資金源の減少は、運営費用、財務義務、または他の義務を履行する能力に悪影響を及ぼす可能性があり、私たちの財務および/または契約義務に違反したり、連邦または州賃金および労働時間法違反を招いたりする可能性がある。上記のいずれかの影響、または上記の要因または他の関連または同様の要因に起因する任意の他の影響は、我々の流動資金、私たちの現在および/または予想される業務運営、ならびに財務状態および運営結果に重大な悪影響を及ぼす可能性がある。
また、マクロ経済や金融サービス業のいずれのさらなる悪化も、私たちと業務を展開している当事者との損失や違約を招く可能性があり、さらに、現在および/または予想される業務運営および運営結果および財務状況に重大な悪影響を及ぼす可能性がある。
私たちの候補製品開発に関するリスク
臨床試験は高価で時間がかかり、しかも内在的なリスクが存在し、著者らはその安全性と有効性を証明できず、関連する監督管理機関を満足させることができないかもしれない。
候補製品の臨床開発は高価で、時間がかかり、重大なリスクに関連している。私たちが約束したどんな臨床試験も計画通りに行われたり、計画通りに完成したりする保証はありません。1つまたは複数の臨床試験の失敗は、開発の任意の段階で起こる可能性がある。候補製品の成功を妨げる可能性があり、または臨床開発をタイムリーに完了する可能性があるイベントは、これらに限定されない
46
臨床開発に成功し、規制機関の私たちの候補製品の承認を得ることができないいかなることも、私たちの追加コストを招き、あるいは私たちの収入を創出する能力を弱める可能性がある。さらに、候補製品の製造または処方変更を行う場合、追加の臨床前研究および/または臨床試験を行う必要がある場合があり、またはこれらの新しい処方から得られる結果は、以前に得られた結果と一致しない可能性がある。臨床試験遅延はまた、私たちの候補製品の任意の予想特許独占期間を短縮する可能性があり、競争相手が私たちの前に製品を開発し、市場に出すことを可能にする可能性があり、これは候補製品を商業化することに成功する能力を弱める可能性があり、私たちの業務と運営結果を損なう可能性がある。
我々がとっている合成生物学を用いて新薬を創造して新しい療法を発見·開発する方法は実証されておらず,決して適切な製品が生じない可能性がある。
我々が候補製品の生成と開発に努める基礎を形成する科学的発見は比較的新しい.我々の方法に基づく薬物開発の実行可能性を支持する科学的証拠は初歩的であり、限られている。合成生物技術は新しい治療方法を代表し、私たちはそれらの治療潜在力を最適化するためにもっと多くの研究と努力が必要かもしれないことに成功した。著者らが開発した任意の候補製品は、実験室および他の臨床前研究においてそれらの治療特性を患者に示すことができない可能性があり、それらは予測不可能、無効、甚だしきに至っては有害な方法でヒト生物システムと相互作用する可能性がある。私たちはまだ成功していないし、重要な臨床試験で私たちの現在または任意の未来の候補製品の有効性と安全性を証明することに決して成功しないかもしれない。もし私たちがこの技術方法に基づいて候補製品を開発して商業化することができなければ、私たちは永遠に利益を上げないかもしれません。私たちの株の価値は下がるかもしれません。
我々の合成生物候補製品は比較的斬新な技術に基づいており、これにより、開発およびその後に規制承認を得る時間およびコスト(あれば)を予測することは困難である。
これまで,我々の研究と開発は,我々の合成生物治療プラットフォームに基づく限られた数の候補製品に集中しており,北大に対するSYNB 1934の概念検証,腸源性高草酸尿に対するSYNB 8802の概念検証,HCUに対するSYNB 1353の機序検証が含まれている。私たちの未来の成功は私たちが実行可能な候補製品を開発することにかかっている。我々が候補製品を開発する際に問題や遅延に遭遇しない保証はなく,これらの問題や遅延が予期しないコストを招くことも保証されず,どのような開発問題も解決される保証はない.
FDA、EMAと他の監督機関の臨床試験と製造要求、及びこれらの監督機関が候補製品の安全性と有効性を決定するための標準は、候補製品のタイプ、複雑性、新規性及び期待用途と市場によって大きく異なる。他のより有名あるいはより広く研究されている治療方式と比較して、合成生物技術などの候補新製品の監督審査過程はより高価である可能性があり、所要時間ももっと長い。我々の候補製品が米国やEUで規制部門の承認を得るのにどのくらいの時間がかかるかを決定することは困難であり,我々の候補製品が商業化にどのくらい時間がかかるかを決定することは困難であり,上場を承認してもそうである.EMAまたは国家規制機関の承認は、FDAが何かを承認する必要がある可能性があることを示すものではなく、各司法管轄区域の規制承認を支援するために、異なるまたは追加の臨床前研究または臨床試験が必要である可能性がある。潜在的な候補製品を市場に投入するために必要な監督管理の承認を遅延または獲得できなかった場合、または監督管理の承認を得る意外なコストは、私たちが十分な製品収入を生成する能力を低下させる可能性があり、私たちの業務、財務状況、運営結果、および見通しが損なわれる可能性がある。
47
私たちの開発プラットフォームを使用して拡張して候補製品パイプラインを構築する努力は成功しないかもしれません。
私たちの戦略の重要な要素の一つは、私たちの的確な重点と経験豊富な管理と科学チームを利用して、広範な人類疾病のために配置できる合成生物技術を作成して、候補製品のパイプを構築することである。これまで,我々の研究開発作業は潜在的な候補製品を生成してきたにもかかわらず,より多くの候補製品を認識·開発し続けることができない可能性がある.私たちがパイプラインを構築することに成功しても、私たちが決定した潜在的な候補製品は臨床開発に適していないかもしれない。例えば、これらの潜在的候補品は、有害な副作用または他の特徴を有することが証明される可能性があり、上場承認を得て市場から受け入れられる薬剤である可能性が低いことを示している。私たちの方法に基づいて候補製品の開発に成功して商業化しなければ、将来的に製品収入を得ることができなくなり、私たちの財務状況に大きな損害を与える可能性があります。私たちの臨床前と臨床開発が成功することは保証できませんし、いずれにしても、監督管理の承認を得る過程には大量の時間と財政資源が必要です。
私たちの候補製品は、不良な副作用を引き起こす可能性があり、またはその規制承認を遅延または阻止し、承認ラベルの商業実行可能性を制限したり、上場承認後の重大な負の結果をもたらす可能性がある(ある場合)特性を有する可能性がある。
私たちの候補製品による副作用は、私たちまたは規制機関が私たちの臨床試験を中断、延期、または終了させたり、ラベルが制限されたり、FDAなどの外国機関の規制承認を延期したりする可能性があります。他の適応の副作用や負の結果は,我々が提案した適応候補製品の開発や承認の潜在力に悪影響を及ぼす可能性がある。
さらに、私たちの1つまたは複数の候補製品が発売承認され、私たちまたは他の人が後にこのような製品による副作用を発見したとしても、これらに限定されない潜在的な重大な負の結果をもたらす可能性がある
これらの事件のいずれも、候補製品に対する市場の受け入れ度を達成または維持することを阻止することができ、承認されても、私たちの業務、運営結果、および将来性を深刻に損なう可能性がある。
私たちの製品開発計画は、私たちの候補製品を服用している患者が経験する可能性のあるすべての有害事象を明らかにしないかもしれません。臨床試験中に私たちの候補製品に接触した被験者の数および臨床開発計画における平均曝露時間は、まれな有害事象または偶然発見を検出するのに十分ではない可能性があり、これらの有害事象または偶然発見は、製品がより多くの患者およびより長い時間のために使用された後にのみ検出される可能性がある。
臨床試験には本質的に潜在患者群のサンプルを用いた。しかし,患者数が限られており,曝露時間が限られているため,候補製品のまれまたは重篤な副作用が発見されることを完全に保証することはできない。この副作用は,この薬物に接触した患者数が有意に増加した場合にのみ発見される可能性がある。候補製品の発売後にこのようなセキュリティ問題が発生または発見された場合、FDAは、製品のラベルの修正や製品のリコールを要求する可能性があり、製品の承認を撤回する可能性もある。これらの事件のいずれも、候補製品に対する市場の受け入れ度を達成または維持することを阻止することができ、承認されても、私たちの業務、運営結果、および将来性を深刻に損なう可能性がある。
48
私たちは私たちの候補製品の成功に大きく依存している。これまで、私たちの候補製品のいくつかは臨床前と臨床環境で結果を出してきましたが、すべての必要な臨床試験を完了した候補製品はありません。私たちはどの候補製品のために十分なデータを生成して、私たちが計画した適応の規制承認を得ることができません。これはそれらの商業化前に必要なものになります。
私たちは私たちの候補製品の組み合わせを決定、買収、開発するためにほとんどの努力と財力を投入した。私たちの将来の成功は、規制部門の1つ以上の候補製品の承認を得て商業化する能力にかかっている。私たちは現在どの製品の販売からも収入を得ていません。私たちは候補製品を開発したり商業化することができないかもしれません。
また,われわれが提案した適応については,われわれの候補製品はいかなる重要な臨床試験にも入っておらず,いずれの重要な臨床試験も起動·完成に数年かかる可能性があり,あれば。FDAや同様の外国規制機関の規制承認を得るまで、私たちは私たちの候補製品のマーケティングや普及は許可されておらず、私たちはいかなる候補製品の規制承認も得られないかもしれない。私たちは私たちのすべての候補製品が臨床試験で成功したり、規制部門の承認を受けると確信できない。また,我々の候補製品は臨床試験で成功しても,規制部門の承認を得られない可能性がある。もし私たちの候補製品が規制部門の承認を得られなければ、私たちは運営を続けることができないかもしれない。
もし私たちが特定の製品の孤児薬の独占経営権を獲得したり維持できなかったら、私たちの競争相手は承認されるかもしれません。同じ病気を治療する競争薬を販売すると、私たちの収入は減少します。
我々の業務戦略の一部として,FDAや欧州委員会孤児薬物指定資格に適合する可能性のある候補製品を開発し,将来的にもこれらの候補製品を開発する可能性がある。FDAは2017年10月、PKUを治療する孤児薬としてSYNB 1618を承認した。孤児医薬品法によれば、米国で20万人未満の人に影響を及ぼすまれな疾患または疾患の治療、診断または予防を目的とした製品であれば、FDAは当該製品を孤児薬として指定することができる。EUでは、生命または慢性衰弱疾患の治療、診断または予防のための薬物は、EUでの流行率が10,000人に5人以下である孤児薬の称号を与えることができる。2022年5月、EMAはSYNB 1618をノースカロライナ大学の孤児の治療として許可した。2022年11月、HCU治療の潜在的薬剤として、FDAからSYNB 1353の孤児薬物指定を取得した。関連するまれな疾患の治療のための指定孤児薬をFDAによって初めて承認された会社は、7年以内に疾患に対するこの薬剤の市場排他的使用を獲得した。いくつかの場合、孤児薬物の独占営業権は、FDAが後に指定要求に重大な欠陥があると認定すること、または製造業者が十分な数の薬剤を保証できない場合を含む、失われる可能性がある。似たような規定もEUで施行され、市場排出期間は10年だ。
私たちのいくつかの候補製品の特許保護範囲と範囲は限られている可能性があるため、孤児薬物指定を獲得することは孤児薬物指定を獲得する資格がある任意の製品候補にとって特に重要である。条件に合致した製品については,“孤児薬品法”下の専門期間に依存して競争地位を維持する予定である。もし私たちが広く特許保護されていない候補製品のために孤児薬の称号を獲得しなければ、私たちの競争相手は同じ病気を治療するための競争薬の販売を求めるかもしれないし、私たちの収入はしたがって不利な影響を受けるかもしれない。
私たちは私たちの候補製品のために孤児薬物の称号を獲得し、他の候補製品のために孤児薬物の称号を求めようとしているにもかかわらず、私たちが最初に発売許可を得た特定の珍しい適応となる保証はない。また、私たちがいくつかの候補製品のために孤児薬物指定を受けていても、あるいは他の潜在的な候補製品のために孤児薬物指定を受けても、この指定は、異なる薬物が同じ条件で許可されることができ、同じ薬剤が異なる条件のために承認され、孤立適応症のラベル外で使用される可能性があるので、競争から効果的に保護することができない可能性がある。孤児薬が承認された後であっても、FDAは、FDAが後者の方が安全またはより有効であると結論した場合、または患者ケアに大きく貢献した場合を含むいくつかの理由で競合する薬物を承認することができる。孤児薬物指定は薬物の開発時間や監督審査時間を短縮することもなく、薬物が監督審査或いは承認過程においていかなる優勢を持たせることもない。
私たちは私たちの1つまたは複数の候補製品のために珍しい小児科疾患の称号またはRPDDを求めるかもしれない。しかし、我々の1つまたは複数の候補製品のBLAは、承認された後に優先審査券の資格基準を満たしていない可能性がある。
2023年1月,SYNB 1934はフェニルアセトン尿症を治療するまれな小児科薬名(RPDD)を獲得し,2022年12月にSYNB 1353はホモシステイン尿症を治療するRPDDを獲得した。2012年の“食品と薬物管理局安全と革新法案”の公布に伴い、国会はFDAが法律の規定基準を満たすある稀な小児科疾患製品申請のスポンサーに優先審査クーポン券を授与することを許可した。この規定はある稀な小児科疾患の予防と治療のための新薬と生物製品の開発を奨励することを目的としている。具体的には、この計画によれば、スポンサーが薬物または生物製剤の承認を得た場合、異なる製品の後続マーケティング申請の優先審査に交換することができるクーポン券を得ることができる。優先審査証明書を取得したまれな小児科疾患医薬製品の発起人は、(販売を含む)証明書を別の発信者に譲渡することができる。この証明書を使用する前に、その証明書は、転送の発信者がまだ提出されていない限り、任意の回数転送することができる
49
申請します。クーポンを獲得したまれな小児科疾患薬が承認日後1年以内に米国で発売されていなければ,FDAはいかなる優先審査クーポンも取り消すことができる。
本計画の場合、“稀な小児科疾患”とは、(A)深刻または生命に危害を及ぼす疾患であり、その深刻または生命を脅かす表現は、一般に新生児、乳児、児童および青少年と呼ばれる年齢層を含む出生から18歳までの個人に主に影響を与える;および(B)“孤児薬物法”が指す稀な疾患または状態を指す。FDAは承認後に私たちの1つまたは複数の候補製品のBLAが優先審査券の資格基準を満たしていないと判断するかもしれない。さらに、RPDDおよびクーポン券計画の現在の法定権限のため、FDAは、(I)この薬剤が2024年9月30日に稀な小児科疾患の指定を受け、その後、2026年9月30日にFDAの承認を得ない限り、(I)議会が計画を再認可しない限り、クーポン券を上場申請のスポンサーに付与しない可能性がある。現在の法定締め切り2024年9月30日までにSYNB 1934とSYNB 1353のまれな小児科疾患指定を受けても,2026年9月までに承認されなければ,証明書を受け取ることができない可能性がある。このようなまれな小児科疾患指定薬の承認が承認されなければ優先審査証明書を得る日が立法で延長されても,その日までに承認を得ることができない可能性があり,承認されても優先審査証明書を得ることができない可能性がある。
製品開発は長くて高価な過程に関連し、結果は不確定であり、早期臨床前研究と臨床試験の結果は未来の臨床試験結果を予測できないかもしれない。
候補製品の前臨床研究または早期臨床試験の結果は、後続の対象または候補製品または任意の他の候補製品の後期臨床試験で得られる結果を予測できない可能性がある。臨床試験の進展が良好であるまでは,臨床試験設計における欠陥は明らかにならない可能性がある。著者らの設計臨床試験における経験は限られており、著者らは臨床試験を設計と実行して、監督部門の著者らの候補製品の承認を支持することができないかもしれない。そのほか、臨床前研究と臨床試験データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品が臨床前研究と臨床試験の中で満足できると思っているが、しかし依然として監督管理機構の許可を得られなかった。臨床前研究と臨床試験の中で満足できるように見える候補製品はまだ監督部門の許可を得られないかもしれない。臨床試験を通過した薬物は高い失敗率を示した。製薬やバイオテクノロジー業界の多くの会社は臨床開発において大きな挫折を経験しており,早期の研究でも奮い立つ結果を得ている。これらの挫折は,他にも,臨床試験施行期間中の臨床前発見,あるいは前臨床研究や臨床試験で行われた安全性や有効性観察によるものであり,これまで報告されていない有害事象を含む。著者らの臨床開発におけるいかなるこのような挫折も私たちの業務と経営業績にマイナス影響を与える可能性がある。
もし私たちが臨床試験で患者登録の遅延や困難に遭遇した場合、私たちのコストは予想以上に高くなる可能性があり、必要な規制承認を受けることは延期または阻止される可能性がある。
新製品候補の臨床試験は、十分な数の健康ボランティアまたは製品候補治療を有する疾患または状況を有する患者を登録し、他の資格基準を満たす必要がある。われわれの臨床試験の時間は,合格した被験者を募集する能力と必要な後続評価の完了状況に依存する。患者と健康ボランティアは著者らの臨床試験に参加したくない可能性があり、原因は新しい治療方法に関連する不良事件の負の宣伝、類似患者群の競争的臨床試験、既存の治療方法の存在、或いはその他の原因は、新冠肺炎の大流行による懸念を含む可能性がある。患者の入院率は多くの要素の影響を受け、潜在的な患者群の規模、患者の年齢と状況、疾病或いは状況の段階と重症度、方案の性質と要求、患者と臨床サイトの接近程度、関連疾病或いは状況に対する有効な治療の可用性、感知のリスク、契約研究組織或いは臨床試験サイトの臨床試験管理実践、CRO或いは臨床試験サイトの労働力不足、研究中の候補製品の投与のメリットと利便性、医師の患者紹介やり方、臨床試験サイトの著者らの試験に対する関心程度を含む。著者らの努力とCROは、臨床試験の適時登録を促進するための努力、及び臨床試験の資格基準を促進する。患者登録の遅延或いは困難或いは試験参加者を維持する困難は、現有或いはその他の研究治療の獲得性を含み、コストの増加、開発時間の延長或いは臨床試験の終了を招く可能性がある。
さらに、私たちの成功は、私たちの臨床試験に参加する資格があるか、または私たちが開発した任意の候補製品から利益を得る可能性のある患者を識別する能力にある程度依存する可能性があり、これは、特定の遺伝子配列または臨床的特徴が存在するかどうかを決定するために、これらの潜在的患者にスクリーニング試験を受けることを要求するであろう。一般に,遺伝子定義の疾患,特に我々の現在の候補製品に対する疾患は,罹患率が相対的に低い可能性がある。例えば,米国,ヨーロッパ,アジアでは約15万人の患者がPKUと診断される可能性が予想される。もし私たちが招いた任意の第三者が協力してくれて、これらの疾患を患っている患者を識別することができなかったり、その過程で遅延に遭遇した場合、私たちは私たちが開発した任意の候補製品のすべての商業的潜在力を達成することができないかもしれない。
50
私たちは潜在的な製品責任クレームに直面するかもしれません。もし私たちに対するクレームが成功すれば、大量の責任とコストが発生するかもしれません。もし私たちの候補製品が患者を傷つけたり、患者を傷つけたと考えられたり、このような損傷が私たちの候補製品と関係がなくても、私たちの規制承認が撤回されたり、他の方法で否定的な影響を与える可能性がある場合、私たちは費用の高さと破壊的な製品責任のクレームを受けるかもしれない。私たちが十分な保険を受けられない場合、あるいは私たちの保険範囲から除外されたり、私たちの保険範囲を超えたクレームによって生じる責任の支払いが要求された場合、このような責任は私たちの財務状況に悪影響を及ぼす可能性があります。
臨床試験で私たちの候補製品を使用または誤用し、市場の承認を得る可能性のあるいかなる製品を販売することは、潜在的な製品責任クレームのリスクに直面させます。消費者、ヘルスケア提供者、製薬会社、または他の販売または他の方法で私たちの候補製品および承認された製品に接触した人は、私たちに製品責任を請求する可能性があります。私たちの候補製品は不良事件を引き起こすかもしれない。もし私たちが製品責任クレームに成功的に対抗できなければ、私たちは大量の責任とコストを招くかもしれない。私たちの候補製品が対象とする疾患を有する患者は、すでに深刻かつ末期の疾患段階にある可能性があり、既知および未知の重大な事前存在および潜在的に生命を脅かす健康リスクを有する。治療中,患者は我々の候補品に関連する可能性のある原因で死亡を含む有害事象を受ける可能性がある。このような事件は、怪我をした患者に巨額の費用を支払うこと、遅延、負の影響、または規制部門が私たちの製品を販売することを許可または維持する機会を得ること、または私たちの商業化努力を一時停止または放棄することを要求する費用の高い訴訟に直面する可能性がある。有害事象が我々の候補製品と無関係な場合でも,その場合の調査に時間や不確実性がある可能性がある.これらの調査は、私たちの規制承認過程や影響を遅延させ、候補製品が獲得または維持される規制承認のタイプを制限する可能性があります。これらの要因により、製品責任クレームが弁護に成功しても、我々の業務、財務状況又は運営結果に実質的な悪影響を及ぼす可能性がある。
私たちはアメリカや国際で行われる可能性のある任意の臨床試験を含む製品責任保険を持っていますが、私たちの保険は私たちが受ける可能性のあるいかなる費用や損失を補償するのに十分ではないかもしれません。私たちはまた、私たちが計画している高度な臨床試験の製品責任保険範囲を増加させることを要求されるかもしれない。もし私たちのすべての候補製品が市場の承認を得たら、私たちは商業製品の販売を含むために、私たちの保険カバー範囲を拡大する必要があるだろう。私たちは私たちが引き続き製品責任保険を得ることができるかどうかを知ることができないし、私たちが必要かもしれない拡大保険を獲得して、十分な金額で私たちを責任損失から保護して、受け入れられる条項、あるいは根本的にはできません。私たちは私たちの保険範囲から除外されたり、私たちの保険範囲を超えたクレームによって生じたどんな責任も支払うのに十分な資源がないかもしれません。もし私たちが第三者との合意に基づいて第三者に賠償を提供すれば、これらの第三者も責任を負い、このような賠償に基づいてクレームを出す可能性がある。個人は、私たちのある候補製品または製品がダメージを与えたと主張したり、消費者の使用に適していないことが発見されたりする製品責任クレームを私たちに提出することができる。このような製品責任クレームは、製造欠陥、設計欠陥、製品固有の危険について警告、不注意、厳格な責任と保証違反の告発を含む可能性がある。州消費者保護法によると、クレームも主張することができる。私たちが提起したどの製品に対する責任クレームも、望ましい点があるかどうかにかかわらず、
製品責任クレームは、私たちを上記のリスクと他のリスクに直面させる可能性があり、これらのリスクは、私たちの業務、財務状況、あるいは運営結果に重大な悪影響を及ぼす可能性があります。
51
私たちまたは私たちが依存している第三者は自然災害の悪影響を受ける可能性があり、私たちの業務の連続性と災害復旧計画は深刻な災害から私たちを十分に保護できないかもしれません。
自然災害は私たちの運営を深刻に混乱させ、私たちの業務、運営結果、財務状況、見通しに実質的な悪影響を及ぼす可能性がある。自然災害、停電、または他の事件が発生した場合、本社の全部または大部分を使用することができず、第三者契約メーカーの製造施設のような重要なインフラを破損したり、他の方法で運営を中断したりすることは困難かもしれませんし、場合によっては、長い間私たちの業務を継続することはできません。深刻な災害や同様の事件が発生した場合、我々の既存の災害復旧および業務連続計画は十分ではないことが証明される可能性がある。私たちの災害復旧と業務連続計画の限られた性質のため、私たちは大量の費用を発生する可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性があります。
新しいコロナウイルスのような伝染病の大流行、流行、爆発ロシアとウクライナの武力衝突のような地政学的緊張もあります私たちの業務と財政的業績に実質的な悪影響を及ぼすかもしれない。
過去数年間、コロナウイルスの爆発は全世界経済の各分野に影響を与え、それは引き続き著者らの業務に実質的な影響を与える可能性があり、著者らの臨床試験活動の深刻な中断を招く可能性がある。また、新冠肺炎は、食品·医薬品局または他の衛生当局の業務に持続的な影響を与える可能性があり、これは、我々の候補製品の審査および承認を含む審査·承認の遅延を招く可能性がある。例えば,2021年には食品医薬品局との協力で新冠肺炎に関連した遅延が生じた。また,新冠肺炎の大流行に対するヘルスケアシステム資源の優先順位により,臨床サイトの起動や患者登録が遅れる可能性がある。例えば、2021年には、新冠肺炎のため、現場IRB会議や議定書改正案を審査する能力が延期されている。ウェブサイトは2021年のある時間に潜在患者との対面スクリーニングアクセスを手配できない場合もある。われわれのCROと臨床試験地点は2022年,2021年,2020年にも労働力不足を経験した。もし隔離あるいは彼らの新冠肺炎に対する懸念が患者の行動を阻害したり、医療サービスを中断したりすれば、一部の患者は臨床試験方案を遵守できない可能性がある。同様に,医療提供者として新冠肺炎リスクを開放している可能性のある患者や首席調査者や現場スタッフを募集·維持する能力もわれわれの臨床試験運営に悪影響を与えている。
世界での疫病の持続的な蔓延はまた、私たちの患者の募集と維持、主要な調査者と現場スタッフの能力を含む、アメリカと他の地域での臨床試験運営に実質的な悪影響を及ぼす可能性があり、もし彼らのいる地域で疫病が発生すれば、医療提供者として、彼らは新冠肺炎への接触を増加させたかもしれない。
我々は引き続きコロナウイルス爆発の潜在的影響,およびすでに実施されている旅行や仕事に関するいかなる制限が,われわれの業務および臨床前と臨床試験に及ぼす影響をモニタリングし続けている。コロナウイルスの我々への影響の程度は将来の事態の発展に依存し,これらの事態の推移は不確実であり,予測もできない.私たちは引き続き私たちのすべての臨床試験の状況を慎重に監視し、現地と連邦衛生当局の指導に従う。
新冠肺炎は,我々が臨床試験に依存している影響地域の第三者契約研究機関や契約製造組織の従業員にも影響を与える可能性がある。例えば、2021年、私たちCROは大流行に関連する人員の問題により、患者サンプルの実験室分析が遅延した。また、私たちは予防措置を取っており、私たち従業員に対するウイルスのリスクを最小限に抑えるための追加措置を取ることができるかもしれない。私たちは必要に応じてこれらの措置を調整し続け、将来的に私たちの業務の需要と従業員の安全をバランスさせることができるようになる。
潜在的なビジネス閉鎖および中断が、私たちまたは私たちと接触している任意の第三者が現在計画されている方法およびスケジュールで業務を展開する能力にどの程度影響するか、または制限する可能性があるかどうかは予測できません。このような影響や制限は、私たちの業務および私たちの運営結果および財務状況に実質的な悪影響を及ぼす可能性があります。コロナウイルスの爆発がもたらす可能性のある潜在的な経済的影響や持続時間は評価や予測が困難である可能性があるが、大流行は世界金融市場の深刻な混乱を招く可能性があり、資金を得る能力を低下させる可能性があり、これは将来的に私たちの流動性にマイナスの影響を与える可能性がある。同様に、現在ウクライナとロシア間の衝突は世界資本市場の極端な変動をもたらしており、グローバルサプライチェーンとエネルギー市場の中断を含むさらなるグローバル経済結果が生じると予想される。新冠肺炎の持続的な拡散やロシアとウクライナの地政学的緊張による景気後退や市場コールは、我々の業務および我々の普通株価格に実質的な影響を与える可能性がある。
52
我々の候補製品の規制承認とその他の法的適合性問題に関するリスク
FDAと他の外国の監督管理機関のような規制審査過程は長く、時間がかかり、本質的に予測できず、もし私たちが最終的に私たちの候補製品のために規制承認を得ることができなければ、私たちの業務は実質的に損害を受けるだろう。
新しい治療製品がFDAおよび他の類似の外国規制機関から発売承認を得るのに要する時間は予測不可能であるが、通常は臨床試験開始後数年が必要であり、監督機関のかなり大きな適宜決定権を含む多くの要素に依存する。さらに、承認政策、法律または法規、または承認を得るために必要な臨床データのタイプおよび数量は、候補製品の臨床開発過程で変化する可能性があり、管轄区域によって異なる可能性がある。私たちはまだ候補製品の商業化の規制承認を得ていません。私たちの既存の候補製品や私たちが将来開発を求める可能性のあるどの候補製品も決して承認されないかもしれません。
私たちの候補製品は多くの理由で規制部門の承認を得ることができないかもしれません
この長い承認過程と未来の臨床試験結果の予測不可能性は、私たちが監督部門の承認を得ることができず、私たちのすべての候補製品を市場に出すことができなくなり、これは私たちの業務、財務状況、そして運営結果を深刻に損なうことになるかもしれない。FDAおよび他の同様の外国規制機関は、承認過程においてかなりの自由裁量権を有し、いつ、または私たちの任意の候補製品のために規制承認を受けるかどうか、および後述するこのような上場承認に任意の条件を適用するかどうかを決定する。我々の候補製品の臨床試験から収集されたデータが有望であると信じていても,これらのデータはFDAや他の同様の外国規制機関の承認を支持するには不十分である可能性がある。
さらに、私たちが承認されても、規制機関は、私たちの要求された範囲以下またはそれ以上の適応を有する任意の候補製品を承認する可能性があり、彼らは、高価な発売後の臨床試験の表現に基づいて承認されるかもしれないか、または候補製品の商業化に成功するために必要または所望のラベル宣言を含まないか、または承認された製品を商業化またはコストよりも高くする制限的なリスク緩和措置または警告言語または禁忌症を含む可能性がある。上記のいずれの場合も、私たちの候補製品のビジネス見通しに実質的な損害を与える可能性があります。
53
私たちは私たちの1つ以上の候補製品のための画期的な治療指定を求めることができるかもしれないが、このような指定を得ることはできないかもしれません。このような指定を受けても、そのような指定はより速い開発や規制審査や承認過程を招くことはなく、私たちの製品候補が上場承認される可能性も増加しません。
私たちはFDAが私たちのいくつかの候補製品の突破的な治療指定を求めるかもしれない。画期的な治療法は、1つまたは複数の他の薬剤と組み合わせて深刻または生命に危険な疾患または状態を単独でまたは1つまたは複数の他の薬剤と組み合わせて治療することを目的とした薬剤または生物製品として定義され、初歩的な臨床証拠は、1つまたは複数の臨床的に重要な終点において、臨床開発早期に観察される実質的な治療効果のような既存の療法よりも実質的に改善される可能性があることを示す。画期的な療法として指定された薬物や生物製品に対して,FDAと試験スポンサーとの相互作用やコミュニケーションは,臨床開発の最も有効な方法の決定に寄与している。FDAで画期的な治療法に指定されている薬物も加速承認を得る資格がある。
画期的療法に指定されたのはFDAの裁量権である。したがって,我々の候補製品の1つが画期的療法として指定された基準に適合していると考えても,FDAは同意せず,この指定を付与しないことにした可能性がある。いずれの場合も、FDAの従来の手順に従って承認を考慮した薬剤と比較して、候補製品に対する画期的な治療指定を受けることは、より速い開発プロセス、審査または承認をもたらすことはなく、FDAの最終承認を保証することもできない可能性がある。さらに、私たちの1つまたは複数の候補製品が資格に適合し、突破的療法として指定されていても、FDAは後で、薬物または生物学的製品が指定された条件を満たしていないと決定する可能性があり、その指定が取り消される可能性がある。
私たちはすでに私たちの候補製品のために高速チャネル認証を取得し、私たちの1つ以上の他の候補製品のためにそのような認証を求めることができますが、私たちはそのような認証を受けなくても、そのような認証は実際にはより速い開発や規制審査や承認過程をもたらすことができないかもしれません。
候補製品が臨床または臨床データではなく、重篤な疾患の治療に使用される場合、このような疾患が満たされていない医療需要を満たす可能性があることを示す場合、製品スポンサーは迅速なチャネル認証を申請することができる。我々は2018年4月にSYNB 1618高速チャネル認証を取得し,2022年8月にSYNB 1353認証を取得した。高速チャネル指定は、候補製品のマーケティング承認を得るか、または任意の特定の時間範囲で承認されることを保証しません。従来のFDAプログラムと比較して、高速チャネル指定のより速い開発または規制審査または承認プロセスを経験しない可能性があります。また,FDAが我々の臨床開発プロジェクトのデータが高速チャネルの指定をサポートしなくなったと考えた場合,その指定を撤回する可能性がある。高速チャネル指定だけではFDA優先審査プログラムに適合する資格は保証されない。
私たちが規制部門の候補製品の承認を得ても、私たちは持続的な規制要求の制約を受けるだろう。
もし私たちの任意の候補製品が発売を許可された場合、私たちは製造、ラベル、包装、貯蔵、広告、販売促進、サンプリング、記録保存、上場後の臨床試験及び安全性、有効性とその他の承認後の情報を提出し、アメリカ連邦と州の要求及び比較可能な外国監督管理機関の要求を含む持続的な法規要求を遵守する。
メーカーとメーカーの工場は、品質管理および製造プロセスが現在の良好な製造規範(CGMP)法規および対応する外国規制製造要件に適合することを保証することを含む、FDAおよび同様の外国規制機関の要求を継続的に遵守しなければならない。したがって、私たちと私たちの契約製造業者は、cGMPの遵守状況、および任意のBLAまたはマーケティング許可申請で行われた約束の遵守状況を評価するために、持続的な審査および検査を受けるだろう。
候補製品のために得られた任意の規制承認は、候補製品が発売される可能性のある承認指示用途の制限または承認条件によって制限される可能性があり、または第4段階の臨床試験、および候補製品の安全性および有効性を監視するモニタリングを含む可能性の高い上場後試験要件を含む可能性がある。私たちはFDAと似たような外国の規制機関に不良反応と生産問題を報告することを要求されるだろう。薬物安全問題を解決するいかなる新しい立法も、製品開発や商業化の遅延を招き、あるいはコンプライアンスを確保するコストを増加させる可能性がある。もし私たちが最初に候補製品の発売許可を加速するルートを通じて獲得したならば、著者らは成功した発売後の臨床試験を行い、著者らの製品の臨床利益を確認する必要があるかもしれない。発売後の臨床試験は成功しない或いはこのような試験を完成できなかったことは上場承認の撤回を招く可能性がある。
54
規制当局が、予期せぬ深刻度または頻度の不良事象など、製品に以前に未知の問題があることを発見した場合、または製品の製造施設に問題がある場合、または製品の販売促進、マーケティング、またはラベルに同意しない場合、規制機関は、製品の市場からの撤退を要求することを含む、製品または私たちに制限を加えることができる。もし私たちが適用される規制要求を遵守できなければ、規制機関や法執行当局は他の措置を取るかもしれない
政府が違法の疑いのあるいかなる調査にも対応するのに時間と資源がかかり、否定的な宣伝が生じる可能性が予想される。現行の規制要件を遵守しないいかなる行為も、我々が製品を開発·商業化する能力に重大な悪影響を及ぼす可能性があり、私たちの価値や経営業績も悪影響を受けるであろう。
1つの管轄区域で私たちの現在と未来の候補製品のマーケティング承認を獲得し、維持することは、他の管轄区域で私たちの現在および未来の候補治療製品のマーケティング承認を得ることに成功するという意味ではありません。
1つの管轄区域で現在と未来の候補製品の上場承認を獲得し、維持することは、任意の他の管轄区で上場承認を獲得または維持できる保証はなく、1つの管轄区で上場承認を獲得できなかったり、遅延したりすることは、他の管轄区のマーケティング承認の流れにマイナス影響を与える可能性がある。例えば、FDAが候補製品の発売を承認しても、外国司法管轄区の比較可能な外国規制機関は、候補製品のこれらの国での製造、マーケティング、普及を承認しなければならない。承認手続きは司法管轄区域によって異なり、1つの管轄区域で行われる臨床研究は他の管轄区の監督当局に受け入れられない可能性があるので、米国と異なるか、または米国より大きい要求と行政審査期間に関連する可能性がある。米国以外の多くの管轄区では、候補製品は先に精算許可を得てから、その管轄区で販売を許可することができる。場合によっては、私たちが未来の製品のために受け取るつもりの価格も承認されるだろう。
私たちはアメリカ以外の他の国でマーケティング申請を提出するかもしれない。米国以外の管轄区域の監督管理機関は候補製品の承認に要求があり、私たちはこれらの管轄区が発売される前にこれらの要求を守らなければならない。外国市場の承認を得て外国の規制要求を遵守することは、私たちに重大な遅延、困難、コストをもたらす可能性があり、私たちの製品がある国/地域で発売されることを延期または阻止する可能性がある。もし私たちが国際市場の規制要求を遵守できず、および/または適用されたマーケティング承認を得られなければ、私たちの目標市場は減少し、私たちの候補製品の市場潜在力を十分に発揮する能力は損なわれるだろう。
米国食品医薬品局、米国証券取引委員会、その他の政府機関の資金不足は、重要な指導部や他の人員の採用と維持の能力を阻害し、新製品やサービスの適時な開発や商業化を阻止するか、あるいはこれらの機関が私たちの業務運営に依存する可能性のある正常な業務機能を履行することを阻止する可能性があり、これは私たちの業務に負の影響を与える可能性がある。
FDAが新製品を審査·承認する能力は、政府予算と資金レベル、肝心な人員の雇用と維持及びユーザー費用の支払いを受ける能力、及び法律、法規と政策の変化を含む様々な要素の影響を受ける可能性がある。したがって、その機関の平均検討時間は近年変動している。また、政府が米国証券取引委員会や我々の業務に依存する可能性のある他の政府機関に提供する資金は、研究開発活動に資金を提供する機関を含め、政治プロセスの影響を受けており、政治プロセス自体が不安定で予測不可能である。
FDAや他の機関の中断も、新薬が必要な政府機関によって審査および/または承認されるのに要する時間を遅らせる可能性があり、これは私たちの業務に悪影響を及ぼすだろう。例えば、ここ数年間、米国政府は何度か閉鎖されており、食品·薬物管理局や米国証券取引委員会などのいくつかの規制機関は、食品·薬物管理局、米国証券取引委員会、および他の政府従業員を休暇させ、重要な活動を停止しなければならない。コロナウイルスの大流行も
55
必要な政府機関の運営。政府が長期的に停止すれば、FDAが私たちが提出した規制文書を適時に審査して処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。また、将来的に政府の閉店は、適切な資本化と事業継続のために、私たちが公開市場に進出し、必要な資本を得る能力に影響を与える可能性がある。さらに、他社または問題からの相互競争の要求は、FDA審査および我々の規制提出の即時性に影響を与える可能性がある。
医療立法改革措置は、私たちの業務、財務状況、または運営結果に実質的な悪影響を及ぼす可能性がある。
アメリカでは、医療費用を統制するための多くの立法計画が続いているだろう。例えば,2010年3月,2010年に医療·教育協調法案(総称してACAと呼ぶ)が改正された“患者保護·平価医療法案”(Patient Protection And Affordable Care Act)が可決され,政府や民間保険会社が医療保険に融資する方式を大きく変更し,米国製薬業に大きな影響を与えた。その他の事項以外に、ACAは生物製品を低コストの生物模倣薬の潜在的な競争を受けさせ、医療補助薬物還付計画に基づいてメーカーが医療補助薬物還付計画の下で借りたリベートを計算し、医療補助薬物還付計画の下でメーカーが不足している最低医療補助還付を増加させ、税金還付計画を医療補助管理の看護組織に登録された個人に拡大し、あるブランドの処方薬と生物製品メーカーに対する年会費と税収を確立し、新しい医療保険D部分カバーギャップ割引計画を作成し、メーカーは70%を提供することに同意しなければならない(2018年の両党予算法により、50%から増加する)。2019年から発効)の適用ブランド薬品と生物製品は保証期間内に条件を満たす受益者に合意価格の販売時点割引を提供し、メーカーの外来薬品或いは生物製品として連邦医療保険D部分の保証条件に組み入れた。
ACA下のさらなる立法および規制の変化は、どのような変化または任意の法律がどのような形態をとるのか、生物製薬業界全体または私たちの将来の業務にどのように影響を与えるかどうか、または影響を与える可能性があるにもかかわらず、依然として可能である。ACA,連邦医療保険,医療補助計画の変化や増加は,連邦政府が薬品価格の変化,その他の医療改革措置による変化を直接交渉することを可能にするなど,特に医療参入,融資または個別州の他の立法において,米国の医療保健業界に実質的な悪影響を及ぼす可能性が予想される。
さらに、ACAが公布されて以来、他の立法改正も提案され、採択された。2011年8月2日、2011年予算統制法が法律に署名され、他を除いて、提供者への医療保険支払いが前期ごとに2%減少し、2013年4月1日に施行され、その後の立法改正により、国会が追加的な行動を取らない限り、2032年まで有効となる。2013年1月2日、“2012年米国納税者救済法”が法律に署名され、病院やがん治療センターを含むいくつかの医療サービス提供者に支払う医療保険を減らし、政府が医療サービス提供者に多額の支払いを取り戻す訴訟時効を3年から5年に延長した。もう一つの例は、2020年12月27日に法律となった2021年総合支出法案に署名し、連邦医療保険B部分でカバーされているすべての薬品および生物製品メーカーにHHSに製品の平均販売価格、すなわちASPを報告することを要求し、民事罰金によって強制的に施行することを含む広範な医療条項と既存の法律の改正を盛り込んだものである。
そのほか、過去数年間、政府は生物製薬メーカーがその上場製品の価格設定の方式に対して更に厳格な審査を行い、アメリカ議会は数回の調査を行い、そして連邦と州立法を提出し、製品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、政府計画の薬品の精算方法を改革することを提出し、公布した。これらの新たに発表された政策の成功確率及び米国処方薬市場への潜在的な影響はまだ不明であり、その中の多くの政策はすでに連邦裁判所システムで法律挑戦を受けている。現在の想定では、このような改革を実施することは持続的な政治的、法的挑戦をもたらすかもしれない。例えば、2021年7月、バイデン総裁は、製薬や医療保険業界に関するいくつかの任務を含む米国経済競争の促進に関する包括的な行政命令を発表し、HHSに高処方薬価格に対応する包括的な計画を発表するよう呼びかけた。2021年9月,HHSが行政命令に応えるために公表した薬品定価計画は,バイ登政府が上昇している薬品価格に対応するための積極的な行動を支持しており,HHSがMedicare B部分とD部分の薬剤のコストについて交渉することを許可することを含むが,このような重大な変化は国会が新たな立法を通過するか,あるいは時間のかかる行政行動が必要となることを明らかにした。
最近は2022年8月に総裁·バイデンが2022年インフレ削減法案、あるいはアイルランド共和軍に署名した。他の点では、アイルランド共和軍には複数の条項があり、連邦医療保険計画や米国全体に販売されている薬品の価格に影響を与える可能性がある。2023年から,連邦医療保険BやD部分がカバーする薬物や生物製品の価格上昇速度がインフレ率よりも速い場合,メーカーは連邦政府にリベートを支払わなければならない。この計算は薬品をもとに行われており、連邦政府の税金還付額は連邦医療保険B部分またはD部分で支払われた薬品の数に直接依存している。また、2026年からCMSは薬品について交渉する
56
模倣薬または生物学的類似競争の選択された数の単一源D部分薬物の年間価格はない。CMSはまた,選定数のB部分薬の薬品価格を2028年から交渉する。CMSが1つの薬物を選択して交渉すれば,このような薬物による収入は減少すると予想される。“2022年インフレ削減法案”が我々の業務や医療業界全体に与える影響は不明である。連邦政府が患者に薬物治療費を負担しやすい方法については,依然として大きな不確実性がある。
州レベルでは、立法機関は、価格または患者の精算制限、割引、特定の製品参入の制限、マーケティングコスト開示および透明性措置を含む、医薬品および生物製品の価格設定を制御するための法規を立法および実施することが増えており、場合によっては、他の国からの輸入および大量購入を奨励することを目的としている。例えば、カリフォルニア州は、製薬業者に、その製品の卸売調達コスト(WAC)が16%を超える増加を要求した場合、少なくとも増加予定の60日前に、健康保険会社および政府健康計画を含む特定の購入者に通知し、さらに、製品の変更または改善がそのような増加が必要かどうかを説明するように製薬業者に要求する。同様に、バーモント州は製薬業者にいくつかの処方薬の価格情報を開示することを要求し、WACがMedicare Part D特殊薬物の敷居を超える新薬を発売する時にその州に通知する。2020年12月、米国最高裁判所も、連邦法律は各州の薬品福祉マネージャー(PBM)および医療保健と薬品サプライチェーンの他のメンバーの能力を妨げていないと一致しており、この重要な決定は各州のこの分野でのさらなるかつ積極的な努力を招くようである。2022年中に、連邦貿易委員会はPBM業界のやり方に対して全面的な調査を展開し、これはこのような実体の運営、薬局ネットワーク或いは財務手配に対するより多くの連邦と州立法或いは規制提案を招く可能性がある。アメリカに現在存在しているPBM業界を変える重大な努力は薬品サプライチェーンと他の利益関係者の業務に影響を与える可能性があり、著者らのような生物製薬開発業者を含む。法律で規定されている第三者支払者の支払金額の価格制御またはその他の制限は、私たちの業務、運営結果、財務状況、見通しを損なう可能性があります。また,地域医療当局や個別病院では,どの薬品やサプライヤーが処方薬や他の医療計画に含まれるかを決定するために入札プログラムが使用されるようになってきている。承認されれば、これは私たちの候補製品に対する最終的な需要を減少させるか、あるいは私たちの製品価格に圧力を与える可能性があり、これは私たちの業務、運営結果、財務状況、見通しにマイナスの影響を与える可能性がある。
EUでは、似たような政治、経済、規制発展は、私たちが製品を利益にして商業化する能力に影響を及ぼすかもしれない。価格とコスト制御措置に対する持続的な圧力に加えて、EUまたはEU加盟国レベルの立法発展は、著しい追加的な要求や障害を招く可能性があり、これは私たちの運営コストを増加させる可能性がある。
私たちは未来の立法や行政または行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することができない。私たちは将来、より多くの連邦と州医療改革措置を取ることが予想され、そのいずれも連邦と州政府が医療製品やサービスのために支払う金額を制限する可能性があり、これはカバー範囲と精算範囲が限られ、承認されると、私たちの製品に対する需要が減少するか、あるいは追加の価格設定圧力をもたらす可能性がある。
私たちは連邦と州医療詐欺と乱用法律、虚偽クレーム法律、および健康情報プライバシーと安全法律の制約を直接または間接的に受ける可能性がある。もし私たちがこのような法律を遵守できないか、または完全に遵守できなければ、私たちは巨額の処罰に直面するかもしれない。
もし私たちの候補製品がFDAの承認を得て、アメリカでこれらの製品を商業化し始めたら、私たちの運営は連邦反リベート法規、連邦虚偽クレーム法案、医師日光法律法規を含む様々な連邦と州詐欺と乱用法律の制約を受ける可能性があります。このような法律は私たちが提案した販売、マーケティング、そして教育計画などに影響を及ぼすかもしれない。また、私たちは連邦政府と私たちが業務を展開している州の患者プライバシー法規によって制限されるかもしれない。私たちの運営能力に影響を与える可能性のある法律には
57
これらの法律の範囲が広く、法定例外状況と選択可能な避難港が限られていることから、私たちのいくつかの商業活動は1つ以上のこのような法律の挑戦を受けるかもしれない。しかも、最近の医療改革法案はこのような法律を強化した。例えば,ACAなどは連邦反リベートや刑事医療詐欺法規の意図要求を改正している。個人や実体はこの規制やそれに違反する具体的な意図を実際に理解する必要はない。また、“反リベート法”の規定によると、米国政府は、“虚偽請求法”については、連邦反リベート法規違反による物品やサービスを含むクレームが虚偽または詐欺的クレームを構成していると断言できる。
私たちの業務が上記の任意の法律または私たちに適用される任意の他の政府法規に違反していることが発見された場合、民事および刑事罰、損害賠償、罰金、連邦医療保険や医療補助などの政府医療計画から除外され、監禁、および私たちの業務を削減または再編することができ、これらはいずれも私たちの業務運営能力と私たちの運営結果に悪影響を及ぼす可能性があります。
私たちは、私たちが個人情報をどのように収集、使用、開示、保存、処理するかに義務を課している米国連邦と州、および外国の法律と法規に支配されているか、または将来的に制限されるかもしれない。私たちは、実際に、このような義務を履行できなかったことが、責任や名声の損害を招き、私たちの業務を損なう可能性があると考えています。これらの法律の遵守を確保することは、私たちの顧客基盤の維持と拡大の努力を損なう可能性があり、私たちの収入を減らすことができる。
多くの活動では,臨床試験を含め,データのプライバシーや健康に関する情報や他の個人情報の保護に関する法律や法規を遵守している。全世界の収集、使用、保護、共有、譲渡、その他の情報処理の規制枠組みは急速に変化しており、予見可能な未来には依然として不確定である可能性がある。例えば、個人健康データを含むEU個人に関する個人データを収集、使用、開示、移転、または他の方法で処理し、2018年5月25日に欧州経済地域(EEA)のすべての加盟国で施行された一般データ保護条例(GDPR)によって制限される。英国のEU離脱と、その後のこれらの地域のデータ保護制度の分離は、EUとイギリス単独のデータ保護法を遵守しなければならないことを意味し、これは追加のコンプライアンスコストを招き、私たちの全体的なリスクを増加させる可能性がある。同様の法律および法規は、個人データの収集、アクセス、使用、分析、修正、保存、送信、セキュリティホール通知、廃棄、および処置を含む個人データの処理を管理します。個人データ源や処理場所に基づく個人データの国際移転に関する法律や法規を遵守しなければならない。個人データの欧州経済圏(EEA)やスイスから米国への移行を促進する法的メカニズムがあるにもかかわらず、欧州裁判所が安全港枠組みの無効を宣言した裁決は、EUプライバシー法の要求を遵守する不確実性を増加させた。この決定により,個人データを欧州連合から米国実体に移行する法的根拠として安全港証明書に依存することは不可能である。2016年2月、欧州委員会は、失効した安全港枠組みを新たなEU-米国安全港枠組みで置き換えることで商務部(DOC)と合意したと発表した。“プライバシー盾”しかし、2020年7月、欧州連合裁判所(CJEU)は、組織がどのように個人データを欧州経済地域から米国に合法的に移転するかを制限し、国際移転の目的のためにEU-米国プライバシー盾を無効にし、個人データ取得に関する支援国の法律を評価し、個人データ取得に関する法律を評価し、企業にプライバシー影響評価を要求することを含む標準契約条項(SCC)の使用にさらに制限を加えた
58
データ保護レベルが欧州経済圏が提供する保護レベルと実質的に同じであることを保証するために、SCC規定に基づいてプライバシー保護を提供する追加措置を実施する必要がある。欧州委員会はその後、2021年6月に新しいSCCを発表し、CJEUの決定と欧州データ保護委員会が提出した提案を説明し、これらの提案は相対的に更に煩雑である。
個人識別情報の記憶、保守、受信または送信のプライバシーおよびセキュリティは、電子的な方法での記憶、保守、受信または送信を含み、米国および海外では厳しい規制を受けている。我々は、適用されるすべてのプライバシーおよびセキュリティ法律および法規を遵守しようと努力しているが、プライバシーの法的基準は発展しており、遵守されていない、または守られていないと思われるいかなる行為も、政府の実体や他の人が私たちに訴訟を提起したり、行動したり、あるいは名声を損なう可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。
多くの外国、連邦、州の法律と法規管理個人は、州プライバシーおよび秘密法(違反行為の開示を要求する州法を含む)、連邦および州消費者保護および就業法、HIPAA;ヨーロッパおよび他の外国データ保護法を含む健康情報の収集、伝播、使用および秘密を識別することができる。これらの法律法規は複雑さや数量的に増加しており,頻繁に変化する可能性があり,時には衝突することもある.
HIPAAは、保護された健康情報、またはPHIを含む個人識別可能な健康情報を保護するための国家プライバシーおよびセキュリティ基準を確立し、健康計画、いくつかの保証取引を電子的に提出する医療情報交換所および医療保健提供者、またはその“業務パートナー”、すなわち、保証エンティティまたは保証エンティティを代表して特定のサービスを実行する個人またはエンティティであり、PHIの作成、受信、保守、または送信に関連する。私たちは現在、HIPAA下の保証エンティティやビジネスパートナーではありませんが、これらのエンティティから識別可能な情報を受信する可能性があります。このような情報を正確に受け取ることができない場合、私たちは各違反に最高50,000ドルの罰金および/または監禁が含まれている可能性があるHIPAAの刑事罰を受けるかもしれない。また、これらや他の法令違反の疑いのある政府調査に対応し、違反が発見されていないことや罰則が加えられていないと最終的に結論が出ても、会社資源を消費して業務に影響を与える可能性があり、公開されていれば、私たちの名声を損なうことになる。
また、カリフォルニア州やマサチューセッツ州などでも同様のプライバシー法律や法規が施行されており、“カリフォルニア医療情報秘密法”のような、健康情報や他の個人識別情報の使用や開示に制限的な要求が加えられている。違反者に罰金と罰を加えるほか、いくつかの州法は、自分の個人情報が乱用されたと思う個人に個人訴訟権利を提供している。例えば、カリフォルニアの患者プライバシー法では、罰金は最大25万ドルに達し、被害者側が損害賠償を要求することを許可すると規定されている。また、カリフォルニア州は“カリフォルニア消費者プライバシー法”(CCPA)を公布し、2020年1月1日に施行され、2020年7月1日にカリフォルニア総検察長によって強制的に施行され、米国初のGDPRのような法律と呼ばれている。CCPAは、カリフォルニア州住民に、彼らの個人情報をアクセスおよび削除するためのより多くの権利を与え、特定の個人情報共有から退出することを選択し、カバーする会社がカリフォルニアの消費者に新しい開示(例えば、この用語の広義の定義)を提供し、これらの消費者に特定の個人情報販売から撤退することを選択する新しい方法を提供することによって、その個人情報がどのように使用されるかに関する詳細な情報を取得する。CCPAは違反行為に対する民事処罰と,データ漏洩に対する個人訴権を規定しており,データ漏洩訴訟が増加すると予想される。また、カリフォルニア州は最近“カリフォルニアプライバシー権法案”(CPRA)を可決した。CPRAは、追加の消費者権利プログラム、データ使用の制限、高リスクデータの新しい監査要件、および敏感なデータからの退出を選択するいくつかの用途を含む、カリフォルニアでビジネスをしている会社に追加のデータ保護義務を課す。また、新たなカリフォルニア州データ保護機関を作成し、実質的な法規の発行を許可し、プライバシーや情報セキュリティ法執行の強化につながる可能性がある。大部分の規定は2023年1月1日に施行され、追加のコンプライアンス投資と潜在的なビジネスプロセスの変化が必要となる可能性がある。CCPAは現在、臨床試験データを含むいくつかの健康関連情報を免除しているが、CCPAとCPRAは著者らのコンプライアンスコストと潜在的な責任を増加させる可能性がある。他の州(例えば、ネバダ州、バージニア州、コネチカット州、ユタ州、コロラド州)も同様の法律を採択したり、他の州と連邦レベルで同様の法律を提出したりしており、通過すれば、このような法律には潜在的な衝突要求があり、コンプライアンスを挑戦に直面させる可能性がある。
連邦と州法律の相互作用は裁判所と政府機関の異なる解釈を受ける可能性があり、私たちと私たちの顧客に複雑なコンプライアンス問題をもたらし、私たちを追加的な費用、不利な宣伝、責任に直面させるかもしれない。また,規制機関のプライバシー問題への関心が高まっていることや,個人情報保護に関する法律法規が拡大し複雑になっていることにより,我々の業務に対する潜在的なリスクが悪化する可能性がある.
59
また、消費者、健康関連とデータ保護法律の解釈と応用は往々にして不確定で、互いに矛盾し、絶えず変化している。
プライバシーとデータセキュリティの立法と規制構造は引き続き発展し、人々はますます私たちの業務に影響を与える可能性のあるプライバシーとデータセキュリティ問題に注目されている。現在および将来の法律法規を遵守しないことは、政府の法執行行動(重大な処罰の適用を含む)、私たちおよび私たちの上級管理者および取締役の刑事および民事責任、個人訴訟および/または負の宣伝を招き、それによって私たちの業務に否定的な影響を与える可能性がある。
もし私たちが環境、健康、安全の法律法規を守らなければ、罰金や罰金を科されたり、私たちの業務、財務状況、または運営結果に重大な悪影響を及ぼす可能性のあるコストが発生する可能性があります。
私たちの研究開発活動と私たちの第三者メーカーとサプライヤーの活動は、私たちの候補製品の成分と他の危険化合物を含む危険材料の制御された貯蔵、使用と処分に関連している。私たちと私たちの製造業者とサプライヤーはこのような危険な材料の使用、製造、貯蔵、処理、処分に関する法律法規を遵守しなければならない。場合によっては、これらの危険材料とそれらを使用して発生した様々な廃棄物は、私たちと私たちの製造業者の施設に貯蔵され、使用と処分を待っている。私たちは汚染リスクを除去することができません。これは私たちの研究開発作業、商業化努力、業務運営を中断させ、環境破壊をもたらし、高価な清掃作業を招き、これらの材料と特定の廃棄物製品の使用、貯蔵、処理、処理を管理する適用法律と法規に基づいて責任を負う可能性があります。我々と我々の第三者メーカーがこれらの材料を処理·処分する際に使用するセキュリティプログラムは、これらの法律法規が規定する基準にほぼ適合していると信じているが、状況が確かにそうであることは保証されず、これらの材料の意外な汚染や傷害のリスクを除去することもできない。この場合、私たちはそれによって生じるいかなる損害に対しても責任を負うことができ、このような責任は私たちの資源範囲を超える可能性があり、州、連邦、または他の適用機関は、特定の材料の使用を減少させ、および/または私たちの業務運営を中断するかもしれない。また,環境法律法規は複雑で変化が頻繁であり,より厳しくなる傾向にある。私たちはこのような変化の影響を予測することもできないし、私たちの未来のコンプライアンス状況を決定することもできない。私たちが行っている研究と開発の性質を考慮して、私たちは現在生物や危険廃棄物保険を受けていません。
国際業務を管理する法律法規は、米国以外で特定の製品を開発、製造、販売することを阻止し、コストの高いコンプライアンス計画の開発、実施、維持を要求する可能性がある。
アメリカ国外で特定の製品を開発、製造、販売するためには、私たちが運営する各司法管轄区の多くの法律と法規を遵守するために資源を投入しなければならない。反海外腐敗法(FCPA)は、いかなる米国人または企業が、いかなる外国人官僚、政党または候補者に直接的または間接的に支払い、提供、許可支払い、または任意の価値のあるものを提供することを禁止し、その個人または企業が業務を獲得または保留することを支援するために、外国の実体の任意の行為または決定に影響を与えることを目的とする。“海外腐敗防止法”はまた、米国に上場する証券会社にある会計条項を遵守することを要求し、これらの条項は、会社(国際子会社を含む)のすべての取引の帳簿と記録を正確かつ公平に反映し、国際業務のために適切な内部会計制御システムを設計し、維持することを要求する。
“反海外腐敗法”を遵守することは高価で困難であり、特に腐敗は公認問題である国である。また、海外腐敗防止法は、多くの国では病院が政府によって運営されているため、医師や他の病院従業員は政府従業員や外国人官僚とみなされる可能性があるため、製薬業に特別な挑戦をもたらしている。その他の場合,臨床試験やその他の仕事に関連して病院に支払われる何らかの金は,政府関係者に支払われる不正金と考えられ,“海外腐敗防止法”の法執行行動につながっている。
様々な法律、条例、および行政命令はまた、米国国外での使用および伝播を制限するか、または国家セキュリティ目的のために秘密にされた情報を特定の非米国国民と共有すること、および特定の製品およびこれらの製品に関連する技術データを制限する。これらの法律は、私たちがアメリカ以外で特定の製品や候補製品を開発、製造、販売することを阻止するかもしれません。これは、私たちの成長潜在力を制限し、私たちの開発コストを増加させるかもしれません。
国際ビジネス慣行に関する法律を遵守しなければ、重大な民事·刑事罰を受け、政府契約の資格を一時停止または廃止する可能性がある。米国証券取引委員会は、発行者が“海外腐敗防止法”の会計条項や輸出規制法に違反したため、発行者の米国取引所での証券取引を一時停止または禁止する可能性もある。
60
私たちの内部コンピュータシステム、または私たちの協力者や他の請負業者やコンサルタントのシステムは、故障したり、セキュリティホールに遭遇したりする可能性があり、これは、私たちの製品開発計画が実質的に破壊される可能性があります。
私たちの内部コンピュータシステム、ならびに私たちの現在および未来の任意の協力者、ならびに他の請負業者、コンサルタント、または臨床試験場所のコンピュータシステムは、ネットワーク攻撃、コンピュータウイルス、許可されていないアクセス、自然災害、テロ、戦争、および電気通信および電気故障の破壊を受けやすい。ネットワーク攻撃の頻度,複雑さ,強度が増加しており,発見されにくくなってきている.ネットワーク攻撃は、サービスの信頼性に影響を与え、情報の機密性、完全性、および利用可能性を脅かすために、有害なマルウェアの配備、恐喝ソフトウェア、サービス攻撃の拒否、ファイルの不正アクセスまたは削除、社会工学、ネットワーク釣り、および他の手段を含む可能性がある。時間が経つにつれて、このような脅威の数と複雑さは増加している。上記のいずれかのイベントが発生し、当社の運営中断を招く場合、我々のビジネス機密や他の独自情報の損失によるものであっても、他の同様の中断によるものであっても、我々の開発計画および業務運営の実質的な中断を招く可能性がある。例えば、臨床前あるいは臨床試験データの紛失は私たちの監督管理の承認作業を遅延させ、データを回復あるいは複製するコストを著しく増加させる可能性がある。もしどんな中断やセキュリティホールが私たちのデータやアプリケーションを紛失したり、破損したり、機密または独自の情報を適切に開示しない場合、私たちは責任を招く可能性があり、私たちの競争地位が損なわれる可能性があり、私たちの候補製品のさらなる開発と商業化は延期される可能性がある。私たちの安全措置の有効性に対する市場の見方は損なわれる可能性があり、私たちの名声と信頼は損なわれるかもしれない。我々はこれらの事件の発生を防止するためのシステムと制御を開発·維持し、脅威を識別·緩和するプロセスもあるが、これらのシステム、制御、プロセスの開発と保守はコストが高く、技術の変化とセキュリティ対策を克服する努力がますます複雑になり、継続的に監視·更新する必要がある。しかも、私たちが努力したにもかかわらず、このような事件が発生する可能性を完全になくすことはできない。より多くの情報システムをサプライヤーにアウトソーシングし、支払人や患者とより多くの電子取引を行い、クラウドベースの情報システムにより多く依存するにつれて、関連するセキュリティリスクが増加し、私たちの技術および情報システムを保護するために追加の資源が必要になるであろう。さらに、システム障害時の障害、サービス中断、データの悪化または損失から私たちを保護するのに十分なセキュリティおよび制御措置を実施するために、私たちの内部情報技術システムまたは私たちの第三者請負業者のシステム、または私たちのコンサルタントが十分なセキュリティおよび制御措置を実施するための努力を保証することは、ネットワーク攻撃、セキュリティホール、産業スパイ攻撃、または金融、法律、商業または名声被害をもたらす可能性のある内部脅威攻撃時にデータが盗まれたり破損したりすることを防止するのに十分である。
合成生物学や遺伝子工学に対する倫理,法律,社会的懸念は,我々の技術の使用を制限または阻止し,我々の収入を制限する可能性がある。
私たちの技術は合成生物学と遺伝子工学の使用に関するものだ。合成生物学や遺伝子工学の安全や環境被害に対する公衆の見方や,その倫理的問題に対する懸念は,我々の技術,候補製品,プロセスに対する公衆の受け入れに影響を与える可能性がある。もし私たちと私たちの協力者が合成生物学や遺伝子工学に関する倫理、法律、社会的懸念を克服できなければ、私たちの技術、候補製品、そしてプロセスは受け入れられないかもしれない。これらの懸念は、費用の増加、規制審査および規制の強化、合成バイオテクノロジーの輸入に対する貿易制限、私たちの計画の遅延または他の障害、または合成バイオテクノロジーの公衆の受け入れと商業化をもたらす可能性がある。また,我々の技術を用いて製造された合成バイオテクノロジーは,悪影響や他の有害事象を引き起こす可能性があり,負の宣伝につながる可能性もある。我々が設計·生産した候補製品は,制御された実験室で自然に産生される生物や酵素に匹敵する特性や劣勢を持っているが,これらの生物を制御されていない環境に放出することは予期しない結果をもたらす可能性がある。このような漏れによるいかなる悪影響も、私たちの業務、財務状況、または運営結果に重大な悪影響を及ぼす可能性があり、それによって生じるいかなる損害にも責任を負う可能性があります。
私たちの知的財産権に関するリスクは
私たちは合成バイオテクノロジー、候補製品、そして私たちの開発プロセスに必要な権利を買収と許可を通じて得ることができないかもしれない。
現在、私たちは第三者からの許可と私たちが持っている特許と特許出願を通じて、いくつかの知的財産権の権利を持っている。私たちの業務の成長は、私たちと私たちの許可側の知的財産権を獲得、維持、または実行する能力があるかどうか、および追加可能な独占権を得ることができるかどうかにある程度かかっているかもしれません。例えば、私たちの計画は、第三者が所有する他の独占権を使用する必要がある可能性がある他の候補製品または配信システムに関連することができる。
61
さらに、我々の候補製品は、効率的かつ効率的に動作するために特定の配合物を必要とする可能性があり、これらの権利は他の第三者によって保持されている可能性がある。私たちは、私たちが決定した第三者から許可成分、使用方法、プロセス、または他の第三者知的財産権を開発、取得、または取得することができないかもしれません。第三者知的財産権の許可·買収は競争分野であり、他の一部の企業も魅力的であると考えられる第三者知的財産権許可または買収戦略をとっている可能性がある。これらの会社の規模、現金資源、そしてより強い臨床開発と商業化能力のため、それらは私たちより競争優位を持っているかもしれない。
例えば,我々は以前学術機関と協力しており,これらの機関との書面合意に基づいて我々の臨床前研究や開発を加速し続けている可能性がある。一般に、これらの機関は、交渉によって、機関が協力において取得した任意の技術的権利のライセンスを取得することができるオプションを提供する。知的財産権のこのような最初の交渉権にかかわらず、私たちは指定された時間範囲内で、またはそれが受け入れられる条項の下で許可を交渉することができないかもしれない。もし私たちがそれができなければ、その機関は他の当事者たちに知的財産権を提供するかもしれないし、私たちの計画を実行し続けることを阻止するかもしれない。
しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。私たちはまた私たちの投資が適切な見返りを得るための条項の許可や第三者知的財産権を得ることができないかもしれない。もし私たちが第三者知的財産権を成功的に得ることができなければ、私たちの業務、財務状況、成長の見通しは影響を受けるかもしれない。
我々は特許権と我々の候補製品の地位に依存し,承認されれば,生物製品価格競争や革新法(BPCIA)に基づいて独占経営する資格のある生物製品とする予定である。Synlogicがこれらの手法の組合せから排他性を獲得または保持できなければ,Synlogicは我々の市場で効率的に競合できない可能性がある.
私たちは、特許、商業秘密保護、および秘密保護協定の組み合わせによって、私たちの技術および候補製品に関連する知的財産権を保護するか、または依存します。私たちの成功は、私たちと私たちの許可者がアメリカと他の国/地域で私たちの独自技術と製品について規制排他性を獲得し、特許および他の知的財産権保護の能力を維持することに大きくかかっている。
私たちは、私たちの候補製品に関連する特許出願をアメリカと海外に提出することで、私たちの独自の地位を保護しようとしています。これらの候補製品は私たちの業務に非常に重要です。この過程は高価で時間がかかり、私たちはすべての必要または望ましい特許出願を合理的なコストまたはタイムリーに提出して起訴することができないかもしれない。我々は,特許保護を得るのが遅くなるまで,我々の研究開発成果で特許を申請できることを確認できない可能性もある.
バイオテクノロジーと製薬会社の特許地位は通常高度に不確定であり、複雑な法律と事実問題に関連しており、その法律原則はまだ解決されていない。私たちが所有しているまたは許可されている特許出願は、発行された特許を生成できない可能性があり、その声明は、米国または他の国/地域における候補製品をカバーしている。私たちの特許および特許出願に関連するすべての潜在的に関連する既存技術が発見されたことは保証されず、これは、特許を無効にするか、または係属中の特許出願から特許が発行されることを阻止する可能性がある。特許が確実に発行されても、これらの特許が私たちの候補製品をカバーしていても、第三者はその有効性、実行可能性、または範囲に疑問を提起する可能性があり、これは、そのような特許が縮小され、実行不可能または無効であることを発見する可能性がある。また、それらが挑戦されていなくても、私たちの特許や特許出願は、私たちの知的財産権を十分に保護できず、私たちの候補製品に排他性を提供したり、他の人が私たちの声明を中心に設計することを阻止したりすることができない可能性がある。これらの結果のいずれも、第三者の競争を阻止する能力を弱める可能性があり、第三者競争は私たちの業務に悪影響を及ぼす可能性がある。
私たちは、私たちの候補製品の様々な側面に関連して、独立して、または私たちの許可者または協力者と共に複数の特許出願を提出した。私たちは、どのような特許が発行されるか(あれば)、任意のそのような特許の広さ、または発行された任意の特許が無効であることが発見されるか、強制的に実行できないか、または第三者の脅威にさらされるかどうかを保証することはできない。これらの特許または特許発行後に私たちが所有または許可してくれた任意の他の特許への成功反対は、私たちが開発する可能性のある任意の候補製品が商業化に成功するために必要な権利を奪う可能性がある。また、規制承認に遅延が生じた場合、特許保護の下で候補製品を販売する期間が短縮される可能性があります。
私たちが私たちの規制努力と知的財産権から私たちの候補製品に対する排他性の効果的な保護を得ることができなくても、特許保護、データ独占性、または薬物排他性を含む、私たちの候補製品は排他的に保護されると信じています。この排他性は、生物類似製品が米国で承認されることを阻止し、期限は、それが主張する類似性製品が初めて承認された日から12年となる。しかし、2009年の“生物製品価格競争と革新法”、“特許保護·平価医療法”第7章、副題A、公共法律第111-148号、第124 Stat.119条、第7001-02条、2010年3月23日に法律となり、米国法第42編262節(“生物·薬物保護法”)に編纂され、生体模倣薬のための複雑で複雑な特許紛争解決メカニズムが作成され、米国で私たちの候補製品が発売されることを阻止したり、このような製品の発表を大きく遅らせる可能性がある。現在の生体模倣薬訴訟はBPCIAのいくつかの要求を満たしており,BPCIAは作成されている
62
BPCIAのある用語がどのように解釈すべきかの不確実性については,生物製品革新者と生物類似者に不確実性をもたらしている。生物類似申請者に必要なBPCIAメカニズムは、特許侵害訴訟が私たちの活動を混乱させ、費用を増加させ、管理層の注意をそらすというより大きなリスクをもたらす可能性がある。もし私たちの候補製品の生物類似バージョンがアメリカで承認されれば、私たちの業務に負の影響を与えるかもしれません。
私たちは私たちの候補製品に十分な特許期間保護を提供していないかもしれません。それによって、私たちの業務を効果的に保護します。
特許には限られた期限がある。アメリカでは、特許の法定失効期間は一般的に申請後20年だ。様々な延期がある可能性があるにもかかわらず、特許の有効期限とその提供される保護は限られている。私たちの候補製品をカバーする特許を取得しても、候補製品の特許有効期限が満了すると、私たちは競争に開放的になるかもしれない。また,米国で発表された場合,どの特許期間も出願人または米国特許商標局による特定の遅延に応じて調整することができる。
米国の“ハッジ·ワックスマン法案”および欧州補充保護証明書によると、特許期間の延長は、私たちの候補製品の特許またはデータ独占期間の延長に使用することができる。私たちは特許期間の延長を求めることができるかもしれません。私たちは何の保証も提供することができません。このような特許期限を延長することができます。獲得すれば、どのくらい延長されますか。したがって、私たちは規制機関の承認後の長い間、私たちの候補製品の独占経営権を維持することができないかもしれません(もしあれば)、これは私たちの業務、財務状況、運営結果、見通しにマイナスの影響を与えます。もし私たちが私たちの候補製品を保護するために十分な特許条項や規制排他性がなければ、私たちの業務と運営結果は悪影響を受けるだろう。
米国および外国特許法の変化は、特許の全体的な価値を低下させ、製品を保護する能力を弱める可能性があり、最近の特許改革立法は、私たちの特許出願をめぐる起訴や、私たちが発行した特許の実行または保護の不確実性とコストを増加させる可能性がある。
他のバイオテクノロジー会社と同じように、私たちの成功は特許に大きく依存する。生物技術業界で特許を獲得し、実施することは技術上の複雑性と法律上の複雑性にも関連するため、コストが高く、時間がかかり、内在的な不確定性を持っている。また、米国は最近、広範囲な特許改革立法を公布し、実施している。米国最高裁判所の最近の判決は、特定の場合に入手可能な特許保護範囲を縮小し、特定の場合における特許所有者の権利を弱体化させた。我々の将来の特許取得能力に関する不確実性の増加に加えて,このようなイベントの結合は,いったん特許を取得する価値に関する不確実性をもたらしている.米国議会、連邦裁判所、および米国特許商標局の決定によると、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、それにより、私たちが新しい特許を獲得したり、既存の特許および将来獲得可能な特許を実施する能力を弱める可能性がある。
欧州統一特許(UP)と統一特許裁判所(UPC)が2023年6月1日に発効すると、欧州特許実践は変化すると予想される。新制度は欧州保留出願と付与された欧州特許に影響を与えるであろうが,この変化の長期的な影響には不確実性が存在する。UPCは欧州競争相手の能力に対抗するために特許権を保護し実行することに特別な不確実性をもたらすかもしれない。UPCの実施は,欧州全体で特許法執行により多くの確実性と効率を提供するためであるが,我々の欧州特許を集中的に撤回するための新たなフォーラムも我々の競争相手に提供するであろう.UPCが認める特許権の範囲と提供される特許救済措置の力を知るのに数年かかるだろう。私たちは最初の7年間に私たちの特許をUPCシステムから除外することを選択する権利があるだろうが、そうすることは私たちが新しい統一裁判所の利点を達成することを阻止するかもしれない。
私たちの特許保護を獲得し、維持することは、政府特許機関によって適用される様々なプログラム、書類提出、費用支払い、および他の要求に準拠することに依存し、これらの要件を遵守しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
発行された特許の定期維持費は、特許有効期間内にいくつかの段階で米国特許商標局および外国特許代理機関に支払われなければならない。米国特許商標局および各種外国政府特許機関は、特許出願過程において、いくつかのプログラム、文書、費用支払い、およびその他の同様の規定を遵守することを要求する。多くの場合、適用される規則に基づいて、滞納金を支払うことによって、または他の方法で失効を是正することができるが、場合によっては、規定を遵守しないことは、特許または特許出願の放棄または失効を招き、関連法ドメインの特許権の一部または全部を喪失させる可能性がある。特許または特許出願が放棄または失効される可能性のある規定に適合しないイベントには、規定された期間内に公式行動に応答することができないこと、費用が支払われていないこと、および失敗が含まれる
63
適切に合法化されて正式な書類が提出される。この場合、私たちの競争相手が市場に参入する可能性があり、これは私たちの業務に実質的な悪影響を及ぼすだろう。
もし私たちが私たちの候補製品や未来の候補製品のために有効な独占権を維持できなければ、私たちは私たちが提案した市場で効果的に競争することができないかもしれない。
特許提供の保護に加えて、私たちは、特許を出願できない、または特許を出願しないことを選択した独自の技術を保護するために、商業秘密保護および秘密保護協定に依存する。私たちはまた特許が強制的に施行されにくいプロセスを利用する。さらに、我々の製品の他の要素および私たちの候補製品発見および開発過程の多くの要素は、特許がカバーされていないノウハウ、情報、または技術に関するものである。商業秘密は保護するのが難しいかもしれない。私たちは、従業員、コンサルタント、協力者、コンサルタント、独立請負業者、または他の第三者と秘密保護協定を締結することによって、私たちのノウハウおよびプロセスを保護することを求めています。私たちはまた、私たちの場所と私たちの情報技術システムの物理的および電子的安全を維持することを含む、私たちのデータと商業秘密の完全性とセキュリティを保護するために努力している。私たちはこのような個人、組織、そしてシステムに自信がありますが、合意や安全措置は違反される可能性があり、私たちはどんな違反にも対応する十分な救済措置がないかもしれません。また,競争相手は我々のビジネス秘密を他の方法で取得したり,基本的に同じ情報や技術を独立して開発したりする可能性がある.また、一部の国の法律の専有権に対する保護の程度や方式は米国の法律とは異なる。したがって、私たちはアメリカでも海外でも、私たちの知的財産権を保護して守ることで大きな問題に直面するかもしれない。もし私たちが第三者に私たちの知的財産権を不正に開示することを阻止できない場合、あるいは第三者が私たちの知的財産権を流用することを阻止できなければ、私たちの市場で競争優位性を確立したり、維持することができないかもしれません。これは私たちの業務、経営業績、財務状況に大きな悪影響を及ぼす可能性があります。
私たちのすべての従業員とコンサルタントが彼らの発明を私たちに譲渡することを望んでいますが、私たちのすべての従業員、コンサルタント、協力者、コンサルタント、独立請負業者、および私たちのノウハウ、情報、または技術にアクセスできる第三者は、秘密協定を締結するために、このようなすべての合意が正式に実行されているか、または私たちのビジネス秘密や他の機密ノウハウが漏洩しないこと、または競争相手が他の方法で私たちのビジネス秘密や独立開発と実質的に同じ情報および技術を獲得しないことを保証することはできません。私たちのビジネス秘密を流用または不正に開示することは、私たちの競争的地位を損なう可能性があり、私たちの業務、財務状況、または運営結果に大きな悪影響を及ぼす可能性があります。また,我々のビジネス秘密を守るための手順が不十分であると考えられれば,第三者による商業秘密の流用に対抗する十分な追跡権がない可能性がある.
知的財産権侵害に対する第三者のクレームは、私たちの開発と商業化努力を阻害または延期する可能性がある。
私たちのビジネス成功は、私たちの候補製品を開発、製造、マーケティング、販売し、第三者特許権を侵害することなく、当社の独自技術を使用する能力にある程度依存します。合成バイオテクノロジー分野には,第三者米国と非米国が発行した特許や係属中の出願が多く存在する。第三者が所有している米国や外国特許および出願中の特許は,我々が開発している候補製品と類似した治療用途を有していることが知られている。私たちは現在このような特許と特許出願を監視している。私たちは将来的にアメリカと外国特許庁で訴訟を起こし、これらの特許と特許出願の有効性に疑問を提起するかもしれない。さらに、または代替として、そのような1つまたは複数の特許および特許出願によってカバーされる技術的権利を交渉する許可を求めるかどうかを考慮することができる。もし任意の特許または特許出願が私たちの候補製品や技術をカバーしている場合、私たちは計画通りに私たちの候補製品を自由に製造または販売することができないかもしれません。そのような許可がなければ、私たちは商業的に合理的な条項で許可を得ることができないかもしれません。
私たちはまた関連する第三者特許や出願を決定できない可能性がある。例えば、2000年11月29日までに提出された出願と、その日以降に提出された出願とは、特許発行前に米国国外で提出されない出願は秘密にされている。さらに、特許検索は、特許間の用語の違い、データベースが不完全であり、特許特許請求の意味を評価することが困難であるため、私たちを含む業界参加者が、特許候補および技術に関連する可能性のあるすべての第三者特許権を決定することは困難である。私たちは、関連する特許または特許出願を識別することができないか、または潜在的利益の係属中の特許出願を識別することができないかもしれないが、そのような特許が我々の技術に関連する可能性のある宣言を誤って予測している。さらに、私たちは、現在または未来の候補製品の製造、販売、または使用が発行された1つまたは複数の特許を侵害することを知らないかもしれないし、または第三者特許が無効であるか、強制的に実行されないか、または私たちの活動によって侵害されていないという結論を誤って導出する可能性がある。また,すでに提出された未決特許出願
64
特定の制限に適合する場合、発表された製品は、私たちの技術、私たちの候補製品、または私たちの候補製品の使用をカバーするために、後で修正することができます。
バイオテクノロジーおよび製薬業では、特許侵害訴訟、介入、異議および再審、ライセンス後審査、および米国特許商標局および対応する外国特許庁に提起された同等の訴訟を含む、特許および他の知的財産権に関連する第三者訴訟および他の訴訟が多く存在している。我々が候補製品を開発する分野には,第三者が所有する米国や外国から発行される特許や係属中の特許出願が多く存在する.バイオテクノロジーや製薬業界の拡張や特許の発行に伴い、我々の候補製品が第三者特許権侵害の告発を受けるリスクが高まる可能性がある。
我々にクレームをつけた当事者は、禁止または他の公平な救済を受ける可能性があり、これは、私たちの1つまたは複数の候補製品のさらなる開発と商業化を効果的に阻止することができる。これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、当社の業務における従業員資源を大量に移転することになる。もし私たちに対する侵害クレームが成功した場合、私たちは故意に侵害された3倍の損害賠償と弁護士費、特許権使用料の支払い、私たちの侵害製品の再設計、または第三者から1つまたは複数の許可を得ることを含む大量の損害賠償を支払わなければならないかもしれないし、多くの時間とお金の支出が必要かもしれない。
私たちは私たちの業務に重要な特許と特許出願を提出、起訴、維持、弁護、そして実行するために私たちの許可者にある程度依存している。
私たちは通常、私たちの候補製品に関連する特許出願を完全に起訴する権利を求めて獲得していますが、私たちの候補製品の特許出願は私たちの許可者によってコントロールされることがあります。もし私たちの既存または未来のいかなる許可者も、私たちの任意の候補製品をカバーする特許保護を適切かつ広く起訴し、維持できなければ、私たちがこれらの候補製品を開発し、商業化する能力は不利な影響を受ける可能性があり、私たちは競争相手の競争製品の製造、使用、輸入、販売を阻止できないかもしれない。さらに、私たちが今、第三者から許可された特許や特許出願の特許起訴を制御する権利があっても、私たちは、特許起訴を制御する前の訴訟で私たちの許可者が取った行動または行動しない悪影響や損害を受ける可能性があります。
もし私たちが合意の義務を履行できなかった場合、私たちはこれらの合意に従って第三者から知的財産権や他の権利を許可したり、許可者との業務関係が妨害されたりすると、私たちの業務に非常に重要な許可権を失う可能性があります。
私たちは特定の知的財産権ライセンス協定の締約国であり、将来的により多くのライセンス契約を締結することを望んでいます。私たちの既存の合意はいくつかの義務を規定しており、将来のライセンス契約は、私たちが許可側が許可した技術を使用して私たちの製品を販売する収入にマイルストーンと特許使用料を支払うことを含むこれらの義務を規定する可能性もあり、これらの義務は、私たちが商業化を求める可能性のある任意の製品の全体的な収益力に悪影響を及ぼす可能性があります。さらに、私たちは、私たちの許可プロトコルがカバーする候補製品の臨床開発、販売、およびマーケティングの多くの面でアウトソーシングし、第三者に依存する必要があるだろう。これらの第三者の遅延または失敗は、第三者許可者とのライセンスプロトコルの継続に悪影響を及ぼす可能性がある。もし私たちがこれらの合意の下で私たちの義務を履行できなかった場合、あるいは私たちが破産に直面した場合、これらの合意は許可されるかもしれません。これは私たちの業務に実質的な悪影響を及ぼすかもしれません。
私たちは私たちの特許を保護または強制的に執行するかもしれない特許の訴訟に巻き込まれるかもしれないが、これは高価で時間がかかり、成功しないかもしれない。
競争相手は私たちの特許や私たちの許可側の特許を侵害するかもしれない。このような侵害または許可されていない使用を停止するために、私たちまたは私たちの許可パートナーのうちの1つは、高価で、時間がかかり、予測不可能な特許を強制的に実行するために、第三者に特許侵害クレームを提出することを要求される可能性がある。さらに、私たちに対する侵害訴訟または宣言的判決訴訟では、裁判所は、私たちの1つまたは複数の特許が無効または強制執行できないと判断するか、または私たちの特許が関連技術をカバーしないことを理由に、他方の関連技術の使用を阻止することを拒否することができる。任意の訴訟または弁護手続きの不利な結果は、私たちの1つまたは複数の特許を無効が宣言されるか、強制的に実行できない、または狭い解釈される危険に直面させる可能性があり、私たちの特許出願を発表できないリスクに直面させる可能性がある。これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、当社の業務における従業員資源を大量に移転することになる。
もし私たちまたは私たちの許可パートナーのうちの1つが第三者に対して法的訴訟を提起して、私たちの候補製品のうちの1つをカバーする特許を強制的に執行する場合、被告は私たちの候補製品をカバーする特許の無効および/または強制執行ができないと反訴することができる。米国の特許訴訟では、被告が特許の無効および/または強制執行不可能と主張する反訴が一般的であり、第三者は様々な理由に基づいて特許が無効または強制的に実行できないと断言することができる。有効性を疑問視する理由は、新規性の欠如、明確性の欠如、書面記述、明瞭性、または実施できないことを含む、いくつかの法定要求のいずれかを満たしていないと言われているからであるかもしれない。主張を実行できない理由は、特許訴訟に関係する人が米国特許商標局に関連情報を隠したり、誤った声明をしたりすることである可能性がある
65
起訴中です。第三者は、訴訟範囲外であっても、米国や他の管轄区域の行政機関に類似したクレームを提起することができる。このようなメカニズムには、再審査、当事者間の審査、付与後審査、および反対または派生手続きのような外国法ドメインの同等の手続きが含まれる。このような訴訟は、私たちの候補製品をカバーして保護しないように、私たちの特許が撤回または修正される可能性がある。法的に無効と実行不可能と断言された後の結果は予測できない。例えば、私たちの特許の有効性について、私たちは、私たち、私たちの特許弁護士、特許審査員が起訴中に知らなかった無効な以前の技術を確認することができない。もし被告が無効、特許不可、および/または強制執行不可能な法的主張に勝った場合、私たちは私たちの候補製品の少なくとも一部またはすべての特許保護を失うかもしれない。このような特許保護の喪失は私たちの業務に実質的な悪影響を及ぼすかもしれない。
第三者によって引き起こされるか、または我々によって提起されるか、またはUSPTOによって発表される干渉または派生プログラムは、我々の特許または特許出願または我々の許可者の特許または特許出願に関する発明優先権または正しい発明権を決定するために必要である可能性がある。不利な結果は、現在の特許権を失う可能性があり、関連技術の使用を中止したり、勝利の方向から権利を許可しようと試みることを要求することができます。もし勝利者が商業的に合理的な条件で私たちにライセンスを提供しなければ、私たちの業務は損害を受ける可能性があります。私たちの訴訟、派生、または妨害訴訟の弁護は、私たちの利益に不利な決定を招く可能性があり、成功しても、巨額のコストを招き、私たちの経営陣や他の従業員の注意を分散させる可能性があります。また,臨床試験に必要な資金を調達できない可能性があり,我々の研究計画を継続できず,第三者から必要な技術許可を得ることができず,候補製品の市場進出に役立つ開発パートナー関係も達成できない可能性がある。
さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。聴聞、動議、または他の一時的手続き、または事態発展の結果を公開することもできる。どんな機密情報の漏洩も私たちの業務に悪影響を及ぼす可能性がある。もし証券アナリストや投資家がこれらの結果がマイナスだと思っていれば、私たちの普通株の価格に大きな悪影響を及ぼす可能性がある。
私たちは私たちの特許と他の知的財産権発明権のクレームに疑問を受けるかもしれない。
将来的には、私たちは、元従業員、コンサルタント、協力者、コンサルタント、独立請負業者または他の第三者が、発明者または共同発明者として、私たちの特許または他の知的財産権において権益を有するクレーム、または私たちの特許または知的財産権(私たちが許可した特許および知的財産権を含む)の所有権に疑問を提起する他のクレームを受ける可能性がある。したがって、これらの特許に対する私たちの権利は独占的ではない可能性があり、競争相手を含む第三者は、私たちの業務に重要な知的財産権を得ることができるかもしれない。さらに、私たちが許可または譲渡を受けていない共通の所有者は、最終的にその特許家族から発行された特許の在庫または所有権をめぐって請求される可能性があり、これは、既存の許可協定に従って発行された特許を得ることができない可能性がある。また、米国以外の管轄区域では、知的財産権のすべての人が許可に同意または同意しない限り、許可は強制的に実行できない可能性がある。また,我々の候補製品開発に参加するコンサルタントや他の人の義務衝突により在庫紛争が生じる可能性がある.訴訟はこのような他の挑戦と私たちの特許リストの疑いに対抗するために必要かもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭損害賠償を支払うことに加えて、貴重な知的財産権、例えば貴重な知的財産権の独占所有権や使用権を失う可能性がある。このような結果は私たちの業務に実質的な悪影響を及ぼすかもしれない。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。
私たちは、私たちの従業員、コンサルタント、協力者、コンサルタント、独立請負業者、または他の第三者が第三者の機密情報を誤って使用または開示するか、または私たちの従業員がその元の雇用主のいわゆる商業機密を誤って使用または開示するという疑惑の影響を受けるかもしれない。
私たちは第三者から機密と固有の情報を受け取った。また、私たちは、以前大学、学術研究機関、他のバイオテクノロジーや製薬会社に雇われていた個人を、私たちの競争相手や潜在的な競争相手を含めて雇用しています。私たちは書面協定を締結し、私たちの従業員、コンサルタント、協力者、コンサルタント、独立請負業者、または他の第三者が私たちのために働く時に他人の独自の情報や知的財産権を使用しないことを保証するために最善を尽くしていますが、私たちは将来、私たちの従業員、コンサルタント、協力者、コンサルタント、独立請負業者、または他の第三者がこれらの第三者の機密情報を意図的にまたは意図的に使用したり、漏洩したりするというクレームを受ける可能性があります。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭損害賠償の支払いに加えて、貴重な知的財産権や人員を失う可能性があり、これは私たちの業務に悪影響を及ぼす可能性がある。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。
66
私たちは世界各地で私たちの知的財産権を保護できないかもしれない。
私たちのアメリカ以外の知的財産権は限られている。世界のすべての国で候補製品の出願、起訴、特許保護の費用は目を引くほど高く、しかもアメリカ以外のいくつかの国の知的財産権は異なる範囲と実力を持っている可能性があり、アメリカほど広くない。また、いくつかの外国の法律は知的財産権の保護の程度はアメリカの連邦や州法律に及ばない。したがって、我々は、米国以外のすべての国/地域で第三者(競争相手を含む)が私たちの発明を実施したり、米国または他の司法管轄区域で私たちの発明を使用して製造された製品を販売または輸入することを阻止することができないかもしれない。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの技術を使って自分の製品を開発することができ、また、他の侵害製品を私たちが特許保護を持っているが執行権がアメリカに及ばない地域に輸出することもできる。これらの製品は私たちの製品と競争するかもしれませんが、私たちの特許や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。
多くの会社は外国の管轄区域の知的財産権の保護と保護に重大な問題に直面している。一部の国の法律制度、特にいくつかの発展途上国の法律制度は、特許、商業秘密、および他の知的財産権保護の強制執行、特に生物製薬製品に関する保護を支持しておらず、これは、私たちの特許や他の知的財産権の侵害や流用、または私たちの専有権を侵害する方法で競争製品を販売することを阻止したり、流用したりすることを困難にする可能性がある。外国司法管轄区域で私たちの特許や他の知的財産権の訴訟を強制的に執行することは、成功するかどうかにかかわらず、巨額のコストを招き、私たちの努力と関心を私たちの業務の他の側面に移す可能性がある。さらに、このような訴訟は、私たちの特許を無効と宣言され、強制執行できない、または狭義の解釈できないリスクに直面させ、私たちの特許出願を発行できないリスクに直面させ、第三者が私たちに侵害または流用クレームを引き起こす可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。したがって、私たちが世界各地で私たちの知的財産権を実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネスメリットを得るのに十分ではないかもしれません。
もし私たちの商標と商号が十分に保護されていなければ、私たちは興味のある市場で知名度を作ることができないかもしれません。私たちの業務は不利な影響を受けるかもしれません。
私たちは私たちの現在のブランドに関連するいくつかの商標のために商標登録を申請した。もし私たちの商標と商号が十分に保護されていなければ、私たちは興味のある市場で知名度を作ることができないかもしれません。私たちの業務は不利な影響を受けるかもしれません。我々の未登録商標または商号は、挑戦、侵害、回避、または汎用商標として発表されるか、または他の商標を侵害していると判断される可能性がある。私たちはこれらの商標や商品名に対する私たちの権利を保護できないかもしれません。私たちは私たちが関心のある市場の潜在的なパートナーや顧客の中で知名度を確立するために必要です。時々、競争相手は私たちと似たような商品名や商標を採用して、ブランド表示を確立する能力を阻害し、市場の混乱を招く可能性があります。さらに、他の登録商標または商標の所有者は、我々の未登録商標または商号の変異体を含む商号または商標侵害クレームを提起することができる。長期的には、私たちが商標や商号を成功的に登録し、私たちの商標や商号に基づいて名称を確立することができなければ、効果的に競争できない可能性があり、私たちの業務は悪影響を受ける可能性があります。商標、商業秘密、ドメイン名、著作権または他の知的財産権に関連する専有権を実行または保護する努力は無効である可能性があり、大量のコストおよび資源移転を招き、私たちの財務状況または運営結果に悪影響を及ぼす可能性があります。
私たちの第三者への依存に関するリスクは
著者らは、製品調合、研究、臨床前および臨床研究のいくつかの態様を行う第三者に依存し、引き続き依存することが予想されるが、これらの第三者の表現は、締め切り前にこのような処方、研究またはテストを完了できなかったことを含む満足できないかもしれない。
著者らはずっと独立臨床研究者、契約実験室と第三者CROを含む第三者に依存し、計画し、適用された法規要求に基づいて著者らの臨床前研究と臨床試験、及びある製品候補の発見と開発活動を行い、そして著者らが行っている臨床前と臨床プロジェクトのためにデータを監視と管理する。私たちはこれらの方に依存して、私たちの臨床前研究と臨床試験を実行し、そして彼らの活動のいくつかの方面だけを制御する。しかし、私たちは私たちのすべての研究と実験が適用された議定書、法律と法規の要求、そして科学的基準に従って行われることを確実にする責任があり、私たちはこれらの第三者への依存が私たちの規制責任を免除しないだろう。私たちと私たちの第三者請負業者およびCROは、臨床開発中に私たちのすべての製品に対して適用されるFDA、EMAおよび他の類似した外国規制機関が臨床開発中に私たちのすべての製品に対して実行する法規およびガイドラインである、適用された良好な実験室規範(GLP)および良好な臨床規範(GCP)要件を遵守しなければならない。監督管理機関は定期的な検査を通じてGLP研究を行う実験室、臨床試験スポンサー、主要な研究者、CROと試験地点を通じてこれらのGLPとGCPを実行し、同時にGCPコンプライアンスを審査する。もし、私たち、私たちの研究者、または私たちの任意のCROまたは契約実験室が適用されるGLPおよびGCPを遵守できなかった場合(場合によっては)、私たちの臨床前研究および臨床試験で生成されたデータは信頼できないと考えられる可能性があり、FDA、EMAまたは他の同様の外国の規制機関は、私たちの候補治療製品のマーケティング申請を承認する前に追加の臨床前研究または臨床試験を行うことを要求するかもしれない。特定の規制機関が検査を行った後、この監督機関は
67
われわれの任意の臨床前研究又は臨床試験が適用されるGLP又はGCP法規に適合することを確認する。また,われわれの臨床試験は適用されたcGMP法規に適合した製品を用いて行わなければならない。私たちがこれらの規定を守らないには、私たちが臨床前研究や臨床試験を繰り返す必要があるかもしれません。これは規制承認過程を延期します。
また、これらの実験室、研究者、CROは私たちの従業員ではなく、契約を除いて、私たちの候補製品と臨床試験に投入される資源の数を時間を含めて制御することができません。独立した実験室、調査者、またはCROが私たちの候補製品を開発するのに十分な資源を投入できなかった場合、または彼らの表現が基準に達していない場合、私たちが開発した任意の候補製品の承認および商業化の将来性を延期または影響する可能性がある。また、第三者サービス提供者を使用して、これらの側に独自の情報を開示することを要求することは、これらの情報が流用されるリスクを増加させる可能性がある。
我々の業務目標を実現するために必要な専門知識を専門的にあるいは持っている第三者サービスプロバイダの数は限られている.もし私たちがこれらの第三者実験室、CROまたは臨床研究者との任意の関係が終了した場合、私たちは他の実験室、CROまたは研究者と手配を達成できないかもしれない、あるいはタイムリーまたは商業的に合理的な条項でそうすることができないかもしれない。もし実験室、CRO或いは臨床研究者がその契約の職責或いは義務を成功裏に履行できなかった場合、或いは予想された期限内に完成できなかった場合、もし彼らが交換する必要がある場合、或いは彼らが獲得した臨床データの品質或いは正確性が私たちの臨床前或いは臨床規程、法規の要求或いはその他の原因を遵守できなかったために影響を受け、私たちの臨床前研究或いは臨床試験は延長、延期或いは終了される可能性があり、私たちは監督部門の私たちの候補製品の承認或いは商業化に成功することができないかもしれない。したがって、私たちの運営結果と私たちの候補製品のビジネス見通しが損なわれ、私たちのコストが増加する可能性があり、私たちの収入を創出する能力が延期される可能性がある。
より多くの研究室またはCRO(または調査者)を交換または増加させることは、管理時間および重点を必要とする追加のコストに関連する。さらに、新しい研究室やCROが作業を開始すると、自然な過渡期もある。したがって,遅延が生じ,期待される臨床開発スケジュールを満たす能力に実質的な影響を与える可能性がある。私たちは私たちと契約した実験室とCROの関係を慎重に処理しているにもかかわらず、私たちが未来に似たような挑戦や遅延に遭遇しない保証はなく、これらの遅延や挑戦が私たちの業務、財務状況、運営結果に重大な悪影響を与えないことを保証することはできない。
また、臨床研究者は時々私たちの科学顧問や顧問を務めることができ、このようなサービスに関連する現金補償を得ることができる。これらの関係と任意の関連する賠償が知覚的または実際の利益の衝突をもたらす場合、またはFDAは、財務関係が臨床前研究または臨床試験の解釈に影響を及ぼす可能性があると結論した場合、適用される臨床前研究または臨床試験場所で生成されたデータの完全性が問われる可能性があり、臨床前研究または臨床試験自体の効用が脅かされる可能性があり、これはFDAの遅延または拒否を招く可能性がある。そのような遅延または拒否のいずれも、私たちの臨床段階候補製品またはそれが開発される可能性のある任意の未来の治療製品候補製品を商業化することを阻止することができる。
私たちの後期臨床活動は第三者供給と製造パートナーの薬品供給に依存し、私たちの候補製品の任意の商業供給に対して同様のやり方をとる可能性がある。
著者らは第三者供給と製造パートナーに依存して材料とコンポーネントを提供し、末期臨床試験薬物供給を生産する。私たちはまだどんな商業規模の候補製品も作っていないし、私たちの候補製品のためにそうすることもできないかもしれない。私たちは私たちの製造プロセスの開発と最適化に努力して、このプロセスが安全、有効、有効な治療法を産生することを保証することができません。あるいは私たちの商業需要の数を満たすことができません。
例えば,我々はAzzurクリーンルームと合意し,その施設のクリーンルーム空間を必要に応じてレンタルし,我々の臨床試験のためにcGMP材料を製造·調製するために,Azzurに費用を支払い,クリーンルーム空間を改修·アップグレードし,用途を拡大することにも同意した。もしAzzurが私たちの合意を守らなかったり、彼らが私たちにサービスを提供してくれた重大な不利な事件に遭遇しなければ、私たちの臨床操作を行う能力は影響を受けるだろう。
私たちの研究開発、臨床前と臨床開発薬物と他の材料の供給が制限されないこと、中断され、いくつかの地理的区域或いは品質が満足できるか、或いは受け入れ可能な価格で供給を継続することを保証することはできない。特に、私たちが招聘する可能性のあるどの製品調合メーカーの任意の交換も、合格した交換数が限られている可能性があるので、多くの努力と専門知識を必要とする可能性がある。
合成バイオテクノロジーは複雑で製造が難しい。私たちは生産や技術移転の問題に直面し、私たちの開発や商業化スケジュールが遅延したり、他の方法で私たちの業務に悪影響を与えたりする可能性があります。製造過程の問題は、正常過程との微小な偏差であっても、生産量不足、製品欠陥或いは製造失敗を招き、それによってロット故障、在庫不足と製品リコールを招く可能性がある。
68
大多数の生物製品と薬物生産の多くのよく見られる要素はまた、原材料不足、原材料故障、成長媒体故障、設備故障、施設汚染、労働力問題、自然災害、公共サービス中断、テロ、または私たちがコントロールできない神の行為を含む生産中断を引き起こす可能性がある。また、私たちの製造プロセスを操作するために必要な経験豊富な専門家を採用し、維持することに問題がある可能性があり、これは、私たちの生産が遅れたり、適用された法規要件を維持することが困難になる可能性があります。
私たちの製造プロセスや施設のどんな問題も、学術研究機関や他の当事者の魅力の低い協力者になる可能性があり、これは、私たちがより多くの魅力的な開発計画を得ることを制限し、私たちの臨床開発やマーケティング計画を遅延させ、私たちの業務を損なう可能性があります。
候補製品の製造過程はFDAと外国規制機関の審査を受けなければならない。サプライヤーとメーカーは、適用される製造要件を満たし、cGMP法規のような規制基準に適合するために、監督機関の要求された厳格な施設とプロセス検証テストを受けなければならない。私たちの任意のサプライヤーまたは製造業者は、このような要求を遵守できないかもしれないし、品質、時間、または他の側面で私たちの義務を履行できない場合、または私たちの部品または他の材料の供給が他の理由で制限または中断される可能性がある。この場合、私たちは、現在能力や資源がない材料を自分で製造したり、その施設で第三者と協力して製造したり、他の第三者と合意したりすることを選択または強要することができますが、合意が全くなければ、合理的な条件でそうすることはできないかもしれません。場合によっては、私たちの候補製品を製造するために必要な技術的スキルまたは技術は、元の製造業者固有または独自である可能性があり、困難に遭遇する可能性があり、またはそのようなスキルまたは技術を他方に譲渡することを禁止する契約制限が存在する可能性があり、実行可能な代替案が存在しない可能性がある。これらの要素は、私たちのこれらの製造業者への依存を増加させ、あるいは他の第三者が私たちの候補製品を生産するために、これらのメーカーからライセンスを取得することを要求するだろう。もし私たちが任意の理由でメーカーの交換を要求された場合、私たちは新しいメーカーの施設とプログラムが品質基準とすべての適用された法規とガイドラインに適合しているかどうかを検証することを要求され、類似の安全性、純度、および効力を確保するために、過渡的な研究または臨床試験を繰り返すことが要求されるかもしれない。新メーカーの検証に関する遅延は、タイムリーまたは予算内で候補製品を開発する能力に悪影響を及ぼす可能性があります。
もし私たちが規制部門の任意の候補製品の承認を受けたら、私たちは第三者メーカーに依存するかもしれない。私たちが第三者とすでにまたは将来製造手配を達成した範囲内で、私たちはこれらの第三者に依存して、品質管理と保証に関する要求を含む契約と監督管理の要求に適合する方法でその義務を適時に履行する。もし私たちが候補製品の第三者製造を獲得したり維持したり、商業的に合理的な条項でそうすることができなければ、私たちは候補製品の開発と商業化に成功できないかもしれない。もし私たちまたは第三者が私たちの製造要求を実行できなかった場合、様々な態様で私たちの業務に悪影響を及ぼす可能性があります
69
私たちは正常な業務過程で各種契約を締結し、契約の他方を賠償します。もし私たちがこれらの賠償条項に基づいて義務を履行しなければならなければ、私たちの業務、財務状況、運営結果に実質的な悪影響を及ぼすかもしれない。
正常な業務過程で、私たちは定期的に学術、商業、サービス、協力、許可、相談、その他の賠償条項を含む協定を締結します。私たちの学術および他の研究協定については、私たちが許可を得た合意に基づいて製造、使用、販売または実行された製品、プロセスまたはサービスに関するクレーム、および私たちまたは私たちの分が許可された人が合意下の権利を行使することによって生じるクレームを、機関および関係者が補償することが一般的である。私たちの協力協定については、私たちの協力者が製品の生産、使用、または消費によって引き起こされる可能性のある任意の第三者製品責任クレーム、および告発された第三者が任意の特許または他の知的財産権を侵害しているというクレームを補償します。諮問協定については、コンサルタントがそのサービスを誠実に履行したことによるクレームを賠償します。
もし私たちが賠償条項の下での義務が適用された保険範囲を超えた場合、あるいは私たちが保険を拒否された場合、私たちの業務、財務状況、経営結果は悪影響を受ける可能性があります。同様に、私たちが協力者に依存して私たちを賠償し、協力者が保険加入を拒否されたり、賠償義務が適用された保険範囲を超えて、協力者が私たちを賠償する他の資産がなければ、私たちの業務、財務状況、そして経営結果は悪影響を受ける可能性があります。
私たちが協力計画や戦略連合に到達できる限り、私たちはこれらの協力と連合に関するリスクに直面するかもしれない。
バイオテクノロジー会社は協力手配や戦略連盟に依存して候補製品の開発と商業化を達成することがある。もし私たちが協力計画や戦略連合に参加することを選択すれば、これらの手配は私たちの候補製品の開発が私たちの統制されていないようにし、重要な権利を放棄することを要求するか、または他の私たちに不利な条項を要求するかもしれない。
協力計画や戦略連合に依存することは、以下のリスクを含むいくつかのリスクに直面させるだろう
私たちは将来私たちの候補製品と協力しようと試みるかもしれませんが、私たちはそうできないかもしれません。これは開発と商業化計画を変えることにつながるかもしれません。
私たちは、私たちの計画やプラットフォームについて第三者と戦略的協力を確立し、合弁企業を作成したり、許可合意を達成したりすることを試みるかもしれません。これらの計画またはプラットフォームは、私たちの既存の業務を補完または強化すると考えられます。適切な戦略的パートナーを探す上で、私たちは激しい競争に直面する可能性があり、適切な条項を確保する交渉過程は時間がかかり複雑である。私たちは私たちが受け入れられる条項を任意の候補製品と計画のためにこのような戦略的協力関係を作ることができないかもしれない。これは、私たちの候補製品や計画が協力努力の開発初期段階にあると考えられる可能性があり、私たちの研究開発ルートは不足とみなされる可能性があり、競争や知的財産権の構造は激しすぎたりリスクとみなされたりする可能性があり、マクロ経済条件は協力に不利である可能性があり、および/または第三者は、十分な安全性および有効性を有する可能性があることを含む、私たちの候補製品および計画が十分な商業化の潜在力を持っていると思わない可能性があるからかもしれない。
70
適切なパートナーを探し、私たちの候補製品の開発および/または商業化の合意を達成する上でのいかなる遅延も、私たちの候補製品の開発または商業化を遅らせる可能性があり、市場に進出してもそれらの競争力を低下させる可能性がある。戦略的パートナーがいなければ、私たちは自費で開発および/または商業化活動を行う必要があるだろう。もし私たちが自ら援助して開発および/または商業化活動に従事することを選択すれば、私たちはより多くの専門知識と追加的な資本を得る必要があるかもしれないが、これらは私たちが受け入れられない条件や根本的には得られないかもしれない。もし私たちがそれができなければ、私たちは私たちの候補製品を開発したり、それらを市場に出すことができないかもしれません。私たちの業務は実質的な悪影響を受けるかもしれません。
私たちの候補製品の商業化に関するリスクは
もし私たちの候補製品がマーケティングおよび商業化のために承認された場合、私たちは自ら販売、マーケティング、流通能力を開発することができないか、または受け入れ可能な条項でこれらの機能を履行するために第三者と合意できなければ、このような未来の製品を商業化することに成功することはできないだろう。
私たちは現在販売、マーケティング、流通能力や経験を持っていない。もし私たちの候補製品がマーケティングおよび商業化を許可された場合、私たちはこのような製品を商業化するために内部販売、マーケティング、および流通能力を発展させる必要があり、これは高価で時間がかかる、または第三者と協力してこれらのサービスを提供するだろう。もし私たちが私たちの製品を直接マーケティングすることを決定すれば、私たちは大量の財務と管理資源を投入して、技術専門長と流通、管理とコンプライアンスをサポートする能力を持つマーケティングと販売チームを発展させる必要があります。もし私たちがこのような能力を持つ第三者に依存して私たちの製品をマーケティングしたり、協力者と共同で製品を普及させることを決定すれば、私たちは第三者とマーケティングおよび流通手配を確立し、維持する必要があり、私たちが受け入れ可能な条項や根本的にこのような計画を達成することができる保証はありません。第三者のマーケティングまたは流通スケジュールを達成する際に、私たちが得た任意の収入は、第三者の努力に依存し、第三者が十分な販売および流通能力を確立するか、または任意の承認された製品に対する市場の受け入れに成功することを保証することはできない。もし私たちが将来マーケティングや商業化を承認された製品を商業化することに成功しなければ、私たち自身も第三者を通じても、私たちの業務、財務状況、運営結果、見通しは不利な影響を受ける可能性がある。
もし私たちの候補製品の市場機会が私たちが思っているより小さいなら、私たちは私たちの収入予想を達成できないかもしれません。候補製品が承認されたと仮定して、私たちの業務は影響を受ける可能性があります。私たちの候補製品市場での患者数は少ないかもしれないので、私たちは患者の識別に成功し、かなりの市場シェアを獲得して、利益と成長を実現することができなければならない。
私たちの目標疾患を有する患者数が少ないことを考慮すると、私たちの合格患者数と価格設定は、私たちの候補製品が対象とする実際の市場とは大きく異なるかもしれない。適応疾患に罹患している人の数と、私たちの候補製品治療から利益を得る可能性があるこれらの疾患患者のサブセットの予測は、私たちの信念と推定に基づいている。これらの推定は科学文献、患者基礎或いは市場研究を含む様々な源から来ており、不正確であることが証明されている可能性がある。また、新しい研究はこれらの疾病の推定発病率或いは流行率を変える可能性がある。患者数は予想より少ないかもしれない。私たちの各候補製品の潜在的にアドレス指定可能な患者集団は限られている可能性があるか、または私たちの候補製品の治療を受け入れられない可能性があり、新しい患者はますます識別または接触が困難になる可能性があり、これは私たちの業務、財務状況、運営結果、および将来性に悪影響を及ぼすだろう。
私たちは激しい競争に直面しており、私たちの競争相手は私たちよりも早く、あるいは製品の発見、開発、商業化に成功しているかもしれない。
新製品の開発と商業化競争は激しい。私たちは世界各地の主要な製薬会社、専門製薬会社、バイオテクノロジー会社、大学、その他の研究機関からの競争に直面しており、将来開発や商業化を求める可能性のある候補製品に関連している。例えば,BioMarin,Inc.,NestléHealth Science S.A.(Codexis,Inc.,Homology Medicines,Inc.,American gene Technologies International Inc.,Generation Bio Co.,Agios PharmPharmticals,Inc.,SOM Biotech SL,Jnana Treeuticsおよび他の発見段階の会社はPKUを治療するための候補製品を開発または開発している.Novome BioTechnologies,Inc.,Federation Bio,Inc.,Oxdien PharmPharmticals L.C.と他社が腸由来高草酸尿症の候補品を開発している。Aeglea BioTreeuticsとTravere TreateuticsはいずれもHCUのための候補製品を開発している。SELECTA生物科学社,江蘇恒瑞医薬有限会社,地平線製薬会社,Dyve生物科学社,矢印製薬会社,血液せん断治療会社は痛風治療の候補薬を開発している。私たちの競争相手は、私たちが現在開発しているまたは開発可能な候補製品よりも効率的またはコストの低い技術および製品の開発、取得、または許可に成功するかもしれません。これは、私たちの候補製品を時代遅れにし、競争力を持たないかもしれません。競争に加えて、私たちの候補製品に照準を合わせたい疾患に対する代替療法に直面しており、Precigen,Inc.のような工学菌を専門に開発している会社もいくつか知られています。また、他にも似たような製品を開発している会社もいくつかあります。第三者支払者は、政府や民間保険会社を含め、模造品の使用を奨励することも可能である。
71
もし私たちの競争相手が私たちよりも早くFDAや同様の外国規制機関のその候補製品の上場承認を得たら、私たちの競争相手が市場に入る前に強力な市場地位を確立することができるかもしれません。
私たちと比べて、私たちの多くの競争相手はより高い知名度とより多くの財務、製造、マーケティング、研究と薬物開発資源を持っている。バイオテクノロジーと製薬産業のより多くの合併と買収は、私たちの競争相手により多くの資源を集中させるかもしれない。大手製薬会社は特に臨床前と臨床試験及び薬品監督管理の許可を得る方面で広範な専門知識を持っている。また、学術機関、政府機関、その他の研究を行う公的·民間組織は、潜在的な競争力を有する製品や技術について特許保護を求めることができる。このような組織はまた私たちの競争相手と独占的な協力や許可関係を作ることができる。もし私たちの候補製品が既存の治療方案や将来開発されている新製品と効果的に競争できなければ、私たちの業務、財務状況、運営結果、および将来性を損なうだろう。
私たちの現在または未来の任意の候補製品の商業成功は医師、患者、第三者支払人、および医学界の他の人の市場に対する受け入れの程度に依存するだろう。
FDAや同様の外国規制機関の承認を得ても、私たちの製品の商業成功は、医療提供者、患者、第三者支払者が私たちの候補製品を受け入れてくれるかどうかにある程度依存し、医療上有用で、費用効果が高く、安全である。私たちが市場に発売したどの製品も医師、患者、第三者支払人の市場で受け入れられないかもしれません。私たちのどの製品に対する市場の受け入れ度は多くの要素に依存しますが、これらに限定されません
1つの製品が承認された後に良好な治療効果と安全性を示しても、市場のこの製品に対する受け入れ程度はまだ確定していない。教育医療界や第三者支払者が製品のメリットを知る努力には大量の投資や資源が必要かもしれないし,決して成功しないかもしれない。もし私たちの製品が医師、患者、第三者支払者、および他の医療保健提供者の十分な受容度を得ることができなければ、利益を達成したり維持したりするのに十分な収入を生み出すことができないだろう。
私たちは他の候補製品を決定、許可、発見、開発、または商業化するためのどんな努力でも成功しないかもしれない。
私たちの多くの仕事は既存の候補製品の臨床試験、潜在的な承認、商業化に集中するが、私たちの業務の成功はまた、他の候補製品を識別、許可、発見、開発、または商業化する能力にある程度依存すると予想される。新製品候補製品を決定する研究計画には大量の技術、財政、人的資源が必要である。私たちは最終的に成功しないことが証明された潜在的な計画や製品候補に私たちの努力と資源を集中させるかもしれない。私たちの研究計画や許可は、臨床開発と商業化のための候補製品をもっと作ることができないかもしれません
72
上記のいずれかの事件が発生した場合、私たちは、1つ以上の候補製品のための開発作業を放棄することを余儀なくされる可能性があり、または、他の候補製品を識別、許可、発見、開発、または商業化することができない可能性があり、これは、私たちの業務、財務状況、または運営結果に大きな悪影響を与え、運営を停止させる可能性があります。
製品のために十分な精算や保険範囲を得ることができない場合、これらの製品をマーケティングする能力を制限し、収入を創出する能力を低下させる可能性があります。
私たちが承認した製品の定価、保証範囲、精算(ある場合)は、私たちのビジネス努力や他の開発計画を支援するのに十分でなければならず、第三者支払者(政府および個人保険会社を含む)の保証および精算の可用性および十分性は、多くの患者が高価な治療を負担できるようにするために重要である。私たちが承認した製品(ある場合)の販売は、私たちが承認した製品のコスト(ある場合)がどの程度健康維持、管理医療、薬局福祉および同様の医療管理組織または政府支払人および個人支払者によってどの程度支払われるかに大きく依存する。保険や精算を受けることができない場合、あるいは限られた金額だけでは、無料で補助金を提供したり、製品を商業化することに成功しないかもしれません。
また,新たに承認された製品の保険カバー範囲や精算に関する不確実性が大きい。アメリカでは、新薬の保証と精算に関する主な決定は通常医療保険と医療補助サービスセンター(CMS)によって行われ、CMSはHHS内の一つの機関であり、CMSは新薬がMedicareの下で保険と精算を受けるかどうか、及びどの程度精算するかを決定する。個人支払者はCMSが確立した保険精算政策に大きく従う傾向がある。CMSが私たちのような新製品候補者の精算がどのような決定を下すのか、承認されれば、私たちの製品候補者はどのような精算コードを受け取るかもしれないと予測することは難しい。米国では、第三者支払者は薬品の保証と精算に対して統一的な政策を持っていない。そのため、薬品の保証範囲と精算範囲は支払人によって異なる。したがって、保証範囲の決定プロセスは、通常、時間がかかり、高価なプロセスであり、保証範囲および十分な補償が一致するか、または最初に得られることを保証することなく、各支払人にそれぞれ私たちの製品を使用する科学的および臨床的支援を提供する必要がある。また、精算に関する規則や条例は常に変化しており、場合によっては短時間で通知されており、これらの規則や条例が変わる可能性があると考えられる。
第三者支払者は、医薬品およびサービスの課金に挑戦するようになっており、多くの第三者支払者は、同等の模倣薬/生物類似薬またはより安い治療法がある場合に、特定の薬物への保険および精算を拒否する可能性がある。第三者支払者は,我々の候補製品や他の治療法が代替可能であると考え,患者に安い製品のみを精算することを提案する可能性がある。私たちの候補製品が他の利用可能かつ比較可能な製品よりも良い治療効果またはより良い投与利便性を示しても、既存の薬剤の価格は、私たちがその候補製品に対して受け取る費用を制限するかもしれない。これらの支払者は,ある薬品の精算状態を拒否または撤回したり,新製品や既存の上場製品の価格を低すぎるレベルに設定したりして,我々の製品開発投資から適切なリターンを実現できない可能性がある.保険や精算が得られない場合、あるいは限られたレベルに限られていれば、候補製品の商業化に成功できない可能性があり、私たちが開発する可能性のある製品から満足できる財務リターンを得ることができない可能性もあります。
医師の監督下で管理されている製品については、このような薬物が高い価格に関連することが多いため、保険および適切な補償を得ることは特に困難である可能性がある。また,製品自体やその製品を使用した治療やプログラムは単独では精算できない可能性があり,医師の使用に影響を与える可能性がある。例えば、これらの場合、医師は、彼らがどれだけ処方されるか、またはどのような場合に私たちの製品を発行または管理するかを制限することができ、患者はそのような製品を購入することを送達するかもしれない。逆に、これは私たちが製品を商業化することに成功する能力に影響を与え、私たちの収益性、運営結果、財務状況、未来の成功に影響を与えるかもしれない。
73
米国以外では、国際業務は一般的に広範な政府価格制御と他の価格制限法規の制約を受けており、ヨーロッパ、カナダ、その他の国のコスト制御措置に対する日々の重視はすでに重視され、製品の定価と使用に圧力を与え続けると考えられる。多くの国では、国家衛生システムの一部として、製品価格は異なる価格制御メカニズムの制約を受けている。他の国は会社が自ら医療製品を価格設定することを許可しているが、会社の利益を監督してコントロールしている。価格制御や定価規制の他の変化は、私たちが製品に受け取ることができる費用を制限するかもしれません(もしあれば)。したがって、アメリカ以外の市場では、私たちの製品を販売する潜在的な収入は、商業的に合理的な収入と利益を生み出すのに十分ではないかもしれない。
また、米国や海外の政府や個人支払者が医療コストの制限や低減に力を入れていることは、新製品の保険範囲や精算レベルが制限されている可能性があるため、私たちの製品に十分な支払いや十分な支払いを提供できない可能性がある。健康維持組織の影響力の増大や,追加的な立法変化を含む管理型医療の傾向が大きくなっているため,製品に関する価格設定圧力に直面することが予想される。全体的には,医療コスト,特に処方薬の下り圧力が増加し続けることが予想される。したがって、私たちの製品が規制部門の承認を得ても、その収益性はもっと難しいかもしれない。
私たちの業務運営や従業員に関するリスク
私たちは高級管理者と肝心な科学者を誘致し、維持することができず、私たちの候補製品或いは任意の未来の候補製品の開発に成功することを阻害し、私たちの臨床試験を行い、そして任意の製品を商業化することができるかもしれない。
私たちの成功部分は私たちが引き続き高い素質の管理、臨床と科学者を吸引、維持と激励する能力にかかっている。私たちの将来の成功は、私たちの上級管理職、特に私たちの総裁とCEO、そして私たちのベテラン科学者と上級管理チームの他のメンバーの貢献に高く依存していると信じています。これらの人のいずれもサービスを失っても、私たちの製品ラインの成功開発、私たちが計画した臨床試験の完成、あるいは私たちが開発した製品の商業化を遅延または阻止する可能性があります。
私たちは合格従業員を誘致し、維持する上で歴史的に重大な困難に直面したことがないにもかかわらず、私たちは将来このような問題に直面するかもしれない。例えば,バイオテクノロジーや製薬分野では,我々の業界に必要なスキルや経験を持つ個人数が限られているため,適格人材に対する競争は非常に激しい。私たちが臨床開発と商業活動を拡大するにつれて、私たちはもっと多くの人員を募集する必要があるだろう。私たちは受け入れ可能な条件で良質な人材を吸引し、維持することができず、甚だしきに至っては人材を誘致と維持することができないかもしれない。
私たちの従業員、独立請負業者、主要な調査者、CRO、コンサルタントと協力者は監督基準と要求を遵守しないこと、およびインサイダー取引を含む不当な行為またはその他の不適切な活動に従事する可能性がある。
私たちは従業員、独立請負業者、コンサルタント、および協力者が詐欺行為や他の不法活動に従事する可能性があるリスクに直面している。このような当事者の不正行為には、意図的、無謀、および/または不注意な行為、または以下の規定に違反する不正活動が含まれる可能性がある:(1)私たちは、真、完全かつ正確な情報を報告することを要求する法律、(2)法規および基準を製造すること、(3)詐欺および乱用および反腐敗法律、または(4)真実、正確な財務情報およびデータを報告することを要求する法律を含む、我々の候補製品に関連する活動を行っている司法管轄区域の規制機関の法規。特に,医療業界の販売,マーケティング,商業手配は,詐欺,偏見,不正行為,リベート,自己取引,その他の濫用行為を防止するための広範な法律法規によって制約されており,これらの法律は国/地域によって大きく異なる可能性がある。これらの法律法規は、広範な価格設定、割引、マーケティングおよび販売促進、販売手数料、顧客インセンティブ計画、および他のビジネス計画を制限または禁止する可能性があります。これらの活動には、臨床試験の過程で得られた情報の不適切な使用も含まれており、これは規制制裁と私たちの名声に深刻な損害を与える可能性がある。従業員および他の第三者の不正行為を常に識別し、阻止することができるわけではなく、このような活動を発見し、防止するための予防措置は、未知または未管理のリスクまたは損失を効果的に制御することができないか、またはそのような法律や法規を遵守できないことによる政府調査または他の行動または訴訟から私たちを保護することができない可能性がある。もし私たちにこのような訴訟を起こした場合、私たちは自分たちの権利を弁護したり、維持することに成功しませんでした。これらの行動は、重大な民事、刑事および行政処罰、損害賠償、罰金、特定の国/地域から排除される可能性のある補助医療計画の参加、契約損害、名声損害、利益および将来の収益の減少、および私たちの業務削減を含む大きな影響を与える可能性があり、これらはいずれも私たちの業務運営能力と私たちの運営結果に悪影響を及ぼす可能性があります。
74
私たちの普通株に関するリスクは
私たちの主要株主と経営陣は私たちのかなりの割合の株を持っていて、株主が承認した事項に大きな統制を加えることができます。
2023年3月22日現在の我々普通株の実益所有権によると、私たちの役員と取締役は、5%以上の発行済み普通株の所有者とそのそれぞれの関連会社とともに、実益は私たちの普通株の約47.3%を持っています。したがって、これらの株主は、取締役の選挙、合併、または当社のほとんどの資産または任意の他の重大な会社取引を含む、株主の承認を必要とする会社の行動の結果に大きな影響を与える。これらの株主の利益はあなたの利益とは違うかもしれないし、あなたの利益と衝突する可能性もある。例えば、これらの株主は、制御権の変更を延期または阻止することができ、このような制御権変更が私たちの他の株主に利益をもたらすとしても、私たちの株主が会社や私たちの資産を売却する際に普通株のプレミアムを得る機会を奪う可能性があり、私たちの普通株の現行の市場価格に影響を与える可能性がある。投資家は利益衝突が存在または発生する可能性があると考えているため、株式所有権の著しい集中は私たちの普通株の取引価格に悪影響を及ぼす可能性がある。
将来私たちの普通株を売却したり、転換可能または私たちの普通株に交換可能な証券は私たちの株価を押し下げるかもしれません。
もし私たちの既存の株主または私たちのオプションの保有者が公開市場で私たちの普通株を大量に売却するか、または私たちの普通株を大量に売却する意向を示した場合、私たちの普通株の取引価格は低下する可能性がある。このような売却が起こりうるという市場の見方は、私たちの普通株の取引価格を低下させる可能性もある。2023年3月22日まで、私たちは67,728,025株の普通株流通株があります。
私たちの四半期の経営業績は大幅に変動する可能性があり、あるいは投資家や証券アナリストの予想を下回る可能性があり、すべての状況は私たちの株価の変動や下落を招く可能性がある。
私たちの経営業績は四半期変動の影響を受けると予想されています。私たちの純損失と他の経営業績は様々な要素の影響を受けます
もし私たちの四半期の経営業績が投資家や証券アナリストの予想を下回れば、私たちの普通株の価格は大幅に低下する可能性がある。また、私たちの経営業績のどの四半期の変動も、会社の株価を大幅に変動させる可能性があります。私たちの財務業績を四半期比較することは必ずしも意味があるわけではなく、私たちの将来の業績としての指標に依存すべきではないと考えられます。
私たちの定款書類やデラウェア州法律の条項は、買収が私たちの株主に有利であっても、経営陣を変更することを困難にする可能性があります。
私たちが改訂して再記載した会社登録証明書、および私たちの改正および再記載された定款の条項は、株主が有利と考えるかもしれない合併、買収、または他の支配権の変化を阻止、延期、または阻止する可能性があり、私たちの株主がその株式から割増取引を得る可能性があることを含む。また、これらの規定は、取締役会の交換や更迭をより困難にし、株主が現在の経営陣を交代または更迭しようとしていることを挫折または阻止する可能性がある。これらの規定には
75
また、デラウェア州法律では、デラウェア州上場企業が利害関係のある株主と商業合併を禁止しており、当該商業合併が所定の方法で承認されない限り、当該商業合併が所定の方法で承認されない限り、当該株主は、通常、その関連会社と共に当該会社の15%以上の投票権を有する株を所有しているか、または過去3年以内に当該会社の投票権を有する株を所有している者である。したがって、デラウェア州の法律は会社の統制権の変更を阻止、延期、または阻止する可能性がある。
さらに、私たちが改正して再記載した会社登録証明書は、法的に許容される最大範囲内で、デラウェア州衡平裁判所が、私たちを代表して提起された任意の派生訴訟または訴訟、受託責任違反を主張するいかなる訴訟、デラウェア州会社法またはDGCL、私たちが再記載された会社登録証明書を修正すること、または私たちが改正して再説明した定款に基づいて私たちにクレームを提起した任意の訴訟、または私たちのクレームに対する内部事務原則によって管轄された任意の訴訟であることが規定されている。この排他的裁判所条項は,取引法で規定されている義務や責任を実行するための訴訟には適用されない.しかしながら、証券法第22条に規定されているので、専属裁判所条項に列挙された1つまたは複数のカテゴリの訴訟に適用され、証券法第22条に規定されているので、連邦裁判所および州裁判所は、証券法またはその規則および条例で規定されている任意の義務または責任を執行するために提起されたすべての訴訟に対して同時管轄権を有する。裁判所が証券法下のクレームに対してこのような条項を強制的に執行するかどうかには不確実性があり、我々の株主は連邦証券法とその規則や条例の遵守を放棄したとはみなされない。
このような裁判所条項の選択は、株主が司法裁判所において、私たちまたは私たちの任意の取締役、上級管理者、または他の従業員と紛争することに有利であると考えられるクレームを提出する能力を制限する可能性があり、これは、このようなクレームに関連する訴訟を阻害する可能性がある。代替的に、裁判所が、私たちが再記載した会社登録証明書に含まれる裁判所条項の選択が訴訟において適用されないか、または実行できないことを発見した場合、私たちは、他の司法管轄区域でこのような訴訟を解決することに関連する追加費用を生じる可能性があり、これは、私たちの業務、運営結果、および財務状態を損なう可能性がある。
私たちの規約の条項とデラウェア州法律の他の条項は投資家が将来私たちの普通株に支払う価格を制限するかもしれません。
私たちは予測可能な未来に私たちの普通株に現金配当金を支払わないと予想している;したがって、私たちの普通株の資本増加はあなたが予測可能な未来に唯一の収益源になるだろう。
私たちは普通株の現金配当金を発表したり支払ったりしたことがない。私たちは予測可能な未来に私たちの普通株にどんな現金配当金も支払わないと予想している。私たちは現在、すべての利用可能な資金と未来のどんな収益も残して、私たちの運営に資金を提供するつもりだ。さらに、任意の将来の債務融資スケジュールの条項には、私たちの普通株が発表または支払い可能な配当金の金額を禁止または制限する条項が含まれている可能性がある。したがって、私たちの普通株の資本付加価値は(もしあれば)あなたが予測可能な未来に唯一の収益源になるだろう。
証券や業界アナリストが我々の業務に関する研究を発表しない場合、あるいは不正確または不利な研究を発表しなければ、私たちの株価や取引量は低下する可能性がある。
私たちの普通株の取引市場は、証券や業界アナリストが発表した私たちまたは私たちの業務に関する研究と報告にある程度依存するだろう。私たちの1人以上のアナリストを追跡して私たちの普通株格付けを引き下げたり、私たちの業務に関する不正確または不利な研究報告を発表したりすれば、私たちの株価は下落するかもしれない。また、私たちの経営業績がアナリストの予測に達しなければ、私たちの株価は下落する可能性がある。これらのアナリストのうちの1人以上が私たちへの報告を停止したり、私たちに関する報告書を定期的に発表できなかったりすれば、私たちの普通株に対する需要が減少する可能性があり、これは私たちの株価と取引量を低下させる可能性がある。
会計規則や規制の変化や解釈は、不利な会計費用を招いたり、私たちの報酬政策を変更することを要求したりする可能性があります。
バイオ製薬会社の会計方法と政策は、収入確認、研究開発と関連費用及び株式給与会計を含む政策であり、米国証券取引委員会を含む関連会計当局のさらなる審査、解釈、指導を受ける。会計方法または政策の変更または説明は、本定期報告に含まれる財務諸表を含む、当社の財務諸表を再分類、再陳述、または他の方法で変更または修正する必要がある場合があります。
76
もし私たちが引き続きナスダック世界市場の持続的な上場要求を守らなければ、私たちの普通株は買収される可能性があり、私たちの普通株価格と私たちの資本市場に入る能力はマイナスの影響を受けるかもしれない。
私たちの普通株は現在ナスダック世界市場(“ナスダック”)で看板取引されている。私たちはナスダックの持続的な上場の要求を満たさなければなりません。その中には、私たちの普通株の最低入札が1株当たり1.00ドルであることを含めて、そうでなければ退市リスクに直面し、これは私たちの業務に実質的な悪影響を与えます。私たちの普通株がナスダックから撤退すると、私たちの普通株の流動性を大幅に低下させ、私たちの普通株価格はそれに応じて大幅に低下する可能性があります。また、退市は、私たちが受け入れられる条項や全く受け入れられない条項で融資源を代替することで資金を調達する能力を損なう可能性があり、投資家、サプライヤー、顧客、従業員の潜在的な自信喪失、および業務発展機会の減少を招く可能性がある。
2022年12月6日、私たちはナスダックの書面通知を受けて、私たちの普通株の30営業日連続の終値に基づいて、私たちの普通株はナスダック上場規則5450(A)(1)に要求された1株1.00ドルの最低購入価格に達していないことを通知して、180個のカレンダー日の自動猶予期間を開始して、コンプライアンスを再獲得しましょう。
ナスダック上場規則5810(C)(3)(A)によると、最低入札要求の遵守を達成するために、通知の日から、または2023年6月5日まで180暦の期限がある。2023年6月5日までのいつでも、普通株の入札価格が10営業日連続で1株1.00ドル以上になった場合、最低入札価格要求を再遵守します。
私たちは2023年6月5日までにナスダック資本市場に移転し、クレームを回復するために2番目の180日の期限を申請することを選択することができる。もし私たちが割り当てられたコンプライアンス期間内にナスダックが承認する可能性のあるいかなる延期も含めてコンプライアンスを再獲得しなければ、ナスダックは私たちの普通株が取得されるという通知を出すだろう。その時、私たちは退市について公聴会グループに控訴することを決定するかもしれない。
我々は、我々の普通株の入札価格レベルを引き続き監視し、潜在的な逆株式分割を含む180暦のコンプライアンス期間内にコンプライアンスを実現するために、適切な代替案を考慮する予定である。しかし、私たちがこれをすることができるという保証はない。
項目1 B。未解決従業員コメント。
ない。
プロジェクト2.ニュースオペラです。
私たちの会社の本社と運営機関はマサチューセッツ州ケンブリッジ市にあります。私たちは今ケンブリッジのビンニー街三零一号で実験室とオフィススペースを借りています。私たちのビンニー街301号のレンタル契約は2028年に満期になります。私たちは私たちの施設が予測可能な未来に私たちの必要性を満たすのに適していると信じている。
項目3.法律法律手続き。
私たちの日常業務活動では、私たちは時々様々な法的訴訟、クレーム、行政訴訟の影響を受ける。訴訟やクレームの結果は正確には予測できないが、本報告日まで、私たちはいかなるクレーム、訴訟または訴訟の当事者であるとは信じておらず、これらのクレーム、訴訟または訴訟の結果が私たちに不利であれば、その結果が個別または全体が私たちの業務に重大な悪影響を及ぼすことを予想する理由がある。結果にかかわらず,弁護や和解コスト,管理資源分流などにより,訴訟は我々に悪影響を与える可能性がある。
プロジェクト4.地雷安全安全に開示する。
適用されません。
77
パー?パーT II
項目5.登録者普通株·関連株の市場保有者は重要であり,発行者は株式証券を購入する.
市場情報
私たちの普通株は2017年8月28日からナスダック資本市場で看板取引され、取引コードはSYBXであり、これまでの取引コードはMIRNである。
株主.株主
私たちの普通株は2023年3月22日までに約105名の登録株主がいます。
配当をする
設立以来、私たちは株主に何の配当金も発表したり、支払ったりしたことはなく、予測可能な未来に、私たちは現金配当金を発表したり支払うつもりもない。私たちは現在、私たちの業務運営と拡張のために、任意の未来の収益を維持すると予想している。
未登録株式証券販売と収益の使用
ない。
発行人が株式証券を購入する
次の表には、2022年12月31日までの四半期における普通株の買い戻しに関する情報を示しています
発行人が株式証券を購入する
期間 |
|
購入した株式総数(%1) |
|
1株平均支払価格(2) |
|
公開発表された計画または計画の一部として購入した株式総数 |
|
計画や計画によってはまだ購入していないかもしれない株の約ドルの価値 |
2022年11月1日から2022年11月30日まで |
|
4,196,889 |
|
$0.60 |
|
__ |
|
__ |
2022年12月1日から2022年12月31日まで |
|
__ |
|
__ |
|
__ |
|
__ |
第六項です[レイザーRVed]
78
項目7.経営陣の以下の問題の議論と分析財務状況と経営実績。
前向き情報
本10−K年度報告第I部第1 A項におけるリスク要因、本10−K年度報告におけるその他の部分の監査財務諸表及び付記、及び本経営層の財務状況及び経営成果の検討及び分析を併せて読まなければならない。歴史情報に加えて、本議論及び分析には、証券法第27 A条及び取引法第21 E条に示される前向き陳述が含まれている。経営業績は必ずしも財政年度全体や将来の任意の他の時期に起こりうる結果を代表するとは限らない。
概要
Synlogicは臨床末期生物技術会社であり、合成生物学に基づく生物療法の設計と開発に取り組んでいる。合成生物学は工学原理を利用して生物システム、遺伝回路と分子コンポーネントの設計への応用。著者らは合成生物学の力を医学に応用し、精密工学と合理的な薬物開発を結合し、合成生物技術を発展させる。合成生物技術は1種の活性プロバイオティクスであり、遺伝子工学を経て、胃腸(GI)において特定の機能を発揮し、有効な生物を標的とする。合成バイオテクノロジーは経口的であり、よく知られているプロバイオティクスをその送達方法またはシャーシとしているため、潜在的で便利で安全な治療方法を提供する。
私たちの候補薬物パイプラインは
Synlogicが準備中のプロジェクトはSYNB 1934であり,これはフェニルアセトン尿症(PKU)における主導的なプロジェクトであり,この概念の臨床証拠が証明されており,2023年上半期にその鍵となる3期研究であるSynheny−3に入る予定である。我々の製品ラインにはホモシステイン尿症(HCU),腸源性高草酸尿,痛風の治療のための新しい候補薬も含まれている。Synlogic社は羅氏社と研究協力を行っており、重点的に炎症性腸疾患(IBD)を治療するための新しい合成生物製剤を発見し、イチョウ生物工学会社(Ginkgo Bioworks,Inc.)と他の未開示の臨床前資産について協力し、Synlogic社の合成生物技術方法とイチョウ会社のコードバンクと鋳造サービスを結合することである。

代謝性疾患
代謝性疾患は,我々の身体が重要な代謝物や分子の欠陥や変化を分解あるいは産生することによる疾患である。これらの疾患を有する患者では、一部の酵素の欠乏は、代謝物の腸管および全身系への蓄積をもたらす。PKUとHCU患者において、これらの代謝物は有毒レベルに蓄積し、不可逆的な神経機能障害が出現する可能性があることを含む深刻な健康結果を招く。
79
北大
概要
私たちの主要な北京大学プロジェクトSYNB 1934は経口的、非系統的に吸収する候補薬物で、プロバイオティクスに基づく遺伝子工学形式です大腸菌.大腸菌ニスラーです。SYNB 1934はLAADとPAL酵素を産生してアミノ酸フェニルアラニン(Phe)を代謝することを目的とし、フェニルアラニンはPKU或いは高フェニルプロピオン酸血症(Phe代謝中にあまり深刻ではない遺伝性欠陥)患者において神経毒性レベルに達することができる。治療効果と安全性の原因により、現在PKUに対する治療選択は限られており、大多数は依然として治療が必要であり、多くの治療を受ける人は更にPHEを下げる必要がある。今までの研究結果支持はPKUに有効、安全、便利と柔軟な治療方案の潜在力を提供する。同一候補薬物の前世代SYNB 1618はFDAの孤児薬物とFast Trackの称号を獲得し、EMAの孤児称号に対する肯定的な評価を獲得した。2022年10月,SYNB 1618とSYNB 1934の第2段階Synheny-1研究における積極的な結果を発表し,SYNB 1934がSynheny-3に入る候補となることを確認し,鍵となる第3段階研究であり,2023年上半期に開始予定である.2023年1月、FDAはSYNB 1934によるPKUのまれな小児科疾患の治療を許可することを発表した。
SYNB 1618早期液体製剤の1/2 a段階臨床試験を完了し,2018年9月にこの研究で評価された健康ボランティアのトップデータを発表した。2019年7月、私たちが発表したデータは、SYNB 1618が安全かつ耐性が良好であり、健康ボランティアとPKU患者において菌株活性の機序証明を実現したことを示している。液体製剤を用いて検討した後,SYNB 1618の固体経口凍結乾燥製剤を開発した。私たちはすでに健康ボランティアのバイパス研究でこの経口製剤を評価した。このようなより患者と商業化に適したSYNB 1618の提示形態の研究により、SYNB 1618は液体製剤と比較して活性とより良い耐性を有することが示唆された。
著者らは2020年第3四半期にSYNB 1618の第2段階臨床試験を開始し、Synheny-1研究と呼ばれた。Synheny-1試験はSYNB 1618固体経口製剤の安全性と耐性、及びそれが成年PKU患者の血液中のフェニルアラニンレベルを低下させる可能性を評価することを目的とした。2021年9月に行われているSynheny−1研究の中期分析では,SYNB 1618は血液Pheレベルで統計的に有意な低下を示した。
我々が北京大学に開発した第2の候補製品SYNB 1934の設計目的はSYNB 1618よりも高いPHE低減活性を潜在的に提供することである。2021年7月、著者らはSYNB 1934固体経口製剤の臨床研究を開始し、体内と体外の臨床前研究により、SYNB 1618と比べ、SYNB 1934がフェニルアラニンを代謝する能力は約2倍に向上した。
2021年9月、私たちは第1段階健康ボランティア研究におけるSYNB 1618とSYNB 1934活性比較の結果を発表した。フェニルアラニン代謝バイオマーカーによる比較解析では,SYNB 1934の活性レベルはSYNB 1618の約2倍であった。また,SYNB 1934は同等用量でSYNB 1618を含む他の合成バイオテクノロジーと類似した安全性と耐性を示した。
SYNB 1934の健康ボランティアにおける臨床結果は臨床前データと一致し、しかも以前に展望性バイオマーカー駆動のモデリングを提案した。SYNB 1618と比較してSYNB 1934活性の増加は機会を提供し,個別患者のニーズに応じて臨床概況を最適化することができると信じている。そこで,我々が行っているSynheny-1段階2研究にSYNB 1934を評価するARMを追加した.Synheny-1段階1研究の正面頂線データは2022年10月に公表され、2023年3月に先天性代謝疾患学会(SIMD)年会で完全に公表された。
北大計画全体で得られたデータによると、SYNB 1934はSynheny-3に入ることにしました。これは第3段階の重要な研究であり、2023年上半期に開始する予定です。
ホモシステイン尿症
ホモシステイン尿症(HCU)は稀な遺伝性代謝疾患であり、メチオニンの代謝に影響し、メチオニンは1種のタンパク質であり、多くの食物に存在し、肉類、魚類と乳製品を含む。HCUは遺伝的欠陥によるものであり,この欠陥はシスチンβ−合成酵素(CBS)という酵素の欠失を引き起こす。CBSが存在しない場合、ホモシステインと他の有毒化学物質及びその副産物はメチオニンを含み、血液と尿中に蓄積する。ホモシステインレベルの上昇は多系統障害と関係があり、目の損害(異所性水晶体と/或いは深刻な近視)、骨格系の損害(身長が高すぎ、四肢が長すぎ、脊柱側弯、漏斗胸)、血管系(血栓塞栓症)と中枢神経系(発育遅延と知的障害)を含む。
80
2021年11月、我々は、胃腸内でホモシステインの前駆体メチオニンを消費し、それによってHCU患者のtHcyを低下させるように設計された新規な経口吸収非系生体生物治療薬候補薬剤であるSYNB 1353を指名することを発表した。この方法の基本原理はメチオニンによる食事制限を含み、これはHCUのホモシステインレベルの低下と制御の標準的な看護である。SYNB 1353はイチョウとの連携研究の一部として開発された。私たちはSYNB 1353の世界開発権と商業化の権利を持っている。2022年11月,健常ボランティアに対するホモシステイン尿症食モデルを用いた第1段階研究の積極的な結果から,SYNB 1353がメチオニンを代謝し,胃腸での吸収を阻止することにより血漿メチオニンを低下させることが示唆された機序の証拠が得られたと発表した。これまで、FDAはSYNB 1353快速チャネル、孤児薬物と稀な小児科疾患をHCUの潜在的治療法として指定することを許可した。
腸源性高草酸尿
腸源性高草酸尿は慢性進行性疾患であり、尿シュウ酸(Uox)レベルが高いことが特徴であり、これは公認された腎結石再発の原因であるが、承認された治療方案はない。それは脂肪吸収不良によるものであり、通常は手術或いはクローン病などの疾病による胃腸脂肪吸収不良であり、食事シュウ酸吸収の増加を招き、シュウ酸は多くのよく見られる食物に存在し、緑葉野菜、ナッツとチョコレート、及び尿シュウ酸レベルの上昇を含む。循環中吸収の増加は腎臓中結晶の形成を招き,Uoxレベルが危険であることを反映しており,腎結石の再発や進行性腎障害を引き起こす可能性がある。2020年5月,腸由来高草酸尿症の臨床候補薬SYNB 8802が発表された。2021年,Synlogicは1 b期研究からSYNB 8802の積極的な機序証拠を報告し,食事誘導高シュウ酸尿を有する健康ボランティアの尿シュウ酸と糞草酸レベルの低下を証明した。2022年12月、SYNB 8802が概念検証を得たことを発表し、SYNB 8802を使用した用量はUoxの用量依存性低下に関連し、1 x 10治療コースでは、UoxのUoxはプラセボのベースラインより-28%(-37.2,−18.2)低下した11TID用量は,3 x 10治療コースでプラセボと比較してUoxのa−38%(−46.4,−28.7)が低下した11TID投与量。
痛風
SYNB 2081は1種の合成生物製剤であり、胃腸中の尿酸を消費することを目的とし、目標は全身尿酸レベルを下げることであり、痛風の潜在的な治療方法として、痛風は1種の複雑な形式の炎症性関節炎である。現在の治療方案は安全性と有効性の面で局限性が存在する。SYNB 2081はSynlogicと有力な細胞プログラミングレベルプラットフォームイチョウによる研究協力によって作られた新規、経口、非システム吸収の候補薬物を提案した。痛風は体内の尿酸が関節に結晶を形成しすぎることによるものである。関節の激しい痛み,炎症,発赤,尿酸レベルの高すぎによる関節活動制限などの症状が出現する可能性がある。また、痛風は慢性腎臓疾患で公認されている危険因子である。現在の治療方案の局限性のため、著者らは明らかに新しい治療方法が必要であると考えている。2022年8月、私たちはSYNB 2081を私たちの潜在的な痛風治療の候補薬として発表し、著者らとイチョウの協力を通じて、第二の臨床開発に入った候補製品となった。我々の計画はSYNB 2081を我々の薬物開発パイプラインの次の段階に移行させることであり,現在INDイネーブル研究が行われている。
代謝性と免疫疾患の追加配管計画
合成生物学的方法は特に既知の代謝と免疫疾患において有効な生物経路に関連する疾患標的の解決に適している。IBDは胃腸局部炎症を特徴とする疾患であり、通常T細胞、活性化マクロファージと上皮バリア機能損傷によって駆動される。IBDの発症機序は遺伝的要因にも関与しており,環境因子にも関与しており,腸管微生物と腸管免疫系との相互作用の変化による可能性がある。腸管バリア機能損傷は自己免疫性疾患の発病機序においても核心的な役割を果たしている。単層上皮細胞は腸管内容物を宿主循環系と体内の免疫細胞から分離する。上皮層を破壊することは管腔内外来抗原の病理的暴露を招き、自己免疫性疾患の感受性を増加させる。腸管微生物相と宿主間の相互作用は上皮バリアと動的平衡免疫の維持に重要な役割を果たしていると考えられている。そのため、バリア機能を増強し、胃腸炎症を減少させることは自己免疫性疾患を治療或いは予防する潜在的な治療機序である。私たちの合成生物プラットフォームは効率的にプログラミングすることができます大腸菌.大腸菌Nissleはこれらの機能を実行し,代謝が短鎖脂肪酸などの因子を産生してバリア機能を増強することや,タンパク質を分泌するなどの免疫調節サイトカインを含む。
現在IBDを治療する方法は主に免疫系の調節と炎症を抑制する療法に集中している。これらの治療法には,プレドニゾンなどのステロイドや,HUMIRA(アダリズマブ)などの腫瘍壊死因子阻害剤がある。しかし,これらの方法は感染症や癌に対するより大きな感受性を含む全身免疫抑制に関与している。米国疾病コントロール·予防センターのデータによると、2015年、米国では約300万人の成人がIBDと診断されたと報告されている。2021年6月、我々は羅氏社と研究協力協定を締結し、IBDを治療する新しい合成生物製剤を発見した。また,他の全額保有IBDの臨床前研究を進めていく。
81
免疫疾患では,IBDの治療に取り組んでおり,われわれの経口代謝計画から得られた知識や専門知識を利用して,腸管疾患部位に局所的に作用する活性薬剤の開発を可能にしている。我々の方法は全身投与ではなく炎症部位の局所投与に基づいているため,我々の合成バイオテクノロジーはこの治療カテゴリにおいて魅力的な安全プロファイルを提供する可能性が予想され,現在利用可能な選択と比較して安全性は製品プロファイルの中で特に理想的な属性である。
著者らは一連の合成生物技術を開発してある癌を治療し、これらの癌は腫瘍微小環境を変化させ、免疫システムを活性化し、そして腫瘍の減少を招くことを目的としている。これらの合成バイオテクノロジーは、チェックポイント阻害剤のような他の癌治療法と組み合わせて使用することができる。著者らの合成生物臨床免疫腫瘍学(IO)候補薬物はSYNB 1891であり、これは腫瘍内投与の合成生物薬物であり、二重天然と適応性免疫活性化剤の作用を有するように設計されている。2021年11月10日、著者らはSYNB 1891とPD-L 1チェックポイント阻害剤の併用による末期固形腫瘍或いはリンパ腫患者の1期試験がすでに登録を完了したことを発表した。SYNB 1891はこれ以上の研究計画を持っておらず,代謝や免疫疾患の研究·開発に取り組んでいる。
業務の概要
私たちは現在報告可能な業務部門である合成バイオテクノロジーの発見と開発を経営している。これまで、私たちのほとんどの活動は私たちの候補製品の研究と開発に取り組んできました。これまで、私たちの運営資金は、主に優先株、普通株、優先株、株式承認証、羅氏協力とオプション合意によって受け取った支払い、先の協力、投資で稼いだ利息、合併で受け取った現金から来ています。
これまで、私たちは製品販売から何の収入も得ておらず、設立以来大きな運営損失を出しています。2022年12月31日と2021年12月31日までの年間で、それぞれ約6610万ドルと6060万ドルの純損失が発生した。2022年12月31日と2021年12月31日までの累計赤字はそれぞれ約3.57億ドルと2.909億ドルであり、予測可能な未来には、候補製品の開発に伴い、損失が出ることが予想される。私たちは私たちが行っている活動によって、私たちの費用と資本需要が大幅に増加すると予想している
臨床開発に成功し、単独でも第三者と協力しても、規制機関から候補製品の承認を得ない限り、製品収入は生じないと予想される。また,第三者契約研究組織(CRO)と契約製造組織(CMO)を用いてわれわれの臨床開発·製造活動を展開したいが,ビジネス組織はない。もし私たちの候補製品が規制機関の承認を得たら、製品販売、マーケティング、流通に関連した巨額の費用を支援するために、私たちの内部商業化能力を発展させることが予想される。したがって、私たちは、公共またはプライベートエクイティまたは債務融資、協力または許可証、融資リース取引、または他の利用可能な融資取引によって、私たちの運営に資金を提供することを求めていると予想されます。しかし、私たちは必要な時にこれらや他の方法で追加資金を集めることができないかもしれない。製品開発に関連する多くのリスクや不確実性のため、費用を増加させる時間や金額を予測することもできず、いつ実現または利益を維持できるかを予測することもできない。たとえ私たちが製品収入を作ることができても、私たちは利益を上げることができないかもしれない。
82
新冠肺炎の流行がわれわれの業務に与える影響
新冠肺炎の大流行は引き続き世界各地の経済と商業に影響を与える。この影響の程度や持続時間はまだ不確定であり,特にウイルス変種が伝播し続ける場合には予測も困難である.私たちは私たちの反応を積極的に監視し、管理しており、私たちの経営結果と財務状況、そして私たちの業務の発展に対する実際と潜在的な影響を評価しており、これは以下に述べる発展、傾向、予想にさらに影響を与える可能性がある。新冠肺炎の大流行の影響に関連するリスク要素を見て、“伝染病の大流行、流行或いは爆発、例えば新冠肺炎、あるいはロシアとウクライナの間の武力衝突のような地政学的緊張情勢は、私たちの業務と私たちの財務業績に実質的な悪影響を及ぼす可能性がある”は、本年度報告10-K表第I部第1 A項の“リスク要因”に記載されている。
インフレの影響
私たちはインフレが私たちの業務や経営業績に本報告で述べた間に実質的な影響を及ぼすとは思わない。しかし、インフレはすでに合格者を誘致と維持するために著者らが発生した労働力コスト、臨床試験を行うコスト、その他の運営コストに影響を与え続ける可能性がある。インフレコストは私たちの業務、財務状況、そして運営結果に悪影響を及ぼすかもしれない。しかも、インフレが悪化し、金利に影響を与え続ける可能性がある。金利上昇は私たちの借入金利と私たちが任意の潜在的な追加資金を得る能力や条件に悪影響を及ぼすかもしれない。
財務概要
収入.収入
2022年12月31日までの年間収入は羅氏協力とオプション協定から来ている。付記10を参照連携プロトコル: 羅氏協力この計画について全面的に検討するために、本年度報告書の他の表格10-Kの連結財務諸表の付記を参照してください。
研究開発費
研究·開発費用には,我々の候補製品の発見·開発に関する費用が含まれており,臨床前と臨床研究および製品開発を行い,これらの費用は発生時に計上されている。これらの費用は主に
新薬が規制の承認を得るための長い過程を保障するには多くの資源が必要だ。どのような遅延や規制部門の承認を得られなかった場合も、私たちの候補製品開発作業と私たちの全体業務に重大な悪影響を及ぼすだろう。医薬製品開発の内在的不確実性を考慮して、私たちはどの程度の確実性推定で規制部門の許可を得て、私たちの候補製品の可能性、時間或いはコストをマーケティングすることができないので、もしあれば、私たちの候補製品はいつ収入とキャッシュフローを産生するだろうか。
私たちの候補製品の開発成功は高度に不確実であり、多くのリスクの影響を受けている。タイトル下のリスク要因をご覧ください私たちの候補製品開発に関するリスク第2部第1 A項を参照し、本年度報告表格10−Kの他の部分を参照。
私たちが私たちのパイプに投資する時、私たちの開発計画の各後続段階に資金を提供する約束は、明確な支持データを受け取るかどうかにかかっている。各候補製品の科学的および臨床データ、および競争構造と候補製品の商業潜在力の継続的な評価に基づいて、どのような追加計画を継続し、各計画にどれだけの資金を継続的に提供するかを決定すると予想される。私たちは予測可能な未来に、私たちの研究開発コストが高くなると予想する。プロジェクトの臨床試験の進展や新プロジェクトによるINDや開発への進展に伴い,われわれの候補薬物開発に関するコストが増加することが予想される。
83
著者らは直接研究開発費用を追跡し、主に外部コスト、例えば契約研究機関と臨床前と臨床薬物製品の製造に関連するコスト、及びその他のアウトソーシング研究開発費用を含み、特定の製品計画に用いられる。候補製品を選択する際には、特定の候補製品に関するコストを追跡する。これらのコストは、研究開発下の複数の候補製品計画に配置されているため、個別に分類されるため、従業員および相談に関連するコスト、私たちのプラットフォームおよび施設費用に関連するコスト(減価償却または他の間接コストを含む)を特定の候補製品計画に分配しない。以下の表は,我々の期間中の研究開発費(千単位)をコスト別にまとめたものである
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
SYNB 1934 |
|
$ |
4,380 |
|
|
$ |
3,189 |
|
SYNB 1618 |
|
|
3,436 |
|
|
|
3,577 |
|
SYNB 8802 |
|
|
4,197 |
|
|
|
6,499 |
|
SYNB 1353 |
|
|
2,259 |
|
|
|
— |
|
SYNB 1891 |
|
|
1,223 |
|
|
|
2,541 |
|
外部開発前候補者費用と未分配 |
|
|
7,129 |
|
|
|
4,526 |
|
内部研究開発費 |
|
|
29,420 |
|
|
|
26,795 |
|
合計する |
|
$ |
52,044 |
|
|
$ |
47,127 |
|
一般と行政費用
一般と行政費用は主に行政、財務、法律、情報技術、投資家関係、業務発展と人的資源機能者の給与、福祉、その他従業員に関連する費用を含む。その他の一般及び行政費用には、知的財産権のための特許保護を求める法律費用、施設及び情報科学技術インフラ費用、役員及び上級者保険、及び会計、税務、法律及びコンサルティングサービスの専門費用が含まれる。
私たちが支援する候補製品の持続的な研究開発と潜在的な商業化に伴い、私たちの一般的かつ管理費用は将来的に増加すると予想される。また、第1候補製品が規制部門の承認を得る可能性があると考えられる場合には、ビジネス運営に備えているため、特に候補製品の販売やマーケティングに関する準備作業により、賃金や関連費用が増加することが予想される。
その他の収入(費用)
利息と投資収入には投資から得られた収入が含まれている。利息支出には私たちの融資リースに関する費用が含まれています。その他の費用には主に外貨領収書の損益が含まれている。
重要な会計政策と試算
私たちの財務状況と経営結果の討論と分析はアメリカ公認会計原則(GAAP)に従って作成した総合財務諸表に基づいている。これらの財務諸表を作成する際には、報告期間内に報告された資産および負債額、報告書の収入および費用、および関連開示に影響を与えるいくつかの推定および仮定を作成する必要がある。このような推定と仮説は,収入確認や研究開発費に関する推定や仮定を含み,我々が事実や状況の変化を監査·分析するが,このような推定は将来的に重大な変化が発生する可能性がある。このような重要な見積もりや仮定は,我々の歴史的経験,我々の業界傾向の観察,および当時の状況に属すると考えられる様々な合理的な要素に基づいており,資産や負債の帳簿価値を判断する基礎を構成しているが,このような資産や負債の帳簿価値は他の出所から容易に見られるものではない。異なる仮定や条件では、実際の結果は私たちの推定とは異なる可能性がある。
以下の会計政策の適用は、我々が報告した財務結果を全面的に理解し評価するために最も重要であり、各政策は管理職が重大な判断と推定を行う必要があると考えられる。私たちの重要な会計政策は付記2により包括的に説明されています重要会計政策の概要私たちの連結財務諸表は、本年度報告の他の場所のForm 10-Kに現れます。
84
収入確認
FASB ASCトピック808に関する連携プロトコルを評価します協力手配手配の性質と契約条項および我々の業務運営の性質を考慮して、取引の分類を決定します。私たちがこの活動の積極的な参加者であり、協力のビジネス成功によって重大なリスクとリターンに直面している場合、吾らは総合財務諸表に私たちの取引を毛数で記録し、総合財務諸表付記に協力手配下の権利と義務を説明する。
ASC 606によれば、エンティティは、そのクライアントが約束された商品またはサービスの制御権を取得したときに収入を確認し、その金額は、エンティティがこれらの商品またはサービスと交換するために予期される対価格を反映する。エンティティがASC 606の範囲内のスケジュールされた収入確認を決定するために、エンティティは、(I)顧客との契約を決定するステップと、(Ii)契約における履行義務を決定するステップと、(Iii)取引価格を決定するステップと、(Iv)契約内の履行義務に取引価格を割り当てるステップと、(V)エンティティが契約義務を履行するときに収入を確認するステップと、の5つの分析を実行する。顧客に転送された商品やサービスと交換するために、私たちが獲得する権利のある価格を受け取る可能性がある場合にのみ、5段階分析を契約に適用します。契約開始時に、契約がASC 606の範囲内にあると決定されると、各契約において約束された商品またはサービスを評価し、どれが契約義務であるかを決定し、各約束された商品またはサービスが異なるかどうかを評価する。そして,履行義務を履行する際にそれぞれの履行義務に割り当てられた取引価格の金額が収入であることを確認した.
私たちは研究開発サービス協力協定を締結することができ、この合意に基づいて、私たちの候補製品のいくつかの権利を第三者に権限を与えることができます。これらの取り決めの条項は、一般に、返還できない前払い許可料、特定の費用の精算、顧客選択権使用料、開発、規制、および商業マイルストーン支払い、および製品の純売上を許可する印税のうちの1つまたは複数を私たちに支払うことを含む。可変対価格は重大な逆転リスクが存在しないとみなされるまで制限される。
我々がパートナーでもある顧客の各合意の義務を履行する際に確認すべき適切な収入金額を決定する際には、(I)契約で約束された商品またはサービスを決定するステップと、(Ii)契約背景で異なるかどうかを含む約束された商品またはサービスが履行義務であるかどうかを決定するステップと、(Iii)可変対価格の制限を含む取引価格の測定と、(Iv)取引価格を履行義務に割り当てるステップと、(V)(または)各履行義務を履行するときに収入を確認するステップとを実行する。これらの手配に対する会計計算の一部として、a)上記(Ii)ステップに基づいて決定された履行義務の数、b)上記(Iii)ステップでの取引価格、およびc)上記(V)ステップでの契約義務の契約期限および履行方式を決定するために、重大な判断を使用しなければならない。以下に述べるように、特許権使用料以外のマイルストーンまたは他の可変対価格が取引価格に含まれるべきかどうかを決定するために重大な判断を使用する。取引価格は私たちが提供する予定の商品とサービスに割り当てられます。見積数を用いて履行義務を履行する時間を決定し,その中には履行義務を履行するための測定基準としてフルタイム等値時間を用いることが含まれている可能性がある.
収入確認基準を満たす前に受け取った金額は私どもの総合貸借対照表で繰延収入として入金されます。貸借対照表の後日12カ月以内に収入と確認された金額は当期繰延収入に分類されると予想される。貸借対照表後12カ月以内に収入が確認されなかった金額は繰延収入に分類され、当期分を差し引く。
知的財産権許可証
約束や義務の履行が他の約束と異なるかどうかを評価する際には、顧客の研究、製造、商業化能力、関連専門知識の一般市場での可用性などを考慮する。さらに、顧客が残りの承諾を受けずにコミットメントから利益を得ることができるかどうか、コミットメントの価値が未履行のコミットメントに依存するかどうか、残りのコミットメントを提供することができる他のプロバイダがいるかどうか、残りのコミットメントから分離して識別できるかどうかを考える。他の承諾と組み合わせたライセンスについては、合併履行義務が時間の経過とともに履行されるか、ある時点で履行されるかを決定するために、合併履行義務の性質を判断するために利用され、時間の経過とともに収入を確認するための進展を測定するための適切な方法が決定される。私たちは報告期間ごとに進捗指標を評価し、必要に応じて業績指標と関連収入確認を調整する。
85
研究と開発サービス
もし一つの手配が私たちが研究開発サービスを提供する約束や義務を含んでいると判断された場合、私たちはこれらのサービスがその計画内の他の約束とは異なるかどうかを確認しなければならない。これらのサービスが他の承諾と異なるかどうかを評価する際には,クライアントがこれらの同じサービスを実行する能力を考慮する.さらに、顧客が残りの承諾を受けずにコミットメントから利益を得ることができるかどうか、そのコミットメントの価値が未履行コミットメントに依存するかどうか、残りのコミットメントを提供することができる他のサプライヤーがいるかどうか、残りのコミットメントとは別に識別できるかどうかを考える。他の承諾と組み合わせた研究開発サービスについては,合併履行義務が一定期間内かある時点で履行されているかを決定するために,合併履行義務の性質を判断を用いて評価し,時間とともに収入を確認するために進展を測る適切な方法である.契約履行義務を履行するために将来発生する予定のフルタイム相当時間と比較して発生するフルタイム相当時間と比較して、管理職の判断が必要となる入力方法を含む、合併履行義務に関する収入の推定を時間基準で記録する。私たちは報告期間ごとに進捗指標を評価し、必要に応じて業績指標と関連収入確認を調整する。この方法では,業績義務履行に必要な総投入を見積もり,これまでの収入確認にかかる努力を評価しなければならない。このような余剰投入の推定は主観的であり,この研究は新たであるため,成功した努力は貸借対照表日の推定努力とは異なる可能性がある.
顧客オプション
1つのスケジュールが、顧客が追加の商品またはサービスを取得することを可能にする顧客選択権を含むと判定された場合、顧客選択権の基礎となる商品およびサービスは、手配開始時に契約義務とみなされない。我々は、顧客の物質的権利オプション、すなわち追加の商品またはサービスを無料または割引で購入するオプションを評価します。顧客選択権が実質的な権利を表すと決定された場合、実質的な権利は、手配の開始時に別個の履行義務として確認される。貨物またはサービス(I)が契約中の元の貨物およびサービスと類似しており、(Ii)元の契約の条項に従って提供される場合、私たちは別の方法に基づいて取引価格を物質的権利に割り当てる。このような選択の下で,クライアントから受信した対価格総額を,クライアントに提供する予定のすべての商品やサービスに割り当てる.重大な権利に割り当てられた金額は,選択権の行使や義務履行まで収入として確認されない。
一里塚払い
マイルストーン支払いを含む各手配の開始時に、マイルストーンの実現に関連する累積収入が大幅に逆転する可能性があるかどうかを評価し、可能な金額法を用いて取引価格に含まれる金額を推定する。累積収入が大きく逆転しない可能性が高い場合、関連するマイルストーン価値は取引価格に含まれる。規制承認のような我々または被許可者の制御範囲内でのマイルストーン支払いは、これらの承認を受ける前に実現可能であるとは考えられない。他のマイルストーンについては、この評価を行う際に特定のマイルストーンを達成するために、科学、臨床、規制、商業、および他の克服しなければならないリスクなどの要因を評価する。累積収入が大きく逆転しない可能性があるかどうかを決定する際には,かなりの判断が必要である。その後の各報告期間の終了時に、すべての制限されたマイルストーンの実現確率を再評価し、必要に応じて全体の取引価格の推定値を調整します。いずれの調整も累積追跡をもとに記録されており,調整期間中の収入や収益に影響を与える.
印税
販売ベースの特許使用料を含む手配については、販売レベルに基づくマイルストーン支払いを含み、許可が特許使用料に関連する主要項目とみなされる場合には、(I)関連販売が発生した場合、または(Ii)特許使用料の一部または全部が割り当てられた履行義務が履行された(または部分的に履行されている)ときに収入を確認する。今まで、私たちは私たちのいかなる許可手配から発生したいかなる独占権料収入も確認していない。
契約費用
顧客との契約の増分コストを資産として確認し、これらのコストが回収されることが期待できれば。実際の便宜策として、確認すべき資産の償却期間が1年以下であれば、取得契約の増額コストを発生時の費用として確認する。今まで、私たちは顧客との契約のために増加費用を発生させていません。
86
研究開発費
すべての研究と開発費用は発生時に費用を計上する。研究開発費には、研究開発活動を行うことによるコスト、給与、福祉及び他の従業員コスト、権益に基づいて計算される給与支出、実験室及び臨床用品及びその他の直接支出、施設支出、間接支出、臨床前研究、臨床試験、臨床製造及び原材料費用、及びその他の外部支出を含む契約サービス料が含まれる。研究及び開発活動の前払金の払い戻しは、関連サービス中又は貨物及びサービス提供を受けたときに資本化及び支出することができない。第三者サービスプロバイダの請求書条項が私たちの期末と一致しない場合、特定の会計期間中に発生した臨床試験コスト、契約サービスコスト、候補薬物の供給コストを含む第三者への義務を推定し、その期間の終了時に計算すべき費用を記録する必要がある。我々の見積りは,研究開発計画の完了状態と関連する未単コスト見積りに基づいている.
経営成果
以下の議論は、我々の経営陣が我々の総合的な財務結果を理解するために必要な重要な要素をまとめたものである。
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
|
|
(単位:千) |
|
|||||
収入.収入 |
|
$ |
1,180 |
|
|
$ |
1,754 |
|
運営費用: |
|
|
|
|
|
|
||
研究開発 |
|
|
52,044 |
|
|
|
47,127 |
|
一般と行政 |
|
|
16,555 |
|
|
|
15,392 |
|
総運営費 |
|
|
68,599 |
|
|
|
62,519 |
|
運営損失 |
|
|
(67,419 |
) |
|
|
(60,765 |
) |
その他の収入(支出): |
|
|
|
|
|
|
||
利子と投資収入 |
|
|
1,267 |
|
|
|
210 |
|
利子支出 |
|
|
(2 |
) |
|
|
(2 |
) |
その他の費用 |
|
|
7 |
|
|
|
(4 |
) |
その他の収入,純額 |
|
|
1,272 |
|
|
|
204 |
|
純損失 |
|
$ |
(66,147 |
) |
|
$ |
(60,561 |
) |
2022年12月31日までの年度と2021年12月31日現在の年度との比較
収入.収入
|
|
締切り年数 |
|
|
|
|
|
|
|
|||||||
|
|
十二月三十一日 |
|
|
変わる |
|
||||||||||
|
|
2022 |
|
|
2021 |
|
|
$ |
|
|
% |
|
||||
|
|
(単位:千) |
|
|
|
|
|
|
|
|||||||
収入.収入 |
|
$ |
1,180 |
|
|
$ |
1,754 |
|
|
$ |
(574 |
) |
|
|
(33 |
)% |
2022年12月31日現在の年度収入は120万ドルであるが、2021年12月31日現在の年度収入は180万ドルである。この2つの時期の収入は主に2021年6月に達成された羅氏の協力で提供されたサービスと関係がある。
運営費
|
|
締切り年数 |
|
|
|
|
|
|
|
|||||||
|
|
十二月三十一日 |
|
|
変わる |
|
||||||||||
|
|
2022 |
|
|
2021 |
|
|
$ |
|
|
% |
|
||||
|
|
(単位:千) |
|
|
|
|
|
|
|
|||||||
運営費用: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
研究開発 |
|
$ |
52,044 |
|
|
$ |
47,127 |
|
|
$ |
4,917 |
|
|
|
10 |
% |
一般と行政 |
|
|
16,555 |
|
|
|
15,392 |
|
|
|
1,163 |
|
|
|
8 |
% |
総運営費 |
|
$ |
68,599 |
|
|
$ |
62,519 |
|
|
$ |
6,080 |
|
|
|
10 |
% |
87
研究開発費
2022年12月31日までの年間研究開発支出は5200万ドルだったが、2021年12月31日までの年間は4710万ドルだった。490万ドルの増加はSYNB 1353の臨床開発コストが200万ドル増加し,SYNB 1934が30万ドル増加し,製造コストが130万ドル増加し,非臨床コストが80万ドル増加したためである。給与、福祉、その他の従業員に関する支出は130万ドル追加増加し、うち70万ドルは2022年11月にリストラを発表した結果である。また,研究開発支援費は130万ドル増加し,専門サービスは110万ドル増加した。SYNB 8802とSYNB 1891の臨床開発コストはそれぞれ210万ドルと110万ドル減少し、これらの増加を相殺した。
一般と行政費用
2022年12月31日までの1年間は、一般·行政費は1660万ドルだったが、2021年12月31日までの年間は1540万ドルだった。120万ドル増加した主な原因は、給与、福祉、その他の従業員に関する支出が90万ドル増加したことであり、そのうち10万ドルは2022年11月にリストラを発表した結果である。
その他の収入(費用)
|
|
締切り年数 |
|
|
|
|
||||||||||
|
|
十二月三十一日 |
|
|
変わる |
|
||||||||||
|
|
2022 |
|
|
2021 |
|
|
$ |
|
|
% |
|
||||
|
|
(単位:千) |
|
|
|
|
|
|
|
|||||||
その他の収入(支出): |
|
|
|
|
|
|
|
|
|
|
|
|
||||
利子と投資収入 |
|
$ |
1,267 |
|
|
$ |
210 |
|
|
$ |
1,057 |
|
|
|
503 |
% |
利子支出 |
|
|
(2 |
) |
|
|
(2 |
) |
|
|
— |
|
|
|
0 |
% |
その他の費用 |
|
|
7 |
|
|
|
(4 |
) |
|
|
11 |
|
|
|
(275 |
)% |
その他の収入合計,純額 |
|
$ |
1,272 |
|
|
$ |
204 |
|
|
$ |
1,068 |
|
|
|
524 |
% |
2022年12月31日までの1年間、その他の収入は130万ドルだったが、2021年同期は20万ドルだった。他の収入が110万ドル増加したのは、私たちの現金、現金等価物、短期投資残高の収入が増加したためであり、これは主に高い金利と関係がある。
流動性と資本資源
私たちは2014年3月14日の成立以来赤字を続けており、2022年12月31日現在、累計3.57億ドルの赤字を計上しています。これまで、私たちは主に優先株、普通株、優先株、引受権証を売却し、協力協定に基づいて受け取った支払いは、銀杏との技術協力、羅氏協力とオプション協定、および以前の協力、投資利息、合併で受け取った現金を含むことで、私たちの業務に資金を提供してきた。2022年12月31日まで、7760万ドルの現金、現金等価物、短期有価証券を持っています。私たちの現金と現金等価物は通貨市場基金が持っている金額を含み、コストプラス未実現損益表示であり、公平な市場価値に近い。私たちの売却可能な証券には商業手形とアメリカ国債で保有する金額が含まれています。私たちの投資政策によると、私たちは即時需要を超える現金に投資し、これは私たちがどの投資タイプにも投資できる金額を制限し、私たちが持っているすべての投資が主に流動性と保証を達成するために、国家公認の統計格付け機関の最低格付けを維持しなければならないことを要求する。
2022年12月31日までの1年間で、私たちの現金、現金等価物、有価証券残高は5900万ドル減少した。この低下は、主に、私たちの主要な候補薬に投資し、独自のプラットフォームの発展を支援するため、研究および開発および一般および行政費用に関連する支払いを含む、私たちの業務を運営するための現金によるものである。これらの減少幅は有価証券の満期収益によって相殺される。
88
以下の表に、以下の各期間の現金、現金等価物、および限定的な現金の主なソースおよび用途を示す
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
|
|
(単位:千) |
|
|||||
提供された現金の純額、現金等価物、および限定的な現金(使用) |
|
|
|
|
|
|
||
経営活動 |
|
$ |
(56,888 |
) |
|
$ |
(52,198 |
) |
投資活動 |
|
|
58,351 |
|
|
|
(53,253 |
) |
融資活動 |
|
|
(2,040 |
) |
|
|
89,382 |
|
現金、現金等価物、および限定的な現金純減少 |
|
$ |
(577 |
) |
|
$ |
(16,069 |
) |
経営活動のキャッシュフロー
2022年12月31日までの1年間、経営活動で使用されている純現金、現金等価物、制限現金は5690万ドル。現金の主な用途は私たちの純損失6610万ドルですが、70万ドルの資産と負債、850万ドルの非現金プロジェクト(主に減価償却、株式ベースの報酬、使用権資産を含む)の変化によって部分的に相殺されます。我々の資産·負債の変化には、経営リース負債の減少、前払い費用及びその他の流動資産の減少、前払い研究開発費の増加、未払い勘定及び売掛金の増加、及び繰延収入の増加が含まれる。
2021年12月31日までの1年間、経営活動で使用された純現金、現金等価物、制限現金は5220万ドルだった。現金の主な用途は私たちの純損失6060万ドルで、私たちの資産と負債の変化は20万ドルですが、820万ドルの非現金プロジェクトによって部分的に相殺され、非現金プロジェクトは主に減価償却、株式ベースの報酬、使用権資産を含みます。我々の資産と負債の変化には,イチョウ協力作業の完了による前払い研究·開発費の減少,前払い費用やその他の流動資産の減少,経営リース負債の減少,支払すべき帳簿·売掛金の増加,繰延収入の増加がある。
投資活動によるキャッシュフロー
2022年12月31日までの年間、投資活動が提供する現金純額は5840万ドルで、主に1億419億ドルの有価証券の満期収益から来ている。これは8280万ドルの有価証券の購入と70万ドルの財産と設備の購入によって相殺される。
2021年12月31日までの年間で、投資活動のための現金純額は5330万ドルで、主に1億679億ドルの有価証券および70万ドルの財産と設備の購入から来ている。これは1.14億ドルの有価証券満期収益と130万ドルの有価証券償還収益によって相殺される。
融資活動によるキャッシュフロー
2022年12月31日までの年間で,融資活動のための現金純額は合計200万ドルであり,主に我々250万ドルの普通株買い戻しに関連しており,一部はATM発売計画で我々の普通株を売却したことによる30万ドルと,株式オプションの行使と我々の従業員株式購入計画による購入から得られた20万ドルの収益である.
2021年12月31日までの1年間に、融資活動が提供する現金純額は合計8940万ドルであり、主に2021年4月と9月に引受の公開発行で我々の普通株を売却して得られた8100万ドルの純収益、ATM発売計画で我々の普通株を売却して得られた800万ドル、株式オプションとESPPの行使に貢献した40万ドルの収益に関連しており、これらの収益は従業員に支払われる制限的株式奨励に関する源泉徴収税によって相殺される。
資金需要
今まで、私たちはどんな製品も商業化されておらず、利益も達成されていない。私たちは、私たちの候補製品をさらに開発し、独自のプラットフォーム技術に投資し、上場企業として運営し、今後数年間も巨額の純損失を招くと予想しています。
89
私たちは現在、羅氏協力から収入を得ており、以前の協力から歴史的な収入を得ているが、私たちが設立されて以来、私たちの候補製品に対する規制機関の承認を得ない限り、いかなる製品収入も生じていない。私たちは、2022年12月31日まで、私たちの現金、現金等価物、および短期有価証券は、少なくとも申請を提出した日から今後12ヶ月の予想される現金需要を満たすのに十分だと信じている。具体的には、私たちの現在の予測によると、私たちの既存の現金、現金等価物、および短期有価証券残高は、2024年度後半まで延長される現金滑走路を提供する予定だ。私たちの財政資源がどのくらいの時間で私たちの業務をサポートするのに十分な予測は展望的な陳述であり、リスクと不確定要素に関連し、実際の結果は様々な要素によって大きく異なる可能性があり、その中には本年度報告10-K表の“リスク要因”と題する節で議論された要素が含まれている。私たちの推定は間違っていることが証明される可能性があるという仮定に基づいており、私たちは現在予想されているよりも早く私たちが利用できる資本資源を利用することができる。
私たちの候補製品開発に関連する多くのリスクと不確実性のため、私たちの候補製品の開発を完了し、規制部門の承認を得るために必要な資本支出と運営支出金額を正確に見積もることができません。私たちの資金需要は以下の要素を含む多くの要素に依存するが、これらに限定されない
契約承諾と義務
私たちは資金を必要とするいくつかの重大な契約義務と約束を持っている。私たちの運営賃貸に対する約束は、マサチューセッツ州ケンブリッジ市ビンニー街301番地のオフィスと実験室空間のレンタルと、マサチューセッツ州ウォルザムのAzzur Group、LLCから借りたcGMPクリーンルーム空間と関連があります。
2017年7月、マサチューセッツ州ケンブリッジ市ビンニー街301号41346平方フィートの実験室とオフィススペースをレンタルすることで合意しました。年間レンタル料は340万ドル(約340万円)。この10年間の賃貸契約は2018年1月から始まり、無料賃貸期間、年間賃貸料上昇、テナント改善補助金の条項が含まれている。さらに、私たちは290万ドルのテナント改善投資を支払った。契約と同時に、私たちは100万オーストラリアドルの信用状を発行しました。
2018年12月7日,会社の完全子会社Synlogic Operating Company,Inc.は,会社とAzzurの間で2018年9月8日に締結された主契約サービス協定(プライマリサービスプロトコル)に基づき,Azzur Group,LLC(Azzur)と作業説明書(最初のSOW)を締結した.
最初のSOWによると、Azzurは、マサチューセッツ州ウォルザムで建設される約700平方フィートのクリーンルーム空間(Azzur Suite)に会社を進出させ、2019年5月1日から2022年12月31日まで44ヶ月間使用することに同意した。Azzurは会社にストレージと人員サポートを提供することにも同意しました
90
Azzurスイートルームです。初期期間中,クリーンルームや貯蔵室および人員支援やその他のサービスに進出·使用するプロジェクトの総費用は480万ドルと見積もられていた。
2021年4月、SynlogicはAzzurと最初のSOWの代わりに新しいプロトコル(第2のSOW)を締結した。2番目のSOWにより,SynlogicはAzzur Suiteへのアクセスと使用を許可され,期限は20カ月であり,2021年5月から2022年12月までである(2期目).同社は、同社がAzzur Suiteの使用を制御しているため、このプロトコルに埋め込みテナントが含まれていると判断した。したがって、協定下の固定および実質固定対価は、第2のSOW発効日の使用権資産およびリース負債を再計量するために使用される。
2022年1月21日、会社はAzzurと2つの作業声明を締結した。1頭目の雌豚(3頭目の雌豚)によると、会社は会社の拡大使用のためにAzzurのクリーンルーム空間を改修してアップグレードするためにAzzurに70万ドルを支払うことに同意した。その中で2頭目の雌豚(4頭目の雌豚)は会社が2021年4月29日にAzzurと締結した2頭目の雌豚に代わった。第4のSOWは、2022年1月から2023年3月(第3期)までレンタル期間を延長した。第3頭と第4頭の母豚は経営賃貸使用権資産と対応する経営賃貸負債に対して180万ドルの調整を行った。
2022年11月、当社はAzzurと新しい協定(第5のSOW)を締結し、レンタル期間を2023年4月から2023年12月(第4期)に延長した。第5のSOWは、2つのリース延長オプションを含み、第1のオプションは2024年6月まで、第2のオプションは2024年12月まで続く。2022年12月31日現在、会社は賃貸借延長行使の第一選択権を合理的に確定できると確定した。第5のSOWは、経営リース使用権資産と対応する経営リース負債を100万ドル調整した。2022年12月31日現在、Azzurリースに関する余剰負債総額は約290万ドル。
当社は期限内の任意の時間に三ヶ月前に五番目のSOWを書面で終了することができます。さらに、いずれの一方も、他方が約束を破ったか、または治癒できなかったために、主サービスプロトコル(雌豚を含む)を終了することができる。当社は2024年6月までに終了選択権を行使しないことを決定した理由があります。
私たちは臨床段階会社なので、私たちの最も重要な臨床試験支出はCROとCMOになると予想されています。これらの契約は通常キャンセルすることができますが、別途通知する必要があり、キャンセル処罰を受けません。
スタートアップ企業として、私たちは他の生命科学会社と同じリスクに直面しています。追加資本を調達する能力、私たちの競争相手の新技術革新の開発、臨床前研究失敗のリスク、私たちの候補製品の臨床試験における安全性と有効性、規制承認過程、私たちの製品を効率的に生産する能力、私たちの製品が承認された後の市場の私たちに対する受け入れ度、マーケティングと販売の歴史の不足、キーパーソンへの依存、そしてノウハウの保護を含む。我々の治療計画は現在商業化前にあり、早期開発から発見まで、広範な臨床前と臨床試験、および任意の候補製品の商業化前に監督部門の承認を得ることを含む多くの追加の研究と開発努力が必要となる。このような努力は多くの追加資本、十分な人員インフラ、そして広範囲なコンプライアンス報告能力を必要とする。私たちの研究と開発が成功することは保証できません。私たちの知的財産権は十分に保護され、開発された任意の製品は必要な規制承認を得ますか、あるいは承認された製品は商業的可能性があります。私たちの製品開発努力が成功しても、いつ(あれば)製品販売から収入を得ることができるかは定かではありません。私たちは利益を達成しなければ、私たちは決して利益を達成しないかもしれないが、私たちは引き続き追加資本を集めたり、戦略的協力や連合のような他の資金源から融資を得る必要があるだろう。もし私たちが十分な資本が不足して私たちの業務を拡大したり、他の方法で私たちのビジネスチャンスを利用することができなければ、私たちの業務、財務状況、そして経営結果は実質的な悪影響を受けるかもしれない。
表外手配
我々は、表外手配または他の契約上の狭いまたは限られた目的を促進することを目的とした構造的融資または特殊な目的エンティティと呼ばれるエンティティなど、合併されていないエンティティまたは金融パートナーシップ企業とは何の関係もない。したがって、もし私たちがこのようなタイプの関係に参加すれば、私たちはいかなる融資、流動性、市場、または信用リスクにも直面しないだろう。私たちは正常な業務過程で私たちの業績と私たちの子会社の業績を保証します。
最近の会計公告
注釈2を読んでください重要会計政策の概要本年度報告書の他の表格10−Kに記載されている連結財務諸表について。
91
第七A項。数量化と高質市場リスクの開示について。
金利リスク
取引法第12 b-2条の定義によると、我々は小さな報告会社であり、本プロジェクトに要求される情報を提供する必要はない。
プロジェクト8.連結財務ST図面と補足データ。
我々の総合財務諸表は、独立公認会計士事務所の報告とともに、本年度報告のF-1~F-26ページにそれぞれ掲載されている。
項目9.Accouとの変更と分岐会計と財務情報開示の専門。
ない。
第9条。制御するプログラムがあります
開示制御の定義と制限
我々の開示制御および手順(例えば、1934年証券取引法(改正取引法)下の第13 a-15(E)および15 d-15(E)規則によって定義される)は、取引法に従って提出された報告(本報告のような)において開示を要求する情報が、米国証券取引委員会の規則および表に指定された期間内に記録、処理、集計および報告されることを保証するための制御および他の手続きである。開示制御および手続きは、開示すべき決定をタイムリーに行うために、我々の最高経営責任者および最高財務官を含む、このような情報の蓄積を確保し、適切に私たちの管理層に伝達することを目的としている。私たちの経営陣はこのような統制と手続きを評価し続けている。
どんな開示統制と手続き制度の有効性には固有の限界がある。これらの制限には,人為的誤りの可能性,制御やプログラムを回避または凌駕すること,および合理的な資源制限がある.また,我々の制御システムは,将来のイベントの可能性について合理的であると考えられるいくつかの仮定に基づいて設計されているため,我々の制御システムは将来すべての可能性が期待される目的を達成できない可能性がある.したがって、私たちの開示制御と手続きはその目標を達成するために合理的な保証を提供するが、絶対的な保証ではない。
情報開示制御とプログラムの評価
我々の最高経営責任者および最高財務責任者は、本10-Kテーブルがカバーされている期間終了までの間の開示制御および手順(取引所法案規則13 a-15(E)および15 d-15(E)で定義されているような)の有効性を評価した後、この評価に基づいて、取引法に基づいて提出または提出された報告書において開示を要求する情報が米国証券取引委員会規則および表に指定された期間内に記録、処理、集計、報告され、我々の管理層に蓄積されて伝達されることを保証するために有効であると結論した。必要な開示について速やかに決定するために、我々の主要行政官および主要財務官、または同様の機能を実行する者を含む。また、我々の最高経営責任者および最高財務官は、新冠肺炎の流行の影響は、財務報告や開示制御プログラムの内部統制能力を維持する能力に影響を与えていないと結論した。
内部制御の変化
2022年12月31日までの財政年度内に、当該等の内部統制の評価に関連する財務報告内部統制には重大な影響や合理的に財務報告内部統制に重大な影響を与える可能性のある変化は生じていない。
財務報告の内部統制に関する経営陣の報告
我々の経営陣は、改正された取引法第13 a-15(F)及び15 d-15(F)条の規定に基づいて、財務報告の十分な内部統制を確立·維持する責任がある。その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。将来的にどのような有効性評価を行うかの予測には,条件の変化により制御が不十分になる可能性がある,あるいは政策やプログラムを遵守する程度が悪化する可能性があるというリスクがある.
92
私たちの経営陣は、2022年12月31日までの財務報告書の内部統制に対する我々の有効性を評価した。この評価を行う際には、管理層は、トレデビル委員会後援組織委員会(COSO)が“内部統制--総合枠組み(2013)”で提案した基準を用いた。我々の評価によると、経営陣は、これらの基準に基づき、2022年12月31日現在、財務報告書の内部統制に有効であると考えている。
制御措置の有効性の内在的制限
発想や運用がどのように整備されていても、絶対的な保証ではなく、合理的な保証しか提供できず、規制の目標が達成されることを確保する管制システム。すべての制御システムに固有の限界があるため,どの制御評価も社内のすべての制御問題や誤った陳述(あれば)が検出されていることを絶対に保証することはできない.したがって、私たちの制御と手続きは、私たちの制御システムの目標が達成されることを確実にするために、絶対的な保証ではなく、合理的な保証を提供することを目的としている。将来的にどのような有効性評価を行うかの予測には,条件の変化により制御が不十分になる可能性がある,あるいは政策やプログラムを遵守する程度が悪化する可能性があるというリスクがある.
プロジェクト9 B。他にも情報です。
ない。
プロジェクト9 Cです。法規を開示する検査が禁止されている外国司法管轄区域。
適用されません。
93
部分(三)
プロジェクト10.役員·役員職ICERSと会社管理。
このプロジェクトに対する応答は、会社2023年株主総会依頼書の“管理と会社管理事項”、“第16条(A)実益所有権報告コンプライアンス”、“行為と道徳基準”などのタイトル下での議論に引用されている。
プロジェクト11.行政官イー補償します。
このプロジェクトへの対応は,当社の2023年株主総会依頼書における役員と役員報酬に関する検討を参考にした。
プロジェクト12.特定の受益者の保証所有権従業員と経営陣と関連した株主問題。
当社は、2023年株主総会の委託書のうち、“若干の実益所有者及び経営陣の担保所有権”というタイトルで、本プロジェクトに対応し、参考にしています。
このプロジェクトに対する応答は,当社の2023年株主総会依頼書における“何らかの関係者取引”と“管理と会社ガバナンス”の2つのタイトルでの議論に引用的に組み込まれている。
第14項.元金口座TING料金とサービスです。
当社は2023年株主総会の依頼書に“独立公認会計士事務所”というタイトルでこのプロジェクトへの対応を参考にこのプロジェクトの検討に取り入れている。
94
部分IV.IV
プロジェクト15.展示品、資金エーアールアイチェックリストです。
第15(A)項以下の書類は、本年度報告の10-Kフォームの一部として提出される
第十五条(A)(1)及び(2)項本年度報告表格10-K第8項の“連結財務諸表と補足データ”を参照。他の財務諸表明細書は含まれていません。それらは適用されないため、あるいはこれらの情報は財務諸表または付記に登録されています。
第十五条第三項以下の証拠は,本年度報告の10−K表の一部として提出されるか,または引用により本年度報告に組み込まれる。
展示品索引
展示品 番号をつける |
展示品説明 |
保存済み これがあった 届ける |
以下の会社が合併する ここで引用する 表や スケジュール |
保存する 日取り |
アメリカ証券取引委員会 ファイル/レジストリ 番号をつける |
|
|
|
|
|
|
2.1^ |
2017年5月15日現在、Mirna Treeutics,Inc.,Meerkat Merge Sub,Inc.およびSynlogic,Inc.によって署名された合併および再構成協定および計画。 |
|
8-K (添付ファイル2.1) |
05/16/2017 |
001-37566 |
|
|
|
|
|
|
3.1 |
改訂および再予約された会社登録証明書 |
|
8-K (添付ファイル3.1) |
10/6/2015 |
001-37566 |
|
|
|
|
|
|
3.2 |
2017年8月25日に改訂·再登録された会社登録証明書(逆株式分割) |
|
8-K (添付ファイル3.1) |
08/28/2017 |
001-37566 |
|
|
|
|
|
|
3.3 |
改訂及び再登録された会社登録証明書の改訂(名称変更) |
|
8-K (添付ファイル3.2) |
08/28/2017 |
001-37566 |
|
|
|
|
|
|
3.4 |
付例を改訂および再制定する |
|
8-K (添付ファイル3.2) |
10/6/2015 |
001-37566 |
|
|
|
|
|
|
4.1 |
普通株の書式 |
|
S-1/A (添付ファイル4.2) |
09/18/2015 |
333-206544 |
|
|
|
|
|
|
4.2 |
事前出資引受権証 |
|
8-K (添付ファイル4.1) |
06/12/2019 |
001-37566 |
4.3 |
証券説明書 |
|
10-K (添付ファイル4.3) |
03/12/2020 |
001-37566 |
|
|
|
|
|
|
10.1# |
2015年度株式インセンティブ奨励計画 |
|
10-K (添付ファイル10.1) |
03/20/2018 |
001-37566 |
|
|
|
|
|
|
10.2# |
2015年株式インセンティブ奨励計画下の株式オプション付与通知及び株式オプションプロトコルフォーマット。 |
|
S-1/A (証拠10.9(B)) |
09/11/2015 |
333-206544 |
|
|
|
|
|
|
10.3# |
2015年持分奨励計画の下で制限株式奨励協定と制限株式単位奨励通知のフォーマット。 |
|
S-1/A (添付ファイル10.9(C)) |
09/11/2015 |
333-206544 |
|
|
|
|
|
|
10.4# |
2017年度株式インセンティブ計画 |
|
10-K (添付ファイル10.4) |
03/20/2018 |
001-37566 |
|
|
|
|
|
|
10.5# |
2017年株式インセンティブ計画下での株式オプション付与通知と株式オプション合意フォーマット。 |
|
10-Q (添付ファイル10.17) |
11/13/2017 |
001-37566 |
|
|
|
|
|
|
10.6# |
非従業員役員報酬計画。 |
|
8-K (添付ファイル10.1) |
01/31/2020 |
001-37566 |
|
|
|
|
|
|
95
展示品 番号をつける |
展示品説明 |
保存済み これがあった 届ける |
以下の会社が合併する ここで引用する 表や スケジュール |
保存する 日取り |
アメリカ証券取引委員会 ファイル/レジストリ 番号をつける |
10.7# |
会社と各役員·上級管理者との賠償協議フォーマット |
|
S-1/A (添付ファイル10.13) |
09/11/2015 |
333-206544 |
|
|
|
|
|
|
10.8# |
SynlogicとAoife M.ブレナン,MB,Bch,Bao,MMScの間の招待状は2016年6月22日 |
|
8-K (添付ファイル10.6) |
08/28/2017 |
001-37566 |
|
|
|
|
|
|
10.9# |
第1修正案-SynlogicとAoife M.ブレナン,MB,Bch,Bao,MMSc間の要項,日付は2016年11月7日 |
|
8-K (添付ファイル10.7) |
08/28/2017 |
001-37566 |
|
|
|
|
|
|
10.10# |
第二修正案、SynlogicとAoife M.ブレナン、MB、Bch、Bao、MMSc間の要項、日付は2017年5月8日 |
|
8-K (添付ファイル10.8) |
08/28/2017 |
001-37566 |
|
|
|
|
|
|
10.11# |
第3修正案は,2018年6月5日現在,Synlogic,Inc.とAoifeブレナン,MB,BCH,Bao,MMScとの間の要項である |
|
10-Q (添付ファイル10.1) |
08/9/2018 |
001-37566 |
|
|
|
|
|
|
10.12# |
Synlogic,Inc.とAoife M.ブレナン,MB,Bch,Bao,MMScとの間の改訂と再署名の手紙プロトコルは,2018年10月1日である |
|
10-Q (添付ファイル10.1) |
11/13/2018 |
001-37566 |
|
|
|
|
|
|
10.13 |
2022年1月24日までの雇用協定は、SynlogicとMichael Jensenが署名した |
|
8-K (添付ファイル10.1) |
03/03/2022 |
001-37566 |
|
|
|
|
|
|
10.14.1 |
2018年11月28日に署名された招聘契約は、SynlogicとAntoine Awadによって署名されました |
|
10-K (添付ファイル10.14.1) |
03/25/2021 |
001-37566 |
|
|
|
|
|
|
10.14.2 |
アントワン·アワードの昇進状は2020年7月21日 |
|
10-K (添付ファイル10.14.2) |
03/25/2021 |
001-37566 |
|
|
|
|
|
|
10.15^ |
AbbVie S.á.r.l.,Suffolk Merge Sub,Inc.,Synlogic IBDCo,Inc.,Synlogic,LLC,Synlogic,Inc.とその中に列挙された創始者間の合併合意および計画は、2015年7月16日であり、日付は2015年12月14日の合意および合併計画第1修正案である |
|
8-K (添付ファイル10.12) |
08/28/2017 |
001-37566 |
|
|
|
|
|
|
10.16^ |
AbbVie S.≡.r.l.,Synlogic IBDCo,Inc.とSynlogic Operating Company,Inc.の間の合意と合併計画の第2修正案は,2018年9月27日である |
|
10-Q (添付ファイル10.2) |
11/13/2018 |
001-37566 |
|
|
|
|
|
|
10.17^ |
AbbVie S.≡.r.l.,Synlogic IBDCo,Inc.とSynlogic運営会社との間の合意と合併計画第3修正案および許可協定第1修正案は,2018年12月18日である |
|
10-K (添付ファイル10.25) |
03/12/2019 |
001-37566 |
|
|
|
|
|
|
10.18 |
Synlogic,Inc.とSynlogic IBDCo,Inc.の間のライセンスプロトコルは,2015年7月16日である |
|
8-K (添付ファイル10.13) |
08/28/2017 |
001-37566 |
|
|
|
|
|
|
96
展示品 番号をつける |
展示品説明 |
保存済み これがあった 届ける |
以下の会社が合併する ここで引用する 表や スケジュール |
保存する 日取り |
アメリカ証券取引委員会 ファイル/レジストリ 番号をつける |
10.19 |
登録者とジェフリー有限責任会社の間の販売契約、日付は2021年7月23日です |
|
10-Q (添付ファイル1.1) |
11/10/2021 |
001-37566 |
|
|
|
|
|
|
10.20 |
契約フォーマットを承認し、日付は2018年4月6日で、Synlogic、Inc.とある投資家によって署名された。 |
|
8-K (添付ファイル10.1) |
04/6/2018 |
001-37566 |
|
|
|
|
|
|
10.21 |
2018年9月8日現在、Synlogic,Inc.とAzzur Group間のマスター契約サービスプロトコル(d/b/a Azzur of New England LLC)がある。 |
|
10-K (添付ファイル10.29) |
03/12/2019 |
001-37566 |
|
|
|
|
|
|
10.22 |
Synlogic,Inc.とAzzur Group間のマスター契約サービスプロトコル(ニューイングランド有限責任会社のd/b/a Azzur)により,2018年9月10日の作業説明書である. |
|
10-K (添付ファイル10.30) |
03/12/2019 |
001-37566 |
|
|
|
|
|
|
10.23 |
Synlogic,Inc.とAzzur Group間のマスター契約サービスプロトコル(ニューイングランド有限責任会社のd/b/a Azzur)により,2018年12月7日の作業説明書である. |
|
10-K (添付ファイル10.31) |
03/12/2019 |
001-37566 |
|
|
|
|
|
|
10.24 |
当社とイチョウ生物工学会社が2019年6月11日に調印した引受協定。 |
|
8-K (添付ファイル10.1) |
06/12/2019 |
001-37566 |
|
|
|
|
|
|
10.25 |
代行サービス条項の契約日は2019年6月11日で、Synlogic運営会社とGinkgo Bioworks,Inc.が署名または締結します。 |
|
10-Q (添付ファイル10.2) |
08/08/2019 |
001-37566 |
|
|
|
|
|
|
10.26 |
改訂された会社とダフォスコンサルタント会社との間の諮問協定は、2019年10月13日から発効します |
|
8-K (添付ファイル10.1) |
10/24/2019 |
001-37566 |
|
|
|
|
|
|
10.27 |
Synlogic、Inc.2015年従業員株購入計画、改訂された |
|
8-K (添付ファイル10.1) |
12/20/2019 |
001-37566 |
|
|
|
|
|
|
10.28 |
許可とサービスプロトコルおよび作業説明書は,2021年4月28日にSynlogic Operating Company,Inc.とAzzur Clean-On-Demand-On-Demand-Boston,LLCの間で署名された. |
|
10-Q (添付ファイル10.1) |
08/12/2021 |
001-37566 |
|
|
|
|
|
|
10.29 |
2021年6月16日、Synlogic Operating Company,Inc.とHoffman-La Roche Inc.との間のパイロット連携およびオプションプロトコル。 |
|
10-Q (添付ファイル10.2) |
08/12/2021 |
001-37566 |
|
|
|
|
|
|
10.30 |
Synlogic,Inc.とAzzur Group間のマスター契約サービスプロトコル(d/b/a Azzur of New England LLC),SOW P-10558-01によると,2022年1月21日の作業説明書である |
|
10-K (添付ファイル10.30) |
03/17/2022 |
001-37566 |
|
|
|
|
|
|
10.31 |
Synlogic,Inc.とAzzur Group間のマスター契約サービスプロトコル(d/b/a Azzur of New England LLC),SOW P-10558-2によれば,2022年1月21日の作業説明書である |
|
10-K (添付ファイル10.31) |
03/17/2022 |
001-37566 |
|
|
|
|
|
|
97
展示品 番号をつける |
展示品説明 |
保存済み これがあった 届ける |
以下の会社が合併する ここで引用する 表や スケジュール |
保存する 日取り |
アメリカ証券取引委員会 ファイル/レジストリ 番号をつける |
10.32 |
Synlogic,Inc.とAzzur Group(d/b/a Azzur of New England LLC)との間の主契約サービスプロトコル(d/b/a Azzur of New England LLC),SOW P-10558-01拡張A,2022年11月22日の作業説明書によると |
X |
|
|
|
|
|
|
|
|
|
21.1 |
登録者の子会社 |
X |
|
|
|
|
|
|
|
|
|
23.1 |
独立公認会計士事務所の同意 |
X |
|
|
|
|
|
|
|
|
|
24.1 |
授権書(本文書の署名ページに含まれる) |
X |
|
|
|
|
|
|
|
|
|
31.1 |
ルール13 a~14(A)またはルール15 d~14(A)によって要求される首席実行幹事証明書。 |
X |
|
|
|
|
|
|
|
|
|
31.2 |
細則13 a~14(A)または細則15 d~14(A)によって要求される首席財務幹事証明。 |
X |
|
|
|
|
|
|
|
|
|
32.1 |
米国法典第18編第63章第13 a-14(B)条又は第15 d-14(B)条及び第1350節に要求された証明(“米国法典”第18編第1350節)。 |
X |
|
|
|
|
|
|
|
|
|
32.2 |
米国法典第18編第63章第13 a-14(B)条又は第15 d-14(B)条及び第1350節に要求された証明(“米国法典”第18編第1350節)。 |
X |
|
|
|
|
|
|
|
|
|
101.INS |
XBRLタグがイントラネットXBRL文書に埋め込まれているので、連結されたXBRLインスタンス文書であるインスタンス文書は、相互作用データファイルには表示されない。 |
X |
|
|
|
|
|
|
|
|
|
101.書院 |
インラインXBRL分類拡張アーキテクチャ文書. |
X |
|
|
|
|
|
|
|
|
|
101.カール |
インラインXBRL分類拡張はリンクベース文書を計算する. |
X |
|
|
|
|
|
|
|
|
|
101.def |
XBRLソート拡張を連結してLinkbase文書を定義する. |
X |
|
|
|
|
|
|
|
|
|
101.介護会 |
XBRL分類拡張ラベルLinkbase文書を連結する. |
X |
|
|
|
|
|
|
|
|
|
101.Pre |
XBRL分類拡張プレゼンテーションLinkbaseドキュメントを内部接続する. |
X |
|
|
|
|
|
|
|
|
|
104 |
表紙インタラクションデータファイル(イントラネットXBRL文書に埋め込む) |
X |
|
|
|
S-K条例第601(B)(2)項によれば、本展示品の付表及び展示品は省略されている。任意の漏れたスケジュールおよび/または展示品のコピーは、要求に応じて米国証券取引委員会に提供される。
#契約または補償計画またはスケジュールを管理します。
米国証券取引委員会の規定によると、本展示品の一部の内容(星番号で示す)は省略されている。
項目16.表10-Kの合計エリー。
ない。
98
登録する解決策
1934年“証券取引法”第13又は15(D)節の要求に基づいて、登録者は、正式に許可された次の署名者が代表して本報告書に署名することを正式に手配した.
|
|
Synlogic社 |
|
|
|
|
|
日付:2023年3月29日 |
|
差出人: |
/s/Aoifeブレナン |
|
|
|
オーエフ·ブレンナン |
|
|
|
社長と最高経営責任者 (首席行政主任) |
授権依頼書
このような陳述を通じて、私はすべての人、以下に署名したすべての人がAoifeブレナンとMichael Jensenのすべての真の合法的な事実受権者と代理人を構成して任命し、任意およびすべての身分で、彼または彼女の名義、場所、代理で、任意およびすべての身分で本表の10-K年間報告書の任意およびすべての修正案に署名し、それをすべての証拠物およびこれに関連する他の書類と共に証券取引委員会に提出し、上記の事実上の受権者と代理人を授与することを知っている。これに関連して必要かつ必要なすべてのことおよび事柄を行う権利が完全にあり、その可能性または自ら行うことができるすべての意図および目的を尽くし、ここで上記事実を承認および確認する権利があり、またはその代替者または代替者は、本条例によって合法的にまたは手配されたすべてのことを行うことができる。
本授権書の署名日は本授権書の署名日であることを証明した。
本報告は、1934年の証券取引法の要求に基づいて、以下の者代表登録者によって以下に示す日に次のような身分で署名された。
名前.名前 |
|
タイトル |
|
日取り |
|
|
|
|
|
/s/Aoifeブレナン |
|
最高経営責任者総裁と役員 |
|
2023年3月29日 |
オーエフ·ブレンナン |
|
(首席行政主任) |
|
|
|
|
|
|
|
/s/マイケル·Jensen |
|
首席財務官 |
|
2023年3月29日 |
マイケル·延ソン |
|
(首席財務官と首席会計官) |
|
|
|
|
|
|
|
/s/ピーター·バレット |
|
|
|
|
ピーター·バレット |
|
取締役会議長 |
|
2023年3月29日 |
|
|
|
|
|
/s/マイケル·バージス |
|
|
|
|
マイケル·バーギス |
|
役員.取締役 |
|
2023年3月29日 |
|
|
|
|
|
/s/マイケル·ヘフナン |
|
|
|
|
マイケル·ヘフナン |
|
役員.取締役 |
|
2023年3月29日 |
|
|
|
|
|
/s/パトリシャ·ヘター |
|
|
|
|
パトリシャ·ヘター |
|
役員.取締役 |
|
2023年3月29日 |
|
|
|
|
|
リサ·ケリー-クロスウェル |
|
|
|
|
リサ·ケリー-クロスウェル |
|
役員.取締役 |
|
2023年3月29日 |
|
|
|
|
|
/s/Nick Leschly |
|
|
|
|
ニック·レシュリー |
|
役員.取締役 |
|
2023年3月29日 |
|
|
|
|
|
//エドワード·マーザス |
|
|
|
|
エドワード·マーザス |
|
役員.取締役 |
|
2023年3月29日 |
|
|
|
|
|
/s/Richard P.Shea |
|
|
|
|
リチャード·P·シェイ |
|
役員.取締役 |
|
2023年3月29日 |
|
|
|
|
|
99
Synlogic,Inc.統合財務諸表インデックス
独立公認会計士事務所報告(ピマウェイ会計士事務所、ボストン、マサチューセッツ州、監査役事務所ID:185) |
F-2 |
合併貸借対照表 |
F-4 |
合併経営報告書と全面赤字 |
F-5 |
株主権益合併報告書 |
F-6 |
統合現金フロー表 |
F-7 |
連結財務諸表付記 |
F-8 |
“独立地域裁判所報告書”英国王立会計士事務所
株主や取締役会に
Synlogic,Inc.:
連結財務諸表に対するいくつかの見方
我々はすでにSynlogic社とその子会社(当社)の2022年12月31日と2021年12月31日までの総合貸借対照表、同年度までの関連総合経営表と全面赤字、株主権益と現金流量、および関連付記(総称して総合財務諸表と呼ぶ)を監査した。総合財務諸表は,すべての重要な点において,会社の2022年12月31日と2021年12月31日までの財務状況と,2022年12月31日と2021年12月31日までの年度の経営結果とキャッシュフローを公平に反映しており,米国公認会計原則に適合していると考えられる。
意見の基礎
これらの連結財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいてこのような連結財務諸表に意見を発表することだ。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、連結財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査することを求められておらず、私たちも招聘されて監査を行っていません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性について意見を述べるためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、連結財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであっても詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれる。これらの手続きは、連結財務諸表中の金額および開示に関する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、合併財務諸表の全体列報の評価も含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
以下に述べる重要な監査事項は、監査委員会が監査委員会に伝達または要求する当期総合財務諸表監査によって生じる事項を指すことである:(1)総合財務諸表に対して大きな意味を有する勘定または開示に関するものであり、(2)私たちが特に挑戦的で主観的または複雑な判断に関するものである。重要監査事項の伝達は、総合財務諸表に対する私たちの全体的な意見をいかなる方法でも変えることはなく、以下の重要な監査事項を伝達することによって、重要な監査事項またはそれに関連する勘定または開示について個別の意見を提供することはない。
第三者による研究·開発活動に関する課税費用の評価
総合財務諸表付記2で述べたように、研究及び発展によるコストは、発生した費用に計上される。第三者サービスプロバイダの請求書条項が当社の期末と一致しない場合、当社は、特定の会計期間中に発生する臨床試験コスト、契約サービスコスト、候補薬物供給コストを含む当該等の第三者の義務を推定し、期末に請求項目を記録しなければならない。同社の見積もりは、研究開発計画、その間に提供されるサービス、および第三者サービス契約の予想期限(適用される場合)の理解に基づいている。連結財務諸表付記7で述べたように、2022年12月31日現在、第三者による研究·開発活動に関する課税費用残高は160万ドルである。
第三者による研究·開発活動に関する課税費用の評価を重要な監査事項として決定する。同社が計算すべき費用残高を決定する際に行った見積もりは、研究と開発計画の完成状況、関連する未開書コスト推定を含む、評価が困難な可能性のある要素に基づいている。
F-2
以下は私たちがこの重要な監査問題を解決するために実行した主な手続きだ。いずれの後の修正も含めて第三者サービスプロバイダとの契約をチェックし、契約項目の下で関連する第三者とこれまでに発生した費用を確認し、契約項目におけるこれまでの請求書金額を評価するために領収書をチェックした。また,第三者が2022年12月31日までに提供するすべてのサービスが2022年12月31日の課税費用残高に適切に計上されているかどうかを評価するために,(1)第三者サービスプロバイダの研究開発活動を監督する会社個人に聞いて,それぞれの計画の進捗状況を知る,(2)第三者サービスプロバイダから受信した通信を状態報告を含めてチェックし,計上すべき費用残高の推定数と比較し,(3)第三者サービスプロバイダが2022年12月31日以降に最終的に発行した伝票金額を会社の計上すべき金額と比較する.
/s/ピマウェイ法律事務所
2015年以来、当社の監査役を務めてきました。
ボストン、マサチューセッツ州
2023年3月29日
F-3
Synlogic社そして付属会社
合併B割当書
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
|
|
2022 |
|
|
2021 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
短期有価証券 |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
流動資産総額 |
|
|
|
|
|
|
||
長期有価証券 |
|
|
|
|
|
|
||
財産と設備、純額 |
|
|
|
|
|
|
||
使用権--資産経営リース |
|
|
|
|
|
|
||
制限現金 |
|
|
|
|
|
|
||
研究開発費を前払いし,当期分の純額 |
|
|
|
|
|
|
||
その他の資産 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
負債と株主権益 |
|
|
|
|
|
|
||
流動負債: |
|
|
|
|
|
|
||
売掛金 |
|
$ |
|
|
$ |
|
||
費用を計算する |
|
|
|
|
|
|
||
収入を繰り越す |
|
|
|
|
|
|
||
レンタル負債--レンタル経営 |
|
|
|
|
|
|
||
融資リース義務 |
|
|
|
|
|
|
||
流動負債総額 |
|
|
|
|
|
|
||
長期負債: |
|
|
|
|
|
|
||
賃貸負債--レンタルを経営し、当期分を差し引く |
|
|
|
|
|
|
||
融資リース債務、当期分を差し引く |
|
|
|
|
|
|
||
長期負債総額 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
株主権益 |
|
|
|
|
|
|
||
普通株、$ |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
追加実収資本 |
|
|
|
|
|
|
||
その他の総合損失を累計する |
|
|
( |
) |
|
|
( |
) |
赤字を累計する |
|
|
( |
) |
|
|
( |
) |
在庫株は,コストで計算する |
|
|
( |
) |
|
|
|
|
株主権益総額 |
|
|
|
|
|
|
||
総負債と株主権益 |
|
$ |
|
|
$ |
|
||
連結財務諸表の付記を参照。
F-4
Synlogic社そして付属会社
合併状態経営企業と全面赤字
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
収入.収入 |
|
$ |
|
|
$ |
|
||
運営費用: |
|
|
|
|
|
|
||
研究開発 |
|
|
|
|
|
|
||
一般と行政 |
|
|
|
|
|
|
||
総運営費 |
|
|
|
|
|
|
||
運営損失 |
|
|
( |
) |
|
|
( |
) |
その他の収入(支出): |
|
|
|
|
|
|
||
利子と投資収入 |
|
|
|
|
|
|
||
利子支出 |
|
|
( |
) |
|
|
( |
) |
その他の費用 |
|
|
|
|
|
( |
) |
|
その他の収入,純額 |
|
|
|
|
|
|
||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
|
|
|
|
|
|
||
1株当たり純損失--基本損失と赤字 |
|
$ |
( |
) |
|
$ |
( |
) |
加重平均発行済み普通株式-基本と希釈後の普通株式 |
|
|
|
|
|
|
||
総合的な損失: |
|
|
|
|
|
|
||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
有価証券は純損失を実現していない |
|
|
( |
) |
|
|
( |
) |
総合損失 |
|
$ |
( |
) |
|
$ |
( |
) |
連結財務諸表の付記を参照。
F-5
Synlogic社そして付属会社
合併報告書株主権益について
(単位は千で、シェアは含まれていない)
|
|
普通株 |
|
|
その他の内容 |
|
|
積算 |
|
|
|
|
|
在庫株 |
|
|
|
|
||||||||||||||
|
|
額面0.001ドル |
|
|
支払い済み |
|
|
全面的に |
|
|
積算 |
|
|
|
|
|
合計する |
|
||||||||||||||
|
|
株 |
|
|
金額 |
|
|
資本 |
|
|
収入(損) |
|
|
赤字.赤字 |
|
|
株 |
|
|
金額 |
|
|
株権 |
|
||||||||
2020年12月31日残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
|
— |
|
|
$ |
— |
|
|
$ |
|
|||||
市場発行に関する普通株発行による収益は,発行コストを差し引く |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
普通株を発行して得られる収益は,発行コストを差し引く |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
オプションの行使 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
制限株を発行する |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
従業員株購入計画による普通株の発行 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
株式減納 |
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
制限株を廃止する |
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
株式ベースの報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||
証券が収益を実現しない |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
2021年12月31日の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
|
— |
|
|
$ |
— |
|
|
$ |
|
||||
市場発行に関する普通株発行による収益は,発行コストを差し引く |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
普通株買い戻し |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
オプションの行使 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
制限株を発行する |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
従業員株購入計画による普通株の発行 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
株式減納 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
制限株を廃止する |
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
株式ベースの報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||
証券が収益を実現しない |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
2022年12月31日の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
|
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
連結財務諸表の付記を参照。
F-6
Synlogic社そして付属会社
合併状態キャッシュフロープロジェクト
(単位:千)
|
|
現在までの年度 |
|
|
現在までの年度 |
|
||
|
|
2022 |
|
|
2021 |
|
||
経営活動のキャッシュフロー: |
|
|
|
|
|
|
||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
純損失と経営活動で使用される現金純額の調整: |
|
|
|
|
|
|
||
減価償却 |
|
|
|
|
|
|
||
株式ベースの報酬費用 |
|
|
|
|
|
|
||
投資証券の付加価値/償却 |
|
|
( |
) |
|
|
|
|
経営的リース使用権資産帳簿金額が減少 |
|
|
|
|
|
|
||
経営性資産と負債変動状況: |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
研究開発費を前払いし,当期分の純額 |
|
|
|
|
|
|
||
その他の資産 |
|
|
( |
) |
|
|
|
|
売掛金と売掛金 |
|
|
|
|
|
|
||
収入を繰り越す |
|
|
|
|
|
|
||
リース負債を経営する |
|
|
( |
) |
|
|
( |
) |
**その他の長期負債 |
|
|
|
|
|
( |
) |
|
経営活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
投資活動によるキャッシュフロー: |
|
|
|
|
|
|
||
有価証券を購入する |
|
|
( |
) |
|
|
( |
) |
有価証券の満期収益 |
|
|
|
|
|
|
||
有価証券を償還して得た金 |
|
|
|
|
|
|
||
財産と設備を購入する |
|
|
( |
) |
|
|
( |
) |
投資活動提供の現金純額 |
|
|
|
|
|
( |
) |
|
資金調達活動のキャッシュフロー: |
|
|
|
|
|
|
||
融資リース債務の支払い |
|
|
( |
) |
|
|
( |
) |
従業員の報酬を支払うために差し押さえられた制限的な株式奨励 |
|
|
|
|
|
( |
) |
|
市場発行に関する普通株発行による収益は,発行コストを差し引く |
|
|
|
|
|
|
||
従業員の株式購入と株式オプション行使の収益 |
|
|
|
|
|
|
||
普通株を発行して得られる収益は,発行コストを差し引く |
|
|
|
|
|
|
||
普通株を買い戻す |
|
|
( |
) |
|
|
|
|
融資活動が提供する現金純額 |
|
|
( |
) |
|
|
|
|
現金、現金等価物、および限定的な現金純減少 |
|
|
( |
) |
|
|
( |
) |
期初現金、現金等価物、および限定現金 |
|
|
|
|
|
|
||
期末現金、現金等価物、および制限現金 |
|
$ |
|
|
$ |
|
||
非現金投資活動の追加開示: |
|
|
|
|
|
|
||
売掛金及び売掛金に含まれる財産及び設備購入 |
|
$ |
|
|
$ |
|
||
経営賃貸義務により取得した資産 |
|
$ |
|
|
$ |
|
||
非現金融資活動の追加開示: |
|
|
|
|
|
|
||
融資リース下の購入 |
|
$ |
|
|
$ |
|
||
売掛金と売掛金に含まれる発行コスト |
|
$ |
|
|
$ |
|
||
利子を支払う現金 |
|
$ |
|
|
$ |
|
||
連結財務諸表の付記を参照。
F-7
Synlogic社そして付属会社
総合備考財務諸表
組織する
Synlogic社及びその完全所有と合併の子会社(Synlogic或いは当社)は臨床段階の生物製薬会社であり、合成生物学を合成生物技術の発見と開発に応用している。合成バイオテクノロジーはSynlogicの独自のプラットフォームから生成され、反復可能なモジュール化方法を用いて重要な治療機能を実行または提供する新しい候補薬剤を生成する。合成生物技術は有毒物質を代謝し、欠損或いは損傷した代謝経路を補うこと、或いは治療要素の組み合わせを提供することを目的としている。Synlogicの目標は,合成バイオテクノロジーを発見·開発し,最終的に商業化することである。設立以来,同社はそのほとんどの努力をその候補製品の研究·開発に投入してきた。
流動性
当社の総合財務諸表は、正常業務過程における業務連続性、資産現金化、負債返済状況に基づいて作成されています。2022年12月31日までに同社は
リスクと不確実性
スタートアップ段階にある会社として、会社は他の生命科学会社と同じリスクに直面しており、追加資本の調達、競争相手の新技術革新の開発、臨床前と臨床研究失敗のリスク、その候補製品の臨床試験における安全性と有効性、契約研究機関(CRO)と契約製造機関(CMO)などの外部機関に依存するリスク、監督審査過程、会社製品が一旦承認された後の市場のそれに対する受け入れ程度、マーケティングと販売歴史の不足、キーパーソンへの依存及びノウハウの保護を含むが、これらに限定されない。同社の治療計画は現在商業化前の段階にあり、早期開発から発見まで、広範な臨床前と臨床テスト及び監督部門の承認を含め、大量の追加的な研究と開発が必要となり、その後、任意の候補製品を商業化することができる。このような努力は大量の追加資本、十分な人員、インフラ、そして広範囲なコンプライアンス報告能力を必要とする。企業の研究·開発が成功することは保証されず、会社の知的財産権が十分に保護される保証はなく、開発されたいかなる製品も必要な監督管理承認を得ることが保証されない、またはいかなる承認された製品も商業的に実行可能であることは保証されない。同社の製品開発努力が成功しても、同社がいつ(あれば)製品販売から収入を得るかは定かではない。会社は利益が達成されない限り、追加資本を集めたり、戦略的協力や連合のような他の資金源から融資を得る必要があり続けるかもしれない。
新冠肺炎
会社はまだ新冠肺炎疫病が2022年12月31日まで持続することが実質的な影響をもたらすことを知らないが、新冠肺炎疫病はどの程度直接或いは間接的に当社の業務、運営結果と財務状況に影響し、費用と製造、臨床試験と研究開発コストを含み、現在不確定な未来の発展に依存する。
陳述の基礎
添付されている連結財務諸表は米国が公認する会計原則に基づいて作成されている。(米国公認会計原則または公認会計原則)。
合併原則
付随する連結財務諸表は、Synlogicおよびその完全子会社の勘定を含む。合併では、すべての会社間口座と取引がキャンセルされた。
F-8
Synlogic社そして付属会社
連結財務諸表付記
予算の使用
公認会計原則に基づいて財務諸表を作成することは、連結財務諸表の日付の資産、負債、収入及び費用の報告金額、又は資産及び負債に関する開示及び報告期間内に報告された費用金額に影響を与えるために、管理層に推定及び仮定を要求する。継続的な基礎の上で、会社管理層は、研究および開発すべき計算および前払い、計算すべき費用、または投資に関連する推定を含むその推定値を評価する。当社は過去の経験や合理的と信じられている他の様々な仮定から推定しているが,これらの仮定の結果は資産や負債額面を判断する基礎となっている。実際の結果はこれらの推定とは異なる可能性がある。推定数の変化は既知期間の報告結果に反映されている。
現金等価物
当社は購入時の残り期限の高流動性投資ツールはすべて
信用リスクが集中する
会社を集中的な信用リスクに直面させる可能性のある金融商品には、現金、現金等価物、有価証券、および制限された現金の形態で保有される金額が含まれる。同社は高品質で認可された金融機関を用いて残高を維持しているため,このような基金の信用リスクは最も小さい。当社は当該等の口座で何の損失も被っておらず、経営陣は、当該等の預金を保有する預金機関の財務状況により、当社は重大な信用リスクに直面しないと信じている。当社には表外損失リスクの金融商品はありません。
制限現金
同社は約1ドルの現金を持っている
次の表は、財務状況表内に報告された現金、現金等価物および制限現金を入金し、これらの現金、現金等価物および制限現金の合計は、現金フロー表に示された同じ額の和と同じである(千で計算する)。
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
|
|
2022 |
|
|
2021 |
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
制限現金 |
|
|
|
|
|
|
||
表示されている現金、現金等価物、および制限された現金総額 |
|
$ |
|
|
$ |
|
||
公正価値
当社は、報告された公正価値を決定する際に使用される投入を評価するために、公正価値報告の資産及び負債のすべての資料を開示しなければならない。会計基準編纂(ASC)テーマ820、公正価値計量と開示公正価値を計量するツールのために公正価値階層構造を構築し、区別する
F-9
Synlogic社そして付属会社
連結財務諸表付記
市場データに基づく仮説(観察可能な投入)と会社自身の仮説(観察不可能な投入)との間にある.この階層構造は3つのレベルから構成されている:
当社は各報告期間の終了時に異なるレベル間の移行を評価します。いくつありますか
販売可能な証券
その会社はその投資意向に基づいてそのすべての投資を売却可能に分類した。会社は投資を短期投資に分類しています 残りの契約満期日は貸借対照表の日から1年または1年以下であり、貸借対照表に投資した日から1年以上の残り契約満期日であれば長期とする。売却可能な証券は公正価値に基づいて帳簿に記載されており、損益は他の総合収益(損失)に記載されていない。このような債務証券の償却コストは、割増の償却や満期時の割引増加に応じて調整される。この償却は利息と投資収入に含まれている。売却可能証券の実現損益と価値低下は非一時的と判断され、利子や投資収入に計上される。
証券売却のコストは具体的な識別方法によって決定される。証券を売却可能な利息と配当金に分類して利息と投資収入を計上する。非一時的な減値が存在するかどうかを決定するために,当社は能力の有無や市場価格の回復まで投資を意図しているかどうか,投資コストが回収可能であることを示す証拠が逆の証拠を超えているかどうかを考慮している。
財産と設備
物件及び設備は、賃貸権の改善、コストに応じて入金され、その推定耐用年数に応じて直線減価償却される。メンテナンス·メンテナンス費用は発生時に費用を計上し,重大改善工事は増加した財産や設備を計上する。
減価償却は資産投入時から始まります。減価償却は、以下の推定耐用年数に基づいて提示される
資産分類 |
|
使用寿命 |
コンピュータとオフィス機器 |
|
|
家具と固定装置 |
|
|
実験室装置 |
|
|
賃借権改善 |
|
長期資産減価準備
事件や環境変化が発生して資産の帳簿価値が回収できない可能性がある限り,長期資産は減値が審査される。このような事件が発生した場合、会社は資産の帳簿価値をその割引されていない予想される将来の現金フローと比較する。この比較が減値が存在することを示していれば,減値金額は資産の帳簿価値と公正価値との差額で計算される.今までのところ
賃貸借証書
会社は判断を用いて、一つの手配が契約開始時に賃貸契約であるかどうかを評価する。契約が異なる識別された資産の使用に関連している場合、レンタル者は実質的な代替権を有さず、会社は資産の実質的な経済的利益を獲得し、資産の使用を指導する権利を得ることによって、全期間にわたって資産の支配権を得ることができる場合、手配はレンタルである。経営リースに分類されるレンタルには、経営リース使用権(ROU)資産、現在の経営リースが含まれています
F-10
Synlogic社そして付属会社
連結財務諸表付記
総合貸借対照表における負債と非流動経営賃貸負債。ファイナンスリースは私たちの総合貸借対照表に含まれている財産と設備および融資リース義務に含まれています。
純収益資産とはリース期間内の基礎資産の使用権であり、リース負債とはリースによるリース金の支払い義務である。純収益資産およびリース負債はリース開始日にレンタル期間内のリース支払いの推定現在値に基づいて確認する。同社はその逓増借款金利を利用して賃貸支払いの現在値を決定している。増量借款金利は、類似経済環境下で、可比期限内に、担保に基づいて類似資金を借り入れることによる金利である。
当社は賃貸の賃貸と非レンタル構成要素をすべての関連資産カテゴリの単一構成要素として計算することを選択し、すべての契約対価格をレンタル構成要素のみに分配した。運営リースのリースコストは,リース期間内の直線に基づいて確認し,運営報告書および総合損失の運営費用を計上する。可変レンタル支払いはレンタル運営費用に加算されます。
レンタル期間には
研究開発コスト
研究·開発に発生したコストは発生時に費用を計上する。関連貨物を受け取ったり、関連サービスを完了したりする前に、当社が研究·開発活動に支払う払戻不可能な前払い資本化を延期し、資本化する。
研究開発費用には、賃金および福祉、株式ベースの補償費用、実験室用品およびその他の直接費用、施設費用、管理費用、契約サービス、および他の外部費用が含まれる研究開発活動を行うことによって生じるコストが含まれる。
第三者サービスプロバイダの請求書条項が当社の期末と一致しない場合、当社は、特定の会計期間中に発生する臨床試験コスト、契約サービスコスト、候補薬物供給コストを含む当該第三者への義務を推定し、期末に計上すべき項目を記録しなければならない。同社の見積もりは,研究·開発計画の完成状況および関連する未開請求書コスト見積もりに基づいている。
収入確認
同社はロ氏社と候補製品の開発と商業化について協力とオプション合意を結ぶことで収入を創出している。
同社はFASB ASCトピック808に関する連携プロトコルを評価している協力手配手配の性質と契約条項及びその業務経営の性質を考慮して、取引の分類を決定する。当社がこの活動の積極的な参加者であり、協力によるビジネス成功の重大なリスクとリターンに直面している場合、当社は、総合財務諸表にその取引を毛数で記録し、総合財務諸表付記に協力手配下の権利と義務を説明する。
ASC 606によれば、エンティティは、そのクライアントが約束された商品またはサービスの制御権を取得したときに収入を確認し、その金額は、エンティティがこれらの商品またはサービスと交換するために予期される対価格を反映する。エンティティがASC 606の範囲内のスケジュールされた収入確認を決定するために、エンティティは、(I)顧客との契約を決定するステップと、(Ii)契約における履行義務を決定するステップと、(Iii)取引価格を決定するステップと、(Iv)契約内の履行義務に取引価格を割り当てるステップと、(V)エンティティが契約義務を履行するときに収入を確認するステップと、の5つの分析を実行する。当社は、実体が顧客に譲渡された商品やサービスと交換するために獲得する権利のある対価格を受け取る可能性がある場合にのみ、5ステップ分析を契約に適用する。契約開始時に、契約がASC 606の範囲内にあると決定されると、会社は、各契約において約束された商品またはサービスを評価し、どれが義務を履行しているかを決定し、各約束された商品またはサービスが異なるかどうかを評価する。そして、会社は、履行義務を履行する際にそれぞれの履行義務に割り当てられた取引価格の金額が収入であることを確認する。
F-11
Synlogic社そして付属会社
連結財務諸表付記
会社は研究開発サービスの協力協定を締結することができ、この合意に基づいて、会社はその候補製品のいくつかの権利を第三者に許可することができる。これらの取り決めの条項は、一般に、払戻不可能な前払い許可料、特定の費用の精算、顧客選択権使用料、開発、監督、および商業マイルストーン支払い、および製品の純売上を許可する特許使用料のうちの1つまたは複数を会社に支払うことを含む。可変対価格は重大な逆転リスクが存在しないとみなされるまで制限される。
当社は、パートナーもその顧客の各合意下の義務を履行する際に確認すべき適切な収入額を決定する際に、(I)契約で約束された商品またはサービスを決定するステップと、(Ii)契約で異なるか否かを含む契約義務であるか否かを決定するステップと、(Iii)可変対価格の制限を含む取引価格の計量と、(Iv)取引価格を履行義務に割り当てるステップと、(V)会社が各履行義務を履行する場合(または義務履行時)に収入を確認するステップとを実行する。このような手配の会計手配の一部として、当社は、a)上記(Ii)ステップに基づいて決定された履行義務の数、b)上記(Iii)ステップでの取引価格、およびc)上記(V)ステップで契約義務を履行する契約条項およびモードを決定するために、重大な判断を使用しなければならない。当社は、以下に述べるように、特許使用料を除くマイルストーンまたは他の可変価格が取引価格に含まれるべきかどうかを決定するために重大な判断を使用します。取引価格は会社が提供する予定の商品とサービスに割り当てられます。当社は見積数を用いて履行義務を履行する時間を決定しており、その中には履行義務を履行するための測定基準としてフルタイムの同値時間を使用することが含まれている可能性がある。
収入確認基準を満たす前に受け取った金額は繰延収入として会社の総合貸借対照表に記録されている。貸借対照表の後日12カ月以内に収入と確認された金額は当期繰延収入に分類されると予想される。貸借対照表後12カ月以内に収入が確認されなかった金額は繰延収入に分類され、当期分を差し引く。
知的財産権許可証
約束や義務の履行が他の約束と異なるかどうかを評価する際には、顧客の研究、製造、商業化能力、関連専門知識の一般市場での可用性などを考慮する。さらに、顧客が残りの承諾を受けずにコミットメントから利益を得ることができるかどうか、コミットメントの価値が未履行のコミットメントに依存するかどうか、残りのコミットメントを提供することができる他のプロバイダがいるかどうか、残りのコミットメントから分離して識別できるかどうかを考える。他の承諾と組み合わせたライセンスについては、合併履行義務が時間の経過とともに履行されるか、ある時点で履行されるかを決定するために、合併履行義務の性質を判断するために利用され、時間の経過とともに収入を確認するための進展を測定するための適切な方法が決定される。私たちは報告期間ごとに進捗指標を評価し、必要に応じて業績指標と関連収入確認を調整する。
研究と開発サービス
もし一つの手配が私たちが研究開発サービスを提供する約束や義務を含んでいると判断された場合、私たちはこれらのサービスがその計画内の他の約束とは異なるかどうかを確認しなければならない。これらのサービスが他の承諾と異なるかどうかを評価する際には,クライアントがこれらの同じサービスを実行する能力を考慮する.さらに、顧客が残りの承諾を受けずにコミットメントから利益を得ることができるかどうか、そのコミットメントの価値が未履行コミットメントに依存するかどうか、残りのコミットメントを提供することができる他のサプライヤーがいるかどうか、残りのコミットメントとは別に識別できるかどうかを考える。他の承諾と組み合わせた研究開発サービスについては,合併履行義務が一定期間内かある時点で履行されているかを決定するために,合併履行義務の性質を判断を用いて評価し,時間とともに収入を確認するために進展を測る適切な方法である.契約履行義務を履行するために将来発生する予定のフルタイム相当時間と比較して発生するフルタイム相当時間と比較して、管理職の判断が必要となる入力方法を含む、合併履行義務に関する収入の推定を時間基準で記録する。私たちは報告期間ごとに進捗指標を評価し、必要に応じて業績指標と関連収入確認を調整する。この方法では,業績義務履行に必要な総投入を見積もり,これまでの収入確認にかかる努力を評価しなければならない。このような余剰投入の推定は主観的であり,この研究は新たであるため,成功した努力は貸借対照表日の推定努力とは異なる可能性がある.
顧客オプション
顧客が追加商品やサービスを取得することを許可する顧客選択権を含む手配が決定された場合、その手配開始時に、顧客選択権の基礎となる貨物やサービスは契約義務とはみなされない
F-12
Synlogic社そして付属会社
連結財務諸表付記
なぜならそれらはオプションの行使にかかっているからだ。当社は、顧客の実質的な権利選択、すなわち無料または割引で追加商品またはサービスの選択を得ることを評価する。顧客選択権が実質的な権利を表すと決定された場合、実質的な権利は、手配の開始時に別個の履行義務として確認される。商品またはサービス(I)が契約中の元の貨物およびサービスと同様であり、(Ii)が元の契約の条項に従って提供される場合、当社は別の方法に基づいて取引価格を物質的権利に割り当てる。この代替案によると、会社は顧客から受け取ることが予想される対価格総額を、顧客に提供する予定のすべての商品またはサービスに分配する。重大な権利に割り当てられた金額は,選択権の行使や義務履行まで収入として確認されない。
一里塚払い
マイルストーン支払いを含む各手配の開始時に、当社はマイルストーンの実現に関連する累積収入が大きく逆転する可能性があるかどうかを評価し、可能な金額法を用いて取引価格に含まれる金額を推定する。累積収入が大きく逆転しない可能性が高い場合、関連するマイルストーン価値は取引価格に含まれる。規制承認のような会社または被許可者の統制範囲内にない記念碑的支払いは、これらの承認を受ける前に実現可能であるとは考えられない。他のマイルストーンについて、同社は、この評価を行う際に特定のマイルストーンを達成するために、科学、臨床、規制、商業、および他の克服しなければならないリスクなどの要因を評価する。累積収入が大きく逆転しない可能性があるかどうかを決定する際には,かなりの判断が必要である。その後の各報告期間が終了した時点で、当社はすべての制限されたマイルストーンの実現確率を再評価し、必要に応じて全体の取引価格の推定を調整する。いずれの調整も累積追跡をもとに記録されており,調整期間中の収入や収益に影響を与える.
印税
販売ベースの特許使用料を含む手配については、販売レベルに基づくマイルストーン支払いを含み、許可が特許権使用料に関連する主要項目とみなされる場合、当社は、(I)関連販売が発生した場合、または(Ii)特許権使用料の一部または全部が割り当てられた履行義務が履行された(または部分的に履行されている)ときに収入を確認する。現在まで、同社はそのいかなる許可手配によって生じたいかなる特許権使用料収入も確認していない。
契約費用
株式ベースの報酬
当社は、付与日に基づいて従業員、非従業員及び取締役の持分報酬を計量し、報酬の必要なサービス期間内(通常は帰属期間)に財務諸表において関連費用を確認する。従業員株式購入計画(ESPP)における各オプションおよび購入権の公正価値は、付与日にBlack-Scholesオプション定価モデルを用いて推定される。当社の普通株の予想変動率は、当社の歴史変動率と同種の上場会社の歴史変動率の平均値に基づいて決定されます。従業員に付与されたオプションの期待期限は、オプションの契約期間とオプションの加重平均帰属期限の平均値を表す簡略化方法を用いて計算される。ESPPの期待購入権期限は発行期間の継続時間に基づく.仮定された配当率は、予測可能な未来に配当金を派遣しない会社の予想に基づいている。無リスク金利は、付与または再測定時の予想期限に見合った米国債収益率曲線に基づく。没収行為は発生時に確認します。
当社は,その総合経営報告書において,権益に基づく補償費用と全面損失を分類し,受賞者の賃金コストや受賞者のサービス支払いを分類する方式と同様である。
所得税
当社は、財務諸表または財務諸表で確認された事件の予想される将来の税務結果について繰延税金資産と負債を確認することを要求する貸借対照法を用いて所得税を計算する
F-13
Synlogic社そして付属会社
連結財務諸表付記
会社の納税申告書です。繰延税金項目は、資産及び負債の財務報告及び税収ベース間の差額に基づいて、予想差額を用いて返送される年度内に発効する現行税率を決定する。繰延税金資産と負債の変動を所得税に計上する準備。当社は、その繰延税金資産が将来の課税所得額から回収される可能性を評価し、既存の証拠の重みに基づいて、繰延税金資産のすべてまたは一部が現金化できない可能性があると考え、所得税費用を計上することで推定支出を確立する。予想未来の課税オーバー額及び慎重かつ実行可能な税務計画策略を考慮して、繰延税金資産を回収する潜在力を評価する。
不確実な税収頭寸とは、準備金が確立された税収頭寸を指す。会社は連結財務諸表で確認された所得税の不確実性を会計処理し、2ステップ法を用いて確認すべき税収割引額を決定する。まず、税務機関が外部審査後にこのような状況を維持する可能性を決定するために、税収状況を評価しなければならない。税務状況がより継続する可能性があると考えられる場合、税務状況は、財務諸表で確認されるべき利益金額を決定するために評価される。確認可能な利益額は
1株当たり純損失
1株当たりの基本純損失は,期間内に発行された普通株の加重平均を用いて計算した。1株当たりの純損失は、期間内に発行された普通株の加重平均株式数の和を用いて計算され、希薄化されている場合、非帰属制限普通株、発行された株式オプション、およびESPPに従って発行可能な潜在株式を含む普通株の潜在株式数の加重平均が計算される。
市場情報を細分化する
経営部門は企業の構成要素として定義されており、これらの部門に関する独立財務情報は、首席運営意思決定者または意思決定グループが資源をどのように割り当てるかを決定し、業績を評価する際に評価することができる。同社は以下の地域で運営している
在庫株
在庫株の購入はコスト法により計算され、すなわち購入した在庫株の全コストが在庫株として記録される。
近く発表·採択された会計公告
新しい会計声明は、時々、財務会計基準委員会(FASB)または会社が指定された発効日から採用される他の会計基準作成機関によって発表される。以下で別途議論されない限り、最近発表された当社に適用されるか、または当社に適用される公告は、当社の現在または将来の総合財務諸表または開示に大きな影響を与えないことはないか、または予想される。
下記表は、付記2で述べたように、2022年12月31日まで、2021年12月31日までの公正価値で恒常的に計量された会社資産の情報を提供し、付記2に記載されているように、公正価値を決定するための評価技術の公正価値レベルを示している重要な会計政策の概要.
同社のポートフォリオには多くの固定収益証券が含まれており、これらの証券はいつも毎日取引されているわけではない。そのため、同社が使用する定価サービスは、基準収益率、同種証券の基準、業界グループやマトリックス定価などの過程で他の適用可能な情報を適用して評価を準備している。また、モデルフローを用いて金利の影響を評価し、早期返済案を作成する。これらのモデルは相関を考慮している
F-14
Synlogic社そして付属会社
連結財務諸表付記
信用情報、感知された市場動向、業界ニュース、経済事件。これらのモデルの入力には、基準収益率、報告された取引、ブローカー見積、発行者利益差、および他の関連データが含まれる可能性がある。
当社は、2022年12月31日と2021年12月31日まで、公正価値の恒常的に計量された資産を以下のように分類しています(千計)
|
|
報告日の公正価値計量使用 |
|
|||||||||||||
|
|
十二月三十一日 |
|
|
イベント中の見積もり |
|
|
大切な他の人 |
|
|
意味が重大である |
|
||||
説明する |
|
2022 |
|
|
(レベル1) |
|
|
(レベル2) |
|
|
(レベル3) |
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
商業手形 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
合計する |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
報告日の公正価値計量使用 |
|
|||||||||||||
|
|
十二月三十一日 |
|
|
イベント中の見積もり |
|
|
大切な他の人 |
|
|
意味が重大である |
|
||||
説明する |
|
2021 |
|
|
(レベル1) |
|
|
(レベル2) |
|
|
(レベル3) |
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
商業手形 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
会社債務証券 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
合計する |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
2022年12月31日および2021年12月31日の現金等価物、前払い費用およびその他の流動資産、売掛金および売掛金は、その短期満期日により公正価値に近い金額で入金される。2022年12月31日と2021年12月31日の融資リース義務市場金利に近い金利で金利を計算する公正な価値。
次の表は保有する売却可能な証券をまとめた2022年12月31日および2021年12月31日(単位:千):
2022年12月31日 |
|
原価を償却する |
|
|
未実現総額 |
|
|
未実現総額 |
|
|
公正価値 |
|
||||
商業手形 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
会社債務証券 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
合計する |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
2021年12月31日 |
|
原価を償却する |
|
|
未実現総額 |
|
|
未実現総額 |
|
|
公正価値 |
|
||||
商業手形 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
会社債務証券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
アメリカ国債 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
合計する |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
F-15
Synlogic社そして付属会社
連結財務諸表付記
2022年12月31日までに保有するすべての証券の契約満期日は6ヶ月以下である。2022年12月31日現在、15件の投資が未実現赤字状態にある。2022年12月31日と2021年12月31日に赤字を達成していない状態にある証券の公正価値合計はい$です
売却投資の実現収益と損失総額は会社の総合経営報告書にとって重要ではない。
前払い料金および他の流動資産には、以下のものが含まれている(千計)
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
|
|
2022 |
|
|
2021 |
|
||
前払い保険 |
|
$ |
|
|
$ |
|
||
前払い研究開発 |
|
|
|
|
|
|
||
その他前払い費用 |
|
|
|
|
|
|
||
その他流動資産 |
|
|
|
|
|
|
||
前払い費用とその他の流動資産総額 |
|
$ |
|
|
$ |
|
||
財産と設備、純額は以下の部分からなる(千計)
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
|
|
2022 |
|
|
2021 |
|
||
実験室装置 |
|
$ |
|
|
$ |
|
||
コンピュータとオフィス機器 |
|
|
|
|
|
|
||
家具と固定装置 |
|
|
|
|
|
|
||
賃借権改善 |
|
|
|
|
|
|
||
建設中の工事 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
減価償却累計を差し引く |
|
|
( |
) |
|
|
( |
) |
財産と設備、純額 |
|
$ |
|
|
$ |
|
||
財産と設備の減価償却費用は#ドルです
計算すべき費用には、以下の項目が含まれる
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
|
|
2022 |
|
|
2021 |
|
||
給与関連の |
|
$ |
|
|
$ |
|
||
専門費 |
|
|
|
|
|
|
||
研究開発 |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
費用総額を計算する |
|
$ |
|
|
$ |
|
||
F-16
Synlogic社そして付属会社
連結財務諸表付記
当社の普通株は以下のような特徴を持っている
2019年6月、当社はイチョウ生物工学株式会社(Ginkgo Bioworks,Inc.)に
同社は2017年10月13日、Cowen and Company,LLC(Cowen)と市場(ATM)発売計画について販売合意した。ATM機の発行において、取引所上場会社は指定されたブローカーを通じて現在の市場価格で二級取引市場に新たに発行された株を徐々に売却している。2021年12月31日までの年間で
2021年7月、当社はJefferies,LLC(Jefferies)とATM機について新しい販売協定を締結し、この合意により、当社は時々適宜その普通株の株式を発行及び売却することができ、総販売収益は最高$に達することができる
2021年4月、同社は販売した
2021年9月に会社が販売しました
会社はすでに未来の発行のために以下の普通株を予約しており、これらの普通株は事前融資承認株式証を行使する可能性があり、株式オプションの行使と従業員の株式購入計画と関係がある
F-17
Synlogic社そして付属会社
連結財務諸表付記
|
2022年12月31日 |
|
|
事前融資権証に基づいて発行できる普通株 |
|
|
|
普通株購入オプションを行使することができる |
|
|
|
従業員株購入計画 |
|
|
|
合計する |
|
|
|
株式計画
その会社は現在3つの活発な株式計画を持っている。
2015年持分インセンティブ奨励計画(2015計画)は、会社の主要持分計画である。2015年度計画は、2015年度計画に従って発行可能な普通株式数を毎年増加させることを可能にする“常青樹条項”を含み、2016年度から2025年度までの各財政年度の初日に毎年増加する普通株式数を増加させ、(I)の小さい数に等しくなる
2017年株式インセンティブ計画(2017計画)では、奨励的株式オプション、非制限株式オプション、制限および非制限株式奨励、その他の株式ベースの奨励を付与することが規定されている。
2015年従業員株式購入計画(ESPP)条件を満たす従業員の賃金減額で最高
株式オプション
ブラック·スコアーズオプション定価モデルで使用されている加重平均仮説は、2015年計画と2017年計画に基づいて12月31日までの年度内に従業員と非従業員に株式オプションを発行するためのものである2022年と2021年は
|
|
十二月三十一日までの年度 |
|
||
従業員: |
|
2022 |
|
2021 |
|
所期期限 |
|
|
|
||
加重平均無リスク金利 |
|
|
|
||
予想変動率 |
|
|
|
||
配当率 |
|
|
|
||
|
|
|
|
|
|
F-18
Synlogic社そして付属会社
連結財務諸表付記
次の表は、2015年と2017年に計画された株式オプション活動をまとめました。
|
|
未償還株式オプション |
|
|||||||||||||
|
|
|
|
|
|
|
|
重みをつける |
|
|
|
|
||||
|
|
|
|
|
|
|
|
平均値 |
|
|
|
|
||||
|
|
|
|
|
重みをつける |
|
|
残り |
|
|
骨材 |
|
||||
|
|
|
|
|
平均値 |
|
|
契約書 |
|
|
固有の |
|
||||
|
|
量 |
|
|
トレーニングをする |
|
|
用語.用語 |
|
|
価値がある (a) |
|
||||
|
|
オプション |
|
|
値段 |
|
|
(単位:年) |
|
|
(単位:千) |
|
||||
2021年12月31日現在の未返済債務 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
授与する |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|||
鍛えられた |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
取消·没収 |
|
|
( |
) |
|
|
|
|
|
|
|
|
— |
|
||
2022年12月31日に返済されていません |
|
|
|
|
|
|
|
|
|
|
$ |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
帰属または予想は2022年12月31日に帰属する |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2022年12月31日に行使できます |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
(a)
2022年12月31日および2021年12月31日までの年度内に授与される購入株式の加重平均授受日の1株当たり公平価値は約#ドルである
2022年12月31日まで約一ドルです
制限普通株
2022年12月31日までに年度末2021年には
次の表に制限された普通株式活動を示す
|
|
制限株奨励 |
|
|||||
|
|
|
|
|
重みをつける |
|
||
|
|
量 |
|
|
公正価値 |
|
||
|
|
株 |
|
|
(1株あたり) |
|
||
2021年12月31日に帰属していません |
|
|
|
|
$ |
|
||
授与する |
|
|
|
|
|
|
||
既得 |
|
|
( |
) |
|
|
|
|
没収される |
|
|
( |
) |
|
|
|
|
2022年12月31日に帰属していない |
|
|
|
|
$ |
|
||
2022年12月31日までおよび2021年12月31日まで年度内に帰属する株式の総公平価値は$
2022年12月31日まで約一ドルです
F-19
Synlogic社そして付属会社
連結財務諸表付記
従業員株購入計画
ESPPは補償計画と考えられ、関連補償費用は6ヶ月の提供期間内に確認される。2022年12月31日と2021年12月31日までの年度の補償費用は#ドル未満
持分補償
会社が記録した株式ベースの報酬支出総額は約#ドルである
次の表は、2022年12月31日と2021年12月31日までの年度会社総合経営報告書内の株式ベースの報酬支出と全面赤字(単位:千)をまとめています
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
研究開発 |
|
$ |
|
|
$ |
|
||
一般と行政 |
|
|
|
|
|
|
||
|
|
$ |
|
|
$ |
|
||
次の表は、2022年12月31日と2021年12月31日までの年間持分ベース給与支出(単位:千)を奨励タイプ別にまとめた
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
株式オプション |
|
$ |
|
|
$ |
|
||
制限株奨励 |
|
|
|
|
|
|
||
ESPP |
|
|
|
|
|
|
||
|
|
$ |
|
|
$ |
|
||
羅氏協力
2021年6月、会社はF.Hoffmann-La Roche Ltd(羅氏バーゼル)とHoffmann-La Roche Inc.(羅氏米国、および羅氏バーゼル、羅氏)とパイロット協力とオプション協定(羅氏協力とオプション協定)を締結した。ロ氏の協力とオプション協定の条項によると、同社と羅氏は協力を求め、炎症性腸疾患を治療する未開示の新しい標的(候補製品)を解決するために、合成バイオテクノロジーを研究·臨床前に開発する。
羅氏の協力とオプション協定によると、羅氏は前払いした返却不可能な技術アクセス料$を会社に支払うことに同意した
“羅氏協力とオプション協定”によると、この合意期間内に、双方が“羅氏協力とオプション協定”の下での義務を履行した場合にのみ、双方は、それによって制御される特定の知的財産権及びノウハウの非排他性、譲渡不可、再許可不可、印税免除の権利及び許可を他方に付与している。双方は羅氏協力とオプション合意基本研究計画の実行状況を監督·管理するための共同研究委員会(JRC)を設置する。
羅氏協力とオプション協定には各種の陳述、保証、契約、賠償とその他の習慣条項が含まれている。研究計画のいくつかの部分がいくつかの成功基準に達した場合、羅氏は書面通知の後すぐに羅氏の協力とオプション協定を終了するか、あるいは90日前に書面で通知することができる
F-20
Synlogic社そして付属会社
連結財務諸表付記
連れ立って。いずれの一方も他方が治癒していない実質的な違約が発生した場合,羅氏協力とオプション合意を終了することができる。
この研究と開発は会社が約
当社はASC 606に基づいてこのスケジュールを評価した顧客との契約収入から契約相手の方羅氏は顧客であると結論した。同社は,合意開始時に羅氏社に,(1)非独占的で印税免除の研究·開発許可証,(2)研究計画下の臨床前活動の研究·開発サービス,(3)羅氏社が契約を終了しないことを決定した暗黙的な契約継続オプション,(4)会社がJRCに参加すること,(5)候補製品のさらなる開発と商業化交渉について最終CLAの独占的権利を決定した。当社は、ライセンスと研究開発活動は互いに区別がなく、ライセンスは研究開発活動を行わない場合には価値が限られているためと認定している。当社のJRCへの参加は数量的にも品質的にも無関係であると判断されたため、義務履行から除外されました。そのため,同社は,ライセンスや研究開発サービスに関する承諾を単一の履行義務に統合すべきであることを決定した。
同社は次に、研究計画で定義された3つの研究段階に関する記念碑的支払い、およびCLAの交渉および締結のオプションを評価し、羅氏に実質的な権利を提供しているかどうかを決定した。同社は、このオプションは顕著かつ増分的な割引で発行されていないため、実質的な権利を提供しないと結論している。したがって、それらは最初から契約履行義務から除外された。ロ氏がオプションを行使することを選択した場合,追加の対価格は取引価格に追加され,それによって生じる履行義務に割り当てられる.
これらの評価をもとに、同社は、羅氏協力とオプション協定開始時に、(1)非独占許可と(2)研究·開発活動を含む業績義務を決定した。
手配開始時には、取引価格は$のみとなります
2021年6月、会社は第1段階の研究計画とドルの研究·開発サービスを開始した
ロ氏協力とオプション合意下での業績義務に関する収入は,研究開発サービスが生成されたフルタイム等価物に基づいて入力法を用いて提供されていることが確認された.統制権の移転は時間の経過とともに発生し、経営陣から見れば、履行義務の履行に進展する最適な測定基準である。受け取った収入が確認されていない金額は会社の総合貸借対照表に繰延収入を計上する。
同社は$を確認した
F-21
Synlogic社そして付属会社
連結財務諸表付記
イチョウ協力
2017年、同社はイチョウと技術協力を確立した。2019年6月、イチョウへの発行合計について
以下の表は、1株当たりの基本純損失と償却純損失の計算方法(1株と1株当たりの金額を除く)を示している
|
|
2022 |
|
|
2021 |
|
||
分子: |
|
|
|
|
|
|
||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
分母: |
|
|
|
|
|
|
||
加重平均発行済み普通株式-基本普通株式と希釈普通株 |
|
|
|
|
|
|
||
1株当たり純損失--基本損失と赤字 |
|
$ |
( |
) |
|
$ |
( |
) |
当社の潜在的希薄化株式は、流通株オプション、非帰属制限性普通株、ESPPによって発行可能な潜在株式を含み、普通株等価物とみなされ、その影響が希薄化効果がある場合にのみ1株当たり希薄純損失を計上する。
以下の潜在普通株は期末既発行金額で示されており、指す期間の普通株株主が1株当たりの純損失を占めるべき計算範囲には計上されておらず、これらの株式を計上することで逆償却効果が生じるからである。
|
|
12月31日まで |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
非既得性制限普通株奨励 |
|
|
|
|
|
|
||
普通株購入の未償還オプション |
|
|
|
|
|
|
||
ESPPによって発行可能な潜在株 |
|
|
|
|
|
|
||
2022年12月31日及び2021年12月31日まで、当社は記録しています
F-22
Synlogic社そして付属会社
連結財務諸表付記
繰延税項は、財務諸表の資産と負債ベースと所得税との一時的な差異で確認されている。繰延税金資産は以下の各項目からなる(千計)
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
繰延税金資産: |
|
|
|
|
|
|
||
純営業損失が繰り越す |
|
$ |
|
|
$ |
|
||
税金の繰り越しを免除する |
|
|
|
|
|
|
||
費用を計算する |
|
|
|
|
|
|
||
財産と設備 |
|
|
|
|
|
|
||
賃貸負債 |
|
|
|
|
|
|
||
持分補償 |
|
|
|
|
|
|
||
無形資産の償却が可能である |
|
|
|
|
|
|
||
研究支出を償却できる(1) |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
繰延税項目総資産 |
|
|
|
|
|
|
||
繰延税金負債: |
|
|
|
|
|
|
||
使用権資産 |
|
|
( |
) |
|
|
( |
) |
繰延税金負債総額 |
|
|
( |
) |
|
|
( |
) |
推定免税額 |
|
|
( |
) |
|
|
( |
) |
繰延税項目純資産 |
|
$ |
— |
|
|
$ |
— |
|
(1) “減税·雇用法案”(TCJA)によると、研究·実験(R&D)支出は2021年12月31日以降の納税年度は第174条に基づいて資本化·償却を行う。これらのコストは研究開発が米国で行われて以来、税務目的で5年以内に償却されてきた。これらのコストの未償却残高は上の表に繰延税金資産として示されている。
当社の経営陣はすでに当社の繰延税金資産の現金化に影響を与えるプラスとマイナスの証拠を評価しており、このような繰延税金資産は主に繰延純営業損失からなり、当社は更に繰延税金資産の利益を確認しない可能性があることを確定した。そのため、全額推定手当は約$
法定連邦所得税率と会社の有効所得税率の入金は以下のとおりである
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
|
|
税率.税率 |
|
|
税率.税率 |
|
||
アメリカ連邦法定金利 |
|
|
% |
|
|
% |
||
連邦福祉を差し引いた州所得税 |
|
|
% |
|
|
% |
||
他の恒久的差異 |
|
|
( |
)% |
|
|
% |
|
税金控除 |
|
|
% |
|
|
% |
||
その他のプロジェクト |
|
|
% |
|
|
% |
||
評価免税額の純変動 |
|
|
( |
)% |
|
|
( |
)% |
有効所得税率 |
|
|
— |
|
|
|
— |
|
12月31日の終了年度の推定免税額を前転した2022年と2021年の状況は以下の通り(単位:千):
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
年初残高 |
|
$ |
( |
) |
|
$ |
( |
) |
評価免税額を引き上げる |
|
|
( |
) |
|
|
( |
) |
年末残高 |
|
$ |
( |
) |
|
$ |
( |
) |
F-23
Synlogic社そして付属会社
連結財務諸表付記
2022年12月31日まで会社は連邦純営業損失の繰越があり、将来の課税所得額を減らすことができます$
2022年8月16日、総裁·バイデンは2022年インフレ削減法案に署名し、法律にした。その中に含まれる条項は
1986年国税法(IRC)第382条によると、会社の所有権のいくつかの重大な変化は将来の年度に使用可能な純営業損失(NOL)の繰越と研究開発信用(R&D信用)の繰越金額の制限を招く可能性がある。以前に発生または将来発生する可能性のある所有権変更制限により、IRC第382条によると、NOLと研究開発信用繰越の使用はかなりの年間制限を受ける可能性がある。これらの所有権の変化は、将来の課税収入と税収を相殺するために毎年使用できるNOLと研究開発信用繰越金額を制限する可能性がある。当社では,所有権変更が発生したかどうかを評価する研究や,設立以来何度も所有権変更が発生しているかどうかを評価する研究が完了していないが,このような研究の重大な複雑さと関連コストによるものである.将来的にはより多くの所有権変化がある可能性があり、これはNOL繰越および信用の利用に追加的な制限をもたらす可能性がある。
会社は審査後に会社の税務状況がより持続可能であるかどうかを決定することを要求され、任意の関連訴訟手続を解決することを含め、控訴に基づいている このポストの技術的利点について。より課税点に達する可能性のある税務ヘッドについては、財務諸表で確認された税額から、最終的に関連税務機関と和解した場合に50%以上の最大利益を達成する可能性がある。その会社は所有している
会社はその経営管轄区の税法の規定に従って、実体レベルで納税申告書を提出する
(13)株式買い戻し
当社は2022年11月25日に、私的に協議した買い戻し取引の一部として、一人の株主と最終株式買い戻し契約を締結した
買い戻し株は、退職または再発行まで原価価格で在庫株として保有される。2022年12月31日までの年度内に、在庫株の退役や再発行はない。
賃貸借契約を経営する
二零一七年七月、当社は契約を結び、賃貸契約を結びました
F-24
Synlogic社そして付属会社
連結財務諸表付記
はい現金制限があります。私たちの運営費用部分によると、不動産税と保険が含まれており、可変支払いは発生時に期間費用として記録されています。その会社は期限を延長する権利がある
2018年12月31日までの年度中に、当社はAzzur Group,LLC(Azzur)と合意(初のSOW)を締結し、これによりAzzurは当社に約を提供することに同意した
経営リースに分類されるリースには、当社の総合貸借対照表における経営リースROU資産、当期経営賃貸負債、非流動経営賃貸負債が含まれています。経営リース、使用権、資産、経営賃貸負債はビンニー街レンタルとAzzur Suiteレンタルです。レンタル負債の現在価値を計上するために支払う現金は#ドルである
2022年12月31日までと2021年12月31日までの経営リースコスト構成は以下の通り(千計)
|
12月31日までの年度 |
|
|||||
賃貸借契約を経営する |
2022 |
|
|
2021 |
|
||
リースコストを経営する |
$ |
|
|
$ |
|
||
可変リースコスト |
|
|
|
|
|
||
総賃貸コスト |
$ |
|
|
$ |
|
||
経営リースの使用権資産は合併貸借対照表に開示されている。
経営リースの加重平均残存期間と加重平均割引率は、
|
12月31日までの年度 |
|
|||||
|
2022 |
|
|
2021 |
|
||
加重平均割引率 |
|
% |
|
|
% |
||
加重平均残存賃貸年限(年) |
|
|
|
|
|
||
F-25
Synlogic社そして付属会社
連結財務諸表付記
次の表は、2022年12月31日までの経営リースの未割引キャッシュフローと貸借対照表に記録されている経営リース負債を照合します
賃貸負債満期日 |
賃貸借契約を経営する |
|
|
|
(単位:千) |
|
|
2023 |
$ |
|
|
2024 |
|
|
|
2025 |
|
|
|
2026 |
|
|
|
2027 |
|
|
|
その後… |
|
|
|
賃貸支払総額 |
|
|
|
差し引く:推定利息 |
|
|
|
リース総負債 |
$ |
|
|
流動賃貸負債 |
|
|
|
長期賃貸負債 |
|
|
|
2022年12月31日および2021年12月31日までの年度の融資リースコストおよび2022年12月31日までの融資リース負債はいずれも重大ではない。
正常業務過程において、会社が経営する業界は特許法律要求の影響を受けやすいため、会社は法的訴訟、クレーム、訴訟の影響を受ける可能性がある。法律訴訟やクレームの推定損失が可能かつ推定可能な場合、当社はこのような損失を会計処理します。これらの事項に関連する法的費用は発生時に費用を計上する。当社は現在、いかなる重大な法的手続きの当事者でもありません。
同社は条件を満たす従業員のための固定拠出金401(K)計画を策定した。従業員は雇われた日から計画に参加する資格がある。この計画の条項によると、従業員は補償の割合で自発的に支払いを行うことができる。2019年1月1日から、会社は従業員に合わせて支払いを開始します。会社はそれにマッチしている
2019年6月、同社はイチョウとの協力を拡大し、工学微生物治療製品の研究開発について合意した。2022年12月31日現在、イチョウ所有
協定によると、会社は銀杏に#ドルの前金を支払った
F-26