
アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
本財政年度末まで
あるいは…。
1934年証券取引法第13条又は15(D)条に基づいて提出された |
手数料書類番号
(登録者の正確な氏名はその定款に記載)
(明またはその他の司法管轄権 会社や組織) |
(税務署の雇用主 識別番号) |
(主にオフィスアドレスを実行) |
(郵便番号) |
登録者の電話番号、市外局番を含む:(
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
|
取引 記号 |
|
登録された各取引所の名称 |
|
|
同法第12条(G)により登録された証券:なし
登録者が証券法第405条に規定する有名な経験豊富な発行者である場合は、再選択マークで表示してくださいはい、そうです ☐
登録者がこの法第13又は15(D)条に従って報告書を提出する必要がないか否かを、再選択マークで示すはい、そうです ☐
登録者が(1)過去12ヶ月以内(または登録者がそのような報告を提出する必要があるより短い期間)に1934年の証券取引法第13条または15(D)条に提出されたすべての報告書を再選択マークで示すかどうか、および(2)このような提出要求を過去90日以内に遵守してきた
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
大型加速ファイルサーバ |
|
☐ |
|
ファイルマネージャを加速する |
|
☐ |
|
☒ |
|
規模の小さい報告会社 |
|
||
|
|
|
|
新興成長型会社 |
|
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オキシリー法案”(“米国連邦法典”第15編7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性の評価が、その監査報告を準備または発表する公認会計士事務所によって提出されたことを証明する左舷です
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する☐
これらのエラーのより真ん中に登録者の任意のエンタルピーCER幹部が相関回復期間内に§240.10 D−1(B)に基づいて受信したインセンティブベースの補償に基づいて回復分析を行う必要があるかどうかを再選択マークで示す☐
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)はい、そうです ☐ 違います。
2022年6月30日まで,すなわち登録者が最近完成した第2財期の最終営業日であり,登録者非関連会社が保有する登録者普通株の総時価は約$である
2023年3月21日現在登録者は
引用で編入された書類
登録者は、その2023年株主総会で作成された委託書の一部が、本年度報告の第III部分に引用的に組み込まれており、表10−Kは、本明細書に記載された範囲内である。このような依頼書は,登録者が2022年12月31日までの財政年度の120日以内に米国証券取引委員会に提出される。
カタログ表
|
|
ページ |
第1部 |
|
|
第1項。 |
業務.業務 |
5 |
第1 A項。 |
リスク要因 |
61 |
項目1 B。 |
未解決従業員意見 |
118 |
第二項です。 |
属性 |
118 |
第三項です。 |
法律訴訟 |
118 |
第四項です。 |
炭鉱安全情報開示 |
118 |
|
|
|
第II部 |
|
|
五番目です。 |
登録者普通株市場、関連株主事項及び発行者による株式証券の購入 |
119 |
第六項です。 |
[保留されている] |
121 |
第七項。 |
経営陣の財務状況と経営成果の検討と分析 |
122 |
第七A項。 |
市場リスクの定量的·定性的開示について |
136 |
第八項です。 |
財務諸表と補足データ |
136 |
第九項です。 |
会計と財務情報開示の変更と相違 |
136 |
第9条。 |
制御とプログラム |
136 |
プロジェクト9 B。 |
その他の情報 |
137 |
プロジェクト9 Cです。 |
検査妨害に関する外国司法管区の開示 |
137 |
|
|
|
第三部 |
|
|
第10項。 |
役員·幹部と会社の管理 |
138 |
第十一項。 |
役員報酬 |
138 |
第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
138 |
十三項。 |
特定の関係や関連取引、取締役の独立性 |
138 |
14項です。 |
最高料金とサービス |
138 |
|
|
|
第4部 |
|
|
第十五項。 |
展示·財務諸表明細書 |
139 |
第十六項。 |
表格10-Kの概要 |
140 |
|
サイン |
141 |
i
前向き陳述に関する警告説明
このForm 10-K年次報告書には、当社の業務、運営および財務業績および状況に関する前向きな陳述と、業務、運営および財務業績および状況に対する私たちの計画、目標、および期待が含まれています。本明細書に含まれる任意の非歴史的事実の陳述は、前向きな陳述と見なすことができる。これらの表現は、既知および未知のリスク、および場合によっては私たちの制御範囲を超える他の重要な要素に関連し、私たちの実際の結果、業績または成果は、展望性表現に明示または示唆された任意の未来の結果、業績または業績と大きく異なる可能性がある。
文意が別に指摘されているほか、本年度報告で言及した“Pyxis Oncology”、“Company”、“We”、“Us”、“Our”はいずれもPyxis Oncology,Inc.とその子会社を指す。場合によっては、“可能”、“会議”、“すべき”、“予想”、“計画”、“予想”、“可能”、“意図”、“目標”、“プロジェクト”、“考慮”、“信じ”、“推定”、“予測”、“展望”、“潜在的”または“継続”またはこれらの用語の否定または他の同様の表現によって識別することができる。本年度報告におけるForm 10−Kに関する前向き記述は予測のみであった。これらの展望的な陳述は主に私たちの現在の未来の事件と財務傾向の予想と予測に基づいており、私たちはこれらの事件と財務傾向が私たちの業務、財務状況と経営結果に影響を与える可能性があると考えている。これらの前向き陳述は,本Form 10−K年次報告発表日にのみ発表され,“リスク要因”と題する節および本Form 10−K年次報告の他の部分で述べた一連のリスク,不確実性,仮説の影響を受ける可能性がある。展望性陳述は、リスクおよび不確実性の影響を固有に受けているので、いくつかのリスクおよび不確実性は予測または定量化できないので、未来のイベントの予測として、これらの前向き陳述に依存してはならない。著者らの展望性陳述に反映された事件と状況は実現できない或いは発生できない可能性があり、実際の結果は展望性陳述中の予測結果と大きく異なる可能性がある。実際の結果が私たちの予想と異なる可能性があるいくつかの重要な要素は
また、“私たちは信じる”のような声明や似たような声明は、関連テーマに対する私たちの信念と意見を反映している。これらの陳述は、本年度報告10-K表までの日に我々に提供された情報に基づいており、これらの情報は、このような陳述の合理的な基礎を構成していると考えられるが、このような情報は、限定的または不完全である可能性があり、私たちの陳述は、入手可能なすべての関連情報を徹底的に調査または検討したことを示すものとして解釈されてはならない。しかも、もし私たちの展望的な陳述が不正確であることが証明されたら、この不正確さは実質的である可能性がある。このような展望的陳述の重大な不確実性を考慮して、あなたはこれらの陳述を私たちまたは任意の特定の時間枠内で、または私たちの目標と計画の陳述または保証を完全に達成しないために、私たちまたは他の誰もと見なしてはならない。法的要件が適用されない限り、私たちは、任意の新しい情報、未来のイベント、または他の理由による、本明細書に含まれる任意の前向きな陳述を公開または修正するつもりはありません。
2
リスク要因をまとめる
本10-K表年次報告第II部第1 A項の“リスク要因”項で後述するリスクを慎重に考慮しなければならない。“概要リスク要因”というタイトルのこの節で言及されている“Pyxis Oncology”,“Company”,“We”,“Us”,“Our”はPyxis Oncology,Inc.とその完全子会社を指す.私たちの業務、財務状況、経営業績と将来性に重大な悪影響を及ぼす可能性のあるリスクの概要は以下の通りです
3
4
パー?パーT I
プロジェクト1.ビジネス
概要
我々は臨床段階の腫瘍学会社であり,次世代治療薬のマルチモード組合せの開発に専念し,治療困難な癌に対して患者の生活の質を向上させる。我々が開発した候補製品の目標は、腫瘍細胞を直接殺し、癌が制御できない増殖と免疫逃避を引き起こす潜在的な病理を解決することである。マルチモードは様々な技術であり,独立しても他の技術と組み合わせても,患者のために有効な癌治療法を確立すると考えられる。2019年の発売以来,新規抗体薬物結合体(ADC),候補品,免疫腫瘍学(IO),候補品,モノクロナル抗体(MAb)を含む幅広い組み合わせが開発されており,単一療法として他の療法と組み合わせて使用する臨床前発見計画を開発している。
著者らは腫瘍学の研究開発において豊富な経験と熟練度を持つチームを結成し、著者らのチームの数十年の薬物開発経験を利用して、各種の革新療法を臨床に連れて行く予定である。私たちのグループの集団体験は製薬とバイオテクノロジーの二つの分野を扱っている。
マルチモード方法の多様化は、患者に利益をもたらすために多様な資産を有効に発展させる能力を最適化したと信じている。腫瘍微小環境やTMEに対する我々の専門知識と既定の業務発展記録を利用することにより、様々な経路(図1参照)を通じて癌療法と技術を開発している
5
図1
私たちのポートフォリオは
私たちの製品組み合わせにはADCとIO候補製品が含まれています。私たちのパイプラインは各項目の間でバランスを維持し、固形腫瘍に重点を置いている。我々は2021年3月にファイザーから2つのADC計画の許可を得、2022年3月にBiosion USA,Inc.からIO計画の許可を得た。Thomas Gajesski博士の研究室で働いている追加の臨床前mAb発見計画もあります。我々はすべての候補製品の完全な全世界開発権と商業化権利を保持しているが、大中華区の中国(大陸、香港、マカオ、台湾)のPYX-106は除く。2022年12月、米国食品医薬品局(FDA)は、我々が調査したPYX−201およびPYX−106の新薬申請(IND)を承認したと発表した。われわれは臨床段階の腫瘍学会社への転換に熱心であり,われわれのチームの経験を利用してわれわれの臨床試験を実行することを期待し,患者により安全かつ効率的な治療選択を提供することを目的としている。
以下は私たちの現在の臨床開発の流れです
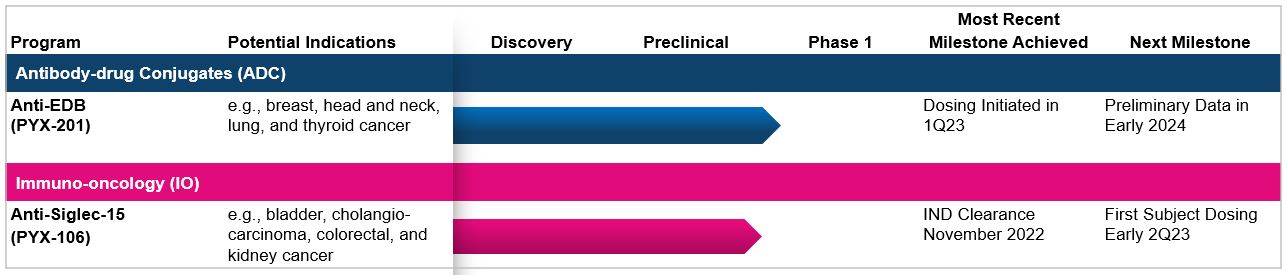
6
PYX-201
我々の主要なADC候補は、免疫グロブリンG 1(またはIgG 1)、抗フィブロネクチン外ドメインB(EDB)、mAbによってカテプシンBを介して黄金色誘導体に切断結合された研究中の新規ADCであるPYX-201である。フィブロネクチンは細胞外基質に存在する糖タンパク質である。フィブロネクチンEDBは血管形態発生を調節し、腫瘍に栄養と酸素を提供し、廃棄物を除去する手段、及び細胞を転移させる経路を提供する。EDBは多くの悪性腫瘍の中で過剰発現し、大多数の正常成人組織の中で最も発現が低く、これはそれを潜在的に魅力的な腫瘍を標的とする手段になり、同時に健康細胞を保留する。患者由来の異種移植やPDXモデルの臨床前モデルでは,単一薬物PYX−201を用いた腫瘍消退を認めた。また,臨床前同遺伝子腫瘍モデルをPYX−201で治療したところ,TMEにおけるT細胞の浸透が増加し,免疫原性細胞死(ICD)のマーカーであり,検査点阻害剤と併用することで相乗活性が得られることが観察された。
2022年12月、FDAは、臨床試験を開始するために、PYX-201のためのINDの使用を承認すると発表しました。2023年第1四半期に、PYX−201第1段階試験の第1被験者の用量をPYX−201−101と呼ぶことを発表した。PYX-201-101は開放ラベル、多中心、用量増加試験であり、PYX-201の安全性、耐性、薬物動態学、薬効学と初歩的な治療効果を評価し、そして更なる研究の推奨投与量を決定することを目的としている。再発或いは難治性固形腫瘍患者は、非小細胞肺癌、ホルモン受容体陽性乳癌、卵巣癌、甲状腺癌、膵臓導管腺癌或いはPDAC、軟組織肉腫或いはSTS、肝細胞癌或いは肝細胞癌及び腎臓癌を含み、すべて参加する資格がある。我々は,バイオマーカーの結果と潜在的な臨床活動の早期兆候を含む2024年初めにこの段階の臨床試験の初歩的なデータを報告する予定である。
PYX-106
我々の主要なIO候補製品はPYX−106であり,これは研究中の完全ヒトIgG 1ホモSiglec−15標的抗体であり,Siglec−15を介したT細胞増殖と機能抑制を遮断することを目的としている。私たちは最初にこの資産を固形腫瘍の治療に開発する予定です。我々はBiosion USA,Inc.から大中華区中国(大陸,香港,マカオ,台湾)を除く世界的にPYX-106の許可を得た.
2022年12月、FDAは、臨床試験を開始するために、PYX-106のためのINDの使用を承認することを発表しました。2023年第1四半期に、私たちは臨床試験サイトの活性化を開始し、現在患者をスクリーニングして第1段階試験、すなわちPYX-106-101に参加している。PYX-106-101はヒト初例1期多中心開放ラベル用量増加試験であり、再発或いは難治性固形腫瘍患者におけるPYX-106の安全性、耐性、薬物動態学、薬効学と初歩的な治療効果を評価することを目的としている。この第1段階用量増加試験は、標準治療によって固形腫瘍と疾病の進展に発展した患者と、生存を獲得できない或いは延長する標準看護治療に適していない患者を含む。この試験は、非小細胞肺癌、乳癌、子宮内膜癌、甲状腺癌、腎臓癌、胆管癌、膀胱癌、結腸直腸癌、頭頸部扁平上皮癌を含む固形腫瘍患者を募集する。私たちは2023年第2四半期初めに患者に薬を投与する予定だ。バイオマーカーの結果と潜在的な臨床活動の早期兆候を含む2023年末にこの第1段階臨床試験の初歩的なデータを報告する予定である。
一時停止/終了の手順
2022年8月、抗CD 123 ADC PYX−203および抗KLRG 1 IOプログラムPYX−102の臨床前開発を休止した。これらの計画は著者らの多様な候補治療方案の組み合わせに依然として重要であるが、現在臨床前開発を一時停止することは、PYX-201とPYX-106の臨床開発を推進するための資源を増加させることができる。さらに、我々は、臨床前毒性研究データの審査および分析、ならびにPYX−202の期待される臨床用途および商業的将来性に基づく、抗DLK 1 ADC PYX−202の継続開発を一時停止することにした。
発見
以上の決定した計画に加えて,モノクロナル抗体,ADC結合,リンカーとペイロード選択および免疫腫瘍学の理解に関する専門知識を用いて,様々な目標の研究·開発を行っている。
7
私たちの戦略
我々の目標は,ADCやモノクロナル抗体免疫療法を含めた優れたバイオマスの組み合わせを構築することにより,難治性癌患者の生活を改善することである。
短期的で長期的な目標を達成するための戦略的要素は
8
腫瘍学では満足されていない需要
いくつかの新薬の承認に伴い、腫瘍学は意義のある進展を得たが、新しい治療方法に対する医療需要はまだ満足されていない。世界保健機関のデータによると、癌は世界2位の死亡原因であり、2020年の死亡者数は1000万人に近い。現在の腫瘍治療の主要な局限性は高毒性、低応答率或いは有限応答率及び再発或いは再発を含む。
化学療法は依然として癌の最もよく見られる治療方法の一つであり、腫瘍の分期とタイプによって、化学療法は通常手術と放射線治療と結合する。癌治療発展における1つの主要な挑戦は腫瘍全体の複雑性と異質性だけでなく、その動態周囲微小環境の複雑性と異質性である。最近の治療法の進展、例えば標的癌発生の特定の腫瘍突然変異或いは患者の免疫系を変化させて腫瘍を除去し、これらの挑戦を解決し始めているが、それらの重点は主に腫瘍細胞である。標的TMEは腫瘍進展、成長と多剤耐性を推進する上で重要な役割を果たしており、腫瘍学で満足されていない需要を解決する新しい方法であると考えられている。例えば,免疫チェックポイント阻害剤の開発は多くの癌の治療パターンを変化させているが,これらの治療に有効な多くの患者は最終的に薬剤耐性を生じ,疾患進展を経験する。TMEの多くの特徴は免疫チェックポイント阻害剤に対する反応と抵抗力に影響することが証明されており、TMEを標的とすることはこれらの制限を克服する可能性がある。我々の開発は,TME生物学に対する我々の深い理解を利用して,特定の部位の結合とカスタマイズされたリンカー−ペイロードの組み合わせ,TME内で発見された適応性と先天性免疫系に対するキーレギュレーターの免疫療法の設計と開発を目指している。
私たちのADC候補品
PYX−201:胎児フィブロネクチンEDBに対する定点研究ADC
概要
我々のADC PYX-201は、非小細胞肺癌、ホルモン受容体陽性乳癌、卵巣癌、甲状腺癌、PDAC、STS、肝癌および腎臓癌を含む固形腫瘍のために、我々が最初に開発したEDBに対する黄金毒素に結合した研究におけるヒトIgG 1アイソフォーム部位である。我々はファイザー社からFACTプラットフォーム上に設立されたPYX−201のグローバルライセンスを取得した。2022年12月、FDAは、臨床試験を開始するために、PYX-201のためのINDの使用を承認すると発表しました。2023年第1四半期に、PYX−201第1段階試験の第1被験者の用量をPYX−201−101と呼ぶことを発表した。PYX-201-101は開放ラベル、多中心、用量増加試験であり、PYX-201の安全性、耐性、薬物動態学、薬効学と初歩的な治療効果を評価し、そして更なる研究の推奨投与量を決定することを目的としている。再発或いは難治性固形腫瘍患者は、非小細胞肺癌、ホルモン受容体陽性乳癌、卵巣癌、甲状腺癌、PDAC、STS、肝癌と腎臓癌を含み、すべて参加する資格がある。
EDB標的の原理とPYX−201の作用機序
フィブロネクチンは細胞外基質の1種の成分であり、その下流シグナル経路は細胞の粘着、遷移、分化と傷口癒合を調節する。EDBはフィブロネクチンの選択的スプライシング形式であり,RNAが再配列して同一のタンパク質の複数の変異体を産生すると,このようなことが起こる。EDBは通常胚発育過程でのみスプライシングされ,健康な成人組織ではあまり発見されない。しかし、癌細胞はEDBを利用して新生血管構造を促進する能力であり、新生血管構造は腫瘍が制御されない成長を栄養と支持するキーポイントである。
EDBは、肺癌、乳癌、卵巣癌、膵臓癌、頭頸部癌、甲状腺癌および脳癌を含むが、これらに限定されない多種類の癌において過剰発現される(図2および図3参照)離体するEDBの引き下げは癌活動の著しい減少を招くことが明らかになった。また,ヒト癌の遠隔転移ではEDBの発現は不変であった。腫瘍組織におけるEDBの厳格な優先発現、及びそれが多くの癌において予後不良な駆動要素のため、EDBは著者らが高度に期待しているADC標的の標準に符合する。
9
図2

EDBは多種の正常組織発現が制限された固形腫瘍において高発現(IHC:免疫組織化学;mRNA:メッセンジャーRNA;TCGA:癌ゲノムマップ門戸;GTEx:遺伝子-組織発現ポータル)。
10
図3
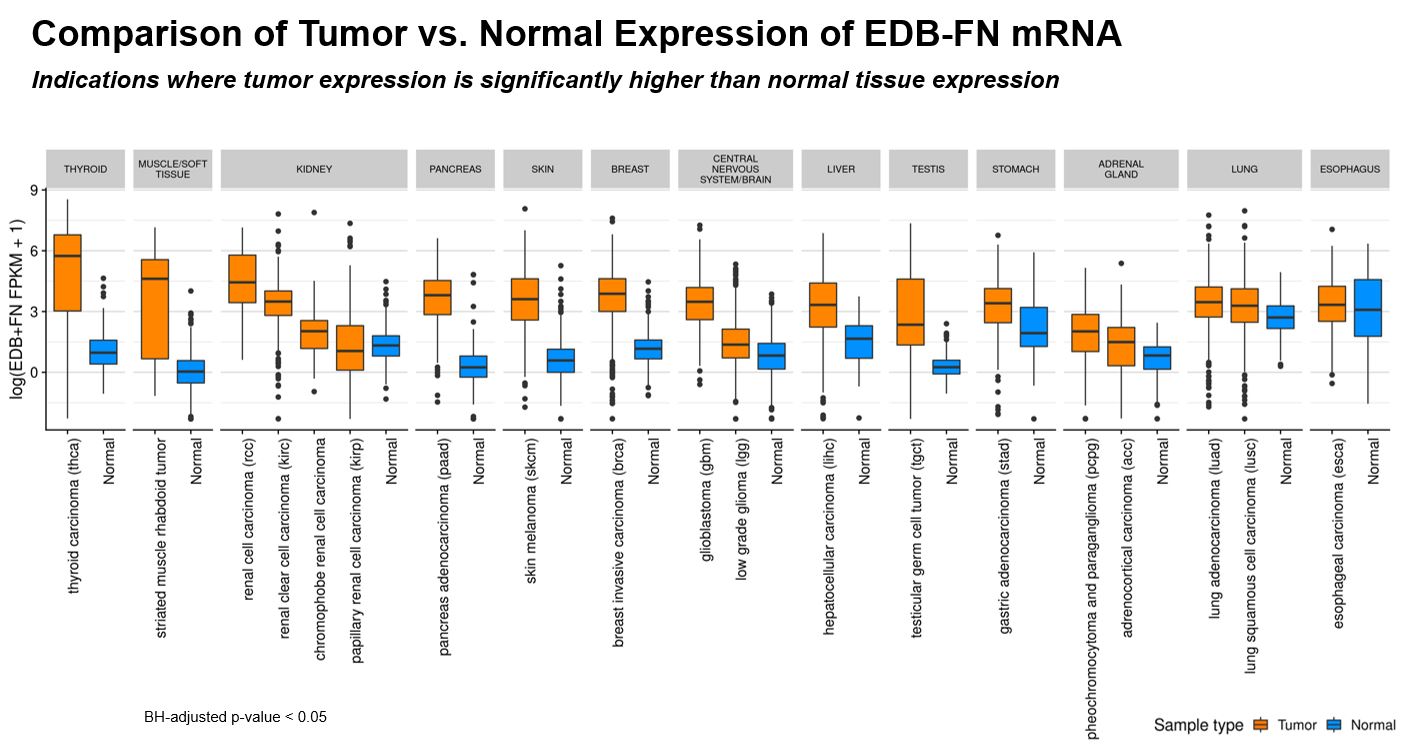
腫瘍と一致した正常サンプル中の差異発現の統計学的有意差が閾値を超える適応を比較した。
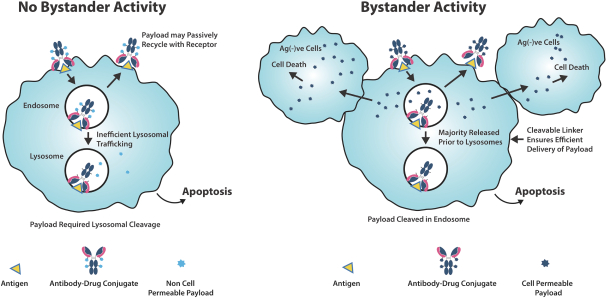
PYX−201はFACTプラットフォームを用いて開発され,高度に安定したADCの生産を目指し,予測可能な薬物対抗体比(DAR)が4を目標としている。PYX−201で使用されているEDB抗体の相補決定領域またはCDRは,EDBへの結合を担う抗体の一部であり,良好な特性を有し,腫瘍イメージングのための放射性結合抗体の形態で臨床試験が行われており−高い腫瘍配向特異性を示している。さらに、PYX−201は、ADCを癌細胞に内在化することなく黄金色素ペイロードを伝達することができるように、接合安定性を最適化することを目的としている。PYX-201は、現在承認されている腫瘍細胞表面に結合するADCとは異なり、腫瘍細胞、間質細胞、および周囲血管からなるTMEに黄金色薬物ペイロードを送達することを意図している。PYX-201は、以下の図4に示すように、3つの異なる作用パターンによって抗腫瘍活性を示すことを意図している。フィブロネクチンの更新により、PYX-201は、コネキシンの細胞外溶解の前に内化され、腫瘍細胞におけるペイロードの放出をもたらす可能性がある。
以上より,PYX−201は癌細胞を直接死滅させ,TMEを調節し,抗腫瘍免疫反応を動員することにより,治療困難な癌に対して多管的な攻撃を生じる可能性が考えられる。
11
図4

PYX-201は周囲基質中のフィブロネクチンEDBに結合し、直接ペイロードによって細胞死、傍観者効果を誘導し、免疫原性細胞死をトリガすることによって腫瘍細胞と支持インフラを殺し、臨床前モデルのデータに基づくことを目的としている。
臨床前発展
PYX−201は有望な臨床前結果を示している。前臨床研究では強いことが観察されました体内にあるNSCLC,PDXとEMT−6同遺伝子マウス乳癌モデルにおける活性。PDXマウスモデルは患者由来の細胞を免疫不全マウスに移植することによって産生され、同遺伝子マウスモデルはマウス由来の腫瘍を移植し、免疫系を完全に維持する。そのため、PDXモデルは臨床前環境の中で最も臨床翻訳可能な治療効果シグナルを提供したが、同遺伝子モデルはPYX-201が免疫反応を産生する能力を評価することを許可した。これらの同遺伝子モデルでは,PYX−201が癌に有効に局在することが証明されており,腫瘍負担を有意に低下させるだけでなく,抗腫瘍免疫反応を動員することも可能である。
例えば,非小細胞肺癌のPDXモデルでは,PYX−201を4日ごとに12日間静注すると,腫瘍負荷は用量依存的に消退し,3 mg/kgで持続的な反応が認められた(図5)。
12
図5
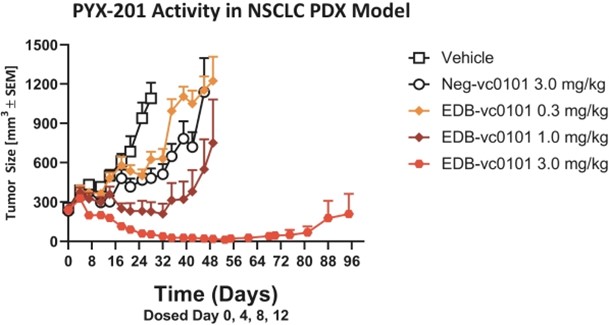
PYX−201は非小細胞肺癌のPDXモデルにおいて高い活性を有することが証明されている。
PYX−201で使用した抗EDBヒト由来モノマブはマウスEDB−フィブロネクチンと交差反応した。そこで,免疫能の良好なマウスで行った同種腫瘍モデルでは,PYX−201は単回用量9 mg/kgで持続的な反応が得られた(図6)。臨床前研究ではCD 3 T細胞の浸潤増加とPD−L 1の上昇が認められ,PYX−201が免疫原性細胞死を誘導する可能性が示唆された。EMT-6モデルでは、二次用量のPYX-201をチェックポイント治療と結合し、協同で腫瘍成長を抑制することができる。したがって,PYX−201は図7に示すように検査点阻害剤と協同作用すると信じられており,PYX−201はわれわれのマウスモデルやラットやカニクイザルにおける毒理学研究においても良好な耐性を有している。カニクイザルに対する探索的毒理学研究では,PYX−201を3週間に3回注射した最高非重篤毒性用量(HNSTD)が12 mg/kgより大きいことが発見された。体重や食物消費量の差は検出されず,観察された毒性タイプ(すなわち線維化,神経病理などはない)により,すべての毒性が可逆的であるか,あるいは可逆的であると予想される。PYX−201の臨床前関連治療指数は16(HNSTDはサルで144 mg/m)が認められた2 9 mg/mのマウスの完全反応に必要な投与量より16倍大きい2)は、異なるADC構造間の相対治療指数を検討した経験から、将来性があると考えられる。
13
図6

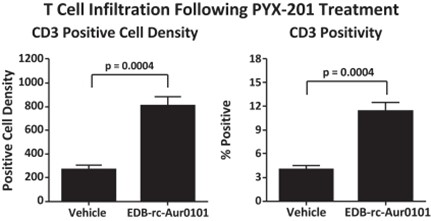
PYX−201体内治療同遺伝子細胞由来の腫瘍モデルはCD 3陽性増加によるT細胞浸潤増加に関与している。(CR:完全応答;rcEDB:逆キメラEDB)
図7

EDB vc 0101 ADCは抗PD-L 1と連合してEMT 6マウスと遺伝子モデルにおいて協同的に腫瘍成長を減少させた。
14
臨床発展計画
2023年第1四半期に、著者らは再発或いは難治性固形腫瘍患者に対するPYX-201の安全性、耐性、薬物動態学、薬効学と初歩的な治療効果を評価し、更なる研究の推奨投与量を決定するために、第1段階、多中心、開放ラベルの投与量増加試験を開始した。この第一段階用量増加試験は、標準治療を通じて固形腫瘍に発展し、疾病の進展がある患者と、生存を獲得できない或いは延長する標準看護治療に適していない患者を含む。この試験は非小細胞肺癌、ホルモン受容体陽性乳癌、卵巣癌、甲状腺癌、PDAC、STS、肝細胞癌、腎臓癌を含む固形腫瘍患者を募集する。
図8に示すように、この試験は、PYX-201の安全性および耐性を調べ、待ち行列拡張のための生物活性用量を決定し、最大耐性用量を決定するために、第1段階単一療法用量増分試験として設計されている。我々は、選択された腫瘍タイプの個々の拡張キューにおいて評価される推奨用量を決定するために、非小細胞肺癌、乳癌、および他のEDB発現頻度の高い腫瘍タイプの患者を最初に募集する予定である。将来の研究では,併用療法としてPYX−201の開発を継続し,看護基準を適切に採用する可能性がある。例えば、非小細胞肺癌および乳癌では、免疫療法は第一線および二線環境において広く使用されており、PYX−201は、臨床前研究においてAuristatinペイロードが免疫原性細胞死をトリガするマーカーが観察されたので、相乗的治療利益を提供する可能性がある。この臨床試験に関する情報は,ClinicalTrials.gov(NCT 05720117)にアクセスしてください。
図8

我々は,バイオマーカーの結果と潜在的な臨床活動の早期兆候を含む2024年初めにこの段階の臨床試験の初歩的なデータを報告する予定である。
15
免疫腫瘍学プロジェクトと目標カタログの概要
現在の免疫腫瘍学的治療の背景
免疫腫瘍学的療法の出現,特に免疫チェックポイント阻害剤の出現は,腫瘍学的治療様式を変えた。免疫系は癌を識別と除去する能力があるが、腫瘍細胞は通常自己免疫を阻止する免疫チェックポイント経路を利用して免疫効果細胞の活動を抑制と逃避する。PD-L 1と細胞毒性Tリンパ球関連蛋白4或いはCTLA-4阻害剤を含むこれらの経路を第一世代に遮断する薬物は、いくつかの患者において持続的な反応を実現できるため、極めて大きな情熱を引き起こした。これらの薬剤は持続的な応答者に有意な治療メリットを提供しているが,多くの患者では応答率が低く,特にT細胞レベルの低い腫瘍である。これらの非炎症性(即ち“冷”)腫瘍はTME内の各種機序を通じて獲得性免疫反応を抑制することができる。
目標カタログの概要
私たちは、Thomas Gajesski博士の実験室で免疫治療標的の仕事を行うために、私たち自身の発見活動とシカゴ大学の独占許可を介して、巨大な独自の標的カタログを持っている。著者らはまたいくつかのヒト腫瘍データベースを利用して、大型の“冷”腫瘍標的発見データベースを構築した。
ターゲットディレクトリは、外部からの体内にあるマウスモデルシステムは、T細胞がサイトカインIL−2を産生する能力に基づいて腫瘍組織の機能や機能障害を検査するT細胞である。また,4−1 BBとLAG 3陽性T細胞はIL−2を分泌しないため,CD 8+T細胞は細胞表面マーカーの発現によって分類される4−1 BBとLAG 3であり,さらに機能や機能障害のT細胞を定義している。遺伝子発現分析は機能障害の細胞の中で上昇した細胞表面分子を発見し、その中にPD 1、CTLA 4とTIM 3などの成熟したマーカーを含み、同時に多くの他の新しい標的を発見し、著者らは生物情報学と深い生物学原理に基づいてその中の1つのサブセットを優先的に選択して発見研究を行った。
著者らの冷腫瘍標的発見データベースはヒト腫瘍データベースのRNA-seq転写群分析を使用して潜在的な新しい標的を識別し、これらの新しい標的はT細胞機能の調節及び/又は冷腫瘍の浸透を招くことを含む。私たちはこのデータベースをより多くの資源で補充しました。私たちは引き続きこれらの資源を発掘して、免疫調節のためのより多くの新しい標的を決定します。これらの冷腫瘍標的は潜在的な主要な免疫抑制因子であり、多種の腫瘍関連細胞に発現し、免疫細胞、腫瘍細胞と間質を含み、新しいIO機序の発見に可能性を提供するだけでなく、著者らのADCプラットフォームに更に多くの新しい標的を提供した。
免疫腫瘍学プロジェクトの概要
著者らはすでに複数のモノクロナル抗体計画を開始し、TME内の重要な免疫調節経路を解決し、他の潜在的な標的を探索している。著者らの計画は、マクロファージ、T細胞およびナチュラルキラー細胞またはNK細胞などの重要な腫瘍浸潤性免疫細胞集団に対して、腫瘍の成長と転移を制限する上で重要な役割を果たしている可能性がある。特定の細胞タイプを抽出するほか,IO計画はT細胞枯渇の機序やT細胞やNK細胞に対するTMEの免疫抑制効果にも関与していると考えられる。これらの効果は,TME内の腫瘍抗原への慢性曝露や,免疫抑制サイトカインや他のシグナル分子への直接曝露による可能性がある。PD−L 1療法はT細胞の立て直しに役立つが,T細胞の枯渇を克服するには不十分であることが多く,多くの患者は標準的なチェックポイント療法に反応しない。いくつかの免疫抑制経路もT細胞活性と他の抗腫瘍免疫反応の重要な成分、例えばNK細胞が直接腫瘍を殺傷することを直接制限することができる。また,T細胞やNK細胞に対してこれらの経路を相殺する療法では,単独でも検査点阻害剤との併用でも免疫エフェクター機能の回復が期待できる。また、MDSCs/TAMsはTMEのもう一つの主要成分であり、顕著な免疫抑制活性を有する。これらの細胞は未成熟髄系細胞の混合集団を代表し,TMEに積極的に募集されており,そこでは免疫活性を広く抑制している。MDSCsと腫瘍関連マクロファージ(TAMs)は他の免疫細胞の免疫機能に必要な重要なアミノ酸を奪うことによって、免疫効果器の機能を遮断するシグナル分子を産生し、そして制御性T細胞の活性化を増強し、腫瘍の成長を支持する。そのほか、MDSCsとTAMSは腫瘍血管新生と成長を促進し、不良予後に関連するマーカーであり、PD-L 1治療反応不良と関係がある。我々のIO計画は、TおよびNK細胞活性を増強することによって、またはTME内のMDSCsおよびTAMの免疫抑制機能を抑制することによって、複数の腫瘍促進機序を克服する可能性がある潜在的な新しい療法を代表すると信じている。
16
PYX−106:標的抗体のIgG 1サブタイプ抗Siglec−15
概要
PYX-106は研究中の全ヒトIgG 1ホモ抗体であり、Siglec-15を介したT細胞増殖と機能の抑制を遮断することを目的としている。PYX-106はマクロファージ、T細胞およびNK細胞などの重要な腫瘍浸潤性免疫細胞集団を解決することを目的としており、これらの細胞は腫瘍の成長および転移を制限する上で重要な役割を果たしている可能性がある。特定の細胞タイプを抽出することに加えて,PYX−106はT細胞枯渇やTおよびNK細胞に対するTMEの免疫抑制作用の機序に関与している可能性が考えられる。われわれが許可された抗Siglec−15モノクロナル抗体PYX−106は,単独でまたは他の療法(他の免疫療法を含む)と組み合わせて使用され,癌患者に追加的な利点を提供する可能性があると信じている。我々はBiosion USA,Inc.から大中華区中国(大陸,香港,マカオ,台湾)以外の地域のPYX−106のグローバルライセンスを取得した。
2022年12月、FDAは、臨床試験を開始するために、PYX-106のためのINDの使用を承認することを発表しました。2023年第1四半期に、私たちは臨床試験サイトの活性化を開始し、現在患者をスクリーニングして第1段階試験、すなわちPYX-106-101に参加している。PYX-106-101はヒト初例1期多中心開放ラベル用量増加試験であり、再発或いは難治性固形腫瘍患者におけるPYX-106の安全性、耐性、薬物動態学、薬効学と初歩的な治療効果を評価することを目的としている。この第1段階用量増加試験は、標準治療によって固形腫瘍と疾病の進展に発展した患者と、生存を獲得できない或いは延長する標準看護治療に適していない患者を含む。この試験では、駆動突然変異/転座のない非小細胞肺癌、乳癌、子宮内膜癌、甲状腺癌、腎臓癌、胆管癌、膀胱癌、結腸直腸癌、頭頸部扁平上皮癌を含む固形腫瘍患者を募集する。
Siglec−15を標的とする原理と我々の標的抗体の作用機序
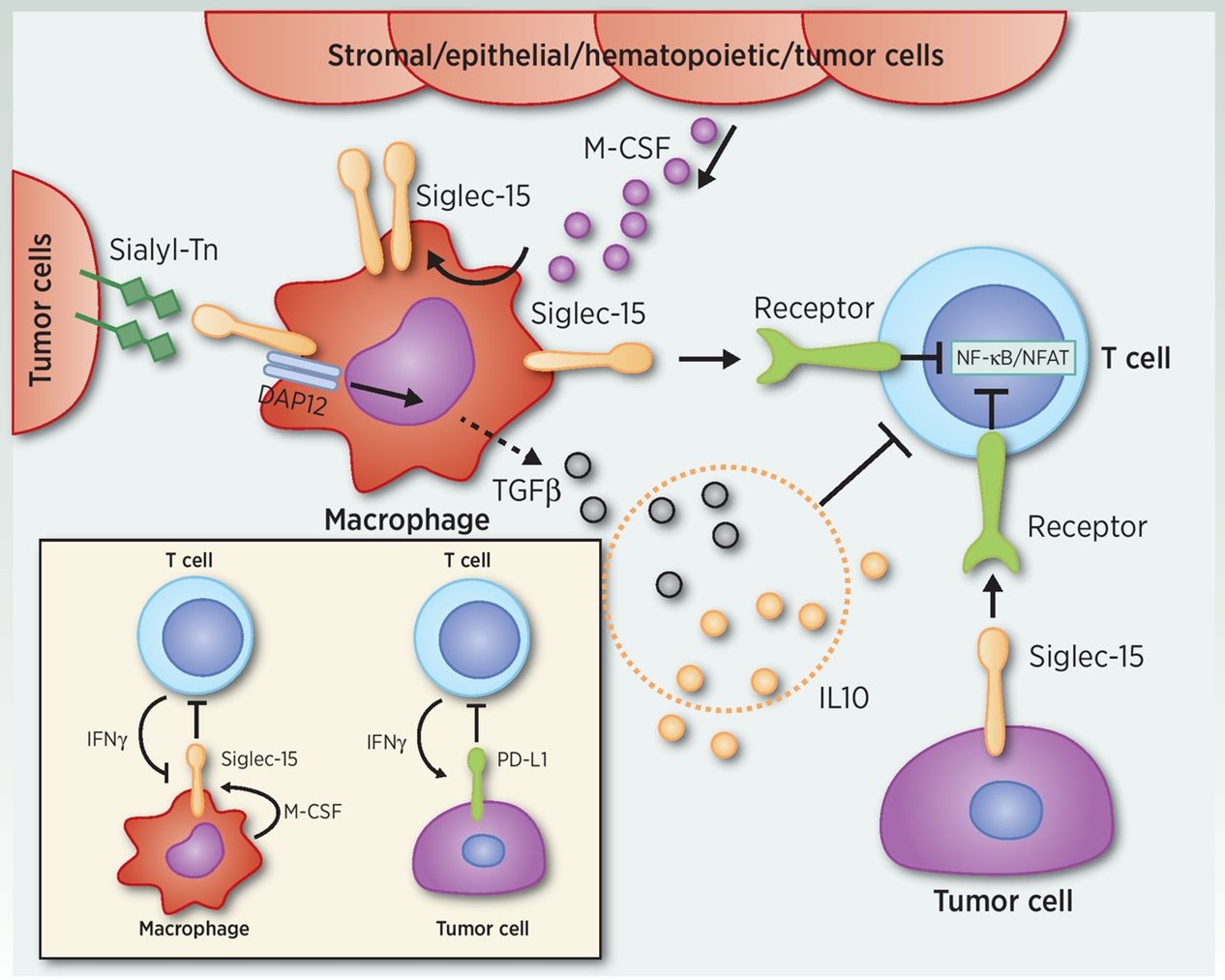
Siglec−15はSiglec(シアル酸結合免疫グロブリンレクチン)ファミリーのメンバーであり,Siglecは免疫調節に関与する免疫グロブリンやIgスーパーファミリー蛋白のユニークな亜群である。Siglecは細胞表面のシアル酸を認識して結合し,この結合は免疫細胞上の細胞シグナルに影響する。Siglec−15は単経路のI型膜タンパク質であり,その膜貫通領域のリジン残基を介して活性化リンカータンパク質DNAX活性化タンパク質(DAP)12およびDAp 10に結合することが証明されており,シグナル分子を活性化する機能を有することを意味している。Siglec-15は正常組織に低レベルで発現しているが、TMEの腫瘍細胞とM 2マクロファージに高発現し、多種の腫瘍タイプは甲状腺癌、HNSCC、肺癌、乳癌と胆管癌を含む。腫瘍内の高度免疫抑制のM 2マクロファージの増加はT細胞の増殖と機能損傷を招き、抗腫瘍免疫反応の低下を招く。また,Siglec−15は腫瘍に関連する髄系細胞相互作用により生存や分化を促進し,この免疫抑制効果を増強し,腫瘍の環境促進を推進する可能性がある。興味深いことに,Siglec−15はPD−L 1経路とは独立して機能しているようであり,選定された腫瘍試料ではほとんど重複して発現していない。したがって,TMEにおけるSiglec−15の標的化は,PD−1/PD−L 1標的治療に対する反応が小さい患者の有望な治療選択である可能性がある。
以下の図9に免疫系の調節と抗腫瘍免疫機能障害の駆動におけるSiglec−15の役割について概説する。
17
図9
資料源:アメリカ癌研究協会定期刊行物-癌臨床研究概要-Siglec-15次世代癌免疫治療の新興標的として、著者:孫経偉、Lu、ミゲル·F·サンマッドと王軍
18
我々のSiglec−15標的抗体は完全ヒト由来のモノクロナル抗体であり,高い親和性を有し,Siglec−15誘導の免疫抑制を遮断し,TMEにおけるT細胞の増殖,機能と抗腫瘍免疫を回復することができる。全体的に,髄系細胞と腫瘍上のSiglec−15活性を結合·遮断することにより,われわれのSiglec−15標的抗体は免疫細胞を介した腫瘍細胞傷害を増強することを目的としている。Siglec−15の広範な腫瘍発現スペクトルを考慮すると,われわれのSiglec−15標的抗体はPD−1/PD−L 1ガイド癌治療が無効なものを含む多くの腫瘍適応を治療する可能性がある。
腫瘍発現が正常組織発現よりも有意に高い組織におけるSiglec−15の発現データを図10に示す。テストの適応では、非小細胞肺癌、乳癌、子宮癌、甲状腺癌、腎臓癌、胆管癌、膀胱癌、結腸直腸癌、肉腫はいずれも、腫瘍におけるSiglec−15の発現が正常組織よりも高いことを示している。試験された正常組織におけるSiglec−15の低発現は,非腫瘍組織におけるSiglec−15に対する治療の作用潜在力が限られていることを示している。多くの腫瘍タイプにおいて有意に高い発現は、このような腫瘍の患者がSiglec-15抗体治療に反応する可能性があることを示している。
[図10]
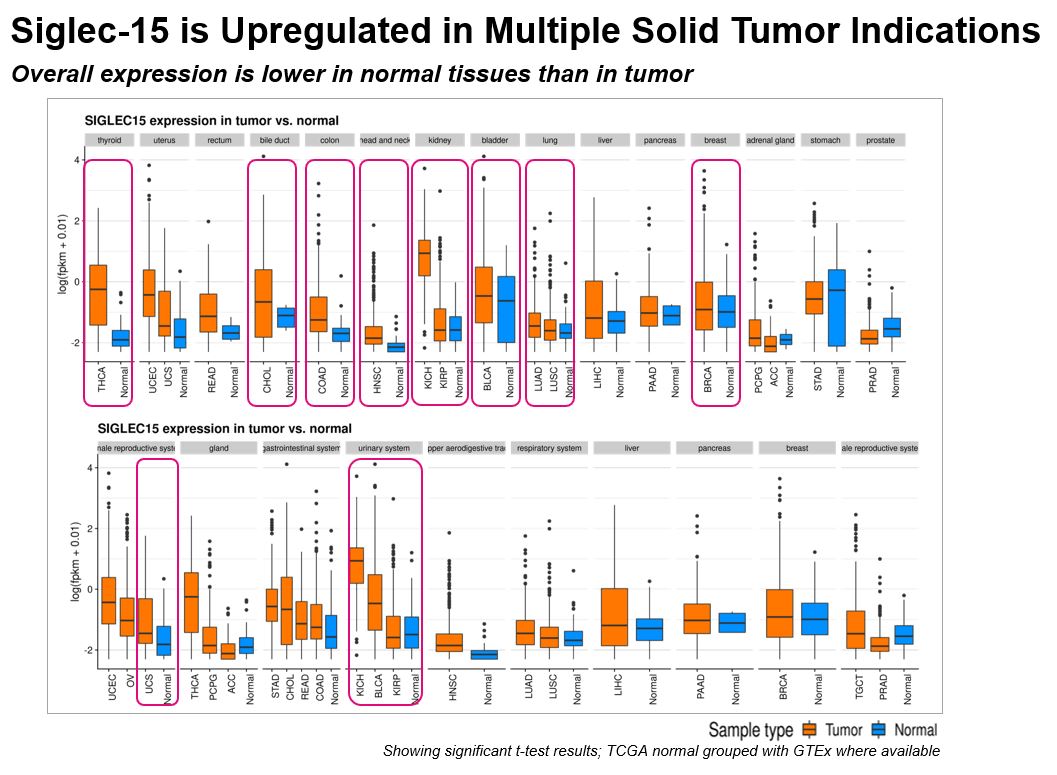
Siglec-15は一連の固形腫瘍に広く発現し、その中で発現レベルが比較的に高い腫瘍は、腎臓癌と甲状腺癌と有意差があった。
19
臨床前発展
PYX−106などの抗体でSiglec−15を遮断することで免疫抑制のTMEを克服し,他の免疫チェックポイント阻害剤の抗腫瘍活性を増強することができる。我々はすでに異なる動物モデルで臨床前研究を行い,Siglec−15遮断効果を評価し,われわれの観察結果は両者に十分な科学的根拠を提供した離体するそして体内にある研究します。
1つは体内にある対照群と比較して,マウス同遺伝子腫瘍モデルでは,PYX−106は15 mg/kgで週2回投与され,6週間連続して腫瘍成長を有意に抑制した。PYX-106は5 mg/kgと15 mg/kgの用量の時、腫瘍体積は約60%減少し、後の用量群の腫瘍体積の減少は統計学的意義に達した。また,腫瘍成長抑制はマウスの行動,体重あるいは脾重量の変化を伴わなかった。
1つは離体する免疫系の重要な構成要素と考えられる血球−正常人末梢血単核細胞を用いた研究により,PYX−106はSiglec−15を介したT細胞増殖抑制とインターフェロン−γ分泌を逆転させることが示唆された。CD_4~+、CD_8~+T細胞数と分泌するIFF-γは明らかに増加したが、陰性対照抗体の増殖抑制作用は明らかではなかった。PBMCsを用いたT細胞増殖に対する単一濃度の抗Siglec−15抗体の影響を図11に示す。抗CD 3抗体の添加はCD 4+とCD 8+T細胞の増殖をもたらし,Siglec−15の添加はこの作用を低下させた。陰性対照抗体の添加はこの増殖抑制に影響しなかった。PYX−106はこの抑制作用を逆転させた。
図11
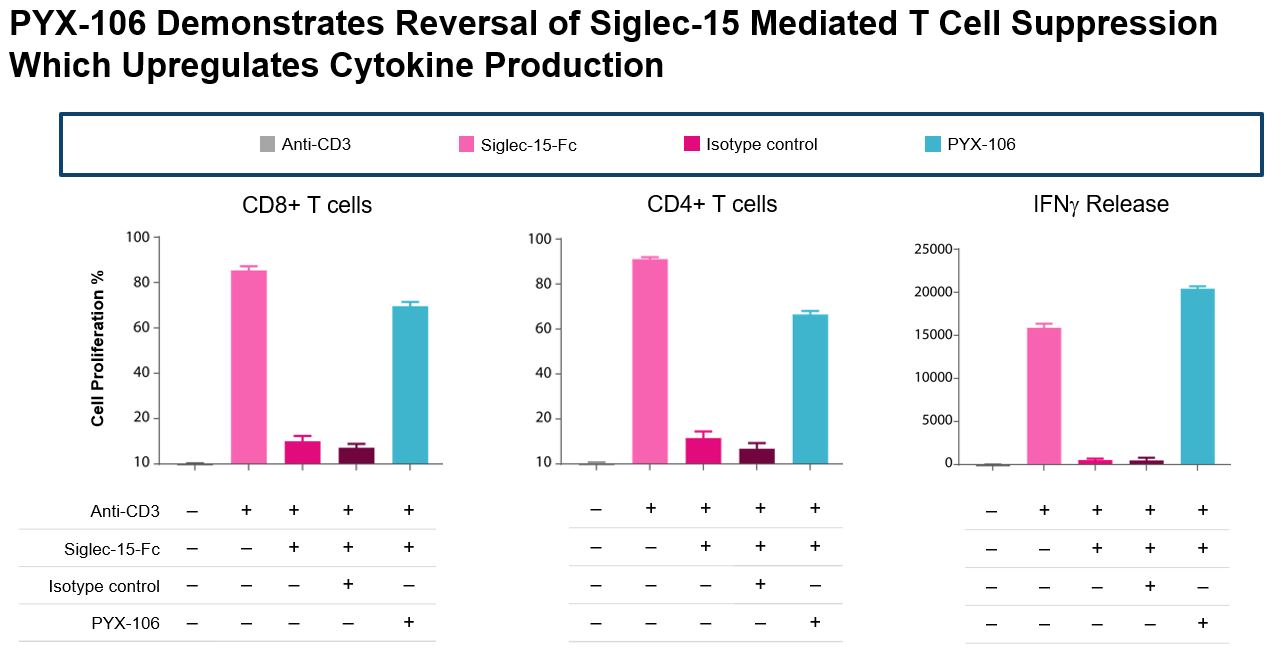
正常健康ドナーからのPBMCsを用いたインビトロ試験を示した。臨床前研究において、PYX-106はSiglec-15を介したT細胞抑制を逆転させ、癌細胞と腫瘍成長を防止するために免疫系を上昇させた。
20
臨床発展計画
2022年12月、FDAは、臨床試験を開始するために、PYX-106のためのINDの使用を承認することを発表しました。2023年第1四半期に、私たちは臨床試験サイトの活性化を開始し、現在患者をスクリーニングして第1段階試験、すなわちPYX-106-101に参加している。PYX-106-101はヒト初例1期多中心開放ラベル用量増加試験であり、再発或いは難治性固形腫瘍患者におけるPYX-106の安全性、耐性、薬物動態学、薬効学と初歩的な治療効果を評価することを目的としている。この第1段階用量増加試験は、標準治療によって固形腫瘍と疾病の進展に発展した患者と、生存を獲得できない或いは延長する標準看護治療に適していない患者を含む。この試験では、駆動突然変異/転座のない非小細胞肺癌、乳癌、子宮内膜癌、甲状腺癌、腎臓癌、胆管癌、膀胱癌、結腸直腸癌、頭頸部扁平上皮癌を含む固形腫瘍患者を募集する。
図12に示すように、この試験は、PYX-106の安全性および耐性を調べ、待ち行列拡大のための生物活性用量を決定し、最大許容用量を決定するための単一療法用量増加試験として設計されている。実験の一部として,参加者から腫瘍試料を収集し,バイオマーカー分析を行った。再発あるいは難治性固形腫瘍患者は,非小細胞肺癌,乳癌,子宮内膜癌,甲状腺癌,腎癌,胆管癌,膀胱癌,結腸直腸癌と頭頸部扁平上皮癌を含み,大量のM 2マクロファージ浸潤とSiglec−15発現を有する患者が参加する資格があることが知られている。この臨床試験に関する情報は,ClinicalTrials.gov(NCT 05718557)にアクセスしてください。
[図12]

私たちは2023年第2四半期初めに患者に薬を投与する予定だ。バイオマーカーの結果と潜在的な臨床活動の早期兆候を含む2023年末にこの第1段階臨床試験の初歩的なデータを報告する予定である。
21
抗体薬物結合体の研究進展
ADCは1種の治療類別であり、その中の細胞毒化学療法分子は標的mAbと接続し、有効に腫瘍殺傷効果を腫瘍病変に伝達し、同時に全身毒性を制限する。全身毒性は化学療法の治療効果を制限し、化学療法は高度な細胞毒性の抗腫瘍薬物である。腫瘍細胞及びその局部環境を標的とし、健康組織を節約することにより、ADCは有毒ペイロードの治療窓を顕著に改善することができ、甚だしきに至っては伝統的な化学療法よりもっと強い細胞毒性を有する。ADCはペイロードをモノクロナル抗体と対にすることでこのレベルの精度を実現しており,モノクロナル抗体はその標的抗原を極めて高い特異性で認識できるタンパク質である。ADCは成熟して急速に成長する生物製品である。これまでに13個のADCがFDAの承認を得ており、そのうち9つは2019年以降に市場に進出している。
[図13]
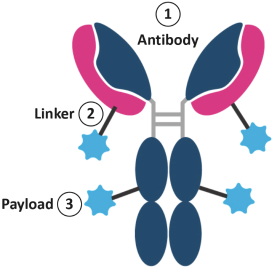
ADC模式図、3つの重要な構成要素を強調した:標的抗体、リンカーとペイロード或いは細胞毒剤(紺色:モノクロナル重鎖;ピンク:モノクロナル鎖)。
ADCの臨床的特性は、3つの構成要素の関数である(上の図13に示す)
理想的なADC標的は通常高度な腫瘍制限性発現を有し、健康組織に影響を与えず、循環抗体にアクセスすることができ、明確な内化動力学を持っているか、或いはTME内に有効に結合することができる。投与されると、ADCは標的抗原に遭遇し、その後有毒なペイロードを放出するまで血液中を伝播する。
ADCの薬物耐性を測定する1つの指標は、安全性を推定するためにデータから計算された臨床前治療指数である(図14参照)。この指標は,サルにおけるHNSTDの臨床前比率とマウス腫瘍モデルにおける最低有効量あるいはMEDの比率である。治療指数はHNSTD(mg/m)と式で定義される2)サル/マウスに対する最小腫瘍消退用量(mg/m2)である。以下の図14に示すように、より広い治療指数は、ADCの潜在的な臨床成功の重要な属性である。
22
図14

治療指数は臨床前研究中のサルの最大耐容量(MTD)とげっ歯類動物のMEDの比率によってADCの臨床耐性を評価する指標である。
次世代ADC設計の重要な革新分野
連結器の最適化
ペイロードを抗体に接続するリンカーは、ペイロードがサイクル中に早期に放出されることを防止し、ペイロードが標的細胞および/またはTMEに有効に放出されることを保証するべきである。一般的には2種類のリンカがあります
私たちは、私たちの切断可能で切断不可能な連結器キットは、各プログラムに適した最適な連結器を選択することを可能にすると信じている。著者らはいくつかの要素に基づいて、標的抗原のレベルと分布及び抗原回転、内化、リソソーム加工と分解の速度を含むが、これらに限定されないリンカーを選択した。
23
特定部位接合
特定部位の結合化学は,次世代ADCが予測可能なDARと改善したADC薬物動態(PK)を有する工程を可能にし,臨床前に観察された。この改良されたPKは、早期のペイロード放出を最大限に減少させ、目標から外れた毒性を低下させ、ADC全体の性能指数を向上させることができる。
DARの定義は,各抗体に付着するペイロード部分の数であり,通常0から8までの範囲である.理想的には、予測可能なPKおよびより予測可能な効果を有するADCを可能にするために、DARの可変性が限られている。DARの可変性と安定性は,主にリンカーと抗体を連結するための技術の結果である。従来のADCカップリング技術で使用される2つの従来のカップリング方法は、リジン残基を利用するか、抗体上に位置する鎖間ジスルフィド結合を利用するかである。以下の図15に示すように、これらの方法は、異なる種類の合成ADCプールからなるランダム共役混合物を生成する。グラフの各々は、ペイロードの数がX軸上に示される1つのADCから構成される。結合体混合物のこれらの部分の各々は、治療効果および毒性に貢献するため、任意の性質を最適化することは困難である。
DARに加えて,結合安定性とそれによるペイロード放出速度は特定の結合部位間で大きく異なることが示唆された。したがって、伝統的な結合体の腫瘍外でのペイロード放出は予測不可能であり、早期であり、非標的毒性を引き起こす。
さらに、著者らの豊富な連結子ツールキットに加えて、著者らの定点結合化学は私たちにTMEにおける薬物の溶解の優勢を微調整と最適化し、同時に腫瘍外放出を制限し、予測可能なDAR分布を許可したと信じている。特定の部位の結合技術は、CMCの製造および一貫性の有効性を促進するために、ADCの予測可能性を改善し、DARの分布が狭い(図15参照)。
[図15]
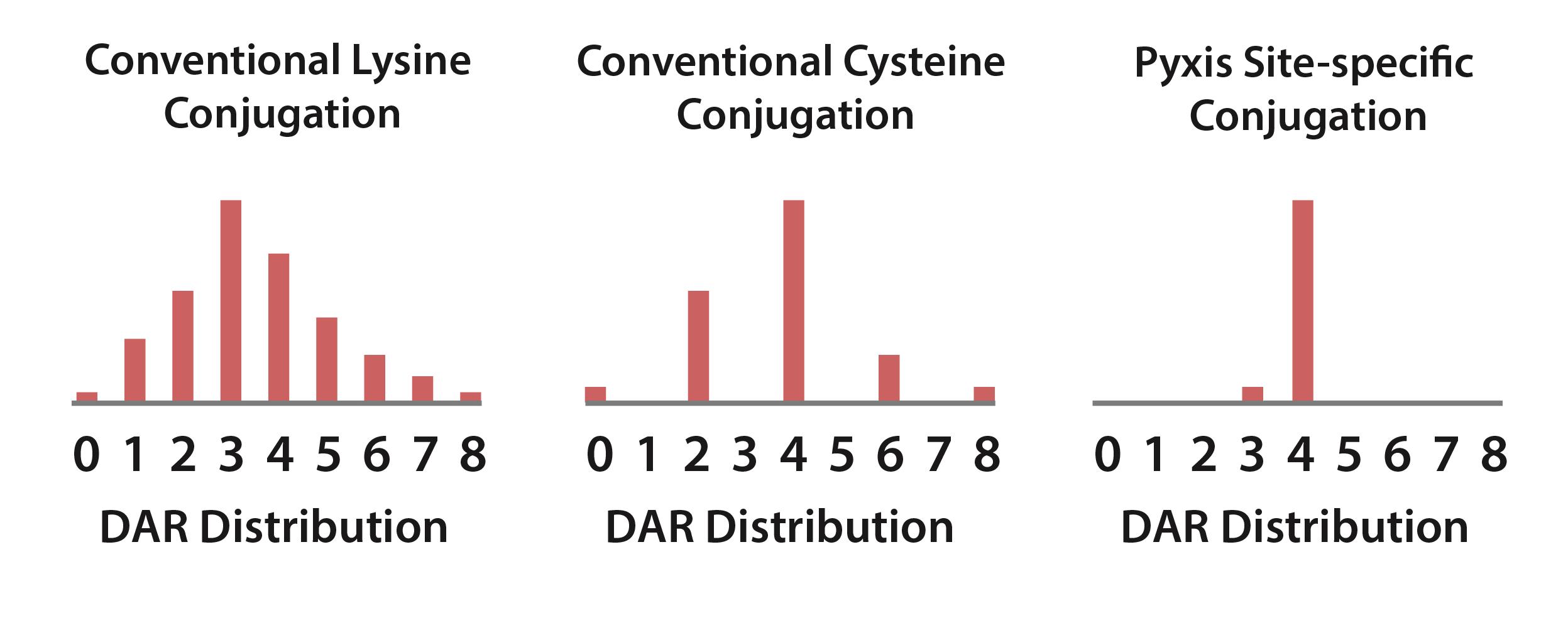
上述したのは、我々のサイト固有の共役技術が、より予測可能なDAR ADCを生成するために、コネクタ/ペイロードが正確に共役することをどのように可能にするかを強調する、異なる共役化学的方法を使用するDAR 4 ADCのDAR分布の例示的な例である。これはまた、CMCの特性を改善し、腫瘍ペイロードの送達を最大限に向上させるために、ADCの安定性を増強する。
24
細胞毒性ペイロードの選択
化学療法ペイロードは高度に有効な毒素であり、そうでなければシステム伝達の単一療法として壊滅的な副作用をもたらす。微小管に作用して細胞分裂を抑制する抗腫瘍黄金色物質と、DNAを破壊するアルキル化または埋め込み剤とを含むいくつかの潜在的ペイロードがある。
ADCの主要な作用モードはペイロードを通じて直接細胞死を誘導するが、ADCは直接細胞毒性以外の多種の抗腫瘍作用経路を利用することができる。例えば,多くの人が興味を持つ分野は,ADCを用いてICDを誘導し,検査点阻害剤を含む免疫療法と協働することである。ADCによる癌細胞の急速な死亡は損傷関連分子パターンや腫瘍抗原の放出を招き,腫瘍特異的免疫反応やT細胞のTMEへの募集を刺激する(図16)。1つの新しい関心分野は、血管新生または腫瘍関連線維芽細胞のようなTMEの様々な態様を妨害するためにADCを使用することである。そのほか、あるペイロード、例えば黄金色グルコサミンは、樹状細胞の成熟と活性化を産生できることが証明されており、樹状細胞は免疫システムの重要な部分であり、先天性と獲得性免疫反応の起動と調節を担当している。
図16
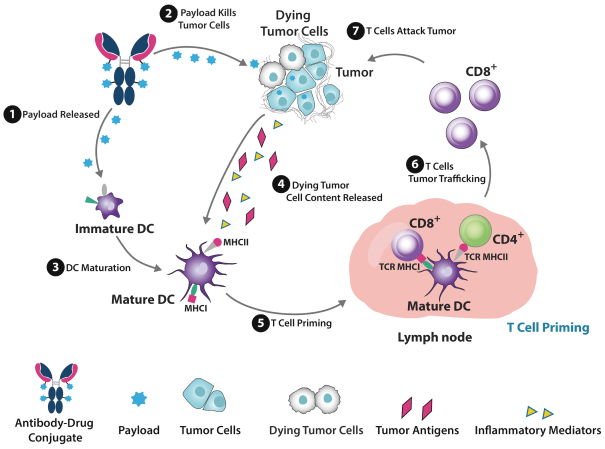
特に特定のペイロードを有するADCは、腫瘍細胞傷害のプロセスを増強するために、免疫原性細胞死の特徴をトリガする可能性があるADCについて概説する。
ADCはいくつかの癌の治療パターンを革命的に変化させ続ける可能性がある。現在、市場のADCはすでに改善を見たが、これらのADCは依然として投与量に影響する限界があり、重大な不良事件と関連している。著者らは伝統的に結合したADCを使用する局限性を克服するために、著者らの候補製品を設計し、患者により安全で、より有効な治療選択を提供することを目的とした。著者らは、著者らのADC設計における専門知識とTME生物学に対する洞察力を結合し、難治性癌患者に多種の機序にまたがる全体治療を提供し、それによって現在のADCの制限を克服し、患者に更に良い結果をもたらす可能性があると信じている。
25
FACTプラットフォーム
我々は,カスタマイズされたコネクタ−ペイロード組合せを用いた次世代ADCを開発しており,これらの組合せは新規であり,FACTプラットフォームからの臨床前データと特定部位結合技術の支持を得ている。これらのペイロードやリンカーは任意のIgG 1抗体に容易に適用でき,我々の部位特異的結合技術を用いて新たな候補製品を効率的に開発できると信じている。我々は最初に2020年12月にファイザーからFACTプラットフォームの許可を得,その後2022年10月にFACTプラットフォームの独占許可を取得し,ファイザーの10年以上の研究と投資から利益を得た。表格10-K本年度報告における項目1“許可と連携プロトコル”を参照.FACTプラットフォームをサポートする特定部位接続技術とADC技術は,臨床前研究に基づいて従来技術よりも有利な薬物特性を有する次世代ADCを開発できると信じている。
ADCは1種の治療カテゴリとして臨床で成功を得たが、多くのADCは依然として伝統的な非部位特異的結合技術を使用し、ADCと異なるDARの異なる混合物を招く。最近、ファイザー社は臨床前研究を展開し、げっ歯動物モデルにおいて結合部位のADC安定性と活性に対する影響を経験的に評価し、結果により、部位特異性結合技術は増強した薬理特性と予測可能なDARを潜在的に治療指数を改善させる可能性があることを表明した。
すでに多くの報告により、部位特異性結合技術はよくadcを臨床前にもっと広範な治療指数、より高い安定性ともっと良い治療効果を持たせることができることを表明した体内にある最適化は、各特定の接続子-ペイロードの組み合わせに依存する経験データ生成に依存するので、従来の不特定、従来のADC共役と比較して、これらの位置をどのように最適化するかについては、より少なく理解されている。FACTプラットフォームは,ADCの多様な生化学パラメータが性能に及ぼす影響を証明していると考えられる体内にあるまた,様々なリンカー−ペイロードの組合せのための結合部位を最適化する枠組みを構築した(図17)。
図17

FACTプラットフォーム技術は、治療指数を改善する潜在力を有する高度な安定性および予測可能なADCを生成するために、コネクタペイロードの最適結合部位を経験的に決定することを目的としている。
26
ファイザー実証研究によるリンカ−ペイロード結合の位置最適化
FACTプラットフォームは、新しいペイロード、切断可能および切断不可能な接続子のセット、および最適化接続部位の深い理解を提供すると信じている。FACTプラットフォームは、我々のADC候補製品PYX−201と臨床前計画PYX−203に基礎を提供し、将来のADCの開発をサポートし、これらのADCは以下の設計要素を最適化し、指導されたと考えられる
27
FACTプラットフォームは、我々のADC候補者の開発において、いくつかの明らかな利点と柔軟性を体現していると信じている
図18

FACTプラットフォームを用いて生成したモデルADCの治療指数が体内PK曝露を最大化しながら潜在的に毒性を軽減するかを模式的に示した。(NHP:非ヒト霊長類)
28
[図19]

臨床前研究では,われわれのPYX−201 ADCと同様のアダプターペイロードと接合部位化学物質を用いたカニクイザルにおけるファイザーNG−HER 2−ADCの安定性と曝露を観察し,同じアダプターペイロードを用いた従来のADCと比較した安定性と耐性(NG:次世代)を改善した。
図20
29
次の表は、現在承認されている従来の共役を使用したADCと比較して、臨床前に最適化されたペイロードとサイト固有の共役を利用する次世代ADCプラットフォームの潜在的な利点をまとめた
|
PYXIS腫瘍学の次世代ADC
|
通常のADC
|
潜在治療指数 |
• 8 – 16 |
• 1 – 5 |
|
|
|
連結器 |
• 特定のDARと高治療指数に対して設計された部位特異的結合リンカー
• ペイロードの早期解放を防止することを目的とした高度に安定したリンク |
• 非部位特異的結合は異種DARをもたらす
• 多くのリンカは不安定で、ペイロードの早期放出と全身毒性を引き起こす |
|
|
|
ペイロード |
• 最適な腫瘍死滅を達成するために腫瘍生物学に適合するために使用できる広範なペイロードアレイ
• ペイロードは微小管阻害剤とDNA損傷剤を含み、その潜在的な作用機序は、免疫治療と結合した臨床前モデルにおいて免疫原性細胞死を誘導することが証明されている |
• リンカの不安定な特性のため、いくつかの伝統的なADCはあまり有効ではない負荷で構築されている |
30
革新的なビジネスモデルと連合を構築する
私たちはパートナーとの連合を構築するために、一連の強力なビジネス発展機会を評価し続けている。この機会は、合弁企業、派生製品、パートナー関係の発見、およびライセンスを含むが、これらに限定されない。しかも、私たちは創造的なビジネスモデルの潜在力を探索する選択権を維持するつもりだ。
Voxall合弁企業
Voxall Treateutics,LLCまたはVoxallは,我々と合金治療会社や合金により設立された各株式の50%を占める合弁企業であり,癌や自己免疫疾患の治療薬の発見と開発を加速させることを目的としている。
我々は我々の目標カタログからVoxallに目標をもたらし,基質や免疫生物学的専門知識を提供し,合金はそのATX−GXトランスジェニックマウスや他の加速抗体薬物発見の発見サービスに貢献した。両方ともフルタイム職員たちとプロジェクト実行とプロジェクト監督を提供する。協力協定によると、双方は共同で合意した研究計画に基づいて研究を行い、最大6つの研究プロジェクトは共同選択の目標に重点を置いている。この第1弾の目標があれば、独占ライセンスを取得する独占的な選択権を保持し、自分の判断に基づいて候補製品をさらに開発·商業化することができる。
他の発見仕事
我々の広範な専門知識は,TMEや他の免疫生物学分野の科学的理解を切り開くことができると信じている。私たちはこれをするための柔軟な方法があり、プラットフォーム開発、創造的なビジネスモデルと連合建設、内部許可と目標カタログを利用することで、治療が困難な癌に対する差別化治療の組み合わせを拡大する。このビジョンを実現するためには、科学とビジネスの観点から迅速な革新と卓越した運営を行わなければならない。そこで,我々の使命は,将来の腫瘍学会社の設立に向けて次の一歩である伝統的な思想や伝統的なやり方に制限されない腫瘍学会社と,治療困難な癌に対応するために台頭する多くの複雑な挑戦に対応する腫瘍学会社と,限られた患者の生活に有効な治療に大きな影響を与える腫瘍学会社である。
競争
生物技術と製薬業界は、腫瘍学部門を含み、迅速に発展する技術、激しい競争及び知的財産権と独自技術に対する有力な保護を特徴とする。私たちが商業化に成功した任意の候補製品は、現在市販されている療法と将来商業化された任意の新しい療法と競争する可能性がある。著者らは著者らの技術、薬物開発の専門知識、指導チーム及び癌標的と生物学に対する強力な科学理解は私たちに一定の競争優勢を提供すると信じているが、著者らは大型製薬会社、生物技術会社、学術機関とその他の公共と個人研究機関を含む多くの源からの潜在的な競争に直面している。
多くの会社は腫瘍学部門の異なる発展段階で活躍し、ADCや免疫療法のような方法を採用した製品をマーケティング·開発している。2023年2月まで、私たちは500以上の業界スポンサーの活発な臨床試験があると推定され、140個以上の独特なADCに関連し、これはこの分野に対する持続的な興味と後続の取引を招く可能性がある。また,いくつかの大小の会社が様々な癌治療の免疫療法を研究している。ADC療法や免疫療法の開発にも多くの会社が参加しており,ADC治療会社,アステラス製薬会社,アスリコン社,第一三共株式会社,遺伝子テーク社,ジリッド科学社,グラクソ·スミスクライン社,ファイザー社,楽天医療会社,レーガン社,NextCURE社,ABCURE社が含まれているがこれらに限定されない。
我々のADCおよび免疫療法候補は、CAR−T療法、二重特異性抗体、および小分子が、パイプライン内の候補で開発されている同じタイプの癌のために開発されているような代替治療からの激しい競争に直面する可能性がある。これらの方法は、我々の技術によって生成された任意の製品よりも効率的で、安全であることが証明されるか、または他の利点を伝達することができる。さらに、我々は、我々のPYX−201候補製品EDBの目標がPhilogen S.p.A.からのものであり、我々のPYX−106製品候補BSI−060 Tの目標がNextCURE,Inc.の主導計画−NC 318からのものであることを含む特定の目標の競争に直面している。2022年11月、NextCUREは、NC 318が患者において安全かつ耐性が良好であることが観察されたにもかかわらず、NC 318が第2段階単一療法臨床試験で進展しないことを発表した。また,腫瘍学的免疫療法の開発には広範な活動があり,われわれの臨床前発見計画と競合する可能性がある。さらに、もし私たちの任意の候補製品が肺癌、血液病および他の癌などの腫瘍学的適応において承認された場合、それらは通常の化学療法、生物製品および標的薬物小分子療法を含む手術、放射線および薬物治療を含む既存の癌治療方法と競争する可能性がある。
31
私たちの競争相手は私たちよりも大きな科学、研究開発能力、そしてより多くの財務、技術、製造、マーケティング、販売、資源または経験を持っているかもしれない。これらの競争相手は臨床試験場所と患者登録、合格した科学と管理者の採用と保留、そして補充或いは必要となる新しい技術を獲得する上で私たちと競争を展開している。私たちの候補製品に対して、私たちのビジネス機会は、私たちが開発する可能性のある任意の製品よりも効果的で、より安全で、またはより安い新製品を開発する能力に依存するかもしれません。私たちの競争相手は私たちの前に私たちと競争する製品の開発に成功し、製品の発売許可を得て、私たちが目標としている同じ市場でこのような製品の受け入れを得ることができるかもしれない。大企業や老舗会社との協力計画を求める小さな会社や早い段階にある会社は重要な競争相手であることが証明される可能性がある。また、私たちの競争能力は政府や他の第三者支払人の精算の影響を受ける可能性がある。承認されれば、私たちの計画の成功に影響を与える競争要因は、それらの安全性と有効性、マーケティング承認の時間と範囲、供給の可用性とコスト、マーケティングと販売能力の深さ、および精算範囲に基づく可能性があります。
化学、製造、制御
私たちのADCとモノクロナル抗体の製造にはかなりの専門知識、技術ノウハウ、資源が必要だと信じています。我々は所有したり経営したりしておらず,cGMP規格に適合した製造施設の計画も確立されていない。我々は現在,外部契約製造組織(CMO)に依存して非臨床·臨床試験を支援する製品の生産と,我々の候補製品が市場承認を得た場合に商業製造を行うことに依存していると予想されている。さらに、私たちの候補製品の原材料と中間体は、場合によっては単一源の供給者から来る可能性がある。我々の候補製品の製造と設計過程の一部として,内部科学と製造技術と商業秘密,および第三者メーカーの技術と商業秘密に依存している.この戦略は、私たち自身の製造施設、設備、人員に投資する必要がなくなり、より効率的なインフラを維持することができ、同時に、私たちの専門知識と資源を現在の候補製品の開発に集中させることができると信じている。私たちは、私たちの候補製品に関連する独自の権利を保護するために、秘密および知的財産権条項を含む私たちのCMOと合意を維持します。私たちは重要な技術、製造、分析、品質(現在の良好な製造規範或いはcGMPを含む)とプロジェクト管理経験を持つ人員を持って、私たちのCMOを監督し、そして製造と品質データと情報を管理して、法規コンプライアンスの目的を達成する。
商業化計画
我々は、独占協力協定によって得られた製品を含むすべての候補製品のすべての商業化権利を保持しているが、大中華区中国(大陸、香港、マカオ、台湾)のPYX-106を除いて、ファイザーはPYX-201の第1の交渉権を付与されている。私たちの候補製品はまだ臨床前あるいは臨床開発段階にあるので、私たちはまだ自分の商業組織あるいは流通能力を確立していない。もし私たちの候補製品がアメリカや他の場所でマーケティング承認または許可を得た場合、承認された候補製品の商業的価値を達成するための計画を立てる必要があるだろう。適切な時期には、承認された候補者の米国での販売商業化を支援するために、私たち自身の専門販売·マーケティング組織を構築する予定です。私たちはまた、私たちの候補製品を米国以外の市場で商業化するために、1つまたは複数の第三者との協力、共同販売促進、流通、および/または他のマーケティング計画を求めることができる。私たちはまた、任意の承認された全額または協力候補製品のすべての商業価値を達成するために、より大きな販売およびマーケティング組織を必要とする場合に、これらの配置を求めることができる。
32
許可と協力協定
ファイザーとのライセンス契約。
2020年12月、ファイザーまたはファイザーと改訂されたライセンス契約を締結し、ファイザーの2つの候補ADC特許製品(現在、PYX−201およびPYX−203と呼ぶ)およびライセンス目標のための他のADC製品のグローバル開発および商業化権利を取得する。ファイザー許可協定は2021年3月に会社に発効した。最初の排他的許可目標はExtraドメインB(フィブロネクチンのEDB)とCD 123であり,他の許可目標を追加するために我々の許容範囲を拡大することを選択することができる。ファイザーはまた,ファイザーのFACTプラットフォーム技術開発が許可されたADCを用いて商業化することを可能にした非独占許可を与えてくれた。2021年3月、私たちはファイザーライセンス協定を改正し、より多くのノウハウを私たちの許容範囲に入れました。ファイザーライセンス契約によると,500万ドルの前払い費用を含む合計2,500万ドルのライセンス料が発生し,2021年にファイザーに12,152,145株のBシリーズ転換可能優先株を発行した。
2022年10月6日、ファイザーと改訂され、再記述されたライセンス契約またはA&Rライセンス契約を締結し、ファイザーライセンス協定を修正し、再確認した。A&Rライセンスプロトコルによれば、ファイザーは、PYX-201およびPYX-203、およびADC候補製品を含むいくつかの許可ターゲットのためのADC候補製品を開発および商業化するために、ファイザーFACTプラットフォーム技術下のグローバル独占権利を付与する。他のADC目標は象徴的な前払いとマイルストーンの許可を得るかもしれない。さらに、EDBおよびCD 123に関する1つまたは複数の国または地域で開発、製造、および商業化許可されたADCおよび認可製品を購入または開発、製造、および商業化する権利をファイザーに独占的に交渉する権利を付与する。A&R許可協定の条項によると、ファイザーに800万ドルを支払うことに同意し、2229,654株の普通株を発行した。また、発効日または発行後180日にその数量の普通株を発行し、500万ドルに相当し、1株当たりの価格は私たちの普通株の発行前の営業日の終値に等しいので、ファイザーの私たちの所有権権益は19.5%(19.5%)を超えてはいけません。株式価値と500万ドルの差額は現金で支払うことができます。
最初の4つの許可されたADCへの合計6.65億ドルのマイルストーン費用を含む、将来的または支払い可能な特許権使用料の支払い義務があります。さらに、我々は、開発、規制、およびビジネスマイルストーンを含む、FACTプラットフォームを介して開発および商業化された最初の4つの許可ADC目標を超える各追加の許可目標に将来の支払いを支払う必要がある。また,ADCライセンス製品が発売されれば,ライセンス製品の純売上高の階層印税をファイザーに支払い,印税料率は低い1桁から10代程度まで様々である。我々のライセンス使用料義務は、第1の商業販売から最近発生したライセンス製品および国/地域ごとに適用される:(1)第1の商業販売から12年、(2)すべての法規またはデータ排他性満了、および(3)ライセンス製品に関する国/地域のライセンス特許の最後の有効な権利要件が満了したとき。ファイザーに一定の割合の分配可収入を支払う義務があり,割合は20%から下位2桁まで様々であり,具体的には適用分許可に入った場合の許可製品の開発段階に依存する。
ファイザー許可協定によると,目標が許可目標になってから4年以内に,商業的に合理的な努力で臨床候補者を指名する義務がある。また、米国および少なくとも1つの他の主要市場国(フランス、ドイツ、イタリア、日本、スペイン、イギリス)の各許可目標について少なくとも1つの許可製品を開発し、規制承認を求め、規制承認を得た後、各国で任意の許可製品を商業化するように商業的に合理的な努力が求められている。私たちは任意の独占ライセンス特許の起訴と実行を統制し、もし私たちがこのような権利を行使しないことを選択すれば、ファイザーは起訴と実行権を持つ。
ファイザー許可協定は、(1)いずれか一方が指定された治癒期間内に相手の重大な違約行為を是正できなかった場合、(2)いずれか一方が他方で何らかの倒産事件が発生した場合、または(3)許可製品の最初の規制承認を受ける前に、または許可製品の最初の規制承認を受けた後、90日前に書面通知を出したり、許可製品の最初の規制承認を受けた後、1年前に書面通知を出したりすることなく、最終期限の許可使用料の期限が満了するまで有効である。
33
シカゴ大学とのライセンス契約
2020年4月、私たちは、私たちの科学創始者Thomas Gajesski博士が行った研究によって生成されたいくつかの特許の一部の独占的許可と、特定の技術および材料の非独占的許可とを得るために、シカゴ大学または大学とライセンス契約を締結した。許可条項によれば、私たちは、許可された技術および材料と組み合わせて、または特定の特定の生物学的標的の活性を評価、調節、または利用することができる独占的な世界的権利を有し、許可された特許の有効な権利要件によってカバーされる製品を開発および商業化することができる。
大学の許可を得た一部の代償として,2020年に大学に48,919株の普通株を発行した。大学ライセンス協定によると、発効日、潜在開発、ビジネスマイルストーンの3周年から大学に年間10,000ドルの維持費を支払う責任があり、総額770万ドル以下、許可製品の純売上高が1%未満から低い桁までの異なるレートで特許権使用料を運用し、ライセンス製品の初商業販売後のある年には、最低年間使用料は100万ドルから300万ドルとなる。私たちのライセンス使用料義務は、(1)特定の国/地域ライセンス特許の有効な権利要件がカバーされるライセンス製品について、そのような有効な権利要件が満了するまで、(2)他のすべてのライセンス製品について、ライセンス製品が指定された国/地域で初めて商業販売されてから10年以内になるまで、ライセンス製品および国/地域に基づいている。適用される再許可を締結した日に応じて,一定割合の再許可収入を大学に支払い,範囲の低い青少年にする義務がある。
大学許可協定によると、私たちは商業上の合理的な努力で開発し、許可製品を市場に投入し、特定の日前にある臨床前と臨床開発マイルストーンを達成し、そして監督管理の許可を受けた後に許可製品を普及と販売する義務があるが、ある無料と支払いに基づく延期を守らなければならない。大学制御ライセンス特許の起訴は、費用は私たちが負担し、私たちはライセンス特許を優先的に実行する権利がありますが、大学の予備執行権の制約を受けなければなりません。
大学ライセンス契約は、次の規定に従って終了しない限り、ライセンス製品に関連するすべてのライセンス製品の印税義務が満了するまで有効である。(1)大学が任意の未治癒支払違反に対して30日前に書面通知を出すか、または他のすべての未治癒違反行為に対して90日前に書面通知を発行するか、(2)何らかの倒産事件や吾等または任意の付属会社の解散が発生した場合、私たちまたは任意の付属会社が大学ライセンス契約を発行するか、または(3)書面終了通知を提供する場合、カレンダー四半期終了時に特定のライセンス製品の全部または特定のライセンス製品についてライセンス契約を終了する。
Voxallと合金治療会社の合弁企業。
2021年3月、PYXIS腫瘍学の現場特定目標目録と合金会社のATX-GXを利用するために、合金会社と協力して設立された合弁会社であるVoxallを援助·運営するための最終取引契約を合金会社と締結したプラットフォームや抗体発見サービスです
VoxallはPYXIS腫瘍学会社と合金会社のVoxallに投票権のある会員単位の50%を授与し、いくつかの初期寄付金と交換した。私たちの最初の貢献には、以下に述べるように、Voxallとの協力を達成するために、50,000ドルと、私たちが所有または制御するいくつかの知的財産権の非独占的全額支払い許可が含まれています。合金の最初の貢献には,以下に述べるように,Voxallとの連携を実現するための50,000ドルの署名とライセンス契約とサービスプロトコルがある.Voxallは取締役会で管理され、取締役会は私たちの代表と合金の代表で構成されている。私たちは私たちの取締役会代表として私たちの最高経営責任者Lara Sullivan M.D.を指定しました。Voxallの運営プロトコルにおける保護条項は,Voxallが何らかの行動をとる前に,PYXIS腫瘍学社や合金会社の承認を得なければならないことを要求している。
Voxallの形成過程において、著者らは合金会社とVoxall社と3年間の研究協力を行い、ある生物標的を確定と選択し、そしてこれらの標的に対してこれらの標的に対する開発候補抗体を創造し、更なる臨床前開発、臨床開発と商業化に用いた。協力協定によると、双方は共同で合意した研究計画と予算に基づいて研究を行い、最大6つの研究プロジェクトは共同選択の目標に重点を置いている。我々とコロラドは,Voxallと単独のサービスプロトコルを締結することで連携に研究支援を提供し,これらのサービスはVoxallが発行した本チケットの形で支払う.Voxallは協力して発生するすべての知的財産権を持つが,合金のATX−GXに関する知的財産権は除外するホームです。
34
研究計画下の開発候補抗体がある双方が合意した選択基準を満たしていれば,Voxallの独占的許可を得,さらにその研究計画で発見されたすべての開発候補抗体を開発して商業化する権利がある。私たちはいくつかの事前に合意された財政条項に基づいて研究プロジェクトを許可するかもしれない。他のすべての許可内の研究項目については、第三者評価によって決定された公平な市場価値を支払う義務がある。私たちが許可していないどんな研究プロジェクトもVoxallによって第三者に許可される可能性がある。
レゴ化学生物科学と署名した協定です
2020年12月、私たちはレゴ化学生物科学会社またはレゴ化学とライセンス契約またはレゴ化学ライセンス契約を締結し、加入、投資、および追加の価格協定を選択し、または加入プロトコルを選択した。レゴ化学ライセンスプロトコルによれば、LCB 67の世界(韓国以外)の開発権および商業化権利を取得し、LCB 67はDLK-1のためのADC候補製品、およびライセンス化合物を含む製品である。私たちは2021年3月にレゴケミカルに900万ドルを支払い、このお金は研究開発費として記録された。さらに、私たちはレゴ化学からいくつかの最初の許可製品を購入する可能性があり、コストは700万ドルと予想され、開発、規制、商業マイルストーン、およびライセンス製品の純販売に対して異なる比率で印税を徴収することを含む未来の支払いまたは支払い義務があるかもしれない。2022年第3四半期に、著者らは毒性研究データの回顧と分析に基づいて、LCB 67の継続開発を停止し、抗DLK 1 ADCの臨床応用と商業将来性を予測した。
さらに、レゴ化学は、いくつかの事件が発生したときに960万ドルのマイルストーン支払いまたは追加マイルストーン支払いを得る権利と引き換えに、800万ドルの選択権を私たちに支払う権利を行使し、これらのイベントは、普通株が初めて公開された定価または要約日、または私たちが支配権が取引を変更する標的である場合を含む。2021年10月に初めて公募した時、追加のマイルストーン支払い事件が触発され、2022年1月にレゴケミカルに960万ドルを支払いました。
Biosion USA,Inc.と締結されたライセンスプロトコル.
2022年3月28日、我々はBiosion USA,Inc.またはBiosionとライセンス契約または生物ライセンス契約を締結し、これにより、BSI-060 T、標的抗体Siglec-15、IO候補製品(現在、PYX-106と呼ばれる)および許可化合物を含む製品の開発、製造および商業化のための大中国(大陸、香港、マカオ、台湾を除く)の独占的な世界的許可を得た。生物学的許可協定の条項によれば、各方向の他方は、任意の二重特異性または多重特異性抗体、一方またはその付属会社によって制御される、予期される作用機序として阻害、調節、または結合したSiglec-15の抗体-薬物結合体を開発、製造および商業化するために、他方の領土(Biosionの大中国、およびPYXISの世界の他の地域)で独占的に許可されるように優先的に契約権を付与する。
Biosionライセンス協定によると、私たちは10,000,000ドルの前払い費用を支払うことに同意し、開発、規制、ビジネスマイルストーンを含む将来の支払いを支払う義務があり、正常に承認された場合、最大2.175億ドル、承認を加速させた場合、最大2.225億ドルに達する。また,製品が発売されれば,ライセンス製品の純売上高に応じてBiosion分級印税を支払い,印税料率は低い1桁から低い青少年まで様々である。我々のライセンス使用料義務は、第1の商業販売から最近発生したライセンス製品および国/地域ごとに適用される:(1)第1の商業販売から12年、(2)すべての法規またはデータ排他性満了、および(3)ライセンス製品に関する国/地域のライセンス特許の最後の有効な権利要件が満了したとき。Biosionに一定割合の分割可収入を支払う義務があり,範囲は−2桁から低い−2桁まで様々であり,適用される許可に入った場合の許可製品の開発段階に依存する。
Biosionライセンス協定によると、少なくとも1つのライセンス製品をライセンス地域で臨床開発し、規制部門の承認を求め、規制部門の承認を受けた後に商業化する商業的合理的な努力を使用する義務がある。私たちは許可区域内での許可特許の起訴と実行を統制している。
Biosionライセンス契約は、以下の規定で終了しない限り、適用されるライセンス使用料の期限が満了するまで、ライセンス製品および国/地域ライセンス製品に基づいて有効に維持される:(1)いずれか一方が他方の重大な違約により指定された治癒期間内にそのような違約を是正できなかったこと、(2)他方の何らかの倒産事件によって任意の一方が終了すること、(3)科学的または安全な理由で終了すること、(4)便宜上、ライセンス製品の最初の臨床試験を完了した後の任意の時間、または(5)特定の時間内に許可製品の開発および商業化活動を停止する場合は、Biosionが負担するが、いくつかの例外は除外する。
35
知的財産権
私たちの知的財産権は私たちの業務に重要であり、私たちはアメリカと国際的に私たちの候補製品、新しい治療方法、潜在的な適応、および私たちの業務に重要な他の発明を通じて特許保護を獲得し、維持することを含むそれを保護するために努力している。私たちはまた、私たちの業務が特許保護から保護されているか、または特許保護に適していないと思う側面を、商業秘密およびノウハウに依存して保護します。
私たちの特許組み合わせには、シカゴ大学、ファイザー、およびBiosionから独占的に許可された特許および特許出願、ならびに私たちが所有する特許出願が含まれている。我々の特許組み合わせは、我々の候補製品PYX−201、PYX−203、PYX−106およびPYX−102をカバーする特許および特許出願を含み、いくつかの地域ではこれらの候補製品を治療目的に使用する。我々のノウハウは,主に学術機関,ファイザー,Biosion,契約研究組織との関係で開発されている。
私たちの候補製品については、一般的に、私たちはまず物質組成と使用方法をカバーする特許保護を求めるつもりだ。我々の候補製品の開発過程全体において、私たちは、他の使用方法、製造プロセス、調製、および用量スキームに関するクレームを含む商業的成功を向上させる可能性がある特許保護を得る他の方法を決定することを求めている。
すべての特許出願について、私たちは具体的な状況に基づいて特許請求戦略を決定するつもりだ。私たちはいつも弁護士の提案と私たちのビジネスモデルと需要を考慮するつもりだ。我々が提出した特許出願は、我々の独自技術および任意の製品の有用なアプリケーションの保護権利と、これらのアプリケーションおよび用途が戦略的価値を有すると仮定して、既存技術および製品のために発見された我々の新しいアプリケーションおよび/または用途とを含む。我々は、既存の特許庁規則及び規定に基づいて、我々のプロセス及び成分が最大のカバー範囲及び価値を得ることを確実にするために、特許出願の数及びタイプ、並びに既存の特許権利要件を絶えず再評価する。また、特許訴訟中には、私たちの知的財産権および業務ニーズを満たすためにクレームが修正される可能性がある。
特許保護を得る能力およびこのような保護の程度は、従来技術の範囲、発明の新規性および非顕著性、および特許法実施要件を満たす能力を含む多くの要因に依存することを認識している。我々のような免疫腫瘍学会社の特許地位は通常不確定であり,複雑な法律,科学,事実問題に関連している。さらに、特許出願において要求されるカバー範囲は、特許発行前に大幅に縮小することができ、特許発行後であっても、その範囲を再解釈またはさらに変更することができる。したがって、私たちは私たちの未来の任意の候補製品や私たちのプラットフォーム技術のために十分な特許保護を獲得または維持することができないかもしれない。私たちが現在求めている特許出願が任意の特定の司法管轄区域で特許として発行されるかどうか、または任意の発行された特許の権利主張が競争相手の影響を受けないように十分な特許保護を提供するかどうかを予測することはできない。私たちが持っているどんな特許も第三者によって挑戦され、回避され、無効に発表される可能性がある。
既存の特許出願において求められているカバー範囲にかかわらず、製品またはプロセスの変更が、侵害クレームを回避するために競争相手に十分な基礎を提供する可能性があるというリスクが常に存在する。また,特許出願において要求されるカバー範囲は特許発表前に大幅に縮小することができ,裁判所は特許発行後に特許範囲を再解釈することができる.さらに、米国を含む多くの司法管轄区域は、第三者が行政訴訟において許可または発行された特許に挑戦することを可能にし、特許主張のさらなる縮小またはキャンセルを招く可能性がある。さらに、私たちは、私たちの未決または任意の未来の出願から任意の特許が付与されること、または現在または未来に発行される任意の特許が私たちの製品を十分に保護することを保証することはできない。
全体的に、私たちの特許の組み合わせは、シカゴ大学、ファイザー社、およびBiosion社によって許可された特許と、世界の異なる司法管轄区域で提出された26個の異なる特許ファミリー、抗体および抗体薬物結合体に対する物質からなるファミリー、抗体および抗体薬剤結合体の製造、使用および構成されたファミリー、ならびに癌の治療および潜在的な標的を識別する方法を含むファミリーを含む。私たちの2022年12月31日までの特許組合の概要は以下のとおりである。
36
物質特許の構成
PYX−201抗EDB抗体−薬物結合体。我々は、追加のフィブロネクチンBドメインスプライシング変異体と結合した一連の抗体および抗体薬物結合体をファイザー社から独占的に許可しており、オーストラリア、ブラジル、カナダ、中国、ヨーロッパ、香港、インド、イスラエル、日本、メキシコ、シンガポール、南アフリカおよび米国の4つの発行された特許を含み、これらの出願はPYX-201の物質組成およびいくつかの使用方法を主張している。このシリーズの特許有効期間は2037年まで20年であり,利用可能な特許期間の調整または延長は何もない。
PYX−203抗CD 123抗体−薬物結合体我々は、韓国および台湾で取得された2つの特許と、オーストラリア、ブラジル、カナダ、中国、コロンビア、ヨーロッパ、香港、インド、インドネシア、イスラエル、日本、韓国、マレーシア、メキシコ、ニュージーランド、ペルー、フィリピン、ロシア、サウジアラビア、シンガポール、南アフリカ、台湾および米国で待っている出願を含むCD 123に特異的に結合する一連の抗体および抗体薬物結合体の一連の特許をファイザー社から独占的に許可しており、これらの国はPYX-203に関連する物質組成およびいくつかの使用方法を主張している。このシリーズの特許有効期間は2038年まで20年であり,利用可能な特許期間の調整または延長は何もない。
PYX−106抗Siglec−15抗体。我々は、最近PCTの国家段階に入り、オーストラリア、ブラジル、カナダ、エジプト、ヨーロッパ、インド、インドネシア、イスラエル、日本、マレーシア、メキシコ、ニュージーランド、フィリピン、ロシア、サウジアラビア、シンガポール、南アフリカ、韓国、アラブ首長国連邦、米国で出願されているヒトSiglecc 15に特化した一連の単一抗特許をBiosion USA,Inc.から独占的に許可している。このシリーズの特許有効期間は20年であり,2041年まで利用可能な特許期間の調整または延長はない。
GPNMB抗体及びその使用方法それは.糖タンパク質非転移性黒色腫タンパク質Bに対する結合特異性を有する組成物を有するPCT出願が行われている。このファミリーの特許有効期間は、2042年まで20年であり、可能な特許期間の延長は含まれていない。もし私たちがそうすることを選択すれば、私たちは2023年までにこの特許家族に国家段階に入った申請を提出することができる。
37
ADC特許権
Spliceostatin類似体いくつかの保持の場合、我々は、新規細胞毒性スティッチング阻害素類似体および誘導体、ならびにオーストラリア、ベルギー、ブラジル、カナダ、中国、ドイツ、デンマーク、スペイン、フィンランド、フランス、イギリス、香港、アイルランド、インド、イタリア、日本、韓国、メキシコ、オランダ、ロシア、スウェーデン、トルコ、および米国で取得された27件の特許が発行されている、一般的な抗体薬物結合体のための組成物、使用方法および/または製造方法のための特許シリーズをファイザー社から独占的に許可している。このシリーズの特許有効期間は2033年まで20年であり,利用可能な特許期間の調整または延長は何もない。
チューブリン類似体及びその製造方法いくつかの保持の場合、我々は、細胞毒性チューブリン類似体および誘導体、ならびに米国で発行された特許を含む、細胞毒性チューブリン類似体および誘導体、ならびに米国で発行された特許を含む抗体-薬物結合体の組成物、使用方法および/または製造方法のための特許シリーズをファイザー社から独占的に許可している。このシリーズの特許有効期間は2037年まで20年であり,利用可能な特許期間の調整や延長は何もない。
ヘテロアリール系カップリングハンドル、それらの製造方法、および抗体医薬結合体の合成におけるそれらの使用いくつかの保持の場合、我々は、カナダで発行された特許と、米国、ヨーロッパ、および日本で出願されている特許とを含む抗体-薬物結合体のための組成物、使用方法および/または製造方法のための特許シリーズをファイザー社から独占的に許可している。このシリーズの特許有効期間は2037年まで20年であり,利用可能な特許期間の調整または延長は何もない。
リン酸ナトリウム勾配で抗体薬物結合体を精製したいくつかの保持の場合、我々は、抗体薬物結合体反応混合物の製剤をヒドロキシアパタイト樹脂と接触させ、リン酸ナトリウムを含む勾配を用いて樹脂からADCを選択的に溶出させることによって、抗体薬物結合体製剤から高分子量種、特に凝集体を除去するための特許シリーズを取得し、フランス、ドイツ、アイルランド、イタリア、スペイン、およびイギリスで発行された6つの特許、および米国における未解決の出願を含む、抗体薬物結合体のための組成物、使用方法および/または製造方法のためのファイザーから独占的に許可されている。このシリーズの特許有効期間は2036年まで20年であり,利用可能な特許期間の調整または延長は何もない。
CTI薬効団を含む二機能性細胞毒剤いくつかの保留の場合、私たちは、独立した薬物として使用可能な二機能性CTI-CTIおよびCBI-CTI二量体、およびオーストラリア、ベルギー、中国、デンマーク、ドイツ、フィンランド、フランス、香港、アイルランド、イスラエル、インド、イタリア、日本、メキシコ、オランダ、ノルウェー、ロシア、シンガポール、韓国、スペイン、スウェーデン、トルコ、トルコを含む、一般的な抗体薬物結合体のための組成物、使用方法および/または製造方法に適した特許シリーズをファイザー社から独占的に許可している。台湾、連合王国、アメリカ、そしてブラジルとカナダで処理される申請。このシリーズの特許有効期間は2036年まで20年であり,利用可能な特許期間の調整または延長は何もない。
Calicheamicin誘導体およびその抗体薬物結合体いくつかの保持の場合、私たちは、抗体薬物結合体のペイロードとして使用されるCalicheamicin誘導体について、カナダおよび日本で発行されている2つの特許、および米国およびヨーロッパで行われている出願を含む抗体薬物結合体のための組成物、使用方法および/または製造方法のための特許シリーズをファイザー社から独占的に許可している。このシリーズの特許有効期間は2038年まで20年であり,利用可能な特許期間の調整または延長は何もない。
システイン工学抗体薬物結合体いくつかの保持の場合、我々は、工学的システイン残基を有する抗CD 33抗体-薬剤結合体について、工学的システイン残基を有する抗CD 33抗体-薬剤結合体について、抗体-薬物結合体のための組成物、使用方法および/または製造方法のための特許系列をファイザー社から独占的に許可しており、係属中の出願がない米国で発行された特許を含む。このシリーズの特許有効期間は2038年まで20年であり,利用可能な特許期間の調整または延長は何もない。
38
プラットフォーム特許権(管理するPYXIS腫瘍学)
Trop−2に対する抗体及びその用途いくつかの保持の場合、我々は、trop-2に対する癌治療のための抗体および抗体結合体を対象としたファイザーFACTプラットフォームに関連する組成物、使用方法および/または製造方法の特許シリーズを独占的に許可しており、米国および日本で発行されている4つの特許を含み、出願中の特許を有さない。このシリーズの特許有効期間は2032年まで20年であり,利用可能な特許期間の調整または延長は何もない。
トランスグルタミナーゼ結合を用いた工学的ポリペプチドいくつかの保持の場合、特定のアシル供与体グルタミンタグおよびアミン供与体エージェントを含む工学的ポリペプチド結合体について、カナダ、フランス、ドイツ、アイルランド、イタリア、日本、スペイン、イギリスおよび米国で取得された12の特許、および米国で出願されている特許を含む、ファイザーFACTプラットフォームに関連する組成物、使用方法および/または製造方法の特許シリーズをファイザーから独占的に許可している。このシリーズの特許有効期間は2034年まで20年であり,利用可能な特許期間の調整または延長は何もない。
抗体薬物結合体と共に使用する安定性調節リンカーいくつかの保持の場合、私たちは、これらの安定性調節抗体-薬物結合体を製造するための安定性調節リンカーコンポーネントを開発することを目的として、ファイザーFACTプラットフォームに関連する組成物、使用方法および/または製造方法の特許シリーズを独占的に許可しており、オーストラリア、オーストリア、ベルギー、ブルガリア、カナダ、中国、チェコ共和国、デンマーク、フィンランド、フランス、ドイツ、ギリシャ、香港、ハンガリー、インド、アイルランド、イスラエル、イタリア、日本、ルクセンブルク、メキシコ、オランダ、ポーランド、ポルトガル、ルーマニア、ロシア、スロバキア、スロベニア、韓国、スペイン、スウェーデン、スイス、トルコ、イギリス、アメリカ、そしてブラジル、メキシコ、ロシア、アメリカの未定申請。このシリーズの特許有効期間は20年であり,2035年まで利用可能な特許期間の調整または延長はない。
共同作用のAuristatinの組み合わせですいくつかの保持の場合、AuristatinまたはADC系ADCと、米国および日本で取得されている2つの特許と、カナダおよびヨーロッパで行われている出願とを含む第2の活性薬剤(PI 3 K/mTOR阻害剤、MEK阻害剤、タキサンまたは他の抗癌薬を含む)との組み合わせをファイザーFACTプラットフォームに関連する組成物、使用方法および/または製造方法の特許シリーズをファイザーから独占的に許可している。このシリーズの特許有効期間は20年であり,2035年まで利用可能な特許期間の調整または延長はない。
抗体-薬物結合におけるエンドキャッピング抗体システインおよび非末端抗体システインおよびそれらの使用いくつかの保持の場合、私たちは、ファイザーFACTプラットフォームに関連する組成物、使用方法および/または製造方法の特許シリーズをファイザーから独占的に許可しており、この一連の特許は、ペアリングされていないシステイン残基を翻訳して修飾し、特定の化学物質でキャップし、封入された抗体が、オーストラリア、オーストリア、ベルギー、ブルガリア、中国、チェコ共和国、デンマーク、フィンランド、フランス、ドイツ、ギリシャ、香港、ハンガリー、インド、アイルランド、イスラエル、イタリア、日本、ルクセンブルク、メキシコ、オランダ、オランダ、ドイツ、ギリシャ、香港、ハンガリー、アイルランド、イスラエル、イタリア、日本、ルクセンブルク、ベルギー、ブルガリア、中国、チェコ共和国、デンマーク、フィンランド、フランス、ドイツ、ギリシャ、香港、ハンガリー、アイルランド、イスラエル、イタリア、日本、ルクセンブルク、メキシコ、オランダ、オランダを含む、さらなる特定の部位連結工程に非常に適している。ポーランド、ポルトガル、ルーマニア、ロシア、スロバキア、スロベニア、韓国、スペイン、スウェーデン、スイス、トルコ、イギリス、アメリカ、ブラジル、メキシコ、ロシアおよび米国の保留申請、ならびにブラジル、カナダ、ヨーロッパ、ロシア、米国の未定申請。このシリーズの特許有効期間は2036年まで20年であり,利用可能な特許期間の調整または延長は何もない。
エンドキャッピング抗体システインと非キャッピング抗体システインの大規模化生産及び治療用蛋白結合における応用いくつかの保持の場合、我々は、カナダ、日本、ロシア、および韓国で発行されている4つの特許と、オーストラリア、ブラジル、中国、ヨーロッパ、香港、インド、イスラエル、メキシコおよび米国で発行されている4つの特許とを含む、細胞増殖条件を制御することによって、抗体上の選択的キャップおよび非キャップシステインの生産を最適化することを目的とした、ファイザーFACTプラットフォームに関連する組成物、使用方法および/または製造方法の特許シリーズを独占的に許可している。このシリーズの特許有効期間は2038年まで20年であり,利用可能な特許期間の調整または延長は何もない。
39
プラットフォーム特許権(ファイザー管理)
細胞傷害性ペプチド及びその抗体薬物結合体いくつかの保留の場合、私たちは、細胞毒性ペンタペプチドおよびその抗体医薬結合体について、現在ファイザーによって管理されているファイザーFACTプラットフォームに関連する組成物、使用方法および/または製造方法の特許系列を輝瑞から独占的に許可しており、その中には、オーストラリア、オーストリア、ベルギー、ブルガリア、カナダ、中国、コロンビア、チェコ共和国、デンマーク、フィンランド、フランス、ドイツ、ギリシャ、香港、ハンガリー、アイスランド、インドネシア、アイルランド、イスラエル、イタリア、日本、ルクセンブルク、マレーシア、メキシコ、オランダ、ニュージーランド、ノルウェー、ペルー、フィリピン、ポーランド、ポルトガル、ルーマニア、ロシア、サウジアラビア、シンガポール、スロバキア共和国、スロベニア、南アフリカ、韓国、スペイン、スウェーデン、スイス、台湾、トルコ、イギリス、米国、ならびにアルゼンチン、ブラジル、インド、ペルー、ベネズエラの未定申請。このシリーズの特許有効期間は2032年まで20年であり,利用可能な特許期間の調整または延長は何もない。
二機能性細胞毒剤いくつかの保留の場合、我々は、CBIおよび/またはCPIに基づくサブユニットを含む細胞毒性二量体、このような二量体を含む抗体医薬結合体について、現在ファイザーによって管理されているファイザーFACTプラットフォームに関連する組成物、使用方法および/または製造方法の特許シリーズをファイザーから独占的に許可しており、オーストラリア、オーストリア、ベルギー、ブラジル、ブルガリア、カナダ、中国、コロンビア、チェコ共和国、デンマーク、フィンランド、フランス、ドイツ、ギリシャ、香港、ハンガリー、アイスランド、インド、インドネシア、アイルランド、イスラエル、イタリア、日本、ルクセンブルク、マレーシア、メキシコ、オランダ、ニュージーランド、ノルウェー、ペルー、フィリピン、ポーランド、ポルトガル、ルーマニア、ロシア、サウジアラビア、シンガポール、スロバキア共和国、スロベニア、南アフリカ、韓国、スペイン、スウェーデン、スイス、台湾、トルコ、イギリス、米国、ならびにアルゼンチンとベネズエラの係属中の出願。このシリーズの特許有効期間は20年であり,2035年まで利用可能な特許期間の調整または延長はない。
抗体および抗体断片は部位特異的結合に用いられる我々は、中国、コロンビア、日本、ロシア、南アフリカ、韓国およびロシアで取得された8つの特許と、アルゼンチン、オーストラリア、ブラジル、カナダ、欧州、香港、インド、インドネシア、イスラエル、マレーシア、メキシコ、ニュージーランド、ペルー、フィリピン、サウジアラビア、シンガポール、台湾、米国で出願されている特許を含む部位特定結合のための代替システインを含むポリペプチド、抗体およびその抗原結合断片について、現在ファイザーによって管理されているファイザーFACTプラットフォームに関連する組成物、使用方法および/または製造方法の特許シリーズを独占的に許可している。ベネズエラもありますこのシリーズの特許有効期間は2036年まで20年であり,利用可能な特許期間の調整または延長は何もない。
部位特異的結合のための工学的抗体定常領域およびその方法および使用いくつかの保持の場合、私たちは、抗体およびその抗原結合部分に対して、カナダ、フランス、ドイツ、アイルランド、イタリア、日本、スペイン、イギリスおよび米国で発行されている9つの発行された特許、およびカナダ、ヨーロッパ、および日本で発行されている特許を含む、現在ファイザーによって管理されているファイザーFACTプラットフォームに関連する組成物、使用方法および/または製造方法の特許シリーズをファイザーから独占的に許可している。このシリーズの特許有効期間は2032年まで20年であり,利用可能な特許期間の調整または延長は何もない。
工程ポリペプチド結合体及びそれを用いたトランスグルタミナーゼの製造方法いくつかの保持の場合、我々は、現在ファイザーによって管理されているファイザーFACTプラットフォームに関連する組成物、使用方法および/または製造方法の特許シリーズをファイザーから独占的に許可しており、この一連の特許は、アシル供与体グルタミンタグおよびアミン供与体試薬を含む工学的ポリペプチド結合体であり、カナダ、日本、および米国で発行されている6つの特許、ならびにヨーロッパおよび米国で行われている出願を含む。このシリーズの特許有効期間は20年であり,2031年まで利用可能な特許期間の調整または延長はない。
抗体−薬物は高負荷量に結合しているいくつかの保留の場合、私たちは、現在ファイザーによって管理されているファイザーFACTプラットフォームに関連する組成物、使用方法および/または製造方法の特許シリーズを輝瑞から独占的に許可しており、この一連の特許は、トランスグルタミナーゼ媒介抗体-薬物結合体を対象としており、オーストラリア、オーストリア、ベルギー、ブルガリア、カナダ、中国、チェコ共和国、デンマーク、フィンランド、フランス、ドイツ、ギリシャ、香港、ハンガリー、インド、アイルランド、イスラエル、イタリア、日本、ルクセンブルク、メキシコ、オランダ、ポーランド、ポルトガル、ルーマニア、ロシア、スロバキア共和国、スロバキア、韓国、スペイン、スウェーデン、スウェーデン、オランダ、ポーランド、ポルトガル、ルーマニア、ロシア、スロバキア共和国、スロバキア、韓国、スペイン、スウェーデン、日本、ルクセンブルク、メキシコ、オランダ、ポーランド、ポルトガル、ルーマニア、ロシア、スロバキア共和国、スロバキア、韓国、スペイン、スウェーデン、スウェーデン、オランダ、ポーランド、ポルトガル、ルーマニア、ロシア、スロバキア共和国、スロバキア共和国、スペイン、スウェーデン、日本、ルクセンブルク、メキシコ、オランダ、ポーランド、ポルトガル、ルーマニア、ロシア、スロバキア共和国、スロバキア、韓国、スペイン、スウェーデン、スウェーデン、スイス、トルコ、イギリス、アメリカ、そしてブラジル、韓国、アメリカの未定申請。このシリーズの特許有効期間は20年であり,2035年まで利用可能な特許期間の調整または延長はない。
40
免疫腫瘍学における方法
T細胞活性に関連する方法および組成物我々は、米国で発行された2つの特許と、ヨーロッパおよび米国で行われている出願とを含む一連の特許をシカゴ大学から独占的に許可しており、これらの特許は、患者が様々な遺伝子の発現レベルを測定した後に非無能T細胞を有する患者として決定された患者に基づいて免疫治療方法を治療することができると主張している。このシリーズの特許有効期間は20年であり,2034年3月まで利用可能な特許期限の調整や延長はない。
癌免疫治療におけるβ-カテニン阻害剤の使用我々は、シカゴ大学から、固形腫瘍癌の治療が可能であると主張する発行された特許と、出願中の米国特許とを含む一連の特許を独占的に許可している。このシリーズの特許有効期間は2036年3月まで20年であり,利用可能な特許期間の調整や延長は何もない。
腫瘍微小環境における機能不全の抗原特異的CD 8+T細胞我々は、シカゴ大学から一連の特許を独占的に許可しており、カナダ、中国、ヨーロッパ、日本、香港、および米国における6つの係属中の出願を含み、これらの出願は、癌の治療方法が、機能不全に特化した腫瘍抗原特異的CD 8剤を管理することを含むと主張している+T細胞です。このシリーズの特許有効期間は20年であり,2038年1月まで利用可能な特許期間の調整または延長はない。
特許期限と期限延長
個別特許には異なる期限があり,特許出願の提出日又は特許発行日及び特許を取得した国の特許の法的期限に依存する。一般に,米国で提出された出願のために発行された実用新案特許の有効期限は,非臨時特許出願の最初の有効出願日から20年である。さらに、場合によっては、米国特許の期限は、米国特許商標局またはUSPTOの一部、すなわち特許の発行時間の一部を遅延させ、FDA規制審査期間によって実際に失われた期間の一部を再取得するために延長することができる。しかし,FDAコンポーネントでは,回復期間は5年を超えることはできず,回復期間はFDA承認日から14年以上延長することはできない。さらに、承認された薬物に適用される1つの特許のみが資格延長を有し、承認された薬物、その使用方法、または製造方法に関する権利要件のみを延長することができる。欧州や他の外国司法管区にも同様の規定があり、承認された薬物をカバーする特許の期限を延長する。一般的には、米国および特許期間の延長を提供する外国司法管轄区で利用可能な特許期間の延長を求めるが、米国のFDAを含む適用当局が保証することはできず、このような延長を承認すべきかどうか、承認された場合、そのような延長の長さの評価に同意する。米国特許商標局および各外国司法管轄区の要求によると、特許のすべての税金、年金または維持費は、特許がこの期間内に有効に維持されるようにタイムリーに支払われなければならない。
1つの特許によって提供される実際の保護は、各国の異なる製品によって異なる可能性があり、特許のタイプ、そのカバー範囲、特定の国で規制に関連する延長および法的救済方法を得ることができるかどうか、ならびに特許の有効性および実行可能性を含む多くの要素に依存する可能性がある。
私たちの特許と特許出願は他の人たちの手続きや法的挑戦を受けるかもしれない。私たちは業務を展開するために必要な知的財産権を獲得、維持、保護することができないかもしれません。私たちは他人の知的財産権を侵害したり、他の方法で侵害されたりする可能性があり、これは私たちの業務に実質的な損害を与える可能性があります。より多くの情報については、“リスク要因-私たちの知的財産権に関連するリスク”というタイトルの節を参照してください
41
商標とノウハウ
私たちの候補製品のアメリカと各国際司法管轄区域での発展と進歩に伴い、私たちは商標の保護を創造し、使用可能かつ適切な状況で商標を追求することでその価値を高めることを求めている。特許や商標保護に加えて、ビジネス秘密や技術ノウハウ、持続的な技術革新に依存して、私たちの競争地位を発展させ、維持しています。私たちは、私たちのビジネスパートナー、協力者、従業員、コンサルタントとの秘密協定、および私たちの従業員および選択されたコンサルタントとの発明分配プロトコルを部分的に使用するために、私たちの独自の情報を保護することを求めています。また、私たちは私たちのビルの実体安全と、私たちの情報科学技術システムの実体と電子安全を維持して、私たちの資料と商業秘密の完全性とセキュリティを維持することに力を入れています。私たちはこのような個人、組織、そしてシステムに自信がありますが、合意やセキュリティ措置が違反される可能性があり、私たちのビジネス秘密や他の固有の情報が漏洩する可能性があります。私たちはどんな違反に対しても十分な救済措置がなく、そのような違反のために私たちの商業秘密と他の固有の情報を失う可能性があるかもしれない。しかも、私たちのビジネス秘密は競争相手に知られたり独立して発見されるかもしれない。私たちのコンサルタント、請負業者、または協力者が、私たちのために働いているときに他人が所有している知的財産権を使用する場合、関連またはそれによって生じる商業秘密、ノウハウ、および発明の権利について議論が生じる可能性がある。
私たちの商業的成功はまた第三者の所有権を侵害しないことにある程度依存するだろう。また、私たちは、将来の製品やサービスの特定の側面を開発、製造、商業化することができる第三者独自技術のライセンス権を持っています。いかなる第三者特許を発行することが、私たちの開発または商業戦略を変更し、私たちのプロセスを変更し、許可証を取得したり、いくつかの活動を停止することを要求するかどうかはまだ確定されていません。第三者が許可した特許または特許出願が満了したか、または任意のライセンス契約に違反したり、将来の技術を開発または商業化するために必要な専有権許可を得ることができなかったりすることは、私たちに大きな悪影響を及ぼす可能性がある。もし第三者が米国で準備して提出した特許出願も権利の技術を要求する場合、私たちは発明の優先権を決定するために、米国特許商標局の介入手続きに参加しなければならないかもしれない。
私たちの知的財産権に関連するリスクに関するより多くの情報は、“リスク要因-私たちの知的財産権に関するリスク”を参照してください
42
政府の監督管理
私たちの候補製品の研究、開発、テスト、製造、品質管理、承認、ラベル、包装、貯蔵、記録保存、系列化と追跡、販売促進、広告、流通とマーケティング、承認後或いは許可証の監視と報告及び輸出入などは、すべてアメリカと他の国家政府当局の広範な監督管理を受けている。米国では,FDAは連邦食品,薬物と化粧品法案(FDCA)とその実施条例,公衆衛生サービス法(PHSA)とその実施条例に基づいて生物製品を規制している。適用された米国の要求を遵守しないことは、FDAが生物製品ライセンス申請またはBLA、警告状、製品リコール、製品差し押さえ、生産または流通の完全または部分的な一時停止、禁止、および/または刑事起訴を拒否するような行政または司法制裁を受ける可能性がある。
生物製品の承認と許可手続き
FDAの事前許可や他国の同様の規制機関の承認や許可がなければ,我々のような生物製品は商業販売に使用することはできない。米国では、このようなライセンスを取得する過程は長く、高価でリスクがあり、以下のステップを含む
FDAはガイドラインで,抗体−薬物結合体は生物製品−薬物の組合せ製品であり,BLAに提出すべきであると指摘している。FDAはガイドラインで,モノクロナル抗体を血中乳酸を必要とする生物製品として規制することを示している。FDAはすでに,用量および臨床薬理学的試験推奨を含む抗体−薬物結合体製品の開発考慮に関するガイドラインを発表している。
米国や外国政府当局の規制は、任意の製品を商業化する能力に影響を与える重要な要素であり、このような商業化のタイミングと我々が行っている研究開発活動である。医薬品の商業化は商業化前に政府機関の規制承認を受ける必要がある。各種の法律法規は著者らの製品の研究開発、非臨床と臨床試験、製造、加工、包装、検証、安全性、ラベル、保存、記録保存、登録、上場、流通、広告、販売、マーケティングと発売後の要求および/または約束に影響を与える。このような承認を求める長い過程と、その後の適用法律と法規の遵守には、多くの資源が必要だ。
43
臨床前試験の結果は、製品の化学、調合、毒性と発ガン性の実験室評価、製品及びその調合の潜在的安全性、純度と有効性を評価する動物研究、薬物製造過程及びその制御に関する詳細、及び提案された臨床試験方案とその他の情報を含み、INDの一部としてFDAに提出しなければならず、臨床試験が開始される前にそれを審査して発効しなければならない。臨床試験中の患者の研究方案とインフォームドコンセント情報もまた、その承認のために独立したIRBに提出しなければならず、この委員会は臨床試験を行う各機関をカバーする。スポンサーがINDを提出すると、スポンサーは30個のカレンダーを待たなければならない。FDAがこの30日以内に意見や問題があれば,臨床試験が開始される前に,これらの問題はFDAが満足して解決しなければならない。また,FDAやIRBが臨床試験がFDAの要求に沿って行われていないと考えたり,臨床試験患者にとって受け入れられない重大なリスクとなったりする場合,FDAやIRBは行っている臨床試験に臨床保留を適用することができる。FDAが臨床保留を強制的に実施すれば,臨床試験はFDAが許可した条項の下でしか行われない。もし適用されれば、著者らの臨床前と臨床研究はそれぞれFDAの良好な実験室実践(GLP)と良好な臨床実践(GCP)の要求に符合しなければならず、これらの要求は提出データの質と完全性を確保し、研究患者の権利と福祉を保護することを目的としている。ある臨床試験の情報も一定の期限内に国家衛生研究院(NIH)が維持する臨床試験登録と結果データベース上で公開しなければならない。
一般に、臨床試験は3段階のプロセスに関連するが、これらの段階は重複または組み合わせられる可能性がある:
臨床試験で研究されている候補治療製品は、場合によっては個別患者、中規模患者集団の治療、または拡大獲得レジメン下での広範な治療使用に使用することができる可能性がある。2016年12月に法律に署名された21世紀治療法によれば、1つまたは複数の重篤な疾患または疾患を診断、監視または治療するための1つまたは複数の研究製品の製造業者は、例えば、そのウェブサイト上に掲示することによって、個々の患者がそのような研究製品を得る要求に評価および応答することに関する政策を提供しなければならない。
また、2018年5月30日、2017年Trickett Wendler、Frank Mongiello、Jordan McLinn、Matthew Bellina Right to Trial Actが法律に署名しました。他の事項以外に、この法律はある患者に連邦フレームワークを提供し、彼らが第一段階の臨床試験を完成し、FDAの許可を得た研究用新薬製品を獲得するために調査を行っていることを許可した。場合によっては、条件に適合する患者は、臨床試験に参加することなく、FDA拡大参入計画に従ってFDAの許可を得ることなく治療を求めることができるが、現在の連邦試用権法によれば、メーカーは試験的新薬製品を提供する義務がない。
臨床試験情報の開示
FDA規制製品のある臨床試験のスポンサーはいくつかの臨床試験情報を登録し、開示しなければならない。そして,登録の一部として,製品,患者群,調査段階,試験地点と調査者および臨床試験の他に関する情報が公開されている。スポンサーも完成後にその臨床試験の結果を開示する義務がある。競争相手はこれらの公開された情報を用いて開発計画の進捗状況を知ることができる.
44
孤児薬
孤児医薬品法によれば、FDAは、米国で20万人未満に影響を与えるか、または米国で20万人を超える影響を与える稀な疾患または疾患を治療するための候補治療薬(薬物または生物製品)に孤児薬の称号を付与することができ、米国でそのような疾患または疾患の治療候補薬を開発および提供するコストが、米国での治療候補薬の販売から回収されることを合理的に予想することができない。特定の疾患または状態に対する候補治療薬のマーケティング申請を提出する前に、指定孤児薬を申請しなければならない。FDAが孤児薬物指定を承認した後、FDAは、治療薬の識別およびその潜在的な孤児の使用を開示する。指定孤児薬物は、規制審査および承認過程においていかなる利点も伝達されず、規制審査および承認過程の持続時間を短縮することもない。孤児薬物を指定する他の利点は、いくつかの研究の税金相殺およびBLA申請料の免除を含む。FDAは孤児薬の指定を撤回するかもしれないが、もしそれがそうすれば、それはこの製品がもはや孤児薬として指定されていないことを公開するだろう。
孤児薬物指定を有する製品がその後、このような指定された疾患に対するFDAの最初の承認を得た場合、この製品は、孤児薬物排他性を得る権利があり、これは、FDAが完全なBLAを含む任意の他の申請を承認することができないことを意味し、限定された場合、例えば孤児薬物に対する排他性を有する製品に対する臨床的利点を示さない限り、7年以内に同じ適応の同一の生物製品を販売することができ、またはFDAは、指定された生物製品の疾患または状態を有する患者の需要を満たすのに十分な量の孤児薬が使用可能であることを証明することができない。孤児薬物の排他性は、FDAが同じ疾患または状態に対する異なる生物製品、または異なる疾患または状態に対する同じ生物製品を承認することを妨げるものではない。
指定された孤児薬物の使用許可範囲が孤児が指定された適応を得るよりも広い場合,その薬物は孤児薬物の排他性を得ることができない。さらに、FDAが指定された要求に重大な欠陥があると後に判断した場合、または製造業者がこのような稀な疾患または疾患患者の需要を満たすのに十分な数の製品を保証できない場合、米国での独占営業権を失う可能性がある。
45
開発と審査計画を加速する
FDAは候補製品の開発と検討を加速させるための多くの計画を持っている。これらの計画には、高速チャネル指定、画期的な治療指定、優先審査指定、加速承認が含まれています。高速チャネル指定は、いくつかの基準に適合する新しい生物製品の審査を加速または容易にすることを目的としている。具体的には,新たな生物製品が重篤あるいは生命に危険な疾患の治療を目指し,このような疾患が満たされていない医療需要を解決する潜在力を示した場合,迅速なチャネル指定を受ける資格がある。高速チャネル指定は,製品と研究中の特定の適応の組合せに適している.新生物製品のスポンサーは、この生物製品の臨床開発過程のいつでも、FDAにこの生物製品を迅速追跡製品として指定することを要求することができる。Fast Track製品の場合、FDAは、完全な出願を提出する前に、マーケティング申請の部分を検討することをスクロールしてもよく、スポンサーが申請部分の提出スケジュールを提供した場合、FDAは、申請の部分を受け入れることに同意し、スケジュールが許容可能であると判断し、スポンサーは、申請の第1の部分を提出する際に必要な使用料を支払うことができる。FDAが迅速チャネルの指定が臨床試験中に出現したデータの支持を得なくなったと考えた場合,その指定を取り消す可能性がある。
画期的な治療計画によると、深刻または生命に危害を及ぼす疾患または状態を治療するための製品は、突破的治療指定を受ける資格がある可能性があり、その中には、迅速チャネル計画の利点を享受する資格があることが含まれており、前提は、このような製品が1つまたは複数の臨床顕著な終点に対して既存の療法よりも実質的に改善される可能性があることを初歩的な臨床証拠であることを示している。また,FDAは,治療法製品を突破したスポンサーがタイムリーなアドバイスやインタラクションを受けることを確保し,スポンサーが可能な限り効率的に開発計画を設計·実施することを支援するように努力している。
1つの製品が重篤な疾患の治療を意図しており、承認または許可を得た場合、安全性または有効性の面で顕著な改善を提供する場合、製品は優先審査を受ける資格がある。FDAは優先審査マーケティング申請を受けてから6ヶ月以内に行動する予定ですが、通常審査で提出された申請は受け取ってから10ヶ月以内に行動する予定です。
さらに、1つの製品が重篤または生命に危険な疾患または状態を治療することを意図し、既存の治療方法よりも有意な治療利益を提供する場合、製品は加速承認を得る資格がある可能性がある。加速承認は、十分かつ良好に制御された臨床研究に基づいて、この製品が臨床利益を合理的に予測する可能性のある代替終点に有効であるか、または非可逆発病率または死亡率よりも早く測定することができ、不可逆発病率、死亡率、または他の臨床的利益を合理的に予測することができる臨床終点を決定するのに有効である。承認の条件として、FDAは、承認を加速させた生物製品のスポンサーに、十分かつ制御された臨床研究を勤勉に行い、臨床的利益を証明することを要求することができ、これらの研究は、2022年の食品·薬物総合改革法案(FDORA)によって承認される前に行われることが要求されるかもしれない。また、FDAは承認を加速させる条件として宣伝材料を事前に提出することを求めており、製品商業発売の時期に悪影響を及ぼす可能性がある。高速チャネル指定、画期的な治療指定、優先審査、承認の加速は許可基準を変更することはありませんが、審査過程を加速させる可能性があります。
46
小児科研究と排他性
2003年の“小児科研究公平法”によれば、BLAまたはそのサプリメントは、すべての関連する小児科亜群において生物学的製品が適応と主張される安全性および有効性を評価するのに十分なデータを含み、安全で有効な各小児科亜群に対する製品の用量および投与をサポートしなければならない。2012年の食品と薬物管理局の安全と革新法案の公布に伴い、スポンサーはまたデータを評価する前に小児科研究計画を提出しなければならない。
これらの小児科研究計画は、研究目標および設計、任意の延期または免除請求、および法規要件の他の情報を含む、提案された小児科研究または出願人計画による研究の大綱を含まなければならない。そして,申請者,FDA,FDAの内部審査委員会は提出された情報を審査し,相互に協議し,最終計画について合意しなければならない。FDAまたは出願人はいつでも計画の修正を要求することができる。
FDAは、製品が成人使用許可を得るまで、または小児科データの要求を完全にまたは部分的に免除するまで、申請者の要求に応じて、または一部の小児科データの提出を延期することができる。延期要求および延期要求に関する他の要求および手続きは、“連邦延期審査法”に記載されている。法律または法規に別の要求がない限り、小児科データ要件は孤児の称号を有する製品には適用されない。
小児科専門権は、米国では非特許マーケティング専門権であり、承認された場合、任意の既存の規制専有権の条項に追加の6ヶ月間の市場保護を追加することができる。BLAスポンサーから提出された小児科データがこのようなデータに対するFDAの書面要求に応じていれば,この6カ月の排他性を与えることができる。これらのデータは,この製品が研究されている小児科群で有効であることを証明する必要はなく,逆に臨床試験がFDAの要求に公平に応答していると考えられれば,追加的な保護が得られる。要求された小児科研究報告が法定期限内にFDAに提出され、FDAによって受け入れられた場合、製品の法定または規制排他性または特許保護期間にかかわらず6ヶ月間延長される。これは特許期間の延長ではないが、FDAが別の出願を承認できない規制期間を効果的に延長する。
47
FDAによるBLASの審査
必要な臨床試験が完了した後,BLAを用意してFDAに提出する。この製品が米国で発売される前に、FDAがBLAを承認する必要がある。BLAはすべての臨床前、臨床および他のテストの結果、および製品の薬理、化学、製造および制御に関するデータアセンブリを含まなければならない。BLAの準備と提出のコストはかなり高い。BLAの多くを提出するには巨額の申請使用料が必要であり,現在BLASには2023年度の臨床データが必要であり,費用は3,242,026ドルであり,承認されたBLAによると,メーカーやスポンサーは計画年会費を支払う必要があり,現在の処方薬1個あたりの年会費は393,933ドルである。これらの費用は通常毎年増加します。孤児薬の指定を受けた薬物を申請した発起人はこれらの使用料を免除することができる。
FDAは、BLAを受信した日から60日があり、申請が実質的な審査を可能にするために十分に完全であることを機関に基づいて決定し、申請が届出を受けるかどうかを決定する。FDAはBLAの届出を受け入れるのではなく、より多くの情報を提供することを要求するかもしれない。この場合、申請は追加情報と共に再提出されなければならない。再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。提出された申請が受け入れられると、FDAは深い検討を始めた。FDAはBLAS審査におけるいくつかの業績目標に同意し、適時性を奨励した。生物製品の標準審査の申請は10ヶ月以内に審査され、優先審査薬の申請は6ヶ月以内に審査される。優先審査は、FDAが治療において重大な進展を得ることを決定するか、または適切な治療方法が存在しない治療を提供する生物学的製品に適用することができる。FDAは、いくつかの遅延された情報を考慮するために、または提出中に提供された情報の情報を明らかにするために、標準審査および優先審査の審査手続きをさらに3ヶ月延長することができる(“重大な改訂”)。
FDAは、新しい生物製品の申請を諮問委員会に提出すること、またはなぜそのような推奨がないのかを説明することを要求されている。諮問委員会は通常、臨床医と他の専門家からなるグループであり、審査、評価、提案が申請を承認すべきかどうかを担当する。FDAは諮問委員会の提案によって制限されていないが、それは一般的にそのような提案に従っている。
BLAを承認する前に、FDAは、通常、GCPに適合することを確実にするために、1つまたは複数の臨床場所を検査する。さらに、FDAは薬物を製造する1つ以上の施設を検査するだろう。FDAは、cGMPが満足できる要件を満たし、アプリケーションが適切な基準に適合しない限り、この製品を承認しないであろう。BLAは生物製品が安全で純粋で有効であることを証明するデータを含まなければならない。
FDAがBLAとその付帯情報と製造施設を評価した後、それは承認状または完全な返信を発行する。承認書は、製品の商業マーケティングを許可し、特定の適応に関する具体的な処方情報を提供する。完全な応答文は、一般に、提出中の不足点を概説し、FDAが出願を再検討するために、大量の追加のテストまたは情報を必要とする可能性がある。またはこれらの欠陥がBLAの再提出時にFDAによって満足的に解決された場合、FDAは承認書を発行するであろう。FDAは、含まれる情報タイプに応じて、そのような再提出された出願を2ヶ月または6ヶ月以内に検討することを約束している。この補足情報を提出しても、FDAは最終的にその申請が承認された規制基準を満たしていないと決定する可能性がある。
48
批准書は生物製品の商業販売を許可し、特定の適応の具体的な処方情報を提供することができるかもしれない。BLA許可の条件として、FDAは、生物学的製品の利点が潜在的リスクよりも大きいことを保証するために、リスク評価および緩和戦略、またはREMSを必要とする可能性がある。REMSは、薬物ガイドライン、医療専門家のコミュニケーション計画、および安全な使用を確保する要素、またはETASUを含むことができる。ETASUは、処方または調剤のための特別なトレーニングまたは認証、場合によっては調剤、特殊な監視、および患者登録簿の使用を含むことができるが、これらに限定されない。REMSに対する要求はこの薬物の潜在的な市場と収益力に重大な影響を与える可能性がある。さらに、製品許可は、薬物の安全性または有効性を監視するために、大量の承認後の試験および監視を必要とする可能性がある。一旦付与された場合、規制基準が遵守されていない場合、または初期マーケティング後に問題が発見された場合、製品ライセンスは取り消される可能性がある。
FDAが製品を承認した場合、製品承認の適応を制限することができ、製品ラベルに禁忌症、警告または予防措置を含むことを要求すること、許可を得た後に薬剤の安全性をさらに評価するための第4段階の臨床試験を含む発売後の研究を要求すること、製品の商業化後に製品を監視するための試験および監視計画を要求すること、または製品の潜在的な市場および利益に大きな影響を与えることができるREMSを含む流通制限または他のリスク管理メカニズムを含む他の条件を適用すること。FDAは発売後の研究或いはモニタリングプロジェクトの結果に基づいて、製品の更なるマーケティングを阻止或いは制限することができる。適応、ラベル、または製造プロセスまたは施設の変更を含む承認申請において決定されたいくつかの条件を変更するには、適用されるような新しいBLAまたはサプリメントを提出し、その後、変更を実施することができるように、FDAの承認を得る必要がある。新しい適応に対するBLAサプリメントは通常,オリジナル出願のような非臨床およびCMCデータを必要とし,FDAはサプリメントを審査する際にBLASを審査するのと同様の手順および行動を使用する。
49
生体模倣薬と参考製品の排他性
2009年の“生物製品価格競争と革新法案”(BPCIA)は、FDA許可の基準生物製品と高度に類似しているか、またはそれと高度に類似しているか、または交換可能であることが証明されたバイオ製品候補製品のための短い承認方法を開いた。生物類似性は通常、分析研究、動物研究と1つ以上の臨床研究によって証明することができ、即ち製品は参考製品と高度に類似していることが要求され、臨床では活性成分のわずかな差がないにもかかわらず、生物製品と参考製品は安全性、純度と効力の面で臨床的に意義のある差異がない。互換性は、製品が基準製品生物と類似していることを必要とし、製品は、任意の所与の患者において、参照製品と同じ臨床結果を生成することが期待できることを証明しなければならず、複数回投与された製品の場合、以前の投与後、交換可能な生物学的類似体および参照生物物品は、安全リスクを増加させることなく、または参照生物製品の独占的使用と比較して治療効果のリスクを低下させることなく、交互にまたは交換することができる。FDAによって承認された参照生物製品と類似しているか、または交換可能であることが証明された製品は、FDAが以前に承認された参照製品の安全性および有効性の決定に部分的に依存する可能性があり、これは、承認された製品を市場に投入するのに要するコストおよび時間を減少させる可能性がある。生物製品のより大きく、しばしばより複雑な構造に関する複雑さ、及びこのような製品を製造するプロセスは、重大な障害を構成し、FDAのBPCIAの実施を緩和した。
BPCIAによると,生物類似製品の申請は,参考製品が初めてFDA許可を得た4年後にFDAに提出されなければならない。また,FDAによる生物類似製品の承認は,参考製品が初めて許可された日から12年後に発効する可能性がある。この12年間の参照製品専門期間内に、FDAが競合製品の完全なBLAを承認した場合、出願人自身の臨床前データおよび十分かつ良好に制御された臨床試験からのデータを含み、その製品の安全性、純度および有効性を証明し、別の会社はFDAの許可を得て、参照製品の競合バージョンを販売する可能性がある。BPCIAはまた、交換可能な製品として承認された生物模倣薬のためのいくつかの排他的期限を設けている。この節では,FDAが“交換可能”と考えている製品が本当に州薬剤法に管轄されている薬局に取って代わられるかどうかは不明である。
生物製品は米国でも小児科市場の排他性を得ることができる。以上のように,小児科専有権が付与されれば,既存の専有期間と特許条項を6カ月増加させる。この6カ月間の排他性は,他の排他的保護や特許期間終了時から,FDAが発表したこのような研究の“書面請求”によって小児科研究を自発的に完成させることができる。
BPCIAは複雑であり、FDAによって解釈され、実施され続けている。また、国会が12年間の参考製品専門期間を短縮すべきかどうかも検討した。BPCIAの他の面では,そのいくつかがBPCIAの排他的条項に影響を与える可能性があり,最近の訴訟のテーマでもある.したがって、生物多様性条約の最終実行には大きな不確実性がある。
許可を得てFDAの要求
FDAライセンスに従って生産または流通される生物製品は、記録保存、定期報告、製品サンプリングおよび流通、広告および製品販売促進に関連する要件を含むFDAの普遍的かつ持続的な規制を受けなければならない。許可を得た後、新たな適応または他のラベル宣言を追加するなど、承認された製品の大多数の変更は、FDAの事前審査および許可を経なければならない。どのような上場製品やそのような製品を製造する機関に対しても,継続的な年間使用料要件と,臨床データ補充応用に対する新たな出願料がある。
通常、生物製品がFDAの販売許可を得ていても、FDAは追加の臨床研究を含むいくつかの許可後の要求を満たすことを要求する可能性がある。承認後の要求を満たさなければ,FDAはこの生物製品の許可を撤回する可能性がある。さらに、生物製品ライセンスの保持者は、いくつかの副作用をFDAに報告し、その製品に関する広告および販売促進ラベルのいくつかの要求を遵守し、承認された後も引き続き品質管理および製造プロセスがcGMPに適合するように維持しなければならない。また,生物製品メーカーとその下請け業者はFDAと州政府機関に彼らの工場を登録し,FDAとこれらの州機関の定期的な抜き打ち検査を受け,cGMP要求や他の法規遵守性に適合しているかどうかを把握しなければならない。製造プロセスの変更は厳しく規制されており,通常FDAが事前に承認して実施する必要がある。FDAの規定はまた、cGMPとのいかなる偏差も調査·是正し、スポンサーやスポンサーが使用を決定する可能性のある任意の第三者メーカーに報告や文書要求を行うことを要求している。そのため、メーカーはcGMPコンプライアンスを維持するために、生産や品質管理の分野で時間、お金、労力をかけ続けなければならない。
50
BLA許可の条件の1つは,製造作業がcGMPに適合し続けることを要求することである.CGMPを守るためには、私たち自身の組織内と私たちの契約製造施設内の訓練、生産、品質管理に時間、お金、労力をかけなければなりません。FDAの生産施設の成功検査は通常,生物製品が最終的に許可を得るための前提条件である。BLAが許可を得た後,我々と我々のメーカーは引き続きFDAの定期検査を受け,cGMP要求や許可条件を遵守し続けるかどうかを評価する.私たちはまた外国の規制機関によって調整された似たような検査に直面するつもりだ。FDAはスポンサーの安全報告および/または生産施設に関する記録を定期的に検査し、後者の仕事はcGMPのコンプライアンスを評価することを含む。そのため、メーカーはcGMPコンプライアンスを維持するために、生産と品質管理に時間、お金、労力をかけ続けなければならない。
許可を得ると,規制要求や基準の遵守が保たれていない場合,あるいは製品発売後に問題が発生した場合,FDAは許可を撤回する可能性がある。その後、製品には、予期されない深刻度または頻度の不良事象、または生産プロセス、または規制要件を遵守できなかったことを含む、以前に未知の問題が存在することが発見され、新しいセキュリティ情報を追加するために承認されたラベルの改訂を招く可能性がある;新しい安全リスクを評価するために発売後研究または臨床試験を実施すること、またはREMS計画に従って流通または他の制限を実施することが可能である。他の他の潜在的な結果には
FDAは市場に投入された製品のマーケティング、ラベル、広告、販売促進を厳格に規制している。生物製品は許可の適応にしか利用できず,承認されたラベルの規定に基づいて宣伝される。FDAや他の機関はラベル外用途の普及を禁止する法律法規を積極的に実行しており,ラベル外用途の普及が不適切であることが発見された会社は重大な責任を負う可能性がある。これらの要求を守らないことは、否定的な宣伝、警告状、改正広告、および潜在的な民事と刑事罰を招く可能性がある。医師は、製品ラベルに記載されていない使用のための合法的に入手された製品の処方、および我々が試験およびFDAによって承認された用途とは異なる使用を行うかもしれない。このようなラベル外の使用は医学専門科でよく見られる。医師は,異なる場合,このような非ラベル使用が多くの患者の最適な治療法であると考えるかもしれない。FDAは医者が治療を選択する時の行動を規範化しない。しかし、FDAは製品ラベルの外使用問題に対する製造業者のコミュニケーションを制限した。
そのほか、処方薬製品の流通は、多くの処方を必要とする生物製品を含み、“処方薬販売法”(PDMA)の制約を受け、この法案は連邦レベルの薬品サンプルの分配を規定し、各州の薬品流通業者の登録と監督管理に最低基準を設定した。PDMA,州法ともに処方薬製品サンプルの分配を制限し,分配における責任の確保が求められている。
51
特許期間を延長する
米国では,BLA許可を得た後,関連薬物特許の所有者は最大5年間の特許延期を申請することができ,これはFDA規制過程で失われた特許期間の補償として特許期間の回復を可能にしている。許可された特許期間延長は、通常、INDの発効日と出願延長された特許の発行日とBLAの提出日との間の時間の2分の1として計算され、BLA提出日とBLA承認日との間の時間を加えると、最長5年以下である。FDAが、出願人が職務調査を行っていないと判断した場合、時間を短縮することができる。展示期間後の総特許期間は製品許可日から14年以下である。生物製品の許可を許可するために適用される特許は1つのみ延期する資格があり、その製品、その使用方法、または製造方法に関連する権利要件のみを延期することができ、延期出願は関連特許が満了する前に提出されなければならない。しかし,テスト段階や規制審査中に職務調査が行われていないこと,適用の最終期限内に出願できなかったこと,関連特許が満了する前に出願できなかったこと,適用要求を満たしていなかったことなどの理由で延期が得られなかった可能性がある。一部の(しかし、すべてではない)外国司法管轄区域は、米国のメカニズムよりも多かれ少なかれ厳格で包括的である可能性がある特許期間延長または他の追加の特許排他的メカニズムを有する。
FDAによる診断に伴う承認と規制
1つの治療製品は体外培養ペアリング診断は,この療法により有効である可能性のある患者を選択するために用いられる。治療薬を安全かつ効果的に使用すれば体外培養診断では、FDAは通常、FDAが治療製品を承認すると同時に、その診断、いわゆる随伴診断を承認または承認することを要求する。FDAは2014年8月に最終指導意見を発表し、承認治療製品への適用と体外培養診断を伴う。ガイドラインによれば、新薬のための、キット診断装置およびその対応する治療装置は、製品ラベルにおいて示される治療のためのFDAの承認または承認を同時に得るべきである。FDAは2016年7月、薬物治療および治療のスポンサーを支援するためのガイドライン草案を発表した体外培養製品の共同開発に関する問題に関するキット診断装置。
FDAがキット診断装置を決定することが新しい治療製品または適応を安全かつ有効に使用するために不可欠である場合、セット診断装置がこの適応のための承認または承認を同時に得られない場合、FDAは通常、治療製品または新しい治療製品の適応を承認しないが、高度に満たされていない医療需要は除外される。キット診断装置の承認または許可は、装置が十分に評価され、ターゲット集団において十分な性能特徴を有することを保証するであろう。の回顧です体外培養したがって,我々の候補治療製品の審査に伴い,FDAの生物製品評価と研究センターおよびFDAの設備や放射線健康センターの審査を調整する必要があるかもしれない。
FDCAのもとでは体外培養診断は,随伴診断を含め,医療機器として規制されている。アメリカでは、医療機器の設計と開発、臨床前と臨床試験、発売前の承認或いは承認、登録と発売、製造、ラベル、貯蔵、広告と販売促進、販売と流通、輸出と輸入及び発売後の監督などの事項は、すべてアメリカ食品薬品監督管理局及びその実施条例及びその他の連邦と州法規と条例によって管理されている。適用免除が適用されない限り、診断テストは商業流通の前に市場許可またはFDAの承認を得る必要がある。医療機器に適したFDAマーケティング許可の3つの主要タイプは、510(K)承認、販売前承認、またはPMAおよびDe Novo分類要求とも呼ばれる販売前通知を含む。FDAは通常体外培養診断に伴い癌治療に反応する患者を選択し,薬物承認とともにその診断のためのPMAを得ることを目的としている。
臨床および臨床前データの収集、およびFDAおよびFDAへの審査の提出を含むPMAプロセスは、数年またはそれ以上を要するかもしれない。これは、厳格な上場前審査を含み、その間、出願人は、装置の安全性および有効性に関する合理的な保証と、装置設計、製造、およびラベルを含む装置およびその構成要素に関する情報とをFDAに準備して提供しなければならない。2023年度の多くのPMAの場合、PMA出願の出願料は441,547ドルである。医療機器機関は年次登録費を納付する必要があり,現在6,493ドル,第3種機器定期報告の年会費は現在15,454ドルである。FDAは設備や放射線健康センターで認証された企業に小企業としての費用金額が低いことを求めている。さらに、いくつかのデバイスのPMASは、一般に、FDA承認を求める各適応に対する装置の安全性および有効性を決定するために、広範な臨床前および十分かつ制御された良好な臨床試験の結果を含まなければならない。特に,診断には,PMAアプリケーションは通常,分析や臨床検証研究に関するデータが必要である。PMA審査の一部として、FDAは通常、メーカーの施設が品質体系法規(QSR)に適合しているかどうかを検査し、この法規は詳細なテスト、制御、文書、その他の品質保証要求を規定している。
52
PMAの承認は保証されず、FDAは最終的に申請中の欠陥に基づいてPMA提出を承認できない決定を下し、追加の臨床試験または他のデータを要求する可能性があり、これらのデータの生成は高価で時間がかかる可能性があり、承認を大幅に遅らせる可能性がある。PMA出願に対するFDAの評価が有利である場合、FDAは、一般に、PMAの最終承認を保証するために、ラベルの変更などの特定の条件に同意すること、または最終ラベルを提出するような特定の条件に同意することを要求する承認可能な書簡を発行する。PMAまたは製造施設に対するFDAの評価があまり有利でない場合、FDAは、PMAの承認を拒否するか、または承認できない書簡を発行する。承認できない手紙は、申請の不足点を概説し、可能な場合には、PMAを承認するために必要な条件を決定する。FDAは追加の臨床試験が必要であることも確定している可能性があり,この場合,PMAの承認は数ヶ月または数年遅れて同時に試験を行い,PMAの修正案にデータを提出する可能性がある。FDAが適用基準を満たしていると結論すれば,FDAは承認された適応にPMAを発行し,申請者が最初に求めた適応よりも限られている可能性がある。PMAは、ラベル、販売促進、販売および流通の制限、および承認後の研究および上場後の監視の要求を含むFDAが、装置の安全性および有効性を確保するために必要と考えられる承認後条件を含むことができる。承認されると、承認後の要求、承認条件、または他の規制基準が遵守されていない場合、または予備マーケティング後に問題が発見された場合、FDAはPMA承認を撤回する可能性がある。
設備が市場に投入された後、それは依然として厳格な規制要求を受けている。医療機器の販売は,その許可や承認の用途や適応にしか利用できない。デバイス製造業者はまたFDAに登録とデバイスリストを確立しなければならない。医療機器製造業者およびその供給者の製造プロセスは、医療機器の設計、テスト、生産、プロセス、制御、品質保証、ラベル、包装および輸送の方法および文書を含むQSRの適用部分に準拠しなければならない。メーカーはまた,医療機器報告要件や,設備改正や解体に関するいくつかの要求を遵守しなければならない。米国食品医薬品局は定期的に国内工場の記録や製造プロセスを不定期に検査する。FDAはまた、米国に製品を輸出する外国施設を検査する可能性がある。
外国監督管理
アメリカの法規のほかに、アメリカ以外でどんな製品を販売するかを選択すれば、私たちは様々な外国法規の制約を受けて、これらの法規は私たちの候補製品の臨床試験と商業販売と流通を管理しています。我々の製品がFDAの承認を得ているか否かにかかわらず,臨床試験を開始する前に規制機関の必要な承認を得なければならず,必要であれば外国独立倫理委員会の承認を得,これらの国で候補製品を販売する前に規制部門の承認を得なければならない。承認手続きは国によって異なり,FDA承認に要する時間よりも長いか短い可能性がある.臨床試験、製品許可、定価と精算を管理する要求は国によって異なる。米国と同様に、承認後の規制要求、例えば、製品製造、マーケティング、または流通に関する要求は、米国国外で承認された任意の製品に適用される。
53
他の医療保険法
その中で、FDA、アメリカ衛生·公衆サービス部(HHS)、監察長事務室、医療保険と医療補助サービスセンター(CMS)及び州と地方司法管轄区及び他の国と地域の類似規制機関は著者らが開発している薬物の臨床前と臨床開発、製造、マーケティングと流通に参与する会社に対して大量の激務の要求を提出した。これらの機関と他の連邦、州と地方実体は、私たちの候補製品の研究開発、テスト、製造、品質管理、安全、有効性、ラベル、貯蔵、記録保存、承認、販売、商業化、マーケティング、広告と販売促進、流通、承認後の監視と報告、サンプリング、輸出入などの活動を監督する。我々が開発したどの候補薬も米国の合法的な発売前にFDAの承認を得なければならず,これらの国が合法的に発売される前に適切な外国規制機関の承認を得なければならない。一般的に、私たちの他の国での活動は、重要な違いがあるかもしれないにもかかわらず、米国と類似した性質と範囲の規制を受けるだろう。さらに、欧州連合やEUの規制のいくつかの重要な側面は集中的に処理されているが、具体的な国に対する規制は多くの面で依然として不可欠である。
私たちは現在何の製品も発売されていないにもかかわらず、FDAの薬品マーケティングの制限以外に、私たちは医療保健法律と規制要求の制約、アメリカ連邦と州政府の法執行を受けている。私たちのような製薬会社は連邦政府およびそれらが業務を展開している州と外国司法管轄区域当局の追加医療監督と法執行を受けている。このような規制は、私たちが研究、開発し、最終的に販売、マーケティング、流通を制限し、マーケティングの許可を得た任意の製品の財務配置と関係を制限するかもしれません。これらの法律には限定されません
54
これらの法律または任意の他の適用可能な法律または法規に違反することは、違反疑惑を解決するために、行政、民事および刑事罰、損害賠償、罰金、返却、削減または再構成業務、誠実な監督および報告義務を含むが、連邦および州医療保健計画(例えば、MedicareおよびMedicaid)から除外されること、および監禁を含む重大な処罰をもたらす可能性がある。ビジネス手配が適用される医療保険法に適合していることを確保し,政府当局が行う可能性のある調査に対応することは,時間や資源がかかる可能性があり,会社の業務への関心を分散させる可能性がある。
55
保証と精算を請け負う
いかなる薬品の販売はある程度第三者支払者のこの製品に対する保証範囲、例えば連邦、州と外国政府医療保健計画、商業保険とホスト医療組織、及び第三者支払人のこの製品に対する精算レベルに依存する。提供される補償範囲と金額に関する決定は、支払者ごとに行われる。これらの第三者決済者は、医療プロジェクト(医薬品を含む)やサービスのカバー範囲や精算をますます減少させている。また,医師の監督下で管理されている製品では,このような薬物は価格が高いことが多いため,保険や適切な補償を得ることは特に困難である可能性がある。しかも、製品自体が単独で精算されるかもしれないし、できないかもしれない。逆に、病院や主管医は、私たちの製品を使った治療や手続きを提供することで精算されるかもしれません。
また、米国政府、各州、外国政府は価格制御、保険と精算の制限、低コストあるいは模造薬の代替を要求するコスト制御計画を継続して実施している。既存の規制·措置の管轄範囲内で価格制御とコスト制御措置を講じ、より厳しい政策をとることで、任意の薬品の販売をさらに制限する可能性がある。いかなる薬品の第三者精算または第三者支払人が製品を保証しないことを決定することは、医師の使用と患者のその製品に対する需要を減少させ、販売に実質的な悪影響を与える可能性がある。
また、ある連邦医療計画に参加して製品を入れる条件として、例えば連邦医療保険と連邦医療補助は、製薬業者に医療補助薬品バックオフ計画下の平均メーカー価格やAMP、連邦医療保険の平均販売価格、340 B上限価格、退役軍人事務部に報告する非連邦AMP、および医療補助についてメーカーが使用する製品について法定リベートを支払うことを含むいくつかの価格指標を製薬業者に計算し、政府に報告することを要求するかもしれない。このような法律と法規を遵守するためには大量の資源が必要であり、私たちの収入に実質的な悪影響を及ぼすかもしれない。
56
医療改革
2010年3月、米国では、政府や民間保険会社が医療を支援する方式を大きく変えた“医療·教育和解法案”によって改正された“患者保護·平価医療法案”が公布された。他の我々の業務に影響を与える可能性のある方法の中で、ACAはこのような研究の協調と発展に努力するために、臨床有効性研究の優先順位を監督と確定するために、患者を中心とした新しい結果研究所を設立した;全国支払いバンドル試験計画を含む支払いシステム改革を実施し、病院、医師および他の医療保健提供者がバンドル支払いモードを通じてある医療サービスの協調性、質と効率を高めることを奨励するために、連邦医師支払陽光法案を実施し、医療補助計画の資格基準とリベートを拡大する。
公布以来、行政、司法、立法部門はACAのいくつかの方面に挑戦した。例えば、国会はまだ全面的な廃止立法を通過していないが、2019年1月1日からACAが1年の全部または一部で合格医療保険を維持できなかった個人の税金ベースの分担責任支払いを廃止することを含むACAの次の特定の税収実施に影響を与える2つの法案に署名しており、これは一般に“個人強制”と呼ばれる。また、2021年6月17日、米国最高裁はいくつかの州がACAに対して提出した最新の司法挑戦を却下したが、ACAの合憲性を具体的に裁くことはなかった。最高裁が裁決を下す前に、総裁·バイデンは、ACA市場を通じた医療保険獲得のための障害を設ける政策を含む、特定の政府機関に医療保健の獲得を制限する既存の政策やルールを審査·再考するよう指示した行政命令を発表した。国会がバイデン政府が実施した医療改革措置,あるいは他の挑戦,廃止または代替ACAの努力がACAにどのように影響するかは不明である。
ACAが公布されて以来、アメリカはまた他の立法改正を提案し、採択した。これらの変化には、2011年の予算制御法案が含まれており、この法案には、他の変化を除いて、2013年4月からプロバイダに支払われる医療保険支出が前期当たり合計2%減少し、後続立法により2031年まで続く(新冠肺炎大流行期間に自動減額を一時停止し、2020年5月1日から2022年3月31日まで、その後2022年4月1日から2022年7月1日まで削減幅が1%に低下する)。新冠肺炎の流行期間の一時停止を補うために、2030年、連邦医療保険の支払いは上半期に2.25%、下半期に3%減少する。また、2021年米国救援計画法案は2024年1月1日から薬品メーカーのMDRP還付責任の法定上限を撤廃した。現行法によると,ACAの一部として,医薬品メーカーがカバーする外来薬に対するMDRPリベート責任上限はAMPの100%である。しかし,2024年から保険外来価格の増加速度がインフレ率よりも速ければ,薬品メーカーのMDRPリベート責任はAMPの100%を超える可能性がある。
また、2022年8月、総裁·バイデンは2022年インフレ削減法案、あるいはアイルランド共和軍と署名し、薬品定価改革と新たな医療保険インフレ税還付の創設を含む医療保険計画を重大な改革を行った。すなわち,アイルランド共和軍は(1)連邦医療保険BとD部分で精算された製品に対してインフレリベートを実施し,これらの製品の価格増加がインフレよりも速いことを前提としている,(2)連邦医療保険D部分の福祉を改正し,2025年から受益者の年間自己負担支出上限を2,000ドルとするとともに,製薬メーカーに新たな割引義務を課す,(3)2026年からCMSと価格交渉を行った後,連邦医療保険BとD部分がカバーする固定数の高支出薬品とバイオ製品のための“最高公平価格”を構築する。
57
2022年2月2日、バイデン政府は2016年に開始されたがん月面着陸計画に引き続き取り組むと表明した。政府は声明で、このイニシアティブの新しい目標は、より広範な先端癌療法の獲得を確保し、強力な新しい治療ルートに投資するための不平等問題の解決を含むと指摘した。また、2022年10月14日、バイデン総裁は米国人の処方薬コストの低減に関する行政命令を発表し、CMS革新センターテストのために新しい医療支払いと交付モードを選択するかどうかをHHS長官に指示し、これらのモデルは薬品コストを低減し、連邦医療保険と医療補助計画に参加する受益者が革新的な薬物療法を獲得することを促進する。最近、HHSは2023年2月14日に、CMS革新センターによって試験された3つの潜在的な薬物負担性および可獲得性モデルを選択することを含む2022年10月14日の行政命令に応答する報告書を発表した。具体的には,(1)D部分スポンサーが“高価値薬物リスト”を作成することを許可し,いくつかのよく見られる模倣薬の最高共同支払い金額を2ドルに設定すること,(2)医療補助に重点を置いたモデルは,CMS,メーカーと州医療補助機関との間に協力パートナーシップを構築し,複数の州結果に基づく合意や特定の細胞や遺伝子治療薬を生成すること,(3)連邦医療保険B部分承認計画薬の支払い金額を調整して新療法開発を進めるモデルに関する。
同時に、アメリカ各州はますます立法を通じて、価格或いは患者の精算制限、割引、ある製品への参入とマーケティングコスト開示の制限及び透明性措置を含む薬品と生物製品の定価を制御するための法規を実施している。ある場合、各州はまた他の国から薬品と生物製品を輸入することを奨励し、大量購入を採用する。第三者支払者の支払金額に対する法律で規定されている価格制御又はその他の制限は、私たちの業務の見通し、財務状況、運営結果に悪影響を及ぼす可能性があります。また,地域医療当局や個別病院では,どの薬品やサプライヤーが処方薬や他の医療計画に含まれるかを決定するために入札プログラムが使用されるようになってきている。これは私たちの候補製品に対する最終的な需要を減らすか、あるいは私たちの製品の価格設定に圧力を与えるかもしれない。
私たちは将来、医薬品の価格設定および/または利用可能性、連邦政府および州政府、および他の第三者支払者が医療製品およびサービスのために支払う金額、および/または私たちが収入を創出し、収益性を達成または維持し、または将来規制の承認を得る可能性のある製品を商業化する能力に影響を与える可能性がある、より多くの州および連邦医療改革および/または薬品価格設定措置をとることが予想される。
58
データのプライバシーとセキュリティ
多くの州、連邦、外国の法律·法規は、個人情報(健康に関する情報を含む)の収集、伝播、使用、取得、プライバシー、セキュリティを管理している。このような法律および法規は、医療情報プライバシーおよびセキュリティ法(例えば、HIPAA)、連邦および州消費者保護法律および法規(例えば、連邦貿易委員会法第5条)、州プライバシー法(例えば、カリフォルニア消費者プライバシー法またはCCPA、2023年にカリフォルニア州消費者プライバシー法案またはCPRA改正;バージニア州消費者データ保護法2023年に発効)、データ漏洩通知法、およびEU一般データ保護法規を含む、私たちの業務または私たちのパートナーのビジネスに適用される可能性がある。プライバシーとセキュリティ法律、法規、その他の義務は絶えず変化し、互いに衝突し、コンプライアンス作業を複雑化させ、調査、訴訟あるいは行動を招き、重大な民事および/または刑事罰およびデータ処理の制限を招く可能性がある。
従業員と人的資本管理
概要
私たちは“賢く”“奉仕する”“勇敢に”“粘り強く”“自分になる”などの価値観を卓越を追求する使命と見なしており、私たちの成功の基礎でもあります。これらの価値観は科学と患者に重点を置いた文化を創造し、信頼、チームワーク、祝賀を促進した。2023年3月22日までに75人の常勤永久従業員を雇用した。これらの従業員のうち,約49%が博士,医学博士,工商管理修士,法学博士を含む高度な学位を有し,約69%が研究·開発活動に従事している。私たちの労働力の半分以上と行政指導部は女性で構成されている。私たちの職員たちは労働組合代表もなく、集団交渉協定のカバー範囲もない。私たちは私たちが従業員と仲がいいと思う。
私たちは従業員の知的資本が私たちの業務の重要な駆動力だと思う。著者らは2022年度に従業員チームを拡大した;新入社員を招聘して著者らの臨床と臨床前の仕事を支持し、著者らの研究、臨床開発、臨床運営及び一般と行政機能を増加させた。私たちは、私たちの業務ニーズと機会を評価し、内部の専門知識と能力とアウトソーシングの専門知識と能力のバランスを取るように努力しています。
戦略.戦略
私たちの人的資本資源目標には、私たちの従業員の識別、採用、維持、激励、統合が含まれている。私たちは私たちの持続的な成功が私たちの職員たちの献身、尊敬、そして表現のおかげだと信じている。著者らは経験豊富なオペレータ、腫瘍学専門家、臨床医師と生物製薬ベテランを誘致と維持するために努力し、彼らは深い市場知識と洞察力を持ち、そして患者に解決方案を提供する堅固なビジョンを持っている。この目標を達成するために、多様性、公平、包容に基づく文化を育成し、業績やリーダーシップを奨励し、競争力のある報酬や福祉計画を提供することにより、包容的かつ認可された労働環境を提供する。
文化と従業員の敬業度
私たちはまた私たちのチームの多様性を重視して、性別、背景、専門知識を含めて、私たちの革新文化を育成します。私たちの従業員は、業務行動のための基本的な要求を設定し、私たちの政策、手続き、ガイドラインの基礎として、これらすべてが予期される従業員の行動に追加的な指導を提供する私たちの行動基準に従っている。
59
報酬と福祉
薬物開発は複雑な仕事であり、広範な学科を越えた深い専門知識と経験が必要である。バイオテクノロジーや製薬会社は、大きさにかかわらず、限られた数の合格申請者を争って専門職を埋めている。私たちの全面的な奨励理念の一部として、私たちはトップレベルの人材を誘致し、維持するために競争力のある報酬と福祉を提供する。私たちは各レベルの職員たちに対する私たちの給与と福祉を公平で公平に扱うために努力している。私たちが提供するすべての奨励は、年間業績インセンティブ機会、退職貯蓄計画、健康と福祉、有給休暇と柔軟な勤務時間スケジュールを含む一連の従業員の財務健康を支援する計画を含む。私たちはまた従業員たちが株式奨励計画に参加することを許可する。私たちの株式激励計画の主な目的は私たちの株主と奨励資格のある人の利益を一致させ、幹部、取締役、従業員と他のサービスプロバイダを維持し、激励し、そして彼らが私たちの長期最適な利益に従って行動することを奨励することである。
持続的専門発展
我々は優秀な人材を引きつけ,引き留め,育成することを重視している.私たちは、技術訓練、能力に基づくセミナー、リーダーシップ開発計画を含む、従業員に精算と専門発展コースに参加する時間を提供します。直接マネージャーはまた個性化発展計画を確定する上で積極的な役割を発揮し、その従業員がその潜在力を十分に発揮することを助け、そして責任の向上と増加のために機会を創造し、従業員の参加度を強化し、従業員を維持する。私たちの発展に伴い、私たちは従業員への投資を維持し、増加させることに力を入れて、私たちの雇用、発展、激励、そして従業員の維持方法を改善することを含む。
安全と福祉
職場の職員たちの健康と安全は私たちの主な優先順位の中の一つだ。新型肺炎の流行が新たな挑戦をもたらしたため、私たちは遠隔作業への移行と柔軟な労働時間の提供を含む、私たちの従業員を支援するための様々な措置を取った。同時に,健康環境を確保するための厳しい合意を策定することにより,我々の研究開発活動を維持するために必要な従業員を含め,施設に依存する従業員を保護した。
取締役会監督
我々の取締役会、あるいは取締役会と呼ばれ、我々チームの重要な重要性を認識し、価値観に基づく文化を中心とした多様化、包摂性及び革新的な作業環境を確保する必要性を認識している。私たちの取締役会は定期的に経営陣と会議を開き、私たちの従業員に影響を与える問題を討論し、私たちの従業員をどのように支援するかに集中しています。私たちの文化に対する関心は私たちの取締役会から来て、会社全体を貫いている。私たちの最高経営責任者と管理チームを評価する時、私たちは彼らの私たちの全体文化への貢献を非常に重視している。
我々の取締役会の報酬委員会は、従業員の発展、管理、敬業度、報酬公平、および私たちの人口統計、多様性、包摂性に関する事項を含む経営陣と共に私たちの人的資源活動を検討する責任があります。
我々の取締役会の指名と会社管理委員会は、環境、社会、管理問題を含む会社の責任と持続可能性に関連する任意の会社計画を策定し、取締役会に推薦する責任があります。
企業情報
私たちは2018年6月11日にデラウェア州で登録設立され、2019年7月に1人目の従業員と最初の融資とともにスタートしました。私たちの主な実行事務室はマサチューセッツ州ケンブリッジ市ケンブリッジ路150号にあります。郵便番号:02140、電話番号は(617221-9059)。私たちのサイトの住所はwww.pyxisoncology.comです。
我々のサイトにおける情報や我々のサイトを介してアクセス可能な情報は,本Form 10-K年次報告の一部ではない.我々の10-Kフォーム年次報告、10-Qフォーム四半期報告、8-Kフォームの現在の報告、および取引法第12(A)または15(D)節に提出または提出されたこのような報告の改正は、米国証券取引委員会に電子的に提出または提出された後、合理的で実行可能な範囲内でできるだけ早く無料で私たちのウェブサイトでまたは私たちのウェブサイトを通じて無料で取得することができる。米国証券取引委員会は、Sec.govで提出された文書に関する報告書、依頼書、情報声明、その他の情報を含むインターネットサイトを維持している。これらのサイトの内容は本ファイルに含まれていません。また,これらのサイトのURLへの参照は,非アクティブテキスト参照にのみ用いられる.
60
第1 A項。RISK因子です。
私たちの業務は高い危険と関連がある。以下に説明するすべてのリスクおよび不確実性要因、ならびに本10-K年次報告書に含まれる他の情報は、本10-K年度報告書の最後の財務諸表および関連説明を考慮してよく読まなければならない。以下に説明する危険は私たちが直面している唯一の危険ではない。以下のいずれかのリスク又はその他のリスク及び不確定要因の発生は、我々の業務、財務状況又は経営業績に重大な悪影響を及ぼす可能性がある。この場合、私たちの普通株の取引価格は下がる可能性があり、あなたは投資の全部または一部を失うかもしれません。このForm 10-K年次報告書はまた、リスクおよび不確実性要因に関する前向きな陳述と推定を含む。特定の要因、以下に説明するリスクおよび不確定要因を含むため、私たちの実際の結果は、前向き陳述で予想される結果とは大きく異なる可能性がある。
私たちの財務状況と追加資本需要に関連するリスク
私たちは臨床段階の生物製薬会社で、運営の歴史は限られており、設立以来ずっと重大な損失を受けている。私たちは少なくとも今後数年以内に損失が出て、永遠に利益を達成したり維持したりしないかもしれないと予想している。
私たちは臨床段階の生物製薬会社で、運営の歴史は限られている。設立以来、私たちは重大な運営損失を受けた。2022年12月31日と2021年12月31日までの年間純損失はそれぞれ1兆207億ドルと7600万ドルと報告した。2022年12月31日までの累計赤字は2兆124億ドルだった。今まで、私たちは製品販売から何の収入も得ていません。主に私たちの株式を売ることで、私たちの運営に資金を提供します。したがって、私たちはまだ数年かかると予想され、もしあれば、規制許可と商業化のための製品候補製品を準備することができる。私たちはこのような活動で決して成功しないかもしれないし、たとえ私たちが成功しても、利益を達成するのに十分な収入が生まれないかもしれない。利益を維持するためには、マーケティング許可証の開発、取得に成功し、大量の収入を生み出した製品を商業化しなければならない。これは、私たちが一連の挑戦的な活動で成功することを要求し、これらに限定されないが、臨床および商業規模の製造を調達し、私たちの候補製品の臨床前研究および臨床試験を成功させ、第三者と臨床試験を行う手配を確立し、私たちの候補製品のためのマーケティング許可を獲得し、マーケティング許可を得る可能性のある任意の製品を製造、販売し、より多くの候補製品の権利を発見または獲得し、パートナーを決定して、私たちが決定した候補製品または既存の候補製品の他の用途を開発し、パートナー候補製品の開発を成功させることを要求する。
少なくとも今後数年以内に、巨額の費用と増加する運営損失を招き続けることを予想している。私たちの費用は大幅に増加すると予想されています
61
米国食品医薬品局(FDA)、欧州医薬品局(European Medicines Agency)または欧州医薬品局(EMA)または他の同様の規制機関が、現在予想されている試験以外に試験を行うことを要求する場合、または私たちが計画している臨床試験または任意の候補製品の臨床開発を完了するために適切な製造スケジュールを確立する上で何らかの遅延が生じた場合、私たちの費用は私たちの予想を超える可能性がある。
医薬品開発に関連する多くのリスクや不確実性のため、私たちが発生する費用の増加時間や金額、あるいはいつ利益を達成できるかを正確に予測することはできません。たとえ私たちが確実に利益を達成したとしても、私たちは四半期や年度の収益性を維持したり向上させることができないかもしれない。もし私たちが実現して利益を維持できなければ、会社の価値を下げ、資金を調達し、業務を拡大し、研究開発努力を維持し、あるいは運営を継続する能力を弱める可能性がある。わが社の価値や私たちの普通株の価値が低下し、投資家が投資の全部または一部を失う可能性もあります。
もし私たちが開発した1つ以上の候補製品が商業販売のために承認されれば、これらの承認された候補製品の商業化に関連する巨額のコストが発生すると予想される。たとえ承認された製品の販売から収入を得ることができても、利益を得ることができない可能性があり、運営を継続するために追加の資金が必要かもしれない。
私たちは私たちの運営に資金を提供するために多くの追加資本が必要になるだろう。もし私たちが必要な時や受け入れ可能な条件下でこのような資金を調達できなければ、私たちは私たちの1つまたは複数の研究および製品開発計画または将来の商業化努力を延期、減少または廃止することを余儀なくされるかもしれない。
生物製薬製品の開発は、臨床前研究と臨床試験を含み、非常に時間がかかり、高価と不確定な過程であり、完成するのに数年かかる。設立以来,我々の業務は大量の現金を消費しており,我々が行っている活動に関連する費用が増加することが予想され,特に候補製品PYX−201とPYX−106の臨床試験や他の臨床前研究·開発計画の推進を継続している場合である。例えば,開発努力と支出を我々の2つの最先端プロジェクトPYX−201およびPYX−106に再集中させるために,PYX−203およびPYX−102の臨床前開発を一時停止することにした。私たちが開発した1つ以上の候補製品が商業販売のために承認されても、販売、マーケティング、製造、流通活動に関連した巨額のコストが生じることが予想される。FDA、EMAあるいは他の類似の規制機関が現在予想されている基礎の上で臨床試験あるいは臨床前研究を行うことを要求すれば、私たちの費用は予想を超える可能性がある。他の予期せぬコストもまた現れるかもしれない。私たちの計画と期待される臨床試験の設計と結果は高度に不確定であるため、私たちが開発に成功した任意の候補製品の開発と商業化に必要な実際の資源と資金の数量を合理的に見積もることができない。したがって、私たちは私たちの行動を続けるために多くの追加資金を得る必要があるだろう。
2022年12月31日現在、私たちは約1億793億ドルの現金と現金等価物を持っている。現在の運営計画によると、既存の現金と現金等価物は、2025年上半期の運営費と資本支出需要に資金を提供できると予想されています。私たちが私たちの業務に資金を提供し続けることができると予想される時間の推定は、間違っていることが証明される可能性があるという仮定に基づいており、私たちは現在予想されているよりも早く私たちが利用できる資本資源を使用するかもしれない。変化する状況-その中のいくつかは私たちの制御を超えているかもしれない--私たちの資本消費速度は私たちの現在の予想よりも大きく速く、私たちは計画よりも早く追加資金を求める必要があるかもしれない。
62
私たちの現金と現金等価物を、私たちの候補製品、発見計画、業務開発活動、他の一般会社の目的に関する開発と規制活動に利用する予定です。私たちの候補製品の開発を進めるには多くの資金が必要になるだろう。私たちの現金と現金等価物は規制許可によって私たちの任意の候補製品に資金を提供するのに十分ではないだろう。任意の単一候補製品の成功した研究および開発に関連する時間および活動の長さは非常に不確定であるため、開発、マーケティング許可、商業化活動のためにどれだけの実際の資金が必要かを見積もることはできない。私たちの運営費の時間と金額は主に見られるだろう
もし私たちが適時あるいは受け入れ可能な条件下で資金を得ることができなければ、私たちは私たちの研究開発計画と臨床前研究あるいは臨床試験を延期、減少または中止しなければならないかもしれません(もしあれば)、戦略的機会を制限したり、リストラや他の会社の再編活動を行ったりしなければなりません。私たちはまた、協力者や他の人との手配を通じて資金を求めることを要求されるかもしれません。これらの手配は、私たちが自分で追求していた技術や製品候補製品の権利を放棄することを要求するかもしれません。私たちの候補製品が臨床試験を経て、商業化許可を得てマーケティングに成功しない限り、予測可能な未来に製品販売収入やライセンス製品の印税収入は実現されないと予想される。これまで、私たちは主に株式証券を売却することで私たちの運営に資金を提供してきた。私たちは未来に追加資金を求めることを要求されるだろうし、私たちが追加資金を調達する能力は金融、経済、そして他の要素に依存するだろう。その中の多くの要素は私たちがコントロールできない。私たちは受け入れ可能な条項や追加的な資金を得ることができないかもしれない。例えば、世界経済発展、政治不安、高インフレ、新冠肺炎疫病などの要素による市場変動は、私たちが必要な時に資本を得る能力に不利な影響を与える可能性がある。もし私たちが株式証券を発行することで追加資金を調達すれば、私たちの株主は希釈され、どの融資条項も私たちの株主の権利に悪影響を及ぼす可能性がある。また、私たちに追加資金を提供する条件として、将来の投資家は、既存の株主よりも高い権利を要求し、付与される可能性がある。
63
流動性、金融機関又は取引相手の違約又は不履行に係る事件又は懸念を含む金融サービス業の不利な事態の発展に影響を与え、我々の業務、財務状況又は運営結果に悪影響を及ぼす可能性がある。
金融機関、取引相手または金融サービス業他社の限られた流動性、違約、不良業績または他の不利な事態の発展に影響を与える事件、またはそのような事件または他の同様のリスクへの懸念または噂は、過去に発生し、将来的に市場全体の流動性問題を引き起こす可能性がある。最近では2023年3月10日にシリコンバレー銀行(SVB)がカリフォルニア金融保護·革新部によって閉鎖され,後者は連邦預金保険会社(FDIC)を担当者に任命した。同様に,2023年3月12日,Signature BankとSilvergate Capital Corp.はそれぞれ破産管理プログラムに巻き込まれた.私たちの銀行と顧客関係を必要または適切に評価する必要があると思いますが、私たちが獲得した資金源や他の信用手配は、私たちの現在および予想される将来の業務運営に資金または資本化を提供するのに十分であり、私たち、金融サービス業、または全体経済に影響を与える要因の深刻な損害を受ける可能性があります。他にも、これらの要因には、流動性の緊張または失敗、様々な金融、信用または流動資金協定または手配された義務を履行する能力、金融サービス業または金融市場の中断または不安定、または金融サービス業会社の将来性に対する懸念または否定的な予想が含まれる可能性がある。
さらに、米国または国際金融システムに対する投資家の懸念は、より高い金利またはコスト、より厳しい財務および運営契約、または信用および流動性源を得るための体系的な制限を含む、あまり有利ではない商業融資条項を招く可能性があり、それによって、私たちは融資を受けにくく、さらには融資を受けることができない。他のリスクに加えて、利用可能な資金または現金および流動性資源の減少は、運営費用、財務義務、または他の義務を履行する能力に悪影響を及ぼす可能性があり、契約義務違反、または連邦または州賃金および労働法違反を招く可能性がある。上記のいずれの影響、または上記の要因または他の関連または同様の要因に起因する任意の他の影響は、我々の流動資金および我々の業務、財務状態または経営結果に重大な悪影響を及ぼす可能性がある
私たちの限られた経営の歴史は、私たちの業務のこれまでの成功度を評価することを困難にするかもしれませんし、私たちの将来の生存能力を評価することも難しいかもしれません。
我々は2018年に登録が成立し,2019年中に人員整備と有意義な運営を開始し,これまで開発と臨床前研究に専念し,2023年第1四半期以来,候補製品の第1段階臨床試験を開始してきた。これまで、私たちは臨床試験を成功させ、マーケティング許可証を取得し、直接あるいは第三者を通じて商業規模の製品を製造したり、成功した製品の商業化に必要な販売、マーケティング、流通活動を行う能力があることを証明していません。したがって、もし私たちがもっと長い運営歴史を持っているなら、あるいは私たちがいくつかまたはすべてのタイプの活動を成功的に達成したなら、あなたは私たちの未来の成功または生存能力に対するいかなる予測もそんなに正確ではないかもしれません
また,臨床期生物製薬会社としては,予見できない費用,困難,合併症,遅延などの既知と未知の挑戦に遭遇する可能性がある。私たちはある時点で研究開発に集中している会社からビジネス活動を支援できる会社に転換する必要がありますが、私たちはこの転換に成功できないかもしれません。
様々な要因により、私たちの財務状況と経営業績は四半期ごとと毎年大幅に変動し続け、その多くの要素はコントロールできないと予想しています。したがって、今後の経営業績の指標として、いかなる四半期や年度の業績にも依存してはいけません。
64
我々の候補製品の発見と開発に関するリスク
私たちは、PYX-201およびPYX-106の成功に大きく依存しており、PYX-201および/またはPYX-106が臨床試験で成功しない場合、または規制の承認または許可を得ていない場合、または商業化に成功していない場合、私たちの業務は実質的かつ不利な影響を受けるであろう。
これまで、私たちはPYX-201とPYX-106の開発に多くの努力と財政資源を投入してきた。私たちの将来の成功は、私たちが臨床開発を成功的に開始し、完成し、規制許可を得て、PYX-201とPYX-106を商業化する能力に大きく依存しているが、これは決して起こらないかもしれない。私たちは現在承認や許可を得て商業販売を行っていない製品で、決して適切な製品を開発できないかもしれません。今後数年間、私たちの大部分の努力と支出はPYX-201とPYX-106に使用されることが予想され、これは臨床開発、臨床と製造活動の管理、規制許可、商業規模製造および重大な販売、マーケティング、流通の確立を必要とし、その後、私たちは任意の商業販売から任意の収入を得ることができる。これらの活動のいずれかを成功させることができるかどうか、またはPYX−201およびPYX−106が規制許可を得ても、これらの製品は、他社が提供する療法および技術との競争に成功することができるかどうかを決定することはできない。
生物製品の研究、テスト、製造、ラベル、許可、販売、包装、マーケティングと流通はすべてFDAと他の国の類似規制機関の広範な監督管理を受けている。このような候補製品に対するFDAのバイオ製品ライセンス申請またはBLAの許可が適切に得られるまで、PYX−201およびPYX−106の米国での販売は許可されていない。さらに、私たちは、これらの国から必要な許可または承認を得るまで、どの外国でもPYX-201およびPYX-106を販売することは許可されていない。私たちはまだBLAをFDAに提出しておらず、PYX−201およびPYX−106の同様の出願も他の同様の規制機関にも提出されていない。もしあれば、私たちは数年以内にこれをすることができない。1つの国/地域で必要なPYX-201およびPYX-106の規制許可または承認を得ることができない場合、私たちはその国/地域でこのような候補製品を商業化することができないだろう。したがって、私たちの財務状況は重大な悪影響を受けるだろうし、私たちは私たちの業務を継続するのに十分な収入を生むことができないかもしれない。
私たちの候補製品は開発に失敗したり、遅延に遭遇したりして、その商業生存能力に実質的かつ不利な影響を与える可能性がある。もし私たちまたは私たちの既存または未来のパートナーが私たちの候補製品の臨床開発を開始し、完成させることができなければ、規制部門の承認や許可を得ることができない、あるいはそれを商業化できない、あるいはそうする上で重大な遅延が発生すれば、私たちの業務は実質的に損害を受けるだろう。
私たちは製品が発売されていません。私たちの候補製品の中で二つだけが現在臨床開発の初期段階にあります。したがって、彼らは失敗する危険が高い。私たちが利益を達成し維持する能力は、規制許可を得て、単独でも第三者と協力しても、私たちの候補製品を商業化することに成功している。私たちの候補製品の商業流通の監督管理許可を得る前に、私たち或いは未来のパートナーは広範な臨床前研究と臨床試験を行い、私たちの候補製品の人体における安全性、純度と有効性を証明しなければならない。また,新しい抗体の開発は複雑で困難である。我々の発見と臨床前計画は最初に潜在的な候補製品の決定に希望を示す可能性があるが、様々な理由で、我々が使用した標的選択方法を含む臨床開発の候補製品に変換できない可能性があり、適用可能な候補抗体を生成できないからである。また、私たちのいくつかの候補製品は許可されており、許可や買収を得るために他の候補製品を探し続けます。私たちの臨床前研究または臨床試験は、私たちが許可を得たり、候補製品を獲得する前に完成した研究計画および臨床前研究の結果を複製または推進しない可能性がある。
65
候補製品の規制許可を遅延させたり、商業化したりする問題に遭遇した場合、候補製品の開発を継続したり、既存の候補製品を修正したり、新たな協力を行う財力がないかもしれません
上記のいずれかの状況が発生した場合、私たちは重大な遅延に遭遇したり、私たちの候補製品を商業化することに成功できなくなり、これは私たちの業務に実質的な損害を与える可能性があります。しかも、もし私たちが規制部門の承認を得なければ、私たちは運営を続けることができないかもしれない。
臨床試験を完了したりBLAやNDAを提出したりした会社としては,経験がなく,PYX−201とPYX−106を成功させることができない可能性がある。
臨床試験の進行は長く、高価で、複雑で、高度な監督管理の過程である。私たちの従業員は過去に他の会社で働いていた時、多くの治療分野で成功した臨床試験を行い、規制申請を提出したにもかかわらず、会社としては何の臨床試験も完成していない、あるいはBLAや新薬申請やNDAを提出したため、私たちが予想していたよりも多くの時間とより大きなコストが必要かもしれない。私たちの臨床試験や計画における規制提出を開始または完了できなかったか、または提出を遅延させ、規制部門の承認を得ることを阻止し、PYX-201およびPYX-106を商業化することは、私たちの財務業績に悪影響を及ぼすだろう。大規模な臨床試験は大量の追加の財政と管理資源を必要とし、第三者の臨床研究者と顧問に依存する。第三者臨床研究者、契約研究組織あるいはCROとコンサルタントに依存することは、私たちがコントロールできない遅延に遭遇する可能性がある。さらに、我々の臨床前研究または臨床試験において第三者に依存することは、我々が規制機関に提出する予定の任意の研究または試験に必要な良好な実験室実践またはGLPまたは良好な臨床実践またはGCPを十分に遵守できない可能性があるというリスクに直面する可能性がある。私たちは私たちが受け入れられる条項で決定し、十分な調査者、CRO、コンサルタントと契約することができないかもしれません。
66
INDを提出して予想される時間内に追加的な臨床試験を開始することはできないかもしれないが,たとえ我々ができてもFDAは継続を許可していないかもしれない。
私たちは未来にもっと多くのINDを提出するかもしれない。INDを支援する研究では、私たちは製造遅延や他の遅延に遭遇する可能性がある。また,INDの提出によりFDAが臨床試験の開始を許可しているか,あるいは開始すると,われわれの臨床試験の一時停止や終了を招くことはないとは判断できない。また、適用される規制機関が私たちINDに規定されている臨床試験の設計と実施に同意しても、これらの規制機関が将来彼らの要求を変えないこと、あるいはFDAや他の規制機関が私たちの臨床試験を一部または全部一時停止する可能性があることは保証できない。これらの考慮は上記INDに適用され,将来既存のINDの修正案や新しいINDの一部として提出される可能性のある新たな臨床試験にも適用可能である。私たちが予想していた期間内にINDを提出することができなかったり、私たちの試験を継続する許可を得ることができなくても、直ちに臨床試験を完成させたり、私たちの製品を商業化することを阻止することができます。
著者らの臨床前研究と臨床試験は著者らの任意の候補製品の安全性、純度と有効性を十分に証明できない可能性があり、これは開発、監督管理許可或いは許可と商業化を阻害或いは延期する。
私たちの任意の候補製品(PYX-201およびPYX-106を含む)の商業販売が規制許可を得る前に、私たちは長い、複雑で高価な臨床前研究および臨床試験を通じて、私たちの候補製品が安全で純粋で効果的であり、BLAの要求に適合していることを証明しなければならない。臨床前や臨床試験は費用が高く,完成までに数年かかる可能性があり,これらの活動の結果自体も確定していない。失敗は臨床前研究と臨床試験過程中のいつでも発生する可能性があり、著者らの候補製品は早期開発段階にあり、人体でテストを行ったことがないため、失敗のリスクが高い。さらに、類似候補製品の他の開発者が臨床前または臨床試験で見た任意の失敗または不良結果は、我々の計画の成功に実質的な影響を与える可能性がある。私たちは絶対に適切な製品を開発することに成功しないかもしれない。
著者らの候補製品の臨床前研究と早期臨床試験の結果も後期臨床試験の結果を予測できない可能性がある。候補製品は臨床前研究および早期臨床試験において有望な結果を示す可能性があるが、その後の臨床試験では有効ではないことが証明されている可能性がある。たとえば,動物でテストを行う条件は人間でテストを行う条件とは異なるため,動物研究の結果は人間の体験を正確に予測できない可能性がある.臨床前研究と臨床試験による候補製品の失敗は通常極めて高い流出率を招く。臨床前研究と/或いは初歩的な臨床試験で成功したにもかかわらず、臨床試験後期段階の候補製品は必要な安全性、純度と効力概況を示すことができないかもしれない。同様に、早期の比較的に小規模な臨床試験は大規模な肝心な臨床試験の最終安全性、純度と効力を予測できない可能性がある。生物製薬業界の多くの会社は高級臨床試験において重大な挫折を受け、原因は効力不足、効力持続性不足或いは受け入れられない安全性問題であり、早期試験で人を奮い立たせる結果を得たにもかかわらず。臨床前研究と臨床試験を開始した候補製品の大多数は商業化承認或いは許可を得たことがない。さらに、我々が行った前臨床研究または臨床試験は、私たちが許可を得るか、または候補製品を獲得する前に完成した研究計画および臨床前研究の結果と矛盾し、破壊または他の方法で複製または推進されない可能性があり、これは私たちの業務、運営結果、および将来性に実質的な悪影響を与える可能性がある。
また,我々の候補製品の第1回臨床試験はオープンラベル研究である可能性が予想され,患者も研究者も,患者が研究製品候補か既存の許可生物製品かを受け入れていることを知っている。最も典型的なのは,オープンラベル臨床試験は候補の研究製品のみをテストし,異なる用量レベルでテストを行う場合がある。開放ラベル臨床試験は様々な制限を受けており,これらの制限は任意の治療効果を誇張する可能性があり,開放ラベル臨床試験中の患者が治療を受ける際に知られているからである。また,オープンラベル臨床試験は,“調査者偏見”の影響を受ける可能性があり,すなわち臨床試験の生理結果を評価·審査する人は,どの患者が治療を受けているかを知り,その知識を知っている場合に治療群の情報をより有利に解釈することが可能である。FDAもオープンラベル臨床試験がBLA許可を支持するのに十分かつ良好な制御試験であるとは考えていない可能性がある。
67
著者らが行う可能性のある任意の臨床前研究或いは臨床試験は、監督管理を獲得することが著者らの候補製品をマーケティングすることができる必要な安全性、純度と効力を証明できないかもしれない。もし私たちが行っているあるいは未来の臨床前研究および臨床試験の結果が私たちの候補製品の安全性、純度および有効性に対して定説がなければ、もし私たちが統計的かつ臨床的意義のある臨床終点に達していなければ、あるいは私たちの候補製品に安全問題がある場合、私たちはこれらの候補製品のマーケティング許可を得ることを阻止または延期するかもしれない。ある場合、多くの要素のため、同一候補製品の異なる臨床前研究と臨床試験の安全性、純度と効力結果は著しい差が存在する可能性があり、方案中に規定された試験プログラムの変化、患者群の大きさとタイプの差異、臨床試験方案の変化と臨床試験方案に対する遵守及び臨床試験参加者の退学率を含む。私たちの実験結果は、これらまたは他の副作用の重症度と流行度を明らかにする可能性があり、これは受け入れられない。このような状況が発生した場合、または他の類似製品の開発者が、彼らの候補製品の副作用が許容できない程度または普遍的に存在することを発見した場合、私たちの実験は一時停止または終了する可能性があり、FDAまたは同様の外国規制機関は、私たちの候補製品に対する任意またはすべての目標適応の許可をさらに停止または拒否するように命令することができる。製品に関連する副作用は、患者が進行中の試験を完了する能力を募集または組み入れたり、潜在的な製品責任クレームを招いたりする可能性もある。このようなどんな状況でも、私たちの業務、財政状況、そして見通しを深刻に損なうかもしれない。
また,我々の候補製品は標的毒性に関する臨床試験で副作用を引き起こす可能性がある。標的毒性が観察される場合、または私たちの候補製品が予期しない特性を有する場合、それらの開発を放棄する必要があるかもしれないし、より狭い用途またはサブ集団に開発を制限する必要があるかもしれないが、これらの用途またはサブ集団では、リスク−収益の観点から、副作用または他の特性がそれほど一般的ではなく、それほど深刻ではない、またはより容易に受け入れられるかもしれない。多くの化合物は最初は癌治療の早期テストで希望を示したが,その後副作用が生じることが発見され,この化合物のさらなる発展を阻止した。
私たちの臨床前計画は遅延または永遠に臨床試験に入らない可能性があり、これは私たちが適時あるいは根本的に監督管理の承認や許可を得ることができない、あるいはこれらの計画を商業化する能力に不利な影響を与える。
FDA、欧州委員会(EMAヒト医薬製品委員会の意見に基づく)または他の同様のライセンスを取得して新しい生物製品を販売するためには、ヒトにおける安全性、純度および効力または効果を証明しなければならない。これらの要求を満たすためには,十分かつ良好な制御の臨床試験が必要である。候補製品の臨床試験を開始する前に、海外で計画されているINDあるいは類似の応用を支持するために、広範な臨床前研究を完成させなければならない。われわれの臨床前研究が速やかに完成したかどうかや結果を決定することはできず,FDAや他の類似した外国当局や独立倫理委員会が我々が提案した臨床計画を受け入れるかどうか,あるいはわれわれの臨床前研究結果が最終的に我々の計画のさらなる発展を支持するかどうかを予測することもできない。したがって、私たちは、私たちが予想しているスケジュールでINDまたは同様の臨床前計画申請を提出できることを保証することができず、INDまたは同様の申請の提出がFDAまたは他の規制機関または独立倫理委員会に臨床試験の開始を可能にすることを保証することもできない。
臨床前研究を行うことは長く、時間がかかり、高価な過程である。時間長は、プログラムのタイプ、複雑さ、および新規性によって大きく異なることができ、各プログラムは、一般に数年またはそれ以上の時間であってもよい。私たちまたは潜在的な未来のパートナーによる臨床前研究のどのような遅延も、追加の運営費用をもたらす可能性があります。候補製品の臨床前研究の開始および完成速度は、例えば、多くの要因によって遅延する可能性がある
また,臨床前評価の基準が発展しているため,急速に変化する可能性があり,IND前の提案についてFDAと合意しても,FDAはIND提出文書を受け入れない可能性がある。われわれの臨床前プロジェクトが本当に臨床試験を開始しても,われわれの臨床試験や開発努力は成功しない可能性がある。
68
臨床試験と製品開発は長くて高価な過程であり、結果は不確定である。臨床試験の完了や候補製品の開発と商業化の過程で予期しないコストや遅延が生じたり、最終的には完成できないかもしれません。
臨床試験費用が高く、設計と実施が困難であり、完成まで数年かかる可能性があり、しかも時間と結果はまだ確定していない。1つまたは複数の臨床試験の失敗は、そのプロセスの任意の段階で発生する可能性がある。臨床試験中または臨床試験の結果として、私たちは、マーケティング許可を得ることを延期したり、私たちの候補製品を商業化することを延期または阻止する可能性がある多くの予見不可能な事件に遭遇する可能性があります
69
もし私たちの候補製品に対して、現在予想されている以上の追加の臨床試験または他の試験を行うことを要求された場合、もし私たちが候補製品の臨床試験または他の試験を成功させることができなければ、これらの試験または試験の結果が陽性でないか、または軽微な陽性である場合、または安全問題がある場合、私たちは:
例えばFDAはプロジェクト オプティマス2021年、腫瘍学薬物開発における用量最適化と用量選択パラダイムを改革するイニシアティブとして、FDAは現在の用量選択パラダイムが分子標的治療の用量とスケジュールがキー試験を開始する前に十分に表現されていない可能性を懸念している。生物製薬業界、学術界とその他の利益関係者との協力を通じて、FDAというイニシアティブの目標は腫瘍学的投与量の発見と用量最適化例を推進し、治療効果及び安全性と耐性を最大限に高める用量選択を強調することである。このイニシアティブを支持するために、FDAは腫瘍学候補製品のスポンサーに承認前または承認後の用量最適化研究を要求する可能性がある。FDAは指導文書の作成と最終決定を継続し,腫瘍学候補製品の開発と臨床研究に関するイニシアティブを実施している。
もし私たちが臨床前研究あるいは臨床試験あるいはマーケティング許可証の取得に遅延があれば、私たちの製品開発コストも増加します。われわれのいかなる臨床前研究や臨床試験が計画通りに開始されるかどうか,再構成が必要かどうか,あるいは予定通りに完成するかどうか,あるいは全く知られていない。重大な臨床前研究または臨床試験遅延は、候補製品を商業化する独占的な権利を有する任意の期限を短縮することも可能であり、または私たちの競争相手が私たちの前に製品を市場に出すことを可能にし、候補製品を商業化することに成功する能力を弱める可能性があり、これは私たちの業務、運営結果、財務状況、および将来性を損なう可能性がある。
さらに、癌治療は、1線、2線、または3線として記述されることがある。FDAは通常,新たな腫瘍学的療法のみを承認することが可能であるかもしれないが,最初は三線または後にのみ使用され,2つ以上の他の治療に失敗した後に使用されることを意味する。癌が早期に発見された場合,第一線の療法は,通常,ホルモン療法,手術,放射線療法,免疫療法あるいはこれらの療法の組み合わせであり,治癒を必要とせずに癌を治癒あるいは延命するのに十分な場合がある。以前の治療が無効であった場合、患者に対して二線と三線治療を行う。我々の臨床試験は,1つ以上の以前の治療を受けた患者で行われ,最初にこれらの候補製品を二線または三線治療として使用するための規制許可が求められる予定である。その後、十分に有益であることが証明された製品(あれば)については、第一線の治療として許可を求めることが予想されるが、我々が開発した任意の候補製品は、二線または三線治療のために承認されても、一次治療のために承認されない可能性があり、および/または一次治療の任意の許可を得る前に、追加の臨床試験を行わなければならないかもしれない。
FACTプラットフォームに関連するいかなる失敗や挫折も、不良イベントを含めて、著者らの研究ルートと未来の成功に悪影響を与える可能性がある。
私たちは私たちのADC製品候補製品PYX-201にFACTプラットフォームを使用します。FACTプラットフォームに関連するいかなる失敗や挫折も、不良イベントを含めて、著者らの研究ルートと未来の成功に悪影響を与える可能性がある。例えば、FACTプラットフォームに関連する以前の未知のリスク、または現在考えられているよりも問題がある可能性のある他の問題が発見される可能性があり、これは、規制承認を得るために必要な観察期間を延長し、追加の臨床試験を行う必要があるか、または規制許可を得ることができない可能性がある。私たちのADC候補製品で使用されるFACTプラットフォームまたはその任意のコンポーネントが安全でない場合、私たちは、FATプラットフォームを介して開発された他の候補製品を放棄または再設計することを要求されます。これは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。
70
FACTプラットフォームを利用して製品候補チャネルを構築し、適切な製品を開発し続けることは成功しないかもしれない。
我々はFACTプラットフォームを用いてPYX−201を開発し,我々の候補製品パイプラインを構築し続けている。私たちの業務は、私たちの候補製品の開発に成功し、規制許可を得て商業化する能力だけでなく、私たちのプラットフォームを通じて新しい候補製品を生産し続ける能力にも依存します。私たちが引き続き私たちのパイプを構築し、現在の候補製品の開発をさらに進めることに成功しても、いかなる追加の候補製品も臨床開発に適していない可能性があり、有害な副作用、製造問題、有効性が限られている、またはそれらが臨床開発で成功する可能性がないこと、マーケティング許可を得ること、または市場に承認されていることを示す他の製品を含むかもしれない。もし私たちが候補製品を商業化することに成功して私たちの技術プラットフォームを検証できなければ、私たちは将来的に製品、許可、または協力収入を得ることができないかもしれません。これは私たちの業務、財務状況、運営結果、および見通しに悪影響を与えます。
私たちは、より利益的またはより成功する可能性の高い候補製品を利用することなく、特定の候補製品を追求することに資源を使うかもしれない。
私たちの財務と管理資源が限られているため、私たちはどの目標と候補製品を追求するかについて戦略的決定を下し、他の目標や候補製品の追求を放棄または延期したり、後により大きな商業潜在力を持っていることが証明された他の兆候を提示しなければならない。私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。潜在的な候補製品を正確に評価できなかったことは、市場潜在力の低い候補製品に重点を置くことになる可能性があり、これは私たちの業務、財務状況、運営結果、将来性を損なうことになる。私たちの現在および未来の特定の目標または適応に対する研究、候補製品、および発見計画上の支出は、いかなる商業的に実行可能な製品も生じないかもしれない。もし私たちが特定の候補製品の商業的潜在力や目標市場を正確に評価しなければ、私たちは協力、許可、または他の印税手配によって候補製品に価値のある権利を放棄するかもしれないが、この場合、候補製品の独占開発権と商業化権利を維持することは私たちに有利だ。
もし私たちが開発したすべての候補製品の市場機会が私たちが思っているより小さいなら、私たちの収入は不利な影響を受けるかもしれません。私たちの業務は影響を受けるかもしれません。
私たちの現在の計画または未来の製品候補の潜在的にアドレス指定可能な患者集団は限られている可能性があり、私たちの目標癌を患っている患者の数は予想を下回る可能性がある。私たちの製品候補製品の潜在的にアドレス指定可能な患者集団は推定数字だけです。これらの推定は不正確であることが証明される可能性があり,米国や他の地域の潜在患者の推定数は予想を下回る可能性がある。このような患者は、私たちの候補製品治療を受け入れることができないかもしれないし、または患者はますます識別および接触が困難になる可能性があり、いずれの場合も、私たちの業務、財務状態、運営結果、および成長の見通しに実質的な悪影響を及ぼす可能性がある。
我々が我々の候補製品のために推定した潜在市場と市場機会は、第三者が発表したデータ、私たち自身の市場洞察と内部市場情報、内部生成されたデータと仮定を含む様々な入力に基づいている。私たちはいかなる第三者情報も独立して確認していないし、その正確性や完全性を保証することもできない。市場機会推定は、第三者源から取得または派生しても、内部から作成されたものであっても、重大な不確実性の影響を受け、不正確であることが証明される可能性のある仮説および推定に基づく。私たちは私たちの市場機会推定が合理的だと信じているにもかかわらず、そのような情報は本質的に不正確だ。また,様々な要因により,我々の市場機会の仮定や推定は必ず高度な不確実性やリスクの影響を受けるが,これらの要因には,本年度報告Form 10−Kで述べた要因が含まれているがこれらに限定されない。これらの第三者または内部で生成されたデータが不正確であることが証明された場合、またはこれらのデータに基づく仮定で誤っている場合、私たちの実際の市場は私たちが推定したものよりも限られている可能性がある。さらに、これらの不正確またはエラーは、資本および他の重要な業務リソースを誤って構成することを招き、これは私たちの業務を損なう可能性があります。
71
市場は私たちの新しい治療方法に基づいているので、私たちの候補製品を受け入れないかもしれません。そして私たちは候補製品の販売や許可から未来の収入を生成しないかもしれません。
候補製品の規制許可を得ても,製品が競争力のあるコストで販売できるかどうか,製品が市場に受け入れられているかどうかなどにより,製品の販売から収入を生じたり維持したりすることができない可能性がある。我々が開発しているいくつかの製品候補はFACTプラットフォームに基づいており,新たな技術と治療法である。私たちの未来の成功はこの新しい治療法の成功にかかっている。また、我々のような新製品候補製品の規制許可過程は、他のより有名あるいは広く研究された製品候補製品よりもコストが高く、時間がかかる可能性がある。規制機関はFACTプラットフォームを用いたいかなる治療も許可していない。これらの要因により,製品候補開発の時間やコストを予測することは困難であり,FACTプラットフォームがどの製品の開発やマーケティング許可につながるかも予測できない.私たちが未来に遭遇する私たちのどのプロジェクトに関連するいかなる開発問題も、重大な遅延や意外なコストを招く可能性があり、あるいは商業的に実行可能な製品の開発を阻害する可能性がある。私たちの製品をアップグレードすることは私たちに大きな挑戦をもたらしました
これらの要素のいずれも、私たちがタイムリーまたは利益的に開発する可能性のある任意の候補製品を商業化することを阻止することができる。
新しい治療法の受け入れに重大な影響を与える市場参加者、例えば医師や第三者支払者は、プラットフォームおよび技術に基づく製品または治療を採用しない可能性があり、医学界および第三者支払人が、私たちまたは私たちの既存または未来のパートナーが開発した任意の候補製品を受け入れて使用するように説得することができないか、またはそれに割引の精算を提供することができないかもしれない。私たちの候補製品に対する市場の受け入れ度は他の要素に依存するだろう
もし私たちが商業化した任意の候補製品が市場の受け入れを得られなければ、私たちの業務、財務状況、運営結果、将来性に実質的な悪影響を及ぼす可能性がある。
72
私たちの開発はまだ初期段階にあります。我々の主要候補製品PYX−201およびPYX−106は臨床開発の初期段階にある。臨床前研究と早期臨床試験の結果は未来の研究或いは試験の結果を予測できないかもしれない。臨床試験の初歩的な成功はこれらの試験の完成後或いは後期臨床試験で得られた結果を代表しないかもしれない。
臨床前研究の結果は臨床試験の結果を予測できない可能性があり,どのような進行中の早期臨床試験の結果も後期臨床試験の結果を予測できない可能性がある。そのほか、臨床試験の初歩的な成功はこれらの試験が後期臨床試験で完成した後に得られた結果を代表しないかもしれない。特に,われわれが計画している早期臨床試験では患者数が少なく,これらの試験の結果がその後の臨床試験の結果をあまり予測できない可能性がある。例えば、成功しても、我々の候補製品PYX−201およびPYX−106および他の候補製品の第1段階臨床試験結果は、これらの候補製品または任意の他の候補製品のさらなる臨床試験結果を予測することができない可能性がある。そのほか、臨床前と臨床データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品が臨床前研究と臨床試験で満足できると考えているが、しかし依然としてその製品のマーケティング許可を得られなかった。私たちの未来の臨床試験は最終的に成功しないかもしれないし、私たちのいかなる候補製品の更なる臨床開発も支持しないかもしれない。臨床試験を通過した候補製品は高い失敗率を示した。製薬やバイオテクノロジー業界の多くの会社は臨床開発において大きな挫折を経験しており,早期の研究でも鼓舞的な結果を得ている。著者らの臨床開発におけるいかなるこのような挫折はすべて私たちの業務、運営結果、財務状況と将来性に実質的な損害を与える可能性がある。
さらに、私たちは時々私たちの計画中の臨床試験の中期、トップ、または予備データを発表するかもしれない。著者らが完成する可能性のある臨床試験の中期データは、患者登録の継続とより多くの患者データの獲得に伴い、1つ以上の臨床結果が実質的に変化する可能性があるというリスクに直面している。初期または最も重要なデータはまだ監査と確認手続きを受ける必要があり、これは最終データが私たちが以前に発表または公表した予備データと大きく異なる可能性がある。したがって、最終データが利用可能になる前に、中間データ、営業データ、および予備データは慎重に見られなければならない。初期、営業、または中間データと最終データとの間の不利な差は、私たちの名声およびビジネスの将来性を深刻に損なう可能性があります。
もし私たちが臨床試験で患者を募集する時に遅延や困難に遭遇した場合、私たちは申請を提出し、必要なマーケティング許可を得る(あれば)時間が遅れたり阻止されたりする可能性がある。
FDAや米国以外の類似規制機関の要求に応じて、十分な数の合格患者を見つけて募集することができなければ、私たちの候補製品のために臨床試験を開始することができないかもしれない。個々の臨床試験に十分な数の患者を募集できると信じているが,一般に,特に新冠肺炎が大流行している間に,これらのまれな適応の患者を募集して試験に参加することがどの程度困難であるかは確実には予測できない。該当する患者を識別·募集する能力は限られていることが証明される可能性があり,あるいはこれらの試験を募集する速度は予想よりも遅い可能性がある。また,我々の競争相手の中には,我々の候補製品と同じ適応を持つ候補製品の臨床試験を行っているため,本来われわれの臨床試験に参加する資格のある患者は,競争相手の候補製品の臨床試験に参加することを選択する可能性がある。患者登録が臨床試験に参加することも他の要素の影響を受けている
73
私たちが計画した臨床試験のために十分な数の患者を募集することができない、あるいは私たちは適時にそうすることができなくて、重大な遅延を招き、1つ以上の臨床試験を完全に放棄することを要求するかもしれない。私たちが計画している臨床試験の登録遅延は、私たちの候補製品の開発コストを増加させる可能性があり、これはわが社の価値を低下させ、追加融資を受ける能力を制限します。
私たちの候補製品は不良と予見できない副作用を招く可能性があり、或いは他の安全性に影響する特性を持っており、その臨床開発を阻止し、その監督管理許可を遅延或いは阻止し、その商業潜在力を制限し、或いは重大な負の結果を招く可能性がある。
私たちの候補製品による不良副作用は、規制機関の臨床試験の中断、延期、または停止を招き、より厳しいラベルをもたらす可能性があり、またはFDAまたは他の規制機関の規制許可または承認を延期または拒否する可能性がある。腫瘍薬の場合のように、それらを使用することは副作用があるかもしれない。私たちの実験結果は、これらまたは他の副作用の重症度と流行度を明らかにする可能性があり、これは受け入れられない。この場合、私たちの実験は一時停止または終了される可能性があり、FDAまたは同様の外国規制機関は、私たちの候補製品の任意またはすべての目標適応の許可または承認をさらに停止または拒否するように命令することができる。このような副作用はまた、患者が試験を完了する能力を募集したり、組み入れたり、あるいは潜在的な製品責任クレームを招く可能性がある。これらのいずれも、私たちの業務、財務状況、経営結果、および見通しに重大な悪影響を及ぼす可能性があります。
また,臨床試験の本質は潜在的な患者集団のサンプルを利用することである。患者数と暴露時間が限られているため、私たちの候補製品は希で深刻な副作用は候補製品に接触する患者数が著しく増加した時にのみ暴露される可能性がある。
もし私たちの候補製品が規制許可または承認を得て、私たちまたは他の人が私たちの製品の1つによる副作用を発見した場合、以下のいずれかの不良事件が発生する可能性があり、これは私たちの重大な収入損失を招き、私たちの運営と業務結果に実質的で不利な影響を与える可能性がある
74
もし私たちが発表し、予想された時間枠内で予想された開発目標を達成できなければ、私たちの製品の商業化は延期される可能性があり、したがって、私たちの株価は下落する可能性がある。
私たちは時々各種の科学、臨床、法規と他の製品開発目標が達成される予定の時間を見積もり、私たちはそれをマイルストーンと呼ぶことがある。これらのマイルストーンは、臨床前研究と臨床試験の開始または完成、および監督文書の提出を含む可能性があり、協力者の支払いと関連している可能性がある。私たちは時々いくつかのマイルストーンの予想時間を公開するかもしれない。このすべてのマイルストーンは今と未来に無数の仮定に基づいている。私たちの推定と比較して、このようなマイルストーンの実際の時間は大きく異なる可能性があり、場合によっては、原因は私たちの統制を超えている。もし私たちが公開発表されたこれらのマイルストーンに達していない、あるいは全く達成されていなければ、私たちの収入は予想を下回るかもしれないし、私たちの製品の商業化は延期されるかもしれないし、永遠に実現されないかもしれないので、私たちの株価は下落するかもしれない。
私たちは、新しい治療法や技術プラットフォームを開発する会社を含め、癌候補製品を開発または開発する可能性のある実体からの競争に直面している。もしこれらの会社が技術や候補製品を開発する速度が私たちよりも速い場合、あるいは彼らの技術がより効果的であれば、私たちが候補製品を開発し、成功させる能力は悪影響を受ける可能性がある。
治療用生物製品の開発と商業化競争は激しい。我々は,様々な多国籍生物製薬会社や専門バイオテクノロジー会社や大学や他の研究機関が開発している技術と競争している。私たちの競争相手はすでに開発していて、開発していますか、あるいは私たちの候補製品と競争する候補製品とプロセスを開発します。競争的治療には、医学界によって承認または許可され、受け入れられた治療法と、市場に進出する任意の新しい治療法とが含まれる。かなりの数の製品が現在開発中であり、将来的に商業的に使用される可能性があり、製品候補の開発を試みる可能性のある条件を治療するために使用される可能性があると信じています。生物技術と製薬業界は、腫瘍学部門を含み、迅速に発展する技術、激しい競争及び知的財産権と独自技術に対する有力な保護を特徴とする。私たちが商業化に成功したどの候補製品も、現在市販されている治療法や将来商業化された任意の新しい療法と競争できないかもしれない。
いくつかの会社が癌免疫療法とADCを開発していることを知っている。私たちと違って、これらの会社の多くの会社は資本が十分で、豊富な臨床経験を持っていて、私たちの既存あるいは未来の協力者を含むかもしれません。また,これらの会社は科学や管理人材の募集や臨床試験に参加可能な患者の募集に競争している。
私たちの成功は競争製品よりも安全で純粋で効果的な治療法を開発して保護する能力にある程度依存するだろう。私たちが開発した治療薬よりも安全で効率的で安価な競争製品が商業化されれば、私たちのビジネス機会と成功は減少または解消されるだろう。
もし私たちの候補製品が許可されれば、彼らは一連の開発または現在発売されている治療法と競争するだろう。多くの会社は腫瘍学部門の異なる発展段階で活躍し、ADCや免疫療法のような方法を採用した製品をマーケティング·開発している。2021年4月までに、全世界で約275個のADCが臨床或いは臨床前開発段階にあり、その大多数は各種癌適応の治療に開発されている。また,いくつかの大小の会社が様々な癌治療の免疫療法を研究している。ADC療法や免疫療法のマーケティングにも多くの会社が参加しており、ADC治療会社、アステラス製薬会社、アスリコン社、第一三共株式会社、遺伝子テーク社、ジリッド科学社、グラクソ·スミスクライン社、ファイザー社、楽天医療会社、レーガン社、NextCURE社、ABCURE社が含まれているが、これらに限定されない。
75
我々の臨床前ADCおよび免疫治療候補は、CAR−T療法、二重特異性抗体、および小分子が、パイプライン内の候補者のために開発されている同じ癌タイプのような代替治療からの激しい競争に直面している可能性がある。これらの方法は、我々の技術によって生成された任意の製品よりも効率的で、安全であることが証明されるか、または他の利点を伝達することができる。さらに、我々のPYX−201候補製品EDBの目標はPhilogen S.p.A.から、PYX−203製品候補CD 123の目標はImmunoGen,Inc.,Vincerx Pharma,Inc.,MacroGenicsおよびByondis B.V.,我々のPYX−106製品候補BSI−060 TはNextCure,Inc.Lead Program−NC 318,および我々のPYX−102候補製品の目標,反KLRG 1,Abcuro,Inc.からの特定の目標での競争に直面している。腫瘍学的免疫療法の開発には広範な活動があり,われわれの臨床前発見計画と競合する可能性がある。さらに、もし私たちの任意の候補製品が肺癌、血液病および他の癌などの腫瘍学的適応において承認された場合、それらは通常の化学療法、生物製品および標的薬物小分子療法を含む手術、放射線および薬物治療を含む既存の癌治療方法と競争する可能性がある。
私たちと比較して、私たちの多くの競争相手はより強力な科学、研究開発能力、そしてより多くの財務、技術、製造、マーケティング、販売と資源あるいは経験を持っている。もし私たちが任意の候補製品の許可を得ることに成功したら、私たちは私たちの製品の安全性、純度と効力、私たちの製品の管理の容易さ、患者が比較的新しい投与経路を受け入れる程度、これらの製品の規制許可の時間と範囲、製造、マーケティングと販売能力の可用性とコスト、価格、精算範囲、特許地位を含む多くの異なる要素に基づく競争に直面するだろう。私たちと競争する製品は、私たちが開発する可能性のある任意の製品よりも効率的で、より安全で、より安価で、またはより効率的なマーケティングおよび販売を含む、より良い治療代替案を提供することができます。我々が候補製品を開発·商業化する費用を回収する前に、競争力のある製品は、私たちが開発したいかなる製品も時代遅れまたは競争力を持たないかもしれない。これらの競争相手はまた私たちの従業員を募集する可能性があり、これは私たちの専門レベルと業務計画を実行する能力にマイナスの影響を与える可能性がある。
76
私たちが許可を申請しようとしているバイオ製品候補製品は予想よりも早く競争に直面するかもしれません。
平価医療法案、またはACAは、2009年の生物製品価格競争および革新法案、またはBPCIAと呼ばれるサブタイトルを含み、FDA許可の基準生物製品と類似しているか、または交換可能な生物製品のための短い許可経路を作成する。BPCIAによると,生物類似製品の申請は,参考製品が初めてFDA許可を得た4年後にFDAに提出されなければならない。また,生物類似製品の許可は,参考製品が初めて許可を得た日から12年後にFDAによって発効する可能性がある。この12年間の独占期間内に、FDAが競合製品の完全なBLAを承認した場合、スポンサー自身の臨床前データおよび十分かつ良好に制御された臨床試験からのデータを含み、その製品の安全性、純度および有効性を証明するために、別の会社は依然としてこの参照製品の競合バージョンを販売する可能性がある。この法律は複雑であり、FDAはまだ説明して施行している。したがって、その最終的な影響、実施、そして意味には不確実性がある。FDAがBPCIAを実施するためのこれらのプロセスをいつ完全に採用できるかは不明であるが、どのようなプロセスも、我々の候補製品の将来のビジネス見通しに悪影響を及ぼす可能性がある。
我々が開発する可能性のあるBLAによってバイオ製品の許可を得た候補製品は、12年の排他期を得る資格がない可能性があり、または国会の行動や他の理由で、この排他性が短縮される可能性があるか、またはFDAは、予想よりも早く競争機会を創出する可能性がある競合製品の参考製品として、我々が開発する可能性のある候補製品を考慮しないかもしれない。BPCIAの他の面では,その中のいくつかはBPCIAの排他的条項に影響を与える可能性があり,ACAの合憲性を問う訴訟を含む最近の訴訟のテーマでもある。
例えば、2018年12月、ある連邦地域裁判所は、“個人強制”処罰(減税や雇用法案の一部として国会で廃止された)のACAは完全に違憲であると判断した。2019年12月、米国第5巡回控訴裁判所は、個人権限条項が違憲である地域裁判所の裁決を維持し、これらの条項がACAの残りの部分から分離できるかどうかをさらに分析するために、事件を地域裁判所に返送した。2021年6月17日、米最高裁はこの事件を却下したが、ACAの合憲性を具体的に裁くことはなかった。しかし、未来にはACAに挑戦、廃止、または代替するための他の努力があるかもしれない。我々は、ACAおよびその可能な廃止および代替が、我々の業務およびBPCIAにおける独占経営権に与える(または生じる可能性がある)影響を評価し続ける。このような変化が私たちの業務や財務状況にどの程度影響を及ぼす可能性があるかはまだ確定されていない。
私たちの業務には重大な製品責任リスクがあり、もし私たちが十分な保険範囲を得ることができなければ、このような故障は私たちの業務、財務状況、運営結果、将来性に重大で不利な影響を与える可能性があります。
もし私たちの候補製品と製品が承認されれば、私たちは開発、テスト、製造過程に固有の重大な製品責任リスクに直面すると予想される。製品責任クレームは候補製品開発計画の完了を延期または阻止する可能性がある。もし私たちがマーケティング製品の面で成功した場合、このようなクレームは、FDAが私たちの製品、第三者メーカーの製造プロセスおよび施設、または私たちのマーケティング計画の安全性と有効性を調査し、私たちの製品をリコールするか、または私たちの製品候補製品が使用する可能性のある承認適応を制限すること、または許可証を一時停止または停止することを含む、より深刻な法執行行動を取る可能性がある。是非曲直や最終結果にかかわらず、責任クレームは、私たちの製品に対する需要の減少、私たちの名声が損なわれたこと、関連訴訟の弁護コスト、管理職の時間と私たちの資源が移転されたこと、実験参加者や患者への巨額のお金の奨励、そして私たちの株価の下落を招く可能性がある。また,既存や将来の協力者がFACTプラットフォームを用いて製品を開発する際の行為によって責任を負う可能性がある.しかも、臨床試験と製品責任保険はますます高くなっている。したがって、私たちは製品責任クレームによる損失から私たちを保護するために、合理的なコストで十分な保険を維持することができないかもしれません。これらの損失は、私たちの業務、財務状況、運営結果、見通しに実質的な悪影響を及ぼす可能性があります。
77
規制許可または承認その他の法的コンプライアンス事項に関連するリスク
FDAや他の同様の規制機関の規制許可と承認過程は長く、時間がかかり、本質的に予測できず、もし私たちの候補製品のためにマーケティング許可や承認を得ることができなければ、私たちの業務は深刻な損害を受けるだろう。
FDAや他の類似した規制機関が承認または許可を得るのに要する時間は予測できないが、通常は臨床試験開始後数年後に必要であり、監督機関のかなり大きな適宜決定権を含む多くの要素に依存する。また、承認と許可政策、法規、あるいは承認または許可を得るために必要な臨床データのタイプと数量は候補製品の臨床開発過程で変化する可能性があり、司法管轄区域によって異なる可能性がある。私たちはどの候補製品のマーケティング承認や許可も得ていません。私たちの既存の候補製品や私たちが将来開発を求める可能性のあるどの製品もマーケティング承認や許可を得ないかもしれません。
私たちの候補製品はアメリカでマーケティング許可を得ることができないかもしれません。理由はたくさんあります
この長い許可過程と未来の臨床試験結果の予測不可能性は、私たちの業務、運営結果、財務状況、将来性を深刻に損なう可能性がある候補製品の市場規制許可を得ることができない可能性がある。FDAは許可過程でかなりの自由裁量権を持ち、いつ、または私たちの任意の候補製品のために規制許可を得るかどうかを決定する。我々の候補製品の臨床試験から収集したデータが有望であると信じていても,これらのデータはFDAの許可を支持するには不十分である可能性がある。
さらに、私たちが許可を得ても、規制機関は、私たちの任意の候補製品の適応が私たちの要求範囲以下であるか、または私たちが要求した範囲を超えることを承認する可能性があり、私たちが製品に徴収しようとしている価格を承認しないかもしれないし、高価な発売後の臨床試験の表現に基づいて許可を与えるかもしれないし、あるいは候補製品を承認できるかもしれないラベルには、候補製品の商業化に必要または必要なラベル宣言が含まれていないかもしれない。上記のいずれの場合も、私たちの候補製品のビジネス見通しに実質的な損害を与える可能性があります。
78
たとえ私たちがアメリカで私たちの候補製品のためにFDAの許可を得ても、私たちはいかなる他の管轄区域でも承認されたり、許可されたり、商業化されない可能性があり、これは私たちがそのすべての市場潜在力を達成する能力を制限するだろう。
任意の特定の司法管轄区域で任意の製品を販売するためには、各国に基づいて、安全性、純度、効力および効果に関する多くのおよび異なる規制要件を確立し、遵守しなければならない。
米国FDAの許可は、他の国または管轄区域の規制機関の承認または許可を確保していない。しかし、一つの管轄区域で承認または免許を取得できなかったことは、私たちが他の場所で承認または免許を得る能力に否定的な影響を及ぼすかもしれない。また、一国で行われる臨床試験は、他国の規制機関によって受け入れられない可能性があり、一国の規制承認又は許可は、いかなる他国での規制承認又は許可も保証されていない。
承認の流れは国/地域によって異なるかもしれませんが、追加の製品テストと検証、および追加の行政審査期間に及ぶ可能性があります。外国の監督管理機関の承認或いは許可を求めることは私たちに困難とコストをもたらす可能性があり、追加の臨床前研究或いは臨床試験が必要であり、これは高価で時間がかかる可能性がある。各国の規制要求には大きな違いがある可能性があり、私たちの製品がこれらの国で発売されることを延期または阻止する可能性がある。私たちは国際市場を含めてどの司法管轄区でも承認されて販売できる候補製品は何もありませんし、国際市場で規制の承認や許可を得た経験もありません。もし私たちが国際市場の規制要求を遵守できなかったり、必要な承認や許可を得られなかったり、あるいは国際市場の規制承認が延期されるかもしれなければ、私たちの目標市場は減少し、私たちが開発したどの製品も市場の潜在力を十分に発揮する能力は達成できないだろう。
79
私たちがどんな候補製品の規制許可を得ても、私たちは持続的な規制義務と持続的な規制審査の制約を受けることになり、これは多くの追加費用を招く可能性があり、もし私たちが規制要求を遵守できなかったり、意外な製品候補問題に遭遇した場合、私たちは処罰を受けるかもしれない。
もし私たちの任意の候補製品が監督機関の許可或いは許可を得たら、それらは製造、ラベル、包装、貯蔵、広告、販売促進、サンプリング、記録保存、上場後の研究、追跡と追跡、系列化、上場後の不良事件報告及び安全性、純度、効力、治療効果とその他の発売後の情報を含む持続的な監督管理要求を遵守し、アメリカ連邦と州の要求及び類似外国監督機関の要求を含む。また,われわれが許可を得た後に行ったいずれの臨床試験についても,cGMPやGCP要求を遵守していく。
メーカーおよびメーカーの工場は、品質管理および製造手順がcGMP法規に適合することを確保することを含む、FDAおよび同様の外国規制機関の広範な要求を遵守することが求められている。したがって、私たちと契約製造業者は、cGMPの遵守状況、および任意のBLAまたは他のマーケティング申請で行われた約束および以前の検査意見に対する応答の遵守状況を評価するために、持続的な審査および検査を受けるであろう。したがって、私たちと私たちと協力している他の人たちは、製造、生産、品質管理を含むすべての規制コンプライアンス分野で時間、お金、エネルギーをかけ続けなければならない。
私たちの候補製品のために得られた任意の規制許可は、製品が発売される可能性のある承認指示用途の制限を受ける可能性があり、あるいは第4段階の臨床試験および候補製品の安全性、純度および効力を監視するモニタリングを含む可能性の高い発売後試験要求を含むかもしれない。FDAはまた、REMS計画を我々の候補製品の許可条件とすることを要求する可能性があり、これは、患者の長期フォローアップ、投薬ガイドライン、医師コミュニケーション計画、または配布方法の制限、患者登録、および他のリスク最小化ツールのような安全な使用を確保する他の要素の要求を必要とする可能性がある。似たような外国の規制機関もまたREMSと似たような計画を持っているかもしれない。また、FDAまたは同様の外国規制機関が私たちの候補製品を許可または承認した場合、安全および他の上場後の情報の提出、報告および登録を含む要求を遵守しなければなりません。
我々の候補製品の臨床試験は,臨床試験に入ることに同意した詳細に定義された患者サブセットで行わなければならない。したがって,我々の臨床試験あるいは任意の未来のパートナーの臨床試験は,候補製品の明らかな積極効果が実際の積極効果よりも大きい(あれば),あるいは不良副作用を識別できないことを示唆している可能性がある。もし私たちの1つ以上の候補製品がマーケティング許可を得て、私たちまたは他の人がこの生物製品の効果が以前に考えられていたほど効果的ではないことを発見した場合、または以前に確定されていなかった不良副作用を引き起こし、いくつかの潜在的な重大な負の結果を引き起こす可能性がある
これらの事件のいずれも、私たちの運営および業務に実質的な悪影響を及ぼす可能性があり、私たちの株価に悪影響を及ぼす可能性がある。
80
FDAはまた、REMSの採用と実施を含む製品の安全性、純度或いは効力を監視するために、高価な発売後の研究或いは臨床試験とモニタリングを要求する可能性がある。FDAおよび米国司法省(DoJ)を含む他の機関は、許可された適応のみを確保し、承認されたラベルの規定に基づいて生物製品の販売および流通を保証するために、生物製品のライセンス後のマーケティングおよび販売活動を密接に規制し、監視する。FDAと米司法省は、ラベル外使用に関するメーカーのコミュニケーションに厳しい制限を加えており、承認された適応だけでなく、私たちの製品を販売すれば、ラベル外マーケティングの法執行行動を受ける可能性がある。虚偽請求法案を含む連邦食品、薬物および化粧品法案(FDCA)および他の法規に違反し、処方薬の普及および広告に関連して、連邦および州医療詐欺および乱用法律、および州消費者保護法違反を告発する調査および法執行行動を引き起こす可能性がある。
私たちまたは彼らがマーケティング許可を得た任意の候補製品に対して、私たちとどの協力者も広告と販売促進に関する要求を守らなければならない。処方生物製品に関する販売促進情報は,様々な法律や法規によって制限されており,製品が承認されたラベル中の情報と一致しなければならない。したがって,我々といかなる協力者も,生物製品の許可されていない適応や用途のために開発したいかなる製品も普及させることはできないであろう。FDAは市場に投入された製品のマーケティング、ラベル、広告、販売促進を厳格に規制している。製品は承認された適応と承認されたラベルの規定でしか販売促進できません。FDAや他の機関はラベル外用途の普及を禁止する法律や法規を積極的に実行しており,ラベル外用途の普及が不適切であることが発見された会社は重大な責任を負う可能性がある。しかし、医師はその独立した医学判断に基づいて、ラベル外で使用される合法的に使用可能な製品のために処方することができる。FDAは医師が治療を選択する行為を規範化していないが,FDAはラベル外でその製品を使用するメーカーのコミュニケーションを制限している。FDAと同様の外国規制機関の政策は変わる可能性があり、私たちの候補製品に対する規制許可を阻止、制限、または延期するための追加の政府法規が公布されるかもしれない。米国や海外の将来の立法や行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することはできない。もし私たちが既存の要求の変化に適応できない場合、あるいは新しい要求や政策を採用することができない場合、あるいは私たちがコンプライアンスを維持できない場合、私たちは私たちが獲得する可能性のあるマーケティング許可証を失う可能性があり、利益を達成したり維持することができないかもしれない。
さらに、その後、我々の製品またはその製造業者または製造プロセスには、以前に未知の副作用または他の問題が存在したり、法規要件を遵守できなかったりすることが発見され、様々な結果が生じる可能性がある
81
規制要件や基準を遵守していない場合、または製品発売後に問題が発生した場合、FDAや同様の外国当局は、同意法令を強制的に実施したり、許可証を撤回したりする可能性がある。その後、私たちの候補製品には、予期されない深刻度または頻度の有害事象、または私たちの第三者製造業者または製造プロセス、または規制要件を遵守できなかったことが、新たなセキュリティ情報を追加するために承認されたラベルの改訂につながる可能性があることが発見された;発売後の研究または臨床試験を実施して新しいセキュリティリスクを評価するため、またはREMS計画に従って流通制限または他の制限を実施する。他の他の潜在的な結果には
EUの安全モニタリング或いは薬物警戒に関する要求、及び小児科人のための製品開発に関する要求(以下に更に説明する)を守らないことは、巨額の経済処罰を招く可能性もあり、小児科要求を守らないことは監督管理部門の承認を阻止する可能性がある。同様に、個人情報保護に関するEUとイギリスの要求を守らないことは深刻な処罰と制裁を招く可能性がある。
FDA付与の画期的な療法指定は、私たちのどの候補製品に付与されても、より速い開発や規制審査が可能な過程を招くことはなく、私たちの製品候補製品がマーケティング許可を得る可能性を増加させることもありません。
私たちは私たちの候補製品と未来の候補製品の一部または全部のための突破的な治療法の称号を求めるかもしれない。画期的な治療法は、単独または1つまたは複数の他の薬物または生物製品と組み合わせて重篤または生命を脅かす疾患または状態を治療することを目的とした薬物であり、初歩的な臨床証拠は、薬物または生物製品が、1つまたは複数の臨床的に重要な終点において、既存の療法よりも実質的に改善された効果を示す可能性があり、例えば、臨床開発早期に観察される実質的な治療効果を示す可能性があることを示している。画期的な治療法として指定された候補製品に対して,FDAと試験スポンサーとの相互作用やコミュニケーションは,臨床開発の最も有効な方法を決定するのに役立つとともに,無効な制御案中の患者数を最小限に抑えることができる。FDAによって突破療法に指定された薬物も,これらの他の計画の要求を満たしていれば,加速承認を含む他の迅速承認計画を得る資格がある可能性がある。
FDAはそれを画期的な治療法に指定する権利がある。したがって,我々の候補製品の1つが画期的療法として指定された基準に適合していると考えても,FDAは同意せず,このような指定をしないことにする可能性がある。いずれの場合も、非迅速FDA審査プログラムにより許可を得ることを考慮した候補製品と比較して、候補製品に対する突破療法指定を受けることは、より速い開発過程、審査または許可を招くことはない可能性があり、FDAが最終的に許可を得ることを保証することもできない。さらに、私たちの1つまたは複数の候補製品が突破療法の条件を満たしていても、FDAは後でその製品が資格条件を満たしていないと決定する可能性がある。例えば,FDAは2022年6月にガイドライン草案を発表し,FDAが治療法指定要求を突破しない製品を廃止する際の考慮事項について概説した。したがって,将来的に様々な癌を治療する候補品の一部またはすべてのためにブレークスルー療法認証を求めようとしても,ブレークスルー療法認証が得られる保証はない。
82
FDAの高速チャネル指定は、他の現在または将来の候補製品を付与しても、より速い開発や規制審査、許可プロセスを招くことなく、私たちの候補製品がマーケティング許可を得る可能性を増加させることはない。
私たちは私たちの未来の1つ以上の候補製品のための迅速なチャネル認証を求めるかもしれない。1つの医薬または生物学的製品が、深刻なまたは生命に危険な疾患または状態を治療することが意図されており、そのような疾患または状態を解決するための満たされていない医療需要の可能性を示す場合、薬物スポンサーは、特定の適応のためのFDA迅速チャネル指定を申請することができる。私たちは私たちの候補製品のための高速チャネル指定を求めるかもしれませんが、FDAがこの指定を私たちが提案した任意の候補製品に与える保証はありません。FDAが提供するポリシーやプログラムによると、Fast Track開発製品のスポンサーが提出したマーケティング申請には資格優先審査が可能であるが、Fast Trackの指定はFDAのどのような資格または最終的なマーケティング許可も保証していない。FDAは広範な裁量権を持ち,高速チャネル認証を付与するかどうかを持っているため,特定の候補製品がこのような認証を受ける資格があると考えても,FDAが付与を決定する保証はない。Fast Track認証を確実に取得しても,従来のFDAプログラムや経路と比較して,より速い開発過程,審査や許可を経験することはなく,Fast Track認証を得ることは最終的にFDA許可を得ることは保証されない可能性がある。また,FDAが我々の臨床開発プロジェクトのデータが高速チャネルの指定をサポートしなくなったと考えた場合,その指定を撤回する可能性がある。また,FDAはいつでもFast Trackの指定を取り消すことができる.
もし私たちが現在または未来の任意の候補製品のために孤児薬物指定を求めることを決定すれば、私たちは市場排他性の可能性を補完することを含む、孤児薬物指定に関連するメリットを維持できないかもしれない。
私たちは私たちの現在または未来の1つまたは複数の候補製品のための孤児薬物の称号を求めるかもしれない。米国やヨーロッパを含むいくつかの管轄区の規制機関は、患者数が比較的少ない薬物や生物製品を孤児薬に指定する可能性がある。孤児医薬品法によれば、FDAは、米国では患者数が20万人未満、または米国では患者数が20万人を超える疾患または疾患であると定義されているまれな疾患または疾患の治療のための医薬または生物製品を孤児として指定することができ、合理的な期待がなければ、米国で医薬または生物製品を開発および提供するコストは、医薬または生物製品の米国販売から回収される。米国では,孤児薬物指定は,臨床試験費用,税収割引,ユーザ費用減免のために贈与資金を提供する機会など,一方が財政的インセンティブを得る権利がある。FDAが孤児薬物指定を承認した後、FDAは、薬物または生物製品の識別およびその潜在的な孤児の使用を開示する。孤児薬物指定は、規制審査及び許可過程においていかなる利点も伝達されず、規制審査及び許可過程の持続時間を短縮することもない。
孤児薬物指定を有する製品がその後、このような指定された疾患の特定の活性成分を有するFDAの最初の承認または許可を得た場合、この製品は、孤児製品の排他性を得る権利があり、これは、FDAがNDAまたはBLAを含む他の出願を承認しない可能性があり、同じ適応の同じ医薬または生物学的製品が7年以内に販売される可能性があることを意味する。限定された場合でなければ、孤児薬物排他性を有する製品に対する臨床的優位性を示すように、またはFDAは、指定された生物製品の疾患または状態を有する患者の需要を満たすために十分な数の孤児薬を得ることができることを証明していないことを発見する。したがって、私たちの候補製品が孤児の独占特許を取得しても、FDAは、同じ適応または疾患の治療に使用するために、異なる有効成分を有する他の医薬または生物学的製品を承認することができるかもしれない。しかも、もし私たちが十分な製品供給を生産できなければ、FDAは孤児の排他性を放棄することができる。
私たちは他の孤児適応の中で私たちの候補製品に孤児薬の指定を求めるかもしれないが、もしこれらの候補製品の使用に医学的に信頼できる基礎があれば。孤児薬物指定を受けても,孤児指定適応よりも広い適応の許可を求めると,米国での独占営業権が制限される可能性があり,FDAが後に指定要求に重大な欠陥があると判断した場合,あるいはまれな疾患や疾患を有する患者の需要を満たすのに十分な数の製品がメーカーによって保証されていない場合には,独占営業権を失う可能性がある。しかも、私たちは他の候補製品のための孤児薬物の称号を求めるつもりだが、私たちは決してこのような称号を受けないかもしれない。
国会では最近の第11巡回裁判所の裁決に応えるために、FDCAの孤児薬物条項の更新も考えられている。孤児薬物条項のいかなる変化も私たちが孤児薬物の独占経営権を獲得する機会或いは成功の可能性を変える可能性があり、そして私たちの業務、財務状況、経営業績、キャッシュフローと将来性に重大な不利な影響を与える。
83
FDAの加速承認は、承認されても、より速い開発や規制審査や承認過程をもたらすことがなく、私たちの候補製品がマーケティング許可を得る可能性を増加させることはありません。アメリカの加速承認計画を通じて私たちの製品の許可を得ることができなければ、現在予想以上の他の非臨床研究や臨床研究や試験を行う必要があるかもしれません。これは、許可を得る費用を増加させ、必要な市場許可を得る可能性を低下させ、および/または許可を得る時間を遅らせる可能性があります。承認計画を加速することでFDAの加速承認を得ても,我々の検証的上場後の試験が臨床的利益を検証していない場合,あるいは厳格な上場後の要求を守らなければ,FDAは承認撤回の加速を求める可能性がある。
我々は、PYX−201およびPYX−106の加速承認を求め、FDAの加速承認経路を使用して将来の候補製品の承認を求めることができるように計画している。生物製品を販売するいかなる許可証についても、我々はFDAおよび外国の監督管理機関に臨床データを提供し、製品の安全性、純度および効力がNDAまたはBLAまたは他の対応する規制文書で申請された適応に適合していることを十分に証明しなければならない。承認計画の加速はFDAが使用するいくつかの方法の一つであり、目的は処方薬或いは生物製品をより迅速に生命に危害を及ぼす疾患の治療に応用することである。アメリカ食品薬品監督管理局第506条(C)条は、FDAは“深刻或いは生命に危害を及ぼす疾患の製品を許可することができ、条件はこの製品が臨床利益を合理的に予測できる代替終点に有効であるか、或いは不可逆的な発病率或いは死亡率よりも早く測定できる臨床終点に有効であることを決定することであり、しかもこの疾患の深刻性、希少性又は流行率及び使用可能又は代替治療の不足を考慮して、不可逆的な発病率又は死亡率又は他の臨床利益への影響を合理的に予測することができる”と規定している。しかし、許可計画を加速することによる許可の条件は、スポンサーは十分かつ良好に制御された発売後の臨床試験を行わなければならず、薬物の臨床利益を検証と記述し、その中で代替終点と臨床効果の関係或いは観察された臨床終点と最終結果の関係に不確定性が存在することである。通常、発売後の臨床試験により生物製品が臨床上意義のある積極的な治療効果を提供することを表明した時、即ち患者の感覚、機能或いは生存方式への影響は臨床メリットを検証する。これらの検証的試験は,職務調査の場合に完了しなければならず,FDORAによる承認前にこのような研究が要求される可能性がある。もしこのような検証性が発売された後、試験がこの製品の臨床特徴或いはリスクと利益を確認できなかった場合、FDAはこの製品の加速承認を撤回する可能性がある。
FDAは承認計画の加速による許可の面で広範な裁量権を有しており,承認計画が我々の製品に適用されると考えても,FDAが最終的に同意することを保証することはできない.また,承認計画を加速させることで許可を得ても,従来のFDAプログラムよりも速い開発過程,審査,許可を経験しない可能性がある.
FDAが加速承認を求めるBLAを審査しても,ライセンスが速やかに発行されるか,あるいはまったく保証されない保証はない。FDAは私たちの研究設計や結果が加速承認を支持することに同意しないかもしれない。さらに、FDAは、承認計画を加速させることによる許可が不適切であることを決定することを含む、任意のタイプの許可を付与する前にさらなる研究または試験を行うことを要求することができ、我々の臨床試験は、従来のアプローチによる許可を支援するために使用されない可能性がある。私たちはFDAの要求をタイムリーに満たすことができないかもしれないが、これは遅延を招くかもしれないし、私たちが提出した材料がFDAによって不完全とされて許可を得ることができないかもしれない。FDAからのその後のフィードバック後も,承認計画を加速させることで許可を求めていく保証はない.承認計画を加速させることで許可を得ることができなかったことは、私たちの製品が許可される時間を長くする可能性があり、その開発コストを増加させる可能性があり、製品を商業化する能力を遅らせる可能性があり、財務状況や市場競争の地位を深刻に損なう可能性がある。
承認計画を加速することで私たちの製品の許可を得ても、FDAが要求する可能性のある1つ以上の検証的な発売後の試験を完了して、この製品の臨床的利益を検証し、配布前にFDAにすべての宣伝材料を提出することを含む厳格な発売後の要求を受ける。このような要求は製品の商業発表時間に悪影響を及ぼす可能性がある。たとえ私たちがより速い承認を得たとしても、私たちはより速い開発、規制審査が可能かもしれない過程を経験しないかもしれない。また,加速承認を得ることは,最終的にFDAの完全な許可を得る保証はない。
FDAは様々な理由で承認の撤回を求めることができ、もし私たちが何の必要な職務調査の検証性上場後の試験を行っていない場合、著者らの検証性上場後の試験は予想される臨床利益を確認していない、他の証拠は製品が使用条件下で安全でないこと、不純或いは非有効であることを示し、あるいは著者らはFDAが発見した虚偽と誤った宣伝材料を伝播することを含む。
承認計画を加速させることによって許可を得ることができないか、または加速承認計画によって得られた承認を維持する上でのいかなる問題も、当社製品の商業化を延期または阻止し、私たちの業務、財務状況、運営結果、キャッシュフロー、および将来性に大きな悪影響を及ぼすだろう。
84
もし外国の規制機関が上場承認された候補製品の生物類似バージョンを承認した場合、あるいはこれらの規制機関が私たちの候補製品の模造バージョンを承認する前に私たちの候補製品に適切なデータ期間や市場排他性を与えなかった場合、私たちの候補製品の販売は悪影響を受ける可能性がある。
EUおよびイギリスでは、革新的な医薬製品は、完全なマーケティング許可申請および条件付き許可(以前に承認された別の医薬製品のマーケティング許可プロファイル内のデータに依存するマーケティング許可申請に依存するのではなく)に従って許可される。革新的な医薬製品の発売許可申請書は含まれていなければなりませんその他を除いて医薬試験、前臨床試験、および販売許可を求める薬品に対する臨床試験の結果(適用の場合、免除または延期が得られない限り--以下に述べる)。
販売許可は集中手続きや国家手続きによって得ることができる。集中的な手続きは、欧州経済地域(EEA)全体で有効である欧州委員会(EMAの意見に基づく)によって発表された単一のマーケティング許可をもたらし、EEAはEU、アイスランド、リヒテンシュタイン、およびノルウェーからなる。以下のヒト薬物の場合、集中手順は強制的である:(1)遺伝子工学のようなバイオテクノロジープロセスに由来する、(2)HIV/エイズ、癌、糖尿病、神経変性疾患、自己免疫および他の免疫機能障害、およびウイルス性疾患などの特定の疾患の治療に使用可能であることを示す新しい活性物質を含む;(3)指定された孤児薬、(4)遺伝子治療、体細胞治療または組織工学薬などの高度治療薬。他の場合には、出願人の要求に応じて、集中手順を使用することもできる。したがって、私たちが開発している候補製品にとって、集中化手続きは強制的になるだろう。
もし上場許可の申請者が完全なファイルを提出した場合、その中に自分の薬物、臨床前試験と臨床試験データが含まれており、しかもこの申請は現有の医薬製品の“全世界マーケティング許可”に属していない場合、上場許可を承認した後、候補参考製品は8年間のデータ独占権と他の2年間の市場独占権を得ることができる。承認された場合、データ排他期間内に、生物模倣薬の承認申請者は、許可された候補製品または参照製品として提出された上場許可ファイルに含まれるデータに依存して、彼らの出願をサポートすることができない。市場排他期は、EUが最初のマーケティング許可を得た10年後まで、その製品をEUで商業化することを成功させた生物類似申請者を禁止しているが、その間に生物類似マーケティング許可申請を提出することができる。この10年の最初の8年間に、マーケティング許可所有者が1つまたは複数の新しい治療適応の許可を得た場合、10年間の市場専門期間をさらに1年間延長することができ、許可前の科学的評価では、これらの適応は、既存の治療法と比較して有意な臨床的利益をもたらすことができると考えられる。しかしながら、1つの化合物が新しい活性物質であると考えられていても、イノベーターはデータ期間および市場排他性を得ることができ、他の知的財産権または規制排他性が適用されない限り、他の関係のない会社もマーケティング許可を申請することができ、同じ治療適応のために別の競争相手の医薬製品をマーケティングすることができ、その会社がその申請をサポートする完全な独立した科学データパケットに基づいて、単独のマーケティング許可申請に従って自分のマーケティング許可を得ることができることを前提とする。
EUでは、生物模倣薬に対して特殊な制度があり、即ち生物模倣薬、即ち参考薬用製品と類似しているが、模倣薬製品の定義に符合しない生物薬用製品であり、例えば、原材料或いは製造技術の違いによる。このような製品については,適切な臨床前試験や臨床試験の結果を提供しなければならず,EMAのガイドラインは異なるタイプの生物製品に提供される補足データのタイプを詳細に説明している。遺伝子や細胞療法薬物製品などの複雑な生物製品に対するこのようなガイドラインは現在のところないため,短期的にはこれらの製品の生体模倣薬がEUで承認される可能性は低い。しかし,EMAのガイドラインは,将来的には当時得られた科学的知識や規制経験に基づいてこれらの提案を考慮することを指摘している。
EUでは、新しい医薬製品の識別許可申請は、EMAの小児科委員会またはPDCOと合意された小児科調査計画またはPIPに適合する小児科集団で行われた臨床試験の結果を含まなければならない。場合によっては、PDCOは、これらの要件を免除または延期することができる(例えば、このような状況が成人人口でのみ発生する場合、免除を得ることができる)。必要があれば、小児科研究は既存と新しい適応、薬理形式と投与経路の小児科人口のすべてのサブグループをカバーしなければならない。生物学的に似たアプリケーションを含む限られた適用をさらに排除する。小児科研究を完成するにはいくつかのインセンティブがあるかもしれない。例えば、すべての欧州連合加盟国で販売許可が得られ、研究結果が製品情報に含まれると、否定的な場合であっても、その製品は、6ヶ月間の追加保護証明書の延期(承認時に有効であれば)を得る資格があるか、または、孤児医薬製品については、孤児市場固有権を2年間延長することが許可される。
85
EUでは、“孤児薬品”を指定する基準は原則として米国と似ている。以下の場合、医薬製品は、孤児として指定することができる:(1)生命または慢性衰弱を脅かす疾患の診断、予防または治療のための、(2)(A)申請時に、そのような疾患のEUでの影響が10,000人中5人以下であるか、または(B)EUで投資が合理的であることを証明するのに十分な見返りを生じない孤児のアイデンティティによるメリットがない場合、(3)満足できる診断、予防または治療方法がEUに発売されることを許可していない場合、またはそのような方法が存在する場合、この製品は、その疾患の影響を受けている人に大きな利益をもたらすであろう。孤児薬物指定申請は発売許可申請の前に提出しなければならない。孤児薬物指定は、一方が費用の低減や費用の免除などの財政的奨励を受ける権利があり、マーケティング許可を得た後、10年間の市場排他性を得る権利がある。10年間の市場専門期間内に、環境保護庁は、同様の医薬製品について別のマーケティング許可申請を受け入れることができないか、または同じ治療適応について別のマーケティング許可を承認するか、または既存のマーケティング許可を延長する申請を受け入れることができない。孤児医薬品はまた、EUの小児科研究において追加的な2年間の市場排他性を得ることができる。いかなる補充保護証明書も孤児の症状に関する小児科研究によって延期してはならない。孤児薬物指定は、規制審査および承認過程においていかなる利点も伝達されず、規制審査および承認過程の持続時間を短縮することもない。
5年目の終了時に当該製品が孤児薬物指定の基準を満たしていないと判断された場合、例えば、その製品の利益が十分に高く、市場独占経営を維持することが合理的であることを証明するのに十分でない場合、10年間の市場独占経営権は6年に減少することができる。以下の場合、同じ適応について類似製品にマーケティング許可を随時付与することができる
イギリスはEUから離脱したにもかかわらず、その規制法の枠組みは類似した保護期間(すなわち規制データ排他性、マーケティング保護、市場排他性)を規定している。
私たちの候補製品は、候補製品の生物類似バージョンからの競争に直面する可能性があり、これは、私たちの将来の収入、収益性、およびキャッシュフローに実質的な悪影響を与え、これらの候補製品の投資から利益を得る能力を大きく制限する可能性がある。私たちの将来の収入、収益力、キャッシュフローも実質的で不利な影響を受ける可能性があり、もし私たちの候補製品が承認された後に適切な非特許特定期間を得られなければ、私たちがこれらの候補製品から投資収益を得る能力は大きく制限されるかもしれない。
私たちが行う可能性のある任意のセットの診断テストについては、必要な規制許可や承認が得られなければ、任意の候補製品の承認を阻止または延期する可能性があります。さらに、セット診断を必要とする候補製品の任意の商業的成功は、任意の必要な規制許可または承認を得るかどうか、およびそのような試験の持続可能性に依存する。
私たちのいくつかの適応候補製品の臨床開発については、私たちの製品候補に適した患者を決定するために、パートナーと協力して開発またはセット診断テストを得ることができるかもしれない。私たちは、これらのセット診断の開発、テスト、および製造、必要な任意の規制許可または承認、およびこれらのセット診断の商業的供給を申請し、受信するために第三者に依存する可能性がある。FDAや外国規制機関は診断に伴う医療機器としての規制を行い,これらの設備は候補製品の臨床試験とともに臨床試験を行う可能性があり,商業化前に単独の規制承認や承認が必要である。このプロセスは、前会議の提出、研究装置免除またはIDEの要求の提出など、衛生当局との追加の会議を含むことができる。“重大リスク機器”として指定された随伴診断については,この診断を対応する候補製品の臨床試験と組み合わせて使用する前に,IDEに対するFDAおよびIRBの承認を得る必要がある。私たちまたは私たちの第三者パートナーは、必要な規制許可や承認を得ることができないかもしれません。これは、私たちの候補製品の承認を阻止または延期するかもしれません。さらに、セット診断を必要とする候補製品の商業的成功は、必要な規制許可または承認を得るかどうかに依存し、そのような第三者が関連地域で合理的な条項でセット診断の持続的な能力を提供してくれる。
86
もし私たちが未来にそうすることを要求され、もし私たちが私たちの候補製品の開発にそのようなテストのセット診断テストを必要とすることに成功できなければ、あるいはそうする過程で重大な遅延に遭遇すれば、私たちはこれらの候補製品のすべてのビジネス潜在力を達成できないかもしれない。
FDAは私たち自身またはパートナーと私たちの製品候補製品のためのいくつかの適応のセット診断テストを開発することを要求するかもしれない。成功するためには、私たちまたは私たちの協力者はいくつかの科学、技術、監督と後方勤務方面の挑戦を解決しなければならない。私たちはこれまで医療設備や診断テスト開発の経験がありません。FDAの承認や承認を自ら開発·求めて診断テストを行うことを選択すれば,医療機器の知識や専門知識を持つ者がより多く必要となる。私たちは第三者が私たちの候補治療製品のための診断テストを設計、開発、製造することに依存するかもしれません。これらの製品はこのようなテストが必要です。これらの候補治療製品の開発に成功した場合、あるいは開発を遅延させることができない場合、私たちは現在および計画されている臨床試験のために十分な患者を募集することができない可能性があり、これらの候補治療製品の開発は不利な影響を受ける可能性があり、これらの候補治療製品はマーケティング承認を得ることができない可能性があり、私たちはマーケティング許可を得たこれらの治療薬のすべての商業的潜在力を実現できないかもしれない。このセットの診断プログラムの開発に成功しなかった場合は、本試験の登録遅延を招く可能性があり、私たちの候補製品が市場の承認を得ることを支持するために、さらなる臨床試験を開始或いは完成できない可能性がある。したがって、私たちの業務、運営結果、そして財務状況は実質的な被害を受ける可能性がある。
私たちと顧客、医師、第三者支払者との関係は、連邦と州医療詐欺と乱用法律、虚偽クレーム法律、その他の医療法律法規の制約を直接あるいは間接的に受けている。もし私たちがこのような法律を遵守できないか、または完全に遵守できなければ、私たちは巨額の処罰に直面するかもしれない。
米国や他の地域の医療提供者、医師、第三者支払者は、マーケティング許可を得た任意の候補製品を推薦し、処方する上で主な役割を果たす。私たちは現在と未来の医療専門家、主要な調査者、コンサルタント、顧客と第三者支払者との間の手配により、私たちは様々な連邦と州詐欺と乱用法律、その他の医療保健法律の制約を受けて、もし私たちがマーケティング許可証を取得すれば、これらの法律は私たちの研究、開発、販売、マーケティング、流通、私たちの候補製品の業務あるいは財務手配と関係を制限するかもしれません。特に、私たちの候補製品の研究、および医療製品やサービスの普及、販売とマーケティング、および医療業界のいくつかのビジネス配置は、詐欺、リベート、自己取引、および他の乱用を防止するための広範な法的制約を受けている。これらの法律法規は、幅広い価格設定、割引、マーケティングおよび販売促進、構造および手数料、特定の顧客インセンティブ計画、および他のビジネスまたは財務スケジュールを制限または禁止する可能性があります。
私たちが第三者の業務手配と適用される医療法律や法規に適合することを確保することは、コストが高くなる可能性があります。持続的に変化するコンプライアンス環境、および異なるコンプライアンスまたは報告要件を有する複数の司法管轄区域の必要性に適合するために、穏健かつ拡張可能なシステムを確立し、維持することは、医療会社が1つまたは複数の要件と衝突する可能性を増加させる。政府当局は、私たちの業務実践が現在または未来に適用される詐欺および乱用または他の医療保健法律および法規に関連する現行または未来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。もし私たちの運営がこれらの法律または任意の他の私たちに適用される可能性のある政府法規に違反していることが発見された場合、私たちは重大な民事、刑事と行政処罰、損害賠償、罰金、返還、監禁、またはMedicareおよびMedicaidのような政府援助に参加する医療計画から除外される可能性があり、もし私たちが会社の誠実な合意または同様の合意の制約を受けて、これらの法律違反に関する告発、契約損害、名声損害、および/または私たちの業務の削減または再編を解決するために、追加の報告要件と監督に直面する可能性があります。
もし私たちがそれと業務を展開することを期待している医師または他の提供者または実体が適用法律を遵守していないことが発見された場合、彼らは政府の援助された医療計画から除外されることを含む重大な刑事、民事または行政制裁を受ける可能性がある。解決策が私たちに有利であっても、医療法令に関連する訴訟や他の法的手続きは、巨額の費用を発生させる可能性があり、私たちの技術や管理者の正常な責任を分散させる可能性があります。さらに、公聴会、動議、または他の一時的な手続き、または事態の発展の結果が発表される可能性がある。もし証券アナリストや投資家がこれらの結果がマイナスだと思っていれば、私たちの普通株の価格に大きな悪影響を及ぼす可能性がある。このような訴訟または訴訟は、私たちの運営損失を大幅に増加させ、開発、製造、販売、マーケティング、または流通活動に使用できる資源を減少させる可能性があります。適用される医療法律法規に関連する訴訟または他の訴訟の開始および継続による不確実性は、市場での競争能力に悪影響を及ぼす可能性がある。
87
私たちの候補製品のアメリカや他の地方での成功した商業化は、政府当局と個人健康保険会社を含む第三者支払者にある程度依存し、保険と十分な補償レベルをどの程度提供するか、私たちの候補製品に有利な定価政策を実施する。もし私たちの候補製品がカバー範囲を獲得または維持し、十分な精算を得ることができなければ、承認されれば、これらの製品をマーケティングする能力を制限し、収入を創出する能力を低下させる可能性がある。
私たちが規制許可を得る可能性のある任意の製品のカバー範囲と精算状態には、重大な不確実性がある。米国や他の国では,治療を受けた患者は通常,第三者支払者によって彼らの治療に関連する費用の全部または一部を精算する。第三者支払者(政府医療計画(例えば、Medicare、MedicaidまたはTRICARE)、管理型ヘルスケア提供者、個人健康保険会社、健康維持組織、および他の組織を含む)は、私たちの製品の保証範囲および精算の十分性を含み、多くの患者が医療サービスおよび医薬品(例えば、私たちの候補製品)を負担することができるために重要である。第三者支払者は彼らがどのような薬を支払い、清算水準を確立するかを決定する。
私たちが私たちの候補製品を商業化することに成功できるかどうかは、マーケティングの許可を得た任意の製品の保証範囲と十分な精算範囲がどの程度第三者支払者から得られるかにかかっている。米国では、第三者支払者の間に統一された薬品保険や精算政策はない。第三者支払者は精算戦略を設定する際には通常Medicare保証政策と支払い制限に依存するが,Medicare保証と精算確定以外にも,彼ら自身の方法や承認の流れがある.したがって、マーケティングの許可を得ることができる製品の保証範囲と精算範囲は支払人によって異なる可能性があります。1人の支払人が1つの製品に保険を提供することを決定し、他の支払者もその製品に保険と補償を提供することを保証することはできない。支払人は、新しい製品を保証するかどうかを決定する際に、例えば、その製品がその健康計画下の保険福祉に属するかどうか、安全で、有効で、医学的に必要であるかどうか、特定の患者に適している、費用効果、および試験的でも研究的でもない一連の要素を考慮する。第三者支払者は、承認されたリストまたは処方表上の特定の製品に保証範囲を制限することもでき、FDAによって承認された特定の適応のすべての製品を含まない可能性がある。
また、支払者が製品に保険を提供することを決定することは、十分な返済率を承認することを意味するものではない。支払者が製品に保険を提供するかどうかを決定するプロセスは、支払者が製品のために支払う支払率を設定するプロセスと分離することができる。保険を提供しても、承認された精算金額が十分に高くない可能性があり、十分な投資リターンを実現するために十分な価格設定を確立したり維持したりするのに十分ではないかもしれません。第三者支払者が保証を受けないか、単独で清算しないかを決定すれば、マーケティングの許可を得る可能性のあるいかなる製品も、医師のこのような製品の使用を減少させる可能性がある。保険や十分な精算が得られない場合、あるいは限られたレベルに限られていれば、候補製品を商業化することに成功できないかもしれません。私たちは、私たちの現在または未来の候補製品、または私たちの現在または未来の候補製品を使用するいかなる手続きも、米国で保険と精算を受けることができ、得られる可能性のあるいかなる精算も十分ではないかもしれない、あるいは将来的に減少またはキャンセルされる可能性があることを確実にすることはできない。また,医師の監督下で管理されている製品では,このような薬物は価格が高いことが多いため,保険や適切な補償を得ることは特に困難である可能性がある。しかも、製品自体が単独で精算されるかもしれないし、できないかもしれない。逆に、病院や主管医は、私たちの製品を使った治療や手続きを提供することで精算されるかもしれません。
また、米国や海外の第三者支払者が医療コストの制限や低減に力を入れていることは、支払人組織が新承認製品の保証範囲や精算レベルを制限する可能性があるため、候補製品の支払いや十分な支払いを提供できない可能性がある。販売が許可される可能性のあるすべての製品の保証範囲と精算を確保するためには、我々の製品の医療必要性および費用効果を証明し、FDAなどのマーケティング許可または承認を得るのに必要なコストを証明するために、高価な薬物経済学的研究を行う必要があるかもしれない。また、購入者、個人健康計画、または政府医療計画に許可された割引を提供する必要があるかもしれない。それにもかかわらず、私たちの候補製品は医学的に必要で費用効果的だと思われないかもしれない。第三者支払者が、製品が他の利用可能な療法と比較して費用対効果があると思わない場合、彼らは、マーケティング許可または承認後に製品をその計画の下の福祉としてカバーしないかもしれない、または、支払いレベルが会社に利益的な方法でその製品を販売させるのに十分ではない可能性がある。私たちは、私たちの任意の候補製品を潜在的に販売する時、第三者支払人からの定価圧力に遭遇すると予想しています。
88
最後に、いくつかの外国の国では、薬物の提案価格は合法的に発売されるために承認されなければならない。各国の薬品定価に対する要求は大きく異なる。例えば、欧州連合加盟国の国は、その国の健康保険制度が精算を提供する医療製品の範囲を制限することができ、人が使用する医療製品の価格を制御することができる。精算または定価の承認を得るために、いくつかの加盟国は臨床試験の完成を要求する可能性があり、特定の候補製品のコスト効果を現在利用可能な治療法と比較する。欧州連合加盟国は医薬製品の具体的な価格を承認するか、あるいは医薬製品を市場に投入する会社の収益力を直接または間接的に制御する制度をとることができる。連合加盟国間の接近法には食い違いがある。例えば,フランスでは,有効な市場参入は病院との合意によって支援され,製品は社会保障基金が精算する可能性がある。薬品価格は保健品経済委員会(CEP)と協議した。薬品に対して価格制御や精算制限を実行することを保証できない国は、私たちの任意の候補製品に対して有利な精算と定価手配を許可する。歴史的に見ると、EUで発売された製品は米国の価格構造に従わず、通常EUの価格は米国の価格よりもはるかに低いことが多い。
今後の医療立法は、臨床計画を推進し、マーケティング許可を得たり、候補製品を承認したり、それを商業化する難しさやコストを増加させ、設定可能な価格に影響を与える可能性があります。
米国や他の管轄地域では,医療システムの複数の立法や規制面の改革や提案中の改革が継続されることが予想され,将来の運営結果に影響を与える可能性がある。特に,米国連邦や州レベルでは,医療コストの低減と医療の質の向上を図る取り組みが継続されている。最近我々の運営に影響を与える可能性のある医療改革努力の詳細については,第1項,第1項,政府法規−その他医療法−医療改革を参照されたい。
私たちは将来、より多くの州と連邦医療改革措置を取ることが予想され、そのいずれも州と連邦政府が特定の医療製品やサービスをカバーする程度を制限し、連邦と州政府が医療プロジェクトやサービスのために支払う金額を制限する可能性がある。これは、私たちが開発した任意の候補製品に対する需要の減少、または追加の価格設定圧力を招く可能性がある。
米国以外の市場では,精算や医療保険支払い制度は国によって異なり,多くの国で特定製品や療法に価格上限が設定されている。米国以外の価格規制法規は特定市場の収益力に大きな影響を与える可能性があり、これらの法律が変化すれば、さらなる不確実性をもたらす。例えば、カナダでは、特許薬品の価格規制立法が現在大きな変化を経験しており、カナダで製品を販売している会社の収益性に大きな影響を与える可能性がある。
私たちは米国や任意の他の司法管轄区域の将来の立法や行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することができない。もし、私たちまたは私たちが接触する可能性のある任意の第三者が、既存の要求の変化や新しい要求や政策の採用に緩やかに適応できない場合、または私たちまたはこれらの第三者が規制適合性を維持できない場合、私たちの候補製品は、取得された可能性のある任意の規制許可または承認を失う可能性があり、私たちは利益を達成または維持することができないかもしれない。
89
実際または予想されるように、適用されるデータ保護、プライバシーおよびセキュリティ法律、法規、基準、および他の要件を遵守できないことは、私たちの業務、運営、および財務状況に悪影響を及ぼす可能性があります。
全世界のデータ保護構造は急速に発展しており、私たちは多くの連邦、州と外国の法律法規、規制ガイドラインの制約または影響を受ける可能性があり、これらの法規と法規は個人情報の収集、使用、開示、伝送、安全と処理を管理しており、例えば、私たちが収集した臨床試験に関連する参加者と医療保健提供者の情報を管理している。予測可能な未来には、基準および法執行慣行を実施することはまだ不確定である可能性があり、これは、私たちの業務に不確実性をもたらし、私たちまたは私たちのサービスプロバイダがいくつかの司法管轄区域で個人データを収集、保存、移転、使用、および共有する能力に影響を与え、私たちが責任を負うか、または追加のコンプライアンスまたは他のコストを適用することをもたらすかもしれない。私たちは連邦、州、または外国の法律または自律基準を遵守できなかったか、または遵守できなかったと考えられ、否定的な宣伝、管理時間と精力の移転、および政府の実体または他の人が私たちに訴訟を提起する可能性がある。
私たちの運営と業務の増加に伴い、私たちは新しいまたは追加のデータ保護法律と法規の制約または影響を受け、規制機関のより厳しい審査や関心に直面する可能性がある。米国では、ほとんどの医療提供者は、患者の健康情報を得ることができるいくつかの研究機関を含み、1996年に公布された“健康保険携帯性·責任法案”に基づいて公布されたプライバシーおよび安全法規に制約されており、この法案は、“健康情報技術促進経済·臨床健康法案”によって改正され、総称してHIPAAと呼ばれている。私たちは現在HIPAA下の保証実体や業務パートナーではないため、HIPAAの直接監督管理を受けない。しかし、誰でもHIPAAの刑事条項に基づいて、直接、または協力および教唆または共謀の原則に従って起訴されることができる。したがって、事実および状況に基づいて、私たちが知らずにHIPAAによってカバーされているヘルスケア提供者または研究機関から個人識別可能な健康情報を受信し、その医療提供者または研究機関がHIPAAが許可されていないまたは許可されていない方法で個人識別可能な健康情報を開示する場合、重大な刑事罰に直面する可能性がある。さらに、将来的には、臨床試験全体にわたって、私たちの研究協力過程において、および/または個人(またはそれらのヘルスケア提供者)から直接取得された敏感な個人情報を、健康に関する情報を含むことができ、これらのタイプの計画を実施することを選択すれば、これらの個人情報は、患者支援計画を登録することができる。したがって,州法が個人情報が漏洩した場合に影響を受けた個人や州規制機関に通知することを要求することを含むデータプライバシーやセキュリティ法によるこのような情報の保護を受けることが可能であり,HIPAAが保護する健康情報よりも広い情報種別である.
90
連邦貿易委員会または連邦貿易委員会と多くの州総検察長は、健康に関連する個人情報および他の個人情報の収集、使用、伝播、および安全実施の変化する基準のために、既存の連邦および州消費者保護法を説明している。プライバシー法は,個人情報をどのように扱うか,個人がその個人情報を処理する可能性のある方法の選択を記述する声明を出すことを要求している.プライバシー権を侵害し、セキュリティに関する虚偽や誤った情報を発表したり、個人情報の安全を保護する適切なステップを取ることができず、連邦貿易委員会法第5条に違反する不公平または詐欺的な行為ややり方を構成する可能性がある。さらに、連邦貿易委員会は最近、“商業監視”およびデータセキュリティに関する提案ルールの作成に関する事前通知を発表し、(2)消費者データを収集、集約、使用、分析および保持する方法、および(2)不公平または詐欺的な方法でこれらのデータを移転、共有、販売するか、または他の方法でこれらのデータを金銭化することに関連する新しい貿易規制ルールまたは他の規制代替案を実施すべきかどうかについて意見を求めている。連邦規制機関、州総検事、原告弁護士はこの分野で活躍し続けている可能性があり、患者の健康情報に関連する既存または新しい法律法規を遵守しなければ、刑事または民事制裁を受ける可能性がある。また、2018年にカリフォルニア消費者プライバシー法案(CCPA)が2020年1月に施行され、消費者のための個人データプライバシー権を創出し、消費者または家庭の特定の個人情報を処理するエンティティにより多くのプライバシーとセキュリティ義務を課すことを含む会社の運営要求を制定した。このような要求は私たちのコンプライアンス費用と潜在的な責任を増加させるかもしれない。CCPAはカリフォルニア住民により大きな権利を与え,彼らの個人情報にアクセスして削除し,ある個人情報を共有しないことを選択し,彼らの個人情報がどのように使用されるかに関する詳細な情報を受信することができる.CCPAは違反行為に対する民事処罰と,データ漏洩に対する個人訴権を規定しており,データ漏洩訴訟が増加すると予想される。現在、商業パートナー或いは保証実体によって維持されている保護された健康情報及び臨床試験データは例外であるが、CCPAは著者らのいくつかの業務活動に影響を与える可能性がある。また,カリフォルニアでは最近“カリフォルニアプライバシー権法案”(California Privacy Rights Act,略称CPRA)が可決され,CCPAが改正された.CCPA(CPRA改訂)は、カバーされたトラフィックに、追加の消費者権利プログラム、データ使用の制限、より高いリスクデータの新しい監査要件、および敏感なデータを選択するいくつかの使用を含む追加のデータ保護義務を適用する。それはまた、実質的な法規の発行を許可し、プライバシーと情報セキュリティ法執行の強化につながる可能性がある新しいカリフォルニア州データ保護機関を作成した。大部分の条項は2023年1月1日に施行され、追加のコンプライアンス投資と潜在的なビジネスプロセスの変化が必要になる可能性があります。他の州でも同様の法律が採択されたり、他の州や連邦レベルで類似の法律が提出されたりしており、採択されれば、これらの法律には潜在的な衝突要求がある可能性があり、これはコンプライアンスを挑戦に直面させるだろう。たとえば,2021年3月2日,“ネバダ州インターネット消費者情報収集プライバシー法案”が2021年10月1日に施行され,“バージニア州消費者データ保護法”が2023年1月1日に施行され,“コロラド州プライバシー法案”が2023年7月1日に施行され,“コネチカット州データプライバシー法案”が2023年7月1日に施行され,“ユタ州消費者プライバシー法案”が2023年12月31日に施行される。もし私たちがHIPAA、CCPA、CPRA、または他のプライバシーおよびデータ保護法律の制約または影響を受けた場合、これらの法律の要求を遵守できなかったために負う任意の責任は、私たちの財務状況に悪影響を及ぼす可能性があります。
また、EU一般データ保護条例(EU GDPR)は2018年5月に施行され、欧州経済圏内の個人の個人データ(すなわち、個人のアイデンティティを識別するデータまたは個人のアイデンティティを識別可能なデータ)を処理することを厳しく要求し、個人の各種データ保護権利(例えば、個人データを削除する権利)を付与する。
EU GDPRの企業がより多くのコンプライアンス義務に直面していることを遵守しなければなりませんその他を除いて(I)責任性および透明性規定、および有効な同意を得るためのより高い要求、(Ii)任意の新製品またはサービスを開発する際にデータ保護を考慮する義務、処理された個人資料の数を制限すること、(Iii)適切な技術および組織措置を講じて個人資料を保護する義務、および特定の個人資料の違反を規制当局に報告する義務(いずれの場合も、実行可能な場合は72時間遅れてはならない)、および(Iv)特殊種別個人資料(健康資料および遺伝子資料を含む)の処理に関する追加的に煩雑な規定。
91
EU GDPRはまた、具体的な保障措置が実施されない限り、EU GDPRによって拘束されている個人データを、このような個人データを十分に保護することが発見されていない第三国に移すことを禁止している。歴史的に見て、米国会社が欧州経済区から個人データを輸入することを許可する主な保障措置は、EU-米国プライバシー盾枠組みの認証である。しかし、欧州連合裁判所(CJEU)は2020年7月に、EU-米国プライバシーの盾の枠組みをデータ転送メカニズムとして宣言する裁決を発表した(Schrems II)と、他の事項に加えて、個人データの取得に関する支援国の法律を評価し、標準契約条項の下で提供されるプライバシー保護以外の追加措置を実施する必要があるかどうかを考慮して、データ保護レベルがEUが提供する“基本的に等”と同じであることを確実にするために、会社にプライバシー影響評価を要求することを含む標準契約条項の使用にさらに制限を加える。その後、欧州委員会は2022年6月に#年の決定を説明するために新しいEU SCCを発表しましたSchrems IIこれは契約締結当事者たちに重い義務をもたらした。現在、標準制御プログラムの代わりに実行可能なプログラムはほとんどない。しかし、2022年10月7日、米国の総裁は、失効したプライバシーの盾の後継者となる新たな米国-EUデータプライバシー枠組みを促進する行政命令を提出した。2022年12月13日、欧州委員会も、新たな行政命令と米国-EUデータプライバシーの枠組みが解決できると考えていることを反映する十分性決定案を公表したSchrems IIそれは.十分な決定草案が承認され、実施される場合、この合意は、個人データの大西洋横断的な流れを促進し、EUから米国に個人データを転送する会社のデータ転送メカニズム(EU SCCおよび拘束力のある会社ルールを含む)に追加的な保障を提供する。しかし、各当事者が新しい枠組みに依存する前に、米国とEUは立法と規制手順を取らなければならない。
EU GDPRの制約を受けた会社はデータ保護要求の強力な監督管理を受ける可能性があり、規定を守らなければ、2000万ユーロに達するか、規定会社の世界年商の4%を満たしていないか、金額の大きい者を基準とする可能性がある。また、EU GDPRはデータ主体と消費者協会に対して個人訴訟を提起する権利を付与し、監督管理当局に苦情を提出し、司法救済措置を求め、EU GDPR違反による損害について賠償を受けることができる。
また,2021年1月1日から会社はEU GDPRおよびイギリスGDPRまたはイギリスGDPRを遵守しなければならない可能性があり,後者は改正されたイギリス2018年データ保護法とともにイギリス国内法でEU GDPRを保持している。イギリスのGDPRはEU GDPRでの罰金、すなわち最高2000万ユーロ(1750万ポンド)または世界の年商の4%の罰金を反映している。欧州委員会は、追加的な保障措置を必要とすることなく、英国に有利な十分な決定を採択し、EU加盟国からイギリスへのデータの移転を可能にした。しかし、英国の充足率決定は、欧州委員会がこの決定を再評価·更新·延長し、その間にEU委員会の審査を継続しない限り、2025年6月に自動的に失効することになった。このような変化は追加的な費用を招き、私たちの全体的なリスクを増加させるかもしれない。イギリス政府は“国際データ転送協定”と新しいEU SCCSの国際データ転送付録と呼ばれる独自の形式のSCCSを公表していることも指摘すべきである。英国情報コミッショナー事務室も、各エンティティがEUやイギリス風の譲渡影響評価を選択する可能性があるにもかかわらず、そのバージョンの譲渡影響評価と改訂された国際譲渡ガイドラインを発表した。英米間の国際データ伝送では、英国と米国が“データブリッジ”と呼ばれる十分な合意について交渉しているという。
適用される法律、法規、基準、私たちの契約義務、および他の法的義務を遵守するために努力しているにもかかわらず、これらの要求は、異なる司法管轄区域で不一致な方法で修正、解釈、適用される可能性があり、お互いまたは私たちが遵守しなければならない他の法的義務と衝突する可能性がある。私たちまたは私たちの従業員、代表、請負業者、コンサルタント、協力者、または他の第三者は、そのような要求を遵守できなかったか、またはプライバシーおよびセキュリティ問題を十分に解決できなかったと考えられ、根拠がなくても、私たちの追加コストおよび責任を招き、私たちの名声を損なうことができ、私たちの業務および運営結果に悪影響を及ぼす可能性があります。
92
もし私たちまたは私たちの第三者製造業者とサプライヤーが環境、健康、安全の法律法規を遵守できなかった場合、私たちは罰金や処罰を受けたり、私たちの業務の成功に悪影響を及ぼす可能性のあるコストが発生する可能性があります。
私たちは多くの環境、健康と安全法律と法規の制約を受けて、それらの研究室の手続きと危険材料と廃棄物の処理、使用、貯蔵、処理と処理を管理する法律と法規を含む。我々の研究·開発活動は,生物や危険材料を使用し,危険廃棄物製品を生成することに関するものである。私たちは一般的に第三者と契約を結び、このような材料と廃棄物を処理する。これらの材料の汚染や傷害リスクを除去することはできません。これは、私たちの商業化努力、研究開発努力、業務運営の中断、高価な清掃作業の環境破壊を招き、これらの材料と指定廃棄物の使用、貯蔵、処理、処理を管理する適用法律と法規に規定された責任を招く可能性があります。我々の第三者メーカーがこれらの材料を処理·処分する際に使用するセキュリティプログラムは,全体的にこれらの法律法規に規定されている基準に適合していると信じているが,状況が確かにそうである保証はなく,これらの材料の意外な汚染や傷害のリスクを解消することもできない。このような事件が発生した場合、私たちはそれによって引き起こされた任意の損害に責任を負う可能性があり、この責任は私たちの資源範囲を超える可能性があり、州、連邦、または他の適用機関は、いくつかの材料の使用を制限し、および/または私たちの業務運営を中断するかもしれない。また,環境法律法規は複雑で変化が頻繁であり,より厳しくなる傾向にある。私たちはこのような性質のどんな変化の影響も予測できないし、私たちの未来のコンプライアンスを決定することもできない。また、現在または未来の環境、健康、安全法律法規を遵守するためには、巨額のコストが生じる可能性がある。このような現行または未来の法律法規は私たちの研究、開発、または生産努力を損なうかもしれない。このような法律法規を遵守しないことはまた巨額の罰金、処罰、または他の制裁につながる可能性がある。
私たちは私たちのコストと支出を支払うために労働者補償保険を維持していますが、危険材料の使用や他の仕事に関する傷害により従業員が負傷する可能性がありますが、潜在的な責任に十分な保険を提供できない可能性があります。私たちは生物学的または危険な廃棄物の暴露または汚染による損害と罰金を含む特定の生物廃棄物または危険廃棄物保険、労働者補償または財産と死傷者および一般責任保険を保証しない。
私たちはアメリカとある外国の輸出入規制、制裁、禁輸、反腐敗法律、反マネーロンダリング法律法規の制約を受けている。このような法律基準を遵守することは国内と国際市場での私たちの競争能力を弱めるかもしれない。私たちは違反によって刑事責任と他の深刻な結果に直面するかもしれないし、これは私たちの業務を損なうかもしれない。
我々は、米国輸出管理条例、米国税関条例、米国財務省外国資産規制弁公室によって実施された様々な経済·貿易制裁条例、1977年に改正された米国反海外腐敗法、米国連邦法典第18編201節に含まれる米国国内賄賂法規、米国旅行法、2001年米国愛国者法、および私たちが活動している国の他の州と国の反賄賂および反マネーロンダリング法を含む輸出規制と輸入法律の制約を受けている。腐敗防止法の解釈は広く、会社とその従業員、代理人、請負業者、および他の協力者が直接または間接的に許可、約束、不正な支払いまたは任意の他の価値のあるものを公共または民間部門の受給者に提供することを禁止している。私たちは第三者を招いてアメリカ以外で臨床試験を行い、商業化段階に入ると、私たちの製品を海外に販売し、および/または必要な許可、許可証、特許登録、その他の規制承認を得るかもしれない。また、政府機関や政府付属病院、大学、その他の組織の役人や従業員と直接または間接的なインタラクションを行うことも可能である。私たちは、私たちが明確に権限を持っていなくても、または実際にこれらの活動を理解していなくても、従業員、代理、請負業者、および他の協力者の腐敗や他の不法活動に責任を負わなければならないかもしれない。上記の法律および条例に違反するいかなる行為も、重大な民事と刑事罰金と処罰、監禁、輸出入特権の喪失、資格取り消し、税額の再評価、契約違反および詐欺訴訟、名誉損害、およびその他の結果を招く可能性がある。
93
従業員事務に関するリスク、私たちの成長を管理し、私たちの業務に関連する他のリスク
もし私たちが合格した高級管理職と重要な科学人材を誘致して維持することができなければ、私たちの業務は実質的な悪影響を受ける可能性がある。
著者らの成功はある程度著者らが引き続き高素質の管理者及び臨床と科学者の能力を吸引、維持と激励することにかかっている。私たちは医学博士Lara Sullivan、私たちのCEO社長と最高経営責任者Pamela Connealy、私たちの最高財務責任者兼最高経営責任者Pamela Connealy、私たちの首席医療官Jay Feingold博士、そして私たちのベテラン科学者と高級管理チームの他のメンバーを含む私たちの上級管理職のメンバーに非常に依存している。これらの人のいずれかがサービスを失った場合、私たちの製品ラインの成功した開発、私たちが計画した臨床試験の開始と完成、または候補製品または任意の未来の候補製品の商業化を遅延または阻止する可能性がある。
製薬、生物製薬、バイオテクノロジー分野の合格人材に対する競争は非常に激しく、私たちの業界に必要な技能と経験を持つ個人の数が限られているからである。私たちが臨床開発を拡大し、ビジネス活動を開始する際には、より多くの人員を募集する必要があるだろう。私たちは受け入れ可能な条件で良質な人材を吸引し、維持することができず、甚だしきに至っては人材を誘致と維持することができないかもしれない。さらに、もし私たちが競争相手から人員を雇用すれば、私たちは彼らが不当に要求されたり、彼らが独自または他の機密情報を漏らしたり、彼らの元雇用主が彼らの研究成果を持っているという告発を受けるかもしれない。
もし私たちの候補製品が臨床試験に入ったら、私たちは私たちの成長と業務の拡大を管理する上で困難に直面するかもしれない。
2023年3月22日までに75人のフルタイム従業員がいます。私たちの発展と商業化計画と戦略の発展に伴い、私たちは上場企業として運営を継続し、管理、運営、財務、その他の資源における私たちの従業員基盤を拡大することが予想される。しかも、製品開発での私たちの経験は限られている。私たちの候補製品が臨床前研究と臨床試験に入って進展することに伴い、私たちは私たちの開発、監督と製造能力を拡大し、あるいは他の組織と契約を締結し、これらの能力を提供する必要がある。将来的に、私たちは協力者やパートナー、サプライヤー、および他の組織とのより多くの関係を管理しなければならないと予想される。私たちが運営と未来に成長する能力を管理することは、私たちの運営、財務、管理制御、報告システム、手続きを引き続き改善することを要求するだろう。私たちは、我々の管理情報および制御システムを効果的またはタイムリーに改善することができず、既存のシステムおよび制御における不足点を発見することができるかもしれない。私たちは私たちの成長を管理し、私たちの業務を拡大することに成功できず、私たちの業務、財務状況、運営結果、将来性に実質的な悪影響を及ぼすかもしれない。
私たちは現在マーケティング、販売、流通インフラを持っていません。販売とマーケティングインフラを構築したり、この機能を第三者にアウトソーシングしたりするつもりです。この二つの商業化戦略は私たちに大きな危険をもたらす。
私たちは現在マーケティング、販売と流通能力がありません。私たちのすべての候補製品はまだ臨床前開発段階にあるからです。もし私たちの任意の候補製品が臨床開発を完了して承認された場合、私たちは技術的な専門性と流通を支持する能力を持つ販売とマーケティング組織を構築し、合法的なコンプライアンス方式で私たちの候補製品を商業化するか、あるいはこの機能を第三者にアウトソーシングするつもりだ。私たちが自分の販売やマーケティング能力を確立したり、第三者と手配してこれらのサービスを提供することを決定すれば、リスクに関連します。私たちがマーケティング、販売、または流通について達成した協力合意について、私たちの製品収入は、私たちが直接マーケティングしたり、許可された製品を販売したりするよりも低いかもしれません。パートナーとのこのような協力は、私たちの製品の商業化を私たちの統制されていないようにし、私たちのパートナーが私たちの製品に投入する資源の数や時間を制御できない可能性があること、または私たちの協力者がその義務を履行する意志または能力、および私たちの手配の下での義務が、業務合併や私たちの協力者の業務戦略の大きな変化によって悪影響を受ける可能性があることを含む多くのリスクに直面する可能性があります。
もし私たちが受け入れ可能な条件やこれらの計画を根本的に達成できなければ、私たちはどんな承認された製品も商業化することに成功できないかもしれない。もし私たちがいかなる承認された製品を商業化することに成功できなければ、私たち自身でも、1つ以上の第三者との協力によっても、私たちの将来の製品収入は影響を受け、私たちは重大な追加損失を受ける可能性があり、これは私たちの業務、財務状況、運営結果に実質的な悪影響を及ぼすだろう。
94
我々の内部コンピュータシステムまたは私たちの既存または将来の任意のCRO、製造業者、他の請負業者、コンサルタント、または協力者のコンピュータシステムは、障害を生じたり、セキュリティまたはデータプライバシーの漏洩を受けたり、当社の独自および機密データ、従業員データまたは個人データの他の無許可または不正なアクセス、使用または破壊を受ける可能性があり、これは、追加のコスト、重大な責任、私たちの名声を損なう、および私たちの運営が実質的に破壊される可能性があります。
私たちの通常のビジネスプロセスでは、当社または他の当事者によって所有または制御される個人情報(健康情報を含む)、知的財産権、ビジネス秘密、および独自の商業情報を含む独自、機密および敏感な情報を収集し、処理し、格納します。
セキュリティ対策が実装されているにもかかわらず、我々の内部コンピュータシステムおよび我々の現在および未来のCRO、製造業者、他の請負業者、コンサルタント、既存または将来の協力者および他の第三者サービスプロバイダのシステムは、ネットワークセキュリティ攻撃、侵入、意図的または意外なエラーまたはエラー、または他の技術障害を含む様々な方法の被害を受けやすく、コンピュータウイルス、盗難または推定されたアクセスシステム、恐喝ソフトウェア攻撃、サービス拒否攻撃、ネットワーク釣り試行、サービス中断、自然災害、火災、テロ、戦争、および電気通信および電気故障を含む不正アクセス試行を含む。ネットワーク脅威構造の変化に伴い、これらの攻撃の頻度、複雑性、強度は増加しており、検出が難しくなってきている。さらに、不正アクセスまたはシステム破壊のための技術はしばしば変化し、一般に目標に対して起動されるまで識別されるため、これらの技術を予測したり、十分な予防措置を実施することができない可能性がある。私たちはまたセキュリティホールに遭遇する可能性があり、このような抜け穴は長い間発見されないかもしれない。
このような事件が発生し、私たちの運営を中断させたり、個人識別情報や個人身分の健康情報を不正に取得またはアクセスさせたりする場合、候補製品開発計画および私たちの業務運営が実質的な妨害を受ける可能性がありますが、これらに限定されず、私たちの薬物開発計画の中断、私たちの規制承認作業の遅延、規制調査または法執行行動、訴訟、賠償義務、負の宣伝、および財務損失と重大な責任です。さらに、システム障害は、私たちの従業員、独立請負業者、または私たちと協力して、あるいは私たちの仕事を代表する他の人に意外、誤り、または汚職を発生させ、貴重な臨床試験データの紛失、盗難、暴露、または許可されていないアクセスまたは使用を招き、あるいは他の方法で私たちの臨床活動を混乱させ、修復するには高価で時間がかかる可能性がある。連邦、州、および外国政府のいくつかの法律要件は、会社が個人に特定の個人識別情報に関連するセキュリティホールを通知する義務があることを含み、これらの脆弱性は、私たちまたは私たちのサプライヤー、請負業者、または私たちと戦略関係を構築する組織が経験した脆弱性によるものかもしれない。セキュリティホールに関する通知や後続行動は、私たちの名声に影響を与える可能性があり、法的費用と救済費用を含む巨額のコストを発生させる可能性があります。例えば、私たちの候補製品の完了、進行中、または将来の臨床試験に関する臨床試験データの損失は、私たちの規制許可作業を遅延させる可能性があり、損失データを回復または複製するコストを著しく増加させる可能性がある。我々の計算機システムのいかなる破壊も,我々が異なる開発段階の多くのプログラムでデータを失ったり,データの完全性を損なう可能性がある.
適用されるデータ保護法律、プライバシーポリシー、データ保護義務を遵守するためには、リソースをかけて、私たちの業務活動や実践を修正したり、私たちの運営(私たちの開発計画活動を含む)や情報技術を修正したりする必要があるかもしれません。
我々はセキュリティホールを防止するためのセキュリティ対策を実施しているが、私たちまたは私たちのサービスプロバイダ、パートナー、および他の第三者のセキュリティ対策が、すべてのセキュリティホールおよびこのような脆弱性が私たちの業務に及ぼす可能性のある重大な悪影響を効果的に防止することは保証されません。当社のプラットフォーム、システム、ネットワーク、および物理施設に統合された回復システム、セキュリティプロトコル、ネットワーク保護機構、および他のセキュリティ対策は、サービス中断、システム障害、またはデータ損失を防止、検出、および最大限に低減するのに十分ではない可能性があります。
私たちはまた、第三者に依存して私たちの候補製品を生産し、彼らのコンピュータシステムに関連する類似イベントも私たちの業務に大きな悪影響を及ぼす可能性があります。もしどんな中断やセキュリティホールが私たちのデータを紛失したり破損したり、あるいは機密または独自の情報を適切に開示しない場合、私たちは訴訟や政府調査に直面する可能性があり、私たちの候補製品のさらなる開発と商業化は延期される可能性があり、私たちは特定の州、連邦、または国際プライバシーとセキュリティ法律に違反することで巨額の罰金または処罰を受ける可能性がある。
私たちの保険証書は、このような中断、故障、またはセキュリティホールによって生じる私たちの潜在的な損失を補償するのに十分ではないかもしれません。しかも、私たちは未来に経済的に合理的な条項でこのような保険を受けないかもしれないし、根本的にできないかもしれない。さらに、私たちの保険には、私たちに対するすべてのクレームが含まれていない可能性があり、どんな場合でも高い免責額がある可能性があり、訴訟を弁護することは、その是非にかかわらず、費用が高く、経営陣の注意を分散させる可能性がある。
95
私たちまたは私たちが依存している第三者は、地震、野火、または他の自然災害の悪影響を受ける可能性があり、私たちの業務の連続性と災害復旧計画は、深刻な災害から私たちを十分に保護できないかもしれない。
洪水、火災、爆発、地震、極端な天気状況、医療流行病または流行病、電力不足、電気通信故障または他の自然または人為的事故または事件のような計画外の事件は、私たちの施設や私たちの第三者契約製造業者の製造施設を十分に利用できず、特に日常生活において、私たちの財務および運営状況に大きなマイナス影響を与える可能性がある。これらの施設を使用できないとコスト増加、候補製品の開発遅延、私たちの業務運営が中断する可能性があります。地震、野火、またはその他の自然災害は、私たちの運営をさらに混乱させ、私たちの業務、財務状況、運営結果、見通しに実質的で不利な影響を与える可能性がある。もし自然災害、停電、または他の事件が私たちが本社の全部または大部分を使用することを阻止し、私たちの研究施設や私たちの第三者契約製造業者の製造施設のような重要なインフラを破損したり、他の方法で運営を中断したりすれば、私たちは難しいかもしれません。場合によっては、かなり長い間私たちの業務を継続することはできません。深刻な災害や同様の事件が発生した場合、我々の既存の災害復旧および業務連続計画は十分ではないことが証明される可能性がある。私たちの災害復旧と業務連続計画の限られた性質のため、私たちは大量の費用を発生する可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性があります。私たちのリスク管理政策の一部として、私たちは私たちの業務に適していると思うレベルに保険範囲を維持します。しかし、これらの施設で事故や事件が発生した場合、保険金額がどんな損害や損失を補うのに十分な保証はできません。もし私たちの工場や私たちの第三者契約メーカーの製造工場が事故や事件あるいは他のどんな理由でも作動できなければ、短い時間であっても、私たちのすべての研究開発と発見計画は損害を受ける可能性があります。どの業務中断も、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。
私たちの業務は国際業務の展開に関連する経済、政治、規制、その他のリスクの影響を受けている。
私たちはアメリカ以外のところで私たちの候補製品のために規制の承認や許可を求めるかもしれない。したがって、必要な承認やライセンスを取得すれば、外国での経営に関連する追加のリスクに直面することが予想されます
私たちの国際業務に関連するこれらのリスクや他のリスクは、収益性ビジネスの能力を実現または維持することに大きな悪影響を及ぼす可能性があります。
96
私たちは新冠肺炎の大流行を含む衛生流行病と疫病に関連するリスクに直面しており、これは私たちの臨床前研究と臨床試験を深刻に乱す可能性があるため、必要な監督許可や承認を得ることは延期または阻止される可能性がある。
2019年12月、武漢で新型コロナウイルス病である新冠肺炎が発見され、中国。その時から、新冠肺炎は世界に広がった。2020年3月、世界保健機関は新冠肺炎を全世界大流行と発表し、アメリカは新冠肺炎が全国緊急状態に入ることを発表した。新冠肺炎の流行に対応するために、アメリカの大部分の地区は“避難所着席”命令と他の公衆衛生指導措置を実施し、私たちのオフィスおよび主要なサプライヤーとパートナーのオフィス所在地を含む。新冠肺炎の大流行或いは類似の流行病、及び関連する“シェルター到着”命令と他の公衆衛生指導措置により、私たちは将来中断に遭遇する可能性があり、私たちの臨床前研究と開発、私たちがその後開始した任意の臨床試験、そして私たちの業務、財務状況と運営結果に実質的な不利な影響を与えるかもしれない。私たちの臨床前開発に対する潜在的な妨害は含まれていますがこれらに限定されません
新冠肺炎または同様の流行病が引き起こす可能性のある臨床活動の中断は、これらに限定されない
97
新冠肺炎は全世界の大流行が持続的に急速に変化した。ヨーロッパや米国のある国を含む多くの国が再開放されているにもかかわらず,新たな症例の増加により一部の国で制限措置が再起動された。疫病は私たちの臨床前研究、臨床試験、業務、財務状況および業務結果にどの程度影響を与える可能性があり、未来の事態の発展に依存し、これらの事態の発展は高度に不確定であり、現在は予測できない、例えば疾病の最終的な地理的伝播、疫病の持続時間、旅行制限、疫病の制御または治療の影響の行動、例えばアメリカと他の国での社会的距離と隔離または封鎖、企業閉鎖または業務中断、およびアメリカと他の国が取った疾病の制御と治療行動の有効性。また、異なる大流行が私たちの業務、財務状況、株価に類似または異なる影響を与えるかどうかを予測することはできない。これらの分野と他の分野の将来の発展は私たちの臨床試験、業務、財務状況と運営結果に重大な不確定性とリスクをもたらした。
資金不足や世界的な健康懸念による食品·医薬品局、米国証券取引委員会、その他の政府機関の中断は、重要な指導部や他の人員の雇用·保持能力を阻害し、新製品やサービスのタイムリーな開発や商業化を阻止したり、これらの機関が私たちの業務運営に依存する可能性のある正常な業務機能を履行することを阻止したりする可能性があり、これは私たちの業務に負の影響を与える可能性がある。
FDAの新製品の審査と承認能力は、政府予算と資金レベル、肝心な人員の雇用と維持及びユーザー費用の支払いを受ける能力、及び法定、監督と政策の変化を含む様々な要素の影響を受ける可能性がある。したがって、その機関の平均検討時間は近年変動している。また、政府が米国証券取引委員会(Securities and Exchange Commission、単に米国証券取引委員会と略す)や、我々の業務が依存する可能性のある他の政府機関に提供する資金は、研究開発活動に資金を提供する機関を含め、政治過程の影響を受け、政治過程自体が不安定で予測不可能である。
FDAや他の機関の中断も、新薬が必要な政府機関によって審査および/または承認されるのに要する時間を遅らせる可能性があり、これは私たちの業務に悪影響を及ぼすだろう。例えば、近年、2018年や2019年を含め、米国政府は何度も閉店しており、米国食品·医薬品局や米国証券取引委員会などのある規制機関は、キー従業員を休暇させ、キー活動を停止せざるを得ない。政府が長期的に停止すれば、FDAが私たちが提出した規制文書を適時に審査して処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。また、将来的に政府の閉店は、適切な資本化と事業継続のために、私たちが公開市場に進出し、必要な資本を得る能力に影響を与える可能性がある。
また、新冠肺炎の流行に対応するため、2020年3月10日、アメリカ食品薬品監督管理局は外国製造施設の大部分の検査を延期するつもりであると発表し、2020年3月18日、アメリカ食品薬品監督管理局は国内製造施設の定例監督検査を一時的に延期した。FDAは2020年7月10日,国内製造施設のある現場検査を再開し,リスクに基づく優先制度を遵守しようとしていると発表した。FDAは、このリスクに基づく評価システムを使用して、特定の地理的地域内で発生する可能性のある規制活動種別を決定し、重要な任務検査からすべての規制活動を回復することを意図している。さらに、FDAは、2021年4月15日に、ある薬物製造施設および臨床研究場所の自発的な遠隔インタラクション評価を行う計画を記述した指導文書を発表した。ガイドラインによると,FDAは対面検査が優先されず,キータスクとされている場合や,直接検査が旅行制限を受けている場合であるが,FDAが遠隔評価が適切であると判断した場合には,このような遠隔インタラクション評価を要求する予定である.
それ以来,FDAはその検査活動を調整し,進行中の新冠肺炎の大流行に対応している。2021年12月29日、機関はその検査活動を一時的に変更し、その従業員と規制されている会社の安全を確保した。FDAは2022年2月2日、全製品分野の国内監督検査を2022年2月7日に再開すると発表した。FDAがいつ新冠肺炎のせいで検査の一時停止または回復を決定するかどうかを予測することはできない。
新冠肺炎疫病に対して、アメリカ以外の規制機関は類似した制限或いは他の政策措置をとる可能性がある。政府が長期的に停止している場合、または世界的な健康問題がFDAまたは他の規制機関の定期検査、審査または他の規制活動を阻害し続けている場合、FDAまたは他の監督管理機関が私たちの規制提出を適時に審査し、処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。
98
私たちの第三者への依存に関するリスク
もし私たちがいかなる許可、協力、または他の合意の下での義務を履行できなかった場合、私たちは損害賠償金の支払いを要求され、私たちの候補製品の開発と保護に必要な知的財産権を失う可能性があり、あるいは再許可を付与するいくつかの権利を失う可能性がある。
私たちはファイザーやファイザー、Biosion USA、Inc.またはBiosion、シカゴ大学とライセンス契約を締結し、これらの合意に基づいて、私たちのいくつかの候補製品に特許および技術許可を付与し、合金治療会社およびVoxall Treateutics、LLCまたはVoxallと協力協定を締結し、これらの合意に基づいて、将来の候補製品に特許および技術を許可することができます。私たちの現在の許可協定と私たちの協力協定、そして私たちが未来に締結した任意の許可協定または協力協定は、様々な開発、商業化、資金、マイルストーン、印税、勤勉、再許可、保険、特許起訴および強制執行および/または他の義務を私たちに課すことができます。もし私たちがこれらの義務に違反したり、許可された知的財産権を無許可で使用したりすれば、損害賠償金の支払いを要求される可能性があり、許可者は許可を終了する権利があるかもしれません。これは、許可技術がカバーする製品を開発、製造、販売できない、あるいは競争相手が許可技術を得ることができるようにすることができます。さらに、私たちの許可者は私たちに許可されていない知的財産権を所有したりコントロールしたりする可能性がありますので、その是非にかかわらず、私たちは私たちが許可者の権利を侵害しているか、または他の方法で侵害しているというクレームを受ける可能性があります。また、将来の製品の販売にどのくらいの印税義務が支払われるかは確定できませんが、金額は大きいかもしれません。私たちの将来の印税義務の金額は、開発や商業化に成功した製品で使用される技術や知的財産権にかかっています(あれば)。したがって、私たちが製品を開発して商業化することに成功しても、私たちは利益を達成したり維持することができないかもしれない。
私たちはすでに第三者と協力して、私たちが開発する可能性のある候補製品の研究、開発、商業化を行っている。私たちは将来的により多くの協力や戦略的同盟を形成したり、より多くの許可手配を達成したりするかもしれない。もしこれらの協力、戦略連合、または他の許可手配のいずれも成功しなければ、私たちはこれらの候補製品の市場潜在力を利用できないかもしれない。
私たちは合金会社と3年間の協力協定を締結し、Voxallを援助·運営し、私たちの特定の場所の標的カタログと合金会社のATX-GXプラットフォームと抗体発見サービスを利用することを目的とした合金会社と協力して設立した合弁会社である。また、現在または将来の候補製品の研究、開発、商業化のために他の第三者パートナーを探すことも可能である。合金やVoxallとの協力および私たちが達成した任意の他の協力合意は、私たちの協力者が開発または商業化のために使用することを制限するかもしれません。私たちは、彼らと開発した任意の候補製品の資源の数量と時間の制御を求めることができます。私たちがこのような計画から収入を創出する能力は、私たちの協力者がこれらの手配の中で彼らに割り当てられた機能を成功的に履行する能力に依存するだろう。私たちは私たちが加入したか、加入可能などんな協力の成功も予測できない。
私たちは将来的に戦略的同盟を形成したり、合弁企業や協力を作成したり、第三者と追加の許可手配を達成したりすることができ、これらの手配は、私たちの候補製品と私たちが開発する可能性のある任意の未来の製品の開発と商業化に関する私たちの努力を補完または強化すると信じています。これらの関係のいずれも、非日常的な費用と他の費用を発生させ、私たちの短期的かつ長期的な支出を増加させ、私たちの既存の株主を希釈したり、私たちの管理と業務を混乱させたりする証券を発行することを要求することができるかもしれない。
また、適切な戦略的パートナーを探す上で激しい競争に直面しており、このような取引の交渉過程は時間がかかり複雑で、コストも高い。また、私たちの候補製品のための戦略的パートナーシップまたは他の代替手配を確立する努力は成功しないかもしれない。なぜなら、彼らは協力努力の開発段階が早すぎると考えられるかもしれないので、第三者は私たちの候補製品が安全性、有効性、純度、有効性を示し、市場の承認を得るために必要な潜在力を持っているとは思わないかもしれない。さらに、我々の既存のパートナーは、腫瘍療法を開発した他の会社との買収または協力を決定する可能性があり、これは、私たちの業務の将来性、財務状況、および運営結果に悪影響を及ぼす可能性がある。
したがって、もし私たちが追加的な協力協定と戦略的パートナー関係を締結すれば、私たちの候補製品かもしれません。もし私たちがこれらの取引を私たちの既存の業務と会社文化と組み合わせることに成功できなければ、私たちはこれらの取引の利点を達成できないかもしれません。これは、私たちのスケジュールを延期したり、他の方法で私たちの業務の将来性、財務状況、運営結果に悪影響を及ぼすかもしれません。戦略的取引や許可証の後に、最初に取引に入ったことを証明する収入または特定の純収入が達成されるかどうかも確認できません。私たちの候補製品に関連する新しい協力または戦略的パートナーシップ協定の任意の遅延は、いくつかの地域での候補製品の開発および商業化を延期する可能性があり、これは、私たちの業務の将来性、財務状況、および運営結果を損なうことになる。
99
私たちは私たちの候補製品を生産するために第三者に依存する。第三者製造業者は、許容可能な原材料または候補製品を生産することができなかったか、またはFDAまたはこれに関連する同様の外国規制機関の許可を得ることができず、臨床試験の開始または完了、規制許可または承認を得るか、または承認された製品を商業化する能力を遅延または弱める可能性がある。
私たちは第三者契約メーカーに依存して、私たちの臨床前試験製品供給と臨床製品供給を生産し、もし私たちが私たちの候補製品を販売することを許可されたら、商業供給に使用します。私たちはこのような供給品を生産する製造施設を持ったり経営したりしない。私たちの臨床前と臨床開発製品の供給が制限されないこと、中断されないこと、あるいは品質が満足できること、あるいは受け入れ可能な価格で供給を続けることを保証することはできない。特に、私たちのメーカーのどの交換にも大量の仕事や専門知識が必要である可能性があります。合格した交換数は限られている可能性があり、完成するのに時間がかかるかもしれません。
候補製品の製造過程はFDAと外国規制機関の審査を受けなければならない。サプライヤーと製造業者は、適用された製造要求を満たし、cGMPのような規制基準に適合するために、監督機関の要求された厳格な施設とプロセス検証テストを受けなければならない。もし私たちのどの製造業者もこのような要求を遵守できなかった場合、または品質、時間、または他の側面での私たちの義務を履行できなかった場合、または私たちの部品や他の材料の供給が他の理由で限られたり中断されたりした場合、私たちは自分で材料を製造することを余儀なくされるかもしれませんが、私たちは現在能力や資源を持っていない、あるいは他方と合意している場合、私たちは合理的な条項でそうすることができないかもしれません。場合によっては、我々の候補製品を製造するために必要な技術的スキルまたは技術は、元の製造業者固有または独自である可能性があり、私たちは、そのようなスキルまたは技術を別の第三者に譲渡することが困難である可能性があり、実行可能な代替案が存在しない可能性がある。これらの要素は、私たちの製造業者への依存を増加させ、あるいは他の第三者が私たちの候補製品を生産するために、その製造業者のライセンスを取得することを要求するだろう。もし私たちが何らかの理由でメーカーの交換を要求された場合、新しいメーカーの施設とプログラムが品質基準とすべての適用された法規とガイドラインに適合しているかどうかを確認するように要求されます。新メーカーの検証に関する遅延は、タイムリーまたは予算内で候補製品を開発する能力に悪影響を及ぼす可能性があります。
もし私たちが任意の候補製品の規制許可を得たら、私たちは引き続き第三者メーカーに依存すると予想される。私たちが第三者とすでにまたは将来製造手配を達成した範囲内で、私たちはこれらの第三者に依存して、品質管理と保証に関する要求を含む契約と監督管理の要求に適合する方法でその義務を適時に履行する。もし私たちが候補製品の第三者製造を獲得したり維持したり、商業的に合理的な条項でそうすることができなければ、私たちは候補製品の開発と商業化に成功できないかもしれない。私たちまたは第三者が私たちの製造要件を実行し、cGMPまたは同様の海外要件を遵守できなかった場合、様々な態様で私たちのビジネスに悪影響を及ぼす可能性があります
私たちは第三者契約製造組織やCMOと合意を作ることができないかもしれないし、受け入れ可能な条項でそうすることができないかもしれない。CMOと合意できても、それらに依存することは追加的なリスクをもたらす
100
私たちの候補製品は限られた技術移転協定しかありません。これらの手配は商業供給には適用されません。場合によっては、臨床供給にも適用されません。私たちは注文を購入した上で多くの重要な材料を得た。したがって、私たちは私たちの候補製品と他の材料に対する長期的な約束計画を持っていない。もし私たちが任意の候補製品のマーケティング許可を得たら、第三者と商業生産協定を確立する必要があるだろう。
我々が保持しているCMOはcGMP法規や米国以外の類似した法規要件を遵守できない可能性がある。私たちまたは私たちのCMOが適用された法規を遵守できなかったことは、臨床差し押さえ、罰金、禁止、民事処罰、遅延、一時停止または販売許可証、販売許可証、候補製品または製品の差し押さえまたはリコール、運営制限、刑事起訴を含む私たちへの制裁をもたらす可能性があり、いずれも私たちの製品供給に重大な悪影響を及ぼす可能性があります。
私たちの契約製造業者が私たちの候補製品を生産するための施設は、FDAまたはEU加盟国がEMAと調整して承認しなければなりません。検査は、私たちがFDAに私たちのBLAを提出するか、またはEMAに私たちのマーケティング許可申請を提出した後に行われます。私たちは私たちの契約製造パートナーの製造過程のすべての側面を完全にコントロールできず、cGMP製造規定を遵守することに依存している。第三者メーカーは、cGMP法規や米国以外の類似した法規要件を遵守できない可能性がある。私たちの契約メーカーが私たちの規格やFDAなどの外国規制機関の厳格な規制要件に適合した材料を生産することに成功しなければ、彼らはその製造施設のマーケティング許可を確保および/または維持することができないだろう。しかも、私たちの契約製造業者が十分な品質管理、品質保証、合格者の能力を維持することを完全にコントロールすることはできません。FDA、EU加盟国、EMAまたは他の同様の規制機関が、これらの施設が私たちの候補製品を生産するために承認されていない場合、または将来的にそのような許可を撤回する場合、私たちは、承認または許可が得られれば、私たちが開発、販売許可、または候補製品をマーケティングする能力に深刻な影響を与えるかもしれない。
私たちまたは私たちの第三者製造業者が適用された法規を遵守できないことは、罰金、禁止、民事処罰、遅延、一時停止または許可証の取り消し、ライセンスの取り消し、製品または候補製品の差し押さえまたはリコール、運営制限、刑事起訴を含む制裁を加える可能性があり、いずれも候補製品の供給に重大な悪影響を与え、私たちの業務と運営結果を損なう可能性があります。私たちの候補製品と私たちが開発する可能性のあるどの製品も他の候補製品や製品と適切な製造施設を競争する可能性があります。したがって、私たちはこのような施設を優先的に使用することができないかもしれないし、全く使用できないかもしれない。CGMP法規の下で運営されているメーカーの数は限られており、私たちのために製品を製造する能力があるかもしれない。
私たちの既存または未来の製造業者のどんな性能障害も、臨床開発またはマーケティング許可を延期する可能性があります。私たちは現在大量の麻薬物質のための余分な供給や二番目の供給源の手配を提供していない。もし私たちの既存のCMOが約束通りに実行できなければ、私たちはこれらのメーカーの交換を要求されるかもしれない。いくつかの潜在的な代替メーカーが私たちの候補製品を生産できると考えているにもかかわらず、任意のそのような代替製造業者を決定し、同定したり、任意の代替製造業者と合意することができる場合には、追加のコストおよび遅延が生じる可能性がある。
私たちの現在と期待されている未来は、私たちの候補製品や製品を生産することに依存して、私たちの将来の利益率と、マーケティング許可をタイムリーかつ競争力的に獲得する製品の商業化能力に悪影響を及ぼすかもしれません。
私たちの候補製品の一部の生産は中国で第三者メーカーを通じて行われています。これらのメーカーの運営に重大な中断が生じた場合、私たちの業務、財務状況、運営結果に大きな悪影響を及ぼす可能性があります。
私たちは現在製造業務を第三者にアウトソーシングしています。私たちの大量の候補製品はこれらのアメリカ以外の第三者によって生産されており、中国に含まれています。生産が中断されたり、私たちのメーカーが私たちの需要を満たすのに十分な数の製品を生産できない場合、私たちの日常的な業務を運営し、私たちの候補製品を開発し続ける能力を損なう可能性があります。また、これらのメーカーのいくつかが中国に位置しているため、米国や中国政府の政策が変化し、政治的動揺や中国の経済状況が不安定であれば、製品供給中断やコスト増加の影響を受ける可能性がある。例えば、貿易戦争は、私たちが使用している中国製の化学中間体に関税をかける可能性がある。これらの事項のいずれも私たちの業務と経営結果に実質的な悪影響を及ぼす可能性がある。臨床試験で使用される我々の候補製品の任意の生産ロットまたは同様の行動の任意のリコールは、試験を延期するか、または試験データの完全性を損なう可能性があり、および将来の規制申告文書におけるその潜在的な使用を損なう可能性がある。
101
私たちのCMOは私たちの候補製品の生産を遅延または阻止し、承認された製品を商業化するのに十分な品質と数量で私たちの候補製品の生産を拡大することに成功できないかもしれない。
私たちの候補製品の臨床試験を行うためには、それらを大量に生産する必要がある。拡張活動中に品質の問題が発生する可能性がある。限られた数のCMOへの依存、薬物製造の複雑さ、生産プロセスの拡大の難しさは、私たちの候補製品の臨床試験、監督提出、必要な許可或いは商業化の遅延を招き、私たちにより高いコストを発生させ、私たちの候補製品の商業化に成功することを阻止するかもしれない。さらに、私たちのCMOが必要な商業品質および数量の材料を商業的に合理的な価格でタイムリーに提供できず、実質的に同じコストでタイムリーに生産できる1つ以上の代替CMOを確保できない場合、候補製品の試験および臨床試験は延期または実施できない可能性があり、いかなる最終製品の規制許可や商業発表を延期または得ることができない可能性があり、これは私たちの業務を深刻に損なう可能性がある。
新型肺炎疫病の影響により、私たちのいくつかのサプライヤーはそれぞれのサプライチェーンの中断に遭遇する可能性があり、これは私たちの開発或いは商業化努力を延期、阻止或いは損害する可能性がある。
著者らは新冠肺炎疫病の影響を受けた国家/地区の候補製品の合成過程において、ある化学或いは生物中間体を獲得した。これらの化学または生物中間体の十分な数をタイムリーに得ることができなければ、候補製品の開発、テスト、および臨床試験は延期または不可能になる可能性があり、任意の最終製品の規制許可または商業発表が延期または入手できない可能性があり、これは私たちの業務を深刻に損なう可能性がある。
もし私たちが十分な原材料と中間材料をタイムリーに得ることができなければ、あるいは他の製造や供給困難に遭遇した場合、私たちの業務は悪影響を受ける可能性がある。
私たちのいくつかの候補製品の生産は十分な量の原材料と中間材料をタイムリーに渡さなければならない。私たちは供給の連続性を確保するためにサプライヤーと密接に協力しているが、このような努力が常に成功することは保証されない。また、私たちの原材料と中間材料の供給源を多様化するために努力していますが、場合によっては単一サプライヤーから原材料と中間材料を取得します。独占的なサプライヤー関係に依存した場合、代替供給源が存在すると信じているが、特定の材料のための追加または代替源を迅速に確立できる保証はない。供給の減少や中断、およびそのような供給の代替源を開発できないことは、タイムリーまたは費用対効果のある方法で私たちの候補製品を生産する能力に悪影響を及ぼす可能性がある。
第三者に依存して臨床試験を行う予定であるが,これらの第三者の表現は締め切りまでに完了できないことを含めて満足できない可能性がある。
私たちは第三者CROに依存して、私たちのバイオ製品候補製品の臨床試験を行います。私たちは現在独立してどんな臨床試験も行うつもりはない。これらのCROとのプロトコルは、彼らが履行できなかった理由を含む様々な理由で終了する可能性がある。必要であれば、代替手配に参加することは、私たちの製品開発活動を大幅に延期する可能性があります。
我々のこれらのCROに対する研究や開発活動への依存は,これらの活動に対する我々の制御を減少させるが,我々の責任を軽減することはない.例えば,われわれの個々の臨床試験が適用されるINDにおける一般的な研究計画や案に沿って行われることを確保していきたい。さらに、FDAは、データおよび報告の結果が信頼性および正確であることを保証し、試験参加者の権利、完全性、および機密性を保護するために、臨床試験結果を進行、記録、および報告する基準を遵守することを要求する。
もしこれらのCROが法規の要求または私たちが規定した規程に従って契約責任を成功裏に履行し、予想された期限内に私たちの臨床試験を完了したり、私たちの臨床試験を行うことができなければ、私たちは私たちの候補製品のマーケティング許可証の取得を得ることができないか、遅延する可能性があり、候補製品を商業化する努力を成功させることができないか、遅延する可能性がある。
102
私たちの知的財産権に関するリスクは
もし私たちが候補製品のために特許保護を獲得して維持することができない場合、または取得された特許保護範囲が十分に広くない場合、または私たちの特許が十分な時間で私たちの候補製品を保護するのに十分でない場合、または私たちの独自技術のために十分な保護を受けることができない場合、私たちは私たちの市場で効果的に競争することができないかもしれない。
私たちは、特許、ノウハウ、商業秘密保護、および秘密保護協定の組み合わせによって、私たちの候補製品および発見計画に関連する知的財産権を保護します。私たちの成功は、私たちが現在および任意の未来の候補製品に対する特許保護を獲得し、維持する能力に大きく依存している。私たちは、現在および将来の製品候補および発見計画に関する特許出願の米国および海外での許可および提出を含む、私たちの独自の地位の保護を求めています。特許起訴過程は高価で時間がかかり、私たちはすべての必要または望ましい特許出願を合理的なコストでまたはタイムリーに提出して起訴することができないかもしれない。私たちはまた、ビジネス秘密、技術ノウハウ、持続的な技術革新に依存して、私たちの独自と知的財産権の地位を発展させ、維持することができます。
私たちは、それらの物質組成および疾患の治療におけるこれらの候補製品の使用のための知的財産権の地位の確立に努力するために、私たちの候補製品のための特許出願を許可して提出する。私たちの知的財産権には、私たちが所有する特許と特許出願、そして私たちが許可している特許と特許出願が含まれている。例えば、ファイザーおよびBiosionとのライセンス契約は、私たちの候補製品に関連するいくつかの特許および特許出願の独占的権利を付与します。
私たちまたは私たちの許可者は、私たちが製品を販売する可能性のあるすべての国または地域で私たちの候補製品のために特許保護を求めたり維持したりしていません。承認されれば、将来的にもこれらの保護を求めたり維持したりすることはありません。さらに、私たちは、私たちの任意の係属中の特許出願が発行されるか、または発行された場合、それらが私たちに有利な形で発表されるかどうかを決定することはできない。米国特許商標局またはUSPTO、国際特許庁または司法機関は、私たちの特許出願に基づいて提出された特許請求を否定または大幅に縮小する可能性があり、私たちが発行した特許は、成功的に挑戦される可能性があり、設計される可能性があり、または私たちの商業製品の保護のために十分な範囲がない可能性がある。
特許保護を受ける前に、私たちの研究開発成果で特許を申請できることを確認できない可能性があります。さらに、特許訴訟は長いプロセスであり、その間、米国特許商標局が最初に審査を提出した特許請求の範囲は、発行時に大幅に縮小されるか、または全く発表されない可能性がある。我々が発行した特許または特許出願の請求項は、発行時に現在または将来の候補製品を含まない可能性があり、またはこれらの特許がカバー範囲を提供しても、得られたカバー範囲はいかなる競争優位性も提供しない可能性がある。私たちが所有しているまたは許可されている特許出願は、発行された特許を生成することができない可能性があり、その声明は、現在または将来米国または他の国/地域における任意の候補製品をカバーしている。我々の特許および特許出願に関連するすべての潜在的に関連する既存技術が発見されていることは保証されず、これらの技術は、特許を無効にするか、または係属中の特許出願から特許が発行されることを阻止する可能性がある。特許が確実に発行されても、これらの特許が現在または任意の未来の候補製品をカバーしていても、第三者はその有効性、実行可能性、または範囲に疑問を提起する可能性があり、これは、そのような特許が縮小され、無効にされ、または実行できない可能性がある。これらの特許または私たちが所有または許可してくれた任意の他の特許に対する成功的な反対は、私たちが開発する可能性のある任意の候補製品の商業化に必要な権利を奪う可能性がある。また、規制許可や承認に遅れが生じた場合、特許保護の下で候補製品を販売できる期間が短縮される可能性があります。
もし私たちが私たちの候補製品や発見計画に関連する特許出願を持っているか許可していない場合、それらの保護の広さや強度が脅かされている場合、またはそれらが私たちの現在または任意の未来の候補製品に意味のある排他性を提供できなければ、会社が私たちと協力して候補製品や未来の薬剤を開発し、商業化することを阻止し、私たちの将来の薬物商業化の能力を脅かすかもしれない。どんなそのような結果も私たちの業務に否定的な影響を及ぼすかもしれない。
103
バイオテクノロジーと製薬会社の特許地位は通常高度に不確定であり、複雑な法律と事実問題に関連しており、近年多くの訴訟のテーマとなってきた。しかも、外国の法律はアメリカの法律のように私たちの知的財産権を保護しないかもしれない。例えば,欧州特許法は米国法よりも人体治療法に対する特許性制限が多い。さらに、他の当事者は、我々の技術に関連しているか、または我々の技術と競合する可能性のある技術を開発しているか、または特許出願を提出している可能性があるか、または特許を受信しているか、または受信している可能性があり、これらの発明が我々の特許出願または発行された特許で主張されている発明と重複または衝突している可能性があると主張しており、これらの発明に依存して市場における我々の製品の排他性を確立することができる。科学文献で発表された発見は往々にして実際の発見に遅れており、米国と他の司法管轄区の特許出願は通常、申請18ヶ月後に発表され、時には全く発表されないこともある。したがって、私たちは、私たちが所有または許可している特許または出願中の特許出願において最初に要求された発明であるか、またはそのような発明のための最初の特許保護であるかを正確に知ることができない。したがって,我々の特許権の発行,範囲,有効性,実行可能性,商業的価値は高い不確実性を持っている.私たちの未解決および未来の特許出願は、私たちの技術または薬物の全部または一部を保護するために、または他社が競争相手の技術および薬物を商業化することを効果的に阻止するために、特許の発行を招くことができないかもしれない。米国や他の国の特許法や特許法解釈の変化は,我々の特許の価値を低下させたり,我々の特許保護の範囲を縮小したりする可能性がある。
私たちの係属中の特許出願では、第三者が既存技術を米国特許商標局または他の特許庁に提出する必要があるかもしれない。このような提出は、私たちに任意の係属中の特許出願を付与することを阻止するか、または狭い権利要件を有する特許の付与をもたらす可能性があり、これは、他人が同様または同じ技術および製品を使用することを阻止するか、またはそれを商業化する能力を制限する可能性がある。特許の発行は,その発明性,範囲,有効性または実行可能性に対して決定的ではなく,我々が所有し許可している特許権は,米国や海外の裁判所または特許庁で挑戦される可能性がある。例えば、私たちは、反対、派生、再審査、当事者間の審査、認可後の審査に参加するか、または私たちの特許権または他の人の特許権の介入手続きに挑戦することができます。このような訴訟または訴訟において不利な裁決を下すことは、私たちの特許主張の範囲を縮小し、私たちの特許権の全部または一部が無効または強制的に実行できない、または私たちの技術および製品の特許保護期間を制限し、第三者が私たちの技術または製品を商業化し、私たちに支払うことなく直接私たちと競争することを可能にしたり、第三者の特許権を侵害することなく製品を製造または商業化することができなくなる可能性がある。さらに、私たちの特許および特許出願によって提供される保護の広さまたは強度が脅かされている場合、会社が私たちと協力し、現在または任意の未来の候補製品を許可、開発、または商業化することを阻止するかもしれない。
しかも、特許の寿命は限られている。米国では、特許は通常、非臨時特許出願の最初の提出日から20年後に満了する。様々な延期があるかもしれない;しかし、特許の有効期限とその提供される保護は限られている。もし私たちが現在または未来の候補製品に特許保護がない場合、私たちはそのような製品からの模倣および/または生物学的に類似したバージョンの競争に直面するかもしれない。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちが所有し許可している特許権は、他の人が私たちと似ているか、同じ薬物を商業化することを排除するために十分な権利を提供してくれないかもしれない。
私たちの特許権が挑戦されなくても、私たちが発行した特許と私たちの未解決の特許出願(発行された場合)は、私たちの特許主張をめぐる競争相手の設計を阻止し、類似または代替技術または製品を非侵害的に開発することによって、私たちの所有する特許権を回避することができます。例えば、第三者は、我々の1つまたは複数の候補製品と同様の利点を提供する競争力のある製品を開発することができるが、製品は、我々の特許保護範囲内ではなく、異なる構成を有する。候補製品に対して提供される特許権保護が十分に広くなく、そのような競争を阻害するのに不十分である場合、または候補製品または任意の未来の製品候補製品に対して提供される特許権保護の広さ、強度または期限(任意の拡張または調整を含む)が成功的に挑戦された場合、候補製品を商業化することに成功した能力が負の影響を受ける可能性があり、これは私たちの業務を損なうことになる。また、臨床試験で遅延に遭遇すれば、私たちの候補製品または将来特許保護された任意の候補製品を販売することができる時間は短縮されるだろう。
104
私たちの特許権を獲得し、維持することは、政府特許機関が提出した様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
米国特許商標局および各種外国政府特許機関は、特許出願過程において、いくつかのプログラム、文書、費用支払い、およびその他の同様の規定を遵守することを要求する。さらに、私たちが所有および許可している特許および/または出願、ならびに私たちが将来所有または許可する可能性のある任意の特許権の有効期間内に、米国特許商標局および米国以外の様々な政府特許機関に定期維持費、継続費、年会費、および特許および/または特許出願に関する様々な他の政府費用を支払わなければならない。私たちは私たちのサービス提供者たちに頼ってこのような費用を支払うことができるかもしれない。米国特許商標局および様々な非米国政府特許機関は、特許出願中にいくつかのプログラム、文書、費用支払い、および他の同様の条項を遵守することを要求する。私たちは私たちの遵守を助けるために信用の良い法律事務所や他の専門家を招いて、私たちが許可を得た知的財産権に関するこれらの要求を守るために必要な行動をとることにも依存しています。特許または特許出願の放棄または失効をもたらす可能性のある規定を遵守しないイベントは、規定された期限内に公式行動に応答できなかったこと、費用を支払わなかったこと、および適切に合法化されず、正式な文書を提出することができなかったことを含むが、これらに限定されない。もし私たちまたは私たちの許可者が私たちの製品や技術をカバーする特許および特許出願を維持できなかった場合、私たちは競争相手が私たちの候補製品と同じまたは似た製品を販売することを阻止できないかもしれません。これは私たちの業務に大きな悪影響を与えます。多くの場合、不注意は、滞納金を支払うことによって、または規則を適用する他の方法によって救済することができる。しかしながら、場合によっては、規定を遵守しないことは、特許または特許出願が放棄または失効される可能性があり、それにより、関連法ドメインの特許権の一部または全部が失われる可能性がある。この場合、潜在的な競争相手が市場に参入する可能性があり、この場合は私たちの業務を損なう可能性がある。
また、適用される特許期間の延長や調整を申請していない場合には、付与された特許権を実行するためのより限られた時間を有することになる。また,我々が特許訴訟と我々に付与された特許権の維持を担当していれば,上記のいずれも適用される特許所有者に責任を負わせることが可能である.
特許条項は候補製品に対する私たちの競争地位を十分に長く保護するのに十分ではないかもしれない。
PYX-201およびPYX-106などの新製品候補製品の開発、テスト、および監督審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。私たちはアメリカで特許期間の延長を求めたいと思っていますが、もしあれば、特許権を所有または取得する他の国/地域でも特許期間の延長を求めます。米国では、1984年の“薬品価格競争と特許期限回復法”は、特許が許可された日から14年以内に強制的に実行できないことを条件として、特許が正常満了後最大5年間延長されることが許可されており、これは承認された適応(または延長期間内に承認された任意の他の適応)に限定される。しかし、適用当局は、米国のFDAおよびUSPTO、および他の国/地域の任意の同等の規制機関を含み、このような延期が利用可能かどうかの評価に同意しない可能性があり、私たちの特許延期の承認を拒否するか、または私たちが要求したよりも限られた延期を承認する可能性がある。このような状況が発生すれば、私たちの競争相手は私たちの臨床と臨床前データを参考にすることで、私たちの開発と臨床試験への投資を利用し、他の場合よりも早く彼らの薬物を発売することができるかもしれない。
105
知的財産権は私たちの業務が直面しているすべての潜在的な脅威を解決できるとは限らない。
私たちの知的財産権が提供する未来の保護の程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できない可能性がある。以下の例は例示的である
第三者は私たちが彼らの知的財産権を侵害したことを告発する法的訴訟を提起する可能性があり、その結果は不確定であり、私たちの業務成功にマイナスの影響を与える可能性がある。
私たちのビジネスの成功は、私たちの現在および任意の未来の候補製品を開発、製造、マーケティング、販売し、第三者の独自の権利および知的財産権を侵害することなく、私たちの独自の技術を使用するために、私たちの能力と、私たちが協力する可能性のある他の人の能力にある程度依存します。バイオテクノロジーと製薬業界の特徴は特許と他の知的財産権に関する訴訟が広くて複雑だということだ。私たちは将来、妨害手続き、許可後の審査、USPTOへの当事者間審査を含む、私たちの現在および任意の未来の候補製品や技術の知的財産権に関連する対抗手続きや訴訟に参加または脅かされる可能性があります。第三者は、既存の特許または将来付与される可能性のある特許に基づいて、その是非曲直にかかわらず、侵害請求を私たちに提起するかもしれない。第三者が彼らの特許権を強制的または他の方法で主張するために私たちと訴訟を行うことを選択する可能性があるリスクがある。このような主張に法的根拠がないと考えても、管轄権のある裁判所は、これらの第三者特許が有効で、強制的かつ侵害されていると判断する可能性があり、これは、現在および将来の候補製品を商業化する能力に悪影響を及ぼす可能性がある。連邦裁判所でこのような米国特許の有効性に挑戦することに成功するためには,有効性推定を克服する必要がある。この負担が重いため,このような米国特許主張の無効について明確で納得できる証拠を提出することが求められているため,管轄権のある裁判所がこのような米国特許の主張の無効を宣言する保証はない.さらに、私たちの技術分野に大量の特許があることを考慮して、私たちは私たちが既存の特許を侵害していないか、あるいは私たちが将来付与される可能性のある特許を侵害しないと確信できない。他の会社および研究機関はすでに提出されており、将来的に抗体または抗体-薬物結合体およびその治療用途に関連する特許出願を提出することが可能である。その中のいくつかの特許出願は許可または発行されており、他のいくつかは未来に発行される可能性がある。将来的に訴訟を開始し、これらまたは他の特許の有効性に挑戦することを決定する可能性があるが、私たちは成功しない可能性があり、米国および海外の裁判所または特許庁は、このような特許の有効性を維持することができる。さらに、特許出願は、発行されるまでに長年の時間を要する可能性があり、提出後18ヶ月以上秘密にされている可能性があり、未解決の特許声明は発行前に修正することができるので、処理されている出願が、私たちの候補製品の製造、使用、または販売によって発行された特許を侵害する可能性がある可能性がある。いつ提出されても、私たちは関連する第三者特許または特許出願を識別できないかもしれないし、第三者特許が無効であるか、または私たちの候補製品や活動によって侵害されていないと誤って結論を出す可能性がある。もし特許保有者が私たちの候補製品がその特許を侵害したと思っていれば、私たちの技術が特許保護を受けても、特許所有者は私たちを起訴することができる。また、私たちは非執行実体の特許侵害請求に直面する可能性があり、これらの実体は関連する薬品収入がないため、私たち自身の特許の組み合わせは彼らに抑止力がないかもしれない。特許侵害訴訟が脅かされたり、私たちに訴訟が提起されたりすると、実際または脅威訴訟の標的となる候補療法または製品の研究、開発、製造、または販売を停止または延期させることを余儀なくされる可能性がある。
106
もし私たちが第三者の有効かつ強制的に実行可能な知的財産権を侵害していることが発見された場合、私たちは、当社の候補製品や技術の開発、製造、マーケティングを継続するために、第三者からライセンスを取得することを要求される可能性があります。このような許可に基づいて、私たちは様々な種類の費用、マイルストーン、印税、あるいは他の金額を支払うことを要求される可能性が高い。しかも、私たちは商業的に合理的な条項や必要な許可証を得ることができないかもしれない。
第三者知的財産権の許可または取得は競争分野であり、より多くの老舗企業は、魅力的または必要と考えられる第三者知的財産権許可または取得戦略をとることも可能である。これらの老舗会社はその規模、資本資源及び更に強い臨床開発と商業化能力のため、私たちより競争優位を持っているかもしれない。しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。私たちはまた私たちの投資を適切なリターンを得るための条項で許可したり、第三者の知的財産権を取得することができないかもしれないし、全くできないかもしれない。もし私たちが必要な第三者知的財産権を得ることに成功したり、私たちの既存の知的財産権を維持することができなければ、関連する計画や候補製品の開発を放棄しなければならないかもしれません。これは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。また,我々が許可を得ることができても,我々の競争相手や他の第三者が我々に許可された同じ技術にアクセスできるように非排他的である可能性があり,大量の許可と印税を支払う必要があるかもしれない.私たちは裁判所の命令、開発、製造、商業化侵害技術または候補製品の開発を中止することを強要されるかもしれない。さらに、私たちが特許や他の知的財産権を故意に侵害していることが発見された場合、私たちは3倍の損害賠償と弁護士費を含む金銭損害賠償責任を負われる可能性がある。私たちは協力者や請負業者のこのようなクレームを賠償するように要求されるかもしれない。権利侵害の発見は、私たちが現在または任意の未来の候補製品を製造して商業化することを阻止するか、または私たちに業務運営の一部または全部を停止させる可能性があり、これは私たちの業務に実質的な損害を与える可能性がある。私たちがこのようなクレームを弁護することに成功しても、訴訟は高価で時間がかかり、経営陣のコア業務への注意をそらす可能性がある。第三者の機密情報や商業秘密を盗用したと主張することは、私たちの業務、財務状況、運営結果、将来性に類似した負の影響を与える可能性があります。
私たちは、私たちの従業員、コンサルタント、またはコンサルタントが、彼らの現在または前任雇用主たちのいわゆる商業機密を誤って使用または開示したと主張するか、または私たち自身の知的財産権を持っていると主張するかもしれない。
私たちの一部の従業員、コンサルタント、またはコンサルタントは、現在または以前、私たちの競争相手または潜在的な競争相手を含む大学または他のバイオテクノロジーまたは製薬会社に雇われている。私たちは、私たちの従業員、コンサルタント、およびコンサルタントが、私たちのために働いているときに他人の固有情報またはノウハウを使用しないことを保証するために努力しているが、私たちは、これらの個人が、そのような個人の現職または前任雇用主の知的財産権(商業秘密または他の固有情報を含む)を使用または開示することによって告発される可能性がある。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭的損害賠償を支払う以外に、私たちは貴重な知的財産権や人員を失う可能性がある。このようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣の注意を分散させる可能性がある。
さらに、私たちは将来、私たちの特許または特許出願の所有権を主張する元従業員やコンサルタントのクレームを受けるかもしれません。これは、彼らが私たちを代表して仕事をしている結果です。私たちの政策は、知的財産権の概念や開発に参加する可能性のある私たちの従業員と請負業者が、このような知的財産権を私たちに譲渡する協定に署名することを要求しているにもかかわらず、私たちは実際に私たちが自分の知的財産権を構想したり開発したりするすべての側とこのような合意を実行することができない可能性があり、このような当事者との合意が潜在的な挑戦に直面したときに維持または違反されないかどうかを決定することはできず、これらの挑戦に対して十分な救済措置がないかもしれない。知的財産権の譲渡は自動的に実行されない可能性があり、あるいは譲渡協定が違反される可能性があり、私たちは第三者にクレームをつけさせられたり、私たちが私たちの知的財産権の所有権だと思うことを決定するために、私たちが提起する可能性のあるクレームを弁護したりすることができます。
107
知的財産権許可は複雑な法律、商業、そして科学的な問題に関する私たちの業務に重要だ。もし私たちがシカゴ大学、ファイザー社、Biosionライセンス契約、または任意の他の合意に違反した場合、私たちはこれらの合意に基づいて私たちの候補製品をカバーする知的財産権を獲得したり、私たちが関連する候補製品を開発し、商業化し続ける能力を失う可能性があります。
知的財産権の許可は、私たちの業務と私たちの現在と未来の候補製品に重要であり、私たちは今後もこのような合意をより多く締結することが予想される。
特に、PYX−201をカバーする知的財産権はファイザーから許可されており、我々の候補製品PYX−106をカバーする知的財産権はBiosionから許可されている。私たちは他の第三者許可者から未来の候補製品の知的財産権を得るかもしれない。
ファイザーライセンスプロトコル(A&Rライセンスプロトコルの改訂および再説明を経て)またはBiosionライセンスプロトコルを含む、当社の任意のライセンス内プロトコルの義務を履行できない場合、ライセンスパーティは、ライセンスプロトコルを終了することができる。もし私たちの材料許可プロトコルのうちの1つが終了すれば、私たちはそのライセンスプロトコルに含まれる候補製品の開発と商業化を継続する権利を失うだろう。私たちは私たちが得ることができるすべての権利と救済措置を行使することを望んでいますが、私たちは私たちの任意の違反を是正することを求め、他の方法で許可内の合意の下で私たちの権利を維持することを求めていますが、私たちはタイムリーに、許容可能な費用で、あるいはそれを根本的にすることができないかもしれません。
私たちは私たちの特許、ライセンシーの特許、または私たちの他の知的財産権を保護または強制する訴訟に巻き込まれるかもしれません。これは高価で、時間がかかり、成功しないかもしれません。
競争相手は私たちの特許、私たちの許可側の特許、または私たちの他の知的財産権を侵害または他の方法で侵害する可能性がある。権利侵害や不正使用に対抗するために、私たちは高価で時間がかかる可能性があり、私たちの技術や管理者の正常な責任を分散させることを含む、私たちのコア業務から大量の資源を分流することができる法的請求を要求される可能性がある。さらに、侵害訴訟では、裁判所は、私たちまたは私たちのライセンシーの特許が無効であるか、または強制的に実行できないと判断することができ、または、私たちの特許が関連技術をカバーしないことを理由に、他方の関連技術の使用を阻止することを拒否することができる。任意の訴訟または弁護手続における不利な結果は、私たちが所有または許可している1つまたは複数の特許を無効または狭義に解釈されるリスクに直面させ、私たちの所有または許可された特許出願を発行できないリスクに直面させる可能性がある。第三者へのクレームは、私たちの特許権が無効または強制的に実行できないと主張するような第三者からの反クレームを引き起こす可能性もあります。米国の特許訴訟では,被告が無効または実行不可能と主張する反訴が一般的である。有効性を疑問視する理由は、新規性の欠如、明らかな、実施できない、または法定テーマの欠如を含む、いくつかの法定要求のいずれかを満たすことができなかったと言われている可能性がある。主張を実行できない理由は,特許訴訟に関連する人が起訴期間中に米国特許商標局に重要な情報を隠蔽したり,重大な誤解を持つ声明をしたりしたからであろう.第三者はまた、一方的な再審、当事者間の再審または付与後の再審、または米国国外での反対または同様の手続が、訴訟と同時に行われ、さらには訴訟の範囲外で行われるように、付与後の手続において米国特許商標局に同様の有効性要件を提出することもできる。法的に無効と実行不可能と断言された後の結果は予測できない。私たちは無効な以前の技術がないかないかを確認することはできないが、私たちと特許審査員は起訴中にこれを知らない。私たちが許可した特許および特許出願については、第三者の挑戦を防ぐために、限られた権利またはいかなる許可特許の弁護に参加する権利もないかもしれない。もし被告が無効または強制不可能な法的主張に勝った場合、私たちは少なくとも部分的、さらにはすべて、私たちの現在または未来の候補製品の任意の未来の特許保護を失うだろう。このような特許保護の喪失は私たちの業務を損なうかもしれない。
私たちは、特に米国のようにこれらの権利を十分に保護していないかもしれない国では、私たちの知的財産権の盗用を単独でまたは許可者と一緒に防ぐことができないかもしれない。訴訟で勝訴した側が私たちに許可を提供しなければ、私たちの業務は損害を受ける可能性があり、このような許可は商業的に合理的な条項に適合しない可能性がある。私たちの知的財産権を強制的に執行する訴訟や他の手続きは失敗する可能性があり、成功しても巨額のコストを招き、私たちの経営陣や他の従業員の注意を分散させる可能性があります。
さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。聴聞、動議、または他の一時的手続き、または事態発展の結果を公開することもできる。もし証券アナリストや投資家がこれらの結果がマイナスだと思ったら、私たちの普通株の価格に悪影響を及ぼすかもしれない。
108
私たちはそのような訴訟や法的手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らがより多くの財政資源とより成熟して発展した知的財産権の組み合わせを持っているので、このような訴訟や法的手続きの費用を私たちよりも効率的に負担するかもしれない。したがって、私たちは努力したにもかかわらず、私たちは第三者が私たちの知的財産権を侵害したり、流用したり、私たちの知的財産権に成功したりすることを防ぐことができないかもしれない。特許訴訟または他の訴訟の開始と継続によって生じる不確実性は、市場での競争能力に重大な悪影響を及ぼす可能性がある。
米国特許法または他の国または管轄区域特許法の変化は、特許の全体的な価値を低下させ、現在および任意の将来の候補製品を保護する能力を弱める可能性がある。
米国は最近、広範囲な特許改革立法を公布し、実施した。近年、米国最高裁判所はいくつかの特許事件に対して裁決を下し、場合によっては利用可能な特許保護範囲を縮小するか、場合によっては特許所有者の権利を弱めるかを決定している。我々の将来の特許取得能力に関する不確実性の増加に加えて,このようなイベントの結合は,いったん特許を取得する価値に関する不確実性をもたらしている.米国議会、連邦裁判所、および米国特許商標局の行動によれば、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、新しい特許を取得したり、私たちが所有している、許可されている、または将来獲得可能な特許を強制的に執行する能力を弱める可能性がある。同様に、他の国または管轄区域の特許法および法規の変化、それらを実行する政府機関の変化、または関連政府当局が特許法律または法規を実行する方法の変化は、私たちが新たな特許を取得したり、私たちが所有しているか、または将来獲得可能な特許を実行する能力を弱める可能性がある。
私たちは世界各地で私たちの知的財産権を保護できないかもしれないが、これは私たちの業務に否定的な影響を及ぼすかもしれない。
世界のすべての国で私たちの現在と未来の候補製品をカバーする特許は、私たちの現在と未来の候補品をカバーする特許を申請、起訴、保護するのは目を引くほど高いだろう。競争相手は、私たちまたは私たちの許可者が特許保護を受けていない司法管轄区域で私たちの技術を使用して彼ら自身の製品を開発することができ、また、私たちは特許保護を受ける可能性がありますが、特許執行力はアメリカの地域に比べて侵害製品を輸出することができます。これらの製品は、私たちが発行できるかもしれない特許を発行していない司法管轄区域で私たちの製品と競争することができるかもしれません。将来のいかなる特許主張や他の知的財産権も、それらの競争を効果的にまたは阻止するのに十分ではないかもしれません。
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの商業と競争の地位は損なわれるだろう。
私たちの候補製品のために特許や商標保護を求めるほか、特許を取得していないノウハウ、技術、その他の独自の情報を含む商業秘密に依存して、私たちの競争地位を維持することも可能です。私たちは、従業員、会社協力者、外部科学協力者、契約製造業者、コンサルタント、コンサルタント、および他の第三者のような、これらの秘密にアクセスする権利のある当事者と秘密協定を締結することによって、私たちのビジネス秘密を保護することを求めています。私たちはまた私たちの従業員、コンサルタント、コンサルタントと秘密と発明または特許譲渡協定を締結します。このような努力にもかかわらず、どちらも合意に違反し、私たちのビジネス秘密を含む私たちの固有の情報を漏洩する可能性がある。私たちの知的財産権の無許可使用と開示を規制することは困難であり、私たちは私たちの知的財産権を保護するために私たちが取った手順が有効かどうかも分からない。しかも、私たちはこのような違反のすべてについて十分な救済措置を得ることができないかもしれない。強制執行側が商業秘密を不正に開示したり流用したりする主張は困難であり、高価で時間がかかり、結果は予測できない。しかも、米国国内外のいくつかの裁判所は商業秘密をあまり望んでいないか、または保護したくない。
また,我々の競争相手は,我々のビジネス秘密に相当する知識,方法,ノウハウを自主的に開発する可能性がある.競争相手は私たちの製品を購入し、特許保護のない技術を開発する際に得られた競争優位性の一部または全部をコピーすることができる。もし私たちの任意の商業秘密が競争相手によって合法的に取得または独立して開発された場合、私たちは彼らまたは彼らが情報を伝達する人がその技術や情報を使用して私たちと競争することを阻止する権利がないだろう。もし私たちのビジネス秘密が競争相手に漏れたり、競争相手によって独立して開発されたりすれば、私たちの競争地位は損なわれるだろう。
109
我々はまた,我々のオフィスの実体セキュリティと我々の情報技術システムの実体と電子セキュリティを維持することで,我々のデータや他の機密情報の完全性とセキュリティの維持に努めている.我々はこれらの個人,組織,システムに自信があるが,プロトコルやセキュリティ措置が違反される可能性があり,機密情報の漏洩や流用行為を検出し,一方が機密情報を不正に開示したり流用したりすることは困難であり,高価で時間がかかり,結果的には予測できない.しかも、私たちはどんな違反についても十分な救済措置を得ることができないかもしれない。さらに、私たちの機密情報は競争相手に知られたり、独立して発見される可能性があり、この場合、私たちは彼らや彼らが情報を伝達する人がその技術や情報を使用して私たちと競争することを阻止する権利がないだろう。
第三者に依存して私たちのビジネス秘密を共有することを要求し、これは、競争相手がこれらの秘密または私たちのビジネス秘密が流用または開示されていることを発見する可能性を増加させる。
私たちは第三者に依存しているので、私たちが現在および任意の未来の候補製品を発見、開発、製造、または商業化するのを助けてくれるから、あるいは私たちが第三者と協力して現在または任意の未来の候補製品を開発、製造、商業化する場合、私たちは時々彼らとビジネス秘密を共有しなければならない。共同研究開発計画を行うことも可能であり、研究開発パートナーシップや同様の合意の条項に基づいてビジネス秘密を共有することが求められるかもしれない。独自の情報の研究または開示を開始する前に、当社のコンサルタント、従業員、第三者請負業者およびコンサルタントと秘密協定、材料譲渡協定、コンサルティング協定、または他の同様の合意を締結することによって、当社のノウハウを部分的に保護することを求めています。これらの協定は、一般に、私たちの商業秘密を含む、第三者が私たちの機密情報を使用または開示する権利を制限する。第三者と協力する際に契約条項が採用されているにもかかわらず、商業秘密および他の機密情報を共有する必要は、そのような商業秘密が私たちの競争相手に知られ、無意識に他の人の技術に組み込まれているか、またはこれらの合意に違反した場合に開示または使用されるリスクを増加させる。私たちの独自の地位が私たちのノウハウおよびビジネス秘密にある程度基づいていることを考慮すると、競争相手は、私たちのビジネス秘密または他の許可されていない使用または開示が、私たちの業務および運営結果に悪影響を及ぼす可能性があることを発見します。
さらに、これらの合意は、通常、私たちのコンサルタント、従業員、第三者請負業者、およびコンサルタントが、私たちのビジネス秘密に関連する可能性のあるデータを発行する能力を制限します。私たちは私たちのビジネス秘密を保護しようと努力しているにもかかわらず、私たちはこれらの合意の当事者が私たちの技術的ノウハウや他の商業秘密を不正に開示したり、使用したりすることを防ぐことができないかもしれない。さらに、私たちは私たちが可能であるか、または私たちの機密情報またはノウハウおよびプロセスに接触したすべての当事者とそのような合意に到達したことを保証することができない。許可されていない使用と開示を監視することは困難であり、私たちはまた私たちのノウハウを保護するために私たちが取った段階が有効かどうか分からない。これらの合意当事者である任意の協力者、科学コンサルタント、従業員、請負業者、およびコンサルタントがこれらの合意に違反または違反する任意の条項であれば、このような違反または違反に対応するための十分な救済措置がない可能性があり、したがって、私たちの商業機密を失う可能性がある。さらに、私たちのパートナー、協力者、または他の人が私たちに許可または開示した機密情報が意図的に漏洩されたり、違反されたり、違反された場合、私たちはその機密情報の所有者に責任を負うかもしれない。特許訴訟のように、第三者に不正または不法取得を強要し、私たちの商業秘密を使用することは高価で時間がかかり、結果は予測できない。しかも、アメリカ以外の裁判所は商業機密を保護することをあまり望まないこともある。
私たちが獲得する可能性のあるどの商標も侵害されたり、成功的に挑戦されたりして、私たちの業務に損害を与える可能性があります。
私たちは商標に依存して、私たちが発売を許可された候補製品を私たちの競争相手の製品と区別したい。私たちはまだ私たちの候補製品のために商標を選択していないし、私たちの現在または未来の候補製品のための登録商標を申請する過程も始まっていない。私たちが商標を選択して登録を申請すると、私たちの商標申請は承認されないかもしれない。第三者は私たちの商標申請に反対したり、他の方法で商標の使用に挑戦したりするかもしれない。もし私たちの商標が成功的に挑戦されれば、私たちは私たちの製品ブランドを再形成することを余儀なくされるかもしれません。これはブランド認知度の喪失を招き、広告と新しいブランドをマーケティングするために資源を投入する必要があるかもしれません。私たちの競争相手は私たちの商標を侵害するかもしれないが、私たちは私たちの商標を実行するのに十分な資源がないかもしれない。
さらに、私たちが登録または申請して商標として登録したかどうかにかかわらず、私たちが米国で使用する予定の任意の独自名または任意の他の候補製品名は、FDAの承認を受けなければならない。FDAは通常、他の製品名と混同される可能性を評価することを含む、提案された製品名を検討する。FDAが私たちが提案した任意の独自製品名に反対する場合、私たちは、適用商標法に適合し、第三者の既存の権利を侵害せず、FDAのために受け入れられる適切な独自製品名を決定するために、多くの追加リソースを必要とするかもしれません。
110
私たちの普通株に関するリスクは
私たちの経営業績は大幅に変動する可能性があり、あるいは投資家や証券アナリストの予想を下回る可能性があり、すべての状況は私たちの株価の変動や下落を招く可能性がある。
私たちの経営業績は年度と四半期変動の影響を受けると予想されています。私たちの純損失と他の経営業績は様々な要素の影響を受けます
もし私たちの経営業績が投資家や証券アナリストの予想を下回れば、私たちの普通株の価格は大幅に低下する可能性がある。しかも、私たちの経営業績のどんな変動も私たちの株価を大幅に変動させる可能性があります。
私たちの株価の変動は大きくて、あなたは投資の全部または一部を失うかもしれません。
私たちの株価は変動が大きい。このような変動により、投資家は普通株を購入する価格または普通株を購入した価格よりも高い価格で普通株を売ることができない可能性がある。私たちの普通株の市場価格は、10-K表の年次報告書の“リスク要因”と題するこの節で述べた他のリスクと以下を含む多くの要因の影響を受ける可能性がある
111
また,株式市場,特に製薬,生物製薬,バイオテクノロジー株式市場は極端な変動を経験しており,この変動は発行者の経営業績とは無関係であることが多い。これらの広範な市場と業界要素は、私たちの経営業績にかかわらず、私たちの普通株の市場価格を深刻に損なうかもしれない。
将来的に株式を発行したり、株式に変換できる債務証券を発行したりすることは、私たちの株式を希釈するだろう。
私たちは未来にもっと多くの資金を集める必要があるだろう。もし私たちが将来株式または転換可能な債務証券を発行することで追加資本を調達すれば、私たちの既存の投資家の権益は希釈され、これらの証券の条項は清算または他の私たちの株主権利に悪影響を及ぼす特典を含む可能性がある。将来的に私たちの普通株や他の株式証券の発行、あるいはこのような売却が発生する可能性があるとの見方は、私たちの普通株の取引価格に悪影響を与え、将来的に株式や株式証券を発行することで資金を調達する能力を弱める可能性がある。市場状況や戦略的考慮のため、現在または将来の運営計画のために十分な資金があると考えても、株式や転換可能な債券を発行することで追加資本を調達することを選択する可能性がある。将来の普通株の売却や将来売却可能な普通株が我々の普通株の取引価格に影響を与えるかどうかは予測できない。
証券や業界アナリストが私たちの業務に関する研究や報告を発表しない場合、または彼らが私たち、私たちの業務、または私たちの市場に関する不利または誤った研究または報告を発表した場合、私たちの株価および取引量は低下する可能性がある。
私たちの普通株の取引市場は、業界または証券アナリストが発表した、私たちの業務、または私たちの市場に関する研究と報告の影響を受けています。証券や業界アナリストが私たちへの報告を始めたり維持したりしなければ、私たちの株の取引価格はマイナスの影響を受けるだろう。もし私たちのどのアナリストも、私たち、私たちのビジネスモデル、私たちの知的財産権、私たちの株式表現、あるいは私たちの市場の不利または誤った研究または報告を発表した場合、または私たちの経営業績がアナリストの予想に達しなかった場合、私たちの株価は下落するかもしれない。もし一人以上のアナリストが私たちの報告書を停止したり、私たちに関する報告書を定期的に発表できなかった場合、私たちは金融市場での可視性を失う可能性があり、これは逆に私たちの株価や取引量を低下させる可能性がある。
112
私たちの主要株主と経営陣は私たちのかなりの割合の株式を持っていて、株主が承認した事項に大きな制御を加えることができ、彼らの利益はあなたが私たちの普通株式所有者としての利益と衝突する可能性があります。
2023年3月22日現在、私たちの役員と取締役は、5%以上の発行済み普通株式保有者とそのそれぞれの関連会社とともに、私たちが発行した普通株式の約56.2%を持っています。したがって、これらの株主が一緒に行動すれば、株主の承認を必要とする会社の行動の結果に大きな影響を与え、取締役の選挙、任意の合併、合併、私たちの所有またはほとんどの資産の売却、および任意の他の重大な会社取引を含む。このような株主の利益は私たちの他の株主の利益とは違うかもしれないし、それと衝突する可能性もある。例えば、これらの株主は、わが社の支配権の変更を延期または阻止することができ、たとえこのような制御権の変更が私たちの他の株主に利益をもたらすとしても、私たちの株主が私たちの会社や私たちの資産を売却する際に普通株のプレミアムを得る機会を奪い、私たちの普通株の現行の市場価格に影響を与える可能性がある。投資家は利益衝突が存在または発生する可能性があると考えているため、株式所有権の著しい集中は私たちの普通株の取引価格に悪影響を及ぼす可能性がある。
公開市場で私たちの大量の普通株を売ることは私たちの株価を下落させるかもしれない。
普通株を大量に売却したり、これらの売却が発生する可能性があると考えられるため、私たちの普通株価格は低下する可能性があります。これらの売却、あるいはこれらの売却が起こりうる可能性は、将来的に私たちが適切だと思う時間と価格で株式証券を売却することをより困難にする可能性もある。
我々の持分インセンティブ計画又は当該等計画に基づいて付与された将来奨励に基づいて、株式オプション及び引受証の行使により発行された株式は、適用される帰属スケジュール、市場対峙協定及び/又はロック協定の規定及び証券法第144及び701条の許可された範囲内で公開市場で販売される。
私たちは“新興成長型会社”と“小さな報告会社”であり、新興成長型会社やより小さい報告会社に適した情報開示要求が低下し、私たちの普通株の投資家への吸引力を低下させる可能性がある。
“雇用法案”の定義によると、私たちは“新興成長型会社”です。(I)財政年度の最終日まで(A)初公募(IPO)完了5周年後、(B)私たちの年間総収入が少なくとも12.35億ドル、または(C)前年6月30日現在、非関連会社が保有する普通株の時価が7億ドルを超え、(Ii)前3年間に10億ドルを超える転換不可能債券を発行したことを意味する新興成長型会社である。
新興成長型会社は特定の低減報告要求や他の一般的に上場企業の負担に適用される可能性がある。これらの規定には
113
私たちは、いくつかの減少した開示義務を利用することを選択し、将来の届出文書で他の減少した報告要件を利用することを選択する可能性がある。したがって、私たちの投資家に提供する情報は、あなたが株式を持っている非新興成長型会社の他の公開報告会社から得られた情報とは異なる可能性があります。雇用法案は、新興成長型企業は、これらの基準が民間企業に適用されるまで、上場企業に適用される新しい会計基準または改正された会計基準を遵守することができる長い過渡期を利用することができると規定している。私たちはこの延長された移行期間を利用しないことを撤回できないので、他の上場企業で新たなまたは改訂された会計基準を採用する必要がある関連日にこのような基準を採用します。
私たちも“小さな報告会社”であり、(I)非関連会社が保有するわが株の時価が2.5億ドル以下であれば、または(Ii)我々の最近終了した会計年度の年収が1億ドル未満であり、非関連会社が保有するわが株の時価が7億ドル未満であれば、より小さな報告会社であり続ける。もし私たちが当時小さな報告会社だったら、私たちはもう新興成長型会社ではなく、私たちは小さな報告会社が得ることができるいくつかの開示要求の免除に依存し続けるかもしれない。具体的には、小さな報告会社として、最近の2つの会計年度の監査財務諸表を当社のForm 10-K年報にのみ示すことを選択し、役員報酬に関する開示義務を削減し、また、新興成長型企業と同様に、年収が1億ドル未満の小さな報告会社であれば、独立公認会計士事務所が発行する財務報告内部統制認証報告を得る必要はないであろう。
私たちの定款文書やデラウェア州法律によると、反買収条項は私たちの買収をより困難にし、これは私たちの株主に有利になる可能性があり、私たちの株主が現在の経営陣を交換または更迭しようとすることを阻止するかもしれない。
当社の登録証明書の改訂及び再記載並びに私たちの改正及び再記述の定款における条項は、私たちの買収又は私たちの経営陣の変動を延期または阻止することができます。また、これらの規定は、株主が取締役会のメンバーを交代させることをより困難にし、株主が現在の経営陣を交代または罷免しようとしていることを挫折または阻止する可能性がある。私たちの取締役会は責任を持ってチームのメンバーを管理するように命じているので、これらの規定は逆に私たちの株主が私たちの管理チームの既存のメンバーを交換するいかなる試みにも影響を与える可能性があります。これらの規定には
また、私たちはデラウェア州に登録して設立されたので、改正されたデラウェア州会社法第203条の規定に管轄されています。この条項は、規定された方法で合併または合併が承認されない限り、私たちが発行した議決権を有する株の15%以上を保有している人が取引日後3年以内に私たちと合併または合併しないことを禁止しています。提案された合併や買収が一部の株主によって有益であると考えられても、これらの規定は適用されるだろう。
114
上場企業としては、私たちの運営コストが増加し、私たちの経営陣は、新しいコンプライアンス措置やコーポレートガバナンス実践を実施するために多くの時間を投入する必要があります。また、適切かつ効率的な内部統制を維持できなければ、正確な財務諸表をタイムリーに作成する能力が損なわれる可能性がある。
上場企業として、特に新興成長型企業や規模の小さい報告会社ではなくなった後、巨額の法律、会計、その他の費用が発生し、これらの費用は民間会社としては発生していません。サバンズ-オクスリ法案、ドッド-フランクウォール街改革と消費者保護法、ナスダックの上場要求とその他の適用される証券規則と法規は上場企業に対して様々な要求を提出し、有効な開示、財務制御と会社管理のやり方を確立と維持することを含む。また、取引法は、私たちの業務と経営業績に関する年度、四半期、現在の報告書を提出することを要求しています。私たちの経営陣と他の人たちはこのようなコンプライアンス計画を実施するために多くの時間を投入する必要があるだろう。さらに、このような規則と法規は増加し、私たちの法律と財務コンプライアンスコストを増加させ続け、いくつかの活動をより時間的で高価にするだろう。例えば、これらの規則および条例は、取締役および高級管理者責任保険をより難しく、より高価に得ることができ、低減された保険限度額および保証範囲を受け入れることを要求されるか、または同じまたは同様の保証範囲を維持するために巨額の費用を発生させる可能性がある。これらの規則はまた、私たちが合格した取締役会や取締役会のメンバーや幹部を引き付けて維持することをより難しくするかもしれない。しかし,これらの規則や条例は異なる解釈を受けることが多く,特殊性に欠ける場合が多いため,規制機関や理事機関が新たな指導意見を提供するにつれて,実践における適用は時間とともに変化する可能性がある。これは遵守事項に関する持続的な不確実性と、開示と統治慣行を絶えず修正するために必要なより高いコストをもたらす可能性がある。
また、上場企業として、サバンズ·オクスリ法案第404条を実施する米国証券取引委員会規則を遵守するための追加コストと義務を発生させる。これらの規則によれば、私たちの財務報告の内部統制の有効性を正式に評価しなければなりません。一旦、新興成長型企業や規模の小さい報告会社でなくなると、私たちの独立公認会計士事務所が発行した財務報告内部統制に関する認証報告書を含むことが要求されます。規定された時間内に第404条を遵守するために、私たちは費用が高く挑戦的な財務報告書に対する私たちの内部統制を記録して評価する過程を行っている。この点では、外部コンサルタントを招聘し、財務報告の内部統制に対する十分性を評価·記録するために詳細な作業計画を採用することが可能な内部資源を引き続き投入し、制御プログラムを適宜改善し、試験により制御措置の設計と効率的な動作を検証し、継続的な報告を実施し、財務報告の内部統制を改善するプログラムを実施する必要がある。
管理管理層は、財務報告の内部統制に達成しなければならない基準を評価するルールは複雑であり、規則下の詳細な基準を満たすためには、大量の文書、テスト、および可能な救済措置が必要である。テスト過程で、私たちの経営陣は重大な欠陥や欠陥を発見する可能性があり、これらの欠陥または欠陥は適時に修復できない可能性があり、“サバンズ-オックススリー法案”に規定されている期限内に完成できない可能性がある。私たちの財政報告書に対する内部統制はすべてのミスと詐欺を阻止したり発見したりしないだろう。
もし私たちがサバンズ-オキシリー法案404条の要求を適時に守ることができなければ、あるいは適切で効果的な内部統制を維持できなければ、私たちはタイムリーで正確な財務諸表を作成できないかもしれない。このような状況が発生すれば、私たちの株式の市場価格は下落する可能性があり、私たちは普通株のある証券取引所、米国証券取引委員会、または他の規制機関の制裁や調査を受ける可能性がある。しかも、もし私たちがこのような要求を満たし続けることができなければ、私たちはナスダックで上場し続けることができないかもしれない。
私たちの開示統制と手続きはすべてのミスや詐欺を阻止したり検出できないかもしれない。
私たちは“取引所法案”の定期報告書の要求事項を守らなければならない。我々の開示制御および手続きは、取引所法案に基づいて提出または提出された報告書に開示されなければならない情報が蓄積され、管理層に伝達され、米国証券取引委員会規則および表に指定された期間内に記録、処理、まとめ、報告されなければならないことを合理的に確保するために設計されている。任意の開示制御およびプログラムまたは内部制御およびプログラムは、発想および動作がどのように完全であっても、絶対的な保証ではなく、合理的な保証を提供することしかできず、制御システムの目標が達成されることを確保することしかできないと信じている。
これらの固有の制約は,意思決定における判断が誤りである可能性があり,簡単な誤りや誤りによって故障が発生する可能性があるという事実を含む.さらに、ある人の個人的な行動、2人以上の結託、または無許可超越制御は、制御を回避することができる。したがって,我々の制御システム固有の制限により,誤りや詐欺による誤り陳述が発生し,発見されない可能性がある.
115
私たちは予測可能な未来に私たちの株に現金配当金を支払わないと予想されているので、あなたが投資リターンを達成できるかどうかは私たちの普通株の価値増加にかかっています。
私たちは私たちの株の現金配当金を発表したり支払ったりしたことがない。私たちは現在、私たちの未来のすべての収益(もしあれば)を維持し、私たちの業務の成長と発展に資金を提供し、予測可能な未来に現金配当金を発表したり、支払ったりするつもりはない。したがって、株主へのいかなる見返りも私たちの普通株式価値の任意の付加価値に限定されることは確実ではない。
私たちは証券訴訟の影響を受けるかもしれないが、これは高価で、私たちの経営陣の注意をそらすかもしれない。
過去、証券市場の価格変動を経験した会社は証券集団訴訟の影響を受ける。私たちは未来にこのような訴訟の目標になるかもしれない。このような訴訟の是非や最終結果にかかわらず、私たちに対する証券訴訟は巨額の費用を招く可能性があり、私たちの経営陣の注意を他の業務からそらすことができます。
当社の登録証明書と定款は、デラウェア州衡平裁判所を、私たち株主が開始する可能性のある特定のタイプの訴訟および訴訟の唯一および独占フォーラムに指定し、これは、私たちまたは私たちの役員、上級管理者、または従業員との紛争において有利な司法フォーラムを得るための株主の能力を制限することができます。
私たちが書面で代替裁判所を選択することに同意しない限り、私たちが書面で代替裁判所を選択することに同意しない限り、私たちが修正して再記載した会社登録証明書規定は、デラウェア州衡平裁判所(または、デラウェア州衡平裁判所に管轄権がない場合、デラウェア州域内に位置する別の州裁判所、またはデラウェア州地域の連邦地域裁判所)は、以下のタイプの訴訟の唯一かつ独占的な裁判所になるであろう:(1)デラウェア州法律に基づいて私たちが提起した任意の派生訴訟または訴訟、(2)私たちのいかなる取締役も受託責任に違反すると主張する訴訟、(3)デラウェア州会社法又は当社が改正及び再記載した会社登録証明書又は定款の任意の規定に基づいて引き起こされる任意の訴訟、(4)内部事務原則によって管轄されるクレームを主張する任意の他の訴訟、又は(5)デラウェア州会社法第115条に定義された“内部会社クレーム”に基づく他の訴訟を主張する。この規定は、証券法、取引法、または米国連邦裁判所が排他的管轄権を有する任意の他のクレームを実行するための訴訟には適用されない。また、証券法第22条は、連邦裁判所と州裁判所は、このようなすべての“証券法”訴訟に対して同時に管轄権を持っていると規定している。したがって、州裁判所と連邦裁判所はこのようなクレームを受理する管轄権を持っている。私たちの改正と重述の付例はさらに、アメリカ合衆国連邦地域裁判所は、法律によって許容される最大の程度が、証券法または取引法によって提起された任意の訴因を解決する唯一のフォーラムとなると規定している。このような裁判所条項の選択は、株主が司法裁判所において、私たちまたは私たちの役員、上級管理者、または他の従業員と紛争することに有利であると考えるクレームを提出する能力を制限する可能性があり、これは、私たちと私たちの役員、上級管理者、および従業員に対するこのような訴訟を阻止する可能性がある。あるいは、裁判所が、私たちが改正および再記載した会社登録証明書および改正および再記載された法律におけるこれらの条項が、1つまたは複数の指定されたタイプの訴訟または法的手続きに適用されないことを発見した場合、または1つまたは複数の指定されたタイプの訴訟または訴訟手続きについて実行できない場合、私たちは、他の司法管轄区域において、そのような問題の解決に関連する追加費用を発生させる可能性があり、これは、私たちの業務および財務状況に悪影響を及ぼす可能性がある。いかなる者又は実体が当社の株式株式を購入又はその他の方法で取得する任意の権益は、当社が改訂及び再記載された会社登録証明書及び上記改正及び再記載された会社細則の規定に同意したとみなされなければならない。
116
純営業損失の繰越や他の税務属性を使用する能力が制限される可能性があります。
私たちの歴史の中で、私たちは大きな損失を受けて、近い将来に利益を達成することを期待していないし、永遠に利益を達成しないかもしれない。我々が課税損失を引き続き発生させた場合、未使用損失は、一定の制限(以下に述べる制限を含む)の制限の下で、当該等の未使用損失が満期(あれば)まで将来の課税収入(あれば)を相殺するために繰り越される。2022年12月31日現在、米国の連邦と州での純運営損失(NOL)はそれぞれ1010万ドル(税引前4800万ドル)と300万ドル(税引き前4810万ドル)だった。米国で繰り越した連邦純営業損失は無期限に繰り越すことができるが、我々の所有権に何らかの大きな変化が生じた場合には、年間使用制限を受ける可能性がある。繰り越しの国の純営業損失は2039年に満期になる。また、2022年12月31日までに、250万ドルと120万ドルの連邦と州信用繰越があり、2039年に満期が始まる。このような損失と貸手の繰り越しは適切な税務機関の審査と可能な調整を受ける可能性がある。
私たちのNOLと信用繰越はアメリカ国税局と州税務当局の審査と可能な調整を受けています。改正された1986年の国内税法第382節によると、わが社の所有権に何らかの累積的な変化が生じた場合、私たちの連邦NOLと貸記繰越は年間制限を受ける可能性がある。“規則”第382条の規定によると、1つ以上の会社の株式の少なくとも5%を保有する株主又は株主団体が3年間のスクロール期間内の持株比率がその最低持株割合より50ポイント以上増加した場合、“所有権変更”が発生する。IPOに関連する潜在的な変更を含む所有権変更のため、将来の課税収入または税務負債を相殺するNOL繰越および他の税務属性を利用する能力が制限される可能性があります。似たような規則は州税法に適用されるかもしれない。最初の公募株や他の取引による私たちの所有権の累積変化金額はまだ確定されていません。NOL繰越や他の税務属性を利用する能力に与える制限も何も確定されていません。しかも、私たちの株がその後変化したので、私たちは未来に所有権の変化を経験するかもしれない。その中のいくつかは私たちがコントロールできないからだ。もし私たちが課税収入を稼げば、この制限は私たちの将来の所得税負担を増加させる可能性があり、私たちの未来のキャッシュフローは悪影響を受けるかもしれない。我々のNOL繰延資産や他の繰延税金資産が最終的に将来の収益を実現する不確実性により、このような資産に関する全額推定準備が記録されている。
117
項目1 B。未解析STaffコメント。
ない。
プロジェクト2.プロパティ。
私たちの本部はマサチューセッツ州ケンブリッジ市剣橋路150号、郵便番号02140にあります。2023年3月31日に終了したレンタル契約によると、約8,955平方フィートのオフィスと実験室空間を借りました。2021年9月29日、マサチューセッツ州ボストンハリソン通り321号、郵便番号02118、約31,659平方フィートのオフィスと実験室空間を購入する新しいレンタル契約を締結しました。リースは2032年12月31日に終了し、追加の5年間選択権が2032年12月31日以降に延長される。既存の施設は現在の需要に対応するのに十分であると信じていますが、後日も商業的に合理的な条項で、適切な追加用地を提供します。
I項目3.法律規定バラの綿。
時々、私たちは正常な業務過程で発生した法的訴訟に巻き込まれるかもしれない。本年度報告Form 10−Kの日付まで、吾らはいかなる重大な法律事項や請求にも関与しておらず、吾等は吾等の未解決又は脅威に対する法的訴訟を一切知らないが、吾等は当該等の訴訟が吾等の業務、経営業績又は財務状況に重大な悪影響を及ぼす可能性があると信じている。
プロジェクト4それは.炭鉱の安全情報開示。
適用されません。
118
パ.パRT II
項目5.登録者Cの市場普通株、関連株主事項、発行者が株式証券を購入する。
市場情報
私たちの普通株はナスダック世界の精選市場に上場しています。取引コードはPYXSです。
私たち普通株保有者
Broadbridge Corporation Issuer Solutions,Inc.は我々の普通株式の譲渡エージェントと登録者である.2023年3月21日終値までに、約34人の普通株保有者が登録されている。これらの数字は私たちの株主記録から来ていて、私たちの普通株の実益所有者は含まれていません。彼らの株は様々な取引業者、決済機関、銀行、ブローカー、その他の受託機関で“街頭”の名義で保有しています。
配当をする
私たちは私たちの株の現金配当金を発表したり支払ったりしたことがない。私たちは現在、すべての利用可能な資金と未来の収益(あれば)を維持して、私たちの業務運営に使用し、予測可能な未来に私たちの普通株に現金配当金を支払わないと予想しています。将来の配当金の発表と支払いの任意の決定は、私たちの取締役会が適宜決定し、適用される法律、私たちの経営結果、私たちの財務状況、私たちの資本要求、一般業務状況、私たちの将来の見通し、および私たちの取締役会が関連すると思うかもしれない他の要素を含む様々な要素に依存するだろう。私たちが将来配当金のために現金配当金を支払う能力は、私たちが発行する可能性のある任意の優先証券の条項や、私たちが発生する可能性のある任意の追加債務を管理する協定によって制限されるかもしれない。
最近売却された未登録持分証券
2023年3月16日または発効日に、ファイザー社またはファイザー社と、2022年10月14日に改正され、再署名されたライセンス契約またはA&Rライセンス契約の第1の修正案である修正案1を締結し、A&Rライセンス契約のいくつかの条項を修正し、再確認した。A&Rライセンス協定は、ファイザーと当社が以前に署名した2020年12月8日のライセンス契約を改訂し、再確認し、2021年3月12日から発効し、現在まで改訂されている。修正案1によれば、私たちは、私たちの普通株のナスダック全世界ベスト市場での終値1株2.46ドル以上の営業日に相当する1株価格で500万ドル相当の普通株式数を発行し、発効日から2022年10月6日またはA&R発効日後180日以内のいずれかに発行することに同意します。もしA&R発効日後180日以内に、私たちの普通株の株価が1株2.46ドルを受け取っていない場合、発効日または発行後180日に500万ドル相当の普通株を発行することに同意します。1株当たりの価格は発行前の営業日の終値に等しいので、ファイザーは私たちの所有権権益が19.5%(19.5%)を超えてはならず、株式価値と500万ドルの差額はすべて現金で支払うことに同意します。
2023年3月17日、我々普通株のナスダック世界精選市場における終値は1株2.76ドルであった。第1号改正案の条項によると、ファイザー社に1,811,594株の普通株を発行し、金額は500万ドルに相当する。
第1号改正案により発行された普通株は、改正後の1933年の“証券法”又は“証券法”に基づいて登録されず、証券法第4(A)(2)条に規定する免除登録に基づいて登録される。
第1号修正案の写しは、本年度報告の添付ファイル10.24としてテーブル10-Kの形でアーカイブされている。第1号改正案の全文を参照することにより、第1号改正案の条項の前述の記述は完全に限定される。
119
初めて公募して得た金の使用
我々の初公募株である初公募株は,米国証券取引委員会が2021年10月7日に発効を発表したS-1表登録声明(文書番号333-259627)の影響を受けた.私たちは全部で10,500,000株の普通株を発行·販売しました。公開発行価格は1株16ドルで、引受割引、手数料、その他の発行コストを差し引いて1,570万ドルの純収益は1兆523億ドルです。米国銀行証券会社、Jefferies LLC、スイス信用証券(米国)有限会社、William Blair&Company、L.L.C.とLifeSci Capital LLCが今回発行の引受業者を務めている。引受割引および手数料または発売費用は、私たちの役員または上級管理者または彼らの連絡先に発生していないか、または私たちの普通株式の10%以上を所有している人、または私たちの任意の連合会社に支払われています。私たちはIPOの純収益を通貨市場基金に投資した。我々は、証券法第424(B)(4)条に基づいて2021年10月8日に米国証券取引委員会に提出された最終募集説明書に記載されている初公募による資金純額の計画用途に実質的な変化はない。
株式インセンティブ計画に基づいて発行された証券
下表は2022年12月31日現在の我々の持分インセンティブ計画の情報をまとめたものである。すべての未完成の報酬は私たちの普通株と関連がある。
|
|
証券数量 以下の期日に発送します 演習をする 卓越した オプション·株式承認証 権利があります (a) |
|
|
重みをつける 平均運動量 値段 卓越した 選択肢は、 株式引受証及び 権利.権利(1) (b) |
|
|
証券数量 使えるようにする 未来への発行 公平な条件の下で 報酬計画 (証券は除く) (A)欄に反映する (c) |
|||
|
|
|
|
|
|
|
|
|
|||
証券所有者が承認した持分補償計画: |
|
|
|
|
|
|
|
|
|
|
|
2021年持分インセンティブ計画(一)(三) |
|
|
5,117,872 |
|
|
$ |
14.28 |
|
|
360,924 |
|
2019年株式インセンティブ計画(1) |
|
|
2,745,402 |
|
|
$ |
6.22 |
|
|
657,484 |
|
2021年従業員株購入計画(4) |
|
|
— |
|
|
$ |
— |
|
|
|
752,524 |
単独合意により発行される限定普通株式(5) |
|
|
151,286 |
|
|
$ |
0.01 |
|
|
|
— |
証券保有者の承認されていない持分補償計画: |
|
|
|
|
|
|
|
|
|
|
|
2022年誘導計画(1)(2) |
|
|
721,242 |
|
|
$ |
1.95 |
|
|
|
678,758 |
合計する |
|
|
8,735,802 |
|
|
$ |
9.43 |
|
|
|
2,449,690 |
(1) 2022年誘導計画、2021年株式激励計画、2019年株式激励計画の下で発行された行権価格のない制限株式単位は、加重平均行権価格の計算には含まれていない。
(2) 2022年7月1日、会社取締役会は“2022年株式インセンティブ計画”(“2022年計画”)を承認し、当日発効した。
(3) 2021年配当インセンティブ計画によれば、発行予定の普通株式数は、各事業年度の初日に自動的に増加し、2021年12月31日までの事業年度から、2031年12月31日まで継続し、(I)前期12月31日に発行された普通株式総数の5%と(Ii)取締役会が決定する可能性のある株式数との間の小さい値を差し引く。2021年計画の常青樹条項により、2022年1月1日現在、会社は2021年計画で1,639,643株の普通株を追加追加した。
120
(4) 2021年9月27日、私たちの取締役会と株主は、私たちの登録声明が米国証券取引委員会によって発効する前日に発効する2021年の従業員株式購入計画を承認した。2021年にESPPは、計画に参加した従業員に合計424,595株の普通株式を発行することを最初に保留し、許可した。ESPPによると発行保留の普通株式数は会計年度ごとに自動的に増加し、2022年12月31日までの会計年度から2031年12月31日までの会計年度まで続き、(I)前会計年度12月31日に発行された普通株式総数の1%と(Ii)取締役会が決定可能な株式数との小さな値を差し引く。2021年ESPPの常青樹条項により、2022年1月1日現在、会社は2021年ESPPに327,929株の普通株を追加追加した。
(5) 2019年、私たちは従業員共同創業者と特定の非従業員コンサルタントに一定数の制限的な普通株式を発行した。制限的普通株は、2019年計画とは独立した独立した制限株式購入プロトコルに従って発行される。制限普通株の買い取り価格は1株0.01ドルに相当する。
発行人が株式証券を購入する
適用されません。
プロジェクト 6. [保留されている].
121
プロジェクト7.経営陣の議論と財務状況及び経営業績分析。
本年度報告のForm 10-Kに含まれる以下の財務状況および経営結果の議論および分析、ならびに監査された総合財務諸表および関連する付記を読むべきである。文意が別に指摘されているほか、本年度報告で言及した“Pyxis Oncology”、“Company”、“We”、“Us”、“Our”はいずれもPyxis Oncology,Inc.とその子会社を指す。
我々は臨床段階の腫瘍学会社であり,次世代治療薬のマルチモード組合せの開発に専念し,治療困難な癌に対して患者の生活の質を向上させる。我々が開発した候補製品の目標は、腫瘍細胞を直接殺し、癌が制御できない増殖と免疫逃避を引き起こす潜在的な病理を解決することである。マルチモードは様々な技術であり,独立しても他の技術と組み合わせても,患者のために有効な癌治療法を確立すると考えられる。2019年の発売以来,新規抗体薬物結合体(ADC),候補品,免疫腫瘍学(IO),候補品,モノクロナル抗体(MAb)を含む幅広い組み合わせが開発されており,単一療法として他の療法と組み合わせて使用する臨床前発見計画を開発している。
著者らは腫瘍学の研究開発において豊富な経験と熟練度を持つチームを結成し、著者らのチームの数十年の薬物開発経験を利用して、各種の革新療法を臨床に連れて行く予定である。私たちのグループの集団体験は製薬とバイオテクノロジーの二つの分野を扱っている。
私たちの製品組み合わせにはADCとIO候補製品が含まれています。私たちのパイプラインは各項目の間でバランスを維持し、固形腫瘍に重点を置いている。我々は2021年3月にファイザーから2つのADC計画の許可を得、2022年3月にBiosion USA,Inc.からIO計画の許可を得た。Thomas Gajesski博士の研究室で働いている追加の臨床前mAb発見計画もあります。我々はすべての候補製品の完全な全世界開発権と商業化権利を保持しているが、大中華区の中国(大陸、香港、マカオ、台湾)のPYX-106は除く。2022年12月、米国食品医薬品局(FDA)は、我々が調査したPYX−201およびPYX−106の新薬申請(IND)を承認したと発表した。われわれは臨床段階の腫瘍学会社への転換に熱心であり,われわれのチームの経験を利用してわれわれの臨床試験を実行することを期待し,患者により安全かつ効率的な治療選択を提供することを目的としている。
以下は私たちの現在の臨床開発の流れです

122
PYX-201
我々の主要なADC候補は、免疫グロブリンG 1(またはIgG 1)、抗フィブロネクチン外ドメインB(EDB)、mAbによってカテプシンBを介して黄金色誘導体に切断結合された研究中の新規ADCであるPYX-201である。フィブロネクチンは細胞外基質に存在する糖タンパク質である。フィブロネクチンEDBは血管形態発生を調節し、腫瘍に栄養と酸素を提供し、廃棄物を除去する手段、及び細胞を転移させる経路を提供する。EDBは多くの悪性腫瘍の中で過剰発現し、大多数の正常成人組織の中で最も発現が低く、これはそれを潜在的に魅力的な腫瘍を標的とする手段になり、同時に健康細胞を保留する。患者由来の異種移植やPDXモデルの臨床前モデルでは,単一薬物PYX−201を用いた腫瘍消退を認めた。また,臨床前同遺伝子腫瘍モデルをPYX−201で治療したところ,TMEにおけるT細胞の浸透が増加し,免疫原性細胞死(ICD)のマーカーであり,検査点阻害剤と併用することで相乗活性が得られることが観察された。
2022年12月、FDAは、臨床試験を開始するために、PYX-201のためのINDの使用を承認すると発表しました。2023年第1四半期に、PYX−201第1段階試験の第1被験者の用量をPYX−201−101と呼ぶことを発表した。PYX-201-101は開放ラベル、多中心、用量増加試験であり、PYX-201の安全性、耐性、薬物動態学、薬効学と初歩的な治療効果を評価し、そして更なる研究の推奨投与量を決定することを目的としている。再発或いは難治性固形腫瘍患者は、非小細胞肺癌、ホルモン受容体陽性乳癌、卵巣癌、甲状腺癌、膵臓導管腺癌或いはPDAC、軟組織肉腫或いはSTS、肝細胞癌或いは肝細胞癌及び腎臓癌を含み、すべて参加する資格がある。我々は,バイオマーカーの結果と潜在的な臨床活動の早期兆候を含む2024年初めにこの段階の臨床試験の初歩的なデータを報告する予定である。
PYX-106
我々の主要なIO候補製品はPYX−106であり,これは研究中の完全ヒトIgG 1ホモSiglec−15標的抗体であり,Siglec−15を介したT細胞増殖と機能抑制を遮断することを目的としている。私たちは最初にこの資産を固形腫瘍の治療に開発する予定です。我々はBiosion USA,Inc.から大中華区中国(大陸,香港,マカオ,台湾)を除く世界的にPYX-106の許可を得た.
2022年12月、FDAは、臨床試験を開始するために、PYX-106のためのINDの使用を承認することを発表しました。2023年第1四半期に、私たちは臨床試験サイトの活性化を開始し、現在患者をスクリーニングして第1段階試験、すなわちPYX-106-101に参加している。PYX-106-101はヒト初例1期多中心開放ラベル用量増加試験であり、再発或いは難治性固形腫瘍患者におけるPYX-106の安全性、耐性、薬物動態学、薬効学と初歩的な治療効果を評価することを目的としている。この第1段階用量増加試験は、標準治療によって固形腫瘍と疾病の進展に発展した患者と、生存を獲得できない或いは延長する標準看護治療に適していない患者を含む。この試験は、非小細胞肺癌、乳癌、子宮内膜癌、甲状腺癌、腎臓癌、胆管癌、膀胱癌、結腸直腸癌、頭頸部扁平上皮癌を含む固形腫瘍患者を募集する。私たちは2023年第2四半期初めに患者に薬を投与する予定だ。バイオマーカーの結果と潜在的な臨床活動の早期兆候を含む2023年末にこの第1段階臨床試験の初歩的なデータを報告する予定である。
一時停止/終了の手順
2022年8月、抗CD 123 ADC PYX−203および抗KLRG 1 IOプログラムPYX−102の臨床前開発を休止した。これらの計画は著者らの多様な候補治療方案の組み合わせに依然として重要であるが、現在臨床前開発を一時停止することは、PYX-201とPYX-106の臨床開発を推進するための資源を増加させることができる。さらに、我々は、臨床前毒性研究データの審査および分析、ならびにPYX−202の期待される臨床用途および商業的将来性に基づく、抗DLK 1 ADC PYX−202の継続開発を一時停止することにした。
発見
以上の決定した計画に加えて,モノクロナル抗体,ADC結合,リンカーとペイロード選択および免疫腫瘍学の理解に関する専門知識を用いて,様々な目標の研究·開発を行っている。
123
設立以来、私たちのほとんどの資源は研究と開発活動を行い、臨床前研究と臨床試験を行い、組織と会社のために人員、業務計画、資金を配備し、私たちの知的財産権の組み合わせを確立と維持し、潜在的な候補製品を確定することに集中している。私たちは販売を許可された製品もなく、製品販売や他の供給源からも何の収入も得ていない。設立以来、私たちは深刻な運営損失が発生した。2022年12月31日と2021年12月31日までの年間純損失はそれぞれ1兆207億ドルと7600万ドルと報告した。2022年12月31日までの累計赤字は2.124億ドル、純株式は1.608億ドル、現金と現金等価物は1.793億ドルだった。私たちは予測可能な未来に巨額の費用と運営損失が続くと予想している。私たちは私たちが行っている活動のために、私たちの費用と資本支出が大幅に増加すると予想する。これまで、私たちの業務は主に転換可能な優先株の売却と株式証券の売却によって資金を調達し、将来の臨床および臨床前活動を支援するために追加の資金が必要になるかもしれない。
許可と協力協定
ファイザーとのライセンス契約。
2020年12月、私たちは、PYX-201およびPYX-203、およびADC候補製品を含むいくつかの許可目標に対する候補抗体薬物結合製品の世界的な開発および商業化権利を得るために、ファイザーまたはファイザー社と改訂されたライセンス契約またはファイザー許可プロトコルを締結した。ファイザーが所有または制御するいくつかの特許については、許可されたADCを含む、私たちの権利は独占的である。ファイザーはまた、ファイザーADC技術またはFACTプラットフォームを用いた非独占的な許可を与えてくれた。最初の許可標的はCD 123および追加ドメインB(フィブロネクチンのEDB)を含み、第三者またはファイザーADC開発計画テーマではない他の許可標的を追加するために、その許容範囲を拡大することを選択することができる。ファイザー許可協定は2021年3月に発効し,500万ドルの前払い現金を含めて2500万ドルの許可料を支払い,2021年10月の初公募で1,911,015株の普通株に変換し,2000万ドルをファイザーに発行した12,152,145株のB系列転換可能優先株を発行した。
2022年10月6日、ファイザーと改訂され、再記述されたライセンス契約またはA&Rライセンス契約を締結し、ファイザーライセンス協定を修正し、再確認した。A&Rライセンスプロトコルによれば、ファイザーは、PYX-201およびPYX-203、およびADC候補製品を含むいくつかの許可ターゲットのためのADC候補製品を開発および商業化するために、ファイザーFACTプラットフォーム技術下のグローバル独占権利を付与する。他のADC目標は象徴的な前払いとマイルストーンの許可を得るかもしれない。さらに、EDBおよびCD 123に関する1つまたは複数の国または地域で開発、製造、および商業化許可されたADCおよび認可製品を購入または開発、製造、および商業化する権利をファイザーに独占的に交渉する権利を付与する。A&Rライセンス契約の条項によると、ファイザーに800万ドル(2023年1月に支払う)を支払うことに同意し、私たちの普通株2,229,654株を発行した。また、発効日または発行後180日にその数量の普通株を発行し、500万ドルに相当し、1株当たりの価格は私たちの普通株の発行前の営業日の終値に等しいので、ファイザーの私たちの所有権権益は19.5%(19.5%)を超えてはいけません。株式価値と500万ドルの差額は現金で支払うことができます。
開発、規制、ビジネスマイルストーンなど、最初の4つの許可されたADCに将来の支払いを支払う義務があり、総額は6.65億ドルに達する。さらに、我々は、ADCの開発、規制、および商業マイルストーンを含む、FACTプラットフォームを介して開発および商業化された最初の4つの許可されたADC以外の各追加の許可取得目標に支払いを要求される。また,ADCライセンス製品が発売されれば,ライセンス製品の純売上高の階層印税をファイザーに支払い,印税料率は低い1桁から10代程度まで様々である。我々のライセンス使用料義務は、第1の商業販売から最近発生したライセンス製品および国/地域ごとに適用される:(1)第1の商業販売から12年、(2)すべての法規またはデータ排他性満了、および(3)ライセンス製品に関する国/地域のライセンス特許の最後の有効な権利要件が満了したとき。ファイザーに一定の割合の分配可収入を支払う義務があり,割合は20%から下位2桁まで様々であり,具体的には適用分許可に入った場合の許可製品の開発段階に依存する。
124
シカゴ大学とのライセンス契約
2020年4月、私たちはシカゴ大学や大学と許可協定、すなわち大学許可協定と賛助研究協定を締結した。許可条項によれば、私たちは、許可された技術および材料と組み合わせて、または特定の特定の生物学的標的の活性を評価、調節、または利用することができる独占的な世界的権利を有し、許可された特許の有効な権利要件によってカバーされる製品を開発および商業化することができる。
大学の許可を得た一部の代償として,2020年に大学に48,919株の普通株を発行した。大学ライセンス契約によると、合計770万ドルの潜在開発とビジネスマイルストーンの支払いと、1%未満の低い桁以下のライセンス製品の純売上高で特許権使用料を運用する義務があり、ライセンス製品の初商業販売後のある年には、最低年間特許権使用料は300万ドルを超えてはならない。適用される再許可を締結した日に応じて,一定割合の再許可収入を大学に支払い,範囲の低い青少年にする義務がある。
レゴ化学生物科学と署名した協定です
2020年12月、私たちはレゴ化学生物科学会社またはレゴ化学とライセンス契約またはレゴ化学ライセンス契約を締結し、加入、投資、および追加の価格協定を選択し、または加入プロトコルを選択した。レゴ化学ライセンスプロトコルによれば、LCB 67の世界(韓国以外)の開発権および商業化権利を取得し、LCB 67はDLK-1のためのADC候補製品、およびライセンス化合物を含む製品である。私たちは2021年3月にレゴケミカルに900万ドルを支払い、このお金は研究開発費として記録された。さらに、私たちはレゴ化学からいくつかの最初の許可製品を購入する可能性があり、コストは700万ドルと予想され、開発、規制、商業マイルストーン、およびライセンス製品の純販売に対して異なる比率で印税を徴収することを含む未来の支払いまたは支払い義務があるかもしれない。2022年第3四半期に、著者らは毒性研究データの回顧と分析に基づいて、LCB 67の継続開発を停止し、抗DLK 1 ADCの臨床応用と商業将来性を予測した。
さらに、レゴ化学は、いくつかの事件が発生したときに960万ドルのマイルストーン支払いまたは追加マイルストーン支払いを得る権利と引き換えに、800万ドルの選択権を私たちに支払う権利を行使し、これらのイベントは、普通株が初めて公開された定価または要約日、または私たちが支配権が取引を変更する標的である場合を含む。2021年10月に初めて公募した時、追加のマイルストーン支払い事件が触発され、2022年1月にレゴケミカルに960万ドルを支払いました。合意に加入することを選択することによって、私たちにはこれ以上の義務がない。
Biosion USA,Inc.と締結されたライセンスプロトコル.
2022年3月28日、我々はBiosion USA,Inc.またはBiosionとライセンス契約または生物ライセンス契約を締結し、これにより、BSI-060 T、標的抗体Siglec-15、IO候補製品(現在、PYX-106と呼ばれる)および許可化合物を含む製品の開発、製造および商業化のための大中国(大陸、香港、マカオ、台湾を除く)の独占的な世界的許可を得た。生物学的許可協定の条項によれば、各方向の他方は、任意の二重特異性または多重特異性抗体、一方またはその付属会社によって制御される、予期される作用機序として阻害、調節、または結合したSiglec-15の抗体-薬物結合体を開発、製造および商業化するために、他方の領土(Biosionの大中国、およびPYXISの世界の他の地域)で独占的に許可されるように優先的に契約権を付与する。
Biosionライセンス協定によると、私たちは1,000万ドルの前払い費用を支払い、開発、規制、ビジネスマイルストーンを含む未来の支払いまたは支払いを義務化し、通常の承認の場合、最大2.175億ドル、承認を加速した場合、最大2.225億ドルに達する。また,製品が発売されれば,ライセンス製品の純売上高に応じてBiosion分級印税を支払い,印税料率は低い1桁から低い青少年まで様々である。我々のライセンス使用料義務は、第1の商業販売から最近発生したライセンス製品および国/地域ごとに適用される:(1)第1の商業販売から12年、(2)すべての法規またはデータ排他性満了、および(3)ライセンス製品に関する国/地域のライセンス特許の最後の有効な権利要件が満了したとき。Biosionに一定割合の分割可収入を支払う義務があり,範囲は−2桁から低い−2桁まで様々であり,適用される許可に入った場合の許可製品の開発段階に依存する。
125
Voxallと合金治療会社の合弁企業。
2021年3月、PYXIS腫瘍学の現場特定目標目録と合金会社のATX-GXを利用するために、合金会社と協力して設立された合弁会社であるVoxallを援助·運営するための最終取引契約を合金会社と締結したプラットフォームや抗体発見サービスですVoxallはPYXIS腫瘍学会社と合金会社のVoxallに投票権のある会員単位の50%を授与し、いくつかの初期寄付金と交換した。
Voxallの形成過程において、著者らは合金会社とVoxall社と3年間の研究協力を行い、ある生物標的を確定と選択し、そしてこれらの標的に対してこれらの標的に対する開発候補抗体を創造し、更なる臨床前開発、臨床開発と商業化に用いた。協力協定によると、双方は共同で合意した研究計画と予算に基づいて研究を行い、最大6つの研究プロジェクトは共同選択の目標に重点を置いている。我々とコロラドは,Voxallと単独のサービスプロトコルを締結することで連携に研究支援を提供し,これらのサービスはVoxallが発行した本チケットの形で支払う.Voxallは協力して発生するすべての知的財産権を持つが,合金のATX−GXに関する知的財産権は除外するホームです。
研究計画下の開発候補抗体がある双方が合意した選択基準を満たしていれば,Voxallの独占的許可を得,さらにその研究計画で発見されたすべての開発候補抗体を開発して商業化する権利がある。私たちはいくつかの事前に合意された財政条項に基づいて研究プロジェクトを許可するかもしれない。他のすべての許可内の研究項目については、第三者評価によって決定された公平な市場価値を支払う義務がある。私たちが許可していないどんな研究プロジェクトもVoxallによって第三者に許可される可能性がある。
126
私たちの運営結果の構成要素は
運営費
研究と開発費
研究開発費には,我々の研究活動によるコスト,我々の発見努力と我々のプロジェクト開発が含まれている.これらの費用には
私たちは発生した費用に応じて研究と開発費用を支払う。私たちが将来受け取る研究·開発活動のための商品やサービスのために支払われる払い戻し不可能な前金は前払い費用として記録されています。前払い金額は、関連する貨物がサービスを提供するか、または貨物がサービスを提供することがもはや予期されていない場合に支出される。
私たちの直接外部研究開発費用には、コンサルタント、請負業者、CMOおよびCROに支払う費用、精算材料、および私たちの臨床前および臨床活動に関連する他のコストが含まれている。従業員コスト、私たちの探索作業に関連するコスト、実験室用品および施設費用(減価償却または他の間接コストを含む)を特定の製品開発計画に割り当てることはありません。これらのコストは複数の計画と私たちのプラットフォームに配置されているため、個別に分類されていません。
臨床開発後期段階にある候補製品は通常,臨床開発早期段階の候補品よりも高い開発コストを有しており,これは主に後期臨床試験の規模と持続時間が増加しているためである。近い将来,われわれが進行·計画中の臨床前·臨床開発活動により,われわれの研究·開発費用が大幅に増加することが予想される。私たちの候補製品が開発に成功するかどうかは大きな不確実性を持っている。現在、私たちは私たちの任意の候補製品の臨床前と臨床開発を完成するために必要な努力の性質、時間、コストを正確に推定または知ることができず、私たちはいかなる候補製品の監督管理許可を得ることに成功することができないかもしれない。
一般と行政費用
一般および行政費用には、主に賃金と人事関連費用が含まれ、株式ベースの補償、行政、法律、財務、会計、人的資源、その他の行政機能者の解散費が含まれる。一般および行政費用には、監査、税務、法律サービスの専門費用、保険、取締役会報酬、コンサルティングおよびその他の行政費用、研究開発費に含まれていない施設コストも含まれる。
その他の収入(費用)
利息収入は主に私たちが投資した現金と現金等価物残高で稼いだ利息からなります。
派生負債の公正価値変動は、レゴ化学と引受、投資及び追加代価合意を達成したことによって記録された派生負債公正価値の増加である。
127
所得税
当社は設立以来、当社で発生した純損失や毎年度および中期に稼いだ研究開発税収控除について所得税割引を確認していません。既存の証拠の重さによると、当社のすべての純運営損失、あるいはNOL、繰越および税務控除は、実現できない可能性が高いと信じています。
2022年12月31日現在、米国の連邦と州での純運営損失はそれぞれ1010万ドル(税引き前4800万ドル)と300万ドル(税引き前4810万ドル)だった。米国で繰り越した連邦純営業損失は無期限に繰り越すことができるが、我々の所有権に何らかの大きな変化が生じた場合には、年間使用制限を受ける可能性がある。繰り越しの国の純営業損失は2039年に満期になる。また、2022年12月31日までに、250万ドルと120万ドルの連邦と州信用繰越があり、2039年に満期が始まる。このような損失と貸手の繰り越しは適切な税務機関の審査と可能な調整を受ける可能性がある。
経営成果
2022年と2021年12月31日終了年度比較
次の表は、2022年12月31日と2021年12月31日までの年間運営結果(単位:千)をまとめたものです
|
|
十二月三十一日までの年度 |
|
|
|
|
||||||
|
|
2022 |
|
|
2021 |
|
|
変わる |
|
|||
運営費用: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
$ |
86,129 |
|
|
$ |
51,054 |
|
|
$ |
35,075 |
|
一般と行政 |
|
|
37,352 |
|
|
|
18,663 |
|
|
|
18,689 |
|
総運営費: |
|
|
123,481 |
|
|
|
69,717 |
|
|
|
53,764 |
|
運営損失 |
|
|
(123,481 |
) |
|
|
(69,717 |
) |
|
|
(53,764 |
) |
その他の収入(支出): |
|
|
|
|
|
|
|
|
|
|||
利子収入 |
|
|
2,764 |
|
|
|
23 |
|
|
|
2,741 |
|
関連側手数料収入 |
|
|
— |
|
|
|
181 |
|
|
|
(181 |
) |
派生負債の公正価値変動 |
|
|
— |
|
|
|
(6,231 |
) |
|
|
6,231 |
|
その他収入合計 |
|
|
2,764 |
|
|
|
(6,027 |
) |
|
|
8,791 |
|
合営企業権益法投資損失 |
|
|
— |
|
|
|
(231 |
) |
|
|
231 |
|
純損失 |
|
$ |
(120,717 |
) |
|
$ |
(75,975 |
) |
|
$ |
(44,742 |
) |
研究と開発費
次の表は、2022年12月31日と2021年12月31日までの年間研究開発費(単位:千)をまとめたものです
|
|
十二月三十一日までの年度 |
|
|
|
|
||||||
|
|
2022 |
|
|
2021 |
|
|
変わる |
|
|||
研究·開発計画費 |
|
$ |
60,967 |
|
|
$ |
40,363 |
|
|
$ |
20,604 |
|
株式ベースの報酬を含む人事関連費用 |
|
|
18,975 |
|
|
|
7,836 |
|
|
|
11,139 |
|
他の研究や開発費 |
|
|
6,187 |
|
|
|
2,855 |
|
|
|
3,332 |
|
研究開発費総額 |
|
$ |
86,129 |
|
|
$ |
51,054 |
|
|
$ |
35,075 |
|
研究開発費は3,510万ドル増加し、2021年12月31日現在の5,100万ドルから2022年12月31日現在の8,610万ドルに増加した。計画支出が2060万ドル増加したのは,主に契約製造コストが1370万ドル増加し,臨床前コストが620万ドル増加し,主に毒性研究やPYX−201やPYX−106に関するIND申告の臨床前作業に関連して臨床試験コストが300万ドル増加したが,許可費が230万ドル減少したことで相殺された。人事に関する支出は860万ドル増加し、株ベースの報酬は250万ドル増加したが、これは主に我々の研究·開発活動を支援するために増加した従業員数によるものである。その他の研究·開発費は330万ドル増加し、主な原因は実験室用品の増加、実験室空間分配費用および専門とコンサルティング費用の増加である。
128
一般と行政費用
次の表は、2022年12月31日と2021年12月31日までの年度の一般と行政費用(単位:千):をまとめています
|
|
十二月三十一日までの年度 |
|
|
|
|
||||||
|
|
2022 |
|
|
2021 |
|
|
変わる |
|
|||
株式ベースの報酬を含む人事関連費用 |
|
$ |
18,732 |
|
|
$ |
8,622 |
|
|
$ |
10,110 |
|
プロおよび顧問料 |
|
|
13,061 |
|
|
|
7,606 |
|
|
|
5,455 |
|
施設、保険、その他の費用 |
|
|
5,559 |
|
|
|
2,435 |
|
|
|
3,124 |
|
一般と行政費用総額 |
|
$ |
37,352 |
|
|
$ |
18,663 |
|
|
$ |
18,689 |
|
一般·行政費は1,870万ドル増加し、2021年12月31日現在の1,870万ドルから2022年12月31日までの3,740万ドルに増加した。株式給与を含む人事関連支出が1010万ドル増加したのは、人事関連支出が330万ドル増加し、株ベース報酬が680万ドル増加したことが主な原因であり、両者とも従業員数が増加したためである。専門家や相談費が540万ドル増加したのは、主に私たちの成長と運営を支援する法律、専門、相談費が全面的に増加したからです。施設、保険、その他の費用が310万ドル増加したのは、主にレンタル料支出が160万ドル増加したことと、役員や上級管理職の保険費用が150万ドル増加したためだ。
その他の収入(費用)
2022年と2021年12月31日までの年間の利息収入はそれぞれ280万ドルと2.3万ドルで、投資現金と現金の同値残高で稼いだ利息を含む。2022年12月31日までの年度内に増加した主な原因は、我々の預金·通貨市場基金の金利上昇である。
その他の支出には、合意への加入を選択した結果として、2021年12月31日までの年間派生負債の公正価値変動620万ドルが含まれている。関連側からのサービス料収入には,2021年12月31日までの年度にVoxall合弁企業に提供されるサービス収入20万ドルが含まれている。
129
流動性と資本資源
2022年12月31日現在、私たちは1億793億ドルの現金と現金等価物を持っている。2022年12月31日と2021年12月31日までの純損失はそれぞれ1兆207億ドルと7600万ドルだった。2022年12月31日までの累計赤字は2兆124億ドルだった。
我々が行っている活動に関する費用は大幅に増加することが予想され,特に我々が開発している候補製品の臨床試験を進めた場合である。私たちが必要な時間と金額は、多くの要素に依存するだろう
これまで、相当な製品収入を生み出すことができれば、株式発行、債務融資、協力、戦略連合、マーケティング、流通、または許可手配を通じて、私たちの運営に資金を提供する予定です。私たちが株式または転換可能な債務証券を売却することによって追加資本を調達する場合、あなたの所有権資本は希釈され、これらの証券の条項は清算または他の特典を含む可能性があり、普通株主としての権利に悪影響を及ぼす可能性がある。債務融資および優先株融資に関与する可能性のある協定は、追加債務を招く、買収、買収、合併または協力取引に従事する、我々の資産を売却することができるかもしれない、資本支出を行う、株式を償還する、特定の投資を行う、または配当を発表するなど、私たちの特定の行動をとる能力を制限または制限する契約を含む。
もし私たちが第三者との協力、戦略連合またはマーケティング、流通または許可手配を通じてより多くの資金を調達するならば、私たちは私たちの技術、将来の収入源、研究計画、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になる可能性のある条項でライセンスを付与しなければならないかもしれない。もし私たちが必要な時に株式や債務融資を通じてより多くの資金を調達できない場合、私たちは私たちの製品開発や将来の商業化努力を延期、制限、減少または終了することを要求されるかもしれません。あるいは私たちは自分で開発とマーケティングをより望んでいた候補製品の権利を与えます。
棚登録表と“市場で”計画
2022年11月1日、我々は普通株式、優先株、権利証、債務証券、権利と単位の発行に使用され、総額2.5億ドルに達するS-3表登録声明を米国証券取引委員会に提出した。2022年11月14日、登録声明は米国証券取引委員会によって発効が発表された。登録声明には、市場で最高1.25億ドルの普通株式売却計画を提供するATMが含まれている。本報告の日現在、普通株は販売されておらず、ATM発売計画から何の収益も得られていない。
130
キャッシュフロー
次の表は、2022年12月31日と2021年12月31日までの年間のキャッシュフロー情報(単位:千)を提供しています
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
経営活動のための現金純額 |
|
$ |
(89,335 |
) |
|
$ |
(35,326 |
) |
投資活動のための現金純額 |
|
|
(6,399 |
) |
|
|
(590 |
) |
融資活動が提供する現金純額 |
|
|
183 |
|
|
|
304,044 |
|
現金、現金等価物、および制限的現金純増加 |
|
$ |
(95,551 |
) |
|
$ |
268,128 |
|
経営活動
2022年12月31日までの年間で、経営活動のための現金純額は8930万ドルで、その中には1兆207億ドルの純損失が含まれており、2690万ドルの非現金費用と460万ドルの営業資産と負債の純変化によって部分的に相殺されている。非現金料金には、普通株での決済と対応するファイザーFACTプラットフォーム許可料に関する930万ドル、株式ベースの補償費用1580万ドル、110万ドルの経営リース使用権資産の償却がある。私たちの経営資産や負債の変化は、主に新本部の経営リースによる経営リース負債の430万ドルの増加と、主に前払い臨床と契約製造コストおよび取締役や高級管理者の保険料に関する前払い費用と他の流動資産が340万ドル増加したが、支出と他の流動負債の1200万ドルの増加と、定例仕入先支払いのスケジュールにより支払いすべき帳簿が830万ドル減少したことで相殺された。
2021年12月31日現在,運営活動で使用されている現金純額は3,530万ドルであり,純損失7,600万ドルを含むが,3,400万ドルの非現金費用および670万ドルの運営資産および負債純変動部分で相殺されている。3400万ドルの非現金費用は、主にファイザー社がBシリーズ転換可能優先株で決済した2000万ドルの研究と開発許可費、620万ドルの派生負債公正価値変化、640万ドルの株式ベース補償によるものである。私たちの営業資産と負債の変化は主に売掛金、課税費用、その他の流動負債が970万ドル増加しましたが、前払いと他の流動資産の変化と300万ドルの賃貸支払い部分はこの増加を相殺しました。これらはすべて私たちの業務の増加、私たちの研究計画の推進、サプライヤーの領収書と支払いの時間スケジュールによるものです。
投資活動
2022年12月31日と2021年12月31日までの年度の投資活動のための現金純額はそれぞれ640万ドルと60万ドルであり、主な原因は財産と設備の購入だ。購入した財産と設備には実験室設備、家具と固定装置、そして私たちの新しい本社の賃貸内装が含まれています。2021年12月31日までの1年間に、私たちはまた、私たちの合弁企業Voxall Treateutics、LLCに10万ドルの投資を行った。
融資活動
融資活動が提供する現金純額は2022年12月31日までの1年間で20万ドルで、主に株式オプション行使の純収益を含む。
2021年12月31日までの年間で、融資活動が提供する現金純額は3.04億ドルで、主に初公募株で売却された普通株の純収益1兆523億ドルと、私たちのBシリーズ転換可能優先株を売却する純収益1兆516億ドルを含む。
2022年12月31日現在の既存現金残高1.793億ドルと研究開発·業務発展計画によると、2025年上半期の運営費と資本支出需要に資金を提供できると予想されています。しかし,我々のこの推定は誤りであることが証明される可能性があるという仮定に基づいており,我々の運営計画は現在我々が知らない多くの要因によって変化する可能性がある.しかも、私たちは期待よりも早く利用可能な資本資源を利用することができる。
131
契約義務と約束
以下では,2022年12月31日までの契約義務と,これらの義務が将来的に予想される流動性とキャッシュフローに及ぼす影響について概説する。
私たちはマサチューセッツ州ボストンでオフィスと実験室を借りた。賃貸支払いは2022年12月22日から2032年12月31日まで続き、毎年3%の賃料上昇が計画されている。賃貸期間内に残った契約固定賃貸支払いはテナント改善手当を差し引くと総額3180万ドルとなる。
しかも、私たちは正常な業務過程で許可と関連協定を締結した。協定によると、他の項目を除いて、将来の支払い、または支払い、特許権使用料、および将来の再許可収入を支払う義務があります。これらの合意の下での支払い義務は、将来の事件に依存するため、これらの許可および協力協定の下で将来満期になる可能性のある支払いを契約義務に入れていません。
ファイザーとのA&Rライセンス契約によると、開発、規制、ビジネスマイルストーンを含む最初の4つの許可されたADCに将来的または支払いを支払う義務があり、総額は6.65億ドルに達する。さらに、我々は、ADCの開発、規制、および商業マイルストーンを含む、FACTプラットフォームを介して開発および商業化された最初の4つの許可されたADC以外の各追加の許可取得目標に支払いを要求される。また,製品が発売されれば,製品純売上高の分級印税をファイザーに支払い,印税料率は低い1桁から10代程度まで様々である。我々のライセンス使用料義務は、第1の商業販売から最近発生したライセンス製品および国/地域ごとに適用される:(1)第1の商業販売から12年、(2)すべての法規またはデータ排他性満了、および(3)ライセンス製品に関する国/地域のライセンス特許の最後の有効な権利要件が満了したとき。ファイザーに一定の割合の分配可収入を支払う義務があり,割合は20%から下位2桁まで様々であり,具体的には適用分許可に入った場合の許可製品の開発段階に依存する。
また、ファイザーとのA&R許可協定によると、2023年4月にこの数量の普通株を発行することに同意し、1株当たりの価格は私たちの普通株の発行日の前の営業日の終値に相当するため、ファイザーの私たちの所有権権益は19.5%(19.5%)を超えてはならず、株式価値と500万ドルの間のいかなる差額も現金で支払うことになった。
レゴ化学ライセンスプロトコルによると,ある開発,規制,販売マイルストーンが実現すれば,レゴ化学に合計2.845億ドルと,中央値から上位数までのライセンス製品の純売上高の階層的特許権使用料を支払うことが義務付けられている。我々がパイプライン優先順位を再決定する一部として,レゴ化学から許可を得たPYX−202の継続開発を停止することを選択した。
シカゴ大学との大学ライセンス契約によると、合計770万ドルの潜在開発とビジネスマイルストーンと、1%未満の低い桁以下のライセンス製品の純売上高で特許権使用料を運用する義務があり、ライセンス製品が初めて商業販売された後のある年には、最低年間特許権使用料は最高300万ドルに達する。適用される再許可を締結した日に応じて,一定割合の再許可収入を大学に支払い,範囲の低い青少年にする義務がある。
Biosionライセンス協定によると、私たちは開発、規制、ビジネスマイルストーンを含む未来の支払いまたは支払いを義務化し、正常に承認された場合には最高2億175億ドル、承認を加速した場合には最高2.225億ドルを支払う義務がある。また,製品が発売されれば,ライセンス製品の純売上高に応じてBiosion分級印税を支払い,印税料率は低い1桁から低い青少年まで様々である。我々のライセンス使用料義務は、第1の商業販売から最近発生したライセンス製品および国/地域ごとに適用される:(1)第1の商業販売から12年、(2)すべての法規またはデータ排他性満了、および(3)ライセンス製品に関する国/地域のライセンス特許の最後の有効な権利要件が満了したとき。Biosionに一定割合の分割可収入を支払う義務があり,範囲は−2桁から低い−2桁まで様々であり,適用される許可に入った場合の許可製品の開発段階に依存する。
著者らはまた正常な業務過程中にCMO、CROとその他の第三者と臨床前仕事と臨床開発関連仕事について契約を締結した。このような契約には最低購入約束は含まれておらず、私たちは事前に書面で契約をキャンセルすることを通知することができる。キャンセル時に支払うべき金額には、キャンセル日までに提供されるサービスの支払いまたは発生した費用のみが含まれており、当サービス提供者のキャンセル不可義務が含まれています。これらの支払いは、このような支払いの額や時間がわからないため、上記の契約義務には含まれていない。
132
表外手配
ここ数年、私たちは持っていませんし、現在もありません。アメリカ証券取引委員会の規則と規定によると、私たちは何の表外手配もありません。
重要な会計政策と重大な判断と見積もり
私たちの総合財務諸表はアメリカで認められている会計原則に基づいて作成されています。我々の連結財務諸表および関連開示を作成する際には、総合財務諸表における資産、負債、コストおよび費用に影響を及ぼす報告金額、または資産および負債の開示に影響を及ぼす推定および判断を行う必要がある。我々は歴史的経験,既知の傾向や事件,および当時の状況では合理的と考えられる様々な他の要因に基づいて推定し,これらの要因の結果は資産や負債の帳簿価値を判断する基礎を構成しているが,これらの資産や負債の帳簿価値は他の源からは明らかではないように見える。私たちは持続的な基礎の上で私たちの推定と仮定を評価する。異なる仮定や条件の下で、私たちの実際の結果はこれらの推定とは異なる可能性がある。
我々の主な会計政策は、本年度報告10-K表の他の部分の審査総合財務諸表付記2により詳細に記載されているが、以下の会計政策は、我々の総合財務諸表を作成する際に使用する重要な会計政策であり、重大な見積もりと判断が必要であると考えられる。
研究と開発費
連結財務諸表作成過程の一部として、我々が計上すべき研究·開発費用を見積もる必要がある。このプロセスは、請求書を受信していない場合、または他の方法で実際のコストを通知する場合に、提供されるサービスのレベルおよびそのサービスのために生成される関連コストを推定することに関する。私たちのほとんどのサービスプロバイダは予定のスケジュールに従って、あるいは契約マイルストーンに達した時に私たちに借金の領収書を発行しますが、前金が必要なものもあります。私たちは、当時私たちが知っている事実と状況に基づいて、連結財務諸表において、各貸借対照表の日付までの課税費用を推定します。各期間の終了時に,サービスプロバイダとこれらの推定の正確性を確認し,必要に応じて調整する.計算すべき研究および開発費用の推定例には、以下の者に支払われる費用に関する費用が含まれる
私たちは、私たちが受け取ったサービスと費用の見積もりに基づいて、契約研究および製造に関連する費用および計算項目を記録し、多くの要素を考慮して、私たちの研究、開発、製造活動の進展に対する理解、契約項の下でこれまでの領収書、CRO、CMOおよび他の会社の期間中に発生した請求書が発行されていない任意の実際のコストに関するコミュニケーション、および契約および調達注文に含まれるコストを含む。サービス料を受け取る際に、サービスを提供する期間と各期間の努力度を見積もります。サービス実行の実際の時間や努力の程度が見積もり値と異なる場合、計算すべき費用または前払い費用の金額を調整します。私たちが計算した研究と開発費用の以前の見積もりには何の大きな調整もありません。
133
株に基づく報酬
私たちは従業員、コンサルタント、役員への長期的なインセンティブとして株式インセンティブ計画を維持しています。我々は、従業員および非従業員に付与された公正な価値に基づいて、従業員および非従業員に付与されたすべての株式奨励を計算し、必要なサービス期間(通常は対応する報酬の帰属期間)内の補償費用を確認する。階層的に帰属する株式報酬の付与日公正価値は、必要なサービス期間内に直線的に確認される。私たちは発生時に株式補償奨励に関する没収を確認し、発生時に以前に確認された没収報酬に関連する補償コストを没収します。
我々はブラック·スコアーズオプション定価モデルを用いてサービス条件を持つ株式オプションを推定した。ブラック·スコアーズオプション定価モデルは様々な入力を使用しており、普通株の公正価値、普通株の予想変動率、株式オプションの期待期限、私たちの普通株式オプションの期待期限に近い一定期間の無リスク金利、および私たちの期待配当収益率についていくつかの仮定をした。以下に使用する投入と仮定をまとめる
予想変動率-私たちは会社に特定された歴史と隠れた変動率情報が不足している。そこで、上場同業者のグループの履歴変動率に基づいて予想株価変動率を推定し、我々が取引する株価変動性に関する十分な履歴データを得る前に、引き続きそうする予定である。
予想期間--米国証券取引委員会第107号スタッフ会計公告に記載されている簡略化方法を用いて、株式支払オプション付与の期待寿命を決定するために(“SAB 107”)。
無リスク金利-無リスク金利は、奨励付与時に発効した米国債収益率曲線を参考にして決定され、期間は奨励の予想期限にほぼ等しい。
配当金-期待配当率はゼロです。私たちはまだ普通株について現金配当金を支払っていないので、予測可能な未来にいかなる現金配当金も支払うことは期待できません。
普通株式公正価値の決定
2021年10月8日に初公開される前に、我々の普通株の推定公正価値は、各オプション付与日に取締役会によって決定され、最新に利用可能な第三者普通株式推定値と、他の客観的かつ主観的要因の評価を考慮して、最近の推定日から付与日まで変化した可能性があると考えられる。これらの第三者評価は、米国公認会計士協会会計·評価ガイドラインで概説された指導に基づいて行われている補償として発行された個人持株会社株式証券の推定値それは.我々の普通株式推定値はオプション定価方法やOPMまたは混合方法を用いて用意されており,いずれも市場手法を用いて我々の企業価値を推定している.
これらの推定値に基づく仮定は、固有の不確実性と経営陣判断の応用に関連する管理職の最適な推定を表している。したがって、もし私たちが全く違う仮定や推定を使用すれば、私たちの普通株の公正価値と株式ベースの報酬支出は大きく異なるかもしれない。
初めて公募した後、著者らは普通株のナスダック全世界精選市場でのオファーに基づいて普通株の公正価値を確定した。
134
最近の会計公告
最近発表された会計基準およびこれらの基準が我々の連結財務諸表に及ぼす影響に関する情報は、当社の連結財務諸表における本年度報告Form 10-K第2部第8項の“付記2--重要会計政策概要”を参照されたい。
私たちの起業法を開始します
JumpStart Our Business Startups Act of 2012,またはJOBS Actは,“新興成長型会社”が延長された過渡期間を利用して新たな会計基準や改訂された会計基準を遵守することを可能にしている。“雇用法案”の定義によると、私たちは“新興成長型会社”です。したがって、新興成長型企業は、これらの基準が民間会社に適用されるまで、いくつかの会計基準の採用を延期することができる。私たちはこの延長された移行期間を利用しないことを撤回できないので、他の上場企業で新たなまたは改訂された会計基準を採用する必要がある関連日にこのような基準を採用します。
私たちも“小さな報告会社”であり、これは私たちの非関連会社が保有している株の時価が7億ドル未満であることを意味し、最近終了した会計年度では、私たちの年収は1億ドル未満である。私たちは規模の小さい報告会社に適用されるいくつかの開示要求の免除に依存するかもしれない。具体的には、小さな報告会社として、最近の2つの会計年度の監査財務諸表を当社のForm 10-K年報にのみ示すことを選択し、役員報酬に関する開示義務を削減し、また、新興成長型企業と同様に、年収が1億ドル未満の小さな報告会社であれば、独立公認会計士事務所が発行する財務報告内部統制認証報告を得る必要はないであろう。
135
第七A項。量市場リスクに関する定性的で定量的な開示。
米国証券取引委員会の規則によると、我々は“規模の小さい報告会社”とされているため、本報告書でこの要求された情報を提供する必要はない。
プロジェクト8.財務報告書と補足データ。
項目8に要求される財務資料は本報告F−1ページから始まる。
項目9.変更と撤回会計士と会計と財務開示について合意した。
ない。
第9条。会社制御とプログラムです
情報開示制御と手続きの評価。
吾等は“開示制御及び手続”(定義は1934年証券取引法(改正又は取引法)下の第13 a−15(E)及び15 d−15(E)条規則)を維持し、吾等が取引法に基づいて提出又は提出した報告において開示すべき情報が米国証券取引委員会の規則及び表で指定された期限内に記録、処理、まとめ及び報告することを確保することを目的としている。
開示制御及び手続きは、取引所法案に基づいて提出又は提出された報告書において開示を要求する情報が蓄積され、我々の経営陣に伝達されることを保証するために、合理的な保証を提供するための制御及び手続に限定されるものではないが、我々の最高経営者及び最高経営責任者(場合によっては)を含めて、速やかに開示を要求する決定を下すことができる。
我々の経営陣は、最高経営責任者と財務官の参加の下、2022年12月31日まで、すなわち本年度報告がカバーする期間が終了したときの、我々の開示制御プログラム及びプログラムの有効性を評価した。この評価に基づき、我々の最高経営責任者と最高財務責任者は、2022年12月31日まで、我々の開示制御及び手続きが合理的な保証水準で有効であると結論した。
財務報告書の内部統制に関する経営陣の報告書。
私たちの経営陣は私たちの財務報告書に対する十分な内部統制を確立して維持する責任がある。“取引法”に基づいて公布された規則13 a-15(F)および15 d-15(F)における財務報告の内部統制は、財務報告の信頼性と米国公認会計原則に基づいて外部目的の財務諸表を作成するための合理的な保証を提供するために、我々の主要幹部および主要財務官が設計またはその監督の下で、財務報告に関する信頼性を提供するために、我々の取締役会、管理層および他の人員によって実施されるプログラムと定義される。私たちの財務報告書に対する内部統制は以下の政策と手続きを含む
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。将来的にどのような有効性評価を行うかの予測には,条件の変化により制御が不十分になる可能性がある,あるいは政策やプログラムを遵守する程度が悪化する可能性があるというリスクがある.
私たちの経営陣は、2022年12月31日までの財務報告書の内部統制に対する我々の有効性を評価した。この評価を行う際には,経営陣はトレデビル委員会後援組織委員会(COSO)が2013年の内部統制−総合枠組みで提案した基準を用いた。我々の評価によると、我々の経営陣は、2022年12月31日現在、財務報告に対する内部統制がこれらの基準に基づいて有効であると結論している。
136
財務報告書の内部統制の変化。
2022年12月31日までの財政四半期において、財務報告の内部統制(“取引法”規則13 a-15(F)および15 d-15(F)の定義に基づく)には何の変化もなく、財務報告の内部統制に大きな影響を与えたり、合理的な可能性が大きな影響を与えたりしている。
内部統制の有効性の内在的限界。
私たちの経営陣は、私たちのCEOやCEOを含めて、私たちの開示制御や手続き、または私たちの内部統制がすべてのミスやすべての詐欺を防ぐことができることを期待していません。発想や動作がどんなに良くても、絶対的な保証ではなく、合理的な保証しか提供できず、制御システムの目標が実現されることを確保する制御システム。また,制御システムの設計は,資源制約が存在し,そのコストに対する制御の利点を考慮しなければならないという事実を反映しなければならない.すべての制御システムに固有の限界があるため,いずれの制御評価もわが社内のすべての制御問題や不正事件が検出されたことを絶対に保証することはできない.
公認会計士事務所の認証報告。
本年度報告には,我々の独立公認会計士事務所の財務報告内部統制に関する証明報告は含まれておらず,雇用法案が“新興成長型会社”として免除が確立されているためである
プロジェクト9 B。Oもっと情報があります。
2023年3月16日または発効日に、ファイザー社またはファイザー社と、2022年10月14日に改正され、再署名されたライセンス契約またはA&Rライセンス契約の第1の修正案である修正案1を締結し、A&Rライセンス契約のいくつかの条項を修正し、再確認した。A&Rライセンス協定は、ファイザーと当社が以前に署名した2020年12月8日のライセンス契約を改訂し、再確認し、2021年3月12日から発効し、現在まで改訂されている。修正案1によれば、私たちは、私たちの普通株のナスダック全世界ベスト市場での終値1株2.46ドル以上の営業日に相当する1株価格で500万ドル相当の普通株式数を発行し、発効日から2022年10月6日またはA&R発効日後180日以内のいずれかに発行することに同意します。もしA&R発効日後180日以内に、私たちの普通株の株価が1株2.46ドルを受け取っていない場合、発効日または発行後180日に500万ドル相当の普通株を発行することに同意します。1株当たりの価格は発行前の営業日の終値に等しいので、ファイザーは私たちの所有権権益が19.5%(19.5%)を超えてはならず、株式価値と500万ドルの差額はすべて現金で支払うことに同意します。
2023年3月17日、我々普通株のナスダック世界精選市場における終値は1株2.76ドルであった。第1号改正案の条項によると、ファイザー社に1,811,594株の普通株を発行し、金額は500万ドルに相当する。
第1号改正案により発行された普通株は、改正後の1933年の“証券法”又は“証券法”に基づいて登録されず、証券法第4(A)(2)条に規定する免除登録に基づいて登録される。
プロジェクト9 Cです。検査妨害に関する外国司法管区の開示
適用されません。
137
パー?パーT III
プロジェクト10.取締役、執行役上級管理者と会社が管理する。
本プロジェクトに必要な情報は、我々が米国証券取引委員会に提出した2023年年次総会に関する我々の最終委託書に含まれ、引用により本明細書に組み込まれる。
プロジェクト11.E役員報酬。
本プロジェクトに必要な情報は、我々が米国証券取引委員会に提出した2023年年次総会に関する我々の最終委託書に含まれ、引用により本明細書に組み込まれる。
プロジェクト12.証明書の保証所有権N実益所有者および管理職および関連株主について。
本プロジェクトに必要な情報は、我々が米国証券取引委員会に提出した2023年年次総会に関する我々の最終委託書に含まれ、引用により本明細書に組み込まれる。
第13項.ある関係PS及び関連取引は、取締役が独立している。
本プロジェクトに必要な情報は、我々が米国証券取引委員会に提出した2023年年次総会に関する我々の最終委託書に含まれ、引用により本明細書に組み込まれる。
第14項主要口座NTING費用とサービスです。
本プロジェクトに必要な情報は、我々が米国証券取引委員会に提出した2023年年次総会に関する我々の最終委託書に含まれ、引用により本明細書に組み込まれる。
138
パ.パRT IV
プロジェクト15.展示品、財務諸表別表。
展示品 番号をつける |
|
説明する |
表 |
書類番号. |
展示品 |
提出日 |
同封アーカイブ |
3.1 |
|
PYXIS腫瘍学社の登録証明書の改訂と再署名。 |
10-Q |
001-40881 |
3.1 |
2021年11月15日 |
|
3.2 |
|
PYXIS腫瘍学定款を改訂·再制定した。 |
10-Q |
001-40881 |
3.1 |
2021年11月15日 |
|
4.1 |
|
登録者の証券説明 |
10-K |
001-40881 |
4.1 |
2023年3月22日 |
|
10.1 |
|
投資家権利協定の改正と再署名は2021年3月5日 |
S-1/A |
333-259627 |
10.1 |
2021年10月1日 |
|
10.2+ |
|
合意の形式を達成する |
S-1/A |
333-259627 |
10.2 |
2021年10月4日 |
|
10.3+ |
|
PYXIS腫瘍学会社とLara Sullivan医学博士との雇用協定 |
S-1/A |
333-259627 |
10.3 |
2021年10月4日 |
|
10.4+ |
|
PYXIS腫瘍学社2019年持分インセンティブ計画 |
S-8 |
333-260441 |
4.3 |
2021年10月22日 |
|
10.5+ |
|
PYXIS腫瘍学社2021年持分インセンティブ計画 |
S-8 |
333-260441 |
4.4 |
2021年10月22日 |
|
10.6+ |
|
PYXIS腫瘍学会社員株式購入計画 |
S-8 |
333-260441 |
4.5 |
2021年10月22日 |
|
10.7 |
|
Pyxis Oncology,Inc.がファイザーと締結したライセンス契約は,2020年12月8日である |
S-1 |
333-259627 |
10.7 |
2021年9月17日 |
|
10.8 |
|
Pyxis Oncology,Inc.とファイザー間のライセンス契約に対する第1号修正案は,2021年3月22日とした |
S-1 |
333-259627 |
10.8 |
2021年9月17日 |
|
10.9 |
|
シカゴ大学とPyxis腫瘍学癌免疫治療技術の独占的許可プロトコルは,2020年4月16日である |
S-1 |
333-259627 |
10.9 |
2021年9月17日 |
|
10.10 |
|
Pyxis Oncology,Inc.とLegoChem Biosciences Inc.との間のライセンス契約は,2020年12月1日である |
S-1 |
333-259627 |
10.10 |
2021年9月17日 |
|
10.11 |
|
Pyxis Oncology,Inc.とLegoChem Biosciences Inc.との間のライセンス契約の第1の修正案は,2021年2月25日である |
S-1 |
333-259627 |
10.11 |
2021年9月17日 |
|
10.12 |
|
Pyxis Oncology,Inc.とLegoChem Biosciences,Inc.の間の選択加入,投資,追加価格協定は,2020年12月1日である |
S-1 |
333-259627 |
10.12 |
2021年9月17日 |
|
10.13 |
|
2021年8月2日,Pyxis Oncology,Inc.とLegoChem Biosciences,Inc.の間の選択加入,投資,および追加対価格協定の修正案 |
S-1/A |
333-259627 |
10.13 |
2021年10月1日 |
|
10.14 |
|
PYXIS腫瘍学社,合金治療会社とVoxall治療会社との協力協定は,2021年3月30日である |
S-1/A |
333-259627 |
10.14 |
2021年10月1日 |
|
10.15 |
|
B 9 LS Harrison&Washington LLCとPyxis Oncology,Inc.の間のレンタルは2021年9月29日である。 |
S-1/A |
333-259627 |
10.15 |
2021年10月1日 |
|
10.16+ |
|
PYXIS腫瘍学社とPamela Connealyとの間の雇用合意。 |
S-1/A |
333-259627 |
10.16 |
2021年10月4日 |
|
10.17+ |
|
PYXIS腫瘍学社とJay Feingold医学博士との雇用協定。 |
S-1/A |
333-259627 |
10.17 |
2021年10月4日 |
|
10.18 |
|
登録者とBiosion USA,Inc.が2022年3月28日に署名したライセンス契約。 |
10-Q |
001-40881 |
10.1 |
2022年5月13日 |
|
10.19 |
|
Pyxis Oncology,Inc.とファイザーによるライセンス契約の改訂と再署名は,2022年10月6日である |
10-Q |
001-40881 |
10.1 |
2022年11月1日 |
|
10.20 |
|
Pyxis Oncology,Inc.とファイザー間の書簡プロトコルは,2022年10月14日である |
10-Q |
001-40881 |
10.2 |
2022年11月1日 |
|
139
10.21+ |
|
改正PYXIS腫瘍学社とLara Sullivan医学博士との雇用協定。 |
|
|
|
|
X |
10.22+ |
|
修正されたPYXIS腫瘍学社とPamela Connealyとの雇用協定 |
|
|
|
|
X |
10.23+ |
|
修正されたPyxis Oncology,Inc.とJitu Wadhaneとの雇用契約 |
|
|
|
|
X |
10.24 |
|
Pyxis Oncology,Inc.とファイザーの間で2022年10月14日に改正·再署名されたライセンス契約の2023年3月16日第1号改正案 |
|
|
|
|
X |
10.25 |
|
登録者とBiosion USA,Inc.が2022年3月28日に署名したライセンス契約第1号改正案。 |
|
|
|
|
X |
10.26 |
|
登録者とBiosion USA,Inc.が2022年3月28日に署名したライセンス契約第2号改正案。 |
|
|
|
|
X |
10.27 |
|
登録者とBiosion USA,Inc.が2022年3月28日に署名したライセンス契約修正案3。 |
|
|
|
|
X |
21.1 |
|
付属会社名簿 |
|
|
|
|
X |
23.1 |
|
独立公認会計士事務所安永会計士事務所の同意を得ました。 |
|
|
|
|
X |
24.1 |
|
授権書(本年報の署名ページに表格10-K形式で掲載) |
|
|
|
|
X |
31.1 |
|
2002年のサバンズ·オキシリー法第302節で可決された1934年の証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて発行された首席執行幹事証明書。 |
|
|
|
|
X |
31.2 |
|
2002年サバンズ−オキシリー法第302節で可決された1934年証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて首席財務幹事が認証された。 |
|
|
|
|
X |
32.1* |
|
2002年にサバンズ·オクスリ法案第906条で可決された“米国法典”第18編1350条に規定されている最高経営責任者証明書。 |
|
|
|
|
X |
32.2* |
|
2002年の“サバンズ-オックススリー法案”906節で採択された“米国法典”第18編1350条による首席財務官の認証。 |
|
|
|
|
X |
101.INS |
|
連結されたXBRLインスタンス文書−インスタンス文書は、XBRLタグがイントラネットXBRL文書に埋め込まれているので、相互作用データファイルには表示されない。 |
|
|
|
|
X |
101.書院 |
|
イントラネットXBRL分類拡張アーキテクチャ文書 |
|
|
|
|
X |
101.カール |
|
インラインXBRL分類拡張計算リンクライブラリ文書 |
|
|
|
|
X |
101.def |
|
インラインXBRL分類拡張Linkbase文書を定義する |
|
|
|
|
X |
101.介護会 |
|
XBRL分類拡張ラベルLinkbase文書を連結する |
|
|
|
|
X |
101.Pre |
|
インラインXBRL分類拡張プレゼンテーションLinkbaseドキュメント |
|
|
|
|
X |
104 |
|
表紙インタラクションデータファイル(イントラネットXBRL文書に埋め込む) |
|
|
|
|
X |
*本契約添付ファイル32.1および32.2に提供される証明は、本年度報告と共にForm 10-Kフォーマットで提出されたものとみなされ、証券取引法第18条の目的に基づいて“既存”とみなされるものとみなされないか、またはこの条項の責任を受けず、参照によって証券法または“取引法”に従って提出された任意の文書に組み込まれているものとみなされてはならない。
+管理契約または補償計画を示します。
本展示品に含まれるいくつかの機密情報を保護し、[***]ただし,S-K規則601(B)(10)(Iv)項により省略されている.
プロジェクト16M 10-Kの概要
ない。
140
サイン
改正された1934年証券取引法第13又は15(D)節の要求に基づいて、登録者は、正式に許可された次の署名者がその代表を代表して本報告書に署名することを正式に手配した.
|
|
PYXIS腫瘍学社は |
|
|
|
|
|
日付:2023年3月22日 |
|
差出人: |
/s/Lara Sullivan |
|
|
|
ララ·サリヴァン医学博士 |
|
|
|
社長と最高経営責任者 |
授権依頼書
このような身分で知っているすべての人は、以下の署名の1人当たり構成され、Lara Sullivan,M.D.およびPamela Connealyおよびそれらがそれぞれその人の真および合法的な代理人および代理人であり、その人の代わりに任意およびすべての身分でその人の名前、職位および代理を委任し、本Form 10-K年次報告の任意およびすべての改訂(発効後の改訂を含む)に任意およびすべての身分で署名し、そのすべての証拠物および関連する他の文書とともに証券取引委員会に提出し、上述した代理弁護士および代理人および彼ら一人一人に完全な権力および許可を付与し、そのような代理弁護士および関係処に関連して必要および必要なすべてのことを行い、その人が可能または自ら行うことができるすべての意図および目的に完全に沿って行動し、本明細書でそのようなすべての代理弁護士および代理人、または彼らのいずれか、またはそのような者の1人または複数の代替者を承認および確認し、本条例によってなされたすべてのことを合法的にまたは手配することができる。
本報告書は、改正された1934年の証券取引法の要求に基づき、以下の登録者によって登録者として指定日に署名された。
名前.名前 |
|
タイトル |
|
日取り |
|
|
|
|
|
/s/Lara Sullivan |
|
取締役最高経営責任者総裁 |
|
2023年3月22日 |
ララ·サリヴァン医学博士 |
|
(首席行政主任) |
|
|
|
|
|
|
|
/s/Pamela Connealy |
|
首席財務官兼首席運営官 |
|
2023年3月22日 |
パメラ·コネリー |
|
(首席財務官) |
|
|
|
|
|
|
|
/s/Jitendra Wadhane |
|
首席会計官 |
|
2023年3月22日 |
ギテンドラ·ワドハネ |
|
(首席会計主任) |
|
|
|
|
|
|
|
/s/John Flavin |
|
取締役会議長 |
|
2023年3月22日 |
ジョン·フレヴィン |
|
|
|
|
|
|
|
|
|
/s/Thomas Civik |
|
役員.取締役 |
|
2023年3月22日 |
トーマス·シビック |
|
|
|
|
|
|
|
|
|
/s/ダレン·クライン |
|
役員.取締役 |
|
2023年3月22日 |
ダレン·クライン |
|
|
|
|
|
|
|
|
|
フレダ·ルイス·ホール医学博士 |
|
役員.取締役 |
|
2023年3月22日 |
フレダ·ルイス·ホール医学博士 |
|
|
|
|
|
|
|
|
|
レイチェル·ハンフリー医学博士 |
|
役員.取締役 |
|
2023年3月22日 |
レイチェル·ハンフリー医学博士 |
|
|
|
|
141
PYXIS腫瘍学社は
連結財務諸表索引
|
|
|
ページ |
|
|
独立公認会計士事務所報告 |
F-2 |
|
|
2022年と2021年12月31日までの連結貸借対照表 |
F-3 |
|
|
2022年と2021年12月31日までの総合経営報告書と全面赤字 |
F-4 |
|
|
2022年12月31日と2021年12月31日までの転換可能優先株と株主権益(赤字)連結報告書 |
F-5 |
|
|
2022年12月31日と2021年12月31日までの統合現金フロー表 |
F-6 |
|
|
連結財務諸表付記 |
F-7 |
F-1
独立登録所の報告公認会計士事務所
Pyxis Oncology,Inc.の株主と取締役会へ
財務諸表のいくつかの見方
PYXIS腫瘍学社(当社)の2022年12月31日と2021年12月31日までの総合貸借対照表,同年度までの関連総合経営表と全面赤字,転換可能優先株と株主権益(赤字)とキャッシュフロー,および関連付記(総称して“総合財務諸表”と呼ぶ)を監査した。総合財務諸表は、すべての重要な面で、2022年12月31日と2021年12月31日までの会社の財務状況と、2022年12月31日と2021年までの経営結果と現金流量を公平に反映しており、米国公認会計原則に適合していると考えられる。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査することを求められておらず、私たちも招聘されて監査を行っていません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性について意見を述べるためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
/s/
2021年以来、私たちは会社の監査役を務めてきた。
2023年3月22日
F-2
PYXIS腫瘍学社は
統合された貸借対照表
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
|
|
|
|
|
|
||
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
制限現金 |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
流動資産総額 |
|
|
|
|
|
|
||
財産と設備、純額 |
|
|
|
|
|
|
||
経営的リース使用権資産 |
|
|
|
|
|
|
||
他の非流動資産 |
|
|
— |
|
|
|
|
|
総資産 |
|
$ |
|
|
$ |
|
||
負債と株主権益 |
|
|
|
|
|
|
||
流動負債: |
|
|
|
|
|
|
||
売掛金 |
|
$ |
|
|
$ |
|
||
費用とその他の流動負債を計算しなければならない |
|
|
|
|
|
|
||
賃貸負債を経営し、今期の部分 |
|
|
— |
|
|
|
|
|
流動負債総額 |
|
|
|
|
|
|
||
賃貸負債を経営し,当期分を差し引く |
|
|
|
|
|
— |
|
|
総負債 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
株主権益: |
|
|
|
|
|
|
||
優先株、額面$ |
|
|
|
|
|
|
||
普通株、$ |
|
|
|
|
|
|
||
追加実収資本 |
|
|
|
|
|
|
||
赤字を累計する |
|
|
( |
) |
|
|
( |
) |
株主権益総額 |
|
|
|
|
|
|
||
総負債と株主権益 |
|
$ |
|
|
$ |
|
||
付記はこれらの連結財務諸表の構成要素である。
F-3
PYXIS腫瘍学社は
連結業務報告書減価償却と包括損失
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
|
十二月三十一日までの年度 |
|
|
|||||
|
|
2022 |
|
|
2021 |
|
|
||
運営費用: |
|
|
|
|
|
|
|
||
研究開発 |
|
$ |
|
|
$ |
|
|
||
一般と行政 |
|
|
|
|
|
|
|
||
総運営費 |
|
|
|
|
|
|
|
||
運営損失 |
|
|
( |
) |
|
|
( |
) |
|
その他の収入(支出): |
|
|
|
|
|
|
|
||
利子収入 |
|
|
|
|
|
|
|
||
関連側手数料収入 |
|
|
— |
|
|
|
|
|
|
派生負債の公正価値変動 |
|
|
— |
|
|
|
( |
) |
|
その他収入合計 |
|
|
|
|
|
( |
) |
|
|
合営企業権益法投資損失 |
|
|
|
|
|
( |
) |
|
|
純損失と総合損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
普通株1株当たり純損失--基本損失と赤字 |
|
$ |
( |
) |
|
$ |
( |
) |
|
発行済み普通株の加重平均株式−基本と希釈− |
|
|
|
|
|
|
|
||
付記はこれらの連結財務諸表の構成要素である。
F-4
PYXIS ONCOLY社
変換可能Pre統合レポート株と株主権益を繰延する
(単位:千、共有データを除く)
|
|
転換可能優先株 |
|
|
普通株 |
|
|
その他の内容 |
|
|
積算 |
|
|
合計する |
|
|||||||||||||
|
|
株 |
|
|
金額 |
|
|
株 |
|
|
金額 |
|
|
資本 |
|
|
赤字.赤字 |
|
|
権益(赤字) |
|
|||||||
2020年12月31日残高 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|||||
ファイザーにBシリーズ転換優先株を発行する。(付記3参照) |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
||
Bシリーズ転換優先株を発行し、発行コストを差し引く$ |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
||
普通株の初公開発行により、引受手数料と発行コストを差し引いて$ |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
AシリーズとBシリーズは優先株を普通株に変換できます |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
行使した株式オプション |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
制限された普通株の帰属 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
株に基づく報酬 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2021年12月31日の残高 |
|
|
— |
|
|
$ |
— |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||
ファイザーに普通株式を発行する。(付記3参照) |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
行使した株式オプション |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
制限された普通株の帰属 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
株に基づく報酬 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2022年12月31日の残高 |
|
|
— |
|
|
$ |
— |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||
付記はこれらの連結財務諸表の構成要素である。
F-5
PYXIS腫瘍学社は
合併状態キャッシュフロープロジェクト
(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
経営活動 |
|
|
|
|
|
|
||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
純損失と経営活動で使用される現金純額の調整: |
|
|
|
|
|
|
||
減価償却および償却 |
|
|
|
|
|
|
||
株に基づく報酬 |
|
|
|
|
|
|
||
非現金研究開発費 |
|
|
|
|
|
|
||
非現金レンタル費用 |
|
|
|
|
|
|
||
合営企業権益法投資の非現金損失 |
|
|
— |
|
|
|
|
|
派生負債の公正価値変動 |
|
|
— |
|
|
|
|
|
経営性資産と負債変動状況: |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
( |
) |
|
|
( |
) |
売掛金 |
|
|
( |
) |
|
|
|
|
費用とその他の流動負債を計算しなければならない |
|
|
|
|
|
|
||
リース負債を経営する |
|
|
|
|
|
( |
) |
|
経営活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
投資活動 |
|
|
|
|
|
|
||
財産と設備を購入する |
|
|
( |
) |
|
|
( |
) |
合弁企業への投資 |
|
|
— |
|
|
|
( |
) |
投資活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
融資活動 |
|
|
|
|
|
|
||
Bシリーズを発行して優先株を転換して得られた金は,発行コストを差し引くことができる |
|
|
— |
|
|
|
|
|
普通株を初公開して得られた収益は,発行コストを差し引く |
|
|
— |
|
|
|
|
|
株式オプションを行使して得られる収益 |
|
|
|
|
|
|
||
融資活動が提供する現金純額 |
|
|
|
|
|
|
||
現金、現金等価物、および制限的現金純増加 |
|
|
( |
) |
|
|
|
|
年初の現金、現金等価物、制限現金 |
|
|
|
|
|
|
||
期末現金、現金等価物、および制限現金 |
|
$ |
|
|
$ |
|
||
キャッシュフロー情報の追加: |
|
|
|
|
|
|
||
受け取った利息現金 |
|
$ |
|
|
$ |
— |
|
|
税金の現金を納める |
|
$ |
|
|
$ |
— |
|
|
非現金投資と融資活動: |
|
|
|
|
|
|
||
売掛金及び売掛金のうち財産及び設備 |
|
$ |
|
|
$ |
|
||
経営リース賃貸負債の経営と引き換えに使用権資産 |
|
$ |
|
|
$ |
— |
|
|
現金、現金等価物、および制限現金の入金: |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
制限現金 |
|
|
|
|
|
|
||
レポートに表示されている現金、現金等価物、および限定的な現金総額 |
|
$ |
|
|
$ |
|
||
付記はこれらの連結財務諸表の構成要素である。
F-6
PYXIS腫瘍学社は
合併の備考D財務諸表
1.説明商務部
業務的性質
PYXIS腫瘍学社はデラウェア州の会社であり,2018年6月に設立され,2019年7月に運営を開始した。同社は臨床段階の会社で、治療困難な癌を打ち破ることに集中している。同社は次世代療法を効率的に製造しており,これらの療法は単一療法と併用療法の潜在力を有している。同社の候補治療薬は腫瘍細胞を直接殺し、癌による制御できない増殖と免疫逃避の潜在的な病理を解決することを目的としている。同社の抗体−薬物結合体(“ADC”)と免疫腫瘍学(“IO”)は新たな戦略と新興の戦略を採用し,現在の治療基準に抵抗力を有する様々な固形腫瘍を狙う計画である。
その会社は所有している
2.重要な会計政策の列報根拠と概要
陳述の基礎
同社の財政年度は12月31日に終了し、前3四半期はそれぞれ3月31日、6月30日、9月30日に終了する。添付されている総合財務諸表は米国公認会計原則(“GAAP”)に従って作成されている。本付記内の適用指針に対するいかなる言及も財務会計基準委員会(“FASB”)の会計基準編纂(“ASC”)及び会計基準更新(“ASU”)に掲載されている権威公認会計原則を指す。総合財務諸表には当社とその完全子会社が含まれています。合併後、すべての会社間の残高と取引はすでに売却された。
当社は2012年のJumpStart Our Business Startups Act(“JOBS法案”)の定義に基づく“新興成長型会社”です。雇用法案第107条(B)によると、新興成長型企業は、延長された過渡期を利用して新たな又は改正された会計基準を遵守することができる。したがって、新興成長型企業は、これらの基準が民間会社に適用されるまで、いくつかの会計基準の採用を延期することができる。当社はこの延長された移行期間を利用しないことを撤回できないため、当社は他の上場企業で新たなまたは改訂された会計基準の関連日を採用する必要があるなどの基準を採用する。
流動性
2022年12月31日までの同社の累計損失は$
これまで、同社はいかなる収入も発生しておらず、開発を成功させ、規制機関の現在または将来の候補製品の承認を得るまで、いかなる収入も発生しないと予想されている。同社は,会社が研究·開発計画を拡大し,その候補製品を開発し続けるにつれて,その運営損失と負のキャッシュフローが予見可能な未来に続くと予想している。
同社は現在、既存の現金と現金等価物を#ドルと予想している
同社は引き続き公共またはプライベートエクイティ、転換可能または債務融資またはその他の出所を通じて、その運営損失と資本融資需要に資金を提供する計画だ。会社が十分な追加資金を得られない場合、会社は支出を削減し、サプライヤーとの支払い期限を延長し、可能な限り資産を清算したり、計画の計画を一時停止または削減したりすることを余儀なくされる可能性がある。これらの行動のいずれも、会社の業務、経営結果、将来の見通しに実質的な損害を与える可能性がある。
F-7
予算の使用
公認会計原則に従って連結財務諸表を作成することは、管理層に資産、負債、費用及び関連開示報告金額に影響を与える推定と仮定を行うことを要求する。当社は、資産、負債、株式に基づく補償、経営賃貸、物件や設備の使用寿命評価、繰延税金資産と負債推定値、合弁企業の投資減価および研究·開発コストに関する推定と仮定を定期的に評価している。当社は過去の経験とその当時の状況に基づいて部下が合理的であると考えている様々な他の要素を推定·仮定しているが、このような要因の結果は資産や負債の帳簿価値を判断する基礎を構成しており、当該などの資産や負債の帳簿価値は他の出所から容易に見られるわけではない。
実際の結果はこれらの見積もり数とは異なる可能性があり,経営陣の推定数は将来的に変化する可能性がある。
リスクと不確実性
同社は、製品の商業化、規制承認、有効成分の主要サプライヤーおよび第三者サービスプロバイダ(例えば、契約研究および製造機関)への依存、知的財産権保護、および任意の許可、協力または供給協定に従ってマイルストーン、特許権使用料または他の支払いを支払う能力に関する不確実性を含むが、バイオ製薬業界の初期会社によく見られるリスクに直面している。
信用リスクが集中する
会社に高度な集中信用リスクを受けさせる金融商品には主に現金と現金等価物が含まれている。同社の現金と現金等価物は認可された金融機関に保管されており、同社は同社などの口座に何の損失も出ていない。同社は現金を銀行預金口座に入金し、連邦保険の限度額を超える場合がある。同社の現金等価物には主に認可された金融機関が保有する短期通貨市場基金が含まれている。同社は、現金と現金等価物には何の重大なリスクも存在しないと考えている。
現金と現金等価物
当社はすべての原始満期日が90日以下の短期·高流動性投資を現金等価物と見なしている。現金等価物は、2022年12月31日と2021年12月31日まで、主に通貨市場基金で構成されている。
公正価値計量
公認会計原則に基づき、ある資産と負債は公正価値に基づいて帳簿に記入される。公正価値は、計量日市場参加者間の秩序ある取引において、資産または負債が原則または最も有利な市場で負債を移動させるために課金または支払いされる交換価格として定義される。公正価値を計量するための推定技術は,観察可能な投入を最大限に利用し,観察不可能な投入を最大限に減少させなければならない。公正価値に基づいて勘定された金融資産と負債は、公正価値レベルの次の3つのレベルのうちの1つで分類され、開示されなければならず、最初の2つのレベルは可視とされ、最後のレベルは見えないとみなされる
水平 1−計量日に取得可能な同じ非制限資産または負債のアクティブ市場の未調整オファー;
水平 2−アクティブな市場のオファーとはみなされない、またはすべての重要な投入された金融商品推定値を直接または間接的に観察することができる;および
水平 3−価格または推定値は、公正な価値計量に重大な意味を有し、観察されない投入を必要とする。
ある程度、推定値は市場で観察または観察できないモデルや投入に基づいており、公正価値の決定にはより多くの判断が必要である。したがって,第3レベルに分類されたツールについては,当社が公正価値を決定する際に行使される判断度が最も大きい.
F-8
財産と設備、純額
財産と設備はコストから減価償却と償却を差し引いて入金される。減価償却と償却費用は、直線法で関連資産の推定耐用年数内で以下のように確認される
|
|
役に立つと思う |
実験室装置 |
|
|
家具と事務設備 |
|
|
賃借権改善 |
|
|
建設中の工事 |
|
適用されない |
減価償却償却費用には研究開発費と一般管理費用が含まれています。重大な増加とアップグレードは資本化されている;メンテナンスと修理はそれぞれの資産の使用寿命を改善または延長することはなく、発生時に費用を計上する。廃棄又は売却時には,処分資産のコスト及び関連する減価償却及び償却はそれぞれの口座から差し引かれ,それによって生じる収益又は損失はいずれも運営収益(損失)に計上される。
長期資産減価準備
当社は少なくとも毎年長期資産(物件および設備および経営賃貸使用権資産を含む)の減値を評価し、イベントや状況変化が資産の帳簿価値が回収できない可能性があることを示した場合に減値を評価する。当社がいつ減値審査を行うかを決定する際に考慮する要素は、業務パフォーマンスが予想に関連する重大な不良、業界或いは経済傾向の重大なマイナス影響、及び資産用途の重大な変化或いは計画変更を含む。そして、管理層は、主に、将来の未割引キャッシュフローが資産回収をサポートするのに十分であるかどうかに基づいて、残りの使用年数が適切であるかどうか、または長期資産を減値したかどうかを決定する。これらの資産の回収可能能力は、資産の帳簿価値と資産予想による将来の未割引キャッシュフローとを比較することで測定される。この資産が減値とみなされる場合、いずれの減値金額も、減値資産の帳簿価値と公正価値との差額で計量される。会社は認識しています
賃貸借証書
運営中ですリース使用権(“ROU”)資産代表会社がリース期間内に対象資産を使用する権利は、リース負債代表会社がリースにより発生したリース金を支払う義務を有する。レンタルROU資産と賃貸負債の初歩的な確認と計量を経営するのは、開始日の予想レンタル期間内の未来の固定賃貸支払いの現在値に基づいて、レンタル資産に適用される当社の逓増借入金利計算であり、隠れた金利が簡単に決定できない限りである。当社は借入期間を賃貸借契約の取消不可期限と決定し、当社がその選択権を行使することを合理的に確定した場合には、賃貸借契約の延長または終了の選択権を含むことができる。レンタルROU資産を経営することは、レンタル開始日または前に生成された任意の初期直接コストおよび任意のレンタル支払いをさらに含み、受信されたレンタルインセンティブを減算する。経営リースROU資産は、その後、レンタル期間全体にわたってリース負債の帳簿価値に初期直接コストを加え、任意の前払い(未払い)賃貸支払いを加え、受信したリースインセンティブの未償却残高を減算して計量する。12ヶ月以下の賃貸契約は、総合貸借対照表では確認されません。最低レンタル料金のレンタル料金はレンタル期間内に直線的に確認します。公共エリアコストや他の運営コストのようなレンタルコストを変動させ、発生時に料金を計上する。当社はリースと非レンタル部分をそのすべての施設レンタルの単一レンタル部分として会計処理を行っている。その会社は所有している
合弁企業への投資
当社の合弁企業への投資には権益会計方法で入金します。会社が被投資先に重大な影響を与えるが権益をコントロールしない場合、会社は投資に対して権益会計方法を採用する。権益法投資への影響程度の判断は所有権利益、取締役会代表、意思決定参加と重大な会社間取引などの重要な要素を考慮することを含む。権益法の下で、投資は最初にコストで入金され、配当金、分配された収益と未分配の収益と損失及び追加投資によって調整される。合弁企業の累積損失に占める会社のシェアが会社の投資残高を超えると、残高は比例して発生した収益が損失を超えるまでゼロ値で報告されるイベントや状況の変化が投資の帳簿価値が回収できない可能性があることを示した場合、当社は投資の減値を評価します。
F-9
事件があったり
当社は時々正常な業務活動による様々なトラブルやクレームの側になる可能性があります。当社は、不利な結果が損失または合理的な可能な損失につながるかどうかを決定するために、それによって生じる訴訟を含めて紛争やクレームを評価し続けており、これらの損失は推定可能である。当社では、負債が発生した可能性のある最初の日にすべてまたは有事を計上し、その負債の金額を合理的に見積もることができると考えています。可能な損失の推定が1つの範囲であり、その範囲内の任意の金額が他の範囲よりも可能である場合、当社は、その範囲内の最小値を言及すべきである。当社が合理的な可能性のある損失が存在すると考えている場合には、当社が開示又は事項のある事実及び状況を、可能であれば推定可能な範囲を含む。
研究と開発費
当社は発生した研究と開発費用を負担します。当社の研究開発費には、主に無形資産の定義に適合しない知的財産権の取得許可料と研究開発活動を行うことによるコストが含まれており、賃金、株式給与、福祉、施設コスト、減価償却、当社を代表して研究開発活動(製造を含む)を行う第三者の外部コストなど、人員に関する費用が含まれている。同社は,契約に基づいて手配された条項に基づいて受けるサービスやかかる努力の評価に基づいて,第三者による開発活動に関する費用を計上すべきである。その中のいくつかの契約下での支払いは臨床前および/または臨床試験マイルストーンに依存する。場合によっては、会社のサプライヤーに支払われるお金は、提供されたサービスレベルを超え、前払い費用をもたらす可能性がある。サービス料を計算する際には、当社はサービスを提供する時間帯と時間ごとにかかる努力度を見積もる。実際にサービスを実行する時間または努力の程度が推定値と異なる場合、会社はそれに応じて計算または前払い費用を調整する。
株に基づく報酬
会社は従業員、コンサルタント、取締役への長期的なインセンティブとして株式インセンティブ計画を維持している。当社は従業員と非従業員に付与された公正価値をもとに、従業員と非従業員に付与されたすべての株式奨励に対して会計計算を行い、必要なサービス期間内に当該等の奨励の補償費用を確認し、このサービス期間は通常相応の奨励の帰属期間である。階層的に帰属する株式報酬の付与日公正価値は、必要なサービス期間内に直線的に確認される。同社は株式補償奨励に関する没収が発生した場合に確認し、発生中に没収補償に関連する任意の以前に確認された補償コストを打ち消した。当社が経営報告書において株式による補償費用を分類する方式は,受賞者の賃金コストを分類する方式や受賞者のサービス支払いを分類する方式と同様である。
同社はブラック·スコアーズオプション定価モデルを用いて、サービス条件に応じて株式オプションを推定している。ブラック·スコアーズオプション定価モデルによると、会社はいくつかの仮定を使用して、報酬の期待寿命、対象株式の変動性、無リスク金利、期待配当率、および会社普通株の公正価値を含む株式オプションの公正価値を決定する。当社は十分な歴史的オプション行使データが不足しており、合理的な基礎を提供して期待期限を見積もることができないため、当社は米国証券取引委員会第107号従業員会計公告で紹介された簡略化方法を採用している株式支払オプション付与の期待寿命を決定するために(“SAB 107”)。会社は会社の歴史と隠れた変動率情報を特定するのに十分な不足がある。そのため、当社は一連の上場同業者会社の歴史変動率に基づいて予想される株式変動率を推定した。無リスク金利は、米財務省が発行した証券支払いに基づく金利であり、期限は株式奨励の期待寿命に近い。同社は支払ったことがなく、普通株に現金配当金を支払うことも期待されていないため、期待配当収益率はゼロとされている。
会社普通株の初公開(“IPO”)が完了する前に、Black-Scholesオプション定価モデルの重要な投入の一つである会社普通株の公正価値は、経営陣の意見に基づいて取締役会によって決定され、いくつかの仮定に基づいて決定され、これらの仮定は、イベントの確率重み付け、業務と市場状況、変動性、清算時間、無リスク金利、および市場流動性の欠如の割引仮説を含む。同社は409 A推定値を用いて普通株の公正価値を決定した。409 aの推定値は、米国公認会計士協会と一致する方法、方法、および仮定を用いて行われた補償として発行された個人持株会社株式証券の会計と評価ガイドラインそれは.2021年10月8日のIPO完了後、当社はその上場普通株の公正価値を使用して、付与日の公正価値を決定する。
F-10
所得税
当社はFASB ASC 740に従って所得税を計算している所得税(米国会計基準第740条)。貸借対照法を用いて所得税の計算が求められている。この方法によれば、繰延税金資産及び負債は、総合財務諸表中の金額と資産及び負債の課税基礎との差額に基づいて決定され、この差額は、予想差額が打ち消される年度の現行税率を採用する。税率変動が繰延税項資産と負債に及ぼす影響は、連結経営報告書における所得税(福祉)費用と公布日を含む期間の全面赤字で確認された。
当社が繰延税金資産を確認する程度は、これらの資産がより顕在化する可能性があると考えられる程度に依存しています。この決定を下す際には、当社は、既存の課税の一時的な差異の将来の輸出、将来の課税収入の予想、税務計画策、および最近の経営の結果を含む、得られるすべてのプラスおよび負の証拠を考慮する。当社が将来的に繰延税金資産がその記録純額を超えることができると判断した場合、当社は繰延税金資産の推定値を調整する予定で、所得税の支出を減らすことになります。確認や計測の変化は変化が発生したと判断した期間に反映される.
不確定な税収頭寸が存在する場合、当社は税収頭寸の税収割引を確認し、より実現可能にする。税収優遇がより実現可能かどうかの決定は,税収状況に基づく技術的利点および既存の事実や状況への考慮である。
1株当たり純損失
普通株株主が1株当たりの基本純収入(損失)を占めるべき計算方法は、普通株株主が純収益(損失)を当期発行普通株で割った加重平均を占めるべきである。普通株株主の1株当たり償却純収益(損失)の計算方法は、普通株株主の希薄純収益(損失)を当期に発行された普通株の償却加重平均で割ったものであり、潜在的な希釈性普通株を含む。
会社が純損失を報告している間、すべての普通株等価物は反償却、すなわち1株当たりの普通株の基本純損失と希釈後の普通株当たりの純損失は等しいとみなされている。これらの証券は会社の純損失による1株当たり純損失に対して逆希釈作用があるため、潜在的な希釈性普通株はすべての届出期間の1株当たり純損失計算から除外されている。基本と希釈後の1株当たり純損失データについては,発行済み普通株総加重平均を計算するための台帳項目はなかった。
最近発表された会計公告
2016年6月、FASBはASU 2016-13を発表した金融商品·信用損失(主題326):金融商品信用損失の測定“ASU 2016-13”では、余剰コストで保有されている金融資産の予想される信用損失の計測および確認が求められている。ASU 2016-13は、既存の発生した損失低減モデルを期待損失モデルで置き換え、このモデルは前向き情報を使用して信用損失推定を計算する必要がある。また、非一時的減価の概念を解消し、証券償却コストベースの減少として記録するのではなく、債務証券を売却できる信用損失を信用損失準備によって記録することを要求している。“新興成長型企業”については、ASU 2016−13年度は、2022年12月15日以降の事業年度およびこれらの年度内の移行期間で有効である。当社は現在、新指針を評価しており、当社の総合財務諸表や関連開示に実質的な影響を与えないと予想されています。
2020年8月にFASBはASU 2020-06を発表しました債務--転換可能な債務および他のオプション(主題470-20)と派生ツールおよびヘッジ--エンティティ自身の権益の契約(主題815-40)(“ASU 2020-06”)。ASU 2020-06は、1株当たりの収益を計算するガイドラインを改訂し、すべての変換可能なツールに対してIF変換方法を使用し、実体が現金または他の資産で決済可能なツールの株式決済推定を覆す能力を廃止することを要求している。ASU 2020-06は、“新興成長型会社”として、2023年12月15日以降の会計年度に当社を発効させ、早期採用を許可する。当社は現在、新指針を評価しており、当社の総合財務諸表や関連開示に実質的な影響を与えないと予想されています。
財務会計基準委員会、米国公認会計士協会、および米国証券取引委員会が最近発表した権威ある指導意見(会計基準委員会の技術修正を含む)は、会社が監査した総合財務諸表および関連開示に実質的な影響を与えないか、または予想されない。
F-11
3.ライセンス契約
“シカゴ大学協定”
2020年4月、会社はシカゴ大学(“大学”)とライセンス契約(“大学ライセンス契約”)と賛助研究協定を締結した。ライセンス条項によれば、同社は、許可された特許の有効な権利要件によってカバーされる製品の開発および商業化、許可技術および材料の導入または使用、または特定の特定の生物学的標的の活性を評価、調節または利用する既知の世界的権利を有する。大学から許可証を取得する代償の一部として,会社は大学に発行した
プスア大学ライセンス契約によると、会社は潜在的な開発とビジネスマイルストーン、最高で$を支払う義務があります
会社は大学許可協定に基づいて2022年12月31日までのマイルストーンと特許権使用料事件を評価した2021年には
ファイザー。同意
2020年12月、同社はファイザー社(以下、“ファイザー社”)とライセンス契約(改訂された“ファイザーライセンス契約”)を締結し、PYX-201およびPYX-203、候補ADC製品を含む候補ADC製品のグローバル開発および商業化権利を付与した。ファイザーが所有または制御するいくつかの特許については、同社の権利は独占的であり、これらの特許は、許可されたADCをカバーする。ファイザーはまた、ファイザーADC技術(“FACT”)プラットフォームを用いた非独占的な許可を同社に付与した。最初の許可標的はCD 123およびExtra Bドメイン(フィブロネクチンのEDB)を含み、同社は、第三者またはファイザーADC開発計画テーマではない他の許可標的を増加させるために、その許容範囲を拡大することを選択することができる。ファイザー許可協定は2021年3月に発効し,同社は合計で支払いを行った
2022年10月6日、当社はファイザーライセンス協定を改訂して再記載したライセンス契約(“A&Rライセンス契約”)を締結した。A&Rライセンスプロトコルによれば、ファイザーは、PYX-201およびPYX-203、およびADC候補製品を含むいくつかの許可ターゲットのためのADC候補製品を開発および商業化するために、ファイザーFACTプラットフォーム技術の下で同社にグローバル独占権を付与する。他のADC目標は象徴的な前払いとマイルストーンの許可を得るかもしれない。さらに、同社は、EDBおよびCD 123に関する1つまたは複数の国または地域でEDBおよびCD 123に関する1つまたは複数の国または地域でのADCおよび認可製品の購入または開発、製造、および商業化のための第1の交渉権をファイザーに付与する。A&Rライセンス契約の条項によると、同社は$の支払いに同意します
同社には開発、規制、ビジネスマイルストーンを含む将来の支払い義務があり、総額は#ドルを超えない
同社は、A&Rライセンス契約下の2022年と2021年12月31日までのマイルストーンと特許使用料事件を評価し、決定した
F-12
レゴ化学生物科学社の合意
当社は2020年12月にレゴ化学生物科学有限公司(“レゴ化学”)とライセンス契約(“レゴ化学ライセンス契約”)および選択加入、投資および追加対価協定(“選択加入合意”)を締結した。レゴ化学ライセンスプロトコルによれば、同社は、LCB 67、DLK-1に対するADC候補製品、およびライセンス化合物を含む製品の世界(韓国以外)の開発および商業化権利の許可を得た。その会社は$を支払った
同社は開発、規制、ビジネスマイルストーン、特許製品の純販売に対して異なるレートで印税を徴収することを含む将来の支払いまたは支払いを義務化している。2022年12月31日までの年度内に、同社は毒性研究データの回顧と分析に基づいて、LCB 67の継続開発を停止し、抗DLK 1 ADCの臨床応用と商業将来性を期待した。LCB 67の臨床開発が中止されたため
また、選択加入プロトコルの一部として、レゴ化学は$を支払う権利がある
Biosion USA,Inc.と締結されたライセンスプロトコル.
当社は2022年3月28日にBiosion USA,Inc.(“Biosion”)とライセンス契約(“Biosionライセンスプロトコル”)を締結し,これにより当社は世界(大中国を除く)(内地中国,香港,マカオおよび台湾を除く)のBSI−060 T,Siglec−15標的抗体,IO候補製品(現在PYX−106と呼ぶ)および許可化合物を含む製品の開発,製造および商品化権利を独占的に許可した。生物学的許可協定の条項によれば、各方向の他方は、任意の二重特異性または多重特異性抗体、一方またはその付属会社によって制御される、予期される作用機序として阻害、調節、または結合したSiglec-15の抗体-薬物結合体を開発、製造および商業化するために、他方の領土(Biosionの大中国、およびPYXISの世界の他の地域)で独占的に許可されるように優先的に契約権を付与する。
Biosionライセンス契約によると、会社は許可料$を前払いします
同社は2022年12月31日までのBiosionに関するマイルストーンと特許使用料事件を評価し、決定した
4.合弁経営
2021年3月、会社は合金治療会社(“合金”)とVoxall治療有限責任会社(“Voxall”)と最終取引合意に達し、Voxallに資金を提供し、Voxallを運営し、会社の技術と合金のATX-GXを利用する合金と協力して設立した合弁会社である プラットフォームや抗体発見サービスですVoxallは会社と合金を授与し
当社のVoxallへの投資は権益会計方法で入金されています。Voxallは設立以来損失が発生しており,Voxall損失における当社のシェア合計は $
F-13
Voxallへの投資とVoxallが発行した本チケットの合計は $
5.公正価値計測
次の表は公正な価値で経常的に入金された金融商品である2022年12月31日および2021年12月31日には、FASB ASC 820階層構造(千単位):
|
|
公正価値に応じて計量する |
|
|||||||||||||
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
|
合計する |
|
||||
資産 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
公正価値に応じて計量する |
|
|||||||||||||
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
|
合計する |
|
||||
資産 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
同社の現金等価物とは、市場オファーを活発にしている短期米国債市場通貨基金における預金であり、1級資産に分類されている。いくつありますか
会社は派生負債#ドルを記録した
いくつありますか
6.前払い料金およびその他の流動資産
前払い料金および他の流動資産には、以下のものが含まれている(千計)
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
|
|
|
|
|
|
|
||
研究開発 |
|
$ |
|
|
$ |
|
||
保険 |
|
|
|
|
|
|
||
受取利息を計算する |
|
|
|
|
|
— |
|
|
他にも |
|
|
|
|
|
|
||
前払い費用とその他の流動資産総額 |
|
$ |
|
|
$ |
|
||
F-14
7.財産と設備、純額
財産と設備の純額は以下の部分からなる(千計)
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
実験室装置 |
|
$ |
|
|
$ |
|
||
家具と事務設備 |
|
|
|
|
|
|
||
賃借権改善 |
|
|
|
|
|
|
||
建設中の工事 |
|
|
|
|
|
— |
|
|
|
|
|
|
|
|
|
||
減算:減価償却累計と償却 |
|
|
( |
) |
|
|
( |
) |
財産と設備の合計 |
|
$ |
|
|
$ |
|
||
2022年12月31日までと2021年12月31日までの年度の減価償却·償却費用は $
8.課税費用およびその他の流動負債
計算すべき費用および他の流動負債には、以下の項目が含まれる(千で計算)
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
研究開発費 |
|
$ |
|
|
$ |
|
||
従業員補償と福祉 |
|
|
|
|
|
|
||
弁護士費と弁護士費 |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
費用とその他の流動負債総額を計算しなければならない |
|
$ |
|
|
$ |
|
||
F-15
9.レンタル契約を経営する
2021年9月、会社はマサチューセッツ州ボストンのオフィスと実験室スペースのために、2022年4月1日、すなわち会社がその物件を制御する日付を契約した経営賃貸契約を締結した。レンタル支払いは2022年12月21日から、2020年まで継続されます
レンタル開始日には、レンタルROU資産を$とします
レンタル料金の構成は以下の通り(千で計算)
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
レンタル料 |
|
|
|
|
|
|
||
リースコストを経営する |
|
$ |
|
|
$ |
|
||
短期賃貸コスト |
|
|
|
|
|
|
||
可変リースコスト |
|
|
|
|
|
|
||
総賃貸コスト |
|
$ |
|
|
$ |
|
||
その他の情報 |
|
|
|
|
|
|
||
経営性リース新経営権と引き換えに使用権資産 |
|
$ |
|
|
$ |
— |
|
|
賃貸負債に含まれる金額を計量するために支払う現金、 |
|
|
— |
|
|
$ |
|
|
加重平均残存賃貸年限(年) |
|
|
|
|
|
|
||
加重平均割引率 |
|
|
% |
|
|
% |
||
可変レンタルコストは主に公共地域コストとその他のビジネスコストに関連します当社が賃貸物件の当該等コストに占める割合に基づいて決定されます。リース総コストは営業費用として会社の総合経営報告書と総合損失に計上される。
賃貸負債満期日2022年12月31日、状況は以下の通り(単位:千):
12月31日までの年度 |
|
賃貸借契約を経営する |
|
|
2023 |
|
$ |
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 |
|
|
|
|
その後… |
|
|
|
|
未割引払い合計 |
|
|
|
|
差し引く:テナント改善手当 |
|
|
( |
) |
減算:現在値調整 |
|
|
( |
) |
将来支払いの現在価値 |
|
|
|
|
差し引く:経営リース負債の当期分 |
|
|
— |
|
賃貸負債を経営し,当期分を差し引く |
|
$ |
|
|
F-16
10.転換可能優先株式
Aシリーズ転換優先株
2019年6月、当社は複数の投資家と証券購入協定(改正“Aシリーズ合意”)を締結し、Aシリーズ転換可能優先株(“Aシリーズ”)の株式を$で売却する
Bシリーズ転換可能優先株
二零二一年三月五日、当社はいくつかの投資家と証券購入協定(“Bシリーズ合意”)を締結し、Bシリーズ転換可能優先株(“Bシリーズ”)株式を$で売却した
11. 株主権益
優先株
初公募の終了について、当社は改訂された会社登録証明書とライセンスを提出しました
普通株
初公募株
2021年10月8日、当社は初公募株を完成し、発行し、販売します
同社は最も多くの発行を許可されている
普通株式保有者の投票権、配当、清算権は優先株保有者の権利、権力と優先権に支配され、その制約を受ける。
投票する.-普通株式流通株のすべての所有者に権利がある
保留株—
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
未償還株式オプション |
|
|
|
|
|
|
||
無帰属限定株奨励 |
|
|
|
|
|
|
||
発行可能株式オプション |
|
|
|
|
|
|
||
従業員株購入計画 |
|
|
|
|
|
|
||
合計する |
|
|
|
|
|
|
||
F-17
12.株ベースの報酬
2022年株式インセンティブ計画
2022年7月1日、会社取締役会は“2022年株式インセンティブ計画”(“2022年計画”)を承認し、当日発効した。2022年計画は、会社がナスダック上場規則第5635(C)(4)条または誘因両立奨励に関連する任意の後続規則に基づいて、株主の承認なしに上級管理者および従業員に持分インセンティブ報酬を発行することを可能にする。取締役会が早期に終了しない限り、2022計画は施行日の四回目の記念日。その会社はすでに初歩的に保留した
2021年株式インセンティブ計画
2021年9月27日、会社取締役会と株主は、2021年10月7日に会社登録書が米国証券取引委員会によって発効することを宣言した“2021年株式インセンティブ計画”(“2021年計画”)を採択した。2021年には、会社が役員、従業員、役員、コンサルタントに対して株式および現金に基づくインセンティブ奨励を可能にする計画だ。その会社はすでに初歩的に保留した
2019年持分インセンティブ計画
2019年、会社は“2019年株式インセンティブ計画”(“2019計画”)を策定し、会社が従業員と非従業員にオプションと制限株を付与することを許可した。“2019年計画”によると予約発行された普通株の最高株式数は
IPO前に、2019計画に基づいて付与される奨励の行使価格、帰属及びその他の制限は取締役会によって決定されるが、付与日普通株の公正時価又は期限が10年を超える方法で株式オプションを発行してはならない。2019年の計画に基づいて付与されたオプションは、帰属後の任意の時間に全部または部分的に行使することができる。2022年12月31日現在、購入オプション
2021年従業員株購入計画
2021年9月27日、会社取締役会と株主は、2021年10月7日に会社登録書が米国証券取引委員会によって発効を宣言された“2021年従業員株購入計画”(略称“2021年従業員株購入計画”)を採択した。2021年にESPPが最初に発行を保留し、発行されたのは合計で最大
F-18
株式オプション
“2022年計画”、“2021年計画”、“2019年計画”(総称して“計画”)に基づいて従業員に付与される株式オプション一般
下表に本年度までの株式オプション活動をまとめた2022年12月31日(千、1株当たりおよび1株当たりの金額を除く):
|
|
量 |
|
|
重みをつける |
|
|
重みをつける |
|
|
骨材 |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
2022年1月1日に返済されていません |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
授与する |
|
|
|
|
|
|
|
|
|
|
|
|
||||
鍛えられた |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
没収される |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
期限が切れる |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
2022年12月31日に返済されていません |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2022年12月31日に行使可能なオプション |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2022年と2021年12月31日終了年度付与オプションの加重平均付与日公正価値はい$です
♪the the the同社はブラック·スコアーズオプション定価モデルを用いて、次の表の一連の仮定を適用し、付与日ごとのオプションの公正価値を推定した
|
|
十二月三十一日までの年度 |
|
||
|
|
2022 |
|
2021 |
|
予想変動率 |
|
|
|
||
無リスク金利 |
|
|
|
||
期待配当収益率 |
|
|
|
||
予想期限(年単位) |
|
|
|
||
|
|
|
|
|
|
財務諸表に記録されている株式オプションに関する株式報酬費用は以下の通り(単位:千):
|
|
十二月三十一日までの年度 |
|
|
|||||
|
|
2022 |
|
|
2021 |
|
|
||
研究開発 |
|
$ |
|
|
$ |
|
|
||
一般と行政 |
|
|
|
|
|
|
|
||
合計する |
|
$ |
|
|
$ |
|
|
||
その会社には集合がある $
F-19
制限株式賞
2019年計画、2021年計画、2022年インセンティブ計画によると、会社は発表
また、会社は2019年に従業員連合創業者及びいくつかの非従業員顧問にいくつかの制限的普通株式(“RSA”)を発行した。制限的普通株は独立した制限的株式購入協定によって発行され、これらの協定は2019年計画、2021年計画、2022年インセンティブ計画から独立している。独立制限株式購入プロトコルにより保有する制限的普通株の購入価格は#ドルに相当する
限定株式買い戻し協議の条項によると、当社は買い戻し選択権を有しており、これにより、当社は、購入終了時に、(I)自社普通株が買い戻し当日に相当する公平時価および(Ii)元購入価格のうち小さい者の1株当たり価格でいずれも帰属していない株式を買い戻す権利がある。会社連合創業者及び非従業員コンサルタントに発行された限定普通株株は、所定数の株式に基づいて付与される。
株式に帰属していない場合は終了時に買い戻しが必要であるため、当社は発行時に独立制限株式購入プロトコルにより発行された制限株式奨励に基づいて、奨励金の購入価格に基づいて関連付金責任を確認する。限定的な株式帰属の奨励として、会社は預金負債を追加実収資本に再分類する。補償コストは,関連普通株の公正価値から制限された普通株の購入価格を引いて計測され,会社は必要なサービス期間内の補償コストを確認した。
下表は今年度までの限定株式活動をまとめたものである2022年12月31日:
|
|
株式数 |
|
|
重みをつける |
|
||
2021年12月31日現在帰属していません |
|
|
|
|
$ |
|
||
授与する |
|
|
|
|
|
|
||
仲直りしなければならない |
|
|
( |
) |
|
|
|
|
没収される |
|
|
( |
) |
|
|
|
|
2022年12月31日現在の未帰属·未決済 |
|
|
|
|
$ |
|
||
会社は制限株に関する株式ベースの報酬支出#ドルを記録した
F-20
13.所得税
2022年12月31日まで及び2021年12月31日まで年度会社は記録しました
連邦法定所得税率を用いて計算された期待所得税収益と会社の実際の所得税税率との入金は以下のとおりである2022年12月31日および2021年12月31日:
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
連邦法定税率で計算される所得税% |
|
|
% |
|
|
% |
||
連邦福祉を差し引いた州税 |
|
|
|
|
|
|
||
評価免除額を変更する |
|
|
( |
) |
|
|
( |
) |
研究開発信用繰り越し |
|
|
|
|
|
|
||
恒久的差異 |
|
|
( |
) |
|
|
( |
) |
実際の所得税率% |
|
|
% |
|
|
% |
||
その会社の実際の税率は
繰延所得税は、財務報告目的のための資産および負債の帳簿金額と所得税目的のための金額との間の一時的な差異の純税影響を反映する。
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
繰延税金資産: |
|
|
|
|
|
|
||
純営業損失 |
|
$ |
|
|
$ |
|
||
無形資産 |
|
|
|
|
|
|
||
税金の繰り越しを免除する |
|
|
|
|
|
|
||
導関数 |
|
|
|
|
|
|
||
株に基づく報酬 |
|
|
|
|
|
|
||
準備金と課税項目 |
|
|
|
|
|
|
||
資本化研究支出 |
|
|
|
|
|
— |
|
|
許可証料 |
|
|
|
|
|
|
||
リース負債を経営する |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
推定控除前の繰延税金資産総額 |
|
|
|
|
|
|
||
減算:推定免税額 |
|
|
( |
) |
|
|
( |
) |
繰延税金資産総額 |
|
$ |
|
|
$ |
|
||
繰延税金負債: |
|
|
|
|
|
|
||
リースROU資産を経営する |
|
|
( |
) |
|
|
( |
) |
繰延税項目純資産 |
|
$ |
— |
|
|
$ |
— |
|
2022年12月31日現在、同社の米国連邦と州での純営業損失(NOL)は$
F-21
当社は既存のプラスおよび負の証拠に基づいて繰延税金資産の決算日ごとの現金化可能度を評価し、最も現金になる可能性のある金額を特定し、必要に応じて推定値を算出して準備している。会社の累積赤字が克服困難な重大なマイナス証拠を提供したため、会社は#ドルの推定準備金を計上した
減税·雇用法案には、2022年1月1日以降に発生した年から発生した第174条のコストを資本化することを求める条項が含まれている。第174項の費用とは、製品、プロセス、配合、発明、コンピュータソフトウェア又は技術の開発又は改善に関連する研究及び開発費用をいう。この規定は、第174項の費用の処理方法を変更し、これらの支出が直ちに差し引かれることが許されなくなるように、資本化及び償却しなければならない。私たちはこの準備金の影響を計上しており、これにより繰延税金資産は約#ドルになる
会社が1回または複数回の所有権変更が発生した場合、米国の税収属性は、1986年の“国内税法”(以下、“コード”と略す)第382節の年間制限、および同様の州規定を受ける可能性があり、これは、将来の課税所得額を相殺するために使用することができる税収属性を制限することになる。一般的に、第382条に規定する所有権変更とは、3年以内に、ある株主又は公共団体の会社株における所有権が50ポイントを超える取引の結果を意味する。もし未来に所有権変更が発生すれば、純運営損失と研究開発信用の繰越を除去或いは制限することができる。廃止された場合、関連資産は繰延税金資産表から削除され、それに応じて推定免税額が減少する。推定免税額の存在により、将来の所有権変更による制限は当社の実際の税率に影響を与えません。
同社は納税が必要で、引き続きアメリカとある州と地方司法管轄区で連邦所得税申告書を提出する。当社は2019年12月31日以降のすべての適用所得税司法管区の税務年度に税務審査を受けなければなりません。税務監査と検討は複雑な問題、説明、そして判断と関連があるかもしれない。問題の解決は数年に及ぶ可能性があり、特に訴訟や交渉が必要な場合には。当社は、当社は合理的な見積もりと仮定を用いてその税務状況を適切に記録していると信じているが、潜在的な税務優遇は清算、決済期間、あるいは訴訟時効が満了した時の経営業績やキャッシュフローに影響を与える可能性がある。同社には現在、未確認の税収割引に関する準備金は何もない。
F-22
14.1株当たり純損失
1株当たり基本と償却純損失は以下のように計算される(千計で、1株および1株当たりの金額は含まれていない)
|
|
十二月三十一日までの年度 |
|
|
|||||
|
|
2022 |
|
|
2021 |
|
|
||
分子: |
|
|
|
|
|
|
|
||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
分母: |
|
|
|
|
|
|
|
||
加重平均発行済み普通株式、基本普通株式、希釈後普通株 |
|
|
|
|
|
|
|
||
1株当たり基本と希釈して純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
当社の潜在的希薄化証券は、限定株と株式オプションを含み、1株当たり純損失の計算から除外されており、1株当たり純損失を減少させるためである。したがって、普通株株主が基本純損失と希釈後の1株当たり純損失を占めるべき加重平均既発行普通株数は同じである。
以下の希釈可能な証券は、その逆希釈作用により、普通株1株当たりの純損失の計算から除外されている
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
未償還株式オプション |
|
|
|
|
|
|
||
無帰属限定株奨励 |
|
|
|
|
|
|
||
発行可能株式オプション |
|
|
|
|
|
|
||
従業員株購入計画 |
|
|
|
|
|
|
||
合計する |
|
|
|
|
|
|
||
F-23
15.関連するパーティ
同社はシカゴ大学トーマス·ガジェスキー博士の研究室で設立された。2020年、同社はシカゴ大学とライセンス契約を締結し、賛助研究協定に署名した。その会社は$を生み出した
ファイザーが所有している株式は
同社と合金会社は,会社の技術と合金のATX−GX?プラットフォームと抗体発見サービスを利用するための合弁会社Voxall Treateutics,LLC(“Voxall”)を設立した。同社と合金会社は$を貢献した
16.支払いの引受および事項
法律訴訟
当社は通常の業務過程で出現する様々な法的手続きに時々巻き込まれる可能性があります。当社は現在、いかなる重大な法的手続きの一方でもなく、いかなる懸案や脅かされた法的手続きが当社の業務、経営業績、財務状況に悪影響を及ぼす可能性があるかも知りません。
支払いを引き受ける
正常な業務過程において、当社は各種の第三者と臨床試験、臨床前研究とテスト、製造と運営目的のための他のサービスと製品について合意を締結し、これらのサービスと製品は一般的に当社が随時キャンセルすることができるが、拘束力のある調達注文項目の下の残りの債務を支払う必要があり、場合によっては、象徴的な早期終了費用を支払わなければならない。このような約束は重要ではないと考えられている。
17.後続のイベント
賃貸借証書
2023年2月2日、当社は分譲協定を締結した
F-24