カタログ表
アメリカ証券取引委員会
ワシントンD.C.,郵便番号:20549
表
|
1934年の証券取引法第13条又は15(D)条 に基づいて提出された年次報告。 本財政年度末まで | |
|
1934年“証券取引法”第13節又は第15(D)節に基づいて提出された移行報告。 そこからの過渡期について
|
依頼書類番号:
(登録者の正確な氏名はその定款に定められている)
|
(明またはその他の司法管轄権 会社や組織) |
アメリカ国税局の雇用主は 識別番号) |
(主な執行機関住所と郵便番号)
(登録者の電話番号、 市外局番を含む)
同法第12条(B)に基づいて登録された証券:
| クラスごとのタイトル | 取引コード | 登録された各取引所の名称 |
| ♪the the the資本市場 | ||
| |
♪the the the |
同法第12条(G)に基づいて登録された証券:
ありません
登録者が証券法第405条に規定する有名な経験豊富な発行者であるか否かをチェックマーク
で示す:YES☐
登録者が当該法第13条又は第15条(D)に基づいて報告を提出する必要がないか否かを再選択マーク
で示す:はい☐
再選択マーク
は、登録者が(1)過去12ヶ月以内(または登録者がこのような報告の提出を要求されたより短い期間)に、1934年の証券取引法第13条または15(D)節に提出されたすべての報告を提出したかどうか、および(2)過去90日以内にこのような届出要件に適合しているかどうかを表す
再選択マーク
は、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間内)に、第
S−T条例(本章232.405節)規則405に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す:
登録者が大型加速申請者,加速申請者,非加速申請者,小さい報告会社か新興成長型会社かを再選択マーク で示す。取引法第12 b-2条規則における“大型加速申告会社”、“加速申告会社”、“小報告会社”、“新興成長型会社”の定義を参照されたい。
| 大型加速ファイルサーバ | ☐ | ファイルマネージャを加速する | ☐ |
| ☒ | 規模の小さい報告会社 | ||
| 新興成長型会社 |
新興成長型会社である場合、登録者が、取引法第13(A)節に従って提供された任意の新しいまたは改正された財務会計基準を遵守するために、延長された移行期間を使用しないことを選択したかどうかをフックで示す
登録者が報告書を提出したか否かを再選択マーク で示し、その管理層が“サバンズ-オキシリー法案”第404(B)条に基づいて、その内部財務報告制御の有効性を評価しており、この内部統制は、 またはその監査報告を作成する公認会計士事務所によって行われている
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する
これらのエラーのより真ん中に再記載があるかどうかをチェックマーク で表すことは、登録者の任意の幹部が関連回復中に受信したインセンティブベースの報酬を§240.10 D−1(B)に従って回復分析する必要があるかどうかを表す[1]
登録者が空殻会社であるかどうかをチェックマークで表す(取引法ルール12 b-2で定義されている):
はい,いいえ
2022年6月30日まで,すなわち登録者の最近の完全な第2四半期の最後の営業日,登録者の非関連会社が保有する投票権と無投票権普通株の総時価
は,登録者普通株のナスダック資本市場における終値に基づいて0.8ドル,約$である
2023年3月10日現在の登録者の普通株流通株数は.
引用で編入された書類
Form 10−K第3部(第10,11,12,13,14項)に必要な情報に応答するために,登録者の最終依頼書,情報説明書,または2023年株主総会のForm 10−K年度報告修正案の一部を統合する。登録者は,登録者が2022年12月31日までの財政年度終了後120日以内に,最終依頼書,情報声明,または本年度報告に対する修正案をForm 10−K形式で証券取引委員会に提出する予定である。
___________________________
[1]米国証券取引委員会の指導意見によると、この空白チェック枠は本表紙に含まれているが、関連証券取引所上場基準が通過して発効するまでは、何も開示してはならない。
Xenetic生物科学社です
表格10−Kの2022年年次報告
表の内容
| 第1部 | 1 | ||
| プロジェクト1 | 業務.業務 | 1 | |
| 第1 A項 | リスク要因 | 23 | |
| プロジェクト1 B | 未解決従業員意見 | 55 | |
| プロジェクト2 | 属性 | 56 | |
| 第3項 | 法律訴訟 | 56 | |
| プロジェクト4 | 炭鉱安全情報開示 | 56 | |
| 第II部 | 57 | ||
| 第5項 | 登録者普通株市場、関連株主事項及び発行者による株式証券の購入 | 57 | |
| プロジェクト6 | [保留されている] | 57 | |
| 第七項 | 経営陣の財務状況と経営成果の検討と分析 | 58 | |
| 第七A項 | 市場リスクの定量的·定性的開示について | 66 | |
| プロジェクト8 | 財務諸表と補足データ | 67 | |
| プロジェクト9 | 会計と財務情報開示の変更と相違 | 68 | |
| 第9 A項 | 制御とプログラム | 68 | |
| プロジェクト9 B | その他の情報 | 69 | |
| プロジェクト9 C | 検査妨害に関する外国司法管区の開示 | 69 | |
| 第三部 | 70 | ||
| 第10項 | 役員·幹部と会社の管理 | 70 | |
| プロジェクト11 | 役員報酬 | 70 | |
| プロジェクト12 | 特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 | 70 | |
| 第13項 | 特定の関係や関連取引、取締役の独立性 | 70 | |
| プロジェクト14 | 最高料金とサービス | 70 | |
| 第4部 | 71 | ||
| プロジェクト15 | 展示品と財務諸表の付表 | 71 | |
| プロジェクト16 | 表格10-Kの概要 | 71 | |
| i |
前向き陳述に関する警告説明
本年度報告(以下、“年次報告”と略す)は、改正後の1934年証券取引法(“取引法”)第21 E節と改正後の1933年証券法第27 A節に適合する前向きな記述を含む。歴史的事実の陳述を除いて、本年度報告に含まれるすべての陳述は、私たちの未来の経営結果と財務状況、私たちの業務戦略と計画、未来の収入、予想コスト、見通しと私たちの未来の経営目標に関する陳述を含み、すべて前向き陳述 である。これらの前向き表現は、コロナウイルスまたは新冠肺炎の全世界大流行の影響およびその対応措置を含むが、これらに限定されない:大流行が全体的な経済と市場状況に与える影響および私たちの業務、経営業績および財務状況への影響、ロシアとウクライナ間の衝突、および米国(以下“米国”)が実施する関連制裁を含む地政学的事件の予想影響を含む。協力計画の性質、時間、範囲;協力計画に基づいて規制機関の承認を得る計画を含む協力計画の予想結果に基づいて。私たちの候補薬物商業化計画の結果;私たちはある市場の計画に対して、第三者メーカーと協力して、後続の商業開発の他の候補薬物、そして私たちの候補薬物に対する競争の可能性と程度を評価し、私たちは革新的な免疫腫瘍学技術を推進し、治療困難な腫瘍適応を解決することを計画している。我々のデオキシリボヌクレアーゼ(“DNase”)プラットフォームへの期待br},例えば、好中球細胞外トラップ(“Net”)を標的とした免疫療法を含む既存の治療の結果を改善することを目的とした固形腫瘍の治療のためのDNaseプラットフォームのような開発されているDNaseプラットフォーム、およびこの新たに許可された技術上の我々の努力と資源を優先することを目的としている;XCARTの開発 ™キメラ抗原受容体(“CARR”)T細胞(“XCART”)技術は、単一患者の悪性腫瘍細胞表面を標的とするユニークなB細胞受容体brによるB細胞リンパ腫の治療のための細胞ベース療法 の開発を計画している;および我々のPolyXenに対する期待である®次世代バイオ医薬の半減期および他の薬学的特性を改善するために、バイオ技術および製薬会社と協力して、このプラットフォームを利用する計画brおよびタンパク質またはポリペプチド療法へのそれらの使用を含むプラットフォーム。
場合によっては、これらの 陳述は、“可能”、“将”、“将”、“可能”、“すべき”、“ ”、“予想”、“計画”、“予想”、“信じ”、“推定”、“求める”、“約”、“意図”、“予測”、“潜在”、“項目”または“継続”などの用語によって識別することができる。 またはそのような用語および他の類似用語の否定。著者らは本文に含まれる展望性 陳述に反映される予想は合理的であると信じているが、著者らは未来の結果、活動レベル、業績或いは成果を保証することができない。これらの 陳述は既知と未知のリスクと不確定要素に関連し、私たち或いは私たちの業界の結果、活動レベル、業績或いは業績は展望性陳述中の明示或いは暗示と大きく異なる可能性がある。
実際の結果が大きく異なる可能性があるいくつかの要因は、これらに限定されない
| · | CLS治療株式会社(“CLS”)との取引およびDNaseプラットフォームの許可による意外なコスト、課金または支出; | |
| · | CLSとの取引やDNaseプラットフォームの許可を完了した後、会社が予想する財務業績の不確実性 | |
| · | DNase、XCART、PolyXen技術の期待可能性を実現できなかった | |
| · | ビジネス戦略を実施する能力は | |
| · | ナスダック資本市場の持続的な上場要求には達していない | |
| · | 私たちは将来的により多くの運営資金を集めて、私たちのパイプラインをさらに発展させ、持続的に経営する企業として続ける必要がある | |
| · | 私たちがビジネスに資金を調達する能力は | |
| · | 私たちは重要な買収と統合を実行し管理し統合する能力を成功させました | |
| · | DNase技術の開発に成功した能力を含む製品開発と商業化リスク |
| II |
| · | 有害な安全結果と臨床試験結果が私たちの療法に与える影響 | |
| · | 私たちは私たちの技術を確保して維持することができるメーカーを持っている | |
| · | 新しい治療法と既存療法の新しい用途が競争環境に及ぼす影響 | |
| · | 私たちは現在と未来の候補薬を商業化する能力に成功しました | |
| · | 私たちは私たちの現在と未来の共同開発協力と戦略計画に関するマイルストーンと他の支払いを達成することができます | |
| · | 私たちはコンサルタント、コンサルタント、サプライヤー、ビジネスパートナーに依存して仕事を展開しています | |
| · | 新しい技術が候補薬と私たちの競争に及ぼす影響は | |
| · | 政府機関の法律や法規が変わりました | |
| · | 既存の契約を中断またはキャンセルすること | |
| · | 競争的製品と価格設定の影響 | |
| · | 製品の需要と市場受容度とリスク | |
| · | より多くの財力を持つ競争相手の存在 | |
| · | 製造に使用される物品または材料は、現在の価格で提供され続ける | |
| · | 経営陣が計画を実行し、これらの計画を実行する能力を奨励する | |
| · | 私たちは重要な人材を引きつけて維持する能力を持っています | |
| · | 私たちの製品や会社自体に関する否定的な宣伝 | |
| · | 私たちの知的財産権に関する不利なクレーム | |
| · | 新しい会計基準を採用したり、会計基準を変更したりする | |
| · | 2002年の“サバンズ-オキシリー法案”のように、公的報告会社に適用される法規や条例に適用される固有のコストを遵守する | |
| · | 会社が将来参入する可能性のある他の新しい事業 | |
| · | 一般的な経済やビジネス状況、インフレ傾向や金融市場の不安定さや銀行システムの倒産により中断されている | |
| · | 新冠肺炎のような自然災害や突発的な公衆衛生事件、およびロシアのウクライナ侵攻などの地政学的事件、ならびに関連する制裁および他の経済中断または懸念、私たちの財務状況および経営業績への影響; | |
| · | 私たちの年次報告書10-K表のリスク要因の部分と、その後に米国証券取引委員会(“米国証券取引委員会”)に提出された文書に記載されている他の要因。 |
これらの要因は,必ずしも実際の結果が本年度報告の前向き陳述で表現されているものとは大きく異なるすべての重要な要素をもたらす可能性があるとは限らない。他の未知または予測不可能な要因は、“リスク要因”の節で議論される要因を含むが、これらに限定されないが、我々の将来の結果に実質的な悪影響を及ぼす可能性がある。本年度報告における前向き陳述は,本年度報告の発表日にのみ行われ,後続の事件や状況を反映するためにいかなる前向き 陳述を公開更新する義務も負わない。私たちはすべての前向きな陳述に“1995年個人証券訴訟改革法”の安全港条項を遵守させるつもりだ。
本年度報告書で使用されるように、他の説明がない限り、本報告書で言及された“Xenetic”、“会社”、“私たち”または“私たち”は、Xenetic Biosciences,Inc.およびその完全子会社を意味する。
私たちのブランドと製品名は含まれていますが、XCARTに限られません™OncoHist™PolyXen®エレpoXen™ とImuXen™本年度報告書には、米国の子会社(米国または米国)におけるXenetic Biosciences,Inc.および/またはその商標、登録商標またはサービスマークが含まれている。他の国もあります他のすべての会社と製品名は関連会社の商標である可能性がある。
| 三、三、 |
リスク要因をまとめる
私たちの業務は多くの危険に直面している。以下の要約に加えて、本年度報告書10-K表の“リスク要因”の部分を慎重に検討しなければなりません。 私たちは他のリスクおよび不確定要素の影響を受ける可能性があり、これらのリスクおよび不確定要素は現在私たちに知られていないか、または現在、これらのリスクと不確定要因は無関係だと考えています。これらのリスクは、本年度報告の10-K表の他の情報と共に読まなければならない。私たちの業務に関連するいくつかの主なリスクは、:
| · | 私たちは決して利益を上げたことがなく、絶対に利益を達成したり維持したりしないかもしれない。もし私たちが私たちの運営から費用を支払うのに十分な収入を生むことができない場合、あるいは私たちが商業的に合理的な条項で追加的な融資を得ることができなければ、私たちの業務、財務状況、運営結果は重大で不利な影響を受ける可能性がある。 | |
| · | 私たちは私たちの目標を達成するために多くの追加資金が必要になるだろう。必要なときに受け入れ可能な条件で必要な資本を得ることができない場合、あるいは資金を全く得ることができない場合、私たちの製品開発努力、他の運営、または商業化努力を延期、制限、または終了させることができないかもしれません。 | |
| · | 追加資本の調達は私たちの株主に希釈し、私たちの運営を制限するか、あるいは私たちの技術または候補薬物の権利を放棄することを要求するかもしれない。 | |
| · | 私たちはナスダック資本市場の持続的な上場要求を満たすことができず、私たちの普通株が銘柄を取られる可能性があります。ナスダック上場規則を再遵守できなければ、私たちの普通株の市場価格と流動性に影響を与え、私たちの融資能力を低下させるかもしれない。 | |
| · | 我々の業務はDNase腫瘍学プラットフォームの成功に大きく依存している。 | |
| · | 私たちは競争が極めて激しい環境で運営されており、競争技術が私たちの業務発展を損なわない保証はありません。 | |
| · | 私たちは候補薬物や技術を含む医薬製品を開発する初期の会社です。このような発展の不確実性を考慮して、私たちの業務運営は決して完全に実現されず、投資家のために価値を創造するかもしれない。 | |
| · | もし私たちが私たちの協力者や戦略的パートナーと衝突すれば、これらの当事者たちは彼ら自身の利益のために行動するかもしれないし、これは私たちが戦略を実施する能力を制限するかもしれない。 | |
| · | 私たちは第三者に依存して、私たちの臨床研究を行い、監督して、これらの第三者の表現が満足できなければ、私たちの業務を損なう可能性があります。 | |
| · | 私たちの協力者や戦略パートナーは、代替技術を採用することを決定したり、私たちの技術を利用して商業的に実行可能な製品を開発できない可能性があり、これは私たちの収入と私たちがこれらの製品を開発する戦略にマイナスの影響を与えるだろう。 | |
| · | 私たちはより多くの協力を求めるかもしれませんが、ビジネス的に合理的な条件下で協力を作ることができなければ、私たちの開発と商業化計画を変えなければならないかもしれません。 | |
| · | もし私たちが1つ以上の協力を達成すれば、私たちは私たちの候補薬物開発に対する重要な権利と統制権を放棄すること、または他の方法で不利な条項の制約を受けることを要求されるかもしれない。 |
| 四 |
| · | 患者を臨床研究に組み込むことは困難であることが発見されるかもしれませんが、これは私たちの薬物製品の臨床研究を延期あるいは阻止する可能性があります。 | |
| · | 私たちは臨床試験の開始、登録、あるいは完成に重大な遅延に遭遇する可能性があり、あるいは関連する規制機関を満足させる安全性と有効性を証明できない可能性があり、これは私たちが現在と未来の候補薬物をタイムリーに商業化することを阻止するかもしれない(もしあれば)。 | |
| · | もし私たちが必要な臨床前と臨床研究を完成すれば、いつ、あるいは規制部門の承認を得るかどうか、候補薬物を商業化するか、あるいは承認された適応が私たちが予想していたよりも狭いかもしれない。 | |
| · | もし私たちが候補薬物の規制承認を受けたら、私たちの候補薬物は引き続き規制されて審査されるだろう。 | |
| · | いかなる現在または未来の薬品の商業成功は医者、患者、第三者支払人と医学界の他の人の市場に対する受け入れの程度に依存する。 | |
| · | 開発中の候補薬物の商業的潜在力は予測が困難である。もし新しい候補薬や技術の市場規模が私たちが予想していたよりはるかに小さい場合、それは私たちの収入、運営結果、および財務状況に重大な負の影響を及ぼすかもしれない。 | |
| · | もし私たちの候補薬が十分な保険や精算を得られなかった場合、承認されれば、これらの製品をマーケティングする能力を制限し、収入を創出する能力を低下させる可能性がある。 | |
| · | 私たちは、より利益的または成功可能性の高い計画または候補薬を利用するのではなく、特定の研究計画または候補薬を追求するために、私たちの財力および人的資源を使用するかもしれない。 | |
| · | 私たちはもっと多くの医薬製品を識別したり発見することに成功しないかもしれない。 | |
| · | 私たちの候補薬物の市場機会は、以前の治療に適合していないか、または通過できなかった患者に限定される可能性があり、小さい可能性がある。 | |
| · | 私たちは製造、販売、マーケティング、流通能力がありません。私たちはこれらの能力を開発するために大量の資源を投入しなければならないかもしれません。 | |
| · | 私たちの第三者への依存は、私たちのビジネス秘密を共有することを要求し、これは、競争相手がこれらの秘密や私たちのビジネス秘密が流用または開示されていることを発見する可能性を増加させる。 | |
| · | もし私たちが私たちの知的財産権を十分に保護したり実行できなかったら、私たちは効果的に作動できないかもしれない。 | |
| · | 法廷で疑問視された場合、私たちの候補薬物に対する発行された特許は無効または実行不可能と認定される可能性がある。 | |
| · | 私たちは世界各地で私たちの知的財産権を保護できないかもしれない。 | |
| · | もし私たちが他人の知的財産権を侵害すれば、私たちの業務と収益性は不利な影響を受けるかもしれない。 |
| v |
| · | もし私たちが第三者に知的財産権を許可する合意の義務を履行できなかった場合、あるいは私たちとライセンス者との業務関係が妨害された場合、私たちは私たちの業務に非常に重要な許可権を失う可能性があります。 | |
| · | 私たちは、私たちの従業員、コンサルタント、または独立請負業者が第三者の機密情報を誤って使用または開示したか、または私たちの従業員がその前の雇用主によって言われた商業秘密を誤って使用または開示したという疑惑の影響を受けるかもしれない。 | |
| · | 私たちは私たちの特許と他の知的財産権の発明権または所有権のクレームに疑問を受けるかもしれない。 | |
| · | 私たちは私たちの機密情報と商業秘密を保護することができず、これは私たちの業務と競争地位を損なうだろう。 | |
| · | 私たちの未来の成功は私たちが幹部チームの主要なメンバー、顧問と顧問、及び合格者を吸引、維持と激励する能力を維持できるかどうかにかかっている。 | |
| · | 私たちは私たちの組織を拡大する必要があり、私たちはこのような成長を管理することに困難に直面するかもしれないし、これは私たちの運営を混乱させるかもしれない。 | |
| · | 私たちは協力協定や他の重要な合意の側であり、これらの協定は複雑なビジネス条項を含み、紛争、訴訟または賠償責任を招く可能性があり、私たちの業務、運営結果、財務状況に悪影響を及ぼす可能性がある。 | |
| · | 私たちの証券の市場価格は大きく変動するかもしれません。あなたは私たちの証券を売ることができないかもしれません。 | |
| · | 私たちの優先株保有の権利、優先権、特権は私たちの普通株主の権利ではなく、普通株主の権利よりも優先しており、これは私たちの優先株保有者の利益が私たちの普通株主の利益とは異なることをもたらす可能性がある。 |
| VI |
第1部
プロジェクト1--ビジネス
概要
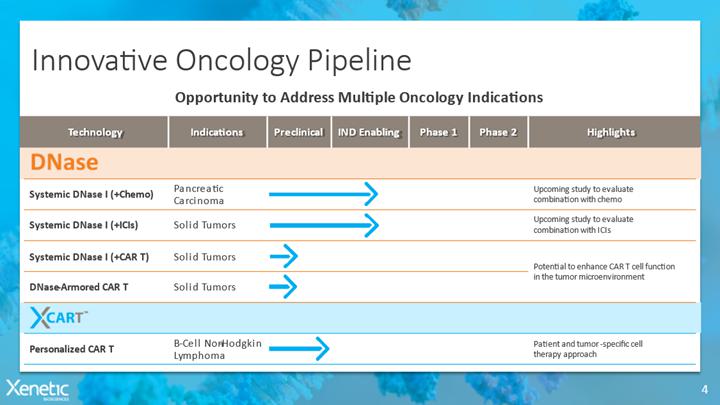
私たちは生物製薬会社で、革新的な免疫腫瘍学技術を推進し、治療が困難な癌を解決することに専念しています。我々独自のDNaseプラットフォームは,Netに対する免疫療法を含めた既存の治療の結果を改善することを目指しており,Netは癌の進行や癌治療に対する耐性に関与している。我々は2022年4月にDNase腫瘍学プラットフォームの許可を得,我々の努力と資源をこの新買収技術の開発に用いた。
DNaseプラットフォームはNetを標的に設計されており,Netはヒストンや他のタンパク質を被覆した細胞外クロマチンからなるメッシュ構造である.Netが活性化された好中球排出は,微生物や炎症促進挑戦に対応する。しかし、Netの過剰生産或いは減少の除去は炎症と自己免疫病理の加重を招くことができ、及び癌の成長と転移の情況下で腫瘍を促進する利基環境を形成することができる。
著者らは局部末期或いは転移性固形腫瘍被験者の中で初のヒト、多中心、用量増加とIV型組換えヒトデオキシリボヌクレアーゼIの用量拡張研究を行う予定である。我々の全身性DNA酵素計画の最初の目標は膵癌を含む数十億ドルの適応であった。膵臓癌の早期診断率は低く、死亡率は高く、5年間の生存予後は悪い。症状は通常非特異的であるため,膵癌は通常末期まで診断される。病気が他の臓器に転移したり拡散したりすると、それは特に治療が困難になる。世界では毎年約185,000人がこの疾患と診断されており,2021年には米国国立癌研究所のモニタリング,疫学,最終結果計画(SEER)が推定され,米国では約60,000人が膵癌と診断される。膵臓癌患者の総5年生存率は7-8%であり、固形腫瘍悪性腫瘍の中で死亡率が最も高い;転移性疾患と診断された患者の中で、総5年生存率はわずか3%である。最近の進展は多くの癌型の生存率を向上させているが,膵癌患者には有効ではなく,新たな治療案の開発の必要性が示唆されている。
また,転移性疾患と診断された二線患者では治療選択はほとんどなかった。二線患者に承認された唯一のレジメンは,5 FUとLVの併用であった。これらの二次治療を受けたIV期患者では,中位総生存期間はわずか4.7カ月(Macarulla ら,膵2020)であった。
大量の科学文献により、NETsは癌の発病機序と癌治療(化学療法、放射線治療と免疫治療、例えばCheckpoint阻害剤と細胞治療を含む)の薬剤耐性と関係があることを表明した。Netレベルの上昇は,多くの癌患者の予後不良に関連するバイオマーカーであることが報告されている。また,既存の治療薬に対する抵抗はNetの免疫抑制シグナル因子の放出,あるいはNetが産生する物理的バリアに関与する可能性があり,腫瘍微小環境における細胞毒性T細胞の浸透,活性,生存を阻害する可能性がある。発表された前臨床モデルは,Netの除去および腫瘍成長および転移防止におけるDNaseの系統的単独または他の薬剤との併用の有効性を証明している。われわれは現在,膵癌や局所末期あるいは転移性固形腫瘍の補助治療として,われわれの全身性DNA ase計画を臨床に進めることに集中している。
過継転移CAR T細胞はすでに癌免疫治療における最も将来性のある進展の一つになっている。固形腫瘍の治療に成功するためには、CAR T細胞は自身が免疫抑制に得意であり、腫瘍細胞の生存に有利な敵対腫瘍微小環境中で浸透、持続し、抗腫瘍機能を維持できなければならない。最近CAR Tを設計する方法は、“装甲”CAR-T細胞を含み、命名されたのは、ネットワークを含む免疫抑制または腫瘍細胞外マトリックスの物理成分の分解に抵抗するための追加の因子を発現することができるからである。われわれは臨床前研究を行う予定であり,メチルCAR T細胞を装着してDNA酵素を分泌することで固形腫瘍適応に対する深さと持続性反応を支持できることを証明することを目的としている。癌関連抗原を識別するために設計された工学的CAR T細胞は腫瘍細胞を持続的かつ選択的に死滅させ、腫瘍負担を大幅に低下させることができる。CAR−T療法は血液系悪性腫瘍の治療に顕著な臨床成功を得たが,これまで固形腫瘍では成功していなかった。すでに発表された証拠は,免疫抑制因子に加えてNetが形成した機械的バリアがT細胞の透過を阻止し,T細胞と腫瘍細胞との接触を遮断することを示している。
| 1 |
著者らはベルギーWilition SARL株式会社(“Wilition”) との協力は早期探索的計画であり、WilitionのNu.Q技術とXeneticのDNase−Armored CAR Tプラットフォームの潜在的な組み合わせを評価し、多様な実体癌に対する特許を用いた細胞療法を開発することを目的とし、現在のCAR T細胞療法はこれらの腫瘍に対して限られた効果を示すか、あるいは効果がない。協力協定の条項によると、Wilitionは1つの研究プロジェクトに資金を提供し、双方は協力によるいかなる製品の商業化も収益できるかもしれないことを共有する。エピジェネティクス修飾核小体は,多様なタイプの固形癌の腫瘍細胞表面や腫瘍微小環境に存在するため,これらの核小体は単一癌タイプに限定されない汎化可能な腫瘍抗原を代表している可能性がある。WilitionのNu.Q 技術はエピジェネティック修飾の核小体を特異的に識別と標的化することができ、著者らのDNase-Armored CAR Tプラットフォームは固形腫瘍微小環境中のCAR T細胞の機能を増強することを目的としている。
XCARTは著者らの個性化CAR Tプラットフォーム技術であり、患者の特定の腫瘍新しい抗原に対して、B細胞リンパ腫における作用機序を証明することを目的としている。br}XCART技術プラットフォームは、成熟したスクリーニング技術を利用して、単一リンパ腫患者の悪性B細胞クローン表面の唯一のB細胞受容体(BCR)に選択的に結合するポリペプチドドメインを識別することを目的としている。brというBCR選択性標的ドメインはCARの抗原結合ドメインに改造され、それによって所与の患者の悪性B細胞クローンのCAR Tのみを識別するCAR T治療の可能性を創造した。われわれの個性化CAR T療法は,癌患者に既存の看護基準や現在承認されているCAR T療法よりも大きな利点を提供する可能性があると信じている。DNaseプラットフォームを優先し,DNaseプログラムの開発に資源を集中させる予定であるため, XCARTプラットフォームの開発を一時停止した.
また,バイオテクノロジーや製薬会社と協力して独自の薬物送達プラットフォームPolyXenを開発し,血液凝固障害領域の独占ライセンス手配に基づいて特許使用料を徴収している。PolyXenはタンパク質とポリペプチド薬物輸送のイネーブルプラットフォーム技術 である。バイオポリマーポリシアル酸(“PSA”)を使用して薬物の半減期を延長し、治療用ポリペプチドおよびタンパク質の安定性を潜在的に向上させる。PSA鎖の付着位置と長さはすべて分子の見かけ流体動力学半径を変化させることによって治療性 に影響することができ、これは逆に治療性の多くの生物学的特性を増強することができる。小分子薬にも使えます
我々の特許とノウハウbrを現在バイオテクノロジーや製薬業界と協力して開発されている候補薬剤に統合し,brが既存療法よりも薬理学的特性を有すると信じている次世代バイオ薬剤を創出する。我々の候補薬物は,我々の研究活動や我々の協力者の研究活動によって生成され,開発段階にある。したがって、私たちは、私たちの研究開発活動のために大量の資源を投入し続け、近い将来にこのように続ける予定です。これまで、私たちの候補薬は、米国や他のどの国や地域でも、どのような適用機関の規制マーケティング許可や承認を得ていませんでした。
我々は広範な特許の組合せを持っているにもかかわらず,2022年の内部努力の重点は,我々のDNaseプラットフォームの許可と進歩と我々のXCARTプラットフォーム技術の開発である.
私たちは2011年8月にネバダ州法に基づいて登録された。私たちは直接または間接的に私たちの完全子会社Hesperix S.A.(“Hesperix”)と Xenetic Biosciences(イギリス)を通じてXenetic UK(“Xenetic UK”)およびその完全子会社Xenetic UK、Lipoxen Technologies Limited(“Lipoxen”)、Xenetic Bioscience、InCorporation and SymBioTec、GmbH(“SymBioTec”)は、XCART、OncoHist、 Polyxen、EporeXenおよびImuxenを含む様々な米国連邦商標登録および出願、ならびに未登録商標およびサービスマークを有するが、これらに限定されない。
私たちの戦略
2022年4月、DNaseプラットフォームを承認しました。 DNaseプラットフォームは、Net(技術の説明については、“概要” および“我々の技術および候補薬”を参照)によって、免疫療法を含む既存の治療の結果を改善することを目的としています。われわれの現在の主な努力は,全身性DNA ase計画を臨床に進め,膵癌や他の局所末期あるいは転移性固形腫瘍の補助治療としてのbr}である。我々の目標は,反応を改善し,検査点阻害剤,化学療法,その他の標準看護治療に対する耐性を克服することにより,固形腫瘍の治療に解決策を提供することである。業界連携や潜在的な許可 を求めて,他の用途や適応のためのDNA aseを開発する予定である。
| 2 |
米国やヨーロッパでは孤児薬物指定と関連腫瘍学的適応の加速承認経路を適宜求める予定である。もし私たちの孤児腫瘍薬の候補薬物が孤児薬物の称号を与えられたら、私たちはある市場の排他性を含む孤児の地位のいくつかの重要な優勢から利益を得るかもしれない。
我々は,主に契約製造と契約研究組織(“CRO”)を用いて我々のDNase プラットフォームの開発を進め,我々の資源を効率的に管理する予定である.許可された候補薬物の持続的なパイプライン成長と進展は、私たちが十分な資本を調達し、私たちの既存の共同開発協力と戦略計画を推進し、新しいこのような手配を達成する能力にある程度依存する。
業務が発展する
排他的再許可協定
2022年4月26日,吾らはCLSと独占的なbr}再許可協定(“再許可協定”)を締結し,これにより,吾らはCLSが所有または制御するいくつかの 特許権およびノウハウ下の独占許可を取得し,br}デオキシリボヌクレアーゼを含む癌治療用医薬製品および方法(“再許可製品”)の開発および商業化を行った。再許可協定の条項によると、他の事項を除いて、私たち は独自に責任を負い、商業的に合理的な努力を尽くし、米国およびいくつかのヨーロッパ市場の再許可製品のマーケティング承認を研究、開発、獲得し、マーケティング承認を得た後、関連市場でこのような再許可製品 を商業化する。
CLSとEirGenとの間のいくつかの第三者契約 義務を履行するために、CLSからOPKOの完全子会社EirGen Pharma Ltd.(“EirGen”)に間接的に譲渡するのではなく、ライセンス契約に基づいて我々の許可および他の権利を付与する代償として、CLSに375,000株の普通株式(“分ライセンス契約株式”)を発行した。さらに、いくつかの臨床および規制マイルストーンを達成するために、潜在的マイルストーンとしてCLSに13,000,000ドルまでの現金を支払う義務があり、特定の規制マイルストーンの実現に基づいて950,000株の普通株式をCLSに追加発行することが義務付けられている。また,許可期間内のライセンス製品の純売上高(再ライセンス契約で定義されているような)には,中桁から下位2桁までの等級別印税を支払い,任意の分被許可者brから受け取った一定の対価格のミドルクラスパーセントシェアを支払うことが義務付けられている.
独占許可協定
2022年4月26日、CLSと独占ライセンス契約(“ライセンス契約”)を締結し、この合意に基づいて、DNaseをCAR T療法 と組み合わせた医薬品および方法(“ライセンス製品”)を開発および商業化するために、CLSが所有または制御するいくつかの特許 権利およびノウハウの独占的許可を得た。ライセンス契約の条項によると、私たちは独占的に責任を負い、他の事項を除いて、米国およびいくつかのヨーロッパ市場でのライセンス製品の販売許可を研究、開発、獲得し、販売許可を得た後、関連市場でこのようなライセンス製品を商業化する。
ライセンス契約が私たちの許可と他の権利を付与することを考慮して、私たちはCLSに500,000ドルの現金費用を一度に支払い、CLSに500,000株の私たちの普通株式brを発行し、潜在的なマイルストーンとして13,000,000ドルの現金を支払う義務があり、各ライセンス製品のいくつかの臨床的および規制マイルストーンを達成する。また,ライセンス範囲内のライセンス製品の純売上高に対して,印税期限内( ライセンス契約で定義されるように)に2桁から低い2桁までの階層印税を支払い,どの 分のライセンシーから得られた一定の対価の低い2桁にも一定の割合のシェアを支払うことが義務付けられている.
| 3 |
特許譲渡と意志協力
2022年10月4日,WilitionとCLSと連携した特許譲渡 を完了した。特許譲渡については,2022年10月12日にデラウェア州有限責任会社CLS Treeutics,LLCと引受協定を締結し,これにより,吾らはCLS LLCに普通株を発行することに同意したが,CLS LLCは850,000株の普通株を承認することに同意し,CLS とその関連方向としてCLSとその関連側が所有しているある特許権の対価を譲渡した。
2022年8月2日私たちはWilitionとの研究開発協力を発表し,癌の治療のためのNets標的細胞療法の開発を発表した。この協力は,WilitionのNu.Q 技術テストと我々のDNase−Armored CAR Tプラットフォームとの潜在的な組み合わせを評価し,多様な固形癌に対する特許に細胞療法 を用いることが可能な開発を目的とした早期探索的計画である。協力協定の条項によると、意志力は1つの研究プロジェクトに資金を提供し、双方は協力によって生じる任意の製品の商業化を共有することができるかもしれない
Catalent
2022年6月30日、Catalent Pharma Solutions LLC(“Catalent”)と作業説明書(以下、“SOW”と略す)を締結し、全体の作業範囲、スケジュール、価格を概説し、これにより、Catalentは、組換えタンパク質であるヒトデオキシリボヌクレアーゼIを製造するためのいくつかのサービスを提供する。
スクリップス研究会社は
2023年3月17日、私たちはスクリプス研究会社と研究援助とオプション協定(“合意”)を締結し、この協定によると、私たちはスクリプス研究会社に総額938,000ドルの資金を提供し、br社のDNase腫瘍プラットフォーム技術の臨床前開発に関連する研究を援助することに同意した。研究経費は、協定調印日に約78,000ドルの頭金を支払い、12ヶ月以内に月ごとに約78,000ドルを支払うことを規定した予算に基づいてスクリプス研究会社に月ごとに支払います。“合意”によれば、我々は、“合意”に規定されている研究計画の実施中に、我々の内部研究目的のためにのみ“TSRI技術”を作成し、使用するために、スクリプス研究会社の技術または特許権(“合意”で定義されるような)における権利のグローバル独占許可、および非独占的、印税免除、br}譲渡不可の許可を得る権利がある。
事前に終了しない限り、本プロトコルの有効期限は、本プロトコルの日から15(15)ヶ月継続する。本協定は私たちが30日前にScripps Researchに書面で通知することができます。発効日(プロトコルの定義)から6(6)ヶ月後から、あるいは私たちが合意に規定された満期金を適時に支払うことができなかった場合、Scripps Researchによって終了しますが、30日前に書面で通知して、このような の不払い状況を是正しなければなりません。他方が合意項目のいずれかの義務を履行できなかった場合、または他方が破産した場合、いずれか一方はさらに合意を終了することができる。
私たちの技術と薬物候補者は
技術
私たちは、私たちの特許とノウハウを多くの候補薬に統合しており、これらの薬剤は現在、私たちの内部または私たちのバイオテクノロジーや製薬パートナーと開発されています。私たちの目標は、私たちが次世代の生物学的薬物や療法であると考えられる薬剤を作ることです。我々は主に腫瘍薬の研究と開発に集中しているが,我々はパートナーが開発している他の疾患を治療する薬剤の所有権や他の経済的利益も持っている。
2022年12月31日までの1年間,我々の内部開発の重点はDNase腫瘍学プラットフォームの許可と発展,および我々のXCART 技術の開発である。私たちはPolyXenや私たちの他のどんな技術の開発にも積極的に取り組んでいない。
| 4 |
| DNA酵素 |
DNaseプラットフォームはNetに対して設計されており,Netはヒストンや他のタンパク質を被覆した細胞外クロマチンからなるメッシュ構造である.Netが活性化された好中球排出は,微生物や炎症促進挑戦に対応する。しかし、Netの過剰産生或いは減少は炎症と自己免疫病理の加重を招き、癌の成長と転移の情況下で腫瘍を促進する利基環境を産生する。
計画の要点: | |
| · | CLSと独占的許可と再許可合意を達成し,免疫療法を含めた既存治療の結果を改善するための介入性DNA酵素プラットフォームを開発した | |
| · | 人類研究の第一歩に向かって、2024-2025年を目標としています | |
| · | 最初の目標は、膵臓癌および他の局所進行または転移性固形腫瘍を含む数十億ドルの適応である全身性DNA酵素計画; | |
| · | DNase−装甲車T計画は臨床前の早期開発段階である。 | |
| XCART | XCART技術プラットフォームはその発起人によって設計され、1種の成熟したスクリーニング技術を利用して、単一患者の悪性腫瘍細胞表面の独特なBCRと特異的に結合するポリペプチドリガンドを識別することを目的としている。その後、ポリペプチドをCAR T細胞の抗原結合領域に挿入し、その後の形質導入/トランスフェクションプロセスは、患者のT細胞を腫瘍を攻撃するように誘導するCAR T形態に改造するために使用される。本質的に、XCARTスクリーニングプラットフォームは典型的なCAR Tスクリーニング方案とは逆に、CAR Tスクリーニング方案の中で、所与の標的に対して高特異性抗体ドメインのライブラリーをスクリーニングする。XCARTスクリーニングの場合,標的自体が抗体ドメインであるため,その性質は高い特異性を有する。XCART技術はリンパ腫を個性的に治療する可能性を創出し,抗原結合ドメインを有するCARを用いて,特定の患者のB細胞リンパ腫のユニークなBCRのみを認識して認識すべきである。XCARTの期待結果は限られた腫瘍外毒性、例えばB細胞再生不良性貧血である。我々は現在,我々のDNase技術プラットフォームに努力と資源を集中させるため,XCARTのさらなる開発を一時停止している |
| PolyXen | ポリシアリル化と呼ばれるプロセスによってPSAを薬物分子に化学的に付着させ、それによって人体内の薬物分子の循環時間を延長し、それによって潜在的に優れた次世代候補治療薬を創出することを目的とする、実現可能なバイオプラットフォーム技術。PSAはバイオポリマーであり、シアル酸分子鎖から構成され、人体の天然成分であり、著者らのPSAは細菌由来であるにもかかわらず |
研究、外部サービス、協力
パートナーの努力により,我々は我々のDNase,XCART,PolyXen特許技術に基づいて我々の次世代生物療法と新規腫瘍学薬brパイプラインを開発している。 我々の管理費用を効率的に管理しながらこれを行うために,契約メーカー,CROのサービスと我々の戦略協力 に依存している。私たちは現在このような計画を推進するための内部研究施設を持っていない。したがって、私たちの技術と候補薬物の持続的なパイプライン成長と進歩は、私たちと以下の方面の手配を含むいくつかの重要な協力と戦略的配置に依存する
| · |
Catalentは、生物製薬、細胞、遺伝子、消費者健康パートナーが多様な方法でより良い患者治療方法を開発、発売し、供給することができる世界的なリーディング企業を提供する
| |
| · | Pharmsynzは、当社の約2.9%の普通株式の実益所有者である完全子会社SynBio LLC(“SynBio”)を含む | |
| · | スクリップス研究所(“スクリプス研究所”)は、世界最大の民間非営利研究機関の一つである。 |
そこで、DNA酵素技術の開発を続けるほか、私たちの協力者が開発している他の疾患を治療するための候補薬にも興味があります。私たちはこれらの協力に基づいて記念碑的な支払いと印税の組み合わせを受け取るかもしれません。これらの薬剤の開発とマーケティングに成功することを前提としています。しかし、武田子の許可下での印税支払いや、Pharmsynzとの協力合意での潜在印税支払い を除いて、短期的にマイルストーンや印税支払いはないと予想されています。 については、より詳細な情報がありますので、次の“重要な協力と戦略手配”というタイトルの部分を読んでください。
| 5 |
私たちの候補薬物パイプラインは
我々の製品ラインには,内部と我々のバイオテクノロジーや製薬パートナーと開発されている候補薬br}が含まれている。次の表は,我々の現在の候補薬に関する重要な情報 :
エレpoXen
ErepoXen、またはポリシアル酸エリスロポエチン(“PSA-EPO”)、 は著者らのPolyXenプラットフォーム技術を用いて慢性腎臓病(“CKD”)患者の貧血を治療する。体内での治療薬の循環半減期を延長することで投与頻度を減少させることを目的としている。私たちはErepoXenの臨床開発を行っているのではなく、私たちの許可地域で候補薬物に許可外の機会を提供し続けている。
我々はPharmsynz や血清研究所と協力協定を結び,限られた市場でErepoXenを開発·発売し,合意に基づき,これらの努力で成功すれば印税 を徴収する。
| 6 |
Pharmsynzは規制部門の承認を得て,2020年にロシアでErepoXen(Epolongとも呼ばれる)の第2(B)/第3段階ヒト臨床試験を開始し,患者募集を完了する。2020年12月,Pharmsynzはこの臨床試験の陽性データを報告し,2021年2月にロシアの承認を得るために登録ファイルを提出することでEpolongの登録段階を開始したと報告している。Pharmsynzはそのプレスリリースで,ロシア段階の登録活動が2021年に完了し,早ければ2022年第1四半期に生産を開始できると報告している。Pharmsynzは、ファイルのいくつかの欠陥が指摘され、訂正後に登録を再提出しようとする返信を受信したことを通知しました。
血清研究所では95名の被験者でErepoXenのI期とII期臨床試験を行った。これらの安全性試験には重大な薬物関連有害事象はなく,brは第二段階反復投与を開始するデータを提供し,br人は薬物技術を用いて国際会議の調整を求めてオーストラリア,ニュージーランド,南アフリカで透析を受けていないCKD患者に対してErepoXenのコンプライアンス臨床試験を行った。私たちはこの研究の三つの列を完成させ、研究を終了した。
また,血清研究所はインドでセンター透析患者に対するErepoXenのI/II期臨床試験を完了した。血清研究所はPharmsynzの試験データと潜在的なロシアマーケティング許可を利用して、インドで行われた第三段階臨床試験の免除を要求する可能性があるが、現地の監督管理機関の許可を得る必要がある。
パイプ拡張の機会
われわれの許可の下で,われわれの協力者はその本土市場 内で運営することができ,広範な治療領域においてわれわれの技術に関する臨床前と臨床データを潜在的に生成することができる。このような合意に基づいて、私たちは主要市場のすべての権利を維持し、臨床データを共同で所有する。したがって、私たちは主要市場の開発と商業化決定過程でこれらのデータを利用する機会がある。
重要な協力と戦略的手配
武田さん
2017年10月、武田に当社のPolyXen技術に関するいくつかの特許の非排他的再許可の権利を武田に付与し、これらの特許はこれまで武田に独占的に許可されていた血液や出血疾患の治療に関連する製品であった。合意に基づき、武田(Br)(I)は2017年11月に750万ドル(7,500,000ドル)を一度に支払い、(Ii)は対象製品の全期間の純売上高に応じて1桁の印税を支払うことに同意した。純売上高の特許権使用料支払いは2019年末から です。会社は2022年と2021年12月31日までの年間で、それぞれ約170万ドルと120万ドルの特許権使用料を収入とし、ある保証製品の純売上高の1桁の特許権使用料に基づいている。
Catalent
2022年6月30日、Catalent とSOWを締結し、一般的な作業範囲、スケジュール、および定価を概説し、この合意に基づいて、Catalentは、組換えタンパク質ヒトDNA IのcGMP製造を実行するためのいくつかのサービスを提供する。双方は、SOWの予期される項目を管理する条項と条件を含み、Catalent標準条項および条件を含むSOW付録の代わりに、主サービス協定(MSA) を締結することに同意する。さらに、SOWとMSAに規定されている項目固有の条項と条件との間に何らかの衝突がある場合には、MSA条項および条件を基準とする。プロジェクト期限内に SOW計画の製造サービスプロジェクトの推定総コストは約500万ドル(いくつかの費用や潜在的な代替案を含まない)に達すると予想され,プロジェクトの各段階はこの 段階の開始に関連して個別に伝票を発行する.これまでに改訂や終了が行われていない限り、SOW計画の製造サービスは2024年上半期に完成することを目標としている。会社は30日前にCatalentに書面で通知した場合、いつでもSOWを終了することができます。SOWには機密性,保証,知的財産権,賠償などに関する慣行条項も含まれている.
| 7 |
スクリップス研究会社は
2020年5月15日、私たちはスクリプス研究会社と研究援助とオプション協定(“スクリプス協定”)を締結し、この合意に基づいて、スクリプス研究会社にXCART臨床前開発の推進に関する研究を支援するための合計300万ドルの資金を提供することに同意した。研究経費 は、スクリプス協定締結日に約300,000ドルの頭金を支払い、その後27ヶ月以内に四半期ごとに約300,000ドル を支払うことを規定している予算に基づいてスクリプス研究会社に四半期ごとに支払います。スクリプス協定によると、スクリプス研究会社は、スクリプス研究会社と2019年2月25日に締結された2019年3月1日に割り当てられた特定のライセンス契約の条項に基づいて、この分野内の任意の 特許権または技術(スクリプス協定で定義されているような)の許可を付与している。また、スクリプス協定が予期する研究計画の実行中に、社内研究目的のみでスクリプス研究技術(例えば、スクリプス協定の定義)を製造し、使用するために、br}スクリプス研究会社が私たちに許可されていない技術または特許権におけるグローバル独占許可、および非独占的、印税免除、br}譲渡不可の許可を得ることを選択することができる。2022年第2四半期に、双方はスクリプス協定の下での追加資金を終了することに共同で同意した。そのため、スクリプス研究会社は、以前に前払いした資金がなくなるまで、合意に基づいて仕事を続けることに同意した。
PJSC薬物合成
2009年11月、我々はPharmsynzと共同開発許可協定(“Pharmsynz手配”)を締結し、これにより、ある地域でのPolyXenおよびImuXen技術に基づく6種類の候補製品 の開発、商業化、マーケティングを許可する独占許可をPharmsynzに付与した。交換として,Pharmsynzは我々に独占的な許可を与え,Pharmsynz手配の範囲でPharmsynzによって開発された任意の臨床前および臨床データを使用し,ある地域以外で自費で候補薬物のさらなる研究,開発,商業化を行うことを許可した。
Pharmsynzはロシアの研究と臨床開発活動に資金を提供し、独自の研究と臨床開発を完全に担当している。Pharmynzプロトコルは、特許権使用料に加えて、マイルストーンまたは他の研究関連支払い を規定していない。Pharmynzスケジュールは、その中で規定された条項と条件に基づいて、終了するまで実行し続けるべきである。
2011年8月、Pharmsynzの完全子会社SynBioと株式引受 と協力開発協定(“共同開発協定”) を締結し、これによりSynBioに基づく技術およびロシアと独体でのPolyXen、OncoHistおよびImuXenプラットフォーム技術の分子 の開発、マーケティング、商業化のいくつかの候補薬の利用を許可し、ここでは総称してSynBio市場と呼ばれる。SynBio同などの許可権を付与した交換として,SynBioはSynBioによる任意の臨床前と臨床データの使用を許可し,共同開発プロトコルによりSynBio市場以外のどの地域でも協力して発生可能な商業候補製品の開発を許可してくれた.
Synbioはロシアでの研究と臨床開発活動を完全に援助し、展開している。共同開発協定によると、双方がそれぞれの技術に基づいてそれぞれの研究用品を提供する費用以外に、brに規定されているマイルストーンや他の研究に関する支払いはない。br}がこれらの費用を提供するのは、双方の便宜によるものであり、研究用品の継続的または恒常的な義務を提供することを意味するものではない。brは、任意の最終製品の商業化に成功した後、ある地域の販売で10%の特許使用料brを得る権利があり、共同開発協定条項によって制限されたある地域以外の販売の特許使用料をSynBioに支払う権利がある。2022年12月31日と2021年12月31日までの 年度には,共同開発プロトコルに関する供給サービス収入 は存在しない。共同開発プロトコルは,プロトコルに規定されている条項や条件によって終了するまで有効である. SynBioは2021年12月20日に共同開発プロトコルをその親会社Pharmynzに譲渡する.
付記4を参照重要な戦略的協力 はPharmsynzの米国における株式所有権である.
| 8 |
血清研究所
2011年8月、私たちは血清研究所と協力研究開発協定(“血清プロトコル”)を締結し、私たちのPolyXen技術を用いて潜在的な商業製品であるポリ唾液分析エリスロポエチン(“PSA-EPO”)の独占許可brを血清研究所に提供した。br}血清研究所はいくつかの所定の領域で規制許可を得るために必要なすべての臨床前と臨床試験を行い、費用は血清研究所が自費で行う。サミュエル研究所は私たちに印税を支払い、サミュエル研究所の販売エリア内のある顧客に純売上を行うために使われています。私たちが許可期間内に受け取った純売上については、血清研究所に特許使用料を支払います。協力計画によると、マイルストーンや研究に関連する他の支払い期限はありません。合意に規定された条項や条件によって終了するまで、血清プロトコルは有効になり続けています。2022年12月31日現在、私たちはいかなる商業製品も開発しておらず、この手配に関連する印税収入や費用も確認されていません。Serve 研究所が保有する株式は2022年12月31日現在、我々が発行した普通株式総数の1%未満である。
我々の知的財産権は
我々は、内部開発であっても、我々の協力者または他の第三者から許可を得たものであっても、我々の業務に重要なビジネス的意義を有するノウハウ、発明、および改善を保護し、強化するために努力している。私たちの政策は、私たちの独自の地位を保護することを求めています。その中には、アメリカとアメリカ以外の管轄地域での特許出願を含めて、私たちのノウハウ、発明、改善、候補製品をカバーしています。これらは、私たちの業務の発展と実施に非常に重要です。著者らはまた、私たちの独自技術と製品候補に関連する商業秘密と技術ノウハウ、持続的な革新と許可内の機会に依存して、腫瘍学領域における私たちの独自の地位を強化し、維持している。データ独占性,市場独占性,特許期間の延長(利用可能であれば)に依存することも計画されている。私たちのビジネス成功は、私たちの技術、発明、および改善のために特許および他の固有保護を取得し、維持する能力があるかどうか、私たちの商業秘密の機密性を保護すること、第三者が所有する知的財産権を使用するライセンスを取得して維持すること、私たちの将来可能な任意の特許を含む私たちの固有の権利を擁護し実行すること、および効果的かつ強制的に実行可能な特許および第三者の他の独自の権利を侵害することなく運営されるだろう。
我々の候補薬物は異なる開発段階にあり,各薬物は米国特許商標局(“USPTO”)の特許保護と米国特許商標局(“USPTO”)の未解決特許出願によって保護されており,いくつかの他の先進国にある。私たちの最初の特許は2021年に満期になり、私たちのPolyXenとOncoHist技術の既存特許の大部分は2025年から2030年の間に満了するだろう。
我々の特許戦略は、世界の主要な薬品市場又は薬品を生産する可能性のある場所を構成する司法管轄区域内に革新及び改善に関する特許出願を提出することである。私たちの主な特許の組み合わせについては、これらの司法管轄区域は、通常、米国、イギリス、オーストラリア、日本、カナダ、韓国、中国、インド、ロシア、EU(“EU”)のいくつかの他の国/地域を含むが、必ずしも各司法管轄区域で各特許家族のために特許出願を提出するとは限らないが、これらに限定されない。
2023年1月23日現在、私たちは、我々の完全子会社HesperixおよびXenetic UK、ならびにXenetic UKの完全子会社Lipoxen、XTIおよびSymBioTecを通じて、私たちの技術の様々な側面をカバーする170件以上の米国および国際特許および進行中の特許出願を、私たちの完全子会社HesperixおよびXenetic UK、ならびにXenetic UKの完全子会社Lipoxen、XTIおよびSymBioTecを通じて直接または間接的に所有している。この数字には、私たちが取得または出願した特許および特許出願が含まれており、私たちのDNaseおよびXCARTプラットフォーム技術の様々な態様に関連して、私たちの世界各地におけるすべての権利および特許出願、“癌個人化治療のための文章および方法”に関連する特許および特許出願、ならびに私たちのPolyXenプラットフォーム技術は、それぞれポリシアリル化 および先進ポリマー共役技術、ならびに私たちの他の候補製品をカバーする。より具体的には、我々の特許および特許出願は、癌治療、使用方法、ポリマー構造、医薬結合体、調製、ポリマーおよびポリマー結合体の製造方法、およびポリマー結合体の管理方法を含む。
| 9 |
我々はすでにいくつかの療法の特許保護を得ており,これらの療法は我々のPolyXen技術を用いて特定の療法をPSAに関連付けている。これらはPSA−EPO,PSA−インスリンおよびPSA−インスリン様蛋白,次世代第VIII因子候補蛋白SHP 656(PSA−rFVIII),PSA−DNase IおよびPSA−顆粒球コロニー刺激因子(PSA−GCSF)を含むがこれらに限定されない。より多くの特許は、PSAに連結されたタンパク質を製造する方法に関する。これらの方法特許は、高pH溶液中でPSAをタンパク質に結合させる特許と、シアル酸単位を開放および酸化することによってシアル酸アルデヒド誘導体を製造するプロセスを使用する特許とを含む。たとえば,N側にPSAリンクが可能な特許保護 がある.
我々はPSAの生産と精製過程でエンドトキシンの除去について特許保護を受けた。PSAに高pH 溶液を添加することと,多分散したイオン電荷多糖(例えばPSA)を異なる 平均分子量の画分に分離する過程によりエンドトキシンを除去する。これは、異なるイオン強度および一定のpHを有するカラムおよび溶出緩衝液を用いることにより実現され、相対分子質量多分散度が1.1以下の分級多糖を得る。
我々のDNase技術も特許保護されており,DNaseを用いた癌治療や癌治療に関連する副作用 を含む。DNA酵素は単独で使用可能であり,癌治療薬と併用することも可能である。この製品の組み合わせおよびXCART製品の組み合わせはまた、DNA酵素を添加するか否かにかかわらず、癌を治療するためのいくつかのタイプのCAR−T細胞の使用をカバーする。製品の組み合わせbrは、免疫チェックポイント阻害剤または調節剤と共に使用して癌を治療するために使用されるDNA酵素を有するか、または有さないCAR-T細胞の使用をさらに含む。
発行された特許は、特許出願の提出日、特許発行日、および特許が取得された国/地域における特許の法定期限に応じて異なるbr期限保護を提供することができる。一般に、米国で出願された出願に発行された特許は、最初の発効日から20年の排他的権利 を提供することができる。さらに、場合によっては、FDA承認された製品をカバーする発行された米国特許の期限は、FDA規制審査期間によって実際に失われた期間の一部を再取得するために延長されてもよく、これは、特許期限延長と呼ばれる。回復期は5年を超えてはならず,回復期を含む総特許期はFDA承認後14年を超えてはならない。米国国外特許の期限は外国司法管轄区の法律によって異なるが,通常は最初の有効出願日から20年である。しかしながら、 特許によって提供される実際の保護は、製品によって異なり、特許のタイプ、カバー範囲、規制に関連する延期の利用可能性、特定の国の合法的な救済措置の可用性、および特許の有効性および実行可能性を含む多くの要因に依存する。
場合によっては、1つ以上の特許によって保護された薬物 を使用する場合、私たちが技術を開発し、商業化する能力は、これらの特許薬物を取得することによって制限される可能性がある。私たちが特許薬を自由に使用できると信じていても、私たちは私たちが第三者の権利を侵害されたり、その薬の使用が禁止されたり、br損害賠償に責任があることが発見されないという保証はありません。このようなアクセス権限または損害賠償責任の制限は、私たちの業務、運営結果、および財務状況に実質的な悪影響を及ぼすだろう。
私たちのような製薬とバイオテクノロジー会社の特許地位は不確実であり、複雑な法律と事実の問題に関連している。発行された特許が法廷で効果的で強制的に施行されることを保証することはできない。有効かつ強制的に実行可能な特許を保持する場合であっても、このような判決の取得に関連する法的手続きは時間的かつ高価である。さらに、発行された特許は、反対または他のbr訴訟手続きの影響を受ける可能性があり、これは、特許が撤回されるか、または修正された形態で(特許が商業的関連性および/または広くカバーされない形態で)維持される可能性がある。さらに、私たちの競争相手は私たちの特許設計を迂回したり、他の方法で迂回することができるかもしれない。医薬品の開発および商業化が大きな遅延を受ける可能性があるため、発行され、強制的に特許を実行することができても、特許は早期に満了する可能性があり、私たちの特許がカバーする製品の商業化後、特許は短期的な保護のみを提供する可能性がある(あれば)。私たちは、特許損失および/または私たちの巨額のコストをもたらす可能性がある米国特許商標局が発表した介入手続きに参加しなければならないかもしれない。さらに、私たちのいかなる未解決特許出願も発行されていない場合、 または発行後に無効とみなされる場合、私たちは貴重な知的財産権保護を失う可能性があることを理解している。
| 10 |
第三者が所有する米国および外国特許権および他の独自の権利が存在し、医薬組成物および試薬、医療機器および装置、ならびに医薬組成物の製造、包装および送達方法に関する。私たちは、これらの権利のうちのどれが、これらの権利が存在する各司法管轄区域の当局を私たちの技術に関連していると見なすかを決定することができず、これらの権利のうちのどれが第三者によって主張されるか、または第三者によって主張される可能性があるかを決定することもできない。私たちはこのようなすべてのクレームに対して自分と私たちのパートナーを弁護する時に大きなコストを発生させるかもしれない。さらに、このようなクレームを出した当事者は、禁止または他の公平な救済を得ることができる可能性があり、これは、米国および他の国/地域で私たちの製品の一部または全部を開発または販売することを効果的に阻止し、巨額の損害賠償を得る可能性がある。権利侵害請求が発生した場合、私たちまたは私たちのパートナー は、第三者から1つまたは複数のライセンスを取得する必要がある可能性がある。私たちが必要とする任意の技術のライセンスを合理的な条項で得ることができる保証はありませんし、代替技術を開発または他の方法で獲得できる保証もありません。必要であれば、許可を得ることができないことは、私たちの業務、運営結果、財務状況に大きな悪影響を及ぼす可能性があります。 また、私たちは商業的に合理的に基づいて、私たちの候補薬物の開発に関する知的財産権の許可を得ることができないかもしれません。 があれば。
私たちの政策は、私たちの従業員とbrコンサルタント、外部科学協力者、賛助研究者、そして私たちから機密情報を取得した他のコンサルタントに、私たちとの雇用やコンサルティング関係を開始する際に秘密協定を実行することを要求します。これらのプロトコルは、個人が私たちとの関係中に開発または開示されるすべての機密情報を秘密にすべきであり、特定の場合でなければ、第三者に開示してはならないと規定している。協定は従業員たちによって構想されたすべての発明が私たちの財産でなければならないと規定している。しかしながら、これらのプロトコルが、そのような情報を無許可に使用または開示する場合に、私たちの商業秘密に意味のある保護または十分な救済措置を提供することは保証されない。
製造と供給
私たちは私たちの候補薬物開発計画を支援するために必要な私たち自身の材料を生産する能力がなく、この能力を私たちの現在の業務戦略の一部とするつもりもありません。我々は現在,Catalentと血清研究所と合意を締結しており,この合意に基づき,Catalentと血清研究所は臨床材料を生産し,それぞれ我々のDNaseとPolyXen技術を用いた候補薬の開発に用いられており,我々のパートナーが開発した候補薬を含めている。我々は我々のXCART技術を開発するための臨床材料を生産するための合意に達しておらず,必要であれば,我々の臨床供給需要を満たすための第三者メーカーを探す。
政府の監督管理
一般情報
その他の事項を除いて、アメリカ連邦、州と地方及びその他の国の政府当局は私たちが開発している製品の研究、開発、テスト、製造、品質管理、承認、ラベル、包装、貯蔵、記録保存、販売促進、広告、流通、マーケティングと輸出及びbr}輸入などの方面に対して広範な監督管理を行った。通常,新薬はNDAプロセスでFDAの承認を得なければならず,新しい生物製剤は生物製品許可証申請(BLA)プロセスでFDAの許可を得なければならず,その後米国で合法的に販売されることができる。
アメリカの監督管理機関
薬物開発の流れ
米国では,FDAは“連邦食品,薬物と化粧品法”(FDCA)に基づいて薬品を規制し,生物製品についても“公衆衛生サービス法”(PHSA)及びその実施条例に基づいて規制を行っている。規制の承認を得て、その後適切な連邦、州、地方、外国の法規と条例を遵守する過程には多くの時間と財力が必要だ。製品開発過程,承認過程または承認後の任意の時間に適用を遵守できなかった米国の要求 申請者は行政または司法制裁を受ける可能性がある.これらの制裁には、FDAによる承認保留申請の拒否、承認撤回、免許取り消し、臨床封印、警告状または無タイトル状、製品リコール、製品差し押さえ、br}の生産または流通の完全または部分的な一時停止、禁止、罰金、政府契約の拒否、原状回復、復帰、または民事または刑事罰が含まれる可能性がある。どんな機関や司法法執行行動も私たちに実質的な悪影響を及ぼすかもしれない。
| 11 |
FDAが1つの薬物または生物学的薬物が米国で発売される前に必要なプログラムは、一般に以下を含む:
| · | “良好な実験室規範”(“GLP”)の規定とその他の適用規定に基づいて臨床前実験室テスト、動物研究と調合研究を完成した | |
| · | ヒト臨床試験が開始される前に有効でなければならないINDをFDAに提出する | |
| · | “良好な臨床実践”(“GCP”)規定に基づいて、その期待される用途に使用される薬剤の安全性と有効性を決定するために、十分かつ制御されたヒト臨床試験を行う | |
| · | NDAやBLAをFDAに提出します | |
| · | 施設、方法、および薬物の特性、強度、品質、および純度を維持するのに十分な制御を保証するために、現行の良好な製造仕様(“cGMP”)要件に適合するかどうかを評価するために、医薬品を製造するための1つまたは複数の製造施設の検査を良好に完了させること; | |
| · | NDAまたはBLAに対するFDAの審査と承認。 |
薬物やバイオメーカーも承認された規制要求を受ける可能性がある。開発する候補薬が決定されると,臨床前試験段階 に入る。臨床前試験には、製品の化学、毒性、処方の実験室評価、および動物研究が含まれている。INDスポンサーは、臨床前試験の結果および生産情報と分析データをINDの一部としてFDA に提出しなければならない。スポンサーはまた、臨床試験の第1段階の目標、安全性を監視するためのパラメータ、および評価されるべき有効性基準を詳細に示すプロトコルを含むであろう(第1段階が治療効果評価に適している場合)。IND提出後も,いくつかの臨床前試験が継続される可能性がある。INDはFDAが30日以内に臨床試験を停止しない限り、FDAが受信してから30日後に自動的に発効する。この場合、INDスポンサーおよびFDAは、臨床試験が開始される前に未解決の問題を解決しなければならない。FDAはまた、進行中または提案されている臨床試験の安全懸念があること、またはFDAの特定の要求に適合していないことから、FDAがスポンサーに棚上げが解除されるまで、試験が開始または継続されない可能性があるため、臨床試験の前または期間の任意の時間に臨床保留を実施することもできる。
すべての臨床試験はGCP規定に符合する1人以上の合格研究者の監督の下で行わなければならない。それらは,評価のために,試験目標,投与手順,被験者の選択と排除基準,および安全·有効性基準を詳細に説明するプロトコルの下で行われなければならない。各シナリオはINDの一部としてFDAに提出されなければならず,何らかの深刻かつ予期しない有害事象が発生した場合,直ちにFDAに安全報告を提出しなければならない。臨床試験に参加する各機関の機関審査委員会(IRB)(場合によっては独立したIRB)は、その機関の臨床試験が開始される前に、各案を審査して承認しなければならない。審査の一部として、IRBはまた、試験に関する情報および各試験対象またはその法律代表に提供されなければならない同意書を承認し、研究が完了するまで監視しなければならず、そうでなければIRBの規定を遵守しなければならない。
人体臨床試験は通常3つの連続段階に分けて行われ、これらの段階は重複或いは合併する可能性がある:
| · | 第一段階:この候補薬物は最初に健康な人体に導入され、安全性、用量耐性、吸収、代謝、分布、および排泄テストを行った。いくつかの深刻または生命に危害を及ぼす疾患(例えば癌)を治療する製品の中で、特に製品がその固有の毒性のために道徳的に健康ボランティアに服用できない可能性がある場合、最初の人体試験は通常患者で行われる。 | |
| · | 第二段階:この段階は限られた患者集団で臨床試験を行い、可能な副作用と安全リスクを決定し、特定の目標疾患に対するこの製品の治療効果を初歩的に評価し、用量耐性と適切な用量を決定することに関連する。 | |
| · | 第3段階:臨床試験は地理分散の臨床研究地点で患者群を拡大し、投与量、臨床治療効果と安全性を更に評価するためである。これらの臨床試験は、候補薬物の全体的なリスク-収益比を決定し、適切な場合に製品ラベルに十分な基礎を提供することを目的としている。 |
| 12 |
承認後の実験は, 第4段階研究と呼ばれることがあり,最初の市場承認後に行われる可能性がある.これらの試験は,期待される治療適応患者の治療からより多くの経験を得るために用いられている。場合によっては、FDAは、NDAまたはBLAを承認する条件として、第4段階臨床試験を強制的に実行することができる。
FDAまたはスポンサーは、研究対象が受け入れられない健康リスクに直面していることを発見することを含む様々な理由で臨床試験を一時停止することができる。br}同様に、臨床試験がIRBの要求に従って行われていない場合、または薬物が患者の意外な深刻な傷害に関連している場合、IRBはその機関の臨床試験の承認を一時停止または終了することができる。また、いくつかの臨床試験はスポンサー組織の独立した合格専門家グループによって監督され、このグループはデータ安全モニタリング 委員会と呼ばれる。その規約によると、当該グループは、試験のあるデータへのアクセスに基づいて、試験が指定されたチェックポイントで行えるか否かを決定することができる。
臨床試験と同時に,スポンサーは薬物化学や物理特性に関するより多くの情報を開発し,cGMP要求に基づいて商業量産製品のプロセスを決定しなければならない。製造プロセスは品質が安定した候補薬物ロットを持続的に生産できる必要があり,また,メーカーは最終薬物の特性,強度,品質,純度を試験する方法を開発しなければならない。また,適切な包装を選択·試験し,候補薬物が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
INDが活動状態にあり、承認を得る前に、スポンサーは、前回の進展報告以来行われた臨床試験および非臨床研究結果をまとめた進展報告を少なくとも毎年FDAに提出しなければならず、深刻かつ意外な副作用を含むIND安全報告をFDAに提出しなければならない。他の研究結果は、同じまたは類似の薬物に曝露された人体に重大なリスクがあることを示し、動物または体外試験結果は、ヒトに重大なリスクがあることを示し、方案または研究者マニュアルに記載された結果と比較して、任意の臨床上重要な深刻な疑わしい副作用の発生率が増加する。
また,行っている臨床試験と完成した試験結果を公的登録機関に報告することが求められている。FDA規制製品のいくつかの臨床試験のスポンサーは、www.Clinicaltrials.gov上で公開して得ることができる特定の臨床試験情報を登録して開示しなければならない。製品,患者群,調査段階,試験地点と調査者および臨床試験の他に関する情報 はその後登録の一部として公開されている。スポンサーは完成後に臨床試験結果を検討する義務があります。これらの試験の結果は、研究中の新製品や新適応が承認されてから発表されるまで延期することができます。
アメリカ市場の承認プロセス
製品開発、臨床前研究とその他の非臨床研究と臨床試験の結果、及び製造技術の記述、薬物化学成分の分析テスト、提案のラベルとその他の関連情報は、NDA或いはBLAの一部としてFDAに提出し、 にこの製品の発売許可を要求する。セキュリティプロトコルまたはBLAの提出には使用料を支払う必要があり、いくつかの限られた場合には、そのような費用の免除を得ることができる。FDAは、報告を受ける前に実質的な審査が十分に完全であることを確実にするために、提出されたすべてのNDAおよびBLAを審査する。FDAは、NDAやBLA を受け入れるのではなく、より多くの情報を提供することを要求するかもしれない。この場合,秘密保持プロトコルやBLAおよび付加情報を再提出しなければならない.再提出された申請はFDAが届出を受ける前にも 審査を経なければならない。
提出された申請が受け入れられると、FDAは深い実質的な審査を開始する。FDAは、NDAまたはBLAを諮問委員会に提出して、審査、評価、および提案 が申請を承認すべきかどうか、およびどのような条件下で承認すべきかを提案することができる。FDAは諮問委員会の提案によって制限されていないが、それは一般的にそのような提案に従っている。承認プロセスは長く、しばしば困難であり、適用される規制基準に適合しない場合、または追加の臨床または他のデータおよびbr情報が必要とされる可能性がある場合、FDAはNDAまたはBLAの承認を拒否する可能性がある。このようなデータや情報を提出しても,FDAはNDAやBLAが承認基準 を満たしていないことを最終的に決定する可能性がある.FDAは機密協定を審査し、他の事項に加えて、製品がその期待用途に対して安全かつ有効であるかどうか、およびその製造がcGMPに適合するかどうかを決定し、製品の特性、強度、品質、および純度を確保し、維持する。FDAは、製品が安全で、純粋かつ有効であるかどうか、および製品の製造、加工、包装、または製品を保持する施設が、製品の持続的な安全、純度および効力を保証するための基準に適合しているかどうかを決定するためにBLAを審査する。FDAは、セキュリティプロトコルまたはBLAを承認する前に、製品を製造する1つまたは複数の施設を検査する。
| 13 |
FDAはセキュリティプロトコルやBLAを評価した後, を承認手紙または完全な返信を発行する.この薬剤の商業マーケティングを承認し、特定の適応の処方情報を提供する。完全な返信は、申請の審査期間が終了し、現在の申請が承認されないことを示している。完全な返信は、一般に、FDAによって決定されたNDAまたはBLAにおける特定の欠陥を記述し、追加のキーIII期試験または臨床試験、非臨床研究または生産に関連する他の重要かつ時間の要件などの追加の臨床データを必要とする可能性がある。完全な返信が発行された場合、スポンサーは、手紙で決定されたすべての不足点を解決するために、または申請を撤回するために、セキュリティプロトコルまたはBLAを再提出しなければならない。このようなデータや情報を提出しても,FDAはNDAやBLAが承認基準を満たしていないと判定する可能性がある.
製品が規制部門の承認を受けた場合、br承認は特定の疾患および用量に限定される可能性があり、または使用適応が制限される可能性があり、これは製品の商業的価値を制限する可能性がある。さらに、FDAは、NDAまたはBLAの承認後に薬物の安全性および有効性をさらに評価することを目的とした臨床試験を含む第4段階試験をスポンサーに要求することができ、商業化された承認製品の安全性を監視するための試験および監視計画を要求する可能性がある。FDAはまた、薬物の安全な使用を確保するために、リスク評価および緩和戦略(“REMS”)を要求することを含む、承認時に他の条件を追加することができる。FDAがREMSが必要であると結論した場合,NDAまたはBLAのスポンサーは提案したREMSを提出しなければならない。必要に応じて、FDAは、承認されたREMSなしにNDAまたはBLAを承認しない。REMSは、制限された配布方法、患者登録、および他のリスク最小化ツールのような薬物ガイドライン、医師のコミュニケーション計画、または安全な使用を保証する要素を含むことができる。承認またはマーケティングに関するこれらの制限は、製品の商業普及、流通、処方、または配布を制限する可能性がある。 が法規要件に適合していない場合や初期マーケティング後に問題が発生した場合、マーケティング承認を撤回する可能性があります。
“孤児薬物法案”
孤児医薬品法は、孤児の薬物指定を申請する際に米国で200,000人未満のまれな疾患または状態に影響を与えるか、または米国の患者数が200,000人を超え、合理的な予想がなく、米国の販売から回収されると予想されていない薬物または生物製品を開発および販売するために、製造業者brにインセンティブを提供する。FDAマーケティングの許可を得た最初の孤児薬物開発者は、米国での7年間の独占販売期間を得る権利がある。しかしながら、FDAは、他の承認された孤児薬よりも臨床的に優れているか、または異なる薬剤は、同じ適応であっても、7年間の独占営業期間内に米国で承認される可能性があると考えている。さらに、孤児薬物の独占所有者は、患者の需要を満たすために十分な数の孤児薬物が使用可能であることを確保しなければならない。そうしなければ、この薬の市場独占経営権が撤回される可能性がある。
小児科情報
2007年の“小児科研究平等法”(“PREA”)によれば、NDAまたはBLASまたはNDAまたはBLASのサプリメントは、すべての関連する小児科亜群において主張される適応の安全性および有効性を評価し、その安全かつ有効な各小児科亜群に対する薬剤の用量および投与をサポートするためのデータを含まなければならない。FDAはデータの提出を延期することを許可するか、またはすべてまたは部分的な免除を与えることができる。規制が別途要求されない限り、PREAは、孤児薬として指定された適応が承認されたいかなる薬物にも適用されない。“児童最優秀薬品法”(BPCA)は、NDAのスポンサーに追加の6ヶ月の市場独占期間 を提供し、スポンサーがBPCAによって特別に要求された小児科研究結果を所定の時間内に提出することを条件とする。“生物製品価格競争と革新法案”は、BPCAが規定する条件を満たしていれば、BLASのスポンサーは、BPCAが規定する活性部分を含むすべての未満期の非特許市場独占経営権を得るために、6ヶ月間追加的に延長することができると規定している。
| 14 |
2012年7月9日に法律となった“食品·薬物管理局安全·革新法案”(FDASIA)に署名してFDCAを改正した。FDASIAは、新しい活性成分、新適応、新剤形、新投与レジメンまたは新投与経路を含む医薬または生物製品のマーケティング申請を計画することを要求するスポンサーは、第2段階会議終了後60日以内に、またはスポンサーとFDAとの間で合意した場合に、予備小児科研究計画(PSP)を提出しなければならない。初期PSPは、研究目標および設計、年齢グループ、関連終点および統計方法、またはそのような詳細な情報を含まない理由、および小児科研究データおよび支援情報の提供の要件を遅延または完全または部分的に免除することを含む、スポンサー計画によって実施される1つまたは複数の小児科研究の概要を含まなければならない。FDAとスポンサーはPSPについて合意しなければならない 。非臨床研究,早期臨床試験および/または他の臨床開発計画から収集したデータに基づいて小児科計画の変更を考慮する必要があれば,スポンサーは合意した初期PSPに対する修正案を随時提出することができる。
開発と審査計画を加速する
FDAは特定の標準に符合する新薬と生物製品の審査過程を加速或いは促進することを目的とした迅速なチャンネル計画を持っている。具体的には,新薬や生物製品が重篤あるいは生命に危険な疾患の治療を目的とし,その疾患が満たされていない医療需要を解決する潜在力を示す場合,迅速チャネル指定を受ける資格がある。高速チャネル指定は 製品と検討中の特定の適応の組合せに適している.新薬または生物学的製剤のスポンサーは、製品の臨床開発中の任意の時間に、FDAが医薬または生物学的製剤を迅速チャネル製品として指定することを要求することができる。高速チャネル指定製品の場合、FDAは、完全な出願を提出する前に、マーケティング申請の部分を検討することをスクロールして考慮することができ、 スポンサーが出願部分のスケジュールを提供した場合、FDAは、申請の一部 を受け入れることに同意し、スケジュールが許容可能であると判断し、スポンサーは、申請の第1の部分を提出する際に任意の必要な使用料を支払う。
迅速チャネル計画を含むFDA発売製品を提出する任意のものは、開発および審査を加速するために設計されたFDAの他のタイプの計画の資格、例えばbr}優先審査および承認の加速を目的としている可能性がある。迅速な指定、優先審査、承認の加速は承認基準 を変更することはありませんが、開発や承認の流れが加速する可能性があります。満足できる代替療法がない場合,あるいは市販製品と比較して治療,診断または予防疾患が有意に改善された場合には,いずれの製品も安全で有効な治療を提供することが可能であり,優先審査の資格がある。FDAは、審査を促進するために、指定された優先審査の新薬または生物製品の出願を評価するために追加のリソースを使用しようと試みるであろう。また, 製品は加速承認を得る資格がある可能性がある.深刻または生命を脅かす疾患の治療における研究された薬物または生物製品の安全性および有効性、ならびに既存の治療よりも意味のある治療効果を提供する薬物または生物製品は、十分かつ制御された臨床試験から製品br}が臨床的利益を合理的に予測する可能性のある代替終点に有効であるか、または臨床 終点への影響(生存または不可逆的な発症率ではなく)に基づいて承認される可能性があることを意味する。承認の一つの条件として,FDAは承認を加速させた薬物や生物製品のスポンサーに十分かつ良好に制御された発売後臨床試験を要求することができる。また,FDAは現在 が承認を加速する条件として販促材料の早期承認を要求しており,製品の商業発表時間に悪影響を与える可能性がある.FDAが、有効であることが証明された薬物は、流通または使用が制限された場合にのみ安全に使用できると結論した場合、薬物の安全な使用を確保するために、必要と考えられる発売後制限を実施することが要求されるであろう。例えば、(I)特定の機関または特殊な訓練または経験を有する医師に限定された流通、または(Ii)特定の医療プログラムを実行するための条件での流通。
FDASIAは新しい薬物と生物製品のカテゴリーを確立し、“突破的療法”と呼ばれ、突破的な治療法の称号を得る資格がある可能性がある。スポンサーbrは、深刻なまたは生命に危険な疾患または状態の治療のために単独または1つまたは複数の他の製品と組み合わせて使用されることが意図されていることを前提として、薬物または生物学的候補薬物を“突破的療法”として指定することをFDAに求めることができ、初歩的な臨床証拠 は、製品が1つまたは複数の臨床的に重要な終点(例えば、臨床開発早期に観察される顕著な治療効果)において既存の治療法よりも著しく改善される可能性があることを示す。この指定には,Fast Track計画のすべての機能, およびより密なFDAインタラクションと指導が含まれる.画期的な治療指定は加速承認や優先審査とは異なり,関連基準を満たしていれば,同一の薬物優先審査を付与することも可能である。1つの製品が画期的な療法として指定された場合、FDAはこのような薬物の開発と審査を加速させるだろう。すべての画期的な治療指定の出願は、受信後60日以内に検討され、FDAは、この要求を承認または拒否するであろう。
| 15 |
2016年に公布された“21世紀治療法”は,細胞や遺伝子療法を含む再生医学製品のための新たな迅速承認計画を構築した。再生医学高度療法(“RMAT”)計画は再生医学 療法の開発と審査を促進し、病状の深刻な患者が満足されていない医療需要を満たすための迅速な審査計画を構築した。研究薬物が再生医学療法(例えば細胞療法或いは遺伝子療法)の定義に符合する場合、(2)深刻な疾病を治療、修正、逆転或いは治癒することを目的としている;及び(3)初歩的な臨床証拠により、再生医学療法はこのような疾病が満たされていない医療需要を解決する可能性があることを表明し、rmat 指定を得る資格がある。RMAT指定の利点は、FDAとの早期相互作用を含む迅速チャネルおよび画期的な治療指定計画のすべての利点を含む。
承認後に要求する
承認されると、規制要求や基準を守っていない場合、あるいは製品発売後に問題が発生した場合、FDAは承認を撤回する可能性があります。 後にある製品に以前未知の問題があることが発見されれば,その製品が制限され,さらには完全に市場から撤退する可能性がある.承認後、承認された製品のいくつかのタイプの変更、例えば、新しい適応、いくつかのbr}生産変更、および追加のラベル宣言を追加するなど、FDAのさらなる審査および承認を受ける必要があります。薬品と生物製品メーカー 及び生産と流通許可に参与する薬品と生物製品の他の実体はFDAとある州機関にその機関を登録し、FDAとある州機関の定期的な抜き打ち検査を受けて、cGMP法規と他の法律法規を遵守することを保証しなければならない。
米国特許期限は市場との排他性を回復する
生物製品価格競争と革新法案、あるいはBPCIAは、公衆衛生サービス法を改正し、FDAに類似のバージョンの革新生物製品を承認することを許可し、一般に生物模倣薬と呼ばれる。生物類似体の承認を求める競争相手は、その分子が承認されたイノベーター生物と高度に類似していること、および他の要件を決定するために、申請を提出しなければならない。しかし、BPCIAはFDAが革新者生物製品が初歩的なマーケティング許可を得てから12年以内に同社のデータに基づいて生物類似申請を許可することを禁止した。もしFDAが革新者会社にこの製品に対する小児科臨床研究を要求すれば、この12年のデータ独占期間は6ケ月、合計12年半延長できる。
FDAが私たちの候補薬物の期間、持続時間、および具体的な状況を承認したことによると、私たちのいくつかの米国特許は、1984年の“薬品価格競争および特許期限回復法”(一般に“ハッジ-ワックスマン修正案”と呼ばれる)に基づいて限られた特許期間の延長を受ける資格がある可能性がある。Hatch−Waxman修正案 は,製品開発やFDA規制審査の過程で失われた特許期間の補償として,最長5年間の特許回復期間を許可する。しかし,特許期間の延長は,特許の残存期間を製品承認日から合計14年を超えることはできない.特許期間延長期間は,一般にIND発効日から秘密協定またはBLA提出日までの時間の半分であり,NDAまたはBLA提出日からその出願承認までの時間を加えると,最長5年延長できる。承認された薬物に適用される特許は1つのみ延期する資格があり,延期出願は特許満了前に提出しなければならない。米国特許商標局はFDAと協議し,任意の特許期間の延長または回復の出願を審査·承認する。将来、私たちは、臨床試験の期待長および関連する秘密協定またはBLAの提出に関連する他の要因に基づいて、合理的に取得された場合に、現在の満期日後まで特許寿命を延長するために、現在所有または許可されている特許の特許期間 を延長する予定である。
| 16 |
FDCA 中のマーケティング排他的条項はまた、いくつかのマーケティング申請の提出または承認を遅延させる可能性がある。FDCAは新しい化学実体秘密協定の承認を得た最初の申請者に5年間の非特許マーケティングの米国での独占経営権を提供した。FDAが以前に同じ活性部分を含む任意の他の新薬を承認していない場合、薬物は新しい化学物質 であり、この活性部分はその薬物物質の作用を担当する分子またはイオンである。排他期間内に、FDAは、同じ活性部分に基づいて別の薬剤のために提出された簡略化された新薬出願 (ANDA)または505(B)(2)NDAを受け入れない可能性があり、その薬剤 が元の革新薬と同じ適応であるか、または別の適応のためのものであるかにかかわらず、出願人が許可を参照するために必要なすべてのデータの合法的な権利を所有していない場合、または許可するために必要なすべてのデータの合法的な権利を有さない。しかしながら、出願がイノベーターNDA所有者がFDAに記載された特許のうちの1つを含む特許が無効または未侵害証明である場合、4年後に出願することができる。FDAが、出願人が行っているまたは後援する新しい臨床研究(バイオアベイラビリティ研究を除く)がbr}出願の承認に重要である(例えば、既存の薬剤の新しい適応、用量または強度)と考えている場合、FDCA はまた、NDAまたは既存のNDAの補充物に3年間の市場排他性を提供する。この3年間の専門権は,この薬物が新たな臨床研究に基づいて承認された修正 のみをカバーしており,原適応や使用条件の活性成分を含む薬物のANDA をFDAが承認することは禁止されていない。5年と3年の排他性は完全な秘密協定の提出や承認を延期したり承認したりしないだろう。しかしながら、完全なセキュリティプロトコルを提出する申請者は、安全性および有効性を証明するために、すべての臨床前研究および十分かつ良好に制御された臨床試験を参照する権利を行うか、または得ることを要求されるであろう。
小児科専門権はアメリカBPCAが規定したもう一つの規制市場専門権である。もしスポンサーが上記の“小児科情報”の節で述べたように児童に臨床試験を行う場合、小児科専門権は追加6ケ月の市場独占経営権を提供する。さらに、上述したように、孤児薬物専門権は、7年間の市場専門期間を提供する可能性があるが、場合によっては除外される。
外国監督管理
アメリカの法規以外に、私たちは他の司法管轄区の各種法規を遵守し、その中には臨床試験と任意の商業販売 及び私たちの候補薬物の流通を含む。
私たちの候補薬がFDAの承認を得ているかどうかにかかわらず、私たちはこれらの国/地域でbr臨床試験を開始したり、発売される前に外国の規制機関の必要な承認を得なければならない。米国以外のある国/地域にも類似した流れがあり,すなわち はヒト臨床試験開始前に臨床試験申請(“CTA”)の提出を要求しており,INDと非常に類似している。例えば、EUでは、CTAはFDAとIRBのような各国の国家衛生当局と独立した倫理委員会に提出されなければならない。CTAが一国の要求に応じて承認されると,臨床研究開発を継続することができる。
臨床試験、製品審査と許可、定価と精算を管理する要求と流れは国/地域によって異なる。いずれの場合も,臨床試験はGCPおよび“ヘルシンキ宣言”からの適用法規要求と倫理原則に従って行われた。
EUの監督管理システムの下で規制機関が研究しているbr薬物或いは生物製品の許可を得るためには、私たちは上場許可申請を提出しなければならない。米国における機密協定またはBLAの提出のための出願 は、EU要求の出願と同様であるが、国/地域固有の文書要件が含まれている点で異なる。連合はまた市場排他性に機会を提供する。例えば、欧州連合では、マーケティング許可を得た後、新しい化学物質は、通常、8年間のデータ独占権および追加の2年間の市場独占権を取得する。承認されれば、データ排他性は、EU規制機関がイノベーターのデータを参照して汎用アプリケーションを評価することを阻止する。追加の2年間の市場独占期間内に、汎用マーケティング許可を提出することができ、革新者のデータを参照することができるが、市場独占権が満了するまで、いかなる模造製品も販売することはできない。しかし,製品がEU規制機関 によって新たな化学実体とみなされることは保証されず,製品はデータ独占を得る資格がない可能性がある。EUで孤児の称号を獲得した製品は10年間の市場排他性を獲得することができ、その間、同じ適応の類似医薬製品は市場に投入されてはならない。孤児製品はまた、EUで追加2年間の小児科研究市場排他性を得ることができる。いかなる補充保護証明書も孤児適応の小児科研究に基づいて延期することはできない。
| 17 |
EUが“孤児薬品”を指定する基準は原則として米国と類似している。(EC)141/2000号条例第3条によると、(1)1つの薬品が生命または慢性衰弱に危害を及ぼす疾患の診断、予防または治療に使用されていれば、その薬品を孤児に指定することができる。(2)(A)申請時に、この場合、EU 10,000人当たり5人以下の影響を与えるか、または(B)孤児身分の利点がなければ、製品は、投資が合理的であることを証明するためにEUで十分なリターンを生じないこと、および(3)満足できる診断、予防または治療の方法がEUで販売されていないこと、またはそのような方法が存在する場合、製品は、(EC)847/2000条例で定義されているように、このような状況に影響を受ける人に大きなメリットを有するであろう。孤児医薬製品は減費や費用減免などの財政奨励を受ける資格があり、マーケティング許可を得た後、許可された治療適応の10年間市場独占経営権を得る権利がある。孤児薬品指定申請は発売申請前に提出しなければなりません。 孤児薬物指定が承認された場合、出願人は、上場許可申請の費用減免を受けるが、上場許可を提出したときにその指定が待っている場合は、そうではない。孤児薬物指定は監督管理審査と審査過程中にいかなる優勢を伝達することもなく、監督管理審査過程の持続時間を短縮することもない。
5年目の終了時に製品が孤児指定の基準に適合しないと判断された場合、例えば、製品の利益が十分に高く、市場独占性を維持するのが合理的であることを証明するのに十分でない場合、10年間の市場独占権は6年間減少する可能性がある。また、以下の場合、同じ適応の類似製品に対してマーケティング許可を随時付与することができる
| · | 第2の出願人は、その製品は類似しているが、より安全で、より効果的であり、または臨床的に優れていることを証明することができる | |
| · | 出願人は,孤児の薬品の第2次出願に同意する | |
| · | 申請者たちは十分な孤児薬を提供することができない。 |
EU以外の他の国/地域、例えば東欧、ラテンアメリカまたはアジアの国/地域では、臨床研究、製品許可または承認、定価、精算を行う要求は国/地域によって異なる。繰り返しますが,すべての場合,臨床研究はGCPおよび“ヘルシンキ宣言”からの適用法規要求と倫理原則に基づいて行われています。
もし私たちが適用された外国監督管理要求を遵守できなかった場合、私たちは罰金、規制承認の一時停止または撤回、製品のリコール、製品の差し押さえ、経営制限、刑事起訴などの処罰を受ける可能性がある。
その他の規制事項
製品承認後の製造、販売、普及とその他の活動はFDAの監督を受ける以外に、アメリカの医療保険と医療補助サービスセンター、衛生と公衆サービス部の他の部門、薬品法執行管理局、消費財安全委員会、連邦貿易委員会、職業安全と健康管理局、環境保護局及び州と地方政府を含む多くの監督管理機関の監督を受ける可能性がある。米国では、販売、マーケティング、科学/教育プロジェクトはまた、州および連邦の反リベート、虚偽声明、データプライバシー、および安全および医師支払い透明性法律を含む州および連邦の詐欺および乱用法律を遵守しなければならない。定価と返却計画は,1990年の“米国総合予算調節法”の連邦医療計画(例えば,医療補助)の帰点要求と,2010年に“医療·教育調節法”で改正された“患者保護法案”と“平価医療法案”の最新の要求に適合しなければならず,総称して“平価医療法案”,“br}および2022年の”インフレ低減法案“と呼ばれる。総務省連邦供給スケジュールの許可されたユーザに製品を提供する場合は、他の法律および要求が適用される。いかなる制御物質の処理も米国の“制御物質法”と“制御物質輸出法”を遵守しなければならない。製品はアメリカの“毒物防止包装法”に適用される児童保護包装要求に適合しなければならない。製造、販売、販売促進、および他の活動は、連邦および州消費者保護および不正競争法によって制限される可能性もある。
| 18 |
医薬製品の流通は他の要求と法規の制約を受け、許可されていない医薬製品の広範な記録保存、許可、貯蔵と安全要求を防止することを含む。
規制要求を遵守できなかったことは、私たちを可能な法律や規制行動に直面させる可能性がある。具体的な状況によると、適用される法規要件を満たすことができないことは、刑事起訴、罰金または他の処罰、禁止、リコールまたは製品の差し押さえ、生産の完全または部分的な一時停止、製品の承認の拒否または撤回、または政府契約 を含む会社の供給契約の締結を許可することを引き起こす可能性がある。さらに、1つの会社がFDAおよび他の要求を遵守していても、製品の安全性または有効性に関する新しい情報は、FDAが製品承認を修正または撤回することをもたらす可能性がある。私たちが販売している未来の製品 の販売を禁止または制限または撤回することは、不利な方法で私たちの業務に大きな影響を与える可能性があります。
規制の変更または既存の規制の解釈は、(I)私たちの製造スケジュールの変更、 (Ii)製品ラベルの追加または修正、(Iii)私たちの製品のリコールまたは生産停止、または(Iv)追加の記録保存 要件のような、私たちの将来の業務に影響を与える可能性があります。このような変化を強制的に実施すれば、私たちの業務運営に悪影響を及ぼす可能性がある。
精算する
国内及び海外市場では、任意の承認された製品の販売及び精算は、政府医療計画、商業保険、信託医療機関などの第三者支払人がこのような製品コストを支払う程度にある程度依存する。これらの第三者決済者は、医療製品およびサービスの価格に挑戦し、コストを管理するための制御を実施することが増えている。医療コストの抑制は連邦と州政府の優先事項となっており、薬品価格はずっとこの努力の重点である。各国政府はコスト制御計画の実施に大きな興味を示し、価格制御、精算に対する制限及び非特許製品に対する代替要求を含む。例えば、アメリカでは、議会は最近数回の調査を行い、連邦と州立法を提出し、公布し、薬品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、連邦医療保険下の薬品のコストを下げ、政府計画薬品精算方法を改革する。また、2018年5月、トランプ政権は、メーカーの競争を増加させ、ある連邦医療計画の交渉能力を増加させること、メーカーにその製品の価格を下げるよう奨励すること、消費者が支払う薬品の自己負担コストを低減することを奨励する追加提案を含む薬品価格の低減と薬品自己負担コストの低減の“青写真”を策定した。バイデン政府はこの努力を続けなかったが、それは他の行動を取る権利がある。2020年12月、トランプ政権は臨時最終規則を発表し、重点的に薬品価格を下げることを試み、カナダからある薬品の輸入を許可し、連邦医療保険B部分に基づいてある薬品カテゴリーに対して最恵国定価 を行い、アメリカ連邦反バックル法規を改正することによって連邦医療保険D部分の薬品リベート計画を修正した。B部分最恵国ルールは2021年1月4日に連邦裁判官に発効を阻止され,このルールはあまりにも性急であり,“行政手続き法”の要求に基づいて公衆に時間を提供しなかったとコメントされている.そして,2021年12月29日にCMSは最終ルール を発表し,最恵国ルールを正式に廃止した。未解決の訴訟ではMedicare Part D薬品 リベート計画とリベート法規の変更を見送ることが求められている。2021年1月30日、コロンビア特区地裁は双方のbr規定請求を承認し、D部分帰点ルールの発効日を2023年1月1日に延期した。2022年8月7日、連邦医療保険Dの一部の薬品還付計画と米国連邦反リベート法規の改正を2032年1月に延期する“2022年インフレ削減法案”が国会で可決された。
また,2022年のインフレ低減法案は,処方薬をカバーする既存の医療保険計画に影響を与える可能性がある。他の関連条項を除いて、2022年の“インフレ低減法案”は、連邦医療保険計画がある“最高公平価格”を設定することを許可し、それぞれ2028年と2026年から、連邦医療保険B部分とD部分がカバーするいくつかの高支出処方薬の価格を直接協議する。また、2022年の“インフレ削減法案”は、連邦医療保険計画がカバーするある薬品の価格上昇速度がインフレ率よりも速い場合、メーカーは連邦政府にリベートを支払わなければならないと要求している。
| 19 |
州レベルでは、立法機関はますます立法を通じて、価格または患者のbr精算制限、割引、ある製品参入の制限、マーケティングコスト開示と透明性措置を含む薬品と生物製品の価格設定を制御するための法規を立法し、実施しており、場合によっては、他の国からの輸入と大量購入も奨励されている。
アメリカ国内で、もし私たちが将来適切な承認を得て私たちの任意の候補製品を販売すれば、私たちはMedicaid、Medicareと公衆衛生サービス(PHS)薬品定価計画に基づいてこれらの製品の承認と保証範囲を求め、また製品を連邦機関に販売することを求めるかもしれない。医療補助brは連邦と州の共同計画であり、各州が管理し、低収入と障害受益者に向けている。Medicaid薬品返却計画によると、メーカーは州Medicaid計画で精算した単位製品ごとに返却点を支払う必要がある。各製品の返金金額は法律で規定されており、ある価格の上昇幅がbrを超えてインフレすれば、追加の割引を受ける可能性がある。連邦医療保険は連邦政府が管理する連邦計画であり、65歳以上の個人及びある障害者をカバーする。連邦医療保険D部は,登録された連邦医療保険患者に自己管理薬(すなわち医師が管理する薬剤を必要としない)の保険を提供する。Medicare Part Dは米国政府が承認した個人処方薬計画によって管理されており,各薬物計画は処方薬の保証範囲と定価のために独自のMedicare Part D処方を確立し,薬物 計画は時々これらの処方を修正する可能性がある。連邦医療保険B部は,入院環境で使用されている注射薬の多くと,病院外来部や医師室の免許を有する医療提供者が管理するいくつかの薬剤をカバーしている。Medicare B部分はMedicare 行政請負者が管理し,通常は保証決定を担当する.いくつかの支払い調整および制限に基づいて、Medicareは、一般に、製造業者によって報告された平均販売価格のパーセンテージに基づいて、B部分保証薬の費用を支払う。薬品br}は連邦機関が連邦供給スケジュール(FSS)で購入すれば、割引定価を受けることができる。ある連邦機関が保証と支払いを受ける薬品、及びMedicaid、Medicare Part B とPHS薬品定価計画によって保証を受ける薬品は、すべてFSS に参加する必要がある。FSS定価は定期的に退役軍人事務部と交渉している。FSS定価 は、メーカーがその製品に対してその最恵国非連邦顧客から徴収した価格を超えないことを目的としている。また、退役軍人管理局、国防部(軍人とその家族がTRICARE小売薬局で計画して購入した薬品を含む)、沿岸警備隊、小霊通が購入した薬品の価格 は定価上限(“連邦最高価格”と呼ばれる)によって制限され、価格上昇幅がインフレを超えると追加割引を受ける可能性がある。医療補助薬品返却計画での薬品カバー範囲を維持するためには,メーカーはPHS薬品定価計画に基づいてある購入者に割引 を提供する必要がある。割引を受ける資格のある購入者には、経済的に困難な患者に不比例サービスを提供する病院、コミュニティ衛生診療所、その他のPHSから衛生サービスの贈与を受ける実体がある。
2010年3月、米国議会は、政府医療計画下の薬品の保険範囲および支払い方法の変更を含む“患者保護·平価医療法案”および“医療·教育調整法案”、または“平価医療法案”を公布した。公布以来、“平価医療法案”の多くの内容は司法と国会の挑戦を受けており、連邦政府の行政と立法部門は“平価医療法案”のいくつかの面の廃止または代替に努力している。例えば、トランプ前総裁は、“平価医療法案”のいくつかの条項の実施を延期したり、“平価医療法案”に規定されているいくつかの医療保険要求を回避したりするための行政命令に署名した。また,米国議会は“平価医療法案”の全部または一部を廃止または廃止し,代替する立法を検討している。国会ではまだ全面的な廃止立法は成立していないが、2019年1月1日から“平価医療法案”の個人医療保険購入規定に適合しない処罰を廃止し、特定の強制料金の実施を延期し、連邦医療保険部分に参加する製薬業者が不足している販売時点割引を増加させるなど、“平価医療法案”を改正するためのいくつかの条項を制定している。テキサス州の地方裁判所裁判官は、2017年の減税と雇用法案または税法の一部として、“個人強制令”が国会で廃止されたため、“平価医療法案”全体が違憲と判断した。最高裁が2021年6月に原告に資格がないと判断したにもかかわらず、“すべてまたは一部の”平価医療法案“を”廃止し、代替する“など、他の行政、立法、または司法行動は、医療製品やサービスのために政府機関が支払う金額を制限する可能性があり、これは、私たちの製品に対する需要の減少や追加の価格設定圧力を招く可能性があり、あるいは規制の大幅な緩和を招く可能性があり、競争製品や技術の導入を容易にする可能性がある。
“平価医療法案”条項の将来にかかわらず、国会は一連の政策を議論し続け、これらの政策は製薬会社が製品に対して受け取る価格brや彼らが獲得した精算金額に影響を与える可能性がある。
| 20 |
環境規制
FDAによって広く規制されていることに加えて、私たちは私たちまたは私たちの候補薬物に適用される環境法規を守らなければならない。製薬会社のような環境法規に適用されるコストはわずかであり,現在のところ候補薬物の生産にも従事するつもりはないからである。我々は現在,我々のすべての候補薬物材料を生産するために非関連メーカーを使用しており,これらのメーカーから最終材料を受け取っているが,我々はこのプロセスのどの段階でも製造プロセスに関与していない。
候補薬物材料の使用,処理,貯蔵,処置の安全手順 は州や連邦法律法規に要求される環境基準に適合していると信じているが,これらの材料による意外な汚染や傷害のリスクを完全に除去することはできない。私たちは私たちや環境に対する危険を軽減するために特定の保険証書を提供しない。
従業員
2022年12月31日現在、4人のフルタイム従業員を雇用しています。私たちは私たちの従業員たちとどんな集団交渉協定も締結していないし、私たちの誰の従業員もどんな労働組合のメンバーでもない。
著者らの専門人員を補充するために、著者らは監督事務、薬物警戒、プロセス工事、製造、品質保証、臨床前と臨床開発、会計と業務発展方面の専門家を招聘した。この人たちは科学顧問と独立顧問を含む。
競争
生物技術と製薬業界の特徴は技術進歩が迅速で、競争が激しく、そして独自製品を高度に重視していることである。私たちは私たちの技術、開発経験と科学知識が私たちに競争優位を提供してくれると信じているが、私たちは主要な製薬、専門製薬とバイオテクノロジー会社、学術機関と政府機関、そして公共と個人研究機関を含む多くの異なる源からの潜在的な競争に直面している。我々が開発·商業化に成功したどの候補製品も,既存療法や将来発売される可能性のある新しい療法と競争するであろう。
私たちの多くの競争相手は研究開発、製造、臨床前テスト、臨床試験を行い、監督管理の許可を得て、 とマーケティング許可を得た製品の面で私たちよりずっと多くの財力と専門知識を持っているかもしれない。製薬、バイオテクノロジー、診断業界の合併と買収は、私たちの数の少ない競争相手により多くの資源を集中させる可能性がある。これらの競争相手はまた合格した科学と管理人員を募集と維持し、臨床試験のために臨床試験場と患者登録 を確立し、そして著者らの計画と相補的或いは必要な技術を獲得する上で私たちと競争を展開する。小さいか早い段階にある会社 も重要な競争相手となり、特に大手や成熟会社との連携で手配される可能性がある。
私たちのすべての候補製品の成功を影響する重要な競争要素 が承認されれば、それらの治療効果、安全性、副作用、利便性、価格、模造薬の競争レベル、br}及び政府と他の第三者支払人が精算できるかどうかである可能性がある。
もし私たちの競争相手が私たちが開発する可能性のある任意の製品よりも安全で、より効果的で、副作用が少ない、より便利で、より安価な製品を開発および商業化すれば、私たちのビジネス機会は減少または消失するかもしれない。私たちの競争相手も私たちよりも早くFDAや他の規制機関のその製品の承認を得ることができ、これは私たちの競争相手が私たちが市場に入る前に強力な市場地位を確立できるようにするかもしれない。さらに、我々の競争能力は、多くの場合、保険会社または他の第三者支払者の影響を受ける可能性があり、非特許製品の使用を奨励しようとしている。現在,我々が求めている適応に適合した模倣薬が多く市販されており,今後数年でより多くの模倣薬が発売されることが予想される。もし私たちの候補治療製品が承認されたら、私たちはそれらの価格が競争相手の模造薬よりも著しく高いと予想する。
| 21 |
癌患者を治療する最もよく見られる方法は手術、放射線治療と薬物治療であり、化学療法、ホルモン治療、免疫治療と標的薬物治療を含む。市場には多くの癌治療の薬物療法がある。多くの場合、これらの薬物は治療効果を向上させるために併用される。ある程度、私たちの候補製品が最終的に既存の薬物または他の療法と組み合わせて使用されるか、またはその補助として使用される場合、私たちの候補製品 はそれらと競合することができないだろう。現在承認されているいくつかの薬物療法はブランド薬物であり、特許保護を受け、他の薬物 は模倣薬である。その中の多くの承認された薬物は有名な治療法であり,医師,患者,第三者支払者に広く受け入れられている。全体的に言えば、過去数十年来癌の治療は著しい進歩を得たにもかかわらず、現在市場での治療方法も多くの患者にメリットを提供したが、これらの治療方法はある程度治療効果と不良事件の発生率の制限を受け、しかもすべての患者の治療に成功する方法はない。そのため、癌の発病率と死亡率は依然として高い。
膵臓癌と固形腫瘍のDNA酵素
膵癌領域では,膵管腺癌(PDAC)を含む現在承認されている数少ない膵癌治療法と競合する。1行目の設定では、ゲムシタビンとアブラシャニが連携しています®あるいはFOLFIRINOXレジメンが現在の治療基準である。二線転移患者に対して、腫瘍学者の現有の治療方法の選択は限られている。FDAが承認した唯一の二線薬はOnivydeです®ゲムシタビンはフルオロウラシル(5 FU)と亜葉酸カルシウム(LV)を併用して治療した。化学療法を除いてメルクのKEYTRUDAは®MSI-H癌(全例の約1%)とLynparzaへの使用が許可されている®BRCA変異の維持に承認された膵癌(全例の約7%)である。
過去数年間に、末期PDAC用化合物はすでにいくつかの末期臨床失敗が出現した。これらの失敗した実験の多くは有望なゴールに基づいている.PDAC開発の後期段階では,化合物 は少ない。
他の固形腫瘍に対しては,多くの会社で承認された免疫療法(免疫チェックポイント阻害剤を含む)と組み合わせて使用するための治療法が開発されており,様々な固形腫瘍適応を治療している。
B細胞リンパ腫のXCART治療
米国とEUはノファ社のKymriah(Tisagenlecleucel);GIlead Sciences,Inc.とKite PharmaのYescarta (Axicabagene Cilolucel)とTecartus(Brexucagene Autolucel);百時美施貴宝(Bristol Myers Squibb)のBreyanzi(Lisocabagene Maraleucel)とAbecma(Idecabagene Vicleucel);およびJanssenのCarvykti(Cilanzene Autucolel)を含む多くのCAR T療法を承認した。また,100種類以上のCAR T療法製品が開発されており,その多くは同種異体や既製細胞療法である。また,われわれのCAR T療法による疾患は,CAR T療法と他のbr法(例えば小分子や抗体)からの興味適応の競争に直面している可能性がある。CAR TやTCR療法などのT細胞に基づく癌治療は,最近学術機関やバイオ製薬会社の重要な研究と開発分野になっている。
薬物送達のためのPSA
現在相互競争のプラットフォームはペグ化、FC融合、アルブミン融合、HES化、PAS化とCTP融合、及び学術機構とその他の薬物開発に従事する比較的に小さい製薬会社のプラットフォームを含む。薬物製品,療法,技術,プロセスの開発において大学や他の研究機関と競合するほか,大学から製品や技術を獲得する権利について他社と競合する可能性がある。
| 22 |
利用可能な情報
私たちのサイトの住所はwww.xeneticBio.comです。我々のサイト上の情報や我々のサイトを介してアクセス可能な情報は,本10-Kフォーム年次報告の一部ではない.私たちの10-Kフォームの年間報告、10-Qフォームの四半期報告、および8-Kフォームの現在の報告、およびこれらの報告の改訂は、私たちが電子的にアメリカ証券取引委員会に提出または提出した後、可能な場合にできるだけ早く無料でbrで、または私たちのサイトで取得することができます。米国証券取引委員会は、www.sec.govで提出した文書に関する報告書、依頼書、情報声明、その他の情報を含むインターネットサイトを維持している。
取引法第13または15(D)節に基づいて現在の情報 を開示し、FD法規が我々の米国証券取引委員会届出文書によって開示されることを要求する情報報告 に加えて、我々の投資家関係サイト、プレスリリース、公開会議 電話会議およびネットワーク放送によって、このような最新の情報を開示する予定である。
プロジェクト1 A--リスク要因
私たちの業務は多くのリスクに直面しています。本年度の報告書に含まれる他の情報と、米国証券取引委員会に提出された他の公開文書に加えて、以下に説明するリスクおよび不確実性をよく考慮しなければなりません。以下のいずれのリスクも、私たちの業務、財務状況、運営結果、見通しに重大な悪影響を与え、私たちの普通株の取引価格を下落させる可能性があります。
私たちの財務状況と資本金要求に関連するリスク
私たちは決して利益を上げたことがなく、絶対に利益を達成したり維持したりしないかもしれない。もし私たちの運営から十分な収入を発生させて費用を支払うことができない場合、あるいは商業的に合理的な条項で追加的な融資を得ることができない場合、私たちの業務、財務状況、および運営結果は実質的なbrと不利な影響を受ける可能性がある.
私たちは臨床段階の生物製薬会社で、運営の歴史は限られている。医薬製品と技術開発は投機性の強い仕事であり、大きなリスクに関連している。私たちは製品が商業販売に使用されることを許可されておらず、今まで限られた収入しか生まれなかった。2022年4月までに,われわれは主にわれわれのXCART技術に関する臨床前開発に注目している。2022年4月にCLSからDNase腫瘍学プラットフォームの許可を得ることに伴い,我々は現在,協力機会や規制承認と商業化によるこの技術の発展を推進することに重点を置いている。私たちは引き続き大量の研究開発費と私たちの持続的な運営に関する他の費用が発生すると予想している。したがって、私たちは利益を達成したことがなく、予測可能な未来には利益が達成できないかもしれない。 があれば。私たちが将来利益を得ることができるかどうかは、多くの要素による
| · | 私たちの候補薬物や技術の研究開発、規制承認、商業化、販売およびマーケティングに関連するコストを援助する | |
| · | 市場は私たちの候補薬や技術を受け入れています | |
| · | 新しい候補薬や技術を獲得し開発するコストは | |
| · | 私たちの候補薬を市場に出す能力は | |
| · | 私たちの業務に関する一般的で行政的な費用は | |
| · | 私たちの研究開発コストを増やし | |
| · | 技術または他の資産の購入に関連する費用; | |
| · | 知的財産権を確立し、維持し、保護する | |
| · | 合格した人材を引きつけ、採用し、引き留める | |
| · | 私たちは追加資本を調達する能力を持っている。 |
| 23 |
2022年12月31日までの累計赤字は約1兆891億ドル。我々の研究開発活動の拡大や商業化、マーケティング、販売努力に伴い、追加の重大な運営損失が生じることが予想される。私たちはまた、予見できない費用、困難、合併症、br}遅延、および私たちの業務に悪影響を及ぼす可能性のある他の未知の要素に直面する可能性がある。さらに、医薬品開発に関連する多くのリスクおよび不確実性 のため、現在の候補薬が適用試験の臨床終点 に到達できない可能性があることを含めて、増加費用の時間または金額を予測することができず、私たちがいつ達成または利益を維持するかどうかを予測することはできません。 もし私たちが運営から十分な収入を生み出して費用を支払うことができない場合、あるいは商業的に合理的な条項で追加融資を得ることができない場合、私たちの業務、財務状況、運営結果は実質的な悪影響を受ける可能性があります。
私たちは私たちの目標を達成するために多くの追加資金が必要になるだろう。必要なときに受け入れ可能な条項で必要な資金を得ることができなかったり、資金を得ることができなかったりすると、私たちの製品開発作業、他の運営、または商業化作業を延期、制限、または終了させる可能性があります。
候補薬の開発は高価でリスクがあり,長い過程であり,特に候補薬の研究·開発を継続し,臨床試験を開始し,上場承認を求める場合には,我々が行っている活動に関連する費用が増加することが予想される。
2022年12月31日現在、私たちは約1,310万ドルの現金を持っている。私たちは、臨床試験を開始し、完成し、規制部門の私たちの候補薬の承認を得て、私たちの他の臨床前候補薬と未来の候補薬を含む追加の資金が必要になると予想しています。しかし、私たちの現在知られていない多くの要素のため、私たちの運営計画は変化する可能性があり、私たちは計画よりも早く追加資金を求める必要があるかもしれない。追加の資金は、公共またはプライベート·エクイティまたは債務融資、第三者融資、マーケティングおよび流通手配、または他の協力、戦略的連合および許可配置(またはこれらの方法の組み合わせ)によって得ることができる。いずれにしても、私たちは臨床前と臨床活動を展開し、監督部門の承認を求め、私たちの長期候補薬を商業化するために、より多くの資金が必要になるだろう。 私たちが現在または未来の運営計画のために十分な資金を持っていると思っても、市場条件が有利であるか、または特定の戦略的考慮があれば、追加の資本を求めることができるかもしれない。
私たちが追加資金を調達する能力は、金融、経済、政治、市場状況、そして私たちがコントロールできないかもしれないまたは限られた他の要素に依存するだろう。最近のロシアのウクライナ侵攻を含む地政学的緊張のような持続的な新冠肺炎の流行または他の要因、およびそれによって生じる任意の制裁、輸出規制、または他の制限的な行動による市場変動は、必要なときに資本を得る能力にも悪影響を及ぼす可能性がある。私たちが追加資金が必要な時、私たちは私たちが受け入れられる条項とコストでこのような資金を得ることができないかもしれない。
任意の追加的な資金調達活動は、私たちの管理層を日常活動から気晴らしさせる可能性があり、これは、私たちの候補薬物の開発と商業化の能力に悪影響を及ぼす可能性があります。また、将来の融資が十分な金額または受け入れられる条項で提供されることを保証することはできません。br}また、任意の融資条項は、私たちの株主の持株や権利に悪影響を及ぼす可能性があり、私たちはbr証券(株式でも債務でも)を追加発行したり、そのような証券を発行したりする可能性があり、株式の市場価格を下落させる可能性があります。債務の発生は固定支払義務の増加をもたらす可能性があり、私たちは追加債務を発生させる能力の制限、私たちが知的財産権を獲得し、販売する能力の制限、および私たちの業務展開能力に悪影響を及ぼす可能性のある他の運営制限のようないくつかの制限的なbr契約に同意する必要があるかもしれない。
もし私たちがタイムリーに資金を得ることができなければ、私たちは私たちの臨床前開発計画または任意の候補薬物の商業化を大幅に削減、延期、または停止する必要があるかもしれない。私たちはまた予想通りに私たちの業務を拡大したり、他の方法で私たちのビジネスチャンスを利用することができないかもしれません。これは私たちの業務、財務状況、そして運営結果を損なう可能性があります。
| 24 |
追加資本の調達は私たちの株主に希釈し、私たちの運営を制限するか、あるいは私たちの技術または候補薬物の権利を放棄することを要求するかもしれない。
これまで、相当な製品収入を生み出すことができれば、株式と債務融資を組み合わせた方法で私たちの現金需要を満たし、協力、戦略連合、許可手配を選択的に継続する予定です。私たちは現在約束された外部資金源 を持っていない。私たちが株式または転換可能な債務証券を売却することによって追加資本を調達する場合、株式は希釈され、これらの証券の条項は、清算または私たちの株主権利に悪影響を及ぼす他の特典を含む可能性がある。 債務融資は、特定の行動をとる能力を制限または制限する契約、例えば、追加債務を生成し、資本支出を行う、または配当を宣言するいくつかの合意に関連する可能性がある。このような債務資金調達はまた私たちの資産の全部または一部によって保証されることができる。
もし私たちが第三者との協力、戦略連合、または許可手配を選択的に継続することによって資金を調達する場合、私たちは、私たちの技術、将来の収入流、研究計画、または候補薬物の追加的に価値のあるbr権利を放棄しなければならないかもしれない、または私たちに不利になる可能性のある条項でライセンスを付与しなければならないかもしれない。もし私たちが必要な時に株式や債務融資を通じてより多くの資金を調達できない場合、私たちは私たちの製品開発や将来の商業化努力を延期、制限、減少、または中止することを要求されるかもしれません。あるいは私たちが自分で開発し、マーケティングすることをより望んでいた候補薬剤の権利を与えてくれます。もし私たちが協力、戦略連合、または許可手配を通じてより多くの資金を調達できない場合、私たちは製品開発や将来の商業化努力を中止したり、運営を完全に停止する必要があるかもしれない。
我々の医薬製品の発見と開発に関するリスク
我々の業務はDNase腫瘍学プラットフォームの成功に大きく依存している。
著者らの業務はDNase腫瘍学プラットフォームの成功した臨床開発、監督管理と商業化に大きく依存する。私たちが商業化を開始することが許可される前に、もしあれば、大量の臨床開発と規制承認作業が必要になるだろう。我々は学術と戦略協力を継続して我々の臨床開発戦略を実施していく予定である。もし私たちがこれらの協力や他の学術協力を維持、獲得できない、または計画通りに獲得できない場合、進行中または計画中の任意の臨床開発を延期、制限、または終了する必要があるかもしれません。これは私たちの業務に悪影響を与えます。DNaseおよび任意の他の候補製品の臨床試験およびbrの製造およびマーケティングは、米国、EUおよび他の司法管轄区域の多くの政府機関の広範かつ厳格な審査と規制を受けることになり、私たちはこれらの司法管轄区で私たちの候補製品 をテストし、販売する予定である。任意の候補製品の商業化販売の規制承認を得る前に、各目標適応に安全かつ有効に使用でき、特定の患者群に使用可能であることを、br前臨床試験および臨床試験によって証明しなければならない。この過程には数年の時間がかかるかもしれないが、上場後の研究と監督を含むかもしれないが、これは現在調達している収益以外の大量の資源支出を必要とするだろう。アメリカとEUが承認した大量に開発されている薬物 の中で、一部の薬物のみがFDA或いはヨーロッパ薬品管理局の監督管理審査プロセス を完成し、商業化されている。したがって、必要な資金を得ることができたり、学術パートナーや戦略パートナーを見つけることができても、研究、開発、臨床プロジェクトに資金を提供し続けることができても、DNase または私たちの任意の他の候補製品が開発または商業化に成功することを保証することはできません。
| 25 |
私たちは候補薬物や技術を含む医薬製品業務を開発した初期会社です。このような発展の不確実性を考慮して、私たちの業務 は決して完全に実現され、投資家のための価値を創造しないかもしれない。
私たちは私たちの製品を開発するためにほとんどの精力と財力を投入していますが、今のところ市場の承認を得た製品は何もありません。これまで、私たちの収入は主に単一パートナーの協力と印税収入から来ていて、製品販売収入ではありません。私たちが製品収入を作る能力は、私たちの候補薬物の開発成功と最終商業化にかかっており、これは数年以内には起こらないかもしれない。私たちは現在、ライセンス契約によって印税収入が発生していますが、br薬品の販売収入は何もなく、私たちは永遠に適切な薬を開発あるいは商業化することができないかもしれません。私たちが薬品販売からいかなる収入を得る前に、私たちのすべての候補薬物は開発、管理 開発と製造活動、複数の司法管轄区で上場許可を得て、製造供給を獲得し、商業組織を創立し、大量の投資と重大なマーケティング仕事を必要とする。我々brは,会社 が新たかつ急速に発展する分野でしばしば遭遇する多くのリスクや不確実性,特に製薬分野での克服に成功していることは証明されていない。例えば、私たちの業務計画を実行するためには、成功 :
| · | 臨床試験の成功と完成を含む候補薬物の開発活動を実行します | |
| · | 候補薬の開発と商業化のために必要な市場承認を得ることができます | |
| · | 私たちの候補薬物の特許と商業秘密保護または規制の排他性を獲得して維持する | |
| · | 知的財産権の組み合わせを保護し、利用し、拡大する | |
| · | 臨床および商業製造能力を確立し、維持するか、または第三者製造業者と臨床および商業製造の手配を行う | |
| · | もし私たちの候補薬が承認され、強力な販売、流通、マーケティング能力を確立し、維持すれば、私たち自身も戦略的パートナーとも協力する | |
| · | もし私たちの候補薬が患者、医学界、第三者支払者の承認を得たら受け入れられる | |
| · | 他の治療法と効果的に競争し | |
| · | 医療保険と適切な精算を受けて維持します | |
| · | 私たちの候補薬が承認された後、許容可能な安全性を維持し続ける | |
| · | 私たちが加入することを選択した任意の戦略的関係を発展させて維持する | |
| · | 知的財産権とクレームを実行して擁護し、 | |
| · | 臨床前開発、臨床試験、市場承認と商業化によるコストと費用の増加に伴い、著者らの支出を管理する。 |
われわれの臨床研究で患者を募集することは困難であることが発見される可能性があり,われわれの薬品の臨床研究を延期または阻止する可能性がある。
患者を確定し、著者らの薬品の臨床研究に参加する資格を持たせることは著者らの成功に非常に重要である。私たちの臨床研究の時間は私たちが患者を募集して私たちの薬品のテストに参加する速度に依存する。私たちは遅延に出会うかもしれない。もし患者が生物製薬業界の不良事件の負の宣伝或いは他のbrの原因(類似患者集団の競争的臨床研究を含む)のために著者らの臨床研究に参加したくない場合、患者を募集し、研究を行い、監督部門の潜在製品に対する承認を得る時間が遅れる可能性がある。これらの遅延はコスト増加を招き,我々の製品開発の推進を遅延させ,我々の技術の有効性を遅延させたり,臨床研究を完全に終了させたりする可能性がある。
| 26 |
十分な数の患者を識別、募集、募集することができないかもしれない、または研究において多様性を達成するために必要または所望の特徴を有する患者は、直ちに我々の臨床研究を完成させることができる。患者の入選は多くの要素の影響を受けている
| · | 調査中の病気の重症度は | |
| · | 代替治療の実際または知覚的獲得可能性; | |
| · | 患者集団の規模と性質 | |
| · | 試験の資格基準と設計について | |
| · | 研究を受けた候補薬のリスクと利益を感知し | |
| · | 潜在患者に臨床場所の近似性と可用性を提供する | |
| · | 潜在競争薬の臨床試験が行われています | |
| · | 既存の治療法や他の開発中の製品に対する私たちが研究している候補薬の潜在的な優位性に対する医師と患者の見方 | |
| · | CROと私たちの試験サイトは臨床試験のタイムリーな登録を促進するために努力しています | |
| · | 医師の患者への転職方法と | |
| · | 治療期間と治療後に患者をモニタリングし,患者データを十分に収集する必要がある。 |
FDAや他の規制機関が要求する臨床研究に十分な数の条件を満たす患者を募集できなければ、臨床研究を開始したり、継続したりすることができない可能性がある。どの国·地域でも臨床研究の開始、登録、完成に成功する能力は、海外で業務を展開することに特有の多くのリスクに直面する
| · | CROや医師との関係を構築したり管理したりすることは困難です | |
| · | 臨床研究を行う異なる基準; | |
| · | 地元のコンサルタントや医師やパートナーを見つけることはできません | |
| · | 各種の外国法律、医療標準と監督管理要求の潜在的負担を遵守し、薬品と生物技術製品と治療に対する監督管理を含む。 |
計画通りに臨床研究を行うのに十分な数の患者を募集することが困難であれば、進行中または計画中の臨床研究を延期、制限または終了する必要があるかもしれず、どのような研究も私たちの業務に悪影響を及ぼす。
| 27 |
私たちは臨床試験の開始、登録或いは完成に重大な遅延に遭遇する可能性があり、あるいは安全性と有効性を証明できず、関連する監督管理機関を満足させることができない可能性があり、これは私たちが直ちに現在と未来の候補薬物を商業化することを阻止するかもしれない(もしあれば)。
規制部門の承認を得て、私たちの現在と未来の候補薬を販売する前に、候補薬の安全性と有効性を証明するために、広範な臨床試験を行わなければならない。いずれの臨床研究も計画的あるいは計画的に完了する保証はなく, があれば。1つまたは複数の臨床研究の失敗は、テストの任意の段階で起こる可能性がある。成功を妨げたり、臨床開発をタイムリーに完成させたりする可能性のある事件は、
| · | 遅延と規制機関は研究設計について合意した | |
| · | 遅延は将来のCROと臨床研究場所と受け入れ可能な条項について合意した | |
| · | 各臨床研究場所で必要なIRBまたは独立倫理委員会の承認を得ることを遅延させる | |
| · | 適切な患者の臨床研究への参加を遅延させました | |
| · | 私たちの臨床研究操作や研究場所を検査した後、監督機関が臨床一時停止を実施した | |
| · | 私たちのCRO、他の第三者、または臨床研究の要求を守ることができませんでした | |
| · | FDAのGCPまたは他の国/地域に適用される法規の要件に従って実行されていない | |
| · | 私たちの候補薬のテスト、検証、製造、臨床場所への配送の遅延 | |
| · | 患者を1つの研究に完全に参加させるか、治療後のフォローアップに戻すことを遅延させる | |
| · | 臨床研究場所や患者は研究から撤退した | |
| · | 臨床試験結果は候補薬物の安全性および/または有効性を証明できない可能性がある | |
| · | 候補薬物に関連する深刻な有害事象の発生は、その潜在的な利点を超えていると考えられている | |
| · | 新しい臨床案の法規要件とガイドラインの変化を修正または提出する必要がある。 |
臨床前研究と臨床試験を成功させることができないいかなる状況も私たちの追加コストを招く可能性があり、あるいは製品販売、規制 及び商業化マイルストーンと版税から収入を得る能力を弱める可能性がある。さらに、私たちが私たちの候補薬を生産または処方変更した場合、私たちの修正された候補薬をより早いバージョンに接続するために、追加の研究が必要かもしれません。臨床試験の遅延はまた、候補薬物を商業化する独占的な権利を持つ可能性のある任意の期限を短縮することができ、あるいは私たちの競争相手が私たちの前に製品brを市場に出すことを可能にすることは、候補薬物を商業化することに成功する能力を弱める可能性があり、私たちの業務、財務状況、運営結果、将来性を損なう可能性がある。
もし私たちの臨床研究結果がbrを確定していない場合、あるいは私たちの薬品に関連する安全問題や有害事象であれば、
| · | もし私たちが本当にそれらを獲得したら、私たちの候補薬の発売承認や許可証の取得を延期します | |
| · | 承認された適応や患者集団は期待や期待ほど広くない | |
| · | 重大な使用または流通制限または安全警告を含むラベルの承認を得ること; | |
| · | 製品の使い方が変わる可能性があります | |
| · | 追加の臨床研究を行うことを要求され、追加の上場後のテスト要求を承認または受け入れることを支持する | |
| · | 規制部門に製品の承認を撤回させたり、修正されたリスク評価と緩和戦略の形で流通に制限を加えたりする | |
| · | 警告や禁忌症のようなタグ付け説明 | |
| · | 起訴される | |
| · | 私たちの名声は損なわれた。 |
以上のように,上記のいずれの事件も我々が我々の薬品に対する市場の受容度を実現または維持することを阻止し,収入を創出する能力を弱めることが可能である。
| 28 |
もし私たちが必要な臨床前研究と臨床研究を完成すれば、いつ、あるいは規制部門の許可を得て候補薬物を商業化するかどうか、あるいは承認の範囲が私たちが予想しているより狭いかもしれない。
候補薬物はbr関連規制部門の審査と承認まで商業化できない。我々の候補薬物が臨床研究において安全性とbr}の有効性を示しても、監督管理機関は適時に審査過程を完成できない可能性があり、あるいは私たちは監督部門の承認を得ることができない可能性がある。FDA諮問委員会または他の規制諮問グループまたは機関が承認または制限を提案しない場合、追加の遅延を招く可能性がある。さらに、製品開発、臨床研究、審査過程において、将来の立法または行政行動における追加の政府規制または規制機関の政策の変化によって遅延または拒否を受ける可能性がある。規制機関も要求された適応よりも少ないあるいは限られた候補薬物を承認することができ、発売後の研究の表現に基づいて承認することもできる。さらに、規制機関は、私たちの候補薬物の商業化に必要または必要なラベル宣言を承認しないかもしれない。候補薬物を商業化する規制承認を得ることができなかったか遅延することは、私たちの収益能力を弱化させ、私たちのbr業務の将来性を損なう。
もし私たちが候補薬物の規制承認を受けたら、私たちの候補薬物は引き続き規制されて審査されるだろう。
もし私たちの候補薬物が承認されたら、それらは生産、ラベル、包装、貯蔵、広告、販売促進、サンプリング、記録保存、br}の発売後研究及び安全性、有効性とその他の発売後情報を提出することを含む持続的な法規要求を遵守し、アメリカの連邦と州のbr要求及び類似の外国監督機関の要求を含む。
製造業者および製造施設は、品質管理および製造手順がcGMP法規に適合することを確保することを含む、FDAおよび同様の外国規制機関の広範な要求を遵守しなければならない。したがって、我々は、cGMPの遵守状況および任意のセキュリティプロトコル、BLAまたはマーケティング許可申請(MAA)におけるコミットメントの遵守状況を評価するために、持続的な審査および検査を受ける。したがって、私たち と私たちの協力者とサプライヤーは製造、生産、品質管理を含むすべてのコンプライアンス分野に時間、お金、エネルギーを投入し続けなければならない。
私たちまたはパートナーのbrが私たちの候補薬物のために得た任意の規制承認は、製品上場の承認指示用途の制限または承認条件によって制限される可能性があり、または候補薬剤の安全性および有効性 を監視するために、コストの高い追加の臨床試験および監視の要件を含む可能性がある。私たちはFDAと同様の外国規制機関にいくつかの副作用と生産問題を報告することを要求されるだろう。医薬品の安全または規制審査および承認に関連する他の問題に関連する任意の新しい立法は、製品開発または商業化の遅延をもたらすか、またはコンプライアンスを確保するコストを増加させる可能性がある。私たちは製品広告と販売促進に関する要求を守らなければならないと思います。処方 薬品に関する販促情報は様々な法律や法規によって制限されており,製品 が承認したラベル中の情報と一致しなければならない.したがって、私たちは承認されていない適応や用途のために私たちの製品を普及させることを許さない。もし私たちの候補薬が承認された場合、私たちは新しい申請または補充申請を提出し、承認されたbr製品、製品ラベル、または製造プロセスをいくつか変更しなければならない。著者らはまた、一般或いは特定の患者亜群におけるわが製品の安全性と有効性を検証するために、発売後の臨床試験を要求されることができる。成功しなかった上場後の研究やこのような 研究が完成できなかったことは、マーケティング承認の撤回につながる可能性がある。
| 29 |
規制当局が承認された製品に以前未知のbr問題、例えば意外な深刻性または頻度の不良事象または私たちの製造施設に問題があることを発見した場合、 または規制機関が製品の販売促進、マーケティングまたはラベルに同意しない場合、規制機関は製品の市場からの撤退を要求することを含む、製品または私たちに制限を加える可能性がある。もし私たちが適用される規制要件を遵守できなければ、規制機関や法執行機関は:
| · | 無見出しと警告状を出す | |
| · | 民事または刑事罰を加える者 | |
| · | 規制承認を一時停止したり、許可証を取り消したりした | |
| · | 私たちが行っている臨床試験を一時停止したり一時停止したりします | |
| · | 私たちが提出した保留申請または承認された申請を承認する補充申請を拒否する; | |
| · | 私たちの製造施設を閉鎖することを含む私たちの業務に制限を加える;または | |
| · | 製品を差し押さえたり差し押さえたり、製品のリコールを要求したりします。 |
政府は違法の疑いのあるいかなる調査にも対応するために多くの時間と資源を必要とする可能性があり、否定的な宣伝が生じる可能性がある。持続的な規制要求を遵守できなかったいかなる行為も、製品を商業化し、そこから収入を得る能力に深刻な影響を与える可能性がある。規制制裁を実施したり、監督管理の承認を撤回したりすれば、会社の価値と私たちの経営業績はマイナスの影響を受ける。
いかなる現在または未来の薬品の商業成功は医者、患者、第三者支払人と医学界の他の人の市場受け入れの程度に依存する。
必要な承認を得ても、私たちの薬品の商業成功は医学界、患者と第三者支払人が私たちの薬品br製品を受け入れるかどうかにある程度依存し、医療用途、費用効果と安全性を持っている。私たちまたは私たちのパートナーが市場に発売したいかなる薬品も医師、患者、第三者支払人、または医学界の他の人の市場で受け入れられないかもしれない。これらの薬品の市場受容度は、商業販売のために許可されれば、多くの要素に依存する
| · | 私たちが承認した候補薬の現在利用可能な製品と比較した有効性 | |
| · | 患者が現在の治療法の代わりに承認された候補薬を採用したいかどうか | |
| · | 私たちは受け入れられる安全性と有効性の証拠を提供する能力; | |
| · | 相手が便利で管理しやすい | |
| · | どんな副作用の流行率や重症度も | |
| · | 他の製品との共同使用を制限する | |
| · | 代替治療法があるかどうか | |
| · | 私たちの候補薬物と目標市場の概要に基づいて、競争力または潜在的なプレミアム要件、定価と費用効果があると仮定する | |
| · | 私たちまたはパートナーの販売とマーケティング戦略の有効性 | |
| · | 私たちは十分な第三者保険や補償を受けることができる | |
| · | 潜在的な製品責任クレーム。 |
1種の潜在的な製品が臨床前と臨床研究において良好な の治療効果と安全性を示しても、市場のこの製品に対する受容度はこの製品 が発売された後に確定することができる。我々の教育医療界や第三者支払者が薬品のメリットを知る努力には大量の資源が必要である可能性があり,決して成功しないかもしれない。もしこれらの製品が十分な受容度に達していなければ、私たちは著しい製品収入が生じないかもしれませんし、利益を上げることができないかもしれません。
| 30 |
開発中の候補薬物の商業的潜在力は予測が困難である。もし新しい候補薬物或いは技術の市場規模が私たちの予想した市場規模より明らかに小さければ、私たちの収入、運営結果と財務状況に重大なマイナス影響を与えるかもしれない。
他の利用可能な技術或いは治療方法と比較した安全性と有効性などの重要な要素のため、絶えず変化する看護標準、第三者支払人の精算基準、患者と医師の選好、長い薬物開発過程中或いは商業導入後に出現する可能性のある競争代替品の可用性、及び私たちの成功した候補薬物の政府衛生部門で承認された後発薬バージョンの可用性を含む薬品の商業潜在力を推定することは困難であり、規制排他性満期に基づいて、或いは私たちは私たちの特許を主張することによって模倣薬バージョンの発売を阻止することができない。もしこれらの要素または他の要素によって、医薬製品の市場潜在力が私たちの予想を下回った場合、その医薬製品の任意の潜在的なパートナー関係の商業条項に重大なマイナス影響を与える可能性があり、あるいは、もし私たちがすでにその医薬製品について協力した場合、印税とマイルストーン支払いの収入潜在力は著しく減少する可能性があり、これは私たちの業務、財務状況、および運営結果に負の影響を与えるだろう。
もし私たちの候補薬が十分な保険や精算を得られなかった場合、承認されれば、これらの製品をマーケティングする能力を制限し、収入を創出する能力を低下させる可能性がある。
私たちの候補薬の成功は、承認されれば、第三者支払者が十分な保険と精算を提供するかどうかに依存する。また、私たちの候補薬物はある疾患を治療する新しい方法を代表するため、私たちの候補薬物が保証と精算を受けることを保証することができない、あるいは私たちの候補薬物の潜在的な収入を正確に推定することはできず、私たちが開発する可能性のあるどの製品も保証と精算を得ることは保証されない。
その病態に医療サービスを提供する患者は、通常、第三者支払者によって、その治療に関連する費用の全部または一部を精算する。政府医療保健計画(例えばMedicareとMedicaid)と商業支払者の十分なカバーと精算は新製品の受容度に重要である。
政府当局と第三者支払者、例えば個人健康保険会社や健康維持組織は、保険を受ける薬物や治療および精算金額を決定する。第三者支払人の保証範囲と精算は多くの要素に依存する可能性があり、第三者支払人の製品の使用が以下の条件を満たすかどうかの決定を含む
| · | 健康計画の下で保障された福祉 | |
| · | 安全で効果的で医学的に必要なものです | |
| · | 特定の患者に適しています | |
| · | 費用対効果があります | |
| · | 実験的でも調査的でもない。 |
米国では,第三者支払者とその契約した薬局福祉マネージャーのためにこのような支払人の処方br福祉を管理する第三者支払者とその契約した薬局福祉マネージャーの間に統一的な保証と製品精算政策はない。そのため、政府や他の第三者から製品の保証と精算承認を得ることは時間がかかり、高価な過程であり、各支払先に科学的、臨床的、費用効果を支援するデータを提供し、支払者ごとに私たちの製品を使用する必要があるかもしれないが、保証され、十分な精算が得られる保証はないかもしれない。特定の製品の保証を受けても、発生した精算支払率は、収益性を実現したり維持したりするのに十分ではない可能性があり、あるいは患者が受け入れられない高い支払い額を発見する必要があるかもしれない。さらに、第三者支払者およびその薬局福祉マネージャーは、私たちの遺伝子修正製品を使用するために必要な長期的な後続評価に保険を提供するか、または十分な精算を提供しない可能性がある。患者が私たちの候補薬を使用する可能性はあまりなく、保険を提供し、候補薬を支払うのに十分な費用の大部分を清算しなければならない。新たに承認された製品の保険範囲や精算に関する不確実性が大きい。 第三者決済者が我々の候補薬の保証範囲や精算についてどのような決定を下すか予測することは困難である。
| 31 |
また、アメリカと海外の政府と第三者支払人が医療コストの制限或いは低減を強化することは、このような組織が新承認製品の保証範囲と精算レベルを制限する可能性があるため、彼らは私たちの候補薬物brの保証或いは十分な支払いを提供できない可能性がある。Br管理型ヘルスケアの傾向、ヘルスケア組織の影響力がますます大きくなり、コスト制御措置、追加の立法変化により、私たちは私たちの任意の候補薬物の販売に関連する価格設定圧力に直面することが予想される。
私たちはアメリカと選定された外国司法管轄区で私たちの候補薬を販売することの承認を求めるつもりです。もし私たちの候補薬物が1つ以上の外国の管轄区域で承認されたら、私たちはこれらの管轄区域の規則によって制限されるだろう。海外では、薬品br製品の定価は政府規制と他の市場規制を受けており、これは私たちの候補薬品の定価と使用に圧力をかけるかもしれない。 これらの国/地域では,候補薬の発売承認を得た後,政府当局との定価交渉にかなりの時間を要する可能性がある。また,候補薬物の市場受容度と販売量は,候補薬物が十分な保証範囲と第三者支払者の精算があるかどうかに大きく依存し,既存と将来の医療改革措置の影響を受ける可能性がある。もし私たちの候補薬が十分な保証範囲と精算を得られなかった場合、承認されれば、これらの製品を販売する能力を制限し、収入を創出する能力を低下させる可能性がある。
私たちは、より利益になるかもしれない計画または候補薬を利用することなく、特定の研究計画または候補薬剤を追求するために、私たちの財務および人的資源 を使用することができるかもしれない。
私たちの資源が限られているので、私たちはいくつかの計画、候補薬、または後により大きな商業的潜在力を有する適応を証明する機会の追求を放棄したり、延期したりする可能性がある。私たちの資源配分決定は、実行可能な商業製品や利益の市場機会を利用できない可能性があります。 候補薬物の現在および未来の研究開発計画への支出は、いかなる商業的に可能な製品も生成しないかもしれません。もし、特定の候補薬物の商業的潜在力や目標市場を正確に評価していなければ、戦略的協力、許可、または他の印税手配を通じて、候補薬物に価値のあるbr権利を放棄する可能性がありますが、この場合、候補薬物の独占的な開発と商業化の権利を保持することが有利になります。あるいは、私たちは、ある治療領域の候補薬に内部資源を割り当てることができ、その分野で協力した方が有利になるかもしれない。br}は、より大きな商業的潜在力を有する機会を求めることができないか、または候補薬剤を放棄する貴重な権利を求めることができず、私たちの業務、運営結果、および将来性に悪影響を及ぼす可能性がある。
私たちは他の薬品の識別や発見に成功できないかもしれません。
私たちの業務の成功は主に私たちが医薬製品を識別して開発する能力にかかっている。様々な理由から,われわれの研究計画では臨床開発のための潜在薬物 を決定できない可能性がある。私たちの研究方法は潜在的な薬品 の識別に成功しない可能性があり、あるいは私たちの潜在的な薬品は有害な副作用を有することが証明されている可能性があり、あるいは製品が販売できないか、または発売許可を得ることができない可能性がある他の特徴を持っている可能性がある。
上記のいずれかの事件が発生した場合、私たちは、1つまたは複数の計画のための開発作業を放棄することを余儀なくされる可能性があり、これは、私たちの業務に大きな悪影響を与え、運営を停止させる可能性があります。新医薬製品を識別する研究プロジェクトは大量の技術、財政と人力資源を必要とする。私たちは私たちの努力と資源を最終的に成功しないことが証明された潜在的な計画や薬品に集中するかもしれません。もし私たちが他の薬品の識別や発見に成功しなかった場合、私たちの業務、運営結果、および将来性に悪影響を及ぼすかもしれません。
| 32 |
私たちの候補薬物の市場機会は、以前の治療を受ける資格がない患者または以前の治療を通過できなかった患者に限定される可能性があり、小さい可能性がある。
癌療法は1線,2線または3線に分類されることがあり,FDAは通常最初に三線使用の新しい療法のみを承認する。癌が十分に早く発見された時、第一線の治療は治愈を必要とすることなく、癌を治癒したり、生命を延長するのに十分であることがある。一次治療(通常、化学療法、ホルモン治療、手術またはこれらの療法の組み合わせを含む)が成功しないことが証明された場合、二次治療が施行される可能性がある。二線治療は、一般に、より多くの化学療法、放射線、抗体薬物、腫瘍標的小分子、またはこれらの薬剤の組み合わせを含む。三線治療は骨髄移植、抗体と小分子標的治療、より侵襲性のある手術形式と新しい技術を含むことができる。治療が承認された市場では,最初に他の承認治療に失敗した患者の後期治療として承認候補薬 を求める予定である。その後,十分有益であることが証明された薬剤(あれば)に対しては,二次療法や潜在的な一次療法としての承認を求める予定であるが,候補薬剤が承認されても二次療法や一次療法として承認されることは保証されない。また,二線または一線治療の承認を得る前に,追加の臨床試験が必要である可能性がある。
我々が狙う癌を有する人数,および後期治療を受けることができるこれらの癌患者のサブセットおよび我々の候補薬物治療から利益を得る可能性のある人数の予測は,我々の信念と推定に基づいている。これらの推定 は様々な源から来ており,科学文献,診療所調査,患者基金会あるいは市場研究を含めて, は正しくないことが証明されている可能性がある。また,新たな研究はこれらの癌の推定発症率や流行率を変える可能性がある。患者数 は予想を下回る可能性がある。さらに,我々の候補薬物の潜在的にアドレス指定可能な患者集団は限られている可能性があるか,あるいは候補薬物の治療を受けられない可能性がある。私たちの候補薬がかなりの市場シェアを得ても、規制部門の他の適応の承認を得なければ、私たちは一線または二線治療としてのbrを含む利益を永遠に達成できないかもしれません。これは私たちの業務と運営結果に悪影響を及ぼす可能性があります。
臨床試験は著者らの候補薬物の安全性と有効性を証明できない可能性があり、そして監督管理の承認を阻止或いは顕著に延期する可能性がある。
NDAやBLAの承認を得て候補薬物を商業化する前に,候補薬物が安全で有効であるか,あるいは生物が安全で純粋で有効であるかを厳密に制御された臨床試験によりFDAに証明しなければならない。これらの試験や将来の臨床試験が成功しなければ、私たちの業務と名声が損なわれる可能性があり、私たちの株価は不利な影響を受ける可能性があります。
臨床失敗は臨床発展のいかなる段階で発生する可能性がある。臨床試験は陰性または不確定な結果をもたらす可能性があり、私たちまたは私たちの現在と未来の任意の協力者は決定または監督機関が追加の臨床または臨床前試験を行うことを要求するかもしれない。厳格に制御された臨床試験により、特定の患者集団における候補薬物の使用がそれぞれの参考製品と同様に安全かつ有効であることを大量のbr証拠を用いて証明することが求められ、規制機関にその商業販売の承認を求めることができる。早期臨床試験の成功は未来のもっと大規模な登録臨床試験が成功することを意味しない。後期臨床試験の候補薬物は同等の安全性と有効性を証明できない可能性があるため、FDAと外国の監督管理機関を満足させ、初歩的な臨床試験で進展が得られたにもかかわらず。早期臨床試験において有望な結果を示す候補薬剤は、その後の検証性臨床試験で失敗する可能性がある。同様に、前臨床試験と早期臨床試験の結果は後続の臨床試験の成功を予測できない可能性があり、臨床試験の中期結果も必ずしも最終結果を予測できるとは限らない。製薬業界の多くの会社は,我々よりも多くの資源や経験を持つ会社を含め,高度臨床試験で大きな挫折を経験し,早期の臨床試験でも奮い立つ結果を得ている。
また,臨床試験の設計はその結果が製品の承認を支持しているかどうかを決定することができるが,臨床試験設計における欠陥は臨床試験の進展が良好になるまで明らかにならない可能性がある。私たちは規制部門の承認を支持するために臨床試験を設計して実行することができないかもしれない。いくつかの場合、多くの要素により、同じ候補薬物の異なる試験間の安全性または有効性結果に有意差が存在する可能性があり、これらの要素は、試験レジメンの変化、患者集団の大きさとタイプの違い、投与レジメンの堅持、および臨床試験参加者の中退率を含むが、これらに限定されない。
これらのリスクにより、私たちの研究開発努力と私たちのパートナーの努力はどんな商業的に実行可能な製品も生まれないかもしれません。これらの開発作業の大部分が成功していない場合、または私たちまたはパートナーが必要な規制承認を得ていない場合、または任意のbr承認された製品が商業的に成功していない場合、私たちは著しい収入や利益を生じないかもしれない。
| 33 |
FDAから孤立薬物指定 を得ることができない可能性があり,このような指定を得ても潜在的な市場排他性を含めて孤立薬物指定に関する利点を保つことができない可能性がある。
孤児医薬品法によれば、FDAは、米国で発生した患者数が200,000人未満であるか、または米国患者数が200,000人を超えることが定義されており、米国での販売によって薬剤または生物学的薬剤の開発コストを回収することを期待していないまれな疾患または疾患を治療するための薬物または生物学的孤児の薬物名を付与することができる。米国では,孤児薬物指定は一方が臨床試験費用に贈与資金を提供する機会,税収割引,ユーザ費用減免などの財政的インセンティブを得る権利がある。さらに、孤児薬物指定を有する製品がその後、そのような指定された疾患に対するFDAの最初の承認を得た場合、この製品は、孤児薬物独占経営権を得る権利があり、これは、FDAが完全なNDAまたはBLAを含む他の任意の出願を承認しない可能性があることを意味し、7年以内に同じ薬物または生物学的製剤を販売し、限定された場合でなければ、例えば、孤児薬物独占性を有する製品に対する臨床的利点を示すか、または製造業者のbr}が十分な製品数を保証することができないことを意味する。
私たちは、彼らが将来承認される可能性のある任意の合格した適応を得るために、私たちの活性候補薬のための孤児薬名 を得ることを求めることができる。このような指定を受けても,医薬品開発に関する不確実性により,孤児指定適応の候補薬物上場承認を得た最初の会社ではない可能性がある。さらに、孤児が指定した適応よりも広い適応を承認することを求める場合、米国での独占営業権が制限される可能性があり、またはFDAが指定要求 に重大な欠陥があると後に決定した場合、または製造業者がこのようなまれな疾患または疾患患者の需要を満たすのに十分な数の製品を保証できない場合、独占営業権を失う可能性がある。また,我々が製品の孤立薬物排他性を獲得しても,この排他性は異なる活性部分を有する異なる薬物が同じbr条件のために許可されるため,競合からこの製品を効率的に保護できない可能性がある。孤児製品が承認された後であっても、FDAが後者の薬剤がより安全で、より効率的で、または患者ケアに大きな貢献があると結論した場合、FDAはその後、同じ場合について同じ活性部分を有する同じ薬剤を承認することができる。孤児の薬物指定は薬物の開発時間或いは監督審査時間を短縮することもなく、この薬物が監督審査或いは審査過程においていかなる優勢を持たせることもない。しかも、私たちが私たちの候補薬のために孤児薬の指定を求めても、私たちは決してそのような指定を受けないかもしれない。
医療立法改革措置は私たちの業務と運営結果に実質的な悪影響を及ぼす可能性がある。
アメリカとある外国の司法管轄区域では、近年、多くの立法と規制法規がbr方式で医療保健システムを変更し、これは私たちの未来の販売候補薬物の収益能力に影響を与える可能性がある。
また、連邦と州レベルはすでに多くの措置を講じて、医療コストの削減を図るだろう。何よりも、2010年3月、“医療·教育調整法案”(総称して“ACA”と呼ぶ)で改正された“患者保護·平価医療法案” が法律として署名され、政府や民間保険会社の医療融資のあり方を著しく変更する措置が含まれている。2017年1月、国会投票は2017年度予算決議、または予算決議を採択し、ACAの一部を廃止する立法を実施することを許可した。また、2017年1月20日、トランプ前総裁は、ACAに基づいて権力と責任を有する連邦機関がACAの実施を放棄、延期、免除、または延期することを指示する行政命令に署名し、ACAのいずれかが州、個人、ヘルスケア提供者、医療保険会社または薬品または医療機器メーカーに財政的または規制的負担をもたらす条項を提供した。また、2017年10月12日、前総裁のトランプ氏は、HHAおよび労働省と財務省の部長たちに、ACAの要求に制約されず、医療精算スケジュールの可用性を増加させ、通常ACAのbr要求に制約されず、医療精算スケジュールの利用可能性を増加させるために、より多くの雇用主が協会健康計画を形成することを可能にするために、HHAおよび労働省と財務省の部長たちに条例 の提案や既存の指導意見の修正を考慮することを要求する別の行政命令を発表した。2017年10月13日、司法省は、HHSはこれらの支払いが国会で支出されていないと判断したため、保険会社への費用分担減少支払いを直ちに停止すると発表した。また、2017年12月22日、前総裁·トランプ氏は“減税·雇用法案”(TCJA)に署名し、法律とし、連邦税収制度の抜本的な改革に加え、2019年1月1日から個人強制令に関する罰則を廃止した。議会または米国の総裁はまた、ACAの内容を代替、除去、または再確認するために、その後の立法または行政行動を考慮することができる。私たちは、ACAおよび任意の将来のACAの措置が私たちの業務に与える影響を評価し、修正、廃止、置換、または再確認するつもりだ。
| 34 |
また,2022年のインフレ低減法案は,処方薬をカバーする既存の医療保険計画に影響を与える可能性がある。他の関連条項を除いて、2022年の“インフレ低減法案”は、連邦医療保険計画がある“最高公平価格”を設定することを許可し、それぞれ2028年と2026年から、連邦医療保険B部分とD部分がカバーするいくつかの高支出処方薬の価格を直接協議する。また、2022年の“インフレ削減法案”は、連邦医療保険計画がカバーするある薬品の価格上昇速度がインフレ率よりも速い場合、メーカーは連邦政府にリベートを支払わなければならないと要求している。私たちは2022年のインフレ低減法案が私たちの業務に及ぼす影響を評価し続けるつもりだ。
私たちはbrの持続的な医療改革討論がアメリカ総裁の立法、法規、訴訟、あるいは行政行動が私たちの業務に不利にならないことを保証できない。
将来提案され採択される可能性のある法律および他の改革およびコスト制御措置はまだ不確定であるが、製品のための私たちの価格設定の能力を制限する条項が含まれている可能性があり、および/または連邦医療保険および他の医療保険資金のさらなる減少をもたらす可能性があり、これは、将来の顧客および私たちが収入を創出し、利益または製品商業化を達成する能力に実質的な悪影響を及ぼす可能性がある。
私たちの第三者への依存に関するリスク
私たちはより多くの協力関係 を構築することを求めるかもしれませんが、合理的なビジネス条項で協力関係を構築できなければ、私たちの開発と商業化計画 を変えなければならないかもしれません。
私たちの候補薬物開発計画と候補薬物の潜在的な商業化は費用を支払うために多くの追加の現金を必要とするだろう。我々のいくつかの候補薬物については,他の製薬やバイオテクノロジー会社と協力して,これらの候補薬物を開発し,潜在的な商業化を行うことにする可能性がある。
私たちは適切なパートナーを探すことで激しい競争に直面している。我々が任意の他の連携について最終的な合意を達成するかどうかは,協力者の資源や専門知識の評価 ,協力を提案する条項や条件,提案した協力者の多くの要因の評価に依存する.これらの要素は、臨床試験の設計または結果、FDAまたは米国国外の類似規制機関によって承認された可能性、候補薬物の潜在的市場、候補薬物を製造し、それを患者に渡すコストと複雑性、競争薬物の潜在力、技術所有権 に存在する不確実性(挑戦の是非曲直を考慮せずに所有権に挑戦する場合) および全体的な業界と市場状況を含むことができる。協力可能なbr適応のような代替候補薬や技術,このような協力が我々の候補薬物との協力よりも魅力的であるかどうかも考えられる。私たちが設立する可能性のある他の協力や他の計画の条項は私たちに不利かもしれない。
既存の連携協定によれば、潜在的な協力者といくつかの条項について将来の合意を締結することができないように制限される可能性もある。交渉と記録連携は複雑で時間がかかる .また,大手製薬会社間で最近大量の業務合併が発生し,将来の潜在的パートナー数の減少を招いている。
私たちは受け入れ可能な条項でより多くの協力 をタイムリーに協議することができないかもしれないし、交渉することができないかもしれない。もし私たちがそれができなければ、私たちは私たちが協力を求めている候補薬物の開発を削減し、その開発計画または私たちの1つまたは複数の他の開発計画を減少または延期し、brの潜在的な商業化を延期したり、いかなる販売やマーケティング活動の範囲を縮小したり、私たちの支出を増加させ、自費で開発や商業化活動を行わなければならないかもしれない。もし私たちが私たちの支出を増やして開発または商業化活動に資金を提供することを選択すれば、私たちは追加の資本を得る必要があるかもしれません。これらの資本は受け入れ可能な条項で私たちに提供できないかもしれません。もし私たちが十分な資金を持っていなければ、私たちは私たちの候補薬をさらに開発したり、それを市場に出して製品収入を作ることができないかもしれない。
| 35 |
もし私たちが私たちの協力者や戦略的パートナーと衝突すれば、これらの当事者たちは彼ら自身の利益に基づいて行動するかもしれないし、これは私たちの戦略を実施する能力を制限するかもしれない。
もし私たちの会社や学術パートナーや戦略パートナーが私たちと衝突したら、相手は自分の利益で行動するかもしれません。これは私たちのbr戦略を実施する能力を制限するかもしれません。私たちのいくつかの学術協力者と戦略パートナーは私たちが協力している各分野と複数の製品開発を行っています。しかし、我々の協力者または戦略パートナーは、これらの協力の対象製品または潜在的な製品と競争力を有する関連分野の製品を単独でまたは他の人と一緒に開発する可能性がある。協力者または戦略パートナーによって開発された競合製品、または協力者または戦略パートナーがbrの権利を有する競合製品を開発することは、パートナーが私たちの候補薬剤の支援を撤回することにつながる可能性がある。
私たちのいくつかの協力者や戦略的パートナー は未来にも私たちの競争相手になるかもしれない。私たちの協力者や戦略パートナーは競争製品を開発し、私たちの競争相手との協力を阻止し、適時に監督管理の承認を得ることができなかった、私たちとの合意を早期に終了したり、製品の開発と商業化のために十分な資源を投入できなかったりする可能性がある。これらの開発のいずれも、私たちの製品開発努力を損なう可能性があり、これは私たちの業務、運営結果、将来性に悪影響を及ぼす可能性があります。
私たちは第三者に頼って、私たちの臨床研究を監督して、これらの第三者の表現が満足できなければ、私たちの業務を損なう可能性があります。
著者らはCRO、臨床研究者と臨床研究サイトに依存し、著者らの臨床研究が正確かつ適時に行われることを確保する。著者らはCRO、臨床研究者と臨床研究サイトの業績に限られた影響を与え、そして著者らは著者らのCRO活動のいくつかの方面のみを制御する。しかし、私たちは、私たちのすべての臨床研究が適用された合意、法律、brの規制要件および科学的基準に従って行われ、私たちのCROへの依存は私たちの規制責任を免除しないことを確保する責任を負います。また、これらの第三者CRO、臨床研究者、および臨床研究場所で使用される施設は、進行中の新しい冠大流行、テロ、戦争または他の武力衝突、ロシアとウクライナの間の持続的な衝突および関連する制裁および他の経済中断または懸念などの流行病のような悲劇的な事件の負の影響を受ける可能性があります。洪水や火災などの自然災害やそのような施設は、例えば、このような施設の監督検査後に汚染や規制の問題が生じるなど、製造問題に直面する可能性がある。この場合、私たちは適切な代替第三者施設を見つけ、契約関係を確立する必要があるかもしれません。これは、既製または許容可能な条項ではないかもしれません。これは、追加的に必要なFDA承認の結果を含む追加の遅延および追加の費用をもたらし、私たちの業務に実質的な悪影響を及ぼす可能性があります。
われわれ、われわれの臨床研究者とわれわれのCRO は、臨床試験結果をFDAが行い、記録し、報告したGCPを遵守して、br}データと報告の結果が信頼性と正確であることを保証し、臨床試験参加者の権利、完全性と機密性を保護しなければならない。FDAは,研究スポンサー,主要研究者,臨床試験地点の定期検査によりこれらのGCPを実行している。もしわれわれ,われわれのCROや臨床研究者が適用されたGCPを遵守できなかった場合,われわれの臨床試験で生成された臨床データは信頼できないと考えられる可能性があり,FDAは任意の上場申請を承認する前に追加の臨床試験を行うことを要求する可能性がある。検査後,FDAはわれわれの臨床試験がGCPに適合していないことを確認する可能性がある。また,我々の将来の臨床試験には,候補薬物の安全性と有効性を評価するのに十分な数の試験対象が必要となる。したがって,我々のCROや臨床調査者がこれらの規定を遵守できなかったり,十分な数の患者を募集できなかったりした場合,このようなbr臨床試験を繰り返すことが要求される可能性があり,規制承認過程を遅らせることになる。
私たちのCROは私たちの従業員ではないので、彼らが私たちの臨床と非臨床プロジェクトに十分な時間と資源を投入しているかどうかを直接監視することはできません。これらのプロジェクトはそれぞれGCPとGLPに従って行わなければなりません。これらのCROはまた、我々の競争相手を含む他の商業実体と関係がある可能性があり、彼らはまた、これらの実体のための臨床研究または他の薬物開発活動を行っている可能性があり、これは私たちの競争地位を損なう可能性がある。もし私たちのCROがその契約の職責や義務を成功裏に履行できず、期待された期限内に達成できなかった場合、あるいは私たちの臨床方案や法規の要求を遵守できなかった(または任意の他の原因)ために、彼らが獲得した臨床データの品質または正確性が影響を受ける可能性があり、私たちの臨床研究は延長、延期または終了される可能性があり、私たちは監督部門の私たちの薬品の承認を得ることができない、あるいはそれを商業化することに成功できないかもしれない。したがって、私たちの財務業績と薬品のビジネス見通しが損なわれ、私たちのコストが増加する可能性があり、私たちの収益能力が延期される可能性がある。
私たちはまた、他の第三者が私たちの可能な臨床研究のために私たちの製品を保存して配布することに依存するかもしれない。もし私たちのディーラーにどんな業績ミスがあったら、私たちの薬品の臨床開発やマーケティング承認を延期する可能性があります。あるいは承認されれば、私たちの製品は商業化され、それによって追加の損失をもたらし、私たちの潜在的な製品収入を奪うことができます。
| 36 |
私たちの協力者や戦略パートナー は、代替技術を採用することを決定したり、私たちの技術を使用して商業可能性のある製品を開発できない可能性があり、これは私たちの収入とこれらの製品を開発する戦略にマイナスの影響を与えるだろう。
私たちの協力者や戦略パートナーはbr代替技術を採用するかもしれません。これは私たちの製品の即売性を低下させる可能性があります。また,我々の現在または将来の協力者 や戦略的パートナーは複数の開発プロジェクトに従事する可能性があるため,彼らはその資源を我々と協力しているプロジェクト以外のプロジェクト に移行することを選択することができる.もし彼らがそうすれば、これは私たちの技術能力をテストすることを延期し、私たちのプラットフォームに基づく潜在的な製品の開発を延期または終了するだろう。さらに、私たちの協力者および戦略パートナーは、私たちの協力および戦略的協力計画によって生成された製品を開発しないか、またはこれらの製品の開発、製造、マーケティング、または販売に十分な資源を投入しないことを選択することができる。現在または将来のパートナーとの合意 に従って候補薬剤を開発して商業化できなければ、将来のマイルストーンや特許使用料支払いを得ることができず、私たちの収入に悪影響を及ぼすだろう。
もし私たちが1つ以上の協力を達成した場合、私たちは私たちの候補薬物開発の重要な権利および制御権を放棄すること、またはbr}不利な条項の制約を放棄することを要求されるかもしれない。
私たちが未来に行う任意の協力は私たちを多くのリスクに直面させるかもしれない
| · | 私たちの協力者が候補薬の開発や商業化に投入する資源の数やタイミングをコントロールできないかもしれません | |
| · | 協力者は臨床試験を延期し、不足した資金を提供し、臨床試験を中止したり、候補薬物を放棄したり、新しい臨床試験を繰り返したり、あるいは新しいバージョンの候補薬物に臨床試験を要求することができる | |
| · | 協力者は、戦略協力計画によって生成された製品のさらなる開発と商業化を行ってはならない、あるいは研究開発計画を停止することを選択することができる | |
| · | 協力者は候補薬をマーケティングし販売するのに十分な資源を投入していないかもしれませんこれらの製品から得られる潜在的な収入を制限しています | |
| · | 私たちと私たちの協力者との間に紛争が発生し、候補薬剤の研究、開発または商業化の遅延または終了、または高価な訴訟や仲裁を招き、経営陣の注意を分散させ、資源を消費する可能性がある | |
| · | 協力者は財務的な困難に直面するかもしれません | |
| · | 協力者は、私たちの知的財産権を正確に維持したり、守ったりすることができないかもしれないし、私たちの固有の情報を危険にさらしたり、無効にしたり、潜在的な訴訟に直面させたりする可能性がある方法で、私たちの固有の情報を使用することができる | |
| · | 業務合併または協力者の業務戦略の重大な変化は、任意の手配の下でその義務を履行する協力者の意志または能力にも悪影響を及ぼす可能性がある | |
| · | 協力者は、独立して、または私たちの競争相手を含む他の人と協力して競争相手の候補薬を開発することを決定することができる | |
| · | 協力者はこの合意を終了するか、または満了を可能にすることができ、これは開発を延期し、候補薬物の開発コストを増加させる可能性がある。 |
| 37 |
私たちは製造、販売、マーケティング、流通能力がありません。私たちはこれらの能力を開発するために大量の資源を投入する必要があるかもしれません。
私たちには内部製造能力がありません。 したがって、製造面では、私たちは第三者メーカーに依存しています。我々の戦略は,製薬市場での商業化のために連携パートナーの能力を利用して我々の製品を開発·製造することに基づいており,薬物開発·製造協力者との連携に依存する。もし私たちが既存の協力計画を維持したり、商業的に受け入れられる条項で新しい手配を作ることができなければ、私たちは製品製造と開発活動を要求され、費用は私たち自身が負担します。これは私たちの資本要求を増加させ、あるいは私たちに開発活動の範囲を制限することを要求します。また、私たちは全面的な生物学的同等性や他の臨床研究、準備と監督申請の提出、流通とマーケティング薬品の経験が限られているか、または経験がありません。したがって、私たちは契約者に依存してこのような仕事を行うことができません。 私たちは受け入れられる財務条項の下で協力したり、コンサルタントや外部サービスプロバイダを招いて、私たちの販売、マーケティング、および 流通機能を支援することができないかもしれません。
もし私たちの任意の開発パートナーがbr協定に違反したり、私たちとの合意を終了したり、その協力活動をタイムリーに展開できなかった場合、私たちの医薬製品の臨床前および/または臨床開発および/または商業化は延期され、私たちは製品開発および商業化のために追加資源を投入するか、またはいくつかの開発計画を終了することを要求される。また,許可関係 は,我々の協力者が自ら終了を決定したり,契約条項が終了したときに終了したりし,場合によっては限られた通知のみを我々に送信することができる.協力手配の終了は、私たちの業務、財務状況、運営結果に重大な悪影響を及ぼす可能性があります。第三者と開発されたいかなる技術の所有権にも議論が生じない保証はない。協力者との間のこれらおよび他の可能な相違は、私たちの薬品の開発または商業化の遅延をもたらす可能性があり、または訴訟または仲裁を引き起こす可能性があり、これは時間がかかり、高価である可能性があり、私たちの業務、財務状況、および運営結果に重大な悪影響を及ぼす可能性がある。私たちが自分で臨床試験、販売、マーケティング、流通機能を実行することを決めても、私たちはいくつかの追加的なリスクに直面する可能性があります
| · | 私たちは臨床研究者を引き付けることができず、有効な臨床試験を構築することができず、堅固なマーケティング部門や販売チームを構築することもできないかもしれない | |
| · | 内部臨床試験計画、マーケティング部門、または販売チームを確立するコストは、私たちが利用可能な財務資源、および私たちの現在の任意の候補製品(承認された場合)、または私たちが開発、許可または買収する可能性のある任意の他の薬品によって生じる収入を超える可能性がある | |
| · | 私たちの直販とマーケティング努力は成功しないかもしれない。 |
このような活動を展開できなかった行為は、私たちの業務、財務状況、および運営結果に大きな悪影響を及ぼす可能性があります。
私たちの第三者への依存は、私たちのビジネス秘密を共有することを要求し、これは、競争相手がこれらの秘密や私たちのビジネス秘密が盗用または開示されていることを発見する可能性を増加させる。
私たちは第三者に依存して私たちの医薬製品を生産し、私たちは様々な組織や学術機関と協力して私たちの医薬製品を開発しているので、私たちは時々彼らと商業秘密を共有しなければならない。独自の情報の研究または開示を開始する前に、当社の協力者、コンサルタント、従業員およびコンサルタントとセキュリティ協定、材料譲渡協定、共同研究協定、コンサルティング協定、または他の同様の合意を締結することによって、当社のノウハウを部分的に保護することを求めています。これらのプロトコルは、一般に、商業秘密のような第三者が私たちの機密情報を使用または開示する権利を制限する。第三者と協力する際には、商業秘密および他の機密情報を共有する必要があり、これは、私たちの競争相手に知られ、意図せずに他の人の技術に組み込まれたり、開示されたり、これらの合意に違反するリスクが増加するbr}のような商業秘密が増加する。私たちの独自の地位が私たちのノウハウおよび商業秘密にある程度基づいていることを考慮すると、競争相手は、私たちの商業秘密または他の許可されていない使用または開示が私たちの競争的地位を損なうことを発見し、私たちのビジネスに実質的な悪影響を及ぼす可能性がある。
| 38 |
さらに、これらの合意は、通常、私たちの協力者、コンサルタント、従業員、およびコンサルタントが、私たちのビジネス秘密に関連する可能性のあるデータを発行する能力を制限します。我々の学術 協力者は通常,事前に通知し,指定された 時間を延期して,連携による知的財産権を保護することが可能であることを前提としたデータを発表する権利がある.他の場合、出版権は私たちが独占的に統制しているが、場合によっては、私たちは他の当事者たちとこのような権利を共有するかもしれない。私たちはまた共同研究開発計画を行うことができます。これらの計画は、私たちの研究開発パートナーまたは同様の合意の条項に基づいて商業秘密を共有することを要求するかもしれません。私たちの競争相手は、これらの合意に違反して、私たちの商業秘密を含む情報を独立して開発したり、発表したりすることによって、私たちの商業秘密を発見することができます。私たちは発行時に所有権または他の保護された権利を持っていません。競争相手は私たちのビジネス秘密が私たちの競争地位を損なうことを発見し、私たちの業務に悪影響を及ぼすだろう。
私たちの契約メーカーは私たちの製品を生産する時に厳しい規制を受けています。私たちが依存している製造施設は規制要求を満たし続けることができず、生産能力が限られているかもしれない。
私たちは現在数量の限られたbr家のサプライヤーと私たちの薬品を生産する関係を構築した。各ベンダーは、コンポーネント を製造するためにライセンスを必要とする場合があり、そのようなプロセスがプロバイダに属さない場合、または公共分野に属しており、私たちが所有する可能性のあるそのような活動に関連する知的財産権 を譲渡または再許可することができない可能性がある。
臨床研究または商業販売のための薬物br製品の製造に参加するすべての実体は、私たちの既存の候補薬物契約メーカーを含めて、広範な監督管理を受けている。商業販売または末期臨床研究のための完成薬品の成分はcGMPに従って生産されなければならないことが許可された。本規定は,調査製品と承認販売された製品の品質 を制御·確保するために,生産過程とプログラム(記録保存を含む) および品質システムの実施と実行を管理する.生産過程の不良な制御は、外来製剤または他の汚染物質の導入、または無意識に私たちの薬品の性能または安定性を変化させる可能性があり、これらの変化は最終製品試験では検出できない可能性がある。我々の契約 メーカーは、NDAまたはBLAをサポートするすべての必要なファイルをタイムリーに提供しなければならず、FDAがその施設検査計画によって実行されるGLPおよびcGMP規定を遵守しなければならない。私たちの一部またはすべての第三者請負業者の施設および品質システムは、適用される法規に適合するために、承認前に検査されなければならず、規制部門として、私たちの医薬製品または任意の他の潜在的製品の条件を承認しなければならない。さらに、規制当局は、私たちの医薬製品または他の潜在的製品または関連品質システムの製造に関連する製造施設が、進行中の活動に適用される法規に適合しているかどうかを随時審査または検査することができる。これらの施設が事前に承認された工場検査を通過しなければ、FDAはその製品を承認しないだろう。
規制当局はまた、製品の販売を承認した後のいつでも、当社の第三者請負業者の製造施設を監査することができる。もしそのような検査またはbr審査が適用法規に適合していないことが発見された場合、または私たちの製品仕様または適用法規に違反する行為が、そのような検査または審査から独立して発生した場合、私たちまたは関連規制機関は、私たちまたは第三者にとって高価および/または時間がかかる可能性があり、臨床 研究または商業販売を一時的または永久的に一時的または永久的に一時私たちと契約を結んだ第三者に実施されるどのような救済措置も、私たちの業務に実質的な損害を与える可能性があります。
我々の第三者製造業者が規制適合性を維持できない場合、FDAは、処理すべき候補薬物の申請を拒否するか、または以前に存在する承認を撤回することを含む規制制裁を実施することができる。したがって、私たちの業務、財務状況、運営結果は実質的な損害を受ける可能性があります
また、承認された製造業者の供給が中断されると、商業供給が深刻に中断される可能性がある。必要な製造能力を備えたメーカーの数は限られている。また,代替メーカーはNDAやBLAで補完して資格を取得する必要があり,さらなる遅延を招く可能性がある.新しいメーカーに依存して商業生産を行う場合、規制機関は追加的な研究を行う必要があるかもしれない。製造業者の交換は大量のコストを伴う可能性があり、私たちに必要な臨床およびビジネススケジュールの遅延を招く可能性があり、これは私たちの業務および運営結果に実質的な損害を与える可能性がある。
| 39 |
これらの要素は私たちの薬品の臨床研究、監督提出、必要な承認或いは商業化の遅延を招き、及び/或いは私たちにもっと高いbrコストを発生させ、私たちの製品の商業化の成功を阻害する可能性がある。さらに、私たちのサプライヤーが契約要件を満たしておらず、 であり、実質的に同じコストで生産できる1つ以上の代替サプライヤーを得ることができない場合、私たちの臨床研究が遅延する可能性があり、または潜在的な収入を損失する可能性があり、これは私たちの業務および運営結果に実質的な損害を与える可能性がある。
私たちの知的財産権に関するリスクは
もし私たちが私たちの知的財産権を十分に保護したり実行できなかったら、私たちは効果的に作動できないかもしれない。
私たちの成功と競争能力は私たちの特許、独自の処方、そして商標に大きく依存する。私たちの特許および関連商標およびライセンスが挑戦されず、その後、失効および/またはキャンセルされることは保証されない。特許または商標のいずれかまたは全部の無効または廃止は、私たちのビジネスの見通しを深刻に損なうだろう。さらに、私たちは、私たちの権利を強化するために、私たちの特許または商標が可能かもしれない商標の当事者に法的に挑戦する必要があることを発見するかもしれない。いかなる 特許が最終的に有効であるかは保証されず、いかなる特許、商業秘密、ノウハウ、または他の知的財産権を保護する努力が成功することも保証されない。
私たちのような製薬とバイオテクノロジー会社の特許地位は不確実であり、複雑な法律と事実の問題に関連している。私たちは多くのアメリカと外国特許、そしていくつかの出願されている特許出願を持っており、私たちの候補薬物と技術の様々な側面をカバーしている。発行された特許 が法廷で有効かつ強制的に実行されることは保証されない。有効かつ強制的に実行可能と認定された特許についても,このような判決の獲得に関する法的手続きは時間的で高価である.さらに、発行された特許は、反対または他の手続きにさらされる可能性があり、これらのプログラムは、特許が撤回されるか、または修正された形態(および、特許が商業的関連性および/または広いカバー範囲を有さない形態で)維持される可能性がある。また,我々の競争相手は を迂回したり,我々の特許を迂回して設計することができるかもしれない.製薬製品の開発や商業化が大きな遅延を受ける可能性があるため、特許が発行されて強制的に実行されても、特許は早期に満了する可能性があり、我々の特許がカバーする製品が商業化された後には、あれば短い保護期間しか提供できない。私たちは米国特許商標局が発表した介入手続きに参加しなければならないかもしれないが、これは特許損失を招き、および/または私たちに巨額のコストをもたらす可能性がある。
私たちは特許出願を提出し、私たちの候補薬物と技術の様々な側面をカバーするために、より多くの特許出願を提出することを計画している。私たちが出願した特許出願が本当に特許として発行されることが保証されないか、または商業的関連性および/または広いカバー面を有するようにすることはできない。 は、特許発行前に、特許出願において要求されるカバー面を著しく減少させることができる。請求範囲 は,我々が第三者と取引を許可する能力と,我々のパートナー関係から印税を得る権利に重要である .科学または特許文献で発見された発表は、そのような発見の日よりも遅れていることが多いため、私たちが私たちの特許または特許出願によってカバーされている発明の最初の発明者であることを決定することはできない。また、ある発明のための特許出願を最初に提出した会社であることは保証されない。
知的財産権(特許を含む)に関連する任意の司法手続きにおいて不利な結果が生じることは、第三者から論争のある権利許可を得る必要があるか、または論争のある技術の使用を停止することを要求する第三者に重大な責任を負わせる可能性がある。私たちが他人に知的財産権の許可を求める場合、私たちは商業的に合理的に許可を得ることができず、私たちの技術および/または製品を自由に商業化する能力に対する懸念を引き起こす可能性がある。開発および商業化活動中に生成された発明の特許可能な態様 を識別できない場合もあり、そうでなければ、特許保護 を得ることができないかもしれない。さらに、場合によっては、第三者からの許可が第三者に与えられるかもしれない技術をカバーする特許出願の準備、提出、および起訴(または保守特許)を制御する権利がない可能性がある。私たちは私たちの許可側または 許可側に依存している。したがって、これらの特許および出願 は、我々の業務の最適な利益に適合した方法で起訴され、強制されてはならない。現在または将来の許可者または許可者が、そのような特許および他の知的財産権を確立、維持または保護できない場合、そのような権利は減少またはキャンセルされる可能性がある。もし私たちのライセンシーまたはライセンシーがいかなる特許権を起訴、維持または強制執行する上で完全に協力しないか、または私たちの意見に同意しない場合、このような特許権は損害を受ける可能性がある。
私たちの知的財産権 を十分に保護または実行できなければ、私たちの業務、運営結果、および将来性に大きな悪影響を及ぼす可能性があります。
| 40 |
我々の候補薬物に適用された発行済み特許 が法廷で疑問視されれば,無効または実行不可能と認定される可能性がある。
私たちまたは私たちの許可パートナーのうちの1つが第三者に対して法的訴訟を提起して、私たちの候補薬剤のうちの1つをカバーする特許を強制的に実行する場合、被告は、私たちの候補薬剤をカバーする特許が無効および/または実行不可能であることを反訴することができる。米国の特許訴訟では,被告が無効および/または実行不可能と主張する反訴はありふれている。有効性を疑問視する理由は,新規性の欠如,明らかな,または実施できないことを含むいくつかの法定要求のいずれかの を満たしていないといわれている可能性がある。執行的に断言できない理由 は,特許起訴に関連する者が起訴中に米国特許商標局に関連情報を隠蔽したり,誤った声明をしたりした疑いである可能性がある.第三者は米国や海外の行政機関に類似したクレームを出すことも可能であり, は訴訟範囲外でも同様である。このようなメカニズムには、外国司法管区における再審、贈与後審査、同等の手続き(例えば、反対手続)が含まれる。このような訴訟は、私たちの特許が撤回または修正される可能性があり、これらの特許が私たちの候補薬物をカバーしなくなる可能性がある。法律が無効と強制できないと断言した後の結果は予測できない。 例えば、有効性の問題については、無効な以前の技術がないとは判断できないが、私たちと特許審査員は起訴中に知らない。被告が無効および/または実行不可能な法的主張で勝利した場合、私たちは候補薬物の少なくとも一部またはすべての特許保護を失うだろう。このような特許保護の喪失は私たちの業務に実質的な悪影響を及ぼすだろう。
私たちは世界的に私たちの知的財産権を保護できないかもしれない。
世界のすべての国/地域で候補薬物特許を申請、起訴、保護する費用は目を引くほど高く、米国以外のいくつかの国/地域での知的財産権は米国の知的財産権ほど広くないかもしれない。また、一部の国の法律は知的財産権の保護程度は米国の連邦や州法律に及ばない。したがって、私たちは、米国以外のすべての国/地域で第三者が私たちの発明を実施したり、米国または他の司法管轄区で私たちの発明を使用して製造された製品を販売したり、輸入したりすることを阻止することができないかもしれない。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの発明を使用して自分の製品を開発することができ、また、他の侵害製品を私たちの特許保護があるが法執行力がアメリカに及ばない地域に輸出することもできる。これらの製品は私たちの製品と競争するかもしれませんが、私たちの特許や他の知的財産権はそれらの競争を効果的にまたは阻止するのに十分ではないかもしれません。
多くの会社は外国の管轄区域の知的財産権の保護と擁護において重大な問題に直面している。特定の国の法律制度、特に特定の発展途上国の法律制度は、特許、商業秘密、および他の知的財産権保護の実行、特にバイオテクノロジー製品に関する保護を支持しておらず、これは、私たちの特許を侵害したり、私たちの独占権に違反する競争製品の販売を阻止することを困難にする可能性がある。外国の管轄区域で私たちの特許権を強制的に執行する訴訟手続きは、巨額のコストを招き、私たちの努力と注意を私たちの業務の他の側面から移転させる可能性があり、私たちの特許が無効または偏狭に解釈されるリスクに直面する可能性があり、私たちの特許出願が発行できないリスクに直面し、第三者のクレームを引き起こす可能性がある。私たちは私たちが始めたどんな訴訟でも勝てないかもしれないし、何か損害賠償や他の救済措置があれば、ビジネス的な意味がないかもしれない。したがって、私たちが世界各地で知的財産権を実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネスメリットを得るのに十分ではないかもしれません。
私たちの世界各地の知的財産権を十分に保護できなければ、私たちの業務、運営結果、将来性に大きな悪影響を及ぼす可能性がある。
もし私たちが他人の知的財産権を侵害すれば、私たちの業務と収益性は不利な影響を受ける可能性がある。
私たちのビジネス成功はまた、私たちと私たちのパートナーが他人の特許や独自の権利を侵害しないことにある程度依存するだろう。私たちのパートナーが使用または開発し、私たちがマーケティングおよび販売するbr技術および製品がこのような権利を侵害しないことは保証されません。このような侵害が発生し、私たちおよび私たちのパートナーが関連する第三者から許可を得ることができない場合、私たちは、そのような侵害技術または製品の開発、製造、使用、または販売を継続することができないだろう。 が第三者技術の必要なライセンスを提供するかどうか、または商業的に合理的な条項でライセンスを提供するかどうかは保証できない。場合によっては、訴訟または他の手続きによって侵害クレームに対抗または主張するか、または第三者固有の権利の範囲および有効性を決定する必要がある場合がある。どんな潜在的な訴訟も、私たちの資源と資源の大量のコストと移転をもたらす可能性があり、 は私たちに実質的な悪影響を及ぼす可能性がある。このような訴訟または訴訟における不利な結果は、主題技術の使用を停止すること、または第三者から主題技術の許可を得ることを要求する重大な責任を私たちに負わせる可能性があり、これらのすべては、私たちの業務に重大な悪影響を及ぼす可能性がある。
| 41 |
我々が第三者に知的財産権を許可するプロトコルにおける義務 ,あるいは我々とライセンス側との業務関係が中断された場合,我々は我々の業務に非常に重要な許可権を失う可能性がある.
私たちは私たちの業務に非常に重要な知的財産権ライセンス協定の多くの参加者であり、将来的により多くのライセンス契約を締結したいと思っています。私たちの既存のbrライセンス協定は、将来のライセンス契約は、私たちに様々な職務調査、マイルストーン支払い、印税、その他の義務を課すことを予想しています。もし私たちがこれらの合意の下で私たちの義務を履行できなかった場合、あるいは私たちが破産状態にある場合、許可側 は許可を終了する権利がある可能性があり、この場合、許可がカバーする製品を販売することができません。
私たちは時々そうするために、第三者からライセンスを取得して私たちの研究を進める必要があるかもしれない。私たちは合理的なコストやbrの合理的な条項でこれらのライセンスを得ることができないかもしれない。この場合、代替技術 を開発または許可するために、多くの時間およびリソースが必要となる可能性がある。もし私たちがそれができなければ、私たちは影響を受けた候補薬を開発できないかもしれません。これは私たちの業務を深刻に損なう可能性があります。私たちは現在の候補薬や未来の製品に強制的に施行される可能性のある第三者特許が存在しないことを保証できません。販売を禁止したり、販売については、第三者に印税および/または他の形態の賠償を支払う義務があります。
多くの場合、私たちが許可した技術の特許訴訟は完全に許可者によって統制されている。もし私たちの許可者が彼らから私たちが許可した独自のbr知的財産権の特許または他の保護を獲得し、維持できなかった場合、私たちは知的財産権の権利またはこれらの権利に関連する専有権を失う可能性があり、私たちの競争相手は知的財産権マーケティング競争製品を使用することができる。場合によっては、私たちは許可技術によって作られた特許の起訴を統制する。もし私たちがこのような起訴に関連したいかなる義務にも違反すれば、私たちは私たちの許可パートナーに重大な責任を負うかもしれない。知的財産権許可は私たちの業務に重要であり、複雑な法律、商業と科学問題に関連し、そして私たちの業界の科学発見の迅速な発展によって複雑になった。ライセンス契約によると、知的財産権について論争が生じる可能性があります
| · | ライセンス契約に従って付与された権利範囲および解釈に関連する他の問題; | |
| · | 私たちの技術およびプロセスは、ライセンス契約に拘束されていないライセンス側の知的財産権をどの程度侵害しているか | |
| · | 私たちの協力開発関係に基づいて、特許と他の権利を再許可する | |
| · | 私たちのライセンス契約の下での義務と、どのような活動がこれらの職務義務を満たしていますか | |
| · | 私たちの許可者、私たちと私たちのパートナーが知的財産権を共同で創造または使用することによって生成された発明およびノウハウの所有権; | |
| · | 特許技術発明の優先権。 |
もし私たちの許可された知的財産権紛争が、許容可能な条項で現在の許可スケジュールを維持する能力を阻害または弱化させた場合、影響を受ける候補薬剤の開発に成功し、商業化することができない可能性があり、これは私たちの業務に大きな悪影響を及ぼす可能性がある。
私たちは、私たちの従業員、コンサルタント、または独立請負業者が第三者の機密情報を誤って使用または開示したか、または私たちの従業員がその前の雇用主が主張した商業機密を誤って使用または開示したという疑惑の影響を受けるかもしれない。
私たちが雇った個人は以前、私たちの競争相手や潜在的な競争相手を含む大学や他のバイオテクノロジーや製薬会社に雇われていた。私たちは、私たちまたは私たちの従業員、コンサルタント、または独立請負業者が、商業秘密または他の固有の情報を含む、私たちの従業員の任意の元雇用主または他の第三者の知的財産権を無意識に使用または漏洩することを指摘されるかもしれません。 は、これらのクレームに対抗するために訴訟を提起する必要があるかもしれません。もし私たちがこのようなクレームを弁護することができなければ、金銭的損失を支払う以外に、私たちは貴重な知的財産権や人員を失う可能性があり、これは私たちの業務に悪影響を及ぼすかもしれない。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。
| 42 |
私たちは私たちの特許と他の知的財産権の発明権や所有権のクレームに疑問を受けるかもしれない。
私たちはまた、元従業員、協力者、または他の第三者が私たちの特許または他の知的財産権に所有権を持っているというクレームを受ける可能性がある。私たちは将来的にbr所有権紛争、例えば、コンサルタントや私たちの候補薬物の開発に参加する他の人の義務衝突 が生じるかもしれない。訴訟は、これらと他の挑戦在庫または所有権のクレームに対抗するために必要かもしれない。もし私たちがこのようなクレームを弁護することができない場合、金銭的損害賠償を支払うことに加えて、貴重な知的財産権、例えば貴重な知的財産権の独自の所有権または使用権を失う可能性がある。このような結果は私たちの業務に実質的な悪影響を及ぼすかもしれない。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。
私たちは私たちの機密情報と商業秘密を保護することができず、これは私たちの業務と競争地位を損なうだろう。
私たちのいくつかの技術と製品のための特許を申請する以外に、私たちは、非特許の技術的ノウハウ、技術、および他の独自の情報を含む商業秘密に依存して、私たちの競争的地位を維持します。私たちは、これらの商業秘密を保護することを求めています。一部は、これらの商業秘密にアクセスする権利のある当事者と秘密保護および秘密協定を締結することによって、例えば、私たちの従業員、会社協力者、外部科学協力者、契約メーカー、コンサルタント、コンサルタント、および他の第三者です。私たちはまた従業員やコンサルタントと秘密協定と発明または特許譲渡協定を締結します。このような当事者たちのいずれも、私たちのビジネス秘密を含む合意に違反し、私たちの固有の情報を漏洩する可能性があり、私たちはこのような違反について十分な救済措置を得ることができないかもしれない。一方が商業秘密を不正に開示したり流用したりするクレームを実行することは困難であり,高価で時間がかかり,結果として予測できない.しかも、アメリカ国内外のいくつかの裁判所は商業秘密を保護することをあまり望んでいないかもしれない。もし競争相手が私たちのいかなる商業秘密を合法的に獲得または独立して開発した場合、私たちはその競争相手がその技術や情報を使用して私たちと競争することを阻止する権利がなく、これは私たちの競争地位と業務を損なう可能性がある。
私たちは、高価で時間がかかり、成功していない可能性があるbrを保護したり、私たちの特許または私たちの許可側の特許を強制的に執行する訴訟に巻き込まれるかもしれない。
競争相手は私たちの特許や私たちの許可側の特許 を侵害する可能性がある。権利侵害や不正使用に対抗するために、私たちは権利侵害請求を要求されるかもしれません。これは高価で時間がかかるかもしれません。さらに、侵害訴訟では、裁判所は、私たちまたは私たちのライセンシーの特許が無効であると判断することができ、br}が強制的に執行されないこと、および/または侵害されていないこと、または私たちの特許が関連技術をカバーしていないことを理由に、他方の関連技術の使用を阻止することを拒否することができる。任意の訴訟または弁護手続における不利な結果は、私たちの1つまたは複数の特許を無効または狭義に解釈されるリスクに直面させ、私たちの特許出願を発行できないリスクに直面させる可能性がある。
第三者によって誘発されるか、または我々によって提起された干渉訴訟手続は、我々の特許または特許出願または我々の許可者の特許または特許出願に関連する発明の優先度を決定するために必要である可能性がある。不利な結果は、私たちが関連技術の使用を停止したり、勝利者から許可を得ようと試みることを要求するかもしれない。もし勝利者が商業的に合理的な条項に従って許可証を提供しなければ、私たちの業務は損害を受ける可能性があります。私たちの訴訟弁護や介入訴訟は失敗する可能性があり、成功しても巨額のコストを招き、私たちの経営陣や他の従業員の注意を分散させる可能性があります。私たちは単独でまたは私たちの許可者と一緒に私たちの知的財産権の盗用を防ぐことができないかもしれません。特にこれらの権利を保護する法律ではアメリカの十分な国に及ばないかもしれません。
また,知的財産権訴訟に関する大量の発見により,我々のいくつかの機密情報は,このような訴訟中に開示により漏洩する可能性がある.聴聞結果、動議、または他の一時的な手続き、または事態の発展も公表される可能性がある。もし証券アナリストや投資家がこれらの結果がマイナスだと思っていれば、私たちの普通株の価格に実質的な悪影響を及ぼす可能性がある。
| 43 |
米国特許法の変化は特許の全体的な価値を低下させ、製品を保護する能力を弱める可能性がある。
他の生物製薬会社と同様に、私たちの成功は知的財産権、特に特許に大きく依存している。バイオテクノロジー業界で特許を取得し、実施することは、技術的な複雑さと、法律上の複雑さとも関連するため、コストが高く、時間がかかり、本質的に不確実である。また、米国は範囲の広い特許改革立法を制定し、実施し続けることが期待されている。さらに、米国最高裁判所のいくつかの裁決は、場合によっては特許保護の範囲を縮小し、および/または場合によっては特許所有者の権利を弱める。我々の将来の特許取得能力に対する不確実性の増加に加えて,このようなイベントの組合せ は,いったん特許を取得する価値に関する不確実性をもたらしている.米国議会、連邦裁判所、USPTOの決定によると、特許を管理する法律·法規は予測不可能な方法で変化する可能性があり、それにより、私たちが新しい特許を獲得したり、私たちの既存特許と将来獲得可能な特許を強制的に執行する能力を弱めることができる。
特許改革立法は、私たちと私たちの許可側を起訴する特許出願および私たちまたは私たちの許可者が発行する特許を実行または保護することをめぐる不確実性とコストを増加させる可能性があります。2011年9月に成立した“ライシー·スミス米国発明法”(“ライシー·スミス法案”)の条項は、米国特許法をいくつかの重大な改正を行い、その影響はまだ現れている。Leahy-Smith法案とその施行、 に加えて、任意の新しい法規に加えて、私たちの特許出願をめぐる起訴と、私たちが発行した特許の実行または保護の不確実性とコストを増加させる可能性があり、これらはすべて私たちの業務と財務状況に実質的な悪影響を及ぼす可能性がある。
私たちの特許保護を獲得し、維持することは、政府特許機関によって適用される様々なプログラム、書類提出、費用支払い、および他の要件を遵守することに依存し、これらの要件に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
特許および/または出願の定期維持費、継続費、年会費および様々な他の政府費用は、特許および/または出願の有効期間内にいくつかの段階でUSPTOおよび米国以外の様々な政府特許機関に支払われる。米国特許商標局および様々な非米国政府特許代理機関は、特許出願中にいくつかのプログラム、文書、費用支払い、および他の同様の条項を遵守することを要求する。規定を遵守しないことは、特許または特許出願の放棄または失効を招き、関連する管轄区域特許権の一部または全部を喪失させる可能性がある。この場合,我々の競争相手が市場に参入する可能性があり, このような状況は我々の業務に実質的な悪影響を与える.
| 44 |
私たちの業務運営に関するリスク
金融サービス業の不利な事態の発展に影響を与え、例えば流動性、違約又は取引相手の実際の事件又は懸念に関連して、会社の現在及び予想されている業務運営及びその財務状況及び運営結果に悪影響を及ぼす可能性がある。
流動性が限られている、違約、業績不良、または他の不利な発展に関連する実際の事件、金融機関、取引相手または金融サービス業または金融サービス業全体に影響を与える他の会社、または任意のこのような事件または他の同様のリスクに対する懸念またはデマは、過去に発生したことがあり、将来は市場全体の流動性問題を招く可能性がある。例えば、2023年3月10日、シリコンバレー銀行(SVB)はカリフォルニア州金融保護·革新部によって閉鎖され、後者は連邦預金保険会社(FDIC)を担当者に指定した。同様に,2023年3月12日にSignature BankとSilvergate Capital Corp. がそれぞれ破産管理プログラムに巻き込まれた.財務省、FRB、連邦預金保険会社の声明によると、SVBのすべての預金者は、1つの営業後だけでそのすべての資金を抽出することができるが、無保険預金口座内の資金、信用協定下の借り手、信用協定下の借り手、信用状およびいくつかの他の金融商品、署名銀行、または連邦預金保険会社に接収された任意の他の金融機関を含むが、その中で抽出されていない金額を抽出できない可能性がある。我々 のほとんどの現金はSVBに保管されており,その大部分はFDICの保険を受けておらず,保険やFDIC保険限度額を超えていない現金残高 を定期的に維持している。2023年3月13日まで、私たちは私たちの資金を完全に使用することができる。もし私たちの預金資金が使用できない場合、私たちの流動資金および私たちの現在および/または予想されている業務および財務状況と運営結果に重大な悪影響を及ぼす可能性があります。私たちはSVB、署名、または現在接収状態にある金融機関の借り手、またはそのような金融機関の任意の側ではありませんが、私たちの任意の顧客、サプライヤー、または私たちと業務往来のある他の当事者が、そのような金融機関のこのようなツールや融資スケジュールに基づいて資金を得ることができない場合、債務を返済してくれたり、追加金を支払う必要がある新しい商業手配を締結する能力が悪影響を受ける可能性があります。この点で、SVB信用協定や手配された取引相手および信用証受益者などの第三者はSVB閉鎖の直接影響を受ける可能性があり、より広範な金融サービス業の流動性に対する懸念は依然として不確定性がある。同様の影響は、例えば2008-2010年の金融危機の間に過去にも発生したことがある。
インフレと金利の急速な上昇は、以前発行された金利が現在の市場金利よりも低い国債の取引価値を低下させる。米国財務省、連邦預金保険会社、および連邦準備委員会は、このようなツール販売の潜在的損失リスクを軽減するために、金融機関が保有するいくつかのこのような政府証券を保証する金融機関に250億ドルまでの融資を提供する計画を発表しているが、即時流動性に対する金融機関の広範な需要または他の流動性需要は、この計画の能力を超える可能性がある。また、米国財務省、連邦預金保険会社、連邦準備委員会が将来、他の銀行や金融機関が閉鎖された場合に未保険資金の使用を提供する保証はなく、適時にそうする保証もない。
私たちは私たちの銀行関係が必要または適切であると評価しているにもかかわらず、私たちが現在および予想されている将来の業務運営に資金または資本化を提供するのに十分な資金源および他の信用手配の機会を得ることは、会社または金融サービス業全体または全体経済に影響を与える要素が、私たちの融資源および他の信用手配を深刻に損なう可能性がある。これらの要因には、流動性緊張や失敗などの事件、様々な金融、信用または流動性合意または手配に基づいて義務を履行する能力、金融サービス業または金融市場の中断または不安定、または金融サービス業会社の将来性への懸念または負の予想が含まれる可能性がある。これらの要因は、会社と財務や業務関係にある金融機関や金融サービス業界会社に関連する可能性があるが、金融市場や金融サービス業界に関連する要因も含まれている可能性がある。
| 45 |
Brのうちの1つまたは複数の要因に関連するイベントまたは問題の結果は、私たちの現在および予想されるビジネス運営、ならびに私たちの財務状態および運営結果に生じる様々な重大かつ不利な影響を含む可能性がある。これらは、預金または他の金融資産の取得遅延、または未加入預金または他の金融資産の損失、または未加入預金または他の金融資産の損失、または現金管理スケジュールおよび/または現金管理スケジュールによって制限された資金の取得遅延または実際の損失を含むことができるが、これらに限定されない。
米国または国際金融システムに対する投資家の懸念は、より高い金利またはコスト、より厳しい財務および経営契約、または信用および流動性源を得るための体系的な制限を含む、あまり有利ではない商業融資条項を招き、私たちがより受け入れ可能な条項または受け入れ可能な条項で融資を受けないようにする可能性がある。利用可能な資金または現金および流動資金源の任意の減少は、他のリスクに加えて、運営費用、財務的義務、または他の義務を履行する能力に悪影響を及ぼす可能性があり、br}は、私たちの財務および/または契約義務の違反を招き、または連邦または州賃金および労働法違反をもたらす可能性がある。上記の影響の任意の ,または上記の要因または他の関連または同様の要因に起因する任意の他の影響は、我々の流動資金、私たちの現在および/または予想されるビジネス運営および財務状態、ならびに 運営結果に重大な悪影響を及ぼす可能性がある。
さらに、マクロ経済または金融サービス業のいずれのさらなる悪化も、私たちの顧客またはサプライヤーの損失または違約を招く可能性があり、さらに、私たちの現在および/または予想される業務運営および運営結果および財務状況に大きな悪影響を及ぼす可能性がある。例えば、顧客 は、満期時に支払うことができない可能性があり、私たちとの合意に基づいて違約、破産または破産を宣言するか、またはサプライヤー がもはや顧客として私たちと付き合っていないと判断する可能性がある。さらに、顧客またはサプライヤーは、上述した企業に重大な悪影響を及ぼす可能性のある任意の流動性または他のリスクの悪影響を受ける可能性があり、これらに限定されないが、未保険預金を取得する機会を遅延させたり、または既存のクレジットスケジュールを利用する能力を失ったりすることは、苦境または倒産に陥った金融機関に関するものである。いかなる顧客またはサプライヤーが破産したり、債務を相殺しないか、または任意の顧客が満期時に支払うことができなかったか、または顧客またはサプライヤーの任意の違約または違約、または任意の重要なサプライヤー関係を失うことは、会社の重大な損失を招く可能性があり、私たちの業務に重大な悪影響を及ぼす可能性がある。
私たちの未来の成功は私たちの幹部チームの主要なメンバー、顧問と顧問及び合格者を吸引、維持、激励する能力にかかっている。
私たちは私たちの実行チームの主要なメンバーに強く依存しており、彼らのサービスの喪失は私たちの目標の実現に悪影響を及ぼすかもしれない。私たちの業務のために他のbrに合格した従業員、コンサルタントと顧問を募集し、維持することは、科学と技術者を含めて、私たちの成功にも重要である。現在、私たちの産業にはスキルのある幹部が不足しており、このような状況は続く可能性が高い。そのため,技能人材に対する競争は非常に激しく,流出率が高い可能性がある。私たちは多くの製薬とバイオテクノロジー会社が似たような技能を持つ人員を争っているため、受け入れ可能な条件で人材を誘致し、維持することができないかもしれない。そのほか、br}が臨床前或いは臨床研究で成功できなかったことは合格者の採用と保留を更に挑戦的になる可能性がある。役員、コンサルタント、コンサルタントを募集したり失うことができないサービスは、私たちの研究開発目標の進展を阻害する可能性があります。
| 46 |
私たちは私たちの組織 を拡張する必要があり、私たちはこのような成長を管理する際に困難に直面する可能性があり、これは私たちの運営を中断する可能性がある。
2022年12月31日現在、私たちは4人の常勤従業員 がいます。私たちが成熟するにつれて、私たちは私たちの全職員基盤を拡大し、より多くのコンサルタントと請負業者を雇う必要があるかもしれない。我々の経営陣 は,我々の日常活動から不比例な注意を移し,これらの成長活動を管理するために多くの時間 を投入する必要があるかもしれない.私たちは私たちの業務の拡張を効果的に管理できないかもしれません。これは、私たちのインフラの弱さ、運営ミス、ビジネスチャンスの喪失、従業員の流失、余剰従業員の生産性の低下を招く可能性があります。これらは、私たちの業務、運営結果、将来性に実質的な悪影響を及ぼす可能性があります。将来のどのような成長も大量の資本支出を必要とする可能性があり、より多くの候補薬剤の開発など、他のプロジェクトから財源を移転する可能性がある。私たちの経営陣が私たちの成長を効果的に管理できなければ、私たちの費用の増加は予想を超える可能性があり、私たちの収入を創出および/または増加させる能力は低下する可能性があり、私たちは私たちの業務戦略を実施できないかもしれない。私たちの将来の財務業績(Br)と候補薬物を商業化し、効果的に競争する能力は、私たちが将来のどんな成長を効果的に管理する能力にある程度依存するだろう。
私たちは協力協定や他の重要な合意の側であり、これらの合意には複雑なビジネス条項が含まれており、紛争、訴訟または賠償責任 を招く可能性があり、これは私たちの業務、運営結果、財務状況に悪影響を及ぼす可能性がある。
私たちは現在、バイオテクノロジーや製薬会社との協力協定から全部または大部分の収入を得ており、予測可能な未来にこれらの収入を得る予定だ。これらの協調 プロトコルは、以下を含む複雑なビジネス条項を含む
| · | ある商業合理性業績標準に基づく臨床開発と商業化義務は、もし私たちのパートナーの業績の適切性が論争が発生すれば、実行が困難である | |
| · | 協力した候補薬物開発プロジェクトのために割り当てられた人員と他の資源の研究開発業績と精算義務 | |
| · | いくつかのプロトコルは、私たちがパートナーに提供する製品の実際のコスト定価に基づいており、コスト分担式および方法は複雑である | |
| · | 私たちと私たちのパートナーとの間の知的財産権所有権割り当ては、協力過程で開発された改善および新しい発明のために使用される | |
| · | 純売上計算、地理的位置、特許請求の範囲、特許有効期間、模倣薬競争相手、バンドル定価、および他の要因を含む、いくつかの複雑な変数に基づく医薬品販売特許権使用料 | |
| · | 知的財産権侵害、製品責任、そしていくつかの他のクレームの賠償義務。 |
私たちは時々第三者と非公式な論争解決討論を行い、私たちの合意に含まれる複雑なビジネス条項をどのように正確に解釈するかを議論するかもしれません。将来的には、私たちの協力協定、取引文書、または第三者許可協定に関する1つまたは複数の論争が発生またはアップグレードする可能性があり、最終的には高価な訴訟や契約条項の不利な解釈を引き起こす可能性があり、これは私たちの業務、財務状況、および運営結果に大きな悪影響を及ぼすでしょう。
| 47 |
我々は競争が極めて激しい環境で を運営しており,競争技術が我々の業務発展を損なわない保証はない.
私たちは急速に発展する分野に従事している。多くの製薬会社からの競争は非常に激しく、さらに激化すると予想される。巨大で急速に増加する腫瘍治療市場は新しい参入者を引き付けるかもしれない。多くのバイオテクノロジーと製薬会社は癌治療と免疫腫瘍技術の開発に集中している。これらの会社の多くは(すべてでなければ)私たちよりも多くの財務や他の資源や開発能力 を持っている。私たちの多くの競争相手は製品の臨床前と臨床テストを担当し、監督管理の許可を得て、処方薬製品の製造とマーケティングにおいてもより豊富な集合経験を持っている。私たちが開発している候補薬が競争相手の製品よりも効果的あるいはより大きな市場受容度を得ることが保証されないし、私たちの競争相手が私たちが開発している製品や技術よりも効果的な製品や技術の開発に成功しない保証もないし、私たちの製品や技術が私たちの製品や技術競争力を低下させたり時代遅れになる保証はない。また、他社が開発した新薬や改善薬が私たちの薬品を余分にしたり時代遅れにしない保証はありません。
潜在的な新しい会計基準や立法行動は、私たちの将来の財務状況や経営結果に悪影響を及ぼす可能性がある。
将来の財務会計基準の変化 は収入や費用を確認する時間に不利な意外な変動をもたらす可能性があり、私たちの財務状況や経営業績に影響を与える可能性がある。将来的には新しい基準が出現する可能性があり、会計政策の変更を要求する可能性があります。変化するコーポレートガバナンスおよび公開開示法規を遵守することは、追加の費用をもたらす可能性があります。2002年の“サバンズ-オクスリ法案”(あるいは“サバンズ-オクスリー法案”)、新しい“アメリカ証券取引委員会”法規、上場企業会計監督委員会(PCAOBと略す)基準、ナスダック規則を含む、会社のガバナンスと公開開示に関する法律、法規、基準が変化しており、私たちのような会社 に不確実性をもたらしている。このような不確実性と他の要素のため、保険、会計、そして監査コストは高い。
私たちの資本資源は限られていて、現在私たちの財務部門はただ一人の常勤職員しかいない。私たちは外部コンサルタントに依存して私たちの内部専門知識を補完し、高い基準を維持したコーポレートガバナンスと公開開示に取り組んでいます。したがって,発展していく基準を遵守するために必要なすべての合理的な資源を投入する予定であり,この投資は一般や行政費用の増加 を招き,管理時間や注意力を創収活動からコンプライアンス活動に移行させる可能性がある。
私たちの普通株に関するリスクは
私たちはナスダック資本市場の持続的な上場の要求を満たしていません。私たちの普通株が銘柄を取られる可能性があります。ナスダック上場規則を再遵守できなければ、私たちの普通株の市場価格と流動性に影響を与え、私たちの融資能力を低下させるかもしれない。
現在、私たちの普通株はナスダック資本市場で取引されている。2022年6月3日、吾らはナスダック証券市場有限責任会社(“ナスダック”)上場資産部 から書面通知(“通知”)を受け、吾らナスダック普通株の市価が30営業日連続で1.00ドルを下回ったことを通知したため、吾らは“ナスダック上場規則”第5550(A)(2)条を遵守できなかった(2)ナスダック資本市場への最低入札価格要求(“購入価格要求”)を継続した。本通知は、当社の普通株がナスダック資本市場に上場することに直ちに影響を与えません。
ナスダック上場規則によると、180暦の期限があり、通知日から計算して、入札価格要求を再遵守する。したがって,2022年11月30日(“コンプライアンス日”)までに入札価格要求を再遵守しなければならない。2022年12月1日、私たちはナスダックから手紙を受け取り、会社の普通株は1株1.00ドルの最低購入価格要求に適合していないにもかかわらず、ナスダックは180暦を延長する資格があることを確定したか、あるいは2023年5月29日までbrコンプライアンスを回復する資格があることを通知した。ナスダックの決定は,自社が公開保有株式に適合する時価継続上場要求およびナスダック資本市場初上場の他のすべての適用要求(入札価格要求を除く),および吾らからの書面通知に基づいて,第二コンプライアンス期間に逆 株式分割を行うことで不足点を補う意向を示している。
| 48 |
2023年5月29日までの任意の時間に、ナスダック普通株の終値が少なくとも10営業日連続の終値が1株1.00ドル以上に達した場合、ナスダックは、入札要求に達したことを通知する書面通知を出す。
我々は,我々の普通株の終値 を監視し続け,利用可能な選択肢を考慮して不足を解決し,割り当てられたコンプライアンス期間内に入札価格要求 を再遵守する.しかし,入札価格 要求を再遵守したり,他の方法でナスダック上場規則を遵守したりできる保証はない.もし私たちが入札要求を含めてナスダック上場規則を再遵守できなかった場合、私たちはブランドを獲得されるかもしれません。アメリカ証券取引委員会の規定によると、私たちの株は細かい株とみなされるので、私たちの証券を売却するブローカーに追加の販売実践要求を加える規則の制約を受けることになります。これらの要求がブローカーの経営者に与える追加負担は、ブローカーが私たちの普通株での取引を阻害する可能性があります。これは私たちの普通株の市場流動性と株主が二級市場で私たちの証券を販売する能力を深刻に制限する可能性があります。もし私たちの普通株がナスダック資本市場から撤退すれば、私たちの普通株の流動性は大きな影響を受け、これは私たちの普通株の投資家に対する吸引力を低下させ、私たちの普通株の市場価格の下落を招くだろう。しかも、私たちが大型取引所に上場しなければ、私たちは追加的な資本を調達することが難しいかもしれない。
私たちの証券の市場価格は大きく変動するかもしれません。あなたは私たちの証券を売ることができないかもしれません。
株式市場で取引されている会社は一般的に極端な価格や出来高変動を経験しており、これらの変動はこれらの会社の経営業績とは無関係か比例しないことが多い。私たちの実際の経営業績にかかわらず、広範な市場と業界要素は私たちの証券の市場価格にマイナス影響を与える可能性がある。
私たちの証券の市場価格は変動する可能性があります。 私たちの証券の価格は様々な要素によって大幅に変動する可能性があります
| · | 臨床前或いは臨床研究中の不良結果、遅延或いは保留; | |
| · | 追加資金を得ることができません | |
| · | INDまたはBLAへの任意の候補薬剤の提出の任意の遅延、およびINDまたはBLAのFDAの検討に関連する任意の不利な開発、または不利な開発とみなされる; | |
| · | 私たちの候補薬の開発に失敗しました | |
| · | 私たちの既存の戦略的協力関係や新しい協力関係を維持することができなかった | |
| · | 私たちまたは私たちの許可者と戦略的パートナーは私たちの知的財産権を起訴、維持、または実行できなかった | |
| · | 未来の製品に適用される法律や法規の変化 | |
| · | 候補薬に十分な製品を供給することができないか、許容可能な価格でそうすることができません | |
| · | 不利な規制決定; | |
| · | 競争相手は新製品、新サービス、または新技術を導入する | |
| · | 私たちが大衆に提供した財政的予測を達成できなかったか、またはそれを超えることができなかった | |
| · | 投資界の財務予測を達成できなかったか、または超えることができなかった | |
| · | 公衆、立法機関、規制機関、投資界の製薬業に対する見方 | |
| · | 私たち、私たちの戦略的パートナー、または私たちの競争相手は、重大な買収、戦略的パートナーシップ、合弁企業、または資本約束を発表します | |
| · | 特許、訴訟事項、および私たちの技術のための特許保護を得る能力を含む、専有権に関する論争または他の発展; | |
| · | 重要な科学技術者や管理者の増減 | |
| · | 特許または株主訴訟を含む重大な訴訟; | |
| · | 同じ会社の市場予想が変化しています | |
| · | 私たちまたは株主は将来私たちの証券を売却します | |
| · | 公衆衛生問題(例えば、コロナウイルス爆発)および地政学的事件(例えば、ロシアのウクライナ侵攻)および関連する制裁および他の経済的妨害または懸念が一般的な経済活動に及ぼす潜在的な悪影響を含む不利な経済条件; | |
| · | 私たちの証券の取引量 |
| 49 |
私たちの優先株は私たちの普通株主に属さない権利、優先権 と特権を持っており、これは私たちの優先株保有者の利益と私たちの普通株株主の利益とは異なる可能性がある。
私たちの優先株の所有者は清算優先権を獲得し、株主に分配できる資産から支払う権利があるようにして、その後、任意の普通株または任意の系列優先株の所有者に任意の優先株レベルがこのような優先株より低い優先株を支払うことができる。清算優先権の存在は、私たちの普通株の価値を低下させ、将来の発行で普通株を売却したり、制御権の変更を阻止したり、延期したりすることを難しくする可能性がある。また、Aシリーズ優先株とBシリーズ優先株の1株は私たちの普通株に変換することができ、発行可能上限の制限といくつかの調整を受けることができ、これは私たちの普通株株主の大幅な希釈を招く可能性がある。優先株権は優先株保有者と私たち普通株保有者の間の利益の相違を招く可能性がある。
未来に普通株式を発行することは私たちの株主に希釈されないかもしれない。
2023年3月10日まで、私たちは約1,520万株のすでに発行された普通株があり、680万株が発行された優先株、株式承認証、オプション、制限性株と普通株奨励に関連する潜在的な希薄普通株を含まない。
これらの普通株の発行やこれらの普通株の売却、さらにはこのような発行や売却の可能性は、私たちの普通株の市場価格を押し下げる可能性があり、このような普通株の発行は私たちの株主に希釈されるかもしれない。
私たちは証券訴訟の影響を受けるかもしれない。
従来,証券集団訴訟 は会社の証券市場価格の下落に伴い提起されることが多かった。このリスクは、製薬会社が近年著しい株価変動を経験しているため、特に私たちと関連している。もし私たちがこのような訴訟に直面すれば、巨額のコストを招き、経営陣の注意と資源を移す可能性があり、これは私たちの業務を損なう可能性があります。
私たちの普通株式や株式承認証は活発で、流動的で秩序のある市場を形成できないかもしれない。
私たちの普通株式と引受権はナスダックで取引されています。私たちの普通株や引受権証の活発な取引市場は決して発展したり持続したりしないかもしれない。もし私たちの普通株や引受権証の活発な市場が持続的に発展していない場合、投資家は市場価格を低くすることなく株や引受権証を売却することは困難である可能性があり、投資家は株や引受権証を売却することができないかもしれない。活発でない市場はまた、普通株や引受権証を売却することで資金を調達する能力を弱める可能性があり、一般株式または引受権証を他の業務、アプリケーション、または技術を価格買収する能力として弱化させる可能性があり、逆に私たちの業務に重大な悪影響を及ぼす可能性がある。
私たちは私たちの株主といくつかの合意を締結した。
私たちは過去に株主と協定を締結し続ける可能性があり、これは利益の衝突を招くかもしれない。さらに、これらの配置は、私たちと距離を置いて交渉されたものではなく、私たちの最適な利益に適合しない条項と条件が含まれている可能性がある。
| 50 |
私たちは私たちの普通株や優先株に配当金を支払うつもりはないので、どんな見返りも私たちの株の価値に限定されるだろう。
私たちは普通株や優先株の任意の現金配当金を発表したり支払ったりしたことがない。私たちは現在、将来の業務発展、運営、拡張のための収益を維持し、予測可能な未来にはいかなる現金配当金も発表または支払いしないと予想している。したがって、普通株または優先株株主への任意のリターンは、その株式の付加価値に限定される。
私たちの定款、付則、ネバダ州改正条例のいくつかの条項は逆買収の効力があるとみなされる可能性があり、これは私たちの普通株の市場価格を下落させる可能性があります
私たちの会社の定款、附則、ネバダ州改訂案のいくつかの条項は反買収効力があるとみなされるかもしれません。このような条項は、株主がその株主の最適な利益に適合していると考える可能性のある要約買収または買収の試みを遅延、阻止、または阻止する可能性があり、試みを含む、株主の保有株式の市価割増を招き、我々の普通株の市価下落を招く可能性がある。
一般リスク因子
我々の財務状況、経営結果、業務およびキャッシュフローは、不利な米国または世界の経済状況の負の影響を受ける可能性がある。
我々の財務状況、経営結果、業務とキャッシュフローは、世界経済と世界金融市場の全体的な状況及び経済安定性の不確定性の負の影響を受ける可能性がある。世界経済は、公衆衛生流行病や大流行、あるいは新冠肺炎の大流行のような他の伝染病の爆発、およびロシアのウクライナ侵攻、関連制裁および他の経済中断のような国際紛争、テロまたは他の地政学的事件を含む極端な変動と中断を経験している。
例えば、新型コロナウイルス(新冠肺炎)の急速な成長の発生に関連する全世界の大流行はすでに重大な変動、 不確定性と経済混乱をもたらす可能性があり、資本市場の大幅な変動を含む。新冠肺炎疫病の著者らの業務、運営、財務業績と普通株取引価格に対する影響程度は多くの著者らが正確に予測できないかもしれない絶えず変化する要素に依存する。もし全世界が新冠肺炎疫病を抑制する反応が更にエスカレートしたり失敗したりすれば、 あるいは政府が疫病に関連する制限の決定を緩和し、無効、早期或いは逆効果であれば、私たちは私たちの業務、財務状況、運営結果とキャッシュフローに実質的な不利な影響を与える可能性がある。
また、世界経済·金融市場は、軍事衝突、テロ、または他の地政学的事件の現在または予想されている悪影響を受ける可能性もある。米国や他の国がこのような紛争に対応するために実施している制裁は、金融市場や世界経済に悪影響を及ぼす可能性もあり、影響を受けた国や他の国の経済対策は市場や経済の不安定を悪化させる可能性がある。2022年2月下旬、ロシアはウクライナに対して重大な軍事行動を開始した。これに応じて、米国や他の一部の国はロシアに対して重大な制裁と貿易行動を実施し、衝突が継続したり悪化したりすれば、さらなる制裁、貿易制限、その他の報復行動を実施する可能性がある。これに関連する地政学的緊張情勢、および米国および他の国がこの点でとる措置および報復行動、およびロシアがとる可能性のある任意の対応または報復行動を含む紛争のより広い結果を予測することはできず、これらは地域不安定および地政学的転換を招き、世界貿易、通貨為替レート、地域経済、世界経済に実質的な悪影響を及ぼす可能性がある。前述のいずれかのわが社への影響を予測することは困難であるが、衝突に対する衝突や行動は、私たちのコストを増加させ、私たちのサプライチェーンを混乱させ、必要に応じて許容可能な条件で追加資本を調達または獲得する能力を弱めるかもしれない(あれば)、あるいは他の方法で私たちの業務、財務状況、運営結果に悪影響を及ぼす可能性がある。
| 51 |
信用と金融市場のさらなる悪化と経済状況への自信が起こらない保証はない。深刻または長期的な経済低迷は、私たちが開発する可能性のある任意の候補製品に対する需要の減弱、必要に応じて許容可能な条件で追加資本を調達する能力など、私たちの業務に様々なリスクをもたらすかもしれない。経済が疲弊したり下落したりすることは、私たちのサプライヤーに圧力を与え、供給中断を招く可能性もある。株式や信用市場が悪化すれば、任意の必要な債務や株式融資をより困難にし、コストが高く、削減度も高くなる可能性がある。有利な条件で任意の必要な融資をタイムリーに得ることができなければ、成長戦略を実現する能力を弱める可能性があり、私たちの財務業績と株価を損なう可能性があり、臨床開発計画の延期または放棄を要求する可能性がある。さらに、私たちの現在または未来のサービスプロバイダ、製造業者、または他の協力者は、経済的困難な時期を乗り切ることができないかもしれません。これは、私たちの時間通りと予算で運営目標を達成する能力に直接影響を与える可能性があります。私たちは、現在の経済環境や金融市場状況が私たちの業務に悪影響を及ぼす可能性のあるすべての方法を予測することができません。
我々は,潜在的な将来の運営損失や連邦と州NOL繰り越しを用いて,運営や会社の協力による収入の課税収入を相殺する能力が制限される可能性がある。
私たちのNOL繰越を使用することは、将来のいくつかの所有権変更または規則と他の税務機関によって規定された他の要素が制限される可能性がある。TCJAはNOL繰越の連邦繰延税額と連邦NOL繰り越しの使用規則を変更した。
もし私たちのNOL繰越が限られていて、私たちがbrの課税所得額がこの期間の利用可能なNOL繰越を超えている場合、私たちはNOL繰越 が満期前の今後数年に利用できる可能性があっても、所得税の責任を負う。このような所得税負債は、私たちの将来のキャッシュフロー、財務状況、および財務業績に悪影響を及ぼす可能性がある。
税制改革は会社と私たちの株主に大きな影響を及ぼすかもしれない。
税法と法規の潜在的な変化あるいはその解釈の変化、税収法規の曖昧性、事実解釈の主観的 および他の要素のため、私たちの有効税率と所得税資産負債の推定は正しくない可能性があり、私たちの財務諸表は不利な影響を受ける可能性がある。本節の第1文で述べたこれらの要因の影響は時期によって異なる可能性がある
また,我々が納めた所得税金額 は,米国連邦,州,地方税務機関および非米国税務機関の継続監査を受ける。監査による支払いや評価が私たちの準備金と異なる場合、私たちの将来の結果は、私たちの納税負債の不利な調整を含む可能性があり、私たちの財務報告書は悪影響を受ける可能性があります。米国または他の管轄区域税制のさらに大きな変化(以下にさらに説明する国際収入課税の変化を含む)は、我々の財務諸表に悪影響を及ぼす可能性がある。
政府は価格規制を実施するかもしれないが、これは私たちの将来の収益性に悪影響を及ぼすかもしれない。
私たちはアメリカと他の管轄区域で私たちの候補薬を販売することを承認することを求めるつもりだ。一部の外国国家と司法管轄区、特にEUでは、処方薬の価格設定は政府によって規制されている。これらの国/地域では,候補薬の発売承認を受けた後,政府当局との定価交渉にかなりの時間を要する可能性がある。ある国/地域で精算或いは定価の承認を得るためには、臨床試験を行う必要があるかもしれず、著者らの候補薬物のコスト効果をbrの他の利用可能な治療法と比較することは、時間もかかり、高価である。もし私たちの未来の製品が精算を得られない場合、あるいは範囲や金額が制限されている場合、あるいは定価が満足できないレベルに設定されている場合、利益を達成したり維持したりすることができないかもしれません。
| 52 |
私たちの従業員、首席調査者、コンサルタント、ビジネスパートナーは、規制基準と要求およびインサイダー取引を遵守しないことを含む、不適切な行為または他の不適切な活動に従事する可能性があります。
私たちは従業員、主な調査者、コンサルタント、ビジネスパートナーの詐欺や他の不適切な行為のリスクに直面している。これらの当事者の不正行為は、FDAおよび非米国規制機関の規定を故意に遵守しないこと、FDAおよび非米国規制機関に正確な情報を提供すること、米国および海外の医療詐欺および乱用法律法規を遵守し、財務情報またはデータを正確に報告すること、または不正な活動を開示することを含む可能性がある。特に、医療業界の販売、マーケティング、業務配置は、詐欺、不正行為、リベート、自己取引、その他の乱用行為を防止するための広範な法律法規によって制約されている。これらの法律法規は、様々な価格設定、割引、マーケティングおよび販売促進、販売手数料、顧客インセンティブ計画、および他のビジネススケジュールを制限または禁止する可能性があります。このような不正行為はまた、brの臨床研究過程で得られた情報の不適切な使用に関連する可能性があり、これは規制制裁を招き、私たちの名声に深刻な損害を与える可能性があり、あるいは規制機関が私たちの候補薬物を承認しない可能性がある。従業員の不正行為を常に識別し、阻止できるわけではなく、私たちがこのような行為を検出し、防止するための予防措置は、未知または未管理のリスクや損失を効果的に制御できないかもしれないし、これらの法律や法規を遵守できないことによる政府の調査または他の行動や訴訟から私たちを保護することができないかもしれない。もし私たちにこのような行動を取って、私たちが自分の権利を弁護したり、維持することに成功しなかったら、これらの行動は、巨額の罰金や他の制裁を加えることを含む、私たちの業務に重大な影響を及ぼすかもしれない。
私たちの候補薬を使用することは副作用を招く可能性がある。
多くのバイオ製薬製品と同様に,我々を使用する候補薬は副作用や有害事象に関連している可能性があり,これらの副作用や有害事象の重症度や頻度が異なる可能性がある。臨床試験または製品の商業化後に、どのような副作用または有害事象も、私たちの規制部門の承認または販売を得る能力に悪影響を及ぼす可能性がある、我々の候補薬剤の使用に関連する副作用または有害事象がいつでも観察される可能性がある。私たちの候補薬の使用に関連する副作用、例えば毒性や他の安全問題は、追加的な研究を行う必要があるかもしれないし、これらの候補薬物の開発や販売を停止したり、製品責任訴訟に直面させたりして、私たちの業務を損なうかもしれません。
予測できない安全問題や有害事象の出現は、規制機関が候補薬物の安全性と有効性に対して追加の臨床前または臨床試験 を要求する可能性があり、これは私たちが計画していないことや期待されていることである。私たちは、任意の製品に関連する有害事象に関連する問題をタイムリーにまたは永遠に解決し、FDAまたは任意の規制機関を満足させることを保証することはできません。これらの問題は、私たちの業務、将来性、および財務状況を損なう可能性があります。私たちも無意識に所定の 時間範囲で私たちが知っている有害事象を報告できなかったかもしれない。私たちはまた、私たちが報告可能な有害事象を認識していることを認識していないかもしれないが、特にそれが有害事象として報告されていない場合、またはそれが意外な有害事象である場合、または私たちの製品を使用する時に直ちに削除されるかもしれない。もし私たちが私たちの報告義務を遵守できなかった場合、FDAまたは他の外国規制機関は、刑事起訴、民事罰金の適用、私たちの製品の差し押さえ、または将来の製品の承認を延期または承認することを含む行動をとるかもしれない。
| 53 |
私たちは潜在的な製品責任に直面しています。もし私たちに対するクレームが成功すれば、私たちは重大な責任とコストを招くかもしれません。もし私たちの候補薬を使用して患者を傷つけた場合、または患者を傷つけたと考えられ、このような損傷が私たちの候補薬物と関係がなくても、私たちの規制承認が撤回されるか、または他の方法で否定的な影響を受ける可能性があり、私たちはコストが高く破壊的な製品責任クレームを受ける可能性がある。
臨床研究で私たちの候補薬物を使用して、私たちが発売許可を得たどの製品を販売することは私たちを製品責任クレームのリスクに直面させます。製品責任消費者、ヘルスケア提供者、製薬会社、または販売、または他の方法で私たちの製品に接触した他の人は私たちにクレームをつける可能性があります。私たちの候補薬は副作用の危険を引き起こすかもしれない。もし私たちがbr製品責任クレームに対抗できなければ、私たちは巨額の責任とコストを招くかもしれない。さらに、利点または最終結果にかかわらず、製品 責任クレームは、:
| · | 私たちのビジネス的名声を損なうことは | |
| · | 臨床研究者は脱退しました | |
| · | 関連訴訟による費用 | |
| · | 私たちの主な業務に対する管理職の関心を分散させる | |
| · | 患者や他のクレーム者に巨額のお金の報酬を提供し | |
| · | 候補薬を商業化することはできません | |
| · | 私たちの候補薬の需要を減らし商業販売に許可されれば |
これらすべては私たちの業務、運営結果、そして見通しに実質的な悪影響を及ぼす可能性がある。
もし私たちが環境、健康、安全法律法規を守らなければ、罰金や罰金を科されたり、コストが発生したりする可能性があり、これは私たちの業務成功に実質的な悪影響を及ぼす可能性がある。
私たちは多くの環境、健康と安全法律法規の制約を受けて、実験室の手続き及び危険材料と廃棄物の処理、使用、貯蔵、処理と処理を管理する法律法規を含む。私たちの業務は化学品と生物学的材料を含む危険で燃えやすい材料の使用に関するものだ。私たちの業務はまた危険な廃棄物製品を発生させるだろう。私たちは一般的に第三者とこのような材料と廃棄物を処理する契約を締結する。私たちはこのような材料が汚染や傷害をもたらす危険を除去することができない。もし私たちが危険な材料を使用して汚染や損傷をもたらしたら、私たちはそれによるいかなる損害に責任を負うかもしれません。どんな責任も私たちの資源の範囲を超えている可能性があります。私たちは民事や刑事罰金と処罰に関連する巨額の費用を発生させる可能性もあります。
私たちは従業員が危険材料や他の労災を使用して怪我をして発生する可能性のあるコストと費用のために、私たちが維持している労災保険は潜在的な責任に十分な保険を提供できないかもしれません。また、現在または未来の環境、健康、安全法律法規を遵守するためには、巨額のコストが生じる可能性がある。これらの現在または未来の法律法規は、私たちの研究、開発、または生産努力を損なう可能性がある。これらの法律法規を遵守しないことは、巨額の罰金、処罰、または他の制裁を招く可能性もあり、これは私たちの業務や運営結果に実質的な悪影響を及ぼす可能性がある。
株式ベースの支払いのような非現金料金は、私たちの運営結果に悪影響を及ぼす可能性があります。
私たちは株式ベースの支出に関する非現金費用を記録しており、会社は株による支払い奨励を継続する予定であるため、大幅に変動し、私たちの運営業績に悪影響を及ぼす可能性があります。
| 54 |
既存の基準とルールの異なる解釈 がしばしば出現し,これまでに報告した操作結果を再確認せざるを得ない可能性がある.
会計政策の既存の基準や既存取引の会計処理方式の異なる解釈は、先に報告したbrの運営結果を再陳述しなければならない可能性がある。
私たちの開示制御と手続きはすべてのエラーまたは詐欺を阻止または検出できない可能性がある。
私たちは取引法の定期的な報告書の要求事項を守らなければならない。任意の開示制御およびプログラムまたは内部制御およびプログラムは、発想および操作がどんなに綿密であっても、絶対的な保証ではなく、制御システムの目標を達成するために合理的な保証を提供することしかできない。これらの固有の制約には, 意思決定過程における判断が誤っている可能性と,簡単な誤りや誤りによって故障が発生する可能性がある現実がある.さらに、制御は、ある人の個人的な行動、2人または複数の個人の共謀、または許可されていないカバー制御 によって回避されることができる。したがって,我々の制御システムの固有の限界により,誤りや詐欺による誤った陳述が発生し,発見されない可能性があり,我々の業務や運営結果に実質的な悪影響を与える可能性がある.
我々の情報技術システムに障害が発生し,ネットワークセキュリティ攻撃や他のデータセキュリティイベントを含めて,我々の運営を大きく乱す可能性がある.
私たちの運営は私たちの情報技術システムの持続的な表現にある程度かかっている。我々の情報技術システムは,物理的または電子的な侵入,コンピュータウイルス,および同様の中断の攻撃を受けやすい可能性がある.私たちの情報技術システムの故障は私たちの業務、収益性、財務状況に悪影響を及ぼす可能性があります。
成功したネットワークセキュリティ攻撃または他のデータ セキュリティイベントは、機密または個人情報の盗用および/または損失をもたらし、システムの中断 をもたらすか、または我々のシステムを攻撃するマルウェアを配備する可能性がある。ネットワークセキュリティ攻撃は一定期間 に知覚されない可能性がある.ネットワークセキュリティ攻撃またはイベントの発生は、私たちの情報技術システムの中断、または否定的な宣伝が、私たちの臨床試験参加者、顧客、株主、および他の利害関係者の名声の損傷をもたらし、および/またはネットワークセキュリティイベントの予防、対応、または緩和のコストを増加させるために、業務中断を引き起こす可能性がある。さらに、敏感な個人情報または独自または機密情報を不正に伝播することは、私たちまたは他の第三者を規制罰金または処罰、訴訟、および潜在的責任に直面させるか、または他の方法で私たちの業務を損なう可能性がある。
私たちは小さな報告会社であり、小さな報告会社に適用される報告要求の低下は、私たちの普通株の投資家に対する吸引力を低下させる可能性があります。
私たちは小さな報告会社(“SRC”)、 これは、改正された2002年のサバンズ-オキシリー法案404条の監査人認証要件を遵守する必要がないことを含む、他のSRCでない上場企業に適した様々な報告要件の免除を利用することを可能にし、私たちの年間報告および定期報告およびbr}代理報告書における役員報酬に関する開示義務を削減し、私たちの年次報告および定期報告に2年間の監査済み財務諸表のみを提供することを可能にする。(A)最近終了した第2四半期までの最終営業日まで、非関連会社が保有する発行済み普通株の総時価が2.5億ドルを超えるか、または(B)(1)我々の年収が1億ドルを超え、(2)最近終了した第2四半期の最終営業日まで、非関連会社が保有する発行済み普通株の総時価が7億ドルを超えるSRCとする。私たちがこのようないくつかまたはすべての免除に依存すれば、投資家が私たちの普通株の吸引力の低下を発見するかどうかは予測できない。したがって、一部の投資家が私たちの普通株の吸引力が低下していることを発見すれば、私たちの普通株取引市場はそれほど活発ではなくなり、私たちの株価はもっと変動し、下落するかもしれない。
プロジェクト1 B--未解決スタッフの意見
適用されません。
| 55 |
プロジェクト2--財産
私たちはマサチューセッツ州フレミングハンコンコッド街九四五号でオフィススペースを借りています。レンタル契約は12ヶ月で、締め切りは2023年9月です。私たちは、この空間は私たちの現在の需要を満たすのに十分であり、追加の空間が必要なら、近くで商業的な合理的な条件で得ることができると信じている。
また、360平方メートルをレンタルしています。英フィナンシャル·タイムズ紙フロリダ州マイアミのオフィススペースです。レンタルの初期期間は12ヶ月で、2016年12月1日から を開始し、2023年11月30日まで年延長されています。私たちは、この空間が現在の需要 を満たすのに十分であり、追加の空間が必要であれば、既存の空間内または近くで商業的に合理的な条件で得ることができると信じている。
プロジェクト3−法的訴訟
時々,我々 は訴訟の側である可能性があり,正常な業務過程におけるクレーム付きの影響を受ける可能性がある.訴訟やクレームの結果は確定的に予測できないが,これらの一般授業事項の最終結果は我々の業務に実質的な悪影響を与えないと信じている。結果にかかわらず、弁護と和解コスト、管理資源の移転、その他の要因により、訴訟は私たちに悪影響を及ぼす可能性がある。
2022年12月31日現在、経営陣は、私たちの財務状況、経営業績、キャッシュフローに大きな悪影響を及ぼす可能性はないと考えています。
プロジェクト4−鉱山安全開示
適用されません。
| 56 |
第II部
項目5-登録者普通株式市場、関連株主事項、発行者による株式証券の購入
我々の普通株と 普通株引受権証はそれぞれナスダック資本市場(“ナスダック”)に上場し、コードは“XBIO”と “XBIOW”である。
記録保持者
2023年3月10日までに、426人の普通株保有者がいます。
配当をする
私たちは以前普通株式に対するどんな現金配当金も発表したり支払ったりしたことがない。私たちは現在、私たちの業務戦略を支援するために収益と利益を保留するつもりで、予測可能な未来に現金配当金を支払うつもりはありません。将来どのような現金配当金を派遣するかの決定は当社取締役会が自ら決定し、当社の財務状況、当社の経営業績、資本要求、一般業務状況、および取締役会が関連すると考えられる任意の他の要素に依存する。
株式報酬計画情報
第5項に記載された株式補償計画に従って発行された許可証券に関する情報は、本表格10-Kの第3部分第12項を参照して本明細書に組み込まれる。
最近売られている未登録証券
ない。
発行者の株式証券を買い戻す
2022年12月31日までの四半期内に、流通株普通株は何も買い戻していません。
第6項-[保留されている]
保留します。
| 57 |
第七項– 経営陣の財務状況と経営成果の検討と分析
業務の概要
私たちは生物製薬会社で、革新的な免疫腫瘍学技術を推進し、治療が困難な癌を解決することに専念しています。我々のDNaseプラットフォームは,Netに対して免疫療法を含めた既存治療の結果 を改善することを目的としている。我々は2022年4月にDNase腫瘍学プラットフォームの許可を得ており,この新たな買収の技術開発への取り組みと資源を優先することが予想される。我々は現在、膵臓癌と局部末期あるいは転移性固形腫瘍の補助治療として、我々のシステムDNA ase計画を臨床に進めることに集中している。私たちはまた、私たちの個人化キメラ抗原受容体(CAR)Tプラットフォーム技術、XCARTを開発している™B細胞リンパ腫の治療には,単一患者の悪性腫瘍細胞表面にユニークなB細胞受容体に対する細胞療法が開発されている。また,バイオテクノロジーや製薬会社と協力し,独自の薬物送達プラットフォームPolyXenを開発し,血液凝固障害領域の独占的な許可手配に基づいて特許使用料の支払いを得た。
我々の特許とノウハウbrを現在バイオテクノロジーや製薬業界と協力して開発されている候補薬剤に統合し,brが既存療法よりも薬理学的特性を有すると信じている次世代バイオ薬剤を創出する。我々の候補薬物は,我々の研究活動や我々の協力者の研究活動によって生成され,開発段階にある。したがって、私たちは引き続き私たちの研究開発活動のために大量の資源を投入し、近い将来に引き続きそうする予定です。これまで、私たちの候補薬はアメリカや他のどの国や地域でもどのような適用機関の規制マーケティング許可や承認を得ていませんでした。私たちのPolyXen技術を業界パートナーに許可する規定によると、私たちは持続的な印税を受けるだろう。幅広い特許の組合せを持っているにもかかわらず,2022年12月31日までの1年間,我々の内部努力の重点は,我々のDNaseプラットフォームの許可と進歩,および我々のXCART プラットフォーム技術の開発である.
重要な会計政策と試算
アメリカ公認会計原則(“アメリカ公認会計原則”)に従って、私たちの財務諸表を作成することは、財務諸表の日付報告の資産と負債額、および報告期間内の報告の収入、コストと費用に影響を与える推定、判断と仮説を作成することを要求する。継続的な基礎の上で、私たちは歴史的経験と私たちがこのような状況で合理的だと思う様々な他の仮定に基づいて私たちの推定を評価します。これらの評価の結果は,資産や負債の帳簿価値や報告された費用金額を判断する基礎 を構成しており,これらの費用は他のソースからは見られない である.未来のイベントとその影響は確定できないため,実際の結果や結果は我々の見積り,判断,仮定と大きく異なる可能性がある.
経営陣は、以下の会計見積もりは、我々の報告書の財務結果を全面的に理解し評価するために最も重要であり、経営者が最も困難な主観的または複雑な判断を行う必要があると考えている。これは、本質的に不確実な事項の影響を推定する必要があるためである。以下に、これらの重要な会計推定、判断および仮定、および実際の結果がこれらの仮定と異なる場合に生じる影響について説明する。
収入確認
私たちは、承認された商業医薬品の潜在的純売上に基づく印税協定を含む、製薬およびバイオテクノロジーパートナーと供給、許可、および協働協定を締結した。
| 58 |
会計基準編纂(“ASC”)主題606に基づいて収入を確認した取引先と契約した収入(“ASC 606”)。本基準 は、顧客と締結されたすべての契約に適用され、レンタル、保険、連携手配、金融商品など、他の標準範囲に属する契約を除く。ASC 606によれば、エンティティは、そのクライアントがコミットメント商品またはサービスの制御権を取得したときに収入を確認し、その金額は、エンティティがこれらの商品またはサービスと交換することが予期される対価格 を反映する。エンティティがASC 606の範囲内に属するスケジュールの収入確認を決定するために、エンティティは、(I)クライアントとの契約を決定するステップと、(Ii)契約における履行義務を決定するステップと、(Iii)取引価格を決定するステップと、(Iv)契約中の履行義務に取引価格を割り当てるステップと、(V)ある時点または時間とともに履行義務を満たす収入を確認するステップと、の5つのステップを実行する。以下の場合にのみ、 は、顧客に譲渡された商品またはサービスと交換する権利のある対価格を受け取る可能性が高い5段階モデルを契約に適用する。 は、契約開始時に、契約がASC 606の範囲内に決定されると、各契約で約束された商品またはサービスを評価し、どれが履行義務であるかを決定し、各約束された商品またはサービスが異なるかどうかを評価する。そして,履行義務が履行された場合には,それに応じて義務を履行する取引価格に割り当てられた金額を収入 と確認する.
これらの手配に対する会計処理の一部として,a)以上のステップ(Ii)で決定された義務履行数, b)上記ステップ(Iii)での取引価格,およびc)上記ステップ(Iv)で取引価格を割り当てる 契約で決定された義務履行ごとの独立販売価格を決定するために重大な判断を用いなければならない.以下に述べるように,マイルストーンや他の変数 を取引価格に計上すべきかどうかを判断を用いて決定する.取引価格は相対独立販売価格ごとに履行義務 に割り当てられ,収入を契約項下の履行義務を履行する際に確認するか確認する.履行義務の独立価格を策定する際には,適用する市場条件 と関連するエンティティ特定要因を考慮し,クライアントと合意交渉する際に考慮する要因と推定される コストを含む.我々は,独立販売価格を決定するためのキー仮説の変化が複数の履行義務間の取引価格割当てに大きな影響を与えるかどうかを評価することで,契約義務の独立販売価格 を検証する.契約資産または負債は、私たちの業績(すなわち、顧客に転送された貨物またはサービス)と顧客の業績(すなわち、顧客が支払い、無条件に支払うべき対価格)との差によるものであることを確認します。
私たちのライセンス契約条項は、 がパートナーにIPライセンスを渡すことを含むことができます。許可手配により、払い戻しできない前払い領収書、開発と規制目標領収書、およびパートナーが将来製品を販売する印税領収書の組み合わせで補償を受けることができます。私たちは、私たちが許可および協力手配で受け取った払い戻し不可能な前払い許可支払いおよび開発と規制マイルストーン支払い を確認する予定であり、供給義務などの将来の義務が含まれており、それぞれの予定に応じた予想履行期間内に比例して計算されます。著者らは予想される業績義務履行の期限に対して最適な推定を行い、その中に技術移転援助、研究活動、臨床開発活動及び開発から製品商業化までの製造活動 を含む可能性がある。これらの連携スケジュールの不確実性を考慮して,履行期間の継続時間 を決定するためには大きな判断が必要である.
私たちが私たちのいくつかの特許を再許可する手配を達成する時、私たちは収入確認の適切な方法と時間を決定する際に、その配置が単一要素か複数の要素かを決定するために義務を履行することを考慮する。我々は、我々が提供するサービス、特許(Br)の国防コスト、技術支援、マーケティングまたは販売支援などの任意の履行義務に加えて、価格調整または払い戻し条項などの要素を考慮するか、またはそれに渡すことができる追加の が収入確認時間を変更することができる任意の他の要素を構成する可能性があるbrの許可または再許可の条項を考慮する。受け取った返却不可の前払い許可と再許可費用は, 継続履行または将来義務は関連許可技術とは無関係またはいいかげんとみなされ, は技術交付時に収入として確認される。
基本契約条項に基づいて販売期間中の特許権使用料収入を確認する予定であり,報告の売上が確実に測定できることを前提としており,残りのbr履行義務はなく,他のすべての収入確認基準を満たしている。我々が完成した連携プロトコルに関する研究開発サービスの精算は,運営中に毛収入として確認されることが予想される.私たちのライセンス(Br)およびいくつかのパートナーとの協働協定はまた、対応するパートナーが締め切りの延長または指定された販売量を達成することを考慮した場合にのみ、指定された販売量を達成することを考慮した医薬品のみに基づいて、将来のマイルストーン領収書を提供することができる。このような受領書については,適用される契約条項に基づいて履行または比例して合意期限内にこれらの受領書を収入として確認したい.もし私たちの持続的な履行や未来の義務がどうでもいいとみなされたり、適当に処理されたりすれば、これらの領収書も収入として確認されることができる。
| 59 |
研究と開発費
研究開発費とは,報酬福祉,施設費用,管理費用,臨床前開発,臨床試験および関連臨床製造費用,契約研究機関(“CRO”)や契約製造機関(“CMO”)に支払われる費用,その他の外部費用を含む研究開発活動で発生する費用である。我々は発生した ごとに研究開発コストを計上した。私たちは義務が発生したため、研究開発サービスのために払い戻しできない費用を事前に支払っています。買収に帰属するが資本化基準に達していない無形資産の価値 は買収時に研究開発費に計上される。ライセンス契約に基づいて支払われた前金は、許可を受けたときに支出されます。ライセンス契約の下のマイルストーン支払い は、マイルストーンが実現可能であり、関連金額が合理的に評価可能であることが決定された間に累積され、対応する費用が確認される。
私たちは各報告期間の計算すべき研究と開発費用を推定しなければならない。この流れは、未決済契約および購入注文を審査し、私たちの代表的に実行されたサービスを決定するために、私たちの担当者とコミュニケーションを行い、インボイスを受け取っていないとき、または他の方法で実際のコストを通知しているときに、実行されたサービスレベルおよびサービスによって生じる関連コストを推定することを含む。私たちのほとんどのサービスプロバイダ は、予め定められたスケジュールに従って、あるいは契約マイルストーンに達したときに借金の領収書を発行してくれます。しかし、一部は 前金が必要です。私たちは、当時既知の事実と状況に基づいて、財務諸表において、貸借対照表毎の日付までの課税費用を推定する。私たちは定期的にサービスプロバイダに推定の正確性を確認し、必要に応じて調整します。 計算すべき研究および開発費用の推定例には、以下の者に支払われる費用が含まれる
| · | 研究開発と臨床前活動を展開するパートナー | |
| · | 臨床試験全体のプロジェクト管理に関するプロジェクトマネージャー; | |
| · | CGMP製造に関するcMOS; | |
| · | 臨床試験に関するCRO;および | |
| · | 臨床試験に関する調査サイト。 |
著者らは、著者らを代表して臨床試験を行って管理する複数の研究機関、CMOとCROのオファーと契約に基づいて、受け取ったサービスと費用の努力を見積もり、これをもとに研究開発、臨床前活動と臨床試験に関連する費用を支払う。これらのbr協定の財務条項は協議が必要であり、契約によって異なり、支払いの流れが不均一になる可能性があります。 では,サプライヤーへの支払いが提供されたサービスレベルを超えて前払い料金を招く場合がある.サービス料金を計算する際には,サービスを実行する時間帯と各時間内の作業支出レベルを見積もる.サービス実行の実時間または作業レベルが推定値と異なる場合、それに応じて計算または前払い金額を調整する。 我々の推定値は、実際に発生した金額と実質的に差がないと予想されるが、実行されたサービスの状態および時間の理解は、実際の状態および実行されたサービスの時間とは異なる可能性があり、報告された金額 が任意の特定の時期に高すぎたり、過小になったりする可能性がある。これまで,我々のこれまでの対応研究や開発費用の見積りには実質的な調整はなかった.
株式ベースの費用
株式ベースの支出には、従業員および非従業員にオプション および制限株式単位(“RSU”)を付与して我々普通株の株式を購入すること、従業員に共通株式を付与すること、非従業員が提供するサービスと交換するために普通株式を発行することの合意が含まれる。
| 60 |
株式ベースの費用は、オプションの推定公正価値に基づくか、またはBlack-Scholesオプション定価モデルを使用して計算される。適切な公正価値モデルと関連仮定を決定するには、株価変動の推定と期待奨励条項を含む判断が必要である。期待変動率 は会社の履歴変動率から推定される。期待期間全体を奨励する企業データが得られない場合は、オプション期待期間内の過去の変動率と、上場企業同業グループと比較可能な加重平均値 を使用します。期待期間はオプション予想が平倉していない時間を表す.私たちは没収が発生した時に付与ではない時にそれを処理するつもりだ。私たちは配当金を支払っていないし、予測可能な未来に現金配当金を支払うことも期待していないので、私たちが使用する期待配当収益率はゼロだ。無リスク金利は米国債の金利に基づいており、その満期日は奨励の予想期限と一致している。行使時には,新たに発行された普通株が株式オプション を償還する.付与された帰属と受け渡し規定を満たした場合、RSUは私たちの普通株の新規発行株を償還する。
サービス条件のみに応じて付与された従業員オプションの場合、公正価値計量日は、通常、付与された日であり、関連報酬支出は、報酬の必要な帰属期間中に直線的に確認される。会社の運営中に消費された商品やサービスを交換するために発行された非従業員オプション については、公正価値計量日は、サービス完了日または業績承諾達成日の中で早い日である。株式オプションの公正価値は,受けたサービスの公正価値よりも確実に計測できると考えられる.非従業員に付与された株式オプションに関する報酬支出 は、報酬の必要な帰属中に直線ベースで確認される。
株式承認証
いくつかの融資、コンサルティング、協力手配について、私たちは普通株の株式を購入するために引受権証を発行した。未清算持分証明書は独立手形であり、所有者は売却或いは強制償還できず、株式奨励に分類される。私たちはブラック-スコアーズオプション価格モデルを使用して、日付までの報酬の公正価値を測定した。普通株発行とともにパートナーに発行した引受権証は,最初に発行された普通株追加実収資本の減少額を公正価値で計上した。
他のすべての株式承認証は,必要なサービス期間内に公正価値 を料金として直線的に計上し,サービス期間がない場合やサービスを提供している場合は,発行日に公正価値に応じて料金と記す.いくつかの目標に基づいて達成されるホームトリガ要因を含む権証について、我々は、 判断を適用して、これらの目標を達成する確率および時間を推定する。これらの見積りは固有の不確実性に関連しているため, したがって,これらの目標を実現する確率や時間が変化すると,費用に関する引受権証の将来が大きく異なる可能性がある.融資手配に関連して発行された権利証については、奨励と他のツールの相対的な公正価値に基づいて収益を分配する。
無期限-生きている無形資産
業務 合併,許可その他の取引で取得した資産や負担する負債は,買収日にそれぞれの公正価値で確認するのが一般的である. 買収の際には,進行中の研究開発(“IPR&D”)を含む無形資産の公正価値を“収益法”を用いて決定するのが一般的である.買収された知的財産権研究開発無形資産は無期限無形資産とみなされ、関連する研究·開発が完了または放棄されるまで。会社の知的財産権の研究開発が技術実行可能性を達成する前に、大量の追加研究と開発を行う必要があるかもしれない。知的財産権研究開発プロジェクトが完成した後、知的財産権研究開発資産はその予想寿命内に償却を行う。
| 61 |
不確定な生きている無形資産は を販売しないが,少なくとも毎年あるいはビジネス環境のイベントや変化が帳簿価値が減値しない可能性があることを示した場合には,減値を審査する。私たちの年間評価は、その公正な価値が帳簿価値を超える可能性が高いかどうかを決定するために、定性または定量分析を含む可能性がある。定性的手法を実行する際には, イベントや環境の存在が無限に生きている無形資産を決定する可能性が高い(すなわち50%を超える可能性) の減少をもたらすかどうかを決定する.定性的要因を最初に評価し、無形資産減価の可能性が より高くないことを決定することを選択すれば、減値をテストするためにさらなる行動をとる必要はない。私たちはまた、定量的欠陥テストのみを実行するために定性的評価をバイパスすることを選択することができ、他の期間ではなく、いくつかのbrの間に実行することを選択することができる。定性的評価を行うオプションは恒久的な選択ではないため,年次減値審査のたびにこのオプションを再評価する.減価損失(ある場合)は無形資産の帳簿価値がその公正価値を超えて計量される。
無形資産は減価費用の影響を受けやすく、特に知的財産権研究開発のための新しい買収資産。研究開発の高リスク性質と業界が開発化合物を市場に出す成功率を考慮すると、知的財産権研究開発減価費用は今後しばらくの間に発生する可能性がある。潜在的な減値を推定する知的財産権研究開発の公正価値は予測と仮説の変化に高度に敏感であり、仮説の変化 は減少を招く可能性がある。
我々の推定と仮定は合理的であると信じており,他の点では市場参加者がその公正な価値推定に用いる仮説と一致している.しかし,将来の業績 が我々の見積もりや仮定と一致しなければ,我々は減価費用に直面する可能性があり,これは実質的である可能性がある. の異なる推定と判断を使用することは、我々の分析において非常に異なる結果をもたらす可能性があり、非常に異なる資産 の価値や費用をもたらす可能性がある。
新冠肺炎の大流行の影響
2020年3月、世界保健機関は全世界の大流行の発生を発表し、これは1種の新型コロナウイルス株(新冠肺炎)の迅速な爆発と関係がある。疫病はアメリカの経済状況に深刻な影響を与え、2020年3月前半に加速し、2021年と2022年まで持続し、連邦、州と地方政府が公衆衛生危機に対して緩和措置を取ったため、アメリカ経済に重大な不確定性をもたらした。著者らは引き続き新冠肺炎疫病が著者らの業務に与える影響を評価し、社交距離とその他の対応措置を取ったが、今までまだ著者らの業務に重大な影響を与えておらず、新冠肺炎疫病の著者らの運営業績と財務状況に対する最終的な影響は未来の事態の発展に依存し、疫病の持続時間と関連深刻度、ワクチン接種の速度と速度、及び新冠肺炎新毒株の持続的な出現、例えばDeltaとオミック変種及びそのいかなる亜変種を含む。また,マクロ経済 の状況に与える影響は不確実であり,現時点では予測できない.もし全世界が新冠肺炎疫病を抑制する反応が更にエスカレートしたり成功しなかったりしたら、あるいは政府が疫病に関連する制限を緩和する決定が無効で、早すぎたり、逆効果になった場合、私たちは私たちの業務、財務状況、運営結果とキャッシュフローに実質的な悪影響を与えるかもしれない。
ウクライナ紛争がわが軍の作戦に与える影響
ロシアがウクライナに侵入した短期的で長期的な影響は現在予測が難しい。制裁および反制裁を実施することは、経済市場全体に悪影響を及ぼす可能性があり、私たちの業務、財務状況、運営結果に影響を及ぼす可能性がある。
| 62 |
経営成果
次の表は,2022年12月31日までの年度と2021年12月31日までの年度の経営履歴実績を比較したものである。
| 説明する | 2022 | 2021 | 増す (減少) | パーセント 変わる | ||||||||||||
| 収入: | ||||||||||||||||
| 特許権使用料収入 | $ | 1,706,925 | $ | 1,160,692 | $ | 546,233 | 47.1% | |||||||||
| 運営コストと支出: | ||||||||||||||||
| 研究開発 | (4,770,834 | ) | (3,163,485 | ) | 1,607,349 | 50.8% | ||||||||||
| 一般と行政 | (3,653,999 | ) | (3,743,972 | ) | (89,973 | ) | (2.4 | )% | ||||||||
| 総運営コストと費用 | (8,424,833 | ) | (6,907,457 | ) | 1,517,376 | 22.0% | ||||||||||
| 運営損失 | (6,717,908 | ) | (5,746,765 | ) | 971,143 | 16.9% | ||||||||||
| その他の収入(支出): | ||||||||||||||||
| その他の収入(費用) | (1,597 | ) | 1,119 | 2,716 | 242.7% | |||||||||||
| 利子収入,純額 | 167,152 | 100,467 | 66,685 | 66.4% | ||||||||||||
| 純損失 | $ | (6,552,353 | ) | $ | (5,645,179 | ) | $ | 907,174 | 16.1% | |||||||
収入.収入
2022年12月31日までの年間収入は50万ドル増加し、47.1%増となり、2021年12月31日までの年度の約120万ドルから170万ドルに増加した。この増加は,2021年同期に比べて武田の再許可協定に関する印税収入が増加していることを示している。
研究開発費
全体的に言えば、2022年12月31日までの年間研究開発支出は160万ドル増加し、50.8%増加し、2021年同期の320万ドルと比較して50.8%に増加し、主に知的財産権研究開発支出が180万ドルだったためである。2022年12月31日までの年間で,会社のDNaseプラットフォーム許可に関する知的財産権研究開発支出は180万ドルであった。2021年に類似した費用がない次の表には、2022年12月31日と2021年12月31日までの年間における費用別の研究開発コストを示しています
| 十二月三十一日までの年度 | ||||||||
| 費用別 | 2022 | 2021 | ||||||
| 知的財産権研究開発費 | $ | 1,793,750 | $ | – | ||||
| 外部サービスと契約研究組織 | 2,314,513 | 2,497,190 | ||||||
| 給料と給料 | 435,564 | 457,313 | ||||||
| 株式ベースの費用 | 86,305 | 68,208 | ||||||
| 他にも | 140,702 | 140,774 | ||||||
| 研究と開発費用総額 | $ | 4,770,834 | $ | 3,163,485 | ||||
| 63 |
480万ドルの研究開発総支出に180万ドルの知的財産権研究開発支出は含まれておらず、2022年12月31日までの1年間で、研究開発支出は2021年12月31日現在の320万ドルから約20万ドル低下し、減少幅は5.9%、300万ドルとなった。外部サービスや契約研究組織費の減少は主に我々のXCART技術プラットフォームに関する支出の減少によるものであるが,許可に関するコストとDNaseプラットフォームに関する初期開発作業はこの支出を大きく相殺している.我々は2022年4月にDNaseプラットフォームの許可を得て,我々のエネルギーと資源をこの新たな買収の技術開発に利用したいと考えている. そのため,XCART技術プラットフォームの開発を休止した.
一般と行政費用
2022年12月31日までの年度の一般·行政費は370万ドルで、前年同期に比べて約10万ドル、または2.4%減少した。この低下は主にわれわれ知的財産権の組合せに関する相談や法的コストの減少により,2021年同期に比べて2022年12月31日までの1年間にCLSのDNase腫瘍学プラットフォーム許可に関する法的コストが大幅に増加したためである。
その他の収入(費用)
2022年12月31日までの年度の他の支出は約1,600ドルであるのに対し,2021年同期の他の収入は約1,100ドルである。その他の費用の増加 は,主に2022年12月31日までの年度内に,2021年同期に比べて外貨為替レートが不利に変化したためである。
利子収入,純額
2022年12月31日までの1年間で、利息純額は約20万ドルに増加したが、前年同期は約10万ドルだった。この増加は、主に2022年12月31日までの年間で、2021年同期に比べて投資資金の金利が高く、利息収入が増加したためである。この増加はPharmsynz ローン利息収入の低下部分によって相殺される
流動性と資本資源
2022年12月31日までの年間で,約660万ドルの純損失が発生した。2022年12月31日現在の累計赤字は約1兆891億ドルであるが、2021年12月31日現在の累計赤字は約1兆825億ドルである。2022年12月31日現在の運営資本は約1,260万ドルであり,2021年12月31日現在の運営資本は約1,730万ドルである。2022年12月31日までの年間で,我々の運営資本が470万ドル減少したのは,2022年12月31日までの年度の純損失およびDNase腫瘍学プラットフォーム許可証取得のための50万ドルの現金 であった。
私たちの主な流動資金源は現金だ。2022年12月31日まで、私たちは約1310万ドルの現金と110万ドルの流動負債を持っています。2021年12月31日まで、私たちは約1820万ドルの現金と140万ドルの流動負債を持っています。
| 64 |
財務諸表発表日から1年以内に,brの状況や事件があるかどうかを評価し,継続経営企業としての継続経営能力に大きな疑いを抱かせた。私たちが設立して以来、私たちはすでに大量の損失が発生しており、私たちは短期的に運営損失が続くと予想しています。これらの要素は私たちの持続的な経営企業としての能力を大きく疑っています。私たちは、継続的に経営する企業として、可能な公共または私募株式発行、債務融資、企業協力、関連側融資、または他の方法で資本資源を得ることができると信じている。私たちの既存の資源は、これらの財務諸表の日から計算するために、私たちの運営に少なくとも12ヶ月の資金を提供するのに十分だと信じています。しかし、長期的には、私たちの業務計画を実施するために、より多くの資金が必要になるかもしれません。将来のいかなる資金調達の条項、時間と範囲は、私たちの臨床開発計画の進展、私たちが許可或いは他の戦略手配に入る能力、私たちがナスダック株式市場(“ナスダック”)に上場し続ける能力、および金融、経済、地政学、業界と市場状況に関連するbr要素を含む複数の要素に依存し、その多くの要素は私たちがコントロールできない。バイオテクノロジー産業の資本市場は非常に不安定かもしれないが、これは未来のいかなる資金調達の条項、時間、そして範囲を不確実にする。2022年6月3日、吾らはナスダック上場資産部から書面通知(“通知”)を受け、吾らの普通株の収市価が30営業日連続で1.00ドルを下回ったことを通知したため、ナスダック上場規則第5550(A)(2)条(“購入価格要求”)に基づいてナスダック資本市場の最低購入価格要求に引き続き組み入れられなかった。この通知は私たちの普通株のナスダック資本市場への上場に対して直ちに発効しません。 ナスダック上場規則によると、私たちは通知日から180暦の期限があって、入札価格要求 を再遵守します。したがって、2022年11月30日までに入札価格要求を再遵守しなければならず、何らかの他の基準を満たしていれば、180日の規定期間を追加する資格がある。2022年12月1日、私たちはナスダックの手紙を受け取り、私たちの普通株が1株1.00ドルの最低購入価格要求に再達していないにもかかわらず、ナスダック は私たちが180日の日数、すなわち2023年5月29日にコンプライアンスを再獲得する資格があることを決定した。ナスダックの決定は,当社が株式の公開保有株式の時価継続上場要求に適合していることと,ナスダック資本市場が初めて上場した他のすべての適用要求(入札価格要求を除く)と,第2コンプライアンス期間内に必要な逆株式分割を行うことでこの不足を補う予定であることを示す書面通知に基づいている。
2023年3月10日、SVBはカリフォルニア州金融保護·革新部によって閉鎖され、この部門はFDICを担当者に指定した。私たちの現金は主にSVBによって維持されている。2023年3月12日、米国財務省、FRB、FDICはSVBのすべての預金者を全面的に保護し、2023年3月13日にSVBでの預金を完全に使用することができる緊急措置を打ち出した。したがって、私たちはこのような残高に何の損失も起こらないと予想する。
経営活動のキャッシュフロー
2022年12月31日までの経営活動で使用されているキャッシュフロー総額は約460万ドルであり,これは主に我々のこの間の純損失が,買収した知的財産権研究開発や株式ベースの費用に関する非現金費用部分が相殺されたためである。また、流動負債は2022年12月31日までの年間で減少している。2021年12月31日までの1年間で,経営活動に用いられたキャッシュフローは合計約470万ドルであり,これは主に我々のこの間の純損失により,株式ベースの費用に関する非現金費用の一部が相殺されたためである。
投資活動によるキャッシュフロー
2022年12月31日までの投資活動で使用されているキャッシュフローは合計500,000ドルであり,DNase腫瘍学プラットフォームを許可するための現金である。2021年12月31日までの年度には、投資活動からのキャッシュフローはない。
融資活動によるキャッシュフロー
2022年12月31日までの年間融資活動のキャッシュフロー はありません。2021年12月31日までの年間融資活動キャッシュフロー総額は約1,150万ドル,すなわち2021年7月に私募を行って得られた純収益である。
| 65 |
契約義務
契約義務とは、第三者との合意下での将来の現金約束および負債のことであり、将来の支払いを合理的に予測できないまたは負債を含まない。私たちの契約義務はオフィス空間の不動産賃貸に由来している。CMOサービスに義務があるにもかかわらず、次の表には、私たちとCMOとの合意に基づいて支払う可能性がある潜在的な支払いは含まれていません。これらのプロトコルによって、支払い時間と実際に支払うbrの金額が異なる可能性があり、具体的には、貨物またはサービスを受信した時間またはいくつかの義務に応じた合意条項または金額の変更に依存し、これらのプロトコルは、会社の書面通知後にキャンセルすることができるので、長期的なbr債務は発生しません。契約には予測困難な可変コストも含まれている可能性があり,登録された患者や臨床試験場所などによって異なる可能性があるため,次の表にも含まれていない。また、以下に列挙する債務の予想支払時間は、現在の情報に基づいて推定される。
次の表は、2022年12月31日までの契約義務を示しており、タイプ別にまとめています
期限どおりの支払い 2022年12月31日まで | ||||||||||||||||||||
| 合計する | 少ないです 1年 | 1-3 年.年 | 3-5 年.年 | 更に 比 5年間 | ||||||||||||||||
| レンタル義務 | $ | 28,524 | $ | 28,524 | $ | – | $ | – | $ | – | ||||||||||
| 合計する | $ | 28,524 | $ | 28,524 | $ | – | $ | – | $ | – | ||||||||||
最新の会計基準
付記3を参照してください重要会計政策の概要項目8に添付されている財務諸表,
プロジェクト7 A−市場リスクの定量的かつ定性的開示について
私たちは小さな報告会社なので、本プロジェクトに必要な情報を提供する必要はありません。
| 66 |
プロジェクト8−財務諸表 と補足データ
| 独立公認会計士事務所レポート(PCAOB ID) |
F-1 |
| 2022年と2021年12月31日までの連結貸借対照表 | F-3 |
| 2022年12月31日と2021年12月31日までの総合全面損失表 | F-4 |
| 2022年と2021年12月31日までの年度株主権益総合レポート | F-5 |
| 2022年、2022年、2021年12月31日までの統合キャッシュフロー表 | F-6 |
| 連結財務諸表付記 | F-7 |
| 67 |
独立公認会計士事務所報告
以下の株主と取締役会へ:
Xenetic生物科学社です
財務諸表のいくつかの見方
添付したXenetic Biosciences,Inc.(“貴社”)2022年12月31日現在と2021年12月31日までの総合貸借対照表,2022年12月31日までの各年度の全面赤字,株主権益とキャッシュフローに関する総合報告書 および関連付記(総称して“財務諸表”と呼ぶ)を監査した。この等財務諸表は,当社の2022年12月31日および2021年12月31日の財務状況と,2022年12月31日までの2年間の各年度の経営業績およびキャッシュフローを各重大な面で公平に反映しており,米国公認の会計原則に適合していると考えられる。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちのbrはアメリカ上場会社会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、アメリカ連邦証券法及びアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければならない。
我々はPCAOBの 基準に従って監査を行った。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても詐欺によるものであっても、監査を計画し、実行することを要求する。当社は必要とせず、brを招聘してその財務報告の内部統制を監査していません。私たちの監査の一部として、財務報告の内部統制を知る必要がありますが、社内財務報告の内部統制の有効性について意見を述べるためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、財務諸表の重大な誤報のリスク(エラーによるものであっても詐欺によるものであっても)を評価するためのプログラムの実行と、これらのリスクに対応するプログラムを実行することとが含まれる。これらの手続きには、財務諸表中の金額および開示に関する証拠をテストに基づいて検討することが含まれています。私たちの監査には、使用されている会計原則および経営陣による重大な推定の評価、財務諸表の全体的なレポートの評価も含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
以下に述べる重要な監査事項とは、当期に財務諸表を監査して生じる事項であり、これらの事項は、(1)財務諸表に対して重大な意義を有する勘定又は開示に関するものであり、(2)特に挑戦性、主観性又は複雑性に係る判断を監査委員会に伝達又は要求している。重要な監査事項のコミュニケーションは、財務諸表全体に対する私たちの意見 をいかなる方法でも変えることはなく、以下の重要な監査事項を伝達することによって、重要な監査事項またはそれに関連する勘定または開示について単独の意見 を提供することもない。
継続経営評価
関係事項の記述
私たちは会社の持続的な経営能力の評価と関連開示を重要な監査事項として決定した。当社は将来のキャッシュフロー予測 を作成し、その中で重要な変数の判断と推定、例えば発展していくDNA ase技術に関する未来予想収入、特許使用料収益とコストに関する。上記の会社の持続経営評価を審査することは、会社の持続経営分析に用いられるキャッシュフロー予測と他の仮定の合理性を評価するために、原子力数師の高度な判断に関連する。
総合財務諸表付記1に記載されているように、管理職は、当社が当該財務諸表を承認した日に十分な資金を使用することができ、当該財務諸表の日から少なくとも12ヶ月間継続的に経営することができると信じている。この評価を行う際に、管理層は、当社の既存資源を考慮している。
| F-1 |
私たちが監査でどのようにこの問題を解決したのか
モデル で用いた仮説を評価し,経営陣が使用した仮説を歴史的業績,予算,会社の戦略計画と比較することで,我々の意見の日から今後12カ月の将来のキャッシュフローを見積もる.また,収入,特許権使用料収益,キャッシュフロー予測における重大な支払い時間に関する仮定を含む重要な仮定を評価し,これらの仮定を履歴データおよび関連プロトコルと比較する方法である.著者らは未来の予想コストなどの重要な仮定に対して敏感性分析を行い、それらが未来のキャッシュフロー予測に与える影響を決定した。また、私たちは持続経営評価に基づいて会社の開示を評価した。
収入確認特許権使用料収入よりも高い
関係事項の記述
総合財務諸表付記3に記載されているように、当社の収入源は、承認された商業医薬製品の潜在的純売上高に基づいて第三者と締結された特許権使用料協定に基づいて得られた特許権使用料収益を含み、この純売上高は推定可変対価格に基づく。会社は、いつ報告された売上が確実に測定できるかを判断するために重要なbrを使用しなければならない。会社には残りの業績義務がなく、他のすべての収入確認基準を満たしている。会社の政策は、予想される特許使用料が確実に測定できる場合には、それを収入として確認することであり、これは第三者の報告を受けて行われる。同社は通常、実際にライセンス者に分けて販売した後の次の四半期にこれらの報告を受けている。
収入確認に関するプログラムの実行を決定し,特に経営陣が潜在純売上高を予想可変対価格と推定するプログラムに関する主な考慮要因は重要な監査事項であり,予想可変対価格金額の最適推定 を決定する際には,管理層は重大な判断を行う必要がある。これにより、監査人がプログラムを実行し、監査証拠を評価する際に高い判断力、主観性、および努力 を有することは、第三者との特許使用料契約において管理層が予想される可変対価格を決定すること、および管理層が可変対価格を推定するために使用する最適な推定の判断と関係がある。
私たちが監査でどのようにこの問題を解決したのか
この問題を処理することは、連結財務諸表に対する我々の全体的な意見を形成するための実行手順および評価監査証拠に関するものである。これらのプログラムは、可変対価格を決定するために、第三者の潜在的純売上に対する管理層の最適な推定値を評価することを含む。 これらのプログラムは、(I)管理層によって使用される重要な仮定の妥当性を評価およびテストすること、 (Ii)は、予想される 金額に関連する履歴経験が相対的に不足していることを指摘し、(Iii)第三者から受信された報告を含む証拠を取得および保証することを含む。
2015年以来、当社の監査役を務めてきました。
2023年3月22日
| F-2 |
Xenetic生物科学社です
合併貸借対照表
| 2022年12月31日 | 2021年12月31日 | |||||||
| 資産 | ||||||||
| 流動資産: | ||||||||
| 現金 | $ | $ | ||||||
| 前払い費用とその他 | ||||||||
| 流動資産総額 | ||||||||
| その他の資産 | ||||||||
| 総資産 | $ | $ | ||||||
| 負債と株主権益 | ||||||||
| 流動負債: | ||||||||
| 売掛金 | $ | $ | ||||||
| 費用とその他の流動負債を計算しなければならない | ||||||||
| 流動負債総額 | ||||||||
| 総負債 | ||||||||
| 引受金及び又は有事項(付記14) | ||||||||
| 株主権益: | ||||||||
| 優先株授権株 | ||||||||
| Bシリーズ、$額面:2022年12月31日と2021年12月31日までの発行済株式 | ||||||||
| Aシリーズ、$額面:2022年12月31日と2021年12月31日までの発行済株式 | ||||||||
| 普通株、$額面価値そして2022年12月31日と2021年12月31日までに認可された株そして2022年12月31日と2021年12月31日にそれぞれ発行された株そして2022年12月31日と2021年12月31日までの流通株 | ||||||||
| 追加実収資本 | ||||||||
| 赤字を累計する | ( | ) | ( | ) | ||||
| その他の総合収益を累計する | ||||||||
| 在庫株 | ( | ) | ( | ) | ||||
| 株主権益総額 | ||||||||
| 総負債と株主権益 | $ | $ | ||||||
付記はこれらの連結財務諸表の構成要素である。
| F-3 |
Xenetic生物科学社です
総合総合損失表
ここ数年で 十二月三十一日 | ||||||||
| 2022 | 2021 | |||||||
| 収入.収入 | ||||||||
| 特許権使用料収入 | $ | $ | ||||||
| 総収入 | ||||||||
| 運営コストと支出: | ||||||||
| 研究開発 | ( | ) | ( | ) | ||||
| 一般と行政 | ( | ) | ( | ) | ||||
| 総運営コストと費用 | ( | ) | ( | ) | ||||
| 運営損失 | ( | ) | ( | ) | ||||
| その他の収入(支出): | ||||||||
| その他の収入(費用) | ( | ) | ||||||
| 利子収入,純額 | ||||||||
| その他の収入合計,純額 | ||||||||
| 純損失 | $ | ( | ) | $ | ( | ) | ||
| 1株当たりの基本と償却純損失 | $ | ) | $ | ) | ||||
| 加重平均普通株式流通株、基本普通株、希釈普通株 | ||||||||
付記はこれらの連結財務諸表の構成要素である。
| F-4 |
Xenetic生物科学社です
合併株主権益報告書
| 優先株 | 普通株 | 積算 | ||||||||||||||||||||||||||||||||||
番号をつける のです。 株 | パル 価値(0.001ドル) | 番号をつける のです。 株 | パル 価値(0.001ドル) | その他の内容 すでに納めた 資本 | 赤字を累計する | その他の総合 収入.収入 | 在庫株 | 合計する 株主権益 | ||||||||||||||||||||||||||||
| 2021年1月1日現在の残高 | $ | $ | $ | $ | ( | ) | $ | $ | ( | ) | $ | |||||||||||||||||||||||||
| 普通株式と引受権証の発行は,発行コストを差し引く | – | |||||||||||||||||||||||||||||||||||
| 事前出資の引受権証を行使する | – | |||||||||||||||||||||||||||||||||||
| 引受権証を行使する | – | ( | ) | |||||||||||||||||||||||||||||||||
| 株式ベースの費用 | – | – | ||||||||||||||||||||||||||||||||||
| 仕入先に普通株を発行する | – | ( | ) | |||||||||||||||||||||||||||||||||
| 権証買収に関する普通株を発行する | – | ( | ) | ( | ) | |||||||||||||||||||||||||||||||
| 純損失 | – | – | ( | ) | ( | ) | ||||||||||||||||||||||||||||||
| 2021年12月31日現在の残高 | $ | $ | $ | $ | ( | ) | $ | $ | ( | ) | $ | |||||||||||||||||||||||||
| 購入が行われている研究と開発に関する普通株の発行 | – | |||||||||||||||||||||||||||||||||||
| 引受権証を行使する | – | ( | ) | |||||||||||||||||||||||||||||||||
| 株式ベースの費用 | – | – | ||||||||||||||||||||||||||||||||||
| 純損失 | – | – | ( | ) | ( | ) | ||||||||||||||||||||||||||||||
| 2022年12月31日現在の残高 | $ | $ | $ | $ | ( | ) | $ | $ | ( | ) | $ | |||||||||||||||||||||||||
付記はこれらの連結財務諸表の構成要素である。
| F-5 |
Xenetic生物科学社です
統合現金フロー表
| 12月31日までの年度 | ||||||||
| 2022 | 2021 | |||||||
| 経営活動のキャッシュフロー: | ||||||||
| 純損失 | $ | ( | ) | $ | ( | ) | ||
| 純損失と経営活動で使用される現金純額の調整: | ||||||||
| 現在行われている研究と開発を買収する | ||||||||
| 使用権資産の償却 | ||||||||
| 株式ベースの費用 | ||||||||
| 経営性資産と負債変動状況: | ||||||||
| 前払い費用とその他 | ( | ) | ||||||
| その他長期資産 | ( | ) | ||||||
| 売掛金、売掛金、その他の負債 | ( | ) | ||||||
| 経営活動のための現金純額 | ( | ) | ( | ) | ||||
| 投資活動によるキャッシュフロー: | ||||||||
| 現在行われている研究開発のために支払った純現金 | ( | ) | ||||||
| 投資活動のための現金純額 | ( | ) | ||||||
| 資金調達活動のキャッシュフロー: | ||||||||
| 普通株式発行および株式承認証で得られた金の純額 | ||||||||
| 株式承認証を行使して得られた収益 | ||||||||
| 融資活動が提供する現金純額 | ||||||||
| 現金純変動額 | ( | ) | ||||||
| 期初の現金 | ||||||||
| 期末現金 | $ | $ | ||||||
| キャッシュフロー情報の追加: | ||||||||
| 利子を支払う現金 | $ | $ | ||||||
| 非現金投融資活動補足スケジュール: | ||||||||
| 普通株を発行して現在行われている研究と開発を買収する | $ | $ | ||||||
| 仕入先に普通株を発行する | $ | $ | ||||||
| 権証買収に関する普通株を発行する | $ | $ | ||||||
| 無現金行使引受権証で普通株を発行する | $ | $ | ||||||
付記はこれらの連結財務諸表の構成要素である。
| F-6 |
Xenetic生物科学社です
連結財務諸表付記
| 1. | 会社(The Company) |
背景
Xenetic Biosciences,Inc.(“Xenetic”あるいは“会社”)はネバダ州に設立され、本部はマサチューセッツ州フレミンガムに設置され、生物製薬会社であり、革新的な免疫腫瘍技術の推進に集中し、治療困難な癌を解決する。同社独自のデオキシリボヌクレアーゼ(DNase)プラットフォームは,好中球細胞外トラップ(Net)を標的とすることにより免疫療法を含めた既存治療の結果を改善することを目指しており,好中球外トラップ(Net)は癌進展や癌治療に対する耐性に関与している。Xeneticは現在その系統DNA ase計画を臨床に推進し、膵臓癌と局部末期或いは転移性固形腫瘍の補助治療として集中している。XCART™同社の個性化キメラ抗原受容体(“CAR”)Tプラットフォーム技術は患者の特定の腫瘍新しい抗原に対して設計され、B細胞リンパ腫における作用機序を証明した。また、Xeneticは生物技術と製薬会社と協力して独自の薬物送達プラットフォームを開発し、 PolyXen®凝固障害領域の独占許可手配に応じて特許使用料を徴収する。
会社はその完全子会社Hesperix S.A.(“Hesperix”)とXenetic Biosciences(イギリス)を介して直接または間接的にXenetic UK(“Xenetic UK”)、およびXenetic UK、Lipoxen Technologies Limited(“Lipoxen”)、Xenetic Bioscience、InCorporationおよび SymBioTec,GmbH(“SymBioTec”)の完全子会社であり、複数の米国(“U.S.”)を有する。連邦商標登録および出願 および未登録商標およびサービスマークは、XCART、OncoHist、PolyXen、ErepoXen、本年度報告で使用されるImuXenを含むが、これらに限定されない。他のすべての会社と製品名は、関連会社の商標 である可能性があります。
継続経営と経営陣の計画
経営陣評価財務諸表発行日から1年以内に、条件や事件(総合的に考慮)があるかどうかは、当社の継続経営企業としての能力に大きな疑いを抱かせます。当社は設立以来巨額の損失を出しており、近いうちに経営赤字が続く見通しだ。これらの要因は、持続的な経営企業としての能力を大きく疑わせている。当社は、可能な公開または私募株式発行、債務融資、会社協力、関連側融資、または他の方法で資本資源を獲得し、経営を継続できると信じている。当社は、当社の財務諸表の日から、その既存資源は、当社の運営に少なくとも12ヶ月の資金を提供するのに十分であると信じている。しかし、同社は、長期的には、その業務計画を実施するために追加の資本が必要になる可能性があると予想している。将来の任意の融資の条項、時間、範囲は、その製品開発計画の進展、その識別および参入許可または他の戦略計画の能力、ナスダック株式市場(“ナスダック”)に上場し続ける能力、および金融、経済、地政学、業界、市場状況に関連する要素を含むいくつかの要素に依存し、その多くの要素は制御できない。バイオテクノロジー産業の資本市場は非常に不安定かもしれないが、これは未来のいかなる資金調達の条項、時間、そして範囲を不確実にする。2022年6月3日、当社はナスダック上場資産部から書面通知(“通知”)を受け、当社の普通株の終値が30営業日連続で1.00ドルを下回ったことを通知したため、当社は“ナスダック上場規則”第5550(A)(2)条のナスダック資本市場に引き続き組み入れられた最低入札価格要求(“入札価格要求”)を遵守していない。本通知は、当社の普通株がナスダック資本市場に上場することに直ちに影響を与えません。ナスダック上場規則によると、当社は通知日から180暦の期間を持ち、入札価格要求を再遵守することができます。したがって,会社 は2022年11月30日までに入札価格要求を再遵守しなければならず,何らかの他の基準を満たしていれば,180日のコンプライアンス期間 を追加獲得する資格がある.2022年12月1日、当社はナスダックから手紙を受け取り、当社の普通株が1株1.00ドルの最低購入価格要求に適合していないにもかかわらず、ナスダックは当社がさらに180暦延長する資格があることを決定し、あるいは2023年5月29日に遵守を再開する資格があることを通知した。ナスダックのセンチ定 は、当社が株式公開株式の時価継続上場要求およびナスダック資本市場初上場の他のすべての適用要求(購入価格要求を除く)に適合していることと、当社が第2コンプライアンス期間内に逆株式分割を行うことで不足点を補うことを意図していることを示す書面通知 に基づいている。
| F-7 |
| 2. | リスクと不確実性 |
新冠肺炎の大流行の影響
2020年3月、世界保健機関は1種の新型コロナウイルス株或いは新冠肺炎の発生と関連する全世界大流行を発表した。連邦、州と地方政府が緩和措置を取って公衆衛生危機に対応することに伴い、疫病はアメリカの経済状況に重大な影響を与え、2020年3月前半に加速し、そして2021年と2022年まで持続し、アメリカ経済に重大な不確定性をもたらした。当社は引き続き新冠肺炎疫病がその業務に及ぼす影響を評価しており,これまで当社の運営に大きな影響を与えていないが,当社では今回の“br”事件が当社の将来の運営に及ぼす影響は確定していない。新冠肺炎疫病が著者らの業務、運営と財務業績に与える影響程度は多くの絶えず変化する要素に依存し、著者らはこれらの要素を正確に予測できない可能性があり、このような不確定性はしばらく続くと予想される。
ウクライナ紛争が行動に与える影響
ロシアがウクライナに侵入した短期的で長期的な影響は現在予測が難しい。制裁および反制裁を実施することは、経済市場全体に悪影響を及ぼす可能性があり、私たちの業務、財務状況、運営結果に影響を及ぼす可能性がある。
| 3. | 重要会計政策の概要 |
合併原則
当社の総合財務諸表には、Hesperix、Xenetic UK、Xenetic UKの完全子会社Lipoxen、Xenetic Bioscience、InCorporation、br}およびSymBioTecの勘定が含まれています。すべての重要な会社間残高と取引はすでに合併で販売されている。
ある前期金額は今期に該当する列報方式で再分類されている.
予算の使用
総合財務諸表及び付記は米国公認会計原則(“米国公認会計原則”)に基づいて作成された。米国公認会計原則に基づいて財務諸表を作成することは管理層に推定、判断と仮定を要求し、これらの推定、判断と仮定は財務諸表中に報告された資産と負債額、収入、コストと費用及び付記中の開示に影響を与える。実際の結果と結果は,経営陣の見積もり,判断,仮定と大きく異なる可能性がある.
本位貨幣変動
会社の海外子会社のビットコインはドルです。2014年に会社がアメリカに移転した時、会社のイギリス子会社の本位貨幣はポンドからドルに変更された。機能通貨の変化は予想に基づいて適用される。したがって,従来計上されていた他の全面収益を積算したいかなる損益も変わらない。
| F-8 |
外貨取引ペン
為替変動による外貨取引による実現済みと未実現損益は総合全面損失表の“その他収入(費用)”で確認された。本位貨幣以外の通貨建ての貨幣資産と負債を貸借対照表日の為替レートで本位貨幣として再計量し、損益を総合全面損失表に計上する。
金融商品の公正価値
会計基準編纂(“ASC”) 主題820、公正価値計測公正価値は、計量日に市場参加者間の秩序ある取引において資産を売却するか、または負債を移動させることによって課金される価格として定義される。当社は以下の公正価値階層構造を採用し,公正価値を計量するための投入を3つのクラスに分類し,獲得可能かつ公正価値計測に重要な意味を持つ最低クラスの投入に基づいて分類した。一次投入とは、報告エンティティが計量日に取得する能力がある同じ資産または負債のアクティブなbr市場でのオファーを意味する。レベル2非アクティブ市場におけるオファー、ブローカーまたはトレーダーオファー、または合理的な価格透明性レベルを有する代替価格ソースを利用します。第三級投入は資産又は負債の観察できない投入であり、計量日には、当該資産又は負債の市場活動が少ない(あれば)。2022年12月31日および2021年12月31日現在、当社のいくつかの金融商品の帳簿価値は満期日が短いため公正価値に近づいている。注9を参照公正価値計量会社の公正な価値を議論するために。
現金と信用リスクの集中度
当社は購入日から90日以下のすべての高流動性投資 を現金等価物としています。購入日から90日を超えているが貸借対照表の日から1年未満の原始期限が である投資は短期投資に分類され、貸借対照表の日から1年以上の投資は長期投資に分類される。経営者は、購入時にその現金等価物および投資証券の適切な分類を決定し、各貸借対照表の日付にそのような決定を再評価する。これらのツールの短期的な性質により,現金等価物の帳票価値はその公正価値に近い.
会社を信用リスクに直面させる可能性のある金融商品には、主に金融機関に保管されている現金が含まれており、その残高はしばしば連邦保険限度額を超えている。2023年3月10日、シリコンバレー銀行(SVB)はカリフォルニア州金融保護·革新部によって閉鎖され、後者は連邦預金保険会社(FDIC)を担当者に任命した。当社の現金は主にSVBが持つ通貨市場基金です。2023年3月12日、米国財務省、FRB、FDICはSVBのすべての預金者を全面的に保護するための緊急措置を打ち出し、2023年3月13日、私たちはSVBでの預金現金を完全に使用することができる。したがって,会社 はこの等残高に関する損失は何も生じないと予想される.
財産と設備
当社はbrコストから減価償却累計価格を引いて財産と設備を記録しています。資産寿命または使用寿命を延長するための重大な更新および改善支出 を資本化に計上する。通常のメンテナンスやメンテナンスの性質の項目は,発生時に運営費用を直接計上する。会社 直線法を用いて資産の予想使用寿命内の減価償却を計算する:
| 資産分類 | 使用寿命を見込む | |
| オフィス及びコンピュータ装置 | ||
| 賃借権改善 | ||
| 家具と固定装置 |
当社は、廃棄またはその他の方法で処分された資産のコストを対応する減価償却とともに関連口座から除外し、それによる収益やbr}損失を経営結果に反映させる。
| F-9 |
無期限-生きている無形資産
業務 合併,許可その他の取引で取得した資産や負担する負債は,買収日にそれぞれの公正価値で確認するのが一般的である. 買収の際には,進行中の研究開発(“IPR&D”)を含む無形資産の公正価値を“収益法”を用いて決定するのが一般的である.買収された知的財産権研究開発無形資産は無期限無形資産とみなされ、関連研究や開発作業が完了または放棄するまで償却されない。会社の知的財産権の研究開発が技術実行可能性を達成する前に、大量の追加研究と開発を行う必要があるかもしれない。知的財産権研究開発プロジェクトが完成した後、 知的財産権研究開発資産はその使用寿命内に償却される予定である。
知的財産権の研究開発は償却しないが、少なくとも毎年或いは商業環境の事件或いは変化が帳簿価値が減値可能であることを表明した場合、減値を審査する。 会社はまた、事件や状況の存在が、企業が買収した知的財産権研究開発が損害を受ける可能性が高い(すなわち50%を超える) を決定することを決定するために、定性的要素を最初に評価することを選択することができる。もし会社がまず品質要素を評価し、知的財産権の研究開発減値の可能性が未買収よりも大きくないと判断した場合、会社はさらなる行動を取って減値をテストする必要がない。会社はまた、定量的減価テストのみを実行するために定性的評価をバイパスすることを選択することができ、会社は、他の期間に実行することなく、いくつかの期間にテスト を実行することを選択することができる。
減価損失(ある場合)は無形資産の帳簿価値がその公正価値を超えて計量される。
無形資産は減価費用の影響を受けやすく、特に知的財産権研究開発のための新しい買収資産。研究開発の高リスク性質と業界が開発化合物を市場に出す成功率を考慮すると、知的財産権研究開発減価費用は今後しばらくの間に発生する可能性がある。 の潜在的な減値を推定するIPR&Dの公正価値は予測と仮説の変化に非常に敏感であり、仮説の変化は減少を招く可能性がある。当社はその推定および仮定はすべて合理的であると信じており、その他の面では市場参加者が公正な価値を推定する際に採用する仮説と一致している。しかし、将来の結果が会社の見積もりや仮定と一致しなければ、会社は減価費用に直面する可能性があり、これは重大な費用である可能性がある。異なる推定 と判断を使用することは、会社の分析において重大な異なる結果が生じる可能性があり、重大な異なる資産 価値や費用をもたらす可能性がある。
長期資産減価準備
イベントや環境変化が資産や資産グループの帳簿金額 が完全に回収できない可能性があることを示す場合、当社は財産や設備の減値を含む保有·使用すべき長期資産を審査します。
回収可能性評価 は、資産または資産グループの使用およびその最終処分によって生じる未割引将来のキャッシュフローの推定に基づいている。 減値(あれば)は、資産の帳簿価値がその公正価値を超えた金額で計算され、通常、割引されたキャッシュフローを使用して公正価値を決定する。このような減値は,2022年12月31日と2021年12月31日までの年間では記録されていない。
| F-10 |
収入確認
同社は、製薬およびバイオテクノロジーパートナーと供給、許可、および協力協定を締結しており、いくつかの合意は、承認された商業医薬製品の潜在的純売上に基づく特許権使用料協定を含む。
会社はASCテーマ606に基づいて収入を確認した取引先と契約した収入(“ASC 606”)。本基準は、リース、保険、協力手配、金融商品など、顧客と締結されたすべての契約に適用され、 は他の標準範囲に属する契約を除く。ASC 606によれば、エンティティは、そのクライアントがコミットメント商品またはサービスの制御権を取得したときに収入を確認し、その金額 は、エンティティがそのような商品またはサービスと交換されることが予想される対価格を反映する。エンティティがASC 606の範囲内のスケジュールされた収入確認を決定するために、エンティティは、(I)顧客との契約を決定するステップと、(Ii)契約における履行義務を決定するステップと、(Iii)取引価格を決定するステップと、(Iv)契約内の履行義務に取引価格を割り当てるステップと、(V)契約義務を履行した時点または時間的に収入を確認するステップと、の5つのステップを実行する。会社は、顧客に譲渡された商品やサービスと交換するために、獲得する権利のある対価格を受け取る可能性がある場合にのみ、5ステップモードを契約に適用する。契約開始時に、契約がASC 606の範囲内にあると決定されると、会社は、各契約において約束された商品またはサービスを評価し、履行義務に属する商品またはサービスを決定し、各約束された商品またはサービスが異なるかどうかを評価する。そして、当社は、履行義務を履行する際に該当履行義務に割り当てられた取引価格の金額を収入 と確認する。
このような手配の会計手配の一部として、当社は、a)上記(Br)(Ii)ステップの決定によって決定された履行義務の数、b)上記(Iii)ステップでの取引価格、およびc)上記(Iv)ステップ取引価格分配契約で決定された各履行義務の独立販売価格を決定するために、重大な判断を使用しなければならない。当社は、以下に述べるように、マイルストーン や他の可変対価格を取引価格に計上すべきかどうかを判断するために使用します。取引価格は相対独立販売価格で契約履行義務ごとに割り当てられ,会社は契約項下の 履行義務を履行する際に収入を確認する.履行義務の独立価格を策定する際には,会社 は適用する市場条件や関連エンティティ特定の要因を考慮し,顧客と 合意を交渉する際に考慮する要因と見積りコストを含む.当社は,独立販売価格を決定するためのキー仮説の変化が複数の履行義務間の取引価格配分に大きな影響を与えるかどうかを評価することにより,契約義務の独立販売価格を検証する.会社は、会社業績(すなわち、顧客に譲渡された貨物またはサービス)と顧客業績との差額 (すなわち、顧客が支払い、無条件に支払うべき対価格)を補うために、契約資産または負債を確認する。
会社ライセンス契約の条項 は、提携パートナーへの知的財産権ライセンスの交付を含む場合があります。ライセンススケジュールによると、パートナーは、払い戻し不可能な前払い領収書、開発および規制対象領収書、および将来の製品販売の印税領収書の組み合わせによって補償を得ることができる。会社は、企業がライセンスおよび協力手配で受け取った払戻不可能な前払い許可証支払いおよび開発および規制マイルストーン支払い を確認する予定であり、供給義務などの将来の義務が含まれており、会社がそれぞれ手配した予想業績に基づいている間は比例 である。会社は,技術移転援助,br研究活動,臨床開発活動,製品開発から製品商業化までの製造活動を含む会社の業績義務履行予定時間を最適に推定している。これらの連携スケジュールの不確実性を考慮して, 実施期間の継続時間を決定するためには大きな判断が必要である。
会社がその部分特許の再許可の手配を達成した場合,収入確認の適切な方法と時間を決定する際に,会社はその手配が単一要素であるか多要素 であるかを決定する義務履行を考慮する。サービス、特許保護費用、技術支援、マーケティングまたは販売協力などのサービス、特許保護費用、または収入確認時間を変更する可能性のある追加の配信コンテンツを構成する可能性のある任意の他の任意の履行義務を履行することに加えて、会社は、ライセンスの条項 または価格調整または払い戻し条項などの要素に対するサブライセンスを考慮する。受信した払戻不可能な前払い許可とbr分割可能費用は,関連許可技術とは無関係またはいいかげんな継続履行または将来義務とみなされ,技術交付時に収入として確認される。
| F-11 |
会社は基本契約条項に基づいて販売期間内に特許権使用料収入を確認する予定であり,報告された売上高が確実に測定できることを前提としており,会社には残りの履行義務がなく,他のすべての収入確認基準を満たしている。会社は,会社が完成した協力協定に関する研究·開発サービスの精算が運営中に総収入 であることを確認する予定である。会社があるパートナーと締結したライセンス契約や提携協定は,将来的に会社に記念碑的な領収書を提供することも規定されており,それぞれのパートナーが締め切り延長を考慮した場合の表現や承認薬品の指定販売量に完全に基づいている。このような領収書については、会社は、適用契約条項に基づいて履行または比例して合意期限内に稼いだ領収書を収入として確認したいと考えています。会社の継続的な履行や将来の義務がどうでもいいとみなされたり、適当に済ませられたりした場合、これらの領収書も収入として確認される可能性があります。
注4を見る重要な戦略的協力.
研究と開発費
研究開発費とは,報酬福祉,施設費用,管理費用,臨床前開発,臨床試験および関連臨床製造費用,契約研究機関(“CRO”)や契約製造機関(“CMO”)に支払われる費用,その他の外部費用を含む研究開発活動で発生する費用である。当社は発生した研究開発費の支払いに用いられている。当社が研究開発サービスのために支払った前期費用は払い戻しできません。br義務が発生しているからです。買収に帰属するが資本化基準に達していない無形資産の価値は,買収時に研究·開発費用に計上される。ライセンス契約に基づいて支払われた前金は、許可を受けたときに支出されます。マイルストーン は、マイルストーンが実現可能であり、関連金額が合理的に評価されることが決定されている間、許可プロトコルに基づいて支払いを計算し、それに応じた費用を確認しなければならない。
会社は報告期間ごとの検討と開発費用を見積もる必要がある。このプロセスは、未締結契約および調達注文を審査し、会社員とコミュニケーションして、会社を代表して実行されたサービスを決定し、会社が領収書を受信していない場合、または他の方法で会社に実際のコストを通知する場合に、実行されるサービスレベルおよびサービスによって生じる関連コストを推定することを含む。当社のほとんどのサービスプロバイダは、予定されたスケジュールまたは契約マイルストーンに達したときに提供されたサービスのための借金領収書を発行します。しかし、 は前金が必要なものがあります。当社は、当時既知の事実と状況に基づいて、財務諸表において貸借対照表毎の日付までの計上費用を推定する。会社は定期的にサービスプロバイダと推定の正確性を確認し、必要に応じて調整する。計算すべき研究および開発費用の推定例には、以下の者に支払われる費用が含まれる
|
· · |
パートナーは研究開発と臨床前活動を展開している 臨床試験の全体的なプロジェクト管理に関するプロジェクトマネージャー; | |
| · | CGMP製造に関するcMOS; | |
| · | 臨床試験に関するCRO;および | |
| · | 臨床試験に関する調査サイト。 |
当社は,当社を代表して臨床試験を行って管理する複数の研究機関,CMOとCROとのオファーと契約に基づき,受け取ったサービスと費用の見積もりを,研究·開発,臨床前活動,製造,臨床試験に関する費用としている。これらの合意の財務条項は交渉が必要であり、契約によって異なり、不均衡な支払い流量をもたらす可能性がある。場合によっては、供給者に支払われるお金は、提供されたサービスレベルを超える可能性があり、それにより、料金の前払い
をもたらす。サービス料を計算する際には、会社はサービスを提供する時間帯と時間ごとにかかる努力度を見積もる。実際にサービスを実行する時間または努力の程度が推定値と異なる場合、会社はそれに応じて計算すべき金額または前払い金額を調整する。実際に発生した金額と実質的に差はないと予想されるが,実際の状態や時間に対する会社の提供サービスの状態や時間の理解が異なる可能性があり,報告された金額が任意の特定の時期に高すぎるか低すぎる可能性がある.これまで、
社の研究開発費に対する先行予想には何の大きな調整もなかった。2022年12月31日まで、2022年12月31日、2021年12月31日まで、会社が記録した累積計画費用は約$です
| F-12 |
当社はオプションと制限株式単位(“RSU”)の形で従業員と非従業員に株式ベースの支払いを与え、自社の普通株株式を購入し、従業員に株式所有権計画(“JSOP”)の奨励と、非従業員が提供するサービスと引き換えに普通株を発行する協定を付与する。
株式ベースの費用は、オプションの推定公正価値に基づくか、またはBlack-Scholesオプション定価モデルを使用して計算される。適切な公正価値モデルと関連仮定を決定するには、株価変動の推定と期待奨励条項を含む判断が必要である。期待変動率 は会社の履歴変動率から推定される。報酬を得ることができない予想期間全体の企業データの範囲内で、自社と上場企業と比較可能な同業者グループのオプション期待期間内の履歴変動性の加重平均を使用する。予想期限代表オプション未完了の時間を予定しています。 当社は没収が発生した場合、付与ではない場合には没収を会計処理します。当社は配当金を派遣していないし、将来に現金配当金を派遣することも期待されていないため、期待配当率はゼロである。無リスク金利 は、満期日が奨励期待期限と一致する米国債金利に基づく。権利を行使する際には、新しく発行された普通株の株式オプションが償還される。付与された帰属および決済条項を満たした場合、RSUは新たに発行された普通株式を償還する。
サービス条件のみに応じて付与された従業員オプションの場合、公正価値計量日は、通常、付与された日であり、関連報酬支出は、報酬の必要な帰属期間中に直線的に確認される。会社の運営中に消費された商品やサービスを交換するために発行された非従業員オプション については、公正価値計量日は、サービス完了日または業績承諾達成日の中で早い日である。当社では,株式オプションの公正価値 は,サービスを受ける公正価値よりも確実に計測されていると考えられる.非従業員に付与された株式オプションに関する報酬支出 は、報酬の必要な帰属中に直線ベースで確認される。
株式承認証
ある融資、コンサルティング、協力手配について、当社は株式承認証を発行してその普通株株式を購入した。未償還権証は独立ツールであり、所有者は売却または強制償還できず、株式奨励に分類される。当社はBlack-Scholesオプション定価モデルを用いて計量日までの奨励の公正価値を計量する。普通株を発行するとともに、パートナーに発行した引受権証は、最初に発行された普通株の追加実収資本の減少を公正価値で計上する。すべての他の株式承認証は公正価値によって必要なサービス期間内に直線的に費用を計上するか、あるいはサービス期間がない場合、あるいはサービスを提供した場合は発行の日に公正価値に基づいて費用を計上する。進行中の手配に関する授権証 は付記11でより全面的に説明されている株主権益.
所得税
会社はバランスシート法を用いて所得税を計算する。この方法によれば、繰延税金資産と負債は、税務·財務報告目的による項目の処理によって生じる一時的な差異に基づいて決定される。繰延税項資産および負債は税率計量を公布し,このような一時的な差が逆転が期待される年度の課税収入への適用が期待される。また、会社は繰延税金資産が将来の課税所得額から差し引かれる可能性を評価しなければならない。当社はその繰延税金資産の回収可能性を四半期ごとに評価する。
| F-13 |
当社が1株当たり基本純損失を計算する方法は、普通株株主に適用される純損失を期間内に会社が発行した普通株の加重平均株式数 を除く。当社は、期間内に発行された株式オプションの希薄化効果を考慮して、当該等の非参加証券が反償却作用を持たない限り、1株当たりの希薄化純損失を計算する。当社のJSOP 奨励は行使前に当社から在庫株とされているため、当社の1株当たり純損失の計算には影響しません。2022年12月31日と2021年までに毎年約JSOP賞を授与しました。
2022年12月31日と2021年12月31日までの年間では、会社の純損失状況により、1株当たりの基本純損失と希釈後の1株当たり純損失は同じである。潜在的に薄く、参加しない証券 は1株当たりの純損失の計算に計上されておらず、これらの証券を組み入れることは逆薄になるからである。2022年12月31日と2021年12月31日までの約そして潜在希釈証券はそれぞれ逆希釈証券と考えられている。
市場情報を細分化する
経営部門は企業の構成部分 として確定され、その独立した財務情報は経営意思決定者(即ち会社の最高経営責任者)が評価することができ、どのように資源を分配し、業績を評価するかについて決定することができる。会社 は、1つの運営部門でその運営を確認し、その業務を管理する。
賃貸借証書
当社は賃貸行政施設をbrにより経営しています。賃貸契約には、賃貸休暇、賃貸料上昇条項、テナント改善手当が含まれる可能性がある。当社はASU 2016-02に基づいてレンタルを会計処理し、レンタル(テーマ842)それは.ASU 2016-02は、テナントが開始日に短期賃貸を除くすべての賃貸の賃貸負債および使用権資産を確認することを要求しています。付記14を参照支払いを受けることと またはあるより多くの情報を得るために。
買収する
同社は買収取引に従事した歴史があり、これらの取引は当該取引が業務合併の基準を満たしているかどうかを評価することを要求している。取引 が業務合併要求に適合していない場合、取引は資産買収または資本再構成に計上され、営業権が確認されない。買収が企業合併の定義に適合する場合、当社は買収資産とその推定公正価値で負担する負債に買収価格(任意または価格を含む)を割り当て、支払われた買収価格は買収純資産が公正価値を推定する任意の部分を超えて営業権に計上される。br}買収資産と負債を負担する公正価値は、通常、リセットコストまたは現金流量推定値を推定する方法によって決定される。
当社は、買収された有形資産の公正価値を決定する際に、年齢、状況 及び資産の経済使用年数などを考慮して、その資産に代わる新資産のコストを見積もる。買収された無形資産の公正価値を決定する際には、当社は判断を用いて適用される割引率、成長率、および将来のキャッシュフローの時間と金額を推定する。購入された資産と負担する負債の公正価値は、通常、独立第三者専門家の協力の下で決定される。
企業合併に関するコストは,発生コストの間に に計上される.資産買収に関連するコストは、通常、買収された資産コストの一部として資本化される。
| F-14 |
最新の会計基準
2016年6月、FASBはASU 2016-13を発表した金融商品-信用損失(主題326):金融商品の信用損失の計測それは.この指導意見は、大多数の金融資産およびいくつかの他のツールの信用損失の計量および確認を修正した。修正案は、償却コストに応じて金融資産の信用損失を計量·記録するガイドラインを更新し、“発生した損失”モデルを“予想損失”モデルに置き換えた。これは損失準備を早く確認することにつながる可能性がある。ASU 2016-13は、2022年12月15日以降に開始される会計年度の報告公共エンティティに適用されますが、早期採用が許可されています。会社は採用の影響を評価し続けるが、これは私たちの連結財務諸表に実質的な影響を与えないと予想される。
| 4. | 重要な戦略的協力 |
武田薬品工業株式会社(及びその完全子会社武田)
2017年10月、当社は武田に当社のPolyXen技術に関するいくつかの特許の非排他的再許可を付与する権利を付与し、これらの特許はこれまで武田に血液や出血疾患の治療に関連する製品を独占的に付与していた。印税支払い
約$
Catalent Pharma Solutions LLC(“Catalent”)
2022年6月30日、会社はCatalentと一般的な作業範囲、スケジュール、定価を概説する作業説明書(SOW)を締結し、これにより、Catalentは会社の組換えタンパク質ヒトDNA IのcGMP生産を実行するためにいくつかのサービスを会社に提供する。双方の
は、SOWが予期する項目を管理する条項と条件を含み、Catalent標準条項と条件を含むSOW付録の代わりに、主サービス協定(MSA)を締結することに同意する。さらに、SOWに規定されている項目固有の条項および条件とMSAとの間に何らかの衝突がある場合、MSA条項および条件を基準とすべきである。プロジェクト期間中、SOW計画のプロジェクト製造サービスの推定総コストは約500万ドル(いくつかの費用および潜在的な代替案を含まない)に達すると予想され、プロジェクトの各段階はその段階の開始に関連して個別に領収書を発行する。これまでに改訂や終了が行われていない限り、SOW計画の製造サービスは2024年上半期に完成することを目標としている。会社は30日前にCatalentに書面で通知した場合、いつでもSOWを終了することができます。SOWには秘密保持,保証,知的財産権,賠償などに関する慣行条項も含まれている.2022年12月31日までの年間で,会社はCatalent
に約$を支払った
| F-15 |
スクリップス研究会社は
2020年5月15日、会社はスクリプス研究院(“スクリプス研究所”)と研究援助とオプション協定(“スクリプス協定”)を締結し、この協定によると、会社はスクリプス研究所に総額300万ドルの資金を提供し、XCART臨床前開発の推進に関する研究を援助することに同意した。研究経費は当社が合意した予算に基づいて四半期ごとにスクリプス研究会社に支払い、この予算はスクリプス協定締結日にスクリプス研究会社に第1期約300,000ドルを支払い、その後27ヶ月以内に四半期ごとに約300,000ドルを支払うことになっている。スクリプス協定によると、スクリプス研究会社は、スクリプス研究会社と2019年2月25日に締結された、2019年3月1日に当社に譲渡される当該特定ライセンス契約の条項に基づいて、当該分野内の任意の特許権又は技術(定義スクリプスプロトコル)
を当社に付与する。また、会社は、スクリプス研究会社の技術的権利または会社に付与されていない特許権のグローバル独占許可、および非独占的、印税免除、譲渡不可の許可を得ることを選択することができ、スクリプス研究技術(例えば、スクリプス協定の定義)をスクリプズ協定が想定する研究計画を実行する間に社内研究目的にのみ使用させることができる。2022年第2四半期に、双方はスクリプス協定の下での追加資金を終了することに同意した。したがって、スクリプズ研究会社は、以前に前払いした資金がなくなるまで、この合意に基づいて仕事を続けることに同意した。その会社は$を支払った
PJSC薬物合成
2009年11月、当社はPharmsynzと協力開発許可協定(“Pharmsynz手配”)を締結し、これにより、当社はPharmsynzに独占許可を付与し、ある地域で当社のPolyXenとImuXen技術に基づく6種類の候補製品の開発、商業化、マーケティングを許可した。交換として,Pharmsynzは同社に独占許可を付与し,Pharmsynzプロトコルの範囲内でPharmsynzが開発した任意の臨床前と臨床データを使用することを許可し,ある地域以外でさらなる研究,開発,商業化を行い,費用は会社が自費している。
Pharmsynzは直接または間接的にその完全子会社SynBio,LLC(“SynBio”)を介して当社約を保有している
2020年6月12日,当社はPharmsynz とメインサービスプロトコル(“Pharmsynz MSA”)を締結し,当社のB細胞悪性腫瘍に対するXCART技術 の開発を進めている。Pharmsynz MSAによれば、Pharmsynzは、ロシアおよびベラルーシの複数の学術機関との協力の管理を支援するために、ロシアおよびベラルーシの複数の学術機関との協力を支援するために、会社の主要な契約研究機関 として機能するが、これらのサービスを含むが、これらに限定されない、双方が時々合意した作業指示に従ってサービスを提供することに同意する。各伝票に規定されている予算と支払い条件によると、会社はPharmsynzがサービスを提供する際に発生する合理的な費用、支出、伝達コストを支払う必要がある。また、作業注文がマイルストーン支払いを規定している場合、会社は、勤務注文に規定されている条項に基づいて、PharmsynzまたはPharmsynzが指定する第三者サービスプロバイダにこのような支払いを支払わなければならず、このマイルストーン支払いは、会社が現金または会社普通株の株式で支払うことができる。
| F-16 |
当社とPharmsynzは2020年6月12日にPharmsynz MSAにより1つの作業注文(“作業注文”)を実行し、この作業注文に基づいて、Pharmsynzはこの作業注文に規定されている研究計画に基づいて当社のXCART技術の第1段階研究を行うことに同意した。ジョブ注文によって実行されるイベントは、Pharmsynz MSAによって事前に終了しない限り、約20ヶ月かかる予定です。
ジョブ注文規定によると、第三者サイトと契約を結んだ後、Pharmsynzが余分な転送コストのために領収書を発行することになっています。また、ワークシートは、ワークシートが規定する研究計画の早期段階の完成に関するマイルストーンに達した場合、会社は総額1,050,000ドルに達するマイルストーン支払いを支払うか、または会社自身が適宜1,000,000株会社の普通株を支払いまたは発行することを規定している。2022年12月31日現在、約$
2021年10月12日,当社はPharmsynzとPharmsynz MSAの第1号修正案(“MSA修正案”)を締結し,Pharmsynz MSA下のすべてのbr作業書を終了した。したがって,伝票の下でさらなるサービスを実行することはなく,任意の追加の サービスは新しい伝票に含まれる.交換として,会社はMSA修正案と同時に新たな工票(“第二工票”) を締結した。2つ目のワークシートの条項によると、Pharmynzは、会社が時々提出したXCART技術の開発を支援するために、会社の書面要求に応じていくつかの列挙されたサービスを提供する。
MSA改正案と第二工票によると、第二工票に入る際に、会社は一度に$を支払いました
SynBioは2011年8月、自社と株式引受および協力開発協定(“共同開発協定”)を締結した。同社はSynBioにSynBio技術に基づく分子と同社のロシアと独自体での独自技術(PolyXen,OncoHistとImuXen)の開発,マーケティング,商業化のある候補薬物の利用を許可し,ここでは総称してSynBio市場と呼ぶ.見返りとして、SynBioは同社に独占ライセンスを授与し、ある合意された製品の中でSynBioが産生した臨床前と臨床データを使用することを許可し、SynBio市場以外の任意の地区で商業候補製品の開発に従事することを許可した。
Synbioは独自に彼ら自身の研究と臨床開発活動を支援し、展開している。各社がそれぞれの技術に基づいてそれぞれの研究用品を提供する費用を除いて、 共同開発協定は、マイルストーンや他の研究に関する支払いを規定していない。br}が提供された場合、これらの費用は、双方の利便性のためであり、研究用品の提供の継続的または恒常的な義務を表すものではない。br}は、任意の最終製品の商業化に成功した後、ある地域の販売で10%の特許使用料 を得る権利があり、共同開発協定の条項に基づいて、SynBioに当該地域以外での販売の特許権使用料を支払う権利がある。SynBioは2021年12月20日から共同開発プロトコルをPharmsynzに譲渡した.
Pharmsynzは2022年12月31日まで研究·開発活動を継続しており,商業製品は何も発生していない。2020年12月,PharmsynzはEpolongの3期臨床研究の陽性データを報告し,Epolongは同社のPolyXen技術を用いて慢性腎臓疾患患者の貧血を治療する薬剤である。2021年2月、PharmsynzはEpolongの登録段階を開始し、ロシアの承認を得るために登録ファイルを提出したと報告している。Pharmsynzはそのプレスリリースで,ロシア段階の登録活動が2021年に完了し,早ければ2022年第1四半期に生産を開始できると報告している。Pharmsynzは当社に通知しました。ファイルのいくつかの欠陥を指摘し、訂正後に登録を再提出しようとする返信を受け取りました。“会社”ができた
| F-17 |
インド血清研究所有限会社
2011年8月、会社はインド血清研究所有限会社(“血清研究所”)と協力研究開発協定を締結し、会社のPolyXen技術を用いて潜在的な商業製品Polysialylated Erythropoietinを開発する独占許可を血清研究所に授与した。血清研究所は特定の予定区域内で規制承認を得るために必要なすべての臨床前と臨床試験を担当しており,費用は血清研究所が自費である。サミュエル研究所は同社に印税を支払い、サミュエル研究所販売エリアのある顧客に純販売を行うために使用している。協力手配によると、マイルストーンや他の研究に関連する 支払いの満期はありません。
2022年12月31日現在、会社はいかなる商業製品 も開発しておらず、この手配に関連する特許権使用料収入や支出も確認されていない。血清研究所は,2022年12月31日と2021年12月31日まで,それぞれ会社全発行普通株未満の株式 を保有している。
| 5. | カードを配る |
排他的再許可プロトコル
当社は2022年4月26日にCLS治療有限会社(“CLS”)とbr}独占再許可協定(“再許可協定”)を締結し、これにより、当社は独占許可を得て、CLSが所有又は制御するいくつかの特許権及びノウハウに基づいて、DNA酵素を用いて癌を治療するbr医薬製品及び方法(“再許可製品”)を開発及び商業化する。 は再許可協定の条項に基づいて、当社は独占的に責任を負い、商業的に合理的な努力を使用すべきであり、その中には、研究、米国およびいくつかのヨーロッパ市場で再ライセンス製品のマーケティング承認を開発し、マーケティング承認を得た後、関連市場でこのような再ライセンス製品を商業化する。
会社はライセンス契約に基づいて会社の許可及びその他の権利を付与する対価としてCLSに発行する当社普通株株式(“再許可契約株式”)のうち250,000株は、CLSからOPKOに間接的に譲渡された全額付属会社EirGen Pharma Ltd.(“EirGen”)の代わりに、OPKO Health,Inc.(“OPKO”) に直接発行され、CLSとEirGenとの間のいくつかの第三者契約義務を履行する。また、当社は、ある臨床および規制マイルストーンを実現する潜在的なマイルストーンとしてCLSに最大13,000,000ドルの現金を支払い、特定の規制マイルストーンを実現することに基づいてCLSに950,000株の会社普通株を追加発行する責任がある。また, 社は,許可期間内(再許可プロトコルで定義されているように)の許可範囲内の許可製品の純売上高に中央値-1桁から下位-2桁までの等級別使用料を支払い,会社が任意の許可者が受け取った一定の対価格の下位青少年から一定割合の使用料を支払うことが義務付けられている.
独占許可協定
当社は2022年4月26日にCLSと独占ライセンス契約(“ライセンス契約”)を締結し,これにより,当社はCLSが所有または制御するいくつかの特許権およびノウハウ下での独占許可を取得し,DNaseおよびCAR T療法を組み合わせた医薬製品および方法(“許可獲得製品”)を開発および商業化した。ライセンス契約の条項によると、会社 は独占的に責任を負い、他の事項を除いて、米国およびあるヨーロッパ市場でのライセンス製品の販売承認を研究、開発、獲得し、販売承認を得た後、このようなライセンス製品を関連市場で商業化すべきである。
当社はライセンス契約により当社の許可及びその他の権利を付与する対価として、CLSに一度に$を支払います
| F-18 |
再許可とbr許可プロトコルの総対価は約$である
特許譲渡と意志協力
同社は2022年10月4日、ベルギー威力SARL株式会社(“威力”)とCLSとの協力による特許譲渡を完了した。特許譲渡については,当社は2022年10月12日にデラウェア州有限責任会社CLS Treeutics,LLC(“CLS LLC”)と引受合意を締結し,これにより,当社はCLS LLCに自社普通株(“株”)を発行することに同意し,CLS LLCは850,000株自社普通株(“株”)の引受に同意し,CLSとその関連会社としてCLSとその関連会社が持ついくつかの特許権の対価を当社に譲渡した。
2022年8月2日,同社はWilitionとの研究·開発協力を発表し,Netを目指した細胞療法による癌治療の開発を発表した。Collaboration は,意志のNu.Qの潜在的な組合せを評価するための早期の探索的計画である®技術テストとbr社のDNase−Armored Car Tプラットフォームは,多様な固形癌に対する特許が可能な細胞療法を開発した。協力協定の条項によると、Wilitionは1つの研究プロジェクトに資金を提供し、双方は協力によるいかなる製品の商業化も収益できるかもしれないことを共有する。
特許譲渡の総対価は約50万ドルであり,終値で発行された850,000株の普通株の公正価値
社株の締め切りの終値に相当する。特許権には将来他の用途がないため,会社は約$の費用を記録した
その会社は約$を生み出した
| 6. | 財産と設備、純額 |
財産と設備、純額には以下が含まれる
| 2022年12月31日 | 十二月三十一日 2021 | |||||||
| オフィス及びコンピュータ装置 | $ | $ | ||||||
| 家具と固定装置 | ||||||||
| 財産と設備--コストで計算する | ||||||||
| 減価償却累計を差し引く | ( | ) | ||||||
| 財産と設備、純額 | $ | $ | ||||||
あったことがある
| F-19 |
| 7. | その他の資産 |
2016年、会社は血清研究所と合意し、臨床PSA用品を前払いし、交換した同社の普通株それは.2022年12月31日と2021年12月31日までに、会社はドルを
別注15を参照関連する 側取引Pharmsynzローンに関する説明を得るために。
| 8. | 費用を計算する |
計算すべき費用には以下が含まれている
| 2022年12月31日 | 十二月三十一日 2021 | |||||||
| 給与と福祉を計算すべきである | $ | $ | ||||||
| 専門費用を計算する | ||||||||
| 研究費に応じて計算する | ||||||||
| 他にも | ||||||||
| 費用総額を計算しなければならない | $ | $ | ||||||
| 9. | 公正価値計量 |
ASCテーマ820、公正価値計測公正価値は、計量日に市場参加者間の秩序ある取引において資産を売却するか、または負債を移転して受信した価格として定義される。当社では,公正価値クラスを用いて,公正価値を計測するための投入を3つのクラスに分類し,得られる公正価値計測に重要な意味を持つ最低クラスの投入に基づいてクラス内で分類した。一次投入とは、報告エンティティが計量日に取得する能力がある同じ資産または負債のアクティブ市場でのオファー
を意味する。第2レベルは、非アクティブ市場のオファー、ブローカーまたはトレーダーオファー、または合理的な価格透明性を有する代替定価ソースを利用する。第3レベル投入は資産または負債の観察不可能な投入であり、計量日には、資産または負債の市場活動が少ない(あれば)
である。2022年12月31日と2021年12月31日まで、満期期間が短いため、当社の金融商品の帳簿価値は公正価値に近い。いくつありますか
| 10. | 所得税 |
繰延税金資産と負債は、税務·財務報告目的による項目の処理によって生じる一時的な差異に基づいて決定される。繰延税 資産と負債は制定された税率計量を用いており,一過性 差額が逆転する見込みの年度の課税所得額に適用される予定である。また、会社は繰延税金資産が将来の課税所得額から差し引かれる可能性を評価しなければならない。当社は当社の繰延税金資産について全額推定手当brを提供しております。当社は繰延税金資産が現金化できない可能性が高いと考えているからです。当社はその繰延税金資産の回収可能性を四半期ごとに評価する。2022年12月31日までおよび2021年12月31日まで年度に所得税の計上(利益)はなく、当社はこれまで赤字を出してきた。
| F-20 |
所得税前損失構成は以下のとおりである
| 十二月三十一日までの年度 | ||||||||
| 2022 | 2021 | |||||||
| 国内(アメリカ) | $ | ( | ) | $ | ( | ) | ||
| 外国(イギリス) | ||||||||
| 外国(ドイツ) | ( | ) | ( | ) | ||||
| 外国(スイス) | ( | ) | ( | ) | ||||
| 所得税前損失 | $ | ( | ) | $ | ( | ) | ||
所得税優遇と米国会社税率(会社の所在国/地域に適用される税率)と純収益の入金は以下の通り
| 十二月三十一日までの年度 | ||||||||
| 2022 | 2021 | |||||||
| 連邦制 | $ | ( | ) | $ | ( | ) | ||
| 状態.状態 | ( | ) | ( | ) | ||||
| 評価免除額を変更する | ||||||||
| 永久差額,純額 | ||||||||
| 外貨利回り | ( | ) | ( | ) | ||||
| 株式に基づく支払い、純額 | ||||||||
| 強化された研究開発税控除 | ( | ) | ( | ) | ||||
| 為替レートの変化 | ( | ) | ||||||
| その他のプロジェクト | ( | ) | ( | ) | ||||
| 所得税純収益 | $ | $ | ||||||
| F-21 |
繰延税金資産および負債は、財務報告目的のための資産および負債の帳簿価値と所得税目的のための金額との一時的な違いによる税純影響を反映する。同社の繰延税金資産の重要な構成要素は以下の通り
| 十二月三十一日までの年度 | ||||||||
| 2022 | 2021 | |||||||
| 繰延税金資産: | ||||||||
| イギリスの純営業損失が繰り越す | $ | $ | ||||||
| イギリス資本損失繰越 | ||||||||
| アメリカ連邦純営業損失繰越 | ||||||||
| スイスの純営業損失が繰り越す | ||||||||
| 知的財産権研究開発 | ||||||||
| 株式ベースの支払い | ||||||||
| 強化された研究開発税控除 | ||||||||
| ドイツ純営業損失繰越 | ||||||||
| 資本化研究と実験支出 | ||||||||
| アメリカ各州の純営業損失が繰り越す | ||||||||
| 他にも | ||||||||
| リース責任 | ||||||||
| 推定控除前の繰延税金資産総額 | ||||||||
| 繰延税金資産の評価準備 | ( | ) | ( | ) | ||||
| 繰延税項目純資産 | ||||||||
| 繰延税金負債: | ||||||||
| 使用権--資産レンタル | ( | ) | ||||||
| 繰延税金負債総額 | ( | ) | ||||||
| 繰延負債純額 | $ | $ | ||||||
当社のイギリス純営業損失は、2022年12月31日、2022年12月31日および2021年12月31日までに約$に転換します
当社が米国で発生した営業損失の繰越と税収控除を用いて将来の課税収入を相殺する能力 は“米国国内税法”(以下、“準則”と略す)第382節の制限を受けている。規則に記載されているいくつかの所有権変更が発生した場合、これらの制限は、将来の営業損失繰越および税収控除の使用を制限する可能性がある。将来的に株式所有権が変化する可能性があり、これは当社の営業損失繰越と税収控除の使用にさらに制限を与えることになる。この場合、会社は、重大な経営損失の繰越や税収控除があっても所得税の納付を要求される可能性がある。
| F-22 |
会社がイギリスで発生した営業損失の繰越と税収控除の能力を使用する能力 はイギリス税法の制限を受ける。 これらの法規は将来の営業損失の繰越を制限する可能性がある:(I)所有権が変化すれば、会社の経営の業務性質や行為が変化し、(Ii)場合によっては、業務の性質や業務行為が変化した場合にのみ。この場合、繰越はこれ以上未来の収入を相殺するために使用されないだろう。
同社がドイツとスイスで発生した営業赤字の繰越や税収控除を利用する能力もドイツとスイスの税法によって制限されている。所有権が変化すれば、これらの規定は将来の営業赤字の繰越を制限する可能性があります。 この場合、繰越は未来の収入を相殺するために使用できなくなります。
2022年12月31日と2021年12月31日まで、会社
会社はアメリカ連邦税収管区、マサチューセッツ州税収管轄区、ある外国税収管轄区で所得税申告書を提出します。2022年までのカレンダー納税年度内に、純営業損失、繰越赤字、研究開発税収控除が利用できるため、会社は米国連邦、州、外国、地方所得税当局の審査を受ける。2022年12月31日まで、国税局または他の税務機関は、当社にいかなる審査
も通知していません。その会社は所有している
潜在的382制限
当社の純営業損失と税収相殺繰越は米国国税局の審査と可能な調整を受ける。当社がその純営業損失(“NOL”)および研究開発信用(“R&D”)を利用して繰り越す能力は実質的にbrに制限される可能性があり、規則第382節及び国の類似条文の要求により、所有権が発生した可能性や将来発生する可能性のある変更が原因である。これらの所有権変更は、毎年それぞれ未来の課税収入と税収を相殺するために使用できるNOLと研究開発信用繰越金額 を制限する可能性がある。一般に、規則382節の定義によれば、所有権変更とは、ある株主または公衆団体によって3年以内に行われる1つまたは一連の取引であり、所有権変更は、会社が発行した株式の50%を超えることを意味する。
当社は当社が規則382節で定義した赤字会社になって以来、1回または複数回の所有権変更が発生したかどうかを評価する研究は完了していませんが、当社は所有権変更が発生している可能性が高いと考えています。もし会社が所有権変更を経験した場合、NOLと研究開発信用繰越の使用は年間制限され、この制限はまず所有権変更時の会社普通株の価値 に適用される長期免税税率を乗じて決定し、必要に応じて追加調整を行う。このような制限は、NOLまたは研究開発積分の一部が使用前に満了する可能性がある。研究が完了し、いかなる制限も知られるまで、いかなる金額も不確定な税金状況または未確認の税金割引として開示されることはない。このような制限により使用前に満期になった繰越は、繰延税金資産から除外され、推定免税額に応じて調整される。評価準備の存在により、いかなる潜在的な制限も当社の経営業績に大きな影響を与えないと予想される。
主要税務管区(すなわちマサチューセッツ州連邦)は時々同社の利息や罰金を評価する可能性がある。2022年12月31日現在、会社には未確認の重大な税収割引がなく、負債や運営を調整する必要もない
| F-23 |
| 11. | 株主権益 |
普通株
会社普通株の1株当たりの株式保有者は、会社の株主投票に提出したすべての事項について一票を投じる権利を持たせます。普通株主 は取締役会が発表した時に配当金を得る権利がある。当社で任意の自発的または非自発的清算、解散または清算が発生した場合、普通株式保有者は、当社が分配可能な資産を比例的に共有する権利がある。
ライセンスシェア増加
2022年12月21日,会社株主投票で定款改正案が可決され,法定普通株が増加した株式(“株式増資許可”)。当社は2022年12月21日にネバダ州州務卿に当社定款改訂証明書を提出し、ライセンス株式増加を実施します。
市場で提供されています
当社は2021年11月19日に独占販売代理(“ウェインライト”)であるH.C.ウェインwright&Co.,LLCとATM発売プロトコル(“ATMプロトコル”)を締結し,この合意により,当社は随時ウェインライトを通してその普通株株式を提供·販売することができ,額面は$である1株あたり 。株式の発売は、2021年10月12日に米国証券取引委員会に提出され、2021年10月22日に発効を発表したS-3表保留登録声明(第333-260201号文書)と関連募集説明書に基づいて行われ、現在はATM協定により販売可能な400万ドル以下の証券に限られており、当社がS-3表I.B.6に基づいて一般的に販売可能な証券数が制限されている可能性があることを前提としている。
ATMプロトコルによると、Wainwrightは、証券法 が公布した第415条規則の定義に基づいて、直接またはナスダック資本市場での販売を含む“市場別”として発行された株式を販売中に販売することができる。単独の条項合意で合意すれば、当社はウェインwrightと当社が合意した買収価格でウェインwrightに元本として株式を売却することができます。Wainwrightはまた、会社の事前承認を得た場合に、個人的に協議した取引で株式を売却することもできる。Wainwrightによる株式の売却(あれば)は、当社が時々決めた金額と時間に応じて作成されますが、当社はいかなる株式も販売する義務はありません。当社またはWainwrightはいつでも契約下の要約または終了を一時停止することができます。実際の売上高は、市場状況、当社普通株の取引価格、当社の適切な資金源の決定を含む当社が時々決定する様々な要因に依存します。ATMプロトコルによる株式要約および売却は,(A)ATMプロトコルによるすべての 株式の発行および販売または(B)Wainwrightまたは当社がATMプロトコルの条項に従ってATMプロトコルを終了したときに終了する.
株式は2022年12月31日までおよび2021年12月31日まで年度はATM協定により販売された。その会社は$を生み出した
| F-24 |
私募する
2021年7月26日、当社は私募注文証券brについて購入契約を締結し、この合意に基づき、当社はナスダック規則に基づいて市価定価
の私募方式で発行及び販売し、(I)当社普通株、額面$1株当たり,(Ii)株式権証を承認し,購入総額は
Bシリーズ株式承認証は直ちに普通株1株当たり0.001ドルの価格で行使することができる。もしBシリーズ株式証所有者(及びその付属会社)が実益が4.99%以上の株式を所有している場合、Bシリーズ株式証所有者はBシリーズ株式承認証の任意の部分を行使する権利がないBシリーズ株式承認証の条項によると、行権発効後に発行された普通株式数のパーセンテージ
によって決定される。株主は当社に通知した後、実益所有権限度額の規定を増加または減少することができるが、実益所有権限度額は所有者が引受権証を行使した後に普通株を発行した後に発行された普通株式数の9.99%
を超えてはならない。実益保有権限度額を引き上げるいかなる措置も、当社に送付してから61日目に発効します。Bシリーズ株式承認証の内在価値は約#ドルである. 2021年12月31日までの年間でBシリーズ株式購入承認証普通株の株式
が行使され、結果として$
Aシリーズ株式承認証は直ちに普通株1株当たり3.30ドルの価格で行使できる。もしAシリーズ株式証所有者(及びその付属会社)が実益が4.99%以上の株式を所有している場合、Aシリーズ株式証所有者はAシリーズ株式承認証の任意の部分を行使する権利がないAシリーズ株式承認証の条項によると、発行権が発効した直後に当社の普通株式発行株式数の百分率
で決定されます。所有者は当社に通知した後、実益所有権限度額条項を増加または減少させることができるが、実益所有権限度額はいずれの場合もbr所有者が保有する株式証を超えてはならない。普通株発行が発効した直後に発行された普通株式数の9.99%を超えてはならない。実益所有権限度額の任意の引き上げは、当社への通知brの交付後61日目に発効します当社は発行された株式証明書の条項を評価し、当該等株式証を持分ツールに分類することを決定した。これらの株式承認証は授出日の公正価値から$と推定される1株、
,合計約$
Aシリーズ優先株
会社は
を指定しました
清算する任意の解散、清算または清算後、任意であっても非自発であっても、Aシリーズ優先株保有者は、Aシリーズ優先株に相当する1株当たりの声明価値(株式分割、合併、再編などの調整後)の金額 に任意の計算すべき配当と未払いの配当を加えて、その後、普通株またはAシリーズ優先株に対して任意の分配 を行う権利がある。
| F-25 |
配当をする*Aシリーズ優先株の株主
は非累積現金配当金を得る権利があり、年利率は
転換するAシリーズ優先株はいつでもその保有者の選択に応じて随時転換することができ、少なくとも61日前に当社に通知し、Aシリーズ優先株は12株Aシリーズ優先株の1株普通株に対する比率で計算する。
救いを求める30日前の書面通知によると、当社は任意のA系列優先株の保有者に、Aシリーズ優先株12株を普通株に変換する比率で当該Aシリーズ優先株の任意または全部を普通株に変換することを要求することができる。
Aシリーズの優先株には他の条項があり、株式配当と分割、投票権、断片的な株式と基本取引をカバーしている。2022年12月31日と2021年12月31日までに
Bシリーズ優先株
会社は
を指定しました
清算する*任意の解散、清算、または清算後、B系列優先株保有者は、任意であっても非自発であっても、B系列優先株保有者が会社資産からB系列優先株1株当たりの声明価値に相当する分配を得る権利がある(株式分割、組み合わせによる、任意の割り当て前に、普通株または任意のBシリーズ優先株(Aシリーズ優先株を含む)に任意の計算すべきおよび支払われていない配当金を加え、改訂および再記載された指定証明書に基づいてその時点で満期に対応する任意の他の費用または違約金に基づいて、Bシリーズ優先株以下の普通株または任意の系列優先株を分配しなければならない。この権利については,改訂および再記述された指定証明書による基本取引または制御権変更が清算を構成しなければならない.Xeneticは、そのような清算を承認するために、任意の株主会議の少なくとも30日前に、B系列優先株の各保有者に任意の清算の書面通知を発行するか、または会議が開催されない場合、少なくとも清算日の45日前に書面通知を発行する。
配当をするもし当社の取締役会が時々設立した任意の発行された系列優先株の任意の優先権利の制限を受けた場合、当社Bシリーズ優先株の株式保有者は、当社取締役会が時々発表する可能性のある当社の普通株式(換算基準)に関する非累積現金配当金 を利用可能な資金から得る権利がある。Bシリーズ優先株の任意の配当金がまだ支払われていない限り、当社は任意の配当金を直接または間接的に支払ったり発表したりすることができず、いかなる一次証券(Aシリーズ優先株を含む)についてもいかなる割り当てを行うこともできず、Bシリーズ優先株と同等の一次証券または株式を予約または償還するために使用することもできない。
転換する*Bシリーズ優先株は、いつでもその所有者の選択権に応じて、1株優先株から約0.33株普通株への比率で変換することができるが、発行可能上限および以下に説明する調整によって制限される必要がある。いくつありますか2022年と2021年12月31日までの年度のBシリーズ優先株転換。
| F-26 |
後続株式販売 .また,B系列優先株はラチェット価格による逆希釈保護を有し,B系列優先株の規定価値よりも低い1株価格で次の融資を行う場合には,通常の で分割される.組込み変換オプションには,ホスト機器と明確で密接に関連する分岐は存在しない.
Bシリーズの優先株には他の条項があり、株式配当と分割、投票権、断片的な株式と基本取引をカバーしている。2022年12月31日と2021年12月31日までに
協業·諮問協定に関する授権書
当社のいくつかの協力協定とコンサルティング手配について、当社は株式承認証を発行して普通株 株を購入してサービスとして支払いを行っています2021年12月31日までの年度内に、すでに協力或いはコンサルティングサービス承認株式証を付与或いは行使し、すべての期限が切れていない協力株式証 はすでに満期になった。だから、2022年12月31日と2021年12月31日まで、連携やコンサルティングサービス授権書はまだ発効していません。
融資手配に関する引受権証
2021年7月の方向性増発について、同社はAシリーズ株式承認証を発行し、購入した
また、同社はすでに株式購入のための取引承認証
を公開している
同社はまだ未償還の引受権証を持っており,約を購入することができる
株式証明書購入契約
同社は追加の未返済債務とbr持分融資権証を持っていて、合計約を購入します
| F-27 |
| 12. | 株式ベースの費用 |
株式オプション,RSU,普通株奨励に関する株式ベース総費用は約$であるそして$2022年12月31日と2021年12月31日までの年度。 総合全面損失表における株式に基づく費用は以下のように分類される
| 十二月三十一日までの年度 | ||||||||
| 2022 | 2021 | |||||||
| 研究開発費 | $ | $ | ||||||
| 一般と行政費用 | ||||||||
| $ | $ | |||||||
株式オプション
当社は、Xenetic Biosciences,Inc.改訂·回復の持分インセンティブ計画 (“株式計画”)に基づいて、異なる帰属条項で従業員および非従業員に株式オプション奨励およびRSU を付与する。当社はブラック·スコアーズオプション定価モデルを用いて株式オプション報酬の公正価値を計測し、このモデル は、無リスク金利、期待期限、株価変動性、配当収益率、罰金率を含む次の表で述べた仮定を使用する。無リスク金利は,付与時に発効する米国債収益率曲線をもとに, 1つの期限はオプションの期待寿命に近い。2022年と2021年に発行される株式オプションについては、“普通”株式オプションの条件を満たし、期待期限は簡略化方法に基づいている。当社が株式オプションを行使する歴史は限られており、当社が従業員と非従業員の株式オプションを見積もる期待期限に合理的な基礎を提供することはできません。他のすべての株式オプションについて、会社は会社の臨床プロジェクトの予想研究開発マイルストーンおよび会社従業員と非従業員の行動から予想寿命を推定した。非従業員 オプションの期待寿命はオプションの契約寿命である。予想変動率は、会社の実際の変動率に基づいて推定される。 会社データは受賞予想期間全体では利用できないため、会社が使用する価格変動率は、会社の候補薬物と類似した治療分野や非臨床開発段階を有する候補薬との比較可能な上場企業の価格変動に基づいている。当社は従来から配当を宣派しておらず、オプションの期待期間内に配当を宣言しないことが予想されているため、当社はすでに予想配当金 収益率を0%としている。当社は発生した没収行為を計算します。
従業員株式オプション
2022年12月31日と2021年12月31日までの年間で、 そして普通株を購入する全株式オプションはそれぞれ当社が付与します。オプションごとの重み付き平均 付与日公平価値は$そして$それぞれ,である2022年12月31日と2021年12月31日までの年間で、株式オプションが行使され、オプション満期は何もない。
2022年12月31日と2021年12月31日までの年間で、 そしてそれぞれ付与された株式オプション総額,公正価値総額は約$であるそして$ 2022年12月31日現在、約$ 付与される予定の従業員株式オプションに関する未確認株式ベースの支払い。会社は加重平均期間中にこの費用を確認する予定で、時間は約何年もです。
| F-28 |
ブラック·スコアーズオプションが2022年12月31日と2021年12月31日までの年間従業員に付与されるオプション定価モデルで使用される主な仮定は以下のとおりである
| 十二月三十一日までの年度 | ||||||||
| 2022 | 2021 | |||||||
| 加重平均予想配当率(%) | ||||||||
| 加重平均予想変動率(%) | ||||||||
| 加重平均無リスク金利(%) | ||||||||
| オプションの加重平均期待寿命(年) | ||||||||
| 加重平均相場(ドル) | ||||||||
以下は、2022年12月31日と2021年12月31日までの年間従業員株式オプション活動の概要である
| 株式数 | 重み付けの- 平均値 トレーニングをする 値段 | 重み付けの- 平均値 残り 生計 (年) | 骨材 固有の 価値がある | ||||||||||||
| 2021年1月1日現在の未返済金 | $ | $ | |||||||||||||
| 授与する | |||||||||||||||
| 期限が切れる | |||||||||||||||
| 2021年12月31日現在の未返済債務 | $ | $ | |||||||||||||
| 授与する | |||||||||||||||
| 期限が切れる | |||||||||||||||
| 2022年12月31日現在の未返済債務 | $ | $ | |||||||||||||
| すでに帰属していますか、あるいは2022年12月31日に帰属する予定です | $ | $ | |||||||||||||
| 2021年12月31日から行使可能 | $ | $ | |||||||||||||
| 2022年12月31日から行使可能 | $ | $ | |||||||||||||
| F-29 |
当社の2022年12月31日までの非既得社員株式オプション株式状況と2022年12月31日までの年間変化の概要は以下の通りです
| 量 株 | 重み付けの- 平均値 授与日 公正価値 | ||||||
| 2022年1月1日現在の残高 | $ | ||||||
| 授与する | $ | ||||||
| 没収される | $ | ||||||
| 既得 | ( | ) | $ | ||||
| 2022年12月31日現在の残高 | $ | ||||||
限定株 単位
ここにあります2022年12月31日と2021年12月31日までの未償還RSU である。RSUは完全に付与され,付与日の公正価値は$である1株当たり。 RSUは2022年12月31日と2021年12月31日までの年度内に付与または満期となる。
非社員株 オプション
非従業員に付与された株式オプションに関する株式ベース費用 は、サービスが直線的に提供されていることが確認されている。同社は、株式オプションの公正価値は、受けたサービスの公正価値よりも確実に計量されていると認定している。当社は2022年及び2021年12月31日までの年度内に、非従業員に 普通株を購入する購入権を付与していない は2022年12月31日と2021年12月31日までの年度内に非従業員株式オプションを行使した すべての 非従業員株式オプションが2020年12月31日にすべて帰属するため、2022年12月31日および2021年12月31日までの年間非従業員オプションに関する報酬支出。
以下は、2022年12月31日と2021年12月31日までの年間非従業員株式オプション活動の概要である
| 株式数 | 重み付けの- 平均値 トレーニングをする 値段 | 重み付けの- 平均値 残り 生計 (年) | 骨材 固有の 価値がある | ||||||||||||
| 2021年1月1日現在の未返済金 | $ | $ | |||||||||||||
| 授与する | |||||||||||||||
| 期限が切れる | ( | ) | |||||||||||||
| 2021年12月31日現在の未返済債務 | $ | ||||||||||||||
| 授与する | |||||||||||||||
| 期限が切れる | |||||||||||||||
| 2022年12月31日現在の未返済債務 | $ | $ | |||||||||||||
| すでに帰属していますか、あるいは2022年12月31日に帰属する予定です | $ | $ | |||||||||||||
| 2021年12月31日から行使可能 | $ | $ | |||||||||||||
| 2022年12月31日から行使可能 | $ | $ | |||||||||||||
| F-30 |
普通株奨励
会社は提供されたサービスと交換するために非従業員に普通株奨励を付与する。当社は、より信頼性の高い計量可能者を基準として、提供されたサービスの公正価値または付与された奨励の公正価値を使用して、これらの奨励の公正価値を計量する。これらの報酬の公正価値計量日は、一般にサービス完了日である。報酬の公正価値は,直線的に提供されるサービス であることが確認された.2022年12月31日と2021年12月31日までの年間で、会社が付与·発行した普通株奨励の概要は以下の通り
| 株式数 | |||
| 2021年1月1日現在の残高 | |||
| 授与する | |||
| 発表されました | ( | ) | |
| 2021年12月31日現在の残高 | |||
| 授与する | |||
| 発表されました | |||
| 2022年12月31日現在の残高 |
普通株奨励は2022年12月31日と2021年12月31日までの 年内に授与される。2022年12月31日現在、普通株奨励残高はまだ発行されていない。
共同持株計画
2022年12月31日と2021年12月31日まで、約 JSOP賞は上位2人の上級管理職に授与され、まだ完成していない。JSOP計画によると、当社の株式は、参加する役員とJSOP信託の受託者が公平な市価で共同購入し、当該等の株式はJSOP信託が保有する。米国公認会計原則の目的については,従業員オプションとしての評価を奨励し,従業員 が行使されるまで在庫株として株減少を記録した。JSOP奨励金は完全に付与されており、満期日はない。いくつありますか2022年12月31日と2021年12月31日までの年内の給与費用。
| 13. | 従業員福祉計画 |
会社には明確な401(K)貯蓄計画(“401(K)計画”)がある。401(K)計画は、実質的にすべての米国人従業員をカバーし、br}参加者が税引前に年間給与の一部の支払いを延期するか、または税引後貢献を行うことを可能にする。401(K)計画に対する会社の貢献は取締役会が適宜決定することができる。会社
は2022年12月31日と2021年12月31日までの年間で約$を貢献している
| F-31 |
| 14. | 引受金とその他の事項 |
賃貸借証書
会社は開始時に
手配がレンタルかどうかを確定します。開ける
我々の経営リースに関する補完キャッシュフロー情報と非現金 活動は以下のとおりである
現在までの年度 十二月三十一日 | 現在までの年度 十二月三十一日 | |||||||
| 2022 | 2021 | |||||||
| 経営キャッシュフロー情報: | ||||||||
| 賃貸負債の金額を計上するための現金 | $ | $ | ||||||
当社の経営リースに関する補足貸借対照表情報は以下の通りです
| 貸借対照表分類 | 十二月三十一日 2022 | 十二月三十一日 2021 | ||||||||
| 使用権資産-ST | 前払い費用とその他 | $ | $ | |||||||
| 流動賃貸負債 | 費用とその他の流動負債を計算しなければならない | $ | $ | |||||||
当社はASU
2016-02の規定をフロリダ州マイアミのオフィススペースレンタルに適用していません。効き目がある
| 15. | 関係者取引 |
当社は血清研究所やPharmynzと様々な研究,開発,許可,供給プロトコルを締結しており,それぞれ関連側であり,その関係,所有権,取引性質,取引性質はこれらの脚注の他の部分で開示されている。付記4を参照してください重要な戦略的協力は と注7、その他の資産また,関連先でもある連携パートナーとのスケジュールに関する詳細な情報を知る.
| F-32 |
当社は2019年第4四半期にPharmsynzと融資契約(“Pharmsynzローン”)を締結し、これにより当社はPharmsynzに元金総額最高$を提供した
当社は2021年1月23日からPharmsynz、Kevelt、SynBioと融資協定第一修正案と他の融資文書(“Pharmsynz融資延期”)
を締結し、Pharmsynzローンの返済条項と満期日を2022年1月に改正した。
というPharmsynzローン延期条項は2(2)筆の等しい毎月元金支払い$である
当社は2021年8月31日からPharmsynz、Kevelt、SynBioと融資協定第2修正案とその他の融資文書(“第2回Pharmsynzローン延期”)を締結し、Pharmsynzローンの返済条項と満期日を2022年7月に改正した。第二次Pharmsynzローン延期の条項は前払い費用を要求します
当社は2022年10月31日にPharmsynz、Kevelt、SynBioと“ローン契約第3修正案”と他のローン文書(“第3回Pharmsynzローン延期”)を締結し、主にPharmsynzローンの返済条項と満期日を2023年5月31日に改正するためだ。第三次ローン延期の条項は、第三次延期ローンに署名する際に一定の元金、利息、費用を支払うことを要求しています。また、第三次延期ローンは、2022年11月30日から2023年5月31日までの間に7(7)
ヶ月に分けて余剰元金と利息を分割返済すること、その他の条項と条件を要求しています。修正されたPharmsynzローンの他のすべての条項はまだ有効だ。Pharmsynzは第3次ローン延期に応じて何らかの金を支払っているが、第3次ローン延期に必要なすべての元金と利息はまだ支払われていない。そのため、会社は2022年12月31日と2021年12月31日までの受取ローンを長期ローンに分類している。当社は融資の回収可能性
を評価し、PharmynzとSynBioが所有する当社のすべての普通株と優先株を含む当社が米国で保有している担保を決定し、未返済元金残高の返済を支援するのに十分である。2022年12月31日と2021年12月31日まで約$
2022年4月、当社はCLSと付記5で述べたいくつかの 協定を締結しました。当社取締役の一人であるRoger KornbergはCLS科学顧問 取締役会のメンバーです。しかし、KornbergさんはCLSのいかなる株式も所有しておらず、このような合意で行われる取引によっていかなる経済的利益も得られません。当社の取締役の1つAdam Logalさんは、OPKO首席財務官、チーフ会計官、財務担当上級副社長です。
| F-33 |
| 16. | 後続事件 |
当社は、貸借対照表の発行日から財務諸表の発行日までの事項を審査し、以下に述べる事項以外に、財務諸表で確認又は開示する必要がないことを決定する。
スクリップス研究会社は
2023年3月17日、会社はスクリプス研究会社と研究援助とオプション協定(“合意”)を締結し、この協定に基づいて、会社のDNase腫瘍プラットフォーム技術の臨床前開発に関する研究を援助するために、スクリプス研究会社に総額938,000ドルの資金を提供することに同意した。研究経費は、契約締結日に約78,000ドルの頭金を支払い、12ヶ月以内に月ごとに約78,000ドルを支払うこととした当社が合意した予算に基づいてスクリプス研究会社に月ごとに支払います。このプロトコルによれば、同社は、プロトコル定義のような技術または特許権におけるScripps Researchの権利のグローバル独占許可、および合意によって予期される研究計画の実行中に、社内の研究目的のためにのみTSRI技術(例えば、プロトコル定義)を製造および使用するために、非独占的、brフリー、譲渡不可能な許可を得る権利がある。
事前に終了しない限り、本プロトコルの有効期限は、本プロトコルの日から15(15)ヶ月継続する。本協定は、会社が30日前にスクリプス研究会社に書面で通知することができ、発効日(契約中の定義)から6(6)ヶ月後から、またはスクリプス研究会社によって終了することができる。会社が本協定に規定されている満期金を適時に支払うことができない場合は、30日前にこのような不払い問題を解決することを書面で通知しなければならない。 他方が合意項目のいずれの義務を履行していない場合、または他方が破産した場合、いずれか一方はさらに合意を終了することができる。
シリコンバレー銀行
SVBは2023年3月10日にカリフォルニア州金融保護·革新部に閉鎖され、FDICを担当者に任命した。同社は主にSVBと現金を保持している。2023年3月12日、米国財務省、FRB、FDICは緊急措置を打ち出し、SVBのすべての預金者を全面的に保護し、2023年3月13日に、私たちのSVBでの預金現金を完全に使用することができた。そのため、会社はこのような残高に何の損失もないと予想しています.
| F-34 |
項目9−会計·財務開示面の変更と会計士との相違
適用されません。
プロジェクト9 A--制御とプログラム
評価開示制御とプログラム
我々の経営陣は、CEO及び最高財務官の参加の下、1934年に改正された“証券取引法”(以下、“取引法”と略す)下の規則13 a−15(E)及び規則15 d−15(E)によって定義された本年度報告書10−K表がカバーされている期間終了までの開示制御及び手続の有効性を評価した。
この評価に基づき、我々の経営陣は、2022年12月31日までに、我々の開示制御およびプログラミングが合理的な保証レベルであり、合理的な保証を効果的に提供し、取引所法案に基づいて提出または提出された報告で開示を要求した情報が、米国証券取引委員会規則および表で指定された期間内に記録、処理、まとめ、報告され、そのような情報が蓄積されて我々の主要幹部および主要財務官を含む、または状況に応じて類似の機能を実行する者を含む、我々の経営陣に結論を出した。必要な開示に関する決定をタイムリーに下すために
財務報告の内部統制に関する経営陣の報告
我々の経営陣は、“取引法”ルール13 a-15(F)に定義されている財務報告の十分な内部統制の確立と維持を担当している。我々の最高経営責任者と財務責任者の参加の下、経営陣は、本年度報告10-K表に含まれる期末までの財務報告内部統制の設計と有効性を我々の監督の下で評価した。財務報告の内部統制を評価する際に、経営陣は#年にトレデビル委員会協賛組織委員会(“COSO”)が制定した基準を使用した内部統制--統合フレームワーク(2013フレームワーク)それは.この評価に基づき,我々の経営陣は,本Form 10-K年次報告がカバーする期間が終了するまで,我々の財務報告に対する内部統制は#年テレデビル委員会COSO が提案した基準に基づいて有効であると結論した内部統制--総合的な枠組み。
本年度報告には、財務報告の内部統制に関する私の公認会計士事務所の認証報告は含まれていません。“ドッド·フランクウォール街改革·消費者保護法”第989 G節に規定されている非加速申請者の免除によると、経営陣の報告は公認会計士事務所の認証を受けていません。
財務報告内部統制の変化
本10-K年度報告がカバーする四半期期間中、財務報告の内部統制に何の変化も生じていない が財務報告の内部統制に大きな影響を与えたり、合理的な可能性が財務報告の内部統制に大きな影響を与えたりする。
| 68 |
制御とプログラムの有効性に対する制約
開示制御およびプログラムを設計·評価する際、管理層は、任意の制御およびプログラムは、設計および動作がどんなに良くても、予想される制御目標を達成するための合理的な保証しか提供できないことを認識している。企業の財務報告に対する内部統制には、以下の条件を満たす政策と手続きが含まれている
| (1) | 会社の資産を合理的かつ詳細かつ正確に反映した取引と処置の記録を保存することと関係がある |
| (2) | 公認された会計原則に基づいて財務諸表を作成するための合理的な保証を提供し、必要に応じて取引を記録することを保証し、会社の収支は会社の管理層と取締役の許可のみに基づいて行われる |
| (3) | 財務諸表に重大な影響を及ぼす可能性のある無許可買収、使用または処分会社の資産を防止または適時に発見することについて合理的な保証を提供する。 |
経営者は、会社の主要行政者や主要財務者、または同様の機能を遂行する者を含み、社内統制が全ての誤りや全ての詐欺行為を防止または発見することができることを期待していない。どんなに良く設計されていても、どんなによく動作しても、絶対的な保証ではなく、合理的な保証しか提供できず、制御システムの目標が実現されることを保証する制御システム。また,制御システムの設計は,資源制約が存在し,制御の利点がそのコストに対するものとみなされなければならないという事実を反映しなければならない.すべての制御系の固有の限界により,内部制御の評価は絶対保証を提供できない すべての制御問題や不正イベント(あれば)が発見されている.また,将来の 期間の制御の有効性のどの評価も,業務条件の変化によりこれらの内部制御が不十分になる可能性があり,あるいは の政策やプロセスに対する遵守度が悪化する可能性がある.
プロジェクト9 B-その他の資料
スクリップス研究会社は
2023年3月17日、会社はスクリプス研究会社と研究援助とオプション協定(“合意”)を締結し、この協定に基づいて、会社のDNase腫瘍プラットフォーム技術の臨床前開発に関する研究を援助するために、スクリプス研究会社に総額938,000ドルの資金を提供することに同意した。研究経費は、契約締結日に約78,000ドルの頭金を支払い、12ヶ月以内に月ごとに約78,000ドルを支払うこととした当社が合意した予算に基づいてスクリプス研究会社に月ごとに支払います。このプロトコルによれば、同社は、プロトコル定義のような技術または特許権におけるScripps Researchの権利のグローバル独占許可、および合意によって予期される研究計画の実行中に、社内の研究目的のためにのみTSRI技術(例えば、プロトコル定義)を製造および使用するために、非独占的、brフリー、譲渡不可能な許可を得る権利がある。
事前に終了しない限り、本プロトコルの有効期限は、本プロトコルの日から15(15)ヶ月継続する。本契約は、会社が30日前にスクリプス研究会社に終了を通知することができ、終了日は発効日(合意定義)から6(6)ヶ月後から始まります。もし会社が合意によって満期になったお金を適時に支払うことができない場合は、スクリプス研究会社が終了することができますが、30日前に書面で通知して、このような不払い行為を是正しなければなりません。他方がbrプロトコルで規定された任意の義務を履行できなかった場合、または他方が破産した場合、プロトコルはいずれか一方によってさらに終了することができる。
このプロトコルのコピー は、2023年3月31日までの3ヶ月間の10-Q四半期報告書に証拠として提出される。
プロジェクト9 C−検査を阻止する外国司法管区の開示について
適用されません。
| 69 |
第 第3部分
プロジェクト10--取締役、執行幹事、およびコーポレートガバナンス
本プロジェクトに要求される情報は、2022年12月31日までの会社会計年度終了後120日以内に米国証券取引委員会に提出される会社2023年株主総会に関する最終依頼書または情報説明書に記載され、引用により本明細書に組み込まれるか、または本年度報告書の10-K表修正案に含まれる。
プロジェクト11--役員報酬
本プロジェクトに要求される情報は、2022年12月31日までの会社会計年度終了後120日以内に米国証券取引委員会に提出される会社2023年株主総会に関する最終依頼書または情報説明書に記載され、引用により本明細書に組み込まれるか、または本年度報告書の10-K表修正案に含まれる。
プロジェクト12--利益を受けるすべての人および管理職の保証所有権および関連株主事項
本プロジェクトに要求される情報は、2022年12月31日までの会社会計年度終了後120日以内に米国証券取引委員会に提出される会社2023年株主総会に関する最終依頼書または情報説明書に記載され、引用により本明細書に組み込まれるか、または本年度報告書の10-K表修正案に含まれる。
第十三項-特定の関係及び関連取引、並びに取締役独立性
本プロジェクトに要求される情報は、2022年12月31日までの会社会計年度終了後120日以内に米国証券取引委員会に提出される会社2023年株主総会に関する最終依頼書または情報説明書に記載され、引用により本明細書に組み込まれるか、または本年度報告書の10-K表修正案に含まれる。
プロジェクト14--主に課金とbrサービス
本プロジェクトに要求される情報は、2022年12月31日までの会社会計年度終了後120日以内に米国証券取引委員会に提出される会社2023年株主総会に関する最終依頼書または情報説明書に記載され、引用により本明細書に組み込まれるか、または本年度報告書の10-K表修正案に含まれる。
| 70 |
第4部
プロジェクト15--証拠品と財務諸表
| (a) | 以下は、本年度報告書10-K表の一部として提出される | |
| · | 連結財務諸表:報告この項に求められる連結財務諸表及び独立公認会計士事務所報告書は、第二部第八項に含まれている | |
| · | 財務諸表明細書:それらが適用されないか、または必要とされないので、すべての付表は省略される、または要求された情報が連結財務諸表またはその付記に表示されるからである。 | |
| (b) | 展示品:本年度報告と共に表格10-Kの形態で提出または提供された展示品、または参照によって本明細書に組み込まれた展示品は、参照によって本明細書に組み込まれた72ページ目からの“展示品インデックス”に列挙される。 | |
項目16--表格10-Kの概要
適用されません。
| 71 |
展示品索引
| 展示品 違います。 | 添付ファイル インデックス | 表 | 提出日 | 展示品 番号をつける | アーカイブ ここから声明する | |||||
| 3.1 | “会社規約” | S-1 | 11/21/2011 | 3.1 | ||||||
| 3.2 | 会社定款修正書 | 8-K | 02/12/2013 | 3.1 | ||||||
| 3.3 | 会社定款修正書 | 8-K | 02/27/2013 | 3.1 | ||||||
| 3.4 | 会社定款修正書 | 10-Q | 01/10/2014 | 3.1 | ||||||
| 3.5 | NRS 78.209により証明書 を変更する | 10-Q | 01/10/2014 | 3.2 | ||||||
| 3.6 | 会社定款修正書 | 8-K | 09/30/2015 | 3.1 | ||||||
| 3.7 | を改訂して付例を新たに作成する | 8-K | 02/27/2017 | 3.1 | ||||||
| 3.8 | 改正·再決定されたAシリーズ優先株の優先株、権利及び制限指定証明書表 | S-1/A | 10/27/2016 | 3.8 | ||||||
| 3.9 | 第2部Bシリーズ優先株式、権利および制限指定証明書の改訂と再発行 | S-1/A | 10/31/2016 | 3.9 | ||||||
| 3.10 | NRS 78.209により証明書 を変更する | 8-K | 06/24/2019 | 3.1 | ||||||
| 3.11 | 会社定款修正書 | 8-K | 06/24/2019 | 3.2 | ||||||
| 3.12 | 会社定款修正書 | 10-K | 03/16/2021 | 3.12 | ||||||
| 3.13 | 会社定款修正書 | 10-K | 03/16/2021 | 3.13 | ||||||
| 3.14 | 会社定款改訂証明書 | X | ||||||||
| 4.1 | 1934年証券取引法第12条に基づく登録者証券説明 | X | ||||||||
| 4.2 | 登録者普通株式証明書表 | S-1/A | 07/14/2016 | 4.1 | ||||||
| 4.3 | 登録 Xenetic Bioscience,Inc.とOJSC Pharmynzの間の権利協定は、2015年7月1日である | 8-K | 07/08/2015 | 10.3 | ||||||
| 4.4 | 登録権協定第1修正案表 | 8-K | 11/16/2015 | 10.8 | ||||||
| 4.5 | 普通株引受権証表 | 8-K | 06/25/2019 | 4.1 | ||||||
| 4.6 | 普通株引受権証表 | 8-K | 07/22/2019 | 4.2 | ||||||
| 4.7 | Aシリーズ株式証明書表 | 8-K | 07/28/2021 | 4.1 | ||||||
| 10.1† | 改訂されたXenetic Biosciences,Inc.株式激励計画表は,改訂され,2021年12月7日から発効した | DEF 14 A | 10/15/2021 | 付録A | ||||||
| 10.2# | Lipoxen plc、Lipoxen Technologies Ltd、SynBio LLCが2011年8月4日に署名した共同開発契約と独占許可条項 | 10-K/A | 02/18/2015 | 10.18 | ||||||
| 10.3 | Xenetic Biosciences(UK)Limited(前身はLipoxen plc),Lipoxen Technologies Limited,SynBio LLCと上場株式会社Pharmsynzが2021年12月17日に締結した共同開発契約と独占許可条項の更新 | 10-K | 03/22/2022 | 10.3 | ||||||
| 10.4## | Lipoxen Technologies Limitedと上場株式会社Pharmynzの間の独占ライセンス契約は,2021年12月20日 である | 10-K | 03/22/2022 | 10.4 |
| 72 |
| 展示品 違います。 | 添付ファイル インデックス | 表 | 提出日 | 展示品 番号をつける | アーカイブ ここから声明する | |||||
| 10.5# | SynBio LLCとLipoxen plcは2011年8月4日にLipoxen plc株式における普通株の引受協定を締結した | 10-K/A | 02/18/2015 | 10.19 | ||||||
| 10.6# | 協力は、2009年11月11日のPharmsynz ZAOとLipoxen Technologies Ltd.との間の許可および開発協定である。 | 10-K/A | 02/18/2015 | 10.20 | ||||||
| 10.7# | Lipoxen plc、Lipoxen Technologies Ltdとインド血清研究所有限会社との間の独占特許と独自技術許可と製造協定は、2011年8月4日 | 10-K/A | 02/18/2015 | 10.21 | ||||||
| 10.8 | Dmitry Genkin,FDS Pharma,Lipoxen Technologies LimitedとXenetic Biosciences Inc.の間の知的財産権譲渡 | 10-K | 04/15/2015 | 10.1 | ||||||
| 10.9† | Xenetic Biosciences,Inc.とJeffrey Eisenberg間の雇用協定は,2016年12月1日である | 8-K | 12/6/2016 | 10.1 | ||||||
| 10.10† | Xenetic Biosciences,Inc.とCurtis Lockshin間の雇用協定は,2017年1月1日である | 8-K | 01/04/2017 | 10.1 | ||||||
| 10.11† | Xenetic Biosciences,Inc.ジェームズ·F·パスロと2017年3月23日に署名した雇用協定 | 8-K | 04/04/2017 | 10.1 | ||||||
| 10.12† | Xenetic Biosciences,Inc.とその各役員と幹部との間の賠償協定フォーマット | 10-Q | 08/14/2017 | 10.1 | ||||||
| 10.13† | Xenetic Biosciences,Inc.とJeffrey Eisenbergが2017年10月26日に改訂し再署名した雇用協定 | 10-K | 03/30/2018 | 10.45 | ||||||
| 10.14# | Xenetic Biosciences,Inc.,Baxalta Inc.,Baxalta US Inc.とBaxalta GmbHの間で締結された2017年10月27日の再ライセンス契約の権利 | 10-K | 03/30/2018 | 10.46 | ||||||
| 10.15 | Xenetic Biosciences,Inc.とOPKO PharmPharmticals,LLC間の譲渡プロトコルは,2019年3月1日である | 8-K/A | 05/20/2019 | 10.1 | ||||||
| 10.16 | 譲渡協定第1回修正案、日付は2019年6月7日 | 8-K | 06/13/2019 | 10.1 | ||||||
| 10.17 | 譲渡協定第2修正案、日付:2019年6月24日 | 8-K | 06/24/2019 | 10.1 | ||||||
| 10.18 | 3回目の“譲渡協定改正案”、日付は2019年7月15日 | 8-K | 07/16/2019 | 10.1 | ||||||
| 10.19 | Xenetic Biosciences,Inc.とある購入者の間で署名された同意書表は、2019年6月24日である | 8-K | 06/25/2019 | 10.1 | ||||||
| 10.20 | Xenetic Biosciences,Inc.と帝国株式譲渡会社との間の認可代理協定は,2019年7月19日である | 8-K | 07/22/2019 | 10.1 | ||||||
| 10.21 | Xenetic Biosciences,Inc.と一部の購入者との間の同意合意は、2019年7月16日である | 8-K | 07/16/2019 | 10.1 | ||||||
| 10.22† | レタープロトコルフォーマット .非従業員、独立取締役を遺伝生物科学会社に任命する | 10-K | 03/26/2020 | 10.51 | ||||||
| 10.23† | Xenetic Biosciences,Inc.株式オプション付与通知テーブル | 10-K | 03/26/2020 | 10.52 | ||||||
| 10.24† | Xenetic生物科学社株オプション付与通知は,2019年12月4日,Jeffrey EisenbergとXenetic Biosciences,Inc.によるものである. | 10-K | 03/26/2020 | 10.53 | ||||||
| 10.25## | Br社とスクリプス研究所が2020年5月15日に署名した資金とオプション協定を研究する | 10-Q | 08/12/2020 | 10.1 |
| 73 |
| 展示品 違います。 | 添付ファイル インデックス | 表 | 提出日 | 展示品 番号をつける | アーカイブ ここから声明する | |||||
| 10.26## | 会社とPJSC Pharmynzが2020年6月12日に締結したMasterサービス契約 | 10-Q | 08/12/2020 | 10.2 | ||||||
| 10.27## | 当社とPJSC Pharmynzが2021年10月12日に締結したプライマリサービス協定改正案1 | 10-K | 03/22/2022 | 10.28 | ||||||
| 10.28## | 会社とPJSC Pharmynzが2021年10月12日に締結したプライマリサービス契約の2つ目のワークシート | 10-K | 03/22/2022 | 10.29 | ||||||
| 10.29 | 当社及びその他の当事者が2021年7月26日に署名した“証券購入協定”表 | 8-K | 7/28/2021 | 10.1 | ||||||
| 10.30 | 権利協定表を登録し、期日は2021年7月26日で、当社及びその他の各方面が署名します | 8-K | 7/28/2021 | 10.2 | ||||||
| 10.31 | Xenetic Biosciences,Inc.と所持者との間の書簡合意書フォーマットは,2021年11月15日である | 8-K | 11/16/2021 | 10.1 | ||||||
| 10.32 | Xenetic Biosciences,Inc.とH.C.Wainwright&Co.,LLC間の市場発売合意は,2021年11月19日である | 8-K | 11/19/2021 | 1.1 | ||||||
| 10.33## | Xenetic Biosciences,Inc.とCLS治療有限会社との間の独占再許可協定は,2022年4月26日である | 10-Q | 8/11/2022 | 10.1 | ||||||
| 10.34## | Xenetic Biosciences,Inc.とCLS治療有限会社の独占許可協定は,2022年4月26日 である | 10-Q | 8/11/2022 | 10.2 | ||||||
| 10.35 | Xenetic Biosciences,Inc.とCLS治療有限会社の引受プロトコルフォーマットは,2022年4月26日である | 10-Q | 8/11/2022 | 10.3 | ||||||
| 10.36## | Xenetic Biosciences,Inc.とCatalent Pharma Solutions,LLC間の作業宣言は,2022年6月30日である | 10-Q | 8/11/2022 | 10.4 | ||||||
| 21.1 | 付属会社名簿 | X | ||||||||
| 23.1 | Marcum LLPの同意 | X | ||||||||
| 24.1 | 授権書(署名ページに含まれる) | X | ||||||||
| 31.1 | 規則第13 a−14(A)条又は規則第15 d−14(A)条の要件に基づいて幹事を実行する | X | ||||||||
| 31.2 | 細則13 a-14(A)または細則15 d-14(A)の要件に応じて財務首席幹事を認証する | X | ||||||||
| 32.1* | “米国法典”第18編第36章第13 a-14(B)条又は第15 d-14(B)条及び第1350節第13 a-14(B)条又は第15 d-14(B)条に要求される最高経営責任者及び最高財務責任者証明書(“米国法典”第18編第1350節) | X | ||||||||
| 101.INS | XBRLインスタンスドキュメントを連結する. | X | ||||||||
| 101.書院 | インラインXBRL分類拡張 アーキテクチャ文書. | X | ||||||||
| 101.カール | インラインXBRL分類拡張 はリンクベース文書を計算する. | X | ||||||||
| 101.def | インラインXBRL分類拡張 はLinkbase文書を定義する. | X | ||||||||
| 101.介護会 | XBRL分類拡張 タグLinkbase文書を連結する. | X | ||||||||
| 101.Pre | インラインXRBL分類拡張 はLinkbase文書を示す. | X | ||||||||
| 104 | 表紙インタラクティブ データファイル(イントラネットXBRL文書に埋め込む) | X |
|
† #
## |
契約または任意の補償計画、契約または手配を管理することを指す。 証券·取引委員会に申請し,本文書に含まれるいくつかの機密材料の機密処理を要求した.機密処理を要求した漏れ書類は,米国証券取引委員会に単独で提出されている. 改正された“1933年証券法”S-K条例第601(B)(10)項によれば、本展示品において四角括弧及び星番号で表記されている部分は省略されており、それら(I)は実質的ではないため、(Ii)公開開示されていれば登録者に競争損害を与える可能性がある。委員会又はその職員が要求を出した場合、登録者は、編集されていない展示品のコピーを補充として迅速に提供することを承諾する。 |
| * | この証明は、1934年証券取引法(改正)第18節の目的のために提出されたものとみなされないか、又は同節の責任を他の方法で負うものとみなされず、1933年証券法(改正後)又は1934年証券取引法(改正後)に基づいて提出されたいかなる文書に引用されてもみなされてはならない。 |
| 74 |
サイン
1934年“証券取引法”第13又は15(D)節の要求に基づいて、登録者は、以下の署名者がその代表として本報告書に署名することを正式に許可した。
| Xenetic生物科学社です | ||
| 日付:3月22, 2023 | 差出人: | ジェフリー·F·アイゼンバーグ |
| ジェフリー·F·アイゼンバーグ | ||
| 最高経営責任者 |
授権書と署名
私たちは、以下に署名したXenetic Biosciences,Inc.の上級管理者と取締役をそれぞれ構成し、私たちの真の合法的な代理人Jeffrey F.Eisenbergを構成して任命し、彼は私たちの名義で次のようなすべての改正に署名し、全体的に私たちの名義と代表でこのような身分ですべてのことをして、Xenetic Biosciences,Inc.が改正された1934年の証券取引法の条項と証券取引委員会のすべての要求を遵守することができるようにする。
1934年の証券取引法の要求 によると、本報告は2023年3月22日に以下の登録者によって次のような身分で署名された。
| サイン | タイトル | |
| ジェフリー·F·アイゼンバーグ |
取締役CEO兼最高経営責任者 (首席行政官) | |
| ジェフリー·F·アイゼンバーグ | ||
| 日付:3月22, 2023 | ||
| /s/ジェームズ·パスロ | 首席財務官 | |
| ジェームズ·パスロ | (首席財務官および首席会計官) | |
| 日付:3月22, 2023 | ||
| /s/Grigory Borisenko | 役員.取締役 | |
| グレゴリー·ボリーセンコ | ||
| 日付:3月22, 2023 | ||
| /s/ジェームズ·カラヴィ | 役員.取締役 | |
| ジェームズ·カラヴィ | ||
| 日付:3月22, 2023 | ||
| /s/Firdaus JAL Dastoor | 役員.取締役 | |
| フェルダス·ジャール·ダストゥール | ||
| 日付:2023年3月22日 | ||
| ロジャー·コエンバーグ | 役員.取締役 | |
| ロジャー·コエンバーグ | ||
| 日付:2023年3月22日 | ||
| /s/Adam Logal | 役員.取締役 | |
| アダム·ローゲル | ||
| 日付:3月22, 2023 | ||
| /s/Alexey Vinogradov | 役員.取締役 | |
| アレクセイ·ヴィノグラドフ | ||
| 日付:3月22, 2023 |
| 75 |