
アメリカ アメリカ
証券取引委員会
ワシントンD.C.,20549
表
(タグ 一)
締め切りの財政年度について
あるいは…。
過去から現在まで,現在から現在への過渡期について
手数料ファイル番号
(登録者がその定款に明記されている氏名)
(州や他の管轄区域 会社や組織) |
(I.R.S.雇用主 標識 番号) |
| ||
| (主に実行オフィスアドレス ) | (Zip コード) |
登録者の電話番号、市外局番を含む:+
同法第12条(B)に基づいて登録された証券:
| (授業ごとのタイトル) | 取引コード | (登録された各取引所の名称 ) |
同法第12条(G)に基づいて登録された証券:
ありません
登録者が証券法ルール405で定義されている経験豊富な発行者である場合、 は再選択マークで表される。はい。☐ ☒
登録者が当該法第13又は15(D)条に基づいて報告書を提出する必要がない場合は,複選マークで示してください。はい。☐ ☒
登録者が(1)過去12ヶ月以内(または登録者がそのような報告を提出する必要があるより短い期間)に1934年の証券取引法第13条または第15(D)節に提出されたすべての報告を提出したかどうか、および(2) が過去90日以内にそのような提出要件に適合しているかどうかを、再選択マークで示す☒No.no☐
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書を提出する必要があるより短い時間内)に、S−T規則(本章232.405節)第405条の要求に従って提出された各相互作用データファイルを電子的に提出したか否かを示す☒No.no☐
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社, か新興成長型会社かを再選択マークで示した。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小報告会社”、“新興成長型会社”の定義を参照されたい。
| 大型 加速ファイルサーバ | ☐ | 加速した ファイルマネージャ | ☐ | |
| ☒ | 小さな報告会社 | |||
| 新興成長型会社 |
もしbrが新興成長型会社である場合、登録者が延長された移行期間を使用しないことを選択したかどうかを再選択マークで示して、取引法第13(A)節に従って提供された任意の新しいまたは改正された財務会計基準を遵守してください
登録者が報告書を提出したか否かを再選択マークで示し、その経営陣が“サバンズ-オキシリー法案”(“米国法典”第15編7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する
登録者が空殻会社であるかどうかをチェックマークで表す(“取引法”第12 b-2条で定義されている)。はい。☐
2022年6月30日まで,すなわち登録者が最近完成した第2財期の最終営業日であり,登録者の非関連会社が保有する投票権と無投票権普通株の総時価は$である
2023年3月17日現在、登録者普通株流通株数は.
参照により組み込まれた文書
| カタログ表 | ページ | |
| 第1部 | ||
| 第 項1. | 業務.業務 | 7 |
| 1 a項目. | リスク要因 | 23 |
| 項目 1 B. | 未解決従業員意見 | 58 |
| 第 項2. | 属性 | 58 |
| 第 項3. | 法律訴訟 | 58 |
| 第 項. | 炭鉱安全情報開示 | 58 |
| 第II部 | ||
| 第 項5. | 登録者普通株市場、関連株主事項及び発行者による株式証券の購入 | 59 |
| 第 項6. | [保留されている] | 59 |
| 第 項7. | 経営陣の財務状況と経営成果の検討と分析 | 59 |
| 第 7 A項。 | 市場リスクの定量的·定性的開示について | 66 |
| 第 項8. | 財務諸表と補足データ | F-1 |
| 第 項9. | 会計と財務情報開示の変更と相違 | 67 |
| 第 9 A項。 | 制御とプログラム | 67 |
| 第 9 B項。 | その他の情報 | 67 |
| 第 9 C項. | 検査妨害に関する外国司法管区の開示 | 67 |
| 第三部 | ||
| 第 項10. | 役員·幹部と会社の管理 | 68 |
| 第 項11. | 役員報酬 | 72 |
| 第 項12. | 特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 | 72 |
| 第 項13. | 特定の関係や関連取引、取締役の独立性 | 72 |
| 第 項14. | チーフ会計士費用とサービス | 72 |
| 第4部 | ||
| 第 項15. | 展示·財務諸表明細書 | 73 |
| 第 項16. | 表格10-Kの概要 | 74 |
| 2 |
本年度報告について
本10−K表年次報告または我々の年次報告では,“我々”,“Indaptus Treateutics”,“Indaptus”,“The Company”,“Our Company”に言及する場合は,Indaptus Treeutics, Inc.(前身はIntec親会社)を参照されたい。および適切な場合には、その合併した子会社Intec Pharma Ltd.およびDecoy BiosSystems,Inc. が本年度報告に記載された他の場所で説明された馴化合併および合併後である。“Intec Parent”とはIntec Parent,Inc.,Inc.はIntec Pharma Ltd.の帰化合併後の後継者であり,“Intecイスラエル”とはIntec Pharma Ltd.,Indaptusの帰化合併前の前身であり,“Decoy”とはIndaptus合併に関連してIndaptusに買収された実体Decoy BiosSystems,Inc.を意味する。すべて“普通株式”および“株式”と言及すると、Indaptusの普通株式および株式を指す。私たちの歴史的結果は必ずしも未来のどの時期に対する私たちの期待結果を示すとは限らない。いずれの表に列挙された金額の合計と総和とのいずれの違いも 丸め込みによるものである.本年度報告で言及されている特定年度の財務·運営データとは、別途説明又は文脈に別の規定があることを除いて、当社が当該年度12月31日までの会計年度をいう。
説明的 注釈
本年度報告で用いた市場データとある業界データと予測は,市場研究データベース,我々が委託したコンサルタント調査,公開情報,政府機関の報告および業界出版物や調査からである。業界調査,出版物,我々が委託したコンサルタント調査や予測は,一般に,その中に含まれる情報 が信頼できると考えられるソースから得られていることを示している.我々は内部調査、業界予測、市場研究を含む第三者ソースからのいくつかのデータに依存しており、我々の経営陣の業界に対する理解によると、これらのデータは信頼できると考えられる。私たちの市場地位に関する陳述 は最新のデータに基づいている。本年度報告における業界データに関するいかなる誤った陳述も知らないが、我々の推定はリスクおよび不確定要因に関連し、様々な要因によって変化する可能性があり、 は第1部1 A項で議論された要因を含む。本年度報告書の“リスク要因”。
| 3 |
前向き陳述に関する警告的説明
本“年次報告”には、“1995年個人証券訴訟改革法案”が指すいくつかの前向きな陳述が含まれており、管理層が作成する可能性がある。本年度報告10-K表に含まれる歴史的事実以外のすべての陳述は前向き陳述 である.場合によっては、前向き記述は、“信じる”、“予想”、“意図”、“計画”、“可能”、“べき”、“予想”、“可能”、“可能”、“br}”、“求める”、“目標”、“会議”、“プロジェクト”、“予測”、“継続”または といった語または他の類似語の否定または変形を使用することによって識別することができる。これらの陳述は、Decoy 20の第1段階の臨床試験の時間と設計を含む私たちの候補製品開発、私たちが候補製品を開発し、それを商業化する計画、Decoy 20を含む私たちの候補製品の市場潜在力と治療潜在力を含むが、これらの陳述に限定されない私たちの商業化、マーケティング、製造能力、戦略;医療専門家が私たちの候補製品を使用したいかどうかを期待しています私たちの全体的な業務戦略と将来の運営の管理計画と目標; 私たちの研究開発活動とコスト私たちの未来の経営業績と状況;私たちの現金、現金等価物、および有価証券は私たちの持続的な活動に資金を提供するのに十分かどうか;そして私たちの業務に対する新冠肺炎疫病の期待的な影響。
本年度報告の展望的陳述は予測のみであり、主に私たちの現在の未来の事件と財務傾向に対する予想と予測に基づいており、これらの事件と財務傾向は私たちの業務、財務状況、運営結果に影響を与える可能性があると考えられる。これらの展望的陳述は、本年度報告の発表日までの状況のみを代表し、本年度報告の第1部で“要約リスク要因”と題する章で述べたものを含む、多くの既知および未知のリスク、不確実性および仮定の影響を受ける。“リスク要因”と第2部、項目7。“経営陣の財務状況と経営結果の検討と分析”および本年度報告の他の部分。
展望性陳述は固有にリスクと不確実性の影響を受けているため、その中のいくつかのリスクと不確実性は予測または定量化できず、いくつかは私たちが制御できないので、あなたは未来のイベントの予測としてこれらの前向き陳述に依存してはならない。我々の前向き表現に反映されるイベントや状況は実現できないか発生する可能性があり,実際の結果は前向き表現で予測された結果とは大きく異なる可能性がある.しかも、私たちは持続的な環境で運営している。新たなリスク要因や不確定要因が時々出現する可能性があり、経営陣がすべてのリスク要因や不確定要因を予測することは不可能である。
法律要件が適用されるbrに加えて、私たちは、 のいかなる新しい情報、未来のイベント、状況の変化、または他の理由で、本明細書に含まれる任意の前向きな陳述を公開または修正するつもりはない。本年度報告に含まれる前向き陳述を、“1933年証券法”(改正後)第27 A節又は“証券法”、“1934年証券取引法”(改正本)第21 E節又は“取引法”に含まれる前向き陳述の安全港条項に盛り込む予定である。
| 4 |
リスクファクターの概要
私たちの普通株に投資するリスクの主な要素と不確実性は
| ● | 私たちは臨床段階の会社で、運営の歴史が限られている。私たちは現在利益を上げていないし、近い将来に利益を出すことを望んでいないし、永遠に利益を上げないかもしれない。 | |
| ● | 私たちが現在キャッシュフローが不足していることを考慮して、私たちはもっと多くの資金を集める必要があるだろう。もし私たちが必要な時に受け入れ可能な条項や十分な資金を集めることができなければ、私たちは私たちの研究計画、製品開発活動、商業化努力の一部または全部を延期、制限、または廃止することを余儀なくされるかもしれない。 | |
| ● | Brの追加資本の調達は、私たちの既存の株主の株式希釈をもたらし、私たちの運営を制限するか、または私たちの技術または候補製品に対する私たちの権利を放棄することを要求するかもしれません。 | |
| ● | 臨床と臨床前開発は長く高価な過程に関連し、結果は不確定である。現在または計画中の臨床試験の開始または完了、または終了または一時停止の任意の困難または遅延は、私たちのコスト増加、遅延、または私たちの創造能力の制限、または私たちのビジネスの将来性に悪影響を及ぼす可能性があります。 | |
| ● | 我々 は引き続き巨額の研究開発費や他の運営費が発生することが予想され,利益を得ることが困難になる可能性がある。 | |
| ● | 私たち は限られた数の研究計画、候補製品、および特定の適応を展開するために限られた資源を使用するかもしれないが、 はより利益的または成功可能性の高い可能性の高い候補製品または適応を利用できない。 | |
| ● | 私たちのbr候補製品は副作用を招く可能性があり、その規制承認や商業化 を延期または阻止したり、私たちの業務、財務状況、運営結果に他の重大な悪影響を与える可能性があります。 | |
| ● | 私たちの候補製品の商業成功は医者、患者、医療支払者と医学界におけるそれらの市場受容度にかかっている。 | |
| ● | 著者らは第三者に依存して臨床前研究と臨床試験を行い、他の任務を実行した。もしこれらの第三者が契約の職責を成功的に履行し、期待期間内に規制要求を達成または遵守できない場合、私たちは規制機関の私たちの候補製品に対する承認を得ることができないか、またはそれを商業化することができない可能性があり、私たちの業務、財務状況、および運営結果は深刻な損害を受ける可能性がある。 | |
| ● | 我々brは現在,臨床開発中に第三者に依存して我々の候補製品を生産しており,予測可能な未来には引き続き第三者に依存していると予想される。このような第三者への依存は、私たちが十分な数の候補製品を得ることができないか、または許容可能なコストでこのような数を得ることができないリスクを増加させ、これは、私たちの開発 または潜在的な商業化努力を延期、阻止、または損害する可能性がある。 | |
| ● | Decy 20または任意の将来の候補製品の商業化に成功することは、許可された場合、br政府当局および健康保険会社が保険を確立する程度、十分な精算レベル、および割引された価格設定政策にある程度依存するであろう。もし私たちの製品が保証範囲を獲得したり維持したりして十分な精算を得られなかったら、これらの製品を販売する能力を制限し、私たちの収益能力を低下させるかもしれません。 | |
| ● | 最近公布された立法、将来の立法、および医療改革措置は、Decoy 20および将来の候補製品のマーケティング承認を得て、それを商業化することの難しさとコストを増加させ、設定可能な価格に影響を与える可能性があります。 |
| 5 |
| ● | もし私たちの競争相手の候補製品が私たちの候補製品よりも早く、マーケティングがより効果的で、寛容度が良く、より有利な安全状況を持っているか、あるいは私たちの候補製品よりも有効であることが証明された場合、私たちのビジネス機会は不利な影響を受ける可能性があります。 | |
| ● | 私たちは生物製品としての候補製品の承認を求めるつもりはありませんが、予想よりも早く競争に直面するかもしれません。 | |
| ● | 私たち は市場で私たちの独自の可能性を十分に保護できないかもしれない。 | |
| ● | 私たち は買収とライセンス内で私たちの候補製品に必要な権利を獲得または維持できないかもしれません。 | |
| ● | 私たちbrは様々なアメリカ連邦、州、外国の医療保健法律法規に制約されており、これはコンプライアンスコストを増加させる可能性があり、私たちがこれらの法律法規を遵守できないことは私たちの運営結果と財務状況を損なう可能性がある。 | |
| ● | 実際の または適用されるデータ保護、プライバシーおよびセキュリティ法律、法規、基準およびその他の要求 を遵守できないと考えられることは、私たちの業務、運営結果、財務状況に悪影響を及ぼす可能性があります。 | |
| ● | 新冠肺炎などの伝染病の大流行、大流行或いは爆発は私たちの業務と運営に実質的な悪影響を与える可能性がある。 | |
| ● | 情報技術システム障害,ネットワーク攻撃やネットワークセキュリティ欠陥が発生すると,我々の業務や運営が影響を受ける可能性がある. | |
| ● | 私たちの財務統制と上場企業の要求を維持し、改善することは、私たちの資源を緊張させ、経営陣の注意力を移転させ、私たちの適格な取締役会のメンバーを引き付ける能力に影響を与える可能性がある。 | |
| ● | 不利なグローバル経済状況は、私たちの業務、財務状況、または経営結果に悪影響を及ぼす可能性がある。 | |
| ● | 私たちの普通株の市場価格の変動は大きく、あなたの投資は完全な損失を受けるかもしれません。 |
| 6 |
第 部分I
第 項1.業務
概要
私たちは臨床バイオテクノロジー会社で、抗癌と抗ウイルス免疫療法を投与する斬新で特許を取得したシステムを開発しています。私たちは1世紀以上の免疫療法の進歩に基づいて進化してきました。我々の方法は、天然および獲得性免疫細胞および関連する抗腫瘍および抗ウイルス免疫反応の有効な活性化は、安全に静脈内注射することができる多標的の免疫系活性化シグナルパッケージを必要とするという仮定に基づく。我々の特許技術は,静注を減少させることを目的とした弱毒化と殺菌,非病原性,グラム陰性細菌の単一菌株 からなる。毒性であるが、先天性免疫と獲得性免疫の多くの細胞成分を起動或いは活性化する能力に大きく影響しない。この方法は、既存の5種類の異なる種類の薬物(チェックポイント治療、標的抗体治療および低用量化学療法を含む)によって観察された持続的な抗腫瘍反応相乗作用を含む、臨床前モデルにおいて広範な抗腫瘍および抗ウイルス活性を産生している。著者らの技術による腫瘍根絶は先天性と獲得性免疫記憶の活性化を示し、重要なことは、臨床前モデルにおいて腫瘍抗原 を提供或いは標的する必要がないことである。著者らはすでに著者らの主要な臨床候補薬物Decoy 20に対して現在の良好な製造規範(CGMP)の製造を行い、他の研究性新薬応用(IND)の研究を完成した。
多くの競争相手の製品とは異なり,我々の技術は特定抗原や特定抗原に対する標的に依存せず,複数の適応にまたがる広範な潜在力を提供している。我々の製品は,小分子,抗体あるいはヒト細胞による療法と比較して,半減期が短く,産生される全身曝露が少なく,非特異的自己免疫反応のリスクを潜在的に低下させている。著者らの技術はすでにリンパ腫、肝細胞癌、結腸直腸腫瘍および膵臓腫瘍においてbr単一薬物活性および/または連合治療に基づく持続的な反応 を産生し、標準的な臨床前モデルにおいて抗B型肝炎ウイルス(B型肝炎)とHIV感染の活性を示した2022年5月、アメリカ食品医薬品局は我々の研究新薬(IND)の申請を許可し、現在承認されている治療に失敗した末期固形腫瘍患者に対して第一段階の臨床試験を行った2022年12月にこの1期臨床試験を開始しました目標適応は結腸直腸癌,肝細胞(±B型肝炎),膀胱癌,子宮頸癌,膵癌に限らないが,GLOBOCAN 2020のデータによると,これらの疾患は合計世界の年間癌症例の23%を占め,年間癌死亡者数の28%以上を占めている。
歴史的には,我々は経験豊富なコンサルタントとコンサルタントチームを持ち,契約研究機関(CRO)で研究開発を行っている。私たちは34個の許可された特許と16個の出願されている特許を持っている幅広い特許の組み合わせを持っている。私たちのbrが設立されて以来、私たちは主に私たちの株式証券を公開と非公開で発行することで私たちの運営に資金を提供してきた。
背景
Interluekin−2,インターフェロン−α,最近承認された“Checkpoint”やCAR−T療法 のような承認された免疫療法は,100種類以上の異なるタイプの癌のうち約12種類の患者に対して数%から約50%の持続反応を生じる。検査点療法は以前治癒できなかった多くの患者を有効に治癒することができたが,この治療を受けた患者の15%しか反応しなかった。現有の免疫療法の主要な局限性は、それらは先天性或いは獲得性免疫系中の1つ或いは少数の肝心なステップしか活性化できないことであるが、人々は普遍的に、高効率な癌免疫療法 は先天性免疫と獲得性免疫を同時に活性化する必要があると考えられている。もし腫瘍細胞が外来あるいは損傷していると考えられれば、人体の先天性免疫系と獲得性免疫系はすべて細胞介在性によって腫瘍を破壊することができる。先天性と適応反応の活性化も免疫細胞が“危険”を感知する存在に依存する。最も有効な免疫細胞活性化危険シグナル は細菌とウイルスによって感染環境中に放出され、Toll様受容体(Toll-like,TLR)、NODとSTINTなどの免疫細胞受容体アゴニストを含む。細菌危険シグナルはTLRアゴニストを含み、病原体関連分子モード(PAMPs)と呼ばれ、先天性免疫細胞と獲得性免疫細胞を活性化することができ、抗原提示細胞を含み、先天性(NK、マクロファージ)と適応性(T細胞媒介)の腫瘍破壊を促進する。
| 7 |
最も古い癌免疫療法は細菌からの餌危険信号の提供を含む。これは細菌感染を背景に腫瘍が消退した長期観察に基づいている。熱致死細菌(“コリトキシン”)による癌患者の治療は1891年に設立され,70年使用され,著しい成功を収めた。例えば,手術不能肉腫,リンパ腫,黒色腫,癌患者432例では,5年生存率は45%と報告されている。この成功にもかかわらず、いくつかの制限は製薬産業がこの方法を放棄した。コリトキシン静注が最も効果的であることが示唆されているが,この経路で投与すると毒性が大きく,局所投与方法が制限されており,非常に異なる結果が生じる。もう一つの制限は作用機序に対する理解が不足し、生産の最適化と標準化を阻害し、臨床反応のもう一つの変異源を招くことである。この高度な可変性のため、コリーの毒素は1963年にFDAによって薬物として承認されず、放射線と化学療法に置換されたが、これらのより現代的な方法は末期癌患者に持続的な反応を産生することは少ない。科学者たちは現在コリー毒素の作用機序を理解している。グラム陰性菌はTLRアゴニスト、例えばリポ多糖(LPS)などの多種の免疫刺激危険シグナルを含む。細菌および精製または単一特異的TLRアゴニストは、エンドトキシン誘導体を含み、早期癌の予防および治療のために検証および承認されている。しかし、腫瘍内に注射された単一特異性TLRアゴニストが強力な全身性抗腫瘍免疫反応を誘導する能力が限られている可能性がある、安全かつ有効なTLRアゴニストに基づく進行癌の治療方法はなかなか見つからない。また,腫瘍内アプローチはすべての腫瘍タイプや患者には適していない。進行癌に対して有効なTLRアゴニストに基づく免疫療法は、安全な静注を可能にするために改変または減弱されたパッケージ、ポリTLRアゴニストまたは多危険シグナル製品 を発明する必要があると仮定している。行政管理。
私たちの 方法
我々の特許方法は,完全な細菌を使用することにより,天然免疫細胞および後天性免疫細胞の有効な活性化および関連する抗腫瘍免疫反応を達成することができ,これらの完全な細菌は複数のPAMPを含み,これらの細菌は減弱しているため,安全に静脈内投与することができるという仮定に基づいている。リポ多糖は毒性と治療効果の最も重要な貢献者のようであるため、著者らの特許候補製品 は単株致死の非病原性グラム陰性菌であり、処理後にbr細菌を殺すことができ、そして著しく低下するが、細胞表面のリポ多糖-エンドトキシン活性を完全に除去するわけではない。著者らの候補製品brは細菌中の他のPAMPと協同作用し、先天性と適応性免疫経路を有効に起動するために、十分な残留エンドトキシンを増強することを目的としている。この方法はすでに臨床前モデルにおいて広範な抗腫瘍反応を引き起こし、現在の5種類の異なる種類の抗腫瘍薬物との協同回帰と持続反応を含み、チェックポイント治療、標的抗体治療と低用量化学療法を含む。我々の技術により腫瘍を根絶することは先天性と獲得性免疫記憶を産生することを目的としており、重要なことは、外因性腫瘍抗原を提供する必要がないことであり、これはエンドトキシンと他のPAMPがすでに腫瘍抗原を捕獲した樹状細胞 を活性化できるためである可能性がある。
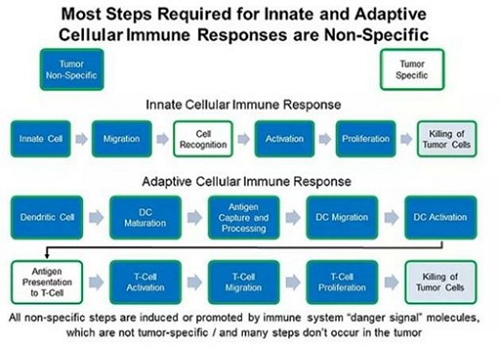
すべての免疫細胞は腫瘍とウイルスの駆除に関与することができる。以下に示すように,現在の治療法は2つの経路のうちの1つまたは一部のみを活性化し,一部の患者のみを治癒している。
| 8 |
しかし、著者らの技術は現有の治療法と協同作用し、天然免疫細胞と獲得性免疫細胞を活性化し、高効率な抗腫瘍免疫反応を誘導し、広範な安全限界を有することを目的としている。適応性抗腫瘍免疫反応と免疫記憶を誘導する著者らの技術は外因性腫瘍抗原を必要としない。
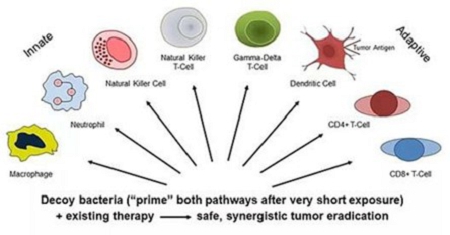
先天性免疫反応と獲得性免疫反応は腫瘍を異体或いは非自己体と識別することを要求する。しかしながら、免疫細胞の遊走および活性化に必要なステップの多くは、腫瘍とは無関係であるか、または腫瘍非特異的である。すべての先天性および適応性の不特定のステップは、私たちの細菌で発見された分子のような免疫系の“危険シグナル”分子によって誘導または推進される。細菌が産生する危険シグナルも腫瘍抗原の処理や認識を増強し,腫瘍抗原はしばしば存在するが,免疫系 には見られない。
結果は…
我々は、非病原性、グラム陰性細菌を減衰および死滅させるための特許治療方法(および関連特許組成物)(発行または許可された34特許)を開発した。臨床前モデルでは,Indaptusで処理された細菌は未処理細菌よりも全身毒性が小さいが,先天性と獲得性免疫反応を活性化することができる。我々の細菌は体内でより低い発熱性とより高い最大耐性を示すにもかかわらず,体外では,我々の細菌はマウスやヒト免疫細胞が多くのサイトカインやケモカインを分泌することを誘導し,そのレベルは未処理細菌に相当する。私たちの細菌はヒト免疫細胞と協働して体外でヒト腫瘍細胞を殺すこともできます
| 9 |
著者らはすでに樹立した非ホジキンリンパ腫及び結腸直腸癌、肝細胞癌と膵臓癌の臨床前同遺伝子移植モデルとヒト腫瘍異種移植モデルにおいて顕著な単剤抗腫瘍活性及び/或いは連合治療を介した回帰を観察し、そして持続的な反応を持っている。臨床前モデルにおいて、著者らの細菌は5種類の異なる種類の承認された薬物のそれぞれと協同作用があり、チェックポイント治療、標的抗体、低用量化学療法、非ステロイド性抗炎症薬(NSAID)と腫瘍消退を誘導するサイトカインを含み、br}は異なるタイプの癌に対して極めて大きな柔軟性を提供した。著者らの技術は先天性(NK細胞)と適応性(CD 4+とCD 8+T細胞)機序を活性化することによって腫瘍を根絶することを目的とし、先天性と適応性免疫記憶を同時に産生することを目標としている。われわれの臨床前研究では,腫瘍はわれわれの細菌の無毒投与量で根絶され,非常に広い(10から33倍) 治療指数を有している。治療した腫瘍の遺伝子発現分析と血漿サイトカイン分析を通じて、著者らはまた顕著な作用情報機序を獲得し、著者らの組み合わせ技術は“冷”腫瘍を“熱”腫瘍に転換し、そして先天性と適応性遺伝子、細胞と経路を誘導、活性化或いは募集する可能性があることを表明した。免疫細胞枯渇前の研究により、天然免疫細胞(NK)と獲得性免疫細胞(CD 4 TとCD 8 T)はすべて腫瘍の除去に参与した。著者らはまた標準的な臨床前モデルにおいて顕著な単剤抗慢性B型肝炎ウイルス(B型肝炎ウイルス)とヒト免疫不全ウイルス(HIV)の活性を示した。
我々 は我々の主要候補製品Decoy 20とcGMP製造と安定性研究に成功した。また,INDを有効にする多用量毒理学的研究は完了しており,サイトカイン放出症候群に関連する因子の持続的誘導は生じていない。
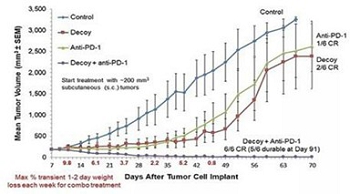
上のグラフは,われわれの細菌が抗PD−1チェックポイント治療と協同作用し,樹立したマウス肝細胞癌(HCC)腫瘍を消退させることを示している。すべてのマウス(全群)が低用量の非ステロイド抗炎症薬(NSAID/インドメタシン)を受け,併用設定における回帰数を増加させた。多くの回帰は持続的であり,91日目の終了 と143日目に終了した反復試験(CR=完全緩和または完全回帰)では,5/6組合せ回帰は安定していた。 反復試験は33倍のIndaptus濃度範囲で各群の5/6または6/6の持続的回帰が生じ,また 安全問題がなく,非常に広い治療指数を示した。マウス非ホジキンリンパ腫モデルでは,われわれの細菌を低用量化学療法と組み合わせることにより,類似した腫瘍根絶結果も得られた。われわれの技術により確立された非ホジキンリンパ腫brヒト腫瘍異種移植を根絶し,天然免疫系の活性化によりも観察された。Toll様受容体(TLR)アゴニストの全身応用による抗腫瘍免疫治療の開発と臨床前治療効果の特徴[要約.要約]それは.参照:第4回CRI-CIMT-EATI-AACR国際癌免疫治療会議論文集:科学を生存に転化する2018年9月30日から10月3日まで、ニューヨーク。フィラデルフィア(ペンシルベニア州):AACR≡癌免疫Res2019≡7(2 Suppl):要約 nr B 178。
| 10 |
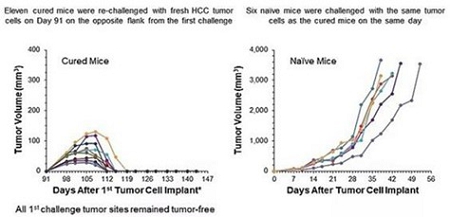
上のグラフは私たちの技術と抗PD-1が腫瘍を併用して免疫記憶を産生していることを示しています上図に示した併用治療により,11匹のマウスで樹立した腫瘍は消退し,新鮮な肝癌腫瘍細胞で再度マウスを攻撃し,さらなる治療は行わなかった。すべての新しい腫瘍は拒絶された。非ホジキンリンパ腫モデルでは,われわれの細菌を低用量化学療法と組み合わせても類似した結果が得られた。
2022年5月、FDAは著者らのIND申請を許可し、現在承認された治療に失敗した末期固形腫瘍患者に対して第一段階の臨床試験を行い、2022年12月、著者らはこの第一段階の臨床試験を開始し、これは開放ラベル、多中心、用量増加 と拡張単腕(単一療法)研究であり、2つの部分に分けて行った。第一段階研究Brは単剤のアップグレードから始まり、その後拡張部分であり、週に1回連続投与する予定である。この研究は承認された治療方案を使い切った末期/転移性固形腫瘍患者を募集しているこの研究の目的はDecoy 20の安全性と耐性を評価し、最大耐性量と推奨される第二段階用量を決定し、Decoy 20の薬物動態学(PK)、薬効学と臨床活性を評価することであるこの研究の主な終点は不良イベントと緊急不良イベントの治療の発生率、相関性と重症度であり、各キュー中の用量制限に基づく毒性不良イベントの被験者数を決定する。副次的終点は治療前後の抗薬物抗体と中和抗体の発生率、Decoy 20 PKパラメータの経時的変化、測定可能な疾患を有する被験者の客観的応答率と反応持続時間を含む。
業務 戦略
著者らの使命は切除不能或いは転移性固形腫瘍とリンパ腫患者に対する根治性癌免疫治療を強化と拡大することであり、これらの腫瘍はすべての癌死亡の約90%を占める。
私たちのビジネス戦略には:
| ● | 当社が計画している製品開発·商業化作業を支援する者、公共報告会社としての報告やその他のコンプライアンス義務を支援する者を含む、br運営、財務·管理情報システムおよび人員を増加させる | |
| ● | Decoy 20≡まで進めた1期臨床試験の拡張部分 | |
| ● | 私たちの細菌製品プラットフォームを拡張して他のタイプの癌や他の感染症に向けて | |
| ● | 私たちの知的財産権の維持、拡大、保護の組み合わせと | |
| ● | 臨床試験を成功させた任意の候補製品のために監督管理の承認を求める。 |
| 11 |
競争優位
著者らの細菌は多種の成分を含み、先天性免疫と獲得性免疫の多くの細胞成分を誘発或いは活性化することができるが、すでに1種の特許技術を通じて弱まり、免疫系を過度に刺激する可能性とそれによる不良自己免疫反応を減少させる。われわれの細菌もすぐに肝臓や脾によって除去される可能性があり,他のタイプの免疫療法と比較して非特異的自己免疫副作用のリスクをさらに低下させる可能性がある。Indaptusに短時間接触すれば単独で機能し,他製品を増強する“プライマー”として十分であると信じられている。また、私たちの製品は費用効果の高いプロセスで製造することができ、発達と発展中の地理的地域でより速い患者訪問を提供することが可能である。
政府規制
他にも、FDAと州と地方司法管轄区および他の国/地域の同様の規制機関は、臨床開発、製造、マーケティング、流通に参加する会社(例えば、私たちが開発している薬物)に対して大量かつ負担の重い要求を提出した。これらの機関と他の連邦、州と地方実体は私たちの候補製品の研究開発、テスト、製造、品質管理、安全、有効性、ラベル、貯蔵、記録保存、承認、広告と販売促進、流通、承認後の監視と報告、サンプリングと輸出と輸入などの面で監督管理を行う。
アメリカの薬品と生物製品の規制
アメリカでは、FDAは“連邦食品、薬物と化粧品法”(FDCA)及びその実施条例に基づいて薬品を監督し、FDCAと公衆衛生サービス法(PHSA)及びその実施条例に基づいて生物製品を監督する。以前承認された薬物の新しい使用を含む未承認新薬または剤形のいずれも、米国で発売されるためにFDA承認が必要である。医薬品と生物製品もまた他の連邦、州、そして地方法規によって制限されている。FDAが候補製品 が米国で発売される前に必要とされるプロセスは、一般に以下を含む:
| ● | 広範な臨床前実験室テスト、動物研究と調合研究を完成し、“良好な実験室規範”(GLP)とその他の適用法規に従って行った | |
| ● | ヒト臨床研究が開始される前に発効し、毎年更新されなければならないINDをFDAに提出する | |
| ● | 各臨床研究を開始する前に、各臨床サイトを代表する独立機関審査委員会(IRB)または倫理委員会によって承認される | |
| ● | 良好な臨床実践(GCP)の十分かつ良好に制御された人体臨床研究の業績に符合し、安全性と有効性の要求を確定し、あるいは生物製品について、各提案の適応に対して候補製品の安全性、純度と効力を確定する | |
| ● | すべての重要な臨床研究を完了した後、新薬申請(NDA)または生物製品許可証申請(BLA)を準備し、FDAに提出する | |
| ● | FDAは、セキュリティプロトコルまたはBLAを受信してから60日以内に審査申請を提出する決定を行う | |
| ● | 適切かつ適用可能な場合には、FDA諮問委員会によって製品申請が審査される可能性がある | |
| ● | CGMPに適合するかどうかを評価して、施設、方法、および薬物の特性、強度、品質および純度を維持するのに十分な制御を確保し、GCPに適合する状況を評価するために、提案された完成品薬物を製造する製造施設に対するFDAの承認前検査を完了することが好ましい | |
| ● | FDA は、米国で任意の商業マーケティングまたは販売を行う前に、NDAまたはBLAの審査および承認を行う。 |
| 12 |
開発する候補製品を決定した後、それは臨床前テスト段階に入る。臨床前試験は製品の化学、毒性と調合に対する実験室評価、及び動物研究を含む。INDスポンサーは,臨床前テストの結果を生産情報や分析データとともにINDの一部としてFDAに提出しなければならない。INDはFDAが研究薬物製品の使用をヒトに許可する要求である。INDはまた、試験が治療効果評価を含む場合、臨床試験の目標、安全性を監視するためのパラメータ、および評価される有効性基準を詳細に説明するレジメンを含むであろう。IND提出後も,いくつかの臨床前試験が継続される可能性がある。INDはFDAが30日以内に臨床試験を保留しない限り,FDAが受領してから30日後に自動的に発効する。この場合,INDスポンサーやFDAは臨床試験開始前に未解決の問題を解決しなければならない。FDAはまた、実施されているか、または提案されている臨床試験またはFDAの特定の要件に適合していない安全を懸念し、FDAがスポンサーに一時停止を解除したことを通知する前に、試験が開始または継続されない可能性があるため、臨床試験の前または間の任意の時間に臨床休止を実施することができる。
すべての臨床試験はGCPに従って1人以上の合格した研究者の監督の下で行われなければならず、すべての研究対象に任意の臨床試験に参加することを書面で提供することを要求するインフォームドコンセントを含む。臨床試験は,試験目標,投与手順,被験者選択と排除基準,評価する安全性と有効性基準を詳細に説明するプロトコルで行わなければならない。各レジメンはINDの一部としてFDAに提出されなければならず、製品開発中に行われる各後続の臨床試験および後続の任意の レジメン修正については、既存のINDに個別に を提出しなければならない。INDが活動状態にある場合には、少なくとも毎年、前回の進捗報告以来行われた臨床試験と非臨床研究結果をまとめた進捗報告、その他の情報をFDAに提出しなければならず、深刻かつ意外な疑わしい有害事象を含むIND安全性報告書をFDAと調査者に提出しなければならず、他の研究結果 は同じ或いは類似の薬物に暴露されたヒトに重大なリスクがあることを示し、動物試験或いは体外試験結果は人類が重大なリスクbrに直面していることを示し、br方案或いは研究者マニュアルに列挙された結果と比較して、任意の臨床上重要な深刻な疑わしい不良反応の発生率は増加する。
また、臨床試験に参加する各機関の独立したIRBは、当該機関の臨床試験が開始される前に各案を審査·承認しなければならず、試験に関する情報および同意書を承認しなければならず、これらの情報および同意書は、各試験被験者またはその法律代表に提供されなければならず、研究が完了するまで監視され、そうでなければIRB規定を遵守しなければならない。FDAまたはスポンサーは、研究対象または患者が受け入れられない健康リスクに曝露されていることを発見することを含む、様々な理由で臨床試験を随時一時停止することができる。同様に,臨床試験がIRBの要求に沿って行われていない場合,あるいは薬物が患者の意外な重篤な傷害に関連している場合,IRBはその機関の臨床試験の承認を一時停止または終了することができる。さらに、いくつかの臨床試験は、スポンサーによって組織された独立した合格専門家グループによって監督され、このグループは、データ安全監視委員会または委員会と呼ばれる。その規約により,このグループは実験のあるデータへのアクセスにより,実験 が指定されたチェックポイントで行えるかどうかを決定することができる.また,行われている臨床研究や臨床研究結果を臨床試験サイトを含めて公的登録機関に報告することも求められている。
薬物の臨床研究は一般的に3段階に分けられる。これらの段階は通常順序で行われるが、それらは重複したり統合されたりする可能性がある。
| ● | 段階br}1.候補製品は、健康なヒト対象または標的疾患または状態を有する患者に最初に導入される。これらのbr研究は、研究製品の人体内での安全性、用量耐性、吸収、新陳代謝と分布、および用量増加に関連する副作用をテストし、可能な場合に有効性の早期証拠を得ることを目的としている。 | |
| ● | 段階2.候補製品は、予備治療効果、最適用量および用量計画を評価し、可能な不良副作用および安全リスクを決定するために、特定の疾患または状態の限られた患者集団のために使用される。より規模が大きく,よりコストの高い3期臨床試験を開始する前に,複数の2期臨床試験を行って情報を得ることが可能である。 | |
| ● | 段階 3.候補製品を拡大した患者群に応用し、更に投与量を評価し、統計学的意義のある臨床治療効果の証拠を提供し、そして更に安全性をテストし、通常複数の地理的に分散した臨床試験地点で行われる。 これらの臨床試験は研究製品の全体的なリスク/収益比率を確定し、そして製品の審査に適切な 基礎を提供することを目的としている。 |
| 13 |
承認後 実験は,第4段階研究と呼ばれる場合があり,最初の市場承認後に行うことができる.これらの試験は,期待される治療適応患者の治療から追加的なbr経験を得るために用いられている。場合によっては,FDAはNDAを承認する条件として4期臨床試験の性能を強制的に要求する可能性がある。
候補製品の開発過程において、スポンサーは特定の時間にFDAと会う機会がある。これらの要件は、INDの提出前、第2段階の終了時、およびセキュリティプロトコルまたはBLAの提出前である可能性がある。他の時間に会議を開催することを要求することができます。 これらの会議はスポンサーに機会を提供し、スポンサーにこれまで収集したデータに関する情報を共有させ、FDAに提案を提供し、スポンサーとFDAのために次の段階の開発について合意することができます。臨床試験と同時に、会社は通常追加の動物研究を完成し、薬物化学と物理特性に関するより多くの情報を開発し、cGMPに基づいて最終的に商業大量生産製品の技術を決定しなければならない。製造過程は高品質の候補製品ロットを継続的に生産できる必要があり,また,メーカー は最終薬物の特性,強度,品質,純度を試験するための方法を開発しなければならない。また,適切な包装を選択·テストし,候補製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
秘密保持プロトコル とBLA審査の流れ
はすべての適用された法規要件に基づいて必要なすべてのテストを成功させると仮定し,製品開発,非臨床研究,臨床試験の結果をNDAまたはBLAの一部としてFDAに提出し,この製品を の1つまたは複数の適応に使用することの承認を要求する。セキュリティプロトコルやBLAは,否定的または不明確な結果や積極的な発見,製品のCMCやアドバイスのラベルに関する詳細な情報など,関連する臨床前研究や臨床試験から得られたすべての関連データを含まなければならない。データは、製品の安全性および有効性を試験するための会社によって支援された臨床研究から来てもよく、研究者によって開始され、賛助された研究を含む多くの代替源から来てもよい。セキュリティプロトコルまたはBLAを提出するには、免除または免除が適用されない限り、FDAに相当な申請使用料を支払う必要がある。
さらに、小児科研究公平法またはPREAによれば、NDAまたはBLAまたはNDAまたはBLAの補充は、すべての関連する小児科亜群において適応が主張される候補生物製品の安全性および有効性を評価し、安全に有効な各小児科亜群に対するこの製品の用量および投与をサポートするためのデータを含まなければならない。“食品および薬物管理安全と革新法案”は、医薬品または生物製品のマーケティング申請を提出する計画のスポンサーが、第2段階会議終了後60日以内にまたはスポンサーとFDAとの間で合意しなければならない場合に、初歩的な小児科研究計画を提出しなければならないことを要求している。規制が別途要求されない限り、PREAは孤児の称号が付与されたいかなる薬物または生物学的製品にも適用されない。
申請が提出されてから60日以内に、FDAは提出されたBLAまたはNDAを審査し、申請がほぼ完了したかどうかを確認し、機関がその提出を受け入れる。FDAは、それが不完全であるか、または提出時に適切に審査できないと考えられる任意のNDAまたはBLAの提出を拒否することができ、より多くの情報の提供を要求する可能性がある。この場合,秘密プロトコルやBLAおよび 付加情報を新たに提出しなければならない.NDAまたはBLAが届出を受けると、FDAの目標は、届出日後 10ヶ月以内に基準申請を審査すること、または、申請が優先審査資格を満たす場合、FDAが届出申請 を受けてから6ヶ月以内に審査することである。標準審査および優先審査では、FDAがより多くの情報を提供すること、または明確にすることを要求することによって、審査プロセスを延長することもできる。FDAがセキュリティプロトコルを検討する目的の1つは、候補製品がその予期される用途に対して安全かつ有効であるかどうかを決定することと、その製造が製品の特性、強度、品質、および純度を確保するのに十分であるかどうかを決定することである。FDAは、候補製品が安全で純粋かつ有効であるかどうかを決定するためにBLAを審査し、製品の製造、加工、包装、または保有施設が、製品の持続的な安全、純度および効力を保証するための基準に適合しているかどうかを決定することである。FDAは諮問委員会を招集し、審査申請について臨床的見解を提供することができます。FDAは諮問委員会の提案に制約されていませんが、決定する際にこれらの提案をよく考慮しています。
| 14 |
FDAは、セキュリティプロトコルまたはBLAを承認する前に、通常、その製品を生産する工場を検査する。FDAは、製造プロセスおよび施設がcGMP要件 に適合していることを決定し、要求された仕様で製品が持続的に生産されることを確実にするのに十分でない限り、申請を承認しない。さらに、NDAまたはBLAを承認する前に、FDAは、通常、GCPに適合することを確実にするために、1つまたは複数の臨床場所を検査する。
FDAがNDAまたはBLAを評価し、研究製品および/またはその医薬物質を生産する製造施設を検査した後、FDAは承認書または完全な返信、またはCRLを発行する可能性がある。承認書は、製品の商業マーケティングを許可し、特定の適応に関する具体的な処方情報を提供する。CRLは、NDAまたはBLAでFDAによって発見されたすべての欠陥を記述するが、FDAが申請をサポートするデータが承認をサポートするのに不十分であると判断した場合、FDAは、最初に必要な検査、テスト提出された製品バッチを行うことなく、および/または提案されたラベルを審査することなくCRLを発行することができる。CRLを発行する際に、FDAは、より多くの情報の提供または明確化を要求することを含む、NDAまたはBLAを承認条件 に置くことを申請者に提案することができる。適用される規制基準が満たされていない場合、追加の試験または情報が要求され、および/または発売後の試験およびbrが製品の安全性または有効性を監視するために監視されていない場合、FDAは、NDAまたはBLAの承認を延期または拒否することができる。
製品が規制部門の承認を受けた場合、そのような承認は、特定の適応に対して承認され、そのような製品が市場の指定用途に使用可能であることが制限される可能性がある。例えば、FDAは、製品の利点がそのリスクよりも大きいことを確実にするために、NDAまたはBLAおよびリスク評価および緩和戦略、またはREMSを承認する可能性がある。RMSは、製品に関連する既知または潜在的な深刻なリスクを管理し、そのような薬剤の安全な使用を管理することによって、患者がこれらの薬剤を継続的に使用することを可能にするための安全戦略であり、制限された分配方法、患者登録および他のリスク最小化ツールのような薬物ガイドライン、医師のコミュニケーション計画、または安全使用を確保する要素を含む可能性がある。FDAはまた,提案されたラベルの変更や適切な制御や仕様の作成などを条件に承認を行うことも可能である.承認されると、発売前と発売後の要求を守らなかったり、製品発売後に問題が発生したりすれば、FDAは製品承認を撤回する可能性がある。FDAは、商品化後の製品の安全性と有効性 をさらに評価し、監視するために、1つまたは複数の第4段階上場後の研究とモニタリングを要求することができ、これらの発売後の研究の結果に基づいて製品のさらなる販売を制限する可能性がある。
開発と審査計画を加速する
FDAは合格した候補製品に一連の迅速な開発と審査計画を提供した。たとえば,Fast Track計画 は,特定の基準に適合した新製品の審査の流れを加速または促進することを目的としている.具体的には、候補製品brが、深刻または生命に危険な疾患または状態を治療し、その疾患または状態が満たされていない医療需要を満たす可能性を示すことが意図されている場合、迅速なチャネル認証を取得する資格がある。高速チャネルは,候補製品と研究中の特定の適応に適した組合せを指定する.Fast Track候補製品のスポンサーは,製品開発期間中に審査チームとより頻繁なインタラクションを行う機会があり,セキュリティプロトコルやBLAを提出すると,申請は優先審査 を得る資格がある可能性がある.高速チャネル候補製品もスクロール審査を行う資格がある可能性があり、完全な出願を提出する前に、FDAは、NDAまたはBLA部分を提出するスケジュール を提供した場合、FDAはNDAまたはBLAの部分を受け入れることに同意し、スケジュール は許容可能であると決定し、スポンサーはNDAまたはBLAの第1の部分を提出する際に必要な使用料を支払うことができる。
深刻または生命に危険な疾患または状態を治療するための候補製品も、その開発および審査を加速するために、画期的な治療指定を得る資格がある。予備臨床証拠が、候補製品が単独でまたは1つまたは複数の他の医薬または生物学的製品と組み合わせて使用されることを示す場合、候補製品は、1つまたは複数の臨床的に重要な終点において、例えば、臨床開発早期に観察される実質的な治療効果を示す可能性がある場合、候補製品は、画期的な治療指定を得ることができる。この指定には、Fast Track計画のすべての機能と、第1段階で開始されたより密集したFDA相互作用および指導、および候補製品開発および審査を加速させる組織約束が含まれており、高度管理者の参加を含む。
| 15 |
迅速なチャネル指定および/または画期的な治療指定を有する候補製品を含むFDA承認に提出された医薬または生物学的上場申請は、優先審査および加速承認のようなFDAの審査および承認プロセスを加速することを意図した他のタイプの計画の資格に適合する可能性がある。候補製品が深刻な疾患または状態の治療、診断または予防において顕著な改善を提供する潜在力を有する場合、優先審査を受ける資格がある。新しい分子実体および元のBLASについて、優先審査指定は、FDAの目標が、60日の届出日の6ヶ月以内にマーケティング申請に行動することであることを意味する(標準審査は10ヶ月)。
さらに、候補製品が深刻または生命に危害を及ぼす疾患または状態の治療における安全性と有効性を決定する際に、候補製品が臨床利益を合理的に予測できる代替終点に影響を与えるか、または不可逆的な発病率または死亡率よりも早く測定された臨床終点に影響を与えるかどうかを決定する場合、すなわち病状の重症度、希少性または流行度、および代替治療があるかどうかを考慮して、不可逆的な発病率または死亡率または他の臨床利益への影響を合理的に予測する可能性があれば、加速承認を得ることができる。承認を加速する条件として、FDAは通常スポンサーに十分かつ良好な制御を行う上場後の臨床研究を要求し、不可逆的な発病率或いは死亡率或いは他の臨床利益に対する期待影響を検証と記述する。スポンサーが必要な上場後研究を適時に行うことができなかった場合、あるいはこのような研究が予測の臨床的利益を検証できなかった場合、加速承認を得た製品は迅速な脱退手続きの制約を受ける可能性がある。また,FDAが現在承認宣伝材料の承認加速を要求している条件の1つは,事前承認であり,製品の商業発表時間に悪影響を与える可能性がある.
迅速な追跡指定、画期的な治療指定、優先審査、承認の加速は承認基準 を変更することはないが、開発や承認過程を加速する可能性がある。製品がそのうちの1つまたは複数の計画の資格に適合していても、FDAは、後で がその製品が資格条件を満たしていないことを決定したり、FDAの審査または承認を決定する期間が短縮されないことができる。私たちは私たちの候補製品のためのいくつかの機会を適切に探索するかもしれない。
| 16 |
承認後に を要求する
FDAによって生産または流通が許可された任意の製品は、記録保存、不良経験報告、定期報告、製品サンプリングおよび流通、ならびに製品広告および販売促進に関連する要件を含むFDAによって普遍的かつ持続的に規制される。承認後、新しいbr適応や他のラベル宣言を追加するなど、承認された製品の多くの変更は、FDAの審査と承認を事前に受ける必要があります。継続的な使用料要求もあり,この要求に応じて,FDAは承認されたセキュリティプロトコルやBLAで決定された各製品の年間計画費用を評価する.医薬品および生物メーカーおよびその下請け業者は、FDAとある州機関にその機関を登録し、FDAとある州機関の定期的な抜き打ち検査を受けてcGMPの適合性を理解しなければならず、これは私たちと私たちの第三者メーカーにいくつかの手続きとファイル要求を加えている。製造プロセスの変更は厳しく規制されており,変更の重要性により,FDAが事前に承認して実施する必要がある可能性がある。FDAはまた、cGMPとのいかなる偏差も調査と是正を要求し、私たちと私たちが使用を決定する可能性のある任意の第三者製造業者に報告を要求している。そのため、メーカーは生産と品質管理の分野で時間、お金、精力をかけ続けて、cGMPや他の方面に合った法規遵守性を維持しなければならない。
規制要求や基準が守られていない場合、あるいは製品発売後に問題が発生した場合、FDAは承認を撤回する可能性がある。予想されていない重症度または頻度の有害事象、または生産プロセス、または法規要件を遵守できないことを含む製品に以前に未知の問題が存在することが後に発見された場合、新しいセキュリティ情報を追加するために承認されたラベルの改訂をもたらす可能性があり、新しい安全リスクを評価するために発売後研究または臨床研究を実施すること、または流通制限またはREMS計画下の他の制限を実施することにつながる可能性がある。他の他の潜在的な結果には
| ● | 製品の販売または製造を制限し、市場から完全に撤退するか、または製品のリコールを制限する | |
| ● | 承認後の臨床研究には罰金、警告状、放置を科す | |
| ● | FDAは、承認されるべき出願または承認された申請の補充を拒否するか、または既存製品の承認を一時停止または撤回する | |
| ● | 製品 は、製品の差し押さえまたは差し押さえ、またはFDAは、製品の輸入または輸出の許可を拒否する | |
| ● | Br法令に同意し、会社の誠実な合意に同意し、連邦医療計画の資格を取り消したり、除外したりする | |
| ● | 宣伝材料とラベルを強制的に修正し、訂正情報を発表する | |
| ● | 安全警報、親愛なるヘルスケア提供者からの手紙、プレスリリース、および製品に関する警告または他のセキュリティ情報を含む他の通信;または | |
| ● | 禁止されたり、民事または刑事処罰が適用される。 |
FDAは薬品と生物製品のマーケティング、ラベル、広告と販売促進を厳格に監督する。1つの会社は、安全性および有効性、純度および効力に関連する、FDAによって承認され、承認されたラベルの規定に適合する声明しか提出できない。FDAと他の機関は非ラベル用途の普及を禁止する法律法規を積極的に施行している。これらの要求を遵守できなかったことは、負の宣伝、警告状、改正広告、および潜在的な民事と刑事罰を招く可能性がある。医師は製品ラベルに説明されておらず、私たちがテストし、FDAによって承認された用途とは異なる合法的に利用可能な製品を処方するかもしれない。このような非ラベル使用は医学専門科でよく見られる。br先生はこのラベル外使用は多くの患者の異なる場合の最適な治療法であると考えているかもしれない。FDAは医者が治療を選択する時の行動を規範化しない。しかし、FDAは製品ラベルの外使用問題に対する製造業者のコミュニケーションを制限した。
| 17 |
薬品製品マーケティング排他性
市場 FDCAによって許可された排他的条項は、いくつかのマーケティング申請の提出または承認を延期する可能性がある。例えば、FDCAは、新しい化学物質のNDA承認を得た最初の出願人に、5年間の米国内の非特許データ独占期間 を提供する。FDAが以前に同じ活性部分を含む他の新薬を承認していない場合、薬物は新しい化学物質である。活性部分は薬物物質の作用を担う分子またはイオンである。排他期間内に、FDAは、505(B)(2)、 または505(B)(2)条に従って提出された別の薬剤が、同じ活性部分に基づいて提出された簡略化された新薬出願またはANDAまたはNDAの審査のための別の会社を承認しないか、または受け入れない可能性があり、その薬剤が元の革新的薬剤と同じ適応であるか、または他の適応のために使用されるかにかかわらず、出願人が参照承認に必要なすべてのデータの合法的な権利 を所有していない場合、または参照承認に必要なすべてのデータを有する場合。しかしながら、出願がイノベーター秘密協定保持者がFDAに記載された特許のうちの1つを含む特許が無効または未侵害証明である場合、4年後に出願することができる。
出願人が行っているか、または賛助する新しい臨床研究(バイオアベイラビリティ研究を除く)が、FDAが既存の薬剤の新しい適応、用量または強度のような承認申請に重要であると考えられている場合、FDCAはまた、NDAに3年間の市場排他性、または既存のNDAの補充を提供することができる。この3年間の専門権は、この薬剤が新しい臨床研究に基づいて承認されたbr改正のみをカバーし、FDAがANDAまたは505(B)(2)NDAを承認することを禁止せず、元の適応または使用条件の有効成分を含む薬剤のためのものである。5年と3年間の独占許可は、完全な秘密協定の提出または承認を遅延させることはない。しかしながら、完全なセキュリティプロトコルを提出する申請者は、安全性および有効性を証明するために必要な任意の臨床前研究および十分かつ制御された臨床試験の参照権を行うか、または得ることが要求されるであろう。
小児科専門権は米国で提供される別のタイプのマーケティング専門権である。スポンサーがFDAの書面要請に応じて児童に臨床試験を行う場合、小児科専門権は別の独占期間内に6ヶ月の市場独占権を追加することを規定する。書面出願の発表はスポンサーに述べた臨床試験を要求しない。さらに、上述したように、孤児薬物独占権は7年間の市場独占期間を提供する可能性があるが、場合によっては除外される。
生体模倣薬と参考製品の排他性
“2009年生物製品価格競争と革新法”(BPCIAと略称する)は、FDAによって承認された参考生物製品と高度に類似しているか、またはそれと高度に類似しているか、または交換可能な生物製品に簡略化された承認方法を開拓した。FDAはすでにいくつかの指導文書を発表し,生体模倣薬の審査·承認方法について概説した。生物類似性、即ち生物製品と参考製品は安全性、純度と効力の面で臨床上意義のある差異が存在しないことが要求され、 は通常分析研究、動物研究と1つ或いは複数の臨床研究によって証明される。互換性は、製品brが基準製品生物と類似していることを必要とし、この製品は、任意の所与の患者において、参照製品と同じ臨床結果を生成することが期待できることを証明しなければならず、複数回投与された製品の場合、生物学的製剤および参照生物製剤は、安全リスクを増加させることなく、または参照生物学的製剤の独占的使用と比較して治療効果のリスクを低下させることなく、以前の投与後に交互にまたは切り替えることができることを証明しなければならない。FDAによって承認された参照生物製品と類似しているか、または交換可能であることが証明された製品は、FDA以前に承認された参照製品の安全性および有効性の決定にある程度依存する可能性があり、これは、承認された製品を市場に発売するのに要するコストおよび時間を減少させる可能性がある。
BPCIAによると,生物類似製品の申請は,参考製品が初めてFDA許可を得た4年後にFDAに提出されなければならない。また,FDAによる生物類似製品の承認は,参考製品が初めて許可された日から12年後に発効する可能性がある。この12年間の独占期間内に、FDAが競合製品の完全なBLAを承認し、競合製品が、その製品の安全性、純度、および効力を証明するために、出願人自身の臨床前データと、その製品の安全性、純度および効力を証明するために、十分かつ良好な臨床試験からのデータとを含む場合、別の会社は、参照製品の競合バージョンを販売している可能性がある。BPCIAはまた、交換可能な製品として承認された生物模倣薬のためのいくつかの排他的期限を設けている。この時点で,FDAに“交換可能”とされた製品が本当に国家薬剤法に管轄されている薬局に取って代わられるかどうかは不明である。
生物製品は米国でも小児科市場の排他性を得ることができる。小児科専門権が付与された場合、既存の専門期間と特許条項に6ヶ月間追加される。この6カ月間の専門権は,他の専有性保護または特許期間終了時から,FDAが発表したこのような研究の書面請求に基づいて,小児科研究を自発的に完成させた上で付与することができる。
| 18 |
その他 医療保険法
製薬会社は連邦政府およびそれらが業務を展開する州と外国司法管轄区当局の追加の医療法規と法執行の制約を受け、私たちがbrによる研究および販売、マーケティング、流通を制限する可能性があり、私たちがマーケティングの許可を得た任意の製品の財務手配と関係を制約する可能性がある。このような法律には、連邦および州のリベート、詐欺および乱用、虚偽声明、データプライバシーおよびセキュリティ、ならびに医師および他のヘルスケア提供者の透明性に関する法律法規が含まれているが、これらに限定されない。もし私たちの業務がこのような法律または任意の他の適用可能な政府法規に違反していることが発見された場合、彼らは行政、民事および刑事罰、損害賠償、罰金、返還、削減または再構成業務、誠実な監督と報告義務、連邦および州医療計画から除外され、監禁されることを含む罰を受ける可能性がある。
保証と精算
任意の製品の販売は、連邦、州と外国政府医療保健計画、商業保険とホスト医療機関、および第三者支払人のこの製品に対する清算レベルなど、第三者支払者のこの製品に対する保証範囲にある程度依存する。提供される補償範囲と金額に関する決定は、個々の計画に基づいて行われる。保証範囲の確定過程は通常時間がかかり、コストの高い過程であり、それぞれの支払人に著者らの製品を使用する科学と臨床支持を提供する必要があるが、保証範囲と十分な精算を保証することはできない。このような第三者決済者たちはますます医療製品、薬品、サービスの精算を減らすようになっている。また、米国政府、州立法機関、外国政府は価格制御、カバー範囲と精算の制限、非特許製品の代替要求を含むコスト制御計画を継続して実施している。価格制御とコスト制御措置、および既存の制御と措置を持つ司法管轄区域でより厳しい政策をとることで、任意の製品の販売をさらに制限する可能性がある。いかなる製品の第三者精算や第三者支払者が製品を保証しないかを決定することは、医師使用量や患者の製品に対する需要を減少させる可能性があり、販売に実質的な悪影響を与える可能性がある。
医療改革
2010年3月、“患者保護·平価医療法案”は、“医療·教育和解法案”の改正後、総称してACAと呼ばれる“患者保護·平価医療法案”が公布され、医療保健が政府や民間保険会社が資金を提供する方式を大きく変え、製薬業に大きな影響を与えた。ACAには、連邦医療計画の登録、精算調整、詐欺や乱用の法律の改正など、多くの条項が含まれている。例えば、ACA:
| ● | ブランド医薬品メーカーが支払うべき医療補助リベートの最低水準をメーカー平均価格の15.1%から23.1%に引き上げた | |
| ● | 必要なbr医療補助管理保健組織が支払う薬品のリベート; | |
| ● | Brメーカーにカバーギャップ割引計画に参加することを要求し、この計画によると、メーカーはそのカバーギャップ期間中に条件を満たす受益者に適用ブランド薬品交渉価格の70%割引を提供することに同意しなければならず、メーカーの外来薬物として連邦医療保険D部分カバーの条件を組み入れなければならない | |
| ● | 連邦政府指定項目に“ブランド処方薬”を販売する薬品メーカーや輸入業者に差し引くことのできない年会費を徴収する。 |
ACAが公布されて以来、アメリカはまた他の立法改正を提案し、採択した。2021年3月11日、“2021年米国救援計画法案”が法律に署名され、2024年1月1日から法定の医療補助薬品還付上限が廃止され、現在の上限は薬品メーカーの平均価格の100%である。最近は2022年8月16日に、“2022年インフレ率低減法案”(IRA)が法律に署名された。他の事項を除いて、IRAはある薬品のメーカーが連邦医療保険と価格交渉を要求し(2026年から)、価格は協議できるが上限がある;連邦医療保険B部分と連邦医療保険D部分に基づいてリベートを実施し、インフレを超えた価格上昇を処罰し(初めて2023年に満期)、D部分のカバーギャップ割引計画の代わりに新しい割引計画を用いる(2025年から)。アイルランド共和軍は,衛生·公衆サービス部(HHS)秘書が最初の数年間,規制ではなく指導によってその多くの規定を実施することを許可した。このような理由と他の理由で、アイルランド共和軍がどのように実施されるのかは不明だ。
| 19 |
また、br政府は最近、メーカーがその市場に製品価格を設定する方式の審査を強化し、これによりいくつかの国会調査を招き、製品定価の透明性の向上、価格決定とメーカー患者計画との関係の審査、政府計画薬品精算方法の改革を目的とした立法と行政命令を提出し、公布した。政府が新冠肺炎の流行に対して追加的な行動を取った可能性もある。アメリカの各州もますます積極的に薬品の価格を制御するための法規を実施しており、価格或いは患者の精算制限、割引、ある製品への参入とマーケティングコスト開示の制限及び透明性措置を含み、場合によっては、他の国/地域からの輸入と大量調達を奨励することを目的としている。
データプライバシーとセキュリティ法
多くの州、連邦、および外国の法律、法規、標準は、健康に関連するbrおよび他の個人情報の収集、使用、アクセス、秘密およびセキュリティを管理しており、現在または将来的には、私たちの運営または私たちのパートナーの運営に適用される可能性があります。米国では、データ漏洩通知法、健康情報プライバシーおよび安全法律、および消費者保護法律法規を含む多くの連邦および州の法律法規が、健康関連および他の個人情報の収集、使用、開示および保護を管理している。また、ある外国の法律は、健康に関連するデータを含む個人データのプライバシーおよびセキュリティを管理している。プライバシーやセキュリティ法律、法規、その他の義務は絶えず変化し、相互衝突し、コンプライアンス作業を複雑化させ、調査、訴訟または行動を招き、データ処理に対する重大な民事および/または刑事罰および制限 を招く可能性がある。
競争
Br製薬と生物技術業界の特徴は技術の迅速な進歩、競争の激しい及び専有製品に対する高度な重視である。私たちは私たちの技術、知識と科学資源が私たちに一定の競争優位を提供すると信じているが、私たちは製薬と生物技術会社、学術機関、政府機関及び公共と個人研究機関を含む多くの源からの競争に直面している。その中の多くの競争相手は私たちよりもっと多くの資本と資源を得ることができるかもしれない。これらの競争相手はまた、合格した科学的·管理職を採用し、維持することで私たちと競争している。私たちが開発と商業化に成功した任意の候補品は、将来出現する可能性のある新しい免疫療法と競争するだろう。私たちの競争相手には、より規模が大きく、資金が豊富なバイオ製薬、バイオテクノロジー、治療会社、特に安進会社、アスリコン、P&G、遺伝子テーク、グラクソ、メルク、ノワ製薬、ファイザー、ロー氏ホールディングス、セノフィ社などの癌免疫治療に集中している会社が含まれています。一方、これらの会社の多くはDecoy 20と一緒に使用可能な免疫療法を開発していますが、この点では、潜在的な無料景品である可能性があると思います。
我々の主要候補Decoy 20では,多くの会社が可能な癌治療法を開発しているが,我々は,先天性および適応免疫系経路を刺激するために,システム注射を用いて殺された非病原性グラム陰性細菌を投与し,エンドトキシンを減少させる唯一の会社であると信じている。
私たちの成功は、Decey 20を商業化することに成功した私たちの能力と、私たちの目標適応において競合製品よりも安全で効率的な治療製品の組み合わせを識別、開発、管理する能力に基づくだろう。もし私たちの競争相手が私たちが開発する可能性のあるどんな療法よりも安全で、より効果的で、副作用が少なく、より便利で、あるいはより安い製品を開発し、商業化すれば、私たちの市場機会は減少または消失するかもしれない。私たちの競争的地位はまた、私たちが合格者を誘致し、維持し、特許保護を得るか、または他の方法で独自製品またはプロセスを開発し、私たちの知的財産権を保護し、技術構想から商業販売までの間に十分な資本資源を確保する能力に依存するだろう。政府や他の第三者支払者の精算も私たちの製品の定価と競争力に大きな影響を与えるだろう。私たちの競争相手も私たちよりも早くFDAや他の規制機関のその製品の承認を得ることができ、これは私たちの競争相手が私たちが市場に入る前に強力な市場地位を確立することをもたらすかもしれない。
| 20 |
知的財産権
私たちの成功は、私たちの独自の技術および知的財産権を保護する能力があるかどうか、および他人の固有の権利を侵害または侵害することなく運営される能力があるかどうかに少なくともある程度依存する。私たちは、特許、商標、商業秘密および著作権法、ノウハウ、知的財産権ライセンス、および他の契約権利(秘密および発明譲渡プロトコルを含む) によって、関連する知的財産権を含む独自技術および知的財産権を保護します。
特許
2023年3月1日現在、私たちは34件のライセンス特許と16件の承認された特許出願を持っており、私たちのビジネス分野で使用することができます。私たちの特許および特許出願は、通常、癌および感染症を治療するための組成物および方法に関するものであり、私たちの特許と我々が出願している特許とは、2033年から2039年の間の異なる日に満了すると予想される。
私たちは私たちが開発したすべての新製品と技術のために特許出願を提出する予定です。私たちのような会社の特許の見通しは通常不確実であり、複雑な法律と事実の問題に関連しているかもしれない。私たちが私たちの技術特許地位を維持し、強化する能力があるかどうかは、私たちが有効なクレームを得ることに成功し、承認された後にこれらのクレームを強制的に執行するかどうかにかかっている。私たちは私たちの任意の特許出願または私たちが許可する可能性のある任意の特許出願が任意の特許の発行につながるかどうか分からない。私たちが発行した特許および将来発行される可能性のある特許、または私たちが独占的に許可する可能性のある特許は、挑戦を受け、範囲を縮小し、brを回避するか、または無効または実行不可能であることが発見される可能性があり、これは、競争相手の関連製品のマーケティングを阻止する能力を制限するか、または製品に対する私たちの特許保護期間を制限する可能性がある。私たちは私たちが最初の発明 が私たち自身の特許または特許出願で主張された発明の会社であることを確認できない。さらに、我々の競争相手は、同様の技術を独立して開発したり、私たちが開発した任意の技術を複製したりすることができ、発行された特許によって付与された権利は、これらの競争相手に対するいかなる意味のある競争優位性も提供してくれない可能性がある。さらに、潜在的な製品の開発、テスト、および規制審査には多くの時間がかかるため、私たちのどの製品が商業化できる前に、どの関連特許も商業化後の短い期間で有効期限が切れたり、有効に維持されたりして、この特許のいかなる利点も弱める可能性がある。
取引秘密と秘密情報
特許に加えて、私たちはビジネス秘密と技術ノウハウに依存して、私たちの競争地位を発展させ、維持しています。ビジネス秘密と技術的ノウハウを保護することは難しいかもしれない。他の事項に加えて、私たちは、秘密および発明譲渡プロトコルによって、私たちのノウハウおよび他の知的財産権を保護し、これらのノウハウおよび他の知的財産権は、特許を申請できない可能性があり、またはbrの開示を必要としない方法で最もよく保護されていると考えられる。例えば、私たちの従業員には、彼らが私たちの雇用関係に関連する秘密協定を実行し、彼らが提供してくれたサービスに関連する発明を開示して分配することを要求します。しかし、このような合意が強制的に実行されることは保証されず、それらが私たちに十分な保護を提供することも保証されない。私たちのオフィスの物理的セキュリティと私たちの情報技術システムの物理的および電子的セキュリティを維持することで、私たちのデータ、ビジネス秘密、技術ノウハウの完全性とセキュリティを維持しようとしています。
私たち は、業務を展開するために必要な知的財産権を獲得、維持、保護することができない可能性があり、私たちの知的財産権を侵害したり、他の方法で他人の知的財産権を侵害したりする疑いを受ける可能性があります。これは、私たちの業務に実質的な損害を与える可能性があります。 私たちの知的財産権に関連するリスクに関するより包括的な概要は、“項目1 A”を参照されたい。リスク要因-私たちの知的財産権に関連したリスクだ“
| 21 |
環境問題
私たちは実験室プログラム、危険材料と廃棄物の処理、使用、貯蔵、処理と処分、汚染された場所の整理を含む様々な環境、健康と安全法律法規を遵守している。私たちの業務は化学品と生物学的材料を含む潜在的な危険と燃えやすい材料の使用に関するものだ。私たちの業務はまた危険な廃棄物製品を発生させるだろう。我々は通常,危険材料や廃棄物の処理に関する作業契約を第三者と締結しているが,これらの材料による汚染や傷害のリスクを解消することはできない。我々が現在把握している情報によると,環境コストや意外な状況 は我々に大きな悪影響を与えないと予想される。しかし,我々は,罰金,処罰,その他の制裁,調査·整理費用,第三者クレームなど,我々の運営や財産に関する環境に違反したり,関連責任を要求したりすることで巨額のコストを発生させる可能性がある.もし私たちが危険な材料を使用して汚染や損傷をもたらしたら、私たちはそれによるいかなる損害に対しても責任を負い、いかなる責任も私たちの資源の範囲を超える可能性がある。“第I項、第1 A項を参照。リスク要因−医療保健法やその他の法律コンプライアンス事項に関するリスク−環境、健康および安全法律法規に違反したり、環境、健康および安全法律法規に基づいて責任を負う場合、私たちは罰金、処罰、または他のコストに直面する可能性があり、これは私たちの業務の成功に実質的な悪影響を及ぼす可能性があります。”
私たちの業務、運営、施設はすべての実質的な面で適用される環境と健康安全法律法規に適合していると信じています。
人的資本管理
2022年12月31日現在、私たちは6人のフルタイム従業員がいます。私たちの職員たちには労働組合代表もなく、集団交渉協定の包括的な範囲もない。
私たちの未来の成功は私たちが引き続き合格した人材を誘致、採用し、維持する能力にある程度依存すると信じている。特に、私たちは高度な管理と研究者のスキル、経験、表現に依存している。我々は他のバイオテクノロジー,医療機器,製薬·ヘルスケア会社,大学や非営利研究機関と合格人材を争っている。
私たちは従業員のニーズを満たすために競争力のある報酬と福祉計画を提供します。これらの計画には、給与に加えて、報酬計画、年金、医療·保険福祉、有給休暇、帰省休暇などが含まれている。私たちはまた、帰属条件を持つ指向性持分贈与を使用して、人員、特に私たちの肝心な従業員を維持することを助けます。
私たちの業務の成功は私たちの従業員の福祉と根本的に関連している。そのため、著者らは新冠肺炎の疫病発生に必要であり、政府法規に符合する安全措置を含む従業員の健康と安全を保障することに取り組んでいる。
私たちは私たちが従業員と仲がいいと思う。
歴史的背景と会社構造
Intec イスラエルまたはIntec Pharma(最初は2000年10月23日にイスラエル民間会社としてイスラエルに設立され、登録設立された) は2010年2月にイスラエルテルアビブ証券取引所(TASE)で初公募株を成功させ、2015年8月に米国で初公募株を完成させた。
Indaptus 治療会社(前身はIntec親会社)は2021年2月24日にデラウェア州に設立され、デラウェア州の民間会社であり、Intecイスラエル社の完全子会社である。
2021年7月27日、Intecイスラエル社、Indaptus治療会社、Indaptusの完全子会社イスラエル社Indaptus Merge Sub Ltd.は、2021年4月27日の合意および合併再編計画(“馴化合併協定”)の条項と条件に基づいて馴化合併(“馴化合併”)を完了し、この合意に基づき、Intecイスラエル社はIndaptusイスラエル社に合併し、Intecイスラエル社はIndaptus 治療会社の馴化、合併、合併時の完全子会社である。Intecイスラエル社は帰化合併直前のすべての資産、権利、権力、財産を引き続き所有し、帰化合併直前のすべての債務、負債、義務を負い続けている。
2021年8月3日,Indaptus Treeutics,Inc.は2021年3月15日の合意と合併再編計画(“合併合意”)によりDecoyとの合併を完了し,DecoyはIndaptus治療会社の存続実体と完全子会社となり,Decoyが展開する業務は合併後会社が展開する業務となる。
また,統合によりIntec Parentは“Intec Parent,Inc.”と改称される.“Indaptus Treeutics,Inc.”へ
| 22 |
合併完了後、私たちの普通株は2021年8月4日にナスダック資本市場で取引を開始し、取引名は“Indaptus Treateutics,Inc.”である。株式コード“INDP”です
利用可能な情報
私たち はhttp://www.indaptusrx.comで会社のサイトを維持しています。我々のサイトに含まれているまたはアクセス可能な情報 は,本年度報告の一部でも本年度報告にも含まれていない.
これらの資料を米国証券取引委員会に提出したり、米国証券取引委員会に提供したりした後、我々は、Form 10-K、Form 10-Q、およびForm 8-Kに関する報告書のコピー をできるだけ早く当社のウェブサイトを通じて無料で取得することができる。 また、米国証券取引委員会は、報告書、依頼書および情報声明、および米国証券取引委員会に電子的に文書を提出する発行者に関する他の情報を含むインターネットサイトを維持することができる。
1 a項目.リスク要因です
私たちの普通株式に投資するかどうかを決定する前に、以下に説明する要因と、F-1ページから始まる監査された総合財務諸表および関連付記を含む本年度報告書に含まれるすべての他の情報をよく考慮しなければなりません。以下で議論するいずれかのリスクが実際に発生すれば、我々の業務、財務状況、経営業績、キャッシュフローは重大な悪影響を受ける可能性がある。これは私たちの普通株の取引価格を下落させるかもしれません。 あなたは投資の全部または一部を失うかもしれません。
私たちの財務状況と資本要求に関連するリスク
私たちは臨床段階の会社で、運営の歴史が限られている。私たちはまだ利益を上げていないし、近い将来利益が出ないと予想され、永遠に利益を上げないかもしれない。
著者らは臨床前生物技術会社であり、主に新しい、特許を獲得したシステムの抗癌と抗ウイルス免疫療法の開発に取り組んでいる。私たちのすべての候補製品は臨床前あるいは早期臨床開発段階にあり、私たちの候補製品は発売が許可されておらず、発売されているか商業化されていない。
したがって、私たちは私たちの業務と将来性を評価する意味のある歴史的運営を持っていないし、私たちの任意の候補製品のために市場の承認を得ることができ、あるいはバイオ製薬業界会社がよく遭遇するリスクと不確実性を克服する能力があることを証明していない。したがって、私たちは設立以来、利益を上げておらず、各報告期間中に重大な運営損失を出してきた。2022年12月31日と2021年12月31日の年度までに,我々が報告した純損失はそれぞれ約1,430万ドルと約770万ドルであり,2022年12月31日までの累計赤字は約3,000万ドルであった。
予測可能な未来には、引き続き損失を被ることが予想され、私たちの開発活動の拡大に伴い、規制機関が私たちの候補製品の承認を求め、商業化を開始し、これらの製品がFDA、欧州医薬品局、EMAなどの外国当局の承認を得たら、損失は歴史的水準から大幅に増加するだろう。また,我々の純損失は四半期間と年度間で大きく変動する可能性があるため,我々の運営結果の期間間比較は我々の将来の業績の良い指示ではない可能性がある。私たちは上場企業の運営に関連した追加費用が発生すると予想している。たとえ私たちが1つ以上の候補製品を開発して商業化することに成功しても、私たちは決して利益を上げないかもしれないし、あるいは未来にbrの利益を達成しても、私たちはその後の時期に収益性を維持できないかもしれない。私たちのこれまでの損失は、予想された将来の損失に加え、私たちの株主権益や運営資本に悪影響を与え続けるだろう。
| 23 |
私たちが現在キャッシュフローが不足していることを考慮して、私たちはもっと多くの資金を集める必要があるだろう。もし私たちがbrが必要な時に受け入れ可能な条項や十分な資金を集めることができない場合、私たちは研究計画、製品開発活動、商業化努力の一部、制限、または廃止を余儀なくされる可能性がある。
予測可能な未来には、私たちは私たちの運営に資金を提供するために十分なキャッシュフローを生成することができないので、私たちは私たちの運営を維持または拡大するために必要な資本を提供するために追加の株式や債務融資を求める必要があるだろう。
は私たちが受け入れ可能な条項で十分な追加資本を調達できるか、または全くできないという保証がない。このような追加融資が満足できる条項で提供できない場合、または十分な資金がない場合、私たちはいくつかのbrまたはすべての研究計画、製品開発活動、商業化努力を延期、制限、または廃止する必要があるかもしれません。私たちは、業務目標を達成する能力、私たちの競争力、および私たちの業務、財務状況、および運営結果が重大な悪影響を受ける可能性があります。 また、私たちは、私たちが開発とマーケティングを望んでいた候補製品を開発およびマーケティングする権利を与えることを要求されるかもしれません。私たちは私たちの業務に資金を提供することができず、あなたの投資損失を招くかもしれません。
私たちの未来の資本需要は多くの要素に依存するだろうが、これらに限定されない
| ● | 著者らの臨床試験、臨床前研究とその他の関連活動の範囲、進捗、結果とコスト | |
| ● | 私たちの現在または未来の任意の候補製品のための規制承認を受ける時間と関連するコスト | |
| ● | 私たちは開発または商業化された候補製品の数量と特徴を求めている | |
| ● | 私たちの候補製品は臨床用品の生産と商業用品の構築のコストです | |
| ● | もし私たちが現在または未来の任意の候補製品が販売を許可されれば、商業化活動のコストは、マーケティング、販売、流通コストを含む | |
| ● | 技術人材の誘致と維持に必要な費用 | |
| ● | 上場企業の利益に関連するコスト | |
| ● | もし私たちの候補製品がbrマーケティングの承認を得たら | |
| ● | 準備、提出、起訴、保守、弁護、および実行可能な特許主張に関連する費用は、訴訟費用および任意のそのような訴訟の結果を含む。 |
もし私たちが現在または未来の運営計画が十分な資金を持っていると思っていても、市場 条件が有利であれば、または特定の戦略に基づいて考えると、追加の資金を求め続けるかもしれない。私たちは受け入れ可能な条項で十分な追加融資を受けることができないかもしれないし、全く得られないかもしれない。もし私たちが適時または優遇条件で十分な資金を得ることができなければ、私たちは私たちの1つまたは複数の研究または製品開発計画および/または商業化努力を大幅に延期、減少、または廃止することを要求されるかもしれない。私たちはまた、私たちの業務を拡大したり、他の方法で必要なビジネスチャンスを利用することができないかもしれない。このような事件のいずれも私たちの財務状況と業務の見通しに重大な悪影響を及ぼす可能性がある。
| 24 |
Brの追加資本の調達は、私たちの株主の株式を希釈し、私たちの運営を制限するか、または私たちのbr技術または候補製品の権利を放棄することを要求するかもしれません。
私たちが相当な製品収入を生み出すことができる前に、私たちは株式および/または債務融資と協力、許可協定、または他の戦略手配の組み合わせによって私たちの現金需要を満たす予定です。私たちは以下のようにより多くの資本を求めるかもしれない:私募と公開、“市場での”発行、株式とリンクと構造化取引、br}債務(直接、転換可能、またはその他)融資、協力、および許可手配。もし私たちが株式または転換可能な債務証券を売却することによって追加のbr資本を調達する場合、あなたの所有権資本は希釈され、条項は清算または株主としての権利に悪影響を及ぼす他の特典を含む可能性がある。例えば2022年6月に市場発売協定を締結しました2022年9月1日に改訂されさらに、2022年12月、私たちはリンカーン公園と購入契約と登録権協定を締結し、リンカーン公園は36ヶ月以内に最大2000万ドルの普通株を時々購入することを約束した(br}が合意条項に従って48ヶ月に延長されない限り)。私たちはまた私たちの普通株より優先的な権利、割引、特権を提供する株式証券を未来に発行することができる。現金に対する私たちの需要と、株式発行は似たような状況にある会社の最も一般的な資金調達タイプであることを考慮して、希釈のリスクは私たちの株主にとって特に大きい。当時の市場流動性によると、任意の所与の時間に登録された株式の追加売却は、我々普通株の取引価格を低下させる可能性がある。債務融資が可能であれば、固定支払義務の増加を招き、債務の発生、資本支出の実施、または配当の宣言など、特定の行動をとる私たちの能力を制限または制限する能力を含むプロトコルに関連することができる。もし私たちが第三者との協力、戦略連合、許可手配を通じてより多くの資金を調達するならば、私たちは私たちの技術、将来の収入流、または候補製品に対する貴重な権利を放棄しなければならないかもしれません。または私たちに不利な条項でライセンスを付与しなければなりません。
我々の候補製品の発見と開発に関するリスク
私たちのbrは私たちの現在の1つ以上の候補製品の成功に依存しており、私たちはそれらのいずれかが規制部門の承認または商業化を受けるとは決定できない。
我々 は我々の主要候補製品Decoy 20を開発するのに多大な時間,お金,労力を費やした.そのため、著者らの業務はDecoy 20の臨床試験の開始と成功を評価することに大きく依存し、著者らは適時にDecoy 20の開発を完成し、監督部門の許可を得て、それを商業化することに成功したかどうかにかかっている。開発、監督部門の許可を得てDecoy 20を商業化する過程は長く、複雑で、コストが高く、しかも結果はまだ確定していない。
これまで、我々のどの候補製品も、安全性、純度、効力または有効性を提供するための実質的な証拠を提供するための臨床試験を完了していない。私たちのすべての候補製品は、臨床試験と更なる臨床前研究を含む更なる開発が必要であり、それらの毒理学を評価し、それらの調合と規制承認を最適化し、その後商業化することができる。早期開発中に得られた積極的な結果は、必ずしも今後の開発が成功するか、あるいは規制承認を得ることを意味するわけではない。私たちの開発努力は商業製品を引き起こさないかもしれません。これは、私たちの候補製品が安全で効果的ではないか、あるいは私たちの候補製品が生物学的製剤、安全、純粋かつ有効であるように規制されているか、または私たちが臨床開発および承認プロセスを通じて私たちの候補製品を推進するのに十分な財政的または他の資源がないからかもしれません。私たちの任意の候補製品が任意の時間または任意の開発段階で安全性、純度、効力または効果を証明できない場合、私たちは潜在的な重大な遅延を経験するか、または候補製品の開発を放棄することを要求される。
我々の現在のどの候補製品もFDA,EMA,あるいは同様の外国機関の規制承認を得る資格がなく,数年以内に商業化が開始されると予想される(あれば).私たちが最終的に規制部門のこれらの候補製品の承認を得ても、私たちまたは私たちの未来の潜在的なパートナーは様々な理由で商業化に成功できない可能性がある。例えば、これらの問題は、代替療法の獲得可能性、費用対効果の欠如、商業規模生産製品のコスト、および他の製品との競争を含む。私たちの候補製品の成功はまたいかなる不良副作用の流行程度と重症度によって制限される可能性があります。もし私たちが現在の1つ以上の候補製品を商業化できなければ、私たちは利益を達成したり維持したりするのに十分な収入を生み出すことができない可能性があり、私たちの財務状況は低下するかもしれない。
| 25 |
臨床と臨床前開発は長く高価な過程に関連し、結果は不確定である。私たちの現在または計画中の臨床試験は、開始、完了、終了、または一時停止において、いかなる困難または遅延に遭遇しても、私たちのコスト増加、遅延、または私たちの創造能力を制限すること、または私たちのビジネスの将来性に不利な影響を与える可能性がある。
Brが監督部門の許可を得て私たちの任意の候補製品を商業化する前に、私たちは広範なbr臨床試験を行い、候補製品の人体上の安全性、純度と有効性を証明しなければならない。臨床前や臨床では薬物開発費用が高く,完成まで数年かかる可能性があり,その結果自体も確定していない。失敗は臨床前研究あるいは臨床試験過程中の任意の時間に発生する可能性がある。臨床前或いは臨床結果の将来性は有望であるが、任意の候補製品は臨床前或いは臨床開発の任意の段階で意外に失敗する可能性がある。私たちの業界では候補製品の歴史的不合格率が高い。
候補製品の臨床前研究或いは早期臨床試験のbr結果はこの候補製品の後続臨床試験の結果を予測できない可能性があり、臨床試験の中期結果は必ずしも最終結果を代表するとは限らない。臨床試験後期段階の候補製品 は必要な安全性と有効性特徴を示すことができないかもしれないが、すでに臨床前研究と初歩的な臨床試験に合格したが。臨床試験では前臨床研究と早期臨床試験に基づく意外な結果が観察されることは珍しくなく、早期結果は非常に有望であるが、多くの候補製品は臨床試験で失敗した。そのほか、臨床前と臨床データはよく異なる解釈と分析の影響を受けやすい。製薬やバイオテクノロジー業界の多くの会社は臨床開発において大きな挫折を経験しており,早期のbr研究においても奮い立つ結果を得ている。
私たちは、任意の候補製品に対して臨床試験を開始することができる前に、INDまたは同様の規制提出の一部として、候補製品の化学成分、製造および制御に関する情報、およびINDまたは同様の規制提出の一部として、FDA、EMAまたは同様の外国規制機関に臨床前研究結果および他の情報を提出しなければならない。FDA、EMA、または同様の外国の監督管理機関は、任意の候補製品に対して追加の臨床前研究を要求する可能性があり、その後、任意のINDまたは同様の規制提出書類に従って臨床試験を開始することを可能にすることができ、これは私たちの臨床前開発計画の遅延とコスト増加を招く可能性がある。また,われわれが臨床試験を開始しても,規制機関のこのような臨床試験の一時停止や中止を招く可能性があるという問題がある。私たちが行っているおよび計画中の候補製品臨床試験の開始または完成にどのような遅延が発生しても、私たちの製品開発スケジュールと製品開発コストに深刻な影響を与え、私たちの財務的地位を損なう可能性があります。
われわれが計画した臨床試験が時間どおりに開始されるかどうか,あるいは予定通りに完了するかどうかは分からない。臨床試験の開始、データ読み取り、完了は様々な原因で遅延する可能性があり、以下に関連する遅延を含む
| ● | 臨床試験の開始または継続を支持するために、十分な臨床前、毒理学または他の体内または体外データを生成することができない; | |
| ● | 監督部門の許可を得ていないか、試験を開始することを許可していない、あるいは監督部門と試験設計について合意した | |
| ● | FDA、EMAなどの外国の監督管理機関は著者らの臨床試験の設計或いは実施に対して異なる意見を持っている | |
| ● | CROおよび臨床試験サイトと合意された任意の 失敗または遅延は、その条項は広範な交渉 を行うことができ、異なるCROと試験サイトとの間に有意差がある可能性がある | |
| ● | 適切な臨床研究者の確定、採用と訓練に遅延が出現した | |
| ● | 臨床試験場所の1つまたは複数のIRBsまたは倫理委員会の承認を得ることができなかった | |
| ● | IRBs は、調査地点の試験の承認、一時停止、または終了を拒否し、より多くの被験者の募集を禁止し、 または試験の承認を撤回する |
| 26 |
| ● | 臨床試験プログラムを変更したり修正したり | |
| ● | 臨床サイトは試験案から外れたり、試験を終了したりした | |
| ● | 我々のCROは、他の国/地域の良好な臨床実践(GCP)の要求または適用された規制規則およびガイドラインに従って操作できなかった | |
| ● | 十分な数の私たちの候補製品を生産できなかったか、またはbr臨床試験の使用のために十分な数の併用療法を得ることができなかった | |
| ● | 被験者は、私たちの試験に滞在できなかったか、または被験者が私たちの試験に残っていなかったことを含む、治療後の後続治療を受けるために戻ってこなかったか、または私たちの試験に残っていなかったことを含む | |
| ● | 患者brは,我々が開発している候補製品の適応のために代替製品を選択したり,競争臨床試験に参加したりしている | |
| ● | 臨床試験を続けるのに十分な資金が足りないか、あるいは私たちの予想よりもコストが高い | |
| ● | 重症または重篤な意外薬の副作用が出現した被験者 | |
| ● | 他社が行った同種の薬物の試験では,我々の候補品に類似していると考えられる深刻な有害事象が発生した | |
| ● | 長時間の臨床観察あるいは結果データの長時間分析を必要とする臨床終点を選択する | |
| ● | 製造プロセスを、契約製造組織(CMO)によって運営されているより大規模な工場に移し、私たちのCMOは、その製造プロセスを必要な変更の遅延または失敗、または私たちのCMOが現在の良好な製造規範(CGMP)、法規または他の適用要件に従って臨床試験材料 の生産を要求することができなかった | |
| ● | 第三者は当方に対する契約義務をタイムリーに履行することを望んでいないか、または履行できません。 |
また,新冠肺炎の流行による中断は,我々が開始,登録,著者らの計画中と行っている臨床試験を開始,登録,完了する際にこのような困難や遅延に遭遇する可能性を増加させる可能性がある。
臨床試験はFDAや他の適用規制機関の法的要求,条例,ガイドラインに従って行い,これらの政府機関や臨床試験を行う医療機関の倫理委員会やIRBsの監督を継続しなければならない。臨床試験が我々,そのような試験を行っている機関のIRBs,そのような試験のデータ安全監視委員会やFDA,EMAなどの外国規制機関によって一時停止または終了されれば,我々も遅延に遭遇する可能性がある。このような機関は、法規の要求或いは適用された臨床試験規程に従って臨床試験を行うことができず、FDA、EMA或いは類似の外国の監督機関が臨床試験場を検査した不良結果、予見できない安全問題或いは副作用、候補製品の使用のメリットを証明できなかったこと、政府法規の変化、行政行為又は十分な資金が不足して臨床試験を継続することを含む、多種の要素のために臨床試験を一時停止又は終了する可能性がある。また、規制要求と政策は変化する可能性があり、私たちはこれらの変化に適応するために臨床試験方案を修正する必要があるかもしれない。修正案は,われわれの臨床試験案を規制機関やIRBsに再提出して再検査することを要求する可能性があり,臨床試験のコスト,時間,あるいは成功したbr}に影響を及ぼす可能性がある。
| 27 |
また,われわれの臨床試験の首席研究員は時々私たちの科学コンサルタントやコンサルタントを務め,このようなサービスに関する報酬 を得る可能性がある。場合によっては、私たちはFDA、EMA、または同様の外国規制機関にいくつかの関係を報告することを要求されるかもしれない。FDA、EMA、または同様の外国の監督管理機関は結論を出すかもしれないが、私たちと主要な研究者との間の財務関係は利益の衝突をもたらし、あるいは他の方法で研究の解釈に影響を与えている。 FDA、EMAまたは同様の外国の監督管理機関は、適用される臨床試験サイトで生成されたデータの完全性を疑問視する可能性があり、臨床試験自体の効用が脅かされる可能性がある。これは、FDA、EMA、または同様の外国規制機関が私たちのマーケティング申請の承認を遅延または拒否することをもたらす可能性があり、最終的には、私たちの1つまたは複数の候補製品が上場承認を拒否する可能性がある。
また、FDA、EMA、および他の規制機関の臨床試験に関する政策はbrを変える可能性があり、追加の政府法規を公布する可能性がある。例えば,EUの臨床試験に関する規制構造が最近変化している。EU臨床試験条例(“CTR”)は2014年4月に採択され、EU臨床試験指令を廃止し、2022年1月31日に施行された。臨床試験指令は,各加盟国で国家衛生当局および独立した倫理委員会に単独の臨床試験申請(“CTA”) を提出することを要求しているが,CTRはすべての関連加盟国に単一の申請を提出することのみを要求する集中的な手続きを導入している。CTRは、スポンサーが各加盟国の主管当局と道徳委員会に文書を提出することを可能にし、各加盟国が決定を下すことを可能にする。CTAの評価手続きも統一されており、すべての関連加盟国による共同評価を含め、各加盟国がその領土に関する具体的な要求について個別に評価し、 道徳基準を含む。各会員国の決定は集中されたEUポータルサイトを通じてスポンサーに伝達される。CTAが承認されると, 臨床研究開発は継続可能である。CTRは3年間の過渡期が予想される。行われている臨床試験と新しい臨床試験がCTRによってどの程度制御されるかはそれぞれ異なる。(I)2022年1月31日までに“臨床試験指令”に基づいて申請を提出した臨床試験、または(Ii)2022年1月31日から2023年1月31日までの間、かつスポンサーが“臨床試験指令”を申請する臨床試験を選択し、2025年1月31日までこの指令によって管轄されている。この日の後、すべての臨床試験(行われている臨床試験を含む)はCTR条項によって制限される。我々と我々の第三者サービスプロバイダ(例えばCRO)がCTR要求を遵守することは、我々の開発計画に影響を与える可能性がある。我々が既存の要求の変化 に緩やかあるいは適応できなければ,臨床試験を管理する新たな要求や政策を採用すれば,我々の開発計画が影響を受ける可能性がある。
さらに、臨床試験の終了または一時停止、または臨床試験の開始または完了遅延をもたらす多くの要因も、最終的に候補製品の規制承認が拒否される可能性がある。我々の臨床試験のどの遅延も,候補製品を商業化する独占的権利を持つ時間を短縮することが可能である。この場合、私たちの競争相手は私たちより先に製品を市場に出すかもしれませんが、私たちの候補製品の商業可能性は著しく低下する可能性があります。このような状況のいずれも私たちの業務、財政状況、そして見通しを損なう可能性がある。
我々 は引き続き巨額の研究開発費や他の運営費が発生することが予想され,収益を実現することが困難になる可能性がある。
私たちは、私たちの候補製品の臨床前研究と臨床試験を含む研究と開発に大量の資金を投入する予定です。もし任意の候補製品が商業販売が許可されたら、私たちはこれらの候補製品を生産し、販売します。私たちはまた、相補的な会社、技術および資産を開発または買収するための追加のbr資金、ならびに運営資金要件および他の 運営および一般会社用途のために必要かもしれない。しかも、私たちが増加する予定の従業員の数は近いうちと長期的に私たちのコストを大幅に増加させるだろう。
我々の候補製品が開発に成功するかどうかは定かではないため,これらの製品を開発し,商業化するために必要な実際の資金が必要であると正確に見積もることはできない。また、私たちは私たちの任意の候補製品 を商業化して利益を達成することができても、十分な収入を生み出すことができないかもしれない。
| 28 |
私たち は、限られた数の研究計画、候補製品、および特定の適応を展開するために限られたリソースを使用する可能性があり、より利益的またはより成功確率が高い可能性のある候補製品または適応を利用することができない。
我々の財務や管理資源が限られているため,限られた数の研究計画や候補製品,および 特定の適応に集中しなければならない。したがって,我々は現在Decoy 20の開発に集中しているしたがって、私たちは他の候補製品を求めるビジネスチャンスや他の指示を放棄したり延期したりするかもしれない抗癌と抗ウイルス免疫治療これはその後、より大きなビジネス潜在力を持っていることが証明された私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。私たちの現在と未来の研究開発計画および特定の適応候補製品への支出はいかなる商業的に実行可能な製品も発生しないかもしれない。
私たちのbr候補製品は不良な副作用を招く可能性があり、その監督審査或いは商業化を遅延或いは阻止する可能性があり、私たちの臨床試験の一時停止或いは停止を招き、候補製品を放棄し、承認された製品の商業イメージを制限し、あるいは私たちの業務、財務状況と運営結果に他の重大な悪影響を与える。
は薬品の一般的な状況であるため,我々の製品に関する副作用や有害事象 候補が使用される可能性が高い.著者らの臨床試験結果は深刻かつ受け入れられない副作用或いは意外な特徴の重症度と流行程度を掲示する可能性がある。例えば,我々の候補製品の作用機序は免疫系の刺激に依存するため,過刺激や不良免疫反応の可能性がある。私たちの候補製品による副作用は、単独使用でも他の療法との併用でも、私たちまたは規制機関の臨床試験の中断、延期または一時停止、またはFDA、EMAまたは同様の外国規制機関の規制承認の遅延または拒否、または、そのような候補製品 が承認された場合、より厳しいラベルおよび他の承認後の要求をもたらす可能性がある。治療に関連する任意の副作用brはまた、患者の募集または入選患者の試験を完了する能力に影響を与える可能性があり、または潜在的な製品責任クレームを引き起こす可能性がある。このようなどんな状況でも、私たちの業務、財政状況、そして見通しに大きな被害を及ぼす可能性がある。
我々の候補製品が臨床前研究または臨床試験において単独で使用されるか、または他の承認された製品または候補製品と組み合わせて使用される場合、副作用または意外な特徴が存在する場合、私たちは、それらの開発を中断、延期または放棄するか、またはその開発をより狭い用途またはサブ集団に制限する必要がある可能性があり、これらの用途またはサブ集団では、副作用または他の特徴があまり一般的ではなく、あまり深刻ではない、またはリスク利益の観点から受け入れやすい。
われわれが計画中のbr臨床試験を行っている患者は,将来われわれの前臨床研究やこれまでの臨床試験では観察されなかった重大な有害事象や他の副作用を受ける可能性がある。私たちの候補製品の治療を受けた患者はまた手術、放射線治療或いは化学療法を受けている可能性があり、これは著者らの候補製品とは関係のない副作用或いは不良事件を招く可能性があるが、依然として著者らの臨床試験の成功に影響を与える可能性がある。重篤な患者を著者らの臨床試験に組み入れることは死亡或いはその他の不良医療事件を招く可能性があり、原因はこのような患者が他の治療法或いは薬物を使用している可能性があり、或いはこのような患者の疾病の深刻性による。現在または未来の任意の臨床試験において、このような深刻な有害事象または他の副作用が観察された場合、臨床試験に患者を募集することが困難である可能性があり、または候補製品の試験または開発を完全に放棄することが要求される可能性がある。我々、FDA、他の同様の規制機関、またはIRBは、そのような試験中の対象が許容できない健康リスクまたは副作用に直面していると考えることを含む、様々な理由で候補製品の臨床試験を随時一時停止することができる。副作用が候補製品の獲得や規制部門の承認を妨げなければ,他の利用可能な療法と比較して不良副作用 は耐性の問題により市場受け入れを抑制する可能性がある。このような状況はどのような発展も私たちの業務、財務状況、そして見通しに実質的な損害を与える可能性がある。
| 29 |
また、もし私たちの任意の候補製品が監督部門の許可を得たら、私たちまたは他の人は後にこのような 製品が不良な副作用をもたらし、いくつかの潜在的な重大な負の結果を招く可能性があることを発見した。例えば、FDAは、そのような候補製品を使用した治療の利点が各潜在的患者のリスクよりも大きいことを保証するために、リスク評価および緩和戦略(REMS)を採用することを要求することができ、医療従事者とのコミュニケーション計画、患者教育、広範な患者監視または分配システムおよびプロセスを含むことができ、これらのシステムおよびプロセスは、高度に制御され、制限され、コストは業界の典型的な よりも高い。もし私たちまたは私たちの協力者が後に私たちまたは私たちの協力者によって開発された任意の製品による副作用 を発見した場合、私たちまたは私たちの協力者はまた、患者のbr}教育、医療専門家認証、または特定の監視のようなREMSまたは同様の操作に参加することを要求される可能性がある。不良事象に関連する他の潜在的に重大な負の結果は、以下の通りである
| ● | 私たちはある製品のマーケティングを一時停止することを要求されるかもしれません。または市場から除去することを決定するかもしれません | |
| ● | 規制部門は製品の承認を撤回または変更することができる | |
| ● | 規制当局は、ラベルに警告を追加すること、または追加の安全報告がある選択的専門センターに製品が入ることを制限することを要求する可能性があり、すべての治療中または一部の治療中にこれらの中心に地理的に近接することを患者に要求することができる | |
| ● | 私たちは患者のために製品のリスクを概説したり、発売後の研究を行うための薬物ガイドラインを作成する必要があるかもしれない | |
| ● | 私たち は製品の管理方法の変更を要求される可能性があります | |
| ● | 私たちは罰金、禁止または刑事または民事処罰が科されるか、または起訴され、被験者または患者に与えられた傷害に責任を負う可能性がある | |
| ● | 製品の競争力は低下するかもしれないし、私たちの名声は損なわれるかもしれない。 |
FDAまたは他の規制機関が承認した場合、これらのすべてのイベントは、私たちの候補製品の使用を減少させたり、他の方法でその商業的成功を制限したり、私たちの候補製品に対する市場の受け入れ度を維持することを阻止したり、私たちの候補製品に対する市場の受け入れを維持したりする可能性がある。
われわれの臨床試験に患者を募集することは困難であることが示唆されるかもしれない。われわれが患者を募集して臨床試験に参加することが困難であれば,われわれの臨床開発活動は延期されたり,他の悪影響を受けたりする可能性がある。
患者登録は臨床試験の時間スケジュールの重要な要素であり、著者らの臨床試験の時間スケジュールはある程度著者らの患者募集試験の速度 及び必要な後続時間の完成状況に依存する。FDAや他の同様の規制機関の要求に基づいて、これらの試験に参加するのに十分な数の合格した患者を見つけて募集することができない場合、私たちは私たちの候補製品のために臨床試験を開始または継続することができないかもしれません。私たちは現在、私たちの候補製品の条件は患者プールの限られた疾患であり、臨床試験に使用できることを評価することを計画しています。われわれの臨床試験の資格基準が決定すると,利用可能なbr試験参加者をさらに制限する可能性がある。
| 30 |
患者が私たちの臨床試験に参加することは他の要素の影響を受ける可能性があります
| ● | ターゲット患者集団の大きさと性質; | |
| ● | 調査を受けた病気や状況の重症度 | |
| ● | 調査を受けた疾患または状況の承認療法の獲得性および有効性; | |
| ● | 患者brレジメンで定義されている問題のある試験の資格基準; | |
| ● | 研究を受けた製品候補製品の知覚可能なリスクと収益; | |
| ● | 研究されている候補製品の他の利用可能な療法に対する潜在的優位性に対する臨床医および患者の見方は、我々が調査中の適応のために承認される可能性のある任意の製品または調査中の任意の候補製品を含む | |
| ● | 臨床試験への参加を促進するために努力しています | |
| ● | 患者br医師の回診方法; | |
| ● | 治療中および治療後に患者の能力を十分に監視する | |
| ● | 潜在患者の臨床試験地点に対する近似性と可用性 | |
| ● | 臨床試験地点で潜在的な患者を募集し続けた | |
| ● | 臨床試験に参加した患者が試験完了前に試験を終了するリスク;および | |
| ● | 新冠肺炎の疫病のため、学生募集と学業完成は遅延或いは困難が出現した。 |
また,これらの疾患に対する他の製薬会社がこれらの患者から臨床試験患者を募集しており,われわれの臨床試験を完全に登録することが困難になる可能性がある。著者らはまたCROと臨床試験サイトに依存し続け、適切かつ適時に著者らの臨床試験と臨床前研究を行うことを確保する。我々は協定を締結して彼らのサービスを管理しているが,彼らの実際の表現への影響は限られている.私たちの臨床試験のために十分な数の患者 を募集することは重大な遅延を招くか、あるいは1つ以上の臨床試験を完全に放棄する必要があるかもしれない。私たちの臨床試験の登録遅延は私たちの候補製品の開発コストを増加させ、監督部門の許可を得て私たちの候補製品を販売する能力を脅かす可能性があります。また,われわれの臨床試験のために十分な数の患者を募集することができても,これらの患者のわれわれの臨床試験における登録者数を保つことは困難である可能性がある。
著者らは時々公表或いは公表した臨床試験と臨床前研究の一時、br}“TOPLINE”と初歩データは更に多くの患者データの出現に従って変化する可能性があり、そして監査と検証プログラムの影響を受け、これは最終データの重大な変化 を招く可能性がある。
私たちは時々私たちの臨床試験と臨床前研究の中期、主要或いは初歩的なデータを公開するかもしれない。これらのデータ は当時利用可能なデータの初歩的な分析に基づいて、特定の研究或いは試験に関連するデータに対してより全面的な審査を行った後、結果及び関連する発見と結論は変化する可能性がある。データ分析の一部として,仮説,推定,計算,結論も行い,すべてのデータを全面的に詳細に評価する機会がないか,または完全に詳細に評価する機会がないかもしれない.したがって、他のデータが受信され、十分に評価されると、私たちの報告の中期、バックライン、または予備結果は、同じ研究または試験の将来の結果と異なる可能性があり、または異なる結論または考慮要因がそのような結果に適合する可能性がある。 バックラインおよび予備データは、まだ審査および検証手順によって制限されており、これは、最終データが私たちが以前に発表したバックラインまたは予備データと実質的に異なることをもたらす可能性がある。したがって、最終データが利用可能になる前に、バックラインおよび予備データは慎重に表示されなければならない。
| 31 |
著者らが完成する可能性のある臨床試験の一時的なbrデータはさらに、患者登録の継続とより多くの患者データの出現に伴い、1つ以上の臨床結果が実質的に変化する可能性があるというリスクに直面している。中間データ、バックラインデータ、または予備データと最終データとの間の不利な差は、私たちのビジネスの将来性に深刻な影響を与える可能性があります。しかも、私たちまたは私たちの競争相手がこのようなデータを開示することは私たちの普通株の価格変動を招くかもしれない。
さらに、 他の人は、規制機関を含み、私たちの仮定、推定、計算、結論または分析 を受け入れないか、またはデータの重要性を異なる方法で解釈またはトレードオフする可能性があり、これは、特定の計画の価値、特定の候補製品または製品の承認可能性または商業化、および当社全体に影響を及ぼす可能性がある。さらに、私たちが公開開示された特定の研究または臨床試験に関する情報を選択することは、一般的に広範な情報に基づいており、あなたまたは他の人は、私たちが決定した重要な情報または他の適切な情報が私たちの開示に含まれていることに同意しない可能性があり、私たちが開示しないことを決定した任意の情報は、最終的に特定の候補製品または私たちの業務に関する将来の決定、結論、観点、活動、または に大きな意味を有すると考えられるかもしれない。もし私たちが報告した中期、バックライン、または予備データが実際の結果と一致しない場合、または規制機関を含む他の人が結論に同意しない場合、私たちが承認を得て私たちの候補製品を商業化する能力が損なわれる可能性があり、これは私たちの業務、運営結果、将来性、または財務状況を損なう可能性がある。
現在の候補製品以外に候補製品を発見する努力は成功しない可能性があり,臨床開発のための任意の候補製品 が本格的に臨床試験を開始しない可能性があることを推奨する。
私たち は既存の核心資産ルートを拡大するつもりだ。しかし、新しい候補製品を研究し開発する過程は高価で、時間がかかり、予測できない。私たちの現在の臨床前計画のデータは私たちの候補製品の臨床開発を支持しない可能性があり、私たちは臨床開発を推薦するのに適した他の製品を確定しないかもしれない。また,臨床開発のための任意の候補製品は,臨床前研究により安全性や潜在的な治療効果を示す兆候がない可能性が推奨され,臨床試験への参加を支援している。このような発見は,我々の臨床開発チャネルを維持または拡大する能力を阻害する可能性がある。我々が新たな候補製品を開発し,臨床開発を進める能力は,研究開発運営に資金を提供するbr能力にも依存しており,受け入れ可能なbr条項で追加資金を得るかどうか,あるいは全くないかどうかは決定できない。
FDA、EMAと外国機関のような監督管理審査過程は冗長で、時間がかかり、本質的に予測できず、 もし私たちが最終的に監督部門の私たちの候補製品に対する承認を得られなければ、私たちの業務は深刻な損害を受けるだろう。
著者らの候補製品の臨床開発、製造、ラベル、記憶、記録保存、広告、販売促進、輸入、輸出、マーケティングと流通はすべてアメリカFDAと国外市場の外国監督管理機関のような広範な監督管理を受けており、例えばヨーロッパのEMAである。アメリカでは、FDAからBLAまたはNDAの承認を受けるまで、米国で私たちの候補製品を販売することは許可されていません。このような監督管理の承認を得る過程はコストが高く、通常臨床試験開始後に何年も必要であり、しかも関連する候補製品のタイプ、複雑性と意外性及び目標適応と患者群によって大きく異なる可能性がある。承認政策や法規は変化する可能性があり、FDA、EMA、および類似の規制機関は、様々な理由で承認候補製品を延期、制限、または拒否することができることを含む、承認過程においてかなりの自由裁量権を持っている。候補製品の臨床開発に大量の時間と費用を投入したにもかかわらず、候補製品が監督管理部門の許可を得ることは常に保証されていない。大量に開発されている薬物のうち、一部だけがFDA、EMAなどの規制承認手続きに成功し、商業化されている。
米国または海外で候補製品を商業化する承認を得る前に、私たちは、良好な臨床試験を十分かつ制御する十分な証拠によって、FDA、EMAまたは同様の外国の規制機関に、brのような候補製品がその予期される用途に対して安全かつ有効であり、生物製品に対して安全で純粋で有効であることを満足させなければならない。非臨床研究と臨床試験の結果は異なる方法で解釈できる。既存の非臨床的または臨床的データが、我々の候補製品の安全性、純度、効力または有効性を支持していると考えても、これらのデータは、FDAおよび同様の外国規制機関の承認を得るのに不十分である可能性がある。FDA、EMA、または同様の外国の規制機関は、承認前または承認後に、私たちの候補製品に対して追加の臨床前研究または臨床試験を行うこと、または私たちの臨床開発計画の要素に反対することを要求する可能性もある。
| 32 |
FDA、EMA、または同様の外国の規制機関は、様々な理由で承認候補製品を延期、制限、または拒否することができる
| ● | このような機関は私たちの臨床試験の設計や実行に同意しないかもしれません | |
| ● | 我々の臨床試験または結果の陰性または不明確な結果は、FDA、EMAまたは同様の外国の規制機関によって承認された統計的意味レベルに適合しない可能性がある | |
| ● | 私たちの臨床試験の参加者または私たちの候補品に類似した薬剤を使用する個人は、深刻なbrおよび予期しない薬物関連副作用を経験するかもしれない | |
| ● | 臨床試験で研究されている人群は十分ではないか、あるいは十分な代表性を持っている可能性があり、私たちが承認を求めるすべての群の安全性を確保することができない | |
| ● | このような当局は、臨床機関または看護基準が自国とは異なる国で行われる可能性のある試験の臨床データを受け入れない可能性がある | |
| ● | 候補製品の臨床的および他の利益がその安全リスクよりも大きいことを証明できないかもしれない | |
| ● | このような機関は前臨床研究や臨床試験データの説明に同意しないかもしれません | |
| ● | このようなbr当局は、私たちの候補製品の臨床試験から収集されたデータが受け入れ可能であるか、またはBLA、NDAまたは他の提出の提出を支持するのに十分であるか、または米国または他の場所の規制承認を得ることに同意しない可能性があり、そのような当局は、追加の臨床前研究または臨床試験を要求する可能性がある | |
| ● | このような権威機関は、私たちの候補製品の配合、ラベル、および/または製品仕様の点で私たちの意見と食い違うかもしれない | |
| ● | 承認 は、私たちが求めているものよりも明らかに限られた適応にしか適用できず、および/または、配布および使用の重大な制限を含む可能性がある; | |
| ● | このようなbr当局は、私たちと臨床および商業用品契約を締結した第三者製造業者の製造プロセスまたは施設に欠陥があることを発見する可能性がある | |
| ● | このような 当局は提出された内容やフォーマットなどの理由で提出を受け入れない可能性がある. |
外国市場については,承認手続きは国/地域によって異なり,上記のリスクに加えて,追加の 製品テスト,行政審査期間,価格主管部門との合意に及ぶ可能性がある。我々が最終的に臨床試験 を完了し、BLA NDAまたは同様の海外マーケティング申請の承認を得ても、FDAまたは同様の外国規制機関は、高価な追加臨床試験のパフォーマンスおよび/またはREMSの実施 によって承認される可能性があり、FDAが承認後の製品の安全な使用を確保する必要があると考えているからである。適用可能な規制承認を得るか得ることができない点でのいかなる遅延 も、候補製品の商業化 を延期または阻止し、私たちの業務および将来性に重大な悪影響を及ぼすであろう。
| 33 |
たとえ私たちの候補製品が米国でFDAの承認を得ても、私たちはいかなる他の管轄区域でも承認されたり、このような 候補製品を商業化することができない可能性があり、これは私たちがそのすべての市場潜在力を達成する能力を制限するだろう。
任意の特定の管轄区域で任意の製品を販売するためには、様々な国/地域で多くの異なる安全および有効性規制要件 を確立し、遵守しなければならない。米国でFDAの承認を得ることは、他の国/地域または管轄区域の規制機関の承認を得ることを保証することはできない。しかし、一つの管轄区域で承認されなかったことは、私たちが他の場所で承認を得る能力に悪影響を及ぼすかもしれない。また、ある国で行われた臨床試験は、他の国の規制機関によって受け入れられない可能性があり、1つの国の規制承認は、他のどの国の規制承認も保証されていない。
承認の流れは国/地域によって異なり、追加の製品テストと検証、および追加の行政審査 周期に関連する可能性があります。外国の監督管理機関の承認を求めることは私たちに困難とコストをもたらす可能性があり、追加の臨床前研究或いは臨床試験が必要であり、これは高価で時間がかかるかもしれない。規制要求は国/地域によって異なる可能性があり、私たちの製品がこれらの国/地域で発売されることを延期または阻止する可能性がある。私たちはどの司法管轄区域(国際市場を含む) で販売された候補製品も承認されておらず、国際市場で規制承認を受けた経験もない。もし私たちが国際市場の規制要求を遵守できなかったり、必要な承認を得られず、あるいはbr}国際市場の規制承認が延期されれば、私たちの目標市場は減少し、私たちが開発した任意の製品のすべての市場潜在力を達成する能力は実現できないだろう。
FDAおよび他の政府機関の資金不足や世界的な健康問題による中断brは、彼らの重要な指導部および他の人員の採用、保持、または配置の能力を阻害する可能性があり、新しいまたは修正された製品がタイムリーまたは根本的に開発、審査、承認または商業化できないようにし、これは私たちの業務に負の影響を与える可能性がある。
FDAと外国の監督管理機関が新製品を審査·承認する能力は、br政府予算と資金レベル、法定、規制と政策の変化、FDAまたは外国の監督管理機関のキーパーソンの雇用と保留、およびユーザー費用の支払いを受ける能力、およびFDAまたは外国の監督管理機関が通常の機能を履行する能力に影響を与える可能性のある他の事件を含む様々な要素の影響を受ける可能性がある。したがって、FDAと外国規制機関の平均審査期間はここ数年で変動している。また,研究や開発活動を援助する他の政府機関への政府の援助は政治プロセスの影響を受けており,政治プロセス自体が不安定で予測不可能である.
FDAおよび他の機関(例えば、アムステルダム移転後のEMA)の中断は、新薬、生物製品、または承認された医薬品および生物製品の修正に必要な政府機関による審査および/または承認に要する時間 を遅らせる可能性もあり、これは私たちの業務に悪影響を及ぼすであろう。例えば、ここ数年間、米国政府は何度か閉鎖されており、FDAのようないくつかの規制機関は、FDAのキー従業員を休暇にし、キー活動を停止しなければならない。
また、brは全世界の新冠肺炎疫病に対応するため、食品薬品監督管理局は異なる場所に位置する国内外の製造施設の大部分の検査を延期した。FDAはすでに実行可能な情況下で国内施設に対する標準検査操作を回復したが、絶えず変化する新冠肺炎疫病に適応する時、FDAは依然としてその検査活動の変化を監視と実施し、その従業員と監督された会社の安全を確保し、いかなるウイルスの灰再発或いは新変種の出現は更なる検査 遅延を招く可能性がある。新型肺炎の流行に対して、アメリカ以外の規制機関は似たような政策措置をとる可能性がある。政府の長時間の停止が発生した場合、または世界的な健康問題がFDAや他の規制機関の定期的な検査、審査、または他の規制活動を阻害し続ける場合、FDAまたは他の規制機関が私たちが提出した規制文書を適時に審査して処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に大きな悪影響を及ぼす可能性がある。
| 34 |
もし私たちがどんな候補製品の規制承認を得ても、私たちは持続的な規制義務と持続的な規制の審査を受けることになり、これは多くの追加費用を招く可能性がある。
私たちが入手可能な任意の候補製品の規制承認は、候補製品の安全性および有効性を監視するために、規制機関に報告書を提出し、監視することを要求し、特定の年齢層の使用制限、警告、予防措置または禁忌症に関連する重大な制限を含む可能性があり、重い承認後の研究またはリスク管理要求を含む可能性がある。例えば、FDAは、私たちの候補製品を承認するためにREMSを必要とすることができ、これは、brの配布方法、患者登録、および他のリスク最小化ツールのような安全な使用を保証する他の要素、例えば、br薬物ガイドライン、医師トレーニングおよびコミュニケーション計画、または安全な使用を保証する他の要素を必要とする可能性がある。さらに、FDAまたは外国規制機関が私たちの候補製品を承認した場合、私たちの候補製品の製造プロセス、ラベル、包装、流通、有害事象報告、貯蔵、広告、販売促進、輸入、輸出、記録保存は広範で持続的な規制要求を受けるだろう。これらの要求 は安全性と他の発売後の情報と報告、登録および著者らが行う可能性のある任意の臨床試験のcGMPとGCPに対する持続的なコンプライアンスを含む。また,医薬品メーカーとその施設はFDAや他の規制機関の持続的な審査と定期的な抜き打ち検査を受け,cGMP法規や基準 に適合することを確保しなければならない。もし私たちまたは監督機関がある製品に以前に知られていない問題、例えば予想されていないbrの重症度または頻度の不良事件、またはその製品の製造施設に問題があることを発見した場合、監督管理機関は、リコールを要求するか、または市場から製品を撤回するか、または生産を一時停止することを含む、製品、製造施設、または私たちに制限を加えることができる。また、FDAや他の類似した外国法規の要求を守らなければ、わが社は行政または司法制裁を受ける可能性がある
| ● | 私たちの製品のマーケティングまたは製造、市場からの製品のリコール、または自発的または強制リコールの制限 | |
| ● | 製品の流通或いは使用の制限、或いは発売後の研究或いは臨床試験の要求を行う | |
| ● | 罰金、賠償、利益または収入の返還、警告状、無見出し状、または臨床試験の一時停止 | |
| ● | FDAは、私たちが提出した係属中の出願または承認された出願を承認することを拒否するか、または承認を一時停止または撤回すること; | |
| ● | 製品brは、私たちの製品の輸出入を許可することを拒否したり、差し押さえたり、差し押さえたり、または許可することを拒否します | |
| ● | 禁止されたり、民事または刑事処罰が適用される。 |
上記のいずれかのイベントや罰が発生すると,候補製品を商業化して収入を創出する能力 を抑制する可能性があり,対応するために多くの時間と資源が必要となり,負の宣伝が生じる可能性がある.
FDAおよび他の規制機関の政策は変わる可能性があり、私たちが開発した任意の候補製品のマーケティング許可を阻止、制限、または延期するための追加の政府法規が公布されるかもしれない。私たちはまた、アメリカや海外の将来の立法や行政行動によって生じる可能性のある政府の規制の可能性、性質、程度を予測することができません。もし私たちが既存の要求の変化や新しい要求や政策の採用に緩やかに適応できない場合、あるいは規制適合性を維持できない場合、私たちは法執行行動の影響を受ける可能性があり、利益を達成したり維持することができないかもしれません。
FDAと他の規制機関は非ラベル用途の普及を禁止する法律法規を積極的に実行している。
FDAは処方薬のマーケティング、ラベル、広告、販売促進を厳格に管理している。これらの規定には、消費者向け広告、業界スポンサーの科学的および教育活動、インターネットに関連する販売促進活動、ラベル外販売促進の基準および制限が含まれる。FDAが付与した任意の規制承認は,FDAが安全かつ有効であると考えられる特定の疾患と適応に限られている。米国の医師は、製品ラベルに記載されていない用途および臨床試験で試験され、監督機関によって承認された用途とは異なる用途を選択し、一般的に処方することができるが、任意の製品を普及させる能力は、FDAによって特に承認された適応brに限定される。
| 35 |
もし私たちがこのようなラベルの外での使用を普及させることが発見されたら、私たちは重大な責任を負うかもしれない。米国連邦政府はすでにラベル外使用の不当な普及の疑いがある会社に対して巨額の民事と刑事罰金を科し、いくつかの会社がラベル外普及に従事することを禁止している。FDAはまた、規定された販売促進行為によって変更または制限された場合に、同意法令または永久禁止を締結することを要求する。もし私たちがどんな候補製品の販売促進活動もうまく管理できなければ、もし が承認されれば、私たちは重大な責任を負うかもしれません。これは私たちの業務や財務状況に大きな悪影響を与えます。
我々の第三者依存に関するリスク
私たちの候補製品の商業成功は医者、患者、医療支払者と医学界におけるそれらの市場受容度にかかっている。
私たちの候補製品が規制部門の承認を得ても、私たちの製品(あれば)は医師、患者、医療支払者、医療界の市場に認められない可能性があります。私たちが承認した任意の候補製品の市場受容度は、いくつかの要因に依存する
| ● | 現在利用可能な製品価格と比較して、私たちが承認した候補製品の有効性 | |
| ● | 患者が現在の治療法の代わりに承認された候補製品を採用したいかどうか | |
| ● | 私たちは許容可能な安全性と有効性の証拠を提供する能力を提供する | |
| ● | 相対 管理顎の利便性と簡単さ | |
| ● | どんな不良副作用の罹患率と重症度も | |
| ● | 他の製品と組み合わせて使用する制限 | |
| ● | 代替治療薬の可用性 | |
| ● | 私たちの候補製品と目標市場の概要に基づいて、競争力あるいは潜在的なプレミアム要求があると仮定して、定価と費用効果を行います | |
| ● | 私たちまたは私たちのパートナーの販売とマーケティング戦略の有効性 | |
| ● | 私たちは十分な第三者保険や精算和を得ることができます | |
| ● | 潜在的な 製品責任クレーム。 |
しかも、私たちの候補製品の潜在的な市場機会を正確に推定することは難しい。私たちの業界知識、業界出版物、第三者研究報告、その他の調査によると、候補製品の潜在的な市場機会の推定にはいくつかの重要な仮定が含まれている。独立したメッセージ源は私たちのすべての仮定を証明しなかった。もしこれらの仮定のいずれかが不正確であることが証明された場合、私たちの候補製品の実際の市場は、潜在的な市場機会の推定値よりも小さくなる可能性がある。もし私たちの候補製品の実際の市場が私たちが予想していたより小さいなら、私たちの製品の収入は制限されるかもしれません。資金を集めることは予想より難しいかもしれません。私たちは利益を達成したり維持したりすることが難しいかもしれません。もし私たちがアメリカと海外で市場の候補製品の受け入れを実現できなければ、私たちの収入は制限され、 利益を実現することはもっと困難になるだろう。
| 36 |
著者らは第三者に依存して臨床前研究と臨床試験を行い、他の任務を実行した。もしこれらの第三者がその契約責任を成功裏に履行し、予想された期限内に規制要求を達成または遵守できない場合、私たちは規制部門の私たちの候補製品の承認を得ることができないか、またはそれを商業化することができず、私たちの業務、財務状況、および運営結果は深刻なbr損害を受ける可能性がある。
我々のbrは第三者CRO、医療機関、臨床研究者、契約実験室に依存して、私たちが行っている臨床前と臨床プロジェクトのデータを監視し、管理している。これらのCRO、調査者、その他の第三者はこれらの実験の進行とスケジュール、その後のデータ収集と分析に重要な役割を果たしている。このようなCRO、調査者、その他の第三者との関係を慎重に処理したいが、将来的に挑戦や遅延に遭遇しない保証はなく、これらの遅延や挑戦が私たちの業務、財務状況、および見通しに実質的な悪影響を与えないことを保証することはできない。また,我々は我々の第三者請負業者の活動を規範化する合意に達しているが,彼らの実際の表現への影響は限られている.それにもかかわらず、私たちはすべての臨床試験と臨床前研究が適用された方案、法律、法規と科学標準に基づいて行われていることを保証する責任があり、私たちはこれらの第三者への依存が私たちの監督責任を免除しない。私たちおよび私たちのCROおよび他のサプライヤーは、cGMPまたは同様の海外要件、GCPおよび良好な実験室仕様またはGLPの要件を遵守しなければならない。これらの要件は、FDA、EMAおよび同様の外国当局が私たちの臨床開発におけるすべての候補製品に対して実行する法律および法規のセットである。監督管理機関 は定期的に臨床前研究と臨床試験スポンサー、主要な研究者、臨床前研究と臨床試験場及びその他の請負業者を検査することによってこれらの規定を実行する。もし私たちまたは私たちの任意のCROまたはサプライヤーが適用された法規を遵守できなかった場合、私たちの臨床前研究および臨床試験で生成されたデータは信頼できないと考えられるかもしれません。FDA、EMAまたは同様の外国機関は、私たちのマーケティング申請を承認する前に追加の臨床前研究と臨床試験を行うことを要求するかもしれません。私たちはあなたに保証することはできません。特定の規制機関が検査を行った後、この監督機関は私たちの任意の臨床 試験がGCP法規に適合していることを確認します。また,われわれの臨床試験はcGMP法規,brなどの海外要求に適合した製品を用いて行わなければならない。これらの規定を遵守しなければ、臨床試験を繰り返す必要があるかもしれませんが、開発と規制承認の流れを遅らせることになります。
私たち はビジネス的に合理的な条項でCROと合意できないかもしれないし、全然できないかもしれません。また,我々のCROは我々のbr従業員ではなく,このようなCROとの合意によって我々に提供された救済措置を除いて,我々が行っている臨床前および臨床計画に十分な時間と資源を投入するかどうかを制御できないであろう。CROがそのbr契約の義務または義務を成功裏に履行できなかった場合、または予想された期限内に達成できなかった場合、もし彼らが交換する必要がある場合、または彼らが得たデータの品質または正確性が私たちの合意、法規の要求または他の理由を遵守できなかった場合、私たちの臨床試験は延長、延期、または終了される可能性があり、私たちは規制部門の私たちの候補製品の承認または商業化に成功することができないかもしれない。CROはまた予想よりも高いコストを発生させる可能性がある。したがって、私たちの業務、財務状況、運営結果 および私たちの候補製品のビジネス見通しは重大な悪影響を受ける可能性があり、私たちのコストが増加する可能性があり、私たちの収益能力が遅れる可能性があります。
さらに、私たちの臨床試験の首席研究者は、時々私たちの科学顧問やコンサルタントを担当することを要求され、このようなサービスによって現金または株式補償を得ることができるかもしれない。これらの関係が任意の関連賠償 と知覚的または実際の利益の衝突をもたらす場合、またはFDAは、財務関係が研究の解釈に影響を及ぼす可能性があると結論した場合、適用される臨床試験場所で生成されたデータの完全性が問われる可能性があり、臨床試験自体の効用が脅かされる可能性があり、これは、FDAが私たちが提出した任意のBLAまたはNDAを延期または拒否する可能性がある。このような遅延や拒否は私たちが候補製品を商業化することを防ぐことができる。
また,未治癒の重大な違約や他の指定されたbrが発生した場合,我々のCROは我々との合意を終了する権利がある。もし私たちがこれらの第三者との任意の関係が終わったら、私たちは商業的に合理的な条項や代替の第三者と合意できないかもしれない。追加のCRO、医療機関、臨床研究者、または契約実験室は追加コストを交換または増加させ、管理時間および重点を必要とする。また,新しいCROが以前のCROの置換を開始すると,自然な過渡期 がある.したがって,遅延が生じ,必要な臨床開発スケジュールを満たす能力に大きな影響を与える可能性がある。私たちが未来に類似した挑戦や遅延に遭遇しないことは保証されず、これらの遅延または挑戦が私たちの業務、財務状況、または運営結果に実質的な悪影響を与えない保証はない。
| 37 |
我々brは現在,臨床開発中に第三者に依存して我々の候補製品を生産しており,予測可能な未来には引き続き第三者に依存していると予想される。このような第三者への依存は、私たちが十分な数の候補製品を得ることができないか、または許容可能なコストでこのような数を得ることができないリスクを増加させ、これは、私たちの開発 または潜在的な商業化努力を延期、阻止、または損害する可能性がある。
我々 は製造施設を所有または運営しておらず,現在のところ自分の臨床や商業規模製造能力を開発する計画 もない。もし私たちの候補製品が規制された場合、私たちは引き続き第三者に依存して私たちの候補製品を生産し、臨床開発と商業生産のための関連原材料に依存すると予想されます。私たちの第三者メーカーが私たちの候補製品を生産するための施設は、FDA、EMA、または任意の同様の外国規制機関の承認を得なければなりません。私たちがFDAに機密協定やBLAを提出したり、外国の監督管理機関に同様のマーケティング申請を提出した後に行われる検査に基づいています。我々は第三者メーカーの製造過程を制御せず、第三者メーカーに完全に依存して我々の候補製品を生産するcGMP要求を満たす。これらの第三者メーカーが、我々の規格およびFDAまたは外国規制機関のような厳格な規制要件に適合する材料の生産に成功しない場合、彼らは、その製造施設の使用の規制承認を確保および/または維持することができないであろう。
また、第三者メーカーが十分な品質管理、品質保証、合格者の能力を維持することを制御することはできない。FDA、EMA、または任意の同様の外国規制機関がこれらの施設を承認して私たちの候補製品を生産しない場合、またはこれらの機関が将来このような承認を撤回すれば、代替製造施設を探す必要があるかもしれません。これは、承認されれば、規制機関の承認を得たり、私たちの候補製品をマーケティングする能力に深刻な影響を与えます。私たちまたは私たちの第三者製造業者が適用された法規に従わないことは、臨床棚上げ、罰金、禁止、民事処罰、遅延、一時停止または承認撤回、差し押さえまたはリコール、運営制限、および刑事起訴を含む制裁を実施する可能性があり、いずれも私たちの財務状況に重大な悪影響を及ぼす可能性があります。
私たちまたは第三者が商業的に合理的な条項およびcGMPまたは他の法規要件に従って私たちの製造要件を実行できない場合、様々な態様で私たちの業務に悪影響を及ぼす可能性があります
| ● | 候補製品の臨床試験をタイムリーに開始または完了することができません | |
| ● | 私たちの候補製品のための監督申請を提出したり、監督管理の承認を得たりする遅延 ; | |
| ● | 第三者製造施設に規制機関の追加検査を受けさせる | |
| ● | 私たちの候補製品の開発を停止またはリコールすることを要求します | |
| ● | いずれの候補製品も上場や商業化が承認された場合、ビジネスニーズを満たすことができない。 |
しかも、私たちはどの第三者製造業者とも長期的な約束や供給協定を締結していない。私たちは、第三者製造業者と任意の長期供給プロトコルを確立することができないか、または許容可能な条項でそうすることができない可能性があり、これは、許容可能なコストで十分な数の候補製品またはそのような数をタイムリーに得ることができないリスクを増加させる。たとえ第三者製造業者とbrプロトコルを確立することができても、第三者メーカーに依存することは、他のリスクをもたらす
| ● | 第三者製造業者は規制要件を遵守し、品質保証を維持できなかった | |
| ● | 第三者は製造協定に違反した | |
| ● | 私たちの規格に基づいて私たちの候補製品を製造できませんでした | |
| ● | 私たちの計画通りに私たちの製品を生産していない、あるいは全然ありません | |
| ● | 私たちのビジネス秘密とノウハウを含めて、私たちの固有の情報を流用します | |
| ● | 第三者は、費用が高い場合、または私たちに不便をもたらした場合、プロトコルを終了または更新しない。 |
| 38 |
私たちの既存または未来の製造業者の任意の性能障害は、臨床開発または上場承認を延期する可能性があり、任意の関連する修復措置が実施されるには、コストが高いか、または時間がかかる可能性がある。私たちは現在私たちの候補製品を製造するために必要なすべての原材料のために余分なbr供給または第2の供給源を配置していません。もし私たちの既存または未来の第三者製造業者が約束通りに動作できなければ、私たちはこれらのメーカーの交換を要求されるかもしれませんし、私たちはそれらをタイムリーにあるいは交換できないかもしれません。これは私たちの財務状況に大きな悪影響を及ぼすでしょう。
私たちが将来達成する可能性のあるどんな協力計画も成功しないかもしれません。これは、現在および潜在的な将来の候補製品を開発し、それを商業化する能力に悪影響を及ぼすかもしれません。
私たちは、私たちの現在と潜在的な未来の候補製品を開発したり、それを商業化したりするために、生物製薬会社との協力手配を求めるかもしれない。我々が連携協定を締結することを決定すれば,適切なパートナーを探すうえで激しい競争 に直面する.さらに、協力スケジュールは複雑で、交渉、実行、実施に時間がかかります。 私たちがこのような計画を達成することを選択すれば、私たちが協力または他の代替手配を確立して実施する努力は成功しないかもしれませんし、手配された条項は私たちに不利になるかもしれません。第三者 と協力して候補製品を開発して商業化すれば,その候補製品の将来成功した制御権の一部または全部を第三者に譲ることが予想される.私たちの協力計画が成功するかどうかは私たちの協力者の努力と活動に大きく依存するだろう。協力者は通常、彼らがこれらの協力の仕事と資源に適用されることを決定する上で大きな裁量権を持っている。
連携手配当事者間の分岐 は,適用候補製品の開発や商業化遅延を招く可能性があり, は互恵的に解決することは困難である.場合によっては、生物製薬会社および他のbrの第三者との協力は、他方によって終了または終了される。このような終了または満了は、私たちの業務、財務状況、および運営結果に悪影響を及ぼすだろう。
もし私たちが自分の商業組織を発展させたり、第三者と合意して私たちの候補製品を販売して普及させることができなければ、私たちは相当な収入を生み出すことができないかもしれない。
私たちは販売とマーケティング組織がありません。会社として、私たちは医薬 製品を販売、マーケティング、流通した経験がありません。もし私たちの任意の候補製品が商業化を許可されれば、私たちは私たちの販売、マーケティング、流通能力を発展させること、または第三者と販売およびマーケティングサービスの実行を手配することを要求されるかもしれない。私たちのすべての候補製品のために販売チームを育成して、高価で時間がかかり、いかなる製品の発表を延期するかもしれません。私たちはタイムリーまたは経済的に効率的な方法で効果的な販売チームを構築して管理することができないかもしれません(もしあれば)、私たちが構築したどの販売チームも私たちの候補製品に十分な需要を与えることができないかもしれません。もし私たちが協力者または他の第三者と合意して販売およびマーケティングサービスを実行すれば、私たちの製品収入は私たちが単独で販売し、候補製品を販売する場合よりも低いかもしれません。もし私たちが独立したり、他社と十分な販売とマーケティング能力を確立できなければ、相当な収入を生み出すことができないかもしれないし、利益を上げることができないかもしれない。
商業化に関するリスク
Decoy 20または任意の将来の候補製品の商業化に成功する(承認された場合)は、政府当局および健康保険会社が保険を確立する程度、十分な精算レベル、および割引された価格設定政策にある程度依存する。もし私たちの製品がカバー範囲を維持し、十分な精算を得ることができなかった場合、これらの製品をマーケティングする能力を制限し、私たちの収益能力を低下させるかもしれません。
医療保険や医療補助などの政府医療計画,個人健康保険会社,他の第三者支払者が提供する保険範囲や精算は,多くの患者にとって重要であり,Decoy 20などの処方薬や任意の未来の候補品(承認されれば)を負担することができる。第三者支払人が私たちの製品のためにカバー範囲と受け入れ可能な精算レベルを実現できるかどうかは、これらの製品を商業化することに成功する能力に影響を与えます。したがって、私たちは任意の承認された候補製品のカバーと精算戦略を成功的に実施する必要がある。第三者支払者が特定の製品の保険を獲得しても,それによる精算支払率が十分高くない場合や,患者が受け入れられないと思う共同支払いが必要となる可能性がある。
| 39 |
もし私たちが医療補助薬品返却計画や他の政府定価計画に参加すれば、場合によっては、私たちの製品はこのような計画によって設定された最高価格によって制限され、これはどのような製品から得られる収入を減少させるかもしれない。このような計画に参加することはまた、重大な民事罰金、制裁、罰金のリスクに直面させ、もし私たちがbrがその計画下のいかなる適用義務にも違反していることが発見されたら。
医師の監督下で管理されている製品では,このような薬剤は通常価格が高いため,保険や適切な精算を得ることは特に困難である可能性がある。また,製品自体やその製品を用いた治療やプログラムは単独での精算が得られない可能性があり,医師の利用率に影響を与える可能性がある。私たちが開発する可能性のあるすべての製品がアメリカ、EU、あるいは他の場所での保険範囲と精算範囲が許容できるレベルに達していることを保証することはできず、将来的には任意の入手可能な精算を減少またはキャンセルする可能性がある。
第三者決済者は生物製薬製品やサービスの価格に挑戦することが増えており、多くの第三者支払者は、同等の後発薬やより安い治療法がある場合に特定の薬に保険と精算を提供することを拒否する可能性がある。br}第三者支払人は、私たちの製品が代替可能であると考え、患者のために安い製品を精算するだけを提案するかもしれない。たとえ私たちの製品が治療効果を高めたり、投与の利便性を改善したことを証明することに成功しても、brの既存の薬物の定価は私たちの製品に対する料金を制限するかもしれない。これらの支払者は、特定の製品の精算状態を拒否またはキャンセルしたり、新製品や既存市場製品の価格を低すぎるレベルに設定したりして、製品開発投資の適切なリターンを実現できない可能性がある。精算が得られない場合や限定精算だけでは、私たちの製品の商業化に成功できないかもしれませんし、私たちが開発する可能性のある製品から満足な財務リターンを得ることができないかもしれません。
新承認製品の第三者支払者カバー範囲と精算に関する重大な不確実性。アメリカでは、 第三者支払人、個人と政府支払人、例えばMedicareとMedicaid計画を含み、新薬の保証範囲を確定する上で重要な役割を果たしている。一部の第三者支払者は、新しいまたは革新的なbr装置または薬物療法の保証範囲を事前に承認してから、このような治療法を使用する医療提供者に精算する必要があるかもしれない。第三者決済者がDecoy 20と任意の未来の候補製品の保証と精算についてどのような決定を下すかを予測することは難しい。
を取得して精算状態を維持するのに時間がかかり、コストが高く、確定していない。連邦医療保険や医療補助計画は,個人支払者や他の政府支払者がどのように薬品保険や精算政策を策定するかのモデルとして利用されるようになってきている。しかし,米国の第三者支払者の間には統一された製品保証や精算政策はない。そのため、製品の保証範囲 と精算金額は支払人によって異なる可能性があります。したがって、保証範囲決定過程は通常、時間がかかり、コストの高いプロセスであり、保証範囲と十分な精算が一貫的に適用または最初に得られることを保証することはできない。また、精算に関する規則や法規は常に変化し、場合によっては短時間で通知され、これらの規則や法規は変更される可能性があると考えられる。
米国以外では、国際運営は一般的に広範な政府価格制御と他の市場によって規制されており、欧州や他の国のコスト制御措置はますます重視されており、これらの司法管轄区域で承認されれば、候補製品の価格設定と使用に圧力を与え続けると考えられる。多くの国では、国家衛生システムの一部として、医療製品の価格は異なる価格制御メカニズムの制約を受けている。他の国は会社が自分の医療製品の価格を固定することを許可しているが、会社の利益を監視し、コントロールしている。追加の外国価格規制や定価法規の他の変化は、私たちが製品に対して受け取ることができる費用を制限するかもしれません。したがって,米国以外の市場(あれば)では,我々の製品の精算金額は米国を下回る可能性があり,商業的に合理的なbr収入や利益を生み出すには不十分である可能性がある。
| 40 |
また、米国や海外の政府や第三者支払者が医療コストの制限や低減を強化することは、新たに承認された製品の保証範囲や精算レベルを制限する可能性があるため、brを保証したり、私たちの製品に十分な支払いを提供できない可能性がある。私たちは私たちのどの製品を販売する時も定価の圧力に直面することが予想されます。 管理式医療の傾向、健康維持組織の影響力の増加、その他の立法改正により。 全体の医療コスト、特に処方薬、手術手順、その他の治療の下振れ圧力は非常に大きい。 そのため、新製品の参入にはますます高い壁が設けられている。
最近公布された立法、将来の立法、および医療改革措置は、Decoy 20および将来の候補製品のマーケティング承認を得て、それを商業化することの難しさとコストを増加させ、設定可能な価格に影響を与える可能性があります。
米国や一部の外国司法管轄地域では、医療システムは、コスト制御措置を含む複数の立法·規制改革を継続しており、これらの措置は、新薬の保証範囲を減少または制限し、上場承認された候補製品を収益的に販売する能力に影響を与える可能性がある。具体的には,米国連邦と州政府は複数の取り組みを継続し,医療コストの低減と医療の質の向上を求めている。
例えば、2010年3月、ACAがアメリカで公布された。ACAは指定ブランドの処方薬や生物製剤を製造または輸入する任意の実体brに対して控除できない年間費用を規定している;メーカーの医療補助リベート責任を連邦医療補助管理に参加する医療機関に割り当てられた個人の保険薬品に拡大した;医療補助計画の資格基準brを拡大した;340 B薬品定価計画に基づいて割引を受ける資格がある実体を拡大した;医療補助薬物リベート計画に基づいてメーカーが支払わなければならない法定最低リベートbrを高めた;患者を中心とした新しい結果研究所を設立し、優先事項を監督、確定し、臨床治療効果比較研究を行い、このような研究に資金を提供した。また はCMSに医療保険と医療補助革新センターを設立し、革新的な支払いとサービス交付モデルをテストし、医療保険 と医療補助支出を下げる。
公布以来、ACAのいくつかの方面はずっと行政、司法、国会の挑戦を受けており、2021年6月17日、アメリカ最高裁判所はいくつかの州がACAに対して提出した最新の司法挑戦を却下したが、ACAの合憲性について具体的な裁決を下しなかった。最高裁が裁決を下す前に,総裁·バイデンはACA市場による医療保険の取得を目的とした2021年2月15日から2021年8月15日までの特殊保険期間を開始する行政命令を発表している。行政命令はまた、特定の政府機関に、医療補助モデルプロジェクトおよび免除計画の再審査、および医療補助またはACAによる医療保険カバー範囲の獲得によって不必要な障害をもたらす政策を含む、医療補助モデルプロジェクトおよび免除計画の再検討、および医療補助またはACAによる医療保険のカバー範囲の獲得を制限する既存の政策および規則を検討するように指示する。医療改革措置が我々の業務にどのように影響するかは不明である。
また、“ACA”が公布されて以来、他の立法改正も提案され、採択された。2021年3月11日、“2021年米国救援計画法案”が法律に署名され、2024年1月1日から医療補助薬品還付の法定上限が撤廃され、現在の上限は薬品AMPの100%となっている。また、処方薬のコストが上昇していることを受けて、米国政府は薬品定価のやり方の審査を強化した。このような審査は最近のいくつかの国会調査を招き,製品定価の透明性の向上,br定価とメーカー患者援助計画との関係の審査,製品の政府計画精算方法の改革を目的とした連邦と州立法を提案·公布した。最近、“2022年インフレ低減法案”(IRA)には多くの重大な薬品定価改革が含まれており、その中で はアメリカ衛生·公衆サービス部(HHS)内に薬品価格交渉計画(2026年から )を確立し、メーカーにいくつかの選択された薬物に対する交渉後の“最高公平価格”を要求し、規定を守らないために消費税 を支払うことを要求し、連邦医療保険BとD部分のメーカーに対して税金還付要求を制定してインフレを超えたbrを懲罰することを要求し、D部分の福祉を再設計する。その一部として,メーカーはD部分薬の割引(2025年から)の提供を求められている。アイルランド共和軍は衛生·公衆サービス部長官が最初の数年に規制ではなく指導によってその多くの規定を実施することを許可した。未来の立法でもっと多くの薬品価格設定提案が現れるかもしれない。また、br政府はより多くの行動を取って新冠肺炎疫病に対応する可能性がある。
| 41 |
州レベルでは、立法機関は、価格や精算制限、割引、ある製品の参入およびマーケティングの制限、他の国/地域からの輸入と大量調達を奨励することを目的とした立法および生物製品の価格設定を制御するための法規を立法し、実施している。場合によっては、他の国/地域からの輸入と大量調達を奨励することが目的である。第三者支払者の支払い金額に対する法的強制価格制御または他の制限は、私たちの業務、運営結果、brの財務状況と将来性を損なう可能性がある。また,地域医療当局や個別病院では,どの薬品やサプライヤーがその処方薬や他の医療計画に含まれるかを決定するために入札プログラムが使用されるようになってきている。承認された場合、これはDecoy 20および任意の未来の候補製品の最終需要を低下させるか、または私たちの製品価格に圧力を与える可能性があり、これは私たちの業務、運営結果、財務状況、および見通しに悪影響を及ぼす可能性がある。
私たち は、将来採用される可能性のあるこれらの新しい法律および他の医療改革措置が、連邦医療保険および他の医療保健資金のさらなる削減、より厳しいカバー基準、新しい支払い方法、および私たちが受け取った任意の承認製品の価格追加の下振れ圧力をもたらす可能性があると予想しています。連邦医療保険や他の政府が計画している精算のいかなる減少も、個人支払者の支払いの同様の減少を招く可能性がある。コスト制御または他の医療改革を実施することは、承認されれば、収入を創出し、利益を達成するか、またはDecoy 20および任意の将来の候補製品を商業化することを阻止する可能性がある。
製品 の私たちに対する責任訴訟は、私たちが大量の責任を負うことを招き、私たちが開発する可能性のある任意の候補製品の商業化を制限する可能性があります。
私たちのbrは、人体臨床試験で私たちの候補製品をテストすることに関連する固有の製品責任リスクに直面し、私たちが開発可能な任意の候補製品を商業的に販売すれば、 はより大きなリスクに直面する。もし私たちが私たちの候補製品にダメージを与えたクレームを自己弁護することに成功できなければ、私たちは重大な責任を招くかもしれない。是非曲直や最終結果にかかわらず、責任クレームは次のようになる可能性がある
| ● | 私たちが開発する可能性のある候補製品の需要を減らしました | |
| ● | 私たちの名声とメディアの大きな否定的な関心を損なう | |
| ● | 高価なリコールや製品修正の規制調査が必要になるかもしれない | |
| ● | 臨床試験参加者は脱退しました | |
| ● | 関連訴訟を弁護するための巨額の費用 | |
| ● | 試験参加者または患者に巨額の金銭的報酬を支給する | |
| ● | 潜在収入損失 ; | |
| ● | 経営陣の関心を私たちの業務を管理することから移すこと | |
| ● | 私たちが開発する可能性のある候補製品を商業化することはできない。 |
私たちは製品責任保険範囲を維持していますが、それは私たちが発生する可能性のあるすべての責任をカバーするのに十分ではないかもしれません。そして、賠償免除額と保証範囲によって制限されています。私たちは私たちがすべての候補製品を商業化することに成功した時、私たちの保険カバー面を増加させる必要があると予想する。保険範囲はますます高くなっています。私たちは合理的な費用または出現可能な責任を支払うのに十分な金額で保険範囲 を維持できないかもしれない。許容可能なbr費用で保険を受けることができない場合、あるいは潜在的な製品責任クレームに対して他の方法で保障を提供することができなければ、私たちは重大な責任に直面し、これは私たちの業務や財務状況に大きな影響を与える可能性があります。このような責任は私たちの商業的な努力を阻害したり妨害したりするかもしれない。
| 42 |
私たち は様々な製造リスクに直面しており、その中でどのリスクも私たちのコストを大幅に増加させ、候補製品の供給を制限する可能性があります。
私たちの候補製品を生産するプロセスは複雑で、厳格な規制を受けており、いくつかのリスクの影響を受けている。例えば、私たちの候補製品を製造するプロセス は、汚染、設備故障、または設備設置または操作の不適切、サプライヤーまたはオペレータのエラーによって製品損失を招きやすい。私たちの任意の候補製品に対して、正常な製造プロセスとの微小な偏差があっても、生産良率の低下、製品欠陥と他の供給中断を招く可能性がある。私たちの候補製品または私たちの候補製品を製造する製造施設で微生物、ウイルス、または他の汚染が発見された場合、汚染を調査および修復するために、そのような製造施設を長時間閉鎖する必要があるかもしれません。また、私たちの候補製品を製造する製造施設は、設備故障、br}労働力不足、自然災害、電力故障、および多くの他の要因の悪影響を受ける可能性があります。
さらに、私たちの候補製品の生産運営に影響を与える不利な発展は、出荷遅延、在庫不足、ロット故障、撤回またはリコール、または私たちの候補製品供給の他の中断を招く可能性があります。私たちはまた、在庫を解約し、規格に適合しない、高価な救済措置をとるか、またはよりコストの高い代替案を製造する候補製品を求めるために、他の費用および支出を発生させる必要があるかもしれない。
競争、鍵となる従業員の維持、管理成長に関するリスク
もし私たちの競争相手の候補製品が私たちの候補製品よりも早く、マーケティングがより効果的で、寛容度が良く、より有利な安全状況を持っているか、あるいは私たちの候補製品よりも有効であることが証明されたら、私たちのビジネス機会は不利な影響を受けるかもしれません。
我々が経営しているbr業界の特徴は,技術の急速な進歩,競争の激しさ,専有品への高度な重視である。私たちは私たちの技術、知識、経験と科学資源がそれに競争優位を提供すると信じているが、私たちは商業生物技術企業、学術機関、政府機関及び私営と公共研究機関を含む多くの異なる源からの潜在的な競争に直面している。我々が開発·商業化に成功したどの候補製品も,既存の免疫療法や将来発売される可能性のある新しい免疫療法と競争する。
私たちと比べ、私たちの多くの競争相手は研究開発、製造、臨床前研究、臨床試験、監督管理許可とマーケティング承認の製品の面でより多くの財務資源と専門知識を持っている。規模が小さいか早い段階にある会社も重要な競争相手になる可能性があり、特に大企業や成熟会社との協力で手配する。私たちの競争相手は、私たちが開発している任意の技術や療法よりも効果的で、耐性がより良い、またはコストの低い技術と療法の開発に成功するかもしれません、あるいは私たちの候補製品を時代遅れにして競争力がありません。たとえ私たちの候補製品が規制部門の承認を得ても、私たちの競争相手は私たちよりも早く規制部門の承認を得ることに成功するかもしれない。私たちはまたこれらの第三者からの競争に直面します:合格した科学と管理者を募集し、維持し、臨床試験場と臨床試験患者登録を確立し、そして私たちの計画と相補的あるいは私たちの業務に有利な技術と製品を獲得する。
もし私たちの各候補製品が承認された場合、その成功に影響を与える重要な競争要素は、その治療効果、安全性、耐性、投与頻度と経路、利便性と価格、ブランドと模倣薬の競争レベル、医師と患者の市場受容度、および政府と他の第三者支払者が保険と補償を提供するかどうかである可能性がある。
私たちは生物製品として承認を求めるすべての候補製品は予想よりも早く競争に直面するかもしれません
患者保護および平価医療法案は、2010年3月23日に法律に署名され、生物製品価格競争および2009年革新法案、またはBPCIAと呼ばれる副題が含まれ、FDA許可の参考生物製品と類似または交換可能な生物製品のための短い承認経路を作成した。BPCIAによると,生物類似製品の申請は,参考製品がFDA許可を初めて得た4年後にFDAに提出される。また、参照製品が初めて許可された日から12年後、FDAは生物類似製品の承認を発効させることができる。brはこの12年間の独占期間内に、FDAがスポンサー自身の臨床前データと十分かつ制御された臨床試験のデータを含み、その製品の安全性、純度、および効力を証明するために、スポンサー自身の臨床前データと十分かつ制御された臨床試験のデータを含む競合製品の完全なBLAを承認した場合、別の会社はこの参照製品の競争バージョンを販売することができる。
| 43 |
我々 は,BLAにより生物製品として承認されたいずれの我々の将来の候補製品も12年の専門期間 を得る資格があると考えている。しかしながら、国会の行動や他の理由により、この独占経営権は短縮される可能性があり、またはFDAは、私たちの候補製品を競合製品の参考製品とみなさず、予想よりも早く模造薬競争の機会を創出する可能性がある。さらに、承認されると、生物類似体がどの程度私たちのいずれかの参照製品を置換することができ、その方法は、非生物製品の伝統的な模倣薬代替に類似しており、これは、まだ発展中のいくつかの市場および規制要因に依存する。
私たちの未来の成功は私たちが肝心な幹部を維持し、合格者を吸引、維持、激励する能力にかかっている。
私たちは私たちの現在の上級管理職に高く依存している。もし私たちが既存の高度管理者や科学者を引き留めることができない場合、あるいはより多くの重要な人員を引き付けて維持することができなければ、私たちは私たちの候補製品の開発に成功したり、商業化することができないかもしれない。私たちの最高経営責任者Jeffrey A.Mecklerと私たちの最高科学官Michael J.Newman博士に高度に依存しています。私たちの成功は私たちが質の高い管理と科学者を引きつけ、維持し、激励する能力にかかっています。しかし 人材競争は激しい。私たちは私たちの現在またはbrの未来のフルタイム雇用需要を満たすために合格者を集めることに成功できないかもしれない、あるいは全くできない。もし私たちが肝心なポストを埋めることができなければ、私たちは開発会社の候補製品を含む、私たちの運営活動や目標を延期する必要があるかもしれません。上場企業としての義務を履行する上で困難に直面する可能性があります。私たちは現在私たちの従業員の誰にも“キーパーソン”保険を提供していません。
しかも、競争相手と他の会社は未来に私たちの従業員を募集しようと試みるかもしれない。私たちのいかなる重要な人員のサービスを失っても、未来の高い素質の人員を引き付けることができない、あるいはそのような人員、特に高級管理者と他の技術者の採用を遅延させることは、私たちの業務、財務状況、および運営結果に実質的な不利な影響を与える可能性がある。さらに、キーパーソンの交換は、多くの時間およびコストに関連する可能性があり、当社のビジネス目標の実現を著しく遅延させたり、阻害したりする可能性があります。私たちの経営陣は時々いくつかの科学顧問とコンサルタントの臨床と法規発展計画、その他の常規事項に対する提案と指導を求めます。これらの科学コンサルタントおよびコンサルタントは私たちの従業員ではなく、他のエンティティと約束、相談、またはコンサルティング契約を締結しているかもしれません。これらのエンティティは私たちに対する彼らの利用可能性を制限するかもしれません。また、私たちの科学コンサルタントは他社と合意し、これらの会社が私たちと競争する可能性のある製品や技術の開発に協力するかもしれません。
私たち は私たちの組織の規模を拡大する必要があり、私たちの成長を管理することに成功できないかもしれない。
私たちのbrは初期臨床段階のバイオテクノロジー会社で、従業員の数が少なく、私たちの現在の管理システム は私たちの未来の成長計画を支持するのに十分ではないかもしれない。私たちが成長を実現し、成長を効果的に管理する能力は、私たちの募集、訓練、維持、管理、そしてより多くの従業員を激励し、私たちの運営、財務と管理システム を実施し、改善することが要求される。これらの要求はまた、より多くの高級管理者を雇う必要があるか、または私たちの高級管理者によってより多くの専門知識を育成する必要があるかもしれない。多くの追加従業員、特に管理職の従業員を募集すると、私たちの支出が著しく増加するだろう。また、私たちの運営、財務、管理システムを拡大して強化することができなければ、私たちの将来の潜在的な成長と組み合わせることができれば、私たちの業務、財務状況、運営結果に実質的な悪影響を及ぼす可能性があります。
| 44 |
私たちの知的財産権に関するリスク
私たち は市場で私たちの独自の可能性を十分に保護できないかもしれない。
私たちは私たちの能力に依存して、私たちの独自の技術と製品、あるいは私たちが許可する可能性のある技術と製品を保護します。私たちは、ビジネス秘密、特許、著作権および商標法、セキュリティ、許可、および従業員および第三者との他の合意に基づいて、私たちの知的財産権を保護するつもりです。私たちの成功は、私たちの能力に大きく依存し、任意の許可者または許可者が、私たちの独自の技術および製品について米国および他の国/地域で特許保護を取得し、維持することができるかもしれない能力に大きく依存する。我々 は、将来の許可者の特許法執行活動が適用される法律法規 に適合しているかどうか、または有効かつ強制的に実行可能な特許または他の知的財産権が生成されるかどうかを決定することはできない。私たちはまた、将来の許可者 が十分なリソースを割り当てるか、または彼らまたは私たちのこのような特許を優先的に実行するかどうかを決定することができない。たとえ私たちがこれらの法的行動の側でなくても、不利な結果は、私たちの業務の運営に必要かもしれない知的財産権を許可することを阻止する可能性があり、これは私たちの業務、財務状況、運営結果に大きな悪影響を与える。
私たちは、私たち自身の技術をカバーする特許出願を起訴することによって、私たちのノウハウのために十分な特許保護を得ることができると信じている。もし、私たちが所有する可能性のある特許および将来の特許を保護または強制するために多くの時間とお金をかけなければならない場合、他人が所有する特許をめぐって設計、または許可または買収を行うことができ、高額な費用、特許または他の他人が持っている独自の権利を支払う必要がある可能性があり、私たちの業務、財務状況、および運営結果は実質的かつ不利な影響を受ける可能性がある。 もし私たちの所有または許可された知的財産権を効果的に保護できなければ、他の会社は同じまたは同様の製品を提供する可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。財務状況と経営結果。私たちは他人から技術許可と、私たちが持っている可能性のある任意の未来の特許を得ることができます。これらの特許は挑戦を受け、範囲を縮小し、無効にしたり、回避されたりする可能性があります。これは、競争相手が同じまたは類似の製品を販売することを阻止する能力を制限したり、私たちの製品に対する特許保護期間を制限したりするかもしれません。
私たち は買収とライセンス内で私たちの候補製品に必要な権利を獲得または維持できないかもしれません。
私たち は第三者 から私たちの現在または未来の候補製品に必要な任意の成分、使用方法、プロセス、または他の知的財産権を取得できないかもしれませんし、許可を与えることもできません。第三者の知的財産権の取得と認可の面で、私たちはいくつかのより成熟した会社からの競争を含む競争に直面する可能性がある。これらの老舗企業は、その規模、現金資源、およびより強い臨床開発と商業化能力のために、私たちに対する競争優位性を持っているかもしれません。また、私たちを競争相手と見なしている会社は、知的財産権の譲渡や許可を望まないかもしれません。私たちはまた、投資から適切な見返りを得ることを可能にする条項に沿って、第三者の知的財産権を得ることができないかもしれません。
私たちは、現在または未来の臨床前候補製品 の開発を加速するために、米国や外国の学術機関と協力協定を締結する可能性がある。一般に、これらのプロトコルは、会社が協力によって生成された機関の知的財産権許可を協議するためのオプションを含む。そのようなオプションがあっても、私たちは指定された時間範囲内で、または私たちが受け入れられる条項の下でライセンスについて交渉することができないかもしれない。もし私たちが協力機関から著作権許可を得ることができなければ、この機関は知的財産権を他の側に提供する可能性があり、これは私たちが必要な計画を実行することを阻止するかもしれない。
もし私たちが必要な第三者知的財産権を得ることができない場合、あるいは私たちの既存の知的財産権 を維持できない場合、私たちは関連計画の開発を放棄する必要があるかもしれません。私たちの業務、財務状況、運営結果は実質的な悪影響を受ける可能性があります。
特許保護の獲得および維持は、政府特許機関によって提出された様々なプログラム、文書提出、費用支払い、および他の要求 を遵守することに依存し、これらの要件に適合しない場合、許可された特許、係属中の特許出願、および潜在的に将来の特許出願および特許に対する特許保護は減少またはキャンセルされる可能性がある。
定期 特許および/または特許出願の維持費、継続費、年会費および様々な他の政府費用は、適用される特許および/または特許出願の有効期間内にいくつかの段階で米国特許商標局(USPTO)および米国以外の様々な政府特許機関に支払われる。米国特許商標局および様々な非政府特許機関 は、特許出願中にいくつかのプログラム、文書、費用支払い、および他の同様の条項を遵守することを要求する。 は、多くの場合、適用される規則に基づいて、滞納金を支払うことによって、または他の方法で不注意を修復することができる。しかし、場合によっては、規定を遵守しないことは、特許または特許出願が放棄または失効され、関連する管轄区域内の特許権の一部または全部を喪失させる可能性がある。もし私たちの許可内の特許または私たちが将来提出する可能性のある特許出願にこのような状況が発生した場合、私たちの競争相手は私たちの技術を使用するかもしれません。これは私たちの業務、財務状況、および運営結果に重大な悪影響を及ぼすでしょう。
| 45 |
製品の特許地位はしばしば複雑で不確実だ。米国および米国以外の多くの司法管轄地域では、特許によって許容される権利請求の広さが一致しない可能性がある。米国および他の国/地域特許法または特許法解釈の変化は、我々が許可または所有している知的財産権の価値を低下させたり、不確実性をもたらしたりする可能性がある。さらに、私たちの現在の候補製品および潜在的製品に関する情報 を発表することは、物質構成特許を含むが、これらの 候補製品および潜在的製品に関連する特許の取得または実行を阻止または実行する可能性があり、これらの特許は、一般に最強の特許保護を提供すると考えられる。
私たちが現在所有または将来所有または許可可能な特許 は、私たちが許可または所有する知的財産権 を保護することを保証するとは限らないが、以下の理由を含むが、これらに限定されない
| ● | これらの特許は、私たちの候補製品と同じまたは同様の他の製品からの競争を阻止するのに十分ではないかもしれません | |
| ● | 特許の有効期間を保証することができない場合は,米国法又は外国類似規定(ある場合)に特許期間を延長する規定により延長することができる | |
| ● | 私たちが現在または将来獲得または許可可能な発行された特許と特許可能なbrは、私たちの候補製品が市場に進出することを阻止しないだろう | |
| ● | 私たちまたは私たちがそれにライセンスするか、またはそれに特許を付与する可能性のある第三者は、1つまたは複数の特許の一部の条項の放棄を要求される可能性があります | |
| ● | 我々が知らない既存技術は、特許請求の範囲の有効性または実行可能な{brに影響を及ぼす可能性があるかもしれない | |
| ● | 我々が知っている従来技術が存在する可能性があり,これらの技術は特許請求の範囲の有効性や実行可能性に影響を与えないと考えられるが,最終的には特許請求の範囲の有効性や実行可能性に影響を与えることが発見される可能性がある | |
| ● | 他の人が獲得した特許は私たちの操作の自由に影響を与える可能性がある | |
| ● | もしこれらの特許が挑戦された場合、裁判所はそれらが無効であるか、または強制的に執行できないと判断することができる | |
| ● | 私たちのライセンス特許または私たちが所有する可能性のある任意の未来の特許の特許性、有効性および侵害性を管理する法律は大きく変化する可能性があり、私たちの特許権の範囲に悪影響を及ぼす可能性がある | |
| ● | 裁判所は競争相手の技術または製品が私たちの特許を侵害していないと判断することができ、あるいは私たちが将来所有する可能性のある和 | |
| ● | 特許は、費用が支払われていないか、または法規に準拠していないために取り返しのつかない無効になる可能性があり、または強制許可を受ける可能性がある。もし私たちが開発や臨床試験で遅延に遭遇すれば、私たちはbr特許保護の下で潜在製品を販売できる時間が短縮される。 |
我々の競争相手は、類似または代替技術または 製品を非侵害的に開発することによって、私たちが所有する可能性のある特許または将来の特許を回避することができるかもしれない。我々の競争相手は、簡略化された新しい出願または生物学的に類似した生物学的製品出願をFDAに提出することによって、任意の承認された製品の模造薬または生物類似バージョンを販売することを求めることができ、これらの出願では、私たちの競合他社は、私たちが許可されたbr特許または私たちが所有する可能性のある任意の未来の特許が無効であり、強制的に実行できない、または侵害されていないと主張することができる。あるいは、私たちの競争相手は、私たちの製品と似ているか、または他の面で私たちの製品と競争している彼ら自身の製品を販売する承認を求めることができる。この場合、私たちは、訴訟を提起することによって特許侵害を告発することを含む、私たちの特許または私たちが所有する可能性のある任意の未来の特許を擁護または維持する必要があるかもしれない。上記の任意のタイプの訴訟では、管轄権のある裁判所または他の機関は、私たちが許可している特許または私たちが所有する可能性のある任意の未来の特許が無効であるか、または強制的に実行できないことを発見することができるかもしれない。私たちはまた、特許保護を受ける前に、私たちの研究および開発の可能な特許態様を決定することができないかもしれない。私たちが許可されていない、有効かつ強制的に実行可能な特許を持っていても、これらの特許は、競合製品またはプロセスに対して、私たちの業務目標を達成するのに十分な保護を提供することができない可能性がある。
| 46 |
特許の発行は、その発明性、範囲、所有権、優先権、有効性、または実行可能性に対して決定的ではない。この点で、 第三者は、米国および海外の裁判所または特許庁において、私たちの特許または将来所有可能な任意の特許に挑戦することができる。このような 挑戦は、排他性または運営自由を失うこと、または特許主張がすべてまたは部分的に縮小され、無効または実行できないことをもたらす可能性がある。 これは、他人が類似または同じ技術および製品を使用することを阻止し、またはそれを商業化する能力を制限することができ、 または我々の技術および潜在的製品の特許保護期間を制限する可能性がある。また、新製品候補製品の開発、テスト、および規制審査に要する時間 を考慮すると、このような候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。
特許(Br)条項は、候補製品における私たちの競争的地位を十分な時間で保護するのに十分ではないかもしれない。
特許は限られており、特許提供の保護も限られている。米国では、すべての維持費が直ちに支払われる場合、特許の自然満了期間は、通常、米国で最初の非臨時出願日から20年である。私たちの候補製品をカバーする特許を取得しても、製品または候補製品をカバーする特許の有効期限が満了すると、競争製品やサービスからの競争を受ける可能性があります。したがって、私たちの特許の組み合わせは、他社が私たちと類似または同じ製品を商業化することを排除するために十分な権利 を提供できないかもしれない。私たちは他人の知的財産権を侵害する可能性があり、これは私たちの製品開発を阻害または延期し、私たちが製品を商業化することを阻止したり、製品の商業化のコストを増加させる可能性があります。
私たちのビジネス成功は、第三者特許や他の知的財産権を侵害することなく運営する私たちの能力に大きく依存している。例えば、私たちは、現在または未来の候補製品が発行された特許 を侵害していることを認識していないかもしれない。私たちは侵害された特許を持っていないと思うかもしれないが、私たちは最終的に侵害を発見されるかもしれない。
さらに、特許出願は、特許が発行されるまで秘密にされている場合がある。科学的発見や特許文献における発見の発表は、通常、基礎発見や特許出願の提出日よりもはるかに遅い。特許の発行には数年かかる可能性があるため、私たちが知らない現在承認されている出願が存在する可能性があり、これらの出願は、私たちの候補製品または潜在的な製品が発行された特許を侵害する可能性がある。例えば、我々の候補製品または潜在的製品侵害テーマを要求する係属中の出願 を要求するか、または修正することができる。競争相手は、特許ファミリーの懸案を維持し、私たちの候補製品をカバーしようとするために、発行された特許よりも優先的な持続的な特許出願を、継続、セクション、または部分的な継続出願の形態で提出することができる。
第三者 は、私たちが彼らのノウハウを不正に使用していると主張し、特許または他の知的財産権 を侵害していることを起訴することができる。これらの訴訟は費用が高く、私たちの業務、財務状況、および運営結果に悪影響を与え、管理と科学者の注意をそらす可能性がある。もし私たちが特許侵害で起訴された場合、私たちは私たちの候補製品、潜在的製品、または方法が関連特許の権利要件を侵害していないか、または特許権利要件が無効であることを証明する必要があり、私たちはそうできないかもしれない。無効性を証明することは難しい。例えば,米国では,特許の無効性を証明するためには,発行された特許が有する有効性推定を覆すために,明確で納得できる証拠を提示しなければならない.私たちがこれらの訴訟で成功しても、私たちは巨額のコストを生む可能性があり、私たちの経営陣と科学者の時間と注意はこれらの訴訟に移される可能性があり、これは私たちに実質的な悪影響を及ぼすかもしれない。さらに、私たちはこのような操作を成功させるために十分な リソースがないかもしれない。もし裁判所が任意の第三者特許が有効で強制的に実行可能であり、 が私たちの製品またはその使用をカバーしていると判断した場合、これらの特許の任意の所有者は、適用された特許からライセンスを取得または取得し、または特許が満了するまで、私たちの製品を商業化することを阻止することができるかもしれない。
| 47 |
私たち は合理的なコストや合理的な条項で許可手配を達成したり、他の手配をすることができないかもしれません。ライセンスや代替技術を取得できないbrは、私たちの製品の発売遅延を招いたり、製品の製造や販売を禁止したりする可能性があります。我々が許可を得ることができても,非排他的である可能性があり,我々のライバル が我々に許可された同じ技術にアクセスできるようにする.私たちは裁判所の命令を含めて権利侵害技術や製品の商業化を停止させることを余儀なくされるかもしれない。また、このような訴訟や訴訟のいずれにおいても、故意に特許侵害が発見された場合、3倍の損害賠償および弁護士費を含む金銭損害賠償責任を負うと判断される可能性がある。権利侵害発見は、私たちの候補製品を商業化することを阻止したり、一部の業務運営を停止させたりする可能性があり、これは私たちの業務、財務状況、運営結果に実質的な悪影響を及ぼす可能性がある。第三者がその機密情報または商業秘密を盗用することに関するいかなるクレームも、私たちの業務、財務状況、および運営結果に類似した重大な悪影響を及ぼす可能性があります。また、いかなる訴訟の開始および継続に生じるいかなる不確実性も、運営を継続するために必要な資金を調達する能力に大きな悪影響を及ぼす可能性があります。
私たちによって提起された、または私たちに対して提出された知的財産権侵害に関するいかなるクレームまたは訴訟も、高価で時間のかかるbrであり、私たちの業務、財務状況、および運営結果に悪影響を及ぼす可能性があります。
私たち は私たちの許可と所有する知的財産権を強制的に執行または保護するために訴訟を提起することを要求されるかもしれない。私たちの知的財産権を保護するために提起された訴訟は非常に時間がかかって費用がかかるかもしれない。バイオ製薬産業では、一般的に特許と他の知的財産権に関する多くの訴訟がある。このような訴訟または訴訟は、私たちの運営費用を大幅に増加させ、開発活動または任意の将来の販売、マーケティング、または流通活動に使用することができるリソースを減少させる可能性がある。
Brのいかなる侵害訴訟でも、私たちが得たいかなる金銭賠償にも商業的価値がない可能性がある。また,知的財産権訴訟に関する発見数が膨大であるため,我々のいくつかの機密情報 は訴訟中に漏洩する可能性がある.さらに、このような侵害クレームを提起し、追及するのに十分な財政的または他の資源があることは保証されず、これらのクレームは通常数年継続して解決される。また,我々 は,侵害者からのいかなるクレームに対しても,これらの当事者に我々に反クレームを促し,我々が彼らの特許を侵害していると主張する可能性がある.私たちのいくつかの競争相手は、彼らがもっと大きな財力を持っているので、私たちよりも効率的にこのような訴訟や訴訟の費用を負担するかもしれない。特許訴訟または他の訴訟の開始および継続によって生じる不確実性 は、市場における我々の競争能力に重大な悪影響を及ぼす可能性がある。
さらに、私たちの特許および特許出願、ならびに将来可能な出願、所有または許可された特許および特許出願は、妨害訴訟、異議訴訟、再審手続き、および他の形態の認可後審査のような他の挑戦に直面する可能性もある。これらの課題のいずれかは、成功すれば、私たちの任意の 特許および特許出願、ならびに将来可能な出願、所有または許可された特許、および特許出願の無効または範囲を縮小させる可能性がある。これらの課題のいずれも、その成否にかかわらず、非常に時間的で高価な防御および解決策である可能性があり、私たちの経営陣と科学者の時間と注意を分散させる。
米国特許法の変化 は特許全体の価値を低下させ,製品を保護する能力を弱める可能性がある。
他のバイオテクノロジー会社の場合のように、私たちの成功は知的財産権、特に特許に大きく依存する。生物技術業界で特許を獲得と実施することは技術上の複雑性と法律上の複雑性にも関連し、しかもコストが高く、時間が長く、しかも内在的な不確定性を持っている。例えば、米国が以前に公布し、実施されている範囲の広い特許改革立法。具体的には、2011年9月16日、“ライシー·スミス米国発明法”または“ライシー·スミス法案”が法律に署名され、米国特許法のいくつかの重大な改正が含まれており、その多くが2013年3月に発効した。しかしながら、裁判所は、“ライシー·スミス法案”の条項を説明するために数年かかる可能性があり、この法規の実施は、私たちの許可および将来の特許出願をめぐる起訴、ならびに私たちの許可および将来の特許の実施または弁護の不確実性およびコストを増加させる可能性があり、これらのすべては、私たちの業務、財務状態、および運営結果に大きな悪影響を及ぼす可能性がある。
| 48 |
さらに、米国最高裁判所は近年、いくつかの特許事件に対して裁決を下しており、場合によっては利用可能な特許保護範囲 を縮小するか、場合によっては特許所有者の権利を弱めるかを決定している。我々の将来の特許取得能力に関する不確実性の増加に加えて,このようなイベントの組合せは,いったん特許を取得する価値に関する不確実性 をもたらしている.米国議会、連邦裁判所、USPTOの決定によると、特許を管理する法律や法規 は予測不可能な方法で変化する可能性があり、新しい特許を獲得したり、将来獲得する可能性のある特許を強制的に執行する能力を弱める可能性がある。
私たち は世界的に私たちの知的財産権を保護できないかもしれない。
世界各地で候補製品特許を申請、起訴、擁護する費用は尻込みするほど高いかもしれない。競争相手は、私たちが許可を得ていない、または特許保護を受けていない司法管轄区域で、私たちのbr許可と所有技術を使用して彼ら自身の製品を開発することができ、また、他の侵害製品を私たちが獲得できるかもしれない特許保護を得ることができるかもしれないが、特許執行力はアメリカの地域に及ばないかもしれない。これらの製品は、私たちが特許を発行できるかもしれない司法管轄区域で私たちの製品と競争する可能性があり、将来のいかなる特許主張や他の知的財産権は彼らが競争するのを効果的に阻止することができないか、または彼らの競争を阻止するのに十分ではないかもしれない。
多くの会社は外国の管轄区域の知的財産権の保護と保護に深刻な問題に直面している。特定の国/地域、特にいくつかの発展途上国の法律制度は、特許の強制執行および他の知的財産権保護を支持しておらず、これは、私たちのライセンス特許および私たちが所有する可能性のある将来の特許の侵害を阻止することを困難にするか、または一般的には私たちの独占権を侵害する方法で競合製品を販売することを困難にするかもしれない。また、特定の国/地域の法律の独自の権利の保護の程度や方法は、米国の法律とは異なる。そのため、米国および海外で許可され、所有されている知的財産権を保護し、守ることに大きな問題が生じる可能性がある。外国の管轄区域で将来の特許権(ある場合)の訴訟手続きを強制的に執行することは、巨額のコストを招く可能性があり、私たちの努力と注意を私たちの業務の他の側面からbrに移します。
我々 は,ビジネス秘密や他の固有情報の漏洩を十分に防ぐことができない可能性がある.
私たちのノウハウとプロセスを保護するために、私たちは、当社のパートナー、従業員、コンサルタント、メーカー、外部科学協力者、スポンサー研究者、および他のコンサルタントと締結されたセキュリティ協定にある程度依存しています。これらのプロトコル は、我々の機密情報の漏洩を効果的に防止できない可能性があり、不正に機密情報が漏洩した場合には十分な救済措置を提供できない可能性がある。また、他の人は、私たちのビジネス秘密および独自の情報を独立して発見する可能性があります。 ビジネス秘密保護を獲得または維持できなければ、私たちの競争ビジネスの地位に悪影響を及ぼす可能性があります。
私たちの特許、私たちが将来所有する可能性のある任意の特許、および他の知的財産権のクレームが問われるかもしれません。
私たちは現在、私たちの特許や私たちが持っている知的財産権に疑問を提起するクレームを受けていませんが、将来的には、元従業員、協力者または他の第三者が発明者または共同発明者として私たちの特許または他の が所有している知的財産権において権利を持つというクレームを受ける可能性があります。例えば、私たちは私たちの候補製品開発に参加するコンサルタントや他の人の義務衝突によって在庫紛争が生じる可能性があります。訴訟を通じてこのようなクレームと他の挑戦在庫のクレームに対抗する必要があるかもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭損害賠償を支払う以外に、貴重な知的財産権、例えば貴重な知的財産権の独占所有権や使用権を失う可能性がある。このような結果は、我々の業務、財務状況、運営結果に実質的な悪影響を及ぼす可能性がある。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。
| 49 |
医療保健法やその他の法律コンプライアンス事項に関するリスク
私たちは様々なアメリカ連邦、州、外国の医療保健法律法規の制約を受けて、これはコンプライアンスコストを増加させる可能性がありますが、私たち はこれらの法律法規を守らなければ、私たちの運営結果と財務状況を損なう可能性があります。
私たちの業務運営と調査者、医療専門家、コンサルタント、第三者支払人、患者組織と顧客との現在と未来の手配は、私たちが広く適用される外国、連邦と州詐欺と乱用、その他の医療法律と法規に直面させます。これらの法律は、私たちがマーケティング、販売、流通をどのように研究、マーケティング、販売、流通するかを含む、私たちが業務を展開する業務または財務配置と関係を制約するかもしれません。これらの法律には
| ● | 個人または実体が直接または間接的に、公開的または隠蔽的に(任意のリベート、賄賂、またはいくつかのリベートを含む)故意に請求、提供、受信、または提供することを禁止する連邦反リベート法規。個人の推薦または購入、レンタルまたは注文に対する見返りとして、または任意の商品、施設、br物品またはサービスを購入、レンタルまたは注文することを手配または推薦する報酬として、現金または実物。連邦医療保険や医療補助のような連邦医療計画の下で全部または一部があります。個人または実体は、連邦反リベート法規を実際に理解または違反する具体的な意図を有する必要がなく、違反を実施することができる | |
| ● | 民事虚偽請求法案と民事罰金法を含む連邦虚偽請求法は、個人または実体が知っている場合に連邦政府に提出または提出することを禁止する。請求支払いまたは承認brは、虚偽または詐欺的であり、虚偽記録の作成、使用、または虚偽記録の使用、または虚偽または詐欺的クレームに関連する陳述を意図的に作成、使用するか、または虚偽陳述を回避するために意図的に作成、または誘導する。連邦政府に資金を支払う義務を減らしたり隠したりする。また、政府は、連邦反リベート法規に違反して発生した物品やサービスを含むクレームは、“民事虚偽請求法”については、虚偽または詐欺的クレームを構成していると断言できる | |
| ● | 1996年“連邦健康保険移行性·責任法案”(HIPAA)は、他の事項に加えて、任意の医療福祉計画をだまし取ろうとする計画を故意に実行または実行しようとする計画に刑事と民事責任を加える。重大な事実を故意に偽造、隠蔽、または隠蔽するか、または医療福祉、br項またはサービスの提供または支払いに関連する重大な虚偽陳述を行うことができる。連邦反リベート法規と類似して、個人或いは実体は実際に法規或いは法規違反の具体的な意図を理解する必要がなく、違反を実施することができる | |
| ● | 連邦医師は日光法案を支払い、いくつかの薬品、機器、生物製品、医療用品のメーカーが連邦医療保険の下で支払いを受けることができることを要求している。Medicaidまたはbr}児童健康保険計画(いくつかの例外を除く)は,医師(医師を含むと定義する)への支払いや他の“価値移転”に関する情報を連邦医療保険·医療補助サービスセンター(CMS)に毎年報告している。歯科医、検眼師、足科医、脊椎マッサージ師)、いくつかの非内科医(医師アシスタント、勤務看護師、臨床看護師専門家、登録看護師麻酔科医、麻酔学アシスタント、登録助産師)。教育病院や他の医療提供者やこのような医療専門家とその直系親族が持っている所有権と投資権益そして | |
| ● | 同様の国および外国の法律、例えば、国の反リベートおよび虚偽クレーム法律は、医療保険プロジェクトまたは個人保険会社を含む非政府第三者支払人によって精算されるサービスに関する販売またはマーケティング手配およびクレームに適用可能である。いくつかの州の法律は生物技術会社に生物技術業界のbr自発的コンプライアンスガイドラインと連邦政府が公布した関連コンプライアンスガイドラインを遵守することを要求し、そして製薬業者に支払いとbrに関する情報を報告することを要求するかもしれない。他の医師や他の医療提供者に価値やマーケティング支出を移転しいくつかの州の法律は生物技術会社がある薬品の定価情報を報告することを要求している;そしていくつかの州と地方の法律は登録或いは薬品販売代表を要求している。 |
我々の現在と将来の第三者の業務配置が適用される医療·プライバシー法律·法規に適合することを確保するための努力は、持続的な巨額のコストに関連する。政府当局は、私たちの業務実践が現在または未来に適合していない可能性があり、詐欺および乱用または他の医療保健法律および法規の適用に関する現行または将来の法律、法規または判例法に適合していない可能性があると結論するかもしれない。もし私たちの運営がこれらの法律のいずれかまたはbrに違反していることが発見された場合、私たちの任意の他の政府法規に適用される可能性があり、私たちは、民事、刑事および行政処罰、損害賠償、罰金、br返還、監禁、MedicareおよびMedicaidのような政府援助に参加する医療計画から除外され、誠実な監督と報告義務、契約損害、名声損害、利益減少および将来の収益、ならびに私たちの業務を削減または再構築することを含む重大な処罰を受ける可能性がある。このような操作を防御するには費用が高く時間がかかる可能性があり、大量の財務と人的資源が必要となる可能性がある。
| 50 |
したがって,我々が我々に提起する可能性のあるいかなる訴訟も防ぐことに成功しても,我々の業務は損なわれる可能性がある.さらに、私たちがそれと業務を行うことが予想される任意の医師または他のヘルスケア提供者またはエンティティが、適用される法律または法規に適合していないことが発見された場合、彼らは、政府が援助する医療計画から除外することを含む重大な刑事、民事または行政制裁を受ける可能性がある。
実際の または適用されるデータ保護、プライバシーおよびセキュリティ法律、法規、基準およびその他の要求 を遵守できないと考えられることは、私たちの業務、運営結果、財務状況に悪影響を及ぼす可能性があります。
世界のデータ保護構造は急速に発展しており、私たちは多くの州、連邦と外国の法律、要求br、および個人情報の収集、使用、開示、保留と安全を管理する法規の制約を受けているか、または影響を受ける可能性がある。予測可能な未来には、実施基準および法執行実践は依然として不確定である可能性があり、私たちはまだ未来の法律、法規、基準、またはその要求に対する見方が私たちの業務に与える影響を決定することができない。このような変化は私たちの業務に不確実性をもたらす可能性があり、brは私たちがある司法管轄区域で業務を展開したり、個人情報を収集、保存、移転し、共有する能力に影響を与え、私たちは契約におけるより重い義務を受け入れなければならず、私たちが責任を負うか、または追加コストをかけなければならない。これらの法律のすべては裁判所と政府機関の異なる解釈を受け、複雑なコンプライアンス問題をもたらしている。もし私たちが適用された法律と法規を遵守できなかった場合、私たちは政府の調査および/または法執行行動、罰金、民事または刑事罰、br}個人訴訟、または否定的な宣伝に直面する可能性があり、これは私たちの業務、財務状況、および運営結果に悪影響を及ぼすかもしれない。例えば、私たちが故意に保証エンティティから個人識別可能な健康情報を取得または開示し、その方法が“健康保険携帯性および責任法案”(“2009年健康情報技術促進経済および臨床健康法案”およびその施行された法規または適用される州法改正)によって許可または許可されていない場合、私たちは刑事罰を受ける可能性がある。
環境、健康および安全法律法規に違反したり、環境、健康および安全法律法規に基づいて責任を負うことは、私たちを罰金、処罰、または他のコストに直面させる可能性があり、私たちの業務成功に実質的な悪影響を及ぼす可能性があります。
私たちbrは多くの環境、健康と安全法律法規の制約を受けて、実験室のプログラムの管理、処理、使用、貯蔵、処理と危険材料と廃棄物の処理と処分及び汚染された場所を整理する法律法規を含む。私たちの業務は化学品と生物学的材料を含む潜在的な危険と可燃性材料の使用に関するものだ。私たちの業務はまた危険な廃棄物製品を発生させるだろう。私たちは、罰金、処罰、その他の制裁、調査と整理費用、および第三者のクレームを含む、私たちの運営や財産に関連する環境要求に違反したり、環境要求に応じて責任を負うことによって巨額のコストを招く可能性があります。br}私たちは通常第三者と契約を結び、私たちの運営中の危険な材料や廃棄物を処理していますが、これらの材料による汚染や傷害のリスクを除去することはできません。危険な材料の使用によって汚染や損傷が生じた場合、私たちはそれによるいかなる損害にも責任を負い、いかなる責任も私たちの資源の範囲を超えている可能性がある。
また,環境法律法規は複雑で変化が頻繁であり,より厳しくなる傾向にある。私たちは法的規制の変更が適用される影響を予測することもできないし、私たちの未来の適合性を決定することもできない。また,現在または将来の環境,健康,安全法律法規を遵守するためには,巨額のコスト が生じる可能性がある。これらの現在または未来の法律法規は、私たちの研究、開発、または生産努力を損なう可能性がある。
危険材料の使用による従業員へのダメージによるコストや支出を支払うために労働者補償保険を維持しているが,潜在的な責任を負うには不十分である可能性がある。私たちは私たちが生物、危険または放射性物質を貯蔵または処分することによって、私たちが提出した環境責任や有毒侵害のためにbr保険を維持することはできません。
| 51 |
私たちの業務に関する他の リスク
新冠肺炎などの伝染病の大流行、大流行或いは爆発は私たちの業務と運営に実質的な悪影響を与える可能性がある。
大流行、流行病あるいは他の伝染病の爆発は過去に発生したことがあり、未来は私たちの業務に負の影響を与えるかもしれない。新冠肺炎疫病はアメリカと世界経済に影響を与え、私たちの業務と私たちが依存している第三者の業務に影響を与え、 は私たちの候補製品の供給中断及び現在と未来の臨床試験の進行を含む。たとえば, の大流行により我々のGMPプロセスにかかる時間は予想よりも長い.また、新冠肺炎の流行は、食品·薬物管理局や他の衛生当局の運営に影響を与え続ける可能性があり、これは、私たちの候補製品の審査と承認を含む審査と承認の遅延を招く可能性がある。また、新冠肺炎疫病による潜在的な経済影響及び持続時間は を評価或いは予測することは困難であるが、新冠肺炎疫病の全世界金融市場への影響は私たちの資本獲得能力を低下させる可能性があり、これは私たちの短期と長期流動性に負の影響を与える可能性がある。新冠肺炎大流行の最終的な影響は非常に不確定であり,変化する可能性がある。これらの条件がどのくらい続くか、私たちの完全な財務影響が何になるかは不明ですが、私たちの融資努力や追加の技術開発はマイナスの影響を受ける可能性があります。
情報技術システム障害,ネットワーク攻撃やネットワークセキュリティ欠陥が発生すると,我々の業務や運営が影響を受ける可能性がある.
我々は,業務展開に必要なデジタル形式の情報を収集·維持し,情報技術システムやインフラに依存して業務を運営するようになってきている.私たちの通常のビジネスプロセスでは、顧客、従業員、および請負業者の知的財産権、独自業務情報、および個人情報を含む大量の機密情報を収集、格納、および送信します。我々は,このような機密情報の機密性と完全性 を守るために安全な方法でそうしなければならない.
我々の情報技術システムおよび我々の第三者サービスプロバイダ、戦略パートナーおよび他の請負業者またはコンサルタントの情報技術システムは、コンピュータウイルスおよびマルウェア(例えば、恐喝ソフトウェア)、悪意コード、自然災害、br}テロ、戦争、電気通信および電気故障、ハッカー攻撃、ネットワーク攻撃、ネットワーク釣り攻撃および他の社会工学計画、br}従業員の窃盗または乱用、人為的エラー、詐欺、拒否またはサービスダウン攻撃、複雑な民族国家および民族国家によって支援されるbr}行為者、または組織内部者が許可されていないアクセスまたは使用の攻撃および破壊または中断を受けやすい。私たちの組織内部システムにアクセスする権利があります我々 はまた,我々の情報技術インフラの要素を外注しているため,多くの第三者サプライヤーが我々の機密情報にアクセスできる可能性や可能性がある.
また, の情報技術システムに対する攻撃は頻度,持続性,複雑性,強度の面で増加しており, は動機や専門知識の広い複雑で組織的な団体や個人によって実施されている.Brの新冠肺炎が大流行したため、私たちはまたより多くのネットワークセキュリティリスクに直面する可能性があります。私たちのインターネット技術への依存と私たちの遠隔作業の従業員の数のために、これはサイバー犯罪者の抜け穴を利用するためにもっと多くの機会を作るかもしれません。さらに, は,不正アクセスやシステム破壊のための技術がしばしば変化し,通常は目標に対して起動する前に を認識できないため,これらの技術を予測したり,十分な予防措置を実施することができない可能性がある.我々はまたセキュリティホールに遭遇する可能性があり,これらの脆弱性は長い間検出できない可能性がある.確認しても、攻撃者がbrの制御を回避し、法医学的証拠の検出および除去または混同を回避するためのツールおよび技術を使用することが多くなっているので、私たちは、事件または違反を調査または修復するのに十分ではない可能性がある。
私たちと私たちの特定のサービス提供者たちは時々ネットワーク攻撃とセキュリティ事件を受けるだろう。私たちは、これまで、重大なシステム障害、事故、またはセキュリティホールに遭遇していなかったと信じていませんが、このような事件が発生し、私たちの運営中断を招く場合、私たちのビジネス秘密、個人情報、または他の独自または敏感な情報の損失、破損、または許可されていない漏洩、または他の同様の中断によって、私たちの開発計画および業務運営が重大な中断を受ける可能性があります。セキュリティホールまたは他のイベントが不正アクセスまたは無許可使用、開示、配布、または他の方法で個人情報を処理する場合、プライバシーおよびセキュリティ法律に基づいて個人、政府当局、規制機関、メディア、および他の当事者に通知する必要がある場合があります。私たちはまた訴訟、処罰、罰金を含む責任を招く可能性があり、私たちは規制行動や調査の対象になるかもしれない。私たちの競争的地位は損なわれる可能性があり、私たちの製品とサービスのさらなる開発と商業化は延期されるかもしれない。私たちはネットワーク責任保険を維持していますが、この保険は、私たちのシステムの中断または破壊によって生じる可能性のある財務、法律、業務、または名声損失をカバーするのに十分ではないかもしれません。
| 52 |
私たち は戦略的取引を行うかもしれません。これらの取引は私たちの流動性に影響を与え、私たちの支出を増加させ、私たちの経営陣を気晴らしさせるかもしれません。
私たちは、買収会社、資産購入、および製品、候補製品または技術の外部許可または内部許可のような戦略的取引を時々考慮するかもしれない。私たちが考慮する可能性のある他の潜在的な取引は、剥離、戦略パートナー関係、合弁企業、再編、資産剥離、業務合併と投資を含む様々な異なるbr業務手配を含む。このような取引は、非日常的な費用または他の費用の発生を要求する可能性があり、短期的および長期的な支出 を増加させ、私たちの管理または業務に重大な統合挑戦または中断をもたらす可能性があり、これは、私たちの業務、財務状況、および運営結果に悪影響を及ぼす可能性があります。例えば、これらの取引は、多くの業務および財務リスクをもたらす可能性がある
| ● | リスクのオープンオープン未知の負債 | |
| ● | 私たちの業務を中断して、私たちの経営陣の時間と注意を移して、買収した製品、候補製品、または技術を開発します | |
| ● | 大量の債務や発行された株式証券を希釈して、これらの取引のいずれかを支払うことができる | |
| ● | 取引と統合コストは予想より高い | |
| ● | 資産減記または営業権または減価費用 | |
| ● | 販売費用が増加しました | |
| ● | 買収された企業または製品ラインの運営および人員を当社の運営および人事と組み合わせることの難しさとコスト | |
| ● | 経営陣および所有権の変化による任意の買収企業または製品ラインの主要サプライヤーまたは顧客との関係減価 | |
| ● | いかなる買収企業の肝心な従業員も引き留めることができない。 |
したがって、 は、上記の性質の任意の取引を行うか、成功するかは保証されないが、私たちが達成した任意の取引は、前述のリスクまたは他のリスクの影響を受ける可能性があり、私たちの業務、財務状況、および運営結果に重大な悪影響を及ぼす可能性がある。
| 53 |
私たちの普通株に関するリスク
私たちの普通株の市場価格の変動は大きく、あなたの投資は完全な損失を受けるかもしれません。
私たちの普通株は現在ナスダック資本市場で取引されています。私たちの普通株の市場価格はずっと不安定で、変動し続ける可能性が高い。私たちの普通株の市場価格は多くの要素によって大幅に変動するかもしれません。その中のいくつかの要素は私たちがコントロールできないことです
| ● | 臨床試験開始に必要なbr承認を得ることができない | |
| ● | 臨床と臨床前研究結果 | |
| ● | 監督管理の承認を発表するか、承認を得られなかったか、または使用された特定のラベル適応または患者集団、または規制審査プロセスにおける変更または遅延 | |
| ● | 私たちまたは他の人の技術革新、新製品、または製品の強化を発表します | |
| ● | 規制当局が私たちの臨床試験、製造サプライチェーン、または販売とマーケティング活動に取った不利な行動 | |
| ● | 私たちの候補製品または特許に適用される法律、法規または決定における変更または発展 | |
| ● | メーカー、サプライヤー、またはパートナーとの関係には何の不利な変化もあります | |
| ● | 私たちの競争相手や製薬やバイオテクノロジー産業に関する公告 | |
| ● | 予想された製品の販売と収益性を達成するか、あるいは私たちは予想を達成できなかった | |
| ● | 私たちは任意の製品責任訴訟または知的財産権侵害訴訟を含むが、これらに限定されない訴訟を開始するか、または訴訟に参加する | |
| ● | 取締役会、管理職、その他の重要な人員のいかなる重大な変動も | |
| ● | 米国、ヨーロッパ、その他の国における医薬品の販売又は定価に関する立法 | |
| ● | 我々は重要な戦略的協力パートナーシップ、外部許可、内部許可、合弁企業、買収または資本約束を発表した | |
| ● | ライセンス、研究契約、または他の連携協定の満了または終了 | |
| ● | 私たち、いかなる許可者、または他の人が開発した治療薬の安全性に対する国民の懸念 | |
| ● | 研究と開発プロジェクトの成功 | |
| ● | 知的財産権や規制承認に関する事態の発展 | |
| ● | 私たちと私たちの競争相手の運営結果は違います | |
| ● | もし私たちの普通株式がアナリストによってカバーされていれば、収益予想の変化または証券アナリストの提案 | |
| ● | 将来的に普通株またはその他の証券を発行する | |
| ● | 一般市場状況には,バイオテクノロジー会社株の一般市場価格変動,その他の要因が含まれており,我々の運営業績とは無関係な要因が含まれている | |
| ● | 政治的および経済的不安定、戦争またはテロ行為または自然災害、大流行の出現、または他の広範な衛生緊急事態(またはそのような緊急事態が発生する可能性のある懸念、例えば新冠肺炎大流行を含む)および | |
| ● | その他の要因はこの“リスク要因”の部分に を記述している. |
これらの要因と任意の対応する価格変動は、私たちの普通株の市場価格に実質的な悪影響を与える可能性があり、これは私たちの投資家に大きな損失をもたらすだろう。
| 54 |
また、株式市場全体、特にナスダック資本市場、特にバイオテクノロジー会社の市場は、極端な価格と出来高変動を経験しており、これらの変動は、それらのような会社の経営業績とは無関係か比例しないことが多い。 別に見られる“-一般的なリスク要因--”不利なグローバル経済状況は私たちの業務、財務状況、または経営結果に悪影響を及ぼす可能性があります“私たちの実際の経営業績にかかわらず、広範な市場と業界要素は私たちの普通株の市場価格にマイナス影響を与える可能性がある。また、金融市場の系統的な下落や私たちのコントロール範囲を超えた関連要因は、私たちの株価を急激に下落させる可能性がある。もし私たちの普通株の取引量が低ければ、私たちの普通株の価格変動はもっと深刻になるかもしれない。過去,市場変動後,株主はしばしば証券 集団訴訟を起こしてきたこのリスクは特に我々に関連しており,生物製薬会社brが近年著しい株価変動を経験しているためである私たちがこのような証券訴訟に直面すれば、巨額のコストと経営陣の資源と注意の分流を招く可能性があり、これは私たちの業務を損なう可能性がある。将来私たちの普通株を売却しても私たちの株の市場価格を下げる可能性があります。
また, 我々の普通株の流動性は限られており,与えられた価格で売買可能な普通株株式数だけでなく,我々の普通株取引を行う時間遅延や証券アナリストやメディアによる我々の報道の減少により が減少する可能性がある.これらの要素は、私たちの普通株の価格が他の場合に得られる可能性のある価格 を下回る可能性があり、私たちの普通株の入札と重要価格の間のより大きな価格差を招く可能性もある。また、大きな流通株がなければ、我々の普通株の流動性は、より広範な公有制会社の株よりも低くなるため、私たちの普通株の取引価格はより不安定になる可能性がある。活発な公開取引市場がない場合、投資家は彼らの私たちの普通株への投資を変えることができないかもしれない。相対的に小さい普通株取引量が私たちの普通株の取引価格に与える影響は私たちの公開流通株よりも大きいかもしれません。私たちは未来の私たちの普通株の取引価格を予測できない。
私たちの普通株の活発な取引市場は持続できないかもしれない。
私たちの普通株の活発な公開取引市場は持続できないかもしれない。活発な市場の不足は、あなたが望む時間に、あるいはあなたが合理的だと思う価格であなたの株を売る能力を弱めるかもしれません。活発な市場の不足はまたあなたの株の公正な価値を下げるかもしれない。不活発な市場はまた、株を売却することで資金を調達して運営に資金を提供し続ける能力を弱める可能性があり、株を対価格で他社や技術を買収する能力を弱める可能性もある。
証券や業界アナリストが研究や報告を発表または停止したり、私たち、私たちの業務または私たちの市場に不利な報告を発表したりしない場合、私たちの株価や取引量はマイナスの影響を受ける可能性がある。
私たちの普通株の取引市場は、業界または証券アナリストが私たち、私たちの業務、私たちの市場、または私たちの競争相手に関する研究と報告を発表する可能性がある影響を受けるかもしれない。私たちはこれらのアナリストに何の統制権もなく、何の保証も提供できないし、アナリストが私たちをカバーしたり、有利なカバー範囲を提供したりすることはできない。もし私たちのアナリストが私たちの普通株に不利な提案をしたり、私たちの競争相手により有利な相対的な提案を提供したりすることができれば、私たちの株価は下落するかもしれない。もし私たちのアナリストが私たちの報告を停止したり、定期的に私たちの報告書を発表できなかったりする可能性があれば、私たちは金融市場での可視性を失う可能性があり、これは逆に私たちの株価や取引量に負の影響を与える可能性がある。
私たちの既存の株主が公開市場で私たちの株を大量に売ることは私たちの株価を下落させるかもしれません。
公開市場で私たちの株を大量に売却したり、これらの売却が発生する可能性があると考えたりすると、私たちの証券の市場価格 を低くし、追加株式証券を売却することで資金を調達する能力を弱める可能性があります。私たちは販売が私たちの証券の現行の市場価格に与える影響を予測できません。
私たちのbrは“小さな報告会社”であり、より小さい報告会社に適用される開示要求の低下は、私たちの普通株の投資家に対する吸引力を低下させる可能性がある。
私たち は“小さな報告会社”とされています。したがって、私たちは、選択された財務データおよび役員報酬情報の提供を免除するなど、いくつかの低減された開示要件に依存する権利がある。私たちは規模の小さい報告会社なので、これらの免除と減少は、私たちがアメリカ証券取引委員会に提出した文書での開示を減少させることは、投資家が私たちの運営結果や財務見通しを分析することを難しくするかもしれない。私たちは投資家がこれらの免除に依存して、私たちの普通株の吸引力が低下していると感じる可能性があるかどうかを予測できません。もし一部の投資家が私たちの普通株の吸引力が低下していることを発見すれば、私たちの普通株取引市場はそんなに活発ではなく、私たちの株価はもっと変動するかもしれません。
| 55 |
私たちの財務統制と上場企業の要求を維持し、改善することは、私たちの資源を緊張させ、経営陣の注意力を移転させ、私たちの適格な取締役会のメンバーを引き付ける能力に影響を与える可能性がある。
上場企業として、私たちは1934年の証券取引法や取引法、サバンズ-オクスリ法案、ナスダック株式市場有限責任会社(“ナスダック”)規則の報告要件を守らなければならない。これらの規則の要求は私たちの法律と財務コンプライアンスコストを増加させ、いくつかの活動を更に困難にし、時間或いはコストを高くし、私たちの人員、システムと資源に圧力を与える。取引法は、私たちの業務および財務状況に関する年間、四半期、および現在の報告書を提出することを要求します。
“サバンズ-オキシリー法”(Sarbanes-Oxley Act)は、私たちが財務報告書に対して効果的な開示制御と手続きおよび内部統制を維持しているかどうかを開示することを要求する。私たちが十分な内部財務と会計制御プログラム を持っていることを確実にすることは高価で時間のかかる仕事であり、常に再評価する必要がある。
私たちは適切な上場会社の経験と技術会計知識を持つ会計と財務者をもっと招聘する必要があるかもしれません。 は私たちの内部統制に任意の適切な変更を実施するには、私たちの役員、高級管理者、従業員に対して特定のコンプライアンス訓練を行う必要があるかもしれません。大量のコストが必要で、かなりの時間がかかります。しかし、このような変化は、私たちの内部統制の十分性を効果的に維持することができない可能性があり、そのため、正確な財務諸表をタイムリーに作成できない場合もあり、私たちの運営コストを増加させ、私たちの業務運営能力を深刻に損なう可能性があります。また、効果的な内部統制は、信頼できる財務報告書を作成するために必要であり、詐欺防止を支援するために非常に重要です。
ナスダック規則によると、独立取締役会の多数の議席を維持することが要求される。上場企業に適用される各種規則(Br)は、私たちの維持役員や高級管理者責任保険をより困難かつ高価にし、より低い保証範囲を受け入れることや、より高いコストを発生させて保証範囲を維持することを要求される可能性があります。もし私たちが十分な役員と高級管理者保険を維持できなければ、私たちは合格した高級管理者と取締役を募集し、維持する能力は大幅に低下します。
上場企業に適用される規則や法規は、大量の法律や財務コンプライアンスコストを発生させることが予想される。これらのコストは、私たちの純収益を減少させたり、私たちの純損失を増加させ、業務の他の分野のコスト を下げることを要求するかもしれません。
2002年のサバンズ-オキシリー法案404条に基づいて財務報告を効率的に内部統制できなければ、我々の株価に大きな悪影響を及ぼす可能性がある。
米国の上場企業として、米国証券取引委員会とナスダック資本市場の要求を遵守するために、第404条やサバンズ·オキシリー法案の他の条項の要求を含むため、多くの会計、法律、その他の費用を発生させている。節404によれば、財務報告の内部統制に関する管理職の報告書を提出しなければならない。しかし、私たちがまだ小さな報告会社である限り、私たちは私たちの独立公認会計士事務所が発行した財務報告の内部統制に関する証明報告書を含むことを要求されません。規定された期間内に第404条を遵守するために、財務報告に対する私たちの内部統制を記録し、評価することは、高価で挑戦的である。財務報告の内部統制基準が時々修正され、補充され、修正されたときに財務報告の内部統制の十分性を維持できなければ、2002年のサバンズ-オキシリー法案404条と米国証券取引委員会の関連規則と法規に基づいて、財務報告に対して有効な内部統制を実施することができることを確実にすることができない可能性がある。もし私たちが将来私たちの財務報告の内部統制の有効性を有利に評価できなければ、投資家の私たちの財務報告の信頼性に対する信頼は悪影響を受ける可能性があり、これは私たちの株価に実質的な悪影響を及ぼすかもしれない。
| 56 |
帰化合併が合併とともに第351(A)条取引の資格を満たしていない場合、イスラエル国際技術会社普通株の前米国保有者は、帰化合併の課税収益を確認する可能性がある。
Intec イスラエルは合併を第351(A)条の交換条件に適合させる予定である。INTECイスラエル社の立場は国税局や裁判所に拘束力がなく、INTECイスラエル社は国税局に合併について裁決を求めるつもりはない。したがって,国税局が第351(A)条取引所としての帰化合併·合併の資格を問わない保証はなく,裁判所がこのような挑戦に耐えないことも保証されない。米国国税局がこのような論争の中で勝利した場合、または他の何らかの理由により、馴化合併が第351(A)条の交換の一部とみなされていない場合、馴化合併は、イスラエル国際技術会社の株を保有する米国の所有者にとって課税事件となる可能性がある。イスラエルの国際技術会社の株の前保有者は、現地化合併の税収結果について自分の税務顧問に相談するように促された。
現地化合併は、第351条交換の資格に適合することが意図されているが、現地化合併は、イスラエル国際技術会社の普通株を有するいくつかの米国保有者にとって課税事件である可能性がある。
は守らなければならない“帰化合併と合併が米国連邦所得税に及ぼす実質的な影響“米国証券取引委員会に提出した改正S-4表登録説明書(第333-255389号文書)又はS-4表に記載されている受動的外国投資会社の出願又は外国投資委員会規則に記載されているように、現地化合併は、合併と共に第351条(A)取引所としての資格を有することを意図する。それにもかかわらず、イスラエル国際技術会社の株のいくつかの元米国保有者 は、イスラエル国際技術会社がPFCに分類される可能性があるため、“規則”のPFICルールに基づいて課税される可能性がある。
一般リスク因子
不利なグローバル経済状況は、私たちの業務、財務状況、または経営結果に悪影響を及ぼす可能性がある。
私たちの運営結果は、世界経済と世界金融市場の全体的な状況の悪影響を受ける可能性がある。例えば、インフレと金利上昇、ロシアとウクライナの間の衝突、および他のマクロ経済要素により、米国と世界市場は資本と信用市場および大口商品価格の極端な変動と中断を経験し続けてきた。現在のマクロ経済環境のような深刻または長期的な経済低迷は、必要に応じて許容可能な条件で追加資本を調達する能力(あれば)を含む、我々の業務に様々なリスクをもたらす可能性がある。Brの疲弊や衰退の経済も私たちの臨床試験候補製品を生産するための原材料サプライヤーを逼迫させ、供給中断を招く可能性がある。また、私たちの株価は低下する可能性があり、一部の原因は株式市場の変動といかなる一般的な経済低迷でもある。
| 57 |
税収法律法規の変化は私たちの業務、財務状況、経営結果に悪影響を及ぼす可能性があります。
新しいbr収入、販売、使用、または他の税金法律、法規、規則、または条例はいつでも公布される可能性があり、これは私たちの将来の任意の収入の税収処理に影響を与える可能性がある。さらに、既存の税金法律、法規、規則、法規または条例は、私たちに解釈、変更、修正、または適用される可能性がある。一般的に、将来適用される税収法法規の変化、あるいはその解釈·適用は、トレーサビリティを有する可能性があり、我々の業務、財務状況、経営業績に悪影響を及ぼす可能性がある。私たちはこのような変化が起こるかどうかを予測できません。もしそうすれば、私たちの業務に最終的な影響を与えます。私たちは投資家に彼らの法律と税務顧問に相談して、税法の潜在的な変化が私たちの普通株への投資に与える影響を理解するように促します。
項目 1 B.未解決の従業員のコメント。
私たち はアメリカ証券取引委員会職員が発表した未解決のコメントはありません。
第 項2.属性
私たちの主な実行事務室はニューヨーク州ニューヨーク市コロンブス環島三号十五階にあります。また、2023年10月31日に満期になる賃貸契約によると、カリフォルニア州サンディエゴで約2,000平方フィートのオフィススペースをレンタルしました私たちの施設は私たちの現在の需要を満たすのに十分で、必要な時に適切な追加空間を提供すると信じています。
第 項3.法的訴訟
私たちは通常の業務過程で発生する様々な訴訟や法的手続きに時々巻き込まれる可能性があります。訴訟 は固有の不確実性の影響を受け,これらや他の事項は時々不利な結果をもたらし,我々の業務を損なう可能性がある.
2022年7月13日、LTS Lohmann治療システム株式会社(“LTS”)は国際商会に仲裁請求(“請求”)を提出し、同社の子会社INTECイスラエルを被告とした。請求書によると,LTSは1つのプロセス開発プロトコル(“PDA”)によって200万ユーロの支払いを得る権利があり,同協定は従来のアコーディオン錠業務を終了した。イスラエルの技術コンサルティング会社は以前このお金と他の関連費用を積算した。2022年8月、イスラエル国際技術会社はいわゆる債務を支払うために約100万ユーロ(約100万ドル)を支払い、2023年2月7日、イスラエル国際技術会社は80万ユーロ(約860,000ドル)を支払い、LTSとの紛争を解決した。
2023年3月17日現在、未解決の重大な法的手続きはなく、私たちは現在、いかなる法的手続きや私たちまたは私たちの財産に対するクレームがあるかを知りません。私たちは私たちの業務、財務状況、または経営業績に大きな影響を与えると思います。私たちの上級管理者や取締役はいかなる法的手続きでも私たちに反対していません。
第br項4.鉱山安全情報開示
は適用されない.
| 58 |
第 第2部分
第 項5.登録者普通株市場、関連株主事項、発行者が株式証券を購入する。
市場情報
INTECイスラエル社の普通株は歴史的にナスダック資本市場で取引されており、コードは“NTEC”である。現地化合併を実現するために、イスラエル国際技術会社の普通株は1対1で私たちの普通株に変換された。合併完了後、私たちの普通株は2021年8月4日にナスダック資本市場に上場し、取引コードは“Indaptus Treateutics,Inc.”である。株式コード“INDP”です
所持者
2023年3月17日まで、私たちの普通株は7人の記録保持者がいます。この数字には、著名人またはマネージャーを通過した“ストリート名”口座におけるその株式の人数は含まれていない。
配当政策
私たちは株主に現金配当金を支払うことを発表したことがありません。予測可能な未来にも現金配当金を支払うつもりはありません。私たちはどんな収益も発展と拡大に投資するつもりです。将来、私たちの配当政策に関連する任意の決定は私たちの取締役会が自ら決定し、多くの要素に依存して、私たちの財務状況、経営の結果、契約制限、資本要求、業務の見通し、私たちの戦略目標と業務拡大の計画、適用されるbr法律、および私たちの取締役会が関連すると思う可能性のある他の要素を含む。
株式補償計画に基づいて発行された証券
当社が株式証券の発行を許可した株式補償計画に関する資料 は、本年報“第III部分-第12項:いくつかの実益所有者及び管理層の保証所有権及び関連株主の件”に掲載されている。
最近販売されている未登録証券
2022年12月31日までの年度内に、リンカーンパーク資本基金有限責任会社に私たちの普通株を売却する以外に、私たちは未登録証券を何も売却していません。これは、2022年12月23日に米国証券取引委員会に提出された現在のForm 8-K報告書で開示されています。
発行者と関連購入者が株式証券を購入
2022年12月31日までの四半期内に、私たちは何の株式証券も買い戻していません。
第 項6[保留されている]
第br項7.経営陣の財務状況と経営成果の検討と分析
あなたは、以下の財務状況と運営結果の検討と分析、ならびに私たちの連結財務諸表と本年度報告書の他の部分に含まれる関連説明および他の財務情報を読まなければなりません。以下の議論には, がリスク,不確実性,仮説の影響を受ける前向き陳述が含まれる.“リスク要因の要約”と第1部1 A項目のタイトルの章を見るべきです。本年度報告における“リスク要因”は, の実際の結果が以下に述べる結果と大きく異なる重要な要因をもたらす可能性があることを検討した
| 59 |
概要
私たちは臨床バイオテクノロジー会社で、抗癌と抗ウイルス免疫療法を投与する斬新で特許を取得したシステムを開発しています。私たちは1世紀以上の免疫療法の進歩に基づいて進化してきました。我々の方法は、天然および獲得性免疫細胞および関連する抗腫瘍および抗ウイルス免疫反応の有効な活性化は、安全に静脈内注射することができる多標的の免疫系活性化シグナルパッケージを必要とするという仮定に基づく。我々の特許技術は,静注を減少させることを目的とした弱毒化と殺菌,非病原性,グラム陰性細菌の単一菌株 からなる。毒性であるが、先天性免疫と獲得性免疫の多くの細胞成分を起動或いは活性化する能力に大きく影響しない。この方法は、既存の5種類の異なる種類の薬物(チェックポイント治療、標的抗体治療および低用量化学療法を含む)によって観察された持続的な抗腫瘍反応相乗作用を含む、臨床前モデルにおいて広範な抗腫瘍および抗ウイルス活性を産生している。著者らの技術による腫瘍根絶は先天性と獲得性免疫記憶の活性化を示し、重要なことは、臨床前モデルにおいて腫瘍抗原 を提供或いは標的する必要がないことである。われわれはすでにわれわれの主要な臨床候補薬Decey 20の現在のcGMP生産に成功し、他のイネーブルINDの研究を完成した。当社の業務·運営に関するより多くの情報は、“第1部項目1.業務”を参照されたい
新冠肺炎の流行とマクロ経済情勢がわれわれの業務に及ぼす影響
新冠肺炎の大流行は引き続きアメリカと世界経済に影響を与え、私たちと私たちが依存している第三者の運営に影響を与える可能性があり、私たちの候補製品の供給中断、現在と未来の臨床試験の進行を含む。例えば,大流行により我々のGMPプロセスにかかる時間は予想よりも長い.また、新冠肺炎の疫病は、食品·薬物管理局や他の衛生当局の運営に影響を与える可能性があり、これは、私たちの候補製品の審査と承認を含む審査と承認の遅延を招く可能性がある。また、サプライチェーンの緊張やインフレ上昇などの関連経済発展は世界金融市場にマイナス影響を与え、私たちの資本獲得能力を低下させる可能性があり、これは私たちの短期的かつ長期的な流動性にマイナスの影響を与える可能性がある。新冠肺炎疫病と経済低迷の最終的な影響は高度不確定 であり、変化する可能性がある。これらの条件がどのくらい続くか、私たちの完全な財務的影響が何になるかは不明だが、私たちの融資努力や追加の技術開発はマイナスの影響を受ける可能性がある。
餌 統合
2021年8月3日、我々はDecoyとの合併(“合併”)を完了し、当社、Decoy、Intecイスラエル社、帰化合併子会社、イスラエル社と当社の完全子会社、およびDillon合併子会社(デラウェア州の会社と当社の完全子会社)との間で2021年3月15日に締結された合併協定に規定されている条件を満たし、免除し、この合意に基づいて、合併子会社がDecoyと合併してDecoyに組み込まれる。おとりが当社の完全子会社として存続するにつれ,おとりによる業務は合併後の会社による業務となっている.
2021年7月27日、吾ら、イスラエル国際技術会社及び帰化合併付属会社は帰化合併協定の条項及び条件に基づいて先に発表した帰化合併を完成し、これにより、帰化合併付属会社はイスラエル国際技術会社と合併して国際技術会社イスラエル会社に合併し、イスラエル国際技術会社は吾などの存続実体及び完全資本付属会社である。帰化合併時には,イスラエル国際技術会社 は帰化合併直前に構成されたすべての資産,権利,権力および財産を継続し,br}は帰化合併直前に構成されたすべての債務,負債および義務を継続している。
また, 合併については,名称を“Intec Parent,Inc.”から“Intec Parent,Inc.”に変更する.“Indaptus Treateutics,Inc.”へ
合併完了後、私たちの普通株は2021年8月4日にナスダック資本市場で取引を開始し、取引名は “Indaptus Treateutics,Inc.”である。株式コード“Indp”です
| 60 |
最近の 事件
リンカーン公園は株式基金を約束した
2022年12月22日、リンカーンパーク資本基金有限責任会社(“リンカーンパーク”)と購入契約を締結しました。この協定によると、リンカーン公園への販売義務はありません。リンカーン公園は購入契約期間内に合計2,000万ドルの普通株(いくつかの制限を受けて)を時々購入する義務があります。また、S-1表登録声明を提出して、購入契約に基づいて発行可能な普通株の転売を支払うことができます。
購入契約調印後、リンカーン公園に142,450株の初期承諾株を発行し、リンカーン公園にbr株普通株を追加発行する義務があり、購入契約に従って計算すると、12.5万ドルの価値があり、購入合意により、私たち は購入契約に従って1,000万ドルを超える普通株を売却し、最大76,220株の普通株(任意の再編、資本再編、非現金配当、株式分割、逆株分割または他の類似 取引によって適切に調整された)。すべての場合、リンカーン公園の撤回できない約束の対価格として、リンカーン公園は購入契約に従って私たちの指示に従って私たちの普通株を購入するだろう。また、私たちは3,781,330株までの普通株を予約して、リンカーン公園にもっと多くの株式を売却することを決定した時、購入契約に従って時々リンカーン公園に発行して販売します。
経営成果の構成要素
研究と開発費
研究開発費は私たちの運営費用の大きな部分を占めている。研究開発費には、主に契約研究組織(CRO)と契約製造組織(CMO)に支払われる費用と、CROおよびCMOの仕事に参加するある従業員の報酬 費用および研究開発活動のための材料が計画、管理、分析されている。私たちは発生した費用に応じて研究と開発費用を支払う。
我々 は,CRO,CMOや他の外部サービスプロバイダと合意した合意に基づき,受信したサービスの見積もりと努力費用に基づいて,第三者が実行する製造,臨床前研究,臨床試験活動に費用を計算しなければならない。これらの見積り数は,完了した作業割合に適した契約金額に基づいて決定され,サービスの進捗や完了段階について内部者や外部サービスプロバイダと分析することで決定される.もしbr}CRO、CMO、または外部サービスプロバイダに前金を支払うと、支払いを前払い資産として記録し、その資産は契約サービスを実行する際に償却または支出される。しかし,これらの活動の実際のコストや時間は高度に不確実であり,リスクの影響を受け,われわれの臨床開発計画を含む多くの要因によって変化する可能性がある。
私たちは予測可能な未来に、私たちの研究開発費用は大幅に増加すると予想されています。私たちは引き続き臨床開発活動を強化し、私たちの研究開発を支援するためのより多くの人員を募集することに関連する費用が発生するからです。私たちの将来の非臨床·臨床開発プロジェクトへの支出は、時間と完成コストの多くの不確実性の影響を受けています。候補製品の臨床前研究、臨床試験と開発の持続時間、コスト、時間は様々な要素に依存する
| ● | 承認の時間と受け入れを規制する | |
| ● | 臨床前研究と臨床試験及びその他の研究と開発活動の範囲、進捗と費用 | |
| ● | 規制当局が要求している潜在的安全監視とその他の研究 | |
| ● | 重要なbrと変化する政府規制。 |
必要な臨床研究を行い、FDAと他の監督部門の許可を得る過程は高価で時間がかかり、候補製品の開発成功も非常に不確定である。我々の研究開発プロジェクトに関するこれらのリスクと不確実性 は“第1部第1 A項”でより全面的に議論されている。リスク要因-巨額の研究と開発費、その他の運営費が引き続き発生することが予想され、利益を実現することが困難になる可能性があります“これらのリスクと不確実性のため、私たちは、私たちの研究開発プロジェクトの持続時間と完成コストをどの程度決定することができないか、または私たちがいつ、またはどの程度規制部門の承認を得た候補製品の商業化および販売から収入を得るかどうかを決定することはできません。私たちは私たちのどんな候補製品も規制部門の承認を得ることに決して成功しないかもしれない。
| 61 |
一般料金 と管理費用
一般費用および行政費用には、役員管理、財務管理、人的資源の報酬、従業員福祉、株式ベースの報酬、施設コスト(賃貸料を含む)、専門サービス料、その他の一般管理費用(減価償却を含む) が含まれ、私たちの運営を支援します。
私たちの予測可能な未来には、私たちの一般的かつ管理費用は大幅に増加すると予想されています。私たちは引き続き従業員数を増やして、私たちの研究開発活動と運営、業務の成長を支援し、もし私たちの任意の候補製品 が市場の承認を得たら、商業化活動を支持します。米国証券取引委員会の規則遵守に関する費用、追加の取締役や上級管理者保険費用、投資家関係活動、その他の行政や専門サービスなど、上場企業として運営 が継続して追加費用が発生することも予想される。
その他 純収入
その他 収入は、純額には預金利息と当社コア業務に付随する他の収入、費用、損益項目 が含まれています。2022年には、他の収入には前年の契約債務を清算する収益が含まれる。
運営結果
2022年12月31日までの年度と2021年12月31日現在の年度との比較
次の表は,2022年12月31日と2021年12月31日までの年間経営実績と,この2年間の相対金額とパーセント変化を示している。
| 十二月三十一日までの年度 | 変わる (2022年~2021年) | |||||||||||||||
| 2022 | 2021 | ($) | % | |||||||||||||
| 運営費用: | ||||||||||||||||
| 研究開発 | $ | 6,324,657 | $ | 2,523,153 | $ | 3,801,504 | 150 | % | ||||||||
| 一般と行政 | 8,586,249 | 5,205,955 | 3,380,294 | 65 | % | |||||||||||
| 総運営費 | 14,910,906 | 7,729,108 | 7,181,798 | 93 | % | |||||||||||
| 運営損失 | (14,910,906 | ) | (7,729,108 | ) | (7,181,798 | ) | 93 | % | ||||||||
| その他の収入、純額 | 588,108 | 17,722 | 570,386 | 3,218 | % | |||||||||||
| 純損失 | $ | (14,322,798 | ) | $ | (7,711,386 | ) | $ | (6,611,412 | ) | 86 | % | |||||
| 普通株株主は純損失,基本損失と希釈損失を占めなければならない | $ | (1.73 | ) | $ | (1.89 | ) | $ | 0.16 | (8.5 | )% | ||||||
| 1株あたりの基本純損失と希釈後の純損失を計算するための加重平均株数 | 8,262,119 | 4,090,599 | ||||||||||||||
研究と開発費
2022年12月31日までの年度の我々の研究開発費は約630万ドルであり,2021年12月31日までの年度の約250万ドルに比べて約380万ドル,あるいは約150%増加しているこの増加は、主に(I)賃金および関連費用が約150万ドル増加したためであり、その中には、約350,000ドルの株式ベースの報酬が含まれており、(Ii)私たちの第1段階臨床試験は、br}IND準備および提出、ならびに私たちの先行臨床候補の製造プロセスに関連するコストを含む約220万ドル増加した。
| 62 |
一般料金 と管理費用
2022年12月31日までの年度は、我々の一般·行政費は約860万ドルであり、2021年12月31日までの年度の約520万ドルに比べて、約340万ドル、または約65%増加している。T.T彼の増加は、主に(I)賃金と関連支出が約110万ドルの株式報酬を含む約180万ドル増加したためであり、これは、我々の役員チームが合併後に従業員数を増加させたことと、(Ii)合併後の役員や上級管理職の保険料、専門費、その他の合併後の上場企業に関する費用が約130万ドル増加したためである.
その他 収入
2021年同期と比較して、2022年他のbr純収入が増加した要因はLTS課税プロジェクトの一部を打ち抜き、約365,000ドルである理由は、私たちが2023年2月にこの債務を決済し、受け取った収益が保有している資産の推定公正価値、預金利息と実現した収益を超えているからです有価証券にあります。
流動資金 と資源
私たちは今のところ承認された製品は何もなく、製品販売から何の収入も得たことがありません私たちの設立以来、私たちは主に私たちの株式証券を公開と非公開で発行することで私たちの運営に資金を提供してきた。
2021年8月、私たちは2,727,273株の私たちの普通株を購入し、2,727,273株の私たちの普通株を購入するために、事前に出資した引受権証を売却した。この株式承認証は1株11.00ドルの行使価格で行使できる。2021年9月、事前資金権証は1株0.01ドルの使用価格で全面的に行使された。事前融資権証はbr権証とともに販売され、合計価格は11.00ドルで、事前融資行使価格を含む。配給代理費と発売費用約270万ドルを差し引いた純収益総額は約2,730万ドルです。予め出資した引受権証と引受権証のより多くの情報については、今年度の報告書の総合財務諸表付記7を参照してください。
2022年6月、吾らはH.C.ウェインwright&Co.,LLCを販売代理(“ウェインライト”)として2022年9月1日に改訂された市場発売協定(“ATMプロトコル”)を締結し、この合意により、吾らは時々Wainwrightを通して普通株を発売·販売することができ、1株当たり額面0.01ドル、総収益は最高630万ドルに達する私たちは合意に基づいて普通株式を発行して販売しますATM協定我々が2022年9月1日に米国証券取引委員会に提出し、2022年9月9日に発効を発表したS-3表に基づく有効な“棚上げ”登録声明による。普通株の株式根拠がないATM協定.
2022年12月、私たちはリンカーン公園と購入契約と登録権協定を締結し、協定によると、リンカーン公園は2000万ドルに達する普通株を購入することを約束した。“--最新の事件−リンカーン公園が約束した持分施設を参照してください。”この計画によると、普通株は販売されていません。
2022年12月31日までに現金と現金等価物を持っています有価証券約2,640万ドルです私たちは信用限度額や保証のような持続的な重大な融資約束を持っておらず、私たちの今後5年間の流動性に影響を及ぼすと予想される。
キャッシュフロー
操作 活動
2022年12月31日までの年度,経営活動で使用されている現金純額は約1,310万ドルであるのに対し,2021年12月31日までの年度では,経営活動で使用されている現金純額は約1,130万ドルである現金使用純額が180万ドル増加したのは主に第1段階の臨床試験と一般事務と行政費を増やす合併後の上場企業と関係がある.
| 63 |
投資 活動
2022年12月31日までの年度の投資活動用現金純額は約1,640万ドルであり,これは主に有価証券への純投資,金額は 約 1,660万ドルは収益の約20万ドルで相殺されます販売待ち資産の入金状況 それは.2021年12月31日までの年度,投資活動が提供する現金純額は約50万ドルであり,主に収益約50万ドルに関係している販売待ち資産を持つ入金
活動に資金を提供する
2022年12月31日まで、融資活動は現金純額を提供していない。2021年12月31日までの年間融資活動が提供する現金純額は約4,830万ドルであり,これは主に合併と2021年8月に完了した私募と,合併発効時に認可投資家に発行された一連の詐欺金庫(将来株式簡単協定) によるものである。
資金需要
私たちの運営費は2022年に大幅に増加し、将来も増加することが予想され、私たちの持続的な活動と関係があり、特にE私たちの研究と開発費用は大幅に増加すると予想されます。私たちは臨床開発活動を強化し、より多くの人を募集して私たちの研究と開発を支援することに関連する費用が発生するからですそれは.また、私たちの候補製品が市場で承認されれば、製品販売、マーケティング、製造、流通に関連した巨額の商業化費用が発生すると予想されています。また、上場企業の運営に関連した巨額のコストが引き続き発生する見通しだ。
私たちは、2022年12月31日まで、私たちの既存の現金と現金等価物と有価証券に加えて、リンカーン公園で持分基金の下で持分証券を売却する能力を加えて、2024年第2四半期まで続く活動に資金を提供するのに十分であると信じている。
私たちの未来の資本需要は多くの要素に依存するだろうが、これらに限定されない
| ● | 臨床前研究と臨床試験の範囲、進展、結果とコスト | |
| ● | 我々の臨床試験と他の研究開発計画の範囲、優先度と数量 | |
| ● | 私たちは将来私たちの候補製品についての許可、協力、開発、商業化の手配に基づいて得られた 収入 | |
| ● | 新冠肺炎の流行が私たちの業務と運営に与える影響は | |
| ● | インフラ施設の開発と拡張のコストは | |
| ● | 私たちの候補製品に対する規制審査のコスト、時間、結果 | |
| ● | 私たちまたは私たちのパートナーは、私たちの将来の潜在的なライセンス契約に基づいて、開発マイルストーン、マーケティング承認、その他のイベントまたは開発を実現する能力を実現します | |
| ● | 特許請求の範囲および他の知的財産権出願の提出、起訴、執行および弁護費用 | |
| ● | 臨床または商業生産工場の製造手配のコストと時間を確保する |
| 64 |
| ● | 第三者との契約は、販売およびマーケティング能力、またはそのような能力を自ら確立するコストを提供してくれます | |
| ● | 任意の未来の製品、候補製品、または技術移転の開発および商業化のコストを取得または負担する | |
| ● | 私たちの一般と管理費用は | |
| ● | 将来的に私たちの1つまたは複数の候補製品に関連する内部および外部許可スケジュールに基づいて、私たちは任意の コストを生成する可能性がある。 |
潜在的な候補製品を決定し、臨床前研究と臨床試験を行うことは時間がかかり、高価で不確定な過程であり、完成するのに数年かかり、しかも私たちは永遠に市場の許可を得て製品販売を実現するために必要なデータ或いは結果を生成できないかもしれない。しかも、私たちの候補製品が承認されれば、商業的成功を得られないかもしれない。私たちの商業収入(あれば)は販売候補製品から来ますが、これらの製品は今後数年以内に商業使用に投入されないと予想されます。もしあれば。したがって、私たちは追加的な資金調達に依存して私たちの業務目標を達成し続ける必要があるだろう。私たちは受け入れ可能な条項で十分な追加的なbr融資を得ることができないかもしれないし、融資を受けることができないかもしれない。例えば、新冠肺炎の流行により、他の生物製薬会社との株の取引価格は非常に不安定になってきた。そのため、受け入れ可能な条件で普通株を売却することで資本を調達する困難(あれば)に直面する可能性がある。本年度報告第I部第1 A項の“リスク要因”を参照。もし私たちが必要な時や魅力的な条件下で資金を調達できない場合、私たちは、利用可能な資源に応じて私たちの支出レベルを下げるために、私たちの研究開発計画や他の業務や将来の商業化努力を延期、縮小または廃止し、利用可能な資源に応じて必要な変更を要求されるかもしれません。
契約義務
Brを運営する賃貸支払いは、カリフォルニア州サンディエゴのオフィスの将来の賃貸料に対する私たちの約束を表しています。これらのレンタル料はキャンセルできないレンタルに基づいて行われます。2022年12月31日現在、私たちの経営賃貸義務の将来の支払い総額は約82,000ドルで、今後12ヶ月以内に満期になります。レンタルの詳細については、本年度報告Form 10-Kに含まれる連結財務諸表付記8を参照されたい。
本報告で述べた期間、私たち はなく、現在のところ米国証券取引委員会 ルールで定義されたいかなる表外手配もありません。
キー会計政策
私たちの財務状況と経営結果の討論と分析は、アメリカ公認会計原則に基づいて作成された当社の連結財務諸表に基づいています。これらの連結財務諸表を作成する際には、資産、負債、費用報告金額に影響を与える推定を行う必要がある。我々が採用した重要会計政策は,見積もりの使用を含め,本年度報告に含まれる年次財務諸表の付記で紹介する。私たちは定期的に私たちの推定を評価します。これらの推定は、歴史的経験と、当時の状況で合理的であると考えられる様々な他の仮定に基づいています。 キー会計政策は、私たちの財務状況と運営結果の記述に最も重要な政策 であり、主観的または複雑な判断が必要であるため、本質的に不確実な事項の影響を推定する必要があります。実績が歴史的経験と異なる場合、あるいは基本的な仮定が変化すれば、私たちの財務状況や運営結果は大きな影響を受ける可能性がある。
| 65 |
私たち は、以下の会計政策が、私たちの合併財務諸表を作成する際に使用する判断と推定に最も重要であると考えています
研究開発コストの計算
我々 はCROとCMOが提供するサービスに関するコストを記録する.作業範囲およびスケジュールは、一般に、署名された合意に基づいているが、支払いプロセスは、常にサービスおよび材料を提供するbr期限に一致するわけではないので、定期料金を決定する際にいくつかの判断が含まれる。したがって、私たちの経営陣は、これらの第三者との合意に基づいて、受信したサービスと費用の努力を期末日ごとに見積もる必要があります。2022年12月31日までの年間で約630万ドルの研究開発費が発生し,そのうち約370万ドルが我々のCROとCMOが提供するサービス に用いられている。2022年12月31日現在、発生したが領収書が発行されていない費用、および約80万ドルの前払い費用および非流動他の資産のための約30万ドルの負債が記録されており、今後の期間に関する支払いのために使用されている。得られたサービスまたは費用を過大評価または過小評価する努力は、報告期間内に発生する研究開発費および関連する計算および前払い費用を過大評価または過小評価する可能性がある。
株に基づく報酬
株式オプションの付与に関する補償 は、付与日に付与された推定公正価値に基づいて計量され、個別に付与された必要なサービス期間(通常は帰属期間に等しい)内で直線的に確認される。我々は,ブレイク-スコアーズ推定モデルを用いて,付与された日における各株式オプションの推定公正価値を決定し,brに関する多くの複雑かつ主観変数に関する仮定を用いた.無リスク金利は米国債収益率をもとに、期限は付与時に発効するオプションの期待期限と一致する。予想変動率は同業会社グループの歴史変動性の分析に基づいている。期待期間は、私たちの株式オプション未償還の期間を表します。 期待期限は、オプション付与日と満期日との間の中間点である米国証券取引委員会従業員会計公告110に規定されている簡略化方法を用いて推定されるものとする。私たちは私たちの普通配当金を発表したり支払ったりしたこともなく、予測可能な未来にそうする計画もない。これらの仮定の変化は、私たちが確認した株式オプションに関する株式報酬支出金額に変化をもたらす可能性がある。
最近会計公告が発表された
最近発表されたある会計声明は、本年報“第8項.財務諸表と補足データ”に列挙された総合財務諸表の付記2“重要会計政策概要”で議論されている。
第 7 A項。市場リスクに関する定量的で定性的な開示。
我々 は取引法ルール12 b-2で定義されている小さな報告会社であり,本プロジェクト7 Aに要求される他の情報を提供する必要はない.
| 66 |
第br項財務諸表および補足データ
INDAPTUS 治療会社
2022年年報
INDAPTUS治療会社は
連結財務諸表
カタログ
|
ページ | |
| 独立公認会計士事務所レポート(PCAOB名:Haskell&White LLPとPCAOB ID: |
F-2 | |
| 合併財務諸表: | ||
| 2022年と2021年12月31日までの連結貸借対照表 | F-3 | |
| 2022年と2021年12月31日までの総合業務報告書 | F-4 | |
| 2022年12月31日までと2021年12月31日まで年度株主権益総合レポート | F-5 | |
| 2022年12月31日と2021年12月31日までの統合現金フロー表 | F-6 | |
| 連結財務諸表付記 | F-7 |
| F-1 |

独立公認会計士事務所報告{br
株主や取締役会に
Indaptus治療会社
連結財務諸表に関する意見
Indaptus Treateutics,Inc.(“当社”)2022年12月31日現在と2021年12月31日までの連結貸借対照表,同年度までの運営と全面赤字,株主権益とキャッシュフローに関する連結報告書,および関連付記 (総称して“合併財務諸表”と呼ぶ)を監査した。総合財務諸表は,すべての重要な面で,会社の2022年,2022年,2021年12月31日までの総合財務状況,およびこれまでの各年度の総合経営実績とキャッシュフローを公平に反映しており,米国公認会計原則に適合していると考えられる。
意見の基礎
これらの連結財務諸表は会社の経営陣が責任を負う。私たちの責任は、私たちの監査に基づいて合併財務諸表に対して意見を発表することです。私たちはアメリカ上場会社会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、アメリカ連邦証券法と適用規則 およびアメリカ証券取引委員会とPCAOBの規定に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って審査を行った。これらの基準は、連結財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても詐欺によるものであっても、監査を計画し、実行することを要求する。当社は必要なく、その財務報告の内部統制を委託されて監査することもありません。私たちの監査の一部として、財務報告の内部統制を知る必要がありますが、社内財務報告の内部統制の有効性について意見を述べるためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、連結財務諸表の重大な誤報リスクを評価するプログラム(エラーによるものであっても詐欺によるものであっても)、これらのリスクに対応するプログラム を実行することが含まれる。これらの手続きは、連結財務諸表中の金額および開示に関する証拠をテストに基づいて検討することを含む。私たちの監査には、経営陣が使用する会計原則の評価と重大な推定、合併財務諸表の全体列報の評価も含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
重要な監査事項とは、連結財務諸表を今期監査する際に生じる、監査委員会に伝達または要求された事項であり、(1)総合財務諸表に対して大きな意義を有する勘定または開示に関連し、および(2)私たちが特に挑戦的、主観的、または複雑な判断に関連する。私たちは重要な監査事項が存在しないと確信する。
| /s/
| |
| Haskell &White LLP |
私たちは2021年以来当社の監査役を務めてきました。
2023年3月16日

| F-2 |
INDAPTUS 治療会社
合併貸借対照表
| 十二月三十一日 | ||||||||
| 2022 | 2021 | |||||||
| 資産 | ||||||||
| 流動資産: | ||||||||
| 現金と現金等価物 | $ | $ | ||||||
| 有価証券 | ||||||||
| 販売待ち資産を保有する | ||||||||
| 前払い費用と他の流動資産 | ||||||||
| 流動資産総額 | ||||||||
| 非流動資産: | ||||||||
| 財産と設備、純額 | ||||||||
| 使用権資産 | ||||||||
| その他の資産 | ||||||||
| 非流動資産総額 | ||||||||
| 総資産 | $ | $ | ||||||
| 負債と株主権益 | ||||||||
| 流動負債: | ||||||||
| 売掛金とその他の流動負債 | $ | $ | ||||||
| 賃貸負債を経営し、今期の部分 | ||||||||
| 流動負債総額 | ||||||||
| 非流動負債: | ||||||||
| 賃貸負債を経営し,当期分を差し引く | ||||||||
| 非流動負債総額 | ||||||||
| 総負債 | ||||||||
| 引受金及び又は有事項(付記8) | ||||||||
| 株主権益: | ||||||||
| 普通株:$額面は2022年12月31日と2021年12月31日までに認可された株そして2022年12月31日と2021年12月31日までの発行済株式 | ||||||||
| 追加実収資本 | ||||||||
| 赤字を累計する | ( | ) | ( | ) | ||||
| その他の総合収益を累計する | ||||||||
| 株主権益総額 | ||||||||
| 総負債と株主権益 | $ | $ | ||||||
連結財務諸表の付記を参照
| F-3 |
INDAPTUS 治療会社
合併経営報告書と全面赤字
| 12月31日までの年度 | ||||||||
| 2022 | 2021 | |||||||
| 運営費用: | ||||||||
| 研究開発 | $ | $ | ||||||
| 一般と行政 | ||||||||
| 総運営費 | ||||||||
| 運営損失 | ( | ) | ( | ) | ||||
| その他の収入、純額 | ||||||||
| 純損失 | $ | ( | ) | $ | ( | ) | ||
| 普通株主1株当たりの基本普通株と希釈した普通株の純損失 | $ | ( | ) | $ | ( | ) | ||
| 1株あたりの基本純損失と希釈後の純損失を計算するための加重平均株数 | ||||||||
| 純損失 | $ | ( | ) | $ | ( | ) | ||
| 他の全面的な収入: | ||||||||
| 純損失を計上した有価証券は収益の再分類調整を実現している | ( | ) | ||||||
| 有価証券の未実現収益 | ||||||||
| 総合損失 | $ | ( | ) | $ | ( | ) | ||
連結財務諸表の付記を参照
| F-4 |
INDAPTUS 治療会社
合併の株主権益報告書
| 系列種を優先的に選択する | 普通株 | その他支払い済み | 積算 | その他の総合 | ||||||||||||||||||||||||||||
| 株 | 金額 | 株 | 金額 | 資本 | 赤字.赤字 | 収入(損) | 合計する | |||||||||||||||||||||||||
| 残高、2021年1月1日 | $ | $ | $ | $ | ( | ) | $ | $ | ( | ) | ||||||||||||||||||||||
| 株に基づく報酬 | - | - | ||||||||||||||||||||||||||||||
| 優先株を転換する | ( | ) | ( | ) | ||||||||||||||||||||||||||||
| 金庫の改装 | - | |||||||||||||||||||||||||||||||
| 合併後に普通株を発行し,おとり会社の取引コストを差し引くと,金額は$となる | - | |||||||||||||||||||||||||||||||
| 源泉融資権証と引受権証を発行し、発行コストを差し引いて#ドルです | - | - | ||||||||||||||||||||||||||||||
| 事前出資の引受権証を行使する | - | |||||||||||||||||||||||||||||||
| 株式オプションの行使 | - | |||||||||||||||||||||||||||||||
| 純損失 | - | - | ( | ) | ( | ) | ||||||||||||||||||||||||||
| 残高2021年12月31日 | ( | ) | ||||||||||||||||||||||||||||||
| 株に基づく報酬 | - | - | ||||||||||||||||||||||||||||||
| 初期承諾株を発行する | - | ( | ) | |||||||||||||||||||||||||||||
| 純損失 | - | - | ( | ) | ( | ) | ||||||||||||||||||||||||||
| 純損失を計上した有価証券は収益の再分類調整を実現している | - | - | ( | ) | ( | ) | ||||||||||||||||||||||||||
| 有価証券は収益変動を実現していない | - | - | ||||||||||||||||||||||||||||||
| 残高2022年12月31日 | $ | $ | $ | $ | ( | ) | $ | $ | ||||||||||||||||||||||||
連結財務諸表の付記を参照
| F-5 |
INDAPTUS 治療会社
統合されたキャッシュフロー表
| 12月31日までの年度 | ||||||||
| 2022 | 2021 | |||||||
| 経営活動のキャッシュフロー: | ||||||||
| 純損失 | $ | ( | ) | $ | ( | ) | ||
| 純損失と経営活動で使用される現金純額の調整: | ||||||||
| 減価償却 | ||||||||
| 株に基づく報酬 | ||||||||
| 保有販売資産の実現収益 | ( | ) | ||||||
| 有価証券の実現収益 | ( | ) | ||||||
| 経営性資産と負債変動状況: | ||||||||
| 前払い費用と他の流動資産 | ( | ) | ||||||
| 売掛金とその他の流動負債 | ( | ) | ( | ) | ||||
| その他の資産 | ( | ) | ( | ) | ||||
| 経営的リース使用権資産と負債純額 | ||||||||
| 経営活動のための現金純額 | ( | ) | ( | ) | ||||
| 投資活動によるキャッシュフロー: | ||||||||
| 販売待ち資産を持って受け取った収益 | ||||||||
| 財産と設備を購入する | ( | ) | ||||||
| 有価証券を購入する | ( | ) | ||||||
| 有価証券満期日 | ||||||||
| 投資活動が提供する純現金(用) | ( | ) | ||||||
| 資金調達活動のキャッシュフロー: | ||||||||
| 合併して得られる収益 | ||||||||
| 餌の取引コスト | ( | ) | ||||||
| 前払い資金の引受権証及び引受権証を発行する | ||||||||
| 指向性増発の発行コスト | ( | ) | ||||||
| 事前出資の引受権証を行使する | ||||||||
| 金庫の収益、純額 | ||||||||
| 株式オプションの行使 | ||||||||
| 融資活動が提供する現金純額 | ||||||||
| 現金および現金等価物の純増加 | ( | ) | ||||||
| 年初現金および現金等価物 | ||||||||
| 年末現金および現金等価物 | $ | $ | ||||||
| 非現金投融資活動 | ||||||||
| 優先株を転換する | $ | $ | ||||||
| 金庫の改装 | $ | $ | ||||||
| 負担した負債は,逆M&Aで受け取った非現金資産を差し引いた純額 | $ | $ | ||||||
| リース開始時の経営的使用権資産とリース負債の初歩的確認 | $ | $ | ||||||
| 非流動資産から流動資産に再分類する | $ | $ | ||||||
| 合併完了時の金の放出 | $ | $ | ||||||
| 初期承諾株を発行する | $ | $ | ||||||
| 有価証券は収益変動を実現していない | $ | $ | ||||||
| 補充キャッシュフロー開示 | ||||||||
| 所得税の現金を納める | $ | $ | ||||||
| 預金利息で受け取った現金 | $ | $ | ||||||
連結財務諸表の付記を参照
| F-6 |
INDAPTUS 治療会社
連結財務諸表付記
注 1:一般情報
| a. | Indaptus Treateutics,Inc.及びその完全子会社Decoy BiosSystems,Inc.とIntec Pharma Ltd.(総称して“会社”と呼ぶ)は生物技術会社であり、切除できない或いは転移性の固形腫瘍とリンパ腫患者の根治性癌免疫治療の強化と拡大に力を入れている。 固形腫瘍とリンパ腫はすべての癌死亡の90%以上を占める。同社は先天性と適応性抗腫瘍と抗ウイルス免疫反応を活性化できる新型の多標的br製品を開発している。 |
Indaptus Indaptus治療会社(“Indaptus”)は、前身はIntec親会社であり、2021年2月24日にデラウェア州に設立され登録され、デラウェア州の民間会社であり、前上場会社Intec Pharma Ltd.(“Intecイスラエル”)の完全子会社でもある。
| b. | 2021年8月3日、Indaptus、Decoy、Intecイスラエル、馴化合併子会社Indaptusの完全子会社IndaptusとIndaptusの完全子会社Dillon Merge Inc.およびIndaptusの完全子会社Dillon Merge Inc.との間の合併合意および計画(期日2021年3月15日)に規定された条件が満たされた後、Indaptus はDecoy BiosSystems,Inc.(“Decoy”)との合併を完了した。この合意により、Merge SubとIndaptusの完全子会社であるDecoy,Inc.,DecoyがIndaptusの完全子会社として存続する(“合併”)にともない,Decoyが行う業務はIndaptusが行う業務となる. |
合併完了後、Indaptus普通株は2021年8月4日にナスダック資本市場で取引を開始し、取引名は“Indaptus Treateutics,Inc.”である。“INDP”の下にあります
| c. | 合併については,Indaptusは2021年7月23日にある機関投資家と証券購入協定(“購入合意”)
を締結し,この合意により,Indaptusは私募で事前資金権証および引受権証を売却·発行することに同意し,総収益純額は約$であった |
リスク と不確実性
Br社は、その業界の他の同様の規模の会社と類似した多くのリスクに直面しているが、これらに限定されないが、製品の開発に成功する必要があること、運営損失を補うために追加の資本または融資が必要であること(後述)、より大きな会社の代替製品およびサービスからの競争、ノウハウ保護、特許訴訟、および重要な個人への依存を含む。
新冠肺炎疫病は引き続きアメリカと全世界経済に影響を与え、会社の運営と会社が依存する第三者の運営に影響を与える可能性があり、候補製品の供給中断及び現在と未来の臨床試験の進行を含む。例えば、大流行により、そのGMPプロセスにかかる時間は、予想されるよりも長い。また、新冠肺炎の流行は、食品·薬物管理局や他の衛生当局の運営に影響を与え続ける可能性があり、これは、同社の候補製品の審査と承認を含む審査·承認の遅延を招く可能性がある。また、サプライチェーンの緊張やインフレや金利上昇などの関連経済発展は世界金融市場にマイナス影響を与え、会社の資本獲得能力を低下させる可能性があり、これはその短期的かつ長期的な流動性 にマイナス影響を与える可能性がある。新冠肺炎疫病と経済低迷の最終的な影響は高度に不確定であり、変化が発生する可能性がある。これらの条件がどのくらい継続するか,会社への完全な財務影響が何であるかは不明であるが,融資努力やその技術の追加開発は負の影響を受ける可能性がある。
| F-7 |
Br注目と経営陣の計画を進めている
会社設立以来運営中に純損失が発生し現金を使用しており,2022年12月31日までの累計赤字は約$である
経営陣は、潜在的な協力、brライセンス、および他の同様の配置を含む、株式および/または債務融資または他の資本源によって追加資本を調達する計画である。しかし,これらの計画は完全にその制御範囲内ではなく,発生可能と評価することはできない.当社の追加資本調達能力は、世界経済状況の潜在的な悪化と、最近のロシアとウクライナ間の衝突および持続的な新冠肺炎の流行が米国と世界の金融市場に与える破壊と変動の悪影響を受ける可能性がある。もし会社が必要な時や魅力的な条件下で資金を調達できない場合、それはその研究開発計画やその他の業務を延期、減少、またはキャンセルさせることを余儀なくされる。上記のようなイベント が発生すると,会社が開発や商業化目標を実現する能力は悪影響を受ける.
これらの総合財務諸表を作成する際には,会社が継続的に経営している企業として,このような不確実性の結果による調整は含まれていないと仮定する。
注 2:重大会計政策
デモベース
当社の総合財務諸表は、米国公認会計原則(“米国公認会計原則”)に基づいて作成されています。
統合原則
連結財務諸表はIndaptusおよびその子会社の勘定を含む。合併後、会社間残高と取引は流された。
見積もりを使った
米国公認会計原則に基づいて財務諸表を作成することは、財務諸表日の資産及び負債及び又は負債の開示、及び報告期間内に報告された費用金額に影響を与えるために、管理層に推定及び仮定を要求する。最も重要な推定数は、株式報酬の公正価値の決定と、いくつかの契約研究組織に対する期末債務の決定に関する。経営陣は、歴史的経験や他の要因(現在の経済環境を含む)を用いてその推定および仮定を継続的に評価し、事実や状況が必要な場合に調整する。これらの推定数は、連結財務諸表の日付までに取得可能な情報に基づいており、したがって、実際の結果はこれらの推定数とは異なる可能性がある。
1株基本と希釈後の損失 は,当期純損失を当期発行普通株の加重平均で割ったものである。1株当たり損失を希釈することは,普通株と普通株等価物の加重平均から計算される。普通株式等価物は、希釈された場合に、これらのオプションおよび引受権証が在庫株式法に含まれる発行された株式オプションおよび引受権証を含む。
| F-8 |
| 12月31日までの年度 | ||||||||
| 2022 | 2021 | |||||||
| 未償還株式オプション | ||||||||
| 株式承認証 | ||||||||
現金 と現金等価物
社は、購入時の元の満期日が3ヶ月以下のすべての高流動性投資を現金等価物と見なしている。 2022年12月31日と2021年12月31日まで、現金と現金等価物は主に小切手と貨幣市場預金からなる。当社の現金残高 は連邦保険の現金残高を超えていますが、当社は現金と現金等価物を持つ預金機関の財務力により、当社は重大な信用リスクに直面しないと考えています。当社ではこれまで未保険残高による損失は何も確認されていません。
販売待ち資産を持つ
合併については、当社は合併前にIntecイスラエル社が保有していたいくつかの設備が当社の運営に役に立たなくなったと認定し、関係者とそのような設備を売却する協定を締結した。当社は推定可変動純資産
を保有していることを確認した。非係り先が2021年11月にある買手から受け取った購入要約によると,デバイスの可変現純値は約#ドルである
有価証券
有価証券への会社の投資は、会計基準に基づいて編纂された(“ASC”)320を含み、証券を売却可能な米国債に分類される“投資--債務証券“と。これらの投資は公正価値で入金され、損益記入は他の全面収益(損失)或いは AOCIに記入されておらず、株主権益の単独構成部分としている。
財産 と設備
財産と設備資産はコストから減価償却累計を引いて申告する。減価償却は資産の予定耐用年数に応じて直線法で計算される。同社が使用している推定寿命は
特許
関連する法的コストを含めて発生した特許コストをBr社が支払い、添付の総合経営報告書にこのようなコストを一般および行政費用 に計上する。
研究開発費
研究および開発費用には、賃金、株式ベースの給与支出、賃金税および他の従業員福祉、下請け業者、および臨床試験および専門サービスを含む研究および開発計画の実施に直接関連するコストが含まれる。研究開発に関するすべてのコストは発生時に費用を計上している。
| F-9 |
当社は契約研究組織(“CRO”)、契約 製造組織(“CMO”)と他の外部サービスプロバイダとの合意義務による費用を計算し、その支払いの流れは当社にサービスや材料を提供する期限 と一致しない。計上額は,CRO,CMOと他の外部サービスプロバイダとの合意に基づいて受信したサービスとかかる作業の見積り数に基づいて記録される.これらの推定数は、一般に、完了した作業割合に適した契約金額に基づいており、サービスの進捗または完了段階を内部者および外部サービスプロバイダと分析することによって決定される。CRO、CMOまたは外部サービスプロバイダに前払いする場合、 支払いは前払い資産として記録され、契約サービスを実行する際に償却または支出される。
一般料金と管理費用
一般費用および行政費用には、会社の運営を支援するための役員管理、財務管理および人的資源の報酬、従業員福祉および株式報酬、施設コスト(賃貸料を含む)、専門サービス料、およびその他の一般管理費用(減価償却を含む) が含まれる。
所得税 税
所得税 は貸借対照法で計算される。繰延税金資産および負債の将来の税金項目での確認brは、既存の資産および負債の帳簿金額とそれぞれの課税ベースとの間の差、および営業損失と税金控除の間の差に起因することができます。繰延税項資産及び負債は、当該等の一時的な差額の回収又は決済が予想される年度の課税所得額の制定税率計測に期待される。税率変動が繰延税金資産や負債に及ぼす影響は、公布日を含む期間の収入で確認されている。もし繰延税金資産の一部または全部が予測可能な将来に現金化できない可能性が高い場合、繰延税金資産の推定値 を計上して到着する。会社は2022年12月31日と2021年12月31日までに、税金資産の全額評価準備を延期している。
所得税の頭寸がさらに持続する可能性がある場合にのみ、会社はこれらの頭寸の影響を確認する。確認された“br}所得税は、50%を超える可能性のある最大金額計量を実現するために使用されます。確認や計測の変化は変化が発生したと判断した期間に反映される.当社は未確認のbr税収割引に関する利息を利息支出に計上し、一般罰金と行政費用を計上している。
会社は、付与された日までにBlack-Scholes-Merton(“Black-Scholes”)オプション定価モデルを用いて決定された株式支払い奨励の公正価値に基づいて、株式支払い奨励に関する費用を計測して記録する。当社は個別授権書に必要なサービス期間(一般に帰属期間に等しい)の株式補償支出を直線 で確認している。
ブラック·スコイルズモデルは、オプションの期待期限と対象株式の価格変動性を含む株式報酬の公正価値を決定するために、高度な主観的かつ複雑な仮定を使用する必要がある。同社はブラック·スコイルモデルを用いて付与されたオプションの公正価値を推定し、以下のように仮定した
期待変動 ·当社は、直前期間の同行会社グループの履歴変動性を評価することによって、オプション付与の変動性を推定し、期限はオプションの予想期限に実質的に等しい。
所期の 期限-当社のオプションの予想期間は、株式ベースの報酬予想が未償還である期間を表します。会社には期待期間を推定するために十分なbr履歴発行権データがないため、従業員株式オプションの簡略化方法を用いて期待期限を推定する。
無リスク金利 ·無リスク金利は、米国財務省ゼロ金利債券が現在利用可能な暗黙的収益率 に基づいており、期間は、付与日におけるオプションの予想期間に等しい。
| F-10 |
配当金 収益率-同社はこれまで配当金を発表または支払いしておらず、配当も発表されないと予想される。したがって,配当 収益率はゼロと見積もられる.
Br社は没収が発生した場合に確認することを選択しました。
公正価値計測
公正なbr}価値は、計量日における市場参加者間の秩序ある取引において、資産または負債が元金または最も有利な市場で負債を移動させるために受信される交換価格または支払いの退出価格として定義される。当社は公正価値計量の既定の枠組みに従い、公正価値計量に関する開示を提供する。
開示を容易にするために、会計基準は公正価値計量を以下の3つに分類した
| レベル1: | 同じ資産や負債の活発な市場オファー。 |
| 第2レベル: | 市場で直接または間接的に観察することができる類似の資産または負債の価格は、レベル1の投入ではない。 |
| 第3レベル: | 観察できない投入、すなわち、市場活動の支援が非常に少ないか、または全くない、および定価モデル、現金フロー方法またはbrのような技術を使用して決定された価値、および公正な価値の決定には、重大な判断または推定を必要とするツールがある。 |
ASC 820,公正価値計量公正価値規定は、すべてのエンティティが資産および負債の金融商品の公正価値を開示することを要求し、金融商品の公正価値を、意思のある当事者間の現在の取引で交換可能な商品の金額として定義する。2022年12月31日と2021年12月31日まで、これらのプロジェクトの短期的な性質のため、現金と現金等価物、販売対象資産、前払い費用、売掛金、その他の流動負債の記録価値はその公正価値 に近い。
以下の金融資産は、2022年12月31日に当社の総合貸借対照表に公正価値に基づいて計量·記録される。2021年12月31日現在、会社の総合貸借対照表にはこのような資産はない。
| 2022年12月31日 | ||||||||||||||||
| 合計する | レベル1 | レベル2 | レベル3 | |||||||||||||
| 有価証券 | $ | $ | $ | $ | ||||||||||||
| 合計する | $ | $ | $ | $ | ||||||||||||
最近は会計公告が採用されている
2019年12月、財務会計基準委員会は、米国会計基準委員会第2019-12号所得税(“ASU 2019-12”)を発表し、ASC 740における期間内税収分配方法、中期所得税算出方法および外部ベース差繰延納税負債確認に関する指導意見のいくつかの例外を解消することにより、所得税の会計処理 を簡略化した。新たなガイドラインはまた、特許経営税の会計計算面を簡略化し、税法や税率の変化を公布し、営業権計税基礎の上昇を招く取引の会計処理を明らかにした。本ガイドラインは,2020年12月31日以降に開始された財政年度と同年度内の移行期間に適用される。当社は2021年1月1日にASU 2019-12を採用し、その連結財務諸表や関連開示に影響を与えません。
| F-11 |
注 3:有価証券
同社の有価証券への投資には、期限が1年未満の米国債が含まれている。これらの投資 は販売可能に分類され,公正価値で記録されており,収益や損失はAOCIに記録されていない.これらの投資 は2段階に分類される.
2022年12月31日現在、有価証券の公正価値は$
2022年12月31日までの年度未実現収益は$
注 4:前払い費用と他の流動資産
前払い料金と他の流動資産は、:
| 12月31日までの年度 | ||||||||
| 2022 | 2021 | |||||||
| 前払い保険 | $ | $ | ||||||
| 前払い研究開発 | ||||||||
| その他売掛金 | ||||||||
| その他前払い費用 | ||||||||
| 前払い費用とその他の流動資産総額 | $ | $ | ||||||
注 5:売掛金とその他の流動負債
支払すべき帳簿およびその他の流動負債は、以下の各項目からなる
| 12月31日までの年度 | ||||||||
| 2022 | 2021 | |||||||
| 売掛金 | $ | $ | ||||||
| 従業員コストを計算する | ||||||||
| 専門費用を計算する | ||||||||
| 計画に応じて研究·開発する | ||||||||
| 取締役会の費用を計算する | ||||||||
| デラウェア州はフランチャイズ税を納めなければならない | ||||||||
| その他の課税費用 | ||||||||
| 売掛金総額及びその他の流動負債 | $ | $ | ||||||
2021年6月、Intecイスラエル株主投票は、従業員、高度管理者、コンサルタント、取締役、および他のサービスプロバイダにbr株権を付与するインセンティブ計画(“2021計画”)を承認し、合併完了後に発効するIndaptus 2021株式インセンティブ計画を承認した。本計画により発行可能な最高株式総数は (“池”)を共有する;ただし前提は.
2021計画“は、非制限株式オプション、奨励株式オプション、制限株式奨励、制限株式単位、非制限株式奨励、株式付加価値権、および他の形態の株式ベース報酬を付与することを規定する。2021年には、会社取締役会が状況の変化に応じて報酬の種類、条項、条件を変更することを可能にする計画です。現在の経済と世界イベントを考慮すると、このような調整によって付与される補償タイプの柔軟性は特に重要である。
| F-12 |
| 加重平均 | ||||||||||||||||
| オプション数 | トレーニングをする 値段 | 残り 契約期限 (単位:年) | 内在的価値 | |||||||||||||
| 2022年1月1日現在の未返済金 | $ | $ | ||||||||||||||
| 授与する | $ | - | $ | - | ||||||||||||
| 没収されキャンセルされました | ( | ) | $ | - | $ | - | ||||||||||
| 2022年12月31日現在の未返済債務 | $ | $ | ||||||||||||||
| 2022年12月31日から行使可能 | $ | $ | ||||||||||||||
| 帰属しており、2022年12月31日に帰属する予定です | $ | $ | ||||||||||||||
以下の表は、連結業務報告書に記載されている各期間の株式ベースの報酬支出総額をまとめたものである
| 12月31日までの年度 | ||||||||
| 2022 | 2021 | |||||||
| 研究開発 | $ | $ | ||||||
| 一般と行政 | ||||||||
| 株式に基づく報酬総支出 | $ | $ | ||||||
2022年12月31日現在,未帰属株式オプションに関する総報酬コストは約$であることが確認されていない100万ドルですが、このうち は加重平均期間内に確認される予定です何年もです。
社はBlack-Scholesオプション定価モデルを用いて日株式オプション付与の公正価値を推定している.ブラック·スコアーズオプション定価モデルは、各株式オプションの公正価値に影響を与える高度な主観的仮定を推定する必要がある。2022年12月31日と2021年12月31日までの年間で付与されたオプション価値を測るための重み
平均投入を以下の表に示す。2022年12月31日までおよび2021年12月31日までに年度発行された購入権の加重平均公正価値は$
| 2022 | 2021 | |||||||
| 株価.株価 | $ | $ | ||||||
| 行権価格 | $ | $ | ||||||
| 予想期限(年単位) | ||||||||
| 波動率 | % | % | ||||||
| 無リスク金利 | % | % | ||||||
| 配当率 | % | % | ||||||
| F-13 |
次の表は、2022年12月31日現在の未償還株式オプションの実行権価格を示しています
| 行権価格 | オプション 卓越した | |||
| $ - $ | ||||
| $ - $ | ||||
| $あるいはそれ以上 | ||||
| 合計する | ||||
注 7:大文字である
| a. | 2018年から2021年にかけて、Decoyは合併前に、認可投資家と一連の種子優先株購入協定(“シリーズ種子SPA”)と将来の株式(“SAFE”)に関する一連の簡単な合意を締結した。2021年8月3日,すなわち合併完了日に,シリーズ種子優先株と金庫により発行されたすべての流通株に変換されたそしてそれぞれ 社普通株である. |
| b. | 前払い資金br承認持分証と引受権証: |
| 1. | 合併直後
,INTECイスラエルのすべての引受権証は株式購入承認証に変換されます | |
| 2. | 2021年7月23日、Indaptusはある機関投資家と購入契約を締結した。これにより,Indaptusはあらかじめ出資した引受権証(1部ごとに,私募で売却·発行することに同意した。“前払い資金株式承認証”と総称して“前払い資金承認株式証”と呼ぶ)購入 |
2022年12月31日までに加重平均行権価格$の未償還株式証加重平均残り契約年限何年もです。
| c. | 当社は2022年6月1日にH.C.ウェインwright&Co.
LLCと2022年9月1日に改訂された市場発売契約(“ATMプロトコル”)を販売代理(“Wainwright”)として締結した。これにより、当社は随時ウェインライトを通じて当社の普通株株式を提供·販売することができ、額面は$となる1株当たりの総収益は最高$に達する |
| F-14 |
| d. | 2022年12月22日当社はリンカーンパーク資本基金有限責任会社(“リンカーンパーク”)と購入契約(“購入契約”)を締結している。購入契約中の条項と条件と制限によって、リンカーン公園が購入を承諾した総金額は |
注 8:引受金とその他の事項
訴訟を起こす
2022年7月13日、LTS Lohmann治療システム株式会社(“LTS”)は国際商会に仲裁請求(“請求”)を提出し、同社の子会社INTECイスラエルを被告とした。請求書によると、LTSはユーロの支払いを受ける権利があるという
2023年2月7日、イスラエルの国際技術会社はユーロ支払いを通じてLTSとの紛争を解決した
会社は通常業務過程で発生する追加紛争や様々な訴訟に時々巻き込まれる可能性がある。これらには、知的財産権、許可、契約法、従業員関係に関する紛争や訴訟 が含まれる可能性がある。会社は、重大事項の状態(存在する場合)を定期的に審査し、その潜在的な財務リスクを評価する。 任意のクレームまたは法的クレームの潜在的損失が可能であると考えられ、金額が推定可能であれば、会社は推定された損失のために責任を負う。法的手続きは不確実な要素の影響を受け、結果は予測が難しい。この不確実性のため, はその時点で利用可能な最適な情報に基づいている.より多くの情報を得るにつれて、会社は未解決クレームや訴訟に関連する潜在的な責任を再評価する。
賃貸借証書
2021年10月1日、当社は取消不能な2年間の経営賃貸契約を締結し、レンタル期間は約
| F-15 |
将来、2022年12月31日まで、会社は経営賃貸項目の最低年度賃貸支払いと会社経営賃貸負債との入金を取り消すことはできません
| 2023年の最低賃貸支払い総額 | $ | |||
| 差し引く:利息を表す額 | ( | ) | ||
| リース負債現在価値を経営する | ||||
| マイナス:現在の部分 | ( | ) | ||
| 賃貸負債を経営し,当期分を差し引く | $ |
同社は賃貸料支出が#ドルであることを確認した
注 9:所得税
2022年12月31日現在、会社は純営業損失の繰越があり、将来年度の課税所得額の削減に利用できる。
Br社の所得税準備金には:
| 12月31日までの年度 | ||||||||
| 2022 | 2021 | |||||||
| 算出された“所期”税収割引 | $ | ( | ) | $ | ( | ) | ||
| 連邦福祉を差し引いた州税 | ( | ) | ||||||
| 差し引かれない項目 | ||||||||
| 繰延税金資産推定値変動準備 | ( | ) | ||||||
| 株に基づく報酬 | ||||||||
| 戻り調整 | ||||||||
| 他にも | ||||||||
| 所得税費用 | $ | $ | ||||||
繰延所得税は、(A)財務報告目的のための資産および負債の帳簿価値と所得税目的のための金額との間の一時的な差異、および(B)営業損失および税収控除の純税の影響を反映する。
2022年12月31日と2021年12月31日現在、会社の繰延税金純資産には、
| 2022 | 2021 | |||||||
| 繰延税金資産: | ||||||||
| 現金を計算する | $ | $ | ||||||
| 株式オプション | ||||||||
| 財産と設備 | ||||||||
| 資本化R&D | ||||||||
| リース負債を経営する | ||||||||
| 純営業損失が繰り越す | ||||||||
| 繰延税項目の総資産総額 | ||||||||
| 繰延税金負債: | ||||||||
| 使用権資産 | ( | ) | ( | ) | ||||
| 繰延税金負債総額 | ( | ) | ( | ) | ||||
| 減算:繰延税金資産推定準備 | ( | ) | ( | ) | ||||
| 繰延税項目純資産総額 | $ | $ | ||||||
| F-16 |
繰延所得税資産および負債は、資産および負債の財務諸表と課税ベースとの間の差額で入金され、これらの差額は、公布された法律および適用される差額に基づいて、課税所得期間の税率に影響を与えると予想される税率が将来的に課税または減税可能金額を生成する。繰延税金資産を予想現金化額に減少させる必要がある場合には、推定免税額が設定される。
2022年から、2017年の減税·雇用法案(TCJA)は、今年度の研究開発支出を差し引く選択肢を廃止し、IRC第174条に基づいて5年または15年以内に研究開発支出を償却するよう納税者に要求した。2022年の間に同社の資本は約$だった
ASC
740は、この実現の“可能性が高い”程度まで、純営業損失、一時的な差異、および融資の繰越の税収割引を資産として記録することを要求する。将来の税務優遇の実現は、会社が繰越期間内に十分な課税収入を生み出す能力にかかっている。当社の最近の経営損失により、管理層は上記のbr未来税優遇による繰延税金資産は現在実現不可能であると考えているため、2022年と2021年12月31日までの年度に全額推定値を提供する準備をしている。2022年12月31日までと2021年12月31日までの年間総推定額純変動はbrドル減少に充てる
開ける2022年12月31日、同社の米国連邦と州の純営業損失(NOL)は$に転換した
国内税法第382及び383節の規定により、会社がその米国の純営業損失を利用する能力が制限される可能性がある。382節で定義した所有権変更が発生した場合には,これらの制限が適用される.通常、ある株主がテスト期間(通常は3年)内にその総持株率をその最低持株率より50ポイント以上増加させると、所有権変更が発生する。当社は第382条の分析を行っていないにもかかわらず、純営業損失の利用が大きく制限される可能性がある。しかも、アメリカ税法はこのような繰越が未来の税金を相殺するために使用できる時間を制限している。したがって、会社は連邦と州の税金目的のためにこのような繰越を活用できないかもしれない。将来の持分の変更も所有権変更をトリガし、第382条の制限を招く可能性がある。
当社は総合財務諸表の申告表で保有または予想されている税務倉位の利益を確認していますが、当局が審査した後、当該等の倉位は維持される可能性が高いです。確認された税務頭寸は最大の収益で評価されています
会社は利息支出に未確認の税収割引に関する利息および一般罰金と行政費用を記録しています。
2022年12月31日と2021年12月31日まで、会社記録
同社はアメリカ連邦と各州の所得税申告書を提出し、それぞれ2019と2018納税年度に連邦と州の審査を受け、その設立以来のNOLは税務機関の調整を受ける可能性があり、法規がまだ審査可能な未来の税務届出でクレームを出す可能性がある。同社はイスラエル納税申告書も提出し、2018年前の納税年度に審査を受けた。同社は現在、米国国税局や他の同様の国、州、地方当局の監査を受けていない。
注: 10:後続事件
会社は、2022年12月31日までの年度の連結財務諸表で確認または開示する必要がある事件について、2022年12月31日(これらの連結財務諸表の日付)から2023年3月17日(連結財務諸表の発行日を代表する)までの後続事件を評価している。当社の結論は,付記8で述べたLTSとの和解合意以外に,総合財務諸表で確認または開示する必要はないと結論した。
| F-17 |
第 項9.会計と財務開示に関する変更と分岐。
ない。
第 9 A項。制御とプログラムです制御と手続きを開示する。
制御とプログラムの有効性に関する制約
我々の開示制御およびプログラムを設計·評価する際に、管理層は、任意の制御およびプログラムが、設計および動作がどんなに良くても、予想される制御目標を達成するために合理的な保証を提供することしかできないことを認識している。さらに、開示制御およびプログラムの設計は、リソース制限が存在し、そのコストに対する可能な制御およびプログラムの利益を評価する際に判断を適用することが要求されるという事実を反映しなければならない。
開示制御とプログラムの評価
最高経営責任者とCEOの参加の下、我々のbr経営陣は、2022年12月31日までの開示制御およびプログラムの有効性(取引法下のルール13 a-15(E)および15 d-15(E)で定義されているような)を評価した。 は、この評価に基づいて、2022年12月31日までに、我々の開示制御および手続きが合理的な保証レベルで有効であると結論した。
経営陣財務報告内部統制年次報告書
私たちの経営陣は、“取引法”の下のルール13 a-15(F)で定義されている私たちの財務報告に対する十分な内部統制の確立と維持を担当しています。
我々の経営陣は、“内部統制-総合枠組み(2013)”に規定されている基準に基づいて、財務報告の内部統制の有効性を評価したトレデビル委員会の後援組織委員会が発行しましたそれは.我々の評価によると、我々の経営陣は、2022年12月31日現在、財務報告の内部統制に有効であると結論している。
独立公認会計士事務所認証報告
本 年次報告には,我々の公認会計士事務所の財務報告内部統制に関する認証報告 は含まれておらず,適用される米国証券取引委員会規則により,“大型加速申告機関”や“加速申告機関”ではない発行者が免除 を得ることができるからである。
財務報告内部統制変更
2022年12月31日までの四半期内に、財務報告の内部統制(取引法第13 a-15(F)および15 d-15(F)規則で定義されているように)に大きな影響を与えなかったか、または合理的に財務報告の内部統制に大きな影響を与える可能性のある変化が発生しなかった。
第 9 B項。他の情報。
ない。
第 9 C項.検査を妨害する外国司法管轄区域を開示する。
は適用されない.
| 67 |
第 第3部分
第br項10.取締役及び行政職
次の表に2023年3月17日現在の私たちの役員や役員に関する情報を示します。
| 名前.名前 | 年ごろ | ポスト | ||
| 執行官 | ||||
| ジェフリー·A·メイクラー | 56 | 最高経営責任者兼取締役 | ||
| マイケル·J·ニューマン博士 | 67 | 首席科学官と役員 | ||
| NIR Sassi | 47 | 最高財務官 | ||
| ウォルター·リンスコット | 62 | 首席ビジネス官 | ||
| 非執行役員 | ||||
| ロジャー·J·ポメランツ博士 | 66 | 取締役会議長 | ||
| ヒラカラハ | 54 | 役員.取締役 | ||
| アンソニー·J·モーター·ルナー | 70 | 役員.取締役 | ||
| ウィリアム·B·ハイエズ | 57 | 役員.取締役 | ||
| マーク·J·ギルバート医学博士 | 62 | 役員.取締役 | ||
| ブライアン·オカラハン | 63 | 役員.取締役 | ||
| ロバート·E·マーテル医学博士 | 60 | 役員.取締役 |
| 以下では,我々の役員と役員に関する履歴情報を提供する. |
我々幹部に関する情報
ジェフリー·A·メイクラー2021年7月以来最高経営責任者を務め、2021年2月の設立以来取締役会のメンバーを務めてきました。これまで、Mecklerさんは設立から2021年7月まで当社の唯一の役員を務め、2017年4月からINTECイスラエル社の取締役会副議長を務め、2017年7月からINTECイスラエル社のCEOを務め、2021年3月からINTEC親会社の総裁や役員を合併まで担当しています。Mecklerさんは、2014年10月から取締役治療会社(ナスダック株式コード:TVTX)に勤務しています。Mecklerさんは、多くの公的およびプライベート会社の取締役会に勤めていました。メイクラーさんは、2015年4月から2016年7月までの間に製薬会社CoCrystal Pharma,Inc.のCEOおよびCEOを務めた。2012年6月から2016年11月までバイオテクノロジー会社QLT,Inc.(ナスダックコード:QLTI)の取締役を務め、2009年から生命科学コンサルティング会社アンドラグループ取締役社長を務めてきた。メイクラー·さんはまた、トレバー治療会社の最高経営責任者(CEO)を2017年1月から2017年7月まで務めている。彼のキャリアの初期に、メイクラー·さんは、製造システム、市場研究、業務開発、戦略計画、および会社の融資に関連して、ファイザーの一連のポストに就いていました。これは、買収と資産剥離の面で重要な役割を果たしています。 メークラーさんは、過去の社長であり、現在もベルヴィュー児童基金の取締役会のメンバーを務めています。この非営利団体は、ベルヴィュー病院の中心部の小児科プロジェクトの提唱と発展に専念しています。Mecklerさんは、カーネギーメロン大学で工業管理の学士号と工業管理の修士号を持っています。また、Mecklerさんはまた、フォーテハム大学法学部の法学博士号を取得しました。我々は、Mecklerさんは、バイオ製薬業界で彼がファイザーのサービスを提供することを含み、上場企業の取締役会で彼のサービス経験を含む豊富な行政リーダーシップの経験を持っているので、私たちの取締役会に勤めている資格があると信じています。
マイケル·J·ニューマン博士2021年8月以来、私たちの首席科学官と取締役会のメンバーを務めてきた。ニューマン博士は製薬/生物技術の幹部であり、微生物学方面の学部生と大学院生の研究と訓練以外に、35年以上の腫瘍学研究と開発を展開と管理する経験を持っている。おとり会社の創業者でCEO兼取締役会のメンバー(2013年8月から2021年8月まで)。彼以前の職には,ブランダイス大学(1984−1987)や羅氏分子生物学研究所(1987−1992)で生化学教員,Sandoz製薬会社で腫瘍学上級アシスタント(癌生物学グローバル担当)を務め,ノワ製薬で腫瘍学執行役員 (米国癌生物学担当)を務めていた。Newman博士はカリフォルニア大学サンディエゴ校で生物学学士号を取得し、ハーバード医学院で細胞と発育生物学博士号(アメリカ国家科学基金会博士後研究員)を取得し、コーネル大学でポストドクター研究に従事している。ニューマン博士は広範な科学と研究背景と、Decoy創始者兼CEOとしての経験を持っているので、私たちの取締役会に勤務する資格があると信じています。
| 68 |
NIR SassiSassiさんは2021年7月から当社のチーフ財務官を務め、2010年3月から合併前のIntecイスラエル会社の首席財務官(2015年1月から2016年8月まで、その間にイスラエルのIntec社の財務副社長を務めた)、2021年3月から合併前のイスラエル会社の社長を務めた。イスラエルのIntecのためにサービスを提供する前に、Sassiさんは、2002年から2010年までの間、普華永道イスラエルの会計士事務所の上級マネージャーを務め、2年間の間、普華永道ニューヨーク事務所に転勤することを含む。Sassiさんはイスラエルの公認会計士で、イスラエルのBe‘er Shevaベングリアン大学で経済学と会計学の学士号を持っています。
ウォルター·リンスコット2021年7月から私たちの首席商務官を務めており、2017年10月にIntecイスラエル社に入社し、2018年7月から合併まで同社の首席商務官を務めています。リンスコット·さんは、2017年10月から2018年7月まで、INTECイスラエルの最高経営責任者(CEO)を務めました。リンスコット·さんは、イスラエルのIntecに勤務する前に、2014年10月にグローバル·コンサルティング·企業を設立し、開発会社に戦略的助言を提供し、最近では2017年3月から2017年10月までトレバー·セラピーの社長兼チーフエコノミストを務めていました。リンスコット·さんはまた、2015年7月から2017年3月までのCcrystore Pharma,Inc.,2011年1月から2015年1月までのCarestream Health,Inc.および2001年から2005年までのSolvay PharmPharmticals,Inc.など、公共および個人用医療機器および製薬会社で上級管理職に就いていた。また、1990年から2001年までThompson Hine LLPのパートナーとパートナーを務め、2005年から2010年までの間に再びパートナーを務め、その間にジョージア州アトランタにある事務所を設立し、同事務所生命科学実践グループの担当パートナーと議長を務めた。リンスコット·さんは、オックスフォード大学の実験および変換治療の修士号、オックスフォード大学の世界的なビジネス大学院生用証書、ケンブリッジ大学の創業大学院生用証書を持っています。彼はシラキュース大学の学士号とデイトン大学法学部の法学博士号を取得した。リン·スコット·さんは法学部に入る前にアメリカ海兵隊の将校として働いていました。
非従業員取締役
ロジャー·J·ポメランツ博士2021年7月から当社の会長を務め、2018年3月から合併までイスラエルインターネット技術会社の取締役会メンバーを務めてきました。ポメラランツ博士は2019年4月からナスダック社(Contrafect Corporation)の会長兼最高経営責任者を務め、2014年5月からContrafect社の副会長を務めてきた。これまで、ポメラランツ博士は2014年から2019年まで旗艦先鋒brのベンチャーパートナーを務めてきた。また、2013年11月から2019年12月にかけて、ボメランツ博士はバイオテクノロジー会社セレス治療会社(ナスダックコード:MCRB)の取締役会長を務め、2014年6月から2019年1月まで同社の総裁兼最高経営責任者を務めた。Seresに加入する前に、ポメランツ博士はメルク社の高級副総裁許可と買収の全世界主管であり、メルク研究実験室のすべての許可と買収を担当し、外部研究、外部許可br地区取引と学術連盟を含む。これまで、メルク社で高級副総裁や感染症グローバル特許経営責任者を務めていた。メルクに加入する前、ポメランツ博士はナスダック社の感染症グローバル主管で、2020年6月からヴィラクタ(VIRX:VIRX)の取締役会メンバーを務め、2020年9月に会長に任命された。2020年2月以来、コルセット植物バイオテクノロジー会社(ナスダック:CLPT)の会長を務め、以前はRubius Treateutics (ナスダック:RUBY)の取締役会メンバーだった。ポメランツ博士はジョン·ホプキンス大学で生化学学士号を取得し、ジョン·ホプキンス医学院で医学博士号を取得した。彼はハーバード医学院マサチューセッツ州総病院で内科実習と入院医師訓練、及び伝染病とウイルス学方面の専門臨床と研究訓練を完成した。彼の分子レトロウイルス学博士後の研究訓練はハーバード医学院とマサチューセッツ工科大学ホワイトウッド研究所(MIT)で獲得された。ポメランツ博士はマサチューセッツ州総合病院の総入院医も務めている。医学科学者の訓練を受けた後、彼はフィラデルフィアトーマス·ジェファーソン大学医学と分子薬理学終身教授と伝染病学部主任となった。ポメランツ博士は国際的に公認されているHIV分子の発病機序と潜伏期の専門家である。彼は10種類のすでに許可された伝染病薬物を開発し、HIV、C型肝炎ウイルス、結核とクロストリジウム感染などの重要な疾病の治療に応用した。私たちは、ポメランツ博士は薬物開発と製薬業において豊富な科学、行政、取締役会の指導経験を持っているため、私たちの取締役会に勤務する資格があると信じている。
| 69 |
ヒラカラハ2021年7月以来、私たちの取締役会に勤めており、2009年12月から合併前までイスラエルのインターネット技術会社の取締役会メンバーを務めてきました。カラハさんは取締役経験豊富な取締役会のメンバーで、2013年以来ずっと私営と上場会社の独立商業顧問を務め、戦略、運営、融資、監督管理と会社管理などの方面についてコンサルティングを提供している。2017年11月から2018年9月まで、Karahさんはイスラエル農業科学技術革新会社FloraFotonica Ltd.の実行主席を務めた。2006年から2013年まで、カラハは家族理財室EuroTrust Ltd.の首席投資官を務め、主に生命科学、インターネットとハイテク会社の投資に集中した。EuroTrustに加入する前に、Karahさんはニューヨークに本部を置くヘッジファンドPerceptive Life Sciences Ltd.の高級アナリストだ。Perceptiveに加入する前に、カラハはコネチカット州の医療保健に集中したヘッジファンド甲骨文パートナー有限会社(Oracle Partners Ltd.)の研究アナリストである。カラハさんは2008年からずっとサイバーセキュリティ会社サイロン株式会社(Cyren Ltd.)の取締役会に勤めており、2014年からダリオ健康会社(Dario Health Corp.)(ナスダック株式コード:DRIO)の取締役会に勤めている。彼女はまだいくつかの民間会社の取締役会に勤めています。Karahさんはカリフォルニア大学バークレー校の分子と細胞生物学の学士号を持ち、カリフォルニア大学バークレー校のUCSB-UCSF共同医療プロジェクトに通っていた。私たちはKarahさんが私たちの取締役会に勤めている資格があると信じている。彼女はIntecイスラエル会社で長期的にサービスしているので、彼女は生命科学会社での投資キャリア、彼女の科学的背景と上場会社の取締役会に勤めた経験がある。
アンソニー·J·モーター·ルナー2021年7月から私たちの取締役会に勤め、2017年12月から合併までイスラエルのインターネット技術会社の取締役会に勤めています。マダルナさんは、製薬業界で40年以上の経験を持ち、工場、地域、および全世界でリーダーシップを担当しています。2011年1月から2016年12月までの間に、マダルーナ·さんはファイザー社で一連のポストを担当し、最近ではファイザー·グローバル·サプライ実行副社長·総裁を務めています。これまで、マダルーナさんは2008年から2011年までファイザーグローバル製造戦略とサプライネットワーク移行の上級副社長を務め、1998年から2008年までファイザーグローバル製造欧州エリア副社長を務めました。マダルーナさんは、2017年8月にケレグループと広東科工集団に買収されるまで、2016年2月からオルバニ分子研究会社の取締役を務めており、現在はこの私営会社の管理委員会のメンバーを務めています。マダルナさんは、東北大学の化学工学の学士号、南イリノイ大学の工商管理修士号を持っています。我々は、マダルーナさんは、製薬業界での彼のサービス経験と、会社の取締役会での彼のサービス経験を含む豊富な経験を持っているので、私たちの取締役会に勤めている資格があると信じています。
ウィリアム·B·ハイエズ2021年7月以来、私たちの取締役会に勤めており、2018年6月から合併までイスラエルのインターネット技術会社の取締役会メンバーを務めています。最近、Hayesさんは、診断実験室会社アメリカの研究所会社(ニューヨーク証券取引所コード:LH)のチーフ財務官兼財務担当総裁さんを務めました。ヘイズさんは1996年にLabCorpに入社し、収入サイクルの機能の日常的な運営を担当しました。一連の昇進で昇進し、2005年にLabCorp首席財務官兼財務担当総裁に任命され、2014年に退職するまで務めてきた。LabCorpに参加する前に、Hayesさんはピマウェイの監査部門で9年間働いていた。Hayesさんは2019年10月以降、建材サプライヤーとメーカーBuilders FirstSource(ナスダック·コード:BLDR)の取締役会に勤めており、現在は監査委員会の議長を務めています。これに先立ち、ヘイズさんは2016年3月から医薬製造会社Patheon N.V.(ニューヨーク証券取引所コード:PTHN)で取締役を務め、2017年末までThermo Fisherに買収された。ヘイズさんはノースカロライナ大学グリンズバーラー校で会計学の学士号を持っています。私たちは ヘスさんは、彼は会計の背景と上場企業の取締役会に勤めている経験があるので、私たちの取締役会に勤めている資格があると信じています。
ブライアン·オカラハン2021年7月から取締役会メンバーを務め、2018年11月から合併までDecoy取締役会メンバーを務めてきました。O‘Callaghanさんは、バイオテクノロジー、大規模な製薬会社、契約研究組織(CRO)の分野で豊富な経験を持つ生命科学の役員です。2020年11月以降、O‘Callaghanさんは、女性の健康を改善するための新しい治療法を開発し、将来の開発、規制申告、製品の発表を完了する臨床段階バイオ製薬会社ObsEvaのCEOを務めています。BrianはObsEvaに加入する前にPetra Pharma(2017年5月~2020年6月)、Sonrgy(2015年5月~2017年5月)、Acucela、Sangart、BioPartnersでCEOを務め、ファイザー、メルク、ノワール、Covance、NPS製薬会社で高級管理職を務めていた。O‘Callaghanさんは、上場企業と非上場企業の管理、合併と買収、初公募株、資金調達、撤退、剥離と戦略連盟の経験を持っています。彼の運営経験も広く、複数の業務やプロジェクトを管理しており、概念から商業化までの多くの治療分野に関連している。彼はまた豊富な取締役会経験を持ち、2021年以来複数のバイオテクノロジー と501(C)(3)非営利取締役会に勤めており、ボルト治療会社(ナスダックコード:BOLT)の取締役会メンバーを含む。O‘Callaghanさんは、レイディング大学ヘンリービジネススクールのビジネスマネジメントの修士号を取得しました。我々は、O‘Callaghanさんは、彼が実行管理経験を豊富に持っているので、私たちの取締役会に勤務する資格があると信じています。
| 70 |
マーク·J·ギルバート2021年11月以来、私たちの取締役会に勤めてきた。Gilbert博士は全世界の医療と臨床研究開発及び医療事務管理において30年以上の経験を持っている。2019年3月から2022年3月まで,ギルバート博士はAcpedia,Inc.で研究開発実行副総裁を務め,同社は臨床段階バイオテクノロジー会社であり,癌看護における空白の解決に取り組んでいる。ギルバート博士は2021年6月以来,同社の完全子会社であるおとり生物システム会社の臨床開発コンサルタントを務めてきた。また、2022年7月からGilbert博士はTune Treateuticsの臨床開発顧問を務め、これはエピジェネティクス編集治療薬に専念するバイオテクノロジー会社であり、2020年10月からGilbert博士はInceptor Bio,LLCの科学諮問委員会議長を務め、これは生物技術会社であり、サービス不足と治療困難な癌のために複数の次世代細胞と遺伝子治療プラットフォームを開発し、2022年11月から取締役会のメンバーを務め、2020年10月からKineticos Venturesの戦略顧問を務め、Kineticos Venturesは新興生命科学会社にコンサルティングサービスと資本を提供する会社である。2020年3月からJW治療会社の首席医療官を務め、細胞免疫治療製品に専念するバイオテクノロジー会社である。これらのポストに就く前に、Gilbert博士は2013年11月から2020年1月までの間に生物製薬会社Juno Treateutics Inc.の首席医療官を務め、そこでいくつかの最初のCAR-T細胞療法の臨床開発を指導した。これまで、ギルバート博士はバイエル先霊製薬会社で指導職を務め、そこで総裁副主任兼治療領域腫瘍学研究会社の全世界臨床開発主管を務め、彼は医療事務、腫瘍学副主任総裁と全世界医学開発グループの責任者を務め、バイエル先霊製薬会社で臨床研究開発高級医学取締役brを務め、2019年5月から2021年5月まで、ギルバート博士はシリコンバレー治療会社の独立取締役を務め、同社は完全に統合された薬物設計と開発会社である。ギルバート博士はアイオワ大学で生化学理学学士号を取得し、アイオワ大学医学院で医学博士号を取得した。彼はそれぞれカリフォルニア大学サンフランシスコ校とワシントン大学で内科、伝染病、腫瘍学の訓練を受けた。Gilbert博士は薬物開発と製薬業界で重要な科学と行政管理能力を持っているため、私たちの取締役会に在任する資格があると信じている。
ロバート·E·マーテル医学博士2023年2月から私たちの取締役会に勤めています。マーテル博士は製薬業界で20年以上の経験を持っている。マテル博士は2018年6月以来、Curis,Inc.の研究開発担当を務めており、癌治療革新療法開発に専念するバイオテクノロジー会社であり、2011年11月から2018年5月まで取締役会のメンバーを務めている。彼は個人持株の早期バイオテクノロジー会社Epi-Cure PharmPharmticalsの共同創業者でもあり、2016年から2018年まで同社の総裁と取締役会メンバーを務めている。マーテル博士は現在もタフツ大学医学センターで主治医を務めており、2009年以来この職を務めている。これらのポストに先立ち,2012年から2015年にかけて,マテル博士は腫瘍学に専念した生物製薬会社Tesaro,Inc.で首席医療官を務め,そこで医療や薬理学的開発を指導した。これまで、2005年から2009年まで、癌治療に専念した上場生物製薬会社メチル化遺伝子会社で首席医療官を務め、臨床戦略と開発の各方面を監督していた。また、2002年から2005年まで生物製薬会社百時美施貴宝腫瘍学グローバル臨床研究部取締役を務め、2000年から2002年までバイエル社製薬事業部で取締役アシスタント/副を務めていた。また、マーテル博士は2001年から2005年までエール大学医学院で腫瘍学臨床補佐教授を務め、1998年から2000年までデューク医学センターで助理教授の職を獲得した複数の学術職を務めたことがある。マーテル博士はカラマ祖学院で化学学士号を取得し、ミシガン大学で薬理学博士号を取得し、ウェイン州立大学で医学博士号を取得した。デューク大学医学センターで内科実習と入院医師の仕事を終え、デューク大学で内科腫瘍学の研究を終えた。マテル博士は研究や開発に豊富な経験を持ち、上場バイオ製薬会社の首席医療官を務めているため、私たちの取締役会に勤務する資格があると信じています。
ビジネス行為と道徳的基準
私たちは私たちの役員、上級管理者、従業員に適用されるビジネス行動と道徳基準を持っています私たちのCEO、財務責任者、会計責任者、主計長、または似たような機能を実行する人を含めてそれは.“商業行為および道徳基準”は、当社のウェブサイトhttp://www.indaptusrx.comで公開されています我々のビジネス行為および道徳規則の改正または免除に関するForm 8-K第5.05項の開示要件、およびナスダックの取締役および役員免除に関する要求を満たす予定です。 は、私たちのサイト上で上記指定された住所および場所を通じてこのような情報を発表します。当サイトに含まれている情報は、本年度報告書に引用されていません。
| 71 |
他にも
本第10項の規定で開示しなければならない残りの資料は、2023年の株主総会のために作成された最終委託書に含まれ、引用して本明細書に組み込まれる。
プロジェクト 11.役員報酬
第11項に必要なbr情報は、2023年株主総会のために作成された最終依頼書に含まれ、参考として本明細書に組み込まれる。
第br項12.ある実益所有者及び管理層の保証所有権及び関連株主事項。
株式補償計画に基づいて発行された証券
次の表は、2022年12月31日までにIndaptus Treateutics Inc.2021年株式インセンティブ計画または2021年計画でオプション を行使する際に発行可能な普通株の情報を提供します
| 計画種別 | 未弁済オプション,株式承認証及び権利を行使する際に発行しなければならない証券数(1) | 未償還オプション、権証および権利の加重平均行権価格 | 株式補償計画に応じて将来発行可能な証券の数(第1欄に反映された証券を除く) | |||||||||
| 証券保有者が承認した持分補償計画(2) | 1,672,873 | $ | 13.01 | 314,494 | ||||||||
| 証券保有者の許可を得ていない持分補償計画 | — | — | — | |||||||||
| 合計する | 1,672,873 | $ | 13.01 | 314,494 | ||||||||
| (1) | 2021年計画によると,株式オプション満期の加重平均残存期間は 8.5年である。 | |
| (2) | 私たちのbr 2021は、2022年1月1日から2024年1月1日まで(2024年1月1日を含む)の年ごとの増加を許可する常青樹条項を計画している。(A)前年の最終日に発行された普通株式総株式数の3%に相当するか、または(B)当社取締役会が決定した少ない株式数に相当する。 |
他にも
本第12項に必要な残りの情報は、2023年株主総会の最終委託書に含まれ、参照して本明細書に組み込まれる。
第 項13.ある関係と関連取引、および取締役独立性。
本項13で要求される情報は、2023年株主総会のために作成された最終依頼書に含まれ、ここで引用して参考にします。
第br項14.総会計士料金とサービス
本第14条に要求される情報は、2023年株主総会のために作成された最終委託書に含まれ、ここで引用して参考とする。
| 72 |
第4部
第 項15.物証、財務諸表付表。
(A)(1) 財務諸表。
本年度報告“財務諸表及び補足データ”には,本プロジェクトに必要な財務諸表が記載されている。
(A)(2) 財務諸表添付表。
財務諸表は、本年度報告“第8項.財務諸表及び補足データ”の下の財務諸表及び付記に適用されていないか、又は必要な資料が記載されていないため、財務諸表の付表を省略する。
(A)(3) 個の展示品.
以下は本年度報告書の一部として提出された展示品リストである。
| 73 |
添付ファイル インデックス
| 添付ファイル 番号: | 添付ファイル 説明 | |
| 2.1++ | 合併及び再編協定及び計画は、期日は2021年3月15日であり、Intec Pharma Ltd.,Intec Parent,Inc.,Dillon Merge Sub Inc.,馴化合併子有限会社及びDecoy BiosSystems,Inc.(Intecイスラエルを参照して2021年3月15日に米国証券取引委員会に提出された8−K表報告添付ファイル2.1を本明細書に組み込む) | |
| 2.2 | 合意及び合併計画は、期日は2021年4月27日であり、Intec Pharma Ltd.,Intec Parent,Inc.と現地化合併子有限会社との間の合意及び計画である(Intecイスラエルを参照して2021年4月30日に米国証券取引委員会に提出された8-K表報告添付ファイル2.1を本明細書に組み込む) | |
| 3.1 | 改訂·再発行されたIndaptus治療会社登録証明書は、2021年7月23日(これに合併し、当社が2021年7月23日に米国証券取引委員会に提出した現在の8-K表報告書の添付ファイル3.1参照) | |
| 3.2 | 改訂·再実施されたIndaptus治療会社規約は、期日は2021年7月23日(これに合併し、2021年7月23日に米国証券取引委員会に提出された会社現在の8-K表報告書の添付ファイル3.2参照) | |
| 3.3 | 改訂·再改訂されたIndaptus治療会社規約の第1号改正案は、期日は2022年7月20日(合併内容は2022年7月20日に米国証券取引委員会に提出された会社現在8-K表報告書の添付ファイル3.1参照) | |
| 3.4 | 2021年8月3日に改訂·再発行されたIntec親会社登録証明書(これに合併し、2021年8月6日に米国証券取引委員会に提出された会社の現在の8-K表報告書の添付ファイル3.1参照) | |
| 4.1 | 第12節に登録された証券説明によれば(会社が2021年3月21日に米国証券取引委員会に提出した10−K年報添付ファイル4.1を参照して本明細書に組み込む) | |
| 4.2 | INTEC親会社普通株式購入承認証表(INTECイスラエル社が2020年5月6日に米国証券取引委員会に提出した8-K表の現在報告書の添付ファイル10.2を参照して本明細書に組み込む) | |
| 4.3 | Intec親会社Aシリーズ普通株引受権証表(これに合わせて、Indaptusが2021年7月29日に米国証券取引委員会に提出した8-K表の現在報告の添付ファイル10.3を参照) | |
| 10.1*+ | Indaptus治療会社2021年株式インセンティブ計画 | |
| 10.1+ | Indaptus治療会社2021年株式インセンティブ計画第1修正案(2021年8月6日に米国証券取引委員会に提出された会社の現在の8-K表報告書の添付ファイル10.7を参照して本明細書に組み込む) | |
| 10.2*+ | オプション奨励プロトコルフォーマット | |
| 10.3+ | 賠償協議表(会社が2021年8月6日に米国証券取引委員会に提出した8-K表添付ファイル10.5を参照して本明細書に組み込む) | |
| 10.4+ | ジェフリー·メイクラーとIndaptus治療会社との間の雇用協定は、2021年8月4日から発効する(合併は、2021年8月6日に米国証券取引委員会に提出された会社の現在の8-K表報告書の添付ファイル10.1参照) | |
| 10.5+ | マイケル·J·ニューマン博士とIndaptus治療会社との間の雇用協定は、2021年8月4日から発効する(合併は、2021年8月6日に米国証券取引委員会に提出された会社の現在の8-Kレポートの添付ファイル10.2参照) | |
| 10.6+ | ウォルター·リンスコットとIndaptus Treateutics,Inc.の間の雇用協定は,2021年8月4日から発効する(これに合併し,2021年8月6日に米国証券取引委員会に提出された会社の現在の8-K表報告書の添付ファイル10.3を参照) | |
| 10.7+ | Nir SassiとIndaptus Treateutics,Inc.との間の雇用協定は,2022年1月1日から発効する(会社が2022年3月21日に米国証券取引委員会に提出したForm 10−K年次報告の添付ファイル10.6を引用した) | |
| 10.8 | Intec親会社と本プロトコル署名ページで指定された各買い手が2021年7月23日に署名した証券購入協定表(これに合併し、2021年7月29日に米国証券取引委員会に提出された会社の現在の8-K表報告書の添付ファイル10.1を参照) | |
| 10.9 | Intec Parent,Inc.と本プロトコル署名ページで指定された購入者1人当たり2021年7月23日に署名された登録権協議表(これに合併し、2021年7月29日に米国証券取引委員会に提出された会社の現在の8-K表報告の添付ファイル10.4参照) | |
| 10.10 | 2022年6月1日にIndaptus治療会社とH.C.Wainwright&Co.,LLCの間で調印された市場発売協定(会社が2022年9月1日に提出したS-3表登録説明書添付ファイル1.2を参考に合併したもの) | |
| 10.11 | Indaptus治療会社とリンカーンパーク資本基金有限責任会社との間の購入契約は、2022年12月22日(2022年12月23日に提出された会社の現在の8-Kレポートの添付ファイル10.1を参照して編入) | |
| 10.12 | Indaptus治療会社とリンカーンパーク資本基金有限責任会社との間の登録権協定は、期日は2022年12月22日(2022年12月23日に提出された会社の現在の8-Kレポートの添付ファイル10.2を参照して編入) | |
| 21.1 | 子会社リスト(会社が2021年3月21日に米国証券取引委員会に提出した10-K表年報添付ファイル21.1を参照して本明細書に組み込む) | |
| 23.1* | 独立公認会計士事務所Haskell&White LLPの同意 | |
| 31.1* | 2002年にサバンズ-オキシリー法第302条に基づいて可決された1934年“証券取引法”第13 a-14条及び第15 d-14(A)条に基づいて発行された最高経営責任者証明書。 | |
| 31.2* | 2002年サバンズ-オキシリー法第302節に基づいて可決された1934年“証券取引法”第13 a-14(A)条及び第15 d-14(A)条に基づく首席財務官の認証。 | |
| 32.1# | 2002年にサバンズ·オクスリ法案第906条で可決された“米国法典”第18編1350条による主要行政官の証明 | |
| 32.2# | 2002年サバンズ·オキシリー法第906条で可決された“米国法典”第18編1350条による首席財務官の証明 | |
| 101.INS* | 相互接続 XBRLインスタンス文書(このインスタンス文書は、そのXBRLタグがイントラネットXBRL文書に埋め込まれているので、対話データファイルには現れない) | |
| 101.Sch* | イントラネット XBRL分類拡張アーキテクチャ文書 | |
| 101.カール* | 連結 XBRL分類拡張計算リンクライブラリ文書 | |
| 101.定義* | 連結 XBRL分類拡張Linkbase文書を定義する | |
| 101.実験所* | 連結 XBRL分類拡張ラベルLinkbase文書 | |
| 101.前期* | インラインXBRL分類拡張プレゼンテーションLinkbase文書 | |
| 104 | 表紙 ページ相互データファイル(添付ファイル101に含まれるイントラネットXBRL形式) |
| * | 同封アーカイブ | |
| # | 同封して提供する | |
| + | 契約または補償計画を管理すること。 | |
| ++ | S-K条例第601(A)(5)項の によれば、プロトコルの付表は省略されている。何か漏れたスケジュールがあれば、要求に応じて米国証券取引委員会にコピーを提供するだろう。 |
第 項16.テーブル10−Kまとめ
ない。
| 74 |
サイン
改正された“1934年証券取引法”第13又は15(D)節の要求に基づいて、登録者は、以下の署名者代表登録者が本報告書に署名することを正式に許可している。
| Indaptus 治療会社 | ||
| 日付: 2023年3月17日 | 差出人: | /s/ ジェフリー·A·メイクラー |
| ジェフリー·A·メイクラー | ||
| CEO | ||
授権書
このような陳述により、私たちは、Jeffrey MecklerとNir Sassiを構成し、任命し、それぞれその実際の代理人として、任意およびすべての身分で彼を完全に代替し、 は、10-K表格本報告の任意およびすべての修正に署名し、添付ファイルおよびこれに関連する他の文書と共に米国証券取引委員会に提出し、ここで、上記の報告の任意およびすべての修正に署名する可能性があるので、私たちの署名を承認し、確認することができる。
改正された1934年の証券取引法の要求によると、本報告は、次の日に登録者として以下の者によって登録者として署名された。
| 名前.名前 | タイトル | 日取り | ||
|
/s/ ジェフリー·A·メイクラー |
最高経営責任者兼取締役 |
2023年3月17日 | ||
| ジェフリー·A·メイクラー | (CEO ) | |||
|
/s/ Nir Sassi |
最高財務官 |
2023年3月17日 | ||
| NIR Sassi | (最高財務会計官 ) | |||
|
/s/ Michal J.Newman博士 |
首席科学官と役員 | 2023年3月17日 | ||
| マイケル·J·ニューマン博士 | ||||
|
ロジャー·J·ポメランツ医学博士 |
取締役会議長 | 2023年3月17日 | ||
| ロジャー·J·ポメランツ博士医学博士 | ||||
|
/s/ ヒラ·カラハ |
役員.取締役 | 2023年3月17日 | ||
| ヒラカラハ | ||||
|
/s/ アンソニー·J.モータールナ |
役員.取締役 | 2023年3月17日 | ||
| アンソニー·J·モーター·ルナー | ||||
|
/s/ Brian O‘Callaghan |
役員.取締役 | 2023年3月17日 | ||
| ブライアン·オカラハン | ||||
|
/s/ Mark J.Gilbert |
役員.取締役 | 2023年3月17日 | ||
| マーク·J·ギルバート | ||||
|
/s/ William B.Hayes |
役員.取締役 | 2023年3月17日 | ||
| ウィリアム·B·ハイエズ | ||||
|
ロバート·E·マーテル医学博士博士 |
役員.取締役 | 2023年3月17日 | ||
| ロバート·E·マーテル医学博士 | ||||
| 75 |