GoSS-20221231誤り2022会計年度0001728117Http://Fasb.org/us-GAAP/2022#Account StandardsUpdate 20 2006メンバーP 2 YHttp://Fasb.org/us-GAAP/2022#AcruedLiabilitiesCurrentHttp://Fasb.org/us-GAAP/2022#AcruedLiabilitiesCurrent0.061600017281172022-01-012022-12-3100017281172022-06-30ISO 4217:ドル00017281172023-03-10Xbrli:共有00017281172022-12-3100017281172021-12-31ISO 4217:ドルXbrli:共有00017281172021-01-012021-12-3100017281172020-01-012020-12-310001728117アメリカ-アメリカ公認会計基準:普通株式メンバー2019-12-310001728117US-GAAP:AdditionalPaidInCapitalMembers2019-12-310001728117アメリカ-公認会計基準:前払いメンバーを保留2019-12-310001728117アメリカ公認会計原則:他の総合収入メンバーを累計2019-12-3100017281172019-12-310001728117アメリカ-アメリカ公認会計基準:普通株式メンバー2020-01-012020-12-310001728117US-GAAP:AdditionalPaidInCapitalMembers2020-01-012020-12-310001728117アメリカ-公認会計基準:前払いメンバーを保留2020-01-012020-12-310001728117アメリカ公認会計原則:他の総合収入メンバーを累計2020-01-012020-12-310001728117アメリカ-アメリカ公認会計基準:普通株式メンバー2020-12-310001728117US-GAAP:AdditionalPaidInCapitalMembers2020-12-310001728117アメリカ-公認会計基準:前払いメンバーを保留2020-12-310001728117アメリカ公認会計原則:他の総合収入メンバーを累計2020-12-3100017281172020-12-310001728117アメリカ-アメリカ公認会計基準:普通株式メンバー2021-01-012021-12-310001728117US-GAAP:AdditionalPaidInCapitalMembers2021-01-012021-12-310001728117アメリカ-公認会計基準:前払いメンバーを保留2021-01-012021-12-310001728117アメリカ公認会計原則:他の総合収入メンバーを累計2021-01-012021-12-310001728117アメリカ-アメリカ公認会計基準:普通株式メンバー2021-12-310001728117US-GAAP:AdditionalPaidInCapitalMembers2021-12-310001728117アメリカ-公認会計基準:前払いメンバーを保留2021-12-310001728117アメリカ公認会計原則:他の総合収入メンバーを累計2021-12-310001728117SRT:累計調整有効期限調整メンバUS-GAAP:AdditionalPaidInCapitalMembers2021-12-310001728117SRT:累計調整有効期限調整メンバアメリカ-公認会計基準:前払いメンバーを保留2021-12-310001728117SRT:累計調整有効期限調整メンバ2021-12-310001728117アメリカ-アメリカ公認会計基準:普通株式メンバー2022-01-012022-12-310001728117US-GAAP:AdditionalPaidInCapitalMembers2022-01-012022-12-310001728117アメリカ-公認会計基準:前払いメンバーを保留2022-01-012022-12-310001728117アメリカ公認会計原則:他の総合収入メンバーを累計2022-01-012022-12-310001728117アメリカ-アメリカ公認会計基準:普通株式メンバー2022-12-310001728117US-GAAP:AdditionalPaidInCapitalMembers2022-12-310001728117アメリカ-公認会計基準:前払いメンバーを保留2022-12-310001728117アメリカ公認会計原則:他の総合収入メンバーを累計2022-12-3100017281172017-10-012022-12-31GoS:市場を細分化する0001728117SRT:最小メンバ数2022-01-012022-12-310001728117SRT:最大メンバ数2022-01-012022-12-310001728117SRT:累計調整有効期限調整メンバ2022-01-010001728117SRT:累計調整有効期限調整メンバUS-GAAP:AdditionalPaidInCapitalMembers2022-01-010001728117SRT:累計調整有効期限調整メンバアメリカ-公認会計基準:前払いメンバーを保留2022-01-010001728117ゴス:2,200人のメンバー2022-01-012022-12-310001728117ゴス:2,200人のメンバー2021-01-012021-12-310001728117ゴス:2,200人のメンバー2020-01-012020-12-310001728117米国-公認会計基準:従業員株式オプションメンバー2022-01-012022-12-310001728117米国-公認会計基準:従業員株式オプションメンバー2021-01-012021-12-310001728117米国-公認会計基準:従業員株式オプションメンバー2020-01-012020-12-310001728117米国-公認会計基準:制限された株式メンバー2022-01-012022-12-310001728117米国-公認会計基準:制限された株式メンバー2021-01-012021-12-310001728117米国-公認会計基準:制限された株式メンバー2020-01-012020-12-310001728117アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001728117アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001728117アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001728117アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001728117アメリカ-公認会計基準:アメリカ政府メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001728117アメリカ-公認会計基準:アメリカ政府メンバーアメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001728117アメリカ-公認会計基準:アメリカ政府メンバーアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001728117アメリカ-公認会計基準:アメリカ政府メンバーアメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001728117米国-GAAP:商業チケットには現金および現金等価物のメンバーは含まれていないアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001728117アメリカ-公認会計基準:公正価値入力レベル1メンバー米国-GAAP:商業チケットには現金および現金等価物のメンバーは含まれていないアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001728117アメリカ-公認会計基準:公正価値入力レベル2メンバー米国-GAAP:商業チケットには現金および現金等価物のメンバーは含まれていないアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001728117アメリカ-公認会計基準:公正価値投入レベル3メンバー米国-GAAP:商業チケットには現金および現金等価物のメンバーは含まれていないアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001728117アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:会社債務証券メンバー2022-12-310001728117アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:会社債務証券メンバー2022-12-310001728117アメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:会社債務証券メンバー2022-12-310001728117アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:会社債務証券メンバー2022-12-310001728117アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001728117アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001728117アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001728117アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001728117米国-GAAP:商業チケットには現金および現金等価物のメンバーは含まれていないアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001728117アメリカ-公認会計基準:公正価値入力レベル1メンバー米国-GAAP:商業チケットには現金および現金等価物のメンバーは含まれていないアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001728117アメリカ-公認会計基準:公正価値入力レベル2メンバー米国-GAAP:商業チケットには現金および現金等価物のメンバーは含まれていないアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001728117アメリカ-公認会計基準:公正価値投入レベル3メンバー米国-GAAP:商業チケットには現金および現金等価物のメンバーは含まれていないアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001728117アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:会社債務証券メンバー2021-12-310001728117アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:会社債務証券メンバー2021-12-310001728117アメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:会社債務証券メンバー2021-12-310001728117アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:会社債務証券メンバー2021-12-310001728117アメリカ-GAAP:前払い費用と他の現在の資産メンバー2022-12-310001728117アメリカ-GAAP:前払い費用と他の現在の資産メンバー2021-12-310001728117アメリカ-公認会計基準:公正価値入力レベル2メンバーゴス:2,200人のメンバー2022-12-310001728117アメリカ-公認会計基準:公正価値入力レベル2メンバーゴス:2,200人のメンバー2021-12-310001728117アメリカ-公認会計基準:アメリカ政府メンバー2022-12-310001728117アメリカ-公認会計基準:会社債務証券メンバー2022-12-310001728117米国-GAAP:商業チケットには現金および現金等価物のメンバーは含まれていない2022-12-310001728117アメリカ-公認会計基準:会社債務証券メンバー2021-12-310001728117米国-GAAP:商業チケットには現金および現金等価物のメンバーは含まれていない2021-12-310001728117モス:TermLoanMembersUS-GAAP:LineOfCreditMember2019-05-020001728117モス:TermLoanMembersUS-GAAP:LineOfCreditMember2019-05-022019-05-02ゴス:バッチ0001728117モス:TermLoanMembersUS-GAAP:LineOfCreditMemberゴス:TranscheOneMembers2019-05-020001728117ゴス:TranscheTwoMembersモス:TermLoanMembersUS-GAAP:LineOfCreditMember2019-05-020001728117モス:TermLoanMembersUS-GAAP:LineOfCreditMember2022-12-310001728117モス:TermLoanMembersUS-GAAP:LineOfCreditMemberゴス:TranscheOneMembers2022-12-310001728117ゴス:TranscheTwoMembersモス:TermLoanMembersUS-GAAP:LineOfCreditMember2022-12-310001728117モス:TermLoanMembersUS-GAAP:LineOfCreditMemberボス:SecuredOvernightFinancingRateSOFRメンバー2019-05-022019-05-02Xbrli:純0001728117モス:TermLoanMembersUS-GAAP:LineOfCreditMemberボス:SecuredOvernightFinancingRateSOFRメンバー2019-05-020001728117モス:TermLoanMembersUS-GAAP:LineOfCreditMemberGoS:PrepaymentOcursThroughFirstAnneveraryOfClosingDateMember2019-05-020001728117GoS:1周年閉鎖日第2周年閉鎖日メンバーモス:TermLoanMembersUS-GAAP:LineOfCreditMember2019-05-020001728117モス:TermLoanMembersUS-GAAP:LineOfCreditMemberGoS:閉鎖日の2回目の記念日と1月までの優先最初の2,000および20,000人のメンバー2019-05-020001728117モス:TermLoanMembersUS-GAAP:LineOfCreditMemberボス:中型企業の財務信頼メンバー2022-01-012022-12-310001728117US-GAAP:LineOfCreditMember2022-12-310001728117ゴス:2,200人のメンバー2020-05-212020-05-210001728117ゴス:2,200人のメンバー2020-05-210001728117Goss:OneHundredThirtyPercentApplicableConversionPriceMemberゴス:2,200人のメンバーSRT:最小メンバ数2020-05-212020-05-21ゴス:取引日0001728117Goss:OneHundredThirtyPercentApplicableConversionPriceMemberゴス:2,200人のメンバーSRT:最大メンバ数2020-05-212020-05-210001728117GoS:価格変更を申請できるメンバーの99%ゴス:2,200人のメンバーSRT:最小メンバ数2020-05-212020-05-210001728117GoS:価格変更を申請できるメンバーの99%ゴス:2,200人のメンバーSRT:最大メンバ数2020-05-212020-05-210001728117ゴス:2,200人のメンバーSRT:最大メンバ数2020-05-212020-05-210001728117ゴス:2,200人のメンバー2022-12-310001728117ゴス:2,200人のメンバー2021-12-310001728117ゴス:2,200人のメンバー2022-01-012022-12-310001728117ゴス:2,200人のメンバー2021-01-012021-12-310001728117ゴス:2,200人のメンバー2020-01-012020-12-310001728117ゴス:PulmokineIncMemberギャス:ライセンス契約のメンバーGoS:InProcessResearchAndDevelopmentSeralutinibメンバ2017-10-022017-10-020001728117ゴス:PulmokineIncMemberギャス:ライセンス契約のメンバーSRT:最大メンバ数GoS:InProcessResearchAndDevelopmentSeralutinibメンバ2017-10-020001728117ゴス:PulmokineIncMemberギャス:ライセンス契約のメンバーGoS:InProcessResearchAndDevelopmentSeralutinibメンバ2017-10-012017-10-310001728117ゴス:PulmokineIncMemberギャス:ライセンス契約のメンバーGoS:InProcessResearchAndDevelopmentSeralutinibメンバ2020-12-310001728117ゴス:PulmokineIncMemberギャス:ライセンス契約のメンバーGoS:InProcessResearchAndDevelopmentSeralutinibメンバ2021-12-310001728117ゴス:PulmokineIncMemberギャス:ライセンス契約のメンバーGoS:InProcessResearchAndDevelopmentSeralutinibメンバ2022-12-310001728117GoS:InProcessResearchAndDevelopmentSeralutinibメンバ2022-01-012022-12-310001728117GoS:InProcessResearchAndDevelopmentSeralutinibメンバ2021-01-012021-12-310001728117GoS:InProcessResearchAndDevelopmentSeralutinibメンバ2020-01-012020-12-310001728117GoS:InProcessResearchAndDevelopmentGBZeroZeroFourMember2022-01-012022-12-310001728117GoS:InProcessResearchAndDevelopmentGBZeroZeroFourMember2021-01-012021-12-310001728117GoS:InProcessResearchAndDevelopmentGBZeroZeroFourMember2020-01-012020-12-310001728117GOSS:InProcessResearchAndDevelopmentOtherProgramメンバ2022-01-012022-12-310001728117GOSS:InProcessResearchAndDevelopmentOtherProgramメンバ2021-01-012021-12-310001728117GOSS:InProcessResearchAndDevelopmentOtherProgramメンバ2020-01-012020-12-310001728117米国-GAAP:国内/地域メンバー2022-12-310001728117アメリカ-公認会計基準:州と地方法律法規のメンバー2022-12-310001728117米国-GAAP:国内/地域メンバー2022-01-012022-12-310001728117アメリカ-公認会計基準:外国人メンバー2022-12-310001728117ゴス:孤児信用のメンバー2022-12-31ゴス:投票00017281172020-05-212020-05-2100017281172020-05-210001728117US-GAAP:PrivatePlacementMembers2022-07-152022-07-150001728117US-GAAP:PrivatePlacementMembers2022-07-150001728117ゴス:FounderSharesMember2015-12-032015-12-030001728117ゴス:FounderSharesMember2015-12-030001728117ゴス:FounderSharesMember2018-01-040001728117ゴス:FounderSharesMember2018-01-042018-01-040001728117ゴス:創始者のメンバー2022-12-310001728117ゴス:創始者のメンバー2018-05-212018-05-210001728117ゴス:創始者のメンバー2018-05-210001728117ゴス:創始者のメンバー2018-09-062018-09-060001728117ゴス:創始者のメンバー2018-09-060001728117ボス:株式制限協定のメンバー2022-01-012022-12-310001728117Goss:2900人の持分インセンティブ計画のメンバー2019-02-060001728117Goss:2900人の持分インセンティブ計画のメンバー2019-02-062019-02-060001728117Goss:2900人の持分インセンティブ計画のメンバー2022-12-310001728117モス:2900人の従業員株式調達計画メンバーSRT:最大メンバ数2019-02-062019-02-060001728117モス:2900人の従業員株式調達計画メンバー2019-02-060001728117モス:2900人の従業員株式調達計画メンバー2019-02-062019-02-060001728117モス:2900人の従業員株式調達計画メンバー2022-01-012022-12-310001728117モス:2900人の従業員株式調達計画メンバー2021-01-012021-12-3100017281172千と7千の株式インセンティブ計画のメンバーは2022-12-3100017281172千と7千の株式インセンティブ計画のメンバーは米国-公認会計基準:制限された株式メンバー2022-12-310001728117米国-公認会計基準:従業員株式オプションメンバーSRT:最小メンバ数2022-01-012022-12-310001728117米国-公認会計基準:従業員株式オプションメンバーSRT:最大メンバ数2022-01-012022-12-310001728117米国-公認会計基準:従業員株式オプションメンバーSRT:最小メンバ数2021-01-012021-12-310001728117米国-公認会計基準:従業員株式オプションメンバーSRT:最大メンバ数2021-01-012021-12-310001728117米国-公認会計基準:従業員株式オプションメンバーSRT:最小メンバ数2020-01-012020-12-310001728117米国-公認会計基準:従業員株式オプションメンバーSRT:最大メンバ数2020-01-012020-12-310001728117米国-公認会計基準:従業員株式オプションメンバー2022-01-012022-12-310001728117米国-公認会計基準:従業員株式オプションメンバー2021-01-012021-12-310001728117米国-公認会計基準:従業員株式オプションメンバー2020-01-012020-12-310001728117SRT:最小メンバ数ゴス:EmployeStockPurche ePlanMember2022-01-012022-12-310001728117SRT:最大メンバ数ゴス:EmployeStockPurche ePlanMember2022-01-012022-12-310001728117SRT:最小メンバ数ゴス:EmployeStockPurche ePlanMember2021-01-012021-12-310001728117SRT:最大メンバ数ゴス:EmployeStockPurche ePlanMember2021-01-012021-12-310001728117SRT:最小メンバ数ゴス:EmployeStockPurche ePlanMember2020-01-012020-12-310001728117SRT:最大メンバ数ゴス:EmployeStockPurche ePlanMember2020-01-012020-12-310001728117ゴス:EmployeStockPurche ePlanMember2022-01-012022-12-310001728117ゴス:EmployeStockPurche ePlanMember2021-01-012021-12-310001728117ゴス:EmployeStockPurche ePlanMember2020-01-012020-12-3100017281172019-01-012019-12-310001728117米国-公認会計基準:従業員株式オプションメンバー2022-12-310001728117米国-公認会計基準:制限された株式メンバー2019-12-310001728117米国-公認会計基準:制限された株式メンバー2020-01-012020-12-310001728117米国-公認会計基準:制限された株式メンバー2020-12-310001728117米国-公認会計基準:制限された株式メンバー2021-01-012021-12-310001728117米国-公認会計基準:制限された株式メンバー2021-12-310001728117米国-公認会計基準:制限された株式メンバー2022-01-012022-12-310001728117米国-公認会計基準:制限された株式メンバー2022-12-310001728117米国-公認会計基準:研究·開発費メンバー2022-01-012022-12-310001728117米国-公認会計基準:研究·開発費メンバー2021-01-012021-12-310001728117米国-公認会計基準:研究·開発費メンバー2020-01-012020-12-310001728117アメリカ-公認会計基準:一般と行政費用メンバー2022-01-012022-12-310001728117アメリカ-公認会計基準:一般と行政費用メンバー2021-01-012021-12-310001728117アメリカ-公認会計基準:一般と行政費用メンバー2020-01-012020-12-310001728117SRT:CEO実行官メンバ米国-公認会計基準:制限された株式メンバー2020-01-012020-12-310001728117米国-GAAP:株式補償計画のメンバーモス:2900人の従業員株式調達計画メンバー2022-12-310001728117米国-GAAP:株式補償計画のメンバーモス:2900人の従業員株式調達計画メンバー2022-01-012022-12-310001728117アメリカ-GAAP:OfficeEquipmentMembersSRT:最小メンバ数2022-01-012022-12-310001728117アメリカ-GAAP:OfficeEquipmentMembersSRT:最大メンバ数2022-01-012022-12-310001728117アメリカ-GAAP:OfficeEquipmentMembers2022-12-310001728117アメリカ-GAAP:OfficeEquipmentMembers2021-12-310001728117US-GAAP:ComputerEquipmentMembers2022-01-012022-12-310001728117US-GAAP:ComputerEquipmentMembers2022-12-310001728117US-GAAP:ComputerEquipmentMembers2021-12-310001728117米国-GAAP:ソフトウェア開発メンバー2022-01-012022-12-310001728117米国-GAAP:ソフトウェア開発メンバー2022-12-310001728117米国-GAAP:ソフトウェア開発メンバー2021-12-310001728117米国-GAAP:デバイス構成員SRT:最小メンバ数2022-01-012022-12-310001728117米国-GAAP:デバイス構成員SRT:最大メンバ数2022-01-012022-12-310001728117米国-GAAP:デバイス構成員2022-12-310001728117米国-GAAP:デバイス構成員2021-12-310001728117アメリカ-公認会計基準:リース改善メンバーSRT:最小メンバ数2022-01-012022-12-310001728117アメリカ-公認会計基準:リース改善メンバーSRT:最大メンバ数2022-01-012022-12-310001728117アメリカ-公認会計基準:リース改善メンバー2022-12-310001728117アメリカ-公認会計基準:リース改善メンバー2021-12-310001728117アメリカ-アメリカ公認会計基準:建設中のメンバー2022-12-310001728117アメリカ-アメリカ公認会計基準:建設中のメンバー2021-12-310001728117GoS:2018年8月にレンタルプロトコルをキャンセルしてメンバに入ることはできません2022-01-012022-12-31 アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
__________________________________________________________________________________
表10-K
__________________________________________________________________________________
| | | | | |
| ☒ | 1934年証券取引法第13項又は15(D)項に基づいて提出された年次報告 |
本財政年度末まで十二月三十一日, 2022
あるいは…。
| | | | | |
| ☐ | 1934年証券取引法第13項又は15(D)項に基づいて提出された移行報告 |
委員会ファイル番号:001-38796
__________________________________________________________________________________
薄紗生物会社です。
(登録者の正確な氏名はその定款に記載)
__________________________________________________________________________________
| | | | | | | | |
| デラウェア州 | | 47-5461709 |
(明またはその他の司法管轄権
会社や組織) | | (税務署の雇用主
識別番号) |
| | |
科学園路3013号 サンディエゴ, カリフォルニア州 | | 92121 |
| (主にオフィスアドレスを実行) | | (郵便番号) |
登録者の電話番号、市外局番を含む:(858) 684-1300
同法第12(B)項に基づいて登録された証券:
| | | | | | | | | | | | | | |
| クラスごとのタイトル | | 取引
記号 | | 登録された各取引所の名称 |
| 普通株、1株当たり0.0001ドル | | ゴス | | ナスダック世界ベスト市場 |
同法第12(G)項により登録された証券:なし
登録者が証券法第405条に規定する有名な経験豊富な発行者であるか否かをチェックマークで示す。:はい違います。 ☒
登録者が法案第13節又は第15(D)節に基づいて報告書を提出する必要がない場合は、複選マークで示してください。:はい違います。 ☒
再選択マークは、登録者が(1)過去12ヶ月以内に(または登録者がそのような報告の提出を要求されたより短い期間内に)1934年の証券取引法第13条または第15条(D)条に提出を要求したすべての報告書を提出したかどうか、および(2)過去90日以内にそのような提出要件に適合しているかどうかを示すはい、そうです☒ううん☐
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示すはい、そうです☒:いいえ。☐
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。1934年の証券取引法第12 b-2条規則における“大型加速申告会社”、“加速申告会社”、“小さい申告会社”、“新興成長型会社”の定義を参照されたい。
| | | | | | | | | | | | | | |
| 大型加速ファイルサーバ | ☐ | | ファイルマネージャを加速する | ☐ |
| 非加速ファイルサーバ | ☒ | | 規模の小さい報告会社 | ☒ |
| 新興成長型会社 | ☐ | | | |
新興成長型企業である場合、登録者が、取引法第13(A)節に従って提供された任意の新しいまたは改正された財務会計基準を遵守するために、延長された移行期間を使用しないことを選択したか否かを再選択マークで示す。☐
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オキシリー法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる☐
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する
これらのエラーのより真ん中に登録者の任意のエンタルピーCER幹部が相関回復期間内に§240.10 D−1(B)に基づいて受信したインセンティブベースの補償に基づいて回復分析を行う必要があるかどうかを再選択マークで示す
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)。*は☐--ありません☒
2022年6月30日まで(登録者が最近完成した第2財期の最終営業日),登録者の非関連会社が保有する登録者普通株の総時価は約$である541.3300万ドル、登録者ベースの普通株のナスダック世界ベスト市場での終値は1株8.37ドル。
2023年3月10日までに登録者は94,984,560普通株式(額面0.0001ドル)を発行した。
引用で編入された書類
登録者が2022年年次総会の最終委託書に関する一部は、第14 A条に従って米国証券取引委員会に提出され、本エントリ10−Kがカバーする本財政年度終了後120日に遅くなく、参照により当テーブル10−Kの第III部分に組み込まれる。
カタログ
| | | | | | | | |
第1部 | | |
| | |
第1項 | 業務.業務 | 3 |
プロジェクト1 A | リスク要因 | 29 |
プロジェクト1 B | 未解決従業員意見 | 79 |
プロジェクト2 | 属性 | 79 |
プロジェクト3 | 法律訴訟 | 79 |
プロジェクト4 | 炭鉱安全情報開示 | 79 |
| | |
第II部 | | |
| | |
プロジェクト5 | 登録者普通株市場、関連株主事項及び発行者による株式証券の購入 | 80 |
プロジェクト6 | 保留されている | 81 |
プロジェクト7 | 経営陣の財務状況と経営成果の検討と分析 | 81 |
プロジェクト7 A | 市場リスクの定量的·定性的開示について | 89 |
プロジェクト8 | 財務諸表と補足データ | 90 |
プロジェクト9 | 会計と財務情報開示の変更と相違 | 90 |
プロジェクト9 A | 制御とプログラム | 90 |
プロジェクト9 B | その他の情報 | 91 |
プロジェクト9 C | 検査妨害に関する外国司法管区の開示 | 91 |
| | |
第三部 | | |
| | |
第10項 | 役員·幹部と会社の管理 | 93 |
プロジェクト11 | 役員報酬 | 93 |
プロジェクト12 | 特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 | 93 |
第13項 | 特定の関係や関連取引、取締役の独立性 | 93 |
第14項 | 最高料金とサービス | 93 |
| | |
第4部 | | |
| | |
第15項 | 展示·財務諸表明細書 | 94 |
| | |
プロジェクト16 | 表格10-Kの概要 | 94 |
| サイン | |
第1部
前向きな陳述と市場データ
この10-K表年次報告書には、1934年“証券取引法”(改正)第21 E節又は“取引法”及び“1933年証券法”(改正)第27 A条又は“証券法”の意味に適合する前向きな陳述が含まれている。本年度報告に含まれるすべての歴史的事実の声明を除いて、私たちの将来の運営と財務状況、業務戦略と計画、研究開発計画、私たちと計画中の臨床前研究および計画中の臨床試験の予想時間、コスト、設計と進行に関する声明、私たちの候補製品の監督申告と承認の時間と可能性、新冠肺炎が私たちの業務に与える影響、成功の時間と可能性、未来の運営の管理計画と目標、および予想製品の将来の結果に関する声明は、すべて前向き声明に属する。これらの表現は既知と未知のリスク、不確定性とその他の重要な要素に関連し、私たちの実際の結果、業績或いは成果は展望性表現中の明示的或いは暗示的な任意の未来の結果、業績或いは成果と大きく異なることを招く可能性がある。このForm 10-K年次報告書には、独立した当事者と私たちによって行われた市場規模と成長に関する推定および他の統計データ、および私たちの業界に関する他のデータも含まれている。このデータは多くの仮定や制限に関連しているので、このような見積もりを過度に重視しないように注意してください。また、私たちの未来表現の予測、仮説と見積もり、そして私たちが経営する市場の未来表現は、必然的に高度な不確実性とリスクの影響を受ける。
場合によっては、“可能”、“会議”、“すべき”、“予想”、“計画”、“予想”、“可能”、“意図”、“目標”、“プロジェクト”、“考慮”、“信じ”、“推定”、“予測”、“潜在的”または“継続”またはこれらの用語の否定または他の同様の表現によって識別することができる。本年度報告書の展望的な陳述はただ予測だけだ。これらの展望的な陳述は主に私たちの現在の未来の事件と財務傾向の予想と予測に基づいており、私たちはこれらの事件と財務傾向が私たちの業務、財務状況と経営結果に影響を与える可能性があると考えている。これらの前向き陳述は、本年度報告が発表された日までの状況のみを代表し、第1部1 A項“リスク要因”で述べられたリスク、不確定要因、仮説を含むいくつかのリスク、不確実性、仮説の影響を受ける可能性がある。著者らの展望性陳述に反映された事件と状況は実現できない或いは発生できない可能性があり、実際の結果は展望性陳述中の予測結果と大きく異なる可能性がある。しかも、私たちは持続的な環境で運営している。新たなリスク要因や不確定要因が時々出現する可能性があり、管理職がすべてのリスク要因や不確定要因を予測することは不可能である。法的要件が適用されない限り、私たちは、任意の新しい情報、未来のイベント、状況の変化、または他の理由による、本明細書に含まれる任意の前向きな陳述を公開または修正するつもりはありません。すべての展望的陳述は1995年の個人証券訴訟改革法の安全港条項に基づいて作られたこの警告的声明である。
本年度報告書には、他の組織の財産に属する商標、商号、サービスマークが含まれている。便宜上、本年度報告で言及された商標および商標名は使用および記号を使用していないが、これらの参照は、適用法に基づいて、これらの商標および商標名に対する私たちの権利を最大限に主張しないこと、または適用されるすべての人がその権利を主張しないことを意味するわけではない。
本年度報告はまた、私たち自身の内部推定および研究、ならびに独立した市場研究、業界および一般出版物および調査、政府機関および公開から得られる情報からの業界、市場、および競争地位データを含む。いくつかの場合、私たちはこのようなデータの出所を明確に言及しなかった。この点で、私たちがどの段落でもこのようなデータの1つまたは複数のソースを言及した場合、他の平文規定または文脈に別の要求がない限り、同じ段落に出現するこのような他のデータは、同じソースからのものであると仮定すべきである。また、本報告に含まれる業界、市場、競争地位データは信頼性があり、合理的な仮定に基づいていると考えられるが、これらのデータは、第1部1 A項“リスク要因”で議論されている要因を含む様々な要因によって変化する可能性がある。これらの要素および他の要素は、結果が独立した当事者または私たちが推定した結果とは大きく異なる可能性がある。
私たちはwww.gosamerBio.comでウェブサイトを維持しています。私たちは定期的に私たちのニュース原稿のコピーと私たちに関するより多くの情報を公開しています。我々が米国証券取引委員会または米国証券取引委員会に提出した文書は、米国証券取引委員会に電子的に提出された後、または米国証券取引委員会に電子的に提出された後、合理的で実行可能な範囲内でできるだけ早く私たちのサイトを介して無料で閲覧することができる。我々のサイトに含まれる情報は、本報告書または米国証券取引委員会に提出された他の文書の一部を構成していません。
プロジェクト1.ビジネス
概要
我々は臨床段階の生物製薬会社であり,免疫学,炎症や腫瘍学などの疾患分野の治療薬の発見,獲得,開発,商業化に専念している。私たちの目標は業界になることです
すべての治療領域でリードし、このような疾病患者の生命を強化し、延長する。この目標を実現するために、著者らは経験豊富で、技術の優れたチームを集め、その中にリーディングバイオテクノロジーと製薬会社からの業界のベテラン、科学者、臨床医師と肝心なオピニオンリーダー、及び世界各地からのリード学術センターを含む。私たちの集団免疫学と翻訳発見と専門知識の開発がわが社の基礎です。著者らは科学的、厳格かつ包容的な企業文化を維持し、従業員は患者により良い治療選択をもたらすように努力するつもりである。
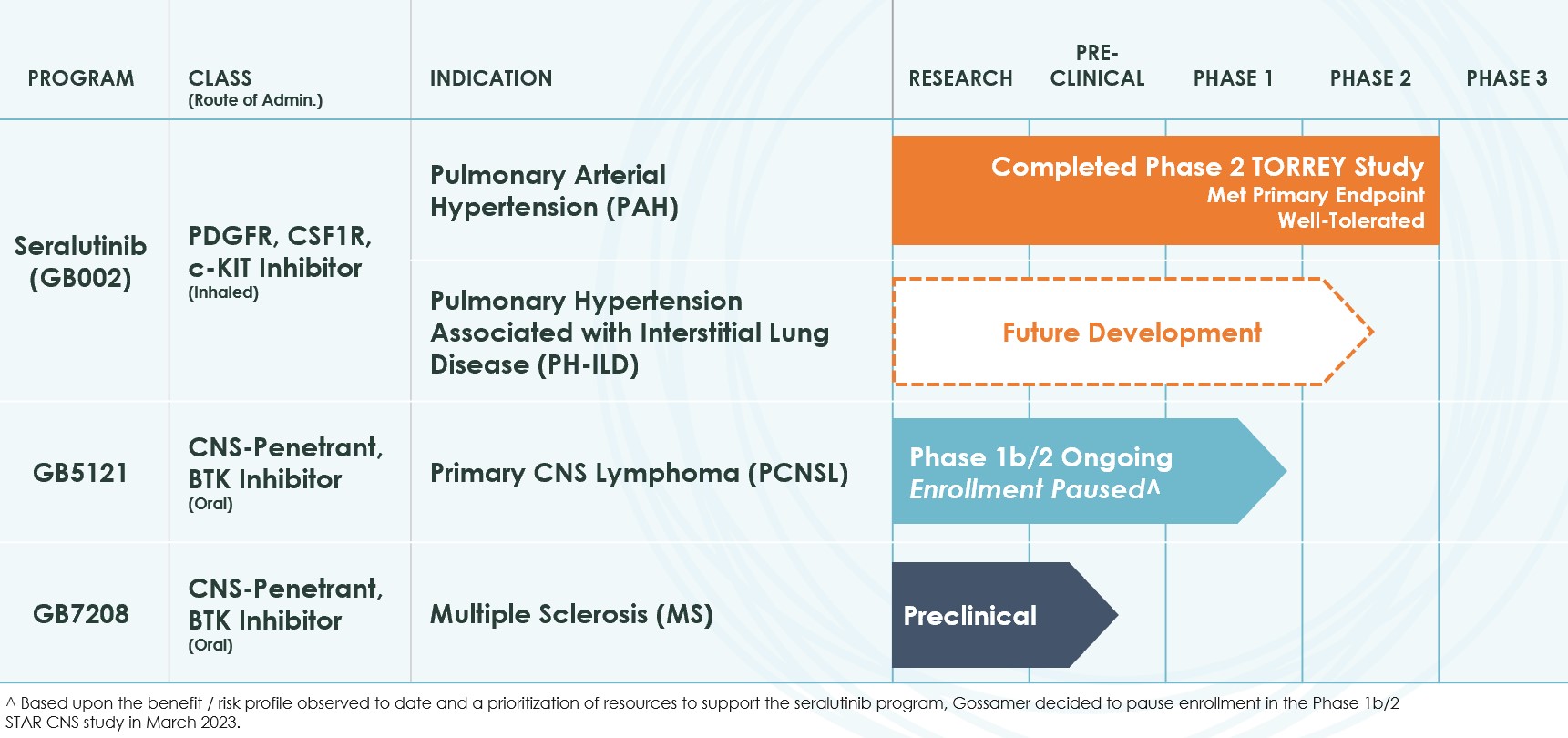
我々は,高度に満足されていない需要があり,一流あるいは一流療法の適応を開発する機会を得るために,強力な科学的根拠を持つ候補製品を探している。著者らは現在2つの臨床段階の候補製品があり、末期臨床前製品候補を除いている。
次の表は私たちの現在の計画をまとめています
セラルチニブ(PDGFR,CSF 1 Rおよびc−kit阻害剤)
Seralutinibは,GB 002とも呼ばれ,吸入性,小分子,血小板由来成長因子受容体(PDGFR),コロニー刺激因子1受容体(CSF 1 R)やc−kit阻害剤であり,肺動脈高圧(PAH)の治療に開発されている。市販の3種類のPAH治療の血管拡張療法と比較して,セラルチニブはPAHの基礎機序を解決することで病理リモデリングを逆転させる可能性が考えられる。吸入セラルチニブはPDGFR,αとβの2種類の異性体,およびCSF 1 Rとc−KIT経路に作用し,多くの動物PAHモデルにおいて肺血管細胞の過成長を抑制·逆転する。2022年12月,86名のPAH患者に対する24週間のTorrey第2段階試験のTOPLINE陽性結果を発表した。これらの良好な治療を受けた患者群では,主要な治療効果の終点,肺血管抵抗あるいはPVRにおいて,セラルチニブ群はプラセボ群より14.3%の統計で有意に改善した。完成したすべての臨床試験において,セラルチニブは全体的に耐性が良好であった。第二段階Torrey研究の24週間盲法部分を完成した後、患者は開放ラベル延長試験に参加することができた。我々は,2023年に行っているオープンタグ拡張実験の結果を報告する予定である.2023年下半期にPAH登録3期臨床試験を開始する予定である。私たちは2017年にPulmokine,Inc.からseralutinibの許可を得て、世界的な権利を保持した。米国食品医薬品局(FDA)と欧州委員会(EC)はセラルチニブをPAH患者を治療する孤児薬として承認した。
GB 5121(中枢神経系透過性BTK阻害剤)
GB 5121は経口、不可逆的な共有結合小分子ブルートンチロシンキナーゼ(BTK)阻害剤であり、臨床開発中であり、原発性中枢神経系リンパ腫(PCNSL)の治療に用いられている。GB 5121はその中枢神経系、透過性とキナーゼ選択性に基づいて選択された。BTKはB細胞や髄系細胞を含むいくつかの免疫細胞に発現しており,これらの細胞では複数の受容体下流のシグナル伝達を介している。BTKを抑制することはいくつかの自己免疫と炎症を推進する細胞活動を直ちに遮断と下方制御を招く。アクティブBTK
シグナル伝達も多くのB細胞悪性腫瘍に存在する。BTK阻害剤は米国で腫瘍適応の治療に許可されている。臨床前マウスモデルでは,選択したBTK阻害剤よりもGB 5121の方が研究用量で良好な中枢神経系透過性を示した。これらの臨床前研究で観察された中枢神経系の浸透は、PCNSLを含む中枢神経系の悪性血液病を治療する潜在的な治療法としてGB 5121を支持すると信じられている。GB 5121は現在再発/難治性PCNSLと他の稀な中枢神経系悪性腫瘍に対して1 b/2期STAR中枢神経系研究を行っている。これまでに観察された報酬/リスクプロファイルおよびseralutinib計画を支援する資源の優先順位から,1 b/2段階STAR CNS研究の登録を一時停止することにした.私たちは次の行動を決定するために、この研究のデータ審査委員会と既存のデータを議論する予定だ。関連する医学会議で行われているオープンラベル臨床試験のデータを報告したい。GB 5121は内部開発されたもので、全額所有です。
GB 7208(中枢神経系透過性BTK阻害剤)
GB 7208は、多発性硬化症、またはMSの治療のための臨床前開発段階にある経口小分子BTK阻害剤であり、GB 5121と同様に、中枢神経系への透過およびキナーゼの選択性に基づいて選択される。免疫細胞はMSの病理において重要な役割を果たしていると考えられ、BTK阻害剤はMSなどの自己免疫適応を治療する後期臨床開発段階にある。臨床前マウスモデルでは、開発中の自己免疫適応用BTK阻害剤(MSを含む)と比較して、GB 7208は検討用量でMS GB 7208を含めて臨床前テストを受けているよりも良い中枢神経系透過性を示している。GB 7208は内部開発されたもので、全額所有です。
私たちの研究能力と臨床前計画は
GB 7208を含めて複数の臨床前開発計画があり,免疫学,炎症,腫瘍学の治療領域に重点を置いており,現在われわれの複数の臨床前開発計画の持続的な発展を評価している
私たちのチームは
私たちの創業者と管理チームはリードするバイオ製薬会社で高級職を務めており、薬物発見、開発、商業化の分野で豊富な経験と専門知識を持っている。
ファシム·ハスナイヌは私たちの共同創業者で、2020年11月以来最高経営責任者を務め、私たちの設立以来会長を務めてきました。Hasnainさんはまた、当社の最高経営責任者として2018年7月から2018年7月までの間に当社のCEOを務め、2018年7月から2019年6月まで当社のCEOを務めます。Gossamer Bioの共同創設に先立ち、ハスナインは2010年11月から2015年8月までReceptos、Inc.最高経営責任者兼取締役を務めた。Receptosは2009年に設立された上場企業で、免疫学と代謝障害の治療法の開発に専念し、2015年8月にCelgene Corporationに買収された。これまでハスネルは取締役社長兼最高経営責任者兼CEOであり,生物指向抗体会社であり,多発性硬化症や腫瘍学に専念していた。彼は2008年12月から同職を務めており、2010年4月までアボットに買収された。
私たちの首席財務官兼首席運営官Bryan Giraudoは豊富なバイオテクノロジーと医療技術金融経験を持ち、Leerink Partnersで取締役高級取締役社長を務め、美林-ピアース-フェンナ-スミス社で取締役マネージャーを務めています。リチャード·アランダ、医学博士、私たちの首席医療官は、百時美施貴宝社、ノとノド社、Receptos、Celeene社で働いていた経験豊富な臨床医と薬物開発者です。著者らの首席科学官ローラ·カーター博士は20年以上の業界経験を持ち、多くの治療領域で第二段階臨床試験による標的識別と検証活動を行い、これまでLycera社、Medimmune、Array BiophmaとWyethで職務を担当したことがある。我々の技術運営と管理を担当する執行副総裁は豊富な管理バイオテクノロジー経験を持ち,Receptosで様々な職を務め,最近では取締役がCelgeneに買収された後に社長を務め,Ardea Biosciences,Inc.で副法律顧問総裁を務めている。カレン·ピーターソン、私たちは監督事務の執行副総裁を担当し、豊富な経験と監督専門知識を持ち、取締役発展と戦略コンサルティング連合会社で取締役社長を務め、Syndax製薬会社とFeRx Inc.で監督事務を指導する管理職を務めた
私たちの戦略
我々は臨床段階の生物製薬会社であり,免疫学,炎症や腫瘍学などの疾患分野の治療薬の発見,獲得,開発,商業化に専念している。我々の目標は,これらの治療分野の業界の先頭者となり,このような疾患患者の生命を向上·延長することである。私たちのビジネス戦略の重要な構成要素は
•私たちの世界的なチームの臨床開発と商業化の専門知識を利用する我々の実行管理チームと主要な科学リーダーはすでに大きさの生物製薬会社で小分子と生物製剤の発見、開発、商業化に成功している。我々は,PAHで計画されているセラルチニブの登録第3段階臨床試験を含め,我々の内部発見·開発戦略を実行する際にこれらの深い広範な専門知識や経験を利用する予定である。
•複数の適応にまたがって開発を拡張することにより,我々の候補製品の影響力を向上させる我々の開発努力を多様な疾患を治療する潜在力のある候補製品に集中させ,必要な場合により多くの適応を開発することを計画している。例えば,WHO第1群と第3群の肺動脈高圧にセラルチニブを開発する予定である。著者らはまた、我々の中枢神経系透過性BTK阻害剤GB 5121とGB 7208の潜在的神経状態を評価し続けた。
•協力、協力、許可取引、または他の取引に参加するGossamerは、その候補製品の開発および商業化を支援するために、追加の財務、臨床または商業資源を得るための取引を求めることができる。
私たちの候補製品
セラルチニブ(PDGFR,CSF 1 Rおよびc−kit阻害剤)
Seralutinibは,GB 002とも呼ばれ,吸入性小分子PDGFR,CSF 1 R,c−kit阻害剤であり,PAHの治療に開発されている。市販の3種類のPAH治療の血管拡張療法と比較して,セラルチニブはPAHの基礎機序を解決することで病理リモデリングを逆転させる可能性が考えられる。吸入セラルチニブはPDGFR,αとβの2種類の異性体,およびCSF 1 Rとc−KIT経路に作用し,多くの動物PAHモデルにおいて肺血管細胞の過成長を抑制·逆転する。2022年12月,86名のPAH患者に対する24週間のTorrey第2段階試験のTOPLINE陽性結果を発表した。これらの良好な治療を受けた患者群では,セラールチニブ群はプラセボ群と比較して主要な治療効果の終点,肺血管抵抗あるいはPVRで統計的に有意な改善を示した。PVRの改善は、すべての予め指定された患者サブグループにおけるセラルチニブの使用に有利である。完成したすべての臨床試験において,セラルチニブは全体的に耐性が良好であった。第二段階Torrey研究の24週間盲法部分を完成した後、患者は開放ラベル延長試験に参加することができた。我々は,2023年に行っているオープンタグ拡張実験の結果を報告する予定である.2023年下半期にPAH登録3期臨床試験を開始する予定である。私たちは2017年にPulmokine,Inc.からseralutinibの許可を得て、世界的な権利を保持した。FDAと欧州委員会はセラルチニブ孤児薬のPAH患者の治療のための指定を許可した。
行動メカニズム
PAHは肺小血管内と周囲の異常細胞増殖によって駆動され,これらの小血管は心臓の右側から肺に血液を輸送する。肺血管系の機能と構造変化は血管再構築と呼ばれ、平滑筋細胞の増殖を招き、血管中層から内層に遷移することができる。これは叢状や新たな内膜病変の進展を招き,血流を阻害する可能性がある。肺血管中の血流の閉塞も,患者がこれらの小さな肺血管内に血栓や血塊を形成しやすくなり,さらに血流を阻止する。このような心臓右側から肺への進行性血流閉塞は右心室不全を招くことができ、それによって深刻な呼吸困難、運動持久力の低下と死亡を招く。セラルチニブはPDGFRα/β、CSF 1 Rとc-Kitを含む肺動脈高圧病理に作用する多種のキナーゼを抑制するように設計されている
PDGFRはチロシンキナーゼ受容体であり,そのアゴニストによって活性化されると細胞増殖を誘導する。PDGFの発現はPAH患者の平滑筋細胞増殖を刺激するために特に重要であることが知られている。PDGFRsとそのリガンドはPAHで上昇した。PDGFRシグナルの上昇は内皮細胞と線維芽細胞の機能障害を招き、そして血管平滑筋細胞の増殖と遷移を招く。この効果は肺内血管の過剰成長と閉塞を招く。PDGFR経路活性を有するキナーゼ阻害剤は,動物モデルにおいてPAHを逆転させる能力を示している。
吸入型サラダルチニブはPDGFRの2種類のサブタイプ,αとβに作用するように設計されている。臨床前動物モデルとPAH患者からのヒト肺組織学的データは,PDGF受容体のこの2つの異性体を抑制することが重要であることを示している。血小板由来増殖因子受容体αは肺小動脈血管平滑筋細胞に高発現する。血管内皮増殖因子受容体αを抑制することは、血管肥厚を引き起こす血管内皮細胞の異常増殖の減少に役立つ可能性がある。Pdgfrβの方が高い
線維芽細胞や筋線維芽細胞に発現し,血管内細胞の異常増殖に関与し,肺小動脈閉塞をきたす。したがって,PDGFRβを抑制することは,これらの細胞型の異常細胞増殖を減少させる上で重要な意義があると考えられる。
C−kit経路は肺血管再構築に関与する重要な成長因子であり,特に血管周囲炎症に関与する細胞でも考えられている。肺や肺小動脈試料の解析でも特発性PAHではc−kit遺伝子発現が増加していることが示された。C−kit陽性の内皮細胞もPDGFを分泌し,血管周囲c−kit陽性の肥満細胞は炎症促進サイトカインやトリプシンを分泌し,PAHの炎症過程をさらに促進する
イマチニブ(Gleevec)は経口チロシナーゼ阻害剤であり,PDGFRやc−KIT経路に対する活性を有することが知られており,PAHの3期臨床試験ではヒトで概念検証を行い,臨床試験ではPDGFRとc−kitキナーゼ阻害剤の機械的検証が観察された。臨床前モデルでは,セラルチニブはイマチニブよりもPDGFFRαサブタイプの抑制作用が強く,PDGFFRβサブタイプやc−KITに対する抑制作用はイマチニブの10倍であった
マクロファージもPAH発生と増悪過程において最も重要な炎症性細胞の一つと考えられている。マクロファージはCSF 1受容体を発現し,現在PAHの病理過程において重要な役割を果たしていると考えられている。活性化されたCSF 1 R陽性マクロファージはPAH患者の肺小動脈周囲に集積しており,陽電子放出断層撮影により生体内に示されている。また,PAHにおけるマクロファージの活性は骨形態発生蛋白受容体IIやBMPR 2のレベルに関与している。PAHのBMPR 2特徴の減少は顆粒−マクロファージコロニー刺激因子(GM−CSF)の誘導とマクロファージの募集をもたらす。BMPR 2ノックアウトマウスでは,組織マクロファージの活性化により著明な肺炎症が認められたことに注意されたい。
また,炎症性マクロファージはPDGFを分泌し,肺動脈平滑筋細胞の遊走や増殖を刺激し,PAHを特徴とする炎症,過増殖,線維化のフィードバック循環を加速させる
PAHにおけるPDGF経路の先行進展−イマチニブのIMPRES 3期臨床試験−
IMPRES試験は,ノファ社のイマチニブ(Gleevec)によるPAH患者の3期臨床試験である。イマチニブはPDGFR,c−kit受容体とAbelsonマウス白血病ウイルス癌遺伝子相同1,あるいはc−ablを含む様々なチロシンキナーゼに対して活性であることが知られている。202名の患者がIMPRES試験に参加し、その41%がプロスタノイド、ホスホジエステラーゼ5またはPDE 5阻害剤の経口投与、およびエンドセリン受容体アゴニストまたはERASの経口治療を受けている。この試験は,24週目にプラセボと比較した6分間歩行距離(6 MWD)の改善であり,統計学的有意差を達成した(p=0.002)。P値は2つのデータセット間の差が偶然性によって生じる確率である.Pの値が小さいほど,この違いは偶然だけではない可能性がある.一般に,p値が0.05以下であれば,結果は統計学的に有意であると考えられる。
イマチニブを服用した患者は肺血管抵抗(PVR)を含む血流動力学指標においても統計的に有意な改善があり,PVRはPAH患者を評価する標準指標である。しかし、全身不良事件、例えば出血と耐性が悪い、及び頻繁に薬を中止し、試験活動腕内の高中退率を招く。試験期間中に抗凝固剤を同時に服用した8名の患者に硬膜下血腫が出現した。ノワール社は2013年にPAHへの追加規制申請を撤回し,適応にイマチニブをさらに開発していないことが知られている
肺動脈高圧の研究概要
PAHはまれな疾患であり,脱酸素血液を心臓右側から肺に輸送する血管に異常高血圧が出現し,進行性であり,通常致命的であることを特徴とする。症状は休憩や少量の運動時の呼吸急を含む。その他の症状は疲労、胸痛、眩暈と失神を含む。この疾患の進行性により右側心臓はより努力して働き,最終的に虚弱あるいは不全になる。
患者は通常,患者が体力活動を行う能力や症状の重症度に応じて患者を分類する機能レベルによって評価される。症状が悪化するため,より高い機能レベルは,より高い死亡率に関連している。以下の表1は世界保健機関設立の4つの機能種別を詳細に説明した。
表1.PAH機能クラス
| | | | | | | | |
機能性
クラス | | 説明する |
| 第I類 | | PAH患者であるが,そのための体力活動は制限されない。通常の体力活動は過度な呼吸困難や疲労、胸痛あるいは失神に近いことを引き起こすことはない。 |
| | |
| 第II類 | | PAH患者の体力活動は軽微に制限された。彼らは楽に休んだ。普通の体力活動は過度の呼吸困難や疲労、胸痛あるいは失神に近い。 |
| | |
| 第III類 | | PAH患者は体力活動が明らかに制限された。彼らは楽に休んだ。運動不足は過度の呼吸困難や疲労、胸痛あるいは失神に近い。 |
| | |
| 第IV類 | | PAH患者は症状なしでは何の体力活動もできない。これらの患者は右心不全の徴候を示した。休憩時には呼吸困難や/または疲労が出現する可能性もある。どんな体力活動でも体調不良が増えます。 |
また,最近の医学会ガイドラインでは,右心不全のバイタルサイン,症状進展率,機能分類,6 MWD,最大酸素消費量,NT−proBNPを含むいくつかの変数からPAHの中高リスク種別が決定されており,心不全のバイオマーカー,右心機能の測定である。完成した第2段階Torrey研究では,その1つのリスク分類ツールであるDisplay 2.0リスクスコアを用いた。
過去20年間に、多環芳香族炭化水素に対する多種の治療方法が相次いで登場したが、多環芳香族炭化水素の発病率と死亡率は依然として非常に高い。REVIEW登録データによると,新たに診断された機能III級とIV患者の5年生存率はそれぞれ60%と44%であったが,先に診断された患者の5年生存率はそれぞれ57%と27%と低かった。
多環芳香族炭化水素市場の概要
PAHは30歳から60歳の女性に最も影響を与える。PAHの真の発病率と流行率はまだ不明である。米国肺協会は,米国で毎年新たに診断された症例は500例から1000例と推定されている。PAHの米国やヨーロッパでの罹患率は7万人と推定されている。確定診断されたPAH患者数が増加し続けており,この増加は本疾患に対する認識や診断の向上による可能性が考えられる。2021年の世界ブランドPAH薬の総売上高は約59億ドル。
パーキンソン病の治療パラダイム
現在承認されたPAH治療は3種類の血管拡張剤を含む:PDE 5阻害剤(とオルニチン環化酵素アゴニスト)、ERASとプロスタグランジン(とプロスタサイクリン受容体アゴニスト)。PDE 5阻害剤はよくERASと併用し、早期治療策略とする。ERAとPDE 5阻害剤の併用治療が無効な患者にプロスタグランジンを添加することはよく見られるやり方である。プロスタグランジンは通常右心不全のエビデンスのある患者の治療にも用いられる。いくつかの現有の治療方法はすでにPAH患者の臨床悪化と他の複合終点の時間を著しく改善したが、潜在的な疾病の病理生理を直接変化させる治療方法はない。承認されたPAH療法の血管拡張作用は,肺を介した血液流動を改善することができるが,最終的には疾患の背後で悪化していく細胞増殖や動脈再構築に置き換えられる可能性がある。病理再構築を安全に逆転させることができる薬物は,機能クラスとリスククラスにまたがる実用的なツールを提供できると信じている。Sotatercept(Merck&Co.,Inc.)のような新しい研究療法は,承認されればPAH治療例に影響する可能性がある。Sotaterceptは皮下投与されたアクチビン受容体IIA-Fc融合蛋白であり、1種の研究候補薬物であり、すでに完成した3期PAH臨床試験で評価を行った。
セラルチニ製品差別化
Seralutinibはイマチニブ試験における有効性の証拠に基づいて、イマチニブで観察された全身安全性と耐性の問題を克服し、イマチニブのキナーゼ阻害スペクトルを改善することを目的とした吸入型キナーゼ阻害剤である。セイラルチニブはイマチニブと比較して異なる選択的特徴を有し,PDGFFRαサブタイプに対する効力が増強し,PDGFFRβ異性体やc−Kitに対する効力が10倍向上し,c−ablやチロシンキナーゼLCKに対して活性がなかった。また,セイラルチニブの方がイマチニブよりもCSF 1 Rに対する作用が強い.セラルチニブはPAH治療薬になる可能性があると信じています
•分化した抗増殖機序は、多環芳香族炭化水素の潜在的機序を解決する
•全身性イマチニブよりも耐性の高い安全性
•便利、簡単、携帯可能な吸入方法及び搬送システムを提供する。
セラルチニブの臨床経過
臨床前研究計画概要
セラルチニブは同時にPGDFRαとβを抑制し、PAHラットモデル肺血管中の細胞の過剰成長を抑制と逆転し、このモデルは肺小血管を遮断できる異常細胞増殖を含むヒトPAHの多くの特徴を複製した。このモデルでは,セラルチニブは小肺血管の閉塞性病変を有意に減少させた。また、セラルチニブはプラセボと比較して右室収縮圧の統計学的有意な低下を示した。単独のPAHラットモデルSU 5416低酸素モデルでは、セラルチニブはプラセボと比較して循環血漿NT−proBNPにおいて統計的に有意な低下を示したが、イマチニブとプラセボとの間の差はこのようなPAHバイオマーカーでは有意ではなかった。Seralutinibはプラセボやイマチニブと比較してラット肺BMPR 2の発現を健康レベルに回復させ,統計的に有意な改善であった。BMPR 2発現異常はPAHに関与している。単独のPAHラットモデルSU 5416低酸素モデルでは,セラルチニブはプラセボと比較して循環血漿NT−proBNPの有意な低下を示したが,PAHバイオマーカーにおけるイマチニブとプラセボの差は有意ではなかった。Seralutinibはプラセボやイマチニブと比較してラット肺BMPR 2の発現を健康レベルに回復させ,統計的に有意な改善であった。BMPR 2発現異常はPAHに関与している。
完成した1 a段階研究の概要
著者らは82名の健康成人ボランティアがセラルチニブを経口投与した1 a期SADとMAD二重盲検、プラセボ対照、無作為研究を完成した。薬物動態またはPKパラメータと安全性を評価した。セラルチニブ耐性は良好であり,用量制限毒性はなかった。深刻な有害事象(SAE)は報告されておらず,検討中止となった有害事象(AEs)も報告されていない。最もよく見られる副作用は咽喉頭刺激と咳であり、重症度は比較的に軽く、発生率はプラセボと類似している。吸入剤を単回と複数回経口投与した後,セラルチニブは速やかに循環吸収·除去された。単用量と多用量投与後,セラルチニブの曝露用量は比例して増加した。
完成した1 b期PAH臨床試験のまとめ
2020年12月、著者らは完成した1 b期無作為、二重盲検、プラセボ対照、多中心のシュラルチニブによる機能性II級とIII級PAH患者に対するTOPLINE試験結果を発表した。8名の患者は2週間の盲法部分試験を完了した。新冠肺炎疫病のため、この試験の登録作業は一時停止したが、2020年第3四半期に再開された。この2週間の試験の主な結果は安全性と耐性だった。PAH患者におけるセラルチニブの耐性は通常良好であり,全8名の患者が2週間の研究を完了した。SAEはなく,最も一般的なAEsは軽度から中等度の咳と軽微な頭痛である。PAH患者における全身性PKの特徴は,全身曝露が低く,薬物除去が速いことであり,健常ボランティア1 a期研究におけるPKデータと一致している。全試験用量レベルにおいて,全血CSF 1 R安定化試験によりPAH患者の標的関与が確認された
2週間の1 b期試験終了後、2名のPAH患者は6ケ月のシュラルチニブ開放ラベル延長試験を登録し、完成することができた。2名の患者とも最大許容量まで滴定でき,1日2回,90 mgであった。試験延期期間中,SAEの報告はなかった。2名の患者のNT-proBNPレベルはすべてベースラインより低下し、6 MWDはベースラインより増加した
完成したPAH 2期臨床試験まとめ(Torrey研究)
2022年12月、著者らは完成した第2段階Torrey研究のTOPLINE陽性結果を発表し、これはPAH患者に対する無作為、二重盲検、プラセボ対照、多中心臨床試験である。背景PAH治療を受けているにもかかわらず,治療目標に達していない機能性II級とIII級PAH患者86名を募集した。入選患者の57%が三重背景PAH治療を受け、40%の患者が二重背景PAH治療を受けた。活動腕上の患者は1日2回セラルチニブ60 mgからの投与量を受け,1日2回90 mgまで滴定した。このプログラムは必要に応じて1日2回滴定を45 mgに低下させることを許可しているが,セラルチニブを服用しているほとんどの患者は1日2回90 mgを達成し維持することができる。全試験中、患者は依然として彼らの背景PAH治療を受けた。
Seralutinibは24週の治療期間中に主要終点で統計的に有意な改善を示し,ベースラインと比較してPVRが変化した。プラセボとセラルチニブ間のPVRの平均改善
検討では96.1 dynes(p=0.0310)が認められ,プラセボ補正後14.3%の改善に相当した。PVRの改善は、すべての予め指定された患者サブグループにおけるセラルチニブの使用に有利である。Torrey研究における重要な副次的な終点は、6 MWDにおいてベースラインから24週目に変更することである。プラセボとセラルチニブの間に6 MWDが認められた平均差は6.5メートルであり,セラルチニブの腕に有利な値であった。あらかじめ指定されたサブグループの多くでは,6 MWDの変化もセラルチニブに有利である.この実験は6 MWDの統計的意味のために設計されたものでもなく,統計的意味のために設計されたものでもない
WHO Function ClassあるいはFCの定義によると,PVRと6 MWDの効果はベースライン疾患のより深刻な患者に増強効果が認められ,2.0のリスクスコアを示した。FC III患者では,プラセボと比較してサラダルチニブを服用した患者のPVRは21%(p=0.0427),6 MWDは37 m改善した(p=0.0476)。ベースラインリスクスコアが6以上の患者では,セラルチニブを服用した患者のPVRはプラセボと比較して23%(p=0.0134),6 MWDは22 m改善した(p=0.2482)。
セラルチニブ投与後,右心圧のバイオマーカーNT−proBNPは12週で有意に低下し,24週でプラセボの平均値と408.3 ng/Lの差があった(p=0.0012)。右心構造と機能の重要な評価では,セラルチニブはプラセボと比較して右心房面積,右心室遊離壁ひずみ,肺動脈コンプライアンスを含むバイオマーカーの変化に伴い,臨床的に相関性と統計学的有意な変化を有していた。
Torreyの研究では,Seralutinibは全体的に耐性が良好であり,プラセボ群とSeralutinib群ではそれぞれ36名(86%)と41名(93%)の患者が緊急不良事象(TEAE)の治療を報告している。研究で報告されているほとんどのTEAEの重症度は軽度から中等度であった。セラールチニブ群では研究薬物に関するSAEが1例報告されているが,プラセボ群では検討薬物に関するSAEの報告はない。この研究で最もよく見られるTEAEは咳嗽であり,それぞれ16名(38%)と19名(43%)がプラセボとセラルチニブを服用した患者が咳嗽を報告している。セラルチニブ服用が報告された19名中17名に軽度の咳嗽が出現し,2名に中等度の咳嗽が出現した。IMPRESのイマチニブによるPAH治療に関する3期研究で最もよく見られるTEAEは,嘔気,周囲浮腫,下痢,嘔吐を含め,Torrey研究で観察される頻度ははるかに低く,報告例では通常シュラルチニブとプラセボとのバランスが良好である。硬膜下血腫症例は認められなかった。
第二段階Torrey研究の24週間盲法部分を完成した後、患者は開放ラベル延長試験に参加することができた。我々は,2023年に行っているオープンタグ拡張実験の結果を報告する予定である
計画中のPAH 3期臨床試験まとめ
2023年下半期にPAHの3期臨床試験を開始する予定である。計画中の第三段階の臨床試験はPAH患者のランダム、二重盲検、プラセボ対照に対する全世界の臨床試験である。患者は、彼らの背景PAH治療を除いて、ランダムにセラルチニブまたはプラセボを受けるであろう。我々は世界の監督管理機関と2023年上半期に接触しており、これらの相互作用は第三段階の臨床試験の最終設計に情報を提供することが予想される。FDAのフィードバックによると、私たちは1日2回90 mgの単回の用量をテストすると予想され、私たちは試験の主要な終点はベースラインに基づいて6 MWDを変化させると予想される;しかし、最終的な試験設計が世界の監督管理機関のさらなるフィードバックに依存する場合。二次と探索的終点に加えて,安全性と耐性も第三段階臨床試験で評価される。
より多くの適応の臨床発展計画
セラルチニブは間質性肺疾患(PH−ILD)に関連する肺動脈高圧の治療に応用できる可能性があると信じている。PH-ILDは世界保健機関の第三グループの肺動脈高圧のサブグループであり、一組の進行性、よく致命的な肺動脈高圧形式であり、肺部の小気道に影響を与える。これらの疾患の特徴は,肺高圧に関連する肺血管病態と,間質性肺疾患による肺間質肥厚と瘢痕形成である。現在FDAが許可したPH-ILDを治療する方法は1つしかなく、EUはまだ承認されていない治療方法である。セラールチニブはPH−ILD患者の生活の質を改善する可能性があると信じている。2023年下半期あるいは2024年上半期にPH−ILDの臨床開発を開始する予定である。
GB 5121(中枢神経系透過性BTK阻害剤)
GB 5121は経口、不可逆、共有結合、小分子のBTK阻害剤であり、臨床開発中でPCNSLの治療に応用されている。GB 5121は中枢神経系に対する透過性とキナーゼに対する選択性に基づいて選択された。BTKはB細胞や髄系細胞を含むいくつかの免疫細胞に発現しており,これらの細胞では複数の受容体下流のシグナル伝達を介している。BTKを抑制することはいくつかの自己免疫と炎症を推進する細胞活動を直ちに遮断と下方制御を招く。活発なBTKシグナルも多くのB細胞悪性腫瘍に存在する。BTK阻害剤は米国で腫瘍適応の治療に許可されている。臨床前マウスモデルでは,選択したBTK阻害剤よりもGB 5121の方が研究用量で良好な中枢神経系透過性を示した。これらの前臨床研究で観察された中枢神経系の浸透は
PCNSLを含む中枢神経系悪性血液病を治療する潜在的療法としてGB 5121の開発を支持する。GB 5121は現在再発/難治性PCNSLと他の稀な中枢神経系悪性腫瘍に対して1 b/2期STAR中枢神経系研究を行っている。これまでに観察された報酬/リスクプロファイルおよびseralutinib計画を支援する資源の優先順位から,1 b/2段階STAR CNS研究の登録を一時停止することにした.私たちは次の行動を決定するために、この研究のデータ審査委員会と既存のデータを議論する予定だ。関連する医学会議で行われているオープンラベル臨床試験のデータを報告したい。GB 5121は内部開発されたもので、全額所有です。
行動メカニズム
BTKは免疫系において鍵と多方面の役割を果たしている。BTKは非受容体タンパク質チロシンキナーゼであり、TECプロテインキナーゼファミリーに属する。B細胞,マクロファージ,好中球,肥満細胞などの造血細胞に存在する。BTKはB細胞受容体(BCR)シグナルと獲得性免疫反応の重要なメディエーターである。BCRの刺激下で、BTK経路は細胞内カルシウムイオンレベルを上昇させ、そしてB細胞の増殖、分化と生存に参与する転写因子を活性化する。この経路も悪性B細胞の増殖、遷移と生存のキーポイントであり、この経路を抑制することはいくつかの血液系悪性腫瘍の治療に有効である。BTK阻害剤はFDAによって多発性B細胞リンパ腫の治療に許可されている
BTK阻害剤は悪性血液病患者に価値のある治療選択を提供したが、現有の治療方法は中枢神経系疾患の治療に適していない可能性がある。血液脳関門,あるいはBBBと呼ばれ,内皮細胞からなり,高度な区分的境界として全身循環と中枢神経系の細胞外液を分離する。BBBは循環病原体から脳を効果的に保護するが,BTK阻害剤の多くを含む薬物が体循環から中枢神経系に入る能力も制限されている。GB 5121は,腫瘍疾患に対する他のBTK阻害剤と比較して,検討用量の臨床前マウスモデルでより良好な中枢神経系透過性を示した。
BTK阻害剤の治療は標的上と標的外AEsと関係があり、例えば皮疹、下痢、感染、出血、細胞減少と心血管AEs、心房細動を含む。BTK阻害剤を受けた患者でも肝酵素の上昇を認めた。適度な選択性を有する分子は、脱標的プロテインキナーゼとの意外な相互作用によって脱標的AEsを増加させる可能性がある。新世代のBTK阻害剤は選択性を増加させることによって非標的毒性を最大限に減少させることに取り組んでいるが、限られた中枢神経系浸透はこれらの分子を中枢神経系疾患を治療する次の最適分子になる可能性がある。前臨床試験ではGB 5121がBTKに対して高い選択性を有することが観察されており,より高い選択性が適度な選択性のBTK阻害剤に関連する有害事象を制限する可能性が考えられる。
原発性中枢神経系リンパ腫の概要
PCNSLは稀な侵襲性非ホジキンリンパ腫であり、脳、目と脳脊髄液から起源し、全身に影響を受けた証拠がない。血液脳関門透過性が悪いため,標準的なリンパ腫治療では不十分である。PCNSLの第一線の治療は通常脊椎上で多種の化学療法、大量のメトトレキサート、その後強固な全脳放射線治療を含む。このような治療選択があるにもかかわらず、50%の患者だけが持続的な緩和を実現した。治療は著明な神経毒性に関与しており,メトトレキサートに耐えられない患者や癌進展患者ではFDA承認されていない治療法である
ヒト試験において、イブルチニブは1種の非選択性BTK阻害剤であり、血液脳関門を越える能力は限られており、高用量で再発或いは難治性PCSPL患者に有効であることが証明された。中枢神経系で治療レベルに達するために必要な高用量、および他のキナーゼに対する親和性は、EGFR、JAK 3とHER 2を含み、イブルチニブ治療に関連するいくつかの毒性反応の原因である可能性が考えられる。臨床前モデルで観察されたBTKの高特異性と高中枢神経系の透過性は,GB 5121を低い全身用量でイブルチニブの臨床効果を得ることが可能であり,耐性を改善する可能性があると信じている。臨床前モデルで観察された有効性,特異性,高脳浸透率に基づいて,GB 5121を選択してヒト臨床試験を行った。
GB 5121臨床発展史
進行中の1期臨床研究概要
2021年第4四半期、著者らは健康な人体ボランティアの中で用量増加の第1段階研究を開始し、GB 5121の人体における安全性、耐性、PKとPDを評価した。この研究にはSADとMAD部分および臨床薬理学的評価がある
PCNSLが行っているSTAR CNS 1 b/2期臨床試験概要
2022年第2四半期、私たちは全世界の1 b/2期臨床試験の1 b期部分を開始した。試験の1 b期部分は再発性或いは難治性原発或いは続発性中枢神経系リンパ腫或いは再発性或いは難治性原発硝子体網膜リンパ腫の患者に組み入れられた。ステップ1 bの主要な終点は、安全性および耐性であり、第2の終点は、全体応答率またはOORおよび反応持続時間を含む。第2段階の用量選択は、1 b段階の結果に基づいて行われる
これまでに観察された報酬/リスクプロファイルおよびseralutinib計画を支援する資源の優先順位から,1 b/2段階STAR CNS研究の登録を一時停止することにした.私たちは次の行動を決定するために、この研究のデータ審査委員会と既存のデータを議論する予定だ。
GB 7208(中枢神経系透過性BTK阻害剤)
GB 7208は経口小分子BTK阻害剤であり、臨床前開発段階でMSの治療に使用されている。GB 5121と同様に、GB 7208は中枢神経系への透過とキナーゼの選択性に基づいて選択される。免疫細胞はMSの病理において重要な役割を果たしていると考えられ、BTK阻害剤はMSなどの自己免疫適応を治療する後期臨床開発段階にある。臨床前マウスモデルでは、開発中の自己免疫適応用BTK阻害剤(MSを含む)と比較して、GB 7208は検討用量でMS GB 7208を含めて臨床前テストを受けているよりも良い中枢神経系透過性を示している。GB 7208は内部開発されたもので、全額所有です
私たちの研究能力と臨床前計画は
GB 7208を含めて複数の臨床前開発計画があり,免疫学,炎症,腫瘍学の治療分野に重点を置いており,これらの計画の持続的な発展を評価している
競争
生物技術と製薬業界の特徴は技術進歩が迅速で、競争が激しく、知的財産権を強調することである。著者らは主要と専門製薬と生物技術会社、学術研究機構、政府機関及び公共と個人研究機関を含む多くの異なる源からの潜在的な競争に直面している。私たちが開発と商業化に成功したどの候補製品も、現在の治療法と将来出現する可能性のある新しい療法と競争するだろう。もし私たちの競争相手が私たちが開発する可能性のあるどの製品よりも安全で、より効果的で、副作用がより少ない、あるいはより便利な製品を開発し、商業化すれば、私たちのビジネス機会は減少または消失するかもしれない。私たちの競争相手はまた私たちよりも早くFDAや他の規制機関からその製品の承認を受けるかもしれない。私たちのすべての候補製品の成功に影響を与える重要な競争要素は、有効性、安全性、利便性、コスト、これらの製品に対する販売促進活動レベル、および知的財産権保護を含むと信じている。
私たちのすべての候補製品について、私たちは既存製品と開発中の製品からの競争に直面することが予想されます。SeralutinibはPDGFR,CSF 1 Rとc−kit阻害剤であり,最初にPAH患者に対して対象となった。このグループの患者の競争には、Orenitram(連合治療会社または連合治療会社)、Uptravi(Janssen)、Tyveso(連合治療会社)、Remodlin(連合治療会社)を含むプロスタグランジン/プロスタサイクリン受容体アゴニストが含まれることが予想される。私たちはまた、Revatio(ファイザー)を含むPDE 5阻害剤の経口投与など、クラスIおよびクラスII患者のための製品からのいくつかの競合に直面する可能性がある。AdCirca(共同治療会社);sGC刺激器Adempas(バイエル株式会社)、および口腔時代は、Tracleer(Janssen)、Letairis(gilead Sciences,Inc.)を含む。オスメット(Janssen)です私たちは承認されれば、セラルーチニブはすべての3種類の承認された治療と一緒に使用できると信じている。PAHも薬物研究の活発な適応であり、将来的にRalinepag(ファイザーと連合治療会社)、Sotatercept(メルク社)、ロダリトエチル社(Enzyant Treateutics GmbH)とMK-5475(メルク社)からの競争に直面する可能性がある。また,PAH治療のための承認されていないにもかかわらず,Tenax治療会社,Aerovate治療会社,Aerami治療会社/Vectura集団の競争を含むイマチニブ製剤からの競争に直面している可能性がある。
GB 5121およびGB 7208は、PCNSLおよびMSを含む腫瘍および免疫学的適応を治療するためのBTK阻害剤であり、現在、FDAまたはECによって承認された難治性または再発性PCNSLの治療法はない。多発性硬化症において承認された場合、GB 7208は、既存の承認および研究療法からの競争に直面する可能性がある。BTK阻害剤は、PCNSLまたはMSの治療のためにFDAまたはECによって承認されていないが、承認された場合、GB 5121および/またはGB 7208は、現在FDAおよび/またはECによって承認されているBTK阻害剤Imbrovica(エバーブ社/Janssen)、Calquence(アスリーカン)、Brukinsa(百済神州、Ltd.)からの競争に直面する可能性があると考えられる。そして/またはJaypirca(礼来会社)。我々のBTK阻害剤はまた、BTK阻害剤Velexbru(小野製薬株式会社)からの競争に直面する可能性があり、後者はすでに日本、韓国、
台湾では再発あるいは難治性原発中枢神経系リンパ腫の治療に用いられている。我々はまた,evobrutinib,tlebrutinib,fenebrutinib,orelabrutinib,rilzabrutinib,remibrutinibを含むBTK阻害剤からの競合に直面する可能性がある
他にも早期臨床プロジェクトがあるかもしれませんが、承認されれば、私たちの候補製品と競争します。私たちの多くの競争相手は私たちよりもっと多くの財力、技術、そして人的資源を持っている。製薬業界のより多くの合併と買収は、私たちの競争相手により多くの資源を集中させるかもしれない。技術の商業適用性の進展や、これらの分野でより多くの投資可能な資本があるため、競争はさらに激化する可能性がある。著者らの成功はある程度私たちが薬物の組み合わせを確立し、積極的に管理する能力に基づいて、これらの薬物の組み合わせは満足されていない医療需要を満たし、患者の治療において価値を創造することができる。
許可協定
Pulmokine
2017年10月、我々は、Pulmokineが所有または制御する特定の知的財産権(PulmokineおよびGilead Sciencesが共同所有する知的財産権を含む)の世界的独占的許可および再許可を取得し、任意およびすべての疾患または疾患の治療、予防および診断のためのサラダルチニブおよび特定の予備化合物を開発および商業化するために、Pulmokine,Inc.とライセンス契約またはPulmokineプロトコルを締結した。いくつかの条件が満たされた場合、私たちはまたPulmokine協定に従って私たちの権利を再許可する権利がある。私たちは、米国と少なくとも2つのEU諸国で少なくとも1つのライセンス製品を開発し、商業化するために、商業的に合理的な努力を要求されている。
Pulmokine協定の条項によると、私たちはPulmokineに550万ドルの前払いと500万ドルのマイルストーン支払いを支払い、将来の開発と規制マイルストーン支払いは5800万ドル、商業マイルストーン支払いは4500万ドル、販売マイルストーン支払いは1億9千万ドルに達する義務がある。2023年には,第3段階臨床試験でseralutinibの使用を開始した場合,1000万ドルの開発マイルストーン支払いが生じると予想される。ライセンス製品の販売ごとに階層印税を支払い、その中のビット数の1桁から高いビット数を支払う義務があります。さらに、私たちがPulmokineプロトコルの下で許可製品の権利を任意の第三者に再許可または譲渡すること、または私たちのseralutinib運営子会社が制御権を変更することを選択した場合、私たちはそのような取引に関連するすべての収入の特定の割合をPulmokineに支払わなければならない。
当社の特許使用料義務及びPulmokine協定は、第1回商業販売日から10年後、又は当該ライセンス製品をカバーする有効な特許主張又は当該ライセンス製品が当該国の指定規制排他性を有しなくなった場合には、個々のライセンス製品及び国/地域に基づいて満了する。Pulmokineプロトコルは、Pulmokineによって、または他方が治癒されていない重大な違約、他方が特定の破産にある場合、債務を相殺しない場合、または場合によっては不可抗力イベントが発生した場合に完全に終了することができる。もし私たちが任意の許可特許の有効性や実行可能性に疑問を提起し始めたら、Pulmokineはこの合意を終了するかもしれない。潜在的な安全または治療効果の問題が許可された製品に影響を与える場合、私たちは製品ごとにプロトコルを完全にまたは1つずつ終了させることができる。
PulmokineとGIlead Sciencesが共有する知的財産権は,PulmokineとGIlead Sciences間の許可プロトコルやGIleadプロトコルによって制約される.ジリッド協定によると、Pulmokineは、少なくとも1つの許可製品を開発および商業化するために商業的に合理的な努力を使用しなければならず、この義務は、Pulmokineプロトコルに要求される開発努力によって履行され、将来の規制マイルストーン支払いおよび特許権使用料をGilead Sciencesに支払うことができる。ジリッド協定がどんな理由でも終了した後、Pulmokine協定の下での私たちの再許可は引き続き有効になるだろう。前提は、私たちが終了の基礎となる重大な違約をもたらしていないことであり、私たちはジリッド協定条項の制約を受けることに同意する。
Pulmokineプロトコルには,Rensselaer翻訳研究センター(Rensselaer Center for Translational Research,Inc.)やRensselaerが所有し,Pulmokineに独占的許可プロトコルまたはRensselaer許可でPulmokineに許可された肺動脈高圧検出方法に関する特許の再許可も含まれている。Rensselaerの許可によると、Pulmokineは、Rensselaer特許権によって保護された少なくとも1つの許可製品を開発および商業化するために商業的に合理的な努力を使用しなければならず、この義務は、我々の開発努力によって履行されることができる。Pulmokineや私たちがその義務を履行していない場合、またはRensselaer許可が何らかの理由で終了した場合、Pulmokineプロトコルでの私たちの再許可は私たちが終了することを選択するか、またはRensselaer承認と私たちがRensselaer許可条項を受け入れた場合、私たちとRensselaerの間の直接許可に変換されるだろう。
Pulmokineプロトコルが何らかの理由で終了した場合、プロトコルに従って私たちに付与されたすべての権利と許可は、Pulmokineに終了して回復され、いくつかの終了イベントが発生した場合、私たちはPulmokineグローバル終了計画の権利を付与する。
製造業
著者らは現在多数の第三者メーカーに依存して著者らの候補製品を生産し、臨床前と臨床テストに応用している。もし私たちの候補製品が市場の承認を得たら、私たちは第三者契約メーカーに依存して商業製造を行うつもりだ。一般的に、私たちの候補製品を製造するために必要なすべての材料には複数の供給源がある。我々の製造戦略は、資源を内部開発製造施設に移すのではなく、候補製品の研究、開発、商業化に財務資源をより効率的に使用することができるようにしている。私たちの候補製品が開発過程で進展するにつれて、主要サプライヤーやメーカーと長期商業供給協定を締結して、私たちの生産需要を満足させ、確保する予定です。
知的財産権
私たちは、内部開発でも第三者から許可を得ても、特許権を求め、維持し、擁護することを含む、当社の業務に重要なビジネス的意義を有するノウハウ、発明、および改善に努めています。私たちはまた、私たちの独自技術および製品候補に関連するビジネス秘密および技術的ノウハウ、および持続的な革新に依存して、私たちの独自の地位を発展、強化、維持しています。データ独占性,市場独占性,特許期間の延長(あれば)に依存する予定である。私たちのビジネス成功は、私たちの技術、発明、および改善のために特許および他の固有の保護を取得し、維持する能力があるかどうか、私たちのビジネス秘密を秘密にすること、私たちの将来所有可能な任意の特許を含む私たちの固有の権利を擁護し、実行すること、および効果的かつ強制的に実行可能な特許および第三者の他の独自の権利を侵害することなく運営されることにある程度依存するだろう。知的財産権は私たちの競争優位が直面しているすべての潜在的な脅威を解決できないかもしれない。
セラルチニ
2022年12月31日現在、seralutinibについては、Pulmokineが発行した2つの発行された米国特許を独占的に許可しており、この2つの特許は2037年前に満了することはなく、追加の特許期間の延長は含まれていない。未解決の米国特許出願は、発行されれば、2037年前に満了することはなく、追加の特許期間延長は含まれていない。メキシコおよびロシアで発行された特許と、オーストラリア、ブラジル、カナダ、中国、欧州特許条約、インド、日本、韓国、およびニュージーランドにおける未解決の出願とを含む他の司法管轄区におけるいくつかの特許および係属中の出願は、発行された場合、2037年までに満了することはなく、追加的な特許期間の延長は含まれない。これらの特許および特許出願は、使用方法請求項を対象としている。我々はまた、PulmokineおよびGIlead Sciences,Inc.が共同で所有する4つの発行された米国特許を独占的に許可しており、これらの特許は、2034年前に満了することはなく、追加の特許期間延長を含まない。2つの出願されている米国特許は、すでに発行されている場合、追加の特許期間延長を含まず、追加の特許期間延長を含まず、オーストラリア、カナダ、中国、欧州特許条約および日本の発行済み特許、ならびにオーストラリア、中国、欧州特許条約および日本の係属中出願を含む他の司法管轄区域の特許および係属中特許出願を独占的に許可している。これらの特許および特許出願は、セラルチニブ組成物、製剤および使用方法のための請求項1~4のいずれか一項に記載の。私たちはまた未解決の特許出願を持っており、発行されれば、セラルチニブ形式の特許出願である2041年までには満了しないだろう。
GB 5121
GB 5121に関しては、2022年12月31日現在、係属中の米国特許出願、10件の対応する外国特許出願、およびGB 5121化合物、製剤、および使用方法に関する請求項の国際出願を有しており、これらの出願が発行された場合、2041年前に満了することはなく、追加の特許期間延長は含まれていない。
GB 7208
2022年12月31日現在、GB 7208については、GB 7208化合物、製剤、および使用方法の請求項に関連する係属中の米国特許出願および対応する国際特許出願を有しており、発行された場合、2042年までに満了することはなく、追加の特許期間延長は含まれていない。
政府の監督管理
アメリカ連邦、州と地方各級及びその他の国の政府当局は、その他を除いて、製品の研究、開発、テスト、製造、品質管理、承認、ラベル、包装、貯蔵、記録保存、販売促進、広告、流通、マーケティングと輸出入を広く管理している
私たちが開発しているもの。新薬は新薬申請或いはNDAプログラムを通じてFDAの承認を得なければならず、それからアメリカで合法的に発売することができる。
私たちのいくつかの候補製品は組み合わせ製品として規制されており、これはそれらが薬物製品も設備製品も含むことを意味する。単独で発売されれば、各成分は異なる監督管理経路を受け、FDA内部の異なるセンターによって審査される。しかし、組み合わせ製品は、組合せ製品の主要な作用モードの決定に基づいて、その規制に対して主要な管轄権を有するセンターに割り当てられ、これは最も重要な治療作用を提供する単一作用モードである。我々の吸入型候補製品が組合せ製品として規制されている場合、主要な行動パターンは製品の薬物成分に起因し、これはFDAの薬物評価と研究センターが発売前の開発、審査と承認の主要な管轄権を持っていることを意味する。そこで,INDフレームワークでこの製品を調査し,NDA経路で承認を求める予定である。FDAはこの装置に対して個別の医療機器の許可を要求しないと予想しているが、私たちが提出する可能性のある任意のマーケティング申請を検討する過程で、これは変わる可能性がある。
アメリカの薬物開発プロセスは
米国では,FDAは連邦食品,薬物と化粧品法案(FDCA)とその実施条例に基づいて薬品を規制している。規制の承認を得て、その後、適切な連邦、州、地方、外国の法規と条例を遵守する過程には、多くの時間と財政資源が必要だ。製品開発過程,承認過程又は承認後のいずれかの場合,出願人が適用される米国の要求を遵守できない場合には,行政又は司法制裁を受ける可能性がある。これらの制裁には、FDAによる未解決申請の承認拒否、承認撤回、臨床一時停止、警告状、製品リコール、製品差し押さえ、生産または流通の完全または部分的停止、禁止、罰金、政府契約の拒否、原状回復、返還、返還、または民事または刑事罰が含まれる可能性がある。どんな機関や司法法執行行動も私たちに実質的な悪影響を及ぼすかもしれない。
FDAが米国で発売される前に必要とされるプログラムは、一般に以下のような態様を含む
•“良好な実験室規範”或いはGLP、法規及びその他の適用法規に基づいて臨床前実験室テスト、動物研究及び調合研究を完成する
•ヒト臨床試験が開始される前に有効でなければならないINDをFDAに提出する
•各臨床試験が開始される前に、各臨床サイトの独立機関は、委員会または倫理委員会の承認を審査する
•良好な臨床実践またはGCP法規に基づいて十分かつ制御されたヒト臨床試験を行い、その予期される用途に使用されるべき薬物の安全性と有効性を決定する
•食品医薬品局に秘密保持協定を提出し
•適用されれば、FDA諮問委員会の審査が満足的に完了する
•GCPの遵守状況を評価するために、施設、方法、および薬物の特性、強度、品質および純度を維持するのに十分な制御を保証するために、現行のGMPまたはcGMP要件に適合しているかどうかを評価するために、医薬品を製造するための1つまたは複数の製造施設の検査を良好に完了させること;
•FDAは、米国で使用される特定の適応の製品の商業マーケティングを可能にするためにNDAを審査および承認する。
開発する候補薬が決定されると,臨床前試験段階に入る。臨床前試験は製品の化学、毒性と調合に対する実験室評価、及び動物研究を含む。INDスポンサーは,臨床前試験の結果を生産情報や分析データとともにINDの一部としてFDAに提出しなければならない。INDはFDAが研究薬物製品の使用をヒトに許可する要求である。スポンサーはまた、臨床試験が適切であれば、臨床試験の目標を詳細に説明するためのパラメータ、安全性を監視するためのパラメータ、および評価される有効性基準を含むプロトコルを含むであろう
治療効果を評価する。IND提出後も,いくつかの臨床前試験が継続される可能性がある。INDはFDAが30日以内に臨床試験を保留しない限り、FDAが受領してから30日後に自動的に発効する。この場合,INDスポンサーやFDAは臨床試験開始前に未解決の問題を解決しなければならない。FDAは、進行中または提案されている臨床試験またはFDAの特定の要求に適合していない安全懸念から、FDAがスポンサーに一時停止を通知する前に、試験が開始または継続されない可能性があるため、臨床試験の前または期間の任意の時点で臨床休止を実施することもできる。したがって,INDの提出はFDA認可による臨床試験の開始につながる可能性があり,そうでない可能性もある。
GCP規定によると、すべての臨床試験は1人以上の合格研究者の監督の下で行われなければならず、すべての研究対象に書面で任意の臨床試験に参加することを要求するインフォームドコンセントを含む。その他の事項以外にも,試験目標,投与手順,被験者の選択と排除基準,評価する安全性と有効性基準を詳細に説明するプロトコルに基づいて行わなければならない。各プロトコルは、INDの一部および任意の後続のプロトコル修正案としてFDAに提出されなければならない。INDは活発であるが、前回の進展報告以来行われた臨床試験と非臨床研究結果の進展報告をまとめ、他の情報以外に、少なくとも毎年1回の書面IND安全報告をFDAに提出しなければならず、その中でFDAと調査者に書面IND安全報告を提出しなければならず、深刻かつ意外な疑わしい不良事件を理解し、他の研究結果は同じ或いは類似の薬物に暴露することは人類に対して重大なリスクがあることを表明し、動物或いは体外試験結果は人類に対して重大なリスクがあることを表明し、及び方案或いは研究者のマニュアルに列挙された深刻な疑わしい不良反応の発生率と比較して、任意の臨床重要な疑わしい副作用の発生率は増加する
また、臨床試験に参加する各機関の独立したIRBは、当該機関の臨床試験が開始される前に各案を審査·承認しなければならず、試験に関する情報および各試験対象またはその法律代表に提供されなければならない同意書を承認し、研究が完了するまで監視しなければならず、そうでなければIRBの規定を遵守しなければならない。FDAあるいはスポンサーは研究対象や患者が受け入れられない健康リスクに直面していることを発見することを含む、様々な理由で臨床試験を一時停止することができる。同様に、1つの臨床試験が委員会の要求に従って行われない場合、または薬剤が患者に予期せぬ深刻な傷害を与えた場合、IRBは、その所在機関の臨床試験の承認を一時停止または終了することができる。さらに、いくつかの臨床試験は、スポンサーによって組織された独立した合格専門家グループによって監督され、このグループは、データ安全監視委員会または委員会と呼ばれる。その規約によれば、当該グループは、試験のあるデータへのアクセスに基づいて、試験が指定されたチェックポイントで行うことができるか否かを決定することができる。また,行っている臨床試験と完成した試験結果を公的登録機関に報告することが求められている。FDA規制製品のいくつかの臨床試験のスポンサーは、www.Clinicaltrials.gov上で公開して得ることができる特定の臨床試験情報を登録して開示しなければならない。そして,登録の一部として,製品,患者群,調査段階,試験地点と調査者および臨床試験の他に関する情報が公開されている。スポンサーも完成後にその臨床試験の結果を開示する義務がある。これらの試験結果の開示は,研究中の新製品や新適応が承認された後に延期することができる。
人体臨床試験は通常3つの連続段階に分けて行われ、これらの段階は重複或いは合併する可能性がある
•ステップ1:この候補製品は、最初に健康なヒトボランティアに導入され、その安全性、用量耐性、吸収、代謝、分布、および排泄を試験し、可能であれば、その有効性の早期兆候を得る。いくつかの深刻または生命に危害を及ぼす疾患(例えば癌)を治療する製品の中で、特に製品がその固有の毒性のために道徳的に健康ボランティアに服用できない可能性がある場合、最初の人体試験は通常患者で行われる。
•第二段階:この段階は限られた患者集団で臨床試験を行い、可能な副作用と安全リスクを決定し、特定の目標疾患に対するこの製品の治療効果を初歩的に評価し、用量耐性と適切な用量を決定することに関連する。
•第3段階:臨床試験は拡大した患者群の中で更に投与量、臨床治療効果と安全性を評価するためであり、通常は地理的に分散した臨床研究地点で行われる。これらの臨床試験は候補製品の全体的なリスク-収益比を確定し、製品ラベルに十分な基礎を提供することを目的としている。
承認後試験は,第4段階研究と呼ばれることがあり,最初の上場承認後に行われる可能性がある。これらの試験は,治療適応が予想される患者の治療から追加的な経験を得るために用いられている。場合によっては,FDAはNDAを承認する条件として4期臨床試験を強制的に実行することができる。
新薬開発期間中、スポンサーはいつかFDAと会う機会がある。これらの要件は、INDを提出する前、第2段階の終了時、および秘密協定の提出前にある可能性がある。他の時間に会議を開催することを要求することができます。これらの会議は,スポンサーにこれまで収集してきたデータに関する情報を共有する機会を提供し,FDAにアドバイスを提供し,スポンサーとFDAに次の段階の開発について合意することができる。スポンサーは通常、第二段階試験終了時の会議を用いて第二段階臨床結果を検討し、新薬承認を支持すると考えられる重要な第三段階臨床試験計画を提出する。
臨床試験と同時に、会社は通常追加の動物研究を完成し、薬物化学と物理特性に関する追加情報を開発し、cGMPの要求に基づいて最終的に商業量産製品のプロセスを決定しなければならない。製造過程は一貫して高品質の候補製品ロットを生産できる必要があり、また、メーカーは最終薬物の身分、強度、品質、純度をテストする方法を開発しなければならない。また,適切な包装を選択·テストし,候補製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
アメリカの組み合わせ製品に対する規制
ある製品は薬品成分と設備成分のような成分から構成される可能性があり、これらの成分は通常異なるタイプの監督管理機関によって監督され、通常FDAの異なる中心によって監督管理される。これらの製品は組合せ製品と呼ばれている。具体的には、FDAが発表した法規によると、組合せ製品は以下のようになる可能性がある
•物理的、化学的、または他の方法で組み合わせまたは混合され、単一の実体として製造される2つ以上の規制された成分からなる製品
•2つ以上の別個の製品、一緒に包装されているか、または1つの単位として、医薬品および器具製品、器具および生物製品、または生物学的および医薬品製品を含む;
•個々に包装された医薬品、器具または生物製品は、その研究計画または提案されたラベルに従って、承認された個別に指定された医薬品、器具または生物物品と共にのみ使用される予定であり、両方が予期される用途、適応または効果を達成するために必要であり、提案された製品が承認された後、承認された製品のラベルは、例えば、予期される用途、剤形、濃度、投与経路の変化または用量の重大な変化を反映するために変更する必要があるであろう
•その提案されたラベルに基づいて個別に包装された任意の研究用医薬、装置または生物製品は、別の個別に指定された研究用薬剤、装置または生物製品と共にしか使用できないが、これら2つの薬剤、装置または生物製品は、所望の使用、適応、または効果を達成する必要がある。
FDCA及びその実施条例によると、FDAは主要な管轄権を持つ中心或いは牽引センターを割り当て、組合せ製品の審査を行う。鉛センターの指定は通常,複数のFDAコンポーネントから組合せ製品の承認を得る必要はないが,鉛センターがFDAの他のコンポーネントと協議することは排除されない。どのセンターが主導センターであるかの決定は,組合せ製品の“主な役割モデル”に基づいている.したがって、薬物-設備組合せ製品の主要な作用モードがこの薬物製品に起因する場合、この薬物製品の発売前の審査を担当するFDAセンターはこの組み合わせ製品に対して主要な管轄権を持つことになる。FDAはまた、共同製品をめぐる問題を解決し、規制審査過程により多くの確実性を提供するために、共同製品事務室を設立した。その事務室は機関審査員と産業組合製品問題の焦点だ。また、組合せ製品の監督管理を明確にし、主要な管轄権を有するFDAセンターの割り当てを担当し、管轄権が明確でない場合、または論争がある場合に組合せ製品を審査するためのガイドラインおよび法規の制定を担当する。
薬物の主要な作用モデルを持つ組合せ製品は一般にFDCA下の薬品承認手続きに基づいて審査と承認を行う。しかしながら、このような製品のNDA申請を審査する際には、医薬品センターのFDA審査員は、製品を組み合わせたデバイスコンポーネントが安全、有効性、耐久性、および性能面の適用要件を満たすことを確実にするために、デバイスセンターの審査員に問い合わせることができる。また,FDAの規定によると,組合せ製品は医療機器に適した品質体系や法規を含む薬品や機器に適したcGMP要求を遵守しなければならない。
守秘契約承認の流れ
製品開発、臨床前と他の非臨床研究と臨床試験の結果、及び製造技術、薬物化学に対する分析テスト、提案のラベルとその他の関連情報の記述は、NDAの一部としてFDAに提出し、この製品の発売許可を要求する。秘密協定の提出には相当な使用料が必要であり、いくつかの限られた場合には、そのような費用を免除することができる。提出されると、FDAは、製品がその予期される用途に対して安全かつ有効であるかどうかを決定し、その製造がcGMPに適合するかどうかを決定して、製品の特性、強度、品質、および純度を保証および維持するためにNDAを検討する。現在発効している処方薬使用料法案(PDUFA)ガイドラインによると,FDAは標準NDA提出日から10カ月以内に新たな分子実体を審査させて提出したNDAに行動させることを目標としている。この審査には通常12カ月の時間が必要であり,NDAがFDAに提出された日から計算すると,FDAは申請提出後約2カ月で“届出”決定を下すためである。FDAは、届出を受ける前に、提出後の最初の60日以内にすべてのNDAを予備審査して、それらが十分に完全であるかどうかを決定し、FDAの実質的な審査を可能にすることは、NDA届出を受け入れるのではなく、より多くの情報を提供することを要求することができる。この場合、秘密協定と追加情報を再提出しなければならない。再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。
FDAは新薬の申請を諮問委員会に提出するかもしれない。諮問委員会は,臨床医や他の科学専門家を含む独立した専門家からなるグループであり,申請を審査,評価し,申請を承認すべきかどうか,どのような条件でアドバイスを提供すべきかを担当する。FDAは諮問委員会の提案に制限されていないが、それは決定を下す時にこれらの提案を慎重に考慮するだろう。秘密協定を承認する前に、FDAは製品を生産する1つまたは複数の施設を検査するだろう。FDAは、製造プロセスおよび施設がcGMP要件に適合していることを決定し、要求された仕様の下で製品が一貫して生産されることを保証するのに十分でない限り、申請を承認しないであろう。さらに、NDAを承認する前に、FDAは、GCP要件に適合することを確実にするために、1つまたは複数の臨床試験地点を検査する可能性がある。
FDAは、NDAを評価した後、承認状または完全な返信を発行する。この薬剤の商業マーケティングを承認し、特定の適応の処方情報を提供する。完全な返信は、申請の審査期間が終了し、申請が現在の形で承認されないことを示している。完全な返信は、一般に、FDAによって決定されたNDAにおける特定の欠陥を記述し、追加の臨床試験、または臨床試験、非臨床研究、または生産に関連する他の重要で時間のかかる要件のような追加の臨床データを必要とする可能性がある。完全な返事が出された場合、スポンサーは、機密協定を再提出するか、手紙で発見されたすべての不足点を解決するか、または申請を撤回しなければならない。このようなデータや情報を提出しても,FDAはNDAが承認基準を満たしていないと認定する可能性がある。
1つの製品が規制部門の承認を得た場合、この承認は、特定の疾患および用量に明らかに限定される可能性があり、または使用の適応が制限される可能性があり、これは、製品の商業的価値を制限する可能性がある。さらに、FDAは、NDA承認後に薬物の安全性および有効性をさらに評価することを目的とした臨床試験を含む第4段階試験をスポンサーに要求することができ、商業化された承認製品の安全性を監視するための試験および監視計画を要求する可能性がある。FDAはまた、薬剤の安全な使用を保証するために、リスク評価および緩和戦略を要求すること、またはREMSを含む承認時に他の条件を設定することができる。FDAがREMSが必要であると結論した場合,NDAのスポンサーは提案したREMSを提出しなければならない。必要であれば、FDAは、承認されていないREMSなしにNDAを承認しないだろう。REMSは、制限された分配方法、患者登録、および他のリスク最小化ツールのような薬物ガイドライン、医師のコミュニケーション計画、または安全な使用を確保する要素を含むことができる。承認またはマーケティングに関するこれらの制限は、製品の商業普及、流通、処方、または配布を制限する可能性がある。規制要件を守らない場合や最初のマーケティング後に問題が発生した場合、マーケティング承認は撤回される可能性がある。
そのほか、“小児科研究公平法”(PREAと略称する)はスポンサーに大多数の薬物、新しい有効成分、新しい適応、新しい剤形、新しい投与方案或いは新しい投与経路に対して小児科臨床試験を行うことを要求する。PREAによれば、元のNDAおよびサプリメントは、スポンサーが延期または免除を受けていない限り、小児科評価を含まなければならない。要求された評価は、すべての関連する小児科亜集団において主張される製品の適応の安全性および有効性を評価し、各安全で有効な小児科亜群に対する製品の用量および投与をサポートしなければならない。スポンサーまたはFDAは、小児科亜群の一部または全部の小児科臨床試験の延期を要求することができる。延期は、小児科臨床試験が完了する前に、成人で使用を許可する準備ができていることを発見するか、または小児科臨床試験が開始される前に追加の安全性または有効性データを収集する必要があることを含むいくつかの理由があるかもしれない。FDAは、必要な評価を提出できなかった、延期された最新の状況を維持し、または小児科処方承認要求を提出できなかった任意のスポンサーに、規定に適合しない手紙を送信しなければならない。
孤児薬名
孤児医薬品法によれば、FDAは、米国で20万人以下に影響を与えるか、またはそれが影響を及ぼす場合、まれな疾患または状態を治療するための薬剤を孤児の称号を与えることができる
米国に200,000人を超える場合、米国でこのような疾患や疾患を治療するための医薬製品の開発および提供のコストを製品の販売から回収できる合理的な期待はない。秘密保護協定を提出する前に、孤児としての指定を要請しなければならない。FDAが孤児の称号を付与した後、FDAは、治療剤の識別およびその潜在的な孤児の使用を開示する。指定孤児は、規制審査および承認過程においていかなる利点も伝達されず、規制審査および承認過程の継続時間を短縮することもない。
孤児として指定された製品がその後、このような指定された疾患または条件を有するFDAの最初の承認を得た場合、この製品は、孤児製品に対して排他的である権利があり、これは、限定された場合、例えば、孤児に対して排他的な製品に対する臨床的優位性を示すか、または十分な数の製品を生産できない限り、FDAが同じ疾患または条件のための任意の他の薬剤の発売の申請を承認しないことを意味する。この薬物の指定は,臨床試験コスト,税収割引,ユーザ費用減免のために贈与資金の機会を提供するなど,一方が財政的インセンティブを得る権利がある。しかし,競争相手は孤児製品に対して排他的な適応を持つことで異なる製品の承認を得るか,あるいは同一製品に対して孤児製品に対して排他的な異なる適応を有することが承認される可能性がある。
また,指定された孤児製品がマーケティング承認を得ており,その適応範囲が指定された範囲よりも広い場合には,孤児排他性を得る資格がない可能性がある。さらに、FDAが後に指定された要求に重大な欠陥があると判断した場合、または上述したように、第2の出願人が、その製品が孤児の排他的な承認製品よりも臨床的に優れていることを証明する場合、または製品を承認する製造業者が、まれな疾患または疾患患者の需要を満たすのに十分な数の製品を保証できない場合、米国における孤児薬の独占営業権を失う可能性がある。
開発と審査計画を加速する
スポンサーは、FDAによる特定基準に適合した新薬や生物製品の審査·承認を加速させるための計画に基づいて、その候補製品の承認を求めることができる。例えば、FDAは、特定の基準に適合する新薬製品の審査プロセスを加速または促進することを目的とした迅速チャネル指定計画を有する。具体的には、候補製品が深刻または生命に危険な疾患または状態を治療することを意図し、その疾患または状態が満たされていない医療需要を満たす潜在力を示す場合、迅速なチャネル指定を得る資格がある。高速チャネルは,候補製品と研究中の特定の適応に適した組合せを指定する.高速チャネル候補製品のスポンサーは,開発過程で適用されるFDA審査チームとより頻繁なインタラクションを行う機会がある。高速チャネル製品については、FDAは、完全な出願を提出する前にNDAを審査する部分をスクロールすることも考慮することができ、スポンサーがNDA部分を提出するスケジュールを提供した場合、FDAはNDAの部分を受け入れることに同意し、スケジュールが許容可能であると判断し、スポンサーはNDAの第1の部分を提出する際に任意の必要な使用料を支払うことができる。
重篤または生命に危険な疾患や状況を治療しようとする候補品も,その開発や審査を加速するための画期的な療法指定を受ける資格がある可能性がある。予備臨床証拠が、候補製品が単独で、または1つまたは複数の他の薬物または生物学的製品と組み合わせて使用されることを示す場合、候補製品は、1つまたは複数の臨床的重要終点において、例えば、臨床開発早期に観察される実質的な治療効果を示す可能性がある場合、候補製品は、画期的な治療指定を得ることができる。この指定は、高速チャネル計画のすべての機能と、第1段階で開始されたより密集したFDA相互作用および指導と、高度管理者の参加を含む候補製品開発および審査を加速する組織約束とを含む。
迅速なチャネル指定または画期的な指定を有する候補製品を含むFDA承認の候補製品を提出する任意のセキュリティプロトコルは、優先的な審査および承認の加速など、FDAの開発および審査を加速するための他のタイプの計画に参加する資格がある可能性もある。候補製品が、深刻なまたは生命を脅かす疾患または状態の治療を意図しており、承認された場合、そのような疾患または状態の既存の代替品と比較して、安全性または有効性の点で有意な改善を提供する場合、NDAは、優先審査を受ける資格がある。FDAは、審査を促進するために、優先審査として指定された申請を評価するために追加のリソースを使用しようと試みるだろう。FDAは,提出日後6カ月以内に優先審査指定を有する出願を審査するように努力しているが,その現在のPDUFA審査目標に基づいて,新分子実体NDAを審査する期間は10カ月である
しかも、候補製品は加速された承認を受ける資格があるかもしれない。深刻または生命に危険な疾患または状態を治療するための医薬製品であって、臨床的利益を合理的に予測する可能性のある代替終点、または不可逆的な発病率または死亡率よりも早く測定可能な臨床終点を決定し、不可逆的な発病率または死亡率または他の臨床的利益への影響を合理的に予測する可能性が高く、病状の重症度、希少性または流行率、および代替治療の利用可能性または不足を考慮して、加速承認を得る資格がある。承認の条件としてFDAは通常薬物のスポンサーに受け入れを要求します
十分かつ良好な制御を行う検証性臨床試験の承認を加速する。また、FDAは現在、承認を加速させる条件として宣伝材料を事前承認することを求めており、製品商業発売の時期に悪影響を及ぼす可能性がある。FDAは、例えば、検証試験が製品の予期される臨床的利益を検証できなかった場合、またはスポンサーがそのような試験をタイムリーに実施できなかった場合、承認を加速させることに基づいて、承認を加速させる薬剤または適応の承認を撤回することができる。
迅速チャネル指定、画期的な治療指定、優先審査、および承認の加速は、承認の基準を変更することはありませんが、開発または承認プロセスを加速させる可能性があります。1人の候補製品が1つまたは複数の計画に参加する資格があっても、FDAは、その候補製品がもはや資格条件に適合していないことを後で決定することができ、またはFDAの審査または承認を決定する期間が短縮されないことができる。
承認後に要求する
承認された後、規制基準に適合した製品や製品の発売後に問題が発生したままでなければ、FDAは承認を撤回する可能性がある。その後、ある製品が以前知られていなかった問題はこの製品が制限され、甚だしきに至っては完全に市場から撤退する可能性があることを発見した。承認後、承認された製品のいくつかのタイプの変更、例えば、新しい適応の増加、いくつかの製造変更、および追加のラベル宣言は、FDAのさらなる審査および承認を受けるであろう。薬品メーカーと生産と流通許可薬品に参与する他の実体はFDAとある州機関にその機関を登録し、FDAとある州機関の定期的な抜き打ち検査を受けて、cGMP法規と他の法律法規を遵守することを保証しなければならない。また、FDAはNDAを承認する条件として承認後の要求を課す可能性がある。例えば、FDAは、第4段階の臨床試験を含む上場後試験を要求し、製品の商業化後の安全性と有効性をさらに評価し、監視するために監視を行う可能性がある。
FDAの承認に基づいて生産または流通される任意の医薬品は、記録保存要件、薬物副作用の報告、FDAへの最新の安全性および有効性情報の提供、医薬品サンプリングおよび流通要件、特定の電子記録および署名要件の遵守、およびFDAの宣伝および広告要件を含むFDAの普遍的かつ持続的な規制を受けるであろう。FDAは、市販製品のラベル、広告、販売促進および他のタイプの情報を厳格に規制し、製品承認されたラベルに記載されていない製品(“非ラベル使用”と呼ばれる)、業界スポンサーの科学的および教育活動、およびインターネットに関連する販売促進活動のための製品の普及を禁止し、製品の承認されたラベルに記載されていない(“非ラベル使用”と呼ばれる)、業界スポンサーの科学的および教育活動、およびインターネットに関する販売促進活動のような医薬品製造業者に要求および制限を加える。
以前未知の問題が発見されたり、適用された規制要求を遵守できなかったりすると、製品の販売を制限したり、その製品を市場から引き揚げたりし、民事または刑事制裁を受ける可能性がある。製品開発プロセス、承認プロセスまたは承認後の任意の時間に適用される米国の要求を遵守できなかった場合、出願人または製造業者は、行政または司法民事または刑事制裁および負の宣伝を受ける可能性がある。FDAの制裁には、承認保留申請の拒否、承認撤回、承認後の臨床試験の臨床棚上げ、警告または命名されていない手紙、製品リコール、製品差し押さえ、生産または流通の完全または部分的な一時停止、禁止、罰金、政府契約の拒否、広告の強制修正、または医師とのコミュニケーション、取り締まり、元の回復、利益の返還または民事または刑事罰が含まれる可能性がある。
マーケティング排他性
FDCAにおける市場排他性条項は、いくつかのマーケティング申請の提出または承認を延期する可能性がある。FDCAは新しい化学実体秘密協定の承認を得た最初の申請者に5年間のアメリカ国内の非特許データ排他期を提供した。FDAが以前に同じ活性部分を含む他の新薬を承認していなければ,薬物は新しい化学実体であり,活性部分は薬物物質の作用を担う分子やイオンである。排他的期間内に、FDAは、第505条(B)(2)条または第505条(B)(2)条に従って提出された別の薬剤が、同じ活性部分に基づいて提出された簡略化された新薬出願またはANDAまたはNDAを審査することを承認しないか、または受け入れない可能性があり、その薬剤が元の革新薬と同じ適応であるか、または別の適応のためのものであるかにかかわらず、出願人が合法的な権利を参照していない場合、または承認に必要なすべてのデータを参照することができない。しかしながら、出願がイノベーターNDA所有者がFDAに記載された特許のうちの1つを含む特許が無効または未侵害証明である場合、4年後に出願することができる。
出願人が行ったり賛助したりする新しい臨床研究(バイオアベイラビリティ研究を除く)がFDAによって承認申請に必要であると考えられている場合、FDCAはまた、NDAに3年間の市場排他性、または新しい適応、用量、または既存のNDAの補充を提供することができる
既存の薬の強度ですこの3年間の排他性は、この薬剤が新しい臨床研究に基づいて承認された修正のみを含み、FDAがANDAまたは505(B)(2)NDAを許可することは禁止されておらず、元の適応または使用条件の有効成分を含む薬剤のために使用される。5年と3年の排他性は完全な秘密協定の提出や承認を延期したり承認したりしないだろう。しかしながら、完全なセキュリティプロトコルを提出する出願人は、安全かつ有効であることを証明するために、必要なすべての臨床前研究および十分かつ良好に制御された臨床試験を参照する権利を行うか、または得ることを要求されるであろう。
小児科専門権は米国で利用可能な別のタイプのマーケティング専門権である。スポンサーがFDAの書面要求に応じて児童に臨床試験を行う場合、小児科排他性規定は別の排他期に加えて6ケ月のマーケティング排他性を追加する。書面出願の発表はスポンサーに述べた臨床試験を要求しない。また、上述したように、孤児薬の独占性は7年間の市場独占期間を提供することができるが、場合によっては除外される。
アメリカの保険と精算
私たちが規制承認を求める可能性のある任意の候補製品のカバー範囲と精算状態には、重大な不確実性がある。米国での販売は、第三者支払者が十分な保険と十分な補償を提供するか否かにある程度依存し、連邦医療保険、医療補助、TRICARE、退役軍人管理局などの政府医療計画、管理型医療組織、個人医療保険会社を含む。私たちあるいは私たちの顧客が私たちの候補製品のために精算を求める価格は第三者支払人の疑問、値下げ、あるいは拒否されるかもしれません。
第三者支払い者が製品に保険を提供するかどうかを決定するプロセスは、一般に、支払者が製品に支払う支払率を設定するプロセスとは分離されている。第三者支払者が製品に保険を提供することを決定することは、十分な販売率が利用可能になるという意味ではない。また、米国では、保険や精算に対して、支払者間に統一された政策はない。第三者支払者が自己の保険·精算政策を設定する際には,通常連邦医療保険引受政策や支払制限に依存するが,独自の方法や承認の流れもある。そのため、製品の保証範囲と精算範囲は支払人によって異なる。カバー範囲や十分な精算がなければ、あるいは限られたレベルだけでは、私たちが開発したどの製品も商業化に成功せず、満足できる財務的見返りを得ることができるかもしれない。
第三者決済者は、価格、医療製品およびサービスの医療必要性および費用効果、ならびにそれらの安全性および有効性を検討することにますます挑戦している。上場が承認される可能性のある製品の保険や補償を得るためには、いかなる製品の医療の必要性や費用効果を証明するために高価な研究を行う必要があるかもしれないが、これは規制の承認を得るのにかかる費用以外の追加費用となる。第三者支払者は、私たちの候補製品が他の利用可能な療法と比較して医療的に必要であるか、または費用対効果があると思わないかもしれないし、有利な保証に必要なリターン率を確保することは、十分なコスト利益率を生成できないかもしれないし、または薬物開発への私たちの投資の適切なリターンを達成するために十分な価格レベルを維持することができないかもしれない。
アメリカの医療改革
米国では、医療保健システムに関するいくつかの立法と規制の変化、提案された変化はすでに存在し、これらの変化は候補製品の上場承認を阻止または延期し、承認後の活動を制限または規範化し、候補製品の利益の販売に影響を与える可能性がある。
米国の政策立案者や支払者の中で,医療システムの変革を推進することに大きな興味があり,医療コストの抑制,質の向上および/または参入拡大を既定の目標としている。米国では、製薬業はこれらの努力の重点であり、重大な立法計画の大きな影響を受けてきた。2010年3月、“患者保護と平価医療法案”(Patient Protection and Affordable Care Act、略称ACA)が可決され、それは政府と民間保険会社が医療保健に資金を提供する方式を大きく変え、アメリカの製薬業に重大な影響を与えた。他の事項を除いて、ACA:(1)メーカーが医療補助薬品リベート計画の下で不足している最低医療補助リベートを増加し、リベート計画を医療補助管理の保健組織に登録されている個人に拡大する;(2)医療補助薬品リベート計画の下でメーカーのリベートに基づいて吸入、点滴、注入、注入または注射されたいくつかの薬物と生物製品のリベートを計算する新しい方法を作成した;(3)ある特定のブランドの処方薬と生物製剤の任意の実体を生産または輸入し、ある政府の医療計画における市場シェアに基づいて、相殺できない年間費用を確立した。(4)340 B薬品定価計画に新たな実体を追加することにより、340 B薬品定価計画の下で低い価格設定の可獲得性を拡大した;(5)医療補助計画の資格基準を拡大した;(6)患者を中心とした新しい結果研究所を作成し、監督、優先順位を決定し、臨床効果比較研究を行い、これらの研究に資金を提供した
研究;(7)新しい連邦医療保険D部分カバーギャップ割引計画を作成し、この計画によると、メーカーはそのカバー間隔期間内に条件を満たす受益者に適用ブランド薬物交渉価格の50%(2019年1月1日から70%に増加)の販売時点割引を提供することに同意しなければならず、メーカーの外来薬物として連邦医療保険D部分カバーの条件に組み入れなければならない;(8)患者を中心とした新しい結果研究所を設立し、優先事項を監督、確定し、臨床有効性比較研究を行い、このような研究に資金を提供する。(9)連邦医療保険·医療補助サービスセンター(CMS)に医療保険·医療補助革新センターを設立し、処方薬を含む可能性がある連邦医療保険·医療補助支出を低減するために、革新的な支払い·サービス交付モデルをテストする。
ACAのいくつかの側面は公布以来、司法と政治的な挑戦を受けてきた。2021年6月17日、米国最高裁はいくつかの州がACAに対して提出した最新の司法挑戦を却下したが、ACAの合憲性を具体的に裁くことはなかった。最高裁が裁決を下す前に,総裁·バイデンはACA市場による医療保険の取得を目的とした2021年2月15日から2021年8月15日までの特殊保険期間を開始する行政命令を発表した。行政命令はまた、特定の政府機関に、医療補助モデルプロジェクトおよび免除計画の再審査、および医療補助またはACAによる医療保険カバー範囲の獲得による不必要な障害をもたらす政策を含む医療補助モデルプロジェクトおよび免除計画の再審査および見直し、医療補助またはACAによる医療保険のカバー範囲を制限する既存の政策およびルールを再検討するように指示する。
ACAが公布されて以来、他の立法改正も提案され、採択された。2011年8月2日、2011年予算統制法は、提供者への医療保険総額の減少を招くことを含む法律に署名し、2013年4月1日に施行され、その後の立法改正により、国会が追加的な行動を取らない限り、2020年5月1日から2022年3月31日まで一時的に一時停止する2032年まで有効となる。2013年1月2日、2012年に米国納税者救済法が法律に署名し、病院を含むいくつかの医療サービス提供者に支払う医療保険を減らし、政府が提供者に多額の支払いを取り戻す訴訟時効を3年から5年に延長した。
また、政府は最近、メーカーが製品価格を販売する方式の審査を強化し、国会で数回の調査を行い、製品定価の透明性の向上、価格設定とメーカー患者計画との関係の審査、政府の薬品に対する計画精算方法の改革を目的とした連邦と州立法を提出し、公布した。最近は2022年8月16日に“2022年インフレ削減法案”(IRA)が署名されて法律となっている。他の事項を除いて、アイルランド共和軍はある薬品のメーカーに連邦医療保険との価格交渉(2026年から)を要求し、価格は交渉できるが上限があり、連邦医療保険B部分と連邦医療保険D部分に基づいてリベートを実施し、インフレを超える価格上昇を処罰し(2023年に初めて満了)、D部分のカバーギャップ割引計画の代わりに新しい割引計画を用いる(2025年から)。アイルランド共和軍は衛生·公衆サービス部長官が最初の数年に規制ではなく指導によってその多くの規定を実施することを許可した。このような理由と他の理由で、アイルランド共和軍がどのように実施されるのかは不明だ。州レベルでは、立法機関は、価格または患者の精算制限、割引、特定の製品参入およびマーケティングコスト開示の制限、および透明性措置を含む薬品の価格設定を制御するための法規を立法し、実施してきており、場合によっては、他の国からの輸入および大量購入を奨励することを目的としている。
将来的にとりうる医療改革措置は、より厳しいカバー基準、新しい支払い方法をもたらす可能性があり、私たちが受け取った任意の承認された製品の価格に追加の下振れ圧力をかけることが予想される。連邦医療保険や他の政府が計画している精算のいかなる減少も、個人支払者の支払いの同様の減少を招く可能性がある。
アメリカの医療詐欺と法律の乱用とコンプライアンス要求
連邦、州、外国の医療保健法律法規は生物製薬業界の商業行為を制限する。このような法律には反リベートと虚偽請求の法律と透明性の法律が含まれている。
連邦反リベート法規は、個人または実体が知られている場合、直接または間接的に、現金または実物の形態で直接または間接的に現金または実物の形態で提供、支払い、請求または報酬を受けて、購入、レンタル、注文または手配または推薦購入、レンタルまたは推奨購入、レンタルまたは注文を誘導または見返りに、Medicare、Medicaid、または他の連邦医療計画に従って精算可能な任意の物品またはサービスを注文することを禁止する。個人または実体は、この法規またはその法規に違反する具体的な意図を実際に知る必要がなく、違反行為を実施することができる。
“民事虚偽請求法案”を含む連邦民事および刑事虚偽クレーム法律は、他の事項に加えて、任意の個人または実体が虚偽クレームの提出を意図的に連邦政府に提出するか、または虚偽クレームの提出を故意に作成、使用するか、または虚偽または詐欺的クレームの作成または使用に重要な意味を有する虚偽記録または陳述を作成または使用することを禁止する。また、政府は、物品やサービスを含むクレームを主張することができる
連邦の“反リベート法規”違反によるクレームは、“民事虚偽請求法”と“民事罰金条例”の規定に基づいて、虚偽または詐欺的クレームを構成する。
1996年の連邦健康保険携帯性·責任法、またはHIPAAは、任意の医療福祉計画を故意かつ故意に詐欺する計画を禁止した追加の連邦民事·刑事法規を制定した。連邦反リベート法規と同様に、個人または実体は、この法規やこの法規に違反する具体的な意図を実際に知る必要がなく、違反を実施することができる。
連邦医師支払い陽光法案は、連邦医療保険、医療補助または児童健康保険計画に従って支払うことができるいくつかの薬品、器具、生物製品および医療用品の製造業者が毎年CMSに医師(医師、歯医者、検眼師、足科医師および脊椎マッサージ師を含む)、ある非医師提供者(医師アシスタントおよび看護師従業員を含む)、教育病院、適用される製造業者および適用される共同購入組織は、医師(法規に基づいて定義されている)およびその直系親族が保有する所有権および投資権益に関する情報をCMSに毎年報告することを要求する。
同様の州、地方、および外国の法律および法規は、州反リベートおよび虚偽請求法のようなバイオ製薬業界の商業的慣行を制限する可能性もあり、これらに限定されないが、研究、流通、販売およびマーケティング配置、および非政府第三者支払者(私営保険会社を含む)または患者自身によって精算された医療項目またはサービスに関するクレームを含むが、これらに限定されない。州法律は、製薬会社に製薬業界の自発的コンプライアンスガイドラインおよび連邦政府が公布した関連コンプライアンスガイドラインを遵守すること、または他の方法で医療保健提供者および他の潜在的な転売源への支払いを制限することを要求する。医薬品製造業者に、価格設定およびマーケティング情報に関連する報告書の提出を要求するか、または医師、他の医療提供者および実体に提供されるプレゼントおよび他の報酬および価値項目を追跡することを要求する州法律および法規、ならびに医薬品販売代表の登録を要求する州および地方法律。
外国の法律·法規の範囲は上記の規定よりも広い可能性があり、支払人にかかわらず適用可能である。これらの法律と法規は大きく異なる可能性があり、コンプライアンス作業をさらに複雑化させる可能性がある。例えば、EUでは、多くのEU加盟国が具体的な反贈与法規を採択し、医療製品の商業行為、特に医療専門家や組織に対する商業行為をさらに制限している。また、最近では、医療専門家や実体への支払いや価値移転の規制を強化する傾向があり、多くのEU加盟国は国家サンシャイン法案を可決し、製薬会社に対して米国の要求(通常は年に1回)のような報告や透明性要件を実施している。いくつかの国はまた、商業コンプライアンス計画を実施することを要求するか、またはマーケティング支出および価格設定情報の開示を要求する。
適用される医療法律と法規を遵守する努力は巨額のコストに関連する可能性があることを確保する。医療保険法違反は、重大な民事、刑事および行政処罰、損害賠償、罰金、返還、個人監禁、Medicare、Medicaidおよび他の米国および外国の医療保健計画への参加から除外される可能性があり、誠実な監督と報告義務、契約損害、名声損害、利益および将来の収益の減少、および業務の削減または再編を含む重大な処罰を招く可能性がある。
アメリカのデータプライバシーとセキュリティ法
多くの州と連邦の法律、法規、標準は、健康に関連する個人情報および他の個人情報の収集、使用、アクセス、秘密およびセキュリティを規定しており、現在または将来的には、私たちの運営または私たちのパートナーの運営に適用される可能性がある。米国では、データ漏洩通知法、健康情報プライバシーおよびセキュリティ法、連邦および州消費者保護法令、健康関連情報および他の個人情報の収集、使用、開示および保護を管理することを含む多くの連邦および州法律法規が、私たちの運営または私たちのパートナーの運営に適用される可能性がある。適用される場合には、これらの法律を遵守しない場合には、重大な民事及び/又は刑事罰及び私的訴訟を引き起こす可能性がある。プライバシーと安全法律、法規とその他の義務は絶えず変化し、互いに衝突し、コンプライアンス仕事をもっと挑戦的にし、調査、訴訟或いは行動を招く可能性があり、それによってデータ処理に対する重大な処罰と制限を招く可能性がある。
外国監督管理
アメリカ国外で任意の製品をマーケティングするために、私たちは他の国と司法管轄区域の品質、安全性と有効性に関する多くの異なる監督管理要求、及び私たちの製品の臨床試験、マーケティング許可、商業販売と流通などの方面の監督管理要求を遵守する必要がある
私たちの製品がFDAの承認を得たかどうかにかかわらず、外国の監督管理機関のような必要な承認を得て、外国や司法管轄区でその製品の臨床試験やマーケティングを開始する必要があります。米国に関する上記の多くの問題は、欧州連合やEUの場合にも同様に適用されるにもかかわらず、承認過程は国および司法管轄区域によって異なり、追加の製品テストおよび追加の行政審査期間に関連する可能性がある。他の国や管轄地域で承認を得るのに要する時間は、FDAの承認を得るのに要する時間とは異なり、さらに長くなる可能性がある。1つの国または管轄区域で規制承認を得ることは、他の国または管轄区域で規制承認を得ることを保証することはできないが、1つの国または管轄区域で規制承認を得ることができなかったか、または遅延して監督管理許可を得ることは、他の国または司法管轄区の規制手続きに悪影響を及ぼす可能性がある。
適用される外国の監督管理要求を守らなければ、他のほかに、罰金、監督管理の一時停止または撤回、製品のリコール、製品の差し押さえ、経営制限、刑事起訴などの処罰を受ける可能性がある。
非臨床研究と臨床試験
アメリカと類似して、EUの非臨床と臨床研究の各段階は重要な監督管理によって制御されている。
非臨床研究を行うのは,新たな化学や生物物質の健康や環境安全性を証明するためである。非臨床(薬物-毒性)研究は、EU指令2004/10/ECに規定されている良好な実験室慣行またはGLPの原則を遵守しなければならない(特定の医薬製品、例えば放射性ラベル目的のための放射性薬物前駆体については、別の正当な理由がない限り)。特に、体外と体内の非臨床研究は必ずGLP原則に従って計画、実行、モニタリング、記録、報告とアーカイブを行い、GLP原則は組織過程の品質体系と非臨床研究の条件として一連の規則と標準を定義した。このようなプロス基準は経済協力と開発組織の要求を反映する。
米国以外のある国にも類似したプログラムがあり,ヒト臨床試験開始前に臨床試験申請を提出することが求められており,INDに似ている。EU医療製品の臨床試験はEUと国家法規、国際協調会議(ICH)のGCPに関するガイドライン及び“ヘルシンキ宣言”からの適用法規の要求と倫理原則に適合しなければならない。臨床試験の発起人がEU内で成立していない場合、それはEU実体をその法定代表者として指定しなければならない。発起人は臨床試験保険証書を購入しなければならず、大多数のEU諸国では、発起人は臨床試験で負傷したいかなる研究対象にも“非のない”賠償を提供する責任がある。
EUの臨床試験に関連する規制構造は最近変化した。EU臨床試験条例,あるいはCTRと呼ばれ,2014年4月に採択され,EU臨床試験指令が廃止され,2022年1月31日に施行された。指示とは異なり、CTRはすべてのEU加盟国に直接適用され、加盟国がそれをさらに国家法律として実施する必要はない。CTRは臨床試験情報システムを通じてEU全体の臨床試験の評価と監督過程を著しく調整し、このシステムは集中したEU門戸とデータベースを含む。
臨床試験指令は,臨床試験を行う各加盟国で主管する国家衛生当局と独立した倫理委員会に単独の臨床試験申請(CTA)を提出することを要求しているが,FDAやIRBのように,CTRは集中的な手続きを導入し,多センター試験の単一申請の提出のみを要求している。CTRは、スポンサーが各会員国の主管当局と道徳委員会に文書を提出することを可能にし、各会員国が決定を下すことを可能にする。他の事項以外に、CTAは試験方案のコピーと被調査薬品の生産と品質情報を含む調査薬品ファイルを含まなければならない。CTAの評価手続きも統一されており、すべての関連加盟国による共同評価を含み、道徳基準を含む各加盟国が個別にその領土に関する具体的な要求を評価する。各会員国の決定は集中されたEUポータルサイトを通じてスポンサーに伝達される。CTAが承認されると,臨床試験開発は継続可能である
CTRは3年間の過渡期が予想される。進行中の臨床試験と新たな臨床試験がCTRによってどの程度制御されるかはそれぞれ異なる。(I)2022年1月31日までに“臨床試験指令”に従って出願された臨床試験、または(Ii)2022年1月31日から2023年1月31日までの間にスポンサーが選択した
EU臨床試験指令の応用は2025年1月31日までこの指令の管轄を受けている。この日以降,すべての臨床試験(行われている臨床試験を含む)はCTR条項に拘束される
臨床試験で使用される薬物は良好な製造規範やGMPに従って生産されなければならない。他の国と連合の範囲の規制要件も適用される可能性がある。
マーケティング許可
EUで医薬製品を販売するためには、私たちはMAというマーケティング許可を得なければならない。EUの監督管理制度の下で研究用薬品の監督管理許可を得るためには、MA申請或いはMAAを提出しなければならない。このようにする過程は,他を除いて医薬製品の性質に依存する。2つのタイプのMAがあります
•“集中MA”はEUが欧州薬品管理局(EMA)人用薬品委員会(CHMP)の意見に基づき、集中プログラムを通じて発行したものであり、EU全体で有効である。特定のタイプの製品の場合、集中手順は、(I)バイオテクノロジー医薬製品由来医薬製品、(Ii)指定された孤児医薬製品、(Iii)高度治療医薬製品、またはATMP、および(Iv)HIV/エイズ、癌、神経変性疾患、糖尿病、自己免疫および他の機能障害、およびウイルス性疾患などの特定の疾患を治療するための新しい活性物質を含む医薬製品のような強制的である。EUで許可されていない新しい活性物質を含む製品、または重大な治療、科学的または技術革新、またはEUの公衆衛生上の利益に適合する製品については、集中手順がオプションである。
•“国家MA”はEU加盟国の主管当局によって発行され、それぞれの領土のみをカバーし、集中プログラムの強制範囲に属さない製品に適用される。1つの製品がEU加盟国で販売されることが許可されている場合、その国MAは、相互認識手順によって別の加盟国で承認を得ることができる。この製品が申請時にどの加盟国でも国MAを取得していない場合、分散手続きによって各加盟国で同時に承認を得ることができる。分権手続きに基づいて、MAを求める各加盟国の主管当局に同じ書類を提出し、申請者はそのうちの1つを参考加盟国として選択する。
上記の手順により、MAを付与する前に、規制当局は、製品の品質、安全及び効能に関する科学的基準に基づいて、製品のリスクと収益バランスを評価する。
集中プログラムにより,環境評価機関が重大な影響評価を評価する最長期限は210日であった。特別な場合、CHMPは、150日以下(クロックポーズを含まない)内でMAAの加速審査を行う可能性がある。満たされていない医療需要に対して、公衆の健康に大きな影響を与えることが期待される革新製品は、優先薬品計画のような迅速な開発と審査計画を得る資格がある可能性があり、この計画は、米国の画期的な治療指定と同様のインセンティブを提供する。EMAは2016年3月、EMAがPrime計画という計画を開始し、未満足な医療需要に対するEMAの薬物開発の支援を強化するための自発的な計画である。その基礎は、有望な薬剤を開発している会社との相互作用と早期対話を増加させ、彼らの製品開発計画を最適化し、より早期に患者に接触するのを助けるために、彼らの評価を加速させることである。Prime指定を受けた製品開発者は加速評価を受ける資格が期待されるが,これは保証ではない。Primeの称号を持つ候補製品のスポンサーは多くのメリットを得ることができ、しかし限らず、早期にEMAと積極的な監督対話を行い、臨床試験設計とその他の開発計画要素を頻繁に討論し、及びファイルを提出した後にMAA評価を加速する。CHMPの専任連絡先と調査委員は、EMA委員会レベルで製品のより多くの理解を促進するために、Prime計画の早期に任命されたことが重要である。最初の会議はこれらの関係を開始し、EMAの多学科専門家チームを含み、全体的な発展と監督戦略に関する指導を提供した
また,EUでは,必要なすべての安全性や有効性データが得られていない場合には,“条件付き”MAが付与される可能性がある.条件付きMAは,失われたデータの生成やセキュリティ対策の増加を確保する条件を満たさなければならない.有効期間は1年で、すべての条件が満たされるまで年に1回更新しなければならない。未完成の研究が提供されると、“標準”のMAとなることができる。しかし,EMAが設定した時間範囲でこれらの条件を満たしていなければ,MAは更新を停止する.また、“特別な場合”では、出願人が、製品が許可され、特定の手順に従った後であっても、正常な使用条件下での有効性および安全性に関する包括的なデータを提供できないことを証明することができれば、MAも承認することができる。これが
特に期待される適応は非常にまれであり,現在の科学的知識状態では網羅的な情報を提供することが不可能である場合や,データを生成することが一般的に受け入れられている倫理原則に違反する可能性がある場合がある.このMAは、重篤な疾患または満たされていない医療需要のために承認されるべき医薬製品を保持し、出願人は、MAに付与するために必要な合法的な完全データセットを有さないので、条件付きMAに近い。しかし,条件付きMAと異なり,申請者は失われたデータを提供する必要もなく,提供する必要もない.“特殊な場合”のMAは最終的に承認されているが,毎年薬品のリスク−収益バランスが審査されており,リスク−収益比が有利でなければMAは撤回される。
MAの初期期限は5年である.この5年後、許可はリスク-収益バランスを再評価した上で無期限に更新することができる。
データとマーケティングの排他性
EUでは、マーケティングを許可された新製品または参考製品は、通常、MAで8年間のデータ独占および追加2年間の市場独占を得る。承認された場合、データ固有期間は、模倣薬または生物類似薬の申請者がEUで模倣薬または生物類似薬を申請することを防止することになり、参照製品がEUで初めて許可された日から8年以内に、参照製品プロファイルに含まれる臨床前および臨床試験データに依存する。追加の2年間の市場独占期間内に、模倣薬または生物学的に類似したMAAを提出することができ、革新者のデータを参照することができるが、EU参考製品の初期MA後10年以内には、いかなる模倣薬または生物類似製品も発売できない。MA保持者が10年の最初の8年間に1つまたは複数の新しい治療適応の許可を得た場合、10年間の市場専門期間は最大11年に延長されることができる。許可前の科学的評価では、これらの適応は既存の治療法と比較して有意な臨床的利益をもたらすと考えられる。しかし、一つの製品がEU規制機関によって新しい化学実体とみなされることは保証されず、製品はデータ排他性を得る資格がない可能性がある。
小児科発展
EUでは、新医療製品のMAAは、EMAの小児科委員会またはPDCOと合意された小児科調査計画またはPIPに適合する小児科群で行われた試験結果を含まなければならない。PIPは,市販認可が求められている薬物の小児科適応を支援するためのデータ生成の時間とアドバイスを規定している。PDCOは、成人における製品の有効性および安全性を証明するのに十分なデータがあるまで、PIPの一部または全ての措置の実施を延期する義務を許可することができる。さらに、小児科臨床試験データが不要または不適切に提供される場合、PDCOは、子供に無効または安全でない可能性があるので、これらのデータを提供する義務を免除することができ、この製品は、治療のために使用される疾患または状態が成人集団でのみ発生することが予想される、または小児科患者の既存の治療に対して有意な治療利益がない場合である。すべてのEU加盟国でMAを取得し、研究結果を製品情報に含めると、否定的な場合であっても、その製品は、6ヶ月間の補充保護証明書の取得延期(任意の補充保護証明書が承認時に有効である場合)、または孤児医薬製品について、孤児市場の独占経営権を2年間延長することを許可する資格がある。
孤児薬名
EUの“孤児薬品”の認定基準は原則的にアメリカと似ている。スポンサーが、(1)生命または慢性衰弱に危害を及ぼす疾患の診断、予防または治療を目的としていることを証明することができる場合、(2)または(A)申請時に、そのような疾患のEUにおける影響が10,000人中5/10以下であることを証明することができる場合、または(B)製品が孤児の身分から得られる利点がない場合、製品はEUで十分な見返りを生じず、必要な投資が合理的であることを証明することができる。ならびに(3)EUで市販されていることが許可されている関連疾患を満足できる診断、予防または治療する方法がない場合、または、そのような方法があれば、製品は、疾患の影響を受けている人に大きな利点を有するであろう
EUでは、孤児製品に指定された申請はMAAが提出される前のいつでも提出することができる。孤児指定は、当事者に費用減免、礼賓援助、集中手続きへの進入などのインセンティブを得る権利がある。MAが承認されると、孤児医薬品は、承認された治療適応の10年間の市場専門期間を得る権利があり、これは、規制当局が別のMAAを受け入れることができないか、またはMAを承認するか、または同じ適応の類似製品のMAを10年間延長する申請を受け入れることができないことを意味する。合意されたPIPにも適合する孤児薬品については,市場専門期間を2年間延長した。いかなる補充保護証明書も孤児の症状に関する小児科研究によって延期してはならない。孤児指定は、規制審査および承認過程においていかなる利点も伝達されず、規制審査および承認過程の継続時間を短縮することもない
しかしながら、製品が指定された孤児の基準をもはや満たしていないと5年目の終わりに決定された場合、例えば、製品の利益が十分に高く、市場排他性が合理的であることを証明するのに不十分であるため、または疾患の流行率が限界値を超えている場合、孤児専門期間は6年に短縮されることができる。以下の場合、(I)第2の出願人は、その製品が許可された製品と類似しているが、より安全で、より効果的であり、または臨床的により良いことを証明することができ、(Ii)出願人は、十分な数の孤児医薬製品を供給することができない、または(Iii)出願人は、第2の孤児医薬製品出願に同意することができる。ある会社は自発的に一つの製品を孤児登録簿から削除することができる。
承認後に要求する
アメリカと同様に、医薬製品の保有者とメーカーはすべてヨーロッパ薬品管理局、欧州委員会及び/又は加盟国主管監督機関の全面的な監督管理を受けている。MAの保有者は、薬物警戒システムを構築し、維持し、個人合格の薬物警戒員、またはQPPVを任命し、このシステムの構築と維持を担当し、医療製品の安全概況と任意の新たに発生した安全問題を監視しなければならない。主な義務は深刻な副作用が疑われる報告を加速させ、安全更新報告書を定期的に提出することを含む。
すべての新しいMAAは、企業が実施するリスク管理システムを記述し、製品に関連するリスクを最小限に防止または低減するための措置を記録するリスク管理計画またはRMPを含まなければならない。規制当局はまた、特定の義務を金融管理専門家の条件として規定することができる。このようなリスク最小化措置または認可後の義務は、追加の安全監視、PSURsのより頻繁な提出、または追加の臨床試験または認可後の安全性研究を含む可能性がある
医薬製品の広告と販売促進も医薬製品の販売促進、医師との相互作用、誤解性と比較広告及び不公平な商業行為に関する法律の制約を受けている。製品のすべての広告および販売促進活動は、承認された製品特性要約と一致しなければならないため、すべてのラベル外の販売促進活動は禁止されます。連合はまた消費者に直接向けた処方薬の広告を禁止する。薬品広告や販売促進の一般的な要求はEU指令に基づいて制定されているが、詳細は加盟国ごとの法規によって管理されており、各国が異なる可能性がある。
上述のEU規則はヨーロッパ経済区、すなわちEEAに一般的に適用され、27のEU加盟国にノルウェー、リヒテンシュタイン、アイスランドを加えて構成されている。
EU及び加盟国が臨床試験の進行、生産承認、医薬製品のM&A及びそのような製品のマーケティングに適用される法律を遵守せず、M&Aを付与する前及び後、医薬製品の製造、法定医療保険、賄賂及び反腐敗、又は他の適用される法規の要求は、行政、民事又は刑事罰を招く可能性がある。これらの処罰は、臨床試験の遅延または許可を拒否すること、またはMAの承認、製品の撤回およびリコール、製品の差し押さえ、一時停止、MAの撤回または変更、生産の完全または部分的な一時停止、流通、製造または臨床試験、経営制限、禁止、免許停止、罰金、および刑事罰を含むことができる。
EUの組合せ製品に対する規制
EUは異なる立法文書を通じて医療機器と医療製品をそれぞれ規制し,適用する要求は薬物−機器組合せ製品のタイプによって異なる。連合は製造業者が正しい規制枠組みを選択するのを助けるための指導意見を発表した。
EUでは、医薬製品を管理するための薬物送達製品が医薬製品として管理されており、前記医薬製品および前記器具は単一の全体製品を構成する。EMAは,集中プログラムによって提出されたMAASの品質,安全性,有効性を評価し,医療製品とともに使用される医療機器の安全性と性能を評価する責任がある。EMAまたはEU加盟国の国家主管当局は、上記の医療製品規則に基づいて製品を評価するが、機器部品は、医療機器法規(添付ファイルIに規定されている一般的な安全および性能要件を含む)に適合しなければならない。MAAは、製造業者のEUデバイス適合性宣言または通知機関によって発行された関連証明書に含まれる医療機器法規に適合するデバイス構成要素の評価結果を含まなければならない。MAAが適合性評価の結果を含まず,かつ設備の適合性評価を単独で使用する場合には,機関の参加を通知する必要があり,主管当局は,申請者に設備適合性に関する通知機関の意見を提供するように要求しなければならない.
対照的に、投与のための薬物送達製品では、器械と医薬製品は単一の全体製品(例えば、共同包装)ではなく、医薬製品は上述した医薬製品の規則に従って規制され、器具部品は医療機器として規制され、“医療機器条例”に規定されているすべての要求に適合しなければならない
投与のための非一体型設備の特性は薬品の品質、安全性と有効性に影響を与える可能性がある。投与装置が薬品と共同包装された範囲内、又は特殊な場合には、医薬品の製品情報において特定のタイプの投与装置を使用することが特に規定されている場合には、医療製品のMAAにおいて医療製品の品質、安全性及び/又は治療効果に影響を与える可能性のある医療機器の特性に関する付加情報を提供する必要がある
医療機器とともに使用する場合,医療製品の品質に関する文書の要求は,単一全体の製品,共同包装,参考製品を含め,2021年7月22日のEMAガイドラインで概説され,2022年1月1日から適用される
上記の連合規則は一般的にヨーロッパ経済地域に適用される。
イギリスの離脱とイギリスの規制枠組み
2021年1月1日の離脱移行期間が終了して以来、イギリス(イングランド、スコットランド、ウェールズ)はEU法律の直接的な制約を受けていないが、アイルランド/北アイルランド議定書の条項によると、EU法律は北アイルランドに一般的に適用されている。英国政府がどの程度その法規をEUと統合することを求めているのかは不明である。二次立法でイギリス法律に転換されたEU法律は依然としてイギリスに適用される。しかし、現在イギリス議会に提出されている“2022年保留EU法律(撤回と改革)法案”によると、いかなる保留EU法律も明確に保留され、国内法によって国内法として吸収されていない場合、または部級法規によって延長され(2026年6月23日より遅くない)、自動的に失効し、2023年12月31日までに撤回される。しかも、(EU)CTRのような新しい立法はイギリスには適用されない。EU-イギリス貿易·協力協定(TCAと略称する)は、医薬品生産施設のGMP検査および発行を相互承認するGMPファイルを含むが、イギリスおよびEUの薬品法規および製品基準の卸売相互承認を含まない。イギリスは将来、EUとは異なる現地要求を持つかもしれないが、これは将来イギリスで発生する臨床と開発活動に影響を与える可能性がある。同様に,イギリスの臨床試験提出はEMA臨床試験情報システム(CTI)でEU諸国の臨床試験提出とバンドルすることができず,イギリスの将来の臨床と開発活動の複雑さ,コスト,潜在リスクをさらに増加させる。英国のEU離脱が英国とEUとの関係をどの程度変えるかは、政治的にも経済的にも大きな不確実性がある
英国政府は、医療製品や医療機器分野の既存の法規を改正または補完するために、国務大臣または適切な機関に権限を付与する新しい“2021年薬品·医療機器法”を可決した。これにより,人間の薬物,臨床試験,医療機器分野の規制格差や将来の変化を解決する上で柔軟性を許すことを目的として,今後二次立法で新たなルールを導入することが可能となる。
2021年1月1日以来、薬品と医療製品規制機関(MHRA)はイギリスの独立した薬品と医療機器監督機関であった。北アイルランド議定書の結果として、北アイルランドはイギリスやGBを含むイングランド、ウェールズ、スコットランドとは異なる規則が適用されるだろう;全体的に、北アイルランドはEUの規制制度に従うだろうが、その国家主管機関はMHRAであるだろう
MHRAは、150日間の評価およびスクロール審査プログラムを含む、患者に利益を得る新薬を優先的に得るプログラムを含む、国家許可プログラムを変更している。2021年1月1日から、中央許可製品のためのすべての既存のEU MAは、MA所有者が脱退を選択しなければならない限り、GBでのみ有効であり、無料である英国MAに自動的に変換またはキャンセルされる。集中プログラムを用いて欧州経済区全体で効果的なM&Aを獲得するためには,欧州経済区に会社を設立しなければならない。したがって,イギリスの離脱後,イギリスで設立された会社はEU集中化プログラムを使用することはできず,ヨーロッパ経済圏実体はいかなる集中式MAを持たなければならない。英国で製品を商業化するためにイギリスMAを得るためには、申請者はイギリスで設立されなければならず、イギリスで製品を商業化するためには、イギリスの国家認可プログラムのうちの1つまたはイギリス離脱後に残りの国際協力プログラムのうちの1つに従ってMAを取得しなければならない。GB許可の申請を決定する際には、MHRAは、新しい(集中プログラム)MAの承認に関する欧州委員会の決定に依存することができ、または、EU加盟国(またはアイスランド、リヒテンシュタイン、ノルウェー)で承認されたMAをGBで付与することができるように、MHRAの分散または相互承認手続きを使用することができる。
MAの元孤児の称号はないだろう。逆に、MHRAは、対応するMA申請を審査しながら孤児指定申請を審査する。これらの基準は基本的に同じであるが,すべて市場のためにオーダーメイドされている,すなわちEUではなくイギリスのこのような疾患の流行率は万分の5を超えてはならない。孤児の称号が付与された場合、期限または市場独占権は、その製品が初めて承認された日からGB単位で設定される
臨床試験に関するイギリスの規制枠組みは、既存のEU立法(二次立法によってイギリス法律に定着された)に由来する。2022年1月17日,MHRAはイギリスの臨床試験立法の見直しについて8週間の諮問を開始した。相談は2022年3月14日に終了し、臨床試験の審査を簡略化し、革新を促進し、臨床試験の透明性を高め、リスク比率を高め、そして患者と公衆の臨床試験への参加を促進することを目的とした。協議の結果は密接に注目されており、イギリスが(EU)CTRと一致するか、規制の柔軟性を維持するために規則から逸脱するかを決定するだろう。
データプライバシーとセキュリティ法
私たちはまた、データプライバシーを管理し、個人データ(健康関連データを含む)を保護する米国の国/地域ではない法律法規によって制約されている。EUおよび他の管轄区域の法律法規は、個人データの収集、使用、保存、開示、処理、安全に広く適用され、時間の経過とともに一般的に厳しくなっている。プライバシーとセキュリティ法律、法規、その他の義務は絶えず変化し、互いに衝突し、コンプライアンス作業を複雑化させ、調査、訴訟あるいは行動を招き、重大な民事および/または刑事罰およびデータ処理の制限を招く可能性がある。
人力資本
著者らは経験豊富、技術の優れたチームを集め、その中にリードバイオテクノロジーと製薬会社からの業界ベテラン、科学者、臨床医師と肝心なオピニオンリーダー、及び世界各地からのリード学術センターを含む。私たちの従業員は高度に尊敬し、情熱的なチームで、彼らは尊重、謙虚、透明、包容、奉仕、協力、そして面白い文化を誇りに思っています。私たちの最終目標は患者の命を向上させて延長することだ。
私たちの理念は、私たちの最大の資産、私たちの従業員、私たちの人的資本目標、例えば適用、識別、吸引、維持、奨励、そして私たちの臨床、科学、他の従業員、コンサルタントを含む、私たちの最大の資産、私たちの従業員、私たちの人的資本目標を支援するための包括的な報酬と福祉プログラムを提供することです。私たちの株式と現金インセンティブ計画の主な目的は、私たちの利益と株主の利益が私たちの従業員とコンサルタントの利益と一致するように、株と現金に基づく報酬奨励を付与することによって、従業員を引き付け、維持し、激励することである。
2023年3月10日現在、178人のフルタイム従業員がおり、アルバイト従業員はいません。この178名の従業員のうち,54人(30%)が博士や医学博士号を持ち,94人(53%)が女性であった。私たちの従業員の中の一人も労働組合代表でもなく、集団交渉協定のカバー範囲もない。私たちは私たちが従業員と仲がいいと思う。
企業情報
我々は2015年10月26日にデラウェア州法律に基づいてFSG,Bio,Inc.に登録し,2017年にGossamer Bio,Inc.と改名した。私たちの主な実行事務室はカリフォルニア州サンディエゴ科学園路3013号にあります。郵便番号:92121、電話番号は(858684-1300)。
利用可能な情報
インターネットの住所はwww.gosamerBio.comです。私たちの投資家関係サイトはhttp://ir.gosamerBio.comにあります。我々は、米国証券取引委員会または米国証券取引委員会にこのような資料を提出または提出した後、当社の投資家関係サイト上の“届出”の項目で、当社の年間報告書Form 10-K、Form 10-Q四半期報告、Form 8-K現在の報告、取締役および上級管理者第16条の報告、およびこれらの報告の任意の修正を無料で提供します。これらはアメリカ証券取引委員会のサイトwww.sec.govで無料で入手することもできます。我々は,重要な非公開情報を開示する手段として我々の投資家関係サイトを用い,FD法規下での開示義務を遵守している.投資家は,我々のニュース原稿,米国証券取引委員会の届出文書,公開電話会議やインターネット放送に注目するほか,このようなサイトにも注目すべきである.コーポレートガバナンスに関する情報は私たちの投資家関係サイトにも含まれています。米国証券取引委員会及び当サイトにおける情報又は当サイトを通じて取得された情報は、本出願に組み込まれることもなく、本出願の一部ともみなされない。また,これらのサイトのURLへの参照は非アクティブテキスト参照のみに用いた.
第1 A項。リスク要因です
普通株の購入または売却の決定を下す前に、以下のリスク要因と、当社の10-K年度報告書に含まれる他の情報、ならびに“経営陣の財務状況および経営業績の検討および分析”を含む他の情報をよく考慮しなければなりません。私たちはあなたに次の危険要素で議論されたどんな事件も起こらないということを保証できません。これらのリスクは、私たちの業務、運営結果、財務状況、および成長見通しに実質的な悪影響を及ぼす可能性がある。もしこのような状況が発生したら、私たちの普通株の取引価格は下がるかもしれない。私たちは今知らないか、あるいは私たちが現在どうでもいいと思っている他のリスクと不確実性はまた私たちの業務運営や財務状況を損なう可能性がある。
リスク要因をまとめる
以下に述べるリスク要因は,米国への投資に関連する主要なリスク要因の概要である。これらは私たちが直面している唯一の危険ではない。あなたはこのような危険要素と、本プロジェクト1 Aに列挙されたリスク要素を慎重に考慮しなければならない
•私たちの経営の歴史は限られており、赤字の歴史があり、未来にはもっと多くの損失が出ることが予想される。
•私たちは私たちの目標を達成するために多くの追加資金が必要になるだろう。
•私たちの業務活動は世界の新冠肺炎の流行と他の流行病の悪影響を受けるかもしれない。
•私たちは臨床開発を通じて私たちの候補製品を成功させる能力に大きく依存している
•臨床薬物開発は長く高価な過程に関連し、結果は不確定であり、臨床前研究と早期臨床試験の結果は必ずしも未来の結果を予測できるとは限らない。
•現在または計画されている臨床試験において患者を募集することの困難または遅延、または現在または計画されている臨床試験の開始または完了、または終了または一時停止は、我々の業務に悪影響を及ぼす可能性がある。
•私たちは、予想外の遅延を招いたり、候補製品の商業化に必要な承認を受けることを阻止したりすることができる高度に規制された業界で運営されている。
•私たちは第三者に依存して臨床前と臨床試験を行う。
•私たちは私たちの臨床前と臨床候補製品を生産するために第三者に依存する。
•私たちは協力、許可、そして他の似たような計画を達成したり維持したりすることに成功できないかもしれない。
•I承認されれば、私たちの候補製品の成功は、持続的な規制義務の履行、市場受容度、そして政府当局と保険会社の十分な保険に依存するだろう。
•私たちは他のバイオテクノロジーや製薬会社からの激しい競争に直面しています。もし私たちが効果的に競争できなければ、私たちの経営業績は影響を受けます。
•私たちの運営結果は大きく変動するかもしれない。
•私たちの業務は私たちの高い素質の管理、臨床と科学者を吸引、維持、激励する能力に依存している。
•私たちに製品責任訴訟を提起すれば、私たちは多くの責任を負い、候補製品の商業化を制限することを要求されるかもしれません。
•私たちの業務は私たちが知的財産権とノウハウを保護する能力に依存する。
•私たちは私たちの許可協定を守らなければならない。そうでなければ、私たちはseralutinibを含むいくつかの候補製品の許可権を失うかもしれない。
•私たちの株価は大きく変動しており、投資家は大きな損失を被る可能性がある。
•私たちは証券集団訴訟に巻き込まれており、将来的に証券集団訴訟の影響を受ける可能性がある
私たちの限られた経営歴史、財務状況、資本要求に関するリスク
私たちの経営の歴史は限られており、設立以来ずっと重大な運営損失を受けており、予測可能な未来に重大な損失が予想されている。私たちはどんな収入も利益を達成しないかもしれないし、あるいは、もし私たちが利益を達成すれば、私たちは持続できないかもしれない。
バイオ製薬製品開発は投機性の強い仕事であり、大きなリスクに関連している。私たちは臨床段階の生物製薬会社で、運営の歴史が限られていますので、それに基づいて私たちの業務と将来性を評価することができます。私たちは2017年に運営を開始し、これまで、私たちは主に私たちの会社、業務計画、資金調達、決定、買収、許可、および臨床前研究と臨床試験に集中してきました。SeralutinibとGB 5121は積極的な臨床開発中であるが,他の複数の開発プロジェクトはまだ臨床前あるいは研究段階にある。私たちは、第2段階以外のいかなる臨床試験を成功させることができ、監督管理の承認を得ることができ、商業規模の製品を製造すること、あるいは第三者代表が私たちにそうすることを手配したり、成功した製品の商業化に必要な販売およびマーケティング活動を行うことができることを証明していない。したがって、もし私たちがバイオ製薬製品の開発と商業化に成功した歴史があれば、私たちの将来の成功や生存能力のいかなる予測もそれほど正確ではないかもしれない。
設立以来、私たちは深刻な運営損失が発生した。もし私たちの候補製品が成功的な開発と承認を受けなければ、私たちは決して何の収入も生じないかもしれない。2022年と2021年12月31日までの年度の純損失はそれぞれ2億294億ドルと2.34億ドルだった。2022年12月31日までの累計赤字は10.322億ドル。我々のほとんどの損失は,我々の研究や開発計画に関する費用と我々の運営に関する一般的かつ行政コストによるものである.私たちのすべての候補製品は、私たちの複数の臨床前候補製品を含み、規制の承認を申請または獲得し、製品販売から収入を得ることができる前に、多くの追加の開発時間と資源が必要になるだろう。私たちは予測可能な未来に損失を被ることが予想され、私たちが開発を継続し、規制機関に私たちの任意の候補製品の承認を求め、それを商業化する可能性があり、識別、評価、買収、許可、または他の候補製品の開発を求めるにつれて、これらの損失は大幅に増加すると予想される。
利益を実現し、維持するためには、大量の収入を生む製品を開発し、最終的に商業化しなければならない。これは、私たちの候補製品の臨床試験と臨床前研究を完成させ、これらの候補製品の規制承認を得ること、および私たちが規制承認を得る可能性のある任意の製品を含む、一連の挑戦的な活動で成功することを要求するだろう。私たちはただこのような活動の大多数の初期段階にいるだけだ。私たちはこのような活動で決して成功しないかもしれないし、たとえ私たちが成功しても、利益を達成するのに十分な収入が生まれないかもしれない。また,我々は,企業が新たかつ急速に発展する分野でしばしば遭遇する多くのリスクや不確実な要因を克服することに成功する能力,特に生物製薬業界ではまだ示されていない。バイオ製薬製品開発に関連する多くのリスクや不確実性のため、費用を増加させる時間や金額、あるいはいつ、または利益を達成できるかどうかを正確に予測することはできない。たとえ私たちが確実に利益を達成したとしても、私たちは四半期や年度の収益性を維持したり向上させることができないかもしれない。もし私たちが実現して利益を維持できなければ、会社の価値を下げ、資金を調達し、業務を拡大し、研究開発努力を維持し、候補製品を多様化し、さらには運営を継続する能力を弱める可能性がある。わが社の価値の低下はあなたの投資損失の全部または一部を招く可能性もあります。
私たちは私たちの目標を達成するために大量の追加資金を必要とし、必要な場合に必要な資金を受け入れられることができない場合、あるいは必要な資金を全く得ることができない場合、私たちの製品開発計画、商業化努力、または他の運営を延期、制限、減少、または終了させることができるかもしれない.
バイオ製薬候補製品の開発は資本集約型である。私たちが行っている活動に関連する費用は増加すると予想され、特に私たちが行っているおよび計画中のseralutinibとGB 5121臨床試験、研究と開発を継続し、GB 7208を含む複数の他の開発計画の臨床試験を開始し、規制機関に現在の候補製品と私たちが開発する可能性のある任意の未来の候補製品の承認を求める時に。また、私たちの候補製品が開発と商業化に進展するにつれて、私たちは必要になります
許可者と他の第三者に記念碑的な支払いを支払い、私たちはseralutinibを含む私たちの候補製品を彼らから獲得または獲得した。さらに、もし私たちが将来的に獲得を求めたり、許可範囲内でより多くの候補製品を獲得したりすれば、私たちは大量の前払い、マイルストーン支払い、および/または支払い可能かもしれない。もし私たちの候補製品が規制部門の承認を得たら、製品製造、マーケティング、販売、流通に関連した巨額の商業化費用が発生することも予想される。いかなる臨床試験或いは臨床前研究の結果はすべて高度に不確定であるため、著者らは著者らの候補製品の開発と商業化に成功するために必要な実際の数量を合理的に見積もることができない。したがって、私たちは私たちの持続的な業務と関連した多くの追加資金を得る必要があるだろう。もし私たちが必要な時や魅力的な条件下で資金を集めることができなければ、私たちは私たちの研究開発計画や将来の商業化努力を延期、減少、または廃止することを余儀なくされるかもしれない。
私たちの既存の現金、現金等価物、および有価証券は、本年度報告書がアメリカ証券取引委員会に提出された日から少なくとも今後12ヶ月間、私たちの運営に資金を提供することができると信じている。特に,これらの資金により,PAHでのサラダルチニブの登録第3段階臨床試験を開始し,我々が行っているGB 5121健常ボランティアの第1段階研究を完成させることができると予想される。私たちは間違っていることが証明される可能性があるという仮定に基づいてこれらの推定をして、私たちは現在予想されているよりも早く私たちの資本資源を使用することができます。私たちの現在知られていない多くの要素のため、私たちの運営計画と私たちの現金資源に対する他の需要は変化する可能性があり、私たちは計画よりも早く追加資金を求める必要があるかもしれません。潜在的な協力、ライセンス、その他の同様の手配を含む、公共または私募株式または債務融資または他の資本源を通じて。また、有利な市場条件や戦略的考慮により、現在または将来の運営計画のために十分な資金があると考えても、追加の資本を求めることができる。例えば、2020年7月、私たちと私たちのいくつかの子会社は、借入者として、代理および融資者MidCap Financial Trustまたは貸手であるMidCapと時々、または貸手MidCapと共に、2019年5月に融資を受ける3,000万ドルの定期融資を提供することに同意し、代理および融資者MidCap Financial Trustまたは融資者であるMidCapと共に、我々のクレジット、保証および保証プロトコルまたは信用スケジュールを修正する。より多くの資金を得ようとすることは、私たちの経営陣の日常活動への注意をそらす可能性があり、これは候補製品を開発する能力に悪影響を及ぼす可能性がある。
私たちの将来の資本需要は多くの要素に依存します
•私たちは将来行われる候補製品の臨床試験および臨床前研究のタイプ、数量、範囲、進展、拡張、結果、コスト、およびタイミングを行っているか、または選択することができる
•我々の候補製品の製造コストと時間は、商業製造を含む(任意の候補製品が承認された場合)
•私たちの候補製品に対する規制審査のコスト、時間、結果
•私たちの特許と他の知的財産権を取得し、維持し、実行するコスト
•財務報告の内部統制を強化することを含む上場企業としての義務を履行するために、運営システムの強化と増任者の強化に努めている
•私たちの臨床活動の増加に伴い、より多くの人員やコンサルタントの雇用に関するコストが増加している
•私たちはライセンシーと他の第三者にマイルストーンまたは他の支払いの時間と金額を支払わなければなりません。私たちは彼らから私たちが得た候補製品の許可を得ました
•任意の候補製品が承認された場合、販売およびマーケティング能力のコストおよび時間を確立または確保する
•私たちは第三者支払者から十分な市場受容度、カバー率、および十分な補償を得ることができ、任意の承認された製品のために十分な市場シェアと収入を得ることができる
•協力、許可、および他の同様のスケジュールの条項および時間を確立して維持すること;
•私たちが許可や買収を受ける可能性のある任意の製品や技術に関連するコスト。
臨床試験と臨床前研究を行うことは時間がかかり、高価と不確定な過程であり、数年の時間をかけて完成する必要があり、しかも著者らは永遠に監督部門の許可を得て製品販売を実現するために必要なデータ或いは結果を生成できないかもしれない。しかも、私たちの候補製品が承認されれば、商業的成功を得られないかもしれない。私たちの商業収入は、もしあれば、何年も商業用途がないと予想される製品を販売します。もしなければ。
したがって、私たちは追加的な資金調達に依存して私たちの業務目標を達成し続ける必要があるだろう。私たちは許容可能な条項で十分な追加融資を得ることができないかもしれないし、金融と信用市場の悪化や不安定、市場全体の流動性不足、地政学的事件、または他の理由を含む十分な追加融資を得ることができないかもしれない。また、有利な市場条件や流動性や戦略的考慮により、現在または将来の運営計画のために十分な資金があると考えても、追加の資本を求めることができる。
私たちの信用手配の条項は私たちの経営と財政的柔軟性を制限する。
2019年5月2日、私たちは信用手配を締結し、2019年9月18日、2020年7月2日、2022年12月7日にさらに改訂しました。2022年12月31日現在、信用手配による未返済元金残高は2420万ドル。
信用手配は私たちに適用される肯定的な契約と否定的な契約を含む。他にも、肯定的な条約には、私たちに合法的な存在と政府の承認の維持、いくつかの財務報告の提出、保険範囲の維持、財産の維持、納税、関連口座のいくつかの要求を満たし、法律法規を遵守することが含まれています。消極的な公約には、担保の譲渡、追加債務の招く、合併または買収、配当金の支払い、または他の分配、投資、留置権の作成、重要な合意と組織文書の修正、資産の売却、コントロール権の変更などの制限が含まれていますが、いずれもいくつかの例外的な状況によって制限されています。
信用手配には、違約事件、違約事件の発生と持続が利息を他の方法で適用される可能性のある金利プラス3.0%で徴収し、MidCapを代理人として私たちに救済措置を行使する権利を持たせることと、私たちの担保信用手配(私たちの現金を含む)に対する財産の停止を含む信用手配の担保品を保証する権利がある。これらの違約事件には、信用手配下の任意の満期金額を支払うことができなかったこと、信用手配下の契約違反、私たちの破産や破産事件の発生、制御権変更の発生、いくつかのFDAと規制事件の発生、当社は、重大な不利な変化が発生した場合を含む、米国証券取引委員会に登録され、ナスダック全世界の精選市場またはナスダックでの上場取引を継続できなかった;重大な合意の違約によって重大な不利な変化を招くことが予想される理由がある;ある他の債務項目の下でいくつかの違約が発生し、金額が250万ドルを超える;および二次債務と転換可能な債務の下でいくつかの違約が発生する。違約事件の発生は私たちの業務と将来性を深刻に損害し、私たちの普通株の価格下落を招く可能性がある。
私たちが計画通りに債務を返済したり、私たちの債務の再融資を行う能力があるかどうかは、私たちの将来のパフォーマンスと追加の現金源を調達する能力にかかっており、これは経済、金融、競争、他の私たちがコントロールできない要素の影響を受ける。もし私たちが債務を返済するのに十分な現金を生成できない場合、私たちは資産の売却、再編債務、または煩雑または高度に希釈される可能性のある条項で追加の株式を得るなどの1つまたは複数の代替案を要求されるかもしれない。もし私たちが私たちの債務を再融資したいなら、私たちはこれをする能力があるかどうかは、当時の資本市場と私たちの金融状況にかかっているだろう。私たちはこのような活動のいずれにも従事できないかもしれないし、理想的な条件でこれらの活動に従事することができないかもしれません。これは私たちの債務不履行をもたらす可能性があります。もし私たちが追加的な債務融資を調達すれば、このような追加債務の条項は私たちの運営と財政的柔軟性をさらに制限するかもしれない。
私たちの負債と負債は、私たちの業務に利用できるキャッシュフローを制限し、私たちの業務、財務状況、運営結果に悪影響を及ぼす可能性のあるリスクに直面し、手形義務を履行する能力を弱める可能性があります。
2022年12月31日現在、私たちは2027年満期の元金総額5.00%の転換可能優先手形2億ドルを売却しており、会社間債務を含まず、私たち(私たちの子会社を含む)は約6470万ドルの追加債務と他の負債を持っており、貿易未払いを含め、約2360万ドルは私たちの信用手配下の保証債務です。私たちはまた未来の資金調達需要を満たすために追加的な債務を発生するかもしれない。私たちの負債は私たちの株主と私たちの業務、経営結果、財務状況に重大なマイナス影響を与えるかもしれません
•不利な経済的条件と産業的条件での私たちの脆弱性を増加させる
•私たちが追加資金を得る能力を制限し
•他の目的で利用可能なキャッシュ量を削減するために、資金フローの大部分を債務返済に使用することが求められている
•私たちの計画や業務の変化に対応する柔軟性を制限する
•第三者が私たちを買収することを難しくしたり高くしたりします
•手形変換時に普通株式を発行し、既存の株主の利益を希釈すること
•私たちはレバレッジ率が私たちより低いか資本を獲得しやすい競争相手に比べて競争劣勢にあるかもしれない。
私たちの業務は十分な資金を生産しないかもしれません。そうでなければ、私たちは手形を含む債務下の満期金額を支払うために十分な現金備蓄を維持できないかもしれません。将来的に現金需要が増加するかもしれません。さらに、私たちの既存の信用計画には、私たちが将来発生する可能性のある任意の債務が、私たちが業務を経営し、資本を調達したり、他の債務を支払う能力を制限する金融および他の制限的な契約を含む可能性があります。もし私たちがこのような条約を遵守できなかったり、期限が切れた時に私たちの債務を返済できなかったら、私たちはその債務の下で約束を破って、それはまたその債務と私たちの他の債務を直ちに全部返済することを招く可能性がある。
追加資本の調達は私たちの株主に希釈して、私たちの運営を制限したり、私たちの技術や候補製品に対する権利を放棄することを要求するかもしれません.
これまで、私たちが相当な製品収入を生み出すことができれば、私たちは、潜在的な協力、許可証、および他の同様の計画を含む、株式発行、債務融資、または他の資本源(例えば、私たちの信用手配)を通じて、私たちの現金需要に融資する予定です。もし私たちが株式または転換可能な債務証券を売却することで追加資本を調達すれば、私たちの株主の所有権権益は希釈され、これらの証券の条項は清算または他の私たちの普通株主の権利に悪影響を及ぼす特典を含む可能性がある。債務融資および優先株融資に関与する可能性のある協定は、追加債務を招く、資本支出を行う、または配当を宣言するなど、私たちが具体的な行動をとる能力を制限または制限する契約を含む。
もし私たちが未来の協力、許可、および他の同様の計画を通じて資金を調達すれば、私たちは私たちの未来の収入源、研究計画、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利である可能性があり、および/または私たちの普通株の価値を低下させる可能性のある条項で許可を付与しなければならないかもしれない。
我々の候補製品の発見,開発,規制承認に関するリスク
私たちの業務は流行病のリスクの影響を受けて、例えば新冠肺炎が大流行しています。
新冠肺炎疫病と政府が取った対応措置は企業と商業に直接と間接的な重大な影響を与えた。新冠肺炎疫病はどの程度著者らの業務に影響する可能性があり、著者らの臨床前研究、臨床試験と財務状況を含み、未来の発展に依存し、これらの発展は高度な不確定性があり、自信に満ちて予測できない。例えば,我々が現在期待しているセラルチニブ3期臨床試験の能力の予想は,臨床試験や医療活動がある程度正常であり,臨床サイトはこのような臨床試験期間中に開放されており,2021年夏のセラルチニブ第2段階臨床試験で新冠肺炎Delta変異体が急増したときに経験したように,登録速度の鈍化を経験していないという仮定に基づいている。可能な範囲で、連邦、州、地方当局の適用指導と一致して、私たちは通常通り業務を展開し、従業員の出張を必要または適切に修正します。私たちは新冠肺炎を監視し続け、連邦、州、あるいは地方当局が要求する可能性のある行動、あるいは私たちの従業員と会社との業務往来のある他の第三者の最適な利益に合致する行動を含む、私たちの運営を変える可能性があります。新冠肺炎の大流行或いは他の高伝染性或いは伝染性疾病の発生或いはその他の健康問題と関連して、私たちは私たちの業務、臨床試験及び製造とサプライチェーンに深刻な影響を与える可能性がある中断に遭遇する可能性がある:
•臨床現場起動の遅延或いは困難は、著者らの臨床試験のための臨床現場調査員と臨床現場スタッフを募集する困難を含む
•私たちの臨床試験で患者を募集することは遅延または困難であり、特にサイトが患者のスクリーニングと募集を継続しなければ、
•医療資源を臨床試験の進行から移し,われわれの臨床試験場所である病院や臨床試験を支援してくれた病院スタッフを他の場所に移すことを含む
•臨床試験場のモニタリングのような重要な臨床試験活動を中断する理由は、連邦または州政府、雇用主および他の人が強要または提案した旅行制限、または臨床試験被験者の訪問および研究プログラムの中断であり、これは被験者のデータおよび臨床研究終点の完全性に影響を与える可能性がある
•人員不足、生産減速または中断、および交付システム中断などの原因により、私たちの契約製造組織から私たちの候補製品の供給中断または遅延を受け取ります
•臨床試験に必要な物資と材料の遅延を受け入れ、臨床試験材料輸送のグローバル輸送中断に影響を与える可能性がある
•従業員資源の制限、そうでなければ、全体的な従業員または従業員またはその家族の疾患の不足、または従業員が大勢との接触を避けることを望むことを含む、私たちの臨床試験の実施に集中するだろう
•FDA、EMA、または他の規制機関の動作中断または遅延は、FDA、EMAまたは他の規制機関から将来の臨床試験または規制提出に関するフィードバックまたは承認を受けることを含む
•新冠肺炎や他の流行病への反応の一部として、現地法規の変化は、意外なコストを招くか、あるいは臨床試験を完全に停止する可能性がある臨床試験の方法を変える必要があるかもしれない
•従業員資源が限られているか、政府従業員が休暇を余儀なくされたため、現地の監督機関、道徳委員会と他の重要な機関と請負業者との必要な相互作用が遅延した
•FDAまたはEMAは、影響を受けた地域の臨床試験データの受け入れを拒否する
•社会的距離協定による医師との接触機会の減少を含む製品の発売や商業化が困難である。
また、新冠肺炎の蔓延は、私たちの普通株の取引価格に影響を与え続け、追加資本を調達する能力がないか、または追加資本を調達する能力がないことに影響を与える可能性がある。
私たちは大きく依存しておりセラルチニ現在,第二段階の臨床開発にある。もし私たちが臨床開発で私たちの候補製品を進めることができなければ、規制部門の承認を得られず、最終的に私たちの候補製品を商業化したり、その過程で重大な遅延に遭遇したら、私たちの業務は実質的に損害を受けるだろう。
私たちの2つの臨床段階の候補製品は現在第1段階または第2段階の臨床開発中である。我々は,2020年に開始したシュラルチニブのPAHにおける第2段階臨床試験の開放ラベル拡張を行い,2023年下半期に登録を開始する予定の第3段階臨床試験を行っている。2022年にGB 5121のPCNSLにおける1 b/2期臨床試験も開始した。
私たちはこれらの候補製品がなぜ未来の開発とこれらまたは任意の適応の潜在的承認に値するのかという仮定について、一部は他社が収集したデータに基づいている。著者らも臨床前候補製品があり、臨床開発前に、外国の司法管轄区でINDを通じて研究或いは類似の研究を進展させる必要がある。私たちが研究している適応については、私たちの候補品は重要な研究段階に入っていない。私たちが製品収入を作る能力は私たちの候補製品の成功と最終商業化に大きく依存します。私たちは製品収入が何年も起こらないと予想しています。私たちの候補製品の成功は以下のいくつかの要素に依存するだろう
•臨床試験への参加に成功し、臨床試験と臨床前研究を完成し、良好な結果を得た
•FDAはINDS或いは類似の外国監督機関の類似規制申請を受けて、著者らの臨床前候補製品の臨床試験と私たちの未来の臨床試験の提案設計を行う
•その安全性と有効性を証明し、適用された規制機関を満足させる
•FDAの新薬申請またはNDAを含む関連監督機関の上場許可を受け、このような承認を維持する
•私たちの第三者製造業者と商業製造能力を手配したり確立したりします
•販売、マーケティング、流通能力を確立し、承認を得た後、単独でまたは他人と協力して私たちの製品の商業販売を開始します
•私たちの候補製品のための特許と商業秘密保護または規制排他性を確立し、維持する
•承認された後、私たちの製品は許容可能な安全状態を維持します
•私たちの製品や技術を開発することができる人員組織を維持して発展させる。
私たちのいくつかの候補製品は、セラルチニブを含み、連合製品として規制されており、これはそれらが薬物製品でも設備製品でも構成されていることを意味する。単独で発売されれば、各成分は異なる監督管理経路を受け、FDA内部の異なるセンターによって審査される。私たちの候補製品は薬物-設備組合せ製品と考えられ、承認前にFDA薬物と設備センターの審査と調整が必要であり、これは承認を延期する可能性がある。FDAの規定によると、組合せ製品は薬品と器械に適用される現行の良好な生産規範或いはcGMP要求を遵守しなければならず、医療機器に適用される品質体系と法規を含む。EUは異なる立法文書を通じて医療機器と医療製品をそれぞれ規制し,適用する要求は薬物−機器組合せ製品のタイプによって異なる。候補組合せ製品の設備構成要素に関連する問題は、承認を延期または阻止する可能性がある。デバイス製品の製造業者が修正された場合、またはデバイスコンポーネントを変更したり、独自のデバイスコンポーネントを開発することを選択した場合、修正されたデバイスコンポーネントを使用する前に、検証テストを実行し、FDAおよび他の規制部門の承認または認証を得る必要がある。FDA、任意の他の規制機関、または通知機関が、これらの改装設備が私たちの候補製品と組み合わせて使用されていることを承認または証明できない場合、または設備コンポーネント製造業者に対して重大な法執行行動をとる場合、私たちは特定の管轄区域でマーケティングを行うことができないか、または私たちの製品のマーケティングを一時停止しなければならない可能性がある。
私たちの事業の成功は、私たちが会社に融資し、未来にどんな収入を創出する能力も含めて、主に私たちの候補製品の成功開発、規制承認、商業化にかかっていますが、これは決して起こらないかもしれません。私たちはまだではなく、臨床試験で任意の候補製品の有効性と安全性を証明することに成功しないかもしれないし、その後発売承認を得ることもできないかもしれない。現在の開発段階を考慮すると、治療の安全性と有効性を証明するには数年かかるかもしれない。もし私たちが私たちの候補製品を開発できない場合、あるいは規制部門の承認を得ることができない場合、あるいは承認されれば、私たちの候補製品を商業化することに成功できない可能性があり、私たちは私たちの業務を継続するのに十分な収入を生み出すことができないかもしれない。
臨床薬物開発は長く高価な過程に関連し、結果は不確定であり、臨床前研究と早期臨床試験の結果は必ずしも未来の結果を予測できるとは限らない。また、私たちの候補製品がなぜ未来の開発と潜在的な承認に値するのかに関するいくつかの仮定は、他社が収集したデータに基づいている。我々の候補製品は,今後の臨床試験で有利な結果(あれば)がないか,あるいは監督部門の承認(あれば)をタイムリーに得る可能性がある。
臨床薬物開発費用は高価であり,完成までに数年かかる可能性があり,その結果自体も定かではない。著者らはいかなる臨床試験が計画通りに或いは予定通りに完成することを保証できず、しかも臨床前研究或いは臨床試験過程中のいかなる時間でも失敗する可能性がある。臨床前或いは臨床結果の将来性は有望であるが、任意の候補製品は臨床前或いは臨床開発の任意の段階で意外に失敗する可能性がある。私たちの産業では、候補製品の歴史的失敗率が高い。
同じ種類の候補製品或いは競争相手候補製品の臨床前研究或いは臨床試験の結果は著者らの候補製品の後続の臨床試験の結果を予測できない可能性があり、臨床試験の中期、背線或いは初歩的な結果は必ずしも最終結果を代表するとは限らない。臨床後期候補製品
臨床前研究と初歩的な臨床試験を通じて進展を得たが、試験は必要な安全性と有効性特徴を示すことができないかもしれない。臨床試験では前臨床研究と早期臨床試験に基づく予期せぬ結果が観察されることは珍しくなく,多くの候補製品は臨床試験で失敗しており,早期結果は非常に有望であるにもかかわらず。例えば,セラルチニブをPAH治療の潜在薬とすることにしたが,部分的にはイマチニブ(Gleevec)の効果に基づいており,イマチニブはチロシンキナーゼ阻害剤であり,抗PDGF活性を有し,腫瘍適応に用いられていることが知られており,ノワールが3期臨床試験で観察されているが,われわれが計画した3期臨床試験では類似した効果は認められない可能性がある。また、これらと任意の未来の臨床前と臨床データは異なる解釈と分析の影響を受ける可能性がある。製薬と生物技術業界のいくつかの会社は臨床開発において重大な挫折を経験し、早期の研究においても奮い立つ結果を得た。また,われわれの臨床前計画が候補同定から第一段階臨床開発に進展することは保証できない。
これらの理由から,われわれが進行·計画中の臨床試験や臨床前研究が成功するかどうかは確認できない。私たちの目標適応のいずれの臨床試験で観察される任意の安全問題も、これらの適応および他の適応において規制部門の承認を得る見通しを制限する可能性があり、これは、私たちの業務、財務状況、および運営結果に大きな悪影響を及ぼす可能性がある。
現在または計画中の臨床試験の開始または完了、または終了または一時停止の任意の困難または遅延は、私たちのコスト増加を招き、収入を創出する能力を遅延または制限し、私たちのビジネスの将来性に悪影響を及ぼす可能性がある。
規制部門の許可を得て私たちの候補製品を販売する前に、私たちは広範な臨床研究を行い、候補製品の人体上の安全性と有効性を証明しなければならない。例えば,われわれは現在,PAH患者におけるサラダルチニブの第2段階臨床試験を開放ラベル拡張し,健常ボランティアでGB 5121の第1段階臨床試験を行い,PCNSL患者でGB 5121の全世界1 b/2段階臨床試験を行っている。2023年下半期にセラルチニブの登録3期臨床試験を開始する予定である
また,臨床前候補製品の臨床開発を開始する前に,候補製品の化学,製造,制御,我々が提案した臨床試験案に関する情報を含む臨床前研究の結果やその他の情報をFDAに提出しなければならず,INDの一部として米国以外で臨床試験を行うための規制文書を外国の規制機関に提出しなければならない。
私たちは私たちの計画中の実験が時間通りに始まるかどうか、あるいは予定通りに完了するかどうか分からない。臨床試験の開始と完成は様々な原因で遅延する可能性があり、以下に関連する遅延を含む
•FDA或いは類似の外国の監督機関は著者らの臨床研究の設計或いは実施に対して異なる意見を持っており、著者らが計画したサラダルーティン3期の臨床試験の投与量と終点を含む
•監督部門の承認を得て試験を開始するか、あるいは監督部門と試験設計について合意した
•契約研究組織またはCROと臨床試験場所との合意の任意の失敗または遅延は、その条項は広範な交渉を行うことができ、異なるCROと試験地点の間に大きな差がある可能性がある
•1つまたは複数の機関審査委員会またはIRBsまたは道徳委員会の承認または肯定的な意見を得ること;
•IRBsは、調査地点で行われた試験の承認、一時停止、または終了を拒否し、より多くの被験者の募集を禁止するか、または試験の承認を撤回する
•臨床試験案を修正し
•臨床サイトが試験案から外れているか、または試験から離脱した者
•臨床試験のための十分な数の候補製品を生産するか、または臨床試験のための十分な数の併用療法を得ること
•被験者は、行動制限、健康原因、または新型コロナウイルス新冠肺炎株による他の原因により、私たちの試験に滞在できなかった被験者を含む、私たちの試験に所望の速度で登録または滞在できなかったか、または治療後の後続治療に戻ることができなかった
•被験者は私たちが開発している候補製品の適応のために代替療法を選択したり競争的な臨床試験に参加したりします
•臨床試験を続けるのに十分な資金が不足しています
•重症または意外な薬物関連副作用が出現した被験者
•他の会社で行われている同種の薬物試験で深刻な有害事象が発生した
•より長い時間を要する臨床観察または結果データ分析の臨床終点を選択する
•経口吸入剤を含む我々の候補製品またはその任意の構成要素を製造する工場セラルチニFDAまたは同様の外国規制機関によって、cGMPまたは同様の外国法規または他の適用要件に違反するか、または製造中に汚染候補製品に感染または交差することによって、一時的または永久的な閉鎖を命じられる
•私たちの製造プロセスの変更が必要か、または望む場合があります
•臨床サイト、メーカー、サプライヤー或いはその他のサプライヤーの運営は新冠肺炎などの衛生流行病或いは人員不足のため中断した
•第三者臨床研究者は、臨床試験を行うために必要な免許または許可を失い、予期されたスケジュールまたは臨床試験規程、良好な臨床実践またはGCPまたは他の法規の要求に適合して臨床試験を行わなかった;第三者請負業者は適時または正確にデータ収集または分析を行っていなかった;または
•第三者請負者は、規制要件違反のためにFDAまたは他の政府または規制機関によって禁止または一時停止または他の方法で処罰される場合があり、この場合、代替請負者を探す必要がある可能性があり、そのような請負者が提供するデータの一部または全部を使用して、私たちのマーケティングアプリケーションをサポートすることができない可能性がある。
この遅延或いは監督機構の著者らの試験設計に対するフィードバックも著者らの臨床試験のコストを著しく増加する可能性があり、著者らが計画したサラダルチニブの第三段階の臨床試験を含む。臨床試験が我々,このような試験を行っている機関のIRBs,そのような試験のデータ安全監視委員会やFDAなどの外国規制機関によって一時停止または終了されれば,我々も遅延に遭遇する可能性がある。このような主管部門は一連の要素のために臨床試験を一時停止または終了する可能性があり、これらの要素は、監督管理要求または著者らの臨床規程に従って臨床試験、FDAまたは同様の外国の監督機関の臨床試験操作または試験場所の検査を行うことができなかったことによる臨床一時停止、予見できない安全問題または副作用、ある種の薬物の使用の利益を証明できなかった、政府法規または行政措置の変化、または十分な資金が不足して臨床試験を継続することを含む。例えば,これまでに観察されてきた収益/リスクプロファイルから,GB 5121の1 b/2段階研究への参加を一時停止し,研究のデータ審査委員会と既存のデータを検討し,次の行動を決定することにした。また、規制要求と政策は変化する可能性があり、私たちはこれらの変化に適応するために臨床試験方案を修正する必要があるかもしれない。修正案は,われわれの臨床試験案をIRBsや倫理委員会に再提出して再検討することを要求する可能性があり,臨床試験のコスト,時間,あるいは成功達成に影響を及ぼす可能性がある。
また,FDAや他の規制機関の臨床試験に関する政策が変わる可能性があり,追加の政府法規が公布される可能性がある。例えば,EUの臨床試験に関する規制構造が最近変化している。EU臨床試験条例,あるいはCTRと呼ばれ,2014年4月に採択され,EU臨床試験指令が廃止され,2022年1月31日に施行された。“臨床試験指令”は,臨床試験を行う各加盟国で主管する国家衛生当局と独立した倫理委員会に単独の臨床試験申請(CTA)を提出することを要求しているが,CTRは集中的な手続きを導入し,多センター試験に関する単一申請の提出のみを要求している。CTRは、スポンサーが各会員国の主管当局と道徳委員会に文書を提出することを可能にし、各会員国が決定を下すことを可能にする。CTAの評価手続きも統一されており、すべての関連加盟国による共同評価を含み、道徳基準を含む各加盟国が個別にその領土に関する具体的な要求を評価する。各会員国の決定は集中されたEUポータルサイトを通じてスポンサーに伝達される。CTAになると
承認を得て,臨床研究開発は継続可能である。CTRは3年間の過渡期が予想される。進行中の臨床試験と新たな臨床試験がCTRによってどの程度制御されるかはそれぞれ異なる。2022年1月31日までに“臨床試験指令”に基づいて申請を提出した臨床試験、または(Ii)は2022年1月31日から2023年1月31日までの間であり、スポンサーはEU臨床試験指令を適用する臨床試験を選択しており、2025年1月31日まではこの指令の管轄を受けている。この日以降,すべての臨床試験(行われている臨床試験を含む)はCTR条項に拘束される。我々と我々の第三者サービスプロバイダ(例えば、契約研究機関やCRO)がCTR要求を遵守することは、我々の開発計画に影響を与える可能性がある
イギリスがどの程度その規制をEUと統合することを求めているのかは不明だ。イギリスでは,臨床試験に関する規制枠組みは既存のEU立法(二次立法によりイギリス法で実施されている)に由来する。2022年1月17日,イギリスのMHRAはイギリスの臨床試験立法の見直しについて8週間の諮問を展開した。相談は2022年3月14日に終了し、臨床試験の審査を簡略化し、革新を促進し、臨床試験の透明性を高め、リスク比率を高め、そして患者と公衆の臨床試験への参加を促進することを目的とした。協議の結果は密接に注目されており、イギリスが(EU)CTRと一致するか、規制の柔軟性を維持するために規則から逸脱するかを決定するだろう。アイルランド/北アイルランドに関する議定書の条項によると,(EU)CTRにおける研究用医薬品と補助薬品の製造·輸入に関する規定は北アイルランドに適用される。イギリス政府は,その法規をEUが採用している新しい方法と密接に一致させないことを決定し,他の国ではなく,イギリスでの臨床試験のコストに影響を及ぼす可能性がある
イギリスの臨床試験提出はEMA CTISではEU諸国の臨床試験提出とバンドルできず、イギリスの将来の臨床と開発活動の複雑さ、コスト、潜在リスクをさらに増加させる。英国のEU離脱が英国とEUとの関係をどの程度変えるかは、政治的にも経済的にも大きな不確実性がある。
もし私たちが既存の要求の変化にゆっくりあるいは適応できない場合、あるいは新しい要求を採用したり、臨床試験を管理する政策を採用すれば、私たちの発展計画も影響を受ける可能性がある。
また、海外で臨床試験を行うことは、現在、私たちの候補製品のために引き続き行う可能性があるように、追加のリスクをもたらし、私たちの臨床試験の完成を遅らせるかもしれない。これらのリスクには、外国に登録された患者が医療サービスや文化的慣習の違いにより臨床案を遵守できなかったこと、外国規制計画に関連する追加行政負担の管理、戦争を含むこれらの外国に関連する政治的·経済的リスクが含まれる。例えば,我々は現在米国以外の場所で何らかの臨床試験を行っており,米国以外の地点でPAHへのセラルチニブの登録第3段階研究を行う予定である。
また,われわれの臨床試験の首席研究員は時々私たちの科学コンサルタントやコンサルタントを務め,このようなサービスに関する報酬を得る可能性がある。場合によっては、私たちはFDAまたは同様の外国規制機関にいくつかの関係を報告することを要求されるかもしれない。FDAや同様の外国の規制機関は結論を出す可能性があり、私たちと主要な研究者との財務関係は利益の衝突をもたらしたり、他の方法でこの研究の解釈に影響を与えたりする。したがって,FDAや同様の外国の規制機関は,適用された臨床試験地点で発生するデータの完全性を疑問視する可能性があり,臨床試験自体の効用が脅かされる可能性がある。これは、FDAまたは同様の外国規制機関が私たちの上場申請の承認を遅延または拒否することを招き、最終的に私たちの1つ以上の候補製品が上場承認を拒否することにつながる可能性がある。
もし私たちが任意の候補製品の臨床試験の完了を遅延または終了すれば、私たちの候補製品のビジネスの将来性は損なわれ、これらの候補製品から製品収入を得る能力は延期されるだろう。また、臨床試験を完成するいかなる遅延も私たちのコストを増加させ、私たちの候補製品の開発と承認過程を緩和し、そして私たちの製品販売と収入を創造する能力を危険にさらす。
さらに、臨床試験の終了または一時停止、または臨床試験の開始または完了遅延をもたらす多くの要因も、最終的に候補製品の規制承認を拒否する可能性がある。私たちは私たちの候補製品の調合や製造面の変更を行うかもしれません。この場合、修正された候補製品を早期バージョンに関連付けるために、追加の臨床前研究を行う必要があるかもしれません。したがって、私たちの臨床試験に生じるどんな遅延も、候補製品を商業化する独占的な権利を持つ可能性のある任意の期限を短縮することができ、私たちの競争相手は私たちの前に製品を市場に出すかもしれません。私たちの候補製品の商業的可能性は著しく低下するかもしれません。このようなどんな状況でも、私たちの業務、財政状況、そして見通しに大きな被害を及ぼす可能性がある。
私たちの臨床試験に参加する患者を募集することは難しいかもしれない。われわれが臨床試験で被験者を募集する困難に遭遇した場合,われわれの臨床開発活動は延期されたり,他の悪影響を受けたりする可能性がある。
FDAや米国以外の同様の規制機関の要求に応じて、これらの試験に参加するのに十分な数の合格者を決定し、募集することができなければ、候補製品の臨床試験を開始または継続できない可能性がある。被験者登録は臨床試験時間の重要な要素であり、それは多くの要素の影響を受け、患者群の大きさと性質、患者と臨床場所の接近程度、試験の資格と排除標準、臨床試験の設計、登録した患者が臨床試験を完成できないリスク、著者らは適切な能力と経験を持つ臨床試験研究者と関連スタッフの能力、競争する臨床試験と臨床医師及び患者が研究している製品候補の他の既存療法に対する潜在的な優勢とリスクに対する見方を含む。我々が調査している適応のために承認される可能性のある任意の新薬および開発中の薬剤を含む。私たちは私たちのすべての臨床試験のために十分な数の被験者を決定し、募集することを要求される。任意の計画された臨床試験の潜在的被験体は、私たちの標的疾患として十分に診断されていないか、またはそのような試験の進入基準に適合していない可能性がある。例えば,PAHやPCNSLの影響を受ける患者数は限られており,PAHはシュラルチニブに対する目標適応であり,PCNSLはGB 5121に対する目標適応であり,従来のPAH患者の臨床試験で患者を募集することは困難であった。われわれが計画した臨床試験に適した疾患段階を有する被験者を決定·募集することや,治療期間や治療後にこれらの被験者を十分にモニタリングすることも困難である可能性がある。FDAや同様の外国の規制機関が要求する臨床試験に参加するのに十分な数の合格者を見つけることができなければ、臨床試験を開始または継続できない可能性がある。また,被験者を発見し診断する過程は高価であることが証明される可能性がある。
著者らの臨床試験の時間スケジュールは著者らが患者を募集して試験に参加する速度、及び必要な後続時期の完成状況にある程度依存する。私たちの候補製品にはセラルチニとPCNSL現在、著者らが評価を計画している状況は孤児或いは稀な疾患であり、臨床試験に供することができる患者池は限られている。われわれの臨床試験の資格基準が確立されると,利用可能な試験参加者をさらに制限する。患者が何らかの理由で私たちの試験に参加したくない場合、類似した患者集団に対する並行臨床試験の存在を含み、プラセボ対照設計を有する臨床試験または承認された治療方法に参加したくない場合、または十分な数の患者を募集することが困難である場合、被験者を募集し、研究を行い、規制部門の私たちの候補製品に対する承認を得るスケジュールが遅れる可能性がある。著者らは臨床試験サイトを活性化し、特定の試験の登録を開放することを決定し、あるいは臨床前研究を開始する能力も新冠肺炎を含む公衆衛生流行病の負の影響を受ける可能性がある。私たちは将来のどの臨床試験のために十分な数の被験者を募集することができず、重大な遅延を招くか、あるいは1つ以上の臨床試験を完全に放棄する必要があるかもしれない。また,CROと臨床試験地点により将来の臨床試験が適切かつタイムリーに行われることが予想され,彼らのサービスについて合意する予定であるが,彼らの実際の表現への影響は限られている。
予想される臨床試験スケジュールを決定するための仮定が正しいか、あるいは登録遅延に遭遇しないことは、このような試験の完了が予想されるスケジュールの後まで遅延することを保証することはできません。
私たちの候補製品の使用は、副作用、有害事象、または他の性質または安全リスクに関連する可能性があり、これは承認を延期または阻止する可能性があり、私たちが臨床試験を一時停止または停止させ、候補製品を放棄し、承認されたラベルの商業イメージを制限すること、または他の深刻な負の結果を招き、私たちの業務、将来性、運営結果、および財務状況を深刻に損なう可能性がある。
薬品の一般的な状況のように、私たちの候補製品の使用に関連する副作用や有害事象がある可能性が高い。われわれの臨床試験結果は副作用や予期せぬ特徴の重症度と流行度を示す可能性がある。私たちの候補製品による副作用は、私たちまたは規制機関の臨床試験の中断、延期、または停止を招く可能性があり、より厳しいラベル、またはFDAまたは同様の外国の規制機関が規制承認を延期または拒否する可能性がある。例えば,これまでに観察されてきた収益/リスクプロファイルから,GB 5121の1 b/2段階研究への参加を一時停止し,研究のデータ審査委員会と既存のデータを検討し,次の行動を決定することにした。薬物に関連する副作用は、患者の募集または患者の試験完了能力に影響を与える可能性があり、あるいは潜在的な製品責任クレームを招く可能性がある。このようなどんな状況でも、私たちの業務、財政状況、そして見通しに大きな被害を及ぼす可能性がある。
さらに、私たちの候補製品が臨床試験において副作用に関連している場合、または予期しない特徴を有する場合、私たちは、それらの開発を放棄することを選択するか、またはそれらの開発をより狭い用途または集団に制限することが可能であり、リスク-利益の観点から、副作用または他の特徴は、それほど一般的ではなく、それほど深刻ではなく、またはより容易に受け入れられ、これは、候補製品の商業的期待を制限する可能性がある。私たちはできる
われわれが行っている臨床試験の結果に基づいてわれわれの研究計画を修正する必要がある。例えば,セラルチニブは完成した臨床試験で全体的に耐性が良好であるにもかかわらず,将来の臨床試験は,われわれがPAH患者で予定しているセラルチニブ3期試験を含めて,不良事象がこれまで観察されてきた安全性発現と一致しない可能性がある。例えば,2013年のイマチニブ(Gleevec)PAHの3期臨床試験の結果,その主な奏効率は統計学的に有意に改善したが,全身毒性も認められた。イマチニブが完成段階の臨床試験で示した全身毒性は認められなかったが,セラルチニブがより大きな第3段階臨床試験で類似した毒性を示さないことは確認できなかった。早期試験で最初に希望を示した化合物の多くは後に副作用が認められ,化合物のさらなる発展を阻害した。さらに、規制当局は異なる結論を出すか、またはこれらの決定を確認するための追加的なテストを要求するかもしれない。
私たちがより大きく、より長く、より広範な臨床試験で私たちの候補製品を試験するとき、またはこれらの候補製品の使用がより広くなるにつれて(規制部門の許可を得た場合)、被験者は、早期試験で観察された疾患、傷害、不快感、および他の有害事象、および以前の試験で発生しなかったか、または検出されなかったことを報告する。これらの副作用が開発後期に、または承認された後に知られていれば、これらの発見は、私たちの業務、財務状況、および将来性に大きな損害を与える可能性がある。
さらに、もし私たちの1つ以上の候補製品が発売許可を得て、私たちまたは他の人が後にこのような製品による不良副作用を発見した場合、多くの潜在的な重大な負の結果を招く可能性がある
•規制部門は、このような製品の承認を撤回、一時停止、または制限することができる
•製品をリコールしたり患者に投与する方法を変更するように要求されるかもしれません
•規制当局は、“ブラックボックス”警告またはタブーのようなラベルに警告を追加することを要求するかもしれない
•私たちは、患者に配布するために、または同様のリスク管理措置のために、そのような副作用のリスクを概説するために、リスク評価および緩和策を実施すること、または薬物ガイドラインを作成することを要求される可能性がある
•私たちは、製品の流通や管理方法を変更し、追加の臨床試験を行うこと、または製品のラベルを変更すること、または追加の発売後の研究または監視を要求されることが要求されるかもしれない
•私たちは起訴され、患者への傷害に責任を負うかもしれない
•製品の販売が大幅に低下する可能性があり、あるいは製品の競争力が低下する可能性がある
•私たちの名声は損なわれるかもしれない。
これらの事件のいずれも、特定の候補製品に対する市場の受け入れ度を達成または維持することを阻止することができ、承認されれば、私たちの業務、運営結果、および将来性を深刻に損なう可能性がある。
セラルチニブを含む複数の候補製品の第2段階臨床試験が完了しているにもかかわらず,組織としては後期臨床試験を完了していないか,秘密協定を提出しておらず,どの候補製品にもそうすることができないかもしれない。
私たちは私たちの候補製品の早期開発を行っています。私たちはFDAなどの外国規制機関の許可を得て、seralutinib、GB 5121、あるいは任意の未来の候補製品を発売するために、後期と重要な臨床試験を成功させる必要があります。後期臨床試験を行い、成功した秘密保持協定または他の同様の外国監督文書を提出することは複雑な過程である。1つの組織として,シュラチジンの2期臨床試験を含む4つの2期臨床試験が完了し,GB 5121の1 b/2期研究が行われており,現在登録を中止している。2023年下半期にサラダルーティンの3期臨床試験を開始する予定である。我々の候補製品に対してはどのような後期あるいは重要な臨床試験も行われておらず,我々の複数の臨床前計画の臨床開発も開始されていない。会社として、規制申告書類の準備、提出、起訴に関する経験も限られており、これまでどの製品候補にも秘密協定や他の類似した外国監督申告書を提出したことはない。今後数年以内に複数の候補製品のための複数の臨床試験を同時に行う予定であり,限られた資源で管理する困難な過程である可能性があり,経営陣の注意を分散させる可能性がある。また,FDAとのインタラクションは限られており,追加的なセラルチニブや任意の他の候補製品の臨床試験がどれだけ必要か,あるいはこれらの試験をどのように設計すべきかは決定できない。だから私たちは
必要な臨床試験を成功的かつ効率的に実行し、完成させることができず、規制部門が私たちの任意の候補製品を提出し、承認することを招く可能性がある。私たちは私たちの競争相手よりも多くの時間とより多くのコストを必要とするかもしれないし、私たちが開発した候補製品の規制承認を得ることに成功しないかもしれない。我々の計画した臨床試験を開始または完了または遅延させることができず、NDAまたは他の同様の外国規制機関が私たちの候補製品を提出して商業化することを阻止または延期する可能性がある。
私たちの候補製品は広範な規制とコンプライアンスを受けなければならない。これは高価で時間がかかり、このような規制は予期せぬ遅延を招き、あるいは私たちの候補製品を商業化するために必要な承認を受けることを阻止する可能性がある。
著者らの候補製品の臨床開発、製造、ラベル、貯蔵、記録保存、広告、販売促進、輸出入、マーケティングと流通はすべてアメリカFDAと国外市場の類似外国監督管理機関の広範な監督管理を受けている。米国では、FDAの規制承認を得るまで、私たちの候補製品の販売は許可されていませんし、同様に、外国規制機関の承認を得るまでは、私たちの候補製品の販売も許可されていません。監督管理の承認を得る過程は費用が高く、通常臨床試験開始後数年後に必要であり、しかも関連する候補製品のタイプ、複雑性と意外性及び目標適応と患者群によって大きく異なる可能性がある。審査政策或いは法規は変化する可能性があり、FDAと外国の監督管理機関は薬品の審査過程中にかなりの自由裁量権を持っており、多種の原因で延期、制限、或いは承認候補製品を拒否する権利がある。候補製品の臨床開発に時間と費用が投入されているにもかかわらず、監督管理部門の承認は永遠に保証されない
承認された候補製品を米国または海外で商業化する前に、十分かつ制御された臨床試験によって大量の証拠を提供し、FDAまたは同様の外国の規制機関に、これらの候補製品がその予期される用途に対して安全かつ有効であることを満足させなければならない。非臨床研究と臨床試験の結果は異なる方法で解釈できる。私たちの候補製品の非臨床的または臨床的データが有望だと信じていても、これらのデータはFDAや同様の外国規制機関の承認を支持するのに十分ではないかもしれない。FDAまたは同様の外国の規制機関は、状況に応じて、承認前または承認後に、私たちの候補製品に対して追加の臨床前研究または臨床試験を行うこと、または私たちの臨床開発計画の要素に反対することを要求する可能性もある。
FDAまたは同様の外国の規制機関は、多くの理由で、承認候補製品を延期、制限、または拒否することができる
•これらの機関は私たちの臨床試験の設計や実施に同意しないかもしれません
•我々の臨床試験または結果の陰性または不明確な結果は、FDAまたは同様の外国規制機関によって承認された統計的意味レベルに適合しない可能性がある
•私たちの臨床試験の参加者や候補品に類似した薬剤を使用した個人は、深刻で予期しない薬物関連副作用を経験するかもしれません
•臨床試験で研究されている集団は、承認を求めるすべての集団の安全性を確保するのに十分な広汎性または代表性がないかもしれない
•これらの当局は、臨床施設または看護基準が米国とは異なる国で行われる可能性のある試験の臨床データを受け入れない可能性がある
•候補製品の臨床的および他の利益がその安全リスクよりも大きいことは証明できないかもしれない
•これらの機関は前臨床研究や臨床試験データの説明に同意しないかもしれません
•これらの機関は、私たちの候補製品の臨床試験から収集されたデータが受け入れ可能であるか、NDAまたは他の提出の提出を支持するのに十分であるか、または米国または他の場所の規制の承認を得ることに同意しない可能性があり、これらの機関は、追加の臨床前研究または臨床試験を要求する可能性がある
•これらの機関は、私たちの候補製品の配合、ラベル、および/または仕様に同意しないかもしれない
•私たちが申請したよりも限られた適応および/または配布および使用において他の重大な制限がある適応のみが承認される
•これらの機関は、私たちが臨床および商業用品契約を締結した第三者製造業者の製造プロセスまたは施設に欠陥があることを発見するか、または政策を承認するかもしれない
•これらの機関の規制は、私たちまたは未来の潜在的協力者の臨床データが承認されるのに十分ではないかもしれません
•その他の理由を除いて、このような主管機関は提出書の内容やフォーマットを受け入れない可能性がある。
海外市場については、審査手続きは国によって異なり、上記のリスクに加え、追加の製品テスト、行政審査期限、価格主管部門との合意に及ぶ可能性がある。また、ある上場薬品の安全性に対する疑問を引き起こす事件はFDAと類似の外国の監督管理機関が安全性、有効性或いはその他の監督管理に基づいて新薬を審査することを考慮する時に更に慎重であり、そして監督管理の許可を得る重大な遅延を招く可能性がある。適用可能な規制承認を得るか得られないかのいずれの遅延も、私たちまたは未来の任意の潜在的パートナーが私たちの候補製品を商業化することを阻止するだろう。
大量に開発されている薬物のうち,一部のみがFDAや外国規制機関の承認手続きに成功し,商業化されている。長い承認過程、そして未来の臨床試験結果の予測不可能性は、私たちが監督部門の承認を得ることができず、私たちの候補製品を市場に出すことができなくなり、これは私たちの業務、財務状況、運営結果、将来性を深刻に損なうだろう。
最終的に臨床試験を完了し、NDAまたは海外マーケティング申請の承認を得ても、FDAまたは同様の外国規制機関は、第4段階臨床試験を含む高価な追加臨床試験の表現に基づいて承認される可能性があり、および/またはREMSまたは同様のリスク管理措置を実施することは、承認後の薬物の安全な使用を確保するために必要である可能性がある。FDAまたは同様の外国規制機関は、私たちが最初に要求したよりも限られた適応や患者数の候補製品を承認することも可能であり、FDAまたは同様の外国規制機関は、製品の商業化に成功するために必要または望ましいラベルであると考えている可能性がある。適用可能な規制承認を得るか得られないかのいずれの遅延も、候補製品の商業化を延期または阻止し、私たちの業務や将来性に大きな悪影響を及ぼすだろう。
私たちは、承認を加速するルートを使用することで、FDAや同様の外国規制機関の承認を得ることを試みるかもしれない。もし私たちがそのような承認を得ることができなければ、私たちは予想以上の追加臨床試験を要求される可能性があり、これは必要な上場承認を得る費用を増加させ、必要な上場承認を遅らせる可能性がある。FDAの承認を得ても検証的な試験が臨床的利益を証明していなければ 私たちは厳格な発売後の要求を守らず、FDAは承認撤回の加速を求めるかもしれない。
私たちは未来に私たちの1つ以上の候補製品の承認を加速させることを求めるかもしれない。加速承認計画によれば、FDAは、代替終点または中間臨床終点に対して臨床的利益を合理的に予測することができる候補製品を決定する場合に、既存の治療法よりも意義のある治療利益を提供する、深刻または生命に危険な疾患の治療のための候補製品の承認を加速することができる。FDAは臨床利益は特定の疾病の背景下で臨床意義のある積極的な治療効果であり、例えば不可逆的な発病率或いは死亡率であると考えている。承認を加速するために、代替終点は1つの標識であり、例えば実験室測定、放射画像、バイタルサイン或いは他の臨床利益を予測できると考えられる指標であるが、それ自体は臨床利益の測定基準ではない。中間臨床終点は不可逆的発病率或いは死亡率への影響の前に測定できる臨床終点であり、それは不可逆的な発病率或いは死亡率或いは他の臨床利益に対する影響を合理的に予測する可能性がある。加速承認経路は既存療法に対する新薬の優位性は直接の治療優位ではないかもしれないが、患者と公衆衛生の観点から見ると臨床上重要な改善状況である。承認された場合、承認を加速することは、一般に、薬剤の予期される臨床的利益を確認および説明するために、勤勉な方法で検証的研究を行うことにスポンサーが同意するかどうかに依存する。もしこのような検証性研究がこの薬物の期待された臨床治療効果を検証できなかった場合、或いは発起人が適時にこのような研究を行うことができなかった場合、FDAは迅速にこの薬物に対する承認を撤回することができる。また、2022年12月、総裁·バイデンは2023年度まで米国政府に資金を提供する総合支出法案に署名した。総合法案には“2022年食品·薬物総合改革法案”が含まれており、この法案は他にも導入されている
FDAによる検証性試験の監視を強化することを含む、承認を加速された製品に対するFDAの監督能力を拡大するが、これらの改革の最終的な影響はまだ不明である。
私たちの任意の候補製品の加速承認を求める前に、FDAのフィードバックを求めるつもりです。そうでなければ、私たちが承認を求めて加速させる能力を評価します。フィードバックや他の要因を評価した後、加速承認または任意の他の形態の加速開発または審査計画を求めるために、機密協定を継続または提出することを決定する保証はありません。また、加速的な承認を求める申請を提出することを決定した場合、このような提出または申請が受け入れられる保証もなく、加速された開発、審査、または承認がタイムリーに、または全く承認されない保証もない。FDAまたは他の同様の外国規制機関も、私たちの申請を考慮したり、任意の種類の申請を承認する前にさらなる検討を要求することができる。もし私たちの候補製品が加速承認または任意の他の形態の加速開発、審査または承認を得られなかった場合、候補製品の商業化の時間がより長くなり(あれば)、候補製品の開発コストを増加させ、市場での競争地位を損なう可能性がある。
また、EUでは、必要なすべての安全性や有効性データが取得されていない場合には、“条件付き”上場許可が付与される可能性がある。条件付きマーケティング許可は、欠落データを生成すること、またはセキュリティ対策を増加させるために必要な条件を保証することに依存する。条件付きマーケティング許可の有効期間は1年であり、すべての関連条件が満たされるまで年に1回更新しなければならない。適用可能な未定研究が提供されると、条件付きマーケティング許可は“標準”マーケティング許可となることができる。しかしながら、EMAによって設定された時間範囲内で条件が満たされていない場合、マーケティング許可は更新を停止する。
また、“特殊な場合”は、出願人が正常な使用条件であることを証明することができれば、製品が許可され、特定のプログラムを採用しなければならない後であっても、その有効性や安全性に関する包括的なデータを提供することができなければ、上場を承認することができる。予想される適応が非常にまれであり,現在の科学的知識状態では網羅的な情報を提供することが不可能である場合や,データを生成することが一般的に受け入れられている倫理原則に違反する可能性がある場合がある。このようなタイプのマーケティング許可は、深刻な疾患または満たされていない医療需要のために承認されるべき医薬品のために保持されているので、条件付きマーケティング許可に近いが、出願人は、マーケティング許可を付与するために必要な法定完全データセットを有していない。しかしながら、条件付きマーケティング許可とは異なり、出願人は、失われたデータを提供する必要はなく、提供する必要はない。“特殊な場合”の上場許可は明確であるが,毎年薬品のリスク−収益バランスを審査しており,リスク−収益比が有利でなければマーケティングライセンスを撤回する可能性がある。
我々は臨床的に複数の候補製品を有し,様々な目標適応を考慮しているため,より利益や成功可能性の高い製品候補や適応を利用することはできない限られた資源をかけて特定の候補製品を追求する可能性がある。
私たちの財務と管理資源が限られているので、私たちは特定の候補製品、適応、開発計画に集中しています。我々は現在2つの候補製品が臨床開発中であり,今後数年以内にseralutinibに関するいくつかの臨床試験を同時に行う予定であり,どの候補製品に集中するかを決定することが困難になる可能性がある。したがって、私たちは他の候補製品を探す機会を放棄したり延期したりするかもしれないが、これらの機会はより大きなビジネス潜在力を持つことができる。例えば,我々はこれまでパートナーがいない場合には,さらなる臨床試験でGB 001やそのバックアップ分子を使用しなくなり,GB 004のさらなる開発を終了することにしてきた。私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。私たちの現在と未来の研究開発計画および特定の適応候補製品への支出はいかなる商業的に実行可能な製品も発生しないかもしれない。もし私たちが特定の候補製品の商業的潜在力や目標市場を正確に評価しなければ、私たちは将来の協力、許可、その他の同様の計画を通じて候補製品に価値のある権利を放棄するかもしれないが、この場合、候補製品の独占的な開発と商業化の権利を維持することは私たちに有利である。
さらに、追加的な許可内や開発段階の資産や計画を買収することが求められる可能性があり、これは私たちに追加的なリスクをもたらすだろう。将来性のある候補製品を確定、選別、獲得するには大量の技術、財政と人的資源の専門知識が必要である。このような努力は、可能な特定の候補製品を実際に獲得することを招くことはなく、何のメリットも生じることなく、私たちの管理職の時間と資源支出の分流をもたらす可能性がある。例えば、最終的に製品の承認につながる計画を決定できなければ、最終的に投資収益を提供できない製品を評価、買収、開発するために大量の資本や他の資源がかかる可能性がある。
私たちは私たちのいくつかの候補製品の孤児薬物指定を獲得または維持することができない可能性があり、私たちは潜在的な市場排他性を含む孤児薬物指定に関する利点を維持できないかもしれない。
米国やEUを含むいくつかの管轄区の規制機関は、患者数が比較的少ない薬物を孤児薬に指定する可能性がある。1983年の“孤児医薬品法”によると、1つの製品がまれな疾患または疾患の治療を目的としている場合、FDAはこの製品を孤児製品に指定することができる。米国では、まれな疾患または疾患は、一般に患者数が20万人未満であると定義されているか、または米国では患者数が20万人を超えると定義されているが、薬物開発のコストは米国の販売から回収されるという合理的な期待はない。EUにおいて、欧州委員会は、EMAの孤児医薬製品委員会の意見に基づいて、以下の製品の開発を促進するために孤児の称号を付与する:(1)生命または慢性衰弱に危険な製品の診断、予防または治療を目的とした製品、および(2)または(A)申請を提出したとき、EUで10,000人を超えない5人に影響を与えるか、または(B)孤児の身分によるメリットがなければ、必要な投資が合理的であることを証明するためにEUで十分な見返りを生じる可能性が低い、および(3)満足できる診断方法がない、EU市場で販売されているこのような疾患の予防または治療を許可するか、または、そのような方法が存在する場合、薬物は、そのような疾患の影響を受ける人に顕著な利益を与えなければならない。私たちはすでにアメリカとEUでPAHを治療するセラルチニブの孤児薬物の称号を得ており、私たちは私たちの他のいくつかの候補製品のために孤児薬物の称号を求めるかもしれない。FDAやECが私たちが申請したいかなる適応にも孤児の称号を与える保証はなく、このような呼称を維持できる保証もない。
米国では,孤児は一方に臨床試験費用,税収割引,ユーザ費用減免のために贈与資金を提供する機会など,財政的インセンティブを得る権利があることを指定している。さらに、孤児指定を有する候補製品がその後、そのような指定された疾患または条件を有するFDAの最初の承認を得た場合、この製品は、孤児薬物排他性を得る権利があり、これは、FDAがNDAを含む他の出願を承認しない可能性があり、限定された場合、例えば孤児薬物に対して排他的な製品に対する臨床的利点を示すことができない限り、または製造業者が十分な製品数を保証することができない限り、7年以内に同じ疾患または条件の同じ薬剤を販売することを意味する。EUがマーケティング許可を得た後、孤児薬品は10年間の市場排他性を有する権利があり、その間、同じ適応の類似薬品は市場に投入されてはならない。5年目の終了時に、製品が市場排他性の維持が合理的であることを証明するのに不十分であること、または疾患の流行率が限界値を超えていることを証明することを含む、孤児指定基準に適合しないと判定された場合、この期間は6年に短縮されることができる。
私たちが製品の孤児薬物排他性を獲得しても、この排他性は、異なる薬物が同じ条件で承認される可能性があるため、競争から製品を効果的に保護することができない可能性がある。孤児薬物が承認された後であっても、FDAまたは同様の外国監督機関はその後、同じ薬物が同じ疾患を治療することを許可することができ、監督機関が結論を出すことを前提としており、後者がより安全で、より有効であることが証明され、或いは患者看護に重大な貢献がある場合、後者は臨床的により優れている。孤児薬物を指定することは薬物の開発時間や監督審査時間を短縮することもなく、監督審査或いは承認過程において薬物にいかなる利点をもたらすこともない。
私たちは現在私たちの製品候補製品のためにアメリカ以外でいくつかの臨床試験を行っており、将来的にも行われる可能性がある。しかし、FDAや他の外国人業者は、このような試験からのデータを受け入れない可能性があり、この場合、私たちの開発計画が延期され、私たちの業務に実質的な損害を与える可能性があります。
私たちは現在進行中であり、将来的にアメリカ以外の候補製品のための1つ以上の臨床試験を行うことができる。FDAや同様の外国規制機関が米国や他の管轄地域以外で行われている臨床試験を受けた研究データは,何らかの条件によって制限される可能性があり,まったく受け入れられない可能性もある。外国の臨床試験からのデータが米国での上場承認の唯一の根拠となることを意図している場合、FDAは通常、(I)データがアメリカの人口とアメリカの医療実践に適用されない限り、外国のデータのみに基づいて申請を承認しない。(Ii)試験は公認能力を有する臨床研究者によって行われ、GCP規定に適合する。および(Iii)データは、FDAによる現場検査を必要とすることなく、有効であると考えることができ、またはFDAがこのような検査を行う必要があると考えた場合、FDAは、現場検査または他の適切な手段によってデータを検証することができる。また,INDに拘束されていないこのような臨床試験については,FDAはこれらのデータを上場承認申請の支援として受け入れず,本研究がGCP要求に基づいて行われない限り,FDAは現場検査により研究データを検証することができる(必要であれば)。多くの外国の規制機関もまた似たような承認要求を持っている。また、このような外国裁判は、裁判を行う外国司法管轄区域に適用される現地法によって管轄される。FDAや同様の外国規制機関が米国や適用司法管轄区域以外で行われた試験データを受け入れることは保証されない。FDAまたは同様の外国の規制機関がこれらのデータを受け入れない場合、追加の試験が必要になり、これは高価で時間がかかる可能性があり、私たちが開発する可能性のある現在または未来の候補製品が適用される司法管轄区域で商業的な承認を得ることができない可能性がある
アメリカ国外で臨床試験を行うことも、以下の方面と関連するリスクを含むより多くのリスクに直面させる
•他の外国の規制要件;
•外国為替変動
•海外の製造、税関、輸送、倉庫の要求を守る
•医療実践と臨床研究における文化的違い
•一部の国では知的財産権の保護力が弱まっている
•私たちの裁判は戦争やテロなどの地政学的事件によって中断されたり遅延されたりする。
私たちが時々発表或いは公表した臨床試験の中期、背線と初歩データはより多くの患者データの獲得に従って変化する可能性があり、そして監査と検証プログラムの制約を受け、これらのプログラムは最終データの重大な変化を招く可能性があり、あるいは私たちは更なる臨床開発を行わなくなる可能性がある。
私たちは時々私たちの臨床研究の初歩的または主要な結論またはデータを公開するかもしれないが、これらのデータは当時利用可能なデータの初歩的な分析に基づいており、結果および関連する発見および結論は、特定の研究または試験に関連するデータをより全面的に検討した後に変化する可能性がある。私たちはまた、私たちのデータ分析の一部として、すべてのデータを全面的かつ詳細に評価する機会がないか、または受け取る機会がないかもしれないという仮説、推定、計算、および結論を出した。したがって、私たちの報告書の初期結果またはバックライン結果は、同じ研究の将来の結果とは異なる可能性があり、またはより多くのデータを受信して十分に評価されると、異なる結論または考慮要因がこれらの結果を合格させる可能性がある。バックラインと初歩データは依然として監査とチェック手続きを受けなければならず、これは最終データが以前公表された予備データと大きく異なる可能性がある。したがって、最終データが利用可能になる前に、バックラインおよび予備データは慎重に表示されなければならない。時々、私たちはまた私たちの臨床研究の中間データを開示するかもしれない。著者らが完成する可能性のある臨床試験の中期データは、患者登録の継続とより多くの患者データの獲得に伴い、1つ以上の臨床結果が実質的に変化する可能性があるというリスクに直面している。バックライン、初期データ、または中間データと最終データとの間の不利な差は、私たちのビジネスの将来性を深刻に損なう可能性があります。
さらに、規制機関を含む他の人は、私たちの仮定、推定、計算、結論または分析を受け入れないか、またはデータの重要性を異なる方法で解釈またはトレードオフする可能性があり、これは、特定の計画の価値、特定の候補製品または製品の承認または商業化、およびわが社の全体的な状況に影響を与える可能性がある。さらに、私たちが開示された特定の研究または臨床試験に関する情報を選択することは、一般に、広範な情報に基づいており、他の人は、私たちが決定した重大な情報または他の適切な情報に同意しない可能性があり、私たちが開示しないことを決定する任意の情報は、最終的に、特定の薬剤、候補薬剤、または私たちの業務に関する将来の決定、結論、観点、活動、または他の側面に対して大きな意味を有すると考えられるかもしれない。もし私たちの報告書の裏線、初歩的または中間データが実際の結果と異なる場合、または規制機関を含む他の人が結論に同意しない場合、私たちが承認を得て私たちの候補製品を商業化する能力が損なわれる可能性があり、これは私たちの業務、運営結果、将来性、または財務状況を損なう可能性がある。
資金不足や世界的な健康懸念によるFDAや他の政府機関の中断は、重要な指導部や他の人員の採用、保留、配置の能力を阻害する可能性があり、あるいは新たなまたは修正された製品の開発、承認、商業化を他の方法で阻止することは、私たちの業務にマイナスの影響を与える可能性がある。
FDAおよび外国規制機関が新製品を審査·承認または承認する能力は、政府予算および資金レベル、法定、規制および政策変化、FDAまたは外国規制機関のキーパーソンの雇用および保留、ユーザ費用支払いを受け入れる能力、およびFDAが通常の機能を履行する能力に影響を与える可能性のある他の事件を含む様々な要素の影響を受ける可能性がある。したがって、FDAと外国規制機関の平均審査時間は近年変動している。また,他の研究開発活動を援助する政府機関への政府の援助は政治過程の影響を受けており,この過程は本質的に不安定で予測不可能である。アムステルダムへの移転後のEMAおよびそれに伴う人員変動のようなFDAおよび他の機関の中断は、新薬または承認または承認された薬物および生物製品の修正が必要な政府機関による審査および/または承認に要する時間を遅らせる可能性もあり、これは私たちの業務に悪影響を及ぼす。例えば、ここ数年間、米国政府は何度か閉鎖されており、FDAのようないくつかの規制機関は、FDAのキー従業員を休暇にし、キー活動を停止しなければならない
また、新冠肺炎の流行に対応するため、アメリカ食品薬品監督管理局は国内外の異なる場所の製造施設の大部分の検査を延期した。FDAはすでに実行可能な情況下で国内施設に対する標準検査操作を回復したが、FDAは依然としてその検査活動の変化を監視と実施し、その従業員及び監督会社の安全を確保し、絶えず変化する新冠肺炎疫病に適応し、ウイルスの灰再発或いは新変種の出現は更なる検査遅延を招く可能性がある。新冠肺炎疫病に対して、アメリカ以外の監督管理機関も類似した制限或いはその他の政策措置を取った。政府が長期的に停止している場合、または世界的な健康問題がFDAまたは他の規制機関の定期検査、審査または他の規制活動を阻害し続けている場合、FDAまたは他の監督管理機関が私たちの規制提出を適時に審査し、処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。
私たちの第三者への依存に関するリスクは
著者らは第三者に依存して多くの臨床前研究と臨床試験を行った。もし第三者がGCPと他の要求に基づいて適時に臨床試験を行うことができない場合、私たちが監督部門の私たちの候補製品に対する承認を求めたり、商業化したりすることを遅延したり阻害したりする可能性がある。
私たちは第三者に依存して私たちの臨床試験と臨床前研究を行い、私たちが行っているあるいは未来に行われる可能性のあるシュラルチニブとGB 5121の臨床試験、および私たちの複数の臨床前開発計画の臨床前研究を含む。具体的には,我々は使用·依存しており,医療機関,臨床研究者,CRO,コンサルタントを継続して使用·依存し,われわれの臨床案や法規の要求に基づいて臨床試験を行う予定である。これらのCRO、調査者、その他の第三者はこれらの実験の進行とスケジュール、その後のデータ収集と分析に重要な役割を果たしている。我々は我々の第三者請負業者の活動を管理する合意があるが,彼らの実際の表現への影響は限られている.しかし、私たちは私たちのすべての臨床試験が適用された方案と法律、法規、科学的基準に従って行われていることを確実にする責任があり、私たちのCROと他の第三者への依存は私たちの規制責任を免除しない。私たちと私たちのCROはGCP要求を遵守しなければならない。これらの要求は、FDAと同様の外国規制機関が私たちの臨床開発におけるすべての候補製品に対して実行する法規とガイドラインである。規制当局は,試験スポンサー,主要調査者,試験地点を定期的に検査することでこれらのGCPを実行している。もし私たちまたは私たちの任意のCROまたは試験サイトが適用されたGCPに従わなかった場合、私たちの臨床試験で生成された臨床データは信頼できないと考えられるかもしれませんが、FDAまたは同様の外国の規制機関は、私たちのマーケティング申請を承認する前に追加の臨床試験を行うことを要求するかもしれません。また,われわれの臨床試験ではcGMPや類似法規により生産された製品を使用しなければならない。私たちがこれらの規定を遵守しないことは、私たちが臨床試験を繰り返す必要があるかもしれないが、これは規制部門の承認過程を遅らせるだろう。
このようなCRO、調査者、または他の第三者が、そのような試験に十分な時間およびリソースを投入するか、または契約要件に従って責務を履行することは保証されない。もしこれらの第三者のいずれかが予想される最終期限内に達成できなかった場合、私たちの臨床方案を遵守したり、規制要求を満たしたり、あるいは他の方法で不合格を示した場合、私たちの臨床試験は延長、延期、または終了される可能性がある。また、私たちと契約した多くの第三者も、私たちの競争相手を含む他の商業実体と関係があるかもしれません。彼らはまた、これらの実体のための臨床試験や他の薬物開発活動を行っているかもしれません。これは、私たちの競争地位を損なう可能性があります。さらに、私たちの臨床試験の首席研究者は時々私たちの科学顧問や顧問を担当し、このようなサービスのために現金または株式補償を得ることができるかもしれない。これらの関係および任意の関連賠償が知覚的または実際的な利益の衝突をもたらし、またはFDAまたは外国の規制当局が、財務関係が研究の解釈に影響を及ぼす可能性があると結論した場合、適用される臨床試験場所で生成されたデータの完全性が問われる可能性があり、臨床試験自体の効用が脅かされる可能性があり、これは、FDAまたは外国規制機関が提出した任意のNDAまたは同様の外国出願を延期または拒否する可能性がある。そのような遅延や拒否は私たちが候補製品を商業化することを防ぐことができる。
もし私たちがこれらの第三者との任意の関係が終了したり、彼らのサービスが遅延したりすれば、私たちは他の第三者と合意できないかもしれないし、商業的に合理的な条項でそうすることができないかもしれない。例えば、著者らは中国のCROに依存してある臨床前研究を行ったが、新冠肺炎の発生前に中国CROと行ったある臨床前仕事を延期した。CRO、調査者、および他の第三者の交換または追加は、管理時間および重点を必要とする追加のコストに関連しています。しかも、新しいCROが仕事を始める時、自然な過渡期がある。したがって,遅延が生じ,必要な臨床や臨床前開発スケジュールを満たす能力に大きな影響を与える可能性がある。CRO、調査者、その他の第三者との関係を慎重に処理しているにもかかわらず、将来的に挑戦や遅延に遭遇しない保証はなく、これらの遅延や挑戦が私たちの業務、財務状況、および見通しに実質的な悪影響を与えないことを保証することはできない。
私たちは第三者に依存して私たちの臨床と臨床前に候補製品を開発し、予測可能な未来にこのようにしていきたい。このような第三者への依存は、許容可能なコストで十分な数の候補製品または製品またはそのような数を得ることができないリスクを増加させ、これは、私たちの開発または商業化努力を延期、阻止、または損害する可能性がある。
私たちは製造施設を所有したり運営したりすることもなく、自分の臨床や商業規模の製造能力を構築する計画もない。私たちは依存し、引き続き第三者が私たちの候補製品と臨床および臨床前開発のための関連原材料を生産し、商業生産に依存することが予想される(任意の候補製品が市場の承認を得たら)。第三者メーカーが我々の候補製品を生産するための施設は、FDAまたは外国規制機関の承認を得なければならず、検査は、我々がFDAに機密協定を提出した後、または外国規制機関に同様の申請を提出した後に行われる。我々は第三者メーカーの生産過程を制御せず、第三者メーカーがcGMPまたは同様の薬品生産要求を遵守することに完全に依存している。これらの第三者製造業者が、効率的な化合物の製造に関連する要求を含む、当社の規範およびFDAまたは他の機関の厳格な規制要件に適合する材料を成功裏に製造できない場合、彼らはその製造施設の規制承認を確保および/または維持することができないであろう。また、第三者製造業者が十分な品質管理、品質保証、合格者の能力を維持することを制御することはできない。FDAや同様の外国規制機関が私たちの候補製品を生産するためにこれらの施設を承認しない場合、または将来的にそのような承認を撤回すれば、代替製造施設を探す必要があるかもしれません。これは、私たちが規制機関の承認を得たり、私たちの候補製品を販売したりする能力に深刻な影響を与えます。私たちまたは私たちの第三者製造業者が適用された法規を遵守できなかったことは、臨床封印、罰金、禁止、民事処罰、遅延、承認の一時停止または撤回、候補製品または製品の差し押さえまたはリコール、運営制限、および刑事起訴を含む制裁を実施する可能性があり、いずれも私たちの製品供給に重大な悪影響を及ぼす可能性があります。
もし私たちまたは第三者が私たちの製造要求を実行せず、商業的に合理的な条項で実行されなければ、アメリカ国外のcGMPや同様の要求に従わなければ、多くの点で私たちの業務に悪影響を及ぼす可能性があります
•臨床試験を開始したり続けたりすることはできませんセラルチニGB 5121または将来開発されている任意の候補製品;
•規制申請の提出を遅延させたり、私たちの製品候補製品の上場承認を遅延させたりします
•第三者製造施設や私たちの製造施設に対して規制部門の追加検査を行う
•私たちの候補製品ロットの開発中止やリコールを要求します
•私たちの候補製品が発売され商業化された場合、私たちの候補製品や他の未来の候補製品の商業的需要を満たすことができません。
しかも、私たちは第三者製造業者と長期的な約束や供給協定を持っていない。私たちは、第三者製造業者と任意の供給プロトコルを確立することができないか、または許容可能な条項でそうすることができない可能性があり、これは、私たちの候補製品または製品をタイムリーに得ること、または許容可能なコストでそのような数を得るのに十分な量のリスクを増加させる。たとえ第三者製造業者と合意できても、第三者メーカーに依存することは、追加的なリスクをもたらす
•第三者製造業者は規制要件を遵守し、品質保証を維持できなかった
•第3者は製造協定に違反した
•私たちの規格に沿って私たちの製品を作っていません
•私たちの計画通りに私たちの製品を生産しなかったか、あるいは全くなかった
•私たちのビジネス秘密とノウハウを含む私たちの固有情報を盗用します
•第三者は、私たちにとって費用が高い場合、または不便な場合に、プロトコルを終了または更新しない。
私たちの候補製品と私たちが開発する可能性のあるどの製品も他の候補製品や製品と製造施設を競争する可能性があります。CGMPや類似の外国法規の下で運営されているメーカーの数は限られており、私たちのために製品を製造する能力があるかもしれない。
私たちの現有或いは未来のメーカーのいかなる表現も臨床開発或いは発売承認を延期する可能性があり、いかなる関連する救済措置の実施はコストが高いか、あるいは時間がかかる可能性がある。私たちはまだ私たちの候補製品を生産するために必要なすべての原材料に余分な供給源や二番目の供給源を提供するように手配していません。さらに、私たちの第三者メーカーは、資源制限や自然災害、労使紛争、不安定な政治環境または公衆衛生流行病(例えば、最近の新冠肺炎流行)によって製造や輸送困難に遭遇する可能性がある。もし私たちの現在の第三者製造業者が合意通りに履行できなければ、私たちはこれらのメーカーの交換を要求されるかもしれません。私たちは彼らをタイムリーにあるいは交換できないかもしれません。
私たちの現在と未来の他人が私たちの候補製品や製品を生産することへの依存は、私たちの将来の利益率と、マーケティングの承認をタイムリーかつ競争力的に獲得する製品の商業化能力に悪影響を及ぼす可能性があると予想されています。
私たちの第三者への依存は、私たちのビジネス秘密を共有することを要求し、これは、競争相手がこれらの秘密や私たちのビジネス秘密が流用または開示されていることを発見する可能性を増加させる。
私たちは現在、他の第三者に依存して私たちの候補製品を生産し、品質テストを行っているため、ビジネス秘密を含む私たちのノウハウと機密情報を彼らと共有しなければならない場合があります。私たちは、独自の情報の研究または開示を開始する前に、当社のコンサルタント、従業員、およびコンサルタントと秘密協定、コンサルティング契約、または他の同様の合意を締結することによって、私たちのノウハウを保護することを求めています。このような協定は一般に第三者が私たちの機密情報を使用または開示する権利を制限する。第三者と協力する際に契約条項が採用されているにもかかわらず、商業秘密および他の機密情報を共有する必要は、そのような商業秘密が私たちの競争相手に知られ、意図的にまたは意図的に他の人に組み込まれているか、またはこれらの合意に違反する方法で開示または使用されるリスクを増加させる。私たちの独自の地位が私たちのノウハウおよびビジネス秘密にある程度基づいていることを考慮すると、私たちはビジネス秘密を保護しようと努力しているにもかかわらず、競争相手は私たちのノウハウや機密情報、または他の許可されていない使用または開示が私たちの競争地位を損なうことを発見し、私たちの業務、財務状況、運営結果、および将来性に大きな悪影響を及ぼす可能性がある。
私たちは協力、許可、他の似たような計画を達成することを求めるかもしれないが、成功しないかもしれないし、私たちが成功しても、私たちはこのような関係の利点を意識していないかもしれない。
候補製品の開発や商業化に要する資本コストや製造制限により、我々は現在、我々の候補製品を開発または商業化するために、協力、合弁企業、ライセンス、その他の同様の手配を求めている。私たちの候補製品のためのこのような協力を確立または維持する努力は成功しないかもしれません。なぜなら、私たちの研究開発ルートが不十分である可能性があり、私たちの候補製品は協力努力の開発段階が早すぎると思われるかもしれません。あるいは第三者は私たちの候補製品が安全性と有効性や重大なビジネス機会を示す必要な潜在力を持っていないと思うかもしれません。また、適切な戦略的パートナーを探す上で激しい競争に直面しており、交渉過程は時間がかかり複雑かもしれない。さらに、そのような任意の手配の一部として、私たちは、私たちの将来の収入源、研究計画、または候補製品に対する貴重な権利を放棄しなければならないか、または潜在的な協力者との追加的な協定の締結を制限する可能性のある条項で許可を付与しなければならないかもしれない。私たちは戦略的取引や許可証の後、私たちが経済的利益を得て、このような取引が合理的であることを証明することを確信できない。
私たちがこのような協力を成功させたとしても、私たちが合意した条項は私たちに不利になるかもしれないが、例えば、候補製品の開発や承認が延期され、候補製品の安全性が疑問視されたり、承認された候補製品の販売が満足できない場合、私たちはこのような協力を維持できないかもしれない。
しかも、どんな潜在的な未来の協力も私たちの戦略的パートナーによって中止されるかもしれないし、私たちはこのような合意の下で私たちの権利を十分に保護できないかもしれない。また、戦略的パートナーは、(承認されれば)候補製品の開発および商業化に関する決定を制御するために、いくつかの権利について交渉することができ、これらの活動を私たちと同じ方法で行わない可能性がある。もし私たちが将来協力を終了したり、私たちの候補製品に関する協力を遅延させたりすれば、候補製品の開発と商業化を延期し、それらの競争力を低下させる可能性があり、それらが市場に進出すれば、私たちの業務、財務状況、運営結果に大きな悪影響を及ぼす可能性がある。
私たちの候補製品の商業化に関するリスクは
私たちがどんな候補製品の規制承認を得ても、私たちは持続的な規制義務と持続的な規制審査の制約を受けることになり、これは多くの追加費用を招く可能性がある。また、私たちの候補製品が承認されれば、ラベルや他のマーケティングによって制限されたり、市場から撤退したりする可能性があり、もし私たちが規制要求を守っていない場合、あるいは私たちの候補製品が予期せぬ問題に遭遇した場合、その中のいずれかの製品が承認された場合、処罰を受けるかもしれません。
我々の任意の候補製品が承認される可能性がある後、FDAまたは外国監督機関は製品の指示用途またはマーケティングに重大な制限を加えるか、あるいは高価で時間がかかる可能性のある承認後の研究、発売後の監視または臨床試験に持続的な要求を提出して、製品の安全性と有効性を監視する。FDAまたは外国規制機関は、私たちの候補製品を承認する条件としてREMSまたは同様のリスク管理措置を要求することも可能であり、薬学的ガイドライン、医師のコミュニケーション計画、または安全な使用を保証する他の要素の要件、例えば、制限された分配方法、患者登録、および他のリスク最小化ツールを含むことができる。さらに、FDAまたは同様の外国規制機関が私たちの候補製品を承認すれば、私たちの製品の製造プロセス、ラベル、包装、流通、有害事象報告、貯蔵、広告、販売促進、輸入、輸出、記録保存は広範で持続的な規制要求を受けるだろう。これらの要件は、安全および他の上場後の情報および報告書の提出、登録、および承認後に行われた任意の臨床試験においてcGMPまたは同様の要件およびGCP要件を継続的に遵守することを含む。その後、予想されていない深刻度や頻度の有害事象、または当社の第三者製造業者または製造プロセス、または規制要件を遵守できなかったことが、以下のような状況を引き起こす可能性がある以前に未知の問題が存在することが分かった
•私たちの製品の販売や製造を制限し、製品を市場からリコールするか、または自発的または強制的に製品をリコールする
•製品の流通或いは使用の制限、或いは発売後の研究或いは臨床試験の要求を行う
•罰金、賠償、利益または収入の返還、警告状、無見出し状、または臨床試験の一時停止
•FDAまたは外国規制機関は、係属中の出願の承認または私たちが提出した承認済み申請の追加を拒否するか、または承認を一時停止または撤回する
•製品を差し押さえたり差し押さえたり、私たちの製品の輸出入を許可することを拒否したり、
•民事または刑事処罰を禁令または適用する。
上記のいずれかの事件や処罰が発生すると、候補製品を商業化し、収入を創出する能力を抑制する可能性があり、対応するために多くの時間と資源が必要となり、負の宣伝が生じる可能性がある。
FDAや他の規制機関の政策は変わる可能性があり、私たちの候補製品に対する規制承認を阻止、制限、または延期するための追加の政府法規が公布されるかもしれない。例えば、欧州委員会が2020年11月に開始した欧州薬品戦略イニシアティブを背景に、EU薬品立法は現在全面的な審査が行われている。欧州委員会は、医薬製品に関するいくつかの立法文書の改正に関する提案(規制排他的期限、迅速通路の資格などを改正する可能性がある)。2023年第1四半期に完成する予定です。提案された改正は,欧州議会と欧州理事会の同意と採択(2024年末または2025年初めまではないと予想される)を得ると,長期的にはバイオ製薬業界に大きな影響を与える可能性がある。米国や海外の将来の立法や行政や行政行動によって生じる可能性のある政府規制の可能性、性質、程度も予測できない。もし私たちが既存の要求の変化に緩やかに適応できない場合、あるいは新しい要求や政策を採用することができない場合、あるいは規制コンプライアンスを維持できない場合、私たちは法執行行動の影響を受ける可能性があり、利益を達成したり維持したりすることができないかもしれない。
私たちの候補製品の商業成功は医師、患者、医療保険支払人と医学界の他の人がこれらの候補製品に対する市場受け入れの程度に依存する。
私たちの候補製品は商業的に成功しないかもしれない。私たちの候補製品が規制部門の承認を得ても、それらは医師、患者、医療支払者、医療者で市場に認められない可能性があります
コミュニティです。私たちの現在または未来の任意の候補製品の商業成功は、承認された適応を得るために、医師および患者の最終製品の広範な採用および使用に大きく依存するであろう。私たちの製品に対する市場の受け入れ度は多くの要素に依存するだろう
•他のより成熟した製品と比較して、臨床治療効果と安全性を示した
•私たちの候補製品が承認された適応
•対象患者数の制限と、FDAまたは外国規制機関によって承認された任意のラベルに含まれる他の制限または警告;
•衛生保健提供者及びその患者は関連適応のための新薬を受け入れた
•私たちの製品の価格と費用効果、代替療法や療法に関する私たちの製品の治療コスト
•私たちは十分な第三者保険を獲得して維持し、政府医療計画(連邦医療保険と医療補助、個人健康保険会社、他の第三者支払者を含む)から十分な補償を受けることができる
•十分な第三者保険と十分な精算がない場合、患者は私たちの製品に関連する自己負担費用の全部または一部を支払うことを望んでいる
•私たちの製品の使用に対する制限や、いかなる悪影響の普遍性と深刻さ
•潜在的な製品責任クレーム
•私たちの製品と薬品が発売されるタイミングを競う
•私たちまたは未来の潜在的なパートナーの販売とマーケティング戦略の有効性;
•この製品に関する不良宣伝。
任意の候補製品が承認されたが、医師、病院、医療支払者、または患者の十分な受容度に達していない場合、私たちはその製品から十分な収入を得ることができず、利益を達成または維持できない可能性がある。私たちの教育医療界と第三者支払者が私たちの製品のメリットを知る努力には大量の資源が必要かもしれませんし、決して成功しないかもしれません。
FDAと他の規制機関は非ラベル使用の普及を禁止する法律法規を積極的に実行している。もし私たちが発見されたり、不正に非ラベル用途を普及させたりすると、私たちは重大な責任を負うかもしれない。
FDAや他の規制機関は、処方薬に対して提示される可能性のある販促声明を厳格に規制し、承認されれば、私たちの候補製品もそうなるだろう。特に、製品は、製品が承認されたラベルに反映されるように、FDAまたは他の規制機関によって承認されていない使用に使用されてはならない。もし私たちがこのようなラベル外の使用を普及させることを発見されたら、私たちは重大な責任を負うかもしれない。連邦政府は不正販売促進の疑いのある会社に巨額の民事と刑事罰金を科し、いくつかの会社がラベル外販売促進に従事することを禁止している。FDAはまた、企業に同意法令または永久禁止を締結し、これらの法令または永久禁止に基づいて、特定の販売促進行為を変更または制限することを要求する。もし私たちが私たちの候補製品の普及を成功的に管理できなければ、承認されれば、私たちは重大な責任を負う可能性があり、これは私たちの業務や財務状況に実質的な悪影響を及ぼすだろう。
私たちの候補製品の商業化に成功し、承認されれば、政府当局と健康保険会社が構築した保険範囲、十分な補償レベル、優遇された価格設定政策にある程度依存する。もし私たちの製品が保証範囲を獲得あるいは維持し、十分な補償を得られなければ、これらの製品をマーケティングする能力を制限し、私たちの収入を創造する能力を低下させるかもしれません.
承認されれば、政府医療計画(例えばMedicareおよびMedicaid)、個人健康保険会社および他の第三者支払者が提供する保険範囲および精算の十分性は、多くの患者が私たちの候補製品などの処方薬を負担することができるために重要である。第三者支払人が私たちの製品のカバー範囲と受け入れ可能な精算レベルを実現する能力は私たちの商業化に成功する能力に影響を与えるだろう
それらの製品です。第三者支払者が特定の製品の保険を獲得したとしても,それによる精算支払率が十分に高くない可能性があり,あるいは患者が受け入れられないほど高い共同支払いが必要である可能性がある。私たちが開発する可能性のあるどんな製品もアメリカ、EU、あるいは他の場所で保険と精算を受けることができることを保証することはできません。将来的にはどんな可能な精算も減らしたりキャンセルしたりする可能性があります。
第三者支払者はますます薬品とサービスの価格に挑戦するようになり、多くの第三者支払者は同等の後発薬あるいはより安い治療法が利用可能な場合、特定の薬物への保険と精算を拒否する可能性がある。第三者支払者は,我々の製品が代替可能であると考え,価格の低い製品のみを患者に精算することを提案するかもしれない。たとえ私たちの製品が治療効果を高めたり、投与の利便性を改善したことを証明したとしても、既存の薬物の定価は製品に対する費用を制限するかもしれません。これらの支払者は、特定の製品の精算状態を拒否またはキャンセルしたり、新製品や既存市場製品の価格を低すぎるレベルに設定したりして、製品開発投資から適切なリターンを実現することができない可能性がある。精算が得られない場合や限られたレベルでしか精算が得られない場合、私たちの製品を商業化することに成功できない可能性があり、私たちが開発する可能性のある製品から満足な財務リターンを得ることができないかもしれません。
第三者支払者のカバー範囲や新承認製品の精算に関する不確実性が大きい。米国では,連邦医療保険や医療補助計画のような第三者支払者,個人や政府支払者を含め,新薬のカバー範囲を決定する上で重要な役割を果たしている。一部の第三者支払者は、新しいまたは革新的な設備または薬物療法の保証範囲を事前に承認し、その後、このような治療法を使用する医療提供者に精算する必要があるかもしれない。第三者決済者が私たちの製品の保証範囲と精算についてどのように決定するか予測するのは難しいです。
獲得とメンテナンスの精算状態は時間がかかり、コストも高く、確定していない。連邦医療保険や医療補助計画は,個人支払者や他の政府支払者がどのように薬品保険や精算政策を策定するかのモデルとして利用されるようになってきている。しかし、米国の第三者支払者では、製品のカバーや精算に統一された政策はない。そのため、製品の保証範囲と精算範囲は支払人によって異なる。したがって、保証範囲の決定過程は通常、時間がかかり、高価なプロセスであり、各支払人にそれぞれ私たちの製品を使用する科学的および臨床的支援を提供する必要があるが、保証範囲と十分な補償が一致するか、または最初に得られる保証はない。また、精算に関する規則や条例は常に変化しており、場合によっては短時間で通知されており、これらの規則や条例が変わる可能性があると考えられる。
米国以外では、国際業務は通常、広範な政府価格制御と他の市場規制を受けており、欧州や他の国のコスト制御措置の日々の重視は、製品の定価と使用に圧力を与え続けると考えられる。多くの国では、国家衛生システムの一部として、医療製品の価格は異なる価格制御メカニズムの制約を受けている。他の国は会社が自ら医療製品を価格設定することを許可しているが、会社の利益を監督してコントロールしている。追加の外国価格規制や定価規制の他の変化は私たちの製品に対する料金を制限するかもしれません。そのため、米国以外の市場では、米国に比べてわが製品の精算が減少する可能性があり、商業的に合理的な収入や利益を生み出すには不十分である可能性がある。
また、米国や海外の政府や第三者支払者が医療コストの制限や低減に力を入れていることは、これらの組織が新承認製品のカバー範囲や精算レベルを制限する可能性があるため、私たちの製品に十分な保険を提供したり、十分なお金を支払うことができない可能性がある。管理型ヘルスケアの傾向、健康維持組織の日々の影響力、および追加の立法変化により、どの製品の販売も価格設定圧力に直面することが予想される。全体的に,医療コストの下振れ圧力は非常に大きくなり,特に処方薬,外科手術,その他の治療が行われている。そのため、新製品の参入にはますます高い壁が設けられている。
私たちは競争に直面しています。もし私たちの競争相手が技術や製品候補を開発することが私たちより速い、あるいは彼らの技術がより効果的であれば、私たちが製品を開発し、商業化する能力は不利な影響を受ける可能性があります。
生物技術と製薬業界の特徴は技術が迅速に進歩し、競争が激しく、特許と新製品及び候補製品を非常に重視していることである。私たちの競争相手は、私たちの候補製品と競争する製品、候補製品、およびプロセスを開発し、開発しているか、または開発しているかもしれません。我々が開発と商業化に成功した任意の候補製品は,既存の療法や将来出現する可能性のある新しい療法と競争するであろう。相当な数の製品が現在開発中であり、将来的に商業的に使用される可能性があり、製品の開発を試みる可能性のある条件を治療するために使用される可能性があると考えられます
候補者です。特に,免疫学,炎症,腫瘍学分野の競争は非常に激しい。私たちの競争相手はより規模が大きく、資金が豊富な製薬、生物製薬、バイオテクノロジー、治療会社を含む。また,大学や他の研究機関と競合する可能性もあり,これらの大学や他の研究機関は我々の目標指標で活躍し,我々と直接競争する可能性がある.また、これらの組織と競合して管理者、科学者、臨床開発者を募集しており、これは私たちの専門レベルや業務計画を実行する能力に悪影響を及ぼす可能性があります。私たちはまた臨床試験場を設立し、臨床試験のための被験者を募集し、新製品の候補を確定と許可する上で競争に直面する。規模が小さいか早い段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。
私たちのすべての候補製品について、私たちは既存製品と開発中の製品からの競争に直面することが予想されます。SeralutinibはPDGFR,CSF 1 Rとc−kit阻害剤であり,最初にPAH患者に対して対象となった。このグループの患者の競争には、Orenitram(連合治療会社または連合治療会社)、Uptravi(Janssen)、Tyveso(連合治療会社)、Remodlin(連合治療会社)を含むプロスタグランジン/プロスタサイクリン受容体アゴニストが含まれることが予想される。私たちはまた、Revatio(ファイザー)を含むPDE 5阻害剤の経口投与など、クラスIおよびクラスII患者のための製品からのいくつかの競合に直面する可能性がある。AdCirca(共同治療会社);sGC刺激器Adempas(バイエル株式会社)、および口腔時代は、Tracleer(Janssen)、Letairis(gilead Sciences,Inc.)を含む。オスメット(Janssen)です私たちは承認されれば、セラルーチニブはすべての3種類の承認された治療と一緒に使用できると信じている。PAHも薬物研究の活発な適応であり、将来的にRalinepag(ファイザーと連合治療会社)、Sotatercept(メルク社)、ロダリトエチル社(Enzyant Treateutics GmbH)とMK-5475(メルク社)からの競争に直面する可能性がある。また,PAH治療のための承認されていないにもかかわらず,Tenax治療会社,Aerovate治療会社,Aerami治療会社/Vectura集団の競争を含むイマチニブ製剤からの競争に直面している可能性がある。
GB 5121およびGB 7208は、PCNSLおよびMSを含む腫瘍および免疫学的適応を治療するためのBTK阻害剤であり、現在、FDAまたはECによって承認された難治性または再発性PCNSLの治療法はない。多発性硬化症において承認された場合、GB 7208は、既存の承認および研究療法からの競争に直面する可能性がある。BTK阻害剤は、PCNSLまたはMSの治療のためにFDAまたはECによって承認されていないが、承認された場合、GB 5121および/またはGB 7208は、現在FDAおよび/またはECによって承認されているBTK阻害剤Imbrovica(エバーブ社/Janssen)、Calquence(アスリーカン)、Brukinsa(百済神州、Ltd.)からの競争に直面する可能性があると考えられる。そして/またはJaypirca(礼来会社)。著者らのBTK阻害剤はまたBTK阻害剤Velexbru(Ono Pharmtics Co.,Ltd.)からの競争に直面する可能性があり、後者はすでに日本、韓国と台湾で許可され、再発性或いは難治性原発性中枢神経系リンパ腫の治療に用いられる。我々はまた,evobrutinib,tlebrutinib,fenebrutinib,orelabrutinib,rilzabrutinib,remibrutinibを含むBTK阻害剤からの競合に直面する可能性がある
他にも早期臨床プロジェクトがあるかもしれませんが、承認されれば、私たちの候補製品と競争します。私たちの多くの競争相手は私たちよりもっと多くの財力、技術、そして人的資源を持っている。製薬業界のより多くの合併と買収は、私たちの競争相手により多くの資源を集中させるかもしれない。技術の商業適用性の進展や、これらの分野でより多くの投資可能な資本があるため、競争はさらに激化する可能性がある。著者らの成功はある程度私たちが薬物の組み合わせを確立し、積極的に管理する能力に基づいて、これらの薬物の組み合わせは満足されていない医療需要を満たし、患者の治療において価値を創造することができる。
もし私たちの製品の市場機会が私たちが思っているより小さいなら、私たちの収入は不利な影響を受けるかもしれません。私たちの業務は影響を受けるかもしれません。
私たちが私たちの製品候補を通じてすべての状況を解決することに努力している正確な発病率と流行率はまだ不明である。これらの疾患を有する人の数と、私たちの候補製品治療から利益を得る可能性があるこれらの疾患患者のサブセットの予測は、私たちの信念と推定に基づいている。これらの推定は様々な源から来ており、科学文献、診療所調査、患者基金会或いは市場研究を含み、正しくないことが証明されている可能性がある。また、新しい試験はこれらの疾病の推定発病率或いは流行率を変える可能性がある。私たちのすべての候補製品の総潜在市場は、最終的には、他の事項を除いて、販売が許可された各候補製品の最終ラベルに含まれる診断基準、代替療法の利用可能性、およびこれらの代替療法に対する私たちの候補製品の安全性、利便性、コストおよび有効性、医学界と患者の受容度、薬品定価と精算に依存する。米国、他の主要市場、および他の地域の患者数は予想を下回る可能性があり、患者は私たちの製品治療を受けることができないかもしれない、あるいは新しい患者はますます識別しにくくなったり、接触したりする可能性があり、これらすべては私たちの運営結果および業務に悪影響を及ぼすであろう。また、私たちの候補製品がかなりの市場シェアを獲得しても、私たちのいくつかの潜在的なターゲット層が非常に少ないので、私たちがこのような巨大な市場シェアを獲得しても、私たちは決して利益を達成しないかもしれない。
私たちは現在マーケティングや販売組織がなく、会社としても製品を商業化した経験がなく、これらの能力を開発するために大量の資源を投入しなければならないかもしれません。マーケティングや販売能力を確立できない場合や、第三者と合意して私たちの製品をマーケティングして販売することができなければ、製品収入を生み出すことができないかもしれません。
私たちは内部販売、マーケティング、流通能力もなく、製品を商業化していない。もし私たちの任意の候補製品が最終的に規制部門の承認を得たら、主要市場でこのような各製品を商業化するために、技術的な専門性と流通能力を支援するマーケティング·販売組織を構築しなければならない。これは高価で時間がかかる、あるいは直接販売チームや流通システムを構築する第三者と協力して、私たち自身の販売チームや流通システムを強化し、あるいは私たち自身の販売チームや流通システムの代わりになるだろう。ある会社として、私たちは以前生物製薬製品のマーケティング、販売と流通の面で経験がなく、販売組織の構築と管理は重大なリスクに関連しており、私たちは合格者を雇用、維持と激励し、十分な販売手がかりを生成し、販売とマーケティング人員に十分な訓練を提供し、異なる地理的な位置に分散した販売とマーケティングチームを効果的に管理する能力を含む。私たちの内部販売、マーケティング、流通能力の発展のいかなる失敗や遅延も、これらの製品の商業化に悪影響を及ぼすだろう。私たちは、受け入れ可能な財務条項で協力することができないか、またはコンサルタントまたは外部サービスプロバイダを招いて、私たちの販売、マーケティング、および流通機能を支援することができないかもしれません。また、私たちが第三者に依存してこれらの機能を実現すれば、私たちの製品の収入と収益力(あれば)は、私たちが開発したどの製品のマーケティング、販売、流通よりも低くなる可能性があります。私たちはこのような第三者に対して支配権がほとんどないかもしれません。彼らのいずれも必要な資源と注意力を投入して、私たちの製品を効果的に販売し、マーケティングすることができないかもしれません。もし私たちが私たちの製品を商業化することに成功できなければ、私たち自身でも、1つ以上の第三者との手配によっても、私たちは未来にどんな製品収入も発生できないかもしれないので、私たちは重大な追加損失を招くだろう。
私たちの将来の成長は海外市場における私たちの運営能力にある程度依存するかもしれませんが、そこでは追加の規制負担や他のリスクや不確実性の影響を受けることになります。
私たちの将来の成長は私たちの候補製品を海外市場で開発し商業化する能力にある程度依存するかもしれない。海外市場適用規制機関の規制承認を得るまで、私たちは私たちの候補製品のマーケティングや普及は許可されておらず、私たちはいかなる候補製品の規制承認も得られないかもしれない。多くの他の国で単独の監督管理許可を得るためには、安全性と有効性、及び私たちの候補製品の臨床試験、商業販売、定価と流通などの方面の監督管理を含む多くの異なる監督管理要求を守らなければならない。もし私たちが規制機関の候補製品の承認を得て、最終的に私たちの製品を海外市場で商業化すれば、私たちは追加のリスクと不確実性に直面します
•国外の薬品審査に対する監督管理要求は異なる
•知的財産権の保護を減らすことです
•私たちのビジネスの潜在的に関連する追加の第三者特許権が存在するかどうか
•関税、貿易障壁、規制要求の意外な変化
•インフレ、特に外国経済と市場の政治的不安定を含む経済的疲弊;居住または海外旅行の従業員は税収、雇用、移民、労働法を遵守する
•外国為替変動は、営業費用の増加と収入の減少、他の国での業務展開に付随する他の義務を招く可能性がある
•海外清算、定価、保険制度
•労働騒乱が一般的に存在する国では労働力の不確実性
•海外の原材料の供給や製造能力に影響を与える事件による生産不足;
•地政学的行動(戦争やテロを含む)、新冠肺炎などの衛生流行病や地震、台風、洪水や火災などの自然災害による業務中断。
私たちの業務運営や業界に関連するリスク
私たちの経営結果は大幅に変動する可能性があり、これは私たちの将来の経営結果を予測することを難しくし、私たちの経営結果が予想を下回ったり、私たちが提供する可能性のある任意の指導を招く可能性があります。
私たちの四半期と年間運営業績は大きく変動する可能性があり、将来の運営業績を予測することは困難です。これらの変動は様々な要因によって引き起こされる可能性があり、その多くの要因は、これらに限定されないが、我々が制御できるものではない
•私たちの候補製品に関する研究、開発、規制承認、商業化活動の時間、コスト、投資レベルは時々変化する可能性がある
•私たちの候補製品についての保険·精算政策(承認されれば)、将来私たちの製品と競争する可能性のある薬
•私たちの候補製品を製造するコストは、生産数量と第三者製造業者と合意した条項によって変化するかもしれません
•マイルストーンまたは他の支払いの時間と金額をライセンシーおよび他の第三者に支払わなければなりません。これらのライセンシーおよび他の第三者から、子会社の支配権変更による満期の支払いを含む当社が買収した候補製品の許可を得ました
•他の候補品や技術の獲得、開発、商業化のための支出が生じる可能性があります
•承認された製品の需要レベルには、大きな差がある可能性がある
•未来の会計宣言や私たちの会計政策の変化;
•我々の候補製品または競合製品の臨床前研究または臨床試験のタイミングおよび成功または失敗、またはわが業界の競争構造における任意の他の変化は、私たちの競争相手またはパートナー間の統合を含む。
これらの要因の累積影響は,我々の四半期や年度運営業績に大幅な変動や予測不可能を招く可能性がある。したがって,時期ごとに我々の業務結果を比較することは意味がないかもしれない.投資家たちは私たちの過去の業績を私たちの未来表現の指標として依存してはいけない。
このような変化性および予測不可能性はまた、業界や金融アナリスト、または投資家の任意の時期に対する期待を満たすことができない可能性がある。もし私たちの収入や運営結果がアナリストや投資家の予想よりも低い場合、または私たちが市場に提供するいかなる予測よりも低い場合、または私たちが市場に提供する予測がアナリストや投資家の予想よりも低い場合、私たちの普通株の価格は大幅に下落する可能性がある。私たちが提供する可能性のある以前に公開された収入や収益案内に到達したとしても、このような株価下落は起こる可能性がある。
私たちは経営陣や他の臨床·科学者のサービスに依存しており、これらの人を引き留めたり、より多くの管理や臨床·科学者を募集することができなければ、私たちの業務は影響を受けるだろう。
私たちの成功部分は私たちが引き続き高い素質の管理、臨床と科学者を吸引、維持と激励する能力にかかっている。私たちは私たちの上級管理職、特に最高経営責任者、そして私たちのベテラン科学者と上級管理チームの他のメンバーに高く依存している。これらの人の誰もがサービスを失っても、私たちの製品パイプラインの成功開発、私たちの計画中の臨床試験の開始または完成、あるいは私たちの候補製品の商業化を遅延または阻止する可能性があります。例えば、2020年11月16日から、ファヒム·ハスナーは、医学博士ヒラ·グジラティの代わりに、私たちの総裁兼CEOに任命され、2021年に3人の新しいメンバーが私たちの実行チームに参加しました。経営陣の交代自体が管理が困難であるため,中断したり,業務を維持したり発展させることが困難になる可能性がある.我々はすでに我々の高度管理チームの各メンバーと雇用協定や招聘書に署名しているにもかかわらず、これらの合意は通知または通知なしに任意に終了することができるため、予想通りに彼らのサービスを保留することができない可能性がある。私たちは現在、私たちの役員や従業員の生命のために“キーパーソン”生命保険を保有していません。保険不足は、私たちがこのような個人のサービス損失を補うために十分な賠償を受けられない可能性があるということを意味する。
私たちの臨床開発と商業化努力に成功するために、私たちの管理、運営、財務、その他の資源を拡大し、効果的に管理する必要があるだろう。私たちは成功しないかもしれません
製薬、生物技術とその他の業界の合格人材に対する競争は日々激しくなっているため、特にサンディエゴ地区では、著者らの独特な会社文化と未来に引き続き合格した管理人員及び科学と臨床人員を誘致或いは維持する。近年、私たちの業界のすべての人たちの流出率が高い。私たちが必要な人員を誘致、統合、維持、激励することができなければ、私たちの業務目標を達成することは、私たちの発展目標の実現、追加資本を調達する能力、そして私たちが業務戦略を実施する能力を深刻に阻害する制限に直面する可能性があります。
私たちは最近私たちの組織の規模を大幅に拡大し、私たちの成長を管理し、私たちの業務を成功的に拡大する上で困難に直面する可能性があります。
我々の組織は2018年1月の11名の従業員から2023年3月10日の178名のフルタイム従業員とパートタイム従業員なしに大幅に増加した。私たちが引き続き私たちの候補製品の潜在的な商業化を開発し、追求することに伴い、上場企業として、私たちの財務、開発、監督、製造、マーケティング、販売能力を拡大または強化し、あるいは第三者と契約を締結し、これらの能力を提供する必要があるかもしれない。私たちの業務の拡大に伴い、様々な戦略パートナー、サプライヤー、他の第三者とのより多くの関係を管理する必要があると予想されます。私たちの将来の財務業績と、私たちの候補製品を開発し、商業化し、効果的に競争する能力は、私たちの最近の大幅な成長と任意の未来の成長を効果的に管理する能力にある程度依存するだろう。
私たちは様々な連邦、州、外国の医療法律と法規の制約を受けて、私たちがこれらの法律と法規を守らなければ、私たちの運営結果と財務状況を損なうかもしれません。
私たちの業務運営と調査者、医療専門家、コンサルタント、第三者支払者と顧客との現在と将来の手配は、広く適用される連邦、州と外国の詐欺と乱用、その他の医療に関する法律と法規に直面させます。これらの法律は、私たちがマーケティング、販売、流通をどのように研究、マーケティング、販売、流通するかを含む、私たちが業務を展開する業務または財務配置と関係を制約するかもしれません。これらの法律には
•他の事項に加えて、個人または実体が直接または間接的に、現金または実物の形態で直接または間接的に現金または実物の形態で直接または間接的に請求され、提供され、受信され、または任意の報酬(任意のリベート、賄賂または何らかのリベートを含む)を提供することを禁止し、個人の推薦または購入、レンタルまたは注文、または任意の商品、施設、物品またはサービスの購入、レンタルまたは注文を手配または推薦して、連邦医療保険および医療補助などの連邦医療計画に従って全部または部分的な支払いを行うことを禁止する連邦反バックル法規。個人や実体は、連邦の“反リベート法規”やこの法規に違反する具体的な意図を実際に知る必要がなく、違反行為を実施することができる。
•他の事項に加えて、個人または実体が故意に連邦政府に虚偽または詐欺的な支払いまたは承認クレームを提出または提出することを禁止し、虚偽または詐欺的クレームに関連する虚偽記録または陳述を故意に作成、使用または使用するか、またはそれを知りながら虚偽陳述をしたり、連邦政府に金銭を支払う義務を回避、減少または隠蔽するための虚偽陳述を行うことを禁止する“民事虚偽申告法”を含む連邦虚偽申告法。また、政府は、民事虚偽請求法については、連邦“反リベート法規”違反による物品やサービスを含むクレームが虚偽または詐欺的クレームを構成していると断言することができる
•1996年の連邦健康保険携帯および責任法案、またはHIPAAは、他にも、詐欺の任意の医療福祉計画を知りながら故意に実行または実行しようとした計画、または医療福祉、プロジェクトまたはサービスの提供または支払いに関連する重大な事実を知りながら、または故意に偽造、隠蔽または隠蔽し、または任意の重大な虚偽陳述を行い、刑事および民事責任を規定する。連邦反リベート法規と同様に、個人または実体は、この法規または法規違反の具体的な意図を実際に理解する必要がなく、違反を実施することができる
•連邦医師は、連邦医療保険、医療補助または児童健康保険計画(いくつかの例外)に基づいて支払い可能な薬品、機器、生物製品および医療用品の製造業者が、医師(医師、歯科医師、検眼師、足科医および脊椎マッサージ師を含む定義)、いくつかの非医師従事者(医師アシスタント、看護師従事者、臨床医師)への支払いおよび他の“価値移転”に関する情報を毎年医療保険および医療補助サービスセンター(CMS)に報告することを要求する陽光法案を支払う
看護師専門医、登録看護師麻酔科医、麻酔学アシスタントおよび登録看護師助産師)と教育病院、および上記医師およびその直系親族が持つ所有権と投資権益;
•同様の州および外国の法律、例えば、州反リベートおよび虚偽クレーム法律は、研究、流通、販売およびマーケティング、および非政府第三者支払者(個人保険会社を含む)または患者自身によって精算される医療項目またはサービスに関するクレームを含むが、これらに限定されないが、これらに限定されないが、州法律は、製薬会社に製薬業界の自発的コンプライアンスガイドラインおよび連邦政府によって発行された関連コンプライアンスガイドラインを遵守すること、または他の方法で医療保健提供者および他の潜在的な転換源への支払いを制限することを要求する。医薬品製造業者に、価格設定およびマーケティング情報に関連する報告書の提出を要求するか、または医師、他の医療提供者および実体に提供されるプレゼントおよび他の報酬および価値項目を追跡することを要求する州法律および法規、ならびに医薬品販売代表の登録を要求する州および地方法律。
我々の内部運営と第三者の業務手配が適用される医療法律や法規に適合していることを確保することは、大量のコストに及ぶ可能性がある。政府当局は、私たちが医師や他の医療保健提供者との諮問および顧問委員会と手配することを含む私たちの業務慣行を結論するかもしれないが、その中の一部の人は、提供されたサービスの補償として株式オプションを取得し、現在または未来に適用される詐欺および乱用または他の医療保健法律および法規に関する現在または将来の法律、法規、機関指導または判例法に適合していない。私たちの業務が上記の任意の法律または私たちに適用可能な任意の他の政府の法律および法規に違反していることが発見された場合、私たちは、民事、刑事および行政処罰、損害賠償、罰金、MedicareおよびMedicaidなどの米国政府の援助から除外された医療計画(例えば、MedicareおよびMedicaid)または他の国または司法管轄区域の同様の計画の外、返還、個人監禁、契約損害、名声損害、追加の報告要求および監督を含む重大な処罰を受ける可能性があり、もし私たちの会社が誠実な合意または同様の合意に制約された場合、これらの法律に違反した疑い、利益の減少、および私たちの業務の削減または再編を解決する。さらに、このような行動を防御するには費用がかかり、時間がかかる可能性があり、大量の財政と人的資源が必要となる可能性がある。したがって、私たちが私たちに提起される可能性のあるいかなる訴訟も防ぐことに成功しても、私たちの業務は損害を受ける可能性がある。もし私たちがそれと業務を展開することを期待している任意の医師または他の提供者または実体が適用されない法律を遵守していないことが発見された場合、彼らは政府の援助された医療計画および監禁から除外されることを含む刑事、民事または行政制裁を受ける可能性がある。上記のいずれかの場合が発生すると、我々の業務運営能力や運営結果に悪影響を及ぼす可能性がある。
私たちは政府の規制とプライバシー、データ保護、情報セキュリティに関する他の法的義務の制約を受けている。実際または予想されているこのような要求を遵守できないことは、私たちの業務、財務状況、または運営結果に重大な悪影響を及ぼす可能性がある。
世界的なデータ保護構造は急速に変化しており、私たちは多くの州、連邦および外国の法律、要求および法規によって制限されているかもしれないが、これらの法律、要求、および法規は、個人情報の収集、使用、開示、保持および安全を管理しており、例えば、私たちが収集する可能性のある臨床試験に関連する情報を管理している。予測可能な未来には、実施基準および法執行実践は依然として不確定である可能性があり、私たちはまだ未来の法律、法規、基準、またはその要求に対する見方が私たちの業務に与える影響を決定することができない。このような変化は私たちの業務に不確実性をもたらす可能性があり、私たちがある司法管轄区域で業務を展開し、あるいは個人情報を収集、保存、移転、使用、共有する能力に影響を与え、私たちの契約でより重い義務を受ける必要があり、私たちが責任を負うか、または追加コストをかける必要がある。これらのデータプライバシーやセキュリティ要求を守ることは厳しくて時間がかかり、私たちの業務コストが増加する可能性があります。私たちは努力したにもかかわらず、実際にまたはこれらの要求を遵守できなかったと考えられることで、罰金や処罰、訴訟、名声被害のリスクに直面する可能性があり、これは私たちの業務、財務状況、運営結果に実質的かつ不利な影響を与える可能性がある
私たちの運営と業務の増加に伴い、私たちは新しいまたは追加のデータ保護法律と法規の制約または影響を受け、規制機関のより厳しい審査や関心に直面する可能性がある。アメリカでは、“経済と臨床健康情報技術法案”によって改正されたHIPAA及びその公布された法規、或いは総称してHIPAAと呼ばれ、保険実体及びその業務パートナーが持つ単独で識別可能な健康情報のプライバシー、安全と伝送に対して関連要求を提出した。我々は現在HIPAA下の保証実体やビジネスパートナーとして行動しているとは思わないため,HIPAAの要求や処罰を直接受けることはない.しかしながら、事実および状況に基づいて、私たちが知らずにHIPAAによってカバーされているヘルスケア提供者または研究機関から個人識別可能な健康情報を受信し、その医療提供者または研究機関が個人識別可能な健康情報の開示に関するHIPAAの要求を満たしていない場合、重大な刑事罰に直面する可能性がある
また、州法は、場合によっては健康情報のプライバシーやセキュリティを管理しており、その多くの法律は互いに大きく異なり、同じ要求がなく、コンプライアンス作業を複雑にしている可能性がある。また、私たちは個人情報のプライバシーを管理し、処理し、保護する他の州の法律によって制限される可能性がある。例えば、カリフォルニア州はCCPAを公布し、2020年1月1日から施行され、カリフォルニアの消費者により大きな権利を与え、彼らの個人情報にアクセスして削除し、特定の個人情報を共有しないことを選択し、彼らの個人情報がどのように使用されるかに関する詳細な情報を得ることができる。CCPAは,違反行為に対する民事処罰と,データ漏洩訴訟の可能性や関連リスクを増加させたデータ漏洩に対する個人訴権を規定している。また、カリフォルニアプライバシー権法案(CPRA)は2023年1月1日に正式に施行され、CCPAの重大な改正が行われた。CPRAは、カリフォルニアでビジネスをしている会社に、追加の消費者権利プログラム、データ使用の制限、高リスクデータの新しい監査要求、および特定の敏感なデータの使用からの選択を含む追加のデータ保護義務を課している。それはまた、実質的な法規の発行を許可し、プライバシーと情報セキュリティ法執行の強化につながる可能性がある新しいカリフォルニア州データ保護機関を作成した。追加的なコンプライアンス投資と潜在的なビジネスプロセスの変更も必要となる可能性があります。
私たちの海外での業務はまたデータ保護部門のより厳しい審査や注目を受ける可能性がある。例えば,ヨーロッパでは,2018年5月に“一般データ保護条例”が施行され,個人データのコントローラやプロセッサに厳しい要求が出されている.GDPRはEUと欧州経済圏加盟国が追加の法律と法規を制定し、遺伝、生物認識、または健康データの処理をさらに制限することを可能にする。GDPRを遵守しなければならない企業は、より強力なデータ保護要件の規制法執行、および不正があれば2000万ユーロまたは不適合会社の世界年収の4%までの罰金が科される可能性があるコンプライアンス義務およびリスクに直面しなければならない。他の要件では、GDPR規制は、GDPRによって拘束された個人データを、このような個人データに対して十分な保護を提供することが発見されていない第三国に移し、2020年7月、欧州連合裁判所(CJEU)は、組織がどのように個人データをEU/欧州経済地域から米国に合法的に転送するかを制限し、方法は、国際移転の目的のためにプライバシー盾の無効を宣言し、標準契約条項の使用にさらに制限を加える。2022年3月、米国とEUは無効な法規の代わりに新たな規制制度を発表したが、バイデン総裁が2022年10月7日に署名した米国の信号情報活動の保障措置の強化に関する行政命令を除いて、この新たなEU-米国データプライバシーの枠組みは実施されていない。2020年7月16日のCJEU裁決の後、欧州裁判所と規制機関の裁決は国際データ転送に制限的なやり方をとった。規制当局が個人資料出力メカニズムについてさらなる指針を出すのに伴い、SCCが使用できない場合、および/または法執行行動を開始すると、追加コスト、クレームおよび/または規制調査または罰金を受ける可能性があり、および/または他の方法で私たちが業務を展開している国と地域の間で個人資料を移転できない場合、これは私たちがサービスを提供する方法、地理的位置、または私たちの関連システムと業務の隔離に影響を与え、私たちの財務業績に悪影響を及ぼす可能性がある
2021年初め以降、GDPR下の義務とは異なるが、1,750万GBまたは違反会社の前会計年度の世界年収の4%までの罰金を含む、同様の義務や同様の罰が加えられている英国データ保護制度の制約も受けている。中国やロシアなどの他の外国司法管轄区はますます自分のプライバシー制度を実施あるいは発展させており、これらの制度は複雑で複雑なコンプライアンス義務と強力な監督法執行権力を持っている。私たちが他の国や司法管轄区域に拡張し続けるにつれて、私たちは追加の法律と法規の制約を受けるかもしれません。これらの法律と法規は私たちが業務を展開する方法に影響を与えるかもしれません。
最近公布された立法、将来の立法、医療改革措置は、候補製品の市場承認を獲得し、それを商業化する難しさとコストを増加させ、設定可能な価格に影響を与える可能性がある。
米国や一部の外国司法管轄地域では、医療システムは、コスト制御措置を含む複数の立法·規制改革を継続しており、これらの措置は、新薬の保証範囲や精算範囲を減少または制限し、上場承認された任意の候補製品を収益性のある方法で販売する能力に影響を与える可能性がある。特に,米国連邦や州レベルでは,医療コストの低減と医療の質の向上を図る取り組みが継続されている。
例えば、2010年3月、アメリカはACAを公布した。ACAの中で私たちの潜在的な製品候補に重要な意義を持つ条項の中で、ACA:任意の指定ブランドの処方薬と生物製剤の実体に対して控除不可能な年間費用を設定する;メーカーの医療補助リベート責任を連邦医療補助管理に参加する医療機関に割り当てられた個人の保険薬品に拡大する;医療補助計画の資格基準を拡大した;公共衛生計画による資格割引を得る実体を拡大した;医療補助薬物リベート計画によってメーカーが支払わなければならない法定最低リベートを高めた;新しい連邦医療保険D部分カバー割引計画を作成した;新しいノッチ患者を中心とした結果研究所を構築し、監督した
臨床有効性研究の優先順位を確定し、比較を行い、このような研究に資金を提供する;そしてCMSに医療保険と医療補助革新センターを設立し、革新的な支払いとサービス交付モードをテストし、医療保険と医療補助支出を下げる。
ACAのいくつかの側面は公布以来、司法と政治的な挑戦を受けてきた。2021年6月17日、米国最高裁はいくつかの州がACAに対して提出した最新の司法挑戦を却下したが、ACAの合憲性を具体的に裁くことはなかった。最高裁が裁決を下す前に,総裁·バイデンはACA市場による医療保険の取得を目的とした2021年2月15日から2021年8月15日までの特殊保険期間を開始する行政命令を発表した。行政命令はまた、特定の政府機関に、医療補助モデルプロジェクトおよび免除計画の再審査、および医療補助またはACAによる医療保険カバー範囲の獲得による不必要な障害をもたらす政策を含む医療補助モデルプロジェクトおよび免除計画の再審査および見直し、医療補助またはACAによる医療保険のカバー範囲を制限する既存の政策およびルールを再検討するように指示する。
さらに、ACAが公布されて以来、他の立法改正も提案され、採択された。2011年8月2日、2011年予算制御法が署名され、提供者への医療保険の支払いを減らすことを含む法律となり、2013年4月1日に施行され、この法規の後続立法改正により、国会が追加的な行動を取らない限り、2020年5月1日から2022年3月31日まで一時的に一時停止される2032年まで有効となる。2013年1月2日、2012年の“米国納税者救済法”が法律に署名し、病院を含むいくつかの医療サービス提供者に支払う医療保険を減らし、政府が提供者に追加金を取り戻す訴訟時効を3年から5年に延長した。また、2021年3月11日には、2024年1月1日から法定の医療補助薬品還付上限が撤廃され、現在の上限は薬品メーカーの平均価格の100%となる“2021年米国救援計画法案”が署名された。
また、処方薬のコストが上昇していることを受けて、米国政府は薬品定価のやり方の審査を強化した。このような審査は,最近の数回の国会調査を招き,製品定価の透明性の向上,価格設定とメーカー患者計画との関係の審査,政府計画の製品精算方法の改革を目的とした連邦や州立法を提案·採択した。2022年8月16日、“2022年インフレ削減法案”(IRA)が正式に発効した。他の事項を除いて、アイルランド共和軍はある薬品のメーカーに連邦医療保険との価格交渉を要求し(2026年から)、連邦医療保険B部分と連邦医療保険D部分に基づいてリベートを徴収し、インフレを超える価格上昇を罰し(初めて2023年に満期)、D部分のカバーギャップ割引計画の代わりに新しい割引計画を採用する(2025年から)。アイルランド共和軍は衛生·公衆サービス部長官が最初の数年に規制ではなく指導によってその多くの規定を実施することを許可した。このような理由と他の理由で、アイルランド共和軍がどのように実施されるのかは不明だ。
今後採用される可能性のある新しい法律や他の医療改革措置は、連邦医療保険や他の医療資金のさらなる減少、より厳しいカバー基準、新しい支払い方法、および私たちが受けた任意の承認製品の価格のより大きな下振れ圧力を招く可能性があると予想される。連邦医療保険や他の政府が計画している精算のいかなる減少も、個人支払者の支払いの同様の減少を招く可能性がある。コスト抑制措置や他の医療改革を実施することは、承認されれば、収入を創出し、利益を達成することができ、あるいは候補製品を商業化することができるかもしれない。
州レベルでは、立法機関は、価格または患者の精算制限、割引、特定の製品参入の制限、マーケティングコスト開示および透明性措置を含む、医薬品および生物製品の価格設定を制御するための法規を立法および実施することが増えており、場合によっては、他の国からの輸入および大量購入を奨励することを目的としている。法律で規定されている第三者支払者の支払金額の価格制御またはその他の制限は、私たちの業務、運営結果、財務状況、見通しを損なう可能性があります。また,地域医療当局や個別病院では,どの薬品やサプライヤーが処方薬や他の医療計画に含まれるかを決定するために入札プログラムが使用されるようになってきている。承認されれば、これは私たちの候補製品に対する最終的な需要を減少させるか、あるいは私たちの製品価格に圧力を与える可能性があり、これは私たちの業務、運営結果、財務状況、見通しにマイナスの影響を与える可能性がある。
EUでは、承認されれば、同様の政治的、経済的、規制的な発展が、候補製品を利益的に商業化または共同商業化する能力に影響を与える可能性がある。例えば,2021年12月13日,衛生技術評価やHTAに関する第2021/2282号条例が可決され,第2011/24/EU指令が改正された。この規定は2022年1月に施行されたが、2025年1月から適用され、その間に実施に関する準備と手順が取られるだけである。この条例は施行されると、関連製品を対象に段階的に実施されるだろう。この規定は、新医薬製品を含む衛生技術の評価におけるEU加盟国の協力を促進し、これらの分野で共同臨床評価を行うEUレベルの協力に基礎を提供することを目的としている。この規制はEU加盟国が一般的なHTAツールを使用することを可能にする
この4つの主要な分野は,患者に最も潜在的な影響を与える革新的な健康技術の共同臨床評価,共同科学相談,開発者がHTA当局にアドバイスを求めることができること,新興の健康技術および早期発見に将来性のある技術を決定すること,および他の分野での自発的な協力を継続することである。個別EU加盟国は、衛生技術の非臨床(例えば、経済、社会、倫理)の評価を引き続き担当し、定価と精算について決定する。
私たちに製品責任訴訟を提起すれば、私たちは重大な責任を負い、私たちの製品の商業化を制限することを要求されるかもしれません。
私たちの候補製品の臨床試験のため、私たちは固有の製品責任リスクに直面して、私たちの候補製品を商業化すれば、私たちはもっと大きなリスクに直面します。例えば、私たちの候補製品が製品テスト、製造、マーケティング、または販売中にダメージを与えたり、不適切なことが発見されたと言われた場合、私たちは起訴されるかもしれない。このような製品責任クレームは、製造欠陥、設計欠陥、候補製品固有の危険について警告、不注意、厳格な責任、保証違反の告発を含む可能性がある。臨床試験参加者、患者、または他の使用、管理、または将来承認される可能性のある製品を販売する人は、私たちにクレームをつけるかもしれない。州消費者保護法によると、クレームも主張することができる。
もし私たちが製品責任クレームで自分を弁護することに成功できなければ、私たちは重大な責任を招いたり、私たちの製品の商業化を制限または停止することを要求されるかもしれません。成功的な防御であっても、多くの財政的で管理的な資源が必要だ。事件がどうであっても最終的な結果がどうであろうと、賠償責任は次のようになるかもしれない
•私たちの製品への需要が減少しました
•私たちの名声とメディアの深刻な否定的な関心を損なう
•臨床試験参加者の脱退
•関連訴訟の弁護費用
•経営陣の時間と資源を移転する
•実験参加者や患者に多額の報酬を与え
•製品のリコール、撤回またはラベル付け、マーケティング、または販売促進制限;
•深刻な財務的負の影響
•候補品を商業化することはできません
•私たちの株価は下落しています。
私たちは現在合計約1,000万ドルの製品責任保険を持っている。私たちの臨床試験の拡大に伴い、あるいは候補製品を商業化し始めたら、私たちの保険カバー範囲を増やす必要があるかもしれません。保険範囲はますます高くなっています。潜在的な製品責任クレームを防止するために、許容可能なコストで十分な製品責任保険を得ることができず、これは私たちの候補製品の商業化を阻止または阻害する可能性がある。私たちはこのような保険を受けますが、私たちに対するいかなるクレームも、裁判所の判決や和解の金額が私たちの保険の全部または一部の保険範囲内ではないか、あるいは私たちの保険範囲を超えてしまう可能性があります。私たちの保険証書も様々な排除があります。私たちは製品責任クレームの影響を受けるかもしれませんが、私たちは保険範囲を持っていません。私たちは私たちの保険範囲の制限を超えたり、私たちの保険カバー範囲内でない裁判所の裁決または和解合意で達成された任意の金額を支払う必要があるかもしれません。私たちはこれらの金額を支払うために十分な資本を持っていないか、または得ることができません。
私たちと私たちの未来の潜在的なパートナーは、私たちが承認した任意の製品が不良医療事件を引き起こしたり、何もしなければ制裁につながると規制機関に報告することを要求され、これは私たちの業務に実質的な損害を与えるだろう。
もし私たちが私たちの任意の潜在的な未来パートナーと私たちの製品を商業化することに成功すれば、FDAと外国規制機関は、もしこれらの製品がこれらの不良事件を引き起こす可能性があれば、私たちと私たちの任意の潜在的な未来パートナーに不良医療事件に関するいくつかの情報を報告することを要求するだろう。私たちの報告義務の時間は私たちが不良事件とイベントの性質を認識した日によって触発されるだろう。私たちと私たちの潜在的な未来のパートナーやCROは規定された時間範囲で不良事件を報告できないかもしれない。もし私たちが
あるいは私たちの将来の任意の潜在的パートナーまたはCROがそのような報告義務を履行できなかった場合、FDAまたは外国規制機関は、刑事起訴、民事罰金の適用、私たちの製品の差し押さえ、または将来の製品の承認を延期または承認することを含む行動をとることができる。
私たちの従業員と独立請負業者は、首席調査者、CRO、コンサルタント、サプライヤーを含み、規制基準と要求を遵守しないことを含む、不当な行為またはその他の不当な活動に従事する可能性がある。
私たちは、首席調査者、CRO、コンサルタント、サプライヤーが不正行為または他の不正活動に従事する可能性があるリスクを含む、私たちの従業員と独立請負業者に直面しています。これらの当事者の不正行為は、(1)FDAの法律および法規および他の同様の規制要件に違反する、意図的、無謀、および/または不注意な行為、または許可されていない活動を開示することを含む可能性があり、(2)cGMPおよび同様の要件を含む製造基準、(3)米国および海外の連邦および州データ、プライバシー、セキュリティ、詐欺および乱用、および他の医療保健法律法規、または(4)財務情報またはデータを真に、完全かつ正確に報告することを要求する法律を含む。これらの法律の制約を受けた活動はまた、不適切な使用或いは歪曲臨床試験過程で得られた情報を含み、著者らの臨床前研究或いは臨床試験において虚偽のデータを作成し、或いは薬物製品を不法に流用し、これは規制制裁を招き、著者らの名声に深刻な損害を与える可能性がある。従業員や他の第三者の不正行為を常に識別し、阻止することができるわけではなく、このような活動を発見し、防止するための予防措置は、未知または管理不可能なリスクや損失を効果的に制御することができないか、またはそのような法律や法規を遵守できないことによる政府調査または他の行動または訴訟から私たちを保護することができない可能性がある。さらに、私たちは、一人や一人の政府が、起こらなくても、このような詐欺や他の不適切な行為を告発する可能性があるというリスクに直面している。もし私たちにこのような訴訟を提起し、私たちが自分の権利を弁護したり、維持することに成功しなかったら、これらの行動は、重大な民事、刑事と行政処罰、損害賠償、罰金、引き渡し、連邦医療保険、医療補助および他の連邦医療保健計画から除外される可能性があり、個人監禁、契約損害、名声損害、利益および将来の収益の減少、これらの法律違反に関する告発を解決し、私たちの業務を削減することを含む、私たちの業務および財務結果に重大な影響を与える可能性があります。いずれも私たちの業務運営能力と運営結果に悪影響を及ぼす可能性があります。
私たちは戦略的取引に従事するかもしれません。これは私たちの流動性に影響を与え、私たちの費用を増加させ、私たちの経営陣に大きな妨害を与えるかもしれません。
私たちは、企業の買収、資産購入および知的財産権、製品または技術の外部許可または内部許可のような戦略取引を時々考慮することができ、内部許可および現在の候補製品を取得する上での方法と同様である。将来のいかなる取引も、私たちの短期的および長期的な支出を増加させ、私たちの株式証券(私たちの普通株を含む)の潜在的な希釈発行、または債務、または負債、償却費用、または過程で得られた研究開発費を発生させる可能性があり、これらのいずれも、私たちの財務状況、流動性、および運営結果に影響を与える可能性がある。私たちが将来考慮する可能性のある他の潜在的な取引は、剥離、戦略パートナー関係、合弁企業、再編、資産剥離、業務合併と投資を含む様々な商業手配を含む。将来の買収はまた私たちが追加的な融資を受ける必要があるかもしれないし、これらの融資は優遇条項で提供されないかもしれないし、全くないかもしれない。このような取引は決して成功しないかもしれないし、多くの時間と経営陣の注意が必要かもしれない。また、私たちが将来買収する可能性のある任意の業務の統合は、私たちの既存の業務を混乱させる可能性があり、複雑で、リスクとコストの高い努力である可能性があり、買収のすべてのメリットを実現することができないかもしれません。したがって、私たちが上記の性質の任意の追加取引を行うか、成功するかは保証できませんが、私たちが実際に達成した任意の追加取引は、私たちの業務、運営結果、財務状況、および見通しに重大な悪影響を及ぼす可能性があります。
私たちの知的財産権に関するリスクは
私たちの成功は私たちの知的財産権とノウハウを保護する能力にかかっている。
私たちのビジネス成功は、私たちの候補製品、ノウハウ、およびその用途のために特許保護および商業秘密保護を取得および維持する能力、および他人の固有の権利を侵害することなく運営する私たちの能力にある程度依存する。もし私たちが私たちの知的財産権を保護できない場合、あるいは私たちの知的財産権が私たちの技術や私たちの候補製品を満たすのに十分でなければ、私たちの競争地位が損なわれる可能性がある。私たちは一般に、私たちの候補製品、ノウハウ、およびその用途に関連する特許出願を米国および海外に提出することによって、私たちの固有の地位を保護しています。これらの特許は、私たちの業務に非常に重要です。我々の特許出願は、そのような出願が特許を取得するまで、またはそのような出願が特許を取得するまで、またはそのような出願で主張される技術を実施する第三者に対して強制的に実行することはできず、発表された特許請求の範囲が技術の範囲をカバーすることに限定される。私たちの特許出願が特許の発行につながることが保証されないか、または発行された特許は競争相手からの攻撃から十分な保護を提供する
類似した技術は,特許が一旦発行されると,第三者に侵害されたり,設計されたり,無効にされない保証もない.発行された特許が後に無効であると認定されたとしても、強制的に執行されないか、または第3の方向の各特許庁または裁判所が提起した訴訟において修正または撤回される可能性がある。未来の私たちの所有権に対する保護の程度は不確実だ。限られた保護だけを提供するかもしれないし、私たちの権利を十分に保護できないかもしれないし、私たちがどんな競争優位性を獲得したり維持したりすることを可能にするかもしれない。これらの不確実性および/または制限は、私たちの財務状況および運営結果に重大な悪影響を及ぼす可能性があり、私たちの候補製品に関連する知的財産権を適切に保護することができる。
私たちは米国および他の国で発行された特許を持っているにもかかわらず、米国の他の係属中の特許出願、対応する国際特許出願、および特定の国の特許出願における私たちの権利要件が、米国特許商標局または米国特許商標局または外国の特許庁および裁判所によって出願可能特許とみなされることを決定することはできず、私たちが発行した特許における権利要件が挑戦されたときに無効または実行不可能であると認定されないことも決定できない。
特許出願プロセスは多くのリスクと不確実性の影響を受けており、私たちまたは私たちの潜在的な未来のパートナーが特許を取得して保護することによって、私たちの候補製品を成功的に保護することを保証することはできない。これらのリスクと不確実性には
•米国特許商標局および様々な外国政府特許機関は、特許プロセス中にいくつかのプログラム、文書、費用支払い、および他の規定を遵守することを要求し、これらの規定を遵守しないことは、特許または特許出願が放棄または失効され、関連する司法管轄区域において特許権の一部または全部を喪失させる可能性がある
•特許出願は特許付与を招いてはならない
•特許は、疑問、無効宣言、修正、撤回、回避、実行不可能が発見されるか、または他の方法ではいかなる競争優位性も提供されない可能性がある
•私たちの競争相手、その多くの人は私たちよりもはるかに多くの資源を持っていて、その中の多くの人は競争技術に重大な投資を行い、彼らは特許を求めたり、獲得したかもしれません。これらの特許は、私たちの候補製品を製造、使用、販売する能力を制限、妨害、または阻止します
•世界的な健康問題に関連する公共政策として、アメリカ政府と国際政府機関は大きな圧力に直面する可能性があり、アメリカ国内と国外の成功が証明された疾病治療方法に対する特許保護範囲を制限することが求められている
•米国裁判所が支持する特許法と比較して,米国以外の国の特許法は特許権者にそれほど有利ではない可能性があり,これにより外国の競争相手により良い機会を与え,競争製品を創造,開発,マーケティングすることができる。
特許訴訟プロセスも高価で時間がかかり、私たちは合理的なコストで、またはタイムリーに、または商業的利点を持つ可能性のあるすべての司法管轄区域を保護して、すべての必要または望ましい特許出願を提出し、起訴することができないかもしれない。我々は,特許保護を得るのが遅くなるまで,我々の研究開発成果で特許を申請できることを確認できない可能性もある.さらに、場合によっては、私たちは、第三者から許可された技術に対する特許出願の準備、提出および起訴、または特許を維持する権利を制御する権利がない。私たちはまた、許可された特許権を実行するために許可側の協力が必要である可能性があり、このような協力は提供されない可能性がある。したがって、このような特許および出願は、私たちの業務の最適な利益に合った方法で起訴され、強制されてはならない。私たちは、私たちのライセンシーの特許起訴および維持活動が適用された法律および法規を遵守しているか、またはそのような特許またはそのような出願が発行される可能性のある任意の特許の有効性および実行可能性に影響を及ぼす可能性があることを確認することができない。もし彼らがそうしなければ、これは私たちが許可している任意の知的財産権を適用する権利を失う可能性がありますので、私たちが製品や候補製品を開発して商業化する能力は不利な影響を受ける可能性があり、競争相手が競合製品を製造、使用、販売することを阻止できないかもしれません。
さらに、私たちは、私たちの従業員、外部科学協力者、CRO、第三者メーカー、コンサルタント、コンサルタント、および他の第三者などと、私たちの研究開発成果を得る権利のある特許に関する当事者と秘密保持および秘密協定を締結したにもかかわらず、いずれもこのような合意に違反し、特許出願を提出する前にそのような成果を開示し、特許保護を求める能力を危険にさらす可能性がある。
もし私たちが第三者に知的財産権を許可する協定に義務を履行できなければ、セラルチニあるいは,我々とライセンス側との業務関係が中断した場合には,我々の業務に対して非常に重要なライセンス権を失う可能性がある.さらに、私たちのいくつかの許可協定には、第三者からの再許可が含まれているセラルチニ私たちは直接許可者たちがその元の許可協定の下の義務を遵守することに依存しなければならない。
私たちは多くのライセンス契約の締約国であり、これらの合意に基づいて、私たちは私たちの業務に重要な知的財産権を付与され、私たちは将来他のライセンス協定を締結する可能性があります。例えば、2017年10月には、特定の知的財産権の独占許可を得るために、Pulmokine,Inc.と独占ライセンス契約を締結し、開発および商業化を行っていますセラルチニ.
これらと私たちの他の既存のライセンス協定は、将来、私たちが知的財産権を許可することを許可するライセンス契約は、様々な開発、規制、および/または商業的な職務調査義務、マイルストーンおよび/または印税の支払い、およびその他の義務を負担することを要求すると予想されます。もし私たちがこれらの合意の下で私たちの義務を履行できなかった場合、あるいは私たちが破産に関連した訴訟を受けた場合、許可側は許可を終了する権利がある可能性があり、この場合、許可がカバーする製品を販売することができません。さらに、私たちの既存のいくつかのライセンス協定は、関連する知的財産権を含まない元のライセンス者の第三者からの従属ライセンスを含むセラルチニそれは.これらの合意によれば、私たちは、知的財産権を適用する権利を得るために、主なライセンスプロトコルの下での義務を私たちの直接ライセンシーに依存しなければならず、私たちは、その権利の元のライセンス者とは何の関係もないかもしれない。もし私たちの許可者がこれらの上流許可協定の下での義務を履行できなかった場合、元の第三者許可者は元の許可を終了する権利がある可能性があり、これは私たちの再許可を終了する可能性がある。このような状況が発生した場合、私たちは関連する権利の所有者から私たち自身の直接許可を得ることができない限り、私たちはこれ以上知的財産権を適用する権利を持たないだろうし、私たちは合理的な条項でそうすることができないかもしれないし、それは関連する知的財産権を含む候補製品の開発と商業化の能力に影響を及ぼすかもしれない。
第三者から許可を得る必要があるかもしれませんが、私たちの研究を進めたり、候補製品の商業化を許可したりすることができ、第三者特許が存在しない保証はありません。そのような許可がなければ、私たちの候補製品に対して強制的に実行されるかもしれません。私たちはもしあれば、商業的に合理的な条項でこのような許可証のいずれかを得ることができないかもしれない。我々が許可を得ることができても,非排他的である可能性があり,我々の競争相手が我々に許可された同じ技術にアクセスできるようにする.この場合、私たちは代替技術を開発または許可するために多くの時間と資源を必要とするかもしれない。もし私たちがそれができなければ、私たちは影響を受けた候補製品を開発したり商業化することができないかもしれません。これは私たちの業務に実質的な損害を与えるかもしれません。このような知的財産権を持つ第三者は、私たちの販売禁止を求めることができますし、私たちの販売に関しては、印税および/または他の形態の賠償を支払う義務があります。知的財産権許可は複雑な法律、商業、そして科学的な問題に関する私たちの業務に重要だ。ライセンス契約によると、私たちとライセンシーとの間で知的財産権紛争が発生する可能性があります
•ライセンス契約に従って付与された権利範囲および解釈に関連する他の問題;
•私たちの技術とプロセスが、ライセンス契約に拘束されていないライセンス側の知的財産権をどの程度侵害しているかどうか
•私たちは特許と他の権利を第三者の権利に再許可する;
•私たちの候補製品の開発と商業化に関するライセンス技術の使用に関する職務義務と、どのような活動がこれらの職務義務を満たしているか
•私たちがライセンスを譲渡または譲渡する権利;および
•私たちの許可者、私たちと私たちのパートナーが共同で知的財産権を創造または使用することによって生成された発明およびノウハウの所有権。
私たちが許可している知的財産権をめぐる紛争が許容可能な条項で現在の許可手配の能力を維持していることを阻害したり、損害を与えたりすれば、影響を受けた候補製品の開発に成功し、商業化することができない可能性があり、私たちの業務に大きな悪影響を及ぼすだろう。
さらに、私たちのいくつかの合意は、私たちがいくつかの取引を完了する能力を制限または延期することができ、これらの取引の価値に影響を与えるかもしれないし、または私たちが特定の活動に従事する能力を制限するかもしれない。例えばもし私たちが
もし私たちがPulmokineとの既存の許可協定の任意の特許製品に関する権利を任意の第三者に再許可または譲渡すれば、私たちはそのような取引に関連するすべての収入の特定の割合をPulmokineに支払う必要があるかもしれない。
もし私たちが獲得した任意の特許保護の範囲が十分に広くない場合、あるいは私たちがいかなる特許保護を失った場合、競争相手が類似または同じ候補製品を商業化する能力が悪影響を受けることを阻止する。
生物製薬会社の特許地位は通常高度に不確定であり、複雑な法律と事実問題に関連しており、近年多くの訴訟のテーマとなっている。したがって,我々の特許権の発行,範囲,有効性,実行可能性,商業的価値は高い不確実性を持っている.私たちの未解決および将来の特許出願は、私たちを保護する候補製品を発行したり、他の競合製品候補製品の商業化を効果的に阻止する特許を発行することにはならないかもしれません。
また,特許出願に要求されるカバー範囲は特許発行前に大幅に縮小することができ,その範囲は特許発行後に再解釈することができる.私たちが現在または未来に所有している特許出願が特許の形で発表されても、それらは私たちに任意の意味のある保護を提供し、競争相手または他の第三者が私たちと競争することを阻止し、または他の方法で私たちにどんな競争優位性を提供してくれるかの形で発表されることはない。私たちが持っている任意の特許は、第三者の挑戦または回避されるかもしれないし、第三者の挑戦のために範囲を縮小したり、無効にしたりする可能性がある。したがって、私たちは私たちの候補製品が保護可能なのか、まだ効果的で実行可能な特許によって保護されているのか分からない。私たちの競争相手または他の第三者は、類似または代替技術または製品を非侵害的に開発することによって、私たちの特許を回避することができ、これは、私たちの業務、財務状況、運営結果、および将来性に大きな悪影響を及ぼす可能性があります。
特許の発行は、その発明性、範囲、有効性、または実行可能性に対して決定的ではなく、私たちの特許は、私たちの候補製品を含まないかもしれない、または米国および海外の裁判所または特許庁で挑戦される可能性がある。私たちは、第3の方向の米国特許商標局が提出した既存技術の発行前に提出されるか、または米国特許商標局または外国特許庁が、私たちの特許権の反対、派生、撤回、再審査、許可後審査またはPGR、当事者間審査またはIPRまたは他の同様の手続きに挑戦することに関与する可能性がある。法的に無効と実行不可能と断言された後の結果は予測できない。例えば、有効性の問題については、私たちは無効な以前の技術がないとは確信できないが、私たちまたは私たちの前任者と特許審査員は起訴中にこれを知らない。私たちの特許および特許出願または私たちのライセンシーの特許および特許出願に関連するすべての潜在的以前の技術が発見されたことは保証されない。私たち、私たちの先輩、またはライセンシーが知られていないことも保証されないが、私たちは、私たちの特許および特許出願が可能かもしれない特許または特許出願における特許請求の有効性または実行可能な以前の技術に影響を与えると信じていないが、最終的には、特許請求の有効性または実行可能性に影響を与えることが発見される可能性がある。このような提出、訴訟、または訴訟における不利な裁決は、私たちの特許権の範囲を縮小したり、私たちの特許権を無効にしたり、実行できなくしたりする可能性があり、第三者が私たちに支払うことなく、私たちの候補製品を商業化し、私たちと直接競争することを可能にします。このような特許権の喪失、排他性の喪失、または特許主張の縮小、無効または実行不可能は、類似または同じ技術および製品の使用を阻止したり、それを商業化する能力を制限したり、候補製品の特許保護期間を制限したりする可能性がある。最終的な結果が私たちに有利であっても、このような手続きは大量のコストを招く可能性があり、私たちの科学者と経営陣に多くの時間がかかる必要がある。さらに、私たちの特許および特許出願によって提供される保護の広さまたは強度が脅かされている場合、結果にかかわらず、会社が現在または将来の候補製品を許可、開発、または商業化することを阻止することができる。
私たちのいくつかの候補製品の特許保護と特許起訴は第三者にかかっているかもしれない。
私たちまたは私たちの許可者は、開発および商業化活動の過程で作られた発明の特許可能な側面を識別できないかもしれません。そうでなければ、遅すぎて、特許保護を受けることができません。したがって、私たちは私たちの特許地位を強化する予想された潜在的な機会を逃すかもしれない。我々の特許または特許出願の準備または提出中に、例えば、適切な優先権請求、リスト、特許請求の範囲、または特許期限調整要求に関して、将来的に生じる可能性のある形態的欠陥が存在するか、または将来的に生じる可能性がある。もし私たちまたは私たちの許可者が、現在であっても将来であっても、そのような特許および他の知的財産権を確立、維持または保護することができなかった場合、そのような権利は減少またはキャンセルされる可能性がある。もし私たちの許可者がいかなる特許権を起訴、維持、または実行する上で完全に協力しないか、または私たちの意見に同意しない場合、これらの特許権は損害を受ける可能性があります。私たちの特許または特許出願が形態、準備、起訴または実行において重大な欠陥がある場合、そのような特許は無効および/または強制的に実行できない可能性があり、そのような出願は有効で強制的に実行可能な特許を決して生成しない可能性がある。これらの結果のいずれも、第三者の競争を阻止する能力を弱める可能性があり、第三者競争は私たちの業務に悪影響を及ぼす可能性がある。
第三者の被許可者として、私たちは第三者に依存して特許出願を提出し、起訴し、特許を維持し、私たちのいくつかの許可協定に従って許可された知的財産権を保護する。私たちの特定の特許や特許出願と他の知的財産権についても、私たちはこれらの活動を主に制御していません
権利です。私たちは、第三者のこのような活動が適用された法律および法規に準拠しているか、または効果的かつ強制的に実行可能な特許または他の知的財産権をもたらすだろうと判断できない。私たちのいくつかのライセンシーと締結されたライセンス契約の条項によると、ライセンス者は、私たちのライセンス特許の実行を制御したり、これらの特許が無効であると主張する任意のクレームを抗弁する権利がある可能性があり、たとえ私たちがこのような強制執行または抗弁を許可されても、私たちライセンシーの協力が必要となるであろう。私たちは、私たちの許可者が十分な資源を割り当てているかどうか、またはそのような特許の実行またはこのような主張の弁護を優先して、許可特許における私たちの利益を保護するかどうかを決定することはできません。たとえ私たちがこれらの法的行動の一方でなくても、不利な結果は私たちの業務を運営するために必要かもしれない知的財産権を許可し続けることを阻止するかもしれないので、私たちの業務を損なう可能性がある。もし私たちの任意のライセンシーまたは私たちの未来の任意のライセンシーまたは未来の協力者が、私たちの任意の候補製品をカバーする特許保護を適切に起訴し、維持することができなければ、私たちがこれらの候補製品を開発し、商業化する能力は悪影響を受ける可能性があり、私たちは競争相手が競合製品を製造、使用、販売することを阻止できないかもしれない。
さらに、私たちが第三者から取得または許可された特許および特許出願に対する特許起訴を制御する権利があっても、私たちの先輩は、私たちが特許起訴を制御する前の人およびその弁護士の行動または不作為の悪影響または損害を受ける可能性がある。
私たちが様々な第三者から取得したり許可したりする技術は保留権利の制約を受けるかもしれない。私たちの先輩は、一般に、非商業的学術および研究用途に基礎技術を使用する権利、その技術に関連する研究の一般的な科学的発見を発表する権利、およびその技術に関連する情報を従来の科学および学術的に開示する権利を含む、彼らと私たちとの合意に従っていくつかの権利を保持することができるかもしれない。私たちの先輩を監視することは難しいかもしれませんが、彼らの技術の使用をこれらの用途に制限しているかどうか、悪用すると、許可技術の権利を強制的に執行するために多くの費用が発生する可能性があります。
取得または許可された技術を利用する能力が制限されている場合、または重要な許可内技術の権利を失った場合、私たちは私たちの製品を開発、許可、マーケティング、販売することに成功できないかもしれません。これは新製品の発売を阻止または延期する可能性があります。私たちの業務戦略はライセンスと買収の技術を商業製品に開発することに成功しています。したがって、これらの技術を利用する能力に対するいかなる制限も、私たちの候補製品を開発、許可、またはマーケティング、販売する能力を損なう可能性があります。
私たちの知的財産権のいくつかは政府が援助したプロジェクトによって発見されたので、“パレード”の権利、いくつかの報告要件、およびアメリカに本部を置く会社の選好のような連邦法規の制約を受ける可能性がある。これらの規定を遵守することは、私たちの独占権を制限し、非アメリカメーカーと契約を締結する能力を制限するかもしれない。
私たちが許可を得たり、または将来獲得または許可する可能性のある知的財産権のいくつかは、米国政府の資金を使用することによって生成される可能性があるため、いくつかの連邦法規の制約を受ける可能性がある。例えばいくつかの研究や開発はセラルチニ政府の研究資金によって支援されていますしたがって、1980年の“ベハ-ドール法案”または“ベハ-ドール法案”によると、アメリカ政府は私たちの候補製品に具現化されたいくつかの知的財産権に対していくつかの権利を持っている可能性がある。米国政府のこれらの権利は、任意の政府の目的のために発明を使用する非排他的、譲渡不可能、撤回不可能な世界的許可を含む。さらに、いくつかの限られた場合、米国政府は、(I)発明を商業化するのに十分なステップを取っていない場合、(Ii)政府は、公衆衛生または安全需要を満たすために行動しなければならない、または(Iii)政府は、公共使用に対する連邦法規の要求を満たすために行動しなければならない、または(Iii)政府が公共使用に対する連邦法規の要求を満たすために、第三者に上述した任意の発明の独占的、部分的または非独占的許可を付与することを要求する権利がある。付与者が政府に発明を開示していない場合、又は所定の期限内に知的財産権登録申請を提出していない場合、米国政府もこれらの発明の所有権を取得する権利がある。政府援助の計画の下で生成された知的財産権もいくつかの報告要求によって制約されており、この要求を守るには大量の資源が必要かもしれない。さらに、米国政府は、これらの発明を含む任意の製品、またはこれらの発明を使用することによって製造された製品のいずれも、実質的に米国で製造されなければならないことを要求する。知的財産権の所有者または譲受人が、合理的ではあるが成功しない努力をしたことを証明することができ、同様の条項で潜在的な被許可者に許可を付与することができ、これらの許可が米国で大量生産される可能性があり、またはこの場合、国内製造が商業的に不可能である場合、資金を提供する連邦機関は、米国工業のこのような選好を放棄する可能性がある。このような米国工業への偏愛は、このような知的財産権がカバーする製品について非米国製品メーカーと契約する能力を制限するかもしれない。もし私たちの未来のどの知的財産権もアメリカ政府の資金を使用することによって生成されれば、“ベハ-ドール法案”の条項も同様に適用されるかもしれない。
知的財産権は必ずしも私たちの競争優位に対するすべての潜在的な脅威を解決できるとは限らない。
私たちの知的財産権が提供する未来の保護の程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できないか、あるいは競争優位性を維持することができるかもしれない。例えば:
•他の人は私たちの候補製品と似たような製品を開発することができるかもしれないが、これらの製品は私たちが所有または許可した特許請求の範囲内ではない
•私たちまたは私たちのライセンス者または先輩は、私たちが所有または許可した発行特許または特許出願によってカバーされた最初の発明をした最初の人ではないかもしれない
•私たちまたは私たちのライセンシーや先輩は、私たちのいくつかの発明をカバーする特許出願を最初に提出した会社ではないかもしれない
•他の人は私たちの知的財産権を侵害することなく、類似または代替技術を開発したり、私たちの任意の技術を複製したりすることができる
•私たちが処理している特許出願は発行された特許を生成しない可能性がある
•競争相手の法的挑戦のため、私たちが所有または許可した発行された特許は、無効または強制執行不可能と認定される可能性がある
•私たちの競争相手は特許権のない国で研究や開発活動を行い、これらの活動から学んだ情報を利用して競争力のある製品を開発し、私たちの主要な商業市場で販売するかもしれません
•私たちは他の特許を申請できるノウハウを開発しないかもしれない
•他の人たちの特許は私たちの業務に悪影響を及ぼすかもしれない。
このような事件が発生した場合、私たちの業務、運営結果、そして将来性を深刻に損なう可能性がある。
私たちのビジネスの成功は、第三者の特許や他の固有の権利を侵害することなく運営する私たちの能力に大きく依存する。第三者は私たちが彼らの専有権を侵害し、損害賠償責任を招いたり、私たちの開発と商業化努力を阻害したり延期したりする可能性があると主張している。
私たちの商業的成功は第三者の特許と固有の権利の侵害を避けることにある程度依存する。しかし、私たちの研究、開発、および商業化活動は、第三者が所有または制御している特許または他の知的財産権を侵害または他の方法で侵害される可能性がある。他のエンティティは、特許または独自の権利を有しているか、または取得することができ、これは、私たちの候補製品および将来承認される可能性のある製品を製造、使用、販売、提供、または輸入する能力を制限し、または私たちの競争的地位を損なう可能性がある。アメリカ国内外で、生物製薬業界は特許とその他の知的財産権に関連する訴訟を大量に持っており、特許侵害訴訟、異議、再審、知的財産権訴訟、アメリカ特許商標局及び/又は外国特許庁に提起されたPGR訴訟を含む。我々が候補製品を開発している分野には,第三者米国や外国から発行された特許や係属中の特許出願が多く存在する.我々の候補製品の使用または製造に関連する第三者特許または材料、配合、製造方法または治療方法の請求項に記載の特許出願が存在する可能性がある。
バイオ製薬業界の拡張や特許の発行に伴い、我々の候補品が第三者特許権侵害の告発を受けるリスクが高まる可能性がある。特許出願は一定期間秘密であるため、関連出願が公表される前に、任意の候補製品の商業化が第三者特許を侵害する可能性があることを知らない可能性があり、製品候補又は技術に関連する特許出願を初めて提出した会社であることも確認できない。さらに、特許出願が発表されるまでに数年かかる可能性があるため、現在処理されている特許出願がある可能性があり、後で私たちの候補製品が発行された特許を侵害する可能性がある。さらに、特許検索は、特許間の用語の違い、データベースの不完全さ、および特許請求の意味を評価することが困難であるため、我々の技術に関連する可能性のある第三者特許権を識別することは困難である。さらに、第三者は将来的に特許を取得し、使用を主張することができる
私たちの技術はこのような特許を侵害した。第三者が主張するいかなる特許侵害請求も非常に時間がかかり、可能性がある
•費用の高い訴訟を引き起こし否定的な宣伝をもたらす可能性があります
•私たちの技術者と管理者の時間と注意力を移します
•開発が遅れています
•主張された特許が満期になるまで、または法廷で最終的に無効または侵害されるまで、私たちの任意の候補製品を商業化することを阻止する
•費用対効果に基づいて非侵害技術を開発することは不可能かもしれません
•私たちは第三者に重大な責任を負わせます
•私たちに特許料またはライセンス契約を締結することが要求され、これらの協定は商業的に合理的な条項では得られないかもしれない、あるいは全く存在しない、あるいは非排他的である可能性があり、これは私たちの競争相手が同じ技術を獲得することをもたらす可能性がある。
本年度報告10-K表の日付まで、第三者が特許を侵害していると主張していないにもかかわらず、他の人は独自の権利を持っている可能性があり、候補製品の発売を阻止する可能性があります。私たちの特許関連法律訴訟に対して損害賠償を要求し、私たちの候補製品やプロセスに関連する活動を禁止しようとするいかなる行為も、故意に侵害されたと判断された場合、3倍の賠償を含む潜在的な損害賠償責任を負う可能性があり、私たちの候補製品を製造または開発する許可証を取得することを要求します。これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、当社の業務における従業員資源を大量に移転することになる。私たちがこのような訴訟に勝つかどうかは予測できませんし、これらの特許に必要ないかなる許可が商業的に受け入れられる条項で提供されるかどうかも予測できません。また,我々や将来の戦略的パートナーが許可を得ることができても,これらの権利は非排他的である可能性があり,これは我々の競争相手に同じ知的財産権を獲得させる可能性がある.さらに、必要であれば、権利侵害を避けるために、私たちの候補製品やプロセスを再設計できるかどうかを決定することはできません。したがって、司法や行政訴訟で不利な裁決を下したり、必要なライセンスを取得できなかったりすることで、私たちの候補製品の開発と商業化を阻止することができ、これは私たちの業務、財務状況、運営結果を損なう可能性があります。
私たちにクレームを出した当事者は、彼らがより多くの資源を持っているので、私たちよりも複雑な特許訴訟の費用を効果的に維持することができるかもしれない。さらに、知的財産権訴訟や行政訴訟に関連する大量の開示要求により、私たちのいくつかの機密情報が開示によって漏洩される可能性がある。さらに、任意の訴訟の開始および継続によって生じる任意の不確実性は、追加資金を調達する能力に重大な悪影響を及ぼすか、または私たちの業務、運営結果、財務状況、および将来性に重大な悪影響を及ぼす可能性がある。
私たちは私たちの特許を保護または強制的に執行する人かもしれない特許の訴訟に巻き込まれるかもしれないが、これは高価で時間がかかり、成功しないかもしれない。さらに、法廷で疑問が提起された場合、私たちが発行した特許は無効または実行不可能と認定される可能性がある。
競争相手は私たちの知的財産権や私たちの許可側の知的財産権を侵害するかもしれない。権利侵害または無許可の使用を防ぐために、私たちおよび/または私たちの許可者は、費用がかかり、時間がかかる可能性がある侵害請求を要求される可能性がある。さらに、特許侵害訴訟では、裁判所は、私たちが所有または許可した特許が無効であること、強制執行できないこと、および/または侵害されていないと判断することができる。もし私たちまたは私たちの任意の許可者または将来の潜在的協力者が第三者に対して法的訴訟を提起して、私たちの候補製品のうちの1つに対する特許を強制的に執行する場合、被告は私たちの特許の全部または一部が無効であり、および/または強制的に実行できないことを反訴することができる。特許訴訟では、被告が無効および/または実行不可能と主張する反訴はありふれたものだ。有効性を疑問視する理由には、新規性の欠如、明らかな、書面的な記述、または実施できないことを含む、いくつかの法定要件のいずれも満たされていないと言われている。主張を実行できない理由には,特許訴訟に関連する者が米国特許商標局に関連情報を隠蔽したり,起訴中に誤った陳述をしたりした疑惑が含まれている可能性がある.
被告が無効および/または強制執行できない法的主張に勝った場合、私たちは候補製品の少なくとも一部またはすべての特許保護を失うだろう。さらに、私たちの特許および特許出願または私たちの許可者が提供する保護の広さまたは強度が脅かされている場合、会社が現在または将来の候補製品を許可、開発、または商業化することを阻止する可能性がある。このような特許保護の喪失は私たちの業務に実質的な悪影響を及ぼすだろう。
解決策が私たちに有利であっても、私たちの知的財産権に関する訴訟や他の法的手続きは、私たちに巨額の費用を発生させ、私たちの技術や管理者の正常な責任を分散させる可能性があります。このような訴訟または訴訟は、私たちの運営損失を大幅に増加させ、開発活動または任意の将来の販売、マーケティング、または流通活動に使用することができるリソースを減少させる可能性がある。私たちはそのような訴訟や訴訟手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らがより多くの財政資源を持っているので、私たちよりもこのような訴訟や訴訟の費用を効率的に負担するかもしれない。特許訴訟や他の訴訟の開始と継続による不確実性は、市場での私たちの競争能力を損なう可能性がある。さらに、知的財産権訴訟または私たちの知的財産権に関連する他の法的手続きが大量の開示を必要とするため、私たちのいくつかの機密情報は、そのような訴訟または他の手続きで開示によって漏洩される可能性がある。
知的財産権訴訟は不利な宣伝を招き、私たちの名声を損ない、私たちの普通株の市場価格を下落させる可能性がある。
任意の知的財産権訴訟において、提訴の公告及び聴聞結果、動議及び裁決及び訴訟における他の臨時手続がある可能性がある。もし証券アナリストや投資家がこれらの声明が否定的だと思うなら、私たちの既存製品、計画、または知的財産権の知覚価値は低下する可能性がある。したがって、私たちの普通株の市場価格は下がるかもしれない。このような声明はまた私たちの名声や私たちの未来の製品の市場を損なう可能性があり、これは私たちの業務に実質的な悪影響を及ぼすかもしれない。
派生または干渉手順は、発明の優先度を決定するために必要である可能性があり、不利な結果は、関連技術の使用を停止すること、または勝利者から権利許可を得ることを試みることを要求する可能性がある。
第三者によって開始されるか、または我々によって提起されるか、または米国特許商標局によって発表される派生または干渉プログラムまたは外国特許庁の同様のプログラムは、我々の特許または特許出願に関連する発明の優先度を決定するために必要である可能性がある。不利な結果は、私たちが関連技術の使用を停止することを要求するか、または勝利者から許可を得ようとすることを要求するかもしれない。もし勝利者が商業的に合理的な条件で私たちにライセンスを提供しなければ、私たちの業務は損害を受ける可能性があります。私たちのこのような訴訟に対する弁護は失敗する可能性があり、成功しても巨額のコストを招き、私たちの経営陣や他の従業員の注意を分散させる可能性がある。さらに、このようなプログラムに関連する不確実性は、私たちの臨床試験を継続するために資金を調達し、私たちの研究計画を継続し、第三者から必要な技術的許可を得たり、パートナー関係を開発または製造する能力に実質的な悪影響を及ぼす可能性があり、これらの資金は、候補製品を市場に出すのを助けるだろう。
最近の特許改革立法は、私たちの特許出願をめぐる起訴や、私たちが発行した特許の実行または保護をめぐる不確実性とコストを増加させる可能性があります。
2011年9月16日、“ライシー·スミス米国発明法”または“ライシー·スミス法案”が法律に署名された。“ライシー·スミス法案”は米国特許法を多くの重大な改正を行った。このような条項は特許出願起訴方式に影響を与える条項を含み、特許訴訟に影響を及ぼす可能性もある。特に、“ライシー·スミス法案”によれば、米国は2013年3月に“最初に特許出願を提出した発明者”制度に移行し、この制度の下で、他の特許性要件を満たすと仮定して、最初に特許出願を提出した発明者は、要求された発明が第三者が最初に発明したものであるか否かにかかわらず、特許を取得する権利がある。したがって、2013年3月以降であるが、我々が以前に米国特許商標局に特許出願を提出した第三者は、たとえ第三者が発明を行う前に本発明を作成したとしても、我々の発明をカバーする特許を付与することができる。これは私たちが発明から特許出願までの提出時間を認識することを要求するだろう。さらに、有効かつ強制的に実行可能な特許を取得して維持する能力は、我々の技術と従来技術との差が、我々の技術が従来技術よりも特許を取得することを可能にするか否かに依存する。米国およびほとんどの他の国/地域の特許出願は、提出後または発行前の一定期間秘密であるため、(1)私たちの候補製品に関連する特許出願または(2)私たちの特許または特許出願に要求される任意の発明を最初に提出した会社であることを確認することはできない。
ライシー·スミス法案には、特許出願の起訴方法に影響を与え、特許訴訟に影響を与える可能性もあるいくつかの重大な変化も含まれている。これらの措置は、特許訴訟中に米国特許商標局が以前の技術を第3の方向に提出することを可能にすることと、PGR、IPRおよび派生プログラムを含む米国特許商標局によって管理された許可後訴訟手続き(PGR、IPRおよび派生プログラムを含む)が特許有効性を攻撃する追加の手続きとを含む。このような提出または手続きにおける不利な裁決は、私たちの特許権の範囲を縮小したり、実行可能にしたり、無効にしたりして、私たちの競争的地位に悪影響を及ぼす可能性があります。
USPTO手続きの証拠基準は、米国連邦裁判所が特許請求の無効を宣言するために必要な証拠基準よりも低いため、第三者は、USPTO手続において、USPTOが権利請求を無効にするのに十分な証拠を提供する可能性があり、同じ証拠が最初に地域裁判所訴訟で提出された場合には、権利請求を無効にするのに十分ではない。したがって,第三者は米国特許商標局の手続きを用いて我々の特許主張の無効を宣言しようとする可能性があり,第三者が地域裁判所訴訟で最初に被告として疑問を提起すれば,我々の特許主張は無効ではない.したがって、Leahy-Smith法案およびその実施は、私たちの特許出願をめぐる起訴および私たちが発行された特許の実行または保護の不確実性およびコストを増加させる可能性があり、これらのすべては、私たちの業務、財務状況、運営結果、および見通しに大きな悪影響を及ぼす可能性がある。
米国特許法や他の国の法律の変化は、特許の全体的な価値を低下させ、候補製品を保護する能力を弱める可能性がある。
他の生物製薬会社と同様に、私たちの成功は知的財産権、特に特許に大きく依存している。生物製薬産業で特許を取得して実施することは高度な技術と法律の複雑さに関連している。したがって、生物製薬特許を取得して実行することは高価で、時間と固有の不確実性だ。米国及び他の国の特許法又は特許法解釈の変化は、我々の知的財産権の価値を低下させる可能性があり、特許出願をめぐる起訴及び特許出願の実行又は保護の不確実性及びコストを増加させる可能性がある。私たちは、私たちの特許または第三者特許で許容または実行される可能性のある特許請求の範囲の広さを予測することができない。しかも、国会や他の外国立法機関は私たちに不利な特許改革立法を通過するかもしれない。
例えば、米国最高裁判所は近年、いくつかの特許事件に対して裁決を下しているが、場合によっては利用可能な特許保護範囲を縮小するか、場合によっては特許所有者の権利を弱めるかである。我々の将来の特許取得能力に関する不確実性の増加に加えて,このようなイベントの結合は,いったん特許を取得する価値に関する不確実性をもたらしている.米国議会、米国連邦裁判所、米国特許商標局または外国司法管轄区の類似機関の決定によると、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、それによって、私たちが新しい特許を獲得したり、私たちの既存特許と将来獲得可能な特許を実施する能力を弱める可能性がある。
私たちは私たちの特許と他の知的財産権の発明権または所有権のクレームに疑問を受けるかもしれない。
私たちはまた、元従業員や他の第三者が私たちの特許や他の知的財産権の所有権を持っているというクレームを受ける可能性がある。訴訟は、これらと他の挑戦在庫または所有権のクレームに対抗するために必要かもしれない。もし私たちがこのようなクレームを弁護することができなければ、お金の損害賠償を支払う以外に、私たちは貴重な知的財産権を失う可能性がある。このような結果は私たちの業務に実質的な悪影響を及ぼすかもしれない。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。
特許条項は候補製品に対する私たちの競争地位を十分に長く保護するのに十分ではないかもしれない。
特許の寿命は限られている。米国では、すべての維持費が適時に支払われる場合、特許の自然失効時間は、通常、米国で最初の非臨時出願日から20年である。様々な延期があるかもしれないが、特許の有効期限とその提供される保護は限られている。私たちの候補製品をカバーする特許を取得しても、特許有効期限が切れたら、競争製品からの競争を受け入れるかもしれません。候補製品の開発、テスト、規制審査に要する時間を考慮すると、候補製品を保護する特許は、候補製品の商業化前または直後に満了する可能性がある。したがって、私たちの特許の組み合わせは、他社が私たちと似ているか同じ製品を商業化することを排除するために、私たちに十分な権利を提供していないかもしれない。
もし私たちが私たちの候補製品のために特許期間を延長しなければ、私たちの業務は実質的な損害を受けるかもしれない。
FDAが我々の候補製品が発売される時間、期限、および詳細を承認することによれば、私たちの1つ以上の米国特許は、1984年の“薬品価格競争および特許期限回復法”または“ハッジ·ワックスマン修正案”に基づいて限られた特許期間を回復する資格がある可能性がある。ハッジ·ワックスマン改正案は、製品開発およびFDA規制審査中に失われた特許期間の補償として、特許回復期間を最長5年とすることを許可している。各FDAが承認した製品は、FDA規制審査中に失われた特許期間の補償として最大1つの特許を延長することができる。特許期間の延長は,製品承認の日から計14年の期限を超えてはならず,その承認に係る薬品,その使用方法又はその製造方法に関する権利要件のみを延長することができる。私たちの候補製品が規制部門の承認を得られれば、特定の国/地域でも特許期間を延長することができる。しかし、例えば、私たちは延期を承認されないかもしれない
適用期間内に出願されていない、関連特許が満了する前に出願されていない、又は適用要件を満たしていないもの。しかも、特許保護の適用期間や範囲は私たちが要求しているものよりも短いかもしれない。もし私たちが特許期間の延長や回復を得ることができない場合、あるいはどのような延長の期限が私たちが要求したよりも短い場合、私たちの競争相手は私たちの特許が満期になった後に競争製品の承認を得ることができ、私たちの収入は減少する可能性があり、実質的である可能性がある。また、このような状況が発生すれば、私たちの競争相手は私たちの開発と試験への投資を利用して、私たちの臨床と臨床前データを参考にして、他の場合よりも早く彼らの製品を発売するかもしれない。
私たちは世界各地で私たちの知的財産権を保護できないかもしれない。
私たちはすでに米国や他のいくつかの国で特許を出願している特許を発行しているが、世界のすべての国で特許を提出、起訴、保護することは目を引くほど高価であり、米国以外のいくつかの国での知的財産権は米国の知的財産権を広く持っていないかもしれない。また、いくつかの外国の法律は知的財産権の保護の程度はアメリカの連邦や州法律に及ばない。したがって、私たちは、米国以外のすべての国/地域で第三者が私たちの発明を実施したり、米国または他の管轄区域で私たちの発明を使用して製造された製品を販売または輸入することを阻止することができないかもしれない。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの技術を使用して自分の製品を開発することができ、また、他の侵害製品を私たちが特許保護を持っているが法執行力がアメリカに及ばない地域に輸出することもできる。これらの製品は私たちの候補製品と競争するかもしれませんが、私たちの特許や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。
多くの会社は外国の管轄区域の知的財産権の保護と保護に重大な問題に直面している。多くの国の法制度は、特許や他の知的財産権保護の強制執行を支持しておらず、これは、私たちの特許の侵害を阻止したり、私たちの独自の権利を侵害する方法で競合製品をマーケティングすることを困難にする可能性がある。外国の管轄区域で私たちの特許権を強制的に執行する訴訟手続きは、巨額のコストを招き、私たちの努力と注意を私たちの業務の他の側面から移転させる可能性があり、私たちの特許が無効または狭義に解釈されるリスクに直面する可能性があり、私たちの特許出願を発表できないリスクに直面させ、第三者からのクレームを引き起こす可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。したがって、私たちが世界各地で私たちの知的財産権を実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネスメリットを得るのに十分ではないかもしれません。
多くの国に強制許可法があり,これらの法律により特許権者は第三者に強制的に許可を付与される可能性がある。また、多くの国は政府機関や政府請負業者に対する特許の実行可能性を制限している。これらの国では,特許権者の救済措置は限られている可能性があり,このような特許の価値を大幅に低下させる可能性がある。もし私たちが第三者に私たちの業務に関連する任意の特許の許可を与えることを余儀なくされた場合、私たちの競争地位は損なわれる可能性があり、私たちの業務、財務状況、運営結果、および将来性は不利な影響を受ける可能性がある。
私たちの特許保護の獲得と維持は、法規および政府特許機関によって提出された様々な手続き、文書、費用支払い、および他の要求に依存しており、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
定期維持費、継続費、年会費、ならびに特許および/または出願に関連する様々な他の政府費用は、我々の特許および/または出願の有効期間内の異なる時間点で米国特許商標局および各外国特許庁に支払われる。私たちは私たちがこの費用を支払うことを想起させるシステムを持っていて、私たちは第三者が満期になった時にこの費用を支払うことに依存している。また、米国特許商標局および各外国特許庁は、特許出願過程において、いくつかのプログラム、文書、費用支払い、および他の同様の条項を遵守することを要求する。私たちは名声の良い法律事務所や他の専門家を招いて私たちの遵守を助け、多くの場合、不注意は滞納金を支払うことによって、あるいは特定の管轄区域に適用される規則に従って他の方法で是正することができる。しかしながら、場合によっては、規定を遵守しないことは、特許または特許出願の放棄または失効をもたらす可能性があり、それにより、関連する管轄区域の特許権の一部または全部が失われる可能性がある。このような事件が発生すれば、私たちの業務に実質的な悪影響を及ぼす可能性がある。
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの商業と競争の地位は損なわれるだろう。
さらに、私たちは、非特許技術的ノウハウ、技術、および他の独自の情報を含む、私たちのビジネス秘密を保護することによって、私たちの競争地位を維持します。第三者との秘密協定の締結や、従業員、コンサルタント、コンサルタントとの秘密情報や発明協定の締結など、当社のビジネス秘密および非特許ノウハウを保護する措置を講じていますが、このようなすべての合意が正式に実行されている保証はありません。これらの当事者のいずれもが合意に違反し、当社のビジネス秘密を含めて独自の情報を漏洩する可能性があり、十分な救済措置を得ることができない可能性があります。強制執行する
一方が商業秘密を不正に開示または流用することは困難であり,高価で時間がかかると主張した結果,予測不可能であった.しかも、米国国内外のいくつかの裁判所は商業秘密をあまり望んでいないか、または保護したくない。
さらに、第三者は、この情報を取得する可能性があり、またはこの情報または同様の情報を独立して取得する可能性があり、私たちは、彼らが技術または情報を使用して私たちと競合することを阻止する権利がない。もしこのような事件が発生した場合、あるいは私たちが私たちのビジネス秘密の保護を失った場合、これらの情報の価値は大幅に低下し、私たちの競争的地位は損なわれるかもしれない。もし私たちが特許発表前に特許保護を申請しない場合、または私たちの独自技術および他の機密情報を他の方法で秘密にすることができない場合、私たちが特許保護または私たちの商業秘密情報を保護する能力が脅かされる可能性がある。
私たちは競争相手から従業員を誤って雇用したか、または私たちまたは私たちの従業員が彼らの前の雇用主の機密情報や商業秘密を間違って使用または開示したという非難を受けるかもしれない。
バイオ製薬業界では、私たちの従業員のほかに、コンサルタントを招いて候補製品の開発を手伝ってくれていますが、これはバイオ製薬業界ではよく見られます。これらのコンサルタントの多く、および私たちの多くの従業員は、以前に他のバイオ製薬会社に雇用されていたか、または以前に他のバイオ製薬会社に提供されていた可能性があり、または現在、競合他社または潜在的な競合他社を含む他のバイオ製薬会社にコンサルティングサービスを提供している可能性がある。私たちは、私たち、私たちの従業員またはコンサルタントが無意識に、またはその前の雇用主またはその前の顧客または現在の顧客の商業秘密または他の固有の情報を使用または漏洩したという疑惑の影響を受けるかもしれない。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭損害賠償の支払いに加えて、貴重な知的財産権や人員を失う可能性があり、これは私たちの業務に悪影響を及ぼす可能性がある。私たちがこれらのクレームを弁護することに成功しても、訴訟は巨額のコストを招き、私たちの管理チームや他の従業員の注意を分散させる可能性がある。
私たちの普通株に関するリスクは
私たちの普通株の活発さ、流動、そして秩序的な市場は維持できないかもしれない。
私たちの普通株は2019年2月にナスダックで取引を開始したばかりで、私たちの普通株が活発な取引市場を維持できる保証はありません。活発な市場の不足は株を売りたいときや合理的だと思う価格で株を売る能力を弱めるかもしれません。不活発な市場は、株を売却することで資金を調達する能力を弱める可能性もあり、株式を犠牲にして他の業務や技術を買収する能力を弱める可能性があり、逆に私たちの業務に大きな悪影響を及ぼす可能性がある
私たちの普通株の取引価格は非常に不安定かもしれません。私たちの普通株を購入した人は大きな損失を受けるかもしれません。
私たちの株価はずっと変動していて、変動する可能性が高い。2019年2月に私たちの初公募株(IPO)でこれらの株を1株16.00ドルで売却して以来、2023年3月10日まで、私たちの普通株の1株当たりの価格は1株当たり1.18ドルと27.15ドルに達しています。一般的な株式市場、特にバイオ製薬会社の株式市場は極端な変動を経験しており、この変動は往々にしてある会社の経営業績とは無関係である。このような変動のため、投資家は彼らが支払った価格で彼らの普通株を売ることができないかもしれない。私たちの普通株の市場価格は、本“リスク要素”の部分的に議論された要素と多くの他の要素の影響を受ける可能性がある
•私たちは進行中と計画中の臨床試験で被験者の能力を募集しています
•私たちの臨床試験と臨床前研究の結果、私たちの競争相手や私たちの市場分野の他社の試験結果
•私たちの候補製品の規制承認、またはそれの使用に対する特定のラベル適応または患者集団の制限、または規制審査中の変化または遅延;
•アメリカや他の国の規制動向は
•医療支払い制度の構造の変化、特に現在の米国の医療制度の改革を考慮する
•私たちが他の候補製品を取得、許可、または開発する努力の成功または失敗
•私たちや競争相手が開発した革新や新製品
•私たちまたは私たちの競争相手は、重大な買収、戦略的パートナーシップ、合弁企業、または資本約束を発表します
•製造、供給、または流通遅延または不足;
•私たちは、任意の製造業者、サプライヤー、許可者、未来のパートナー、または他の戦略的パートナーシップとの任意の変化
•予想される製品販売と収益性を達成する
•私たちの財務業績や私たちと似ていると思われる会社の財務業績の違い
•バイオ製薬業界の市場状況と証券アナリスト報告または提案の発表;
•当社の普通株式出来高
•追加資金を得ることができません
•社内人と株主が自社株を売却する
•一般的な経済、業界と市場状況その他の事件や要素、その多くは私たちがコントロールできない、例えば新冠肺炎の疫病、ロシアとウクライナの軍事衝突、インフレと金利の変化、金融機関の不安定などである
•キーパーソンの増減
•知的財産権、製品責任、または私たちに対する他の訴訟。
また,過去には,バイオ製薬会社の株価が変動した後,これらの会社の株主がこれらの会社に対して集団訴訟を起こしていた。私たちにこのような訴訟を提起すれば、私たちに巨額の費用が発生し、経営陣の注意と資源を移転させる可能性があり、これは私たちの業務、財務状況、運営結果に実質的な悪影響を及ぼす可能性がある。
私たちはナスダックの持続的な上場要求を満たすことができなくて、私たちの普通株が銘柄を取られるかもしれません.
もし私たちがナスダックの継続上場の要求、例えば会社の管理要求や最低終値要求を満たすことができなければ、ナスダックは私たちの普通株を撤退させる措置を取るかもしれない。このような退市は、私たちの普通株の価格にマイナス影響を与える可能性があり、私たちの普通株の売却または購入を希望する時に私たちの普通株を売却または購入する能力を弱める可能性があります。もし退市事件が発生した場合、私たちが上場要求を遵守するために取ったいかなる行動も、私たちの普通株の再上場を許可し、市場価格を安定させたり、私たちの普通株の流動性を高めたり、私たちの普通株がナスダックの最低購入価格要求以下に下落することを防ぐことを保証することができません。あるいは将来ナスダックの上場要求に合わないことを防止します。
もし私たちの役員、役員、主要株主が共同行動を選択すれば、彼らは株主に提出されたすべての事項を制御または顕著に影響する能力がある。しかも、私たちの現在の多くの役員は私たちの主要株主によって任命されている。
2023年3月10日現在、我々の役員、取締役、5%を超える株主は、私たちが発行した普通株の約67.0%を持っています。したがって、これらの者又は彼らが我々取締役会メンバーに任命された者は、一緒に行動し、我々の取締役会又は株主承認に提出されたすべての事項を制御又は著しく影響する能力があり、我々の経営陣の任命、取締役の選挙及び罷免、任意の重大な取引の承認、並びに我々の管理及び商業事務を含む。このような所有権の集中は、制御権の変更を遅延、遅延、または阻止する可能性があり、我々の合併、合併、買収または他の業務合併に関連することを阻害するか、または潜在的な買収者が要約買収を提出することを阻止するか、または他の方法で我々の業務に対する制御権を獲得しようと試みる可能性があり、たとえそのような取引が他の株主に利益をもたらすであろう。
私たちは現在私たちの普通株に配当金を支払うつもりはありませんので、あなたが投資リターンを達成する能力は私たちの普通株の価格が上昇しているかどうかにかかっています。
私たちは普通株式に対するどんな現金配当金も発表したり支払ったりしたことがない。私たちは現在、将来の収益を維持して、私たちの業務を発展、運営、拡大するために、申告や支払いを期待していません
予測可能な未来に現金配当金を送る。しかも、いくつかの例外を除いて、私たちの信用手配条項は私たちが配当金を支払うことを禁止して、私たちが未来に締結したどんな債務協定もそうかもしれない。したがって、株主のどんな見返りもその株の増価に限定されるだろう。私たちの普通株の株が値上がりする保証はなく、株主が株を購入する価格が変わらない保証さえありません。
私たちの既存株主が公開市場で大量の普通株を売ることは私たちの株価を下落させる可能性があります。
公開市場で私たちの普通株の大量の株を売却したり、これらの売却が発生する可能性があると考えたりすることは、私たちの普通株の市場価格を大幅に低下させ、追加株式証券を売却することで十分な資本を調達する能力を弱める可能性がある。
私たちが発行した普通株を保有する11,495,184株、または2023年3月10日までに発行された普通株総数の約12%を保有し、証券法によりその株式を登録する権利がある。証券法に基づくこれらの株式の登録は、証券法第144条に規定されているように、証券法の下で制限されずに自由に取引されることになるが、付属会社が保有する株式を除く。これらの株主がどのような証券を売却したり、このような株を登録したりすることは、私たちの普通株の取引価格に重大な悪影響を及ぼす可能性がある。
私たちの定款やデラウェア州の法律の条項は、株主が有利だと思う買収を阻止し、経営陣の強固につながる可能性があります。
私たちが改訂·再記述した会社登録証明書および改訂·再述の定款に含まれる条項は、私たちの株式の価値を大幅に低下させ、潜在的な買収対象にするか、取締役会の同意を得ていない支配権の変更や経営陣の変更を延期または阻止する可能性があります。私たちの憲章の文書には以下の内容が含まれている
•3年間の任期を交錯させた分類取締役会は、株主が私たちの取締役会の多数のメンバーの能力を変更することを延期する可能性がある
•役員選挙では投票権が蓄積されておらず、小株主が取締役候補を選挙する能力を制限している
•取締役会が株主にこのような権利を付与して、取締役会の拡大や取締役の辞任、死亡、解任による欠員を埋めるために取締役を選出しない限り、株主は私たちの取締役会の空きを埋めることができない
•少なくとも66-2/3%の取締役を罷免する権利のある株式の承認を求め、無断取締役の罷免を禁止する
•私たちの取締役会は、株主の承認を必要とすることなく、優先株や投票権を含む株式の価格および他の条項を決定し、敵意の買収側の所有権を著しく希釈するために使用される可能性がある
•取締役会は、株主の承認なしに改訂および再記載された会社定款を変更することができる
•少なくとも66-2/3%の投票権のある株式の承認を得なければ、私たちの改正および再記載された会社定款を採択、改正または廃止することができ、または私たちの改正および再記載された会社証明書の取締役の選挙および罷免に関する条項を廃止することができる
•株主が書面による同意で行動することを禁止し、株主に年次会議または株主特別会議での行動を強要する
•デラウェア州衡平裁判所は特定の行動と手続きの専属裁判所になると規定している
•株主特別会議が取締役会のみで開催されることを要求することは、取締役の罷免を含む提案または行動能力を強制的に考慮することを遅延させる可能性がある
•株主が遵守しなければならない事前通知手続は、我々の取締役会候補者を指名したり、株主総会で行動すべき事項を提出したりすることで、潜在的な買収者が代理選挙購入者自身の取締役リストを依頼することを阻止または阻止するか、または他の方法で我々に対する制御権を獲得しようと試みる可能性がある。
私たちはまたデラウェア州一般会社法203節に含まれる反買収条項の制約を受けている。第203条によれば、会社は、一般に、その株式の15%以上を保有する株主と業務合併を行ってはならない。当該株式を保有する株主が当該株式を3年間保有している場合を除き、又はその他の例外を除いて、取締役会は、当該取引を承認した。
我々の会社登録証明書規定を改訂し、再記述することは、デラウェア州衡平裁判所は、私たちと私たちの株主との実質的にすべての紛争の独占裁判所となり、私たちの改正と重述の定款規定は、連邦地域裁判所は、証券法に基づいて提出された任意の訴えを解決する独占的なフォーラムであり、これは、私たちの株主が有利な司法フォーラムを獲得することを制限して、私たちまたは私たちの役員、上級管理職、または従業員との紛争を解決することができるかもしれない。
私たちが改正して再説明する会社登録証明書は、デラウェア州衡平裁判所は、私たちが提起した任意の派生訴訟または訴訟、受託責任に違反した任意の訴訟、デラウェア州一般会社法、私たちの改正および再記載された会社登録証明書、または私たちの改正と再記載の定款に基づいて私たちにクレームを提起した任意の訴訟、または私たちが内部事務原則によって管轄されている任意の訴訟を提起する独占裁判所であることを規定しているが、この排他的裁判所規定は、取引法で規定された責任または責任を強制執行するための訴訟、または連邦裁判所が専属管轄権を持っている他のクレームには適用されないことを前提としている。また、私たちの改正と再記述の付例は、私たちが書面で代替裁判所を選択することに同意しない限り、アメリカ連邦地域裁判所は証券法に基づいて提起された任意の訴因を解決する独占的な裁判所になると規定している。これらの裁判所条項の選択は、私たちまたは私たちの役員、役員、または他の従業員とのトラブルに有利だと考える株主のクレームを司法裁判所で提出する能力を制限する可能性があり、これは、私たちと私たちの役員、役員、および他の従業員に対するこのような訴訟を阻止する可能性があります。しかし、この条項に同意することで、株主は連邦証券法とその下の規則と条例の遵守を放棄したとみなされないだろう。また,他社の会社登録証明書の中で類似した場所条項を選択する実行可能性が法的手続きで問われており,裁判所はこれらのタイプの条項が適用されないか実行不可能であると考える可能性がある.もし裁判所が私たちが改正して再記載した会社の登録証明書または改正および再記載の法律の中で選択された裁判所条項が訴訟で適用されないか、または実行できないことが発見された場合、私たちは他の管轄区域でこのような訴訟の解決に関連する追加費用を発生させる可能性があり、これは私たちの業務や財務状況に悪影響を及ぼす可能性がある。
私たちは純営業損失の繰越と他の税務属性を使用する能力が限られている可能性がある。
私たちの歴史の中で、私たちは大きな損失を受けて、近い将来に利益を達成することを期待していないし、永遠に利益を達成しないかもしれない。私たちが税務目的で損失を出し続ける場合、このような損失は、そのような損失が課税収入(あれば)または満期(あれば)を相殺するために使用されるまで、将来の課税収入(ある場合)を相殺するために繰り越されるだろう。2022年12月31日現在、我々の連邦と州の純営業損失繰越(NOL)はそれぞれ約4.946億ドルと160万ドルである。私たちが満期になる連邦と州NOLは以前に使用されたことがない限り、2034年に期限が切れるだろう。私たちが2017年12月31日以降の課税年度に発生した連邦NOLは期限が切れませんが、2020年12月31日以降の課税年度における私たちの課税収入の80%を相殺するためにしか使用できません。同社は2022年12月31日現在、約20020万ドルの外国NOLを保有している。海外のNOLは無期限に続くことができる。2022年12月31日まで、私たちは約3580万ドルの孤児薬物相殺と連邦研究税収免除、そして1060万ドルのカリフォルニア研究税収免除があります。連邦研究税収控除は2038年に満期になるだろう。カリフォルニア研究税収控除は満期になりません。使用するまで無期限に繰り越すことができます。
私たちのNOLと信用繰越はアメリカ国税局と州税務機関の審査と可能な調整を受けます。また、1986年に改正された国内税法第382条及び383条又は同法規によれば、重要株主(又は株主団体)の所有権利益が3年間スクロール期間中のいくつかの累積変化が50ポイントを超える場合、我々の連邦NOL及び信用繰越は年間制限される可能性がある。似たような規則は州税法と外国税法に適用されるかもしれない。2019年2月の私たちの初公募株については、規則第382と383節による所有権変更を経験しました。したがって、2019年2月までに、私たちが発生した連邦NOLと税金控除は年間制限されるだろう。しかし、今後しばらくの間に課税所得額や所得税負債があると仮定すると、私たちのNOLおよび税収控除は、このような年間制限で未使用になることはないと予想されます。追加の所有権変更が発生した場合、または将来的に私たちの株式所有権の変化によって追加の所有権変更が発生し、その多くが私たちの制御範囲内にない場合、NOLおよび信用繰越はさらに年間制限される可能性があります。課税所得を稼ぐとこのような年間制限は
未来の私たちに対する納税義務と私たちの未来のキャッシュフローを増加させることは不利な影響を受けるかもしれない。これらの資産の将来収益の不確実性を最終的に実現するため、我々のNOLや他の繰延税金資産に関する全額推定準備金を記録した。
私たちは証券集団訴訟に巻き込まれており、将来的に証券集団訴訟の影響を受ける可能性がある。
従来、証券集団訴訟は、ある会社の証券市場価格が下落した後に提起されることが多かった。このリスクは、バイオテクノロジーと製薬会社が近年著しい株価変動を経験しているため、特に私たちと関連している。2020年4月3日、私たち、私たちのいくつかの幹部と取締役、そして私たちのIPOの引受業者は、証券集団訴訟と言われる被告に指定された。改訂された起訴状は、2019年2月8日のIPOに基づいて当社の証券を購入したか、または当社のIPOに遡ることができるすべての投資家を代表して提起されたものであり、私たちおよび私たちのIPOのこれらの役員および取締役および引受業者が虚偽および/または誤った陳述をしたことを告発し、私たちの業務、運営、および将来性に関する重大な不利な事実を開示することができませんでした。2022年9月30日、裁判所は集団訴訟和解協定を承認し、慣用的な解放と和解条項と引き換えに約240万ドルを支払うことに同意した。この訴訟と私たちがどちらかになる可能性のある任意の未来の訴訟は固有の不確実性の影響を受け、調査、弁護、解決は高価で時間がかかる可能性があり、私たちの経営陣の関心や財政や他の資源を分散させる可能性がある。訴訟の結果は必然的に不確実であり、私たちは未来の訴訟を弁護するために多くの資源を費やすことを余儀なくされるかもしれないし、私たちは勝てないかもしれない。私たちが一方として参加している任意の訴訟は、重いまたは不利な判決を招く可能性があり、控訴後に覆すことができない場合があり、または巨額の金銭損害賠償または罰金を支払うことができないか、または私たちの業務、財務状況、運営結果、または株価に悪影響を及ぼす可能性がある同様の不利な条項で解決することを決定する可能性があります
私たちは証券法の意味で小さな報告会社であり、より小さい報告会社に適した様々な報告要求のいくつかの免除を利用することを決定すれば、私たちの普通株の投資家に対する吸引力が低下する可能性がある。
私たちは規模の小さい報告会社です。私たちがより小さな報告会社になる資格がある限り、私たちは、当社の定期報告書や委託書で役員報酬に関する開示義務を減らすことを含むが、これらに限定されない他の非小報告会社に適した上場企業の様々な報告書や他の要件のいくつかの免除を選択することができる。また、私たちが大型加速申告機関とも加速申告機関ともみなされない限り、私たちは免除を継続して使用することができ、2002年に改正されたサバンズ-オキシリー法案第404条またはサバンズ-オキシリー法案の監査人認証要件を遵守しないことができる。
私たちは、最近完成した第2四半期の最終営業日に7億ドル以上の公開流通株を持ち、年収が1億ドル未満、または私たちが最近完成した第2四半期の最終営業日までに2.5億ドル以上の公開流通株を持ち、年収が1億ドル以上に達するまで、規模の小さい報告会社と非加速申告会社である。2023年6月30日現在、2023年12月31日現在の会計年度以降も、規模の小さい報告会社と非加速申請者として申請を提出する資格があるかどうかを再評価する必要があります。私たちは投資家が私たちがこのような免除に依存する可能性があるために私たちの普通株の吸引力が低下していることを発見するかどうか予測できない。したがって、一部の投資家が私たちの普通株の吸引力が低下していることを発見すれば、私たちの普通株はそれほど活発ではない取引市場がある可能性があり、私たちの株価はもっと変動するかもしれない。
一般リスク因子
私たちおよび私たちの任意の第三者製造業者またはサプライヤーは、強力な化学剤および危険材料を使用する可能性があり、これらの材料の不適切な処理、貯蔵または処置に関連するいかなるクレームも、非常に時間的または費用がかかる可能性がある。
私たちおよび私たちの任意の第三者製造業者またはサプライヤーは、生物学的材料、有効な化学剤を使用し、化学品および生物学的製剤、ならびにヒトの健康および環境安全に危険な化合物を含む危険材料を使用することが可能である。私たちの業務と私たちの第三者製造業者とサプライヤーの業務も危険な廃棄物製品を発生させます。連邦、州、地方の法律法規はこれらの材料と廃棄物の使用、発生、製造、貯蔵、処理、処理を管理する。適用される環境法律や法規を遵守することは費用がかかる可能性があり、現在または未来の環境法律と法規は私たちの製品開発努力を損なう可能性がある。しかも、私たちはこのような材料や廃棄物が意外なダメージや汚染をもたらす危険を除去することができない。私たちは特定の生物或いは危険廃棄物保険を受けていません。私たちの財産、意外と一般責任保険は生物或いは危険廃棄物の暴露或いは汚染による損害と罰金は明確に含まれていません。はい
汚染や傷害が発生した場合、損害賠償責任を請求されたり、私たちの資源を超えた罰金が科されたりする可能性があり、私たちの臨床試験や監督管理の承認は一時停止される可能性があります。
いくつかのコストと支出に労災賠償保険を提供していますが、私たちは従業員が危険な材料や他の労災を使用して怪我をする可能性がありますが、この保険は潜在的な責任に対応するのに十分ではないかもしれません。私たちは私たちが生物、危険、あるいは放射性物質を貯蔵したり処分したりすることによって、私たちに提起された有毒侵害に対する保険を維持することはできません。
また、現在または将来の環境、健康、安全法律および法規を遵守するためには、時間の経過とともにより厳しくなることが多い巨額のコストが生じる可能性があります。このような現行または未来の法律法規は私たちの研究、開発、または生産努力を損なうかもしれない。これらの法律および法規を遵守しないことは、巨額の罰金、処罰、または他の制裁または責任をもたらす可能性もあり、これは、私たちの業務、財務状況、運営結果、および将来性に重大な悪影響を及ぼす可能性がある。
我々の情報技術システム、または私たちの任意のCRO、製造業者、他の請負業者またはコンサルタント、または将来の潜在的パートナーのシステムは、故障したり、セキュリティホールに遭遇したりする可能性があり、これは、私たちの製品開発計画を深刻に中断させる可能性があり、これは私たちの業績に大きな影響を与える可能性があります。
セキュリティ対策が実施されているにもかかわらず、私たちの情報技術システムおよび私たちの現在および未来のCROおよび他の請負業者、コンサルタントおよび協力者のシステムは、コンピュータウイルスおよびマルウェア(例えば、恐喝ソフトウェア)、ネットワークセキュリティ脅威、不正アクセス、自然災害、テロ、戦争、ならびに電気通信および電気故障の攻撃および破壊を受けやすい。情報技術システムへの攻撃は頻度,持続性,複雑性,強度の面で増加しており,動機や専門長の異なる複雑で組織的な団体や個人によって実施されている.新冠肺炎の流行と持続的な混合作業環境により、私たちのインターネット技術への依存と私たちの遠隔作業の従業員の数により、私たちはまたより大きなネットワークセキュリティリスクに直面する可能性があり、これはサイバー犯罪者の抜け穴を利用するためにより多くの機会を創出するかもしれない。さらに、不正アクセスまたはシステム破壊のための技術はしばしば変化し、一般に目標に対して起動されるまで識別されるため、これらの技術を予測したり、十分な予防措置を実施することができない可能性がある。私たちはまたセキュリティホールに遭遇する可能性があり、このような抜け穴は長い間発見されないかもしれない。発見されても、攻撃者が法医学証拠の検出を回避し、検出を回避し、除去し、または混同するためのツールおよび技術を使用するために、事件または違反行為を十分に調査または修復することができない可能性がある
私たちと私たちの特定のサービス提供者たちは時々ネットワーク攻撃とセキュリティ事件の影響を受ける。これまでに重大なシステム障害、事故、セキュリティホールを経験したとは思いませんが、このような事件が発生し、私たちの運営中断をもたらしたり、個人識別情報や健康関連情報の不正流出やアクセスを招いたりすると、私たちの商業機密損失によるものであっても、他の同様の中断であっても、私たちの開発計画や業務運営が大きな妨害を受ける可能性があります。完成或いは未来の臨床試験中の臨床試験データの紛失は著者らの監督管理の審査作業の遅延を招く可能性があり、そして著者らのデータの回復或いは複製のコストを著しく増加させる。私たちはまた責任を招く可能性があり、私たちの候補製品のさらなる開発と商業化は延期されるかもしれない。また,我々は第三者に依存して我々の候補製品を生産するため,彼らの計算機システムに関連する類似したイベントも我々の業務に実質的な悪影響を与える可能性がある.連邦、州、外国政府のデータプライバシーおよびセキュリティ法律の下でのいくつかの要求は、会社が特定の個人識別情報に関連するセキュリティホールを個人に通知する義務があり、これは、私たちまたは私たちのサービスプロバイダまたは私たちと戦略関係を構築する組織が経験した脆弱性によるものである可能性がある。セキュリティホールに関する通知や後続行動は、私たちの名声に影響を与える可能性があり、法的費用を含む巨額のコストを発生させ、顧客の信頼を損ない、新しい市場への私たちの拡張を損ない、救済費用を発生させたり、既存の顧客を失ったりします。さらに、私たちの保険カバー範囲は、私たちのシステムの中断または破壊によって引き起こされる可能性のある財務、法律、商業、または名声損失をカバーするのに十分ではないかもしれません。もしどんな妨害やセキュリティホールがプライバシーやセキュリティ法律に違反する可能性がある場合、私たちはまた巨額の罰金、罰金、または責任を受ける可能性があり、これは私たちの業務、財務状況、運営結果、および見通しに悪影響を及ぼす可能性があります。
業務中断は私たちの将来の収入と財務状況を深刻に損害し、私たちのコストと支出を増加させるかもしれない。
私たちの業務は地震、電力不足、電気通信故障、水不足、洪水、ハリケーン、台風、火災、極端な天気条件、その他の自然あるいは人為的災害あるいは業務中断の影響を受ける可能性があり、私たちは主に自己保険です。私たちは私たちの候補製品を生産するために第三者メーカーに依存している。これらのサプライヤーの運営が人為的または自然災害または他の業務中断の影響を受ける場合、候補製品の臨床供給を得る能力が妨害される可能性がある。また、当社の本社はカリフォルニア州サンディエゴにあり、主な地震断層と火域に近く、主要地震断層と火域の近くに位置し、ある地理的地域で強固にされていることが私たちに与える最終的な影響は不明です。この事件の発生
このような業務中断は、私たちの運営と財務状況を深刻に損害し、私たちのコストと支出を増加させる可能性がある。
不利なグローバル経済状況や金融機関側の不利な発展及び関連する流動性リスクは、我々の業務、財務状況及び株価に悪影響を及ぼす可能性がある。
彼は、世界の信用と金融市場は現在、流動性と信用供給の深刻な減少、金利とインフレ率の上昇、消費者自信の低下、経済成長の低下、失業率の上昇、経済安定の不確定を含む極端な変動と妨害を時々経験していると述べた。金融市場と世界経済はまた、ロシアとウクライナの間で持続的な衝突、テロ、または他の地政学的事件を含む軍事衝突の現在または予想される悪影響を受ける可能性がある。米国や他の国がウクライナ紛争を含むこのような紛争に対応するために実施している制裁は、金融市場や世界経済に悪影響を及ぼす可能性もあり、影響を受けた国や他の国のいかなる経済対策も市場や経済の不安定を悪化させる可能性がある。最近,シリコンバレー銀行(SVB)や署名銀行(Signature Bank)の倒産や,連邦預金保険会社(FDIC)におけるそれらの倒産管理は,特定の銀行やより広範な金融機関の流動性リスクや懸念を引き起こしている.財政部、FRB、FDICは共同で声明を発表したが、系統的なリスク例外によると、SVBとSignature Bankの預金者は彼らの資金を抽出することができ、標準FDIC保険限度額を超える預金者であっても、未来の特定の金融機関あるいはより広範な金融サービス業の不利な発展は市場全体の流動性不足を招き、会社が短期運営資金需要を獲得する能力を弱めることができ、追加の市場と経済不確定性をもたらす可能性がある。将来の信用や金融市場の不安定さや経済状況への自信悪化が起こらない保証はない。我々の一般的な業務戦略は、このような経済低迷、流動資金不足、不安定なビジネス環境、または持続不可能で不安定な市場状況のいずれかの悪影響を受ける可能性がある。株式や信用市場が悪化した場合、あるいは金融機関が不利な発展を遂げた場合、短期流動性リスクをもたらす可能性があり、任意の必要な債務や株式融資をより困難にし、コストがより高く、財務と運営契約がより重く、希釈度も高くなる。適時に有利な条件で任意の必要な融資を得ることができなければ、私たちの成長戦略、財務業績、株価に実質的な悪影響を与える可能性があり、臨床開発計画の延期または放棄を要求する可能性がある。さらに、我々の現在の1つまたは複数のサービスプロバイダ、金融機関、製造業者、および他のパートナーは、上記のリスクの悪影響を受ける可能性があり、これは、時間通りおよび予算で運営目標を達成する能力に直接影響を与える可能性がある。
私たちはアメリカとある外国の輸出入規制、制裁、禁輸、反腐敗法律、反マネーロンダリング法律法規の制約を受けている。このような法律基準を遵守することは国内と国際市場での私たちの競争能力を弱めるかもしれない。私たちは違反によって刑事責任と他の深刻な結果に直面するかもしれないし、これは私たちの業務を損なうかもしれない。
我々は、米国の輸出管理条例、米国税関条例、米国財務省外国資産規制事務所が管理する様々な経済·貿易制裁条例、改正された米国の1977年の“海外腐敗防止法”、米国“米国法典”第18編201節に掲載された米国国内賄賂法規、米国旅行法、米国愛国者法を含む輸出規制と輸入法律法規に支配されている。そして私たちが活動している国の他の州と国の反賄賂と反マネーロンダリング法。腐敗防止法は広く解釈され、会社およびその従業員、代理人、臨床研究組織、請負業者および他の協力者およびパートナーが直接または間接的に許可、承諾、提供、勧誘、または不当な支払い、または任意の他の価値のある公共または民間部門の受領金を受け入れることを禁止する。私たちは第三者を招いてアメリカ以外で臨床試験を行い、商業化段階に入った後に私たちの製品を海外に販売し、および/または必要な許可、許可証、特許登録、その他の規制承認を得ることができる。私たちは政府機関や政府付属病院、大学、その他の組織の役人や従業員と直接または間接的な相互作用を持っている。私たちは、私たちが明確に権限を持っていなくても、または実際にこれらの活動を理解していなくても、従業員、代理店、臨床研究組織、請負業者、および他の協力者およびパートナーの腐敗または他の不正活動に責任を負わなければならないかもしれない。上記の法律および条例に違反するいかなる行為も、重大な民事と刑事罰金と処罰、監禁、輸出入特権の喪失、資格取り消し、税額の再評価、契約違反および詐欺訴訟、名誉損害、およびその他の結果を招く可能性がある。
また、米国の輸出規制法と経済制裁は、米国の制裁対象国、政府、個人に特定の製品やサービスを提供することを禁止している。他国の軍事衝突により、米国がすでにまたは実施可能な制裁は、このような制裁がカバーする地域内で臨床試験地点で活動し続ける能力に影響を与える可能性がある。例えば、ロシアとウクライナの間の軍事衝突により、米国とその欧州の盟友は最近、ロシアとウクライナのいくつかの業界部門と政党に対する制裁を発表した
ウクライナのドネツクとルガンスク地域、そしてある製品と業界への輸出規制を強化する。これらおよび任意の追加の制裁および輸出規制、ならびにロシアまたは他の管轄区域政府の任意の経済対策は、このような制裁がカバーされる地域内で臨床試験場所で活動し続ける能力に悪影響を及ぼすか、または直接的または間接的に私たちのサプライチェーンを混乱させる可能性がある。もし私たちが輸出入条例とこのような経済制裁を守らなければ、罰金および/または特定の輸出特権の剥奪を含む罰を受けるかもしれない。
上場企業としては、私たちの運営コストが高く、私たちの経営陣は、新たなコンプライアンスを実施するために多くの時間を投入することを求められます。
上場企業として、私たちは巨額の法律、会計、その他の費用を招いた。私たちは、我々の業務および財務状況に関する年間、四半期、および現在の報告書を米国証券取引委員会に提出することを要求する取引法の報告要件を遵守しなければならない。また、サバンズ-オクスリ法案と、米国証券取引委員会とナスダックが後にサバンズ-オクスリ法案条項を実施するために採択された規則は、有効な情報開示と財務制御の確立と維持を要求し、コーポレートガバナンスのやり方を変更することを含む上場企業に重大な要求を提出した。また、2010年のドッド·フランクウォール街改革と消費者保護法によると、米国証券取引委員会はこれらの分野で、私たちの強制的な“報酬発言権”投票要件に適用されるような追加の規則や規定を採択した。株主急進主義、現在の政治環境、および現在の高度な政府介入と規制改革は、大量の新しい法規と開示義務を招く可能性があり、これは追加のコンプライアンスコストを招き、現在予見できない方法で業務を運営する方法に影響を与える可能性がある。
上場企業に適用される規則や条例が増加しており、我々の法律や財務コンプライアンスコストを増加させ続け、いくつかの活動をより時間的で高価にする可能性がある。これらの要求が私たちの経営陣と従業員の注意を他の業務から移すと、それらは私たちの業務、財務状況、運営結果に実質的な悪影響を及ぼす可能性がある。増加したコストは私たちの純収益を減少させ、あるいは私たちの純損失を増加させ、他の業務分野のコストを下げたり、私たちの製品やサービスの価格を向上させることを要求するかもしれません。例えば、これらの規則や条例は、取締役や上級者責任保険を獲得するコストをより高くし、同じまたは同様の保証範囲を維持するために多くの費用を発生させる可能性がある。私たちは、これらの要求に応答して生成される可能性のある追加コストの金額や時間を予測または推定することができない。これらの要求の影響は、私たちの取締役会、取締役会委員会、または役員に参加することをより難しくし、合格した人を引き付け、維持することを可能にするかもしれない。
証券や業界アナリストが私たちの業務に不利な研究報告や報告を発表しなければ、私たちの株価や取引量は低下する可能性がある。
私たちの普通株の取引市場は、証券や業界アナリストが発表した、私たちの業務、私たちの市場、または私たちの競争相手に関する研究と報告にある程度依存するだろう。もしこれらのアナリストがわが社への報道をやめたら、私たちの株の取引価格はマイナス影響を受けるだろう。もし私たちの1人以上のアナリストを追跡して私たちの株の格付けを下げたら、私たちの株価は下落するかもしれない。これらのアナリストのうちの1人以上が私たちの追跡を停止したり、私たちに関する報告書を定期的に発表できなかったりすれば、私たちの株への興味が減少する可能性があり、これは私たちの株価や取引量を低下させる可能性がある。
もし私たちが財務報告に対して適切かつ効果的な内部統制を維持できなければ、私たちが正確かつタイムリーな財務報告を作成する能力が損なわれる可能性があり、投資家は私たちの財務報告に自信を失う可能性があり、私たちの普通株の取引価格は下落する可能性がある.
サバンズ·オキシリー法404条によると、私たちの経営陣は財務報告書の内部統制の有効性を毎年報告しなければならない。しかし、規模の小さい報告会社および非加速申告会社として、2020年に施行される新米国証券取引委員会規則によると、“加速申告会社”または“大型加速申告会社”とみなされない限り、我々の独立公認会計士事務所は、第404条に基づいて財務報告書の内部統制に対する有効性を証明する必要はない。管理管理層が財務報告内部統制を評価するために達成しなければならない基準の規則は複雑であり、大量の文書、テスト、可能な救済措置が必要である。もし私たちまたは私たちの監査人が財務報告の内部統制に有効であると判断できなければ、投資家は私たちの財務報告に自信を失って、私たちの普通株の取引価格が低下する可能性がある。
私たちの財務報告内部統制は2022年12月31日から施行されることが確認されましたが、今後私たちの財務報告内部統制に重大な弱点や重大な欠陥が存在しないことを保証することはできません。財務報告を内部統制できなかったいかなる行為も、財務状況、運営結果、またはキャッシュフローを正確に報告する能力を深刻に抑制する可能性がある。財務報告書の内部統制に有効であると結論できなければ、あるいは独立公認会計士事務所が確定すれば
我々は、財務報告の内部統制に重大な欠陥または重大な欠陥が存在することについて、同社が第404条の審査を開始すると、投資家は私たちの財務報告の正確性と完全性に自信を失う可能性があり、私たちの普通株の市場価格は下落する可能性があり、私たちはナスダック、米国証券取引委員会、または他の規制機関の制裁または調査を受ける可能性がある。財務報告の内部統制のいかなる重大な欠陥を補うことができなかったり、上場企業に必要な他の効果的な制御システムを実施または維持できなかったりすることは、将来的に資本市場に参入する機会を制限する可能性もある。
税法の変化は我々の財務状況、経営業績、キャッシュフローに実質的な悪影響を及ぼす可能性がある。
新しい収入、販売、使用、または他の税金法律、法規、規則、法規または条例は、米国、アイルランド、またはルクセンブルクで随時発行されることができ、または私たちに不利な解釈、変更、修正、または適用を含むことができ、いずれも私たちの業務運営および財務業績に悪影響を及ぼす可能性がある。これらの変化が発生するかどうか,およびこれらの変化が確実に発生すれば,我々の業務への最終的な影響は予測できない.これらの変化が関連する不確実性の結果を含む負の影響を与える場合、これらの変化は、私たちの業務、財務状況、運営結果、およびキャッシュフローに実質的な悪影響を及ぼす可能性がある。
項目1 B。未解決の従業員のコメント。
適用されません。
項目2.財産.
私たちの会社の本社はカリフォルニア州サンディエゴにあり、現在私たちはそこで約63,667平方フィートのオフィス、実験室、コンパートメント空間を借りています。私たちの会社本部は主に会社、研究、開発、臨床、監督、製造と品質機能に使われています。この施設に対する私たちの賃貸契約は2025年1月に満期になるだろう。私たちの施設は現在の需要に対応するのに十分で、必要があれば、後日商業上の合理的な条項に従って、適切な追加空席を提供すると信じています。
項目3.法的訴訟
私たちは現在どんな実質的な法的手続きの当事者でもない。時々、私たちは法的手続きに巻き込まれたり、正常な業務過程で付随するクレームの影響を受ける可能性がある。結果にかかわらず、弁護および和解費用、資源移転、および他の要因により、このような訴訟またはクレームは私たちに悪影響を及ぼす可能性があり、有利な結果が得られる保証はない
第4項鉱山安全情報開示
適用されません。
第II部
第五項登録者普通株式市場、関連株主事項及び発行者が株式証券を購入する。
市場情報
私たちの普通株はナスダック世界の精選市場に発売され、コードは“GoS”です
普通株保有者
2023年3月10日まで、私たちの普通株流通株は94,984,560株で、約33名の私たちの普通株の記録保持者が持っています。この数字は私たちの株主記録から来ていて、私たちの普通株の利益所有者は含まれていません。彼らの株は様々な取引業者、決済機関、銀行、ブローカー、および他の受託者の名義で保有されています。
配当政策
私たちは私たちの株のどんな現金配当金も発表したり支払ったりしたことがない。私たちは将来の収益(あれば)を残して、私たちの業務運営に資金を提供し、予見可能な未来には何の現金配当金も支払わないつもりです。将来の配当政策に関する任意の決定は、当社の取締役会が当社の財務状況、経営結果、資本要求、業務の将来性及び取締役会が関連する他の要素を考慮した後に適宜行い、任意の未来融資ツールに記載されている制限によって制限される。しかも、私たちの信用手配の条項は私たちが配当金を支払う能力を制限しますが、いくつかの例外は除外します。
株式補償計画に基づいて発行された証券
当社の持分補償計画に関する情報は、参照によって本明細書に組み込まれる本年度報告の第III部分表10-Kの第(12)項を参照されたい。
株式表現グラフ
以下の株式表現グラフは、我々の総株式リターンと(I)ナスダック総合指数および(Ii)ナスダックバイオテクノロジー指数の2019年2月8日(ナスダック全世界精選市場での我々の普通株の取引開始日)から2022年12月31日までの総リターンを比較したものである。次の図は2019年2月8日の終値17.94ドルで我々の普通株に投資し、2019年2月8日にナスダック総合指数とナスダックバイオテクノロジー指数に投資し、配当金を普通株に再投資すると仮定している。グラフ中の比較は,我々の普通株の将来可能性を予測したり指示したりするための表現ではない.
株式証券の未登録販売
2022年12月31日までの年度内に、未登録証券は発行または売却していません。
発行者は株式証券を買い戻す
ない。
プロジェクト6.保留
第7項:経営陣の財務状況と経営成果の検討と分析。
以下の財務状況と経営結果の検討と分析、および私たちの総合財務諸表と本年度報告書の他の部分に関する付記を読むべきです。本討論と分析は、私たちの現在の信念、計画と期待に基づく展望的陳述を含み、リスク、不確定性と仮説に関連する。様々な要因により、“リスク要因”や本年度報告の他の部分に列挙された要因が含まれているため、我々の実際の結果は、これらの前向き陳述で予想される結果とは大きく異なる可能性がある。
概要
我々は臨床段階の生物製薬会社であり,免疫学,炎症や腫瘍学などの疾患分野の治療薬の発見,獲得,開発,商業化に専念している。肺動脈高圧(PAH)治療のためのサラダルチニブを開発している。2022年12月,PAH患者で行ったTorrey第2段階研究の陽性背線結果を発表した。24週間のTorrey第2段階研究の盲点部分を終えた後
患者は開放ラベルの延期試験への参加を登録することができる。私たちは報告が行われているこの結果を期待している
2023年中にラベル拡張試験を開放し、2023年下半期にPAHで第3段階計画を開始する予定です。われわれは再発/難治性原発CNSリンパ腫(PCNSL)の治療のためのGB 5121を開発しており,2021年第2四半期から健康ボランティアを募集して一期臨床試験に参加し,2022年第4四半期に再発/難治性PCNSLと他のまれなCNS悪性腫瘍の1 b/2期STAR CNS研究を開始した。これまでに観察された報酬/リスクプロファイルおよびseralutinib計画を支援する資源の優先順位から,1 b/2段階STAR CNS研究の登録を一時停止することにした.私たちは次の行動を決定するために、この研究のデータ審査委員会と既存のデータを議論する予定だ。多発性硬化症の治療のためのGB 7208を開発している。GB 7208は現在臨床前テストを行っている。われわれは免疫学,炎症,腫瘍学の治療領域にも異なる発展段階にある臨床前計画が複数ある。著者らは経験豊富、技術の優れたチームを集め、その中にリードバイオテクノロジーと製薬会社からの業界ベテラン、科学者、臨床医師と肝心なオピニオンリーダー、及び世界各地からのリード学術センターを含む。私たちの従業員は高度に尊敬し、情熱的なチームで、彼らは尊重、謙虚、透明、包容、奉仕、協力、そして面白い文化を誇りに思っています。私たちの最終目標は患者の命を向上させて延長することだ。
私たちは2015年10月に登録設立され、2017年に運営を開始した。今まで、私たちは主に会社の組織と人員の配置、業務計画、資金の調達、確定、買収と許可、及び臨床前研究と臨床試験を行うことに集中している。私たちは主に株式と債務融資を通じて私たちの業務に資金を提供する。2017年10月から2022年12月31日まで、AシリーズとBシリーズの転換可能な優先株融資の売却、転換可能な手形の発行、2019年2月のIPO完了の収益、私たちの信用手配の収益、私たちが同時に引き受けた5.00%2027年満期の転換可能優先手形または2027年債の公開発行収益、2020年5月の普通株と2022年7月の私たちの普通株の私募収益により、10.621億ドルを調達しました。2022年12月31日まで、私たちは255.7ドルの現金、現金等価物、有価証券を持っています。
設立以来,我々はすでに重大な運営損失が発生しており,予見可能な将来も重大な運営損失が続くと予想される。2022年と2021年12月31日までの年間純損失はそれぞれ2億294億ドルと2.34億ドルだった。2022年12月31日までの累計赤字は10.322億ドル。私たちは、進行と計画中の臨床試験、私たちの研究開発活動の継続と臨床前研究の継続、規制機関の私たちの候補製品の承認を求め、より多くの人員を募集し、私たちの知的財産権を保護し、上場企業に関連する追加コストが発生することに伴い、私たちの費用と運営損失は大幅に増加すると予想される。また、私たちの候補製品が開発と商業化の過程で進展するにつれて、私たちは承認者と他の第三者に記念碑的な支払いを支払う必要があり、私たちはseralutinibを含む私たちの候補製品を彼らから獲得または獲得した。われわれの純損失は四半期間と年度間に大きく変動する可能性があり,これは特にわれわれの臨床試験や臨床前研究の時間や他の研究や開発活動への支出にかかっている。
私たちは開発に成功し、規制部門から1つ以上の候補製品の承認を得ない限り、製品販売から何の収入も得られないと予想され、数年かかると予想される。もし私たちの候補製品が規制部門の承認を得たら、製品販売、マーケティング、製造、流通に関連した巨額の商業化費用が発生すると予想される。したがって、私たちは、相当な製品収入を生成して私たちのコスト構造を支援することができる前に、株式発行、債務融資、または他の資本源(潜在的な協力、許可、および他の同様の手配を含む)によって、私たちの現金需要を満たすことが予想される。しかし、私たちは必要に応じて優遇条件で、または追加資金を調達できないか、またはそのような他の計画を達成することができないかもしれない。私たちが必要な時に資金を調達したり、他の手配を達成できなかったりすることは、私たちの財務状況や私たちの業務計画や戦略を実行する能力に悪影響を及ぼすかもしれません。もし私たちが必要な時にもっと多くの資金を集めることができなければ、私たちは私たちの候補製品の開発や将来の商業化努力を延期、制限、減少または終了させることを余儀なくされるかもしれません。たとえ私たちが自分でこれらの候補製品を開発し、マーケティングすることを望んでいたとしても、私たちの候補製品を開発し、マーケティングする権利を与えます。
新冠肺炎が大流行する
著者らは引き続き積極的に著者らの計画を推進することに伴い、著者らは著者らの主要な研究者と臨床場所と密接な関係を維持し、そして行われている新冠肺炎の全世界大流行が著者らの薬品製造、非臨床活動、臨床試験、期待スケジュールとコストに与えるいかなる影響を持続的に評価し続けた。また臨床試験を続けると同時に
全世界各地の臨床試験が行われているため、新冠肺炎予防措置及び各試験場所と主要な供給者の関係者不足は延期されており、例えば、私たちが行っている多環芳香族炭化水素中のセラルチニブの第二段階試験中のいくつかの場所の登録は2020年に一時閉鎖され、引き続き私たちの現在と未来の試験の完成を延期する可能性があり、現在及び未来の臨床試験のデータ読み出し、起動及びモニタリング、データ収集と分析及びその他の関連活動のスケジュールに直接或いは間接的に影響を与える可能性がある。新冠肺炎の大流行を受け、そしてアメリカ食品と薬物管理局が更新した臨床試験業界ガイドラインと一致し、臨床試験は奪われる可能性があり、ウィルスに感染した患者の治療或いはウィルス伝播の防止を支持する。新冠肺炎が私たちの業務に与える直接的かつ間接的な影響は私たちの予測スケジュールを変える可能性があり、これは私たちの業務、運営結果、財務状況に実質的な悪影響を及ぼす可能性がある。私たちは引き続き私たちの業務に及ぼす新冠肺炎の影響を評価するつもりだ。
経営成果の構成部分
収入.収入
私たちは設立以来何の収入も生まれておらず、予測可能な未来にも製品販売から何の収入も生じないだろう。
運営費
研究開発
研究と開発費用は主に著者らの候補製品の臨床前と臨床開発、及び著者らが生産を停止した臨床候補製品に用いられる。研究·開発費用は発生したことが確認され,研究·開発のための貨物またはサービスを受け取る前になされた支払いは,貨物またはサービスを受けるまで資本化されている。
研究開発費には
•研究開発に参加した個人の賃金、賃金税、従業員福祉、株式ベースの給与費用
•契約研究組織あるいはCRO、研究場所とコンサルタントとの合意による外部研究と開発費用に基づいて、著者らの臨床試験、臨床前と非臨床研究を行う
•研究室用品
•第三者製造業者への費用を含む、臨床試験および臨床前研究のための製品の製造に関連するコスト
•規制要件の遵守に関するコスト;
•施設、減価償却、その他の分担費用は、レンタル料、施設メンテナンス、保険、設備、その他の用品の直接と分担費用を含む。
私たちの直接研究開発費用は主に外部コスト、例えば私たちの臨床試験、臨床前と非臨床研究に関連するCRO、研究場所とコンサルタントに支払う費用、臨床試験材料の製造に関するコストを含む。私たちは私たちの人員と施設関連資源を私たちのすべての研究開発活動に配置した。我々は、外部コストおよび人員支出を1つずつ計画し、その計画に割り当てられた人員資源に基づいて、施設に関連する資源のような公共費用を各計画に割り当てる。株式ベースの報酬および特定の計画に属さない人員および一般費用は、割り当てられていない研究·開発費用とみなされる。
予測可能な未来に、私たちは引き続き私たちの候補製品を開発し、私たちの臨床前計画のための発見と研究活動を展開するにつれて、私たちの研究と開発費用は相対的に安定していると予想される。臨床前と臨床開発自体の予測不可能性のため、著者らは著者らの候補製品の現在或いは未来の臨床前研究と臨床試験の開始時間、持続時間或いは完成コストを確定できない。臨床と臨床前の開発スケジュール、成功の確率と開発コストは期待と大きく異なる可能性がある。私たちは進行中と未来の結果に基づいて、どの候補製品を追求するか、そして持続的な基礎の上で各候補製品にどれだけの資金を提供するかを決定する予定です
臨床前研究と臨床試験、監督管理の発展及び著者らは各候補製品の商業潜在力の持続的な評価を行った。私たちは未来に多くの追加資本を調達する必要があるだろう。
以下の要因により,われわれの臨床開発コストは大きく異なる可能性がある
•患者1人当たりの試験コストは
•承認に必要な試験回数
•実験に含まれる場所の数
•どの国で実験を行っていますか
•条件に適合する患者を登録するのに要する時間長;
•実験に参加した患者数
•患者が受ける投与量
•患者の中退率や中途停止率
•規制当局が要求する潜在的な追加的な安全監視;
•患者が試験とフォローアップの持続時間に参加した
•私たちの候補製品を作るコストとタイミング
•臨床試験の遅延を含む新冠肺炎の疫病発生によるコスト
•私たちの候補製品の開発段階
•私たちの候補製品の有効性と安全性。
現在行われている研究と開発
プロセス開発またはIPR&Dでは、費用には、資産買収の一部として取得されたIPR&Dが含まれているか、または将来代替用途がないライセンス内で、発生時に費用が計上される。
知的財産権研究開発費用には、私たちがPulmokine,Inc.に支払ったseralutinibの許可に関連した前払いとマイルストーン支払い、およびいくつかの臨床前計画の買収に関連する前払いおよびマイルストーン支払いが含まれる。
一般と行政
一般および行政費用は、主に株式ベースの報酬を含む行政、財務および他の行政機能者の賃金および従業員に関連する費用を含む。その他の重大なコストには、施設に関する費用、知的財産権や会社の事務に関する法律費用、会計やコンサルティングサービスの専門費用、保険料が含まれています。
予測可能な未来には、我々の一般的かつ行政的費用は、現在のインフラや上場企業としての運営の継続的なコストを支援するために相対的に横ばいに維持されると予想される。これらの費用には、取引所上場や米国証券取引委員会の要求を遵守することに関する監査、法律、規制、税務関連サービス、取締役や役員保険料の維持、上場企業の運営に関連する投資家関係コストが含まれる可能性がある。
その他の収入,純額
その他の収入(支出)、純額には、(1)現金、現金等価物および有価証券の利息収入、(2)転貸収入、(3)我々の信用手配および2027年手形に関する利息支出、および(4)その他の雑収入(支出)が含まれる。
重要な会計政策と試算
私たちの経営陣は私たちの財務状況と経営結果の討論と分析は私たちの合併財務諸表に基づいています。これらの報告書はアメリカで公認されている会計原則やGAAPに基づいて作成されています。これらの財務諸表を作成するには、連結財務諸表に報告されている資産、負債および費用金額、または資産および負債の開示に影響を与えるために、判断および推定を行う必要があります。私たちの推定は、歴史的経験、既知の傾向、および事件、およびこのような場合に合理的と考えられる様々な他の要素に基づいている。異なる仮定または条件では、実際の結果は、これらの推定とは異なる可能性がある(我々の合併財務諸表付記2参照)。
費用を計算する
連結財務諸表作成過程の一部として、貸借対照表毎の日付までの計上費用を見積もる必要がある。このプロセスは、未締結契約および調達注文を審査し、私たちの担当者とコミュニケーションして、私たちに代わって実行されるサービスを決定し、インボイスを受け取っていない場合、または他の方法で実際のコストを通知している場合に、実行されたサービスレベルおよびサービスによって生じる関連コストを推定することを含む。私たちは、私たちが当時知っていた事実と状況に基づいて、各貸借対照表の日付までの課税費用を推定した。私たちは定期的にサービスプロバイダと私たちの推定の正確性を確認し、必要に応じて調整します。私たちの計算すべき研究開発費用の大きな見積もりには、私たちのサプライヤーが提供する研究開発活動に関連するサービスによるコストが含まれていますが、領収書は受け取っていません。
我々は、我々を代表して研究·開発を行っているサプライヤーとのオファーや契約に基づいて、受信したサービスや費用の見積もりに基づいて、研究·開発活動に関する費用を計算する。これらの合意の財務条項は交渉が必要であり、契約によって異なり、支払いの不均衡を招く可能性がある。場合によっては、私たちのサプライヤーに支払われるお金は、提供されたサービスレベルを超え、研究開発費の前払いにつながる可能性があります。サービス料を計算する際に、サービスを提供する時間帯と各時間の努力度を見積もります。サービス実行の実際の時間または努力の程度が私たちの見積もりと異なる場合、私たちはそれに応じて計算すべき費用または前払い費用を調整します。将来の研究開発活動のための貨物およびサービスの前金は、支払い時に支出するのではなく、活動が行われたときまたは貨物を受信したときに支出される。
実際に発生した金額と実質的に差がないと予想されているにもかかわらず、提供されたサービスの状態および時間の推定が提供されたサービスの実際の状態および時間と異なる場合、これは、任意の特定の時期に報告された金額が高すぎたり、低すぎたりする可能性がある。これまで、このような費用の見積もりと実際に発生した金額との間に実質的な差はありませんでした。
2022年と2021年12月31日までの年間経営実績
以下の表に、2022年12月31日と2021年12月31日までの年間業務報告書データを掲載します
| | | | | | | | | | | | | | | | | |
| 2011年12月31日までの数年間 | | 2022年と2021年の変化 |
| 2022 | | 2021 | |
| (単位:千) |
| 運営費用: | | | | | |
| 研究開発 | $ | 170,919 | | | $ | 170,267 | | | $ | 652 | |
| 現在行われている研究と開発 | 65 | | | 75 | | | (10) | |
| 一般と行政 | 47,609 | | | 45,782 | | | 1,827 | |
| 総運営費 | 218,593 | | | 216,124 | | | 2,469 | |
| 運営損失 | (218,593) | | | (216,124) | | | (2,469) | |
| その他の収入(費用) | | | | | |
| 利子収入 | 1,583 | | | 761 | | | 822 | |
| 利子支出 | (13,880) | | | (19,440) | | | 5,560 | |
| その他の収入 | 1,512 | | | 799 | | | 713 | |
| その他の費用の合計 | (10,785) | | | (17,880) | | | 7,095 | |
| 純損失 | $ | (229,378) | | | $ | (234,004) | | | $ | 4,626 | |
運営費
研究開発
2022年12月31日までの1年間の研究·開発費は1兆709億ドルだったが、2021年12月31日までの年間は1億703億ドルで70万ドル増加したが、これは主にGB 5121の前臨床研究と臨床試験に関連するコストが2310万ドル増加し、シェラルチニブの臨床前研究と臨床試験に関連するコストが1650万ドル増加したためである。中止したGB 004計画の臨床前研究と臨床試験に関するコストは2,090万ドル減少し,他の計画の臨床前研究に関するコストは1,030万ドル減少し,他の中止計画の臨床前研究と臨床試験に関するコスト削減770万ドルで相殺された。
次の表は、2022年12月31日と2021年12月31日までの年間の計画別研究開発費を示しています
| | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 |
| (単位:千) |
| セラルチニ | 62,983 | | | 46,490 | |
| GB 5121 | 50,425 | | | 27,365 | |
| GB 004 | 21,449 | | | 42,338 | |
| その他の計画 | 33,378 | | 43,692 |
| 他の中止された計画 | 2,684 | | | 10,382 | |
| 総研究開発 | $ | 170,919 | | | $ | 170,267 | |
現在行われている研究と開発
2022年12月31日まで、2022年と2021年12月31日までの年度に重大な知的財産権研究開発費はない。
一般と行政
2022年12月31日までの年度の一般·行政費は4760万ドルであったが、2021年12月31日までの年度は4580万ドルで180万ドル増加したが、これは主に株式ベースの報酬コストが510万ドル増加したためであり、2021年の未解決証券訴訟の和解に関する課税コストの240万ドルの減少と保険コストの100万ドルの減少によって相殺された。
その他の費用、純額
2022年12月31日までの年度、他の支出純額は1,080万ドルであったが、2021年12月31日までの年度、他の支出純額は1,790万ドルであり、710万ドル減少した。これは主に利息支出が560万ドル減少し、投資増額や償却に関する他の収入が220万ドル増加したことと、私たちの現金、現金等価物、有価証券の利息収入が80万ドル増加したこと、転貸収入が110万ドル減少したこととその他の収入が50万ドル減少したことによって相殺された。
2021年12月31日まで及び2020年12月31日までの経営実績
当社の2020年12月31日までの年度の財務状況及び経営業績の検討、及び2021年と2020年の業績の比較。経営層の当社の財務状況及び経営業績の検討及び分析表格10-Kの年報2021年12月31日までの年度は,2021年に引用により本MD&Aに組み込まれる.
流動性と資本資源
会社設立以来、私たちはすでに大きな運営損失を受けており、予測可能な未来には引き続き重大な運営損失を受け、永遠に利益を上げない可能性がある。2022年、2021年、2021年12月31日までの累計赤字はそれぞれ10.322億ドルと8.115億ドルだった。
私たちの現金の主な用途は運営費に資金を提供し、主に研究開発支出、次いで一般と行政支出である。運営費に資金を提供するための現金は、これらの費用を支払う時間の影響を受けており、これは私たちの未払い売掛金や売掛金の変化に反映されている。公開市場での買い戻しを促進するために、ルール10 B 5-1取引計画を介して、または他の方法で時々買い戻すことを含む、公開市場取引を通じて2027年の債券を買い戻すこともできる。
Pulmokineとのライセンス契約や他のライセンス·買収プロトコルによると、将来のイベントに依存して、例えば、特定の開発、規制、ビジネスマイルストーンが実現されているかどうかに依存し、これらの合意に基づいて開発された製品の販売に関する特許権使用料の支払いが要求されています。2022年12月31日現在、マイルストーンや将来の製品販売が実現する時期や可能性を見積もることはできません。その他の契約義務には、私たちの信用手配による将来の支払い、2027年の手形、既存の経営リースが含まれています。
会社設立から2022年12月31日までの年度まで、私たちの運営資金は主に転換可能な優先株の売却、転換可能な手形の発行、初公募収益、信用融資収益、同時に引受した2027年手形および普通株公開収益および私募普通株収益から得られた毛収入10.621億ドル。2022年12月31日現在、現金、現金等価物および有価証券255.7ドルを持っている。即時需要を超えた現金は主に保証と流動性のために私たちの投資政策に基づいて投資されるだろう。
2019年5月2日、2019年9月18日と2020年7月2日に改正されたクレジット、保証、保証協定を締結し、融資先は、期限までに融資された3,000万ドルの定期融資を含み、残りの1.2億ドル(1ロット当たり6,000万ドル)を2ロットに分けて取得することができ、使用可能期間、特定の臨床開発マイルストーンの実現状況、最低現金要件、その他の慣行条件を指定することができるクレジット、保証、保証協定を締結した。信用機関でもあります2022年12月31日まで、信用手配の下で抽出できる部分はない
2020年4月10日、私たちは、時々発行される普通株式、優先株、債務証券、権利証、単位を含むS-3表登録声明、または2020年4月10日に自動的に発効するS-3表登録声明を提出した。
2020年5月21日に元金総額2.0億ドルの5.00%転換可能優先債券を発行し、2027年に登録公開で満期になります。2027年に発行された債券金利は年利5.00厘とした。利息は半年ごとに支払い、2020年12月1日から毎年6月1日と12月1日に延滞する。引受割引、手数料、その他の発行コストを差し引くと、2027年の債券の純収益総額は約1兆936億ドル。2027年債券の登録包売公開と同時に、9,433,963株普通株の包売公開を完了した。引受割引、手数料、その他の発行コストを差し引いて、私たちは1.171億ドルの純収益を得ました。我々が同時に発行した2027年手形と普通株は2020年の棚登録声明に基づいて登録された。
2022年3月3日、時々発行される普通株式、優先株、債務証券、権証、単位を含むS-3表の登録声明を提出し、この登録声明は2022年3月3日に自動的に発効した
2022年7月15日、16,649,365株普通株の私募を完了しました。発売費用を差し引くまで、私募の総収益は約1億201億ドル。2022年8月9日、2022年8月9日に自動発効した私募発行の普通株式売却を登録する登録説明書をS-3用紙に提出した。
我々の長期借入金に関するより多くの情報は、本リスト10-K第II部分第8項に含まれる連結財務諸表付記5“負債”に掲載されているので、ここで参考にする。
次の表は私たちの毎年のキャッシュフローをまとめています
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| (単位:千) |
| 経営活動のための現金純額 | $ | (187,032) | | | $ | (188,890) | | | $ | (176,360) | |
| 投資活動提供の現金純額 | (1,035) | | | (117,427) | | | 215,342 | |
| 融資活動が提供する現金純額 | 117,090 | | | 3,329 | | | 312,540 | |
| 現金、現金等価物および限定現金に及ぼす為替レート変動の影響 | (517) | | | (165) | | | 9 | |
| 現金、現金等価物、および制限的現金純増加 | $ | (71,494) | | | $ | (303,153) | | | $ | 351,531 | |
経営活動
2022年12月31日までの1年間に、経営活動は約1.87億ドルの現金を使用し、主な原因は純損失2.294億ドルと営業賃貸負債270万ドルの支払いであり、部分的に減少したのは株による報酬支出4260万ドルと営業賃貸使用権資産の償却260万ドルだった。
2021年12月31日までの1年間に、経営活動には約1億889億ドルの現金が使用されており、主な原因は純損失2.34億ドルであり、一部減少したのは株による報酬支出3200万ドル、長期債務割引と発行コスト償却670万ドル、および研究·開発費580万ドルであった。
2020年12月31日までの年間で、経営活動には約1億764億ドルの現金が使用されており、主な原因は純損失2.34億ドルであり、一部減少したのは株式による報酬支出3870万ドル、知的財産権研究開発支出2340万ドル、および長期債務割引と発行コストの償却390万ドルである。
投資活動
2022年12月31日までの年間で、投資活動は約100万ドルの現金を使用し、主に2.38億ドルの有価証券の購入と40万ドルの不動産や設備の購入によるものだが、一部は2億375億ドルの有価証券の満期日によって相殺されている。
2021年12月31日までの年間で,投資活動は約1億174億ドルの現金を使用し,主に1.52億ドルの有価証券を購入したが,一部は3620万ドルの有価証券満期日で相殺された。
2020年12月31日までの年間で,投資活動は主に3.492億ドルの有価証券の販売と満期日から約2億153億ドルの現金を提供したが,一部は1.09億ドルの有価証券購入および我々の臨床·臨床前計画の許可や買収に関連して第三者に支払われた2340万ドルの前金とマイルストーン支払いによって相殺された。
融資活動
2022年12月31日までの年間で、融資活動は1億171億ドルの現金、私募発行からの収益1.199億ドル、株式オプション行使収益170万ドル、および2019年の従業員株式購入計画に基づいて株を購入する収益120万ドルを提供し、一部は長期債務の元金返済580万ドルで相殺された。
融資活動は2021年12月31日までの1年間に330万ドルの現金を提供し、うち200万ドルは株式オプションを行使する収益、130万ドルはESPPによる株購入収益からである。
2020年12月31日までの年間で、融資活動は3億125億ドルの現金を提供し、主に同時に登録された2027年手形と普通株の引受公開から来ており、純収益はそれぞれ1兆936億ドルと1億171億ドルだった。
資金需要
私たちの現在の運営計画によると、私たちは既存の現金、現金等価物と有価証券、そして私たちの信用手配は、2024年第2四半期までの運営に資金を提供するのに十分であると信じている。しかし、私たちの財務資源がどのくらいの間私たちの運営を支持するのに十分な予測は前向きに述べられており、リスクと不確定要素に関連しており、実際の結果はこれとは大きく異なる可能性がある。私たちは間違っていることが証明される可能性があるという仮定に基づいてこの推定をして、私たちは予想よりも早く私たちの資本資源を使用することができる。また,臨床試験で候補製品を試験する過程は高価であり,これらの試験の進展や費用の時間も不確定である。
私たちの将来の資本需要は多くの要素に依存します
•私たちは未来に行われる候補製品の臨床前研究および臨床試験のタイプ、数量、範囲、進展、拡張、結果、コスト、およびタイミングを行っているか、または選択することができる
•私たちの候補製品のためのコストと時間
•私たちの候補製品に対する規制審査のコスト、時間、結果
•私たちの特許と他の知的財産権を取得し、維持し、実行するコスト
•財務報告の内部統制を強化することを含む上場企業としての義務を履行するために、運営システムの強化と増任者の強化に努めている
•私たちの臨床前と臨床活動の増加に伴い、より多くの人員とコンサルタントの雇用に関連するコスト
•私たちはライセンシーと他の第三者にマイルストーンまたは他の支払いの時間と金額を支払わなければなりません。私たちは彼らから私たちが得た候補製品の許可を得ました
•任意の候補製品が承認された場合、販売およびマーケティング能力のコストおよび時間を確立または確保する
•私たちは第三者支払者から十分な市場受容度、カバー率、および十分な補償を得ることができ、任意の承認された製品のために十分な市場シェアと収入を得ることができる
•協力、許可、および他の同様の計画の条項と時間を確立し、維持する
•私たちが許可または買収する可能性のある任意の製品または技術に関連するコスト;
•新冠肺炎の流行や他の流行病による任意の遅延とコストの増加。
私たちは、相当な製品収入を生成して私たちのコスト構造を支援することができる前に、株式発行、私たちの信用手配、債務融資、または他の資本源(潜在的な協力、許可、および他の同様の手配を含む)によって、私たちの現金需要を満たす予定です。
しかし、私たちは必要に応じて優遇条件で、または追加資金を調達できないか、またはそのような他の計画を達成することができないかもしれない。私たちが株式または転換可能な債務証券を売却することによって追加資本を調達する場合、私たちの株主の所有権権益は希釈されるか、または希釈される可能性があり、これらの証券の条項は、清算または他の私たちの普通株主の権利に悪影響を及ぼす特典を含む可能性がある。債務融資および優先株融資に関与する可能性のある協定は、追加債務を招く、資本支出を行う、または配当を宣言するなど、私たちが具体的な行動をとる能力を制限または制限する契約を含む。もし私たちが第三者との協力、許可、および他の同様の計画を通じて資金を調達すれば、私たちは私たちの技術、将来の収入源、研究プロジェクト、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利である可能性があり、および/または私たちの普通株の価値を低下させる可能性のある条項で許可を付与しなければならないかもしれない。私たちが必要な時に資金を調達したり、他の手配を達成できなかったりすることは、私たちの財務状況や私たちの業務計画や戦略を実行する能力に悪影響を及ぼすかもしれません。もし私たちが必要な時にもっと多くの資金を集めることができなければ、私たちは私たちの候補製品の開発や将来の商業化努力を延期、制限、減少または終了させることを余儀なくされるかもしれません。たとえ私たちが自分でこれらの候補製品を開発し、マーケティングすることを望んでいたとしても、私たちの候補製品を開発し、マーケティングする権利を与えます。
最近の会計公告
本年度報告の他の部分に掲載されている総合財務諸表付記2を参照されたい。
第七A項。市場リスクに関する定量的で定性的な開示。
私たちの市場リスクに対する主な開口は金利感受性であり、金利敏感性はアメリカの金利全体の水準変化の影響を受けている。2022年12月31日現在、私たちの現金と現金等価物は現金、通貨市場基金、商業手形を含み、私たちの有価証券は商業手形と会社債務証券を含む。これらのツールの保守的な性質のため、私たちは金利リスクに重大なリスクがあるとは思わない。金利の変化は100ベーシスポイントで私たちのポートフォリオの総価値に大きな影響を与えない。
私たちの信用手配下の未返済債務の利息年利は(I)保証付き隔夜融資金利、あるいはSOFRに等しく、相応の利差を加え、(Ii)7.00%、SOFR下限2.00%、金利上限16%である。金利の上限を考慮すると、市場金利の引き上げは毎年増加します
利息支出とキャッシュフローは最大210万ドル減少した。2022年12月7日、LIBORの代わりにSOFRを借入基準金利とする“信用手配第3改正案”に署名した。
我々が現地通貨で業務を展開している海外業務では、外貨為替レートの変化に関する市場リスクに直面している。私たちはまたアメリカ以外のサプライヤーと契約を結びます。ある領収書は外貨建てです。私たちはこれらの手配に関連した外貨為替レートの変動の影響を受けるだろう。私たちは現在私たちの外貨為替レートに対するリスクを持っていない。2022年12月31日、2022年12月31日、2021年12月31日まで、外貨建ての資産と負債が最も低く、外貨為替レートが直ちに10%変化すると、我々の総合貸借対照表と総合経営表と全面赤字に約30万ドルの純影響が生じる。
インフレは一般的に労働コストと臨床試験費用を増加させることで私たちに影響を及ぼす。2022年12月31日までの1年間でインフレ率は上昇し、近い将来も上昇し続けると予想される。インフレ要因、例えば私たちの材料、用品、管理費用の増加は、私たちの経営業績に悪影響を及ぼす可能性があります。WWEはインフレが我々の業務、財務状況、あるいは経営結果に実質的な影響を与えるとは考えていないが、インフレ率が上昇し続けると、悪影響を及ぼす可能性がある。未来のインフレと物価の重大な不利な変化は大きな損失を招くかもしれない。
項目8.財務諸表と補足データ
本プロジェクトの要求に基づいて作成された連結財務諸表は,本年度報告第.15項に記載された適用資料から参考方式で本報告に組み込まれ,第F−1ページから列報される。
第九項会計及び財務開示に関する変更と相違。
ない。
第9条。制御とプログラムです
開示制御とプログラムの有効性に関する結論
我々は、米国証券取引委員会に提出された定期的かつ現在の報告で開示すべき情報が、米国証券取引委員会規則および表で指定された期間内に記録、処理、集約および報告されることを保証し、必要に応じて必要な開示に関する決定をタイムリーに行うために、必要に応じて私たちの管理層に伝達されることを保証するために、開示制御および手続きを維持する。開示制御およびプログラムを設計および評価する際、管理層は、任意の制御およびプログラムは、設計および動作がどんなに良好であっても、予想される制御目標を達成するために絶対的な保証ではなく合理的な保証を提供することしかできないことを認識している。合理的な保証レベルを達成するためには、管理層は必然的にその判断を用いて可能な制御とプログラムのコスト-利益関係を評価する必要がある。さらに、任意の制御システムの設計も、将来のイベント可能性のいくつかの仮定に部分的に基づいており、任意の設計がすべての潜在的な将来の条件でその目標を成功的に達成する保証はなく、時間の経過とともに、制御が条件の変化によって不十分になる可能性があり、またはポリシーまたはプログラムを遵守する程度が悪化する可能性がある。費用対効果を有する制御システムの固有の制限により、エラーまたは詐欺によるエラー陳述が発生し、発見されない可能性がある。
我々の経営陣は、最高経営責任者及び最高財務官の参加の下、本年度報告書に記載されている期間が終了した時点で、取引所法第13 a−15(E)及び15 d−15(E)規則に基づいて定義された開示制御及び手続の有効性を評価した。同等の評価によると、我々の主要行政総裁及び主要財務官は、当該日までに、我々の開示制御及び手続が合理的な保証レベルで有効であると結論した。
財務報告の内部統制の変化
2022年12月31日までの四半期内に、財務報告の内部統制には何の変化もなく、これらの変化は、私たちの財務報告の内部統制に大きな影響を与えたり、合理的になったりする可能性がある。
経営陣財務報告内部統制年次報告書
我々の経営陣は、取引所法案第13 a-15(F)及び15 d-15(F)条の規定に基づいて、財務報告の十分な内部統制を確立·維持する責任がある。我々の財務報告に対する内部統制は、財務報告の信頼性と公認会計基準に基づいて外部目的の財務諸表の作成に合理的な保証を提供することを目的とした過程である。その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来的にどのような有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムの遵守度が悪化する可能性がある.
我々の経営陣は、2013年の内部統制総合枠組みでテレデビル委員会後援組織委員会が提案した基準に基づき、2022年12月31日までの財務報告書の内部統制に対する有効性を評価した。この評価に基づき、我々の経営陣は、財務報告書の内部統制が2022年12月31日から有効であると結論した。
プロジェクト9 B。他の情報。
ない。
プロジェクト9 Cです。検査を妨害する外国司法管轄区域を開示する。
適用されません。
第三部
プロジェクト10.取締役、行政、企業管理
このプロジェクトに必要な情報は、米国証券取引委員会に提出された2023年株主総会に関する最終依頼書または最終依頼書に含まれ、最終依頼書は、2022年12月31日までの財政年度終了後120日以内に提出される予定で、タイトルは“取締役選挙”、“会社管理”、“私たちの役員”、および“延滞第16条(A)報告書”であり、引用により本明細書に組み込まれる。
ビジネス行為と道徳的基準
私たちは、私たちのウェブサイトwww.gosamerBio.comで見つけることができる、私たちの高級管理者、役員、および従業員に適したビジネス行動と道徳基準を通過しました。“商業行為および道徳規則”は、2002年の“サバンズ-オックススリー法案”第406節およびS-K条例406項の意味での“道徳基準”に適合することを目的とした最高商業道徳基準に従って当社の業務を展開する一般的な基準を含む。さらに、我々は、(1)我々の最高経営責任者、最高財務官、最高会計官または制御者または同様の機能を実行する者に適用される、我々の商業行為および道徳基準の任意の修正の性質、および(2)これらの指定者のうちの1つを付与する私たちの道徳基準条項の任意の免除の性質を迅速に開示するつもりであり、免除の性質、免除を取得した人の名前、および将来私たちのウェブサイトで免除される日を含む。
第11項.行政職報酬
本プロジェクトに要求される情報は、我々の最終委託書における“役員報酬および他の情報”の節で説明され、参照によって本明細書に組み込まれる。
第12項:特定の実益所有者の保証所有権及び管理職及び関連株主事項。
本条項によって要求される情報は、“特定の利益を受けるすべての人および管理職の保証所有権”というタイトルの私たちの最終委託書に記載され、参照によって本明細書に組み込まれるであろう。
法規S−K第201(D)項で要求される情報は、“役員報酬及びその他の情報”と題する当社の委託書の章に記載され、参照により本明細書に組み込まれる。
第十三条特定関係及び関連取引、並びに取締役の独立性。
本プロジェクトに要求される情報は、我々の最終委託書における“いくつかの関係および関係者取引”、“取締役会独立性”、“取締役会委員会”の節で説明され、参照によって本明細書に組み込まれる。
第14項目主要会計費用とサービス
本プロジェクトに要求される情報は、我々の最終委託書における“独立公認会計士費用”の節に記載され、ここに組み込まれて参考となる。
第4部
項目15.物証、財務諸表付表
(1)すべての財務諸表
Gossamer Bio,Inc.の合併財務諸表および独立公認会計士事務所安永会計士事務所の報告は本年度報告に含まれ,表10−KはF−1ページから始まる。
(2)財務諸表明細書
その中に記載されている資料が連結財務諸表または付記に適用または表示されないことが要求されるので、すべての付表は省略される。
(3)陳列品
本年度報告署名ページの前のテーブル10−Kには、“表インデックス”に展示品リストが設定され、参照により本明細書に組み込まれる。
項目16.表格10-Kの概要
ない。
ミント生物株式会社
連結財務諸表索引
| | | | | |
| ページ |
独立公認会計士事務所報告(PCAOB ID:42) | F-1 |
合併貸借対照表 | F-3 |
企業経営と全面赤字合併報告書 | F-4 |
株主権益合併報告書 | F-5 |
統合現金フロー表 | F-6 |
連結財務諸表付記 | F-7 |
独立公認会計士事務所報告
Gossamer Bio,Inc.株主と取締役会へ。
財務諸表のいくつかの見方
Gossamer Bio,Inc.(当社)2022年12月31日現在、2022年12月31日および2021年12月31日までの連結貸借対照表、2022年12月31日までの3年度の関連総合経営表と全面損失表、株主権益とキャッシュフローおよび関連付記(総称して“合併財務諸表”と呼ぶ)を監査した。総合財務諸表は、すべての重要な点において、会社の2022年12月31日、2022年12月31日、2021年12月31日までの財務状況、および2022年12月31日までの3年度の経営結果とキャッシュフローを公平に反映しており、米国公認会計原則に適合していると考えられる。
ASU第2020-06号を採用
連結財務諸表付記1に記載されているように、会計基準更新(ASU)第2020-06号を採用しているため、債務:変換および他の選択された債務(主題470-20)および派生ツールおよびヘッジを有するエンティティ自身の権益を有する契約(主題815-40)(“ASU 2020-06”)2022年1月1日に施行される
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査することを求められておらず、私たちも招聘されて監査を行っていません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性について意見を述べるためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
以下に述べる重要な監査事項とは、監査委員会に伝達または要求が監査委員会に伝達された当期財務諸表監査によって生じる事項である:(1)財務諸表に対して重大な意義を有する勘定または開示に関するものであり、(2)特に挑戦的で主観的または複雑な判断に関するものである。重要監査事項の伝達は、総合財務諸表に対する私たちの全体的な意見をいかなる方法でも変えることはなく、以下の重要な監査事項を伝達することによって、重要な監査事項またはそれに関連する勘定または開示について個別の意見を提供することはない。
| | | | | | | | |
| | 研究と開発費用を計算すべきである |
関係事項の記述 | | 2022年12月31日までに、会社は研究開発費1560万ポンドを計上しなければならない。総合財務諸表付記2に記載されているように、当社は研究開発コストを推定する計算項目を記録し、第三者建設業者、実験室、臨床試験地点及びその他の機関が行った仕事に参与する対応金を含む。その中のいくつかの請負者の毎月の請求書は、実際に提供されるサービスに基づいており、他のいくつかは、いくつかの契約マイルストーンを実装することに従って定期的に課金される。後者については、会社が貨物やサービスを利用または提供する際には費用を計上しなければならない。臨床試験場費用は患者が試験に入り、試験過程で進展することに従って蓄積された
監査管理層は研究開発費用に対応する会計処理は特に挑戦的であり、会社の研究開発プロトコル下の各活動の進展或いは完成段階を評価することは第三者サービス提供者と内部臨床人員からの大量のデータに依存し、これらのデータは電子フォームと他のエンドユーザ計算プログラムで追跡するからである
|
| | |
私たちが監査でどのようにこの問題を解決したのか | | 会社が計算すべき研究と開発費用の完全性,その他のプログラムをテストするために,重大な臨床試験のための研究と開発活動の支援証拠を得た。私たちは会計と臨床プロジェクトマネージャーとの会議を通じて重大な研究と開発活動の状況を確認した。計算すべき研究開発コストの適切な計測を検証するために,サンプル取引のコストを関連伝票や契約と比較し,第三者サービスプロバイダとこれまでに発生した金額を確認した.我々はまた、計算すべき研究と開発費用の完全性を評価するために、後続支払いのサンプルを検討した |
/s/ 安永法律事務所
2018年以来、当社の監査役を務めてきました。
カリフォルニア州サンディエゴ
2023年3月17日
薄紗生物会社です。
合併貸借対照表
(千単位、株および額面を除く)
| | | | | | | | | | | |
| 十二月三十一日 |
| 2022 | | 2021 |
| 資産 | | | |
| 流動資産 | | | |
| 現金と現金等価物 | $ | 111,973 | | | $ | 183,403 | |
| 有価証券 | 143,705 | | | 141,815 | |
| 制限現金 | — | | | 64 | |
| 前払い費用と他の流動資産 | 6,202 | | | 6,498 | |
| 流動資産総額 | 261,880 | | | 331,780 | |
| 財産と設備、純額 | 3,981 | | | 5,320 | |
| 経営的リース使用権資産 | 5,909 | | | 5,477 | |
| その他の資産 | 680 | | | 1,080 | |
| 総資産 | $ | 272,450 | | | $ | 343,657 | |
| 負債と株主権益 | | | |
| 流動負債 | | | |
| 売掛金 | $ | 1,459 | | | $ | 3,244 | |
| 研究と開発費用を計算すべきである | 15,626 | | | 16,205 | |
| 長期債務の当期部分 | 11,613 | | | — | |
| 費用とその他の流動負債を計算しなければならない | 20,532 | | | 20,410 | |
| 流動負債総額 | 49,230 | | | 39,859 | |
| 長期転換可能優先手形 | 195,709 | | | 150,038 | |
| 長期債務 | 11,988 | | | 29,079 | |
| 賃貸負債を経営しています--長期 | 3,446 | | | 3,218 | |
| 総負債 | 260,373 | | | 222,194 | |
| 引受金とその他の事項 | | | |
| 株主権益 | | | |
普通株、$0.0001額面価値700,000,0002022年12月31日と2021年12月31日までに認可された株94,478,405発行済みおよび発行済み株式94,423,1812022年12月31日までに発行された株と、76,470,588発行済みおよび発行済み株式75,752,6642021年12月31日現在の既発行株 | 10 | | | 8 | |
| 追加実収資本 | 1,044,864 | | | 932,944 | |
| 赤字を累計する | (1,032,223) | | | (811,534) | |
| その他の総合収入を累計する | (574) | | | 45 | |
| 株主権益総額 | 12,077 | | | 121,463 | |
| 総負債と株主権益 | $ | 272,450 | | | $ | 343,657 | |
付記はこれらの連結財務諸表の構成要素である。
薄紗生物会社です。
合併経営報告書と全面赤字
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| 運営費用: | | | | | |
| 研究開発 | $ | 170,919 | | | $ | 170,267 | | | $ | 160,854 | |
| 現在行われている研究と開発 | 65 | | | 75 | | | 23,380 | |
| 一般と行政 | 47,609 | | | 45,782 | | | 49,728 | |
| 総運営費 | 218,593 | | | 216,124 | | | 233,962 | |
| 運営損失 | (218,593) | | | (216,124) | | | (233,962) | |
| その他の収入(費用) | | | | | |
| 利子収入 | 1,583 | | | 761 | | | 3,442 | |
| 利子支出 | (13,880) | | | (19,440) | | | (12,666) | |
| その他の収入,純額 | 1,512 | | | 799 | | | (174) | |
| その他の費用の合計 | (10,785) | | | (17,880) | | | (9,398) | |
| 純損失 | $ | (229,378) | | | $ | (234,004) | | | $ | (243,360) | |
| その他総合(赤字)収入: | | | | | |
| 外貨換算 | (544) | | | (329) | | | 441 | |
| 有価証券は赤字を実現していない | (75) | | | (225) | | | (100) | |
| その他総合収入 | (619) | | | (554) | | | 341 | |
| 総合損失 | (229,997) | | | (234,558) | | | (243,019) | |
| 1株当たり基本と希釈して純損失 | $ | (2.71) | | | $ | (3.13) | | | $ | (3.55) | |
| 加重平均発行済み普通株式、基本普通株式、希釈後普通株 | 84,574,869 | | | 74,843,482 | | | 68,510,260 | |
付記はこれらの連結財務諸表の構成要素である。
薄紗生物会社です。
株主権益合併報告書
(単位は千で、シェアは含まれていない)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | 普通株 | | その他の内容
支払い済み
資本 | | 積算
赤字.赤字 | | 積算
他にも
全面的に
収入(損) | | 合計する
株主.株主
株権 |
| | 株 | | 金額 | | | | |
| 2019年12月31日現在の残高 | | 61,635,477 | | | $ | 7 | | | $ | 686,390 | | | $ | (334,170) | | | $ | 258 | | | $ | 352,485 | |
| 公開発行に関する普通株を発行し,引受割引,手数料,発行コストを差し引いた純額 | | 9,433,963 | | | 1 | | | 117,093 | | | — | | | — | | | 117,094 | |
| 転換可能手形発行の権益部分 | | — | | | — | | | 53,635 | | | — | | | — | | | 53,635 | |
| 債券発行コストは変換可能機能に起因することができる | | — | | | — | | | (109) | | | | | | | (109) | |
| 制限株の帰属 | | 2,557,375 | | | — | | | — | | | — | | | — | | | — | |
| 株式オプションの行使 | | 134,803 | | | — | | | 534 | | | — | | | — | | | 534 | |
| 株に基づく報酬 | | — | | | — | | | 38,748 | | | — | | | — | | | 38,748 | |
| 従業員の株式購入計画に基づいて普通株を発行する | | 113,286 | | | — | | | 1,300 | | | — | | | — | | | 1,300 | |
| その他の追加実収資本 | | — | | | — | | | 16 | | | — | | | — | | | 16 | |
| 純損失 | | — | | | — | | | — | | | (243,360) | | | — | | | (243,360) | |
| その他総合収益 | | — | | | — | | | — | | | — | | | 341 | | | 341 | |
| 2020年12月31日の残高 | | 73,874,904 | | | $ | 8 | | | $ | 897,607 | | | $ | (577,530) | | | $ | 599 | | | $ | 320,684 | |
| 制限株の帰属 | | 906,037 | | | — | | | — | | | — | | | — | | | — | |
| 株式オプションの行使 | | 325,494 | | | — | | | 2,014 | | | — | | | — | | | 2,014 | |
| 株に基づく報酬 | | — | | | — | | | 32,008 | | | — | | | — | | | 32,008 | |
| 従業員の株式購入計画に基づいて普通株を発行する | | 160,790 | | | — | | | 1,315 | | | — | | | — | | | 1,315 | |
| 既存限定株式単位に普通株を発行する | | 485,439 | | | — | | | — | | | — | | | — | | | — | |
| 純損失 | | — | | | — | | | — | | | (234,004) | | | — | | | (234,004) | |
| その他総合損失 | | — | | | — | | | — | | | — | | | (554) | | | (554) | |
| 2021年12月31日現在の残高 | | 75,752,664 | | | $ | 8 | | | $ | 932,944 | | | $ | (811,534) | | | $ | 45 | | | $ | 121,463 | |
| 会計原則変動の累積影響調整(付記2参照) | | — | | | — | | | (53,527) | | | 8,689 | | | — | | | (44,838) | |
非公開発行に関する普通株式発行は,発行コストを差し引いて#ドルである184 | | 16,649,365 | | | 2 | | | 119,944 | | | — | | | — | | | 119,946 | |
| 制限株の帰属 | | 662,700 | | | — | | | — | | | — | | | — | | | — | |
| 株式オプションの行使 | | 270,707 | | | — | | | 1,736 | | | — | | | — | | | 1,736 | |
| 株に基づく報酬 | | — | | | — | | | 42,553 | | | — | | | — | | | 42,553 | |
| 従業員の株式購入計画に基づいて普通株を発行する | | 157,858 | | | — | | | 1,214 | | | — | | | — | | | 1,214 | |
| 既存限定株式単位に普通株を発行する | | 929,887 | | | — | | | — | | | — | | | — | | | — | |
| 純損失 | | — | | | — | | | — | | | (229,378) | | | — | | | (229,378) | |
| その他総合損失 | | — | | | — | | | — | | | — | | | (619) | | | (619) | |
| 2022年12月31日現在の残高 | | 94,423,181 | | | $ | 10 | | | $ | 1,044,864 | | | $ | (1,032,223) | | | $ | (574) | | | $ | 12,077 | |
付記はこれらの連結財務諸表の構成要素である。
薄紗生物会社です。
統合現金フロー表
(単位:千)
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| 経営活動のキャッシュフロー | | | | | |
| 純損失 | $ | (229,378) | | | $ | (234,004) | | | $ | (243,360) | |
| 純損失と経営活動で使用される現金純額の調整: | | | | | |
| 減価償却および償却 | 1,832 | | | 1,740 | | | 1,409 | |
| 株に基づく報酬費用 | 42,553 | | | 32,008 | | | 38,748 | |
| 製品研究開発費 | 65 | | | 75 | | | 23,380 | |
| 経営的リース使用権資産の償却 | 2,597 | | | 3,427 | | | 2,859 | |
| 長期債務割引と発行コストの償却 | 1,163 | | | 6,731 | | | 3,857 | |
| 割引を差し引いた投資割引(割増)償却純額 | (1,405) | | | 339 | | | 98 | |
| 投資はすでに純損失を達成した | — | | | — | | | (256) | |
| 財産と設備処分損失 | — | | | 20 | | | — | |
| 経営性資産と負債変動状況: | | | | | |
| 前払い費用と他の流動資産 | 296 | | | 2,631 | | | (1,641) | |
| その他の資産 | 400 | | | (53) | | | 532 | |
| リース負債を経営する | (2,721) | | | (3,580) | | | (2,851) | |
| 売掛金 | (1,813) | | | (4,428) | | | 6,984 | |
| 費用を計算する | (1,659) | | | 736 | | | 1,096 | |
| 研究と開発費用を計算すべきである | (579) | | | 5,774 | | | (8,827) | |
| 報酬と福祉に計上すべきである | 1,618 | | | (278) | | | 1,612 | |
| 利子支出を計算する | (1) | | | (28) | | | — | |
| 経営活動のための現金純額 | (187,032) | | | (188,890) | | | (176,360) | |
| 投資活動によるキャッシュフロー | | | | | |
| 研究開発資産買収、買収現金を差し引いた純額 | (65) | | | (75) | | | (23,380) | |
| 有価証券を購入する | (238,060) | | | (152,031) | | | (108,968) | |
| 有価証券の満期日 | 237,500 | | | 36,225 | | | 265,678 | |
| 有価証券の販売 | — | | | — | | | 83,515 | |
| 財産と設備を購入する | (410) | | | (1,546) | | | (1,503) | |
| 投資活動提供の現金純額 | (1,035) | | | (117,427) | | | 215,342 | |
| 融資活動によるキャッシュフロー | | | | | |
| 普通株で得た金を公開発行し,純額 | — | | | — | | | 117,110 | |
| 転換可能優先手形を発行して得られた純額 | — | | | — | | | 193,596 | |
| 普通株を非公開で発行して得られる収益は,発行コストを差し引く | 119,946 | | | — | | | — | |
| 従業員の株購入計画に基づいて株式を購入する | 1,214 | | | 1,315 | | | 1,300 | |
| 株式オプションを行使して得られる収益 | 1,736 | | | 2,014 | | | 534 | |
| 長期債務返済の元金 | (5,806) | | | — | | | — | |
| 融資活動が提供する現金純額 | 117,090 | | | 3,329 | | | 312,540 | |
| 現金、現金等価物および限定現金に及ぼす為替レート変動の影響 | (517) | | | (165) | | | 9 | |
| 現金、現金等価物、および制限的現金純増加 | (71,494) | | | (303,153) | | | 351,531 | |
| 期初の現金、現金等価物、および限定現金 | 183,467 | | | 486,620 | | | 135,089 | |
| 期末現金、現金等価物、および制限現金 | $ | 111,973 | | | $ | 183,467 | | | $ | 486,620 | |
| | | | | |
| キャッシュフロー情報の追加開示: | | | | | |
| 利子を支払う現金 | $ | 12,712 | | | $ | 12,738 | | | $ | 7,871 | |
| | | | | |
| 非現金投資と融資活動を追加開示します | | | | | |
| 賃貸負債と引き換えに使用権資産 | $ | 3,029 | | | $ | — | | | $ | 3,106 | |
| リース負債と引き換えにROUリース資産の確認を取り消します | — | | | $ | 1,650 | | | $ | — | |
| 有価証券は赤字変動純額を実現していない | $ | (75) | | | $ | (225) | | | $ | (100) | |
| 売掛金及び売掛金その他流動負債に掲げる財産及び設備の購入 | $ | 83 | | | $ | — | | | $ | 15 | |
付記はこれらの連結財務諸表の構成要素である。
Gossamer Bio,Inc.連結財務諸表説明
注1-業務説明
Gossamer Bio,Inc.(その子会社、略称“私たち”、“私たち”、“私たち”あるいは“会社”を含む)は臨床段階の生物製薬会社であり、免疫学、炎症と腫瘍学疾患領域の治療薬の発見、買収、開発に専念し、それを商業化する。同社は2015年10月25日にデラウェア州に登録設立された(最初の名称はFSG Bio,Inc.)本部はカリフォルニア州サンディエゴにあります。
連結財務諸表は、Gossamer Bio,Inc.およびその完全子会社の勘定を含む。合併実体間のすべての会社間残高と取引はすでに合併中に販売されている。
流動性と資本資源
設立以来、同社は重大な経営赤字を発生させた。会社の累計赤字は2022年12月31日、2022年12月31日、2021年12月31日まで1,032.2百万ドルとドル811.5それぞれ100万ドルです
当社は設立から2022年12月31日まで、当社は主に株式及び債務融資を通じてその運営資金を提供しています。その会社は$を集めた1,062.12017年10月から2022年12月31日まで、AシリーズとBシリーズの転換可能優先株の売却、転換可能手形の発行、初公募株(IPO)、信用手配、2027年手形(以下付記5)と、2020年5月と2022年7月に普通株式を発行し、100万ユーロに達した。信用手配と2027年手形に関するより多くの情報は、付記5を参照されたい。
同社は予測可能な未来に引き続き重大な経営損失を計上し、永遠に利益を上げない可能性がある。したがって、会社は株式発行、債務融資、または他の資本源を通じて追加資本を調達する必要があり、潜在的な協力、許可証、および他の同様の手配を含む。経営陣は、その手元に十分な運転資金を持っており、少なくともこれらの連結財務諸表が印刷発行された日から今後12ヶ月以内に業務に資金を提供できると信じている。会社が追加資金を得ることに成功することは保証されず、将来の運営資金需要に対する会社の予測が正確であることが証明されるか、または任意の追加資金が今後数年間運営を継続するのに十分であることが保証されない。
新冠肺炎
著者らは引き続き積極的に著者らの計画を推進することに伴い、著者らは著者らの主要な研究者と臨床場所と密接な関係を維持し、そして行われている新冠肺炎の全世界大流行が著者らの薬品製造、非臨床活動、臨床試験、期待スケジュールとコストに与えるいかなる影響を持続的に評価し続けた。また、私たちは世界各地での臨床試験を継続しているが、新冠肺炎の予防措置および各試験場所と主要な供給者の関係者不足は延期されており、例えば、私たちが行っている多環芳香族炭化水素中のシュラルチニブの第2段階試験では、ある場所の募集作業は2020年に一時閉鎖され、現在と未来の試験の完成を延期し続ける可能性があり、現在および未来の臨床試験のデータ読み出し、起動および監視、データ収集と分析およびその他の関連活動のスケジュールに直接または間接的に影響を与える可能性がある。新冠肺炎の大流行を受け、そしてアメリカ食品と薬物管理局が更新した臨床試験業界ガイドラインと一致し、臨床試験は奪われる可能性があり、ウィルスに感染した患者の治療或いはウィルス伝播の防止を支持する。新冠肺炎が私たちの業務に与える直接的かつ間接的な影響は私たちの予測スケジュールを変える可能性があり、これは私たちの業務、運営結果、財務状況に実質的な悪影響を及ぼす可能性がある。私たちは引き続き私たちの業務に及ぼす新冠肺炎の影響を評価するつもりだ。
注2-重要会計政策の概要
陳述の基礎
会社の総合財務諸表は、米国公認の会計原則(“公認会計原則”)に従って作成されている。ある前期の金額は今期の新聞に合うように再分類された.
予算の使用
公認会計基準に従って連結財務諸表を作成することは、連結財務諸表の日付の資産及び負債額、又は有資産及び負債の開示、並びに報告期間内の費用報告金額に影響を与えるために、管理層に推定及び仮定を要求する。会社の連結財務諸表の中で最も重要な見積もりは計算すべき研究と開発費用と関係がある。これらの推定および仮定は、現在の事実、歴史的経験、および信じられる様々な他の要素に基づいている
この場合は合理的であり、その結果、資産や負債の帳簿価値を判断する基礎を構成し、他のソースからは見えない費用を記録する。実際の結果はこれらの推定とは異なる可能性がある。
細分化市場
経営分部は企業の構成要素として確認され、その独立した離散財務情報は、首席経営決定者が資源配分に関する決定と業績評価を行う際に評価することができる。会社は以下の点でその運営·管理業務を確認します1つは運営部門です。
現金と現金等価物
当社はすべての原始期限が三ヶ月以下の高流動性投資を現金等価物と見なしています。総合貸借対照表で報告されている現金および現金等価物の帳簿金額はコスト値であり,そのコストはその公正価値に近い。
有価証券
当社は未満期日が90日を超える証券を有価証券と見なしている。必要であれば、当社は今後12ヶ月以内の流動資金需要を満たすために、その任意の現金等価物および有価証券を清算することができる。したがって、契約満期日が購入日から1年を超える投資は、付随する総合貸借対照表において流動資産に分類される。同社の有価証券には、米国債や機関証券、商業手形、会社債務証券が含まれる。有価証券は公正価値で計上され、損益計上は他の総合損失を累積することは実現されていない。有価証券の推定公正価値は、同様のツールの市場オファーまたは金利に基づいて決定される。当社は、このような損失(あれば)が信用に関連する要因に起因しているかどうかを決定するために、未実現損失のある証券を評価する。未実現の損失が信用に関する要因によるものである場合、会社は信用損失準備を記録した。実現した損益は特定の確認方法で計算し,利息収入または費用と記す。当社は一般的にこのような投資を売却するつもりはなく、その償却コストベースを回収する前にそのような投資を売却することを要求する可能性もなく、償却コストベースが満期になっている可能性も高い。当社は2022年12月31日まで、その投資の公正価値が信用関連要素によって実質的に低下していないことを確定した。
制限現金
2022年12月31日現在,すべての会社施設レンタルに関する制限された現金が解放され,2021年12月31日現在,これらの施設レンタルは担保として所持している現金である。
信用リスクと表外リスクの集中度
現金、現金等価物、および有価証券は、信用リスク集中の影響を受ける可能性のある金融商品である。同社の現金と現金等価物は大型金融機関の口座に保管されており、金額は連邦保険の限度額を超える可能性がある。当社は、現金と現金等価物を持つ預金機関の財務力により、重大な信用リスクに直面しないと信じている。同社が保有する現金等価物には、期限が3カ月未満の米国債や機関証券や商業手形、米国債や機関証券に投資する通貨市場基金が含まれる。
同社が売却可能な証券は米国債や機関証券にも投資されている。本報告に記載されているいずれの期間においても、当社は当該等の口座が信用リスクにより被ったいかなる損失も確認していない。同社は、その現金、現金等価物、販売可能な証券に重大な信用リスクは存在しないとしている。
財産と設備、純額
財産と設備の純額は、主に実験室設備とレンタル改善から構成され、コストから減価償却累計を引いて入金される。減価償却はそれぞれの資産の推定耐用年数で計算されるのが一般的です二つ至れり尽くせり7年になる直線法を採用した。
転換可能優先手形
ASU 2020−06年度を採用する前に、同社は2027年の手形を負債と権益部分として会計処理した。負債部分の帳票は,変換可能な特徴を持たない類似債務ツールの公正価値を計測することで計算される.権益部分の帳簿金額は
転換オプションは、2027年手形の額面から負債部分の公正価値を減算することによって決定される。権益部分が引き続き権益分類の条件を満たしている限り,それを再計測することはない.負債部分元本金額が帳簿金額を超えた部分(“債務割引”)は2027年手形期限内に償却して利息支出とする。
当社は2027年期手形の相対公正価値に基づいて、発生した発行コストを負債および権益部分に分配する。負債部分の発行コストは2027年債負債部分の減少額として記録され、2027年債期限内に利子支出として償却される。転換選択権を代表する権益部分は発行コストと株主権益中の権益部分を相殺しなければならない
2022年1月1日から、会社はASU 2020-06を採用している。採用後、同社は現在、2027年期手形を償却コストで計量した単一負債として会計処理を行っている。持分部分を単独の部分に分割する必要がなくなったため、当社はこの更新を反映する調整を記録した。最近採択された会計公告を見て、この調整が2027年に付記された場合の影響を知る。
賃貸借証書
会計基準更新(“ASU”)第2016-02号によると、賃貸借証書(主題842)では、会社は、開始時にレンタルであるか否かを判断する。経営性賃貸は貸借対照表において使用権資産と経営性賃貸負債として、既製の暗黙的金利がない限り、自社逓増借入金金利で計算される賃貸支払現在値に計上される。当社は初期賃貸期間が12ヶ月を超えない賃貸約に対して短期賃貸契約を適用して免除を確認し、長期借約の賃貸及び非レンタル部分を分離しないことを選択した。当社はレンタル期間中にレンタル料を直線的に記録しています。
研究と開発
すべての研究と開発費用は発生した費用に計上されている。研究開発コストは主に給料、従業員福祉、臨床前研究と臨床試験に関連するコスト(臨床研究機関と他の専門サービスに支払う金額を含む)を含む。研究·開発のための貨物またはサービスを受け取る前に支払われる金は、貨物またはサービスを受け取る前に資本化される。
同社は、第三者請負業者、実験室、臨床試験場所およびその他の方面の作業支払いを含む、推定された研究と開発コストの計算すべきプロジェクトを記録した。その中のいくつかの請負者は、実際に提供されるサービスに基づいて月ごとに課金され、他のいくつかの請負者は、いくつかの契約マイルストーンを実装することに基づいて定期的に課金される。後者については、会社が貨物やサービスを利用または提供する際には費用を計上しなければならない。患者登録に関する臨床試験地点コストは,患者が試験に入り,試験中に進展するにつれて蓄積される。前期コストは,試験に参加する臨床試験地点の構築に関するコストであれば,発生すると直ちに研究開発費として支出される。
現在行われている研究と開発
研究と開発コストは,Aadi Bioscience,Inc.へのGB 004許可プロトコルの改訂に関する前金の支払いと,サラダルチニブ第二段階臨床試験を開始する記念碑的支払いをPulmokineに支払うことに関連している。
特許費用
このような費用の回収可能性が不確定であるため、提出及び特許出願に関する費用は発生した費用に計上される。これらの費用は一般的で行政的費用に含まれている。
所得税
所得税は財務会計基準委員会(FASB)基準編纂(ASC)740号に従って入金され、所得税繰延税項目は、資産および貸借対照法を使用して抽出される(“ASC 740”)。当社は、繰延税金資産と負債が財務諸表または納税申告書に含まれているイベントの予想される将来の税務結果であることを確認します。繰延税項資産及び負債は、財務諸表と資産及び負債の税ベースとの差額に基づいて、予想差額を用いて返送される年度の現行税率を決定する。既存の証拠の重みに基づいて、繰延税金資産の一部または全部が現金化できない可能性が高い場合には、推定免税額を計上する。
ASC 740の規定によると、当社は不確定な税務頭寸を会計処理する。不確定な税務倉位が存在する場合、当社は税務倉位の税務優遇を確認し、税務機関が審査すれば、当該税務優遇がより実現可能である。税収優遇がより実現可能かどうかの決定は,税収状況に基づく技術的利点および既存の事実や状況への考慮である。
繰延税金資産と負債は、現行の税率を採用した資産と負債の財務報告と課税基礎との差による将来の税務結果を反映している。このような繰延税金資産の現金化能力がより達成可能な閾値を満たしていない場合には、推定準備を計上する。税収優遇を確認するためには、税務機関が審査した後、税収状況がより持続可能でなければならない。同社はアメリカ、カリフォルニア州、アイルランド、ルクセンブルクで税金を払わなければならない。2022年12月31日現在、会社の未使用純営業損失(“NOL”)と税収控除のため、会社設立以来の納税年度は税務機関の審査を受けなければならない。
株に基づく報酬
会社は報酬の推定に基づいて期日公正価値を付与し、必要なサービス期間内に従業員および非従業員に株式ベースの報酬を支払う。経営陣がマイルストーンに達する可能性があると判断した場合、当社は業績マイルストーンの帰属に基づく株式報酬報酬の費用を記録する。管理層は各報告日の業績条件に対する期待満足度に基づいて、いつ業績を基礎とするマイルストーンを実現することが可能かを評価する。当社は、Black-Scholesオプション定価モデルを用いて株式オプション付与を推定し、会社2019年従業員株式購入計画(“ESPP”)に基づいて購入可能な株式の公正価値を推定し、株式の奨励に基づく公正価値を計算する際に使用される仮説が経営層の最適推定を代表し、内在的不確実性と管理層判断の応用に係る。当社は当社の普通株の付与日の終値に基づいて制限株式単位の公正価値を推定します。当社は発生した没収行為を計算します。すべての株式ベースの給与コストは、関連従業員または非従業員の社内の役割に基づいて経営報告書に記録されている。
外貨?外貨
現地通貨環境下で運営される非米国子会社の資産と負債、すなわち現地通貨は機能通貨であり、貸借対照表の日の有効為替レートでドルに換算される。収入と費用口座は今年度内の平均為替レートに換算すると,為替レートは取引日の実際の為替レートとほぼ同じである。これにより生じた換算調整は、当社の総合貸借対照表の累計その他の全面収益に計上される。為替取引損益は、会社総合経営報告書と全面赤字の他の収入(費用)を計上する。
最近の会計公告は
2020年8月、財務会計基準委員会はASU 2020-06を発表し、債務:転換と他のオプションを有する債務(特別テーマ470-20)と実体自己権益派生ツールとヘッジ契約(特別テーマ815-40)(“ASU 2020-06”)を発表し、実体自己資本の中で転換可能なツールと契約の会計処理を簡略化した。本ガイドラインは,2021年12月15日以降に開始された年次報告期間に適用され,これらの年間移行期間を含めて,2020年12月15日以降に開始された年次報告期間のみ早期採用が許可されている
当社は2022年1月1日に改訂された遡及手法を用いてASU 2020−06を採用したため,当社は埋め込みされた変換機能が分離されていないように2027年手形を反映した調整を記録した。総合貸借対照表採用後の影響は約#ドル増加した44.82000万の転換可能な優先手形、純額、ログアウト#ドル9.4繰延所得税負債1.3億ドル、#ドル減少53.5追加的な実収資本は2.5億ユーロだ。また,通過後,調整数は#ドルとした8.7総合貸借対照表上の累積赤字の期初残高を増加させ、先に確認した利息支出のために、発行時に転換機能を組み込んだ帳簿価値に関する債務割引償却に係る。会社の1株当たり純損失の計算に影響はありません。2027年期手形のさらなる資料については、付記5“負債”を参照されたい。
1株当たり純損失
普通株1株当たりの基本純損失の計算方法は、普通株株主が純損失を当期発行普通株の加重平均株式数で割るべきである。会社は2027年期手形を想定したIF−変換法を用いて普通株の加重平均シェアを計算している
既に発行された1株当たりの償却純損失.1株当たり純損失に自社普通株オプションおよび制限株を含まない未帰属株式を希釈する潜在的影響と、2027年手形転換時に発行可能な潜在株式を希釈することは、当社の純損失により、それらの影響が逆薄となるからである。会社は報告の期間ごとに純損失があるため、普通株の基本純損失は希釈後の純損失と同じだ。
次の表で提供される潜在的な希薄化証券は、1株当たりの純損失の計算には含まれていない
| | | | | | | | | | | | | | | | | |
| 十二月三十一日 |
| 2022 | | 2021 | | 2020 |
| 2027年ノート | 12,321,900 | | | 12,321,900 | | | 12,321,900 | |
| 株式オプション行使時に発行可能な株式 | 17,487,165 | | | 9,434,660 | | | 9,401,082 | |
| 制限株付与下の非既得株 | 1,350,035 | | | 2,561,219 | | | 3,330,821 | |
| 潜在的希薄化証券総額 | 31,159,100 | | | 24,317,779 | | | 25,053,803 | |
注3-費用とその他の流動負債を計算しなければならない
計算すべき費用および他の流動負債には、以下の項目が含まれる(千で計算)
| | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 |
| 報酬と福祉に計上すべきである | $ | 13,534 | | | $ | 11,916 | |
| リース負債を経営する | 2,983 | | | 2,902 | |
| 相談料を計算する | 1,104 | | | 956 | |
| 応算利息 | 1,065 | | | 1,066 | |
| 弁護士費を計算する | 380 | | | 202 | |
| 訴訟負債 | — | | | 2,375 | |
| 会計費用を計算する | 521 | | | 154 | |
| その他の措置を講じる | 945 | | | 839 | |
| 費用とその他の流動負債総額を計算しなければならない | $ | 20,532 | | | $ | 20,410 | |
注4-公正価値計量と販売可能な投資
会計指針は公正価値を定義し、公正価値を計量するために一致した枠組みを構築し、公正価値の経常性或いは非日常性に基づいて計量する各主要資産と負債カテゴリの開示範囲を拡大した。公正価値は、市場参加者間の秩序ある取引において資産を売却して受信した金額または移転負債によって支払われた金額を表す脱退価格として定義される。したがって、公正価値は市場に基づく計量であり、資産または負債の定価のために市場参加者が使用する仮定に基づいて決定されるべきである。このような仮定を考慮する基礎として、会計基準は三級公正価値レベルを確立し、公正価値を計量する際に使用する投入の優先順位は以下の通りである
第一段階:活発な市場のオファーのような観察可能な投入;
第2レベル:活発な市場オファーを除いて、直接または間接的に観察可能な投入;および
第三レベル:観察できない投入は、その中で市場データが少ないか全くないか、これは報告エンティティに自分の仮説を立てることを要求する。
同社は、その現金等価物および販売可能な投資をレベル1またはレベル2に分類している。会社の投資レベルの会社債務証券および商業手形の公正価値は、専門評価モデルおよび分析ツールを使用して決定され、これらのモデルおよび分析ツールは、市場定価または同様のツールの価格を利用しており、これらのツールは、客観的であり、また、行列定価または報告された取引、基準収益率、仲介人/取引業者オファー、発行者価格差、二国間市場、基準証券、入札および要約のような開示されている。
公正な価値に応じて恒常的に計量された資産と負債
次の表は、2022年12月31日まで、2021年12月31日までの公正価値で恒常的に計測される資産の階層構造(千単位)を示している
| | | | | | | | | | | | | | | | | | | | | | | |
| 期末公正価値計量使用: |
| 合計する
公正価値 | | 中国株式相場
価格:
まったく同じ資産
(レベル1) | | 意味が重大である
他にも観察されるのは
入力量
(レベル2) | | 意味が重大である
見えない
入力量
(レベル3) |
| 2022年12月31日まで | | | | | | | |
| 貨幣市場基金 | $ | 54,662 | | | $ | 54,662 | | | $ | — | | | $ | — | |
| アメリカ財務省と機関証券は | 31,458 | | | 31,458 | | | — | | | — | |
| 商業手形 | 134,954 | | | — | | | 134,954 | | | — | |
| 会社債務証券 | 8,838 | | | — | | | 8,838 | | | — | |
| 2021年12月31日まで | | | | | | | |
| 貨幣市場基金 | $ | 139,794 | | | $ | 139,794 | | | $ | — | | | $ | — | |
| 商業手形 | 113,939 | | | — | | | 113,939 | | | — | |
| 会社債務証券 | 37,873 | | | — | | | 37,873 | | | — | |
本報告に記載されている間、当社は公正価値階層間の投資再分類を行っていない。
他の金融商品の公正な価値
2022年12月31日、2022年12月31日と2021年12月31日まで、当社の金融商品の帳簿価値は、現金、制限性現金、前払いとその他の流動資産、売掛金、研究開発費、売掛金と売掛金とその他の流動負債を含み、その短期満期日のため、その帳簿価値は公正価値に近い。
あったことがある違います。2022年12月31日までの重大受取利息。2021年12月31日までの受取利息はドルです0.2百万ドルは、連結貸借対照表に前払い費用及びその他の流動資産の構成要素として入金される。
当社は、その信用手配の利息金利は類似の特徴を持つツールの現行市場金利と一致しているため、信用手配の帳簿価値は公正価値に近いと信じている。当社は現在値計算を用いて,観察可能な投入に基づいて現金フローモデルを用いて元金と利息支払いおよびこれらの債務の最終満期日払いを割引している。そして、報告日までの現在の市場金利に基づいて債務ツールを割引する。これらの負債を公正価値で推定するための仮定によれば、債務ツールは、公正価値レベルで第2レベルに分類される。
2022年、2022年、2021年12月31日までの会社の2027年手形の公正価値は61.0百万ドルとドル190.5それぞれ100万ドルです公正価値は同類のツールに見られる市場価格によって決定され、公正価値等級中の第二級とみなされる(付記5参照)。
売却可能な投資
同社はその余分な現金を米国債や機関証券、会社債務証券、商業手形に投資し、これらは売却可能な投資に分類されている。この等投資は公正価値に基づいて帳簿に記載されており、以下の表に並んでいます。当社は損失を実現していない証券があることを評価し、このような損失が信用関連要素によるものかどうかを確認します。損益は特定の確認方法を用いて計算し,会社合併経営報告書と全面赤字の他の収益(費用)を計上した。当社はこのような投資を売却するつもりはありませんが、当社もその余剰コスト基準を回収する前に当該などの投資を売却する必要はありません。
2022年12月31日、2022年と2021年12月31日まで、証券タイプ別の売却可能投資の総時価、コストベースと未実現損益総額は、有価証券と長期投資によって以下のように分類される
| | | | | | | | | | | | | | | | | | | | | | | |
| 償却する
コスト | | 毛収入
実現していない
収益.収益 | | 毛収入
実現していない
損 | | 合計する
公正価値 |
| 2022年12月31日まで | | | | | | | |
| 米国債と機関証券 | $ | 31,445 | | | $ | 13 | | | $ | — | | | $ | 31,458 | |
| 会社債務証券 | 8,876 | | | — | | | (38) | | | 8,838 | |
| 商業手形 | 103,508 | | | — | | | (99) | | | 103,409 | |
| 有価証券総額 | $ | 143,829 | | | $ | 13 | | | $ | (137) | | | $ | 143,705 | |
| 2021年12月31日まで | | | | | | | |
| 会社債務証券を購入する | $ | 37,921 | | | $ | — | | | $ | (48) | | | $ | 37,873 | |
| 商業手形 | 103,942 | | | — | | | — | | | 103,942 | |
| 有価証券総額 | $ | 141,863 | | | $ | — | | | $ | (48) | | | $ | 141,815 | |
2022年、2022年、2021年12月31日まで、会社はドルに分類されている31.5百万ドルとドル10.0それぞれ、元の満期日が90日以下の資産を現金および現金等価物とする。
各報告日には、会社が減価評価を行い、未実現損失があるかどうかが信用関連要因によるものであるかどうかを決定する。未実現の損失が信用に関する要因によるものである場合、会社は信用損失準備を記録した。売却可能な投資減値を評価する際に考慮される要因は、減値の深刻さ、関連信用格付けの変化、発行者の財務状況、所定の現金を継続的に支払う可能性、および当社が償却コストベース回収まで投資を保有する意向および能力である。当社は赤字を達成していないことでその投資を保有し、その余剰コストベースを回収する能力があると考えている。2022年12月31日、2022年12月31日、2021年12月31日まで、会社が売却可能な投資の時価は信用関連要因による実質的な低下は見られなかった。
2022年12月31日現在、債務証券の売却が可能な契約満期日は以下の通り(千単位)
| | | | | |
| 推定数
公正価値 |
| 1年もたたないうちに | $ | 143,705 | |
| 1年を超える | — | |
| 合計する | $ | 143,705 | |
必要があれば、当社は今後12ヶ月の流動資金需要を満たすために、その任意の現金等価物および有価証券を清算する能力がある。
注5-負債.負債
信用手配
当社は2019年5月2日にMidCap Financial Trust(“MidCap”)および時々増加する貸金人(MidCap(“貸金人”)とともに)と2019年9月18日,2020年7月2日および2022年12月7日に改訂されたクレジット,担保および担保協定(“クレジット手配”)を締結し,これにより,融資者(MidCapおよび珪谷銀行を含む連属会社)は,当社に元金が最高$を超えない定期融資を提供し,運営資金および一般業務用途を行うことに同意した150.01ドルを含む2000万ドルの定期融資約束30.0期限までに資金の100万ドルの定期融資を受けて、残りのドルを得ることができます120.02000万インチ二つ他の部分($ごと)60.0規定の使用可能期間、ある臨床開発マイルストーンの実現、最低現金需要とその他の慣例条件の下で。同社は訪問に必要な臨床開発マイルストーンに達していない1つはドルの中で60.03,000万部と他のドルを獲得60.02021年12月31日、100万部の債券が満期になった。当社の完全子会社GB 001,Inc.,GB 002,Inc.およびGB 004,Inc.はクレジット手配の共同借主として指定されているが,当社の完全子会社GB 003,Inc.,GB 005,Inc.,GB 007,Inc.,GB 008,Inc.およびGossamer Bio Services,Inc.は指定されている
保証人です。信用手配は、当社とその国内子会社のほとんどの個人財産(知的財産を含む)を担保としている。
信用手配下の各定期ローンの年利率は(I)担保付き隔夜融資金利(SOFR)と相応の利差プラス(Ii)の和に等しい7.00%は、SOFR下限で2.00%です。借り手は2022年7月1日までのすべての支払日に定期ローンの純利息を支払わなければならない。クレジット手配された定期ローンは2022年7月1日に償却を開始し、会社はこの純利息期間後に月連続分割払いで融資者に等額の元金と利息を支払い、信用手配が2025年1月1日に満期になるまで保証人に支払う。最終的に定期ローンを返済する際には、借り手が支払わなければなりません1.75クレジット手配によって借入金額の%は、以前に支払われた任意の部分脱退費用を差し引く。一部の定期ローンの一部を前払いした後、借り手は一部の脱退費用を支払わなければなりません1.75元金の%は前払いです。借り手の選択の下で、借り手は定期ローンの未返済元金残高を全部または一部前払いすることができますが、前払いは3.00前払金が第2改正案の発効日の1周年に発生した場合、2.00前払額の割合は、前払金が第2改正案施行日の1周年後に発生した場合、第2改正案施行日の2周年(当該2周年を含む)、及び1.00第二改正案の発効日から二周年後から2025年1月1日までに前払いされる任意の金額の%
2022年12月7日、当社は信用手配第3修正案を締結し、信用手配に適用される金利を除いて、元本または返済条項は変わらず、LIBORの代わりにSOFRに基づく前向き定期金利を実施する。ASU 2020-04の規定によると、会社は基準金利の変化を実質的な改正ではないとみなしている
信用手配には、当社とその特定の子会社に適用されるプラスと負のチノが含まれている。他にも、支援条約には、これらの実体がその合法的な存在と政府の承認、特定の財務報告の提出、保険範囲の維持、財産の維持、税金の納付、関連口座のいくつかの要求を満たすこと、法律と法規を遵守することを要求する条約が含まれている。その他を除いて、消極的な条約は、このような実体が担保を譲渡することを制限すること、追加債務を招くこと、合併または買収を行うこと、配当金を支払うこと、または他の分配を行うこと、投資を行うこと、留置権を設立すること、重要な合意と組織文書を修正すること、資産の売却と制御権を変更することを含むが、いずれの場合もいくつかの例外的な状況によって制限される。会社とそのある子会社はまた行われている最低現金財務契約を守らなければならない。この契約では、制限されない現金金額を下回らないようにしなければならない25定期ローンは元金の%を返済していません。当社は2022年12月31日までこれらの条約を遵守している。
信用手配には違約事件も含まれており、違約事件の発生と継続は利息を他の適用金利で印加する可能性がある3.00そして、MidCapを代理人として付与する権利は、当社および/またはそのいくつかの付属会社に救済を行使し、担保信用手配の担保品は、担保信用手配の財産(現金を含む)の償還を含む。これらの違約事件は他の事項以外に、信用手配下の任意の満期金を支払うことができなかったこと、信用手配下の契約違反、債務返済ができなかったこと、債務非相殺事件の発生、コントロール権の変更、あるアメリカ食品薬物管理局(“fda”)及び監督管理事件の発生、米国証券取引委員会に登録し、ナスダックでの上場取引を継続できなかったこと、重大な不利な変動が発生したこと、理由によって重大な不利な変動を招く重大な合意違約、ある他の債務項目の下で発生した金額が#ドルを超えるある違約を含む2.5二次債務と転換可能な債務の下でいくつかの違約が発生した。
債務は以下の部分から構成される(千で計算)
| | | | | |
| 2022年12月31日 |
| 債務、流動部分 | $ | 11,613 | |
| 債務、非流動部分 | 12,581 | |
| 債務総額 | 24,194 | |
| 差し引く:未償却債務割引と発行コスト | (593) | |
| 債務,純額 | $ | 23,601 | |
将来予定の最低元金返済額は以下の通り(千単位)
| | | | | |
| 2022年12月31日 |
| 2023 | $ | 11,613 | |
| 2024 | 11,613 | |
| 2025 | 968 | |
| 合計する | $ | 24,194 | |
2027年に有効期限の5.00%の移行優先チケット
2020年5月21日に会社はドルを発行しました200.0元金総額は1,000万ドル5.002027年に満期の転換可能優先債券パーセンテージ(“2027年債券”)が公開発売される。2027年債は、会社が2020年4月10日に米証券取引委員会に提出したS-3表棚上げ登録声明に基づいて登録されている。2027年発行の債券の金利を5.00年利率です。利息は半年ごとに支払い、2020年12月1日から毎年6月1日と12月1日に延滞する。2027年債は2027年6月1日に満了する。引受割引,手数料,その他の発行コストを差し引いて,次発行で得られた純額は約$である193.6百万ドルです。2027年の手形は現金、会社普通株の株式または両者の組み合わせで決済でき、完全に会社が選択する。2027年手形の予備交換株価は1,000元当たり元金約61.6095株であり、交換株価約1,000元に相当する16.23また、満期日までに発生したある会社の事件や当社が償還通知を出した場合、当社は、保有者が関連償還期間中に当該会社のイベントについて2027年手形を転換することを選択する換算率を向上させる場合があります
2027年手形は当社の優先無担保債務であり、その支払権は当社の任意の債務よりも優先されるが、当社の任意の債務の支払権は2027年手形に明確に従属し、その債務の担保価値を保証する範囲内で、実際には信用手配下のすべての債務を含む当社の既存および将来の有担保債務に従属する。
所有者は、以下の場合にのみ変換手形を選択することができる:(1)2020年9月30日に終了したカレンダー四半期以降に開始される任意のカレンダー四半期期間(かつ、当該カレンダー四半期期間のみ)、企業普通株の最後の報告が1株当たり販売価格を超える場合130換算価格の割合は少なくともそれぞれです20取引日(連続の有無にかかわらず)30前シーズン最後の取引日(この日を含む)までの連続取引日5人次の日の直後の連続営業日10連続取引日期間(例えば10連続取引日期間,すなわち“精算期間”),すなわち1ドルあたりの取引価格である1,000精算期間内の取引日あたりの手形元本金額は以下である98(3)会社普通株にある会社の事件又は分配が発生した場合、(4)会社が当該等の手形の償還を要求した場合、及び(5)2027年3月1日(当該日を含む)から満期日直前の予定取引日収市までのいずれかの時間
同社は2024年6月6日までに2027年期手形を償還する権利がないだろう。2024年6月6日またはその直後および満期直前の50番目の予定取引日または前に、会社は2027年手形の全部または一部を償還することができ、会社の普通株の最終報告販売価格が少なくとも130当時有効な転換価格の割合(1)少なくとも20取引日(連続の有無にかかわらず)30当社が償還通知日の直前の取引日の終了に関する連続取引日を発行し、及び(2)当社が償還通知日の直前の取引日を発行する。もし何か償還する選択があれば,当社は同じになる100償還した当該等債券元金の%を、別途償還日(ただし償還日を除く)の課税及び未償還利息を加算する
もし会社が2027年の債券満期日までに大きな変化が発生した場合、2027年の債券保有者は会社に全部または一部の2027年の債券を現金で買い戻すことを要求することができ、買い戻し価格は100購入した2027年期債券元金の%に、基本変動買い戻し日(ただし含まない)の応計及び未払い利息を加算する。
2027年紙幣を管理する契約は慣例条項と契約を規定し、ある違約事件が発生した時、受託者或いは以上を含む25当時未返済の2027年手形元金総額の%は、2027年手形の未払い元金額及びその計上及び未払い利息(あれば)の即時満期及び支払が必要であることを宣言することができる。2022年12月31日まで、会社はこの条約を遵守した。.の場合
もしある破産、債務返済ができない、あるいは再編事件が発生した場合、2027年期手形の元金はその計算および未払い利息(あればある)と共に自動的に満期と即時支払いとなる
2022年12月31日、2022年、2021年12月31日まで、2027年手形の転換が許可された事件や市況はありません。2027年手形が貸借対照表日から12ヶ月以内に両替できる場合、2027年手形の帳簿価値は短期に再分類されます。
ASU 2020−06年度までに発行された2027年期手形を用いた会計計算では、会社は2027年期手形を負債部分と権益部分に分類している。負債部分の帳簿価値は、変換可能な特徴のない類似債務ツールの公正価値を計量することで計算される。転換オプションの権益部分を代表する帳票金額は#ドルである53.52027年手形の額面から負債部分の公正価値を引いて決定されます。権益部分が権益分類の条件を満たし続ける限り,再計測は行われない.債務を2027年債期内に償却して利息支出とし、実際の金利は11.172027年期手形の契約条項より2%高い。2022年1月1日現在,会社はASU 2020−06を採用しており,2027年付記採用の影響については付記2を参照されたい。
ドルの債務発行コストを計算する際に0.42027年債に関連する1000万ドルのため、当社は2027年債の相対公正価値に基づいて生じた総金額を2027年債の負債と権益部分に分配する。負債部分の発行費用は#ドルです0.32027年手形の契約条項で、実際の利息方法で利息支出を償却します。 権益部分の発行コストは株主権益中の権益部分と相殺すべきである。
2027年期手形負債部分の帳簿純額は以下の通り(千計)
| | | | | | | | | | | |
| 2022年12月31日 | | 2021年12月31日 |
| 元金金額 | $ | 200,000 | | | $ | 200,000 | |
| 未償却債務割引 | (4,021) | | | (49,716) | |
| 未償却債務発行コスト | (270) | | | (246) | |
| 帳簿純額 | $ | 195,709 | | | $ | 150,038 | |
2027年に発行された債券の株式部分の帳簿純価値は以下の通り(千計)
| | | | | | | | | | | |
| 2022年12月31日 | | 2021年12月31日 |
| 転換オプション価値に関する債務割引 | $ | — | | | $ | 53,635 | |
| 起債コスト | — | | | (109) | |
| 帳簿純額 | $ | — | | | $ | 53,526 | |
次の表に2027年手形に関する確認された利息支出(千単位)を示す
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| 契約利子支出 | $ | 10,000 | | | $ | 9,972 | | | $ | 6,139 | |
| 債務割引償却 | 782 | | | 6,364 | | | 3,555 | |
| 債務発行原価償却 | 53 | | | 32 | | | 17 | |
| 2027年発行の債券に関する利子支出総額 | $ | 10,835 | | | $ | 16,368 | | | $ | 9,711 | |
注6-ライセンス、資産買収、または価格の比較があります
以下の購入資産は資産購入に計上され,購入資産の公正価値はほぼすべて1組の類似資産に集中しているため,および/または購入資産は従業員の不足や開発初期のため産出が生じない.この等資産は監督管理機関の承認を得ていないため、この等資産の公正価値は、2022年12月31日、2021年12月31日及び2020年12月31日までの総合経営報告書及び全面損失のうち、製品研究開発(“IPR&D”)支出と記されている。
当社は、当該等の資産買収において何らかの規制、開発又は販売マイルストーンに達したときに支払うべき又は対価を計上し、関連又は事項が満たされた場合に支払う。
Pulmokine,Inc.(Seralutinib)のライセンス
同社は2017年10月2日、Pulmokine,Inc.と、Pulmokineが所有または制御する特定の知的財産権の世界的独占的許可および再許可を取得し、任意およびすべての疾患または状態を治療、予防および診断するためのサラダルチニブおよび特定の予備化合物を開発および商業化するライセンス契約を締結した。ある条件を満たす場合、会社はライセンス契約に基づいてその権利を再許可する権利もある。買収された資産はFDA承認手続きの初期段階にあり,同社はFDAの潜在的承認で得られた資産をさらに開発しようとしており,この契約におけるマイルストーン計画が証明している。会社の大きなコストと努力がなければ、開発活動はできない。協定は、製品および国ごとの許可に基づいて遅くなるまで、早期終了しない限り、発効日から有効に継続されるであろう10年最初の商業販売の日から、または当該国に当該ライセンス製品をカバーする有効な特許権利要件またはそのライセンス製品の特定の規制排他性がもはやない場合。会社は将来の発展と規制マイルストーンの支払いを支払う義務があり、金額は最高$に達する58.0100万ドルの$が含まれています10.0最初の3期臨床試験開始時に100万ドルを支払い、ビジネスマイルストーンの支払いは最高$に達します45.0百万ドルで、販売マイルストーンの支払いは最高ドルに達します190.0百万ドルです。同社にはライセンス製品の販売ごとに等級版税を支払う義務があり、百分率は数桁から高い桁まで様々である。また、会社が合意の下で特許製品の権利を任意の第三者に再許可または譲渡することを選択した場合、または会社のseralutinib経営子会社が制御権変更を行うことを選択した場合、会社は、そのような取引に関連するすべての収入の特定の割合をPulmokineに支払わなければならない。同社は#ドルを前払いした5.52017年10月は100万人。2020年12月に会社は記念碑的な支払いをしました#ドル5.02021年1月に支払われたSeralutinib第1段階2期臨床試験開始費用は100万ドルであった。2022年12月31日と2021年12月31日までに違います。根本的な意外な状況がまだ達成されていないので、他のマイルストーンが蓄積された。
同社は総合経営報告書に以下の知的財産権研究開発費(千計)を記録している
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| セラルチニ | $ | — | | | $ | — | | | $ | 5,000 | |
| GB 004 | — | | | — | | | 15,000 | |
| 他の臨床前プロジェクト | 65 | | | 75 | | | 3,380 | |
| 全過程研究と開発 | $ | 65 | | | $ | 75 | | | $ | 23,380 | |
注7-所得税
2022年12月31日現在、2021年12月31日現在、2020年12月31日までの税前純損失額は以下の通り(単位:千)
| | | | | | | | | | | | | | | | | |
| 十二月三十一日 |
| 2022 | | 2021 | | 2020 |
| (単位:千) |
| アメリカ税前損失 | $ | 175,777 | | | $ | 183,194 | | | $ | 186,888 | |
| 外国の税引き前損失 | 53,593 | | | 50,802 | | | 56,472 | |
| 所得税前損失 | $ | 229,370 | | | $ | 233,996 | | | $ | 243,360 | |
繰延所得税は、財務報告目的のための資産および負債の帳簿金額と所得税目的のための金額との間の一時的な差異の純税影響を反映する。当社の2022年、2022年、2021年、2020年12月31日までの繰延税金資産と負債の重要な構成要素を以下に示す。繰延税項目の純資産が現金化できるかどうかの不確定性のため、当社は同社などの資産の推定値について準備している。当社は繰延資産の回収可能性を定期的に評価している。繰延税金資産がより顕在化する可能性があると判断された場合、推定免税額は減少する。2022年12月31日までの年間推定手当変動は#ドル増加56.3百万ドルです。
| | | | | | | | | | | | | | | | | |
| 十二月三十一日 |
| 2022 | | 2021 | | 2020 |
| (単位:千) |
| 繰延税金資産: | | | | | |
| 純営業損失 | $ | 128,989 | | | $ | 116,425 | | | $ | 77,946 | |
| 税収控除,純額 | 34,193 | | | 24,964 | | | 17,372 | |
| 償却する | 8,487 | | | 9,190 | | | 10,939 | |
| 株に基づく報酬 | 8,445 | | | 4,917 | | | 4,448 | |
| リース責任 | 1,354 | | | 1,285 | | | 2,383 | |
| 補償すべきである | 2,514 | | | 2,238 | | | 2,137 | |
| 第百四十四条 | 21,840 | | | — | | | — | |
| 他にも | 37 | | | 18 | | | 16 | |
| 繰延税項目の総資産総額 | 205,859 | | | 159,037 | | | 115,241 | |
| | | | | |
| 繰延税金負債: | | | | | |
| 転換可能優先手形 | — | | | (9,395) | | | (10,592) | |
| 使用権資産 | (1,244) | | | (1,150) | | | (2,215) | |
| 財産·工場·設備 | (565) | | | (790) | | | (776) | |
| 繰延税金負債総額 | (1,809) | | | (11,335) | | | (13,583) | |
| 推定免税額 | (204,050) | | | (147,702) | | | (101,658) | |
| 繰延税項目純資産 | $ | — | | | $ | — | | | $ | — | |
2022年12月31日現在,同社の連邦と州NOL繰り越し額は約$である494.6百万ドルとドル1.6それぞれ100万ドルです2018年1月1日までに生成された連邦NOL繰り越しは、以前に使用されたことがない限り、2034年に満期になります。2017年12月31日以降に開始された納税年度に発生した連邦NOLは$491.6百万ドルは無期限に繰り越すことができるが,相殺にしか使えない80毎年未来の課税所得額の%を占めています。カリフォルニア州NOL繰越は2036年に満期になる。2022年12月31日現在、会社は約ドルのNOL海外繰り越しも持っている200.2百万ドルです。海外のNOLは無期限に続くことができる
同社は2022年12月31日現在、約ドルの孤児薬物相殺と連邦研究税収免除を持っている35.8百万ドルとカリフォルニア州の研究税控除10.6百万ドルです。連邦研究税収控除は2038年から満期になり、カリフォルニア研究税収控除は満期にならず、使用まで無期限に繰り越すことができる。
連邦法定所得税率と会社の有効所得税率の入金は以下のとおりである
| | | | | | | | | | | | | | | | | |
| 十二月三十一日 |
| 2022 | | 2021 | | 2020 |
| 連邦法定所得税率 | 21.00 | % | | 21.00 | % | | 21.00 | % |
| 連邦福祉を差し引いた州所得税 | — | % | | — | % | | — | % |
| 評価免除額を変更する | (20.25 | %) | | (19.73 | %) | | (19.50 | %) |
| 研究と実験単位 | 3.39 | % | | 2.62 | % | | 2.76 | % |
| 外貨利回り | (1.88 | %) | | (1.95 | %) | | (1.64 | %) |
| 株に基づく報酬 | (0.90 | %) | | (1.56 | %) | | (1.81 | %) |
| 利子を差し引くことができない | (0.99 | %) | | (0.96 | %) | | — | % |
| 他にも | (0.37 | %) | | 0.57 | % | | (0.80 | %) |
| 所得税支給 | — | % | | — | % | | — | % |
もし当社が1回または複数回の所有権変更を経験し、将来の課税所得額と税収控除を相殺するためにそれぞれ使用できるNOLと税収控除繰越金額を制限した場合、NOL繰越は1986年の国税法第382条と383条および同様の国が規定する年間制限を受ける可能性がある。一般的に、第382条及び第383条で定義された所有権変更とは、3年の間に、ある株主又は公共団体の会社株における所有権が50ポイント以上増加した取引所をいう
当社の2019年2月の初公募については、当社は守則第382及び383条の規定により所有権変更を経験しています。所有権変更は、この日前に生成されたNOLまたはクレジットが没収されることをもたらすことはありません。そのため、同社が2019年2月までに発生した連邦と州NOLと税収控除は年間制限される。追加の所有権変更が発生した場合、または追加の所有権変更が発生した場合、繰越のNOLおよび税収控除をキャンセルまたは制限することができる。廃止された場合、関連資産は繰延税金資産表から削除され、それに応じて推定免税額が減少する。推定免税額の存在により、将来の所有権変更による制限は当社の実際の税率に影響を与えません。
同社はアメリカ、カリフォルニア州、フロリダ州、アイルランド、ルクセンブルクで所得税申告書を提出した。当社の損失のため、当社は設立以来主管部門の所得税審査を受けてきました。当社の政策は、所得税に関する利息支出と罰金を税費支出と確認することです。2022年12月31日、2021年12月または2020年12月31日まで、未確認の税収優遇または税収処罰に関する利息には課税項目がない。
2022年、2021年、2020年の税収割引が確認されていない期初と期末金額(利息や罰金は含まれていない)の入金は以下の通り
| | | | | | | | | | | | | | | | | |
| 十二月三十一日 |
| 2022 | | 2021 | | 2020 |
| (単位:千) |
| 年初残高 | $ | 7,551 | | | $ | 5,060 | | | $ | 2,754 | |
| 今年度のポストに関する増加 | 3,021 | | | 2,491 | | | 2,306 | |
| 年末の残額 | $ | 10,572 | | | $ | 7,551 | | | $ | 5,060 | |
2022年12月31日現在の未確認税収割引残高は$を含む10.6当社の繰延税金資産がまだ全額推定手当によって制限されていることを確認すれば、当社の所得税優遇や実際の税率に影響を与えないことが確認された。当社は、今後12ヶ月以内に、当社が確認していない税収割引が大きな増加や減少はないと予想しています。
注8-株主権益
普通株
普通株は1株当たり獲得権がある1つは投票しましょう。資金が合法的に利用可能であり、取締役会によって発表された場合、普通株式所有者は配当金を得る権利がある。
棚登録表と株式発行
2020年4月10日、当社は、時々発行される普通株式、優先株、債務証券、権証、単位を含む汎用棚登録表をS-3表形式で提出し、この登録表は2020年4月10日に自動的に発効する(以下、“棚登録表”と略す)。
2020年5月21日、当社は公募株を完成しました9,433,963普通株の公開発行価格は$です13.25一株ずつです。引受割引と手数料その他の発行コストを差し引いて,次発行で得られた金の純額は約$である117.11000万ドルです。今回発売された株式は当社の棚登録声明に基づいて登録されています。
2022年3月3日、当社は、時々発行される普通株式、優先株、債務証券、権証、単位を含むS-3表の汎用棚登録声明を提出し、この登録声明は2022年3月3日に自動的に発効した。
私募融資
2022年7月15日私たちは16,649,365私たちの普通株の買い取り価格は$です7.21一株ずつです。個人配給の総収益は約$であった120.1300万ドルです。発売費用を差し引く前です。2022年8月9日、2022年8月9日に自動発効したS-3用紙に私募発行の普通株式を登録する登録説明書を提出した。
買い戻しの影響を受けた普通株
2015年12月3日、当社発表9,160,888当社にサービスを提供する創設者株式としての普通株,価値$0.00011株当たりの額面は合計で約$である4,100(“方正株”)。開ける
2018年1月4日、これまでに発行された方正株式に増分帰属条件を設定した50歳これまでに発行された方正株式の30%が2018年1月4日に帰属し、残りの方正株式は一定期間帰属制限されていた5年それは.創設者が当社への雇用やサービスを終了した場合、当社はこれらの株式を買い戻すことができます。
当社の創設者と2018年1月4日に締結された雇用契約によると、当社は、彼等及びその共同会社が保有する普通株式総数(当社が株式奨励を付与する任意の株式を含む)が代表されることを確保するために、創設者毎にいくつかの潜在的な追加発行普通株式(“逆希釈株式”)を提供する15会社が完全に資本の%を薄くして、会社が$を集めるまで300.0株式資本は、最初の融資で調達した資本を含む。
この義務を履行するため、当社は2018年5月21日に発行しました251,547創業者に普通株を発行し、会社に提供するサービスと引き換えに、価値を$とする2.611株に別の分を加える251,547創業者株式と同じ帰属制限および帰属期限を受ける制限株。また、2018年9月6日、会社は1,795,023創業者に普通株を発行し、会社に提供するサービスと引き換えに、価値を$とする9.63一株当たり、追加のを加えます1,795,023創業者株式と同じ帰属制限および帰属期限を受ける制限株
2022年12月31日までの年度違います。雇用関係を中止したため、株は没収された。いずれも当社が買い戻しなければならない株式は、当該等の株式が帰属する前に、会計目的については、発行済み株式とはみなさない。そこで,当社は帰属期間制限株の計量日公允価値が補償費用であることを確認した。2022年12月31日までに55,227普通株式は当社が買い戻すことができます。このような報酬に関連した非帰属株式負債は提出されたすべての期間に関係ない。
注9-持分激励計画
2019年持分インセンティブ計画
2019年1月、会社取締役会と株主は“2019年インセンティブ奨励計画”(“2019年計画”)を承認した。2019年は2019年2月6日に発効、すなわちIPOに関する登録声明が発効する前日に予定されています。2019年計画によると、会社は、当時会社員、上級管理者、取締役またはコンサルタントであった個人および会社子会社の従業員およびコンサルタントに、株式オプション、株式付加価値権、制限株式、制限株式単位、およびその他の株式または現金に基づく報酬を付与することができる。合計する5,750,000普通株式は2019年の計画に基づいて発行のために予備予約されることが承認された。2019年計画発効日までに、2017計画(後述)に基づいても発行可能な株式数は、2019年計画発効日までに、2017計画に基づいて奨励が必要ですが、その後当社にログアウト、没収または買い戻しられた株式は、2019年計画に基づいて予約された株式に加入します。また、2019年に発行可能な普通株式数は、例年の初日に自動的に増加する予定だ10年2019年計画期間は、2020年1月1日から2029年1月1日までで、額は5前年の12月31日に当社の普通株式発行済株式数の%または当社取締役会が決定した低い金額に相当します。2022年12月31日までの合計491,0472019年計画によると、普通株式の発行が可能です16,199,2022019年の計画によると、普通株は未返済奨励金を受けなければならない。
2019年従業員株購入計画
2019年1月、会社取締役会と株主は“2019年従業員株購入計画”(以下、“ESPP”)を承認した。ESPPは2019年2月6日に発効、すなわちIPOに関連する登録声明が発効する前日に発効します。ESPPは参加者が賃金減額で普通株を購入することを許可し、最高で20彼らは条件を満たした補償の%です。合計する700,000普通株式は発行のためにESPPに従って予備的に保留されることが許可された。また,ESPPによって発行可能な普通株式数は,ESPP期限の前10年の例年ごとに自動的に増加し,2020年1月1日から2029年1月1日までに終了し,額に相当する1前年の12月31日に当社の普通株式発行済株式数の%または当社取締役会が決定した低い金額に相当します。2022年12月31日と2021年12月31日までの年間で157,858株と160,790株はそれぞれESPPによって発行された。2022年12月31日までの合計2,450,855普通株式はESPPにより発行できます。
2017持分インセンティブ計画
会社2017年株式インセンティブ計画(“2017計画”)は、奨励的株式オプション、非法定株式オプション、制限株式、制限株式単位、その他の株式ベースの奨励を許可しています。2019年計画を通過した後、2017年計画に基づいて追加の株式奨励を行うことはできません。2022年12月31日までに2,582,771普通株は2017年計画下の未償還オプションの制約を受け、違います。2017年計画に基づいて付与された制限株奨励株は帰属されていない。
株式オプション奨励の公正価値
各従業員および非従業員株式オプション付与の公正価値は、付与された日にBlack-Scholesオプション定価モデルを用いて推定される。当社は、十分な取引履歴があれば、十分な取引履歴がない場合には、同業グループを使用する独自の変動性を使用する。歴史的な行権の歴史が不足しているため、当社従業員の株式オプションの期待期限は“簡略化”奨励方法で決定された。非従業員に付与された株式オプションの期待期限は、オプション付与の契約期間に等しい。無リスク金利は、奨励付与時に発効した米国債収益率曲線を参考にして決定され、時間帯は奨励の期待期限にほぼ等しい。期待配当収益率はゼロ当社は現金配当金を派遣したことがないことから、予測可能な未来には現金配当金は何も発行されないと予想される。
以下の仮定は、本報告に記載されている間に、会社持分インセンティブ計画に従って従業員に付与された株式オプション報酬の公正価値およびESPPに従って購入可能な株式の公正価値を推定するためのものである
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| 従業員株式オプション | | | | | |
| 予想期限(年単位) | 4.4 - 6.1 | | 4.6 - 6.1 | | 4.6 - 6.1 |
| 無リスク金利 | 1.31% - 4.35% | | 0.65% - 1.30% | | 0.22% - 1.67% |
| 波動率 | 71.10% - 89.87% | | 78.80% - 84.79% | | 84.38% - 87.23% |
| 配当率 | — | | — | | — |
| | | | | |
| 従業員株購入計画 | | | | | |
| 予想期限(年単位) | 0.49 - 2.00 | | 0.49 - 2.00 | | 0.49 - 2.00 |
| 無リスク金利 | 0.60% -3.51% | | 0.06% - 0.20% | | 0.12% - 0.95% |
| 波動率 | 64.98% - 121.43% | | 61.29% - 87.13% | | 85.50% - 99.13% |
| 配当率 | — | | — | | — |
株式オプション
次の表は、2022年、2022年、2021年、2020年12月31日までの年間株式オプション活動をまとめています
| | | | | | | | | | | | | | | | | | | | | | | |
| 株は制限されるだろう
未完成オプション | | 重み付けの-
平均値
残り
契約書
命
(年) | | 骨材
内在的価値 |
| 株 | | 重み付けの-
平均値
トレーニングをする
値段 | | |
| | | | | | | (単位:千) |
| 2019年12月31日現在の未返済債務 | 8,538,060 | | | $ | 13.67 | | | 9.0 | | $ | 35,385 | |
| 付与したオプション | 2,712,372 | | | $ | 13.78 | | | | | |
| 行使のオプション | (134,803) | | | $ | 3.96 | | | | | |
| オプションは没収/キャンセルされる | (1,714,547) | | | $ | 15.99 | | | | | |
| 2020年12月31日現在の未返済債務 | 9,401,082 | | | $ | 13.42 | | | 8.1 | | $ | 10,182 | |
| 付与したオプション | 3,159,126 | | | $ | 9.82 | | | | | |
| 行使のオプション | (325,494) | | | $ | 6.19 | | | | | |
| オプションは没収/キャンセルされる | (2,800,054) | | | $ | 14.14 | | | | | |
| 2021年12月31日現在の未返済債務 | 9,434,660 | | | $ | 12.24 | | | 7.4 | | $ | 15,822 | |
| 付与したオプション | 9,271,272 | | | $ | 6.75 | | | | | |
| 行使のオプション | (270,707) | | | $ | 6.41 | | | | | |
| オプションは没収/キャンセルされる | (948,060) | | | $ | 15.69 | | | | | |
| 2022年12月31日現在の未返済債務 | 17,487,165 | | | $ | 9.24 | | | 8.1 | | $ | 47 | |
2022年12月31日現在帰属と予想帰属のオプション | 17,487,165 | | | $ | 9.24 | | | 8.1 | | $ | 47 | |
2022年12月31日までに行使可能なオプション | 6,433,686 | | | $ | 12.19 | | | 6.4 | | $ | — | |
二零二年、二零二年、二零二一年及び二零二年十二月三十一日までの年度中に、購入持分の加重平均授与日当たりの公平価値を$とする4.77, $6.93そして$9.82それぞれ,である.
2022年,2022年,2021年および2020年12月31日までの年度内に帰属する株式オプションの総公平価値は20.7百万、$21.9百万ドルとドル29.9それぞれ100万ドルです
上表の合計内在価値は,当社の普通株価格の公正価値と株式オプション取引価格との差額である.2022年、2021年および2020年12月31日までの年間で、行使された株式オプションの内的価値の合計は1.5百万、$1.6百万ドルとドル1.1それぞれ100万ドルです
2022年12月31日現在、未帰属株式オプション奨励に関する未確認補償総額は$である45.1百万ドル、会社は加重平均期間中に約を確認する予定です2.3何年もです。
制限株
当社の2022年、2022年、2021年および2020年12月31日までの年度限定株式活動の概要は以下の通りです
| | | | | | | | | | | |
| 量
制限される
株式単位
卓越した | | 重み付けの-
平均値
授与日
公正価値 |
| 2019年12月31日現在帰属していない | 4,648,526 | | | $ | 3.98 | |
| 授与する | 2,003,900 | | | 10.96 | |
| 既得 | (2,557,377) | | | 4.00 | |
| 没収/キャンセルされる | (764,228) | | | 8.30 | |
| 2020年12月31日帰属していない | 3,330,821 | | | $ | 7.16 | |
| 授与する | 1,349,885 | | | 10.23 | |
| 既得 | (1,391,476) | | | 5.86 | |
| 没収/キャンセルされる | (728,011) | | | 10.03 | |
| 2021年12月31日現在帰属していません | 2,561,219 | | | $ | 8.67 | |
| 授与する | 572,901 | | | 11.94 | |
| 既得 | (1,592,588) | | | 7.78 | |
| 没収/キャンセルされる | (191,497) | | | 10.68 | |
| 2022年12月31日現在帰属していません | 1,350,035 | | | $ | 10.83 | |
2022年12月31日現在、未帰属制限株式奨励に関する未確認補償総額は$である7.3百万ドル、会社は加重平均期間中に約を確認する予定です1.0何年もです。
株に基づく報酬費用
株式ベースの給与費用は、会社の総合経営報告書と全面赤字で以下のように報告されている(単位:千)
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| 研究開発 | $ | 24,415 | | | $ | 18,943 | | | $ | 18,997 | |
| 一般と行政 | 18,138 | | | 13,065 | | | 19,751 | |
| 株式に基づく報酬総支出 | $ | 42,553 | | | $ | 32,008 | | | $ | 38,748 | |
会社の前総裁とCEOが2020年11月に退職したことについて、会社は#ドルを確認しました5.52020年12月31日までの1年間、役員の既存の制限株式奨励が改正されたため、役員移行協定の条項に基づいて役員に付与された発行制限株式18カ月を加速させることを含め、株式ベースの増額報酬支出が生じた。
2022年12月31日現在、ESPPに関する未確認報酬支出総額は1億ドル1.4百万ドル、会社は加重平均期間中に約を確認する予定です0.7何年もです。
付記10-財産と設備、純額
会社の財産と設備の純額には以下の内容が含まれている(千計)
| | | | | | | | | | | | | | | | | |
| 推定数
使用寿命
(単位:年) | | 十二月三十一日
2022 | | 十二月三十一日
2021 |
| 事務設備 | 3-7 | | $ | 1,097 | | | $ | 1,097 | |
| コンピュータ装置 | 5 | | 123 | | | 123 | |
| ソフトウェア | 3 | | 130 | | | 130 | |
| 実験室装置 | 2-5 | | 6,098 | | | 5,688 | |
| 賃借権改善 | 6-7 | | 2,562 | | | 2,562 | |
| 建設中の工事 | 適用されない | | 83 | | | — | |
| 総資産と設備 | | | 10,093 | | | 9,600 | |
| 減算:減価償却累計 | | | 6,112 | | | 4,280 | |
| 財産と設備、純額 | | | $ | 3,981 | | | $ | 5,320 | |
2022年12月31日現在、2021年と2020年12月31日までの年間減価償却費用は約1億ドル1.8百万、$1.7百万ドルとドル1.4総合経営報告書および全面赤字には、それぞれ一般および行政費用および研究開発費が計上されている。
注11-引受金とその他の事項
賃貸借証書
当社は、2025年1月に満期となる取消不可経営賃貸契約に基づき、いくつかのオフィスおよび実験室スペースを最初の賃貸スペースと、2018年8月に締結された賃貸協定改正によりレンタルされた拡張スペースに分けます。2022年2月、当社は継続選択権を行使し、拡張空間の期限を2025年1月まで延長する。転貸協定には、不動産全体を2028年10月に延長するオプションが含まれている。延長された選択権は元のレンタル協定が終了する前に行使されなければならない。選択権によってカバーされる期間は、実行されるべきであると合理的に決定されることができないので、キャンセル不可能なレンタル期間には含まれない。レンタルは公共エリアの維持費とその他の費用を支払う必要があり、基本レンタル料は年間レンタル料で計算されます3その後毎年%増加します。経営リース負債を計量する際には、可変と判定され、指数または料率に基づいていない費用は計上されない。
毎月のレンタル料支出は直線法でレンタル期間内に確認します。経営リース賃貸支払現在値に貸借対照表を計上し,加重平均割引率は7%は、賃貸が暗黙的な金利を提供していないので、同様の経済環境下での賃貸支払いに相当する%を担保基準として当社が借入基準で借金するために必要な金利で計算されます。加重平均残余レンタル期間は2.0何年もです。
レンタル料金は以下の部分から構成されています(千計算)
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| リースコストを経営する | $ | 3,097 | | | $ | 4,029 | | | $ | 3,615 | |
| 短期賃貸コスト | 45 | | | 53 | | | 78 | |
| 総賃貸コスト | $ | 3,142 | | | $ | 4,082 | | | $ | 3,693 | |
2022年と2021年12月31日現在、賃貸負債を経営して計量した金額に計上して支払う現金は#ドルである3.2百万ドルとドル4.0それぞれ100万ドルです
2022年12月31日までの将来最低年間賃貸料約束総額は以下の通り(千単位)
| | | | | |
| 未割引家賃
支払い |
| 十二月三十一日までの年度 | |
| 2023 | $ | 3,319 | |
| 2024 | 3,419 | |
| 2025 | 144 | |
| 未割引賃貸料支払総額 | $ | 6,882 | |
| |
| 現価割引 | (453) | |
| 賃貸支払いの現在価値 | $ | 6,429 | |
| 賃貸負債を経営する流動部分(計上すべき費用及びその他の流動負債の構成要素として含む) | 2,983 | |
| 非流動経営賃貸負債 | 3,446 | |
| リース総負債を経営する | $ | 6,429 | |
当社は2022年、2022年、2021年および2020年12月31日までに約3.3百万、$4.3百万ドルとドル4.0レンタル料支出はそれぞれ100万ポンドです。
後続事件
当社は申請日までのすべての後続イベントと取引を評価しました。監査された総合財務諸表や開示に重大な事件の影響はない。
展示品索引
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
展示品
番号をつける | | 展示品説明 | | 引用で本明細書に組み込む | | 保存済み
ここから声明する |
| | 表 | | 日取り | | 番号をつける | | |
| 3.1 | | 会社登録証明書の改訂と再予約。 | | 8-K | | 2/12/2019 | | 3.1 | | |
| 3.2 | | 添付例を改訂及び再編成する。 | | 10-Q | | 5/12/2020 | | 3.2 | | |
| 4.1 | | 普通株式証明書フォーマット。 | | S-1/A | | 1/23/2019 | | 4.1 | | |
| 4.2 | | 登録者およびその特定の株主によって2018年7月20日に改正され、再署名された投資家権利協定。 | | S-1 | | 12/21/2018 | | 4.2 | | |
| 4.3 | | 取引法第12条に基づいて登録された証券説明。 | | 10-K | | 2/26/2021 | | 4.3 | | |
| 4.4 | | 契約は,日付は2020年5月21日であり,登録者と全国協会ウィルミントン信託会社との間で署名されている。 | | 8-K | | 5/21/2020 | | 4.1 | | |
| 4.5 | | 最初の補充契約は、日付が2020年5月21日で、登録者と全国協会ウィルミントン信託基金との間で署名された。 | | 8-K | | 5/21/2020 | | 4.2 | | |
| 4.6 | | グローバル手形フォーマットは、(添付ファイル4.5の一部として)2027年に満期された変換可能優先手形の5.00%に相当する。 | | 8-K | | 5/21/2020 | | 4.3 | | |
| 10.1# | | Gossamer Bio、Inc.2017年株式激励計画は、改訂された。 | | S-1 | | 12/21/2018 | | 10.1 | | |
| 10.2# | | Gossamer Bio,Inc.2017年持分インセンティブ計画下での株式オプション付与通知及び株式オプションプロトコルフォーマットは、改訂された。 | | S-1 | | 12/21/2018 | | 10.2 | | |
| 10.3# | | Gossamer Bio,Inc.2017年持分インセンティブ計画下での制限株式付与通知及び制限株式協定のフォーマットは、改訂された。 | | S-1 | | 12/21/2018 | | 10.3 | | |
| 10.4# | | 方正制限株式付与通知書及び制限株式協議フォーマット。 | | S-1 | | 12/21/2018 | | 10.4 | | |
| 10.5# | | Gossamer Bio,Inc.2019年インセンティブ奨励計画およびその下での株式オプション付与通知および株式オプションプロトコルのフォーマット。 | | S-1/A | | 1/23/2019 | | 10.5 | | |
| 10.6# | | Gossamer Bio,Inc.2019年従業員株式購入計画。 | | S-1/A | | 1/23/2019 | | 10.6 | | |
| 10.7# | | 薄紗生物株式会社非従業員役員報酬計画。 | | | | | | | | X |
| 10.8# | | Gossamer Bio,Inc.2019年持分インセンティブ計画に基づいて達成された制限株式単位合意。 | | 10-Q | | 5/12/2020 | | 10.1 | | |
| 10.9# | | 書簡協定は,日付は2020年11月16日であり,Faheem Hasnainと登録者の間で署名されている。 | | 10-K | | 2/26/2021 | | 10.11 | | |
| 10.10# | | 2018年12月4日、Bryan Giraudoと登録者の間の招聘状。 | | S-1 | | 12/21/2018 | | 10.10 | | |
| 10.11# | | 招聘状は、2018年12月4日にChristian Waageと登録者の間で書かれている。 | | S-1 | | 12/21/2018 | | 10.11 | | |
| 10.12# | | Caryn Petersonと登録者の間の招聘状は、2021年4月16日。 | | 10-Q | | 8/9/2021 | | 10.1 | | |
| 10.13# | | ローラ·カーターと登録者の間の招聘状は、日付は2021年5月1日。 | | 10-Q | | 8/9/2021 | | 10.2 | | |
| 10.14# | | リチャード·アランダと登録者の間の招聘状は、2021年6月21日。 | | 10-Q | | 8/9/2021 | | 10.3 | | |
| 10.15# | | 賠償協議形式。 | | S-1 | | 12/21/2018 | | 10.14 | | |
| 10.16 | | 転貸協定は,日付は2017年12月29日であり,製薬会社と登録者の間で締結されている。 | | S-1 | | 12/21/2018 | | 10.15 | | |
| 10.17 | | 医薬品会社と登録者との間の転貸協定第1改正案は,期日は2018年8月24日である。 | | S-1 | | 12/21/2018 | | 10.16 | | |
| 10.18 | | 医薬品会社と登録者との間の転貸協定第2改正案は,期日は2022年6月1日である。 | | 10-Q | | 8/9/2022 | | 10.10 | | |
| 10.19† | | 独占許可協定は,2017年10月2日にGB 002,Inc.,登録者とPulmokine,Inc.が署名した。 | | S-1 | | 12/21/2018 | | 10.17 | | |
| 10.20 | | クレジット,保証,セキュリティ協定は,日付は2019年5月2日であり,GB 001,Inc.が借り手として,Gossamer Bio,Inc.が保証人として,MidCap Financial Trustが代理と貸金者として,時々増加する貸主が署名した。 | | 8-K | | 5/3/2019 | | 10.1 | | |
| 10.21 | | “信用、担保及び担保協定第一修正案”は、2019年9月18日に、GB 001,Inc.が借り手として、登録者を保証人として、その他の保証人が、MidCap金融信託会社、代理及び融資者、及び時々増加する貸金者と随時締結される。 | | 10-Q | | 11/12/2019 | | 10.1 | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
展示品
番号をつける | | 展示品説明 | | 引用で本明細書に組み込む | | 保存済み
ここから声明する |
| | 表 | | 日取り | | 番号をつける | | |
| 10.22 | | クレジット、担保および担保協定の第2の修正案は、2020年7月2日に、登録者、GB 001、Inc.,GB 002、Inc.およびGB 004,Inc.が共通借主として、時々その他の保証人として、MidCap Financial Trustが代理人および融資者として、時々その追加融資者として署名される。 | | 8-K | | 7/2/2020 | | 10.1 | | |
| 10.23 | | “信用、担保および担保協定第三修正案”は、2022年12月7日に、登録者GB 001,Inc.,GB 002,Inc.およびGB 004,Inc.が共同借主として、その他の保証人およびMidCap Financial Trustとして、時々その追加の融資者として署名される。 | | | | | | | | X |
| 10.24 | | 信用、担保および担保協定の第4の修正案は、日付が2023年2月14日であり、登録者GB 001,Inc.,GB 002,Inc.およびGB 004,Inc.が共通借主として、時々その他の保証人およびMidCap Financial Trustとして代理人および融資者として、時々その追加融資者として署名される。 | | | | | | | | X |
| 10.3 | | 株式購入協定は,期日は2022年7月12日であり,登録者とその中で指名された購入者が署名する。 | | 8-K | | 7/13/2022 | | 10.1 | | |
| 21.1 | | 登録者の子会社リスト。 | | | | | | | | X |
| 23.1 | | 独立公認会計士事務所安永会計士事務所の同意を得ました。 | | | | | | | | X |
| 31.1 | | Gossamer Bio,Inc.CEOの認証は、1934年の“証券取引法”ルール13 a-14(A)またはルール15 d-14(A)の要求に基づいている。 | | | | | | | | X |
| 31.2 | | Gossamer Bio,Inc.首席財務官の証明は、1934年の“証券取引法”規則13 a-14(A)または規則15 d-14(A)の要求に基づいている。 | | | | | | | | X |
| 32.1* | | 2002年のサバンズ·オキシリー法第906条に基づいて最高経営責任者証明書が発行された。 | | | | | | | | X |
| 32.2* | | 2002年のサバンズ·オキシリー法第906条に基づいて首席財務官証明書が発行された。 | | | | | | | | X |
| 101.INS | | XBRLレポートインスタンスドキュメント | | | | | | | | X |
| 101.書院 | | XBRL分類拡張アーキテクチャドキュメント | | | | | | | | X |
| 101.カール | | XBRL分類計算リンクライブラリ文書 | | | | | | | | X |
| 101.def | | XBRL分類拡張Linkbase文書を定義する | | | | | | | | X |
| 101.介護会 | | XBRL分類ラベルLinkbaseドキュメント | | | | | | | | X |
| 101.Pre | | XBRLプレゼンテーションリンクライブラリドキュメント | | | | | | | | X |
| | | | | |
| # | 契約または補償計画を管理すること。 |
† | S-K条例第601(B)(10)(Iv)項によると、秘密のため、本展示品の一部の内容(星番号で示す)は省略されている。 |
| * | 米国法第18編第1350節の規定によれば、これらの証明は、1934年“証券取引法”第18節の目的のために提出されるのではなく、本年度報告書と共に提供されるだけであり、引用によって登録者のいずれの文書にも組み込まれず、本文書の日付の前であっても後であっても、当該文書中の任意の一般的な合併言語にかかわらず、記載されている。 |
サイン
1934年“証券取引法”第13又は15(D)節の要求に基づいて、登録者は、次の署名者が代表して本報告書に署名することを正式に許可した。
薄紗生物会社です。
| | | | | | | | | | | |
| 差出人: | /s/Faheem Hasnain | |
| | ファシム·ハスナイヌ | |
| | 社長と最高経営責任者 | |
| 日取り | 2023年3月17日 | |
署名と授権書
本報告は、1934年の証券取引法の要求に基づき、以下の者によって登録者として指定日に署名された。
| | | | | | | | | | | | | | |
| サイン | | タイトル | | 日取り |
| | | | |
| /s/Faheem Hasnain | | CEO兼取締役会長総裁
(首席行政官) | | 2023年3月17日 |
| ファシム·ハスナイヌ | | |
| | | | |
| /s/Bryan Giraudo | | 最高経営責任者と首席財務官
(主な財務と
会計主任) | | 2023年3月17日 |
| ブライアン·ジラウド | | |
| | | | |
| /s/ジョシュア·H·ビレンク | | 役員.取締役 | | 2023年3月17日 |
| ジョシュア·H·ビレンク医学博士 | | |
| | | | |
| /s/クリスティーナ·ブロ | | 役員.取締役 | | 2023年3月17日 |
| クリスティーナ·ブロ | | |
| | | | |
| /s/ラッセル·コックス | | 役員.取締役 | | 2023年3月17日 |
| ラッセル·コックス | | |
| | | | |
| トマス·ダニエル医学博士 | | 役員.取締役 | | 2023年3月17日 |
| トーマス·ダニエル医学博士です | | |
| | | | |
| /s/Renée Gal゚ | | 役員.取締役 | | 2023年3月17日 |
| ルネ·ガララ | | |
| | | | |
| サンドラ·ミリガンM.D.J.D. | | 役員.取締役 | | 2023年3月17日 |
| サンドラ·ミリガン | | |