カタログ表
アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
| (マーク1) | |
| | 1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
本財政年度末まで
あるいは…。
| | 1934年証券取引法第13条又は15条に基づいて提出された移行報告 |
移行期になります 至れり尽くせり
依頼書類番号:
|
| Vaxart,Inc. |
|
|
| (登録者の正確な氏名はその定款に記載) |
|
|
| |
| |
|
|
| (登録設立又は組織の国又はその他の管轄区域) |
| (税務署雇用主身分証明書番号) |
|
|
| |
| ( |
|
|
| (主な執行機関の住所、郵便番号を含む) |
| (登録者の電話番号、市外局番を含む) |
|
同法第12条(B)に基づいて登録された証券:
|
| クラスごとのタイトル |
| 取引記号 |
| 登録された各取引所の名称 |
|
|
| |
| |
| ♪the the the |
同法第12条(G)により登録された証券:なし
登録者が証券法第405条規則で定義されている有名な経験豊富な発行者である場合は、登録者が有名な経験豊富な発行者であるか否かを複選マークで示す。登録者は、そうである
登録者が当該法案の第13節又は第15(D)節に基づいて報告を提出する必要がない場合は,再選択マークで示してください。登録者は,当該法案の第13節又は第15(D)節に基づいて報告を提出する必要がない。登録者が法案第13節又は第15(D)節に基づいて報告を提出する必要がない場合は,登録者が報告を提出する必要がないことを示す。登録者が当該法案の第13節又は第15(D)節に基づいて報告を提出する必要がない場合は,その登録者は報告書を提出する必要がない
再選択マークは、登録者が、(1)過去12ヶ月以内に(または登録者にそのような報告の提出を要求するより短い期間内に)1934年の証券取引法第13条または第15(D)節に提出されたすべての報告書を提出したかどうか、および(2)過去90ヶ月以内にそのような提出要件を満たしてきたかどうかを示す
再選択マークは、登録者が過去12ヶ月以内に(または登録者がそのような文書の提出を要求されたより短い時間以内に)S−T規則405条(本章232.405節)に従って提出されることを要求した各対話データファイルを電子的に提出したかどうかを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社か新興成長型会社かを再選択マークで示した。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
| 大規模加速ファイルマネージャ☐ | ☐中の加速ファイルマネージャ |
| | 規模の小さい報告会社です |
|
| 新興成長型企業: |
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる
証券が同法第12条(B)に基づいて登録されている場合は,登録者が届出中の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示す。☐
これらのエラーのより真ん中に登録者の任意の幹部が関連回復中に§240.10 D−1(B)に従って受信されたインセンティブベースの報酬に基づいて回復分析を行う必要があるかどうかを再選択マークで示す。☐
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条の規則で定義されている)。登録者は空殻会社である
登録者が最近完成した第2財期の最終営業日である2022年6月30日までに,登録者の非関連会社が保有する登録者普通株の総時価は1株当たり3.50ドルであり,これは登録者普通株の最終報告販売価格に基づいて計算される
引用で編入された書類
登録者は,2022年12月31日までの財政年度終了後120日以内に,第14 A条に基づいて最終的な依頼書を提出する予定である。この依頼書の部分内容は、参照によって本テーブルの第III部分:10-Kに組み込まれる。
カタログ
|
|
|
ページ |
|
|
|
前向きに陳述する |
1 |
|
| 第1部 |
|
|
|
| 第1項。 |
業務.業務 |
2 |
|
| 第1 A項。 |
リスク要因 |
48 |
|
| 項目1 B。 |
未解決従業員意見 |
79 |
|
| 第二項です。 |
属性 |
79 |
|
| 第三項です。 |
法律訴訟 |
79 |
|
| 第四項です。 |
炭鉱安全情報開示 |
79 |
|
|
|
|
|
|
| 第II部 |
|
||
| 五番目です。 |
登録者普通株市場、関連株主事項及び発行者による株式証券の購入 |
80 |
|
| 第六項です。 |
[保留されている] |
80 |
|
| 第七項。 |
経営陣の財務状況と経営成果の検討と分析 |
81 |
|
| 第七A項。 | 市場リスクの定量的·定性的開示について | 86 | |
| 第八項です。 |
財務諸表と補足データ |
87 |
|
| 第九項です。 |
会計と財務情報開示の変更と相違 |
112 |
|
| 第9条。 |
制御とプログラム |
112 |
|
| プロジェクト9 B。 | その他の情報 | 113 | |
| プロジェクト9 Cです。 | 検査妨害に関する外国司法管区の開示 | 113 | |
|
|
|
|
|
| 第三部 |
|
||
| 第10項。 |
役員·幹部と会社の管理 |
114 |
|
| 第十一項。 |
役員報酬 |
114 |
|
| 第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
114 |
|
| 十三項。 |
特定の関係や関連取引、取締役の独立性 |
114 |
|
| 14項です。 |
最高料金とサービス |
114 |
|
|
|
|
|
|
| 第4部 |
|
||
| 第十五項。 |
展示品と財務諸表の付表 |
115 |
|
|
|
|
|
|
| 展示品索引 |
|
116 |
|
|
|
|
|
|
| 第十六項。 |
表格10-Kの概要 |
118 |
|
|
|
|
|
|
| サイン |
|
119 | |
前向きに陳述する
この2022年12月31日までの年間10-K表年次報告(本“年次報告”)には、1933年証券法第27 A条(改正された)と1934年証券取引法第21 E条(改正された)が指す展望的な陳述が含まれており、このような条項によって作成された“安全港”の制約を受けて、我々の業務、運営、財務業績及び状況、並びに業務運営及び財務業績及び状況に対する我々の計画、目標、期待が含まれている。本明細書に含まれる任意の非歴史的事実の陳述は、前向きな陳述と見なすことができる。これらの陳述は、未来のイベントおよび未来の傾向の予測または指示である“予想”、“仮説”、“信じ”、“可能”、“推定”、“予想”、“意図”、“可能”、“計画”、“すべき”、“将”、“会議”、および他の同様の表現によって識別することができる。これらの展望的陳述は、将来の業績や発展の保証ではなく、既知および未知のリスク、不確実性、および他の場合によっては私たちの制御範囲を超える要素に関連する、私たちが経営している業務および業界の現在の予想、推定、予測と予測、および経営陣の信念と仮定に基づいている。したがって、私たちの本年度報告書の任意またはすべての展望的陳述は不正確であることが証明されるかもしれない。我々の業務運営、財務業績、および状況に重大な影響を与える可能性のある要因は、本明細書で“第1 A項--リスク要因”に記載されているリスクおよび不確定要因を含むが、これらに限定されない。私たちはあなたが展望的な陳述を評価する時にこれらの要素を慎重に考慮し、展望的な陳述に過度に依存しないように警告することを促します。前向き陳述は,本年度報告書提出日までに我々が把握した情報に基づいている。法的要求がない限り、私たちは新しい情報または未来のイベントまたは他の状況を反映するために、いかなる前向きな陳述も公開的に更新または修正するつもりはない。しかし、本年度報告日後に時々米国証券取引委員会(“米国証券取引委員会”)に提出される報告書に記載されている要因及びリスクを検討しなければなりません。
本年度報告書には我々の業務や業界に関する市場データも含まれている。これらの市場データは複数の仮定に基づく予測を含む。これらの仮説が正しくないことが証明されれば,実際の結果はこれらの仮定に基づく予測とは異なる可能性がある.したがって、私たちの市場はこのようなデータ予測の速度で増加しないかもしれないし、全くそうではないかもしれない。もしこれらの市場がこれらの予想された速度で成長できなければ、私たちの業務、運営結果、財務状況、私たちの普通株の市場価格に損害を与えるかもしれない。49ページのリスク要因の概要を参照してください。
第1部
第1項:国際業務
概要
Vaxart Biosciences,Inc.は最初に2004年3月にカリフォルニア州で登録設立され、名称はWest Coast Biologals,Inc.であり、2007年7月にデラウェア州で再登録された時にVaxart,Inc.(“Private Vaxart”)と改名した。
2018年2月13日,Private VaxartはAviragen Treateutics,Inc.Inc.(Aviragenと略す)との逆合併を完了し,これにより,Private VaxartはAviragenの完全子会社として生存した。合併条項によると、AviragenはVaxart,Inc.,Private VaxartはVaxart Biosciences,Inc.と改称される。別の説明がない限り、本年度報告で言及されている“Vaxart”、“We”、“Our”、または“Company”は、合併後の会社Vaxart,Inc.を意味する。
我々は臨床段階のバイオテクノロジー会社であり,主に我々の担体−アジュバント−抗原標準化技術(“VAAST”)に基づく経口組換えワクチンの開発に焦点を当てており,独自の経口ワクチンプラットフォームである。われわれの経口ワクチンは、広範かつ持続的な免疫反応を産生することを目的としており、各種感染症を予防することができ、慢性ウイルス感染や癌の治療に有用である可能性がある。私たちが研究しているワクチンは注射ではなく室温で安定した錠剤を使用している。
我々は、ノウォーカーウイルス(急性胃腸炎の原因を広く引き起こす)、SARS-CoV-2(2019年のコロナウイルス疾患を引き起こすウイルス(新冠肺炎))、インフルエンザ、季節性インフルエンザを含む一連の伝染病に対する予防ワクチン候補を開発している。私たちのノウォーカーウイルス候補ワクチンのいくつかの第一段階の人体研究は成功した。我々GI.1ノウォーカー候補ワクチンの安全性と臨床有効性を評価する第二段階挑戦研究が現在行われている。我々の二価GI.1およびGII 4候補ノウォーカーウイルスワクチンの安全性および免疫原性を評価する第2段階用量範囲の研究が現在行われている。我々は、第1の新冠肺炎候補ワクチンの第1段階の臨床試験を完了し、この研究が主要かつ副次的な終点に達したと報告している。私たちの第2の新冠肺炎候補ワクチンは、2021年末に開始された第2の段階研究の第1の部分が完了した。著者らはまた新型新冠肺炎ワクチンの臨床前仕事を開始し、SARS-CoV-2と他のベータコロナウイルス(例えばSARS-CoV-1とMERS-CoV)に対応するために、有効なパンベタコロナウイルス候補ワクチンを創造することを求めた。データにより、著者らの単価H 1インフルエンザ候補ワクチンは参加者をH 1インフルエンザ感染から保護し、そして2020年に発表された第二段階挑戦研究でリードした発売注射可能ワクチン(柳葉刀ID)である。また,気道合胞体ウイルス(RSV)(気道感染のよく見られる原因)に対する予防的候補ワクチンと,子宮頸癌とヒトパピローマウイルス(HPV)による異型増殖に対するわれわれの最初の治療的候補ワクチンの臨床前データを生成した。
経口錠剤ワクチン候補品は以下のような重要な利点があると信じています
まず、それらは系統、粘膜とT細胞反応を含む広範かつ持続的な免疫反応を産生することを目的としており、これはノウォーカーウイルス、新冠肺炎、インフルエンザと呼吸器合胞体ウイルスのようないくつかの伝染病に対する保護を強化する可能性があり、そしてヒトパピローマウイルスによるもののようないくつかの癌と慢性ウイルス感染に対して潜在的な臨床利益がある可能性がある。
次に、著者らの候補錠剤ワクチンは、より効率的、より便利な管理方法を提供し、患者の受容度を高め、流通ボトルネックを減少させることを目的としており、ワクチン接種活動の有効性を高めると信じている。例えば,米国疾病コントロール·予防センター(CDC)のデータによると,2021/2022年の季節性インフルエンザシーズンには,米国人口の約51%しかインフルエンザワクチンを接種しておらず,18歳から49歳の成人のワクチン接種率は特に低い。
私たちの製品ライン
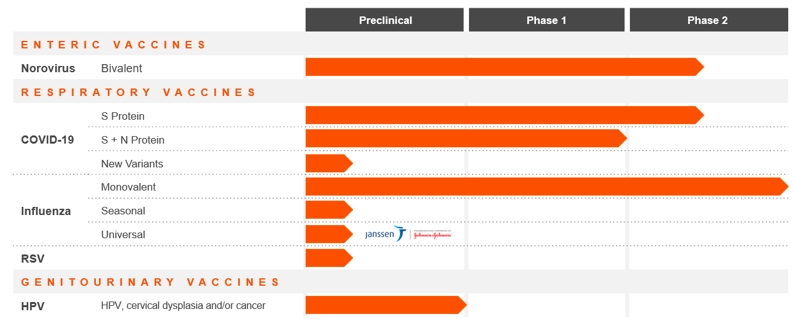
図1.下表は,我々の経口ワクチン開発計画の状況を概説した
私たちは独自のプラットフォームに基づいて次の候補プレートワクチンを開発しています
| ● |
ノウォーカーウイルスワクチンですノルウォーカーウイルスはアメリカのすべての年齢層の人の急性胃腸炎症状の主要な原因であり、例えば嘔吐と下痢である。平均毎年、ノルウォーカーウイルスは1900万から2100万例の急性胃腸炎を招き、そして10.9万人の入院と900人の死亡を招き、その大部分は児童と老人である。典型的な症状としては,脱水,嘔吐,腹部痙攣を伴う下痢,嘔気がある。疾病管理センターとジョン·ホプキンス大学が2016年に発表した研究では、ノルウォーカーウイルス疾患の全世界に対する毎年の経済影響は600億ドルと推定され、その中の340億ドルはアメリカを含む高収入地域で発生している。主要著者の最新報告では、2018年には米国一国の負担だけで105億ドルにのぼると推定されている。ノウォーカーウイルス疾患のほとんどはノウォーカーウイルスのGiとGII遺伝子によるものであり,両ウイルスの予防を目的とした2価ワクチン候補ワクチンを開発している。ノルウォーカーウイルスは小腸上皮細胞に感染する腸管病原体であるため、腸管でノウォーカーウイルス抗体を産生するワクチン、例えば我々の候補錠剤ワクチンは、腸管に直接輸送され、最適な保護を提供する可能性があると考えられる。承認されれば、このワクチンは冬のノルウォーカーウイルス感染ピーク前に年に1回接種され、インフルエンザ季節のようなものになると予想される。 |
2019年、我々は1 b期の臨床試験の活性段階を完成し、私たちの経口錠剤ワクチン候補GI.1とGII 4ノウォーカーウイルス株を獲得した。ノウォーカーウイルスGI.1とGII 4候補ワクチンの内服耐性は良好であり,重篤な有害事象の報告はなかった。大多数の能動と能動の副作用の重症度は比較的に軽く、候補ワクチン群とプラセボ治療群の間に有意差は認められなかった
われわれの二価候補ワクチン(GI.1とGII.4の共同接種)は強い免疫原性を示し,検討した二価行列ではGI.1株とGII.4株のIgA ASC応答率はそれぞれ78%と93%であったのに対し,両一価行列はそれぞれ86%と90%であった。これらの結果は,単独(単価)ワクチン接種と比較して,2つの候補ワクチンの併用接種(第2段階と第3段階試験に入る予想される方法)が菌株間干渉や免疫反応の減少を示さなかったことを示している
2021年初め、1 b期二価研究では、一部の被験者にG 1.1ワクチンの接種(1年以上後の2回目の接種)を開始した。2021年7月に発表された結果では、以前に接種した被験者のG 1.1ノウォーカーウイルス候補ワクチンの免疫応答を増強することに成功したと報告されている。これらの増強された応答には、IgA抗体分泌細胞、およびIgGおよびIgA血清抗体応答が含まれる。2021年に、我々は55歳から80歳までの高齢成人対象において、G 1.1候補ワクチンの高齢者群における安全性および免疫原性を評価するためにプラセボ対照の用量範囲研究を開始した。最も重要な結果は2022年6月に発表された。健康な高齢者(55歳~80歳)では、候補ワクチンに対する免疫反応は、抗体分泌細胞(IgA ASC)および血清抗体の数によって測定された以前の研究の若者と類似している。最後に、若年者においてBoostを使用する最適な時間を評価するための開放ラベル試験を行い、3群の被験者が初回ワクチン接種後1~3カ月の異なる時点で第2剤(Boost)を受けた。この研究の主な結果は2022年6月に発表された。データによると,G 1.1候補ワクチンは抗体反応の増強に成功しており,短い間隔よりも3カ月以内に接種した抗体反応の傾向が良いことが分かった
我々は現在、候補ノウォーカーウイルスワクチンに対して追加の第2段階臨床試験を行っている。第1の試験は2022年初めに開始され、第2段階のノルウォーカーウイルス挑戦研究であり、ノルウォーカーウイルス挑戦後のプラセボ対照と比較して、ノルウォーカーGI.1候補ワクチンの安全性、免疫原性、臨床治療効果を評価する。2023年第1四半期に、Vaxartは進行中の第2段階GI.1ノウォーカーウイルス挑戦研究を拡大し、追加の挑戦行列を取り入れた。Vaxartは、増加したデータセットはワクチン免疫反応の決定とノウォーカーウイルス感染および/または急性胃腸炎リスクを低下させることとの間の保護相関性を高める可能性を高めると考えている。Vaxartは,新たな保護関連性を識別することが3期試験の規模と持続時間を減少させる可能性があると予想している。この試験の主要なデータは2023年第3四半期に発表される予定だ。第2の臨床試験は2023年初めに開始され、第2段階多中心プラセボ対照用量範囲試験であり、Vaxartの二価ノロウイルス候補ワクチンの18歳以上の被験者における安全性と免疫原性を評価する。この第2段階臨床試験の主要なデータは、2023年に発表される予定で、この研究は、第3段階で使用しようとしている用量を選択することを可能にすることを目的としている。投与量選択後、後続の第2段階研究は約500人の被験者を募集し、これらの被験者は、選択された用量で十分な安全なデータを生成し、第2段階会議の終了時にFDAと第3段階の重要な治療効果研究の範囲と設計について一致することができるようになる予定である。
2022年秋、私たちは、私たちの二価ノルウォーカーウイルス候補ワクチンが哺乳期の母親の母乳で抗体を誘導するかどうか、生後6ヶ月の赤ちゃんが母乳でこれらの抗体を得ることができるかどうかを評価するために、ビルとメリンダ·ゲイツ財団の大量の資金と支持を得る研究を発表した。幼児は特にノルウォーカーウイルスの感染を受けやすく、深刻な脱水と潜在死を招き、特に発展途上国である。また、新生免疫系により、最小の子供にワクチンを接種する能力は難しいかもしれない。牛乳で誘導された抗体は乳児に受動的に転移し、母乳で育てられた赤ちゃんを感染病原体から保護する可能性がある。母乳中で抗体を誘発することに成功すれば、次のステップは、これらの抗体が幼児を保護し、家族でのノルウォーカーウイルスの伝播を抑制できることを証明することになる。この研究は2023年に開始される予定だ。Vaxartは,ビルとメリンダ·ゲイツ財団の贈与受給者として,低所得国と中所得国の母乳育児母親にその二価ノウォーカー候補ワクチンを使用することを世界的に承諾し,有効であることが証明され承認されれば同意した。
| ● |
コロナウイルスワクチンです。新冠肺炎はSARS-CoV-2ウイルスによる深刻な呼吸器感染であり、アメリカと世界各地の入院と死亡の主要な原因である。疾病管理センターによると、新冠肺炎は2019年末に武漢で爆発し、中国、そして全世界で急速に伝播している。2023年1月19日までに、全世界で6.67億例を超える新冠肺炎病例が発見され、その中にアメリカを含み、アメリカ疾病管理センターが報告した感染者数は1.01億人を超え、死亡者数は100万人を超えた。在宅注文など,新冠肺炎の制限の多くは中止されているが,新冠肺炎は蔓延し続けており,公共健康を脅かしており,特に新たな変種が続出しているためである。 |
過去数年間、私たちは新冠肺炎候補ワクチンの開発に多くの労力を費やした。私たちは公表されたSARS-CoV-2ゲノムに基づいて複数の候補ワクチンを生成し、臨床前モデルで粘膜と系免疫反応を産生する能力を評価した。特に興味深いのは、コロナウイルスが主に気道に感染しているため、粘膜免疫反応である。最近出現したS蛋白変異のコロナウイルス株は、原始株よりも感染性があると考えられていることから、ワクチンを投与した血清抗体は、これらのSARS−CoV−2変種を予防するのに十分ではない可能性があり、保存的エピトープに対する交差反応粘膜抗体やT細胞を産生することができる新規ワクチンが顕著に優れている可能性がある。
2020年9月14日、私たちはアメリカ食品医薬品局(FDA)が私たちの臨床研究新薬(IND)の申請を許可し、私たちの最初の経口新冠肺炎(SとN蛋白)候補ワクチンVXA-CoV 2-1の臨床試験を開始することを許可したと発表した。2021年2月、私たちは実験の初歩的な結果を発表した。この研究はそれぞれ安全性と免疫原性の主要と副次的な終点を達成した。交差反応を示す粘膜抗体反応の予備結果が発表された科学転換医学より詳細な研究結果と粘膜耐久性データはMedRxiv2022年7月。
われわれは2021年2月に、スパイクタンパク質のみを含む新たな新冠肺炎候補ワクチンと、異なる変異体に対する異なる候補ワクチンを評価することを発表した。臨床前評価(非ヒト霊長類研究を含む)では、スパイクタンパク質のみを発現する新冠肺炎候補ワクチン(VXA−CoV 2−1.1−S)が抗体反応を改善することを示した後、この候補ワクチンを臨床評価に進めることにした
FDAは2021年7月、VXA-CoV 2-1.1-SのINDプログラムを承認した。2021年10月、2つの部分からなる第2段階臨床研究報告で候補薬剤の用量分配を開始し、2つの研究設計を採用し、約896人の参加者を登録する予定である。研究の第1部分は、18歳から55歳の年齢48人の参加者と、56歳から75歳の年齢の参加者48人を募集し、安全性および免疫原性をさらに評価し、最適な用量を評価することを計画している。さらに、試験中の半分の被験体は、VXA-CoV 2-1.1-Sが免疫反応を増強し、変種特異的交差反応を増強する能力を試験するために、事前にワクチン(2剤メッセンジャーリボ核酸ワクチンを接種した)を接種し、半分の被験体は以前のワクチン接種を無邪気にする。この研究の目的は、安全性および免疫原性を評価し、最適な用量を評価することである。第1の部分の用量選択に基づいて、研究の第2の部分(“第2の部分”)は、18歳~75歳の年齢の約800人の被験者を募集することを計画している。第2の部分の目的は、SARS−CoV−2感染を予防するためにワクチンの予備効果を試験することである。研究の第1の部分(“第1の部分”)が完了した。ワクチンの幼稚な個人をタイムリーに識別して登録することができないため、第1の部分の実際の参加者数は計画よりも少ない。この部分の試験の主要なデータは2022年9月に公表され、主要と副次的な端末はすべて要求を満たしていることを表明した。VXA-CoV 2-1.1-Sは、以前に遺伝子ワクチン(ファイザー/バイオテクノロジーまたはModerna)を受けたボランティアの血清抗体応答を向上させることができる。この集団では、SARS-CoV-2(武漢)に対する血清中和抗体応答は、481の幾何平均値から778に向上し、1.6倍に増加した。開始力価の低いボランティアは、力価の高い被験者よりもはるかに増加した。Svnt検出により、これらのボランティアのSARS−CoV−2オミックBA 4/5ウイルスに対する中和抗体応答も有意に増加した。約50%の被験者の粘膜IgA抗体応答(鼻と口腔内の抗体)が増加した。SARS−CoV−2武漢Sに対する粘膜IgA応答が増加した被験者は、SARS−CoV−2オクテュウムBA 4/5、SARS−CoV−1およびMERS−CoVのIgA応答も他のコロナウイルスを含む他のコロナウイルスに対して増加し、これらの免疫読み取りの交差反応性質が示唆された
著者らはすでに新型ワクチン構造の臨床前仕事を開始し、SARS-CoV-2と他のベータコロナウイルス(例えばSARS-CoV-1とMERS-CoV)に反応する有効なパンベタコロナウイルス候補ワクチンの作成を求めた。以上のように,Vaxartの臨床データは,複数のSARS−CoV−2株およびSARS−CoV−1とMERS−CoVに対する交差反応抗体を粘膜表面に誘導する可能性が示唆されている。これは大きな問題です
2022年6月、我々はhVivo Services Limited(“hVivo”)との協力を発表し、SARS-CoV-2人類挑戦研究においてVaxart新冠肺炎候補ワクチンの有効性をテストした。2023年第1四半期、私たちはSARS-CoV-2人類挑戦研究の開始を延期することにした。私たちが新しい候補ワクチンを提案するにつれて、私たちは人類挑戦研究を含むかもしれない最適な開発計画を決定する
| ● |
季節性インフルエンザワクチンですインフルエンザはアメリカと世界範囲内の発病率と死亡率の主要な原因であり、疾病管理センターのデータによると、2021/2022年には約51%の合格アメリカ公民のみがワクチンを接種し、18歳から49歳までの成人のワクチン接種率は特に低い。私たちの経口錠剤ワクチン候補方案は現在利用可能なインフルエンザワクチンの保護効果を高め、インフルエンザワクチンの接種率を高める可能性があると信じている。 |
インフルエンザは全世界で最もよく見られる伝染病の一つであり、軽微な生命に危害を及ぼす疾患、甚だしきに至っては死亡を引き起こすことができる。全世界で毎年約3.5億の季節性インフルエンザ病例が発生し、その中の300~500万例は深刻な病例と考えられ、毎年29万から65万人が死亡した。幼児と高齢者が直面するリスクが最も大きい。米国では,人口の5%から20%がインフルエンザに罹患しており,14万から71万人がインフルエンザ合併症で入院し,毎年1.2万から5.2万人がインフルエンザとその合併症で死亡し,90%に達するインフルエンザ関連死亡は65歳以上の成人で発生している。季節性インフルエンザの総経済負担は871億ドルと推定され、その中には平均毎年104億ドルの医療費用が含まれているが、毎年疾病と生命損失による収入損失は163億ドルに達している。
私たちの候補錠剤ワクチンは、以下のような理由で、注射可能な卵型インフルエンザワクチンによる多くの制限を潜在的に解決することができると信じている:(I)私たちの候補錠剤ワクチンは、広範かつ持続的な免疫反応を産生し、より有効な免疫を提供し、他の株変異を防止することができる;(Ii)私たちの候補ワクチンは室温で安定した錠剤形態で提供され、これはより便利な投与方法を提供し、それによって患者の受容度を高め、配布と管理過程を簡略化することができると信じている;(Iii)私たちの候補錠剤ワクチンの製造速度は卵ベースの方法を用いて組換え方法によって製造されたワクチンよりも速いかもしれない。(Iv)卵ワクチンの代わりに我々の平板ワクチン候補ワクチンを用いることで,卵蛋白に対するアレルギー反応のリスクを解消する
2018年9月、私たちは衛生と公衆サービス部、生物医学高度研究と発展管理局(HHS BARDA)を通じてアメリカ政府と1,570万ドルの契約を完成し、契約に基づいて、私たちは私たちのH 1 N 1インフルエンザ候補ワクチンに対して第2段階の挑戦研究を行った。著者らはH 1インフルエンザに接種し、実験した健康ボランティアの中で、H 1インフルエンザ経口錠剤ワクチン候補がプラセボより39%減少した臨床疾患を発表した。市場をリードする4価インフルエンザワクチンFluzoneは約27%の臨床疾患を減少させた。われわれの候補錠剤ワクチンも良好な安全性を示し,プラセボとあまり変わらなかった
2018年10月、著者らは研究データを公表し、著者らの候補ワクチンは粘膜ホーミング受容体形質母細胞の著しい拡張を誘導し、約60%の活性化B細胞に達することを表明した。著者らはこれらの粘膜形質母細胞は保護性粘膜免疫反応の重要な指標であり、著者らの候補ワクチンの一つの独特な特徴でもあると信じている。このデータは,我々の候補ワクチンが粘膜免疫誘導(インフルエンザ,ノウォーカーウイルス,呼吸器合胞体ウイルスなどの粘膜感染に対する第一の防御線)を誘導することによって保護されており,ワクチン注射に対する重要な利点である可能性が示唆されている。
我々は、従来の季節性インフルエンザ候補ワクチンに加えて、Janssen汎用インフルエンザワクチン計画に対する独自の経口ワクチンプラットフォームを評価するために、2019年7月にヤンソンワクチンおよび予防会社(“Janssen”)と研究協力協定を締結した。プロトコルによると、Janssenの特定の独自抗原を含む非GMP経口候補ワクチンを生産し、臨床前挑戦モデルで製品を試験した。臨床前研究が完了し、Janssenに報告書を提出した
Vaxartは世界各国政府と協力し、要求に応じて大流行単価インフルエンザワクチンを作成し、緊急使用または貯蔵のために開発します。私たちはまた、臨床前の季節性および汎用型インフルエンザ候補ワクチンの開発を続けています。彼は言いました
| ● |
呼吸器合胞体ウイルスワクチンそれは.呼吸器合胞体ウイルスは主要な呼吸器病原体であり、幼児と老人に大きな疾病負担がある。 |
著者らの臨床前の綿マウス研究の積極的な結果に基づいて、著者らは著者らの独自の経口ワクチンプラットフォームはRSVの最適なワクチン送達システムになる潜在力があると信じ、注射ワクチンより顕著な優勢を提供する
同社は依然として監督管理機関、政府、非政府組織、その他の潜在的な戦略各方面と討論を行い、そのRSV計画を推進する最適な方法を決定している。彼は言いました
| ● |
ヒトパピローマウイルス治療用ワクチンそれは.著者らの最初の治療性経口ワクチン候補はHPV 16とHPV 18に対して、この2種類のウイルスは70%の子宮頸癌と癌前子宮頚の非典型的な増殖を招く。 |
子宮頸癌は全世界とアメリカ女性の第四によく見られる癌であり、全国子宮頸癌連盟のデータによると、アメリカでは毎年約1.3万の新しい確定診断例がある
私たちはすでに2種類の異なるHPV-16固形腫瘍モデルでHPV-16候補ワクチンをテストした。HPV 16候補ワクチンはT細胞反応を刺激し、活性化されたT細胞の腫瘍内への遷移を促進し、腫瘍細胞殺傷を招く。われわれのHPV−16候補ワクチンを接種したマウスは、確立された腫瘍体積が有意に減少することを示した
2018年10月、著者らはFDAにIND前会議要求を提出し、HPV 16およびHPV 18に対する最初の治療的候補ワクチンを要求し、その後、IND前報パッケージを提出した。我々は2019年1月にFDAからフィードバックを受け,IND申請の提出を支援し,臨床試験の開始を支援した
同社は依然として監督管理機関、政府、非政府組織とその他の潜在的な戦略各方面と討論を行い、そのHPV計画を推進する最適な方式を確定している
抗ウイルス薬
| ● |
合併を通じて、私たちは二つの印税収入製品を得た:RelenzaとInavir。2期臨床段階の抗ウイルス化合物も3種類得られ,独立した臨床開発を中止した。しかし,そのうちの1つであるVapendavirについて,我々は2021年7月にAltesa Biosciences,Inc.(以下Altesaと略す)と世界的独占ライセンス契約を締結し,Altesaがこのカプシドに結合した広域抗ウイルス薬の開発を許可し,商業化した。2022年5月、Altesaは臨床試験を開始する意向を発表した。 |
| ● |
レラシャとイナビルはインフルエンザ治療の抗ウイルス薬であり,それぞれグラクソ·スミスクライン社(GSK)と第一三共株式会社(Daiichi Sankyo)によって販売されている。私たちは瑞楽沙とイナビルの日本での純売上高から印税を稼いでいます。レラシャの最後の特許は2019年7月に満期になり、イナビルの最後の特許は2029年12月に満期になった。これらの抗ウイルス薬の販売は四半期によって異なり,インフルエンザウイルス活動は強い季節的周期を示すため,インフルエンザ季節の強度と持続時間によって,新冠肺炎は季節性インフルエンザやタミフルやXoflzaなどの他の抗ウイルス薬からの競争に影響を与え続ける可能性がある。 |
私たちのタブレットワクチンプラットフォームは
著者らの独自のVAASTプラットフォームに基づくワクチンは広範かつ持続的な免疫反応を産生することを目的としており、これは広範な伝染病に対応する上で重要な優勢を提供する可能性がある。
プラットフォームコンポーネント
我々のプラットフォーム技術はベクトルに基づく方法を採用しており,以下のコンポーネントからなる
| ● |
ウイルスであって、ワクチン抗原をコードするDNAを送達するためのベクターとして使用され、腸管免疫系を活性化するように選択されたアジュバントであるベクター。具体的には,抗原とアジュバントのDNAを小腸細胞に輸送し,そこでは抗原とアジュバントが共発現している非複製型アデノウイルス5型(Ad 5)を用いた。 |
| ● |
標的病原体に対する免疫反応を刺激するウイルスまたは細菌タンパク質であるタンパク質抗原。臨床候補ワクチンごとに異なる抗原を使用しています |
| ● |
ワクチンの免疫刺激特性を増強する物質であるアジュバント。腸管の免疫系を活性化するために選択されたToll様受容体3(TLR 3)アゴニストを使用した。 |
| ● |
Ad 5担体を小腸に輸送することを目的とした我々の固有腸溶解錠。 |
図2:VAASTプラットフォームです
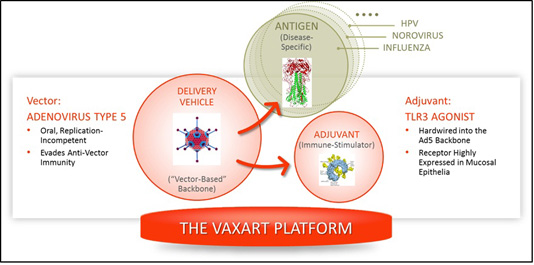
図2.担体−アジュバント−抗原標準化技術プラットフォーム
私たちのプラットフォーム担体送達システムと、担体によって発現される抗原およびアジュバントとの組み合わせ。
アデノウイルス5型ベクター
Ad 5は研究が広く特性の良い担体である。他の人が行った200件以上の臨床試験はAd 5を広く応用しており、私たちの候補プレートワクチンで同じアデノウイルスを使用することは、規制機関がそれを知っているので、規制リスクを低下させると信じている。
組換え抗原
我々のベクターはクローン空間を含み,その中に任意の組換え抗原をコードするDNAを挿入することができる。これまでに行われてきたワクチン計画では,対応する病原体を保護する能力を有する免疫系の重要な標的として知られている組換え抗原が選択されている。Ad 5ベクターアジュバント遺伝子カセットはモジュール化方法を採用することを可能にした。
アジュバント
著者らは補助剤として小セグメント二本鎖RNA(DsRNA)を用いて、候補錠剤ワクチンの免疫原性を増強した。DsRNAはTLR 3アゴニストであり,先天性免疫系によって望ましくないウイルス複製を行っているシグナルとして認識され,免疫反応をトリガして防御する。DsRNAは腸管で使用可能な数少ないシグナルの一つであり,天然の大型細菌貯蔵庫(“微生物群”)により細菌関連シグナルの使用が困難になっている。このアジュバントを選択したのは,経口投与時に非複製型アデノウイルスを補充でき,免疫系がシグナルを認識する経路が小腸に出現することが少ないためである。重要なのは,個々の成分として提供するのではなく,我々のアジュバントを細胞内に発現させ,局所反応を引き起こすことである。
腸溶性錠
錠剤は通常小分子を腸管に輸送するために用いられるが,我々が設計した錠剤はより大きなアデノウイルス粒子を輸送することができる。私たちは候補錠剤ワクチンの組成と配合に関する知的財産権を持っている。我々の錠剤生産には無菌充填や仕上げ加工,例えば注射剤を必要とせず,標準的な打錠装置を用いている。
私たちのタブレットワクチン候補はどのように働いているのか
私たちの錠剤は小腸にワクチンを送るために設計されている。これらの錠剤は胃の低pH環境中で完全に維持され、コアに含まれる有効成分を胃中の酸性環境の影響から保護する保護層で覆われている。このコーティングは小腸の中性pH環境中に溶解することを意図しており、最適な免疫反応を産生することを目標としている。コーティングが溶解すると錠剤が崩壊し,ワクチンは小腸に放出され,そこでは腸管内の粘膜細胞に到達して入ることができる。粘膜細胞に入ると,抗原タンパク質およびアジュバントは細胞によって発現または製造される。アジュバントは本質的に分子であり,常に抗原を産生する全く同じ腸管細胞で産生される。重要なことは,我々の方法で伝達される抗原の産生は,実際の病原体が粘膜に侵入した場合の抗原産生と同様である。また,複製能を持たないAd 5担体ワクチンを錠剤で腸管に直接輸送することにより,血液や筋組織免疫細胞の中和を回避できると考えられる。
図3:経口組換えワクチンプラットフォームです
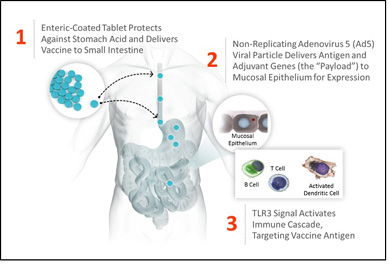
図3.1.腸溶性錠を経口投与する。錠剤コーティングは活性成分を胃酸分解から保護することができる。2.錠剤が小腸に到達すると、活性成分ウイルスベクターを放出し、次いでウイルスベクターを粘膜上皮細胞にトランスフェクトし、2つのペイロード(抗原およびアジュバント)の遺伝子を送達することができる。3.上皮細胞における抗原およびアジュバントの発現は、TLR 3シグナル伝達を低下させ、B細胞およびT細胞を活性化させる。
免疫細胞はタンパク質と接触し,タンパク質が免疫認識シグナルを引き起こすと免疫細胞が活性化される。これは最終的に免疫反応を引き起こし,記憶細胞や大量のキー抗原に結合する抗体を産生する。発現された抗原とそのプラットフォームのアジュバントは、他のワクチンのように、抗原特異的なB細胞やT細胞を誘導することができる。未成熟の樹状細胞(専門の免疫細胞)がAd 5担体から輸送された抗原とアジュバントを同時に発現する上皮細胞を吸収すると,誘導が開始されると考えられている。誘導後,樹状細胞は領域リンパ節に遊走し,そこでは循環する初期B細胞やT細胞と相互作用する。樹状細胞はその表面に抗原断片を提示してT細胞を刺激し、いくつかの抗原はリンパ節に流入してB細胞を刺激する。その特定の抗原が認識されると,小さなBやT細胞は遊走や拡大を停止する。そして,これらの細胞はクローンで増殖し,最終的に組織に循環する。B細胞は抗原を認識する抗体を分泌し,T細胞は表面に抗原を有する細胞を発現するか,提示細胞を殺すか,局所炎症反応を刺激する。B細胞およびT細胞が記憶細胞(その後の曝露時に保護抗原に迅速に反応する細胞)を形成したり,キー抗原に対する抗体を大量に産生して感染を阻止することができれば,成功したワクチン接種が発生する。
粘膜免疫とT細胞反応の意義
新しい免疫システムは、湿潤表面に対する粘膜抗体および病原体感染細胞を死滅させることができるキラーT細胞のような特殊な免疫エフェクターを作成することによって病原体を防御する。今日使用可能なワクチンの多くは,主に血液循環や系B細胞反応を刺激するために開発されている。しかし、ノウォーカーウイルスのような多くの感染があり、現在のところワクチンはありません。これらの病原体や他の病原体は,より強い血清抗体以外の免疫反応を必要とする可能性がある。これらの感染を引き起こす生体は,血液中の血清抗体による抗体免疫反応を大きく避けており,病原体は開口した粘膜細胞を通過することができるため,血液と直接接触することはない。あるいは,血清抗体は病原体に感染した細胞を透過できない。
現在利用可能な注射可能なワクチンは通常粘膜免疫反応を誘導できず、サブユニットワクチンは通常強力な殺傷T細胞免疫反応を誘導できないが、これはいくつかの困難な病原体に対して有効な免疫レベルを産生するために必要である。非粘膜経路によるワクチン接種は粘膜病原体の保護作用が悪いことが多く,主にこのようなワクチンが粘膜表面に遊走する記憶リンパ球を生じないためである。粘膜ワクチンは粘膜ホーミング記憶リンパ細胞を誘導できるが、現在完全な粘膜組換え経口ワクチンは商業用途に応用できると考えられている。弱毒生ワクチンは安全リスクをもたらす可能性があるが,殺された病原体や分子抗原は完全な粘膜に適用される場合には通常弱免疫原である。また,多くの粘膜感染の免疫保護機序はあまり知られていない。
われわれの技術の鍵となる利点の1つは,ワクチンを胃腸に輸送し,ワクチンを腸管の粘膜表面に直接進入させ,腸管の免疫系を活性化させることである。粘膜ワクチン接種は粘膜病原体が侵入した表面に免疫力を産生することによって、このような病原体の保護を強化すると考えられている。我々の錠剤ワクチン候補は,担体ベースの方法により粘膜免疫細胞に対して,より強力な細胞毒性T細胞反応および粘膜抗体反応を創出することを目的としており,特定の疾患により効果的な免疫を提供する可能性がある。強力な粘膜と系抗体反応に加えて,ヒト臨床試験では強力かつ多機能なT細胞反応が観察され,候補錠剤ワクチンがBとT細胞を有効に活性化することが証明された。
非複製Ad 5ベクターの経口投与は逆ベクター問題を迂回することを目的としている
注射したAd 5担体ワクチンは強い抗Ad 5反応を産生し,抗Ad 5中和抗体価は100倍に増加した。対照的に、著者らの経口Ad 5担体ワクチンは抗Ad 5免疫に関連する合併症を回避し、このプラットフォームが多種のワクチンに使用することを許可し、年間とワクチン接種を繰り返し強化することを目的としている。
われわれのH 1インフルエンザ1期と2期研究,および2つのノウォーカーウイルス1期研究では,抗媒介反応が検討されている。12名の被験者の第1回H 1インフルエンザ経口ワクチン候補研究では,免疫後中和抗体力価はAd 5まで有意に上昇しなかった。60人以上の被験者において、同じH 1インフルエンザ経口錠剤候補ワクチンを用いて第2段階挑戦研究を行った。この研究では,プラセボ群の1.1倍の中和抗体価と比較してAd 5の中和抗体価が2.2倍上昇していることが分かった。最後に、ノウォーカーウイルスワクチン候補研究の2段階、即ち研究#101と研究#102において、ワクチン抗病媒免疫反応の上昇をモニタリングした。候補ワクチン接種1回または2回後,中和抗Ad 5抗体価は有意に増加せず,高用量でも同様であった(下図参照)。
図4.免疫前後の抗病媒力価
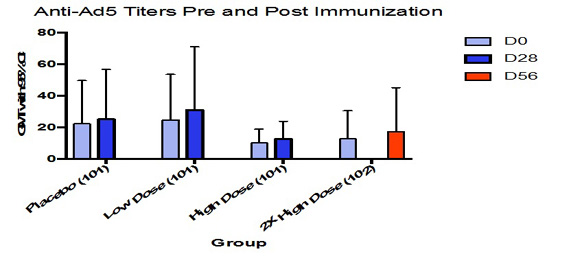
図4.単剤101の研究では、単剤後28日に抗病媒力価を測定した。両剤102回の研究では,これらは2回目の注射後28日に測定した。この2つの研究では,いずれの群もAd 5力価の有意な増加は認められなかった。
また,これまでのすべての研究において,選択された抗原に対する免疫反応は,受容者が以前に存在した抗Ad 5免疫状態とは無関係のようであった。われわれのAd 5ベクターH 1インフルエンザ経口錠剤候補ワクチンの研究では,あらかじめ存在するAd 5抗体価は,候補ワクチンがインフルエンザ中和抗体反応(赤血球凝集素抑制や微量中和試験)を誘導する能力に影響しなかった。著者らのAd 5ベクターノウォーカーGI.1経口錠剤候補ワクチンの2つの完成した第一段階研究において、既存の抗Ad 5抗体価を有する被験者において、候補ワクチンがノルウォーカーウイルス抗体価の上昇或いはノルウォーカーウイルス様粒子力価(“VLP”)(BT 50試験)を特異的に遮断する能力は低下しなかった。これらの結果を以下に示す.つまり,われわれのAd 5ベクターワクチンの経口接種効果は,受容者が以前に存在していた血清抗体状態の悪影響を受けないようである。
図5.抗病媒免疫は、ノロウイルス候補ワクチンがBT 50力価を誘導する能力に影響を与えない。
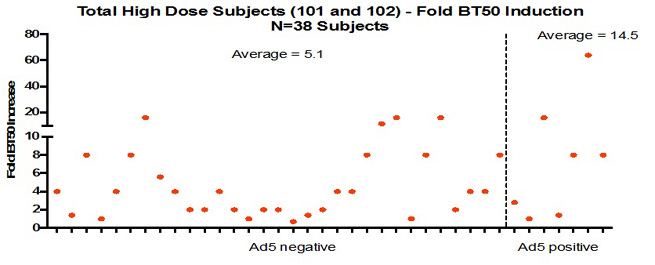
図5.高用量群の被験者を0日目にあらかじめ存在した抗Ad 5力価に基づいてグループ化した。力価100の人はAD 5陽性とされています
ノウォーカー·ウイルス計画は
市場の概要
ノルウォーカーウイルスはアメリカのすべての年齢層の人における急性胃腸炎による嘔吐と下痢の主要な原因である。平均毎年、ノルウォーカーウイルスは1900万から2100万例の急性胃腸炎を招き、そして10.9万人の入院と900人の死亡を招き、その大部分は児童と老人である。典型的な症状は脱水を含み、これは最もよく見られる合併症であり、嘔吐、下痢に腹部痙攣、および吐き気を伴う。アメリカでは、ノウォーカーウイルスワクチンは5歳以下の乳幼児、高齢者と高齢者、ならびに食品と観光業界のスタッフ、医療保健、児童看護と老年看護者、救急人員、軍隊、およびレジャーとビジネス旅行者のようなハイリスク群に有益であると信じている。ジョンホプキンス大学と疾病管理センターが2016年に発表した研究では、世界の年間ノルウォーカーウイルスの総経済負担は600億ドルと推定されている。そのうち340億ドルは米国を含む高所得国で発生している。2020年7月に“感染症誌”に発表された更新された衛生経済学研究では、毎年米国への経済影響は105億ドルと推定されている。2021年1月に“米国予防医学誌”に発表された記事では、ノルウォーカーウイルスワクチンが5歳以下の子供は毎年500ドル節約され、65歳以上の成人には年間75ドル節約されていると推定されている。現在は承認されていないワクチンや治療法はノーウォーカーウイルス感染を予防または治療している。
ノロウイルス候補ワクチンは
我々はVP 1に基づく2価経口錠剤候補ワクチンを開発しており,ノロウイルスGI.1ノウォーカー株とノロウイルスGIIシドニー株を標的とすることにより,ノロウイルスGIとノルウォーカーウイルスGIIの2つのヒトに影響を与えるノルウォーカーウイルスゲノムから人々を保護している。ノルウォーカーウイルスは小腸上皮細胞に感染する腸管病原体であることから,腸管局所でノウォーカーウイルス抗体を産生するワクチン,例えば我々の候補錠剤ワクチンは,腸管に直接輸送され,最適な感染予防効果が生じる可能性が考えられる。
臨床前結果
私たちはマウスとミンクでノウォーカーウイルス候補ワクチンについて多くの臨床前研究を行っています全体的に、VP 1タンパク質ワクチン候補マウスとの対面研究では、我々のノウォーカーウイルス候補ワクチンは、VP 1注射可能タンパク質ワクチン候補ワクチンに相当する血清抗体レベルおよびより高いレベルの粘膜抗体を産生する。
臨床試験
一価シート状ノウォーカーウイルスGI.1候補ワクチンの4つの第1段階研究と、2価シート状候補ワクチンの1つの1 b段階研究(GI.1およびGII.4ワクチンの併用)を完成させた。3つの研究は大流行前に完成し、その中の2つの研究は最近完成し、老年人口の用量間隔と反応を評価する。すべての研究において,主終点は安全性であり,副次的終点は免疫原性である。二価研究では、私たちはまた連合投与の潜在的な妨害を評価した。
私たちは2つの研究を行っており、もう1つの研究は2023年に始まる予定だ。
大流行前に完成した研究:101、102、103
101この第一段階研究はノウォーカーウイルス候補ワクチン(VXA-GI.1-NN)を評価することを目的としている。66人の健常成人をランダムに3つのグループに分け、そのうち23人の被験者が1×10の単一小用量を受けた10IIU、23人の被験者が1 x 10の単回大量投与を受けた11IU,20名の被験者は一致したプラセボ対照を受けた。
102.この第1段階開放ラベル用量最適化研究は、60人の被験者におけるノウォーカーウイルスGI.1一価ワクチン候補ワクチン(VXA-GI.1-NN)の使用状況を評価することを目的としており、低用量群の計画が異なる。最初の3つのグループ(各グループ15人)は1 x 10の低用量を使用10感染症単位(“IU”)。A群では0,7日目にVXA−GI.1−NN,B群では0,2,4日目に3剤,C群では0と28日目に2剤を受けた。第4のグループ、Dグループ(N=15)、2つの1 x 10の高用量を評価する11Iuは0日目と28日目に投与された。この研究の主な終点はすべての投与レジメンの安全性と耐性を評価することであり,副次的な終点はBtによる各群間の免疫原性の比較である50力価と抗体分泌細胞(ASC)数。
103この第一段階研究は二価ノウォーカーワクチン候補投与(VXA-GI.1-NNとVXA-GII-4-NS)を評価することを目的としている。80人の健常成人をランダムに4つの治療群のうちの1つに分けた。治療1群は、その後の治療群の開始前に登録された5人の対象からなる開放標識哨戒群である。この5名の歩哨被験者は単価GII.4候補ワクチンを接種し,安全性と免疫原性のモニタリングを受けた。治療1~4群のランダム化はそれぞれ1:1:2:1であり、被験者はランダムに一価GI.1、一価GII.2、二価GI.1/GII.4またはプラセボ群に分けられた。患者は5×10の完全な研究投与量を受けた10一価ワクチン治療アーム内のIuと1×1011Iuは二価治療腕あるいはプラセボ錠にある。
安全な結果
表1.101件の研究(N=66)で募集したAEs要約
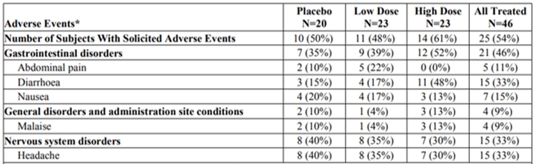
資料源:VXA-G 11-101臨床研究報告2017年8月10日
表2.102件の研究(N=60)で募集した有害事象の要約
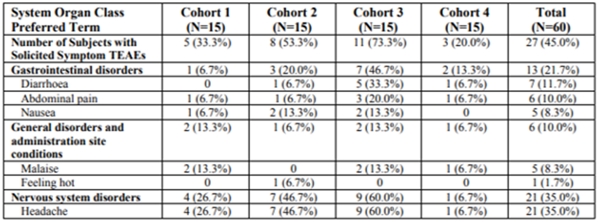
出典VXA-G 11-101臨床研究報告2019年1月9日
表3.103件の研究(N=80)で募集したAEs要約
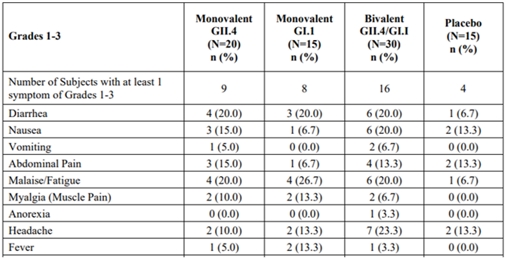
資料源:VXA-NVV-103臨床研究報告2020年9月23日
最初の3つの研究の安全まとめ。
この3つの第1段階研究では,171名の被験者がVaxartのノルウォーカーウイルス候補ワクチン治療を受けた。候補ワクチンは全体的に耐性が良好である。すべての用量の中で、最もよく見られる副作用は頭痛(27.5%)であり、これはプラセボ群頭痛患者の28.6%と類似している。アンケートの副作用(“副作用”)の多くは一過性であり,重症度は軽度または中等度であり,アンケートの副作用による中止はなかった。その中の2つの研究の中で、候補ワクチン治療群が報告した下痢の発生率(20.5%)はプラセボ群(11.4%)より高かった。しかし,102研究中の高用量群では,最高用量の候補ワクチン接種を2回受けた後も,1名の被験者(6.7%)のみが下痢を報告していた。全体的に、これらの結果は、安全性に影響を与える用量依存効果がないことを示している。これらの研究には、深刻な治療に関連する能動的有害事象は何も報告されておらず、関連する重篤な有害事象(SAE)、臨床的に注目すべき顕著な観察(NOCI)、または特に興味のある有害事象(AESIs)もない。
免疫原性結果−研究101
いくつかの異なる検出方法は、ノウォーカーウイルス候補ワクチンの免疫原性を評価するために使用される。これらの結果はキムらの雑誌に発表されましたJCI洞察2018年です。肝心な免疫学結果は以下のように記述される。
BT 50力価。主要な免疫学的終点はノロウイルスVLPと組織群血液抗原(HGBA)の相互作用を抗体が阻止する能力を評価することによって抗体力価を測定することである。この試験はBT 50(遮断力価50%抑制)試験と呼ばれる。Leb合成多糖を被包抗原としてBT 50力価を測定した。候補ワクチン受容者の力価はすべての時点で上昇している(図M 1)。Leb BT 50測定によれば、低用量群の14/23(61%)の対象および高用量群の18/23(78%)の対象が少なくとも2倍に増加した。プラセボ群では,1名の被験者の発症率が2倍以上上昇した。28日目には、低用量候補ワクチン群の幾何平均力価(GMT)は59.0であり、ベースラインの初期GMT 26.2より2.3倍向上した。高用量候補ワクチン群のGMTは98.5であり,ベースライン時の初期GMT 25.8の3.8倍であった。高用量群は28日目にプラセボ群より明らかに高かった(P=0.0003)。次の表(表4)に完全な結果を示す.
Leb BT 50検出GMT
表4.101,Leb BT 50で測定した最小二乗幾何平均力価(LSGMT)を検討した。
| HBGA |
レブ |
|||
| 集団化する |
D 0 LSGMT (95 CI) |
D 28 LSGMT (95 CI) |
LSGMR |
P値* |
| ロー |
26.2 (16.6-41.2) |
59.0 (33.0-105.4) |
2.3 |
0.0459 |
| 高 |
25.8 (18.3-36.2) |
98.5 (64.4-150.7) |
3.8 |
0.0003 |
| プラセボ |
24.6 (15.3-39.3) |
27.4 (17.0-44.2) |
1.1 |
参考までに |
| 全局的意義 |
0.0017 |
|||
*Mann-Whitneyとプラセボの意味;Kruskal-Wallis検定の全体的な意味
抗体分泌細胞(ASC)。ASC法を用いてノロウイルスワクチン候補ワクチンが末梢血中のノロウイルス特異的B細胞を誘導する能力を測定した。この検出は,基本的に免疫後に出現し末梢血中のノウォーカーウイルスを認識するB細胞の数を計算した。免疫前血液中循環の数は低いため,ワクチン特異的効果を有意に評価する方法である。低用量群では23名中16名(70%)が反応したが,高用量群では23名中19名(83%)が7日目にIgAとIg G ASCsに同時に反応した(図M 2)。背景ASCsは0日目に一般的に無視できる。大量の候補ワクチン治療群に対して、平均1 x 10当たり561個のIgA ASCと278個のIg G ASCsがある6低用量候補ワクチン治療群は平均1×10日ごとに372個のIgA ASCsと107個のIg G ASCsがあった67日目にPBMCを発見した。プラセボ群は応答者がなく、平均1 x 10あたり3.3個のIgA ASCs点と2.2個のIg G ASCs点があった6治療群とプラセボ群は7日目に免疫グロブリンGまたは免疫グロブリンA-アスパラギン酸アミノトランスフェラーゼ反応を誘導する能力に有意差があった(P
糞便中のIgA。直接糞便サンプルを観察することによって、ノロウイルスVP 1特異性粘膜IgAを探索した。これらの試料中のIgA含有量の変化が大きいため,総IgAも測定し,試料ごとのVP 1特異的IgA/総IgA間の比率を測定した。IgAレベルが検出下限を下回った試料は分析から除外した。特異的IgA/総IgAの比率の増加は,ベースラインと28日目(およびベースラインと糞便IgAの180日目)の間で測定された。高用量群では,28日目に19検体中9検体(47%)のIgA反応が4倍以上増加し,180日目には21検体中9検体(43%)であった(図M 3)。特異性IgA/総IgA比は平均17.2倍と9.7倍増加した。これらの結果はプラセボ群より有意に高く,28日目と180 d,2/18(11%)および0/16(0%)で4倍以上の増加が認められ(それぞれP=0.029およびP=0.0049),平均1.8%および1.0%増加した(図M 3)。低用量群の反応は高用量群と同様であり、28日目および180日目には、それぞれ7/20(35%)および5/16(31%)の反応が4倍以上増加した。28日目には応答者数はプラセボより高い傾向にあったが,180日目には統計学的有意差が認められた(P=0.13と0.043)。低用量群は28日目に36.2倍,180日目に5.6倍に増加した(図M 3)。
図はM 1である.幾何学的平均力価と時間です
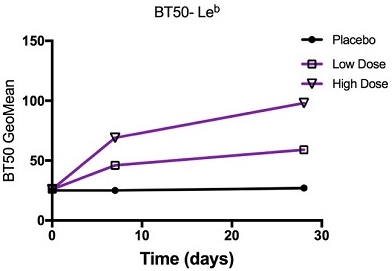
図M 1.Geomean血清BT 50力価は時間の経過とともにLebであった。
図はM 2である.免疫後7日目のASC力価。
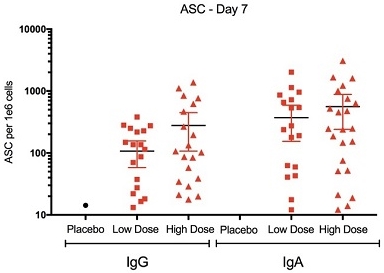
図M 2.7日目にノロウイルスVLPの免疫グロブリンと免疫グロブリンAに対するAASCカウントを示す。この測定方法は免疫後末梢血中の抗原特異性B細胞を測定する。
図はM 3である.ノロウイルス免疫後の糞便特異性IgA反応のフォールディング誘導。
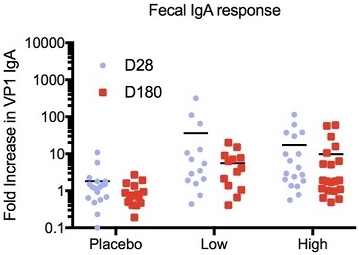
図M 3。候補ワクチンの糞便反応に対して,各被験者(群と各時点別)の特異的IgA/総IgA倍数が増加する様子をプロットした。平均上昇幅は黒い条である.
免疫学的結果102件の研究は
BT 50力価。この研究の目的は,計画と用量による免疫反応の能力を比較し,特にBT 50力価を評価することである。BT 50力価は、複数の用量が投与される場合、複数の時間点で評価される。高用量群では,15名中12名が1回目の注射後にBT 50力価が1倍以上増加し,15名中14名(92%)が2回目の注射後にBT 50力価が1倍以上増加した。GMT力価は当日の21.3から28日目の85.1に上昇し,GMFRは3.8であった。56日目のGMT測定では75.8であり,GMFRはベースラインより3.6高かった。低用量ワクチンを投与した他の群の応答率は低かった。これは統計学的に有意ではなかったにもかかわらず,B群と比較してA群とC群の方が力価上昇幅が大きかった。ANCOVAモデルを用いてGMFR増加の統計学的意義を決定した。ANCOVAモデルの最小二乗平均値(LSM)を指数演算することにより、対数変換後のベースライン力価またはベースライン力価の対数変換後の変化を従属変数、行列を要因、ベースライン対数力価を共変数とする最小二乗幾何平均フォールディング上昇幅(LSGMFR)を算出した。異なるグループ間の糸球体濾過率の増加は統計学的有意差があり(LSGMFR=0.0008)、P=0.0008、0.1224、0.0004と であった
102の研究ですBT 50力価、Leb
表5.研究102、Leb BT 50によって測定された幾何平均力価(GMT)。
| 集団化する |
説明する |
グリニッジ標準時にします |
D 28(またはD 36) |
GMFR |
グリニッジ標準時D 56 |
GMFR D 56 |
| A |
低、2 X、7日間隔 |
32.2 |
64.5 |
2.0 |
66.0 |
2.0 |
| B |
ロー、3 X、2日間隔 |
31.5 |
51.2 |
1.6 |
42.5 |
1.4 |
| C |
低、2 X、28日間隔 |
29.4 |
66.0 |
2.2 |
64.5 |
2.2 |
| D |
高さ2 X 28日ぶり |
21.3 |
85.1 |
3.8 |
75.8 |
3.6 |
エーエスエスエーです。異なる群間のASC応答者を比較することにより,追加的な免疫学的分析を行った。高用量群15名中14名が候補ワクチンに反応し,平均1 x 10人中698人がIgA ASCカウントに反応した6細胞です。2回目の投与後,1回目に反応しなかった被験者のASCカウントは有意に増加したため,全15名(100%)が2回の注射後にASC反応を引き起こすことができた。典型的な場合と同様に,1回目の免疫後のASCカウントが高い被験者は2回目の免疫後の応答が低かった発送する投与量。投与と分析は異なる中間時点で行われるため,全体応答率を検査することで低用量群を比較した。A群の全体応答率が最も高く,14名中12名(86%)が1回または2回の用量後に有意なASC反応を誘導できた。B群の応答率はやや低く,数人の対象のみが第1剤ワクチン後に反応したが,より多くの対象が追加のワクチン投与後に反応した。C群の反応は全群の中で最も変化が大きかった。平均1 x 10個の広告点数は839個6細胞は最初の注射後であったが,これはいくつかの被験者が極めて高いスポット数を有する結果であった(3人の被験者は1 x 10あたり1500個以上であった6)は、全く反応しない被験者が多く混合されている。
免疫原性結果–研究103
BT 50力価。一価GI.1と二価GI.1の血清HBGA遮断抗体価は29日目のBT 50が1日目より有意に増加した。一価GII.4と二価GII.4/GI.1の29日では,血清GII.4 HBGA遮断抗体のGMTが初日より有意に増加した。BT 50などの血清測定では,力価は2倍から3倍に増加し,血清転換率は50%に達した。一価GI.1群と二価GI.2 4/GI.1群の血清GI.1 HBGA遮断抗体BT 50のGMTは有意差がなかった。単価GII.4群と二価GII.4/GI.1群の間では,血清GII.4 BT 50 GMTに有意差はなかった。
抗体分泌細胞(ASC)。ASC法を用いて候補ワクチンが末梢血中のノロウイルス特異的B細胞を誘導する能力を測定した。この検出は,基本的に免疫後に出現し末梢血中のノウォーカーウイルスを認識するB細胞の数を計算した。免疫前血液中循環の数は低いため,ワクチン特異的効果を有意に評価する方法である。
初日に、各治療群のGI.1 IgA ASC平均計数は類似しており、循環中のノウォーカーウイルス特異的B細胞の背景レベルを代表した。しかし,8日目には単価GI.1群のGI.1 IgA ASC平均カウントが統計学的に有意に増加した(p
1日目には,各治療群間のGII.4 IgA ASC平均計数は類似していたが,8日目には二価GII.4/GI.1群のGII.4 IgA ASC平均数は統計学的に有意に増加した(p
1日目には各治療群のAsc GI.1免疫グロブリン平均カウントは類似していたが,8日目には単価GII.4群(p=0.0002),一価GI.1群(p=0.0019),二価GII.4/GI.1群(p )のAsc GI.1免疫グロブリン平均数が有意に増加した
1日目には,各治療群のAsc GII.4免疫グロブリンの平均カウントは類似していたが,8日目には二価GII.4/GI.1群のASC GII.4免疫グロブリンの平均カウントが有意に増加した(p
図M 4.用量成分別の8日目のASC GI.1およびGIII.4免疫グロブリンAおよび免疫グロブリン反応グラフ(PP集団)。
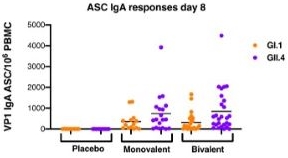
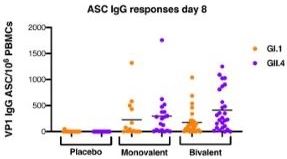
図M 4.研究8日目(免疫後7日)に異なるグループの末梢血中のIgAとIg G ASC計数を評価した。各被験者はGII.4(紫)とGI.1(オレンジ)を評価し,このグループの平均回答を1つのドットでプロットし,中実黒線で表した。 これらの結果は,有意なASC反応が認められなかったプラセボ群と比較して,GI.1とGII.4に対するIgAとIg G反応を誘導できることを示している。 また,単価群と二価群の平均反応は類似しており,2種類のワクチン株を一緒に接種した場合,妨害が乏しいことが示唆された。
最近完成したノウォーカーウイルス実験は
103研究のブースターそれは.この試験は、年間強化ノーウォーカーウイルス候補ワクチンの安全性と免疫原性を評価することを目的としており、方法は、16ヶ月後にG 1.1候補ワクチンまたは二価G 1.1/G 2.4候補ワクチンを受けるボランティアサブセットに追加のG 1.1ワクチン候補用量を与えることである。二価一期試験の活性部分は2019年に完了し、バックライン結果は2019年第3四半期に報告された。2021年初め、ノウォーカーウイルス候補ワクチンの安全性および免疫原性をさらに評価するために、一部の対象のための強化用量が開始された。2021年7月に発表された結果では,以前に接種した被験者のG 1.1ノロウイルス候補ワクチンの免疫反応の増強に成功したと報告されており,これらの反応にはIgA抗体分泌細胞,IgGおよびIgA血清抗体反応が含まれている。投与量を強化した後に請求される副作用は最初の投与量に相当し、頭痛は最もよく見られる症状(25%)である。免疫後AEsの多くは軽度であり、少数は中度であり、深刻な能動或いは能動AEsがない。SAE,AESI,NOCIの報告はない。
104.ノロウイルス1期加齢試験それは.55~80歳の年齢のボランティア66人がVXA-G 1.1-NN錠剤を経口投与し、その安全性および免疫原性を決定するために29日ごとに経口投与した。プラセボ対照研究では、4つの異なるキューが登録され、3つのワクチンキュー:低、中、高(1 X 10)10Iu,3 x 1010Iuと1 x 1011Iu)。毎回免疫後7日に能動症状(SS)を記録し、接種後28日に能動不良事件を記録した。MSD法を用いてVP 1特異的血清中の免疫グロブリンIg GとIg Aを測定し、BT 50法を用いてその機能活性を測定し、系統の体液免疫原性を決定した。抗体分泌細胞(ASC)法を用いて細胞免疫機能を測定した。最後に,被験者の唾液と鼻腔試料中のVP 1特異的な粘膜IgA反応が定量化され,総IgAに分類された。
VXA−G 1.1−NNはすべての群で安全かつ耐性が良好であり,候補ワクチンやプラセボキューに出現する有害事象はほとんど報告されていない。要求される副作用の多くは軽度または中等度であり,どの用量レベルでもワクチンに関連する重篤な副作用はない。活動期間内に名目上の未請求の副作用が記録されており,いずれも無関係であり,最高用量を受けた人では報告されていない。現在1年間の安全追跡が行われている。(表6).
表6.NVV-104の2つの用量のキューおよび年齢別の募集AEs要約(予備データ)
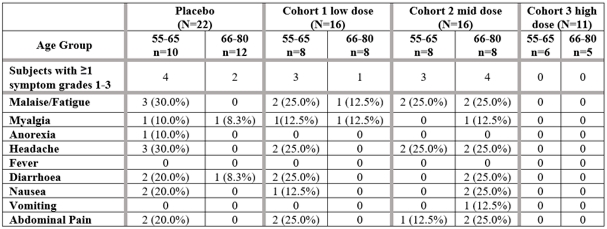
出典:VXA-NVV-104 TLF予備テストT 14.3.1.1.1、実行日11月22日
免疫学的結果
ワクチン接種を受けた列では、血清BT 50力価、VP 1特異的Ig GおよびVP 1特異的Ig Aはいずれも用量依存的に増加し、接種後210日目にベースラインレベル以上に維持され(図M 5)、高用量群の反応は平均20-50倍増加した。すべての候補ワクチン群は強いIgA-ASC反応を産生し、候補ワクチンの投与量が最も高い者は最も強い反応を産生した(図M 6)。免疫後29日に唾液や鼻腔分泌物でVP 1特異的IgAの上昇が検出され(図M 7),この腸管に注射されたNVワクチンが複数の組織で粘膜クロストークを誘導していることが示唆された。
図M 5.経口ワクチンはGI.1 VP 1に対してBT 50活性を有する強健で持続的な循環抗体を誘導する。
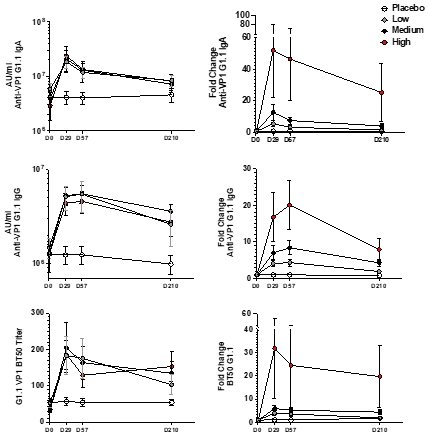
図M 5(A)MSD法を用いて血清抗GI.1 VP 1のIgA反応を測定した。Au/mlはD 0,D 29,D 57およびD 210(左)で測定した。ベースラインレベルと比較してIgA AU/ml応答倍数が変化した(右)(B)血清中の抗GI.1 VP 1の抗体をMSD法で測定した。Au/mlはD 0,D 29,D 57およびD 210(左)で測定した。ベースラインレベルと比較した免疫グロブリンAU/ml反応の倍数変化(右)(C)D 0,D 29,D 57とD 210(左)では,BT 50力価抗GI.1 VP 1であった。ベースライン力価と比較して、血清BT 50反応は倍増した(右)。すべてのデータは平均値+/−走査電子顕微鏡で表された
図M 6:経口免疫誘導GI.1 VP 1特異的循環IgA抗体分泌細胞
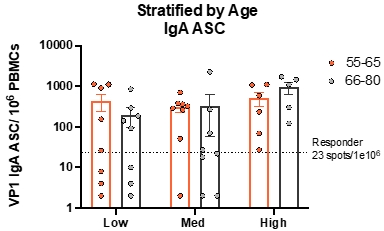
図M 6.免疫後8日目にGI.1 VP 1特異的IgA AscをELISPotを用いて定量測定した。各円は各被験者の平均IgA ASCスポットカウントを表して10に正規化されている6PBMCsは,用量列と年齢別に階層化されている。オレンジ色のバー55-65年、グレーのバー66-80年
図M 7:上気道(および口腔)における小腸送達による遠位粘膜IgAの標的化
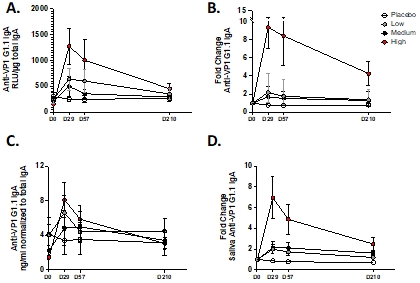
図M 7.(A)MSD免疫後D 0、D 29、D 57とD 210の鼻様において、GI.1 VP 1特異性IgAと総IgAは正常化した。各円はRLU/の平均値+/-SEMを表すµGは列別(黒い開放円プラセボ、灰色円低、黒丸中、およびオレンジ円高)(B)GI.1 VP 1特異的鼻腔IgA折り畳みベースラインレベル変化,+/-走査電子顕微鏡(C)免疫後D 0、D 29、D 57、D 210は酵素結合免疫吸着試験を用いてGI.1 VP 1特異性唾液IgAと総IgAの関係を測定した。各円は平均+/-走査型電子顕微鏡を表しますµG/ml G 1.1特異的IgA(D)GI.1 VP 1特異的唾液IgAベースラインレベル変化,+/-走査電子顕微鏡。
105を研究する。過給期間研究。
この研究の主な目的は,VXA−G 1.1−NNの18~55歳の健常成人における反復投与1日目と異なる強化計画(初期投与後4,8または12週)の免疫原性を評価することである。いくつかのワクチン形態は、最適な用量の増加と強化用量との間の時間が観察される反応を増加させることができることを発見している。これは18歳から55歳までの健康な成人被験者において行われたオープンな研究であり,主要用量と強化用量の間でより長時間待つことがメリットであるかどうかを決定することを目的としている。この研究では30名の被験者を募集し,3つの治療キューのうち1つに分け,10名の被験者が1人当たり2剤1 x 10を受けた10感染単位(IU)は1日目と4週目に10名の被験者が1人当たり2剤1 x 10を受けた101日目と8週目にIIUを行い,10名の被験者が1人当たり2剤1 x 10を受けた101日目と12週目に始まります
VXA−G 1.1−NNはすべての投与レジメンにおいて安全と耐性が良好であった。すべての要求の副作用は軽度または中等度であった。ワクチンに関する能動的副作用は報告されていない(表7)。
表7.NVV-105要求AEの概要
資料源:2022年12月23日VXA-NVV-105臨床研究報告
免疫原性の結果:
VXA−G 1.1−NNはワクチン特異的免疫応答(GI.1特異的血清Ig GおよびIg A、GI.1特異的血清遮断抗体を含む[BT 50]および、1回目および2回目の強化接種後、すべてのキューにおけるGI.1 VP 1特異的Ig GおよびIg A ASC反応)。第2剤VXA−G 1.1−NN投与7日後には,4週強化群よりも8週と12週強化群のGI.1 VP 1特異的IgA ASCの平均値が高い傾向にあった。2回目の投与28日後、異なる強化免疫レジメンのすべての列(または任意の列間)のGI.1 VP 1特異的血清免疫グロブリンの幾何平均濃度および幾何平均倍増(GMFR)は統計学的に有意差はなかったが、4週間強化行列と比較して8週および12週の強化行列のGMFRは上昇傾向にあった。2回目の投与後28日にVP 1血清IgAが3倍(P総=0.19)または4倍(P総=0.72)増加した被験者では,強化群別に有意差が認められたが(フィッシャー確実検定),4週群と比較して8週および12週群で応答者数が有意に増加した。2回目の免疫後28日に,すべての3つの行列において,GI.1 VP 1特異的血清IgAの2倍上昇を示した被験者は,統計学的有意差があった(P_TOUAL=0.04)。8週の強化キュー中の被験者は、4週間の強化キュー中の被験者と比較して、GI.1 VP 1特異的血清レベルが2倍有意に上昇した(P
試験は2023年に完了する予定だ
ノロウイルスGI.1株2期挑戦研究(VXA-NVV-201)我々の単価GI.1ノウォーカーウイルス候補ワクチンの若年成人参加者における第2段階挑戦研究は2022年第1四半期に開始され,現在進行中である。この研究はランダム、二重盲検とプラセボ対照であり、GI.1ウイルス攻撃後のGI.1ノウォーカーウイルス候補ワクチンの安全性と臨床治療効果を評価する。私たちは100~150人の参加者に挑戦し、2023年第3四半期にこの研究の裏線データを報告する予定だ。
第2段階用量測距試験(VXA−NVV−202)それは.この試験は、より大規模な第3段階試験を行う用量の確認を可能にするために、18歳から80歳までの成人において二価候補ワクチンの安全性および免疫原性を評価する。この第2段階臨床試験は,米国の3地点で約135名の健常成人を募集する予定である。最初の10人の被験体は、開放ラベル大量ワクチン接種を受け、残りの被験体は、無作為に高用量または低用量ワクチン(各群50人)またはプラセボ(N=25人)を受ける。2つの異なる用量レベル、各ワクチン5 e 10国際単位、各ワクチン1 e 11および2 e 11国際単位の総二価用量を探索する。この第2段階臨床試験のトップデータは、2023年に発表される予定で、この研究は、第3段階で使用しようとしている用量を選択することを目的としている
承認を得る道それは.用量選択後、後続の第2段階研究は約500人の被験者を募集し、これらの研究は、18歳以上の成人に対する許可された第3段階の重要な治療効果研究の範囲および設計を支援するために、選択された用量で十分な安全なデータを生成し、FDAとの第2段階会議を終了することができると予想される。
追加年齢群と亜群
小児科人口それは.我々の現在の錠剤ワクチン配合は固体剤形で設計されており,腸溶性錠を用いて腸管に輸送されており,成人と8歳以上の小児の最適なワクチン送達システムと考えられている。6カ月から7歳までの小児では,担体ワクチンをそのまま腸管に輸送できるマイクロ製剤の開発を計画している。我々のノウォーカーウイルスワクチンの小児科群における開発は、段階的に低年齢化する方法で行われる(すなわち、8歳から5歳、4歳から2歳、2歳から6ケ月)。
108を研究する。著者らは現在ビルとメリンダ·ゲイツ基金会と協力して、76名の健康な産後哺乳期女性ボランティアの中でノウォーカーウイルス二価ワクチン候補第一段階の研究を行い、著者らが構築したノウォーカーウイルスワクチンが母乳ノウォーカーウイルス特異性IgAに与える影響及び母乳授乳後の乳児糞便サンプル中の潜在的存在を決定する。この研究は、ランダム、二重盲検、およびプラセボ対照となり、プラセボキューおよび2つのワクチンキューの安全性、耐性および免疫原性を評価する:中用量(1×10)11高用量(2×10)11(Iu)。臨床前部分で指摘されているように,粘膜ワクチンは抗体産生に優れている可能性があり,これらの抗体はウイルスの伝播を遮断し,局所伝播を減少させることができる。さらなる研究も必要であるが、これは、幼児にワクチンを接種し、ノーウォーカーウイルス感染から家族全体を保護するための別の方法を提供する可能性がある。この実験は2023年に開始される予定だ。
表8.108の研究の研究設計
| VXA-NVV-108:ノロウイルス二価ワクチン単剤、用量範囲の研究 |
|||||
| 集団化する |
ワクチン |
投与量 |
総用量 |
線量数 |
課題数 |
| 中程度の用量 |
VXA-GII.4-NS+VXA-G 1.1-NN |
5×1010各菌株の単位単位は |
1×10112つの単位 |
1 |
30 |
| 高用量 |
VXA-GII.4-NS+VXA-G 1.1-NN |
1×1011各菌株の単位単位は |
2×10112つの単位 |
1 |
30 |
| プラセボ |
プラセボ |
北米.北米 |
北米.北米 |
1 |
16 |
Iu=感染単位
私たちの新冠肺炎計画は
市場の概要
各国政府はすでに新冠肺炎ワクチンを大規模に購入し、各国国内で大規模に配布している。また、非政府組織(“NGO”)や世界保健機関(World Health Organization)は、国内製造や/または資源が限られていない国を代表して購入を行うCovaxなどの調達組織を設立し、事前購入合意を達成している。2023年、このような中央政府調達は世界の大多数の国で継続される可能性が最も高く、米国は例外であり、2023年以降に民間市場部門に新冠肺炎ワクチンの調達と流通を管理させる意向を示している。第一弾の新冠肺炎ワクチンは原始SARS-CoV-2毒株に対する第三段階試験において有効であるが、これらのワクチンの貯蔵と処理要求のため、配布と管理問題は予想よりはるかに遅い。
第一世代ワクチンは新しく出現したSARS-CoV-2毒株に対して異なる程度の効力を有するようである。従来の菌株適応の選択的圧力は,ワクチン接種や感染の公衆レベルが非常に低い環境で行われていた。ワクチン接種人口の増加に伴い、菌株の変化の速度が速くなる可能性がある。
新冠肺炎ワクチンが公衆に提供される前に、ワクチン接種に大きな躊躇があることが報告されている;いくつかの国では、人口の50%以上が新冠肺炎ワクチンを接種しないことを示している。より多くの人がワクチンを接種するにつれて深刻な有害事象が発生せず,このワクチンへの躊躇が弱まっているようであり,最終的にはインフルエンザワクチンなどの他のワクチンのワクチン遅延率のようになる可能性がある。
SARS−CoV−2流行株の変異性
SARS-CoV-2はRNAウイルスであり、時間の経過とともに自然に遺伝子変異を進化させ、多くのウイルス変異体を産生する。2019年12月以来、世界の協調努力はSARS-CoV-2変種の出現を追跡し、複数の国で発生する頻繁な遺伝子突然変異を確定した。ウイルス変異株は世界の多くの地区で迅速に出現し、いくつかのゲノム変化を有し、アミノ酸配列と蛋白質構造の著しい変化を招く。2020年後半には,イギリス起源のAlpha(B.1.1.7),南アフリカ起源のBeta(B.1.351),ブラジル起源のGamma(P.1)の3つの異なるSARS−CoV−2変種が急速に伝播した。2021年には,Delta(B.1.617.2)とオミック(B.1.1.529)と呼ばれる2つの変種注目(VOC)が出現し,前者はインド,後者はボツワナに起源した。すべての変種は外層S蛋白のキー領域で変化し,ウイルスはこの蛋白を利用してACE 2という受容体を介してヒト細胞に感染する。これらの変異体中のS蛋白受容体結合部分の構造変化はすでにウイルス伝播を増強し、より高いウイルス負荷とより悪い疾病結果を招く可能性が証明された。最近のデータでは,変異体はワクチンの血清抗体(http://www.nature.com.cn/s 41586.021−04387−1_Reference.pdf)を迂回する能力が強いことが示されている。現在,ワクチン抗原としてS蛋白を用いて抗体反応を開始し,SARS−CoV−2ウイルスの細胞への進入を阻止するワクチン戦略の開発やFDAの承認がなされている。最初のS蛋白からなるワクチン配合は最初の武漢毒株から由来し、最近のいくつかのワクチンはオミック/武漢毒株の二価組み合わせで作られている。現在の注射ワクチンは最新の/現在主導的な流行のSARS-CoV-2毒株に追いつくことは困難である。したがって、これらの注射可能なワクチンは交差保護抗体反応を引き起こさず、新しいウイルス変異体が受容体に結合して細胞に入ることを阻止する可能性がある。最近のオミック爆発により、3種類の遺伝子ワクチンは深刻な疾病と入院に対して一定の保護作用があることを表明したが、感染に対する保護作用はわずか37%(http://www.medrxiv.org/content/10.1101/221.12.30.21268565 v 1.full.pdf)であった。これらの結果は,新しいS蛋白変異株の出現に伴い,現在のワクチン接種方法は,新たなSARS−CoV−2変異株から十分な保護を提供するために更新や修正が必要であることを示している。あるいは,交差反応(汎コロナウイルスや汎βコロナウイルス)抗体やT細胞を産生する新しい方法が必要である。
私たちの新冠肺炎候補ワクチンは
過去数年間、我々は多くの労力をかけて新冠肺炎候補ワクチンを開発した。生成されたデータは以下のページで詳しく紹介され、我々の新冠肺炎候補ワクチンが臨床試験で交差反応を誘導していることを示しており、将来のコロナウイルスの大流行に対応するワクチンの開発に重要な意義を持っている。
著者らの最初の新冠肺炎候補ワクチン(Rad-S-N、Vaxart臨床候補ワクチンVXA-CoV 2-1と呼ばれる)はSARS-CoV-2ウイルスとは異なる2つの遺伝子、刺突起蛋白と核蛋白(N)を発現する。N蛋白はコロナウイルスファミリーの中でより保守的であり、新たに出現したSARS-CoV-2毒株S蛋白に実質的な突然変異が発生しても、著者らの候補ワクチンに含まれるのは1つのT細胞標的を作るためである。マウスの臨床前の結果から、マウスにおいて抗体およびT細胞反応を誘導する能力と、SARS-CoV-2に対する粘膜IgAを肺で誘導する能力とを有するため、ワクチンがこれらの株からSを認識する保護免疫反応を産生する能力を低下させる。我々の候補ワクチンは2020年春に選択された。
著者らの第二の候補ワクチン(Rad-S、Vaxart臨床候補VXA-CoV 2-1.1-Sと呼ばれる)はSARS-CoV-2武漢株のS蛋白のみを発現する。この候補ワクチンは,他の候補ワクチンと比較して,非ヒト霊長類動物(NHP)研究においてより良い抗体免疫反応が得られ,ハムスター伝播実験で伝播を抑制することができる。
著者らはまた他の新冠肺炎候補ワクチンの臨床前仕事を開始し、潜在的なパンベルタコロナウイルスワクチンの開発を目標とした。
臨床前結果
私たちの最初の新冠肺炎候補ワクチンの有効性を評価するために、私たちはロフレス生物医学会社(ニューメキシコ州アルバカーキ)でハムスター挑戦研究を行った。ハムスターは鼻腔経路で感染し、体重減少、呼吸困難、ふさふさなどの臨床症状を得ることができるので、SARS-CoV-2に感染する良いモデルである。また、ヒトと類似した肺問題も出現する。SARS-CoV-2に感染したハムスターに対する顕微コンピュータ断層撮影により、深刻な肺損傷はSARS-CoV-2感染のヒト肺と同じ特徴を有し、深刻な多小葉磨ガラス様混濁と肺実変領域を含むことを示した。彼は言いました
われわれの背線の結果,VXA−CoV 2−1(Rad−S−N)を1 e 9 IUの2回経口投与することでハムスターを感染関連体重減少から実質的に保護でき(図N 1 a),肺CoV−2を介した損傷に関連する肺体重増加を防止し(図N 1 b),攻撃後5日間の肺における高ウイルス力価(図N 1 b)を有意に防止した(図N 1 b)。VXA-CoV 2-1ワクチンの経口投与は肺のウイルス力価を4~5つの対数まで低下させることができる(図N 1 C)。未治療動物とVXA−CoV 2−1経口免疫動物の肺組織病理学的比較は有意差を示した。すべての未治療動物の多くは中等度(8匹中6匹)から有意(8匹中2匹)の混合細胞炎症であり,中心細胞領域は軽度(8匹中1匹)~中等度(8匹中2%)の上皮肥大/増殖であり,軽微(8匹中5頭)から軽度(8匹中3頭)の肺胞出血が多く,VXA-CoV 2-1ワクチンを接種したすべての動物は、軽微な混合細胞炎症を持っている。これらの動物は、中心細胞領域の上皮肥大/増殖、肺胞出血または気管支上皮肥大/増殖を示す証拠はない。*鼻腔接種ワクチン(I.N.)VXA−CoV 2−1の注射も経口と同様の保護レベルを生じた
ワクチンは動物血清中で抗体反応を誘導し、S 1に結合する免疫グロブリンG抗体もあれば、経口または鼻腔免疫後に測定した中和抗体(図2)もある。代替中和試験(Genscript)を用いて中和抗体価を測定した。研究4週目には、免疫強化後に動物の免疫グロブリンG抗体価が上昇した。
第二の臨床候補は、2021年の非ヒト霊長類動物とハムスター研究で探索された。ビルとメリンダ·ゲイツ財団が援助し、デューク大学が管理している研究では、ハムスターはワクチン突破後のエアロゾル伝播をシミュレートするために使用されている。ワクチンを完全に接種した人でも最新の懸念される変種に感染し、他の人に感染することができ、SARS-CoV-2伝播に影響を与える戦略が有益である可能性がある。米国指数ハムスターはr-Ad-Sを2剤接種した(VXA-CoV 2-1.1-S)。粘膜刺激の対照として鼻腔(“IN”)r−Ad−S,蛋白質対照として筋内刺突起蛋白(IM S)を用い,シミュレーション対照としてPBSを経口投与した。インデックス動物はその後、IN分娩を通じて高力価SARS-CoV-2を有する動物に感染し、ワクチン接種後の突破的な感染を複製する。ウイルス挑戦後の日には,インデックスハムスターをワクチン幼稚ハムスターの上流に置き,エアロゾル運動を可能にするが直接接触や原虫伝播ができない部屋に置いた。重要なのは、指数免疫動物の鼻スワブに大量のウイルスRNAが存在するにもかかわらず、経口とIN r-Ad-Sワクチン接種がSARS-CoV-2のエアロゾル伝播を著しく減少または遅延させ(図N 3 A)、ワクチンを接種していない幼稚動物の肺炎症や体重減少などの疾患指標を減少させた(図N 3 E-G)。これらのデータは、r-Ad-Sワクチンの経口投与により臨床前モデルにおける疾患減少やSARS-CoV-2伝播が減少し、未接種ワクチン/保護されていない動物であっても同様であることを示している
NHP研究は、異なる候補ワクチン間の免疫原性を測定するために使用される。すべてのNHPは、5 x 10のrAd 5免疫を鼻腔注射によって免疫する10 Iuは1日目と30日目にある。各群4頭のカニクイザルはワクチン試験を受けた:ED 88(rAd 5−S−N武漢),ED 90(Rad−S武漢),ED 94(Rad−S Beta),稚群は生理食塩水を投与した。第4群は精製したS蛋白(NIHから)を1日目に筋注し,その後30日目にED 88を増強した。ED 88と比較して,ED 90を接種した動物は,より高いレベルの抗SARS−CoV−2武漢,Beta,Delta S蛋白の血清抗体応答を誘導する(図N 4)。ED 90で免疫した動物は全長三量体S蛋白と受容体結合領域(RBD)に対する特異的免疫グロブリンを誘導し、武漢蛋白、Delta蛋白とBeta S蛋白に対する特異的免疫応答を測定することができる。ED 90免疫動物はED 88免疫動物に比べて武漢株,Delta株,Beta株に対する免疫応答が高かった。ED 90のBeta変異体S蛋白に対する反応はED 94と類似しているが、武漢変異体とDelta変異体に対する反応はED 94より優れている。以上より,ED 90候補ワクチンは他の候補ワクチンよりもNHPにおいて重要なSARS−CoV−2変異株に対してより良い血清と粘膜抗体応答を誘導し,臨床候補ワクチンとしてVXA−CoV 2−1.1−Sが臨床に入る。
図1
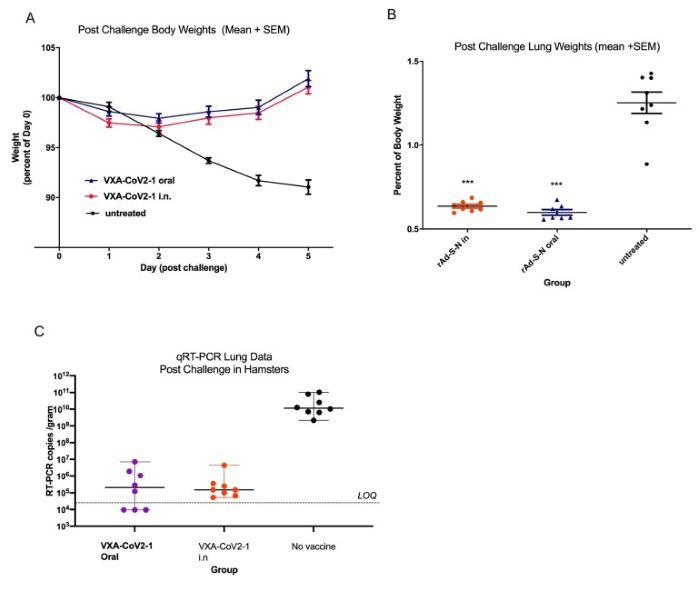
図N 1.0週目と4週目にハムスターを免疫し、8週目にSARS-CoV-2ウイルスに経鼻感染した。各ハムスターは1 e 9 IUでrAd-S-Nを経口投与または注射した。未処理の動物はワクチンを接種しなかったが、ワクチン群と同時に攻撃した。各グループ8匹。Aグループの動物は挑戦後5日以内に体重モニタリングを行った。各グループの平均値(+/-SEM)は示した。B.5日目の肺重量を取り、実際の動物の体重で正規化して体重のパーセンテージを計算した。各グループの平均値(+/-SEM)を示した
図3:n 2
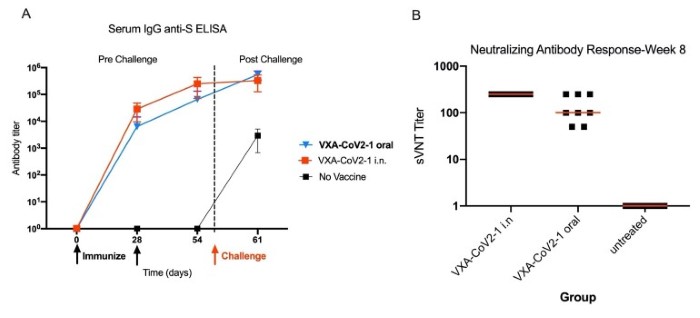
図20週目と4週目に1または2剤のワクチンを接種した後の血清中の抗体反応。8週目の攻撃後。時間の経過とともに、A.免疫グロブリン血清ELISA抗体価はS 1タンパク質に達した。B.8週目に抗体反応(SVNT)を中和した。
図-N 3
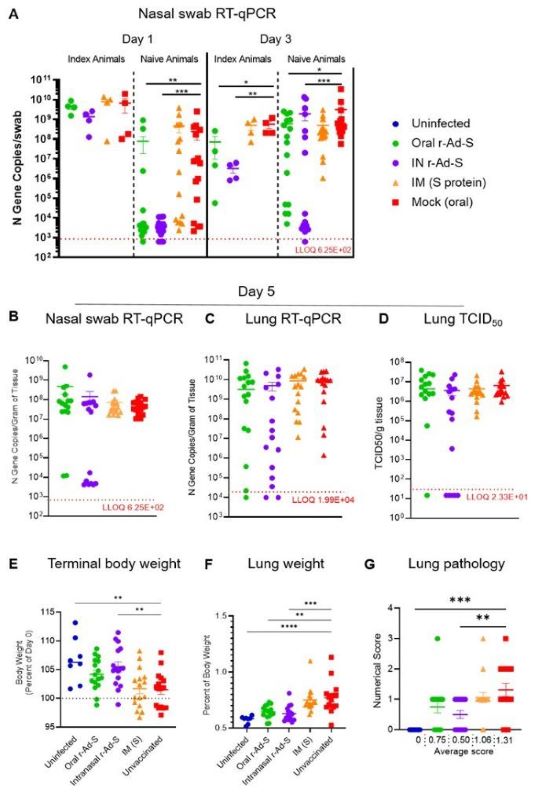
図N 3.SARS−CoV−2ワクチンの経口接種および鼻内接種は、SARS−CoV−2の伝播および疾患の臨床指標を減少させた。以下(A)ナイーブ動物は、指数感染ハムスターに曝露した後の1、3および(B)5日間、エアゾールカプセルで鼻スワブを収集した。N遺伝子の定量逆転写ポリメラーゼ連鎖反応(qRT-PCR)を用いてウイルスRNA負荷量を測定した。(C)剖検(5日目)に肺組織を収集し、N遺伝子のqRT-PCRによるSARS-CoV-2の検出および(D)TCIDによる感染性ウイルス力価の検出のためのRNAを抽出する工程50それは.(A-D)破線はLODを表し,検出限界を下回るデータは1/LODにプロットされる.データ解析には単因子分散分析とDunnett多重比較を用いた。(E)終末体重は、0日目のパーセンテージによって決定される(SARS-CoV-2接種に対して)。(F)末梢肺重量および(G)肺病理スコアを測定する工程。肺部赤色変色の重症度は0~4点で全肺が影響を受けるパーセンテージを表す:無(無)、軽度(1)、軽度(2)、中度(3)、顕著(4)であり、それぞれ0、1-25、26-50、51-75と76-100%と相関している。(E-G)資料分析は単因子分散分析とTukey多重比較を用いた。(A-G)エラーバーは走査電子顕微鏡を表す。*P
図4
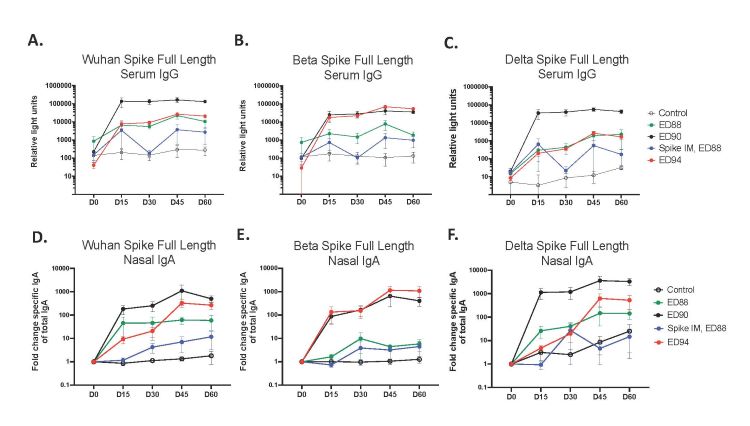
図N 4. ED 90粘膜免疫後、血清と鼻腔免疫グロブリンAは比較的に強い交差反応を産生する。D 1とd 30にそれぞれベクター対照群(開放輪),ED 88(緑輪),ED 90(黒輪),ED 94(赤輪)で免疫したり,d 1にSpike蛋白を筋注し,d 30でED 88増強免疫(青圏)を行った。それぞれ免疫後D 0,D 15,D 30,D 45とD 60で全長三重穂(A),武漢(B),Beta変異体(B.1.351)とDelta変異体家系(B.1.617.2)の血清免疫グロブリンを測定した。MSD相対光単位;走査電子顕微鏡(n=4)。各試料時点で,全長三量体ピーク(D),武漢(E),Beta変異体(B.1.351)とDelta変異体(B.1.617.2)の鼻腔特異的IgAを定量し,総IgAに正規化した。鼻腔免疫グロブリンAの発現レベルはベースラインレベルより2倍高かった。
臨床試験
ステージ1−VXA−COV 2−101
第一段階研究は開放標識、用量範囲設計を用いて、経口健康成人ボランティアのVXA-CoV 2-1の安全性と免疫原性を評価する。第1段階レジメンによれば、35人の参加者が入選し(2020年10月~11月)、低用量(n=20)または中用量(n=15)のVXA−CoV 2−1ワクチン接種を受けた。低用量群の5人の被験者は、最初のワクチン接種後4週間で免疫接種を受けた。研究参加者は最後のワクチン接種後に周囲の安全性と免疫原性を追跡され,その後安全追跡期間に入り,1年間の最後のワクチン接種を継続した。最初の35名の参加者のうち7名が別のBoost延期子研究に参加し,2021年10月から11月までの間に初回接種後約12カ月にVXA−CoV 2−1.1−Sの増強接種を受けた。Boost亜研究参加者はその後、Boostワクチン接種後4週間追跡され、もう1年間の安全フォローアップ期間を加えた。
男性または女性ボランティアは,年齢18歳から54歳の間,体重指数(BMI)は17歳から30キロ/mの間である2スクリーニング時には,SARS−CoV−2に曝露された低リスクを含み,スクリーニング時のSARS−CoV−2感染スクリーニングは陰性であり,病歴,健康診断,バイタルサインと臨床実験室(完全な血球計数,化学と尿分析)に基づく全体的な健康状態が良好で,重大な疾患がない人は,この研究に参加する条件に適合している。合格を確認した後、5人の歩哨被験者がキュー1に入り、少量(1 X 10)を接種した10Iu±0.5 log)VXA−CoV 2−1経口ワクチン。
主な目的はVXA−CoV 2−1の安全性を決定することである。安全性および耐性は、反応性症状(各接種後7日)、要求されていない副作用(最終接種後28日目(29日目)、キュー1の57日目)、SAE、MAAE(新冠肺炎の証拠を含む)、およびワクチン増強疾患(360日まで)の要求症状を検出および記録することによって評価される。臨床実験室(血液化学、血液学と尿分析)結果、体格検査とバイタルサイン結果も評価した。
すべての3つのキューの条件を満たす被験者は、VXA-CoV 2-1.1-S 1 x 10用量の増強治療を受けるように招待された11初回接種VXA−CoV 2−1は約12カ月後,Iu±0.5 logであった。資格再確認後,7名の被験者が新たに同意し,追加のBoost拡張子研究に参加した。最初にVXA-CoV 2-1ワクチンを接種した後に追加の新冠肺炎接種を受けなかった被験者と、研究期間中(積極的かつ安全なフォローアップ)に症状が報告されなかった新冠肺炎および/またはSARS-CoV 2-1感染検出が陽性であった被験者のみが、BOOST分研究に参加する資格がある。
安全結果
VXA-COV 2-101-137%の研究参加者(35人中13名)が要求された症状を報告し、より多くの被験者が低用量(20%)ではなく中間用量(60%)の症状を報告した。最もよく見られる誘引症状は,嘔気(27%),嘔吐(27%),下痢(27%)である。誘引症状の多くは中等度まで重篤であり,治療を必要とせずに緩和され,また,有害事象の誘引により中止された被験者はいない。
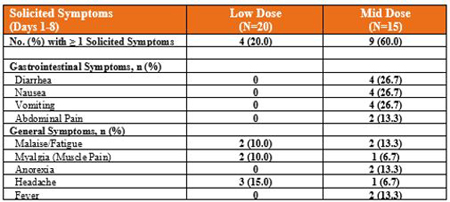
5人の被験者(14%)は57日目に能動的副作用を経験した。他の2人の被験者は、研究治療に関連すると考えられる能動的副作用:中咽頭痛(2日目~5日目)1級と7日目の1級身震いを経験した。*招待されていないすべての副作用の重症度は軽く、治療を必要とせずに緩和された。主に研究した35人の被験者のうち4人は主な研究安全追跡期間を終えていなかった。この間,SAEは報告されていない.
VXA-COV 2-101 Boost-Boost亜研究では,Boost被験者7名中3名(43%)がBoost接種後にAEsを獲得したと報告している。報告された副作用は、頭痛(29%)、不快感/無力(29%)、および吐き気(14%)を含む。増強用量後に募集したすべての急性脳炎の重症度は軽微または中等度であった。重篤な副作用もなく,薬物を併用する必要もない。
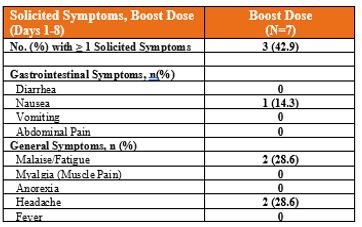
BOOSTグループ研究活動期には,無関係な能動AE(創傷)の1例が報告されている。関連する副作用や副作用はなかった。7名の被験者全員が推進活動期を完了した。4人の被験者は強化用量後12ヶ月間フォローアップし、3人の被験者は180日間の安全フォローアップ後に退出した。Boostセキュリティ追跡の間、SAEやMAEsの報告はない。
免疫原性結果
T細胞の分極とT細胞誘導。抗ウイルス免疫反応の一部として、T細胞は特定の“キラー”として機能することができるので、ウイルス感染細胞を探して破壊し、感染および疾患の重症度を制御することができる。新冠肺炎候補ワクチン(例えばVxA-CoV 2-1)を接種することは、SARS-CoV-2感染細胞を認識するT細胞の増加を誘導するはずである。しかし、T細胞が活性化すると保護性(Th 1)やアレルギー反応(Th 2)を産生することができる。この臨床研究の主要な免疫学的終点は、保護性Th 1反応に向かってもアレルギーTh 2反応にも、SARS-CoV-2特異的T細胞の分極を測定することである。これは再刺激試験により測定し,接種前後に採取した末梢血単核細胞(PBMC)とS蛋白(S)または核蛋白(N)由来のSARS−CoV−2ポリペプチドを培養し,Th 1/Th 2応答を測定した。1日目および8日目からのPBMC試料26対は、単剤の前および後の研究から評価することができる。残りの試料は、使用可能でも品質差もなく、いずれの測定対象においても、スパイク(S)または核タンパク質(N)に対するTh 2応答(CD 4 T細胞から放出されるIL 5/IL 4/IL 13と定義される)の有意な増加は認められず、0/26はワクチン接種後8日目に2倍増加し、Nに対する平均応答率はそれぞれ0.09/0.02/0.04%およびSに対する0.02/0.09/0.1であった(図N 5 C)。
多くの被験者はTh 1応答が増加し,IFNG/TNFa/CD 107 a,特にCD 8+T細胞のSペプチドに対する応答と定義した(図N 5 A−B)。Sペプチドに対する反応では,26名中13名(50%)のTh 1サイトカインの放出が1倍以上増加したか,CD 107 aの場合,CD 8 T細胞の発現が1.5倍以上増加したのに対し,26名中17名(65%)のTh 1サイトカイン放出が1.5倍以上増加した。26人の被験者のうち19名(73%)がベースラインよりもCD 8 T細胞応答を高かった。免疫後8日目、IFNG/TNFa/CD 107 aは接種前のベースラインより平均1.5/4.6/1.95ポイント増加した。26人の対象中5名(19%)のCD 4 T細胞が1倍以上増加し、14名(54%)の対象のCD 4 T細胞反応がベースラインレベルよりも高かった。IFNG/TNFa/CD 107 aのCD 4 T細胞の平均パーセンテージは0.6/1.0/0.9増加した。Nペプチドに対する反応では、26人中9人(35%)のCD 8 T細胞のTh 1反応が接種前よりも2倍以上増加し、そのうち11人(42%)が測定可能なCD 8 T細胞反応を示した。26人の被験者のうち1名のみのTh 1 CD 4 T細胞のNに対する応答は、Nの2倍以上であり、そのうち9名(35%)はNに対して測定可能なCD 4 T細胞応答を有した。IFNG/TNFa/CD 107 aに対して、CD 8の平均パーセントは0.1/0.2/0.6増加し、CD 4中の平均パーセントはそれぞれ0.08/0.08/0.2増加した。Th 2応答がない場合、Th 1 CD 8 T細胞のSに対する高振幅応答は、VXA-CoV 2-1を接種した被験者が、Th 2応答の潜在的有害事象を発生させることなく保護抗ウイルス応答を増強したことを示した。
図:N 5
| A | B | C |
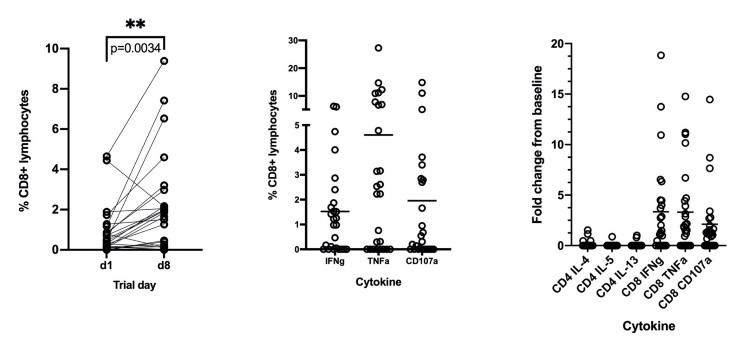
図N 5.T細胞の分極と特徴。A.免疫後8日目にCD 8 T細胞を産生するインターフェロン-γは1日目より増加した。ペアT検定を用いて接種前後の頻度を比較したVXA−CoV 2−1免疫の対象では,CD 8 T細胞のCD 107 aパーセントが背景免疫後より増加した。
B細胞反応。ワクチン接種の主な目標は感染或いは疾病に対する保護を調節するために免疫反応を誘導することである。Bリンパ球は,B細胞とも呼ばれ,感染性病原体を特異的に認識·抑制できる抗体を産生することにより,この目標を実現する上で重要な役割を果たしている。B細胞は異なる形式の抗体を産生することができ、タイプごとに異なる特徴と作用がある。A型同型(“IgA”)抗体を持つB細胞は粘膜表面に優先的に分泌される細胞,例えば気道であり,そこでは異物のヒトへの侵入を阻止する。われわれの候補ワクチンは,高レベルの抗体を産生できる特定のB細胞(“形質母細胞”と呼ぶ)を促進する能力をフローサイトメトリーによる測定とElispotによる抗体分泌細胞(ASC)分析により試験した。フローサイトメトリーは細胞が発現するタンパク質を測定することができ,細胞表面でも細胞内部でも。この分析は,ワクチン接種後8日で血漿母細胞総数が有意に増加したことを示している(p
図6
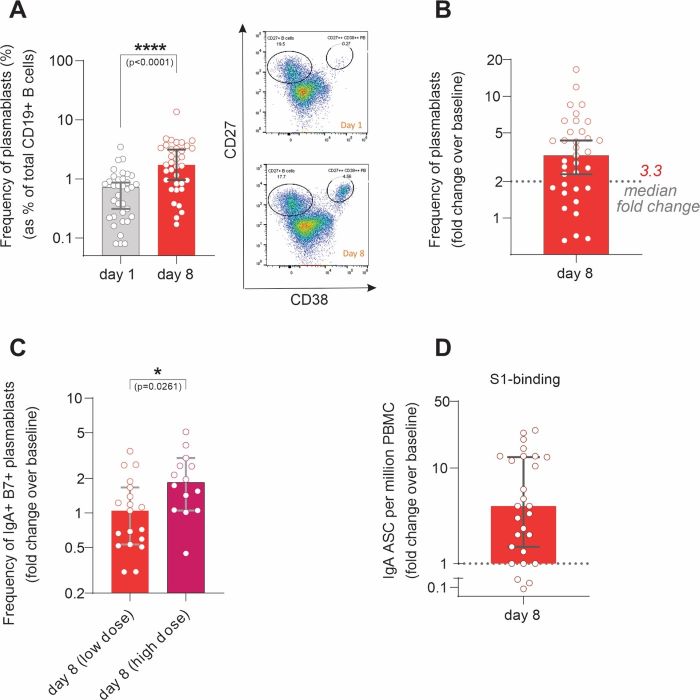
図N 6.接種前(1日目)と接種後(8日目)の末梢血中のCD 27++CD 38++形質母細胞の頻度をフローサイトメーターで測定した。ストリップは中間値を表し、誤差ストリップは95%の信頼区間に対応する。接種前後の頻度をWilcoxon検定で比較した;B.血漿母細胞頻度の変化(8日目は1日目と比較)。35人の被験者のうち、24人(69%)が2倍以上の増加(全体的な中央値変化3.3倍)を示し、低用量および高用量ワクチンキューにおいてIgAおよびB 7を発現する形質母細胞の変化(8日目は初日と比較して)が2倍以上増加した。2つの異なる用量群間の頻度をMann−Whitney検定で比較したところ,1日目と比較して8日目に免疫グロブリンA陽性抗体分泌細胞(ASC)の数が2倍に増加した
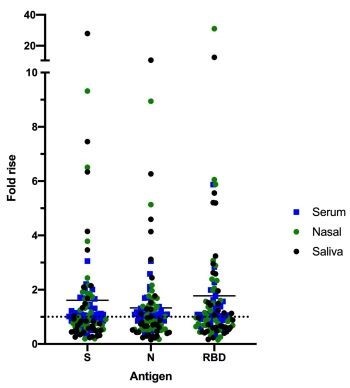
抗体反応。血清試料を中和抗体検査した。29日目(低用量を2回服用した5人の被験者では、56日目)の血清に中和抗体は認められなかった。数名の被験者の血清のみで免疫グロブリンG応答の増加が検出された。局所免疫反応はウイルスの侵入を阻止する能力を有するため、感染部位の局所免疫反応は特に興味深いが、IgAは多くの粘膜組織の第1の防御線と考えられている。粘膜中の免疫反応を測定するために、鼻腔や唾液試料も採取された。また、血清試料も採取された。AS血清にはIgAも含まれていてもよい.SARS−CoV−2 S蛋白,N蛋白とSpike受容体結合ドメイン(RBD)に対する抗体レベルを中スケール発現プラットフォーム上で多重分析法を用いて測定した。このプラットフォームは、従来のELISAフォーマットよりも低い試料量を用いて、複数の抗原に対する特異的抗体を一度に捕捉することを可能にする。予備分析では,ワクチン接種29日後,35名の被験者のうち18名(52%)が接種前試料で各区画で発見されたSARS−CoV−2特異的IgAの1倍以上の増加が検出された。S蛋白二重反応11例(32%)、N蛋白二重反応13例(37%)、RBD二重反応16例(46%)、2種類或いは2種類以上の抗原二重反応14例(40%)。列1において、被験者は2剤ワクチンを服用し、5人中4人(80%)のSARS-CoV-2 IgA反応は2倍以上であり、5人(100%)の5人(100%)の1つ以上の区画における反応は1.5倍以上であった。これらの結果はすべての被験者を含む。その中でいかなるIgAを欠いている可能性のあるサンプルが特異的な抗体反応を示す可能性が低いため、将来の仕事はIgA総量でサンプルを正規化し、分析中にIgAを含まないサンプルを破棄する。
図7
図N 7.図2血清,鼻腔,唾液試料中のIgAは2倍に増加した。
中尺度(MSD)SARS-CoV-2 V-plex平板を用いて血清、鼻腔と唾液中のS蛋白、核蛋白と受容体結合ドメイン(RBD)を測定した。血清は1:100の希釈度で測定し,鼻腔と唾液試料は1:10の希釈度で測定し,8日目を1日目(ベースライン)MSDの任意の単位で割って倍数を計算した。
フェーズ2 a VXA-COV 2-201の研究:第1部分:成人用量最適化
2021年10月の2 a期研究の第1部で用量測定を開始し、開放ラベル、用量範囲設計(第1部)を用いて健康成人ボランティアに経口投与されたVXA−CoV 2−1.1−Sの安全性および免疫原性を評価した。2021年10月の第2段階臨床研究で投与を開始し、約896名の参加者が2つの研究設計を用いて登録することを計画した。研究の第1部(第1部)では、18歳から55歳の年齢48名と56歳から75歳の年齢48名の参加者を募集し、安全性および免疫原性をさらに評価し、最適用量を評価する予定である。さらに、試験中の半分の被験者は、Vaxart新冠肺炎候補ワクチンが免疫反応を増強し、変種特異的交差反応を増強する能力を試験するために、事前にワクチン(メッセンジャーリボ核酸ワクチンを2剤接種した)を接種し、半分の被験者は以前のワクチン接種に無邪気であることを試験した。第1の部分から用量を選択した後、研究の第2の部分(“第2の部分”)は、18歳から75歳の年齢の約800人の被験者を募集することを計画している。第2の部分は、SARS−CoV−2感染を予防するための予備ワクチン効力を試験することを目的としている。研究の第1部(“第1部”)の活動期は,最後の服薬後4週間以内に被験者を追跡し,完成した。12ヶ月間の安全な追跡期間が進行中です。ワクチン初心者を識別できないため、第1部の実際の参加者数は計画より少ない(表N 1)。また、研究地点のグループ規模が小さいことや、SARS-CoV-2の大規模爆発期間中の登録者数が少なく、分析に利用可能な対象が少ないため、多くの分析に低用量群と高用量群が結合されている。この部分の試験のトップデータは2022年9月に発表され、主要かつ副次的な終点が要求を満たしていることが示された。大流行と新たな新冠肺炎株の出現により、華泰は第2部を継続しない
表N 1.VXA-COV 2-201第1部:実科目募集
| ワクチン接種状況 |
治療班 |
投与量 |
年ごろ |
違います。線量のどのくらいですか |
違います。主題の数 |
| 無邪気なテーマ |
キュー1 a |
ロー、1 e 10 |
18-55歳 |
2 |
12 |
| キュー1 b |
高、1 e 11 |
18-55歳 |
2 |
9 |
|
| キュー1 c |
ロー、1 e 10 |
56-75年 |
2 |
8 |
|
| 事前にワクチンを接種したことがある |
キュー2 a |
ロー、1 e 10 |
18-55歳 |
2 |
13 |
| キュー2 b |
高、1 e 11 |
18-55歳 |
2 |
12 |
|
| キュー2 c |
ロー、1 e 10 |
56-75歳 |
2 |
12 |
安全結果
VXA-COV 2-201-登録対象66人のうち65人が2剤のワクチンを同時に接種した(1日目と29日目)。1人の被験者は第2剤ワクチン接種前に妊娠検出が陽性で、第2剤ワクチン接種を受けなかった。選択的に妊娠を中止し、安全フォローアップ期間を継続した。もう1人の被験者は第2剤ワクチン接種後に同意を撤回した。研究参加者の52%(66人中34人)が自発的な副作用を報告した。最もよく見られる副作用は、頭痛、不快感、疲労、筋肉痛です。すべての報告の症状は軽微から中等度です。重篤な副作用はなく,有害事象(AE)による中止もなかった。
66名の被験者のうち14名(21%)が研究活動期(~57日目)に能動的副作用を起こした。4名の被験者は、研究治療に関与している可能性があるか、または可能性があると考えられる能動的副作用を6回経験した:1人の被験者は2日目に軽度のめまいが出現し、1人の被験者は2回の服薬後に軽度の口渇が加重し、1人の被験者は服用後2~3日に無症状の低ナトリウム血症が出現し、1人の被験者は服用後2~3日に中度の上腹部痛が出現した。すべての関連する非被験者のAEsの重症度は軽度または中等度であり、治療を必要とせずに緩和した。活動期間中には,関連しないSAEが1例あり,特殊に注目した有害事象(AESI)は報告されていなかった。12カ月間の安全追跡期間は,SAE,AESI,医療参加の有害事象(MAAE)の研究対象後に行われており,2023年第2四半期に終了する予定である
多くの人がメッセンジャーリボ核酸ワクチンを接種しているので、Vaxartはメッセンジャーリボ核酸ワクチン接種後に抗体反応を向上させる能力が特に重要である。Vaxartの経口ワクチン候補は、錠剤免疫前の6ヶ月以上前に2つのメッセンジャーリボ核酸ワクチン(ファイザー/バイオテクノロジーまたはModerna)を接種したボランティアの血清抗体応答を向上させることができる。既知の感染対象を分析から除外し、18~55歳の年齢群の高用量群および低用量群に結合した後、SARS−CoV−2(公認保護相関)に対する血清中および抗体応答は、481個の幾何平均値から778 AU/mlに増加する。2021年に採取された武漢ウイルスに感染する可能性のある未接種回復期被験者の血清を測定したところ、幾何平均力価は84であった。これは、感染と比較して、メッセンジャーリボ核酸ワクチンの接種が中和抗体価を著しく向上させ、Vaxart経口投与錠がこれらの力価をさらに向上させることができることを示している。力価の低い被験者の方が感染しやすいことから、データは力価が200より高い被験者と力価が200未満の被験者に分類される。200 AU/mlのしきい値は、回復期幾何平均の約2倍であった。NAB力価が200未満の被験者は、2.5倍の幾何学的増加を有し、経口錠剤後の50%から200 AU/mlから80%までの人口を有した(図N 8)。これらの力価は最初の武漢毒株に対するものである。svnt検定によると、これらのボランティアは最近流行しているSARS-CoV-2オミックオスミウムBA 4/5株に対する中和抗体応答も大幅に増加している(図N 9)。単一被験者の抑制率は最低から最高抑制率(左から右)までである。Limらによると、68%の抑制が保護に関連しているという。35%の被験体が経口ワクチンの前に“血清保護”を受け、70%が接種後に“血清保護”を受けた。約50%の被験体が粘膜免疫グロブリンA抗体応答(鼻および口腔内の抗体)を増加させた。SARS-CoV-2武漢Sに対する粘膜免疫応答が増加した被験体は、SARS-CoV-2オミックBA 4/5およびSARS-CoV-1を含む他のコロナウイルスに対する免疫応答も増加し、これらの免疫読み取りの交差反応特性を示した(図N 10)
図N 8.経口免疫後の武漢に対する中和抗体反応
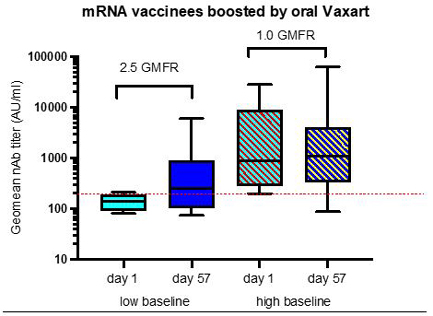
図N 8. 合格した擬似ウイルス検出方法を用いて、以前にメッセンジャーリボ核酸ワクチンを接種した被験者に対して中和抗体反応の検出を行った。経口免疫前(1日目)および経口免疫後(57日目)に力価を測定した。力価が200 AU/ml未満の被験者は幾何平均力価が増加し、開始力価の高い被験者は増加しなかった
SARS−CoV−2に対するN 9オミックBA 4/5の中和抗体反応
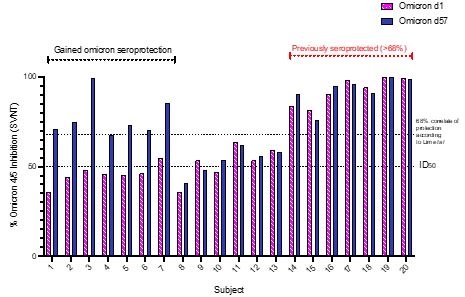
図N 9. Vaxart経口錠剤ワクチンを接種した被験者のオミックBA 4/5接種後1日目と57日目の中和抗体応答を代替中和抗体試験(SVNT)を用いて検出した。Limおよび彼の同僚に記載された方法(Lim,et al,Vaccines,2021)によると、血清保護閾値は68%の阻害に設定されている。この閾値を使用して、35%の被験者が初日に血清保護を受け、70%の被験者が57日目に“血清保護”に達した
図10.SARS−CoV−2と他のb型コロナウイルスS蛋白との粘膜反応
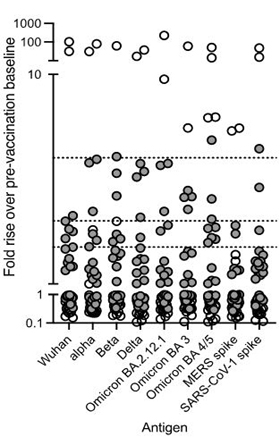
[図10]. 異なるSARS−CoV−2変種およびMERSとSARS−CoV−1に対して,個体のS蛋白に対する粘膜免疫反応をプロットした。開放円は鼻腔免疫グロブリンA応答を表し、閉鎖円は唾液免疫グロブリンA応答を表す。被験者の約50%は武漢に対する応答が1.5倍以上であり、55%の被験者がオミックBA 4/5に応答している
次のステップ
新しい毒株の出現速度と、私たちのプラットフォームを用いて見た実質的な交差反応粘膜免疫反応を考慮すると、Vaxartはすでに新しいワクチン構築の臨床前作業を開始し、SARS-CoV-2と他のベタコロナウイルス(例えばSARS-CoV-1とMERS-CoV)に対応できる有効なパンベタコロナウイルス候補ワクチンの創造を求めている。成功すれば、これは毎年の毒株の変化を否定することになる
2022年6月、我々はhVivo Services Limited(“hVivo”)との協力を発表し、SARS-CoV-2人類挑戦研究においてワトソン新冠肺炎候補ワクチンの有効性をテストした。2023年第1四半期、SARS-CoV-2ヒト挑戦研究の開始を延期することにした。私たちが新しい候補ワクチンを提出するにつれて、私たちは人類挑戦研究を含む可能性がある最高の発展計画を決定する。
私たちの季節性インフルエンザ計画は
市場の概要
インフルエンザは最もよく見られる全世界の伝染病の一つであり、軽いから生命に危害を及ぼす疾病を引き起こし、症状は喉の痛み、鼻水、発熱、甚だしきに至っては死亡を含む。全世界で毎年少なくとも3億5千万人の季節性インフルエンザ症例が発生し、その中の300万から500万例は深刻な病例と考えられ、全世界で毎年29万から65万人が死亡していると推定されている。幼児と高齢者が直面する死亡リスクが最も大きい。アメリカでは、人口の5%から20%がインフルエンザに感染し、14万人から71万人がインフルエンザ合併症で入院し、毎年1.2万~5.2万人がインフルエンザとその合併症で死亡し、90%に達するインフルエンザ関連死は65歳以上の成人で発生している。季節性インフルエンザの総経済負担は871億ドルと推定され、その中には平均年間104億ドルの医療費が含まれているが、毎年疾病や生命損失による収入損失は163億ドルに達している。
疾病管理センターは一般的に6ケ月以上の人は毎年インフルエンザワクチンを接種することを提案している。アメリカでは、これは3億人以上がインフルエンザワクチンを接種することを提案することを意味する。2021/2022年のインフルエンザシーズンには,米国では約1.75億剤のインフルエンザワクチンが提供された。アメリカ市場の差別化インフルエンザワクチンは引き続き、それらが公共健康に提供する追加価値によって価格を要求する能力があることを表明した。全世界範囲内で、市場成長の主要な駆動力は認識の向上、新興国のワクチン接種カバー率の向上、政府の季節性インフルエンザワクチン接種に対する支持の増加、製品差別化による価格上昇及びワクチン治療の生産と改善に対する更なる関心を含むと考えられる。
現在の季節性インフルエンザワクチンの限界は
米国で確定診断されたインフルエンザ症例数は多いにもかかわらず,疾病管理センターのデータによると,2021/2022年の季節性インフルエンザシーズンには,米国総人口の約51%しかインフルエンザワクチンを接種しておらず,18歳から49歳の成人のワクチン接種率は特に低い。この集団の低接種率はワクチン接種の以下の制限によるところが大きいと考えられる
提供者への制限
| ● |
供給者の製造、輸送、運搬時間はもっと長い |
| ● |
流通チェーン全体の冷蔵要求 |
| ● |
ワクチン接種中と接種後に医療専門の監督が必要である |
| ● |
針刺しの可能性 |
| ● |
医療廃棄物です |
ユーザーへの制限
| ● |
診療所や薬局でワクチンを得るのに必要な不便さと時間的約束 |
| ● |
針への恐怖 |
| ● |
注射部位が痛みます |
| ● |
ワクチン中の卵成分にアレルギー反応を起こす可能性がある。 |
季節性インフルエンザワクチン候補ワクチンは
私たちは健康な成人に季節性インフルエンザワクチンを接種するための候補錠剤ワクチンを開発している。今日の商業季節性インフルエンザワクチンは、3種類(3価)または4種類(4価)の毒株からなるか、またはB型インフルエンザウイルスと2種類のA型インフルエンザウイルス株、またはそれぞれ2種類を有する。我々の季節性インフルエンザ候補ワクチンは、2種類の流通A型インフルエンザウイルス(H 1 N 1とH 3 N 2)および2種類の流通B型インフルエンザウイルスを含む4種類の毒株または4価の季節性インフルエンザワクチンをカバーするように設計されており、FDAの季節的更新の提案と一致している。私たちの錠剤ワクチン候補処方を各菌株1錠、または4価ワクチンの合計4錠にすることを想定している。私たちはこのモジュール化が柔軟性を強化することを可能にすると信じている。例えば、季節後期に毒株が変化した場合、古い毒株を含む錠剤を容易に交換することができ、正確にマッチしたワクチン錠剤を3枚捨てる必要がない。あるいは、すべての4つの菌株を1錠に調製することを選択することができる。このフォーマットは最も管理が容易であるが、独立タブレットに耐えられるいくつかの柔軟性の利点を奪うことになる。市場研究を行って商業化に近い市場受容度を評価した後,我々の平板ワクチン候補の最終処方を評価する。
私たちの平板ワクチン候補は現在卵に基づく季節性インフルエンザワクチンの多くの限界を解決する可能性があると信じている。まず、著者らの候補錠剤ワクチンは広範かつ持続的な免疫反応を産生することを目的としており、これはより有効な免疫力を提供し、より多くの菌株の変異を防止する可能性がある。第二に、より便利な管理方法を提供することにより、患者の受容度を向上させ、分配と管理を簡略化することである。最後に,組換え法を用いることにより,我々の候補錠剤ワクチンは卵法を用いて製造したワクチンよりも早く製造され,卵蛋白アレルギー反応のリスクを除去し,哺乳動物ウイルス卵適応による問題を緩和する可能性があると信じられている。
季節性インフルエンザ臨床試験
これまで、私たちは2つの第1段階試験を完成し、私たちのH 1 N 1インフルエンザ候補ワクチンの第2段階挑戦試験の活発な部分を行った。B型インフルエンザ候補ワクチンの第1段階試験も完了しました。そこで、季節性インフルエンザワクチンに必要な少なくとも3つの毒株のうちの2つの臨床結果がありました。
第1段階試験,VXA 02−001,H 1 N 1インフルエンザ候補ワクチン,109そして1010Iu投与量
第一段階A型H 1 N 1インフルエンザ試験の投与量は1×109そして1 x 1010Iu。二回の投与間隔は一ヶ月です。候補錠剤ワクチンは良好な安全性と耐性を産生した。この試験は,強力なT細胞反応と適度な凝固抑制試験(HAI)反応を示し,いずれの反応も用量レベルに依存する。
第1段階試験VXA 02−003,H 1 N 1インフルエンザ候補ワクチン,1011Iu投与量
第二のH 1 N 1試験は1種の錠剤ワクチン試験であり、投与量は1×10である11IUは,単一投与で提供した。この用量レベルでは,良好な安全性と耐性が観察された。ワクチン群のHAI血清転換率は75%であったが,プラセボ群では0%であった。92%の被験者は、錠剤単一服用後、微中和試験(MN)力価が4倍に増加した。HAI血清転換率とMN反応率はいずれも試験VXA 02-001で観察された低用量の対応比率より明らかに高かった。ワクチンまたはプラセボの注射後7日前の副作用は軽微であり、重篤な副作用はなかった。接種後の最初の7日間に,ワクチン群とプラセボ群で計8回求めた副作用(各群4回)が報告された。すべてのAEsの重症度は1級であった。最もよく見られる急性脳脊髄炎は頭痛である(2例はプラセボ服用,1例はワクチン接種)。1年間のフォローアップ期間中,アジュバントに関連するSAEや新たな慢性疾患は記録されていなかった。
以下の表は著者らの2つのプラセボ対照のA型H 1 N 1期臨床試験の試験設計と結果(血清抗体反応)をまとめた。
表7.概要:H 1インフルエンザ1期プラセボ対照研究。
| 取り調べ番号/ |
試行設計 |
研究グループの投与量/スケジュール |
重要な免疫原性発見 |
| ステップ1 試用版VXA 02-001 N = 36 |
投与量の増加、プラセボ対照、二重盲腸溶解カプセル |
109, 1010VXA-A 1.1(H 1)ワクチンまたはプラセボを0日目および28日目に経口投与する、錠剤形態 |
109投与量レベル: ·中国にはHAI血清変換がない
1010投与量レベル: ·HAI血清転換率27% ·MNが6.4%(4倍増) |
| ステップ1 試用版VXA 02-003 N = 24 |
プラセボ対照、二重盲検、腸溶性錠剤 |
1011Iu VXA-A 1.1(H 1)ワクチンまたはプラセボ、0日目、表形式で単回接種 |
·HAI血清転換率75%を超える ·MNが9.92%(4倍増) |
第一段階実験。B型インフルエンザ
2015年と2016年、著者らはランダム、二重盲検、プラセボ対照の第一段階試験を行い、B型インフルエンザ錠剤ワクチンの安全性と免疫原性を試験した。18歳から49歳までの健常成人54名が参加し,そのうち38人がワクチン接種を受け,16人がプラセボ治療を受けた。この試験に参加するために、被験者は、1:20以下の初期HAI測定を有することが要求された。試験の活性段階は28日目まで続き、安全監視の後続段階は1年間継続された。ワクチンを受けたすべての被験者は単剤1 x 10を受けました10Iuまたは1 x 10110日目のIu。
安全問題それは.ワクチンまたはプラセボの服薬後7日前の副作用は一般に軽く,重篤な有害事象はなかった。安全性と耐性では,活性用量群とプラセボの間に有意差は認められなかった。
海.海それは.プラセボ群では,HAI GMTは投与後28日目にほぼ不変であった(1:33)。投与後28日で、両活性治療群のHAI力価のGMFRは約2倍であり、用量とは無関係であった。1×10接種のワクチン群では10IU×1または1×1011 IU,血清陰性化率はそれぞれ26.3%(5/19)と15.8%(3/19)であった。プラセボ群では血清転換はなかった。
抗体分泌細胞(ASC)。免疫後0日目と7日目に,末梢血中のインフルエンザウイルスHAを認識する循環B細胞の数をASC法で測定し,HAに対する総抗体応答を測定した。その結果、ワクチン治療群は7日目に確実にASCsを測定できることが分かった。背景ASCsは0日目に一般的に無視できる。免疫グロブリンASCにより1×10の68%が10 Iu用量の被験者は反応し、84%の被験者は1×10であった11Iu用量群は反応があった。1×10の場合11IU用量ワクチン治療群、平均1×10個の免疫グロブリンA SCs(95%信頼区間:7-35)と73個の免疫グロブリン免疫グロブリンASCs(95%信頼区間:35-111)67日目に末梢血単核細胞(PBMC)を発見した10 Iu用量ワクチン治療群は、7日目に平均16個のIgA ASCs(95%信頼区間:2-29)と44個のIg G ASCs(95%信頼区間:21-66)を発見した。プラセボ群は7日目に応答者がおらず、平均斑点数(1個以下)は無視できる(95%信頼区間:-0.6-2)。
H 1 N 1インフルエンザ第2段階挑戦研究はHHS BARDAによって援助されている
2015年、HHS傘下のBARDAは1390万ドルの契約を与えてくれた。この2年間の契約は、季節性および大流行インフルエンザの準備を改善するために、より効果的なインフルエンザワクチンの高度な開発を支援することを目的とした広発機関が発表した公告に基づいて付与された。この契約は主に人類ボランティアに対する第二段階の挑戦研究を援助し、著者らのH 1 N 1シート状ワクチン候補ワクチンが現在市販されている注射可能ワクチンよりも広く持続的な保護を提供しているかどうかを評価することを目的としている。HHS BARDAとの契約はその後1570万ドルに増加し、契約期間は2018年9月に延長された。
この第2段階の研究では,ボランティアはランダムに3つのグループに分類された。1群はH 1 N 1インフルエンザ候補錠剤ワクチンの経口投与を受け、第2群は商業許可の筋肉注射不活化インフルエンザワクチンを受け、第3群はプラセボを受けた。免疫3ケ月後、ボランティアはH 1 N 1(A/H 1 N 1 pdm 09)生ウイルス挑戦(意図的な実験薬品使用)を鼻腔注射した。プラセボ群を対照群とし,ワクチン未接種ボランティアがどれだけ感染しているか,インフルエンザ症状がどの程度深刻になっているかを確認した。この挑戦研究では、各ワクチンの有効性を決定するために、私たちのワクチン候補グループおよび商業的に許可された不活化ワクチングループからのデータをプラセボと比較する。重要なのは、この2つのワクチンも対面比較を行っているということだ。この研究の目標は,免疫3カ月後に,我々のワクチンがH 1 N 1インフルエンザ挑戦による疾患からボランティアを保護することの有効性をワクチン注射とプラセボと比較することである。
VXA−CHAL−201臨床試験結果
第2段階挑戦研究は2016年から2017年の間に行われた。その間、179人のスクリーニングによって要求された被験者は、私たちの錠剤ワクチン、商業注射ワクチン、またはプラセボの単剤注射をランダムに受けた。この179名の被験者のうち、143名の被験者は服用90~120日後にその後、A型H 1 N 1インフルエンザ生ウイルスの攻撃を受けた。
| ● |
安全ですワクチンとプラセボの接種後7日前の副作用は一般に軽微であった。服薬後の7日前には,ワクチン群とプラセボ群で報告された有害事象の多くは重症度1級であり,2級を超えていなかった。最もよく見られる有害事象はわれわれの錠剤ワクチン群の頭痛(7%),商業許可不活化ワクチン群の注射部位圧痛(26%)とプラセボ群の頭痛(19%)であった。研究の追跡期間中,重篤な有害事象は記録されておらず,われわれのワクチンアジュバントに関連する新たな慢性疾患も記録されていない。以下のグラフは,局所と系統的有害事象の経時的分布と重症度を示している(図15と16). |
[図15]要求された局所症状の最大重症度。
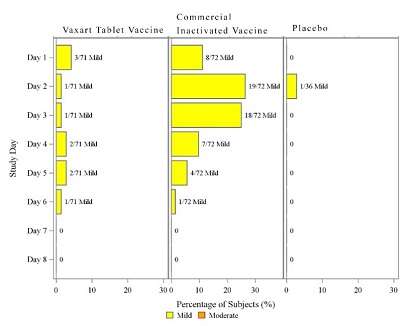
[図15]免疫後7日間に収集された局所症状を示す図である。時間の経過とともに,各治療群は要求症状の重症度を示した。すべての事件は穏やかである
図16.要求される全身症状の重症度。
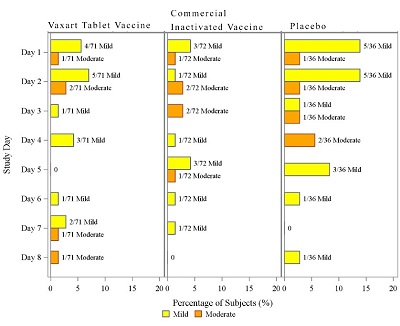
図16.ワクチン接種後7日以内に収集された全身症状。時間の経過とともに,各治療群は要求症状の重症度を示した
治療効果−ポリメラーゼ連鎖反応で確認されたインフルエンザ疾患を減少させた
主要な効力目標は野生型A型H 1 N 1ウイルス株(A/H 1 N 1 pdm 09)に挑戦した後の私たちの錠剤ワクチンのワクチン効力を決定することである。主な治療効果の終点は病気である。われわれのシート状ワクチンの発症率は29%,商業不活化インフルエンザワクチンの発症率は35%,プラセボ群の発症率は48%であった。著者らのシート状ワクチンの発病率は商業ワクチンより低い(-6%の発病率の差は著者らのワクチンに有利である)、研究規模が小さいことを考慮すると、これらの差は統計学的に顕著ではない。同様に、われわれのシート状ワクチンとプラセボとの間の発症率の差(−19.1%)と商業注射ワクチンとプラセボとの間の差(−13.2%)は保護される傾向にあるが、統計学的に有意差はなかった。これらの結果は,我々のワクチンはこれ以上悪くないことを示しており,保護面では商業ワクチンよりも良い傾向にあることを示している。これらの結果を下表にまとめる。
表8.H 1インフルエンザの第二段階挑戦研究:発病率*。
| Vaxart |
商業広告 |
Vaxart-ビジネス版 |
プラセボ |
|||
| n |
%(95%CI) |
n |
%(95%CI) |
レート差(95%CI) |
n |
%(95%CI) |
| 58 |
29.3 (18.1, 42.7) |
54 |
35.2 (22.7, 49.4) |
-5.9 (-24.3, 12.5) |
31 |
48.4 (30.2, 66.9) |
*疾患は、患者報告結果ツール(Flu-Pro)で報告された症状の組み合わせとして定義されるTM)および定量的逆転写ポリメラーゼ連鎖反応(qRT-PCR)は、ハウスインフルエンザウイルスを検出することができる。
効果−インフルエンザ前症状スコア
商業不活化インフルエンザワクチンとわれわれのシート状ワクチンはFlu−Proアンケートで統計的有意差はなく,Flu−Proアンケートはコミュニティインフルエンザ臨床試験で用いられた検証された患者記録結果ツールである。しかし、私たちのワクチンの全体的な症状の重症度は低下傾向にある。VXA−A 1.1群の被験者は低いFlu−Pro総得点中央値(2.0)を示した[0, 72])QIV群より高い(5.0[0, 59])またはプラセボ群(5.0[0, 52]).
薬効が減退する
脱落は感染後に鼻スワブで検出されたインフルエンザウイルスを表し、ウイルス感染と複製を代表する。この研究では,VXA−A 1.1では44.8%の被験者が少なくとも1日脱毛陽性であったのに対し,市販ワクチンの脱落率は53.7%,プラセボ被験者の脱落率は71.0%であった。われわれのシート状ワクチンと商用インフルエンザ不活化ワクチンの曲線下でのウイルス脱落面積(AUC)との間に統計的有意差は認められなかった。しかしながら、AUCは標準的な対数台形法を用いて計算され、脱落持続時間の5日前に検出可能な脱落のみを含み、インフルエンザに感染していない5日間の被験者を分析から除外した(ゼロ値は対数計算および積分のために使用できない)。これはプラセボに対して,この2つのワクチンのウイルス脱落への影響を過小評価している可能性がある。したがって、脱皮に対するワクチンの影響をよりよく決定するために、ボランティアが挑戦後36時間以内の任意の時間にウイルス脱出が検出された場合に、感染と定義される別の方法が使用される。この方法は、計算(ゼロ値の対数計算)および大量チャレンジウイルス(最初の36時間はインフルエンザを複製するのではなく通過する可能性がある)に関連する可能性のある問題を除去する。ベイズ分析では,プラセボと比較して両ワクチンとも脱落の可能性が有意に低下した(われわれのシート状ワクチンのベイズ事後p=0.001,商業不活化インフルエンザワクチンのp=0.009)。われわれのワクチンもより効果的な傾向にあり,事後確率は約80%であった(表9)。
表9.H 1インフルエンザ第二段階挑戦研究:感染率*.
| 治療腕 |
N |
感染者数 |
パーセント(95%CI) |
事後P |
| プラセボ |
31 |
22 |
71% (55-85%) |
- |
| 商業広告 |
54 |
24 |
44% (32-58%) |
0.009 |
| Vaxartワクチン |
58 |
21 |
36% (24-49%) |
0.001 |
*感染は、ウイルス攻撃36時間後の任意の日に任意の陽性を検出することができる定量的逆転写ポリメラーゼ連鎖反応(qRT-PCR)脱落インフルエンザウイルスとして定義される。ベイズ解析では,両ワクチンとも統計的に有意な感染予防を提供している。われわれのワクチンもより良い効果を示す傾向にあり,事後確率は約80%である。
免疫原性
Haiは答えた。HAIは血清抗体がインフルエンザウイルスと赤血球との結合を破壊する能力を測定した。歴史的に見ると、HAIはインフルエンザワクチン注射の保護と関係がある。免疫後30日後にHAI反応を測定して、血清変換ボランティアの数およびパーセンテージを決定した。私たちの錠剤ワクチン群では,32%のボランティアが血清変換を実現した。商業インフルエンザ殺活ワクチン群では,84%のボランティアがワクチン接種後30日でHAI血清変換を実現した。この差は統計的に有意であった(P
表10.血液凝固抗体抑制(HAI)幾何平均力価(GMT)と幾何平均フォールディング上昇(GMFR)の結果は菌株、研究日および治療群の用量後95%信頼区間であった。
| 全面分析集−ワクチン接種段階 |
|||||||
|
|
ベースライン(投与前) |
薬を飲んでから30日 |
|||||
| 治療班 |
N |
グリニッジ時間 (95%信頼区間) |
N |
グリニッジ時間 (95%信頼区間) |
GMFR (95%信頼区間) |
%4倍の上昇 (95%信頼区間) |
血清転化率 (95%信頼区間) |
| 毒株:オーストラリア/カリフォルニア州/7/2009 |
|||||||
| Vaxart錠剤ワクチン |
70 |
11.13 (9.55, 12.96) |
69 |
29.99 |
2.72 |
36.2 |
31.9 |
| 商業化インフルエンザ不活化ワクチン |
72 |
9.84 |
70 |
273.13 |
27.50 |
90.0 |
84.3 |
| プラセボ |
35 |
10.49 |
35 |
10.40 |
0.99 |
0.0 |
0.0 |
IgA抗体は細胞を分泌する免疫前と免疫後8日目にインフルエンザウイルスHA特異性B細胞(IgA抗体分泌細胞或いはIgA ASCs)を測定し、B細胞のワクチンに対する応答を確定した。ワクチン接種後8日で,商業不活化インフルエンザワクチン群の被験者の10スポットあたりの平均数は有意に高かった6細胞(p 6細胞)(p 6細胞は平均10個あたり116個と比較して6Vaxart錠剤ワクチン用細胞。また,商業不活化インフルエンザワクチン群の応答率は96%であったのに対し,Vaxart錠剤ワクチン群の応答率は71%であった。これらのデータを下表にまとめた。
表11.研究日および治療群によって分類された免疫グロブリンAおよび免疫グロブリンによって検出されたASC反応であるワクチン接種段階。
|
|
ワクチン接種段階 |
||||||||
|
|
ベースライン(投与前) |
8日目(服薬後) |
|||||||
| 化学分析をする |
治療班 |
N |
平均する |
中央値[射程距離] |
少なくとも8人の定員 |
N |
平均する |
中央値[射程距離] |
少なくとも8人の定員 |
| IGA ASC |
Vaxart錠剤ワクチン |
70 |
2.0 |
0.0 [0, 18] |
6 (8.6) |
70 |
116.0 |
32.0 [0, 3251] |
50 (71.4) |
| 商業化インフルエンザ不活化ワクチン |
71 |
1.5 |
0.0 [0, 13] |
8 (11.3) |
71 |
286.4 |
153.0 [3, 1753] |
68 (95.8) |
|
| プラセボ |
36 |
2.8 |
0.0 [0, 26] |
6 (16.7) |
36 |
16.3 |
1.0 [0, 256] |
8 (22.2) |
|
Vaxart錠剤ワクチン中のIgA-ASCsと疾病の相関性。以上のように、商業不活化インフルエンザワクチン群のASCsの絶対平均数はより高い(10点あたり286個)6Vaxart錠剤ワクチンではありません(10細胞あたり116個の斑点があります6セル)。しかし、罹患していないボランティアにおける2種類のワクチンのIgA ASCs比率を比較したところ、Vaxart錠剤ワクチン群の比率は4.7であり、商業注射ワクチンの比率は1.4であった。疾病と非疾病の比較を結果とし、Ig A Ascを引数とするLogistic近似モデルにおいて、Vaxart錠剤ワクチンIg A AsCは疾病と非疾病を予測できるが、商業インフルエンザ不活化ワクチンのLogisticフィッティングモデルはできない(著者らのワクチンp=0.0005、商業注射ワクチンp=0.3066)。これらのデータは,われわれの経口ワクチンではIgA ASCがインフルエンザ予防に重要であることを示しているが,注射された商業ワクチンにとっては重要ではない。これらのデータは定性的の異なる方法で免疫後のB細胞の差異を誘導する。私たちはこのような質的な違いを積極的に探索している。
図17.Vaxart錠剤ワクチンのIGA ASCsは疾病と関連している。
*商業不活化ワクチン
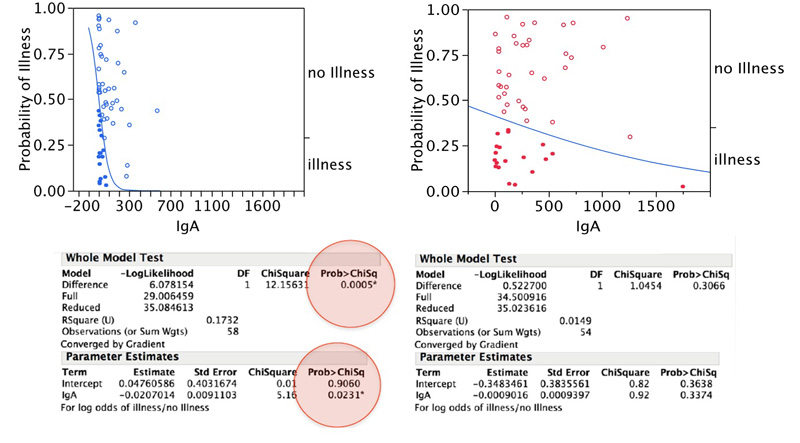
図17.Logistic Fit回帰分析により、Vaxart錠剤ワクチンはIgA ASCsと疾病の符合程度に対して統計的に顕著な意義があることを表明した。比較的に高いASCsと低い発病率の間の相関性が観察された。商業不活化ワクチンは同じモデルが一致することが観察されなかった。
この仕事の資金の全部または一部は、HHS、準備と反応を担当する国務長官補佐官室、BARDAの連邦資金から来ている。
臨床前結果
私たちはインフルエンザに対するいくつかの動物挑戦研究を終えた。1つのH 1 N 1インフルエンザ挑戦研究において、未免疫対照動物と比較して、著者らの候補錠剤ワクチンを経口投与したマウスは疾病と死亡から保護された。同様に、ワクチンが鳥インフルエンザHA構築物を発現する時、私たちの経口H 5 N 1候補ワクチンは未免疫動物ではなく、シイタクとマウスを致命的な鳥インフルエンザ挑戦から保護する。
シイタチ挑戦モデルにおけるVaxart四価季節性インフルエンザワクチンの交差保護作用
最近のシイタチ挑戦実験は2017年に完成し、著者らが設計した経口四価ワクチンと商業ワクチンFluzoneの保護作用を比較し、1種の強毒鳥インフルエンザ毒株に対抗した。鳥インフルエンザウイルスから製造された季節性インフルエンザワクチンにはHAにマッチする成分がないため,このウイルスはワクチンとウイルスのミスマッチの深刻な例を代表している。私たちの四価ワクチンは4つの組換えアデノウイルスを混合して作られ、各組換えアデノウイルスは異なるHAを発現し、挑戦中のHAではなく、商業ワクチン中のHAと一致する。2種類の異なる用量を評価した;高用量はVaxart人体用量の1:10で使用し(Vaxart Quad)、低用量(Vaxart Quad Low)は人体用量の1:100で使用した。フルオロケトン群(QIV)をヒト用量の1:10で投与し、直接Vaxart四価高用量群と比較した。Vaxart動物と陰性対照(PBS)動物は内視鏡的にワクチンを注射した。QIV動物は筋肉注射された。動物はそれぞれ0日目と28日目にワクチンを接種する。56日目に動物たちは約10回挑戦されました2.69野生型A/ベトナム/1203/2004のTCID 50/ml(A/VN)。その結果、Vaxart四価ワクチンはA/VN不整合に対して保護作用があり、傾向はFluzoneより優れていることが分かった。高用量グループはすべてのシイタチを死亡から保護することができ、低用量Vaxartグループは75%のシイタチを保護することができる。
図18.季節性インフルエンザワクチンを接種し、H 5 N 1ベトナムに挑戦したシイタチの生存率を示す。
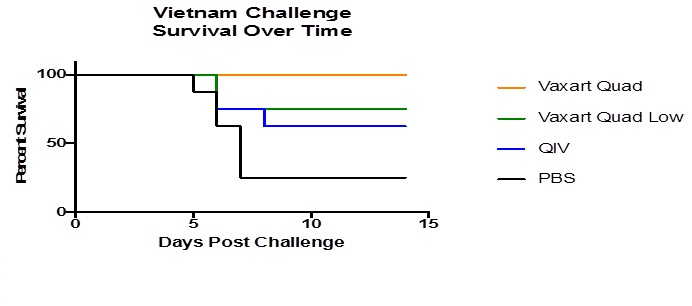
図18.各時点で各グループの患者の生存率を測定する。生存評価の14日以内に、Vaxart四連ワクチングループは一致しない鳥インフルエンザに対して100%の保護作用を持っている。他のグループはよく保護されなかった。
この仕事の資金の全部または一部はHHS BARDAの連邦資金から来た。
HPV治療用ワクチン候補は
これまでのH 5インフルエンザ候補ワクチンの臨床研究では,アジュバントワクチンや弱毒化生ウイルスワクチンを含む他のインフルエンザワクチンの公表結果に匹敵するような強力なT細胞反応が観察された。具体的には,我々の候補ワクチンは,慢性ウイルス感染および癌において治療的利益を得るために必要である可能性がある高レベルの多機能細胞毒性CD 4およびCD 8細胞を産生する。これらの観察に基づいて,HPV関連異型増殖と子宮頸癌に対する最初の治療的候補ワクチンの開発を開始した。
医療需要、ビジネスチャンス
HPVは120種類以上のウイルスからなる家族であり、全世界で非常によく見られる。少なくとも13種類のHPVタイプが発癌している。ヒトパピローマウイルスは主に性接触を通じて伝播し、性行為開始後の感染は非常に一般的である。ほとんどの子宮頸癌症例はHPV感染と関係があり、その中の2種類のHPVタイプであるHPV-16とHPV-18--は70%の子宮頸癌と前癌病変を引き起こした。全国子宮頸癌連盟のデータによると、子宮頸癌は全世界で最もよく見られる女性癌であり、アメリカでは毎年約1.3万の新しい確定診断例がある。米国では男性と女性が生涯にHPVに感染する可能性が高いことが示されており,少なくとも80%の女性と男性が45歳までにHPVに感染していることが示唆されている。アメリカ疾病コントロールと予防センターの推定によると、現在8,000万人のアメリカ公民がHPVに感染し、総人口の25%を占め、毎年約1,400万人が新たに感染している
女性では,多くの子宮頚HPV感染は2~3年以内に自発的に消退·消失するが,持続感染している女性は子宮頚上皮内腫瘍(CIN)と呼ばれる細胞異常のリスクが高く,時間の経過とともに浸潤性癌に進展する可能性がある。アメリカでは、毎年40万人を超える女性がCINと診断され、CIN 1とCIN 2/3の年間発病率はそれぞれ1000人当たり1.6と1.2人と推定されている。
現在、HPV感染或いは癌を治療するための治療性ワクチンはまだ承認されていない。現在HPVに感染した女性(以下に述べる)に対する治療選択はCIN状態のモニタリング、感染組織を切除する外科手術及び局部或いは転移性子宮頸癌を治療する化学療法或いは放射線治療を含む。したがって、医学的には依然としてHPV関連CINおよび/または子宮頸癌を有する女性を治療するための治療用ワクチンが必要である。
HPV治療用ワクチン候補は
われわれはHPV−16とHPV−18に対する二価HPVワクチンを開発している初期段階であり,両ウイルス株は約70%の子宮頸癌症例をきたしている。我々は各菌株に対するE 6とE 7遺伝子産物を計画しており,これはCIN段階から浸潤性子宮頸癌への進展を担当する主要な発癌蛋白である。臨床前研究では,われわれのHPV−16とHPV−18候補ワクチンが免疫原性を有することが証明された。具体的には,われわれにHPV−16またはHPV−18ワクチンを投与したマウスがHPVに対するT細胞の反応を誘導し,これをインターフェロンガンマELISPTにより測定した。また,われわれのHPV−16ワクチンは,ロバストなマウスHPV腫瘍モデルにおいて腫瘍成長抑制と生存率向上作用を示した。現在のCINや子宮頸癌を治療する方法と比較して,われわれのHPVワクチンにはいくつかの利点があると信じている。現在CINの治療選択は侵襲的であり、深刻な妊娠禁忌症を招く可能性がある。また,CINの外科的治療は潜在的HPVを治療するのではなく,感染組織を切除するものである。したがって,現在のCIN治療レジメンには著明な失敗率があり,子宮頸癌に進展するリスクが増加する可能性がある。これまで,われわれのワクチンは臨床被験者において良好な安全性と耐性を示してきた。現在、子宮頸癌の治療方案、例えば化学療法と放射線治療は多種の副作用があり、例えば脱毛、食欲不振と深刻な吐き気がある。
HPV−16に対するT細胞の応答は、形質転換HPV由来固形腫瘍を縮小させることができる
固形腫瘍増殖モデルにおいて、HPV−16に対するT細胞の反応が治療反応を産生する能力を試験した。TC−1細胞(HPV−16形質転換細胞系)をB 6マウスの後側皮下に注射し,数日間培養した後にワクチンまたは対照群でマウスを免疫した。研究1では,マウスはそれぞれ7,14,21日目に免疫した。第4群と第5群では,ワクチンはHPV−16抗原E 6/E 7(Ad−HPV)を発現した。第5群はワクチン接種と同時にチェックポイント阻害因子(PD−1抗体),第4群はチェックポイント阻害因子の同型対照(Iso)を用いた。第1群と第2群はAd−HPVと同様であるがHPV抗原(Ad−nr)を発現しない組換えRadベクターを用い,非特異的効果を対照した。未治療の動物は何のワクチンも接種しなかった。
研究の結果、Ad-HPV群は腫瘍の成長を阻止でき、甚だしきに至っては腫瘍を縮小できることが分かった。チェックポイント抑制器の使用の有無にかかわらず,このようなことが発生する.チェックポイント阻害剤だけでは腫瘍の発展を阻止することはできず,最終的にこれらの動物はすべて死亡した。他にヒトパピローマウイルスを担持していない対照動物も生存していない。Ad−HPVワクチンとともにチェックポイント阻害剤を用いた生存率はやや向上したが(10/10比9/10),有意ではなかった。
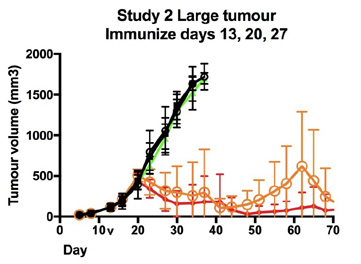
研究2では,TC−1腫瘍は以前のように移植されたが,免疫が発生する前により長く成長させた。免疫接種は13、20、27日目に発生した。この研究では,Ad−HPVワクチンやチェックポイント阻害剤を受けたマウスは,40日目までいくつかのマウスが死亡し始めた。80日目の実験終了時には70%以上の動物が生存していましたチェックポイント阻害剤がない場合,Ad−HPV免疫マウスでも60日以内(最終免疫後33日)に腫瘍をほぼコントロールすることができ,その後何匹かの動物が死亡した。Ad−HPVがない場合には,対照群はいずれの腫瘍もコントロールできず,すべてのマウスが40日目前に死亡した。
図19.小型腫瘍ワクチン研究。
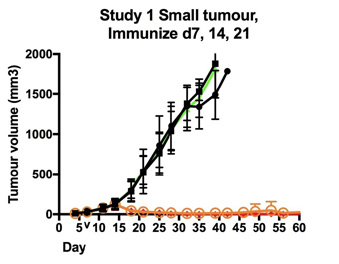 |
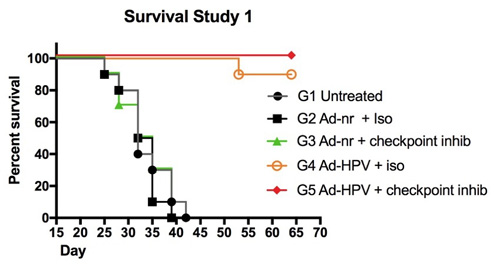 |
図19.小型腫瘍ワクチン研究(研究1)では,免疫計画を開始する前に腫瘍の成長が7日間許可されている。Vaxart HPVワクチン(Ad-HPV)を投与した動物は保護され、腫瘍の成長の影響を受けず、生存状況はもっと良い。チェックポイント抑制器の使用の有無にかかわらず、状況はそうである。
図20.大型腫瘍ワクチン研究。
 |
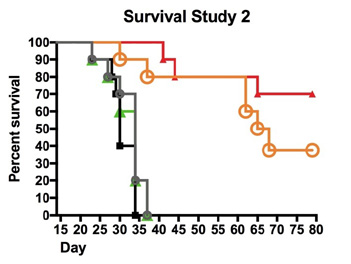 |
図20.大型腫瘍ワクチン研究(研究2)では,ワクチン投与前に腫瘍の成長が13日間許可されている。同様に、Ad-HPVを投与した動物は腫瘍成長に対してより良い保護がある。チェックポイント阻害剤の添加は生存率を向上させた。
腫瘍モデルでは,免疫後に誘導されたT細胞は固形腫瘍を輸送して癌細胞を攻撃·破壊すると考えられている。これは別の腫瘍モデル実験で検証されました腫瘍は従来のように移植し,13日目と21日目に免疫接種を行った。実験24日目に腫瘍を取り、フローサイトメトリーを用いて腫瘍内のT細胞の数量を計数した。HPV−16ワクチン群(チェックポイント阻害剤または同種型対照抗体を用いて)は,CD 4およびCD 8陽性T細胞のT細胞浸透を有する。Ad-HPV群のCD 8 T細胞数は対照群より明らかに良かった。CD 4 T細胞はAd-HPV+検査点群で対照群より明らかに良く、Ad-HPV+同型対照群で上昇傾向があった。
図21.Ad-HPVワクチンはT細胞の腫瘍への遊走を誘導する。
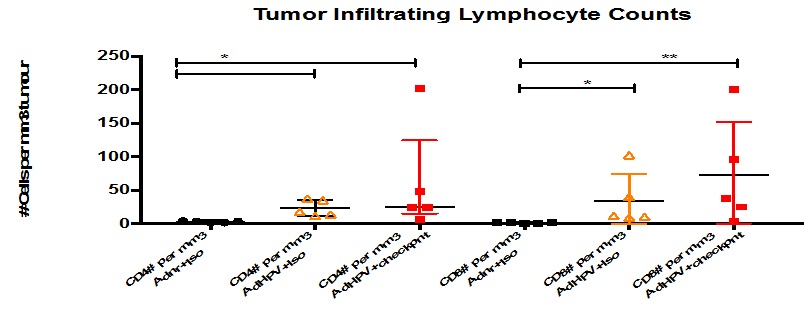
図21.腫瘍内に発見されたCD 4およびCD 8 T細胞の数をフローサイトメーターで分析した。研究により、Ad-HPVワクチン単独接種と比べ、Ad-HPV群はT細胞の腫瘍への転移を誘導できるが、Ad-HPVプラスチェックポイント阻害剤によるT細胞転移はやや多い。
最近のHPVワクチン発展戦略
臨床.臨床
候補HPVワクチンの臨床試験を継続するために規制書類を提出する必要がある。我々の臨床計画は、HPV-16またはHPV-18に関連する子宮頚異形成患者において候補ワクチンをテストし、候補ワクチンのHPV感染除去、子宮頚異型増殖スコアの低下、HPV除去に重要であることが知られているT細胞を誘導する能力を評価することである。T細胞はフローサイトメーターとインターフェロン−g−ELISPOTにより検出される。T細胞反応を検査することにより,主な終点は安全であり,副次的な終点は免疫原性である。臨床反応は追跡されるが、第一の研究は統計学的意義のある治療効果の読み取り値を得ることができない可能性が予想される。
他適応症
著者らは現在RSV、基底孔ケン雅、B型肝炎とHSV-2などの適応の動物モデルの初歩的なデータがある。
製造業
我々の経口錠剤ワクチンの製造は、主に2段階、原料薬の製造及び製剤及び錠剤(医薬品)を含む。生薬の生産は主に有効成分の生産と精製を含む。次に,我々が開発した特許配合と打錠プロセスを用いて,原料薬を凍結乾燥,調製し,打錠とコーティングを行った。
大口ワクチン生産(薬用物質)
最初から,第三者契約メーカーと内部施設を組み合わせることで,我々の錠剤ワクチン候補薬の臨床cGMP原料薬を生産してきた。2017年から私たちは、カリフォルニア州にある会社本社に小型cGMP量産工場を設立し、第1段階と小型第2段階のためにcGMP製品を試験生産することを目指して独自のバルクワクチン製造プロセスを投資開発しました。2021年11月に別のGMP製造工場を転貸することで拡張し、この工場を用いて内部同じ量産プロセスを実行します。2022年4月、我々は凍結乾燥技術会社とより大規模な薬物凍結乾燥協定を締結した。
ワクチン錠剤生産
最初から、第三者契約メーカーと契約して、私たちの薬品の製造、ラベル、包装、貯蔵、流通を担当しています。2016年、私たちは会社本部に薬品製造能力を設立しました。我々の工場はカリフォルニア州公共衛生部食品·薬物支部の許可を得て,臨床試験用の薬物製品を生産することができる。2022年7月、私たちはAttwillと、医薬品生産(錠剤プレスおよびコーティング)をさらに拡大し、カリフォルニア州に新しいGMP施設を設立し、私たちの候補ワクチンのプレス、コーティング、および包装のための協定に署名した。
CGMP法規と監督管理記録文書によると、著者らは技術開発及び薬物物質と薬物製品の製造、テスト、品質放出、貯蔵と流通方面の経験が限られている。CGMP条例には、人員、建物および施設、設備、構成要素および薬品容器および閉鎖的な制御、生産およびプロセス制御、包装およびラベル制御、保有および分配、実験室制御、記録および報告、ならびに製品の返品または回収に関する要件が含まれる。私たちの工場と私たちの第三者メーカーはFDAと地方当局の定期検査を受けています。これらの検査には、私たちが適用される法規に適合しているかどうかを評価するために、私たちの候補ワクチンをテストして製造する際に使用されるプログラムや操作が含まれています。もし私たちまたは私たちの第三者製造業者が法律と法規の要求を遵守できなかった場合、私たちと彼らは警告状、製品の差し押さえまたはリコール、禁止、製造業務に重大な制限または一時停止の同意法令、民事と刑事処罰を含む法律または規制行動に直面する可能性がある。このような行動は私たちの候補平板ワクチンの供給に実質的な悪影響を及ぼすかもしれない。契約メーカーと同様に、私たちは過去にも生産量、品質管理と品質保証の困難に直面したことがあり、もし私たちが十分な数量の薬物製品或いは薬物物質を生産できなければ、私たちは臨床試験を行い、私たちの候補錠剤ワクチンを商業化する能力は損害を受け、もし承認されたら。
研究と開発
正常な業務過程において、著者らは第三者と協議、例えば臨床研究機構、医療機関、臨床研究者と契約実験室を締結し、著者らの臨床試験及び著者らの研究と臨床前試験の各方面を行う。これらの第三者は、プロジェクト管理と監視サービス、規制コンサルティングと調査サービスを提供する。
競争
製薬とワクチン業界の特徴は、新技術と独自製品の開発競争が激しいことである。一般的に、薬品間の競争は、製品の効果、安全性、信頼性、獲得性、価格、特許地位に部分的に基づいている。
独自の平板ワクチン候補が競争優位を提供していると信じていますが、バイオテクノロジーや製薬会社を含む多くの異なる源からの競争に直面しており、学術機関、政府機関、公共および民間研究機関からの競争に直面している可能性もあります。私たちが商業化する可能性のあるどの製品も、既存の製品や療法、将来出現する可能性のある新製品や療法と競争しなければならない。
既存の治療法、ワクチンまたは送達方法を改善するために、またはその選択された適応のための新しいワクチン、療法または送達方法を開発するために、他の組織が取り組んでいる。これらの努力の成功度によると、承認されれば、候補ワクチンの採用と成功の障害を増加させる可能性がある。
新しいワクチンの市場進出と先進技術の出現に伴い、私たちは激しい競争と日々の競争に直面することを予想しています。我々が開発·商業化した任意の錠剤または他の経口送達ワクチン候補薬は、有効性、安全性、投与および送達の利便性、価格、治療薬の利用可能性、模倣薬競争レベル、および政府および他の第三者支払者が精算できるかどうかに基づいて競争することが予想される。
もし私たちの競争相手が私たちが開発する可能性のあるどの製品よりも安全で、より効果的で、副作用が少なく、より便利で、より安い製品を開発し、商業化すれば、私たちのビジネス機会は減少または消失するかもしれない。私たちの競争相手も候補ワクチンの承認を得るよりも早くFDAや他の規制機関の承認を得ることができ、これは私たちの競争相手が私たちが市場に入る前に強力な市場地位を確立することをもたらすかもしれない。また、多くの場合、私たちの競争能力は、後発薬の使用を奨励することを求める保険会社または他の第三者支払者の影響を受ける可能性がある。
我々は比較的小さな会社からの競争に直面しており,これらの会社は我々のように投資家に研究開発に資金を提供し,大手や老舗製薬会社と共同開発と許可を争う機会に依存している。協力の機会を求めたり、他社、金融投資家、企業から戦略的買収を行う上でも、激しい競争に直面する可能性があり、これらの会社の資本コストは私たちよりも低い可能性があります。私たちの市場では、将来のパートナーシップや資産買収機会の競争が非常に激しく、このような資産に支払う価格の引き上げを迫られる可能性があります。
私たちはまた、私たちが合格者を誘致し、維持し、特許保護を得ること、または他の方法で独自製品やプロセスを開発する能力、および私たちの製品を開発し商業化するのに十分な資本資源を確保することに依存します。
ノウォーカーウイルス候補ワクチン
現在、全世界で販売されているノウォーカーウイルスワクチンは承認されていない。HilleVax,Inc.はノーウォーカーウイルスワクチン(最初に武田社が開発した)を開発しており、注射で配信されると考えられています。ノウォーカーウイルス候補ワクチンを開発しているもう一つの会社は安徽智飛龍康生物製薬有限公司です。他にも知らない開発計画があるかもしれません。
新冠肺炎候補ワクチン
新冠肺炎ワクチン市場の競争は激しい。ファイザー-生物科学技術の新冠肺炎ワクチンとModernaの新冠肺炎ワクチンはすでにアメリカと世界の多くの国で承認された。ジョンソンの新冠肺炎ワクチンとアスリコンの新冠肺炎ワクチンはすでに全世界の多くの国で承認され、全世界に大部分の“西洋剤”を供給した。世界各国で新冠肺炎ワクチンの承認を得た他のワクチン会社には、セノフィ社とノワール社が含まれている。
季節性インフルエンザワクチン候補ワクチン
私たちの季節性インフルエンザ候補ワクチンは、注射または鼻腔投与による非組換えおよび組換え製品を含む市場で承認されたワクチンと直接競争しないと信じている。全世界の主要な非組換え注射ワクチンの競争相手はAstellas Pharma Inc.,アボット、アスリーカンイギリス有限会社、百特国際会社、大阪大学微生物疾患研究基金会、CSL Sequirus、グラクソ史克、サイノフィ、ファイザーと武田製薬有限会社を含む。非組換え鼻腔内競争にはMedImmune,Inc.,可能な他社がある。再注射可能な競争相手は、セノフィ社およびNovavax,Inc.を含む。多くの他のグループは、新しいまたは改善されたインフルエンザワクチンまたは送達方法を開発している。
候補HPV治療用ワクチン
現在まだ承認されていないHPV治療用ワクチンは全世界で販売されている;しかし、いくつかのワクチンメーカー、学術機関と他の組織は現在すでにこのようなワクチンを開発する計画がある。いくつかの会社はInovio製薬会社、Advaxis、Genexine、その他のいくつかの会社を含むHPV治療用ワクチンを開発する異なる段階にあると考えられる。
イナビル
他の抗インフルエンザ薬はタミフルやレラシャを含む日本でも販売されている。2018年2月23日、大阪に本社を置く製薬業者Shionogiは、日本でインフルエンザを治療する新薬Xofluzaの発売許可を得た。この薬剤はA型やB型インフルエンザウイルスに対する投与が許可されており,年齢にかかわらず単剤でよい。発売以来、Xofluzaは日本でInavirからかなりの市場シェアを獲得し、第1 3回の日本でのInavir販売数を大幅に減少させた。これは、私たちがDaiichi Sankyoから受け取った特許使用料支払いに大きな負の影響を与え、将来の特許使用料収入に大きなマイナス影響を与え続ける可能性がある。
知的財産権
私たちは、特許権を求め、維持し、守ることを含む、私たちの業務に重要なビジネス的意義を持つノウハウ、発明、改善の保護と強化に努めています。私たちはまた私たちのプラットフォームに関連する商業秘密と技術ノウハウに依存して、絶えず技術革新を行い、ワクチン分野における私たちの独自の地位を発展、強化し、維持している。また,データ独占性,市場独占性,特許期間延長(可能であれば)による規制保護に依存している。私たちはまた、私たちの会社名に商標保護を使用し、製品および/またはサービスが発売された時にそうすることを期待しています。
私たちのビジネス成功は、私たちの業務に関連する重要な商業技術、発明およびノウハウの特許および他の独自保護を取得して維持する能力があるかどうか、私たちの特許を擁護し実行する能力があるかどうか、私たちのビジネス秘密を秘密にし、第三者が効果的に強制的に実行可能な特許および独自の権利を侵害することなく運営されるだろう。私たちが第三者が私たちのタブレットワクチン候補ワクチンを製造、使用、販売、提供、または輸入することを阻止する能力は、これらの活動をカバーする有効かつ実行可能な特許または商業秘密に基づいて私たちが権利を持っている程度に依存するかもしれない。会社が持っている知的財産権については、私たちの任意の未解決特許出願または将来提出される可能性のある任意の特許出願が特許を得ることを保証することはできません。私たちはまた、私たちの既存のいかなる特許または将来私たちに付与される可能性のある任意の特許が、私たちの商業製品を保護し、これらの製品を製造する方法において商業的用途を持つことを保証することはできません。
私たちは大量の特許と特許出願を開発し、私たちのプラットフォームや錠剤候補ワクチンに関する多くの技術的ノウハウと商業秘密を持っている。
| ● |
ワクチンプラットフォーム技術それは.2022年12月31日現在、私たちは、私たちのプラットフォーム技術に関連するライセンス権利の3つの米国特許を持っています。そのうちの2つの米国特許は私たちの季節性インフルエンザ候補ワクチンに関連する権利主張を含む。これらの特許は2027年に満期になり、特許期限が延長された場合、より遅い時間に満期になる。2022年12月31日現在、私たちは、発行された50件以上の外国特許と、私たちのプラットフォーム技術および/または私たちの候補ワクチンに関連して出願されている外国特許を持っています。これらの特許は2027年に満期になり、特許期限が延長された場合、より遅い時間に満期になる。 |
| ● |
錠剤ワクチン配合それは.私たちはアメリカ、EU、日本、中国、シンガポール、オーストラリア、ロシア、イスラエル、韓国、南アフリカで相当な技術ノウハウと特許を持っています。私たちはアメリカや世界各地でも私たちの平板ワクチン調合技術に関する未定の申請をしている。これらの出願が発行する特許は2035/2036年に満了し、出願が特許期限を延長した場合、より遅い時間に満了する。 |
| ● |
新冠肺炎候補ワクチンそれは.2022年12月31日現在、私たちは米国で私たちの新冠肺炎候補ワクチンに関する臨時申請を提出した。これらの出願から発行されるいずれの特許も2041年に満了し、特許期限が延長された場合、より遅い時間に満了する。 |
| ● |
インフルエンザ、ノロウイルス、呼吸器合胞体ウイルス候補ワクチンそれは.2022年12月31日現在、私たちは南アフリカ、EU、そしていくつかの国で特許を持っており、米国と世界各地で私たちのノウォーカーウイルスおよびRSV候補ワクチンに関連する出願が行われている。これらの出願から発行されるいずれの特許も2036年に満了し、特許期限が延長された場合、より遅い時間に満了する。私たちは現在のH 1 N 1インフルエンザ候補ワクチンに関連する13件の外国特許を取得した。これらの特許は2030年に満期になり、特許期限が延長されれば、より遅い時間に満期になるだろう。 |
| ● |
リエンザそれは.2022年12月31日現在、私たちは、GSKによって独占的に発行され、2019年7月に満了したRelenzaに関連する最後の日本特許であるRelenza特許を所有していません |
| ● |
イナビルそれは.2022年12月31日現在、私たちはDaiichi Sankyoに独占的にライセンスされているInavir関連の日本特許を持っています。日本のInavir関連の最後の特許は2029年12月に満了し、印税収入が停止されます。しかし、ラニミウェシン酸エステル化合物をカバーする特許は2024年に満了し、後発薬競争が市場に参入する可能性があり、受信した特許使用料を減少または除去する可能性がある |
そのほか、著者らは臨床前の研究開発、製造と製造技術の拡大、品質管理、品質保証、監督管理事務及び臨床試験設計と実施などの領域で専門知識と開発能力を確立した。私たちは私たちの集中と専門知識が私たちの自主知的財産権に基づく製品を開発するのを助けると信じている。
個別特許の期限は特許を取得した国の法的期限に依存する。私たちが出願を提出したほとんどの国では,特許期間は非仮出願を提出した日から20年である。米国では、特許期間の延長は、特許期限調整によって延長することができ、これは、特許権者が特許付与時の特許付与時の行政遅延による特許権者の損失を補償することができ、または、1つの特許が以前に提出された特許によって最終的に放棄された場合、特許期間を短縮することができる。
FDAによって承認された薬物をカバーする特許期限も特許期間を延長する資格がある可能性があり、これにより、FDA規制審査中に失われた特許期限の補償として、米国特許の特許期限の回復が可能となる。ハッジ-ワックスマン法は特許期間を特許満了後最大5年間延長することを許可している。特許期間の延長の長さは,薬物が規制審査を受ける時間の長さと関係がある。1つの特許の残り期間を延長することは、製品承認日から合計14年間を超えることができず、承認された薬物に適用される特許を延長することしかできない。また、1つの特許は1回しか展示期間がないので、1つの特許が複数の製品に適用される場合は、1つの製品展示期間に基づくしかない。欧州や他の外国司法管区にも同様の規定があり、承認された薬物をカバーする特許の期限を延長する。可能であれば,臨床試験の長さや新薬出願(NDA)の提出に関わる他の要因に基づいて,我々の候補ワクチンとその使用方法をカバーする特許出願のために特許期間を延長する予定である。
商業秘密
場合によっては、私たちはビジネス秘密に依存して私たちの技術を保護する。しかし、商業秘密は保護するのが難しいかもしれない。私たちは、従業員、コンサルタント、科学コンサルタント、請負業者と秘密保持協定を締結することで、私たちのノウハウやプロセスを保護することを求めています。また、私たちは私たちのビルの実体安全と、私たちの情報科学技術システムの実体と電子安全を維持して、私たちの資料と商業秘密の完全性とセキュリティを維持することに力を入れています。私たちはこのような手続きに自信がありますが、合意や安全措置は違反される可能性があり、私たちはどんな違反にも対応する十分な救済措置がないかもしれません。しかも、私たちのビジネス秘密は競争相手に知られたり独立して発見されるかもしれない。私たちのコンサルタント、請負業者、または協力者が、私たちのために働いているときに他人が所有している知的財産権を使用する場合、関連するまたはそれによって生じるノウハウおよび発明の権利について論争が生じる可能性がある。
政府規制と製品審査
アメリカとその他の国の連邦、州と地方政府当局は生物と医薬製品の研究、開発、テスト、製造、品質管理、承認、ラベル、包装、貯蔵、記録保存、販売促進、広告、流通、承認後のモニタリングと報告、マーケティング及び輸出入などの方面に対して広範な監督管理を行った。私たちの候補ワクチンはFDAの承認を得なければならず,米国で合法的に販売され,適切な外国規制機関の承認を得てこそ,海外で合法的に販売することができる。一般的に、私たちの他の国での活動は、いくつかの点で異なるかもしれないが、米国と類似した性質と範囲の規制を受けるだろう。規制マーケティングの承認を得る過程とその後、適切な連邦、州、地方、外国の法律法規を遵守する過程には、多くの時間と財力が必要だ。我々の業務に適用される規制は変化する可能性があり,これらの変化が我々の業務に影響を与えるかどうか,我々の業務にどのように影響するか,あるいはいつ我々の業務に影響を与えるかを予見することは困難である.
アメリカ製品開発プロセス
米国では、FDAは連邦食品、薬物と化粧品法案、公衆衛生サービス法案或いはPHSA及びその実施条例に基づいて薬品と生物製品を規制している。製品はまた他の連邦、州、そして地方法律法規によって制限されている。規制の承認を得て、その後、適切な連邦、州、地方、外国の法規と条例を遵守する過程には、多くの時間と財政資源が必要だ。製品開発過程,承認過程又は承認後のいずれかの場合,出願人が適用される米国の要求を遵守できない場合には,行政又は司法制裁を受ける可能性がある。FDAの制裁には、承認保留申請の拒否、承認撤回、臨床一時停止、警告状、製品のリコールまたは市場からの撤回、製品の差し押さえ、生産または流通禁止の完全または部分的な一時停止、罰金、政府契約の拒否、原状回復、返還、返還、または民事または刑事罰が含まれる可能性がある。どんな機関や司法法執行行動も私たちに実質的な悪影響を及ぼすかもしれない。FDAが医薬品または生物製品が米国で発売される前に必要とされるプログラムは、一般に以下のような態様を含む
| ● |
非臨床実験室試験および動物研究は、GLPおよび適用された実験動物の人道使用要件または他の適用法規に基づいて達成される |
| ● |
ヒト臨床試験が開始される前に有効でなければならないINDをFDAに提出する |
| ● |
FDAが一般的に良好な臨床実践(GCP)と呼ばれる法規及び人体研究対象及びその健康情報を保護する任意の追加の要求に基づいて、十分かつ制御された人体臨床試験を実行して、提案された生物製品の期待用途に対する安全性と有効性を決定する |
| ● |
非臨床試験および臨床試験の結果に基づいて、BLAの標的となる製品の持続的な安全性、純度、効力を確保するために、適用要件に適合する生物製品ライセンス申請書(BLA)をFDAに提出する |
| ● |
施設、方法、および生物製品を維持するのに十分な特性、強度、品質、および純度を保証するために、cGMPの遵守状況を評価するために、バイオ製品を製造するためのFDAの1つまたは複数の製造施設の検査を満足的に完了させる |
| ● |
FDAは、BLAを支持するデータを生成する非臨床研究および臨床試験場所を監査することができる |
| ● |
FDAによるBLAの審査および承認、またはライセンス。 |
人体で任意の生物ワクチン候補ワクチンをテストする前に、著者らの平板ワクチン候補ワクチンを含み、ワクチン候補は臨床前試験段階に入る。臨床前試験は、非臨床研究とも呼ばれ、製品化学、毒性と調合に対する実験室評価、及び動物種に対する毒理学と薬理学研究を含み、候補ワクチンの潜在的安全性と活性を評価する。臨床前試験の実施はいくつかの動物研究のGLPと農業部によって実行される動物福祉法を含む連邦法規と要求に適合しなければならない。臨床試験スポンサーは臨床前試験の結果を生産情報、分析データ、任意の利用可能な臨床データ或いは文献、提案された臨床方案と共にFDAに提出し、INDの一部としなければならない。IND提出後も,いくつかの臨床前試験が継続される可能性がある。候補製品の安全性および有効性を評価するために米国で臨床試験を賛助する任意の個人またはエンティティは、そのような試験を開始する前にFDAにIND申請を提出しなければならず、この出願は、FDAが人体上で製品を試験するのに十分な基礎を提供していると結論する。INDはFDAが提案された臨床試験に対して懸念または問題を提起し、30日以内に臨床試験を一時停止しない限り、FDAが受信後30日以内に自動的に発効する。この場合,INDスポンサーやFDAは臨床試験開始前に未解決の問題を解決しなければならない。FDAはまた、臨床試験の前または期間のいつでも、安全考慮または規定に適合しない理由で、候補生物製品に臨床的制限を加えることができる。FDAが臨床一時停止を強制した場合、試験はFDA許可なしに再開されず、その後、FDA許可の条件下でのみ再開される可能性がある。したがって,INDの提出によりFDAが臨床試験の開始を許可するか,あるいは開始すると,このような試験を一時停止または終了するという問題はないとは判断できない。
臨床試験は,合格した調査者の監督の下で健康なボランティアや患者に候補生物製品を服用することに関連し,これらの調査者は通常,試験スポンサーに雇用されたりコントロールされていない医師である。臨床試験は,いくつかの有害事象発生時に臨床試験が停止されることを確保する停止ルールを含む,臨床試験の目標,投与手順,被験者選択と排除基準,および被験者の安全性を監視するためのパラメータを詳細に説明するプロトコルで行われる。各スキームおよびスキームの任意の修正は、INDの一部としてFDAに提出されなければならない。臨床試験は広範な監督管理を受けている。臨床試験はFDAの生物研究モニタリング法規とGCP要求を構成する法規に基づいて行わなければならず、すべての研究対象にインフォームドコンセントを提供することを含む。さらに、各臨床試験は、臨床試験を行う各機関に位置するか、またはサービスする独立した機関審査委員会またはIRBによって審査および承認されなければならない。IRBは試験参加者の福祉や権利の保障を担当し,臨床試験に参加する個人のリスクが最低に低下するかどうか,期待利益と比較して合理的かどうかなどの項目を考慮している。IRBはまた、各臨床試験対象またはその法律代表によって署名されなければならないインフォームドコンセントの形態および内容を承認し、完成まで臨床試験を監視しなければならない。
INDの下で行われる外国研究は,米国で行われている研究に適用されるのと同じ要求に適合しなければならない。しかしながら、外国の研究がINDに従って行われていない場合、研究がGCPに従って行われ、FDAがデータを検証することができる場合、データは依然として製品申請を支援するためにFDAに提出されることができる。
臨床試験のスポンサーまたはスポンサーが指定した責任者は、試験に関するいくつかの情報を登録し、Clinicaltrials.govのようないくつかの結果を政府または独立登録サイト上で開示することを要求される可能性がある。
人体臨床試験は通常3つの連続段階に分けて行われ、これらの段階は重複或いは合併する可能性がある
| ● |
ステップ1それは.このようなバイオ製品は、最初に少量の健康なヒト対象に導入され、その薬理および薬物動態学的作用の詳細なプロファイルを作成し、用量増加に関連する副作用を決定し、可能な場合に有効性の早期証拠を得るための安全性試験を行う。深刻または生命に危険な疾患を治療するいくつかの製品、特に製品自体の毒性が大きすぎて健康ボランティアに道徳的に服用できない場合、最初の人体テストは一般に被験者に行われる。 |
| ● |
第二段階それは.生物製品は限られた患者集団で評価を行い、可能な副作用と安全リスクを確定し、特定の目標疾患に対するこの製品の治療効果を初歩的に評価し、用量耐性、最適用量と用量計画を確定する。 |
| ● |
第3段階それは.臨床試験は地理的に分散した臨床試験地点で患者群を拡大し、投与量、臨床治療効果、効力と安全性を更に評価するためである。これらの臨床試験は、製品の全体的なリスクと利益の概況を決定し、製品ラベルに十分な基礎を提供することを目的としている。第3段階のデータは、通常、FDAが製品の応用を考慮する際に候補製品の安全性と有効性を評価する核心的な基礎を構成する。 |
承認後の臨床試験は,4期臨床試験と呼ばれることがあり,最初の上場承認後に行われる可能性がある。これらの臨床試験は,治療適応が予想される患者の治療から追加的な経験を得るためのものであり,特に長期安全なフォローアップのためである。
臨床開発のすべての段階において、監督管理機関はすべての臨床活動、臨床データと臨床試験調査人員に対して広範なモニタリングと監査を行うことを要求している。臨床試験結果を詳細に説明する年次進展報告はFDAに提出しなければならない。深刻かつ予期しない有害事象については、他の研究からの任意の発見、実験室動物試験または体外試験により、ヒト被験者に重大なリスクがあることを示す任意の発見、または任意の臨床上の深刻な疑わしい副作用の発生率は、方案または研究者マニュアルに記載されているよりも増加し、迅速にFDAと調査者に書面IND安全報告を提出しなければならない。スポンサーは15日以内にINDセキュリティ報告書を提出し,スポンサーがその情報有資格報告を確定した後でなければならない。スポンサーはまた、スポンサーが初めて情報を受け取ってから7日以内に、任意の意外、致命的、あるいは生命に危害を及ぼす疑いのある副作用をFDAに通知しなければならない。第1段階、第2段階、および第3段階の臨床試験は、もしあれば、任意の指定された時間で成功しない可能性がある。FDAまたはスポンサーまたはそのデータ安全監視委員会は、研究対象が受け入れられない健康リスクに直面していることを発見することを含む、様々な理由で臨床試験を一時停止または終了することができる。同様に、臨床試験がIRBの要求に従って行われない場合、または生物製品が対象に予期せぬ深刻な傷害を受けることに関連している場合、IRBは、その機関の臨床試験の承認を一時停止または終了することができる。
臨床試験と同時に、会社は通常追加の研究を完成しなければならず、生物製品の物理的特徴に関する追加情報を開発し、cGMP要求に基づいて商業大量生産製品のプロセスを最終的に決定しなければならない。PHSAは,生物製品を用いた外来製剤導入のリスク低減を支援するために,属性が正確に定義できない製品の製造制御の重要性を強調した。生産過程は一貫して高品質の候補製品ロットを生産することができなければならず、他の標準以外に、スポンサーは最終生物製品の特性、強度、品質、効力と純度をテストする方法を制定しなければならない。また,適切な包装を選択·試験し,候補生物製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
アメリカの審査と承認の流れ
生物製品の臨床試験が完了した後,生物製品の商業販売の前に,FDAによるBLAの承認を得なければならない。BLAには,製品開発,実験室と動物研究,人体試験の結果,製品製造と成分の情報,アドバイスのラベル,その他の関連情報が含まれていなければならない。FDAはデータの提出を延期することを承認するか、またはすべてまたは部分的な免除を与えるかもしれない。テストや承認過程には多大な時間と労力が必要であり,FDAがBLAの届出を受ける保証はなく,届出してもどの承認もタイムリーに承認される保証はない.
改正された処方薬使用料法案(PDUFA)によると、各BLAは相当な使用料を伴わなければならない。FDAは毎年PDUFAユーザ料金を調整する。PDUFAは生物製品に年間計画費も徴収している。場合によっては、小企業が初めて出願した出願料を免除することを含む、費用を免除または減免することができる。
出願提出後60日以内に、FDAは、機関が提出を受け入れる前に実質的に完了したかどうかを決定するために、提出されたBLAを審査する。FDAは、不完全であるか、または提出時に適切に審査できないと考えられる任意のBLAの提出を拒否することができ、より多くの情報の提供を要求することができる。この場合,BLAおよび付加情報を再提出しなければならない.再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。提出された申請が受け入れられると,FDAはBLAの深い実質的な審査を開始する。FDAの業績目標は、通常、提出後12ヶ月以内にBLAに対して行動することを規定している。場合によっては、FDAがより多くの情報を提供することを要求することを含む、この期限を延長することができる。FDAは、提案された製品が安全であるかどうか、有効であるかどうか、および/またはその予期される用途に有効であるかどうか、許容可能な純度プロファイルを有するかどうか、および製品がcGMPに従って製造されているかどうかを決定して、製品の特性、安全性、強度、品質、効力および純度を確保および保存するためにBLAを審査する。FDAは、新規な生物製品または安全性または有効性の問題を提起する生物製品の申請を諮問委員会に提出することができ、一般に、申請を承認すべきかどうか、およびどのような条件下で承認すべきかを審査、評価および提案するための臨床医および他の専門家を含むグループである。FDAは諮問委員会の提案に制限されていないが、それは決定を下す時にこれらの提案を慎重に考慮するだろう。バイオ製品の承認過程において、FDAはまた、バイオ製品の安全な使用を確保するために、リスク評価および緩和戦略、またはREMSを策定する必要があるかどうかを決定する。FDAがREMSが必要であると結論した場合,BLAのスポンサーは提案したREMSを提出しなければならない。必要であれば、FDAはREMSのないBLAを承認しないだろう。
BLAを承認する前に、FDAはこの製品を生産する施設を検査する。FDAは、製造プロセスおよび施設がcGMP要件に適合していることを決定し、要求された仕様の下で製品が一貫して生産されることを保証するのに十分でない限り、この製品を承認しないであろう。さらに、BLAを承認する前に、FDAは通常、IND試験要求およびGCP要求に従って臨床試験が行われることを確実にするために、1つまたは複数の臨床場所を検査する。CGMPとGCPに適合することを確保するためには,申請者は訓練,記録保存,生産,品質管理に多大な時間,お金,労力をかけなければならない。
関連データおよび情報が提出されたにもかかわらず、FDAは、BLAがその承認の規制基準を満たしていないことを最終的に決定し、承認を拒否する可能性がある。臨床試験から得られたデータはつねに決定的ではなく,FDAのデータ解釈は我々の同じデータに対する解釈とは異なる可能性がある。FDAが現在の形態のBLAを承認しないと決定した場合、FDAは、FDAによって決定されたBLA内のすべての特定の欠陥を記述する完全な返信を発行するであろう。決定された欠陥は微小である可能性があり、例えば、ラベル変更が必要であるか、または重大であり、例えば、追加の臨床試験が必要である。さらに、完全な返信状は、出願人がとり得る、申請を承認条件に置くための提案行動を含むことができる。完全な返信はまた、FDAが出願を再検討するために、追加の臨床前または臨床データを含む追加の情報を提供することを要求する可能性がある。完全な返信が発行された場合、出願人は、BLAを再提出し、手紙で決定されたすべての不足点を解決するか、または出願を撤回することができる。
1つの製品が規制部門の承認を得た場合、この承認は、特定の疾患および用量に明らかに限定される可能性があり、または使用の適応が制限される可能性があり、これは、製品の商業的価値を制限する可能性がある。
さらに、FDAは、いくつかの禁忌症、警告、または予防措置を製品ラベルに含めることを要求する可能性がある。FDAは、リスク管理計画の形態で製品流通、処方または調剤に制限および条件を適用することができ、または他の方法で任意の承認範囲を制限することができる。そのほか、FDAは発売後の臨床試験を要求する可能性があり、時々第四段階の臨床試験と呼ばれ、生物製品の安全性と有効性を更に評価することを目的とし、そして商業化された承認製品の安全性を監視するためにテストと監督計画を要求する。
さらに、“小児科研究公平法”によれば、BLAまたは補足BLAは、すべての関連する小児科亜群において主張される適応の安全性および有効性を評価し、各安全で有効な小児科亜群に対する製品の用量および投与をサポートするためのデータを含まなければならない。FDAはデータの提出を延期することを許可するか、またはすべてまたは部分的な免除を与えることができる。
承認を得るには数年かかる可能性があり、大量の資源が必要であり、標的疾患或いは状況の深刻さ、代替治療の獲得性及び臨床試験で示されたリスクと利点を含む多くの要素に依存する。
承認後に要求する
FDAによって承認されたどの製品も、記録保存要件、製品副作用報告、FDAへの最新の安全および治療効果情報の提供、製品サンプルおよび流通要件、およびFDAの宣伝および広告要件の遵守を含むFDAの持続的な規制を受けなければならない。これらの要件は、消費者向けの広告基準を含むが、患者集団において製品承認用途に記載されていない製品(“非ラベル”使用と呼ばれる)の普及を制限すること、業界支援の科学的および教育活動の制限、およびインターネットに関連する販売促進活動の要求を含むが、これらに限定されない。医師が合法的な製品を非ラベル用途のために処方する可能性があるが、医師が彼らの専門的な医学的判断が適切であると思っている場合、メーカーはこのようなラベル外用途をマーケティングしたり、普及させたりしない可能性がある。持続的な規制要求を満たしていない場合、あるいは製品が市場に入った後に安全問題が発生した場合、FDAは制限、一時停止、さらには承認を撤回することを含む製品発売の条件を変更する行動をとる可能性がある。
また、品質管理及び製造プロセスは、製品の長期安定性を確保するために、承認された後も適用される製造要件に適合し続けなければならない。CGMP条例では,他の事項のほかに,品質管理と品質保証およびそれに応じた記録やファイルの保守が要求され,cGMPから外れた状況を調査·是正する義務がある.承認製品の製造·流通に参加するメーカーおよび他のエンティティは、FDAおよびある州機関にその工場を登録し、cGMPおよび他の法律を遵守することを確実にするために、FDAおよび特定の州機関の定期的な抜き打ち検査を受けなければならない。そのため、メーカーはcGMPコンプライアンスを維持するために、生産と品質管理に時間、お金、労力をかけ続けなければならない。承認後に製品が発見された問題は、製品のリコールまたは市場からの撤回を含む製品、製造業者、または承認されたBLA所有者の制限をもたらす可能性がある。また,製造プロセスの変更は厳しく規制されており,変更の重要性に応じてFDAの承認を得て実施する必要がある可能性がある。新たな適応や声明を増やすなど、製品の他のタイプの変化を承認するには、FDAのさらなる審査と承認も必要である。
製品が以前に未知の問題を発見し、あるいは適用されたFDA要求を遵守できなかったことは、負の結果をもたらす可能性があり、負の宣伝、司法或いは行政法執行、FDAの警告状、強制要求の是正広告或いは医師とのコミュニケーション、民事或いは刑事罰などを含む。新たに発見または開発された安全性または有効性データは、新たな警告や禁忌症を増加させることを含む製品承認のラベルを変更する必要がある場合があり、他のリスク管理措置を実施する必要がある可能性もある。さらに、新しい立法によって生成された要求を含む新しい政府要求が確立される可能性があり、またはFDAの政策が変更される可能性があり、これは、規制部門が私たちが開発している錠剤候補ワクチンの承認を延期または阻止する可能性がある。
他のアメリカの医療保険法やコンプライアンスの要求は
米国では、FDAに加えて、私たちの活動は、医療保険や医療補助サービスセンター(CMS)、監察長事務室、米国司法省(DoJ)、司法省内の個別連邦検事室、州や地方政府など、様々な連邦、州、地方当局によって規制されている可能性があるが、これらに限定されない。例えば、販売、マーケティング、科学/教育補助計画は、“社会保障法”、“虚偽申告法”、“医師支払い透明性法”、“健康保険携帯性·責任法”(HIPAA)の反詐欺·乱用条項、改正された“健康情報技術·臨床健康法案”(HITECH)のプライバシー·安全条項、および同様の州法を遵守しなければならない。
他の事項に加えて、連邦反リベート法規は、任意の個人またはエンティティが、購入、レンタル、注文または購入の手配、レンタルまたは購入の手配、レンタルまたは注文として、Medicare、Medicaid、または他の連邦医療計画に従って精算可能な任意の物品またはサービスの見返りとして、故意に、または故意に現金または実物で直接または間接的に、公開または隠蔽的に提供、支払い、請求または任意の報酬を受けることを禁止する。報酬という単語は価値のあるものを含むと広く解釈されている。逆リベート法規は、一方では、医薬品メーカーと処方者、購入者、および処方管理者との間の配置に適用されると解釈される。いくつかの法的例外と規制避難所がいくつかの一般的な活動を保護することは起訴されない。例外や安全港の範囲は狭く,処方,購入または推奨の報酬を誘導するために告発される可能性があるやり方に関連しており,例外や安全港の資格を満たさなければ審査される可能性がある。私たちの接近はすべての場合、法定例外や安全港保護を規制するすべての基準を満たしていないかもしれない。しかし、特定の適用された法定例外または安全港を規制するすべての要求を満たすことができず、“反リベート法規”に基づいて、このような行為自体が不法であることを意味するわけではない。代わりに、そのすべての事実と状況の累積審査に基づいて、この手配の合法性を逐案的に評価する。
また、“平価医療法”は“反リベート条例”下の意図基準を修正し、より厳格な基準に達し、個人或いは実体が実際に法規を理解する必要がなく、或いは法規違反の具体的な意図があれば違反行為を実施できるようにした。また,“平価医療法案”は連邦“虚偽クレーム法案”(以下“FCA”)について,連邦の“反リベート法規”違反による物品やサービスを含むクレームが虚偽や詐欺的クレームを構成する判例法を編纂している。
民事罰金法規は、任意の個人またはエンティティに処罰を加え、その個人またはエンティティは、連邦健康計画へのクレームを出したか、または結果として判断され、そのクレームがクレームに従って提供されていないプロジェクトまたはサービス、または虚偽または詐欺的であることを知っているか、または知るべきである。
他の事項に加えて、連邦FCAは、任意の個人または実体が虚偽支払いまたは承認をもたらす虚偽請求を意図的に連邦政府に提出するか、または虚偽請求材料を作成、使用、または作成または使用して、連邦政府に虚偽または詐欺的クレーム材料を提供することを禁止する。2009年の“詐欺法執行·回収法案”の改正により、クレームには、米国政府に提出された金銭又は財産に対する“任意の請求又は要求”が含まれている。最近,いくつかの製薬や他のヘルスケア会社がこれらの法律により起訴されており,顧客に無料製品を提供し,顧客がその製品の連邦計画に課金することを期待しているといわれている。他社も起訴され、これらの会社は未承認の用途に製品を販売し、虚偽の声明を提出したため、精算できなかった。
HIPAAは、計画を故意に実行または実行しようとすることを禁止し、虚偽または詐欺的な言い訳、陳述または承諾の方法で任意の医療福祉計画(個人第三者支払者を含む)が所有または制御または保管している任意の金銭または財産を詐欺または取得し、トリック、計画または装置、重大な事実、または任意の重大な虚偽、架空または詐欺的陳述によって、医療福祉、プロジェクトまたはサービスの提供または支払いに関連する情報を故意に偽造、隠蔽、または隠蔽することを禁止する新しい連邦刑法を制定する。連邦反リベート法規と同様に、個人または実体は、この法規やこの法規に違反する具体的な意図を実際に知る必要がなく、違反を実施することができる。
また、多くの州で同様の詐欺や法律や法規の乱用があり、医療補助や他の州が計画して精算するプロジェクトやサービスに適用されたり、いくつかの州では支払者にかかわらず適用されている。
私たちは連邦政府と私たちが業務を展開している州のデータプライバシーと安全規制によって制限されるかもしれない。HIPAAはHITECH法案の改正後、個人が識別できる健康情報のプライバシー、安全と伝送に対して要求を提出した。他の事項に加えて、HITECHは、HIPAAのプライバシーおよびセキュリティ基準を、カバーエンティティが保護された健康情報を受信または取得する商業パートナー、独立請負業者、またはエージェントに直接適用するようにする。HITECHはまた4つの新しい民事罰金等級を作成し、HIPAAを改訂し、民事と刑事処罰を商業パートナーに直接適用し、州総検察長に新しい権力を与え、連邦裁判所に民事訴訟を提起し、損害賠償または禁止令を要求して連邦HIPAA法を執行し、連邦民事訴訟に関連する弁護士費と費用を求めることができる。また,州法は特定の場合に健康情報のプライバシーやセキュリティを管理しており,その多くの法律は互いに大きく異なり,コンプライアンス作業を複雑化させている。
また、“合理的医療費用法”に規定されている“連邦医師支払陽法”及びその実施条例によると、連邦医療保険、医療補助又は児童健康保険計画に基づいて支払われる特定の薬品、器械、生物及び医療用品の製造業者は、ある例外的な場合を除いて、医師及び教育病院、又は医師及び教育病院に要求すべきか、又はその指定された実体又は個人を代表して行われる何らかの支払い又はその他の価値移転に関する情報を報告し、医師及びその直系親族が保有するある所有権及び投資権益を毎年報告する。必要な情報をタイムリーかつ正確かつ完全に提出できなかったことは、年間150,000ドルの民事罰金と、“失敗を承知で”年間100万ドルに達する民事罰金を招く可能性がある。いくつかの州はまた、医薬品製造業者のマーケティング行為に制限を加え、および/または医療提供者およびエンティティに提供されるプレゼント、補償、および他の報酬を追跡および報告することを要求するコンプライアンス計画を強制的に実施する。
製品を商業的に流通させるためには、州の薬品および生物製品の製造業者および卸売業者に登録を要求する州の法律を遵守しなければならない。これらのメーカーまたはディーラーが州に営業場所がなくても、いくつかの州で製品を州のメーカーおよびディーラーに輸送することを含む。一部の州はまた、メーカーと流通業者が流通チェーン中で製品の系統を確立することを要求しており、いくつかの州はメーカーと他の州に流通チェーン中の製品の流れを追跡し追跡できる新しい技術を採用することを要求している。いくつかの州はすでに立法を公布し、製薬と生物技術会社にマーケティングコンプライアンス計画を確立し、州政府に定期報告を提出し、販売、マーケティング、定価、臨床試験およびその他の活動を定期的に公開し、および/またはその販売代表を登録し、薬局および他の医療保健実体が製薬および生物技術会社にいくつかの医師処方データを販売およびマーケティングのために提供することを禁止し、いくつかの他の販売およびマーケティング行為を禁止する。私たちのすべての活動は連邦と州消費者保護と不正競争法によって制限されるかもしれない。
もし私たちの運営が上記のいずれかの連邦および州医療保健法律または私たちに適用される任意の他の政府法規に違反していることが発見された場合、私たちは、MedicareとMedicaid、禁止、個人通報者が政府の名義で提起した個人訴訟、または政府契約、契約損害、名声損害、名声損害、行政負担、利益減少と将来の収入減少、および私たちの業務の削減と再編を含む、民事、刑事および/または行政処罰、損害賠償、罰金、返還、MedicareおよびMedicaid、禁止、個人通報者が政府名義で提起された個人訴訟を含むが、処罰を受ける可能性がある。いずれも私たちの業務運営能力と運営結果に悪影響を及ぼす可能性があります。
保証範囲·定価·精算
著者らが監督部門の許可を得た任意の錠剤ワクチン候補ワクチンのカバー範囲と精算状態に対して、重大な不確定性が存在する。米国や他の国·地域の市場では、規制機関の承認を得て商業販売を行う任意の製品の販売は、第三者支払者が保険を提供する程度にある程度依存し、そのような製品のための十分な補償レベルを確立する。米国では,第三者支払者には連邦や州医療計画,個人管理のヘルスケア提供者,医療保険会社,その他の組織が含まれている。第三者支払者が製品に保険を提供するかどうかを決定するプロセスは、製品価格を決定するプロセス、または第三者支払者が製品のために支払うべき支払率を決定するプロセスから分離することができる。第三者支払者は、承認リスト上の特定の製品に保証範囲を制限することができ、処方表とも呼ばれ、FDAによって承認された特定の適応のすべての製品を含まない可能性がある。第三者決済者は、価格に挑戦し、医療の必要性を審査し、医療製品、療法、サービスの費用対効果を審査するとともに、それらの安全性と有効性を疑問視するようになっている。私たちは、私たちの錠剤ワクチン候補の医療必要性と費用効果、およびFDA承認を得るのに必要なコストを証明するために、高価な薬物経済学的研究を行う必要があるかもしれない。私たちの候補錠剤ワクチンは医学的に必要あるいは費用効果があると思われないかもしれない。支払者が製品に保険を提供することを決定することは、十分な返済率を承認することを意味するものではない。また、支払者が1つの商品に保険を提供することを決定し、他の支払者もその商品に保険を提供することを保証することはできない。製品開発への投資の適切な見返りを実現するために、十分な価格レベルを維持することができる十分な第三者精算がないかもしれない。
他の国もまた違う価格設定と精算プログラムを持っている。いくつかの法域はプラスリストとネガティブリスト制度を実行し、補償価格を合意した後にのみ、製品を販売することができる。精算または定価の承認を得るために、その中のいくつかの国は臨床試験の完成を要求する可能性があり、特定の候補製品のコスト効果を現在利用可能な治療法と比較する。他の国は会社が自分の薬品価格を固定することを許可しているが、会社の利益を監視する。医療コストの下振れ圧力は非常に大きくなっている。そのため、新製品の参入にはますます高い壁が設けられている。また、一部の国では、低価格市場からの国境を越えた輸入が一国国内の定価に商業圧力をかけている。
政府や第三者支払者が十分な保険や精算を提供できない場合、商業販売のための錠剤ワクチン候補薬物の適切性が影響を受ける可能性がある。また,米国では管理型医療への重視度が向上しており,医療定価の圧力が増加し続けることが予想される。保証政策と第三者精算料率は随時変化する可能性がある。規制部門の承認を得た1つまたは複数の製品が有利な引受·精算状態を獲得しても、将来的にはあまり有利ではない引受政策や精算料率が実施される可能性がある。
アメリカの医療改革
現在と将来の米国の立法医療改革は、私たちが受け取った任意の承認された製品の価格を追加の下ぶれ圧力に直面させる可能性があり、カバーすれば、私たちの業務を深刻に損なう可能性があると予想しています。医療保険や他の政府が計画している精算のどの減少も、個人支払者の支払いのような減少を招く可能性がある。コスト抑制措置や他の医療改革を実施することは、私たちが収入を創出し、利益を達成することを阻止し、あるいは私たちの平板ワクチン候補製品を商業化することを阻止するかもしれない。さらに、さらなる立法や規制がある可能性があり、私たちの業務、財務状況、運営結果を損なう可能性があります。
外国監督管理
アメリカ国外で任意の製品をマーケティングするために、私たちは他の国と司法管轄区域の品質、安全性と有効性に関する多くの異なる監督管理要求、及び私たちの製品の臨床試験、マーケティング許可、商業販売と流通などの方面の監督管理要求を遵守する必要がある。私たちの製品がFDAの承認を得たかどうかにかかわらず、外国の監督管理機関のような必要な承認を得て、外国や司法管轄区でその製品の臨床試験やマーケティングを開始する必要があります。米国に関する上記の多くの問題は欧州連合にも同様に適用されるにもかかわらず、承認手続きは国および司法管轄区域によって異なり、追加の製品テストおよび追加の行政審査期限が関連する可能性がある。他の国や管轄地域で承認を得るのに要する時間は、FDAの承認を得るのに要する時間とは異なり、さらに長くなる可能性がある。1つの国または管轄区域で規制承認を得ることは、他の国または管轄区域で規制承認を得ることを保証することはできないが、1つの国または管轄区域で規制承認を得ることができなかったか、または遅延して監督管理許可を得ることは、他の国または司法管轄区の規制手続きに悪影響を及ぼす可能性がある。
情報のプライバシーとセキュリティ
私たちはいくつかのタイプの個人データの使用、安全、開示を規制する連邦と州の法律法規の制約を受けている。その中には1996年の“健康保険携帯性と責任法案”(HIPAA)の許可によって公布された連邦法規が含まれており、この法規は私たちが個人の健康情報に一定の保護を提供することを要求している。HIPAAプライバシーと安全法規は、保護された健康情報(“PHI”)の使用と開示を広く規制し、カバーするエンティティ(ヘルスケア提供者および健康計画を含む)およびその業務パートナーに、このような情報の安全を保護するために、行政、物理および技術保障措置を実施し、維持することを要求する。他のセキュリティ要求は電子PHIに適用される.このような規制はまた個人にその健康情報に関する実質的な権利を提供する。
HIPAAプライバシー·セキュリティ規制は、私たちの保証実体顧客と私たちの下請け業者(ビジネスパートナーとも呼ばれる)と書面協定を締結し、PHIを開示することも求められています。ビジネスパートナーがエンティティをカバーするエージェントであることが発見された場合,カバーエンティティはビジネスパートナーがHIPAAに違反したことで処罰される可能性がある.HIPAAの何らかのプライバシーやセキュリティ規定により,ビジネスパートナーも直接責任を負い,そのエージェントの違反行為に責任を負うことが発見される可能性がある.
HIPAAは保証実体が不合理に遅延したが、違反行為が発見されてから60日以内に影響を受けた個人の安全保障のないPHI違反を通知することを要求した。業務パートナーが保証エンティティのエージェントである場合、その保証エンティティは、業務パートナーが違反を発見した時間に基づいて、必要な通知を個人に提供しなければならない。また、衛生·公衆サービス部民権事務室(“OCR”)に報告しなければならず、1つの州または司法管区500人以上の住民に関連する安全な公衆衛生施設の違反については、メディアにも報告しなければならない。保証実体または事業パートナーが公衆衛生施設が被害を受ける可能性が低いと判断しない限り、安全でない公衆衛生施設の使用または開示は違反と推定される。様々な州の法律法規は、情報が漏洩する可能性を考慮することなく、個人情報に関するデータ漏洩事件において、影響を受けた個人や州規制機関に通知することも要求される可能性がある。州法と標準的なやり方では通常規定されているデータ漏洩報告時間はHIPAA要求よりも短い。
HIPAAプライバシーやセキュリティ条例に違反すると刑事罰を招く可能性があり、違反のたびに実質的な民事罰を受ける。民事処罰は毎年消費者物価指数の更新に応じて調整される。OCRは、違反苦情および報告に応答するために、コンプライアンス監査を実行し、HIPAAコンプライアンスを調査することを要求される。OCRの法執行を除いて、州総検察長は民事訴訟を提起し、禁止または損害賠償を求め、HIPAAのプライバシーや安全法規に違反する行為に対応する権利があり、これらの行為は州住民のプライバシーを脅かす。OCRは、例えば、保証エンティティが是正行動計画を実施することを許可するなど、HIPAA違反を非公式的に解決することができるが、OCRは、直接行動して罰金を科す権利があり、故意の不注意による違反行為を処罰することが要求される。私たちが調査の対象にならないことは保証できません(報告可能な違反事件、監査、または他の原因によって引き起こされます)、私たちはPHIを維持する際にHIPAA規定に適合していないと告発します。
連邦貿易委員会法案(FTCA)はまた、連邦貿易委員会が個人情報を保護するルールや法規を実行することを許可し、データが不公平または詐欺的な行為ややり方を構成する場合には法執行行動をとる。連邦貿易委員会は定期的に法執行行動をとり、医療や他の医療情報の保護を法執行優先事項とすることを強調している。米国最高裁のドーブス判決と特定の医療保健手続きを犯罪とする可能性のある州法の後、伝統的に健康関連とは考えられない個人情報タイプは現在、医療決定に関する個人情報を漏洩し、政府のデータプライバシーに対する審査を強化する可能性がある。
HIPAAとFTCAのほか、多くの州と連邦法律法規は、医療情報の収集、伝播、使用、プライバシー、機密性、安全性、可用性、完全性、作成、受信、送信、ストレージ、およびその他の処理を管理している。医療、雇用、消費者、そして他の個人情報。プライバシーやデータセキュリティ法規は州によって異なり,これらの法律法規はFTCAやHIPAAやその実施規則よりも限定的な場合が多く,通常先制されることはない.これらの法律および法規は、個人情報の使用、販売、および共有を規範化し、そのデータに関する個人に実質的な権利を付与し、データ処理者は、これらの権利を尊重する義務があり、厳格な情報セキュリティおよびデータ漏洩義務を適用し、場合によっては、政府の法執行によって生じるコストおよび責任に加えて、違法行為の疑いのある影響を受けた個人に個人訴訟権利を提供する
私たちはまた、従業員データの収集、送信、記憶、および使用に関連する国際データ保護法規の制約を受ける可能性がある。例えば、2018年5月25日に施行された“一般資料保護規則”(以下、“一般資料保護規則例”と呼ぶ)は、個人資料の収集、使用、保留、警備、処理、移転及び削除に厳格なコンプライアンス義務を加え、健康及びその他の敏感な資料の処理に関する通知、場合によっては資料に関する個人の同意を得ること、個人に資料処理活動に関する資料を提供すること、個人資料の安全及び秘密を保障するための保障措置を実施し、資料違反事項について通知すること、及び個人資料に接触可能な第三者プロセッサを採用する際に何らかの措置をとることを含む、2018年5月25日に施行される“一般資料保護規則”(以下、“一般資料保護規則例”と呼ぶ)である。GDPRは,欧州経済圏以外の国への個人データの移転に対しても,米国やイギリスを含む厳しいルールを実施している。GDPR要求を守らないエンティティは、2000万ユーロまたは世界の年収4%に達する可能性のある罰金を含む非常に深刻な処罰を受ける可能性がある。
政府の監督管理機関、プライバシー擁護者、集団訴訟弁護士も、会社が他のタイプの個人データをどのように収集、処理、使用、保存、共有、転送するかを検討している。例えば、2020年1月1日に施行される2018年カリフォルニア消費者プライバシー法(CCPA)と、2023年1月1日に全面的に施行されるカリフォルニアプライバシー権法案(CPRA)は、カリフォルニア州の任意の家庭や個人に関する情報を識別または特定するために広く適用され、個人の個人情報に関する要求に応答する流れを含むいくつかの操作変化を実施することが求められている。CPRAはまた、CCPAおよびCPRAを実行するための新しい実行機関を設立し、プライバシーリスク評価、監査、およびデータ共有、ライセンス、およびアクセス手配のサプライヤー契約要件を含む他の要求を提出した。CCPAとCPRAは違反行為に対する民事処罰を規定し、データ漏洩行為に対する個人訴訟を許可している。バージニア州、コロラド州、コネチカット州、ユタ州でも全面的なプライバシー立法が採択され、2023年に施行され、他のいくつかの州や連邦議員も追加の消費者プライバシー立法を提出した。プライバシー、データ保護、情報セキュリティに関する複雑で動的な法律環境は、私たちに重大なコンプライアンス挑戦をもたらし、潜在的に私たちがデータを収集、使用、開示する能力を制限し、私たちを追加的な費用に直面させ、もし私たちが適用された法律を適時または根本的に遵守できなければ、また不良な宣伝が生じ、私たちの名声を傷つけ、責任を負うことになる。
従業員と人的資本
著者らの管理と科学チームはワクチンと抗感染研究、臨床開発と監督管理事務の面で豊富な経験を持っている。我々の研究開発チームには,粘膜免疫学,T細胞,ウイルスベクター,ウイルス学において専門的な知識を持つ博士レベルの科学者が含まれている。一般的かつ行政には、財務、人的資源、行政、商業、および一般管理が含まれる。2022年12月31日までの1年間、私たちの従業員は次のように転転した(実習生を含まない)
| 研究と開発 |
一般と行政 |
合計して |
||||||||||
| 2021年12月31日 |
90 | 20 | 110 | |||||||||
| 合流する |
82 | 16 | 98 | |||||||||
| 終了しました |
35 | 9 | 44 | |||||||||
| 2022年12月31日 |
137 | 27 | 164 | |||||||||
2022年12月31日現在、私たちは13人のフルタイム同等請負業者も持っている。私たちは従業員たちと集団交渉合意に到達しなかったし、何の中断も経験しなかった。私たちは私たちが従業員と仲がいいと思う。
私たちの人的資本目標には、私たちの既存の従業員と新入社員を識別、採用、訓練、維持、奨励することが含まれている。私たちは株式報酬奨励を与えることで従業員を吸引、維持、奨励することを主な目的としており、これらの従業員を激励してできる限りのことをし、私たちの目標を実現することで、株主価値と会社の成功を増加させることを目的としている。2022年第4四半期、私たちは従業員の株式購入計画を提供し始めました。その主な目的は、従業員がVaxartの株式を獲得することを助け、これらの従業員が彼らの未来の安全保障を提供し、Vaxartの継続的なVaxart滞在を奨励することです。*人材を誘致し、維持するために、私たちはVaxartを安全でリターンのある職場にするために努力し、私たちの従業員は彼らのキャリアの中で成長し、発展し、競争力のある報酬、福祉、健康計画の支持を得て、従業員間に連絡を結ぶ計画を通じて支持することができます。
また,新冠肺炎の流行により,従業員の健康と安全を守るための措置がとられており,全体的にカリフォルニア州や関連自治体の指令や疾病管理センターの指導に一致した在宅勤務政策がとられている。現場活動はある必要な施設と実験室支援機能に限られ、各種の安全規程を実行した。
項目1 A:リスク要因
以下のリスク要因と、当社の財務諸表や関連説明や“経営陣の財務状況や経営結果の検討·分析”や、当社の他の公開文書を含む本年度報告書の他の情報を慎重に考慮すべきです。以下のいずれのリスクが発生しても、私たちの業務、財務状況、運営結果、および/または成長の見通しを損なう可能性があり、または、当社の実際の結果は、本年度報告書で行われた展望的陳述および私たちが時々行う可能性のある展望的陳述に含まれる結果とは大きく異なる可能性がある。私たちの業務を評価する時、あなたは説明されたすべての危険要素を考慮しなければならない。
私たちは急速に変化する環境で運営されており、その中には多くのリスクが含まれており、その中のいくつかのリスクは私たちがコントロールできない。この討論は未来の経営業績に影響を及ぼす可能性のあるいくつかのリスクを強調する。これらは私たちが最も考慮しなければならないと思う危険と不確実性だ。私たちは私たちがこのような危険に成功的に対応するということを確信できない。これらのリスクに対応できなければ、私たちの業務は成長できないかもしれません。私たちの株価は影響を受けるかもしれません。私たちは経営を続けることができないかもしれません。私たちは現在、どうでもいい追加のリスクや不確定要因、あるいは私たちの業界や業務の他の会社が直面しているようなリスクや不確定要因と、私たちの業務運営に影響を与える可能性があると考えています.
リスク要因の概要
私たちの業務は多くの危険と不確実性に直面しているもっと詳しく議論した次の節です。他にも、これらのリスクには以下の主なリスクが含まれている
私たちの業務、財務状況、資本要求に関連するリスク
| ● |
私たちの運営の歴史を見ると、私たちが作った製品の収入は限られている。 |
| ● |
私たちは設立以来大きな損失を受けており、予測可能な未来には引き続き重大な損失を受け、永遠に利益を実現したり維持したりすることはないかもしれない。 |
| ● |
私たちはノウォーカーとコロナウイルス感染を予防するための候補錠剤ワクチンの成功に大きく依存している。 |
| ● |
私たちはまだ商業的に実行可能なワクチンを生産していないかもしれないし、私たちは永遠に生産できないかもしれない。 |
| ● |
私たちは私たちの運営に資金を提供するための追加的な資本が必要だ。 |
| ● |
私たちは私たちの組織を拡大し、成長を管理する上で困難に直面する可能性があるだろう。 |
| ● |
我々の業務は,進行中のコロナウイルスの大流行や他の変種の出現など,大流行,流行病,あるいは感染症爆発の悪影響を受ける可能性がある。 |
臨床開発,規制承認,商業化に関するリスク
| ● |
コロナウイルスワクチンの制御経路は変化しており、新しい変種のランダム出現も同様であり、これは思わぬ或いは予見できない挑戦を招く可能性がある。 |
| ● |
臨床試験は非常に高価で、時間がかかり、設計と実施が困難であり、しかも不確定な結果に関連している。 |
| ● |
私たちは他のバイオテクノロジーと製薬会社からの激しい競争に直面している。 |
| ● |
私たちの候補錠剤ワクチンは副作用を引き起こす可能性があり、アメリカ食品と薬物管理局(FDA)の承認及び/又は私たちに対する製品責任訴訟を得ることができない。 |
| ● |
私たちは私たちの持続的な需要を満たすのに十分なバルクワクチンを生産できないかもしれない。 |
| ● |
私たちは第三者に依存して生産と臨床試験を行う。 |
| ● |
私たちは私たちの知的財産権と関連した多くの危険に直面している。 |
第三者に依存するリスク
| ● |
私たちの第三者への依存は、候補製品の開発、承認、製造、または任意の最終的な商業化を遅延または阻止する可能性があります。 |
| ● |
私たちの製造過程のいくつかの部分は第三者契約製造業者に依存する。もし第三者契約製造業者が私たちの規範に従って生産していない場合、あるいは厳格な政府法規を遵守していない場合、私たちの臨床前研究や臨床試験は不利な影響を受ける可能性があり、私たちの候補製品の開発は延期または終了される可能性があり、あるいは大量の追加費用が発生する可能性がある。 |
私たちの業務、財務状況、資本要求に関連するリスク
私たちの運営の歴史を見ると、私たちが作った製品の収入は限られている。
著者らは著者らの商業化インフルエンザ製品のイナビルから特許権使用料収入を獲得したが、著者らはまだ臨床開発過程の初期段階にあり、まだ大規模な肝心な臨床試験を成功しておらず、まだマーケティングの許可を得ておらず、まだ商業規模で私たちの候補錠剤ワクチンを生産しておらず、まだ私たちの候補製品を商業化するために必要な販売とマーケティング活動を展開していない。したがって、もし私たちが候補製品の開発と商業化に成功した歴史があれば、私たちの将来の成功や生存能力の予測はそれほど正確ではないかもしれない。
私たちが相当な収入を生産し、利益を達成し、維持できるかどうかは、ノウォーカーウイルス、SARS-CoV-2、インフルエンザと呼吸器合胞体ウイルス(RSV)感染の予防およびヒトパピローマウイルス(HPV)と他の伝染病による子宮頸癌と異形成を治療するための候補錠剤ワクチンの開発に成功し、必要な監督管理許可を得ることができるかどうかにかかっている。本文書の他の部分に開示されているように、会社はいくつかのプロジェクトの最適な方法を評価している。
私たちのどの候補製品の販売が規制部門の承認を得ても、私たちはいつ相当な収入を生み出すことができるのか分からない。私たちが相当な収入を作る能力は私たちが能力があるかどうかを含む多くの要素にかかっている
| ● |
私たちの候補製品のために受け入れ可能な価格を設定し、第三者支払者から保険と十分な補償を受ける |
| ● |
私たちの製品と候補製品にはイナビルの販売に関する費用が含まれています |
| ● |
販売、マーケティング、製造、流通システムを構築し |
| ● |
私たちの臨床、製造、計画を支援する将来の臨床開発および商業化努力を支援する人員を含む、私たちの運営、財務および管理情報システムおよび人員規模を増加または拡大し続ける |
| ● |
他の人と協力して、大口材料の製造能力を発展させ、受け入れ可能なコストレベルで私たちの候補製品の商業ロットを生産する |
| ● |
私たちの製品を医療界や第三者支払者や消費者に広く市場受容度を得ることができます |
| ● |
経験豊富な管理とコンサルティングチームを引き付けて維持します |
| ● |
私たちの候補製品の商業販売を開始し、単独販売でも他社と協力しても |
| ● |
開発、許可、または私たちが開発および商業化に成功できると考えられる候補製品または商業段階製品; |
| ● |
私たちの知的財産権の組み合わせを維持し、拡大し、保護する。 |
ワクチン開発や製造に関連する多くのリスクや不確実性により,開発費用を増加させる時間や金額を予測することもできず,いつ利益を達成または維持できるか(あれば)も予測できない。FDAまたは同様の非米国規制機関が現在予想されている基礎の上で研究や臨床試験を行うことを要求すれば、私たちの費用は予想を超える可能性がある。私たちの候補製品が商業販売に承認されても、私たちの候補製品の商業発売や関連ビジネス規模製造要求に関連する巨額のコストが生じることが予想されます。もし私たちが上に列挙されたすべての要素を成功的に実行できなければ、私たちの業務は成功しないかもしれない。
私たちは設立以来大きな損失を受けており、予測可能な未来には引き続き重大な損失を受け、永遠に利益を実現したり維持したりすることはないかもしれない。
私たちは限られた製品収入しか生じていません。私たちが私たちの業務戦略を推進し続けるにつれて、大量かつ増加していく損失を招き続けることが予想されます。私たちの候補製品はまだアメリカで発売されることが承認されておらず、決してそのような承認を得られないかもしれない。したがって、私たちはいつ、あるいは利益を達成できるかどうか、もしそうであれば、私たちは利益を維持できるかどうか分からない。私たちが相当な収入を創出し、利益を達成する能力は、私たちが開発を完了し、必要な規制承認を得て、私たちの候補製品を製造し、成功させる能力にかかっている。私たちが私たちの候補製品を商業化することに成功しても、私たちは私たちが利益を得るかどうかを確信できない。もし私たちが規制部門の承認を得ることに成功し、私たちの候補タブレットワクチンを市場に投入すれば、私たちの収入は規制によって承認された地域の市場規模、これらの市場における競争相手の数、私たちが私たちの候補製品の価格を提供することができ、私たちがその地域の商業権を持っているかどうかにある程度依存するだろう。規制部門が承認した適応が私たちが予想していたより狭い場合、あるいは治療群が競争、医師の選択、または治療ガイドラインによって縮小された場合、承認されても、候補製品の販売から著しい収入を得ることができない可能性がある。たとえ私たちが確実に利益を達成したとしても、私たちは四半期や年度の収益性を維持したり向上させることができないかもしれない。もし私たちが達成して利益を維持できなければ、私たちの普通株の市場価格と私たちの資金調達と運営継続能力は不利な影響を受けるだろう。
いずれの錠剤ワクチンの全体的な研究開発費も、ノウォーカーウイルス、SARS-CoV-2、インフルエンザおよびRSV感染を予防するワクチン、およびこれらのコストの大部分に資金を提供しようとしているにもかかわらず、HPV関連異型増殖および癌を治療するワクチンを大幅に増加させることが予想される。また,規制部門の承認を得ても,候補錠剤ワクチンを商業化するには大量の販売とマーケティング費用が必要である。そのため、予測可能な未来には、重大かつ増加していく運営損失と負のキャッシュフローを受け続けることが予想される。このような損失はすでに私たちの財務状況と運営資本に悪影響を与え続けるだろう。2022年12月31日までの累計赤字は3兆271億ドル。
私たち:錠剤ワクチンの成功予防に大きく依存しています ノロウイルスとコロナウイルス 感染はまだ初期臨床開発段階にありますもしこの2つのシート状ワクチンの一方または両方が存在すれば 規制部門の承認を得ていないか、商業化に成功していない場合、私たちの業務は損なわれる可能性があります.
私たちの候補製品の中には後期臨床開発または商業販売が許可されているものは一つもありません。私たちは決して適切な平板ワクチン候補製品を開発できないかもしれません。私たちは今後数年間、私たちの努力と支出の大部分が私たちのノウォーカーとコロナウイルス候補錠剤ワクチンのために使用されると予想している。私たちは財政資源を投入してノーウォーカーワクチンとコロナウイルスワクチンを開発していますが、これは私たちの他の開発計画の遅延や他の方法で負の影響を与える可能性があります。また,我々の管理と科学チームは,我々のノウォーカーワクチンとコロナウイルスワクチンの開発に取り組んでいる。そのため、著者らの業務は現在、ノウォーカーウイルスとコロナウイルス錠剤ワクチンの成功開発、監督管理許可と商業化に大きく依存している。規制部門の承認を得ても、これらのタブレットワクチンは規制部門の承認を得ず、商業化に成功しない可能性がある。候補錠剤ワクチンの研究、テスト、製造、ラベル、承認、販売、マーケティングと流通は現在、FDAとアメリカとその他の国家監督管理機関の広範な監督管理を受けており、これらの国と地区はそれぞれ異なる監督管理規定がある。FDAまたは任意の外国の生物製品許可申請書(BLA)の承認を得るまで、またはこれらの国の必要な承認を得るまで、米国で私たちの錠剤ワクチンを販売することは許可されていない。これまで、著者らはノウォーカーウイルス候補ワクチンと新冠肺炎候補ワクチンの早期臨床試験しか完成しなかった。したがって、私たちはまだFDAにBLAを提出していないし、他の規制機関にも似たような申請を提出しておらず、予見可能な未来でもそうすることはできない。BLAの承認を得ることは、広く、長く、高価で、内面的に不確定なプロセスであり、FDAは、多くの理由で、我々の錠剤ワクチンの承認を延期、制限、または拒否する可能性がある
| ● |
FDAが満足できる錠剤ワクチンが安全で効果的であることは証明できないかもしれません |
| ● |
FDAは完成したノウォーカーウイルスワクチンの第1段階臨床試験および新冠肺炎ワクチンの第1段階および第2段階臨床試験がFDAの要求を満たすことに同意しない可能性があり、追加の試験を要求するかもしれない |
| ● |
著者らの臨床試験結果はFDA要求の上場承認の統計或いは臨床意義レベルに符合しない可能性がある |
| ● |
FDAは、私たちの1つまたは複数の臨床試験の数、設計、規模、実施、または実施に同意しないかもしれない |
| ● |
臨床試験を行う契約研究組織やCROを招聘することは、私たちの臨床試験に実質的な悪影響を与える可能性がある |
| ● |
FDAは、私たちの臨床前研究および臨床試験のデータが、私たちの錠剤ワクチンの臨床および他の利点が安全リスクを超えていることを証明するのに十分であるとは思わないかもしれない |
| ● |
FDAは前臨床研究と臨床試験データの解釈に同意しないかもしれない |
| ● |
FDAは私たちの臨床試験サイトで生成されたデータを受け入れないかもしれません |
| ● |
私たちのNDAまたはBLAが諮問委員会によって検討された場合、FDAは諮問委員会会議をタイムリーに手配することが困難である可能性があり、または諮問委員会は、承認の条件として、承認されたラベルまたは流通および使用制限を制限するために、追加の臨床前研究または臨床試験を要求することを提案する可能性がある |
| ● |
FDAは承認の条件としてリスク評価と緩和戦略の制定を要求する可能性がある |
| ● |
FDAは私たちの製造プロセスや施設に存在する欠陥を発見するかもしれません |
| ● |
FDAはその承認政策を変えたり、新しい規制を採用したりするかもしれない。 |
新しいものを開発しましたノーウォーカーウイルス候補ワクチンはまだ初期段階にある。私たちは効果的なワクチンを生産できず、ヒトを免疫させることに成功しているかもしれないノーウォーカーウイルスもしあれば、適時でもあります。
注射ではなく錠剤で投与される経口ワクチンを開発しています。ノウォーカーウイルスワクチンの開発はまだ初期段階であり、有効なワクチンを生産できないかもしれませんが、もしあれば、ヒトをノウォーカーウイルスに免疫することに成功しています。
CROやCMOなどの重要な第三者との関係を維持することに成功しなければ,我々の経口ノルウォーカーウイルス候補ワクチンを開発し,市場で競争する能力が損なわれる可能性があり,我々の運営結果が影響を受ける可能性がある。私たちが成功しても、これらの関係が私たちの経口ノウォーカー候補ワクチンの開発と商業化につながることを保証することはできません。私たちまたはそのようなパートナーまたは潜在的なパートナーが適用された法規を遵守できなかったことは、臨床保留、遅延、臨床調査の承認、免許取り消し、操作制限、および刑事起訴を含む臨床的保留、遅延、一時停止または撤回を含む制裁を実施することをもたらす可能性があり、これらのいずれも、私たちの潜在的なノルウォーカーウイルスワクチンの供給に重大な悪影響を及ぼす可能性がある。
組換え技術を用いて、私たちの5型アデノウイルスに基づくワクチンのような任意の医薬製品を生産することは、技術的挑戦に直面している。私たちの製造パートナーは、私たちのVAASTプラットフォームを使用してどんなワクチンの製造にも成功できないかもしれないし、cGMP、法規、または同様の規制要件を遵守できないかもしれません。私たちが生産できる潜在的なワクチンの投与量は、私たちが製造能力を成功させ、迅速に拡大する能力にかかっています。私たちが生産できる用量は、患者に接種する必要があるワクチンの用量にも大きく依存し、これは私たちの臨床試験で決定される。ビジネスプロセスを適切に拡大し、開発するためには、大量の資源、専門知識、資本が必要かもしれません。
規模拡大は生産コストと生産量、品質管理(候補製品の安定性と品質保証テストを含む)、合格者或いは肝心な原材料不足及び厳格に執行された連邦、州と外国法規の遵守などの問題をもたらす可能性がある。私たちの契約製造業者は約束通りに契約を履行しないかもしれない。いずれかのメーカーがこれらまたは他の困難に遭遇した場合、臨床試験において候補製品を患者に提供する能力が脅かされる可能性がある。
私たちは私たちの運営に資金を提供するために追加の資金が必要であり、必要な融資を得ることができなければ、私たちは候補錠剤ワクチンの開発と商業化を達成できないかもしれない。
私たちは大量の資金をかけて私たちの平板ワクチン候補薬の開発を完成させ、規制部門の承認を求め、商業化する予定だ。私たちはノウォーカーウイルス、SARS-CoV-2、インフルエンザ、RSVおよびHPVに対する候補錠剤ワクチンの開発と潜在的商業化、および他の候補製品の開発を達成するために大量の追加資金が必要である。もし私たちが必要な時や受け入れ可能な条項の下で資金を調達したり、適切なパートナーを見つけることができなければ、私たちは私たちの1つまたは複数の開発計画または任意の未来の商業化努力を延期、減少、またはキャンセルすることを余儀なくされるかもしれない。また、より多くの資金を得ようとすることは、私たちの経営陣の日常活動に対する時間と注意力を移し、私たちの発展努力を損なう可能性がある。
2022年12月31日まで、私たちは9570万ドルの現金、現金等価物、制限された現金と有価証券を持っています。私たちは高品質、認可された金融機関と私たちの現金、現金等価物、制限された現金と有価証券を維持しています。しかしながら、いくつかのアカウントは連邦保険の限度額を超えており、会社はこれらのホスト機関または投資の財務力のために重大な信用リスクに直面しないと考えているが、そのうちの1つまたは複数のホスト機関の倒産やこれらの投資の違約は、これらの資産を回収する能力に重大な悪影響を与え、および/または私たちの財務状況に実質的な損害を与える可能性がある。
これらの現金、現金等価物、制限された現金、および有価証券は、本年度報告書が発表された日から少なくとも1年以内に、私たちの現在の運営計画に従って私たちの運営に資金を提供するのに十分だと信じているが、私たちが達成できる目標の推定は、不正確であることが証明される可能性のある仮定に基づいており、私たちは現在予想されているよりも早く私たちの利用可能な資本資源を枯渇させるかもしれない。我々の候補製品の開発成功に関連する時間や活動の長さは非常に不確定であるため、開発および承認されたマーケティングおよび商業化活動のためにどれだけの実際の資金が必要かを推定することはできない。私たちの将来の短期的かつ長期的な資金需要は多くの要素に依存するだろうが、これらに限定されない
| ● |
私たちが協力と協力合意に到達する能力は |
| ● |
私たちが計画している臨床試験の開始、進捗、時間、コスト、結果 |
| ● |
FDA、欧州薬品管理局(EMA)と他の類似外国監督管理機関が制定した監督管理要求の結果、時間とコストを満たす |
| ● |
私たちの特許主張と他の知的財産権の提出、起訴、弁護、そして実行のコスト |
| ● |
現在または将来的に第三者が私たちに提起した任意の特許侵害訴訟を含む潜在的な知的財産権紛争を弁護するコスト |
| ● |
競争の技術と市場発展の影響 |
| ● |
私たちの候補製品を商業化することを選択した地域で販売、マーケティング、流通能力を確立するコスト |
| ● |
私たちの候補製品の商業化の開始、進捗、時間、結果は、承認されれば、商業販売に使用されます。 |
追加的な資金は受け入れ可能な条項では得られないかもしれないし、全く得られないかもしれない。もし私たちが受け入れられる条件で十分な追加資本を調達できなければ、私たちは候補製品の開発や商業化を大幅に延期、削減、あるいは停止しなければならないかもしれない。
証券発行による追加資金調達は既存の株主の権益を希釈する可能性があり、貸借や許可手配による資金調達は、私たちの運営を制限したり、所有権の放棄を要求したりする可能性があります。
私たちは未来に私たちが計画した業務を継続するために多くの追加資本が必要になると予想する。これまで、相当な製品収入を生み出すことができれば、株式発行、特許使用料、債務融資、戦略連合、および任意の協力に関する許可および開発協定によって、私たちの現金需要を満たす予定です。私たちは現在約束された外部資金源を持っていない。私たちが株式証券を発行することで追加資本を調達する場合、私たちの既存株主の所有権は大幅に希釈される可能性があり、これらの証券の条項は清算または他の私たちの普通株主の権利に悪影響を及ぼす特典を含む可能性がある。債務融資および優先株融資に関与する可能性のある合意には、追加債務を招く、資本支出を行う、配当金を発表する、留置権を作成する、株式を償還する、または投資を行うなど、特定の行動をとる能力を制限または制限する契約が含まれる。
もし私たちが第三者との協力、戦略連合またはマーケティング、流通または許可手配を通じてより多くの資金を調達するならば、私たちは私たちの技術、将来の収入源、研究計画、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になる可能性のある条項でライセンスを付与しなければならないかもしれない。もし私たちが必要な時に株式や債務融資を通じてより多くの資金を調達することができない場合、あるいは第三者と受け入れ可能な条項で協力し、戦略連合またはマーケティング、流通または許可手配を行うことによって、私たちは私たちの製品開発または将来の商業化努力を延期、制限、減少または終了することを要求されるか、または私たちが本来自分で開発およびマーケティングする候補製品の権利を付与することができる。
私たちの普通株の価格はずっと不安定で、変動が大きく、これは株主に大きな損失を与える可能性があります。
私たちの株価は過去ずっと、将来も、大きな変動を受けていたかもしれない。このような変動のため、私たちの株主は大きな損失を受けるかもしれない。総じて,株式市場,特にバイオ製薬会社の市場は,極端な変動を経験しており,この変動は特定の会社の経営業績とは無関係であることが多い。このような変動のため、初期購入価格あるいは初期購入価格以上で普通株を売ることができないかもしれません。
私たちの普通株式の市場価格は、私たちの製品や競争相手の臨床試験結果、規制または法律の発展、発展、紛争、特許出願、発行された特許または他の固有の権利に関する他の事項、重要な人員を募集し、維持する能力、私たちまたは私たちの戦略パートナーの私たちの候補開発プロジェクトの進展に関する公開公告、私たちの競争相手の類似した公開公告、および本四半期報告および米国証券取引委員会に提出された他の報告書に記載されている他の要素を含む多くの要素の影響を受ける可能性がある。
もし私たちの四半期や年間業績が投資家や証券アナリストの予想を下回れば、私たちの普通株の価格は大幅に低下する可能性がある。さらに、私たちの業績のどの四半期や年間の変動も、私たちの株価を大幅に変動させる可能性があります。我々の業績を経時的に比較することは必ずしも意味があるとは限らず,我々の将来の業績としての指標に依存すべきではないと考えられる.
また、私たち、政府機関、メディア、あるいはSARS-CoV-2疫病に関する他の公開声明(新冠肺炎の新ワクチン開発に関する努力を含む)は過去に招き、将来も私たちの株価を大幅に変動させる可能性がある。世界がコロナウイルス爆発に注目していることから、公共舞台上のこの話題に関するいかなる情報も、正確であるか否かにかかわらず、我々の株価に過大な影響を与える可能性がある(積極的でも消極的であっても)。我々の候補ワクチンの開発·製造·流通に関する情報、あるいは競争相手がその潜在的ワクチンに関するこのような努力に関する情報は、我々の株価にも影響を与える可能性がある。
私たちの株価は引き続き変動し、市場と他の要素の重大な価格と出来高変動の影響を受ける可能性があり、これらの要素は、私たちが本明細書で引用した文書または未来の定期報告で議論された他の要素を含む;私たちの四半期の経営業績は私たちの期待や証券アナリストや投資家の予想との違い、証券アナリストの推定の引き下げ、および私たちまたは私たちの競争相手は重大な買収、戦略的パートナー関係、合弁企業または資本約束を発表する。
歴史的に見ると、初期段階にある製薬、バイオテクノロジー、その他の生命科学会社の証券市場価格の変動は特に大きい。普通株市場の価格変動を引き起こすいくつかの要因は
| ● |
私たちは候補製品を開発して臨床試験を行い、候補製品が安全かつ有効であることを証明することができます |
| ● |
私たちはイナビルを含む候補製品の販売について交渉し、印税を受け取ることができる |
| ● |
私たちは私たちの候補製品のために規制された承認を得る能力と、そのような承認を得るための遅延または失敗 |
| ● |
私たちのすべての候補製品は安全性と有効性を証明できず、監督部門の許可を得られず、商業成功を得られなかった |
| ● |
既存の第三者ライセンス、製造、供給協定を維持することができませんでした |
| ● |
私たちまたは私たちの許可者たちは私たちの知的財産権を起訴、維持、または実行できなかった |
| ● |
私たちの候補製品に適用される法律や規制の変化 |
| ● |
十分な候補製品供給が得られないか、または許容可能な価格で製品を提供することができない |
| ● |
規制当局の不利な決定 |
| ● |
私たちの競争相手は新しい製品や競争相手の製品を発売します |
| ● |
私たちが大衆に提供する可能性のある財務と発展予測を達成できなかったか、またはそれを超えることができなかった |
| ● |
公衆、立法機関、規制機関、投資界の製薬業に対する見方 |
| ● |
私たちまたは私たちの競争相手は、重大な買収、戦略的パートナーシップ、合弁企業、または資本約束を発表します |
| ● |
特許、訴訟事項、および私たちの技術のための知的財産権保護を得る能力を含む、特許権に関連する紛争または他の発展 |
| ● |
キーパーソンの増減 |
| ● |
知的財産権または株主訴訟を含む重大な訴訟; |
| ● |
もし証券や業界アナリストが私たちの研究や報告書を発表しなかった場合、または彼らが私たちの業務および株に不利または誤った意見を発表した場合 |
| ● |
同じ会社の市場予想が変化しています |
| ● |
一般的な市場やマクロ経済状況 |
| ● |
既存株主は将来的に当社の普通株を売却する |
| ● |
当社の普通株式出来高 |
| ● |
このような市場の他の製品および潜在的な製品の否定的な宣伝を含む、私たちの市場への一般的な否定的な宣伝 |
| ● |
医療支払い制度の仕組みを変え |
| ● |
私たちの財務業績の周期的な変動。 |
私たちの普通株はナスダック資本市場に上場していますが、私たちは引き続き持続的な上場の要求と規則を満たすことができないかもしれません。1株当たりの最低購入価格要求及び私たちの株主権益、上場証券の時価、或いは持続的な運営純収益に関連するいくつかの財務指標を含むかもしれません。もし私たちがナスダック資本市場が上場を維持する基準を満たすことができなければ、私たちの証券はカードを取られるかもしれない。もしナスダック資本市場が私たちの証券を取得すれば、私たちは重大な結果に直面するかもしれません
| ● |
私たちの証券の市場オファーは限られています |
| ● |
証券の流動性が減少しています |
| ● |
私たちの普通株が“細価格株”であることを確認することは、私たちの普通株を取引するブローカーに、より厳しい規則を遵守し、取引を減少させる可能性があることを要求する |
| ● |
私たちの普通株式二級取引市場の活動 |
| ● |
ニュースとアナリストの報道の数は限られている |
| ● |
将来的により多くの証券を発行したり、より多くの融資を得る能力が低下する。 |
また、一定数の独立取締役を有し、他社のガバナンス基準を満たすことを要求するルールを含め、ナスダック資本市場ルールの制約を受けなくなる。
我々の業務は,進行中のコロナウイルスの大流行や他の変種の出現など,大流行,流行病,あるいは感染症爆発の悪影響を受ける可能性がある。
我々の業務は,臨床試験場所や他の業務活動が集中している地域で衛生流行病の悪影響を受ける可能性があり,我々が依存している第三者契約メーカーや契約研究組織の運営や,患者を募集して臨床試験を行う能力が深刻に妨害される可能性がある。例えば,進行中のコロナウイルス(新冠肺炎)の大流行は引き続き世界社会,経済,金融市場,世界各地のビジネス実践に予測不可能な影響を与えている。現在行われているコロナウイルスの大流行が著者らの業務、運営結果と未来の成長見通しに与える影響の程度は様々な要素と未来の発展に依存し、これらの要素と未来の発展は高度に不確定であり、大流行の持続時間、範囲と深刻度、特にウイルス変種が伝播し続ける場合を含む、把握できない。
疫病の発生および政府または私たちがどのような流行病に対して取る可能性のあるいかなる予防または保護行動も、一定期間の商業中断および業務減少をもたらす可能性がある。現在、それによるいかなる財務影響も合理的に見積もることはできないが、私たちの業務、財務状況と運営結果に重大な影響を与える可能性がある。疫病が私たちの結果に与える影響の程度は未来の事態に依存し、これらの事態の発展は高度に不確定で、発生する可能性のある疫病の重症度に関する新しい情報や、疫病のコントロールやその影響を治療する行動などを含む予測もできない。メーカーが正常な業務運営や出荷を継続できないため、私たちのサプライチェーンは中断される可能性がある。また、感染症の深刻な爆発は、広範な健康危機を招く可能性があり、これは世界経済や金融市場に悪影響を与え、経済低迷を招き、私たちの業務、財務状況、運営結果に影響を与える可能性があります。私たちは現在、会社のオフィス感染時に従業員を保護する措置や、潜在的な強制的な隔離に対応することを含む、当社の業務連続性計画の強化に努めています。
ロシアとウクライナ間の持続的な軍事衝突は、地政学的不安定、経済的不確実性、金融市場の変動、資本市場の混乱を招く可能性があり、これは私たちの収入、財務状況、または運営結果に悪影響を及ぼす可能性がある。
ロシアとウクライナの間の現在の軍事衝突は、私たちの行動と私たちが依存している第三者の行動に妨害や他の方法で悪影響を及ぼすかもしれない。米国、EU、またはロシアなどの国によって開始される可能性のある関連制裁、輸出規制または他の行動(例えば、潜在的なネットワーク攻撃、エネルギー流動中断など)が、すでにまたは将来的に開始される可能性がある。私たちの業務、私たちの契約研究組織、契約製造組織、私たちと業務を展開している他の第三者に悪影響を及ぼすかもしれません。これによる信用や金融市場の変動、破壊または悪化は、任意の必要な債務や株式融資をさらに困難にし、コストをより高くする可能性がある。適時に有利な条件で任意の必要な融資を得ることができなければ、私たちの成長戦略、財務業績、株価に実質的な悪影響を与える可能性があり、臨床開発計画の延期または放棄を要求する可能性がある。さらに、現在の1つまたは複数のサービスプロバイダ、製造業者、および他のパートナーは、悪化しつつある経済状況の悪影響を受ける可能性があり、これは、運営目標を達成し、将来の業務活動を正確に予測し、計画する能力に直接影響を与える可能性がある。
もし私たちが私たちの候補製品のために十分な精算と保険を獲得したり維持できなければ、私たちが大量の収入を作る能力は制限されるかもしれない。
政府と個人支払者の獲得性と精算範囲は大多数の患者が高価な治療費用を負担できる鍵である。私たちの任意の候補製品がマーケティング承認を得た販売は、私たちの候補製品のコストがどの程度健康維持、管理医療、薬局福祉、および同様の医療管理組織によって支払われるか、または政府衛生行政当局、個人健康保険会社、および他の第三者支払者によってどの程度精算されるかに大きく依存する。精算が得られない場合や、限られた基礎の上で精算を提供するだけでは、候補製品の商業化に成功できない可能性がある。保険を提供しても、承認された精算金額は、十分な価格設定を確立したり、維持したりするのに十分ではない可能性があり、私たちの投資は十分なリターンを実現することができる。
アメリカ以外では、国際業務は通常広範な政府価格制御と他の市場監督管理を受けており、私たちはヨーロッパ、カナダ、その他の国のコスト制御措置が日々重視されており、候補製品に対する定価条件が現在予想されている条件を下回ってしまう可能性があると考えている。多くの国、特に欧州連合国では、国家衛生システムの一部として、医療製品の価格は異なる価格制御メカニズムの制約を受けている。これらの国では、製品の発売許可を受けた後、政府当局との定価交渉にかなりの時間がかかる可能性がある。一部の国で精算或いは定価の承認を得るためには、私たちの候補製品のコスト効果を他の利用可能な治療法と比較する臨床試験を行う必要があるかもしれない。一般的に、この制度での製品価格はアメリカよりずっと低い。他の国は会社が製品に価格を設定することを許可しているが、会社の利益を監視する。追加の外国価格規制や定価規制の他の変化は、私たちの候補製品に受け取ることができる費用を制限するかもしれません。そのため、米国以外の市場では、米国に比べてわが製品の精算水準が低下する可能性があり、商業的に合理的な収入や利益を生み出すのに十分ではない可能性がある。
また、米国や国際では、政府や第三者支払者が医療コストの制限や低減に力を入れており、これらの組織が新承認製品の精算範囲や精算レベルを制限する可能性があるため、候補商品に保険を提供したり、十分な支払いを提供できない可能性がある。私たちは、管理型ヘルスケアの傾向、ヘルスケア組織のますます増加する影響力、および追加的な立法変化により、私たちの任意の候補製品の販売に関連する価格設定圧力に直面すると予想される。全体的に,医療コストの下振れ圧力は非常に大きくなり,特に処方薬,外科手術,その他の治療が行われている。その結果,新製品のヘルスケア市場への参入への壁が高まっている。
私たちの未来の成功は私たちが行政人員を維持できるかどうか、そして合格した人員を吸引、維持、激励できるかどうかにかかっている。
私たちは私たちの執行官たちと実行と科学チームの他の主要な会員たちに依存する。私たちの上級管理者は勝手に採用して、私たちの高級管理者はいつでも彼らの採用を終了することができます。私たちの高度管理者を失ったサービスは、私たちの研究、開発、商業化目標の達成を阻害する可能性がある。私たちはどんな幹部や従業員にも“キーパーソン”保険を提供しない。
合格した科学、臨床及び販売とマーケティング人員を募集と維持することも著者らの成功のキーポイントである。多くの製薬とバイオテクノロジー会社の間の類似者に対する競争を受けて、私たちは受け入れ可能な条件でこれらの人員を吸引し、維持することができないかもしれない。私たちはまた、大学や研究機関から科学や臨床人を募集する競争に直面している。近年,我々の業界の管理者や科学者の流出率が上昇している。また、私たちは科学や臨床コンサルタントを含むコンサルタントやコンサルタントに依存し、私たちの研究開発と商業化戦略の設計を手伝ってくれます。私たちのコンサルタントやコンサルタントは第三者に雇われ、他のエンティティとの相談やコンサルティング契約に基づいて約束する可能性があり、これは彼らが私たちの戦略目標を推進することを制限するかもしれない。このようなコンサルタントやコンサルタントのいずれかがこれ以上私たちのために十分な時間を投入できなければ、私たちの業務は損なわれる可能性がある。
私たちは私たちの組織を拡張する必要があり、このような成長を管理する時に困難に直面する可能性があり、これは運営を混乱させるかもしれない。
私たちの将来の財務業績と私たちが候補製品を商業化し、特許権使用料と効果的な競争を稼ぎ続ける能力は、私たちが将来どのような成長する能力を効果的に管理するかにある程度依存するだろう。2022年12月31日現在、私たちは約164人の常勤従業員を持っており、これは私たちの候補ワクチン製品を商業化するのに十分ではないと信じている。私たちは新しい人員を確定し、採用し、統合することで業務困難に直面するかもしれない。今後の成長は、決定、採用、維持、インセンティブ、より多くの従業員、コンサルタント、請負業者を含む、私たちの経営陣により多くの責任を負わせるだろう。また、私たちの経営陣は、私たちの日常活動から不比例な注意を移し、これらの成長活動を大量に管理する必要があるかもしれない。私たちは私たちの業務の拡張を効果的に管理できない可能性があり、これは私たちのインフラが弱く、操作ミスを招き、ビジネス機会を失い、従業員を失い、残りの従業員の生産性を低下させる可能性がある。私たちの予想成長は大量の資本支出を必要とし、私たちの候補製品を開発するなど、財務資源を他のプロジェクトから移すことができるかもしれない。もし私たちが私たちの成長を効果的に管理できなければ、私たちの支出は予想よりも増加するかもしれないし、私たちの収入を創出および/または増加させる能力は低下する可能性があり、私たちは私たちの業務戦略を実施できないかもしれない。
私たちと適格な人材やコンサルタントを競争している多くの他の製薬会社は、より多くの財務や他の資源、異なるリスク状況、および私たちよりも長い業界の歴史を持っている。彼らはまたより多くの違う機会とより良い職業昇進の機会を提供するかもしれない。このような特徴の中のいくつかは私たちが提供できるより素質の高い候補者と顧問を引き付けることができるかもしれない。もし私たちが引き続き高い素質の人員と顧問を引き付けることができなければ、私たちは私たちの候補製品と業務の速度と成功率を選択して発展させることができます。
私たちは何らかの法的手続きの影響を受ける可能性があり、追加の法的手続きの影響を受ける可能性があり、これは巨額の費用を招き、経営陣の注意をそらし、私たちの業務、財務状況、運営結果に大きな悪影響を及ぼす可能性があります.
この報告書で述べたように、私たちは現在いくつかの未解決の法的手続きの制約を受けている。我々は,上記の事項に関する追加の法的手続きに巻き込まれたり,上記の事項とは無関係な法的手続きに巻き込まれたりする可能性がある.法的手続き自体に不確実性があるため、私たちはその最終結果を正確に予測することができない。私たちの株価は非常に不安定で、未来に私たちはもっと多くの証券集団訴訟に巻き込まれるかもしれない。このような法的手続きは、その是非にかかわらず、巨額のコストを招き、私たちの業務を成功させるために必要な管理職の注意と資源を移転する可能性があり、会社が取締役、高級管理者および他のキーパーソンを募集および維持する能力を弱める可能性があり、融資、保険および他の取引(または任意のこのような融資、保険または他の取引の条項)を獲得する能力に影響を与える可能性があり、これらおよびその他の理由により、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
証券や業界アナリストが我々の業務に関する研究報告を発表しない場合、あるいは不正確または不利な報告を発表しなければ、私たちの株価や取引量は低下する可能性がある。
私たちの普通株の取引市場は、証券や業界アナリストが時々発表している私たちまたは私たちの業務に関する独立した研究と報告の影響を受けています。私たちの1人以上のアナリストを追跡して私たちの株式格付けを下げたり、私たちの業務の見通しに対する彼らの見方を変えたりすれば、私たちの株価は下落するかもしれません。
新冠肺炎を参照する 大流行中、1つまたは複数の政府エンティティは、私たちのいくつかの権利または機会を奪う直接的または間接的な行動をとるかもしれない。もし私たちが新しい肺炎ワクチンを開発するなら、このワクチンは私たちに対する経済的価値が限られているかもしれない。
米国政府を含む複数の政府エンティティは、商業組織がコロナウイルスの予防および治療薬への追加投資を奨励するために、報酬、贈与および契約を提供しており、これは、競争相手の数を増加させ、および/または既知の競争相手に利点を提供する可能性がある。したがって,われわれの新冠肺炎ワクチンが競争力のある市場シェア(あれば)の構築に成功することは保証されない。
私たちは小さな報告会社であり、より小さい報告会社に適用される情報開示要求の低減は、私たちの普通株の投資家に対する吸引力を低下させる可能性がある。
“取引法”の定義によると、私たちは現在“小さな報告会社”です。規模の小さい報告会社は、米国証券取引委員会に提出された文書に簡略化された役員報酬開示を提供し、米国証券取引委員会に提出された文書において他の何らかの減少した開示義務を負うことができる。私たちは投資家が私たちが小さい報告会社の免除に依存して私たちの普通株の吸引力が低下していることを発見するかどうか予測できない。したがって、一部の投資家が私たちの普通株の吸引力が低下していることを発見すれば、私たちの普通株はそれほど活発ではない取引市場がある可能性があり、私たちの株価はもっと変動するかもしれない。
臨床開発,規制承認,商業化に関するリスク
ノウォーカー関連疾患を予防するためのワクチンはまだ承認されていない。したがって、ノルウォーカーウイルスワクチンの任意の承認された規制経路は完全に解明されておらず、予期せぬ挑戦または予見できない挑戦を招く可能性がある。
現在、FDA或いは他の監督管理機関が許可したノウォーカーウイルス予防ワクチンがまだないため、ノウォーカーウイルスワクチンを承認するいかなる監督管理経路も完全に解明されておらず、予期せぬ或いは予見できない挑戦を招く可能性がある。ノウォーカーウイルスワクチンの発見と開発に成功したことは高度に不確定であり、多くの要素に依存し、その中の多くの要素は私たちが制御できない。私たちのワクチン開発はまだ初期段階で、もし本当にあれば、ウイルスの適時治療に成功したワクチンを生産できないかもしれません。臨床試験の結果は新しい問題を提起する可能性があり、提案された終点を修正すること、または新しい臨床試験場所または被験者の列を増加させることを含む提案された臨床試験を再設計することを要求する。また,FDAの臨床データの分析はわれわれの解釈とは異なる可能性があり,FDAは追加的な分析を要求する可能性がある。
中国の監督管理ルートは何ですか新冠肺炎:ワクチンは進化しており、予期せぬ挑戦や予見できない挑戦をもたらすかもしれない。
各方面が新冠肺炎のために治療方法とワクチンを創造とテストする速度は異例であり、しかもアメリカ食品と薬物管理局の内部で絶えず変化或いは変化する計画或いは優先事項は、新冠肺炎に対する新しい知識及びこの疾患がどのように人体の変化に影響するかに基づいて、VXA-CoV 2-1或いはVXA-CoV 2-1.1-Sの監督スケジュールに顕著に影響する可能性がある。臨床試験の結果は新しい問題を提出する可能性があり、提案の終点を修正したり、新しい臨床試験場所或いは被験者の列を増加させることを含む提案された臨床試験を再設計することを要求する。私たちのワクチン(他の新冠肺炎と)試験の結果は、私たちの候補ワクチンを推進するために、追加の臨床前研究を行う必要があるかもしれない。FDAとVXA−CoV 2−1あるいはVXA−CoV 2−1.1−Sの期待第2期と第3期研究の設計について検討しているが,試験設計の重要な面はまだ決定されておらず,組み込む患者数,試験の具体的な終点,試験中に試料を取得·試験する方法を含む。著者らが研究を行う可能性のあるコミュニティにおける新冠肺炎の発病率は場所によって異なる。もしこれらの地区の新冠肺炎の全体的な発病率が低い場合、著者らは被験者を募集することが困難かもしれないし、プラセボを受けた研究参加者とVXA-CoV 2-1あるいはVXA-CoV 2-1.1-Sを受けた研究参加者との間の感染率の差を証明することも困難であるかもしれない。他の許可されたワクチンの獲得性は著者らの未来の試験に参加したい臨床試験被験者の数を減少させる可能性がある。
FDAは、十分な、承認および利用可能な代替品がない場合に、緊急時に許可されていない医療製品を使用して、深刻または生命に危険な疾患または状態を診断、治療または予防することを可能にする緊急使用許可を付与する権利がある。VXA−CoV 2−1またはVXA−CoV 2−1.1−Sの緊急使用許可を取得すれば,FDA承認前に候補ワクチンを商業化することができるであろう。さらに、FDAは、基本的な衛生緊急事態がもはや存在しないか、またはそのような許可が必要であると判断した場合に緊急使用許可を取り消すことができ、緊急使用許可がどのくらい維持されるか(あれば)予測できない。このような撤回は、候補ワクチンがFDAの承認を得ていない場合、および緊急用途許可に応じて候補ワクチンを提供するために、私たちと私たちの製造パートナーがサプライチェーンに投資した場合を含む、様々な方法で私たちの業務に悪影響を及ぼす可能性がある。
また、新冠肺炎候補ワクチンの臨床前試験で観察されたいかなる成功も、後期ヒト臨床試験の結果を予測できない可能性がある。臨床試験では、治療効果、免疫原性、有害事象などの不利な要素が随時出現する可能性があり、私たちの臨床試験を行う能力に不利な結果を与える可能性がある。他の要素、例えば、製造挑戦、原材料の可用性、グローバルサプライチェーンの減速など、候補ワクチンが規制部門の承認を得ることを延期または阻止する可能性があり、あるいは、もし私たちが監督部門の許可を得たら、製品の成功発表を阻止するかもしれない。私たちはワクチンの開発に成功しないかもしれないし、もう一方はより効果的なワクチンの生産に成功するかもしれないし、新しい冠肺炎を治療する他の方法に成功するかもしれない。
私たちの候補平板ワクチンの調合を開発し、改善できなければ、規制部門の承認を得ることができないかもしれません。承認されても、私たちの候補平板ワクチンの商業的受容度は制限されるかもしれません。
H 1 N 1インフルエンザの第2段階試験では約1.5 x 10のワクチン錠剤を使用しました10国際機関のワクチンですしたがって、この試験では、被験者は、1×10の総用量に達するために、1回の設定で7錠の服用を要求された11IU、この試験の目標用量。我々の季節性インフルエンザ候補ワクチンの商業成功を十分に実現するためには,我々の配合を改善し,インフルエンザワクチン錠剤を開発し,各ワクチン株に必要な用量を1錠に含ませ,4錠以下のワクチン接種制度を実現する必要があると考えられる。ワクチン片の効力を高めることはワクチンの安定性に影響する可能性があり、私たちは1種のインフルエンザウイルス株のワクチン接種方案を1枚に減らすことができないか、あるいは4種類のインフルエンザウイルス株を1枚のワクチンに合併することができないかもしれない。そのほか、ワクチン錠剤の効力を高める或いは四価ワクチンを製造するために必要なインフルエンザ株を結合することは生産生産量に不利な影響を与える可能性があり、そしてこのような錠剤の生産コストが高すぎて、商業規模の生産を実現できない。著者らはノウォーカーウイルスと呼吸器合胞体ウイルス候補錠剤ワクチンを開発する努力は類似した調合挑戦に直面している。私たちの候補平板ワクチンの処方をさらに開発して改善することができなければ、FDAや他の規制機関の規制承認を得ることができないかもしれません。承認されても、私たちの候補平板ワクチンの商業的受容度は限られている可能性があります。
臨床試験は非常に高価で、時間がかかり、設計と実施が困難であり、しかも不確定な結果に関連しており、もしそれらがFDA或いは類似の監督管理機関を満足させる安全性と有効性を証明できなければ、著者らは著者らの錠剤候補ワクチンを商業化することができない。
著者らはノウォーカーウイルスと季節性インフルエンザに対する候補錠剤ワクチンはまだ早期臨床開発段階にある。規制部門の承認のために適応または任意の他の治療案をBLAに提出する準備ができる前に、この2つの方法は広範な追加の臨床試験を必要とする。このテストは高価で時間がかかり、専門知識と専門知識が必要だ。現在臨床開発中の任意の錠剤候補ワクチンを規制部門の承認のためにBLAに提出する可能性があるか、またはそのようなBLAがFDAの承認を得るかどうかを確実に予測することはできない。人体臨床試験は非常に高価で、設計と実施が困難であり、一部の原因はそれらが厳格な監督管理要求を受けているからである。例えば,FDAはわれわれが提案したいかなる臨床試験の終点にも同意しない可能性があり,われわれの臨床試験の開始を遅らせる可能性がある。臨床試験過程も時間がかかる。著者らは著者らのノウォーカーウイルス、季節性インフルエンザとRSV候補錠剤ワクチンのBLASを提出するために必要な臨床試験を行う必要があり、完成には数年かかると予想される。また,試験のどの段階でも失敗する可能性があり,臨床試験の放棄や重複を招く問題に遭遇する可能性がある。著者らの候補ワクチンは臨床試験の後期段階で期待される安全性と有効性の特徴を示すことができないかもしれないが、著者らはすでに臨床前研究と初歩的な臨床試験を通じて進展を得たが。そのほか、ノウォーカーウイルス、季節性インフルエンザ、インフルエンザとRSV候補錠剤ワクチンの早期臨床試験結果は後続の臨床試験の結果を予測できない可能性がある。また、FDAはHPV治療性ワクチン候補を推進するために臨床前研究を要求する可能性があり、これは第一段階研究の開始を遅らせる可能性がある。生物製薬業界のいくつかの会社は高級臨床試験で重大な挫折を受け、早期の試験で良好な結果を得たが、治療効果或いは副作用が乏しいためである。
そのほか、臨床前と臨床データはよく多種の解釈と分析の影響を受けやすい。多くの会社は彼らの候補ワクチンが臨床前研究と臨床試験で満足できると思っているが、彼らの製品はまだ市場の承認を得られなかった。また,前臨床試験と早期臨床試験の成功は以降の臨床試験での成功を確保することはできず,後者はより多くの被験者に関連しており,インフルエンザに対しては,われわれがこれまで第1段階の臨床試験で研究してきた1種の毒株ではなく,すべての4種類の毒株が同様である。したがって、後の臨床試験の結果は、以前の臨床試験と前臨床試験の結果を複製しないかもしれない、あるいは上場承認を得るのに十分ではない可能性がある方法で解釈される可能性がある。
臨床試験期間中あるいは臨床試験の結果において、私たちは多くの予見できない事件に遭遇する可能性があり、これらの事件は私たちの発売許可を得ることを延期または阻止することができ、あるいは私たちの錠剤候補ワクチンを商業化することを含む
| ● |
規制機関または機関審査委員会(“IRBs”)は、私たちまたは私たちの研究者が予想される試験場所で臨床試験を開始するか、または臨床試験を開始することを延期または許可することができる |
| ● |
予想される試験場所やCROと受け入れられる臨床試験契約または臨床試験案との合意が遅れたり、合意に達しなかったりする可能性がある |
| ● |
私たちの候補錠剤ワクチンの臨床試験は否定的または不確定な結果をもたらす可能性があり、私たちは決定したり、監督機関が追加の臨床試験を要求したり、製品開発計画を放棄することを要求するかもしれない |
| ● |
私たちのシート状ワクチン候補臨床試験に必要な被験者の数は私たちの予想よりも多いかもしれません。これらの臨床試験の登録速度は私たちが予想していたよりも遅いかもしれません。あるいは参加者がこれらの臨床試験から退出する速度は私たちが予想していたよりも高いかもしれません |
| ● |
私たちの第三者請負業者は、法規の要求を適時に遵守したり、私たちに対する契約義務を履行できなかったり、全く守らなかったりする可能性があります |
| ● |
規制機関またはIRBsは、規制要件を遵守しないこと、または参加者が受け入れられない健康リスクに直面していることを含む、様々な理由で、私たちまたは私たちの研究者に臨床研究の一時停止または終了を要求する可能性がある |
| ● |
私たちの候補錠剤ワクチンの臨床試験コストは予想以上に高いかもしれません |
| ● |
我々の候補錠剤ワクチンあるいは臨床試験を行うために必要な他の材料の供給または品質が不足または不十分である可能性がある。 |
私たちが現在考えている候補錠剤ワクチンの追加の臨床試験または他の試験を要求された場合、候補錠剤ワクチンの臨床試験または他の試験を成功させることができなければ、これらの試験または試験の結果が陽性でないか、またはわずかに陽性である場合、または安全な問題がある場合、私たちは:
| ● |
私たちの錠剤ワクチン候補品の発売承認を遅延させます |
| ● |
市場の承認を得ていません |
| ● |
承認された適応や患者集団は期待や期待ほど広くない |
| ● |
重大な使用または配布制限または安全警告(ボックス警告を含む)を含むラベルによって承認される; |
| ● |
追加の上場後のテスト要求を受ける |
| ● |
発売承認を得た後にこの製品を市場から撤退させます。 |
もし私たちがテストやマーケティング承認を得る上で遅延に遭遇すれば、製品開発コストも増加するだろう。いずれの臨床試験が計画通りに開始されるかどうか,再構成が必要かどうか,予定通りに完成するかどうか,あるいは全く知られていない。重大な臨床試験遅延は、候補平板ワクチンを商業化する独占的な権利を持つ可能性のある任意の期限を短縮する可能性があり、私たちの競争相手が私たちの前に製品を市場に出すことを可能にし、候補平板ワクチンを商業化することに成功する能力を弱めるかもしれません。これらはいずれも私たちの業務と運営結果を損なう可能性があります。
新冠肺炎は著者らの臨床前研究と臨床試験に不利な影響を与える可能性がある。
2019年12月に中国で初めて新型コロナウイルス株SARS-CoV-2が報告されて以来、新冠肺炎は米国を含む多くの国に伝播してきた。われわれは米国で積極的かつ計画された臨床前研究と臨床試験点を持っている
新冠肺炎の継続伝播に伴い、著者らは中断に遭遇する可能性があり、これは著者らの計画と行っている臨床前研究と臨床試験に深刻な影響を与える可能性があり、VXA-CoV 2-1とVXA-CoV 2-1.1-Sの臨床前と臨床研究と製造、及びGI.1とGII 4ノロウイルス株候補ワクチンに対する臨床試験を含む。我々の前臨床研究と臨床試験計画への影響は含まれているが、これらに限定されない
| ● |
私たちの臨床前研究では被験者の調達を遅延させました |
| ● |
患者を臨床試験に参加させるのは遅延したり困難です |
| ● |
臨床前と臨床場所の開始の遅延或いは困難は、臨床前と臨床場所で適切かつ安全な社会距離とその他の保障措置を確立する上での困難を含む |
| ● |
医療資源を前臨床試験と臨床試験の進行から移し,われわれの臨床試験場所である病院や臨床試験を支援してくれた病院スタッフを他の場所に移転することを含む |
| ● |
大流行のプロセス、連邦または州政府、雇用主および他の人が強要または提案した貨物輸送および/または旅行制限であるため、重要な臨床前研究および臨床試験活動を中断し、例えば、臨床前および臨床試験場所のモニタリング、被験者募集および被験者テスト |
| ● |
従業員資源の制限、そうでなければ、従業員または彼らの家族が病気になったため、現場訪問および他の必要な旅行の遅延または困難、および従業員が多くの人との接触を避けることを望むことを含む、私たちの臨床前研究および臨床試験に集中する |
| ● |
私たちが計画した臨床前研究と臨床試験を開始または継続するために、現地の監督機関の許可を得ることを遅延させる |
| ● |
アメリカや他の国の規制や法律の発展; |
| ● |
競争力のあるワクチン製品或いは新冠肺炎治療及び関連技術の成功。 |
もし私たちの臨床試験の1つに参加した患者が新冠肺炎に感染した場合、これはこれらの試験のデータ読み取りに負の影響を与える可能性がある;例えば、患者は私たちの臨床試験にさらに参加できない可能性がある(または参加を制限しなければならない可能性がある)、患者は患者が感染していない場合とは異なる治療効果評価を示すかもしれない、あるいは患者は私たちの薬物製品によって引き起こされる可能性のある有害事象を経験する可能性がある。
新冠肺炎の全世界的な発生は持続的に急速に変化している。新冠肺炎はどの程度私たちの臨床前研究と臨床試験に影響を与える可能性があり、これは未来の事態の発展に依存し、これらの事態の発展は非常に高い不確定性を持っており、例えば疾病の最終地理伝播、疫病発生の持続時間、アメリカと他の国の旅行制限と社会距離、企業閉鎖或いは商業中断、およびアメリカと他の国が疾病をコントロールと治療するための行動の有効性を自信に満ちて予測することができない。
私たちのプラットフォームは新しいワクチンアジュバントを含み、私たちの現在のすべての候補錠剤ワクチンはこのような新しいアジュバントを含み、これは、錠剤ワクチン開発の時間およびコスト、およびFDAまたは他の規制機関が錠剤ワクチン候補ワクチンの安全性を証明するために適用される可能性のある要件を予測することを困難にする可能性がある。
私たちのいくつかの候補シート状ワクチンに含まれる新しいワクチンアジュバントは、患者の安全リスクを増加させる可能性がある。アジュバントは、ワクチンの活性を増強し、ワクチンの免疫応答及び効力を向上させるためのワクチン抗原に添加された化合物である。新しいアジュバントを有するワクチンの開発は、治療薬の典型的な場合と比較して、承認前により多くの患者で評価する必要がある。FDAおよび他の規制機関および専門家委員会は、新規アジュバントを有するワクチンを評価するガイドラインを作成している。新規アジュバントを含むノウォーカーウイルスを含む現在の候補シート状ワクチンであって、将来の候補ワクチンが1つ以上の新規ワクチンアジュバントをさらに含む可能性がある。いずれのワクチンも、アジュバントの存在により副作用がある可能性があり、患者にとって大きなリスクとされており、ワクチンの承認は保証されていない。伝統的に、監督当局は新型アジュバントに対して広範な研究を要求している。ワクチンは通常健康な人、特に乳児、児童と老人に接種されるため、疾病患者ではない。このような広範な研究は、通常、多くの一般の人々の安全を長期的に監視することを含み、時には10,000人を超える被験者を含む。これは,新療法の承認に通常数千人の被験者が必要とするのとは対照的である。これまで、FDAおよび他の主要な規制機関は、5種類のアジュバントを含むワクチンのみを承認してきたが、これにより、我々の平板ワクチン候補が米国または他の場所で規制承認を得るのにどのくらいの時間またはどのくらいの費用がかかるかを決定することは困難である。
臨床試験で被験者を登録·保持することは高価で時間のかかるプロセスであり,我々が制御できない様々な要因によりより困難になったり不可能になったりする可能性がある。
私たちは私たちの任意の臨床試験を完了するのに十分な数の参加者を登録する時に遅延に遭遇したり、登録できないかもしれません。登録されると、私たちは私たちのどんな実験も完了するのに十分な数の参加者を維持できないかもしれない。著者らのノウォーカーウイルスとSARS-CoV-2候補錠剤ワクチンの後期臨床試験は大量の被験者を登録と保留する必要がある。被験者の臨床試験における登録と保留は、被験者群の規模、試験方案の性質、研究薬物に関連する既存の安全性と有効性データ、同一適応の競争的治療と行われている臨床試験の数量と性質、被験者と臨床場所の接近程度及び研究の資格基準を含む多くの要素に依存する。また、信頼できるノウォーカーウイルス感染の動物モデルがないため、ヒト挑戦研究は、感染を防止するために、ウイルス活性と可能な免疫相関性を理解するために使用されており、実験は動物ベースの研究よりも高価である。
さらに、候補錠剤ワクチンの臨床試験で報告された任意の負の結果は、同一の候補錠剤ワクチンの他の臨床試験の参加者を募集および維持することを困難または不可能にする可能性がある。計画中の被験者の登録または保留の遅延または失敗は、コスト増加、計画遅延、または両方をもたらす可能性があり、これは、候補錠剤ワクチンを開発する能力に有害な影響を与える可能性があり、またはさらなる開発を不可能にする可能性がある。また,CROと臨床試験地点で将来の臨床試験の適切かつタイムリーな進行を確保する予定であり,彼らのサービスについて合意する予定であるが,適用法規による実際の表現を強制する能力が制限される。これらの第三者に対して提起された法執行行動は、私たちの臨床開発計画に関連するさらなる遅延と費用をもたらす可能性がある。
私たちは他のバイオテクノロジーや製薬会社からの激しい競争に直面しています。もし私たちが効果的に競争できなければ、私たちの経営業績は影響を受けます。
ワクチン開発競争は激しく、迅速かつ重大な技術進歩に支配されている。私たちは、より規模が大きく、資金がより余裕のある製薬、専門製薬とバイオテクノロジー会社、学術機関、政府機関、公共および民間研究機関を含む様々な源からの競争に直面している。特に、私たちの候補インフルエンザワクチンは、既存で市場で受け入れられている標準治療レジメンの製品と競争するだろう。さらに、将来的には、私たちが狙っている疾患の治療に使用できるより多くの薬物や他の治療法がある可能性が高い。
錠剤ワクチンについては,承認されたワクチンからの競争に直面しており,新しい錠剤ワクチンは有効性,利便性,耐性,安全性の面で納得できる優位性を示さなければならず,特許,発見,開発あるいは商業化に取り組む競争相手から,シート状ワクチンに対して同様のことをすることができる。
私たちの多くの既存または潜在的な競争相手は、私たちよりも多くの財政、技術、人的資源を持っており、疾病を治療する製品の発見と開発、および米国と外国のこれらの製品に対する監督管理の承認を得る上でも明らかに多くの経験がある。私たちは現在、潜在的な未来の競争相手とより多くの経験を持っており、発売が承認された薬物を商業化している。製薬とバイオテクノロジー産業の合併と買収は、私たちの少数の競争相手により多くの資源を集中させる可能性がある。
技術の商業適用性の進歩やこれらの業界に投資する資本の増加により、競争はさらに激化する可能性がある。私たちの競争相手は、私たちが開発する可能性のある任意の錠剤ワクチン候補薬よりも効果的または安価な薬剤の開発、獲得、または許可に独占的に成功するかもしれない。
現在行われている他の感染症の治療については、現在承認されているか、または将来承認される他の薬剤からの競争に直面する。したがって、私たちが競争に成功する能力は私たちの能力に大きく依存するだろう
| ● |
市場の他のワクチンよりも優れた錠剤候補ワクチンを開発し商業化することです |
| ● |
私たちの臨床試験を通じて、私たちの錠剤ワクチン候補薬物は既存と未来の治療方法と異なることが証明された |
| ● |
合格した科学、ワクチン開発、商業人材を誘致する |
| ● |
私たちの候補錠剤ワクチンの特許や他の独自の保護を受けています |
| ● |
必要な規制の承認を得る |
| ● |
第三者支払者から保険と適切な補償を受け、競争力のある価格設定を第三者支払者と交渉すること |
| ● |
独立またはパートナーと新しい錠剤候補ワクチンの開発と商業化に成功した。 |
競争相手のワクチン供給は、私たちが開発した任意の平板ワクチン候補ワクチンの需要と私たちが受け取ることができる価格を制限するかもしれません。既存またはその後に発売されたワクチンと競争できないことは、私たちの業務、財務状況、および将来性に悪影響を及ぼすだろう。
古い製薬会社は、新しい化合物の発見と開発を加速し、あるいは許可を得るために投資する可能性があり、これらの新しい化合物は私たちの任意の錠剤候補ワクチンの競争力を低下させる可能性がある。また、承認されたワクチンと競争するいかなる新しいワクチンも、効力、利便性、耐性と安全性の面で納得できる優位性を示し、価格競争を克服し、商業的に成功しなければならない。したがって、私たちの競争相手は、私たちの前に特許保護を成功させ、FDAの薬品の承認、または商業化を発見、開発、獲得することに成功する可能性があり、これは私たちの業務および運営結果に悪影響を及ぼすだろう。
バイオテクノロジーと製薬業界の特徴は,新技術と独自製品の開発競争が激しいことである。私たちは私たちの独自平板ワクチン候補が競争優位を提供すると信じているが、私たちはバイオテクノロジーと製薬会社、学術機関、政府機関、そして公共および個人研究機関を含む多くの異なる源からの競争に直面している。私たちが商業化する可能性のあるどの製品も、既存の製品や療法、将来出現する可能性のある新製品や療法と競争しなければならない。
既存の治療法、ワクチンまたは送達方法を改善するために、またはその選択された適応のための新しいワクチン、療法または送達方法を開発するために、他の組織が取り組んでいる。これらの努力の成功度によると、承認されれば、候補ワクチンの採用と成功の障害を増加させる可能性がある。
新しいワクチンの市場進出と先進技術の出現に伴い、私たちは激しい競争と日々の競争に直面することを予想しています。我々が開発·商業化した任意の錠剤または他の経口送達ワクチン候補薬は、有効性、安全性、投与および送達の利便性、価格、治療薬の利用可能性、模倣薬競争レベル、および政府および他の第三者支払者が精算できるかどうかに基づいて競争することが予想される。
もし私たちの競争相手が私たちが開発する可能性のあるどの製品よりも安全で、より効果的で、副作用が少なく、より便利で、より安い製品を開発し、商業化すれば、私たちのビジネス機会は減少または消失するかもしれない。私たちの競争相手も候補ワクチンの承認を得るよりも早くFDAや他の規制機関の承認を得ることができ、これは私たちの競争相手が私たちが市場に入る前に強力な市場地位を確立することをもたらすかもしれない。また、多くの場合、私たちの競争能力は、後発薬の使用を奨励することを求める保険会社または他の第三者支払者の影響を受ける可能性がある。
現在、全世界で販売されているノウォーカーウイルスワクチンは承認されていない。HilleVax,Inc.はノーウォーカーウイルスワクチン(最初に武田社が開発した)を開発しており、注射で配信されると考えられています。ノウォーカーウイルス候補ワクチンを開発しているもう一つの会社は安徽智飛龍康生物製薬有限公司です。他にも知らない開発計画があるかもしれません。
新冠肺炎ワクチン市場の競争は激しい。ファイザー-生物科学技術の新冠肺炎ワクチンとModernaの新冠肺炎ワクチンはすでにアメリカと世界の多くの国で承認された。ジョンソンの新冠肺炎ワクチンとアスリコンの新冠肺炎ワクチンはすでに全世界の多くの国で承認され、全世界に大部分の“西洋剤”を供給した。世界各国で新冠肺炎ワクチンの承認を得た他のワクチン会社には、セノフィ社とノワール社が含まれている。
我々の候補錠剤ワクチンは、悪影響をもたらす可能性があり、またはその規制承認を遅延または阻止するか、または任意の承認のラベルまたは市場受容度の範囲を制限することを遅延または阻止する特性を有する可能性がある。
著者らの候補錠剤ワクチンによる不良事件は審査実体、臨床試験場所或いは監督機関の中断、臨床試験の延期或いは停止を招く可能性があり、そして監督部門の承認拒否を招く可能性がある。もし私たちの候補錠剤ワクチンの臨床試験で報告された有害事象の頻度或いは重症度が受け入れられなければ、私たちは監督部門の許可を得てこれらの候補錠剤ワクチンの能力は負の影響を受ける可能性がある。
さらに、もし私たちの任意の錠剤ワクチンが承認され、その後深刻または予期しない副作用を招く場合、いくつかの潜在的な重大な負の結果を引き起こす可能性がある
| ● |
規制部門は、錠剤ワクチン候補ワクチンの承認を撤回したり、その分布または他のリスク管理措置に制限を加えたりすることができる |
| ● |
規制当局は警告や禁忌症のようなラベル宣言の追加を要求するかもしれない |
| ● |
候補錠剤ワクチンの投与方法を変えることや追加的な臨床試験を行うことが求められるかもしれません |
| ● |
私たちは起訴され、患者が受けた傷害に責任を負うかもしれない |
| ● |
ワクチン傷害賠償計画の制約を受けるかもしれません |
| ● |
候補錠剤ワクチンの販売を中止することができます |
| ● |
私たちの名声は損なわれるかもしれない。 |
これらの事件のいずれも、影響を受けた錠剤候補ワクチンに対する市場の受容度を達成または維持することを阻止することができ、商業化コストを大幅に増加させる可能性がある。
もし私たちが必要な規制承認を得ることができない場合、あるいは規制承認を得ることに遅延が生じた場合、私たちは私たちの平板ワクチン候補を商業化することができない、あるいは商業化を遅らせることになり、私たちが大量の収入を創出する能力は損なわれるだろう。
著者らの候補錠剤ワクチン及びそれらの開発と商業化に関連する活動は、それらの設計、テスト、製造、安全性、有効性、記録保存、ラベル、貯蔵、承認、広告、販売促進、販売と流通を含み、すべてアメリカFDAとその他の監督機関及び他の国の類似機関の全面的な監督管理を受けている。もし錠剤候補ワクチンが発売許可を得られなければ、私たちは錠剤候補ワクチンを商業化することができない。私たちはまだいかなる司法管轄区域の監督管理機関の許可を得ていません。私たちの任意の候補錠剤ワクチンを販売することができます。私たちは市場承認を得るために必要な申請の提出と支援に必要な経験が限られており、この過程でCROに依存することが予想される。監督管理部門の許可を得るためには、各監督機関に広範な臨床前と臨床データ及び支持情報を提出し、錠剤ワクチン候補薬物の安全性と有効性を確定する必要がある。監督管理の承認を得るためには、製品の製造過程に関する情報を関連監督機関に提出し、関連監督機関が製造施設を検査する必要がある。私たちの候補錠剤ワクチンは効果がない可能性があり、中程度の有効である可能性があり、あるいは不良或いは意外な副作用、毒性或いは他の特徴があることが証明される可能性があり、私たちの上場承認を阻止するか、或いは商業使用を阻止或いは制限する可能性がある。
アメリカと他の地方では、発売許可を得る過程はすべて高価で、何年もかかるかもしれないし、各種の要素によって大きく異なるかもしれない。関連する錠剤候補ワクチンのタイプ、複雑性と新規性を含む。私たちは私たちがどんな司法管轄区でもどんなマーケティング承認を受けるかどうかを確信できない。開発中の市場承認政策の変化、追加法規または法規の変化、または各提出された製品申請に対する規制審査の変化は、承認の遅延または申請の拒否を招く可能性がある。FDAや他国の類似機関は承認過程において大きな裁量権を有しており,任意の申請を拒否することができ,我々のデータが承認を得るのに不十分であり,追加の臨床前や他の研究や臨床試験が必要であることを決定することもできる。そのほか、臨床前試験と臨床試験から得られたデータの異なる解釈は候補錠剤ワクチンの発売許可を延期、制限或いは阻止する可能性がある。さらに、私たちが最終的に得たどのマーケティング承認も限られているかもしれないし、制限されたり、承認された後の約束は、承認された製品が商業的に実行できないようにすることができる。
米国でFDAの承認を得ても、私たちはいかなる他の管轄区域でも私たちの平板ワクチン候補薬の承認を得たり、商業化したりすることはできないかもしれません。これは、私たちがすべての製品のすべての市場潜在力を達成する能力を制限するだろう。
特定の司法管轄区で私たちの任意の候補錠剤ワクチンを販売するために、私たちは各国の安全性と有効性の面での多くのと異なる監督管理要求を確立し、遵守しなければならない。米国FDAの承認は、他の国や管轄区域の規制機関も承認することを確保していない。また、一国で行われた臨床試験は他の国の規制機関に受け入れられない可能性があり、一国の規制承認は他の国の規制承認を保証することはできない。審査手続きは国によって異なり、追加の錠剤ワクチン候補テストと検証及び追加の行政審査期限に関連する可能性がある。外国の監督管理機関の承認を求めることは私たちに困難とコストをもたらす可能性があり、追加の臨床前研究或いは臨床試験が必要であり、これは高価で時間がかかるかもしれない。各国の監督管理要求は大きく異なる可能性があり、私たちの錠剤候補ワクチンのこれらの国での発売を延期或いは阻止する可能性がある。私たちは国際市場でも、国際市場で監督管理の承認を受けた経験もありませんでした。もし私たちが国際市場の規制要求を遵守できなかった場合、あるいは必要な承認を得て維持できなかった場合、あるいは国際市場の規制承認が延期されれば、私たちの目標市場は減少し、私たちが開発したいかなる錠剤ワクチン候補薬のすべての市場潜在力は達成できないだろう。
私たちが監督部門の承認を得ても、私たちは広範な持続的な規制要求に直面し、私たちの平板ワクチン候補は将来の開発と規制困難に直面する可能性がある。
著者らは発売許可を得た任意の候補錠剤ワクチン、及びこの候補錠剤ワクチンの製造プロセス、承認後の臨床データ、ラベル、包装、流通、不良事件報告、貯蔵、記録保存、輸出、輸入、広告と販売促進活動などは、すべてFDAとその他の監督機関の広範かつ持続的な要求と審査を受ける。これらの要求は安全性、有効性とその他の発売後の情報と報告の提出、機構登録と薬品の発売要求、引き続き現在の良好な製造規範或いはcGMPの要求を遵守し、製造、品質管理、品質保証と記録と書類の相応の維持に関連する要求、医師へのサンプルの配布と記録の保存に関する要求、及び著者らが承認後に行った任意の臨床試験の要求を含む。錠剤ワクチン候補が上場承認を得ても、承認は錠剤ワクチン候補上場の指定用途の制限或いは承認条件の制限を受ける可能性がある。候補錠剤ワクチンが発売承認されれば,付随するラベルが当該錠剤ワクチンの承認使用を制限する可能性があり,販売を制限する可能性がある。
FDAはまた、著者らの候補錠剤ワクチンの安全性及び/又は有効性を監視するために、高価な発売後の研究或いは臨床試験とモニタリングを要求する可能性がある。FDAは薬品の承認後のマーケティングと販売促進を密接に監督し、薬品が承認された適応のみに対する販売を確保し、承認されたラベルの規定に基づいて販売する。FDAはメーカーの非ラベル使用に関するコミュニケーションに厳しい制限を加えており、承認された適応に基づいて候補錠剤ワクチンを販売しなければ、ラベル外マーケティングの法執行行動を受ける可能性がある。処方薬の促進に関連する連邦食品、薬物、化粧品法案の違反は、FDAが法執行行動を取り、連邦と州医療保健詐欺と法律乱用、州消費者保護法に違反していることを告発する可能性がある。
さらに、その後、私たちの錠剤ワクチン候補、製造業者または製造プロセスは、以前に未知の有害事象または他の問題が出現したり、監督要求を遵守できなかったりして、様々な結果が生じる可能性があることを発見した
| ● |
このような候補錠剤ワクチンの生産を制限し |
| ● |
候補錠剤ワクチンのラベルやマーケティングの制限; |
| ● |
錠剤ワクチンの配布や使用を制限し |
| ● |
発売後の研究や臨床試験が求められている |
| ● |
警告状 |
| ● |
市場から候補錠剤ワクチンを撤退させ |
| ● |
私たちが提出した保留申請または承認された申請を承認する補充申請を拒否する; |
| ● |
このような候補錠剤ワクチンをリコールし |
| ● |
罰金、利益または収入の返還、 |
| ● |
上場承認の一時停止または撤回 |
| ● |
候補錠剤ワクチンの輸入または輸出を許可することを拒否し; |
| ● |
候補錠剤ワクチンてんかん発作 |
| ● |
民事または刑事処罰を禁令または適用する。 |
FDAの政策は変わる可能性があり、政府は追加の法規を公布する可能性があり、これは規制部門の私たちの任意の錠剤候補ワクチンの承認を阻止、制限、または延期する可能性がある。もし私たちが既存の要求の変化に緩やかに適応できない場合、あるいは新しい要求や政策を採用することができない場合、あるいは私たちがコンプライアンスを維持できない場合、私たちは私たちが得る可能性のあるマーケティング承認を失う可能性がある。
たとえ我々の平板ワクチン候補が市場の承認を得ても,医師,患者,第三者支払人あるいは医学界の他の商業成功に必要な人の受け入れを得ることができない可能性がある。
もし私たちの候補錠剤ワクチン、著者らのコロナウイルスとノウォーカーワクチンが発売許可を得た場合、それらは依然として医者、患者、第三者支払人と医学界の他の人の十分な市場受容度を得ることができないかもしれない。もし彼らが十分な受容度に達していなければ、私たちは相当な収入を得られないかもしれないし、利益も得られないかもしれない。市場受容度は、商業販売のために許可された場合、多くの要因に依存するが、これらに限定されない
| ● |
代替療法と比較した治療効果と潜在的優位性 |
| ● |
販売とマーケティングの有効性 |
| ● |
代替治療に関連する治療費 |
| ● |
私たちは競争力のある価格で候補タブレットワクチンを販売することができます |
| ● |
代替療法と比較して投与の利便性と簡易性 |
| ● |
対象患者群が新たな療法を試みる意欲と,医師がこれらの治療法を処方する意欲 |
| ● |
医学界はワクチンの代替または補充のために、私たちの錠剤ワクチン候補選択を顧客に提供することを望んでいる |
| ● |
有力なマーケティングと流通支援 |
| ● |
第三者保険と適切な補償を提供する |
| ● |
どんな副作用の流行や重症度も |
| ● |
私たちの錠剤ワクチンを他の薬と一緒に使用することに制限はありません |
承認されれば、我々のコロナウイルスおよび/またはノウォーカー候補錠剤ワクチンの販売は、予測可能な未来にほぼすべての収入を生成することが予想されるので、これらの錠剤ワクチンは、市場の受け入れを得ることができず、私たちの業務を損なうことになり、他の計画よりも早く追加的な融資を求めることが要求されるかもしれない。
もし私たちが州と連邦医療規制法を守らなければ、私たちは重大な処罰、損害賠償、罰金、返還、政府医療計画への参加から除外され、私たちの業務削減に直面する可能性があり、これらはすべて私たちの業務を損なう可能性があります。
医療サービスを提供したり、第三者精算請求を提出したりしていないにもかかわらず、連邦や州政府の医療詐欺や規制乱用や法執行の影響を受けており、業務に深刻な影響を与える可能性がある。私たちの運営能力に影響を与える可能性のある法律は含まれているが、これらに限定されない
| ● |
連邦反リベート法規は、他の事項に加えて、個人および実体がインフォームドコンセントおよび故意の場合、直接または間接的に現金または実物で報酬を請求、受け入れ、提供または報酬を支払うことを禁止し、個人の紹介または購入、レンタル、任意の商品、施設、物品またはサービスを注文または推薦することを禁止し、これらの商品、施設、物品またはサービスは、連邦医療保険および医療補助などの連邦医療計画に従って全部または部分的に支払うことができる。個人または実体は、この規制について実際に理解したり、それに違反したりする具体的な意図を持つ必要はない |
| ● |
個人またはエンティティが虚偽または詐欺的な連邦医療保険、医療補助または他の第三者支払人の支払い請求を意図的に提出または提出させることを禁止する民事虚偽請求法案、またはFCAであって、政府の支払いまたは承認された虚偽または詐欺的クレームを得るために虚偽記録または報告書を作成、使用または作成または使用すること、または連邦政府への支払いの義務を回避、減少または隠蔽するための虚偽記録または報告書の作成、使用または使用を意図的に作成、使用または誘導すること; |
| ● |
刑事FCAは、このような虚偽、虚構または詐欺的だと主張することを知りながら政府に提出または主張した個人または実体に刑事罰金または監禁を科す |
| ● |
1996年の連邦健康保険携行性と責任法案、またはHIPAAは、連邦刑法を制定し、詐欺の任意の医療福祉計画の実行を禁止し、あるいは医療事項に関する虚偽陳述を行うことを禁止した |
| ● |
連邦民事罰金法規は、他の事項に加えて、受益者に影響を与える可能性があることを知っているか、または知っているべきか、連邦または州政府が計画して精算することができる物品またはサービスを選択した受益者への提供または報酬の提供を禁止する |
| ● |
“平価医療法案”の連邦医師陽光要求では、いくつかの薬品、設備、生物製品および医療用品メーカーが、医師、他の医療保健提供者および教育病院への支払いおよび他の価値移転に関する情報、ならびに医師および他の医療保健提供者およびその直系親族が所有する所有権および投資権益を毎年米国衛生·公衆サービス部に報告することを要求する |
| ● |
州法律は、商業保険会社を含む任意の第三者支払者によって償還される可能性のあるプロジェクトまたはサービスに適用可能な反リベートおよび虚偽クレーム法律のような上記の各連邦法律に相当し、州法律は、製薬会社にデバイス業界の自発的コンプライアンスガイドラインおよび連邦政府によって発行された関連コンプライアンスガイドラインを遵守することを要求するか、または医療保健提供者および他の潜在的な転換源に支払う可能性のあるお金を他の方法で制限すること;およびデバイス製造業者に医師および他の医療保健提供者またはマーケティング支出への支払いおよび他の価値移転に関する情報を報告することを要求する州法律に相当する。 |
また、“平価医療法案”は他にも、連邦“反リベート法規”と何らかの医療詐欺を管理する刑事法規の意図要件が改正された。現在、個人又は実体が法規に違反する具体的な意図を実際に理解していない場合には、有罪判決を受けることができる。また、“平価医療法案”では、政府は、“反リベート法”については、連邦“反リベート条例”違反による物品やサービスを含むクレームが虚偽や詐欺的クレームを構成していると断言できる。また、クレームを提出することもありませんが、お客様は最終的にクレームをどのように提出するかを決定しますが、時々お客様に精算指導を提供することができます。もし政府当局が私たちが顧客に不適切な提案をしたと判断したり、虚偽の精算申請の提出を奨励したりすれば、政府当局の私たちに対する訴訟に直面する可能性がある。これらの法律に違反するいかなる行為も、私たちのこれらの法律に違反するいかなる行為も、私たちが防御に成功しても、私たちの名声、業務、運営結果、財務状況に実質的な悪影響を及ぼす可能性がある。
私たちは医師や他の医療提供者と相談と科学顧問委員会の手配を達成した。その中のいくつかの計画の補償は株式オプションを提供することを含む。私たちは適用された法律を遵守するために私たちの手配を構築しようと努力してきたが、これらの法律の複雑かつ深遠な性質のため、規制機関はこれらの取引を再構成または終了しなければならない禁止された手配と見なすか、あるいは私たちはそれによって他の重大な処罰を受けるかもしれない。規制当局が私たちの製品の発注や使用に影響を与えるサプライヤーとの財務関係を適用法違反と解釈すれば、悪影響を受ける可能性がある。
これらの法律の範囲も執行も不確定であり,現在の医療改革環境下では,特に適用の前例や法規が乏しい場合には,急速な変化が生じる可能性がある。連邦と州法執行機関は最近、医療保険会社と医療保健提供者の間の相互作用の審査を強化し、医療保健業界の一連の調査、起訴、有罪判決と和解を招いた。
調査への対応に時間と資源がかかる可能性があり、経営陣の業務への関心を分散させる可能性がある。また,これらの調査の結果として,医療提供者や実体は,法令や会社の誠実な合意の一部である余分な煩雑なコンプライアンスや報告要求に同意しなければならない可能性がある。このような調査や和解は、私たちのコストを増加させるか、または他の方法で私たちの業務に悪影響を及ぼす可能性がある。
私たちの製品に対する責任訴訟は私たちに重大な責任を負わせる可能性があり、私たちが開発する可能性のある任意の錠剤候補ワクチンの商業化を制限する可能性がある。
我々は我々の平板ワクチン候補が人体臨床試験でテストを行うことに関連する固有製品責任暴露リスクに直面しており、承認後に開発可能な任意の製品を商業化販売すれば、より大きなリスクに直面する。例えば,我々のノウォーカーウイルス錠剤挑戦研究は健康なヒトボランティアで行われているため,どのような副作用もこれらの傷害のクレームを引き起こす可能性があり,大きな法的責任を招く可能性がある。是非曲直や最終結果にかかわらず、賠償責任は
| ● |
開発可能な錠剤ワクチン候補の需要を減らすことができます |
| ● |
私たちの名声とメディアの深刻な否定的な関心を損なう |
| ● |
臨床試験参加者の脱退 |
| ● |
関連訴訟を正当化するための巨額の費用 |
| ● |
被験者や患者に巨額の金銭的報酬を与え |
| ● |
収入の損失 |
| ● |
私たちが開発する可能性のあるどんな製品も商業化できない。 |
私たちは製品責任保険の保証金額を1件当たり1,000万ドルに維持していますが、全体的に、これは私たちが発生する可能性のあるすべての責任をカバーするのに十分ではないかもしれません。また、季節性インフルエンザは国家ワクチン傷害賠償計画の保証ワクチンであり、承認されれば、私たちがこの計画に参加するには時間と資源が必要であり、製品の吸収を阻害する可能性がある。私たちが臨床試験を続けるにつれて、どんな製品を商業化することに成功すれば、私たちの保険カバー範囲を増やす必要があると予想されます。保険範囲はますます高くなっています。私たちは可能などんな責任にも対応するために、合理的な費用や十分な金額で保険範囲を維持することができないかもしれない。
もし製品責任クレームが私たちに未保険の責任を提起することに成功した場合、あるいはこのようなクレームが私たちの保険範囲を超えていれば、巨額の損害賠償金を支払うことを余儀なくされる可能性があり、これは私たちの業務に実質的な損害を与える可能性があります。
臨床試験で私たちの既存または未来の任意の候補製品を使用し、任意の承認された薬品を販売することは、私たちを重大な製品責任クレームに直面させるかもしれない。私たちは現在私たちが行っている臨床試験に500万ドルの製品責任保険を提供している。また,これらの試験に使用されている材料を試験したり生産したりする臨床研究や製造組織への協力を求め,このようなクレームに製品責任保険を提供する。この保険範囲は私たちを未来に私たちが提起するかもしれないすべての製品に対する責任クレームから保護できないかもしれません。私たちは潜在的な損失から身を守るために、商業的に合理的なコストまたは十分な金額または範囲で十分な製品責任保険を購入または維持することができないかもしれない。もし私たちに製品責任を請求すれば、私たちは法律と他の費用を払ってクレームを弁護することと、私たちに成功したクレームによるカバーされていない損害賠償を要求されるかもしれません。もし私たちの候補製品がFDAや他の国の同様の規制機関によって販売され商業化された場合、私たちの製品責任保険金額を大幅に増加させる必要があるかもしれない。いずれか1つまたは複数の製品責任クレームを弁護するためには、私たちの業務に悪影響を及ぼす可能性がある大量の財務および管理資源を必要とする可能性があります。
もし私たちが単独でまたは第三者と協力して販売、マーケティング、および流通能力を確立することができなければ、承認されれば、私たちの錠剤候補ワクチンを商業化することに成功できないかもしれない。
私たちは、候補錠剤ワクチンを販売、マーケティング、または配布するためのインフラを持っていません。そのような組織を確立し、維持するためのコストは、そのような費用効果を超える可能性があります。承認される可能性のある任意の錠剤候補ワクチンをマーケティングするためには、私たちの販売、流通、マーケティング、管理および他の非技術的能力を確立するか、または第三者とこれらのサービスを実行するように手配しなければならない。私たちが市場で承認された任意の錠剤ワクチン候補薬を商業的に成功させるためには、販売とマーケティング組織が必要である。私たちは季節性インフルエンザやRSVタブレットワクチンと協力したいと思っていますが、承認されれば、私たちの他の候補タブレットワクチンをアメリカでマーケティングするために、販売、流通、およびマーケティングに集中したインフラを構築したいと思います。私たち自身の販売、マーケティングと流通能力を確立することは、私たちが合格者を募集し、維持し、適切に激励し、十分な販売手がかりを生成し、販売とマーケティング人員に十分な訓練を提供し、異なる地理的位置に分散した販売とマーケティングチームを効果的に管理する能力を含む大量の費用とリスクに関連する。私たちの内部販売、マーケティングおよび流通能力の発展のいかなる失敗または遅延は、任意の錠剤ワクチン候補製品の発売を延期する可能性があり、これは商業化に不利な影響を与える。
私たちの錠剤候補ワクチンの商業化を阻害するかもしれません
| ● |
十分な数の効果的な販売とマーケティング担当者を募集し、訓練し、維持することはできません |
| ● |
販売員は医師に触れたり、私たちの錠剤ワクチンを管理するのに十分な数の医師を得ることができない |
| ● |
独立した販売およびマーケティング組織の作成に関連する予測不可能なコストおよび費用。 |
承認されれば、私たちはいくつかの国際市場での錠剤候補ワクチンの販売とマーケティングについて協力するつもりだが、このような協力計画を確立または維持することはできないかもしれないが、そうすることができれば、私たちの協力者は効果的な販売を行うことができないかもしれない。私たちが第三者に依存してマーケティングと流通を行う程度では、私たちが得たどんな収入もこれらの第三者の努力にかかっており、これらの努力が成功することを保証することはできません。
もし私たちがアメリカで自分の販売チームを作ることができない場合、あるいは私たちの錠剤候補ワクチンのアメリカ以外での商業化交渉について協力関係を結ぶことができなければ、潜在的な商業化を延期したり、私たちの販売やマーケティング活動の範囲を縮小したりすることを余儀なくされる可能性がある。私たちは私たちが選択したよりも早い段階で第三者と合意しなければならないかもしれないし、私たちの知的財産権の権利を放棄すること、または他の方法で私たちに不利な条項に同意することを要求されるかもしれません。いずれも私たちの業務、経営業績、および見通しに悪影響を及ぼす可能性があります。
私たちは多くの会社と競争しているかもしれません。これらの会社は現在広範で資金的なマーケティングと販売業務を持っています。マーケティングおよび販売機能を実行するための内部チームまたは第三者の支援がなければ、これらのより成熟した会社との競争に成功することができないかもしれない。
いずれかの錠剤候補ワクチンを米国以外の地域で商業化することが承認されれば、国際運営に関連する様々なリスクが私たちの業務を損なう可能性がある。
もし私たちの錠剤候補ワクチンが商業化を許可されたら、私たちは第三者と合意して、アメリカ以外のいくつかの司法管轄区でそれらを販売するつもりです。私たちは、国際運営や国際業務関係の構築に関する追加的なリスクに直面すると予想しています
| ● |
国外の薬品審査に対する異なる監督管理要求と薬品商業化規則 |
| ● |
知的財産権の保護を減らすことです |
| ● |
関税、貿易障壁、規制要求の意外な変化 |
| ● |
インフレ、特に外国経済と市場の政治的不安定を含む経済的疲弊 |
| ● |
外国に住んだり旅行したりする従業員は税収、雇用、移民、労働法を遵守する |
| ● |
海外清算、定価、保険制度 |
| ● |
外国の税金 |
| ● |
外国為替変動は、営業費用の増加と収入の減少、他の国での業務展開に付随する他の義務を招く可能性がある |
| ● |
労働騒乱がアメリカよりも一般的な国では労働力の不確実性 |
| ● |
米国の“海外腐敗防止法”、イギリスの2010年反賄賂法、その他の管轄区域に類似した反賄賂と反腐敗法に違反する可能性がある |
| ● |
海外の原材料の供給または製造能力に影響を与えるいかなる事件による錠剤ワクチン接種不足; |
| ● |
地政学的行動(戦争やテロを含む)や地震、台風、洪水、火災などの自然災害による業務中断。 |
私たちは以前このような分野で経験がなかった。また、EUとヨーロッパの多くの個別国家は複雑な規制、税金、労働者、その他の法律要求を提出しており、私たちはこれらの要求を遵守する必要がある。
政府の介入は私たちの平板ワクチン候補製品の商業成功を制限するかもしれない。
米国政府を含む複数の政府エンティティは、商業組織がコロナウイルスおよびインフルエンザ予防の予防および治療薬への追加投資を奨励するために、報酬、贈与および契約を提供しており、これは、競争相手の数を増加させ、および/または既知の競争相手に優位性を提供する可能性がある。したがって,我々のコロナウイルスやインフルエンザワクチンの競争力のある市場シェアを確立することに成功する保証はない。
さらに、現在のインフルエンザワクチンは、一般に3価(3種類の毒株を含む)または4価(4種類の毒株を含む)である。FDAがインフルエンザワクチンの重大な修正を要求または提案している場合、例えば、単価ワクチンの使用、または現在ヒト集団で伝播されていない株を使用する場合、私たちが商業的に合理的な速度でそのようなワクチンを製造または製造することができるかどうかは不明である。
我々の目標適応の季節性,特にインフルエンザ,新製品からの競争は,イナビルの特許使用料収入が予測できず,我々の経営業績に大きな変動を招く可能性がある。
インフルエンザは季節性があり、現在ワクチンの販売は主に例年の第1四半期と第4四半期に発生している。また,ノウォーカーウイルスや呼吸器合胞体ウイルスの爆発は通常冬季に発生する。製品販売のこのような季節的集中は四半期と四半期の経営結果の大きな差を招く可能性があり、そしていかなる製造或いは供給遅延、いかなる在庫の突然の損失、いかなる製品の需要を満たすことができない、返品とリベートを推定できない影響、顧客購入モードの正常或いは異常な変動、或いは販売季節中のいかなる成功しない販売或いはマーケティング戦略による結果を誇張する可能性がある。
私たちはInavirの純売上高から特許権使用料収入を稼いでおり、この製品は私たちの許可側が販売しています。我々ライセンシーが我々に支払う特許権使用料は,ライセンサーがこれらの製品の純売上高の一定割合に固定しているにもかかわらず,これらの特許権使用料から得られる定期的および年間収入は従来可変であり,季節性インフルエンザの発症率と重篤性に基づく変動の影響を受けている。インフルエンザの季節性は主に冬に発生し、これにより、Healthcare Royalty Partners III、L.P.(Healthcare Royalty Partners III、L.P.の財務諸表付記7参照)と合意した合意は、確認された総収入に影響を与えないため、主に冬に発生し、収入確認後の四半期の純キャッシュフローにのみ影響を与える。特定の年間で私たちの特許使用料収入がどの程度になるかを確定的に予測することはできない
しかも、私たちの許可された人たちはInavirの模造薬を含む市場に進出した新製品の競争に遭遇する可能性があり、これは私たちの特許使用料収入に悪影響を及ぼすかもしれない。イナビルに関連する最後の特許は2029年12月に日本で満期になり、特許使用料収入は停止される。しかし、ラニミウェシン酸エステル化合物をカバーする特許は2024年に満了し、後発薬競争が市場に参入する可能性があり、受信した特許使用料を減少または除去する可能性がある。2018年2月23日、大阪に本社を置く製薬業者Shionogi&Co.,Ltd.は日本でインフルエンザを治療する新薬Xofluzaの発売許可を得た。この薬剤はA型やB型インフルエンザウイルスに対する投与が許可されており,年齢にかかわらず単剤でよい。Xofluzaは日本でイナビルからかなりの市場シェアを獲得し、イナビルの販売を大幅に減少させた。これは第一三共株式会社から得た特許権使用料を大幅に削減するだろう。
私たちの成功は私たちが薬物開発の各段階で私たちの候補製品を推進する能力に大きく依存している。もし私たちが私たちの候補製品を推進したり開発したりできなければ、私たちの業務は実質的な損害を受けるだろう。
著者らは著者らの2種類の商業化インフルエンザ製品から特許権使用料収入を得たが、著者らのすべての残りの候補製品はまだ早期開発段階にあり、それらの商業実行可能性は依然として未来の臨床前研究、臨床試験、製造技術、監督管理許可の成功結果及び候補薬物製品の開発過程に普遍的に存在する固有のリスクに依存する。私たちの1つ以上の候補製品の開発を進めることができなかったことは、私たちの業務に実質的な悪影響を及ぼす可能性があります。例えば,Aviragenとの合併により得られた製品teslexivirの第2段階試験コストが高く,我々の他の候補製品の資源を移動させ,主要な治療効果の終点に達しず,開発活動を断念した。私たちの業務の長期的な成功は、最終的には、臨床前研究と臨床試験を通じて私たちの候補製品の開発を推進し、厳格な規格と法規に基づいてそれらを適切に制定し、一致して製造し、FDAや他の国/地域の同様の規制機関による私たちの候補製品の販売承認を得て、最終的に私たちの候補製品を商業化することに依存します。私たちが行っている研究、または将来の研究、臨床前研究、または臨床試験の結果が、私たちの候補製品の継続開発が合理的であることを支持または証明することはできません。あるいは、最終的には、私たちの候補製品の開発を進めるために、fdaや他の国の同様の規制機関の承認を得ることになります.
私たちの候補製品は厳格な安全、治療効果、製造規制基準を満たさなければなりません。その後、私たちはそれらの開発を推進または完成することができ、それらはFDAや他の国の同様の規制機関によって販売を許可することができます。これらの基準を満たすために、私たちは高価で長い研究と臨床試験を行い、受け入れられ、費用効果の高い製造技術を開発し、監督部門の私たちの候補製品に対する承認を得なければならない。このような努力にもかかわらず、私たちの候補製品はないかもしれません
| ● |
治療を受けていない患者と比較して、または同じ患者集団を治療するための既存の薬剤または開発されている他の候補製品を超えることを証明することは、臨床的意義のある治療または他の医療的利益を有する |
| ● |
将来の前臨床研究や臨床試験で安全かつ有効であることが証明されている |
| ● |
期待される治療や医療効果があります |
| ● |
悪い副作用や予期しない副作用を容認することができます |
| ● |
適用される規制基準に適合している |
| ● |
適切な配合および製造を商業的に適切な数量または規模で許容可能なコストで行うことができる;または |
| ● |
私たちや私たちの許可者や協力者たちによって商業化に成功した。 |
われわれが前臨床研究や早期臨床試験で良好な結果を示しても,後期臨床試験の結果が候補製品の継続開発を支援するのに十分であることは確認できない。製薬やバイオ製薬業界の多くの会社は,大多数の会社でなければ,発展のすべての段階で,後期臨床試験を含めて重大な遅延,挫折,失敗を経験しており,前臨床試験や早期臨床試験がエキサイティングな結果を得た後も同様である。したがって、著者らの候補製品の完成した臨床前研究と早期臨床試験の結果は、将来の後期試験で得られる可能性のある結果を予測できないかもしれない。さらに、我々の任意の候補製品に関連する臨床前研究および臨床試験から収集されたデータが満足できる安全性、耐性、および有効性を示しても、これらの結果は、米国FDAまたは他の管轄地域の他の同様の規制機関の規制承認を得るのに十分ではない可能性があり、これは、製品のマーケティングおよび販売に必要である。
私たちの任意の候補製品の実際または予想される治療効果、または安全性または耐性特徴が、販売または臨床開発が許可された他の競合薬に等しいか、またはそれよりも優れていない場合、私たちは任意の候補製品の開発を随時終了する可能性があり、私たちの業務の将来性および潜在的な収益性は損なわれる可能性がある。
私たちの知る限り、複数の会社が様々な抗感染候補製品またはHPVおよびRSV患者の治療のための製品をマーケティングまたは開発しており、これらの製品は販売が許可されているか、臨床開発において私たちよりも進んでいるため、彼らが承認および商業化される時間は私たちの候補製品よりも短い可能性がある。
小児科、老人と免疫機能が損傷した人群の中で、RSV感染の有効な治療方法は非常に限られている。現在、リバビリン(リバビリン)のみが呼吸器合胞体ウイルスによる深刻な下気道感染の治療に応用されている。乳児呼吸器合胞体ウイルス感染を予防するための薬物は、メド免疫会社のモノクロプリビズマブ(Synagis)と、セノフィとアスリコンのニセビズマブ(Beyfortus)を含むことが許可されている。現在、いくつかのワクチンは臨床開発中の呼吸器合胞体ウイルス感染を予防することを目的としている。後期開発段階にある臨床候補製品はファイザーのRSVP preFワクチン、ModernaのmRNA 1345、ノワールのRSVFワクチン、およびグラクソのGSK 3844766 Aワクチンを含む。
もしいつでも、私たちの任意の候補製品が意味的または差別的な治療効果を提供できない可能性があると考え、知覚的にも真実であっても、競争相手の製品または候補製品と同じまたはそれ以上であってもよく、または私たちの候補製品に潜在的な競合相手の化合物が有利な安全性または耐性を有していない可能性があると考え、任意の候補製品の将来の開発を延期または終了する可能性がある。私たちの任意の候補製品の将来の開発が、現在承認されている販売または開発されている潜在的競争相手化合物よりも有意な治療的利益を示すか、または許容可能な安全性または耐性が、それらが開発を継続することの合理性を証明するのに十分であることを保証することはできない。
私たちの候補製品を単独で使用するか、または他の承認された薬品と組み合わせて使用する場合、副作用が生じる可能性があり、これは、その開発または規制承認を延期または排除するか、または(承認された場合)その使用を制限する可能性がある。
薬物開発過程全体において、私たちは絶えず私たちの候補製品の活性、安全性と耐性を証明して、監督部門の許可を得て、その臨床開発を更に推進し、あるいは最終的にそれを市場に押し出さなければならない。我々の候補製品が十分な生物活性および明らかな臨床的利益を示していても、単独で使用するか、または他の薬剤と一緒に使用する場合、任意の許容できない副作用または有害事象は、これらの潜在的な利点を超える可能性がある。候補製品の臨床前研究または臨床試験において、不良または深刻な有害事象または薬物と薬物との相互作用が観察される可能性があり、これは、それらの開発遅延または終了、規制部門の承認を阻止するか、またはそれらの市場受容度を制限し、最終的に承認されれば、それらの市場受容度を制限する可能性がある。
もし私たちの候補製品の臨床前研究または臨床試験の結果が不利であれば、既存または将来の許可または協力協定によって制限された製品を含む場合、私たちの候補製品のさらなる開発または商業化が延期または禁止される可能性があり、これは私たちの業務に実質的な損害を与える可能性がある。
候補製品の開発をさらに進め、最終的に私たちの候補製品を販売する市場承認を得るために、私たちは広範な臨床前研究と臨床試験を行い、その安全性と有効性を証明し、FDAあるいは他の国/地域の類似規制機関(状況に応じて)を満足させなければならない。臨床前研究と臨床試験は高価で複雑であり、完成するには何年もかかるかもしれず、しかも高度に不確定な結果を持っている。遅延、挫折または失敗は、安全性、耐性、毒性、証明の欠如を懸念する生物活性、または販売またはより高度な開発段階にある類似製品と比較して、より高い治療効果、悪い研究または試験設計の欠如、および試験を行うための材料の配合または製造プロセスに関連する問題である可能性があり、任意の時間および臨床前または臨床試験の任意の段階で発生する可能性がある。従来の前臨床研究や早期臨床試験の結果は,われわれが後期臨床試験で観察可能な結果を予測することはできなかった。多くの場合、臨床開発中の候補製品は必要な耐性、安全性と有効性の特徴を表現できない可能性があり、臨床前研究或いは早期臨床試験でこれらの特徴を示したにもかかわらず。
さらに、前臨床研究および臨床試験の過程において、私たちは、候補製品の開発の推進、マーケティングの承認または商業化を進める能力を遅延または阻害する可能性があるが、これらに限定されない多くの予見不可能なイベントに遭遇する可能性がある
| ● |
FDAまたは異なる国の同様の規制機関と1つまたは複数の試験の範囲または設計についてコミュニケーションするか、候補製品の開発を臨床的に保留するか、または問題または問題が満足できる解決されるまで、追加の研究を行って彼らの質問に答えることを含む開発の次の段階を延期する |
| ● |
規制機関やIRBsは予想される試験場所で臨床試験を開始または実施することを許可していません |
| ● |
私たちの臨床試験の登録は延期されたり、進行速度が予想より遅くなったりします。参加者や参加者を募集して予想以上の速度で臨床試験を終了することは難しいからです |
| ● |
私たちの第三者請負業者、私たちは彼らの臨床前研究、臨床試験、私たちの臨床試験材料の製造に依存して、監督要求を適時に遵守したり、私たちに対する契約義務を履行することができませんでした |
| ● |
参加者が受け入れられない健康または安全リスクに直面している場合、臨床試験を一時停止または最終的に終了しなければならない |
| ● |
規制機関またはIRBsは、規制要求を遵守しないことを含む様々な理由で、私たちの臨床前研究と臨床試験を一時停止、一時停止または終了することを要求する;および |
| ● |
著者らの臨床前研究或いは臨床試験を行うために必要な材料の供給或いは品質不足、不適切或いは使用できない。 |
我々の候補製品に関連する臨床前研究または臨床試験から収集されたデータが満足できる耐性、安全性、および有効性を示しても、これらの結果は、米国FDAまたは他の外国司法管轄区の他の同様の規制機関の承認を得るためにBLAまたはNDAの提出を支持するのに十分ではない可能性があり、これは、私たちの候補製品をマーケティングおよび販売するために必要である。
著者らは臨床試験を管理する能力が限られており、これは私たちの適時に候補製品の臨床試験を開始或いは完成する能力を遅延或いは弱める可能性があり、そして著者らの業務に実質的な損害を与える可能性がある。
FDAや他の国の同様の規制機関の私たちの候補製品に対する承認を得るために必要なすべての臨床試験を募集して管理しています。対照的に、大きな製薬と生物製薬会社は通常大量の従業員或いは部門を持っており、複数の適応の複数の候補製品に対して臨床試験を行い、各国の監督部門の許可を得る上で豊富な経験を持っている。また、これらの会社は、臨床試験のために募集しようとしている同じ臨床研究者、場所、患者を競争するためのより多くの財政資源を持っているかもしれない。したがって、私たちは競争的に不利になる可能性があり、これは私たちの臨床試験の開始、採用、時間と完成、そして私たちの候補製品の発売承認を遅らせることができるかもしれない(もし本当に得られたら)。
私たちの産業競争は激しく、迅速な技術変化の影響を受けやすい。したがって、私たちは競争に成功できないかもしれませんし、革新や差別化された製品を開発することもできません。これは私たちの業務を損なう可能性があります。
私たちの産業競争は激しく、技術は日進月歩だ。私たちの業界における重要な競争要素は、臨床前と臨床試験を通じて候補製品開発を成功的に推進する能力、製品または候補製品の治療効果、毒理学、耐性、安全性、薬剤耐性または交差耐性、相互作用または用量プロファイル、上場承認の時間と範囲、競合製品と製薬製品の販売率と平均販売価格、候補製品を生産する原材料と合格した契約製造と製造能力、相対製造コスト、私たちの知的財産権と特許の確立、維持と保護、および販売とマーケティング能力を含む。
候補薬品の開発は競争が激しく、コストが高く、リスクが大きく、商業周期が長い活動である。多くの組織は、市場上の既存製品や臨床開発において私たちの候補製品と競争する可能性のある大型製薬と生物製薬会社を含み、私たちよりも多くの資源を持っており、私たちよりも強力な能力と経験を持っており、これらの能力と経験は、私たちが研究と発見、設計、臨床前研究と臨床試験を行うこと、高度に規制された環境での運営、薬物物質の調合と製造、製品と設備およびマーケティングと販売の面よりはるかに強力である。私たちの競争相手は私たちよりも規制機関の候補製品の承認を得ることに成功し、承認された後に幅広い市場で受け入れられるかもしれない。私たちの競争相手の製品または候補製品は、私たちまたは将来の潜在的許可者またはパートナーが開発または商業化する可能性のある任意の製品よりも有効で、副作用が少なく、管理を容易にし、より有利な抵抗力を有するか、またはより効率的にマーケティングおよび販売される可能性がある。競争相手からの新薬や薬物カテゴリーは、私たちの候補製品が私たちが開発に成功する前に時代遅れになったり、競争力を失ったりするかもしれないし、承認されれば、開発と商業化の費用を回収することができる前に。新薬と薬物カテゴリーが市場に進出し、先進技術或いは新薬標的の出現に伴い、私たち或いは私たちの潜在的な未来の許可証獲得者或いはパートナーは激しく日々激しい競争に直面することを予想している。もし私たちの候補製品が既存製品、新製品、あるいは候補製品と比較して有意な競争優位性を示さなければ、私たちはいつでも候補製品の開発や商業化を中止することができます。
私たちの競争相手は、単独でも彼らとのパートナーでも、私たちよりも効率的で、より安全で、より安く、または管理しやすい候補製品または製品の開発に成功することができる。したがって、私たちの競争相手は私たちよりも早く規制部門のその候補製品の承認を得ることに成功するかもしれない。競争相手の前に臨床試験を完了し、必要な市場承認を得て、その製品を商業化することができる会社は、いくつかの特許およびマーケティング排他権を含む顕著な競争優位を得る可能性があり、これは、競争相手が特定の製品を販売する能力を遅らせる可能性がある。
私たちはまた、(I)より大きな製薬·生物製薬会社を誘致して、私たちの候補製品を買収、許可、または共同開発するために、他の分野で他社からの激しい競争に直面し続けることが予想され、(Ii)より多くの臨床段階開発計画を決定して獲得し、私たちのチャネルを支援し、(Iii)臨床試験を行うことができる研究者や臨床サイトを誘致し、(Iv)患者を募集して臨床試験に参加する。私たちの研究開発作業や私たちの潜在的な将来の許可者やパートナーと共同で努力して生成された候補製品が、私たちの競争相手の既存製品や開発中の候補製品との競争に成功することは保証されません。
もし私たちの許可者または協力者がその契約義務を履行または履行していない場合、候補製品の開発を延期したり、合意を終了したりすると、既存または将来のライセンス契約または連携のテーマである候補製品の開発に成功できない可能性があります。
我々は、将来的には、我々の既存および将来の候補製品の一部または全てをさらに開発および/または商業化するために、許可および連携協定、または第三者との他の同様の業務配置を継続して締結し、依存することが予想される。たとえ私たちが合意規定の義務を履行したとしても、これらのライセンシーまたはパートナーは、約束または予期された方法で実行されないかもしれないし、厳格な規定を遵守しないかもしれないし、または彼らが私たちの候補製品の開発または商業化の努力を延期または終了することを選択する可能性がある。
我々が既存および将来の任意の許可および協力から得る可能性のある潜在的収入の大部分は、開発または規制マイルストーンを達成するために受信された支払い、および承認された製品を販売するために支払われるべき印税などのマイルストーン支払いを含むか、またはマイルストーン支払いを含むことができる。これらの許可と協力によって、私たちが得ることができるマイルストーンと印税収入は、主に私たちの許可者または協力者が私たちの候補製品の開発に成功し、それを商業化する能力に依存するだろう。さらに、私たちのライセンシーまたは協力者は、私たちの技術を使用して、私たちの既存または未来の協力開発された製品を商業化することを決定することができ、これは、私たちが得る可能性のあるマイルストーンおよび特許使用料収入を減少させる可能性がある。多くの場合、私たちは許可または協力に制約された候補製品の開発や商業化に直接または密接に参加しないため、私たちの候補製品を開発または商業化するために、私たちの許可者または協力者に大きく依存する。私たちのライセンス保有者は市場に進出した新製品からの競争に遭遇する可能性があり、これは私たちの特許権使用料収入に悪影響を及ぼす可能性がある。私たちの許可者や協力者は、候補製品を開発したり、効果的に商業化することができないかもしれません
| ● |
内部制限により、必要な科学的専門知識を有する者が限られている場合、資本資源が限られている場合、または他の候補製品または内部計画が規制承認を得る可能性がより高い可能性があると考えられ、またはより大きな投資リターンが生じる可能性があるため、必要な資源は割り当てられない |
| ● |
臨床開発、規制承認、商業化によって候補製品を十分に支援するための十分な必要な資源がない |
| ● |
必要な規制の承認を得ることができない; |
| ● |
経営陣、業務運営、または戦略の変化により、他の計画を優先し、私たちの候補製品または製品の開発および/またはマーケティングに対する彼らの支援を減らすことを優先します。 |
上記のいずれかのイベントが発生した場合、私たちは、私たちの許可または協調スケジュールのすべての潜在的または予期された利点を十分に認識できず、私たちの運営結果が悪影響を受ける可能性があります。また,許可側や協力者は,我々と彼らとの合意以外に開発された候補競合製品の開発を継続することを決定することができる.候補製品の臨床開発または商業化、マイルストーン金額の実現と支払い、手配過程で開発された知的財産権の所有権または他の許可協定条項に関連する進展または他の活動に論争があれば、衝突する可能性もある。許可者またはパートナーが上記のいずれの理由でも私たちの候補製品を開発したり、効果的に商業化できなかった場合、私たちの候補製品を同様の条項で開発し、商業化したい他の第三者(もしあれば)を使用することができないかもしれません。同様に,どちらが新規開発や共同開発の知的財産権を所有しているかについては,許可側や協力者と意見が異なる可能性がある.もし合意が論争によって修正または終了された場合、私たちは、この計画の予想される利点を達成する前に、私たちが期待しているいくつかの開発支援や収入を得ることができないかもしれない。私たちはまた、私たちが受け入れられる条項に従って、私たちが候補製品を開発し続け、それを商業化するために必要または有用な任意の知的財産権の許可をそのパートナーから得ることができないかもしれない。任意の候補製品が、任意の候補製品のために締結される可能性のある任意の既存または将来の許可または協調プロトコルから来る保証はない。
政府や第三者支払者が私たちの製品や許可や協力によって開発された製品に十分な精算や保険を提供できない場合、私たちの収入や利益の潜在力が損なわれる可能性があります。
アメリカと大多数の外国市場では、製品収入あるいは関連印税収入、および私たちの製品の内在的価値は、第三者支払者がこのような製品のために制定した販売率に大きく依存する。第三者支払者には、政府衛生行政部門、管理医療組織、個人健康保険会社、その他の類似組織が含まれる。第三者支払者はますます多くの医療製品、サービスと薬品のコスト効果を審査し、これらの製品とサービスの価格に挑戦を提出している。また,新たに承認された薬品の精算状態(あれば)には大きな不確実性がある。また,既存療法に対する新製品の比較有効性や他の非臨床結果の評価が支払者によって販売率決定の決定に考慮されるようになってきている。私たちまたは私たちの許可者あるいは協力者(適用すれば)も発売後の臨床試験を要求されて、私たちの製品の費用効果を証明する可能性があります。そのような研究は私たちが多くの管理時間と財政資源を投入する必要があるかもしれない。私たちまたは私たちの許可者または協力者が開発に成功する可能性のあるいかなる製品も、どの国/地域の任意の第三者支払者の一部または全部の精算を受けることが保証されません。
多くの政府は,医薬品を含む医療保健のカバー範囲を拡大するとともに,コストを低減するための立法を継続して提案している。多くの海外市場で、政府機関は処方薬の定価をコントロールしている。アメリカでは、連邦医療保健政策の重大な変化は過去数年間に承認され、そして引き続き変化し、未来の多くの薬品の販売率の低下を招く可能性がある。連邦と州政府は引き続き提案を提出し、政府の薬品販売率の制御を強化することを予想している。また,管理式医療への日々の重視と政府の米国医療システムへの関与は引き続きそこの薬品定価に下振れ圧力をもたらすことが予想される。最近発生した事件は公衆と政府が薬品コストの審査を強化し、特に会社がある薬品権利を買収した後の価格上昇を招いた。特に,米国連邦検事は最近ある製薬会社に伝票を発行し,その薬品定価のやり方に関する情報の提供やその他の問題を要求し,米国議会議員もある製薬会社に買収後の薬品価格上昇に関する情報を提供することを求めている。これらの調査が立法や規制提案につながり、将来承認される可能性のある製品価格を制限する能力を向上させれば、私たちの収入や将来の収益力は負の影響を受ける可能性がある。私たちの候補製品が販売を許可される前に、薬品の価格設定に影響を与える法律と法規が変化する可能性があり、これはその販売率をさらに制限または廃止する可能性がある。また,社会や患者権利団体の目標は医療コスト,特に医薬製品の価格を下げることであり,これらの団体もこれらの製品の価格に下振れ圧力を与える可能性があり,我々の製品の価格低下を招く可能性がある。
もし私たちが独立して開発したり、許可者や協力者によって開発された候補製品が承認されたりすれば、その予想市場で有意な受け入れが得られなければ、相当な収入を生み出すことはあまりできない。
私たちの候補製品が開発に成功し、私たちまたは許可者または協力者が将来的に市場に投入するために必要な規制承認を得たとしても、これらの製品は医師、患者、または第三者支払者で市場で認められたり、広く使用されたりすることができない可能性がある。私たちのどの製品も獲得可能な市場受容度は多くの要素に依存します
| ● |
既存の治療法と比較して、製品の治療効果または期待される臨床的利益(もしあれば); |
| ● |
後発薬の存在を含む市場承認された時間および競合薬品の既存市場 |
| ● |
第三者支払者が患者の製品コストを支払うための精算レベル; |
| ● |
ユーザーまたは第三者支払者に対する製品の純コスト |
| ● |
製品の利便性と管理しやすさ |
| ● |
この製品は既存または代替療法に対する潜在的な利点である |
| ● |
同類製品の実際または知覚の安全性 |
| ● |
不良影響の実際或いは予想存在、発生率と重症度 |
| ● |
販売、マーケティング、流通能力の有効性; |
| ● |
FDAまたは他の管轄区域は、規制機関によって承認された製品ラベルの範囲に似ている。 |
医師が私たちの製品を私たちの製品に処方したり管理したりすることを選択する保証はありません。承認されれば、予想される患者集団に。もし私たちの製品が意味のある市場に認められなかったら、あるいは私たちの製品の市場が予想より小さいことが証明されたら、私たちは決して相当な収入を発生しないかもしれません。
食品·薬物管理局、米国証券取引委員会および他の政府機関の資金の変化は、重要な指導部や他の人員の採用と維持の能力を阻害し、新製品やサービスの適時な開発や商業化を阻止し、あるいは他の方法でこれらの機関が私たちの業務運営に依存する可能性のある正常な機能を履行することを阻止し、それによって私たちの業務に負の影響を与える可能性がある。
FDAが新製品を審査·承認する能力は、政府予算と資金レベル、肝心な人員の雇用と維持及びユーザー費用の支払いを受ける能力、及び法律、法規と政策の変化を含む様々な要素の影響を受ける可能性がある。したがって、その機関の平均検討時間は近年変動している。また、政府が米国証券取引委員会や我々の業務に依存する可能性のある他の政府機関に提供する資金は、研究開発活動に資金を提供する機関を含め、政治プロセスの影響を受けており、政治プロセス自体が不安定で予測不可能である。
FDAや他の機関の中断も、新薬が必要な政府機関によって審査および/または承認されるのに要する時間を遅らせる可能性があり、これは私たちの業務に悪影響を及ぼすだろう。例えば、過去数年間、最近の2018年12月22日を含めて、米国政府は何度も少なくとも部分的に閉鎖されており、食品·医薬品局や米国証券取引委員会などのいくつかの規制機関は、食品·医薬品局、米国証券取引委員会、および他の政府従業員を休暇にし、重要な活動を停止しなければならない。政府が長期的に停止すれば、FDAが提出した規制文書をタイムリーに審査·処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に重大な悪影響を及ぼす可能性がある。また、将来的に政府の閉店は、適切な資本化と事業継続のために、私たちが公開市場に進出し、必要な資本を得る能力に影響を与える可能性がある。
第三者に依存するリスク
私たちの第三者への依存は、候補製品の開発、承認、製造、または任意の最終的な商業化を遅延または阻止する可能性があります。
私たちの候補製品の研究、開発、製造、そして任意の最終商業化の過程で、私たちは第三者協力者、サービスプロバイダ、他の人に依存しています。我々は,薬物開発や製造活動の多くの点でこれらの側に大きく依存しており,我々の候補製品のテストを含めた第三者による商業化活動に依存することが予想される。このような活動の多くの側面に対する私たちの統制は非常に限られている。1つまたは複数の第三者協力者、サービスプロバイダ、または他の人は、計画的に、または私たちの期待通りに活動を完了することができなかったか、または私たちに対する契約または他の義務を履行することができなかった;1つまたは複数の当事者は、適用される法律または法規に準拠できなかった。あるいは、私たちとこれらの当事者との関係の任意の中断は、私たちの候補製品の開発、承認、製造、または任意の最終的な商業化を遅延または阻止する可能性があり、私たちを次善的なサービス配送または配送品質に直面させ、予想された締め切りまたは他の適時性の問題、誤ったデータおよび供給中断などの結果を逃し、法律や法規の要求や業界基準を遵守しない、あるいは私たちに名声の損害を受ける可能性があり、これらはすべて私たちの製品ルートや業務に潜在的な負の影響を与える可能性がある。
私たちの製造過程のいくつかの部分は第三者契約製造業者に依存する。私は…もし第三者契約製造業者が私たちの規範に従って生産していない場合、あるいは厳格な政府法規を遵守していない場合、私たちの臨床前研究または臨床試験は不利な影響を受ける可能性があり、私たちの候補製品の開発は延期または終了される可能性があり、あるいは重大な追加費用が発生する可能性がある。
ある程度、私たちは第三者契約メーカー(場合によっては独占的な源である可能性がある)に依存しており、私たちは多くのリスクに直面しており、その中のどのリスクも、私たちの臨床前研究や臨床試験の完成を延期または阻止することができ、あるいは私たちの候補製品が規制の承認や商業化を得て、コスト上昇を招き、あるいは私たちの将来の潜在的な製品収入を奪う可能性がある。リスクの中にはこれらに限定されないリスクがあります
| ● |
私たちの潜在的な契約製造業者は、私たちの候補製品の後期臨床試験または商業化を支援するために許容可能な処方を開発することができなかった |
| ● |
私たちの潜在的な契約製造業者は、彼ら自身の基準、私たちの規格、cGMPまたは規制ガイドラインに基づいて私たちの候補製品を生産することができなかったか、または私たちまたは監督機関が私たちの臨床試験や商業使用に適していないと思う材料を生産することができませんでした |
| ● |
私たちの潜在的な契約メーカーは私たちの候補製品の規模や生産能力を増加させることができません。あるいは私たちの候補製品の形を再設定することは、私たちが供給不足を経験したり、私たちの候補製品を製造するコストを増加させる可能性があります。私たちの潜在的な契約製造業者が適切な商業規模で私たちの候補製品を生産できるか、あるいは私たちが受け入れられる代替メーカーを見つけることができるという保証はありません |
| ● |
私たちの潜在的な契約メーカーは、私たちの製品ではなく、他の顧客や彼ら自身の製品を製造することを優先しています |
| ● |
私たちの潜在的な契約メーカーは、約束通りに契約を履行したり、契約製造業務を脱退することができませんでした |
| ● |
私たちの潜在的な契約製造業者の工場は規制制裁や自然災害で閉鎖されている。 |
薬品メーカーはFDA、アメリカ薬品監督管理局(DEA)及び相応の州とその他の外国機関の持続的な定期検査を受けて、FDAが規定したcGMP、その他の政府法規と相応の外国標準を厳格に遵守することを確保する。私たちの第三者契約製造業者がこれらの法規や基準を遵守しているかどうかをコントロールすることができませんので、もし私たちの第三者メーカーや私たちが適用された法規を遵守しなければ、私たちまたは私たちの製造業者に対する制裁を招く可能性があり、これは私たちの業務に大きな悪影響を及ぼす可能性があります。
もし第三者契約製造業者を変更する必要があれば、私たちの臨床前研究または臨床試験、および私たちの候補製品の商業化は延期され、不利な影響を受けたり、終了したりする可能性があり、あるいはそのような変更は、私たちの業務に実質的な損害を与える可能性がある大幅に高いコストをもたらす可能性がある。
アメリカと他の多くの国の様々な規制制限と、私たちの業界で時々発生する潜在的な製造能力制限のため、私たちの候補製品の製造過程における各ステップはある契約メーカーによって独占的に調達されている。CGMPによると、製造業者を交換するには、製造プロセスおよびプログラムを再検証する必要がある可能性があり、異なるメーカーによって製造された材料間の比較可能性を証明するために、さらなる臨床前研究または臨床試験が必要である可能性がある。契約製造業者を交換することは困難かもしれませんし、非常に高価で時間がかかるかもしれません。これは、長い間私たちの候補製品を生産できず、候補製品の開発遅延、コストの増加を招く可能性があります。
私たちはそれを商業化するのに十分な数の候補製品を生産できないかもしれない。
私たちの候補製品に対するFDAの承認を得るためには、私たちはこのような候補製品を大量に生産する必要がある。私たちは私たちの候補製品の製造能力をタイムリーにあるいは経済的に向上させることができないかもしれないし、根本的にできないかもしれない。もしFDAが承認すれば、私たちは私たちの候補製品の生産量を増加させる必要があるだろう。大規模な製造には追加的な検証研究が必要かもしれず、FDAは審査して承認しなければならない。もし私たちが候補製品の製造能力を高めることに成功できなければ、私たちの候補製品の臨床試験、監督管理許可と商業発売は遅れるかもしれません。あるいは供給不足が発生する可能性があります。私たちの候補製品は精密で高品質な製造が必要だ。製造ミスの発生率を含む高品質の製造を実現し、維持できなかったことは、患者の負傷または死亡、テストまたは交付遅延または失敗、コスト超過または他の我々の業務、財務状況および運営結果を損なう可能性のある問題を招く可能性がある。
CGMP法規に符合する薬品生産は先進的な製造技術と技術制御の開発を含む大量の専門知識と資本投資が必要である。
薬品メーカーは生産過程中に常に困難に直面し、生産コストと生産量方面の困難、品質管理方面の困難、候補製品の安定性と品質保証テスト、或いは合格者の不足を含む。これらの困難に遭遇したり,適用法規下の義務を履行できなかったりすれば,臨床試験で研究材料を提供する能力が脅かされる。臨床試験材料の供給のいかなる遅延或いは中断も著者らの臨床試験の完成を遅らせる可能性があり、著者らの臨床試験計画の維持に関連するコストを増加させ、遅延した時間帯に基づいて、新しい試験を開始し、大量の追加費用を支払うか、或いは研究と試験を完全に終了することを要求する。
私たちはFDAがその施設検査計画を通じて実行したcGMP要求を守らなければならない。これらの要求は、他にも、品質管理、品質保証、および記録およびファイルの維持を含む。我々のコンポーネント材料のメーカーは,これらのcGMP要求やFDA,州,外国の他の規制要件を遵守できない可能性がある。FDAまたは同様の外国規制機関は、製品の製造、包装、または試験のために、新しい基準を随時実施したり、既存の標準の解釈および実行を変更したりすることもできる。私たちは私たちの製造業者がこのような規制と基準を遵守しているかどうかをほとんど統制できない。これらの要件を遵守しないことは、罰金や民事処罰、生産停止、製品承認の一時停止または延期、製品の差し押さえまたはリコール、または製品承認の撤回につながる可能性があります。もし私たちが適用法を遵守できなかった場合、あるいは他の理由で、私たちが提供したいかなる製品の安全が損なわれた場合、私たちは規制機関の私たちの製品の承認を得ることができないか、または商業化に成功できない可能性があり、それによるいかなるダメージにも責任を負わなければならないかもしれない。これらの要因のいずれも、将来開発または買収される可能性のある任意の候補製品の臨床試験、規制提出、承認または商業化の遅延、またはより高いコストをもたらすか、または私たちの名声を損なう可能性がある。
私たちは現在単一源の供給者に依存して重要な錠剤ワクチンコンポーネントと私たちの錠剤候補ワクチンを提供するために必要ないくつかの菌株を提供し、これは私たちの錠剤ワクチン候補ワクチンを製造し供給する能力を弱める可能性がある。
私たちは現在単一源の供給者に依存して、私たちの候補錠剤ワクチンを生産するためのいくつかの原材料を提供する。関連原材料の供給に影響する生産量の不足は、私たちの業務、財務状況、経営業績に重大な悪影響を及ぼす可能性がある。これらのサプライヤーから製品を調達し続けることができないのは、サプライヤーの監督管理措置や要求、サプライヤーが経験した不利な財務または他の戦略発展、労使紛争または不足、意外な需要または品質の問題に影響を与える可能性があり、私たちの経営業績または私たちの臨床試験を行う能力に実質的な悪影響を与える可能性があり、そのいずれも私たちの業務を深刻に損なう可能性がある。
私たちは第三者に依存して、私たちの臨床試験を監督して、これらの第三者の表現が満足できなければ、私たちの業務を損なうかもしれません。
われわれはCROと臨床試験地点に依存してわれわれの臨床試験の適切かつタイムリーな進行を確保しており,彼らの実際の表現への影響は限られていると予想される。
私たちはまたCROに依存して、私たちの臨床プロジェクトのデータ、そして未来の非臨床研究の実行を監視し、管理する。私たちは私たちのCRO活動のいくつかの側面だけを統制することを望んでいる。しかし、私たちは私たちのすべての研究が適用された合意、法律、法規、そして科学的基準に従って行われ、私たちのCROへの依存が私たちのこのような規制責任を免除しないことを確実にする責任を負うつもりだ。
私たちと私たちのCROは良好な実験室操作規範とGCPを遵守しなければならない。これらはFDAによって実行された法規とガイドラインであり、欧州経済圏加盟国の主管当局および同様の外国規制機関もまた、国際調整ガイドライン会議の形で、臨床前および臨床開発段階にある任意の候補製品を要求する。監督管理当局は定期的に試験スポンサー、主要な調査者と臨床試験地点を検査することによってGCP規定を実行する。もし私たちまたは私たちのCROがGCPに従わなかった場合、私たちの臨床試験で生成された臨床データは信頼できないと考えられるかもしれませんが、FDAまたは同様の外国の規制機関は、私たちのマーケティング申請を承認する前に追加の臨床試験を行うことを要求するかもしれません。したがって,われわれのCROがこれらの規定を遵守できなかった場合や十分な被験者を募集できなかった場合には,臨床試験の重複が要求される可能性があり,規制承認過程を遅らせることになる。
私たちのCROは私たちの従業員ではなく、私たちは彼らが私たちの臨床と非臨床プロジェクトに十分な時間と資源を投入するかどうかを制御できない。これらのCROはまた、私たちの競争相手を含む他の商業実体と関係がある可能性があり、彼らはまた、これらの実体のための臨床試験または私たちの競争地位を損なう可能性のある他の薬物開発活動を行っているかもしれない。私たちはCROが私たちの知的財産権を不正に開示または流用する可能性があるというリスクに直面しており、これは私たちのビジネス秘密保護を低下させ、私たちの潜在的な競争相手が私たちのノウハウを訪問して利用することを可能にするかもしれない。もし私たちのCROがその契約の義務または義務を成功裏に履行できず、予想された期限内に達成できなかった場合、または彼らが得た臨床データの品質または正確性が私たちの臨床方案や法規の要求または任意の他の理由を遵守できなかった場合、私たちの臨床試験は延長、延期、または終了される可能性があり、私たちは規制部門の私たちが開発した任意の候補製品の承認を得ることができないか、または商業化に成功できないかもしれない。したがって、私たちの財務業績と私たちが開発した任意の候補製品のビジネス見通しは損なわれ、私たちのコストは増加する可能性があり、私たちが相当な収入を創出する能力は延期されるかもしれない。
もし私たちがこのようなCROとの関係が終わったら、私たちは他のCROと合意できないかもしれないし、商業的に合理的な条項でそうすることができないかもしれない。追加のCROを交換または増加させることは、多くのコストに関連し、管理時間と重点を必要とする。しかも、新しいCROが仕事を始める時、自然な過渡期がある。したがって,遅延が生じ,期待される臨床開発スケジュールを満たす能力に実質的な影響を与える可能性がある。我々はCROとの関係を慎重に管理しようと努力しているが、将来挑戦や遅延に遭遇しない保証はなく、これらの遅延や挑戦が私たちの業務、財務状況、将来に悪影響を与えないことも保証されない。
私たちは選択的に協力関係を構築することを求めているかもしれませんが、ビジネスの合理的な条件で協力関係を築くことができなければ、私たちの開発と商業化計画を変えなければならないかもしれません。
私たちの製品開発計画と候補製品の潜在的な商業化には費用を支払うために多くの追加の現金が必要になります。私たちのいくつかの候補製品について、私たちの季節性インフルエンザおよびRSV錠剤を含めて、私たちは政府の実体または他の製薬およびバイオテクノロジー会社と協力して、これらの候補製品を開発し、商業化を達成することを決定するかもしれない。
私たちは適切な協力者を探すことで激しい競争に直面している。私たちが協力について最終的な合意に到達するかどうかは、他に加えて、パートナーの資源と専門長の評価、提案された協力の条項と条件、提案されたパートナーのいくつかの要因の評価に依存する。これらの要因は、臨床試験の設計または結果、FDAまたは米国国外の類似規制機関の承認の可能性、候補研究製品の潜在市場、そのような候補製品の製造と患者への配送のコストと複雑性、競争製品の潜在性、私たちの技術所有権の不確実性を含む可能性があり、挑戦の利点および業界および市場条件を考慮せずにこのような所有権に挑戦すれば、このような不確実性が存在する可能性がある。協力者は,類似した連携可能な指示を得るために代替製品候補を考えることや,このような連携が製品候補との連携よりも魅力的であるかどうかを考えることができる.
顧客と第三者支払者との関係は、刑事制裁、民事処罰、契約損害、名声損害、利益および将来の収益の減少に直面する可能性がある反リベート、詐欺および乱用、および他の医療法律法規の制約を受けることになります。
医療保健提供者、医師、第三者支払人は、私たちが市場の承認を得た任意の候補製品の推薦と処方の中で主要な役割を果たしている。私たちの将来の第三者支払者や顧客との手配は、広く適用される詐欺や乱用、その他の医療法律や法規に直面する可能性があり、これらの法律と法規は、私たちがマーケティング、販売、流通を制限する可能性があり、私たちが上場許可を得た薬品の業務または財務手配と関係を制限するかもしれない。適用される連邦と州医療法律法規の制限は
| ● |
他の事項に加えて、連邦反バックル法規は、個人の転転または購入を誘導または奨励し、任意の商品またはサービスを注文または推薦するために、故意に、または故意に現金または実物で直接または間接的に報酬を請求、提供、受け入れ、または提供することを禁止し、これらの商品またはサービスは、連邦および州医療保険および医療補助のような連邦医療計画に従って支払うことができる |
| ● |
連邦FCAは、連邦政府に故意に連邦政府に虚偽または詐欺的な支払い要求を提出するか、または虚偽陳述によって連邦政府への支払いの義務を回避、減少または隠蔽する個人または実体に対して、民事告発者またはQui tam訴訟を含む刑事および民事罰を適用する |
| ● |
“経済·臨床健康情報技術法”改正された連邦HIPAAは、詐欺の任意の医療福祉計画を実施する計画に刑事と民事責任を適用し、個人が健康情報を識別できるプライバシー、安全、伝送を保護する上での義務を規定し、強制的な契約条項を含む |
| ● |
連邦虚偽陳述法は、医療福祉、プロジェクトまたはサービスの交付または支払いに関連する重大な事実を知り、故意に偽造、隠蔽、または隠蔽することを禁止し、または任意の重大な虚偽陳述を行うことを禁止する |
| ● |
“平価医療法案”の下の連邦透明性は、医薬品、器具、生物製品および医療用品の製造業者が、医師の支払いおよび他の価値移転、ならびに医師の所有権および投資利益に関する情報を衛生公衆サービス部に報告することを要求する |
| ● |
州反リベートおよび虚偽請求法のような同様の州法律法規は、非政府第三者支払者(個人保険会社を含む)によって精算される医療項目またはサービスの販売またはマーケティング手配およびクレームに関連することに適用される可能性があり、いくつかの州法律は、製薬会社に製薬業界の自発的コンプライアンスガイドラインおよび連邦政府によって発行された関連コンプライアンスガイドラインを遵守することを要求し、また、ワクチン製造業者に医師および他の医療保健提供者への支払いまたはマーケティング支出に関する情報を報告することを要求する。 |
私たちはより多くの戦略的パートナーシップの構築と維持に成功することができないかもしれませんが、これは私たちの製品を開発·商業化する能力に悪影響を与え、私たちの経営業績に悪影響を及ぼす可能性があります。
私たちは引き続き戦略的に私たちのパートナーシップを評価し、適切な場合、私たちは将来、主要なバイオテクノロジーやバイオ製薬会社と構築可能なパートナーシップを含むより多くの戦略的パートナーシップを構築することが予想される。私たちは私たちの候補製品のために適切なパートナーを探す上で激しい競争に直面しており、交渉過程は時間がかかり複雑である。私たちが候補製品との協力を成功させるためには、潜在的なパートナーは、私たちが求めている条項や他の会社が許可することができる製品に基づいて、これらの候補製品が魅力的だと思う市場で経済的価値があると考えなければならない。たとえ私たちが戦略的パートナーシップの構築に成功したとしても、私たちが合意した条項は私たちに不利になる可能性があり、例えば、候補製品の開発や承認が延期または承認された製品販売に失望した場合、私たちはこのような戦略的パートナー関係を維持できないかもしれない。我々の候補製品に関連する戦略的パートナーシップ協定のいかなる遅延も、このような候補製品の開発および商業化を遅延させ、市場に進出しても競争力を低下させる可能性がある。もし私たちが私たちの期待や業界アナリストの予想に基づいて私たちの戦略的パートナーシップの下で収入を得ることができなければ、この失敗は私たちの業務を損なう可能性があり、直ちに私たちの普通株の取引価格に悪影響を及ぼすかもしれない。
もし私たちが私たちの非協力候補製品に関連する追加の戦略的パートナーシップを確立し、維持することができない場合、私たちはそのような候補製品の開発に関連するすべてのリスクとコストを負担し、追加の融資を求め、追加の従業員を雇用するか、または他の方法で規制専門知識など、予算のない専門知識を発展させる必要があるかもしれない。もし私たちが追加的な融資を求めたり、より多くの従業員を雇用したり、より多くの専門知識を開発することに成功しなければ、私たちの現金消費率が増加するか、あるいは候補製品開発速度を下げる措置が必要になるだろう。これは協力していない候補製品の開発に否定的な影響を及ぼすかもしれない。
私たちがすでにまたは可能な戦略的協力や買収は成功的ではないことが証明されるかもしれない。
私たちの戦略の一部として、私たちは私たちの株主のために価値を創造すると信じている戦略的パートナー関係や買収機会を監視し、分析します。私たちは、私たちの既存業務の会社、業務、製品、技術を補完または強化する可能性がありますが、このような買収は多くのリスクに関連する可能性があり、これらのリスクは、私たちが予想しているこのような取引の利点を十分に達成することを阻止するかもしれません。
買収した事業を統合したり、買収した事業を収益的に運営することができない可能性があります。また、どの新たに買収された業務を統合することは高価で時間がかかり、管理、運営、財務資源に大きな圧力を与える可能性があり、私たちが予想していたよりも困難または高価であることが証明される可能性がある。私たちの経営陣の関心の移転や、私たちが将来達成する可能性のあるいかなる買収に関連したいかなる遅延や困難は、私たちが行っている業務中断や基準と制御面の不一致を招く可能性があり、これは私たちが第三者関係を維持する能力にマイナスの影響を与えるかもしれない。
規制部門の承認を得られなかった場合や製品が承認されると貨幣化が実現できないことや、成功した結果が保証されていない場合には製品開発時間が長く、前期開発コストが高いリスクがあることなど、買収された技術、製品、知的財産権からいかなる商業価値も得られない可能性がある。取得された技術および/または知的財産権に関連する特許および他の知的財産権は取得されない可能性があり、取得されても、技術または知的財産権を十分に保護するのに不十分である可能性がある。私たちはまた事故の訴訟費用を含めて責任を負うことができます。これらの費用は私たちが受ける可能性のある賠償保護の範囲内ではありません。私たちが戦略取引を行う際には、買収された会社やパートナーに対して誤った推定値を行うことができ、私たちの資産多様性の増加に伴い、私たちの運営をうまく管理することができず、買収や統合の過程で予測不可能なコストがかかったり、他の予見できないリスクや挑戦に遭遇したりする可能性がある。我々は、組合企業または買収の価値を正確に評価することができない可能性があり、我々の合併財務諸表において適切な会計処理を行うか、それを剥離することに成功したり、私たちの最初の予想またはその後に我々の合併財務諸表に反映される価値を実現したりすることができる。
さらに、我々は、株主の希釈または債務の発生をもたらす可能性のある任意の業務または製品を買収するために、公的または個人債務または株式融資によって追加資金を調達するか、または追加株式を発行する必要があるかもしれない。
私たちが戦略的パートナーシップや買収を実施する際に、このようなリスクを効果的に制限できなければ、私たちの業務、財務状況、または運営結果に重大な悪影響を与え、私たちの純収入にマイナスの影響を与え、私たちの証券価格の下落を招く可能性がある。
イベントtでは第三者契約メーカーは適時に十分なバルクワクチンを供給することができなくて、私たちはワクチン錠剤、臨床前研究或いは臨床試験を生産することができますs私たちの候補製品の商業化は延期され、不利な影響を受けたり、終了されたりする可能性があり、あるいは私たちの業務に実質的な損害を与える可能性があります.
アメリカと他の多くの国の様々な規制制限と、私たちの業界で時々発生する潜在的な製造能力制限のため、私たちの候補製品の製造過程における各ステップはある契約メーカーによって独占的に調達されている。CGMPによると、製造業者を交換するには、製造プロセスおよびプログラムを再検証する必要がある可能性があり、異なるメーカーによって製造された材料間の比較可能性を証明するために、さらなる臨床前研究または臨床試験が必要である可能性がある。契約製造業者を交換することは困難かもしれませんし、非常に高価で時間がかかるかもしれません。これは、長い間私たちの候補製品を生産できなくなり、候補製品の開発を延期することになるかもしれません。また、第三者契約メーカーが変化した場合に我々の開発スケジュールを維持するために、より高い生産候補製品のコストを発生させる可能性がある。
第三者サプライヤー(私たちは第三者サプライヤーに依存して臨床前研究または臨床試験を行う)が実行されないか、または厳格な法規を遵守できない場合、これらの研究または試験は延期、終了または失敗する可能性があり、または私たちは大量の追加費用を発生する可能性があり、これは私たちの業務に実質的な損害を与える可能性がある。
著者らは著者らの臨床前研究と臨床試験の資源が限られていることを設計、実施と管理に力を入れている。著者らは従来、臨床研究機構、顧問と主要な研究者を含む第三者に依存し続け、著者らの臨床前研究と臨床試験のデータの設計、管理、実施、監視と分析を助けるつもりであった。著者らはこれらのサプライヤーと個人代表に依存して著者らが臨床開発プロセスを実行する多くの方面に依存し、臨床前研究を行い、著者らの臨床試験に参加する地点と患者を募集し、臨床地点と良好な関係を維持し、これらの地点が試験方案と適用法規に従って著者らの試験を行うことを確保する。もしこれらの第三者が私たちが彼らと合意した条項の義務を満足に履行できなかった場合、私たちは不適切な遅延や追加支出なしに代替手配を達成できないかもしれないので、私たちの候補製品の臨床前研究および臨床試験は延期されるか、または成功しないことが証明されるかもしれない。
さらに、FDAまたは他の国の同様の規制機関は、私たちの臨床試験に参加するいくつかの臨床サイトまたは当社の第三者サプライヤーサイトを検査して、私たちの臨床試験がGCPまたは同様の法規に従って行われているかどうかを決定するかもしれない。もし私たちまたは監督機関が私たちの第三者サプライヤーが適用された法規に基づいて私たちの臨床試験を行っていないと判断した場合、私たちは試験結果からいくつかのデータを排除したり、このような臨床試験を延期、繰り返し、または中止させられたりする可能性がある。
知的財産権に関するリスク
もし私たちが経口ワクチンプラットフォーム技術と候補製品のために特許保護を獲得し、維持することができなければ、あるいは獲得した特許保護範囲が十分に広くなければ、私たちは私たちの市場で効果的に競争することができないかもしれない。
私たちの成功は私たちがアメリカや他の国で特許保護を取得して維持する能力に大きくかかっている。我々は,我々の開発計画や候補品に関する特許出願を米国や海外に提出することで,我々の独自の地位を保護することを求めている.特許訴訟プロセスは高価で時間がかかり、私たちは、必要または望ましいすべての特許出願を合理的なコストまたはタイムリーに提出して起訴することができないか、またはこれらの特許出願によって発行される可能性のある任意の特許を維持して実行することができないかもしれない。
我々は,特許保護を得るのが遅くなるまで,我々の研究開発成果で特許を申請できることを確認できない可能性もある.私たちが持っている特許出願は、発行された特許が、米国または他の国/地域での任意の候補製品をカバーすることを招くことはないかもしれない。我々の特許および特許出願に関連する潜在的に関連する既存技術全体が見つかったことは保証されず、これらの技術は、特許を無効にするか、または係属中の特許出願から特許が発行されることを阻止する可能性がある。特許が確実に発行されたとしても、第三者はその有効性、実行可能性、または範囲に疑問を提起する可能性があり、これは、このような特許が縮小され、無効にされ、または強制的に実行されない可能性がある。これらの特許または私たちが所有または許可してくれた任意の他の特許に対する任意の成功的な挑戦は、私たちが開発する可能性のある任意の候補製品またはセット診断プログラムを商業化することに成功するために必要な権利を奪う可能性がある。また、規制承認に遅延が生じた場合、特許保護の下で候補製品と診断セットを販売する期間が短縮される可能性があります。
もし私たちが持っているプラットフォーム技術および候補製品に関連する特許出願が発表されなかった場合、それらの保護の広さや強度が脅かされている場合、またはそれらが私たちの候補製品に意味のある排他性を提供できなければ、会社が私たちと協力して候補製品を開発することを阻止し、私たちの将来の薬物商業化の能力を脅かすかもしれない。そのような結果は私たちの業務を損なう可能性がある。
最近の特許改革立法は、私たちの特許出願をめぐる起訴や、私たちが発行した特許の実行または保護をめぐる不確実性とコストを増加させる可能性があります。2011年9月16日、“ライシー·スミス米国発明法”または“ライシー·スミス法案”が法律に署名された。ライシー·スミス法案には、米国特許法のいくつかの重大な改正が含まれている。このような条項は特許出願起訴方式に影響を与える条項を含み、特許訴訟に影響を及ぼす可能性もある。米国特許庁は最近,Leahy−Smith法案の管理を管理するための新たな法規や手続きを制定し,Leahy−Smith法案に関連する特許法の多くの実質的な改正,特に最初の提出条項の改正が2013年3月16日に施行された。したがって,Leahy-Smith法案が我々の業務運営にどのような影響を与えるかは不明である(もしあれば).しかし、Leahy-Smith法案とその実施は、私たちの特許出願をめぐる起訴と、私たちが発行した特許の実行または保護の不確実性とコストを増加させる可能性があり、これらはすべて私たちの業務と財務状況に悪影響を及ぼす可能性がある。
さらに、我々は、第3の方向の米国特許商標局(USPTO)が既存技術の発行前に提出するか、または派生、再審査、当事者間の審査、付与後審査または妨害手続きに参加し、私たちの特許権または他の人の特許権に挑戦する可能性がある。他の国では、私たちの特許権や他人の特許権に挑戦する反対手続きを受けたり、巻き込まれたりする可能性があります。このような提出または手続きにおける不利な裁決は、私たちの特許権の範囲を縮小したり、無効にしたりして、第三者が私たちの技術または候補製品を商業化し、私たちと直接競争することを可能にし、または第三者特許権を侵害することなく候補製品を製造または商業化することができなくなる可能性がある。さらに、私たちの特許および特許出願によって提供される保護の広さまたは強度が脅かされている場合、会社が私たちと協力し、現在または将来の候補製品を許可、開発、または商業化することを阻止するかもしれない。
特許の発行は、その発明性、範囲、有効性、または実行可能性に対して決定的ではなく、私たちが所有し、許可している特許は、米国および海外の裁判所または特許庁で挑戦される可能性がある。このような挑戦は、特許主張の全部または部分的な縮小、無効または実行不能をもたらす可能性があり、これは、他人が同様の技術および錠剤ワクチンを使用または商業化することを阻止する能力を制限するか、または我々の技術および候補製品の特許保護期間を制限する可能性がある。しかも、特許の寿命は限られている。アメリカと他の国では、特許の自然消滅時間は一般的に提出後20年だ。様々な延期があることができるが、特許の有効期間およびその提供される保護は限られている。もし私たちが現在または未来の平板ワクチン候補に特許保護がなければ、私たちはそのような候補製品からの模造薬の競争に直面するかもしれない。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちが持っている特許と許可された特許の組み合わせは、他の会社が私たちと似ているか同じ候補製品を商業化することを排除するために十分な権利を提供してくれないかもしれない。
私たちは私たちの特許、ライセンシーの特許、または私たちの他の知的財産権を保護または強制する訴訟に巻き込まれるかもしれません。これは高価で、時間がかかり、成功しないかもしれません。
競争相手は私たちの特許、私たちの許可側の特許、または私たちの他の知的財産権を侵害または他の方法で侵害する可能性がある。権利侵害や不正使用に対抗するために、私たちは高価で時間がかかるかもしれない法的請求を要求されるかもしれない。さらに、侵害訴訟では、裁判所は、私たちまたは私たちのライセンシーの特許が無効であるか、または強制的に実行できないと判断することができ、または、そのような特許が関連する技術をカバーしないことを理由に、他方の関連する技術の使用を阻止することを拒否することができる。任意の訴訟または弁護手続における不利な結果は、私たちの1つまたは複数の特許を無効または狭義に解釈されるリスクに直面させ、私たちの特許出願を発行できないリスクに直面させる可能性がある。第三者へのクレームは、私たちの特許が無効または強制的に実行できないと主張するような第三者からの反クレームを引き起こす可能性もある。米国の特許訴訟では,被告が無効または実行不可能と主張する反訴が一般的である。有効性を疑問視する理由は、新規性の欠如、明らかな、実施できない、または法定テーマの欠如を含む、いくつかの法定要求のいずれかを満たすことができなかったと言われている可能性がある。主張を実行できない理由は,特許訴訟に関連する人が起訴期間中に米国特許商標局に重要な情報を隠蔽したり,重大な誤解を持つ声明をしたりしたからであろう.第三者は、例えば、訴訟と同時に、当事者間の審査、許可後の審査、または反対または米国国外の同様の手続きのような訴訟の範囲外でも、認可後手続きにおいて米国特許商標局に同様の有効性要件を提出することもできる。法的に無効と実行不可能と断言された後の結果は予測できない。私たちは無効な以前の技術がないことを確認することができず、私たちと特許審査員は起訴中にこれを認識しなかった。私たちが許可した特許および特許出願については、第三者の挑戦を防ぐために、限られた権利またはいかなる許可特許の弁護に参加する権利もないかもしれない。もし被告が無効または強制不可能な法的主張に勝った場合、私たちは少なくとも部分的、さらにはすべて、私たちの現在または未来の候補製品の任意の未来の特許保護を失うだろう。このような特許保護の喪失は私たちの業務を損なうかもしれない。
私たちは、特に米国のようにこれらの権利を十分に保護していないかもしれない国では、私たちの知的財産権の盗用を単独でまたは許可者と一緒に防ぐことができないかもしれない。訴訟で勝訴した側が商業的に合理的な条項でライセンスを提供しなければ、私たちの業務は損害を受ける可能性があります。私たちの知的財産権を強制的に執行する訴訟や他の手続きは失敗する可能性があり、成功しても巨額のコストを招き、私たちの経営陣や他の従業員の注意を分散させる可能性があります。
さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。聴聞、動議、または他の一時的手続き、または事態発展の結果を公開することもできる。もし証券アナリストや投資家がこれらの結果がマイナスだと思ったら、私たちの普通株の価格に悪影響を及ぼすかもしれない。
もし第三者が私たちがその知的財産権を侵害していると主張すれば、私たちは巨額の費用を発生させたり、私たちの候補製品のさらなる開発を阻止されたり、商業化されたりする可能性があり、これは私たちの業務に実質的な損害を与える可能性がある。
私たちの成功は、第三者特許や他の独自知的財産権を侵害することなく運営する私たちの能力に大きく依存するだろう。これは一般的に“経営の自由”と呼ばれる。しかし、私たちの研究、開発、および商業化活動は、第三者が所有または制御している特許または他の知的財産権を侵害または他の方法で侵害される可能性がある。我々が開発候補を求めている分野には,第三者が所有する米国や外国から発行された特許や係属中の特許出願が多く存在する.バイオテクノロジーや製薬業界の拡張や特許の発行に伴い、現在または将来の候補製品が第三者特許権侵害のクレームを受けるリスクが増加する可能性がある。バイオテクノロジーと製薬産業の特徴は特許と他の知的財産権に関する広範な訴訟だ。米国と国際では、知的財産権主張の弁護と起訴、介入訴訟および関連する法律と行政訴訟は複雑な法律と事実問題に関連している。そのため,このような訴訟手続きは冗長であり,費用が高く,時間がかかり,その結果はきわめて不確実である.私たちは、他人の固有の権利の実行可能性、範囲、有効性を決定するために、長期的で費用の高い訴訟に巻き込まれるか、または私たちが他人の知的財産権に関連する運営自由を持っているかどうかを決定するかもしれない。
ほとんどの場合、米国の特許出願は、特許出願が提出されてから約18ヶ月後まで秘密にされている。科学または特許文献における発見の発表は、通常、潜在的発見の日よりもはるかに遅い。したがって、私たちと類似した候補製品の特許出願は、私たちが知らずに他の人によって提出されたかもしれない。第三者が私たちの候補製品または他の権利要件をカバーする特許出願も提出した場合、私たちは、発明の優先度を決定するために、米国特許商標局の対抗プログラム(干渉プログラムと呼ばれる)または他の国/地域の同様の手順に参加しなければならないかもしれない。もし私たちに権利侵害請求をすれば、私たちはこのようなクレームを弁護するために巨額の弁護士費と他の費用を支払うことを要求されるかもしれません。もし私たちがクレーム弁護に失敗した場合、私たちは候補製品の開発と商業化を阻止される可能性があり、禁止および/または損害賠償の制約を受ける可能性があります。
将来、米国特許商標局または外国特許庁は、私たちの候補製品に特許権を付与するか、または第三者に他の権利要件を付与する可能性がある。これらの将来の特許の発表に伴い、適切な自由があり、それらをさらに開発または商業化するために、これらの権利の許可または再許可を得る必要があるかもしれない。私たちは許容可能な条項で必要な許可証を得ることができないかもしれない(もしあれば)。もし私たちがこのような許可や再許可を得る必要があるが、それができなければ、候補製品の開発過程で遅延に遭遇したり、私たちの候補製品を開発、製造、商業化することができないかもしれない。私たちが発行された特許を侵害し、経営の自由がないと判断された場合、私たちは禁止令の制約を受け、および/または懲罰的損害賠償を含む重大な損害賠償の支払いを余儀なくされる可能性がある。私たちが許可内の知的財産権を持っている場合、私たちがこのような合意の条項や条件を守らなければ、私たちの業務を損なう可能性があります。
開発に成功した候補製品や承認された薬物の特許主張に第三者が疑問を提起することが一般的になってきている。もし私たちまたは私たちのライセンシーや協力者が任意の特許訴訟、妨害、または他の法的手続きに巻き込まれた場合、私たちは巨額の費用を発生する可能性があり、私たちの技術と管理者の努力と注意力は著しく分散するかもしれない。このような訴訟または訴訟の負の結果は、私たちの独自の地位を失ったり、重大な責任を負うことに直面したり、商業的に許容可能な条項で第三者から取得できない可能性があるライセンスを求めることを要求する可能性があります(あれば)。司法または行政訴訟で不利な判決が下された場合、または必要な許可証を取得できなかった場合、私たちは私たちの候補製品の開発、製造、販売を制限または阻止される可能性がある。
私たちの特許保護の獲得と維持は、政府特許機関によって提出された様々な手続き、書類の提出、費用の支払い、および他の要求に依存しており、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
発行された特許の定期維持費は、特許有効期間内にいくつかの段階で米国特許商標局および外国特許代理機関に支払われなければならない。米国特許商標局と各種外国政府特許機関は,特許出願過程においていくつかのプログラム,文書費用の支払い,その他の規定を遵守することを要求している。多くの場合、適用規則に従って滞納金を支払うか、または他の方法で不注意を是正することによって失効を是正することができるが、場合によっては、規定を遵守しないことは、特許または特許出願が放棄または失効され、関連法ドメインの特許権の一部または全部の喪失をもたらす可能性がある。特許または特許出願の放棄または失効をもたらす可能性のある規定を遵守しないイベントは、規定された期限内に公式行動に応答できなかったこと、費用を支払わなかったこと、および適切に合法化されず、正式な文書を提出することができなかったことを含むが、これらに限定されない。もし私たちと私たちの許可者たちが私たちの候補製品をカバーする特許と特許出願を維持できなかったら、私たちの競争地位は不利な影響を受けるだろう。
米国特許法の変化は特許の全体的な価値を低下させ、候補製品を保護する能力を弱める可能性がある。
米国は最近、広範囲な特許改革立法を公布し、実施した。近年、米国最高裁判所はいくつかの特許事件に対して裁決を下し、場合によっては利用可能な特許保護範囲を縮小するか、場合によっては特許権者の権利を弱めるかを決定している。我々の将来の特許取得能力に関する不確実性の増加に加えて,このようなイベントの結合は,いったん特許を取得する価値に関する不確実性をもたらしている.米国議会、連邦裁判所、および米国特許商標局の行動によれば、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、それにより、私たちが新しい特許を獲得したり、私たちが許可または将来獲得可能な特許を実行する能力を弱める可能性がある。
私たちは世界各地で私たちの知的財産権を保護できないかもしれないが、これは私たちの業務を損なうかもしれない。
世界中で私たちの候補製品のための申請、起訴、そして特許保護の費用は目を引くほど高いだろう。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの技術を使用して彼ら自身のワクチンを開発するかもしれませんし、特許保護を受ける可能性がありますが、特許執行力はアメリカの地域よりも他の侵害ワクチンを輸出するかもしれません。これらのワクチンは、可能な特許が発行された可能性のある司法管轄区域で私たちの候補製品と競争することができないかもしれません。将来の任意の特許主張または他の知的財産権は、それらのような競争を効果的にまたは阻止するのに十分ではないかもしれません。
私たちは、私たちの従業員、コンサルタント、または独立請負業者が第三者の機密情報を誤って使用または開示したか、または私たちの従業員がその前の雇用主によって言われた商業秘密を誤って使用または開示したという疑惑の影響を受けるかもしれない。
私たちの多くの従業員は、私たちの高級管理者を含めて、大学や他のバイオテクノロジーや製薬会社に雇われていた。これらの従業員たちは一般的に彼らの以前の仕事と関連した所有権、秘密、そして競技禁止協定を実行する。私たちは、私たちの従業員が私たちのために働いているときに他人の固有情報やノウハウを使用しないことを保証するために努力しているが、私たちは、その従業員またはこれらの従業員が、商業秘密または他の固有情報を含む、そのような任意の元雇用主の知的財産権を使用または開示しているという疑惑を受ける可能性がある。私たちはこれらの問題に関連した脅威や未解決のクレームがあることを知らないが、将来的にはこのようなクレームに対抗するために訴訟を提起する必要があるかもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭的損害賠償を支払う以外に、私たちは貴重な知的財産権や人員を失う可能性がある。このようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣の注意を分散させる可能性がある。
私たちの第三者への依存は、私たちのビジネス秘密を共有することを要求し、これは、競争相手がこれらの秘密や私たちのビジネス秘密が流用または開示されていることを発見する可能性を増加させる。
私たちは、当社のノウハウをある程度保護し、独自情報の研究または開示を開始する前に、適用されれば、当社のコンサルタント、従業員、第三者請負業者およびコンサルタントと材料譲渡協定、コンサルティング契約、または他の同様の合意を締結することを求めています。これらの協定は、一般に、私たちの商業秘密を含む、第三者が私たちの機密情報を使用または開示する権利を制限する。第三者と協力する際に契約条項が採用されているにもかかわらず、商業秘密および他の機密情報を共有する必要は、そのような商業秘密が私たちの競争相手に知られ、無意識に他の人の技術に組み込まれているか、またはこれらの合意に違反した場合に開示または使用されるリスクを増加させる。私たちの独自の地位が私たちのノウハウおよびビジネス秘密にある程度基づいていることを考慮すると、競争相手は、私たちのビジネス秘密または他の許可されていない使用または開示が私たちの競争地位を損なうことを発見し、私たちの業務および運営結果に悪影響を及ぼす可能性があります。
さらに、これらの合意は、一般に、私たちの合意がいくつかの限られた発行権を含む可能性があるにもかかわらず、私たちのコンサルタント、従業員、第三者請負業者、およびコンサルタントが、私たちのビジネス秘密に関連する可能性のあるデータを発行する能力を制限します。私たちは私たちのビジネス秘密を保護しようと努力しているにもかかわらず、私たちの競争相手は、私たちと第三者との合意、独立開発、または私たちの任意の第三者協力者が情報を発表することによって、私たちのビジネス秘密を発見するかもしれない。競争相手は私たちのビジネス秘密が私たちの競争地位を損なうことを発見し、私たちの業務に悪影響を及ぼすだろう。
特許条項は候補製品に対する私たちの競争地位を十分に長く保護するのに十分ではないかもしれない。
特許の寿命は限られている。米国では、すべての維持費が適時に支払われれば、特許の自然満了期間は、通常、その最初の米国非臨時出願日から20年である。様々な延期があるかもしれないが、特許の有効期限とその提供される保護は限られている。私たちの候補製品をカバーする特許を取得しても、候補製品の特許有効期限が満了すると、模造薬を含む競争相手からのワクチンと薬物の競争に直面する可能性がある。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちが持っている特許と許可された特許の組み合わせは、他の会社が私たちと似ているか同じ候補製品を商業化することを排除するために十分な権利を提供してくれないかもしれない。
FDAが私たちの候補製品の上場承認の時間、持続時間、条件に基づいて、私たちの1つ以上の米国特許は、1984年の“薬品価格競争と特許期限回復法”(“ハッジ-ワックスマン修正案”と略称する)とEUの同様の立法によって限られた特許期間延長を受ける資格がある可能性がある。Hatch-Waxman修正案は、製品開発およびFDA規制審査中に失われた有効特許期間の補償として、承認された製品をカバーする特許を最大5年間延長することを可能にする。しかしながら、試験段階または監督審査中に職務調査を行うことができず、適用の最終期限内に出願を提出できなかった場合、関連特許の満了前に出願を提出できなかった場合、または適用の要求を満たすことができなかった場合、延期されない可能性がある。しかも、延期の長さは私たちが要求したものより短いかもしれない。各承認された製品は1つの特許しか延長できず,展示期間は承認された日から総特許期間を14年以上延長することはできず,承認された薬物,その使用方法または製造方法に関する権利要件のみを延長することができる。もし私たちが特許期間の延長を得ることができない場合、あるいはこのような延長の期限が私たちが要求したものよりも短い場合、適用される候補製品に対して特許権を行使することができる期限が短縮され、競争相手は市場競争製品の承認をより早く得ることができるかもしれない。したがって、私たちは適用された製品からの収入が減少するかもしれない。また、もしこのような状況が発生したら、私たちの競争相手は私たちの臨床と臨床前データを参考にして、私たちの開発と試験への投資を利用して、他の場合よりも早く彼らの製品を発売して、私たちの競争地位、業務、財務状況、運営結果、将来性は深刻な損害を受ける可能性があります。
我々は現在,Vaxartの欧州特許に対する欧州特許庁での反対手続きを行っており,現在控訴段階である。もし私たちがこのような訴訟で成功しなければ、私たちはヨーロッパの他の人たちが私たちの候補製品をコピーすることを阻止できないかもしれないし、もしヨーロッパ特許が支持されたら、私たちはそうすることができないだろう。
私たちは現在、欧州特許庁(EPO)で私たちの欧州特許に反対訴訟を起こしている。欧州特許第3307239号は、ノウォーカーウイルスおよび呼吸器合胞体ウイルスに対するワクチン組成物を要求しているが、欧州特許庁では反対されている。反対党は欧州3307239号特許の有効性を疑問視し、欧州特許庁はこの特許を維持し、最初の独立した権利要求を保持し、従属権利請求書のいくつかの標的を取り消した。反対者はこの決定に上訴したため、反対の最終結果はまだ確定していない。Vaxartが控訴において最終的に勝訴しない場合、それは、他の会社が一部またはすべてのヨーロッパ諸国でそのノウォーカーウイルスおよびRSV製品を複製することを阻止できない可能性があり、反対控訴において特許の有効性を維持すれば、会社はこれらの国で特許を待つことができる。反対されたヨーロッパ特許が欧州特許庁によって部分的または全部撤回された場合、競争相手は、それと競合するノウォーカーウイルスまたはRSVワクチンをより早く販売することができるかもしれないが、Vaxartはそれらに対して特許を主張することができない。VaxartはヨーロッパにはノーウォーカーウイルスとRSV製品をカバーする別の特許があるが、反対派控訴で成功しなければ、私たちは特許保護を2036年に延長することができないだろう。
私たちは私たちの特許と他の知的財産権発明権のクレームに疑問を受けるかもしれない。
私たちまたは私たちの許可者は、元従業員、協力者または他の第三者が、発明者または共同発明者として、私たちの特許、商業秘密、または他の知的財産権において権益を有するクレームの制約を受ける可能性がある。例えば、私たちまたは私たちの許可者は、私たちの候補製品の開発に参加している従業員、コンサルタント、または他の人の義務紛争によって発明権紛争を生じる可能性があります。これらまたは他の挑戦発明権または私たちまたは私たちのライセンシーによる私たちの特許、商業秘密または他の知的財産権の所有権のクレームについては、訴訟を提起する必要があるかもしれない。もし私たちまたは私たちの許可者がこのようなクレームを弁護できなかった場合、金銭損害賠償を支払うことに加えて、私たちの候補製品に非常に重要な知的財産権の独占所有権や使用権のような貴重な知的財産権を失う可能性があります。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。上記のいずれも、当社の業務、財務状況、経営結果、見通しに重大な悪影響を及ぼす可能性があります。
一般リスク因子
証券発行による追加資金調達は既存の株主の権益を希釈する可能性があり、貸借や許可手配による資金調達は、私たちの運営を制限したり、所有権の放棄を要求したりする可能性があります。
私たちは未来に私たちが計画した業務を継続するために多くの追加資本が必要になると予想する。これまで、相当な製品収入を生み出すことができれば、株式発行、特許使用料、債務融資、戦略連合、および任意の協力に関する許可および開発協定によって、私たちの現金需要を満たす予定です。私たちは現在約束された外部資金源を持っていない。私たちが株式証券を発行することで追加資本を調達する場合、私たちの既存株主の所有権は大幅に希釈される可能性があり、これらの証券の条項は清算または他の私たちの普通株主の権利に悪影響を及ぼす特典を含む可能性がある。債務融資および優先株融資に関与する可能性のある合意には、追加債務を招く、資本支出を行う、配当金を発表する、留置権を作成する、株式を償還する、または投資を行うなど、特定の行動をとる能力を制限または制限する契約が含まれる。
もし私たちが第三者との協力、戦略連合またはマーケティング、流通または許可手配を通じてより多くの資金を調達するならば、私たちは私たちの技術、将来の収入源、研究計画、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になる可能性のある条項でライセンスを付与しなければならないかもしれない。もし私たちが必要な時に株式や債務融資を通じてより多くの資金を調達することができない場合、あるいは第三者と受け入れ可能な条項で協力し、戦略連合またはマーケティング、流通または許可手配を行うことによって、私たちは私たちの製品開発または将来の商業化努力を延期、制限、減少または終了することを要求されるか、または私たちが本来自分で開発およびマーケティングする候補製品の権利を付与することができる。
既存の株主は将来株式を売却することで私たちの株価を下落させるかもしれない。
もし私たちの既存株主が公開市場で私たちの普通株を大量に売却する意向を示したり、私たちの普通株の取引価格が低下する可能性があります。私たちの普通株の大量株を公開市場で売却したり、売却が発生する可能性があると考えたりすると、私たちの普通株の市場価格を下げ、追加株式証券を売却することで資金を調達する能力を弱める可能性があります。私たちは売却が私たちの普通株の現在の市場価格に及ぼす影響を予測できない。
税収法や経営活動の変化は、私たちの有効税率に影響を与え、私たちの業務、財務状況、経営業績に悪影響を及ぼす可能性があります。
私たちが経営している任意の司法管轄区の税法の変化、あるいは私たちがこのような司法管轄区域で受けた任意の税務監査の不利な結果は、私たちの将来の有効税率に悪影響を及ぼす可能性があり、これは私たちの業務、財務状況、および経営業績に悪影響を及ぼす可能性があります。
デラウェア州法律の反買収条項は買収をより困難にする可能性があり、私たちの株主が私たちの経営陣を交換または罷免しようとすることを阻止するかもしれない。
私たちはデラウェア州に登録して設立されたので、DGCL 203節の条項によって管轄されています。この条項は、15%以上の発行会社が議決権を持っている株を持っている株主が会社と合併または合併することを禁止しています。これらの条項は,潜在的な買収者に我々の取締役会との交渉を要求することで,より高い入札を受ける機会を提供していると信じているが,一部の株主が要約が有益であると考えていても,これらの条項は適用されるであろう.また、これらの規定は、株主が管理職メンバーを任命する取締役会メンバーを交代させることを困難にし、株主が管理層を交代または更迭するいかなる試みも挫折または阻止する可能性がある。
もしシステムが故障したら、私たちの業務と運営は影響を受けるだろう。
私たちの業務と運営は、私たちのCRO、および私たちの運営に必要なエンティティの運営とシステムを含み、情報技術システムに大きく依存しており、障害、自然災害、セキュリティホール、攻撃、中断、または他のネットワークセキュリティイベントが発生した場合に被害を受けやすい。これらのイベントは、これらのシステムの安定性、完全性またはセキュリティ、およびこれらのシステムによって格納または処理された情報の正確性および完全性を危険にさらし、私たちが業務を成功させる能力を著しく中断する可能性がある。恐喝ソフトウェア攻撃は、すべての業界部門でますます一般的になり、医療市場で運営されている企業の一部に集中し、進行中の業務運営を混乱させ、完全に停止する可能性がある。ロシアとウクライナとの間の持続的な軍事衝突など、ますます激化する世界的な緊張は、サイバーセキュリティ事件の頻度を増加させる可能性がある。
我々のクライアントシステムは,フィールドデータセンターやクラウドサービスを含め,規模や複雑さを増加させ続け,障害や他の割込みの影響を受けやすいようにしている.中断には日常的なメンテナンス、アップグレード、補修の問題が含まれている。これらのシステムは,我々の業務においてより重要で統合され続けており,特に遠隔作業を継続している場合には,それらへの依存が増加し続ける.我々はクラウドサービスに内蔵されている高可用性と冗長性に依存しているが,これらのシステムはプロバイダによって管理されており,我々の直接制御を受けていない.新しいまたは既存のシステムの実装、サポート、セキュリティ、可用性、保守または保守に関連する任意の潜在的な問題および中断は、私たちの運営効率を妨害または低減し、開発計画の交付に大きな影響を与える可能性があります。
釣りや魚叉釣り攻撃,分散拒否サービス攻撃,中間者攻撃,恐喝ソフトウェアや他のマルウェア挿入を含むネットワークセキュリティイベントは,より複雑で頻繁になっている.これらの機密または独自の情報を流用または漏洩したり、企業のITシステムを浸透または破壊しようとしたりする場合、データやアプリケーションの損失や破損、さらには基本業務運営中断および財務損失、および法律および規制責任のリスクを招く可能性がある。したがって、私たちの候補製品のさらなる開発は大幅に延期されるかもしれない。データ漏洩は,キータスクデータや他の知的財産権を暴露したり,会社員や臨床試験に参加した者のPII(個人識別情報)やPHI(保護された健康情報)を暴露したりする可能性がある。成功したネットワーク釣りの試みは、さらなるハッカーの試み、許可されていない電子メールアクセスまたはメッセージ伝達、または他の垂直/水平ネットワーク攻撃をもたらす可能性があります。これらのイベントは、いずれの事件も、私たちのビジネスに影響を与える深刻な名声被害をもたらす可能性があります。
.
もし私たちが効果的な内部統制システムを維持できなければ、私たちは私たちの財務結果を正確に報告したり、不正を発見することができないかもしれない。したがって、投資家は私たちの財務報告に自信を失う可能性があり、これは私たちの普通株の取引価格に負の影響を与えるかもしれない。
サバンズ·オキシリー法404条によると、財務報告書の内部統制に対する私たちの有効性を含む四半期報告書を管理職が提出しなければならない。この評価には、財務報告書の内部統制で発見された私たちの経営陣の重大な弱点を開示する必要がある。私たちは四半期ごとに財務報告書の内部統制の変化を開示することを要求された。また、2022年6月30日の公開流通株は7億ドル未満であり、2022年12月31日から、より規模の小さい報告会社としての資格を再取得したため、財務報告内部統制の監査人認証要求に制約されなくなった
私たちは信頼できる財務報告書を提供するために効果的な開示と内部統制を維持しなければならない。私たちは改善されなければならない分野を決定するために私たちの統制措置を評価してきた。我々の2022年12月31日までの評価によると,2022年12月31日までに我々の内部統制や手続きは有効であるが,過去の大きな弱点を発見し,将来的に再びそうする可能性があると結論した。“重大な欠陥”とは、我々の年度または中期財務諸表の重大な誤報が合理的な可能性があり、タイムリーに防止または発見できないように、財務報告の内部統制に欠陥または欠陥が存在する組み合わせを意味する。有効なこのような制御システムを維持するために必要な制御改善を維持できなければ、経営業績を正確に報告する能力を損なう可能性があり、投資家が私たちが報告した財務情報に自信を失う可能性がある。このような信頼の喪失は私たちの普通株の取引価格に否定的な影響を及ぼすだろう。
私たちのほとんどの施設は既知の地震断裂帯の近くに位置している。地震、火災、または任意の他の悲劇的な事件の発生は、私たちの運営を混乱させたり、重要な支援機能を提供してくれる第三者の運営を混乱させる可能性があり、これは私たちの業務や財務状況に大きな悪影響を及ぼす可能性があります。
私たちは停電、自然災害、テロ、そして私たちの制御範囲を超えた似たような意外な事件のような悲劇的な事件の破壊を受けやすい。私たちの業務の大部分は旧金山湾区にあり、この地域は過去に深刻な地震と火災を経験したことがある。
私たちは災害復旧と事業継続計画を持っていない。地震、洪水、ハリケーン、または他の自然災害は、私たちの運営を深刻に混乱させ、私たちの業務、運営結果、財務状況、将来性に大きな悪影響を及ぼす可能性があります。
自然災害、停電、または他の事件が発生した場合、本社の全部または大部分を使用することができなくなり、私たちの金融システムや製造施設のような重要なインフラを損傷させたり、他の方法で私たちの運営を混乱させたりすることは、困難かもしれませんし、場合によっては、かなり長い間業務運営を継続することはできません。
私たちの環境、社会、または管理責任のより厳しい審査は、追加のコストとリスクを招き続ける可能性があり、私たちの名声、従業員の維持、そして顧客とサプライヤーが私たちとビジネスをする意欲に悪影響を及ぼす可能性があります。
いくつかの顧客、消費者、従業員、および他の利害関係者は、企業市民権および持続可能性を含む環境、社会およびガバナンス(“ESG”)問題にますます注目している。また、上場企業のESG実践に関連する公衆利益と立法圧力は増加し続けている。もし私たちのESG実践が責任ある企業市民に対する規制要件または利害関係者の絶えず変化する期待と基準を満たしていない場合、これらの分野には環境管理、地域コミュニティへの支援、取締役会と従業員の多様性、人的資本管理、従業員の健康と安全実践、会社の管理と透明性、および私たちの運営においてESG戦略が採用されている場合、私たちのブランド、名声、従業員の保持率は負の影響を受ける可能性があり、顧客とサプライヤーは私たちとビジネスをしたくないかもしれない。
また、私たちのESG実践を業界基準と一致させるために努力するにつれて、私たちはこれらの分野での私たちの開示範囲を拡大しており、将来も拡大し続けるかもしれません。私たちは時々環境問題、責任ある調達、社会投資などのいくつかのイニシアティブについて意思疎通を行い、目標を含む。私たちはまた、追加のコストが発生することが予想され、我々の様々なESG慣行を監視、報告、および遵守するために追加のリソースが必要である。ESGに関する事項の追跡と報告基準は比較的新しく、統一されておらず、発展し続けている。私たちは、それと一致する開示フレームワーク(あれば)が時々変化する可能性があり、異なる時期に一致または意味のある比較データが不足する可能性があることを選択する。様々なESG追跡および報告義務を遵守するために適切なシステムおよびプロセスが保証されることは、管理時間および費用を必要とするであろう。さらに、我々のプロセスおよび制御は、常に識別、測定、および報告ESG指標の持続的な発展の基準に適合しているわけではなく、報告基準の解釈は、時間の経過とともに変化する可能性があり、いずれも、これらの目標の達成における我々の目標の重大な修正または報告の進展をもたらす可能性がある。
ESG基準または実践を利害関係者の意思で迅速に採用できなかった場合、そのような計画または目標を達成する過程で失敗または失敗とみなされたり、そのような計画および目標における私たちの進展を全面的に正確に報告することができなかった場合、私たちの名声、サービス、財務業績、および成長は悪影響を受ける可能性がある。しかも、私たちはこのような計画や目標の範囲で批判されるかもしれないし、このような問題について責任感を持って行動していないと思われるかもしれない。私たちの業務はこのような事件の否定的な影響を受けるかもしれない。このような問題、または関連する企業市民および持続可能な開発の問題は、私たちの業務に実質的な悪影響を及ぼす可能性がある。
項目1 B:未解決スタッフの意見
ない。
第2項目:ホテル物件
私たちには不動産は何もありません。2022年12月31日まで、私たちのレンタル施設は以下の通りです
| 位置 |
|
平方フィート |
|
主な用途 |
|
賃貸条項 |
|
|
|
|
|
|
|
|
| サンフランシスコ南部とカリフォルニア州バーリントン |
|
47,453平方フィート |
|
実験室製造工場オフィス |
|
5つのレンタル契約は2025年5月から2029年3月までで満期となります |
| カリフォルニア州サンフランシスコ南部 |
|
23,790平方フィート |
|
製造業と事務業 |
|
2022年、2029年3月に満期になった賃貸契約を締結します |
私たちは私たちの既存の施設が私たちの現在の必要性を満たすのに十分だと信じている。
第三項:その他の法的手続き
連結財務諸表第二部第八項“付記9.支払及び又は有事項、第-(C)節訴訟”に列挙された資料は、参照により本項目に組み込まれる。
当社も当社の業務に関する法的訴訟に時々巻き込まれる可能性があります。現在把握されている情報によると、それに対する任意の未解決の行動に関連する合理的な損失可能な金額または範囲は、全体的に確立された準備金を超えており、その総合財務状況やキャッシュフローには重要ではないと考えられる。しかしながら、現在または将来の任意の紛争解決または法的手続きは、どのような手続きの是非にもかかわらず、巨額のコストを招き、経営陣が業務を成功させるために必要な注意や資源を移転し、私たちの業務、財務状況、運営結果に大きな悪影響を及ぼす可能性がある。
第2項第4項:炭鉱安全情報開示
適用されません。
第II部
第五項:登録者普通株、関連株主事項及び発行者が株式証券を購入する市場
取引情報
私たちの普通株はナスダック資本市場に上場しています。取引コードはVXRTです。
2023年3月14日現在,我々普通株の記録保持者は約2355人である。
株式と補償計画に基づいて発行された証券
次の表には、2022年12月31日までの持分給与計画情報が含まれています。
| 計画種別 |
未償還オプション,制限株式単位,権利を行使する際に発行される証券数(A)(1) |
未平倉オプションの加重平均行権価格(2) |
株式補償計画に基づいて将来発行可能な証券の数((A)欄に反映されている証券を含まない)(3) |
|||||||||
| 証券保有者が承認した持分補償計画 |
15,533,571 | $ | 4.48 | 13,874,692 | ||||||||
| 証券保有者の許可を得ていない持分補償計画 |
— | $ | — | — | ||||||||
| 合計する |
15,533,571 | $ | 4.48 | 13,874,692 | ||||||||
(1)新株は、2016年持分インセンティブ計画および2019年持分インセンティブ計画に従って購入株権を行使せずに発行可能な株式14,725,261株と、2019年の持分インセンティブ計画に基づいて発行された定期制限株式単位の帰属および支払いに基づいて発行可能な株式を含む。
(2)これらの報酬は、これらの報酬が価格を行使していないので、上記脚注1に記載された時間ベースの制限株式単位を含まない。
(3)これには、2019年の株式インセンティブ計画に従って将来発行可能な12,074,692株の普通株と、2022年の従業員購入計画に従って購入可能な1,800,000株の普通株が含まれる。2016年株式インセンティブ計画によると、新たな奨励を与えてはならない。
.
株式表現グラフ
改正後の1934年の“証券取引法”第18節または“取引法”については、以下の業績グラフは“募集材料”とみなされてはならず、米国証券取引委員会に“届出”されていると見なすべきでもなく、Vaxart,Inc.が“証券法”または“取引法”に基づいて提出されたいかなる文書にも引用されているとみなされてはならない。
次の図は我々の普通株であるナスダックバイオテクノロジー指数とナスダック総合指数-2017年12月31日から2022年12月31日までの累積総リターンの比較を示しており、この2つの指数はいずれも初期投資を100ドルと仮定し、すべての配当金に再投資を行っている。このリターンは歴史的価格に基づいており、2018年2月13日の合併までは、歴史的価格はAviragen治療会社の価格であり、将来の業績を指示できない可能性がある。
配当政策
私たちは普通株の配当金を発表したり支払ったりしたことがない。私たちは将来の収益(あれば)を残して、私たちの業務発展を支援するつもりですので、予測可能な未来に現金配当金は支払われません。将来の配当金の支払い(あれば)は、現在の財務状況、経営業績、および現在と予想される現金需要を含む、我々の取締役会が様々な要素を考慮して適宜決定する。
第6項:[保留されている]
プロジェクト七、財務管理部門の財務状況と経営成果の検討分析
以下では、当社の総合財務諸表とその付記を含む、当社の財務状況及び経営結果の検討及び分析を本年度報告の他の部分と一緒に読まなければならない。この討論は私たちの計画、推定、そして信念を反映する多くの展望的な陳述を含む。私たちの実際の結果は展望的陳述で議論された結果と大きく違うかもしれない。これらの差異をもたらすか、または促進する可能性のある要因は、以下および年次報告の他の部分的に議論される要因、特に項目1 Aの“リスク要因”で議論される要因を含む。本年度報告で行った前向きな陳述は,本報告の発表日にのみ行われた。
会社の概要
著者らは臨床段階のバイオテクノロジー会社であり、主に著者らの担体-アジュバント-抗原標準化技術(VAAST)に基づく専用経口ワクチンプラットフォームに焦点を当てて経口組換えワクチンを開発している。われわれの経口ワクチンは、広範かつ持続的な免疫反応を産生することを目的としており、各種感染症を予防することができ、慢性ウイルス感染や癌の治療に有用である可能性がある。私たちの研究ワクチンは注射ではなく室温で安定した錠剤を用いて注射されている
我々は、ノウォーカーウイルス(急性胃腸炎の原因を広く引き起こす)、SARS-CoV-2(2019年のコロナウイルス疾患を引き起こすウイルス(“新冠肺炎”))、インフルエンザ、季節性インフルエンザを含む一連の伝染病に対する予防ワクチン候補を開発している。私たちのノウォーカーウイルス候補ワクチンのいくつかの第一段階の人体研究は成功した。我々GI.1ノウォーカー候補ワクチンの安全性と臨床有効性を評価する第二段階挑戦研究が現在行われている。我々の二価GI.1およびGII 4候補ノウォーカーウイルスワクチンの安全性および免疫原性を評価する第2段階用量範囲の研究が現在行われている。我々は、第1の新冠肺炎候補ワクチンの第1段階の臨床試験を完了し、この研究が主要かつ副次的な終点に達したと報告している。私たちの第2の新冠肺炎候補ワクチンは、2021年末に開始された第2の段階研究の第1の部分が完了した。著者らはまた新型新冠肺炎ワクチンの臨床前仕事を開始し、SARS-CoV-2と他のベータコロナウイルス(例えばSARS-CoV-1とMERS-CoV)に対応するために、有効なパンベタコロナウイルス候補ワクチンを創造することを求めた。データにより、著者らの単価H 1インフルエンザ候補ワクチンは参加者をH 1インフルエンザ感染から保護し、そして2020年に発表された第二段階挑戦研究でリードした発売注射可能ワクチン(柳葉刀ID)である。また,われわれは呼吸器合胞体ウイルス(RSV)(気道感染のよく見られる原因)に対する予防的候補ワクチンおよびわれわれが初めて子宮頸癌とヒトパピローマウイルス(HPV)による異型増殖に対する治療的候補ワクチンについて臨床前データを生成した
Vaxart Biosciences,Inc.は2004年3月にカリフォルニア州で最初に登録成立し,名称はWest Coast Biologals,Inc.,Inc.である。合併条項により,AviragenはVaxart,Inc.,Private VaxartはVaxart Biosciences,Inc.と改称される.
当社は2022年12月31日から比較的小規模な報告会社(SRC)資格を新たに備えているため、財務報告内部統制監査師認証(ICFR)の要求を受けなくなった。
財務運営の概要
収入.収入
顧客サービス契約収入
2021年12月31日までの1年間に、修正された固定価格顧客サービス契約から13,000ドルの収入を得て、契約は現在完了しています
2016年4月、Aviragenは日本市場でInavirに関する特定の特許権使用料を2000万ドルでHealthcare Royalty Partners III、L.P.(以下HCRP)に販売した。私たちはHCRPに最初の300万ドルを支払い、3月31日までの年間期間に稼いだ100万ドルの印税の15%を加えた。合併時,HCRPの推定将来収益は公正価値で再計量され,1,590万ドルと推定され,負債として入金され,実際の利子法を用いて手配された残存推定寿命内に償却された。2022年12月31日および2021年12月31日に,公正価値がそれぞれ500万ドルおよび1,060万ドルと推定された場合,推定された将来収益が再計量され,2022年12月31日までの年度および2021年12月31日までの年度のリスコアリング収益はそれぞれ700万ドルおよび380万ドルとなった。たとえ取引項目の関連特許権使用料を保持しなくても、金額がHCRPに送金されるため、関連負債と関連利息の金額が完全に償却されるまで、これらの特許権使用料に関する収入を記録し続ける。
研究と開発費
研究開発費とは,我々の平板ワクチンプラットフォームの開発や,我々の平板ワクチン候補の臨床前と臨床開発活動を支援するなど,研究所を行うコストである。私たちはそれらが発生したので、すべての研究と開発コストを確認した。研究と開発費用には主に以下の項目が含まれている
| ● |
従業員に関する費用には、賃金、福祉、株式給与が含まれる |
| ● |
私たちを代表して臨床試験を行っている契約研究機関(“CRO”)との合意による費用 |
| ● |
臨床試験のための製品を生産する契約製造組織(“CMO”)との合意に基づいて生成される費用 |
| ● |
臨床試験のための候補ワクチンの製造に必要な材料および試験サービスの分析および放出の費用; |
| ● |
大口ワクチンと錠剤の生産活動の効率と生産量を高めるために内部と外部で発生する技術開発費用 |
| ● |
臨床前研究活動に関連する実験室用品とサプライヤー費用; |
| ● |
私たちの臨床、規制、製造活動を支援するサービスの顧問料 |
| ● |
施設、減価償却、分配された間接費用。 |
私たちは内部費用を特定のプロジェクトに割り当てないつもりだ。私たちの従業員や他の内部資源はいずれの研究計画とも直接縛られておらず、通常は複数のプロジェクトに配置されている。内部研究と開発費用を総額として列記する。
我々を代表して臨床試験を行ったCROと我々の候補錠剤ワクチンを生産するCMOにとっては,2022年には低下しているにもかかわらず,これらのコストは2022年には低下しているにもかかわらず,現在の生産活動の大部分は内部で行われているからである。私たちはすべてのワクチンプロジェクトのこのような外部費用を統計した。臨床前研究や過程開発の外部コストを特定の項目に割り当てることはない。
次の表は,我々の一定期間の研究開発費を示し,各ワクチン計画および臨床前研究と過程開発単独で発生する外部コスト(千単位)を決定した
| 十二月三十一日までの年度 |
||||||||
| 2022 |
2021 |
|||||||
| 外部計画コスト: |
||||||||
| ノーウォーカー·ウイルス計画 |
$ | 11,066 | $ | 4,449 | ||||
| 新冠肺炎計画 |
6,758 | 9,847 | ||||||
| 他のすべての計画は |
129 | — | ||||||
| 臨床前研究 |
1,414 | 2,199 | ||||||
| プロセス開発 |
2,262 | 2,235 | ||||||
| 外部総コスト |
21,629 | 18,730 | ||||||
| 内部コスト |
59,425 | 30,019 | ||||||
| 研究開発総コスト |
$ | 81,054 | $ | 48,749 | ||||
われわれは2023年以降にわれわれの平板ワクチン候補を臨床試験に推進し,規制機関によるわれわれの平板ワクチン候補の承認を求め,可能な商業発売に備えて,これらすべてを製造と在庫に関するコストに大量に投資する必要があると予想している。私たちが許可、協力、または協力協定を締結する範囲では、このようなコストの大部分は第三者が負担する可能性がある。
必要な臨床試験を行って監督管理の承認を得る過程は高価で時間がかかる。私たちは私たちの平板ワクチン候補を市場承認を得ることに絶対に成功しないかもしれない。著者らの候補錠剤ワクチンの商業化に成功する可能性は未来の試験で得られた臨床データ、競争、製造能力と商業実行可能性を含む多くの要素の影響を受ける可能性がある。そのため、私たちの研究開発プロジェクトの持続時間と完成コストを決定することができず、私たちがいつ、そしてどの程度私たちの錠剤候補ワクチンの商業化と販売から収入を得るかを決定することもできない。
一般と行政費用
一般および行政費用には、人件費、保険、分担費用、外部専門サービス費用が含まれ、法律、監査、会計、公共関係、市場研究、その他のコンサルティングサービスが含まれる。人件費には賃金、福祉、株式ベースの給与が含まれている。分担費用には賃貸料、減価償却、その他の施設関連費用が含まれている。
経営成果
以下の表は、連結業務表と総合損失報告書の中で選定された項目の期間変動状況(百分率を除く)を示しています
| 十二月三十一日までの年度 |
||||||||||||
| 2022 |
変更率 |
2021 |
||||||||||
| 収入.収入 |
$ | 107 | (88 | )% | $ | 892 | ||||||
| 運営費 |
114,694 | 56 | % |
73,644 | ||||||||
| 営業損失 |
(114,587 | ) | 58 | % |
(72,752 | ) | ||||||
| 営業外純収入 |
6,896 | 189 | % |
2,389 | ||||||||
| 所得税前損失 |
(107,691 | ) | 53 | % |
(70,363 | ) | ||||||
| 所得税支給 |
67 | (37 | )% | 107 | ||||||||
| 純損失 |
$ | (107,758 | ) | 53 | % |
$ | (70,470 | ) | ||||
総収入
下表は、12月31日までの年間収入の期間変化(百分率を除く千単位)をまとめた
| 2022 |
変更率 |
2021 |
||||||||||
| 顧客サービス契約収入 |
$ | — | (100 | )% | $ | 13 | ||||||
| 将来の特許使用料の販売に関する非現金特許権使用料収入 |
107 | (88 | )% | 879 | ||||||||
| 総収入 |
$ | 107 | (88 | )% | $ | 892 | ||||||
顧客サービス契約収入
2021年12月31日までの1年間に、顧客サービス契約から13,000ドルの収入を得ました。これらの収入は2019年7月に署名された改訂された固定価格契約から確認され、総額は617,000ドルであり、現在この契約を完了しています。2022年12月31日までの1年間、顧客サービス契約からの収入は何も確認されていません。
将来の特許使用料の販売に関する非現金特許権使用料収入
2022年12月31日までの年度では,将来の特許使用料の販売に関する非現金特許権使用料収入は10.7万ドルであるのに対し,2021年12月31日現在の年度は87.9万ドルである。3月31日までの毎年、非現金特許権使用料収入は330万ドルに達する可能性がある。2021年12月31日までの年度では,2021年3月31日までの年度の493,000ドルと,2022年3月31日までの年度に関する386,000ドルを確認した。
総運営費
下表は、12月31日までの年度の営業費用の期間変動(百分率を除く千計)をまとめた
| 2022 |
変更率 |
2021 |
||||||||||
| 研究開発 |
$ | 81,054 | 66 | % |
$ | 48,749 | ||||||
| 一般と行政 |
29,386 | 34 | % |
21,890 | ||||||||
| 無形資産減価準備 |
4,254 | 42 | % | 3,005 | ||||||||
| 総運営費 |
$ | 114,694 | 56 | % |
$ | 73,644 | ||||||
研究と開発
2022年12月31日までの年間で、研究開発費は8,110万ドルで、2021年12月31日までの年間4,870万ドルより約3,230万ドル増加し、66%に増加した。増加の要因は、従業員の増加に関連する株式給与や施設配分、より高い内部製造コスト、我々のノウォーカー候補ワクチンに関する臨床試験費用の増加およびより高い減価償却であるが、新たな冠肺炎候補ワクチンに関する首席営業職コストの低下部分はこの増加を相殺している
2023年には,我々のノウォーカーウイルス候補ワクチンに関する製造や臨床試験に大量の研究開発費が生じることが予想される。
一般と行政
2022年12月31日までの年度の一般および行政費は2,940万ドルであり,2021年12月31日までの年度の2,190万ドルより約750万ドル増加し,34%増加した。2022年の業績増加の要因は、株主訴訟を解決するコストの増加、人件費の増加、非現金株補償や裁決改正とは無関係な費用、法律やその他の非専門費の増加を含むが、前取締役会議長に付与された未償還オプションの条項を修正するための一度の非現金費用がないため、増加した費用を相殺している。
無形資産減価準備
無形資産減額430万ドルと300万ドルはそれぞれ2022年12月31日と2021年12月31日までの日本Inavirインフルエンザワクチンの特許権使用料収入に関する開発技術の部分減値を代表し、主に新冠肺炎の大流行による隔離、社会距離、マスク着用が増加したと予測を下方修正した
その他の収入(費用)
下表は、12月31日までの年間営業収入の期間変動(百分率を除く千単位)をまとめたものである
| 2022 |
変更率 |
2021 |
||||||||||
| 利子収入 |
1,247 | 1,440 | % |
81 | ||||||||
| 将来の特許使用料の販売に関する非現金利息支出 |
(1,305 | ) | (12 | )% | (1,480 | ) | ||||||
| 将来の特許権使用料責任の収益を再計量する |
6,960 | 84 | % |
3,789 | ||||||||
| 純為替損失 |
(6 | ) | 500 | % |
(1 | ) | ||||||
| 営業外純収入 |
$ | 6,896 | 189 | % |
$ | 2,389 | ||||||
2022年12月31日までの1年間で、純営業外収入は690万ドル、2021年の純営業外収入は240万ドルを記録した。
2022年の将来の特許権使用料責任の再測定収益は,HCRPに支払う金額に対応する予測を引き下げたためであり,新冠肺炎の大流行が始まって以来,日本のイナビルインフルエンザワクチンの特許権使用料が低下したため,主に新冠肺炎による隔離,社会的距離,マスク着用が増加したためであると予測した。
非現金利息支出は将来の特許使用料の販売に関連しており,Inavirから得られた特許使用料収入を債務としてHCRPに支払うことに関連しており,2022年は130万ドル,2021年の150万ドルを下回っており,HCRPへの未返済残高が返済され再計量されているためである。私たちはHCRPに対する私たちの負債を再評価した後、2023年にさらに減少すると予想する。
所得税支給
下表は、12月31日までの年間所得税支給の期間変動(単位千、百分率を除く)をまとめたものである
| 2022 |
変更率 |
2021 |
||||||||||
| 特許使用料収入の外国源泉徴収税 |
$ | 5 | (89 | )% | $ | 44 | ||||||
| 会社間利子払いの外国税金 |
59 | (5 | )% | 62 | ||||||||
| 州所得税 |
3 | 200 | % | 1 | ||||||||
| 所得税支給 |
$ | 67 | (37 | )% | $ | 107 | ||||||
それぞれ2022年12月31日と2021年12月31日までの年度において、所得税の大部分は、2022年の会社間利息で支払うべき外国税と、2022年と2021年に日本でInavirを販売したことで得られた特許使用料収入の外国源泉徴収税である。私たちは日本でInavirを販売して得られた特許使用料収入に徴収された外国源泉徴収税は、外国税収として回収できるかもしれませんが、繰延税金資産に100%の推定免税額を記録したため、費用を計上しました。記録された所得税支出額はInavir特許使用料に比例し,HCRPに移行する部分を含め,特許使用料収入の減少に伴い低下した。しかも、私たちはアメリカで州所得税の費用を発生させた。
流動性と資本資源
我々の主な融資源は,公開発売中に普通株と普通株承認権証を売却·発行し,引受権証を行使する収益である.2020年10月には、我々の普通株の株式を売却することができ、総発行価格は最大2.5億ドルに達する公開市場販売協定(“2020年10月現金自動支払機”)に合意した。2020年10月のATM機により合計1.334億ドルの総収益で普通株を売却した後、2021年9月に2020年10月のATM機を終了し、制御対象株式発売および販売協定(“2021年9月ATM”)を締結し、この合意により、吾らは時々販売代理を通じて総発行価格が1億ドルに達する普通株を発売·販売することができる。私たちは直接費用を発生させ、2021年9月のATMで株式を売却して得られた利益の3.0%までの販売手数料を支払う。
2022年12月31日まで、現金等価物、制限現金、有価証券を含む9570万ドルの現金を持っています。それ以来,2023年3月14日現在,2021年9月のATMでの普通株の売却により150万ドルの純収益を得ている。2021年9月のATM機によると、約7900万ドルの純収益が使用できる。最近のシリコンバレー銀行(SVB)の影響を受け、連邦預金保険会社のSVB買収を含め、同社は以前SVBで保有していた任意の現金や他の預金をより大きな金融機関に移転することにした。私たちは以前SVBで持っていたすべての現金と他の預金を使用することができる。私たちはSVBの状況が私たちの財務状況や運営に実質的な影響を与えないと予想する。
2023年第1四半期、同社は主にそのノウォーカーウイルス経口ワクチン候補案の推進に集中し、SARS-CoV-2ヒト挑戦研究の開始を延期することを決定した。同社はまた、運営コストを低減し、従業員チームがその業務ニーズにより良く適応できるように再編計画を実施している。その計画はその会社の約27%のリストラを招いた。
私たちは、本年度報告書が発表された日から、私たちの既存の資金(2023年に受け取った資金を含む)が、少なくとも1年間の資金を提供するのに十分だと信じている。その後運営を継続するためには、より多くの証券を売却することや他の方法でより多くの資金を調達する必要があると予想される。私たちの運営ニーズには、運営資本や資本支出に資金を提供するのに必要な金額を含む運営業務の計画コストが含まれています。私たちの未来の資本需要と利用可能な資金の十分性は多くの要素に依存し、最も明らかなのは私たちが製品とサービスを商業化する能力に成功していることだ。
私たちは協力と協力協定を通じて私たちが行っている業務に大きな資金を提供するかもしれません。これは、私たちのリスクを下げ、私たちの現金滑走路を拡大すると同時に、候補ワクチンから得られる最終収入のシェアを減少させます(もしあれば)。政府プロジェクトの助けを借りて、私たちは特定の活動に資金を提供することができるかもしれない。追加株式の売却は私たちの株主の持分をさらに希釈させるだろう。私たちはまた、債務融資を通じて私たちの業務に資金を提供することができ、これは債務返済義務につながり、このような債務を管理するツールは、私たちの業務の運営と融資契約を制限することができます。もし私たちが十分な金額や許容可能な条件で追加資金を調達できない場合、私たちは私たちの製品開発または将来の商業化努力を延期、制限、減少または終了することを要求されるか、または私たちが自分で開発し、マーケティングすることをより望んでいた候補ワクチンの権利を付与することができるかもしれない。このような行動のいずれも私たちの業務、運営結果、そして将来性を損なう可能性がある。
私たちの未来の支出需要は多くの要素に依存するだろう
| ● |
私たちは候補製品計画の臨床前研究の時間とコストについて |
| ● |
私たちが計画した候補製品の臨床試験の時間とコストは |
| ● |
私たちの製造能力は、契約製造組織の可用性を含み、合理的なコストで候補製品を供給する |
| ● |
イナビアの販売で受け取った特許使用料の額と時間 |
| ● |
私たちが求めている候補製品の数量と特徴は |
| ● |
規制承認の結果、時間、コストを求める |
| ● |
私たちの未来の製品の商業販売から得られる収入は、監督部門の承認が必要になるだろう |
| ● |
私たちが将来達成する可能性のある任意の協力、許可、相談、または他の計画の条項と時間 |
| ● |
任意の特許または特許出願または他の知的財産権の許可、提出、起訴、維持、抗弁および実行に関連する任意の支払いの額および時間; |
| ● |
私たちは他の製品と技術の程度を許可したり獲得したりする。 |
キャッシュフロー
次の表は、示す期間のキャッシュフロー(千単位)をまとめた
| 十二月三十一日までの年度 |
||||||||
| 2022 |
2021 |
|||||||
| 経営活動のための現金純額 |
$ | (94,779 | ) | $ | (59,832 | ) | ||
| 投資活動のための現金純額 |
(20,415 | ) | (49,097 | ) | ||||
| 融資活動が提供する現金純額 |
17,462 | 125,804 | ||||||
| 現金、現金等価物、および制限的現金純増加 |
$ | (97,732 | ) | $ | 16,875 | |||
経営活動に使われている現金純額
2022年と2021年、私たちの経営活動はマイナスキャッシュフローを生み出し、それぞれ9480万ドルと5980万ドルだった。2022年の経営活動で使用される現金は、純損失107.8ドルと運営資本の470万ドル増加の現金部分の援助に使用されるが、減価償却や償却に関連する非現金収入純額、株式報酬、無形資産減価、将来の特許権使用料負債の再計量の収益、および将来の特許権使用料の販売に関する非現金利息支出の調整部分によって相殺される。2021年の経営活動のための現金減少は、7,050万ドルの純損失を援助するための現金および210万ドルの運営資本の増加が原因であるが、減価償却や償却に関する非現金収入、株式ベースの報酬、将来の特許権使用料の販売に関する非現金利息支出、および将来の特許権使用料の販売に関する非現金収入の調整部分によって相殺され、これらの収入総額は1,280万ドルである
投資活動のための現金純額
2022年、私たちは1080万ドルを使って有価証券(満期日を除く)を購入した。2021年には480万ドルで商業買収を行い、3910万ドルで有価証券を購入した(満期日を除く)。2022年12月31日と2021年12月31日までの会計年度では、それぞれ960万ドルと520万ドルを使って不動産や設備を購入した
融資活動が提供する現金純額
2022年には、2021年9月のATMでの普通株販売から1720万ドル、株式オプション行使から20万ドルを獲得した。2021年には,2020年10月のATMでの普通株式販売から122.2ドル,一般権証の行使により190万ドル,株式オプションの行使により170万ドルを獲得した
契約義務と商業承諾
2022年12月31日現在、以下の契約義務と商業的約束(千単位)があります
| 契約義務 |
合計する |
|
1-3年 |
3-5年 |
>5年 |
|||||||||||||||
| 長期債務、HCRP |
$ | 9,695 | $ | 95 | $ | 1,878 | $ | 3,415 | $ | 4,307 | ||||||||||
| 賃貸借契約を経営する |
29,392 | 4,072 | 8,569 | 10,102 | 6,649 | |||||||||||||||
| 購入義務 |
7,879 | 7,879 | — | — | — | |||||||||||||||
| 合計する |
$ | 46,966 | $ | 12,046 | $ | 10,447 | $ | 13,517 | $ | 10,956 | ||||||||||
長期債務、HCRP。2016年に調印された協定によると、HCRPに前300万ドルを支払う義務があり、3月31日までの毎年Inavirを販売して稼いだ100万ドルの特許使用料収入の15%を支払う義務がある。詳細は第2部第8項連結財務諸表付記7を参照。
賃貸借契約を経営する。経営リース金額には、契約されたレンタルの合計1300万ドルを含む、当社の初期期限が1年を超えた取消不可能な経営リース項目の将来の最低レンタル支払いが含まれています。詳細は第2部第8項連結財務諸表付記8を参照。
購入義務。*これらの額には、貨物またはサービスを受けていない契約製造業者およびサプライヤーとの約束を含む、通常のビジネスプロセスにおけるすべての未締結購入注文および契約義務の推定値が含まれています。私たちは、一般的に強制的に実行可能で法的拘束力のあるすべての未締結注文を約束と見なしています。条項は、貨物を渡す前に、またはサービスを履行する前に、私たちの業務ニーズに応じてキャンセルを選択することを可能にするかもしれません。
重要な会計政策と試算
我々の経営陣の財務状況と経営結果の検討と分析は、私たちの総合財務諸表に基づいており、これらの報告書はアメリカで公認されている会計原則に基づいて作成されている。これらの財務諸表を作成する際には、資産、負債、費用報告金額に影響を与える見積もりと判断を行う必要があります。持続的な基礎の上で、私たちはこのような推定と判断を評価する。私たちの推定は歴史的経験と私たちがこのような状況で合理的だと思う様々な仮定に基づいている。これらの見積りと仮定は,資産や負債の帳簿価値を判断する基礎を構成し,他のソースからは見えない費用を記録している。実際の結果はこれらの推定とは大きく異なる可能性がある。以下で議論する会計政策は、これらの政策がより重要な分野に関連しているため、経営陣の判断と見積もりに関連しているため、我々の歴史や将来の業績を知るために重要であると考えられる。
研究と開発費用を計算すべきである
臨床前研究と臨床試験および契約製造活動を含む第三者サービスプロバイダによる研究と開発活動の見積もりコストの計上費用を記録した。我々は,提供されたサービスの推定金額に基づいて研究開発活動の推定コストを記録し,合併貸借対照表には発生したが他の計上すべき負債に計上されていないコストと,総合経営報告書および総合損失に研究開発費を計上するコストを計上した。このような費用は私たちの研究開発費の重要な構成要素かもしれない。
内部者や外部サービス提供者とサービスの進捗や完了段階,およびそのようなサービスに支払う取り決め費用を検討することで,完了した作業量を想定している.私たちは各報告期間の計算すべき残高を決定する時に重要な判断と推定をする。実際のコストの理解に伴い、私たちは計算計算量を調整した。
無形資産
合併で買収された無形資産は当初,推定公正価値2,030万ドルで記録されており,このうちInavirに関する開発技術はリスコアリング前に,今後11.75年の特許使用料の推定期間内に直線的に償却され,Relenzaに関する開発技術は2022年と2021年12月31日に180万ドルが完全に償却された。2022年12月31日現在,Inavirに関する開発技術は500万ドルに再評価され,記録された減値損失は430万ドルであった。これらの推定値は,独立第三者が推定した将来の収入ストリームの割引キャッシュフローに基づいて作成されており,主観的である.2022年12月31日までに再評価された公正価値は、今後6.9年間の特許使用料の残り期間内に直線的に償却される。
株に基づく報酬
著者らは、付与日に従業員、非執行役員と顧問に与えられたすべての株式オプション奨励の公正価値を測定し、これらの奨励の公正価値を記録し、推定された没収を差し引いて、サービス期間内の補償費用とした。オプションの公平価値はBlack-Scholes推定モデルを用いて推定され,記録された費用はいくつかの変数に関する主観的仮定の影響を受け,以下のようになる
所期期限−これは、我々が付与した株式報酬の予期される未償還期間であり、簡略化された方法(その元の契約期間および平均帰属期間の算術平均値)を使用して決定される。私たちの歴史的情報は非常に限られており、私たちの株式奨励のために未来の行使モデルと付与後の雇用終了行為に対する合理的な期待を立てることができない。2022年に付与されたオプションに適用される加重平均によれば、期待期限名目で10%減少することは、公正価値と関連補償費用を約2.2%減少させる。
予想変動率−これは、我々の普通株式価格が変動したか、または変動すると予想される振幅の測定である。2020年初め以降、測定日オプションの期待期限に対応するトレーサビリティ期間内の自株の履歴変動率に基づいて変動率を測定した。2022年に付与されたオプションに適用される加重平均値によれば、予想変動率は名目で10%(126%から113%に低下)低下し、公正価値および関連補償費用を約4.3%減少させる。
無リスク金利−これは、株式ベースの報酬の予想期限に対応する測定日に基づく米国債収益率曲線である。
配当を期待する*-私たちは配当金を支払いませんし、予測可能な未来に配当金を支払うつもりもありません。したがって、私たちが使用する期待配当収益率はゼロだ。
罰金率−これは、付与されないと予想される報酬金額の測定であり、四半期ごとに再評価される。ペナルティ率の増加はサービス期間初期関連補償費用の小幅な減少を招くと予想されるが,各奨励の最終費用は付与オプションの数に付与日の公正価値を乗じたものであるため,記録の総費用に影響はない.
最近発表された会計公告
2022年の新会計基準の発表に関する情報は、連結財務諸表第2部付記2の“最近の会計公告”を参照してください。項目8は、私たちの連結財務諸表に実質的な影響を与えるか、または実質的な影響を与えることが予想されています。
第七A項。市場リスクの定量的·定性的開示について
金利感度
私たちの投資活動の主な目標は元本を維持することであり、同時にリスクを著しく増加させることなく、投資から得られる収入を最大化することだ。私たちのすべての現金はドルで計算して、銀行口座に保管するか、通貨市場基金に保管して、現在稼いでいる利息は少ないです。金利は十分低く、借入基準金利が1%増加するごとに、現金預金から稼いだ利息はわずかに増加すると信じています。
為替レート感度
私たちの特許使用料収入はドルで計算され、円で計算される売上に基づいているため、ドル対円の強さが1%増加するごとに、特許権使用料収入は1%減少する。私たちのすべての他の収入とほとんどの支出、資産、負債はドル建てであるため、私たちは最近重大な外貨収益を経験したことがなく、近い将来外貨収益や損失はそれほど大きくないと予想される。
8項:財務諸表と補足データ
| 連結財務諸表索引 |
| ページ |
|
|
|
|
| PCから独立した公認会計士事務所WithumSmith+Brownの報告 |
| 88 |
| 合併貸借対照表 |
| 92 |
|
|
|
|
| 合併経営報告書と全面赤字 |
| 93 |
|
|
|
|
| 株主権益合併報告書 |
| 94 |
|
|
|
|
| 統合現金フロー表 |
| 95 |
|
|
|
|
| 連結財務諸表付記 |
| 97 |
独立公認会計士事務所報告
株主や取締役会に
Vaxart,Inc.
連結財務諸表と財務報告書内部統制の見てみよう
Vaxart,Inc.(“当社”)2022年12月31日と2021年12月31日までの連結貸借対照表,2022年12月31日までの2年間の各年度の関連総合経営報告書と全面赤字,株主権益と現金流量,および関連付記(総称して“総合財務諸表”と呼ぶ)を監査した。また、中で確立された基準に基づいて、2022年12月31日までの財務報告内部統制を監査した内部制御—フレームワークを統合するテレデビル委員会(COSO)は組織委員会(COSO)が発表した(2013)を後援している。
上記の総合財務諸表は、米国公認の会計原則に従って、当社の2022年、2022年および2021年12月31日の財務状況、および2022年12月31日までの2年間の各年度の経営業績およびキャッシュフローを各重大な面で公平に反映していると考えられる。同様に、当社はすべての実質的な面で2022年12月31日までの財務報告に対する有効な内部統制を維持しており、その根拠は内部制御--統合フレームワーク(2013)COSOによって発表されます。
意見の基礎
当社経営陣は、これらの総合財務諸表の作成を担当し、財務報告の有効な内部統制を維持し、財務報告の内部統制に関する報告書に含まれる財務報告の内部統制の有効性を評価する。私たちの責任は、会社の連結財務諸表について意見を発表し、私たちの監査に基づいて会社の財務報告内部統制に意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、連結財務諸表に重大なミスがないかどうか、エラーによるものであっても詐欺であっても、すべての重大な点で財務報告に対する有効な内部統制が維持されているかどうかを決定するために、合理的な保証を得るために監査を計画し、実行することを要求する。
我々の連結財務諸表の監査には、連結財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであっても詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれる。これらの手続きは、連結財務諸表中の金額および開示に関する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、合併財務諸表の全体列報の評価も含まれています。我々の財務報告の内部統制の監査には、財務報告の内部統制を理解すること、重大な弱点があるリスクを評価すること、評価されたリスクテストに基づいて内部統制の設計と運営有効性を評価することが含まれる。私たちの監査はまた、私たちがこのような状況で必要だと思う他の手続きを実行することを含む。私たちは私たちの監査が私たちの意見に合理的な基礎を提供すると信じている。
財務報告の内部統制の定義と限界
会社の財務報告に対する内部統制は、アメリカ合衆国で一般的に受け入れられている会計原則に基づいて、財務報告の信頼性と外部目的の財務諸表の作成に合理的な保証を提供することを目的としたプログラムである。会社の財務報告に対する内部統制には、(1)会社の資産の取引および処分を合理的かつ詳細かつ正確に反映した記録を保存することに関する政策および手順、(2)アメリカ合衆国で一般的に受け入れられている会計原則に従って財務諸表を作成するための必要な取引記録を提供すること、および会社の収支が会社の経営陣および取締役の許可のみに基づいて行う政策および手順、および、会社の収支が会社の管理職および取締役の許可のみに基づいて行われる政策および手順が含まれる。(三)財務諸表に重大な影響を及ぼす可能性のある不正買収、使用又は処分会社の資産の行為を防止又はタイムリーに発見し、合理的な保証を提供する。
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来的にどのような有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムの遵守度が悪化する可能性がある.
重要な監査事項
以下に述べる重要な監査事項は、監査委員会が監査委員会に伝達または要求する当期総合財務諸表監査によって生じる事項を指すことである:(1)総合財務諸表に対して大きな意味を有する勘定または開示に関するものであり、(2)私たちが特に挑戦的で主観的または複雑な判断に関するものである。重要な監査事項の伝達は、総合財務諸表に対する私たちの全体的な意見をいかなる方法でも変えることはなく、以下の重要な監査事項を伝達することによって、重要な監査事項またはそれに関連する勘定または開示について個別の意見を提供することもない。
臨床と製造費用を計算すべき−合併財務諸表付記2を参照−
重要な監査事項の説明
同社は,第三者サービスプロバイダの特定のタスク達成における進捗状況の評価に基づき,臨床前研究,臨床試験,製造活動で発生するコストを研究開発費として確認した。支払い時間はコストが料金として確認されている期間とは大きく異なるかもしれません。まだ支払われていないサービス料金は課税料金であることが確認されました。
特定のタスクを達成するためのサプライヤーの進展を推定する際に、同社は、患者登録、臨床サイト活性化、またはサプライヤー情報などの実際に発生するコストのようなデータを使用する。これらのデータは,会社員や第三者サービスプロバイダの試験進捗や完了状態やサービス完了状況に関する報告や検討によって得られたものである.
進行中の臨床前研究と臨床試験活動の数量及び臨床試験と製造費用を推定することに関連する主観性を考慮し、監査累積の臨床と製造費用は特に主観的判断に関連し、それによって著者らはこの事項が重要な監査事項であることを確定させた。
監査で重要な監査事項をどのように処理するか
私たちが蓄積した臨床前研究、臨床試験、製造費用に関する監査プログラムは以下の内容を含む
| ● | 著者らは累積した臨床前研究、臨床試験と生産費用見積もりの制御の設計、実施と有効性をテストした。 |
| ● | 私たちは研究、協力、そして製造協定と契約とその修正案のサンプルを得て読んだ。 |
| ● | 著者らは臨床前研究、臨床試験と生産活動状況に関する公開情報(例えばニュース原稿と投資家陳述)と取締役会材料を評価した。 |
| ● | 選定した契約と契約について,前期末の計上額と今年度の活動を比較し,会社の見積もり方法の正確性を評価した。 |
| ● | 当社の第三者サービスプロバイダから臨床試験と生産状態の書面確認を受けました。 |
| ● | 研究開発費の確認および計上すべき費用と確認された具体的な金額を選択し、管理層のサプライヤー進捗の見積もりを評価し、以下の手順を実行した |
| o | 会社の臨床運営や製造運営者と確実な問い合わせを行った。 |
| o | 関連する作業説明書、購入注文、または他のサポート文書(例えば、会社と第三者サービスプロバイダとの間の通信)を読む。 |
| o | 経営陣の判断を得られた証拠と比較することで経営陣の判断を評価する。 |
| o | 研究開発費に関するすべての契約リストを取得し、計算すべき項目の完全性を評価する。 |
| o | 経営陣が合併財務諸表の中で臨床試験と生産活動の計算すべき項目を計算する数学正確性をテストした。 |
将来の特許権使用料販売に関する無形資産減額と負債再評価–連結財務諸表の付記2、5、7を参照
重要な監査事項の説明
管理層は将来のキャッシュフローモデルを有限年の無形資産減価を評価する重要な参考資料として利用し、HCRPと締結した特許権使用料権益買収協定(“HCRP協定”)に基づいて未来の特許権使用料の売却に関する負債を決定する。これらのモデルは、将来の経済状況を含む将来のキャッシュフローや他の要因の推定に依存する。同社の将来の特許権使用料販売に関する有限寿命無形資産と負債の総合残高は、2022年12月31日現在、それぞれ500万ドルと570万ドルである。当社は2022年12月31日までの年間限定期無形資産を評価し、減価費用430万ドルを確認した。また、同社は将来の特許使用料の売却に関する負債の収益700万ドルの見直しも記録している。減値およびリスコアリングは,負債関連無形資産の減値評価に用いた改訂特許権使用料予測および割引率仮定から計算される。
無形資産の減価評価と将来の特許使用料の売却に関する負債の再評価を重要な監査事項として決定する。重要な監査事項の主な考慮要因は、無形資産減額評価と関連負債再評価における経営陣の重大な判断であると考えられる。プログラムを実行し、管理層が“フルオロカーボン削減案協定”の有効期間内に予想される将来の特許使用料の現在の推定に対する管理層の仮定を実行するためには、高度な監査人の判断力、努力、主観が必要である。
監査で重要な監査事項をどのように処理するか
この問題を処理することは、統合財務諸表に対する私たちの全体的な意見を形成するための実行手順および評価監査証拠に関するものである。これらのプログラムには,無形資産推定に関する制御措置の有効性をテストすることと,公正な価値を推定するための仮定を作成することがある.他にも、これらのプログラムは、無形資産および将来の特許権使用料のキャッシュフロー予測に関連する方法の適切性を評価するステップと、推定に使用される基礎データの完全性および正確性をテストするステップと、将来の売上高、長期成長率、および将来の経済状況を含む重大な仮定の合理性を評価するステップとを含む、管理層が推定を作成するプロセスをテストすることを含む。これらの仮定の評価に用いる仮説が合理的であるかどうかについては,(1)歴史的業績,(2)業界や経済予測,および(3)これらの仮定が監査の他の分野で得られた証拠と一致するかどうかを考慮する.
/s/
2019年以来、当社の監査役を務めてきました。
2023年3月15日
PCAOB ID番号
Vaxart,Inc.そして付属会社
合併貸借対照表
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
| 2022年12月31日 | 2021年12月31日 | |||||||
| 資産 | ||||||||
| 流動資産: | ||||||||
| 現金、現金等価物、および限定現金 | $ | $ | ||||||
| 短期投資 | ||||||||
| 売掛金 | ||||||||
| 前払い費用と他の流動資産 | ||||||||
| 流動資産総額 | ||||||||
| 長期投資 | ||||||||
| 財産と設備、純額 | ||||||||
| 使用権資産、純額 | ||||||||
| 無形資産、純額 | ||||||||
| 商誉 | ||||||||
| その他長期資産 | ||||||||
| 総資産 | $ | $ | ||||||
| 負債と株主権益 | ||||||||
| 流動負債: | ||||||||
| 売掛金 | $ | $ | ||||||
| 贈与収入を繰延する | ||||||||
| その他流動負債 | ||||||||
| 賃貸負債の当期部分を経営する | ||||||||
| 将来の特許使用料の販売に関する現在の負債部分 | ||||||||
| 流動負債総額 | ||||||||
| 賃貸負債を経営し,当期分を差し引く | ||||||||
| 将来の特許使用料の売却に関する負債は,現在の部分を差し引く | ||||||||
| その他長期負債 | ||||||||
| 総負債 | ||||||||
| 引受金及び又は有事項(付記9) | ||||||||
| 株主権益: | ||||||||
| 優先株:ドル額面価値ライセンス株;2022年12月31日までまたは2021年12月31日まで発行および未返済 | ||||||||
| 普通株:ドル額面価値そして2022年12月31日と2021年12月31日までの認可株式そして2022年12月31日と2021年12月31日までの発行済株式 | ||||||||
| 追加実収資本 | ||||||||
| 赤字を累計する | ( | ) | ( | ) | ||||
| その他の総合損失を累計する | ( | ) | ( | ) | ||||
| 株主権益総額 | ||||||||
| 総負債と株主権益 | $ | $ | ||||||
付記はこれらの連結財務諸表の構成要素である。
Vaxart,Inc.そして付属会社
合併経営報告書と全面赤字
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
| 十二月三十一日までの年度 |
||||||||
| 2022 |
2021 |
|||||||
| 収入: |
||||||||
| 顧客サービス契約収入 |
$ | $ | ||||||
| 将来の特許使用料の販売に関する非現金特許権使用料収入 |
||||||||
| 総収入 |
||||||||
| 運営費用: |
||||||||
| 研究開発 |
||||||||
| 一般と行政 |
||||||||
| 無形資産減価準備 |
||||||||
| 総運営費 |
||||||||
| 営業損失 |
( |
) | ( |
) | ||||
| その他の収入(支出): |
||||||||
| 利子収入 |
||||||||
| 将来の特許使用料の販売に関する非現金利息支出 |
( |
) | ( |
) | ||||
| 将来の特許権使用料責任の収益を再計量する |
||||||||
| 純為替損失 |
( |
) | ( |
) | ||||
| 所得税前損失 |
( |
) | ( |
) | ||||
| 所得税支給 |
||||||||
| 純損失 |
$ | ( |
) | $ | ( |
) | ||
| 1株当たり純損失--基本損失と赤字 |
$ | ( |
) | $ | ( |
) | ||
| 1株当たり純損失株式の算出−基本株式と希釈株式− |
||||||||
| 総合的な損失: |
||||||||
| 純損失 |
$ | ( |
) | $ | ( |
) | ||
| 売却可能投資の未実現損失,税引き後純額 |
( |
) | ( |
) | ||||
| 総合損失 |
$ | ( |
) | $ | ( |
) | ||
付記はこれらの連結財務諸表の構成要素である。
Vaxart,Inc.そして付属会社
株主権益合併報告書
(単位は千で、シェアは含まれていない)
| 積算 | ||||||||||||||||||||||||
| その他の内容 | 他にも | 合計する | ||||||||||||||||||||||
| 普通株 | 支払い済み | 積算 | 全面的に | 株主の | ||||||||||||||||||||
| 株 | 金額 | 資本 | 赤字.赤字 | 損 | 権益 | |||||||||||||||||||
| 2021年1月1日現在の残高 | $ | $ | $ | ( | ) | $ | $ | |||||||||||||||||
| 2021年10月にATMが普通株式を発行することにより、発行コストを差し引く$ | ||||||||||||||||||||||||
| 普通株式承認証の行使時に普通株式を発行する | ||||||||||||||||||||||||
| オプション行使時に普通株式を発行する | ||||||||||||||||||||||||
| 株に基づく報酬 | — | |||||||||||||||||||||||
| 売却可能投資の未実現損失 | — | ( | ) | ( | ) | |||||||||||||||||||
| 純損失 | — | ( | ) | ( | ) | |||||||||||||||||||
| 2021年12月31日現在の残高 | $ | $ | $ | ( | ) | $ | ( | ) | $ | |||||||||||||||
| 2021年9月にATM機が普通株式を発行することにより、発行コストを差し引く$ | ||||||||||||||||||||||||
| 普通株式承認証の行使時に普通株式を発行する | ||||||||||||||||||||||||
| オプション行使時に普通株式を発行する | ||||||||||||||||||||||||
| 株に基づく報酬 | — | |||||||||||||||||||||||
| 売却可能投資の未実現損失 | — | ( | ) | ( | ) | |||||||||||||||||||
| 純損失 | — | ( | ) | ( | ) | |||||||||||||||||||
| 2022年12月31日現在の残高 | $ | $ | $ | ( | ) | $ | ( | ) | $ | |||||||||||||||
付記はこれらの連結財務諸表の構成要素である。
Vaxart,Inc.そして付属会社
統合現金フロー表
(単位:千)
| 十二月三十一日までの年度 |
||||||||
| 2022 |
2021 |
|||||||
| 経営活動のキャッシュフロー: |
||||||||
| 純損失 |
$ | ( |
) | $ | ( |
) | ||
| 純損失と経営活動で使用される現金純額の調整: |
||||||||
| 減価償却および償却 |
||||||||
| 投資割増の増加 |
( |
) | ||||||
| 無形資産減価準備 |
||||||||
| 株に基づく報酬 |
||||||||
| 将来の特許権使用料責任の収益を再計量する |
( |
) | ( |
) | ||||
| 将来の特許使用料の販売に関する非現金利息支出 |
||||||||
| 将来の特許使用料の販売に関する非現金収入 |
( |
) | ( |
) | ||||
| 営業資産と負債の変動: |
||||||||
| 売掛金 |
||||||||
| 前払い費用と他の資産 |
( |
) | ( |
) | ||||
| 売掛金 |
||||||||
| 贈与収入を繰延する |
||||||||
| 負債を計算すべきである |
( |
) | ( |
) | ||||
| 経営活動のための現金純額 |
( |
) | ( |
) | ||||
| 投資活動によるキャッシュフロー: |
||||||||
| 財産と設備を購入する |
( |
) | ( |
) | ||||
| 買収業務は,現金買収後の純額を差し引く |
( |
) | ||||||
| 購入投資 |
( |
) | ( |
) | ||||
| 投資満期で得られた収益 |
||||||||
| 投資活動のための現金純額 |
( |
) | ( |
) | ||||
| 資金調達活動のキャッシュフロー: |
||||||||
| 市場融資で普通株の純収益を発行する |
||||||||
| 普通株式承認証を行使する際に普通株式取得金を発行する |
||||||||
| 株式オプション行使時に普通株で得られた金を発行する |
||||||||
| 融資活動が提供する現金純額 |
||||||||
| 現金、現金等価物、および制限的現金純増加 |
( |
) | ||||||
| 期初現金、現金等価物、および限定現金 |
||||||||
| 期末現金、現金等価物、および制限現金 |
$ | $ | ||||||
付記はこれらの連結財務諸表の構成要素である。
Vaxart,Inc.そして付属会社
合併現金フロー表(継続)
(単位:千)
| 十二月三十一日までの年度 |
||||||||
| 2022 |
2021 |
|||||||
| 非現金投資と融資活動を追加開示します |
||||||||
| 使用権資産の取得による経営リース負債 |
$ | $ | ||||||
| リースに関連する資産や負債は、リースの早期終了および修正時には確認されません |
$ | $ | ||||||
| 売掛金と売掛金に計上された財産と設備を購入する |
$ | $ | ||||||
以下の表は、会社合併貸借対照表における現金、現金等価物、および制限現金の入金状況を示す
| 十二月三十一日 |
||||||||
| 2022 |
2021 |
|||||||
| 現金と現金等価物 |
$ | $ | ||||||
| 制限現金 |
||||||||
| 現金フロー表に表示されている現金、現金等価物、および限定的な現金 |
$ | $ | ||||||
付記はこれらの連結財務諸表の構成要素である。
Vaxart,Inc.そして付属会社
連結財務諸表付記
注:1.*ビジネスの組織および性質
総司令官
Vaxart生物科学社は最初に#年にカリフォルニア州に登録設立されました三月一日2004,会社はVaxart,Inc.(“プライベートVaxart”)と改称した七月です2007,デラウェア州に再登録されましたはい2018年2月Private VaxartはAviragen Treateutics,Inc.(“Aviragen”)との業務合併を完了し、これにより、AviragenはPrivate Vaxartと合併し、Private VaxartはAviragenの完全子会社として存続する(“合併”)。合併条項によると、AviragenはVaxart,Inc.(その子会社、“会社”または“Vaxart”)と改称され、Private VaxartはVaxart Biosciences,Inc.と改称される。
開ける2020年10月13日当社は公開市場販売協定を締結している“2020年10月ATM機“)これにより,つねに販売エージェントを介してその普通株の株を提供·販売することができ,総発行価格は最高$に達する
本年度末までに十二月三十一日2021,*同社は追加で販売しています
開ける2021年9月13日♪the the the2020年10月ATM機は終了し、オンになりました2021年9月15日当社は制御持分発売および販売プロトコル(The Ctrl Equity Offering And Sales Agreement)を締結した“2021年9月ATM機は,この規定に基づいている5月.時々販売代理を通じてその普通株の株を提供·販売することで、総発行価格は最高$に達する
同社の主な業務はカリフォルニア州サンフランシスコ南部に本部を置き、1つは経口組換えタンパク質ワクチンの発見と開発であり,その独自の経口ワクチンプラットフォームに基づく報告が可能である。
注:2.“重要会計政策の概要”
列報基礎、流動性、持続経営-会社が継続経営の企業となると仮定し、添付されている総合財務諸表は、米国公認会計原則(“米国公認会計原則”)および米国証券取引委員会(“米国証券取引委員会”)の会計·開示規則および規定に従って作成されている“と述べた
その会社は臨床段階のバイオテクノロジー会社で違います。製品の販売量。その主要な資金源は普通株式と普通株式承認証の売却と発行である。はい2022年12月31日同社は現金、現金等価物、制限現金、投資#ドルを持っている
その後に2022年12月31日同社はノウォーカーウイルス経口ワクチン候補ワクチンを優先的に開発することにしました15)である。優先順位の結果,同社は滑走路に十分な現金が入ると予想している二番目1/42024.
同社は、その普通株、手形または他の証券、借金、または戦略パートナーとパートナーシップを構築することによって、その事業計画を実施するために追加の資本を調達することに依存するであろう。あるかもしれない違います。企業が継続的に経営する企業として追加資本の調達に成功することを確実にする。
財務諸表はできます注釈会社が経営を継続できない場合に必要かもしれないどんな調整も含まれています。
連結ベースである連結財務諸表には、Vaxart,Inc.およびその子会社の財務諸表が含まれる。Vaxart,Inc.とその子会社との間のすべての重大な取引および残高は合併中に除去されている。
見積もり数を使用して--米国公認会計原則に基づいて財務諸表を作成する場合、管理層は、本付記された資産、負債(計算すべき臨床および製造業を含む)の計算すべき項目に影響を与える推定および仮定を行う必要がある2)、収入および支出、ならびに財務諸表および付記または資産および負債の開示。実際の結果と結果は,これらの推定や仮定とは異なる可能性がある.
外貨-会社非米国子会社がドルを本位貨幣とする資産と負債の為替損益を為替損益に計上し、その他の収入と(費用)純額を会社の総合経営報告書と全面赤字に計上する。その会社は所有している違います。現地通貨を本位貨幣とする子会社。
現金、現金等価物、および制限的現金--会社はすべての原始期限を考慮しています三つ現金等価物として購入された場合、6ヶ月以下である。現金等価物、すなわち5月.貨幣市場基金、社債、商業手形に投資する金額からなり、公正価値に応じて列報する。*会社の制限された現金には現在が含まれている2022年12月31日そして2021合計ドルになる
投資--超過現金残高5月.有価証券に投資する。いつでも既知の金額に変換することができ、期限が大きいことが規定されています三つ購入した月は投資に分類される
同社は、有価証券を購入する際にその有価証券投資の適切な分類を決定し、各貸借対照表日にその指定を再評価する。取引可能な債務証券は、売却可能な債務証券として分類され、記録される。同社の保本目標および流動資金要求を考慮したところ、5月.このような債務証券は規定された期限までに売却される。これらの証券は公正価値別に計上されており、未実現損益(税引き後純額)は株主権益の構成要素として報告されているが、非一時的な未実現損失は除外されており、これらの未実現損失は他の収入(費用)に記録されている。有価証券を売却した実現損益を特定の確認方法で決定し、当該等損益を他の収入(費用)の構成要素に計上する。売却可能投資は、各ツールの有効期限および当社が一定期間を超える投資を意図的かつ保有する能力があるか否かに応じて、流動または非流動資産に分類される12何ヶ月になりますか。残りの期限は126ヶ月以下は流動資産として分類され、総合貸借対照表では短期投資として報告される。残り期限が1ドルを超える有価証券12当社はこの投資が5ヶ月を超える月を保有する意欲と能力があります12これらの月は非流動月に分類され、総合貸借対照表に長期投資が計上されている。
証券は各報告期間の終了時に減値評価を行う。減値を評価する時に多種の要素を考慮し、公正価値が余剰コスト基礎まで低下する以下は信用関連要素か非信用関連要素か、発行者の財務状況と最近の見通し、及び期待公平価値回収を実現するための投資の意図と能力を含む。クレジットに関する減値は貸借対照表上で準備していることを確認し,収益に応じた調整を行う。どのような欠陥も注釈信用に関する項目は適用税項を差し引いた他の総合損失で確認されます。
Vaxart,Inc.そして付属会社
連結財務諸表付記
会社を深刻な信用リスクに直面させる可能性のある集中信用リスクの金融商品は、主に現金、現金等価物、制限された現金、および販売可能な投資を含む。同社は、その現金、現金等価物、制限された現金備蓄、および販売可能な投資を、管理層が高い信用品質を有すると考えている金融機関に置いている。現金、現金等価物、制限された現金を持つ金融機関が違約すれば、当社は連邦保険の限度額を超える限り信用リスクに直面する。発生した損失やこのような資金が得られないことは、会社の財務状況、経営業績、キャッシュフローに重大な悪影響を及ぼす可能性がある。
会社の投資戦略の主な重点は資本の保存と流動性の要求を満たすことだ。その会社の投資政策は制限によって任意のものに集中している1つは企業発行者或いは業界に対して格付けを行い、最低許容信用格付けを確立する。
売掛金--売掛金は、会社が販売しているInavirの売掛金特許使用料収入から、推定収益を控除し、将来の期間に予定されている金額で報告します。不良債権準備は、歴史的経験、帳簿年齢分析、特定口座の情報に基づいて記録され、関連金額は収入が確認された準備金として記録される。口座残高はすべての入金手段が尽き、取り戻す可能性がわずかとされた後、手当から解約されます。その会社はすでに提供した不良債権準備完了2022年12月31日そして2021.
財産と設備--財産と設備はコストから減価償却累計を引いて入金されます。減価償却は直線法で計算され、各資産の推定耐用年数を計算する。減価償却は資産投入時から始まります。メンテナンスとメンテナンス費用は発生時に作業費用を計上する。資産を売却又は廃棄する際には、コスト及び関連する減価償却が貸借対照表から差し引かれ、それによって生じる収益又は損失は、現金化期間中の他の収入及び(費用)に反映される。
財産と設備の使用年数は以下のとおりである
| 実験室設備(年) | ||||
| オフィスとコンピュータ機器(年) | ||||
| 賃借権改善 | 残りのレンタル期間や見積もり耐用年数が短い |
営業権とその他の無形資産--営業権は、買収価格が買収資産の公正価値を超えていることを代表する部分である注釈償却。無形資産には発達した技術と知的財産権が含まれている。無形資産はコストから累積償却勘定を差し引く。償却は直線法で計算されます
長寿資産の減価準備--当社は年内にその長寿資産を審査し、財産と設備、営業権及び有限年限無形資産の減価を含む第四に毎年四半期ごとにイベントや状況の変化がこれらの資産の帳簿価値を示す場合には5月.注釈取り戻すことができます。これらの資産の回収可能性は、各資産の帳簿価値と、資産がその残存寿命内に生じると予想される将来の未割引キャッシュフローとを比較することによって測定される。減値の兆候が生じ、当該等の資産を用いて生じる推定未割引将来のキャッシュフローが当該等資産の帳簿価値よりも少ない場合には、関連資産は公正価値に減記される。同社が開発した技術は年末までの年間で被害を受けたと評価している2022年12月31日そして2021(付記参照)7).
臨床と製造費用を計算すべき−会社が計算すべき研究開発活動の見積もりコスト−第三に−前臨床研究および臨床試験、ならびに契約製造活動を含む締約国サービスプロバイダ。当社は、提供されたサービスの見積もり金額に基づいて、研究·開発活動の見積もりコストを記録し、発生コストを含むが、注釈しかしながら、貸借対照表中の他の計算すべき負債および合併経営報告書における研究および開発費用および全面赤字で領収書を発行しなければならない。このような費用は会社の研究開発費の重要な構成要素かもしれない。
当社は,内部者や外部サービスプロバイダとサービスの進捗や完了段階およびこのようなサービスに支払う取り決め費用について検討することにより,提供されるサービス量を見積もる.当社は各報告期間ごとの計上残高を決定する際に重大な判断と見積もりを行います。実際のコストの理解に伴い、それは計算すべき見積もりを調整した。会社がそうしたにもかかわらず注釈その見積もりは,実際に発生した金額,提供されるサービスの状態や時間,登録されている科目数や登録率を知ることと大きく異なると予想される5月.見積もりと違って、会社が特定の時期に報告した金額が高すぎたり低すぎたりする可能性があります。当社の課税費用部分は契約研究組織やその他の機関から受けたタイムリーかつ正確な報告にかかっている第三に--党のサービス提供者。これまで会社は注釈計算すべきコストと実際に発生するコストの間には実質的な差がある。
リース-当社は、その連結貸借対照表において、すべての条項を超える1つは一年です。使用権資産とは、行使される延期オプションを合理的に決定することと、リース金を支払うための経営リース負債とを含む、リース期間内に標的資産を使用する権利である。使用権資産と経営性賃貸負債はレンタル期間内にリース支払いの現在値を確認します。賃貸契約の範囲内で注釈隠れ金利を提供する場合、当社はレンタル開始日に利用可能な情報に基づく逓増借入金金利を用いてレンタル支払いの現在値を決定します。経営リース支払い費用はリース期間中に直線的に確認し、会社総合経営報告書と総合損失中の経営費用を計上する。当社はすでに選択しました注釈施設レンタルのレンタル部分と非レンタル部分とが分離されているが、設備レンタルの非レンタル部分はレンタル部分とは別に入金される。
Vaxart,Inc.そして付属会社
連結財務諸表付記
収入確認--会社が承諾した商品やサービスの支配権を顧客に譲渡すると、会社は収入を確認し、その金額は、会社がこれらの商品やサービスと交換することを期待している対価格を反映している。収入確認のため、会社は以下の操作を実行します5人手順:
| (i) | 契約で約束された貨物またはサービスの表示; |
| (Ii) | 契約範囲内で異なるかどうかを含む、約束された貨物またはサービスが契約義務であるかどうかを決定する |
| (Iii) | 可変対価格の制約を含む取引価格の計量; |
| (Iv) | 推定販売価格に基づいて取引価格を契約履行義務に割り当てること |
| (v) | 会社として各業績義務を履行した場合に収入を確認する。履行義務は,契約中に独自の商品やサービスを顧客に移転することを承諾する承諾であり,課金単位である. |
特許権使用料収入が売上高に占める割合は、特定の販売水準に達したマイルストーン支払いに基づいて、許可が特許権使用料に関連する主要項目とみなされる場合には、(I)関連販売が発生した場合、又は(Ii)販売及び使用量に基づく特許権使用料例外要件に基づいて、特許権使用料に割り当てられた履行義務の一部又は全部が履行された(又は部分的に満たされている)場合には、収入として確認される。
当社が顧客に提供する承諾サービスの金額は、当社がこれらのサービスから得ると予想されている対価格を反映しているため、顧客との契約の収入は発生したコストに比例して確認されます。
はい2022,同社は、ビルとメリンダ·ゲイツ財団(BMGF)の研究·開発のための贈与を受けた。*契約が実行され、契約価格が固定または確定可能な場合にのみ、会社は研究契約下の収入を確認する。BMGF贈与の収入は,関連コストの発生と関連サービス提供期間中に確認され,契約下の適用条件が満たされていることを前提としている.契約収入コストは合併業務表と総合損失表に業務費用の構成要素として入金される。会社は認識しています違います。今年度末までのBMGF補助金からの収入2022年12月31日前の3か月
返済可能なコストの契約により、会社は収入が許容コストが発生したことを確認し、固定費用を稼いだ。契約下の精算可能コストには、主に直接人工、下請けコスト、材料、設備、出張、および承認された管理費用と間接コストが含まれる。費用償還可能契約項下の固定費用は,仕事履行時に発生する許容費用の契約推定総費用に対する割合で稼いでおり,発生した費用は完了した仕事を比例して合理的に測定されている。
コスト返済可能契約(本契約のような)に応じて会社に支払う金は仮払いであり、政府の年次監査に応じて調整することができる。経営陣は期間の収入は注釈しかし、監査された記録された金額は、最終監査および決済時に現金化される予定だ。任意の年の許容費用について最終決定を下した後、収入と請求書5月.対応する調整は、既知の調整の間に行われなければならない。
研究·開発コスト−研究·開発コストは発生時に費用を計上する。研究開発費には、主に給料と福祉、株式ベースの給与、相談費、第三に−臨床試験を実施し、臨床試験材料を製造するための当事者費用、いくつかの施設費用、および臨床試験に関連する他の費用。他のエンティティへの支払いは、当社が通常キャンセル可能な合意に基づいて支払われます。研究·開発活動の前金は前払い費用と記されている。前払い額は関連サービスを実行する際に料金を計上する。
株式ベースの報酬-株式ベースの報酬スケジュールには、我々の株式インセンティブ計画下の株式オプション付与および制限株式単位(RSU)報酬と、我々の従業員株式購入計画(ESPP)に基づいて発行された株とが含まれ、従業員はこれらの株を介して5月.私たちの普通株は、市場価格より低い価格で購入されます。株式ベースの報酬は、付与日に従業員と非従業員に対して行われたすべての株式ベースの奨励の計算に基づいて、奨励の公正価値から推定された没収を差し引くものです。ESPPにより調達された補償費用は見積日の奨励公正価値に基づいて確認されます。これらの報酬の公正価値はブラック·スコアーズ推定値モデルを用いて推定された。各オプションの予想期間は、その元の契約期間とその平均帰属期限との算術平均値をとることによって推定される。
1株当たり純収益(損失)−基本1株当たり純収益(損失)の計算方法は,純収益(損失)を期内に発行された普通株の加重平均数で除し,潜在的な普通株は考慮しない。
1株当たりの普通株式の純収益(損失)はすべての潜在的な薄普通株で計算され、株式オプションの行使、株式承認証、RSU及びESPPの時に発行できる普通株を含む。同社は在庫株方法を用いてその株式オプション、株式承認証、RSUとESPPの1株当たり損益(損失)を計算した。この計算については、普通株を購入するオプション、株式承認証、RSU、ESPP計画は潜在とされる普通株を付与し、それらの影響が希薄化である場合にのみ、希釈後の1株当たり純収益に含まれる。純損失が発生した場合、すべての潜在的希薄化株式の影響は、赤字計上されるため、1株当たりの純損失計算から除外される。
最近の会計公告
当社は新たに発表されたすべての重要な会計声明を審査しました。これらの声明には注釈しかしまだ効果的で結論を出しています注釈その運営やその採用に適しています注釈その財務状況や経営業績に実質的な影響を及ぼす。
Vaxart,Inc.そして付属会社
連結財務諸表付記
注.注3.**ビジネス統合
開ける2021年11月30日同社はKindred生物科学会社のカリフォルニア州バーリンガムにあるGMP生産ラインの買収を完了し、ドルと交換した
買収に関する買収資産の公正価値は以下のとおりである(千で計算)
| 財産と設備 | $ | |||
| 商誉 | ||||
| 購入総価格 | $ |
また、会社は#ドルを支払った
商業権、すなわち購入価格が獲得された資産の公正な価値を超える部分は、主に予想される内部製造のコスト節約、アウトソーシングではなく、集まった労働力によるものである。すべての営業権は所得税の面で控除されると予想される。
注.注4.金融商品の公正価値
公正価値会計は、財務諸表において価値確認または開示されたすべての金融資産および負債、ならびに非金融資産および負債に適用される。金融商品には、現金および現金等価物、有価証券、売掛金、売掛金、および満期日が相対的に短いために公正価値に近い売掛金が含まれる。
総合貸借対照表では,公正価値が恒常的に記録されている資産と負債は,その公正価値を計測するための投入に関する判断レベルに基づいて分類される.公正価値会計基準は公正価値を計量する枠組みを提供し、公正価値がどのように確定したかに関するいくつかの情報を開示することを要求した。公正価値は、報告日の市場参加者間の秩序ある取引において資産を売却するために請求されるか、または負債を移転するために支払われる価格(脱退価格)と定義される。“会計基準”は三つ公正価値を計量するための評価技術の入力が観察可能であるか観察不可能であるかに基づいて、これらの入力の優先順位を決定する一次推定階層構造。観察可能な投入は独立源から得られた市場データを反映しており,観察できない投入は報告実体による市場仮定を反映している。
♪the the the三つ-評価技術投入の階層構造の概要は、以下のとおりである
レベル:1未調整の投入とは、計量日と同じ資産または負債の活発な市場で調整されていないオファーを意味する
レベル:2短期投入とは、アクティブ市場における同様の資産または負債の観察可能な未調整オファー、同じまたは同様の資産または負債の市場における未調整オファーを意味する注釈資産または負債に関して実質的に全期間にわたって観察可能であるか、または観察可能な市場データによって確認された他の投入;
レベル:3**これらの資産または負債は、より少ないまたは少ないものから、計量資産または負債の公正な価値に大きな意味を持つ観察できない投入違います。市場データです。
以下の表に当社の金融資産の公正価値を示し、これらの資産は経常的な基礎に基づいて計量されている2022年12月31日何度も何度も2021(単位:千):
| レベル1 | レベル2 | レベル3 | 合計する | |||||||||||||
| 2022年12月31日 | ||||||||||||||||
| 金融資産: | ||||||||||||||||
| 貨幣市場基金 | $ | $ | $ | $ | ||||||||||||
| アメリカ国債 | ||||||||||||||||
| 商業手形 | ||||||||||||||||
| 会社債務証券 | ||||||||||||||||
| 総資産 | $ | $ | $ | $ | ||||||||||||
| レベル1 | レベル2 | レベル3 | 合計する | |||||||||||||
| 2021年12月31日 | ||||||||||||||||
| 金融資産: | ||||||||||||||||
| 貨幣市場基金 | $ | $ | $ | $ | ||||||||||||
| アメリカ国債 | ||||||||||||||||
| 商業手形 | ||||||||||||||||
| 会社債務証券 | ||||||||||||||||
| 総資産 | $ | $ | $ | $ | ||||||||||||
その会社は持っている
Vaxart,Inc.そして付属会社
連結財務諸表付記
注:5.*貸借対照表の構成要素
(a) 現金、現金等価物、制限現金、および投資
現金、現金等価物、限定的な現金および投資には、以下が含まれる(千単位)
| 償却する | 未実現総額 | 推定数 | 現金·現金等価物 | 短期.短期 | 長期の | |||||||||||||||||||||||
| コスト | 収益.収益 | 損 | 公正価値 | 制限された現金 | 投資する | 投資する | ||||||||||||||||||||||
| 2022年12月31日 | ||||||||||||||||||||||||||||
| 銀行の現金 | $ | $ | — | $ | — | $ | $ | $ | — | $ | — | |||||||||||||||||
| 貨幣市場基金 | — | — | — | — | ||||||||||||||||||||||||
| アメリカ国債 | — | ( | ) | — | ||||||||||||||||||||||||
| 商業手形 | — | — | — | |||||||||||||||||||||||||
| 会社債務証券 | — | ( | ) | — | — | |||||||||||||||||||||||
| 合計する | $ | $ | — | $ | ( | ) | $ | $ | $ | $ | ||||||||||||||||||
| 償却する | 未実現総額 | 推定数 | 現金と現金 | 短期.短期 | 長期の | |||||||||||||||||||||||
| コスト | 収益.収益 | 損 | 公正価値 | 等価物 | 投資する | 投資する | ||||||||||||||||||||||
| 2021年12月31日 | ||||||||||||||||||||||||||||
| 銀行の現金 | $ | $ | — | $ | — | $ | $ | $ | — | $ | — | |||||||||||||||||
| 貨幣市場基金 | — | — | — | — | ||||||||||||||||||||||||
| アメリカ国債 | — | ( | ) | — | ||||||||||||||||||||||||
| 商業手形 | — | — | — | — | ||||||||||||||||||||||||
| 会社債務証券 | — | ( | ) | — | ||||||||||||||||||||||||
| 合計する | $ | $ | — | $ | ( | ) | $ | $ | $ | $ | ||||||||||||||||||
(b) 売掛金
売掛金には売掛金使用料#ドルが含まれています
(c) 財産と設備、純額
財産と設備、純額は以下の部分からなる(千計)
| 2022年12月31日 | 2021年12月31日 | |||||||
| 実験室装置 | $ | $ | ||||||
| オフィス及びコンピュータ装置 | ||||||||
| 賃借権改善 | ||||||||
| 建設中の工事 | ||||||||
| 総資産と設備 | ||||||||
| 減算:減価償却累計 | ( | ) | ( | ) | ||||
| 財産と設備、純額 | $ | $ | ||||||
減価償却費用は$
Vaxart,Inc.そして付属会社
連結財務諸表付記
(d)--使用権を使用する資産、純額
使用権資産、純額にはドルの施設が含まれています
(e) 無形資産、純額
無形資産には、開発された技術、知的財産権、完全な減価とされる前に行われている研究·開発が含まれている。無形資産はコストから累積償却勘定を差し引く。償却は直線法で計算されます
| 2022年12月31日 | 2021年12月31日 | |||||||
| 発達した技術 | $ | $ | ||||||
| 知的財産権 | ||||||||
| 総コスト | ||||||||
| 差し引く:累計償却 | ( | ) | ( | ) | ||||
| 無形資産、純額 | $ | $ | ||||||
無形資産の償却費用は#ドルです
自分から2022年12月31日年ごとに推定される将来の償却費用は以下の通り(単位:千):
| 十二月三十一日までの年度 | 金額 | |||
| 2023 | $ | |||
| 2024 | ||||
| 2025 | ||||
| 2026 | ||||
| 2027 | ||||
| その後… | ||||
| 合計する | $ | |||
(f)**親善
営業権とは、購入価格が取得された資産の公正価値を超える部分であり、#ドルを含む
(g) その他計上すべき資産負債
その他の負債には、以下の(千計)が含まれる
| 2022年12月31日 | 2021年12月31日 | |||||||
| 補償すべきである | $ | $ | ||||||
| 臨床と製造費用を計算しなければならない | ||||||||
| 専門とコンサルティングサービス | ||||||||
| その他の費用を計算しなければならない | ||||||||
| 合計する | $ | $ | ||||||
Vaxart,Inc.そして付属会社
連結財務諸表付記
注:6.**収益
特許権使用料協定
Aviragenが第一三共株式会社(“第一三共”)と締結した協力及び許可協定によると、会社は日本でInavirを販売することにより特許使用料収入を発生させる2009.はい2010年9月カプリル酸ラネミビルは日本厚生労働省の許可を得て成人と小児インフルエンザの治療に用いられ,第一三共株式会社はイナビルと命名した。協定によると,その会社は現在1部を受け取っている
注:7.将来の特許使用料の販売に関する責任
はい2016年4月AviragenとHealthcare Royalty Partners III,L.P.(“HCRP”)は特許権使用料権益買収プロトコル(“RIAA”)を締結した。RIAAでHCRPは$を稼いだ
関連する会計指導によると、HCRPがRIAAによって稼ぐことができる特許権使用料額が制限されているため、この取引は負債とみなされ、有効利息方法を用いて手配された有効期限内に償却されている。その会社は所有している違います。HCRPには任意の額を支払う義務があるが、第一三共から徴収した印税シェアをHCRPに転嫁する。債務の償却を記録するためには、会社はライセンス契約によって将来受信される特許権使用料の支払い総額と、本契約の有効期間内にHCRPに転嫁される支払総額を推定する必要がある。そこで、当社は負債の未償却部分に利息を計上し、推定実金利を用いて非現金支払利息を記録している。HCRPに渡される各期間に得られる特許使用料は、任意の超過があれば、将来の特許使用料の販売に関連する非現金特許権使用料収入として記録される注釈伝達記録は特許使用料収入として記録されなければならない。振込特許使用料が次の四半期にHCRPに支払われる場合、将来の特許使用料の販売に関連する帰属責任はそれに応じて減少する。当社は、予想される特許使用料支払いを定期的に評価し、当該等支払いが最初の推定以上またはそれ以下の範囲で、貸借対照償却および金利を調整する。この会計計算の結果として、たとえ会社が注釈特許使用料におけるHCRPのシェアを保持するために、関連利息を含む関連債務額が完全に償却されるまで、これらの特許権使用料に関する非現金収入を記録し続ける。
下表に同年度までの負債口座内の活動を示す2022年12月31日(千単位):
| 将来の特許使用料の販売に関する総負債、年明け | $ | |||
| HCRPに支払われる非現金特許使用料収入 | ( | ) | ||
| 非現金利息支出が確認されました | ||||
| 収益の再評価を確認する | ( | ) | ||
| 将来の特許使用料の販売に関する総負債、年末 | ||||
| 現在の部分 | ( | ) | ||
| 長期部分 | $ |
COVID爆発以来の特許使用料の低下-19,一次保健プログラムに支払われるべき額は#年に再評価されることが予想される2022年12月31日と…2021,それによって発生した債務は#ドルをリスコアリングします
Vaxart,Inc.そして付属会社
連結財務諸表付記
注:8.3つの賃貸借契約
当社は事務及び製造施設の使用権を取得しました
年、同社はカリフォルニア州サンフランシスコ南部にある不動産使用権を獲得しました2020年11月当初の予定によると2025年9月30日それは延長されました2029年3月31日使用違います。その他の拡張オプション。年、同社はカリフォルニア州サンフランシスコ南部にある別の物件の使用権を獲得しました2015年6月この協定は8月1日に終了する予定だった2020年4月30日1つを使う-会社が年に行使する年間延長オプション2019年7月賃貸借契約を延長する2025年4月30日さらに延長されました2029年3月31日追加の延長を選択することができます何年もです。さらにここでは2021年9月同社はカリフォルニア州サンフランシスコ南部にある施設レンタルに署名しました。初期期限は2029年3月31日追加の延長を選択することができます何年もです。本レンタルは、基本的に改善されたコンポーネントを含む三つ現在までの月2022年9月30日会計目的の場合、その構成要素のレンタルはいつ開始されたとみなされる。テナント改善のためのコンポーネントも含まれており、費用は約$と推定されています
自分から2022年12月31日初期期限以上1つは一年が過ぎました
下表は会社の経営リース負債に対する未割引現金支払い義務をまとめており、初期期限を超えています12個2018年までの月2022年12月31日(単位:千):
| 十二月三十一日までの年度 | 金額 | |||
| 2023 | $ | |||
| 2024 | ||||
| 2025 | ||||
| 2026 | ||||
| 2027 | ||||
| その後… | ||||
| 未割引合計 | ||||
| 差し引く:推定利息 | ( | ) | ||
| 将来の最低返済額の現在価値 | ||||
| 賃貸負債の当期部分を経営する | ( | ) | ||
| 賃貸負債を経営し,当期分を差し引く | $ | |||
その会社の将来の債務はドルです
いくつかの施設の経営リース協定には、公共エリアメンテナンスのような非レンタル費用が含まれており、これらの費用は可変レンタルコストとして記録されている2022年12月31日と…2021,以下の項目を含む可変レンタルコスト1つは借約がある注釈会計目的がまだ始まっていないため(以下参照)、現在以下のように概説する(千計である)
| 十二月三十一日までの年度 | ||||||||
| 2022 | 2021 | |||||||
| レンタル料 | ||||||||
| リースコストを経営する | $ | $ | ||||||
| 短期賃貸コスト | ||||||||
| 可変リースコスト | ||||||||
| 転貸収入 | ( | ) | ||||||
| 総賃貸コスト | $ | $ | ||||||
Vaxart,Inc.そして付属会社
連結財務諸表付記
注.注9.*支払いの引受および事項
(a) 購入承諾
自分から2022年12月31日同社は約$を持っています
(b) 完済する
通常業務の過程において、当社は以下の協定を締結します5月.賠償条項が含まれています。同等の合意に基づき,当社は5月.補償された側が受けた損失を賠償し、無害にし、それを弁護する。その中のいくつかの条項は損失を次のような原因による損失に制限する第三に--党の行動。場合によっては、補償は合意の終了後に継続されるだろう。これらの規定によると、会社が将来支払う可能性の高い潜在金額は注釈確定できます。当社もすでにいくつかの上級職員及び取締役と合意を締結しており、当該等の協定は他の事項を除いて、当社は当該等の高級職員又は取締役が規定する状況及び範囲内で、その支出、損害賠償、判決、罰金及び和解について当該等の高級職員又は取締役に補償及び立て替えを行い、当該等の行動、訴訟又は法律手続に関連する支出を支払うことを規定している5月.彼か彼女か彼女の訴訟や法的手続きで支払いを要求されます5月.デラウェア州法律と当社定款で許可されている最大範囲では,取締役,当社幹部あるいはその他の代理として当事者とされている。その会社には現在役員と上級管理職保険があります。
(c)*訴訟
時々会社は5月.その業務に関連した法的訴訟に参加する。現在把握している情報によると、当社は、それに対する任意の未解決行動に関連する合理的な損失可能な金額や範囲が確立された準備金を超えており、全体的にその総合財務状況やキャッシュフローに基づいて決定することはできないと考えている。しかしながら、現在または将来の任意の紛争解決または法的手続きは、どのようなプログラムの是非にも、巨額のコストを招く可能性があり、経営陣を移して自社の運営に必要な関心や資源を成功させ、その業務、財務状況、運営結果に重大な悪影響を及ぼす可能性がある。
開ける2020年8月4日株主派生訴訟はサンマテオ県カリフォルニア州高等裁判所に提出されましたGodfreyはLatourらの事件を訴えた。*修正された訴えは2020年9月4日事件は新たに命名されたエンニス訴ラトゥールらの事件. A 二番目改訂された訴えは2020年11月25日A 第三に改訂された訴えは2021年6月11日♪the the the第三に修正後の起訴状はある現職と前任Vaxart取締役を被告とし、彼らは受託責任、不当な利益と浪費に違反し、そして裁決が指定されていない損害賠償、ある平衡法救済及び弁護士費と費用などを求めると主張した。♪the the the第三に改訂された起訴状は、受託責任違反や停戦資本有限責任会社(“停戦”)への受託責任違反のクレームを協力·教唆することも主張している。♪the the the第三に修正された起訴状は,会社のある役員と役員に付与されたある株式オプションに疑問を提起した2020年6月当社の2020年4月24日依頼書と二つ停戦組織が持っている授権証は2020年6月8日♪the the the第三に改正された起訴状は,会社を代表して会社の利益に由来する訴訟を提起することを目的としており,会社を対象としている違います。損害賠償を求めました。開ける2021年8月31日当社とそのいくつかの取締役(“Vaxart被告”)および他のすべての被告に第三に修正された起訴状。日付別の注文2022年11月17日裁判所は修正許可なしに被告の抗弁を維持し,それ以来原告が控訴する時間が経過した。
はい8月そして2020年9月二つ米国カリフォルニア州北区地方裁判所も基本的に似たような証券集団訴訟を起こした。♪the the the1つ目は動作、タイトルはヒメルバーグはVaxart,Inc.らを訴えた。提出されました2020年8月24日♪the the the二番目動作、タイトルはHovhannisyanはVaxart,Inc.らを訴えている提出されました2020年9月1日(合計は,“推定された集団訴訟”)2020年9月17日♪the the the二つ訴訟は関連があると考えられている。開ける2020年12月9日裁判所は首席原告と首席原告弁護士を任命した。
開ける2021年1月29日月曜日、主な原告は彼らの合併の修正された起訴状を提出した2021年7月8日すべての被告は合併後の修正された訴えを却下するために行動した。開ける2021年5月14日裁判所は合併後の改訂された起訴状の改正に関する主原告の請求を承認し、意味がないと被告の却下動議を却下した。開ける2021年6月10日主要原告は1部提出した1つ目は改正された合併申し立てと2021年8月9日主な原告は訂正を提出した1つ目は修正された合併訴え。♪the the the1つ目は改正された合併起訴状は訂正され、Vaxartの一部の現職と前任幹部と役員および停戦を被告とした三つ連邦民事証券法に違反する10(B)“取引法”及び“米国証券取引委員会”規則10b-5,当社とすべての個別被告に対して20(A)交流法に違反し,停戦協定及びすべての個別被告に違反する20A“停戦反対の交流法”1つ目は修正された改正合併起訴状は、被告が証券法違反、会社がノウイルスワクチンの開発、業務相手側のワクチン製造能力および会社が反り速度行動(“OWS”)に参加したことに関する情報を誤って陳述および/または見落とし、Vaxart株価の上昇計画に参加した。♪the the the1つ目は改正された合併申し立ては、似たような立場にある株主であることを証明する集団訴訟を求め、額不明の損害賠償および弁護士費と費用を賠償することを求めた2021年7月8日すべての被告が行動して却下した1つ目は修正された合併訴え。日付別の注文2021年12月22日裁判所は停戦却下動議を承認し、修正を許可した。そうでなければ、動議を却下する。開ける2022年7月27日主な原告は、当社およびその現または前任上級管理者および/または取締役に対して当社の上級管理者および/または取締役(“和解被告”)として提起されたすべてのクレームを解決するために、部分和解(“部分和解”)が達成されたことを宣言する通知を提出する。部分和解協定によると、会社は和解金額を#ドルと同意した
開ける2020年10月23日米ニューヨーク南区地方裁判所に提訴しましたRothは停戦資本有限責任会社らの事件を訴えた。*起訴状は、停戦および停戦に関するいくつかの当事者を被告とし、取引法の一部に違反していると主張しています16(B)短期利益の返還を求める。起訴状は、会社を代表して会社の利益のために訴訟を提起し、会社を利益損害賠償を求める“名義被告”とした。
開ける2021年1月8日いわゆる株主のフィリップ·チャンは専門家アメリカカリフォルニア州北区地方裁判所が起こした訴訟はChanはVaxart,Inc.らを訴えた.(“脱退訴訟”を選択).この訴えは推定された集団訴訟で提出された訴えの早いバージョンとほぼ同じであるため,推定された集団訴訟保留期間中に脱退訴訟を選択することは保留されている
Vaxart,Inc.そして付属会社
連結財務諸表付記
注:10.*株主資本
(a) 優先株
当社は発行を許可されている
(b) 普通株
自分から2022年12月31日同社は発行を許可されています
会社が自動的または非自発的に清算、解散、資産分配または清算を行う場合、優先株保有者の権利が満たされた後、普通株式保有者は、株主に分配可能なすべての資産の同等額の1株当たり収益を得る権利がある。ここにあります違います。補償基金は普通株式に適用される。
同社が発行のために予約した普通株式は以下の通り
| 2022年12月31日 |
2021年12月31日 |
|||||||
| 既発行と未償還のオプション |
||||||||
| 発行済みと未返済のRSU |
||||||||
| 将来に使える持分奨励付与 |
||||||||
| 普通株式引受証 |
||||||||
| 2022年従業員株購入計画 |
||||||||
| 合計する |
||||||||
(c) 株式承認証
現在、当社は以下の未償還持分証を持っています2022年12月31日その後の配当、株式分割、株式配当、または他の非常配当または会社の普通株式または資本構造の他の同様の変化の場合、これらのすべては、標準的な逆希釈保護を含むありません彼らにはどんな損失にも参加権があります
| 株式承認証転換可能な証券 |
未弁済持分証 |
行権価格 |
期日まで |
||||||
| 普通株 |
$ | 2024年4月 |
|||||||
| 普通株 |
$ | 2024年4月 |
|||||||
| 普通株 |
$ | 2025年3月 |
|||||||
| 普通株 |
$ | 2025年2月 |
|||||||
| 普通株 |
$ | 2024年3月 |
|||||||
| 普通株 |
$ | 2026年12月 |
|||||||
| 合計する |
|||||||||
会社の制御範囲内で基本的な取引(株式承認証によって定義された会社の所有権移転)が発生した場合、行使されていない一般株式証所有者は1ドルの価格で行使することができる
注:11.*持分インセンティブ計画
開ける2019年4月23日会社の株主が承認した2019持分インセンティブ計画(“2019計画“)は,この計画に基づき,会社は奨励的株式オプション,非制限株式オプション,株式付加権,制限株式奨励,制限株式単位(RSU),その他の株式奨励,業績奨励を発行する権利がある5月.現金、株、その他の財産で決済する。♪the the the2019計画は従業員、取締役と顧問のサービスを確保し、維持し、会社の従業員、取締役と顧問を激励し、会社とその関連会社の成功に最大の努力を尽くし、従業員、取締役と顧問に1種の手段を提供することを目的としている5月.会社の普通株価値の増加から利益を得る機会がある2019計画、以前の計画はすべて凍結されて、没収、キャンセル、満期になると、これらの計画の下での奨励は注釈から2019計画してみます。
発行を許可した普通株式総株式数2019最初の計画は
年末までに年度を終える2022年12月31日その会社は授与された
株式オプションとRSU取引の概要二つ締切り年数2022年12月31日と…2021具体的には以下のとおりである
| 重みをつける | 重みをつける | |||||||||||||||||||
| 株 | 量 | オプション平均値 | 量 | RSU平均値 | ||||||||||||||||
| 使用可能である | オプション | トレーニングをする | RSU | 授与日 | ||||||||||||||||
| グラントのために | 卓越した | 値段 | 卓越した | 公正価値 | ||||||||||||||||
| 2021年1月1日の残高 | $ | — | $ | |||||||||||||||||
| 2019年計画改正案によると許可 | — | $ | — | — | $ | — | ||||||||||||||
| 授与する | ( | ) | $ | — | $ | |||||||||||||||
| 鍛えられた | — | ( | ) | $ | — | $ | ||||||||||||||
| 没収される | ( | ) | $ | — | $ | |||||||||||||||
| キャンセルします | ( | ) | $ | — | $ | |||||||||||||||
| 2021年12月31日の残高 | $ | — | $ | |||||||||||||||||
| 2019年計画改正案によると許可 | — | $ | — | — | $ | — | ||||||||||||||
| 授与する | ( | ) | $ | $ | ||||||||||||||||
| 鍛えられた | — | ( | ) | $ | $ | |||||||||||||||
| 没収される | ( | ) | $ | ( | ) | $ | ||||||||||||||
| キャンセルします | ( | ) | $ | — | $ | |||||||||||||||
| 2022年12月31日の残高 | $ | $ | ||||||||||||||||||
自分から2022年12月31日何人いますか
合計内面的価値とは現在までのことです2022年12月31日、会社普通株終値$に基づいて
会社はすでに$を受け取りました
Vaxart,Inc.そして付属会社
連結財務諸表付記
この年度までに付与されたオプションの加重平均授出日公正価値2022年12月31日そして、2021,はい$です
| 十二月三十一日までの年度 | ||||||||
| 2022 | 2021 | |||||||
| 無リスク金利 | % | % | ||||||
| 予想期限(年単位) | ||||||||
| 予想変動率 | % | % | ||||||
| 配当率 | — | % | — | % | ||||
同社は授与日にすべての株式奨励の公正価値を測定し、これらの奨励の公正価値を記録し、推定された没収を差し引いて、サービス期間内の補償費用を計上する。オプション確認の株式報酬総額は以下の通り(千で計算)
| 十二月三十一日までの年度 | ||||||||
| 2022 | 2021 | |||||||
| 研究開発 | $ | $ | ||||||
| 一般と行政 | ||||||||
| 株に基づく報酬総額 | $ | $ | ||||||
効き目がある2021年6月16日会社はウォルト·W·ラトゥール前取締役会長に付与された未償還オプションを付与する条項を修正しました
自分から2022年12月31日,期待付与された未償還株式オプションとRSUに関する未確認株式ベース報酬コストは$である
開ける2022年8月4日♪the the the2022従業員株購入計画(“2022ESPP“)会社の株主の承認を得ます。”会社は保留します
ESPP株の公正価値はブラック·スコアーズオプション定価モデルを用いて推定した。われわれのESPP発行株の会計年度の加重平均に基づいて公正価値を推定する2022はい$です
注:12.退職福祉計画
同社は税務条件に適合した従業員の貯蓄と退職計画を提供し、一般的には401(K)会社合資格従業員の計画(“計画”)をカバーする。この計画によると従業員は5月.現在の給与をアメリカ国税局の年間納付上限に延期することを選択しました$20,500例年上の2022そして$19,500例年の数字に使われる2021.従業員年齢50またはそれ以上5月.余分な資金を寄付することを選択する$6,500年に1回2022そして、2021.
従業員は彼らの資金を一連の共同基金に直接投資し、これらの資金は直ちに付与された。ここ数年で2022年12月31日そして、2021,会社は従業員の支払いを一致させた
Vaxart,Inc.そして付属会社
連結財務諸表付記
注:13.所得税を免除する
所得税準備金は以下の部分からなる(千計)
| 十二月三十一日までの年度 | ||||||||
| 2022 | 2021 | |||||||
| 現在: | ||||||||
| 連邦制 | $ | $ | ||||||
| 状態.状態 | ||||||||
| 外国.外国 | ||||||||
| 総電流 | ||||||||
| 延期: | ||||||||
| 連邦制 | ||||||||
| 状態.状態 | ||||||||
| 外国.外国 | ||||||||
| 遅延合計 | ||||||||
| 所得税支給 | $ | $ | ||||||
繰延税金資産の構成は以下のとおりである(千計)
| 2022年12月31日 | 2021年12月31日 | |||||||
| 繰延税金資産: | ||||||||
| 純営業損失が繰り越す | $ | $ | ||||||
| 研究開発税収控除 | ||||||||
| 資本化研究と開発 | ||||||||
| 将来の特許使用料を販売します | ||||||||
| リース責任 | ||||||||
| 課税項目、準備金その他 | ||||||||
| 繰延税金資産総額 | ||||||||
| 推定免税額 | ( | ) | ( | ) | ||||
| 繰延税項資産から推定免税額の純額を差し引く | ||||||||
| 繰延税金負債: | ||||||||
| 無形資産 | ( | ) | ( | ) | ||||
| 使用権資産 | ( | ) | ( | ) | ||||
| 財産と設備の減価償却 | ( | ) | ( | ) | ||||
| 繰延税金負債総額 | ( | ) | ( | ) | ||||
| 繰延税項目純資産 | $ | $ | ||||||
所得税支出と#年連邦法定所得税税率の適用により計算された期待所得税の計上との入金21%所得税控除準備前の純損失:
| 十二月三十一日までの年度 | ||||||||
| 2022 | 2021 | |||||||
| 法定税率で徴収されるアメリカ連邦税 | % | % | ||||||
| 州税(連邦福祉を差し引いた純額) | ||||||||
| 外貨利回り | ( | ) | ||||||
| 永久控除不能項目 | ( | ) | ||||||
| 税金控除 | ||||||||
| 評価免除額を変更する | ( | ) | ( | ) | ||||
| 制御変更による税務属性のログアウト | ||||||||
| 前年の調整 | ( | ) | ||||||
| 他にも | ( | ) | ( | ) | ||||
| 所得税支給 | ( | )% | ( | )% | ||||
会社の実際の税収支出は法定の連邦所得税支出と異なり、税率は
自分から2022年12月31日そして、2021,会社は純営業損失(“NOL”)が#ドルに繰り越している
Vaxart,Inc.そして付属会社
連結財務諸表付記
自分から2022年12月31日また、同社は#ドルの税金損失を累計している
締め切り:2022年12月31日-そして、2021,同社には連邦用途のための税収控除の研究開発があり、金額は$です
始まったのは2022年1月1日年に公布された“減税·雇用法案”(以下“法案”)2017年12月今期の研究開発支出を差し引く選択を取り消し、納税者にアメリカと非アメリカの研究開発支出を資本化と償却するよう要求した5人そして15歳それぞれ数年です。この法律は完成しました注釈会社の現在の納税義務に影響を与える。
セグメント化する382そして383国税法では,ある所有権変更が当社がこれらの繰り越し能力を利用することを制限する可能性がある後,NOLと税収控除繰越の年間使用を制限することが規定されている。同社はこのような所有権変更が発生したかどうかを決定するための分析を完了し、より可能性が高いと結論した注釈所有権が変化した。全額推定免税額が存在するため,第382そして383意志注釈会社の実際の税率に影響を与える。財務諸表中の任意の損失または貸出のメリットを確認する前に、さらなる分析が行われる。
もし会社が以下の状況を超える可能性が高い場合は、その会社は推定手当によって繰延税金資産を減らすことを要求する注釈繰延税金資産の一部または全部は注釈実現されました。経営陣は、評価手当の潜在的需要を評価する際に判断を使用しなければならず、これには負の証拠と正の証拠の評価が必要である。消極的と積極的な証拠に対する潜在的影響の重視度は客観的に確認できる程度に相応すべきである。見積もり準備の需要と金額(あればある)を決定する時、当社は過去の収入レベル、未来の収入の見積もり及び税務計画策略に基づいて、繰延税金資産を回収する可能性を評価する。過去累計損失のため、同社は、すべての既存の証拠に基づいて、今後の期間に記録された繰延税金純額を回収するかどうかに大きな不確実性があると認定している。そこで、当社は以下の日にそのすべての繰延税項純資産計の推定値を準備します2022年12月31日そして2021.推定手当総額の純変化は約#ドル
当社は、適切な場合には、すべての不確定な所得税頭寸に未確認の税収割引を記録する。-会社が不確定な税収頭寸記録の未確認税収割引は約ドルである
税収割引が確認されていない期初と期末金額の入金は以下の通り(千計)
| 十二月三十一日までの年度 | ||||||||
| 2022 | 2021 | |||||||
| 期初残高 | $ | $ | ||||||
| 本年度に関連する納税状況に基づいて計算される増加額 | ||||||||
| 数年前の納税状況に関する増加(減少) | ( | ) | ||||||
| 期末残高 | $ | $ | ||||||
同社の政策は、いかなる未確認の税収割引の課税利息と罰金を所得税費用の構成要素として確認することである。年末までに年度を終える2022年12月31日そして2021,会社は認識しています
当社はアメリカ、オーストラリアおよびアメリカ各州に所得税申告書を提出します。当社はすべての所得税申告書を提出した司法管轄区で税務監査を受けています。税務監査自体の性質はしばしば複雑で、完成するのに数年かかるかもしれない。今のところ違います。任意の司法管轄区域で所得税申告書に対する税務監査が開始された。
国税法に適用される税務訴訟の時効によると、会社とその米国子会社は、独立しても合併グループの一部としても違います。アメリカ国税局は税引き前年度にアメリカ連邦所得税の審査を受ける時間がもっと長いです2017.ほとんどの州の所得税法に適用される訴訟時効によると会社は違います。税務機関は前納税年度に対して国家所得税審査を行わなくなったそれがすでに所得税申告書を提出した州で。しかし、会社は純営業損失や由来などの所得税の属性を繰り越しているため2004そして、より早い納税年度は、これらの属性を将来提出された申告書で使用する際にも監査を行うことができます。当社は本会計年度税務機関の異郷税務審査を受けます2015前へ行ってください。
Vaxart,Inc.そして付属会社
連結財務諸表付記
注.注14.*普通株主の1株当たり純損失
次の表は、1株当たりの基本純損失と償却純損失の計算方法(単位は千、1株と1株当たりの金額は含まない)を示している
| 十二月三十一日までの年度 |
||||||||
| 2022 |
2021 |
|||||||
| 純損失 |
$ | ( |
) | $ | ( |
) | ||
| 1株当たり純損失を計算するための株、基本的かつ希釈された |
||||||||
| 普通株主は1株当たり基本損失と希釈して1株当たり純損失を占めるべきである |
$ | ( |
) | $ | ( |
) | ||
以下の希釈可能な加重平均証券は、逆希釈されるので、発行された加重平均株式の計算から除外される
| 十二月三十一日までの年度 |
||||||||
| 2022 |
2021 |
|||||||
| 普通株購入オプション |
|
|||||||
| 限定株単位で普通株を購入する |
||||||||
| 普通株購入引受権証 |
|
|||||||
| 従業員株購入計画 |
||||||||
| 1株当たりの収益を希釈して計算した分母から除外した潜在的希薄化証券総額 |
||||||||
注:15.*後続のイベント
以来2022年12月31日会社は発行されました
以来の訴訟地位の変化2022年12月31日付記に含まれる9.(C)訴訟“と約束したり、事項があったりする。
中国では1つ目は1/42023,同社は主にノウォーカーウイルス経口ワクチン候補案の推進に集中し、SARS-CoVの起動を延期することを決定した2人間は研究に挑戦する。同社はまた、運営コストを低減し、従業員チームがその業務ニーズにより良く適応できるように再編計画を実施している。この計画は約減少した27%会社員チームの一部です
最近のシリコンバレー銀行(SVB)の影響を受けて、連邦預金保険会社がSVBを買収することを含め、会社は以前にSVBで保有していた任意の現金または他の預金をより大きな金融機関に移転することを決定した。私たちは以前SVBで持っていたすべての現金と他の預金を使用することができる。私たちは注釈SVBの状況は私たちの財務状況や運営に実質的な影響を及ぼすと予想される。
第9項:会計·財務開示における会計担当者との変更と相違
2021年7月15日独立公認会計士事務所WithumSmith+Brown,PC(Withum)は、Oum&Co.LLPのある資産を買収しましたオム)、独立公認会計士事務所会社“(”取引記録“)”この取引の結果、Oumは2021年7月15日に独立公認会計士事務所を辞任した。会社を辞めるとともに、当社はその監査委員会の承認を得て、Withumを当社の新たな独立公認会計士事務所に招聘することに同意し、2021年7月15日から発効します。
取引前に、当社は会計原則についていかなる完成或いは行う予定の具体的な取引に適用したり、Withumが当社の財務諸表について提出した監査意見のタイプについてWithumと協議する可能性があり、Withumもいかなる書面或いは口頭意見も提供しておらず、このような意見は当社が任意の会計、監査或いは財務報告問題について決定する時に考慮する重要な要素である。
Oum独立公認会計士事務所報告書監査報告書“)当社の2020年及び2019年12月31日までの財政年度の財務諸表には何の不利な意見や免責声明もなく、不確実性、監査範囲または会計原則に保留または修正はありません。
二零二年及び二零一年十二月三十一日まで、及び最近完成した財政年度終了から辞職日二零一年七月十五日までの移行期間内に、OUMとは、任意の会計原則又は実務、財務諸表開示又は監査範囲又は手順等の事項に“相違”がなく(S−K条例第304(A)(1)(Iv)項及び第304項の関連指示を参照)であり、これらの相違が解決されなければ、OUMがその報告書にこの等の相違を言及することになる。2020年12月31日と2019年12月31日までの財政年度内、およびその後2021年7月15日までの移行期間内には、“報告すべき事項”は発生していない(S-K条例第304(A)(1)(V)項で定義されている)。
項目9 A:管理制御とプログラム
開示制御とプログラムの有効性に関する結論
我々は、我々の取引所法案報告において開示すべき情報が、米国証券取引委員会規則および表で指定された期間内に記録、処理、集計および報告され、これらの情報が蓄積されて、必要な開示に関する決定を迅速に行うために、我々の経営陣に伝達されることを保証するために、開示制御および手続きを維持する。
米国証券取引委員会規則13 a-15(B)の要求に基づき、本Form 10-K年次報告がカバーする期間終了までの開示制御プログラムおよびプログラムの設計および実行の有効性を、CEOおよび最高財務官を含む管理層の監督の下で評価した。上記に基づき、我々の最高経営責任者及び最高財務官は、我々の開示制御及び手続が合理的な保証レベルで有効であると結論した。
内部統制の内在的限界
私たちの経営陣は、私たちの社長やCEOを含めて、私たちの開示制御や手続き、または私たちの内部統制がすべてのミスやすべての詐欺を防ぐことができることを期待していません。発想や動作がどんなに良くても、絶対的な保証ではなく、合理的な保証しか提供できず、制御システムの目標が実現されることを確保する制御システム。また,制御システムの設計は,資源制約が存在し,そのコストに対する制御の利点を考慮しなければならないという事実を反映しなければならない.すべての制御システムの固有の限界により,どの制御評価もVaxart内部のすべての制御問題や不正イベントが検出されていることを絶対に保証することはできない.
財務報告の内部統制に関する経営陣の報告
我々の経営陣は、“取引法”ルール13 a-15(F)に定義されている財務報告の十分な内部統制の確立と維持を担当している。我々の経営陣(最高経営責任者や財務責任者を含む)の監督と参加の下、トレデビル委員会後援組織委員会が発表した“内部統制-総合枠組み(2013)枠組み”に基づいて、財務報告の内部統制の有効性を評価した。“内部統制-統合枠組み(2013)”の枠組みでの我々の評価結果によると、我々の経営陣は、財務報告に対する内部統制が2022年12月31日に発効すると結論した。
2022年12月31日現在、財務報告の内部統制の有効性を独立公認会計士事務所が監査しており、その報告は以下のとおりである。
財務報告の内部統制の変化
2022年12月31日までの四半期内に、財務報告の内部統制には何の変化もありません。これらの変化は、財務諸表の内部統制に大きな影響を与えているか、または合理的に影響を与えている可能性があります。
第9 B項:その他の資料
本部が移転する
2021年12月、私たちの取締役会は会社本部と主要執行オフィスの南フロリダ州への移転を許可した。会社本部と主要執行オフィスを南フロリダ州に移転する決定は様々な考えからです。その他の考慮事項の中で、会社の執行役員の大多数と会社の取締役の大多数は、会社の最高経営責任者アンドレ·フロロ約を含む東海岸に住んでいる。私たちの取締役会は、会社管理チームと取締役会が会議を開くために、会社を東海岸に本部を置くことにしました。また、我々の取締役会は、東海岸に会社本部を設立することで、ワシントンD.C.の政府機関、ニューヨークの投資銀行や金融サービスプロバイダ、米国、ヨーロッパ、中東の潜在的な顧客や戦略パートナーとのコミュニケーションやその他の相互作用を促進できると考えている。私たちはまだ南フロリダ州でオフィススペースを購入していません。私たちが行動した後に会社本部の最新の位置を提供するつもりです。
トッド·C·デイビスは取締役会を辞めた
2023年3月14日、会社取締役会長、取締役会社取締役トッド·C·デイビスは、取締役会長兼取締役を辞任することを会社に通知し、2023年3月17日から発効した。デービスさん氏が辞任した日から、マイケル·J·フェンニが取締役会長に就任した。Davisさんの辞任決定は、会社がその運営、政策、または接近に関連するいかなる問題でも意見の相違があるからではない。
プロジェクト9 C.検査妨害を開示する外国司法管区
ない。
第三部
プロジェクト10.役員、役員、および企業管理
このプロジェクトに要求される情報は、会社が財政年度終了後120日以内に証券取引委員会に提出する依頼書に記載され、参照によって本明細書に組み込まれる。
第11項:役員報酬
このプロジェクトに要求される情報は、会社が財政年度終了後120日以内に証券取引委員会に提出する依頼書に記載され、参照によって本明細書に組み込まれる。
第十二項:特定実益所有者及び経営陣の保証所有権及び関連株主事項
このプロジェクトに要求される情報は、会社が財政年度終了後120日以内に証券取引委員会に提出する依頼書に記載され、参照によって本明細書に組み込まれる。
第13項:特定の関係及び関連取引の確立、並びに取締役独立性
このプロジェクトに要求される情報は、会社が財政年度終了後120日以内に証券取引委員会に提出する依頼書に記載され、参照によって本明細書に組み込まれる。
第14項目:主な課金とサービス
このプロジェクトに要求される情報は、会社が財政年度終了後120日以内に証券取引委員会に提出する依頼書に記載され、参照によって本明細書に組み込まれる。
第4部
項目15.各種展示品と財務諸表明細書
財務諸表明細書
これらは適用されないので、または必要な資料が財務諸表または付記に記載されているので、すべての財務諸表の添付表は省略される。
陳列品
以下のファイルは、本年度報告とともにアーカイブされます。
| (1) |
財務諸表(第8項“財務諸表および補足データ”を参照し、参照によって本明細書に組み込まれる)。 |
| (2) |
財務諸表付表(財務諸表添付表は省略されている。本明細書に記載されている情報は、添付の財務諸表または付記に適用または表示されていないことが要求されるので)。 |
| (3) |
展示品です。 |
展示品索引
|
|
引用で編入する |
||||
| 展示品 |
書類説明 |
別表/表 |
ファイル.ファイル |
展示品 |
提出日 |
| 2.1 |
Aviragen Treateutics,Inc.,Vaxart,Inc.およびAgora Merge Sub,Inc.が2017年10月27日に署名した合併および再構成協定および計画。 |
8-K |
001-35285 |
2.1 |
2017年10月30日 |
| 2.2 |
Aviragen Treateutics,Inc.,Vaxart,Inc.とAgora Merge Sub,Inc.が2017年10月27日に署名した合併再編協定と計画の第1号修正案は,2018年2月7日である. |
8-K |
001-35285 |
2.1 |
2018年2月7日 |
| 3.1 |
Aviragen治療会社登録証明書を再記述します。 |
10-K |
001-35285 |
3.1 |
2016年9月13日 |
| 3.2 |
Aviragen治療会社は証明書の改訂証明書を再登録した。 |
8-K |
001-35285 |
3.1 |
2018年2月20日 |
| 3.3 |
Vaxart,Inc.証明書の改訂証明書を再登録する. |
8-K |
001-35285 |
3.2 |
2018年2月20日 |
| 3.4 | 再改訂された改訂証明書Vaxart,Inc.登録証明書 | 8-K | 001-35285 | 3.1 | 2019年4月24日 |
| 3.5 | Vaxart,Inc.証明書の改訂証明書を再登録する. | 8-K | 001-35285 | 3.1 | 2020年6月9日 |
| 3.6 | Vaxart,Inc.証明書の改訂証明書を再登録する. | 表格10-Q | 001-35285 | 3.3 | 2022年8月8日 |
| 3.7 | Vaxart,Inc.改正と再制定された定款は,2021年4月7日から発効する | 8-K | 001-35285 | 3.1 | 2021年4月13日 |
| 4.1 |
添付ファイル3.1~3.7を参照 |
|
|
|
|
| 4.2 |
普通株式証明書サンプル |
S-3 |
333-228910 |
4.2 |
2018年12月20日 |
| 4.3 | 前払い資金株式証明書表(2019年4月) | S-1 | 333-229536 | 10.25 | 2019年2月6日 |
| 4.4 | 普通株式承認証表(2019年4月) | S-1/A | 333-229536 | 4.4 | 2019年4月8日 |
| 4.5 | 代表依頼書表(2019年4月) | S-1/A | 333-229536 | 4.5 | 2019年4月8日 |
| 4.6 | 前払い資金株式証明書表(2019年9月) | S-1 | 333-233717 | 4.3 | 2019年9月11日 |
| 4.7 | 普通株式承認証表(2019年9月) | S-1 | 333-233717 | 4.4 | 2019年9月11日 |
| 4.8 | 代表依頼書表(2019年9月) | S-1/A | 333-233717 | 4.5 | 2019年9月24日 |
| 4.9 | 普通株式承認証表(2020年3月) | 8-K | 001-35285 | 4.1 | 2020年3月2日 |
| 4.10 | 配給代理人授権書表(2020年3月) | 8-K | 001-35285 | 4.2 | 2020年3月2日 |
| 4.11 * | 登録者の証券説明 | ||||
| 10.1 + |
2003年9月29日、Biotaホールディングスと三共株式会社との間の協力と許可協定。 |
10-Q |
001-35285 |
10.5 |
2013年5月10日 |
| 10.2 + |
2005年6月30日Biota Holdings LimitedとBiota Science Management Ptyとの間の協力および許可プロトコル修正案1。和三共株式会社です。 |
10-Q |
001-35285 |
10.6 |
2013年5月10日 |
| 10.3 |
2009年3月27日、Biota Holdings LimitedとBiota Science Management Ptyとの間の協力および許可プロトコル修正案2。第一三共株式会社と |
10-Q |
001-35285 |
10.7 |
2013年5月10日 |
| 10.4 + |
2009年3月27日、Biota Holdings LimitedとBiota Science Management Ptyの間の商業化協定。第一三共株式会社と。 |
10-Q |
001-35285 |
10.8 |
2013年5月10日 |
| 10.5 + |
2011年3月31日、Biota Science Management Pty.有限会社とアメリカ衛生·公衆サービス部が準備と反応を担当する補佐部長事務室内の生物医学高度研究と発展許可事務室 |
10-Q |
001-35285 |
10.9 |
2013年5月10日 |
| 10.6 + |
1990年2月21日の研究·許可協定は,生物群科学管理会社と生物群科学管理会社が締結した。BIOTAホールディングス株式会社、グラクソ·スミスクラインオーストラリア有限会社。グラクソ·スミスクライングループ有限公司と |
10-K |
001-35285 |
10.6 |
2013年9月27日 |
| 10.7 # |
2007総合株式および奨励計画(委託書付録Aを含む) |
定義14 A |
000-04829 |
- |
2007年04月12日 |
| 10.8 # |
2007年総合株式およびインセンティブ計画下の従業員株式オプション協定フォーマット |
8-K |
001-35285 |
10.1 |
2013年12月10日 |
| 引用で編入する | |||||
| 展示品 番号をつける |
書類説明 | 別表/表 | ファイル.ファイル 番号をつける |
展示品 | 提出日 |
| 10.9 + |
Aviragen Treateutics,Inc.,Biota Holdings Pty Ltd,Biota Science Management Pty間の特許使用料権益買収協定。LTDとHealthcare Royalty Partners III,L.P.,2016年4月22日 |
8-K |
001-35285 |
10.1 |
2016年4月26日 |
| 10.10 |
Aviragen Treateutics,Inc.とHealthcare Royalty Partners IIIとの間の保護権協定,L.P.,2016年4月22日 |
8-K |
001-35285 |
10.2 |
2016年4月26日 |
| 10.11 # |
2016年度持分インセンティブ計画従業員株式オプション契約フォーマット |
10-Q |
001-35285 |
10.1 |
2017年5月8日 |
| 10.12 # |
2016持分インセンティブ計画(委託書付録Aに含まれる) |
定義14 A |
001-35285 |
- |
2016年9月27日 |
| 10.13 # |
取締役株式オプション協定 |
S-4 |
333-222009 |
10.22 |
2017年12月12日 |
| 10.14 |
Vaxart,Inc.とその役員と役員との間の賠償協定フォーマット |
8-K |
001-35285 |
10.3 |
2018年2月20日 |
| 10.15 # |
Vaxart,Inc.2007年株式インセンティブ計画、株式オプション協定、株式オプション付与通知フォーマット、オプション付加条項と条件フォーマット、および株式オプション行使プロトコルを改訂し、再改訂した |
S-4/A |
333-222009 |
10.24 |
2017年12月29日 |
| 10.16 # |
2011年5月25日の招待状と、2011年10月1日にVaxart,Inc.とWoutW.Latour,M.D.の間の招待状とオプション付与協定修正案。 |
S-4/A |
333-222009 |
10.25 |
2017年12月29日 |
| 10.17 |
工業賃貸日は2013年10月28日で、Vaxart,Inc.とOyster Point LLCによって締結され、両者の間で締結された |
S-4/A |
333-222009 |
10.26 |
2017年12月29日 |
| 10.18 |
Vaxart,Inc.とCRP Edgewater,LLCの間で締結されたリース契約日は2015年4月17日である |
S-4/A |
333-222009 |
10.27 |
2017年12月29日 |
| 10.19 # |
解散費福祉計画及び離職福祉計画参加通知 |
8-K |
001-35285 |
10.1 |
2018年6月6日 |
| 10.20 |
Vaxart,Inc.とB.Riley FBR,Inc.の間の2018年12月19日の販売プロトコルフォーマット. |
S-3 |
333-228910 |
1.2 |
2018年12月2日 |
| 10.21 |
改訂と再発行された引受権証は、オックスフォード金融有限責任会社に発行され、日付は2018年2月13日です |
8-K |
001-35285 |
10.2 |
2018年2月20日 |
| 10.22 | 契約書は、日付は2019年1月25日で、Vaxart,Inc.とH.C.Wainwright&Co.,LLCの間で署名され、改訂された | 8-K | 001-35285 | 10.2 | 2019年3月20日 |
| 10.23 | 配給代理人授権書表(2019年3月) | 8-K | 001-35285 | 10.3 | 2019年3月20日 |
| 10.24 # | 改訂された2019年持分インセンティブ計画 | 8-K | 001-35285 | 10.1 | 2021年6月21日 |
| 10.25 # | 株式オプション付与通知、株式オプション協定及び2019年持分インセンティブ計画下の行使通知のフォーマット | S-8 | 333-239727 | 10.2 | 2020年7月7日 |
| 10.26 # | “2019年株式奨励計画限定株授出通知書及び限定株奨励協定”フォーマット | 8-K | 001-35285 | 10.3 | 2019年4月24日 |
| 10.27 + | Vaxart,Inc.とLonza Houston,Inc.の間で2019年7月17日に署名された製造サービス協定。 | S-1/A | 333-233717 | 10.30 | 2019年9月24日 |
| 10.28 | 賃貸契約第1修正案は、日付が2019年9月17日で、Vaxart,Inc.とHCP Inc.である。 | 8-K | 001-35285 | 10.1 | 2019年9月19日 |
| 10.29 | Vaxart,Inc.とその中に列挙された買い手との間の証券購入プロトコルフォーマットは,日付は2020年2月27日である | 8-K | 001-35285 | 10.1 | 2020年3月2日 |
| 10.30 # | 会社とショーン·タッカー博士の招聘状は2006年5月1日です | 10-Q | 001-35285 | 10.2 | 2020年5月12日 |
| 10.31 # | 会社とマーガレット·エチェードの間の招聘状は、2018年3月26日です | 10-Q | 001-35285 | 10.3 | 2020年5月12日 |
| 10.32 # | 2018年12月27日、会社からマーガレット·エチェードへの手紙 | 10-Q | 001-35285 | 10.4 | 2020年5月12日 |
| 10.33 # | 別居協議は、期日は2020年6月14日であり、Vaxart,Inc.とWoutW.Latour,M.Dからなる。 | 8-K | 001-35285 | 10.1 | 2020年6月15日 |
| 10.34 # | Vaxart,Inc.とAndrei Floroiu間の書簡プロトコルは,2020年6月14日である | 8-K | 001-35285 | 10.2 | 2020年6月15日 |
| 10.35 | 販売契約は,日付は2020年7月8日であり,SVB Leerink LLC,B.Riley FBR,Inc.とVaxart,Inc.によって署名されている。 | S-3 ASR | 333-239751 | 1.2 | 2020年7月8日 |
| 10.36 + | Vaxart,Inc.とKindred Biosciences,Inc.の間で2020年4月17日に締結されたメインサービスプロトコル. | 10-Q | 001-35285 | 10.4 | 2020年11月12日 |
| 10.37 + | Vaxart,Inc.とKindred Biosciences,Inc.の間で2020年4月17日に締結されたプライマリサービス協定によると,ワーキングステート003の日付は2020年9月11日である. | 10-Q | 001-35285 | 10.5 | 2020年11月12日 |
| 10.38 + | Vaxart,Inc.とKindred Biosciences,Inc.の間で2020年4月17日に締結されたプライマリサービス協定によると,ワーキングステート004の日付は2020年9月11日である. | 10-Q | 001-35285 | 10.6 | 2020年11月12日 |
| 10.39 | 公開市場販売協定は,2020年10月13日にVaxart,Inc.,Jefferies LLCとPiper Sandler&Co.が署名した。 | 8-K | 001-35285 | 1.1 | 2020年10月14日 |
| 10.40 | Vaxart,Inc.とVera Treateutics,Inc.の間で2020年11月16日に締結された転貸プロトコル. | 10-K | 001-35285 | 10.40 | 2021年2月25日 |
| 引用で編入する | ||||||
| 展示品 番号をつける |
書類説明 | 別表/表 | ファイル.ファイル 番号をつける |
展示品 | 提出日 | |
| 10.41 | 当社、Cantor Fitzgerald&Co.とB.Riley Securities,Inc.が2021年9月15日に署名した制御持分発行と販売協定。 | 8-K | 001-35285 | 1.1 | 2021年9月16日 | |
| 10.42 | Vaxart,Inc.とBritannia Pointe Grand Limited Partnershipとの間で締結されたレンタル契約は,2021年9月17日である | 8-K | 001-35285 | 10.1 | 2021年9月21日 | |
| 10.43 | Vaxart,Inc.とHealthPeak Properties,Inc.の間で2021年9月17日に署名された賃貸協定第2修正案。 | 8-K | 001-35285 | 10.2 | 2021年9月21日 | |
| 10.44 # | 会社とジェームズ·カミングス医学博士の招待状は2021年8月16日です | 10-Q | 001-35285 | 10.4 | 2021年11月4日 | |
| 10.45 + | 資産購入協定は,2021年11月24日にVaxart,Inc.とKindred Biosciences,Inc.によって署名された. | 10-K | 001-35285 | 10.45 | 2022年2月24日 | |
| 10.46 # | 会社とEdward Bergの間の招待状は、2022年1月18日です | 10-K | 001-35285 | 10.46 | 2022年2月24日 | |
| 10.47 # | 非従業員役員報酬計画は、2022年4月1日から施行される | 10-Q | 001-35285 | 10.2 | 2022年5月9日 | |
| 10.48 # | 会社とFLG Partners,LLC間の守秘諮問協定は,2022年4月20日から発効する | 8-K | 001-35285 | 10.1 | 2022年5月11日 | |
| 10.49 # | 会社がエチェードさんと締結したコンサルティングサービス協定は、2022年5月11日から発効します | 8-K | 001-35285 | 10.2 | 2022年5月11日 | |
| 10.50+ | 当社とhVIVOサービス株式会社が2022年6月29日に締結したチャレンジ前研究サービス契約 | 10-Q | 001-35285 | 10.4 | 2022年8月8日 | |
| 10.51# | 2019年持分インセンティブ計画 | 8-K/A | 001-35285 | 10.1 | 2022年8月5日 | |
| 10.52# | 2022年従業員株購入計画 | 8-K/A | 001-35285 | 10.2 | 2022年8月5日 | |
| 10.53#* | 会社とフィリップ·リーの間の招待状は2022年12月5日です | |||||
| 21.1 * |
登録者の子会社 |
|
|
|
|
|
| 23.1 * |
独立公認会計士事務所スミス+Brown PCは同意しました |
|
|
|
|
|
| 24.1 * |
授権書。こちらのサインページをご参照ください |
|
|
|
|
|
| 31.1* | 2002年サバンズ-オキシリー法302節で採択された取引法規則13 a-14(A)および15 d-14(A)による最高経営責任者の認証 | |||||
| 31.2 * |
2002年サバンズ-オキシリー法第302節で可決された取引法規則13 a-14(A)および15 d-14(A)に基づいて首席財務官の認証を行う |
|
|
|
|
|
| 32.1 § |
1934年改正証券取引法第13 a−14条及び2002年“サバンズ·オックススリー法案”第906条に基づいて可決された“米国法”第18編第1350条による首席行政·財務官の認証 |
|
|
|
|
|
| 101.INS* |
連結されたXBRLインスタンス文書-インスタンス文書は、そのXBRLタグがイントラネットXBRL文書に埋め込まれているので、対話データファイルには表示されない |
|
|
|
|
|
| 101.Sch* | イントラネットXBRL分類拡張アーキテクチャ文書 | |||||
| 101.カール* | インラインXBRL分類拡張計算リンクライブラリ文書 | |||||
| 101.定義* | インラインXBRL分類拡張Linkbase文書を定義する | |||||
| 101.実験所* | XBRL分類拡張ラベルLinkbase文書を連結する | |||||
| 101.前期* | インラインXBRL分類拡張プレゼンテーションLinkbaseドキュメント | |||||
| 104 | 表紙相互データファイル(添付ファイル101に含まれるイントラネットXBRLのフォーマット) | |||||
| * |
同封アーカイブ |
| # |
契約または報酬計画またはスケジュールを管理する |
| + |
“取引法”公布の規則24 b-2に基づく機密待遇は、本展示品の機密部分が漏れており、単独で欧州委員会に提出された |
| § |
S−K規制第601(B)(32)(Ii)項及び米国証券取引委員会が発行した第33−8238号及び第34−47986号最終規則によれば、財務報告の内部統制に関する管理層の報告及び取引所法定期報告における開示情報の証明によると、本プロトコル添付ファイル32.1で提供された証明は、本年度報告と共にForm 10−K形式で提供されるものとみなされ、取引法第18節において“アーカイブされた”とはみなされない。このような証明は、登録者が参照によって明示的に組み込まれない限り、“証券法”または“取引法”に基づいて提出された任意の文書に引用によって組み込まれているとはみなされない |
第16項:表格10-K要約
ない。
サイン
1934年“証券取引法”第13又は15(D)節の要求に基づいて、登録者は、次の署名者が代表して本報告書に署名することを正式に許可した。
|
|
|
Vaxart,Inc. |
|
|
|
差出人: |
/s/アンドレイFLOROIU |
|
| 日付:2023年3月15日 |
|
アンドレ·フロイル |
|
|
|
|
総裁と行政長官将校.将校 |
|
授権依頼書
これらの陳述を通じて、私はすべての人が、以下に署名したすべての人がAndrei FloroiuとPhillip Leeを彼または彼女の事実代理人として構成し、任命することを知っている。彼らのすべての人は、彼または彼女が本報告書の任意の修正に署名し、証拠物とこれに関連する他の文書と共に米国証券取引委員会に提出し、ここで上述のすべての事実代理人を承認し、確認する権利があり、彼らまたは彼または彼女の代替者は、本報告の規定に従って合法的に行動することができる。
本報告は、1934年の証券取引法の要求に基づき、以下の者によって登録者として指定日に署名された。
| サイン |
|
タイトル |
|
日取り |
| /s/アンドレイFLOROIU |
|
|
|
|
| アンドレ·フロイル
|
|
社長と最高経営責任者兼CEO (CEO兼CEO) |
|
2023年3月15日 |
| /s/PhillipLee |
|
|
|
|
| フィリップ·リー
|
|
首席財務官 (首席財務会計官) |
|
2023年3月15日 |
| トッド·C·デイビス |
|
|
|
|
| トッド·C·デイビス |
|
取締役会議長 |
|
2023年3月15日 |
| エレーン·J·ヘレン博士 | ||||
| エレーン·J·ヘレン | 役員.取締役 | 2023年3月15日 | ||
| /s/マイケルJ.Finney |
|
|
|
|
| マイケル·J·フェニー博士 |
|
役員.取締役 |
|
2023年3月15日 |
| /s/David WHeon M.D |
|
|
|
|
| デイビッド·ウェリントン医学博士です |
|
役員.取締役 |
|
2023年3月15日 |
| /s/マーク·ワトソン |
|
|
|
|
| マーク·ワトソン | 役員.取締役 | 2023年3月15日 | ||
| ロバート·A·エディッド | ||||
| ロバート·A·エディッド |
|
役員.取締役 |
|
2023年3月15日 |