
アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
本財政年度末まで
あるいは…。
手数料書類番号
(登録者の正確な氏名はその定款に記載)
(明またはその他の司法管轄権 会社や組織) |
(税務署の雇用主 識別番号) |
(主にオフィスアドレスを実行) |
(郵便番号) |
登録者の電話番号は市外局番を含んでいます(
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
|
取引 記号 |
|
登録された各取引所の名称 |
|
|
同法第12条(G)により登録された証券:なし
登録者が証券法規則405で定義されている経験豊富な発行者である場合は、再選択マークで示してください。はい、そうです☐
登録者が当該法第13条又は第15条(D)に従って報告書を提出する必要がないか否かを、再選択マークで示す。はい、そうです☐
登録者が(1)過去12ヶ月以内(または登録者がそのような報告の提出を要求されたより短い期間)に、1934年の証券取引法第13または15(D)節に提出されたすべての報告を提出したかどうか、および(2)このような提出要求を過去90日以内に遵守してきたかどうかを、再選択マークで示す
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
大型加速ファイルサーバ |
|
☐ |
|
ファイルマネージャを加速する |
|
☐ |
|
|
|
|
|||
|
☒ |
|
規模の小さい報告会社 |
|
||
|
|
|
|
|
|
|
新興成長型会社 |
|
|
|
|
|
|
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する☐
これらのエラーのより真ん中に登録者の任意のエンタルピーCER幹部が相関回復期間内に§240.10 D−1(B)に基づいて受信したインセンティブベースの補償に基づいて回復分析を行う必要があるかどうかを再選択マークで示す☐
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)。はい、そうです
2022年6月30日現在、すなわち登録者が最近完成した第2四半期の最終営業日、登録者の非関連会社が保有する普通株のナスダックグローバル市場における2022年6月30日の終値によると、登録者が保有する普通株の総時価は約1ドルである
2023年3月2日までの登録者の発行済み普通株式数は
引用で編入された書類
登録者は,登録者が2022年12月31日までの財政年度終了後120日以内に,2022年株主総会に関する最終依頼書を第14 A条に基づいて提出する予定であるそれは.この最終依頼書の内容の一部は、本明細書に記載された範囲内の本年度報告のForm 10−Kの第3の部分に参照されて組み込まれる。
カタログ表
|
|
ページ |
第1部 |
|
|
第1項。 |
業務.業務 |
1 |
第1 A項。 |
リスク要因 |
48 |
項目1 B。 |
未解決従業員意見 |
91 |
第二項です。 |
属性 |
91 |
第三項です。 |
法律訴訟 |
91 |
第四項です。 |
炭鉱安全情報開示 |
91 |
|
|
|
第II部 |
|
|
五番目です。 |
登録者普通株市場、関連株主事項及び発行者による株式証券の購入 |
92 |
第六項です。 |
保留されている |
92 |
第七項。 |
経営陣の財務状況と経営成果の検討と分析 |
93 |
第七A項。 |
市場リスクの定量的·定性的開示について |
104 |
第八項です。 |
財務諸表と補足データ |
105 |
第九項です。 |
会計と財務情報開示の変更と相違 |
105 |
第9条。 |
制御とプログラム |
105 |
プロジェクト9 B。 |
その他の情報 |
106 |
プロジェクト9 Cです。 |
検査妨害に関する外国司法管区の開示 |
106 |
|
|
|
第三部 |
|
|
第10項。 |
役員·幹部と会社の管理 |
107 |
第十一項。 |
役員報酬 |
107 |
第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
107 |
十三項。 |
特定の関係や関連取引、取締役の独立性 |
107 |
14項です。 |
チーフ会計士費用とサービス |
107 |
|
|
|
第4部 |
|
|
第十五項。 |
展示品と財務諸表の付表 |
108 |
第十六項。 |
表格10-Kの概要 |
108 |
このForm 10-K年次報告では、他に説明や文脈が要求されない限り、言及された“Fulcrum”、“Fulcrum Treateutics”、“The Company”、“We”、“Us”、“Our”および同様の提案法は、Fulcrum Treatetics,Inc.およびその合併子会社を指す。Fulcrum Treateutics標識、FulcrumSeekおよびFulcrum Treateutics,Inc.本Form 10-K年次報告に登場する他の商標またはサービスマークは、Fulcrum Treateutics,Inc.の財産である。本Form 10-K年次報告には、他社の登録商標、商標、商品名も含まれている。本明細書に記載されている他のすべての商標、登録マーク、および商号は、それぞれの所有者の財産である。
前向き陳述に関する警告説明
このForm 10-K年度報告書には前向きな陳述が含まれており、これらの陳述は現在の私たちの運営や財務業績などに対する私たちの見方を反映している。本Form 10-K年度報告に含まれる歴史的事実に関する陳述のほか、我々の戦略、将来の運営、将来の財務状況、将来の収入、予想コスト、見通し、計画、管理目標、および予想される市場成長に関する陳述はすべて前向きな陳述である。“予想”、“信じる”、“継続”、“可能”、“推定”、“予想”、“予定”、“可能”、“可能”、“展望”、“計画”、“潜在”、“予測”、“プロジェクト”、“すべき”、“目標”、“会議”であり、これらの語の否定バージョンおよび他の類似表現は、前向き陳述を識別することを目的としている。すべての前向き陳述がこれらの識別語を含むわけではないが、他の陳述に加えて、以下の記述が含まれている
i
私たちは私たちの展望声明で開示された計画、意図、または予想を実際に達成できないかもしれません。あなたは私たちの展望的声明に過度に依存してはいけません。実際の結果や事件は私たちが展望的声明で開示した計画、意図、そして予想とは大きく違うかもしれない。私たちはこの10-K表の年次報告書の警告声明に重要な要素を含んでおり、特に“リスク要因”の部分では、これらの要素は実際の結果や事件をもたらす可能性があり、私たちが行った前向き声明とは大きく異なると考えられる。私たちの前向きな陳述は、私たちが行う可能性がある任意の未来の買収、合併、処置、協力、合弁、または投資の潜在的な影響を反映していない。
Form 10-K年次報告書と私たちがForm 10-K年度報告書として提出した文書を完全に読み、私たちの将来の実際の結果が私たちが予想していたものと大きく異なる可能性があることを理解しなければなりません。本年度報告に含まれるForm 10−Kに含まれる前向き陳述は、本年度報告におけるForm 10−Kにおいて行われ、法的要件が適用されない限り、新たな情報、未来のイベント、または他の理由でいかなる前向き陳述を更新する義務も負わない。
このForm 10-K年度報告書には、業界出版物と研究、第三者による調査と研究から得られた統計および他の業界および市場データ、および潜在市場機会の私たち自身の推定が含まれている。本年度報告で用いたForm 10−Kのすべての市場データは複数の仮説や制限を扱っており,このようなデータを過度に重視しないように注意されたい。業界出版物および第三者研究、調査および研究は、一般に、それらの情報は、そのような情報の正確性または完全性を保証しないにもかかわらず、信頼できるソースから得られることを示している。候補製品の潜在的な市場機会の推定には、私たちの業界知識、業界出版物、第三者研究とその他の調査に基づくいくつかの重要な仮定が含まれており、これらの調査は小サンプルに基づいている可能性があり、市場機会を正確に反映できないかもしれない。私たちは私たちの内部仮定が合理的だと信じているが、これらの仮定を証明する独立したメッセージ源はない。
リスク要因をまとめる
私たちの業務は多くのリスクの影響を受けており、これらのリスクを実現すれば、私たちの業務、財務状況、運営結果、キャッシュフロー、流動性に大きな影響を与える可能性があります。これらのリスクは,本年度報告の10−K表における“リスク要因”の部分でより包括的に検討されている。私たちの主なリスクは以下の点を含む
II
三、三、
パー?パーT I
プロジェクト1.BU無邪気ですね。
概要
我々は臨床段階の生物製薬会社であり,高度に満たされていない医療需要地域の遺伝定義を改善するまれな疾患患者の生活の改善に専念している。我々の最先端の臨床候補製品であるlosmapimodは開発中であり,肩肩周囲筋ジストロフィー(FSHD)の治療に利用可能である。私たちのもう一つの臨床候補はFTX−6058であり、鎌状細胞病またはSCDを含むいくつかのヘモグロビン疾患を潜在的に治療するために開発されている。私たちは Initiated REACHは無作為,二重盲検,プラセボ対照,多国のロモパモント3期臨床試験であり,2022年第2四半期に行われ,2023年下半期に募集を完了する予定である。2023年1月、SCDにおけるFTX−6058の臨床試験の1 b段階データを発表した。私たちは6 mgと2 mgの用量列の登録を終えて、私たちはこの列でもっと多くの科目を募集するつもりはない。12 mg用量列の登録を開始したにもかかわらず,FDAは2023年2月23日にSCD用FTX−6058のIND臨床試験を全面的に一時停止した。私たちはFTX-6058 1 b期試験の登録と用量を一時停止し、ベータ地中海貧血に対するFTX-6058単独のINDを撤回し、FDAと勤勉に協力して、一時停止問題をできるだけ早く解決しようとしている。
我々はすでに特許製品エンジンFulcrumSeekを開発し、それを用いて細胞薬物標的を系統的に識別し、検証し、これらの標的は遺伝子発現を潜在的に調節して、既知の遺伝子定義疾患の根本的な原因を治療することができる。著者らの製品エンジンは患者由来の組織と疾病関連細胞モデルを統合し、著者らは著者らの薬物多様性と高度に注釈された小分子化合物ライブラリー及びカスタマイズされたCRISPRとRNAiライブラリを用いて検索を行った。これらの画面は数千万のデータ点と高コンテンツ画像を生成する。そして,計算生物学と分析を用いて特異性と選択性を持つ目標を識別し,開発を著しく加速させる包括的なデータセットを伴った。この方法は、FSHDのためのlosmapimodとヘモグロビン疾患のためのFTX−6058の識別、および強力な発見導管をもたらす。
我々の最先端の候補製品であるlosmapimodは、グラクソ·スミスクラインまたはグラクソ·スミスクラインの付属会社から許可を得た小分子薬物であり、FSHDの治療のために開発されており、FSHDはまれな進行性と障害性疾患であり、筋肉変性と脂肪浸透であることを特徴とする。疾患の進展により障害の蓄積が生じ,多くの患者が最終的に車椅子に依存し独立性を失うようになり,日常生活活動を行う能力が低下したためである。Losmapimod選択的標的p 38α/?有糸分裂原活性化プロテインキナーゼ、またはp 38?α/?私たちの製品エンジンを利用して、p 38α/?が低下していることを発見しましたDUX 4FSHD患者の筋細胞中の遺伝子。この遺伝子の異常発現はDUX 4遺伝子は既知のFSHDの根本的な原因である。現在承認されていないFSHDの治療法は,FSHDが最もよく見られる筋ジストロフィーの一つであり,米国の患者数は1.6万から3.8万人,世界では30万から78万人と推定されている。LosmapimodはFSHDの治療のためにFDAとヨーロッパ薬品管理局(EMA)の孤児薬物指定を獲得し、2021年5月にFDAの迅速チャネル指定を獲得した。
著者らはランダム、二重盲検、プラセボ対照、多中心、国際2 b期の臨床試験を行い、ReDUX 4と呼ばれ、80名のFSHD患者の中でロモパモントを評価した。この2 b期の臨床試験では主な終点はDUX 4遺伝子発現、実験的分子バイオマーカー。第2の終点は、安全性および耐性の評価、血液中の薬物動態またはPK、ならびに筋肉脂肪浸透(MFI)、到達可能な作業空間(RW)、および患者報告の結果を含む筋肉健康、構造および機能の測定を含む。同時に、著者らは単中心開放ラベル第二段階の臨床試験を開始し、FSHD患者の慢性治療に対するロスパモートの安全性と耐性を調査した。現在行われているオープンラベル試験の拡張では,筋機能,筋力,患者報告の生活の質の指標も評価されている。
2021年6月のReDUX 4試験48週間のデータを示した。主な終点には達していないが,48週ではプラセボと比較して筋健康や機能の複数の指標および患者報告の結果において,プラセボと比較して臨床的に有用であることが示唆された。ロスパモート治療の参加者が中間筋で測定したMFIの進展は遅く,これらの筋はすでに疾患の影響を受けており,疾患進展の兆候を示している可能性が最も高い。プラセボ群と比較して,ロスラパモ群の正常筋は保護されているようであった。ロスパモントの治療は,RWSの低下速度の鈍化とRWSの接近可能な表面積の改善が証明されており,上肢活動範囲を評価する機能指標であり,独立性を測る重要な指標であることが証明されている。また,患者のグローバル変化印象(PGIC)評価により,プラセボよりもlosmapimod治療の方が感覚が良いと報告されている。PGICは自己報告の患者の感覚と機能変化の指標である。ロスパモントの耐性は一般に良好であり,薬物に関する重篤な副作用の報告はない。
1
ReDUX 4のデータに基づき,FDAを含む米国やEU規制機関と接触し,3期試験設計の鍵の面で一致した。私たちは2022年第2四半期に第3段階試験REACHを開始し、2023年下半期に登録を完了する予定です。REACHはランダム、二重盲検、プラセボ対照の多国試験であり、ロモパモントによるFSHD治療の治療効果と安全性を評価することを目的としている。この実験では,FSHDを有する成人約230名を募集する予定である。患者はランダムに1:1に分けてロスパモート(15ミリグラム錠剤を1日2回経口投与)またはプラセボを受け、48週間の治療期間内に評価した。研究の主な終点はRWSとベースラインの絶対的な変化である。副次的終点はMFI、PGICと上肢神経性疾患の生活の質、あるいは神経QOL UEを含む。この試験には,患者を中心としたヘルスケア利用評価も含まれている。
2022年10月、ReDUX 4オープンタグ拡張部96週のデータを公表しました。提供されたデータによると,最初の治療群でロスコパモート治療を継続した患者は96週間の治療効果維持を経験し,RWSとベースラインの平均変化から測定した。また,最初の48週の試験期間後,プラセボからロモパモドに移行した患者は,疾患進展の改善と緩和を示し,RWSの平均変化によって測定した。ロスモスは引き続き鼓舞的な安全状況を示し、全体的に耐性が良好であった。
我々のもう1つの候補製品FTX−6058は研究中の経口胎児ヘモグロビンまたはHBF誘導剤であり,SCDや他の何らかのヘモグロビン疾患の治療に有用である可能性が開発されている。FTX-6058は胚外胚葉発育(EED)と結合し、ポリコーム抑制複合体2(PRC 2)の転写サイレンシング活性を抑制することを目的としている。EED抑制はBCL 11 Aを含む重要な胎児グロブリン抑制因子の有効な低下を招き、HbFの増加を招く。EEDはPRC 2複合体のメンバーであり,PRC 2複合体もEZH 2を含む。EZH 2系薬剤には承認された製品があり,その承認されたラベルは悪性腫瘍のリスク増加を含む安全リスクを概説している。
SCDは遺伝性血液疾患であり、オスミウムサブユニット遺伝子変異によるもの、またはHBBジーン。この変異により異常ヘモグロビンやHBSが形成され,赤血球が円形から鎌状に変化し,その機能を著しく損なう。私たちが設計したFTX-6058は2つのγ-グロブリン遺伝子の発現を誘導することでヘモグロビン疾患の根本的な原因を補うことができますHBG 1/2彼の表情は通常、生まれて間もなく沈黙する。♪the the theHBG 1/2γ−グロブリンをコードする遺伝子はHBFの成分であり,SCD中の異常な赤血球形状を修復し,SCD中のHBSの存在を補償することが知られている。私たちは観察しました体外培養そして体内にある活性化するHBG 1/2FTX-6058臨床前研究における遺伝子。FTX−6058は強いHbF上昇レベルを示し,重要な細胞健康マーカーに悪影響は認められなかった。CD 34+由来細胞を追加的に臨床前分析し,FTX−6058治療によりHbFレベルを総ヘモグロビンの30%程度に増加させることが観察され,質量分析,高速液体クロマトグラフィー,高速タンパク質液体クロマトグラフィー技術により測定した。細胞モデルでは,HbFの上昇はわれわれが観察したヒドロキシウレアより有意に大きかった。
我々のFTX−6058のSCD(ヒドロキシウレア服用および非服用)被験者における1 b期試験は現在全面的な臨床休止状態にある。我々は2023年1月にSCD患者が6ミリグラムFTX-6058治療を受けたデータを発表し、6 mgと2 mg用量キューの登録を完了し、12 mg用量キューの登録を開始したが、FDAは2023年2月にSCD FTX-6058のIND臨床試験を全面的に一時停止した。したがって,我々はFTX−6058 1 b期試験の登録と用量を一時停止し,FDAと共同で努力し,一時停止問題をできるだけ早く解決しようとしている。
評価可能な対象からの1 b期データは,2022年12月21日現在,6 mg用量列(n=10)におけるデータ遮断値は,HbFがベースラインより9.5%増加しており,水酸基尿素(n=3)を服用している被験者とヒドロキシル尿素(n=7)を服用していない被験者の反応に差がないことを示している。6 mgの評価可能な被験者では溶血のバイオマーカーの改善も認められた。次の図は,6 mg用量列において,評価可能な対象(n=7)の高速液体クロマトグラフィーで測定したHbF増加率とHbF変化の絶対パーセンテージ(星番号を有する対象の水酸基尿素服用)を示した。
2
私たちは6 mgおよび2 mg用量キューの登録を完了しており、臨床保留を解決できるまで、これらのキューまたは他の任意のキューでより多くの被験者を募集するつもりはありません。2 mg用量列の被験者(n=2)からのデータでは,HbFはベースラインより4.6%増加した。
3

1 b期試験が臨床的に放置されているため、12 mg用量列(ヒドロキシウレアを服用している対象および服用していない対象を含む)の登録および用量は現在一時停止されている。試験休止前の12 mg用量列(n=3)の対象からのデータは、治療42日後にHBFの絶対値がベースラインより10.0%増加したことを示している。星号を有する被験者はヒドロキシウレアを服用した。
4

HbFの増加は、血管閉塞危険像或いはVOC、貧血、疼痛、感染、脳卒中とその他を含む一連のSCD症状の頻度或いは重症度を低下させることが証明されている。大量の遺伝、臨床と観察証拠により、もっと高いレベルのHBFのSCD患者への影響を表明し、HBFの誘導はベースラインより5-10%高いことは疾病負担の軽減と臨床結果の改善と関係があるかもしれない。これらの予備データは,FTX−6058がHbFレベルを9.5%向上させ,承認されればSCD患者の変革療法となることを示していると信じている。
FTX-6058は、2023年3月3日までのデータ締め切りを全体的に受け入れることができる。これまでに14種類の治療緊急副作用が報告されており,そのうち2種類は研究薬(頭痛,唇麻痺)に関与している可能性があり,いずれも重篤ではなく,重篤でないとも考えられていない。TEAEによる生産停止は現在のところない。14個のTEAEのうち3つはVOCsとして特徴付けられ,FTX−6058とは無関係と考えられ,そのうちの1つは重篤な有害事象またはSAEと報告され,不コンプライアンス対象に急性胸部症候群を伴った。
2022年12月、我々はFDAからSCDを治療するFTX−6058の迅速な追跡指定を取得した。
米国国立衛生研究院(NIH)のデータによると,約7000種類のまれな遺伝子で定義されたヒト疾患があり,その多くは十分でないか承認されていない治療法である。現在の薬物標的認識と開発はまれな筋肉、血液、神経疾患に集中している。FulcrumSeekを用いて他の治療領域や他の疾患において遺伝子定義疾患に対する薬物標的の発見が期待される。国内開発のための薬物目標を優先するほか,パートナーシップによる開発を考慮した他の薬物目標を決定することも可能である。例えば,百時美施貴宝社の完全子会社MyoKardiaとの協力と許可プロトコルによると,FulcrumSeekを用いて何らかの遺伝性心筋疾患を潜在的に治療する薬物標的を発見している。
5
私たちのパイプは
FulcrumSeekを用いて,まれな遺伝病の既知の根本的な原因を解決する一連の潜在的疾患修正療法が生じている。以下のグラフは,われわれの臨床段階と臨床前計画に関する重要な情報をまとめたものである。

私たちの戦略
我々は,我々の特許製品エンジンの広範な適用を用いて,高度に満たされていない医療需要分野における遺伝子定義のまれな疾患の既知の根本原因を解決するために遺伝子発現を調節する小分子療法を発見·開発している。私たちは、私たちが最初にFSHDとSCDを治療するために使用された候補製品は、これらの虚弱疾患を治療する患者の潜在力を持っている可能性があり、場合によっては、生命を脅かす可能性もあると信じている。私たちの戦略の主な構成要素は
6
私たちの主な候補製品
私たちは私たちの独自製品エンジンとスクリーニング作業を使用して、私たちの主要な候補製品の薬物標的を決定しました。私たちは二つの候補製品が臨床試験を行っている。次の表にこれらの主要候補製品に関する重要な情報をまとめた。

ロス·マプト
顔面肩上腕骨筋ジストロフィー症の概要
顔面肩腕筋ジストロフィーはまれな進行性と障害性疾患であり,現在のところ承認されていない治療法である。FSHDは最もよく見られる筋肉栄養不良形式の一つであり、男女患者の影響は均等であり、通常青少年と若者に発病する。FSHDの特徴は進行性骨格筋喪失であり,最初に顔面,肩,腕,体幹の筋無力をきたし,下肢や骨盤ベルトの筋無力に進展する。骨格筋無力による深刻な身体制限は,顔面筋への衝撃,コミュニケーション問題,腕を用いた日常生活活動が困難であること,離床が困難であることなど,独立性の喪失を含め,多くの患者が最終的に車椅子による日常活動に依存するようになった。多くのFSHD患者も慢性疼痛,不安,抑うつを経験したと報告している。FSHD患者の診断と治療は通常神経科医によって行われる。
FSH協会は,米国におけるFSHDの罹患率は約20,000人に1人と推定している。最近オランダで行われた研究では,罹患率がより高く,8,333人に1人が報告されている。これらの推定と米国の人口3.2億人から,米国の患者数は16,000から38,000人と推定されている。より多くの患者が正式に診断されていない可能性があり,診断が困難であり,承認されていない治療法が考えられるからであると考えられる。約3分の2の症例は常染色体優性遺伝の家族性遺伝であり,3分の1の症例は散発的であった。FSHDはすべての民族に影響し、その発病率と流行率は類似している。
現在のところFSHDを治療する方法は承認されていない。現在の治療は対症治療に限られており,物理/職業治療,患者の虚弱分布に応じてオーダーメイドされた低強度有酸素運動,一般的な疼痛治療など,限られた有益な効果を提供する可能性がある。肩帯の活動範囲制限は肩周囲筋無力による肩甲翼に起因する可能性があり、この場合、手術肩甲骨固定術はある患者にいくつかの機能改善をもたらすことができる。
ロスパモントはFSHD患者の開発において他の治療法からの競争に直面している可能性がある。羅氏は成人FSHDの第二段階試験において、ミオスタチン阻害剤RO 7204239を評価している。親和性は、成人FSHDの1/2期臨床試験において、AOC 1020、siRNA抗体-オリゴヌクレオチド複合体を評価している。FSHDの臨床開発にロスパミと同様の作用機序を有する候補製品があるかどうかは不明である。
7
FSHD生物学
FSHDはFSHDの異常発現によるものであるDUX 4骨格筋に不適切な存在を引き起こすDUX 4タンパク質は、他の遺伝子の発現を引き起こす転写因子である。正常な場合,DUX 4が駆動する遺伝子発現は早期胚発育に限られ,その後DUX 4ジーンは黙っていた。FSHD患者では異常が生じていますDUX 4骨格筋中のタンパク質は他のタンパク質をコードする遺伝子を調節し,その中のいくつかは筋肉に有毒である。異常な結果DUX 4FSHDの発現は筋死であり,脂肪に置換され,骨格筋無力や進行性障害をきたす。私たちの考えではDUX 4遺伝子及びその下流転写プログラムはFSHDの治療に新しい治療経路を提供する可能性がある。発表された臨床前とヒトのデータを除いて体外培養私たちが行った実験によるとどんな減少もDUX 4発現は患者に有益かもしれない。臨床前研究では,筋細胞死(アポトーシス)とDUX 4表現して減少しましたDUX 4それに伴うアポトーシスの減少を招く。次の図に示すように動物モデルではDUX 4骨格筋では,ホルモンレベルの増加に伴い,対応する機能喪失が認められたDUX 4表情。この低レベルの動物モデルではDUX 4活動性評価では,これらの動物の表現は健康動物と類似していることが示唆されたDUX 4減税は機能的福祉の必要条件ではない。人間の筋組織検査からのデータも同様にDUX 4活動は筋病理の悪化に関与している。

すべてのFSHD患者ではDUX 42つの異なるタイプの遺伝子変化のうちの1つにより、FSHD 1またはFSHD 2をもたらし、遺伝子はサイレンシングまたは解除抑制されない。約95%の患者がFSHD 1を有し、約5%の患者がFSHD 2を有する。FSHD 1は,D 4 Z 4反復と呼ばれるDNAのセットが10個以上の反復単位から9単位以下に収縮したことによるものである。この収縮はDUX 4それは.FSHD 2を有する患者はD 4 Z 4の重複収縮は有意ではなかったが,1つはSmchd 1遺伝子は通常抑制に役立ちますDUX 4遺伝子はDNAによりメチル化される。
Fulcrum SeekはFSHDの薬物標的を決定しました
患者由来のFSHD 1筋細胞、すなわち筋管を用いて、私たちの小分子プローブバンクを用いてそれらをスクリーニングし、減少を決定したDUX 4表情。著者らはいくつかの潜在的な薬物標的を確定したが、大多数の標的の調節は筋肉細胞の健康或いは分化に不利な影響を与えた。我々がスクリーニング作業から決定した1つの薬物標的はp 38α/?であり,他の疾患で広く研究されてきたが,それとの関連は報告されていないDUX 4スクリーニング作業を行うまで表現やFSHDを行います種々の小分子p 38α/?阻害剤を評価し,両者の持続的な減少が認められたDUX 4各p 38α/オスミウム阻害剤との発現およびDUX 4駆動遺伝子転写産物。さらに検証実験を行い,小干渉RNAやCRISPR単誘導RNAなどの遺伝的手法を用いてp 38αを抑制することも可能であることを確認したDUX 4表情。また,セントルイス大学の研究者は独立して,losmapimodを含むp 38α/?阻害剤が抑制されていると結論した研究結果を発表したDUX 4細胞や動物FSHDモデルでの発現。
ロスパモントの概要
P 38α/?を潜在的な薬物標的として決定した後,複数のp 38?α/?の小分子阻害剤を評価した。これらの阻害剤のいずれも様々な疾患を治療する臨床試験で評価されているが,筋疾患で評価されたことはない。我々の評価の結果として,他のp 38α/オスミウム阻害剤に対して,大量かつ魅力的な臨床前および臨床データからロムパモントが第一選択の開発候補薬であることが決定され,これらのデータは安全性,pk,標的に関与している
8
抑制力とその発展の高度な段階。ロスパモントは当初,グラクソ·スミスクラインが複数の国·地域で行った複数の適応の臨床試験で3500名近くの被験者を評価した。グラクソ·スミスクラインはFSHDまたは任意の他の筋肉疾患におけるロスパミの治療効果を評価しなかった。グラクソ·スミスクラインは評価の適応について監督部門の承認を求めていないにもかかわらず、ロスパモントは慢性投与量の面を含む魅力的なPK、PD、安全性と耐性を示した。また,ロスパモントを用いた臨床前研究では,p 38α/?経路の抑制の減少が認められたDUX 4発現と下流遺伝子発現。Losmapimodを同定した後,安全性と薬理学的歴史が我々の開発計画を大幅に加速し,将来の規制提出を強化すると信じているため,グラクソ·スミスクラインからこの分子の許可を得た。
2021年6月,無作為,二重盲検,プラセボ対照の多中心国際2 b期臨床試験(ReDUX 4)の全データを報告した。主な終点に達していないにもかかわらず,プラセボと比較してロスコパモート治療のFSHD患者の疾患進展は遅く,機能改善が認められた。2022年10月,ReDUX 4開放ラベル延長部96週のデータを公表し,最初の治療ARMでlosmapimod治療を継続した患者は96週間の奏効率維持を経験し,RWS平均値とベースラインの変化で測定した。また,最初の48週間の試験期間後,プラセボからロモパモドに移行した患者は,疾患進展の改善と緩和を示し,RWSの平均変化によって測定した。
2022年第2四半期に、私たちはREACHを開始し、これは48週間の無作為、二重盲検、プラセボ対照の3期試験であり、ロムパモントによるFSHDの治療効果を評価し、2023年下半期に登録を完了する予定である。
2020年1月、FDAはFSHDの孤児薬物指定のためのロスパモートの使用を許可した。2020年3月,EMAはlosmapimodによるFSHD治療の孤児薬物指定を承認した。FDAは2021年5月、losmapimodによるFSHD治療の迅速チャネル指定を承認した。
臨床試験:2 b期(ReDUX 4)
2021年6月、FSHD国際研究大会で、著者らは著者らのランダム、二重盲検、プラセボ対照の多中心国際2 b期臨床試験ReDUX 4試験の完全なデータを展示し、この試験は80名のFSHD 1患者に対して研究を行い、臨床重症度採点はRicci尺度で2から4までであった。この試験では,24週間または48週の間に15ミリグラムのパスパモートまたはプラセボを1日2回服用した治療を評価した。学生募集は2020年2月に完成する。患者はランダムに治療群とプラセボ群に分けられ、割合は1:1であった。FDAは2019年6月にlosmapimodのINDを受け,我々も2019年に異なる日にCTAを提出し,ヨーロッパとカナダで実験を行ったが,これらはすべて受け入れられた。私たちは2020年8月に予め指定された中期分析データを提供した。著者らは2021年1月にReDUX 4試験を完成し、2021年6月24日にFSHD国際研究大会で試験の完全なデータを展示した。
主な終端点はDUX 416週または36週で影響を受けた骨格筋で駆動される遺伝子発現を実験バイオマーカーとした。この試験は、安全性、目標参加度、およびPKデータに加えて、FSHD進展に関連する広範なデータを捕捉することを目的としている。副次的終点はFSHD患者の安全性と耐性の評価,血液中のPK,骨格筋生検中のロモパモント濃度,血液と筋生検における標的参加度,および全身骨格筋MRIバイオマーカーによる治療効果である。全身MRIスキャンによりMFI、筋肉脂肪分率と赤身体積の変化を評価した。試験で評価された筋は,外観が正常(疾患の影響を受けない),中等(疾患の影響を明らかに受けるが,重篤な脂肪代替には至らずすべての機能を喪失する)や終末期(重篤な脂肪代替ではなく,すべての機能を失っている場合)に分類される。研究の終点は,RWS,Timed Up and Go,あるいはTUG試験,FSHDまたはFSHD TUGの最適化Timed Up and Go試験,手持ち測力計による筋力測定,他の筋機能測定,患者報告の結果である。
ReDUX 4の最初の設計は,24週目の治療期間中に16週目に筋活組織検査を行い,開放ラベルを延長することを含む。試験した80名の被験者のうち,16名が24週間の治療期を完了し,試験の開放ラベル拡張部に移行した。持続的な新冠肺炎の大流行により,われわれは修正案によりReDUX 4の治療期間を24週から48週に延長し,被験者の安全を確保し,16週目と36週目の生検を許可した。試験のランダム部分では,最初の24週間の治療期を終えていない約64名の被験者が48週間の治療期を継続した。24週から48週に延長することはまた、プラセボ対照設計において、骨格筋MRIの副次的終点および様々な探索的臨床終点のより長時間の評価、例えば、RWS、最適化FSHD TUGテスト、筋肉機能測定、および患者報告の結果を可能にする。
2020年8月,ReDUX 4実験の主要終点のあらかじめ指定された中期分析の結果を発表したが,これはベースラインからであるDUX 4被験者がロスコパミルまたはプラセボ治療を受けた後、影響を受けた骨格筋で駆動される遺伝子発現。副次的かつ探索的な終点はこの分析の一部として評価されていない.最初の29人のランダム被験者の中期分析の結果DUX 416週では,遺伝子発現によって駆動される遺伝子発現はプラセボとの分離を示さなかった。しかし,あらかじめ指定された感度分析では,それらの前処理が最も高いものは
9
DUX 4彼らの筋肉生検サンプルで駆動される遺伝子発現はDUX 4ロスパモントとプラセボ治療後の遺伝子発現の比較。最高発現筋生検はベースライン評価による生検の最高4分の1を表しているDUX 4遺伝子発現が駆動する。
中期結果は、16週の生組織検査を完了した上位29人の被験者の分析を含む。PK,人口統計学,主要終点を評価した。中期分析には統計的意義がなく,個別患者レベルのデータも含まれていない。被験者はランダムに2群に分けられ、ロスタット15 mg(n=15)またはプラセボ(n=14)を1日2回経口投与した。しかし結果によるとDUX 4ベースライン生検で最も高いレベルを示す筋生検における遺伝子発現DUX 4遺伝子発現(ロスパモント38倍,n=3,プラセボ5.4倍,n=5減少),個体群レベルデータ解析DUX 4全29名の被験者の遺伝子発現はロスパモントとプラセボの分離を示さなかった(ロスパモントは3.7倍,n=15,プラセボは2.8倍,n=14)増加した。その結果,筋生検は高い範囲であることが分かったDUX 4減少を観察するためにはベースライン時に遺伝子発現を駆動する必要があるかもしれない。
2021年6月、私たちはReDUX 4の完全な結果を報告した。実験は主要な終端点に達しておらず,中のベースラインとは異なるDUX 416週目または36週目に影響を受けた骨格筋における遺伝子発現。構造と機能FSHD進展と患者報告の48週間の結果の多くの指標において、ロモパモント治療群はプラセボ群と比べ、二次と探査終点は臨床相関と名目上の統計学的有意なメリットを示した。主要終点に達していないため,すべての比較分析は名義統計p値を用いて報告する.
48週で、疾患の進行および機能を測定する他の二次および探索的終点は、ロスマプトとプラセボの間に差があることを示した。特別な分析では,筋力を測定する力測定法は,プラセボと比較してロモパモート群の参加者が進行が遅い非統計学的有意な傾向を示し,両側肩外転筋と足首背屈筋の力が有意に改善し(12%−27%),この2つの筋群がFSHDに特に影響を受けたことを示した。機能採点はRWSとTUG表示肢体機能の改善と測定結果と一致した。最近設計された2つの尺度(FSHD TUGとFSHD Health Index)はいずれも両群の患者のベースラインの変化を示しておらず、ロスパモ群とプラセボ群との差も示されておらず、これらのテストは48週間以内の変化に敏感ではないことが示唆された。運動機能テストでも,48週間で両群のいずれの群も変化せず,群間の差もなかった。48週の中間筋では,ロスパメット群とプラセボ群との間の筋脂肪分率や赤身筋体積に差はなかった。
安全性と耐性データはこれまでに報告された結果と一致しており,薬物に関するSAEは報告されていない。ロスパモートの一般的な耐性は良好であり,多くのTEAEが研究薬物に関与している可能性や無関係ではないと考えられている。Losmapimod群の参加者2名のうち,3種類のSAE(術後創部感染,アルコール中毒,自殺未遂)が報告されており,いずれもlosmapimodとは無関係と評価されている。誰も死んでいません
10
不良事件で中止になりました。ロスパモントは現在,FSHDを含む複数の適応の臨床試験で3600名以上の被験者で評価されている。
REACH、第3段階登録試験
我々は2022年第2四半期にREACHを開始したが,これはFSHDのlosmapimodの第3段階試験であり,2023年下半期に登録を完了する予定である。ReDUX 4ステージ2 b研究のデータに基づき,FDAを含む米国やEU規制機関と接触し,完全登録承認を支援するための第3段階試験設計の鍵を得ることで一致した。REACHはランダム、二重盲検、プラセボ対照の多国試験であり、ロモパモントによるFSHD治療の治療効果と安全性を評価することを目的としている。この実験では,FSHDを有する成人約230名を募集する予定である。患者は、ロスパモート(15ミリグラム錠剤を1日2回経口投与)またはプラセボを1:1の割合でランダムに受け、48週間の治療期間内に評価する。研究の主な終点はRWSとベースラインの絶対的な変化である。二次端末は、MFI、PGIC、およびNeuro QOL UEを含む。この試験には,患者を中心としたヘルスケア利用評価も含まれる。
臨床試験:ReDUX 4オープンタグ拡張
2020年2月にReDUX 4試験の開放ラベル延長を開始し,ReDUX 4でlosmapimodまたはプラセボを用いて24週間または48週間の治療期間を達成した患者がlosmapimodの長期治療を受けることができるようにした。このオープンタグ拡張には,3カ月ごとの安全性と有効性の臨床評価,6カ月ごとの全身筋骨格核磁気共鳴検査,最初の96週間の治療6カ月後に筋針刺し生組織検査がある。96週目以降は,患者の安全性と耐性のみをモニタリングした。この試験は,薬物が承認されて商業的に使用されるか,あるいはFSHDにおけるロスパモートの臨床開発が終了するまで継続されることが予想される。
2022年10月、ReDUX 4オープンタグ拡張部96週のデータを公表しました。提供されたデータによると,最初の治療群でロモパモント治療を継続した患者は96週間の治療効果維持を経験し,RWSとベースラインの平均変化から測定した。また,最初の48週の試験期間後,プラセボからロモパモドに移行した患者は,疾患進展の改善と緩和を示し,RWSの平均変化によって測定した。ロスモスは引き続き鼓舞的な安全状況を示し、全体的に耐性が良好である。最初の48週の研究では,97%の参加者が96週でもオープンラベル拡張に残っていた.次の図は500 g重量の主と非主要可達表面積(RSA)の百分率変化を示している。

臨床試験:第二段階開放ラベル研究試験
ReDUX 4 2 b期臨床試験と同時に、著者らは2019年8月にロスコパモントの開放ラベル単センター2期臨床試験を開始し、16名のFSHD患者に関連し、臨床重症度採点はRicci尺度で2~4点であった。試験の第1部では,15ミリグラムのパスパモートを含む錠剤を1日2回受け,52週間と持続した。治療前に8週間の治療前評価を行い、筋肉骨格MRIバイオマーカーと臨床結果評価のベースラインを確立した。ウェアラブルセンサを用いて外来移動性評価を行った。52週の治療期間後,参加者は研究を継続することを選択することができ,この研究が行われている。私たちはオランダの中心で実験を行った。
主な目的はFSHD患者の慢性投与におけるロスコパモートの安全性と耐性を研究することである。主な終点は52週間の間の安全性と耐性の評価である。副次的な端点は変更です
11
投与期間中のp 38α/?に対する抑制作用は,ベースラインのPHSP 27および血液と筋肉中のPHSP 27と総HSP 27の比率から評価した。この実験では,以下の変化に関する初期データを提供することを目的としているDUX 4遺伝子発現駆動、核磁気共鳴バイオマーカー、客観的な臨床結果評価、患者報告の結果から、これらの結果は、治療前と比較してロスパモート治療開始後の異なる時間に発生する可能性がある。これらのデータを用いて,FSHDにロスパモートを応用した臨床開発戦略をさらに指導する予定である。
52週間の治療の間にDUX 4治療前および治療期間中に,筋針組織生検を用いて影響を受けた筋で遺伝子発現を駆動する。すべての患者が治療前の生組織検査を受け,治療4または8週間後に各患者に2回目の筋針刺し生組織検査を行う。最初の試験設計には48週の慢性治療期間の追加生検が含まれていたが,ReDUX 4の開放ラベル拡張には慢性治療期間の生検を含むため,この評価は試験レジメンから削除された。
2 b期臨床試験中の評価と類似して、著者らはロストモント治療が肩と上腕機能と活動/歩行に与える潜在的な影響、及び筋肉力と機能、生活の質と日常生活能力への影響を測定した。臨床結果評価はRWS,FSHD−TUG,筋力,運動機能能力及び汎用的かつFSHDに特定された患者の生活の質と日常生活能力の報告である。その他の探索的評価には,6分間歩行試験,肺活量測定,筋超音波がある。また,ウェアラブルセンサを用いて日常移動性の評価を行った.
自然歴史研究を解決する
臨床試験はFSHD中の薬物開発障害、或いはRESOLE研究を解決するために準備され、アメリカ国立衛生研究院が援助した持続的な自然歴史研究であり、未来のFSHD薬物試験の患者群、治療効果バイオマーカーと臨床結果の評価を確定することを目的としている。この研究はロチェスター大学とカンザス大学医学センターが調整し、2018年4月に最初の被験者を募集した。この研究は、8つのアメリカ臨床センターからなるネットワークにおいて160人の被験者に対して24ヶ月間の追跡調査を行い、臨床試験に適している可能性のある複数のバイオマーカーおよび臨床結果を評価し、遺伝、人口統計学または臨床特徴に基づいて患者選択基準を評価する。EUの3つのサイトはすでにRESOLEプログラムに参加し、60人の被験者に対して24ヶ月の追跡調査を行う。この自然歴史研究の結果は臨床試験の設計と実施に情報を提供し、監督機関との討論に情報を提供すると信じている。この研究は,治療が乏しい場合のFSHDの疾患進展や機能変化のスケジュールに価値のある知見を提供している可能性も考えられる。
RESOLE研究では,臨床結果評価を追加し,RWSと呼ぶ。RWSは上肢の活動範囲を評価する機能指標である。具体的には,3 D運動センサ技術を用いて肩部および近位腕の全体的な活動能力を評価する。RWSの評価によると,保存機能は自立能力や他の生活の質に直接影響する日常生活活動能力を保持するために重要である。公表された結果より,RWSは独立性を測る重要な指標である。RWS評価は中央リーダが分析した.米国RESOLE研究の8つのサイトとヨーロッパの3つのサイトのRWSを評価するために、標準化されたハードウェア、ソフトウェア、およびテスト条件を提供した。また,RWS評価は米国,カナダ,ヨーロッパで医療機器として登録されている。
最近の第三者研究は18名のFSHD患者の5年間にわたるRWS変化を評価した。以下の図に示すように,RWS測定はFSHD被験者で1年以内に上肢機能の緩徐な低下が検出されたと結論した。最も有意なRWS低下は肩レベル象限に認められたが,下象限には有意な変化はなく,被験者の手首に体重500グラムを装着するとRWSの低下が有意であった。
12

上の図に4つの象限におけるRWを示す.最適化されたRWSは、次の図に示すように、追加のバック領域を含む。

13
グラクソ·スミスクラインによるロモパミ治療の臨床研究進展
グラクソ·スミスクラインは慢性閉塞性肺疾患、COPD、急性冠症候群とその他の心血管疾患、神経性疼痛、深刻な抑うつ障害、局所性分節性糸球体硬化と関節リウマチの患者を含む複数のロスコパモートの1期と2期臨床試験及び1項目の3期臨床試験を行った。24個の試験の中で、3500名近くの被験者はロスパモントを服用し、単回投与量は60 mgに達し、繰り返し経口投与量は15 mgに達し、毎日2回、52週間に持続した。われわれはFSHD治療の臨床試験に15 mgを1日2回使用した。グラクソ·スミスクラインはFSHDまたは任意の他の筋疾患を有する患者に対してロスパモートの臨床試験を行っていない。
グラクソ·スミスクラインによるロスパモート臨床試験では,ロスコパモートを服用した被験者とプラセボを服用した被験者では有害事象や副作用の頻度に有意差は認められなかった。グラクソ·スミスクラインでは,ロスコパモートを服用した患者とプラセボを服用した患者の重篤な有害事象やSAEおよび死亡の頻度はほぼ同じであることが観察された。これらの試験はロスタット心血管リスクプロファイルの広範な評価を含み、補正QTを延長する潜在力の評価を完成させることを含む。グラクソ·スミスクラインは,プラセボと比較して,ロスコパモートはプラセボと比較して,ベースライン後の心電図異常やバイタルサインの発生に臨床的に差はないと報告している。これらすべての実験において、グラクソ·スミスクラインにはロスパモントの安全信号は発見されなかった。この24項目のロスパモントの臨床試験のうち,SAEの報告は14件であった。
グラクソ·スミスクラインが行った最大規模のプラセボ対照臨床試験は心臓発作後の急性冠症候群を治療する3期臨床試験であり、1700人以上の患者は毎日7.5 mgのロスパモント或いはプラセボを服用し、12週間持続し、その後12週間フォローアップした。この試験では,グラクソ·スミスクラインではプラセボ群とlosmapimod群の副作用とSAEの割合が類似していることが認められた。
プラセボ群でも10個の死亡SAEがあり,losmapimod群では13個の死亡SAEがあった。プラセボ群では,致死性SAEはそれぞれ感染と感染(2例),全身疾患と管理部位疾患(2例),呼吸系,胸部と縦隔疾患(3例),心臓疾患(1例),胃腸疾患(1例)と腫瘍(1例)であった。Losmapimod群では,死亡原因は感染と感染(4例),全身疾患と管理部位疾患(3例),呼吸系,胸部と縦隔疾患(2例),心臓疾患(1例),損傷中毒と操作合併症(1例),胃腸疾患(1例)と腫瘍(1例)の順であった。
ロスコパモート1期および2期プラセボ対照臨床試験の14試験(ロスコパモート服用N=1327例;プラセボ服用N=735例)では,ロスコパモートを服用した被験者とプラセボを服用した被験者のSAE分布は類似していた。最も一般的なSAEは、心臓疾患(2%のプラセボ;3%のロスパモッド)および呼吸器系、胸部および縦隔疾患(1%のプラセボ;2%のロスパモド)である。この14試験のうち11試験でSAEが報告され,3試験報告でSAEはなかった。
グラクソ·スミスクラインが行った24項目の試験のほか、もう一つのスポンサーはロスコパモートのプラセボ対照第二段階臨床試験を行い、73名の心血管症状を有するCOPD患者73名が7.5ミリクロスポモート或いはプラセボを16週間服用した。ロスコパモ群は36名,プラセボ群は37名であった。この試験において、ロモパモート群は全部で6例(17%)のSAEであり、COPDと肺炎の加重を含むが、プラセボ群は1例(3%)のSAEであった。
FTX-6058
ヘモグロビン病は遺伝性疾患であり、赤血球の重要な構成部分であるヘモグロビンに影響を与える。ヘモグロビンの機能は酸素を組織に輸送することであり,赤細胞が肺を循環するとヘモグロビンは酸素を吸収し,酸素を組織に排出し,正常に動作するようにする。ヘモグロビン疾患は異常な(変異した)ヘモグロビンあるいは低レベルのヘモグロビンを引き起こすが,いずれも有意な発症率と死亡リスクに関与している。我々は,HBFレベルを向上させ,SCDを含む何らかのヘモグロビン疾患の治療に用いるFTX−6058を開発している。いくつかのタイプのβ−地中海貧血を有する人は,FTX−6058の治療から利益を得る可能性も考えられる。
鎌状細胞病の概要
鎌状細胞病は遺伝性赤血球疾患である。SCDの根本的な原因は低酸素条件下で重合した変異ヘモグロビンである。この重合は異常な,細長いあるいは鎌状の赤血球を産生し,最終的に溶血や血管損傷を招き,重篤な疾患を招き,SCD患者の寿命を著しく制限する。SCD患者は通常深刻な臨床結果を受け、血管閉塞危険像、貧血、疼痛、感染、脳卒中、心臓病、肺高圧、腎不全、肝疾患と期待寿命の短縮を含む可能性がある。アメリカ医学会が発表した研究によると、約32.5%の成人SCD患者は毎年疼痛危機のため3回以上入院している。SCDは期待寿命を約20歳から30歳に短縮することが報告されている。SCD患者は主に血液科の医者によって治療された。
14
米国では新生児SCDスクリーニングは強制的であり,罹患率は約10万人と推定されている。ヨーロッパでは罹患率は約5万人と推定されている。世界保健機関のデータによると、全世界の毎年の発病率は約30万人の新生児と推定される。ポリオはアフリカと中東で最も流行している。
承認されたSCD薬物治療は主に苦痛な血管閉塞発症の管理と減少、溶血性貧血の改善に集中している。アメリカで承認された4種類の薬物治療はヒドロキシウレア、Voxelotor、CrizanlizumabとL-グルタミンである。ヒドロキシウレアは、苦痛発症の頻度を減少させ、輸血の必要性を減少させるために、SCDの治療のために許可されている。ヒドロキシウレアにはブラックボックスがあり、骨髄抑制と悪性腫瘍を警告する。一般に,その副作用,反応不一致,薬物細胞毒作用に対する懸念に制限されている。ファイザー社が販売しているVoxelotorはヘモグロビンを増加させるためのヘモグロビン重合阻害剤として加速的に承認されている。この方法は、SCD中の変異ヘモグロビンであるHBSの総量をより長時間酸素を維持または増加させることによって維持または増加させる。クリザンリズマブはノワ製薬或いはノワール社が販売している全ヒト型モノクロナル抗体P-セレクチン阻害剤であり、血管閉塞発症の頻度を減少させることが許可されている。L−グルタミンは、このような疾患の急性合併症を減少させるために承認されている。
現在、深刻なSCDの治療に唯一許可されている潜在的な治療方法は造血幹細胞移植(HSCT)である。しかし、HSCTのより一般的なのは、利用可能なHLA適合同胞ドナーを有する小児科個体に提供されることである。この若いSCD群では5年生存率はかなり高いが,高齢者(>16歳)では生存率がはるかに低い可能性がある。不妊症と移植片対宿主病を含む造血幹細胞移植に関連するリスクも大きい。
SCDを治療する多種の実験方法は臨床試験中に探索されているが、多くの方法は症状の緩和或いは最後の第一線の遺伝子治療の接近に集中している。研究中の対症方法は細胞粘着、鎌状、血栓形成と鉄恒常性に関連する問題を影響することを目的としている。変異ヘモグロビンによる病理重合を抑制することによりSCDの根本的な原因に影響し,SCDの標準看護となる可能性が予想される新規経口HBF誘導剤である。ノバ社とファイザー社はSCDの特定成分(それぞれ低ヘモグロビンとVOCs)を緩和するための治療法を承認している。SCDを治療するいくつかの遺伝子治療方法は主にHBFの向上に集中しているが、まだ遺伝子治療SCDの方法は承認されておらず、その有効性、安全性と持続性はまだ確定されていない。遺伝子治療は造血幹細胞移植による入院中の施行が必要である。移植過程の一部として,患者は清髄化学療法を受け,骨髄中の細胞を死滅させ,遺伝子治療を支持する。SCDの遺伝子療法の開発に努めているにもかかわらず,低分子経口療法でHBFを増加させることでより良好に治療できる高度なニーズがあると考えられる。
SCD生物学
SCDは遺伝子変異によるものだHBBジーン。この遺伝子はヘモグロビンの重要な成分であり、ヘモグロビンは体内で酸素を輸送する機能を有するタンパク質複合体であるタンパク質をコードする。成人のヘモグロビンは4種類のタンパク質からなる複合体であり,2つのヘモグロビンサブユニットと2つのヘモグロビンαサブユニットを含む。全身性エリテマトーデス患者では,ヘモグロビンは2つの変異Bサブユニットと2つのαサブユニットからなり,その結果HBsを形成する。変異の結果,酸素輸送効率が低下し,鎌形状を有する赤血球が形成された。これらの鎌状の細胞は健康細胞よりも柔軟性がはるかに悪く,血管(血管閉塞)や破裂細胞(溶解)を閉塞し,痛み,貧血,不可逆的な臓器障害,さらには死亡を招く。
胎児発育過程におけるヘモグロビンの主な形式はHbFである。成人のヘモグロビンと類似しており,HbFも4種類のタンパク質,2つのαサブユニット,2つのγサブユニットからなる複合体である。生後間もないγサブユニットをコードする遺伝子HBG 1そしてHBG 2遺伝子は沈黙していますHBB遺伝子が活性化されています上述したように、SCDはHBBこの遺伝子は突然変異?サブユニットを産生することができる。
追加の遺伝子変異を遺伝するため、一部の鎌状細胞変異を有するヒトは、HBFの遺伝的持続性、またはHPFと呼ばれる高レベルのHBFを産生し続ける。HbFが上昇した個体は軽微なSCD臨床所見を示した。HPFがない個体では、治療干与或いは他の遺伝特徴の遺伝により、HbFレベルはベースラインレベルの3%まで低く、この病気の臨床表現の減少を招くことができる。
15
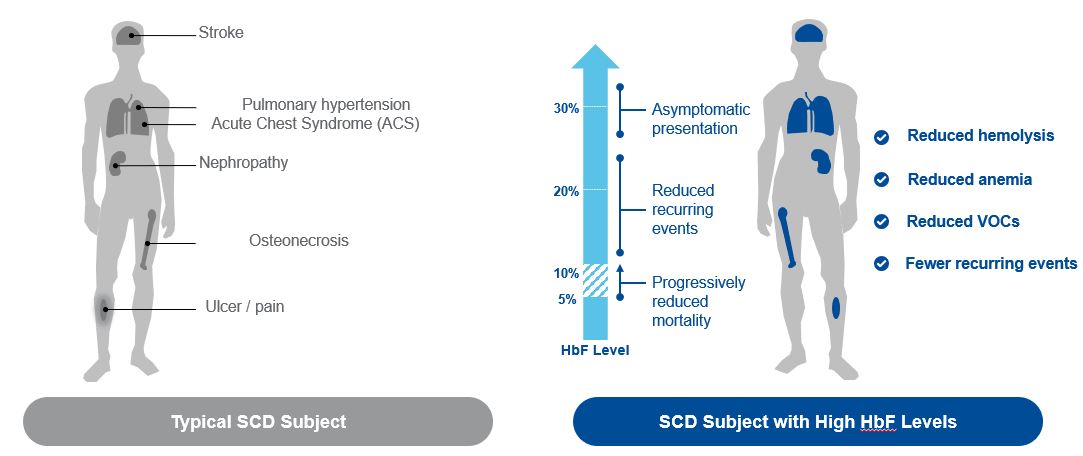
SCDの根本的な原因を解決する方法は
著者らはSCDの根本的な原因を解決する策略はHBF発現を誘導する薬物機序を決定することである。FTX−6058は,この作用機構によりSCDの根本的な原因を解決する可能性が考えられる。
概要ß-地中海貧血
地貧は1種の稀な血液疾患であり、成人ヘモグロビンを構成する2種類の蛋白質の一つであるグロブリンの欠損或いは産生減少と関係がある。これはヘモグロビンレベルの異常な低さや,赤血球破壊をきたすα−グロブリン鎖の過剰を招く。表現型の重症度はα-と非α-グロブリン鎖合成の不平衡程度と関係がある。貝珠蛋白質の欠損はHBB遺伝子欠損は0地中海貧血。他にもHBB遺伝子の変化はいくつかの貝珠蛋白を産生するが、数は減少する。減少した鉱化グロブリンは+地中海貧血。低ヘモグロビンレベルによる重篤な貧血により,多くの患者が長期輸血を必要とし,輸血依存患者と呼ばれている。全世界で毎年40,000人の乳児が出生時にβ-地中海貧血を患っており,そのうち25,000名の乳児は輸血が必要であると推定されている。貝氏地中海貧血患者は主に血液科医師によって治療された。
貝氏地中海貧血は疾患の重症度によって3種類のタイプに分けられる:重症、中間型と軽型。最も深刻な形式は重度地貧(クリー貧血とも呼ばれる)であり,通常出生直後に診断され,患者は生命にかかわる貧血を有している。小児科患者は典型的な速度で成長し、体重が増加することはなく、しかもよく肝臓、心臓と骨格の問題がある。多くの貧困患者は深刻な貧血を予防するために頻繁に輸血する必要があり、このような治療自体は体内の鉄の蓄積による長期的な問題を招く。中間地中海貧血はあまり重篤ではない疾患であり,軽度から中等度の貧血をきたす。これらの患者は輸血が必要な場合があり,具体的には症状の重症度に依存する。軽度貧困患者は非常に軽微な貧血を有し,一般に治療を必要としない。次のいずれかを持つ0 or ß+地中海貧血は必ずしも臨床疾患の重症度を示唆しているとは限らず,両タイプとも重症地中海貧血と中間型地中海貧血と診断されているからである。
現在、多くの輸血依存型貝氏地中海貧血患者に対する看護標準は頻繁に輸血して貧血を治療することである。これらの頻繁な輸血は鉄過負荷の合併症をきたす可能性があり,鉄キレート療法で治療しなければならない。同種造血幹細胞移植は潜在的に地中海貧血を治療する方法であるが、このような干与措置の使用は制限されており、死亡を含む合併症のリスクがあるため、適切なHLA一致同胞ドナーを探すことは困難である。2019年6月、欧州委員会はZYNTEGLOの条件付きマーケティング許可を承認し、ZYNTEGLOはブルーバード生物会社(Bluebird Bio,Inc.)が開発した遺伝子療法であり、ヨーロッパで輸血依存性β-地中海貧血およびある遺伝子タイプを有する成人および青少年患者の治療のための遺伝子療法である。しかし、2021年8月、ブルーバードは欧州での商業運営を終了する計画を発表し、米国市場に重点を置くことにした。ZYNTEGLOは2022年8月にFDAによって定期輸血を必要とする成人と小児β地中海貧血患者の治療に許可された。Reblozyl(Luspatercept)は1種の赤系成熟剤であり、成人患者の治療に許可され、これらの患者は伯-地中海貧血に関連する貧血を有し、頻繁な輸血が必要である。最近これらの遺伝子療法や小分子が承認されているにもかかわらず,依然として高い需要が満たされておらず,小分子経口療法によりHBFを増加させることで疾患を治療できると考えられる。
16
貝型地中海貧血の生物学的研究
β−地中海貧血は遺伝子変異によるものであるHBBジーン。これらの突然変異は鉱化グロブリンの生産を妨害した.いくつかの突然変異は貝珠蛋白を産生しないことを招き、もういくつかの突然変異は貝珠蛋白の産生を減少させる。
貝型地中海貧血の根本的な原因を解決する方法は
あるタイプのβ-地中海貧血はHBFを上昇させる療法で治療できると考えられている。血液中にHBFが含まれているため,出生時に重症地中海貧血に罹患した乳児は通常出生直後には何の症状も出現しない。出生後HbFレベルの低下やβ−グロブリンの増加に伴い貧血が出現し,?地中海貧血を有する乳児に疾患が出現し始めた。HbFレベルの高い中間地中海貧血患者の症状はHbFレベルが低い患者より少なかった。FTX−6058は非患者の治療のための臨床開発に適している可能性が考えられる0輸血に依存する人です
Fulcrum Seekはヘモグロビン疾患の薬物標的を特定しました
FulcrumSeekを用いて、著者らはヒト臍帯血由来の赤系前駆細胞2(HUDEP 2)をモデルとして標的識別と検証活動を行い、HBFの再活性化を研究した。HUDEP 2細胞は未成熟赤血球である。私たちの小分子プローブバンクとCRISPRライブラリーをスクリーニングすることで、いくつかの活性化を決定しましたHBG 1/2遺伝子はHbFの上昇を引き起こす。いずれのスクリーニング方法も同じタンパク質複合体を決定しており,これらのタンパク質複合体はHBF産生を担う遺伝子の発現に重要な役割を果たしていると考えられる。追加的な実証実験を行い,この複合体のいくつかの成分を抑制することにより必要なHbFの上昇が観察された。これらの成分を抑制することは重要な細胞健康マーカーに悪影響を与えないことも観察された。
このタンパク質複合体の薬物標的としての操作性を評価した後,このタンパク質複合体のメンバーを選択して薬物発現活動を行い,HBF薬物標的と呼ぶ。HBF薬物標的の正常な生理作用は翻訳後の蛋白質修飾を促進することであり、著者らの薬物化学計画の目標はHBF薬物標的の抑制剤を最適化することである。我々は開発しました体外培養そして体内にある標的結合分析およびエネルギーX線結晶学を用いて、FTX-6058、新規なHBF薬物標的の小分子阻害剤を発見し、開発した。
FTX-6058
FTX−6058は経口HBF誘導剤であり,SCDを含むいくつかのヘモグロビン疾患に開発されている。FTX−6058は、EEDに結合し、PRC 2の転写サイレンシング活性を阻害するように設計されている。EED抑制はBCL 11 Aを含む重要な胎児グロブリン抑制因子の有効な低下を招き、HbFの増加を招く。EEDはPRC 2複合体のメンバーであり,PRC 2複合体もEZH 2を含む。EZH 2系薬剤には承認された製品があり,その承認されたラベルは悪性腫瘍のリスク増加を含む安全リスクを概説している。著者らは2021年第4四半期にSCD患者におけるFTX-6058の1 b期臨床試験を開始した。FDAは2022年2月、SCDを治療する孤児薬としてFTX−6058を承認した。FDAは、2022年12月、SCDのためのFTX−6058の高速チャネルを承認した。2023年2月23日、FDAは、SCD潜在的治療のためのFTX−6058のINDを全面的に臨床的に保留した。そこで,FTX−6058の1 b期試験の登録と用量を一時停止し,ベータ地中海貧血に対するFTX−6058の単独INDを中止した。FDAの臨床保留は,2022年4月,10月,12月にINDに提出されたマウスと犬の毒理学研究データと,2023年2月中旬に我々が提出したFDA 2023年2月初めのこれらの毒理学研究に関する情報要請への対応を参考にした。
我々のFTX−6058開発計画の一部として、良好な実験室実践(GLP)の下で行われた研究を含む多くの非臨床毒理学研究が行われた。これらの毒理学研究は反復投与量の最大耐性量と用量範囲の発見研究を含む;ラット28日、13週、17週と26週の研究;および犬の28日、13週と39週の研究を含む。臨床試験についてFDAは,FTX−6058の毒理学的研究で観察された血液悪性腫瘍の特徴は他のPRC 2阻害剤で観察されたものと類似していることを指摘しており,他のPRC 2阻害剤を用いた血液悪性腫瘍が臨床的に報告されている。私たちは勤勉に働き、FDAが臨床保留を解決するために必要な情報をできるだけ早く提供するつもりだ。
17
臨床試験:1 b期
2023年1月、私たちは、6 mg用量キュー内の対象の1 b段階データ、および6 mgおよび2 mg用量キューの登録完了を発表し、12 mg用量キューの登録を開始した。臨床的に放置される前に,1 b期試験はヒドロキシウレアを服用および服用しない被験者を評価した。
6 mg用量列(n=10)の被験者からの1 b期データでは,HbFの絶対値はベースラインより9.5%増加した。これらのデータは,ヒドロキシウレア(n=3)とヒドロキシウレア(n=7)を服用しない被験者では反応に差がないことを示している。6 mgの評価可能な被験者では溶血のバイオマーカーの改善も認められた。次の図は,6 mg用量列において,評価可能な被験者(n=7)の高速液体クロマトグラフィーで測定したHbF増加率とHbFのベースラインに対する絶対パーセンテージの変化を示している。
私たちは6 mgおよび2 mg用量キューの登録および用量分配を完了しており、臨床的放置問題を解決できれば、これらのキューに登録またはそれ以上の用量の被験者を登録するつもりはない。2 mg用量列の被験者(n=2)からのデータでは,HbFはベースラインより4.6%増加した。
18
12 mg用量列中の追加の対象(ヒドロキシウレアを服用している対象および服用していない対象を含む)の登録および用量は現在放置されている。試験休止前の12 mg用量列(n=3)の対象からのデータは、治療42日後にHBFの絶対値がベースラインより10.0%増加したことを示している。星号を有する被験者はヒドロキシウレアを服用した。
19
HbFの増加は、VOC、貧血、疼痛、感染、脳卒中、その他を含む一連のSCD症状の頻度或いは重症度を低下させることが証明されている。大量の遺伝、臨床と観察証拠により、もっと高いレベルのHBFのSCD患者への影響を表明し、HBFの誘導はベースラインより5-10%高いことは疾病負担の軽減と臨床結果の改善と関係があるかもしれない。これらの初歩的なデータは,FTX−6058がHBFレベルを9.5%向上させたことを示しており,SCD患者の変革療法となる可能性を支持していると信じている。
2023年3月3日までのデータ締め切りは,FTX−6058の耐性は全体的に良好であった。これまでに14例のTEAEが報告されており,そのうち2例は検討薬(頭痛,唇麻痺)に関与している可能性が報告されており,いずれも深刻ではなく,深刻ではないと考えられている。TEAEによる生産停止は現在のところない。14個のTEAEのうち3つはVOCsとして特徴付けられ,FTX−6058とは無関係と考えられ,そのうちの1つは重篤な有害事象またはSAEと報告され,不コンプライアンス対象に急性胸部症候群を伴った。
現在,いつ1 b段階実験に回復できるかは不明であり,できれば.
臨床試験:第1段階健康ボランティア
2020年第4四半期、著者らは健康成人ボランティアにおけるFTX-6058の臨床試験を開始した。第1段階ランダム、二重盲検、プラセボ対照試験は、FTX-6058漸増用量の安全性、耐性、およびPKを評価することを目的としている。SADキューでは、健康ボランティアは、プラセボまたは2、4、10、20、30、40、または60 mgのFTX−6058の剤を受ける。狂った列では、健康ボランティアは毎日1回のプラセボを服用するか、または2、6、10、20または30 mgのFTX−6058を14日間連続して服用する。各狂った列には6人の被験者が薬を服用し、2人はプラセボを服用していた。食物効果も単独の20 mg用量列で検討された。MAD行列には探索的措置が含まれており、標的参与、HBG mRNAとHBFを含む網状赤血球(F網状赤血球)の変化を評価する。
2021年8月の健常ボランティア2,4,10,20,30および40 mg SADキューおよび2,6および10 mg MADキューのデータを報告し,健常ボランティア60 mg SADキューと20および30 mg MADキューのデータ,および2021年12月に食事効果を評価した20 mgキューデータを報告した。
20
FTX−6058は全体的に耐性が良好であり,SAE報告もなく,すべてのSADとMADキュー中のTEAEによる中止もなかった。データは用量に比例したPKを示し続け,MADキューにおける平均半減期は約6−7時間であり,1日1回の投与をサポートしており,FTX−6058では食物効果は認められなかった。MAD列からのデータは引き続き強力な標的関与を示し,治療14日後にH 3 K 27 me 3のレベルはベースラインより約75%~95%低下した。われわれの臨床前研究によると,このレベルの標的参加は強力な誘導をもたらすことが予想されるHBG 1/2HBFの生産量を増加させました
MADキューからのデータは時間と用量依存のHBG mRNA誘導も示し,次の図に示すように生物学的証拠が証明された。治療後にHBG mRNAの持続誘導7~10 dを観察した。FTX−6058の臨床前研究では,HBG mRNAの増加はHBF蛋白の同倍数の増加に変換されてきた。ヒト遺伝学的には,SCD患者ではHbFが典型的なベースラインレベル(5−10%HbF)より2−3倍増加することが予後の有意な改善,さらには機能的治癒に関与していることが示唆された。それは.
FTX−6058 HBG mRNAの平均フォールディング誘導はプラセボと比較して
|
2 mg* |
6 mg* |
10 mg* |
20 mg |
30 mg |
|||||
|
平均折り畳み誘導法 |
P値 |
平均折り畳み誘導法 |
P値 |
平均折り畳み誘導法 |
P値 |
平均折り畳み誘導法 |
P値 |
平均折り畳み誘導法 |
P値 |
7日目 |
1.28 |
0.3494 |
1.94 |
0.0135 |
2.08 |
0.0063 |
2.06 |
0.0072 |
2.29 |
0.0025 |
14日目 |
1.20 |
0.5122 |
2.45 |
0.0025 |
3.54 |
5.63 |
6.15 |
|||
安全フォロー(第21-24日目) |
1.21 |
0.3736 |
2.75 |
3.22 |
6.45 |
6.13 |
||||
FTX-6058活性は臨床前研究で
私たちは観察しました体外培養そして体内にある活性化するHBG 1/2FTX-6058臨床前研究における遺伝子。FTX−6058はHBFレベルを向上させ,重要な細胞健康マーカーへの悪影響が最も小さいことが観察された。図に示すように観察されています体外培養7日間の薬物治療後、初代ヒトCD 34+細胞HbF発現は上昇し、その中に9例の健康ドナー、4例のSCDドナーと1例の鎌状細胞特性ドナーを含む。FTX−6058は,これら14個のドナー細胞株において,いずれのHBFもベースラインレベルより有意に高いことを示している。CD 34+由来細胞を追加的に臨床前分析し,FTX−6058治療によりHbFレベルが総ヘモグロビンの30%程度まで増加することが観察され,質量分析,高速液体クロマトグラフィー,高速タンパク質液体クロマトグラフィー技術により測定した。注目すべきは、他の機序データの回顧に基づいて、CD 34+細胞におけるHBFフォールディングの誘導が確実に臨床に応用されていることである。
FTX−6058治療効果観察
分化した初代CD 34+細胞では

21
また,CD 34+細胞におけるFTX−6058とヒドロキシウレアの作用を比較した。HbF上昇に対する水酸基尿素の影響は小さかったが,FTX−6058ではHbFの有意な上昇が認められた。FTX−6058とヒドロキシウレアを組み合わせて処理した細胞では,いずれかの化合物に対して単独で作用する増強効果が観察された。
また,Townesマウスモデルと呼ばれるSCDマウスモデルにおけるFTX−6058の役割も検討した。このモデルでは,マウスのグロブリン遺伝子はすでにヒトのグロブリン遺伝子に置換されており,ヒトヘモグロビン遺伝子の発現を調節する可能性のある機序を検討することができる。TownesマウスモデルはすでにSCDの潜在的な治療方法の研究に広く用いられている。以下の図に示すように,FTX−6058の1日5 mg/kg投与13日後にはHbF発現細胞とHbF蛋白レベルが有意に増加し,水酸基尿素によりF細胞とHbFがやや増加することが観察された。
TownesマウスにおけるF細胞の百分率 FTX-6058で治療します |
TownesマウスにおけるHBFタンパク質レベルの研究 FTX-6058で治療します |
|
|
左のグラフでは、3つの治療条件下でマウス血液中のF細胞が総細胞に占めるパーセンテージ(%F細胞)を定量化し、賦形剤単独で治療したSCDマウスのパーセンテージとして示した。右のグラフでは,3つの処理条件下でヒトHbF蛋白のレベルを測定し,総ヘモグロビンに占めるHbF蛋白のパーセンテージを定量化した。各値は、治療13日後の各治療群の8匹のマウスの平均値を表す。これらの研究では,一方向分散分析,あるいはANOVAと呼ばれる従来の統計的有意性を評価する手法を用いている.両研究ともFTX−6058のp値は0.001未満,右側研究では水酸基尿素のp値は0.01%未満であった。
私たちの探索プラットフォームは
FulcrumSeekは私たちの高スループット発見プラットフォームであり、私たちは私たちの珍しい疾患の組み合わせの薬物標的を識別し、検証するためにこのプラットフォームを設計した。米国国立衛生研究院(NIH)のデータによると,約7000種類のまれな遺伝子で定義されたヒト疾患があり,その多くは十分でないか承認されていない治療法である。われわれの方法では,患者由来の組織関連細胞系や他の疾患関連細胞系を獲得し,目的疾患の病理を概説·聴取するために分化した。
これらの細胞株は大規模に質問され,我々独自の自動化システムを用いて化学およびゲノム摂動が疾患表現型や転写群に及ぼす影響を検討した。我々は生物多様性を最適化するために、私たちが高度に注釈した独自の小分子化合物化学ゲノムプローブライブラリーを応用した。化学多様性を最適化する他の小分子スクリーニング手法とは異なり,我々が高度に注釈したライブラリーは,標的を迅速に認識·検証し,これらの目標を調節する潜在的先導分子である。
機械学習アルゴリズムのような計算生物学を用いて薬物標的の選択を指導しています私たちの反復とシステム方法は私たちが比較的に定義の少ない遺伝的関連を持つ疾病を探索することができ、逆に根本原因生物学を調節する標的をスクリーニングすることができる。したがって、私たちは私たちがFulcrumSeekで潜在的に聞くことができる病気の数を大きく拡大できると信じている。
22
スクリーニング計画を発見する
FulcrumSeekを用いてFSHDやSCDのために探している小分子標的を発見した。我々は,FulcrumSeekの広範な適応を利用して,筋肉,血液,神経疾患の他にまれな遺伝子定義疾患のための薬物標的を発見している。

私たちの目標識別戦略と方法は発展し続けている。単一の根本原因遺伝子の発現を調節する標的を識別するためのスクリーニングを行うほか、複数(約10個)の根本原因遺伝子を同時に尋ね、単一のスクリーニングで細胞の健康への影響を監視することができる(約10個)即..多重化フィルタリング).私たちは、この新しい方法が生産性の面で著しい効率を提供し、複数の仮説を並列にテストすることを可能にすると信じている。重要なことは,FulcrumSeekの拡張はRNAseqと細胞イメージングを含む高含有量の分子プロファイルを用いて,8,000−10,000個の遺伝子の発現を同時に測定し,細胞健康や生物学に関連する重要な測定を統合することができ,スクリーニング能力や生産性を拡張することができることである。我々の小分子プローブライブラリーと我々の機能ゲノミクス能力を用いることにより,大幅に増加した規模とコスト効果で標的認識を行うことを目標としている。また,我々は検証モードと仮説生成モードで我々の製品エンジンを使用していると仮定しており,我々のポートフォリオやパートナーと協力する際に魅力的な目標を決定する可能性が増加すると予想される.
ライセンス契約と連携
グラクソ·スミスクラインと締結された参考権と許可協定
2019年2月、GSKは、GSKまたはその共同経営会社によって制御されたlosmapimodに関連するINDの権利を参照してFDAに提出され、losmapimodに関連するいくつかの特許権に基づいて独自のグローバルライセンスを付与するグラクソ·スミスクラインの連属会社と参照権利およびライセンス契約を締結した。このプロトコルはまた、GSKのいくつかのlosmapimodの臨床前および臨床データに関する世界的な独占的許可を提供している。参照権利とプロトコルにより付与された許可の部分対価格として,参照と許可契約を締結する際にGSKに12,500,000株のB系列優先株を発行した.この協定では,商業的に合理的な努力を用いて,FSHD治療のための許可製品を開発し,商業化することが義務付けられている。
ライセンス特許権およびデータ権に基づいて、このプロトコルは、losmapimodまたはlosmapimodを含む任意の製品を原料薬として研究、開発および商業化するために、独占的、再許可可能な許可を付与し、ヒト疾患の治療のための許可製品と呼ぶ。グラクソ·スミスクラインは、特許およびデータを許可する権利の下で非臨床研究を行う権利を保持しているが、再許可を付与する権利はなく、GSKは、我々の同意の下で、特定の予防用途に関連する許可製品の使用に関連するいくつかの開発活動に従事することができる。グラクソ·スミスクラインはまた、既存のロストパジン生産供給を私たちに移管することに同意した。
この合意に基づき,吾らは,このようなマイルストーンに初めて到達した認可製品が指定された開発および規制マイルストーンに達したときに,2022年12月31日までのREACH 3期臨床試験開始時のGSKへの500万ドルのマイルストーン支払いと,2019年12月31日までにReDUX 4を開始した年度内向きGSKへの250万ドルのマイルストーン支払いと,初めてライセンス製品に達した一度の年間世界純販売合計マイルストーンの時点でGSKに最高6,000万ドルを支払う責任がある。印税の支払いも義務付けられております
23
中間ビット数のパーセントから低い二桁ですが十代未満のパーセントは、私たちと私たちの任意の関連会社とライセンス保有者のライセンス製品の年間純売上に基づいています。特許使用料はライセンス製品や国/地域によって支払われ、特定の場合には減少する可能性がある。
私たちは、ライセンス製品の模造バージョンがその国/地域に適用される規制機関の承認を得るまで、またはそのライセンス製品が国/地域で初めて商業販売される10周年(特許権使用料条項と呼ぶ)の前まで、特許権使用料を支払う義務を国/地域の許可製品に適用する。規制機関が各国·地域の任意のライセンス製品に付与された任意の独占営業権またはデータ独占権(特許権を除く)が満了した後、その国/地域に適用される印税料率が低下する。また、我が国または関連会社または分割許可者が、任意の特許権に基づいて第三者から許可を得る必要があると判断した場合、国/地域で許可製品を使用することができる場合には、当該国/地域でGSKに支払われるべき使用料から、第三者に支払うべき許可料の割合を第三者に支払うことができる。
協定がその条項に従って早期に終了しない限り、協定は、各国/地域のライセンス使用料の期限が満了するまで、国/地域およびライセンス製品/ライセンス製品に基づいて継続し、その国/地域におけるライセンス製品の合意が満了し、その国/地域のライセンス製品に関するライセンス特許権およびデータ権利の全額納付、印税免除、永久ライセンスを有することになる。他方が合意項目の義務を履行する際に深刻に違約し,その違約が適用された救済期間内にも是正されていない場合には,いずれも合意を終了する権利がある.
百時美施貴宝社の完全子会社MyoKardiaとの協力と許可協定
2020年7月、著者らはMyoKardiaと協力と許可協定を締結し、ある遺伝子定義の心筋症に関連する興味のある遺伝子を調節できる生物学的標的を決定した。協定条項によると、著者らはある知的財産権の下でMyoKardiaグローバル独占許可を付与し、その研究、開発、製造、製造、使用、使用、販売、要約販売、輸入、輸入、輸出、輸出、流通、流通、マーケティング、普及、普及、あるいは他の方法で私たちが決定した特定の生物標的のための製品を開発することを可能にし、これらの製品はある遺伝子定義の心筋症に関連する特定の遺伝子を調節することができる。
双方が合意した研究計画に基づいて、著者らはMyoKardiaの更なる研究、開発、製造と商業化のために、最も多くの特定数量の潜在心筋症遺伝子標的、或いは確定された標的を確定と検証するために、化学スクリーニングと関連研究活動を行う。私たちはMyoKardiaと一緒に努力し、どのように研究計画下の研究活動の各段階で最適な進展を得るかを決定し、どのような確定された目標(あれば)が研究計画或いは候補心筋症目標に規定された基準に符合するかを決定する。研究計画完了後,双方は最終的なパケットを共同で用意し,MyoKardiaはプロトコルによりMyoKardiaのさらなる開発に何らかの心筋症目標候補,あるいは心筋症目標を指定する可能性がある。MyoKardiaが指定された期間内に心筋症標的を指定していなければ,プロトコルは自動的に終了する。MyoKardiaが1つ以上の心筋症の標的を指定した場合、MyoKardiaは、規制部門の承認を求め、特定の指定された国/地域で決定された目標に対して製品を商業化するための商業的合理的な努力を使用する義務がある。
我々が研究計画や研究期間に応じて研究活動を行っている期間や,研究期間後の特定の時間内にMyoKardiaが心筋症目標を指定していれば,このような研究活動によるデータをプロトコルに基づいてMyoKardiaにしか利用できない。研究期間およびその後の特定の期間において、我々は、(A)プロトコルに従って、心筋症標的候補に対して治療、予防または診断を行う化合物または製品、または(B)心筋症標的によって特定の調節された特定の遺伝子に関連することが証明された任意の遺伝子定義心筋症を治療するための化合物または製品を研究、開発、製造、商業化、使用、または他の方法で開発してはならない。
協定によると、MyoKardiaは2020年7月に私たちに1000万ドルの前払い金と250万ドルの前払い研究資金を支払った。MyoKardiaは,前払い研究資金にカバーされていない研究活動の費用を精算し,最高限度額は研究資金総額(前払い研究資金を含む)である。特定の臨床前、開発、販売に基づくマイルストーンを達成した後、いくつかの決定された目標については、臨床前マイルストーン支払い、開発マイルストーン支払い、販売ベースマイルストーン支払いを得る権利があり、いくつかの決定された目標については、各目標の合計2.985億ドルを得る権利があり、いくつかの他の決定された目標については、各目標の合計1.5億ドルを得る権利がある。今まで、私たちは指定された250万ドルの臨床前マイルストーンに到達した。MyoKardiaはまた、MyoKardiaとその任意の付属会社とライセンス所有者が合意に基づいて、製品の年間世界純売上高の等級別使用料を支払い、中間の1桁パーセントから低い2桁の数百分比まで様々である
24
目標です。特許使用料は、指定された特許使用料期間内に製品毎に支払われ、特定の場合には減少する可能性がある。
このプロトコルは、製品の最後の特許使用料期限が満了するまで、個々の国および個々の製品に基づいて継続され、この場合、その合意がその条項に従って早期に終了しない限り、製品はその国の合意で終了する。他方がその義務を履行する際に深刻に違約し,その違約が適用された救済期間内にも是正されていない場合には,いずれも合意を終了する権利がある.MyoKardiaはまた,あらかじめ書面で通知されている場合には,便宜上,プロトコルを完全に終了するか,ターゲットごと,製品ごと,または分子ごとにプロトコルを終了する権利がある.
知的財産権
我々は、内部開発でも第三者から許可を得ても、特許権を求め、維持し、擁護することを含む、我々の業務発展に重要なビジネス的意義を有するノウハウ、発明、改善を保護し、強化するために努力している。私たちはまた、ビジネス秘密、技術ノウハウ、持続的な技術革新、許可内の機会に依存して、私たちの分野での私たちの独自の地位を強化し、維持しています。
私たちの将来のビジネス成功は、私たちの能力にある程度依存します:私たちの業務に関連する重要な商業技術、発明およびノウハウの特許および他の独自保護の獲得と維持;私たちの知的財産権、特に私たちの特許権の擁護と実行、私たちのビジネス秘密の秘密、および第三者が効果的かつ強制的に実行可能な特許および独自の権利を侵害、流用または侵害することなく運営されます。私たちが第三者が私たちの製品を製造、使用、販売、提供、または輸入することを阻止する能力は、これらの活動をカバーする効果的かつ強制的に実行可能な特許または商業秘密に基づいて私たちが権利を持っている程度に依存するかもしれない。
私たちのようなバイオテクノロジーと製薬会社の特許地位は通常不確定であり、複雑な法律、科学、そして事実の問題に関連しているかもしれない。私たちが現在求めている特許出願が任意の特定の司法管轄区域で特許として発行されるかどうか、または任意の発行された特許の権利主張が競争相手の影響を受けないように十分な特許保護を提供するかどうかを予測することはできない。私たちまたは私たちの許可者が将来提出する可能性のあるどの特許出願も特許が発行されることを保証することもできませんし、私たちが所有したり許可したりする任意の特許または将来の特許が、私たちの候補製品を保護し、これらの特許を製造する方法に商業的な用途を持つことを保証することもできません。また,特許出願で要求されるカバー範囲は特許発行前に大幅に縮小される可能性があり,その範囲は特許発行後に再解釈でき,挑戦することも可能である.したがって、私たちは私たちのどの製品も強制的に施行可能な特許によって保護されるか、または引き続き保護されることを保証することはできない。しかも、私たちが持っているどんな特許も第三者の挑戦、回避、または無効にされる可能性がある。私たちの知的財産権に関するリスクのより全面的な説明については、“リスク要因-私たちの知的財産権に関するリスク”を参照されたい。
私たちは一般的に私たちの重要な計画に対して特許出願を提出して、これらの計画に対する私たちの知的財産権の地位を確保するために努力している。2023年3月2日現在,我々は11件の米国特許,7件の米国係属中の非仮特許出願および関連する未解決外国特許出願,および係属中の米国仮特許出願3件を所有または許可している。
2023年3月2日現在、我々の最先端計画の知的財産権の組み合わせの概要は以下の通りです。起訴は長い過程であり、その間、最初に米国特許商標局によって提出された審査の権利請求の範囲は、もし本当に発表されれば、発表前に大幅に縮小される可能性がある。私たちは以下に言及されたいくつかの未解決特許出願にこのような状況が発生する可能性があると予想する。
ロス·マプト
Losmapimodについては、losmapimodを用いてFSHD患者を治療する方法と、他の臨床段階のp 38阻害剤を用いてFSHD患者を治療する2つの米国特許とを含む米国特許を有しており、各特許は2038年に満了する予定であり、カナダとメキシコ、ヨーロッパ、アフリカ、オーストラリア、ニュージーランド、南米およびアジアの関連特許および出願中の特許の有効期間は2038年に満了する。我々はまた、losmapimodを用いてFSHDおよび他の疾患を治療する方法に関する3つの関連する米国未決仮出願、係属中のPCT出願および係属中の米国仮出願を有しており、発行された特許が生成された場合、2038年から2043年の間に満了する予定である。グラクソ·スミスクラインが付与したlosmapimodは、物質組成物および医薬組成物としての特許が満了している。
FTX-6058
現在、私たちのFTX−6058関連特許の組み合わせには、物質組成に対する2つの発行された特許(2040年に満了する予定)、米国の非一時的出願、カナダの関連未解決特許2つが含まれている
25
メキシコ、ヨーロッパ、アフリカ、オーストラリア、ニュージーランド、南米、アジアでは、発行すれば2039年から2040年の間に満期になる見通しだ。我々はまた、3つの係属中のPCT出願およびFTX−6058使用方法および処方のための2つの係属中の米国仮出願を有しており、発行された特許が生成された場合、2042年から2043年の間に満了すると予想される。
個別特許の期限は特許を取得した国の法的期限に依存する。私たちが出願したほとんどの国では,特許期間は非臨時特許出願が提出された最初の日から20年である。
米国では、場合によっては、1984年の“医薬品価格競争および特許期限回復法”によれば、FDAによって承認された薬物をカバーする特許期限は、FDA規制審査中の特許期限損失の補償として延長される可能性がある。延長期間は最長5年に達するが、特許の残り期間を製品承認日から合計14年間延長することはできない。延期する資格のある特許のうち、1つの特許のみを延期することができ、承認された薬物、その使用方法、または製造方法に関する権利要件のみを延期することができる。ヨーロッパおよび他のいくつかの管轄区域にも同様の規定があり、承認された薬物をカバーする特許の期限を延長する。ロスパモントの使用に関する米国特許と私たちの知的財産権からの製品は、特許期間の延長を受ける権利がある可能性がある。候補薬剤の使用または候補薬剤自体がFDAによって承認された場合、承認された用途または候補薬剤をカバーする特許期間を延長するために、特許期間の延長を申請する予定である。私たちはまた、利用可能な司法管轄区で特許期間の延長を求める予定ですが、FDAを含む適用当局が、このような延長を承認すべきかどうか、そのような延長の長さを承認すべきかどうかの評価に同意する保証はありません。
特許保護に加えて、私たちは非特許の商業秘密と機密技術、そして持続的な技術革新に依存して、私たちの競争地位を発展させ、維持しています。しかし、商業秘密と機密技術は保護することが難しい。私たちは、私たちの固有の情報を保護し、任意の協力者、科学コンサルタント、従業員、コンサルタントとの秘密協定、および私たち従業員との発明分配協定を部分的に使用することを求めています。私たちはまた、選定されたコンサルタント、科学コンサルタント、協力者と発明の分配を要求する協定を締結した。このような合意は意味のある保護を提供しないかもしれない。このような合意はまた違反される可能性があり、私たちはこのような違反に対応するための十分な救済策を持っていないかもしれない。さらに、私たちの商業秘密および/または機密技術は、第三者によって知られたり、独立して開発されたり、またはそのような情報を開示する任意の協力者によって悪用される可能性がある。私たちの知的財産権を保護するための任意の措置が取られているにもかかわらず、許可されていない当事者は、私たちの製品のいくつかの態様をコピーしようとしたり、私たちが独自と考えている情報を取得したり、使用したりするかもしれない。私たちは私たちの固有の情報を保護するための措置を取っているにもかかわらず、第三者は同じまたは同様の独自の情報を独立して開発したり、他の方法で私たちの固有の情報にアクセスすることができる。したがって、私たちは私たちの商業秘密と固有の情報を意味的に保護することができないかもしれない。私たちの知的財産権に関するリスクのより全面的な説明については、“リスク要因-私たちの知的財産権に関するリスク”を参照されたい。
製造業
私たちには何の生産施設もありません。私たちは私たちが行っているFSHD第3段階臨床試験を完成させるために、契約製造機関から十分な数のロスパモントを得ました。
私たちはすでに契約製造機関から十分な数のFTX-6058を獲得し、現在臨床で保留されている1 b期の臨床試験を完成させた。
我々は、引き続き第三者生産FTX−6058に依存して任意の将来の臨床試験を行い、臨床前および臨床試験のための任意の将来の候補製品を生産し、私たちの候補製品が市場の承認を得た場合に商業生産に使用する予定である。私たちの主要な候補製品は小分子であり、信頼性があり、再使用可能な合成プロセスによって既製の原料から生産することができる。契約製造施設で高コストで生産できる候補製品の開発を継続する予定である。
競争
生物技術と製薬業界の特徴は技術進歩が迅速で、競争が激しく、そして独自製品を高度に重視していることである。私たちは私たちの技術、知識、経験と科学資源が私たちに競争優位を提供してくれると信じているが、私たちは主要な製薬、専門製薬とバイオテクノロジー会社、学術機関と政府機関、そして公共と個人研究機関を含む多くの異なる源からの競争に直面している。我々が開発と商業化に成功した任意の候補製品は,既存の療法や将来出現する可能性のある新しい療法と競争するであろう。
26
私たちと比較して、私たちが競争しているか、あるいは将来競争する可能性のある多くの会社は、研究開発、製造、臨床前テスト、臨床試験を行い、監督管理許可とマーケティング承認製品を獲得する上で、より多くの財務資源と専門知識を持っている。製薬とバイオテクノロジー産業の合併と買収は、私たちの数の少ない競争相手により多くの資源を集中させる可能性がある。規模が小さいか早い段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。これらの競争相手はまた、合格した科学と管理者を募集と維持し、臨床試験場と臨床試験の患者登録を確立し、そして私たちの計画と相補的或いは必要な技術を獲得する上で私たちと競争している。
もし私たちのすべての候補治療製品が承認された場合、その成功に影響を与える重要な競争要素は、それらの有効性、安全性、利便性、価格、診断に伴う関連療法の使用指導における有効性、後発薬競争のレベル、および政府および他の第三者支払者が精算できるかどうかである可能性がある。
もし私たちの競争相手が私たちが開発する可能性のあるどの製品よりも安全で、より効果的で、副作用が少なく、より便利で、より安い製品を開発し、商業化すれば、私たちのビジネス機会は減少または消失するかもしれない。私たちの競争相手も私たちよりも早くFDAや他の規制部門の承認や製品の緊急使用許可を得ることができ、これは私たちの競争相手が市場に入る前に強力な市場地位を確立することをもたらすかもしれない。また、多くの場合、私たちの競争能力は、後発薬の使用を奨励することを求める保険会社または他の第三者支払者の影響を受ける可能性がある。もし私たちの候補製品が市場の承認を得たら、私たちはそれらの価格が競争相手の模造薬より著しく高いと予想する。
我々の主要候補製品が現在行われている臨床試験の適応に承認されれば,以下に検討する療法や現在市販されている薬物と競合する。
FSHD
現在のところFSHDを治療する方法は承認されていない。現在の治療は対症治療に限られており,物理/職業治療,患者の虚弱分布に応じてオーダーメイドされた低強度有酸素運動,一般的な疼痛治療など,限られた有益な効果を提供する可能性がある。肩帯の活動範囲制限は肩周囲筋無力による肩甲翼に起因する可能性があり、この場合、手術肩甲骨固定術はある患者にいくつかの機能改善をもたらすことができる。
ロスパモントはFSHD患者の開発において他の治療法からの競争に直面している可能性がある。羅氏はミオスタチン阻害剤RO 7204239を評価しており,成人FSHDに対する2期研究である。親和性は、成人FSHDの5.2期臨床試験において、AOC 1020、siRNA抗体-オリゴヌクレオチド複合体を評価している。FSHDの臨床開発にロスパミと同様の作用機序を有する候補製品があるかどうかは不明である。
SCD
承認されたSCD薬物治療は主に苦痛な血管閉塞発症の管理と減少、溶血性貧血の改善に集中している。アメリカで承認された4種類の薬物治療はヒドロキシウレア、Voxelotor、CrizanlizumabとL-グルタミンである。ヒドロキシウレアは、苦痛発症の頻度を減少させ、輸血の必要性を減少させるためにSCDの治療のために許可されている。ヒドロキシウレアには後発薬とブランド薬の2種類があり,百時美施貴宝社製DROXIAとAddMedica社製Siklosがある。ファイザーにより販売されているVoxelotor,商標名OXBRYTAは,ヘモグロビンを増加させるためのヘモグロビン重合阻害剤として加速承認されている。Crizanlizumabはノ華社が販売している全ヒト型モノクロナル抗体p-セレクチン阻害剤であり、VOCsの頻度を減少させることが許可されている。L−グルタミン,ブランド名ENDARIは,Emmaus生命科学社により販売され,この疾患の急性合併症を減少させるために承認されている。
SCD治療の1種の支持性看護選択は赤血球輸血であるが、これは同種異体免疫、輸血反応と鉄過負荷によって複雑になる可能性がある。
現在、深刻なSCD群の唯一の潜在的治癒方法は造血幹細胞移植であることが許可されている。しかし、HSCTのより一般的なのは、利用可能なHLA適合同胞ドナーを有する小児科個体に提供されることである。この若年群では5年生存率はかなり高いが,年齢の大きい個体(>16歳)では生存率がはるかに低い可能性がある。不妊症と移植片対宿主病を含む造血幹細胞移植に関連するリスクも大きい。
FTX−6058は、SCD患者のために開発されている多くの異なる治療法からの競合に直面する可能性がある。ノドA/S社は,HBF産生を増加させるための小分子薬であり,2022年夏に第2段階臨床試験を開始するndec(ジシタビン−テトラヒドロウロシド)を評価している。Novoもエトピットを評価しています
27
2/3期臨床試験にあるPKRアゴニスト。Agios製薬会社はPKRアゴニストMitapivatに対して2/3期臨床試験を行い,SCD患者に用いている。ファイザー社はHBS重合阻害剤GBT−601を評価しており,2023年に第3段階臨床試験を開始し,2つの第3段階臨床試験でP−セレクチン阻害剤Clacumabを評価し,第1段階臨床試験で抗E−セレクチン阻害剤PF−07209326を評価する予定である。武田製薬有限会社はTAK−755を評価しており,これは組換えADAMTS 13蛋白であり,ベースライン健康なSCDとSCD合併急性VOCsの参加者で第1段階臨床試験を行っている。Vertex製薬会社,あるいはVertexは,2023年第1四半期にexagamlobgene autemcel(EXA−cel)に関するBLAをスクロール提出する予定であり,SCDを潜在的に治療する遺伝子療法である。Vertex社は,米国とEUの3.2万人の重篤なSCDとオスミウム−地中海貧血を有する患者に最初の商業化努力を集中させようとしていると発表した。Sangamo治療社(Sangamo Treateutics Inc.)は,細胞を修正し,HBFを用いて機能性赤血球を産生し,1/2期臨床試験を行う遺伝子編集細胞療法であるSAR 445136を開発している。ブルーバードはlovo−celという遺伝子療法を評価しており,3期臨床試験を行っており,2023年第1四半期にBLAを提出する予定である。Intellia Treateutics,Inc.(ノファ社と提携),Editas Medicine,Inc.,Graphite Bio,Beam Treeuticsも他のいくつかの遺伝子編集手法を評価している.
貝氏−地中海貧血
現在、多くの輸血依存型貝氏地中海貧血患者に対する看護標準は頻繁に輸血して貧血を治療することである。これらの頻繁な輸血は鉄過負荷の合併症をきたす可能性があり,鉄キレート療法で治療しなければならない。同種造血幹細胞移植は潜在的に貝氏地中海貧血を治療する方法であるが,この介入措置の使用は死亡,HLA一致の同胞の識別が困難である合併症のリスクがあるため制限されている。2019年6月、欧州委員会はZYNTEGLOの条件付きマーケティング許可を承認し、ZYNTEGLOはヨーロッパで輸血依存性β-地中海貧血と特定の遺伝子タイプを有する成人と青少年患者の治療のために開発された遺伝子療法である。しかし、2021年8月、ブルーバードは欧州での商業運営を終了する計画を発表し、米国市場に重点を置くことにした。ZYNTEGLOは2022年8月にFDAによって定期輸血を必要とする成人と小児β地中海貧血患者の治療に許可された。Reblozyl(Luspatercept)は1種の赤系成熟剤であり、成人患者の治療に許可され、これらの患者は伯-地中海貧血に関連する貧血を有し、頻繁な輸血が必要である。最近これらの遺伝子療法や小分子が承認されているにもかかわらず,依然として高い需要が満たされておらず,小分子経口療法によりHBFを増加させることで疾患を治療できると考えられる。
FTX−6058は,輸血依存型オスミウム−地中海貧血患者の治療に開発されている多くの異なる治療法からの競争に直面する可能性がある。Agios製薬会社はPKRアゴニストMitapivatを評価しており,2段階の3期臨床試験では非輸血依存型と輸血依存型?地中海貧血患者に用いられている。NovoはPKRアゴニストetavopivatを評価しており,非輸血と輸血依存型β−地中海貧血に対する2期臨床試験である。エオニス製薬会社はSapablursen(前Ionis TMPRSS 6−LRx)を評価しており,TMPRSS 6に対するASO療法であり,非輸血依存型β−地中海貧血中間型の第二段階臨床試験である。Disk MedicineはTMPRSS 6に対するモノクロナル抗体MWTX−003を開発しており,2023年下半期に一期臨床試験を開始する予定である。Silent TreeuticsはTMPRSS 6に対するsiRNA療法の健康ボランティア研究でSLN 124を調査している。Vertexは2023年第1四半期にexagamlobgene autemcel(exa−cel)に関するBLA提出を終了する予定であり,輸血依存型β−地中海貧血の潜在的な遺伝子療法である
政府の監督管理と製品審査
その他の事項以外に、アメリカ連邦、州と地方各級及びEUを含む他の国と司法管轄区の政府当局は生物製薬製品の研究、開発、テスト、製造、定価、精算、品質管理、承認、包装、貯蔵、記録保存、ラベル、広告、販売促進、流通、マーケティング、承認後の監視と報告及び輸出入などの方面に対して広範な監督管理を行っている。米国や他の国や管轄地域で上場承認された手続きや、適用される法律や法規、その他の規制機関を遵守するには、多大な時間と財力が必要である。
アメリカの薬品審査と規制
米国では,薬品は“連邦食品,薬物と化粧品法”(FDCA)および適用される実施条例とガイドラインの規制を受けている。申請者が、非臨床試験、臨床試験、承認プロセス、または承認後のプロセスを含む、製品開発中の任意の時間に適用される法規要件を遵守できない場合、研究、規制審査および承認および/または行政または司法制裁の進行遅延を招く可能性がある。
28
米国での新薬の販売および流通の承認を求める出願人は、通常、以下の各ステップを満足して達成しなければならず、その後、候補製品がFDAの承認を得ることができる
臨床前研究
申請者が潜在的な治療価値を有する候補製品のテストを開始する前に、候補製品は臨床前試験段階に入り、体外培養薬物の安全性及び活性を評価するための動物研究と、ヒトで予備試験を行い、治療使用の理論的基礎を確立するための動物研究とを備える。臨床前テストは製品の化学、調合と安定性の実験室評価、及びその他の候補製品の毒性を評価する研究を含む。臨床前試験の進行と試験に使用する化合物調合はGLP法規と標準を含む連邦法規と要求に符合しなければならない。臨床前試験の結果は,製造情報,分析データ,任意の利用可能な臨床データや文献,臨床試験計画などとともにINDの一部としてFDAに提出される。IND提出後、生殖不良事象や発ガン性の動物試験や長期毒性研究などの長期的な臨床前試験が継続される可能性がある。
INDとIRBプロセス
臨床試験は、GCP要求に従って合格した研究者の監督の下でヒト対象に研究製品を服用することを含み、すべての研究対象に任意の臨床試験に参加する前に書面で自発的なインフォームドコンセントを提供することを含む。臨床試験は書面による研究案に基づいて行い,その中で組み入れと排除の基準,研究の目標,安全性モニタリングのためのパラメータ,評価すべき有効性基準を詳細に説明した。INDの一部として,各臨床試験の案と任意の後続の案修正案をFDAに提出しなければならない。
INDはFDCAの免除であり、未承認候補製品が州間商業で臨床研究のために輸送されることを許可し、FDAにこのような研究製品をヒトに使用することを許可することを要求する。承認された機密協定の対象に属さない候補製品を州間輸送·管理する前に、このような許可を得なければならない。IND申請を支援するためには,申請者は各臨床試験に1つの案を提出しなければならず,どの後続の案修正もINDの一部としてFDAに提出されなければならない。FDAは各IND提出後30日間の待機期間を求め,臨床試験を開始することが求められている。この待機期間は,人間の研究対象が受けるかどうかを決定するためにFDAがINDを審査することを可能にすることを目的としている
29
不合理な健康リスク。この30日間のいつでも、FDAはINDで概説された試験の進行に対して懸念または問題を提起し、臨床保留または一部の臨床保留を強制的に実施する可能性がある。これらの場合,INDスポンサーやFDAは臨床試験開始前に未解決の問題を解決しなければならない。
IND下での臨床試験開始後,FDAもこの試験を臨床放置あるいは一部の臨床放置を実施することができる。臨床、非臨床及び/或いは化学、製造と制御領域の新しいデータ、新しい発見或いは新しい発展が患者の安全問題を招く可能性がある時、FDAはすべて臨床一時停止を強制的に実施する。臨床保留はFDAがスポンサーに発表した命令であり,提案された臨床研究の延期や進行中の研究の一時停止が要求されている。一部の臨床保留はIND要求の一部の臨床仕事を遅延或いは一時停止することである。例えば、特定のプロトコルまたはプロトコルの一部は継続が許可されない可能性があり、他のプロトコルは許可される可能性がある。臨床保留或いは一部の臨床保留を実施した後30日を超えない後、FDAはスポンサーに棚上げ根拠に関する書面解釈を提供する。臨床保留或いは一部の臨床保留を発表した後、FDAがスポンサーに通知した後にのみ、臨床試験は回復することができる。FDAは、スポンサーによって提供された以前に引用された欠陥を修正する情報に基づいて、またはFDAが臨床試験を継続することができるようにする他の方法でこの決定を決定するであろう。
スポンサーは選択可能であるが,必要ではなく,IND下で海外臨床研究を行っている。ある国外の臨床研究がIND下で行われる時、放棄しない限り、FDAのすべてのIND要求を満たさなければならない。外国の臨床研究がINDの下で行われていない場合、スポンサーは、この研究をINDまたは上場承認申請の支援として使用するために、GCP要件を含むFDAのいくつかの法規要件に適合することを保証しなければならない。GCPは臨床研究の倫理とデータ完全性基準を含むことが要求される。FDAの規定は、非IND外国臨床研究に参加したヒト被験者の保護、及び結果データの質と完全性を確保することを目的としている。これらはまた、非IND外国研究がIND研究に匹敵する方法で行われることを確保するのに役立つ。
上述したIND要件に加えて、臨床試験に参加する各機関を代表するIRBは、機関が任意の臨床試験を開始する前に計画を審査および承認しなければならず、IRBは少なくとも年に1回の継続的な審査および再承認を行わなければならない。他の事項以外にも,IRBは研究対象に提供される研究案とインフォームドコンセント情報を審査·承認しなければならない。IRBの運営はFDAの規定に適合しなければならない。臨床試験がIRBの要求、手順、または他の要求に従って行われていない場合、または候補製品が患者の予期しない深刻な損傷に関連している場合、IRBは、その機関またはその代表機関の臨床試験の承認を一時停止または終了することができる。
さらに、いくつかの試験は、データ安全監視委員会または委員会と呼ばれる試験スポンサーによって組織された独立した合格専門家グループによって監督される。このグループは,そのグループのみが保守している研究から利用可能なデータへのアクセスに基づいて,許可試験が指定されたチェックポイントで行えるかどうかを判定する.
臨床試験の任意の段階で開発を一時停止または終了するには、FDA、IRB、データ安全監視委員会を含む様々な理由があるかもしれない、または参加者または患者が受け入れられない健康リスクに直面していると判断する。我々は、変化するビジネス目標および/または競合環境などの要因に基づいて、一時停止または終了の他の原因を提示する可能性がある。
臨床試験に関する情報は,そのClinicalTrials.govサイト上で公開伝播するために,特定の時間枠内でNIHに提出されなければならない。臨床試験情報を発表する類似要求は欧州連合(EudraCT)サイト:https://eudract.ema.Europa.eu/と他の国/地域にも存在する。
治療のための薬を研究する機会を広げる
使用を拡大することは、“同情的使用”と呼ばれることがあり、登録臨床試験以外に研究新薬製品を使用し、比較可能または満足できる代替治療案がない場合には、深刻または直ちに生命に危害を及ぼす疾患または条件を有する患者を治療する。参入拡大に関する規則と条例は、登録試験と衝突しない研究療法から利益を得る可能性がある患者が薬物を研究する機会を得ることを改善することを目的としている。FDAの法規は、個別患者(緊急時および非緊急時に治療する単患者IND申請);中規模の患者集団;および治療レジメンまたは治療INDに従って薬物の使用を申請するより大きな集団のための、会社または治療医がケースに基づいてIND項下の研究薬物を得ることを可能にする。
30
IND申請の拡大が研究製品を獲得することを審議するとき、FDAは、以下のすべての基準が適用される場合に、その適合性を決定するであろう:患者は、深刻なまたは直ちに生命を脅かす疾患または状態を有し、この疾患または状態を診断、監視または治療するための比較可能または満足できる代替療法がない;潜在的患者の利益は、治療の潜在的リスクが合理的であり、潜在的リスクが治療すべき背景または条件下で不合理ではないことを証明する。要求された治療に対して、研究薬物の使用を拡大することは、製品の発売許可を支持する臨床研究の起動、進行または完成、あるいは他の方法で製品の潜在的な開発を損害する可能性がある。
スポンサーは、その医薬製品をより広範な使用に提供する義務はないが、スポンサーは、2期または3期の臨床試験が開始されるより早い時間に、または薬物または生物学的製剤が突破的療法、迅速チャネル製品または再生医学高度療法として指定された15日後に、その参入拡大政策を公開しなければならない。
NDAを支持するヒト臨床試験
臨床試験は、GCP要求に従って合格した研究者の監督の下でヒト対象に候補研究製品を提供することを含み、すべての研究対象に任意の臨床試験に参加する前に書面でインフォームドコンセントを提供することを含む。臨床試験は書面臨床試験案に基づいて行われ,その中で研究の目標,組み入れと排除基準,安全性モニタリングのためのパラメータと評価すべき有効性基準が詳細に説明されている。
人体臨床試験は通常3つの連続段階に分けて行われるが、これらの段階は重複あるいは組み合わせられる可能性がある。承認された後、追加的な研究が必要かもしれない。
ステップ1臨床試験は最初に限られた人の中で行われ、候補製品の安全性をテストし、副作用、用量耐性、吸収、代謝、健常人或いは患者における分布、排泄と薬効学を含む。第一段階臨床試験期間中に、薬物製品のPKと薬理作用に関する情報を得ることができ、科学的に有効な第二段階臨床試験の設計を可能にする。
第二段階臨床試験は通常限られた患者集団で行われ、可能な副作用と安全リスクを決定し、特定の標的適応に対する候補製品の治療効果を評価し、用量耐性と最適用量を決定する。スポンサーは、より規模が大きく、コストの高い3期臨床試験を開始する前に情報を得るために、複数の2期臨床試験を行うことができる。2期臨床試験は非常に良いコントロールと密接なモニタリングを得て、そして限られた患者群で行った。
第3段階第二段階の臨床試験が候補製品の一定量の範囲が潜在的に有効であり、許容可能な安全性を有することを証明した場合、臨床試験は引き続き行われる。第三段階の臨床試験は拡大した患者群で行われ、更に投与量を評価し、臨床治療効果の実質的な証拠を提供し、そして複数の地理的に分散した臨床試験地点で拡大と多様化した患者群に対して更なる安全性テストを行う。コントロールが良好で、統計的に穏健な3期臨床試験は“キー”と呼ばれ、規制機関が承認されたかどうかを決定し、承認された場合、どのように適切に薬物にラベルを貼るかを決定するためのデータを提供することを目的としている
場合によっては、FDAは候補製品の秘密協定を承認する可能性があるが、承認後の候補製品の安全性および有効性をさらに評価するために、スポンサーに追加の臨床試験を行うことが要求される。この承認後の試験は通常4期臨床試験と呼ばれる。これらの研究は、予想される治療群におけるより多くの患者の治療から追加の経験を得るために使用される。
臨床試験結果を詳細に説明するIND年次報告はFDAに提出しなければならず、IND安全性報告はFDAに以下のいずれかに提出しなければならない:深刻かつ意外な疑わしい副作用;他の研究または動物または体外培養この製品に接触する人体に重大なリスクがあることを表明したテスト;及び方案或いは研究者マニュアルに記載されている場合と比較して、深刻な副作用が疑われる場合にはいかなる臨床上重要な増加が出現した。第1段階、第2段階、および第3段階の臨床試験は、任意の指定された時間内に成功しないか、または全く成功しない可能性がある。FDAは、通常、GCPおよび提出された臨床データの完全性を保証するために、1つまたは複数の臨床場所を検査する。
臨床試験と同時に、会社は通常追加の動物研究を完成させる。彼らはまた,薬物の化学的および物理的特性に関するより多くの情報を開発し,cGMPの要求に応じて最終的に商業量産製品のプロセスを決定しなければならない。製造過程は一貫して高品質の候補薬物ロットを生産することができなければならず、特に最終薬物の識別、強度、品質、純度と効力をテストするための方法を開発しなければならない。また,適切な包装を選択·試験し,候補薬物が賞味期限内に受け入れられない変質が生じていないことを証明するために安定性研究を行う必要がある。
31
小児科研究
2003年の“小児科研究公平法”またはPREAによれば、NDAまたはその付録は、すべての関連する小児科亜集団において主張される適応の安全性および有効性を評価し、安全かつ有効な各小児科亜群に対する製品の用量および投与をサポートするための十分なデータを含まなければならない。スポンサーはまた、研究目標および設計、任意の延期または免除請求、および法規要件の他の情報を含む、提案された小児科研究または申請者計画による研究の大綱を含む小児科研究計画を提出しなければならない。そして、出願人、FDA、FDAの内部審査委員会は、提出された情報を審査し、相互に協議し、最終計画について合意しなければならない。FDAまたは出願人はいつでも計画の修正を要求することができる。
FDAは、成人のために製品が承認されるまで、または小児科データ要件の完全または部分的免除を承認するまで、出願人の要求に能動的にまたは対応することができ、承認は、小児科データの一部または全部の提出を延期することができる。FDAはPREA要求を免除する疾病リストを保持しているが、児童人口中の疾病罹患率が比較的に低いため、孤児薬物指定を獲得した候補製品は通常PREA要求の制限を受けないが、ある分子標的癌適応の治療に使用される孤児指定薬物は免除を受ける資格がない。
機密協定の審査と承認
米国での上場承認を得るためには,FDAにマーケティング申請を提出し,提案された薬物製品の安全性と有効性を証明し,その期待される適応を決定するのに十分なデータを提供しなければならない。この出願は,陰性または不明確な結果および陽性結果,および製品の化学,製造,制御および提案されたラベルなどに関する詳細な情報を含む関連する前臨床試験および臨床試験から得られたすべての関連データを含む。データは、製品の安全性および有効性を試験することを目的とした会社のスポンサーからの臨床試験から来ることができ、独立した調査者によって開始された研究を含む多くの代替源から来ることもできる。上場審査を支持するために、提出したデータは品質と数量で十分でなければならず、薬物製品の安全性、純度と効力を確定し、FDAを満足させる。
NDAはツールであり、申請者はそれを通じてFDAに新薬製品の米国での発売と販売を許可し、1つ以上の適応を得ることを正式に提案する。すべての新しい非生物学的薬剤候補製品は、米国で商業化されることができる前に、承認されたNDAの対象でなければならない。BLASは生物製品の承認に用いられている。連邦法によると、多くのNDAの提出には申請使用料が必要であり、2023年度、臨床データを必要とする申請の使用料は3,242,026ドルである。承認された守秘契約のスポンサーには年度計画費が必要であり,2023年度の年会費は393,933ドルである。その中のいくつかの費用には、特定の例外および免除、例えば、孤児指定製品を有する申請料例外があり、特定の財政年度内に薬物生産に従事しないことが計画されている場合、計画費用例外、およびいくつかの小企業の免除がある。
FDAは申請の予備審査を行い,一般に申請を受けてから60日以内にスポンサー申請が十分であるかどうかを74日以内に通知し,実質的な審査を行うことができるように努力している。FDAは届出申請を受け入れるのではなく、より多くの情報を提供することを要求することができる。この場合、申請は追加情報と共に再提出されなければならない。再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。場合によっては、FDAは、申請が完全ではないと認定し、実質的な審査を許可することができず、提出拒否状を発行する可能性がある。提出された申請が受け入れられると、FDAは深い実質的な審査を開始する。FDAはNDAの審査過程で具体的な業績目標を設定することに同意した。このプロトコルによれば、新規分子実体(NME)の承認を求める申請の90%は、FDAが申請を受けた日から10ヶ月以内に審査され、90%が優先審査に指定されたNME申請は、提出日から6ヶ月以内に審査される。FDAは、出願人によって提供された新しい情報を考慮して、元の提出においてFDAによって発見された不足点を解決するために、または他の理由で、審査プロセスおよび処方薬使用料法案(PDUFA)の目標日を延長することができる。
申請を承認する前に、FDAは通常、製品を生産しているか、または製品を生産する1つまたは複数の施設を検査する。これらの承認前検査は、部品製造、完成品製造、制御試験実験室を含むNDA提出に関連するすべての施設をカバーすることができます。FDAは、製造プロセスおよび施設がcGMP要件に適合していることを決定し、要求された仕様の下で製品が一貫して生産されることを保証するのに十分でない限り、申請を承認しないであろう。さらに、NDAを承認する前に、FDAは、GCPに適合することを確実にするために、通常、1つまたは複数の臨床場所を検査する。
32
FDAは、新製品の申請を諮問委員会に提出したり、なぜそのような推薦がないのかを説明するかもしれない。通常,諮問委員会は臨床医や他の科学専門家を含む独立した専門家からなるグループであり,申請を審査,評価し,申請を承認すべきかどうか,どのような条件でアドバイスを提供すべきかを担当する。FDAは諮問委員会の提案に制約されていないが,FDAは決定を下す際にこれらの提案を慎重に考慮する。
高速道路、画期的な治療法、優先レビュー
FDAには候補製品の開発と審査を加速するための計画があり、これらの候補製品は深刻な或いは生命に危害を及ぼす疾病或いは条件の治療において満たされていない医療需要を解決することを目的としている。これらの計画には、迅速チャネル指定、突破的治療指定、優先審査指定、再生医学高度治療指定が含まれる。スポンサーは開発過程の適切な時点でこのような名前の指定を要求しなければならない。
FDAは、深刻なまたは生命に危険な疾患または状態を治療するために、1つまたは複数の他の製品と単独でまたは組み合わせて使用することが意図されている場合、迅速チャネル審査のための製品を指定することができ、そのような疾患または状態を解決するための満たされていない医療需要の可能性を示す。Fast Track製品については,スポンサーがFDAとより多くのインタラクションを行う可能性があり,FDAは申請完了前にFast Track製品申請部分の審査を開始する可能性があり,この過程をスクロール審査と呼ぶ.スポンサーはまた、残りの情報を提出するスケジュールを提供しなければならず、FDAの承認を得なければならず、スポンサーは適用された使用料を支払わなければならない。しかしながら、FDAが高速チャネル申請のPDUFA目標を検討することは、申請の最後の部分が提出されてから開始される。また,FDAが高速チャネルの指定が臨床試験中に出現したデータや他の原因の支持を得なくなったと考えた場合,その指定を撤回する可能性がある。
1つの製品が単独で、または1つまたは複数の他の製品と組み合わせて、深刻または生命に危険な疾患または状態を治療するために使用され、予備臨床証拠が、1つまたは複数の臨床的重要終点において、例えば、臨床開発早期に観察された実質的な治療効果のような既存の療法よりも有意な改善を示す可能性があることを示す場合、製品は突破的療法として指定することができる。画期的な治療認証を取得した製品には,迅速チャネル認証のすべての機能を取得する資格があり,また,開発過程全体で密な指導を受ける資格があり,高度な従業員の参加を約束している。
FDAは、重篤な疾患を治療する場合、承認された場合、安全性または有効性の面で顕著な改善を提供する製品を優先的に審査することができる。FDAは具体的な状況から,他の利用可能な療法と比較して,提案された製品が有意な改善を表すかどうかを決定している。顕著な改善は,疾患治療の有効性の増加,治療を制限する産物反応の除去あるいは大幅な減少,記録されている患者のコンプライアンスの向上が重篤な結果の改善をもたらす可能性があること,新たな亜群における安全性と有効性の証拠であることが示唆された。優先審査を指定する目的は、全体的な注意とリソースをこのような申請の評価に誘導し、FDAがマーケティング申請に行動する目標を10ヶ月から6ヶ月に短縮することである。
承認ルートを加速する
FDAは、患者に既存の治療よりも意義のある治療優位性を提供する深刻または生命に危険な疾患の承認を加速する可能性があり、その基礎は、この製品が代替終点に影響を与えることを決定することであり、疫学、治療学、病理生理学または他の証拠に基づいて、この終点は臨床利益を合理的に予測する可能性がある。中間臨床終点に対する製品の影響が不可逆的な発病率または死亡率またはIMMへの影響よりも早いことができ、このような状況の重症度、希少性または流行率、および代替治療が利用可能または不足している場合を考慮すると、IMMまたは他の臨床的利益への影響を合理的に予測することが可能である場合、FDAはこのような状況の加速承認を許可することもできる。加速された承認を受けた製品は、従来承認された製品と同じ安全性と有効性法定基準に適合しなければならない。
承認を加速するために、代替終点は、実験室測定、放射線画像、バイタルサイン、治療効果バイオマーカー、または臨床的利益を予測できると考えられる他のマーカーであるが、それ自体は臨床的利益の測定基準ではない。代替終点は通常、臨床終点よりも容易または迅速に測定される。
加速承認経路は疾病の病気経過が比較的に長く、製品の期待される臨床利益を測定するために時間を延長する必要がある環境に最もよく用いられ、代用或いは中間臨床終点への影響は非常に速く発生した。承認を加速させる利点は、優先順位審査のように、FDAの承認スケジュールを明確に短縮することで承認を得るのではなく、臨床的または生存終点を有する試験について、代替終点に基づく承認をより迅速に得ることができることである。
33
承認を加速する方法は、製品の臨床的利益を検証および説明するために、勤勉な方法で追加的な承認後の検証的研究を行うことにスポンサーが同意することに依存する。そのため、この基礎の上で承認された候補製品は必ず厳格な発売後のコンプライアンス要求を守らなければならず、4期或いは承認後の臨床試験を完成し、臨床終点への影響を確認することを含む。2022年食品·薬物総合改革法案によると、FDAは現在、このような試験を承認前または承認加速日後の特定の期間内に行うことを適宜要求されている。FDORAによれば、FDAは、例えば、検証試験が製品の予期される臨床的利益を検証することができない場合、製品の承認を撤回することができるように、手続きを加速する権限を増加させる。さらに、機関が別途通知しない限り、FDAは通常、製品の商業発売時間に悪影響を及ぼす可能性がある販売促進材料の事前承認を要求する。
FDAの秘密保持協定に関する決定
FDAによる出願の評価及び付随する情報によれば、製造施設の検査結果を含めて、FDAは承認状又は完全な返信を発行する可能性がある。承認書は、製品の商業マーケティングを許可し、特定の適応に関する具体的な処方情報を提供する。完全な応答文は、一般に、提出文書の不足点を概説し、FDAが出願を再検討するために、多くの追加のテストまたは情報を必要とする可能性がある。NDAを再提出する際に,これらの不足点がFDAによって満足的に解決されれば,FDAは承認書を発行する。FDAは、含まれる情報タイプに応じて、そのような再提出された出願を2ヶ月または6ヶ月以内に検討することを約束している。この補足情報を提出しても、FDAは最終的にその申請が承認された規制基準を満たしていないと決定する可能性がある。
FDAが新製品を承認した場合、製品の承認適応を制限する可能性があり、製品ラベルに禁忌症、警告または予防措置を含むか、または承認後の薬物の安全性をさらに評価するための第4段階臨床試験を含む承認後の研究を要求する可能性がある。この機関はまた、製品が商業化された後にそれを監視するために、または製品の利益が潜在的リスクよりも大きいことを保証するために、流通制限または他のリスク管理メカニズムを含む他の条件を適用することをテストおよび監視計画に要求することができる。REMS計画は、薬物ガイドライン、衛生保健専門家のコミュニケーション計画、安全使用を確保する要素、またはETASUを含むことができる。ETASUは、処方または調剤に関する特別なトレーニングまたは認証、場合によっては調剤、特殊監視、および特許登録所の使用を含むことができるが、これらに限定されない。FDAは発売後の研究或いはモニタリングプロジェクトの結果に基づいて、製品の更なるマーケティングを阻止或いは制限することができる。FDAが製品の使用に関連する深刻なリスクを認識している場合、それは承認の前または後にREMSを要求する可能性がある。REMSへの要求は製品の潜在的な市場と収益力に大きな影響を与える可能性がある。承認後、承認製品の多くのタイプの変更、例えば、新しい適応の増加、製造プロセスの変更、ラベル宣言の追加など、さらなるテスト要件とFDAの審査と承認を受けなければなりません。
承認後の規則
監督管理機関の許可を得た新製品の発売或いは既存製品の新適応のスポンサーは、多くの審査後の監督管理要求の制約を受ける。スポンサーは、いくつかの副作用や製造問題をFDAに報告し、最新の安全性と有効性情報を提供し、広告や販売促進ラベル要求に関する要求を遵守し、NDA年度報告書を提出することが求められる。製造業者およびその特定の下請け業者は、製品、成分およびコンポーネントを提供する下請け業者を含み、FDAおよびいくつかの州機関にその工場を登録し、製造業者に特定の手順および文書要件を適用するcGMP法規を含む持続的な法規要件を遵守することを確実にするために、FDAおよび特定の州機関の定期的な抜き打ち検査を受けなければならない。製造プロセスの変更は厳しく規制されており,通常FDAが事前に承認して実施する必要がある。したがって、スポンサーおよびその第三者メーカーは、cGMP法規および他の法規要件の遵守を維持するために、時間、お金、エネルギーをかけ続けなければならない。
規制要求を遵守していない場合や、製品発売後に問題が発生した場合、FDAは承認を撤回する可能性がある。その後、製品には、予期されない重症度または頻度の不良事象、または生産プロセスを含む以前に未知の問題が存在することが発見され、新しいセキュリティ情報、発売後の研究または臨床試験の要求を追加して安全リスクを評価するために承認されたラベルの改訂をもたらす可能性があり、またはREMS計画に従って流通または他の制限を実施する可能性がある。他の潜在的な結果には
34
FDAは市場に投入された処方薬製品のマーケティング、ラベル、広告、販売促進を厳格に規制している。他の事項に加えて、この規定には、消費者向け直接広告、未承認用途に関する通信、業界スポンサーに関する科学的および教育活動、ならびにインターネットおよびソーシャルメディアに関連する販促活動の基準および規定が含まれる。薬物が承認されるまで,薬物の安全性や有効性の宣伝は禁止されている。承認された後、医薬品は一般にFDAの承認されていない使用に使用されてはならず、これはこの製品の処方情報に反映される。米国では、医療専門家は、FDAが医薬品の使用を規制していないので、医薬品ラベルに記載されていないこのような用途のための処方、いわゆるラベル外用途を許可されている。しかし、FDAの規定はメーカーのコミュニケーションに厳しい制限を加え、ラベル外の使用を禁止している。非常に特殊な場合、製造業者は、ラベル外情報に関する非販促、非誤解的伝播、例えば、科学または医学定期刊行物情報の配信に従事することを可能にすることができる。
もしある会社がラベル外の使用を促進したことが発見された場合、それはFDA、司法省、米国司法省、衛生·公衆サービス部監察長事務室、および州当局の行政と司法執行を受ける可能性がある。これは、民事および刑事罰金、および会社の薬品の宣伝または流通を実質的に制限する方法の合意を含む一連の重大な商業的影響を及ぼす可能性のある一連の処罰を企業に受ける可能性がある。連邦政府は、不当な販売促進の疑いのある会社に対して巨額の民事と刑事罰金を科し、企業に同意法令または永久禁止令を締結し、これらの法令または永久禁止に基づいて、特定の販売促進行為を変更または制限することを要求する。
また、処方薬製品の製造業者および他の薬品サプライチェーンに参加する各当事者は、製品追跡と追跡要求を遵守し、偽、移転、窃盗、故意に混入した製品または本来米国で流通するのに適していない製品をFDAに通報しなければならない。
第五百五十五条第二項新開発区
多くの新薬製品のNDAは2つの全面的な臨床研究に基づいており、この2つの研究は提案されて使用された新製品の安全性と有効性を提案する大量の証拠を含まなければならない。これらの出願はFDCA第505(B)(1)条に基づいて提出された。しかしながら、FDCA第505(B)(2)条によれば、FDAは、代替タイプのNDAを承認することを許可される。このようなタイプの出願は、出願人が以前に承認された医薬製品(市販薬とも呼ばれる)の安全性および有効性の発見または文献にある程度依存することを可能にする。具体的には、第505条(B)(2)条は、薬物が安全で有効であるか否かを証明するために行われるいくつかの調査“出願人又は出願人のために行われたものではなく、出願人が調査を行った者又はそれのために調査を行った者から転介又は使用の権利を得ていない”場合に適用される
第505条(B)(2)条に従って提出された国家医薬品監督管理局は、以前に承認された製品の新しいまたは改善された処方または新しい用途のために、FDAの承認を得るために、より迅速である可能性のある別の方法を提供する。505(B)(2)出願人がFDAの以前の承認に依存することが科学的に適切であることを証明することができる場合、出願人は、新しい製品のいくつかの臨床前または臨床研究の必要性を除去することができる。FDAはまた、承認された製品からの変更をサポートするために、追加の研究または測定を行うことを企業に要求する可能性がある。次に、FDAは、市販薬が承認されたすべてまたは部分的なラベル適応の候補新薬を承認することができるが、上場薬物の任意の規制排他性または特許(以下に述べる)、および505(B)(2)条の出願人が求める任意の新しい適応または使用を遵守しなければならない。
後発薬の略新薬申請
1984年、FDCAに対するHatch-Waxman修正案の採択に伴い、国会は短い規制計画を確立し、FDA承認がNDAによって以前に承認された薬物と同じ有効成分を含み、生物学的同等性を有する模倣薬を参照市販薬、またはRLDと呼ばれることが証明された。新薬申請、またはANDAを簡単に書くと、通常、安全性と有効性を証明する臨床前および臨床データは含まれていない。逆に、申請者は情報とデータを提供しなければならず、その提案された模倣薬は有効成分、投与経路、剤形、強度と使用条件の面でRLDと同じであることを示した。FDAはまた,後発薬がRLDと生物学的同等性を有するかどうかを確認しなければならない。この法規によると、模倣薬の吸収速度と程度が市販薬物の吸収速度と程度と有意差がなければ、この模倣薬は生物的にRLDと同等である。ANDAが承認された後FDAは
35
後発薬は治療上RLDとその出版物“治療同等性評価を有する承認薬物製品”における“治療同等性”であり,“オレンジ書”とも呼ばれる。州法によると,治療上等価な後発薬は,処方者の介入を必要とすることなく,調剤薬剤師がRLDの処方を自動的に代替できることが発見された。
Hatch−Waxman修正案によれば、FDAは、RLDの任意の適用可能な非特許固有期間が満了するまでANDAを承認しない可能性がある。FDCAは新しい化学実体を含む新薬に5年間の非特許データ排他性を提供した。本条項の場合、新しい化学物質またはNCEは、FDAが任意の他のNDAで以前に承認された活性部分を含まない薬剤を意味する。活性部分は薬物物質の生理的あるいは薬理作用を担う分子またはイオンである。このようなNCE排他性が付与された場合、ANDAは、提出された書類に第4項の証明が添付されていない限り、5年の満了前にFDAに提出することができず、この場合、出願人は、原製品の承認後4年以内に出願を提出することができる。
Hatch-Waxman特許認証と30ヶ月の滞在
NDAまたはその付録が承認された後、NDAスポンサーは、各特許をFDAに列挙し、その権利が出願人の製品または承認された製品使用方法をカバーすることを要求しなければならない。NDAスポンサーにリストされたすべての特許はオレンジ色の本に発表されている。ANDA出願人がFDAに出願を提出する場合,出願人はオレンジマニュアルに記載されている参照製品の任意の特許をFDAに証明しなければならないが,ANDA出願人が承認を求めていない使用方法の特許は除外される。第505条(B)(2)の出願人が承認された製品の研究に依存している場合,出願人は,ANDA出願人と同様に,オレンジブックに記載されている承認製品の任意の関連特許をFDAに証明しなければならない。
具体的には、出願人は、各特許証明について:
新製品が承認された製品の上場特許又はそのような特許を侵害しないか、又は強制的に実行できない認証を第4項認証と呼ぶ。出願人が列挙された特許に疑問を提起していない場合、参照製品を必要とするすべての特許(出願人が承認を求めていない適応に関連する使用方法特許を除く)が満了するまで、その出願は承認されないであろう。
ANDA又は第505条(2)のNDA出願人が第4項の認証をFDAに提供した場合,FDAが出願の届出を受けると,出願人はまた,NDA及び特許所持者に第4項の認証の通知を送信しなければならない。そして、国家特許庁及び特許所有者は、第4項の通知の通知に対して特許侵害訴訟を提起することができる。第四項の認証を受けてから45日以内に特許侵害訴訟を提起することは、FDAがANDA又は505(B)(2)の出願を承認することを自動的に阻止し、第四項の通知、特許満了又は侵害事件においてANDA出願人に有利な裁決を受けてから30ヶ月前の者までである。
したがって、オレンジブックに記載された参照製品の任意の非特許排他性、例えば、新しい化学物質の承認を得る排他性が期限切れになるまで、参照製品のすべてのリストされた特許が満了するまで、条項505(B)(2)NDAまたはANDAの承認が延期される可能性があり、または第4段落の認証およびその後の特許侵害訴訟の場合、訴訟または侵害事件において出願人に有利な裁決のより30ヶ月前に和解が成立するまでである。
小児科排他性
小児科排他性は、承認された場合、既存の規制排他性または特定の特許の期間にわたって6ヶ月のマーケティング保護を追加的に提供することができる米国の別の非特許マーケティング排他性である。NDAスポンサーから提出された小児科データがこのようなデータに対するFDAの書面要求に公平に応答すれば,この6カ月の排他性を与えることができる。これらのデータは,この製品が研究されている小児科群で有効であることを証明する必要はなく,逆に臨床試験がFDAの要求に公平に応答していると考えられれば,追加的な保護が得られる。
36
孤児薬の指定と排他性
孤児医薬品法によれば、1つの医薬製品がまれな疾患または疾患の治療を目的としている場合、FDAは、それを“孤児薬”として指定することができ、これは、一般に、米国で影響を及ぼす個人が20万人未満であることを意味するか、または合理的な予想がない場合、このような疾患または疾患を治療するための製品を米国で開発および生産するコストが製品の販売から回収される場合、医薬製品は“孤児薬”として指定されることができる。候補製品の秘密保持協定を提出する前に、会社は孤児薬物指定を求めなければならない。この要求が承認された場合、FDAは、治療薬の識別情報およびその潜在的用途を開示するであろう。孤児薬物指定は、税金優遇およびPDUFA申請料の免除などのいくつかの利点を確実に伝達しているにもかかわらず、PDUFA規制審査および承認プロセスの目標日を短縮しない。
孤児として指定された製品が、このような指定された疾患または状態を有するFDAの最初の承認を受けた場合、またはまれな疾患または状態に対する適応または使用として指定された場合、製品は、通常、孤児薬物排他性を得る。孤児薬物排他性とは、FDAが、いくつかの限られた場合を除いて、同じ疾患に対する別のスポンサーの同じ薬物の発売申請を7年以内に承認しない可能性があることを意味する。孤児の排他性は同一の稀な疾病或いは疾病の異なる製品に対する承認を妨げることはなく、同一製品の異なる状況に対する承認を阻止することもない。孤児薬として指定された薬物が最終的に市販承認された場合,その適応範囲は孤児薬物申請で指定された範囲よりも広く,排他性を得る資格がない可能性がある。
特定の場合、孤児薬独占性も、より良い治療効果または安全性を有するため、または患者ケアに大きな貢献を果たしているため、同じ場合に同じ薬物を使用する後続製品が臨床的に承認された製品よりも良いことが証明された場合、または患者ケアに重大な貢献をした場合、または孤児薬独占性を有する会社が市場ニーズを満たすことができないことを含む、別の製品の承認を阻止することもできない。
特許期限の回復と延長
ハッジ·ワックスマン法案によると、新薬製品を持つ特許は、FDA規制審査中に失われた特許期間が5年間に及ぶ特許を回復することを可能にする限られた特許期間延長を受ける資格がある可能性があると主張している。製品をカバーする特許付与の回復期は、通常、IND発効日と出願提出日との間の時間の半分であり、出願提出日と最終承認日との間の時間を加える。特許期間回復は特許の残存期間の延長には利用できず,製品承認日から合計14年を超える。承認された製品に適用される特許は1つのみ延期する資格があり、承認された製品、その使用方法、または製造方法に関する権利要件のみを延期することができる。しかも、延期申請は関連特許が満期になる前に提出されなければならない。複数の製品をカバーする特許は、そのうちの1つの承認に関連して延期することしかできない。米国特許商標局は,FDAと協議した後,任意の特許期間の延長または回復の出願を審査·承認する。
医療保健法と法規
医療保健提供者と第三者支払人は推薦と処方が発売承認された薬品の面で主要な役割を果たしている。提供者、コンサルタント、第三者支払者および顧客との配置は、詐欺および乱用、リベート、虚偽申告法、患者プライバシー法律および法規、ならびに業務および/または財務手配を制限する可能性のある他の医療法律および法規の制約を広く適用される。適用される連邦と州の医療法律と規制によると、
37
また、いくつかの州の法律は、製薬会社が製薬業界の自発的コンプライアンスガイドラインと連邦政府が公布した関連コンプライアンスガイドラインを遵守することを要求し、また、メーカーに医師および他の医療保健提供者への支払いまたはマーケティング支出に関する情報を報告することを要求している。また、いくつかの州と地方の法律はこの司法管轄区に薬品販売代表を登録することを要求している。
州法はまた、健康情報を含む個人情報のプライバシーとセキュリティを管理する。多くの州の法律は大きく異なり、コンプライアンス作業を複雑にしている。例えば、カリフォルニア消費者保護法(CCPA)は、カリフォルニアに位置する個人のためのデータプライバシー権を確立し、企業がこのような個人の個人情報をどのように収集して使用するかを要求している。2023年1月1日に施行されたカリフォルニアプライバシー権法案(CPRA)は、立法がカバーする会社に追加的な義務を課し、特定の敏感な個人情報に対する消費者の権利を拡大し、CCPAを実行する権利を有する州機関を構築することを含むCCPAの重大な修正を行った。CCPA(CPRA改訂により)は、他の州に類似した全面的なプライバシーとデータ保護立法を公布することを促した。例えば、バージニア州は2021年3月に消費者データ保護法(CDPA)を公布し、2023年1月1日に施行された。2021年7月、コロラド州では2023年7月1日に施行される“コロラド州プライバシー法案”(CPAと略す)が可決された。また、ユタ州は2022年3月に2023年12月31日に施行される“ユタ州消費者プライバシー法”(UCPA)を公布した。また、2022年5月、コネチカット州は2023年7月1日に発効するコネチカット州データプライバシー法案(CTDPA)に署名した。また,米国の他のいくつかの州でも同様のプライバシーやデータ保護立法が提案されており,いくつかの提案が採択される可能性がある.多くの既存の州プライバシー法はHIPAAが管轄する臨床試験情報や健康情報を免除しているにもかかわらず,将来のプライバシーやデータ保護法の範囲が広くなる可能性がある。さらに進む, 外国司法管轄区域のデータプライバシーおよびセキュリティ法律法規は、EUの一般的なデータ保護条例またはGDPR、およびイギリスのこの条例の実施のような米国の義務よりも厳しいまたは異なる可能性がある個人情報の収集、使用、および他の処理に追加的な義務を課す可能性がある。
38
医薬保険カバー面と医療改革
米国や他国の市場では,自分の病状に応じて処方治療を受けている患者や処方サービスを提供する提供者は,通常第三者支払者に依存してすべてまたは一部の関連医療費を精算している。FDAや他の政府当局が承認した製品のカバー範囲や精算状態には大きな不確実性がある。したがって、候補製品が承認されても、製品の販売は、MedicareおよびMedicaid、商業健康保険会社および管理型医療機関などの米国の政府医療計画を含む第三者支払者にある程度依存し、その製品に保険を提供し、十分な補償レベルの程度を確立する。支払者が商品に保険を提供するか否かを判定するプロセスは、保険が承認された後に支払者が製品に支払う価格または販売率を設定するプロセスと分離することができる。第三者支払者は、徴収された価格に挑戦し、医療の必要性を審査し、医療製品およびサービスの費用対効果を審査し、コストを管理するために制御を実施することが増えている。第三者支払者は、保証範囲を承認リスト上の特定の製品に制限することができ、特定の適応のすべての承認製品を含まない可能性がある配合表とも呼ばれる。
販売が許可される可能性のある任意の製品の保険·精算を確保するためには、企業は、製品の医療必要性および費用効果、およびFDAまたは他の同様の市場承認を得るために必要なコストを証明するために、高価な薬物経済学的研究を行う必要があるかもしれない。それにもかかわらず、候補製品は医学的に必要または費用効果があると考えられないかもしれない。製品が承認されると、第三者支払人による不保証製品の決定は医師の利用率を低下させ、販売、運営結果、財務状況に実質的な悪影響を与える可能性がある。また、支払者が製品に保険を提供することを決定することは、十分な返済率を承認することを意味するものではない。また、1人の支払人が1つの製品に保険を提供することを決定することは、他の支払者もその製品に保険や精算を提供することを保証することはできず、また、支払者によって保険や精算レベルが大きく異なる可能性がある。
医療費の抑制も連邦,州,外国政府の優先順位となっており,製品価格はこの努力の重点となってきた。各国政府はコスト制御計画の実施に大きな興味を示し、価格制御、精算制限と代替後発薬の要求を含む。価格制御及びコスト制御措置を講じ、既存の制御及び措置を有する司法管区においてより限定的な政策をとることにより、任意の承認された製品の販売から得られる収入をさらに制限することができる。保証政策と第三者精算料率は随時変化する可能性がある。1つの会社またはその協力者がマーケティング承認を得た1つまたは複数の製品が有利な保証·精算状態を獲得しても、将来的にはあまり有利ではない保証政策および精算料率を実施することが可能である。
過去数年間に、連邦と州政府は多くの提案を提出し、薬品と生物製薬製品の定価制限、薬品、生物製品とその他の医療製品のカバー範囲と精算範囲、政府制御及びアメリカ医療保健システムの他の改革に関連した。
2010年、米国議会は政府医療計画下の薬品のカバー範囲の変更と支払い方法を含むACAを公布した。ACAにおいて私たちの潜在的な製品候補製品に重要な意味を持つ条項は、以下のことを含む
ACAが公布されて以来、アメリカはまた他の立法改正を提案し、採択した。2011年の“予算制御法案”(Budget Control Act)などの法案は国会の支出削減に向けた措置を策定した。これには、毎年度にプロバイダに支払われる連邦医療保険総額が2%まで減少することが含まれている。その後の立法は2%の有効期間を2030年に延長する。2012年の米国納税者救済法は医療保険支出をさらに減少させた
39
いくつかのタイプの提供者が、政府が提供者に多額の金を取り戻す訴訟時効期間を3年から5年に延長する。これらの法律は、連邦医療保険および他の医療資金のさらなる減少をもたらし、他の方法で、規制によって承認される可能性のある任意の候補製品の価格、またはそのような任意の候補製品の処方または使用頻度に影響を与える可能性がある。
2022年の“インフレ率低減法案”(IRA)中のいくつかの条項は私たちの業務に異なる程度の影響を与える可能性があり、その中には2025年から連邦医療保険D部分の受益者の自己負担上限を2,000ドルに下げることを含む;連邦医療保険D部分下のある薬物に新しいメーカー財務責任を適用する;アメリカ政府はある高コストの薬物と生物製品のB部分とD部分の価格上限について交渉することを許可し、模造薬や生物類似競争が存在しない;会社にインフレより速い価格増加の薬品の連邦医療保険へのリベートを要求し、薬局福祉マネージャーが費用を徴収できるリベート規則を延期することを要求する。また,IRAによると,孤児薬は連邦医療保険薬品価格交渉計画の影響を受けないが,まれな疾患の名称があることを前提としており,唯一承認された適応はこの疾患や条件に対するものである。1つの製品が複数のまれな疾患の指定または複数の承認された適応を得た場合、孤児薬免除を受ける資格がない可能性がある。Ireland共和軍が私たちの業務と医療産業全体に及ぼす影響はまだ明確ではない。
アメリカでも、処方薬のコストはかなり議論されてきた。これまで、アメリカ議会は最近数回の調査を行い、連邦と州立法を提出し、公布し、薬品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、連邦医療保険下の薬品のコストを下げ、政府の薬品に対する計画精算方法を改革することを目的とした。バイデン総裁はすでに複数の行政命令を発表し、処方薬のコストを削減しようとしている。いくつかの措置および他の提案された措置は発効するために追加の立法によって許可される必要があるかもしれないが、バイデン政府はこれらの措置を撤回または他の方法で変更する可能性があるが、バイデン政府と国会は薬品コストを制御するための新しい立法措置を求め続けると表明している。
州レベルでは、各州はますます積極的に、価格または患者の精算制限、割引、ある製品への参入の制限、マーケティングコストの開示および透明性措置を含む薬品および生物製品の価格設定を制御するための法規を立法と実施し、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。また、地域衛生保健当局や個別病院は、どの薬品やサプライヤーが彼らの処方薬や他の医療計画に含まれるかを決定するために入札プログラムを使用することが増えている。これらの措置が承認されると、私たちの製品に対する最終的な需要を下げたり、私たちの製品の価格設定に圧力をかけるかもしれません。私たちは将来、より多くの州と連邦医療改革措置を取ることが予想され、そのいずれも連邦と州政府が医療製品やサービスのために支払う金額を制限する可能性があり、これは私たちの候補製品に対する需要の減少または追加の価格設定圧力を招く可能性がある。
EUの医薬製品の審査と承認
アメリカ国外で任意の製品をマーケティングするために、会社はまた他の国と司法管轄区域の品質、安全性と有効性及び製品に対する臨床試験、マーケティング許可、商業販売と流通などの方面の多くの異なる監督管理要求を守らなければならない。FDAによる製品の承認を得るか否かにかかわらず、出願人は、非米国規制機関のような必要な承認を得る必要があり、その後、これらの国または司法管轄区で製品の臨床試験またはマーケティングを開始することができる。承認過程は最終的に国と司法管轄区によって異なり、追加の製品テストと追加の行政審査期限に関連する可能性がある。他の国や管轄地域で承認を得るのに要する時間は、FDAの承認を得るのに要する時間とは異なり、さらに長くなる可能性がある。1つの国または管轄区域で規制承認を得ることは、他の国または管轄区域で規制承認を得ることを保証することはできないが、1つの国または管轄区域で規制承認を得ることができなかったか、または遅延して監督管理許可を得ることは、他の国または司法管轄区の規制手続きに悪影響を及ぼす可能性がある。しかし、具体的には、EU(European Union)の医薬製品の承認の流れはアメリカとほぼ同じである。製品の安全性と有効性の各提案の適応を決定するために、臨床前研究と十分かつ良好に制御された臨床試験を満足できるように完成させる必要がある。また、関連主管当局にマーケティング許可申請、またはMAAを提出し、これらの主管部門からマーケティング許可を付与し、その後、製品をEUで販売·販売することができるように要求されている。
臨床試験許可
2014年4月,EUは新たな臨床試験条例を採択し,(EU)第536/2014号は,2022年1月31日に臨床試験指令2001/20/ECに代わった。臨床試験条例はすべてのEU加盟国に直接適用され、これは各EU加盟国で国家立法を制定する必要がないことを意味する。新しい臨床試験条例はEUの臨床試験の審査を簡略化と簡素化することを目的としている。 新しい臨床試験承認調整プログラムによると、臨床試験の発起人は必ず臨床試験承認申請を提出しなければならない
40
欧州連合ポータルサイトを通じて報告書を提出したEU加盟国に裁判を行う。臨床試験は一つのEU加盟国でも複数のEU加盟国でも行われ、提出手続きは同じだ。
EUのトップクラスの称号
2016年3月,EMAは適応候補の開発を促進するイニシアティブを開始したが,これらの適応はまれであり,現在のところ治療法はほとんどない。優先薬品計画は満たされていない医療需要領域の薬物開発を奨励し、重大な革新を代表する製品に加速評価を提供することを目的としており、これらの製品の中で、上場許可申請は集中プログラムによって行われる。条件に適合する製品は、満たされていない医療需要が存在する条件(EUには満足できる診断、予防または治療方法がない、または、ある場合、新薬は重大な治療優位性をもたらす)に対して必要であり、それらは満たされていない医療需要を満たす潜在力を示さなければならない。中小企業の製品は大企業よりも早くPrime計画に参加する資格があるかもしれません。Primeの称号を持つ候補製品のスポンサーは多くのメリットを得ることができ、しかし限らず、早期にEMAと積極的な監督対話を行い、臨床試験設計とその他の開発計画要素を頻繁に討論し、及びファイルを提出した後にMAA評価を加速する。重要なのは、ヒト医薬製品委員会(CHMP)または高級治療委員会の専任EMA連絡先と調査委員がPrime計画の早期に任命され、EMA委員会レベルで製品のより多くの理解を促進することである。会議を開始してこれらの関係を開始し、EMAを含む1つの多学科専門家チームは、全体発展と監督戦略に関する指導を提供する。研究開発過程において、もし1種の薬物が資格標準に符合しなくなったら、Prime計画下の援助を取り消すことができる。
マーケティング許可
EU規制制度の下で製品のマーケティング許可を得るためには,出願人はMAAを提出しなければならないか,EMAによって管理されている中央手続きか,EU加盟国主管部門が管理する手続きのうちの1つ(分散手順,国家手続き,または相互承認手続き)でなければならない。マーケティング許可は連合に設立された申請者にのみ付与されることができる。(EC)第1901/2006号条例は、EUの上場許可を得る前に、出願人は、(1)特定の製品免除、(2)カテゴリ免除、または(3)PIPに含まれる1つまたは複数の措置を延期しない限り、EMAによって承認された小児科人口のすべてのサブクラスをカバーする小児科調査計画(PIP)に含まれるすべての措置を証明しなければならないと規定している。
中央手続きは、EU全体および欧州経済圏の他の加盟国アイスランド、リヒテンシュタイン、ノルウェーに有効な単一マーケティング許可を欧州委員会によって付与することを規定している。(EC)第726/2004号条例によれば、特定の製品については、いくつかのバイオテクノロジーによって生産された薬剤、孤児薬として指定された製品、高度な治療薬製品(遺伝子治療、体細胞治療または組織工学薬)、およびHIV、エイズ、癌、神経変性疾患、糖尿病、自己免疫および他の免疫機能障害、ならびにウイルス性疾患を治療するための新しい活性物質を含む製品は、集中手順を強制的に実行しなければならない。EUで許可されていない新しい活性物質を含む製品、または重大な治療、科学的または技術革新、またはEUの公衆衛生上の利益に適合する製品については、集中手順がオプションである。私たちは私たちが開発している候補製品について集中化手続きが義務的になると予想する。
中央手続きの下で、CHMPは、製品の予備評価を担当し、既存のマーケティング許可の修正または拡張を評価するなど、いくつかの許可後および保守活動を担当する。EUの中央手続きによると、環境評価機関が重大な影響評価を評価する最長期限は210日であり、タイミングポーズは含まれておらず、申請者はCHMPからの質問に答えるために追加的な書面または口頭情報を提供する。タイミングを停止することは、MAAを評価する時間フレームを210日を超えるように大幅に延長する可能性がある。CHMPが肯定的な意見を与えた場合、それは、EMAの提案を受けて67日以内にマーケティング許可の最終決定を発行する支援ファイルと共にEU委員会に提供される。特殊な情況下で、公共衛生の角度から、特に治療革新の角度から、ある医薬製品が重大な利益を持つことを期待すれば、CHMP許可によって加速的に評価することができる。CHMPがこの要求を受信すると、210日のタイムリミットは150日に減少し、クロック停止を含まないが、CHMPが加速評価に適していないと考えた場合、中央プログラムの標準タイムリミットに回復することもできる。
41
欧州委員会はいわゆる“特別な場合のマーケティング許可”を承認するかもしれない。このような許可は、製品に関する適応が非常にまれであるため、出願人が正常な使用条件下で治療効果および安全性に関する包括的なデータを提供できないことを証明することができる製品に適用され、申請者が包括的な証拠を提供することを合理的に期待することができない、または現在の科学的知識状態では、全面的な情報を提供することができない、またはそのような情報を収集することは、公認された医学道徳原則に違反するであろう。したがって、特別な場合には、いくつかの特定の義務の制約の下でマーケティング許可を承認することができ、これらの義務は、以下を含むことができる
特別な場合、上場許可は年度見直し手続きでリスクと収益のバランスを再評価するために年間審査を受けなければならない。ライセンスの継続は年間再評価と関連があり、否定的な評価はマーケティング許可が一時停止または撤回される可能性がある。しかし、特殊な場合、医薬製品の発売許可の継続は“正常”販売許可と同じ規則に従う。したがって、特別な場合のマーケティング許可は最初は5年であり、その後のライセンスは、EMAがセキュリティ理由を決定しなければ、さらに5年間延長する価値がある限り、無期限に発効する。
欧州委員会はまた、包括的なマーケティング許可を申請するために必要な包括的な臨床データを得る前に、いわゆる“条件付きマーケティング許可”を承認することができる。(I)候補製品のリスク-利益バランスが積極的である場合、(Ii)出願人が許可後に必要な全面的な臨床試験データを提供することができる可能性が高い場合、(Iii)この製品は、満たされていない医療需要を満たし、(Iv)関連医薬製品の即時発売による公衆健康への利点が、依然として追加のデータを必要とする固有のリスクを超えている場合、深刻な衰弱または生命に危険な疾患(孤児薬として指定された薬剤を含む)の治療、予防または診断のために使用される候補製品に、このような条件付きマーケティング許可を付与することができる。条件付き販売許可には,現在行われているあるいは新たな研究の完了や薬物警戒データの収集義務など,販売許可保持者が履行しなければならない具体的な義務が含まれていることができる。条件付きマーケティング許可の有効期間は1年であり、リスク−収益バランスが正のままであり、追加または修正された条件および/または特定の義務の必要性が評価された後、毎年更新することができる。上記の集中プログラムのスケジュールは、条件付きマーケティング許可申請の審査にもCHMPに適用される。上場許可所有者が規定の義務を履行し、完全なデータが薬品の利益がそのリスクよりも引き続き大きいことを確認すると、条件付きマーケティング許可は標準的な集中マーケティング許可に変換することができる(これ以上特定の義務の制約を受けない)。
EU薬品規則は、EU加盟国が国家立法を通じて、胚性幹細胞のような特定のタイプのヒトまたは動物細胞を含む任意の医療製品の販売、供給、または使用を制限、供給または使用することを明確に許可する。我々が開発している製品は胚性幹細胞を使用していないが、いくつかのEU加盟国の法律は、EUのマーケティング許可を得ていても、製品を商業化することを禁止または制限する可能性がある。
集中認証プログラムとは異なり、分散マーケティング許可プログラムは、製品販売が存在する各EU加盟国の主管当局に個別に申請し、それによって単独で承認する必要がある。この申請は,中央プログラムを介して環境管理機関に提出して許可する申請と同様である。EUは加盟国が有効な申請を受けてから120日以内に評価草案と関連材料の草稿を作成することを参考にする。このようにして生成された評価報告書は関係EU加盟国に提出され、これらの加盟国は評価報告および関連材料を受け取ってから90日以内にその報告書および関連材料を承認するかどうかを決定しなければならない。もしEU加盟国が公衆健康の潜在的な深刻なリスクに対する懸念から評価報告書や関連材料を承認できない場合、論争内容はすべてのEU加盟国に対して拘束力を持つことを決定する欧州委員会に提出することができる。
相互承認手続きの基礎は、同じくEU加盟国の主管当局がEUの他の加盟国の主管当局によるある医薬製品の販売許可を受け入れることである。♪the the the
42
国家マーケティング許可の所有者は、EU加盟国の主管当局に申請を提出することができ、その主管部門に、他のEU加盟国の主管当局が提供するマーケティング許可を認めるように要求することができる。
データと市場排他性
EUでは、2001/83/EC指令に基づいて、完全な独立パケットによって承認された革新医薬製品は8年間のデータ独占権と他の2年間の市場独占権を獲得する資格がある。(EC)726/2004条例は、集中認可手順に従って許可された医薬品に対してこの権利を繰り返している。データ排他性は、これらの革新製品の模倣薬または生物模倣薬の許可申請者がEUで模倣薬または生物類似薬のマーケティング許可を申請する際に、参照製品が初めてEUで許可を得た日から8年以内に、この参考製品のファイルに含まれる革新者の臨床前および臨床試験データを参照する。追加の2年間の市場排他期間内に、模造薬または生物に類似したMAAを提出し、革新者のデータを参照することができるが、市場排他性が満了するまで、いかなる模倣薬または生物に類似した薬品もEU市場に投入することができない。この10年の最初の8年間に、マーケティング許可保持者が1つまたは複数の新しい治療適応の許可を得た場合、10年全体の期間は最大11年に延長され、許可前の科学的評価では、これらの適応は既存の療法と比較して有意な臨床的利益をもたらすことができると考えられる。1つの製品がEMAによって革新的な医薬製品とみなされることは保証されず、しかも製品はデータ独占性を得る資格がない可能性がある。1つの製品が革新的な医薬製品とされていても、革新者は所定のデータ独占期を獲得した, しかしながら、別の会社は、会社がMAAベースのマーケティング許可を取得した場合、薬物試験、臨床前試験、および臨床試験を含む完全な独立したデータパケットを有する製品の別のバージョンを販売することもできる。
授権期間と継続期間
上場授権書の初期有効期限は原則として5年である。マーケティング許可は、5年後に欧州市場管理局またはEU加盟国の主管当局が国家ライセンス製品のリスク-収益バランスを再評価することによって更新することができる。その後最終的に更新されると、欧州委員会または国家主管当局が薬物警戒に関する正当な理由に基づいて、追加の5年間の継続を決定しない限り、上場許可は無期限に有効でなければならない。任意の認可の後、認可が失効してから3年以内に医薬製品が実際にEU市場に投入されない場合(集中手続きの場合)、またはEU加盟国の国家認可製品の市場に許可されていない場合、その許可は無効になる(いわゆる日没条項)。
小児科研究と排他性
EUのマーケティング許可を得る前に、出願人は、EMAが特定の製品の免除、カテゴリ免除、またはPIPに含まれる1つまたは複数の措置を承認しない限り、EMA承認に適合するすべての小児科集団のサブセットをカバーするPIPに含まれるすべての措置を証明しなければならない。すべての販売許可手続のそれぞれの要求は(EC)第1901/2006号条例、いわゆる小児科条例に規定されている。会社が認可された薬物のために新たな適応,薬物形式あるいは投与経路を増加させようとした場合にも,この要求は適用される。EMAの小児科委員会、またはPDCOは、ある薬物の開発を延期することを承認する可能性があり、会社が成人に対する有効性および安全性を証明するのに十分な情報があるまで、小児薬の開発を延期することを可能にする。小児薬の開発を必要としない場合、または適切でない場合、PDCOは、高齢者人口のみに影響を与える疾患のような免除を与えることもできる。MAAを提出するか、または既存のマーケティング許可を修正する前に、EMAは、会社が各関連PIPに列挙された合意された研究および措置を実際に遵守していると判断する。出願人がすべてのEU加盟国で販売許可を取得した場合、または欧州委員会が集中手続きで付与された販売許可を取得し、その薬物個体群に関する研究結果が製品情報に含まれている場合、否定的な場合であっても、補充保護証明書またはSPCの期限を延長することによって6ヶ月の合格特許保護期間を追加的に取得する資格があり、製品のSPC出願を提出しながら、またはSPCが満了する前のいつでも、またはSPCが満了する前のいつでも、その製品のSPC出願を提出することを前提としている, 実験結果が陰性であっても。孤児薬品の場合、孤児市場の独占権は2年間延長することができる。この小児科奨励は特定の条件の制約を受け,PIPに適合したデータを開発·提出する際に自動的に獲得されない。
孤児薬の指定と排他性
EUで孤児に指定された製品は10年間の市場排他性を得ることができ,その間,“類似した医薬製品”を市場に投入してはならない。類似医薬製品“の定義は、許可された孤児医薬製品に含まれる1つ以上の類似活性物質を含む医薬製品であり、
43
同じ治療適応に用いられている。孤児製品はEUで追加2年間の市場排他性を得ることもでき,EUでは小児科研究の取り決め小児科調査計画が遵守されている。いかなる補充保護証明書も孤児の症状に関する小児科研究によって延期してはならない。
(EC)第847/2000条例により施行された(EC)第141/2000条例では、(1)生命又は慢性衰弱に危害を及ぼす疾患の診断、予防又は治療を目的としている薬物のスポンサーが証明できる場合、欧州委員会により孤児薬として指定することができる。(2)(A)申請時、このような疾患は、EU 1万(10,000)人当たり5(5)人以下の影響を与えるか、または(B)孤児身分によるメリットがない場合、その製品は、その開発に必要な投資が合理的であることを証明するためにEUで十分な見返りを与える可能性が低い、(3)このような疾患を満足できる診断、予防または治療方法がEUで販売されていないか、または、そのような方法が存在する場合、その疾患の影響を受けている人に大きな利益をもたらすであろう。孤児医薬製品は費用を下げたり、費用を免除したりするなど、財政的奨励を受ける資格がある。発売許可を申請する前に、孤児薬物指定申請を提出しなければならない。孤児薬物指定が承認された場合、出願人はMAA費用減免を受けるが、上場許可の提出時にその指定が待っている場合は、そうではない。孤児薬物の指定は、監督審査と承認過程においていかなる利点も伝達されず、規制審査と承認過程の持続時間を短縮することもない。
しかし、10年以内に、元孤児薬品販売授権者の同意、あるいは元孤児薬品生産企業が十分な数量を供給できない場合、同じ孤児の適応を持つ同類薬品の発売を許可することができる。同じ孤児の適応を有する類似薬品も発売が許可されており、もしこの製品が原始孤児薬品より安全で、より有効であれば、あるいは臨床的にもっと良い。さらに、5年目の終了時に、製品が指定された孤児の基準をもはや満たしていないと判定された場合、例えば、製品の利益が十分に高く、市場排他性を維持するのが合理的であることを証明するのに十分でない場合、市場排他性期間は6年に短縮されることができる。
営業許可を得た後の規制要件
もし医薬製品がEUで許可された場合、マーケティング許可の保持者は、医薬製品の製造、マーケティング、普及、販売に適した一連の要求を守らなければならない。これらの措置には
上記のEU規則は、EU加盟国にノルウェー、リヒテンシュタイン、アイスランドを加えた欧州経済地域(EEA)に一般的に適用されている。
一般資料保障規程
個人健康データを含むEU個人に関する個人データを収集、使用、開示、移転、または他の方法で処理し、2018年5月25日に施行されたEU GDPRの制約を受ける。英国のEU離脱や離脱後、EU GDPRは英国法、または英国GDPR(EU GDPR、GDPRとともに)に組み込まれている。GDPRの範囲は非常に広く、個人データを処理する会社に対して多くの要求を提出し、健康や他の敏感なデータの処理、個人データに関する個人の同意の取得、個人にデータ処理活動に関する情報を提供すること、個人データの安全と機密性を保護するための保障措置の実施、データ違反行為の通知の提供、ある責任措置の確保、第三者プロセッサの使用時に何らかの措置をとることなどの要求を含む。GDPRはまた、EUやイギリス以外の国(米国を含む)への個人データの移転に厳しいルールを実施し、データ保護当局が2000万ユーロ(1750万ポンド)や世界年収の4%に達する可能性のある罰金を含むGDPR違反行為に巨額の罰金を科すことを許可している。♪the the the
44
GDPRはまた,データ主体や消費者協会に対して個人訴訟を提起する権利を与え,監督当局に苦情を申し立て,司法救済を求め,GDPR違反による損害について賠償を受けることができる。GDPRを守ることは厳格で時間のかかる過程であり,ビジネスコストを増加させたり,会社にそのビジネスやり方を変更して完全な遵守を確保したりする可能性がある.
イギリスの離脱とイギリスの規制枠組み
イギリスは2020年1月31日に正式にEUを離脱し、EUはイギリスと貿易·協力協定を締結し、TCAと略称し、2021年1月1日から暫定的に適用され、2021年5月1日から正式に適用される。TCAには薬品に関する具体的な条項が含まれており,その中にはGMP相互承認,薬品の製造施設の検査,発表されたGMP文書が含まれているが,イギリスとEUを大規模に相互承認する薬品法規は規定されていない。現在、イギリスは“2012年人類薬品条例”(改正された)(北アイルランド議定書に基づき、EU規制枠組みは引き続き北アイルランドに適用されている)を採択し、医薬製品のマーケティング、普及、販売に関するEUの立法を実施している。そのため,新しいEU臨床試験規則を除いて,イギリスの規制制度は現在EUの規制制度とほぼ一致しているが,イギリスの規制制度はEUから独立しているため,“薬物規制法”もイギリスとEUの薬剤業法の相互承認について規定されていないため,これらの制度は後日より大きく異なる可能性がある。
イギリスが離脱したにもかかわらず、EUとイギリスのGDPRはほぼ一致している。現在、最も影響力のある分岐点は、EUやイギリスの規制機関が承認した様々な異なる契約条項を実施することが求められているため、移転メカニズム(すなわちEU/イギリス社が米国を含む第三国に個人情報を移転する能力)に関連している。今後は行政負担の面も含めてさらなる食い違いがあるかもしれない。英国はそのデータ改革法案で、EU GDPR導入の大きな変化となる同国のデータ保護法の枠組みを改革する計画を発表した。
製品の定価決定を承認する
連合では、様々な国の価格設定と補償プログラムの差が大きい。一部の国では、補償価格を合意した後にのみ、製品を販売することができると規定されている。いくつかの国は、特定の候補製品のコスト効果を現在利用可能な治療法またはいわゆる衛生技術評価と比較して、精算または価格設定の承認を得るために、追加の研究を達成することを要求するかもしれない。例えば、EU加盟国は、その国の健康保険制度が精算を提供する製品範囲を制限し、人が使用する医療製品の価格を制御することを選択することができる。EU加盟国は製品の具体的な価格を承認することができ、その製品を市場に投入する会社の収益力を直接または間接的に制御する制度をとることもできる。他のEU加盟国は会社が自分の製品価格を固定することを許可したが、処方量を監視と制御し、医師に指導を発表して処方を制限した。最近,EUの多くの国が薬品要求の割引額を増加させ,各国が医療支出を管理しようとしているに伴い,これらの努力は継続する可能性があり,特にEUの多くの国が深刻な財政危機や債務危機を経験している場合である。全体的には,医療コスト,特に処方薬の下り圧力が大きくなっている。
そのため、新製品の参入にはますます高い壁が設けられている。政治、経済、規制面の事態は定価交渉をさらに複雑化させる可能性があり、補償を受けた後、定価交渉が継続される可能性がある。EU加盟国が使用する参考価格と平行貿易は即低価格と高価なEU加盟国の間で裁定を行うことで、価格をさらに下げることができる。特別な価格と精算規則は孤児薬品に適用されるかもしれない。いかなる薬物と同様に、孤児薬物を精算制度に入れることはよく患者と医療保健システムの医療有用性、需要、品質と経済利益に重点を置いている。いずれの医療製品の精算を受けるかはコスト,用途,通常の数量制限を伴う可能性があり,これらの制限も国によって異なる可能性がある。しかも、成果に基づく精算規則が適用される可能性がある。薬品に対して価格規制や精算制限を実施することが保証されていない国は、これらの国で承認されれば、任意の製品に対して有利な精算と定価手配を許可する。
EUで医療サービスを提供することは、医療サービスの確立と運営、薬品の定価と精算を含み、ほぼ完全に国家の法律と政策の問題であり、EUの法律や政策ではない。この点で,各国政府や保健サービス提供者は,保健サービスの提供や製品の定価や精算に異なる優先順位や方法を持っている。しかし、全体的に、ほとんどのEU加盟国の医療予算制限は、関連医療サービス提供者の薬品の定価と精算の制限を招いている。
45
人的資本管理
私たちの使命と従業員は
私たちは遺伝子で定義された病気を根本的に治療することでプロセスを変える大胆なビジョンを立ち上げました我々の薬物発見方法は疾患生物学に対する重大な知見を生み出し,調節と平衡遺伝子発現の最適な方法を創造的に考えることができるようになった。著者らは患者を中心とした製品エンジンFulcrumSeekは細胞薬物標的を系統的に識別と検証することを目的とし、これらの標的は遺伝子発現を調節し、既知の遺伝子定義疾患の根本的な原因を治療することができる。目的のある患者パートナーとして,患者のためだけでなく,患者のためにもしていることを誇りに思っている。
私たちは職員たちが使命を履行する最も価値のある資産の中の一つだと思っている。私たちは私たちの未来の成功が才能のある従業員たちを引きつけ、激励し、維持することにかかっていると信じている。私たちは従業員とその家族の健康と健康を重視する。私たちの目標は、私たちの従業員がその中で彼らのキャリアを成長させ、向上させることができるように、公平、包容、そして付与された労働環境を創出することであり、全体的な目標は、私たちの現在のルートと未来の業務目標を支持するために、私たちの従業員チームを発展、拡大、維持することである。私たちの成功はまた私たちが違う従業員たちを引き付け、吸引し、維持する能力にかかっている。
私たちの行動は私たちの使命を支持します
私たちと私たちの従業員が私たちの使命と一致している時、成功が来ると信じています。私たちの使命は、高度に満たされていない医療需要の分野で遺伝子定義を改善する珍しい疾患患者の生活を改善することです。私たちはこの柱の周りに団結しているFULCrew
わが国の人的資本管理
私たちの従業員を有効に利用して管理するために、私たちの採用需要が私たちの戦略と直接一致することを確保し、私たちはFulcrumの間に彼らの発展と旅行に集中している従業員に投資し、最も重要なのは、私たちが彼らを維持することに集中していることを確実にするために、私たちの重要な人材を決定することだ。著者らは内部で重要な人材指標を追跡と報告し、総人数と機能、求人指標、職業発展(昇進など)、離職傾向と従業員人口統計(人種、性別、民族を含む)に重点的に注目することを含む。著者らの高級管理者はこれらの指標を使用して、資源計画、採用と維持計画及び給与と福祉計画の設計を含む従業員をめぐって熟慮の決定を行った。これらの指標を四半期ごとに上級管理者や取締役会と共有し、(A)企業報酬理念の確立、(B)当社の報酬計画の管理、(C)我々の役員·キーパーソンのパフォーマンスの評価、および(D)経営陣の発展·後継計画の審査·監督の役割を果たすことを支援する。
2023年3月2日現在、医学博士号を持つ36人の従業員を含む89人の常勤従業員がいる。これらの常勤従業員のうち、52人の従業員が研究開発に従事している。私たちの従業員の中の一人も労働組合代表でもなく、集団交渉協定のカバー範囲もない。私たちは私たちが従業員と仲がいいと思う。
多様性、公平、そして包括性に対する私たちの約束
私たちは多様な職場で、私たちのすべての従業員が差別、嫌がらせ、偏見、偏見がなく、包容的な環境ですくすくと成長できると信じている。私たちの目標はすべての人を尊重し、尊重し、すべての従業員に平等な機会と利点に基づく公平な待遇を提供することだ。多様性と包括性を抱擁することで、私たちの使命を支援し、私たちの価値観と一致するように、革新的な解決策を協力して開発するための組織を作りました。私たちは、異なる背景や観点が尊重され、耳を傾けるだけでなく、抱擁とお祝いを得る文化と環境を育成した。多様化、公平と包容の心理状態と文化は積極的に参加し、責任を果たす職場に重要であるだけでなく、著者らが助けを求める患者の薬物需要を理解し、満足させるためにも重要である。
我々の成長と成熟に伴い,多様性,公平性,包摂性,帰属感を支援する計画/指標の構築が期待される。Fulcrumにより多くの多様な人材をもたらし続け、知名度(講演者など)を創出することで包容的な環境を創出していきたいと考えています。私たちの代表的な不足している職員たちが正しい職業機会を得ることを確実にする。
46
2023年3月2日現在、女性は私たちの常勤従業員の約60%を占めている。2023年3月2日現在、私たちの従業員の記録によると、フルタイムのアメリカ人従業員の約38%が非白人と考えている。
私たちの報酬と福祉は
私たちの業界の高度な競争性質と従業員の採用と維持が私たちの成功に対する重要性を考慮して、私たちは私たちの従業員に、報酬、福祉、サービスを含む、非常に競争力があると考えられる包括的な報酬プランを提供するように努力しています。このプランには競争的な報酬や株式報酬も含まれています 医療保険および歯科ケア、視力保険、障害保険、有給病気休暇、有給休暇、有給帰省休暇、401(K)計画のマッチング支払い、従業員株式購入計画、有給休暇(休暇、祝祭日、焦点日を含む)および従業員支援サービスを含む、医療保険および歯科ケア、視力保険、障害保険、有給病気休暇、有給休暇、401(K)計画のマッチング支払いを管理するためのリソースを提供する包括的な福祉計画。
新型肺炎の流行に対応するための私たちの努力は
従業員の安全と福祉はどの年も私たちにとって重要であり、現在の新冠肺炎疫病を考慮して、2021年の従業員の安全と福祉は特に注目されている。疫病に対応するため、著者らは安全、コミュニケーションと投資を通じて従業員が新冠肺炎の疫病を抑制することを支持し、その中には:
しかも、大流行の間、私たちは時々私たちの従業員を支援するための追加的な計画を立てた。私たちは毎週の会社会議を利用して、私たちの従業員に疫病期間中に発生したこと(予想を管理し、彼らの需要に耳を傾ける)を理解させ、従業員の連絡を確立する(関係を作る/連絡する精神に基づいて面白い突破を行う)。また、家庭オフィスの設置に補助金を提供し、勤務先/時間を柔軟に決定し、従業員支援サービス(包括的なメンタルヘルス、ワークライフ、管理サービス)を強化しました。
企業情報
私たちの主な実行事務室はマサチューセッツ州カンブリッジランドトン街二十六号にあります。郵便番号:02139、電話番号は6176518851です。私たちのインターネットサイトはwww.fulcrumtx.comです。我々のサイトに含まれている,あるいは我々のサイトを介してアクセスできる情報は,本Form 10-K年次報告の一部ではない.
47
第1 A項。RISK因子です。
以下に述べるリスクと不確実性のため、当社の将来の経営業績は、本10-K表年次報告に記載されている結果とは大きく異なる可能性がある。私たちの業務を評価する時、あなたは以下のリスクに関する情報を慎重に考慮しなければならない。もし実際に以下のいかなるリスクが発生すれば、私たちの業務、財務状況、経営業績、未来の成長見通しは重大な不利な影響を受ける可能性がある。この場合、私たちの普通株の市場価格は下落するかもしれない。しかも、私たちは私たちの仮定と期待が正しいことが証明されることを投資家に保証することはできない。重要な要素は私たちの実際の結果が展望性陳述で表明したり暗示したりする結果と大きく異なる可能性がある。これらのリスク要因によって制限されたいくつかの前向き陳述の議論については、本年度報告(Form 10−K)の1ページ目の“前向き陳述に関する戒め”を参照されたい。このような差異をもたらすか、または促進する可能性のある要因は、以下に説明する要因を含む。
私たちの財務状況と追加資本需要に関連するリスク
設立以来、私たちは大きな損失を受けた。私たちは今後数年間赤字になると予想していて、決して達成されたり利益を維持したりしないかもしれない。
設立以来、私たちは重大な運営損失が発生した。2022年12月31日までの年度の純損失は1.099億ドルで、2021年12月31日までの年度の純損失は8080万ドルだった。2022年12月31日までの累計赤字は4億123億ドルだった。今まで、私たちの運営資金は主に私たちの株式株式の売却と私たちの協力と許可契約に基づいて受け取った前金から来ています。我々はほとんどの財政資源と努力を臨床試験と臨床前研究を含む研究と開発に投入した。私たちはまだ候補製品開発の初期段階にあり、候補製品の開発はまだ完了していない。私たちは今後数年間巨額の費用と運営損失が発生すると予想している。私たちの純損失は四半期ごとと毎年大きく変動するかもしれません。私たちの費用は大幅に増加すると予想されています
48
利益を維持するためには、大量の収入を生み出す1つまたは複数の製品を開発し、最終的に商業化することに成功しなければならない。この成功を達成するためには、私たちの候補製品の臨床前テストと臨床試験を完成させ、より多くの候補製品を発見し、これらの候補製品の監督管理許可を得ること、および私たちが規制承認を得る可能性のある任意の製品を製造、マーケティング、販売することを含む、一連の挑戦的な活動において機能する必要がある。私たちはただこのような活動の大多数の初期段階にいるだけだ。私たちはこのような活動で決して成功しないかもしれないし、たとえ私たちが成功しても、利益を達成するのに十分な収入が生まれないかもしれない。医薬品開発に関連する多くのリスクや不確実性のため、費用を増加させる時間や金額、あるいはいつ、または利益を達成できるかどうかを正確に予測することはできない。他の事項を除いて、私たちの支出は増加するだろう
たとえ私たちが確実に利益を達成したとしても、私たちは四半期や年度の収益性を維持したり向上させることができないかもしれない。もし私たちが実現して利益を維持できなければ、会社の価値を下げ、資金を調達し、業務を拡大し、研究開発努力を維持し、候補製品ルートを多様化し、甚だしきに至っては運営を継続する能力を弱める可能性がある。わが社の価値の低下は私たちの株主に彼らの投資の全部または一部を損失させる可能性もあります。
私たちは多くの追加資金が必要になるだろう。もし私たちが必要な時に資金を集めることができなければ、私たちは私たちの製品開発計画や商業化努力を延期、減少、またはキャンセルさせることを余儀なくされるかもしれない。
私たちは、特に私たちが進行して計画中のロスパモントとFTX−6058の臨床試験、これらおよび他の候補製品の臨床試験の研究開発を継続し、規制機関の承認を求める場合に、私たちが行っている活動および計画中の活動に多くの財源を投入する予定だ。われわれが行っている活動に関する費用は大幅に増加することが予想され,特にわれわれが臨床前活動や臨床試験を進める際には。また、いずれかの候補製品が規制部門の承認を得た場合、製品の製造、販売、マーケティング、流通に関連した巨額の商業化費用が発生すると予想される。したがって、私たちは私たちの持続的な業務と関連した多くの追加資金を得る必要があるだろう。現在の資本市場の不確実性と他の要素を考慮して、このような融資は私たちに有利な条項で提供されないかもしれないし、根本的にはないかもしれない。もし私たちが必要な時や受け入れ可能な条件で資金を調達できなければ、私たちは私たちの研究開発計画や将来の商業化努力を延期、減少、または廃止することを余儀なくされるかもしれない。
私たちの将来の資本需要は多くの要素に依存します
49
2022年12月31日現在、私たちは約2.029億ドルの現金、現金等価物、有価証券を持っている。2022年12月31日までの現金、現金等価物、有価証券に、2023年1月に完成した公募株の純収益に加え、2025年中までの運営費と資本支出需要に資金を提供できると信じています。しかし,我々のこの推定は誤りであることが証明される可能性があるという仮定に基づいており,我々の運営計画は現在我々が知らない多くの要因によって変化する可能性がある.したがって、私たちは現在予想されているよりも早く私たちの資本資源を枯渇させるかもしれない。
潜在的な候補製品を確定し、臨床前テストと臨床試験を行うことは時間がかかり、高価かつ不確定な過程であり、数年を要して完成する必要があり、しかも著者らは監督部門の許可を得て製品販売を実現するために必要なデータ或いは結果を永遠に生成できないかもしれない。しかも、私たちの候補製品が承認されれば、商業的成功を得られないかもしれない。商業収入は、あれば得られません。製品の販売が実現できない限り、私たちは何年もこれを期待していません。したがって、私たちは追加的な資金調達に依存して私たちの業務目標を達成し続ける必要があるだろう。私たちは許容可能な条件で十分な追加融資を得ることができないかもしれないし、十分な追加融資を得ることができないかもしれないし、金利上昇と現在の米国資本市場とバイオテクノロジー産業全体の低迷により、より困難になるかもしれない。また、有利な市場条件や戦略的考慮により、現在または将来の運営計画を実行するのに十分な資金があると考えても、追加の資本を求めることができる。もし私たちが十分な資金をタイムリーに得ることができない場合、私たちは、私たちの1つまたは複数の候補製品または発見段階計画の臨床前研究、臨床試験または他の開発活動の遅延、制限、減少、または終了を要求されるかもしれない、または私たちの候補製品を商業化するために必要な活動である可能性がある。私たちはまた、最近発表された戦略調整よりも高い運営効率を実現するために、私たちの業務をさらに調整することを選択することも可能です。
追加資本の調達は私たちの株主に希釈し、私たちの運営を制限したり、私たちの技術や候補製品に対する権利を放棄することを要求するかもしれません。
これまで、相当な製品収入を生み出すことができれば、株式発行、債務融資、協力、戦略連合とマーケティング、流通、または許可手配の組み合わせで私たちの現金需要を満たす予定です。私たちは約束された外部資金源を持っていない。もし私たちが株式または転換可能な債務証券を売却することで追加資本を調達すれば、私たちの株主の所有権権益は希釈され、これらの証券の条項は清算または他の特典を含む可能性があり、普通株主としての私たちの権利に悪影響を及ぼす。債務融資および優先株式融資に関与する可能性のある協定は、追加債務の発生、我々の資産の売却、資本支出の実施、または配当の発表など、私たちが具体的な行動をとる能力を制限または制限する契約を含む。
もし私たちが第三者との協力、戦略連合またはマーケティング、流通または許可手配を通じてより多くの資金を調達するならば、私たちは私たちの技術、将来の収入源、研究計画、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になる可能性のある条項でライセンスを付与しなければならないかもしれない。もし私たちが必要な時に株式や債務融資を通じてより多くの資金を調達できない場合、私たちは私たちの製品開発や将来の商業化努力を延期、制限、減少または終了することを要求されるかもしれません。あるいは私たちは自分で開発とマーケティングをより望んでいた候補製品の権利を与えます。
従来,我々は市場のATMによる普通株の発売にある程度依存していた.株式証券、特に我々の普通株の市場価格の変動の増加及び低下は、ATM発売計画による我々の普通株の継続売却の意思及び/又は能力に悪影響を及ぼす可能性がある。これらの売上高の減少は株式資本のコストまたは獲得性に影響を与える可能性があり、更に現在の運営、未来の成長、収入、純収入、および私たちの普通株の市場価格を含む私たちの業務に悪影響を及ぼす可能性がある。
2022年5月、私たちは普通株の株を売却するための新しいATM発行計画を立ち上げ、総発行価格は時々5000万ドルに達した。2023年1月、我々が引き受けた普通株式公開発行を開始する前に、ATM発行計画に関する目論見補充部分の使用を停止し、終了した。新しい募集説明書の付録を提出しない限り、ATM機の発売計画に基づいてどんな証券販売も行いません。しかし、資本市場の全体的な変動を考慮して、新たな目論見書が補充されても
50
申請を提出すると、私たちは私たちのATM発行計画を通じて株式を調達したくないか、続けることができないかもしれません。したがって、私たちは他の資金源に転換する必要があるかもしれません。これらの資金源の条項は私たちに不利かもしれません。あるいは資本制限によって私たちの業務運営を減らすことができます。
代替融資スケジュールは、普通株、優先株、転換可能債券、普通株買収、または他の証券の引受権証を含む1つまたは複数の証券の発行に関連することができる。これらの証券の発行価格は当時の私たちの普通株の現行の市場価格に等しいか、以下にすることができる。また、もし私たちが債務証券を発行した場合、元金、課税利息、未払い利息、およびいかなるプレミアムまたは補償支払いが完了する前に、債務保有者の私たちの資産に対する権利は株主の権利よりも高くなるだろう。また、吾等が付属会社を通じて資金及び/又は債務証券を借入及び/又は発行すれば、当該等の債務証券の貸手及び/又は所持者が、吾等の付属会社の権益所有権よりも実際に優先する支払いを得る権利があることは、吾等の権益証券保有者及び(あれば)吾等の債務及び債務証券保有者の権利に悪影響を与える。
新たに発行された債務証券及び/又は新たに発生した借金の利息は、我々の運営コストを増加させ、我々の純収入を減少させることができ、これらの影響は実質的である可能性がある。もし新証券の発行が私たちの普通株保有者の権利を減少させれば、私たち普通株の市場価格は実質的な悪影響を受ける可能性がある。もし私たちが必要な時に運営資金の需要を維持するために必要な融資を得ることができない場合、あるいは融資コストが目を引くほど高く、その結果は私たちの業務、経営業績、財務状況、見通しに実質的な悪影響を与える可能性がある。
私たちの限られた経営の歴史は、株主が私たちの業務のこれまでの成功度を評価することを困難にし、私たちの将来の生存能力を評価することも困難になるかもしれない。
私たちは2015年に活動を開始し、臨床段階のバイオテクノロジー会社です。これまで、私たちの業務は、組織と私たちの会社のための人員、業務計画、資金調達、私たちの知的財産権の構築、私たちの発見プラットフォームの構築、薬物目標と潜在的な候補製品の決定、許可資産、臨床試験のための薬物物質と薬物製品材料の生産、および臨床前研究と臨床試験を行うことに限られてきた。私たちは、どんな候補製品の開発に成功し、規制の承認を得たり、ビジネス規模の製品を製造したり、第三者代表がそうしてくれたり、成功した製品の商業化に必要な販売やマーケティング活動を行う能力があることを証明していません。したがって、もし私たちがより長い経営歴史や成功して製品を開発·商業化した歴史があれば、株主は私たちの将来の成功や生存能力に対するいかなる予測もそれほど正確ではないかもしれない。
また、業務の発展に伴い、私たちは予見できない費用、困難、複雑な状況、遅延、および他の既知および未知の要素に直面する可能性がある。私たちはある時点で研究開発に集中している会社からビジネス活動を支援できる会社に転換する必要があります。そのような移行で、私たちは成功しないかもしれない。
様々な要因により、私たちの財務状況と経営業績は四半期と年度の間に大幅に変動し、その多くの要素はコントロールできないと予想しています。そのため、株主はどの四半期や年度の業績にも依存して将来の経営業績の指標とすべきではない。
私たちの業務は持続的な新冠肺炎疫病の負の影響を受け、未来はいかなる未来の疫病の影響を受けるかもしれない。また、この大流行は継続する可能性があり、将来のどの大流行も世界各地の経済に悪影響を及ぼす可能性があり、これは私たちの業務や運営に悪影響を及ぼす可能性がある。
新冠肺炎の流行により,われわれのReDUX 4臨床試験の臨床試験地点は試験に関する活動を一時的に延期したため,ReDUX 4臨床試験の登録遅延に遭遇した。私たちのケンブリッジ研究機関の労働力が一時的に減少したため、他の業務活動も一時的に中断されたことが見られた。私たちの従業員はすでに職場に復帰し、いくつかのワクチンが使用可能であり、多くの制限はすでに廃止されたが、新冠肺炎が私たちの業務に与える全体的な影響、及びそれが全世界経済に与える持続的な影響に対して、依然として不確定性が存在している。未来に流行病が出現する可能性があり、それらは新冠肺炎のように、私たちの会社、私たちのCMOと契約研究組織(CRO)に影響を与え、中断をもたらし、私たちの臨床前研究或いは臨床試験を開始と完成する能力に影響を与え、私たちの研究開発活動のサプライチェーンを乱し、そして各種の原因で当時計画中あるいは行われていた臨床試験を混乱させる。いかなる未来の大流行も同様に臨床試験の患者募集或いは維持に影響を与え、あるいは資源が再分配されることを招き、それによって私たちの監督管理の承認と私たちの知的財産権を保護する能力を推進することに不利な影響を与える可能性がある。また、新冠肺炎の大流行と同様に、今後の任意の大流行における対面インタラクションを制限するための安全対策が提案されているため、規制会議や承認の障害に直面する可能性がある。
現在行われている新冠肺炎の大流行はすでに金融市場に大きな妨害を与えており、それは継続する可能性があり、未来のどの大流行も同様にこのような妨害を引き起こす可能性があり、これは私たちのより多くの資金を調達する能力に影響を与える可能性がある
51
株式を公開することで、私たちの株価や株式取引の変動性にも影響を与える可能性があります。私たちは発生している新冠肺炎の大流行あるいは未来のいかなる大流行が私たちの業務にどのような全体的な影響を与えるかを確定することができません。新冠肺炎疫病と未来のいかなる疫病が著者らの業務、財務状況、運営結果と将来性に与える影響程度は不確定な未来の発展に依存する。
税法またはその実施または解釈の変化は、私たちの業務および財務状況に悪影響を及ぼす可能性がある。
米国連邦、州、地方所得税に関する規則は立法過程に参加する人員およびアメリカ国税局とアメリカ財務省の審査を受け続けている。税法の変化(これらの変化はトレーサビリティを有する可能性がある)であり、純営業損失や研究開発税収控除に関する変化を含めて、私たちまたは私たち普通株の保有者に悪影響を及ぼす可能性がある。近年、このような変化は多く発生しており、未来も変化し続けるかもしれない。将来の税法の変化は、私たちの業務、キャッシュフロー、財務状況、または運営結果に実質的な悪影響を及ぼす可能性があります。私たちは投資家に税法の潜在的な変化が私たちの普通株に投資する影響について彼らの法律と税務顧問に相談することを促します。
私たちは純営業損失と研究開発税収控除を使って未来の課税収入を相殺する能力は一定の制限を受ける可能性があります。
2022年12月31日まで、我々の連邦と州の純営業損失はそれぞれ2.751億ドルと2.726億ドルに転換し、2036年に満期になる。約2兆515億ドルの連邦純運営損失は無期限に繰り越すことができる。2022年12月31日まで、私たちは1460万ドルの連邦孤児薬物免除があり、これらの相殺は2040年に満期になります。2022年12月31日までに、連邦と州の研究開発税収の繰越免除はそれぞれ690万ドルと400万ドルで、それぞれ2035年と2030年に満期になる。これらの純営業損失と税収控除は満期時には使用されていない可能性があり、将来の所得税負債を相殺するためには使えない。
一般的に、改正された“1986年米国国税法”第382条又はこの法典及び州法律の対応条項によると、ある会社が3年間に経験した“所有権変更”は、通常、ある株主の株式の価値上の変化が50%を超えると定義され、その利用変更前の純営業損失と研究開発税収を将来の課税収入を相殺する能力が制限される。私たちは、2021年12月31日までの所有権履歴変化が制限されているかどうか、または他の方法で、変更前の純営業損失と研究開発税収の繰越を利用して将来の課税収入を相殺する能力を利用するかどうかを決定するために、第382条に基づいて分析を行った。分析の結果、私たちは私たちの純営業損失と研究開発税収の繰越を利用して、未来の課税収入を相殺する能力に重大な制限があるとは思わない。しかし、私たちは未来にそのような所有権の変化を経験するかもしれない(これは私たちがコントロールできるものではないかもしれない)。したがって,課税収入の純額を稼ぐと,変動前の純営業損失や研究開発税収を用いてこのような課税収入を繰返す能力が制限される可能性がある。州法によると、私たちの純営業損失や信用も減価される可能性があります。
私たちは累積赤字の歴史があり、予測可能な未来に私たちは引き続き重大な損失が発生すると予想されています。そのため、私たちは私たちの純運営赤字や研究開発税収を利用して繰越を免除するために必要な課税収入が発生するかどうかを知りません。
我々の候補製品の発見と開発に関するリスク
私たちの開発はまだ初期段階にあり、私たちは二つの臨床段階の候補製品しかありません。もし私たちの候補製品を商業化できない場合、あるいはそうする過程で重大な遅延に遭遇すれば、私たちの業務は実質的な損害を受けるだろう。
我々の開発は初期段階であり,我々は2種類の候補製品のみが臨床試験に入った,すなわちFSHD治療のためのlosmapimodとSCD治療のためのFTX−6058は,後者の計画は現在臨床的に棚上げされているにもかかわらず。著者らはすでにほとんどの努力と財力を著者らの独自の製品エンジンに投入し、細胞薬物標的を識別と検証し、これらの標的は遺伝子発現を調節し、遺伝子定義の稀な疾病の根本的な原因を解決する可能性がある。私たちが製品収入を作る能力は、私たちの候補製品の成功した開発、規制承認、最終商業化に大きく依存します。製品収入は何年も起こらないと予想されます。私たちの候補製品の成功は以下のいくつかの要素に依存するだろう
52
もし私たちがこれらの要素のうちの1つまたは複数をタイムリーにまたは根本的に実現できなければ、私たちは重大な遅延に遭遇したり、私たちの候補製品を開発して商業化できなかったりする可能性があり、これは私たちの業務に深刻な損害を与えるだろう。
私たちは候補製品チャンネルを構築するために私たちの製品エンジンを使用することに成功しないかもしれない。
著者らの戦略の1つの重要な要素は著者らの特許製品エンジンを用いて細胞薬物標的を識別と検証することであり、これらの標的は遺伝子発現を潜在的に調節し、遺伝子定義の稀な疾病の根本的な原因を解決することができ、最初の重点は識別した細胞標的の小分子を識別することである。私たちが薬物標的と潜在的な候補製品を決定することに成功したとしても、有害な副作用や他の特徴が証明されたため、上場承認を得ることができず、市場で承認される可能性が低いことを含む、これらの候補薬剤は臨床開発に適していない可能性がある。より多くの候補製品の監督管理承認を確定、開発、獲得し、それを商業化するには大量の追加資金が必要であり、製品開発固有の失敗リスクが発生しやすい。MyoKardiaとの協力を含め、私たちの製品エンジンで他の候補製品を成功的に識別できることを株主に保証することはできません。開発過程で任意の他の候補製品を推進したり、そのような他の候補製品を商業化することに成功しました。規制当局は承認過程でかなりの裁量権を持ち、承認や申請拒否の遅延を招く可能性がある。これらの要因により,製品候補開発の時間やコストを予測することは困難である.私たちが未来に遭遇する自社製品エンジンや私たちの任意の研究開発計画に関連するいかなる開発問題も重大な遅延や意外なコストを招くことは保証されず、このような開発問題が解決される保証はない。もし私たちが認識し開発できなければ, 私たちの技術的方法によって規制機関の候補製品の承認を得て商業化すれば、製品収入を生み出すことができません。
臨床薬物開発は長くて高価な過程に関連し、結果は不確定である。臨床前研究と早期臨床試験の結果は未来の結果を予測できないかもしれない。私たちは私たちの候補製品の開発と商業化を完了したり、最終的に完成できなかったりする過程で追加のコストが発生したり、遅延が生じる可能性があります。
私たちは臨床開発に2種類の候補製品を持っている。私たちはすべての候補製品の失敗の危険が高い。私たちの候補製品がいつ、人体で有効または安全を証明するか、あるいは規制部門の承認を受けるかどうかを予測することはできない。監督部門の許可を得て任意の候補製品を販売する前に、私たちは臨床前開発を完成し、それから広範な臨床試験を行い、私たちの候補製品の人体における安全性と有効性を証明しなければならない。私たちはまだ候補製品の重要な臨床試験を終えていない。臨床試験は私たちの候補製品が人類にとって安全であり、指定用途に有効であることを証明できないかもしれない。例えば、SCDを治療する臨床試験段階候補薬FTX−6058はEEDIである。EEDはPRC 2複合体のメンバーであり,PRC 2複合体もEZH 2を含む。EZH 2系薬剤には承認された製品があり,その承認されたラベルは悪性腫瘍のリスク増加を含む安全リスクを概説している。FTX−6058が他のPRC 2薬物と同様の安全リスクを有する場合、これはその受容度に影響を与える可能性がある。臨床試験が成功しても、開発期間中の上場承認政策の変化、付加法規、法規或いはガイドラインの制定或いは公布の変化、或いは各提出された製品申請に対する監督審査の変化は、申請の承認或いは拒否の遅延を招く可能性がある。
候補製品の臨床試験を開始する前に、私たちは米国と海外で提出される予定のINDおよび他の規制文書を支持するために、広範な臨床前試験と研究を完成させなければならない。確かではありません
53
我々の臨床前テストと研究の適時な完成或いは結果はFDA或いは他の監督機関が私たちの提案した臨床計画を受け入れるかどうか、あるいは私たちの臨床前テストと研究の結果が最終的に私たちの現在または未来の候補製品のさらなる開発を支持するかどうかを予測できない。したがって、私たちは、私たちが予想しているスケジュールでINDまたは同様の臨床前計画申請を提出できることを保証することができず、INDまたは同様の申請の提出がFDAまたは他の規制機関に臨床試験の開始または継続を可能にすることを保証することもできない。FDAは2023年2月23日,SCDにFTX−6058を使用したINDの臨床休止を行った。棚上げ問題をできるだけ早く解決するように努力するつもりであるが,FDAが試験の速やかな回復や根本的な回復を許可する保証はない。また,候補製品は持続的な臨床前安全性研究を受け,これらの研究はわれわれの臨床試験と同時に行われる可能性がある。これらの安全性研究の結果は,将来の臨床試験の開始や登録を遅らせる可能性があり,臨床試験を継続する能力に影響する可能性がある。
臨床試験は費用が高く,設計と実施が困難であり,完成まで数年かかる可能性があり,結果はまだ確定していない。いかなる臨床試験が計画通りに行われるか,あるいは計画通りに完了するか,あるいは全く保証されない保証はない。例えば,我々のFTX−6058臨床試験は現在放置されており,いつ回復するかは不明であり,もし本当に回復すれば。1つまたは複数の臨床試験の失敗は、研究設計の欠陥、用量選択問題、プラセボ効果、患者登録基準、および良好な安全性または有効性特徴を証明できなかったことを含むが、これらに限定されない、試験の任意の段階で発生する可能性がある。臨床前試験と早期臨床試験の結果は後の臨床試験の成功を予測できない可能性があり、臨床試験の初歩的或いは中期結果も必ずしも最終結果を予測できるとは限らない。例えば,われわれの候補製品は臨床開発において期待される安全性と有効性を示すことができない可能性があり,臨床前研究で積極的な結果が得られたにもかかわらず,あるいは予備臨床試験に成功している。臨床的メリットの不足は投与量不足或いはその他の原因による可能性がある。製薬と生物技術業界の多くの会社は後期臨床試験で重大な挫折を経験し、臨床前試験と早期臨床試験で人を奮い立たせる結果を得ても、私たちは類似した挫折に直面しないことを確定できない。そのほか、臨床前と臨床データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品が臨床前研究と臨床試験で満足できると考えているが、しかし依然としてその製品のマーケティング許可を得られなかった。さらに何かがある, 私たちの任意の候補製品が任意の臨床試験において安全性および有効性を証明できない場合、私たちの他の候補製品の感想に負の影響を与える可能性があり、および/またはFDAまたは他の規制機関が、私たちの任意の候補製品を承認する前に追加的な試験を要求する可能性がある。
本年度報告Form 10−Kの第1項“ビジネス−ライセンスおよび協力--グラクソ·スミスクラインとの参照権およびライセンス契約”で述べたように、GSKの関連会社と改訂された参照権およびライセンス契約を締結した。グラクソ·スミスクラインは最初に3,600人近くの被験者でロムパモントを評価したが、グラクソ·スミスクラインはFSHDまたは任意の他の筋ジストロフィーにおける治療効果を評価しなかったが、これらの試験の多くの被験者は、私たちが1日2回15 mgの計画用量よりも低い用量を投与した。したがって,グラクソ·スミスクパスパモット臨床試験による安全性データは,われわれの臨床試験結果を予測あるいは指示できない可能性がある。同様に,グラクソ·スミスクライン臨床試験の安全性データはlosmapimodの安全性データベースをある程度支持している可能性があると考えられるにもかかわらず,グラクソ·スミスクラインは15 mgの限られた数の被験者を1日2回評価した。
臨床試験中や臨床試験の結果では、私たちは多くの予見できない事件に遭遇する可能性があり、これらの事件は、私たちの上場承認を延期したり、私たちの候補製品を商業化することを阻止したりする可能性があります
54
例えば,持続的な新冠肺炎に対応するために,ReDUX 4試験の臨床試験地点は試験に関する活動を一時的に延期し,われわれの臨床試験実行計画に影響を与え,将来的に他の延期や類似の困難に直面しないことは確認できない。また,FDAは2023年2月23日,SCDにFTX−6058を用いたINDの臨床休止を行った。
もし私たちの候補製品に対して、現在予想されている以上の追加の臨床試験または他の試験を行うことを要求された場合、もし私たちが候補製品の臨床試験または他の試験を成功させることができなければ、これらの試験または試験の結果が陽性でないか、または軽微な陽性である場合、または安全問題がある場合、私たちは:
55
もし私たちがテストや市場承認を得る上で遅延に遭遇したら、私たちの製品開発コストも増加するだろう。われわれのいかなる臨床前研究や臨床試験が計画通りに開始されるかどうか,再構成が必要かどうか,あるいは予定通りに完成するかどうか,あるいは全く知られていない。私たちはまた、より多くの患者または腕を増加させることを含む、1つまたは複数の臨床試験の設計または計画を変更することを決定することができ、これは、コストおよび費用の増加および/または遅延をもたらす可能性がある。重大な臨床前研究または臨床試験遅延は、候補製品を商業化する独占的な権利を有する私たちの任意の期限を短縮するか、または私たちの競争相手が私たちの前に製品を市場に出すことを可能にし、候補製品を商業化することに成功する能力を弱化させ、私たちの業務と運営結果を損なう可能性がある。
我々は臨床経験の限られた疾患を治療するための候補製品を開発しているため、場合によっては、新しい終点または方法を使用して、FDAまたは他の監督機関は、臨床的に意義のある結果を予測または提供するために、私たちの臨床試験の終点を考慮しないかもしれない。
現在のところFSHD治療のための治療法は承認されておらず,われわれが解決しようとしているあるいは将来解決可能な疾患の根本的な原因を治療するための治療法も承認されていない可能性がある。したがって、これらの疾患を治療する新規性の一部として、これらの疾患を治療するための1つまたは複数の候補製品の設計および実施のための臨床試験は、より長い時間、より高いコスト、またはより低い効率を必要とする可能性がある。場合によっては、RWSのような新しいエンドポイントまたは新しいエンドポイントまたは方法を使用することができ、我々の知る限り、これらのエンドポイントまたは方法は登録可能であることが証明されていない。FDAや他の規制機関は,RWSを主要終点とし,副次的終点から他の適切な支持的データを得ることを支持することを示している。しかし,このような規制機関は,このような結果が臨床的に意義があると考えても,われわれの臨床試験の終点を考慮して臨床的に意義のある結果を提供しない可能性がある。例えば、私たちはすでに規制機関と会って、FSHDのロスパモッドのためのREACH試験設計と登録戦略を検討しましたが、規制機関はRWS機能の主要な終点をサポートして、FSHDのためのロスパモッドを承認するために追加のデータを必要とする可能性があります。
FDAが我々の主要な終点が十分な検証と臨床的意義を得ていることを確実に発見しても、私たちが候補製品のために行う可能性のあるいかなる重要な或いは他の臨床試験においても、私たちは予め指定された終点の規模、持続時間或いは統計的意義に達することができない可能性がある。われわれが確実に主要ゴールに到達したとしても,われわれの実験では予測不可能な結果が生じるか,あるいは試験中の他のより伝統的な治療効果の終点の結果と一致しない可能性がある。FDAは臨床試験データを解釈する際にかなりの重みを他の治療効果の終点に与えることも可能であり,主要終点で統計的に有意な結果が得られても,FDAはわれわれの副次的治療効果の終点に統計的に有意な影響を示さなかったと考えられる可能性があり,薬物治療効果への疑問である。FDAは製品の利益とリスクも考慮し,FDAは安全性を背景に治療効果結果を承認を支持していないと見なす可能性がある。ヨーロッパと他の国の他の規制機関はこのような端末に対して似たような結論を下すかもしれない。
もし私たちが臨床試験の患者登録過程で遅延や困難に遭遇した場合、必要な規制承認を受けることは延期または阻止される可能性がある。
患者を確定し、参加させ、著者らの候補製品の臨床試験を完成させることは著者らの成功に非常に重要である。臨床試験の成功とタイムリーな完成には,試験が終了するまで十分な数の患者を募集する必要がある。例えば,我々のFTX−6058 1 b期試験(現在臨床保留状態)では,初期キューで6人の被験者を募集したにもかかわらず,初期データ締め切りまでに3名の被験者のみが評価可能であった。その後,被験者のコンプライアンスを監視するために研究案を修正した。しかし,このようなシナリオが実験回復後にコンプライアンスやコンプライアンス(本当であれば)を向上させることができなければ,意味のあるデータを生成できない可能性がある.また,FDAや米国以外の同様の規制機関が要求するこれらの試験に十分な数の合格者を見つけて募集することができなければ,候補製品の臨床試験を開始または継続できない可能性がある。著者らは主に遺伝子定義の稀な疾患に注目しているため、著者らは十分な数の合格患者を募集することが困難かもしれない。
患者の入選は様々な他の要素の影響を受けている
56
著者らは十分な数の患者を発見し、著者らの臨床試験に参加することができず、重大な遅延を招き、1つ以上の臨床試験を完全に放棄し、必要な監督管理許可を得ることを延期或いは阻止する必要があるかもしれない。
私たちの臨床試験の登録遅延は私たちの候補製品の開発コストを増加させる可能性があり、これはわが社の価値を低下させ、追加融資を受ける能力を制限する。
もし私たちの候補製品の開発過程で深刻な有害事象や受け入れられない副作用が発見された場合、私たちは私たちのいくつかの候補製品の開発を放棄または制限する必要があるかもしれない。
私たちの候補製品が臨床試験における深刻な有害事象または副作用に関連している場合、または臨床試験または前臨床試験において予期しない特徴を有する場合、それらの開発を放棄するか、またはその開発をより狭い用途またはサブ集団に制限する必要があるかもしれないが、これらの集団では、リスク効果の観点から、重篤な有害事象、不良副作用、または他の特徴がそれほど一般的ではなく、それほど深刻ではない、またはより受け入れやすい。
例えば、2023年2月23日、FDAは非臨床毒理学研究で観察された血液悪性腫瘍に基づいて、著者らのFTX-6058のINDを臨床放置した。FDAがFTX−6058のさらなる臨床開発の適切な利益−リスクプロファイルを有するSCD患者集団に関する情報を提供することを要求することを含むFDAの懸念を可能な限り解決することを意図しているが、FDAは、健康なボランティアで行われ得る任意のさらなる研究の潜在的リスクを決定するために情報を提供することを要求しているが、FDAがFTX−6058の臨床開発を回復させることを可能にする保証はない。FDAが臨床制限を解除し、FTX-6058の臨床研究を回復することを許可しても、著者らはFTX-6058治療を受けた患者が将来血液系悪性腫瘍或いは他の不良事件に発展しないことを保証することができない。著者らも悪性血液病の臨床前研究において追加的な観察結果或いは他の不良事件が出現しないことを保証することはできない。このような追加的な有害事象が発生した場合、私たちの臨床研究のさらなる進展は停止または延期される可能性があり、私たちは規制部門のFTX−6058の承認を得ることができないかもしれない。私たちが規制部門のFTX-6058の承認を得ても、私たちのラベルは制限される可能性があり、および/または私たちの製品に対する市場の受容度は低下する可能性があり、私たちFTX-6058計画の商業潜在力は実質的な否定的な影響を受けるかもしれない。
薬物開発では,早期あるいは臨床試験で最初に希望を示した化合物の多くが後に発見され副作用を引き起こし,化合物のさらなる発展を遅延または阻止する。
さらに、我々の臨床試験結果が許容できない副作用を示す場合、我々、FDAまたは我々の研究機関のIRBsは、私たちの臨床試験を一時停止または終了することができ、またはFDAまたは同様の外国の規制機関は、臨床試験を停止するか、または私たちの任意またはすべての目標適応の候補製品を承認することを拒否するように命令することができる。治療に関連する副作用はまた、患者の募集または入選患者が任意の臨床試験を完成する能力に影響する可能性がある。もし私たちが候補製品の任意の臨床試験を一時停止または終了させることを選択または強制された場合、その候補製品の商業的将来性は損なわれ、候補製品から製品収入を生成する能力は延期またはキャンセルされるだろう。このような状況のいずれも私たちの業務に実質的な被害を及ぼす可能性がある。
57
もし私たちの候補製品が発売承認された場合、私たちまたは他の人は後にその薬の効果が以前に考えられていたほど効果的ではないことを発見したり、以前に発見されなかった副作用を招いたりして、私たちがその薬を販売する能力が影響を受ける可能性がある。
我々の候補製品の臨床試験は,入念に定義された臨床試験に同意した患者のサブセットで行われた。したがって,われわれの臨床試験では,候補製品の有意なプラス効果が実際のプラス効果(あれば)よりも大きいか,あるいは不良副作用を認識できない可能性がある。もし私たちの1つ以上の候補製品が監督部門の承認を得た場合、私たちまたは他の人はその後、それらの効果が以前に考えられていたほど効果的ではないことを発見し、あるいは不良な副作用をもたらし、多くの潜在的な重大な負の結果を招く可能性がある
これらの事件のいずれも、特定の候補製品に対する市場の受容度を達成または維持することを阻止することができ、承認されれば、私たちの業務、財務状況、および運営結果を深刻に損なう可能性がある。
私たちは、より利益的または成功可能性の高い候補製品または適応を利用することなく、特定の候補製品または適応を追求するために限られたリソースを費やす可能性がある。
私たちの財務と管理資源が限られているため、著者らは稀な神経筋肉、筋肉、血液と中枢神経系疾患の研究と開発に集中している。したがって、私たちは他の候補製品を探す機会を放棄または延期するか、または後により大きな商業潜在力を有することが証明された他の兆候を探す機会を放棄または延期するかもしれない。私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。私たちの現在と未来の研究開発計画および特定の適応候補製品への支出はいかなる商業的に実行可能な製品も発生しないかもしれない。もし私たちが特定の候補製品の商業的潜在力や目標市場を正確に評価しなければ、私たちは協力、許可、または他の印税手配によって候補製品に価値のある権利を放棄するかもしれないが、この場合、候補製品の独占開発権と商業化権利を維持することは私たちに有利だ。資源の配置や利用戦略に成功しなければ,我々の業務に悪影響を与える.
我々はヨーロッパ,イギリス,カナダでFSHD患者のロスパモントの臨床試験を行っており,現在米国以外の地点で我々の候補製品のより多くの臨床試験を行う予定であり,FDAはこれらの地点での試験データを受け入れていない可能性がある。
われわれは現在,3期臨床試験,われわれの2 b期臨床試験の開放ラベル拡張,および我々がヨーロッパ,イギリス,カナダで行ったFSHD患者におけるロスパモットの第二段階開放ラベル臨床試験の開放ラベル拡張を行っている。私たちはまたアメリカ以外でより多くの臨床試験を行うかもしれない。FDAは米国国外で行われた臨床試験のデータを受け入れることができるが,これらのデータの受け入れはFDAが適用した条件に依存する。例えば、臨床試験は入念に設計と進行し、合格した研究者が道徳原則に従って行わなければならない。試験群はまたアメリカ人口を十分に代表しなければならず、データはFDAが臨床的意義があると考えられる方式でアメリカ人口とアメリカの医療実践に適用しなければならない。また,これらの臨床試験は適用された現地法に制限されているが,FDAがデータを受け入れるかどうかは,試験もすべての適用された米国の法律や法規に適合しているかどうか,良好な臨床実践,FDAがデータを検証する能力を含むかどうかに依存する。もしFDAがアメリカ国外で行ったどんな実験のデータも受け入れなければ
58
米国であれば、これはより多くの実験を必要とする可能性があり、これは高価で時間がかかり、適用可能な候補製品の開発を延期または永久に停止する可能性がある。
私たちの候補製品の商業化に関するリスク
たとえ私たちのすべての候補製品が市場の承認を得ても、医者、患者、第三者支払人と医学界の他の人が商業成功に必要な市場受容度を達成できない可能性があり、しかも私たちの任意の候補製品の市場機会は、承認されれば、私たちが予想しているより小さいかもしれない。
もし私たちのすべての候補製品が市場の承認を得たら、それはまだ医者、患者、第三者支払人と医学界の他の人の十分な市場受容度を得ることができないかもしれない。教育医療界や第三者支払者が我々の候補製品のメリットを知るには大量の資源が必要であり,成功しない可能性がある。もし私たちの候補製品が十分な受容度に達していなければ、私たちは著しい製品収入が生じないかもしれません。私たちは利益を上げることができないかもしれません。もし私たちの候補製品が商業販売に使用されることが許可されれば、市場の受け入れ度は多くの要素に依存するだろう
候補製品の潜在的な市場機会の評価は,業界出版物や研究,調査,第三者による研究から得られた業界と市場データに基づいており,そのうちの1つは我々が依頼したものである.業界出版物および第三者研究、調査および研究は、一般に、それらの情報は、そのような情報の正確性または完全性を保証しないにもかかわらず、信頼できるソースから得られることを示している。私たちはこれらの業界の出版物と第三者研究、調査と研究が信頼できると信じているが、私たちはこれらのデータを独立して確認していない。候補製品の潜在的な市場機会の推定は、著者らの業界知識、業界出版物と第三者研究、調査と研究に基づくいくつかの重要な仮定を含み、これらの仮定は小サンプルに基づく可能性があり、市場機会を正確に反映できなかった。私たちは私たちの内部仮定が合理的だと信じているが、これらの仮定を証明する独立したメッセージ源はない。もし私たちの任意の仮定や推定、あるいはこれらの出版物、研究、調査、または研究が不正確であることが証明された場合、私たちの任意の候補製品の実際の市場は私たちが予想していたより小さいかもしれないので、私たちの製品収入は制限されるかもしれません。私たちは利益を達成したり維持したりすることがもっと難しいかもしれません。
もし私たちが販売、マーケティング、流通能力を確立できない場合、あるいは第三者と販売、マーケティング、流通合意を達成できなければ、私たちの候補製品が承認されたら、商業化に成功できないかもしれません。
私たちは医薬製品を販売したり、マーケティングしたり、流通した経験もありません。私たちが発売許可を得た任意の製品を商業的に成功させるためには、販売、マーケティング、流通組織を構築する必要があり、私たち自身であるか、第三者との協力やその他の手配を通じて行う必要がある。
59
将来、私たちのいくつかの候補製品が承認されれば、私たちはこれらの製品をアメリカでマーケティングするために、集中的で専門的な販売、マーケティングインフラを構築したいと思います。私たち自身の販売、マーケティング、そして流通能力を確立することはリスクに関するものだ。例えば、販売チームの採用と訓練は高価で時間がかかり、どんな製品の発表も延期される可能性がある。販売チームを募集し、マーケティング能力を確立する候補製品の商業発表が何らかの理由で延期または発生していない場合、これらの商業化費用を早期または不必要に発生させる。これらの努力は費用が高いかもしれません。もし私たちが私たちの販売とマーケティング担当者を維持したり、再配置できなければ、私たちの投資は損失します。
私たち自身が製品を商業化するのを阻害するかもしれません
もし私たちが自分の販売、マーケティング、流通能力を確立することができなければ、私たちは第三者と合意してこれらのサービスを提供し、私たちの製品収入と私たちの収益力(もしあれば)は私たち自身が開発したいかなる製品もマーケティング、販売、流通を下回るかもしれません。さらに、私たちは第三者との販売、マーケティング、流通に成功できないかもしれません。あるいは私たちが受け入れられる条項でそうすることができないかもしれません。私たちはこのような第三者に対して支配権がほとんどないかもしれません。彼らのいずれも必要な資源と注意力を投入して、私たちの製品を効果的に販売し、マーケティングすることができないかもしれません。もし私たちが販売、マーケティング、流通能力を成功的に確立できなければ、私たち自身も第三者と協力しても、私たちの候補製品を商業化することに成功しないだろう。
私たちは激しい競争に直面しており、これは他の人たちが私たちよりも製品の発見、開発、商業化に成功する可能性がある。
新薬製品の開発と商業化競争は激しい。私たちの現在の候補製品は競争に直面しており、将来的に開発または商業化を求める可能性のある任意の候補製品も、世界各地の主要な製薬会社、専門製薬会社、バイオテクノロジー会社からの競争に直面するだろう。現在,多くの大手製薬やバイオテクノロジー会社が製品をマーケティング·販売しているか,あるいは我々が開発している製品候補の多くの疾患適応を治療するための製品が開発されている。競争力のある製品および療法のいくつかは、私たちの方法と同じまたは類似した科学的方法に基づいており、他のいくつかは完全に異なる方法に基づいている。潜在的な競争相手はまた学術機構、政府機関とその他の公共と個人研究組織を含み、これらの組織は研究を展開し、特許保護を求め、研究、開発、製造と商業化のための協力手配を確立する。
例えば、臨床開発におけるいくつかの候補製品は、私たちが開発および商業化に成功する可能性のある候補製品と競争力がある可能性があることが知られている。本年度報告書における表10-Kの第1項“ビジネス競争”を参照されたい。
60
もし私たちの競争相手が私たちが開発する可能性のあるどの製品よりも安全で、より効果的で、副作用が少なく、より便利で、より安い製品を開発し、商業化すれば、私たちのビジネス機会は減少または消失するかもしれない。私たちの競争相手も私たちよりも早くFDAや他の規制機関のその製品の承認を得ることができ、これは私たちの競争相手が私たちが市場に入る前に強力な市場地位を確立することをもたらすかもしれない。また、多くの場合、私たちの競争能力は、後発薬の使用を奨励することを求める保険会社または他の第三者支払者の影響を受ける可能性がある。もし私たちの候補製品が市場の承認を得たら、私たちはそれらの価格が競争相手の模造薬より著しく高いと予想する。
私たちと比較して、私たちが競争しているか、あるいは将来競争する可能性のある多くの会社は、研究開発、製造、臨床前テスト、臨床試験を行い、監督管理許可とマーケティング承認製品を獲得する上で、より多くの財務資源と専門知識を持っている。
製薬とバイオテクノロジー産業の合併と買収は、私たちの数の少ない競争相手により多くの資源を集中させる可能性がある。規模の小さい会社や他のスタートアップ会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配されている。これらの第三者は合格した科学と管理者を募集と維持し、臨床試験場と臨床試験の患者登録を確立し、そして私たちの計画と相補的或いは必要な技術を獲得する上で私たちと競争している。
もし私たちの候補製品の市場機会が私たちが思っているより小さいなら、私たちの収入は不利な影響を受けるかもしれません。私たちの業務は影響を受けるかもしれません。私たちの候補製品のいくつかの目標患者集団は小さいため、アドレス指定可能な患者集団はさらに少なく、私たちは患者を成功裏に識別し、利益と成長を達成するためにかなりの市場シェアを占めることができなければならない。
我々の研究と製品開発は主に遺伝子定義のまれな疾患の治療に集中している。我々が目標としているまれな疾患を有する患者数が少ないことを考慮して,これらのまれな疾患を有する患者の識別に成功し続けており,我々の成長と収益性に重要である。これらの疾患を有する人の数と、私たちの候補製品治療から利益を得る可能性があるこれらの疾患患者のサブセットの予測は、私たちの信念と推定に基づいている。これらの推定は、科学文献、診療所調査、患者基金、あるいは私たちが行った市場研究を含む様々な源から来ており、不正確であるか、あるいは誤りを含むことが証明される可能性がある。新しい研究はこれらの疾病の推定発病率或いは流行率を変えるかもしれない。患者数は予想より少ないかもしれない。我々が治療を求めている疾患を識別する患者の努力はまだ早期段階であり,治療が得られる可能性のある患者数を正確に予測することはできない。さらに、私たちの各候補製品の潜在的にアドレス指定可能な患者集団は限られている可能性があるか、または私たちの候補製品の治療を受け入れられない可能性があり、新しい患者はますます識別または接触が困難になる可能性があり、これは私たちの運営結果およびトラフィックに悪影響を及ぼすであろう。また、私たちが候補製品のためにかなりの市場シェアを得ていても、私たちが評価している多くの指標の潜在的なターゲット層が非常に少ないので、私たちはこのような巨大な市場シェアを獲得しているにもかかわらず、決して利益を達成しないかもしれない。
われわれが評価しているいくつかの適応の対象患者群は比較的少なく,現在のところFSHDのようなわれわれの目標適応に対する看護治療基準はない。したがって、私たちの候補製品の定価と精算が承認されれば不確実だが、商業インフラを支援するのに十分でなければならない。もし私たちが十分な清算水準を得ることができなければ、私たちが候補製品をマーケティングして販売する能力は不利な影響を受けるだろう。
私たちは依存して、私たちの候補製品を生産するためにCMOに依存し続けると予想される。もし私たちが予想された計画を達成できない場合、あるいはこれらの組織が私たちの供給要求を満たすことができなければ、私たちの候補製品の開発および/または商業化は延期されるかもしれない。
私たちは何の製造施設もなく、依存し、引き続き第三者が私たちの候補製品を生産する臨床用品に依存することが予想され、適用された規制機関の承認を得て発売されれば、第三者が私たちの製品を生産する商業用品、包装、滅菌、貯蔵、流通、その他の生産物流に依存すると予想される。もし私たちが予想していた条項やスケジュールに従ってこのような計画を達成できなければ、私たちの候補製品の開発および/または商業化は延期されるかもしれない。
これらの第三者が規制要件に従って契約責任を履行し、予想される期限内に候補製品を完成または製造することができなかった場合、これらの当事者との間に食い違いがある場合、または市場で承認された任意の候補製品の商業化を支援する能力を拡張することができない場合、供給要求を満たすために十分な候補製品の生産を満足または遅延させることができない可能性があり、または私たちは自分で材料を製造することを余儀なくされる可能性があり、能力や資源がないか、または他の製造業者と合意することができない可能性があり、合理的な条項でこれを行うことができないかもしれない。もし本当にあれば。いずれの場合も臨床試験の供給が著しく遅れる可能性があります代替案を確立したからです
61
供給源。場合によっては、私たちの製品または候補製品を製造するために必要な技術的スキルは、元の製造業者固有または独自である可能性があり、困難に遭遇する可能性があり、またはそのようなスキルを予備または代替サプライヤーに譲渡することを禁止する契約制限が存在する可能性があり、またはそのようなスキルを全く譲渡できない可能性があります。
また、何らかの理由でメーカーの交換を要求された場合、新しいメーカーが品質基準およびすべての適用法規に適合する施設やプログラムを持っているかどうかを確認することを要求されます。我々はまた、例えば比較可能な研究を製造することによって、任意の新しい製造プロセスが、以前にFDAまたは他の規制機関に提出された仕様に基づいて、私たちの候補製品を生産することを検証する必要がある。新メーカーの検証に関連する遅延は、私たちがタイムリーにまたは予算内で候補製品を開発したり、製品を商業化する能力に悪影響を及ぼす可能性があります。また、メーカーは、当該メーカーが独立して所有する我々の候補製品の製造に関する技術を持つことができる。これは私たちのこれらのメーカーへの依存を増加させ、あるいは他のメーカーが私たちの候補製品を生産するために、これらのメーカーからライセンスを取得することを要求するだろう。また、メーカーの変化は通常、製造プロセスやプロセスの変化に関連しており、これは、臨床試験で使用されている以前の臨床供給と任意の新しいメーカーの供給との間の過渡的な研究が要求されるかもしれない。臨床用品の比較可能性の証明には成功しない可能性があり,追加の臨床試験が必要かもしれない。これらの施設はまた、洪水や火災、公衆衛生問題(例えば、新冠肺炎などの感染症の発生)などの自然災害の影響を受ける可能性があり、あるいはそのような施設は、汚染やそのような施設の監督検査後の規制懸念などの製造問題に直面している可能性がある。
私たちの第三者メーカーはFDAの検査と承認を受けて、その後、私たちの任意の候補製品の製造と販売を開始し、その後、時々FDAの検査を受けることができます。もし私たちの第三者製造業者がこのような検査を通過し、他の方法でFDAの私たちの候補製品に対する承認案を円満に完成させなかった場合、FDA Form 483の観察通知、警告状、または禁止または運営許可証の取り消しなどの規制行動を引き起こす可能性がある。
我々の競争相手や他社がこのような原料または原料薬を購入することによる不足を含む生産能力制限または原料または原料薬市場の遅延または中断により、我々または我々の第三者メーカーはまた、我々の候補製品を生産するために必要な原材料または原料薬の不足に遭遇する可能性があり、これらの原料または原料薬の数は、私たちの臨床試験に必要な数を必要とし、または、候補製品が承認された場合、商業化または需要の増加のために十分な数を使用することができる。もし私たちまたは私たちの第三者メーカーが私たちの候補製品を生産するのに必要な原材料や原料薬を十分な数得られなかったら、私たちの業務に重大な悪影響を及ぼすかもしれません。
第三者への私たちの依存は、許容可能なコストまたは品質で十分な数の候補製品または製品またはそのような数を得ることができないリスクを増加させ、私たちの開発または商業化努力を延期、阻止、または損害する可能性がある。
私たちには何の生産施設もありません。グラクソ·スミスクラインから十分なlosmapimod錠剤が得られたと信じていますが、FSHD治療のためのlosmapimodの臨床試験を完了していると信じていますが、私たちの医薬製品と原料薬の需要を正確に推定しているか、あるいはその医薬製品や原料薬が使用しようとするまで有効期限が切れていないとは判断できません。私たちはまたCMOを招いて私たち自身の原料薬を準備し、ロスパモ錠を生産した。私たちは私たちが計画した第3段階の登録試験を完了するのに十分なロスパモ錠を生産したと信じているが、私たちは私たちの薬物製品と原料薬の需要を正確に推定したか、あるいはこれらの薬物製品または原料薬は私たちが使用しようとするまで期限が切れないと確信できない。また,臨床的放置問題を解決できれば,我々のSCD 1 b期臨床試験を完成させるのに十分な数のFTX−6058がCMOから得られていると信じているにもかかわらず,我々の薬物製品需要を正確に見積もることはできず,開発努力を延期,阻止,または損なう可能性がある。
著者らは未来のいかなる臨床試験及び臨床前と臨床試験候補製品の生産は第三者のFTX-6058生産に依存すると予想している。 私たちはまた、私たちまたは私たちのパートナーが市場の承認を得た任意の他の候補製品の商業供給を生産するために、第三者製造業者または第三者パートナーに依存したい。このような第三者への依存は、許容可能なコストまたは品質で十分な数の候補製品または製品またはそのような数を得ることができないリスクを増加させ、これは、私たちの開発または商業化努力を延期、阻止、または損害する可能性がある。
私たちの候補製品と私たちが開発する可能性のあるどの製品も他の候補製品や製品と製造施設を競争する可能性があります。CGMP法規の下で運営されているメーカーの数は限られており、私たちのために製品を製造する能力があるかもしれない。
私たちの既存または未来のメーカーのどんな業績失敗も、臨床開発やマーケティング承認を延期する可能性がある。私たちは現在余分な供給の手配もなく、大量の麻薬物質の供給源もない。もし私たちの未来の契約メーカーが約束通りに契約を履行できなければ、私たちはこれらのメーカーの交換を要求されるかもしれない。いくつかの潜在的な代替メーカーが私たちの候補製品を生産できると信じていますが、そのような代替製品を決定して同定する際に、追加のコストと遅延が生じる可能性があります。
62
私たちの現在と未来の他人が私たちの候補製品や製品を生産することへの依存は、私たちの将来の利益率と、マーケティングの承認をタイムリーかつ競争力的に獲得する製品の商業化能力に悪影響を及ぼす可能性があると予想されています。
任意の候補製品を商業化することができても、これらの製品は、不利な価格設定法規、第三者保険、清算やり方、医療改革措置の制約を受ける可能性があり、これは私たちの業務を損なう可能性があります。
新薬製品の発売審査、定価、カバー範囲と精算を管理する規定は国によって異なる。現在と未来の立法は承認要求を大幅に変更する可能性があり、これは追加のコストに関連し、承認の遅延を招く可能性がある。一部の国は薬品の販売価格が承認されてから発売されることを要求している。多くの国で、定価審査期間はマーケティングまたは製品許可を承認した後から始まる。一部の外国市場では、処方薬の定価は初歩的な承認を得た後も、政府の持続的なコントロールを受けている。したがって、特定の国/地域での製品のマーケティング承認を得ることができるかもしれませんが、その後、価格法規の制約を受けて、これらの法規は私たちの製品の商業発表を延期し、長い時間遅延し、その国/地域で製品を販売することによって生じる収入に悪影響を与える可能性があります。不利な価格設定制限は、私たちの候補製品が市場承認を得ても、1つ以上の候補製品への投資を回収する能力を阻害する可能性がある。本年度報告表格10-K中の第1項“企業-政府監督と製品審査--薬品保険カバー範囲と医療保健改革”を見た。
私たちが任意の候補製品を商業化することに成功した能力はまた、政府衛生行政当局、個人健康保険会社、および他の組織がこれらの製品および関連治療に提供する保険範囲と十分な補償にある程度依存するだろう。政府当局と第三者支払者、例えば個人健康保険会社や健康維持組織は、どのような薬剤を支払うかを決定し、精算レベルを確立する。アメリカの医療業界と他の地域の主な傾向の一つはコストコントロールだ。政府当局と第三者支払者は,特定の薬物のカバー範囲や精算金額を制限することでコストを抑制しようとしている。ますます多くの第三者支払人は製薬会社に価格に基づいて所定の割引を提供し、医療製品の定価に挑戦することを要求している。私たちが商業化しているどの製品も保険や精算を受けることができないかもしれません。あっても、精算レベルは満足できないかもしれません。精算は私たちが市場で承認された任意の候補製品の需要や価格に影響を及ぼすかもしれない。私たちの製品のために十分な精算を得て維持することは難しいかもしれない。私たちは、カバー範囲と精算または他の治療法に対する精算レベルを証明するために、高価な薬物経済学的研究を要求されるかもしれない。カバー範囲や十分な精算がない場合、あるいは精算が限られたレベルに限られている場合、マーケティングの承認を得た任意の候補製品を商業化することに成功しない可能性があります。第三者支払者の製品使用状況の決定を含む第三者支払人の保証範囲と精算は多くの要素に依存する可能性がある
新たに承認された薬物については,保険獲得や精算に重大な遅延がある可能性があり,保険範囲はFDAや米国以外の同様の規制機関がこの薬剤を承認する目的よりも限られている可能性がある。また、保険や精算を受ける資格があるということは、すべての場合に薬物の費用を支払うことを意味するのではなく、研究、開発、製造、販売、流通費用を含む私たちのコストを支払うことを意味するわけではない。もし適用されれば、新薬の一時精算レベルも私たちのコストを支払うのに十分ではないかもしれないし、永久的にならないかもしれない。販売率は薬物の使用や臨床環境によって異なる可能性があり,すでに低コスト薬物のために設定された精算レベルに基づいている可能性があり,他のサービスの既存支払いにも組み込まれている可能性がある。医薬品の正味価格は、政府医療計画または個人支払者が要求する強制的な割引またはリベート、および将来、米国価格よりも低い価格で販売される可能性のある国から医薬品を輸入することを制限する法律の緩和によって低下する可能性がある。第三者支払者が自分の精算政策を設定する際には,通常連邦医療保険カバー政策と支払制限に依存する。私たちは政府の資金援助と個人支払人から私たちが開発してくれた任意の承認された製品から保険と十分な販売率を迅速に得ることができません。これは私たちの経営業績、製品の商業化に必要な資金を調達する能力、そして私たちの全体的な財務状況に実質的な悪影響を及ぼすかもしれません。
63
私たちの候補製品がアメリカや他の国/地域で販売されることが承認されても、私たちの候補製品が医学的に合理的で必要であることは保証されず、特定の適応や第三者支払者にとって費用対効果的であり、私たちの保険範囲と十分な精算レベル、あるいは第三者支払者の精算政策が私たちの候補製品を販売する収益力に悪影響を与えない保証はない。
私たちの将来の成長は海外市場に進出する能力にある程度依存しており、そこでは追加の規制負担や他のリスクや不確実性の影響を受け、これらのリスクや不確実性が現実になれば、私たちの業務を損なう可能性があります。
私たちの将来の収益性は、アメリカとEU以外の市場で私たちの候補製品を商業化する能力にある程度依存するだろう。もし私たちの候補製品を海外市場で商業化すれば、私たちは追加のリスクと不確定要素に直面します
これらの不確実性のいずれかに関連するリスクが現実的になれば、我々の業務に実質的な悪影響を及ぼす可能性がある。
私たちの臨床試験と製品責任訴訟は私たちの資源を移転し、私たちが重大な責任を負い、私たちが開発する可能性のある任意の製品の商業化を制限する可能性があります。
私たちは、人体臨床試験で私たちの候補製品をテストすることに関連する固有の臨床試験と製品責任暴露のリスクに直面しており、私たちが開発可能な任意の製品を商業的に販売すれば、より大きなリスクに直面する。現在、商業販売のための製品は承認されていませんが、現在と将来の臨床試験で使用される候補製品、将来の任意の承認された製品の販売は、私たちを責任クレームに直面させるかもしれません。これらのクレームは、その製品を使用する患者、ヘルスケア提供者、製薬会社、またはそのような製品を販売する他の人によって提起される可能性がある。もし私たちが私たちの候補製品や製品に被害を与えるクレームを自己弁護することに成功できなければ、私たちは大きな責任を招くだろう。是非曲直や最終結果にかかわらず、賠償責任は
64
私たちは現在全部で1,000万ドルの臨床試験責任保険を持っています。各事件の上限は1,000万ドルで、私たちが生じる可能性のあるすべての責任を支払うのに十分ではないかもしれません。私たちの臨床試験の拡大に伴い、あるいは候補製品を商業化し始めたら、私たちの保険カバー範囲を増やす必要があるかもしれません。保険範囲はますます高くなっています。私たちは可能などんな責任にも対応するために、合理的な費用や十分な金額で保険範囲を維持することができないかもしれない。成功した臨床試験または製品責任クレームまたは一連のクレームが、保険されていない負債または保険を超える負債によって私たちに提起された場合、私たちの資産はそのようなクレームを支払うのに十分ではない可能性があり、私たちの業務運営は損害を受ける可能性がある。
私たちの第三者への依存に関するリスク
我々は第三者に依存して我々の臨床試験を継続することが予想され,これらの第三者の表現は締め切りまでにこのような試験を完了できないことを含めて満足できない可能性があり,我々の業務を損なう可能性がある。
我々は現在第三者契約CROによる臨床試験を行っている。我々は第三者CROあるいは第三者研究協力に依存して任意の未来の臨床試験を行う予定である。私たちは私たちの他の候補製品に対して独立した臨床試験をするつもりはない。CRO,臨床データ管理組織,医療機関,臨床研究者など第三者に依存して臨床試験を行う予定である。これらのプロトコルは、第三者が履行できなかったことを含む様々な理由で終了する可能性がある。もし私たちが代替計画を達成する必要があれば、私たちの製品開発活動は延期されるかもしれない。
私たちのこれらの第三者の研究開発活動への依存は、これらの活動に対する私たちの統制を減少させるだろうが、私たちの責任を軽減することはない。
もしこれらの第三者が法規の要求や私たちが規定した規程に従って契約の職責を成功裏に履行し、期待された期限内に私たちの臨床試験を完成または行うことができない場合、私たちは私たちの候補製品のマーケティング承認を得ることができないか、遅延する可能性があり、私たちの候補製品の開発と商業化に成功した努力を遅延させることができないかもしれない。さらに、これらの第三者は、他のエンティティと関係がある可能性もあり、その中のいくつかは私たちの競争相手である可能性がある。
私たちはまた依存し、引き続き他の第三者に依存して私たちの臨床試験のための薬品の貯蔵と配布を提供することが予想される。私たちのディーラーのどんな業績ミスも、私たちの候補製品の臨床開発やマーケティング承認、あるいは私たちの製品の商業化を延期し、追加の損失を生じ、私たちの潜在的な製品収入を奪う可能性があります。
私たちは未来に第三者と協力して、私たちの候補製品を発見、開発、または商業化することができます。もし私たちの協力が成功しなければ、私たちはこれらの候補製品の市場潜在力を利用できないかもしれないし、私たちの業務は不利な影響を受けるかもしれない。
私たちはMyoKardiaと協力して許可プロトコル(ある遺伝子で定義された心筋症)を持っている。本年度報告書の表10-Kの項目1“ビジネスライセンス契約および連携”を参照してください。現在の候補製品のすべての権利を保持し、独自に開発していますが、私たちは将来、既存または未来の候補製品について開発、流通、またはマーケティング計画を第三者と達成するかもしれません。このような販売、マーケティング、流通、開発、許可、またはより広範な協力手配のいずれかにおいて可能なパートナーは、大中型製薬会社、地域的および全国的な製薬会社、およびバイオテクノロジー会社を含む。もし私たちが将来どの第三者ともこのような計画を達成すれば、私たちの協力者が私たちの候補製品の開発や商業化に投入する資源の数と時間を制限する可能性が高い。私たちがこれらの計画から収入を創出する能力は、私たちの協力者がこれらの計画の中で彼らに割り当てられた機能を成功的に履行する能力と努力にかかっているだろう。
65
私たちが参加した協力は、MyoKardiaとの協力を含めて、成功しないかもしれませんが、どんな成功もこれらの協力者の努力と活動に大きく依存します。協力は以下のようなリスクを含む多くのリスクをもたらす
66
協調プロトコルは、候補製品の開発や商業化を最も効果的な方法で招くことができないか、または全くそうではない可能性がある。もし私たちが行ったいかなる協力が製品の開発と商業化に成功しなかった場合、または私たちの協力者が私たちとの合意を終了した場合、私たちはその協力によって未来の研究資金、マイルストーン、または印税支払いを得ることができないかもしれない。もし私たちがこれらの合意に基づいて期待した資金を受け取っていなければ、私たちの候補製品の開発は延期されるかもしれません。私たちは私たちの候補製品を開発するために追加の資源が必要かもしれません。ここで説明した製品開発、規制承認、商業化に関するすべてのリスクは、私たちの協力者の活動にも適用されます。
さらに、私たちの契約義務に対する制約の下で、私たちの協力者が業務合併に参加する場合、その協力者は、私たちの許可された任意の候補製品の開発または商業化を淡水化または終了する可能性がある。例えば、2020年11月、MyoKardia協力協定を締結した後、MyoKardiaは百時美施貴宝社に買収された。百時美施貴宝社は、MyoKardiaの開発計画の優先順位を再決定し、私たちの計画の開発を中止し、および/またはMyoKardiaと私たちとの間の合意を終了させることを決定する可能性がある。もし私たちの協力者が私たちとの合意を終了したら、私たちは新しい協力者を引き付けることがもっと難しいことを発見するかもしれないし、ビジネスや金融界での私たちの見方は不利な影響を受けるかもしれない。
もし私たちが協力を構築したり維持したりできなければ、私たちの開発と商業化計画を変えなければならないかもしれません。私たちの業務は悪影響を受ける可能性があります。
私たちのいくつかの候補製品については、製薬会社やバイオテクノロジー会社と協力して、これらの候補製品を開発し、潜在的な商業化を行うことを決定するかもしれません。例えば、2020年7月、著者らはMyoKardiaと協力と許可協定を達成し、いくつかの遺伝子定義の心筋症の潜在的治療のための潜在的な生物標的を決定し、検証する。私たちは適切なパートナーを探す上で激しい競争に直面しており、いくつかのより成熟した会社も私たちが魅力的だと思う第三者知的財産権をライセンスしたり獲得したりする戦略を求めているかもしれない。これらの老舗会社はその規模、財務資源及び更に強い臨床開発と商業化能力のため、私たちより競争優位を持っているかもしれない。しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。私たちが協力について最終的な合意に到達するかどうかは、他に加えて、パートナーの資源と専門長の評価、提案された協力の条項と条件、提案されたパートナーのいくつかの要因の評価に依存する。これらの要因は、臨床試験の設計または結果、FDAまたは米国国外の類似規制機関の承認の可能性、候補研究製品の潜在的市場、そのような候補製品の製造と提供のコストと複雑性、競争製品の潜在性、技術所有権の不確実性を含む可能性があり、挑戦の利点を考慮せずにこのような所有権に挑戦すれば、不確実性が存在する可能性がある, 一般的な産業や市場状況を見ています協力者はまた、同様の協力可能な指示を得るための代替候補製品または技術を考慮することができ、そのような連携が、私たちと私たちとの連携よりも私たちの候補製品に魅力的であるかどうかを考慮することができる。
既存または将来のライセンス契約によれば、私たちも制限される可能性があり、潜在的な協力者と特定の条項の合意を締結することはできません。たとえば,我々の現在のGSKとのライセンスプロトコルにより,我々はGSK第1交渉権に制限されており,Acceleronとの連携も制限されている.MyoKardiaとの協力の下で、我々は、(A)任意の適応の治療、予防または診断を行う研究活動において決定された特定の標的に対するプロトコルにおいて決定された特定の標的の化合物または製品、または(B)任意の遺伝子定義心筋症の治療のための薬剤、およびその後の指定された時間内に、任意の遺伝子定義心筋症を治療するための医薬、または(B)任意の遺伝子定義心筋症の治療のための医薬のいずれかの化合物または製品の利用が制限されている。
協力の交渉と記録は複雑で時間がかかる。また,最近では大手製薬とバイオテクノロジー会社間の業務合併数が大きく,将来の潜在的パートナー数の減少を招いている。
もし私たちが適時に、受け入れ可能な条項によって、または適切なパートナーと合意できない場合、私たちは候補製品の開発を削減し、その開発計画または私たちの1つまたは複数の他の開発計画を減らしたり、その潜在的な商業化を延期したり、いかなる販売またはマーケティング活動の範囲を縮小したり、または私たちの支出を増加させ、自費で開発または商業化活動を行わなければならないかもしれない。もし私たちが自分で援助して開発や商業化活動に従事することを選択すれば、私たちはより多くの専門知識と追加的な資本を得る必要があるかもしれないが、これらは私たちが受け入れられない条件や根本的には得られないかもしれない。もし私たちが協力できず、必要な開発や商業化活動を展開するのに十分な資金や専門知識がなければ、私たちの候補製品をさらに開発したり、市場に出したり、私たちの製品エンジンを開発し続けることができないかもしれません。
67
私たちの知的財産権に関するリスクは
もし私たちの技術や候補製品のために特許保護を獲得し、維持し、実行し、保護することができない場合、または取得された特許保護範囲が十分でなければ、私たちの競争相手は、私たちと同様または同じ技術および製品を開発し、商業化する可能性があり、私たちの技術および候補製品の開発と商業化に成功する能力は不利な影響を受ける可能性がある。
私たちの成功は、私たちが開発した任意の独自技術および候補製品について、私たちが単独で他人と共同で所有する可能性のある知的財産権の保護、または他の人から許可、特にアメリカおよび他の国/地域での特許を取得し、維持する能力があるかどうかに大きく依存する。我々は,米国や海外で我々の業務に重要な候補製品に関する特許出願を提出し,我々の技術や候補製品に関連する知的財産権を付与することで,我々の独自の地位を保護することを求めている.私たちがどんな独自技術または候補製品の特許保護を獲得または維持できない場合、私たちの業務、財務状況、運営結果、および見通しは深刻な損害を受ける可能性があります。
特許訴訟過程は高価で、時間がかかり、複雑であり、私たちは合理的なコストまたはタイムリーな提出、起訴、維持、弁護、またはすべての必要または望ましい特許出願を許可することができないかもしれない。我々は,特許保護を得るのが遅くなるまで,我々の研究開発成果で特許を申請できることを確認できない可能性もある.さらに、場合によっては、私たちは、第三者から許可された技術を含む特許出願の準備、提出、起訴、または特許を維持、実行、擁護する権利がない。したがって、これらの許可内の特許および出願は、私たちの業務の最良の利益に適合する方法で準備、提出、起訴、維持、弁護、実行されてはならない。
製薬とバイオテクノロジー会社の特許地位は通常高度に不確定であり、複雑な法律と事実問題に関連しており、近年多くの訴訟のテーマとなってきた。また、米国以外の特許保護範囲は不確定であり、外国法律は米国法律のように私たちの権利を保護しない可能性があり、その逆も同様である。例えば,欧州特許法は米国法よりも人体治療法に対する特許性制限が多い。所有およびライセンス内の特許権については、我々および我々のライセンシーが現在求めている特許出願が任意の特定の司法管轄区域で特許として発行されるか否か、または任意の発行された特許の権利主張が競争相手から十分な保護を提供するかどうかを予測することはできない。しかも、私たちは私たちの候補製品に関連する可能性のあるすべての第三者知的財産権を知らないかもしれない。さらに、科学文献に発表された発見は、実際の発見よりも遅れがちであり、米国および他の管轄区域の特許出願は、通常、出願18ヶ月後に発表されるか、または場合によっては全く発表されない。したがって、私たちまたは私たちのライセンシーは、私たちまたは私たちのライセンスが、私たちが現在または将来可能な特許および特許出願に要求された最初の発明であるか、または私たちまたは私たちのライセンシーが最初にそのような発明のために特許を出願して保護されたものであることを確実に知ることはできない。したがって、私たちが所有していると許可されていない特許権の発行、範囲、有効性、実行可能性、商業的価値は高い不確実性を持っている。さらに、私たちが持っているおよび許可されていない保留および将来の特許出願は、私たちの技術および候補製品、全部または一部を保護するための特許発行につながらないかもしれない, あるいは他の人が競争力のある技術や製品を商業化することを効果的に阻止する。米国や他の国の特許法または特許法解釈の変化は、私たちの特許の価値を低下させ、私たちの特許権を獲得、保護、維持、擁護、実行する能力を低下させ、私たちの特許保護の範囲を縮小し、より広く言えば、私たちの特許権の価値や範囲に影響を与えたり縮小したりする可能性がある。我々の特許組合に関する情報は、本年度報告表格10−Kの第1項“商業−知的財産権”を参照されたい。
さらに、私たちまたは私たちの許可者は、第三者が既存技術を米国特許商標局(USPTO)に提出する第三者によって事前発行されるか、または反対、派生、撤回、再審査、当事者間の審査、付与後審査、または私たちの特許権または他の人の特許権に挑戦する介入手続きに参加する可能性がある。このような提出、訴訟、または訴訟における不利な裁決は、私たちの特許権の範囲を縮小したり、無効にしたりして、第三者が私たちの技術または候補製品を商業化し、私たちに支払うことなく、直接競争することを可能にしたり、第三者特許権を侵害することなく薬物を製造または商業化することができなくなる可能性があります。もし私たちの特許および特許出願によって提供される保護の広さまたは強度が脅かされている場合、結果にかかわらず、会社が私たちと協力し、現在または将来の候補製品を認可、開発、または商業化することを阻止することができる。例えば、我々の米国特許に含まれる特定および汎用声明は、losmapimodを使用してFSHDを治療する方法の保護を提供していると考えられているが、我々が発行および係属中の米国の非一時的および一時的出願に含まれている特定および汎用声明は、FTX−6058の医薬成分および使用方法を保護していると考えられるが、第三者はこのような声明に疑問を提起することができる。もしこのような任意の理由で無効または実行できないと宣言された場合、私たちは貴重な知的財産権を失い、他人が私たちと競争することを阻止する能力も損なわれるだろう。
68
また、特許出願において要求されるカバー範囲は、特許発行前に大幅に縮小することができ、その範囲は特許発行後に再解釈することができる。私たちが所有して許可された特許出願が特許の形で発表されても、それらの発表形態は私たちに何の意味のある保護も提供してくれず、競争相手が私たちと競争することを阻止したり、他の方法で私たちにどんな競争優位性を提供してくれたりしない。特許の発行は、その発明性、範囲、有効性、または実行可能性に対して決定的ではなく、私たちが持っている特許および許可されていない特許は、米国および海外の裁判所または特許庁で挑戦される可能性がある。このような挑戦は、独占的または経営の自由を失うこと、または特許主張の全部または部分的な縮小、無効、または実行不能をもたらす可能性があり、それにより、他人が類似または同じ技術および製品を使用することを阻止するか、またはそれを商業化する能力を制限するか、または我々の技術および候補製品の特許保護期間を制限することができる。最終的な結果が私たちに有利であっても、このような訴訟は巨額のコストを招く可能性があり、私たちの経営陣や従業員に時間がかかる必要がある。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。さらに、私たちの競争相手は、同様の技術または代替技術または製品を非侵害的に開発することによって、私たちが所有または許可している特許を迂回することができるかもしれない。したがって、私たちが所有し、許可された特許の組み合わせは、他社が私たちの任意の候補技術や製品と似ているか、または同じ技術および製品を商業化することを排除するために、十分な権利を提供してくれないかもしれない。
特許条項は候補製品に対する私たちの競争地位を十分に長く保護するのに十分ではないかもしれない。
特許の寿命は限られている。米国では、すべての維持費が適時に支払われる場合、特許の自然失効時間は、通常、米国で最初の非臨時出願日から20年である。様々な延期があるかもしれないが、特許の有効期限とその提供される保護は限られている。私たちの候補製品が特許を取得しても、特許有効期限が切れると、私たちは模造薬や生体模倣薬を含む競争製品からの競争に直面するかもしれない。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちが持っている特許と許可された特許の組み合わせは、他社が私たちと似ているか同じ製品を商業化することを排除するために十分な権利を提供してくれないかもしれない。例えば、グラクソ·スミスクラインによって許可されたlosmapimodをカバーする物質組成物特許は期限切れであり、私たちの他の特許および特許出願がカバーしていない任意の新しい用途の進入障害ではない。
商業的に合理的な条項で第三者からライセンスを取得できない場合、またはそのような合意の下での義務を履行できない場合、私たちの業務は損害を受ける可能性があります。
第三者の特許やノウハウを用いて私たちの製品を商業化する必要があるかもしれませんが、この場合、これらの第三者から許可を得ることが求められます。もし私たちがこのような技術を許可できない場合、あるいは私たちが不利な条項でそのような技術を許可することを余儀なくされた場合、私たちの業務は実質的な損害を受ける可能性がある。もし私たちが必要な許可を得ることができない場合、私たちは影響を受けた候補製品を開発したり商業化することができなくなり、これは私たちの業務に実質的な損害を与える可能性があり、このような知的財産権を持つ第三者は、私たちの販売禁止または私たち側が印税および/または他の形態の賠償を支払う義務を求めることができる。我々が許可を得ることができても,非排他的である可能性があり,我々の競争相手が我々に許可された同じ技術にアクセスできるようにする.
もし私たちが必要な第三者知的財産権を得ることができない場合、あるいは私たちの既存の知的財産権を維持することができない場合、私たちは私たちの技術、候補製品、またはそれらを製造する方法を再設計するために多くの時間と資源を必要とするかもしれません、または代替技術を開発または許可することは、技術的にも商業的にも不可能かもしれません。もし私たちがそれができなければ、影響を受けた技術や候補製品を開発または商業化することができないかもしれません。これは、私たちの業務、財務状況、運営結果、将来性を深刻に損なう可能性があります。
さらに、許可プロトコルの下での義務を履行できない場合、私たちの取引相手は、これらのプロトコルを終了する権利がある可能性があり、この場合、私たちは、開発、製造、またはマーケティングを行うことができない場合があり、または、これらのプロトコルによってカバーされる任意の製品の開発、製造、またはマーケティングを停止させることを余儀なくされる可能性があり、またはそのようなプロトコルの下での他の処罰に直面する可能性がある。このような状況は、任意のこのようなプロトコルに従って開発された候補製品の価値に実質的な悪影響を及ぼす可能性がある。これらのプロトコルを終了するか、またはこれらのプロトコルの下での私たちの権利を減少または廃止するか、または私たちの業務の利益に適合した場合に、私たちのこのような合意の下での私たちの権利を自由に譲渡または再許可することを制限することは、重要な知的財産権または技術に対する私たちの権利、またはそのようなプロトコルに依存する1つまたは複数の候補製品のさらなる開発または商業化を阻害または遅延または禁止することを含む、これらの合意の下での私たちの権利を失ってしまう可能性がある。
69
現在のライセンス契約によると、私たちには最終的または唯一の決定権がないかもしれませんが、2023年に全面的に承認される予定で、私たちが最近設立されたEU統一特許裁判所(UPC)の中から、私たちのいくつかのライセンスから離脱するヨーロッパ特許および特許出願を選択できるかどうかを決定することができます。私たちの許可側はUPCからの脱退を選択しないことを決定する可能性があり、これは私たちの許可範囲内のヨーロッパ特許と特許出願をUPCの管轄にするだろう。また,我々の許可側がUPCからの脱退を決定しても,我々の許可側がUPCからの脱退を適切に選択する法的手続きや要求を遵守する保証はない.したがって、私たちは私たちの許可内のヨーロッパ特許と特許出願がUPCの管轄に属さないということを確認することができない。UPCによると、欧州特許は多くのヨーロッパ諸国で効果的で強制的に施行されるだろう。UPCによる欧州特許の有効性に挑戦すれば、成功すれば、多くのヨーロッパ諸国が特許保護を失う可能性があり、これは、私たちの業務および私たちの技術および候補製品を商業化または許可する能力に大きな悪影響を及ぼす可能性がある。
もし私たちが“ハッジ·ワックスマン法案”に基づいてアメリカと外国で類似した立法によって特許期間を延長しなければ、私たちの業務は実質的な損害を受ける可能性がある。
米国では,FDAが承認した薬物をカバーする特許期限は,特許満了後最大5年間限定的に延長する資格がある可能性がある。その他の要因を除いて、特許展示期間の長さは薬物が監督審査を受ける時間の長さと関係があるが、この特許展期間は特許の残り期限を製品承認日から合計14年間延長することができず、条件に合った特許を延長することしかできない。ヨーロッパとアメリカ以外のいくつかの他の管轄区域にも似たような規定がある。我々の候補製品がFDAの承認を得た場合,適用された場合には特許期間の延長を申請する予定であるが,適用される保証のない政府当局は,このような延長を承認すべきかどうか,そのような延長が承認されてもその長さの評価に同意することに同意するであろう.例えば、試験段階または規制審査中に職務調査を行うことができなかったため、適用された最終期限内に出願を提出できなかったため、関連特許の満了前に出願を提出することができなかったか、または適用要件を満たすことができなかったため、米国または他のどの国/地域でも特許期間延長の許可を得ることができない可能性がある。もし私たちがいかなる特許期間の延長を得ることができない場合、あるいはどのような延長の期限が私たちが要求したものよりも短い場合、私たちの競争相手は私たちの特許権が満期になった後に競争製品の承認を受ける可能性があり、私たちの業務、財務状況、運営結果、見通しは大きな損害を受ける可能性があります。
また、私たちのライセンス特許については、米国特許商標局に特許期間を延長する請願書を提出することを含む起訴を制御する権利がない可能性がある。したがって、私たちのライセンス特許が特許期間を延長する資格がある場合、米国特許商標局に特許期間を延長するための請願書を提出または取得するか否かを制御することができない可能性がある。
特許に関する詳細な規則と要求があり、これらの特許はオレンジ本に列挙するためにFDAに提出される可能性がある。私たちはオレンジブックの発売要件を満たす1つ以上の特許を含む私たちの候補製品をカバーする特許を得ることができないかもしれない。私たちがオレンジブックに特許を提出しても、FDAはその特許のリストを拒否するかもしれないし、模倣薬製造業者は発売に疑問を提起するかもしれない。私たちの候補製品のうちの1つが承認され、候補製品をカバーする特許がオレンジマニュアルに記載されていない場合、ANDA出願人は、その特許に関する通知を私たちに提供する必要はないであろう。特許法及び特許保護に関する他の情報は、本年度報告における表10−Kの第1項“商業−知的財産権”を参照されたい。
法廷または米国特許商標局で疑問が提起された場合、私たちの候補製品に関連する発行された特許は、無効または実行不可能と認定される可能性がある。
私たちのヨーロッパ特許と特許出願は2023年に全面的に承認されると予想されるUPCで挑戦されるかもしれません。私たちは私たちのヨーロッパ特許と特許出願をUPCから除外することを決定するかもしれない。しかし、いくつかの手続きや要求を満たさなければ、私たちのヨーロッパ特許および特許出願は、規定に適合していないことで疑問視され、UPCの管轄内に置かれる可能性がある。もし私たちがUPCから脱退することを決定したら、私たちのヨーロッパ特許と特許出願がUPCの管轄に陥ることを避けるかどうかを確認することはできない。UPCによると、付与された欧州特許は多くのヨーロッパ諸国で効果的で強制的に施行されるだろう。この特許権は多くのヨーロッパ諸国に適用されるが,UPCによる欧州特許の成功的な挑戦により,多くのヨーロッパ諸国が特許保護を失う可能性がある。したがって、統一商標法による欧州特許の有効性や侵害問題に対する単一訴訟は、このような特許が常に裁かれているため、検証された各国で単独で行われるのではなく、多くのヨーロッパ諸国の特許保護喪失を招く可能性がある。このような特許保護の喪失は、私たちの業務および私たちの技術および候補製品を商業化または許可を得る能力に実質的な悪影響を及ぼす可能性がある。
70
米国や他の管轄地域の特許法の変化は,特許の全体的な価値を低下させ,製品を保護する能力を弱める可能性がある。
“Leahy-Smith America発明法”や“Leahy-Smith Act”のような特許改革立法を含む米国特許法または特許法解釈の変化は、私たちの所有および認可内の特許出願をめぐる起訴および私たちの所有および認可内の認可された特許をめぐる不確実性およびコストを増加させる可能性がある。“ライシー·スミス法案”は米国特許法を多くの重大な改正を行った。これらの変化には、特許出願起訴方式に影響を与える条項、既存技術を再定義し、競争相手に特許の有効性を疑問視するためのより効果的かつ費用対効果的な経路を提供し、特許起訴中に米国特許商標局が以前の技術を第3の方向に提出することを可能にすることと、付与後審査を含む米国特許商標局が管理する付与後プログラムにおいて特許有効性を攻撃する追加プログラムとが含まれる各方面間審査と派生手続き。特許性の他の要件が満たされていると仮定すると,2013年3月までに,米国では,まず発明により保護された発明を発明した者が特許を取得する権利があり,米国以外では,最初に特許出願を提出した者が特許を取得する権利がある。2013年3月以降、“ライシー·スミス法案”(Leahy-Smith Act)によれば、米国は先に出願を提出する制度に移行し、この制度の下で、特許性の他の法定要件が満たされたと仮定すると、最初に特許出願を提出した発明者は、第三者が最初にその発明を発明した者であるか否かにかかわらず、発明の特許を取得する権利がある。したがって、Leahy-Smith法案およびその実施は、私たちの特許出願をめぐる起訴および私たちが発行された特許の実行または保護の不確実性およびコストを増加させる可能性があり、これらのすべては、私たちの業務、財務状況、運営結果、および見通しに大きな悪影響を及ぼす可能性がある。
また,生物製品や薬品の開発や商業化における会社の特許地位は特に不確定である。米国最高裁判所の最近の裁決は、場合によっては入手可能な特許保護範囲を縮小し、場合によっては特許所有者の権利を弱める。この一連の事件は特許取得後の有効性と実行可能性の面で不確実性をもたらしている。米国議会、連邦裁判所、および米国特許商標局の将来の行動によれば、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、私たちの特許権および私たちの将来の特許権の保護、擁護、執行能力に実質的な悪影響を及ぼす可能性がある。
私たちまたは私たちのライセンシーは現在何の訴訟にも参加していないにもかかわらず、私たちは私たちの特許や他の知的財産権を保護または実行する訴訟に巻き込まれる可能性があり、これは高価で、時間がかかり、成功しないかもしれない。
競争相手および他の第三者は、私たちまたは私たちの許可者が発行した特許または他の知的財産権を侵害、流用または他の方法で侵害する可能性がある。したがって、私たちまたは私たちの許可者は、費用がかかり、時間がかかるかもしれない侵害、流用、または他の知的財産権に関するクレームを提起する必要があるかもしれない。私たちは侵害者と思われるいかなるクレームに対しても、私たちが侵害、流用、あるいは他の方法で彼らの知的財産権を侵害していると主張するように、このような当事者に反訴を促す可能性がある。さらに、特許侵害訴訟では、このような当事者は、私たちまたは私たちのライセンシーが主張する特許を無効または強制的に実行できないことを反訴することができる。米国の特許訴訟では,被告が無効または実行不可能と主張する反訴が一般的である。有効性を疑問視する理由は、新規性の欠如、明らかな、または実施できないことを含む、いくつかの法定要件のいずれかを満たしていないと言われているからかもしれない。主張を実行できない理由は,特許起訴に関連する者が起訴中に米国特許商標局に関連情報を隠蔽したり,誤った声明をしたりしたためかもしれない.第三者は、訴訟範囲外であっても、米国や海外の行政機関にこのようなクレームを提起することができる。この仕組みには再審査、贈与後審査、各方面間審査、妨害訴訟、派生訴訟、及び外国司法管轄区における同等の訴訟(例えば:,手続きに反対する).法的に無効と実行不可能と断言された後の結果は予測できない。
このような訴訟のいずれかの不利な結果は、私たちが所有または許可している1つまたは複数の特許が無効または狭義の解釈を宣言されるリスクに直面し、私たちが所有または許可している任意の特許出願が発行された特許を生成できないリスクに直面させる可能性がある。裁判所はまた、私たちが所有しているまたは許可内の特許がこのような技術を含まないため、第三者が訴訟で論争のある技術を使用することを阻止することを拒否することができる。さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟では、私たちのいくつかの機密情報または商業秘密が開示によって漏洩される可能性がある。上記のいずれの条項も、第三者が技術および製品を開発および商業化することを可能にし、当社の業務、財務状況、運営結果、および将来性に重大な悪影響を及ぼす可能性があります。
第三者によって引き起こされるか、または我々によって提起されるか、または米国特許商標局によって発表される干渉または派生プログラムは、我々の特許または特許出願に関連する発明の優先権を決定するために必要である可能性がある。不利な結果は、私たちが関連技術の使用を停止することを要求するか、または勝利者から許可を得ようとすることを要求するかもしれない。もし勝利者が商業的に合理的な条項で私たちに許可を提供しない場合、あるいは私たちに許可を全く提供しない、あるいは非独占的な許可を提供し、私たちの競争相手が同じ技術を獲得した場合、私たちの業務は損害を受ける可能性がある。私たちの訴訟、介入、または派生訴訟の弁護は失敗する可能性があり、成功しても巨額のコストを招き、私たちの経営陣や他の従業員の注意を分散させる可能性がある。また,訴訟に関連する不確実性は実質的な悪影響を及ぼす可能性がある
71
これは、私たちの臨床試験を継続し、私たちの研究計画を継続し、第三者から必要な技術許可を得たり、開発パートナー関係を構築して、候補製品を市場に出す能力を支援するための資金調達に影響を与えるだろう。
さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。聴聞、動議、または他の一時的手続き、または事態発展の結果を公開することもできる。もし証券アナリストや投資家がこれらの結果がマイナスだと思っていれば、私たちの普通株の価格に実質的な悪影響を及ぼす可能性がある。
第三者は、私たちが知的財産権を侵害、流用、または他の方法で侵害したことを告発する法的訴訟を提起する可能性があり、その結果は不確実であり、私たちの業務成功に実質的な悪影響を及ぼす可能性がある。
私たちのビジネス成功は、私たちと私たちの協力者が、私たちの候補製品を開発、製造、マーケティング、販売し、当社の独自技術を使用して、第三者の知的財産権および独自の権利を侵害、流用、または他の方法で侵害する能力に依存します。製薬とバイオテクノロジー産業では、かなり多くの特許と他の知的財産権訴訟がある。私たちは、妨害訴訟、認可後の審査、各方面間米国特許商標局の審査及び派生手続、並びに欧州特許庁の反対意見のような外国司法管轄区域の類似手続。我々が開発候補を求めている分野には,第三者が所有する米国や外国から発行された特許や係属中の特許出願が多く存在する.バイオテクノロジーや製薬業界の拡張や特許の発行に伴い、第三者特許権侵害の疑いを受ける可能性のある技術または製品候補が増加する可能性がある。
提訴や論争のある訴訟手続きの法的敷居が低いため,勝訴確率の低い訴訟や訴訟手続きが提起される可能性があり,弁護には大量の資源が必要である.訴訟と論争のある訴訟手続きはまた高価で時間がかかるかもしれないが、私たちのこのような訴訟手続きの中の相手は私たちよりも多くの資源を投入してこのような法的行動を起訴する能力があるかもしれない。私たちの製品候補が商業化に近づいていれば、上場企業に関するより大きな知名度を得るにつれて、このような訴訟や訴訟に巻き込まれるリスクが高まる可能性があります。第三者は、既存の特許または将来付与される可能性のある特許に基づいて、是非曲直を考慮することなく、侵害請求を私たちに提起する可能性がある。我々は、我々の技術および製品候補およびその用途に関連する可能性のあるすべてのこのような知的財産権を知らないかもしれない、または第三者知的財産権が無効であるか、または私たちの活動および製品候補がそのような知的財産権を侵害していないという誤った結論を誤って導出する可能性がある。したがって、私たちの技術と製品候補、あるいは私たちの開発と商業化は、いかなる第三者の知的財産権を侵害、流用、あるいは他の方法で侵害することもないことを確実に知ることはできない。
第三者は私たちが許可されていない状況で彼らのノウハウを使用していると主張するかもしれない。我々の技術に関連する候補製品の発見、使用または製造に関連する第三者特許または特許出願が存在する可能性があり、これらの特許または特許出願には、製造方法または治療方法のような材料、処方または方法が存在する可能性がある。特許出願は、発行するのに数年かかる可能性があるので、現在処理されている特許出願が存在する可能性があり、これらの出願は、私たちが決定する可能性のある候補製品が侵害される可能性のある発行された特許をもたらす可能性がある。さらに、第三者は将来的に特許を取得し、私たちの技術を使ってこれらの特許を侵害したと主張するかもしれない。さらに、上述したように、私たちが知らない既存の特許、または私たちが誤って導出した無効または私たちの活動によって侵害されていない特許が存在する可能性がある。管轄権のある裁判所が、我々が識別する可能性のある候補製品の製造プロセス、製造プロセス中に形成された任意の分子または任意の最終製品自体をカバーするような任意の第三者特許を所有している場合、そのような特許の所有者は、適用特許に基づいてライセンスを取得したか、またはそのような特許が満了するまで、その候補製品を商業化する能力を阻止することができるかもしれない。
我々にクレームをつけた当事者は、禁止または他の公平な救済を受ける可能性があり、これは、私たちが決定する可能性のある候補製品をさらに開発し、商業化する能力を効果的に阻止することができるかもしれない。これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、当社の業務における従業員資源を大量に移転することになる。もし私たちに対する侵害クレームが成功した場合、私たちは故意に侵害された3倍の損害賠償と弁護士費、特許権使用料の支払い、私たちの侵害製品の再設計、または第三者から1つまたは複数の許可を得ることを含む大量の損害賠償を支払わなければならないかもしれないし、多くの時間とお金の支出が必要かもしれない。
私たちは許可を受けることを選択することができます。あるいは、私たちが第三者の知的財産権を侵害していることが発見され、流用された場合、私たちはまた、当社の技術および候補製品の開発、製造、およびマーケティングを継続するために、第三者から許可を得ることを要求される可能性があります。しかし必要なものは何も得られないかもしれません
72
商業的に合理的な条項や根本的に存在しない条件で許可を得る。私たちが許可を得ることができても、それは非排他的である可能性があり、私たちの競争相手や他の第三者が私たちに許可された同じ技術にアクセスできるようにし、大量の許可と印税の支払いを要求することができるかもしれない。私たちは裁判所の命令によって、権利侵害技術や製品の開発、製造、商業化を中止することを余儀なくされるかもしれない。さらに、特許や他の知的財産権を故意に侵害していることが発見され、顧客や協力者に賠償を余儀なくされる可能性がある場合、3倍の損害賠償と弁護士費を含む重大な金銭損害に責任を負うと判断される可能性があります。権利侵害の発見は、候補製品を商業化することを阻止したり、いくつかの業務運営を停止させたりする可能性があり、これは私たちの業務に実質的な損害を与える可能性がある。また、私たちは私たちの候補製品を再設計し、新しい規制承認を求め、契約協定に基づいて第三者を賠償することを余儀なくされる可能性がある。第三者の機密情報や商業秘密を盗用したと主張することは、当社の業務、財務状況、運営結果、見通しに類似した重大な悪影響を及ぼす可能性があります。
知的財産権訴訟や他の知的財産権に関する法律手続きは、私たちに大量の資源を費やし、私たちの人員のその正常な職責に対する関心を分散させる可能性がある。
解決策が私たちに有利であっても、知的財産権クレームに関連する訴訟や他の法的手続きは、巨額の費用を発生させ、私たちの技術や管理者の正常な責任を分散させる可能性があります。また、公聴会、動議、または他の一時的な手続きまたは事態の発展の結果が公表される可能性があり、証券アナリストや投資家がこれらの結果がマイナスだと思う場合、私たちの普通株の価格に大きな悪影響を及ぼす可能性がある。このような訴訟または訴訟は、私たちの運営損失を大幅に増加させ、開発活動または任意の将来の販売、マーケティング、または流通活動に使用することができるリソースを減少させる可能性がある。私たちはそのような訴訟や訴訟手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らがより多くの財政資源を持っているので、私たちよりも効率的にこのような訴訟や訴訟の費用を負担するかもしれません。そして、彼らのより成熟して発展した知的財産権の組み合わせのために、彼らはこのような訴訟においても優勢である可能性があります。知的財産権訴訟または他の訴訟の開始と継続によって生じる不確実性は、市場での競争の能力を損なう可能性がある。
特許保護の獲得と維持は、政府特許機関が提出した様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要件に適合しない場合、私たちの特許保護は減少またはキャンセルされる可能性がある。
私たちが所有し許可されている特許および特許出願の有効期間内には、定期的なメンテナンス、更新および年金費用、ならびに発行および処理された特許出願の任意の様々な他の政府費用を、いくつかの段階に分けて、または毎年米国特許商標局および外国特許代理機関に支払わなければならない。米国特許商標局および各種外国政府特許機関は、特許出願過程において、いくつかのプログラム、文書、費用支払い、およびその他の同様の規定を遵守することを要求する。場合によっては、私たちは、私たちの許可パートナーに依存して、関連する特許エージェントにこれらの費用を支払うか、またはその手続きおよび文書ルールを遵守します。私たちの特許については、私たちは年金サービス、外部会社、外部弁護士に依存して納期を注意し、私たちが彼らにそうするように指示した後に費用を支払う。多くの場合、適用規則に従って滞納金を支払うか、または他の方法で不注意を是正することによって失効を是正することができるが、場合によっては、規定を遵守しないことは、特許または特許出願が放棄または失効され、関連法ドメインの特許権の一部または全部の喪失をもたらす可能性がある。特許または特許出願が放棄または失効される可能性のある規定を遵守しないイベントは、規定された期限内に公式行動に応答することができなかったこと、費用を支払わなかったこと、および適切に合法化されず、正式な文書を提出することを含む。この場合、潜在的な競争相手は、同様または同じ製品または技術で市場に参入することができる可能性がある。もし私たちまたは私たちの許可者が私たちの候補製品をカバーする特許および特許出願を維持できなかった場合、私たちの業務、財務状況、運営結果、および将来性に大きな悪影響を与えます。
知的財産権の許可や第三者との融資手配における私たちの義務を履行できなかった場合、あるいは許可側との業務関係が妨害された場合、私たちは私たちの業務に重要な知的財産権を失う可能性があります。
私たちはGSKとの協定のような許可と融資協定の当事者であり、私たちは第三者と追加の許可と融資手配を締結し、勤勉、開発と商業化スケジュール、マイルストーン支払い、特許権使用料、保険、その他の義務を私たちに強要するかもしれない。私たちの既存の許可と助成協定によると、候補製品または関連技術の製品純売上高に合意が含まれている範囲内の印税を支払う義務があります。もし私たちが現在または未来の許可と融資協定の下でこのような義務を履行できなかった場合、私たちの取引相手はこれらの合意を終了する権利があるか、または私たちに特定の権利を与えることを要求する権利があるかもしれない。このような状況は、任意のこのようなプロトコルに従って開発された任意の候補製品の価値に実質的な悪影響を及ぼす可能性がある。これらの合意を終了したり、これらの合意の下で私たちの権利を減少させたり、除去したりすることができる
73
これは、私たちがあまり有利でない条項で新しい合意または回復された合意を交渉しなければならないこと、または重要な知的財産権または技術に対する私たちの権利を含むこれらの合意の下の権利を失うことをもたらす可能性があり、これは私たちの業務、財務状況、運営結果、および見通しに大きな悪影響を及ぼすだろう。私たちはまた私たちの製品エンジンのいくつかの技術のための許可証と合意を持っています。これらはすべて非独占的です。私たちはまだこれらの合意に関連したすべてのリスクに直面しているが、私たちは第三者がこれらの技術にもアクセスすることを防ぐことはできない。しかも、私たちの免許は私たちの未来のビジネス機会に制限を加えるかもしれない。例えば、我々のグラクソ·スミスクラインとの許可によれば、GSK許可の任意の特許またはデータ権利を第三者に再許可して米国以外で使用することを望む場合、GSKはいくつかの優先交渉権を有する。これは、ある取引を阻止または延期する可能性があり、これは、losmapimodの開発および商業化、および私たちの業務に悪影響を及ぼす可能性がある。
ライセンス契約によると、知的財産権に関する論争が発生する可能性があります
また,我々が現在第三者から知的財産権や技術許可を得ているプロトコルは複雑であり,このようなプロトコルのいくつかは様々な解釈の影響を受ける可能性がある.可能性のある契約解釈の相違の解決は、関連する知的財産権または技術に対する私たちの権利と考えられる範囲を縮小するか、または関連協定の下での私たちの財務または他の義務を増加させる可能性があり、いずれも、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。また、我々が許可している知的財産権をめぐる紛争が、商業的に許容可能な条項で現在の許可スケジュールの能力を維持していることを阻害したり、損害を与えたりすれば、影響を受けた技術や候補製品の開発および商業化に成功できない可能性があり、これは、私たちの業務、財務状況、運営結果、および見通しに大きな悪影響を及ぼす可能性がある。
私たちの現在または未来の許可者は、第三者コンサルタントまたは協力者または第三者からの資金に依存する可能性があり、したがって、私たちの許可者は、私たちが許可した特許および特許出願の唯一および独占所有者ではない。他の第三者が私たちが許可した特許または特許出願の所有権を持っている場合、彼らはこれらの特許を私たちの競争相手に許可することができ、私たちの競争相手はそれと競争する製品や技術を販売することができるかもしれない。これは私たちの競争地位、業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼすかもしれない。
私たちが最善を尽くしたにもかかわらず、私たちの許可側は、許可協定に深刻に違反している可能性があるので、許可協定を終了し、これらの許可協定がカバーする候補製品や技術を開発し、商業化することができないかもしれません。これらの許可が終了した場合、または基礎知的財産権が予想される排他性を提供できなかった場合、競争相手は規制部門の承認を求め、私たちと同じ製品や技術を市場に出す権利があるだろう。これは私たちの競争地位、業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼすかもしれない。
私たちは世界各地で私たちの知的財産権と固有の権利を保護できないかもしれない。
世界のすべての国で候補製品の申請、起訴、特許を守る費用は目を引くほど高く、外国の法律はアメリカの法律のように私たちの権利を保護できないかもしれない。また、一部の国の法律は知的財産権の保護程度は米国の連邦や州法律に及ばず、名目上このような保護があっても、このような知的財産権に対する司法や政府の法執行が不足している可能性がある。したがって、私たちは、米国以外のすべての国/地域で第三者が私たちの発明を実施したり、米国または他の管轄区域で私たちの発明を使用して製造された製品を販売または輸入することを阻止することができないかもしれない。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの技術を使用して自分の製品を開発することができ、また、私たちに特許保護や許可証を持っているが、法執行力がアメリカの地域に比べて他の侵害製品を輸出することもできる
74
各州です。これらの製品は私たちの製品と競争するかもしれませんが、私たちの特許や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。
多くの会社は外国の管轄区域の知的財産権の保護と保護に重大な問題に直面している。特定の国の法律制度、特に特定の発展途上国およびロシアの法律制度は、特許、商業秘密、および他の知的財産権保護、特にバイオテクノロジー製品に関する保護の実行に賛成せず、私たちの特許の侵害や私たちの知的財産権および独自の権利に違反する競争製品の販売を阻止することを困難にする可能性がある。また、ある司法管轄区域は新しい治療方法を構成する発明に対して同程度或いは完全な保護を与えていない。
外国の管轄区域で私たちの知的財産権と独自の権利を強制的に執行する訴訟手続きは巨額のコストを招く可能性があり、私たちの努力と注意を私たちの業務の他の側面から移すことは、私たちの特許を無効または狭義に解釈されるリスクに直面させる可能性があり、私たちの特許出願を発表できないリスクに直面させ、第三者が私たちにクレームを提起する可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。したがって、私たちの知的財産権および独自の権利を世界各地で強制的に実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネス的優位性を得るのに十分ではないかもしれません。
多くの国に強制許可法があり,これらの法律により特許権者は第三者に強制的に許可を付与される可能性がある。また、多くの国は政府機関や政府請負業者に対する特許の実行可能性を制限している。これらの国では,特許権者の救済措置は限られている可能性があり,このような特許の価値を大幅に低下させる可能性がある。もし私たちまたは私たちの任意の許可者が第三者に私たちの業務に関連する任意の特許の許可を付与することを余儀なくされた場合、私たちの競争的地位が損なわれる可能性があり、私たちの業務、財務状況、運営結果、および見通しは不利な影響を受ける可能性があります。
私たちは私たちの特許と他の知的財産権の発明権または所有権のクレームに疑問を受けるかもしれない。
私たちまたは私たちの許可者は、元従業員、協力者または他の第三者が、発明者または共同発明者として、私たちの所有または許可内の特許、商業秘密、または他の知的財産権において権益を有するというクレームを受ける可能性がある。例えば、私たちまたは私たちの許可者は、私たちの候補製品の開発に参加している従業員、コンサルタント、または他の人の義務紛争によって発明権紛争を生じる可能性があります。これらのまたは他の挑戦在庫または私たちまたは私たちの許可者による私たちの所有または認可内の特許、商業秘密、または他の知的財産権の所有権のクレームに対抗するために、訴訟を提起する必要があるかもしれない。もし私たちまたは私たちの許可者がこのようなクレームを弁護できなかった場合、金銭損害賠償を支払うことに加えて、私たちの候補製品に非常に重要な知的財産権の独占所有権や使用権のような貴重な知的財産権を失う可能性があります。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。上記のいずれも、当社の業務、財務状況、経営結果、見通しに重大な悪影響を及ぼす可能性があります。
75
私たちは、私たちの従業員、コンサルタント、または請負業者が第三者の機密情報を誤って使用または開示したと主張する第三者のクレームを受けるかもしれないし、私たちは彼らの現在または前任雇用主のいわゆる商業機密を誤って使用または開示したり、私たちが彼らの知的財産権を流用したと主張したり、私たちが私たち自身の知的財産権を所有していると主張したりするかもしれない。
私たちの多くの従業員、コンサルタント、および請負業者は、以前、私たちの競争相手または潜在的な競争相手を含む大学または他の製薬またはバイオテクノロジー会社に雇われていた。私たちは、私たちの従業員、コンサルタント、および請負業者が、私たちのために働いているときに他人の固有情報またはノウハウを使用しないことを保証するために努力しているが、私たちは、商業秘密または他の固有情報を含む任意のそのような個人の現職または前任雇用主の知的財産権を使用または開示していると告発される可能性がある。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。
また、私たちの政策は、知的財産権開発に参加する可能性のある私たちの従業員、コンサルタント、請負業者が合意に署名し、このような知的財産権を私たちに譲渡することを要求しているにもかかわらず、実際に私たちが自分の知的財産権を開発しているすべての側とこのような合意を実行することができないかもしれません。私たちと彼らの知的財産権譲渡協定は自動的に実行されないかもしれないし、違反される可能性があります。私たちは第三者にクレームを出させたり、私たちが私たちの知的財産権の所有権だと思うことを確認するために、私たちが提起したクレームを弁護することを余儀なくされるかもしれません。このようなクレームは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。
もし私たちがこのような任意のクレームを起訴または弁護できなければ、金銭損害賠償の支払いに加えて、貴重な知的財産権や人員を失う可能性があり、これは私たちの競争業務の地位と将来性に実質的な悪影響を及ぼす可能性がある。このような知的財産権は第三者に付与することができ、私たちは、私たちの技術または製品を商業化するために、第三者から許可を得る必要があるかもしれません。この許可は、商業的に合理的な条項では得られないかもしれません、または全く存在しないかもしれません。このようなクレームの起訴や抗弁に成功しても、訴訟は巨額のコストを招き、経営陣や従業員の注意を分散させる可能性がある。
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの商業と競争の地位は損なわれるだろう。
私たちの候補製品のための特許を申請することに加えて、私たちは、私たちの独自製品エンジンのいくつかの態様を含む、私たちの競争的地位を維持するために、ビジネス秘密および秘密協定に依存して、私たちの非特許ノウハウ、技術、および他の固有情報を保護します。私たちは、私たちの従業員、会社協力者、外部科学協力者、CRO、CMO、コンサルタント、コンサルタント、および他の第三者のような、これらの秘密にアクセスする権利のある当事者と秘密協定を締結することによって、私たちのビジネス秘密および他のノウハウを保護することを求めています。私たちはまた、私たちの従業員やコンサルタントと秘密および発明または特許譲渡協定を締結していますが、私たちが私たちの商業秘密またはノウハウに接触している可能性があるか、または接触したことがあるすべての当事者とこのような合意を締結した保証はありません。このような努力にもかかわらず、どちらも合意に違反し、私たちのビジネス秘密を含む私たちの固有の情報を漏洩する可能性があり、私たちは十分な救済措置を得ることができないかもしれない。商業秘密の漏洩や流用を検出し、当事者に商業秘密の不法開示或いは流用を要求することは困難であり、高価で時間がかかり、結果は予測できない。しかも、米国国内外のいくつかの裁判所は商業秘密をあまり望んでいないか、または保護したくない。もし私たちの任意の商業秘密が競争相手または他の第三者によって合法的に取得または独立して開発された場合、私たちは彼らまたは彼らが情報を伝達する人がその技術や情報を使用して私たちと競争することを阻止する権利がないだろう。もし私たちの任意の商業秘密が競争相手または他の第三者に漏洩された場合、または競争相手または他の第三者によって独立して開発された場合、私たちの競争地位は実質的かつ不利な損害を受けるだろう。
知的財産権はすべての潜在的な脅威を解決できるとは限らない。
私たちの知的財産権が提供する未来の保護の程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できないか、あるいは競争優位性を維持することができるかもしれない。例えば:
76
これらの事件が発生すると、それらは私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
我々の候補製品の規制承認とその他の法的適合性問題に関するリスク
たとえ著者らが必要な臨床前研究と臨床試験を完成しても、上場審査過程は高価で、時間と不確定であり、著者らは一部或いはすべての候補製品の商業化審査を得ることができないかもしれない。もし私たちが必要な規制承認を得ることができない場合、あるいは規制承認を得る上で遅延が生じた場合、私たちの候補製品を商業化することができなくなり、私たちの収入を作る能力は実質的に損なわれるだろう。
医薬品の米国での発売承認はFDAに新薬申請やNDAを提出する必要があり,NDAの承認を得るまで米国での候補薬物の販売は許可されていない。NDAは大量の臨床や臨床前データ,薬理学,化学,製造,制御に関する大量の情報によって支持されなければならない。私たちはまだアメリカや他の任意の司法管轄区で私たちの任意の候補製品の申請を提出したり、マーケティングの許可を得ていません。
我々は,上場承認を得るために必要な申請の提出と支援には経験が限られており,この過程で第三者臨床研究組織や他の第三者コンサルタントやサプライヤーに依存することが予想される。上場承認を得るためには、候補製品の安全性と有効性を決定するために、広範な臨床前と臨床データ及び支持情報を監督機関に提出する必要がある。私たちの候補製品は効果がないかもしれないし、中程度の効果しかないかもしれないし、あるいは不良または意外な副作用、毒性、または他の特徴があることが証明される可能性があります。これらは私たちの上場承認を阻止するか、または商業用途を阻止または制限するかもしれません。もし私たちの候補製品が発売承認されれば、付属のラベルは私たちの薬物の承認使用を制限するかもしれません。これは製品の販売を制限するかもしれません。
77
米国でも海外でも、市場承認を得る過程は高価であり、承認されれば何年もかかる可能性があり、関連する候補製品のタイプ、複雑性、新規性を含む様々な要因によって大きく異なる可能性がある。開発期間中の市場承認政策の変化、追加法規又は法規の変化、又は各提出製品申請に対する規制審査の変化は、出願の承認又は拒絶の遅延を招く可能性がある。監督管理機関は審査過程中にかなりの自由裁量権を持っており、いかなる申請を受け入れることを拒否することができ、著者らのデータが承認を得るのに十分ではなく、追加の臨床前、臨床或いはその他の研究を行う必要があることを決定することもできる。そのほか、臨床前と臨床試験から得られたデータの異なる解釈は候補製品の上場承認を延期、制限或いは阻止する可能性がある。私たちが最終的に得たどのマーケティング承認も限られているかもしれないし、制限されたり、承認された後の約束は、承認された製品が商業的に実行できないようにすることができる。
FDAおよび他の機関の中断は、規制審査提出書類および/または必要な政府機関が新薬を承認するのに要する時間を延長する可能性があり、これは私たちの業務に悪影響を及ぼすだろう。例えば、ここ数年間、米国政府は何度か閉鎖されており、FDAのようないくつかの規制機関は、キー従業員を休暇にし、キー活動を停止しなければならない。政府が長期的に停止すれば、FDAが私たちが提出した規制文書を適時に審査して処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。
また、2020年3月以来、アメリカ食品薬品監督管理局は常規のモニタリング、生物研究モニタリングと審査前検査の回復に努力してきた。当時、新冠肺炎の疫病のため、国内外の施設の検査は基本的に保留された。FDAが承認を得るために検査を行う必要があると判断し、旅行制限のために審査期間内に検査を完了することができない場合、FDAは、遠隔相互作用評価が不十分であると判断した場合、場合に応じて、検査を完了することができるまで、申請に完全な返信または行動を延期することを意図していることを示している。新冠肺炎突発公共衛生事件の期間中、アメリカ食品薬品監督管理局はその申請に対する規定検査を完成できなかったため、多くの会社は完全な回復状を受け取ることを発表した。米国以外の規制機関は、進行中の新冠肺炎の大流行に対応するために、類似した制限や他の政策措置をとる可能性があり、規制活動に遅延が生じる可能性がある。
もし私たちが承認を得る上で遅延に遭遇した場合、あるいは私たちが私たちの候補製品の承認を得られなかったら、私たちの候補製品の商業的な見通しが損なわれる可能性があり、私たちの収入を作る能力は実質的に損なわれるだろう。
私たちは私たちの候補製品の孤立した薬物称号や排他性を得ることができないかもしれないし、私たちがそうしても、この排他性はFDAやEMAが他の競争製品を承認することを阻止しないかもしれない。
米国やヨーロッパを含むいくつかの管轄区の規制機関は、患者数が比較的少ない薬物を孤児薬に指定する可能性がある。FDAとEMAはロスパモッドにFSHDを治療する孤児薬物の称号を与えた。私たちは私たちの現在と未来の他の候補製品のために孤児薬物の称号を求めるかもしれない。
一般に、孤児薬物名を有する製品がその後、そのような名称を有する適応の最初の発売承認を得た場合、製品は市場排他期間を有する権利があり、これは、FDAまたはEMAが特定の期間内に同じ薬物の別の上場許可申請を承認することを阻止するであろう。適用期間はアメリカでは7年、EUでは10年だ。薬物が指定された孤児薬物の基準に適合しなくなった場合、その薬剤が市場排他性を合理的でなくするのに十分な利点がある場合を含む場合、5年目の終了時に欧州連合の専門期間を6年に短縮することができる。FDAまたはEMAが、指定された要求に重大な欠陥があると判断した場合、または製造業者が稀な疾患または疾患を有する患者の需要を満たすのに十分な数の薬剤を保証できない場合、孤児薬の排他性を失う可能性がある。
我々が製品の孤児薬物排他性を獲得しても,この排他性は異なる活性成分を含む競合薬が同じ条件で許可されるため,競合から製品を効率的に保護することができない可能性がある。さらに、FDAが、より安全、より効果的、または患者ケアに大きな貢献を果たしていることが証明されたので、後者の薬剤が孤児薬物排他性を獲得する最初の薬剤よりも臨床的に優れていると結論した場合、FDAはその後、同じ薬剤を同じ疾患のために承認することができる。また,同一製品に対する別の適応の承認を求めて獲得し,孤児薬物の排他性を得る権利がない場合には,我々の孤児排他期は,第三者が同じ有効成分を含む競合薬の承認を得ることを阻止せず,この他の孤児適応に用いられる。もしこのような状況が発生したら、私たちが孤児から排他的に得た保護は不利な影響を受けるかもしれない。
78
迅速なチャネルや画期的な療法のようなFDAの特別な指定は、より速い開発や規制審査や承認過程を招くことがなく、私たちの候補製品が発売承認される可能性も増加しないかもしれない。
FDAは、FSHDを治療するためのlosmapimodおよびSCDを治療するためのFTX−6058の高速チャネル指定を承認し、私たちの他のいくつかの候補製品のための高速チャネル指定、およびlosmapimodを含む画期的な治療指定を求めるかもしれない。重症または生命に危険な疾患の治療に使用される薬剤が使用され、そのような疾患が満たされていない医療ニーズを解決する可能性を示す場合、薬物スポンサーはFDA迅速チャネル指定を申請することができる。FDAは広範な裁量権を持ち,この称号を付与するかどうかを持っているため,ある特定の候補製品がこの称号を得る資格があると考えても,FDAがそれを付与することを株主に保証することはできない。高速チャネル指定があっても、従来のFDAプログラムと比較して、より速い開発過程、審査、または承認を経験しない可能性があります。FDAが我々の臨床開発プロジェクトのデータが高速チャネルの指定をサポートしなくなったと考える場合,その指定を撤回する可能性がある。
画期的な治療法は、1つまたは複数の他の薬剤と単独でまたは1つまたは複数の他の薬剤と組み合わせて重篤または生命を脅かす疾患または状態を治療することを意図した薬剤として定義され、初歩的な臨床証拠は、1つまたは複数の臨床的に重要な終点において、臨床開発早期に観察される実質的な治療効果のような既存の療法よりも有意な改善を示す可能性があることを示す。
画期的療法に指定されたのはFDAの裁量権である。したがって,我々の候補製品の1つが画期的療法として指定された基準に適合していると考えても,FDAは同意せず,このような指定をしないことにする可能性がある。我々が画期的な治療指定を得ても,FDAの従来のプログラムによる承認を考慮した薬物と比較して,候補製品のこのような指定を受けることは,より速い開発過程,審査または承認につながる可能性がなく,FDAの最終承認を確保することはできない。さらに、我々の1つまたは複数の候補製品が画期的な療法の条件に適合していても、FDAは、これらの製品が資格条件を満たしていないことを後で決定したり、FDAの審査または承認を決定する期間が短縮されない可能性がある。
FDAがNDA提出の加速を求めることができることに同意しても、NDAの承認を保証することはできず、承認NDAの提出加速も保証されず、候補製品のより速い開発や規制審査過程が確保されることはない。
適用されれば、FDAの加速承認手続きを使用して、私たちの候補製品の承認を求めることができます。製品が深刻な疾患を治療した場合、一般に、既存の療法よりも有意な利点を提供し、臨床的利益を合理的に予測する可能性のある代替終点への影響、または不可逆的な発症率または死亡率またはIMMよりも早く測定することができる臨床終点への影響を証明する場合、製品は、IMMまたは他の臨床的利益への影響を合理的に予測する可能性がある加速承認を得る資格がある可能性がある即中間臨床終点)。
加速承認を求める前に、FDAからのフィードバックを求め、このような加速承認を求める能力を他の方法で評価します。
FDAフィードバック後、私たちが最初にそうすることを決定しても、承認の加速、または任意の他の形態の加速開発、審査または承認を求めたり申請したりすることは保証されない。さらに、加速承認または他の迅速な規制指定された申請を提出することを決定した場合、そのような提出または申請が受け入れられるか、または任意の迅速な審査または承認がタイムリーに承認されるか、または全く保証されない保証はない。
また,承認を加速させる条件として,FDAは十分かつ良好に制御された発売後臨床試験を行い,本製品の臨床的利益を確認することが求められる可能性がある。このような検証的な実験は職務調査の方法で行われなければならない。2022年食品·薬物総合改革法案(FDORA)によれば、FDAは、承認前または承認が加速された製品が承認された日後の特定の期間内に、1つまたは複数の承認後の検証的研究を行うことを適宜要求することが許される。FDORAは,登録目標の実現を含めて180日ごとにFDAにこのような研究の最新状態を送信することも求められており,FDAはこれらの情報を迅速に公開しなければならない。FDORAはまた、スポンサーがこのような研究をタイムリーに行うことができなければ、必要な更新をFDAに送信するか、またはそのような承認後の研究が薬剤の期待される臨床的利益を検証できない場合、FDAは、加速的な承認を得た薬物または生物学的製剤の承認を迅速に撤回することができる。FDORAによれば、FDAは、職務調査を行っていない会社に対していかなる承認後の検証的研究を行うか、または速やかに当該機関に進捗報告を提出した会社に罰金を科すように行動する権利がある。さらに、FDAは、通常、承認を加速することを考慮している製品の販売促進材料の事前承認を要求しており、これは、製品の商業発売時間に悪影響を及ぼす可能性がある。したがって,候補製品への承認加速経路の使用を求めても,より速い開発や規制審査や承認を経験しない可能性がある
79
この製品の流れ。また,加速承認を得ることは,製品の加速承認が最終的に従来の承認に変換される保証はない.
外国の管轄区域でマーケティングの許可を得られなければ、私たちの候補製品は海外で販売できないだろう。
EUや他の多くの外国司法管轄地域で私たちの製品をマーケティングして販売するためには、私たちまたは潜在的な第三者パートナーは、単独のマーケティング承認を得て、多くの異なる法規要件を遵守しなければなりません。承認手続きは国によって異なり、追加的なテストが含まれるかもしれない。承認を得るのに要する時間は、FDA承認を得る時間とは大きく異なる可能性がある。米国以外の規制承認過程には、通常、FDA承認の取得に関するすべてのリスクが含まれる。また、米国以外の多くの国では、製品がその国での販売を許可される前に、精算承認を受けなければならないことが求められている。私たちまたは私たちの潜在的な第三者協力者は、もしあれば、条件付き許可を含めて、米国以外の規制機関から直ちに承認を得ることができないかもしれない。FDAの承認は、他国又は管轄区域の規制機関の承認を確保するものではなく、米国以外の1つの規制機関の承認も、他の国又は司法管区の規制機関又はFDAの承認を確保することができない。しかし、1つの国で規制承認を得ることができなかったり、遅延したりすることは、他の国の規制過程に悪影響を及ぼす可能性がある。私たちはマーケティング承認を申請できないかもしれないし、どの市場でも私たちの製品を商業化するために必要な承認を得ることができないかもしれない。
しかも、イギリスはもはやEUの一部ではないので、私たちの製品はイギリスとEUの規制承認を得るために単独の申請と手続きが必要になるだろう。どんなマーケティング承認を得ても得られない点でのいかなる遅延も、イギリスおよび/またはEUで任意の候補製品を商業化し、収入を創出し、利益を達成し、維持する能力を制限することを阻止する可能性がある。
私たちが発売許可を得たどの候補製品も発売後に市場から制限されたり撤退したりする可能性があります。もし私たちが規制要求を守っていない場合、あるいは私たちの製品が予期せぬ問題に遭遇した場合、その中のどの製品が承認された場合、私たちは重大な処罰を受けるかもしれません。
私たちが発売許可を得た任意の候補製品、及びその製品の製造プロセス、承認後の臨床データ、ラベル、広告と販売促進活動は、FDAと他の監督管理機関の持続的な要求と審査を受ける。候補製品の上場が承認されても,承認は再生可能エネルギー管理システムの実施要求を含めて,製品が上場可能な指定用途の制限や承認条件の制限を受ける可能性がある。もし私たちの候補製品が発売承認されれば、付属のラベルは私たちの薬物の承認使用を制限するかもしれません。これは製品の販売を制限するかもしれません。
FDAはまた、REMSの採用と実施を含む製品の安全性または有効性を監視するために、高価な発売後の研究または臨床試験とモニタリングを要求する可能性がある。FDAおよび司法省を含む他の機関は、承認のみの適応を確保し、承認されたラベルの規定に基づいて薬物の販売および流通を確保するために、医薬品承認後のマーケティングおよび販売促進を密接に監視している。“虚偽請求法”を含むFDCAおよび他の処方薬販売促進および広告に関する法規に違反することは、連邦および州医療詐欺および法律乱用および州消費者保護法違反を告発する調査および法執行行動につながる可能性がある。
さらに、その後、私たちの製品、製造業者あるいは製造プロセスに以前未知の不良事件や他の問題が発生したり、監督管理要求を遵守できなかったりすることが発見され、様々な結果が生じる可能性がある
80
欧州連合の安全モニタリングや薬物警戒に関する要求や,小児科群のための製品開発に関する要求を守らないことも,重大な経済的処罰を招く可能性がある。同様に、EUや連合王国の個人情報保護に関する要求を守らないことも重大な処罰や制裁につながる可能性がある。
また,承認された製品のメーカーとその工場は,品質管理や製造プロセスが医薬品メーカーに適したcGMPに適合することを確保することを含むFDAの広範な要求を遵守しなければならない。さらに、FDORAによれば、承認された薬物および生物製品のスポンサーは、市場状態が任意に変化したときに、薬物の撤回などの6ヶ月の通知をFDAに提供しなければならず、そうしなければ、FDAは製品を生産停止製品リストに登録する可能性があり、これは製品の発売能力をキャンセルするであろう。著者らはまた、安全とその他の上場後の情報と報告の提出、登録と上場要求、臨床医師へのサンプル配布の要求及び記録保存を含む他の法規要求を遵守する。
医療提供者、医師、第三者支払者との関係は、違反が発生した場合、刑事制裁、民事処罰、契約損害、名声損害、行政負担、利益および将来の収入減少に直面する可能性があるアンチバックル、詐欺および乱用、虚偽声明、透明性、健康情報プライバシーと安全、および他の医療保健法律および法規との制約を受ける。
私たちが規制部門の承認を得て任意の製品を商業化すれば、医療提供者、医師、第三者支払者は、私たちが市場の承認を得た任意の候補製品の推薦と処方で主な役割を果たすだろう。私たちの将来の医療保健提供者、医師、第三者支払者との手配は、私たちがマーケティング、販売、流通、マーケティングの承認を得た任意の製品の業務または財務配置と関係を制限するかもしれない広範に適用される詐欺や乱用、および他の医療法律法規に直面するかもしれません。また、米国連邦と州政府および私たちが業務を展開している外国司法管轄区域政府の透明性法律と患者プライバシー法規の制約を受ける可能性がある。本年度報告表格10-K中の第1項“企業--政府監督と製品審査--保健法律と監督管理”を参照。
医薬製品の流通は広範な記録保存、許可、貯蔵と安全要求を含む追加の規定と条例を遵守し、許可されていない医薬製品の販売を防止しなければならない。
これらの法律の範囲も執行も不確定であり,現在の医療改革環境下では,特に適用の前例や法規が乏しい場合には,急速な変化が生じる可能性がある。私たちが第三者の業務配置と適用される医療法律や法規に適合するように努力することは、多くのコストに関連することを確実にします。政府当局は、私たちの業務実践が現在または未来に適用される詐欺および乱用または他の医療保健法律および法規に関連する現行または未来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。もし私たちの業務が、私たちの販売チームによる予想される活動を含めて、これらの法律または任意の他の私たちに適用される可能性のあるいかなる政府法規に違反していることが発見された場合、私たちは重大な民事、刑事と行政処罰、損害賠償、罰金、監禁、製品を政府が援助する医療計画(例えばMedicareおよびMedicaid)から除外し、私たちの業務を削減または再構成することができます。もし医者や他の人が
81
もし私たちがそれと業務往来がある医療提供者や実体が適用法に適合していないことが発見されることが予想される場合、彼らは政府の援助に参加する医療計画から除外されることを含む刑事、民事または行政制裁を受ける可能性がある。
グローバルなプライバシーやデータセキュリティ要件を遵守することは、追加的なコストと責任をもたらしたり、グローバルでデータを収集して処理する能力を抑制したりする可能性があり、これらの要求を守らないことは、私たちを巨額の罰金と処罰に直面させる可能性があり、これは、私たちの業務、財務状況、または運営結果に大きな悪影響を及ぼす可能性があります。
世界各地で情報を収集、使用、保護、共有、譲渡、および他の処理情報の立法および規制の枠組みは急速に変化しており、予測可能な未来にはまだ不確定である可能性がある。世界的に、私たちが業務を展開しているほとんどの管轄区域は、自分のデータセキュリティとプライバシーの枠組みを構築しており、これらの枠組みを守らなければならない。例えば、個人健康データを含むEU個人に関する個人データを収集、使用、開示、移転、または他の方法で処理し、2018年5月に欧州経済地域(EEA)のすべての加盟国で発効するEUの一般データ保護条例またはEU GDPRの制約を受ける。英国のEU離脱や離脱に伴い、EU GDPRは英国法に組み込まれ、EU GDPRとともに英国法に組み込まれている。イギリスが離脱したにもかかわらず、EUとイギリスのGDPRはほぼ一致している。現在、最も影響力のある分岐点は、EUまたはイギリスの規制機関が承認した様々な異なる契約条項の実施を要求しているため、移転メカニズム(すなわちEUまたはイギリスの会社が米国を含む第三国に個人情報を移転する能力)に関連している。このような複雑さと追加的な契約負担は私たちの全体的な危険を増加させる。今後は行政負担の面も含めてさらなる食い違いがあるかもしれない。英国はそのデータ改革法案で、EU GDPR導入の大きな変化となる同国のデータ保護法の枠組みを改革する計画を発表した。これは追加のコンプライアンスコストを招く可能性があり、私たちの全体的なリスクを増加させる可能性があります。私たちはEUと連合王国全体で統一的な方法を取ることができないかもしれません, 私たちは新しい枠組みと一致するために私たちの手続きと手続きを修正する必要があるだろう。
似たようなデータ保護法はアメリカですでに準備されているか、施行されている。様々なデータ保護およびセキュリティ法律法規は、健康関連および他の個人情報の収集、使用、開示および保護を管理する活動に適用可能である(例えば、州データ漏洩通知法、健康情報および/または遺伝プライバシー法、連邦貿易委員会法案第5条、HIPAAおよびカリフォルニア消費者保護法またはCCPAを含む連邦および州消費者保護法を含む)。州と連邦の2レベルの広範な法執行機関は,一般消費者保護法に基づいて会社のプライバシーやデータセキュリティ問題を審査することができる.連邦貿易委員会と州総検察長は消費者のプライバシーとデータ安全保護を積極的に検討している。州と連邦の二級も新しい法律を考慮している。例えば、カリフォルニア州では、2020年1月に発効したCCPAは、データの使用、共有、および透明性に対していくつかの要求を提出し、カリフォルニア州住民にその個人情報に関するいくつかの権利を提供し、他の多くの州では同様の立法が制定されているか、または制定されることが考えられている。GDPR、CCPA、その他の法規に関するより多くの情報は、本年度報告10-K表の第1項“企業-政府法規と製品承認”を参照されたい。
プライバシー、データ保護、および消費者保護義務の変化の広さと深さを考慮して、これらの要求を準備し、遵守することは厳格で時間がかかり、大量の資源が必要であり、私たちの技術、システムおよび実践、ならびに個人データを保存、処理、または送信する任意の第三者協力者、サービスプロバイダ、請負業者またはコンサルタントを代表する技術、システム、および実践を検討する必要がある。これらの法律の多くは互いに大きく異なり,解釈や適用において司法管轄区域によって一致しない可能性があり,契約遵守作業を複雑化させる可能性がある。GDPRおよびいくつかのタイプの敏感なデータの保護を強化することに関連する他の同様の法律または法規、例えば、私たちの臨床試験からの医療データまたは他の個人情報を遵守することは、私たちの業務慣行を変更し、追加のコンプライアンスメカニズムを確立する必要があるかもしれません。私たちの開発、規制、および商業化活動を中断または延期し、私たちの業務コストを増加させるかもしれません。私たちがこのような法律および法規を遵守できなかったか、または遵守できなかった場合、政府の法執行行動、個人訴訟、および私たちへの巨額の罰金および処罰を招き、私たちの業務、財務状況、または運営結果に重大な悪影響を及ぼす可能性がある。また,これらの法律や個人データ全体の保護に関する消費者集団訴訟の脅威もある。私たちがこれらの法律に違反していると判断されなくても、政府のこれらの問題の調査には通常、大量の資源がかかり、否定的な宣伝が生じ、私たちの名声や業務を損なう可能性がある。
最近公布された立法および将来の立法は、候補製品のマーケティング承認を得て商業化することの難しさとコストを増加させ、米国や外国の管轄地域で承認される可能性のある任意の製品の価格に影響を与える可能性がある。
米国や一部の外国司法管轄地域では、医療システムに関する立法や規制変更、提案された変更は、候補製品の上場承認を阻止または延期し、承認後の活動を制限または規制し、マーケティング承認された候補製品を収益的に販売する能力に影響を与える可能性がある。製薬業界はこのような努力の特別な焦点であり顕著になってきました
82
立法計画の影響を受ける。現行法、および将来取られる可能性のある他の医療改革措置は、より厳しいカバー基準をもたらす可能性があり、私たちが受けた任意のFDA承認製品の価格に追加の下振れ圧力をもたらす可能性がある。もし私たちの製品が清算や範囲が限られていなければ、私たちの業務は実質的な損害を受けるかもしれません。本年度報告表格10-K中の第1項“企業-政府監督と製品審査--薬品保険カバー範囲と医療保健改革”を見た。
2022年8月に“2022年インフレ低減法案”が可決され、その中で医療保険と医療補助サービスセンターが連邦医療保険B部分とD部分によって精算されるいくつかの単一由来薬物と生物製品の価格について交渉し、2026年の一部の高コスト薬物から始まる。この立法規定は、薬品メーカーが提供する価格が法律で規定された交渉価格以下でない場合、あるいは値上げ幅がインフレを超えた場合、民事罰金と潜在的な消費税に直面する。この立法はまた、メーカーに連邦医療保険D部分の価格上昇幅がインフレを超えた薬品にリベートを支払うことを要求している。また,この立法は連邦医療保険受益者の年間自己払い薬品費用を2,000ドルに限定している。“2022年インフレ削減法案”が我々の業務や医療業界全体に与える影響は不明である。
我々は,これらの医療改革,および将来とりうる他の医療改革措置は,連邦医療保険や他の医療資金のさらなる減少,より厳しいカバー基準,新たな支払い方法,および任意の承認された製品のために得られる価格および/または医師が市場に進出する可能性のある任意の承認された製品を管理することによって得られる補償レベルの追加的な下振れ圧力をもたらす可能性があると予想している。精算レベルの低下は私たちが受け取った価格や私たちの製品の処方や管理頻度に悪影響を及ぼす可能性があります。連邦医療保険や他の政府が計画している精算のいかなる減少も、個人支払者の支払いの同様の減少を招く可能性がある。
米国以外の政府は厳しい価格制御を実施する傾向があり、これがあれば私たちの収入に悪影響を及ぼす可能性がある。
米国以外の国、特にEU諸国では、処方薬の定価は政府によってコントロールされている。これらの国では、製品の発売許可を受けた後、政府当局との定価交渉にかなりの時間がかかる可能性がある。一部の国で精算或いは定価の承認を得るためには、私たちの候補製品のコスト効果を他の利用可能な治療法と比較する臨床試験を行う必要があるかもしれない。もし私たちの製品が精算を受けられない場合、あるいは範囲や金額が制限されている場合、あるいは定価が満足できないレベルに設定されている場合、私たちの業務は損害を受ける可能性があり、実質的かもしれません。
もし私たちまたは私たちが現在または将来雇用している任意の第三者製造業者が環境、健康、安全の法律法規を遵守できなかったら、私たちは罰金や処罰を受けたり、私たちの業務を損なう可能性のあるコストや責任を生じるかもしれません。
私たちと私たちが今従事している第三者製造業者、そして私たちが将来従事する可能性のあるいかなる第三者メーカーも、研究室手続きおよび危険材料と廃棄物の処理、使用、貯蔵、処理、処理を管理する法律法規を含む多くの環境、健康、安全な法律法規の制約を受けるだろう。私たちの行動は化学物質と生物学的材料を含む危険で燃えやすい材料の使用に関するものだ。私たちの業務はまた危険な廃棄物製品を発生させるだろう。私たちは一般的に第三者と契約を結び、このような材料と廃棄物を処理する。私たちはこのような材料が汚染や傷害をもたらす危険を除去することができない。もし私たちが危険な材料を使用して汚染や損傷をもたらしたら、私たちはそれによるいかなる損害に責任を負い、いかなる責任も私たちの資源の範囲を超える可能性がある。危険材料の放出や清掃を管理するいくつかの環境法によると,責任は連携しており,非を考慮せずに加えることができる。私たちはまた、民事または刑事罰金や処罰に関連する巨額の費用を招いたり、このような法律や法規を遵守できなかったために、私たちの活動を制限または禁止する禁止令の制約を受ける可能性があります。
危険材料の使用により従業員が負傷したコストや支出を支払うために一般責任保険と労働者補償保険を維持しているが、この保険は潜在的な責任に対応するには不十分である可能性がある。私たちは私たちが生物、危険または放射性物質を貯蔵したり処分したりすることによって、私たちが提起した環境責任や有毒侵害に対して保険を維持することはできません。
また、現在または未来の環境、健康、安全法律法規を遵守するためには、巨額のコストが生じる可能性がある。このような現行または未来の法律法規は私たちの研究、開発、または生産努力を損なうかもしれない。私たちがこのような法律を守らないことはまた巨額の罰金、処罰、または他の制裁につながるかもしれない。
さらに、私たちの現在および将来の任意の第三者契約製造業者の運営について、彼らが適用される環境、健康および安全法律法規に準拠していない場合、または私たちの製品に関連する廃棄物を適切に処理することができない場合、私たちは、それによって生じるいかなる損害、名声損害、または私たちの候補製品または製品の製造と供給中断に責任を負わなければならないかもしれない。さらに、私たちの第三者契約製造業者が環境、健康、安全法律法規を守らないことで禁止または他の制裁を受けた場合、私たちのサプライチェーンは悪影響を受ける可能性があります。
83
私たちは反腐敗法、そして輸出規制法、税関法、規制法、その他私たちの業務を管理する法律を守らなければならない。もし私たちがこれらの法律を遵守しなければ、私たちは民事または刑事処罰、他の救済措置、法律費用を受け、米国以外で特定の製品の開発と販売を禁止されたり、費用の高いコンプライアンス計画の制定と実施が要求される可能性があり、これは私たちの業務、運営結果、財務状況に悪影響を及ぼすかもしれない。
私たちの業務は、イギリスの“2010年収賄法”、米国の“海外腐敗防止法”、および私たちが業務を展開し、将来業務を展開する可能性のある国/地域に適用される他の反腐敗法律を含む反腐敗法を遵守しなければならない。収賄法、海外腐敗防止法、およびその他の法律は、一般に、業務を獲得または保留し、またはいくつかの他の商業的利益を得るために、私たち、私たちの職員、および中間者が政府官僚または他の人に贈賄、贈賄され、または他の他の禁止された金を他人に支払うことを禁止している。“反海外腐敗法”を遵守することは特に高価で困難であり、特に腐敗は公認問題である国である。また、海外腐敗防止法は製薬業に特別な挑戦をもたらしており、多くの国では病院が政府によって運営されているため、医師や他の病院従業員は外国人官僚とされている。臨床試験やその他の仕事に関連して病院に支払われた何らかの金は、政府関係者に支払われた不正金と考えられ、“海外腐敗防止法”の法執行行動につながった。
私たちは将来的に“収賄法”や“海外腐敗防止法”に違反する可能性のある高リスク司法管轄区域で業務を展開する可能性があり、私たちは第三者との協力や関係に参加する可能性があり、これらの第三者の行為は私たちに“反収賄法”、“海外腐敗防止法”あるいは現地反腐敗法に規定された責任を負わせる可能性がある。また、将来の規制要求の性質、範囲、影響を予測することはできず、私たちの国際業務はこれらの要求によって制約される可能性があり、現行の法律が管理または解釈される可能性がある方式を予測することもできない。私たちの業務をアメリカ以外の地域に拡張すれば、私たちが各管轄区域で業務を展開することを計画している多くの法律と法規を遵守するために、追加の資源を投入する必要があります。
私たちはまた、私たちの国際業務を管理する他の法律と法規、イギリスとアメリカ政府及びEU当局が管理する法規を含み、適用される輸出規制法規、国と人員に対する経済制裁、税関要求と貨幣両替法規を含み、総称して貿易規制法と呼ばれる。さらに、様々な法律、法規、および行政命令は、米国国外での使用および伝播を制限するか、または国家セキュリティ目的のために秘密にされた情報、および特定の製品およびこれらの製品に関連する技術データを特定の非米国国民と共有することも制限される。もし私たちがアメリカ以外での業務を拡大すれば、これらの法律を遵守するためにもっと多くの資源を投入する必要があります。これらの法律は、私たちがアメリカ以外で特定の製品や候補製品を開発、製造、販売することを阻止するかもしれません。これは、私たちの成長潜在力を制限し、私たちの開発コストを増加させるかもしれません。
私たちが貿易統制法を含む“反収賄法”、“海外腐敗防止法”または他の法律要件を含むすべての適用された反腐敗法律を遵守することを完全かつ効果的に保証することはできない。私たちが“反収賄法”、“海外腐敗防止法”および他の腐敗防止法または貿易統制法を遵守しなければ、私たちは刑事および民事処罰、返還および他の制裁および救済措置、および法的費用を受ける可能性があり、これは私たちの業務、財務状況、運営結果、流動性に悪影響を及ぼす可能性がある。米国証券取引委員会は、発行者が海外腐敗防止法の会計規定に違反したため、発行者の米国取引所での証券取引を一時停止または禁止する可能性もある。英国、米国または他の当局は、“反収賄法”、“反海外腐敗法”、その他の腐敗防止法または貿易制御法に違反する可能性のあるいかなる調査も、私たちの名声、業務、運営結果、財務状況に悪影響を及ぼす可能性がある。
私たちの従業員、独立請負業者、コンサルタント、サプライヤーは、規制基準と要求およびインサイダー取引を含む不適切な行為や他の不適切な活動に従事する可能性があり、これは私たちに重大な責任を与え、私たちの名声を損なう可能性があります。
私たちは従業員、独立請負業者、コンサルタント、そしてサプライヤーの詐欺または他の不適切な行為のリスクに直面している。これらのパートナーの不正行為は、FDA法規または外国規制機関と比較することができる類似の法規を故意に遵守しないこと、FDAまたは外国規制機関よりも正確な情報を提供すること、製造基準を遵守すること、連邦および州医療詐欺および法律法規を乱用すること、および外国規制機関よりも制定および実行可能な類似の法律法規、財務情報またはデータを正確に報告すること、または許可されていない活動を開示することを含むことができる。従業員の不当行為はまた臨床試験過程で得られた情報を不当に使用する可能性があり、これは規制制裁と著者らの名声に深刻な損害を与える可能性がある。これには、HIPAA、他の米国連邦、州法律、およびEU GDPRを含む非米国司法管轄区域の要求が含まれる可能性がある。私たちはまた従業員や私たちと関連のある他の人たちがインサイダー取引に違反するリスクに直面している。従業員の不正行為を常に識別し、阻止できるわけではなく、このような行為を発見し、防止するための予防措置は、未知または未管理のリスクまたは損失を効果的に制御することができない可能性があり、または、このような法律、基準、法規、ガイドラインまたは行動基準に準拠できないことによる政府の調査または他の行動または訴訟から私たちを保護することができるかもしれない。もし私たちにこのような行動を取って、私たちが自分の権利を弁護したり、維持したりすることに成功しなかったら、これらの行動は、巨額の罰金や他の制裁を加えることを含む、私たちの業務および業務結果に大きな影響を及ぼすかもしれない。
84
我々の内部コンピュータや情報技術システムおよびインフラ、または私たちの協力者または他の請負者またはコンサルタントのシステムおよびインフラは、故障したり、セキュリティ脅威や脆弱性を受けたりする可能性があり、これにより、私たちの製品開発計画が実質的に破壊される可能性があります。
我々の内部コンピュータおよび情報技術システムおよびインフラ、ならびに私たちの業務に依存するCRO、協力者および他の請負業者またはコンサルタントのシステムおよびインフラは、コンピュータウイルス、許可されていないアクセス、データ漏洩、ネットワーク釣り攻撃、サイバー犯罪者、システム故障、自然災害(ハリケーンおよび地震を含む)、テロ、戦争、電気通信、および電気故障の破壊または破壊を受けやすい。このようなシステムおよびインフラストラクチャは、我々の従業員、CROまたは他の第三者サプライヤー、請負業者、コンサルタント、および/またはビジネスパートナーまたは他の第三者の不注意または意図的な行為によるサービス中断またはセキュリティ被害または破壊、または悪意のある第三者のネットワーク攻撃を受けやすい。ネットワーク攻撃の頻度,複雑さ,強度が増加しており,発見されにくくなってきている.ネットワーク攻撃には、内部従業員またはサプライヤーの不法行為、敵対する外国政府、産業スパイ活動、電気通信詐欺および他の形態のネットワーク詐欺またはネットワーク攻撃が含まれる可能性があり、有害マルウェアの配備、恐喝ソフトウェア、サービス拒否攻撃、ファイルの不正アクセスまたは削除、ネットワーク釣り攻撃および社会工学、商業電子メール漏洩、およびサービスの信頼性および情報セキュリティ、完全性および可用性に影響を与える他の手段を含むことができる。したがって、私たちまたは私たちのサービスプロバイダのネットワークセキュリティ対策が、不正なアクセス、攻撃、危害、または私たちの従業員または請負業者のデータの不適切な処理を防ぐことができない場合、私たちの名声、顧客信頼、業務, 運営結果と財政状況は不利な影響を受けるかもしれない。我々のコンピュータシステムまたは我々の協力者または他の請負者またはコンサルタントのコンピュータシステムを浸透および破壊しようと試みる可能性のある脅威参加者によって使用される技術は、しばしば変化し、ターゲットに攻撃を開始する前に識別できない可能性があるため、これらの技術を予測することができない可能性がある。例えば、クラウドベースのストレージシステムを広く使用していますが、2018年10月、このようなシステムが破られたことを経験しました。この抜け穴は、私たちの重要な情報が永久的に失われたり、盗まれたり、他の重大な結果をもたらしていませんが、多要素認証の確立や私たちのデータセキュリティ協定の改善などの救済措置を取っているかもしれませんが、私たちが今まで取ってきた措置と、私たちが将来取る可能性のある行動は、任意の未来の違反を救済するのに十分な措置をとることはできません。
これまで、私たちは重大なシステム障害、事故、ネットワーク攻撃、またはセキュリティホールに遭遇していませんが、このような事件が発生して私たちの運営が中断された場合、私たちのビジネス機密や他の固有情報の損失によるものであっても、他の同様の中断によるものであっても、私たちの開発計画や業務運営の実質的な中断を招く可能性があります。例えば、完成した或いは未来の臨床試験の臨床試験データの紛失は著者らの監督管理の承認作業を遅延させ、著者らのデータの回復或いは複製のコストを著しく増加させる可能性がある。任意の中断またはセキュリティ被害または脆弱性が、私たちのデータ、システム、インフラまたはアプリケーションの損失、破損、許可されていないアクセスまたは乱用、または機密または独自の情報を適切に開示しない場合、私たちは責任(訴訟や政府の調査および法執行行動に関連しているか、またはそれによるものを含む)を招く可能性があり、私たちの競争的地位が損なわれる可能性があり、私たちの候補製品のさらなる開発および商業化は延期される可能性があり、私たちの業務は他の悪影響を受ける可能性がある。
従業員の事務と管理成長に関するリスク
私たちの未来の成功は私たちが肝心な幹部を維持する能力、及び合格した人材を吸引、維持、激励する能力にかかっている。
私たちは私たちの幹部と私たちの管理、科学と臨床チームの他の主要なメンバーの研究開発、臨床、財務、運営とその他の業務の専門知識に高度に依存している。私たちは私たちの幹部と招聘状を締結したが、彼らの誰もが私たちとの雇用関係をいつでも終わらせることができる。私たちは私たちの役員や他の職員たちに“キーパーソン”保険を提供しない。合格した科学、臨床、製造、会計、法律及び販売とマーケティング人員を募集と維持することも著者らの成功の鍵となる。
85
我々は最近,最高経営責任者,研究開発部門の総裁,首席科学者,首席医療官を含む幹部交代を行った。私たちは未来の幹部交代の可能性、タイミング、あるいは影響を予測できない。幹部や他の重要な従業員を失ったサービスは、私たちの研究開発と商業化目標の実現を阻害し、業務戦略を成功させる能力を深刻に損なう可能性がある。また、幹部やキーパーソンを交換することは困難かもしれませんし、私たちの業界では開発に成功し、規制承認や製品商業化に必要なスキルや経験を持つ個人の数が限られているので、長い時間がかかるかもしれません。この限られた人材バンクから募集する競争は非常に激しく、多くの製薬と生物技術会社の間の類似人員に対する競争を考慮して、私たちは受け入れ可能な条件でこれらの肝心な人員を採用、訓練、維持或いは激励することができないかもしれない。私たちはまた、大学や研究機関から科学や臨床人を募集する競争に直面している。また、私たちは、科学と臨床コンサルタントを含むコンサルタントとコンサルタントに依存して、私たちの研究開発と商業化戦略の制定を助けてくれます。2022年8月に研究開発部門のリストラを発表しました, これは私たちの雇用主が未来の求職者にそんなに魅力的ではないようにするかもしれない。私たちのコンサルタントやコンサルタントは、私たち以外の雇用主に雇われる可能性があり、他のエンティティと締結された相談または相談契約に基づいて約束することができ、これは、私たちが彼らを得る機会を制限するかもしれない。上場企業として、私たちの成功はまた内部統制の実施と維持と私たちの財務報告の正確性と即時性にかかっている。もし私たちが引き続き高い素質の人材を誘致し、維持することができなければ、私たちが成長戦略を推進する能力は制限されるだろう。
私たちは私たちの開発と規制能力を拡大し、販売、マーケティング、流通能力を実施する可能性があると予想されているので、私たちは私たちの成長を管理することが困難になるかもしれません。これは私たちの運営を混乱させるかもしれません。
私たちは、特に薬物開発、臨床、規制、および私たちの任意の候補製品がマーケティング承認、販売、マーケティング、流通を獲得すれば、私たちの従業員の数と業務範囲が大幅に増加することを予想しています。私たちが予想している将来の成長を管理するためには、私たちの管理、運営、財務システムを継続して実施し、改善し、私たちの施設を拡大し、より多くの合格者を募集し、訓練し続けなければならない。私たちの財務資源が限られていることと、私たちの管理チームがこのような成長を期待している会社を管理する上での経験が限られているため、私たちの業務の拡張を効果的に管理したり、より多くの合格者を募集したりすることができないかもしれません。私たちの業務の拡張は巨大なコストを招き、私たちの管理と業務発展資源を移転する可能性があります。成長を管理できないどんな状況も、私たちの業務計画の実行を延期したり、私たちの運営を妨害したりする可能性がある。
私たちの普通株に関するリスクは
もし私たちの役員、役員、主要株主が共同行動を選択すれば、彼らは株主に提出されたすべての事項を制御または顕著に影響する能力がある。
2023年3月2日現在、私たちの役員と取締役、および私たちが発行した普通株の5%以上の株主実益を持っている株式総数は、私たちの株式の約57.1%を占めています。したがって、これらの株主が共同行動を選択すれば、彼らは私たちの株主に提出されたすべての事項と、私たちの管理と事務を制御または顕著に影響することができるだろう。例えば、この人たちが一緒に行動することを選択した場合、彼らは取締役の選挙と、私たちのすべてまたはほとんどの資産の任意の合併、合併、または売却の承認を制御するだろう。
このような所有権制御の集中可能性:
86
わが社の定款書類やデラウェア州法律の条項は、わが社の買収をより困難にする可能性があり、これは私たちの株主に有利になる可能性があり、私たちの株主が現在の取締役や経営陣のメンバーを交換または罷免しようとすることを阻止するかもしれません。
会社登録証明書及び定款における条項は、株主がその株式から割増取引を得る可能性があることを含む、株主が有利であると考えられる会社の合併、買収又はその他の制御権変更を阻害、延期又は阻止する可能性がある。これらの条項はまた、投資家が将来私たちの普通株に支払いたいかもしれない価格を制限し、それによって私たちの普通株の市場価格を下げる可能性がある。また、我々の取締役会が責任を持って我々の管理チームのメンバーに命じているため、これらの規定は、株主が取締役会のメンバーを交換する難しさを増やすことで、現在の経営陣の任意の試みを交換または罷免することを阻害または阻止する可能性がある。他にもこれらの条項には
また、私たちはデラウェア州に登録して設立されたので、私たちはデラウェア州会社法第203条の規定によって管轄されています。この条項は、規定された方法で合併または合併が承認されない限り、私たちが発行した議決権のある株を15%以上保有している人が取引日後3年以内に私たちと合併または合併することを禁止しています。
証券アナリストが研究報告書を発表したり停止したりしなければ、私たちの業務の誤解性、不正確または不利な研究報告を発表したり、彼らが私たちの株に対する否定的な評価を発表したりすると、私たちの株式の価格や取引量が低下する可能性がある。
私たちの普通株の取引市場は、業界や金融アナリストが発表した私たちまたは私たちの業務に関する研究と報告にある程度依存している。既存のアナリストが私たちを追跡し続ける保証はなく、新しいアナリストが私たちを追跡し始めるという保証もない。どんなカバーアナリストも有利な報告書を提供するという保証はない。私たちはアナリストの報告を得ていますが、1人以上の私たちの業務を追跡しているアナリストが私たちの株の評価を下方修正したり、私たちの業務の不正確または不利な研究報告を発表したり、私たちの競争相手に関するより有利な相対的な提案を提供したりすれば、私たちの株価は下落する可能性があります。もしこのようなアナリストの一人以上が私たちの株を追跡しなければ、私たちは市場での私たちの株の可視性を失うかもしれません。これは逆に私たちの株価と取引量を低下させる可能性があります。
私たちの普通株の価格は大きく変動する可能性があり、これは私たちの株主に大きな損失をもたらすかもしれない。
私たちの普通株の取引価格はずっと、変動し続ける可能性が高く、様々な要素の広範な変動を受ける可能性があり、その中のいくつかの要素は私たちがコントロールできない。総じて言えば、株式市場、特に小型バイオ製薬会社の市場は極端な変動を経験しており、この変動はしばしば
87
特定会社の経営実績。私たちの普通株の市場価格は多くの要素の影響を受けるかもしれません
過去には、ある会社の証券市場価格が変動した後、同社に対して証券集団訴訟が提起されることが多かった。私たちは一方としてのいかなる訴訟も、正当な理由の有無にかかわらず、不利な判決を招く可能性がある。私たちはまた不利な条件で訴訟を解決することに決定するかもしれない。このような負の結果は、巨額の損害賠償または罰金の支払い、私たちの名声を損なうこと、または私たちの製品またはビジネス実践に悪影響を及ぼす可能性があります。このような訴訟は、このようなクレームを弁護し、経営陣の注意と資源を移転させるために、他の巨額の費用を発生させる可能性もある。また、公聴会、動議、または他の一時的な手続きや事態の発展結果に対する負の公開公告は、私たちの普通株の市場価格に負の影響を与える可能性がある。
私たち総流通株の大部分は市場で売る資格があります。これは私たちの普通株の市場価格を大幅に低下させる可能性があります。たとえ私たちの業務が良好であっても。
公開市場で私たちの普通株の大量株を売ったり、市場で大量の株を持っていると思っている人が株を売却しようとしたりすると、私たちの普通株の市場価格が下がる可能性があります。私たちが初めて公募するまで私たちの株主だった人は私たちの普通株の大量の株式を持ち続けました。もしこの人たちが公開市場で私たちの普通株を大量に売却したり、意図的に私たちの普通株を売却したりすると、私たちの普通株の取引価格が下がるかもしれません。
また、我々は、一般棚登録声明(販売時に決定された価格及び条項に基づいて1つ以上の製品に応じて証券を随時提供及び売却することを可能にすることを許可する)と、当社の持分補償計画又は株式補償計画以外の新入社員に基づいて取得した持分奨励に基づいて発行可能なすべての普通株の登録声明とを提出又は提出することを意図している。この等登録株式は発行されると公開市場で自由に販売することができるが,連属会社の適用数量制限に制限される必要がある。
88
私たちは“新興成長型会社”であり、新興成長型会社に適用される開示要求を下げることは、私たちの普通株の投資家への魅力を低下させる可能性があります。
私たちは“新興成長型会社”やEGCで、2012年のJumpStart Our Business Startups ActやJOBS Actの定義に基づいています。私たちは2024年12月31日までEGCになるかもしれませんが、それまでの任意の6月30日に、非関連会社が保有している私たちの普通株の時価が7億ドルを超えている場合、または任意の会計年度の年間毛収入が12.35億ドルに達した場合、適用年度の12月31日からEGCになることを停止します。もし私たちが3年以内に10億ドルを超える転換不能債券を発行すれば、私たちはまたEGCではないだろう。私たちがまだEGCである限り、私たちは他のEGCではない上場企業に適用されるいくつかの開示要求の免除に依存することを許可され、意図されている。これらの免除には
私たちは利用可能な免除の一部または全部を利用することを選択することができる。私たちはもし私たちがこのような免除に依存すれば、投資家が私たちの普通株の吸引力の低下を発見するかどうか予測できない。したがって、一部の投資家が私たちの普通株の吸引力が低下していることを発見すれば、私たちの普通株はそれほど活発ではない取引市場がある可能性があり、私たちの株価はもっと変動するかもしれない。
また、雇用法案は、EGCが、これらの基準が民間企業に適用されるまで、上場企業に適用される新しい会計基準または改正された会計基準を遵守することを延長する移行期間を利用することを可能にする。私たちは、“選択脱退”延長の移行期間を選択したが、これは、基準が発表または改正された場合、それが公共または民間会社に対して異なる適用日を有する場合、私たちは、民間会社が新しい基準または改正基準を採用する際に、私たち(I)が“選択脱退”延長の移行期間を撤回できなくなるまで、または(Ii)EGCの資格に適合しなくなるまで、このようにしていくことを意味する。
上場企業として、コストを増加させ続けており、我々の経営陣は、新たなコンプライアンス措置やコーポレートガバナンス実践を実施するために多くの時間を投入する必要があるだろう。
上場企業として、私たちはすでに発生しています。特に私たちがEGCでなくなった後、私たちは引き続き大量の法律、会計、その他の費用を発生させます。これらの費用は私たちが個人会社として発生していません。2002年のサバンズ-オキシリー法案、ドッド-フランクウォール街改革と消費者保護法、ナスダック世界市場の上場要求、その他の適用された証券規則と法規は上場企業に対して様々な要求を提出し、有効な開示、財務制御と会社管理やり方を確立と維持することを含む。私たちの経営陣と他の人たちはこのようなコンプライアンス計画を実施するために多くの時間を投入する必要があるだろう。また、これらの規則および法規は、特に上場企業内部統制および財務報告要件を満たすために追加の財務および会計従業員を雇用した場合に、いくつかの活動をより時間とコストを高くするために、我々の法律および財務コンプライアンスコストを増加させる。例えば、これらの規制は、私たちが取締役や上級管理職責任保険を獲得することをより困難かつ高価にする可能性があり、逆に、合格した取締役会メンバーを引き付け、維持することが困難になる可能性があると予想されます。
私たちはこのような規則と規定を評価しており、私たちが発生する可能性のある追加コスト金額やそのようなコストの時間を予測または推定することはできない。これらの規則や条例は往々にして異なる解釈を持ち,多くの場合特殊性に欠けるため,規制機関や理事機関が新たな指導意見を提供するにつれて,実践における適用は時間とともに変化する可能性がある。これは遵守事項に関する持続的な不確実性と、開示と統治慣行を絶えず修正するために必要なより高いコストをもたらす可能性がある。
89
2002年サバンズ-オキシリー法404条または404条によると、私たちは財務報告書の内部統制に関する管理職報告書を提出しなければならない。しかし、私たちはまだEGCまたは収入が1億ドル未満の小さな報告会社であるにもかかわらず、私たちの独立公認会計士事務所が発行した財務報告内部統制の認証報告を含むことは要求されません。規定された期間内に第404条の規定を遵守するために、私たちは毎年、費用が高く挑戦的な財務報告に対する私たちの内部統制を記録して評価する過程を行っている。この点では、より多くの財務·会計担当者を雇用することを含む内部資源を提供し続ける必要があり、外部相談者を招聘することが可能であり、財務報告内部統制の十分性を評価·記録することにより、財務報告内部制御プログラムを適宜改善し、制御措置が文書に規定された方法で動作しているか否かをテスト検証することにより、継続的な報告を実施し、財務報告内部制御プログラムを改善する必要がある。私たちは努力したにもかかわらず、私たちは規定された時間枠内で結論を出すことができないかもしれない、すなわち私たちは財務報告書の内部統制に有効であり、404条の要求に適合する可能性がある。財務報告書の内部統制に1つまたは複数の重大な弱点があることが発見されれば、財務諸表の信頼性への自信を失った金融市場の不良反応を招く可能性がある。
私たちは予測可能な未来に私たちの株に何の現金配当も支払わないと予想されるので、資本付加価値(あれば)が私たちの株主の唯一の収益源になるだろう。
私たちは私たちの株の現金配当金を発表したり支払ったりしたことがない。私たちは現在、私たちの未来のすべての収益を維持するつもりで、もしあれば、私たちの業務の成長と発展に資金を提供します。したがって、私たち普通株の資本増価(あれば)は私たちの株主が予測可能な未来に唯一の収益源になるだろう。
私たちの会社登録証明書は、デラウェア州の州裁判所が私たちの株主のために起こしうるいくつかのタイプの訴訟および訴訟の唯一のおよび独占フォーラムを指定し、これは、会社と私たちの役員、上級管理者、および従業員に対する訴訟を阻止するかもしれない。
私たちの会社登録証明書は、私たちが書面で別のフォーラムを選択することに同意しない限り、デラウェア州衡平裁判所(または、デラウェア州衡平裁判所に管轄権がない場合、デラウェア州連邦地域裁判所)は、(1)私たちが提起した任意の派生訴訟または法的手続きを代表して、(2)私たちのいかなる取締役、上級管理職、従業員または株主のわが社または私たちの株主に対する受託責任に違反すると主張する任意の唯一かつ独占的なフォーラムとなる。(3)当社の登録証明書または別例(各ケースにおいて、随時改訂することができる)の任意の条文によって引き起こされる任意の訴訟、または(4)当社の会社登録証明書または別例(各ケースにおいて、随時改訂することができる)の任意の条文によって生成される申立の任意の訴訟、または(4)当社の会社登録証明書または附例のいずれかの条文に従って生成された請求書を主張する訴訟、または(4)当社の会社登録証明書または添付例のいずれかの条文に基づいて生成された請求書を主張する訴訟。この排他的法廷条項は、証券法や1934年に改正された証券取引法に基づいて提起された訴訟には適用されない。
この排他的な裁判所条項は、私たちの株主が司法裁判所でこのような株主が、私たちまたは私たちの役員、上級管理者、または従業員と紛争することに有利だと思うクレームを提起する能力を制限する可能性があり、これは、私たちと私たちの役員、上級管理者、および従業員に対するこのような訴訟を阻止する可能性がある。あるいは、裁判所が私たちの会社登録証明書に含まれる裁判所条項の選択が訴訟で適用されないか、または実行できないことを発見した場合、私たちは、他の司法管轄区でこのような訴訟の解決に関連する追加費用を発生させる可能性があり、これは、私たちの業務、財務状況、および経営業績に大きな悪影響を及ぼす可能性があります。
90
項目1 B。取消解析Dスタッフがコメントした。
適用されません。
プロジェクト2.専門家ペルテスです。
私たちの主な施設はオフィスと実験室を含む。現在2028年6月に満期になる賃貸契約によると、私たちはマサチューセッツ州ケンブリッジ市に約28,731平方フィートのオフィススペースを持っている。
項目3.法律法律手続き。
私たちは現在どんな実質的な法的手続きの当事者でもない。
プロジェクト4.地雷安全安全に開示する。
適用されません。
91
部分第2部:
項目5.登録者普通株·関連株の市場保有者は重要であり,発行者は株式証券を購入する.
市場情報
我々の普通株のナスダック世界市場での取引コードはFULCであり、2019年7月18日から公開取引される。その前に、私たちの普通株は市場を公開しなかった。
私たち普通株保有者
2023年3月2日現在、約17名の普通株式保有者が登録されている。この数字には、“著名人”や“街”の名で株を持っている株主は含まれていない。
配当をする
私たちは私たちの株のどんな現金配当金も発表したり支払ったりしたことがない。私たちは現在、すべての利用可能な資金と任意の将来の収益を維持し、私たちの業務運営のために、予測可能な未来に私たちの普通株にいかなる配当も支払わないと予想している。将来配当を発表する任意の決定は、私たちの取締役会が適宜決定し、私たちの財務状況、経営業績、資本要求、一般業務状況と私たちの取締役会が関連する他の要素に依存するかもしれません。
株式証券の未登録販売
ない。
プロジェクト6.Re料理が出ました。
適用されません。
92
項目7.経営陣の以下の問題の議論と分析財務状況と経営実績。
以下、当社の財務状況と経営結果の検討と分析は、本年度報告末にForm 10−K形式で表示された連結財務諸表と関連付記とともに読むべきである。本議論および分析に含まれるいくつかの情報または本Form 10−K年次報告における他の情報は、リスクおよび不確実性に関する前向きな陳述を含む、我々の業務計画および戦略に関する情報を含む。本年度報告10-K表の“リスク要因”の部分に列挙された要素を含む多くの要因の影響により、我々の実際の結果は、以下の議論および分析に含まれる前向き陳述に記載または示唆された結果とは大きく異なる可能性がある。
概要
我々は臨床段階の生物製薬会社であり,高度に満たされていない医療需要地域の遺伝定義を改善するまれな疾患患者の生活の改善に専念している。我々の最先端の臨床候補製品losmapimodは開発中であり,潜在的なFSHD治療に用いられている。我々のもう1つの臨床候補はFTX−6058であり,SCDを含むいくつかのヘモグロビン疾患を潜在的に治療するために開発されている。私たちは Initiated REACHは無作為,二重盲検,プラセボ対照,多国のロモパモント3期臨床試験であり,2022年第2四半期に行われ,2023年下半期に募集を完了する予定である。2023年1月、SCDにおけるFTX−6058の臨床試験の1 b段階データを発表した。私たちは6 mgと2 mgの用量列の登録を終えて、私たちはこの列でもっと多くの科目を募集するつもりはない。12 mg用量列の登録を開始したにもかかわらず,FDAは2023年2月23日にSCD用FTX−6058のIND臨床試験を全面的に一時停止した。私たちはFTX-6058 1 b期試験の登録と用量を一時停止し、ベータ地中海貧血に対するFTX-6058単独のINDを撤回し、FDAと勤勉に協力して、一時停止問題をできるだけ早く解決しようとしている。
我々はすでに特許製品エンジンFulcrumSeekを開発し、それを用いて細胞薬物標的を系統的に識別し、検証し、これらの標的は遺伝子発現を潜在的に調節して、既知の遺伝子定義疾患の根本的な原因を治療することができる。著者らの製品エンジンは患者由来の組織と疾病関連細胞モデルを統合し、著者らは著者らの薬物多様性と高度に注釈された小分子化合物ライブラリー及びカスタマイズされたCRISPRとRNAiライブラリを用いて検索を行った。これらの画面は数千万のデータ点と高コンテンツ画像を生成する。そして,計算生物学と分析を用いて特異性と選択性を持つ目標を識別し,開発を著しく加速させる包括的なデータセットを伴った。この方法は、FSHDのためのlosmapimodとヘモグロビン疾患のためのFTX−6058の識別、および強力な発見導管をもたらす。
設立以来、私たちの業務の重点は私たちの会社を組織と配備し、業務計画を制定し、資金を調達し、私たちの知的財産権を構築し、私たちの発見プラットフォームを構築し、私たちの独自化合物バンクと製品エンジンを含み、薬物標的と潜在的な候補製品を識別し、許可資産、臨床試験のための薬物物質と薬物製品材料の生産、および臨床前研究と臨床試験を行うことである。今まで、私たちの運営資金は主に私たちの株式株式の売却と私たちの協力と許可契約に基づいて受け取った前金から来ています。
2022年8月、私たちは1株7.82ドルの公開発行価格で11,029,410株の普通株を公開発行し、販売業者が公開発行価格で追加株式の選択権を購入する際に発行した1,438,618株を全面的に行使し、引受割引と手数料を引いた。引受割引と手数料および発売費用を差し引くと、次発行の純収益は8,080万ドルです。
2023年1月、私たちは1株13.00ドルの公開発行価格で9,615,384株の普通株を発行し、引受割引と手数料を引いた。引受割引と手数料および発売費用を差し引くと、次発行の純収益は1億173億ドル。
93
私たちが設立して以来、私たちはすでに重大な運営損失が発生しており、私たちは予測可能な未来に重大な運営損失が続くと予想している。私たちは利益のある製品収入を達成するのに十分な能力を創出し、もしあれば、私たちの1つ以上の候補製品の成功開発と最終商業化に大きく依存するだろう。2022年12月31日と2021年12月31日までの年度の純損失はそれぞれ1.099億ドルと8080万ドルだった。2022年12月31日までの累計赤字は4億123億ドルだった。私たちは今後数年間、私たちの持続的な活動に関連する費用と運営損失が大幅に増加すると予想している
したがって、私たちは私たちの持続的な運営を支援し、私たちの成長戦略を実施するために多くの追加資金を必要とするだろう。私たちが製品販売から相当な収入を得ることができる前に、もしあれば、株式発行、債務融資、協力、戦略連合、マーケティング、流通、または許可手配を通じて、私たちの運営に資金を提供する予定です。私たちは必要に応じて追加資金を優遇条項で調達したり、そのような他の合意や手配を達成することができないかもしれません。もし私たちが必要な時に資金を調達したり、合意に達しなかったら、私たちは私たちの1つ以上の候補製品の開発と商業化を大幅に延期、削減、または停止しなければならないかもしれないし、たとえ私たちが自分でこのような候補製品を開発し、マーケティングすることを望んでいたとしても、私たちの候補製品を開発し、マーケティングする権利を与えなければならないかもしれない。
薬物開発に関連する多くのリスクや不確実性により,費用の増加時間や金額を予測することはできず,いつあるいは利益を得ることができるかどうかを予測することもできない。たとえ私たちが製品販売を作ることができても、私たちは利益を上げることができないかもしれない。もし私たちが利益を上げることができない場合、または持続的に利益を上げることができない場合、私たちは計画通りに運営を継続できず、私たちの運営を減少または終了させることができないかもしれない。
2022年12月31日現在、私たちは2.029億ドルの現金、現金等価物、有価証券を持っている。私たちは既存の現金、現金等価物、有価証券に加え、2023年1月に公募株で私たちの普通株を売却した純収益に加えて、2025年中までの運営費と資本支出需要に資金を提供できると信じている。私たちは間違っていることが証明される可能性があるという仮定に基づいてこの推定をして、私たちは予想よりも早く利用可能な資本資源を枯渇させるかもしれない。“流動性と資本資源”を見てください
94
経営成果の構成部分
収入.収入
私たちは製品販売から何の収入も得ていないし、数年以内に製品販売から収入を得ることも望んでいません。もしあれば。現在または将来の候補製品の開発に成功し、市場の承認を得られれば、将来的に製品販売から収入を得ることができるかもしれない。私たちは私たちの候補製品の商業化と販売からどの程度収入を得るか、いつ、あるいはどの程度収入を得るか予測できない。私たちのどんな候補製品も規制部門の承認を得ることができないかもしれない。
2019年12月、私たちはAcceleronと協力と許可協定を締結し、肺疾患領域における標的適応に関連する特定の経路を調節するための生物学的標的を決定した。この協定は2022年10月1日に終了し、これまでAcceleronは2022年6月に便宜上合意終了を決定することを通知した。
2022年と2021年12月31日までの年度では,Acceleron連携プロトコルによりそれぞれ100万ドルと960万ドルの連携収入を確認した。2022年12月31日現在、私たちはAcceleron連携協定に関連した繰延収入を記録していない。2021年12月31日現在、私たちはAcceleron連携協定に関連する60万ドルの繰延収入を記録しており、収入予想に基づいて確認されている期間に、この収入は私たちの総合貸借対照表において現在の部分または現在の部分の純額に分類されている。2022年12月31日現在、490万ドルのコスト精算支払いと200万ドルのAcceleron協力協定マイルストーン支払いを受け取りました。2022年12月31日現在、Acceleron協力協定下の精算可能な研究開発コストに関する未開請求書の売掛金は記録されていません。
2020年7月20日、私たちはMyoKardiaと協力と許可協定を締結し、この協定に基づいて、私たちはある知的財産権に基づいてMyoKardiaに世界的な独占許可を授与し、その研究、開発、製造、製造、使用、使用、販売、販売、要約、販売、輸入、輸入、輸出、輸出、流通、マーケティング、マーケティング、普及、普及、あるいは他の方法で私たちが決定した特定の生物標的に対する製品を開発することを可能にし、これらの製品はある遺伝子で定義された心筋症に関連する特定の数量の関心遺伝子を調節することができる。MyoKardiaはその後、2020年11月に百時美施貴宝社に買収された。協力の主要な目標は更なる研究の潜在的な生物標的を確定と検証し、MyoKardia社がある遺伝性心筋症を潜在的に治療する候補製品の開発、製造と商業化を支持することである。
MyoKardia協力協定の条項によると、私たちは2020年7月に1000万ドルの前払いと250万ドルの前払い研究資金を受け取った。MyoKardiaは,前払い研究資金にカバーされていない研究活動の費用を精算し,最高限度額は研究資金総額(前払い研究資金を含む)である。特定の臨床前、開発と販売マイルストーンを実現した後、ある潜在的な心筋症遺伝子目標に対して、私たちは各目標の合計2.985億ドルの臨床前マイルストーン支払い、開発マイルストーン支払いと販売マイルストーン支払いを獲得する権利があり、いくつかの他の潜在心筋症遺伝子目標に対して、私たちは各目標の合計1.5億ドルに達する支払いを得る権利がある。今まで、私たちは指定された250万ドルの臨床前マイルストーンに到達した。MyoKardiaはまた、MyoKardia協力協定により、任意の決定された目標製品に対する年間グローバル純売上高MyoKardiaとその任意の付属会社と、中間位数百分から下位2桁まで数百分の段階的使用料を支払う。特許使用料は、指定された特許使用料期間内に製品毎に支払われ、特定の場合には減少する可能性がある。
2022年と2021年12月31日までの年度では,MyoKardia協力協定によりそれぞれ530万ドルと960万ドルの協力収入を確認した。2022年12月31日と2021年12月31日までに、それぞれ90万ドルと410万ドルのMyoKardia協力協定に関連する繰延収入を記録し、収入予想で確認された期間に応じて、現在または非現在に分類され、私たちの総合貸借対照表から現在の部分を差し引く。MyoKardia協力協定によると、2022年12月31日現在、560万ドルのコスト返済と250万ドルのマイルストーン支払いを受けています。2022年12月31日現在、MyoKardia協力協定下の返済可能な研究開発コストに関する未開請求書20万ドルを記録し、2022年12月31日までの3ヶ月間の活動に使用しています。
将来的には、私たちの業績義務と、MyoKardia協力協定によるコストの精算のため、1000万ドルの前金と2021年12月に実現された250万ドルの臨床前マイルストーンに関する追加収入を確認します。将来的には次のような側面から追加収入を得ることも可能です
95
MyoKardia協力協定の下でマイルストーンと特許使用料が支払われる。私たちの収入は、MyoKardia協力協定における私たちの表現パターンと、MyoKardia協力協定によるマイルストーンとコスト精算の時間、金額、実現状況に基づいて、四半期と四半期と年度と年度の間で変動すると予想されます。
私たちはまた、将来的に私たちの候補製品や知的財産権のための追加の許可または協力協定を締結することができ、私たちは将来、そのような許可または協力協定によって生成された支払いから収入を得ることができるかもしれない。
運営費
研究と開発費
研究開発費とは、私たちの候補製品を発見、開発、製造するためのコストのことです
私たちは発生した費用に応じて研究と開発費用を支払う。我々は,患者登録やサプライヤーから提供された他の情報などのデータを用いて特定のタスク達成の進捗を評価し,臨床試験や製造などのいくつかの開発活動の費用を確認した。これらの活動の支払いは個別合意の条項に基づいており、これらの条項は発生した費用モデルとは異なる可能性がある。将来受けた研究·発展活動のための貨物又はサービスの前金は払い戻しできず、前払い費用と表記する。これらの金額は、貨物が関連サービスを提供するときに費用として確認されるか、または貨物がサービスを提供することが予期されなくなるまで確認される。
外部コストは我々の研究開発費の大きな部分を占めており,開発候補者を指名した後,1つずつ計画して追跡している.私たちの内部研究開発費は主に株式ベースの給与費用を含む人員関連の費用です。資源が複数のプロジェクトに配置されているので、私たちは計画通りに内部研究開発費を追跡しない。
下表は、我々が開発候補に指名された後、2022年12月31日と2021年12月31日までの年間で計画的に区分された外部研究開発費をまとめたものである。予備研候補費用、未分配費用、内部研究開発費は単独で分類される。Losmapimodの外部費用は,2022年12月31日までの年間で,2022年第2四半期にREACH試験を開始した後,GSKとの参考権やライセンス契約により実現した500万ドルのマイルストーンを含む。
|
|
現在までの年度 |
|
|||||
(単位:千) |
|
2022 |
|
|
2021 |
|
||
Losmapimod外部費用 |
|
$ |
26,260 |
|
|
$ |
19,128 |
|
FTX-6058外部費用 |
|
|
15,133 |
|
|
|
14,041 |
|
開発前候補者費用と未分配費用 |
|
|
14,964 |
|
|
|
16,100 |
|
内部研究開発費 |
|
|
20,425 |
|
|
|
20,432 |
|
研究開発費総額 |
|
$ |
76,782 |
|
|
$ |
69,701 |
|
96
私たちの候補製品が開発に成功するかどうかは大きな不確実性を持っている。現在、私たちの候補製品の余剰開発を完成させるために必要な仕事の性質、時間、見積もりコストを合理的に推定または知ることはできません。私たちの候補製品が承認されれば、いつ(あれば)大量の現金純流入が始まることも予測できない。これは,我々の候補製品の開発に関連する多くのリスクや不確実性が,以下の点に関連する不確実性を含むためである
我々の任意の候補製品の開発に関連するこれらの変数のいずれかの結果の変化は、候補製品の開発に関連するコストおよび時間を著しく変化させ、他の候補製品の開発に関連する時間およびコストを変更することが可能である。
97
研究開発活動は私たちの運営費用の大きな部分を占めている。我々の業務戦略の継続に伴い、将来的には、FSHDを治療するためのlosmapimodの推進、現在の臨床問題を解決できれば、いくつかのヘモグロビン疾患(SCDを含む)を治療するためのFTX-6058を推進し、より多くの人員を招いて私たちの研究開発努力を支援し、臨床試験を成功させた私たちの候補製品のための規制承認を含む研究開発努力を拡大することが予想される。また,臨床開発後期にある候補製品は通常,臨床開発早期段階の候補製品よりも高い開発コストが生じるのは,主に後期臨床試験の規模と持続時間が増加しているためである。したがって,我々の候補製品が臨床開発の後期段階に入るにつれて,我々の研究·開発費が増加することが予想される。しかし、私たちは現在、商業化によって特定の計画の総費用を正確に予測することは不可能だと思う。私たちの任意の候補製品の商業化成功に関連する要素はたくさんあり、未来の試験設計と各種法規要求を含み、その中の多くの要素は現在私たちの開発段階に基づいて正確に確定できない。
一般と行政費用
一般及び行政支出は人事に関連するコストを含み、行政、財務及び会計、人力資源、業務運営及びその他の行政職能者の賃金、福祉及び株式給与支出、特許、知的財産権及び会社事務に関する法律費用、会計及び税務サービス費用、相談費及び研究開発費に計上されていない施設関連費用を含む。
将来的には,我々の一般的かつ行政的費用が増加し,我々のインフラの拡大を支援し,我々の業務の拡大や上場企業としての運営コストの増加を予想している.これらの増加には、会計、監査、法律、規制、税務に関する費用の増加、取引所上場や米国証券取引委員会の要求を遵守することに関連する費用、取締役や高級職員保険料、上場会社の運営に関連する投資家関係コストが含まれる可能性がある。
再編成費用
2022年8月、我々は2つの臨床プロジェクトの優先順位を決定するために、内部投資と運営を再調整する戦略計画を実施することを発表した。この決定に関連して、私たちは25%のリストラを発表し、リストラ計画は2022年8月に完了した。再構成費用には解散費と他の従業員と関連した費用が含まれている。
その他の収入、純額
他の収入を除いて、純額には主に現金等価物と有価証券投資に関する利息収入が含まれる。
経営成果
2022年と2021年12月31日終了年度比較
以下は、2022年12月31日までと2021年12月31日までの経営実績概要と、これらの項目のドル変動である
|
|
現在までの年度 |
|
|
変わる |
|
||||||
(単位:千) |
|
2022 |
|
|
2021 |
|
|
$ |
|
|||
協力収入 |
|
$ |
6,342 |
|
|
$ |
19,163 |
|
|
$ |
(12,821 |
) |
運営費用: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
|
76,782 |
|
|
|
69,701 |
|
|
|
7,081 |
|
一般と行政 |
|
|
41,694 |
|
|
|
30,516 |
|
|
|
11,178 |
|
再編成費用 |
|
|
427 |
|
|
|
— |
|
|
|
427 |
|
総運営費 |
|
|
118,903 |
|
|
|
100,217 |
|
|
|
18,686 |
|
運営損失 |
|
|
(112,561 |
) |
|
|
(81,054 |
) |
|
|
(31,507 |
) |
その他の収入、純額 |
|
|
2,690 |
|
|
|
207 |
|
|
|
2,483 |
|
純損失 |
|
$ |
(109,871 |
) |
|
$ |
(80,847 |
) |
|
$ |
(29,024 |
) |
98
協力収入
協力収入は1280万ドル減少し、2021年12月31日現在の1,920万ドルから2022年12月31日現在の630万ドルに低下した。AcceleronとMyoKardia連携プロトコルでの各収入が,それぞれが決定した業績義務に関するパフォーマンスモデルに基づいていることを確認し,各プロトコルに基づいて研究サービスを提供する期限である.2022年と2021年12月31日までの年間で,Acceleron連携プロトコルによりそれぞれ100万ドルと960万ドルの連携収入を確認し,MyoKardia連携プロトコルによりそれぞれ530万ドルと960万ドルの連携収入を確認した.
研究と開発費
次の表は、2022年12月31日と2021年12月31日までの年間研究開発費をまとめています
|
|
現在までの年度 |
|
|
変わる |
|
||||||
(単位:千) |
|
2022 |
|
|
2021 |
|
|
$ |
|
|||
外部研究開発 |
|
$ |
44,944 |
|
|
$ |
36,934 |
|
|
$ |
8,010 |
|
従業員報酬 |
|
|
20,425 |
|
|
|
20,432 |
|
|
|
(7 |
) |
実験室用品 |
|
|
4,242 |
|
|
|
5,115 |
|
|
|
(873 |
) |
施設コスト |
|
|
5,593 |
|
|
|
5,552 |
|
|
|
41 |
|
他にも |
|
|
1,578 |
|
|
|
1,668 |
|
|
|
(90 |
) |
研究開発費総額 |
|
$ |
76,782 |
|
|
$ |
69,701 |
|
|
$ |
7,081 |
|
研究開発支出は710万ドル増加し、2021年12月31日現在の6970万ドルから2022年12月31日現在の7680万ドルに増加した。研究と開発費が増加した要因は以下のとおりである
一般と行政費用
次の表は、2022年12月31日と2021年12月31日までの年度の一般と行政費用をまとめたものである
|
|
現在までの年度 |
|
|
変わる |
|
||||||
(単位:千) |
|
2022 |
|
|
2021 |
|
|
$ |
|
|||
従業員報酬 |
|
$ |
23,488 |
|
|
$ |
14,801 |
|
|
$ |
8,687 |
|
専門サービス |
|
|
13,278 |
|
|
|
12,488 |
|
|
|
790 |
|
施設コスト |
|
|
2,206 |
|
|
|
960 |
|
|
|
1,246 |
|
他にも |
|
|
2,722 |
|
|
|
2,267 |
|
|
|
455 |
|
一般と行政費用総額 |
|
$ |
41,694 |
|
|
$ |
30,516 |
|
|
$ |
11,178 |
|
一般および行政支出は2021年12月31日現在の3,050万ドルから2022年12月31日までの4,170万ドルに増加し、1,120万ドルに増幅された。一般費用と行政費用が増加した要因は以下のとおりである
99
再編成費用
2022年12月31日までの1年間、再編費用は40万ドルだったが、2021年12月31日までの1年間は再編費用はゼロだった。2022年8月、我々は2つの臨床プロジェクトの優先順位を決定するために、内部投資と運営を再調整する戦略計画を実施することを発表した。この決定に関連して、私たちは25%のリストラを発表し、リストラ計画は2022年8月に完了した。
その他の収入、純額
その他の収入純額は250万ドル増加し、2021年12月31日までの年度の20万ドルから2022年12月31日までの年度の270万ドルに増加した。その他の純収入増加の主な原因は収益率の増加である。
流動性と資本資源
流動資金源
設立以来、我々は重大な運営損失が発生しており、予測可能な未来には引き続き重大な運営損失が発生し、永遠に利益を上げない可能性がある。私たちはまだ私たちの候補製品を商業化していません。これらの製品は臨床前と臨床開発の異なる段階にあります。もしあれば、数年以内にいかなる製品の販売からも収入が生じないと予想されます。2022年12月31日現在、私たちの運営資金は主に私たちの株式株式の売却と、私たちの協力と許可契約に基づいて受け取った前金によって得られた合計5.875億ドルの毛収入から来ています。2022年12月31日現在、私たちは2.029億ドルの現金、現金等価物、有価証券を持っている。
2022年8月、私たちは普通株の公開発行を完了し、1株7.82ドルの公開発行価格で11,029,410株の普通株を発行·販売し、引受割引と手数料および発行費用を差し引いた純収益は8,080万ドルだった。
2023年1月、私たちは普通株の公開発行を完了し、1株13.00ドルの公開発行価格で9,615,384株の普通株を発行·販売し、引受割引と手数料および発売費用を差し引いた純収益は1.173億ドルだった。
キャッシュフロー
次の表は、2022年12月31日と2021年12月31日までの年間キャッシュフロー情報を提供します
|
|
現在までの年度 |
|
|||||
(単位:千) |
|
2022 |
|
|
2021 |
|
||
経営活動のための現金純額 |
|
$ |
(97,050 |
) |
|
$ |
(78,478 |
) |
投資活動提供の現金純額 |
|
|
12,413 |
|
|
|
(129,669 |
) |
融資活動が提供する現金純額 |
|
|
84,323 |
|
|
|
186,507 |
|
現金、現金等価物、および限定的な現金純減少 |
|
$ |
(314 |
) |
|
$ |
(21,640 |
) |
経営活動に使われている現金純額
2022年12月31日までの年間では、経営活動用の現金純額は9710万ドルだったが、2021年12月31日までの年間では、経営活動に用いられた現金純額は7850万ドルだった。経営活動のための現金純額が1,860万ドル増加したのは,主に我々の先行計画の推進に伴い,外部研究開発コストの増加,従業員報酬コストの増加,および我々の組織成長を支援する一般的かつ行政コストの増加である。
投資活動による現金純額
2022年12月31日までの1年間で、投資活動が提供した純現金は1240万ドルだったが、2021年12月31日までの1年間で、投資活動に用いられた現金純額は1兆297億ドルだった。投資活動が提供する現金純額が1.42億ドル増加したのは、主に2022年12月31日までの1年間に証券を販売可能な純満期日であったが、2021年12月31日までの1年間に証券の純購入量が販売されたためである。
100
融資活動が提供する現金純額
2022年12月31日までの1年間で、融資活動が提供した現金純額は8430万ドルだったが、2021年12月31日までの1年間で、融資活動が提供した現金純額は1兆865億ドルだった。2022年12月31日までの1年間に、融資活動が提供する純現金には、2022年8月に我々の普通株が公開発売された純利益8080万ドルが主に含まれている。融資活動が提供する現金純額は、2021年12月31日までの1年間に、主に2021年1月と2021年8月に普通株式公開を完了して得られた1兆829億ドルの純収益を含む。
資金需要
我々が行っている研究開発活動に関する費用は大幅に増加することが予想され,特に我々が研究開発を継続し,臨床試験を開始し,候補製品のマーケティング承認を求めている場合である。しかも、私たちはまた私たちの組織の発展を支援するための追加的な費用を発生すると予想される。そのため、予測可能な未来には巨額の運営損失と負の運営キャッシュフローが予想される。
私たちの現在の運営計画によると、2022年12月31日までの既存の現金、現金等価物、有価証券に加え、2023年1月に公募株で売却された普通株の純収益に加え、2025年中までの運営費と資本支出需要に資金を提供できると信じている。しかし、私たちのこの推定は間違っていることが証明される可能性があるという仮定に基づいており、私たちは予想よりも早く私たちの資本資源を枯渇させるかもしれない。
私たちの資金需要と運営支出の時間と金額は大きく依存するだろう
我々の任意の候補製品の開発に関連するこれらまたは他の変数のいずれの結果も変化し、候補製品の開発に関連するコストおよび時間を著しく変化させることができる。私たちは私たちの業務目標を達成するために追加的な資金調達に依存し続ける必要があるだろう。
上記の変数以外に、もし私たちの任意の候補製品が開発に成功すれば、他の商業コスト以外に、私たちは監督管理記録、マーケティング許可、発売後の要求、私たちの知的財産権の維持と監督管理保護に関連する大量の追加コストを発生する。現在、私たちはこのような費用を合理的に見積もることができない。
101
これまで、相当な製品収入を生み出すことができれば、株式発行、債務融資、協力手配、戦略連合とマーケティング、流通、または許可手配の組み合わせで現金需要を満たす予定です。私たちは現在信用計画もなく、約束された資金源もない。もし私たちが将来株式証券を売却することでより多くの資本を調達すれば、私たちの株主の所有権権益は希釈され、これらの証券の条項は清算または他の優遇を含む可能性があり、私たちの既存の普通株主の権利に悪影響を及ぼす。もし私たちが債務証券を発行することでより多くの資金を調達すれば、これらの証券には私たちの運営を制限する契約が含まれるかもしれない。私たちは私たちが現在予想している金額よりも多くの追加資本が必要かもしれないが、追加資本は合理的な条項で得られないかもしれない、あるいは全くないかもしれない。もし私たちが未来に協力計画、戦略連合またはマーケティング、流通、または許可手配を通じてより多くの資金を調達すれば、私たちは私たちの技術、将来の収入源、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になる可能性のある条項で許可証を授与しなければならないかもしれない。もし私たちが必要な時に株式や債務融資を通じてより多くの資金を調達できない場合、私たちは開発または将来の商業化努力を延期、制限、減少または終了する必要があるかもしれないし、私たちが自分で開発し、マーケティングすることをより望んでいた候補製品の権利を与える必要があるかもしれない。
重要な会計政策と試算
私たちの経営陣の財務状況と経営結果の討論と分析は、アメリカで公認されている会計原則に基づいて作成された私たちの総合財務諸表に基づいています。これらの連結財務諸表を作成する際には、報告中に報告された資産および負債額、または有資産および負債の開示および報告期間内に報告された費用金額に影響を与える判断および推定を行う必要がある。我々の見積りは,我々の歴史的経験,既知の傾向,事件,および当時の状況では合理的であると考えられる様々な他の要因に基づいており,これらの要因の結果は,資産や負債の帳簿価値や他のソースからは見えにくい確認された費用金額を判断する基礎となっている.異なる仮定または条件では、実際の結果は、これらの推定値とは異なる可能性がある。私たちは持続的な基礎の上で私たちの推定と仮定を評価する。重大な改訂が予想される場合、その影響は推定変動の日から連結財務諸表に反映されることになる。
これらの政策は、経営陣の判断と見積もりの最も重要な分野に関連しているため、以下で議論する会計政策は、我々の歴史や将来の業績を知るために重要であると考えられる。我々の他の重要会計政策の説明については、本年度報告に他の箇所のForm 10−Kに含まれる連結財務諸表の付記2を参照されたい。
収入確認
財務会計基準委員会会計基準に基づいてASC,606を編纂したり取引先と契約した収入それは.私たちの顧客が約束された商品またはサービスの制御権を得ると、私たちはASC 606に従って収入を確認し、この金額は、これらの商品またはサービスと交換することが予想される対価格を反映している。
契約開始時に、契約がASC 606の範囲内にあると決定されると、契約で約束された商品またはサービスを評価し、それらが義務を履行していると判断する。そして私たちは取引価格を決定し、確定された履行義務に割り当てる。これらの手配の会計処理の一部として、マイルストーンまたは他の可変対価格を取引価格に計上すべきかどうかを決定することを含む、契約義務の数量および取引価格を決定するために重大な判断を使用しなければならない。
私たちは取引価格にマイルストーンまたは他の可変対価が含まれるべきかどうかを決定するために判断を使用する。経営陣の取引価格評価の一部として、マイルストーンの実現が私たちのコントロールを超えているかどうか、他の人の努力にかかっているか、成功リスクの影響を受けているかなど、多くの要因が考えられています。もし私たちが結論を出せば、重大な収入逆転が起こらない可能性が高い場合、関連するマイルストーン支払いは取引価格に含まれる。我々が制御できないイベント(例えば、規制承認のような)に基づいて発生するマイルストーン支払いは、通常、基礎イベントが発生したり、関連承認が受信されるまでは実現可能ではないと考えられる。報告期間が終了するごとに,吾らは取引価格に含まれる推定変動の価格および任意の関連制限を再評価し,必要に応じて全体の取引価格の推定を調整する.いずれの調整も調整期間内に累積追跡方式で記録されている.変動による価格制限の変化は当期に確認された収入額に実質的な影響を与える可能性がある。
契約が単一履行義務を含む場合、取引価格全体をその単一履行義務に割り当てる。複数の履行義務を含む契約要求は,相対的に独立した販売価格に基づいて取引価格を個々の履行義務に割り当てるが,すべて単一の履行義務または単一の履行義務の一部を構成する異なるサービスにすべて割り当てることに該当する任意の可変対価格は除外する.
102
義務履行時(または義務履行として)にそれぞれの義務を履行する取引価格に割り当てられた金額が収入であり,ある時点でも一定時間内であっても確認する.履行義務が時間の経過とともに履行されれば,産出や投入方法の使用確認収入に応じて収入を確認する。推定進捗状況は複雑であり、重大な判断に関連し、発生する内部人員費用及び外部費用総額を含む義務履行に要する総費用の推定値の影響を受ける。このような推定値の変化は私たちの収入確認に実質的な影響を及ぼすかもしれない。エンティティが一定期間義務を履行していない場合、ある時点で、約束された貨物またはサービスの制御権を顧客に移すことによって、関連する履行義務を履行する。
我々の収入確認政策のさらなる説明については、本年度報告Form 10-Kに含まれる他の部分に含まれる監査済み総合財務諸表の付記2“重要会計政策概要-収入確認”を参照されたい。ASC 606がAcceleronおよびMyoKardia連携協定に適用されるより多くの情報については、本年度報告書10-K表に含まれる他の部分に含まれる我々が監査した合併財務諸表の付記10、“協力および許可協定”を参照されたい。
研究と開発費用を計算すべきである
連結財務諸表作成過程の一部として、貸借対照表毎の日付までの計上費用を見積もる必要がある。このプロセスは、未締結契約および調達注文を審査し、私たちの担当者とコミュニケーションして、私たちに代わって実行されるサービスを決定し、インボイスを受け取っていない場合、または他の方法で実際のコストを通知している場合に、実行されたサービスレベルおよびサービスによって生じる関連コストを推定することを含む。私たちのほとんどのサービスプロバイダは毎月私たちが提供してくれるサービスや契約マイルストーンに達した時に私たちに借金の領収書を発行してくれます。私たちは、私たちが当時知っていた事実と状況に基づいて、各貸借対照表の日付までの課税費用を推定した。私たちは定期的にサービスプロバイダと私たちの推定の正確性を確認し、必要に応じて調整します。私たちの計算すべき研究開発費用の大きな見積もりには、私たちのサプライヤーが提供する研究開発活動に関連するサービスによるコストが含まれていますが、領収書は受け取っていません。
我々は、我々を代表して研究·開発を行っているサプライヤーとのオファーや契約に基づいて、受信したサービスや費用の見積もりに基づいて、研究·開発活動に関する費用を計算する。これらの合意の財務条項は交渉が必要であり、契約によって異なり、支払いの不均衡を招く可能性がある。場合によっては、私たちのサプライヤーに支払われるお金は、提供されたサービスレベルを超え、研究開発費の前払いにつながる可能性があります。サービス料を計算する際に、サービスを提供する時間帯と各時間の努力度を見積もります。サービス実行の実際の時間または努力の程度が私たちの見積もりと異なる場合、私たちはそれに応じて計算すべき費用または前払い費用を調整します。将来の研究·開発活動のための貨物·サービスの前金は払い戻しできず、活動が行われた場合や貨物を受け取った場合には、支払い時ではなく費用を計上する。
実際に発生した金額と実質的に差がないと予想されているにもかかわらず、提供されたサービスの状態および時間の推定が提供されたサービスの実際の状態および時間と異なる場合、これは、任意の特定の時期に報告された金額が高すぎたり、低すぎたりする可能性がある。これまで、このような費用の見積もりと実際に発生した金額との間に実質的な差はありませんでした。
株に基づく報酬
私たちは、付与日奨励の公正価値に基づいて、すべての制限的な株式奨励、制限株式単位、および株式オプションに関する株式ベースの報酬支出を測定する。私たちはこれらの奨励が必要なサービス期間内の補償費用であることを確認し、これは通常、相応の奨励の帰属期間である。一般に,サービスの付与条件に基づく報酬のみを発行し,直線的な方法を用いてこれらの報酬の費用を記録する.私たちはまた業績の付与条件に基づいたいくつかの株式奨励を与えた。経営陣が業績条件が実現可能であると判断した場合には、加速帰因法を用いて、残りのサービス期間内の業績帰属条件に基づく報酬の補償費用を確認する。報告日ごとに、パフォーマンス条件に対する期待満足度に基づいて、パフォーマンスベースのマイルストーンを実現することが可能かどうかを評価する。
103
私たちは付与日の私たちの普通株の推定公正価値から任意の適用可能な購入価格を引いて、制限株式奨励と制限株式単位の公正価値を決定します。私たちはBlack-Scholesオプション定価モデルを使用して、付与された株式オプションの公正価値を推定する。オプション定価モデルを用いて株式オプション付与日の公正価値を決定することは主に著者らの普通株の推定公正価値の影響を受け、そしてオプションの期待期限、対象株式の推定変動率、無リスク金利と期待配当を含む多くの他の仮定を管理層に要求する。付与日を決定する際に使用する仮想株式オプションの公正価値は、管理層の計量時の最適な推定を表す。私たちの普通株が初回公募株終了前に公開市場が不足していることと、会社に特定された歴史と隠れた変動率データが不足していることを考慮して、著者らは代表的な上場会社の歴史変動率(取得可能な履歴情報)に基づいて期待変動率を推定した。履歴変動率は,期待期限に用いた仮定に見合った一定時間で計算される.私たちは単純化された方法を使用してすべての株式オプションの期待期間を計算する。私たちがこの方法を使用したのは、予想期間を推定するための合理的な基礎を提供するために十分な履歴演習データがないからである。無リスク金利は、株式オプションの予想期限と一致する米国債ツールに基づいている。期待配当収益率はゼロと仮定しています。私たちは配当金を支払ったことがないので、今のところ普通株の配当金を支払う計画もありません。
将来的には、私たちの既存の未確認株式ベースの報酬支出と、従業員を引き付け、維持し続けるために、株式ベースの報酬支出が増加することが予想される。
所得税
連結財務諸表または我々の納税申告書で確認されたイベントの将来の税務結果の繰延税金資産および負債を確認することを要求する貸借対照法を用いて所得税を会計処理する。この方法によると、繰延税項資産及び負債は、財務諸表の帳簿額面と資産及び負債の課税基礎との差額に基づいて、予想差額を用いて返送される年度の現行税率を決定する。既存の証拠の重みに基づいて、繰延税金資産の一部または全部が現金化できない可能性が高い場合は、繰延税金資産の推定準備に計上しなければならない。繰延税金資産の回収の潜在力を評価する時に多くの要素を考慮し、未来に予想される課税オーバー額の推定、現有の課税の一時的な差額の推定、将来の引越期間の課税オーバー額を考慮すること、及び慎重かつ実行可能な税務計画策略を考慮することを含む。
不確定な税収状況を確認して解決するために、より可能な敷居を使用して、不確定な税収状況を考慮する。不確定税務状況の評価は、法律の変化、納税申告書において採用されたまたは予想される税収状況の測定、監査すべき事項の効果的な解決、新しい監査活動、および税収状況に関連する事実または状況の変化を含むが、これらに限定されない様々な要因に基づく。すべての貸借対照表の日付まで、私たちは何の不確定な税務頭証もありません。
最近発表された会計公告
最近発表された我々の財務状況及び経営結果に影響を与える可能性のある会計声明の記述は、本年度報告書10−K表の他の場所の監査された総合財務諸表の付記2に開示される。
新興成長型会社の地位
2012年のJumpStart Our Business Startups Actは、これらの基準が非上場企業に適用されるまで、私たちのような“新興成長型企業”が、延長された移行期間を利用して、上場企業に適用される新しいまたは改訂された会計基準を遵守することを可能にした。私たちは、“脱退”延長を選択しない移行期間を選択したが、これは、基準が発表または改正された場合、上場企業または民間企業に対して異なる適用日があれば、民間会社が新たな基準または改正基準を採用する際に新たな基準または改訂基準を採用し、我々(I)が“選択脱退”延長の移行期間を取り消すことができなくなるまで、または(Ii)新興成長型企業の資格に適合しなくなるまでそうすることを意味する。
第七A項。定量と合格市場リスクの開示について。
私たちの市場リスクに対する主な開口は金利感受性であり、金利敏感性はアメリカ金利の全体的なレベルの変化の影響を受けており、特に私たちの現金等価物は通貨市場基金の形でアメリカ国債に投資されているため、私たちの投資は短期有価証券、例えば社債や商業手形のようなものです。2022年12月31日現在、私たちは2.029億ドルの現金、現金等価物、有価証券を持っている。利子収入
104
私たちは一般金利水準の変動に非常に敏感だが、これらの投資の性質のため、金利が直ちに10%変動することは、私たちのポートフォリオの公平な市場価値に実質的な影響を与えない。
私たちはまた外貨為替レートの変化と関連した市場リスクに直面している。私たちはアメリカ以外のサプライヤーと契約を結びます。ある領収書は外貨建てです。私たちはこれらの手配に関連した外貨為替レートの変動の影響を受けるだろう。私たちは現在私たちの外貨為替レートに対するリスクを持っていない。2022年12月31日現在、私たちの外貨建ての債務は微々たるもので、ありません。
インフレは一般的に労働コストと臨床試験費用を増加させることで私たちに影響を及ぼす。2022年12月31日と2021年12月31日までの年間では、インフレは我々の業務、財務状況や運営結果に実質的な影響を与えないと考えられる。
プロジェクト8.財務諸表Sと補足データ。
我々の総合財務諸表および独立公認会計士事務所の報告は,本年度報告のF−1ページからForm 10−Kフォーマットを採用した。
項目9.Accouとの変更と分岐会計と財務情報開示の専門。
ない。
第9条。制御するプログラムがあります
情報開示制御とプログラムの評価
私たちの経営陣は、臨時最高経営責任者と最高財務官(それぞれ私たちの最高経営責任者とCEO)の参加の下で、2022年12月31日までの開示統制と手続きの有効性を評価しました。1934年に改正された“証券取引法”又は“取引法”の下の第13 a−15(E)及び15 d−15(E)条の規則で定義された“開示制御及び手続”という言葉は、取引法に基づいて提出又は提出された報告において開示を要求する企業の情報が米国証券取引委員会規則及び表で指定された期間内に記録、処理、集計及び報告されることを確実にするための会社の制御及びその他の手続を意味する。開示制御及び手続は、会社が“取引法”に基づいて提出又は提出された報告書において開示を要求する情報が蓄積され、その主要幹部及び主要財務官を含む会社経営者に伝達されることを保証することに限定されるものではないが、必要な開示に関する決定をタイムリーに行うために、同様の機能を果たす者の制御及び手続を適宜行う。私たちの経営陣は、どんな制御やプログラムも、どんなに設計や操作が良くても、その目標を達成するために合理的な保証を提供するしかないことを認識しており、私たちの経営陣は、可能な制御とプログラムの費用対効果関係を評価する際にその判断を適用しなければならない。2022年12月31日までの開示統制及び手続の評価によると、我々の臨時最高経営責任者及び最高経営責任者は、その日までに、我々の開示制御及び手続が合理的な保証レベルで有効であると結論した。
経営陣財務報告内部統制年次報告書
私たちの経営陣は会社の財務報告書の十分な内部統制の確立と維持に責任があります。財務報告の内部統制は、取引法第13 a-15(F)及び15 d-15(F)条において、会社の主要執行者及び主要財務官又は同様の機能を果たす者が設計又はその監督の下で行われ、公認会計原則に基づいて財務報告の信頼性及び外部目的の財務諸表を作成するために合理的な保証を提供するために、会社の取締役会、管理層及びその他の人員によって実施されるプログラムとして定義されている
105
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。したがって,有効と判定されたシステムであっても,財務諸表の作成や列報に合理的な保証を提供することしかできない.将来的にどのような有効性評価を行うかの予測には,条件の変化により制御が不十分になる可能性がある,あるいは政策やプログラムを遵守する程度が悪化する可能性があるというリスクがある.
最高経営責任者と最高財務責任者の監督·参加の下、我々の経営陣は、テレデビル委員会(COSO)後援組織委員会が内部統制-総合枠組み(2013枠組み)で提案した基準に基づいて、2022年12月31日までの財務報告書の内部統制に対する有効性を評価した。この評価に基づき、経営陣は、財務報告書に対する内部統制は2022年12月31日から有効であると結論した。
“雇用法案”による“新興成長型会社”の免除により、本10-K表年次報告には、我々の独立公認会計士事務所の認証報告は含まれていない。
財務報告の内部統制の変化
2022年12月31日までの3ヶ月間、財務報告の内部統制(“外国為替法案”規則13 a-15(F)および15 d-15(F)の定義に基づく)に大きな影響を与えなかったか、または合理的に財務報告の内部統制に大きな影響を与える可能性の高い変化が発生しなかった。
プロジェクト9 B。他にも情報です。
ない。
プロジェクト9 Cです。検査を妨害する外国司法管轄区域を開示する。
適用されません。
106
第三部
プロジェクト10.役員·役員職ICERSと会社管理。
以下に提供される範囲を除いて、本第10項で要求される情報は、2023年年次総会に関する米国証券取引委員会に提出された最終委託書に含まれ、参照により本明細書に組み込まれる。
私たちは、当社サイトwww.fulcrumtex.comの“投資家関係”部分(ir.fulcrumtex.com)の“コーポレート·ガバナンス”部分の“コーポレート·ガバナンス”部分に、私たちのCEO、最高経営責任者、最高財務責任者、首席会計官または主計長、または同様の機能を実行する人を含む、私たちの役員、高級管理者、従業員に適したビジネス行動と道徳基準を掲示しています。私たちは、表格8-K第5.05項の開示要件に基づいて開示される必要がある“商業行為および道徳基準”の任意の改正または免除を、私たちのウェブサイトで開示する予定です。
プロジェクト11.行政官イー補償します。
第11項に要求される情報は、2023年年次総会に関する我々が米国証券取引委員会に提出した最終委託書に含まれ、参照により本明細書に組み込まれる。
プロジェクト12.特定の受益者の保証所有権従業員と経営陣と関連した株主問題。
第12項に要求される情報は、2023年年次総会に関する我々が米国証券取引委員会に提出した最終委託書に含まれ、参照により本明細書に組み込まれる。
第13条に要求される情報は、2023年年次総会に関する我々が米国証券取引委員会に提出した最終委託書に含まれ、引用により本明細書に組み込まれる。
第14項.元金口座弁護士代とサービス料です。
第14条要求された情報は、2023年年次総会に関する我々が米国証券取引委員会に提出した最終委託書に含まれ、引用により本明細書に組み込まれる。
107
第4部
プロジェクト15.ExhITSと財務諸表明細書。
(1)連結財務諸表
以下の文書は、本文書に添付されているF−1~F−31ページに含まれ、本年度報告の10−Kテーブルの一部として提出される。
|
ページ |
独立公認会計士事務所報告 (PCAOB ID:42) |
F-2 |
合併貸借対照表 |
F-3 |
合併経営報告書と全面赤字 |
F-4 |
株主権益合併報告書 |
F-5 |
統合現金フロー表 |
F-6 |
連結財務諸表付記 |
F-7 |
(2)財務諸表添付表
これらは、適用されない、必要ではない、または要求される情報が連結財務諸表またはその付記に表示されるので、すべての財務諸表の添付表は省略される。
(3)展示品
S-K条例第601項および本年度報告テーブル10-K第15(B)項で要求される証拠物は、本年度報告テーブル10-K署名ページ前の添付ファイルインデックスに記載される。展示品索引に記載されている展示品は、引用されて本明細書に組み込まれる。
項目16.表10-Kの概要
ない。
108
連結財務諸表索引
|
ページ |
独立公認会計士事務所報告(PCAOB ID: |
F-2 |
合併貸借対照表 |
F-3 |
合併経営報告書と全面赤字 |
F-4 |
株主権益合併報告書 |
F-5 |
統合現金フロー表 |
F-6 |
連結財務諸表付記 |
F-7 |
F-1
“独立公認会計士報告書”アイレード会計士事務所
会社の株主と取締役会を支点に治療する。
財務諸表のいくつかの見方
Fulcrum治療会社(当社)の2022年12月31日現在と2021年12月31日までの連結貸借対照表、同年度までの関連総合経営報告書と全面赤字、株主権益と現金流量および関連付記(総称して“総合財務諸表”と呼ぶ)を監査した。総合財務諸表は、すべての重要な面で、2022年12月31日と2021年12月31日までの会社の財務状況と、2022年12月31日と2021年までの経営結果と現金流量を公平に反映しており、米国公認会計原則に適合していると考えられる。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査することを求められておらず、私たちも招聘されて監査を行っていません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性に対する意見を表明するためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
/s/
2017年以来、当社の監査役を務めてきました。
March 9, 2023
F-2
支点治療会社
合併B割当書
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
有価証券 |
|
|
|
|
|
|
||
売掛金 |
|
|
|
|
|
|
||
未開売掛金 |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
流動資産総額 |
|
|
|
|
|
|
||
財産と設備、純額 |
|
|
|
|
|
|
||
経営的リース使用権資産 |
|
|
|
|
|
|
||
制限現金 |
|
|
|
|
|
|
||
その他の資産 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
負債と株主権益 |
|
|
|
|
|
|
||
流動負債: |
|
|
|
|
|
|
||
売掛金 |
|
$ |
|
|
$ |
|
||
費用とその他の流動負債を計算しなければならない |
|
|
|
|
|
|
||
繰延収入,当期分 |
|
|
|
|
|
|
||
賃貸負債を経営し、流動 |
|
|
|
|
|
|
||
延期レンタル奨励、今期の部分 |
|
|
|
|
|
|
||
流動負債総額 |
|
|
|
|
|
|
||
当期分は含まれていない賃貸負債を経営しております |
|
|
|
|
|
|
||
他の負債は流動部分は含まれていません |
|
|
|
|
|
|
||
繰延賃貸料は現在の部分は含まれていません |
|
|
|
|
|
|
||
現在の部分は含まれていないレンタル延期インセンティブ |
|
|
|
|
|
|
||
総負債 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
株主権益: |
|
|
|
|
|
|
||
優先株、$ |
|
|
|
|
|
|
||
普通株、$ |
|
|
|
|
|
|
||
在庫株は、コストで計算する |
|
|
|
|
|
|
||
追加実収資本 |
|
|
|
|
|
|
||
その他の総合損失を累計する |
|
|
( |
) |
|
|
( |
) |
赤字を累計する |
|
|
( |
) |
|
|
( |
) |
株主権益総額 |
|
|
|
|
|
|
||
総負債と株主権益 |
|
$ |
|
|
$ |
|
||
付記はこれらの財務諸表の不可分の一部だ。
F-3
支点治療会社
業務所合併報告書損失と全面的損失
(単位は千、1株当たりのデータは除く)
|
|
現在までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
協力収入 |
|
$ |
|
|
$ |
|
||
運営費用: |
|
|
|
|
|
|
||
研究開発 |
|
|
|
|
|
|
||
一般と行政 |
|
|
|
|
|
|
||
再編成費用 |
|
|
|
|
|
|
||
総運営費 |
|
|
|
|
|
|
||
運営損失 |
|
|
( |
) |
|
|
( |
) |
その他の収入、純額 |
|
|
|
|
|
|
||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
1株当たり基本と希釈して純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
加重平均発行済み普通株式、基本普通株式、希釈後普通株 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
総合的な損失: |
|
|
|
|
|
|
||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
その他の全面的な損失: |
|
|
|
|
|
|
||
有価証券は赤字を実現していない |
|
|
( |
) |
|
|
( |
) |
その他総合損失合計 |
|
|
( |
) |
|
|
( |
) |
総合損失 |
|
$ |
( |
) |
|
$ |
( |
) |
付記はこれらの財務諸表の不可分の一部だ。
F-4
支点治療会社
合併報告書株主権益
(単位は千で、シェアは含まれていない)
F-5
|
|
普通株 |
|
|
在庫株 |
|
|
その他の内容 |
|
|
積算 |
|
|
積算 |
|
|
合計する |
|
||||||||||||||
|
|
株 |
|
|
金額 |
|
|
株 |
|
|
金額 |
|
|
資本 |
|
|
損 |
|
|
赤字.赤字 |
|
|
権益 |
|
||||||||
2020年12月31日残高 |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
( |
) |
|
|
( |
) |
|
|
|
||||
公開発行に関する普通株発行は、発行コストを差し引く |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
従業員福祉計画に基づいて普通株を発行する |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
制限株奨励の帰属 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
未帰属限定株の買い戻し奨励 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
在庫株の廃棄 |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
有価証券は赤字を実現していない |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2021年12月31日の残高 |
|
|
|
|
$ |
|
|
|
— |
|
|
$ |
— |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
公開発行に関する普通株発行は、発行コストを差し引く |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
従業員福祉計画に基づいて普通株を発行する |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
制限株奨励の帰属 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
有価証券は赤字を実現していない |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2022年12月31日の残高 |
|
|
|
|
$ |
|
|
|
— |
|
|
$ |
— |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
付記はこれらの財務諸表の不可分の一部だ。
F-6
支点治療会社
合併状態現金流動額
(単位:千)
|
|
現在までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
経営活動 |
|
|
|
|
|
|
||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
純損失と経営活動で使用される現金純額の調整: |
|
|
|
|
|
|
||
減価償却費用 |
|
|
|
|
|
|
||
株に基づく報酬費用 |
|
|
|
|
|
|
||
有価証券の割増純償却 |
|
|
|
|
|
|
||
経営性資産と負債変動状況: |
|
|
|
|
|
|
||
売掛金 |
|
|
|
|
|
( |
) |
|
未開売掛金 |
|
|
|
|
|
( |
) |
|
前払い費用と他の流動資産 |
|
|
( |
) |
|
|
( |
) |
リース資産と負債を経営する |
|
|
( |
) |
|
|
( |
) |
その他の資産 |
|
|
( |
) |
|
|
|
|
売掛金 |
|
|
( |
) |
|
|
|
|
費用とその他の負債を計算すべきである |
|
|
|
|
|
|
||
収入を繰り越す |
|
|
( |
) |
|
|
( |
) |
経営活動のための現金純額 |
|
$ |
( |
) |
|
$ |
( |
) |
投資活動 |
|
|
|
|
|
|
||
有価証券を購入する |
|
|
( |
) |
|
|
( |
) |
有価証券の満期日 |
|
|
|
|
|
|
||
財産と設備を購入する |
|
|
( |
) |
|
|
( |
) |
投資活動提供の現金純額 |
|
|
|
|
|
( |
) |
|
融資活動 |
|
|
|
|
|
|
||
公開発行に関する普通株発行による収益は,発行コストを差し引く |
|
|
|
|
|
|
||
資本賃貸債務の元金支払い |
|
|
|
|
|
( |
) |
|
福祉計画の下で普通株を発行して得た金,純額 |
|
|
|
|
|
|
||
融資活動が提供する現金純額 |
|
|
|
|
|
|
||
現金、現金等価物、および限定的な現金純減少 |
|
|
( |
) |
|
|
( |
) |
期初現金、現金等価物、および限定現金 |
|
|
|
|
|
|
||
現金、現金等価物、制限された現金、期末 |
|
$ |
|
|
$ |
|
||
キャッシュフロー情報を補充する |
|
|
|
|
|
|
||
賃貸負債経営のための現金 |
|
$ |
|
|
$ |
|
||
非現金投資と融資活動: |
|
|
|
|
|
|
||
期末未払いの財産と設備購入 |
|
$ |
|
|
$ |
|
||
以下の表は、上記各期間の現金、現金等価物、および制限された現金残高の入金状況を示す
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
制限現金 |
|
|
|
|
|
|
||
現金総額、現金等価物、制限された現金 |
|
$ |
|
|
$ |
|
||
付記はこれらの財務諸表の不可分の一部だ。
F-6
支点治療会社
総合備考財務諸表
1.ビジネスの性質とレポートの根拠
Fulcrum治療会社(“会社”または“Fulcrum”)は2015年8月18日にデラウェア州に登録設立された。同社は高度に満たされていない医療需要地域の遺伝性まれな疾患患者の生活改善に注力している。
同社は、臨床前研究と臨床試験失敗のリスク、キーパーソンへの依存、ノウハウの保護、第三者組織への依存、開発可能な任意の候補製品の規制承認を得るリスク、競争相手の技術革新の開発、政府法規の遵守及び追加融資の需要を含む、生物技術業界の他の会社と類似した多くのリスクに直面している。現在開発されている候補製品は、広範な臨床前と臨床試験、商業化前に監督部門の許可を得ることを含む、大量の追加的な研究と開発作業が必要となる。このような努力は多くの追加資本、十分な人員インフラ、そして広範囲なコンプライアンス報告能力を必要とする。同社の開発努力が成功しても、同社がいつ(あれば)製品販売から相当な収入を得るかは定かではない。
陳述の基礎
添付されている総合財務諸表は、米国公認会計原則(“公認会計原則”)に従って作成されている。本付記内の適用指針に対するいかなる言及も財務会計基準委員会(“FASB”)の会計基準編纂(“ASC”)及び会計基準更新(“ASU”)に掲載されている権威公認会計原則を指す。
普通株販売
2021年1月22日,会社は普通株の公開と販売を完了した
2021年8月16日,会社は普通株の公開と販売を完了した
2022年8月,会社は普通株の公開と販売を完了した
2023年1月、会社は普通株の公開と販売を完了した
流動性
同社は設立以来、運営中に経常赤字と負のキャッシュフローが発生しており、その運営資金は主にその株式を売却する収益と、メルク社(“メルク”)や百時美施貴宝社(Bristol Myers Squibb Company)の完全子会社Acceleron Pharma Inc.(“Acceleron”)とMyoKardia,Inc.(“MyoKardia”)との協力と許可契約で受け取った前払いによるものである。2022年12月31日現在の会社の累計損失は$
F-7
同社は、その現金、現金等価物、および有価証券は、これらの財務諸表の発行日から少なくとも12ヶ月の運営費用および資本支出需要を支払うのに十分であると予想している。しかし,同社のこの推定は,誤りであることが証明される可能性があるという仮定に基づいており,その運営計画は現在未知の多くの要因によって変化する可能性がある。したがって、その会社は現在予想されているよりも早くその資本資源を枯渇させるかもしれない。会社が必要なときに株式または債務融資によってより多くの資金を調達できない場合、開発または将来の商業化努力を延期、制限、減少または終了するか、または本来より開発およびマーケティングを望む候補製品を開発およびマーケティングする権利を付与する必要がある可能性がある。
2.主な会計政策の概要
合併原則
予算の使用
現金と現金等価物
現金等価物は高流動性の投資であり、購入時に元の満期日が3ヶ月以下の現金に変換しやすい。現金等価物には、米国債に投資する通貨市場基金と商業手形が含まれる。その会社は主要金融機関でその銀行口座を維持している。
制限現金
金融商品の公正価値
当社の金融資産及び負債の公正価値は、計量日に市場参加者間の秩序ある取引において当該等の資産を売却することにより当社が当該等の負債を譲渡することにより支払われた金額の推定を反映している。その資産と負債の公正な価値を計測する際には、会社は観察可能な投入(会社以外の独立したソースから得られた市場データ)を最大限に使用し、観察できない投入を最大限に減らすことを求めている(会社が市場参加者がどのように資産や負債の価格を設定しているかの仮定)。以下の公正価値レベルは、資産および負債を推定するために、観察可能な投入および観察できない投入に基づいて資産および負債を分類するために使用される
第1レベル:同じ資産または負債の活発な市場でのオファー。資産または負債の活発な市場とは、資産または負債の取引が発生する頻度および数が定価情報を継続的に提供するのに十分な市場を意味する。
レベル2:レベル1入力以外の観察可能な入力.第2レベル投入の例は、アクティブ市場における同様の資産または負債の見積もりと、非アクティブ市場における同じ資産または負債の見積もりとを含む。
レベル3:資産または負債の価格設定で市場参加者が使用するという会社の仮定に基づく評価では、観察できない入力。
♪the the the会社の現金等価物と有価証券は公正価値に記載されており、上記公正価値等級別に分類されている(付記3)。現金等価物と有価証券の初期推定値は
F-8
取引記録各報告期間が終了した時点で、第三者価格設定サービスを利用して再評価する。定価サービスは業界標準推定モデルを利用して、収入と市場に基づく方法を含めて公正価値を決定する。
有価証券
同社は3カ月以上の残存期限を超える証券を有価証券に分類している。同社の有価証券には、2022年12月31日現在、米国債、政府機関証券、社債、商業手形への投資が含まれている。有価証券が現金に変換されて現在の業務に資金を提供することができる場合、有価証券は、合併貸借対照表において流動資産に分類される。
有価証券は公正価値に基づいて入金され、損益が株主権益構成部分の累積他の総合損失に計上されておらず、この損益が実現するまで実現していない。購入時に発生する任意の割増は、最も早い償還日の間に利子支出(他の収入の一部、純額)に償却され、購入時に発生した任意の割引は、手形の有効期間内の利息収入(他の収入の一部、純額)に計上される。損益は特定の確認方法で決定され,他の収入,純額に計上されている。
公正価値の任意の調整が投資価値の低下を反映していれば、当社はこの低下の程度をすべての利用可能な証拠を考慮して評価し、そうであれば、会社の経営報告書と全面損失に費用を計上することで投資を市価に計上する。
財産と設備
財産と設備は減価償却累計を差し引いたコストで入金されます。資産の維持·メンテナンスがその使用寿命を改善または延長できない場合は、運営費用を計上する
|
使用寿命(年)を見込む |
実験室装置 |
|
家具と固定装置 |
|
コンピュータ装置 |
|
ソフトウェア |
|
賃借権改善 |
長期資産減価準備
賃貸借証書
2022年1月1日から、当社はASU 2016-02号を通過したレンタル(テーマ842)(“アリゾナ州立大学2016-02”)。発効日又は後に締結された契約については、契約開始時に、当社は、手配に存在する独自の事実及び状況に基づいて、手配がリース契約であるか否かを決定する。期限付き借約
F-9
12ヶ月を超える資産は、貸借対照表上で使用権資産および流動および非流動賃貸負債であることが確認された(場合によっては)。当社は貸借対照表上の12ヶ月以下のレンタルを確認していません。当社が賃貸契約を更新することを合理的に把握していない限り、更新賃貸契約の選択は当社の最初のレンタル期間評価には含まれていません。
レンタルは融資リースか経営的賃貸に分けられます。リースは、(I)レンタル期間の終了時に資産所有権を譲渡すること、(Ii)レンタルは、行使されるべき購入資産の選択権を合理的に決定すること、(Iii)賃貸期間が資産残使用年数の主要部分であるか、または(Iv)賃貸支払いの現在値が資産の全公正価値に等しいか、またはそれを超える場合のいずれかの基準に適合するように分類される。レンタルが上記のいずれの基準を満たしていない場合、経営的賃貸に分類される。
すべての経営リースについて、レンタル負債とそれに応じた使用権資産を確認します。賃貸負債とは、残りのレンタル期間内にレンタル支払いの現在値を予想し、レンタル暗黙的な金利を用いて割引したり、その金利が容易に確定できない場合には、当社の逓増借款金利を割引し、この金利は、当社が類似した経済環境下で担保ベースで同じ通貨を借り入れた賃貸支払い金額の固定金利を反映している。当社の増量借入金利を推定するために、当社は格付け機関による信用格付けを行っていないため、総合信用格付けと収益率曲線分析を用いて当社に適した信用格付けを推定しています。使用権資産とは、リース期間内のリース資産の使用権のことです。使用権資産は、最初にコストで計量され、主にレンタル負債の初期金額を含み、生成された任意の初期直接コスト(例えば、ある)を加えて、受信された任意のレンタル報酬を減算する。
賃貸負債計量に計上された賃貸支払いは、(I)固定不能賃貸支払いと、(Ii)継続期間が行使される選択可能な継続期間の支払いを合理的に決定するステップと、(Iii)賃貸契約が早期に終了しないと適切に決定されない限り、オプションの支払いを早期終了するステップと、を含む。経営的レンタルのレンタル料金には、レンタル支払いに任意の初期直接コストが含まれており、レンタル期間内に直線原則で確認されます。レンタル料金には、初期リース負債が計上されていない期間内に発生した任意の可変リース支払いと、初期期限が12ヶ月以下の任意のリース期間に発生したリース支払いとが計上される。当社はリースと非リース構成要素を単一リース構成要素として会計処理を行っている。
収入確認
ASC 606では取引先と契約した収入エンティティは、その顧客が約束された貨物またはサービスの制御権を取得したときに収入を確認し、その額は、エンティティがこれらの貨物またはサービスの交換から得られると予想される対価格を反映する。ASC 606を適用すると、会社は以下の5つのステップを実行する
1)お客様との契約の決定
(I)会社が顧客と強制的に実行可能な契約を締結した場合、当該契約は、譲渡する商品またはサービスに対する各当事者の権利を規定し、関連する支払い条項を決定し、(Ii)契約が商業的実質を有し、(Iii)会社が顧客の意図および支払い承諾の対価能力に基づいて、譲渡された商品およびサービスのほぼすべての対価格を徴収する可能性があると決定した場合、顧客との契約が存在する。
2)契約における約束と義務の決定
契約において約束された履行義務は、顧客に譲渡される貨物およびサービスによって決定され、これらの貨物およびサービスは、顧客が単独でまたは他のいつでも利用可能なリソースと共に貨物またはサービスから利益を得ることができ、契約の文脈で異なることができるので、貨物またはサービスの譲渡は、契約内の他の約束とは別に識別することができる。1つの契約に複数の承諾された商品及びサービスが含まれている場合、会社は、約束された商品及びサービスが契約の背景で区別できるか否かを決定するために判断を適用しなければならない。承諾した商品やサービスがユニークであるかどうかを評価する際には,会社は顧客の研究,製造と商業化能力,市場に関する専門知識の可用性などを考慮する.約束された貨物またはサービスが契約中の他の約束とは別に識別できるかどうかを評価する際に、会社はまた、契約の予想利益を考慮する。これらの基準を満たしていなければ、約束された貨物とサービスは総合履行義務として入金されるだろう。
F-10
3)出来高決定
取引価格は、商品やサービスを顧客に移転することと引き換えに、会社が獲得する権利のある対価格によって決定される。契約で約束された対価格に可変金額が含まれている場合、会社は、約束された貨物またはサービスを顧客に移転するために、獲得する権利のある対価格金額を推定する。当社は期待値法または最大可能値法を用いて可変対価金額を決定します。同社は制限されない推定可変対価金額を取引価格に計上している。取引価格に含まれる金額は,確認された累積収入が大きく逆転しない可能性の高い金額に制限される.その後報告期間ごとに終了すると,当社は取引価格に含まれる推定変動コストおよび任意の関連制限を再評価し,必要に応じて全体の取引価格の見積りを調整する.いずれの調整も調整期間内に累積追跡方式で記録されている.変動による価格制限の変化は当期に確認された収入額に実質的な影響を与える可能性がある。
計画に研究開発マイルストーン支払いが含まれていれば、当社はマイルストーンが達成可能であると考えられているかどうかを評価し、最も可能な金額法を用いて取引価格に含まれる金額を推定する。大きな収入逆転が起こらない可能性が高い場合、関連するマイルストーン価値は取引価格に含まれるだろう。規制承認など、企業がコントロールできない事件発生に基づくマイルストーン支払いは、関連イベントが発生したり、関連承認が受領されるまでは実現不可能であると考えられるのが一般的である。
知的財産権ライセンス取り決めについては、販売水準に基づくマイルストーン支払いを含む販売ベースの使用料を含み、ライセンスが使用料に関連する主要項目とみなされ、当社は、(I)関連販売が発生した場合、又は(Ii)分配された使用料の履行義務が履行された場合に、使用料収入及び販売に基づくマイルストーンを確認する。
取引価格を決定する際に、支払時間が当社に重大な融資利益を提供していれば、当社は貨幣時間価値の影響を対価格調整する。同社はその創設計画を評価し、重要な融資構成要素が存在するかどうかを決定する。
4)取引価格を契約に割り当てる履行義務
契約が単一履行義務を含む場合、取引価格全体をその単一履行義務に割り当てる。複数の履行義務を含む契約要求は,相対的に独立した販売価格に基づいて取引価格を個々の履行義務に割り当てるが,すべて単一の履行義務または単一の履行義務の一部を構成する異なるサービスにすべて割り当てることに該当する任意の可変対価格は除外する.
5)会社が契約履行義務を履行した場合または義務履行時に収入を確認する
契約履行義務の性質により、会社は時間の経過やある時点で履行義務を履行する可能性がある。顧客がエンティティ業績によって提供される利益を同時に受け取り、消費する場合、エンティティ業績が資産を作成または強化する際に顧客制御資産を作成または強化する場合、またはエンティティ業績が代替エンティティ用途に代替可能な資産を作成しておらず、エンティティがこれまでに完了した実績を強制的に支払う権利を有する場合、収入は時間の経過とともに確認される。エンティティが一定期間義務を履行していない場合、ある時点で、約束された貨物またはサービスの制御権を顧客に移すことによって、関連する履行義務を履行する。
企業が一定期間内に確認した収入について、企業は、投入または産出方法が業績義務の履行状況に関する進展の適切な測定基準であるかどうかを評価する。進捗を測る適切な方法を決定する際には,会社は顧客に譲渡することを承諾した商品やサービスの性質を考慮する.産出法は,これまで顧客に譲渡されてきた貨物またはサービスが契約に対して承諾した残りの貨物またはサービスの価値の直接計量に基づいて収入を確認する。投入方法は,エンティティが履行義務を履行するための努力や投入確認収入に基づいている.投入方法に基づいて履行義務の履行に関する進捗状況に固有の推定数を測定することは、関連履行義務を履行する推定総費用を含む。
ASC 606がAcceleronとの連携およびライセンスプロトコル(“Acceleron連携プロトコル”)およびMyoKardiaとの連携およびライセンスプロトコル(“MyoKardia連携プロトコル”)に適用されることについては、付記10“連携およびライセンスプロトコル”を参照されたい。
F-11
研究と開発費
契約コストと計上項目を検討する
特許関連費用
株に基づく報酬
当社は付与日の公正価値をもとに株式奨励を計量します。これらの賠償金に関する補償費用は必要なサービス期間内に確認され、サービス期間は通常各賠償金の授権期間である。一般に、会社が発行する報酬は、サービスに基づく帰属条件のみを含み、直線法を用いてこれらの報酬の費用を記録する。
各制限株式奨励の公正価値は、日会社普通株に付与された公正価値から任意の適用可能な買収価格を減算することに基づく。各株式オプションの公正価値は、付与日にブラック·スコアーズオプション定価モデルを使用して推定され、このモデルは、予想される株価変動、予想される付与期限、無リスク金利、および予期される配当を含むいくつかの主観的仮定に基づく投入を必要とする。予想変動率は代表的な上場企業の報告変動率データから計算され、これらの会社は歴史情報を持っている。履歴変動率は,期待期限に用いた仮定に見合った一定時間で計算される.無リスク金利は、付与時に有効な米国債収益率曲線に基づいており、期待期限の仮定に見合っている。当社は簡略化手法を用いて,期待期限を帰属日と契約期限終了との間の中点と推定した。歴史的な行権データや株式奨励の簡単な性質が乏しいため、当社はこの方法を採用した。期待配当収益率は
F-12
所得税
当社は、連結財務諸表または当社納税申告書で確認された事件の将来の税務結果を予想する繰延税金資産と負債の確認を要求する貸借対照法を用いて所得税を計算する。この方法によると、繰延税項資産及び負債は、財務諸表の帳簿額面と資産及び負債の課税基礎との差額に基づいて、予想差額を用いて返送される年度の現行税率を決定する。既存の証拠の重みに基づいて、繰延税金資産の一部または全部が現金化できない可能性が高い場合は、繰延税金資産の推定準備に計上しなければならない。繰延税金資産の回収の潜在力を評価する時に多くの要素を考慮し、未来に予想される課税オーバー額の推定、現有の課税の一時的な差額の推定、将来の引越期間の課税オーバー額を考慮すること、及び慎重かつ実行可能な税務計画策略を考慮することを含む。
再編成費用
当社は、会計基準アセンブリ420に従って、脱退および処置活動に関連するコストおよび負債を記録する脱退または処分費用債務それは.このようなコストは、負債発生期間の公正価値推定に基づいて計算される。より多くの情報を得ることに伴い、当社は状況の変化に応じてこれらのコストを適切に評価·調整する。
総合損失
総合損失は、企業が一定期間内に非所有者由来の取引や他のイベントや状況によって発生する権益変化と定義される。2022年12月31日と2021年12月31日までの年度、全面赤字投資純損失と未実現損失からなります。
1株当たり純収益
表外リスクと信用リスク集中度
当社には外国為替契約、オプション契約、その他の海外ヘッジ手配などの重大な表外リスクはありません。会社を集中的な信用リスクに直面させる可能性のある金融商品は、主に現金、現金等価物、有価証券、および制限された現金を含む。会社の現金、現金等価物、および限定現金は大型金融機関の口座に保管されている。当社は、現金、現金等価物、制限された現金を持つ預金機関の財務力により、当社は重大な信用リスクに直面しないと信じている。同社はその通貨市場基金の現金等価物を維持し、これらの基金は米国債、米国債、商業手形に投資している。会社の有価証券には米国債、社債、商業手形が含まれており、会社を集中的な信用リスクに直面させる可能性がある。会社はどの投資タイプにも投資できる金額を制限する投資政策をとっている。当社はいかなる信用損失も経験しておらず、そのような基金に重大な信用リスクがあるとも信じていない。
市場情報を細分化する
運営中です部門は企業の構成要素として定義され、これらの構成要素に関する独立した離散財務情報は、首席運営意思決定者が関連資源配分を行うことができる
F-13
新興成長型会社の地位
最近の会計公告は採択されるだろう
2016年6月、FASBはASU第2016-13号を発表した金融商品·信用損失(主題326):金融商品信用損失の測定それは.この基準は、現在使用されている発生した損失モデルではなく、期待損失モデルを使用して信用損失を報告することを要求し、信用リスクに関連する追加開示を確立する。未実現損失のある売却可能債務証券については、本基準は、投資の償却コストを削減するのではなく、準備を計上することを要求する。新基準は2023年1月1日から当社に対して施行されます。同社は現在、この基準がその総合財務状況と経営結果に潜在的な影響を与える可能性があることを評価している。
2019年12月、FASBはASU第2019-12号を発表した所得税--所得税の会計計算の簡素化それは.この基準は、期間内の税収分配方法、中期所得税計算方法、および外部ベース差を確認する繰延税金負債に関するいくつかの例外を除去する。新たなガイドラインはまた、特許経営税の会計処理を簡略化し、税法や税率の変化を公布し、営業権計税基礎の上昇を招く取引の会計処理を明らかにした。新基準は2023年1月1日から当社に対して施行されます。同社は現在、この基準がその総合財務状況と経営結果に潜在的な影響を与える可能性があることを評価している。
F-14
最近の会計公告は
2016年2月、FASB ASB 2016-02は、複数の機関の改訂を経て、その後、ASUを発表した。この基準は、テナントが貸借対照表上の期限が1年を超える経営リースを、賃貸支払いの現在値で測定するために、使用権資産および対応する賃貸負債として確認することを要求する。テナントはリースを融資リースや経営的賃貸に分類することを要求されている。レンタルが実際にテナントが融資して購入した場合は融資リースに分類され、そうでなければ経営的賃貸に分類される。この分類は,レンタル料金が有効利子法に基づいているかレンタル期間に基づく直線ベースで確認されるかを決定する.2018年7月、FASBもASU 2018-11を発表したレンタル(テーマ842):的確な改善これは、エンティティがASC 840にレガシー·ガイダンスを適用し続けることを可能にする賃貸借証書このエンティティが新しいレンタル基準を採用したその年に提示された比較期間内に、その開示要求を含む。この移行方法によると,最初に適用された改訂されたASU 2016−02年度の累積効果は,初回適用日を含む年次報告期間開始時の留保収益や累積赤字期初期残高の調整であることが確認された。
そこで,会社は改訂後のASU 2016−02が許可する移行方法を採用し,2022年1月1日から発効した。新基準を採用する際に、会社はある既存の便宜的な計を利用することを選択し、ASU 2016-02年度に許可された実際の便宜的な移行方案を選択し、会社が再評価する前に賃貸借契約、既存または満期賃貸借契約の分類、および新基準に基づいて資格資本化された以前の初期直接コストをどのように処理するかに関する会計結論を再評価しないことを許可した。当社は実際の便宜的な方法を新借約および改訂された借約に適用することを選択して借約および非借約部分を分離しません。同社はまた、初期期限が12カ月以下の賃貸を貸借対照表から除外する会計政策選択を行った。
改訂されたASU 2016−02年度を採用した後、会社は以前ASC 840項目で記録されていたレガシー繰延賃貸料残高を削除し、#ドルの経営リース使用権資産を構築した
|
|
2022年1月1日 |
|
|||||||||
|
|
ASUを採用する前に2016年に改訂されました |
|
|
養子縁組の効果 |
|
|
改訂されたASU 2016-20の後 |
|
|||
|
|
(単位は千、1株当たりのデータは除く) |
|
|||||||||
経営的リース使用権資産 |
|
$ |
|
|
$ |
|
|
|
|
|||
賃貸負債を経営し、流動 |
|
|
|
|
|
|
|
|
|
|||
費用とその他の流動負債を計算しなければならない |
|
|
|
|
|
( |
) |
|
|
|
||
延期レンタル奨励、今期の部分 |
|
|
|
|
|
( |
) |
|
|
|
||
賃貸負債を経営し,当期分を差し引く |
|
|
|
|
|
|
|
|
|
|||
繰延賃貸料は現在の部分は含まれていません |
|
|
|
|
|
( |
) |
|
|
|
||
現在の部分は含まれていないレンタル延期インセンティブ |
|
|
|
|
|
( |
) |
|
|
|
||
F-15
改訂されたASU 2016−02年度を採用することは,2022年12月31日までの総合業務および全面損益表あるいは総合キャッシュフロー表に大きな影響を与えないそれは.改訂されたASU 2016-02が当社の既存オフィスおよび実験室空間運営賃貸に適用される詳細については、付記12、“レンタル”を参照されたい。
3.公正価値計測
以下の表に当社の公正価値によって日常的に計量された金融資産の資料を記載し、この等公正価値の分類を以下のように示す2022年12月31日および2021年12月31日(単位:千):
|
|
公正価値に応じて計量する |
|
|||||||||||||
|
|
合計する |
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
||||
現金等価物: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
||
商業手形 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
有価証券: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
政府機関証券 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
商業手形 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
社債 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
合計する |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
|||
|
|
公正価値に応じて計量する |
|
|||||||||||||
|
|
合計する |
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
||||
現金等価物: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
||
有価証券: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
商業手形 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
社債 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
合計する |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
|||
いくつありますか
4.現金等価物および有価証券
現金等価物および有価証券には以下のものが含まれる2022年12月31日および2021年12月31日(単位:千):
|
|
公正価値に応じて計量する |
|
|||||||||||||
|
|
償却する |
|
|
毛収入 |
|
|
毛収入 |
|
|
公正価値 |
|
||||
現金等価物: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
商業手形 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
||
現金等価物合計 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
||
有価証券: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
||
政府機関証券 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
||
商業手形 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
||
社債 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
有価証券総額 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
現金等価物と有価証券総額 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
F-16
|
|
公正価値に応じて計量する |
|
|||||||||||||
|
|
償却する |
|
|
毛収入 |
|
|
毛収入 |
|
|
公正価値 |
|
||||
現金等価物: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
現金等価物合計 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
有価証券: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
商業手形 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
||
社債 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
有価証券総額 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
現金等価物と有価証券総額 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
いくつありますか
その会社は確かにやったと確信しています
5.財産と設備、純額
財産と設備、純価値は以下の通り(千で計算)
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
実験室装置 |
|
$ |
|
|
$ |
|
||
家具と固定装置 |
|
|
|
|
|
|
||
コンピュータ装置 |
|
|
|
|
|
|
||
ソフトウェア |
|
|
|
|
|
|
||
賃借権改善 |
|
|
|
|
|
|
||
建設中の工事 |
|
|
|
|
|
|
||
総資産と設備 |
|
|
|
|
|
|
||
減算:減価償却累計 |
|
|
( |
) |
|
|
( |
) |
財産と設備、純額 |
|
$ |
|
|
$ |
|
||
2022年12月31日までと2021年12月31日までの年度減価償却費用はい$です
6.追加の貸借対照表の詳細
前払い料金および他の流動資産には、以下のものが含まれている(千計)
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
前払い費用 |
|
$ |
|
|
$ |
|
||
帰属条項に制限された前払いでボーナスにサインする |
|
|
|
|
|
|
||
受取利息収入 |
|
|
|
|
|
|
||
前払い費用とその他の流動資産総額 |
|
$ |
|
|
$ |
|
||
計算すべき費用および他の流動負債には、以下の項目が含まれる(千で計算)
F-17
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
外部研究開発 |
|
$ |
|
|
$ |
|
||
賃金総額と福祉 |
|
|
|
|
|
|
||
専門サービス |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
費用とその他の流動負債総額を計算しなければならない |
|
$ |
|
|
$ |
|
||
7.優先株
2022年12月31日と2021年12月31日まで,
8.普通株式
2022年12月31日と2021年12月31日まで,
自分から2022年12月31日と2021年12月31日、会社は将来の発行のために以下の数の普通株式を予約しています
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
発行済み株式オプション行使のために保留された株式 |
|
|
|
|
|
|
||
制限された株式単位に帰属する株式を予約する |
|
|
|
|
|
|
||
2019年株式インセンティブ計画に基づいて将来のために予約された株式を発行する |
|
|
|
|
|
|
||
2019年の従業員の株式購入計画によると、将来のために予約された株式を発行する |
|
|
|
|
|
|
||
2022年インセンティブ株式インセンティブ計画に基づいて将来の株式発行のために予約された株式 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
9.株ベースの報酬費用
2016年度株式インセンティブ計画
2016年7月、会社は2016年度株式インセンティブ計画(“2016計画”)を採択し、会社が条件を満たす従業員、上級管理職、取締役、コンサルタント、コンサルタントに制限株式奨励、制限株式単位、奨励株式オプション、非法定株式オプション、その他の株式ベースの奨励を付与することを規定した。2019年株式インセンティブ計画(“2019年計画”)発効日から、2022年、2021年12月31日まで,
F-18
2019年株式インセンティブ計画
2019年7月2日、会社株主は2019年計画を承認し、2019年7月17日に発効した。2019年には、会社役員、従業員、取締役、コンサルタント、コンサルタントに奨励的株式オプション、非法定株式オプション、株式付加価値権、制限株式奨励、制限株式単位、その他の株式ベースの奨励を付与する予定です。2019年の計画によると発行のための予備予約株式数は
当社は、2016年計画または2019年計画満了、終了またはその他の方法で提出、ログアウト、没収、または買い戻しに係る任意の奨励に係る普通株式に基づいて、2019年計画下で発行可能な普通株式を再計上します。2019年7月17日から、2016年度計画は奨励を与えない。
2022年インセンティブ株式インセンティブ計画
2022年2月、会社取締役会は“2022年インセンティブ株式インセンティブ計画”(以下、“インセンティブ計画”と略す)を採択し、会社はその計画に基づいて付与することができるまた、インセンティブ計画及びナスダック規則条項に該当する場合には、非法定株式オプション、株式付加権、制限株式奨励、制限株式単位等の株式ベースの奨励を行う。その会社は最初は全部保留した
制限株式賞
従業員または非従業員が解雇または当社との雇用またはサービス関係を終了した場合、当社は元の購入価格で帰属していない制限的な株式奨励を買い戻すことができる。従業員と非従業員から買い戻す普通株とは、会社の倉庫が保有する株式(“在庫株”)のことである。取締役会は、許可されているが発行されていない普通株に在庫株を返却することを適宜許可することができる。
制限株式奨励の基礎となる普通株は、通常、
下表に当社の今年度までの限定株式奨励活動をまとめた2022年12月31日:
|
|
量 |
|
|
重みをつける |
|
||
2021年12月31日に帰属していません |
|
|
|
|
$ |
|
||
授与する |
|
|
|
|
|
|
||
既得 |
|
|
( |
) |
|
|
|
|
すでに買い戻した |
|
|
|
|
|
|
||
2022年12月31日に帰属していない |
|
|
|
|
$ |
|
||
株式オプション
会社が付与した株式オプションは通常授与される
F-19
株です。2019年計画と2016計画に基づいて付与された株式オプションのほか、当社は、ナスダック上場規則第5635(C)(4)条に基づいて株式オプションを付与し、雇用の実質的なインセンティブとして、2019年計画及び2016計画を除いて付与されている
|
|
量 |
|
|
重みをつける |
|
|
重みをつける |
|
|
骨材 |
|
||||
2021年12月31日現在の未返済債務 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
授与する |
|
|
|
|
|
|
|
|
|
|
|
|
||||
鍛えられた |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
キャンセルします |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
2022年12月31日に返済されていません |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2022年12月31日に行使できます |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
行権価格が会社普通株公正価値を下回るオプションについては、株式オプションの内在価値合計は、株式オプションの行権価格とアセットバランスシート日までの会社普通株公正価値との差額として計算される。
2022年12月31日まで及び2021年12月31日まで年度に付与された株式オプションの加重平均授受日公正価値はい$です
年内に付与される株式オプションの公正価値2022年と2021年12月31日終了年度は、以下の重み付き平均仮定に基づいて付与日に算出される
|
|
年.年 |
|
|
年.年 |
|
||
無リスク金利 |
|
|
% |
|
|
% |
||
期待配当収益率 |
|
|
% |
|
|
% |
||
所期期間(年) |
|
|
|
|
|
|
||
株価の変動を予想する |
|
|
% |
|
|
% |
||
限定株単位
その会社はまた制限株式単位を付与した。限定株式単位の基礎となる普通株の株式は通常4年以内に帰属する。普通株式の株式は帰属時に株主権益に計上される。
下表は会社の年内の限定株式単位活動をまとめたものである2022年12月31日までの年度:
|
|
量 |
|
|
重みをつける |
|
||
2021年12月31日に帰属していません |
|
|
|
|
$ |
|
||
授与する |
|
|
|
|
|
|
||
既得 |
|
|
|
|
|
|
||
キャンセルします |
|
|
( |
) |
|
|
|
|
2022年12月31日に帰属していない |
|
|
|
|
$ |
|
||
2022年12月31日まで及び2021年12月31日まで年度内のすべての制限株式単位及び制限株式奨励の合計内的価値はい$です
F-20
株に基づく報酬費用
財務諸表で確認した総補償コスト会社が付与したすべての株式補償に関する総合損失は以下の通り(千単位)
|
|
現在までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
一般と行政 |
|
$ |
|
|
$ |
|
||
研究開発 |
|
|
|
|
|
|
||
株式に基づく報酬総支出 |
|
$ |
|
|
$ |
|
||
2022年12月31日まで会社の総金額は$です
2019年従業員株購入計画
2019年7月2日、株主は2019年7月17日に発効する2019年従業員株購入計画(ESPP)を承認した。合計する
10.連携とライセンスプロトコル
Acceleron連携プロトコル
2019年12月20日,当社は肺疾患領域における標的適応(“適応”)に関する特定の経路を調節するために生物標的を決定する加速器協力協定を締結した。Acceleron協力協定の条項によると、当社は、特定の知的財産権に基づいて、当社が決定したいくつかの生物学的標的の分子および製品を製造、製造、使用、販売、輸入、輸出、流通および流通、販売、マーケティング、マーケティング、普及、普及、または他の方法で開発、治療、予防または診断適応のために開発、または表現するためにAcceleronグローバル独占ライセンスを付与する。
Acceleronは2022年6月3日、会社に通知を受けて120日後、2022年10月1日に発効するAcceleron便利協力協定を終了したことを会社に通知した。
双方が合意した研究計画によると、同社は化学分析スクリーニングと関連研究活動を行い、更なる研究の潜在生物目標を確定と検証し、Acceleron社の候補製品の開発、製造と商業化を支持する。研究活動完了後,同社はAcceleronに治療,予防または診断適応に関する研究活動で決定した生物標的に関するパケットを提供した。Acceleronは、プロトコル開始時のグローバル独占ライセンス規定に基づいて、当社が決定した特定の数の生物標的を指定する権利があり、これらの目標に対するAcceleronの製品または分子の研究、開発、製造および商業化のために、治療、予防または診断適応(“目標”)のために使用される。Acceleronが終了通知を出す前に、Acceleronは何の目標も指定しなかった。
会社は払い戻しできない前金を受け取りました#
F-21
会計分析
契約的識別
同社はAcceleron連携プロトコルを評価し,ASC 606の範囲内の顧客との契約を代表していると結論した。
約束と義務を履行する
当社は、Acceleron協力協定は、(I)肺疾患領域における特定の数の生物標的を治療、予防または診断するための特定の数の生物標的を治療、予防または診断するための権利(“許可”)、(Ii)潜在的生物標的を識別および検証する研究サービス(“研究サービス”)、および(Iii)共同指導委員会(“JSC”)への参加を含むいくつかの知的財産権の下でのグローバル独占許可を含むことを決定した。
当社は上記の承諾を評価し,ライセンスが研究サービスを履行していない場合には限られた価値があることから,研究サービスの専門性により,研究サービスは当社でしか実行できないため,ライセンスは研究サービスと区別できないと結論した。そのため、同社はライセンスと研究サービスは単一の総合的な履行義務であると結論した。
同社はJSCへの参加状況も評価し、加速器協力協定を背景に、この約束は数量的にも品質的にも無関係であると結論した。したがって、当社はJSCへの参加を義務履行とは考えていません。
成約価格の決定
会社は払い戻しできない前金を受け取りました#
同社は取引価格に研究サービス費の精算額も含まれている。
Acceleronの終了通知を受け取る前に、当社は、各報告期間の終了時および不確定イベントが解決された場合や他の状況が変化した場合に取引価格を再評価し、必要に応じて取引価格の推定を調整する。あったことがある
取引価格と履行義務の分配
以上のように,同社はアクセラレータ連携協定に関する単一履行義務を決定した。そこで,会社は取引価格の全金額を確定した単一履行義務に割り当てる。
収入の確認
研究サービスの提供にともない,同社は時間の経過とともにAcceleron連携プロトコルに関する収入を確認した.同社の結論は,入力法は“アクセラレータ協力協定”に基づいてサービスを譲渡する代表的な記述である.Acceleronの終了通知を受信する前に、履行義務を履行することが予想される内部および外部総コストに対して、実際に累積された内部および外部コストを含む配送サービスの進捗状況を測定する方法。総コストを見積もる期間は、会社が研究サービスを提供している期間の見積もりを反映している。発生すると予想される内部と外部費用総額推定数の変化は変動期間中に累積追跡調整であることが確認された。
F-22
2022年12月31日までの年度内、会社は$を確認しました
MyoKardia協力協定
2020年7月20日、当社はMyoKardia協力協定に調印し、この協定に基づき、当社はある知的財産権に基づいてMyoKardiaに世界的に独占的な許可を与え、その研究、開発、製造、使用、使用、販売、要約販売、輸入、輸入、輸出、輸出、流通、流通、マーケティング、マーケティング、普及、普及、あるいは他の方法で当社が決定したある生物標的に対する製品を開発することを許可し、これらの製品はある遺伝子定義の心筋症に関連する特定の数量の興味のある遺伝子を調節することができる。
双方が合意した研究計画によると、同社はMyoKardiaの更なる研究、開発、製造と商業化のために、最も多くの特定数の潜在心筋症遺伝子標的(“確定した標的”)を確定と検証するために、検査スクリーニングと関連研究活動を行う。同社はMyoKardiaと共同で努力し,研究計画下の研究活動の各段階でどのように最適な進展を得るかを決定し,どのような決定された目標(あれば)が研究計画に規定されている基準(“心筋症目標候補対象”)に適合するかを決定する。研究計画完了後,双方は最終的なパケットを共同で用意し,MyoKardiaはMyoKardia連携プロトコル(“心筋症目標”)によりMyoKardiaのさらなる開発に何らかの心筋症目標候補を指定する可能性がある。MyoKardiaが指定された期間内に心筋症標的を指定していなければ,MyoKardia連携プロトコルは自動的に終了する。MyoKardiaが1つ以上の心筋症の標的を指定した場合、MyoKardiaは、規制部門の承認を求め、特定の指定された国/地域で決定された目標に対して製品を商業化するための商業的合理的な努力を使用する義務がある。
会社が研究計画に基づいて研究活動を行っている期間(“研究期間”)や,研究期間を超えた特定の期間において,MyoKardiaが心筋症目標を指定していれば,会社はMyoKardia協力プロトコルに従ってMyoKardiaにその研究活動から発生したデータしか使用できない。研究期間およびその後の特定の期間内に、会社は、MyoKardia協力プロトコルの下で心筋症標的候補に対して任意の適応を治療、予防または診断するための化合物または製品、または(B)心筋症標的調節が証明された特定の特定の遺伝子に関連する任意の遺伝子定義の心筋症の治療のための任意の化合物または製品を研究、開発、製造、商業化、使用または他の方法で開発してはならない。
MyoKardia協力協定によるとMyoKardiaは$を稼いだ
F-23
MyoKardia協力協定によると、任意の決定された目標に対して、MyoKardiaおよびその任意の付属会社およびライセンス所有者の製品の世界的な年間純売上高の2桁から数百ポイント低い。特許使用料は、指定された特許使用料期間内に製品毎に支払われ、特定の場合には減少する可能性がある。
MyoKardia協力協定は、製品の印税期限が最終的に満了するまで、個々の国および個々の製品に基づいて継続され、このとき、MyoKardia協力協定は、製品に関する国/地域の協力協定が満了する。どちらか一方がMyoKardia連携プロトコルの下の義務を履行する際に深刻に違約し,適用された救済期間内に是正されなければ,どちらもMyoKardia連携プロトコルを終了する権利がある.便宜上、MyoKardiaはまた、MyoKardia協調プロトコルを終了するか、またはターゲットごと、製品毎、または分子毎に終了する権利がある。
会計分析
契約的識別
同社はMyoKardia協力協定を評価し、ASC 606の範囲内の顧客との契約を代表していると結論した。
約束と義務を履行する
同社は、MyoKardia協力協定は、(I)会社が決定した特定の数の潜在的心筋症遺伝子標的の権利を含む特定の知的財産権下での独占的なグローバル許可、さらに研究、開発、製造および商業化手配の開始時に伝達されるいくつかの遺伝子定義の心筋症の治療、予防または診断(“MyoKardiaライセンス”)、(Ii)潜在的生物標的を識別および検証する研究サービス(“MyoKardia研究サービス”)、および(Iii)共同指導委員会(“MyKardia JSC”)に参加する約束を含むことを決定した。
同社は上記の承諾を評価し,MyoKardiaライセンスはMyoKardia研究サービスと区別できないが,MyoKardia研究サービスの履行がなければMyoKardiaライセンスの価値は限られており,MyoKardia研究サービスはその専門性のため当社でしか実行できないと結論した。そこで,同社はMyoKardiaライセンスとMyoKardia研究サービスが単一の共同履行義務を代表していると結論した。
F-24
同社はまた,MyoKardia JSCの参加状況を評価し,MyoKardia協力協定を背景に,この約束は数量的にも品質的にも無関係であると結論した。そこで,当社はMyoKardia JSCへの参加を履行義務としている。
成約価格の決定
会社は払い戻しできない前金を受け取りました#
同社はまた、MyoKardia研究サービスの予想精算費用金額を取引価格に含まれており、#ドルを含む
当社は、各報告期間末および不確定イベント解決や状況が他に変化した場合に取引価格を再評価し、必要に応じて取引価格の見積もりを調整する。2022年12月31日までの年間で、拘束されている可変対価格金額は変化していない。
販売マイルストーン支払い(特許使用料を含む)に関連する任意の対価格は、関連販売が発生したときに確認され、これらの金額は、主にMyoKardiaに付与されたライセンスに関連すると決定されているので、関連販売が発生したときまたは義務を履行したときに確認される。
取引価格と履行義務の分配
以上のように,同社はMyoKardia連携協定に関する単一履行義務を決定している.そこで,会社は取引価格の全金額を確定した単一履行義務に割り当てる。
収入の確認
MyoKardia研究サービスの提供にともない,同社は時間の経過とともにMyoKardia連携プロトコルに関する収入を確認した.同社の結論は,入力法はMyoKardia連携プロトコルでのサービス移転の代表的な記述である.サービス提供の進捗状況を測定する方法は、契約義務を履行するために発生すると予想される内部および外部費用の総額に対して、実際に累積された内部および外部費用を含む。総コストを見積もる時間帯は,同社がMyoKardia研究サービスを提供している期間の推定を反映している。発生すると予想される内部と外部総費用推定数の変化は変動期間内に累積追跡調整であることが確認された。
2022年12月31日までの年度内、会社は$を確認しました
F-25
のです$
11. 引用権と許可協定
2019年2月、当社はグラクソ·スミスクラインの付属会社(総称して“グラクソ·スミスクライン”)と改訂された参考権利及びライセンス契約(“GSKプロトコル”)を締結し、これにより、当社はロスコパモトの開発及び商業化のグローバル独占許可を授与した。グラクソ·スミスクライン協定によると、同社はまた、関連法規および生産文書、ならびにグラクソ·スミスクラインの既存のロモパジン薬物物質および製品供給を参照する権利を獲得した。ある条件を満たす場合、会社はライセンス契約に基づいてその権利を再許可する権利もある。同社は商業的に合理的な努力を用いてlosmapimodを開発·商業化し,その全費用を支払う義務がある。当社はライセンス特許権の提出とメンテナンスに関する費用も担当しています。
GSK協定に基づき,同社は発行した
GSKプロトコルは、いずれか一方が他方の実質的な違約によって終了することができるが、通知および救済条項を遵守しなければならない。以前に終了しない限り、グラクソ·スミスクライン協定は、当社の特許使用料義務が満了するまで有効であり、このような特許権使用料義務は、(I)同国での初の商業販売後10年または(Ii)適用規制機関がlosmapimodの模造薬を承認した後、より遅い時間に国ごとに満了する。
基本的な意外な状況が解決され、対価格または対価格を支払う時、会社は臨床と監督管理マイルストーン支払いを確認する。確認された日までの関連資産の性質に基づいて、マイルストーン支払いを資本化または支出する。会社は販売マイルストーン支払いと特許権使用料を、それに応じた販売期間中に関連製品の販売が発生した追加料金として記録する。
12.レンタル証書
賃貸借契約を経営する
ランデーン街26番地
二零一七年十一月、当社は既存の会社本社について契約を締結しました
改訂されたASU 2016−02年度に行われた会計
改訂されたASU 2016-02を採用したため、会社は2022年1月1日に総合貸借対照表にLandsdown Street 26号の賃貸借に関する使用権資産とそれに応じた賃貸負債を記録し、2022年12月31日現在となっている。26 Landsdown Street賃貸契約には暗黙的なレートがないため、同社はその増量を推定している
F-26
借金をする総合信用格付けと収益率曲線分析に基づく金利。この分析によると、同社が算出した割引率は
改訂されたASU 2016-02によると、2022年12月31日までの年度、本レンタルに関する経営リース費用と可変レンタル費用約$です
26 Landsdown Streetリースに関する将来最低レンタル料2022年12月31日、状況は以下の通り(単位:千):
2023 |
|
$ |
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
その後… |
|
|
|
|
最低賃貸支払総額 |
|
|
|
|
差し引く:推定利息 |
|
|
( |
) |
リース総負債 |
|
$ |
|
ASC 840での会計計算
改正ASU 2016−02年度を採用する前に,ASC 840におけるレガシー指導原則に基づき,当社は賃貸料支出を賃貸期間終了まで直線法で記録し,繰延賃貸料を総合貸借対照表に記録した。当社は総合貸借対照表で賃貸改善インセンティブを繰延賃貸インセンティブと表記し、賃貸期間内に賃貸料を比例的に減少させることで繰延リースインセンティブを償却している。
ASC 840,Reによると2021年12月31日までの年度本レンタルに関する新台湾ドル支出約$です
2022 |
|
$ |
|
|
2023 |
|
|
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
その後… |
|
|
|
|
最低賃貸支払総額 |
|
|
|
シドニー通り125番地
2000年11月,当社は契約を含めて賃貸契約を締結した
改訂されたASU 2016−02年度に行われた会計
改訂されたASU 2016-02を採用したため、当社は2022年1月1日にシドニー通り125番地の賃貸に関する使用権資産とそれに応じた賃貸負債を総合貸借対照表に記録した。シドニー街125号のレンタル契約には隠れた金利がないため、同社はその増量借金を推定している総合信用格付けと収益率曲線分析に基づく金利。この分析によると、同社が算出した割引率は
F-27
改訂されたASU 2016-02によると、本レンタルに関する2022年12月31日までの年度約$です
2023 |
|
$ |
|
|
2024 |
|
|
|
|
最低賃貸支払総額 |
|
|
|
|
差し引く:推定利息 |
|
|
( |
) |
リース総負債 |
|
$ |
|
ASC 840での会計計算
改正ASU 2016−02年度を採用する前に,ASC 840におけるレガシー指導原則に基づき,当社は賃貸料支出を賃貸期間終了まで直線法で記録し,繰延賃貸料を総合貸借対照表に記録した。
ASC 840によると,2021年12月31日までの年度本賃貸契約に関する賃貸料支出約$です
2022 |
|
$ |
|
|
2023 |
|
|
|
|
2024 |
|
|
|
|
最低賃貸支払総額 |
|
$ |
|
13.支払いの引受およびまたは事項
賠償協定
正常な業務過程において、当社は売り手、レンタル者、業務パートナー及びその他の当事者と当社との間の関係によって生じるいくつかの事項について、売り手、レンタル者、業務パートナー及びその他の当事者に異なる範囲及び条項の賠償を提供することができる。また、当社は取締役会メンバーおよび上級管理者と合意を締結し、取締役または上級管理者としての身分やサービスとして生じる可能性のあるいくつかの法的責任について補償することを当社に要求している。これらの賠償協定によると、会社が将来支払う必要がある可能性のある最大潜在金額は多くの場合無制限である。当社はこれまで、当該等の賠償により重大なコストを発生させていません。その会社は賠償手配のクレームがあることを知りませんでした。そしてそれはすでに
F-28
法律訴訟
当社は現在、いかなる重大な法的手続きの当事者でもありません。報告日ごとに、当社は、潜在損失金額又は潜在損失範囲が権威的な案内処理又は事項の規定によって可能かつ合理的に評価されているか否かを評価する。当社はその法的訴訟に関する費用を支払います
14.所得税
米国連邦法定所得税率と会社の有効所得税率の入金は以下の通りである
|
|
現在までの年度 |
|
|
現在までの年度 |
|
||
法定税率で徴収される連邦所得税 |
|
|
% |
|
|
% |
||
恒久的差異 |
|
|
( |
) |
|
|
( |
) |
連邦と州の研究開発信用限度額 |
|
|
|
|
|
|
||
連邦孤児薬品信用限度額 |
|
|
|
|
|
|
||
州所得税、連邦福祉を差し引いた純額 |
|
|
|
|
|
|
||
所有権変更の影響 |
|
|
( |
) |
|
|
|
|
他にも |
|
|
( |
) |
|
|
( |
) |
評価免除額を変更する |
|
|
( |
) |
|
|
( |
) |
有効所得税率 |
|
|
% |
|
|
% |
||
同社は2022年12月31日と2021年12月31日までの年間で帳簿·税務赤字が発生し、その繰延税純資産に対して全額推定準備金を保持しているため、連邦や州所得税費用や福祉は確認されていない。
同社の繰延税金資産と負債には、以下の内容が含まれている(千計)
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
繰延税金資産: |
|
|
|
|
|
|
||
純営業損失が繰り越す |
|
$ |
|
|
$ |
|
||
資本化研究開発コスト |
|
|
|
|
|
|
||
孤児薬品信用繰り越し |
|
|
|
|
|
|
||
研究開発信用繰り越し |
|
|
|
|
|
|
||
無形資産 |
|
|
|
|
|
|
||
費用その他を計算する |
|
|
|
|
|
|
||
リース負債を経営する |
|
|
|
|
|
|
||
収入を繰り越す |
|
|
|
|
|
|
||
延期リースインセンティブ |
|
|
|
|
|
|
||
賃料を繰延する |
|
|
|
|
|
|
||
繰延税項目総資産 |
|
|
|
|
|
|
||
推定免税額 |
|
|
( |
) |
|
|
( |
) |
繰延税項目純資産 |
|
|
|
|
|
|
||
繰延税金負債 |
|
|
( |
) |
|
|
( |
) |
繰延税項目純資産 |
|
$ |
|
|
$ |
|
||
当社は繰延税項目の純資産能力を実現するためのプラスとマイナスの証拠を評価した。同社は,設立以来累計純損失の歴史や,設立以来何の製品も商業化していないことを考慮して,繰延税項純資産のメリットを実現できない可能性があると結論した。そこで,2022年12月31日と2021年12月31日までの繰延税項純資産について全額推定準備を設けたそれは.推定免税額は#ドル増加した
F-29
本年度の純損失、資本化研究開発コストの増加及び研究開発と孤児薬品税収控除の発生繰越。会社は各報告期間に肯定的で否定的な証拠を再評価する。
2022年12月31日まで同社が繰り越した連邦純営業損失は約$
2022年12月31日まで同社の連邦孤児薬物信用限度額は約$です
以前に発生または将来発生する可能性のある所有権変更により、繰り越しの純営業損失と研究開発税収控除繰越は、1986年に改正された“国税法”(以下、“国税法”と呼ぶ)第382節及び州法律相応条項の年間制限を受ける可能性がある。これらの所有権の変化は、将来の課税所得額を相殺するために毎年使用できる繰越金額を制限する可能性がある。一般に、第382条の定義によれば、所有権変更とは、3年以内にある株主又は公衆団体の会社株における所有権を50%以上増加させた取引の結果をいう。当社は、2021年12月31日までの所有権履歴変化が、当社がその変更前の純営業損失と研究開発税収控除を利用して将来の課税収入を相殺する能力を制限または制限するかどうかを決定するために、第382条に基づいて分析を行った。分析の結果、当社は純営業損失及び研究開発税項目相殺繰越を利用して将来の課税収入を相殺する能力に重大な制限があるとは考えていないことを示した。当社は2021年12月31日以降、制御権が変更されたかどうかや制御権が複数回変更されているかどうかを評価する研究は行っていません。会社が2021年12月31日以降に第382条に定義された支配権変更を経験した場合、第382条の規定により、繰越の純営業損失又は研究開発税収繰越控除の使用は、まず所有権変更時の会社株の価値に適用される長期免税率を乗じて決定される第382条に規定される年次制限を受ける, そして必要に応じて追加的な調整を行うことができる。いかなる制限も繰越の一部の純営業損失や研究開発税収相殺が使用前に満期になる可能性がある。しかも、ある研究が完了して制限があることを知るまで、不確定な税金状況として記載されていない額はない。
会社はその経営管轄区域の税法の規定に従って納税申告書を提出します。通常業務の過程において、当社は連邦と州司法管轄区の審査を受けなければならない(適用されるように)。現在未解決の税務審査はありません。2022年12月31日まで会社の納税年度は法律に基づいて開放されています今までです。
同社の政策は、所得税に関する罰金と利息支出を所得税として支出することの構成要素である。2022年12月31日と2021年12月31日まで会社が所有しています
15.供給計画を定義する
当社は国税法第401(K)条に基づいて固定払込貯蓄計画を策定した
F-30
16.再構成活動
2022年8月、会社は戦略計画の実施を発表し、会社の優先順位を優先するために内部投資と運営を再調整する
2021年12月31日までの課税再編成費用 |
|
|
|
|
期間内に発生する再編成費用 |
|
|
|
|
その期間内に支払われる金額 |
|
|
( |
) |
2022年12月31日までの課税再編成費用 |
|
|
|
17.1株当たり純損失
これらの等価物を計上することは、逆希釈効果をもたらすので、以下の普通株等価物は、これらの期間の希釈1株当たり純損失の計算には含まれない
|
|
現在までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
未償還株式オプション |
|
|
|
|
|
|
||
未帰属限定株式単位 |
|
|
|
|
|
|
||
合計する |
|
|
|
|
|
|
||
F-31
展示品索引
展示品 番号をつける |
|
説明する |
3.1 |
|
登録者の再記載登録証明書(添付ファイル3.1を参照して登録者に組み込まれ、2019年7月22日に証券取引委員会の8−K表の現在の報告書に提出される)。 |
3.2 |
|
登録者の別例(参考登録者が2019年7月22日に証券取引委員会に提出した8−K表の現在の報告書の添付ファイル3.2)を改訂および再改訂する。 |
4.1 |
|
普通株式株式を証明する株式サンプル証明書(2019年6月21日に米国証券取引委員会に提出された登録者S-1表登録説明書(第333-232260号文書)の添付ファイル4.1を参照して編入)。 |
4.2 |
|
取引法第12条に基づいて登録された登録者証券の説明(2020年3月5日に米国証券取引委員会に提出された登録者年次報告書10−K表の添付ファイル4.2を参照して編入)。 |
10.1 |
|
登録者と協議の他の当事者が2018年8月24日に改訂及び再署名した投資家権利協定(参照登録者が2019年6月21日に米国証券取引委員会に提出したS−1表登録声明(第333−232260号文書)添付ファイル10.1により編入)。 |
10.2 |
|
登録者とその他の当事者との間で2020年6月9日に締結された“登録権協定”(登録者が2020年6月10日に米国証券取引委員会に提出した8−K表の現在報告書の添付ファイル10.2を引用して統合された)。 |
10.3# |
|
改訂された2016年株式インセンティブ計画(2019年6月21日に米国証券取引委員会に提出された登録者S-1表登録説明書(第333-232260号文書)添付ファイル10.2を参照して編入)。 |
10.4# |
|
2016年株式インセンティブ計画下のインセンティブ株式オプション契約表(2019年6月21日に米国証券取引委員会に提出された登録者S-1フォーム登録説明書(第333-232260号ファイル)添付ファイル10.3参照). |
10.5# |
|
2016年株式インセンティブ計画下の非法定株式オプション協定表(2019年6月21日に米国証券取引委員会に提出された登録者S-1登録説明書(第333-232260号文書)添付ファイル10.4参照)。 |
10.6# |
|
2016年株式インセンティブ計画下の限定株式契約表(2019年6月21日に米国証券取引委員会に提出された登録者S-1表登録説明書(第333-232260号文書)添付ファイル10.5を参照して編入)。 |
10.7# |
|
2019年株式インセンティブ計画(2019年7月8日に米国証券取引委員会に提出されたS-1表登録者登録説明書修正案第1号添付ファイル10.6(第333-232260号書類)を参照)。 |
10.8# |
|
2019年株式インセンティブ計画下の株式オプション協定表(2019年7月8日に米国証券取引委員会に提出された登録者S-1表登録説明書修正案第1号添付ファイル10.7(第333-232260号文書)を参照して編入)。 |
10.9# |
|
誘因株式オプションプロトコル表(参照登録者が2021年11月4日に米国証券取引委員会の10-Q表四半期報告書の添付ファイル10.1に提出する)。 |
10.10# |
|
2019年株式インセンティブ計画下の限定株式単位プロトコル表(添付ファイル10.10を参照して登録者に組み込まれ、2022年3月3日に米国証券取引委員会の10-K表年次報告書に提出される)。 |
10.11# |
|
2019年従業員株購入計画(2019年7月8日に米国証券取引委員会に提出されたS-1表登録者登録声明修正案第1号添付ファイル10.8(第333-232260号文書)を参照)。 |
10.12# |
|
2022年インセンティブ株式インセンティブ計画(添付ファイル10.12を参照して登録者に組み込まれ、2022年3月3日に証券取引委員会の10-Kフォーム年次報告書に提出される)。 |
10.13*# |
|
“2022年株式インセンティブ計画第1修正案”。 |
10.14# |
|
2022年インセンティブ株式インセンティブ計画下の非法定株式オプション協定表(添付ファイル10.13を参照して、2022年3月3日に証券取引委員会に提出された登録者年次報告Form 10-Kに組み込まれる)。 |
10.15# |
|
2022年インセンティブ株式インセンティブ計画下の制限株式単位プロトコル表(添付ファイル10.14を参照して、2022年3月3日に証券取引委員会に提出された登録者年次報告Form 10-Kに組み込まれる)。 |
10.16*# |
|
非従業員役員報酬プログラムの概要。 |
10.17# |
|
上級管理者採用契約表(2019年7月8日に米国証券取引委員会に提出された登録者登録説明書第1号修正案添付ファイル10.12(第333-232260号文書)を参照)。 |
10.18*# |
|
手紙協定は,期日は2023年1月3日であり,登録者とロバート·J·グールドが署名した。 |
10.19*# |
|
別居協定は,期日は2023年1月2日であり,登録者とブライアン·スチュアートが署名した。 |
10.20# |
|
登録者とカーティス·ウルトラマンとの間で2020年10月29日に締結された雇用協定(添付ファイル10.16を参照して登録者に組み込まれ、2021年3月4日に米国証券取引委員会の10−K表年次報告書に提出される)。 |
10.21*# |
|
登録者とジュディス·ダンとの別居合意は、2022年10月14日。 |
10.22# |
|
登録者とMel Hayesとの間の雇用協定は、日付が2021年9月7日である(2022年11月8日に証券取引委員会に提出された登録者10−Q四半期報告の添付ファイル10.1を参照して格納される)。 |
10.23# |
|
登録者とEsther Rajaveluとの間で2022年1月3日に締結された雇用協定(2022年3月3日に証券取引委員会に提出された登録者10−K表四半期報告の添付ファイル10.1を参照して編入される)。 |
10.24*# |
|
登録者とサンディエゴ·アロヨの間で2022年11月7日に締結された雇用協定。 |
10.25# |
|
登録者とその執行者及び取締役との間の賠償協定表(登録者を参照して2019年6月21日に米国証券取引委員会に提出されたS−1表登録声明(第333−232260号文書)添付ファイル10.15を参照して編入)。 |
10.26 |
|
登録者、グラクソ·スミスクライン知的財産権(第2号)有限会社、グラクソ·スミスクライン有限責任会社とグラクソ·スミスクライン集団有限公司の間で2019年2月8日に署名された参照権及び許可協定(2019年6月21日に米国証券取引委員会に提出された登録者S-1表登録声明(第333-232260号文書)添付ファイル10.10を参照して合併)。 |
10.27 |
|
登録者、グラクソ·スミスクライン知的財産権(第2号)有限会社、グラクソ·スミスクライン有限責任会社とグラクソ·スミスクライン集団有限公司(参考登録者が2020年11月10日に米国証券取引委員会に提出した10-Q四半期報告書(第001-38978号文書)添付ファイル10.2を統合した)は、2020年9月23日に登録者、グラクソ·スミスクライン知的財産権(第2号)有限会社とグラクソ·スミスクグループ株式会社が署名した“参照権とライセンス契約第1修正案”である。 |
10.28 |
|
協力及び許可協定は、登録者と百時米施貴宝社の完全子会社MyoKardia,Inc.によって締結され、日付は2020年7月20日である(登録者を引用して2020年11月10日に米国証券取引委員会に提出された10-Q表四半期報告(文書番号001−38978)の添付ファイル10.1により編入)。 |
10.29 |
|
UP 26 Landsdown,LLCと登録者(2019年6月21日に米国証券取引委員会に提出された登録者S−1登録説明書(文書番号333−232260)添付ファイル10.11を参照して合併することにより)、日付は2017年11月22日のLandsdown Street 26の借約である。 |
10.30 |
|
登録者とPiper Sandler&Co.との間の持分割当協定は、2022年5月9日(登録者を参照して2022年5月10日に米国証券取引委員会に提出された8-K表の現在の報告書の添付ファイル1.1に組み込まれる)である。 |
21.1* |
|
登録者の子会社。 |
23.1* |
|
独立公認会計士事務所安永会計士事務所の同意を得ました。 |
31.1* |
|
2002年のサバンズ·オキシリー法第302節で可決された1934年の証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて発行された首席執行幹事証明書。 |
31.2* |
|
2002年サバンズ−オキシリー法第302節で可決された1934年証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて首席財務幹事が認証された。 |
32.1+ |
|
2002年にサバンズ·オクスリ法案第906条で可決された“米国法典”第18編1350条に規定されている最高経営責任者証明書。 |
32.2+ |
|
2002年の“サバンズ-オックススリー法案”906節で採択された“米国法典”第18編1350条による首席財務官の認証。 |
101.INS |
|
XBRLインスタンスドキュメント |
101.書院 |
|
XBRL分類拡張アーキテクチャドキュメント |
101.カール |
|
XBRL分類拡張計算リンクライブラリ文書 |
101.def |
|
XBRL分類拡張Linkbase文書を定義する |
101.介護会 |
|
XBRL分類拡張タグLinkbaseドキュメント |
101.Pre |
|
XBRL分類拡張プレゼンテーションLinkbaseドキュメント |
104 |
|
表紙インタラクションデータファイル(添付ファイル101に含まれる適用分類拡張情報を含むイントラネットXBRL形式) |
#は、管理契約または任意の補償計画、契約、またはスケジュールを表します。
*登録者が実質的ではなく、登録者が個人または機密とみなされる情報タイプであることが確認されたので、本展覧会のいくつかの部分は漏れています。
*アーカイブをお送りします。
+メールで提供されます。
サイン
改正された1934年“証券取引法”第13又は15(D)節の要求に基づいて、登録者は、正式に許可された次の署名者が代表して本報告書に署名することを正式に手配した.
|
|
支点治療会社 |
|
|
|
|
|
日付:2023年3月9日 |
|
差出人: |
ロバート·J·グールド |
|
|
|
ロバート·J·グールド博士 |
|
|
|
臨時総裁兼最高経営責任者 |
本報告書は、改正された1934年の証券取引法の要求に基づき、次の日に以下の者によって登録者として署名された。
名前.名前 |
|
タイトル |
|
日取り |
|
|
|
|
|
ロバート·J·グールド |
|
臨時総裁兼最高経営責任者 |
|
March 9, 2023 |
ロバート·J·グールド博士 |
|
役員(最高経営責任者) |
|
|
|
|
|
|
|
/s/Esther Rajavelu |
|
首席財務官(首席財務官) |
|
March 9, 2023 |
エスター·ラジャヴェール |
|
|
|
|
|
|
|
|
|
/s/Gregory Tourangeau |
|
財務総監(首席会計主任) |
|
March 9, 2023 |
グレゴリー·トゥランゴ |
|
|
|
|
|
|
|
|
|
ケイト·ハビラン |
|
取締役会議長 |
|
March 9, 2023 |
ケイト·ハビラン |
|
|
|
|
|
|
|
|
|
/s/Sonja Banks |
|
役員.取締役 |
|
March 9, 2023 |
Sonja Banks |
|
|
|
|
|
|
|
|
|
ジェームズ·J·コリンズ |
|
役員.取締役 |
|
March 9, 2023 |
ジェームズ·J·コリンズ博士 |
|
|
|
|
|
|
|
|
|
カティナ·ドートン |
|
役員.取締役 |
|
March 9, 2023 |
カティナ·ドートン |
|
|
|
|
|
|
|
|
|
/s/エレン·イゼコヴィッツ |
|
役員.取締役 |
|
March 9, 2023 |
エレン·イゼコヴィッツMBCHB D.Phil |
|
|
|
|
|
|
|
|
|
ジェームズ·ジェラティ |
|
役員.取締役 |
|
March 9, 2023 |
ジェームズ·ジェラティ |
|
|
|
|
|
|
|
|
|

