nlnk-2022123100011262342022会計年度誤り.1111P 3 YP 1 Y00011262342022-01-012022-12-3100011262342022-06-30ISO 4217:ドル00011262342023-03-03Xbrli:共有00011262342022-12-3100011262342021-12-31ISO 4217:ドルXbrli:共有0001126234US-GAAP:ライセンスとサービスメンバー2022-01-012022-12-310001126234US-GAAP:ライセンスとサービスメンバー2021-01-012021-12-310001126234US-GAAP:RoyaltyMember2022-01-012022-12-310001126234US-GAAP:RoyaltyMember2021-01-012021-12-3100011262342021-01-012021-12-310001126234アメリカ-アメリカ公認会計基準:普通株式メンバー2020-12-310001126234米国-公認会計基準:財務省株式公開金メンバー2020-12-310001126234US-GAAP:AdditionalPaidInCapitalMembers2020-12-310001126234アメリカ-公認会計基準:前払いメンバーを保留2020-12-310001126234アメリカ公認会計原則:他の総合収入メンバーを累計2020-12-3100011262342020-12-310001126234US-GAAP:AdditionalPaidInCapitalMembers2021-01-012021-12-310001126234アメリカ-アメリカ公認会計基準:普通株式メンバー2021-01-012021-12-310001126234米国-公認会計基準:財務省株式公開金メンバー2021-01-012021-12-310001126234アメリカ-公認会計基準:前払いメンバーを保留2021-01-012021-12-310001126234アメリカ-アメリカ公認会計基準:普通株式メンバー2021-12-310001126234米国-公認会計基準:財務省株式公開金メンバー2021-12-310001126234US-GAAP:AdditionalPaidInCapitalMembers2021-12-310001126234アメリカ-公認会計基準:前払いメンバーを保留2021-12-310001126234アメリカ公認会計原則:他の総合収入メンバーを累計2021-12-310001126234US-GAAP:AdditionalPaidInCapitalMembers2022-01-012022-12-310001126234アメリカ-アメリカ公認会計基準:普通株式メンバー2022-01-012022-12-310001126234米国-公認会計基準:財務省株式公開金メンバー2022-01-012022-12-310001126234アメリカ公認会計原則:他の総合収入メンバーを累計2022-01-012022-12-310001126234アメリカ-公認会計基準:前払いメンバーを保留2022-01-012022-12-310001126234アメリカ-アメリカ公認会計基準:普通株式メンバー2022-12-310001126234米国-公認会計基準:財務省株式公開金メンバー2022-12-310001126234US-GAAP:AdditionalPaidInCapitalMembers2022-12-310001126234アメリカ-公認会計基準:前払いメンバーを保留2022-12-310001126234アメリカ公認会計原則:他の総合収入メンバーを累計2022-12-310001126234NLNK:プライベートLumos株主メンバーアメリカ-アメリカ公認会計基準:普通株式メンバー2020-03-182020-03-180001126234アメリカ-アメリカ公認会計基準:普通株式メンバー2020-03-182020-03-18Xbrli:純0001126234米国-GAAP:シリーズAPReferredStockMembers2020-03-182020-03-180001126234アメリカ-アメリカ公認会計基準:シリーズBPferredStockMember2020-03-182020-03-1800011262342020-03-180001126234NLNK:元株主のメンバー2020-03-192020-03-190001126234NLNK:プライベートLumos株主メンバーNLNK:新しいリンク遺伝子のメンバー2020-03-192020-03-1900011262342020-03-182020-03-180001126234US-GAAP:ComputerEquipmentMembersSRT:最小メンバ数2022-01-012022-12-310001126234SRT:最大メンバ数US-GAAP:ComputerEquipmentMembers2022-01-012022-12-310001126234米国-公認会計基準:従業員株式オプションメンバー2022-01-012022-12-310001126234米国-公認会計基準:従業員株式オプションメンバー2022-12-310001126234米国-GAAP:制限株式単位RSUメンバー2022-01-012022-12-310001126234アメリカ公認会計基準:従業員ストックメンバー2022-01-012022-12-310001126234NLNK:AmmonettMember2018-01-012018-12-310001126234NLNK:AmmonettMember2018-12-310001126234NLNK:MerckSharpeand DohmeCorpMembersアメリカ-公認会計基準:連携性手配メンバー2022-12-310001126234NLNK:MerckSharpeand DohmeCorpMembersSRT:最小メンバ数アメリカ-公認会計基準:連携性手配メンバー2022-01-012022-12-310001126234SRT:最大メンバ数NLNK:MerckSharpeand DohmeCorpMembersアメリカ-公認会計基準:連携性手配メンバー2022-01-012022-12-310001126234NLNK:MerckSharpeand DohmeCorpMembersアメリカ-公認会計基準:連携性手配メンバー2014-11-300001126234NLNK:MerckSharpeand DohmeCorpMembersアメリカ-公認会計基準:連携性手配メンバー2020-03-180001126234US-GAAP:DisposalGroupeldForSaleOrDisposedOfBySaleNotDisContinedOperationsMemberNLNK:PRV転送プロトコルのメンバーNLNK:MerckSharpeand DohmeCorpMembersアメリカ-公認会計基準:連携性手配メンバー2020-07-27NLNK:分割払い0001126234NLNK:MerckSharpeand DohmeCorpMembersアメリカ-公認会計基準:連携性手配メンバー2020-07-270001126234US-GAAP:DisposalGroupeldForSaleOrDisposedOfBySaleNotDisContinedOperationsMemberNLNK:PRV転送プロトコルのメンバーNLNK:MerckSharpeand DohmeCorpMembersアメリカ-公認会計基準:連携性手配メンバー2020-07-012020-09-300001126234US-GAAP:DisposalGroupeldForSaleOrDisposedOfBySaleNotDisContinedOperationsMemberNLNK:PRV転送プロトコルのメンバーNLNK:MerckSharpeand DohmeCorpMembersアメリカ-公認会計基準:連携性手配メンバー2021-01-110001126234NLNK:MerckSharpeand DohmeCorpMembersUS-GAAP:ライセンスとサービスメンバー2022-01-012022-12-310001126234NLNK:MerckSharpeand DohmeCorpMembersUS-GAAP:ライセンスとサービスメンバー2021-01-012021-12-310001126234NLNK:MerckSharpeand DohmeCorpMembersUS-GAAP:RoyaltyMember2022-01-012022-12-310001126234NLNK:MerckSharpeand DohmeCorpMembersUS-GAAP:RoyaltyMember2021-01-012021-12-310001126234アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ公認会計基準:MoneyMarketFundsMembers2022-12-310001126234アメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ公認会計基準:MoneyMarketFundsMembers2022-12-310001126234アメリカ公認会計基準:MoneyMarketFundsMembers2022-12-310001126234アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-公認会計基準:会社債務証券メンバー2022-12-310001126234アメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-公認会計基準:会社債務証券メンバー2022-12-310001126234アメリカ-公認会計基準:会社債務証券メンバー2022-12-310001126234アメリカ-公認会計基準:公正価値入力レベル1メンバー米国-公認会計基準:外国会社債務証券メンバー2022-12-310001126234アメリカ-公認会計基準:公正価値入力レベル2メンバー米国-公認会計基準:外国会社債務証券メンバー2022-12-310001126234米国-公認会計基準:外国会社債務証券メンバー2022-12-310001126234アメリカ-公認会計基準:公正価値入力レベル1メンバー2022-12-310001126234アメリカ-公認会計基準:公正価値入力レベル2メンバー2022-12-310001126234米国-GAAP:ビジネス紙のメンバーアメリカ-公認会計基準:公正価値入力レベル1メンバー2022-12-310001126234米国-GAAP:ビジネス紙のメンバーアメリカ-公認会計基準:公正価値入力レベル2メンバー2022-12-310001126234米国-GAAP:ビジネス紙のメンバー2022-12-310001126234アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-公認会計基準:アメリカ政府機関債務証券メンバー2022-12-310001126234アメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-公認会計基準:アメリカ政府機関債務証券メンバー2022-12-310001126234アメリカ-公認会計基準:アメリカ政府機関債務証券メンバー2022-12-310001126234アメリカ公認会計基準:現金等価物メンバーアメリカ公認会計基準:MoneyMarketFundsMembers2022-12-310001126234アメリカ-公認会計基準:会社債務証券メンバーアメリカ公認会計基準:現金等価物メンバー2022-12-310001126234アメリカ公認会計基準:現金等価物メンバー米国-公認会計基準:外国政府債務証券メンバー2022-12-310001126234アメリカ公認会計基準:現金等価物メンバー2022-12-310001126234米国-GAAP:ビジネス紙のメンバー米国-GAAP:ShortTermInvestmentsメンバー2022-12-310001126234アメリカ-公認会計基準:アメリカ政府債務証券メンバー米国-GAAP:ShortTermInvestmentsメンバー2022-12-310001126234米国-GAAP:ShortTermInvestmentsメンバー2022-12-310001126234SRT:最小メンバ数2022-12-310001126234SRT:最大メンバ数2022-12-310001126234NLNK:A 2009株式インセンティブ計画メンバー2022-01-012022-12-310001126234NLNK:A 2009株式インセンティブ計画メンバー2019-05-092019-05-090001126234NLNK:A 2009株式インセンティブ計画メンバー2022-12-310001126234米国-公認会計基準:従業員株式オプションメンバー2021-12-310001126234米国-公認会計基準:従業員株式オプションメンバー2021-01-012021-12-310001126234米国-公認会計基準:従業員株式オプションメンバーSRT:最小メンバ数2022-01-012022-12-310001126234SRT:最大メンバ数米国-公認会計基準:従業員株式オプションメンバー2022-01-012022-12-310001126234米国-公認会計基準:従業員株式オプションメンバーSRT:最小メンバ数2021-01-012021-12-310001126234SRT:最大メンバ数米国-公認会計基準:従業員株式オプションメンバー2021-01-012021-12-310001126234米国-公認会計基準:制限された株式メンバー2021-12-310001126234米国-公認会計基準:制限された株式メンバー2022-01-012022-12-310001126234米国-公認会計基準:制限された株式メンバー2022-12-310001126234米国-公認会計基準:制限された株式メンバー2021-01-012021-12-310001126234NLNK:A 2010非従業員株式オプション計画メンバー2022-12-310001126234NLNK:A 2010従業員株式調達計画メンバー2022-12-310001126234NLNK:A 2010従業員株式調達計画メンバーアメリカ公認会計基準:従業員ストックメンバー2022-01-012022-12-310001126234米国-公認会計基準:研究·開発費メンバー2022-01-012022-12-310001126234米国-公認会計基準:研究·開発費メンバー2021-01-012021-12-310001126234アメリカ-公認会計基準:一般と行政費用メンバー2022-01-012022-12-310001126234アメリカ-公認会計基準:一般と行政費用メンバー2021-01-012021-12-310001126234アメリカ-GAAP:LoansPayableメンバーNLNK:アイオワ州経済発展部融資2005年2005-03-310001126234アメリカ-GAAP:LoansPayableメンバーNLNK:アイオワ州経済発展部融資2005年2022-12-310001126234米国-GAAP:国内/地域メンバー米国-公認会計基準:資本損失繰越メンバー2022-12-310001126234アメリカ-公認会計基準:研究メンバー2022-12-3100011262342021-10-012021-10-310001126234NLNK:IncomeTaxBenefitMember2021-10-012021-10-310001126234アメリカ公認会計基準:その他の収入メンバー2021-10-012021-10-310001126234アメリカ-公認会計基準:一般と行政費用メンバー2021-10-012021-10-31NLNK:投票00011262342022-08-160001126234NLNK:制御持分要約メンバー2020-12-302020-12-3000011262342020-12-300001126234NLNK:制御持分要約メンバー2020-12-300001126234NLNK:制御持分要約メンバー2022-01-012022-12-310001126234米国-公認会計基準:従業員株式オプションメンバー2022-01-012022-12-310001126234米国-公認会計基準:従業員株式オプションメンバー2021-01-012021-12-310001126234米国-公認会計基準:制限された株式メンバー2022-01-012022-12-310001126234米国-公認会計基準:制限された株式メンバー2021-01-012021-12-310001126234NLNK:首席医療官のメンバー2021-02-042021-02-0400011262342021-01-012021-03-310001126234SRT:首席財務官メンバー2021-04-162021-04-160001126234SRT:首席財務官メンバー2021-04-012021-06-300001126234SRT:首席財務官メンバー2021-12-31 アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表10-K
☒ 1934年証券取引法第13条又は15(D)条に基づいて提出された年次報告。
本財政年度末まで十二月三十一日, 2022.
あるいは…。
☐ 1934年証券取引法第13条又は15(D)条に基づいて提出された移行報告。
For the transition period from to .
手数料書類番号001-35342
ルモス製薬会社
(登録者の正確な氏名はその定款に記載) | | | | | | | | |
デラウェア州 | | 42-1491350 |
| (登録設立又は組織の国又はその他の管轄区域) | | (国際税務局雇用主身分証明書番号) |
| | |
| 4200マラソン大通り200号 | | |
オースティン, テキサス州 | | 78756 |
(CES主要行政官住所) | | (郵便番号) |
| | |
登録者の電話番号は市外局番を含んでいます (512) 215-2630 |
同法第12条(B)に基づいて登録された証券: | | | | | | | | |
| クラスごとのタイトル | 取引コード | 登録された各取引所の名称 |
| 普通株 | ルモ | ナスダック株式市場 |
同法第12条(G)により登録された証券:なし
登録者が証券法規則405で定義されている経験豊富な発行者である場合は、再選択マークで示してください。はい、そうですo 違います。 ☒
登録者が当該法第13条又は第15条(D)に従ってLE報告書を提出する必要がないか否かを再選択マークで示す. はい、そうですo 違います。 ☒
再選択マークは、登録者が(1)過去12ヶ月以内(または登録者が承認を要求されたより短い期間)に、1934年の証券取引法第13または15(D)条に基づいてイによって指導されなければならないすべての報告を指導したかどうか、および(2)このようなLing要求を過去90日以内に遵守してきたことを示すはい、そうです ☒ No o
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出および掲示を要求されたより短い時間)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示すはい、そうです ☒ No o
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい | | | | | | | | |
大型加速ファイルサーバo | | ファイルマネージャを加速する o |
| | |
非加速ファイルサーバ ☒ | | 規模の小さい報告会社☒ |
| | |
新興成長型会社☐ | | |
| | |
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守するo |
登録者がその監査報告を作成または発行したか否かを示す登録公共会計簿RMは、“サバンズ·オクスリ法案”(“米国法典”第15編7262(B)節)第404(B)節に基づいて財務報告の内部統制の有効性を評価した報告書を再選択マークで示し、その経営層が財務報告の内部統制の有効性を評価していることを証明している☐
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)。はい、そうです☐ No ☒
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用するo
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に第240.10 D−1(B)条に従って受信されたインセンティブベースの補償に従って回復分析を行う必要があるかどうかを再選択マークで示すo
登録者の非関連会社が保有する投票権と無投票権普通株の総時価に基づいて、登録者普通株の終値に基づいて、2022年6月30日、♪the the theナスダックの世界市場は1ドルです46.4百万ドルです
2023年3月3日までに8,219,292登録者の普通株は、1株当たり額面0.01ドルで、発行された。
引用で編入された書類
登録者が2023年5月10日に開催する2023年株主総会の最終依頼書は、2022年12月31日以降120日以内に提出され、一部は引用によって本年度報告書に組み込まれたForm 10-K第3部に提出される.
カタログ表
前向きな陳述に関する警告的声明 | | | | | | | | | | | |
| | | ページ |
| 第1部 | |
| 第1項。 | 業務.業務 | 5 |
| 第1 A項。 | リスク要因 | 25 |
| 項目1 B。 | 未解決従業員意見 | 64 |
| 第二項です。 | 属性 | 64 |
| 第三項です。 | 法律訴訟 | 64 |
| 第四項です。 | 炭鉱安全情報開示 | 64 |
| 第II部 | |
| 五番目です。 | 登録者普通株市場、関連株主事項及び発行者による株式証券の購入 | 65 |
| 第六項です。 | [保留されている] | 65 |
| 第七項。 | 経営陣の財務状況と経営成果の検討と分析 | 66 |
| 第七A項。 | 市場リスクの定量的·定性的開示について | 75 |
| 第八項です。 | 財務諸表と補足データ | 76 |
| 第九項です。 | 会計と財務情報開示の変更と相違 | 98 |
| 第9条。 | 制御とプログラム | 98 |
| プロジェクト9 B。 | その他の情報 | 98 |
| プロジェクト9 Cです。 | 検査妨害に関する外国司法管区の開示 | 97 |
| 第三部 | |
| 第10項。 | 役員·幹部と会社の管理 | 99 |
| 第十一項。 | 役員報酬 | 99 |
| 第十二項。 | 特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 | 99 |
| 十三項。 | 特定の関係や関連取引、取締役の独立性 | 99 |
| 14項です。 | 最高料金とサービス | 99 |
| 第IV部 | |
| 第十五項。 | 展示·財務諸表明細書 | 100 |
| 第十六項。 | 表格10-Kの概要 | 101 |
サイン | |
前向きに陳述する
本2022年12月31日現在の10-K表年次報告(“本年度報告”)には、改正後の1933年証券法(“証券法”)第27 A条と改正後の1934年証券取引法(“取引法”)第21 E条の定義に適合するいくつかの前向き記述が含まれている。これらの展望的陳述はリスクと不確定要素に関連し、現在の未来の事件と私たちの財務業績に対する私たちの見方を反映している本報告で用いた場合,これらの語は“信じる”“可能”“可能”“将”“予想”“継続”“予想”“予定”“期待”“求める”“求める”“すべき”“将”“および類似した表現すべての前向き陳述がこれらの識別語を含むわけではないにもかかわらず、前向き陳述を識別することが意図されているこれらの展望的陳述は歴史的事実ではなく、私たちの業界の現在の予想、推定と予測、経営層の信念と経営陣に対するいくつかの仮定に基づいており、その中の多くの仮説は本質的に不確定であり、私たちのコントロールを超えている。したがって、このような前向きな陳述はいずれも将来の業績の保証ではなく、予測困難なリスク、仮説、見積もり、不確実性の影響を受ける可能性があることを想起させます。本年度報告が発表された日までに,これらの前向き陳述に反映される予想は合理的であると考えられるが,実際の結果は前向き陳述で明示的あるいは示唆された結果とは大きく異なる可能性がある
実際の結果が前向き陳述における結果と大きく異なることをもたらす可能性のある重要な要素は、以下の概要を含むが、これらに限定されない
•我々のLum−201試験の最終結果は、2022年11月14日に発表されたこのような試験の中期結果と実質的に異なる可能性がある
•新冠肺炎(“新冠肺炎”)を招く新型コロナウイルスSARS-CoV-2株及び任意の関連する低迷、政府法規或いは制限は著者らの研究、臨床試験、製造と財務状況を含むわが業務にどの程度不利な影響を与える可能性があるか
•ロシアとウクライナ間の軍事衝突、およびいかなる関連する経済低迷、政府法規または制限は、私たちの研究、臨床試験、製造、財務状況への影響を含む、私たちの業務に悪影響を及ぼす可能性がある程度である
•マクロ経済環境の弱まり、高インフレ率、及び私たちの業務への影響、私たちの運営コストと財務状況への影響を含む
•私たちの候補製品である成長ホルモン分泌促進剤イブナモレン(“Lum−201”)の開発計画
•候補製品の潜在的な利益、活性、有効性、および安全性に対する私たちの期待
•私たちの既存のパイプの開発計画と潜在的なパートナーシップと対外許可の機会
•計画中の臨床前研究と臨床試験の時間およびこのような臨床試験の臨床データの有用性;
•私たちの候補製品のための規制承認のタイミングと能力
•私たちの候補製品の臨床的実用性は
•私たちは既存の技術を利用してより多くの候補製品を発見し開発する予定です
•知的財産権の地位は
•私たちは戦略的協力、許可、または他の計画を達成する能力を達成する
•私たちは協力パートナーに依存して規制の承認を得て、協力中の候補製品を商業化します
•費用、将来の収入、資本需要、追加融資需要の推定
•私たちの候補製品を開発し商業化する計画です
•私たちは私たちの業務のために追加的な資金を得ることができる
•承認された候補製品の市場受容率と程度
•承認された候補製品の商業化
•私たちの業務、技術、候補製品に対して、私たちの業務モデルと戦略計画を実施します
•私たちは第三者に依存して臨床前研究や未来の臨床試験を行います
•合格した重要な管理と技術者の能力を吸引し、維持する
•株式買い戻しの金額と時間(ある場合);
•私たちは第三者供給と製造パートナーに依存して、私たちの研究開発、臨床前、臨床試験製品の供給に材料とコンポーネントを提供し、製造します
•私たちの競争相手や私たちの産業と関連した発展
我々の実際の結果が我々の予測結果とは異なる既知の重要要素をもたらす可能性のあるより多くの情報については,(1)第1部,第1 A項を読んでください。本年報の“リスク要因”、“2)我々が時々米国証券取引委員会(”米国証券取引委員会“)に提出した報告書及び登録声明、および(3)我々が時々発表する他の公開公告。このような不確実性を考慮して、あなたはこのような前向きな陳述に過度に依存してはいけない。法的要求がない限り、私たちは未来に新しい情報があっても、これらの前向きな陳述を任意の理由で更新または修正する義務がない。
第1部
プロジェクト1.ビジネス
概要
Lumos製薬会社は臨床段階の生物製薬会社である。本年度報告では“私たち”“私たち”“私たちの”““会社“又は”Lumos“とは、Lumos Pharma,Inc.及びその完全子会社を意味する。私たちの主な実行事務室はテキサス州オースティンに位置し、他の実行と行政事務室はアイオワ州エイムズに位置し、著者らは著者らの臨床計画を推進することに力を入れ、そして全世界範囲内で稀な疾病を有する患者のために新製品と新療法を識別、獲得、開発、商業化することに集中し、現在安全で有効な治療に対する巨大な需要はまだ満足されていない。我々の普通株はナスダック世界市場(“ナスダック”)に上場し、株式コードは“LUMO”である
我々は我々の唯一の候補製品である成長ホルモン分泌促進剤イブナモロン(“Lum−201”)の開発に集中し,特発性児童成長ホルモン欠乏症(“PGHD”)や他のまれな内分泌疾患の治療に用いる潜在的な経口療法である。
PGHDはまれな内分泌疾患であり,約3500名から17歳まで生まれた人に1人が発生する。PGHDの原因は,先天性(このような疾患を有する小児),後天的(脳腫瘍,頭部損傷あるいは他の原因),医原性(医療による)または特発性(原因不明)であってもよい。未治療のPGHD児童は顕著な成長発育障害があり、潜在的な成人の身長は5フィートより明らかに低く、そして身体成分の異常があり、骨鉱物質が減少し、痩せ体重が減少し、脂肪の質が増加する可能性がある。
PGHDの主要な治療目標は成長を回復させ、小柄な児童を正常な身長に達成させ、そして代謝異常、認知欠陥と生活の質低下に関連する合併症を防止することである。現在、PGHD治療の標準は毎日組換えヒト成長ホルモンを皮下注射することに限られ、治療周期は平均7年に達する。治療期間中、組換えヒト成長ホルモンを毎日注射する依存性が悪いことは成長に不利な影響を与える可能性がある。2021年8月、FDAは、Skytrofa、週1回の注射は、治療中の患者の注射回数を減少させる新しい治療法を承認した;しかし、私たちの市場研究によると、多くの提供者と患者は経口治療に傾向があると信じている
二零二年三月十八日、当社はNewLink Genetics Corporation(“NewLink”)、NewLink全資子会社Cyone Merger Sub,Inc.(“Merge Sub”)とLumos Phとの業務統合(“合併”)を完了したArma,Inc.(“個人ルモス”)、後に“Lumos Pharma Sub,Inc.”と改名しました。合併子会社はPrivate Lumosと合併してPrivate Lumosに合併し、Private Lumosは当社の完全子会社として存続する。合併完了直前、NewLink普通株の株式は9株1株の逆分割比率で調整された。合併条項によると、Lumosプライベート株主は合計4,146,398株のNewLink普通株を獲得した(逆分割実施後)。後に続く逆株式分割と合併が完了した時点で、私たちの普通株は8,292,803株の流通株があり、その約50%はPrivate LumosとNewLink証券所有者がそれぞれ持っている。今回の合併は逆資産買収とみなされている
戦略.戦略
我々の戦略は,世界規模でまれな疾患を治療する新製品を識別,取得,開発して商業化し,選定された市場で直接商業化することを優先し,米国から始まり,他の市場でのパートナー関係や許可手配を求めることである。私たちのビジネス戦略の重要な構成要素は
•治療の選択が限られているか、または選択されていない、まれな疾患に注目し続けた
•疾病と治療を重視し、明確な病理生理と作用機序がある
•私たちの経験と関係を利用して、学術機関、珍しい患者基金会、および/または他の製薬会社とのパートナーに将来性のある製品候補許可を提供する
•創造性、適応性、迅速な臨床的および規制的実行を重視し、
•可能な場合には、長期価値の最大化を達成するために、候補製品を保持する世界的または広範な商業化権利が求められる。
希少病患者、彼らの家族、そして希少病コミュニティに対する責任感の推進の下で、私たちの目標は先進的な希少病薬物会社になることである。
LUM−201成長ホルモン分泌促進剤
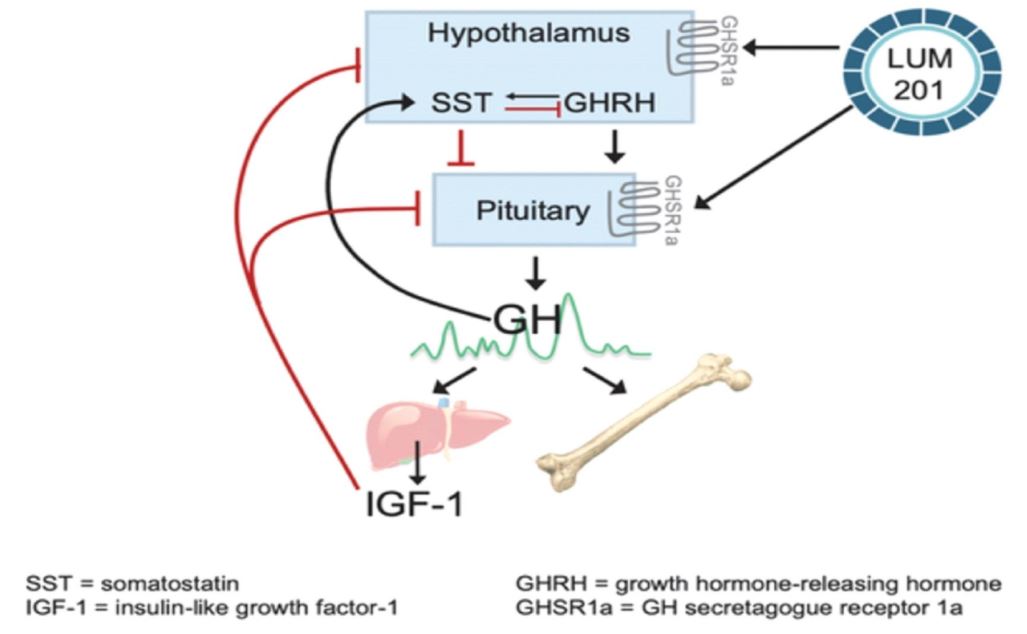
著者らの研究の重点は1種の経口小分子Lum-201を開発することであり、それは成長ホルモン(GH)分泌促進剤であり、ibuamorenとも呼ばれ、稀な内分泌疾患の治療に用いられ、現在組換えヒト成長ホルモン(“rhGH”)の注射が許可されている。Lum-201は錠剤製剤であり、1日1回投与する
Lumosは2018年7月にAmmonett Pharma LLC(“Ammonett”)からLum-201を買収した。Lum−201は2017年に成長ホルモン欠乏症(GHD)により米国とEUで孤児薬物名(“ODD”)を獲得した。米国特許“成長ホルモン欠乏症の検査と治療”は2036年に満期になった。関連特許はすでにEU(検証中)、オーストラリア、イスラエル、日本、韓国、香港、ウクライナで発行されており、関連特許出願は他の複数の司法管轄区で決定されている。承認されれば,Lum−201は最初に承認された経口成長ホルモン分泌促進剤となる可能性があり,PGHDから成長ホルモン欠乏に関連するまれな内分泌疾患の治療に用いられ,現在の組換え成長ホルモン製品注射の標準案に代替案を提供している。分泌促進剤は別の物質の分泌あるいは放出を刺激する物質である。LUM−201は成長ホルモンの放出を刺激し,成長ホルモン分泌促進剤と呼ばれる
Lum-201はGH分泌促進受容体(GHSR 1 A)を通じてGHを刺激し、グレリン受容体とも呼ばれ、そしてソマトスタチンの放出を抑制し、それによって1種の差別化された作用機序を提供し、内因性、脈動性GH分泌の幅を増加させることによっていくつかの稀な内分泌疾患(GH欠乏に関連する)を治療する。LUM-201の刺激作用はGH循環レベル及び下流メディエーターインスリン様成長因子(IGF-1)によって調節され、IGF-1は超生理レベルでフィードバック或いは負方向に下垂体の更なるGH放出を調節し、それによって下垂体を過剰刺激から保護する
PGHD群は、器質性PGHD(より深刻なGH欠損症)および特発性PGHD(あまり深刻ではないGH欠損症)と診断された患者を含む。Lum−201は、機能が正常であるが視床下部GH軸が低下する患者の内因性GH分泌を刺激することが観察されており、中等度または特発性PGHD患者とも呼ばれる。特発性PGHD患者(すなわちそれらの機能は正常であるが視床下部GH軸が低下した患者)はPGHD患者の約60%を占めており,Lum−201に反応すると考えられている
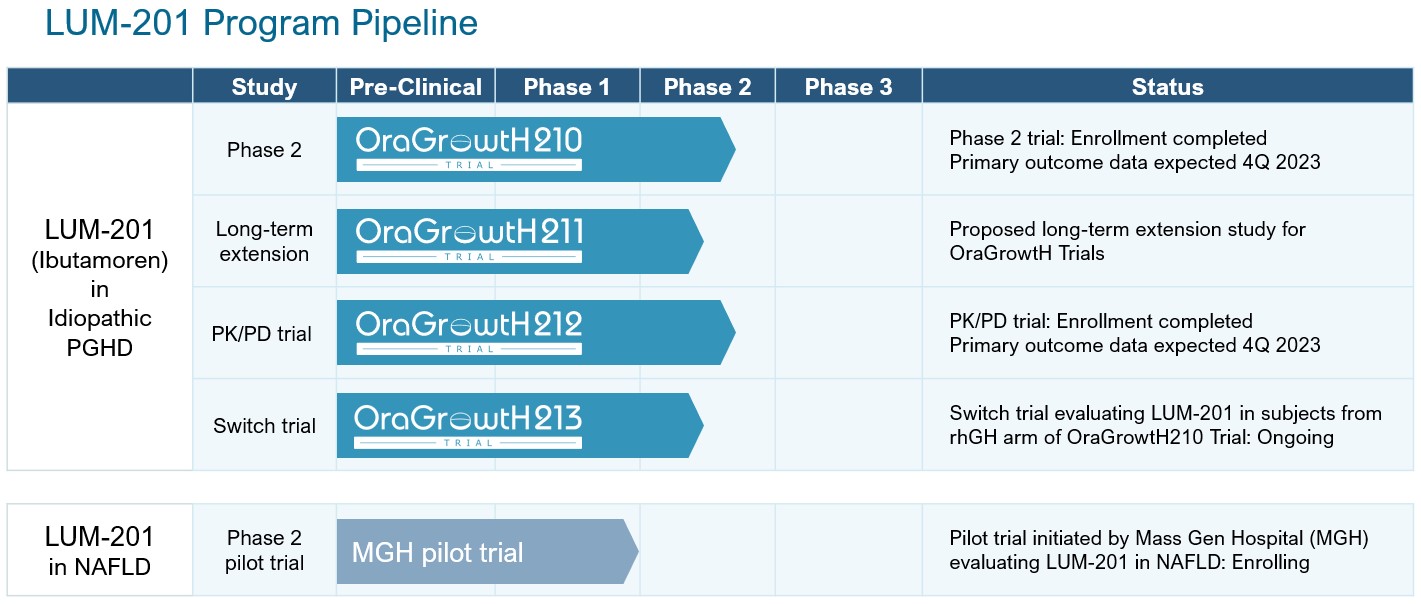
2020年第4四半期に、著者らはPGHDにおけるLum-201の作用を研究する計画を開始し、著者らの第2段階臨床試験(OraGrowth H 210試験あるいは“第2段階試験”)を開始し、この研究に参加する初期サイトを開放した。OraGrowth H 210試験は世界的な多地点ランダム試験であり、特発性PGHDと診断された約80名の被験者が3種類の用量レベル(0.8、1.6と3.2 mg/kg/日)を経口投与したLum-201と毎日標準用量を注射した組換え成長ホルモンの治療効果を評価した。
この研究の主な終点は,われわれの予測的濃縮マーカー(“PEM”)患者の選択戦略の初歩的な検証であり,Lum−201に反応する選択された患者のパーセンテージから証明された。著者らの第二段階の臨床試験では、各患者は1日0.8 mg/kgの単用量のLUM-201を投与され、彼らが登録のカットオフ基準、すなわちベースラインインスリン様成長因子-1>30 ng/mlと刺激成長ホルモン5 ng/mlに適合するかどうかを決定した。主要な治療効果の終点は年身長速度である。二次終点は、将来の研究のための小児科用量のLum-201を選択することを含み、第三段階を含み、PEM陽性患者がOraGrowth H 210スクリーニングに参加するPEM選択過程の再現性の程度を決定することを含む
我々は2023年2月28日にOraGrowth H 210試験の登録を完了し、82名の被験者を発表し、2023年第4四半期の全82名の被験者の完全な6ヶ月の主要な結果データを予定している。
PGHDにおけるLum-201の第2の同時試験(“OraGrowth H 212試験”)は2021年第2四半期に開始し、成長ホルモン脈動性分泌の拡大におけるLum-201の新しい作用機序の効果を探索することを目的とした。PGHDで行ったOraGrowth H 212試験はOraGrowth H 210試験と並行して行った。OraGrowth H 212試験は単一地点、開放ラベル試験であり、Lum-201の2つの異なる用量レベル(1.6と3.2 mg/kg/日)で24名のPGHD被験者に及ぶ薬物動態学と薬効学(“PK/PD”)効果を評価した。OraGrowth H 212試験の目的は以前の臨床データを実証することであり、LUM-201特有の内因性成長ホルモンパルス式放出増加及びその対の潜在力を説明し、この作用機序は治療効果の向上に役立つ。我々のOraGrowth H 212試験は専門の小児科センターで行われ、このような臨床試験に必要なより頻繁なサンプル採集とモニタリングを行うことができる。OraGrowth 212試験のデータは、将来の規制申告文書でサポートされる可能性があるが、この試験は、規制部門がLum-201を承認するために必要なものではない。この実験の主な終点は6カ月のPK/PDと身長速度である
24人もの被験者のデータです我々が2023年2月28日に発表したように,我々はこの試験の22名の被験者の登録を完了しており,全22名の被験者の予備データは2023年第4四半期に発表される予定である
2021年7月16日のFDAの手紙の要求に応えるために、私たちは2021年7月に、FDAがLum−201の治療を12ヶ月以下に制限したと発表した。その際,我々が最初に計画していた長期延長試験(“OraGrowth 2 11試験”)を開始する前に,PGHDにおけるLum−201の追加効果データを収集するために,OraGrowth H 210とOraGrowth H 212試験の治療期間を6カ月から12カ月に延長した。2022年5月10日に発表されたように,OraGrowth H 210とOraGrowth 212試験の初歩的な安全性と有効性データを審査した後,FDAは部分保留をキャンセルし,現在Lum−201による12カ月以上の治療を許可している。そのため、OraGrowth H 210試験は24ケ月まで延長し、被験者が途切れることなくLum-201治療を継続できるようにした。また,2022年第2四半期にOraGrowth 212試験の案が改正され,被験者が女性14歳と男性16歳の骨齢に達するまで治療を許可され,成人に近い身長を反映していた
治療期間の延長はOraGrowth H 210やOraGrowth H 212試験の主要な結果データに影響を与えず,これらのデータは治療前6カ月の経年化データに基づく。我々は引き続きOraGrowth H 210試験を延期した場合にLong−Term Expansion Studyの開始時間を評価し,適切な時期に延期研究を開始する予定である。これらの案の変化は起こらず,Timeを単独で延長してOur3臨床試験を開始する予定である。
2022年第1四半期に、我々のOraGrowth H 213試験(“OraGrowth H 213試験、およびOraGrowth H 210試験、OraGrowth H 211試験およびOraGrowth H 212試験、すなわち”OraGrowth H試験“)を開始し、開放的で多中心的な第2段階研究であり、OraGrowth H 210試験を完成した20名までの特発性PGHD患者に対するLum-201の成長効果と安全性を評価した。被験者はLum-201を3.2 mg/kg/日の用量レベルで服用し、最長12カ月に達する
次の図はLUM-201の臨床発展計画を示している
LUM-201の潜在的拡張は他の適応に
2022年5月、マサチューセッツ州総病院(MGH)との臨床協力を発表し、研究者による試験において、経口Lum-201による非アルコール性脂肪性肝疾患(NAFLD)の治療を評価した。この試験はNAFLDを有する男性10名と女性1日25 mgのLum−201用量を評価する。試験の登録が始まり,最初の被験者は投与量を服用していた。GHは脂肪分解の重要な刺激因子であり,臨床前データではGH分泌の増加が肝臓脂肪変性を減少させ,NAFLDの進展を防止する可能性が示唆された。短期臨床研究により、成長ホルモンの自然脈動放出を促進することは脂肪分解を誘導する上で成長ホルモンの持続注入よりも有効であることが示唆された。主な終点は,成長ホルモンの増加に伴う肝内脂肪含量,肝臓炎症,線維化の変化をH−MRSと透視社のLiverMultiScanにより決定することである。一部の被験者に生検を行い,この適応における遺伝的および細胞レベルの追加情報を得る。
私たちはこの研究の能動的な支出申請を承認し、この試験試験にLUM-201を提供する。Lumosは、NAFLDにおけるLUM−201の使用方法特許を出願しており、この適応におけるLUM−201の知的財産権を保持している
私たちはLUM-201の他の適応に拡大するために私たちの発展の道を探索し続けた。我々は、他の潜在的適応の影響を受ける患者のサブセットにおけるLum−201の作用機序を積極的に検討している。我々のこれまでの予備検討によると,特発性低さ,アジア市場,Prader Willi症候群に重点を置いた次の適応への注目範囲を縮小し,魅力的な世界的なチャンスを見た。PGHD用資源の優先順位を決定し,OraGrowth 210のデータ読み出し後にPrader Willi症候群と特発性低身長の指標をあらかじめ計画しておく予定である
LUM−201の作用機序
メルク社が最初に開発したLum-201はGH分泌促進剤であり、GHSR 1 a、特に下垂体前葉と視床下部に選択的に作用し、GHの過剰放出を刺激する。Lum−201はマウス、ラット、犬、豚、ヒトの経口投与後に刺激性成長ホルモン反応を示した。LUM−201の作用機序は以下のとおりである。
Lum−201に結合したGHSR 1 a活性化によりGH放出が誘導され,体外ラット下垂体細胞(Lum−201 EC 50 1.3 nM)で証明されているように。また、Lum-201で下垂体細胞を処理することは外因性GHRHのGH分泌に対する影響を増強することができ、この2種類の化合物は協同でラット下垂体細胞GHの放出を刺激し、異なる作用機序を示した。
珍しい病気の焦点
患者を中心としたまれな疾患薬物開発がLumosの基本的な注目点である。我々は,最も緊急かつ最も慎重な方法で潜在的な治療法の開発に取り組んでおり,早期かつ常に患者と接触することで,候補製品を承認することで,薬物開発過程の連続過程で彼らの観点をよりよく知ることができる。我々は,我々のパートナーや利害関係者において患者中心の理念を促進することに取り組んでおり,疾患意識の向上,より良い診断方式や経路の実現,患者や医療コミュニティの支援など,患者を中心とした連携プロジェクトを開始している。
市場需要
米国では,約3500人の子供に1人が出生時にPGHDを患っている。PGHDを有する児童の特徴は小柄、代謝異常、認知欠陥と生活の質が悪いことである。現在のPGHDの看護基準は
組換えヒト成長ホルモンを毎日皮下注射することは,1985年にさかのぼり,1950年代以来ドナー由来の成長ホルモンを使用している。2021年8月、FDAは、治療中の患者の注射回数を減少させることができる競争相手の治療方法Skytrofaを承認した;しかし、私たちの市場研究は、多くの提供者と患者が毎日経口治療を行う傾向があることを示している。
毎日注射治療方案を完全に遵守したGH欠陥児童は成人後にその家族と国家標準に相当する身長に達する可能性がある。組換えヒト成長ホルモン治療は利点を示しているにもかかわらず、コンプライアンスは依然として挑戦であり、毎日組換え人成長ホルモン治療を受けている患者は通常数年の過程で数千回の注射を受けるからである。長年組換え成長ホルモンを毎日注射しなければならない可能性のある幼児や青少年の介護者にとって,針疲労の問題−日常治療の痛み,鬱傷あるいはその他の影響で注射できない−依然として日常治療を守らない重要な原因の1つである。
製薬業は多種の方法を用いて組換えヒト成長ホルモン製品を開発し、患者の毎日注射の負担を軽減し、そして患者の用量方案に対する依存性を高め、比較的に少ない頻度注射を必要とする長時間効果成長ホルモン治療、例えばSkytrofaを含む。経口治療は特発性PGHD患者が現在の看護標準ではなく、より良い治療コンプライアンスを通じてより良い治療結果を得ることを助けると信じている。
Lum−201は、特発性であり、機能性を有するが視床下部GH軸が減少するPGHD患者の内因性GHの放出を刺激するためのものである。これらの基準を満たす割合は全PGHD患者の約60%を占めていると考えられる。承認されれば、Lum-201は最初に承認された経口成長ホルモン分泌促進剤となる可能性があり、PGHDから成長ホルモン欠乏に関連する稀な内分泌疾患の治療に用いられ、現在の標準毎日注射方案に代替方案を提供した。
PGHD以外に、アメリカ食品と薬物管理局(FDA)は以前Prader Willi症候群と特発性低身長を含むrhGH治療の多種の他の適応を許可した。約15,000人に1人がブレード·ウィリー症候群を患っている。FDAは児童低さをこの年齢層の平均身長より2.25個低い標準偏差と定義し,対応する人口の1.2%に相当する。60%から80%の人が特発性低さと定義されていると推定されています1つまり2022年には米国では約39万人から52万人の子供が推定される。Prader Willi症候群や特発性低身長を含むいくつかの他の適応におけるLUM−201の安全性と有効性を会社の優先順位と資金源に基づいて調査する予定である
商業化戦略
市場独占経営権や特許保護を得ることができる市場でPGHD用LUM−201を商業化する予定であり、規制マーケティング許可(MA)を取得し、必要な費用が合理的であることを証明するのに十分な強力な製品販売を前提としている。Lum-201の初期市場には米国とEUが含まれると予想され、両国とも承認された孤児疾患製品に独占マーケティングを提供する。私たちはこの二つの地域で奇数を受け取ったが、承認されれば、これはこのような排他性を得るために必要な構成要素だ。私たちはまた中国、日本、韓国を含む他の市場を狙うかもしれない。私たちは適切な時にもっと多くの国でODDを求めるつもりだ。中国のようないくつかの地域は、奇妙な排他性を提供しない。後発医薬品市場がこれらの市場に侵入することを防ぐために,我々の既存のPEM特許と将来可能な特許出願により特許保護を求める.
私たちは現在販売、製造、生産、または流通能力を持っていない。我々は、LUM−201および任意の他の候補製品を1つまたは複数の地域で製造、生産、マーケティング、および販売することで、第三者と合意したい。私たちの1つ以上の候補製品が規制部門の承認を得た場合、我々の候補製品を共同で普及および/または商業化するために、技術的専門性と流通支援能力を有する専門販売組織を構築する予定である。直接販売チームを持ち、製造、生産、流通システムを構築した第三者と協力して、自分たちの販売チームやシステムを強化したり、自分たちの販売チームやシステムの代わりにしたりすることを選択することができます
まれな疾患を有する患者は通常少数の専門医によって治療される。したがって、私たちのビジネス構造は適度であり、患者の識別、診断、患者と医療提供者への援助を加速し、治療と精算支援に関連する市場参入を支援するための支援プロジェクトに重点を置いていると予想される。
1 インザジら、“小児科雑誌”2019;92:71-83
競争
新しい治療製品の開発と商業化競争が激しい。私たちはLum-201方面で競争に直面しており、将来、開発または商業化を求める可能性のある任意の候補製品は、世界各地の主要な製薬会社、専門製薬会社、バイオテクノロジー会社からの競争に直面する可能性がある。現在いくつかの大手製薬やバイオテクノロジー会社が私たちの目標患者集団に再編ヒト成長ホルモン療法をマーケティング·販売している。これらの会社は通常成熟しており、私たちの目標適応の範囲で豊富な経験を持っている。潜在的な競争相手には、学術機関、政府機関、および他の研究、特許保護を求め、研究開発、製造および商業化のための協力手配を確立する公的および個人研究機関、および化合物または他の適応のためのLum−201化合物を不正に販売する可能性のある製造業者および販売業者も含まれる。これらの競争相手の多くは私たちの目標適応のための療法を開発しようとしている。
私たちは、1日1回の体重ベースの経口用量レジメンに基づいてPGHD患者のサブセットを治療するための候補製品Lum−201を開発している。米国では,現在の成長療法患者に対する看護基準は,組換えヒト成長ホルモンを毎日皮下注射することである。現在多くの現在市販されている組換えヒト成長ホルモン療法は、成長ホルモン欠乏症の治療に毎日皮下注射されており、主にノードロピン(ノとノードA/S社)、Humtrepe(礼来社)、Nutroin-AQ(F.Hoffman-La Roche Ltd./遺伝子テーク社)、ジノロピン(ファイザー)、サイザン(メルクSerono S.A.)、Tev-troin(Teva製薬工業株式会社)、Omnitrop(Sandoz GmbH)、Valtroin(LG生命科学と生物製薬会社)、Zomacton(Ferring)、Skytrofa(Sandoz GbH)、Valtroin(LG生命科学と生物製薬会社)、Zomacton(Ferring)、Skytrofas(Sandoz GmbH)、Valtroin(LG生命科学と生物製薬会社)、Zomacton(Ferring)、Skytrofa(Sandoz GbH)、Valtroin(LG生命科学と生物製薬会社)、Zomacton(Ferring)、Skyrofroa(Sandoz GbH)、Valtroin(LG生命科学と生物製薬会社)、Zomacton(Ferring製薬)、Sknitrop(Sandoz GbH)、Valtroin(LG生命科学と生物製薬会社)、Zomacton毎週皮下注射で投与する。これらの組換えヒト成長ホルモン薬は,万托品やSkytrofaを除いて古くから知られている療法であり,医師,患者,介護者,第三者支払人,薬局福祉マネージャーに広く受け入れられ,成長ホルモン欠乏症治療の標準看護となっている。医師、患者、第三者支払者、およびPBMは、Lum-201を使用することによる患者への追加的な利点が限られていると考えられること、または現在利用可能なrhGH治療と比較して長期安全性データが限られていることを含む、様々な潜在的理由のために、Lum-201を彼らの現在の治療レジメンに追加することを受け入れない可能性がある。
現在承認·販売されている日常組換えヒト成長ホルモン療法のほか,すでに組換えヒト成長ホルモン市場に参加している会社や潜在的な新規参入者には,臨床開発の異なる段階にある様々な実験的療法や設備があり,主にノボノード社,OPKO Health,Inc.(ファイザー社と協力)とGenexine Inc.である。
知的財産権
米国特許番号9763919,10898472,10105352および11234969およびヨーロッパ特許番号3352752“成長ホルモン欠乏症の検出および治療”を取得した。これらの特許は2036年までに米国やヨーロッパでは満期にならず、オーストラリア、イスラエル、日本、韓国、香港、ウクライナも関連特許出願を発表し、関連特許出願は他の複数の管轄区域で懸案されている。より具体的には、Ammonettはすでにブラジル、カナダ、中国、欧州特許庁、ニュージーランド、およびシンガポールで関連特許出願を提出している(このような特許出願は現在Lumos所有)。LUM−201の物質組成特許は満了しており,LUM−201の化学構造は公有分野にある。しかし,成長ホルモン欠乏症に対する米国の使用方法特許(その他の地域で待っている類似出願)を取得している。
米国特許9763919号、10105352号、および11234969号の請求項は、小児成長ホルモン欠乏症の治療方法におけるLUM-201(従来のMK-0677)の使用に関する。これらの特許は、一部の機能の視床下部-下垂体オーキシン軸に関連するいくつかのPEMを患者に満たすことを要求する。
また、特許出願PCT/US 19/017964の独占的権利を有しており、“非アルコール性脂肪性肝炎および非アルコール性脂肪性肝炎を治療する組成物”というタイトルである。米国の申請は2019年2月に非臨時申請に転換された。私たちは協力を求め、将来的にこれらの適応のためのLUM-201を開発することを選択するかもしれない。
2022年11月、“イブラモロンの圧縮性経口製剤”と題する特許出願PCT/US 22/050700を提出した。申請は現在待っており、その中には、第3段階試験で使用し、最終的に商業化しようとしているいくつかの改善された処方に対するクレームが含まれている。承認された場合、この特許は、Lum−201の商業化バージョンを2042年11月の物質組成保護に提供するであろう。
許可と資産購入協定
2018年7月、吾らはAmmonettと資産購入プロトコル(“APA”)を締結し、Ammonettを買収して二零一三年十月にMerck Sharpe and Dohme Corp.(“Merck”)(“Lumos Merckプロトコル”)から許可を得たLum-201に関するほとんどの資産を取得した。
Lumos Merckプロトコルは、Lumos(Ammonettの権益継承者として)に世界範囲内で独占的、再許可可能(米国、ヨーロッパの主要国と日本でのメルク社の同意を得なければならず、このような同意は無理に抑留されてはならない)の特定の特許とノウハウ権利を付与し、“精神疾患診断と統計マニュアル”第5版で定義された自閉症スペクトラム障害を含まない任意およびすべての適応のために開発、製造および商業化することができる
2020年8月12日、メルク社とLumos Merck協定第1号修正案(“Lumos Merck合意修正案”)を締結した。 Lumos Merckプロトコル修正案によると,Merckから世界的に排他的で再許可可能な(Merckの米国,指定された主要ヨーロッパ諸国と日本の同意を得る必要があり,このような同意は無理に抑留されてはならない)許可を得ており,これらの許可は我々が診断目的のために開発,製造,商業化Lum-201の独占的に許可された標的であり,自閉症スペクトラム障害を含まない
APAによると、私たちは2018年にAmmonettに350万ドルの前金を支払いました。私たちが追求している最初の特定のマイルストーンの実現には、総額1,700万ドルの発展マイルストーン支払いを生成することも可能であり、私たちが追求している第2の兆候の特定のマイルストーンの達成については、以下に述べるように、1,400万ドルまでの販売マイルストーン支払い、世界製品販売の販売マイルストーン支払い総額5,500万ドル、および世界製品売上に基づく特許権使用料支払いを生じる可能性がある。
Lumos Merckプロトコルによると,我々はMerckに大量の開発マイルストーン支払いを支払い,第1および第2の適応に関連する特定のマイルストーンを実現することを要求される.私たちが追求している最初の指標については、潜在的な発展マイルストーンの支払い総額は最高1,400万ドルに達し、私たちが追求している第2の指標については、最高850万ドルに達する。世界の純製品売上高は10億ドルを超えず、合計8,000万ドルにのぼる分級販売マイルストーン支払いが必要であり、製品販売が実現すれば、製品売上高に応じて大量の印税を支払う必要がある
製品販売が実現すれば,APAとLumos Merckプロトコルに基づいて特許使用料を支払わなければならず,総額は年間製品純売上高の10%から12%であり,模造薬侵食の標準減幅制限を受けなければならない。Lumos Merckプロトコル下の特許使用料義務は、製品および国/地域に基づいており、その製品をカバーする最後のライセンス特許が、国/地域の排他的規制が満了したときに継続される。APAに規定される特許使用料義務は、メルク許可項下の特許使用料義務期間と、その後、“APA”によって割り当てられた当該製品が当該国/地域をカバーする最後の特許が満了するまで、製品及び国/地域に基づくものである。
Lumos Merckプロトコルは、個々の国および製品の印税義務が満了するまで、または180日前にMerckに書面通知を提出するか、他方が治癒されていない重大な違約や特定の破産事件によって任意に終了しない限り有効である。特許使用料義務が満了すると、Lumos Merckプロトコルは全額支払いの永久非独占許可に変換される。
もしLumos Merckプロトコルが終了し、Merckの書面要求に応じて、私たちは合理的で勤勉な努力をして、私たちが以前に与えた任意の再許可をMerckに譲渡する義務があります。
製造業
私たちは現在、私たちの唯一の候補製品Lum-201の臨床または商業生産施設を持つつもりもないし、所有するつもりもない。著者らは現在Lumos Merckプロトコルに関連するLum-201活性薬物成分(“原料薬”)の供給を有しており、著者らはOraGrowth H 210試験を含む現在のLum-201臨床試験を満たすことができると信じている。私たちは第三者と技術評価と最適化を行い、より多くの原料薬を生産し、私たちの臨床試験をさらに支援している。著者らはある契約メーカーとすでにOraGrowth H 210試験の臨床薬物の生産に供給する既存の手配を持っている
エボラワクチン
2014年11月,NewLinkはメルク社とNewLink Merckプロトコルを締結し,PHACから許可を得たエボラワクチンrVSVG−ZEBOVを開発し,商業化することが可能である。食品·医薬品局の承認を得た場合、RVSVG−ZEBOVもPRVを取得する資格があり、会社はPRVを販売、譲渡、または他の方法で処分することで得られたPRV価値の60%を得る権利がある。2019年12月20日、メルク社は、米国食品薬品監督管理局が18歳以上の個人によるザイールエボラウイルス感染予防のための(ザイールエボラ生ワクチン)の申請を許可したと発表した。資産購入契約によると、メルク社は2回に分けてPRV費用を支払うことに同意した。合意の要求によると、2020年9月30日までの3ヶ月間、メルク社は成約時に3,400万ドル、2021年1月11日に2,600万ドルを支払った。
私たちは特定の国のワクチン販売から特許使用料を稼ぎ続ける可能性があると受け取っている。しかし,ワクチンの市場は主に特許権使用料の支払いから除外された発展途上国,あるいはワクチンが低利益率やゼロ利益率で寄付または販売されている地域に限られると考えられるため,将来的にはメルク社の巨額の特許権使用料支払いを受けることはないと予想される。
腫瘍学候補者
私たちは二つの候補の小分子製品があります。この二つの製品は合併でNewLinkから買収しました。これらの候補製品、インドキシモッド、NLH 802(インドキシモッドのプロドラッグ)およびNLG 919(直接IDO 1酵素阻害剤)はインドール−2,3−ジオキシゲナーゼ経路阻害剤
2017年8月15日と2019年2月19日に、インドキシモ塩およびプロドラッグ配合を含む2つの米国特許がそれぞれ米国で発行され、少なくとも2036年の排他性を提供した。私たちはいくつかの国でこのような処方のための国際特許カバーを求め続けている。私たちはこれらの候補製品のさらなる開発と許可機会の潜在力を探るかもしれないが、私たちは現在、これらの獲得された小分子候補製品に対するいかなる積極的な計画も持っていない。
政府規則
米国-FDAプロセス
アメリカではFDAは薬品を規制している。“連邦食品、薬品と化粧品法”及びその他の連邦と州法規と条例は、他の事項以外に、薬品の研究、開発、テスト、製造、貯蔵、記録保存、承認、ラベル、販売促進とマーケティング、流通、承認後のモニタリングと報告、サンプリングと輸出入を管理する。薬物臨床試験を開始する前に,新薬(IND)の申請下で継続するためにFDAの許可を得なければならず,米国が開始する前にFDAは候補薬剤ごとの臨床試験案を審査する。アメリカで発売される前に、FDAの承認を受けなければならない。規制承認を得る過程と、その後適切な連邦、省、州、地方、外国の法律法規を遵守する過程には多大な時間と財力がかかり、必要な規制承認を得ることができない可能性がある。
審査の流れ
FDAは任意の新薬や以前に承認された薬物を何らかの修正した薬物を承認しなければならず,その後メーカーは米国で発売されることができる。ある会社が適用された米国の要求に従わない場合、FDAが係属中の申請、警告または無見出し手紙の承認を拒否する、臨床封印、薬品リコール、薬品差し押さえ、生産または流通の完全または部分的な一時停止、禁止、罰金、民事処罰、および刑事起訴のような様々な行政または司法制裁を受ける可能性がある。薬を販売する前に達成しなければならないステップは
•適用された良好な実験室規範(“GLP”)とその他の規定に基づいて臨床前実験室テスト、動物研究と調合研究を完成した
•FDAにIND人体臨床試験申請を提出し、この申請は人体臨床試験開始前に発効しなければならない;スポンサーは毎年INDを更新しなければならない
•各臨床試験が開始される前に、試験は、機関審査委員会(“IRBs”)または各臨床場所を代表する倫理委員会によって承認される
•適用された現在の良好な生産規範(“cGMP”)と現在の良好な臨床実践(“GCP”)に基づいて十分かつ制御された人体臨床試験を行い、各適応に対する薬物の安全性と有効性を確定し、FDAを満足させる
•FDAに新薬申請(“NDA”)を提出する
•薬物申請は、適切かつ適用可能な場合にFDA諮問委員会によって検討される可能性がある
•ライセンス要件および州規制当局によって公布された法規を含むcGMPまたは他の法規に適合するかどうかを評価するために、FDAの1つまたは複数の製造施設の検査が満足的に完了する;
•NDAに対するFDAの審査と承認。
会社は通常FDAの承認要求を満たすために数年を必要とするが、薬物或いは疾病のタイプ、複雑性と新規性によって、この要求は大きく異なる。臨床前試験は薬物の化学、調合と毒性に対する実験室評価、及び薬物特性と潜在的安全性と有効性を評価する動物研究を含む。臨床前試験の進行はGLPを含む連邦法規と要求に適合しなければならない。同社は臨床前試験の結果をFDAに提出し,INDの一部,その他の情報として,当該製品薬物の化学,製造と制御に関する情報,提案された臨床試験案を含む。初期IND提出後,生殖毒性や発ガン性の動物試験など,長期的な臨床前試験を継続することができる。
FDAは各IND提出後30日間の待機期間を要求し,会社が人体での臨床試験を開始することができる。FDAは30日以内に1つまたは複数の提案された臨床試験に対して懸念または問題を提出し、試験を保留することができる。この場合,会社やFDAは会社が臨床試験を開始する前に未解決の問題を解決しなければならない。したがって,INDを提出することはFDAがスポンサーに臨床試験を開始させるのに十分ではないかもしれない。同社はまた,薬物開発期間中に行われたすべての後続臨床試験のために既存のINDに単独で文書を提出しなければならない。
臨床試験と同時に、会社は通常追加の動物研究を完成し、薬物の物理特性に関する追加情報を開発し、GMP要求に基づいて最終的に商業大量生産製品の技術を決定しなければならない。生産過程は一貫して高品質の候補製品ロットを生産できなければならず,他の要求以外にもスポンサーは最終薬物の品質を確保する方法を策定しなければならない。また,適切な包装を選択·試験し,候補薬物がその標識された棚期に受け入れられない変質が生じていないことを証明するために安定性研究を行わなければならない。
NDAを承認する前に、FDAは、それらがcGMP要件に適合しているかどうかを決定するために、新製品の製造施設を承認前に検査する。FDAは、製造プロセスおよび施設がcGMP要件に適合していることを決定し、要求された仕様の下で製品が一貫して生産されることを保証するのに十分でない限り、この製品を承認しないであろう。
候補製品の開発に伴い、前臨床試験から後期臨床試験まで承認と商業化まで、開発計画の各方面、例えば製造方法と調合、この過程で変更を行い、製品、技術と結果の最適化に努力することは非常に一般的である。このような変化はこのような期待された目標を達成できない可能性がある。これらの変化のいずれも、我々の候補製品の表現が異なり、計画中の臨床試験または変更された材料を用いた他の将来の臨床試験の結果に影響を与える可能性がある。これは臨床試験の完成を遅らせる可能性があり、移行臨床試験を行うか、または1つ以上の臨床試験を繰り返し、臨床試験コストを増加させ、私たちの候補製品の承認を延期し、および/または私たちまたは私たちの協力者が製品販売を開始し、収入を創出する能力を脅かす必要がある。
臨床試験
臨床試験には,合格した研究者の監督の下で,健康なボランティアや患者に対してINDを行うことが含まれている。同社は臨床試験を行わなければならない
•連邦法規に適合しています
•GCP国際標準に符合し、患者の権利と健康を保護し、臨床試験発起人、管理者と監督者の役割とIRBの要求を定義することを目的としている
•実験目標,安全モニタリングパラメータ,有効性基準を詳細に説明するプロトコルに基づいている。
IND申請の一部として、同社は、米国患者の試験に関する各案およびその後の案修正案をFDAに提出しなければならない。FDAがスポンサーがFDAの要求に応じて臨床試験を行っていないと考えている場合,あるいは臨床試験患者に受け入れられないリスクとなっている場合,FDAはいつでも臨床試験の一時的または永久的な停止を命じたり,他の制裁を加えたりすることができる。スポンサーは,臨床試験における患者の試験案やインフォームドコンセント情報をIRB承認に提出しなければならない。IRBは、IRBの要求に従わなかったために、臨床試験を一時的または永久的に停止することができ、または他の条件を適用することができる。
会社は通常1種の薬物の臨床研究を3~4段階に分けている。会社は通常これらの段階を順番に実行しているが、それらは重複したり合併したりすることがある。
•ステップ1それは.同社は、健康なヒト被験体または標的疾患または疾患を有する患者において、その薬剤を評価する。これらの試験は、通常、ヒトにおけるINDの安全性、用量耐性、新陳代謝および薬理作用、および用量の増加に関連する有害事象を評価し、可能であれば、有効性に関する早期証拠も得られる。
•第二段階です。同社はこの薬物を限られた患者集団に応用し、用量耐性と最適な投与量を評価し、可能な不良事件と安全リスクを識別し、そして初歩的に治療効果を評価する。
•第三段階です。同社は通常地理的に分散した臨床試験地点でより多くの患者群にこの薬物を管理し、十分なデータを生成して投与量、臨床有効性と安全性に対して統計評価を行い、薬物の全体的な利益-リスク関係を研究し、そして製品の承認に十分な基礎を提供する。
•第四段階。場合によっては、FDAは、承認後に追加の臨床試験を行うことに会社が同意することを条件に、薬物のNDAを承認する可能性がある。他の場合、スポンサーは、承認を得た後に自発的に追加の臨床試験を行うことができ、その薬剤に関するより多くの情報を得ることができる。われわれは通常,この承認後試験を4期臨床試験と呼ぶ。
重要な試験は監督管理機関の要求を十分に満たす臨床試験であり、薬物の有効性と安全性を評価し、この薬物の承認が合理的であることを証明する。通常、肝心な試験は3期試験であるが、試験設計が臨床利益に対する良好な制御と信頼性評価を提供すれば、特に満たされていない医療需要が存在し、結果が十分に穏健である場合、FDAは2期試験の結果を受ける可能性がある。
FDA、IRB或いは臨床試験スポンサーはいつでも様々な理由で臨床試験を一時停止或いは中止することができ、研究対象が受け入れられない健康リスクに直面していることを発見することを含む。そのほか、臨床試験スポンサーが組織した合格専門家からなる独立グループ、即ちデータ安全監視委員会は、いくつかの臨床試験を監督する可能性がある。このグループは,実験のあるデータへのアクセスにより,許可試験が指定されたチェックポイントで行えるかどうかを決定する.我々はまた,変化する業務目標や競争環境に応じて臨床試験を一時停止または中止することも可能である。
新冠肺炎突発公共衛生事件のため、私たちは追加の臨床試験政策とプログラムおよび/または利益リスク分析を制定し、実施して、被験者を新冠肺炎ウイルスの影響から保護することを助ける必要があるかもしれない。例えば、2020年3月、アメリカ食品と薬物管理局は疫病期間中の臨床試験に関する指導意見を発表し、この指導意見はその後更新を行い、その中に疫病の影響を受けた臨床試験スポンサーのいくつかの考慮要素を述べ、臨床試験報告には臨床試験を管理するための応急措置、及びいかなる新冠肺炎の大流行による臨床試験の中断などの要素を含む。2020年6月、アメリカ食品薬品監督管理局はまた薬品生産従業員の新冠肺炎感染に対応する良好な生産規範注意事項ガイドラインを発表し、薬品汚染を防止する生産制御提案を含む。アメリカ食品薬品監督管理局はまた、これまでのガイドラインの更新、薬品製造と生物研究モニタリング施設の遠隔相互作用評価、薬品製造とサプライチェーン検査などを含む他の新冠肺炎に関連する業界ガイドラインを発表した。新冠肺炎疫病の著者らの業務運営に対する最終的な影響は高度に不確定であり、変化が発生する可能性があり、未来の発展に依存し、新しい監督管理要求と現有の法規に対する変化を含む。最近、総裁·バイデン総裁は、政府が2023年5月11日に新冠肺炎国家と公衆衛生突発事件を終了する予定であることを発表した。突発公共衛生事件の終了がFDAとその他の監督管理政策と運営に与える全面的な影響はまだ不明である。
守秘契約を提出する
必要な臨床試験が完了した後,NDAを準備してFDAに提出することができ,FDAはまずNDAを承認しなければならず,米国での販売を開始することができる。セキュリティプロトコルには,陰性または曖昧な結果および陽性結果,薬物の化学,製造,対照,提案ラベルなどに関する詳細な情報を含む関連する臨床前および臨床試験から得られたすべての関連データが含まれていなければならない。データは、会社が提供する薬物臨床試験から来ることができ、研究者によって開始された試験を含む多くの代替源から来ることもできる。NDAを支援するためには,研究薬物の安全性と有効性を決定し,FDAを満足させるために,我々が提出したデータは品質的にも数量的にも十分でなければならない。
秘密協定を準備して提出する費用は巨大だ。NDAの多くは高額な申請使用料を支払う必要があるが,承認されたNDA下のメーカーや/またはスポンサーも年間計画使用料を支払う必要がある。FDAは通常毎年このような費用を増加させる。ODDは一方に財政インセンティブを獲得させ、臨床試験コストに寄付資金の機会、税収優遇、ユーザー費用の減免を提供する権利がある。
FDAは,NDAを受信した日から60日で当該機関の届出申請を受け入れるか否かを決定しており,これは,この機関が申請が十分に完全であり,実質的な審査が可能であることを決定した敷居に基づいている。FDAが申請を受けると、FDAは深い検討を始めた。FDAはNDAのレビューでいくつかのパフォーマンス目標を設定することに同意した。処方薬使用料法案によると,FDAは60日の届出審査期間後10カ月以内に標準審査NDAに対応することを目標としているが,この期限は延長されることが多い。FDAは、10~12ヶ月以内にほとんどの標準審査薬の申請を審査し、6~8ヶ月以内に優先審査薬の申請の大多数を審査する。優先審査は、FDAが治療において大きな進展を得た薬剤を決定するか、または適切な治療方法がない場合に治療を提供する薬剤に適用することができる。
FDAは安全性や有効性の問題を提起した新薬申請を諮問委員会に提出することも可能である。これは一般に、FDAが申請を承認すべきかどうかを審査、評価、提案する臨床医および他の専門家を含むグループである。FDAは諮問委員会の提案によって制限されていないが、それは一般的にそのような提案に従っている。NDAを承認する前に、FDAは、通常、GCPに適合することを保証するために1つまたは複数の臨床場所を検査し、薬物を製造する1つまたは複数の施設を検査する。FDAは、cGMPに適合しない限り、薬剤を承認しないであろうし、NDAに含まれるデータは、研究された適応において安全かつ有効であることを証明する証拠を提供するであろう。
FDAの秘密保持協定に関する決定
FDAがNDAと製造施設を評価した後、それは承認状または完全な返信を発行するだろう。完全な返信は、FDAがこの出願の審査を完了したことを示し、機関は現在の形態の出願を承認しないことを決定した。完全な返信状は、一般に、大量の追加の臨床データ、および/または臨床試験、臨床前研究および/または製造に関連する他の重要、高価および時間の要件を必要とする可能性がある提出文書中の不足点を列挙する。FDAは、そのような欠陥を解決するために、そのような欠陥を解決するために、2ヶ月または6ヶ月以内に再提出されたNDAを検討することを約束しており、特に含まれる情報タイプに依存する。我々がこのようなデータを提出しても,FDAはNDAが承認基準を満たしていないことを最終的に決定する可能性がある。さらに、政府は、新しい立法によって生じる要求、またはFDAの政策が変わる可能性がある追加の要求を制定する可能性があり、これは、私たちが開発している薬物が規制部門の承認を得ることを延期または阻止する可能性がある。
この薬物の商業マーケティングを許可し、特定の適応の具体的な処方情報を提供する。NDA承認の条件として、FDAは、薬物の利点が潜在的なリスクよりも大きいことを保証するために、リスク評価緩和戦略(“REMS”)を要求する可能性がある。REMSは、薬物ガイド、医療専門家のコミュニケーション計画、処方または調剤の特殊トレーニングまたは認証、特定の場合のみの調剤、特殊モニタリング、および患者登録簿の使用を含むことができる。REMSに対する要求はこの薬物の潜在的な市場と収益力に重大な影響を与える可能性がある。また,FDAは大量の承認後の試験やモニタリングを条件として,薬物の安全性や有効性をモニタリングする可能性がある。承認されると、同社が規制基準を遵守できなかった場合、または初歩的なマーケティング後に問題が発見された場合、FDAは薬物承認を撤回する可能性がある。
承認申請において確立されたいくつかの条件の変更は、適応、ラベルまたは生産プロセスまたは施設の変更を含み、新しいNDAまたはNDA付録を提出し、FDAの承認を得て、変更を実施する必要があります。新適応のNDAサプリメントは通常,オリジナル出願と類似した臨床データが必要であり,FDAがNDAサプリメントを審査する際に使用するプログラムや行動は,新たなNDAを審査する場合と同様である。新しいNDAと同様に、FDAは、より多くの情報の提供または明確化を要求することによって、審査プロセスを著しく延長することが多い。
承認後に要求する
FDA監督管理はFDAによって製造或いは流通を許可した薬物を許可し、そして記録保存、定期報告、薬物サンプリングと流通、広告と販売促進及び報告薬物の副作用に対して具体的な要求がある。承認後、FDAは、承認された薬剤の大多数の変更に対して、新しい適応または他のラベル宣言を追加するなどの審査および承認を提供しなければならない。任意の市販薬やその薬剤を生産する機関に対しては,持続的な年間使用料要件,および臨床データ補充出願に対する新規出願料がある。
場合によっては、FDAは、スポンサーが承認後に追加の臨床試験を行うことに同意することを条件に、薬物のNDAを承認する可能性がある。他の場合、スポンサーは、承認を得た後に自発的に追加の臨床試験を行うことができ、その薬剤に関するより多くの情報を得ることができる。この承認後の試験は通常4期臨床試験と呼ばれる。
医薬品メーカーはFDAと州政府機関からcGMP要求の定期抜き打ち検査を受けなければならない。製造過程の変更には厳格な規定があり,それに基づいて
この変化の重要性のため、私たちはFDAが事前に承認して施行する必要があるかもしれない。FDAの法規はまた、cGMPから外れた任意の状況の調査と是正を要求し、私たちと私たちが使用を決定する可能性のある任意の第三者メーカーに報告と文書要求を提出します。そのため、メーカーは生産と品質管理の分野で時間、お金、精力をかけ続け、cGMPやその他の法規遵守性を維持しなければならない。
ある会社が規制要求を遵守して基準を維持していない場合、あるいは薬物発売後に問題が発生した場合、FDAは承認を撤回する可能性がある。ある企業またはFDAが、予期しない深刻または頻度の有害事象、生産プロセスの問題、または同社が規制要件を遵守していないことを含む以前に未知の問題が存在することを発見した場合、FDAは、新しいセキュリティ情報を追加するために承認されたラベルを修正する可能性があり、発売後試験または他の臨床試験を実施して新しい安全リスクを評価するか、またはREMS計画に従って流通または他の制限を実施する可能性がある。他の潜在的な結果には
•薬品の販売や製造を制限し、薬品や薬品のリコールを完全に市場から撤退させる
•承認後の臨床試験には罰金、警告状、一時停止を科す
•FDAは、承認されるべき新薬または承認された新薬の補充剤の承認を拒否するか、または医薬品許可証の承認を一時停止または取り消し;
•麻薬を押収したり押収したり麻薬の輸出入を許可することを拒否した者は
•民事または刑事処罰を禁令または適用する。
FDAは発売薬品のマーケティング、ラベル、広告と販売促進活動を厳格に監督する。承認の適応と承認のラベルの規定に基づいてのみ薬物を普及させることができる。FDAと他の機関は非ラベル用途の普及を禁止する法律法規を積極的に施行している。もし私たちがこのような法律に違反したら、私たちは重大な責任を負うかもしれない。
孤児薬名
FDAは、米国で20万人未満の影響を与える珍しい疾患または疾患の治療を目的とした薬剤に奇数を付与する可能性があり、または米国で20万人を超える影響を与え、合理的な期待がない場合、この疾患または疾患の薬物開発および製造コストは米国の販売から回収されるであろう。
ODDは一方に財政インセンティブを獲得させ、臨床試験コストに寄付資金の機会、税収優遇、ユーザー費用の減免を提供する権利がある。さらに、1つの薬剤が孤児指定適応を有するFDAの承認を得た場合、この薬剤は、孤児薬物排他性を得る権利がある可能性があり、これは、FDAが、限定された場合、例えば孤児に対して排他性を有する薬物に対する臨床的利点を示さない限り、7年以内に同じ適応に対して同一の薬剤を販売する他のいかなる出願も承認しないことを意味する。
はいCatalyst Pharms,Inc.Becera事件を訴える、1299“連邦判例編”第14巻(第11巡回法廷)2021年)には,裁判所はFDAの長期的な立場,すなわち孤児薬物排他性が条件を満たす疾患内の承認用途または適応にのみ適用されることに同意しない。特に,巡回裁判所は,Catalyst薬物の孤児薬物排他性は,Catalystの薬物が成人LEMSの治療に許可されていても,Catalystの薬物が同一孤児指定疾患またはLambert−Eaton筋無力症候群(LEMS)のすべての用途または適応に使用されることをFDAが承認したことを排他的に阻止していると考えている。したがって、裁判所はFDAに児童LEMSのための薬物の承認を保留するように命令した。この決定は孤児薬の独占権の適用に不確実性をもたらす。FDAは2023年1月24日に連邦紀要紙に通知を発表し、機関が裁判所を遵守しているにもかかわらず、触媒.触媒FDAは、この条例の長期的な解釈をこの条例の範囲外に適用し続けることを意図している触媒.触媒命令-すなわち、機関は、孤児薬物の排他的範囲を、薬物が承認された用途または適応と一緒にバンドルし続け、これにより、他のスポンサーが、同じ孤児指定疾患または条件下で、承認されていない薬物の新しい用途または適応の承認を得ることを可能にする。未来の訴訟、立法、機関決定と行政行動がどのように孤児薬物の独占経営の範囲に影響するかはまだ不明である。
小児科情報
“小児科研究公平法”によると、新薬または新薬補充剤は、すべての関連小児科亜群で薬物が主張する適応の安全性と有効性を評価し、用量を支持するためのデータを含まなければならない
そして薬物に対して安全かつ有効な個々の小児科亜群の管理。FDAは、提出されたデータの全部または一部を免除または延期することができる。規制が別途要求されない限り、PREAは、FDAが孤児として指定された適応として承認されたいかなる薬剤にも適用されない。
医療改革
アメリカと他の司法管轄区域では、立法構造が変化し続けている。医療システムの立法や規制面のいくつかの変化は、その将来の運営結果に影響を与える可能性がある。特に,米国連邦と州の各レベルでは,医療資金調達方式の改革,医療コストの低減を図る取り組みが継続されている。2010年3月、“衛生保健·教育負担能力調整法”(総称して“PPACA”)により改正された“患者保護·平価医療法”が公布され、その中に含まれる措置が政府や民間保険会社の医療資金調達を著しく変化させた。他の以外に、PACAは製薬と生物技術業界に対して重要な意義を持つ条項は以下の通りである
•特定のブランドの処方薬代理を製造または輸入する任意のエンティティに徴収される控除不可能な年間費用は、特定の政府医療計画におけるこれらのエンティティの市場シェアに基づいてこれらのエンティティ間で分担される
•医療補助薬品還付計画によると、メーカーが支払わなければならない税金還付はそれぞれブランドと後発薬メーカーの平均価格の23.1%と13%に増加した
•新しい連邦医療保険D部分の保証不足割引計画は、メーカーはその保証間隔期間内に条件を満たす受益者に適用ブランド薬品交渉価格の50%(2019年1月1日から70%)の販売時点割引を提供することに同意しなければならず、メーカーの外来薬物として連邦医療保険D部分で保険を受ける条件である
•メーカーの医療補助税還付責任を、340 B薬品割引計画に基づいて割引しない限り、医療補助管理保健組織に参加する個人に配布される保険薬に拡大する
•医療補助計画の資格基準を拡大し、他を除いて、各州がより多くの個人に医療補助を提供することを許可し、連邦貧困レベル133%以下の収入のある個人に新しい強制資格種別を増加させ、それによってメーカーの医療補助リベート責任を潜在的に増加させる
•公共衛生サービス薬品の定価プランの下で割引を受ける資格のある実体を拡大する
•連邦民事虚偽申告法と連邦反リベート法案、新しい政府の権力調査と違反行為への懲罰の強化など、医療詐欺と乱用法を拡大する
•連邦“医師支払い陽光法案”の適用を要求する薬品メーカーは、毎年、医師(医師、歯科医師、検眼師、足科医師および脊椎マッサージ師を含むと定義される)、一部の非医師保健専門家(例えば、医師アシスタントおよび看護師従業員など)および教育病院、および医師およびその直系親族が所有する所有権または投資権益に関する情報を含む前の保険受給者への支払いおよび他の価値移転に関する情報を報告することを要求する
•“健康情報技術促進経済·臨床健康法”(“HITECH”)及びその実施条例が改正された1996年の“健康保険携帯性と責任法”(“HIPAA”)も、健康計画、医療情報交換センターと特定の医療保健提供者である強制契約条項を含む保険実体に対して義務を規定しており、これらの条項はHIPAA及びそのそれぞれの商業パートナーによって定義され、個人が識別できる健康情報のプライバシー、安全及び伝送を保護することに関連している
•新しい規定は、製造業者および流通業者が所有する医師、病院薬局、または他の医療機関に提供されるいくつかの薬物サンプルを毎年報告する
•同様の州および外国の法律および法規、例えば、州反リベートおよび虚偽請求法は、個人保険会社を含む非政府第三者支払者によって返済される保健プロジェクトまたはサービスの販売またはマーケティング手配およびクレームに適用可能であり、州法律は、製薬会社に製薬業の自発的コンプライアンスガイドラインおよび連邦政府によって公布された関連コンプライアンス条例を遵守することを要求し、医師および他の保健提供者への支払いおよび他の方法での価値移転、マーケティング支出または薬品定価に関する情報を医薬品メーカーに報告することを要求する可能性がある
医薬品販売および医療代表登録を要求する法律;2018年カリフォルニア消費者プライバシー法(CCPA)およびカリフォルニアプライバシー権法案(CPRA)のような健康情報プライバシーおよび安全を管理する州法律は、多くの法律が互いに大きく異なり、HIPAAによって先制されず、コンプライアンス作業を複雑化させることが多い。
PPACAは引き続きアメリカ製薬業に重大な影響を与えている。PACAのいくつかの側面は公布以来、司法と国会の挑戦を受けてきた。2021年6月、米最高裁はテキサス州と他の挑戦者がPPACAの法的地位に挑戦していないと判断し、手続きを理由にこの事件を却下したが、PPACAの合憲性を具体的に裁くことはなかった。したがって、PACAは現在の形で継続的に有効になるだろう。PPACAは未来に司法や国会で挑戦される可能性がある。バイデン政府が公布した未来の挑戦と医療保健措置がPPACA、著者らの業務、財務状況と運営結果にどのように影響するかはまだ不明である。任意の新しい立法を遵守したり、PACAによって実施された変化を逆転させることは、多くの時間とコストを費やし、私たちの業務に実質的な悪影響を及ぼす可能性があります。PPACAに含まれる条項は薬品の収益性を低下させる可能性があり,医療補助計画で精算された薬品のリベートを増加させ,医療補助リベートを医療補助管理保健計画に拡大し,ある連邦医療保険D部分の受益者に強制的な割引を提供することと,製薬会社の連邦ヘルスケア計画における販売シェアに基づいて年会費を徴収することである。医療補助薬品還付計画は、製薬業者がアメリカ衛生と公衆サービス部部長或いはHHS部長と全国税金還付協定を締結し、発効することを要求し、各州が医療補助患者に提供する外来薬物の連邦マッチング資金を獲得する条件とする。PACAは医療補助薬品リベート計画をいくつか修正し、メーカーが医療補助薬品リベート計画の下で不足している最低医療補助リベートを増加させることを含む, 税金還付計画を医療補助管理保健組織に登録されている個人に拡大する。PPACAはまたあるブランドの処方薬のメーカーに対して年間費用と税収を制定し、そして新しいMedicare Part D引受切欠き割引計画を作成し、メーカーは保証不足期間中に条件を満たす受益者に70%の販売時点割引を提供することに同意しなければならない(2018年の両党予算法により向上し、2019年から発効)、メーカーの外来薬物としてMedicare Part Dの条件に入れなければならない。
また,PACAが公布されて以来,米国では他の医療改革措置が提案され採択されている。例えば、改正2011年予算制御法により、提供者は2030年前の財政年度ごとに2%の連邦医療保険支払い削減を受けるが、各種新冠肺炎救済立法に基づいて2020年5月1日から2022年3月31日まで実施される臨時停止は、追加の国会行動をとらない限り除外される。現在の立法によると、医療保険支出の実際の減少幅は2022年の1%から本自動減額の最終年度の4%まで様々になる。また、2012年の“米国納税者救済法”は、いくつかの医療サービス提供者への医療保険支払いを減少させ、政府が提供者に多額の支払いを取り戻す訴訟時効期間を3年から5年に延長した。
アメリカでは、特殊薬品の価格設定に関する立法と法執行の関心が増加している。具体的には,米国議会は最近いくつかの調査を行い,薬品定価の透明性の向上,連邦医療保険下の処方薬のコスト低減,定価とメーカー患者計画との関係の審査,政府計画の薬品精算方法の改革を目的とした連邦と州立法を提案した。連邦レベルでは,2021年1月1日に発効する“2021年米国救援計画法案”によると,メーカーが州医療補助計画に支払う医療補助薬品還付計画還付の法定上限が撤廃される。この上限を撤廃することは、販売製品よりも多くのリベートの支払いを製薬業者に要求する可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。2022年8月、国会は“2022年インフレ低減法案”を可決し、その中には製薬業と連邦医療保険受益者に重大な影響を与える処方薬条項を含み、連邦政府がある高価な単一源連邦医療保険薬物の最高公平価格について交渉することを許可し、薬品価格交渉要求を守らないメーカーに罰と消費税を加え、すべての連邦医療保険B部分とD部分の薬物がインフレリベートを獲得することを要求し、もしその薬品価格の増加がインフレより速い場合、限られた例外、及び連邦医療保険D部分を再設計して受益者の自己払い処方薬コストを下げるなどの変化を含む。これらの立法や行政の影響は, バイデン政府が我々と製薬業全体に対する行政行動や将来実施されるどの医療措置や機関規則も不明である。コスト抑制措置や他の医療改革を実施することは、収入を創出し、利益を達成すること、または候補製品を商業化することを阻止するかもしれない(承認されれば)。
州レベルでは、立法機関は、価格または患者の精算制限、割引、特定の製品参入およびマーケティングコスト開示の制限、および透明性措置を含む、価格または患者の精算制限、割引、特定の製品参入およびマーケティングコスト開示の制限および透明性措置を含む医薬品および生物製品の定価を制御するための法規を立法および実施することが増えており、場合によっては、他の国からの輸入および大量購入を奨励することが目的である。例えば、いくつかの州は州の薬品価格の透明性と報告法を考慮しているか、最近公布しています。これは私たちのコンプライアンス負担を大幅に増加させ、私たちを直面させるかもしれません
私たちが規制部門の任意の製品の承認を得て商業化を開始すると、このような州法律によって、私たちはより大きな責任を負うことになります。また、地域衛生保健当局や個別病院は、どの薬品やサプライヤーが彼らの処方薬や他の医療計画に含まれるかを決定するために入札プログラムを使用することが増えている。これらの措置が承認されると、私たちの製品に対する最終的な需要を下げたり、私たちの製品の価格設定に圧力をかけるかもしれません。私たちは将来、より多くの州と連邦医療改革措置を取ることが予想され、そのいずれも連邦と州政府が医療製品やサービスのために支払う金額を制限する可能性があり、これは私たちの候補製品に対する需要の減少または追加の価格設定圧力を招く可能性がある。
EU-欧州医薬品局(♪the the the “EMA”)プロセス
連合では、私たちの候補製品もまた広範囲な規制要求を受ける可能性がある。アメリカのように、医薬製品は主管監督機関が発行したMAを獲得した後にのみ販売することができる。
アメリカと類似して、EUの臨床前と臨床研究の各段階は重要な監督管理によって制御されている。EU医療製品の臨床試験はEUと国家法規および国際協調会議のGCPに関するガイドラインに沿って行わなければならない。EU臨床試験指令2001/20/ECはEU臨床試験監督管理枠組みを調整し、EU臨床試験の制御と許可のために共通規則を制定したが、EU加盟国は異なる方法でこの指令の条項を交換と応用した。これは会員国制度の大きな変化を招いた。現行制度を改善するため、2014年4月16日にヒト用薬品の臨床試験に関する(EU)第536/2014号条例(“臨床試験条例”)が採択され、この条例は第2001/20/EC号指令を廃止し、2014年5月27日にヨーロッパ公式定期刊行物で発表された。“臨床試験条例”は2022年1月31日から実施され、現行の臨床試験許可規則と業績標準を簡略化することを目的としている。例えば、申請手続きは、単一の入口点、欧州連合ポータル、およびデータベースを介して簡略化される。新しい臨床試験門戸とデータベースはヨーロッパ医学協会がヨーロッパ委員会とヨーロッパ連合加盟国と協力して維持する。新法規の目標には、EU全体で試験を行う一致規則、一致したデータ基準と有害事象リスト、および許可状態に関する一致情報が含まれる。また,EUで行われている個々の臨床試験の進行と結果に関する情報が公開される。
現行制度では、臨床試験を開始する前に、各EU加盟国の承認を得なければならず、これらの加盟国では、試験は2つの異なる機関によって行われる:国家主管機関(NCA)と1つ以上の道徳委員会(ECs)。現行制度によると,臨床試験期間中に発生するすべての予期せぬ深刻な副作用が疑われ,これらの反応が発生した加盟国の国家薬品監督管理局と欧州薬品監督管理局に報告しなければならない。
審査の流れ
中央手続きによると,EMAが意見を発表した後,欧州委員会はEUおよびアイスランド,リヒテンシュタイン,ノルウェーで有効な単一MAを発表した。以下のヒト薬物の場合、集中手順は強制的である:遺伝子工学のようなバイオテクノロジープロセスに由来する;HIV/エイズ、癌、糖尿病、神経変性疾患または自己免疫疾患および他の免疫機能障害などの特定の疾患の治療に使用可能であることを示す新しい活性物質;および公式に指定された孤児薬を含む。これらのカテゴリーに属さない薬剤については、関連する薬剤が重大な治療、科学的または技術的革新である限り、またはその許可が公衆の健康に有利である限り、出願人は、中央MAの出願を環境管理協会に提出することを選択することができる。
他にもEUの医薬製品を許可するための3つの可能な方法がありますこれらの経路は集中プログラムの範囲に属さない製品に適用されます
•国家手続きです。欧州経済圏加盟国の主管当局が発行した国家地域があるが、これらの地域はそれぞれの領土しかカバーしていない
•分散したプログラム。分散プロセスを使用して、出願人は、1つ以上のEU諸国において、どのEU諸国でも承認されていない医薬製品を同時に承認することができる
•プログラムを相互認識する。相互認識手続きでは、欧州連合加盟国の国家手続きに基づいて、まずその国で許可を得る。その後,さらなるMAを求めることができる
プログラムでは,関係国はオリジナルの国のMAの有効性を認めることに同意した.
私たちの現在のどの候補製品も、集中プログラムの強制的な基準に属するため、全国MAに適していないと予想される。したがって、私たちの候補製品は集中承認手続きを通過するだろう。
2021年1月から、医薬品·保健製品監督局(MHRA)は、EMA以前の汎EU規制手続きがイギリスに適用されなくなるため、イギリスで販売されている医療製品に対して追加の規制責任を負う。MHRAは最近,イギリス体制下の規制についてこの業界に新たな指導意見を発表した。新しいMHRAガイドラインで提案されている提案は、議会で承認された立法改革によって発効し、イギリスで規制承認を得るのに必要な資源と時間を増加させ、私たちの臨床開発と商業化を遅らせる可能性がある。イギリスの離脱が私たちの業務に及ぼす全面的な影響はまだ明確ではない
(EC)第1901/2006号条例によれば、すべてのMA新薬出願は、薬物が延期または免除によって免除されない限り、規制当局および出願人によって合意された小児科調査計画(“PIP”)に記載された試験結果を含まなければならない(例えば、関連疾患または状態が成人でのみ発生するため)。EMAが集中プログラムMA申請の評価を開始する前に,申請者が合意した小児科調査計画を遵守しているかどうかを検証する。出願人およびEMAは,十分な理由がある場合には,検証に協力するために小児科調査計画を修正することに同意することができる。修正は常に可能ではない;同意は有効許可期間よりも長い時間を必要とする可能性があり、依然として出願人にその上場許可申請(MAA)を撤回し、追加の非臨床試験および臨床試験を行うことを要求する可能性がある。PIPによる小児科臨床試験に基づいてMAを取得した製品は,補充保護証明書(承認時に有効であれば)により6カ月の保護延長を得る資格があるか,あるいは孤児医薬製品に対して孤児市場排他性を2年間延長する資格がある。この小児科奨励は特定の条件の制約を受け,PIPに適合したデータを開発·提出する際に自動的に獲得されない。
孤児薬名
欧州連合では、医薬品のスポンサーが証明できれば、改正(EC)第141/2000条例が規定されている
•申請を行う際には、欧州連合において万分の5を超えない生命の脅威または慢性衰弱に影響を与える疾患の診断、予防または治療に使用されるか、または欧州連合における生命を脅かす、深刻な虚弱または深刻かつ慢性的な疾患の診断、予防または治療のために使用されることが意図されており、インセンティブ措置がない場合、必要な投資が合理的であることを証明するために、欧州連合での販売が十分な見返りをもたらすことは不可能である
•欧州連合によって許可されていない満足できる診断、予防または治療方法、または、そのような方法があれば、薬剤は、その疾患の影響を受けている人に大きな利益をもたらすであろう。
(EC)条例847/2000は、薬物を孤児薬として指定する基準を施行することをさらに規定している。孤児薬物として薬物を指定する出願は、薬物開発の任意の段階で提出することができるが、MA申請を提出する前に提出しなければならない。
孤児薬物のための集中プログラムMAが(EC)726/2004条例に従って承認された場合、規制当局は、通常、同様の薬物について別のMA出願を受け入れるか、またはMAを承認するか、または既存のMAの治療適応を延長する出願を10年以内に受け入れない。しかし,5年目の終了時に関連薬物が奇数基準を満たさなくなったこと,換言すれば,既存の証拠からその製品の利益が十分に高く,市場排他性を維持することが合理的であることを証明するのに不十分であれば,この期間は6年に短縮できる。MAAが合意した小児科調査計画の試験結果を含めると,排他期は12年に増加する可能性がある。上記の規定にもかかわらず、以下の場合、類似薬の同じ治療適応についてMAを付与することができる
•元孤児薬物授権書所持者は、第2の出願人に同意した
•元孤児薬物のMA保持者は、十分な量の薬剤を供給することができない;または
•第2の出願人は、第2の薬剤は、許可された孤児薬と類似しているが、より安全で、より効果的であり、または臨床的に良好であることを出願で証明することができる。
(EC)条例847/2000には、“類似薬”および“臨床的優位性”などの概念の定義が規定されている。欧州連合が孤児薬に提供する他のインセンティブには、費用の削減や費用の免除、プログラム援助などの財政的インセンティブが含まれている。ODDは規制承認過程の継続時間を短縮しない。
良好な製造規範
FDAと同様に、欧州薬品管理局、欧州連合加盟国の主管当局と他の監督機関は、1種の薬物を承認する前に、薬品生産に使用する設備、施設、技術に対して監督と検査を行う。
もしある会社が規制機関の承認を得た後、製造設備、場所、あるいはプロセスを重大に変更した場合、追加の監督審査と承認が必要になる可能性がある。私たちまたは私たちのパートナーが薬品を商業化すると、私たちはcGMPと、薬品の承認後に欧州委員会、欧州薬品管理局、EU加盟国の主管当局によって実行される薬品に対する法規を遵守することが要求されるだろう。FDAと同様に、EMA、EU加盟国の主管当局および他の規制機関も定期的に訪問して、薬物が初歩的な承認を得た後に設備、施設、およびプロセスを再検査する。これらの検査の結果、規制機関が、私たちまたは私たちのパートナーの設備、施設、またはプロセスが適用される薬品承認法規および条件に適合していないと判断した場合、彼らは、私たちの製造業務を一時停止すること、または私たちの薬物を市場から撤退させることを含む、私たちの民事、刑事または行政制裁および/または救済措置を求めることができる。
承認後にコントロールする
ヨーロッパMAの保有者は薬物警戒システムを構築·維持し,資格のある個人に薬物警戒を担当させ,このシステムの監督を担当しなければならない。主な義務は深刻な副作用の疑いの報告の加速と定期的な安全更新報告(“PSURs”)の提出を含む。
すべての新しいMAAは、企業が実施するリスク管理システムを記述し、製品に関連するリスクを予防または最小化するための措置を記録するリスク管理計画(RMP)を含まなければならない。規制当局はまた、特定の義務を金融管理専門家の条件として規定することができる。このようなリスク最小化措置または認可後の義務は、追加の安全監視、PSURsのより頻繁な提出、または追加の臨床試験または認可後の安全性研究を含む可能性がある。RMPおよびPSURsは通常,アクセスを要求する第三者に利用可能であるが,限られた編集を行う必要がある.製品のすべての広告および販売促進活動は、承認された製品特性要約と一致しなければならないため、すべてのラベル外の販売促進活動は禁止されます。連合はまた消費者に直接向けた処方薬の広告を禁止する。医療製品の広告や販売促進の一般的な要求はEU指令に基づいて制定されているが,詳細はEU加盟国ごとの法規によって管轄されており,各国が異なる可能性がある。
データと市場排他性
アメリカと同様に、EUはまた革新的な薬物模倣薬を許可する手続きを持っている。データ排他性が満了した場合、模倣薬競争者は、簡略化された申請を提出することができ、革新者のデータを参照し、参照薬との生物学的同等性などを証明する集中手順によって、EMAによって許可された模倣薬バージョンを許可することができる。新しい活性物質を含む医薬製品にMAを付与すれば,この製品は8年間のデータ排他性から利益を得,その間,その製品データに関連する後発薬申請は規制機関に受け入れられない可能性があり,他の2年間の市場排他性であり,その間,このような後発薬は市場に投入されてはならない。最初の8年以内に新たな治療適応が承認され,既存療法と比較して有意な臨床的メリットがあれば,2年間の期間を3年に延長することができる。この制度は一般的に“8+2”と呼ばれる。生物模倣薬についても、参考医薬製品と類似しているが、模倣薬製品の定義に適合しない生物医薬製品、例えば、原材料または製造技術の違いによる特殊な制度がある。このような製品については,適切な臨床前または臨床試験の結果を提供しなければならず,EMAのガイドラインは異なるタイプの生物製品に提供される補足データのタイプを詳細に説明している。
その他の国際市場である薬品承認手続き
いくつかの国際市場(例えば、中国または日本)では、米国またはEU試験で生成されたデータがMAAを支持するために提出される可能性があるが、規制機関は、ホスト国で追加の臨床試験を行うことを要求するか、またはその国内でMAを提出または承認する前に、ホスト国領土の民族を研究することを要求する可能性がある。
定価と精算
私たちが規制承認を受ける可能性のある任意の薬物のカバー範囲と精算状態には、大きな不確実性がある。米国や他の国·地域の市場では、規制部門の承認を得て商業販売のための任意の薬物の販売は、第三者支払者が保険や精算を提供するか否かに依存する。第三者支払者には、政府当局、医療計画の管理、個人健康保険会社、その他の組織が含まれる。支払人が薬剤に保険を提供するかどうかを決定するプロセスは、支払者が薬剤のために支払うべき送達率を設定するプロセスと分離することができる。第三者支払者は、承認されたリストまたは処方セット上の特定の薬剤にカバー範囲を制限することができ、FDAによって承認された特定の適応のすべての薬剤を含まない可能性がある。また、支払人が薬品に保険を提供することを決定することは、適切な販売率を承認することを意味するものではない。また,支払者によって薬品カバー範囲や精算状況が大きく異なる可能性がある。第三者支払者は、特定の薬剤を保証することを決定し、他の支払者もその薬剤に保険を提供することを保証することができないか、または適切な販売率で保険を提供することを決定する。十分な第三者精算が得られない可能性があり、薬物開発への投資の適切なリターンを実現するために十分な価格レベルを維持することができる。
第三者支払者は、価格、医薬品およびサービスの医療必要性および費用効果、ならびにそれらの安全性および有効性を検討することにますます挑戦している。販売が許可される可能性のある任意の薬物の保険や補償を得るためには、私たちの薬物の医療必要性と費用効果を証明するために、高価な薬物経済学的試験を行う必要があるかもしれない。このような試験は規制の承認を受けるために必要な試験の補完になるだろう。第三者支払者が、1つの薬剤が他の利用可能な療法と比較して費用対効果があると考えない場合、彼らは、承認後にその薬剤をその計画の下の福祉としてカバーしないかもしれない、または、もし彼らがそう思う場合、支払いレベルは、会社が利益的な方法でその薬剤を販売するのに十分ではないかもしれない。
アメリカ政府、州立法機関と外国政府はコスト制御計画の実施に大きな興味を示し、政府が支払う医療コストの増加を制限し、価格制御、精算制限とブランド処方薬の代わりに模造薬を要求することを含む。例えば、PACAには、医薬品の収益性を低下させる可能性のある条項、例えば、連邦医療補助計画に販売されている薬品のリベートを増加させ、医療補助リベートを医療補助管理保健計画に拡大し、ある連邦医療保険Dの一部の受益者に強制的に割引し、連邦医療計画における製薬会社の販売シェアに基づいて年会費を徴収することが含まれる。政府の統制と措置、既存の統制と措置の司法管轄区域で制限的な政策を強化することは、私たちの薬品の支払いを制限する可能性がある。
欧州共同体では,各国政府はその定価と精算規則や国家医療保健システムの制御により薬品価格に影響を与え,これらのシステムは消費者が支払う薬品費用の大部分に資金を提供している。一部の司法管轄区域はプラスリストとネガティブリスト制度を実施しており、この制度の下で、薬品は政府が価格を精算することに同意した後にのみ販売することができる。精算或いは定価の承認を得るために、その中のいくつかの国は臨床試験の完成を要求する可能性があり、特定の候補薬物の費用効果を現在利用可能な治療法と比較する。他の会員国たちは会社が自分の薬品価格を固定することを許可するが、会社の利益を監視する。医療コスト,特に処方薬の下り圧力が非常に大きくなっている。その結果,新薬の市場進出への壁が高まっている。また、一部の国では、低価格市場からの国境を越えた輸入が一国国内の定価に商業圧力をかけている。
政府や他の第三者支払者が保険や十分な補償を提供しない場合、商業販売のための任意の薬物の適正性が影響を受ける可能性がある。また,米国や他の国ではコスト制御措置への関心が増加しており,薬品価格の圧力を増加させ続けることが予想される。保証政策と第三者精算料率は随時変化する可能性がある。Lumosが私たちの監督管理の許可を得た1つ以上の薬物に対して有利な保証と精算状態を獲得したとしても、将来的にはあまり有利ではない保証政策と精算料率を実施する可能性がある。
販売、マーケティング、その他の会社の活動に影響を与える他の医療法
FDAを除く多くの規制機関は、米国におけるCMS、HHSの他の部門、米国司法省、および同様の外国、州、地方政府機関を含み、医薬品メーカーの販売、販売促進、およびその他の活動に適した法律および法規を管理し、実行する。承認されると、これらの法律と法規は、私たちの臨床研究計画、提案された販売とマーケティングおよび教育活動、および私たちの未来の候補製品処方者との財務および業務関係に影響を与える可能性がある。これらの法律および法規には、以下に説明するように、米国連邦、アメリカ各州、および外国の反リベート、虚偽声明、データプライバシーおよびセキュリティ法律、および私たちの現在および将来の運営に影響を与える可能性のある他の法律要件が含まれています。
FDAはその管轄内の薬品のすべての広告と販売促進活動を規制し、承認前も後も規制する。薬物が承認されると、FDAによって承認された安全性および有効性に関連する宣言のみがラベルに使用されることができる。医師は、薬物ラベルに記載されていない使用のための合法的に利用可能な薬剤、および私たちが試験およびFDAによって承認された用途とは異なる使用を処方するかもしれない。このような非ラベル使用は医学専門科でよく見られ,通常医師の信念を反映しており,すなわち非ラベル使用は患者にとって最も良い治療である。FDAは医師が治療を選択する行為を規範化していないが,FDAの規定は確かにメーカーの非ラベル使用に関するコミュニケーションに厳しい制限を加えている。もし私たちが適用されたFDAの要求に従わなければ、私たちは不利な宣伝、FDAの法執行行動、広告の是正、同意法令、およびFDAが利用可能なすべての民事と刑事罰に直面するかもしれない。医薬品の非ラベル使用を促進することは、以下に説明する虚偽宣言法にも関連する可能性がある。
反リベート法は、連邦医療保険および医療補助などの政府医療計画に基づいて精算可能な物品およびサービスに適用される連邦反リベート法規を含むが、これらに限定されないが、個人または実体が知られている場合、直接または間接的に報酬を請求、受信、提供または支払いして、購入、レンタル、注文または推薦、購入、レンタル、または連邦医療計画の下で全部または部分的に精算可能な任意の商品、施設、物品またはサービスを誘導または推薦することが違法である。法律規定の広範性、限られた法定例外状況と安全港の監督管理、および業界実践に対する法規、機関コンサルティング意見、次監督管理指導と司法裁決形式の指導が不足しているため、私たちのやり方は反リベートまたは同様の法律の挑戦を受ける可能性がある。しかも、最近の医療改革法案はこのような法律を強化した。例えば、PACAは、他に加えて、個人または実体がこれらの法規またはこれらの法規に違反する特定の意図を実際に理解する必要がないことを明らかにするために、連邦反バックトラック法規および刑事医療詐欺法規の意図要件を修正する。また、PACAは、政府は、連邦民事虚偽クレーム法案の目的に基づいて、連邦反リベート法規違反による物品やサービスのクレームが虚偽または詐欺的クレームを構成することを含むと断言できることを明らかにした。
虚偽請求法は、連邦民事虚偽請求法案に限定されるものではないが、他の事項に加えて、インフォームドコンセントおよび自発的な場合に連邦政府(連邦医療保険および医療補助を含む)に虚偽または詐欺的医薬品またはサービスの精算請求を提起すること、または請求に応じて提供されていない項目またはサービスのクレーム、または医療上不必要な項目またはサービスに対するクレームを禁止する。我々の薬品販売やマーケティングに関する活動は,これらの法律の審査や,民事罰金法やHIPAAの一部として制定された刑事医療詐欺条項を受ける可能性がある。
HIPAAは、任意の医療福祉計画を詐欺または意図的に実行または実行しようと意図的および意図的に、または医療福祉、プロジェクトまたはサービスの提供または支払いに関連する重大な事実を故意および故意に偽造、隠蔽または隠蔽または隠蔽するか、または任意の重大な虚偽陳述を行うなどに刑事および民事責任を課す。アメリカ連邦反リベート法規と類似して、個人或いは実体は実際にこの法規或いはこの法規に違反する具体的な意図を理解する必要がなく、違反を実施することができる。
“健康情報技術促進経済と臨床健康法案”改正されたHIPAA及びその実施条例はある電子医療取引の進行を管理し、そしてHIPAAカバーの実体及びその商業パートナーの保護された健康情報の安全とプライバシーを保護することについて要求を提出し、これらの実体はこのようなカバー実体にHIPAA保護された健康情報に関連するサービスを提供する。
連邦医師は、連邦医療保険、医療補助または児童健康保険計画(いくつかの例外を除く)に基づいて支払うことができる薬品、器具、生物製品および医療用品のいくつかのメーカーに、医師(医師、歯科医師、光線師、足科医および脊椎マッサージ師を含む)、いくつかの非医師保健専門家(医師アシスタントおよび看護師従業員などを含む)および教育病院支払い金または他の“価値移転”に関する情報、および上述した医師およびその直系親族が所有する所有権および投資権益に関する他の情報を毎年政府に報告することを要求する
さらに、私たちは、これらに限定されないが、研究、流通、販売およびマーケティング計画、および任意の第三者支払人(商業保険会社を含む)の精算に関連する医療項目またはサービスの請求を提出することを含むが、これらに限定されないが、これらに限定されないが、研究、流通、販売、およびマーケティング配置、および任意の第三者支払人(商業保険会社を含む)の清算に関する医療項目またはサービスの請求を提出することを含む、上述した連邦法律と同等の州法律に制限されることができる。医薬品の価格設定および/またはマーケティング情報に関する報告書を州に提出することを製薬業者に要求する州法律、例えば、医療専門家および実体に提供されるプレゼント、報酬およびその他の報酬および価値項目の追跡および報告;州および地方の法律要件
医薬品販売代表の登録;および場合によっては健康情報のプライバシーと安全を管理する州法律であり、その中の多くの法律は互いに大きく異なり、それによってコンプライアンス仕事を複雑化させる。
これらの法律の広範性と利用可能な法定例外状況と安全港の規制の範囲が限られているため、私たちのいくつかの業務活動は、私たちを代表して行動したり、サービスを提供したりする独立請負業者または第三者の行為、および候補製品が発売を許可された後の任意の販売およびマーケティング活動を含み、法律の挑戦と法執行行動を受ける可能性がある。私たちの業務が上記の任意の連邦および州法律または私たちに適用される任意の他の政府法規に違反していることが発見された場合、私たちは重大な民事、刑事および/または行政処罰および不利な行動を受ける可能性があり、いずれも私たちの業務運営能力および私たちの運営結果に悪影響を及ぼす可能性があります。これらの法律に違反することは、罰金および民事罰金を含む刑事、民事、行政制裁をもたらす可能性があり、連邦医療保険および医療補助を含む連邦医療計画から除外される可能性があり、返還、契約損害、名声損害、および政府エンティティとの会社誠実協定または他の同様の合意を実施することは、会社に厳しい運営および監督要求を加える可能性がある。執行幹事や従業員にも同様の制裁や処罰および個人監禁を加えることができ、いわゆる“責任ある会社幹事”の原則に基づいて執行幹事に刑事制裁を科すことを含め、執行幹事が意図しない違法や不正行為があることを知らない場合であっても同様である。もし有罪になったら、会社と個人は重罰と罰金を受ける可能性があります, 調査された会社や個人が何の不正もないことを認めても、このような違反に対する告発は和解につながることが多い。和解にはしばしば重大な民事制裁と追加的な会社の誠実な義務が含まれる。もし政府が私たちや私たちの幹部たちがこのような法律に違反したことを告発したり有罪にしたりすれば、私たちの業務は損なわれるかもしれない。
EUや他の国では、医薬品の普及とマーケティングにも同様の硬性制限が加えられている。私たちが薬品の普及やマーケティングを直接担当していないかもしれない国でも、潜在的な国際流通パートナーが不適切な活動をしていれば、私たちに悪影響を及ぼすかもしれません。
施設
私たちの会社の本社はテキサス州オースチンにあります。2023年11月に満期になった賃貸契約によると、私たちはそこで約5,000平方フィートのオフィススペースを借りました。2026年3月に満期になったレンタル契約によると、私たちはアイオワ州エイムズで約6,000平方フィートの追加行政と行政事務スペースも借りました。2023年1月、私たちはアイオワ州のエイムズで賃貸契約を更新した。契約更新期限は2026年3月31日です。私たちは合理的な条件の下で、私たちが未来の需要を満たすために追加的な空間を便利に得ることができると信じている。
従業員と人的資本
2022年12月31日まで、私たちは32人の従業員がいる。私たちの職員たちの中で誰も集団交渉協定を代表していない。私たちは私たちが職員たちと良い関係を維持していると信じている。
私たちは最も優秀な人材を誘致して維持するために努力している。私たちの人的資本目標には、私たちの既存と新しい従業員、コンサルタント、コンサルタントを識別、採用、維持、激励、統合が含まれている。私たちの株式と現金インセンティブ計画の主な目的は、株式と現金に基づく報酬奨励を付与することで、これらの従業員を激励してできる限りのことをし、私たちの目標を実現することで、株主価値と会社の成功を増加させることです。
利用可能な情報
アメリカ証券取引委員会に年度、四半期、現在の報告書、依頼書、その他の情報を提出します。インターネットサイト:www.Lumos-pharma.comもあります。我々は、これらの資料を米国証券取引委員会に電子的に提出または提供した後、我々のインターネットサイト上で、我々の年間報告書(Form 10-K)、Form 10-Q四半期報告、Form 8-Kの現在の報告、取引法第13(A)または15(D)条に基づいて提出または提供された報告書の修正案、および我々の委託書を無料で提供する。あなたはまたアメリカ証券取引委員会のウェブサイトからこれらの書類のコピーを得ることができます。
第1 A項。リスク要因
リスク要因の概要
以下は,我々の普通株投資に投機的あるいはリスクを持たせる要因の概要である.この結論は私たちが直面しているすべての危険を解決していない。我々の普通株に対する投資決定を行う前に、本リスク要因要約でまとめられたリスクおよび我々が直面している他のリスクのより多くの議論を以下の“リスク要因”のタイトルで見つけることができ、これらの議論は、本年度報告および米国証券取引委員会の他の文書の他の情報と共に慎重に考慮すべきである。
私たちの財務状況と資本要求に関連するリスク
•私たちの経営の歴史は限られており、設立以来ずっと大きな損失を受けており、予測可能な未来には、引き続き大量かつ増加していく損失を被ることが予想される。私たちはただ一つの候補製品があり、商業販売がなく、私たちの限られた運営履歴に加えて、私たちの業務と将来の生存能力を評価することは難しい。
•私たちは現在製品収入源がなく、永遠に利益を上げないかもしれない
•私たちは私たちの運営を支援するための追加の資金が必要になるだろうが、これらの資金は受け入れ可能な条項で私たちに提供できないかもしれないし、全く得られないかもしれない。これは、私たちの研究開発計画と他の運営または商業化努力を延期、減少、または一時停止させるだろう。追加資本の調達は、私たちが不利な条項の影響を受け、私たちの既存の株主の株式が希釈され、私たちの運営を制限したり、私たちの候補製品や技術に対する権利を放棄することを要求するかもしれません。
•私たちの経営業績は大幅に変動する可能性があり、これは私たちの将来の経営業績を予測しにくくし、私たちの経営業績が予想を下回ったり、私たちの指導を招いたりする可能性があります。
•私たちの純営業損失繰越およびいくつかの他の税務属性を使用する能力は、1986年に改正された国税法(以下、国税法)第382条および383条によって制限されている。
私たちの候補製品の開発と商業化に関するリスク
•著者らの臨床試験の中期、初歩或いは主要なデータは、著者らが2022年11月14日に発表したLum-201試験の中期データを含み、更に多くの患者データの獲得に従って変化し、そして監査と検証プログラムの影響を受ける可能性があり、これは最終データの重大な変化を招く可能性がある。
•私たちの成功は私たちの唯一の候補製品Lum-201の成功した開発、規制承認、商業化に大きく依存する。
•PGHDのためのLUM−201の開発を支援する基礎的な分析は,マク社が1990年代に行った3つの臨床試験のデータと,そのうちの1つの試験の事後分析からである。このような試験と分析に関連する様々な問題は著者らのLum-201臨床試験設計と著者らの未来の発展計画に実質的な悪影響を与える可能性がある。
•臨床前試験あるいは早期臨床試験の結果が必ずしも将来の結果を予測できるとは限らないため、Lum-201は今後の臨床試験で有利な結果がない可能性があり、監督部門の承認も得られない可能性がある。
•もし私たちの任意の候補製品を変更すれば、追加の臨床試験が必要になり、追加のコストと遅延を招くかもしれません
•私たちは、より利益的または成功可能性の高い候補製品または適応を利用することなく、特定の候補製品または適応を追求するために限られたリソースを費やす可能性がある。
私たちの業務運営に関するリスク
•私たちの未来の成功は私たちがCEO総裁と管理チームの他の重要なメンバーを引き留める能力と、合格した人材を誘致、維持、激励する能力にかかっている
•私たちは私たちの開発、規制、販売とマーケティング能力を拡大することを望んでいますので、私たちは私たちの成長を管理する上で困難に直面するかもしれません。これは私たちの運営を乱すかもしれません。
•業務中断は私たちの臨床試験、未来の収入と財務状況を深刻に損害し、私たちのコストと支出を増加させる可能性がある。
•もし私たちがアメリカ以外の場所でLUM-201を商業化することを許可されれば、私たちは追加のリスクに直面するだろう。
•我々の内部コンピュータシステム、または私たちの契約研究組織(“CRO”)または他の請負業者またはコンサルタントのシステムは、故障したり、セキュリティホールが発生したりする可能性があり、これは、私たちの薬物開発計画が実質的に破壊される可能性がある。
私たちの知的財産権に関するリスクは
•もし私たちが私たちの技術や候補製品のために有効な知的財産権を獲得して維持することができなければ、あるいは知的財産権保護の範囲が十分でなければ、私たちの技術や製品を商業化することに成功する能力は重大な悪影響を受ける可能性がある。
•私たちはLUM-201の物質成分に関する特許保護を持っていない
•私たちは私たちの知的財産権を保護したり実行したりする法的手続きに巻き込まれるかもしれないが、これは高価で時間がかかり、成功しないかもしれない。
•第三者は、私たちが彼らの知的財産権を侵害したことを告発する法的訴訟を提起する可能性があり、その結果は不確実であり、私たちの業務成功に実質的な悪影響を及ぼす可能性がある。
•もし私たちがその商業秘密の機密性を保護できなければ、私たちの技術的価値は実質的な悪影響を受け、私たちの業務と競争地位を損なうかもしれない。
政府の規制に関連するリスク
•規制承認過程は高価で、時間がかかり、不確定であり、私たちまたは私たちの協力パートナーが私たちの候補製品を商業化する承認を得ることを阻止する可能性がある。
•私たちが候補製品の規制承認を得ても、持続的な規制義務と持続的な規制審査の制約を受けることになり、これは多くの追加費用を招く可能性があり、適用される規制要求を遵守できなければ処罰されるだろう。
•外国の管轄区域の規制承認を得られなければ、私たちは国際的に私たちの製品を販売することができないだろう。
•医療改革措置は私たちの候補製品の商業的成功を阻害したり阻止したりするかもしれない。
•私たちは医療専門家、臨床研究者、CRO、第三者支払者との関係は、私たちの現在と未来の業務活動に関連しており、連邦および州の医療詐欺と乱用法律、虚偽クレーム法律、透明性法律、政府価格報告、医療情報プライバシーと安全法律の制約を受ける可能性があり、これは私たちを刑事制裁、民事処罰、契約損害、政府医療計画から排除された名声損害、行政負担、利益、将来の収入減少などのリスクに直面させる可能性がある。もし私たちが医療法規を守らなければ、私たちは巨額の処罰に直面する可能性があり、私たちの業務、運営、財務状況は不利な影響を受けるかもしれない。
私たちの普通株式所有権に関連するリスク
•私たちの普通株の取引価格は大きく変動する可能性があり、様々な要素によって大幅に変動する可能性があり、その多くの要素はコントロールできません
•私たちの主要株主と経営陣は私たちのかなりの割合の株を持っており、株主の承認が待たれる事項に大きな影響を与えることができるだろう。
•我々が改正して再記述した定款(“定款”)は、デラウェア州の州裁判所を指定するか、または、デラウェア州に位置する州裁判所が管轄権を有していない場合には、デラウェア州地域の連邦裁判所を唯一かつ独占的なフォーラムとして指定する。
•私たちは予測可能な未来に、私たちはどんな現金配当金も支払わないと予想している。
•私たちの会社の登録証明書、私たちの定款あるいはデラウェア州の法律の条項はわが社の支配権の変更、あるいは私たちの経営陣の変更を阻止、延期、阻止する可能性があり、それによって私たちの普通株の取引価格を下げることができます。
リスク要因
私たちの普通株に投資することは重大な危険と関連があり、その中のいくつかの危険は以下のように説明される。私たちの業務を評価する際に、投資家は以下のリスク要因を慎重に考慮しなければならない。これらのリスク要因には歴史的情報のほかに、重大なリスクと不確実性に関する前向きな陳述が含まれている。私たちの実際の結果は展望的陳述で議論された結果と大きく違うかもしれない。このような差異をもたらすか、または促進する可能性のある要因は、以下の説明に限定されるものではないが、これらに限定されない。以下のリスクの提示順序は、そのリスクの大きさを反映するためではない。以下のリスクの発生は、当社の業務、財務状況、経営結果、および将来性に重大な悪影響を及ぼす可能性があります。この場合、私たちの普通株の取引価格は下がるかもしれません。あなたはあなたの全部あるいは一部の投資を失うかもしれません
私たちの財務状況と資本要求に関連するリスク
私たちの経営の歴史は限られており、設立以来ずっと大きな損失を受けており、予測可能な未来には、引き続き大量かつ増加していく損失を被ることが予想される。私たちはただ一つの候補製品があり、商業販売がなく、私たちの限られた運営履歴に加えて、私たちの業務と将来の生存能力を評価することは難しい。
私たちは臨床段階の生物製薬会社で、運営の歴史は限られている。私たちは販売を許可された製品は何もありません。現在私たちは私たちの候補製品Lum-201の開発に集中しています。私たちの業績、生存能力、または未来の成功を評価することは、私たちがより長い運営歴史や市場で承認された製品を持っている場合よりも難しいだろう。私たちは引き続き私たちの運営に関する多くの研究と開発、一般と管理費用を発生させ続けている。生物製薬製品開発への投資は非常に高い投機性があり、それは大量の前期資本支出を必要とし、重大なリスクが存在するため、即ち任意の潜在的な候補製品は十分な効果或いは受け入れ可能な安全性を証明できず、監督部門の許可を得られない、或いは商業上実行可能ではない。私たちが設立して以来、私たちは毎年重大な運営損失が発生しており、予測可能な未来には大量かつ増加する損失が予想される。2022年12月31日現在、私たちの累計赤字は1兆275億ドルです。
我々はこれまで,臨床試験を含めて基本的にすべての努力を研究·開発に投入してきたが,候補製品の開発はなされていない。私たちの費用は大幅に増加すると予想されています
•私たちの候補製品LUM-201と任意の未来の候補製品の研究開発を続けてください
•我々のOraGrowth H試験を含むLum−201の臨床試験を行った
•より多くの候補製品の許可を得て、これらの候補製品を開発するための任意の将来のコストを生成することを求める
•Lum-201と将来臨床試験に成功した任意の候補製品に対する監督部門の承認を求める
•販売、マーケティング、および流通インフラを確立し、規制部門の承認を得た場合に、Lum-201または任意の将来の候補製品を商業化するための製造能力を拡大し、Lum-201または任意の将来の候補製品を商業規模で製造するためのプロセスを改善すること;および
•LUM−201および任意の将来の候補製品の開発を支援する人員を含むより多くの人員を雇用し、候補製品が承認された場合には、その商業化努力を支援する業務、財務、および情報管理システムを強化する。
将来的に利益を得るためには、LUM-201や他の巨大な市場潜在力を持つ製品の開発に成功し、最終的に商業化しなければならない。これは、Lum-201および任意の未来の候補製品を推進し、これらの候補製品の臨床試験を完了し、これらの候補製品の規制承認を得ること、および規制承認を得る可能性のある製品の製造、マーケティング、および販売を含む一連の活動で成功することを要求するであろう。私たちはただその中のいくつかの活動の初期段階にいるだけだ。私たちはこのような活動で成功しないかもしれないし、未来に利益を得るのに十分な収入を得られないかもしれない。私たちが利益を出しても、私たちは四半期や年間の収益性を維持したり向上させることができないかもしれない。私たちが持続的な利益を達成できなければ、私たちの価値を低くし、資金を調達し、業務を拡大し、私たちの候補製品を多様化し、候補製品をマーケティングする(承認されれば)、あるいは運営を継続する能力を弱めるかもしれない。
私たちは現在製品収入源がなく、永遠に利益を上げないかもしれない
今まで、私たちは商業製品販売から何の収入も得ていない。規制部門のLum-201または任意の未来の候補製品の承認を得ることに成功したとしても、これらの製品がいつ製品販売から収入を生むのか分からない。私たちが製品販売から収入を得て利益を達成する能力は、LUM-201または将来的に開発、許可または買収する可能性のある任意の候補製品を含む、私たち単独または任意の未来のパートナーと製品を商業化することに成功する能力に依存するだろう。LUM-201または任意の将来の候補製品の製品販売から収入を得る能力は、私たちまたは任意の未来のパートナーの能力を含む多くの他の要素にも依存します
•OraGrowth試験とLum-201の第三段階臨床試験を含む開発活動を完成し、成功し、適時であった
•Lum-201の安全性と有効性を証明し、FDAを満足させ、Lum-201および将来商業市場がある候補製品の監督部門の承認を得た
•申請を記入して外国の監督機関に提出し、監督部門の許可を得る
•私たちの製品に商業的に可能な価格を設定します
•信頼できる第三者と供給および製造関係を確立し、維持し、このような供給を維持するために、バルク薬物および医薬製品を十分かつ合法的に製造することを保証する
•私たちが独立して商業化しようとしている市場で販売、マーケティング、流通することができるビジネス組織を設立し、マーケティングの承認を得た任意の製品を販売することができます
•適切な流通パートナーを探して、私たちが承認した製品を他のマーケティング、販売、流通を支援してくれます
•政府と個人支払者を含む第三者支払者から保険と適切な補償を受ける
•私たちが承認した製品を市場に承認させます
•第三者の特許干渉または特許侵害クレームを回避するために、私たちの知的財産権を確立、維持、保護すること
•人材を引きつけ、採用し、引き留める。
さらに、Lum−201または任意の将来の候補製品を含む医薬品開発に関連する多くのリスクおよび不確実性のため、開発中に進展することができないか、または適用臨床試験の終点に達することができない可能性があり、費用を増加させる時間または金額、またはいつ、または利益を達成または維持することができるかどうかを予測することができない。さらに、FDAや外国規制機関に現在予想されている基礎の研究や実験を要求されたり、決定されたりすると、私たちの費用は予想を超える可能性があります。LUM−201や任意の将来の候補製品の開発や規制過程を完成させることができても、これらの製品の商業化に関連した巨額のコストが生じることが予想される。
Lum-201の販売または承認される可能性のある任意の将来の候補製品から収入を得ることができても、私たちは利益を得ることができず、運営を継続するために追加の資金が必要かもしれない。もし私たちが利益を上げることができない場合、または持続的に利益を上げることができない場合、私たちは計画通りに運営を継続できず、私たちの運営を減少または閉鎖させることができないかもしれない。
私たちは私たちの運営を支援するための追加の資金が必要になるだろうが、これらの資金は受け入れ可能な条項で私たちに提供できないかもしれないし、全く得られないかもしれない。これは、私たちの研究開発計画と他の運営または商業化努力を延期、減少、または一時停止させるだろう。追加資本の調達は、私たちが不利な条項の影響を受け、私たちの既存の株主の株式が希釈され、私たちの運営を制限したり、私たちの候補製品や技術に対する権利を放棄することを要求するかもしれません。
LUM−201および任意の将来の候補製品の開発および潜在的商業化を完了し、それらが承認されれば、大量の資金が必要となる。私たちの未来の資金調達需要は多くの要素に依存し、その中のいくつかの要素は私たちがコントロールできない
•私たちの臨床試験の進捗とコスト
•FDAと他の規制機関が承認した時間と関連するコストを求める
•適用される規制機関に必要な上場後の承認承諾の程度を決定する
•適切なパッケージが販売のための完成品を得るために、第三者と商業的に実行可能な供給および製造関係を確立および維持することを含む、LUM-201および任意の将来の候補製品のための高効率、費用対効果、および拡張可能な製造プロセスを開発すること;
•LUM−201または任意の将来の候補製品が承認された場合、製品販売、マーケティング、製造および流通を含む商業化活動のコスト;
•私たちまたは未来のパートナーが発売した任意の製品の市場受容度と受容率
•任意の上場承認後の持続可能な安全プロファイル;
•任意の特許請求書および他の知的財産権の費用の提起、起訴、弁護、および実行;
•私たちは追加的な協力、許可、商業化、または他の計画の能力、およびそのような計画の条項と時間を達成する
•競争的な技術や他の不利な市場発展が現れました
•合格した人材を引きつけ、採用し、維持するコスト。
私たちは私たちが計画した開発のために約束したいかなる物質、外部資金源、または他の支援も持っていない。私たちが十分な製品収入を生成して私たちの現金需要を満たすことができる前に、私たちは公開または私募株式発行、債務融資、協力、戦略連合、許可手配、その他のマーケティングと流通手配を通じて、未来の現金需要に資金を提供する予定です。私たちが必要な時、私たちは追加的な融資を受けることができないかもしれないし、これらの追加的な融資は優遇された条項で提供されないかもしれない。もし私たちがマーケティングおよび流通手配、または第三者との他の協力、戦略連合、または許可手配によって追加の資本を調達する場合、私たちはLUM-201または任意の潜在的な将来の候補製品、技術、将来の収入フロー、または研究計画のいくつかの価値のある権利を放棄しなければならないか、または私たちに不利になる可能性のある条項に許可を付与しなければならないかもしれない。公開または私募株式発行によってより多くの資本を調達すれば、私たちの既存株主の所有権権益は希釈され、これらの証券の条項には、清算またはその株主権利に悪影響を及ぼす他の特典が含まれる可能性がある。もし私たちが債務融資を通じて追加資本を調達すれば、私たちは追加債務を負担し、資本支出を行ったり、配当を発表するなど、条約によって制限されたり、私たちが具体的な行動を取る能力を制限するかもしれない。もし私たちが必要な時に十分な資金を得ることができなければ、私たちは私たちの1つ以上の臨床試験や研究開発計画、または私たちの商業化努力を延期、縮小、または一時停止しなければならないかもしれない。
私たちの経営業績は大幅に変動する可能性があり、これは私たちの将来の経営業績を予測しにくくし、私たちの経営業績が予想を下回ったり、私たちの指導を招いたりする可能性があります。
私たちの四半期と年間経営業績は将来的に大きく変動する可能性があり、将来の経営業績を予測することは困難です。私たちは時々他の会社と協力協定を締結するかもしれません。開発資金と重要な前払いとマイルストーン支払いおよび/または印税を含む。したがって、私たちの収入は、開発資金と、任意の潜在的な未来の協力および許可協定によって達成される開発および臨床マイルストーン、およびその候補製品の販売(承認されれば)に依存する可能性がある。これらの前金とマイルストーン支払いは時期によって大きく異なる可能性があり、このような違いは、私たちの経営業績が異なる時期の間に大きな変動をもたらす可能性があります。また,ブラック·スコアーズオプション定価モデルを用いて付与日の公正価値とそれによる株式ベースの報酬費用を推定し,コストを従業員に必要なサービス期間内の費用として確認した。これらの賞を評価するための変数が時間とともに変化するにつれて,我々が認識しなければならない費用の大きさが大きく異なる可能性がある.また、私たちの経営業績は様々な他の要素によって変動する可能性があり、その中の多くの要素は私たちがコントロールできないもので、以下の要素を含む予測が困難かもしれません
•LUM−201および任意の将来の候補製品に関連する研究および開発活動の時間、コスト、および投資レベルは、時々変化するであろう
•患者の臨床試験に参加する能力と募集時間を募集します
•LUM−201および任意の将来の候補製品を製造するコストは、FDAのガイドラインおよび要求、生産数量、および私たちが製造業者と達成した合意条項によって異なるかもしれない
•他の候補製品や技術を取得または開発するための支出を獲得または生成することができます
•LUM−201および任意の将来の候補製品または競合候補製品の臨床試験の時間および結果;
•業界競争構造の変化は、私たちの競争相手またはパートナー間の統合を含む
•LUM−201または将来の任意の候補製品の規制審査または承認における任意の遅延;
•LUM−201および任意の将来の候補製品の需要レベルは、それらが承認されれば、大きく変動する可能性があり、予測が困難である
•私たちの候補製品のリスク/収益プロファイル、コストおよび精算政策(承認されれば)、および私たちの候補製品と競争する既存および潜在的な未来の薬剤について;
•Lum−201または私たちの将来の任意の候補製品と競合する既存および潜在的な将来の薬剤からの競争;
•私たちは独立して、または第三者と協力して、アメリカ境内外でLUM-201または任意の未来の候補製品を商業化することができる
•私たちは協力、許可、または他の手配の能力を確立し、維持する
•私たちは未来の成長能力を十分に支持しています
•潜在的な予測不可能な業務中断は、私たちのコストや支出を増加させる
•未来の会計宣言や私たちの会計政策の変化;
•絶えず変化して不安定な世界経済環境。
これらの要因の累積影響は我々の四半期や年度経営業績に大きな変動と予測不可能を招く可能性がある。したがって、異なる時期に私たちの経営業績を比較することは意味がないかもしれない。投資家は私たちの過去の業績をその未来表現の指標として依存してはいけない
私たちの純営業損失の繰越といくつかの他の税務属性を使用する能力は規則382と383節に制限されています。
この規則382および383条は、会社が任意の将来の課税収入または税金を相殺するために、その純営業損失の繰越およびいくつかの他の税務属性(研究控除を含む)を利用する能力を制限し、会社が任意のスクロールの3年間の間に50%を超える所有権変更を経験したことを前提としている。国の純営業損失繰越(ある他の税務属性と)も同様の制限を受ける可能性がある。したがって、第382条の所有権変更は、このような変更がない場合には会社が生じる納税義務よりもはるかに大きくなる可能性があり、いかなる増加した負債も、会社の業務、経営業績、財務状況及びキャッシュフローに悪影響を及ぼす可能性がある。
2020年3月18日までの第382条所有権変更分析によると,合併の結果,歴史上のNewLinkとPrivate Lumosはいずれも2020年3月18日に第382条所有権変更を経験した
これらの所有権変更は、連邦純営業損失の繰越能力と、私たちと私たちの子会社のそれぞれの所有権が変更される前に蓄積された他の税務属性を利用して、将来このような属性を利用する能力を制限し続ける可能性があります。
その後の分析によると、2020年3月19日から2021年12月31日まで、第382条の所有権変更を経験していない。私たちが少ないまたはコントロールできない事件のため、将来的には、私たちの5%の株主が私たちの株式を購入して売却すること、新しい5%の株主が出現すること、追加の株式発行または償還、または私たちの5%の株主のいずれかの所有権がいくつか変化することを含む追加の所有権変化が生じる可能性がある。
会計宣言は私たちが報告した経営結果と財政状況に影響を及ぼすかもしれない。
米国公認の会計原則(“米国公認会計原則”)及び関連する実施ガイドラインと解釈は非常に複雑であり、主観的判断に関連する可能性がある。これらの規則またはその解釈の変化、新しい声明を採用するか、または既存の声明を当社の業務に適用する変化は、私たちが報告した財務諸表および運営結果を著しく変更する可能性があります。
上場企業として、私たちの運営コストは高く、私たちの経営陣はコンプライアンス義務を履行するために多くの時間を投入する必要があります。
上場企業として、私たちは取引所法案、2002年のサバンズ-オクスリ法案(“サバンズ-オキシリー法案”)および米国証券取引委員会とナスダックがその後施行した規則の報告要求を守るために、大量の法律、会計、その他の費用を発生させた。これらの規則と条例の要求を満たすには巨大な法律と財務コンプライアンスコストが必要であり、いくつかの活動を更に困難にし、時間或いはコストを高くし、また私たちの人員、システムと資源に不必要な圧力をもたらす可能性がある。私たちの経営陣と他の人たちはこのようなコンプライアンス要求に多くの時間を投じた。さらに、これらの規制は、取締役および上級者責任保険をより難しく、より高価にする可能性があり、私たちは、同じまたは同様の保険を得るために、低減された保険限度額および保険範囲を受け入れることを要求されるかもしれません。したがって、私たちは合格した人を私たちの取締役会、私たちの取締役会、または役員に引き付けることがもっと難しいかもしれない。
サバンズ·オキシリー法第404条に基づいて効果的な内部統制を実現し、維持することができなかったことは、正確な財務諸表を作成する能力と我々の株価に実質的な悪影響を及ぼす可能性がある。
サバンズ·オキシリー法404条によると、私たちの経営陣は、私たちの財務報告書の内部統制に関する報告書を発表しなければならない。第404条の規定に適合するために、私たちは高価で挑戦的な財務報告書に対する私たちの内部統制を記録して評価する過程を展開した。コンプライアンスを継続的に維持するためには,専門的な内部資源が必要であり,外部コンサルタントを招聘し,詳細な作業計画を行う必要がある。私たちが努力したにもかかわらず、私たちは第404条で要求されたように、財務報告書の内部統制に有効であるという結論を出すことができない可能性がある。これは、私たちの財務諸表の信頼性に自信を失ったため、金融市場に不利な反応をもたらす可能性がある。
私たちの有効所得税率の変化は私たちの将来の経営業績に悪影響を及ぼすかもしれません。
私たちの有効な所得税率、そして私たちの相対的な国内と国際納税義務は、未来の任意の収入の異なる管轄区域間の分配にある程度依存するだろう。さらに、様々な要因は、個別の管轄区域または全体的な有効所得税税率に有利または不利な影響を及ぼす可能性がある。これらの要因には、税務機関が現行税法の解釈、株式オプションおよびその他の株式ベースの報酬の任意の必要な会計処理、税法および税率の変化(最近公布された米国連邦所得税法の変化を含む)、私たちの将来の研究開発支出レベル、会計基準の変化、私たちが業務を展開する可能性のある各税収管轄区域の将来の収入組み合わせの変化、米国国税局または他の税務機関の任意の審査結果が含まれる。確認されていない税収利益と繰延税金資産の実現および税前収益の全体レベル変化の推定の正確性。例えば、今回の政府はアメリカ企業の所得税税率を高め、アメリカの国際商業運営に対する税収を増加させ、世界の最低税を徴収する税改正立法を提出し、これは企業の限界税率の引き上げを招く可能性がある。いくつかの国や経済協力開発機構などの組織は, 世界的な最低税率計画を支持する。これらの国や組織も、現行税法の改正を積極的に検討しており、新たな法律を提出したり公布したりしており、これらの法律は、事業所の国での納税義務を増加させたり、業務運営方式を変更したりする可能性がある。上記の要因やその他の要因がわれわれの所得税負債に与える影響は、われわれの経営業績に重大な悪影響を及ぼす可能性がある。
私たちの候補製品の開発と商業化に関するリスク
著者らの臨床試験の中期、初歩或いは主要なデータは、著者らが2022年11月14日に発表したLum-201試験の中期データを含み、更に多くの患者データの獲得に従って変化し、そして監査と検証プログラムの影響を受ける可能性があり、これは最終データの重大な変化を招く可能性がある。
2022年11月14日、我々はLum−201試験の中間データを公表し、他の臨床試験の中期、予備、または背線データを時々公表するかもしれない。著者らの臨床試験のこれらの一時データは患者登録の完成とより多くの患者データの出現に伴い実質的に変化する可能性がある。臨床試験の中期或いは初歩的なデータは、私たちの2022年11月14日の臨時データを含み、試験の最終結果を指示できないかもしれない、あるいは非決定的である可能性があり、そして患者登録の完了とより多くの患者データの獲得に伴い、1つ以上の臨床結果が実質的に変化する可能性があるリスクに直面している。したがって、有利な中期結果は、必ずしも有利な最終結果またはFDAまたは他の規制機関の承認をもたらすとは限らない。我々が2022年11月14日に発表したLum−201試験の中期結果は鼓舞的であったが,制御群のベースライン特徴に不平衡があり,このような試験の最終結果は中期結果と大きく異なる可能性がある。中期結果も不確実である可能性があり,臨床試験や試験を継続すべきかどうかの最終結果が何であるかの不確実性をもたらす。一時データまたは予備データは依然として監査とチェック手続きを遵守しなければならず、これは最終データが一時データまたは予備データと大きく異なるか、大きく異なる可能性がある。したがって、最終データが利用可能になる前に、任意の中期または予備データを慎重に見なければならない。中期と中期の不利な違い, 予備データまたはバックラインデータおよび最終データは、私たちの名声とビジネスの将来性を深刻に損なう可能性がある。私たちが行う可能性のある任意の臨床試験が一致または十分な有効性と安全性を証明するかどうか、マーケティング承認を得て私たちの候補製品を販売するのに十分であるかどうかはわかりません。
さらに、開示された特定の研究または臨床試験に関する情報は、一般に、より広い情報に基づいており、あなたまたは他の人は、私たちが開示に含めるべき材料または他の適切な情報を決定することに同意しない可能性がある。私たちが開示しないことを決定したいかなる情報も、最終的には、特定の候補製品または私たちの業務に関する未来の決定、結論、観点、活動、または他の側面に対して重要な意味を持つと考えられるかもしれない。同様に,我々の現在の開発スケジュールに基づいて我々の候補製品の計画や進行中の臨床前研究や臨床試験を完成させることができても,我々の候補製品のこのような臨床前研究や臨床試験の積極的な結果は後続の臨床前研究や臨床試験結果に複製されない可能性がある。そのほか、臨床前、非臨床と臨床データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品が臨床前研究と臨床試験で満足できると考えているが、しかし依然としてFDA或いはその他の監督管理機関の許可を得られなかった。
私たちの成功は私たちの唯一の候補製品Lum-201の成功した開発、規制承認、商業化に大きく依存する。
私たちは規制部門の承認を受けた製品は何もない。著者らの現在の臨床段階候補製品Lum-201は1種の経口GH刺激性療法であり、部分PGHD患者と潜在的な他の内分泌疾患に適している。私たちの最近の見通しは資金調達と収入創出のための能力も含めて
これは、私たちが規制部門の承認を得る能力があるかどうか、そして承認されれば、LUM-201の商業化にタイムリーに成功できるかどうかに大きく依存する。
FDAの規制承認が得られないまで、米国でLum-201または任意の将来の候補製品を商業化することはできず、外国規制機関よりも規制の承認を得ずに、米国以外の地域でLum-201または任意の将来の候補製品を商業化することもできない。FDAの承認過程は一般的に完成するのに数年かかり、承認される保証は決してない。規制機関の承認を得てLUM−201を標的PGHD適応または任意の将来の候補製品に使用する前に、我々は、通常、臨床前および制御された良好な臨床試験で収集された大量の証拠によって、候補製品が目標適応に使用されることが安全かつ有効であり、製造施設、プロセス、および制御が十分であることを証明しなければならない。我々は、Lum−201のために同じ制御経路を求めており、その後、多くの承認された組換えヒト成長ホルモン製品および開発されている長時間効果GH製品であり、Lum−201は、以前に診断されたPGHD患者のサブセットに集中している。著者らは著者らのOraGrowth H 210用量発見試験と主要な終点が12ケ月の平均身長速度である3期臨床試験を含む幼稚な患者の治療を研究する予定であり、監督部門の許可を支持することを目的としている。以前の組換えヒト成長ホルモン製品よりも多くのまたは異なる試験を行わなければならない場合、規制部門がLum-201を承認するのに要する時間と費用(あれば)を増加させる可能性がある。また,発表された承認された組換えヒト成長ホルモン治療製品の研究から得られた成長データは,6カ月と12カ月の平均身長速度に良い相関があることを示しているにもかかわらず,その独自の特性のため,Lum−201は, 様々な結果が出ますOraGrowth H 210試験で観察されたLum−201の6カ月平均高度速度が、可能な任意の第3段階臨床試験で最終的に観察された12カ月の平均高度速度と相関しない場合、Lum−201は第3段階臨床試験で必要な主要な終点に到達できない可能性があり、Lum−201は規制部門の承認を得ない可能性がある。また、1つの国でLUM-201上場の規制承認を得ることは、他の国で規制承認を得ることができることを保証することはできないが、1つの国で規制承認を得られなかったり、遅延したりすることは、他の国の規制プロセスに悪影響を及ぼす可能性がある。
LUM−201または将来の任意の候補製品がFDAおよび同様の外国規制機関の承認を得ることに成功したとしても、任意の承認は、特定の年齢層の使用制限、警告、予防措置または禁忌症に関連する重大な制限を含むことができ、または重い承認後の研究またはリスク管理要件の影響を受ける可能性がある。私たちが1つ以上の司法管轄区域でLUM-201の規制承認を得ることができない場合、または任意の承認に重大な制限が含まれている場合、私たちはその運営に資金を提供し続けるために十分な資金を得ることができないか、または十分な収入を生成することができないかもしれない。さらに、Lum−201または任意の将来の候補製品に対する規制部門の承認が得られると、撤回される可能性がある。さらに、私たちが規制部門のLum-201の承認を得ても、Lum-201の商業成功は以下の要素を含む多くの要素に依存するだろう
•私たち自身のビジネス組織を発展させたり商業インフラとのビジネス協力を構築したり
•商業的に実行可能な価格設定を制定し、第三者と政府支払者から適切な補償の承認を得る
•当社の第三者製造業者は、予想される需要を満たし、私たちの製造コストを低減し、FDAのcGMPに適合する規模で大量のLum−201を生産するのに十分な能力で、商業的に実行可能なプロセスを使用する
•私たちはLUM−201の利点、管理、および使用に関する医師および患者を教育することに成功した
•代替療法と競争療法の獲得性、知覚優位性、相対コスト、相対安全性と相対的有効性
•私たち自身または私たちの潜在的な戦略パートナーのマーケティング、販売、流通戦略と運営の有効性
•患者、看護者、および医学界がLUM-201を受け入れることは安全で有効である
•承認された後も、LUM−201のセキュリティ状態は許容される
•知的財産権許可における私たちの義務を優遇条項と第三者で履行し続けます。
その中の多くの要素は私たちがコントロールできることではない。もし私たちや私たちの商業化パートナーがLum-201を商業化することに成功しなければ、私たちは私たちの業務を続けるのに十分な収入を得ることができないかもしれない。
PGHDのためのLUM−201の開発を支援する基礎的な分析は,マク社が1990年代に行った3つの臨床試験のデータと,そのうちの1つの試験の事後分析からである。このような試験と分析に関連する様々な問題は著者らのLum-201臨床試験設計と著者らの未来の発展計画に実質的な悪影響を与える可能性がある。
OraGrowth H 210実験が成功する確率の高さは,実験設計の十分性に依存する.このような試験を設計する際に、20世紀90年代にメルク社が完成したLum-201に関する3つの研究(“メルク試験”)のデータと分析を振り返り、著者らのメルク社の臨床データの分析結果をOraGrowth H 210試験の設計に組み入れた。しかし、私たちはこれらのデータを曲解したり、これらのデータを欠陥的に分析したりするかもしれない。メルク実験の解釈や分析に影響を与える可能性がある要素は
•20世紀90年代にメルク試験を行って以来、臨床試験プログラムと統計分析方法は変化した可能性があり、これは著者らが試験設計の変化がOraGrowth H 210試験結果にどのように影響する可能性があるかを効果的に予測する能力を制限した
•メルク社の2つの試験は完成する前に治療効果の不足で中止された
•その中の1つのメルク試験は小児患者の治療試験において薬物の処方を部分的に変化させ、もう1つの以前に治療した患者試験では、製剤の変化は試験全体で行われ、その後、変更後の処方のバイオアベイラビリティが30%~40%低下したことが決定された
•メルク実験のいくつかのオリジナル文書を含むメルク実験のいくつかの関連情報は、まだ提供されていないかもしれないので、私たちの分析およびOraGrowth H 210実験設計の参考にすることができません
•小サンプル量のばらつきや,我々がOraGrowth H 210テスト設計に依存しているメルク実験の事後解析に固有の他の制約は,このような事後解析が信頼できない可能性がある.
これらの要素および他の要素により、OraGrowth H 210試験の設計に欠陥がある可能性があり、その失敗を招く可能性があり、これは私たちの業務、将来の発展計画、および将来に実質的な悪影響を与えるだろう
臨床前試験や早期臨床試験の結果が必ずしも将来の結果を予測できるとは限らず,他の適応にも変化しない可能性があるため,Lum−201は以降の臨床試験では有利な結果が得られない可能性があり,規制部門の承認も得られない可能性がある。
臨床前試験と早期臨床試験の成功は今後の臨床試験が十分なデータを産生することを確保せず、研究薬物の有効性と安全性を証明する。製薬やバイオテクノロジー業界のいくつかの会社は,より多くの資源や経験を持つ会社を含め,臨床試験で大きな挫折を経験し,比較的早い臨床試験でも有望な結果を見ている。私たちが行っているか、あるいは可能な臨床試験が十分な有効性と安全性を証明するかどうかはわかりません。それによって、監督管理機関はLum-201の発売を許可します。規制部門が候補製品の上場申請を承認するのに十分なデータがあると考えても、FDA、EMA、または他の適用される外国の規制機関は同意しない可能性があり、追加の臨床試験を要求するかもしれません。後期臨床試験に有利な結果が生じなければ,規制部門の承認を得たLum−201の能力は悪影響を受ける可能性がある。
Lum−201は,これまでに行ったメルク試験と比較してOraGrowth H 210試験で新たな安全リスクや増加した安全リスクを示さないか,あるいはOraGrowth H 210試験を完了すれば,計画された3期臨床試験で新たなあるいは増加した安全リスクは認められない。他の適応の試験は成功しない可能性があり,あるいは新たなあるいは増加した安全リスクを示す可能性がある。我々のOraGrowth H 210実験の中期結果は鼓舞的であったが,制御群のベースライン特徴に不平衡があり,我々の最終結果は大きく異なる可能性がある。そのほか、臨床前と臨床データはよく異なる解釈と分析の影響を受けやすく、多くの他の会社は彼らの候補製品が臨床前研究と臨床試験の中で満足できると思っているが、しかし依然として監督管理部門のその製品マーケティングに対する許可を得られなかった。
また,2022年11月14日に発表された仮データに基づいて1.6 mg/kg/日のLum−201用量を進める予定であるが,これはLum−201の最適用量ではない可能性があり,OraGrowth H 210試験の最終データによれば,より理想的な用量が存在すれば,余分なコストや遅延が生じる可能性があると結論した。OraGrowth H 210試験で現在投与されている3つの用量レベルが有効であるかどうかは保証されないし、またはそれらが有効である場合、いずれかが最適な用量であることも保証されない。OraGrowth H 210試験は、我々のPEM戦略の成功した患者選択を成功的に示すことができないか、または将来の開発および潜在的市場承認に適したLum-201用量または用量レジメンを決定することができるかもしれない。1つの適応において最適な用量が決定されても、異なる用量は、そのような最適な用量を決定するために追加の臨床試験を必要とする他の適応において最適である可能性がある。
もし私たちの任意の候補製品を変更すれば、追加の臨床試験が必要になり、追加のコストと遅延を招くかもしれません。
私たちは、私たちのいくつかの候補製品の配合または化学成分を修正することによって、その効力、治療効果、および/または安全性を向上させる潜在的な機会を調査するための研究計画を持っている。この研究計画は、私たちの将来の臨床作業を支援するために、製造能力を確立することを目的としたより広範な技術移転計画の一部である。この技術移転計画によると、この過程で同じ結果が生じる可能性はあまりないが、最終製品が我々の許可側が製造または獲得した製品と同等かそれ以上であることを確保するために努力している。これらの努力は成功しないかもしれない。新しい配合物または組成物が有望に見える場合、既存の候補製品が臨床試験において許容可能な安全性および有効性を示しても、そのような配合または組成物の臨床開発を行うことが決定される可能性がある。新しい処方または新しい組成物に必要とされる可能性のある追加の臨床試験の性質および範囲は、多くの要因に依存するであろう。候補製品に対して材料変更を行い、製造方法の変更を含め、一致した純度、特性、品質、効果と結果を実現できないリスクをもたらす可能性がある。これらの変化のいずれも、私たちの候補製品の表現の違いをもたらす可能性があり、改善された製造方法、材料およびプロセスを用いて製造された候補製品を用いて行われる計画または他の臨床試験に影響を与える可能性がある。これは臨床試験の完成を遅らせる可能性があり、非臨床的または臨床的接続と比較可能な研究が必要となる可能性があり、これはコストを増加させ、候補製品の承認を延期し、候補製品を商業化する能力を危険にさらす可能性がある(承認されれば)。新しい処方や新しい組成物の臨床開発を行うことにしたら, 私たちは追加的な費用を招き、潜在的な商業化のスケジュールが延期されるだろう。任意の新しい配合物または組成物が安全または有効であることが証明されるか、または既存の候補製品よりも優れていることは保証されない。新しい処方または組成物の商業化のいかなる遅延も、私たちの競争地位に悪影響を及ぼす可能性がある。
私たちは、より利益的または成功可能性の高い候補製品または適応を利用することなく、特定の候補製品または適応を追求するために限られたリソースを費やす可能性がある。
私たちの財務と管理資源が限られているため、私たちは研究プロジェクトと候補製品に重点を置いて、私たちが最も科学的価値と商業の将来性があると考えられる特定の適応を探しなければならない。したがって、私たちは過去に、IND申請が非活動状態に失効することを可能にすることを含む、私たちのいくつかの開発プロジェクトをアイドル状態に維持することを決定し、私たちは将来、他の候補製品を求めるか、または後により大きな科学的または商業的潜在力を有することが証明された他の指示を放棄または延期する機会を決定するかもしれない。私たちの資源配分決定は私たちが実行可能な科学的または商業的な製品や利益のある市場機会を利用できないようにするかもしれない。さらに、私たちは貴重な時間、管理、および財務資源を特定の適応の研究計画および候補製品に費やすかもしれないが、これらの特定の適応は最終的に科学的または商業的可能性のある製品を生成しない。さらに、我々の資源配分決定および任意の特定の候補製品をどのように開発または商業化するかに関する決定は、候補製品の科学的および商業的潜在力または目標市場の評価に基づく可能性があり、これらの評価は後に実質的に不正確であることが証明される。もし私たちが特定の候補製品を開発または商業化するために協力、許可、または他の印税手配を達成すれば、独占的な開発および商業化の権利を保持することがより有利な場合、私たちはその候補製品に対する貴重な権利を放棄するかもしれない。
新冠肺炎を引き起こす新型コロナウイルスSARS-CoV-2の爆発はすでに著者らの計画した臨床試験を含め、著者らの業務に悪影響を与え続ける可能性がある。
大流行や流行のような公衆衛生危機はすでに私たちの業務に悪影響を与え続けている可能性がある。2019年12月、2019年のコロナウイルス病を引き起こす新型コロナウイルス株であるSARS-CoV-2あるいは新冠肺炎が武漢で浮上し、中国。その時から、新冠肺炎は全世界的に伝播し、その後多種の新しい変異体が出現した。新冠肺炎の伝播に対応するために、私たちの多くの従業員はオフィスの外で彼らの仕事を続けている。
新冠肺炎の発生或いは類似の流行病のため、私たちはすでに私たちの業務、製造、臨床前開発活動、臨床前研究と計画に深刻な影響を与える可能性のある臨床試験の中断を経験し続ける可能性がある:
•患者が臨床試験に参加する遅延或いは困難;
•政府が米国食品医薬品局、他の機関、および同様の外国規制機関の運営を閉鎖、中断、または遅延させることは、監督管理提出、試験開始、規制承認のスケジュールに影響を与える可能性がある
•臨床前開発活動、臨床前研究と計画の臨床試験に関連するCROと協力者は予想される期限内に法規要求の中断或いは遅延を完成或いは遵守する
•人員不足、生産減速或いは生産停止及び輸送システムの中断のため、著者らの臨床試験に必要な材料(例えば、Lum-201と組換えヒト成長ホルモン)の受信中断或いは遅延;
•IRBの承認を得ること、臨床サイト調査員および臨床サイトスタッフを募集することの困難を含む、任意の臨床サイト起動計画の遅延または困難
•患者が新冠肺炎に感染したり、隔離されたりしたため、任意の計画中の臨床試験から退出する比率が増加した
•病院を任意の潜在的な臨床試験場所とすることと,我々が計画した臨床試験の進行を支援する病院スタッフの移転を含む,我々の臨床前開発活動,臨床前研究と計画された臨床試験の進行から医療資源を移行する
•連邦または州政府、雇用主および他の人が強要または提案した旅行制限、または臨床試験被験者のアクセスおよび研究プログラム(特に不要と考えられる可能性のある任意のプログラム)の中断により、被験者のデータおよび計画の臨床研究終点の完全性に影響を与える可能性があり、臨床試験現場データ監視のような計画の重要な臨床試験活動中断;
•従業員または協力者資源の制限、そうでなければ、従業員または彼らの家族の病気による、従業員は大勢との接触を避けたい、在宅勤務または公共交通中断への日々の依存を含む、我々の臨床前開発活動、臨床前研究および計画における臨床試験の実施に集中する
•通年予定の会議を中止することにより、医学界や投資界と接触する能力が低下した
•大流行期間中、臨床現場のプログラムと要求及び臨床試験を行う監督管理要求の変化。
私たちは対象を新冠肺炎ウイルスから保護するために、追加の臨床試験政策とプログラムを制定し、実施する必要があるかもしれない。例えば,2020年3月,FDAは大流行期間中の臨床試験に関する指導意見を発表し,その後更新を行った。その他の米国食品と薬物管理局が発表した新冠肺炎業界指導とこれまでの指導文書の更新は薬品製造と生物研究モニタリング施設の遠隔相互作用評価、及び製造、サプライチェーン及び薬品と生物製品検査などに関連している。最近、総裁·バイデン総裁は、政府が2023年5月11日に新冠肺炎国家と公衆衛生突発事件を終了する予定であることを発表した。突発公共衛生事件の終了がFDAとその他の監督管理政策と運営に与える全面的な影響はまだ不明である。
新冠肺炎の大流行によるこれらとその他の要素は疫病が部分的にコントロールされている国/地域に悪化或いは再出現する可能性があり、その中のいずれの要素も更に私たちの臨床前開発活動、臨床前研究と計画中の臨床試験を行う能力、及び私たちの業務に不利な影響を与える可能性があり、そしてすでに私たちの運営、財務状況及び業績に重大な不利な影響を与える可能性がある。
2021年7月20日、われわれの臨床試験のサイトの起動と登録速度は予想より遅く、主に新冠肺炎の制限によるものであることを発表した。この影響は,患者登録速度がより速いと予想される国際地点で特に顕著である。我々は現在OraGrowth H 210試験の全面登録を実現しており,2023年第4四半期にデータがあると予想される。コロナウイルスの大流行は続いており,医療提供者や臨床研究施設の獲得性,政府や民間組織が実施している様々な保護措置など,予測不可能な状況が予想されている。この状況は引き続き私たちの業務に悪影響を与え、さらなる遅延をもたらす可能性があると予想される。
疫病が引き続き私たちの業務、臨床前研究、臨床試験に影響を与える可能性の程度は未来の事態の発展に依存し、これらの事態の発展は高度に不確定であり、例えば疫病の持続時間、新しい変種、旅行制限と疫病の抑制、あるいはその影響を治療する行動、例えばアメリカと他の国における社会的距離と隔離または封鎖、企業閉鎖または業務中断、およびアメリカと他の国が疾病をコントロールおよび治療するための行動の有効性などを把握することができない
組織としては,これまで3期臨床試験や新薬申請(NDA)を行ったことがなく,Lum−201に成功できない可能性がある。
われわれは2020年第4四半期にOraGrowth 210試験を開始し,計画中の第3段階臨床試験を開始する前に,より多くの臨床試験を行う必要があるかもしれない。OraGrowth H 210の実験が成功すれば独立して行うつもりです
Lum−201の3期臨床試験。3期臨床試験を行い、成功したNDAを提出することは複雑な過程である。組織としては,3期臨床試験を行ったことがなく,規制文書の準備,提出,起訴の経験が限られており,これまで守秘協定も提出されていなかった。著者らはFDAと限られた相互作用を行い、FDAと提案された3期臨床試験の設計或いは実施を検討していない。したがって,OraGrowth H 210試験が成功しても,必要な臨床試験を成功かつ効率的に行うことができず,NDAの提出とLum−201の承認につながる可能性がある。私たちが計画した臨床試験や遅延を開始または完了できず、Lum-201を商業化することを阻止または延期する。
われわれのいずれの臨床試験においても,患者募集の遅延はわれわれの開発コストを増加させ,試験の完了を遅らせる可能性がある。
同時に競争するPGHD臨床試験がある可能性があり,将来の任意の第3段階臨床試験における登録を抑制または緩和するであろう。登録遅延に遭遇すると,計画された臨床試験を遂行する能力が損なわれる可能性があり,このような試験を行うコストが増加する可能性があり,いずれもわれわれの業務に実質的な悪影響を及ぼす可能性がある。
私たちの業務はロシアがウクライナに侵入した結果の不利な影響を受けるかもしれない
ロシアのウクライナ侵攻による経済、政治と社会条件は、サプライチェーン中断、インフレと臨床サイト閉鎖を含み、私たちの臨床試験を実質的に混乱させ、私たちのコストを増加させ、計画中の臨床開発活動を乱す可能性がある。例えば、私たちはドイツの供給者に依存しており、ウクライナとロシアの間の軍事衝突が私たちのサプライヤーが私たちのOraGrowth H 210実験に必要な物資を生産して流通する能力に悪影響を及ぼす場合、あるいはこのような流通が適時に完了できなければ、私たちがOraGrowth H 210実験を完了する時間は悪影響を受ける可能性がある。また、米国、連合王国、欧州連合などの政府は、ロシアまたはロシアとの商業的利益および(または)政府との関連が指摘された個人および実体、またはロシアの軍事活動に参加する個人および実体に対する包括的な金融制裁を含む、今回の侵攻に対して様々な制裁および輸出規制措置を実施している。各国政府はまた、ロシアの輸入品に対する輸出規制や貿易制裁を強化している。現在、この衝突の持続時間と強度とそれが私たちのヨーロッパ業務に与える経済的影響はまだ確定していないが、私たちの業務、運営結果、財務状況は実質的な悪影響を受ける可能性がある。
Lum−201および任意の将来の候補製品の臨床試験が、FDAまたは米国以外の同様の規制機関を満足させる安全性および有効性を証明できなかった場合、または積極的な結果が生じなかった場合、Lum−201または将来の候補製品の開発および商業化の完了を遅延させ、または最終的には達成できない追加のコストが生じる可能性がある。
任意の候補製品販売の規制承認を得る前に、私たちの候補製品の人体における安全性と有効性を証明するために、広範な臨床試験を行わなければならない。臨床試験は高価で、設計と実施が困難であり、完成には何年もかかるかもしれないし、結果はまだ確定していない。私たちの1つ以上の臨床試験はテストのどの段階でも失敗する可能性がある。
私たちはOraGrowth H 210試験プログラムのいくつかの側面を決定しており、これらの側面は、私たちが規制承認を受けたり、Lum-201を商業化する能力を延期したり阻止したりする可能性がある。例えば、Lum−201を用量レベルで使用することができ、その治療効果および/または安全性は、他の組換えヒト成長ホルモン療法よりも低い。OraGrowth H 210試験はLum-201の用量を試験し、これらの用量は小児科多用量メルク試験で測定された最高用量に相当し、それぞれ2倍と4倍高い。メルク試験の多用量試験において、これらの比較的に高い用量は成人或いは児童にテストを行ったことがなく、試験はこれらの比較的に高い用量が治療効果を高めたことを証明できても、このような比較的に高い用量はメルク試験中にテストした用量のように安全ではないかもしれない。そのため,実験期間中に頻繁なセキュリティ評価を行う必要があるかもしれない
FDAまたは他の規制機関は、私たちの臨床試験プログラムまたは研究設計に同意しない可能性があり、私たちの研究の参入基準に適合する患者を識別するための臨床研究プログラムまたは実験室プログラムを変更することを要求する可能性があり、これは、臨床実験室または私たちの臨床試験に参加するサプライヤーとの既存のスケジュールに悪影響を与える可能性があり、登録を延期したり、研究プログラムを修正したりする可能性があり、これらすべてが、私たちの臨床開発計画を延期し、規制部門がLUM-201を承認するのに要する時間および費用を増加させるか、または私たちが提案する適応または対象患者の集団の範囲に負の影響を与える可能性がある。例えば、2021年7月、FDAは、OraGrowth H 210試験を6カ月から12カ月に延長し、より多くの治療効果データが検討されるまで、Lum−201の治療を12カ月以下に制限することを要求している。われわれのOraGrowth H 210とOraGrowth H 212試験の初歩的な安全性と有効性データを検討したところ,FDAは一部の臨床放置を中止し,現在Lum−201による12カ月以上の治療を許可している。その結果,OraGrowth H 210試験は24カ月に延長し,われわれのOraGrowth 212試験は成人身長に近づくまで治療被験者まで延長した。これらの治療期間を延長することは、これらの試験に要する時間と費用を増加させるだろう。実験設計以外にも実験期間中や
臨床試験の結果として、規制の承認を得るか、またはLum-201または任意の将来の候補製品を商業化する能力を遅延または阻止する可能性がある
•臨床試験は否定的または不確定な結果をもたらす可能性があり、私たちは決定または監督機関が追加の臨床試験を要求したり、製品開発計画を放棄したりする可能性がある
•臨床試験に必要な患者の数は私たちが予想していたよりも多いかもしれません。私たちの基準に適合している被験者のこれらの臨床試験における登録人数は、私たちが予想していたよりも遅いかもしれません。または患者がこれらの臨床試験から退出する割合は、私たちが予想していたよりも高いかもしれません
•現在供給されているLum−201原料薬は、20年以上前に製造されており、試験を行い、その供給が臨床使用に適していると信じているが、意外に使用不可能になる可能性があり、または供給に関する文書が第三者によって所有され、時間の経過とともに利用できなくなる可能性がある
•臨床試験や候補製品の製造コストは予想以上に高いかもしれません
•私たちの第三者請負業者は、法規の要求を適時に遵守したり、私たちに対する契約義務を履行できなかったり、全く守らなかったりする可能性があります
•様々な理由で、私たちは、私たちの候補製品に予期せぬ深刻な有害事象または他の予期しない特徴があることを発見すること、または患者が受け入れられない健康リスクに直面していることを含む、候補製品の臨床試験を一時停止または終了しなければならないかもしれない
•規制当局は私たちが提案した臨床開発計画を承認しないかもしれません
•監督機関のフィードバックによると、私たちはすでに未来に私たちの臨床試験方案や設計を修正して、私たちと臨床試験サプライヤーや試験場所の手配を修正することを要求される可能性があります
•規制機関や機関審査委員会は、私たちまたは私たちの研究者が予想される試験場所で臨床試験を開始したり、臨床試験を行ったりすることを許可してはならない
•規制機関または機関審査委員会は、規制要件を満たしていないことを含む、様々な理由で臨床研究を一時停止または終了することを私たちまたは私たちの研究者に要求するかもしれない
•私たちの候補製品の供給または品質、あるいは私たちの候補製品の臨床試験に必要な他の材料が不足しているか、不十分である可能性がある。
LUM−201または任意の将来の候補製品に対して、予想以上の追加の臨床試験または他の試験を行うことが要求された場合、臨床試験または他の試験を成功させることができない場合、またはこれらの試験または試験の結果が陽性でないか、またはわずかに陽性である場合、または安全上の問題がある場合、私たちは:
•LUM-201または他の候補製品の発売承認を得る上で重大な遅延が発生した
•市場の承認を得ていません
•承認された適応は予期されたものや的確なものほど広くありません
•承認された後、製品を下積みする
•追加の上場後のテスト要求を受ける
•製品をどのように配布したり使用したりするかについて制限されている。
もし私たちの製品がテストや承認に遅延があったら、私たちの製品開発コストも増加します。いずれの臨床試験が計画通りに開始されるかどうか,再構成が必要かどうか,予定通りに完成するかどうか,あるいは全く知られていない。
重大な臨床試験遅延は、候補製品を商業化する独占的な権利を持つ可能性のある任意の期限を短縮するか、または私たちの競争相手が私たちの前に製品を市場に出すことを可能にし、候補製品を商業化する能力を弱化させ、私たちの業務と運営結果を損なうことになるかもしれない。
たとえ私たちがLum-201の発売許可を得ても、いくつかの要素はLum-201の市場を制限する可能性があり、これは私たちがこのような製品から収入を創造する能力を深刻に弱める可能性がある。
私たちが規制部門のLum-201の承認を得ても、いくつかの要素はLum-201の市場を制限したり、代替療法に対して競争的に不利になったりする可能性がある。例えば,この治療はPGHD患者の約60%にのみ有効であり,SGAやTurner症候群を有する患者の約50%に有効であり,実際の割合ははるかに低い可能性が考えられる。ある司法管轄区、例えばオーストラリアとEUは、PGHDの診断に対して異なる診断標準があるため、LUM-201はこれらの司法管轄区における市場は比較的に小さい。また,医師の受け入れと使用を得るためには,Lum−201は多くの課題に直面する。医師は、彼らの患者がLum-201治療を受ける資格があることを決定するために、追加のテストを行う必要があるだろう。Lum−201と競合する承認された製品は、数十年または数十年使用されており、非常に良い安全性を有している。Lum-201の結果はいくつかの医者と患者家族の要求を満たすために必要な快適さを提供するのに数年かかる。一部の医者は経口製品のメリットが限界を超えていないと思うかもしれない。例えば、試験中にLum−201治療を受けた患者を含む平均年増加速度は、非劣勢研究の要求に適合しているにもかかわらず、rhGH治療を受けているすべてのPGHD患者の平均年増加速度よりもはるかに低い可能性がある。これらの要因は、Lum-201が解決しようとしている市場規模と市場受け入れ率を制限する可能性があり、これは私たちの収入創出能力を深刻に弱める可能性がある
LUM-201または将来の候補製品は、深刻な不良事象を引き起こす可能性があり、またはその規制承認を遅延または阻止し、承認ラベルの商業イメージを制限するか、または任意の上場承認後に重大な負の結果をもたらす可能性があるという特性を有する可能性がある。
私たちの候補製品Lum-201はまだ臨床開発が完了していない。臨床開発に失敗するリスクが高い。このようなまたは任意の未来の候補製品がいつまたは十分に安全であるかどうかを予測することはできず、規制部門の承認を得ることができる。Lum-201または任意の将来の候補製品によって引き起こされる不良事象は、私たちまたは規制機関の臨床試験の中断、延期、または停止をもたらす可能性があり、より厳しいラベルまたはFDAまたは他の同様の外国規制機関の規制承認遅延または拒否をもたらす可能性がある。
以前メルク試験で測定した用量によると,Lum−201は小児では全体的な耐性が良好であり,最もよく報告されている副作用は食欲増加を含む消化器系イベントである。肝酵素は軽度上昇したが,ビリルビンの変化を伴わないことも報告されている。われわれの知る限り,これまでLum−201治療を用いた小児では薬物関連の重篤な有害事象は報告されていない。しかし、現在または将来の臨床試験において、Lum−201からの有害事象がLum−201の開発中断を促進しないことを保証することはできない。同様に、私たちの将来の候補製品は、深刻な有害事象を引き起こすかもしれないし、その規制承認を延期または阻止する可能性がある他の特性を持っているかもしれない。これらの有害事象または将来臨床試験で遭遇する可能性のあるさらなる安全または毒性問題のため、我々は発売Lum-201または任意の未来の候補製品の承認を得ることができない可能性があり、これは私たちの収入の発生を阻止したり、利益を達成したりすることができるかもしれない。我々の実験結果は,有害事象の深刻さや流行率が受け入れられないほど高いことを示している可能性がある.この場合、我々の実験は一時停止または終了される可能性があり、FDAまたは同様の外国規制機関は、その任意またはすべての目標適応の候補製品の開発を停止または拒否するように命令することができる。薬物に関連するいかなる有害事象も、患者募集または被験者が試験を完了する能力に影響を与えるか、または潜在的な製品責任クレームを引き起こす可能性がある。これらのいずれも、私たちの業務、経営結果、財務状況、キャッシュフロー、および将来の見通しに重大な悪影響を及ぼす可能性があります。
さらに、LUM-201または将来の任意の候補製品が発売承認された場合、私たちまたは他の人は、この製品によって引き起こされる不良事件を後に発見し、多くの潜在的な重大な負の結果をもたらす可能性がある
•私たちはこのような製品の販売を一時停止させられるかもしれません
•規制部門はこのような製品に対する私たちの承認を撤回することができる
•規制当局は、そのような製品の使用を減少させるために、または他の方法でその商業的成功を制限するために、ラベルに警告を追加することを要求することができる
•FDAまたは他の規制機関は、セキュリティ警報、親愛なる医療提供者レター、プレスリリース、またはこのような製品に対する警告を含む他の通信を発行することができる
•FDAは、REMSの確立または修正を要求することができ、または同様の外国の規制機関は、例えば、私たちの製品の流通を制限し、私たちに煩雑な実行要件を適用することができるように、同様の戦略の確立または修正を要求することができる
•私たちは製品の投与方法の変更や追加の臨床試験を要求されるかもしれません
•私たちは起訴され被験者や患者への傷害に責任を負うかもしれません
•私たちは訴訟や製品責任クレームの影響を受ける可能性があります
•私たちの名声は損なわれるかもしれない。
これらの事件のいずれも、承認されれば、特定の候補製品に対する市場受容度を達成または維持することを阻止することができる。
著者らの臨床試験が証明しても、1日1回の経口投与方案に基づいて、Lum-201はPGHD患者の成長に対して許容可能な安全性と有効性があり、FDA或いはアメリカ以外の類似規制機関はLum-201の発売を許可しないかもしれない、あるいはそれを承認してラベルを制限する可能性があり、これは私たちの業務、財務状況、運営結果、成長の将来性に重大な悪影響を与える可能性がある。
著者らの臨床試験が成功したと仮定すると、著者らはすべての主要な司法管轄区で監督管理機関にPGHD患者のサブセットの治療にLum-201を許可することを求め、その基礎は毎日1回の体重に基づく投与方案である。しかしながら、FDA、EMA、または他の国の規制機関は、我々の臨床試験結果がLUM−201のこの適応を承認するのに十分ではないと考えているかもしれない。一般に,FDAはスポンサーが2つの十分かつ制御された良好な臨床試験を完成させ,有効性を証明することを提案しているが,2つの説得力のある試験に基づく結論は,1つの試験に基づく結論よりも説得力があるからである。OraGrowth H 210試験あるいはその後の3期臨床試験で良好な結果を得ても,Lum−201が新しい化学実体であることを考慮すると,FDAは追加の臨床試験を要求する可能性があり,異なる臨床試験設計を使用する可能性がある。
さらに、FDAまたは他の規制機関が、1日1回の体重ベースの用量レジメンに基づいてPGHD患者のサブセットを治療するためのLum−201を承認しても、承認は、Lum−201の医師および患者に対する吸引力を、より広い適応のために承認される可能性のある他の製品よりも低くすることができ、Lum−201の潜在的販売を制限する可能性がある。
もし私たちがFDAまたは他の規制機関のLUM-201の承認を得られなかった場合、または承認の範囲が私たちが求めているより狭い場合、私たちの業務、財務状況、運営結果、および成長の見通しに実質的な悪影響を及ぼす可能性がある。
Lum-201または任意の未来の候補製品が監督部門の許可を得ても、それらは医師、患者、看護者、医療保険支払者、および医学界の他の人が商業成功に必要な市場受容度を達成できない可能性がある。
Lum-201または任意の未来の候補製品が規制部門の承認を得た場合、それらはまだ医師、病院管理者、患者、医療保険支払人、および医学界の他の人の十分な市場受容度を得ることができない可能性がある。もし私たちの候補製品が商業販売のために承認されたら、市場受容度は以下の要素を含む多くの要素に依存するだろう
•どのような有害事象の発生率や重症度も
•代替療法と比較して治療効果と潜在的利点があります
•Lumosが候補製品に受け取る価格
•医師が現在の治療法を変えたいかどうか
•代替療法よりも便利で投与しやすい
•対象患者群が新たな療法を試みる意欲と,医師がこれらの治療法を処方する意欲
•マーケティングや流通支援の実力
•第三者保険の利用可能性や十分な精算。
例えば、多くの会社は、PGHD患者を治療するための毎日の注射レジメンに基づく治療法を提供しており、医師、患者、または彼らの家族は、毎日の注射用量を除去することができても、Lum−201を使用することを望まない可能性がある。LUM-201または任意の未来の候補製品が承認され、十分な受容度に達していなければ、顕著な製品収入が生じない可能性があり、持続的に利益を上げることができず、利益を上げることもできないかもしれない。
また,2021年8月,FDAはPGHD患者の治療に用いられる競争相手の治療法,すなわち週1回のSkytrofaの治療法を承認し,世界の大手製薬会社を含む他社も現在PGHDに毎週注射治療を提供する製品を開発している。週1回の注射の承認に伴い、医師、患者、および彼らの家族は、Lum-201の毎日治療レジメンではなく、週1回の治療レジメンを好むかもしれない(もし彼らが使用可能であれば)。
Lum−201は商業規模の製造を行ったことがなく,製造規模を商業規模に拡大するリスクがある。我々は第三者製造業者によるLum-201の生産を手配したが、これは成功しない可能性があり、これはLum-201の規制承認と商業化を延期する可能性がある。
我々の既存のLum-201原料薬はLumosとAmmonettから得られ、LumosとAmmonettの間のAPAと2014年11月にメルク社と締結されたLumos Merckプロトコルと関係があり、これらの原料薬は私たちのOraGrowth H 210試験を満たすのに十分であると信じている。Lum-201原料薬はメルク社以外の生産基地で商業規模の生産を行ったことがなく、しかも製造技術を商業規模に拡大することに関するリスクはコスト超過、技術拡大の潜在問題、プロセス再現性、安定性問題、ロット一致性及び原材料の適時可用性などを含む。私たちがLUM-201の規制承認を他の方法で得ることができても、私たちが手配したメーカーがFDAまたは他の規制機関が許容できる仕様に従って承認された製品を生産して、製品が発売される可能性のある要求または将来の潜在的な需要を満たすのに十分な数の製品を生産することができる保証はない。メーカーが商業化のために十分な数の承認された製品の生産をタイムリーかつ効率的に開始できなければ、私たちの商業化努力は損なわれ、これは私たちの業務、財務状況、運営結果、および成長の見通しに悪影響を及ぼすだろう。
私たちはより多くの製品や候補製品を識別、獲得、開発、商業化することに成功できず、私たちの成長能力を弱めるかもしれない。
私たちの多くの努力は私たちの候補製品Lum-201の持続的な臨床テストと潜在的な承認に集中するが、私たちの長期成長戦略の重要な要素の1つは、より多くの製品および候補製品を獲得、開発、および/またはマーケティングすることである。候補製品を決定する研究計画は、最終的に任意の候補製品が決定されたか否かにかかわらず、大量の技術、財政、人的資源を必要とする。私たちの内部研究能力が限られているので、私たちは製薬とバイオテクノロジー会社、学術科学者、他の研究者に依存して製品や技術を販売したり許可したりするかもしれません。この戦略の成功はある程度私たちが有望な候補薬品と製品を識別、選択し、獲得する能力に依存する。候補製品や承認された製品を提案、交渉、実施し、許可する過程は長く複雑である。他の会社には、より多くの財務、マーケティング、販売資源を有する会社が含まれており、候補製品や承認された製品の許可または買収を競合する可能性がある。私たちは、第三者製品、業務、および技術の買収または許可を識別して実行し、それらを現在のインフラに統合するために限られたリソースしかありません。また、未完成の潜在的買収に資源を投入する可能性があるかもしれませんが、そうでなければ、このような努力の予想されるメリットを実現できないかもしれません。我々が得た任意の候補製品は、商業販売の前に、広範な臨床試験およびFDAおよび適用される外国規制機関の承認を含む追加の開発作業が必要となる可能性がある。すべての候補製品は典型的な薬品開発失敗リスクが発生しやすい, 候補製品が規制機関の承認のために十分に安全で効果的であることが証明されない可能性を含む。また、私たちが開発したどんな製品も、私たちが買収した承認された製品が利益のある方法で生産されたり、市場に認められたりする保証はありません。
私たちは現在販売や流通者がいません。マーケティング能力は限られています。もし私たちが単独でまたはパートナーまたは他のマーケティングパートナーを通じて販売、マーケティング、および流通能力を発展させることができなければ、LUM-201または他の未来の製品を商業化することに成功しないだろう。
私たちは販売やマーケティングインフラもなく、治療製品を販売、マーケティング、流通した経験もありません。任意の承認された製品を商業的に成功させるためには、販売およびマーケティング組織を発展させるか、またはこれらの機能を第三者にアウトソーシングしなければならない。Lum-201が承認された場合、私たちは現在、アメリカ、EU、他の地域で私たち自身の専門販売チームを使用して商業化するつもりです。
私たち自身の販売とマーケティング能力の確立、および第三者とこれらのサービスを実行する計画を達成することはリスクに関連している。例えば、販売チームの採用と訓練は高価で時間がかかり、どんな製品の発表も延期される可能性がある。最近の労働市場の動きは、雇用の穴埋めに利用できる候補者の減少を招き、より高い賃金を提供しなければならない。したがって、従業員の募集と維持はより困難になり、場合によっては受け入れ可能な条件で適切な候補者を得ることができず、欠員を埋めることができない可能性がある。販売チームを募集してマーケティング能力を確立する候補製品の商業発表が延期されたり、
どんな理由も起こらず、私たちはこのような商業化費用を早くまたは不必要に発生させるだろう。これは費用が高いかもしれません。もし私たちが私たちの販売とマーケティング担当者を維持したり再配置できなければ、私たちの投資は損失します。
私たちはまた第三者と私たちの候補製品の販売とマーケティングの手配を成功的に達成できないかもしれません。あるいは私たちに有利な条項でそうすることができないかもしれません。私たちはこれらの第三者に対して支配権がほとんどない可能性が高く、彼らのいずれも必要な資源と注意力を投入して、私たちの製品を効果的に販売し、マーケティングすることができず、私たちの名声を損なう可能性があります。もし私たちが販売とマーケティング能力を確立することに成功できなければ、私たち自身も第三者と協力しても、私たちの候補製品を商業化することに成功しません。
私たちは激しい競争に直面しており、これは他の人たちが私たちよりも製品の発見、開発、商業化に成功する可能性がある。
新しい治療製品の開発と商業化競争が激しい。私たちはLum-201方面で競争に直面しており、将来、開発または商業化を求める可能性のある任意の候補製品は、世界各地の主要な製薬会社、専門製薬会社、バイオテクノロジー会社からの競争に直面する可能性がある。現在いくつかの大手製薬やバイオテクノロジー会社が私たちの目標患者集団に再編ヒト成長ホルモン療法をマーケティング·販売している。これらの会社は通常、市場シェアを獲得または維持し、支払者に提供できる可能性のある価値主張を破壊するために、競争相手の薬品価格を低下させるより大きな能力を持っている。潜在的な競争相手には、学術機関、政府機関、および他の研究、特許保護を求め、研究開発、製造および商業化のための協力手配を確立する公的および個人研究機関、および化合物または他の適応のためのLum−201化合物を不正に販売する可能性のある製造業者および販売業者も含まれる。これらの競争相手の多くは私たちの目標適応のための療法を開発しようとしている。
私たちは、1日1回の体重ベースの経口用量レジメンに基づいてPGHD患者のサブセットを治療するための候補製品Lum−201を開発している。米国では,現在の成長療法患者に対する看護基準は,組換えヒト成長ホルモンを毎日皮下注射することである。現在多くの現在市販されている組換えヒト成長ホルモン療法は、成長ホルモン欠乏症の治療に毎日皮下注射されており、主にノードロピン(ノとノードA/S社)、Humtrepe(礼来社)、Nutroin-AQ(F.Hoffman-La Roche Ltd./遺伝子テーク社)、ジノロピン(ファイザー)、サイザン(メルクSerono S.A.)、Tev-troin(Teva製薬工業株式会社)、Omnitrop(Sandoz GmbH)、Valtroin(LG生命科学と生物製薬会社)、Zomacton(Ferring)、Skytrofa(Sandoz GbH)、Valtroin(LG生命科学と生物製薬会社)、Zomacton(Ferring)、Skytrofas(Sandoz GmbH)、Valtroin(LG生命科学と生物製薬会社)、Zomacton(Ferring)、Skytrofa(Sandoz GbH)、Valtroin(LG生命科学と生物製薬会社)、Zomacton(Ferring)、Skyrofroa(Sandoz GbH)、Valtroin(LG生命科学と生物製薬会社)、Zomacton(Ferring製薬)、Sknitrop(Sandoz GbH)、Valtroin(LG生命科学と生物製薬会社)、Zomacton毎週皮下注射で投与する。これらの組換えヒト成長ホルモン薬は,万托品やSkytrofaを除いて古くから知られている療法であり,医師,患者,介護者,第三者支払人,薬局福祉マネージャーに広く受け入れられ,成長ホルモン欠乏症治療の標準看護となっている。医師、患者、第三者支払者、およびPBMは、Lum−201に関連する潜在的な追加コストを心配すること、Lum−201を使用することによる患者への追加的な利点が限られていると考えること、または現在利用可能なrhGH治療と比較して長期安全性データが限られていることを含む、様々な潜在的理由のために、Lum−201を彼らの現在の治療レジメンに追加することを受け入れない可能性がある。
現在承認·販売されている日常組換えヒト成長ホルモン療法のほかに,すでに組換えヒト成長ホルモン市場に参加している会社や潜在的な新規参入者には,臨床開発の異なる段階にある様々な実験的療法や設備があり,主にノボノード社,Genexine Inc.とOPKO Health,Inc.(ファイザー社との協力)である。2021年第4四半期、OPKO Health Inc.のNGENLA(成長ホルモン)は、週に1回注射する長時間作用型ヒト成長ホルモン分子であり、ヨーロッパ、日本、オーストラリアとカナダの監督管理の許可を得て、2022年第1四半期にカナダで発売された。
私たちの多くの競争相手は、私たちと直接競争する大型製薬会社を含み、研究開発、製造、臨床前試験、臨床試験を行い、監督管理許可とマーケティング許可を得た製品の面で私たちより多くの財務資源と専門知識を持っている。製薬、バイオテクノロジー、診断業界の合併と買収は、より多くの資源を私たちの数の少ない競争相手に集中させる可能性がある。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。これらの第三者は合格した科学と管理者を募集と維持し、臨床試験場と臨床試験の患者登録を確立し、そして私たちの計画と相補的或いは必要な技術を獲得する上で私たちと競争している。
私たちは未来に戦略的連合を構成するかもしれないが、私たちはこのような連合の利点を認識しないかもしれない。
私たちは戦略同盟を結成し、合弁企業や協力関係を構築したり、第三者とライセンス合意に達したりする可能性があり、私たちの業務を補完したり拡大したりすると思います。これらの関係や同様の関係は、非日常的な費用や他の費用を発生させ、私たちの短期的および長期的な支出を増加させ、私たちの既存の株主を希釈したり、私たちの管理や業務を混乱させたりする証券の発行を要求する可能性がある。また、適切な戦略的パートナーを探す上で激しい競争に直面しており、交渉過程は時間がかかり複雑である。さらに、LUM-201または任意の未来の候補製品および計画のための戦略的パートナーシップまたは他の代替スケジュールを構築するための努力は成功しない可能性があり、私たちの研究開発チャネルが不足している可能性があるので、私たちの候補製品および計画は、協力努力の開発段階が早すぎると考えられる可能性があり、第三者は、私たちの候補製品および計画が安全性および有効性を証明するために必要な潜在力を持っているとは思わないかもしれない。もし私たちが製品や業務を許可すれば、私たちがそれらを既存の運営や会社文化と組み合わせることに成功できなければ、私たちはこのような取引のメリットを実現できないかもしれません。私たちは戦略的取引または許可証の後に、この取引の合理的な収入または特定の純収入を証明することを達成することを確認することができない。私たちの候補製品に関連する新しい戦略パートナー協定のいかなる遅延も、私たちの候補製品の開発と商業化を延期し、市場に進出してもそれらの競争力を低下させる可能性がある。
LUM-201または任意の将来の候補製品を商業化することができれば、これらの製品は、不利な価格設定法規、第三者精算慣行、または医療改革措置の制約を受け、私たちの業務を損なう可能性がある。
新治療製品の発売承認、定価と精算を管理する法規は国によって異なる。一部の国は製品の販売価格が発売前に承認されることを要求しています。多くの国で、定価審査期間はマーケティングまたは製品許可を承認した後から始まる。一部の外国市場では、処方薬の定価は初歩的な承認を得た後も、政府の持続的なコントロールを受けている。したがって、特定の国で製品の規制承認を受ける可能性がありますが、その後、製品の商業発表を延期し、その国/地域で製品を販売することによる収入に悪影響を及ぼす価格規制の制約を受ける可能性があります。不利な価格設定制限は、私たちの候補製品が規制部門の承認を得ても、1つ以上の候補製品への投資を回収する能力を阻害する可能性がある。
私たちはLUM-201あるいは任意の未来の製品を商業化することに成功する能力があるかどうかは、政府衛生行政当局、個人健康保険会社、その他の組織によるこれらの製品と関連治療の清算程度にも依存する。政府当局と第三者支払者、例えば個人健康保険会社や健康維持組織は、どのような薬剤を支払うかを決定し、精算レベルを確立する。アメリカの医療業界と他の地域の主な傾向の一つはコストコントロールだ。政府当局とこれらの第三者支払人は,特定の薬物のカバー範囲や精算金額を制限することでコストを抑制しようとしている。ますます多くの第三者支払人は会社に価格を基礎に所定の割引を提供し、医療製品の定価に挑戦することを要求している。私たちは商業化されたどの製品も精算できるかどうかを確認することができません。もし精算できれば、清算レベルはいくらですか。精算は私たちが市場で承認されたどんな製品の需要や価格に影響を及ぼすかもしれない。私たちの製品のための精算は特に難しいかもしれません。医者の監督下で管理されている製品は通常価格が高いからです。精算が得られない場合や限られたレベルに限られていれば、私たちが開発に成功した任意の候補製品を商業化することに成功できないかもしれません。
承認された製品の精算には大きな遅延が生じる可能性があり、カバー範囲はFDAや他の国·地域規制機関がこの製品を承認する目的よりも限られている可能性がある。さらに、精算を受ける資格があるということは、どの製品もすべての場合に支払われることを意味するわけではありません。または支払いの費用率は、研究、開発、製造、販売、流通を含む私たちのコストをカバーします。新製品の仮払い(適用すれば)も私たちのコストを支払うのに十分ではない可能性があり、恒久的な支払いにならない可能性があります。支払率は、製品の使用や臨床環境によって異なる可能性があり、精算された低コスト製品によって許容される支払いに基づく可能性があり、他のサービスの既存の支払いに組み込まれる可能性もある。製品の正味価格は、政府医療計画または個人支払者が要求する強制的な割引またはリベート、および将来の法律の任意の緩和によって低下する可能性があり、これらの法律は、現在、製品が米国より低い価格で販売されている国からの製品の輸入を制限している。第三者支払者が自分の精算政策を設定する際には,通常連邦医療保険カバー政策と支払制限に依存する。私たちは政府援助と個人支払者から私たちが開発した新製品の保証範囲と利益の支払率を迅速に得ることができず、これは私たちの経営業績、製品の商業化に必要な資金を調達する能力、そして私たちの全体の財務状況に実質的な悪影響を及ぼす可能性がある。一部の外国諸国では、EUや日本の主要市場を含め、処方薬の定価は政府によってコントロールされている。これらの国では, 製品の規制マーケティング承認を受けた後、政府当局との定価交渉には9~12ヶ月以上かかる可能性がある。特定の国·地域で精算や定価の承認を得るためには
臨床試験を行い,われわれの製品の費用対効果を他の利用可能な療法と比較する必要がある。もし私たちが承認した製品が精算されない場合、あるいは範囲や金額が限られているか、あるいは定価レベルが満足できなければ、私たちの業務は実質的な損害を受ける可能性があります。
私たちに対する製品責任訴訟は、私たちが重大な責任を負い、私たちが開発する可能性のある任意の製品の商業化を制限する可能性があります。
我々は人体臨床試験におけるLum−201および任意の未来候補製品の試験に関連する固有製品責任リスクに直面しており、私たちが開発可能な任意の製品を商業化販売すれば、より大きなリスクに直面する。もし私たちが私たちの候補製品や製品に被害を与えるクレームを自己弁護することに成功できなければ、私たちは大きな責任を招くだろう。是非曲直や最終結果にかかわらず、賠償責任は
•すべての候補製品や私たちが開発する可能性のある製品の需要が減少した
•私たちの名声とメディアの深刻な否定的な関心を損なう
•患者は臨床試験から撤退したり、試験を中止したりした
•関連訴訟の巨額の抗弁費用
•患者に相当なお金の報酬を与える
•収入の損失
•私たちが開発する可能性のあるどんな製品も商業化できない。
私たちが未来に得ることができるどんな製品責任保険も私たちが発生する可能性のあるすべての責任をカバーするのに十分ではないかもしれない。保険範囲はますます高くなっています。私たちは可能などんな責任にも対応するために、合理的な費用や十分な金額で保険範囲を維持することができないかもしれない
皮膚疾患領域やパーキンソン病,ハンチントン病,ALS疾患分野のいずれの製品も開発または商業化しないことに同意した。
我々がAvicena Group,Inc.およびその最高経営責任者と合意した和解合意の条項によると、私たちは、2037年11月19日までに、皮膚病領域またはパーキンソン病、ハンチントン病および筋萎縮性側索硬化症領域の任意の物質、治療、診断、または他の方法を開発、商業化、販売、許可、譲渡、または他の方法で利用しないことに同意する。したがって、これらの分野で製品を開発または協力する能力は制限される可能性があり、予想される貴重な未来の機会を逃す可能性があり、私たちの財務業績、業務、および業務の将来性に悪影響を及ぼす可能性があります。
2014年11月のNewLinkとメルクの間のグローバルライセンス·コラボレーション協定(“NewLink Merckプロトコル”)によると、将来の特許使用料義務は、将来のERVEBO販売の限られた収入(あれば)を超える可能性があります。
現在ERVEBOが承認されていても、ERVEBOの販売から得られる収入には限りがあり、このような製品の商業販売やNewLink Merckプロトコルでの将来の収入に悪影響を及ぼす可能性があります。例えば、ウイルスワクチンに対する理解の欠如、およびワクチン接種に関連する潜在的な有害事象は、医師および患者のこのような製品に対する認知および吸収に悪影響を及ぼす可能性がある。また,ワクチンが政府準備計画に承認されることは保証されず,候補製品の米国や海外でのビジネス成功に重要な意義を持つ可能性がある。最後に、場合によっては、カナダ公共衛生局(“PHAC”)に特許使用料を支払う義務が、メルク社から得られた特許使用料を超える可能性があります。
私たちの候補製品のいくつかは、私たち以外の組織や機関が共同で後援する臨床試験で研究されているか、または研究者によって開始された臨床試験で研究されている可能性があり、このような試験の進行はほとんどコントロールされていないことを意味します。
私たちは過去も現在もインドキシモートを提供しており、第二段階の研究者による臨床試験を支持している。私たちのエボラ候補ワクチン製品は西アフリカで臨床試験を行った。さらに、将来的に研究者によって開始される臨床試験にNLG 919およびNLG 802を提供することにも同意する。著者らはマサチューセッツ州総病院と臨床協力を行い、非アルコール性脂肪性肝疾患における経口Lum−201の作用を評価した。私たちは未来に似たような実験を提供したり、他の方法で支援し続けるかもしれない。しかし、私たちはこれらの試験のスポンサーではないため、患者のフォローアップや治療後の継続的なデータ収集、および、これらの試験の計画、管理、実施を制御していない
したがって,特に何らかの問題が発生した場合,これらのタイプの実験の進め方はリスクの影響を受ける.これらのリスクには、調査者や管理者とのコミュニケーションの困難や遅延、プログラムの遅延、および他の時間の問題、およびデータを解釈する上での困難や分岐が含まれる。
私たちの業務運営に関するリスク
私たちの未来の成功は私たちがCEO総裁と管理チームの他の重要なメンバーを引き留める能力と、合格した人材を誘致、維持、激励する能力にかかっている
私たちは私たちの最高経営責任者、社長、そして私たちの管理チームの他の会員たちに強く依存している。彼らの雇用条項によると、私たちの管理者はいつでも私たちとの雇用関係を終わらせることができる。この人たちのいずれかがサービスを失ったことは、私たちが研究、開発、商業化目標を達成することを阻害する可能性がある。
合格した科学、臨床、製造及び販売とマーケティング人員を募集と維持することも著者らの成功の鍵となる。多くの製薬とバイオテクノロジー会社の間の類似者に対する競争を受けて、私たちは受け入れ可能な条件でこれらの人員を吸引し、維持することができないかもしれない。最近の労働市場の動きは、雇用の穴埋めに利用できる候補者の減少を招き、より高い賃金を提供しなければならない。したがって、従業員の募集と維持はより困難になり、場合によっては受け入れ可能な条件で適切な候補者を得ることができず、欠員を埋めることができない可能性がある。私たちはまた、大学や研究機関から科学や臨床人を募集する競争に直面している。また、私たちは科学や臨床コンサルタントを含むコンサルタントやコンサルタントに依存し、その研究開発と商業化戦略の策定を支援してくれます。私たちのコンサルタントやコンサルタントは、私たち以外の雇用主に雇われる可能性があり、他のエンティティと締結された相談または相談契約に基づいて約束することができ、これは、私たちが彼らを得る機会を制限するかもしれない。
私たちは私たちの開発、規制、販売とマーケティング能力を拡大することを望んでいますので、私たちは私たちの成長を管理する上で困難に直面するかもしれません。これは私たちの運営を乱すかもしれません。
2022年12月31日まで、私たちは32人の従業員がいる。次の数年間、私たちの従業員の数と業務範囲は大幅に増加し、特に薬物開発、法規制、商業開発、販売とマーケティングの分野で大幅に増加することが予想される。私たちが予想している将来の成長を管理するためには、私たちの管理、運営、財務システムを継続して実施し、改善し、私たちの施設を拡大し、より多くの合格者を募集し、訓練し続けなければならない。私たちは私たちの業務の拡張を効果的に管理できないかもしれないし、より多くの合格者を募集して訓練することもできないかもしれない。私たちの業務の実体拡張は巨大なコストを招き、私たちの管理と業務発展資源を移転する可能性があります。今後の成長は、経営陣のメンバーにより多くの重大な責任を負わせるだろう
•私たちの臨床試験を効果的に管理し、多くの臨床場所で行われる予定です
•私たちに必要かもしれない専門知識と経験を持つより多くの従業員を確定し、採用し、統合して、私たちの未来の成長を支援します
•私たちの内部開発作業を効果的に管理しながら、ライセンス者、被許可者、請負業者、その他の第三者に対する契約義務を遵守します
•将来の様々な戦略的パートナー、サプライヤー、および他の第三者との任意の追加関係を管理すること
•私たちの管理、開発、運営、そして財務報告システムと手続きを改善する。
もし私たちがこのような任務のいずれも達成できなければ、私たちの成功的な発展を阻害するかもしれない。成長を管理できないどんな状況も、私たちの業務計画の実行を延期したり、私たちの運営を妨害したりする可能性がある。
業務中断は私たちの臨床試験、未来の収入と財務状況を深刻に損害し、私たちのコストと支出を増加させる可能性がある。
新冠肺炎疫病によるかもしれない中断以外に、私たちの運営或いは私たちのサプライヤーの運営或いは臨床試験はまた地震、電力不足、電気通信故障、洪水、ハリケーン、台風、火災、極端な天気条件、医療流行病及びその他の自然災害或いは人為的災害或いは業務中断の影響を受ける可能性がある。このような業務中断の発生は、私たちの運営と財務状況を深刻に損害し、私たちのコストと支出を増加させる可能性がある
もし私たちがアメリカ以外の場所でLUM-201を商業化することを許可されれば、私たちは追加のリスクに直面するだろう。
もし私たちが承認されたすべての承認された製品をアメリカ以外の地域で商業化すれば、国際業務に関連する様々なリスクは、私たちの業務に実質的な悪影響を及ぼす可能性があります
•国外の薬品の審査、定価と精算制度に対する異なる監督管理要求
•知的財産権の保護を減らすことです
•関税、貿易障壁、規制要求の意外な変化
•インフレや特に外国経済と市場の政治的不安定を含む経済的疲弊
•外国に住んだり旅行したりする従業員は税収、雇用、移民、労働法を遵守する
•源泉徴収賃金税を含む外国税
•外国為替変動は、経営費の増加と収入の減少、他の国での業務展開に付随する他の義務を招く可能性がある
•米国の“海外腐敗防止法”や同様の外国法規に基づいて負う可能性のある責任
•労働騒乱がアメリカよりも一般的な国では労働力の不確実性
•海外の原材料の供給や製造能力に影響を与える事件による生産不足;
•地政学的行動(戦争やテロを含む)や地震、台風、洪水、火災などの自然災害による業務中断。
我々の内部コンピュータシステム、または私たちのCROまたは他の請負業者またはコンサルタントのシステムは、故障したり、セキュリティホールが発生したりする可能性があり、これは、我々の薬物開発計画が実質的に破壊される可能性がある。
セキュリティ対策が実施されているにもかかわらず、我々の内部コンピュータシステムおよび私たちのCROおよび他の請負者およびコンサルタントのコンピュータシステムは、コンピュータウイルス、許可されていないアクセス、自然災害、テロ、戦争、および電気通信および電気故障の破壊を受けやすい。我々の知る限り,これまでこのようなシステム故障,事故,セキュリティホールを経験したことはないが,このような事件が発生して我々の運営が中断されると,我々の薬物開発計画が実質的に破壊される可能性がある。例えば、候補製品の完了または行われている臨床試験における臨床試験データの損失は、我々の規制承認作業を遅延させる可能性があり、データを回復または複製するコストを著しく増加させる可能性がある。任意の中断またはセキュリティホールが、私たちのデータやアプリケーションを紛失したり、破損したり、機密または独自の情報を適切に開示しない場合、私たちは責任を招く可能性があり、任意の候補製品のさらなる開発が延期される可能性があります。
システム障害、セキュリティホール、またはネットワーク攻撃が発生した場合、私たちの業務と運営は影響を受けます。
CROおよび他の請負業者、コンサルタント、および法律および会計会社を含む、我々のコンピュータシステムおよび我々が依存する様々な第三者のコンピュータシステムは、コンピュータウイルス、不正アクセス、データ漏洩、ネットワーク釣り攻撃、サイバー犯罪者、自然災害、テロ、戦争、および電気通信および電気故障の破壊を受ける可能性がある。我々は、第三者プロバイダに依存して有効なセキュリティ措置を実施し、そのような任意の故障、欠陥、または違反を識別して修正する。世界各地からの未遂攻撃と侵入の数、強度と複雑性の増加に伴い、セキュリティホールや破壊のリスクは普遍的に増加し、特にコンピュータハッカー、外国政府とネットワークテロリストを含むネットワーク攻撃或いはネットワーク侵入を介している。私たちは将来的に重大なシステムの故障やセキュリティホールに遭遇する可能性があり、これは私たちの運営が中断したり、私たちの薬物開発計画が実質的に破壊されたりする可能性がある。例えば、完了した、行われている、または計画されている試験における非臨床または臨床試験データの損失は、我々の規制承認作業を遅延させ、データを回復または複製するコストを著しく増加させる可能性がある。任意の中断またはセキュリティホールが、私たちのデータやアプリケーションを紛失したり、破損したり、個人、機密、または独自の情報を適切に開示しない場合、私たちは責任を招く可能性があり、私たちの候補製品のさらなる開発が延期される可能性があります。
私たちの従業員、独立請負業者、コンサルタント、首席調査者、CRO、CMO、および他のサプライヤー、および任意の未来のビジネスパートナーは、規制基準および要求を遵守しないことを含む不当な行為または他の不適切な活動に従事する可能性があり、これは私たちに重大な責任をもたらし、私たちの名声を損なう可能性があります。
我々は、従業員、独立請負業者およびコンサルタント、主要調査者、CRO、契約マーケティング組織(“CMO”)および他のサプライヤー、および将来のビジネスパートナーが、FDA法規または同様の外国規制機関の類似法規を意図的に遵守しないことを含む、任意の将来のビジネスパートナーが詐欺行為または他の不正行為に従事する可能性があり、FDAまたは同様の外国規制機関に正確な情報を提供し、私たちの製造基準またはcGMP要件の基準を遵守し、連邦遵守および州医療詐欺および法律法規の乱用、および同様の外国規制機関によって制定され実行される類似の法律法規を含むリスクに直面している。財務情報やデータを正直に報告するか、または許可されていない活動を開示する。私たち従業員と他のサービスプロバイダの不当な行為は、臨床試験の過程で得られた情報の不適切な使用に関連する可能性があり、これは規制制裁と私たちの名声に深刻な損害を与える可能性がある。私たちはすでに商業道徳と行為準則を実施しているが、常にこのような不正行為を識別し、阻止することができるわけではなく、私たちがこのような行為を発見し、防止するための予防措置、例えば品質システムを実施し、品質専門家にサプライヤーの審査を要求し、未知または未管理のリスクや損失を効果的に制御できない可能性があり、あるいはこのような法律や法規を遵守できないことによる政府の調査または他の行動または訴訟から私たちを保護する。もし私たちにこのような行動を取って、私たちが自分の権利を弁護したり、維持したりすることに成功しなかったら、これらの行動は、巨額の罰金や他の制裁を加えることを含む、私たちの業務および業務結果に大きな影響を及ぼすかもしれない。例えば, もし私たちの製造パートナーが同意法令の下に置かれたら、私たちが臨床または商業用品を生産する能力が阻害されるかもしれない。
もし私たちが契約約束の義務を履行できなかった場合、私たちの取引相手は適用された合意を終了したり、私たちにクレームをつけたりする可能性があり、これは私たちに実質的な悪影響を及ぼすかもしれない。
メルク社とのライセンス契約とAmmonett社とのAPAプロトコルによると、ビジネス上の合理的かつ勤勉な努力を用いてLum-201を開発·商業化する義務がある。私たちはメルクとAmmonettに大量の記念碑的な支払いと特許権使用料を支払う義務があり、これは私たちの将来の収益性とマーケティング協力合意を達成する能力を制限するかもしれない。もし私たちがメルク、Ammonett、または任意の他の取引相手に対する私たちの契約約束を履行できなければ、取引相手は2014年11月に締結された独占的なグローバル許可および協力協定Lumos Merck協定を終了することができ、あるいはこの2つの合意に基づいて私たちにクレームを出すことができ、これは私たちの業務、運営結果、将来性に大きな悪影響を及ぼす可能性がある。
我々は第三者に依存して臨床試験を行っているが,これらの第三者の表現は締め切りまでにこのような試験を完了できなかったことを含めて満足できない可能性がある。
私たちは独立して臨床試験を行わない。我々はCRO、臨床データ管理組織、医療機関、臨床調査者などの第三者に依存してこの機能を履行している。これらの第三者の臨床開発活動への依存は,これらの活動の制御を減少させたが,われわれの責任を軽減していない。著者らはまだ著者らのすべての臨床試験が試験の全体的な研究計画と方案に従って行われることを保証する責任がある。さらに、FDAは、データおよび報告の結果が信頼性および正確であることを保証し、臨床試験における患者の権利、完全性、およびセキュリティを保護するために、臨床試験結果を行い、記録し、報告する基準を遵守することを要求する。さらに、これらの第三者は、他のエンティティと関係がある可能性もあり、その中のいくつかは私たちの競争相手である可能性がある。これらの第三者が規制要件や私たちが規定した規程に従ってその契約責任を成功的に履行し、予想される期限内に私たちの臨床試験を完了または行うことができない場合、私たちは候補製品の規制承認を得ることができないか、遅延する可能性があり、候補製品の商業化に成功する努力を遅らせることができないか、または遅延する可能性がある。
私たちはまた他の第三者に私たちの臨床試験のための貯蔵と配布に依存している。私たちの既存または未来の流通業者のどんな業績ミスも、私たちの候補製品の臨床開発や規制承認を延期したり、私たちの製品の商業化を延期したり、追加の損失をもたらし、私たちの潜在的な製品収入を奪う可能性があります。
私たちは現在、LUM-201を製造および供給するために第三者CMOに依存し続けている可能性がある。もし私たちのメーカーとサプライヤーが私たちの需要を十分に履行または満足できなければ、私たちは巨額のコストを負担し、新しいサプライヤーやメーカーを探すために多くの努力をすることが要求されるかもしれない。私たちの候補製品の開発と商業化の面で、私たちはまた遅延に直面する可能性がある。
著者らは現在著者らの唯一の候補製品Lum-201の臨床規模生産における経験が限られており、著者らも施設がなく、著者らは現在単一の第三者CMOに依存し続けて、著者らのLum-201臨床試験のために薬物製品を製造と供給する可能性がある。FDAのcGMP標準に従って薬品を生産するには大量の専門知識と資本投資が必要であり、先進的な製造技術と技術制御の開発を含む。薬品メーカーは生産中によく困難に直面し、生産コスト、生産量と品質管理方面の困難を含み、候補製品の安定性と品質保証テスト、合格者不足、及び厳格に実行したcGMP要求、その他の連邦と州監督管理要求及び外国法規を含む。私たちと契約した任意のメーカーがこれらの困難に遭遇したり、私たちへの義務や適用法規を履行できなかったりすれば、臨床試験で薬物を研究する能力が脅かされる。臨床試験材料供給のいかなる遅延または中断もわれわれの臨床試験の完成を遅らせる可能性があり、著者らの臨床試験計画の維持に関連するコストを増加させ、遅延した時間帯に応じて、顕著な追加費用で新しい試験を開始するか、または完全に試験を終了することを要求する。
私たちの候補製品のすべてのメーカーはFDAが私たちの施設検査計画を通じて実行したcGMP要求に従わなければなりません。これらの要求は、他にも、品質管理、品質保証、および記録およびファイルの維持を含む。私たちの候補製品のメーカーはこれらのcGMP要求やFDA、州、外国の他の規制要求を遵守できないかもしれない。FDAまたは同様の外国規制機関は、新しい基準を随時実施したり、製品製造、包装、またはテストの既存の基準の解釈および実行を変更したりすることもできる。私たちは私たちの製造業者がこのような規制と基準を遵守しているかどうかをほとんど統制できない。これらの要件を遵守しないことは、罰金および民事処罰、生産停止、臨床試験および製品承認の一時停止または遅延、製品の差し押さえまたはリコール、または製品承認の撤回をもたらす可能性がある。もし私たちのメーカーが適用された法律や他の理由を遵守できなかった場合、提供されたいかなる製品の安全が損なわれた場合、私たちは規制機関の私たちの製品の承認を得ることができないか、または商業化に成功できない可能性があり、それによるいかなるダメージにも責任を負わなければならないかもしれない。これらの要素のいずれも、私たちの候補製品の臨床試験、規制提出、承認または商業化の遅延を招き、コスト上昇または私たちの名声を損なう可能性がある。
必要な製造·規制の専門知識や施設を持つ第三者メーカーの数は限られており、代替サプライヤーを配置するコストが高い可能性があり、多大な時間を要する可能性があり、これは我々の業務に実質的な悪影響を及ぼす可能性がある。任意の候補製品の新しい製造業者は、適用される法規要件に基づいて資格を取得することを要求され、適用される知的財産権法に基づいて候補製品を製造する方法に対して十分な権利を有する必要がある。適用される法規要件に基づいて必要なFDA承認または他の資格を取得し、第三者の知的財産権を侵害しないことを保証することは、供給の深刻な中断を招く可能性があり、新しいメーカーが私たちに転嫁される可能性のある大量の追加コストを負担する必要があるかもしれない。
私たちがLUM-201または任意の他の候補製品のために締結する可能性のある任意の未来の協力協定は、Lum-201または他の候補製品の開発が私たちの制御を受けないようにし、重要な権利を放棄することを要求するか、または他の私たちに不利な条項を要求するかもしれない。
我々は、LUM-201の米国内または海外での商業化について第三者と協力したり、将来的に米国内または海外で商業化された製品について協力したりする可能性がある。私たちの流通、マーケティング、許可、または他の協力パートナーには、大中型製薬会社、地域的、全国的な製薬会社、バイオテクノロジー会社が含まれています。私たちは、私たちの協力者が私たちの候補製品の開発または商業化のために投入する資源の数と時間、そして私たちの協力に関連するいくつかの知的財産権と、私たちの協力者の他の協力要素の限られた制御に依存することを制限します。私たちがこのような計画から収入を創出する能力は、私たちの協力者がこれらの手配の中で彼らに割り当てられた機能を成功的に履行する能力に依存するだろう。いずれの協力の終了や中断も、候補製品開発の遅延を招き、候補製品開発のコストを増加させたり、候補製品の開発を終了したりする可能性がある。
私たちは絶対に達成されないか失敗しないかもしれない戦略的協力を探索することを計画している。
私たちの戦略の一部として、私たちは様々な可能な戦略協力を探索し、より多くの候補製品、地理的地域、あるいは資源を獲得することを計画している。今のところこのような形の
戦略的協力が必要かもしれない(あれば)。適切な戦略的パートナーを探す過程で、私たちは激しい競争に直面するかもしれないが、この協力は複雑かもしれないし、交渉や記録に時間がかかるかもしれない。私たちは受け入れ可能な条件で戦略的協力を交渉することができないかもしれないし、根本的にできないかもしれない。私たちは、これらの協力の確立に関連する多くのリスクと不確実性のため、いつ(もしあれば)私たちが追加的な戦略的協力に参加するか予測できない。
NewLink Merckプロトコルによると,我々は候補製品開発の重要な権利と制御権を我々の協力者に譲らなければならないが,そうでなければ不利な条項に制約される.
私たちの協力は、私たちが未来に達成したどんな戦略的協力も、私たちを多くのリスクに直面させる可能性があります
•私たちは多くの業務、財政、そして管理資源の支出を負担する必要があるかもしれない
•私たちは既存の株主の持株比率を希釈する株式証券の発行を要求されるかもしれない
•私たちは相当な実際的または負債を負担することを要求されるかもしれない
•私たちの戦略パートナーが私たちの候補製品の開発や商業化に投入される資源の数とタイミングをコントロールできないかもしれません
•戦略的パートナーは、臨床試験を延期すること、不足した資金を提供すること、臨床試験を終了すること、または候補製品を放棄すること、新しい臨床試験を繰り返すか、または候補製品の新しいバージョンを要求することを臨床試験を行うことができる
•戦略パートナーは、戦略協力計画による製品のさらなる開発と商業化を行ってはならない、あるいは研究開発計画を停止することを選択することができる
•戦略的パートナーは、これらの製品から得られる潜在的な収入を制限するために、私たちの候補製品をマーケティングして配布するのに十分な資源を投入していないかもしれない
•私たちと私たちの戦略パートナーとの間で紛争が発生し、私たちの候補製品の研究、開発または商業化の遅延または終了、または高価な訴訟や仲裁を招き、経営陣の注意を分散させ、資源を消費する可能性がある
•戦略的パートナーは財務的な困難に直面するかもしれない
•戦略的パートナーは、私たちの知的財産権を正確に維持したり、守ったりすることができないかもしれないし、私たちの固有の情報を危険にさらしたり、無効にしたり、潜在的な訴訟に直面させる可能性がある方法で私たちの固有の情報を使用する可能性があります
•業務合併または戦略協力者の業務戦略の重大な変化はまた、戦略協力者が任意の手配の下でその義務を履行する意志または能力に悪影響を及ぼす可能性がある
•戦略的パートナーは、競合製品候補製品を独自に開発するか、または我々の競争相手を含む他の人と協力して競合製品候補製品を開発することを決定することができる
•戦略的パートナーは合意を終了したり、期限を許可したりすることができ、これは開発を延期し、私たちの候補製品を開発するコストを増加させる可能性がある。
私たちは業務で危険材料を使用し、環境法律と法規を守らなければならないが、これは高価かもしれない。
我々は、FDA、医薬品執行局、外国衛生当局によって施行された法律および法規、ならびに“職業安全と健康法”、“環境保護法”、“有毒物質制御法”、“食品、薬品および化粧品法”、“資源保護および回収法”、および他の現行および潜在的な連邦、州、地方、外国の法律と法規を含む他の法規、これらの法律と法規、これらの法律と法規は、私たちの製品を管理し、私たちの候補製品を開発し、製造するための材料、それによって生成された廃棄物の使用、製造、貯蔵、処理、および処分を守らなければならない。私たちはこのような材料を処理して処分するセキュリティ手順と、それらを処理する前に未使用の微生物を殺すセキュリティプログラムは、州と連邦法規に規定されている基準に適合していると信じていますが、事故のリスクは
このような材料の汚染や傷害は完全に除去されることはできない。もしこのような事故が発生した場合、私たちはそれによるいかなる損害に対しても責任を負う可能性があり、このような責任は私たちの資源範囲を超える可能性がある。
私たちの知的財産権に関するリスクは
もし私たちが私たちの技術や候補製品のために有効な知的財産権を獲得して維持することができなければ、あるいは知的財産権保護の範囲が十分でなければ、私たちの技術や製品を商業化することに成功する能力は重大な悪影響を受ける可能性がある。
私たちの成功は、私たちの独自技術と製品に関連して、米国や他の国で特許や他の知的財産権保護を取得し、維持する能力にかかっている。
生物技術と製薬会社の特許地位は通常高度に不確定であり、複雑な法律と事実問題に関連し、その法律原則はまだ解決されていない。近年、特許権は重大な訴訟のテーマとなってきた。したがって,我々が依存する特許権の発行,範囲,有効性,実行可能性,商業的価値は高い不確実性を持っている.未解決および将来の特許出願は、我々の技術または製品を保護する特許を発行すること、または他社が競争技術および製品を商業化することを効果的に阻止する特許をもたらすことができないかもしれない。米国や他の国の特許法または特許法解釈の変化は,我々が依存する特許の価値を低下させたり,我々の特許保護の範囲を縮小したりする可能性がある。外国の法律はアメリカの法律のように私たちの権利を保護しないかもしれない。科学文献で発表された発見は往々にして実際の発見に遅れており、米国と他の司法管轄区の特許出願は通常、申請18ヶ月後に発表され、時には全く発表されないこともある。したがって、私たちがすでにまたは入手可能なキー特許の発明者が、私たちのライセンス特許または係属中の特許出願に要求された最初の発明を提出した人であるか、または私たちがそのような発明のために特許を保護した最初の人であることを確認することはできない。特許可能性の他の要件が満たされていると仮定すると,2013年3月16日までに,米国では,最初に保護を要求した発明を提出した者が特許を取得する権利があり,米国国外では,2013年3月16日またはその後,最初に特許出願を提出した者が特許を取得する権利がある。
私たちが依存している未解決の特許出願が特許の形で発表されても、それらは私たちに何の意味のある保護を提供することができ、競争相手が私たちと競争することを阻止し、または他の方法で私たちにどんな競争優位性を提供することができる形で発表されないだろう。私たちの競争相手は、非侵害的に類似または代替技術または製品を開発することによって、私たちの特許を迂回することができるかもしれない。特許の発行はその範囲、有効性、または実行可能性に対して決定的ではなく、私たちが依存する特許は、米国または海外の裁判所または特許庁で挑戦される可能性がある。このような挑戦は、特許主張の縮小、無効、または実行不能をもたらす可能性があり、これは、他人の同様の技術および製品の使用または商業化を阻止または阻止する能力を制限するか、または我々の技術および製品の特許保護期間を制限する可能性がある。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちの特許の組み合わせは、他社が私たちと似ているか同じ製品を商業化するか、または他の方法で競争優位性を提供することを排除するために、十分な権利を提供してくれないかもしれない。
私たちはLUM-201の物質成分に関する特許保護を持っていない
Lum−201を使用する特定の方法に関するいくつかの特許および特許出願を有しており、Lum−201がこれらの市場で承認されておらず、GHD治療のODDが得られているので、Lum−201がこれらの市場で承認されていないので、FDAおよびEMAから市場独占経営権を得ることができる。しかし、アメリカと他の場所では、私たちはLum-201の物質構成保護をカバーしていない。LUM−201の物質組成特許がなく、LUM−201の化学構造が開示されているため、別の会社が別の適応のためにLUM−201を開発し、治療法を承認せずに市場に出すことが可能であるか、またはFDAおよびEMAがその孤児が指定した適応を承認する場合、市場排他性である。Lum-201が承認された場合、私たちの製品がFDA承認されたラベルと一致する使用が私たちの特許主張範囲に属していなければ、私たちがFDAのオレンジブックに私たちの特許を列挙する能力は制限されるかもしれない。さらに、これらの競合他社が、使用方法特許を要求する特許を含む任意の他の特許を侵害しない限り、我々のライバルは、製品を提供して販売することができるかもしれない。一般に、使用方法特許は、例えば、FDAが使用方法特許がカバーされていない主題化合物の代替用途を承認する可能性があり、他の人が主題化合物のラベル外販売または使用に従事する可能性があるので、物質組成物特許よりも実行が困難である。医師は製品ラベルに記載されていない用途のために承認された製品を発行することを許可された。ラベル外の処方は私たちの使用方法の特許を侵害する可能性がありますが, このようなやり方は医学専門によく見られ、このような権利侵害行為は防止或いは起訴することが困難である。FDAが私たちの特許が含まれていない用途を承認した場合、商業販売のために承認された場合、Lum-201の販売から収入を得る能力を制限する。
私たちは私たちの知的財産権を保護したり実行したりする法的手続きに巻き込まれるかもしれないが、これは高価で時間がかかり、成功しないかもしれない。
競争相手は私たちが依存している特許や私たちの他の知的財産権を侵害したり、他の方法で侵害するかもしれない。権利侵害や不正使用に対抗するために、私たちは費用が高く時間がかかるかもしれない侵害請求を要求されるかもしれない。私たちは侵害者からの任意のクレームに対しても、私たちが彼らの知的財産権を侵害していると主張するように、これらの当事者に反クレームを促す可能性があります。さらに、侵害訴訟では、裁判所は、私たちが主張する特許が無効または強制執行できないと判断することができ、または、私たちが主張する特許が関連技術をカバーしていないことを理由に、相手が関連する技術の使用を阻止することを拒否することができる。いずれの訴訟手続きにおける不利な結果も、1つまたは複数の特許が無効が宣言されるか、または狭義に解釈されるリスクに直面する可能性がある。さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。
第三者によって開始されるか、または米国特許商標局(“USPTO”)または任意の外国特許当局によって提起された干渉または派生プログラムは、特許および特許出願に関する発明または他の発明権事項の優先権を決定するために必要である可能性がある。私たちまたは私たちの許可者は、ライセンス後訴訟、異議、妨害、派生訴訟、当事者間の審査、特許無効訴訟または再審査、私たちの特許権または他の人の特許権に挑戦することを含む訴訟手続きに巻き込まれる可能性があり、そのような訴訟の結果は非常に不確定である。このような訴訟における不利な裁決は、重要な特許権の範囲を縮小したり、無効にしたりして、第三者が私たちに支払うことなく、または第三者の特許権を侵害することなく製品を製造または商業化することができないように、第三者が私たちの技術または製品を商業化し、私たちと直接競争することを可能にする可能性がある。もし勝利者が商業的に合理的な条項で私たちに許可を提供しなければ、何か許可を提供すれば、私たちの業務は損害を受ける可能性があります。訴訟や他の手続きは失敗する可能性があり、成功しても巨額のコストを招き、我々の経営陣や他の従業員の注意を分散させる可能性がある。私たちはまた知的財産権に関する他の人たちとの紛争に巻き込まれる可能性がある。例えば,我々のキー特許と特許出願の基礎を構成するデータは,メルク社が行ったいくつかの臨床試験の結果であるため,このような関係に基づいて開発された任意の知的財産権の所有権や有効性に食い違いが生じる可能性がある.もし私たちがこのような紛争を解決できなければ、私たちは貴重な知的財産権を失うかもしれない。
解決策が私たちに有利であっても、知的財産権クレームに関連する訴訟や他の法的手続きは、巨額の費用を発生させる可能性があり、私たちの技術および/または管理者の正常な責任を分散させる可能性があります。また、公聴会、動議、または他の一時的な手続きや事態の発展の結果が公表される可能性があり、証券アナリストや投資家がこれらの結果がマイナスだと思う場合、私たち普通株の市場価格に大きな悪影響を及ぼす可能性がある。このような訴訟または訴訟は、私たちの運営損失を大幅に増加させ、開発活動または任意の将来の販売、マーケティング、または流通活動に使用することができるリソースを減少させる可能性がある。知的財産権訴訟または他の訴訟の開始と継続によって生じる不確実性は、市場での競争能力に重大な悪影響を及ぼす可能性がある。
第三者は、私たちが彼らの知的財産権を侵害したことを告発する法的訴訟を提起する可能性があり、その結果は不確実であり、私たちの業務成功に実質的な悪影響を及ぼす可能性がある。
私たちのビジネス成功は、私たちと私たちの協力者が、私たちの候補製品を開発、製造、マーケティング、販売し、当社の独自技術を使用して、第三者の独自の権利または知的財産権を侵害、流用、または他の方法で侵害する能力に依存します。私たちは私たちの製品や技術に関連する知的財産権の将来的な対抗訴訟や訴訟の側になったり、脅かされたりする可能性がある。第三者は既存または未来の知的財産権に基づいて私たちに権利侵害を請求するかもしれない。もし私たちが第三者の知的財産権を侵害していることが発見された場合、私たちは第三者からライセンスを取得して、私たちの製品や技術の開発とマーケティングを継続することを要求される可能性があります。私たちはまた、懸案または脅かされた訴訟を解決するために、そのようなライセンスを締結することを選択することができる。しかし、私たちは商業的に合理的な条項や必要な許可証を得ることができないかもしれない。たとえ我々が許可を得ることができても,我々の競争相手が我々に許可してくれた同じ技術を使用できるように非排他的である可能性があり,巨額の印税や他の費用の支払いを要求する可能性がある.私たちは裁判所の命令を含めて権利侵害技術や製品の商業化を停止させることを余儀なくされるかもしれない。しかも、私たちはお金の損失の責任を負うと判断されるかもしれない。権利侵害の発見は、候補製品を商業化することを阻止したり、いくつかの業務運営を停止させたりする可能性があり、これは私たちの業務に実質的な損害を与える可能性がある。いくつかのLumos従業員とコンサルタントは以前、私たちの競争相手や潜在的な競争相手を含む大学や他のバイオテクノロジーや製薬会社に雇われていた。私たちの従業員がLumosのために働く時に他人の独自の情報やノウハウを使用しないことを確実にしようと努力していますが, 私たちは、私たちまたはこれらの従業員が、商業秘密または他の固有情報を含む任意のそのような従業員の前雇用主の知的財産権を使用または開示しているという疑惑を受けるかもしれない。これらは私たちが流用した他のクレームと
第三者の機密情報や商業秘密は,我々の業務に上記の侵害クレームと類似した負の影響を与える可能性がある.
知的財産権クレームに対する抗弁に成功しても、このようなクレームに関連する訴訟や他の法的手続きは、巨額の費用を発生させる可能性があり、私たちの技術や管理者の正常な責任を分散させる可能性があります。また、公聴会、動議、または他の一時的な手続きまたは事態の発展の結果が公表される可能性があり、証券アナリストや投資家がこれらの結果がマイナスだと思う場合、私たちの普通株の価格に大きな悪影響を及ぼす可能性がある。このような訴訟や訴訟は、私たちの運営損失を大幅に増加させ、開発活動に利用できる資源を減少させる可能性がある。私たちはそのような訴訟や法的手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らの財政資源がはるかに大きいので、私たちよりもこのような訴訟や訴訟の費用を効果的に負担するかもしれない。訴訟や他の知的財産権関連法律手続きの開始と継続による不確実性は、市場での競争能力に重大な悪影響を及ぼす可能性がある。
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの技術的価値は実質的な悪影響を受け、私たちの業務と競争地位を損なうかもしれない。
私たちの製品と特許技術に加えて、私たちは、ビジネス秘密、非特許技術、技術、および他の独自の情報を含む機密の固有情報に依存して、私たちの競争地位を発展させ、維持します。私たちの機密固有情報を第三者に開示したり盗用したりすることは、競争相手が私たちの技術的成果を迅速にコピーしたり、超えたりして、市場での私たちの競争地位を侵食する可能性がある。私たちは私たちの従業員と私たちの協力者とコンサルタントと秘密協定を締結することによって、私たちの機密固有の情報を保護することを求めている。私たちはまた、私たちの従業員と選定されたコンサルタントと協定を締結し、彼らの発明を私たちに譲渡する義務があることを規定した。このような合意は私たちの固有の情報を保護することを目的としている;しかし、私たちのビジネス秘密および他の機密情報が漏洩しないか、または競争相手が他の方法で私たちのビジネス秘密を取得しないか、または私たちの業務に関連する技術が、そのような合意当事者ではない人によって独立して開発されないことを確認することはできない。さらに、これらの合意当事者である従業員、コンサルタント、または協力者がこれらの合意の条項に違反または違反した場合、私たちは、そのような違反または違反を補うための十分な救済措置がない可能性があり、私たちは、そのような違反によって私たちのビジネス秘密を失う可能性があります。さらに、私たちのビジネス秘密は漏れたり、流用されたり、他の方法で私たちの競争相手に知られたり、独立して発見される可能性がある。また、外国の知的財産権法は、米国の法律のように商業秘密や機密情報を保護しない可能性がある。もし私たちの技術に関連する知的財産権を第三者に開示することを阻止できなければ, 私たちは私たちの市場で競争優位性を確立したり維持することができないかもしれないが、これは私たちの権利を保護する能力を損ない、私たちの業務に実質的な悪影響を及ぼすだろう。
私たちは世界各地で私たちの知的財産権を保護および/または実行できないかもしれない。
世界各地で訴訟を提起し、起訴し、私たちの知的財産権を守ることは、私たちと私たちの許可者にとって、目を引くほど高いかもしれない。競争相手は、私たちまたは私たちの許可者が特許保護を受けていない司法管轄区域で私たちの技術を使用して私たちまたは彼ら自身の製品を開発することができ、また、他の侵害製品を私たちが特許保護を持っているが、法執行力がアメリカに及ばない地域に輸出することもできる。これらの製品は、私たちまたは私たちの許可者が特許を発行した司法管轄区域で私たちの製品と競争していないかもしれませんが、私たちの特許声明や他の知的財産権は、彼らがこのような競争を行うのを効果的にまたは阻止するのに十分ではないかもしれません。多くの会社は外国の管轄区域の知的財産権の保護と保護に重大な問題に直面している。特定の国の法律制度、特に特定の発展途上国の法律制度は、特許や他の知的財産権保護、特に医薬品や生物製薬に関連する特許保護の強制執行に賛成しておらず、これにより、私たちの特許の侵害を阻止したり、私たちの独占権を侵害する方法で競争製品を販売することを阻止することが困難になる可能性がある。外国の管轄区域で私たちの特許権を強制的に執行する訴訟手続きは巨額のコストを招く可能性があり、私たちの努力と注意を私たちの業務の他の側面から移すことができます
私たちは許可された知的財産権に依存している。もし私たちが知的財産権を許可する権利を失ったら、私たちは私たちの唯一の候補製品Lum-201を開発し続けることができないだろう。もし私たちが第三者に私たちの唯一の候補製品や技術の使用、開発、商業化を許可する合意に違反したり、いくつかの開発または支払い締め切りまでに完了できなかった場合、私たちは私たちの業務に非常に重要な許可権を失うかもしれません。
APAについては、Lumos Merckプロトコルが割り当てられており、このプロトコルによると、私たちは私たちの業務に重要な知的財産権を付与されており、将来的に追加のライセンス契約を締結する必要があるかもしれません。私たちの既存のライセンス協定は、将来のライセンス協定が、様々な開発、規制、および/または商業的職務調査義務、費用の支払い、マイルストーンおよび/または特許使用料およびその他の義務を規定すると予想されています。もし私たちが合意に規定された義務を履行できなければ、許可側メルク社は許可証を終了する権利があるかもしれない
私たちはライセンスがカバーされているかもしれない製品を開発したり販売できない事件になるだろう。例えば、現在または将来のライセンスが終了した場合、許可者がライセンスの条項を遵守できなかった場合、許可された特許または他の権利が無効または強制的に実行できないことが発見された場合、または許容可能な条項で必要なライセンスを締結できない場合、私たちの業務は影響を受ける可能性がある。
私たちが以前にしたように、私たちは第三者からライセンスを取得して、私たちの研究を進めたり、唯一の候補製品の商業化を許可する必要があるかもしれませんし、このようなライセンスなしにLUM-201または未来の製品に対して強制的に施行される可能性のある第三者特許が存在しないという保証はありません。私たちはもしあれば、商業的に合理的な条項でこのような許可証のいずれかを得ることができないかもしれない。我々が許可を得ることができても,非排他的であり,競争相手が我々に許可してくれた同じ技術にアクセスできるようにする可能性がある.この場合、私たちは代替技術を開発または許可するために多くの時間と資源を必要とするかもしれない。もし私たちがそれができなければ、影響を受けた候補製品を開発したり、商業化することができないかもしれません。これは私たちの業務に実質的な損害を与える可能性があり、このような知的財産権を持つ第三者は、私たちの販売禁止を禁止したり、私たちの販売について印税および/または他の形態の賠償義務を支払うことを求めることができます。
知的財産権許可は複雑な法律、商業、そして科学的な問題に関する私たちの業務に重要だ。ライセンス契約によると、私たちとライセンシーとの間で知的財産権紛争が発生する可能性があります
•ライセンス契約に従って付与された権利範囲および解釈に関連する他の問題;
•私たちの技術とプロセスが、ライセンス契約に拘束されていないライセンス側の知的財産権をどの程度侵害しているかどうか
•私たちは協力開発関係の下で特許と他の権利を第三者に再許可する権利;
•私たちの候補製品の開発と商業化に関するライセンス技術の使用に関する職務義務と、どのような活動がこれらの職務義務を満たしているか
•私たちの許可者、私たちと私たちのパートナーが共同で知的財産権を創造または使用することによって生成された発明およびノウハウの所有権。
私たちが許可している知的財産権をめぐる紛争が許容可能な条項で現在の許可手配の能力を維持していることを阻害したり、損害を与えたりすれば、影響を受けた候補製品の開発に成功し、商業化することができない可能性がある。私たちは私たちの業務に必要または有用な第三者知的財産権の追加許可を締結するかもしれない
私たちの現在のLUM-201ライセンスと私たちが入る可能性のある任意の未来のライセンスは、様々な印税、マイルストーン、その他の義務を強制的に支払うことを要求します。例えば、ライセンス者は、ライセンス契約に従って特許起訴およびメンテナンスの制御権を保持することができ、この場合、特許起訴に十分な影響を与えることができない場合や、保守費の支払いができないことによる予期せぬカバーミスを防止することができない可能性がある。現在または将来のライセンス契約のいずれかの義務を履行できない場合、私たちのライセンス側は、私たちがライセンス契約に違反していると主張し、それに応じて許可の終了を求めることができるかもしれません。しかも、未来の許可者たちは私たちの許可を終わらせることを自由に決定することができる。現在または将来の許可を終了することは、許可知的財産権を使用する権利を失う可能性があり、これは、承認された場合、候補製品または製品を開発および商業化する能力に重大な悪影響を与え、競争業務の地位および業務の将来性を損なう可能性がある。
知的財産権は必ずしも私たちの競争優位に対するすべての潜在的な脅威を解決できるとは限らない。
私たちの知的財産権が提供する未来の保護の程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できないか、あるいは競争優位性を維持することができるかもしれない。以下の例は例示的である
•他の人は、私たちの候補製品と同様であるが、私たちが所有する特許請求の範囲内ではない製品を製造および/または使用することができるかもしれない
•私たちが所有している特許の発明者は、発表された特許または係属中の特許出願がカバーされた発明をした最初の人ではなく、および/または発明に関連する特許出願を最初に提出した人ではないかもしれない
•他の人は、私たちの知的財産権を侵害することなく、類似または代替技術を独立して開発したり、私たちまたは私たちの許可者の任意の技術をコピーしたりすることができる
•係属中の特許出願は、中国における潜在的な重要市場LUM−201を含む特許発行を招くことはないかもしれない
•発行された特許は、私たちにいかなる競争優位性を提供してくれないかもしれないし、私たちの競争相手の法的挑戦のために無効または実行不可能と認定されるかもしれない
•私たちの競争相手は特許権のない国で研究や開発活動を行い、これらの活動から学んだ情報を利用して競争力のある製品を開発し、私たちの主要な商業市場で販売するかもしれません
•他の特許を申請できるノウハウを開発しないかもしれません
•他人の特許は私たちの業務に悪影響を及ぼすかもしれないs.
このような事件が発生した場合、私たちの業務、運営結果、そして将来性を深刻に損なう可能性がある。
特許保護の獲得および維持は、政府特許機関によって適用された様々なプログラム、書類提出、費用支払い、および他の要件を遵守することに依存し、これらの要件に適合しない場合、私たちまたは私たちのライセンシーの特許保護は減少またはキャンセルされる可能性がある。
我々および/または我々のライセンス者は、許可された特許および/または出願の有効期間内に、USPTOおよび米国以外の様々な政府特許代理機関に、定期維持費、継続費、年会費、および特許および/または出願に関する様々な他の政府費用をいくつかの段階に分けて支払う。米国特許商標局および様々な非政府特許機関は、特許出願中にいくつかのプログラム、文書、費用支払い、および他の同様の条項を遵守することを要求する。多くの場合、不注意は、滞納金を支払うことによって、または規則を適用する他の方法によって救済することができる。しかしながら、場合によっては、規定を遵守しないことは、特許または特許出願の放棄または失効をもたらす可能性があり、それにより、関連する管轄区域の特許権の一部または全部が失われる可能性がある。この場合、私たちの競争相手は私たちの技術と私たちに許可された技術を使用することができるかもしれません。この場合は私たちの業務に実質的な悪影響を及ぼすでしょう。
特許改革立法は、特許出願をめぐる起訴および私たちが発行した特許の実行または保護の不確実性およびコストを増加させる可能性がある。
2013年3月,米国の発明法(“AIA”)により,米国は先行出願制度に移行し,その特許法について何らかの他の改正が行われた。これらの変化の全面的な程度はまだ完全には明らかではないが、例えば、裁判所は“友邦保険”の多くの条項を処理していない。したがって,この法案と本稿で検討した具体的な特許や特許出願に関する新条例の適用性は未定であり,審査が必要である。そのため,友邦保険が我々の業務運営にどのような影響を与えるかは不明である(あれば).しかしながら、友邦保険およびその実施は、特許出願起訴および発行された特許の実行または弁護をめぐる不確実性およびコストを増加させる可能性があり、これらすべては、私たちの業務および財務状況に重大な悪影響を及ぼす可能性がある。
もし私たちが“ハッジ-ワックスマン法案”に基づいてアメリカと外国で類似した法律によって特許期間の延長を得ることができなければ、私たちの候補製品のマーケティング排他的期間を延長する可能性があり、私たちの業務は実質的な損害を受ける可能性がある。
FDAによる我々の候補製品の上場承認の時間、期限、および詳細(ある場合)によれば、私たちが承認した製品またはその使用をカバーする1つまたは複数の米国特許は、“ハッジ·ワックスマン法”に基づいて5年にわたる特許期間を回復する資格がある可能性がある。ハッジ-ワックスマン法案は各FDAによって承認された製品が最大1つの特許を延長することを許可する。私たちの候補製品が規制部門の承認を得られれば、特定の国/地域でも特許期間を延長することができる。しかしながら、私たちまたは私たちのライセンシーは、適用の最終期限内に出願を提出できなかったか、関連する特許が満了する前に出願を提出できなかったか、または適用された法的要件を満たすことができなかったため、米国または他のどの国/地域でも特許期間を延長する許可を得ない可能性がある。また、政府当局が提供する延期期限およびそのような延期期間のいずれかの特許保護範囲は、私たちが要求しているものよりも短い可能性がある。
もし私たちが特許期間の延長や回復を得ることができない場合、あるいはそのような延長の期限が要求されたものよりも短い場合、私たちがその製品を独占販売する権利がある期間は短縮され、競争相手はその特許が満了した後に競争製品の承認を得る可能性があり、私たちの収入は大幅に減少する可能性がある。
政府の規制に関連するリスク
規制承認過程は高価で、時間がかかり、不確定であり、私たちまたは私たちの協力パートナーが私たちの候補製品を商業化する承認を得ることを阻止する可能性がある。
薬品の研究、テスト、製造、ラベル、承認、販売、輸入、輸出、マーケティングと流通はすべてアメリカとその他の国家FDAとその他の監督管理機関の広範な監督管理を受けており、これらの監督管理規定は国によって異なる。私たちがFDAの機密協定の承認を得るまで、私たちと私たちのパートナーは私たちの候補製品をアメリカで販売することを許可しなかった。私たちと私たちの協力パートナーは、LUM-201または任意の未来の候補製品に申請を提出したり、マーケティング承認を得たりしていません。秘密協定の承認を得ることは長くて高価で不確実な過程になるかもしれない。さらに、FDAおよび他の適用される米国および外国の法規要件を遵守しなければ、私たちは行政または司法制裁を受ける可能性がある
•警告状
•民事または刑事罰と罰金
•禁令
•規制承認の一時停止または撤回;
•進行中の臨床試験を一時停止します
•自発的または強制的な製品のリコールと宣伝要求
•わが社が提出した新薬の発売承認申請の受理または承認を拒否した
•コストの高い新しい製造要件を含む業務への制限;
•私たちの製品を差し押さえたり差し押さえたり輸入を禁止したりします。
私たちの任意の候補製品を米国または海外で商業化することが承認される前に、私たちとパートナーは、厳格に制御された臨床試験を通じて大量の証拠を提供し、FDAおよび他の外国規制機関に満足させ、これらの候補製品がその期待用途に対して安全かつ有効であることを証明しなければならない。臨床前研究と臨床試験の結果は異なる方法で解釈できる。私たちと私たちのパートナーが私たちの候補製品の臨床前または臨床データが有望であると信じていても、これらのデータはFDAおよび他の規制機関の承認を支持するのに十分ではないかもしれない。私たちの候補製品を人間に使用することは、私たちの候補製品の臨床試験を中断、延期、または一時停止し、FDAまたは他の規制機関が私たちの任意またはすべての目標適応の候補製品を承認することを拒否する可能性がある不良事象を発生させる可能性がある。
規制機関の機密協定の承認は保証されておらず、承認過程がコストが高く、数年かかるかもしれない。FDAは承認過程でも大きな裁量権を持っている。大量の時間と費用がかかったにもかかわらず、失敗は任意の段階で発生する可能性があり、私たちはいくつかの問題に直面し、私たちは臨床試験を放棄或いは繰り返し、あるいは追加の臨床前研究と臨床試験を行う可能性がある。FDAの承認に必要な前臨床研究および臨床試験の数は、候補製品、候補製品が対象とする疾患または状況、および任意の特定の候補製品に適用される法規に依存する。FDAは、以下の理由を含むが、これらに限定されないが、多くの理由で承認候補製品を延期、制限、または拒否することができる
•候補製品は、市場の承認を得ることができない、または商業用途を阻止または制限することができないように、安全または有効であると考えられないかもしれない
•FDA関係者は臨床前研究と臨床試験からのデータが不十分であると考えているかもしれないし、あるいは前臨床研究或いは臨床試験データの解釈に同意しないかもしれない
•FDAは私たちまたは私たちの第三者製造業者のプロセスまたは施設を承認しないかもしれない
•FDAは私たちの臨床試験の設計、実施、または結果に同意しないかもしれない
•臨床試験で研究された集団は、承認を求めるすべての集団の有効性と安全性を保証するのに十分な広汎性または代表性がないかもしれない
•私たちの候補薬物の臨床試験から収集されたデータは、機密保持協定の提出をサポートするのに十分ではないかもしれない
•われわれが提案した適応候補薬のリスク−収益比が受け入れられることをFDAに証明することはできないかもしれない。
Lum−201または任意の将来の候補製品が臨床試験において安全性および有効性を証明できなかった場合、または規制部門の承認を得られなかった場合、私たちの業務および運営結果は実質的かつ不利な損害を受けるであろう。
私たちが候補製品の規制承認を得ても、持続的な規制義務と持続的な規制審査の制約を受けることになり、これは多くの追加費用を招く可能性があり、適用される規制要求を遵守できなければ処罰されるだろう。
規制の承認を受けると、承認された製品およびその製造業者は、FDAおよび/または非米国規制機関の持続的な審査を受けるだろう。LUM-201または任意の将来の候補製品に対して得られる任意の規制承認は、製品が発売される可能性のある指定された用途によって制限される可能性があり、または製品の安全性および有効性を監視するために、コストの高い発売後の後続試験の要求を含む可能性がある。さらに、FDAおよび/または非米国規制機関がLUM−201または任意の将来の候補製品を承認した場合、我々は、FDAおよび他の規制機関の製品に関するラベル、包装、有害事象報告、貯蔵、広告、販売促進および記録の広範かつ持続的な規制要件を受ける
監督管理部門は薬品の承認後の販売と販売促進を密接に監督し、薬品が承認の適応のみを確保し、承認されたラベルの規定に基づいて販売を行うことを確保する。規制部門は、ラベル外使用に関するメーカーの通信に厳格な制限を実施しており、規制部門がこれらの制限に違反していると判断すれば、ラベル外マーケティングの法執行行動を受ける可能性がある。米国連邦食品、薬物、化粧品法および外国司法管轄区域の他の処方薬の普及に関する類似法規に違反することは、FDA、司法省、州総検察長、その他の外国監督機関が法執行行動を取り、調査を行い、米国連邦と州医療保健詐欺と乱用法律、および外国司法管轄区の州消費者保護法および類似法律に違反していることを告発する可能性がある。
また、私たちの薬品メーカーはcGMP法規を遵守しなければならず、その中には品質管理と品質保証に関する要求と、それに応じた記録とファイルの維持が含まれている。また、規制部門は、これらの生産施設を承認してから、私たちの薬品を生産するために使用されなければならず、これらの施設は、cGMP法規に適合することを確実にするために、FDAおよび他の規制機関の持続的な審査および定期的な検査を受ける。もし私たちまたは第三者がある製品に以前に未知の問題、例えば意外な深刻性や頻度の不良事件、またはその製品の生産施設に問題があることを発見した場合、監督管理機関は、製品の市場からの撤退を要求すること、または生産を一時停止することを含む、製品、製造業者、または私たちに制限を加える可能性がある。もし私たち、私たちの候補製品、または私たちの候補製品の製造施設がFDAおよび/または他の非米国規制機関の規制要件を遵守できない場合、私たちは以下の制裁を含む行政または司法制裁を受ける可能性がある
•警告状
•民事または刑事罰と罰金
•禁令
•規制承認の一時停止または撤回;
•進行中の臨床試験を一時停止します
•自発的または強制的な製品のリコールと宣伝要求
•私たちが提出した新薬、生物製品または承認申請を受理または承認したサプリメントの上場承認申請の受理または承認を拒否する
•コストの高い新しい製造要件を含む業務への制限;
•私たちの製品を差し押さえたり差し押さえたり輸入を禁止したりします。
規制要件や政策は変化する可能性があり、追加的な政府法規が公布される可能性があり、私たちも遵守を要求される可能性がある。米国や他の国の将来の立法や行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することはできない。もし私たちが規制コンプライアンスを維持できなければ、私たちは私たちの未来の製品の販売を許可されないかもしれません。私たちの業務は影響を受けるかもしれません。
外国の管轄区域の規制承認を得られなければ、私たちは国際的に私たちの製品を販売することができないだろう。
私たちはLUM-201のアメリカ以外の流通とマーケティングパートナーを探し、将来の製品を国際市場で販売するかもしれないと考えている。ヨーロッパ経済区、アジア太平洋地域、他の多くの外国司法管轄区で私たちの未来の製品を販売するためには、単独の監督管理許可を得なければならない。
例えば、ヨーロッパ経済地域では、医薬製品は“MA”を獲得した後にのみ商業化できる。MAを付与する前に、EMAまたはEEA加盟国の主管当局は、製品の品質、安全性、有効性に関する科学的基準に基づいて製品のリスク-利益バランスを評価する。日本では,衛生·労働·福祉省の薬品·医療機器庁は“薬学事務法”に基づいて申請を承認しなければならず,日本で新薬を販売することができる。
私たちは外国の規制機関との相互作用が限られている。承認手続きは国によって異なり,追加の臨床試験に関与する可能性があり,承認を得るのに要する時間はFDAの承認を得るのに要する時間とは異なる可能性がある。また、一国で行われた臨床試験は、他の国の規制機関に受け入れられない可能性がある。FDAの承認は他国の規制機関の承認を確保しておらず、1つ以上の外国規制機関の承認は他国の規制機関またはFDAの承認を確保していない。しかし、1つの国で規制承認を得ることができなかったり、遅延したりすることは、他の国の規制過程に悪影響を及ぼす可能性がある。外国規制機関の承認過程には、FDA承認の取得に関連するすべてのリスクが含まれる可能性がある。もしあれば、私たちは外国の規制部門の承認をタイムリーに得られないかもしれない。私たちは規制承認を申請できないかもしれません。申請を提出しても、どの市場でも私たちの製品を商業化するために必要な承認を得ることができないかもしれません。
医療改革措置は私たちの候補製品が商業的に成功することを阻害または阻止するかもしれない.
米国では,医療システムが複数の立法や規制改革を継続していることが予想され,これらの改革は,将来の収入や収益性,潜在顧客の将来の収入や収益性に影響を与える可能性がある。連邦と州立法者は定期的に立法を提出し、時々立法を公布し、医療保健システムに重大な変化を招き、その中のいくつかは医療製品とサービスのコストを制御或いは低減することを目的としている。例えば、PACAは数十年来最も重要な医療改革措置の一つであり、2010年に公布された。PPACAには,連邦医療計画の登録,精算変化,詐欺や乱用措置を管理する条項が多く含まれており,これらは既存の政府医療計画に影響を与え,新たな計画の発展を招くであろう。PACAを含むPPAA
•“ブランド処方薬”を販売する製薬業者または輸入業者に差し引くことのできない年会費を徴収する
•ブランド医薬品メーカーが支払うべき医療補助税還付の最低水準を15.1%から23.1%に引き上げ、2011年から発効した
•禁止令の強制執行につながる可能性がある
•医療補助管理保健機関が支払った薬品のリベートを要求する
•メーカーに保険切欠き割引計画に参加するように要求し、この計画によると、彼らは現在、保険不足中に条件を満たす受益者に適用ブランド薬品交渉価格の70%の販売時点割引を提供し、メーカーの外来薬として連邦医療保険D部分の条件に組み込むことに同意しなければならない
•承認された生物学的製品と類似または同じ生物療法を承認するためのプロセスを作成する。
PPAAの一部または全部を廃止、置換、または変更するために立法と司法的努力が行われた。2021年6月、米最高裁はテキサス州と他の挑戦者がPPACAの法的地位に挑戦していないと判断し、この事件を却下したが、PPACAの合憲性を具体的に裁くことはなかった。したがって、PACAは現在の形で依然として有効である。バイデン政府が将来公布した訴訟と医療保健措置がPPACAの実施、著者らの業務、財務状況と運営結果にどのように影響するかはまだ不明である。PACAに関する訴訟と立法は継続される可能性があり、結果は予測不可能で不確実だ。任意の新しい立法を遵守したり、PACAによって実施された変化を逆転させることは、多くの時間とコストを費やし、私たちの業務に実質的な悪影響を及ぼす可能性があります。現在公布または将来改訂されたPACAが私たちの業務や財務業績に悪影響を与えないことを保証することはできませんし、将来の医療改革に関連する連邦や州立法や行政変化が私たちの業務にどのように影響するかを予測することもできません。
また、PACAが公布されて以来、他の立法改正も提案され、採択された。例えば、2011年の“予算制御法”では、他にも赤字削減合同特別委員会が設置され、提案されている
2013年から各年度に提供者に支払われる連邦医療保険総額を含めて国会に提出された支出削減提案は、2031年まで有効となるが、国会がさらなる行動をとらない限り、各種新冠肺炎救済立法に基づいて2020年5月1日から2022年3月31日まで実施を休止する。現在の立法によると、医療保険支出の実際の減少幅は2022年の1%から本自動減額の最終年度の4%まで様々である。2013年1月、オバマ総裁は“自動減額法案”に署名し、2011年の“予算制御法案”の自動減額条項で要求された予算削減をさらに2カ月延期した。他にもATRAは病院を含むいくつかの医療サービス提供者に支払う医療保険を減少させ,政府が提供者に多額の支払いを取り戻す訴訟時効期間を3年から5年に延長した。私たちは追加的な立法変化がその業務に影響を及ぼすかどうかを予測できない
最近、国会は数回の調査を行い、薬品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、政府計画の薬品精算方法を改革するための立法を提出し、公布した。例えば、2021年1月、総裁·バイデンは、PPACA市場を介して医療保険を取得することを目的とした特別な保険加入期間を開始する行政命令を発表し、特定の政府機関に医療保健の獲得を制限する既存の政策やルールを検討し、再検討するよう指示した。2021年1月1日に施行される“2021年米国救援計画法案”によると、メーカーが州医療補助計画に支払う医療補助薬品還付計画還付の法定上限が撤廃される。この上限を撤廃することは、販売製品よりも多くのリベートの支払いを製薬業者に要求する可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。2022年8月、国会は“2022年インフレ低減法案”を可決し、その中には製薬業と連邦医療保険受益者に重大な影響を与える処方薬条項を含み、連邦政府がある高価な単一源連邦医療保険薬物の最高公平価格について交渉することを許可し、薬品価格交渉要求を守らないメーカーに罰と消費税を加え、すべての連邦医療保険B部分とD部分の薬物がインフレリベートを獲得することを要求し、もしその薬品価格の増加がインフレより速い場合、限られた例外、及び連邦医療保険D部分を再設計して受益者の自己払い処方薬コストを下げるなどの変化を含む。これらの立法や行政の影響は, バイデン政府が私たちと製薬業界全体にとった行政行動や将来実施される任意の医療措置や機関規則は不明であり,我々の業務,財務状況,運営結果に実質的な悪影響を及ぼす可能性がある。コスト抑制措置や他の医療改革を実施することは、収入を創出し、利益を達成すること、または候補製品を商業化することを阻止するかもしれない(承認されれば)。
州レベルでは、立法機関は、価格または患者の精算制限、割引、特定の製品参入の制限、マーケティングコスト開示および透明性措置を含む、医薬品および生物製品の価格設定を制御するための法規を立法および実施することが増えており、場合によっては、他の国からの輸入および大量購入を奨励することを目的としている。多くの州では、州薬品価格透明性と報告法が検討されているか、最近公布されており、これは私たちのコンプライアンス負担を大幅に増加させ、規制部門の承認を得て商業化を開始した後、このような州法律に基づいてより大きな責任を負わせる可能性がある
連邦や州レベルで医療コストを制御または低減するための立法と規制提案が継続される可能性がある。私たちは未来に取られる可能性のある計画やそのすべての影響を予測できない。政府、保険会社、医療組織を管理し、他の医療サービス支払者が医療コストのコントロールあるいは低減に努力し続けることは、以下の点に悪影響を及ぼす可能性がある
•私たちは私たちの製品に公平だと思う価格を設定することができます
•私たちは収入を作り利益を達成したり収益性を維持したり
•資金の入手可能性。
また、監督管理要求とガイドラインは変化する可能性があり、著者らはこれらの変化を反映するために臨床試験方案を修正する必要があるかもしれない。改正案は,臨床試験案を機関審査委員会に再提出して再審査することを要求する可能性があり,臨床試験のコスト,時間,あるいは成功達成に影響を及ぼす可能性がある。ある薬品の安全リスクに関する事件が広く宣伝されていることから、監督管理当局、国会議員、政府会計事務室、医療専門家と公衆は潜在的な薬品安全問題に対して懸念を提出した。これらの事件は薬品のリコールと撤回を招き、薬品ラベルを改訂し、更に薬品の使用を制限し、そしてリスク管理計画を構築し、例えば、薬品の流通を制限したり、安全モニタリング及び/又は患者教育を要求したりする可能性がある。薬物安全問題に対する日々の関心はFDAが臨床試験と薬物審査過程においてもっと慎重な方法をとることを招く可能性がある。臨床試験のデータは、安全性の面でより厳しい審査を受ける可能性があり、これは、FDAまたは他の規制機関が、完了前に臨床試験を終了または一時停止する可能性が高いか、またはより長時間以上の臨床試験を要求する可能性があり、これは、多くの追加費用をもたらす可能性があり、承認または承認を得ることが最初に求められたより限られた適応よりも遅延または失敗をもたらす可能性がある。
いくつかの薬品の高度な不良安全事象に深刻な公共健康リスクが存在することを考慮すると、FDAは、安全監視、配布および使用の制限、患者教育、強化ラベル、特殊包装またはラベル、特定の有害事象の報告を加速する、販売促進材料の事前承認、および消費者向け直接広告を制限することができる承認条件のうちの1つとして高価なREMSを要求する可能性がある。
私たちは医療専門家、臨床研究者、CRO、第三者支払者との関係は、私たちの現在と未来の業務活動に関連しており、連邦および州の医療詐欺と乱用法律、虚偽クレーム法律、透明性法律、政府価格報告、医療情報プライバシーと安全法律の制約を受ける可能性があり、これは私たちを刑事制裁、民事処罰、契約損害、政府医療計画から排除された名声損害、行政負担、利益、将来の収入減少などのリスクに直面させる可能性がある。もし私たちが医療法規を守らなければ、私たちは巨額の処罰に直面する可能性があり、私たちの業務、運営、財務状況は不利な影響を受けるかもしれない。
医療提供者と第三者支払者は,我々が上場承認を得た任意の候補薬の推薦や処方において主要な役割を果たしている。私たちの現在と未来の医療専門家、臨床研究者、CRO、第三者支払者、顧客との手配は、私たちがマーケティング許可を得た製品の業務または財務配置と関係を制限するかもしれない広範に適用される詐欺と乱用、その他の医療法律と法規に直面する可能性があります。適用される連邦および州医療法律法規の制限は、以下のものを含むが、これらに限定されない
•連邦医療計画は、他の事項に加えて、個人の転転または購入を交換または誘導し、連邦医療保険および医療補助計画のような連邦医療計画に従って支払うことができる任意の商品またはサービスを交換または誘導するために、直接または間接的に報酬を提供、請求、受け入れ、または提供することを禁止する
•他の事項に加えて、個人またはエンティティが意図的に提出したり、虚偽声明の提出を招いたり、連邦政府から支払いを得るために虚偽声明を意図的に使用することを禁止する連邦虚偽クレーム法案であって、私たちのように顧客にコード化および課金提案を提供するエンティティに適用可能である連邦虚偽クレーム法案
•連邦刑法は、詐欺の任意の医療福祉計画の計画を実行したり、医療事項に関する虚偽陳述を行うことを禁止する
•“陽光法案”下の連邦透明性要求は、一部は、医療保険、医療補助または児童健康保険計画の下で支払いを受けることができ、前年にカバーされた受給者に行われたいくつかの支払いおよび他の価値移転に関する情報をCMSに報告し、医師(医師、歯科医師、検眼師、足科医師および脊椎マッサージ師を含む)、ある非医師の医療専門家(例えば、医師アシスタントおよび看護師従業員など)および教育病院、ならびに医師および他の医療保健提供者およびその直系親族の所有権および投資権益に関する情報を含む、医薬品、器具、バイオ製品および医療用品の適用メーカーに要求される
•他の事項に加えて、HIPAAは、任意の医療福祉計画を詐欺または実行しようとする計画の実行または実行を禁止するか、または医療に関する虚偽の陳述を行うことを禁止する
•HITECHとその実施条例改正されたHIPAAはまた、強制的な契約条項を含む個人が識別できる健康情報のプライバシー、安全、伝送を保護する義務を規定している
•同様の州および外国の法律、例えば州反リベートおよび虚偽請求法は、非政府第三者支払者(私営保険会社を含む)によって精算される医療項目またはサービスの販売またはマーケティング手配およびクレームに関連する場合がある。
いくつかの州法律は、生物技術会社が生物技術業界の自発的コンプライアンスガイドラインと連邦政府が公布した関連コンプライアンスガイドラインを遵守することを要求し、医師および他の医療保健提供者への支払いと他の価値移転またはマーケティング支出に関する情報を報告することを製薬業者に要求する可能性がある。いくつかの州の法律は生物技術会社がある薬品の定価情報を報告することを要求し、ある州と地方の法律は薬品販売代表の登録を要求する。州法や外国法は健康情報のプライバシーやセキュリティを管理する場合もあり、多くの法律は互いに大きく異なり、HIPAAに先を越されず、コンプライアンス作業を複雑化させることが多い。例えば、欧州連合の衛生データの収集と使用は、ある条件下で欧州連合データ保護法の地理的範囲を非欧州連合実体に拡大する“一般データ保護条例”(GDPR)の管轄を受けている
既存のEUデータ保護原則を引き締め、会社のために新たな義務を創造し、個人のために新たな権利を創出する。GDPRを守らないことは巨額の罰金と他の行政処罰を招く可能性がある。GDPRは個人データを処理する上での私たちの責任と責任を増加させる可能性があり、GDPRの遵守を確保するために追加的なメカニズムの確立が要求されるかもしれません。これは深刻かもしれないが、もし私たちがGDPRや他の適用されたEUの法律や法規を遵守する努力が成功しなければ、EUでの私たちの業務に悪影響を及ぼすかもしれない。イギリスではすでにGDPRのような立法が施行されており、イギリスのデータ保護法とGDPRのような立法、すなわちイギリスのGDPRは、最高1750万ポンドまたは会社の世界売上高の4%の罰金を科すことが規定されており、金額の高い者を基準としている。データ保護法,イギリスGDPR,他のイギリスデータ保護法律や法規が中長期的にどのように発展する可能性があるかを完全に予測することはできず,イギリスに出入りするデータ転送をどのように規制するかに関する異なる法律や指導の影響を完全に予測することはできない。私たちは、EU加盟国とイギリスのGDPRとデータ保護法を遵守するために、私たちの政策と慣行を必要かつ適切に修正する必要があり、これらの法律とやり方を遵守するために私たちが取った任意の措置で責任、費用、コスト、および他の運営損失を招く可能性があることを発見するかもしれない
我々の現在と将来の第三者の業務配置が適用される医療法律や法規に適合することを確保する努力は、持続的な巨額のコストに関連する。政府当局は、私たちの業務実践が現在または未来に適用される詐欺および乱用または他の医療保健法律および法規に関連する現行または未来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。もし私たちの運営がこれらの法律または任意の他の私たちに適用される可能性のあるすべての政府法規に違反していることが発見された場合、私たちは民事、刑事および行政処罰、損害、罰金、返還、個人監禁、政府援助の医療計画(例えばMedicareおよびMedicaid)から除外された参加、誠実な監督と報告義務、契約損害、名声損害、利益および将来の収益の減少、および私たちの業務の削減または再編を含む重大な処罰を受ける可能性がある。このような行動を防御するには費用がかかり、時間がかかる可能性があり、大量の財政と人的資源が必要かもしれない。したがって、私たちが私たちに提起される可能性のあるいかなる訴訟も防ぐことに成功しても、私たちの業務は損害を受ける可能性がある。さらに、私たちがそれと業務を展開することを期待している任意の医師または他の医療提供者またはエンティティが、適用された法律に適合していないことが発見された場合、彼らは、政府の援助された医療計画から除外されることを含む刑事、民事または行政制裁を受ける可能性がある。
製品の販売とマーケティングを承認する以外に、生物製薬業界はアメリカでも厳格な監督と監督を受けている;私たちがこれらの法律を守らなければ、私たちの運営結果と財務状況を損なう可能性がある。
バイオ製薬製品のマーケティングに対するFDAの制限を除いて、私たちの業務は直接または間接的に私たちと医療保健提供者、顧客、第三者支払者との関係を通じて行われる可能性があり、連邦反バックル法規を含むが、これらに限定されない様々な連邦と州詐欺と乱用法律の制約を受ける。これらの法律は、生物製薬業界のいくつかのマーケティング行為を制限するために適用されている我々が提案した販売や教育計画などに影響を与える可能性がある。また、アメリカ連邦政府と私たちが業務を展開している州の患者プライバシー法規の制約を受ける可能性があります。私たちの運営能力に影響を及ぼす可能性のある法律には以下のものがある
•他の事項に加えて、連邦反バックル法規は、購入、レンタル、注文または購入、レンタル、またはMedicare、Medicaid、または他の連邦によって援助された医療計画に従って精算可能な任意の医療項目またはサービスの報酬を誘導または発注するために、故意に提供、支払い、請求または報酬を受けることを禁止する。この法規は一方では薬品メーカーであり,他方では処方者,購入者,処方マネージャーの間の手配に適用されると解釈されている。いくつかの法定例外状況と規制安全港保護のいくつかの一般的な活動は起訴されないが、例外状況と安全港の範囲は狭い。報酬に関するアプローチは、例外または避難港の資格に適合していない場合、処方、購入、または推薦を誘導するために告発される可能性があるため、審査される可能性がある。私たちの接近はすべての場合、安全港が反リベート責任から保護されるすべての基準を満たしていないかもしれない。また,個人や実体は,法規や法規違反の具体的な意図を実際に知る必要がなく,違反行為を実施することができる.また、連邦民事虚偽請求法については、連邦反リベート法規違反による物品やサービスを含むクレームが虚偽または詐欺的クレームを構成している。
•連邦民事虚偽請求法は、任意の個人または実体が連邦政府に故意に提出するか、または虚偽または詐欺的な支払いまたは承認クレームの提出をもたらすか、または連邦政府への支払いの義務を回避、減少または隠蔽するために故意に虚偽陳述を行うことを禁止する。これらの法律により,いくつかの製薬や他のヘルスケア会社が起訴され,顧客に製品を無料で提供する疑いがあるため,顧客がその製品のために連邦計画に請求書を発行したいと考えている。他の会社もラベル外販売促進による虚偽声明の提出で起訴された。私
当事者は連邦民事虚偽請求法案に基づいて政府の名義で任意の個人または実体に対して訴訟を提起し、訴訟収益を共有することができる。
•HIPAAは、医療福祉、プロジェクトまたはサービスの提供または支払いに関連する重大な事実を故意および故意に、または任意の医療福祉計画を意図的かつ故意に実行または実行しようとしている計画、または故意および故意に偽造、隠蔽または隠蔽し、または任意の重大な虚偽陳述を行い、刑事および民事責任を規定する;連邦反バックル法規と同様に、個人または実体は、法規または法規違反の具体的な意図を実際に理解することなく、違反行為を実施することができる。
•HITECHおよびその実施条例によって改正されたHIPAAは、適切な許可なしに、個人識別健康情報の使用または開示に関連するいくつかのサービスを提供する保証エンティティ、例えば、健康計画、医療チケット交換所、医療提供者およびその業務パートナーに対して、個人識別健康情報のプライバシー、安全および送信を保護するための強制的な契約条項を含むいくつかの義務を課す。
•FDCAは他を除いて,医薬品や医療機器に偽や誤ったブランドを貼り付けることを禁止している。
•連邦医師支払陽光法案及びその実施条例の一部は、Medicare、Medicaidまたは児童健康保険計画に従って精算可能な薬品、設備、生物製品および医療用品の適用メーカーが、前年に保険受給者に行った支払いおよび他の価値移転に関する情報を毎年CMSに報告し、医師(医師、歯科医師、視光師、足科医師および脊椎マッサージ師を含む)、特定の非医師の医療専門家(例えば、医師アシスタントや看護師従業員など)や教育病院、および医師および他の医療保健提供者およびその直系親族が持つ所有権と投資利益に関する情報を含む
•同様の州の法律および法規は、州反リベートおよび虚偽クレーム法律は、研究、流通、販売およびマーケティング手配、および任意の第三者支払人(個人保険会社を含む)の精算に関連する医療項目またはサービスに関するクレームを含むが、これらに限定されないが、製薬会社に製薬業界の自発的コンプライアンスガイドラインおよび米国連邦政府によって発行された関連コンプライアンスガイドラインを遵守することを要求しているか、または他の方法で医療提供者および他の潜在的な転換源に支払う可能性のあるお金を制限することを含む。州の法律と法規は薬品メーカーに価格とマーケティング情報に関する報告を提出することを要求し、医療専門家と実体に提供されるプレゼント及びその他の報酬と価値項目を追跡することを要求する;州と地方の法律は薬品販売代表の登録を要求する;そしてある場合の健康情報のプライバシーと安全を管理する州法律は、その中の多くの法律は互いに重大な違いがあり、しかも往々にしてHIPAAに先を越されず、コンプライアンス仕事を複雑化させる。
将来的に第三者の業務手配と適用される医療法律や法規に適合することを確保し、巨額のコストが及ぶ可能性がある。私たちの運営が、上記の任意の法律または任意の他の私たちに適用される可能性のある政府の法律および法規に違反していることが発見された場合、私たちは、民事、刑事および行政処罰、損害賠償、罰金、返還、個人監禁、政府援助から除外された医療計画(例えばMedicareおよびMedicaid)から除外され、追加の報告義務および監督(これらの法律を遵守しない疑いを解決するために会社の誠実な合意または他の合意に拘束されている場合)、および私たちの業務を削減または再構成することができます。私たちのいくつかの業務活動は、私たちの業務、財務状況、および運営結果に重大な悪影響を及ぼす可能性がある1つまたは複数の法律の挑戦を受ける可能性がある。
米国または国外の将来の訴訟、立法または行政または行政行動によって生じる可能性のある政府規制の可能性、性質、または程度を予測することはできない。任意の新しい立法を遵守したり、PACAによって実施された変化を逆転させることは、多くの時間とコストを費やし、私たちの業務に実質的な悪影響を及ぼす可能性があります
アメリカ議会は最近数回の調査を行い、連邦と州立法を提出し、薬品定価の透明性を高め、連邦医療保険下の処方薬のコストを下げ、定価とメーカー患者計画との関係を審査し、政府計画の薬品精算方法を改革することを目的とした。例えば、国会は2022年8月に“2022年インフレ低減法案”を可決し、その中には製薬業と連邦医療保険受益者に重大な影響を与える処方薬条項を含み、連邦政府がある高価な単一源連邦医療保険薬の最高公平価格の交渉を許可し、薬品価格交渉要求を守らないメーカーに罰と消費税を適用し、すべての連邦医療保険B部分とD部分の薬物がインフレリベートを獲得することを要求し、もしその薬品価格の増加がインフレより速い場合、連邦医療保険D部分を再設計して受益者の自己払い処方薬コストを下げるなどを含む
変えます。これらの立法、行政と行政行動、バイデン政府が将来実施する任意の医療措置と機関規則が私たちと製薬業全体に与える影響はまだ不明である。州レベルでは、一部の州では、州薬品価格透明性と報告法が検討されているか、最近公布されており、これは私たちのコンプライアンス負担を大幅に増加させ、規制部門の承認を得て商業化を開始した後、このような州法律に基づいてより大きな責任を負わせる可能性がある。これらの立法、行政と行政行動、バイデン政府が将来実施する任意の医療措置と機関規則が私たちと製薬業全体に与える影響はまだ不明である。コスト抑制措置や他の医療改革を実施することは、収入を創出し、利益を達成すること、または候補製品を商業化することを阻止するかもしれない(承認されれば)。
米国連邦や州医療立法の将来の行方を予測することはできません。これらの立法は、医療の獲得性を拡大し、医療コストをコントロールまたは低減することを目的としています。これらや法律や規制枠組みの任意のさらなる変化は、私たちの収入を減らしたり、コストを増加させたりして、私たちの業務、財務状況、運営結果に重大で不利な影響を与える可能性もあります。
私たちの普通株式所有権に関連するリスク
私たちの普通株の市場価格は非常に不安定かもしれないし、大幅に下落するかもしれない。
我々普通株の取引価格はずっと大きく変動しており、様々な要素の影響を受けて大幅に変動する可能性があり、その多くの要素は本年度報告の“リスク要因”の部分と以下に述べる要素を含む制御できない
•新冠肺炎の疫病または他の要素による臨床試験の実際または予想結果または任意の遅延、ならびに私たちの候補製品の承認に関連する監督審査結果;
•新冠肺炎の大流行の影響
•我々の戦略的パートナーまたは競合他社が発売または発表した新製品、候補製品または既存製品の新しい使用、およびこれらの発売または発表の時間;
•私たちの臨床研究組織および臨床試験場所の請求書発行時間および他の請求書実践に関連する費用を含む、私たちの任意の候補製品または臨床開発計画に関連する費用レベルの変化
•商業販売のための任意の将来の製品の十分な供給を承認することに関連する費用、または十分な供給を受ける能力があると考える能力があるか、または十分な供給を得ることができると考える
•私たちは他の候補製品や製品の結果を発見し、開発し、得るために努力している
•特許、訴訟事項、および私たちの技術のための特許保護を得る能力を含む、専有権に関する論争または他の発展;
•私たちまたは私たちの競争相手は重大な買収、戦略協力、合弁企業、資本約束を発表します
•私たちのパートナー(メルク社を含む)の商業的または臨床的成功または失敗、または知覚の成功または失敗;
•重要な科学技術者や管理者の増減
•バイオテクノロジーとバイオ製薬産業の状況や傾向
•メディアの関心、またはメディアの関心の変化、癌および癌治療への関心、最近のエボラ疫病、ならびにエボラ治療方法およびワクチンの開発の努力、または私たちが開発している候補製品が治療する任意の他の疾患または疾患;
•収益推定、発展スケジュール、または証券アナリストが提案した実際または予想変化;
•私たちの四半期の経営業績の実際と予想は変動している
•私たちが大衆に提供する可能性のある財務的予測と、これらの予測のいかなる変化も、私たちはこれらの予測を満たすことができなかった
•私たちの普通株を追跡する証券アナリストは、証券アナリストの推定または他のアナリストの格付け引き下げの影響から外れている
•流行病、政治的不確実性、戦争、テロ事件、自然災害、またはこれらの事件に対する反応などの公衆衛生危機による事件または要因を含む他の事件または要因;
•会計原則の変化
•財経や科学メディアやオンライン投資家コミュニティによる私たちの株価の議論
•一般的な経済や市場状況、および類似会社の市場評価の変化を含む、我々の経営業績や競争相手の経営業績とは無関係な要素を含む可能性がある
•私たちまたは私たちの株主は未来に普通株と、私たちの普通株の総取引量を売却します。
また,株式市場全体,特にバイオテクノロジーやバイオ製薬会社の市場は,極端な価格や出来高変動を経験しており,これらの変動はこれらの会社の経営業績に関係なくあるいは比例しないことが多い。これらの広範な市場と業界要素は、私たちの経営業績にかかわらず、私たちの普通株の市場価格を深刻に損なうかもしれない。過去、市場が一定期間の変動を経験した後、証券会社は集団訴訟を起こすことが多い。このような訴訟は将来的に我々に提起される可能性があり,巨額のコストや経営陣の注意や資源の移転を招く可能性があり,我々の業務や財務状況に重大かつ不利な影響を与える可能性がある。
私たちの主要株主と経営陣は私たちのかなりの割合の株を持っており、株主の承認が待たれる事項に大きな影響を与えることができるだろう。
2022年12月31日現在、我々の役員、役員、主要株主、およびそれらのそれぞれの関連会社は、2022年12月31日以降60日以内に行使可能な未償還オプションの株式を含む約44.1%の普通株を保有している。これらの株主は、私たちの取締役会の選挙、私たちの普通株または他の証券の将来の発行、私たちの普通株の配当を発表し、他の重大な会社取引を承認することを含む、私たちの管理と事務、株主の承認を必要とする事項に大きな影響を与えることができるだろう。このような所有権の集中は、私たちの支配権の変化を遅延または阻止するか、または他の方法で潜在的な買収者が私たちの支配権を獲得しようとすることを阻止する可能性があり、これは逆に私たちの普通株の公平な市場価値に実質的な悪影響を及ぼす可能性がある。また、役員や取締役およびその関連会社の実益が持つ株を売却することは第三者にマイナスとみなされ、私たちの株価にマイナス影響を与える可能性がある。しかも、私たちはあなたにこの株式がどのように分配され、その後投票することができるかを保証することができません。
我々の規約は、デラウェア州の州裁判所を指定しているか、または、デラウェア州に位置する州裁判所が管轄権を有していなければ、デラウェア州地域の連邦裁判所は、我々の株主が開始する可能性のある特定のタイプの訴訟および訴訟の唯一のおよび独占フォーラムとして、私たちまたは私たちの役員、上級管理者または従業員に対する訴訟を阻止することができる。
私たちの定款は、私たちが書面で代替裁判所を選択することに同意しない限り、デラウェア州衡平裁判所(または、デラウェア州衡平裁判所に標的管轄権がなければ、デラウェア州国内に位置する任意の州裁判所、またはそのようなすべての州裁判所に標的管轄権がない場合のみ、デラウェア州連邦地方裁判所)が法律によって許容される最大範囲内で、デラウェア州成文法または一般法の下で次のタイプの訴訟または手続の唯一かつ独占的裁判所となるべきである。(1)会社を代表して提起された任意の派生訴訟または訴訟;(2)会社の現職または前取締役、高級職員、他の従業員または株主が、会社または会社株主に対する信頼責任に違反していると主張する任意の訴訟、(3)DGCL、我々が改正および再記載した会社登録証明書または定款によって生成されたクレームを主張する任意の訴訟、またはDGCLがデラウェア州衡平裁判所に司法管轄権を付与する任意の訴訟、または(4)内部事務原則によって管轄されているクレームを主張する任意の訴訟。このような裁判所条項の選択は、証券法又は“取引法”に規定された義務又は責任を執行するための訴訟にも適用されず、連邦裁判所が排他的管轄権を有するいかなるクレームにも適用されない
これらの裁判所条項の選択は、株主が司法裁判所において、私たちまたは私たちの役員、上級管理者、または他の従業員と紛争することに有利であると考えるクレームを提出する能力を制限し、このような訴訟を阻止する可能性がある。さらに、裁判所が私たちが改正して再記載した会社登録証明書に含まれる裁判所条項の選択が訴訟で適用されないか、または実行できないことが発見された場合、私たちは他の管轄区域でそのような訴訟の解決に関連する追加費用を発生させる可能性がある。
私たちは予測可能な未来に、私たちはどんな現金配当金も支払わないと予想している。
現在の予想は、私たちは将来の収益を維持し、もしあれば、私たちの業務の発展と成長に資金を提供することだ。したがって、予測可能な未来には、私たち普通株の資本付加価値(あれば)が投資家の唯一の収益源となる
私たちの会社の登録証明書、私たちの定款あるいはデラウェア州の法律の条項はわが社の支配権の変更、あるいは私たちの経営陣の変更を阻止、延期、阻止する可能性があり、それによって私たちの普通株の取引価格を下げることができます。
当社の登録証明書、当社の定款またはデラウェア州法律の条項は、能動的な買収を阻止する効果があるか、あるいはわが社の支配権の変更を遅延または阻止する効果があるかもしれません。私たちの株主は、その株式によって現在の市場価格よりも高い割増取引を得る可能性があります。また、これらの規定は、株主が自分の利益に最も合っていると思う取引を承認する能力を制限する可能性がある。これらの規定には
•私たちの取締役会は3つのレベルに分かれていて、任期が3年交差しています
•株主提案と指名の事前通知要求;
•株主は特別会議を開くことができない
•株主が取締役を罷免したり、定款を改正したりする能力を制限する
•当社取締役会は、株主承認を必要とせずに条項を指定して新シリーズ優先株を発行する能力を有しており、その中には、買収または我々の支配権の他の変更を承認する権利、または権利計画を策定するために使用される可能性があり、潜在的敵意買収側の株式を希釈することを目的とした毒丸計画とも呼ばれ、当社取締役会の承認されていない買収を阻止する可能性がある。
また、デラウェア州会社法第203条は、当該商業合併が所定の方法で承認されない限り、株式を公開しているデラウェア州会社が利害関係のある株主と商業合併を行うことを禁止している。
上記の条項と反買収措置の存在は、投資家が将来私たちの普通株に支払うことを望む可能性のある価格を制限する可能性がある。また、潜在的な買収者がわが社を買収することを阻止し、買収で普通株の割増を得る可能性を下げることができます。
証券や業界アナリストが我々の業務に関する研究報告を発表しない場合、あるいは不正確または不利な研究報告を発表しなければ、私たちの株価や取引量は低下する可能性がある。
私たちの普通株の取引市場は、証券や業界アナリストが発表した私たちまたは私たちの業務に関する研究と報告にある程度依存するだろう。私たちの1人以上のアナリストを追跡して私たちの株式格付けを引き下げたり、私たちの業務に関する不正確または不利な研究報告を発表したりすれば、私たちの株価は下落するかもしれない。これらのアナリストのうちの1人以上が私たちへの報告を停止したり、私たちに関する報告書を定期的に発表できなかったりすれば、私たちの株に対する需要が減少する可能性があり、これは私たちの株価や取引量を低下させる可能性がある。
私たちの株主の保有量は希釈される可能性があり、私たちの証券価格は発行された株式オプションや将来発行される証券の行使によって低下する可能性があります。
我々は、追加の普通株、優先株、制限株式単位(RSU)、または普通株に変換することができ、または普通株に交換可能な証券を発行することができる。また、発行済み株式オプションを行使することができる普通株のほとんどは、購入されると、直ちに公開市場で販売する資格がある。追加の普通株、優先株、RSU、または私たちの普通株に変換することができ、または私たちの普通株に交換可能な証券を発行するか、または株式オプションを行使することは、既存の投資家の権益を希釈し、私たちの証券の価格に悪影響を及ぼす可能性がある。さらに、このような証券の権利は、既存の投資家が保有する証券の権利よりも優先される可能性がある。
項目1 B。未解決従業員意見
ありません
項目2.財産
以下に述べるレンタル施設で主な操作を行う | | | | | | | | | | | | | | | | | | | | |
| 位置 | | 行われた操作 | | 2平方フィートに近い | | レンタル期日 |
| テキサス州オースティン | | 行政事務室 | | 5,000 | | 2023年11月30日 |
| エイムズアイオワ州 | | 行政事務室と研究開発 | | 6,000 | | March 31, 2023 |
2023年1月、私たちはアイオワ州のエイムズで賃貸契約を更新した。契約更新期限は2026年3月31日です。私たちは私たちの行政事務空間が予測可能な未来の私たちの需要を満たすのに十分だと信じている
項目3.法的手続き
正常な業務過程で、私たちは時々様々な訴訟、訴訟、紛争、あるいはクレームの影響を受ける可能性があります。私たちの接近法はこのようなクレームが発生した時に調査することだ。クレーム自体は予測できませんが、私たちに不利であれば、単独でまたは統合して、その業務、財務状況、運営結果、またはキャッシュフローに大きな悪影響を及ぼすことは、現在のところ何も知られていません。
プロジェクト4.鉱山安全開示
適用されません。
第II部
項目5.登録者普通株式市場、関連株主事項、発行者による株式証券の購入
私たちの普通株はナスダックの世界市場でオファーされ、コードは“LUMO”です。
2023年2月28日までに、私たちは69人の普通株主がいます。株主の実際の数は,実益所有者である株主を含むこの記録保持者の数を超える可能性があるが,その株式は仲介人や他の被指名者が街頭名義で保有する.この数の登録所有者は、その株式が他のエンティティが信託形式で保有する可能性のある株主も含まれていない。
配当政策
私たちは現金配当金を送ったことがありません。私たちは近い未来に私たちの普通株のいかなる現金配当金も発表したり支払わないと予想している。私たちはすべての収益を維持するつもりで、もしあれば、私たちの業務に投資します。将来の配当金の支払いは私たちの取締役会が適宜決定し、私たちの未来の収益(あれば)、私たちの資本要求、財務状況、その他の関連要素に依存するだろう。
発行人が株式証券を購入する
次の表は、2022年12月31日までの3ヶ月間の私たちの普通株の買い戻し状況をまとめています。
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| 期間 | | 購入株式総数 | | 1株平均支払価格 | | 公開発表された計画又は計画の一部として購入した株式総数(1) | | 計画や計画によってはまだ購入していないかもしれない株の約ドルの価値 |
| 2022年10月1日-2022年10月31日 | | — | | — | | — | | $2,808,000 |
| 2022年11月1日-2022年11月30日 | | 25,382 | | $4.87 | | 25,382 | | $2,685,000 |
| 2022年12月1日2022年12月31日 | | 90,037 | | $3.78 | | 90,037 | | $2,345,000 |
| 合計する | | 115,419 | | $4.02 | | 115,419 | | $2,345,000 (2) |
1.本四半期のすべての購入は我々の取締役会の許可の下で公開市場取引によって行われ、2022年8月16日に発表された最大300万ドルの普通株を購入することができます。
2それは.この金額には、2022年12月31日現在、2022年8月に査定された300万ドルの残り部分が含まれている。この買い戻し計画に基づいて購入した商品は満期日がありません。
第六項です[保留されている]
適用されません。
プロジェクト7.経営陣の財務状況と経営成果の検討と分析
以下の議論及び分析は、本年度報告に含まれる監査された総合財務諸表及びその付記とともに読まなければならない。本年度報告書には、1933年証券法第27 A条(改正)、1934年証券取引法第21 E条(改正)、および1995年個人証券訴訟改革法の定義に適合する展望的陳述が含まれており、このような陳述は、これらの条項によって創出された“安全港”に制約されているリスクと不確実性にも触れています. 前向きな陳述は,我々の経営陣の信念と仮定と,本稿の発表日までに我々の経営陣が把握している情報に基づいている多くの要因のため,以下に述べるような要因本年度報告の第I部第1 A項“リスク要因”では、我々の実際の結果は、これらの前向き陳述で予想される結果と大きく異なる可能性があるので、これらの前向き陳述に過度に依存してはならない。法的要件がない限り、私たちは、これらの前向き陳述を公開的に更新する義務がないか、または、未来に新しい情報があっても、これらの前向き陳述で予想される結果と大きく異なる理由で、実際の結果を更新する義務がない。新冠肺炎疫病及び私たちの業務と全世界経済に対する潜在的な影響はこれらの要素を増幅する可能性がある。
概要
Lumos製薬会社は臨床段階の生物製薬会社である。本年度報告では“私たち”“私たち”“私たちの”““会社“又は”Lumos“とは、Lumos Pharma,Inc.及びその完全子会社を意味する。私たちの主な実行事務室はテキサス州オースティンに位置し、他の実行と行政事務室はアイオワ州エイムズに位置し、著者らは著者らの臨床計画を推進することに力を入れ、そして全世界範囲内で稀な疾病を有する患者のために新製品と新療法を識別、獲得、開発、商業化することに集中し、現在安全で有効な治療に対する巨大な需要はまだ満足されていない。我々の普通株はナスダック世界市場(“ナスダック”)に上場し、株式コードは“LUMO”である
我々は我々の唯一の候補製品である成長ホルモン分泌促進剤イブナモロン(“Lum−201”)の開発に集中し,特発性児童成長ホルモン欠乏症(“PGHD”)や他のまれな内分泌疾患の治療に用いる潜在的な経口療法である。
PGHDはまれな内分泌疾患であり,約3500名から17歳まで生まれた人に1人が発生する。PGHDの原因は,先天性(このような疾患を有する小児),後天的(脳腫瘍,頭部損傷あるいは他の原因),医原性(医療による)または特発性(原因不明)であってもよい。未治療のPGHD児童は顕著な成長発育障害があり、潜在的な成人の身長は5フィートより明らかに低く、そして身体成分の異常があり、骨鉱物質が減少し、痩せ体重が減少し、脂肪の質が増加する可能性がある。
PGHDの主要な治療目標は成長を回復させ、小柄な児童を正常な身長に達成させ、そして代謝異常、認知欠陥と生活の質低下に関連する合併症を防止することである。現在、PGHD治療の標準は毎日組換えヒト成長ホルモンを皮下注射することに限られ、治療周期は平均7年に達する。治療期間中、組換えヒト成長ホルモンを毎日注射する依存性が悪いことは成長に不利な影響を与える可能性がある。2021年8月、FDAは、Skytrofa、週1回の注射は、治療中の患者の注射回数を減少させる新しい治療法を承認した;しかし、私たちの市場研究によると、多くの提供者と患者は経口治療に傾向があると信じている
二零二年三月十八日、当社はNewLink Genetics Corporation(“NewLink”)、NewLink全資子会社Cyone Merger Sub,Inc.(“Merge Sub”)とLumos Phとの業務統合(“合併”)を完了したArma,Inc.(“個人ルモス”)、後に“Lumos Pharma Sub,Inc.”と改名しました。合併子会社はPrivate Lumosと合併してPrivate Lumosに合併し、Private Lumosは当社の完全子会社として存続する。合併完了直前、NewLink普通株の株式は9株1株の逆分割比率で調整された。合併条項によると、Lumosプライベート株主は合計4,146,398株のNewLink普通株を獲得した(逆分割実施後)。後に続く逆株式分割と合併が完了した時点で、私たちの普通株は8,292,803株の流通株があり、その約50%はPrivate LumosとNewLink証券所有者がそれぞれ持っている。今回の合併は逆資産買収とみなされている
LUM−201成長ホルモン分泌促進剤
著者らの研究の重点は1種の経口小分子Lum-201を開発することであり、それは成長ホルモン(GH)分泌促進剤であり、ibuamorenとも呼ばれ、稀な内分泌疾患の治療に用いられ、現在組換えヒト成長ホルモン(“rhGH”)の注射が許可されている。Lum-201は錠剤製剤であり、1日1回投与する
Lumosは2018年7月にAmmonett Pharma LLC(“Ammonett”)からLum-201を買収した。Lum−201は2017年に成長ホルモン欠乏症(GHD)により米国とEUで孤児薬物名(“ODD”)を獲得した。米国特許“成長ホルモン欠乏症の検査と治療”は2036年に満期になった。関連特許はすでにEU(検証中)、オーストラリア、イスラエル、日本、韓国、香港、ウクライナで発行されており、関連特許出願は他の複数の司法管轄区で決定されている。承認されれば,Lum−201は最初に承認された経口成長ホルモン分泌促進剤となる可能性があり,PGHDから成長ホルモン欠乏に関連するまれな内分泌疾患の治療に用いられ,現在の組換え成長ホルモン製品注射の標準案に代替案を提供している。分泌促進剤は別の物質の分泌あるいは放出を刺激する物質である。LUM−201は成長ホルモンの放出を刺激し,成長ホルモン分泌促進剤と呼ばれる
Lum-201はGH分泌促進受容体(GHSR 1 A)を通じてGHを刺激し、グレリン受容体とも呼ばれ、そしてソマトスタチンの放出を抑制し、それによって1種の差別化された作用機序を提供し、内因性、脈動性GH分泌の幅を増加させることによっていくつかの稀な内分泌疾患(GH欠乏に関連する)を治療する。LUM-201の刺激作用は、GH循環レベルおよびその下流媒体インスリン様成長因子によって調節され、後者は、超生理レベルで下垂体からのGHの追加放出をフィードバックまたは負方向に調節し、それによってGH放出の過剰刺激を防止する。Lum−201は、機能は正常であるが視床下部GH軸が減少する患者の内因性GH分泌を刺激することが観察されている
PGHD患者は、器質性PGHD(より深刻なGH欠損症)および特発性PGHD(あまり深刻ではないGH欠損症)と診断された患者を含む。特発性PGHD患者(すなわちそれらの機能は正常であるが視床下部GH軸が低下した患者)はPGHD患者の約60%を占めており,Lum−201に反応すると考えられている
2020年第4四半期に、著者らはPGHDにおけるLum-201の作用を研究する計画を開始し、著者らの第2段階臨床試験(OraGrowth H 210試験あるいは“第2段階試験”)を開始し、この研究に参加する初期サイトを開放した。OraGrowth H 210試験は世界的な多地点ランダム試験であり、特発性PGHDと診断された約80名の被験者が3種類の用量レベル(0.8、1.6と3.2 mg/kg/日)を経口投与したLum-201と毎日標準用量を注射した組換え成長ホルモンの治療効果を評価した。
この研究の主な終点は,われわれの予測的濃縮マーカー(“PEM”)患者の選択戦略の初歩的な検証であり,Lum−201に反応する選択された患者のパーセンテージから証明された。著者らの第二段階の臨床試験では、各患者は1日0.8 mg/kgの単剤Lum-201を投与され、彼らが入選カットオフ基準に符合するかどうか、即ちベースラインインスリン様成長因子-1>30 ng/mlと刺激GH 5 ng/mlを決定した。主要な治療効果の終点は年化身長速度である。二次終点は、将来の研究のための小児科用量のLum-201を選択することを含み、第三段階を含み、PEM陽性患者がOraGrowth H 210スクリーニングに参加するPEM選択過程の再現性の程度を決定することを含む
我々は2023年2月28日にOraGrowth H 210試験の登録を完了し、82名の被験者を発表し、2023年第4四半期の全82名の被験者の完全な6ヶ月の主要な結果データを予定している。
PGHDにおけるLum-201の第2の同時試験(“OraGrowth H 212試験”)は2021年第2四半期に開始し、成長ホルモン脈動性分泌の拡大におけるLum-201の新しい作用機序の効果を探索することを目的とした。PGHDで行ったOraGrowth H 212試験はOraGrowth H 210試験と並行して行った。OraGrowth H 212試験は単一地点、開放ラベル試験であり、Lum-201の2つの異なる用量レベル(1.6と3.2 mg/kg/日)で24名のPGHD被験者に及ぶ薬物動態学と薬効学(“PK/PD”)効果を評価した。OraGrowth H 212試験の目的は以前の臨床データを実証することであり、LUM-201特有の内因性成長ホルモンパルス式放出増加及びその対の潜在力を説明し、この作用機序は治療効果の向上に役立つ。我々のOraGrowth H 212試験は専門の小児科センターで行われ、このような臨床試験に必要なより頻繁なサンプル採集とモニタリングを行うことができる。OraGrowth 212試験のデータは、将来の規制申告文書でサポートされる可能性があるが、この試験は、規制部門がLum-201を承認するために必要なものではない。この試験の主な終点は,24名の被験者に及ぶ6カ月のPK/PDと身長速度データである。我々が2023年2月28日に発表したように,我々はこの試験の22名の被験者の登録を完了しており,全22名の被験者の予備データは2023年第4四半期に発表される予定である
2021年7月16日のFDAの手紙の要求に応えるために、私たちは2021年7月に、FDAがLum−201の治療を12ヶ月以下に制限したと発表した。その際,我々が最初に計画していた長期延長試験(“OraGrowth 2 11試験”)を開始する前に,PGHDにおけるLum−201の追加効果データを収集するために,OraGrowth H 210とOraGrowth H 212試験の治療期間を6カ月から12カ月に延長した。2022年5月10日に発表されたように,OraGrowth H 210とOraGrowth 212試験の初歩的な安全性と有効性データを審査した後,FDAは部分保留をキャンセルし,現在Lum−201による12カ月以上の治療を許可している。そのため、OraGrowth H 210試験は24ケ月まで延長し、被験者が途切れることなくLum-201治療を継続できるようにした。また#年第2四半期には
2022年には,OraGrowth 212試験のプログラムが改正され,被験者が女性14歳と男性16歳の骨齢に達し,成人に近い身長を反映するまで治療を受けることが認められた
治療期間の延長はOraGrowth H 210やOraGrowth H 212試験の主要な結果データに影響を与えず,これらのデータは治療前6カ月の経年化データに基づく。我々は引き続きOraGrowth H 210試験を延期した場合にLong−Term Expansion Studyの開始時間を評価し,適切な時期に延期研究を開始する予定である。これらの案の変化は起こらず,Timeを単独で延長してOur3臨床試験を開始する予定である。
2022年第1四半期に、我々のOraGrowth H 213試験(“OraGrowth H 213試験、およびOraGrowth H 210試験、OraGrowth H 211試験およびOraGrowth H 212試験、すなわち”OraGrowth H試験“)を開始し、開放的で多中心的な第2段階研究であり、OraGrowth H 210試験を完成した20名までの特発性PGHD患者に対するLum-201の成長効果と安全性を評価した。被験者はLum-201を3.2 mg/kg/日の用量レベルで服用し、最長12カ月に達する
2022年11月14日、OraGrowth H 210およびOraGrowth H 212試験の中期分析が期待されたことを発表した。OraGrowth H 210の中期分析は,41名の患者をランダムに4つの治療群に分け,約10名の患者で6カ月の治療を完了した後に行った。Lum−201の6カ月の年化身長速度(AHV)は1.6 mg/kg/日であり,我々の成長予想に合致していた。4つの治療群の各群の6ケ月の平均(中央値)AHVは以下の通りである
•7.26(7.71)cm/年、投与量は0.8 mg/kg(n=11)
•8.57(8.61)cm/年(n=10)1.6 mg/kg投与
•3.2 mg/kg群(n=10)7.77(8.11)cm/年
•成長ホルモンアーム11.05(10.48)cm/年(n=10)
1.6 mg/kg投与群で観察された6カ月平均AHVは8.6 cm/年であり,われわれが予想していた8.3 cm/年AHVと一致し,組換え成長ホルモン(RhGH)治療12カ月後に観察された大型20年期礼来社Genesisデータベースからの中等度単純治療PGHD患者群であった。2 これは,他の3つの大型歴史データベースで観察された組換えヒト成長ホルモン治療を受けた中等度PGHD患者の1年目の身長速度にも相当する。3,4,5
組換えヒト成長ホルモンコントロール群では6カ月で11.05 cmのAHVが産生され,特発性PGHD群では思わぬ成長反応であった。この予期しない成長は、既知の組換えヒト成長ホルモン列の中の2人の最年少被験者が強い成長反応(15.6 cm/年と12.7 cm/年)を示し、いくつかのベースライン特徴における他の不均衡が、組換えヒト成長ホルモンのより大きな成長反応に対する予測因子としても記録されているためである可能性がある。3,5,6 組換え成長ホルモン対照群で治療を受けた特発性PGHD患者で発見された予想以上のAHVは,類似した特徴の群で行われた複数の歴史的試験と一致せず,これらの試験予測の増加範囲は8.3−8.6 cm/年であった。3-6年齢を除いて、治療中により大きな成長を予測するベースライン特徴は、身長(低い身長)、低い身長とIGF-1標準偏差スコア(SDS)、遠い親からの高さ(MPH)、およびより高い身体質量指数標準偏差スコア(BMI SDS)を含む。1.6 mg/kg Lum-201群と組換えヒトオーキシン群の不均衡ベースラインパラメータを以下の表に示す
2 Blum et al Jes 2021
3 Lechuga-Sanchoら2009年JPEM
4 Rankeらの2010年のJCEM
5 ブラントらのジャース2021
6 ヤンたち。自然科学代表2019
5つのベースラインパラメータの不平衡は、組換えヒト成長ホルモンのより高い成長を示唆している
| | | | | | | | |
| ベースライン指標 | 1.6 mg LUM-201平均値(SD) N=10 | 組換え人成長ホルモン 平均値(SD) N=10 |
| 年齢は月単位である | 99.3 (28.3) | 90.3 (26.7) |
| 身長(センチ) | 114.6 (9.6) | 111.6 (11.9) |
| IGF-1ドデシルチオ酸エステル | -1.17 (0.72) | -1.37 (0.48) |
| マイル毎時(センチ単位) | 166.98 (7.15) | 168.78 (8.85) |
| BMI SDS | -0.35 (0.79) | +0.31 (1.05) |
年齢の不均衡は,年齢が階層的要因であるため,入学者数の増加とともにバランスになると考えられる。5歳以下の3名の被験者のうち,2名は成長ホルモン列中の成長異常値であった。より高い登録数に伴い、制御グループにおけるより速い成長をサポートする予測要因の不均衡が解決され、すべてのキュー間のより大きなバランスをもたらす可能性があると信じている
仮発表時には,試験に参加した被験者の約75%のベースライン特徴に関する追加情報,すなわち合計58名の被験者がいた。以下に示すベースライン特徴はキュー間のより良いバランスを表しており,実験が完全に登録されるにつれて異常値の影響が弱まることが予想される.
| | | | | | | | | | | | | | |
| LUM-201 0.8 mg | LUM-201 1.6 mg | LUM-201 3.2 mg | 組換え人成長ホルモン |
平均値(SD) | 平均値(SD) | 平均値(SD) | 平均値(SD) |
N=14 | N=15 | N=14 | N=15 |
年齢(月) | 99.1 (28.3) | 98.4 (28.6) | 92.9 (22.6) | 94.1 (23.7) |
身長(センチ) | 115.1 (12.5) | 114.6 (11.2) | 112.4 (9.2) | 113.4 (10.6) |
高度標準偏差 | -2.32 (0.3) | -2.31 (0.5) | -2.32 (0.4) | -2.25 (0.4) |
最大高さSD | -1.76 | -1.66 | -1.57 | -1.73 |
IGF-1ドデシルチオ酸エステル | -1.43 (0.67) | -1.30 (0.67) | -1.35 (0.57) | -1.32 (0.46) |
最大IGF-1 SDS | -0.3 | -0.3 | -0.6 | -0.7 |
毎時マイル(センチ) | 165.5 (7.1) | 164.3 (7.2) | 166.1 (7.0) | 168.5 (7.9) |
MPH-SDS | 1.43 (0.66) | 1.70 (0.54) | 1.92 (0.73) | 1.75 (0.63) |
BA遅延(年) | 1.89 (1.02) | 1.91 (0.53) | 2.20 (0.86) | 1.68 (0.9) |
BMI SDS | -0.47 (1.09) | -0.38 (0.91) | -0.55 (0.79) | +0.14 (1.08) |
OraGrowth H 210中期分析9ヶ月と12ヶ月のポイント
一部の被験者が得られる9カ月と12カ月の中期データは、患者数が少ないにもかかわらず、これらの遅い治療間隔において、Lum−201の成長反応が持続的であることを示している。LUM−201 1.6 mg/kg群(6カ月8.57 cm/年から12カ月8.14 cm/年)と比較して,組換えヒトグレリン群のAHV率は時間の経過とともに有意に低下した(6カ月で11.05 cm/年,12カ月で9.93 cm/年)。
LUM-201は12ヶ月間の持続的な成長率を示しています
| | | | | | | | | | | | | | | | | | | | |
| OraGrowth H 210 AHV(中央値) | 6か月 | 9ヶ月です | 12か月 |
| Cm/年 | n | Cm/年 | n | Cm/年 | n |
| 0.8 mg/kg/日LUM-201 | 7.26 | 11 | 6.17 | 5 | 6.74 | 4 |
| 1.6 mg/kg/日LUM-201 | 8.57 | 10 | 8.48 | 6 | 8.14 | 4 |
| 1日3.2 mg/kg LUM-201 | 7.77 | 10 | 6.80 | 6 | 6.94 | 3 |
| 34μg/kg/日組換えヒト成長ホルモン | 11.05 | 10 | 10.46 | 7 | 9.93 | 4 |
OraGrowth 212中期分析のポイント
OraGrowth H 212試験は単地点、開放ラベル試験であり、1.6と3.2 mg/kg/日の2つの用量レベルで24名の治療を受けていないPGHD被験者における経口Lum-201の薬物動態学(PK)と薬効学(PD)効果を評価した。OraGrowth H 212試験ではいずれの被験者もPEM陽性であったため,Lum−201に対する反応性が増強した
OraGrowth H 212試験の中期分析は,2つのLum−201治療群の1つにランダムに割り当てられた10名の被験者が6カ月の治療を完了した後に行った。各腕のAHVはOraGrowth H 210試験で観察されたAHVに相当する。また,被験者の数が限られているにもかかわらず,この増加は12カ月まで持続可能であることが示唆された。この単独の研究はまた、対象が治療12カ月近くで治療されたので、OraGrowth H 210試験で見られたLum−201とrhGHとの間のAHV差の縮小を支持した
GraGrowth H 212のLUM-201は、OraGrowth H 210と同様の成長率を示す:
| | | | | | | | | | | | | | | | | | | | |
| OraGrowth H 212 | 6か月 | 9ヶ月です | 12か月 |
| Cm/年 | N | Cm/年 | n | Cm/年 | n |
| 1.6 mg/kg/日LUM-201 | 7.14 | 5 | 6.85 | 4 | 7.21 | 2 |
| 1日3.2 mg/kg LUM-201 | 8.60 | 5 | 8.00 | 4 | 7.78 | 3 |
OraGrowth 212試験の他のデータは2023年3月5日に国際小児科内分泌学会議で口頭報告の形で提出された。中間分析以来15人の被験者または他の5人の被験者(1.6 mg/kg治療群3名,3.2 mg/kg治療群2名)の分析結果を報告した。その結果,1.6から3.2 mg/kg/dの用量範囲ではLum−201の6カ月連続耐性が良好であり,用量依存性を産生する成長ホルモンAUCが有意に増加した0-12hそれは.ベースラインと比較した成長ホルモンパルス幅の増加は身長速度の改善に関与しており,年化身長速度への影響は12カ月間持続したと報告されている。
OraGrowth H 210とOraGrowth 212の合同事後分析
3期試験の最適投与量を確定するために、特殊な後分析を行い、OraGrowth H 210とOraGrowth H 212試験の成長データを結合し、サンプル量を増加させた。この分析は,2つの高い用量のLum−201の6カ月,9カ月,12カ月の平均AHVを示した。研究間にはいくつかのベースライン特徴の差があるが,これまでに登録されているすべての被験者がPEM陽性であったため,より広範な中等度PGHD群を代表していた。この2つの試験の1.6 mg/kgと3.2 mg/kgの組み合わせの事後分析は、最大2種類のLum-201用量間の成長率が非常に類似しており、第3段階試験の最適用量として1.6 mg/kgを選択することを支持することを確認した。歴史的研究に基づき,投与量反応は0.8から1.6 mg/kgではなく1.6 mg/kgから3.2 mg/kgの間で予想され,以前正常健常ボランティアで生成されたPK/PDデータに基づいてPDプラットフォームは2.8 mg/kgであることが示された。7
| | | | | | | | | | | | | | | | | | | | |
| OraGrowth H 210+OraGrowth H 212 | 6か月 | 9ヶ月です | 12か月 |
| Cm/年 | N | Cm/年 | n | Cm/年 | n |
| 1.6 mg/kg/日LUM-201 | 8.09 | 15 | 7.83 | 10 | 7.83 | 6 |
| 1日3.2 mg/kg LUM-201 | 8.05 | 15 | 7.28 | 10 | 7.36 | 6 |
安全性とフォールトトレランスのポイント
Lum−201はこれまで2つのOraGrowth試験のデータが,安全性と耐性の面で試験中の組換えヒト成長ホルモン被験者に相当することを示してきたため,良好な安全性を示すと信じている。中期データでは,治療に関する重篤な有害事象(SAE)はなく,SAEによる退出はなく,実験室値,有害事象データ,心電図値に有意なセキュリティ信号は認められなかった。OraGrowth 212試験のセキュリティデータはOraGrowth H 210試験のデータと一致した。
7 メルク001研究
エボラワクチン
2014年11月,NewLinkはメルク社とNewLink Merckプロトコルを締結し,PHACから許可を得たエボラワクチンrVSVG−ZEBOVを開発し,商業化することが可能である。食品·医薬品局の承認を得た場合、RVSVG−ZEBOVもPRVを取得する資格があり、会社はPRVを販売、譲渡、または他の方法で処分することで得られたPRV価値の60%を得る権利がある。2019年12月20日、メルク社は、米国食品薬品監督管理局が18歳以上の個人によるザイールエボラウイルス感染予防のための(ザイールエボラ生ワクチン)の申請を許可したと発表した。資産購入契約によると、メルク社は2回に分けてPRV費用を支払うことに同意した。合意の要求によると、2020年9月30日までの3ヶ月間、メルク社は成約時に3,400万ドル、2021年1月11日に2,600万ドルを支払った。
私たちは特定の国のワクチン販売から特許使用料を稼ぎ続ける可能性があると受け取っている。しかし,ワクチンの市場は主に特許権使用料の支払いから除外された発展途上国,あるいはワクチンが低利益率やゼロ利益率で寄付または販売されている地域に限られると考えられるため,将来的にはメルク社の巨額の特許権使用料支払いを受けることはないと予想される。
腫瘍学候補者
私たちは合併でNewLinkから買収した小分子製品候補製品があります。これらの候補製品、インドキシモッド、NLH 802(インドキシモッドのプロドラッグ)およびNLG 919(直接IDO 1酵素阻害剤)はインドール−2,3−ジオキシゲナーゼ経路阻害剤。我々は、トポイソメラーゼ1阻害剤カンプトテシンに結合したシクロデキストリンポリマー骨格からなり、2019年12月17日からEllses Pharma Limitedの許可を得るナノ粒子-薬物結合体である追加の小分子製品候補NG 207を有する。
2017年8月15日と2019年2月19日に、インドキシモ塩およびプロドラッグ配合を含む2つの米国特許がそれぞれ米国で発行され、少なくとも2036年の排他性を提供した。私たちはいくつかの国でこのような処方のための国際特許カバーを求め続けている。私たちはこれらの候補製品のさらなる開発と許可機会の潜在力を探るかもしれないが、私たちは現在、これらの獲得された小分子候補製品に対するいかなる積極的な計画も持っていない。
財務概要
収入.収入
私たちは商業販売を許可された製品もなく、製品販売から何の収入も得ていない。将来、私たちがどんな協力または許可合意に達したら、私たちは製品販売、許可料、マイルストーン、または他の前払いから収入を得ることができるかもしれない。私たちはまた製品販売の印税から収入を得ることができるかもしれない。私たちは、多くの理由で、今後の収入が四半期ごとに変動し、このような支払いや販売の不確定な時間と金額を含むと予想している。
研究と開発費
研究開発費は主に私たちの候補製品Lum-201を推進する費用を含む。私たちの研究開発費には、内部人員支出と、契約研究·製造機関、コンサルタント、私たちの科学コンサルタントなどの第三者との合意の下で発生する外部研究開発費が含まれています。
私たちは発生した費用に応じて研究と開発費用を支払う。将来の研究開発活動のための貨物およびサービスの払戻不可能な前払いを資産として資本化し、サービスを提供するか、または貨物を受信したときに支出する。私たちは、予測可能な未来に、私たちの候補製品のための臨床試験計画を継続し、私たちのパイプラインを開発し、規制部門が私たちの候補製品の承認を求めるにつれて、私たちの研究開発費が増加すると予想している。
一般と行政費用
一般と行政費用は主に法律、監査、税務と商業コンサルティングサービスの専門費用、人件費と出張費を含む。私たちは、私たちの経営活動の拡大に伴い、将来の一般的かつ行政的費用が増加すると予想している
新冠肺炎
新冠肺炎の発生による疫病はすでに深刻な国と世界経済の中断を招き続け、私たちの運営に悪影響を与え続ける可能性がある。私たちは新しい冠肺炎(あれば)がある資産の帳簿価値と私たちの持続的な運営に与える潜在的な影響を積極的に監視している。今まで、私たちは臨床試験の進展に関連する遅延を経験して、臨床サイトは彼らのプログラムを調整して、大流行中に患者の面倒を見た;しかし、私たちは新冠肺炎のためにいかなる資産減少値も産生しなかった。これらの事件はどの程度私たちの業務、臨床開発、監督管理努力及び私たちの普通株の価値に影響する可能性があり、これは未来の発展に依存し、これらの発展は高度な不確定性を持っており、現在予測できない。これらの影響およびそれによって我々の運営に与える干渉の持続時間および強度はまだ不明であり、これらのイベントが2023年度の当社の運営、財務状況、運営結果、およびキャッシュフローに及ぼす可能性のある影響を評価し続ける。
経営成果
2022年12月31日までと2021年12月31日までの年次比較 | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | 十二月三十一日までの年度 | | |
| |
| | 2022 | | 2021 | | ドル単位の変化 | | 変動率% |
| | (単位:千) | | (単位:千) | | |
| 収入: | | | | | | |
| 許可と協力収入 | | $ | — | | | $ | 10 | | | (10) | | | (100) | % |
| 特許権使用料収入 | | 1,523 | | | 220 | | | 1,303 | | | 592 | % |
| 総収入 | | 1,523 | | | 230 | | | | | |
| 運営費用: | | | | | | | | |
| 研究開発 | | 17,857 | | | 16,246 | | | 1,611 | | | 10 | % |
| 一般と行政 | | 15,706 | | | 15,331 | | | 375 | | | 2 | % |
| 総運営費 | | 33,563 | | | 31,577 | | | | | |
| | | | | | | | |
| その他の収入、純額 | | 965 | | | 281 | | | 684 | | | 243 | % |
| | | | | | | | |
| 所得税割引 | | 13 | | | 636 | | | (623) | | | (98) | % |
| | | | | | | | |
| 純損失 | | $ | (31,062) | | | $ | (30,430) | | | | | |
収入を得る。2021年同期と比較して,2022年12月31日までの1年間で許可と連携収入が10,000ドル減少したのは,主にメルクの下請けとしてERVEBOに関する作業が完了したためである。2021年同期と比較して,2022年12月31日までの1年間で特許使用料収入が130万ドル増加したのは,ERVEBO販売に関する特許使用料収入が原因であった
研究と開発費それは.2021年同期と比較して,2022年12月31日までの1年間に研究開発費が160万ドル増加したのは,主に臨床試験や契約製造費が110万ドル増加し,相談費用が50万ドル増加し,人員関連費用が30万ドル増加したが,株式報酬費用が20万ドル減少し,用品,減価償却,レンタル料の運営費が10万ドル減少したためである
一般と行政費用です。2021年同期と比較して、2022年12月31日までの1年間、一般と行政費用は40万ドル増加し、主な原因は特許使用料が90万ドル増加し、出張費用が40万ドル増加し、その他の費用が30万ドル増加したが、人事関連費用は40万ドル減少し、株式報酬費用は40万ドル減少し、相談費用は30万ドル増加し、用品、減価償却、レンタル料の運営費用は10万ドル減少した
他の収入、純額。2022年12月31日までの1年間で、2021年同期に比べて純他収入が70万ドル増加したのは、主に2022年の利息収入の増加によるものだ。
所得税の割引2021年同期と比較して、2022年12月31日までの年間所得税優遇が60万ドル減少したのは、主に2021年の不確定納税義務の放出により2021年に確認された60万ドルの当期税収割引によるものである。
流動性と資本資源
私たちはこれまで研究開発に取り組んできましたが、私たちの製品の商業販売から収入を得たことはありません私たちは予測可能な未来には研究開発計画や私たちの運営に関する一般的かつ行政的コストそれは.しかし私たちは現在の現金、現金等価物、短期投資残高を信じています近似値2022年12月31日現在、テリーは6740万ドルです2024年第3四半期の運営に資金を提供するのに十分です特発性(中等度)PGHD患者のOraGrowth H 210とOraGrowth H 212試験本年度報告書の提出日から少なくとも12か月.
もし私たちの利用可能な現金と現金等価物が私たちの流動性要件を満たすのに十分でない場合、または私たちがもっとそうする機会を開発した場合、私たちは追加の株式や債務証券を売却したり、時々信用手配を得ることを求めるかもしれない。追加的な株式と債務証券の売却は、私たちの株主の所有権をさらに希釈させる可能性がある。もし私たちが債務証券や優先株を発行することでより多くの資金を調達すれば、これらの証券は私たちの普通株より優先する権利を持っている可能性があり、私たちの運営を制限する契約を含む可能性がある。私たちは現在予想されている金額よりもっと多くの資金が必要かもしれない。このような必要な追加資本は、もしあれば、合理的な条項では得られないかもしれない。もし私たちが追加的な融資を受けることができなければ、私たちは私たちの計画の一部または全部の研究開発活動の縮小、延期、または廃止を要求される可能性があり、これは私たちの業務を損なうかもしれない。
私たちの候補製品の研究や開発に関連する多くのリスクと不確実性のため、私たちの運営資金需要の正確な金額を見積もることができません。私たちの将来の資金需要は多くの要素にかかっていますが、これらに限定されません:
•私たちの候補製品の臨床試験の範囲、進捗、結果とコスト、そして新候補製品に関する発見と開発活動
•私たちの候補製品のための規制承認の時間と関連したコスト
•もし私たちの候補製品が販売を許可された場合、マーケティング、販売、施設、流通コストを含む商業化活動のコスト
•私たちの候補製品や商業化された製品を作るコストは
•私たちは、戦略的協力、許可または他の手配、およびそのような合意の財務条項の能力を確立し、維持する
•政府が提供した未返済ローンをどの程度返済する必要があるか
•請求項の準備、提出、起訴、維持、弁護、および執行に関連する費用は、訴訟費用および訴訟結果を含む
•新冠肺炎の大流行やウクライナの軍事衝突による持続的な悪影響、労働力不足、サプライチェーンの中断とインフレを含む国内と全世界の商業またはマクロ経済状況の変化
•私たちの未来の製品(あれば)の販売時間、領収書、販売金額、あるいは印税です。
2020年12月30日に制御対象株式発行協定を締結しましたSMエージェント(“エージェント”)Cantor Fitzgerald&Co.と販売プロトコル(“販売プロトコル”)を締結し,このプロトコルにより,吾らはエージェントを通して最大5,000,000,000ドルまでの普通株式(“株式”)を随時提供および販売することができる.株式の発行と売却は証券法に基づいて登録されている。販売協定によれば、代理人は、証券法第415条(A)(4)条に定義された“市場で”発売された任意の方法で株式を販売することを法律で許可され、またはナスダックによって、株式の任意の他の既存の取引市場で、販売時の市価またはそのような現行の市価に関連する価格および/または法律によって許可されている任意の他の方法で行われる取引取引を含むことができる。吾らは、発行された株式数、売却を要求する時間帯、任意の日以内に売却可能な株式数のいかなる制限、及び下回ってはならない任意の最低売却価格を代理人に通知する。今回発行した純収益を運営資金や一般会社用途に利用する予定であり,我々の候補製品の臨床開発機会を潜在的な他の適応に拡大すること,一般と行政費用を含むがこれらに限定されない。純収益の一部を将来の戦略取引に投資することも可能であり、買収によって私たち自身と相補的な候補製品や技術を買収することで、私たちの製品ラインを拡大し、多様化することができるかもしれない。私たちは代理店に販売総価格の3.0%の手数料を支払うつもりだ
販売契約に基づいて同社を通じて売却された株式。また、私たちは今回の発売によって発生したいくつかの費用を精算することに同意した。販売プロトコルに記載されているように、代理店または吾等は、他方に通知した後にいつでも販売プロトコルを終了することができ、または場合によっては、吾等を含む業務または財務状況に重大かつ不利な変化が生じ、非現実的または株式販売契約を実行することができない場合には、代理店はいつでも販売契約を終了することができる。2022年12月31日現在、販売契約に基づいて株式を発行していない。
我々の公開流通株が7,500万ドルを下回る限り,S-3表の一般指示I.B.6に記載されている制限を受け,これらの制限は,販売プロトコル下での市場計画下での発行を含む表S-3登録声明による初公開発行能力を制限している.これらの制限により、いかなる12ヶ月の間も、総時価が私たちの公衆流通株の三分の一を超えるS-3表証券を売ってはいけません。2023年3月3日現在,表S-3の一般命令I.B.6から計算した我々の公開現存金は2340万ドルである.
2022年8月16日、我々の取締役会は株式買い戻し計画を承認し、この計画によると、最大300万ドルの発行済み普通株を購入することができると発表した。2022年12月31日までの1年間に、137,526株を約675,000ドルで買い戻しました。これらすべての購入は公開市場取引によって行われ、株は買い戻し時に実際にログアウトした。
キャッシュフロー
次の表は、以下の各期間の主要な現金源および用途(千計)を示しています | | | | | | | | | | | | | | | | | |
| | 十二月三十一日までの年度 | |
| | 2022 | | 2021 | |
| 経営活動のための現金純額 | $ | (26,623) | | | $ | (29,648) | | |
| 投資活動提供の現金純額 | (11,358) | | | 25,970 | | |
| 融資活動のための現金純額 | (821) | | | (192) | | |
| 現金および現金等価物の純減少 | $ | (38,802) | | | $ | (3,870) | | |
我々の経営活動は、2022年と2021年12月31日までの年間で、それぞれ2660万ドルと2960万ドルの現金を使用している。減少の主な原因は、計上費用の増加と前払い費用の減少、運転資金の変化により現金純流入が440万ドル増加したが、業務損失が60万ドル増加したことと、純損失と経営活動で使用された現金純額を調整する調整減少によって相殺され、株式補償費用が60万ドル減少し、減価償却と償却が20万ドル減少したことである
我々の投資活動は、2022年と2021年12月31日までに、それぞれ現金1,140万ドルと現金2,600万ドルを使用している。減少の原因は,2021年12月31日までの年度内にPRV売却最終期から受け取った現金2600万ドルと,2022年12月31日までの年度内に短期投資を購入するための現金1140万ドルである。
我々の融資活動は、2022年と2021年12月31日までの年間で、それぞれ80万ドルと20万ドルの現金純額を使用している。この伸びは、主に2022年12月31日までの年間、普通株買い戻しのための現金が70万ドルだったが、2022年12月31日までの1年間で、既存株奨励のための源泉徴収税の現金が減少し、その増加を部分的に相殺したためである。
重要な会計政策と重大な判断と見積もり
私たちの経営陣は財務状況と経営結果の討論と分析は私たちの連結財務諸表に基づいています。私たちの連結財務諸表はアメリカ公認会計原則に基づいて作成されています。これらの財務諸表を作成する際には、資産、負債、費用報告金額に影響を与える推定、仮説、判断を行う必要があります継続的にこれらの推定を評価していますそして判断力。私たちの見積もりは歴史的経験と私たちが当時の状況で合理的だと思う様々な仮定に基づいています。これらの見積りと仮定は,資産や負債の帳簿価値を判断する基礎を構成し,他のソースからは見えない費用を記録している。したがって,異なる仮定や条件では,実際の結果はこれらの推定値と大きく異なる可能性がある.
我々の主要会計政策は、本年度報告その他の部分の総合財務諸表付記2により詳細に説明されているが、我々の主会計政策は、総合財務諸表の付記2においてより詳細に説明されている。以下の会計政策が総合財務諸表の作成に用いる判断と見積もりが最も重要であると考えられる。
研究開発コスト
研究·開発コストは発生時に費用を計上する。研究開発支出は、主に従業員に関連する支出を含み、その中には、賃金、ボーナス、福祉、株式ベースの給与、製造に関連するコスト、臨床試験を行う契約研究機関、研究場所およびコンサルタントとの合意の下で生じる支出、施設、固定資産およびその他の分配された支出、研究施設および設備の賃貸料およびメンテナンスの直接および分配支出、ライセンス製品および技術に関連する許可料およびマイルストーン支払い、および非臨床活動および規制承認に関連するコストが含まれる。将来の研究開発活動のための貨物やサービスの前払いが延期され,前払い資産として入金される。繰延金額は、関連貨物の引渡しまたはサービス提供時に料金を計上します。
第三者との契約に基づいて費用を計上する
私たちは、未決済契約および購入注文を審査し、実行されたサービスを決定するために担当者とコミュニケーションし、実行されたサービスレベルを評価し、プロバイダから請求書を発行していない可能性のあるサービスから発生する関連コストを評価するプロセスによって、計算すべき費用を推定する。資産負債表日あたりの課税費用推定数は、当時知られていた事実と状況に基づいている。このような推定は、正確性を確認するために、定期的にサービスプロバイダに確認されるであろう。
我々は,我々を代表して臨床試験を行って管理している複数の研究機関や契約研究組織と締結した契約に基づいて,受信したサービスや費用を推定し,臨床試験に関連する費用を決定する。第三者請負業者が提供するサービスのために発行された領収書は数ヶ月遅れる可能性がある。我々は,契約に規定されている管理費,現場管理とモニタリング費用およびデータ管理費用の見積もりに基づいて,第三者請負業者活動に関するサービス費用を計上しなければならない。実際の臨床試験コストと推定臨床試験コストとの差は,手術によりそれらが分かった時間帯に調整されている。
第七A項。市場リスクの定量的·定性的開示について
私たちは金利の変化と関連した市場リスクに直面している。2022年12月31日と2021年12月31日まで、私たちの現金、現金等価物、短期投資はそれぞれ6740万ドルと9480万ドルで、主に通貨市場基金、商業手形、会社債務証券、アメリカ政府と機関証券を含む。私たちの市場リスクに対する主な開口は金利感受性であり、これは米国金利の全体的な水準の変化の影響を受けている。2021年の大部分の期間、金利は歴史的に比較的低い水準を維持し、FRBは連邦基金目標区間を0.0%~0.25%に維持した。しかし、2022年全体で、FRBは金利を4.25%から4.50%の目標範囲に引き上げ、インフレ兆候が増加していることを考慮して、2023年と2024年にはさらに利上げが予想されると述べた。私たちのポートフォリオの存続期間が短く、私たちの投資リスクが低いため、金利が直ちに10%上昇することは、私たちのポートフォリオの公平な市場価値に実質的な影響を与えない。
項目8.財務諸表と補足データ | | | | | |
| ルモス製薬会社 | |
| |
| 連結財務諸表索引 | |
| |
| ページ |
| |
独立公認会計士事務所報告 (ピマウェイ会計士事務所, テキサス州オースティンPCAOB ID番号:185) | 77 |
2022年と2021年12月31日までの連結貸借対照表 | 78 |
2022年12月31日と2020年12月31日までの総合経営報告書と全面赤字報告書21 | 79 |
2022年12月31日までと2021年12月31日まで年度株主権益変動表 | 80 |
2022年12月31日と2022年12月31日までの連結キャッシュフロー表1 | 81 |
連結財務諸表付記 | 82 |
独立公認会計士事務所報告
株主や取締役会に
Lumos製薬会社:
連結財務諸表に対するいくつかの見方
我々は、Lumos Pharma社とその子会社(当社)が2022年12月31日と2021年12月31日までの総合貸借対照表、同年度までの関連総合経営表と全面赤字、株主権益変化と現金流量、および関連付記(総称して総合財務諸表と呼ぶ)を監査した。総合財務諸表は,すべての重要な点において,会社の2022年12月31日と2021年12月31日までの財務状況と,2022年12月31日と2021年12月31日までの年度の経営結果とキャッシュフローを公平に反映しており,米国公認会計原則に適合していると考えられる。
意見の基礎
これらの連結財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいてこのような連結財務諸表に意見を発表することだ。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、連結財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査することを求められておらず、私たちも招聘されて監査を行っていません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性について意見を述べるためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、連結財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであっても詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれる。これらの手続きは、連結財務諸表中の金額および開示に関する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、合併財務諸表の全体列報の評価も含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
重要な監査事項とは、連結財務諸表を当期監査する際に生じた、伝達された、または監査委員会に伝達された事項である:(1)総合財務諸表に対して重大な意義を有する勘定または開示に関連する;(2)私たちが特に挑戦的で、主観的または複雑な判断に関連する。私たちは重要な監査事項が存在しないと確信する。
/s/ピマウェイ法律事務所
2015年以来、当社の監査役を務めてきました。
テキサス州オースティン
March 7, 2023
ルモス製薬会社
合併貸借対照表
(千単位で1株当たりおよび1株当たりのデータは含まれていない) | | | | | | | | | | | | | | |
| | 十二月三十一日 |
| | 2022 | | 2021 |
| 資産 | | | | |
| 流動資産: | | | | |
| 現金と現金等価物 | | $ | 56,007 | | | $ | 94,809 | |
| 短期投資 | | 11,352 | | | — | |
| 前払い費用と他の流動資産 | | 4,427 | | | 4,740 | |
| その他売掛金 | | 223 | | | 128 | |
| | | | |
| | | | |
| 流動資産総額 | | 72,009 | | | 99,677 | |
| 非流動資産: | | | | |
| 財産と設備、純額 | | 53 | | | 79 | |
| 使用権資産 | | 230 | | | 556 | |
| 非流動資産総額 | | 283 | | | 635 | |
| 総資産 | | $ | 72,292 | | | $ | 100,312 | |
| 負債と株主権益 | | | | |
| 流動負債: | | | | |
| 売掛金 | | $ | 275 | | | $ | 612 | |
| 費用を計算する | | 6,200 | | | 4,166 | |
| 賃貸負債の当期分 | | 233 | | | 352 | |
| | | | |
| | | | |
| 流動負債総額 | | 6,708 | | | 5,130 | |
| 長期負債: | | | | |
| アイオワ州経済発展局に支払う特許権使用料義務 | | 6,000 | | | 6,000 | |
リース責任 | | — | | | 205 | |
| | | | |
| 長期負債総額 | | 6,000 | | | 6,205 | |
| 総負債 | | 12,708 | | | 11,335 | |
| 引受金とその他の事項 | | | | |
| | | | |
| | | | |
| | | | |
| 株主権益: | | | | |
非指定優先株式、$0.01額面:ライセンス株式-5,000,0002022年12月31日および2021年12月31日;発行済および発行済み株式-02022年12月31日と2021年12月31日に | | — | | | — | |
普通株、$0.01額面:ライセンス株式-75,000,0002022年と2021年12月31日に8,283,708そして8,366,8192022年と2021年12月31日にそれぞれ流通株-8,267,968そして8,357,3912022年12月31日と2021年12月31日にそれぞれ | | 82 | | | 83 | |
国庫株は、原価で計算する15,740そして9,4282022年と2021年12月31日に保有する株式 | | (170) | | | (114) | |
| 追加実収資本 | | 187,164 | | | 185,429 | |
| 赤字を累計する | | (127,483) | | | (96,421) | |
| その他の総合損失を累計する | | (9) | | | — | |
| 株主権益総額 | | 59,584 | | | 88,977 | |
| 総負債と株主権益 | | $ | 72,292 | | | $ | 100,312 | |
| 連結財務諸表の付記を参照。 |
ルモス製薬会社
合併経営報告書と全面赤字
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
| | | | | | | | | | | | | | | | | |
| | 十二月三十一日までの年度 | |
| | |
| | 2022 | | 2021 | |
| 収入: | | | | | |
許可と協力収入 | | $ | — | | | $ | 10 | | |
| 特許権使用料収入 | | 1,523 | | | 220 | | |
| 総収入 | | 1,523 | | | 230 | | |
| 運営費用: | | | | | |
| 研究開発 | | 17,857 | | | 16,246 | | |
| 一般と行政 | | 15,706 | | | 15,331 | | |
| 総運営費 | | 33,563 | | | 31,577 | | |
| 運営損失 | | (32,040) | | | (31,347) | | |
| 他の収入や支出: | | | | | |
| その他の収入、純額 | | 91 | | | 269 | | |
| 利子収入 | | 874 | | | 12 | | |
| | | | | |
| | | | | |
| その他の収入、純額 | | 965 | | | 281 | | |
| 税前純損失 | | (31,075) | | | (31,066) | | |
| 所得税割引 | | 13 | | | 636 | | |
| 純損失 | | $ | (31,062) | | | $ | (30,430) | | |
| | | | | |
| 1株当たり純損失: | | | | | |
| 基本的希釈の | | $ | (3.71) | | | $ | (3.65) | | |
| | | | | |
| | | | | |
| | | | | |
| 発行済み普通株式加重平均 | | | | | |
| 基本的希釈の | | 8,373,821 | | | 8,334,516 | | |
| | | | | |
| その他の全面的な損失: | | | | | |
| 短期投資の未実現損失 | | (9) | | | — | | |
| 全面損失総額 | | $ | (31,071) | | | $ | (30,430) | | |
| | | | | |
| | | | | |
| 連結財務諸表の付記を参照。 |
ルモス製薬会社
合併株主権益変動表
(単位:千、共有データを除く)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | 普通株 | | | 在庫株 | | その他の内容 実収資本 | | 赤字を累計する | | その他の総合損失を累計する | | 合計する 株主権益 |
| | 株 | | 金額 | | | | 株 | | 金額 | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| 2020年12月31日残高 | | 8,305,269 | | | $ | 83 | | | | | — | | | $ | — | | | $ | 182,480 | | | $ | (65,991) | | | $ | — | | | $ | 116,572 | |
| 株式ベースの報酬 | | — | | | — | | | | | — | | | — | | | 2,879 | | | — | | | — | | | 2,879 | |
| 株式オプションの行使 | | 26,922 | | | — | | | | | — | | | — | | | 64 | | | — | | | — | | | 64 | |
| 帰属制限株式単位時に発行される株 | | 33,933 | | | — | | | | | — | | | — | | | — | | | — | | | — | | | — | |
| 源泉徴収金の奨励のために出された株式 | | (9,428) | | | — | | | | | 9,428 | | | (114) | | | — | | | — | | | — | | | (114) | |
| 株購入計画に基づいて株式を売却する | | 695 | | | — | | | | | — | | | — | | | 6 | | | — | | | — | | | 6 | |
| 純損失 | | — | | | — | | | | | — | | | — | | | — | | | (30,430) | | | — | | | (30,430) | |
| 2021年12月31日の残高 | | 8,357,391 | | | $ | 83 | | | | — | | 9,428 | | | $ | (114) | | | $ | 185,429 | | | $ | (96,421) | | | $ | — | | | $ | 88,977 | |
| 株式ベースの報酬 | | — | | | — | | | | | — | | | — | | | 2,320 | | | — | | | — | | | 2,320 | |
| 株式オプションの行使 | | 17,288 | | | — | | | | | — | | | — | | | 40 | | | — | | | — | | | 40 | |
| 帰属制限株式単位時に発行される株 | | 25,771 | | | — | | | | | — | | | — | | | — | | | — | | | — | | | — | |
| 源泉徴収金の奨励のために出された株式 | | (6,312) | | | — | | | | | 6,312 | | | (56) | | | — | | | — | | | — | | | (56) | |
| 株購入計画に基づいて株式を売却する | | 11,356 | | | — | | | | | — | | | — | | | 49 | | | — | | | — | | | 49 | |
| 普通株買い戻し | | (137,526) | | | (1) | | | | | — | | | — | | | (674) | | | — | | | — | | | (675) | |
| その他総合損失 | | — | | | — | | | | | — | | | — | | | — | | | — | | | (9) | | | (9) | |
| 純損失 | | — | | | — | | | | | — | | | — | | | — | | | (31,062) | | | — | | | (31,062) | |
| 2022年12月31日の残高 | | 8,267,968 | | | $ | 82 | | | | | 15,740 | | | $ | (170) | | | $ | 187,164 | | | $ | (127,483) | | | $ | (9) | | | $ | 59,584 | |
| 連結財務諸表の付記を参照。 |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | |
ルモス製薬会社
統合現金フロー表
(単位:千) | | | | | | | | | | | | | | | |
| | 十二月三十一日までの年度 | |
| | 2022 | | 2021 | |
経営活動のキャッシュフロー | | | | | |
| 純損失 | | $ | (31,062) | | | $ | (30,430) | | |
| 純損失と経営活動で使用される現金純額の調整: | | | | | |
| 株式ベースの報酬 | | 2,320 | | | 2,879 | | |
| 減価償却および償却 | | 49 | | | 217 | | |
| その他の収入、純額 | | (26) | | | — | | |
| 経営性資産と負債変動状況: | | | | | |
| 前払い費用と他の流動資産 | | 492 | | | (1,086) | | |
| その他売掛金 | | (92) | | | 136 | | |
| 売掛金と売掛金 | | 1,696 | | | (1,364) | | |
| | | | | |
| | | | | |
| | | | | |
| 経営活動のための現金純額 | | (26,623) | | | (29,648) | | |
| 投資活動によるキャッシュフロー | | | | | |
| | | | | |
| 優先審査クーポン券の最終号支払いを販売しております | | — | | | 26,000 | | |
| 有価証券を購入する | | (11,337) | | | — | | |
| 設備を購入する | | (21) | | | (30) | | |
| 投資活動提供の現金純額 | | (11,358) | | | 25,970 | | |
| 融資活動によるキャッシュフロー | | | | | |
| 株式オプションの行使 | | 40 | | | 64 | | |
| 株購入計画に基づいて株式を売却する | | 49 | | | 6 | | |
| 既得賞与源泉徴収税の支払い | | (56) | | | (114) | | |
| 普通株買い戻し | | (675) | | | — | | |
支配持分発行下の普通株発行コストSM | | (179) | | | (148) | | |
| 融資活動のための現金純額 | | (821) | | | (192) | | |
| 現金と現金等価物の純減少 | | (38,802) | | | (3,870) | | |
| 年初現金および現金等価物 | | 94,809 | | | 98,679 | | |
| 年末現金および現金等価物 | | $ | 56,007 | | | $ | 94,809 | | |
| | | | | |
| | | | | |
| 連結財務諸表の付記を参照。 | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
1. D業務のeScription
業務の組織と性質
Lumos製薬会社は臨床段階の生物製薬会社である。 本年度報告書で言及されている“私たち”、“会社”または“Lumos”は、Lumos Pharma,Inc.およびその完全子会社を意味する。私たちの主な執行事務所はテキサス州オースチンにあり、他の執行と行政事務所はアイオワ州のエイムズにあり、私たちは力を入れています私たちの臨床計画と集中して 世界的にまれな疾患患者のために新製品と新療法を識別、取得、開発し、商業化する現在安全で有効な治療には満足されていない大量の需要がある. 私たちの普通株はナスダック世界市場(“ナスダック”)に上場し、取引価格は株式コードは“LUMO”である
会社(The Company)業務合併を達成する(“合併”)同社の前身はNewLink Genetics Corporation(“NewLink”)、NewLinkの完全子会社Cyclone Merge Sub,Inc.(“Merge Sub”)およびLumos Pharma,Inc.(“Lumos Pharma Sub,Inc.”)である。(“プライベートルモス”)合併は2020年3月18日に完了した合併子会社はPrivate Lumosと合併してPrivate Lumosに合併し、Private Lumosは会社の完全子会社として存続する合併が完了する直前に、NewLink普通株の株式は逆分割比率に調整された1‑for‑9. 合併条項によると、個人Lumos株主は全部で4,146,398株NewLink普通株式(逆分割実施後)適用することができます1株当たり普通株、Aシリーズ優先株、Bシリーズルモス優先株を発行しており、株式交換割合は0.1308319305, 0.0873621142そして0.1996348626お別れしますそれは.株式の逆分割と合併が完了した直後に現れた8,292,803会社は普通株式の株式を発行したそのうちの約1割は50%は、Private LumosおよびNewLink証券所有者によってそれぞれ保有されています。今回の合併は逆資産買収とみなされている
合併が完了した後、合併後の会社はPrivate Lumos唯一の候補製品の開発に集中した分泌促進剤Ibuamoren(“Lum-201”)は特発性児童成長ホルモン欠乏症(“PGHD”)と他の稀な内分泌疾患を治療する潜在的経口療法である。
流動性とリスク
同社は従来,そのほとんどの努力を研究·開発に投入しており,その製品の商業販売から収入を得たことがない。経営陣は、予見可能な将来において、会社の研究·開発活動により、会社はより多くの大きな被害を受け続けると予想している。しかし、会社は既存の現金、現金等価物、短期投資は約#ドルだと考えている67.42022年12月31日までに、Lum-201の第2段階臨床試験(“OraGrowth H 210試験”)とOraGrowth H 212試験(以下“OraGrowth H 212試験”)の初歩的な読み取りは、2024年第3四半期までその運営に資金を提供することができ、各試験は2023年第4四半期に行われる予定である。もし満期の会社の債務を流動資金で支払うのに十分でない場合、私たちの将来の業務は追加の株式または融資スケジュールに依存するだろう。会社が追加融資を必要とする場合、これらの融資は会社に有利な条項で提供されるか、または全くできないことは保証されない。会社が将来の運営資金需要を満たすためにより多くの資金を調達できない場合、それはその研究計画の範囲を延期または縮小し、および/またはその運営を制限または停止させることを余儀なくされる。
新冠肺炎(“新冠肺炎”)は新型コロナウイルスSARS-CoV-2の爆発による大流行であり、すでに深刻な国家と世界経済の混乱を招き続け、会社の運営に悪影響を与え続ける可能性が高い。新冠肺炎の制限は著者らの臨床試験中のサイト起動と患者登録の速度が比較的に遅く、それによって著者らのOraGrowth H 210試験読み出しの6ケ月の主要な結果データの遅延を招いた。臨床試験の遅延を経験しましたが私たちは新冠肺炎のためにどんな資産減価も発生していない。これらの事件はどの程度会社の業務、臨床開発、監督管理努力及び普通株の価値に更に影響する可能性があり、これは未来の発展に依存し、これらの発展は高度な不確定性を持っており、現在予測できない。これらの影響の持続時間と強度、それによる会社運営への干渉は不確定である会社は、これらの事件が運営、財務状況、および運営およびキャッシュフロー結果に及ぼす可能性のある影響を評価し続ける。
2. 重要会計政策の概要
列報と合併の基礎
添付されている総合財務諸表は、Lumosとその完全子会社の勘定を含み、米国公認会計原則(“米国公認会計原則”)に基づいて作成されている。重要なのです統合で会社間口座と取引が解消されたn. 経営陣は、公正新聞に必要とされるすべての正常かつ恒常的な調整が、添付の連結財務諸表に含まれていると考えている。
使用推定数
準備作業米国公認会計原則に基づいて作成された連結財務諸表は、会社の財務諸表および付記に報告された資産、負債および費用金額、または有資産および負債の開示に影響を与えるために、管理層に推定および仮定を要求する。報告された資産および負債金額に影響を与える重大な管理職推定には、株式に基づく報酬、臨床試験に基づく課税項目、および繰延税金資産が含まれる。現在の事件と理解可能な行動の理解に基づいて、私たちの連結財務諸表を作成する際に使用される見積もりと仮定に基づいていると考えられます未来の柯氏 適切であれば、実際の結果はこれらの推定値とは異なる可能性があり、どのような違いも実質的である可能性がある。
金融商品は価値と信用リスクを公正に許容する
当社の金融商品の公正価値は、計量資産や負債の際に使用する投入の主観的程度に応じて、階層開示フレームワーク記録を採用している。これら3つのレベルの記述は以下のとおりである
•第1級:計量日には、同じ資産又は負債の投入が調整されておらず、活発な市場でオファーされる.
•第2レベル:第1レベル以外に直接または間接的に観察可能な投入、例えば資産や負債の見積もりやあまり活発でない市場のオファー。
•第3レベル:資産や負債の投入は観察できず、その際に入手可能な最適な情報に基づいて制定されており、その中には会社自身のデータが含まれている可能性がある。
現金と現金等価物
現金および現金等価物には、現金預金、預金、通貨市場基金、および購入時の元の満期日が90日以下の債務証券投資が含まれる。
投資する
経営陣は、購入時に売却可能な債務証券投資の適切な分類を決定する。一般に、購入日の元の満期日が90日を超える投資は、経営陣がこれらの投資を利用して現在の業務に資金を提供すること、または現在の業務に利用できるようにすることを意図しているので、短期投資として分類される。売却可能な証券に対する投資を公正価値で報告し、未実現収益と損失(税項控除)は総合貸借対照表の中で他の全面的な損失を累計する構成部分と表記した
当社は、特定の確認方法に基づき、各報告期間終了までの売却可能投資を審査し、公正価値が低下しているかどうかを決定します。公正価値の低下が信用に関連する要因である場合、当社は信用損失準備金に計上する。1つの投資が減値するかどうかを決定する際に、当社は、減値の深刻さ、関連信用格付けの変動、予想の回復、その売却意向、あるいは予想時価が回復する前にこの投資の売却を要求される可能性、および所定の現金支払いを継続する可能性を含む様々な要因を考慮する。当社が信用に関する減価が発生したと判断した場合、当社は、その証券を売却しようとしているかどうか、または回収前に証券の売却を要求される可能性が高いかどうかを評価します。この2つの条件のいずれかが満たされた場合、会社は純損失の中で1つの費用を確認し、その費用は証券の余剰コストベースとその公正価値との間のすべての差額に等しい。もし会社が証券を売却しようとせず、回収前に証券の売却を要求される可能性が低い場合、損失は実現されない
信用損失を代表する金額に分け,純損失で確認し,他のすべての要因に関する金額を累積他の総合収益損失に記録した。
財産と設備
当社はリース改善や設備が将来的に他の用途があると信じており,コスト別に計上しているため,資本化している。すべての賃貸改善と設備の減価償却は、資産のレンタル期間または推定耐用年数の短い時間で直線法で計算される。コンピュータ装置の使用寿命は三つ至れり尽くせり5年.
状況が変化する限り、資産の帳簿価値が回収できない可能性があることを示し、長期資産の減価審査を行う。保有·使用する資産の回収可能性は,資産群の帳簿金額と資産群が予想する将来の未割引現金流量純額(主に売却資産の収益に関連する)との比較により測定した。このような資産は減値とみなされ,確認すべき減値は資産の帳簿価値が資産公正価値を超える金額で計測される.処分すべき資産を帳簿または公正価値から売却コストの中の低いものを引いて申告する。
賃貸借証書
♪the the the会社は最初から賃貸であるかどうかを確認しています。リース使用権(“ROU”)資産代表会社がリース期間内に対象資産を使用する権利を経営し、リース負債代表会社がリースにより発生したリース金を支払う義務を負う。経営リースROU資産および負債は、開始日にレンタル期間内のリース支払いの現在値によって確認されます。会社(The Company)’レンタル条項は、会社が選択権を行使することを合理的に決定したときに、リース契約を延長または終了する選択権を含むことができる。レンタル支払いのレンタル料金はレンタル期間内に直線法で確認します。(付記6を参照。)
第三者との契約に基づいて費用を計上する
同社は、未締結契約および購入注文を審査し、提供されたサービスを決定し、提供されたサービスのレベルを推定し、サプライヤーから請求書を発行していない可能性のあるサービスによって生じる関連コストを決定するプロセスによって、その計算すべき費用を推定する。資産負債表日あたりの課税費用推定数は、当時知られていた事実と状況に基づいている。このような推定は、正確性を確認するために、定期的にサービスプロバイダに確認されるであろう。
同社の臨床試験に関する費用は,代表会社と臨床試験を行って管理する複数の研究機関と契約研究組織が締結した契約に基づいて受信したサービスや費用の努力を推定している。第三者請負業者が提供するサービスのために発行された領収書は数ヶ月遅れる可能性がある。当社は、契約による管理費、現場管理·監視費用及びデータ管理費用の見積もりに基づいて、第三者請負業者活動に関するサービス費用を計上しなければならない。実際の臨床試験コストと推定臨床試験コストとの差は,手術によりそれらが分かった時間帯に調整されている。
所得税
所得税は貸借対照法で計算される。繰延税金項目の資産と負債は現有の資産と負債の帳簿額面及びそれぞれの課税基礎と営業損失及び税項相殺の間の差異による将来の税務項目の結果を確認することができる。税金資産及び負債を繰延して税率計量を策定し、その等の一時的な差額を回収又は決済する予定の年度の課税収入に適用されることが予想される。税率変動が繰延税金資産や負債に及ぼす影響は、公布日を含む期間の経営実績で確認されている。経営陣は繰延税金資産の現金化能力を評価し、繰延税金資産のすべてまたは一部が現金化できない可能性が高い場合に評価値を記録して準備する。
当社はいかなる不確定な税務倉位の影響についても会計処理を行い、根拠となる敷居は、税務機関審査を適用する税務倉位の技術的利点に基づいて当該等の税務倉位を維持確認することである可能性が高い。1つ以上の税務頭寸がこれらの税務頭寸の不確実性をもたらしたと考えられる場合、未確認税収頭寸は、すべての不確定推定税金負債をまとめた累積確率評価に基づいて推定される
税収頭寸。評価された利息及び罰金(ある場合)は、総合経営報告書の利息支出及びその他の支出に記入する。
株式ベースの報酬
株式オプションと業績株価オプション
当社は、付与日に従業員と非従業員の株式オプションを付与する推定公正価値に基づいて、従業員及び非従業員オプションの付与に関する補償コストを確認する。会社はブラック·スコアーズオプション定価モデルを使用して、付与日の公正価値とそれによって生じる株式ベースの補償費用を推定する。その会社は発生時に記録を没収した。日株式オプションを付与する公正価値は,直線原則に従って適用される帰属期間内に列支するのが一般的である4年. 業績株式オプションの公正価値確認は補償費用であり、業績条件が実現可能であると考えられたときから、残りの必要なサービス期間内であるブラック·スコアーズオプション定価モデルで使用される仮定は以下のとおりである
•行権価格それは.インセンティブ株式オプションを付与すれば10%の株主、執行権価格は下回ってはいけません110普通株は付与された日の公平時価の%である。
•普通株式の公正時価合併後、付与日の公平市場価値は会社普通株の見積市場価格である。
•所期期限それは.株式オプションの期待期限とは、株式オプションが未償還状態を維持すると予想される期間であり、オプションの帰属条項、行権期限、契約期間に基づく。期待期間は簡略化方法をもとに,加重平均帰属期限と付与日までの満了時間の平均値で推定する.
•予想変動率それは.同社の普通株の取引量が低いことから、同社は自分の歴史データと比較可能な上場生物製薬会社の普通株の取引履歴に基づいて、混合変動率を使用している。従業員の株購入計画(“ESPP”)株の変動率は会社の6か月入札期間。
•無リスク金利それは.無リスク金利は米国債収益率をベースとしており、その満期日は付与時に有効な株式オプションの期待期限に等しい。
•配当率それは.期待配当金はゼロ同社は配当金を支払ったことがないため、その普通株について何の配当も支払う計画もない。
制限株式単位
サービスに基づく制限株式単位は我々普通株の付与日の市場価格を用いて推定します。制限株式単位の付与日公正価値は、適用される帰属期間中に直線的に計算され、一般に4年.
従業員株購入計画
私たちのESPPは社員が15発行期間の初日または最終日には、普通株終値の割引率が低い。現在の発売期間は2023年1月1日から2024年12月31日まで。我々は,上記の仮定を含むブラック·スコイルモデルを用いて公正価値を決定した.ESPPの付与日公正価値は,適用される帰属期間内に直線的に計算され,一般に6か月.
1株当たり純損失
1株当たり基本純損失の計算方法は,普通株株主に適用した純損失を当期発行普通株の加重平均で割ったものであり,普通株等価物は考慮しない。1株当たりの純損失は在庫株方法で潜在的な希薄化を反映している。
収入確認
販売レベルに基づくマイルストーン支払いを含む販売ベースの特許使用料を含む可能性のある手配については、(I)関連販売が発生した場合、または(Ii)特許使用料の一部または全部が割り当てられた履行義務が履行されたときに収入を確認する。
研究開発コスト
研究·開発コストは発生時に費用を計上する。研究開発支出は、主に従業員に関連する支出を含み、その中には、賃金、ボーナス、福祉、株式ベースの給与、製造に関連するコスト、臨床試験を行う契約研究機関、研究場所およびコンサルタントとの合意の下で生じる支出、施設、固定資産およびその他の分配された支出、研究施設および設備の賃貸料およびメンテナンスの直接および分配支出、ライセンス製品および技術に関連する許可料およびマイルストーン支払い、および非臨床活動および規制承認に関連するコストが含まれる。将来の研究開発活動のための貨物やサービスの前払いが延期され,前払い資産として入金される。繰延金額は、関連貨物の引渡しまたはサービス提供時に料金を計上します。
特許
会社は一般的に工芸と製品に対して特許保護を申請している。特許出願費用は、このような費用の回収可能性が不確定であるため、一般および行政費用の一部として発生した費用に計上される。
3. ライセンス契約と資産購入プロトコル
ライセンスとLUM-201資産購入プロトコル
当社は2018年7月、Ammonettと資産購入プロトコル(“APA”)を締結し、2013年10月にMerckから許可を得たLum-201に関するほとんどの資産(“Lumos Merckプロトコル”)を買収した。
Lumos MerckプロトコルはLumos(Ammonettの権益相続人として)に世界的に独占的、再許可可能(メルク社のアメリカ、ヨーロッパの主要国と日本での同意を得なければならず、このような同意は無理に抑留されてはならない)権利を付与し、特定の特許とノウハウに基づいて任意とすべての適応のためにLum-201を開発、製造、商業化し、第5版“精神疾患診断と統計マニュアル”で定義された自閉症スペクトラム障害を含まない
2020年8月12日、メルク社とLumos Merck協定第1号修正案(“Lumos Merck合意修正案”)を締結した。 Lumos Merckプロトコル修正案によると,Merckから世界的に排他的で再許可可能な(Merckの米国,指定された主要ヨーロッパ諸国と日本の同意を得る必要があり,このような同意は無理に抑留されてはならない)許可を得ており,これらの許可は我々が診断目的のために開発,製造,商業化Lum-201の独占的に許可された標的であり,自閉症スペクトラム障害を含まない
行政手続法により,会社はAmmonettに#ドルの前払い費用を支払った3.52018年は研究開発費の100万ドルとして記録された。同社は合計$までの発展マイルストーンの支払いも発生する可能性がある17.0Lumosが追求した最初の指示によると、指定されたマイルストーンの100万ドルを実現し、最高で$に達する14.0Lumosが追求した第2の指示上の指定されたマイルストーンの達成について、販売マイルストーンの支払い総額は$に達しています55.0グローバル製品販売の百万ドルと、グローバル製品販売に基づく特許権使用料の支払いは、以下のとおりである。
Lumos Merckプロトコルによると、Lumosは、第1および第2の適応に関連する特定のマイルストーンを実現するために、メルク社に大量の開発マイルストーン支払いを要求される。潜在的な発展マイルストーンの支払い総額は$まで必要です14Lumosが追求した最初の指示に100万ドルを提供しました8.5100万ドル、これはルモスが追求している二番目の指示だ。段階的販売マイルストーン支払い総額は$に達する80.0世界の純製品の売上高は最高100万ドルに達します1.010億ドル、製品販売が実現すれば、製品販売に応じて大量の特許使用料を支払う必要がある
製品販売が実現すれば、LumosはAPAとLumos Merckプロトコルに基づいて印税を共同で支払わなければならない10%から12年間製品純売上高総額の%は,後発薬侵食の基準減少額を基準とした。Lumos Merckプロトコル下の特許使用料義務は、製品および国/地域に基づいており、その製品をカバーする最後のライセンス特許が、国/地域の排他的規制が満了したときに継続される。APAに規定される特許使用料義務は、メルク許可項の下での特許使用料義務期間であり、その後、“APA”によってLumosに割り当てられた当該製品が当該国の最後の特許が満了するまで、製品および国/地域に基づく。
Lumos Merckプロトコルは、個々の国および製品の印税義務が満了するまで、またはLumosが180日前にMerckに書面通知を提出するか、または他方が治癒されていない重大な違約または特定の破産事件によって任意に終了しない限り有効である。特許使用料義務が満了すると、Lumos Merckプロトコルは全額支払いの永久非独占許可に変換される。
もしLumos Merckプロトコルが終了し、Merckの書面要求に応じて、Lumosは合理的かつ勤勉な努力をして、Lumosが以前に与えた任意の再許可をMerckに譲渡する義務がある。
ライセンスとPRV資産購入プロトコル
2014年11月、NewLinkはメルク社と世界的な許可と協力協定(“NewLink Merckプロトコル”)を締結し、そのエボラワクチンrVSVG-ZEBOVを開発し、商業化する可能性があるPHAC“)”米国食品医薬品局の許可を得ていれば、RVSVG-ZEBOVもPRVを取得する資格があり、同社は権利がある60メルク社は残りを得る権利がある40PRVを売却、譲渡、または他の方法で処理することによって得られるPRV価値の%。2019年12月20日、メルク社は、米国食品医薬品局が、18歳以上の個人でザイールエボラウイルスによる疾患を予防するエボラワクチン(エボラザイル生ワクチン)の申請を承認し、PRVを付与したと発表した
2020年7月27日、LumosとMerckは資産購入協定(“PRV資産購入協定”)を締結し、LumosとMerckはそれぞれ同意したそれはメルクは同社からPRVを購入する。メルク社は同社に合計$を支払うことに同意した60百万インチ二つ分割払いです。これは1ドルです35.7百万ユーロの負債、すなわちMerckが獲得する権利のあるPRV価値部分も、PRV資産購入プロトコルによって解除される。初回分割払い$34.0二零年九月三十日までの三ヶ月間、当社は決済時に百万元を受け取り、第二期は$です26.02021年1月11日に100万部を受け取った
改訂されたNewLink Merck協定によると、同社はすでに、ある国でワクチンを販売する特許権使用料を稼ぎ続ける可能性がある。しかし,ワクチンの市場は主に特許権使用料の支払いから除外された発展途上国,あるいはワクチンが低利益率やゼロ利益率で寄付または販売されている地域に限られると考えられるため,将来的にはメルク社の巨額の特許権使用料支払いを受けることはないと予想される。同社が確認した収入は、2022年12月31日と2021年12月31日までの年度0そして$10,000企業がメルク下請けとして行っているERVEBOに関する仕事と$にそれぞれ用いられている1.5百万ドルとドル220,000ワクチンの商業販売に関する特許権使用料にそれぞれ使用される
また、PHACとのライセンス契約条項によると、当社はPHACに稼いだ特許権使用料を支払う義務があります。当社は2022年12月31日及び2021年12月31日までにPHACに支払います1.0百万ドルとドル146,000ワクチンの商業販売に関する特許権使用料にそれぞれ使用される。特許使用料費用は、総合業務報告書の一般費用及び行政費用に含まれる。
4. 金融商品
公正価値
以下に同社の金融商品の評価概要(単位:千)を示す.これらの表は、手元の現金も、歴史的コストまたは公正な価値以外の任意の基準で計量された資産および負債も含まない。2021年12月31日現在、同社の現金等価物は完全に通貨市場基金(1級)で構成され、公正な価値に近いコストで入金されている。その会社は所有している違います。2021年12月31日までの短期投資。
| | | | | | | | | | | | | | | | | |
| 2022年12月31日まで |
| レベル1 | | レベル2 | | 合計する |
| 現金等価物: | | | | | |
| 貨幣市場基金 | $ | 52,045 | | | $ | — | | | $ | 52,045 | |
| 会社債務証券 | — | | | 2,497 | | | 2,497 | |
| 非アメリカ債務証券 | — | | | 800 | | | 800 | |
| 現金等価物合計 | $ | 52,045 | | | $ | 3,297 | | | $ | 55,342 | |
| | | | | |
| 短期投資: | | | | | |
| 商業手形 | $ | — | | | $ | 2,909 | | | $ | 2,909 | |
| アメリカ政府と機関証券は | 2,451 | | | 5,992 | | | 8,443 | |
| 短期投資総額 | $ | 2,451 | | | $ | 8,901 | | | $ | 11,352 | |
| | | | | |
| 合計する | $ | 54,496 | | | $ | 12,198 | | | $ | 66,694 | |
2022年12月31日と2021年12月31日現在、会社には3級資産や負債がない。2022年12月31日と2021年12月31日までの年度では,1級,2級または3級測定の間に移行はなかった
当社の他の金融商品には、現金、売掛金、売掛金が含まれており、納期が短いため、その公正価値に近い金額で入金されています。同社は将来の製品販売状況に基づいてアイオワ州経済発展局の特許権使用料義務の公正価値を推定することができず、支払いの時間(あれば)が不確定であるためである
信用リスクが集中する
会社を集中的な信用リスクに直面させる可能性のある金融商品には、主に現金と現金等価物、短期投資が含まれる。現金及び現金等価物及び有価証券投資は、当社の現金投資政策に基づいて投資を行い、主な目標は保本及び流動資金の維持である。現金投資政策には、金融商品の品質に関するガイドラインが含まれており、会社が信用リスク集中リスクを最小限に抑えることができると考えられる許容投資を定義している。当社はその現金と現金等価物および短期投資を高信用品質の金融機関に置くことで、直面している信用損失を制限している。
投資契約満期日
2022年12月31日現在、同社のすべての販売可能な投資は1年以下で満了しなければならない
売却可能な投資
以下の表は、会社が販売可能な証券を証券タイプ別にまとめた
| | | | | | | | | | | | | | | | | | | | |
| 2022年12月31日まで |
| 原価を償却する | | | 未実現損失 | | 公正価値 |
| 現金等価物: | | | | | | |
| 貨幣市場基金 | $ | 52,045 | | | | $ | — | | | $ | 52,045 | |
| 会社債務証券 | 2,498 | | | | (1) | | | 2,497 | |
| 非アメリカ債務証券 | 800 | | | | — | | | 800 | |
| 現金等価物合計 | $ | 55,343 | | | | $ | (1) | | | $ | 55,342 | |
| | | | | | |
| 短期投資: | | | | | | |
| 商業手形 | $ | 2,909 | | | | $ | — | | | $ | 2,909 | |
| アメリカ政府と機関証券は | 8,451 | | | | (8) | | | 8,443 | |
| 短期投資総額 | $ | 11,360 | | | | $ | (8) | | | $ | 11,352 | |
| | | | | | |
| 合計する | $ | 66,703 | | | | $ | (9) | | | $ | 66,694 | |
2022年12月31日現在の未実現損失総額は主に市場金利の変化によるものである。投資市場価値の低下が信用に関連する要因である場合、当社は信用損失準備金に計上する。1つの投資の減値を評価する際に、当社は、減値の深刻さ、関連信用格付けの変化、予想の回復、当社の売却の意向、あるいは予想時価が回復する前にその投資を売却する可能性、および所定の現金支払いを継続する可能性など、様々な要因を検討する。2022年12月31日現在、売却可能な投資の時価は信用関連要因によって実質的に低下していない。
2022年12月31日現在、会社は売却可能な投資に関する重大な未実現収益を達成していない。
5. 費用を計算する
計算すべき費用は以下の部分からなる(千計)
| | | | | | | | | | | |
| 2022年12月31日 | | 2021年12月31日 |
| 補償と関連福祉 | $ | 3,729 | | | $ | 2,812 | |
| 臨床·契約製造費 | 1,800 | | | 508 | |
| 他にも | 671 | | | 846 | |
| 費用総額を計算する | $ | 6,200 | | | $ | 4,166 | |
| | | |
6. 賃貸借証書
その会社はいくつかのキャンセルできない条項の施設賃貸契約を持っていて、条項の範囲は1つはそして2年.いくつかの更新オプションがあります
当社は賃貸期間内の賃貸支払いの現在価値に基づいて賃貸負債を記録し、逓増借入金金利を用いてそのレンタル負債を割引しているが、レンタルに隠れている金利は通常確定しにくいためである。賃貸負債の現在値を計算するために,同社は加重平均割引率を用いている3%です。いくつかのレンタル協定には、会社の統制下の更新選択権が含まれている。当社は、当社がその選択権を行使することを合理的に決定した場合にのみ、レンタル期間内に選択可能な継続期間を含む。2022年12月31日までの加重平均残存レンタル期間は0.7何年もです
当社はレンタル構成要素を非レンタル構成要素と分離していません。可変レンタル支払いには、レンタル者に支払われる税金、メンテナンス、保険、および他の運営コスト、および指数またはレートに応じて調整された支払いが含まれる。その会社の賃貸契約にはどんな残存価値保証や制限的な契約も含まれていない。
2022年12月31日までの経営リース(初期または残りのレンタル期間が1年を超える)の将来の満期日は以下の通り(千で計算)、オプションの更新は含まれていません
| | | | | |
| 2022年12月31日まで: | |
| |
| 2023 | $ | 236 | |
| その後… | — | |
| 賃貸支払総額 | 236 | |
| 差し引く:推定利息 | (3) | |
| 合計する | $ | 233 | |
レンタルを運営するレンタルコストは#ドルです0.32022年と2021年12月31日までの毎年。
7. 株式に基づく報酬と従業員福祉計画
株式オプションと業績ストックオプション
2012年に二等兵ルーモスは通過しました2012株式インセンティブ計画(“2012計画”)は、2016年に2016年株式計画(“2016計画”)を採択し、♪the the the2012年計画、“計画”)。合併については,計画下のすべての未償還オプションが仮定されており,この仮定の選択権は行使されることができる会社合併後の普通株を購入する。統合された後、未来の報酬に関する計画は終了された.
合併に関連して、会社はNewLinkの2009年の株式インセンティブ計画を担当した2009年7月から施行され、その後5月9日に改訂された, 2019 (the “2019 Plan”)それは.2019年の計画は1つ10取締役会成立日2019年3月22日からの年次任期及び毎年1月1日から2029年1月1日まで、“常青樹条項”によると、3前年12月31日発行された普通株式総数のパーセントまたは取締役会が承認した当該より小さい額の株式(または株式なし)は、2019年に保留予定の株式に追加される。2019年には、会社の上級管理者、従業員、取締役会メンバー、コンサルタント、コンサルタントに奨励的株式オプション、非法定株式オプション、制限株式奨励および株式付加価値権を付与することを規定する予定ですそれは.2022年12月31日までに426,4372019年計画に基づいて付与可能な株式
次の表に在庫状況をまとめた選択権活動して市場と業績条件を持つオプションも含めて2022年12月31日までの年度:
| | | | | | | | | | | | | | | | | | | | |
| | 番号をつける 選択肢の数 | | 加重平均 トレーニングをする 値段 | | 加重平均 余剰契約 期限(年) |
期初未返済債務 | | 1,201,209 | | | $ | 10.77 | | | 6.1 |
付与したオプション | | 306,986 | | | 9.41 | | | |
行使のオプション | | (17,288) | | | 2.31 | | | |
没収されたオプション | | (25,960) | | | 13.36 | | | |
オプションは期限が切れた | | (15,760) | | | 20.09 | | | |
期末未済債務 | | 1,449,187 | | | $ | 10.43 | | | 6.9 |
満期に行使可能なオプション | | 842,557 | | | $ | 10.19 | | | 5.9 |
ブラック·スコアーズオプション定価を用いた株式オプションの推定値の加重平均は以下のように仮定される:
| | | | | | | | | | | | | | |
| | 十二月三十一日までの年度 |
| | 2022 | | 2021 |
無リスク金利 | | 1.32%から3.01% | | 0.56%から1.05% |
| 期待配当収益率 | | —% | | —% |
| 予想変動率 | | 83.7%から96.1% | | 77.2%から90.3% |
| 予想期限(年単位) | | 5.5至れり尽くせり6.1 | | 5.4至れり尽くせり7.7 |
加重平均付与日1株当たり公正価値 | | $6.75 | | $10.65 |
限定株単位
下表は2022年12月31日までの年度の限定株式単位活動をまとめたものである:
| | | | | | | | | | | | | | |
| | 番号をつける 販売制限株 | | 加重平均 付与日公正価値 |
| 期初は帰属しなかった | | 81,015 | | | $ | 8.91 | |
| 授与する | | 14,550 | | | 9.18 | |
| 既得 | | (25,771) | | | 8.87 | |
| 期末未帰属 | | 69,794 | | | $ | 8.98 | |
以下に当日に付与された加重平均公正価値の要約を示す:
| | | | | | | | | | | | | | |
| | 十二月三十一日までの年度 |
| | 2022 | | 2021 |
| 制限株式単位を付与するたびに | | $ | 9.18 | | | $ | 10.32 | |
2010年度非従業員取締役株式奨励計画
合併に関する問題また、当社は二零一年十一月十日からNewLink 2010年度非従業員取締役株式奨励計画(“取締役計画”)を実施しています。2022年12月31日までに5,624役員の規定により,依然として株式を授与することができる’計画してみます。
2010年従業員株購入計画
合併に関連して、当社はNewLinkが改訂した二零一零年従業員株購入計画(“二零一零年株購入計画”)が二零一一年十一月十日に発効したと仮定している。2022年12月31日までに48,6442010年の購入計画によると、株はまだ発行されることができる。2021年7月22日、取締役会は二零一零年購入計画(“A&R ESPP”)の改訂と再記述を許可し、A&R ESPPに基づいて特別発売期間を設立し、2021年9月1日から2023年6月30日までであるが、A&R ESPPに記載されている再開条項の制限を受けなければならない。A&R ESPP項の特別発売期限は、株主が2022年株主総会でA&R ESPPを承認することに完全に依存する。A&R ESPPは,A&R ESPPにより発行予約のための株式数が増加することを規定している60,000株式です。2022年5月4日、2022年株主総会でA&R ESPPが承認された。2022年6月30日、再開条項が触発され、新たな発売期限が2022年7月1日から2024年6月30日までとなった。2022年12月30日、再起動条項がトリガされ、新たな提供期限が2023年1月1日から2024年12月31日までとなった。
株式ベースの給与費用
当社の今年度までの総合経営報告書に記載されている株式報酬支出2022年12月31日2021年とは(千計):
| | | | | | | | | | | | | | |
| | 十二月三十一日までの年度 |
| | 2022 | | 2021 |
| 研究開発 | | $ | 661 | | | $ | 809 | |
| 一般と行政 | | 1,659 | | | 2,070 | |
| 合計する | | $ | 2,320 | | | $ | 2,879 | |
2022年12月31日現在、未確認の補償コストがあります$4.3100万ドルで確認された加重平均期間は2.4何年もです。
従業員福祉計画
同社は、固定された年間雇用主納付を規定する401(K)計画を開始した。同社の固定資金は#ドルです0.32022年12月31日と2021年12月31日までの毎年同社はまた、この計画に#ドルの適宜寄付を提供した0.12021年12月31日までの年度.
8. 長期債務と特許権使用料義務への転換
2005年3月NewLinkは$を締結しました6.0アイオワ州経済発展部(“IDED”)と締結した百万円ローン免除協定。協議により、違約がない場合には、プロジェクト完了日までに元金又は利息を支払う必要がない。2012年3月26日にアイオワ州経済発展局と締結された和解協定条項によると、この融資は特許権使用料義務に転換され、IDEDの利息継承者となる。今後12ヶ月以内には何の支払いもないと予想されるため、特許権使用料全体の義務は#ドルとなる6.02022年12月31日現在、会社が合併により負担する100万ドルは長期負債に分類されている。
9. 所得税
当社は2022年12月31日および2021年12月31日までに所得税割引$を記録しました13,000そして$0.6それぞれ100万ドルです所得税の割引は以下の通り(千単位): | | | | | | | | | | | | | | |
| | 十二月三十一日までの年度 |
| | 2022 | | 2021 |
| | | | |
| 現在の税金優遇--州と地方 | | $ | 13 | | | $ | 636 | |
| | | | |
| | | | |
| 所得税優遇総額 | | $ | 13 | | | $ | 636 | |
その会社は所有している違います。2022年と2021年12月31日までの各年度の繰延税金負債2022年12月31日、2022年12月31日、2021年12月31日に大量の繰延税金資産の一時的な差異が生じる税収影響は以下の通り(千計) | | | | | | | | | | | | | | |
| | 12月31日まで |
| | 2022 | | 2021 |
| 繰延税金資産: | | | | |
| 純営業損失が繰り越す | | $ | 35,236 | | | $ | 32,281 | |
| 連邦研究開発税控除 | | 39,190 | | | 37,953 | |
| 資本化研究と開発 | | 4,018 | | | — | |
| 株式ベースの報酬 | | 678 | | | 627 | |
| 資本損失繰越 | | 41,144 | | | 41,144 | |
| 補償すべきである | | 221 | | | 192 | |
| レンタル改善と設備 | | 1,205 | | | 1,292 | |
| 他にも | | 2 | | | 26 | |
| 繰延税項目総資産 | | 121,694 | | | 113,515 | |
| 減算:推定免税額 | | (121,694) | | | (113,515) | |
| 繰延税金資産総額 | | $ | — | | | $ | — | |
繰延税金資産の現金化能力を評価する際には、管理層は、繰延税金資産の一部または全部が現金化できない可能性が高いかどうかを考慮する。繰延税金資産の最終的な現金化は、これらの一時的差額控除可能期間中に生成された将来の課税所得額に依存する。繰延税金資産収益を実現する会社の能力には不確定性があるため、繰延税項目純資産は2022年12月31日と2021年12月31日に推定支出によって完全に相殺される。推定免税額は#ドル増加した8.2百万ドルとドル8.22022年12月31日と2021年12月31日までの年間で、それぞれ100万ドル。
2020年3月18日までの第382条所有権変更分析によると,合併の結果,歴史上のNewLinkとPrivate Lumosはいずれも2020年3月18日に第382条所有権変更を経験した。これらの所有権変更は、連邦純営業損失の繰越能力と、私たちと私たちの子会社のそれぞれの所有権が変更される前に蓄積された他の税務属性を利用して、将来このような属性を利用する能力を制限し続ける可能性があります。その後の分析によると、2020年3月19日から2021年12月31日まで、第382条の所有権変更を経験していない。2022年12月31日現在,同社の連邦運営損失は約ドルに繰り越ししている141.5百万ドル連邦資本損失は約$に転換しました164.6百万ドル連邦研究信用繰越$39.2百万ドルです。一部の繰り越しは2023年から2042年まで満期であり、いくつかの繰越には無限の生命がある
法定連邦所得税率で計算される所得税と付随する合併経営報告書に含まれる所得税純額との台帳を以下の表に示す | | | | | | | | | | | | | | | | | | | | |
| | 十二月三十一日までの年度 |
| | 2022 | | | 2021 | |
| 法定税率でアメリカ連邦所得税の割引を受ける | | (21.0) | | % | | (21.0) | | % |
| 州所得税、連邦税引き後の純額 | | (2.1) | | | | (1.4) | | |
| 連邦税収控除 | | (6.3) | | | | (8.9) | | |
| 評価免除額を変更する | | 26.3 | | | | 26.5 | | |
| 他にも | | 3.1 | | | | 2.7 | | |
| 有効所得税率 | | 0.0 | | % | | (2.1) | | % |
当社はいかなる不確定な税務倉位の影響についても会計処理を行い、根拠となる敷居は、税務機関審査を適用する税務倉位の技術的利点に基づいて当該等の税務倉位を維持確認することである可能性が高い。1つ以上の税務頭寸が等頭寸の不確実性をもたらすとみなされる場合、確認されていない税務頭寸は、すべての不確定税収頭寸の推定税務負債をまとめた累積確率評価に基づいて推定される。評価された利息と罰金(ある場合)は、それぞれ利息支出または雑支出を計上し、総合経営報告書に記入しなければならない
その会社は所有している違います。2022年12月31日までの不確定税収状況準備金は、2022年12月31日までの年度に利息や罰金が確認されていない。2021年10月、州不確定税収状況に関する訴訟時効が満了し、全額準備金は#ドルとなる1.2計算された費用から100万ドルが差し引かれ、課税利息と罰金を含めて#ドルが発生しました0.6百万の所得税の割引、一ドル0.3他の収入は100万ドル増えました0.3一般的と行政費用の中の雑役費用は100万ドル減少する。2018年から2021年までの納税年度は、当社が経営する主要税務管区審査に供することができます。
10. 株主権益
普通株
同社の普通株はナスダックで取引され、コードは“LUMO”。私たちの株主は権利がある1つはすべての事項について保有する普通株ごとに投票し、株主投票で投票する。私たちは75,000,000普通株式、額面$を許可する0.01一株ずつです。普通株保有者には権利がある1つは会社の株主によって投票されたすべての事項に対して1株当たり投票する
2022年8月16日、会社は取締役会が株式買い戻し計画を承認し、この計画によると、会社は最高$を購入できると発表した3.0百万株はすでに普通株を発行している。会社の買い戻し137,526株価は約$675,0002022年12月31日までの年間で。これらすべての購入は公開市場取引によって行われ、株は買い戻し時に実際にログアウトした。
当社は2020年12月30日に制御持分発行協定を締結したSMエージェント(“エージェント”)であるCantor Fitzgerald&Co.と締結された販売プロトコル(“販売プロトコル”)により,当社はつねにエージェントを通して最高$まで提供および販売することができる50.0百万株の会社普通株、$0.01額面(“株”)。株式の発行·売却は、改正された1933年の証券法(“証券法”)に基づいて登録されている。販売協定によれば、代理人は、証券法第415条(A)(4)条に定義された“市場で”発売された任意の方法で株式を販売することを法律で許可され、またはナスダックによって、株式の任意の他の既存の取引市場で、販売時の市価またはそのような現行の市価に関連する価格および/または法律によって許可されている任意の他の方法で行われる取引取引を含むことができる。会社は、発行された株式数、販売を要求する時間帯、どの日に販売可能な株式数の制限、および下回ってはならない任意の最低価格を代理人に通知します。会社は代理店に最高可達を支払います3.0販売契約に基づいて、その売却された株式の販売総価格の%を取得する。また、会社は代理店が今回の発売による何らかの費用を精算することに同意した。販売契約に記載されているように、代理店または当社は、他方に通知した後、いつでも販売プロトコルを終了することができ、または場合によっては、当社の業務または財務状況が重大かつ不利に変化し、非現実的または株式を売却する契約を実行することができない場合には、代理店はいつでも販売プロトコルを終了することができる。2022年12月31日までに違います。株式は販売契約に基づいて発行された。
優先株
当社の改訂および再記載された会社の登録証明書の発行5,000,000優先株、額面$0.01一株ずつです。当社取締役会は、株主の承認なしに配当金、清算、転換、投票権または他の権利を有する優先株を発行する権利を有しており、これらの権利は、普通株式保有者の投票権または他の権利に悪影響を及ぼす可能性がある。2022年12月31日までに会社は違います。優先株を発行した。
11. 普通株1株当たり純損失
1株あたりの基本損失は,期内に発行された普通株の加重平均をもとに,普通株等価物は考慮しない.1株当たり償却損失は、当期に発行された普通株の加重平均に、希薄化期間に影響を与える追加加重平均潜在薄普通株等価物を加えて算出した。
以下の表に、普通株1株当たりの基本損失および希釈損失の計算(株および1株当たりのデータを含まない)と、通常株等価物として未行使株式オプションおよび制限株式単位の数を示す。これらの株式オプションおよび制限株式単位は、これらの影響が全ての期間にわたって逆希釈されているので、希釈純損失計算から除外されている | | | | | | | | | | | | | | | | | | |
| | 十二月三十一日までの年度 | | |
| | 2022 | | 2021 | | | | |
| 純損失 | | $ | (31,062) | | | (30,430) | | | | | |
| | | | | | | | |
| 加重平均流通株−基本と希釈 | | 8,373,821 | | | 8,334,516 | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| 1株当たり純損失--基本損失と赤字 | | $ | (3.71) | | | $ | (3.65) | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| 逆希釈株オプション | | 1,449,187 | | 1,201,209 | | | | | |
| 逆希釈制限株式単位 | | 69,794 | | | 81,015 | | | | | |
総逆希釈普通株等価物は含まれていません | | 1,518,981 | | | 1,282,224 | | | | | |
12. 再編成と分割払い
2021年2月4日,医学博士ユージン·P·ケネディは,2021年3月6日から会社首席医療官を辞任することを会社に通知した。ケネディ博士の雇用協定における支配権福祉の変化に関する条項によると、会社は約#ドルを支払った0.73年間で確認された解散費は百万ドルです
2021年3月31日までの数ヶ月間、非既得持分奨励金の帰属を加速した。2021年3月31日までの3ヶ月間、会社は追加株式報酬支出が約$であることを確認した0.7百万ドル、理由はケネディ博士が持っているすべての非既得持分奨励が帰属を加速させるからです。
カール·W·ラングレンは2021年4月16日、2021年6月30日から会社首席財務官を辞任することを会社に通知した。ラングレンは退職のため退職し、発効日は2021年6月30日。ラングレンさんの雇用契約における支配権の福祉変更に関する条項に基づき,会社は約#ドルを支払った0.9解散費として、すべての非既存持分奨励の帰属を加速し、任意の既得持分奨励の行使期間を延長した24か月別居の日から計算します。2021年6月30日までの3ヶ月間、追加株式報酬支出が約$であることを確認しました0.4ラングレンさん所有のすべての非既得持分への報酬の帰属が加速されているので。最後に累算解散費#ドルを支払います0.92021年第4四半期に100万ドルを支払いました違います。解散費に関する費用は2022年12月31日までの年度内に確認された。
項目9.会計·財務開示面の変化と会計士との相違
ない。
第9条。制御とプログラム
情報開示制御とプログラムの評価
我々は、最高経営責任者及び最高財務官の監督の下、取引所法案の要求の下で、2022年12月31日までに取引所法案第13 a-15(E)条に規定する我々の開示制御及びプログラムの設計及び操作の有効性を評価した。この評価に基づき、我々の最高経営責任者およびCEOは、2022年12月31日まで、取引所法案に基づいて提出または提出された報告書で開示を要求する情報が米国証券取引委員会規則および表で指定された期間内に記録、処理、まとめ、報告されることを確実にし、必要に応じて合理的な保証を提供し、必要な開示について決定するために、これらの情報が蓄積されて私たちの管理層に伝達されることを保証するために、2022年12月31日までに有効であると結論した。
財務報告の内部統制に関する経営陣の報告
経営陣は、取引法規則13 a-15(F)によって定義されたように、財務報告の十分な内部統制の確立と維持を担当する。経営陣は、テレデビル委員会後援組織委員会(COSOフレームワーク)が発表した“内部統制-総合枠組み(2013)”で確立された基準に基づき、2022年12月31日現在の財務報告書の内部統制に対する有効性を評価した。この評価の結果、経営陣は、2022年12月31日現在、財務報告の内部統制が財務報告の信頼性を効果的に保証し、公認された会計原則に基づいて外部目的の財務諸表を作成すると結論した。
我々の財務報告に対する内部統制には、(1)一般的に受け入れられた会計原則に基づいて総合財務諸表を作成するために、一般的に受け入れられた会計原則に基づいて取引を記録するための合理的な保証を提供し、一般的に受け入れられた会計原則に基づいて総合財務諸表を作成し、私たちの収入および支出が我々の管理層および取締役の許可のみに基づいて行われる政策および手順、および(3)総合財務諸表に重大な影響を与える可能性のある不正な買収、使用、または処分について合理的な保証を提供するための政策および手順が含まれる。
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来的にどのような有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムの遵守度が悪化する可能性がある.
財務報告の内部統制の変化
私たちの財務報告の内部統制は2022年第4四半期に大きな影響を与えなかったか、または合理的に私たちの財務報告の内部統制に大きな影響を与える可能性のある変化が発生しなかった。
コントロールに対する制限
経営陣は私たちの開示統制と手続き、または財務報告に対する私たちの内部統制がすべてのミスや詐欺を防止または発見することを期待していない。いかなる規制制度も,その設計や運営がどのように整備されても,何らかの仮定に基づいて,絶対的な保証ではなく,その目標が達成できることを確保するために合理的な保証しか提供できない。また,制御の評価は,誤りや不正による誤り陳述が発生しないことや,すべての内部制御問題や不正イベント(あれば)が発見されていることを絶対に保証することはできない.
プロジェクト9 B。その他の情報
ない。
プロジェクト9 Cです。検査を妨害する外国司法管轄区域を開示する。
ない。
第三部
プロジェクト10.役員、役員、および企業管理
本プロジェクトに必要な情報は2023年の依頼書を参考に本明細書に組み込まれ、依頼書は2022年12月31日以降120日に米国証券取引委員会に提出される。
プロジェクト11.役員報酬
本プロジェクトに必要な情報は2023年の依頼書を参考に本明細書に組み込まれ、依頼書は2022年12月31日以降120日に米国証券取引委員会に提出される。
プロジェクト12.特定の実益所有者の保証所有権及び管理職及び株主に関する事項
本プロジェクトに必要な情報は2023年の依頼書を参考に本明細書に組み込まれ、依頼書は2022年12月31日以降120日に米国証券取引委員会に提出される。
項目13.特定の関係および関連取引、および取締役の独立性
本プロジェクトに必要な情報は2023年の依頼書を参考に本明細書に組み込まれ、依頼書は2022年12月31日以降120日に米国証券取引委員会に提出される。
プロジェクト14.主な課金とサービス
本プロジェクトに必要な情報は2023年の依頼書を参考に本明細書に組み込まれ、依頼書は2022年12月31日以降120日に米国証券取引委員会に提出される。
第4部
プロジェクト15.証拠品、財務諸表の添付表
(A)本報告の一部として、以下の書類を提出した
(1)財務諸表:総合財務諸表及び関連付記は、独立公認会計士事務所ピマウェイ会計士事務所の報告とともに、本年報第II部第8項財務諸表及び補足データに掲載される。
(2)財務諸表明細書:米国証券取引委員会関連会計条例が規定している全ての明細書は、関連指示に要求または適用されていないため省略される。
(3)展示品:以下展示品インデックスに登録されている展示品は,本年度報告の一部としてアーカイブまたは格納を参考にする.
| | | | | | | | | | | | | | | | | | | | |
| 展示品索引 |
| | | | | | |
| | | 引用で編入する | |
展示品番号 | | 説明する | 表 | 提出日 | 番号をつける | 同封アーカイブ |
| | | | | | |
| 2.1 | † | 合併·再編協定と計画は、2019年9月30日、NewLink Genetics Corporation、サイクロン連結子会社、Lumos Pharma,Inc. | 8-K | 9/20/2019 | 2.1 | |
| 2.2 | | 2019年11月19日NewLink Genetics Corporation,Cyone Merge Sub,Inc.とLumos Pharma,Inc.の間の統合と再構成プロトコルと計画の修正案第1号。 | 8-K | 11/20/2019 | 2.1 | |
| 3.1 | | 2011年11月16日に提出された改正会社登録証明書 | 10-K | 3/9/2021 | 3.1 | |
| 3.2 | | 付例を改訂および再制定する | 8-K | 9/30/2019 | 3.2 | |
| 4.1 | | 登録者普通株式証明書フォーマット | 8-K | 3/18/2020 | 4.1 | |
| 4.2 | | 証券説明書 | 10-K | 3/9/2021 | 4.2 | |
| 10.1 | † | 被支配持分発行SM登録者とCantor Fitzgerald&Co.との販売合意は,2020年12月30日である。 | 8-K | 12/30/2020 | 10.1 | |
| 10.2 | † | 登録者とメルク、シャープ、ドルム社との間で署名された、期日は2020年7月27日のPRV譲渡協定。 | 10-Q | 8/14/2020 | 10.1 | |
| 10.3 | † | メルク·シャープ·ドム社とAmmonett Pharma LLC間のライセンス契約は、2013年10月22日から発効した | 8-K/A | 5/29/2020 | 10.1 | |
| 10.4 | † | 2020年8月12日からLumos MerckとMerckが合意した改正案第1号 | 10-Q | 8/14/2020 | 10.2 | |
| 10.5 | † | Lumos Pharma,Inc.,Ammonett Pharma LLCと各上場個人間の資産購入協定は,2018年7月26日に発効した | 8-K/A | 5/29/2020 | 10.2 | |
| 10.6 | * | 2009年の株式インセンティブ計画の改訂と再策定 | S-1 | 12/21/2010 | 10.6 | |
| 10.7 | * | 2009年株式インセンティブ計画下の株式オプションプロトコルフォーマット | S-1 | 12/21/2010 | 10.7 | |
| 10.8 | * | 2009年株式インセンティブ計画の下で株式オプション通知書を付与するフォーマット | S-1 | 12/21/2010 | 10.8 | |
| 10.9 | * | 修正された2009年株式インセンティブ計画下の制限株式単位奨励協定フォーマット | 10-Q | 8/5/2014 | 10.6 | |
| 10.10 | * | 制限株式単位授権書のフォーマット[4年に1回の年次帰属]改正された2009年の株式インセンティブ計画によると | 10-Q | 8/5/2014 | 10.7 | |
| 10.11 | * | 制限株式単位授権書のフォーマット[直ちに帰属する]改正された2009年の株式インセンティブ計画によると | 10-Q | 8/5/2014 | 10.8 | |
| 10.12 | * | 2010年従業員株購入計画 | 8-K | 5/14/2013 | 10.2 | |
| 10.13 | * | 2010年に改訂された非従業員取締役株奨励計画 | 10-Q | 11/8/2016 | 10.1 | |
| 10.14 | * | 2010年に改訂された非従業員取締役株奨励計画下の制限株式単位奨励協定のフォーマット | 10-Q | 8/5/2014 | 10.4 | |
| 10.15 | * | 改訂された2010年非従業員取締役株式奨励計画下の制限株式単位付与通知のフォーマット | 10-Q | 8/5/2014 | 10.5 | |
| 10.16 | * | Lumos Pharma,Inc.2012持分インセンティブ計画 | 8-K | 3/18/2020 | 10.1 | |
| | | | | | | | | | | | | | | | | | | | |
| 10.17 | * | “2012年株式インセンティブ計画インセンティブ株式オプション協議表” | 8-K | 3/18/2020 | 10.2 | |
| 10.18 | * | Lumos Pharma,Inc.2016持分インセンティブ計画 | 8-K | 3/18/2020 | 10.3 | |
| 10.19 | * | 2016年度株式オプションプロトコルフォーマット | 8-K | 3/18/2020 | 10.4 | |
| 10.20 | * | 登録者とその役員及び上級者との間の合意のフォーマット | 10-K | 3/9/2021 | 10.20 | |
| 10.21 | * | 登録者とリチャード·ホーキンス間の雇用協定、期日は2020年3月27日 | 8-K | 4/2/2020 | 10.1 | |
| 10.22 | * | 登録者とJohn McKewの間の雇用協定は2020年3月27日です | 8-K | 4/2/2020 | 10.2 | |
| 10.23 | * | 登録者とカール·ラングレンとの雇用契約は,2019年9月30日となっている | 8-K | 9/30/2019 | 10.3 | |
| 10.24 | * | 登録者とユージーン·ケネディ間の雇用契約は,2019年9月30日となっている | 8-K | 9/30/2019 | 10.4 | |
| 10.25 | * | 登録者とローリー·ローリー間の雇用契約は、2019年9月30日となっている | 8-K | 9/30/2019 | 10.5 | |
| 10.26 | * | 登録者とブラッド·ボルスとの間で2019年9月30日に締結された雇用契約 | 8-K | 9/30/2019 | 10.6 | |
| 10.27 | * | 登録者とDavid·カルプフの間の雇用協定は2021年8月3日です | 10-Q | 11/5/2021 | 10.1 | |
| 10.28 | * | 登録者とローリー·ローリーとの間で2021年6月30日に署名された雇用協定改正案第1号 | 10-Q | 8/6/2021 | 10.1 | |
| 10.29 | * | 登録者とJohn McKewの間で2021年8月1日に署名された雇用協定改正案第1号 | 10-Q | 8/6/2021 | 10.2 | |
| 21.1 | | 子会社情報 | | | | X |
| 23.1 | | 独立公認会計士ピマウェイ会計士事務所の同意 | | | | X |
| 24.1 | | 授権書(本文書署名ページに含まれる) | | | | X |
| 31.1 | | ルール13 a-14(A)/15 d-14(A)によって要求される主実行幹事の証明 | | | | X |
| 31.2 | | 第13 a-14(A)/15 d-14(A)条に規定する主要財務幹事の証明 | | | | X |
| 32.1 | # | 第1350節認証 | | | | X |
101.INS | ‡ | XBRLインスタンス文書−インスタンス文書は、そのXBRLタグがイントラネットXBRL文書に埋め込まれているので、対話データファイルには表示されない。 | | | | X |
101.書院 | ‡ | XBRL分類拡張アーキテクチャドキュメント | | | | X |
101.カール | ‡ | XBRL分類拡張計算リンクライブラリ文書 | | | | X |
101.def | ‡ | XBRL分類拡張Linkbase文書を定義する | | | | X |
101.介護会 | ‡ | XBRL分類拡張タグLinkbaseドキュメント | | | | X |
101.Pre | ‡ | XBRL分類拡張プレゼンテーションLinkbaseドキュメント | | | | X |
| 104 | | 表紙相互データファイル(添付ファイル101に含まれるイントラネットXBRLのフォーマット) | | | | X |
____________________ | | | | | |
| |
| |
# | 本10-K表年次報告書に添付された添付ファイル32.1に添付された証明は、米国証券取引委員会に提出されたものとはみなされず、参照によってLumos Pharma,Inc.が1933年の“証券法”(改正)または1934年の“証券取引法”(改正)に従って提出された任意の文書とみなされてはならない。この文書は、この書類に含まれる任意の一般的な登録言語にかかわらず、本表の10−K日付の前または後に提出されてはならない。 |
‡ | 同封は電子的に提出します。 |
| * | 契約または補償計画を管理すること。 |
| † | S-K規則601(A)(5)項によれば、いくつかの付表および証拠物は省略されている。任意の漏れたスケジュールおよび/または証拠品のコピーは、米国証券取引委員会に提供することを要求しなければならない。 |
| |
項目16.表格10-Kの概要
ない。
サイン
1934年の証券取引法の要求に基づいて、登録者は正式に正式に許可された署名者がそれを代表して本報告に署名することを手配した。
| | | | | |
| ルモス製薬会社 |
|
差出人: | リチャード·J·ホーキンス |
| リチャード·J·ホーキンス |
| 最高経営責任者 |
| (首席行政主任) |
| 日付:2023年3月7日 |
|
差出人: | /s/Lori D.Lawley |
| ローリー·D·ローリー |
| 首席財務官兼秘書 |
| (首席財務会計官) |
| 日付:2023年3月7日 |
授権依頼書
このような陳述を通じて、以下の署名のすべての人が、リチャード·J·ホギンズとローリー·D·ローリーをその真の合法的な事実代理人と代理人として構成し、任命し、その任意およびすべての身分で、その名前、場所、代理人として、本表の10-K年度報告の任意およびすべての修正案に署名し、この表をそのすべての証拠品および他のすべての関連文書と共に証券取引委員会に提出し、上記の事実代理人と代理人を付与することを知っている。すべての必要およびしなければならないことおよびしなければならないことを行う権利が完全にあり、その可能性または自ら行うことができるすべての意図および目的を尽くし、ここで、上述したすべての代理弁護士および代理人、ならびに彼らのいずれかまたはその代替者を承認および確認することは、本条例によって行われたことを合法的に行うことができるか、または手配することができる。
本報告書は、1934年に改正された証券取引法の要求に基づき、登録者として指定された日に署名された
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| 名前.名前 | | | | タイトル | | 日取り |
| | | | |
| リチャード·J·ホーキンス | | 最高経営責任者 | | March 7, 2023 |
| リチャード·J·ホーキンス | | (首席行政主任) | | |
| /s/Lori D.Lawley | | 首席財務官兼秘書 | | March 7, 2023 |
| ローリー·D·ローリー | | (首席財務会計官) | | |
| トーマス·A·ラフィン | | 役員.取締役 | | March 7, 2023 |
| トーマス·A·ラフィン | | | | |
| ジョー·マクラケンDVm MS | | 役員.取締役 | | March 7, 2023 |
| ジョー·マクラケンDVM MS | | | | |
| /秒/ロタ·ゾース | | 役員.取締役 | | March 7, 2023 |
| ロタ·ゾス | | | | |
| チャド·A·ジョンソンJD | | 役員.取締役 | | March 7, 2023 |
| チャド·A·ジョンソンJD | | | | |
| ヴァエズ·ジョンソン | | 役員.取締役 | | March 7, 2023 |
| 一台のファンエスジョンソン | | | | |
| /s/Kevin Lalande | | 役員.取締役 | | March 7, 2023 |
| ケビン·ラランド | | | | |