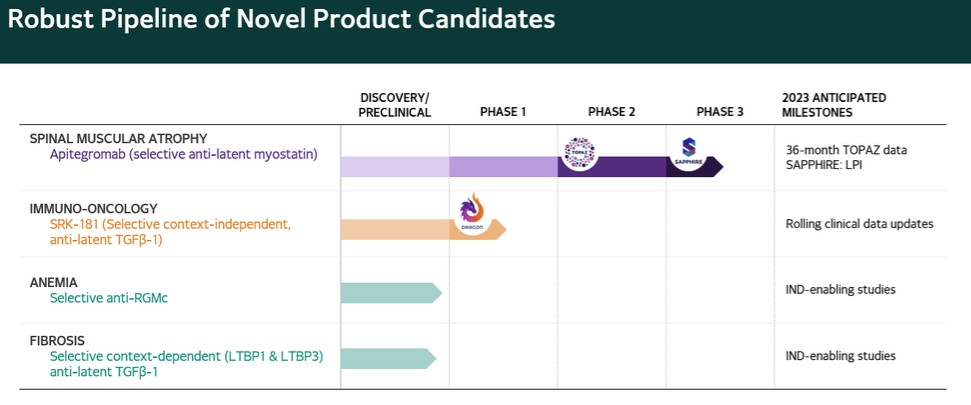
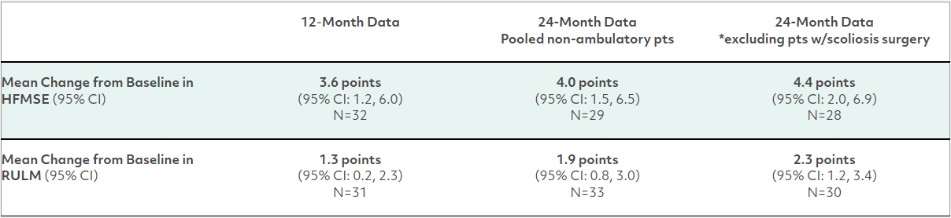
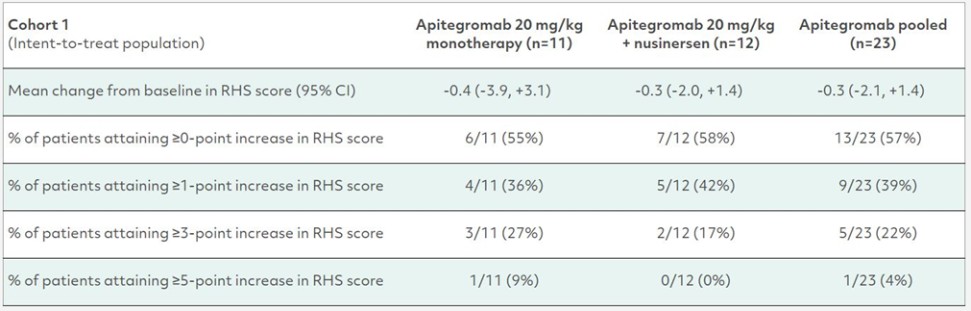
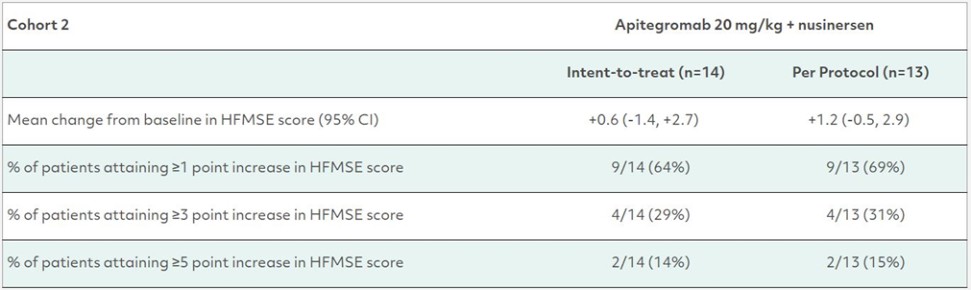
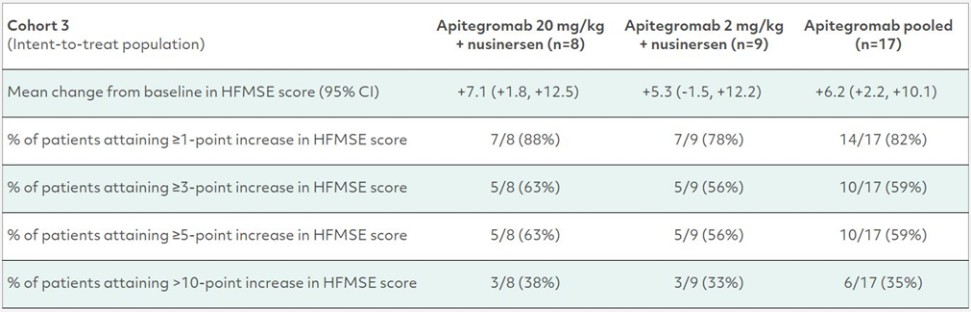
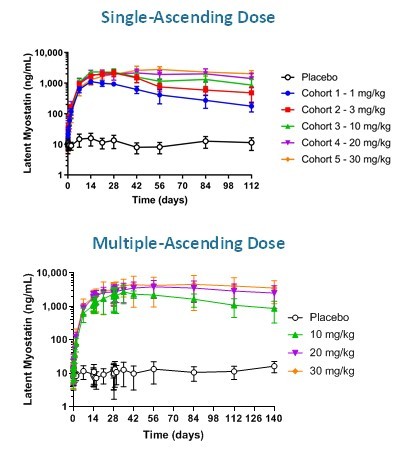
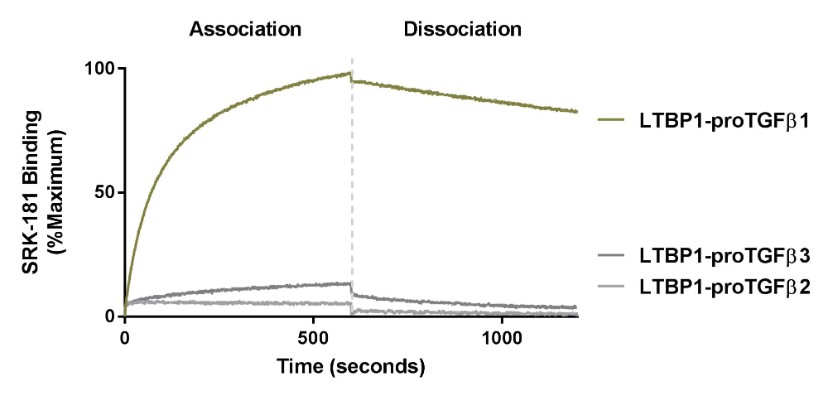
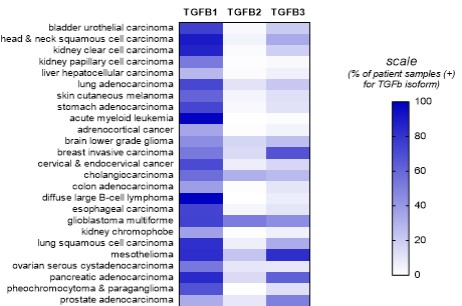
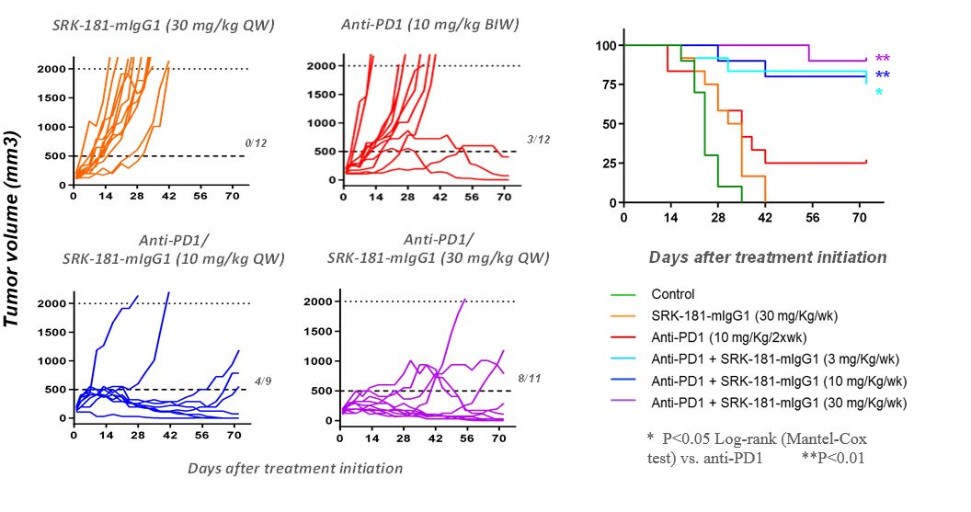
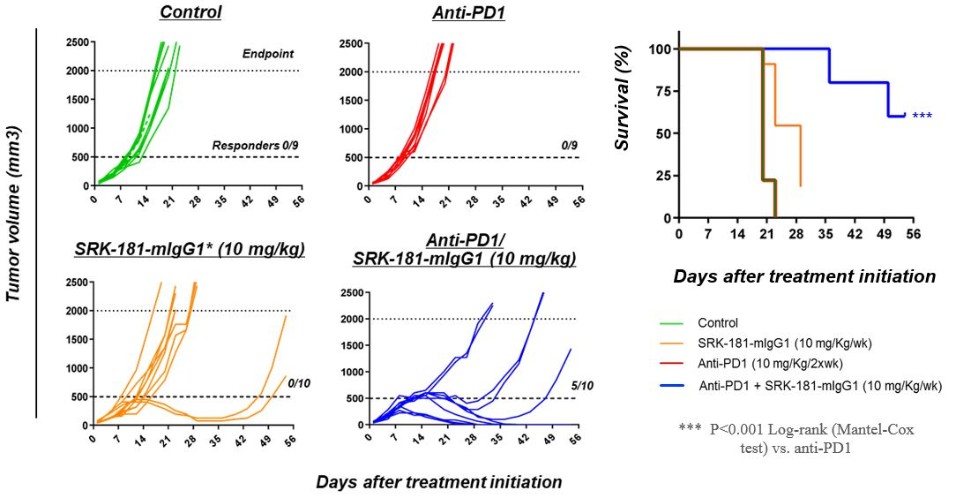
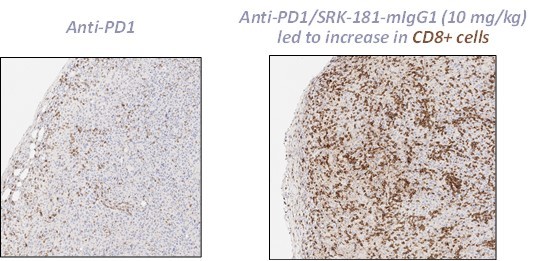
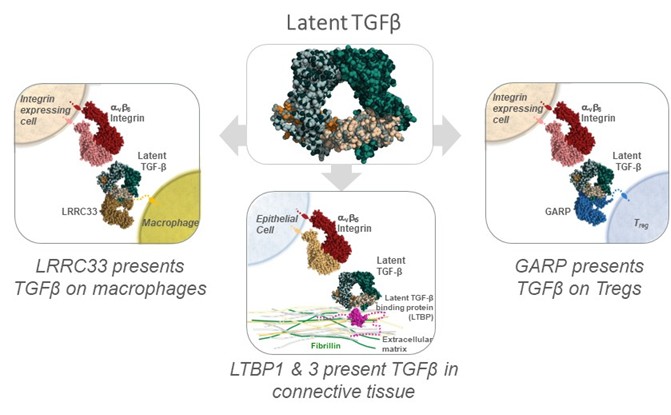
カタログ表
成長因子が基本的な役割を果たす疾患では,活性化された成長因子やその受容体を標的として全身系で行われ,様々な欠陥を受けている
| ● | 経路選択性の欠如--複数の増殖因子はしばしば同じまたは重複する関連受容体を介してシグナルを発し、1つの経路を特定の調節することが困難である |
| ● | 標的選択性の欠如--同じ成長因子スーパーファミリーのメンバーは構造的にかなり類似しており、これは標的成長因子の特定の抑制を達成することが困難である;これは広範な系統的抑制を招く可能性があり、それによって不良、多くの場合有毒な副作用を招く可能性がある; |
| ● | 疾病微環境定位の不足-系統性と非選択的に成長因子を抑制することは疾病過程における成長因子の作用を阻止することができるが、同時にその他の正常な生理作用を妨害することもできる。 |
成長因子標的薬を発見·開発する方法は根本的に新たであり,従来法とは異なる。著者らの成長因子の前駆体或いは潜在形式に対する方法は著者らの共同創立者、ハーバード医学院とボストン児童病院の博士ティモシー·A·スプリンガーの実験室で得られた画期的な発見に基づいている。
成熟あるいは活発な細胞から産生され分泌される他の多くのタンパク質とは異なり、多くの増殖因子は細胞によって潜伏の形で発現される。例えば、形質転換増殖因子β1は、細胞から産生される単一タンパク質であり、その後、細胞酵素によって2つの異なる物理的に分離されたドメインである成熟増殖因子および原始タンパク質の残りの部分に処理され、原ドメインと呼ばれ、それらは依然として複合体の一部である。この分泌複合体は潜伏または不活発であり,まず活性化されなければならず,高度に局在化した組織や疾患微小環境でその正常な機能を実行することができる。2011年に発表された先駆的な同業者評議文章の中で、スプリンガー博士は転化成長因子β1という潜在形式の高分解能X線結晶構造(以下の図に示す)を解決することを通じて、転化成長因子βスーパーファミリーメンバー中の潜在成長因子複合体活性化機構に対する新しい理解を解明した。
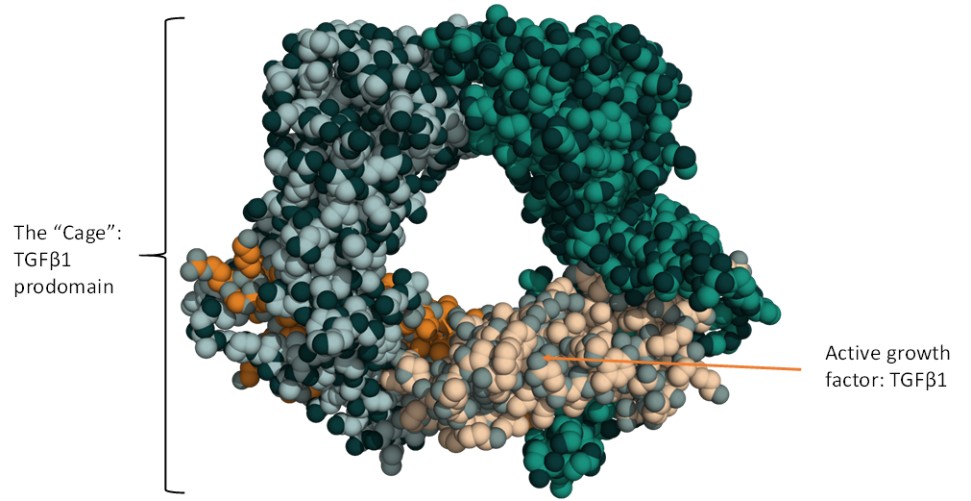
トランスフォーミング成長因子β潜伏期の構造表現1 その中で前ドメインは活性成長因子を被覆している
この研究は,なぜ分泌型形質転換成長因子β1が活性でないのかを分子レベルで説明している。原ドメインは成熟した成長因子ドメインから物理的に分離されているが,形質転換成長因子β1の活性形態の周囲にケージが形成され,遮断されている
7