
l
アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
本財政年度末まで
あるいは…。
1934年証券取引法第13条又は15(D)条に基づいて提出された |
手数料書類番号
(登録者の正確な氏名はその定款に記載)
(明またはその他の司法管轄権 会社や組織) |
(税務署の雇用主 識別番号) |
(主にオフィスアドレスを実行) |
(郵便番号) |
登録者の電話番号は市外局番を含んでいます
(
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
|
取引 記号 |
|
登録された各取引所の名称 |
|
|
同法第12条(G)に基づいて登録された証券:ありません
登録者が証券法第405条に規定する有名な経験豊富な発行者である場合は、再選択マークで表示してください
登録者がこの法第13又は15(D)条に従って報告書を提出する必要がないか否かを、再選択マークで示す。はい。☐
登録者が(1)過去12ヶ月以内(または登録者がそのような報告を提出する必要があるより短い期間)に1934年の証券取引法第13条または15(D)条に提出されたすべての報告書を再選択マークで示すかどうか、および(2)このような提出要求を過去90日以内に遵守してきた
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
大型加速ファイルサーバ |
|
☐ |
|
ファイルマネージャを加速する |
|
☐ |
|
|
|
|
|||
|
☒ |
|
規模の小さい報告会社 |
|
||
|
|
|
|
|
|
|
|
|
|
|
新興成長型会社 |
|
|
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する
これらのエラーのより真ん中に登録者の任意のエンタルピーCER幹部が相関回復期間内に§240.10 D−1(B)に基づいて受信したインセンティブベースの補償に基づいて回復分析を行う必要があるかどうかを再選択マークで示す
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)。はい、そうです
2022年6月30日の普通株の終値によると、登録者の非関連会社が保有する普通株の総時価は約#ドルである
2023年3月1日現在、登録者が発行する普通株式数は
引用で編入された書類
登録者の最終委託書のうち2023年株主総会に関連する部分は,本年度報告の10−K表第III部分に引用的に組み込まれており,範囲は本明細書で述べた範囲内である。最終委託書は登録者が2022年12月31日までの財政年度の120日以内に提出する。引用により本10-Kテーブルに明示的に含まれる情報を除いて,依頼書は本10-Kテーブルの一部として提出されるとはみなされない.
前向き陳述に関する警告説明
このForm 10−K年次報告書には、1934年“証券取引法”(改正)第21 E節又は“取引法”及び“1933年証券法”(改正)第27 A条又は“証券法”の意味に適合する前向きな陳述が含まれている。歴史的事実の陳述を除いて、本年度報告に含まれるすべての陳述は、私たちの将来の運営結果と財務状況、業務戦略、市場規模、潜在的成長機会、非臨床および臨床開発活動、私たちの候補製品の治療効果と安全性、私たちの候補製品の潜在的な治療効果と経済的価値、公開された純収益の使用、候補製品が受けた特定の指定された利益を維持し、確認する能力、非臨床研究および臨床試験の時間と結果、第三者とのビジネス協力、商業化協定のマイルストーンおよび特許権使用料支払いの能力、グローバル業務またはマクロ経済状況の潜在的影響、本プレスリリースに含まれる展望性表現は新冠肺炎疫病、インフレと金利上昇による展望性表現、及び潜在監督管理指定、承認と商業化候補製品の受信と時間スケジュールを含み、すべて展望性表現に属する。“信じる”、“可能”、“そうなる”、“可能”、“推定”、“継続”、“予想”、“予測”、“目標”、“計画”、“可能”、“プロジェクト”、“計画”、“予想”、および未来のイベントまたは結果の不確実性を表す同様の表現は、これらの識別可能な言葉を含むわけではないが、前向き表現を識別することを意図している。
これらの展望的陳述は、第1 A項“リスク要因”および本年度報告の他の部分で説明されたリスク、不確定要素、および仮説を含む多くのリスク、不確定要素、および仮説の影響を受ける。また、私たちは競争が非常に激しく、変化が迅速な環境で運営されており、新たなリスクが時々発生している。私たちの経営陣はすべてのリスクを予測することはできませんし、すべての要素が私たちの業務に与える影響を評価することもできません。あるいは任意の要素や要素の組み合わせは、実際の結果が私たちが行う可能性のある任意の前向きな陳述に含まれる結果と大きく異なる程度をもたらす可能性があります。これらのリスク、不確定性と仮定を考慮して、本年度報告で議論された前向き事件と状況は発生しない可能性があり、実際の結果は展望性陳述中の予想或いは示唆の結果と大きく異なる可能性がある。
あなたは未来の事件の予測として前向きな陳述に依存してはいけない。私たちは展望性陳述に反映された予想は合理的であると考えているが、私たちは展望性陳述に反映された未来の結果、活動レベル、業績或いは事件と状況が実現或いは発生することを保証できない。法律の要件を除いて、本報告書の発行日後に、これらの陳述が実際の結果または私たちが予想する変化に適合するように、任意の理由で任意の前向き陳述を公開更新する義務はありません。あなたが本年度報告書を読む時、私たちの未来の実績、活動レベル、業績、事件、状況は私たちの予想とは大きく違うかもしれません。
文脈が別に説明されていない限り、本Form 10−K年次報告で使用される用語“初日”、“会社”、“私たち”、“私たち”および“私たち”は、別の説明がない限り、最初の日にバイオ製薬会社、デラウェア州の会社およびその合併子会社を全体として意味する。“初日”およびすべての製品候補名は、私たちの一般法商標です。本年度報告書には,他社の他の商号,商標,サービスマークが含まれており,これらはそれぞれの所有者の財産である。私たちは、これらの他社との関係、またはこれらの他の会社の私たちへの支援または賛助を示唆するために、他社の商標、商標またはサービスマークを使用または展示するつもりはありません。
カタログ表
|
|
ページ |
第1部 |
|
|
第1項。 |
業務.業務 |
1 |
第1 A項。 |
リスク要因 |
46 |
項目1 B。 |
未解決従業員意見 |
117 |
第二項です。 |
属性 |
117 |
第三項です。 |
法律訴訟 |
118 |
第四項です。 |
炭鉱安全情報開示 |
118 |
|
|
|
第II部 |
|
|
五番目です。 |
登録者普通株市場、関連株主事項及び発行者による株式証券の購入 |
119 |
第六項です。 |
保留されている |
120 |
第七項。 |
経営陣の財務状況と経営成果の検討と分析 |
121 |
第七A項。 |
市場リスクの定量的·定性的開示について |
135 |
第八項です。 |
連結財務諸表と補足データ |
135 |
第九項です。 |
会計と財務情報開示の変更と相違 |
135 |
第9条。 |
制御とプログラム |
136 |
プロジェクト9 B。 |
その他の情報 |
137 |
プロジェクト9 Cです。 |
検査妨害に関する外国司法管区の開示 |
137 |
|
|
|
第三部 |
|
|
第10項。 |
役員·幹部と会社の管理 |
138 |
第十一項。 |
役員報酬 |
138 |
第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
138 |
十三項。 |
特定の関係や関連取引、取締役の独立性 |
138 |
14項です。 |
最高料金とサービス |
138 |
|
|
|
第4部 |
|
|
第十五項。 |
展示·財務諸表明細書 |
139 |
第十六項。 |
表格10-Kの概要 |
141 |
i
第1部
情報技術EM 1.ビジネス。
概要
設立初日は重要な未満足の需要を解決するためだった:癌児童は抗癌薬開発革命で後ろに振られた。私たちの名前のインスピレーションは、医師と患者とその家族が初歩的な癌診断と治療計画について行った“初日トーク”に由来している。われわれの目標は,1日目から抗癌剤の開発を再想定し,年齢にかかわらずすべての癌患者の可能性を再定義することである。
我々は臨床段階の生物製薬会社であり,生命にかかわる疾患を有するすべての年齢の患者のための標的治療の開発と商業化に取り組んでいる。当初,癌を有する小児科患者に臨床開発努力を集中させたが,これは脆弱な集団であり,最近の標的治療や免疫腫瘍学革命では十分なサービスが得られていなかった。
私たちの主要な候補製品tovorafenib(DAY 101)は経口、脳透過、高選択性II型PAN急速加速型繊維肉腫、あるいはPAN-RAF、キナーゼ阻害剤である。Tovorafenib(DAY 101)は325人以上の患者で研究されており、単一療法として全体的な耐性が良好であることが証明されている。Tovorafenib(DAY 101)は特定の遺伝子変化を有する児童と成人群の中で鼓舞的な抗腫瘍活性を示し、このような変化はRAS/マイトジェン活性化プロテインキナーゼ(MAPK)経路の過剰活性化を招き、細胞成長の暴走を招く。
Tovorafenib(DAY 101)はすでに2020年8月にアメリカ食品と薬物管理局(FDA)に突破的治療称号を授与され、児童低レベルグリオーマ(PLGG)の治療に応用され、その第一段階試験の初歩的な結果により、pLGG患者は迅速な抗腫瘍活性と持続的な反応を有することを示した。児童低レベルグリオーマは児童の中で診断される最もよく見られる脳腫瘍であり、標準的な治療法もなく、大多数の患者に対して承認された治療方法もない。著者らは2020年9月にFDAから悪性グリオーマ治療の孤児薬物指定を獲得し、2021年5月にEUグリオーマ治療委員会から悪性グリオーマ治療の指定薬物を獲得した。さらに、FDAは2021年7月にtovorafenib(DAY 101)の活性化RAF変化が存在する低レベルグリオーマ(LGG)の存在する稀な小児科疾患の治療を許可した。
我々はすでにtovorafenib(DAY 101)の重要な2期試験あるいはFirefly-1を開始し、完全に募集し、児童再発或いは進行性低レベルグリオーマ患者の単一療法として、これらの患者は活性化BRAF変化を有する。最初の患者は2021年5月にFirefly−1の用量治療を受け,2022年5月にARM登録を完了した。Firefly-1試験はまた、(A)主要なキューが登録されているので、合格患者のアクセス範囲を拡大するために、他の2つの研究分枝に拡張された;および(B)6ケ月から25歳までの再発または進行性頭蓋外固形腫瘍を有し、RAF融合を活性化した患者におけるtovorafenib(DAY 101)の初歩的な治療効果を評価する。2022年6月のFirefly−1試験中期分析の予備データと2023年1月の全患者のトップデータを報告した。2023年1月のTOPLINEデータによると、69項目の神経腫瘍学的反応評価(RANO)評価可能な患者のうち、総応答率(OOR)は64%であり、3名の確認された完全応答(CR)および41名の部分応答(PR)(31名の確定診断された部分応答および10名の未確認部分応答(UPR))を含む。著者らは他の19名の安定期(SD)患者を観察し、臨床受益率は91%(CR+PR/UPR+SD)であった。77名の治療を受けた患者の安全性データによると,ボルラフェニ(DAY 101)の単一療法は全体的に耐性が良好であった。腫瘍の減少や安定はpLGG患者に臨床的意義があると考えられ,多くの患者が承認された治療法が乏しいことから,両者とも有益であると考えられている。2023年第2四半期の医学会議でFirefly−1試験からのより多くのデータを提出し、新薬申請前(NDA)にFDAとともに研究データの重要な部分を審査する予定です, 私たちは2023年上半期に秘密協定を提出する前に会議を開催する予定です。
2022年6月,われわれはpLGGの第一線治療としてtovorafenib(DAY 101)のキー3期試験あるいはFirefly−2を開始した。2023年3月,1人目の患者はFirefly−2を服用した。
1
私たちの第二の候補製品pimasertibは経口的、高選択性のマイトジェン活性化プロテインキナーゼ1と2或いはMEKの小分子阻害剤であり、MEKはMAPK経路中の特徴が明確な重要なシグナルノードである。Pimasertibは850名を超える異なる腫瘍タイプの患者の10個以上の1/2期臨床試験で検討されており,単一療法としても併用標準看護療法としても検討されている。すでに発表された臨床前研究により、Pimasertibは他のMEK阻害剤と比較して高い中枢神経系(CNS)浸透率を有することが示唆された。
我々は、2つのサブ研究を含む開放ラベル、マルチセンター、1 b/2 a期の傘式主試験、または火-1、トレフラフィニ単一療法または併用療法を開始した。サブ研究1はトヴォラフェニ(DAY 101)の第2段階試験であり、12歳以上のRAF変化腫瘍を有する患者の単一治療に用いられ、1人目の患者は2021年11月に服用した。第二の研究はtovorafenib(DAY 101)とpimasertibの1 b/2期連合試験であり、12歳以上の各種MAPK変化を有する固形腫瘍の患者に用いられ、1人目の患者は2022年5月に治療を受けた。臨床前モデルでは,RAFとMEKの同時抑制は協同抗腫瘍活性を引き起こすことが証明されている。この組み合わせは、NRAS変異黒色腫および肺癌、クラスII BRAF変化によって駆動される腫瘍、BRAF野生型融合を有する腫瘍、およびKRAS変化によって駆動される腫瘍を含む、MAPK変化によって駆動される様々な成人固形腫瘍において増強された抗腫瘍活性を示す可能性がある。
著者らの業務開発能力は、腫瘍学薬物開発における著者らの豊富な経験と、研究と患者権益提唱コミュニティにおける深いつながりに加え、特に小児科環境において、すべての年齢の患者のための識別、獲得と治療方法の開発においてリードしていると信じている。私たちはすべての治療分野におけるtovorafenib(DAY 101)とpimasertibの世界的な独占権利を持っているが、いくつかのマイルストーンと特許権使用料を支払わなければならない。
毎年,米国では約15,500人の18歳以下の児童が癌と診断され,世界には約300,000名がいる。また、癌は依然としてアメリカの児童に最もよく見られる疾病の死亡原因であり、毎年1700人以上が癌で死亡している。児童癌はもっと安全、もっと有効な治療方法が必要であるが、児童患者に応用される新薬は非常に少ない。1997年から2017年までの間にFDAの許可を得た117種類の非ホルモン腫瘍学薬物の中で、6種類のみが初歩的な承認を得ており、その中に児童を含む。通常,小児の薬品テストは成人試験が臨床開発に達する後期段階に延期される。そのため、腫瘍学製品候補の第1回小児科試験は通常成人初臨床試験後約6年後に開始される。
また,小児科腫瘍学でアドレス可能な亜群を定義するために必要な大規模分子マップデータセットの産生は最近発生している。著者らは小児科癌生物学に対する理解が進展し、薬物遺伝変化を有する患者群を掲示した。著者らの管理チームは豊富な小児科腫瘍学と稀な疾病薬物開発経験を持っており、彼らはtovorafenib(DAY 101)のような標的療法を信じ、小児科癌の中で大量に満足されていない需要を満たすために、もっと早く小児で研究を行う可能性があり、これらの癌の中で、腫瘍の特定の遺伝駆動要素に対する新しい薬物は長期予後を有効に改善することができる。
著者らのチームの小児科腫瘍学における広範な能力と経験、及び著者らと小児科医学界のすべての肝心な利益関係者との関係は、著者らが小児科薬物開発の挑戦とニュアンスを有効に制御できるようにした。児童の臨床発展は簡単に小成人の臨床発展と見なすべきではないことを理解している。有利な監督管理経路の潜在力、即ち突破的治療と孤児薬物指定を考慮して、著者らは著者らの独特な専門知識を利用して著者らの初歩的な開発努力を小児科患者に集中した。
私たちは児童やその家族が癌と戦うのを助けるとともに、長期にわたって満たされていない医療ニーズを解決するように駆り立てられている。著者らは小児科患者の中で新しい腫瘍候補製品を開発することは多くの独特な優勢があると信じている
2
著者らは高需要癌に対する高価値発癌駆動要素の候補製品の識別、獲得と開発を求め、最初の重点は小児科患者である。次の表に私たちの候補製品ルートをまとめました。
著者らの主要な候補製品tovorafenib(DAY 101)は経口、脳透過、高選択性II型汎RAFキナーゼ阻害剤であり、単量体RAFキナーゼを抑制することもでき、ダイマーRAFキナーゼも抑制できる。Vemurafenibやencorafenibなどの承認されたBRAF製品は,I型RAF阻害剤と呼ばれ,RAFモノマーのみを抑制するため,BRAF V 600改変のための腫瘍に限られている。I型RAF阻害剤とは異なり,トレフラフィニ(DAY 101)は臨床有効量でRAF野生型細胞の矛盾した活性化を引き起こすことが証明されていない−このような現象では,MAPKシグナルの意外な増加が腫瘍再成長を引き起こす可能性がある。Tovorafenib(DAY 101)のRAF単量体と二量体に対する抑制はその潜在的な臨床応用を拡大し、一連のRAF或いはRAF変化の固形腫瘍の治療に用いられる。また,tovorafenib(DAY 101)は他のMAPK経路阻害剤よりも高い脳浸透率,分布,曝露を有することが示唆された。以上より,tovorafenib(DAY 101)はpLGGの高影響標的治療薬となる可能性があり,その半数以上のpLGGはRAF変化による異常シグナルによって駆動されていると考えられる。
3
この理論的基礎は,Dana−Farber癌研究所の研究者がpLGGにおいてtovorafenib(DAY 101)の開発を開始した基礎である。1つの第1段階用量増加研究において、9名の小児科患者(
私たちの第二の候補製品pimasertibは経口的、高選択性のマイトジェン活性化プロテインキナーゼ1と2或いはMEKの小分子阻害剤であり、MEKはMAPK経路中の特徴が明確な重要なシグナルノードである。いくつかのMEK阻害剤は、BRAF V 600変異腫瘍のI型RAF阻害剤と共に使用することができる規制部門の承認を得ている。臨床前実験により、MEK阻害剤とII型RAF阻害剤の併用の潜在的利益はもっと大きい可能性があり、II型阻害剤の下流シグナル伝達に対する矛盾した影響が乏しいためである。Tovorafenib(DAY 101)がRAF単量体と二量体を選択的に抑制する能力はその潜在的な臨床応用を広げる可能性があり、MEKと結合してRAS変化、非BRAF V 600突然変異とRAF融合によって駆動される固形腫瘍を抑制する。
著者らは生物製薬会社の設立に成功記録を持つ指導チームと、小児科薬物開発において独特な経験と能力を持つ薬物開発チームを結成した。我々の最高経営責任者Jeremy Bender,Ph.D.,M.B.A.は15年以上のバイオ製薬リーダー経験を会社にもたらした。彼は以前ジリード科学会社で企業発展副総裁を務め、指導チームはギリッド社の買収、協力と株式投資を担当し、そして40余りの取引を監督し、前払い取引価値は100億ドルを超え、その中には477、Inc.を含む。著者らの共同創業者で首席医療官サミュエル·ブラックマン博士は小児科血液学/腫瘍学と神経腫瘍学の訓練を受けた内科科学者であり、10種類以上の新型癌療法の早期臨床開発を指導し、ダプラファニーの小児科開発を担当した。それによって、業界が賛助した最初の小児科腫瘍学“バスケット試験”を産生した。私たちの首席運営と財務官Charles York IIは以前、Aeglea BioTreeuticsの首席財務官と企業発展主管を務め、20年以上の戦略資本形成と指導経験を持っていた。われわれの首席開発官,PharmD,Davy Chiodinは,成人と小児腫瘍薬物開発においてAcerta(現在のアスリカン)でacalabrutinibを開発し,羅氏/遺伝子技術会社で小児科腫瘍学の全世界監督管理担当者を務めている15年以上の経験を有している。マイク博士、私たちの技術運営総監は、製品開発において25年以上の経験を持ち、アレイでCMC担当者を10年以上務め、20以上の研究新薬申請、またはINDを提出しました, ビニミチニブやツカチニブなどの市販薬の開発を支援している。Jaa Roberson、私たちの首席人事官は、バイオ製薬、医療保険、小売業界で20年間の人材経験を持っています。彼女は以前,Bellicum製薬会社の人的資源担当を務めていたが,細胞免疫腫瘍学に専念した臨床段階バイオ製薬会社である。私たちの総法律顧問Adam Du博は20年以上の業界経験を持ち、最近では百時美施貴宝の首席コンプライアンスと道徳官を務め、アメリカ、アジア、ヨーロッパで様々な法律と政策職を務めている。
4
私たちの戦略
私たちはサービス不足の患者群を解決するために、治療薬を識別、開発と商業化することによって、差別化された全世界の生物製薬会社を構築する使命駆動の戦略があり、最初の重点は小児科患者である。私たちの戦略の主な内容は
5
私たちの方法:小児癌や他の高度に満たされていない需要分野を優先する
わが社は小児科患者のために新たな標的治療薬を優先的に開発することに集中している。歴史的には,多くの製薬会社が新たな癌療法の発見と開発の努力を成人腫瘍型に集中させている。したがって,1997年から2017年の間にFDAが初歩的に承認された腫瘍学的適応の126薬剤については,初回成人試験と初回小児試験の間の中位期間は6.5年であり,化学療法薬,生物製剤,標的治療薬であった。
小児科腫瘍学的薬物開発に関する歴史的仮定を見直し,是正するのに適したタイミングであると考えられる。このようにすると同時に、小児科患者の中で新しい腫瘍候補製品を開発することは独特の優位性があり、成人適応と並行して、成人適応よりも先になると信じている
6
わが社は小児科腫瘍患者に緊急の標的治療を提供する上で恵まれた優勢を持っている。著者らはこれらの患者の面で広範な能力と経験を持っており、著者らと小児科医学界のすべての重要な利益関係者との信頼できる関係は、小児科薬物開発の挑戦とニュアンスを有効に制御することができる。小児科腫瘍学の機会を成功的に識別し、利用することができる主な利点は以下の通りである
これらの能力は著者らが的確な治療方法を開発し、小児科患者に利益を得ることができる。我々はこの発展分野のリーダーであると信じており,この地位をさらに推進するために,バイオ製薬会社,学術小児科腫瘍学者や科学者および患者権益団体と相談と戦略協力を継続し,小児科腫瘍学で満足されていない領域を決定し,影響力の高い資産を買収し,これらのサービス不足患者を解決する予定である。われわれは最初は小児科患者に重点を置いていたが,成人群にも同等の強度の標的治療の臨床開発を求めている。
7
私たちの候補製品
著者らは高需要癌に対する高価値発癌駆動要素の候補製品の識別、獲得と開発を求め、最初の重点は小児科患者である。われわれの臨床開発は,小児科腫瘍学環境における我々の独自の専門知識の利用から始まったが,成人癌患者に対する標的治療を同等の強度で進めることに取り組んでいる。次の表に私たちの候補製品ルートをまとめました。

Tovorafenib(DAY 101)
我々の主要候補製品tovorafenib(DAY 101)は経口、脳透過、高選択性II型汎RAFキナーゼ阻害剤である。Tovorafenib(DAY 101)はすでに325人以上の患者で研究されており、単一療法として、特定のMAPK経路の変化を有する児童と成人群で良好な耐性と鼓舞的な抗腫瘍活性を示した。Tovorafenibのキー2期Firefly−1試験(DAY 101)を開始し,完全に募集したが,pLGGの単一療法としてpLGGは小児で最もよく見られる脳腫瘍であり,現在承認されていない治療法も認められておらず,多くの患者の看護基準も認められていない。Firefly-1試験はまた、(A)主要なキューが登録されているので、合格患者のアクセス範囲を拡大するための他の2つの研究分枝を含み、(B)6ヶ月~25歳までの再発または進行性頭蓋外固形腫瘍を有し、RAF融合を活性化する患者におけるtovorafenib(DAY 101)の初歩的な治療効果を評価する。われわれは2022年6月にこの試験の中期分析の予備データを報告し,2023年1月に全患者の主なデータを報告した。2023年第2四半期に開催される医学会議でFirefly−1試験からより多くのデータを提出し,2023年上半期にNDAを提出する予定のNDA前会議でFDAとともに本研究のキーデータ部分を検討する予定である。私たちの業界の慣例のように, 私たちはまた2023年の医学や科学会議で私たちのデータを報告する予定だ。Tovorafenib(DAY 101)は、pLGG治療のための突破療法指定がFDAによって承認されており、これは、pLGG患者が迅速な抗腫瘍活性と持続的な反応を有することを示す第1段階試験の初歩的な結果に基づく。Tovorafenib(DAY 101)単独で他の薬剤との併用も検討されており,これらの薬剤はMAPK経路中の重要なシグナルノードに対して,患者集団において各種RASやRAF変化が疾患駆動に重要な役割を果たしていると考えられている。
8
RAFキナーゼ駆動細胞増殖と癌化
細胞の成長、生存と分化などの機能は下落などのシグナルイベントの調節を受け、RAFキナーゼはその中の重要な構成部分である。RAFはプロテインキナーゼであり、通常RASによって活性化され、RASは活性化シグナルを細胞外受容体からRAFに伝達するタンパク質である。RAFの活性化はMEKキナーゼやMAPK下流経路の活性化を引き起こす。この経路の過剰活性化を招く遺伝子改変、例えばRAS或いはRAF変化は、長い間ずっと発ガンと考えられてきた。

図1.RAFキナーゼ(ARAF、BRAF、CRAF)はMAPK経路の重要な構成部分である。BRAF V 600 Eは、モノマーとしてシグナルを発することができ、I型およびII型RAF阻害剤に感受性である。野生型RAFダイマーはII型RAF阻害剤にのみ敏感である。原作はソレットとローソン癌発見2014です
この経路の中で最もよく見られる改変遺伝子の一つはBRAFであり、それはヒト細胞中の三つのRAF遺伝子の一つであり、RAFが最もRASによって活性化されやすい形式でもある。BRAFの多くの変化はV 600と呼ばれる変異である。V 600の変異は、非変異型または野生型BRAFをBRAFの一形態に変換し、この形態のBRAFはシグナル活性を増加させ、RASに依存しなく活性化する。V 600変異体BRAFの豊富さ及び腫瘍成長における核心作用はそれを歴史上の薬物発見努力の焦点にした。
もう一つの重要な発癌BRAF変化はBRAF野生型遺伝子融合である。BRAFに関する遺伝子融合は染色体内や染色体間の再編成によって発生し,再編成では無関係タンパク質の遺伝子が物理的に結合し,キメラタンパク質の合成を招く。BRAFはBRAF活性を調節する制御ドメインと1つの触媒活性化領域からなり、触媒活性化領域は下流シグナルを活性化し、それによって細胞成長を促進する。BRAF融合において、BRAFの調節ドメインは異なる配列で置換され、BRAFはRAS活性化とは独立してシグナルを発することができる。BRAF調節ドメインと触媒ドメインのこのような脱共役は重要な結果がある:そこから産生された新しい癌遺伝子は異常に発現し、組成或いは常に活性化された活性化ドメインも示した。このキナーゼ活性は下流の発癌シグナルの活性化を招き、腫瘍の成長を悪化させることができる。BRAF遺伝子融合はすでに前立腺癌、黒色腫、放射線誘導甲状腺癌およびpLGG患者に観察されている。
9
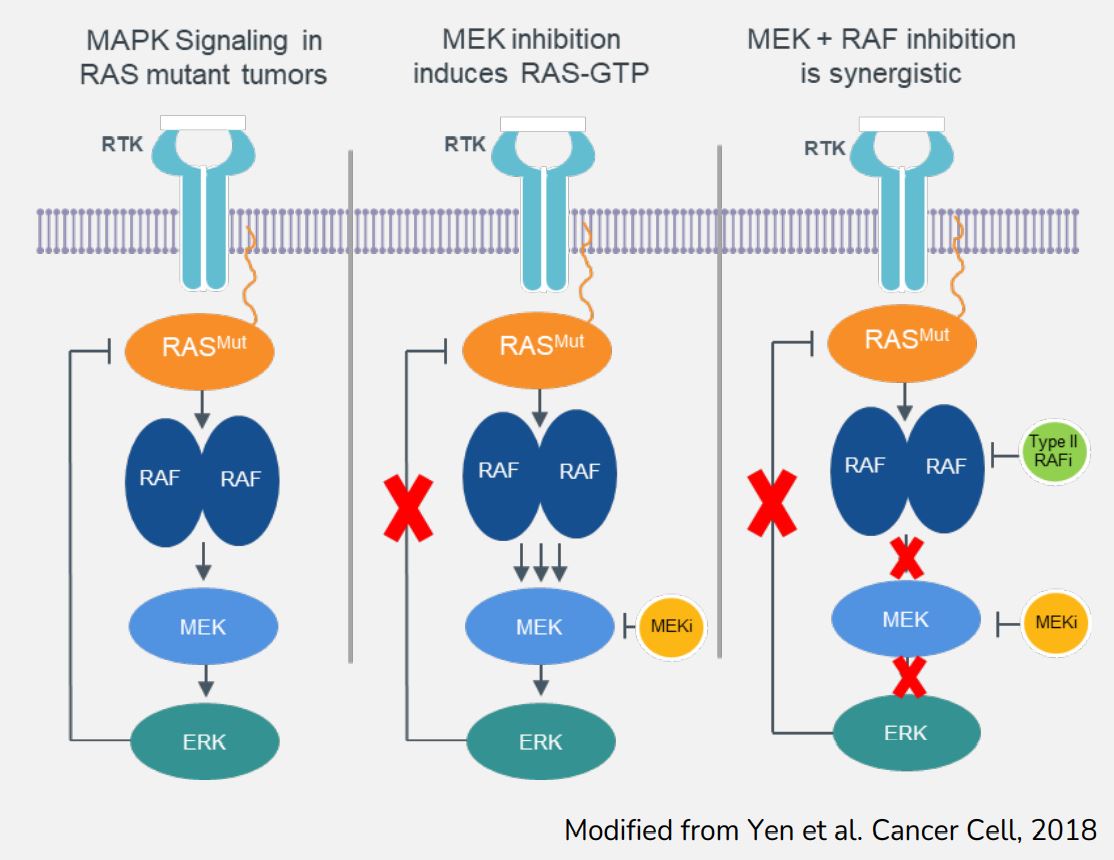
FDAは、黒色腫、非小細胞肺癌、間変性甲状腺癌および結腸直腸癌を含むBRAF V 600 EまたはV 600 K変異のみを含むいくつかの固形腫瘍の治療に3種類のBRAF阻害剤を許可した。これらの第1世代BRAF阻害剤は,I型RAF阻害剤であり,ベモラファニーであり,遺伝子テークによってZelborafの名称で販売されていること,ノワール社による名称で販売されているダプラファニ,およびファイザー社がBraftoviの名称で販売していることがより知られている。しかし,単一治療1型RAF阻害剤は最初に臨床反応があったにもかかわらず,多くの患者は治療開始後1年以内に再発した。
I型RAF阻害剤に対する耐性の一つの様式は,細胞における正常RAF活性化の機序に関与している。モノマー活性であるV 600 EやV 600 K変異体とは異なり,正常なRAF機能はRAFの二量体を形成する必要がある。許可されたV 600 E/K BRAF阻害剤は、RAFダイマーまたは他の非V 600 BRAF変異の活性を阻害することができない。実際、これらのインヒビターのいくつかがV 600 E/K BRAFに結合することは、二量体の形成を刺激し、RAF野生型細胞における矛盾した活性化(MAPキナーゼシグナルの意外な増加)をもたらす-この現象は新しい腫瘍成長を引き起こす可能性がある。野生型RAFの矛盾活性化は非腫瘍組織でも発生する。これはこれらの薬物に関連するよく見られる不良イベントである増殖性、癌前と悪性皮膚病変の発展を招く。薬剤耐性や矛盾の活性化を避けるためには,I型RAF阻害剤はMEK阻害剤と併用する必要があることが多いが,同様にBRAF V 600 E/K変異患者にのみ適用される。
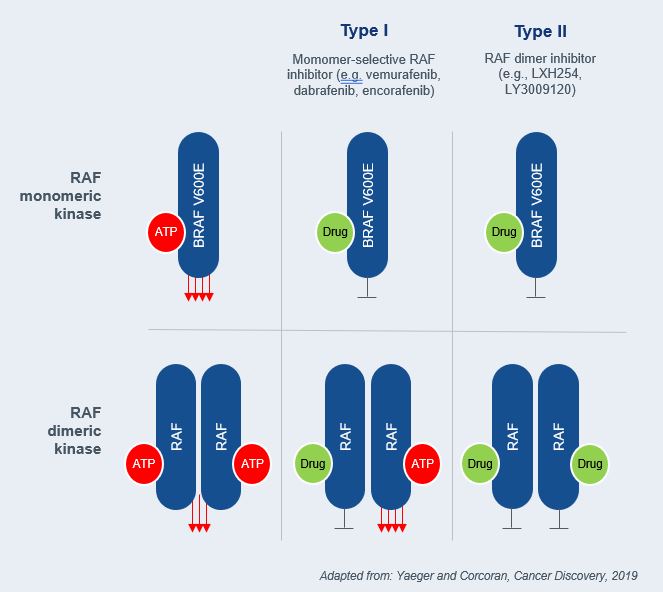
[図2]モノマーRAFキナーゼ(すなわち、BRAF V 600 E;上部)またはダイマーRAFキナーゼ(下部)に対する異なるRAF阻害剤の影響を示す模式図である。ERK活性化はBRAF V 600 E下流で強く活性化され,ダイマーRAFキナーゼシグナルよりも強い。単量体選択的I型RAF阻害剤はBRAF単量体中のATP部位に結合し,下流シグナル伝達を抑制する。RAFダイマーキナーゼでは,薬物の結合が結合したRAF原型を抑制したが,二量体中のもう一つの原型の立体構造変化とこの原型の強いトランス活性化を招き,ERKの全体活性化(矛盾活性化)を招いた。II型RAF阻害剤は,変異RAF単量体および二量体に同じ用量で結合することができるため,変異RAF単量体および二量体を同じ用量で抑制することができる。イェーガーとコクランが原作で、“癌発見”、2019年。
V 600 E/K改変に対するI型RAF阻害剤はKIAA 1549−BRAF遺伝子融合における野生型RAF作動域を抑制できないため,この融合による過活性化シグナル伝達を効果的に抑制することができなかった。また,矛盾した活性化の可能性があるため,これらのRAF阻害剤はBRAF遺伝子融合患者では禁忌である。
10
DAY 101(Tovorafenib)の役割メカニズム
Tovorafenib(DAY 101)は、野生型RAF、BRAFおよびCRAF融合タンパク質、二量体としての変異体(クラスII変異)、およびBRAF V 600 Eおよび非V 600 E変異(クラスI変異)などのモノマーの変異体としてのRAF活性を阻害することができる選択的小分子RAF阻害剤である。Tovorafenib(DAY 101)は単量体と二量体RAFキナーゼを同時に抑制できるためII型RAF阻害剤と考えられている。DAY 101(Tovorafenib)のRAF単量体と二量体に対する抑制はその潜在的な臨床応用を拡大し、一連のRAF変化の腫瘍の治療に用いられる。
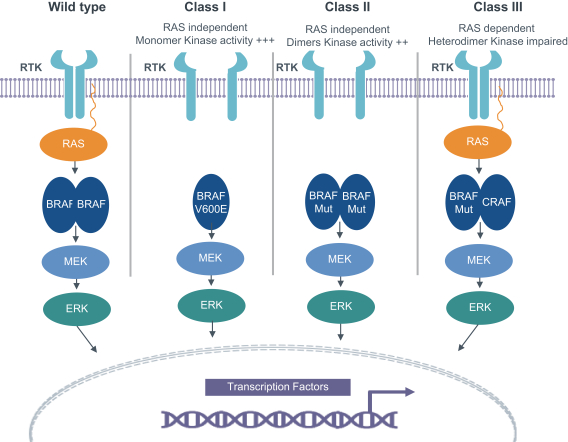
[図3]異なるタイプのBRAF変異におけるシグナル経路。BRAF V 600突然変異(第I類)はRASシグナルとは独立し、単体の形式で作用を発揮する。BRAF非V 600第II類突然変異もRASに依存しないが、構造的二量体としてシグナルを発する。クラスII変異はBRAF野生型融合を含む。非V 600 IIIクラスBRAF変異は低いまたはないキナーゼ活性を有し、RASの活性化に依存してRASシグナル経路の増幅器として機能する。Tovorafenib(DAY 101)は、BRAF野生型融合および非V 600 E/K変異を含むクラスIおよびクラスII RAF変化を抑制することができる。FontanaとValeri、臨床癌研究、2019年から修正された。
児童低レベル脳グリオーマ疾患と治療概要
児童低レベルグリオーマは児童に最もよく見られる脳腫瘍であり、すべての中枢神経系腫瘍の30%-50%を占める。ほとんどの場合、このような腫瘍の成長は遅く、慢性的で非情だ。PLGGsの悪性転化と拡散は希であるが、この疾患は多くの長期的な結果がある。PLGGの成長は高度に病態であり,pLGG腫瘍は占拠性病変であるため,脳中の重要な神経血管構造を圧迫する可能性がある。腫瘍の位置や周囲組織にかかる圧力の大きさによって患者の症状が異なる可能性がある。これらの症状は、特に腫瘍の位置に依存する頭痛、吐き気、嘔吐、傾眠、第六脳神経麻痺、てんかん発作、および行動変化を含む可能性がある。PLGGを有する大多数の児童は長期の生存者であり、成人まで生きている;しかし、児童グリオーマの生存者は通常彼らの疾病および/または治療のために長期的な機能、神経と内分泌合併症を受ける。これらの患者はもっと有効な治療策略が必要であり、長期発病率と治療関連毒性を最小限に下げる。
11
PLGG患者は歴史上手術、放射線治療と化学療法を受けたことがある。手術によるpLGG切除は90%以上の10年総生存率と関係があるが、大多数の児童は完全切除を受けることができず、場合によっては、このような手術は顕著かつ長期的な発病率と関係があるかもしれない。不完全切除或いは切除できないpLGGは疾病の進展或いは再発率と高い相関がある。大部分切除患者の10年無進展生存率は55%であった。より現代的な放射線治療法は無進展生存率を改善できることが証明されているが,歴史的には,放射線治療は小児の神経認知結果が著しく低下するリスク,内分泌機能障害,二次性悪性腫瘍と脳卒中リスクの増加に関与している。したがって,現代放射線治療技術であっても,他のすべての治療法が失敗した場合に使用するために保持され続けている。
大多数の進行性pLGG患者は最初の系統治療が必要であり、カルボプラチン/ビンクリスチンなどの連合化学療法を採用し、ある国では、ビンクリスチンは単一薬物として治療を行う。新たに診断されたpLGG児に対する最大規模のランダム3期研究の結果,ビンクリスチン/カルボプラチンの5年無事象生存率は47%であった。神経線維腫症と関係のないpLGG患者のグループは、BRAF変化を有する患者を含み、結果は比較的に悪く、39%の5年無事象生存率を示した。新たに診断されたpLGG患者の化学療法に対する全体応答率は30%−35%であったことに注意されたい。化学療法の治療効果の制限以外に、治療に関連する発病率も非常に高く、95%を超える患者は少なくとも1回の3級或いは4級の不良事件を経験したことがある。これらの併用治療失敗後に腫瘍が進展した患者では,標準的な治療法もなく,このような患者に対する標的治療も承認されていない。
図4.pLGGの治療例を示す。
多くのpLGGは患者の年齢が20歳に達した時に老化が出現するため、治療の目標は腫瘍を最大限にコントロールすることであり、同時に手術、化学療法と放射線治療に関連する毒性を最小限に下げることである。そのため、大量のpLGG患者は彼らの疾病過程中に複数の一連のシステム治療を受ける。
学術誌に発表された発症結果によると,毎年約1100名の25歳以下の患者がBRAF改変pLGGと新たに診断されていると推定される。2017年1月1日現在,米国の25歳以下の患者のSEER罹患率は約130,000名であり,脳や他の神経系腫瘍を示す患者は約26,000名であり,そのうち26,000名はBRAF変化のpLGGを示していると推定されている。
12
過去10年間に50%から60%のpLGGsがRAF変化による異常シグナルによって駆動されることが発見され,その約85%から90%がKIAA 1549−BRAFと呼ばれる遺伝子融合である。このような遺伝子改変は、その正常な調節ドメインではなく、野生型BRAF触媒ドメインの発現をもたらし、BRAFに構造的活性を持たせる。また,pLGGsを有する小児では5%から17%の腫瘍がBRAF V 600 E活性化変異を有していた。PLGGを標的とする薬剤は現在のところpLGGの治療のために承認されておらず,RAF変化を有する小児患者の治療−pLGG患者の最大サブセットも承認されていない。
KIAA 1549-BRAF遺伝子改変を間接的に標的とすることは可能であり、MEKを標的とする治療のような、承認された下流RAFシグナル経路構成要素を標的とする薬剤をタグ外で使用する。BRAF V 600 Eを標的とする変異は可能であり、タグ外にI型RAF阻害剤を使用することにより、黒色腫などの成人適応への使用が許可されている。最近,研究者が後援したMEK阻害剤selumetinibの臨床試験が発表された。この研究はKIAA 1549-BRAF融合またはBRAF V 600 E変異患者25名を含む。25例の患者の中で9例は持続的に部分的に緩和した。16%の患者はグレード3のクレアチンホスホキナーゼが上昇し、8%の患者は3級ざ瘡様皮疹があった。25人の患者のうち10名(40%)は治療に関連する有害事象のために用量を減少させる必要があり、1名(4%)は2回の用量を減少させる必要がある。同様に,最近18名の患者にMEK阻害剤トリメチニブを用いた回顧性分析が発表され,6例の部分反応,2例の軽微な反応,10例の安定疾患が最適な全体反応であった。89%の患者が治療に関連する有害事象を発生し、44%の深刻な(レベル3またはレベル4)有害事象を含み、33%の患者が用量を減少させる必要があり、11%の患者が服薬を中止する必要がある。最近、業界が後援したBRAF V 600 E変異を有するpLGG患者110名に対するダプラファニー1/2 a期研究を発表した結果、確認された客観緩和率(OOR)は47%であり、その中に1つの完全緩和と13個の部分緩和を含み、中位緩和期間は26ケ月であった。28%の患者が治療に関連するレベル3またはレベル4の有害事象を報告し、そのうちの25%の患者が新たなまたは増大したメラニン細胞母斑を認めたが、扁平上皮癌症例はなかった。10人の患者(31%)は、用量中断または減少をもたらす有害事象を有する, 患者の6%が治療中止につながる有害事象が発生した。最後に、最近、業界によって後援されたトリメチニブ単独治療またはダプラファニブ/トリメテニブ併用治療BRAF V 600変異再発/難治性pLGG患者の第二段階研究が発表された。この研究は,トリメチニブ単独での客観的有効率は15%(n=13),併用投与の客観的有効率は25%(n=36)であることを示している。併用治療を受けた患者の25%に重篤な副作用が発生した。16%の患者が用量減少をもたらす有害事象を発生し、44%の患者が用量中断をもたらす有害事象を発生し、11%の患者が投与中止をもたらす有害事象を発生した。最もよく見られる副作用は発熱(50%)、皮膚乾燥(41%)、ざ瘡様皮疹(39%)、無力(39%)と皮疹(36%)である。
以上より,これらの研究から,いくつかの既存のMEKやI型RAF阻害剤は小規模試験でpLGGに活性を示すが,頻繁なレベル3や4次有害事象に伴い,用量の減少や中断が必要であることが示唆された。6歳以上のBRAF V 600 E/K変異の再発/進行期腫瘍を有する患者には,ダプラファニブ/トリメテニブが用いられているほか,薬剤の使用が許可されていないため,臨床試験やラベル外処方でしか得られない。ラベル外使用は小児科腫瘍学環境によく見られるが,小児を潜在的なリスクに曝露し,長期安全モニタリングや薬物警戒活動などの包括的な臨床開発活動に伴う保障措置を伴わない副次的な方法と考えられている。PLGGに対する脳透過療法の開発は,これらの患者の予後改善に重要であり,特にBRAF融合を有する患者,BRAF V 600 E変異を有する患者の開発が信じられている
13
PLGGの臨床試験結果
Tovorafenib(DAY 101)は現在,Dana−Farber癌研究所がPNOCと協力し,再発/難治性グリオーマ(高レベルと低レベル)や他の腫瘍患者で行われている多中心研究(PNOC 014,NCT 03429803)で評価されている。この試験はまだ開放されているが、新しい患者に対する費用は閉鎖されている。2022年11月までに,44名の患者がこの第1段階用量逓増試験のB部分(A部分9名,B部分35名)に参加し,PNOCネットワーク内の複数の施設で行った。週に1回トヴォラフェニ(DAY 101)、420 mg/mまでの投与量2/週耐性良好、患者150万2530 mg/mまでの投与量です2/週は患者における耐性が良好であることが発見された2それは.最近、2022年神経腫瘍学会会議でB部分研究の35名の患者のデータが発表されました。このグループのうち、2名の完全反応、7名の部分反応、15名の病状が安定している患者、8名の病状が進行している患者があります。6種類の用量制限毒性があり、いずれも530 mg/m2/週は,いずれもグレード3,および5つの既知の副作用(2疲労,3つの皮疹,1カ月経過多)であった。
以下の図5に示すように、最初に2018年2月に開始された第1段階試験は、小児科患者の最大耐容量を決定することを目的としている。この試験のA部分は,単一療法の初期用量として3+3で設計されたトレフラフィニ(DAY 101)を用いて増加した。280 mg/平方メートルの開始用量は、成人によって推奨される第2段階用量の80%、すなわち、身体表面積調整後に600 mgを週1回経口投与する第2段階用量である。この試験に参加した患者は2年間にわたる治療を受けた。この試験は2019年12月に修正され、適応性設計を採用し、用量制限毒性或いはDLTS或いはMTDが観察されるまで用量が増加し続けた。
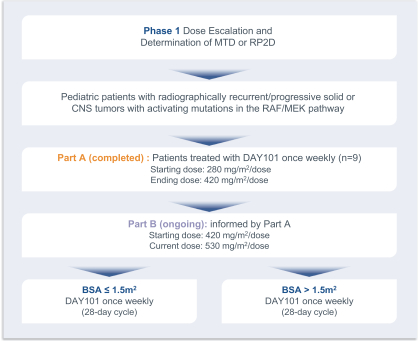
図5.pLGGにおけるtovorafenib(DAY 101)の1期実験設計
14
Tovorafenib(DAY 101)は研究者による第1段階試験(PNOC 014;NCT 03429803)で行われた研究であり、この試験では、Tovorafenibは経口速放錠として週1回、LGGsと他のRAS/RAF/MEK/ERK経路活性化腫瘍を有する再発/難治性腫瘍患者に用いられる。この研究A部分のデータは2020年11月に発表され、トレフラフィニは3つの異なる用量レベルで評価され、280 mg/m 2、350 mg/m 2、および420 mg/m 2であり、各用量レベルに3人の患者がいる。Tovorafenibは試験のすべての用量で耐性が良好であり、420 mg/m 2以下の用量を受けた患者は用量の減少または中断を有さなかった。これらの患者の中にDLTを経験した人は一人もいなかった。ほとんどの治療で発生する有害事象(TEAE)はレベル1またはレベル2である。眼科や心臓不良は観察されていない。A部分のすべての用量列の中で、最もよく見られるTEAEはすべて1級或いは2級であり、皮疹(89%)、髪灰白(無毛症)(78%)、色素母斑(78%)、貧血(67%)と掻痒(67%)を含む。1例の患者は単一の3級不良事件(クレアチンホスホキナーゼ上昇)が出現し、4級不良事件の報告がなかった。これらの副作用は可逆的で制御可能であることが発見された。
420 mg/平方メートルは最初はRP 2 Dと考えられていたが、A部分にすべての用量レベルの抗腫瘍活性が認められたため、本研究のB部分では引き続き用量が増加し、MTDの決定を試みた。この試験のB部分回復用量増加部分の後、用量増加は、身体表面積に応じて、530 mg/m 2以上の用量レベルで、所与の用量レベルで成人MTDを超える可能性があり、より小さい子供はそうではない可能性があることを考慮するために、2つのサブグループに分けられる。本研究B部分のデータは2022年11月に公表された。他に35名の患者がB部分PNOC 014:21名KIAA 1549:BRAF融合遺伝子を持つグリオーマ患者9名,BRAFV 600 E変異遺伝子を持つ腫瘍患者9名,新たなRAF遺伝子を持つ患者4名,FGFR 1遺伝子変異を有する腫瘍患者1例に参加した。組織学的行列は30個のLGG、4つの高レベルグリオーマと1つの軟組織肉腫を含む。6つのDLT:1個体当たり表面積またはBSA亜群が3個あり,いずれも530 mg/m 2/用量であり,いずれも3級,および5つの既知の副作用(2つの疲労,3つの皮疹,1カ月経過多)であった。この35名の患者のうち,トレフラフィニ(DAY 101)の毎週経口投与耐性は良好であった。Tite−Boin持続再評価モデルはBSA 1.5 m 2の患者に週530 mg/m 2またはPOの経口投与を推奨しているが,BSA患者がDLTを受ける可能性がある
全体的には,現在完成しているA部分からのデータであり,患者は2年間にわたる連続治療を受け,B部分のデータ支持を得ており,tovorafenib(DAY 101)の耐性曲線は420 mg/m 2であり,tovorafenib(DAY 101)の長期使用の可能性を支持している。
FDAが受けた脳腫瘍治療効果の標準客観的な測定標準はRANOと呼ばれる放射線学的測定標準である。Rano基準は、治療または疾患の進行に対する反応を追跡するために、腫瘍次元の様々な測定を考慮する。PNOC 014 A部分患者からのデータは,独立した神経放射線科医がRANO基準を用いて審査した。9名の患者のうち8名の患者はRAF融合pLGG(7名はKIAA 1549-BRAFを融合し、1名はSRGAP 3-CRAFを融合した)が存在し、1名の患者の神経線維腫症1(NF 1)遺伝子は機能喪失変異を起こした。以下の図6に示すように、RAF融合を受けた8名の患者のうち5名は、RANO基準に従って完全に緩和または部分的に緩和され、ベースラインと比較して50%低下すると定義され、すべての測定可能な強化病変の垂直直径積の合計は少なくとも4週間持続する。RAF融合を受けた8名中2名の患者の病態は長期的に安定していた。RAF融合患者1例はtovorafenibに対して反応しなかった(DAY 101)。NW 1関連pLGGを有する1人の患者は、tovorafenibに反応しなかった(DAY 101)。体積画像分析や最近発表された臨床的に検証されていないRAPNO基準,あるいは小児科神経腫瘍学的反応評価など,探索的イメージング法を用いた放射線学的反応は,RANOスコアとほぼ一致していると考えられる。
15
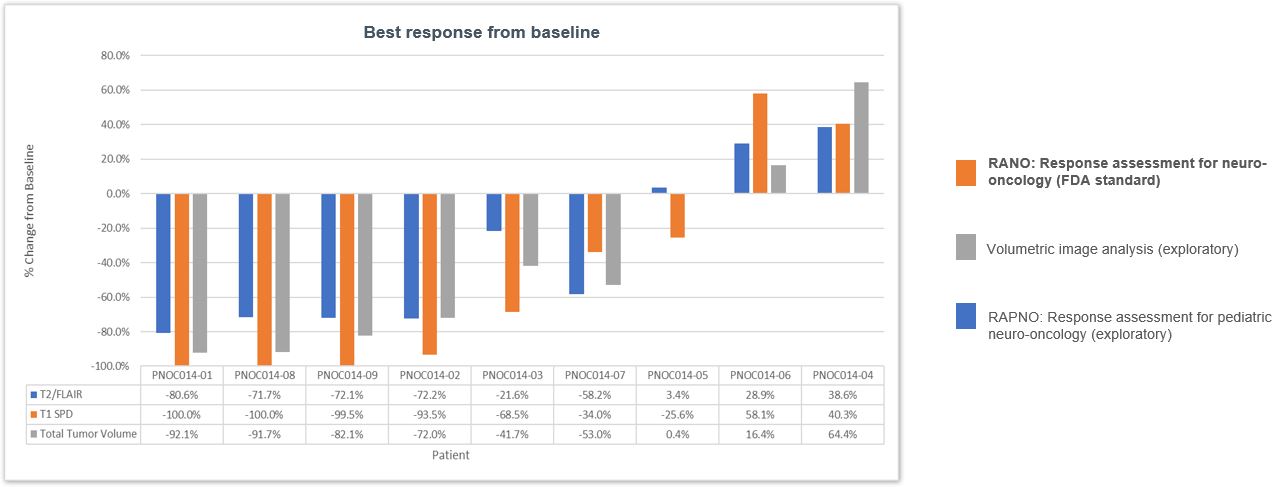
図6.pLGGのtovorafenib(DAY 101)の第1段階試験では、9名の患者のうち5名が完全(100%減少)または部分的に緩和した(腫瘍の2次元測定減少>50%)。
治療に対する反応を評価する以外に、スクリーニングと以前の放射線学画像からベースライン成長動力学を計算する過程において、すべての患者は目標皮膚損傷を識別した。9名中8名については,試験登録前に腫瘍成長歴が記録されていた。図7に示すように,トロフラフィニ(DAY 101)の服用開始後に得られた1枚目の放射線画像では,9名中6名に病変の大きさの縮小を認めた。中位有効時間は10.5週間であり,pLGGは不活性で成長の遅い腫瘍であるため注目すべき所見である。2名の患者は完全に緩和され,2年間の投与期間は不変であった。3名の患者は部分的に緩和し、2名の患者の病状は長期的に安定し、2名の患者は反応しなかった。この試験は最長2年間の治療を可能にする。
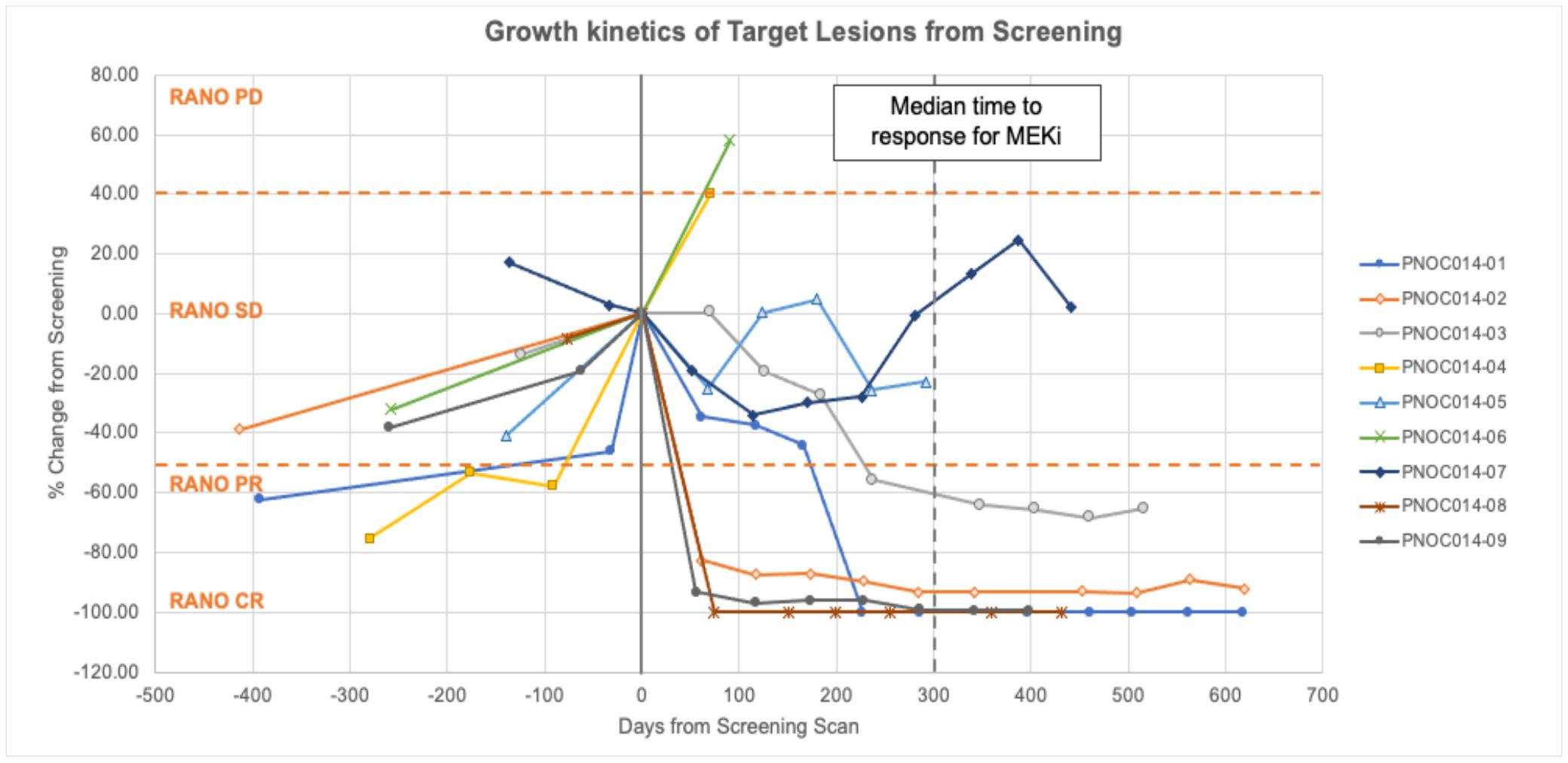
図7.pLGGにおけるtovorafenib(DAY 101)1期試験における患者の個体反応を示す。
PNOC 014 A部分の結果によれば、tovorafenib(DAY 101)は、活性化RAF変化が存在するpLGG小児患者の治療のための画期的な治療資格をFDAに付与されており、これらの患者は、システム的治療を必要とするか、または以前の治療後に病状が悪化するか、または好ましい代替治療選択を有さない。Tovorafenib(DAY 101)はFDA指定の悪性グリオーマ治療孤児薬も獲得している。
16
PLGGの臨床発展計画
著者らはすでに6ケ月から25歳までの再発或いは進行性pLGG患者においてtovorafenibの重要な2期Firefly-1試験(DAY 101)を開始し、これらの患者はV 600 Eのような活性化BRAF変化、例えばKIAA 1549-BRAF融合或いはBRAF活性化変異を有する。これはオープンラベルで世界的に登録されている単一アーム経口トロヴォラフェニ試験(DAY 101)であり,週1回投与され,420 mg/m 2である。患者は、放射線学的証拠が治療研究者によって決定されたRANO標準疾患の進行、許容できない毒性、患者の同意撤回または死亡を示すまで、tovorafenib(DAY 101)を服用し続けるであろう。最初の患者は2021年5月にFirefly−1の用量治療を受け,2022年5月にARM登録を完了した。Firefly-1試験はまた、(A)主要なキューが登録されているので、合格患者のアクセス範囲を拡大するための他の2つの研究分枝を含み、(B)6ヶ月~25歳までの再発または進行性頭蓋外固形腫瘍を有し、RAF融合を活性化する患者におけるtovorafenib(DAY 101)の初歩的な治療効果を評価する。この実験では,既存のセキュリティデータベースと組み合わせて,規制承認の基礎となるデータセットが生成されることが予想される.主な終点は総緩解率であると予想され,最適な総確認緩解率を有する患者の割合(RANO基準による完全緩解率と部分緩解率)と定義し,独立審査により決定した。副次的および探索的終点は、RAPNOおよび容量分析に基づく全体応答率、無イベント生存、安全性、機能結果、および生活の質測定を含む。
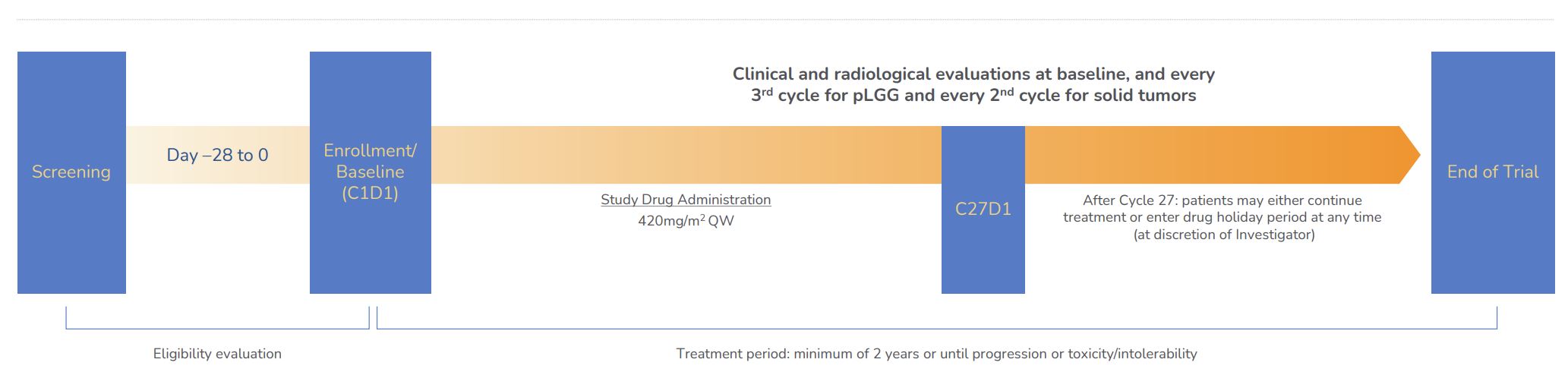
図8.pLGGにおけるtovorafenib(DAY 101)2期実験の設計
トヴォラフェニ(DAY 101)は現在速放錠として服用されている。私たちはすでに6ヶ月の子供の経口投与に適した小児科処方を開発し、私たちの重要な第2段階Firefly-1試験で患者にこの小児科処方を服用した。
CLIA/アメリカ病理学者学会或いはCAP、認可された病院実験室或いは第三者商業サプライヤーを利用して、新しい診断或いは再発/進展性pLGGに対して全面的なゲノムマップ分析を行うことは全米小児科神経腫瘍学プロジェクトの標準的なやり方である。また、2021年に改訂されたWHO中枢神経系腫瘍分類は現在、LGGs診断の一部としてBRAF変異或いは融合状態の評価を含む。したがって,新たに診断·再発したBRAF変化を伴うpLGG患者の多くが確認されることが予想される。個別の研究者が現在使用しているBRAF V 600 E突然変異とBRAF野生型融合を同定するための技術プラットフォームと解決方案は臨床試験登録標準を満たすために使用され、同時に著者らは引き続き監督機関と協力して、任意のセットの診断分析或いは設備の要求を満たすことを保証し、そのため、著者らはすでにFoundation Medicine,Inc.と協力して開発した
17
われわれはすでにtovorafenibのキー3期Firefly−2試験(DAY 101)を開始し,pLGGの第一線の治療とした。1人目の患者は2023年3月に薬物治療を受けた。患者が多輪毒性化学療法を受ける前に治療を行うことは,tovorafenib(DAY 101)の効果を高め,現在使用されている細胞毒剤の使用に関する全体的な治療負担や関連毒性を減少させる可能性が考えられる。
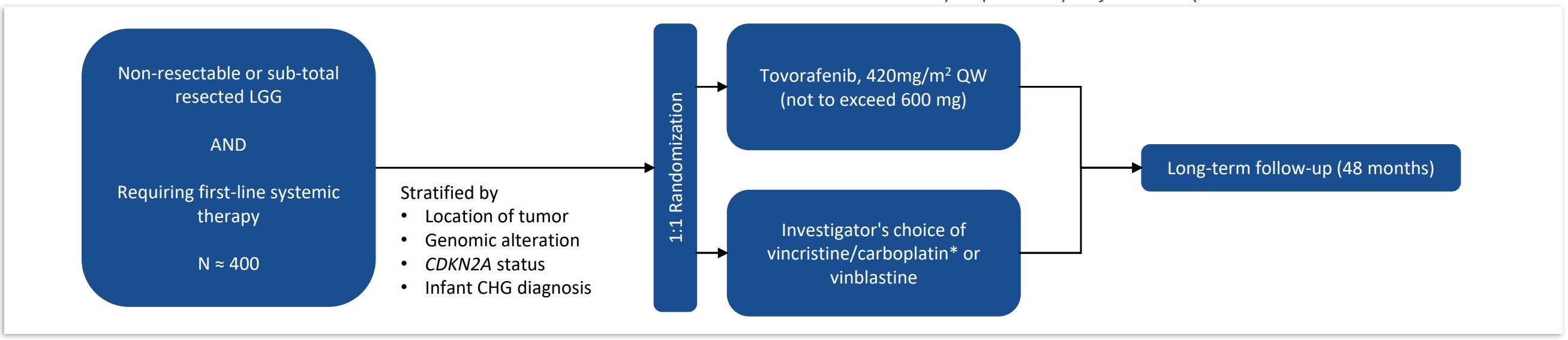
図9.pLGGにおけるtovorafenib(DAY 101)の3期実験設計
PLGGにおけるTovorafenib(DAY 101)の潜在的市場機会
脳腫瘍は児童に最もよく見られる固形腫瘍である。PLGGは最もよく見られる脳腫瘍であり、すべての児童脳腫瘍の約30%を占めるが、アメリカでは、pLGGの年間発病率は1.3‰から2.1‰と推定され、約2015年に新診断された1000から1600例を占める。このような疾患の発症率を考慮して、我々のチームは、この患者集団においてtovorafenib(DAY 101)を開発する市場機会を認識している
承認されればtovorafenib(DAY 101)はpLGG治療の標準的なケアになる可能性があると信じている。長期投与の需要により、長年を必要とする可能性があり、看護の標準は有効で、長期的な治療であるべきであり、同時に耐性概況を提供し、長期発病率と治療関連毒性を最低に下げる。Tovorafenib(DAY 101)は、より伝統的な慢性稀な疾患の有効な治療と類似したpLGG患者に長期的な利益を提供する可能性があると信じている。第一段階試験で観察されたtovorafenib(DAY 101)の概要から、tovorafenib(DAY 101)は迅速かつ持続的な抗腫瘍活性を招く高中枢神経系浸透率と良好な耐性を潜在的にバランスさせることができ、これらの小児科患者に深刻な不良事件がなく、及び毎週tovorafenib(DAY 101)を長期服用する2年間の臨床経験があることを表明した。私たちはまたtovorafenib(DAY 101)の週1回の経口投与レジメンが医者、患者、そして彼らの両親を引き付けると信じている。
18
他のMAPK駆動腫瘍におけるTovorafenib(DAY 101)の潜在的応用
小児科患者における著者らの初歩的な臨床開発を拡大するために,青少年や成人患者群におけるtovorafenib(DAY 101)の他の適応を探索する予定であり,これらの患者において,様々なMAPK経路の変化が疾患の推進に重要な役割を果たしていると考えられる。武田以前に行った2つの独立した1期試験では,225名を超える成人患者がtovorafenib(DAY 101)を服用しているデータが支持されている。これらの試験はまた、進行中のpLGG肝心な段階2試験を含む未来の臨床試験に開始用量レベルと毎週用量方案を提供する。これらの試験の結果,tovorafenib(DAY 101)は進行癌患者では耐性が良く,単独でも他の抗癌剤との併用も示唆されたが,tovorafenib(DAY 101)の単一療法に反応すると予想されるRAF変化が豊富であったか,あるいは抗腫瘍活性をきたす可能性が高いことが知られている併用研究が行われていなかったため,軽微な奏効率しか認められなかった。
臨床前研究のデータに基づき,武田率いる第一段階試験の初期データに基づいて,オープンタグ,マルチセンター,1 b/2 a期Firelight−1主試験のtovorafenib単一療法または併用療法を開始し,2つのサブ研究を含む。1回目の研究はトヴォラフェニ(DAY 101)の第2段階臨床試験であり、12歳以上のRAF変化の腫瘍患者の単一治療に用いられ、1人目の患者は2021年11月に治療を受けた。第二の研究はtovorafenib(DAY 101)とpimasertibの1 b/2期連合試験であり、12歳以上の各種MAPK変化を有する固形腫瘍の患者に用いられ、1人目の患者は2022年5月に治療を受けた。臨床前モデルでは,RAFとMEKの同時抑制は協同抗腫瘍活性を引き起こすことが証明されている。この組み合わせは、NRAS変異黒色腫と肺癌、クラスII BRAF変化によって駆動される腫瘍、およびKRAS変化によって駆動される腫瘍を含むMAPK変化によって駆動される様々な成人固形腫瘍において増強された抗腫瘍活性を示す可能性がある。
将来的には,tovorafenib(DAY 101)と他のMAPKシグナル経路のキーノードの選択的阻害剤との併用を探索する可能性がある。例えば、II型RAF阻害剤は、ERKまたはSHP 2阻害剤と組み合わせて使用することができ、相乗的利益を提供することができる。Tovorafenib(DAY 101)は、承認されたI型RAF阻害剤によって観察される矛盾活性化の傾向を誘発することなく、野生型RAFおよびRAFダイマーを含む複数の形態のRAF遺伝子変化を抑制し、癌MAPKシグナル伝達を抑制するための併用治療の潜在的バックボーンとしてのイメージを増強することができると信じている。
研究者によるトロヴォラファニ実験(DAY 101)
著者らは学術研究者および小児科腫瘍学協力団体と連合との関係を利用して、他の稀な小児科腫瘍タイプにおけるtovorafenib(DAY 101)の潜在力を探索するつもりである。
再発性ランゲルハンス細胞組織細胞増殖症の2期試験は2022年3月に児童腫瘍学グループによって開始され,国立癌研究所が支援する臨床試験グループであり,世界最大の小児と青少年癌研究に特化した組織でもある。この研究は単腕非ランダム試験であり、28名の参加者が参加すると推定され、彼らはボルラフィニ(DAY 101)の治療を受け、再発或いは難治性ランゲルハンス細胞組織細胞増加症の児童と若年者の総有効率を決定する。
太平洋小児科神経腫瘍学連盟は2022年7月に頭蓋咽頭腫の第二段階試験を開始した。この研究はランダム多群試験であり、56名の患者が参加すると推定され、彼らはnivalumbとtovorafenib(DAY 101)とnivalumbの連合治療を受け、PD-1(Nivolumab)と汎RAF-Kinase(Tovorafenib)阻害剤の連合治療を受けて児童と若者の頭蓋咽頭腫の耐性と有効性を評価する。
19
ピマチニブ
Pimasertibは経口的な高選択性マイトジェン活性化プロテインキナーゼ1および2(MEK)のアロステリック小分子阻害剤である。すでに発表された臨床前研究から,Pimasertibは他のMEK阻害剤と比較して高い中枢神経系浸透率を有することが示唆された。著者らは2021年2月にドイツDarmstadtのMerck KGaAからpimasertibの独占許可を得て、2022年3月にMAPK改変腫瘍に対する1 b/2期傘式主試験を開始し、tovorafenib(DAY 101)とpimasertibの12歳及び以上の患者における潜在的有益な組み合わせを研究する。ドイツダムシュタットのメルクKGaA社はこれまで第二段階で広範な非臨床と臨床開発を行い,日本で行われた固形腫瘍試験やpimasertibを他の薬物と組み合わせた。前向きな無作為第二段階試験では、Pimasertibはダカバヒドラジンと比較して、NRAS変異黒色腫患者において、客観応答率と無進展生存率の改善を含む単一療法の臨床活性を示したが、総生存率の改善ではなかった。Pimasertibの臨床開発過程で観察される主要な有害事象は,他のアロステリックMEK阻害剤と同様に典型的であり,胃腸に関連する有害事象,CPK上昇,皮疹,視力障害を含む。
臨床前研究
MEKはMAPK経路中のRAS下流に位置する重要なシグナル伝達ノードであり、1種の独特な二重特異性蛋白加水分解酵素であり、セリン/トレオニンとチロシン残基をリン酸化することができる。MEKは2つのサブタイプMEK 1とMEK 2からなり,MEK 1とMEK 2はERK 1とERK 2を順次リン酸化する。活性化ERK 1/2は、細胞膜、細胞質、核中に位置する多くの基質(>160)によって一連の異なる細胞過程を制御している。その中の多くは転写因子であり、細胞の増殖、分化、生存、血管新生と遷移に重要な役割を果たしている
以下の図10に示すように、RAS或いはRAFシグナル上昇によって駆動される癌において、MEKを抑制することはRASの遮断を放出することができ、そしてRASを介したシグナルと経路活性化を増加させ、MEK抑制に対する細胞の敏感性を更に低下させることに役立つ。単一療法として、MEK阻害剤はRAS或いはRAFシグナルが上昇する臨床前腫瘍モデルにおいて限られた抗腫瘍活性を示した。Mekiに耐性を産生し増殖を継続する癌の多くは,MAPK経路の再活性化とその後ERKの再活性化によって実現される。ERKの再活性化はRAS、RAF、NF 1またはMEKなどのMAPK経路中のERK上流の分子を変化または変異させることによって実現できる。このような腫瘍モデルにおいてRASシグナルの過剰活性化を回避する1つの方法はRAF阻害剤とMEK阻害剤を結合し、2つの異なる結節でこの経路を抑制することであり、複数のグループは細胞と腫瘍モデルの成長を抑制する上で相乗効果があることを証明した。
20
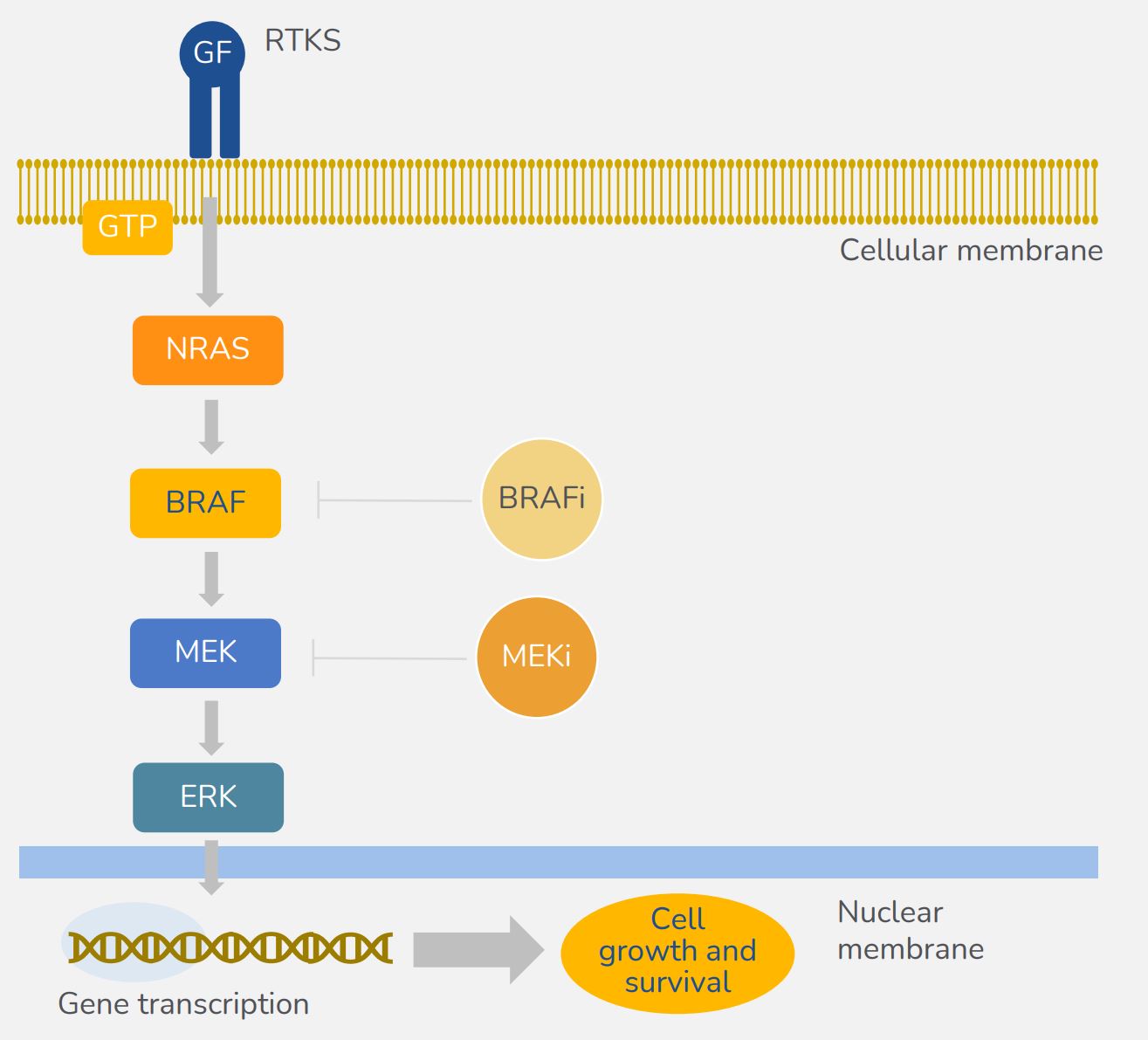
図10:BRAFとMEKの二重抑制はMAPK駆動の腫瘍を解決する重要な策略である。
[図11]RAF阻害剤感受性を誘導する提案機序モデル。左パネル:基礎条件下で、MAPK経路はRas-GTPレベルとRAF活性化を含む負方向に上流経路の活性化を調節するフィードバックループが複数あり、最適な経路信号を確保する。中板:MEK阻害剤治療を受けた後、これらのフィードバックループは無効にされ、RAS-GTP誘導、BRAF/CRAF二量体化、およびRAFキナーゼ活性化を引き起こす。右図:MEK阻害剤とII型RAF阻害剤の併用治療は相乗効果が期待できる。Yenらによって修正された。2018年の癌細胞です
21
この方法と一致したのは,臨床前実験では,II型RAF阻害剤とpimasertibの組み合わせが確実に協同細胞傷害活性を引き起こすことが示唆されたことである。CALU−6細胞はKRAS G 12 C変異を含むヒト肺腺癌細胞系であり,II型RAF阻害剤BGB−283やpimasertibの細胞傷害に感受性が認められた。これらの阻害剤の組み合わせによるCALU−6細胞の処理は、pimasertibの存在下で3ミクロン用量のBGB−283のEC 50が約60倍低下したため、より大きな細胞殺傷率をもたらすことができる。これらの結果は,RAF阻害剤の存在下でのMEK阻害剤の使用は,いずれの阻害剤単独よりも多くの細胞傷害利益が得られることを示している。
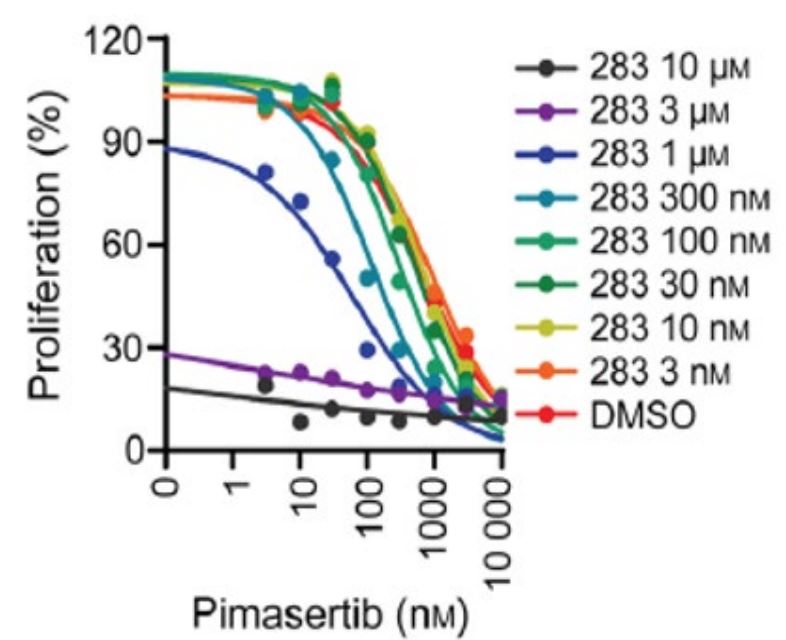
図12.Pimasertibに対するCALU-6細胞の感受性は、細胞をII型RAF阻害剤BGB-283で処理したときに増強される。
同様に,KRAS G 12 C変異を含むCALU−6細胞とNCI−H 1792細胞の実験では,細胞株はtovorafenib(DAY 101)やMeki−1に単一療法として敏感であることが証明されているが,これらの阻害剤の組み合わせはこれら2つの阻害剤単独よりも大きな細胞傷害作用を産生する。これらのデータは、MEK阻害剤を101日目のようなII型RAF阻害剤と組み合わせることが、特定のMAPK駆動成人固形腫瘍の治療戦略としての潜在的追加的利点をさらに支持する。
22
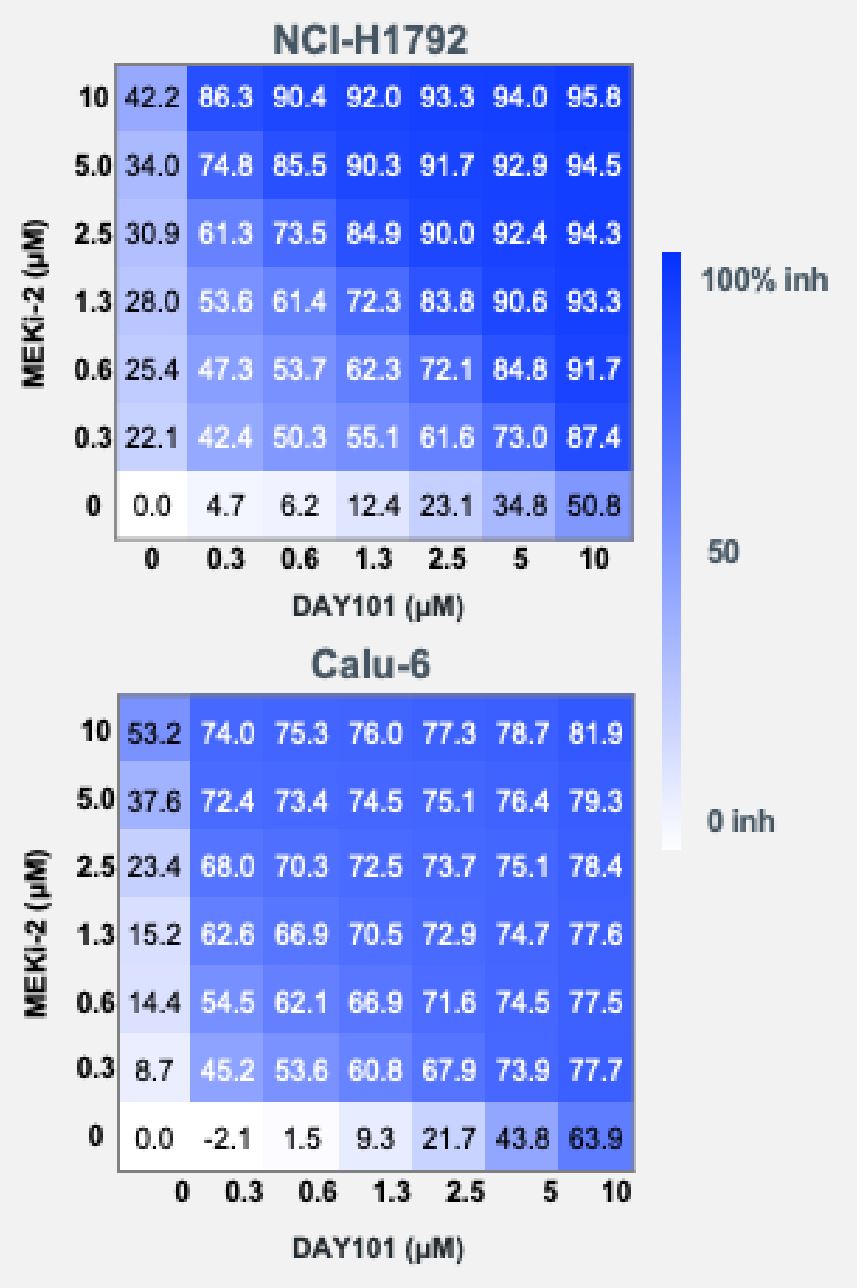
図13:インビトロKRAS G 12 CまたはQ 61変異腫瘍細胞株において、MEK阻害剤と併用した場合、tovorafenib(DAY 101)の相乗作用が観察された。
臨床結果
ドイツのメルクKGaA社が後援した試験では,Pimasertibは850人以上の癌患者に使用されており,単一療法としても標準看護療法の併用としても使用されており,ゲムシタビン,ダカバ津,結腸直腸癌レジメンFOLFIRI,および選定された研究薬(Hdm 2阻害剤SAR 405838,PI 3 K/mTOR阻害剤SAR 245409)である。これまでpimasertibとRAF阻害剤の併用の可能性は臨床試験で検討されていない。
23
Pimasertibの初期1期試験は、結腸直腸癌、黒色腫、前立腺癌、肺および中皮腫を含む様々な固形腫瘍患者における異なるスケジュールと漸増用量のPimasertib単一療法の使用を評価することを目的としている。この試験の主な目標は安全性と薬物動態学を確立し、更なる研究の最適な投与量とスケジュールを決定することである。初歩的な治療効果は腫瘍反応によって評価された。異なる腫瘍タイプではいくつかの安定型疾患の例が認められたが,黒色腫患者では様々な部分反応が認められ,このような腫瘍タイプのさらなる検討が引き起こされた。この試験の用量拡張アームでは、89人の黒色腫患者が、4つの用量レジメンにおいて28 mg~255 mg/日の薬理活性用量を受けた。OORは12.4%であり,その中の1例は完全に軽快し,10例は部分的に軽快し,46例は病状が安定していた。第一段階単一療法試験において、用量制限毒性は主に120 mg/日以上の用量が観察され、皮疹/ざ瘡皮膚炎と眼イベント、例えば漿液性網膜剥離を含む。最もよく見られる薬物関連不良事件は他のMEK阻害剤の治療効果と一致し、下痢、皮膚疾患、眼疾患、虚弱/疲労と末梢水腫を含む。単一療法第1段階臨床試験に参加した黒色腫患者の結果によると,69名中8人がレベル3以上のTEAEを経験していた。TEAEでは皮膚イベント4例,眼部イベント2例,下痢2例であった。すべての4種類の皮膚イベントは1日2回(R 3)群で発生したが,眼と下痢イベントは1日2回連続群と不連続群で報告されている。大量のpimasertibは網膜剥離と関連がある, このようなおよび他のすべての毒性は、支持的ケアおよび治療中断または用量の減少によって制御することができる。
ドイツのダムシュタットのメルクKGaA社はまた194名のNRAS突然変異の局部末期或いは転移性皮膚黒色腫患者の中で1種の多中心、開放ラベル、ランダム2期試験を行い、投与量60 mgの単剤pimasertibとダカルバジドを比較した。治療群の中位無進展生存期間(PFS)は13.0週であり、ダカバヒドラジン群の6.9週より有意に長かった。
臨床発展計画
我々はすでに開放ラベル、多中心、1 b/2 a期火光-1傘式主試験を開始し、この試験は2つのサブ研究:tovorafenib単一療法または併用療法を含む。1回目の研究はトヴォラフェニ(DAY 101)の第2段階臨床試験であり、12歳以上のRAF変化の腫瘍患者の単一治療に用いられ、1人目の患者は2021年11月に治療を受けた。第二の研究はtovorafenib(DAY 101)とpimasertibの1 b/2期連合試験であり、12歳以上のNRAS突然変異、BRAFwt融合と他のBRAF突然変異(V 600 EとV 600 K突然変異を除く)を有する患者に用いられた;第1の患者は2022年5月に治療を受けた。連合亜研究はtovorafenib(DAY 101)とpimasertibを組み合わせて使用する2期用量を決定するための1 b期用量範囲試験である。この用量が決定されると、患者は、NRAS変異またはBRAFwt融合などの遺伝的変異によって定義されるキューに、今回の試験に参加する組み合わせ用量拡張部分を登録する。連合亜研究1 b期部分の主な終点は安全性である。RAFwt融合とRAF 1増幅の単一治療サブ研究と第二段階拡張行列に対して、主要な終点は全体応答率と反応持続時間である。臨床前モデルでは,RAFとMEKの同時抑制は協同抗腫瘍活性を引き起こすことが証明されている。この組み合わせは、NRAS変異黒色腫および肺癌、クラスII BRAF変化によって駆動される腫瘍、BRAF野生型融合を有する腫瘍、およびKRAS変化によって駆動される腫瘍を含む、MAPK変化によって駆動される様々な成人固形腫瘍において増強された抗腫瘍活性を示す可能性がある。
24
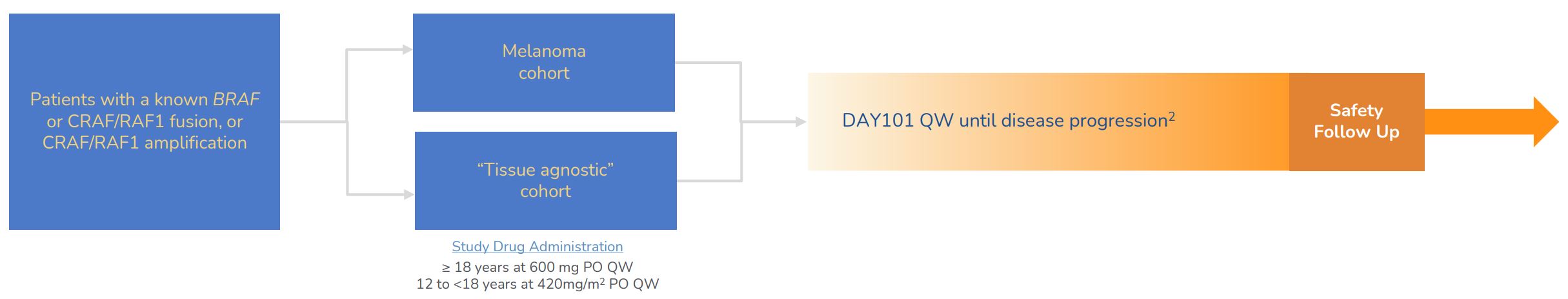
図14.傘形主試験-DAY 101-102(主レジメン)からボルラフィニ(DAY 101)とMAPK経路異常、サブ研究1単一療法(DAY 101-102 a)。
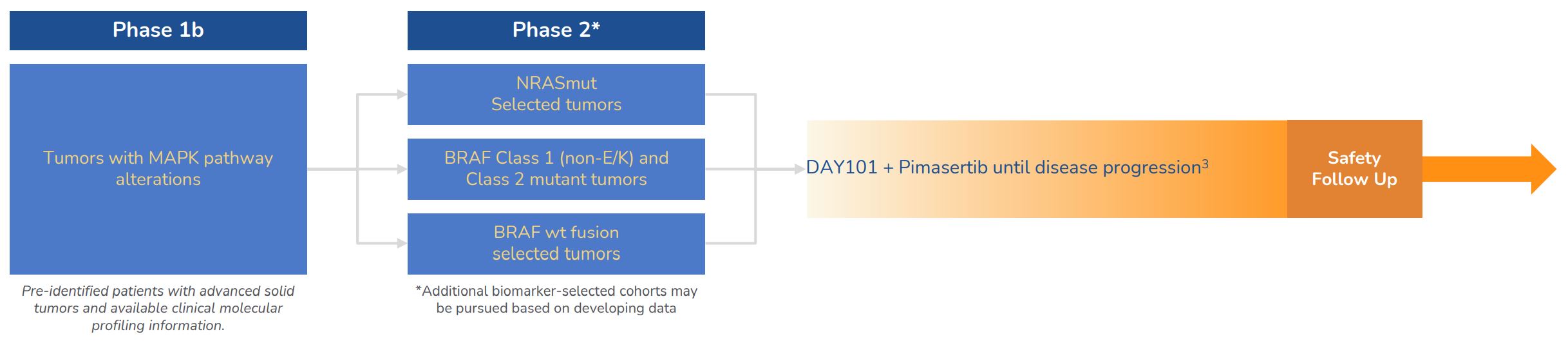
図15.傘形主試験-DAY 101-102(主レジメン)からボルラフィニ(DAY 101)とMAPK経路異常まで、2 MEK組み合わせ(DAY 101-102 B)に分けて検討した。
複数のグループからの臨床前データは、pimasertibとII型RAF阻害剤の組み合わせなどのMEK阻害剤が様々なMAPK駆動腫瘍環境において有益な活性を有することを示しており、これは、予期されるpimasertib濃度未満で単一治療によって産生されるデータから強力な細胞傷害活性が得られることを意味する。適切な用量のpimasertibとtovorafenib(DAY 101)の併用は,MTDでpimasertib単独で観察される有害事象の頻度や重症度を制限する可能性があり,これは生物活性用量の組み合わせを定義できるためと考えられる。成人患者で余分な投与量と安全性データを収集すると,小児科連合試験が探索されることが予想される。
製造業
私たちは所有したり経営したりしないし、現在は製造施設を設立する計画もない。私たちは依存し、引き続き第三者に依存して私たちの候補臨床試験製品を生産し、私たちの任意の候補製品が市場の承認を得た場合、商業製造に使用されることが予想される。私たちはまた依存して、私たちの研究候補製品、および私たちのビジネス製品を包装、マーク、記憶、配布する第三者に依存し続けることが予想されます(マーケティングの承認が得られれば)。この戦略は、私たち自身の製造施設、設備、人員に投資する必要がなくなり、より効率的なインフラを維持することができ、同時に、私たちの専門知識と資源を私たちの候補製品の開発に集中させることができると信じている。
これまで、私たちはそれぞれSTA Pharmtics Hong Kong Limited、Quantient Sciences-Philadelphia,LLC、Experic ServicesとFisher Clinic Servicesから活性薬物成分或いは原料薬、薬品製品と私たちの候補製品の包装/流通契約を獲得し、現在著者らはこれらの会社に単一源契約として組織或いはCMOを製造することに依存している。私たちは合意に基づいて、第三者CMOは私たちの発展需要に応じて、注文に応じて必要な数量の原料薬、薬品、包装/流通を提供する合意があります。Tovorafenibの商業供給について、私たちはこの製品の予想される商業需要を満たすために供給協定について交渉している。私たちが開発を通じて私たちの候補製品を推進することに伴い、私たちはすべての候補製品に原料薬、薬物製品、包装と調合の予備供給者を増加させ、任意の潜在的な供給中断を防止することを探索する。
私たちは一般的に私たちが開発可能などんなセットの診断プログラムを作るために第三者に依存することを望んでいる。
25
競争
製薬と生物技術業界の特徴は技術の迅速な進歩、競争の激しいことと独自製品に対する高度な重視である。私たちは私たちの技術、私たちのチームの専門知識、そして私たちの開発経験と科学知識が私たちに競争優位を提供してくれると信じていますが、私たちは製薬とバイオテクノロジー会社、学術機関、政府機関、そして公共と個人研究機関を含む多くの異なる源からの日々の激しい競争に直面しています。我々が開発·商業化に成功した候補製品は,既存療法や将来出現する可能性のある新しい療法と競争する可能性がある。
私たちの多くの競争相手は、単独でも彼らとのパートナーでも、私たちより多くの財務資源を持って、市場で足場を立ち、そして研究開発、製造、臨床前と臨床テストの方面で専門知識を持って、監督部門の許可を得て、そして精算とマーケティングの許可を得た製品です。これらの競争相手はまた、合格した科学と管理者を募集と維持し、臨床試験場と臨床試験の患者登録を確立し、そして私たちの計画と相補的或いは必要な技術を獲得する上で私たちと競争している。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。より多くの合併と買収は私たちの競争相手により多くの資源を集中させるかもしれない。
もし私たちの競争相手が私たちが開発する可能性のある製品よりも安全かつ効率的で、副作用が少なく、より便利で、より安価な製品を開発·商業化すれば、私たちのビジネス潜在力は減少または解消されるかもしれない。私たちの競争相手も私たちよりも早くFDAや他の規制機関からその製品の承認を得ることができるかもしれません。これは、私たちの競争相手が市場に入る前に強力な市場地位を確立することができ、そうでなければ、私たちの開発をより複雑にするかもしれません。私たちのすべてのプロジェクトの成功に影響を与える重要な競争要素は、有効性、安全性、患者の利便性である可能性があると信じている。
Tovorafenib(DAY 101)はFDAが承認したpLGG治療のための最初のPAN−RAF阻害剤となる可能性があると考えられ,どのような競争の候補製品が開発されているのか分からないからである。しかし、これはtovorafenib(DAY 101)が有効であることが証明されたか、または規制部門の承認を受けることを示していない。
3つのBRAF阻害剤は、V 600 EまたはV 600 K変異を含む腫瘍の治療のためにFDAによって承認されている。これらの第1世代BRAF阻害剤は、より一般的にI型RAF阻害剤と呼ばれ、遺伝子テークによってZelborafの名称で販売されているヴィモラファニーであり、ノワール社によって販売されているダプラファニー、およびBraftoviの名称で輝瑞社によって販売されている。ダプラファニブとトリメチニブを併用し、ノファ社はそれをMekinistと命名し、切除できないまたはBRAF V 600 E変異を有する転移性固形腫瘍を有する6歳の成人および小児患者の治療のために許可されており、これらの患者は以前の治療後に病状が進展し、満足できる代替治療方案がない。これは、tovorafenib(DAY 101)開発計画におけるより大きなRAF変更のpLGG臨床範囲のサブセット(約10%~20%)であるBRAF V 600 E pLGGを含む。
FDAは4種類のMEK阻害剤を承認した。このうち3つは、コビミチニブを含むBRAF V 600 EまたはV 600 K変異を含む腫瘍の治療のために許可されており、遺伝子テーク社によってCotelicの名前で販売されている;トリメチニブは、ノバ社によってMekinistの名前で販売されている;および比ニミチニブは、ファイザー社によってMektoviの名前で販売されている。第4の脳微小アンギオテンシン変換酵素阻害剤セルミチニブは、アスリコン社によってKoselugoと呼ばれ、1型神経線維腫を有する2歳以上の小児患者の治療に許可されており、これらの患者は、症状があり、手術できない叢状神経線維腫を有する。
Erascaは次世代BRAF阻害剤Naporafenib(LXH 2 54)を開発し,様々な薬剤と組み合わせて計画の第1段階と第3段階の臨床試験を行っている。百済神州には2つの次世代BRAF方案がある:リフェラファニー(BGB-283)とBGB-3245であり、前者は現在ミダミニと連合した1/2期試験中であり、後者は現在1期用量増加研究の単剤中にある。HanmiとGenentechはBelvarafenibとcobimetinibを併用して1 b期の臨床試験を開発している。Forre Treateutics社(Forelly NovellusDx)は,コビシタンと併用して1/2期試験を行うRAFダイマー切断剤PLX 8394を開発している。KinnateはKIN−2787を開発しており,単一療法の第1段階臨床試験にある。ブラックダイヤモンド治療会社が臨床前に開発した異なる段階には次世代BRAF阻害剤がある。
26
PLGGの治療については,ノ華社が協賛した無作為第二段階臨床試験で,新たに診断されたBRAF V 600変異pLGG患者を評価し,ダプラファニブとトリメチニブを併用している。ノワール社はすでに彼らの計画を発表し、2023年初めにこの試験のデータに基づいて補充新薬申請を提出する。さらに,いくつかのMEK阻害剤やいくつかのI型RAF阻害剤や他の標的治療が学術研究者による臨床試験で研究されており,一部の地域では非タグ方式で使用されている可能性がある。これらのエージェントは,トレフラフィニ(DAY 101)が市場に進出した際の競争を代表する可能性がある.
重要な合意
武田資産協定と千年証券取引所協定
2019年12月16日、DOT Treateutics-1,Inc.または当社の子会社DOT-1は、武田製薬株式会社の関連先および関連会社ミレニアム製薬会社と資産購入契約または武田資産協定を締結した。武田資産プロトコルによれば、DOT-1は、TAK-580に関連するいくつかの技術的権利および独自技術(現在、TOVORAFenib(DAY 101))を購入し、原発性脳腫瘍または固形腫瘍脳転移患者の治療に新しい方法を提供する。DOT−1はまた,われわれのこのようなRAF−阻害剤の研究·開発活動のための臨床在庫用品を受け取り,指定された調査者臨床試験プロトコルを得た。武田はViracta治療会社(F/k/a Sunesis PharmPharmticals,Inc.)やViractaの独占ライセンスプロトコルやViractaライセンスプロトコルとDOT-1にも割り当てている.武田はまた、武田資産協定に基づいてDOT−1に指定特許項下のグローバル再許可独占許可と、武田資産協定に基づいて生成された他の特許及び独自技術項下の非独占許可とを付与した。DOT-1はまた、武田資産協定で定義されている帰還許可を武田に付与し、武田資産協定が規定する適用時間範囲内で指定された開発マイルストーンを実現できなければ、この許可を自動的に終了またはDOT−1で終了することができる。この武田に付与された帰還許可証は転換時に終了し、ミレニアム証券取引所協定と関係がある。
DOT-1は、資産の売却と譲渡および武田資産協定によるライセンス付与の代償として、100万ドルの現金を前払いし、2019年12月にDOT-1で9,857,143株のAシリーズ償還可能転換優先株を発行した。他の投資家がDOT-1のAシリーズ融資で発行済み株式に支払った価格によると、発行済み株の公正価値は990万ドルと推定される。ミレニアム証券取引所協定の条項によると、武田は転換発効後、2021年5月26日にDOT-1の9,857,143株Aシリーズ償還可能転換可能優先株を我々の普通株6,470,382株に交換した
武田資産協定の有効期限は、各国のすべての譲渡特許権とすべてのライセンス特許権が満期になった後、国ごとに終了します。武田は我々が初めて商品を商業販売する前に武田資産協定を終了する可能性があり、一定期間連続して指定された時間内にいかなる開発活動も停止すれば、この停止は当事者の同意を得ておらず、規制当局の指導に応えるためでもない。また、武田は我々が破産した場合に武田資産協定を終了することができる。武田が我々の開発停止や破産により武田資産協定を終了すれば、譲渡されたすべての特許、ノウハウ、契約(Viractaライセンス契約を除く)が武田に譲渡され、武田は特許やノウハウに基づいて回復許可を得て、これ等で終了したすべての製品を利用する。
DOT-1は2021年12月31日から当社と合併して当社に組み込まれ、当社は既存の会社であり、武田資産購入協定に従ってDOT-1の義務を負う。
Viracta許可協定
2019年12月16日、DOT-1は、武田資産協定に従って譲渡されたViractaライセンス協定を修正し、再確認した。Viractaライセンスプロトコルによると,DOT−1は特定の特許権やノウハウにより世界的に独占的に許可されており,RAFタンパク質ファミリーに結合した化合物を含む製品を開発,使用,製造,商業化することができる。
27
DOT-1はViractaに現金200万ドルを前払いしており,このお金は2019年に研究開発費として記録されている.DOT−1は2021年2月にViractaに300万ドルの記念碑的支払いを支払い,2021年4月にマイルストーンを実現した際に研究開発費として記録された。DOT−1はまた,2つの適応の中で許可製品ごとに特定の開発と規制マイルストーンを実現する際に5400万ドルまでの追加マイルストーン支払いを要求されており,第2の適応は特定のマイルストーン事件を実現するために支払われるマイルストーンは第1の適応のマイルストーンよりも低い。また、DOT−1が許可製品に関する優先審査券を取得し、その優先審査券を第三者に販売するか、またはその優先審査券を使用する場合、DOT−1は、そのような販売のいずれかから受信したすべての純対価格または使用済み優先審査券の価値の特定のパーセンテージをViractaに支払う義務がある。DOT−1は,特許製品の1カ国/地域での初商業販売から,特許製品の純売上高の中央値から1桁パーセントまでの分級印税(あれば)の支払いを義務付けている。印税の支払い義務は、国/地域およびライセンス製品のライセンス製品をもとに、1つの国/地域での最初の商業販売から、(I)我が国/地域でその製品を使用または販売することをカバーするViractaライセンス特許、共通所有の協力特許または指定特許の最後の有効権利主張が満了したとき、(Ii)当該製品とその国/地域での最後の法定独占経営権が満了したとき、または(Iii)当該製品が当該国/地域で初めて商業販売されて10周年になるまで続く。以上のようにしない限り、他のマイルストーンはない, 達成され、2022年12月31日に満了される。
Viractaライセンス契約の期限は,このような製品の当該国/地域での印税に関する義務期間が満了した後,ライセンス製品ごとに国/地域ごとに終了する.DOT-1は、指定された通知期間内に任意またはすべてのライセンス製品に関連するViractaライセンスプロトコルを任意に終了する権利がある。
DOT-1は2021年12月31日からわが社と合併して当社に合併し、わが社は生き残った会社であり、Viracta許可協定に従ってDOT-1の義務を負います。
メルクKGaA社とライセンス契約を結び、ドイツのダムシュタット
2021年2月10日、DOT Treateutics-2,Inc.または我々の子会社DOT-2は、ドイツのダムシュタットに位置するメルクKGaA製薬会社と、MRKDG許可協定であるライセンス契約を締結した。MRKDGライセンス協定によると、ドイツのダムシュタットのMerck KGaAは独占的なグローバルライセンスを付与し、特定の特許権利と技術ノウハウに基づいて、複数のレベルで再許可を付与する権利があり、PimasertibとMSC 2015103 Bの化合物を含む製品を研究、開発、製造し、商業化することができる。私たちはまた、私たちの研究と開発活動のための臨床在庫用品を受け取った。著者らの独占許可はメルクKGaAが付与した非独占許可の制約を受けなければならず、メルクKGaAはドイツの癌研究組織の付属会社であり、ドイツDarmstadtメルクKGaAはいくつかのpimasertibに関連する臨床研究を直接或いは間接的に行う権利を保持する。
MRKDGライセンス協定によると、2029年までに商業的に合理的な努力を使用し、少なくとも2つの指定された主要市場国で少なくとも2つのライセンス製品を開発して商業化する義務がある。
MRKDG許可協定により付与された権利や臨床用品を考慮すると,800万ドルが前払いされており,将来的には代替用途がなく,研究活動に用いられているため,研究開発費として記録されている。また,250万ドルの記念碑的支払いを行い,ライセンス契約の性質と,2022年12月31日までの1年間の初臨床試験における患者初投与に関するマイルストーン事件により,研究開発費として記録された。特定の開発、法規、ビジネスマイルストーンの実現状況、および将来のライセンス製品の純売上高の高ビット数印税パーセント(ある場合)に応じて、3億645億ドルの費用を追加的に支払うことも要求される可能性があります。マイルストーンと特許使用料は未来の事件に依存し、マイルストーンと満期支払いが達成されたときに記録される。
MRKDGライセンス契約の期限は,当該国/地域内の当該ライセンス製品についてライセンス側への印税支払い義務期間が満了した後,個々のライセンス製品と国/地域に基づいて満期となり,すべてのライセンス製品とMRKDGライセンス契約下のすべての国/地域に対する全支払義務期間が満了する。
28
2021年12月31日からDOT−2はわが社と合併し,わが社は生き残った会社であり,MRKDG許可協定によりDOT−2の義務を担っている。
知的財産権
私たちのビジネスの成功は、私たちの候補薬物、技術およびノウハウの独自または知的財産権保護を獲得し、維持する能力があるかどうか、他人の独自または知的財産権を侵害することなく運営できるかどうか、および他の人が私たちの独自または知的財産権を侵害することを防止することができるかどうかにある程度依存する。我々は,米国や米国以外の管轄地域で独自の技術,発明,改善,候補薬物に関する特許保護などの方法を求めて獲得することで,我々の独自および知的財産権の地位を保護することが予想され,これらは我々の業務の発展と実施に重要である。私たちはまた、ビジネス秘密、技術ノウハウ、商標、持続的な技術革新、許可機会に依存して、私たちの独自と知的財産権の地位を発展させ、維持しています。現在、私たちの特許の組み合わせは、私たちによって許可され、所有され、および/または共同所有されている発行された特許および係属中の特許出願を含む。
私たちは現在、私たちの候補薬剤に関連するかもしれない特許出願および発行特許を有し、共同所有し、様々な疾患(例えば、小児科癌)の治療におけるそれらの使用を継続していることが予想される。私たちの候補薬物については、物質の組成、使用方法、製造方法を含む多段階の特許保護が求められている。私たちはより多くの特許出願を通じて私たちの候補薬物と技術の特許保護を強化するつもりだ。
2023年1月1日現在、ドイツ·ダムシュタット·マクKGaAからの3つの独占ライセンス特許シリーズと、武田薬業株式会社からの非独占ライセンス内特許シリーズ1つの特許シリーズとを含む10個の特許シリーズからなる特許組み合わせを所有または共同で所有している。私たちが所有または共同所有する10個の特許シリーズは、物質の成分、薬物成分、合成方法、合成中間体、治療方法、および私たちの候補製品のうちの1つであるtovorafenib(DAY 101)またはpimasertibに関連する併用療法を含む特許出願および発行された特許を含む。武田薬業株式会社の非独占ライセンス特許シリーズは、我々の候補製品tovorafenib(DAY 101)を製造するために使用することができる触媒を含む。ドイツのダムシュタットからのメルクKGaAの3つの独占ライセンス特許シリーズは、私たちの候補製品pimasertibの物質組成と使用方法をカバーしている。私たちがここで議論しているすべての、共同所有または許可された特許の特許条項は、利用可能な特許期間延長を含まない。
2023年1月1日現在、我々が所有または共同所有する特許の組み合わせには、tovorafenib物質の組成および使用方法に対する共通所有特許シリーズ(DAY 101)が含まれており、ドイツ、フランス、イギリス、ベルギー、スイス、デンマーク、スペイン、アイルランド、イタリア、オランダ、オーストラリア、ブラジル、カナダ、中国、インド、日本、韓国、メキシコ、シンガポール、南アフリカ、台湾、香港のライセンス特許を含む4つの発行された米国特許と複数の外国特許とを有しており、これらの特許は2028年~2031年に満了する予定である。
29
私たちが所有または共同所有する特許の組み合わせは、ドイツ、フランス、イギリス、ベルギー、ブラジル、カナダ、スイス、スペイン、アイルランド、イタリア、ルクセンブルク、モナコ、日本、および中国のライセンス特許を含むtovorafenib(DAY 101)の医薬製剤に対する特許シリーズを含み、これらの特許は2035年に満了する予定である。私たちが所有または共同所有している特許の組み合わせには、米国、ヨーロッパ、中国、および日本における係属中の出願を含むtovorafenib(DAY 101)製剤に対する追加の医薬製剤特許シリーズが含まれており、発表されれば2040年に満了する予定である。私たちが所有しているか、または共同所有している特許の組み合わせは、tovorafenib(DAY 101)のための米国仮出願の追加処方を含み、非一時的出願に変換されて発行される場合、2043年に満了する予定である。私たちが所有または共同所有している特許の組み合わせは、合成tovorafenib方法のための特許シリーズ(DAY 101)をさらに含み、発表された米国特許と、オーストラリア、ユーラシア大陸、イスラエル、日本、メキシコの許可された特許とを含み、2038年に満了する予定であり、米国とヨーロッパでの係属中の出願が発表された場合、2038年に満了する予定である。私たちが所有または共同所有する特許の組み合わせは、tovorafenib(DAY 101)とドセタキセルを組み合わせて癌を治療する方法に対する特許シリーズと、2035年に満了する予定の中国、ドイツ、フランス、イギリス、ベルギー、スイス、スペイン、アイルランド、イタリア、ルクセンブルク、およびモナコを含む複数の外国特許をさらに含む。我々が所有または共同所有する特許の組み合わせは、pLGGを治療する方法のための係属中のPCT出願を有する特許シリーズを含み、国有化されて発表される場合, 2041年に満期になる予定です。私たちが所有しているか、または共同所有している特許の組み合わせは、tovorafenib(DAY 101)とpimasertibのようなMEK阻害剤(例えば、pimasertib)とを組み合わせて癌を治療する方法を含むPCT出願が未解決である特許シリーズを含み、それを国有化して発表すれば、2042年に満了する予定である。私たちが所有しているか、または共同所有している特許の組み合わせは、tovorafenib(DAY 101)治療を受ける患者を選択する方法に関する係属中のPCT出願を有する特許シリーズを含み、それが国有化されて発表されれば、2042年に満了すると予想される。私たちが所有しているか、または共同所有している特許の組み合わせは、pimasertibのようなMEK阻害剤に対する米国仮出願の治療方法のうちの1つをさらに含み、非一時的出願に変換されて発表される場合、2043年に満了する予定である。
2023年1月1日現在、私たちの特許組み合わせは、物質の組成およびpimasertibの使用方法をカバーするドイツのダムシュタットメルクKGaAによって独占的に許可された特許シリーズを含む。この特許シリーズは、アルゼンチン、オーストリア、オーストラリア、ベルギー、ブルガリア、ブラジル、カナダ、スイス、中国、キプロス、チェコ共和国、ドイツ、デンマーク、ユーラシア大陸、エストニア、スペイン、フィンランド、フランス、ギリシャ、香港、ハンガリー、アイルランド、イスラエル、インド、アイスランド、イタリア、日本、韓国、リトアニア、ルクセンブルク、ラトビア、モナコ、メキシコ、オランダ、4つの許可された米国特許および/または複数の外国特許および/または出願を含む。ノルウェー、ポーランド、ポルトガル、ルーマニア、ロシア連邦、スウェーデン、シンガポール、スロベニア、スロバキア、トルコ、ウクライナ、南アフリカは、2025年から2028年までの期間が満了する予定だ。私たちの特許の組み合わせは、固体形態のpimasertibを対象としたドイツDarmstadtのMerck KGaAによって独占的に許可された特許シリーズを含む。この特許シリーズは、オーストリア、オーストラリア、ベルギー、カナダ、スイス、チェコ共和国、ドイツ、デンマーク、ユーラシア大陸、スペイン、フランス、イギリス、イタリア、日本、ルクセンブルク、メキシコ、オランダ、ポーランド、ポルトガル、ロシア連邦、スウェーデン、シンガポール、台湾、南アフリカでのライセンス特許を含むライセンスされた米国特許および/または複数の外国特許および/または出願を含み、これらの特許は2033年に満了する予定である。私たちの特許組み合わせは、MSC 2015103 Bの物質組成と使用方法を含むドイツDarmstadtのMerck KGaAによって独占的に許可された特許シリーズと、アルゼンチン、オーストリア、オーストラリア、ベルギー、ブラジル、カナダ、スイス、中国、チェコ共和国、ドイツ、デンマーク、ユーラシア大陸、エストニア、スペイン、フィンランド、フランス、香港、クロアチア、ハンガリー、アイルランド、イスラエル、インド、アイスランド、イタリアを含む2つの発行された米国特許および複数の外国特許および/または出願をさらに含む。韓国、リトアニア、ルクセンブルク、ラトビア、北マケドニア, マルタ、メキシコ、オランダ、ノルウェー、ニュージーランド、フィリピン、ポーランド、ポルトガル、ルーマニア、ロシア連邦、スウェーデン、シンガポール、スロベニア、スロバキア、トルコ、ウクライナ、南アフリカは、2029年に満了する予定だ。
30
個別特許の期限は,特許を付与した国の特許法的期限に依存する。私たちが出願したほとんどの国では、特許期間は通常、非臨時特許出願が提出された最初の日から20年である。米国では、場合によっては、特許期間を特許期限調整によって延長することができ、これは、特許権者が米国特許商標局(USPTO)の特許の審査および付与時の行政遅延による損失を補償するか、または1つの特許が共通所有特許または共通発明者と命名された特許によって最終的に放棄され、有効期限が早い場合には、特許期限が短縮される可能性がある。さらに、1984年の“医薬品価格競争および特許期限回復法”または“ハッジ·ワックスマン法案”は、薬剤の特許有効期間中に規制審査を受ける時間長の部分補償として、米国特許満了後に最大5年間の特許期間の延長を可能にしている。特許期間の延長の長さは,薬物が規制審査を受ける時間の長さと関係がある。1つの特許の残り期間は、製品が承認された日から14年を超えてはならず、各規制審査期間は、1つの特許のみを延長することができ、承認された薬物、その使用方法、またはその製造方法に関する請求項を延長することしかできない。
EUおよび他のいくつかの外国司法管轄区域にも同様の規定があり、承認された薬物をカバーする特許の期限を延長する。将来、私たちの候補薬がFDAや外国規制機関の承認を得たら、これらの製品をカバーする発行された特許の期間(あれば)を延長することを申請する予定です。しかし,米国FDAを含む適用当局が,このような延期を承認すべきかどうか,承認された場合のこのような延期の期限の評価に同意する保証はない.我々の知的財産権に関連するリスクに関するより多くの情報は、“リスク要因-私たちの知的財産権に関連するリスク”というタイトルの部分を参照してください。以上言及した満期日は,我々が入手可能な潜在的特許期間の延長や他の市場排他性とは無関係である。
私たちのような生物製薬会社の特許地位は通常不確定であり、複雑な法律、科学、事実の問題に関連している。私たちのビジネス成功はまた第三者の独占権を侵害しないことにある程度依存するだろう。いかなる第三者特許を発行することが、私たちの開発または商業戦略を変更し、私たちの薬物やプロセスを変更し、許可証を取得したり、いくつかの活動を停止することを要求するかどうかはまだ確定されていません。私たちは、任意のライセンス契約に違反したり、将来の製品を開発または商業化するために必要な独自の権利の許可を得ることができず、私たちに実質的な悪影響を及ぼす可能性があります。もし第三者が米国で準備して提出した特許出願も権利の技術を要求する場合、私たちは発明の優先権を決定するために、米国特許商標局の干渉または派生プログラムに参加しなければならないかもしれない。より多くの情報については、“リスク要因-私たちの知的財産権に関連するリスク”というタイトルの節を参照してください
特許保護に加えて、私たちは、ビジネス秘密、技術的ノウハウ、商標、その他の独自の情報、および持続的な技術革新に依存して、私たちの競争地位を発展させ、維持しています。私たちの商標組合は現在、米国初日、初日バイオ製薬および抗癌薬開発の登録申請および/または登録を含んでいる。私たちは、私たちの業務において特許保護から保護されているか、または特許保護に適していないと考えられる側面を保護し、保護し、独自の情報の機密性を維持することを求めている。私たちは、当社の従業員やコンサルタントと契約を締結することを含む、当社の独自情報およびビジネス秘密を保護する措置をとっていますが、第三者は、実質的に同じ独自の情報および技術を独立して開発したり、他の方法で私たちのビジネス秘密を取得したり、当社の技術を開示したりすることができます。したがって、私たちは私たちの商業秘密を意味的に保護することができないかもしれない。私たちの政策は、私たちの従業員、コンサルタント、外部科学協力者、協賛研究者、および他のコンサルタントに、私たちとの雇用や相談関係を開始する際に秘密協定を実行することを要求します。これらの合意は、個人と私たちとの関係中に開発または開示された当社の業務または財務に関するすべての機密情報は、特定の場合を除いて第三者に開示されてはならないことを規定している。私たちと従業員の合意はまた、従業員が私たちに雇われた過程で構想されたすべての発明または従業員が私たちの機密情報を使用することによって生成されたすべての発明が私たちの固有財産であることを規定している。しかしながら、このようなセキュリティプロトコルおよび発明譲渡プロトコルは違反される可能性がある, このようなどんな違反に対しても、私たちは十分な救済措置を持っていないかもしれない。我々の知的財産権に関連するリスクに関するより多くの情報は、“リスク要因-私たちの知的財産権に関連するリスク”を参照されたい
31
政府の監督管理
その他の事項以外に、アメリカ連邦、州と地方各級及びその他の国家と司法管轄区の政府当局は薬品の研究、開発、テスト、製造、品質管理、承認、包装、貯蔵、記録、ラベル、広告、販売促進、流通、マーケティング、承認後のモニタリングと報告及び輸出入などの方面に対して広範な監督管理を行った。米国や他の国や管轄区域で規制の承認を得る手続きや、その後適用される法規や条例、その他の規制当局の遵守には、多くの時間と財力が必要だ。
FDA承認プロセス
アメリカでは、“連邦食品、薬物と化粧品法”及びその他の連邦と州法規と法規に基づいて、薬品はFDAの広範な監督管理を受け、これらの法規と法規は薬品の研究、開発、テスト、製造、貯蔵、記録保存、承認、ラベル、販売促進とマーケティング、流通、承認後のモニタリングと報告、サンプリング及び輸出入などの方面に対して管理を行った。適用されない米国の要求を遵守しないことは、臨床封印、FDAが未解決のNDAの承認を拒否すること、警告または無タイトル手紙、製品リコール、製品差し押さえ、生産または流通の完全または部分的な一時停止、禁止、罰金、民事処罰、および刑事起訴のような様々な行政または司法制裁を受ける可能性がある。
米国では、新製品の医薬品開発または承認製品のいくつかの変更は、通常、臨床前実験室および動物試験に関連し、IND(臨床試験が開始される前に発効しなければならない)をFDAに提出し、FDA承認を求める各適応に対する薬剤の安全性および有効性を決定するために、十分かつ制御された臨床試験を含む。FDA上場前の審査要求を満たすには通常長年の時間を要し、実際の所要時間は製品或いは疾病のタイプ、複雑性と新規性によって大きく異なる可能性がある。
臨床前試験は製品の化学、調合と毒性に対する実験室評価、及び製品特性と潜在安全性と有効性を評価する動物試験を含む。臨床前試験の進行は必ず良好な実験室実践を含む連邦法規と要求に符合しなければならない。臨床前試験の結果はINDの一部として他の情報とともにFDAに提出され,製品化学,製造,制御に関する情報,提案された臨床試験案が含まれている。IND提出後,生殖毒性や発ガン性の動物試験など,長期的な臨床前試験を継続する可能性がある。ヒト臨床試験を開始する前に,各INDの提出後30日間の待機期間が求められている。FDAがこの30日間INDにコメントもINDにも疑問を提起しなければ,INDで提案された臨床試験が開始される可能性がある。臨床試験は合格した研究者の監督の下で、健康ボランティア或いは患者に研究用新薬を提供することに関連する。臨床試験は、(I)連邦法規に適合する;(Ii)良好な臨床実践またはGCPに適合し、患者の権利および健康を保護し、臨床試験発起人、管理者および監督者の役割を定義するための国際基準であり、(Iii)試験目標を詳細に説明し、安全性を監視するためのパラメータおよび評価すべき有効性基準のプロトコルである。米国患者のテストに関する各々のプログラムおよび後続のプログラム修正案は、INDの一部としてFDAに提出されなければならない。
FDAが臨床試験がFDAの要求に沿って行われていないと考えている場合,あるいは臨床試験患者に受け入れられないリスクとなっている場合,FDAはいつでも臨床試験の一時的または永久的な停止を命じたり,他の制裁を加えたりすることができる。臨床的保留はすべてであってもよく、部分的であってもよい。臨床試験における患者の研究案やインフォームドコンセント情報も機関審査委員会やIRB承認に提出しなければならない。IRBは完成するまで臨床試験も監視する。IRBはまた、IRBの要求を遵守できなかったために、現場の臨床試験を一時的または永久的に停止することを要求することができ、または他の条件を適用することができる。さらに、いくつかの臨床試験は、データ安全監視委員会または委員会と呼ばれる臨床試験スポンサーによって組織された独立した合格専門家グループによって監督される。このグループは,実験のあるデータへのアクセスにより,許可試験が指定されたチェックポイントで行えるかどうかを決定する.
32
ニューノミンの発売承認を支持する臨床試験は通常3つの連続段階で行われるが、これらの段階は重なる可能性がある。第1段階、すなわち、最初に健康なヒト対象または患者に薬物を導入した場合、新陳代謝、薬物動態、薬理作用、用量増加に関連する副作用、および可能であれば有効性を評価するための早期証拠を評価するために、薬物が試験される。第2段階は、一般に、特定の適応、用量耐性、および最適用量での薬剤の有効性を決定し、よく見られる副作用および安全リスクを決定するために、限られた患者集団で試験を行うことに関連する。薬剤が第2段階評価において有効性および許容可能な安全性を証明する場合、第3段階試験は、FDAが薬剤の全体的な利益-リスク関係を評価し、薬剤のラベルに十分な情報を提供することを可能にするために、より多くの患者の臨床治療効果および安全性に関する追加の情報を得るために実施される。多くの場合、FDAはこの薬物の治療効果を証明するために、十分かつ良好にコントロールされた2つの3期臨床試験を必要とする。ごく少数の場合、単一の試験で十分である可能性があり、(1)研究は大型多中心試験であり、内部一致性を表明し、統計学的に非常に説得力があり、死亡率、不可逆的な発病率或いは疾病の予防に臨床的意義のある影響を発見し、潜在的な深刻な結果を持っているが、第二回の試験で結果を確認することは実際に或いは倫理的に不可能であり、あるいは(2)他の確認性証拠と結合している。
これらの段階は、重ねたり、組み合わせたりすることができる。例えば、1/2期臨床試験は、用量増加段階および用量拡張段階を含むことができ、後者は、将来の臨床試験(例えば、従来の1期臨床試験)において推奨される拡張用量の耐性を確認することができ、選択された亜群における研究療法の抗腫瘍効果の洞察を提供することができる。通常,腫瘍学療法の開発過程では,第一段階臨床試験に参加したすべての被験者が疾患の影響を受けた患者であることから,非腫瘍学的療法の第一段階臨床試験よりも,このような試験期間中に臨床活動に関する情報がより多く収集される可能性がある。
深刻または生命に危険な疾患の2期または3期臨床試験を行う研究薬剤の製造業者は、例えば、そのウェブサイト上に掲示することによって、その評価およびアクセス拡大要求に応答するためのその政策を提供しなければならない。
必要な臨床試験が完了した後,NDAを用意してFDAに提出する。この製品が米国で発売される前に、FDAのNDAの承認を得る必要がある。NDAは、すべての臨床前、臨床および他の試験の結果、および製品の薬理、化学、製造および制御に関連するデータアセンブリを含まなければならない。
秘密協定を準備して提出する費用は巨大だ。新開発計画の提出の多くは高額な申請使用料を支払う必要があります。場合によっては、小企業が初めて出願した出願料を免除することを含む、費用を免除または減免することができる。また,孤児薬として指定された製品については,NDAに対して使用料を評価せず,非孤児適応も含まれていない限りである。承認された秘密保持協定によると、申請者は計画年会費を支払う必要がある。FDAは毎年ユーザー料金を調整し、費用は通常毎年増加する。
FDAは、NDAを提出するか否かを決定する前に、各提出されたNDAを審査し、これは、機関が実質的な審査を可能にするのに十分に完全であると判断したことに基づいて、FDAは、より多くの情報の提供を要求する可能性がある。FDAは、受信後60日以内にNDAを提出するか否かについて決定しなければならず、このような決定には、FDAが提出を拒否することが含まれる可能性がある。もし申請が提出されれば、FDAはNDAの深い検討を開始するだろう。FDAはNDAのレビューでいくつかのパフォーマンス目標を設定することに同意した。ほとんどの標準的な審査薬物製品の申請は10~12ヶ月以内に審査され、ほとんどの優先審査薬の申請は6~8ヶ月以内に審査される。優先審査は、FDAが治療において大きな進展を得るか、または適切な治療方法がない場合に治療を提供する薬剤を決定するのに適用することができる。FDAは、いくつかの遅延された情報を考慮するために、または提出中に提供された情報の情報を明確にするために、標準審査および優先審査の審査手続きをさらに3ヶ月延長することができる。FDAは、常に基準および優先NDAの目標日を達成するわけではなく、FDAは、より多くの情報を提供することを要求するか、または明確にすることができ、審査プロセスを延長することができる。
FDAはまた、新薬製品の申請、または安全性または有効性の問題の医薬製品の申請を外部諮問委員会に提出することができる−通常、臨床医および他の専門家を含むグループである−審査、評価を行い、申請を承認すべきかどうか、およびどのような条件下で提案するべきかについて提案することができる。FDAは諮問委員会の提案によって制限されていないが、それは一般的にそのような提案に従っている。
33
NDAを承認する前に、FDAは、それらがcGMP要件に適合しているかどうかを決定するために、新製品の製造施設を承認前に検査する。FDAは、製造プロセスおよび施設がcGMP要件に適合していることを決定し、要求された仕様の下で製品が一貫して生産されることを保証するのに十分でない限り、この製品を承認しないであろう。FDAは、一般に、GCP要件に適合し、安全性および有効性をサポートするデータの完全性を保証するために、1つまたは複数の臨床試験地点を検査する。
FDAがNDAおよび製造施設を評価した後、それは、承認書または完全な返信、すなわちCRLを発行する。CRLは、一般に、提出中の不足点を列挙し、FDAが、追加の臨床データ、追加の重要な臨床試験、および/または臨床試験、臨床前研究、または生産に関連する他の重要かつ時間的な要件のような申請を再検討するために、大量の追加の試験または情報を必要とする可能性がある。CRLを発行すれば,申請者は手紙で決定されたすべての不足点を解決するNDAを再提出し,申請を撤回し,正式な論争解決や聴聞機会を要求することができる.FDAは、含まれる情報の種類に応じて、2ヶ月または6ヶ月以内に再提出された出願を検討することを約束している。このようなデータや情報を提出しても,FDAはNDAが承認基準を満たしていないと認定する可能性がある。
NDAが再提出された場合、またはいつ、CRLで決定された欠陥がFDAによって満足的に解決された場合、FDAは承認書を発行する。この薬物の商業マーケティングを許可し、特定の適応の具体的な処方情報を提供する。NDA承認の条件として、FDAは、薬剤の利点が患者への潜在的リスクよりも大きいことを保証するために、リスク評価および緩和戦略、またはREMSを必要とする可能性がある。REMSは、薬物ガイドライン、医療専門家のコミュニケーション計画、および安全な使用を確保する要素、またはETASUを含むことができる。ETASUは、処方または調剤のための特殊トレーニングまたは認証、特定の場合にのみ調剤、特殊モニタリング、および患者登録簿の使用を含むことができるが、これらに限定されない。REMSに対する要求はこの薬物の潜在的な市場と収益力に重大な影響を与える可能性がある。さらに、製品承認には、薬物の安全性または有効性を監視するために、大量の承認後の試験および監視が必要となる可能性がある。承認されると、規制基準が守られていない場合、または最初のマーケティング後に問題が発見された場合、製品承認は撤回される可能性がある。
承認申請において確立されたいくつかの条件の変更は、適応、ラベルまたは製造プロセスまたは施設の変更を含み、変更を実施する前に、NDA付録を提出してFDAの承認を得る必要があり、場合によっては、新たなNDAを承認する必要がある。新適応のNDAサプリメントは通常,オリジナル申請と類似した臨床データが必要であり,FDAがNDAサプリメントを審査する際に使用するプログラムや行動は,NDAを審査する際に使用するプログラムや行動と同じである。
臨床試験情報の開示
FDAが監督する製品(薬品を含む)の臨床試験スポンサーはいくつかの臨床試験情報を登録し、開示しなければならない。製品、患者集団、調査段階、研究場所と研究者、および臨床試験の他の方面に関する情報は、その後、登録の一部として公開される。スポンサーも完成後に彼らの臨床試験結果を検討する義務がある。場合によっては、これらの裁判結果の開示は、裁判が完了した日から最大2年に延期されることができる。競争相手はこれらの公開された情報を用いて開発計画の進捗状況を知ることができる.
孤児薬
孤児医薬品法によれば、FDAは、米国では通常20万人未満、または米国では20万人を超える影響を与えるまれな疾患または疾患の治療のための薬剤を孤児薬として指定することができるが、このような疾患または疾患のための製品の開発および製造コストは、米国での製品の販売から回収されるという合理的な予想はない。
秘密保持協定を提出する前に、指定孤児薬を申請しなければならない。FDAが孤児薬物指定を承認した後、FDAは、薬物の識別情報およびその潜在的な孤児用途を開示する。孤児薬物の指定は、監督審査と承認過程においていかなる利点も伝達されず、規制審査と承認過程の持続時間を短縮することもない。
34
FDAによって承認された最初の特定の活性部分は、このような指定されたまれな疾患を有するNDA申請者を治療するために使用され、この適応に対して米国で7年間の独占営業期間を取得する権利がある。7年間の独占期間内に、FDAは、限定された場合、例えば、より効果的、より安全、または患者ケアに重大な貢献をすることによって、孤立した薬物に対する独占的製品に対する臨床的優位性を示すか、または薬品供給の問題の場合でなければ、同じ疾患のために同じ薬剤を販売する他のいかなる出願も承認しない可能性がある。孤児薬物排他性は、FDAが同じ疾患または状態に対する異なる薬物、または異なる疾患または状態に対する同じ薬物を承認することを阻止しない。孤児薬を指定する他の利点は、いくつかの研究に対する税金免除と、NDA使用料の免除を含む。
突破的治療指定
FDAはまた、重症または生命に危険な疾患または疾患を治療するための薬物承認申請の開発および検討を加速することを要求されており、初歩的な臨床証拠が示されている場合、薬剤は1つまたは複数の臨床的に重要な終点で既存の治療法よりも実質的に改善されている可能性がある。画期的療法計画によると,新候補製品のスポンサーは,候補製品のIND提出と同時にあるいはその後,特定適応の候補品を画期的療法として指定することをFDAに求めることができる。FDAはスポンサーの申請を受けてから60日以内に候補品が画期的な治療指定を受ける資格があるかどうかを決定しなければならない。FDAは突破的な治療法の面で何らかの行動をとる可能性があり、全体の開発過程中にスポンサーと会議を行い、適時に製品スポンサーに開発と承認に関する提案を提供し、より多くの高級者を審査過程に参加させ、審査チームのために1つの学際的なプロジェクト担当者を指定し、他のステップを取って効率的に臨床研究を設計することを含む。
承認を加速する
加速的に承認された製品は、深刻なまたは生命を脅かす疾患の治療に使用することができ、通常、既存の治療方法よりも患者に有意義な治療利点を提供する。許可を加速する資格のある製品は、臨床利益を合理的に予測する可能性の高い代替終点に基づいて、または不可逆的な発病率または死亡率よりも早く測定できる臨床終点に基づいて、不可逆的な発病率または死亡率または他の臨床利益への影響を合理的に予測することができ、病状の重症度、希少性または流行率、および代替治療を利用可能または不足していることを考慮することができる。加速承認経路は病気経過が比較的に長く、製品の期待される臨床利益を測定するために時間を延長する必要がある環境に最もよく用いられ、代用或いは中間臨床終点への影響は非常に速い。したがって、加速承認は、様々な癌を治療するための製品の開発および承認に使用されており、その中で、治療の目標は、通常、生存率を向上させること、または発症率を低下させることであり、典型的な病気経過の持続時間は、臨床または生存利益を証明するために長い、時には大規模な研究を必要とする。承認経路を加速するには、通常、この製品の臨床的利益を検証および説明するために、追加の承認後の検証的研究を行うことにスポンサーの同意が必要である。これらの検証試験は、職務調査の場合に完了しなければならず、多くの場合、FDAは、承認前に設計、起動、および/または完全に試験に組み込むことを要求する可能性がある。必要な承認後研究を行っていない、あるいは発売後の研究期間中に臨床利益が確認されていない, FDAがこの製品を市場からのリコールを加速させることを可能にするだろう。加速規制によって承認された候補製品のすべての販売促進材料はFDAの事前審査を経なければならない。
小児科情報
“小児科研究公平法”によると、新薬または新薬補充剤は、すべての関連小児科亜群で薬物が主張する適応の安全性と有効性を評価し、薬物に対して安全かつ有効な各小児科亜群の投与量および投与を支持するためのデータを含まなければならない。FDAは、提出されたデータの全部または一部を免除または延期することができる。法規が別途要求されない限り、PREAは、PREAが元のNDA中の新しい活性成分に適用される場合でなければ、孤児指定の適応として承認されたいかなる薬剤にも適用されず、PREAが成人癌を治療するための分子標的癌製品であり、FDAに対して小児癌の増殖または進行に実質的に関連する分子標的が決定された場合、この新しい活性成分は孤児によって指定される。
35
小児最適医薬品法案、またはBPCAは、いくつかの条件が満たされる場合、任意の薬物の排他的-特許または非特許-であるNDA保持者に6ヶ月の延長を提供する。排他的条件は、FDAが小児科集団における新薬の使用に関連する情報がその集団の健康に利益をもたらす可能性があることを決定すること、FDAが小児科研究の書面請求を提出すること、および申請者が法定時間内に要求された研究を行うことに同意し、報告することを含む。BPCA下の出願は優先出願とみなされ,与えられたすべての利点を指定することができる.
承認後に要求する
機密協定が承認されると、製品はいくつかの承認後に要求される制約を受けるだろう。例えば、FDAは薬品の承認後のマーケティングと販売促進を密接に監督し、消費者向けの広告、ラベル外販売促進、業界賛助の科学と教育活動及びインターネットに関連する販売促進活動の基準と法規を含む。薬品は承認された適応でのみ販売され,承認されたラベルと一致するように販売される。
FDAがNDAを承認した後,有害事象報告と定期報告を提出する必要がある。FDAはまた、承認製品の効果を監視するために、いわゆる第4段階試験、REMSおよび監督を要求することができ、またはFDAは、承認時に条件を付加して、その製品の流通または使用を制限する可能性がある。また、品質管理、薬品製造、包装とラベルプログラムは承認された後、現在の良好な生産実践或いはcGMPに引き続き適合しなければならない。医薬品製造業者と彼らのいくつかの下請け業者はFDAといくつかの州機関に彼らの工場を登録することを要求された。FDAの登録要求エンティティはFDAの定期抜き打ち検査を受け,その間,FDAは製造施設を検査し,cGMPの遵守状況を評価する。そのため,メーカーはcGMPの遵守を維持するために,生産や品質管理の分野で時間,お金,精力をかけ続けなければならない。ある企業が規制基準を遵守できなかった場合、初期マーケティング後に問題に遭遇した場合、または後に以前に意識されていなかった問題が発見された場合、監督管理機関は製品の承認を撤回したり、製品のリコールを要求したりすることができる。
まれな小児科疾患指定と優先審査証明書
まれな小児科疾患優先審査クーポン計画によれば、FDAは、承認された珍しい小児科疾患の治療又は予防製品のマーケティング申請のスポンサーに優先審査クーポン券を発行することができる。クーポンはスポンサーに後続のマーケティング申請を優先的に審査する権利を持たせる。許可された珍しい小児科疾患製品の申請だけがクーポン券を得ることができる。まれな小児科疾患製品出願は、深刻または生命に危険な疾患を治療または予防するための薬剤(小分子の場合)に対するNDAであり、このような疾患では、深刻または生命に危険な表現が主に出生から18歳までの人に影響を与える;通常、米国では20万人未満の影響を与えなければならない;NDAは資格優先審査があるとみなされなければならない;NDAは、異なる成人適応(すなわち、異なる疾患/状態に対する)の承認を求めてはならない;この薬剤は、FDAが以前に承認した活性成分を含んではならない;NDAは小児科群の研究で得られた臨床データに依存しなければならず,承認された製品が小児科群に十分にラベルを貼ることができるようにしなければならない。NDAが承認される前に,FDAは開発中の製品をまれな小児科疾患の製品に指定する可能性がある。
まれな小児科疾患優先審査証明書を得るためには,スポンサーはNDAを提出した後にFDAにその申請証明書の意図を通知しなければならない。FDAがNDAがまれな小児科疾患製品適用であると判断した場合、NDAが承認された場合、FDAはNDAの承認後にNDAのスポンサーにクーポン券を発行する。受賞した製品が製品が承認されてから365日以内に米国に発売されなければ、FDAは珍しい小児科疾患優先審査証明書を撤回する可能性がある。この証明書は、他のスポンサーに譲渡することができ、その後のセキュリティプロトコルまたはバイオ製品ライセンス申請(BLA)と共に提出することができ、所有者に付随するセキュリティプロトコルまたはBLAを優先的に審査する権利を持たせることができる。優先審査証明書を提出するスポンサーは、NDAまたはBLAを提出する前に、NDAまたはBLAと共に証明書を提出しようとしていることをFDAに少なくとも90日間通知しなければならず、任意の他の必要なユーザ費用に加えて、優先審査ユーザ料金を支払わなければならない。FDAは、NDAまたはBLAを受信してから6ヶ月以内に優先審査されたNDAまたはBLAに行動しなければならない。
2020年12月,この計画は再認可され,2024年9月30日までにまれな小児科疾患製品に指定されることが許可され,2026年9月30日までに合格したNDAの承認を得た後,まれな小児科疾患優先審査券を取得する資格がある。
36
“ハッジ·ワックスマン修正案”
オレンジ図書リスト
1984年の“医薬品価格競争および特許期限回復法”(一般にハッジ·ウェクスマン修正案と呼ばれる)によれば、NDA申請者は、その特許請求が出願人の薬物または承認された薬物使用方法の各特許をカバーすることをFDAに証明しなければならない。薬物を承認した後、出願人は、国家薬品監督管理局に提出された特許リストを直ちに更新しなければならず、次いで、薬物出願に列挙された各特許を、FDAによって承認された治療同等性評価を有する医薬製品において、一般にオレンジブックと呼ばれる。
逆に、オレンジブックに記載されている薬物は、簡略化された新薬申請、すなわちANDAの承認を支持するために、潜在的な模倣薬競争相手によって参照されることができる。ANDAは市販薬物と同じ有効成分、強度、投与経路と剤形を有することを規定し、生物学的同等性テストを通じて治療上上場薬物と同じ薬物製品のマーケティングを証明した。承認されたANDA製品は治療上列挙した薬剤と同様と考えられている。生物学的同等性テストの要求以外に、ANDA申請者は臨床前或いは臨床テスト或いは提出結果を行う必要がなく、その薬物製品の安全性或いは有効性を証明する。ANDA経路で承認された薬物は通常市販薬の“模倣等価物”と呼ばれ,通常薬剤師が各州の薬物代替に関する法律に基づいて原始的な市販薬の処方に基づいて代替することができる。
ANDA申請者はオレンジブックに記載されている参照薬として決定された任意の特許をFDAに証明しなければならない。具体的には、出願人は、(I)必要な特許情報を提出していないこと、(Ii)特許が満了していること、(Iii)に記載されている特許が満了していないが、特定の日に満了し、特許が満了した後に承認を求めること、または(Iv)に列挙された特許が無効であるか、または新製品の侵害を受けないかのうちの1つで各特許に証明しなければならない。新製品が承認された製品の上場特許又はそのような特許を侵害しない無効な認証を第4項認証と呼ぶ。承認された使用方法を要求する特許については、場合によっては、ANDA出願人は、列挙された使用方法特許を証明するのではなく、その提案されたANDAタグが特許使用方法に関するいかなる言語も含まれていないことを証明するために、第8節の宣言を提出することを選択することができる。出願人が第4段落の認証によって列挙された特許に疑問を提起していない場合、ANDA出願は、参照製品を要求するすべての特許が満了するまで承認されないであろう。ANDA出願人が第4項の認証をFDAに提供した場合、FDAがANDA届出を受けると、出願人はまた、NDA所持者及び特許権者に第4項の認証の通知(“通知状”と呼ぶ)を送信しなければならない。そして、国家特許庁及び特許所持者は、通知状に対して特許侵害訴訟を提起することができる。第四項の証明を受けてから45日以内に特許侵害訴訟を提起し、通知状を受け取った日から30ヶ月、すなわち特許が満了するまで、FDAがANDAを承認することを自動的に阻止する, 裁判所が和解命令又は同意法令に署名して入力した日は,第4項の認証対象となる特許が無効又は侵害されていないこと,又は特許事件においてANDA出願人に有利な裁決を宣言する。
ANDA出願もオレンジブックに記載されている引用製品の任意の適用の非特許排他性が満了するまで承認されないであろう。場合によっては、出願人がANDA処方情報からこのような排他的に保護された情報を省略することを求め、FDAが許可する場合、ANDA出願人は、いくつかの非特許排他性が満了する前に承認を得ることができる。
排他性
NDAが新しい化学物質またはNCEを承認すると、すなわち、FDAが任意の他のNDAで承認された活性部分を含まない薬剤は、5年間の市場排他性を得ることになり、その間、FDAは、その薬剤の模倣薬バージョンの承認を求めるANDAを受け取ることができず、申請が第4の段落の証明を含まない限り、この場合、出願は、NCE排他的有効期限が満了する1年前に提出することができる。Orange Bookに記載されている特許がなければ,第4段落の認証がない可能性があるため,排他期が満了するまでは,その薬剤の後発薬にANDAを申請してはならない。
37
承認薬のいくつかの変化は,新たな適応の承認,新たな強度の承認,新たな使用条件の承認など,承認日からの3年間の排他期に関連しており,その間,FDAはこのような変化を含む後発薬のANDAを承認することはできない。場合によっては、出願人がANDAパッケージ挿入においてこのような排他的に保護された情報を省略することを求め、FDAが許可する場合、ANDA出願人は、3年の排他的満了前に承認を得ることができる。
特許期間を延長する
Hatch Waxman修正案は,FDA規制審査過程で失われた特許期間の補償として特許期間の延長を許可している。しかし,特許期間の延長は,特許の残り期間を製品承認日から合計14年間延長することはできない.国家薬品監督管理局の承認後、関連薬物特許の所有者は延期を申請することができる。許容される特許期間延長は、薬物試験段階の半分(IND出願とNDA提出との間の時間)とすべての審査段階(NDA提出と承認との間の時間)として計算され、最長5年である。FDAが出願人が職務調査を行っていないと判断して承認を求めるいつでも、時間を短縮することができる。
米国特許商標局はFDAと協議し,任意の特許期間の延長または回復の出願を審査·承認する。しかしながら、米国特許商標局は、例えば、試験段階または監督審査中に職務調査を行うことができなかったため、適用された最終期限内に出願を提出することができなかったため、関連する特許の満了前に出願を提出することができなかったか、または適用の要件を満たすことができなかったため、承認延期を決定しない可能性がある。さらに、適用期間または提供される特許保護範囲は、要求されたものよりも短い可能性がある。
延期された特許の総期限は14年を超えてはならず、どの単一製品も1つの特許しか延期できない。特許延期出願は,特許が満了する前に提出されなければならず,出願段階で満了する可能性のある特許については,特許権者は一時特許延期を申請することができる。臨時特許の延期は特許期間を1年間延長し,最大4回延長することができる.暫定特許の承認が延期されるごとに、承認後の特許延期は1年減少する。米国特許商標局の取締役は、特許延期を求めている特許に含まれる薬物が承認される可能性が高いことを確認しなければならない。秘密保持協定を提出していない薬物は一時的な特許延期を受けることができない。
FDAの診断に伴う規定は
インビトロ診断の使用が医薬製品の安全かつ有効な使用に不可欠である場合、FDAは通常、FDAが医薬製品を承認しながらこの診断を承認または承認すること、いわゆる随伴診断を要求する。FDAは通常、薬物承認と同時に発売前の承認またはPMAの診断を得るために、癌治療に反応する患者を選択することを目的とした体外随伴診断を要求する。これらの体外随伴診断の審査は癌治療の審査とともにFDA薬物評価と研究センターおよびFDA装置と放射線健康センターの審査の協調に関連している。キット診断の承認と承認には,異なる会社であれば薬物メーカーと設備メーカーとの高度な協調も必要である。
臨床および臨床前データの収集、およびFDAおよびFDAへの審査の提出を含むPMAプロセスは、数年またはそれ以上を要するかもしれない。これは、厳格な上場前審査を含み、その間、出願人は、装置の安全性および有効性に関する合理的な保証と、装置設計、製造、およびラベルを含む装置およびその構成要素に関する情報とをFDAに準備して提供しなければならない。PMA申請は相当な申請料を支払う必要があり、通常毎年増加する。
さらに、PMASは、一般に、FDA承認を求める各適応に対する装置の安全性および有効性を決定するために、広範な前臨床試験および十分かつ良好に制御された臨床試験の結果を含まなければならない。特に、診断の場合、出願人は、診断が十分な感度および特異性を有し、十分な試料および試薬の安定性を有し、同じ試料が複数の実験室の複数のユーザによって複数回検出されたときに再現可能な結果を生成することを証明しなければならない。PMA審査の一部として、FDAは通常、メーカーの施設が品質体系法規(QSR)に適合しているかどうかを検査し、この法規は詳細なテスト、制御、文書、その他の品質保証要求を規定している。
38
PMAの承認は保証されず、FDAは最終的に申請中の欠陥に基づいてPMA提出を承認できない決定を下し、追加の臨床試験または他のデータを要求する可能性があり、これらのデータの生成は高価で時間がかかる可能性があり、承認を大幅に遅らせる可能性がある。PMA出願に対するFDAの評価が有利である場合、FDAは、一般に、PMAの最終承認を保証するために、ラベルの変更などの特定の条件に同意すること、または最終ラベルを提出するような特定の条件に同意することを要求する承認可能な書簡を発行する。FDAが適用基準を満たしていると結論すれば,FDAは承認された適応にPMAを発行し,申請者が最初に求めた適応よりも限られている可能性がある。PMAは、ラベル、販売促進、販売、および流通の制限を含むFDAが、装置の安全性および有効性を保証するために必要と考えられる承認後条件を含むことができる。
設備が市場に投入された後、それは依然として厳格な規制要求を受けている。医療機器の販売は,その許可や承認の用途や適応にしか利用できない。デバイス製造業者はまた、毎年の機関登録料を支払い、FDAにデバイスをリストすることを含む、彼らの機関を登録しなければならない。医療機器製造業者およびその供給者の製造プロセスは、医療機器の設計、テスト、生産、プロセス、制御、品質保証、ラベル、包装および輸送の方法および文書を含むQSRの適用部分に準拠しなければならない。米国食品医薬品局は定期的に国内工場の記録や製造プロセスを不定期に検査する。FDAはまた、米国に製品を輸出する外国施設を検査する可能性がある。
他の医療保険法
FDAの薬品マーケティングに対する制限以外に、他のいくつかのタイプの州と連邦法律を応用して製薬業界のいくつかの一般的な商業とマーケティングやり方を制限した。これらの法律には、リベート、虚偽声明、透明性、健康情報プライバシー法、および他の医療法律法規が含まれている。
他の事項に加えて、連邦反バックル法規は、Medicare、Medicaid、または他の連邦によって援助された医療計画に従って精算可能な任意の医療項目またはサービスを誘導または見返りとして購入、レンタル、注文または購入、レンタル、またはMedicaid、または他の連邦によって援助された医療計画に従って精算することを誘導または発注することを意図的に提供、支払い、請求または受け取ることを禁止する。“保健と教育和解法案”によって改正された“患者保護と平価医療法案”は、総称してACAと呼ばれ、連邦の“反リベート法規”の意図内容を改正し、個人或いは実体がこの法規或いはこの法規に違反する具体的な意図を実際に理解する必要がなく、違反を実施することができる。この法規は,薬品メーカーと処方者,購入者,処方管理人などの間の手配に適用されると解釈されている。いくつかの法定例外と規制避風港はいくつかの一般的な活動を起訴や他の規制制裁から保護しているが、例外と避風港の範囲は狭く、処方、購入または推薦を誘導するための報酬に関するやり方が例外や避風港の資格に適合していなければ、審査される可能性がある。また,ACAは連邦反リベート法規を改正し,この法規に違反した行為を連邦民事虚偽クレーム法案の規定の責任基盤とすることができるようにした。
連邦民事虚偽請求法案を含む連邦民事および刑事虚偽請求法は、任意の個人または実体が虚偽請求を意図的に連邦政府に提出するか、または虚偽請求を故意に行うか、または虚偽請求を支払うために虚偽陳述を引き起こすことを禁止する。これには,連邦政府が連邦供給スケジュール外で購入した場合のように,連邦政府が直接購入者である項目(例えば連邦医療保険や医療補助など)の精算項目に対するクレームが含まれる.これらの法律によると、製薬会社や他の医療保険会社は起訴されており、価格設定サービス機関に報告された薬品価格をつり上げた疑いがあり、価格設定サービス機関は逆に連邦医療保険や医療補助販売率を設定するために使用されており、顧客に製品を無料で提供する疑いがあり、顧客が製品の連邦計画に料金を請求することが予想される。さらに、ラベル外販売促進を含むいくつかのマーケティング行為は、虚偽クレーム法律に違反する可能性もある。ほとんどの州にも連邦反リベート法規や民事虚偽請求法案のような法規や法規があり、医療補助や他の州で計画されている精算プロジェクトやサービスに適用されたり、いくつかの州では支払者にかかわらず適用されています。
39
医療詐欺および乱用に関連する他の連邦法規は、医療補助または医療保険受益者への報酬の提供または支払いを禁止する民事罰金法規を含み、要人または支払人は、特定のサプライヤーからの精算可能な物品またはサービスの注文または受け入れに影響を与える可能性があることを知っているか、または知るべきであり、1996年に“健康保険携帯および責任法案”(HIPAA)によって作成された追加連邦刑法では、任意の医療福祉計画または虚偽または詐欺的な言い訳によって得られた計画を故意に実行または実行しようとすることが禁止されている。任意の医療福祉計画が、医療福祉、プロジェクトまたはサービスの交付または支払いに関連する任意の金銭または財産を所有または制御することを陳述または承諾する。
また、2009年に発表された“健康情報技術促進経済·臨床健康法案”(HITECH)によって改正されたHIPAAおよびそのそれぞれの実施条例は、2013年1月25日に発表された最終総合規則を含み、特定の医療保健提供者、健康計画およびヘルスケア交換所(カバーエンティティと呼ぶ)およびその業務パートナーおよびその下請け業者が、個人が識別可能な健康情報のプライバシー、安全および伝送を保護する上で義務を負い、これらのサービスは、個人識別可能な健康情報(強制契約条項を含む)を記憶、使用または開示し、特定の個人識別可能な健康情報セキュリティに違反した場合に影響を受けた個人および規制当局に通知することを要求する。HITECHは、実体、商業パートナー、および可能な他の人に適用される可能性のある民事および刑事罰を増加させ、州総検察長に新たな権力を与え、連邦裁判所に民事訴訟を提起し、連邦HIPAA法を実行するために損害賠償または禁令を要求し、連邦民事訴訟の提起に関連する弁護士費と費用を求めることができる。また、多くの州の法律は、場合によっては健康情報のプライバシーやセキュリティを管理しており、その中の多くの法律は互いに大きく異なり、同じ効果が生じない可能性があり、HIPAAに先を越されないことが多い。
さらに、ACAによると、医療保険および医療補助サービスセンター(CMS)は、ある処方薬メーカーが医師(医師、歯科医、検眼師、足科医師および脊椎マッサージ師を含むと定義されている)および教育病院に何らかの価値を支払いまたは移転する情報、および医師およびその直系親族が所有する所有権および投資権益を収集し、毎年報告することを要求する最終規則を発表した。2021年1月1日から,メーカーは,次の年に報告するために,医師アシスタント,勤務看護師,臨床看護師専門家,麻酔科医アシスタント,登録看護師麻酔科医および登録看護師助産師への支払いおよび他の方法での価値移転に関する情報を収集しなければならない。報告されたデータは毎年公共サイト上で検索可能な形で提供される。必要な情報を提出できなかったことは民事罰金を招く可能性がある。
私たちはまた、販売またはマーケティング計画、および非政府第三者支払者(個人保険会社を含む)によって精算される医療項目またはサービスに関するクレームに適用されるか、または支払者にかかわらず適用される可能性がある同様の州および外国の反リベートおよび虚偽請求法律によって制約される可能性がある。また、いくつかの州は現在、処方薬会社に薬品マーケティングや販売促進に関するいくつかの費用を報告し、これらの州の個人保健事業者に支払われたプレゼントと支払いを報告することを要求している。他の州では、いくつかのタイプのプレゼントや食事を提供するなど、マーケティング関連の様々な活動が禁止されている。また、ある州は臨床研究とその結果に関する情報の公表を要求している。一部の州は値上げに関する情報と値上げを弁護する情報を含むいくつかの薬品の定価情報を報告することを要求している。さらに、いくつかの州は、製薬会社にコンプライアンス計画および/またはマーケティングコードを実施することを要求する。他のいくつかの州も似たような提案を考慮している。いくつかの州と地方司法管轄区域もまた薬品販売代表の登録を要求する。また、場合によっては、健康情報のプライバシーや安全を管理する州や外国の法律の制約を受ける可能性もあり、その多くの法律は互いに大きく異なり、HIPAAに奪われず、コンプライアンス作業を複雑化させることが多い。
40
第三者の業務手配と適用される州、連邦、外国の医療保健法律法規に適合するようにする努力は大量のコストに関連している。製薬会社の運営がこのような要求に違反していることが発見された場合、民事、刑事および行政処罰、損害賠償、罰金、返還、監禁、削減または再構成、その業務の削減または再構築、FDAの承認を得た資格を失うこと、連邦医療保険と医療補助、誠実監督と報告義務、監禁、名声損害を含む、民事、刑事·行政処罰、損害賠償、罰金、罰金、または州政府医療保健計画などの重大な処罰を受ける可能性がある。有効なコンプライアンス計画は,これらの法律違反による調査·起訴のリスクを低減することができるにもかかわらず,これらのリスクを完全に解消することはできない。容疑や違反の疑いのあるいかなる行動も製薬会社に巨額の法的費用を招き、このような行動が成功しても経営陣の業務運営への注意を移す可能性がある。
アメリカの医療改革
米国では、連邦政府、州政府、監督機関、第三者支払者は、医療コストの増加を制御または管理するために引き続き提案を提出し、より広く言えば、米国の医療システムを改革する。製薬産業はこのような努力の特別な重点であり、重大な立法計画の重大な影響を受けてきた。例えば、2010年3月、ACAは、医療保険の許容性を拡大し、医療支出の増加を減少または制限し、詐欺や乱用に対する救済措置を強化し、医療保健と医療保険業界の新たな透明性要求を増加させ、医療業界に新たな税費を徴収し、追加の医療政策改革を実施し、医療保健が政府と民間保険会社によって融資される方式を大きく変え、米国製薬業に大きな影響を与えた。ACAは他の事項以外に、(I)後続の生物製品のための許可証フレームを作成することによって、治療性生物製品に低コストの生物模倣薬の潜在的な競争を受けさせ、(Ii)新しい方法を規定し、この方法に基づいて、吸入、輸液、点滴、移植或いは注射した薬物と治療性生物製品に対して、メーカーがMedicaid薬品還付計画の下で不足している税金還付を計算し、(Iii)医療補助薬物還付計画の下でメーカーが不足している最低医療補助税金還付を高め、そして税金還付計画をMedicaidが管理する看護組織に登録されている個人に拡大し、(Iv)あるブランドの処方薬と治療性生物製品メーカーに対する年間控除できない費用と税収を確立する。これらの実体のいくつかの政府医療計画における市場シェアがこれらの実体間で分配されることに基づいて、(V)新しいMedicare Part D Coverage Gap割引計画が確立された, その中でメーカーは、保証間隔期間内に条件を満たす受益者に50%(現在70%)の販売時点割引を提供することに同意しなければならず、メーカーの外来薬物と治療性生物製品が連邦医療保険D部分カバーの条件に組み込まれているとして、(Vi)医療補助計画の資格基準を拡大し、各州がより多くの個人に医療補助保険を提供することを許可し、連邦貧困レベル133%以下の収入を有する個人のために新たな強制資格カテゴリーを増加させ、それによってメーカーの医療補助リベート責任を潜在的に増加させることを含む。(Vii)公衆衛生計画の下で割引を受ける資格のあるエンティティを拡大し、(Viii)監督、優先順位を決定し、臨床的有効性比較研究を行うために、患者を中心とした新たな結果研究所を作成し、そのような研究に資金を提供し、(Ix)CMSに医療保険·医療補助革新センターを設立し、革新的な支払いおよびサービス交付モデルをテストして、処方薬支出を含む可能性がある連邦医療保険および医療補助支出を低減する。
“ACA”の全てまたはいくつかの条項を修正、廃止、または他の方法で無効にするために、行政、立法、および司法努力が行われている。2021年6月17日、米国最高裁は、ACAは全体的に違憲であり、“個人権限”が国会で廃止されたため、プログラム理由に基づく挑戦を却下した。したがって、ACAは現在の形態で継続的に有効であるだろう。ACAは未来に司法や国会で挑戦される可能性がある。米国最高裁の裁決,他のこのような訴訟やバイデン政府の医療措置がACAや我々の業務にどのように影響するかは不明である。
41
ACAの公布以来、米国はまた、医療支出を減少させるために、他の立法改正を提出し、採択した。米国連邦政府機関は現在、潜在的な大幅な支出削減に直面しており、医療支出にさらに影響を与える可能性がある。2011年8月2日、“2011年予算抑制法案”などの法案が国会のために支出削減措置を制定した。赤字削減合同特別委員会の任務は、2013年から2021年までの間に少なくとも1.2兆ドルの赤字削減を提案することであるが、同委員会は必要な目標を達成できず、立法をいくつかの政府プロジェクトに自動的に削減することを触発した。これには、各年度に提供者に支払われる医療保険総額が2%減少することが含まれる。これらの削減は2013年4月1日に施行され、その後の法規の立法改正により、BBAが含まれているため、2020年5月1日から2022年3月31日まで一時停止することを除き、2030年まで有効となる。2022年4月1日から2022年6月30日まで連邦医療保険の削減は1%から段階的に戻り,2022年6月30日以降は完全な2%に増加した。また、2013年1月2日、2012年に“米国納税者救済法”が署名され、病院、画像形成センター、がん治療センターを含むいくつかのタイプの提供者に支払われる医療保険をさらに減少させ、政府が提供者に多額の支払いを取り戻す訴訟時効を3年から5年に延長した。連邦支出がさらに減少すれば、予算不足は食品·医薬品局や国家衛生研究院のような関連機関の能力にも影響を与える可能性がある, 現在のレベルで動作し続けています連邦贈与と契約に割り当てられた金額は減少またはキャンセルされる可能性がある。これらの削減は、関連機関が研究開発、製造、マーケティング活動を適時に審査·承認する能力にも影響を与える可能性があり、私たちが開発、マーケティング、販売する可能性のある任意の製品を開発、販売する能力を遅らせる可能性があります。
最近、政府はメーカーが販売する製品の価格設定方式をより厳格に審査し、いくつかの大統領行政命令、国会調査を招き、製品定価の透明性の向上、定価とメーカー患者計画との関係の審査、政府の薬品の精算方法の改革を目的とした連邦と州立法を提出し、採択した。連邦レベルでは、トランプ政権はいくつかの手段を用いて、連邦予算提案、行政命令、政策措置を含む薬品定価改革を提出または実施している。例えば、2020年7月24日と2020年9月13日、米トランプ政権はいくつかの処方薬の定価に関する行政命令を発表し、政府のいくつかの提案を実施しようとしている。そのため、FDAは2020年9月24日に最終規則を発表し、各州がカナダからの薬物輸入計画の制定と提出に指導を提供した。バイデン政権もトランプ政権も、国内メーカーからの政府調達を支援するための行政命令を発表した。また、トランプ政権は薬品調達に特化した行政命令を発表し、連邦政府に“基本”薬品リストを作成し、原料薬の製造を含む他の米国で製造された医療用品を購入するよう指示した。バイデン政府がこの行政命令や同様の命令を実行するかどうかは不明である。
また、2020年11月20日、アメリカ衛生と公衆サービス部は1つの法規を決定し、薬品メーカーのD部分下でスポンサーの値下げを計画する避風港の保護を取り消し、直接にも薬局福祉マネージャーを通じても、法律が値下げを要求しない限り、。この規定はまた、販売時点での値下げを反映するための新しい安全港を創出し、薬局福祉マネージャーと製造業者との間のいくつかの固定料金手配のための安全港を作成する。裁判所の命令により,上記安全港の除去と増加は延期され,最近の立法は同規則の実施を2026年1月1日に一時停止した。この最終期限は、両党の“より安全なコミュニティ法案”によって2027年1月1日に延期された。2022年のインフレ削減法案、あるいはアイルランド共和軍は、さらにこの規則の実施を2032年1月1日に延期する。また、総裁·バイデンは2021年3月11日に“2021年米国救援計画法案”に署名し、2024年1月1日から単一源と革新多源薬に対する法定医療補助薬品還付上限を廃止し、現在この上限は薬品メーカー平均価格の100%である。
42
最近では2022年8月に総裁·バイデンが“アイルランド共和法”に署名しましたアメリカ衛生公衆サービス部(HHS)はCMSが連邦医療保険B部分とD部分で精算されたいくつかの薬物や生物製品の販売価格に基づいて交渉することを許可しますこれは少なくとも7年間承認された(生物製品は11年)高支出単一由来薬にのみ適用されますが交渉価格は2026年に初めて発効し,2023年10月から法定最高価格を上限とし,インフレ率よりも高い速度で連邦医療保険B部分とD部分の薬物価格を向上させる製薬業者を懲罰する。また,2025年から受益者の最高自己負担コストを大幅に低減することにより,メーカーに新たに設立されたメーカー割引計画により,D一部参加者ブランド薬物処方コストの10%を自己払い最高限度額を下回り,自己最高限度額に達すると20%補助することを求め,連邦医療保険D部分下の“ドーナツ穴”を解消した。アイルランド共和軍は衛生·公衆サービス部長官が最初の数年に規制ではなく指導によってその多くの規定を実施することを許可した。IRAを守らないメーカーは民事罰金を含めて様々な処罰を受ける可能性がある。アイルランド共和軍はまた、ACA市場で医療保険を購入した個人に2025年まで強化された補助金を提供する。このような規定は法的挑戦を受ける可能性があるにもかかわらず、2023年から段階的に施行されるだろう。
州レベルでは、立法機関は、価格または患者の精算制限、割引、特定の製品参入およびマーケティングコスト開示の制限、および透明性措置を含む、価格または患者の精算制限、割引、特定の製品参入およびマーケティングコスト開示の制限および透明性措置を含む医薬品および生物製品の定価を制御するための法規を立法および実施することが増えており、場合によっては、他の国からの輸入および大量購入を奨励することが目的である。
私たちは未来にもっと多くの州と連邦医療改革措置を取ると予想している。
保証と精算を請け負う
米国や他の地域では,患者は通常第三者支払者によって処方薬に関する費用の一部または全部を精算する。そのため、市場の私たちの薬品に対する受け入れ度は、政府衛生行政当局(アメリカのMedicareとMedicaidなどの政府医療保健計画に関連する項目を含む)、個人医療保険会社と他の医療資金調達組織が第三者保険と精算を提供する程度に依存する。私たちが規制承認を受ける可能性のある任意の薬品のカバー範囲と精算状態には、大きな不確実性が存在する。すでにより成熟或いはより低コストの治療代替方案がある時、カバー決定は新薬製品に不利である可能性がある。患者が私たちの薬品の全部あるいは大部分のコストを支払うのに十分でなければ、私たちの製品を使用することはあまりできません。
薬品の保証範囲と精算政策は支払人によって異なるが,米国では第三者支払側の間に統一された薬品保証と精算政策がないためである。保険の獲得と精算に重大な遅延が生じる可能性があり、保険と精算を確定する過程は往々にして時間がかかり、高価であるため、各支払人にそれぞれ私たちの製品を使用する科学的と臨床的支援を提供することを要求し、保険或いは十分な精算を受けることを保証することはできない。政府当局と第三者支払人が私たちの薬品の保険や精算についてどのように決定するか予測するのは難しい。また、私たちは自分たちまたはパートナーと私たちの製品候補製品のためのいくつかの適応のセット診断テストを開発することができます。承認されると、私たちまたは私たちの協力者(ある場合)は、私たちの候補製品のために求められているカバー範囲と精算を除いて、これらのテストのカバー範囲と精算を個別に取得することを要求されます。
我々の候補製品の市場は,第三者支払者の薬物処方や第三者支払者が保険と精算の薬物リストを提供できるかどうかに大きく依存する。このような公式に組み込まれた競争は往々にして定価引き下げの圧力を招く。特に、価格の低い後発薬または他の代替品がある場合、第三者支払者は、その処方に特定の参照リスト薬を含むことを拒否するか、または患者が参照リスト薬を得ることを制限することができる。
アメリカ政府、州立法機関と外国政府実体はコスト制御計画の実施に大きな興味を示し、政府が支払う医療コストの増加を制限し、価格制御、精算とカバー範囲の制限及びブランド処方薬の代わりに模造薬を要求する。既存の制御と措置を持つ司法管轄区域では、政府のコントロールと措置および規制政策を強化し、私たちの薬品を保険範囲から除外したり、そのカバー範囲を制限したり、薬品への支払いを制限したりする可能性がある。
43
また,米国の第三者支払者と政府当局の管理的医療保健とコスト制御措置の日々の重視が継続し,薬品定価やカバー範囲に圧力となることが予想される。保証政策と第三者精算料率は随時変化する可能性がある。規制部門の承認を得た1つ以上の薬品が有利な引受·精算状態を獲得したとしても、将来的にはあまり有利ではない引受政策や精算料率が実施される可能性がある。
従業員と人的資本
私たちは、私たちの従業員が毎日彼らの最高の自己を発揮できるように、多様で包容的で安全な労働環境を創造し、維持するために努力している。多様性に対する私たちの約束は、私たちの採用、維持、学習、そして参加、そして地域社会のパートナーシップによって延長される。私たちは、多様化、公平、包容、帰属感戦略の一部として、学習と参加の機会を求め、異なる背景の人を集めて対話を行う積極的な決定を下した。
2022年12月31日現在、私たちは121人のフルタイム従業員を持っている。これらの従業員のうち,34人が博士,薬学あるいは医学博士号を有し,72人が研究,開発,技術運営に従事している。私たちはまた時々私たちの組織を支援するために独立請負業者を雇う。私たちの従業員の約半分はカリフォルニア州ブリスベンにある本社で働いていて、他の従業員は遠隔で働いています。私たちは停止を経験したことがありません。私たちの従業員の中には労働組合代表や集団交渉合意に含まれている従業員がいません。私たちは長期的な仕事関係を育成するために、適格な従業員を誘致し、維持するために努力しています。
私たちは従業員の発展、従業員の尊敬度及び多様性と包容性に集中して、最も優秀な人材を確定、採用、発展と維持する。我々のインセンティブ株式計画の主な目的は、インセンティブ株に基づく報酬奨励と現金に基づく業績ボーナス奨励を付与することによって、選定された従業員、顧問、取締役を誘致、維持、激励することである。私たちは競争力のある給与と福祉を提供し、従業員の需要と要求のためにカスタマイズして、私たちが合格した人材を誘致、採用し、維持する目標を実現することを目的としています。
施設
私たちの主な行政事務室はカリフォルニア州ブリスベンにあり、私たちはそこで約12,000平方フィートのオフィス空間を借りました。レンタル契約は2024年12月に満期になります。賃貸契約期間を延長する選択もなく、賃貸契約が満期になる前に賃貸契約を終了する選択もない。私たちはこれらの施設が私たちの持続的な需要に対応するのに十分だと信じており、もし私たちがもっと多くの場所が必要なら、私たちは商業的に合理的な条件でより多くの施設を得ることができるだろう。
法律訴訟
時々、私たちは正常な業務過程で法的訴訟に巻き込まれるかもしれない。私たちは現在、経営陣が私たちの業務に重大な悪影響を及ぼすと考えているいかなる法的手続きにも参加していません。結果にかかわらず、訴訟は弁護と和解費用、管理資源の移転、負の宣伝と名声損害などの要素によって私たちに不利な影響を与える可能性がある。
企業情報
我々は2018年11月にデラウェア州法律に基づいて有限責任会社として設立され、名称は英雄治療持株会社である。その後,2018年12月に初日治療ホールディングスに改称し,2020年3月に初日バイオ製薬ホールディングスに改称した。私たちの最初の公募株では、私たちはデラウェア州の有限責任会社からデラウェア州の会社に転換し、私たちの名前をDay One BiopPharmticals、Inc.,またはThe Convertionに変えた。私たちの主な実行オフィスはカリフォルニア州ブリスベン501 Suit 501 Sierra Point Parkway 2000番地にあります。私たちの電話番号は(94005)484-0899です。私たちのサイトの住所はwww.Day oneBio.comです
44
情報を付加する
私たちのサイトはhttp://www.Day oneBio.comです。このような材料を米国証券取引委員会または米国証券取引委員会に電子的に提出または提供した後、このような報告書の改訂を含む、合理的で実行可能な範囲内でできるだけ早く合理的で実行可能な範囲で私たちの年間、四半期、および現在の報告書を無料で提供します。米国証券取引委員会は、米国証券取引委員会に資料を電子的に提出した会社に関する報告書やその他の情報を含むウェブサイトwww.sec.govを保持している。
当社のウェブサイトでは、当社のコーポレート·ガバナンス基準、私たちのビジネス行動および道徳的基準(私たちの取締役、上級管理者、従業員の使用のため)、および私たちの取締役会規約を含む会社管理および取締役会に関する情報も提供されています。書面の要求があれば、上記のいずれかの情報を当社の秘書に無料で提供します。初日はバイオ製薬会社、住所は2000年Sierra Point Parkway、Suite 501、Brisbane、CA 94005です。
我々は,重大な非公開情報を開示する手段として投資家関係サイトを用い,米国証券取引委員会が公布したFD法規規定の開示義務を遵守している.これらの開示内容は、私たちのサイトの“プレスリリース”および“イベントおよびプレゼンテーション”の部分に含まれています。したがって,投資家は我々のニュース原稿,米国証券取引委員会の届出文書,公開電話会議やインターネット放送に注目するほか,我々のサイトのこれらの部分にも注目すべきである.
我々のサイトに含まれる情報は構成されておらず、本10-K年報を構成するもの、または米国証券取引委員会に提出または提出された任意の他の報告書の一部を構成するものとみなされてはならない。我々のサイトURLへの参照は,非アクティブテキスト参照のみに用いられる.
45
情報技術EM 1 A。リスク要因です
私たちの普通株に投資することは高い危険と関連がある。私たちの普通株への投資を決定する前に、以下に述べるリスクおよび不確実性、および当社の財務諸表および関連説明、ならびに“経営陣の財務状況および経営業績の検討および分析”を含む本年度報告書に含まれる他の情報をよく考慮しなければなりません。以下に説明するリスクと不確実性は私たちが直面している唯一の危険と不確実性ではない。私たちは意識していない、あるいは私たちは現在実質的な他のリスクや不確実性ではないと考えており、私たちに影響を与える重要な要素になる可能性もある。私たちはあなたに次のように議論されたどんな事件も起こらないという保証がありません。これらの事件は、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼすかもしれない。もしこのような状況が発生したら、私たちの普通株の取引価格は下がるかもしれません。あなたはあなたの全部あるいは一部の投資を損失するかもしれません。
リスク要因の概要
我々の業務は、本要約に続くリスクおよび不確実性を含むいくつかのリスクおよび不確実性の影響を受ける。いくつかのリスクは
46
私たちの財務状況と追加資本需要に関連するリスク
私たちの経営歴史は限られており、第一段階以降のいかなる臨床試験も完成しておらず、製品が商業販売を許可されておらず、何の収入も生じておらず、投資家が現在の業務および成功と生存の可能性を評価することを困難にする可能性がある。
私たちは臨床段階の生物製薬会社で、運営の歴史が限られていますので、それに基づいて私たちの業務と将来性を評価することができます。私たちは2018年に運営を開始し、いかなる製品も商業販売を許可されておらず、何の収入も生じていない。薬物開発への投資は投機性の高い仕事であり、大きなリスクに関連している。これまで、私たちはほとんどの資源を投入して、私たちの候補製品を決定し、獲得し、開発し、私たちのパイプラインを構築し、私たちの会社を組織し、そのための人員、業務計画、私たちの知的財産権の組み合わせを確立し、維持し、第三者と私たちの候補製品の製造手配を確立し、資金を調達し、これらの業務に一般的かつ行政的な支援を提供してきた。
私たちの設立以来、私たちは私たちのすべての努力と財政資源を私たちの主要な候補製品tovorafenib(DAY 101)と私たちの現在のもう一つの候補製品pimasertibの臨床開発に集中してきて、tovorafenib(DAY 101)は最初に再発または進行性低レベルグリオーマの治療に使用され、pimasertibは経口MEKキナーゼ小分子阻害剤であり、私たちはtovorafenib(DAY 101)と組み合わせて使用し、RASとRAF依存性腫瘍の治療に使用する予定である。これまで、私たちは主に償還可能な転換可能な優先株、転換可能な手形を売却して発行し、私たちの初公開(IPO)を完了し、私たちの普通株の後続公開を完成させ、私たちの業務に資金を提供してきました。
私たちは、第1段階以外の任意の臨床試験を成功させ、市場の承認を得たり、商業規模の製品を製造したり、第三者代表がそうしたり、成功した製品の商業化に必要な販売およびマーケティング活動を行うことができることを証明していません。したがって、あなたは私たちがより長い運営歴史を持っているよりも、私たちの成功と生存の可能性を正確に予測することが難しいかもしれない。
また、私たちは予見できない費用、困難、合併症、遅延と他の既知と未知の要素とリスクに遭遇する可能性があり、これらはすべて臨床段階の生物製薬会社が急速に発展する領域でよく遭遇する。研究開発に専念している会社から、ビジネス活動を支援できる会社に転換する必要もあるかもしれません。私たちはこのような危険と困難を成功的に克服したり、そのような移行を達成する能力があるということを見せていない。これらのリスクや困難に十分に対応できなかったり、そのような転換を成功させたりすることができなければ、私たちの業務は影響を受けるだろう。
47
設立以来、私たちは重大な純損失が発生し、何の収入も生じなかった。私たちは予測可能な未来に損失が続き、永遠に達成されたり利益を維持したりしないかもしれないと予想している。
会社設立以来、各報告期間中に重大な純損失が発生し、これまで何の収入も生じておらず、主に私たちの償還可能な転換可能優先株、私たちの転換可能な手形、私たちの初公開株の完成とその後の私たちの普通株の発行を通じて私たちの運営に資金を提供してきた。2022年12月31日と2021年12月31日の年度までに、それぞれ1億422億ドルと7280万ドルの純損失を報告します。2022年12月31日までの累計赤字は2兆697億ドル。予測可能な未来には,我々の運営損失レベルはますます高くなり,特に臨床開発においてボルラフィニ(DAY 101)とピマスタチオの臨床開発を進めている場合が予想される。私たちのこれまでの損失は、予想された将来の損失に加え、私たちの株主権益や運営資本に悪影響を与え続けるだろう。主要候補製品や他の候補製品のための追加臨床試験を計画しているため,我々が行っている重要な2期Firefly−1試験(DAY 101),pLGGの潜在的第一線療法であるtovorafenibのキー3期Firefly−2試験(DAY 101),我々が行っている1 b/2期firellight−1主試験(DAY 101),tovorafenibを単一療法としてpimasertibと併用し,将来のいずれかの候補製品に対する研究的新薬応用プログラムやINDの開発を行っていると予想される。さらに、tovorafenib(DAY 101)、pimasertib、または他の候補製品の発売承認を得た場合、tovorafenib(DAY 101)、pimasertib、またはそのような他の候補製品の商業化に関連する巨額の販売、マーケティング、およびアウトソーシング製造費用を生成する。私たちもまた継続して, 上場企業の運営に関連する追加コスト。
したがって、予測可能な未来に、私たちは引き続き重大で増加していく純損失を受けることが予想される。医薬品開発に関連する多くのリスクや不確実性により,将来の損失の程度やいつ利益が達成されるかは予測できない(あれば)。私たちが本当に利益を達成したとしても、私たちは四半期や年度の収益性を維持したり向上させることができないかもしれない。また、様々な要因により、当社の財務状況や経営業績は四半期と年度の間に大幅に変動し、その多くの要因は制御できないと予想されています。したがって、今後の経営業績の指標として、いかなる四半期や年度の業績にも依存してはいけません。
48
著者らは著者らの主要な候補製品tovorafenib(DAY 101)の成功に大きく依存し、この薬は現在臨床開発段階にあり、まだ重要な試験を完成していない。
私たちの未来の成功は私たちが適時に成功した臨床試験を完成できるかどうかに依存し、監督部門の許可を得て、それから私たちの候補製品を商業化することに成功した。私たちの開発はまだ初期段階にあり、私たちの主要な候補製品tovorafenib(DAY 101)は現在重要な第二段階の臨床試験にある。私たちの現在のもう一つの候補製品pimasertibは比較的早い開発段階にある。私たちは現在どの管轄区でも販売を許可していない。Tovorafenib(DAY 101)、pimasertib、または私たちが開発した任意の未来の候補製品が、その臨床試験で成功するか、または規制部門の承認を得ることは保証されない。
私たちが製品収入を作る能力は、近い将来には起こらないと予想され、もしあれば、私たちの主要候補製品tovorafenib(DAY 101)の開発成功と最終商業化に大きく依存するだろう。Tovorafenib(DAY 101)の成功は以下の要因に依存する:
49
その多くの要素は私たちがコントロールできないことであり、私たちが多くの時間と資源をかけて規制部門の承認を求めても、私たちの候補製品は決して承認されないかもしれない。もし私たちがこれらの要素のうちの1つまたは複数をタイムリーにまたは根本的に達成できなければ、私たちは重大な遅延に遭遇したり、私たちの候補製品を商業化することに成功しなかったりする可能性があり、これは私たちの業務に深刻な損害を与えるだろう。
私たちが収入を創出し、利益を達成する能力は、私たちの候補製品の発見や識別、開発、商業化に関連するいくつかの目標を達成する能力に大きく依存する。
私たちの業務は候補製品の成功的な発見や識別、開発、商業化に完全に依存する。私たちは商業販売が許可されていない製品で、今後12ヶ月以内に製品販売から何の収入も得られないと予想されます。マーケティングの承認を得てtovorafenib(DAY 101)、pimasertib、または他の候補製品の販売を開始しない限り、私たちは相当な収入を生成しないと予想される。私たちが収入を創出し利益を達成する能力はいくつかの要素に依存しますがこれらに限らず私たちの能力は
50
利益を実現し、維持するためには、適切な収入を生み出す製品を設計、開発し、最終的に商業化しなければならない。これは、私たちの候補製品のための臨床試験を完成させ、より多くの候補製品を設計および/または取得し、第三者と私たちの候補製品を生産する臨床用品について手配を確立し、候補製品および製造のためのマーケティング許可を得ること、知的財産権またはマーケティング排他性を保持すること、およびマーケティングおよび販売がマーケティング承認を得る可能性のある任意の製品(あれば)を提供することを含む、一連の挑戦的な活動で成功することを要求するであろう。私たちはほとんどこのような活動の初期段階にいる。私たちはこのような活動で決して成功しないかもしれないし、たとえ私たちが成功しても、利益を達成するのに十分な収入が生まれないかもしれない。
もし私たちが規制部門の承認を得て、私たちの1つ以上の候補製品を市場に出すことに成功すれば、私たちの収入は、私たちが監督部門の承認を得た地域の市場規模、製品の受け入れ可能な価格、医者が私たちの製品に適していると思う治療時間、医師が採用する速度、保険と補償を受ける能力、そして私たちがその地域の商業権を持っているかどうかにある程度依存するだろう。もし私たちの潜在的な患者数が私たちが推定したほど多くなければ、規制機関が許可した適応は私たちが予想していたより狭い、あるいは治療者たちは競争、医者の選択、支払人の決定、または治療ガイドラインによって縮小され、承認されても、このような製品の販売から相当な収入を得ることができないかもしれない。
FDAまたは他の管轄区域の規制機関によって現在予想されている基礎の上で研究または臨床試験を行うことを要求したり、進行中または計画中の臨床試験を修正したり、または私たちの任意の候補製品のための適切な製造計画を確立し、現在および計画されている臨床試験または開発を開始または完了することに遅延が生じた場合、私たちの費用は大幅に増加する可能性があり、利益はさらに遅れる可能性がある。
私たちが実現して利益を維持できなかったことは、会社の価値を弱める可能性があり、資金調達、業務拡大、研究開発努力の維持、製品供給の多様化、さらには運営を継続する能力を弱める可能性がある。わが社の価値の低下はあなたの投資損失の全部または一部を招く可能性もあります。
51
私たちは私たちの運営と私たちの目標を達成するために多くの追加資本が必要になるだろう。もし私たちが必要な時や私たちが受け入れられる条件で資金を集めることができなければ、私たちは私たちの研究や製品開発計画、将来の商業化努力、または他の運営を延期、減少または廃止することを余儀なくされるかもしれない。
臨床前研究と臨床試験を含む薬物製品を開発することは、非常に時間がかかり、高価かつ不確定な過程であり、完成するのに数年かかる。設立以来、私たちの業務は大量の現金を消費し、特に臨床開発を通じて私たちの主要な候補製品tovorafenib(DAY 101)、pimasertib、および任意の未来の候補製品を推進する際に、私たちが行っている活動に関連する費用が大幅に増加することが予想される。私たちが研究開発を続け、より多くの臨床試験を開始し、私たちの製品ラインの拡大を求め、私たちの主要な計画と未来の候補製品のために市場承認(あれば)、私たちの組織への投資を求めることに伴い、費用が増加すると予想される。また、いずれかの候補製品が市場承認されれば、製品製造、マーケティング、販売、流通に関連した巨額の商業化費用が発生すると予想される。また、経験豊富な人員の獲得と維持、新たな情報技術システムの開発、上場企業に関する他のコストなど、上場企業の運営に関する追加コストを負担し続けている。さらに、私たちは、特許出願の準備と提出、私たちの知的財産権の維持、および私たちのオフィス施設の拡大に関連する持続的かつ追加的なコストに直面することが予想されます。したがって、私たちは私たちの持続的な業務と関連した多くの追加資金を得る必要があるだろう。
私たちは割引された条件で十分な追加融資を受けないかもしれないし、全くないかもしれない。また、有利な市場条件や戦略的考慮により、現在または将来の運営計画のために十分な資金があると考えても、追加の資本を求めることができる。もし私たちが必要な時や有利な条件下で資金を集めることができなければ、私たちは私たちの研究開発計画、商業化計画、または他の業務を延期、減少、またはキャンセルさせることを余儀なくされるかもしれない。私たちの資金調達能力は、世界の経済状況の潜在的な悪化とアメリカと世界各地の信用と金融市場の中断と変動の不利な影響を受ける可能性があり、これらの要素はインフレ、金利の変化、ウクライナ戦争、進行中の新冠肺炎の疫病、あるいはその他の要素を含む地政学的不安定を含む。
2022年12月31日現在、私たちは3.423億ドルの現金、現金等価物、短期投資を持っている。私たちの既存の現金、現金等価物、短期投資は、2025年までの運営費用と資本支出需要に資金を提供できると信じている。私たちは間違っていることが証明される可能性があるという仮定に基づいてこの推定をして、私たちは現在予想されているよりも早く私たちの資本資源を使用することができる。私たちがコントロールできない変化が起こる可能性があり、それまで私たちの薬物開発活動の変化と進展、規制の変化を含めて、私たちの利用可能な資金を使用することができます。私たちの将来の資本需要は多くの要素に依存します
52
私たちは私たちが計画した臨床開発計画を達成するために追加の資金が必要になり、私たちの現在の候補製品は規制部門の承認を得るために、私たちの候補製品の開発と商業化を達成するために追加の資金を調達する必要があると予想される。私たちが追加資金を調達する能力は金融、経済、そして市場状況、そして他の要素に依存し、私たちはこれらの要素をコントロールできないか、あるいは限られているかもしれない。必要に応じて商業的に許容可能な条項で十分な資金を得ることができない場合、私たちは私たちの研究計画や候補製品の開発または商業化の全部または一部を延期、減少または終了させることを余儀なくされるかもしれないし、将来のビジネス機会を利用できないかもしれない。さらに、任意の追加的な資金調達努力は、現在および将来の候補製品を開発および商業化する能力(承認されれば)を含む、私たちのチームの日常活動への関心を移す可能性があります。変化する状況-その中のいくつかは私たちの制御を超えているかもしれない--私たちの資本消費速度は私たちの現在の予想よりも大きく速く、私たちは計画よりも早く追加資金を求める必要があるかもしれない。
私たちは公共または私募株式融資、債務融資、協力協定、許可手配、または他の融資源を通じてより多くの資金を得ることが要求され、これは私たちの株主を希釈したり、私たちの経営活動を制限したりするかもしれない。私たちは約束された外部資金源を持っていない。2022年6月、私たちはPiper Sandler&Co.とJones Trading Institution Services LLCと販売エージェントとして株式分配協定を達成し、市場発行計画(すなわち2022年ATM)に基づいて1.5億ドルまでの総発行価格で私たちの普通株の株式を発行し、売却することを含む。私たちの2022年のATMに基づいて、各投資家の所有権権益が希釈されることを含む、株式または転換可能な債務証券を売却することによって追加資本を調達する範囲内で、条項は、清算または各投資家の株主としての権利に悪影響を及ぼす特典を含むことができる。債務融資は、債務契約の強制実施、固定支払義務の増加、または他の我々の業務の制限に影響を及ぼす可能性がある。もし私たちが第三者との戦略的協力に基づいて前金やマイルストーン支払いによって追加資金を調達すれば、私たちは私たちの候補製品に対する貴重な権利を放棄したり、私たちに不利な条項で許可証を授与しなければならないかもしれない。我々のより多くの資金を調達する能力は、世界経済状況の潜在的な悪化や米国と世界各地の信用と金融市場の中断と変動の悪影響を受ける可能性があり、これらの要素はインフレ、金利変化、ウクライナ戦争、進行中の新冠肺炎の大流行、その他の要素を含む地政学的不安定を含む。
私たちは必要な時や受け入れ可能な条件下で資金を調達することができず、私たちの財務状況と私たちの業務戦略を実施する能力に負の影響を与え、私たちは延期、縮小、一時停止、または私たちの1つまたは複数の研究または薬物開発計画、臨床試験、または将来の商業化努力を延期しなければならないかもしれない。
53
私たちの候補製品の開発と商業化に関するリスク
臨床試験は非常に高価で、時間、設計と実施が困難であり、しかも不確定な結果に関連している。また、早期臨床前研究と臨床試験の結果は未来の臨床前研究或いは臨床試験の結果を予測できない可能性がある。我々の候補製品は今後の臨床試験で有利な結果が得られない可能性があり,もしあれば,あるいは規制部門の承認を得ることができる。
私たちの候補製品には、失敗のリスクが高い。私たちの候補製品がいつ、人体で有効または安全を証明するか、あるいは規制部門の承認を受けるかどうかを予測することはできない。私たちのすべての候補製品を発売し、販売するために必要な監督管理許可を得るためには、広範な臨床前研究と臨床試験を通じて、私たちの候補製品が人体上で各目標適応に使用することが安全かつ有効であることを証明しなければならない。臨床試験費用は高価であり,完成まで数年かかる可能性があり,結果自体も確定していない。臨床試験では,いつでも失敗する可能性がある。
また、臨床前研究と早期臨床試験の結果は後期臨床前研究或いは臨床試験の結果を予測できない可能性がある。私たちの候補製品の臨床データは限られている。臨床前と早期臨床試験で進展が得られたにもかかわらず、臨床試験後期段階の候補製品は類似或いは期待される安全性と有効性特徴を示すことができないかもしれない。
いくつかの場合、多くの要素のため、同一候補製品の異なる臨床試験間の安全性或いは有効性結果は有意差が存在する可能性があり、方案に規定されている臨床試験プログラムの変化、患者群の大きさとタイプの差異、投与方案と他の臨床試験方案に対する堅持、及び臨床試験参加者の中止率を含む。もし私たちが計画した任意の候補製品の臨床試験が積極的な結果を生むことができなければ、私たちの候補製品の開発スケジュール、規制承認と商業化の見通し、そしてそれに応じた私たちの業務と財務の見通しは、実質的で不利な影響を受けるだろう。
我々は規制申告文書において研究者が開始した第二段階臨床試験のデータに依存しており、試験操作や結果報告を制御していない。
われわれの主要候補製品tovorafenib(DAY 101)のキー2期Firefly−1試験は,研究者による再発/難治性pLGG患者に対するマルチセンター試験であり,Dana Farber癌研究所と太平洋小児科神経腫瘍連合会(PNOC)が協力して行った。この実験の最後のデータ報告書は2023年1月だった。より多くのデータは報告時に似たような結果を示さないかもしれない。私たちはこのような臨床データが発表される時間をコントロールできない。また,pLGGで行われたPivotal 2期FIREFELY−1試験は,規制機関との予備的な議論に基づいて十分なデータセットを提供して承認を支援することが予想されるが,FDAがより多くの患者のデータや既存患者の追加的な後続データを要求しないことを保証することはできない。後期臨床試験では,完成した早期臨床試験よりも厳しい統計分析を受ける可能性がある。製薬業界のいくつかの会社は後期臨床試験において治療効果の不足或いは副作用のために重大な挫折を受け、早期試験で良好な結果を得たにもかかわらず、私たちは類似した挫折に直面しないことを確定できない。そのほか、臨床前と臨床データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品が臨床前研究と臨床試験で満足できると考えているが、しかし依然としてその製品のマーケティング許可を得られなかった。
54
また、研究者が後援する試験の設計や管理を制御することも、これらの試験を行うために必要なINDや海外の同種の試験の提出や承認も制御せず、研究者が後援する試験は、これらの第三者の行動によって生じる臨床データの有効性を脅かす可能性があり、私たちの研究結果や臨床試験に影響を及ぼす可能性のある私たちの候補製品に対する重大な懸念を識別し、FDAまたは他の適用可能な規制機関から上場承認を得る能力に悪影響を及ぼす可能性がある。この試験または他の研究者が後援する試験結果が、我々が計画している会社支援試験の結果と一致しないか、または我々の候補製品への懸念を引き起こす場合、FDAまたは外国規制機関は、企業支援試験の結果を疑問視したり、そのような結果を他の場合よりも厳しい審査を行う可能性がある。この場合、FDAやこのような外国規制機関は、候補製品の臨床開発や上場承認を遅らせる可能性がある追加の臨床データの取得と提出を要求する可能性があります。研究者による試験は,我々自身の臨床開発に情報を提供するのに役立つ可能性があるが,研究者が開始した試験のデータやデータの発表時間を制御することはできず,これらの試験のデータを用いて規制部門が候補製品を承認する基盤を形成できる保証もない。
さらに、一部の患者は、商業的承認を得る前に、同情的使用、拡大獲得計画、または権利獲得を試みることによって薬物を獲得し、総称して同情使用計画と呼ばれ、彼らは生命を脅かす疾患を有し、他のすべての利用可能な治療方法を使い切っている。これらの患者群の中で深刻な不良事件が発生するリスクは非常に高く、もしこれらの不良事件が薬物と関係があると決定されたら、もし私たちがこれらの患者にこれらの薬物を提供すれば、私たちの候補薬物の安全性に負の影響を与える可能性があり、これは重大な遅延を招き、或いは私たちの候補薬物を商業化することに成功できず、そして私たちの業務に実質的な損害を与える可能性がある。もし私たちが慈悲使用計画に従って患者に任意の候補薬を提供すれば、私たちの供給能力は、その計画に登録できる患者数を制限する可能性があり、私たちは将来、制御された臨床試験において十分な数の患者を募集するために、任意の同情使用計画を再構成または一時停止する必要があるかもしれない。これは、規制部門が私たちの候補薬を承認し、それを商業化することに成功するために必要であり、このような計画の既存または潜在的参加者に関連する負の宣伝または他の妨害を引き起こす可能性がある。
私たちの臨床試験は私たちの任意の候補製品の安全性と有効性を十分に証明できないかもしれません。これは開発、監督管理の承認と商業化を阻害または延期します。
FDAや同様の外国規制機関から現在の候補製品を販売する市場承認を得る前に、私たちの候補製品が各目標適応において安全かつ有効であることを、長い、複雑で高価な臨床試験によって証明しなければならない。臨床試験は費用が高く,設計や実施が困難であり,完成まで数年かかる可能性があり,その最終結果も確定していない。臨床試験中に随時失敗が発生する可能性があり,また我々の候補製品は開発の初期段階にあるため,失敗のリスクが高く,適切な製品の開発には決して成功しない可能性がある。
臨床試験中または臨床試験の結果では、私たちは多くの予見できない事件に遭遇する可能性があり、これらの事件は、上場承認を得ることを遅延または阻止することができ、あるいは候補製品を商業化する能力を含むかもしれない
55
もし私たちが現在予想している候補製品に対して追加の臨床試験や他の試験を行うことを要求された場合、もし私たちが候補製品の臨床試験または他の試験を適時に成功させることができなければ、これらの試験または試験の結果が陽性でない場合、あるいは軽微な陽性のみである場合、あるいは安全問題がある場合、計画外コストが発生し、上場承認を求めて獲得する際に延期され、もし私たちがこのような承認を得て、より限られたまたは限定的な上場承認を得て、追加の上場後の試験要求の制約を受けたり、上場承認を受けた後に薬物を市場から除去したりする可能性がある。
私たちの候補製品は最初に小児科群を対象としており、規制機関はこれらの人々の安全問題を特別に審査するかもしれない。小児科群に関連した試験は困難である可能性があり,かなり高価である可能性があり,他の臨床試験のように期待した結果が生じない可能性がある。また,小児科研究はより数の少ない専門臨床試験地点に依存することが多く,逆に地点の有用性を制限し,試験の進行コストをより高くする可能性がある。また,“加速治療と公平児童研究法案”や他の市場力の小児科適応への興味の増加に伴い,合格患者への競争により試験募集が困難になる可能性がある。そのほか、児童或いは青少年患者が臨床試験方案を遵守することを確保することは挑戦的であるかもしれない。著者らは十分な数の小児科患者を著者らの臨床試験に参加することができず、重大な遅延を招く可能性があり、私たちは1つ以上の臨床試験を完全に放棄し、私たちの追加資金を集める能力に影響を与え、そして任意の候補薬物製品のために必要な監督管理許可を得ることを延期或いは阻止することを要求する。
さらに、もし私たちの臨床試験結果が確定していない場合、あるいは私たちの候補製品に関連する安全問題や深刻な有害事象が存在すれば、私たちは:
もし私たちがテストや発売承認を得る上で遅延に遭遇すれば、私たちの薬物開発コストも増加するだろう。また、市場に参入する前に競争環境がより激しくなると、市場承認の遅れが商業化コストを増加させる可能性がある。われわれのいかなる臨床前研究あるいは臨床試験が計画通りに開始されるかどうか、再構成が必要かどうか、あるいは計画通りに完成するかどうかは分からない。
56
さらに、私たちまたは私たちの協力者が規制要件(FDAの現在の良好な臨床実践またはGCP法規を含む)に基づいて試験を行うことができない場合、参加者が受け入れられない健康リスクに直面するか、またはFDAが我々の研究NDAまたはこれらの試験の進行に欠陥があることを発見した場合、我々FDAまたは機関審査委員会またはIRBは、いつでも臨床試験を一時停止することができる。したがって,将来の臨床試験の開始と完了スケジュールを確実に予測することはできない。また、海外で臨床試験を行うことは、私たちの候補製品のために行われているかもしれないように、追加のリスクをもたらし、私たちの臨床試験の完成を遅らせるかもしれない。これらのリスクには,外国に登録された患者が医療サービスや文化的慣習の違いにより臨床案を遵守できなかったこと,外国規制計画に関する追加行政負担を管理すること,およびこのような外国に関連する政治的·経済的リスクがある。
また,われわれの臨床試験の首席研究員は時々私たちの科学コンサルタントやコンサルタントを務め,このようなサービスに関する報酬を得る可能性がある。場合によっては、私たちはFDAまたは同様の外国規制機関にいくつかの関係を報告することを要求されるかもしれない。FDAや同様の外国の規制機関は結論を出す可能性があり、私たちと主要な研究者との財務関係は利益の衝突をもたらしたり、他の方法でこの研究の解釈に影響を与えたりする。したがって,FDAや同様の外国の規制機関は,適用された臨床試験地点で発生するデータの完全性を疑問視する可能性があり,臨床試験自体の効用が脅かされる可能性がある。これは、FDAまたは同様の外国規制機関が私たちの上場申請の承認を遅延または拒否することを招き、最終的に私たちの1つ以上の候補製品が上場承認を拒否することにつながる可能性がある。
さらに、臨床試験の終了または一時停止、または臨床試験の開始または完了遅延をもたらす多くの要因も、最終的に候補製品の規制承認を拒否する可能性がある。私たちは私たちの候補製品の調合や製造面の変更を行うかもしれません。この場合、修正された候補製品を早期バージョンに関連付けるために、追加の臨床前研究を行う必要があるかもしれません。もし私たちの臨床試験の開始が遅れたり完成したりすれば、もし私たちが完成する前に臨床試験を終了すれば、私たちの候補製品の商業的将来性は負の影響を受ける可能性があり、候補製品から収入を創出する能力は延期または完全に消失する可能性がある。
進行中または計画中の臨床試験で患者を募集する際に遅延や困難に遭遇した場合、必要な規制承認を受けることは延期または阻止される可能性がある。
FDAや同様の外国規制機関の要求に応じて、これらの試験に参加するのに十分な数の合格者を決定し、募集することができなければ、私たちが行っているまたは計画中の候補製品の臨床試験を開始または継続することができないかもしれない。我々のtovorafenib(DAY 101)計画では,患者腫瘍のゲノムマップを用いて,われわれの臨床試験に適した患者を決定する。(I)私たちの臨床試験に組み込むために必要な変更を行う患者がどれだけいるか、(Ii)各計画に登録された患者数が規制部門の承認を得るのに十分であるかどうか、または(Iii)各特定のBRAF変異が承認された薬物タグに含まれるかどうかを決定することはできない。もし私たちの患者の識別と登録戦略が成功しないことが証明されたら、私たちの製品候補に適した患者を募集したり維持したりすることは困難かもしれません。著者らは現在、著者らの候補製品の条件は孤児或いは稀な疾病であり、臨床試験に供することができる患者池は限られていることを評価する予定である。われわれの臨床試験の資格基準が確立されると,利用可能な試験参加者をさらに制限する。また、私たちのいくつかの競争相手は現在候補製品の臨床試験を行っており、これらの候補製品は私たちの臨床候補製品と同じ患者を治療し、本来私たちの臨床試験に参加する資格のある患者は私たちの競争相手候補製品の臨床試験に参加することができる。また,いずれの競合他社もFDAのある製品の承認を得ていれば,患者が承認された製品を用いて治療を求めることを決定すれば,患者の臨床試験参加能力を制限する可能性がある。例えば、ノバ社は最近、ダプラファニーと曲美チニブの共同使用の承認を得た, これは将来的にtovorafenib臨床試験(DAY 101)に参加する患者を募集する能力を制限するかもしれない。患者の入選は他の要素の影響を受けています
57
十分な数の患者を募集して維持することはできませんが、これは重大な遅延を招き、あるいは1つ以上の臨床試験を完全に放棄する必要があるかもしれません。相互競争の試験、及び小児科腫瘍学機構の運行試験の限られた帯域幅が存在する可能性があり、これはある試験の優先順位を招き、著者らの臨床試験の遅延を招く可能性がある。また,親は彼らの子供をわれわれの臨床試験に参加させたくないか,あるいは他の療法を求めるために子供をわれわれの臨床試験から離脱させることを決定する可能性がある。
私たちは激しい競争に直面しており、これは他の人たちが私たちよりも製品の発見、開発、商業化に成功する可能性がある。
製薬と生物技術業界の特徴は技術が迅速に進歩し、競争が激しく、特許と新製品及び候補製品を非常に重視していることである。私たちの競争相手は、私たちの候補製品と競争する製品、候補製品、およびプロセスを開発し、開発しているか、または開発しているかもしれません。我々が開発と商業化に成功した任意の候補製品は,既存の療法や将来出現する可能性のある新しい療法と競争するであろう。相当数の候補製品が現在開発中であり、将来的に商業的に使用される可能性があり、候補製品の開発を試みる可能性のある条件を治療するために使用される可能性があると信じている。また、私たちの候補製品は、医師がラベルの外で承認を求める適応を治療するための薬物と競争する必要があるかもしれない。これは私たちが既存の治療法を代替するために私たちの候補製品を使用することを難しくするかもしれない。
特に腫瘍学分野の競争は非常に激しい。私たちはアメリカと国際に競争相手がいて、大型国際製薬会社、老舗バイオテクノロジー会社、専門製薬会社、新興とスタートアップ会社、大学とその他の研究機関を含みます。
また、これらの組織と競争して、合格した科学、管理、販売、マーケティング担当者を募集し、維持することは、私たちの専門レベルと業務計画を実行する能力にマイナスの影響を与える可能性がある。臨床試験場所や臨床試験の患者登録の確立や,われわれの計画の補完やわれわれの計画のために必要な技術的競争にも直面する。
58
私たちは各プロジェクトが既存製品と開発中の製品からの競争に直面することを予想しています。V 600突然変異に集中した薬物発見はすでにいくつかの癌の臨床で成功した。3つのBRAF阻害剤は、V 600 EまたはV 600 K変異を含む腫瘍の治療のためにFDAによって承認されている。これらの初代BRAF阻害剤は,I型RAF阻害剤であり,ビモラフェニであり,市場名はZelborafであることがより知られている®Genentech提供;dradfenib,Tafinlarの名称で販売®ノーファ社とアンコラフィニはBraftoviという市場名を持っています®ファイザー社が提供しています。ダプラファニブとトリメチニブを併用し、ノファ社はそれをMekinistと命名し、切除できないまたはBRAF V 600 E変異を有する転移性固形腫瘍を有する6歳の成人および小児患者の治療のために許可されており、これらの患者は以前の治療後に病状が進展し、満足できる代替治療方案がない。これは、tovorafenib(DAY 101)開発計画におけるより大きなRAF変更のpLGG臨床範囲のサブセット(10%~20%)であるBRAF V 600 E pLGGを含む。また,ノワール社が協賛した無作為第二段階臨床試験では,ダプラファニーとトリメチニブの併用を評価し,新たな診断に用いたBRAF V 600変異pLGG患者を評価している。ノワール社はすでに彼らの計画を発表し、2023年初めにこの試験のデータに基づいて補充新薬申請を提出する。
FDAは4種類のMEK阻害剤を承認した。BRAF V 600 EまたはV 600 K変異を含む腫瘍の治療のための3つの薬剤が承認されており、Cobimetinibを含み、市場名はCotelicである®遺伝子テークによって販売されています®ノバ社が提供する;ビニミチニブはMektoviの名前で販売されている®ファイザー社が提供しています。4つ目のMEK阻害剤-selumetinib、市場名はKoselugo®アスリカン-1型神経線維腫症またはNF 1を有する2歳以上の児童患者の治療に許可されており、これらの患者は症状があり、手術できない。
百済神州には2つの次世代BRAF方案がある:リフェラファニー(BGB-283)とBGB-3245であり、前者は現在ミダミニと連合した1/2期試験中であり、後者は現在1期用量増加研究の単剤中にある。Hanmi/GenentechはBelvarafenibとcobimetinibを併用した1 b期臨床試験を開発している。FORE Treateutics社(前身はNovellusDx)はRAFダイマー切断剤PLX 8394を開発しており,1/2期試験であり,Cobicistatと併用している。Kinnateは単一療法の第1段階臨床試験であり,MEK阻害剤ビニチニブと併用した1 b段階臨床試験でもあるKIN−2787を開発している。ブラックダイヤモンド治療会社が臨床前に開発した異なる段階には次世代BRAF阻害剤がある。Jazz製薬会社とRedx社は,汎RAF阻害剤JZP 815が臨床開発段階に入り,第一段階試験にあると発表した。Erascaは最近、それはすでにノワール社とNaporafenibの全世界独占許可協定を達成し、Naporafenibは第2段階の枢軸が整った汎RAF阻害剤であり、NRAS変異黒色腫と他のRAS/MAPK経路によって駆動される腫瘍の治療において潜在的な一流と最適な特徴を持っていることを発表した。
PLGGの治療については,いくつかのMEK阻害剤およびいくつかのI型RAF阻害剤の他の標的治療が学術研究者による臨床試験で行われており,一部の地域ではラベル外で使用されている可能性がある。これらの薬物の非ラベル使用はtovorafenib(DAY 101)が市場に進出した際の競争を表している可能性がある。
私たちの多くの競争相手は、単独でも彼らとのパートナーでも、私たちより多くの財務資源を持って、市場で足場を立ち、そして研究開発、製造、臨床前と臨床テストの方面で専門知識を持って、監督部門の許可を得て、そして精算とマーケティングの許可を得た製品です。
特に大手製薬と生物技術会社は、臨床テスト、監督管理の許可を得、患者の募集と生物技術製品の候補製品の製造において豊富な経験を持っている。これらの企業の研究、マーケティング、販売能力も私たちよりはるかに優れており、承認されたか、開発後期の段階にある候補製品や、ターゲット市場でリーディングカンパニーや研究機関との協力手配も可能である。古い製薬会社やバイオテクノロジー会社も、新しい化合物の発見と開発を加速させたり、私たちが開発した候補製品を時代遅れにする可能性のある新しい化合物の使用を許可したりするために投資する可能性がある。規模が小さい、または初期段階にある企業も、特に大型および成熟会社との協力を通じて、私たちの計画の補完または必要な技術を得る上で重要な競争相手であることが証明される可能性がある。
これらのすべての要因により、私たちの競争相手は、FDAや同様の外国規制機関の承認を得ることに成功したり、私たちの前に私たちの分野の候補製品を発見、開発、商業化したりすることができ、これは、私たちの競争相手が市場に入る前に強力な市場地位を確立したり、私たちの開発をより複雑にしたりする可能性があります。
59
もし私たちの競争相手が私たちが開発する可能性のあるどんな製品よりも安全で、より効果的で、副作用が少なく、より便利で、より広いラベル、より効果的なマーケティング、より広範な精算、またはより安い製品を開発し、商業化すれば、私たちの潜在的なビジネス機会は減少または消滅するかもしれない。私たちが開発した候補製品が市場の承認を得ても、その時までにどんな競争製品も承認されていれば、それらの価格は競争相手の製品よりもはるかに高くなり、競争力が低下する可能性がある。私たちの競争相手が開発した技術の進歩や製品は、私たちの技術や候補製品を時代遅れにし、競争力に欠けたり、経済的ではないかもしれません。もし私たちが効果的に競争できなければ、私たちが開発する可能性のある製品を販売することから収入を得る機会は不利な影響を受けるかもしれない。
Tovorafenib(DAY 101)、pimasertib、または私たちが開発する可能性のある任意の将来の候補製品に関連する不良副作用、他の安全リスクまたは美容副作用は、承認を延期または阻止する可能性があり、臨床試験の一時停止またはさらなる開発の放棄、承認製品の商業イメージの制限、または上場承認後に重大な負の結果をもたらす可能性がある(あれば)。
薬品の一般的な状況と同様に,我々の主要候補製品tovorafenib(DAY 101)と我々の他の候補製品に関連する副作用や有害事象が観察された。これらの副作用は斑丘疹,貧血,頭痛,血筋酸ホスファターゼ(CPK)の上昇,嘔気,皮膚と髪の変色および疲労を含む。
われわれが行って計画中の臨床試験の結果は,副作用や意外な特徴の高さと受け入れられない重症度と流行率を明らかにする可能性がある。私たちの候補製品による不良副作用は私たち或いは監督機関が多種の原因で臨床試験を延期、一時停止或いは中止することを招く可能性がある。また,臨床試験の本質は潜在的な患者集団のサンプルを利用することである。被験者の数および曝露時間が限られているため、私たちの候補製品または競合他社製品のまれかつ深刻な副作用は、薬物に接触した患者数が著しく増加した場合にのみ発見される可能性がある。
さらに、私たちの候補製品を使用して治療された患者は、内科、外科、放射線、および化学療法治療を受けている可能性があり、これらの治療は、私たちの候補製品とは無関係な副作用または有害事象をもたらす可能性があるが、依然として私たちの臨床試験の成功に影響を及ぼす可能性がある。重篤な患者を著者らの臨床試験に組み入れることは死亡或いはその他の不良医療事件を招く可能性があり、原因はこれらの患者は他の治療方法或いは薬物を使用している可能性があり、或いはこれらの患者の病状が深刻であるためである。例えば、私たちの将来の臨床試験に登録される予定のいくつかの患者は、私たちの臨床試験中に、またはそのような試験に参加した後に非治療関連原因で死亡したり、重大な臨床イベントを経験したりすることが予想され、これは、tovorafenib(DAY 101)、pimasertib、または我々の他の候補製品の開発に影響を与える可能性がある。もし私たちが任意の臨床試験を延期、一時停止、または終了することを選択または要求された場合、私たちの候補製品の商業的将来性は損なわれ、候補製品から製品収入を創出する能力は延期またはキャンセルされるだろう。臨床試験で観察された深刻な有害事象やSAEは、市場が私たちの候補製品を受け入れることを阻害または阻止するか、あるいは医師が特定の患者で私たちの製品を使用することを期待する持続時間を短縮する可能性がある。このようなどんな状況でも、私たちの業務、見通し、財政状況、そして経営結果を深刻に損なう可能性がある。
さらに、私たちの候補製品が臨床試験において副作用に関連しているか、または予期しない特徴を有する場合、私たちは、その開発をより狭い用途または集団に制限することを選択することができ、リスク-利益の観点から、副作用または他の特徴は、それほど一般的ではなく、それほど深刻ではない、またはより容易に受け入れられ、これは、承認されれば、候補製品の商業的期待を制限する可能性がある。私たちはまた臨床試験の結果に基づいて私たちの研究計画を修正することが要求されるかもしれない。このような副作用は、患者の募集或いは入選患者が試験を完成する能力にも影響する可能性がある。最初に早期テストで希望を示した薬物の多くはその後副作用が認められ,さらなる開発を阻害した。さらに、規制当局は、これらの決定を確認するための追加のテストを要求し、より厳しいラベルを要求したり、規制部門の候補製品の承認を拒否したりする異なる結論を出すかもしれない。
私たちがより大きく、より長く、より広範な臨床試験で私たちの候補製品を試験する時、異なる用量レジメンの使用、または任意の規制承認後、私たちの候補製品の使用がより広くなり、患者は早期試験で観察された疾患、傷害、不快感、および他の有害事象、および以前の試験で発生しなかったか、または検出されなかったことを報告するかもしれない。これらの副作用が開発後期にまたは承認された後に知られた場合(あれば)、これらの発見は、私たちの業務、財務状況、運営結果、および将来性に深刻な損害を与える可能性があります。
60
また、もし私たちの任意の候補製品が発売許可を得て、私たちまたは他の人が後にこの薬物治療による不良副作用を発見したら、いくつかの潜在的な重大な負の結果を招く可能性がある:
これらの事件のいずれも、私たちの候補製品に対する市場の受け入れ度を達成または維持することを阻止することができ、承認されれば、私たちの業務、財務状況、運営結果、および将来性を深刻に損なう可能性がある。
著者らが時々発表或いは公表した臨床試験の初歩、中期、初期と背線データはより多くの患者データの獲得に従って変化する可能性があり、そして監査と検証プログラムの影響を受け、これは最終データの重大な変化を招く可能性がある。
私たちは時々、私たちのtovorafenib(DAY 101)試験の重要な第2段階の初期中期データおよびバックラインデータ分析のような、私たちの臨床試験の初期、中期またはバックラインデータを開示するかもしれない。これらの更新は,当時入手可能なデータの初歩的な分析に基づいており,特定の研究や実験に関するデータをより網羅的に審査したところ,結果や関連する調査結果や結論が変化する可能性がある.また,我々が達成可能な臨床試験の中期データは,患者登録の継続とより多くの患者データの獲得に伴い,1つまたは複数の臨床結果が実質的に変化する可能性があるというリスクに直面している。したがって,どのような進行中の臨床試験においても,積極的な中期あるいは初歩的な結果は,完成した研究や試験の結果を予測できない可能性がある。私たちはまた、私たちのデータ分析の一部として、すべてのデータを全面的かつ詳細に評価する機会がないか、または受け取る機会がないかもしれないという仮説、推定、計算、および結論を出した。したがって、我々の報告の初期結果またはバックライン結果は、同じ研究の将来の結果と異なる可能性があり、またはより多くのデータを受信して十分な評価を行うと、異なる結論または考慮要因がこれらの結果を合格させる可能性がある。初期データやバックラインデータも監査や確認手続きを受ける必要があり、最終データは以前に公表された予備データと大きく異なる可能性がある。
さらに、規制機関を含む他の人は、私たちの仮定、推定、計算、結論または分析を受け入れないか、またはデータの重要性を異なる方法で解釈またはトレードオフする可能性があり、これは、特定の計画の価値、特定の候補製品または製品の承認または商業化、およびわが社の全体的な状況に影響を与える可能性がある。さらに、私たちが開示する特定の研究または臨床試験に関する開示を選択する情報は、一般に広範な情報に基づいており、あなたまたは他の人は、私たちが開示すべき重大な情報または他の適切な情報を含むことに決定したことに同意しないかもしれないが、私たちが開示しないことを決定した任意の情報は、最終的には、特定の薬剤、候補薬剤、または私たちの業務に関する将来の決定、結論、観点、活動、または他の側面に対して大きな意味を有すると考えられるかもしれない。もし私たちが報告したバックラインデータが実際の結果と異なる場合、あるいは規制機関を含む他の人が結論に同意しない場合、私たちが承認を得て私たちの候補製品を商業化する能力が損なわれる可能性があり、これは私たちの業務、経営業績、見通し、または財務状況を損なう可能性がある。
61
私たちは、より利益的または成功可能性の高い候補製品または適応を利用することなく、特定の候補製品または適応を追求するために限られたリソースを費やす可能性がある。
私たちの財務と管理資源が限られているため、私たちは私たちが確定した特定の適応の研究プロジェクトと製品に重点を置いています。したがって、私たちは他の候補製品を探す機会を放棄または延期するか、または後により大きな商業潜在力を有することが証明された他の兆候を探す機会を放棄または延期するかもしれない。私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。私たちの現在と未来の研究開発計画および特定の適応候補製品への支出はいかなる商業的に実行可能な製品も発生しないかもしれない。もし私たちが特定の候補製品の商業的潜在力や目標市場を正確に評価しなければ、私たちは協力、許可、または他の印税手配によって候補製品に価値のある権利を放棄するかもしれないが、この場合、候補製品の独占開発権と商業化権利を維持することは私たちに有利だ。
私たちが開発した任意の候補製品の市場機会は、承認されれば、いくつかの小さい患者亜群に限られる可能性があり、私たちが推定したものよりも小さいかもしれない。
幼稚および再発/進行性pLGGを治療する方法としてtovorafenib(DAY 101)の承認を求める予定である。私たちの候補製品がどの治療法にも承認されることは保証されませんが、いずれかの承認を得る前に、コストが高く、時間がかかり、リスクがある可能性がある追加の臨床試験を行わなければならないかもしれません。
私たちの目標癌患者数の予測と、特定の治療経路を受けることができ、私たちの候補製品治療から利益を得る可能性があるこれらの癌患者のサブセットは、私たちの信念と推定に基づいている。例えば、pLGGはまれな疾患であり、したがって、このような疾患を有する人数および候補製品治療から利益を得る可能性があるpLGG患者のサブセットの予測は、推定に基づいている。これらの推定は様々な源から来ており、科学文献、診療所調査、患者基金会或いは市場研究を含み、正しくないことが証明されている可能性がある。また、新しい研究は、私たちが対象とする癌の推定発症率や流行率を変えるかもしれない。私たちの候補製品の潜在的にアドレス指定可能な患者集団は限られているかもしれないし、あるいは私たちの候補製品の治療を受け入れられないかもしれない。したがって,候補製品が承認されても,我々の候補品を用いて治療を行う資格がある患者数は予想よりもはるかに少ない可能性がある。また,腫瘍タイプごとに異なる承認療法があれば,治療医が複数の腫瘍タイプのために承認された製品をどのように開発することが期待されるかを決定するための市場研究は行われていない。私たちの製品がかなりの市場シェアを獲得していても、承認されれば、潜在的なターゲット層が少なければ、規制部門のより多くの適応の承認を得なければ、決して利益を達成しないかもしれない。
62
我々の臨床開発活動は,主にゲノム定義された癌患者のための標的療法の開発に集中しており,急速に発展している科学分野であり,我々が講じている薬物の発見·開発方法は斬新であり,承認または販売可能な製品は決して生じない可能性がある。
遺伝子定義癌患者に対する標的療法の発見と開発は新興分野であり,候補製品の発見·確定·開発に努める基礎となる科学的発見が比較的新しい。これらの発見に基づく候補製品の開発可能性を支持する科学的証拠は初歩的であり、限られている。著者らは、著者らの候補製品の臨床前試験結果と著者らの臨床仕事に基づいて、著者らが計画したゲノム変化は発ガン要素であると信じているが、臨床結果はこの仮説を実証しないかもしれない、あるいはある変化或いはある腫瘍タイプに対してのみこの仮説を実証する可能性がある。我々の候補製品の患者集団は,特定の目標変化を有する患者に限られており,完全に定義されていない可能性があるが,一般的な治療の癌集団よりもはるかに少なく,これらの目標変化を有する患者をスクリーニングし識別する必要がある。患者の識別に成功することは、特定の変化が私たちの候補製品にどのように応答するかを決定することと、そのような変化を識別する能力とを含むいくつかの要因に依存する。また,患者の識別に成功しても,各変異による患者集団が十分大きいかどうかを決定することはできず,各変異型の承認を得ることに成功し,候補製品を商業化して利益を得ることができる。また,われわれの方法がRAF駆動癌に対するtovorafenib(DAY 101)計画の臨床的利益を示すことに成功しても,他のMAPK駆動腫瘍においてtovorafenib(DAY 101)に敏感な他の発癌変化を発見することは決して成功しないかもしれない。遺伝子定義癌患者を治療する方法が成功するかどうかはわかりません, もし私たちの方法が成功しなければ、私たちの業務は影響を受けるだろう。
私たちの候補製品は、医師、患者、あるいは彼らの家族、医療支払者、および商業成功に必要な医療界の他の人に十分な市場受容度を得ることができないかもしれない。
私たちの候補製品が規制部門の承認を得ても、それらは医師、患者、あるいは彼らの家族、第三者支払人、医学界の他の人の中で十分な市場受容度を得ることができないかもしれない。私たちが承認した候補製品の市場受容度は多くの要素に依存するだろう
63
もし私たちのすべての候補製品が承認されたが、医師、病院、医療支払者、および患者の十分な程度の受け入れを得られなければ、私たちはその候補製品から十分な収入を得ることができないかもしれず、私たちの財務業績は否定的な影響を受けるかもしれない。
私たちが開発したすべての候補製品は不利な第三者の保証と清算のやり方と定価法規の制約を受けるかもしれません。
第三者支払人は、政府衛生行政当局、個人健康保険会社、管理性医療組織と他の第三者支払人を含み、その保険範囲と範囲及び適切な精算は大多数の患者に高価な治療を負担できることに重要である。私たちの任意の候補製品がマーケティング承認を得た販売は、米国でも国際的にも、これらの候補製品のコストがどの程度第三者支払者によって支払われ、精算されるかに大きく依存する。精算が得られない場合、あるいは限られたレベルに限られていれば、候補製品を商業化することに成功できないかもしれません。保険を提供しても、承認された精算金額が十分に高くない可能性があり、十分な投資リターンを実現するために十分な価格設定を確立したり維持したりするのに十分ではないかもしれません。保証範囲と精算は、マーケティングの承認を得た任意の候補製品の需要や価格に影響を与える可能性があります。保険や精算が得られない場合や、精算が限られたレベルに限られている場合には、マーケティング承認を受けた任意の候補製品を商業化することに成功しない可能性があります。
第三者支払者のカバー範囲や新承認製品の精算に関する不確実性が大きい。米国では,小児科製品の支払者組合は各州特定の医療補助政策と広範な民間保険会社の支離滅裂な組み合わせである。他の価格設定エンティティに通知するための一貫した政策や主要支払者はいない。国家支払者政策は私たちが広範囲な支払いカバーを達成する能力に必須的だと予想される。しかしながら、第三者支払者がある候補商品に保険を提供することを決定することは、他の支払者がその候補商品に保険を提供することを保証することはできない。したがって、カバー範囲を決定するプロセスは、しばしば時間がかかり、高価である。この過程は、各第三者支払者にそれぞれ私たちの製品を使用する科学的かつ臨床的支援を提供することを要求するが、保険と適切な精算を一貫的に適用すること、または最初に十分な精算を得ることを保証することはできない。
連邦政府と州政府が処方薬の定価を下げる措置を含む追加の医療コスト制御措置を実施することに伴い、私たちの製品が承認された場合、個人または公共支払者が保証を受け、保険を受けた場合、精算金額が十分かどうか、あるいは他の市場製品と競争するかどうかを決定することはできない。連邦と州政府および医療計画のこれらおよび他の行動は、薬品の価格設定と医療コストに追加の下振れ圧力をもたらす可能性があり、承認されれば、これは私たちの製品のカバー範囲と清算、私たちの収入、および私たちが他の市場製品と競争し、私たちの研究開発コストを回収する能力にマイナス影響を与えるかもしれない。
ますます多くの第三者支払人は製薬会社に価格に基づいて所定の割引を提供し、医療製品の定価に挑戦することを要求している。また,これらの支払者は価格に挑戦し,医療の必要性を検査し,候補医療製品の費用対効果を審査することが増えている。新たに承認された薬物の保険や補償には,特に重大な遅延が生じる可能性がある。第三者支払者は、承認されたリスト上の特定の候補製品、いわゆる処方表にカバー範囲を制限することができ、FDAによって承認された特定の適応のすべての薬剤を含まない可能性がある。私たちは私たちの製品の医療の必要性と費用効果を証明するために高価な薬物経済学的研究を行う予定だ。それにもかかわらず、私たちの候補製品は医学的に必要で費用効果的だと思われないかもしれない。私たちが商業化したどの製品も保険と精算を受けることができることを確実にすることはできません。もし精算できるなら、清算レベルはいくらですか。
そのほか、セット診断テストは単独で保証と精算を受ける必要があり、しかもそのセットの薬品或いは生物製品の保証と精算を含まない。医薬品や生物製品に適用される保険獲得や精算に適した類似課題は随伴診断にも適用される。さらに、任意のセット診断提供者が精算や精算不足を得ることができない場合、このようなセット診断の可用性を制限する可能性があり、承認されれば、候補製品の処方に負の影響を与える。
64
米国以外では、治療薬の商業化は一般的に広範な政府価格制御と他の市場法規の制約を受けており、ヨーロッパ、カナダ、その他の国のコスト制御措置の日々の重視はすでに私たちの候補製品などの治療薬の定価と使用に圧力を与え続けると信じている。多くの国、特に欧州連合やEU諸国では、国家衛生システムの一部として、医療製品価格は異なる価格制御メカニズムの制約を受けている。これらの国では、製品が発売承認された後、政府当局との定価交渉にはかなりの時間がかかるかもしれない。一部の国で精算或いは定価の承認を得るためには、私たちの候補製品のコスト効果を他の利用可能な治療法と比較する臨床試験を行う必要があるかもしれない。一般的に、この制度での製品価格はアメリカよりずっと低い。他の国は会社が自ら価格を設定することを許可しているが、会社の利益を監視する。追加の外国価格規制や定価規制の他の変化は、私たちの候補製品に受け取ることができる費用を制限するかもしれません。そのため、米国以外の市場では、米国に比べてわが製品の精算が減少する可能性があり、商業的に合理的な収入や利益を生み出すには不十分である可能性がある。
第三者支払者の任意の候補製品のために保証範囲を確立または維持することができず、十分な精算ができない場合、これらの製品の採用および販売収入は悪影響を受け、承認された場合、これは逆にこれらの候補製品をマーケティングまたは販売する能力に悪影響を及ぼす可能性がある。保証政策と第三者支払人の販売率はいつでも変化する可能性があります。規制部門の承認を得た1つまたは複数の製品が有利な引受·精算状態を獲得しても、将来的にはあまり有利ではない引受政策や精算料率が実施される可能性がある。
私たちの業務には製品責任の重大なリスクがあり、十分な保険範囲を得ることができなければ、このような保証ができなければ、私たちの業務や財務状況に悪影響を及ぼす可能性があります。
私たちの業務は治療療法の開発、テスト、製造、マーケティングの過程に固有の重大な製品責任リスクに直面しています。製品責任クレームは私たちの開発計画の完了を延期または阻止する可能性があります。もし私たちがマーケティング製品の面で成功すれば、このような声明はFDAまたは他の規制機関が私たちの製品、私たちの製造プロセス、施設、または私たちのマーケティング計画の安全性と有効性を調査することにつながるかもしれない。FDAまたは他の規制機関の調査は、これらの製品が使用可能な承認適応を制限するか、または承認を一時停止または撤回するために、我々の製品をリコールするか、またはより深刻な法執行行動をとることをもたらす可能性がある。是非曲直あるいは最終結果にかかわらず、責任クレームは私たちの製品に対する需要の減少、私たちの名声に対する損害、関連訴訟の弁護コスト、管理層の時間と資源の分流及び試験参加者或いは患者への巨額の金銭奨励を招く可能性がある。私たちは現在、製品責任保険があります。これは私たちの開発段階に適していて、私たちの候補製品が臨床試験に入ったり、私たちの任意の候補製品をマーケティングしたりする前に、より高いレベルを得る必要があるかもしれません。私たちが持っているか得ることができるどんな保険も潜在的な責任に十分な保険を提供できないかもしれない。しかも、臨床試験と製品責任保険はますます高くなっている。したがって、私たちは製品責任クレームによる損失から私たちを保護するために合理的なコストで十分な保険を得ることができないかもしれません。これらの損失は私たちの業務や財務状況に悪影響を及ぼす可能性があります。
政府の規制に関連するリスク
薬品の開発と商業化は広範な監督管理を受けており、私たちはトロヴォラフィニ(DAY 101)、ピマ司あるいは任意の未来の候補製品の監督管理許可を迅速に或いは根本的に得られないかもしれない。
臨床開発、製造、ラベル、包装、保存、記録、広告、販売促進、輸出、輸入、マーケティング、流通、有害事象報告、安全性および他の発売後の情報と報告の提出、およびtovorafenib(DAY 101)とpimasertibに関連する可能性のある他の活動、および私たちが将来開発する可能性のある他の任意の候補製品は、広く規制されている。米国での医薬品の発売承認はNDAをFDAに提出する必要があり、FDAのNDA承認を得るまで、米国での候補製品の販売は許可されていません。NDAは大量の臨床や臨床前データ,薬理学,化学,製造,制御に関する大量の情報によって支持されなければならない。私たちの候補製品は商業化される前に他の管轄区域のような規制機関の承認を受けなければならない。
65
NDAに対するFDAの承認は保証されているわけではなく、審査および承認プロセスは高価で不確実なプロセスであり、数年かかるかもしれない。米国で大量に開発されている薬物のうち,一部のみがFDAの規制承認手続きに成功し,商業化されている。したがって、私たちのすべての候補製品がアメリカや他の司法管轄区域で規制されるという保証はない。
FDAは承認過程でも大きな裁量権を持っている。NDAの承認に必要な臨床前研究および臨床試験の数量およびタイプは、候補製品、疾患または候補製品設計のための条件、および任意の特定の候補製品に適用される法規によって異なる。例えば,成功すれば,重要な2期Firefly−1試験tovorafenib(DAY 101)は,tovorafenib(DAY 101)のNDAをFDAが承認するのに十分である可能性があると考えられるが,FDAは我々のデータの十分性に同意しない可能性があり,より多くの臨床試験が必要である。また,tovorafenibのキー2期Firefly−1試験(DAY 101)の結果から,tovorafenib(DAY 101)の加速承認が得られる可能性があり,この薬剤の臨床的利益を検証するためには,より多くのデータと潜在的検証試験が必要となる。臨床前研究や臨床試験に関連する時間や費用は高いにもかかわらず,失敗はどの段階でも発生する可能性がある。Tovorafenib(DAY 101)またはpimasertibまたは任意の他の候補製品の臨床前および早期臨床試験結果は、我々の後期臨床試験の結果を予測できないかもしれない。また,競争相手が一線pLGGで行っている臨床試験が知られており,FDAや他の規制機関は,我々の一線計画を評価する際にこれらの試験の設計や結果を考慮する可能性があると信じている。例えば、安定した疾患のような患者のいくつかの結果を信じ、臨床活動を奨励することを示すかもしれないが、安定した疾患は、全体応答率またはOORの終点を評価する調節目的における反応とはみなされない。
さらに、将来の立法や行政行動における追加的な政府規制、または製品開発、臨床試験、審査過程における規制機関の政策の変化による遅延や拒否に遭遇する可能性がある。例えば、2022年5月に、FDA内の腫瘍学卓越センターは最近Project Optimusを提出し、これは腫瘍学薬物開発中の用量最適化と用量選択範式を改革する提案であり、最適な用量を選択すること、即ち薬物治療効果を最大化するだけでなく、安全性と耐性の1つ或いは複数の用量を最大化することを強調する。このような転換は、従来一般的に最大耐性用量を決定する方法とは異なり、ターゲット集団における最適な用量選択を促進するために、候補製品の用量-反応関係をさらに探索するために、スポンサーがより多くの時間と資源を費やす必要があるかもしれない。腫瘍学卓越センターの最近の他の計画は、多くの以前の一連の治療を受けたか、または利用可能な治療選択を使い切った患者を治療するための、より早い末期環境において初期臨床開発のための候補薬剤を決定するための枠組みを制定することを目的とした新しい計画であるProject Foretrunnerを含む。
臨床試験失敗は多種の要素によるものである可能性があり、試験設計、用量選択、プラセボ効果、患者登録標準及び良好な安全性或いは有効性特徴を証明できず、臨床試験失敗は任意の段階で発生する可能性がある。製薬業界の会社は,早期の試験で良好な結果が得られたにもかかわらず,治療効果や不良安全性の欠如により臨床試験の進展に挫折することが多い。陰性または不確定の結果から、私たちは決定することができますか、あるいは監督機関は追加の臨床試験または臨床前研究を要求するかもしれません。また、臨床試験から得られたデータは異なる解釈の影響を受けやすく、監督管理機関は私たちのように私たちのデータを有利に解釈しない可能性があり、これは更に上場承認を延期、制限或いは阻止する可能性がある。FDAは、FDAが可能であるため、多くの理由で承認候補製品を延期、制限、または拒否することができる
66
私たちはまだFDAのいかなる製品に対する承認も得ていない。このような経験の欠如は,我々がFDA承認をタイムリーに得る能力を阻害する可能性があり,もしあれば,我々の臨床製品候補となる。また、FDAの承認を得ても、外国の管轄地域の比較可能な規制機関から同様の承認を得る保証はありません。これは私たちの潜在的な市場を制限し、私たちの業務、将来性、財務状況、運営結果に悪影響を及ぼす可能性があります。
もし私たちが承認を得る上で遅延に遭遇した場合、またはtovorafenib(DAY 101)やpimasertibまたは私たちの将来の候補製品の承認が得られなかった場合、私たちのビジネスの見通しは損なわれ、私たちの収益能力は実質的に損なわれ、これは私たちの業務、見通し、財務状況、および運営結果に悪影響を及ぼすだろう。
私たちの候補製品の承認プロセスの加速は、より速い開発や規制審査や承認過程をもたらすことができない可能性があり、私たちの候補製品が上場承認される可能性を増加させることはありません。
FDAの加速承認計画によれば、FDAは、患者に既存の治療よりも意義のある治療利益を提供する深刻または生命に危険な疾患に対する薬剤を承認することができ、その基礎は、臨床利益を合理的に予測する可能性のある代替終点、または不可逆的な発症率または死亡率よりも早く測定することができる臨床終点であり、不可逆的な発症率または死亡率または他の臨床的利益への影響を合理的に予測することができ、同時に、病状の深刻性、希少性または流行率、および代替療法の利用可能性または不足を考慮することである。私たちはORRによって私たちの1つまたは複数の候補製品の承認を加速することを求めるかもしれないが、OORは臨床的利益を合理的に予測することが可能だと考えられる代替終点である。
加速承認を得た薬物に対して、発売後の検証性試験は不可逆的な発病率或いは死亡率或いは他の臨床利益に対する期待影響を記述する必要がある。これらの検証試験は、職務調査の場合に完了しなければならず、多くの場合、FDAは、承認前に設計、起動、および/または完全に試験に組み込むことを要求する可能性がある。もし私たちの競合他社が加速承認を得る前に加速承認を求めている適応の完全な承認を得た場合、私たちが求めている指示はもはや条件を満たしていないかもしれない、すなわち満たされていない医療ニーズが存在し、候補製品の加速承認がより困難になるか、起こらない可能性がある。また、以下の場合、FDAは、承認経路の加速下で承認された候補製品の承認を撤回する可能性がある
67
最近、承認を加速させる経路はFDA内部でも国会でも審査された。FDAはより多くの重点を責任を果たして検証的な研究を行うことに重点を置き、最終的にこのような研究が利益を実証することを確保した。例えば、FDAは、その腫瘍薬物諮問委員会を招集して、FDAで呼ばれる未解決または遅延の加速承認を審査し、これらの承認は、検証的研究が完了していない場合、または結果として利益が確認されていない場合に行われる。また、2021年、腫瘍学卓越センターは、腫瘍学適応の加速承認に関連する結果の透明性を促進し、承認と発売後のプロセスにおける議論、研究、革新を促進するための枠組みを提供することを目的とした確認プロジェクトを発表し、癌と血液悪性腫瘍患者の利用可能な治療の獲得と利益検証のバランスを強化することを目的としている。また,国会では,このような場合に承認を撤回する可能性を増やす提案を含む,承認経路の変更を加速する可能性のある様々な提案が考えられている.
FDA指定されたpLGG治療の画期的な薬物tovorafenib(DAY 101)を取得しても、この指定はより速い開発や規制審査や承認過程につながらない可能性があり、tovorafenib(DAY 101)が上場承認される可能性は増加しない。
著者らはすでにFDAによる末期pLGG患者のトレフラフィニ(DAY 101)に対する突破的治療指定を獲得した。画期的な治療法は、1つまたは複数の他の薬剤と単独でまたは1つまたは複数の他の薬剤と組み合わせて重篤または生命を脅かす疾患または状態を治療することを意図した薬剤として定義され、初歩的な臨床証拠は、1つまたは複数の臨床的に重要な終点において、臨床開発早期に観察される実質的な治療効果のような既存の療法よりも有意な改善を示す可能性があることを示す。画期的な治療法として指定された薬物に対して,FDAと試験スポンサーとの相互作用やコミュニケーションは,臨床開発の最も有効な経路を決定するのに役立つとともに,無効対照案中の患者数を最小限にすることができる。FDAで画期的な治療法に指定されている薬物は,NDA提出時に臨床データの支持を得ていれば,優先審査を受ける資格もある。
画期的な治療指定や他の加速計画の獲得は開発や承認過程を加速させる可能性があるにもかかわらず,承認の基準を変更することはない。末期pLGGでtovorafenib(DAY 101)の画期的な治療称号を得たにもかかわらず、従来のFDAプログラムと比較して、より速い開発時間を経験したり、より速い審査や承認を達成したりすることはないかもしれない。例えば、製造および制御に関連する問題を識別および解決するのに要する時間は、臨床試験目的のために十分な製品供給を得ること、または追加の非臨床的または臨床的研究を必要とすることは、画期的な治療認証を取得する資格があっても、または任意の他の加速計画を取得する資格があっても、FDAの承認を遅らせる可能性がある。FDAが我々の臨床開発計画のデータがこの指定を支持しなくなったと考える場合,加速計画へのアクセスを撤回する可能性もある。しかも、いかなる迅速な審査手続きの資格も、私たちが最終的にこのような製品に対する規制部門の承認を得ることを保証していない。
私たちは私たちの候補製品の孤立した薬物指定または独占経営権を獲得したり維持できないかもしれない。
われわれは米国とEUでそれぞれ悪性グリオーマとグリオーマの治療のためのトヴォラフェニ(DAY 101)の孤児薬物指定を獲得した。私たちは、より多くの地域または適応においてtovorafenib(DAY 101)のための孤児薬物名を求めるか、またはpimasertibまたは私たちが将来開発する任意の候補製品のために孤児薬物名を求めるかもしれない。米国を含むいくつかの管轄区の規制機関は、患者数が相対的に少ない薬物を“孤児薬”とする可能性がある。孤児医薬品法によれば、1つの薬剤がまれな疾患または疾患の治療を目的としている場合、または米国における疾患または疾患の影響が20万人を超え、米国での製品の販売から開発コストを回収できることが合理的に期待されていない場合、FDAはその薬剤を孤児薬として指定することができる。米国では、まれな疾患または疾患の定義は、通常、患者数が20万人未満である。
68
一般に、孤児薬物名を有する製品がその後、その名称を有する適応の最初の発売許可を得た場合、製品は、一定期間内に市場排他期間を得る権利があり、これにより、FDAは、その間に同じ薬物の同じ適応の別のマーケティング申請を承認することができない。適用期間はアメリカでは7年、EUでは10年だ。薬物が孤児薬物指定の基準に適合しなくなった場合、またはその薬物の利益が十分に高く、市場排他性がもはや合理的でない場合、EUの排他的期限は6年に短縮されることができる。FDAが指定要求に重大な欠陥があると判断した場合、または製造業者がこのようなまれな疾患または疾患を有する患者の需要を満たすのに十分な数の薬剤を保証できない場合、米国における孤児薬の排他性を失う可能性がある。
場合によっては、FDAは、後続製品が臨床的利点を示す場合(すなわち、後続製品が孤児排他性を有する製品よりも安全で、より効果的であるか、または患者ケアに重大な貢献がある場合)のように、排他的期間内に同じ適応で同じ薬剤を販売する後続の出願を許可することができる。しかし,競争相手は孤児製品が排他的な同一適応により異なる製品の承認を得たり,同じ製品の承認を得たりする可能性があるが,その適応は孤児製品と排他的な適応とは異なる。孤児薬物指定は,一方に臨床試験費用,税収割引,ユーザ費用減免のための贈与資金の機会を提供するなど,財政的インセンティブを得る権利がある。
私たちはあなたに未来の他の候補製品に対する孤児薬物指定申請が承認されるということを保証することができません。米国や他の管轄地域の他の候補製品の孤児薬物指定を得ることができなければ,孤児薬物指定による市場排他期を得る資格もなく,孤児薬物指定に関する経済的インセンティブを得る資格もない。
また,第11巡回裁判所は最近,Catalyst PharmPharmticals,Inc.がFDA案で下した裁決を訴え,孤児の適応として承認された薬物の解釈,すなわちその薬物の孤児指定範囲よりも狭い孤児薬物法案に適用される排他的条項に関連しており,このような製品の孤児薬物の排他性の範囲を著しく拡大する可能性がある。Catalyst決定に対するFDAの適用範囲がどれだけ広いかに依存して,会社依存や孤児薬物の排他的解決を求める方式を根本的に変える可能性がある。Catalyst決定を覆す可能性のある立法も提案されているが,この立法はまだ初期段階であり,まだ採択されていない。
私たちは私たちの1つ以上の候補製品のために珍しい小児科疾患の称号を求めるかもしれない。たとえ我々の製品候補が稀な小児科疾患の称号を有する承認を得たとしても、稀な小児科疾患優先審査券計画は承認された時に有効ではなくなる可能性があり、あるいは稀な小児科疾患優先審査券計画の価値を得ることができない可能性がある。
Tovorafenib(DAY 101)は2021年5月にFDAに稀な小児科指定薬物を授与され、低レベルグリオーマ(LGG)の治療に使用され、この薬物は活性化RAF変化を有し、児童への影響は比例しない。我々は,最初のtovorafenib(DAY 101)NDAをまれな小児科指定上場申請として提出する予定であり,FDAは審査後に指定してもこのような申請を指定しない可能性がある。
国会はFDAが特定の基準を満たすいくつかの稀な小児科疾患製品申請のスポンサーに優先審査クーポン券を付与することを許可した。これらのクーポン券はある稀な小児科疾患の予防と治療のための新薬と生物製品の開発を奨励することを目的としている。
具体的には、この計画によれば、スポンサーが薬物または生物製剤の承認を得た場合、異なる製品の後続マーケティング申請の優先審査に交換することができるクーポン券を得ることができる。優先審査証明書を取得したまれな小児科疾患医薬製品の発起人は、(販売を含む)証明書を別の発信者に譲渡することができる。譲渡を行うスポンサーがまだ申請を提出していない限り、その証明書を使用する前に、その証明書は、さらに任意の回数譲渡されることができる。クーポン券は第三者に売却または譲渡することができるが,このようなクーポン券を受け取ることができる保証はないし,クーポン券を受け取って販売しようとすれば,任意の価値を実現することができる.
本計画において、稀な小児科疾患は(I)深刻或いは生命に危害を及ぼす疾患であり、その深刻或いは生命に危害を及ぼす表現は主に新生児、乳児、児童と青少年と呼ばれる年齢層を含む出生から18歳までの個人に影響を与える;および(Ii)“孤児薬物法”が指す稀な疾病或いは状況を含む。FDAは、承認後、私たちの1つまたは複数の候補製品の申請が優先審査券の資格基準を満たしていないと判断することができる。
69
また,2020年9月30日までに指定資格を取得していない会社が優先審査券を取得する機会は満了すべきであるが,国会は2020年12月にまれな小児科疾患優先審査券計画を延長した。現在の法定日没条項によると、FDAは、2024年9月30日以降、スポンサーにまれな小児科疾患薬指定があり、その指定が2024年9月30日までに付与された場合にのみ、承認された珍しい小児科疾患製品申請にクーポン券を発行することができる。2026年9月30日以降、FDAはいかなる稀な小児科疾患優先審査クーポンを付与しない可能性がある。
FDAの迅速なチャネル指定を求めることにした場合、より速い開発や規制審査や承認プロセスを招くことはないかもしれません。
私たちは私たちの1つ以上の候補製品のための迅速なチャネル認証を求めるかもしれない。重症または生命に危険な疾患の治療に使用され、そのような疾患が満たされていない医療需要を解決する可能性を示す薬剤がある場合、製品スポンサーはFDA迅速チャネル認証を申請することができる。FDAは幅広い裁量権を有しており,この称号が付与されているかどうか,したがって,ある特定の候補製品がこの称号を得る資格があると考えても,FDAがそれを付与することを決定することを保証することはできない。私たちが高速チャネル認証を受けたとしても、私たちは従来のFDAプログラムよりも速い開発過程、審査、または承認を経験しないかもしれない。FDAが我々の臨床開発プロジェクトのデータが高速チャネルの指定をサポートしなくなったと考える場合,その指定を撤回する可能性がある。
もし私たちが開発、検証、監督機関のセット診断テストを必要とする候補製品の承認を得て商業化することができなければ、あるいはそうする過程で重大な遅延に遭遇することができなければ、私たちはこれらの候補製品のすべての商業潜在力を実現することができないかもしれない。
診断とは医療機器です通常は体外培養装置、対応する治療薬製品を安全かつ有効に使用するために必要な情報を提供する。随伴診断は、治療製品から利益を得る可能性が最も高い患者を識別するために使用することができる。将来,セット診断を開発する必要があれば,自分やパートナーと私たちの候補製品のために何らかの適応の診断テストを開発する機会を評価する可能性がある。
随伴診断は通常,関連治療製品の臨床プログラムとともに開発される。これまで、FDAは大多数の癌治療のセット診断方法が発売前に承認されることを要求してきた。一般に、診断に伴う診断が医薬製品の安全かつ有効な使用に重要である場合、FDAは、診断に伴う診断が治療製品を承認する前または同時に承認され、製品が商業化される前に必要であることを要求する。診断に伴う診断を治療製品ラベルの一部として承認し,治療製品の使用を診断に伴う検出のための特定の遺伝子変化を発現する患者に制限する。
セット診断の開発には、前会議の提出および調査装置免除申請の提出の要件など、規制当局との追加会議が含まれる可能性がある。“重大リスク機器”に指定された随伴診断については,この診断を対応する候補製品の臨床試験と組み合わせて使用する前に,FDAおよびIRBによる研究装置免除の承認を得る必要がある。
70
開発、検証、承認を得てセット診断を獲得し、それを商業化するために、著者ら或いは著者らの協力者はいくつかの科学、技術、監督管理と後方勤務方面の挑戦を解決する必要がある。私たちはこれまで医療設備や診断テスト開発の経験がありません。もし私たちが独自に開発し、FDAのセット診断テストの承認を求めることを選択すれば、私たちは追加の人員が必要になるだろう。私たちは第三者が私たちの候補治療製品の設計、開発、テスト、検証と製造のためのセット診断テスト、任意の必要な法規の承認、およびこれらのセット診断の商業供給を申請し、受け入れることに依存する可能性がある。これらの候補治療製品の開発に成功した場合、あるいは開発を遅延させることができない場合、私たちは現在および計画されている臨床試験のために十分な患者を募集することができない可能性があり、これらの候補治療製品の開発は不利な影響を受ける可能性があり、これらの候補治療製品はマーケティング承認を得ることができない可能性があり、私たちはマーケティング許可を得たこれらの治療薬のすべての商業的潜在力を実現できないかもしれない。製品候補製品の使用から利益を得る可能性のある患者の候補製品を選択するためにセット診断を必要とする任意の候補製品について、セット診断の開発に成功しなかったいかなる場合も、著者らの臨床試験の遅延登録を招くか、或いは促進する可能性があり、そして著者らが肝心な試験を開始することを阻止する可能性がある。また、, 私たちがセット診断を必要とする候補製品の商業的成功は、必要な規制承認を得るかどうか、およびそのような第三者が関連地域で合理的な条項でセット診断の持続能力を提供してくれるかどうかにかかっている。医師がどのような特定の随伴診断を採用するか,それをどのように使用したいか,どのように補償を得るか,それを患者にどのように説明するか,あるいはスタッフを指定して使用することは保証されない。これができなかったすべてのことは、私たちの業務、運営結果、そして財務状況に実質的な損害を与える可能性がある。
私たちが私たちの候補製品のために市場承認を得ても、承認された条項、私たちの製品の持続的な規制、あるいは他の承認後の制限は、私たちの製品の製造とマーケティングの方法を制限するかもしれません。これらの要求を遵守することは大量の資源に関連する可能性があり、これは私たちの収益能力を深刻に弱めるかもしれません。
FDAによって加速的に承認された任意の候補製品は、1つまたは複数の検証的臨床試験を受けなければならない。もしこのような候補製品がこのような検証性臨床試験においてその安全性と有効性の終点に達しなかった場合、監督管理機関はその条件付き承認を撤回することができる。どのような製品もその検証性臨床試験に成功することは保証されない。したがって,候補製品がFDAの加速承認を得ても,この承認は以降の日に撤回される可能性がある。候補製品が上場承認されても、承認された製品およびその製造業者および営業業者は、持続的な審査および広範な規制を受ける必要があり、その中には、製品の安全性または有効性を監視するために、再生可能エネルギー管理システムの実施または高価な発売後の研究または臨床試験および監視を要求することが含まれている可能性がある。
私たちはまた広告と販売促進に関する要求を守らなければなりません。私たちのすべての候補製品のために、私たちは市場の承認を得ました。処方薬に関する販売促進情報は様々な法律や法規によって制限されており,製品が承認されたラベル中の情報と一致しなければならない。したがって、私たちが開発した未承認の適応や用途のための製品を広めることはできません。
さらに、承認された製品の製造業者およびその工場は、品質管理および製造プロセスが現在の良好な製造実践、すなわちcGMPに適合することを確実にしなければならず、その中には、品質管理および品質保証に関連する要求、および対応する記録および文書の維持および報告要件が含まれている。我々と我々のCMOはFDAの定期的な抜き打ち検査を受け,cGMPのコンプライアンスを監視·確保する可能性がある。
したがって、私たちの1つ以上の候補製品が市場承認を得たと仮定すると、私たちと私たちのCMOは製造、生産、製品監視、品質管理を含むすべてのコンプライアンス分野で時間、お金、エネルギーをかけ続けるだろう。承認後の規制要件を遵守できなければ、規制機関によって当社製品のマーケティング承認を撤回される可能性があり、将来の製品をマーケティングする能力が制限される可能性があり、利益を達成したり維持したりする能力に悪影響を及ぼす可能性があります。そのため、承認後の法規を遵守するコストは、私たちの経営業績や財務状況に悪影響を及ぼす可能性があります。
71
私たちが上場承認を得た任意の候補製品は、規制機関が継続的に実行している上場後要求の制約を受け、もし私たちがすべての規制要求を遵守できなかった場合、あるいは私たちの製品が予期せぬ問題に遭遇した場合、いずれかが承認された場合、私たちの製品を市場から撤回することを含む重大な処罰を受けるかもしれない。
私たちが発売許可を得た任意の候補製品、及びその製品の製造プロセス、承認後の臨床データ、ラベル、広告と販売促進活動は、FDAと他の監督管理機関の持続的な要求と審査を受ける。これらの要件には、承認された製品の普及、安全および他の発売後の情報および報告の提出、登録および上場要件、生産、品質管理、品質保証および記録およびファイルの対応する維持に関するcGMP要件、ならびに薬品の流通および医師へのサンプルの配布および記録の保存に関する要件が含まれるが、これらに限定されない。
FDAおよび他の連邦および州機関は、司法省を含み、処方薬製品に対するすべての要求の遵守を厳格に規制し、承認されたラベル規定による薬物の販売および普及の要求、およびcGMP要求に基づいて製品を生産する要求を含む。例えば,FDAや他の機関はラベル外用途の普及を禁止する法律法規を積極的に実行しており,ラベル外用途の普及が不適切であることが発見された会社は重大な責任を負う可能性がある。これらの要件に違反することは、虚偽請求法案および他の連邦および州医療詐欺および乱用法律、ならびに州消費者保護法を含む連邦食品、薬物および化粧品法案(FDCA)および他の法規に違反することを告発する調査につながる可能性がある。私たちはすべての法規の要求を守ることができず、その後、私たちの製品、製造業者、または製造プロセスに以前に未知の不良事件または他の問題が存在することが発見され、様々な結果が生じる可能性があります
もし私たちまたは任意の未来のパートナーが安全モニタリングまたは薬物警戒、および小児科群の製品開発に関連する要求を含む法規要件を遵守しなければ、重大な経済的処罰を招く可能性もある。
72
もし私たちが外国の管轄区域でマーケティングの許可を得ることができなければ、私たちの候補製品はこれらの管轄区で販売することができません。私たちがアメリカで得たいかなる候補製品の承認も、私たちの候補製品が外国の管轄区域で承認されることを保証できません。
米国以外のいかなる司法管轄区域でも我々の製品をマーケティング·販売するためには、単独のマーケティング承認を得て、多くの異なる監督管理要求を遵守しなければならない。承認手続きは国によって異なり、追加的なテストが含まれるかもしれない。承認を得るのに要する時間は、FDA承認を得る時間とは大きく異なる可能性がある。米国以外の規制承認手続きには、通常、FDA承認の取得に関するすべてのリスクが含まれる。また、米国以外の多くの国では、製品がその国での販売を許可される前に、精算承認を受けなければならないことが求められている。もしあれば、私たちはアメリカ以外の規制機関から直ちに承認されないかもしれない。FDAの承認は、他国又は管轄区域の規制機関の承認を確保するものではなく、米国以外の1つの規制機関の承認も、他の国又は司法管区の規制機関又はFDAの承認を確保することができない。マーケティング承認を提出できないかもしれませんし、どの市場でも私たちの製品を商業化するために必要な承認を得ることができないかもしれません。
私たちの現在と未来の顧客および第三者支払者との関係は、刑事、民事と行政処罰、契約損害、名声損害、利益および将来の収入の減少を含む重大な処罰に直面する可能性があり、詐欺、詐欺、乱用、透明性、健康プライバシー、および他の医療保健法律および法規の制約を受ける可能性がある。
医療提供者は、医師や第三者支払者を含め、マーケティング承認を得た任意の候補製品を推薦·処方する上で主な役割を果たす。私たちの現在と将来の医療保健提供者、第三者支払者、顧客との手配は、私たちがマーケティングの承認を得た任意の製品の業務または財務配置と関係を制限する可能性があり、広範に適用される詐欺と乱用、その他の医療法律法規に直面する可能性があります。私たちの業務に適用される連邦と州医療法律法規の制限は
73
我々の内部業務プロセスと第三者の業務配置に適用される医療法令に適合することを確保する努力は、多くのコストに関連する。政府当局は、私たちの業務実践が現在または未来に適用される詐欺および乱用または他の医療保健法律および法規に関連する現行または未来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。私たちの運営がこれらの法律または他の任意の政府法規に違反していることが発見された場合、私たちは、MedicareおよびMedicaidや他の連邦医療計画、契約損害、名声損害、利益減少および将来の収益減少、追加の誠実な報告および監督義務、ならびに私たちの業務を削減または再構成するなど、重大な民事、刑事および行政処罰、損害賠償、罰金、監禁、MedicareおよびMedicaidおよび他の連邦医療計画、契約損害、名声損害、利益減少および将来の収益減少、および追加の誠実な報告および監督義務、およびこれらのいずれも、私たちの業務運営能力および私たちの運営結果に悪影響を及ぼす可能性がある。もし私たちがそれと業務を展開することを期待している任意の医師または他の医療提供者または実体が適用されない法律を遵守していないことが発見された場合、彼らは政府の援助された医療計画から除外されることを含む重大な刑事、民事、行政制裁を受ける可能性があり、これは私たちの業務、運営結果、財務状況、および見通しに実質的な悪影響を及ぼす可能性がある。
最近公布された立法と将来の立法は、候補製品の上場承認と商業化の難しさとコストを増加させ、私たちが得る可能性のある価格を下げるかもしれない。
米国や一部の外国司法管轄地域では、医療システムに関する立法や規制変更、提案された変更は、候補製品の上場承認を阻止または延期し、承認後の活動を制限または規制し、マーケティング承認された候補製品を収益的に販売する能力に影響を与える可能性がある。
例えば、2010年3月、ACAは法律に署名された。ACAは医療保険の許容性を拡大し、医療支出の増加を減少または制限し、詐欺と乱用に対する救済措置を強化し、医療保健と医療保険業界の新しい透明性要求を増加させ、医療業界に新しい税金を徴収し、追加の医療政策改革を実施することを目的とした全面的な法律である。
ACAの条項の中で、私たちの潜在的な製品候補製品に非常に重要な条項は以下の通りです
74
“海外腐敗防止法”の廃止または代替のいくつかの面では、米国の元大統領総裁政権中の措置を含め、行政、司法、国会面の挑戦が存在してきた。トランプ政権は、ACAのいくつかの条項の実施を延期したり、ACAが規定しているいくつかの医療保険要件を迂回したりするための行政命令や他の指令を発表した。同時に、国会はACAの全部または一部を廃止または廃止し、代替する立法を審議した。議会はまだ立法を全面的に廃止していないが、ACAのいくつかの条項を改正し、例えば2019年1月1日以来、ACA個人が医療保険の購入を許可していないという処罰を取り消し、ACAに規定されているいくつかの費用の実施を廃止し、連邦医療保険Dの一部に参加する製薬メーカーが不足している販売時点割引を増加させた。2020年11月、米国最高裁は米国第5巡回控訴裁判所の判決について口頭弁論を行い、個人ライセンスは違憲と判断した。2021年2月10日、バイデン政府はACA打倒への連邦政府の支持を撤回した。2021年6月、米国最高裁はこの事件を再審に戻し、法的地位の欠如を理由にこの事件を却下するよう指示した。しかし、米国最高裁判所は個人権限の有効性という最終的な問題について裁決を下していない。したがって、個人の権限や挑戦、腐敗防止条約に挑戦したり、廃止したり、代替する他の努力があるかもしれない。米国最高裁の裁決,その他のこのような訴訟,現大統領政府の医療改革措置がACAや我々の業務にどのように影響するかは不明である。
さらに、ACAが公布されて以来、他の立法改正も提案され、採択された。2011年8月2日、赤字削減合同特別委員会を設立し、国会に支出削減提案を提案することを含む“2011年予算制御法案”が法律に署名された。合同特別委員会は的確な赤字削減を実現せず、立法をいくつかの政府プロジェクトに自動的に減少させた。これらの変化には、2013年から提供者に支払われる医療保険総金額を前期ごとに2%削減することが含まれており、その後の法規改正により、これらの削減は2030年まで有効となるが、新冠肺炎の大流行により、国会が追加行動しない限り、2020年5月1日から2021年12月31日まで支払いを停止することが含まれている。2013年1月、“2012年米国納税者救済法”が署名され、いくつかの医療サービス提供者に支払う医療保険を減らし、政府が提供者に多額の支払いを取り戻す訴訟時効が3年から5年に延長された。このような法律は医療保険と他の医療資金のさらなる減少を招くかもしれない。
75
そのほか、政府は最近薬品メーカーがその上場製品の価格設定の方式に対して更に厳格な審査を行い、国会が数回の調査を行い、そして連邦と州立法を提出し、製品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、政府の薬品に対する計画精算方法を改革することを提出し、公布した。連邦レベルでは、前アメリカの総裁は多種の手段を利用して薬品価格改革を提出或いは実施し、連邦予算提案、行政命令と政策計画を含む。例えば、2021年9月9日、バイデン政府は、連邦医療保険交渉価格の許可および価格上昇の抑制、サプライチェーンの強化、生体模倣薬および模倣薬の促進、および価格透明性の向上を含む処方薬価格を低下させるための範囲の広い政策提案リストを公表した。これらの取り組みは最近ピークに達し、2022年8月に“インフレ低減法案”(IRA)が公布され、米国衛生·公衆サービス部(HHS)は、少なくとも7年間(生物製品は11年)を承認する高支出単源薬にのみ適用されるが、CMSが連邦医療保険B部分とD部分で精算されたいくつかの薬物や生物製品の販売価格に基づいて交渉することを許可する。交渉価格は2026年に初めて発効し,2023年10月から法定最高価格を上限とし,インフレ率よりも高い速度で連邦医療保険B部分とD部分の薬物価格を向上させる製薬業者を懲罰する。また、, 2025年からこの法律は,受益者の最高自己負担コストを大幅に低減し,メーカーに新たに設立されたメーカー割引計画により,D部分加入者ブランド薬物処方コストの10%を自己払い最高限度額を下回り,自己最高限度額に達すると20%補助金を要求することで,連邦医療保険D部分下の“ドーナツ穴”を解消した。アイルランド共和軍は衛生·公衆サービス部長官が最初の数年に規制ではなく指導によってその多くの規定を実施することを許可した。IRAを守らないメーカーは民事罰金を含めて様々な処罰を受ける可能性がある。アイルランド共和軍はまた、ACA市場で医療保険を購入した個人に2025年まで強化された補助金を提供する。このような規定は法的挑戦を受ける可能性があるにもかかわらず、2023年から段階的に施行されるだろう。また、衛生·公衆サービス部部長は最近、3種類の定価効率をテストする新しいモードを提案し、その中の1つのモードは食品と薬物管理局と協議し、承認された薬物開発支払い方法を加速するために、適時に検証性試験を完成させ、発売後の安全性と有効性データを得る機会を改善することを奨励し、臨床治療効果が確認されていない薬物への医療保険の支出を減らすことを目標としている。さらに、州レベルでは、価格または患者の精算制限、割引、特定の製品への参入の制限、マーケティングコスト開示および透明性措置を含む医薬品および生物製品の価格設定を制御するための法規が立法および実施されており、場合によっては、他の国からの輸入および大量購入を奨励することが目的である。
私たちは、将来、特に新大統領政府を背景に、より多くの州と連邦医療改革措置をとることを予想している。将来取られる可能性のあるこのような改革措置は、より厳しいカバー基準をもたらし、私たちが受け取った任意の承認された製品の価格に追加的な下振れ圧力をもたらす可能性がある。コスト抑制措置や他の医療改革を実施することは、私たちの収入の創出、利益の実現、あるいは私たちの製品の商業化を阻止するかもしれない。
承認後の要求を拡大し、医薬品の販売や販売促進活動を制限するための立法·規制提案がなされている。私たちは、より多くの立法変化が公布されるかどうか、あるいはFDAの法規、ガイドライン、解釈が変わるかどうか、あるいはこれらの変化が私たちの候補製品の上場承認にどのような影響を与える可能性があるかどうかを決定することはできない。また、米国議会のFDA承認過程に対するより厳格な審査は上場承認を著しく延期または阻止する可能性があり、より厳格な製品ラベルと上場後のテストとその他の要求の影響を受ける可能性がある。政府も、発生している新冠肺炎の大流行に対応するために、より多くの行動をとる可能性がある。
76
米国以外の政府は厳しい価格制御を実施する傾向があり、これがあれば私たちの収入に悪影響を及ぼす可能性がある。
一部の国では、カナダといくつかのEU加盟国を含め、処方薬の価格設定は政府によって統制されている。これらの国では、製品の発売許可を受けた後、政府当局との定価交渉にかなりの時間がかかる可能性がある。一部の国で精算或いは定価の承認を得るためには、私たちの候補製品のコスト効果を他の利用可能な治療法と比較する臨床試験を行う必要があるかもしれない。もし私たちの製品が精算を受けられない場合、あるいは範囲や金額が制限されている場合、あるいは定価レベルが満足できなければ、私たちの業務は損害を受ける可能性があります。政治、経済、規制面の事態は定価交渉をさらに複雑化させる可能性があり、補償を受けた後、定価交渉が継続される可能性がある。EUの各加盟国が使用する参考価格と、低価格と高価な加盟国の間の裁定のような平行貿易は、価格をさらに下げることができる。薬品に対して価格規制や精算制限を実施することが保証されていない国は、これらの国で承認されれば、任意の製品に対して有利な精算と定価手配を許可する。第三者支払者または主管当局が割引を公表することは、公布国または他の国の価格または補償レベルにさらなる圧力を与える可能性がある。また、イギリスがどのEU法律をコピーまたは代替するかを決定することに伴い、イギリスのEU加盟国の資格は、イギリスのさらなる法律と規制の不確実性を招き、処方薬の価格設定に関連する法律や法規を含む、イギリスとEUが異なる法律や法規を採用することを招く可能性がある。もしイギリスが処方薬の価格設定に影響を与える法規を大幅に変更すれば, 私たちは大きな新しい費用に直面するかもしれない。したがって、イギリスの離脱は私たちがEUとイギリスで事業を展開する能力を弱めるかもしれない。
私たちが将来所有する可能性のある任意の国際業務を管理する法律法規は、米国以外でいくつかの候補製品や製品を開発、製造、販売することを阻止し、コストの高いコンプライアンス計画の開発と実施を要求するかもしれない。
私たちの業務をアメリカ以外の地域に拡張すれば、私たちは各管轄区域で事業を展開することを計画している多くの法律と法規を遵守するために追加の資源を投入しなければならない。反海外腐敗法(FCPA)は、個人または企業が業務を獲得または保持するのを助けるために、任意の米国人または企業およびその政党代理人が、任意の外国人官僚、政党または候補者に直接的または間接的に支払い、提供、許可支払い、または任意の価値のあるものを提供することを禁止している。“海外腐敗防止法”はまた、米国に上場する証券会社にある会計条項を遵守することを要求し、これらの条項は、会社(国際子会社を含む)のすべての取引の帳簿と記録を正確かつ公平に反映し、国際業務のために適切な内部会計制御システムを設計し、維持することを要求する。
“反海外腐敗法”を遵守することは高価で困難であり、特に腐敗は公認問題である国である。また、海外腐敗防止法は製薬業に特別な挑戦をもたらしており、多くの国では病院が政府によって運営されているため、医師や他の病院従業員は外国人官僚とされている。
様々な法律、法規、および行政命令はまた、米国国外での使用および伝播を制限するか、または国家セキュリティ目的のために秘密にされた情報と、特定の製品およびこれらの製品に関連する技術データとを特定の非米国国民と共有する。私たちはまた、輸出規制に関する米国の法律法規と、特定の国と個人に対する経済制裁と禁輸を受けている。もし私たちがアメリカ以外での業務を拡大すれば、これらの法律を遵守するためにもっと多くの資源を投入する必要があります。これらの法律は、私たちがアメリカ以外でいくつかの候補製品や製品を開発、製造、または販売することを阻止するかもしれません。これは、私たちの成長潜在力を制限し、私たちの開発コストを増加させるかもしれません。
国際ビジネス慣行に関する法律を遵守しなければ、重大な民事·刑事罰を受け、政府契約の資格を一時停止または廃止する可能性がある。米国証券取引委員会(Securities and Exchange Commission,略称米国証券取引委員会)も、発行者が“反海外腐敗法”の会計規定に違反したため、発行者の米国取引所での証券取引を一時停止または禁止する可能性がある。
77
もし私たちが環境、健康、安全の法律法規を守らなければ、罰金や処罰を受けたり、私たちの業務を損なう可能性のあるコストが発生するかもしれません。
私たちと私たちの第三者請負業者は、実験室手続きおよび危険材料および廃棄物の処理、使用、貯蔵、処理、および処理を管理する法律法規を含む、多くの外国、連邦、州、地方環境、健康および安全法律法規の制約を受けている。私たちの行動は化学物質と生物学的材料を含む危険で燃えやすい材料の使用に関するものだ。私たちの業務はまた危険な廃棄物製品を発生させるだろう。私たちは一般的に第三者と契約を結び、このような材料と廃棄物を処理する。私たちはこのような材料が汚染や傷害をもたらす危険を除去することができない。もし私たちが危険な材料を使用して汚染や損傷をもたらしたら、私たちはそれによるいかなる損害に責任を負うかもしれません。どんな責任も私たちの資源を超えて、任意の利用可能な保険を含むかもしれません。私たちはまた私たちの商業オフィスで発生する可能性のある意外な安全事件に責任を負うことを要求されるかもしれない。
しかも、いくつかの法律や法規によると、私たちの不動産賃貸と運営は私たちに責任を負わせるかもしれない。米国の既存の環境法律や法規によると,危険物質を処分する不動産や実体の現または前任所有者や経営者は,危険物質漏洩による汚染を調査または救済する費用を厳格,連帯,個別に負担することが要求される可能性があり,知らなくても危険物質漏洩に責任を負わない。
私たちはこのような法律や法規を遵守できなかったために重大なコストと責任を招く可能性があり、これらのコストと責任は、民事または刑事罰金と処罰、財産損失および人身傷害クレーム、私たちの施設のアップグレードや操作手順の変更に関連するコスト、または私たちの運営の禁止を制限または変更することを含む私たちの財務状況および経営業績に悪影響を及ぼす可能性があります。
従業員の負傷により発生する可能性のあるコストと費用を支払うために責任保険を維持していますが、潜在的な責任に十分な保険を提供できない可能性があります。私たちは私たちが生物、危険または放射性物質を貯蔵したり処分したりすることによって、私たちが提起した環境責任や有毒侵害に対して保険を維持することはできません。
また、現在または未来の環境、健康、安全法律法規を遵守するためには、巨額のコストが生じる可能性がある。これらの現行または未来の法律法規はますます厳しくなっており、私たちの研究、開発、あるいは生産努力を損なう可能性がある。私たちがこのような法律を守らないことはまた巨額の罰金、処罰、または他の制裁につながるかもしれない。
私たちはアメリカと外国のいくつかの反腐敗、反マネーロンダリング、輸出規制、制裁、その他の貿易法律法規の制約を受けている。私たちは違反によって深刻な結果に直面するかもしれない。
米国および外国の反腐敗、反マネーロンダリング、輸出規制、制裁および他の貿易法律法規は、会社およびその従業員、代理人、CRO、CMO、法律顧問、会計士、コンサルタント、請負業者および他のパートナーの許可、承諾、提供、請求、直接または間接的に公共または民間部門の受取人の腐敗または不当な支払い、または任意の他の価値のあるものを受け入れることを禁止している。これらの法律違反は、大量の刑事罰金と民事処罰、監禁、貿易特権の喪失、資格取り消し、税収の再評価、契約違反および詐欺訴訟、名誉損害、およびその他の結果を招く可能性がある。私たちは政府機関や政府付属病院、大学、その他の組織の役人や従業員と直接または間接的な相互作用を持っている。私たちはまた、時間が経つにつれて、アメリカ以外での私たちの活動が増加すると予想している。我々は、第三者に依存した研究、臨床前研究および臨床試験、および/または必要な許可、ライセンス、特許登録、および他のマーケティング承認を得ることが予想される。私たちは、私たちがそのような活動を明確に許可していなくても、私たちの人員、代理、またはパートナーの腐敗や他の不法活動に責任を負うことを要求されるかもしれない。
上記の法律および条例に違反するいかなる行為も、重大な民事と刑事罰金と処罰、監禁、輸出入特権の喪失、資格取り消し、税額の再評価、契約違反および詐欺訴訟、名誉損害、およびその他の結果を招く可能性がある。
78
我々は現在の候補製品を開発しており,将来の候補製品の開発を継続し,他の療法と組み合わせることが可能であり,追加のリスクに直面するであろう。
現在承認されている1つ以上の癌療法や開発中の治療法に合わせて現在の候補製品を開発している。現在または将来の任意の候補製品が他の既存療法と併用するために発売承認または商業化されていても、FDAまたは同様の外国規制機関が、任意の候補製品との併用療法の承認を取り消す可能性があるか、またはこれらの既存療法が安全性、有効性、製造、または供給の問題になる可能性があるというリスクに直面するであろう。また,我々の候補製品の使用が承認された既存の療法自体も,寵愛を失ったり,より遅い治療経路に降格されたりする可能性がある。これは私たちの候補製品または私たち自身の製品のための他の組み合わせ療法を決定する必要があるかもしれません。これらの製品は市場から撤退されるか、または商業的にはあまり成功しません。
我々はまた、FDAまたは同様の外国規制機関が発売されていない1つまたは複数の他の癌療法を組み合わせて、現在の候補製品を評価することも可能である。私たちは、どんな候補製品も、最終的にマーケティング承認を得ていないこのような承認されていない癌療法と組み合わせてマーケティングおよび販売を行うことができない。
FDAまたは同様の外国の規制機関がこれらの他の療法の承認を承認または撤回していない場合、または私たちが現在または将来の候補製品に関連して評価する療法を選択した場合、安全性、有効性、商業採用、製造、または供給の問題が生じた場合、私たちは、私たちが開発した現在または未来の候補製品のいずれか1つまたはすべての承認または成功したマーケティングを得ることができないかもしれない。さらに、現在または将来の候補製品と組み合わせて使用されている療法または開発中の療法の第三者プロバイダが、臨床試験のために十分な数の薬剤を生産できない場合、または現在または将来の候補製品を商業化するか、または併用療法のコストが懸念されるほど高い場合、私たちの開発および商業化努力は損なわれ、これは、私たちの業務、財務状態、運営結果、および成長の見通しに悪影響を及ぼすであろう。
会社として、私たちはこれまで候補製品を商業化したことがなく、現在は全面的、全員の専門知識、人員と資源が不足しており、単独あるいは適切なパートナーと一緒に任意の製品の商業化に成功することはできない。
会社として、私たちは候補製品を商業化したことがない。私たちは私たちの候補製品に関連するいくつかの権利を協力者に許可し、これらの協力者の助けと指導に依存するかもしれない。私たちが商業化権利とマーケティング承認を保持する候補製品については、私たちは自分の販売、マーケティング、市場参入、商業計画、供給組織を制定したり、これらの活動を第三者にアウトソーシングしなければならないだろう。私たちは規制部門の承認を得て、私たちの製品をアメリカ以外の場所で商業化するための協力を求めている。私たちはどんな協力も会社に短期的または長期的な利益をもたらすという保証がない。
承認されれば、私たちの候補製品の自己商業化に影響を与える可能性のある要素は、十分な数の有効な販売、マーケティング、市場参入者の募集と保持、十分な教育およびマーケティング計画を開発し、作成して、私たちが承認した候補製品に対する公衆の受容度を向上させ、わが社、販売促進分野のすべてのコミュニケーションおよび材料、医療保健法の適用下の従業員および第三者のコンプライアンス、および独立した販売およびマーケティング組織の作成に関連する他の予見できないコストを確保することを含む。販売·マーケティング組織を構築することは高価で時間がかかり、承認された後の私たちの候補製品の発表を延期する可能性がある。私たちは効果的な販売とマーケティング組織を作ることができないかもしれない。あるいは、私たちが直接販売チームと構築された流通システムを持つ第三者と協力することを選択すれば、世界的にも地域ごとに基づいて、私たち自身の販売チームと流通システムを強化するか、または私たち自身の販売チームや流通システムの代わりに、これらの第三者と提案された協力について交渉して合意することを要求され、このような配置は、私たち自身が製品を商業化する利益よりも低いことが証明されるかもしれない。もし私たちが自分の流通やマーケティング能力を確立できない場合、あるいは適切なパートナーを見つけて私たちの候補製品を商業化することができなければ、私たちはこれらの製品から収入を得ることができず、利益を達成したり維持することもできないかもしれない。
79
私たちの第三者への依存に関するリスクは
著者らは引き続き第三者に依存して臨床試験を行い、著者らのいくつかの研究と潜在的な臨床前研究を実行するつもりである。これらの第三者がその契約義務を満足に履行できなかった場合、適用された規制要件を遵守できなかった場合、または予期された期限までに完了できなかった場合、私たちの開発計画は延期されたり、コストが増加したりする可能性があり、または私たちは規制部門の承認を得ることができない可能性があり、これらはすべて私たちの業務、財務状況、運営結果、および見通しに悪影響を及ぼす可能性がある。
私たちは臨床試験のすべての側面を自分で独立して行うことができない。したがって,我々が行っているおよび計画中のtovorafenib(DAY 101)とpimasertibの臨床試験,および任意の将来の候補製品の臨床前研究と臨床試験を第三者に依存して行う。したがって,これらの実験の起動と完了時間はこれらの第三者によって部分的に制御され,我々の開発計画が遅延する可能性がある.これらの第三者部分はこれらの実験の進展を制御しているため,これらの実験に関するデータを取得または承認せずに公表することも可能である.具体的には,CRO,独立した臨床調査者,コンサルタントがこれらの試験の進行とその後のデータ収集と分析に重要な役割を果たすことが予想される。例えば,Dana Farber癌研究所とPNOCが協力して行った一期臨床試験に加え,小児腫瘍学グループは,国立癌研究所が支援する臨床試験グループであり,世界最大の小児や青少年癌研究に特化した組織でもあり,tovorafenib(DAY 101)が再発性ランゲルハンス細胞組織細胞増加症を治療する全群臨床試験を開発している。しかし、このような調査者たち、CRO、そして他の第三者は私たちの従業員ではなく、私たちは彼らの活動のすべての側面を制御することができない。しかし、私たちはすべての臨床試験が適用された方案と法律、法規と科学基準に従って行われることを保証する責任があり、私たちの研究者、CROと他の第三者への依存は私たちの監督責任を免除しない。私たちとCROはGCP要求を守らなければなりません, これらはFDAが臨床開発における候補製品に対して実行する法規とガイドラインである。監督管理機関は定期的に試験スポンサー、臨床試験研究者、臨床試験地点を検査することによって、これらのGCP要求を実行する。もし私たちまたは私たちの任意のCROまたは臨床試験サイトが適用されたGCP要求を遵守できなかった場合、私たちの臨床試験で生成されたデータは信頼できないと考えられる可能性があり、FDAは私たちのマーケティング申請を承認する前に追加の臨床試験を行うことを要求するかもしれない。検査後,FDAはわれわれの臨床試験がGCPに適合していることを確認することは保証できない。また,われわれの臨床試験はcGMP法規により生産された製品を用いて行わなければならない。私たちの失敗または私たちが依存している第三者がこれらの規定を遵守できなかった場合、私たちは臨床試験の停止および/または重複を要求する可能性があり、これは上場承認過程を延期する。
私たちが依存しているどのようなCRO、臨床試験研究者、または他の第三者が、私たちの開発活動に十分な時間および資源を投入するか、または契約の要求に従ってタスクを実行することは保証されない。さらに、これらの第三者はサプライチェーンやインフレ圧力の影響を受ける可能性があり、これらの圧力は彼らが予想されるスケジュールを達成する能力を制限し、あるいは私たちにより大きなコストをもたらす。例えば,臨床前研究に利用可能な非ヒト霊長類の不足は,現在の業務に影響を与えないことが予想されるが,新たな製品開発計画を開始すれば,より長い開発時間や必要な研究を達成することが困難になる可能性があることを認識した。これらの第三者のいずれかが予想される最終期限内に完了できなかった場合、私たちの臨床プログラムを遵守したり、法規要件を満たしたり、他の方法で目標を達成しなかったり、私たちとの協力を終了したりすることができない場合、私たちの開発計画のスケジュールは延長または延期される可能性があり、または私たちの開発活動は一時停止または終了する可能性がある。われわれの臨床試験サイトが何らかの理由で終了した場合,これらの対象を別の合格した臨床試験サイトに移すことができない限り,このような臨床試験に関与している被験者の後続情報を失う可能性がある。
80
さらに、可能な研究者が支援する試験については、これらの試験の設計または実施を制御することはなく、FDAは、試験設計または実行の要素、安全問題、または他の試験結果を含むいずれか1つまたは複数の理由で、これらの研究者が後援する試験は、将来の臨床試験または市場承認に十分な支援を提供すると考えられ、我々によっても第三者によって制御されても、これらの研究者が支援する試験は、将来の臨床試験または市場承認に十分な支援を提供すると考えられる。私たちは、調査員が支援する実験に関するいくつかの情報権を提供してくれることを期待しており、調査員によって支援された実験によって生成されたデータにアクセスし、参考にすることができ、私たち自身が提出した規制材料を含む。しかし,研究者が試験のデータを賛助する時間や報告を制御することはなく,研究者が試験を支援するデータを持つこともない。研究者が後援した試験結果を確認したり複製したりできなければ,陰性結果が得られれば,さらなる臨床開発をさらに延期または阻止する可能性がある。また,研究者や機関が候補製品の臨床開発における義務に違反している場合,あるいは我々が得られる可能性のある第1次知識と比較してデータが不十分であることが証明された場合,我々自身が任意の将来の臨床試験を設計·行う能力が悪影響を受ける可能性がある。研究者はより実現が困難な臨床終点の臨床試験を設計するかもしれない, あるいは他の点では,われわれが自ら設計する可能性のある臨床試験と比較して,臨床試験結果が陰性となるリスクが増加している。研究者が後援する臨床試験の負の結果は、私たちの候補製品のための規制部門の承認を得るための努力と、私たちの候補製品に対する大衆の見方に実質的な悪影響を及ぼすかもしれない。さらに、FDAは、これらの研究者が後援する試験によって生じる臨床前、生産または臨床データの参照権利の十分性、またはこれらの研究者が後援する試験の臨床前、生産または臨床データの説明に同意しない可能性がある。もしそうであれば,FDAは追加の臨床前,生産,または臨床データを取得して提出することを要求するかもしれない。
さらに、これらの第三者は他のエンティティと関係がある可能性もあり、その中のいくつかは私たちの競争相手である可能性があり、彼らはまた、これらの競争相手のための臨床試験または私たちの競争地位を損なう可能性のある他の薬品開発活動を行っているかもしれない。これらの第三者が規制要件または私たちが宣言した規程に従ってその契約義務の履行に成功し、予想される期限内にまたは私たちの臨床試験を行うことができない場合、tovorafenib(DAY 101)、pimasertib、または任意の将来の候補製品の発売承認を得ることができないか、または遅延する可能性があり、私たちの製品の商業化に成功した努力で遅延することはできないかもしれない。
私たちの候補製品の製造は複雑です。私たちの第三者製造業者は生産中に困難に直面する可能性があり、これは、(承認された場合)臨床試験または商業販売のための私たちの候補製品の供給を延期または完全に停止する能力があるかもしれない。
私たちは何の生産施設もなく、現在私たちは中国のいくつかの第三者メーカーと契約を締結している。私たちは依存し、規制申請の提出を支援し、商業製造のために、臨床試験、製品開発目的のために、第三者に当社の候補製品を生産し続けることが予想されます(任意の候補製品が市場承認を得た場合)。また,分析実験室と契約を結び,我々の候補製品の放行と安定性テストを行いたい。このような第三者への依存は、許容可能なコストまたは品質で十分な数の候補製品または製品またはそのような数を得ることができないリスクを増加させ、これは、私たちの開発または商業化努力を延期、阻止、または損害する可能性がある。例えば、新冠肺炎疫病が候補製品を開発するために十分な供給を得る能力に対する影響の程度はウイルス伝播の重症度と持続時間、及び新冠肺炎ウイルスを抑制或いはその影響を治療するための行動に依存する。封鎖措置や制限が実施されるかどうか、そしてもしあれば、生産減少や中断、生産コストの増加、または私たちのサプライチェーンの他の中断を含むが、生産減少や中断、生産コストの増加、または私たちのサプライチェーンの他の中断を含む私たちの地域の施設や運営にどのような影響を与えるかを確認することはできません。また、生産の中断や私たちのメーカー、特に中国のメーカーは、自然災害でも他の理由でも、私たちのニーズを満たすのに十分な数を生産することができず、私たちの日常業務運営能力と候補製品を開発し続ける能力を損なう可能性があります。またこれらのメーカーは中国にあるため, 米国や中国政府の政策が変化し、政治的動揺や中国の経済状況が不安定になれば、製品供給の中断やコスト増加の可能性に直面する可能性がある。これらの事項のいずれも、私たちの業務、財務状況、および経営結果に重大な悪影響を及ぼす可能性がある。しかも、物流経路と輸送能力の中断は私たちのサプライチェーンを混乱させるかもしれない。そして、時間が経つにつれて、私たちの需要が予期せぬ急増を見せたら、必要な供給を使い果たしてしまう可能性があります。
81
私たちは第三者製造業者とどんな合意にも到達できないかもしれないし、優遇条項でそうすることができないかもしれない。たとえ第三者製造業者と合意できても、第三者メーカーに依存することは、追加的なリスクをもたらす
私たちは私たちの候補製品に限られた供給スケジュールしかありません。このような計画は商業供給まで延長されません。私たちは注文を調達した上ですべての重要な材料を獲得した。したがって、私たちは私たちの候補製品と他の材料に対する長期的な約束計画を持っていない。したがって、サプライチェーン問題、例えばある包装材料に関する問題は、効率的に管理できなければ、候補製品を包装·納入する能力に悪影響を及ぼす可能性がある。薬物製造プロセスを開発·拡大し、薬物試験を行い、規制提出を支援するためのデータを生成するために、第三者と1つ以上のプロトコルを確立する必要がある。もし私たちのすべての候補製品が市場の承認を得たら、私たちは第三者と商業生産協定を確立する必要があるだろう。
第三者メーカーは、cGMP法規や米国以外の類似した法規要件を遵守できない可能性がある。FDAが、我々のCMOがFDAの法律および法規に適合していないと判断した場合、cGMPを管理する法律および法規を含む場合、FDAは欠陥が是正されるまでNDAを承認しない可能性があり、または申請中の製造業者を要求に適合する製造業者に置き換える。さらに、私たちまたは私たちの第三者製造業者とサプライヤーが適用された法規を遵守できなかったことは、臨床隔離、罰金、禁止、民事処罰、候補製品または製品の差し押さえまたはリコール、運営制限、および刑事起訴を含む制裁を実施する可能性があり、いずれも私たちの製品供給に重大な悪影響を及ぼす可能性があります。また、承認された製品およびその製造施設は、品質管理および製造プロセスがcGMP要件に適合することを確保することを含む、FDAの広範な要求および他の類似機関の要求に継続的に適合しなければならない。したがって,我々のCMOはcGMPの要求に適合するかどうかを評価するために継続的な審査と定期的な検査を受ける.また,我々のCMOの運営には日常的な制御がないにもかかわらず,cGMPを含めて適用される法律や法規の遵守を確保する責任がある。
さらに、私たちの第三者製造業者と供給者は、廃棄物製品を管理、使用、貯蔵、処理、処分する法律法規を含む多くの環境、健康、安全な法律法規の制約を受けており、これらの法律法規を遵守しなければ、このような第三者の民事または刑事罰金および処罰に関連する巨額のコストをもたらす可能性がある。将来的にこれらの第三者に対して取られる可能性のある規制行動の深刻さに基づいて、私たちの臨床または商業薬品供給および包装および他のサービスは中断または制限される可能性があり、これは私たちの業務を損なう可能性がある。
私たちの候補製品と私たちが開発する可能性のあるどの製品も他の候補製品や製品と製造施設を競争する可能性があります。したがって、私たちはこのような施設を優先的に使用することができないかもしれないし、全く使用できないかもしれない。CGMP法規の下で運営されているメーカーの数は限られており、私たちのために製品を製造する能力があるかもしれない。
82
後期臨床試験と潜在的な商業化に備えて、候補製品の生産規模を増加させる措置が必要である。Tovorafenib(DAY 101)以外にも、候補製品の製造プロセスを拡大しておらず、任意の候補製品の将来の供給需要を支援するために、さらなる規模を拡大する必要があるかもしれません。第三者製造業者は、私たちの任意の候補製品の製造能力をタイムリーにまたは費用効果的に向上させることに成功できないかもしれない、または全くできないかもしれない。また、規模の拡大や商業活動の間に品質の問題が発生する可能性がある。例えば、私たちの候補製品または私たちの候補製品を製造する製造施設において微生物、ウイルス、または他の汚染が発見された場合、そのような製造施設は、汚染を調査および修復するために長い間閉鎖される必要があるかもしれない。
私たちの既存または未来のメーカーのどんな業績失敗も、臨床開発やマーケティング承認を延期する可能性がある。私たちは現在大量の麻薬物質のための余分な供給や二番目の供給源の手配を提供していない。現在臨床試験に用いられているCMOが約束どおりに実行できなければ,このようなCMOを交換する必要があるかもしれない。いくつかの潜在的な代替メーカーが私たちの候補製品を生産できると考えているにもかかわらず、任意のそのような代替製造業者を決定し、同定したり、任意の代替製造業者と合意することができる場合には、追加のコストおよび遅延が生じる可能性がある。さらに、私たちの第三者メーカーは、資源制限や自然災害、労使紛争、不安定な政治環境または公衆衛生流行病(例えば、新冠肺炎)によって製造や輸送困難に遭遇する可能性がある。もし私たちの現在の第三者製造業者が合意通りに履行できなければ、私たちはこれらのメーカーの交換を要求されるかもしれません。私たちは彼らをタイムリーにあるいは交換できないかもしれません。
私たちの現在と未来の他人が私たちの候補製品や製品を生産することへの依存は、私たちの将来の利益率と、マーケティングの承認をタイムリーかつ競争力的に獲得する製品の商業化能力に悪影響を及ぼす可能性があると予想されています。
私たちは限られた数量のサプライヤーに原材料を提供することに依存して、私たちの独占的なサプライヤーによるいかなる中断も私たちの臨床試験の遅延を招く可能性があり、あるいは他の方法で私たちの業務と運営結果に悪影響を与える可能性がある。
私たちは限られた数量のサプライヤーに依存していて、その中のいくつかは私たちのある材料の唯一の源であり、いくつかのサプライヤーは外国司法管轄区に位置している。著者らの数少ないサプライヤーは多くの追加のリスクに関連し、サプライヤーとの生産能力制限、コンポーネント獲得性、価格上昇、適時納品、コンポーネント品質、肝心なサプライヤーが経営を継続できず、市場状況に応じて調整するリスクを含み、インフレと金利変化、自然災害、火災、テロ行為、流行病を含み、新冠肺炎の大流行或いはその他の壊滅的な事件を含む。また、独占サプライヤーの材料がある場合、任意の原材料や他の材料の代わりに代替案を用いることができても、この代替案はコストが高く、生産量が低い、あるいは私たちの目的に合わない可能性があります。しかも、私たちが製品候補を製造するために使用するいくつかの材料は複雑な材料であり、代替がもっと難しいかもしれない。したがって、当社の独占サプライヤーによるいかなる中断も遅延や追加的な規制提出を招く可能性があり、これは私たちの業務や運営結果に悪影響を及ぼす可能性があります。
私たちは私たちの候補製品を開発して商業化するために第三者と協力するかもしれない。もしこのような協力が成功しなければ、私たちはこの候補製品の市場潜在力を利用できないかもしれない。
私たちは選定に基づいて私たちのいくつかの候補製品の開発と商業化のために第三者パートナーを探すかもしれない。今まで、私たちは何の協力も達成していない。私たちの将来の任意の協力パートナーには、大中型製薬会社、地域的、全国的な製薬会社、バイオテクノロジー会社が含まれる可能性がある。私たちは適切な協力者を探すことで激しい競争に直面している。私たちが未来の協力について最終的な合意に到達できるかどうかは、他に加えて、未来のパートナーの資源と専門知識の評価、提案協力の条項と条件、提案パートナーの多くの要素の評価にかかっている。
83
もし私たちがどの第三者ともこのような計画を達成すれば、私たちの将来のパートナーが私たちの候補製品の開発または商業化に投入する資源の数と時間を限られた制御を行う可能性が高い。私たちがこれらの計画から収入を作る能力は、私たちの未来の協力者がこれらの計画の中で彼らに割り当てられた機能を成功的に履行する能力と努力にかかっているだろう。私たちの候補製品に関連する未来のパートナーとの協力は、以下の点を含む多くのリスクをもたらすだろう
もし私たちが1つまたは複数の協力関係を構築すれば、ここで説明される製品開発、規制承認、商業化に関連するすべてのリスクは、このような任意の未来のパートナーの活動にも適用されるだろう。
84
従業員の事務と私たちの運営に関するリスク
私たちの未来の成功は私たちが幹部と肝心な従業員を維持する能力、及び合格者を吸引、維持と激励及び私たちの人力資本を管理する能力に依存する。
私たちが競争の激しい生物技術と製薬業界の中で競争できるかどうかは、私たちが高い素質の管理、科学と医療人員を誘致、激励と維持できるかどうかにかかっている。私たちはJeremy Bender博士、M.B.A.、最高経営責任者Samuel Blackman、M.D.博士、首席医療官及び著者らの管理チームの他のメンバー、その他の重要な従業員と顧問の開発と管理専門知識に高度に依存している。私たちは現在このような個人に重要な人物保険を提供していない。私たちは私たちの幹部と雇用協定を締結したが、彼らの誰もが私たちとの雇用関係をいつでも終わらせることができる。
近年、私たちの産業は高い流出率を経験している。競争の激しい製薬業における競争の能力は科学、臨床、監督管理、製造と管理技能と経験を持つ高技能と経験者の能力を吸引、維持と激励することに依存する。
著者らは主に旧金山湾区で業務を展開し、この地区には他の製薬会社及び多くの学術と研究機関があり、合格人材に対する激しい競争を招いた。薬企業間の限られた数量の合格人材に対する激しい競争のため、私たちは未来に合格した人材を引き付けることができないかもしれない。私たちと競争している多くの他の製薬会社はより多くの財務と他の資源、異なるリスク状況、そして私たちよりも長い業界の歴史を持っている。私たちの競争相手は、より高い報酬、より多様な機会、および/またはより良い職業昇進の機会を提供するかもしれない。また、私たちの業務の変化に伴い、キーパーソンはより大きなビジネス企業のために働きたくないかもしれません。これらすべての競争要因は、私たちが高素質の人材を誘致し、維持し続ける能力を制限する可能性があり、これは、私たちの候補製品の開発と商業化に成功し、現在の想定に沿って私たちの業務と運営を発展させる能力にマイナスの影響を与えるかもしれない。私たちは採用実践の中でより大きな柔軟性を採用して、旧金山湾区以外の応募者を誘致し、採用することを目的としています。これは留任率を増加させることを目的としていますが、従業員の尊敬度に悪影響を与え、より大きな従業員流出率を招く可能性があります。
私たちは私たちの組織の規模と能力を拡大する必要があり、私たちはこのような成長を管理する時に困難に直面するかもしれない。
2022年12月31日までに、121人のフルタイム従業員がいます。私たちは、特に臨床開発、臨床運営、製造、監督管理、および私たちの任意の候補製品がマーケティングの承認、販売、マーケティング、流通の面で、私たちの従業員の数と業務範囲が大幅に増加することを予想しています。私たちが予想している将来の成長を管理するためには、私たちの管理、運営、財務システムを継続して実施し、改善し、私たちの施設を拡大し、より多くの合格者を募集し、訓練し続けなければならない。私たちの財務資源が限られているため、私たちの管理チームは、このような成長を期待している会社や開発販売、マーケティング、流通インフラの管理における経験が限られているため、私たちの業務の拡張を効果的に管理することができず、より多くの合格者を募集し、訓練することもできないかもしれません。私たちの業務の拡張は巨大なコストを招き、私たちの管理と業務発展資源を移転する可能性があります。
さらに、私たちは現在、予測可能な未来において、主にいくつかの第三者契約組織、コンサルタント、およびコンサルタントに依存して、私たちの臨床試験およびtovorafenib(DAY 101)、pimasertib、または任意の未来の候補製品の製造に重大な責任を負うことを含むいくつかのサービスを提供し続けるだろう。必要に応じて、このような第三者契約組織、コンサルタント、コンサルタントのサービスがタイムリーに提供されていくか、あるいは適格な代替者を見つけることができることを保証することはできません。さらに、私たちのアウトソーシング活動を効率的に管理できない場合、または私たちのサプライヤーやコンサルタントが提供するサービスの品質または正確性が何らかの理由で影響を受ける場合、私たちの臨床試験は延長、延期、または終了される可能性があり、tovorafenib(DAY 101)、pimasertib、または任意の将来の候補製品の上場承認を得ることができないか、または他の方法で私たちの業務を促進することができないかもしれません。私たちは、私たちの既存のサプライヤーやコンサルタントを経済的に合理的な条件で適切に管理することができるか、または他の適任な外部サプライヤーとコンサルタントを見つけることができるか、または全くできないことを保証することはできません。
85
もし私たちが成長を効率的に管理し、私たちの組織を拡大することができなければ、私たちはさらなる開発と商業化tovorafenib(DAY 101)、pimasertib、私たちの他の候補パイプライン製品、または任意の未来の候補製品に必要な任務を実行することに成功しないかもしれないので、私たちは私たちの研究、開発、商業化目標を達成できないかもしれない。
私たちの従業員、臨床試験研究者、CRO、CMO、コンサルタント、サプライヤー、および任意の潜在的な商業パートナーは、規制基準と要求を遵守しないこと、およびインサイダー取引を含む不正行為または他の不正活動に従事する可能性がある。
私たちは、従業員、臨床試験調査者、CRO、CMO、コンサルタント、サプライヤー、および任意の潜在的なビジネスパートナーの詐欺または他の不適切な行為のリスクに直面している。これらの当事者の不正行為は、意図的、無謀、および/または不注意な行為、または(I)FDA法規または真実、完全かつ正確な情報の報告を要求する法律を含むFDA法規または同様の外国規制機関の法規、(Ii)製造基準、(Iii)連邦および州の健康およびデータプライバシー、セキュリティ、詐欺および乱用、政府価格報告、透明性報告要件、ならびに米国および海外の他の医療保健法律法規、(Iv)セクハラおよび他の職場での不正行為、または(V)財務情報またはデータを真に、完全かつ正確に報告することを要求する法律を含む可能性がある。このような不正行為はまた臨床試験過程で得られた情報を不当に使用する可能性があり、これは監督部門の制裁を招き、著者らの名声に深刻な損害を与える可能性がある。
我々は、すべての従業員に適用される行動基準および開示計画および他の適用可能な政策および手順を通過したが、常に従業員の不適切な行為を識別し、阻止することができるわけではなく、このような行為を発見し、防止するための予防措置は、未知または未管理のリスクまたは損失を効果的に制御することができないか、またはこれらの法律や法規を遵守できないことによる政府の調査または他の行動または訴訟から私たちを保護することができないかもしれない。もし私たちにこのような訴訟を提起し、私たちが私たちの権利を弁護したり、維持することに成功しなかった場合、これらの行動は、Medicare、Medicaidおよび他の連邦医療保健計画、契約損害、名声損害、利益および将来の収益の減少、追加の誠実な報告と監督義務、および私たちの業務を削減または再構成することを含む、私たちの業務に重大な影響を与える可能性があり、いずれも私たちの業務の運営能力および運営結果に悪影響を及ぼす可能性がある。
もし私たちのセキュリティ対策が破壊された場合、あるいは私たちの情報技術システムや私たちのCRO、CMO、サプライヤー、請負業者、コンサルタント、または他の第三者パートナーのシステムに障害が発生したり、セキュリティホール、ネットワーク攻撃、データ損失や漏洩、および他の中断を受けたりすることは、私たちの開発計画に重大な中断をもたらし、私たちの業務や他の個人情報に関連する敏感な情報を危険にさらしたり、重要な情報へのアクセスを阻止したり、私たちの名声を損なう、あるいは他の方法で私たちの業務に悪影響を及ぼす可能性があります。
通常のビジネスプロセスでは、独自、機密および敏感な情報(知的財産権、商業秘密、固有商業情報、個人情報、および保護された健康情報またはPHIを含むが、これらに限定されない)を収集、処理、記憶および送信することができる。私たちはこのような情報の機密性、完全性、そして利用可能性を維持するために安全な方法でそうしなければならない。我々の業務の重要な構成要素は、情報技術および電気通信システムに依存しており、我々は、人的資源、財務報告および制御、顧客関係管理、規制コンプライアンス、および他のインフラ業務を処理するシステムなど、幅広いビジネスプロセスおよび機能分野に影響を与えるいくつかの企業ソフトウェアシステムを実装し、拡大することが予想される。私たちは、アクセス権限を失うリスク、不適切な使用または開示、不適切な修正、および重要な情報の制御を十分に監視、監査、修正できないリスクを含む、これらの重要な情報の保護に関連する多くのリスクに直面している。このようなリスクは、私たちの重要なビジネスシステムを実行し、機密、独自、および敏感な情報を処理するために、複数の第三者に依存しているので、私たちと協力する第三者に延長される。
86
セキュリティ対策が実施されているにもかかわらず、当社の内部情報技術システムおよび当社のCRO、CMO、サプライヤー、請負業者、コンサルタント、および他の第三者パートナーが維持する独自、敏感かつ機密情報の規模、複雑さ、および数が増加していることを考慮すると、私たちは、故障、サービス中断、システム障害、私たちの人員または第三者パートナーの事故、自然災害、テロ、世界的流行病、戦争および電気通信および電気故障の影響、ならびに私たちの人員または私たちのCRO、CMO、サプライヤー、請負業者、コンサルタント、業務パートナーおよび/または他の第三者パートナーの不注意または意図的な行為によるセキュリティホールを受けやすいかもしれません。または悪意のある第三者のネットワーク攻撃(ウイルス、ワーム、悪意のあるコード、マルウェア、恐喝ソフトウェア、サービス拒否攻撃、社会工学、および他の方法によってサービスの信頼性および情報の機密性、完全性および利用可能性に影響を与えることを含む)は、私たちのシステムインフラ、または私たちのCRO、CMO、サプライヤー、請負業者、コンサルタント、および他の第三者パートナーのシステムインフラに危害を及ぼす可能性があり、またはデータ漏洩を引き起こす可能性がある。
世界各地からの未遂攻撃と侵入の数、強度と複雑性の増加に伴い、セキュリティホール或いは破壊のリスクは普遍的に増加し、特にコンピュータハッカー、ウイルス、外国政府とネットワークテロリストを含むネットワーク攻撃或いはネットワーク侵入を含む。ますます多くの会社や個人がネットで仕事や遠隔作業をすることに伴い、新冠肺炎の流行とそれに伴う“在宅勤務”が増加し、全体的に利用可能な攻撃面が増加しているため、潜在的にサイバーセキュリティ事件が発生するリスクや、このような事件に対するリスク緩和に対応する投資が増加している。例えば,新冠肺炎の流行を利用して自分の利益を図りたい“ハッカー”は,釣りや迷惑メールやソーシャルエンジニアリングへの試みが増加している。私たちはすべての種類の安全脅威を予見できないかもしれないし、これらすべての安全脅威に対して効果的な予防措置を取ることができないかもしれない。サイバー犯罪者が使用する技術は常に変化し,起動前には認識されない可能性があり,外部サービスプロバイダ,組織犯罪分岐機関,テロ組織や敵対する外国政府や機関などの外部団体を含む様々なソースから来ている可能性がある.任意の中断またはセキュリティホールが、私たちのデータまたはアプリケーションまたは私たちのCRO、CMO、サプライヤー、請負業者、コンサルタント、および他の第三者パートナーのデータまたはアプリケーションの損失または破損、または機密、敏感または独自の情報を適切に開示しない場合、私たちは責任および名声の損害を招く可能性があり、tovorafenib(DAY 101)、pimasertib、または任意の将来の候補製品のさらなる開発および商業化が延期される可能性がある。いかなる侵害、損失、または専有、敏感なものは, または機密情報はまた、HIPAAおよび米国の他の関連州および連邦プライバシー法に基づくことを含む民事罰金および処罰を受ける可能性がある。例えば、カリフォルニアプライバシー権法案(CPRA)によって改正された2018年カリフォルニア消費者プライバシー法(CCPA)は、安全違反行為に個人訴権を適用し、規制審査、罰金、個人訴権和解、その他の結果を含む何らかの形の救済措置をもたらす可能性がある。
重大なセキュリティホールや中断に関連するコストは巨大である可能性があり、このようなリスクに対して提供されるネットワークセキュリティ保険の制限を超える可能性がある。もし私たちのCRO、CMO、サプライヤー、請負業者、コンサルタント、および他の第三者パートナーの情報技術システムが中断またはセキュリティホールの影響を受ける場合、私たちはこのような第三者に対する追跡権が不足している可能性があり、私たちはこのようなイベントの影響を軽減するために大量の資源を費やし、将来のこのような事件の発生を防止するために保護措置を制定し、実施しなければならないかもしれない。
87
私たちのデータ保護努力と情報技術への私たちの投資は、私たちのシステムまたは私たちのCRO、CMO、サプライヤー、請負業者、コンサルタント、および他の第三者パートナーの重大な故障、データ漏洩、侵入、または私たちの名声、業務、運営、または財務状況に重大な悪影響を及ぼす可能性のある他のネットワークイベントを防止することを保証することはできません。例えば、このようなイベントが発生して私たちの運営中断を招いたり、第三者CRO、CMO、サプライヤー、他の請負業者、コンサルタントの運営中断を招いたりすると、私たちの計画が深刻に中断される可能性があり、私たちの候補製品の開発が延期される可能性があります。さらに、tovorafenib(DAY 101)、pimasertib、または任意の他の候補製品の臨床試験データの損失は、私たちの市場承認作業を遅延させ、データを回復または複製するコストを著しく増加させる可能性があります。さらに、我々の内部情報技術システムまたは第三者CRO、CMO、サプライヤーおよび他の請負業者およびコンサルタントのシステムが深刻に中断されているか、またはセキュリティホールは、財務、法律、商業および名声をもたらす可能性がある機密情報(商業秘密または他の知的財産権、専有業務情報および個人情報を含む)の損失、流用、および/または無許可アクセス、使用または開示、またはアクセスを阻止する可能性がある。例えば、不正アクセス、使用、または個人情報(私たちの臨床試験対象または人員に関連する個人情報を含む)をもたらすこのような事件は、私たちの名声を直接損なう可能性があり、連邦および/または州が通知法律および外国と同等の法律に違反し、強制是正行動の制約を受けるように強要される可能性がある, そうでなければ、個人情報のプライバシーや安全を保護する法律·法規に基づいて責任を負うことになり、重大な法律や財務リスクや名声被害を招き、私たちの業務に悪影響を及ぼす可能性があります。
私たちは私たちが個人情報の安全を維持することを要求する法律、規則、そして規制を遵守することを要求された。私たちは契約と他の法的義務を持っていて、セキュリティホールを関連する利害関係者に通知するかもしれない。ネットワーク攻撃を予防または緩和することができないことは、不正アクセスの敏感性、機密、または固有の情報をもたらす可能性がある。ほとんどの管轄区域は法律を制定し、会社にあるタイプのデータのセキュリティホールに関連したときに個人、規制機関、その他の人に通知することを要求している。さらに、CRO、CMO、サプライヤー、請負業者、コンサルタント、および他の第三者パートナーとの合意は、セキュリティホールが発生した場合に通知することを要求する可能性があります。このような強制開示はコストが高く、否定的な宣伝を招く可能性があり、私たちの顧客が私たちのセキュリティ対策の有効性に自信を失ってしまう可能性があり、実際または予想されるセキュリティホールによる問題に対応および/または緩和するために大量の資本と他の資源を必要とすることが要求される。
発見される可能性のある任意のセキュリティホールに対応し、および/または緩和するコストは高くなる可能性があり、これらの問題を解決するための私たちの努力は成功しないかもしれません。これらの問題は、中断、遅延、負の宣伝、顧客の信頼を失うこと、私たちの製品の使用、および私たちの業務および競争地位に対する他の損害を減少させる可能性があります。どんな潜在的なセキュリティホールを修復するには多くの時間、資源、そして費用がかかるかもしれない。どんなセキュリティホールも、規制調査、訴訟、または他の調査を招き、私たちの財務と運営状況に影響を及ぼす可能性がある。
セキュリティホールによる訴訟は私たちの業務に悪影響を及ぼすかもしれない。私たちのシステム、ネットワーク、または物理施設への不正アクセスは、私たちの顧客または他の関係者との訴訟を引き起こす可能性があります。これらの訴訟は、私たちにお金をかけて弁護や和解を強要し、経営陣の時間と注意を分散させ、私たちの経営コストを増加させたり、私たちの名声に悪影響を与えたりする可能性がある。
私たちは、罰金、判決、和解、処罰、費用、弁護士費、および事件または違反による他の影響を含む安全事件または違反行為に十分な保険を提供していないかもしれない。利用可能な保険範囲を超える1つまたは複数の多額のクレームの提示に成功したり、保険証券の変化(保険料の増加または多額の免責額または共同保険要件の実施を含む)をもたらしたりすることは、私たちの業務に悪影響を及ぼす可能性がある。さらに、私たちの既存の保険範囲とミスと漏れ保険が受け入れ可能な条項で提供され続けるか、あるいは私たちの保険会社は未来のいかなるクレームも拒否しないと確信できません。私たちが顧客群を拡大し、独自かつ敏感なデータを処理、保存、転送することによって、私たちのリスクが増加する可能性があります。
88
私たちは厳格で変化していく法律、法規、基準、プライバシー、データ保護、データセキュリティに関する契約義務の制約を受けています。実際にまたはそのような義務を履行できなかったと考えられることは、政府の法執行行動(民事または刑事罰を含む可能性がある)、罰金および制裁、個人訴訟および/または負の宣伝を招き、私たちの経営業績および業務に負の影響を与える可能性がある。
我々と協力する第三者は、多くの国内外のデータ保護法律および法規(すなわち、プライバシーおよびデータセキュリティに関連する法律および法規)によって制約されている可能性があり、その範囲は変化しており、異なる適用および解釈の影響を受け、異なる国間で一致しないか、または他の規則と衝突している可能性がある。私たちはプライバシー、データ保護、およびデータセキュリティに関連する契約義務条項の制約を受けているか、または制限されているかもしれない。私たちまたは関連する第三者が実際にまたはこのような義務を履行できなかったことは、私たちのコンプライアンスと運営コストを増加させ、規制審査、行動、罰金、処罰に直面させ、名声損害を招き、顧客の流失を招き、訴訟と責任を招き、他の方法で私たちの業務、財務状況、運営結果に重大な悪影響を与える可能性があると考えている。
米国では、健康関連情報および他の個人情報の収集、使用、開示および保護を管理する多くの連邦および州法律法規、連邦健康情報プライバシーおよびセキュリティ法、連邦および州データ漏洩通知法、州健康情報プライバシー法および連邦および州消費者保護法(例えば、連邦貿易委員会法第5条)は、我々の業務または私たちの協力者の業務に適用される可能性がある。さらに,プライバシーおよびセキュリティ要件に制約された第三者(臨床試験データを取得した研究機関を含む)から保護された健康情報をHIPAA(HITECH改訂)から取得する可能性がある。事実および状況によると、HIPAAによって許可されていない方法でHIPAAによってカバーされているエンティティによって維持されている個別に識別可能な健康情報を取得、使用、または開示する場合、民事および刑事罰を受ける可能性がある。
カリフォルニア州は最近CCPAを公布し、カリフォルニアの消費者のために新しいプライバシー権を創造し、消費者または家庭の個人情報を処理するエンティティに対してより多くのプライバシーとデータ安全義務を規定した。CCPAは2020年1月1日に施行され、最近CPRAによって改正され、現在2023年1月1日に施行され、2023年7月1日から施行され、新たに設立された執行機関カリフォルニア州プライバシー保護局(CPPA)によって発表された規定の制約を受けている。CCPAは、カリフォルニア州住民に、彼らの個人情報の訂正、アクセス、および削除を要求する権利、特定の個人情報を共有しない権利を選択する権利、およびカリフォルニア州住民の雇用主を含む、彼らの個人情報がどのように処理されているかに関する詳細な情報を取得する権利を含む、カリフォルニア住民に拡大されたプライバシー権を与える。CCPAとCPRAはデータ漏洩に対する民事処罰と個人訴権を規定しており,データ漏洩訴訟が増加すると予想される。CCPAとCPRAは私たちのコンプライアンスコストと潜在的な責任を増加させるかもしれない。CCPAはいくつかの新しい連邦と州レベルのプライバシー立法の提案を推進しており、例えばネバダ州、ニューハンプシャー州、オハイオ州、ニューヨーク州、ワシントン州、イリノイ州、ネブラスカ州、バージニア州、バージニア州でバージニア州消費者データ保護法(VCDPA)(2023年1月1日から施行)、コロラド州でコロラド州プライバシー法が公布され、2023年7月1日に施行される。VCDPA、COPA、および他のこのような提案された立法が採択されれば、私たちの潜在的な責任とコンプライアンスコストを増加させ、私たちの業務に悪影響を及ぼす可能性がある。
89
一般データ保護条例(GDPR)と呼ばれる2016/679号法規を含む外国データ保護法は、欧州経済地域(EEA)およびスイスの個人から取得された個人情報(健康関連データを含む)に適用可能である。GDPRとそのEU各地で実施されている立法は,インフォームドコンセントのやり方を変更して臨床試験対象や調査者により詳細な通知を要求し,データ処理を制限し,個人情報を処理する法的基盤を構築し,データ処理義務を通知し,適切なデータ保護機関やデータ主体にセキュリティイベントを通知し,個人情報のセキュリティと秘密を保護し,データ主体がその個人情報に関する権利を行使する手段を構築することを含む企業に厳しい義務を課している。GDPRは,規格外の会社に対して最高2000万ユーロまたはその世界年収4%の罰金を科し,個人情報(臨床試験を含む)の処理や個人訴訟の処理を禁止する可能性がある。適用可能な範囲では、GDPRは個人情報を処理する上での私たちの責任と責任を増加させ、EUデータ保護ルールの遵守を保証するために追加的なメカニズムを構築し、より多くの時間と資源を費やす必要があるかもしれません。また,イギリスでは2018年5月に“データ保護法”が施行され,2019年には実質的にGDPRが施行され,イギリス特有の克減を含むGDPRをどのように適用するかに関する条項が含まれている。これらの法規の変化は追加の複雑性、要求の変化、制限、潜在的な法的リスクを増加させる可能性があり、コンプライアンス計画の資源への追加的な投資が必要だ, 以前有用なデータの戦略および利用可能性に影響を与える可能性があり、コンプライアンスコストの増加および/またはビジネス慣行およびポリシーの変化をもたらす可能性がある。また、欧州経済圏、スイス、イギリスの規制当局がデータ保護立法を実行する状況は一致しておらず、一部の地方司法管轄区にのみ適用される規則や指導を遵守するために追加の資源を使わなければならない可能性がある。
90
また,欧州データ保護法では,欧州経済圏以外の国,イギリス,スイス,米国のような個人情報の移動を禁止するのが一般的であり,欧州委員会はこれらの国は十分なデータ保護を提供できないと考えている。スイスはまた似たような制限措置を取った。個人情報をヨーロッパ経済地域、イギリス、スイスからアメリカや他の国に移すことができる法的メカニズムがありますが、それらは法的挑戦を受けているかもしれません。成功すれば、これらの挑戦はこれらのメカニズムを無効にし、ヨーロッパ以外のヨーロッパ人の個人情報を処理する能力を制限し、私たちの業務に悪影響を与える可能性があります。例えば、2020年7月、EU裁判所は、EU-米国プライバシー盾の無効を宣言し、EU-米国プライバシー盾が米国に移転したEU個人情報に十分な保護を提供できなかったため、プライバシー盾に認証した会社が個人情報をEUから米国に移転することを許可した。CJEUは標準契約条項のような他のデータ転送機構の使用無効を宣言していないが,この決定はこの機構を用いて米国へのデータ転送に関する不確実性を招き,CJEUはすべての場合,標準契約条項だけで十分とは限らないことを明らかにしている.欧州データ保護委員会(EDPB)は,CJEUが2020年11月11日に下した決定について,国境を越えたデータ転送に標準契約条項などのデータ転送機構を使用することにより高い負担をかけている追加指導意見を発表した。2021年6月, 欧州委員会はGDPRに基づいて新たな標準契約条項を採択し、個人データをEU以外の国に移したが、欧州委員会はこれらの国がこのような個人データを十分に保護していないと考えている。もし私たちが新しい標準契約条項に依存して個人資料をEUに移し、EDPBの提案に基づいて、新しい標準契約条項に加えられた義務を履行するために大量の資源を使う必要があるかもしれない;例えば、このような国境を越えたデータ転送の影響評価を行い、追加のセキュリティ措置を実施する必要があるかもしれない。私たちはまた、これらの新しい標準契約条項が管理データ処理と国境を越えた移転のすべての契約に含まれ続けることを確実にするために資源を投入しなければならない。また,英国では国境を越えたデータ転送を管理する新たなモデル条項が決定されることが予想されるため,英国のこの分野での独自の要求を遵守するためにより多くの時間と資源をかけなければならない。潜在的な法的挑戦により、新しいEU標準契約条項が依然として個人情報を欧州経済区に移転する有効なメカニズムであるかどうかにはまだ不確実性が存在する。一部の欧州データ保護監督機関も、標準契約条項を用いて専門的に米国に個人情報を移転する状況を審査している。例えば、ドイツとアイルランドの規制当局は、EU-米国のデータ転送に対する標準契約条項だけの保護が不足していると述べている。現在,データ転送機構の使用状況を逐案的に評価するとともに,目的国が適用する法制度を考慮しなければならない, 特に適用される監督法と個人の権利。これらの要求を遵守するためには、規制機関がさらなる指導を継続するにつれて、欧州から移転されたデータの安全性をさらに強化するための追加の保障措置を実施する必要がある可能性があり、追加的なコスト、クレーム、規制調査、または罰金を受ける可能性があり、業務を展開している国と地域の間で個人情報を移転できなければ、製品やサービスを提供する方法、私たちの関連システムや業務の地理的位置や隔離に影響を与え、私たちの財務業績に悪影響を及ぼす可能性がある。
また、英国が2020年1月31日にEUを離脱したのに続き、GDPRは2020年12月31日の過渡期終了時に英国での適用を停止した。しかし、2021年1月1日現在、イギリスの2018年“EU(離脱)法案”は、GDPR(2020年12月31日に存在するものと同様であるが、あるイギリス特有の改正)をイギリスの法律、すなわちイギリスGDPRに組み入れなければならない。英国GDPRと2018年の英国データ保護法は,EUのデータ保護制度から独立した英国のデータ保護制度を規定しているが,EUのデータ保護制度と一致している。イギリスのGDPR違反は、金額が高い者を基準に、1750万GBまたは世界収入4%の罰金を招く可能性があります。個人データをEUからイギリスに移すことについて、2021年6月28日、欧州委員会は、領土間で個人データを合法的に移転するために、組織に契約や他の措置を要求することなく、EU加盟国から英国へのデータの移転を許可する英国データ保護枠組みに関する十分な決定を発表した。計画は少なくとも4年間継続されているにもかかわらず、欧州委員会はいつでも一方的に充足率決定を撤回することができ、このような状況が発生すれば、追加的なコストを招き、私たちの全体的なリスクを増加させる可能性がある。
91
例えば、中国、ブラジル、オーストラリア、日本を含む他の国は、データのローカルな記憶および処理および個人情報の国境を越えた伝送にいくつかの法的要件を採用しており、これらのすべての要件は、臨床試験および臨床試験を行うか、または私たちの将来の製品(あれば)を提供し、私たちの業務を運営するコストおよび複雑さを増加させる可能性がある。このような義務の解釈と適用は異なる司法管轄区域の間で一致しない可能性があり、他の要求や私たちの接近と衝突する可能性がある。
私たちは、プライバシーおよびセキュリティに関連する外部および内部プライバシーおよびセキュリティポリシー、陳述、認証、および出版物の条項に制限されているか、または制限されている可能性がある。
国内外のプライバシー、データセキュリティ、データ保護の法律、法規、契約およびその他の義務を遵守することは、私たちがデータを収集、使用、開示する能力を制限し、あるいは場合によっては、ある司法管轄区域で運営する能力に影響を与えることを契約の中でより重い義務を負うことを要求するかもしれません。実際または国内外のプライバシー、データプライバシー、およびデータ保護法律法規を遵守しないと考えられることは、政府の法執行行動(民事、刑事および行政処罰を含む可能性がある)、個人訴訟および/または負の宣伝を招く可能性があり、私たちの経営業績および業務に負の影響を与える可能性がある。また,我々または我々の潜在的協力者が情報を取得する臨床試験対象,およびこれらの情報を共有する提供者は,我々の情報の使用や開示能力を契約的に制限する可能性がある。私たちがプライバシー権を侵害し、プライバシー、データセキュリティ、データ保護法を遵守できなかったと主張したり、私たちの契約義務に違反したりして、私たちが責任を負わなくても、弁護の費用が高く時間がかかり、負の宣伝を招く可能性があり、私たちの業務を損なう可能性があります。
私たちまたは私たちが依存している第三者は自然災害の悪影響を受ける可能性があり、私たちの業務の連続性と災害復旧計画は深刻な災害から私たちを十分に保護できないかもしれません。
私たちの現在の業務は主にサンフランシスコ湾区にあります。地震、洪水、火災、爆発、極端な天気条件、医療疫病または大流行、電力不足、電気通信故障または他の自然または人為的事故または事件などの計画外の事件は、私たちの施設や私たちの第三者契約製造業者の製造施設を十分に利用できず、特に日常生活において、私たちの財務および運営状況に大きなマイナス影響を与える可能性がある。また,気候変動が一般経済条件,特に製薬業に及ぼす長期的な影響は不明であり,既存の自然災害リスクを増加あるいは悪化させる可能性がある。これらの施設を使用できないことは、コスト増加、候補製品の開発遅延、当社の業務運営中断を招き、当社の業務、財務状況、運営結果、将来性に大きな悪影響を及ぼす可能性があります。自然災害、停電、または他の事件が発生した場合、本社の全部または大部分を使用することができず、私たちの研究施設や私たちの第三者契約メーカーの製造施設のような重要なインフラを損傷させたり、他の方法で運営を中断したりすることは難しいかもしれません。場合によっては、かなり長い間私たちの業務を継続することはできません。深刻な災害や同様の事件が発生した場合、我々の既存の災害復旧および業務連続計画は十分ではないことが証明される可能性がある。私たちの災害復旧と業務連続計画の限られた性質のため、私たちは大量の費用を発生する可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性があります。私たちのリスク管理政策の一部として, 私たちは私たちの業務に適していると思うレベルで保険範囲を維持する。しかし、これらの施設で事故や事件が発生した場合、保険金額がどんな損害や損失を補うのに十分な保証はできません。もし私たちの工場や私たちの第三者契約メーカーの製造施設が事故や事件あるいは他のいかなる理由でも作動できなければ、短い時間であっても、私たちのいかなる研究開発プロジェクトも損害を受ける可能性があります。どの業務中断も、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。
92
私たちに不利な税金法律または法規の変化は、私たちの業務、キャッシュフロー、財務状況、または経営結果に大きな悪影響を及ぼすかもしれません。
新しい収入、販売、使用、または他の税金法律、法規、規則、法規または条例はいつでも公布される可能性があり、これは私たちの業務運営および財務業績に悪影響を及ぼす可能性があります。さらに、既存の税金法律、法規、規則、法規または条例は、私たちに解釈、変更、修正、または適用される可能性がある。例えば、2017年の減税や雇用法案、あるいは減税や雇用法案と呼ばれ、米国税法には多くの重大な改正が行われている。米国国税局や他の税務機関が将来的に減税や雇用法案についての指導を提供することは私たちに影響を与える可能性があり、減税や雇用法案のいくつかの側面は将来の立法で廃止または改正される可能性がある。例えば、CARE法案は減税と雇用法案のいくつかの条項を修正する。また、各州が減税や雇用法案、CARE法案、または新たに公布された任意の連邦税収立法をどの程度遵守するかどうかは定かではない。会社税率の変化、私たちの業務に関連する繰延税純資産の現金化、外国収益の課税、減税と雇用法案、CARE法案または将来の改革立法による費用の控除は、私たちの繰延税金資産の価値に実質的な影響を与える可能性があり、重大な一次費用を招き、将来のアメリカ税費を増加させる可能性があります。
私たちの純営業損失の繰越と他の税務属性を使用する能力は限られているかもしれません。
私たちは歴史的に大きな損失を受けて、近い将来の利益を期待していませんし、私たちは永遠に利益を達成しないかもしれません。2017年12月31日までの納税年度に発生した未使用損失は、このような未使用損失が満期になるまで、将来の課税所得額を相殺するために繰り越されます。CARE法案により改正された減税と雇用法案によると、2017年12月31日以降の納税年度に発生した未使用の米国連邦純営業損失は満期にならず、無期限繰り越しとなる可能性があるが、2020年12月31日以降の課税年度では、このような連邦純営業損失の控除額は今年度の課税収入の80%を超えてはならない。各州が減税や雇用法案やCARE法案をどの程度遵守するかは定かではない。さらに、もし私たちが“所有権変更”を経験したか、または経験した場合、私たちの現在および未来の未使用損失および他の税務属性は、改正された1986年の国税法第382条および383条の制限を受ける可能性があり、この変更は、通常、特定の株主が3年以内に私たちの株式所有権(価値別)に対する変化が50ポイントを超えると定義される。私たちは、所有権変更が発生したかどうかを評価するために、まだ第382条の研究を完了していない、あるいは私たちの設立以来、このような研究の複雑さとコスト、および将来より多くの所有権変更がある可能性があるという事実から、複数回の所有権変更が発生したかどうかを評価する。したがって、私たちが2017年12月31日までの納税年度に発生した純営業損失繰越は使用前に満期になる可能性があり、2017年12月31日以降の納税年度に生じる純営業損失繰越の控除は限られている可能性があります。もし私たちが所有権変更を経験した場合(あるいは私たちが以前にこのような所有権変更を経験した場合), 私たちはすべての変動前の純営業損失の繰越とその他の変動前の税収項目の属性(例えば税収控除を研究する)を用いて、変動後の収入或いは税務項目を相殺する能力は限られている可能性がある。州税法の似たような規定はまた私たちが累積的な州税収属性を使用することを制限するのに適用されるかもしれない。また、州レベルでは、一定期間使用を一時停止したり、他の方法で純営業損失を制限したりする可能性があり、これは州の課税税を加速または永久的に増加させる可能性がある。したがって、私たちが利益を達成しても、私たちの純営業損失と他の税金属性の全部または大部分を使うことができないかもしれません。これは私たちの将来のキャッシュフローに悪影響を及ぼすかもしれません。
私たちはすでに戦略取引に従事しています。これらの取引は私たちの流動性に影響を与え、私たちの費用を増加させ、私たちの経営陣に大きな妨害をもたらす可能性があります。
武田薬業株式会社、ビラクタ治療会社、メルクKGaA(ドイツダムシュタット)との関連会社などの戦略的取引に従事したことがあり、会社、業務または資産の買収、製品、候補製品または技術の外部許可または内部許可などのさらなる戦略的取引も時々考慮される可能性がある。私たちが考慮する可能性のある他の潜在的な取引には、剥離、戦略パートナー関係、合弁企業、再編、資産剥離、業務合併と投資を含む様々な異なる商業計画が含まれる。このような取引は、私たちが非日常的または他の費用を発生させることを必要とする可能性があり、私たちの短期的または長期的な支出を増加させる可能性があり、重大な統合課題をもたらしたり、私たちの管理や業務を妨害したりする可能性があり、これは私たちの運営や財務業績に悪影響を及ぼす可能性がある。例えば、これらの取引は、多くの業務および財務リスクをもたらす可能性がある
93
私たちの知的財産権に関するリスクは
もし私たちが私たちの製品や技術のために特許保護や他の必要な権利を獲得し、維持することができない場合、あるいは取得された特許保護範囲が十分でない場合、あるいは私たちの特許(所有、共同所有可能かもしれない)による権利が十分に広くない場合、私たちの競争相手は私たちと類似または同じ製品および技術を開発し、商業化する可能性があり、私たちの製品および技術を商業化することに成功する能力は悪影響を受ける可能性がある。
私たちのビジネス成功は、米国や他の国で現在の候補製品や将来の製品の独自または知的財産権保護を獲得し、維持する能力があるかどうか、私たちの製造技術を含む私たちのコア技術にある程度依存しています。私たちは、私たちの知的財産権を求め、維持し、守ることによって、内部開発でも第三者から許可を得ても、私たちの業務発展に重要なビジネス的意義を持つノウハウ、発明、改善の保護と強化に努めています。私たちはまた商業秘密、技術ノウハウ、持続的な技術革新と許可内の機会に依存して、抗癌薬物開発領域における私たちの特許地位を強化し、維持している。また,希少薬物指定,データ排他性,市場排他性および特許期限延長(可能であれば)による規制保護に依存する予定である。
バイオテクノロジーと製薬会社の特許地位は通常高度に不確定であり、複雑な法律と事実問題に関連しており、近年多くの訴訟のテーマとなってきた。私たちが市場で競争に成功するために必要な特許保護の程度は、場合によっては入手できないかもしれないし、深刻に制限されている可能性があり、私たちの権利を十分に保護できないかもしれないし、どんな競争優位性を獲得または維持することを可能にするかもしれません。私たち自身または許可された任意の特許出願が発行された特許に成熟することを保証することはできず、このような特許が発行されると、現在および将来の候補製品を保護するのに十分な、または他の方法で任意の競争優位性を提供するのに十分な権利要件を含むことも保証されない。しかも、特許は発行された特許の管轄区域でのみ強制的に施行されることができる。しかも、特許の寿命は限られている。米国では、特許の自然失効期間は、通常、その初の非一時的な米国出願後20年である。適用される現地法の規定によると、米国国外特許の自然失効時間は異なるが、通常は最初の現地出願日から20年である。様々な延期があるかもしれない;しかし、特許の有効期限とその提供される保護は限られている。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。
94
しかも、私たちの独占ライセンスは分野によって制限され、保留される権利を受ける可能性があり、これは私たちの競争的地位に悪影響を及ぼすかもしれない。“経営陣の財務状況と経営成果の討論と分析--重要な合意”を参照。私たちが許可を得た特許の組み合わせは、他社が私たちの候補製品と同様の製品をそのような製品の模造バージョンを含めて商業化することを阻止するのに十分かつ持続的な特許保護を提供してくれないかもしれない。さらに、私たちに付与された特許の組み合わせは、一定の強制執行権を有する可能性がある許可分野以外の第三者に付与される可能性がある。したがって、私たちに付与された特許は、別の許可者によって提起された、または別の許可者に対して提起された訴訟において、または別の許可者がそのような訴訟または他の理由に応答して提起された行政訴訟において、無効または狭義に解釈されるリスクに直面する可能性がある。
他の当事者は、発明が私たち自身の特許出願または発行された特許において要求された発明と重複または衝突する可能性があると主張する、私たち自身の技術に関連するか、または競争力を有する可能性のある技術を開発し、または特許出願を提出したか、または特許を受信した可能性がある。科学文献で発表された発見は往々にして実際の発見に遅れており、米国と他の司法管轄区の特許出願は通常、申請18ヶ月後に発表され、時には全く発表されないこともある。したがって、私たちの特許及び出願の発明者が、それらの特許又は未解決特許出願で主張された発明を最初にした者であるか、又はこれらの発明のために特許保護を出願した最初の者であるかを決定することはできない。さらに、私たちの特許および特許出願に関連する可能性のあるすべての既存技術が見つかったことを保証することはできません。そのような従来技術が存在する場合、それは、特許を無効にするか、または係属中の特許出願から特許を発行することを阻止することができる。したがって、私たちの特許権の発行、範囲、有効性、商業価値は何の確定的な予測もできません。さらに、私たちの特許および特許出願によって提供される保護の広さまたは強度が脅かされている場合、結果にかかわらず、会社が現在または将来の候補製品を許可、開発、または商業化することを阻止することができる。
さらに、特許起訴過程は高価で時間がかかり、私たちまたは私たちの許可者は、すべての必要または望ましい特許出願を合理的なコストまたはタイムリーに提出して起訴することができないかもしれない。また、最初に審査に提出されたクレームの範囲は、発行時に大幅に縮小する可能性があり、本当にあれば。我々や我々の許可側も,特許保護を得るのが遅くなるまで,我々の研究開発成果の特許可能性を識別できない可能性がある.私たちが私たちの研究開発努力に基づいて追加の特許保護を求めたり、あるいは私たちが生成した任意のこのような特許や他の知的財産権がいかなる競争優位性を提供するかを保証することはできません。さらに、私たちは、第三者から許可された技術を含めて、特許出願の準備、提出、起訴を制御する権利がありません。したがって、これらの特許および出願は、私たちの業務の最良の利益に適合した方法で提出、起訴または維持されてはならない。
競争優位を保ちたい特許保護を得ても,特許の発行はその発明性,範囲,有効性,あるいは実行可能性に関する決定的な結論ではない.第三者は、競争相手を含めて、その発明性、範囲、有効性、または実行可能性に疑問を提起する可能性があり、これは、そのような特許の範囲の縮小、無効、または強制実行をもたらす可能性がある。もし私たちの特許が発表されたら、私たちの特許はアメリカと海外の特許庁で挑戦されるか、または法廷で挑戦されるかもしれない。例えば、私たちは、第三者が既存技術を米国特許商標局(USPTO)に提出し、私たちの1つ以上の特許主張が発表された後の有効性に疑問を提起することに直面する可能性がある。このような提出は、我々の特許出願に基づいて特許を付与することを排除するために、特許が発行される前に提出されてもよい。私たちは反対に巻き込まれ再審されるかもしれません各方面間私たちの特許が発表されると、米国または海外で私たちの特許主張に疑問を提起する審査、許可後の審査、派生、妨害、または同様の手続きが行われる。しかも、特許が発表されると、法廷で挑戦されるかもしれない。競争相手は、彼らが我々の特許発明者の前にそのような特許または特許出願で主張された発明を発明したと主張するか、または我々の特許発明者の前に特許出願を提出した可能性がある。競争相手はまた私たちがその特許を侵害していると主張するかもしれないので、私たちは私たちの特許出願や特許で主張されている技術(発行すれば)を実践することができません。したがって、私たちの1つ以上の特許請求書は縮小または失効する可能性がある。訴訟では、競争相手は、もし私たちの特許が発行されたら、様々な理由で、私たちの特許は無効だと主張するかもしれない。もし裁判所が同意すれば、私たちはこのような疑問視された特許の権利を失うだろう。
95
挑戦されていなくても、私たちの特許および処理されている特許出願(発行されている場合)は、私たちの特許声明をめぐる競争相手の設計を阻止し、類似または代替技術または療法を非侵害的に開発して私たちの特許を回避することができないかもしれません。例えば、私たちが有効かつ強制的に実行可能な特許を持っていても、相手が私たちの出願日前に発明を商業に使用していることを証明したり、相手が強制許可から利益を得たりすることができれば、他の人が私たちの発明を実践していることを排除できないかもしれません。候補製品の保有または出願の特許および特許出願に提供される特許保護が十分に広くなく、このような競争を阻害するのに不十分であれば、候補製品を商業化することに成功する能力は負の影響を受ける可能性があり、これは私たちの業務を損なうことになる。いくつかの規制排他性は利用可能である可能性があるが、このような規制排他性の範囲は変化する可能性があり、他の人が私たちの候補製品と類似した製品を商業化することを排除するために、十分かつ持続的な保護を提供してくれないかもしれない。
もし私たちが獲得した任意の特許保護の範囲が十分に広くない場合、あるいは私たちがいかなる特許保護を失った場合、競争相手が類似または同じ候補製品を商業化する能力が悪影響を受けることを阻止する。
生物製薬会社の特許地位は通常高度に不確定であり、複雑な法律と事実問題に関連しており、近年多くの訴訟のテーマとなっている。したがって,我々の特許権の発行,範囲,有効性,実行可能性,商業的価値は高い不確実性を持っている.私たちの未解決および将来の特許出願および我々の許可側の特許出願は、私たちの候補製品を保護する特許の発行、または他の候補競合製品の商業化を効果的に阻止する特許の発行につながる可能性があります。
また,特許出願に要求されるカバー範囲は特許発行前に大幅に縮小することができ,その範囲は特許発行後に再解釈することができる.私たちが所有しているまたは未来に許可された特許出願が特許の形で発表されても、それらの発表形態は私たちに意味のある保護を提供してくれず、競争相手や他の第三者が私たちと競争することを阻止したり、他の方法で私たちにどんな競争優位性を提供してくれたりしない。私たちが所有または許可している任意の特許は、第三者の挑戦または回避される可能性があり、または第三者の挑戦のために範囲を縮小したり、無効にしたりする可能性がある。したがって、私たちは私たちの候補製品が保護可能なのか、まだ効果的で実行可能な特許によって保護されているのか分からない。我々の競争相手または他の第三者は、同様の技術または代替技術または製品を非侵害的に開発することによって、私たちの特許または私たちの許可者の特許を回避することができ、これは、私たちの業務、財務状況、運営結果、および将来性に大きな悪影響を及ぼす可能性がある。
特許の発行は、その発明性、範囲、有効性、または実行可能性に対して決定的ではなく、我々の特許またはわれわれライセンシーの特許は、米国および海外の裁判所または特許庁で挑戦される可能性がある。私たちは、第3の方向の米国特許商標局が提出した既存技術の発行前提出、または反対、派生、撤回、再審査、付与後審査、および参加を受け入れるかもしれない各方面間審査、または他の同様の手続きは、私たちが所有する特許権に挑戦する。このような提出、訴訟、または訴訟における不利な裁決は、私たちの特許権の範囲を縮小したり、第三者が私たちの候補製品を商業化し、私たちと直接競争することを可能にしたり、第三者の特許権を侵害することなく製品を製造または商業化することができなくなる可能性があります。さらに、我々の特許又は我々ライセンシーの特許は、例えば外国特許庁の反対、我々の発明優先権又は我々の特許及び特許出願及び我々許可者の特許に関する他の特許的特徴に挑戦することができる後の挑戦手続を受けることができる。このような挑戦は、特許権の喪失、排他性の喪失、または特許主張の縮小、無効または実行不能をもたらす可能性があり、これは、他人が類似または同じ技術および製品を使用することを阻止するか、またはそれを商業化する能力を制限するか、または候補製品の特許保護期間を制限する可能性がある。最終的な結果が私たちに有利であっても、このような手続きは大量のコストを招く可能性があり、私たちの科学者と経営陣に多くの時間がかかる必要がある。さらに、我々の特許および特許出願または我々の許可側の特許および特許出願によって提供される保護の広さまたは強度が脅かされている場合、結果にかかわらず、会社が現在または将来の候補製品を許可、開発、または商業化することを阻止することができる。
しかも、特許の発行は私たちに特許発明を実践する権利を与えない。第三者は私たち自身の特許製品の販売を阻止し、私たち自身の特許技術を実践する特許を持っているかもしれない。
96
知的財産権は必ずしも私たちの競争優位に対するすべての潜在的な脅威を解決できるとは限らない。
私たちの知的財産権が提供する未来の保護の程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できないか、あるいは競争優位性を維持することができるかもしれない。例えば:
このような事件が発生した場合、私たちの業務、運営結果、そして将来性を深刻に損なう可能性がある。
私たちのビジネスの成功は、第三者の特許や他の固有の権利を侵害することなく運営する私たちの能力に大きく依存する。第三者は私たちが彼らの専有権を侵害し、損害賠償責任を招いたり、私たちの開発と商業化努力を阻害したり延期したりする可能性があると主張している。
私たちの商業的成功は第三者の特許と固有の権利の侵害を避けることにある程度依存する。しかし、私たちの研究、開発、および商業化活動は、第三者が所有または制御している特許または他の知的財産権を侵害または他の方法で侵害される可能性がある。他のエンティティは、特許または独自の権利を有しているか、または取得することができ、これは、私たちの候補製品および将来承認される可能性のある製品を製造、使用、販売、提供、または輸入する能力を制限し、または私たちの競争的地位を損なう可能性がある。アメリカ国内外には大量の生物製薬業界の特許とその他の知的財産権に関連する訴訟があり、特許侵害訴訟、異議、再審、各当事者間の審査手続きとアメリカ特許商標局及び/又は対応する外国特許庁への授権後審査手続きを含む。我々が候補製品を開発している分野には,第三者米国や外国から発行された特許や係属中の特許出願が多く存在する.我々の候補製品の使用または製造に関連する第三者特許または材料、配合、製造方法または治療方法の請求項に記載の特許出願が存在する可能性がある。
97
特許として発行されれば、私たちに不利になるかもしれない特許出願もあるかもしれない。米国および他の地方の特許出願は、通常、優先権を要求する最初の出願の約18ヶ月後に発行され、このような最も早い出願日は、一般に優先権日と呼ばれる。米国国外で提出されないいくつかの米国特許出願は、特許が発行される前に秘密にすることができる。したがって、私たちの候補製品に関する特許出願は、私たちが知らずに第三者によって提出されたかもしれない。さらに、開示された保留特許出願は、いくつかの制限された場合に、我々の候補製品およびその用途または製造プロセスをカバーするために、後に修正することができる。特許請求の範囲は、法的解釈、特許における書面開示、および特許の起訴履歴によって決定され、専門家の意見のような他の要因に関連する可能性がある。特許または係属中の特許または請求項の関連性または範囲の説明は正しくない可能性があり、これは私たちの候補製品をマーケティングする能力に負の影響を与えるかもしれない。さらに、我々は、我々の候補製品およびその用途および製造プロセスが第三者特許のカバー範囲内にないかどうか、または第三者の係属特許出願が関連範囲の特許請求を提出するかどうかを誤って予測する可能性がある。私たちが関連する特許の満期日の決定は、アメリカまたは海外のどのようなものでも正しくないかもしれません。これは、私たちの候補製品を開発し、マーケティングする能力に悪影響を及ぼす可能性があります。第三者知的財産権者もまた、知的財産権侵害または他の知的財産権に関するクレームを積極的に私たちに提起する可能性がある, たとえ私たちが私たちの候補製品と関連用途とプロセスのために特許保護を受けたとしても。
バイオ製薬業界の拡張や特許の発行に伴い、我々の候補品が第三者特許権侵害の告発を受けるリスクが高まる可能性がある。特許出願は一定期間秘密であるため、関連出願が公表される前に、任意の候補製品の商業化が第三者特許を侵害する可能性があることを知らない可能性があり、製品候補又は技術に関連する特許出願を初めて提出した会社であることも確認できない。さらに、特許出願が発行されるまでに数年かかる可能性があるため、現在処理されている特許出願が存在する可能性があり、これらの出願は、私たちの候補製品が発行された特許を侵害する可能性がある。さらに、特許検索は、特許間の用語の違い、データベースの不完全さ、および特許請求の意味を評価することが困難であるため、我々の技術に関連する可能性のある第三者特許権を識別することは困難である。私たちが知っていない保証もありませんが、私たちの業務とは関係のない既存技術だと思いますが、それにもかかわらず、これらの技術は最終的には、私たちが将来承認される可能性のある製品を製造、使用、販売、提供、または輸入する能力を制限したり、私たちの競争的地位を損なう可能性があります。さらに、第三者は将来的に特許を取得し、私たちの技術を使ってこれらの特許を侵害したと主張するかもしれない。第三者が主張するいかなる特許侵害請求も非常に時間がかかり、可能性がある
98
2022年12月31日現在、第三者が私たちに特許侵害請求をしていないにもかかわらず、他の人は独自の権利を持っている可能性があり、私たちの候補製品の発売を阻止する可能性がある。第三者は私たちの任意の候補製品に対して特許侵害請求をするかもしれない。私たちの特許関連法律訴訟に対して損害賠償を要求し、私たちの候補製品、治療適応、またはプロセスに関連する商業活動を禁止しようと努力しても、故意に侵害されたと判断された場合、3倍賠償し、私たちの候補製品を製造またはマーケティングする許可証を取得することを含む重大な損害賠償責任を負うことができます。これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、当社の業務における従業員資源を大量に移転することになる。私たちがこのような訴訟に勝つかどうかは予測できませんし、これらの特許に必要ないかなる許可が商業的に受け入れられる条項で提供されるかどうかも予測できません。さらに、私たちまたは私たちの現在および/または未来の戦略パートナーが許可を得ることができても、これらの権利は非排他的である可能性があり、これは、私たちの競争相手が同じ知的財産権を獲得することをもたらす可能性がある。さらに、必要であれば、権利侵害を回避するために、私たちの候補製品、治療適応、またはプロセスを再設計できるかどうかを決定することはできません。したがって、司法や行政訴訟で不利な裁決を下したり、必要なライセンスを取得できなかったりすることで、私たちの候補製品の開発と商業化を阻止することができ、これは私たちの業務、財務状況、経営業績を損なう可能性があります。また知的財産権訴訟はその結果にかかわらず, 否定的な宣伝を招く可能性があり、私たちの候補製品や技術をマーケティングまたは他の方法で商業化することを禁止するかもしれません。
私たちにクレームを出した当事者は、彼らがより多くの資源を持っているので、私たちよりも複雑な特許訴訟の費用を効果的に維持することができるかもしれない。さらに、知的財産権訴訟や行政訴訟に関連する大量の開示要求により、私たちのいくつかの機密情報が開示によって漏洩される可能性がある。さらに、任意の訴訟の開始および継続によって生じる任意の不確実性は、追加資金を調達する能力に重大な悪影響を及ぼすか、または私たちの業務、運営結果、財務状況、および将来性に重大な悪影響を及ぼす可能性がある。
私たちの現在のいくつかの候補製品と研究プロジェクトは第三者から許可を得ている。もしこれらの許可プロトコルが終了されたり、私たちの権利を縮小したと解釈された場合、私たちはこれらの技術に基づいて既存の候補製品を開発したり、新製品候補製品を開発する能力が実質的に悪影響を受けるだろう。
私たちは現在、Viracta治療会社、武田製薬有限会社、Dana Farber癌研究所、ミレニアム製薬会社、ドイツダムシュタットのMerck KGaAに少なくともある程度依存しており、Viracta治療会社、武田製薬有限会社、Dana Farber癌研究所、ミレニアル製薬有限会社、メルクKGaA、ドイツDarmstadtのライセンスと転授許可証、および潜在的に第三者との他の戦略関係に依存して、現在の候補製品の研究、開発、製造、商業化を行っている。もし私たちの任意のライセンスまたは関係、または私たちのライセンスに基づく任意のライセンス内のライセンスが終了または破壊された場合、私たちは:
さらに、終了または違反がなくても、私たちの知的財産権許可または再許可は、契約解釈上の相違を受ける可能性があり、これは、関連する知的財産権または技術に対する私たちの権利範囲を縮小したり、私たちの財務的または他の義務を増加させたりする可能性がある。
もし私たちが上記のいずれかの状況に遭遇した場合、私たちの業務に実質的な悪影響を及ぼす可能性があり、運営を停止させ、あなたのすべての投資損失を招く可能性があります。
99
もし私たちが許可協定に違反すれば、私たちの候補製品の商業化努力に実質的な悪影響を及ぼすかもしれない。
もし私たちがどんな合意にも違反したら、私たちはこれらの合意に従って私たちの候補製品または第三者に使用、開発、商業化の権利を許可し、私たちは私たちの業務に非常に重要な許可権を失うかもしれない。あるいは、私たちが第三者に知的財産権を許可する契約の義務を履行できなかった場合、あるいは私たちとライセンス者との業務関係が妨害された場合、私たちは私たちの業務に非常に重要な許可権を失う可能性があります。
私たちの現在の主要候補製品は、Viracta治療会社(F/K/a Sunesis PharmPharmticals,Inc.)によって独占的に許可されている特許および特許出願によって保護されている。私たちの現在の主要な候補製品とパイプライン、ならびに私たちが予想している最近のパイプラインは、例えば、ドイツのダムシュタットのメルクKGaAを含む他の第三者から許可を得る技術を含むことができる。
ライセンス契約によると、開発や商業化義務、潜在的な特許使用料支払いその他の義務など、職務遂行義務を含めた様々な義務を負わなければならない。もし私たちがこのような義務を履行できなかったり、他の方法で私たちの許可協定に違反した場合、私たちの許可者は適用された許可を全部または部分的に終了する権利があるかもしれない。一般的に、私たちの現在のどのライセンスも失われたり、私たちが将来獲得する可能性のある他のライセンスは、私たちの業務、見通し、財務状況、および運営結果を損なう可能性があります。
知的財産権許可は複雑な法律、商業、そして科学的な問題に関する私たちの業務に重要だ。ライセンス契約によると、私たちとライセンシーとの間で知的財産権紛争が発生する可能性があります
私たちが許可している知的財産権をめぐる紛争が、許容可能な条項で現在の許可手配の能力を維持しているか、または全くできない場合、影響を受けた候補製品の開発に成功し、商業化することができないかもしれない。また、知的財産権の所有権を許可する上で議論が生じた場合、特許権を追求または実行する能力が損なわれる可能性がある。もし私たちまたは私たちの許可者がこの知的財産権を十分に保護できなければ、私たちの製品商業化能力は影響を受ける可能性がある。
また,我々とViracta治療会社,武田製薬有限会社,Dana Farber癌研究所,ミレニアム製薬会社,ドイツダムシュタットのMerck KGaA社との許可を含む第三者から知的財産権や技術の許可を得るかどうかに基づいて,これらの合意のいくつかの条項は様々な解釈の影響を受ける可能性がある。可能性のある契約解釈の相違の解決は、関連する知的財産権または技術に対する私たちの権利と考えられる範囲を縮小するか、または関連協定の下での私たちの財務または他の義務を増加させる可能性があり、いずれも、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。また、私たちが許可している知的財産権をめぐる紛争が、商業的に許容可能な条項で許可スケジュールを維持する能力を阻害したり、損害を与えたりすれば、影響を受ける候補製品の開発および商業化に成功することができず、これは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
100
私たちが最善を尽くしたにもかかわらず、私たちの許可側は、許可協定に深刻に違反していると結論するかもしれませんので、許可協定を終了し、これらの許可協定がカバーする製品や技術を開発し、商業化することができません。これらの許可内で終了された場合、または基礎特許が予想される排他性を提供できなかった場合、競争相手は、規制部門の承認を求め、私たちと同じ製品を販売する権利がある。これは私たちの競争地位、業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼすかもしれない。
将来の製品の販売にどのくらいの印税義務が支払われるかは確定できませんが、金額は大きいかもしれません。私たちの将来の印税義務の金額は、開発や商業化に成功した製品で使用される技術や知的財産権にかかっています(あれば)。したがって、私たちが製品を開発して商業化することに成功しても、私たちは利益を達成したり維持することができないかもしれない。
将来的には、私たちは第三者技術の追加許可を得る必要があるかもしれません。これらの許可は私たちに提供できないかもしれないし、商業的に不合理な条項でしか得られないかもしれません。これは、私たちがより高価で不利な方法で私たちの業務を運営することにつながるかもしれません。
私たちは私たちの候補製品パイプラインを拡大することを求めており、一部の方式は重要な技術を付与する権利だ。私たちのビジネスの将来の成長は、私たちが許可する能力があるかどうか、または他の方法でより多くの候補製品または技術を獲得する権利があるかどうかにある程度依存するだろう。私たちは許容可能な条項で、または第三者から任意の候補製品または技術の許可または権利を得ることができることを保証することはできません。
他の会社や学術機関も、私たちの業務に関連する可能性のある特許出願をすでにまたは計画している可能性があります。時々、これらの第三者特許の侵害を避けるために、私たちは、既存または将来の候補製品をさらに開発したり、商業化するために、第三者から技術許可を得ることを要求される可能性があります。もし私たちが既存または未来の候補製品を製造、使用または販売するために必要な任意のそのような特許を含む、任意の第三者技術の許可を得ることを要求された場合、私たちは、商業的に合理的な条項でそのような許可を得ることができないか、またはそのような許可を得ることができないかもしれない。私たちの既存または将来の任意の候補製品を開発または商業化するために必要ないかなる第三者ライセンスも取得できず、関連する努力を放棄する可能性があり、これは私たちの業務と運営を深刻に損なう可能性があります。
これらの技術の内部ライセンスや買収は競争分野であり、一部のより成熟した企業も戦略を実施しており、魅力的と考えられる候補製品や技術を許可または買収することができる。これらの老舗会社は私たちより競争優位を持っているかもしれません。それらの規模、現金資源及びより強い臨床開発と商業化能力のためです。しかも、私たちを競争相手だと思っている会社は私たちに権利を権限を与えたくないかもしれない。さらに、私たちは私たちの重点分野で適切な候補製品や技術を決定できないかもしれない。もし私たちが適切な候補製品や技術の権利を得ることに成功できなければ、私たちの業務、財務状況、および見通しは影響を受ける可能性がある。
101
私たちは私たち自身の特許または私たちの許可者の特許を保護または強制する訴訟に巻き込まれるかもしれないが、これは高価で、時間がかかり、成功しないかもしれない。さらに、法廷で疑問視された場合、私たち自身が発行した特許または私たちの許可者の特許は無効または実行不可能と認定される可能性がある。
競争相手は私たちの知的財産権を侵害するかもしれない。権利侵害や不正使用を防ぐために、私たちは権利侵害請求を要求されるかもしれません。これは高価で時間がかかるかもしれません。さらに、特許侵害訴訟では、裁判所は、私たちが所有しているまたは許可中の特許が無効であること、強制執行できないこと、および/または侵害されていないと判断することができる。もし私たちまたは私たちの任意の協力者が第三者に対して法的訴訟を提起して、私たちの候補製品に対する特許を強制的に執行しなければならない場合、被告は私たちの特許または私たちの許可者の特許の全部または一部が無効であり、および/または強制的に実行できないことを反訴することができる。米国の特許訴訟では,被告が無効および/または実行不可能と主張する反訴が一般的である。有効性を疑問視する理由は、新規性の欠如、明らかな、十分な書面記述の欠如、実施できない、または明らかなタイプの二重特許を含む、いくつかの法定要件のいずれかを満たすことができなかったといわれるものを含む。主張を実行できない理由には,特許訴訟に関連する者が米国特許商標局に関連情報を隠蔽したり,起訴中に誤った陳述をしたりした疑惑が含まれている可能性がある.
第三者は、訴訟の範囲外であっても、米国特許商標局または海外特許庁に類似した無効クレームを提出することができる。このようなメカニズムには,再審,当事者間の再審手続き,付与後再審手続き,派生プログラムと外国法ドメインとの同等のプログラム(たとえば反対手続き)がある.法的に無効および/または実行不可能と断言された後の結果は予測不可能である。例えば、有効性の問題については、私たちは無効な以前の技術がないことを確認することはできないが、私たち、私たちの許可者、特許審査員は起訴中にこれを知らない。我々が知っていることがない保証もないが,我々の特許や特許出願が人間であるかもしれない特許や特許出願におけるクレームの有効性や実行可能な既存技術に影響を与えないと考えられるにもかかわらず,最終的にはクレームの有効性や実行可能性に影響を与える可能性がある.もし第三者が法律上の無効または実行不可能な主張によって勝利すれば、私たちは私たちの技術または私たちが開発する可能性のある任意の候補製品の少なくとも一部またはすべての特許保護を失うだろう。このような特許保護の喪失は、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼすだろう。
さらに、我々の特許および特許出願または我々の許可側の特許および特許出願によって提供される保護の広さまたは強度が脅かされている場合、会社が現在または将来の候補製品を許可、開発、または商業化することを阻止する可能性がある。
解決策が私たちに有利であっても、私たちの知的財産権に関する訴訟や他の法的手続きは、私たちに巨額の費用を発生させ、私たちの技術や管理者の正常な責任を分散させる可能性があります。このような訴訟または訴訟は、私たちの運営コストを大幅に増加させ、開発活動または任意の将来の販売、マーケティング、または流通活動に使用することができるリソースを減少させる可能性がある。私たちはそのような訴訟や訴訟手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らがより多くの財政資源を持っているので、私たちよりもこのような訴訟や訴訟の費用を効率的に負担するかもしれない。特許訴訟や他の訴訟の開始と継続による不確実性は、市場での私たちの競争能力を損なう可能性がある。
さらに、知的財産権訴訟または私たちの知的財産権に関連する他の法的手続きが大量の開示を必要とするため、私たちのいくつかの機密情報は、そのような訴訟または他の手続きで開示によって漏洩される可能性がある。
知的財産権訴訟は不利な宣伝を招き、私たちの名声を損ない、私たちの普通株の市場価格を下落させる可能性がある。
任意の知的財産権訴訟過程において、訴訟開始に関する公告及び公聴会の結果、動議の裁決及び他の仮手続又は訴訟の発展がある可能性がある。もし証券アナリストや投資家がこれらの公告が否定的だと思う場合、私たちの既存の候補製品、承認された製品、計画、または知的財産権の知覚価値は低下する可能性がある。したがって、私たちの普通株の市場価格は下がるかもしれない。このような声明はまた私たちの名声や私たちの候補製品の市場を損なう可能性があり、これは私たちの業務に実質的な悪影響を及ぼすかもしれない。
102
発明の優先度を決定するためには、派生プログラムが必要である可能性があり、不利な結果は、関連技術の使用を停止すること、または勝利者から権利許可を得ようと試みることを要求することができる。
第三者によって開始されるか、または我々によって提起されるか、または米国特許商標局によって発表される派生プログラムは、我々の特許または特許出願または我々の許可者の発明に関連する発明の優先度を決定するために必要である可能性がある。不利な結果は、私たちが関連技術の使用を停止することを要求するか、または勝利者から許可を得ようとすることを要求するかもしれない。もし勝利者が商業的に合理的な条件で私たちにライセンスを提供しなければ、私たちの業務は損害を受ける可能性があります。派生訴訟の弁護は失敗する可能性があり、成功しても巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。また、このようなプログラムに関連する不確実性は、私たちの臨床試験を継続するために資金を調達し、私たちの開発計画を継続し、第三者から必要な技術的許可を得たり、パートナー関係を開発または製造する能力に実質的な悪影響を及ぼす可能性があり、これらの資金は、候補製品を市場に出すのを助けるだろう。
訴訟の費用と不確実性のため、私たちは第三者に対して私たちの知的財産権を強制的に実行できないかもしれない。
訴訟の費用と不確実性のため、第三者が私たちが発表した特許を侵害しても、私たちの未解決または将来の特許出願または他の知的財産権によって発行される可能性のある任意の特許は、そのようなクレームや訴訟を提起して実行するリスク調整コストが高すぎるか、わが社または私たちの株主の最適な利益に適合していないか、または他の方法で特定の第三者に私たちの知的財産権を強制的に実行することは非現実的または望ましくない可能性があると結論するかもしれません。より多くの財力とより成熟して発展した知的財産の組み合わせを持っているので、私たちの競争相手または他の第三者は、複雑な特許訴訟または訴訟の費用を私たちよりも効果的に負担することができる。この場合、私たちは、状況を簡単に監視するか、または他の非訴訟の行動または解決策を開始または求めることで、より慎重なやり方を決定するかもしれない。また、訴訟に関連する不確実性は、私たちの製品開発、許可に必要な技術を継続し、または開発パートナー関係を構築するために必要な資金を調達する能力に影響を与える可能性があり、これらのパートナーは、私たちの候補製品を市場に出すのを助けるだろう。
最近の特許改革立法は、私たちの特許出願および/または私たちのライセンシーの特許出願をめぐる起訴、ならびに私たちが発行した特許および/または私たちのライセンシーの特許の実行または弁護の不確実性およびコストを増加させる可能性がある。
2011年9月16日、“ライシー·スミス米国発明法”または“ライシー·スミス法案”が法律に署名された。“ライシー·スミス法案”は米国特許法を多くの重大な改正を行った。このような条項は特許出願起訴方式に影響を与える条項を含み、特許訴訟に影響を及ぼす可能性もある。特に、“ライシー·スミス法案”によれば、米国は2013年3月に“最初に特許出願を提出した発明者”制度に移行し、この制度の下で、他の特許性要件を満たすと仮定して、最初に特許出願を提出した発明者は、要求された発明が第三者が最初に発明したものであるか否かにかかわらず、特許を取得する権利がある。したがって、2013年3月以降であるが、我々が以前に米国特許商標局に特許出願を提出した第三者は、たとえ第三者が発明を行う前に本発明を作成したとしても、我々の発明をカバーする特許を付与することができる。これは私たちが発明から特許出願までの提出時間を認識することを要求するだろう。さらに、有効かつ強制的に実行可能な特許を取得して維持する能力は、我々の技術と従来技術との差が、我々の技術が従来技術よりも特許を取得することを可能にするか否かに依存する。米国およびほとんどの他の国/地域の特許出願は、提出後または発行前の一定期間秘密であるため、私たちまたは私たちの許可者が、(1)私たちの候補製品に関連する特許出願または(2)発明特許または特許出願に要求される任意の発明を提出した最初の会社であることを確認しない可能性がある。
ライシー·スミス法案には、特許出願の起訴方法に影響を与え、特許訴訟に影響を与える可能性もあるいくつかの重大な変化も含まれている。これらの措置は、特許訴訟中に米国特許商標局が以前の技術を第3の方向に提出することを可能にすることと、PGR、IPRおよび派生プログラムを含む米国特許商標局によって管理された許可後訴訟手続き(PGR、IPRおよび派生プログラムを含む)が特許有効性を攻撃する追加の手続きとを含む。このような提出または手続きにおける不利な裁決は、私たちの特許権の範囲を縮小したり、実行可能にしたり、無効にしたりして、私たちの競争的地位に悪影響を及ぼす可能性があります。
103
USPTO手続きの証拠基準は、米国連邦裁判所が特許請求の無効を宣言するために必要な証拠基準よりも低いため、第三者は、USPTO手続において、USPTOが権利請求を無効にするのに十分な証拠を提供する可能性があり、同じ証拠が最初に地域裁判所訴訟で提出された場合には、権利請求を無効にするのに十分ではない。したがって,第三者は米国特許商標局の手続きを用いて我々の特許主張の無効を宣言しようとする可能性があり,第三者が地域裁判所訴訟で最初に被告として疑問を提起すれば,我々の特許主張は無効ではない.したがって、“ライシー·スミス法案”およびその実施は、私たちを起訴する特許出願および/または私たちのライセンシーをめぐる特許出願を増加させ、私たちが発行した特許または私たちのライセンシーの特許の実行または保護の不確実性およびコストを増加させる可能性があり、これらのすべては、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
米国特許法や他の国の法律の変化は、特許の全体的な価値を低下させ、候補製品を保護する能力を弱める可能性がある。
他の製薬会社と同様に、私たちの成功は知的財産権、特に特許に大きく依存する。生物製薬産業で特許を取得して実施することは高度な技術と法律の複雑さに関連している。したがって、生物製薬特許を取得して実行することは高価で、時間と固有の不確実性だ。米国及び他の国の特許法又は特許法解釈の変化は、我々の知的財産権の価値を低下させる可能性があり、特許出願をめぐる起訴及び特許出願の実行又は保護の不確実性及びコストを増加させる可能性がある。私たちは、私たちの特許または第三者特許で許容または実行される可能性のある特許請求の範囲の広さを予測することができない。しかも、国会や他の外国立法機関は私たちに不利な特許改革立法を通過するかもしれない。
例えば、米国最高裁判所は近年、いくつかの特許事件に対して裁決を下しているが、場合によっては利用可能な特許保護範囲を縮小するか、場合によっては特許所有者の権利を弱めるかである。我々の将来の特許取得能力に関する不確実性の増加に加えて,このようなイベントの結合は,いったん特許を取得する価値に関する不確実性をもたらしている.米国議会、米国連邦裁判所、米国特許商標局または外国司法管轄区の類似機関の決定によると、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、それによって、私たちが新しい特許を獲得したり、私たちの既存の特許と私たちが将来獲得または許可する可能性のある特許を実施する能力を弱める可能性がある。
私たちは私たちの特許と他の知的財産権の発明権または所有権のクレームに疑問を受けるかもしれない。
私たちおよび/または私たちの許可者は、元従業員、協力者または他の第三者が、発明者または共同発明者として、私たちの特許または他の知的財産権において権益を有するクレームの制約を受ける可能性がある。さらに、私たちが一生懸命働いているにもかかわらず、私たちおよび/または私たちの協力者がすべての発明者の身分を確認したか、または確認することを保証することはできません。特許出願上適切な発明者が示されていないことは、その上で発行された特許が強制的に実行できない可能性がある。発明権紛争は、発明者として指定された異なる個人の貢献に関する相互矛盾した意見、外国国民が特許標的開発に参加する外国法律の影響、我々の候補製品の開発に参加する第三者の義務衝突、または潜在的な共同発明の共同所有権に関する問題によるものである可能性がある。訴訟は、これらおよび他の挑戦在庫および/または所有権のクレームを解決するために必要である可能性がある。代替または追加として、私たちはこのような知的財産権上の私たちの権利範囲を明確にするために協定を締結することができる。もし私たちがこのようなクレームを弁護することができなければ、金銭損害賠償を支払うことに加えて、貴重な知的財産権、例えば貴重な知的財産権の独占所有権や使用権を失う可能性がある。このような結果は私たちの業務に実質的な悪影響を及ぼすかもしれない。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。
私たちのライセンス者は第三者コンサルタントや協力者に依存する可能性がありますので、私たちのライセンス者は私たちが許可を得た特許の唯一および独占所有者ではありません。もし他の第三者が私たちが許可した特許の所有権または他の権利を持っている場合、彼らはこれらの特許を私たちの競争相手に許可することができ、私たちの競争相手は競争製品を販売することができるかもしれない。これは私たちの競争地位、業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼすかもしれない。
104
また、私たちの政策は、知的財産権の概念や開発に参加する可能性のある私たちの従業員と請負業者が合意に署名し、このような知的財産権を私たちに譲渡することを要求しているにもかかわらず、実際に私たちが自分の知的財産権と見なしているすべての当事者とこのような合意を実行することができないかもしれない。知的財産権の譲渡は自動的に実行されない可能性があり、あるいは譲渡協定が違反される可能性があり、私たちは第三者にクレームをつけさせられたり、私たちが私たちの知的財産権の所有権だと思うことを決定するために、私たちが提起する可能性のあるクレームを弁護したりすることができます。このようなクレームは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。
特許条項は候補製品に対する私たちの競争地位を十分に長く保護するのに十分ではないかもしれない。
特許の寿命は限られている。米国では、すべての維持費が適時に支払われる場合、特許の自然失効時間は、通常、米国で最初の非臨時出願日から20年である。様々な延期があるかもしれないが、特許の有効期限とその提供される保護は限られている。私たちの候補製品をカバーする特許を取得しても、特許有効期限が切れたら、競争製品からの競争を受け入れるかもしれません。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちの特許の組み合わせは、他社が私たちと似ているか同じ製品を商業化することを排除するために、私たちに十分な権利を提供していないかもしれない。
もし私たちが私たちの候補製品のために特許期間を延長しなければ、私たちの業務は実質的な損害を受けるかもしれない。
FDAが我々の候補製品が発売される時間、期限および詳細を承認することによれば、私たちの1つ以上の米国特許または私たちの許可側の特許は、1984年の“薬品価格競争および特許期限回復法”または“ハッジ·ワックスマン修正案”に従って限られた特許期間を回復する資格がある可能性がある。ハッジ·ワックスマン改正案は、製品開発およびFDA規制審査中に失われた特許期間の補償として、特許回復期間を最長5年とすることを許可している。各FDAが承認した製品は、FDA規制審査中に失われた特許期間の補償として最大1つの特許を延長することができる。特許期間の延長は、製品承認日から計14年の期間を超えてはならず、当該承認された薬品、その使用方法又はその製造方法に関連する請求項のみが延長することができる。私たちの候補製品が規制部門の承認を得られれば、特定の国/地域でも特許期間を延長することができる。しかし,適用の最終期限内に出願を提出できなかったこと,関連特許の満了前に出願を提出できなかったことや適用の要求を満たしていなかったことなどにより延期が得られなかった可能性がある.しかも、特許保護の適用期間や範囲は私たちが要求しているものよりも短いかもしれない。もし私たちが特許期間の延長や回復を得ることができない場合、あるいはどのような延長の期限が私たちが要求したよりも短い場合、私たちの競争相手は私たちの特許が満期になった後に競争製品の承認を得ることができ、私たちの収入は減少する可能性があり、実質的である可能性がある。また、このような状況が発生すれば、私たちの競争相手は私たちの開発と試験への投資を利用して、私たちの臨床と臨床前データを参考にして、他の場合よりも早く彼らの製品を発売するかもしれない。
私たちは世界各地で私たちの知的財産権を保護できないかもしれない。
私たちはアメリカや他の国で未解決の特許出願を持っているにもかかわらず、世界のすべての国で特許を提出、起訴、保護することは恐ろしいほど高価であり、私たちのアメリカ以外のいくつかの国の知的財産権は米国の知的財産権を広く持っていないかもしれない。また、いくつかの外国の法律は知的財産権の保護の程度はアメリカの連邦や州法律に及ばない。したがって、私たちは、米国以外のすべての国/地域で第三者が私たちの発明を実施したり、米国または他の管轄区域で私たちの発明を使用して製造された製品を販売または輸入することを阻止することができないかもしれない。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの技術を使用して自分の製品を開発することができ、また、他の侵害製品を私たちが特許保護を持っている地域に輸出することもできるが、法執行力はアメリカに及ばない。これらの製品は私たちの候補製品と競争する可能性があり、私たちの特許、ライセンシーの特許、または他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれない。
105
多くの会社は外国の管轄区域の知的財産権の保護と保護に重大な問題に直面している。多くの国の法制度は特許や他の知的財産権保護の強制執行を支持しておらず、これは私たちの特許が人かもしれない特許を侵害したり、私たちの独占権を侵害する方法で競争製品をマーケティングすることを阻止することを困難にするかもしれない。外国の管轄地域で私たちの特許権を強制的に執行する訴訟は、大きなコストを招く可能性があり、私たちの努力と注意を私たちの業務の他の側面から移すことは、私たちの特許または私たちのライセンシーの特許が無効または狭義に解釈されるリスクに直面する可能性があり、私たちの特許出願または私たちのライセンシーの特許出願は発行できないリスクに直面し、第三者が私たちにクレームを請求する可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。したがって、私たちが世界各地で私たちの知的財産権を実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネスメリットを得るのに十分ではないかもしれません。
多くの国に強制許可法があり,これらの法律により特許権者は第三者に強制的に許可を付与される可能性がある。また、多くの国は政府機関や政府請負業者に対する特許の実行可能性を制限している。これらの国では,特許権者の救済措置は限られている可能性があり,このような特許の価値を大幅に低下させる可能性がある。もし私たちが第三者に私たちの業務に関連する任意の特許の許可を与えることを余儀なくされた場合、私たちの競争地位は損なわれる可能性があり、私たちの業務、財務状況、運営結果、および将来性は不利な影響を受ける可能性がある。
私たちの特許保護の獲得と維持は、法規および政府特許機関によって提出された様々な手続き、文書、費用支払い、および他の要求に依存しており、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
我々の特許及び/又は出願及び我々のライセンシーの特許及び/又は出願の生命期間内に、定期維持費、継続費、年会費及び各種他の政府費用は、USPTO及び各外国特許庁の異なる時間点で支払われる。私たちはシステムが私たちにこれらの費用を支払うことを注意して、私たちは私たちの外部特許年金サービスが満期になった時にこれらの費用を支払うことに依存します。また、米国特許商標局および各外国特許庁は、特許出願過程において、いくつかのプログラム、文書、費用支払い、および他の同様の条項を遵守することを要求する。私たちは名声の良い法律事務所や他の専門家を招いて私たちの遵守を助け、多くの場合、不注意は滞納金を支払うことによって、あるいは特定の管轄区域に適用される規則に従って他の方法で是正することができる。しかしながら、場合によっては、規定を遵守しないことは、特許または特許出願の放棄または失効をもたらす可能性があり、それにより、関連する管轄区域の特許権の一部または全部が失われる可能性がある。このような事件が発生すれば、私たちの業務に実質的な悪影響を及ぼす可能性がある。
もし私たちの商標と商号が十分に保護されていなければ、私たちは興味のある市場で知名度を作ることができないかもしれません。私たちの業務は悪影響を受けるかもしれません。
私たちは登録または未登録の商標または商号を使用して、私たち自身と私たちの製品をブランドしてマーケティングするつもりです。私たちの商標または商号は、挑戦、侵害、回避、または汎用商標として発表されるか、または他の商標を侵害していると判断される可能性がある。私たちはこれらの商標や商品名に対する私たちの権利を保護できないかもしれません。私たちは私たちが関心のある市場の潜在的なパートナーや顧客の中で知名度を確立するために必要です。時々、競争相手は私たちと似たような商品名や商標を採用して、ブランド表示を確立する能力を阻害し、市場の混乱を招く可能性があります。さらに、他の登録商標または商標の所有者は、我々の登録または未登録商標または商号の変異体を含む商号または商標侵害クレームを提出することができる。長期的には、私たちの商標や商号に基づいて名称を確立することができなければ、効果的に競争できない可能性があり、私たちの業務は悪影響を受ける可能性があります。商標、商業秘密、ドメイン名、著作権または他の知的財産権に関連する専有権を実行または保護する努力は無効である可能性があり、大量のコストおよび資源移転を招き、私たちの財務状況または運営結果に悪影響を及ぼす可能性があります。
106
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの商業と競争の地位は損なわれるだろう。
私たちは、非特許の技術的ノウハウ、技術、および他の独自の情報を含めて、私たちのビジネス秘密をある程度保護して、私たちの競争地位を維持しています。第三者との秘密協定の締結や、従業員、コンサルタント、コンサルタントとの秘密情報および発明協定の締結など、当社のビジネス秘密および非特許ノウハウを保護する措置を講じていますが、そのようなすべての合意が正式に実行されている保証はありません。いずれも契約に違反し、私たちのビジネス秘密を含めて独自の情報を漏洩する可能性があり、このような違反について十分な救済措置を得ることができない可能性があります。強制執行側が商業秘密を不正に開示したり流用したりする主張は困難であり、高価で時間がかかり、結果は予測できない。しかも、米国国内外のいくつかの裁判所は商業秘密をあまり望んでいないか、または保護したくない。
さらに、第三者は、この情報を取得する可能性があり、またはこの情報または同様の情報を独立して取得する可能性があり、私たちは、彼らが技術または情報を使用して私たちと競合することを阻止する権利がない。もしこのような事件が発生した場合、あるいは私たちが他の方法で私たちの商業秘密の保護を失った場合、これらの情報の価値は大幅に低下し、私たちの競争的地位は損なわれるかもしれない。もし私たちが特許発表前に特許保護を申請しない場合、または私たちの独自技術および他の機密情報を他の方法で秘密にすることができない場合、私たちが特許保護または私たちの商業秘密情報を保護する能力が脅かされる可能性がある。
私たちは私たちまたは私たちの職員たちがいわゆる機密情報や商業秘密を誤って使用または漏洩したと告発されるかもしれない。
私たちは、外部科学協力者、CRO、サードパーティ製造業者、コンサルタント、コンサルタント、潜在的パートナー、および他の第三者のような第三者の独自の地位を保護するために、将来的にセキュリティおよびセキュリティ協定を締結することができます。第三者が私たちまたは私たちの従業員が無意識にまたは他の方法で合意に違反し、第三者の独自の商業秘密または他の情報を使用または開示したと主張した場合、私たちは訴訟を受ける可能性がある。このような問題の弁護は、その是非曲直にかかわらず、巨額の訴訟費用がかかり、私たちの業務から従業員資源を大量に移転する可能性がある。私たちは私たちがこのようなどんな行動でも勝つかどうか予測できない。また、知的財産権訴訟は、その結果にかかわらず、負の宣伝をもたらす可能性があり、私たちの候補製品や技術をマーケティングまたは他の方法で商業化することを禁止する可能性があります。このようなクレームに抗弁しなければ、重大な金銭的損失責任を負わせたり、開発や商業化努力を阻害したり、延期したりする可能性があり、これは私たちの業務に悪影響を及ぼす可能性がある。私たちがこれらのクレームを弁護することに成功しても、訴訟は巨額のコストを招き、私たちの管理チームや他の従業員の注意を分散させる可能性がある。
私たちにクレームをつけた当事者は、彼らがより多くの資源を持っているので、私たちよりも複雑な知的財産訴訟の費用を効率的に負担することができるかもしれない。さらに、知的財産権訴訟に関連する大量の開示要求により、私たちのいくつかの機密情報が開示によって漏洩する可能性がある。さらに、任意の訴訟の開始および継続によって生じる任意の不確実性は、追加資金を調達する能力に重大な悪影響を及ぼすか、または私たちの業務、経営業績、財務状況、および見通しに重大な悪影響を及ぼす可能性があります。
107
私たちは競争相手から従業員を誤って雇用したか、または私たちまたは私たちの従業員が彼らの前の雇用主の機密情報や商業秘密を間違って使用または開示したという非難を受けるかもしれない。
バイオ製薬業界では、私たちの従業員のほかに、コンサルタントを招いて候補製品の開発を手伝ってくれていますが、これはバイオ製薬業界ではよく見られます。これらのコンサルタントの多く、および私たちの多くの従業員は、以前に他の製薬会社に雇用されていたか、または以前に他の製薬会社に提供されていた可能性があり、または現在、私たちの競争相手または潜在的な競争相手を含む他の製薬会社にコンサルティングサービスを提供している可能性がある。私たちは、私たち、私たちの従業員またはコンサルタントが無意識に、またはその前の雇用主またはその前の顧客または現在の顧客の商業秘密または他の固有の情報を使用または漏洩したという疑惑の影響を受けるかもしれない。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭損害賠償の支払いに加えて、貴重な知的財産権や人員を失う可能性があり、これは私たちの業務に悪影響を及ぼす可能性がある。私たちがこれらのクレームを弁護することに成功しても、訴訟は巨額のコストを招き、私たちの管理チームや他の従業員の注意を分散させる可能性がある。
私たちのいくつかの候補製品の特許保護と特許起訴は第三者にかかっているかもしれない。
私たちは通常、私たちの候補製品に関連する特許の起訴、維持、および実行を制御する権利を得ることを求めているが、時々私たちの候補製品に関連する特許および特許出願の提出および起訴活動は、私たちの許可者またはパートナーによって制御される可能性がある。もし私たちのいかなる許可者やパートナーも私たちの業務の最適な利益に合った方法でこのような特許および特許出願を起訴、維持、強制実行できなかった場合、私たちの候補製品に適用されるすべての特許費用を支払うことを含めて、私たちはこれらの権利に対する私たちの知的財産権やこれらの権利に対する私たちの排他性を失う可能性があり、私たちがこれらの候補製品を開発し、商業化する能力は悪影響を受ける可能性があり、私たちは競争相手が競合製品を製造、使用、販売することを阻止できないかもしれない。また、第三者から許可された特許および特許出願の特許起訴を制御する権利があっても、特許起訴の日までの私たちの許可者およびその弁護士の行動または不作為の悪影響または損害を受ける可能性がある。
現在、私たちの知的財産権保護には、Viracta治療会社、武田製薬有限会社、ドイツダムシュタットのメルクKGaAから取得した特許と特許出願が含まれています。私たちの独占ライセンスと非独占ライセンスはいくつかの保留権利によって制限される可能性があり、これは私たちの競争地位に悪影響を及ぼすかもしれない。私たちはいくつかのライセンス特許の組み合わせの起訴と維持を制御しません;したがって、私たちは私たちの業務の最適な利益に合った方法で特許シリーズを準備、提出、起訴、または維持することを保証することはできません。“経営陣の財務状況と経営成果の討論と分析--重要な合意”を参照。私たちが許可を得た特許の組み合わせは、他社が私たちの候補製品と類似した製品の商業化を阻止するのに十分で持続的な特許保護を提供してくれないかもしれない。
108
政府が援助したプロジェクトによって発見された知的財産権は、“進行権”、いくつかの報告要件、および米国会社の選好のような連邦法規の制約を受ける可能性がある。これらの規定を遵守することは、私たちの独占権を制限し、非アメリカメーカーと契約を締結する能力を制限するかもしれない。
私たち自身の発行されたいくつかの特許または未解決の特許出願は、米国政府資金を使用することによって生成される可能性があり、私たちは将来、米国政府資金や贈与を使用することによって生じるかもしれない知的財産権を獲得するかもしれない。1980年の“ベハ·ドール法案”によると、米国政府は政府が援助して開発した発明に対して何らかの権利を持っている。米国政府のこれらの権利は、任意の政府の目的のために発明を使用する非排他的、譲渡不可能、撤回不可能な世界的許可を含む。さらに、ある限られた場合、米国政府は、(1)発明を商業化するのに十分なステップが取られていない場合、(2)政府は、公衆衛生または安全需要を満たすために行動しなければならない、または(3)政府は、公共使用に対する連邦法規の要求を満たすために行動しなければならない、または(3)政府は、公共使用に対する連邦法規の要求を満たすように行動しなければならない、という権利を第三者に付与する権利がある。もし米国政府が米国政府の資金や贈与を使用することによって生じる既存または将来の知的財産権の進行権を行使した場合、私たちは私たちが開発した知的財産権を許可または再許可することを余儀なくされるか、または私たちに不利な条項で許可される可能性があり、このような権利の行使によって米国政府の補償を受けることは保証されない。付与者が政府に発明を開示していない場合、又は所定の期限内に知的財産権登録申請を提出していない場合、米国政府もこれらの発明の所有権を取得する権利がある。政府援助の計画の下で生成された知的財産権もまた、特定の報告書によって要求された制約を受けている, これを遵守するためには私たちが多くの資源を使う必要があるかもしれない。さらに、米国政府は、これらの発明を含む任意の製品、またはこれらの発明を使用することによって製造された製品のいずれも、実質的に米国で製造されなければならないことを要求する。知的財産権の所有者または譲受人が、合理的ではあるが成功しない努力をしたことを証明することができ、同様の条項で潜在的な被許可者に許可を付与することができ、これらの許可が米国で大量生産される可能性があり、またはこの場合、国内製造が商業的に不可能である場合、資金を提供する連邦機関は、米国工業のこのような選好を放棄する可能性がある。このような米国工業への偏愛は、このような知的財産権がカバーする製品について非米国製品メーカーと契約する能力を制限するかもしれない。
2023年6月1日より早く、欧州出願は、単一特許裁判所(UPC)の管轄を受ける特許付与後に単一特許となる選択権をすぐに有することになる。
これは欧州特許実践の大きな変化になるだろう。UPCは新しい裁判所制度であるため、裁判所には前例がなく、いかなる訴訟の不確実性も増加している。
米国および外国の地政学的行動は、私たちの特許出願または任意の既存または将来のライセンシーの特許出願をめぐる起訴または維持、ならびに私たちが発行した特許または任意の既存または将来のライセンシーの特許の維持、強制執行または保護の不確実性およびコストを増加させる可能性がある。
例えば、ロシアのウクライナ侵攻に関連する米国と外国政府の行動は、ロシアでの特許出願の提出、起訴、維持を制限または阻止する可能性がある。政府の行動はまたロシアで発行された特許を維持することを阻止するかもしれない。これらの行動は、ロシアにおける特許権の一部または全部の喪失を招く可能性がある私たちの特許または特許出願を放棄または失効させる可能性がある。このような事件が発生すれば、私たちの業務に実質的な悪影響を及ぼす可能性がある。また、ロシア政府は、2022年3月に、ロシアの会社や個人がロシアで友好的でないと考えている米国や他の国で、同意または補償なしに、米国および他のロシアが友好的でないと考えている国を有する公民権または国籍、米国および他の国に登録されている、または主にこれらの国で商業または利益活動を行う特許権者が所有する発明を利用することを可能にする法令を採択した。したがって、私たちは、第三者がロシアで私たちの発明を実践したり、私たちの発明を使用して製造された製品をロシア国内で販売したり輸入したりすることを防ぐことができないだろう。したがって、私たちの競争地位は損なわれる可能性があり、私たちの業務、財務状況、経営業績、見通しは不利な影響を受ける可能性があります。
109
私たちの普通株に関するリスクは
私たちの普通株の活発さと流動性取引市場は永遠に続かないかもしれない。したがって、買い取り価格や買い取り価格以上の価格で普通株を転売することができないかもしれません。
私たちの普通株の活発な取引市場は永遠に続かないかもしれない。私たちの普通株の時価は買い取り価格から下落するかもしれない。これらと他の要素のため、あなたが持っている普通株を買い取り価格以上の価格で転売することができないかもしれません。活発な市場の不足は株を売りたいときや合理的だと思う価格で株を売る能力を弱めるかもしれません。活発な市場の不足はまたあなたの株の公平な市場価値を下げるかもしれない。
また、活発でない市場は、普通株を売却することで資金を調達する能力を弱める可能性があり、戦略的協力を達成したり、私たちの普通株を対価格で会社や製品を買収する能力を弱める可能性があります。
私たちの四半期の経営業績は大幅に変動する可能性があり、あるいは投資家や証券アナリストの予想を下回る可能性があり、すべての状況は私たちの株価の変動や下落を招く可能性がある。
私たちの経営業績は四半期変動の影響を受けると予想されています。私たちの純損失と他の経営業績は様々な要素の影響を受けます
もし私たちの四半期の経営業績が投資家や証券アナリストの予想を下回れば、私たちの普通株の価格は大幅に低下する可能性がある。また、私たちの経営業績のどの四半期の変動も、私たちの普通株の価格を大幅に変動させる可能性があります。私たちの財務業績を四半期比較することは必ずしも意味があるわけではなく、私たちの将来の業績としての指標に依存すべきではないと考えられます。
110
私たちの普通株の市場価格は非常に不安定になる可能性があり、これは私たちの普通株の購入者に大きな損失をもたらすかもしれない。
私たちの普通株の市場価格は引き続き高度に変動し、様々な要素の広範な変動を受ける可能性があり、その中のいくつかの要素は私たちがコントロールできない。このような変動のため、あなたは支払う価格以上で普通株を売ることができないかもしれません。私たちの普通株の市場価格は、本“リスク要因”の部分に記載されている他のリスクと以下を含む多くの要素の影響を受ける可能性がある
111
そのほか、株式市場、特に製薬、生物製薬と生物技術株式市場は、極端な価格と出来高の変動を経験し、新冠肺炎の疫病、インフレ激化と金利変化及びサプライチェーンの中断を含み、これらは往々にして発行者の経営業績と関係がない或いは比例しない。また、我々普通株の取引価格は、第三者が市場価格を押し下そうとする悪影響を受ける可能性がある。もし私たちの株が下落すれば、空売り者や他の人たち--その中の一部の人はソーシャルメディアに匿名で投稿して利益を得るかもしれません。彼らの活動は私たちの株価に悪影響を及ぼすかもしれません。これらの広範な市場と業界要素は、私たちの実際の経営業績にかかわらず、私たちの普通株の市場価格を深刻に損なうかもしれない。本“リスク要因”部分に記載されたリスクを含む、上記の任意のリスクまたは任意の広範な他のリスクを達成することは、私たちの普通株の市場価格に重大かつ不利な影響を与える可能性がある。
過去、証券集団訴訟は上場企業の証券市場価格が下落した後に提起されることが多かった。このリスクは生物製薬会社と特に関連しており,これらの会社は近年大幅な株価変動を経験している。私たちがこのような訴訟に直面すれば、巨額のコストを招き、経営陣の注意と資源を移転させる可能性があり、これは私たちの業務を損なう可能性があります。
私たちは現在、私たちの普通株に配当金を支払うつもりはありませんので、私たちの株主が投資リターンを達成する能力は私たちの普通株の価値増加に依存します。
私たちは普通株の現金配当金を発表したり支払ったりしたことがない。私たちは現在、運営を支援し、私たちの業務の成長と発展に資金を提供するために、すべての利用可能な資金と未来の任意の収益を維持するつもりです。予測可能な未来に、私たちは私たちの株のいかなる現金配当金も発表したり支払うつもりはない。したがって、私たちの普通株のどんな投資収益も私たちの普通株価値の増加にかかっていることは確実ではない。
私たちの普通株を売る大量の株は私たちの普通株の価格を下落させるかもしれない。
公開市場で私たちの大量の普通株を売ることはいつでも起こる可能性がある。もし私たちの株主や市場が私たちの株主が公開市場で私たちの普通株を大量に販売しようとしていると思うなら、私たちの普通株の市場価格は大幅に低下する可能性があります。
2022年12月31日現在、私たちが発行した普通株を保有する合計73,458,176株の保有者は、ある条件に適合した場合に彼らの株式登録声明を提出すること、または彼らの株式を自分または私たちの株主に提出する可能性のある登録声明に含めることを要求する権利があるだろう。私たちはまた登録された普通株を持っていて、私たちは私たちの株式激励計画に基づいて発行することができる。これらの株は発行されると公開市場で自由に取引することができる。
公開市場で私たちの株や売却可能な株が私たちの普通株の市場価格にどのような影響を与えるか予測できません(あれば)。しかし、将来的には、私たちの未償還オプションを行使する際に発行された株を含む、私たちの普通株を公開市場で大量に売却したり、このような売却が発生する可能性があると考えられたりして、私たちの普通株の市場価格に悪影響を及ぼす可能性があります。
私たちはまた、未来に私たちが計画している業務を継続するために多くの追加資本が必要かもしれないと予想している。資本を調達するために、私たちは1回または複数回の取引で時々決定された価格および方法で普通株、転換可能証券、または他の株式証券を販売することができる。株式または他の株に変換可能な証券を売却して発行することで追加資本を調達すれば、我々の株主は希釈されるだろう。これらの売却、あるいは市場で大量の株式保有者が株を売却しようとしているとの見方は、我々の普通株の市場価格を低下させる可能性がある。
112
私たちの主要株主と経営陣は私たちのかなりの割合の株を持っており、株主の承認が待たれる事項に大きな制御を加えることができるだろう。
2022年12月31日現在の私たちの普通株の実益所有権によると、私たちの役員、取締役、5%以上の株式の所有者と、それぞれの関連会社の実益は、私たちの34%の議決権のある株を持っています。このグループの投票権は、彼らが持っている無投票権を普通株に転換する程度に増加する可能性がある。したがって、これらの株主が一緒に行動すれば、取締役の選挙、私たちの組織文書の修正、任意の合併、合併、または私たちのほとんどの資産、および任意の他の重大な会社取引を含む、株主の承認を必要とする会社の行動の結果に大きな影響を与え続ける。これらの株主の利益はあなたの利益とは違うかもしれないし、あなたの利益と衝突する可能性もある。例えば、これらの株主は、わが社の支配権の変更を延期または阻止することができ、たとえこのような制御権の変更が私たちの他の株主に利益をもたらすとしても、私たちの株主が私たちの会社や私たちの資産を売却する際に普通株のプレミアムを得る機会を奪い、私たちの普通株の現行の市場価格に影響を与える可能性がある。
私たちは“新興成長型会社”であり、“小さな報告会社”でもあり、新興成長型会社やより小さい報告会社に適用される報告要求を低減し、私たちの普通株の投資家に対する吸引力を低下させるかどうかを確定することはできません。
“雇用法案”の定義によると、私たちは“新興成長型会社”です。私たちが新興成長型企業であり続ける限り、(I)サバンズ-オキシリー法案を遵守する必要がない監査員認証要求、(Ii)定期報告や依頼書の役員報酬に関する開示義務の削減、(Iii)役員報酬に対する拘束力のない諮問株主投票の免除、および株主承認前に承認されていない金パラシュート支払いの要求を免除することを含む、他の非新興成長型上場企業に適用される様々な報告要件の免除を利用することができる。
IPO完了後、最大5会計年度で新興成長型企業になることができます提供, しかし、場合によっては、もし私たちが“大型加速申告会社”とみなされていれば、非関連会社が保有する普通株の時価が7億ドル以上であれば、もし私たちの年間総収入が12.35億ドル以上であれば、あるいはそれまでの任意の3年以内に10億ドルを超える転換不可能債券を発行した場合、このような状況が発生することを含む場合がある。2023年12月31日から、新興成長型企業の地位を失い、各種報告要件の免除を利用することができなくなり、2024年までに提出される2023年12月31日までの会計年度報告から開始する。
私たちが新興成長型会社になる資格がなくなっても、私たちは“小さな報告会社”になる資格があり、これは、サバンズ-オキシリー法案404条の監査人認証要件の遵守が要求されないこと、および私たちの定期報告書や依頼書で役員報酬に関する開示義務を減らすことを含む、多くの同じ開示要件を利用することができるようになるだろう。私たちは投資家が私たちがこのような免除に依存する可能性があるために私たちの普通株の吸引力が低下していることを発見するかどうか予測できない。したがって、一部の投資家が私たちの普通株の吸引力が低下していることを発見すれば、私たちの普通株はそれほど活発ではない取引市場が出現する可能性があり、私たちの株価はもっと変動するかもしれない。
雇用法案によると、新興成長型企業は、これらの基準が民間企業に適用されるまで、新たな会計基準の採用や改正された会計基準の採用を延期することもできる。私たちはこの過渡期を延長する利点を利用することを選択した。したがって、私たちの財務諸表は、このような新しい会計基準または改訂された会計基準を遵守する会社の財務諸表と比較できない可能性がある。我々が“新興成長型企業”でなくなったり、証券法第7(A)(2)(B)条に規定されている免除の日を明確かつ撤回不可能に選択する前に、我々の合併財務諸表に適用され、上場企業と非上場企業に対して異なる発効日を有する新たなまたは改訂された会計基準が発行された場合には、非新興成長型企業が採用すべき日および最近発表された会計基準を採用する日を開示する。
113
私たちも“小さな報告会社”であり、これは私たちの非関連会社が保有している株の時価が7.0億ドル未満であることを意味し、最近終了した会計年度では、私たちの年収は1.00億ドル未満である。(I)非関連会社が保有するわが株の時価が2.5億ドル未満である場合、または(Ii)最近終了した会計年度中に、我々の年収が1.00億ドル未満であり、非関連会社が保有するわが株の時価が7.00億ドル未満であれば、規模の小さい報告会社であり続ける可能性がある。もし私たちが新興成長型会社でなくなったときに規模の小さい報告会社であれば、私たちは同じ免除に依存し続け、サバンズ-オキシリー法案第404条の監査人認証要件の遵守を要求されないこと、役員報酬に関する開示義務を減少させること、および当社のForm 10-K年度報告書で最近の2つの会計年度の監査済み財務諸表のみを示すことを含む、特定の開示要件を遵守しない可能性がある。私たちは投資家が私たちがこのような免除に依存する可能性があるために私たちの普通株の吸引力が低下していることを発見するかどうか予測できない。したがって、一部の投資家が私たちの普通株の吸引力が低下していることを発見すれば、私たちの普通株はそれほど活発ではない取引市場が出現する可能性があり、私たちの株価はもっと変動するかもしれない。
私たちの定款書類やデラウェア州法律によると、反買収条項は私たちの買収を阻止または延期する可能性があり、これは私たちの株主に有利になる可能性があり、私たちの株主が現在の経営陣を交換または更迭しようとすることを阻止することができるかもしれない。
私たちが再記述した会社登録証明書と私たちの改訂と再記述の定款には、わが社の制御権変更を延期または阻止する可能性のある条項が含まれています。これらの規定は、我々の取締役会の現職メンバーが指名した取締役や他の会社の行動ではなく、株主が我々の経営陣を変動させることを含めて、株主を選挙することを困難にする可能性もある。これらの規定には
また、デラウェア州会社法(DGCL)第203条は、わが社への支配権変更を阻止、延期、または阻止する可能性がある。第203条私たちと15%以上の普通株式を保有する者との間の合併、業務合併、その他の取引には、いくつかの制限が加えられている。
114
私たちの組織文書の独占法廷条項は、司法フォーラムで、私たちまたは私たちの任意の取締役、上級管理者または他の従業員、またはそのようなクレームの発行を引き起こす任意の引受業者と紛争を引き起こす任意のクレームに有利であると考える株主の能力を制限する可能性があり、これは、そのようなクレームに関連する訴訟を阻害する可能性がある。
我々が再記述した会社登録証明書は、法律が許容する最大範囲内で、デラウェア州衡平裁判所は、私たちを代表して提起された任意の派生訴訟または訴訟、受託責任違反を主張する任意の訴訟、DGCL、私たちが再記載した会社登録証明書、または私たちが再説明した定款に基づいて私たちにクレームを提起する任意の訴訟、または内部事務原則に基づいて私たちにクレームを提起する任意の訴訟であることが規定されている。この排他的裁判所条項は、1934年に改正された“証券取引法”または“取引法”に規定された義務または責任を執行するための訴訟には適用されない。しかし、排他的裁判所の規定によって列挙された1つまたは複数のカテゴリに属する訴訟に適用することができる。
このような裁判所条項の選択は、我々または私たちの任意の取締役、上級管理者または他の従業員、またはそのようなクレームを引き起こす任意の発行された引受業者と紛争を引き起こすと考えられる株主のクレームを司法裁判所で提出する能力を制限する可能性があり、これは、そのようなクレームに関連する訴訟を阻害する可能性がある。代替的に、裁判所が、私たちが再記載した会社登録証明書に含まれる裁判所条項の選択が訴訟において適用されないか、または実行できないことを発見した場合、私たちは、他の司法管轄区域でこのような訴訟を解決することに関連する追加費用を生じる可能性があり、これは、私たちの業務、運営結果、および財務状態を損なう可能性がある。
証券法第22条は,連邦裁判所及び州裁判所は,証券法又はその下の規則及び条例に規定されている任意の義務又は責任を執行するために提出されたすべてのクレームに対して同時管轄権を有すると規定されている。私たちが再言及する付例は、法律によって許容される最大範囲内で、アメリカ合衆国の連邦地域裁判所は、訴状に指名された任意の被告に対して提起されたすべての訴因を含む、証券法または連邦フォーラム条項に基づいて提起された訴因を解決する任意の訴えを解決する独占的フォーラムとなることを規定する。疑問を生じないために、本条文は、吾等、吾等の上級職員及び取締役、任意の株式引受業者、及び任意の他の専門実体(その専門が当該者又は実体になされた声明を許可し、目論見の基礎となる文書の任意の部分を準備又は証明した)に恩恵を受け、本条文を強制的に執行することができるようにすることを目的としている。私たちが連邦フォーラムの規定を採択することを決定する前に、デラウェア州最高裁判所は、デラウェア州の法律によると、これらの規定は事実上有効であると判断した。連邦または州裁判所はデラウェア州最高裁判所の裁決に従わないかもしれないし、特定の事件で連邦フォーラム条項を実行することを決定する可能性があるが、連邦フォーラム条項の適用は、私たちの株主が証券法を実行するために生じるいかなる義務や責任のために提起された訴訟は連邦裁判所で提起されなければならず、州裁判所で提起することはできず、私たちの株主は連邦証券法とその下の規則と法規を遵守することを放棄することはできないことを意味する。
取引法第27条は,連邦政府が取引法又はその下の規則及び条例を実行するために生じた任意の義務又は責任に対して提起されたすべてのクレームに対して排他的連邦管轄権を有すると規定されている。また,排他的裁判所条項も連邦裁判所条項も,取引法で規定されているいかなる義務や責任を実行するための訴訟にも適用されない.したがって、私たちの株主は、取引法またはその下の規則と法規によって生じるいかなる義務または責任を実行するために連邦裁判所に訴訟を提起しなければならず、私たちの株主は連邦証券法とその下の規則と法規を遵守することを放棄することはできない。
任意の個人またはエンティティが、私たちの任意の証券の任意の権益を購入または他の方法で取得または保有することは、連邦フォーラム条項を含む、私たちの独占フォーラム条項を通知し、同意したとみなされなければならない。これらの条項は、株主がクレームを出す能力を制限する可能性があり、株主が、私たちまたは私たちの役員、役員または他の従業員、またはそのようなクレームの発行を引き起こす任意の引受業者と紛争した場合、彼らが選択した司法裁判所でそのようなクレームを提出するコストが増加する可能性があり、これは、私たちと私たちの役員、役員、および他の従業員に対する訴訟を阻止する可能性がある。
115
証券や業界アナリストが私たちの業務に関する研究や報告を発表していない場合、または彼らが私たちの会社に不利または誤った意見を発表した場合、私たちの普通株価格と取引量は低下する可能性がある。
私たちの普通株の取引市場は、業界や証券アナリストが発表した私たちまたは私たちの業務に関する研究と報告の影響を受けています。私たちはアナリストや彼らの報告書に含まれている内容と意見に対して何の統制権もない。もし私たちのどのアナリストが私たち、私たちのビジネスモデル、私たちの知的財産権、あるいは私たちの株式表現に否定的または誤った意見を発表した場合、あるいは私たちの臨床前研究と未来の臨床試験と運営結果がアナリストの期待に達しなかったら、私たちの株価は下落するかもしれない。1人以上のそのようなアナリストが私たちへの報告を停止したり、私たちに関する報告書を定期的に発表できなかったりすれば、私たちは金融市場で可視性を失う可能性があり、これは逆に私たちの株価や取引量を低下させる可能性がある
一般リスク因子
上場企業としては、私たちの運営コストが増加し、私たちの経営陣は、新しいコンプライアンス措置やコーポレートガバナンス実践を実施するために多くの時間を投入する必要があります。
上場企業として、特に新興成長型企業ではなくなった後、巨額の法律、会計、その他の費用が発生することは、私たちが民間会社としては起こらなかったことである。サバンズ-オクスリ法案、ドッド-フランクウォール街改革と消費者保護法、ナスダック世界選りすぐりの市場またはナスダックの上場要求、その他の適用される証券規則と法規は、有効な開示、財務制御と会社管理やり方の確立と維持を含む上場企業に対して様々な要求を提出した。私たちの経営陣と他の人たちはこのようなコンプライアンス計画を実施するために多くの時間を投入する必要があるだろう。さらに、私たちはこれらの規則と法規が私たちの法律と財務コンプライアンスコストを大幅に増加させ、いくつかの活動をより時間的で高価にすると予想する。例えば、これらの規制は、取締役や上級管理者責任保険をより難しく、より高価に得ることができ、十分な保険範囲を維持するために多くの費用を発生させることが要求される可能性があると予想されます。私たちは、これらの要求に応答して生成される可能性のある追加コストの金額や時間を予測または推定することができない。これらの要求の影響は、私たちの取締役会、取締役会委員会、または役員に参加することをより難しくし、合格した人を引き付け、維持することを可能にするかもしれない。コストの増加は、他の業務分野のコストを下げる必要があるかもしれません。あるいは商業化すると、私たちの製品の価格を上げる必要があります。また、これらの規則と条例はしばしば異なる解釈を受け、多くの場合、特殊性が不足しているため、, 規制や理事機関が新たな指導を提供するにつれて、それらの実践への応用は時間の経過とともに変化する可能性がある。これは遵守事項に関する持続的な不確実性と、開示と統治慣行を絶えず修正するために必要なより高いコストをもたらす可能性がある。
財務報告書を適切かつ効果的な内部統制を維持することができなければ、正確かつタイムリーな財務諸表を作成する能力が損なわれる可能性がある。
サバンズ·オキシリー法第404条によると、我々の経営陣は、2022年12月31日までのForm 10−K年次報告から、財務報告書の内部統制に対する有効性を報告しなければならない。“新興成長型会社”の地位を失い、“加速申告会社”または“大型加速申告会社”となった場合、独立公認会計士事務所は、財務報告に対する内部統制の有効性を証明することを求めることになる。管理管理層は、財務報告の内部統制に達成しなければならない標準的なルールを評価するには複雑であり、大量の文書、テスト、可能な救済措置が必要である。規定された時間内に第404条を遵守するために、私たちは費用が高く挑戦的な財務報告書の内部統制の過程を記録して評価するつもりだ。この点では、外部コンサルタントを招聘することが可能な内部資源を継続的に提供する必要があり、詳細な作業計画により、財務報告内部制御の十分性を評価して記録し、適宜ステップ改善制御プログラムを採用し、制御措置がファイルのように機能しているかどうかをテストすることにより、財務報告内部制御の継続報告及び改善手順を実施する。この過程は時間がかかり、高価で複雑になるだろう。
116
財務報告を内部統制できなかったいかなる行為も、財務状況、経営結果、あるいはキャッシュフローを正確に報告する能力を深刻に抑制する可能性がある。もし私たちが財務報告の内部統制に有効な結論を出すことができない場合、あるいは私たちの独立公認会計士事務所が私たちの財務報告の内部統制に重大な弱点や重大な欠陥があると判断した場合、投資家は私たちの財務報告の正確性と完全性に自信を失う可能性があり、私たちの普通株の市場価格は下落する可能性があり、私たちはナスダック世界の精選市場、ナスダック、米国証券取引委員会、または他の規制機関の制裁または調査を受ける可能性がある。財務報告の内部統制のいかなる重大な欠陥を補うことができなかったり、上場企業に必要な他の効果的な制御システムを実施または維持できなかったりすることは、将来的に資本市場に参入する機会を制限する可能性もある。
私たちの開示統制と手続きはすべてのミスや詐欺を阻止したり検出できないかもしれない。
私たちは“取引所法案”の定期報告書の要求事項を守らなければならない。我々の開示制御および手続きは、取引所法案に基づいて提出または提出された報告書に開示されなければならない情報が蓄積され、管理層に伝達され、米国証券取引委員会規則および表に指定された期間内に記録、処理、まとめ、報告されなければならないことを合理的に確保するために設計されている。任意の開示制御およびプログラムまたは内部制御およびプログラムは、発想および動作がどのように完全であっても、絶対的な保証ではなく、合理的な保証を提供することしかできず、制御システムの目標が達成されることを確保することしかできないと信じている。
これらの固有の限界は,意思決定過程における判断が誤りである可能性があり,簡単な誤りや誤りによって故障が発生する可能性があるという現実を含む.例えば、私たちの役員または役員は、意図せずに新しい関係または手配を開示できなかった可能性があり、関連するいかなる取引も開示できなかった。さらに、ある人の個人的な行動、2人以上の結託、または無許可超越制御は、制御を回避することができる。したがって,我々の制御システム固有の制限により,誤りや詐欺による誤り陳述が発生し,発見されない可能性がある.しかも、私たちは他の分野での私たちの業務のリスクを識別して解決するための正式なリスク管理計画を持っていない。
私たちは証券訴訟の影響を受けるかもしれませんが、これは高価で、経営陣の注意をそらすかもしれません。
私たちの普通株の市場価格は変動するかもしれない。株式市場全体、特にナスダックとバイオ製薬会社は、極端な価格と出来高変動を経験しており、これらの変動は往々にしてこれらの会社の経営業績に関係なく、あるいは比例しない。過去に、株式市場の価格変動を経験した会社は証券集団訴訟の影響を受ける。私たちは未来にこのような訴訟の目標になるかもしれない。私たちに対する証券訴訟は巨額のコストを招き、私たちの経営陣の注意を他の業務から移す可能性があり、これは私たちの業務を深刻に損なう可能性があります。
情報技術EM 1 B.未解決の従業員のコメント。
ない。
情報技術EM 2.財産.
私たちの主な行政事務室はカリフォルニア州ブリスベンにあり、私たちはそこで約12,000平方フィートのオフィス空間を借りました。レンタル契約は2024年12月に満期になります。賃貸契約期間を延長する選択もなく、賃貸契約が満期になる前に賃貸契約を終了する選択もない。私たちはこれらの施設が私たちの持続的な需要に対応するのに十分だと信じており、もし私たちがもっと多くの場所が必要なら、私たちは商業的に合理的な条件でより多くの施設を得ることができるだろう。
117
イットM 3.法的訴訟
当社は時々正常な業務過程で発生する訴訟の当事者になる可能性があります。当社はいかなる重大な法律手続きの影響も受けておらず、その知る限り、現在のところ重大な法的手続きが待っていることや脅かされていることはない。
情報技術EM 4.鉱場安全を開示する
適用されません。
118
第II部
情報技術EM 5.登録者普通株、関連株主事項、発行者が株式証券を購入する市場
普通株式市場情報
私たちは2021年5月26日に上場企業になった。私たちの普通株はナスダック資本市場で看板取引され、取引コードは“曙光”です
記録保持者
我々の譲渡エージェントが提供する情報によると,2023年3月1日現在,我々普通株の登録株主は約36人である.株主の実際の数は受益者である株主を含む記録保持者の数を超えているが、その株は仲介人や他の被著名人が街頭名義で保有している。この数の登録所有者は、その株式が他のエンティティが信託形式で保有する可能性のある株主も含まれていない。
配当政策
私たちは普通株の現金や株式配当金を発表したり支払ったりしたことがない。私たちは現在、すべての利用可能な資金と任意の将来の収益を維持し、私たちの業務運営のために、予測可能な未来に私たちの普通株にいかなる配当も支払わないと予想している。将来的に普通配当金を発表する任意の決定は私たちの取締役会が適宜決定し、私たちの財務状況、経営業績、資本要求、一般業務状況と私たちの取締役会が関連する他の要素に依存するかもしれない。
株式補償計画に基づいて発行された証券
本プロジェクトに要求される情報は、本10−Kフォーム年次報告の修正案に含まれるか、又は本テーブル10−K年次報告に含まれる財政年度終了後120日以内に第14 A条に基づいて提出される最終依頼書に参照される。
119
最近売られている未登録証券
ない。
登録証券を使って収益を得る
2021年6月1日、私たちはIPOを完了し、1株16.00ドルのIPO価格で1150万株の普通株を売却した。今回の株式募集の全株式は、米国証券取引委員会が2021年5月26日に発効を発表したS-1表登録声明(第333-255754号文書)に基づいて証券法に基づいて登録されている。追加株式は登録されていません。
約1,290万ドルの引受割引と手数料および推定約410万ドルの発売費用を差し引いた後,IPOから約1.67億ドルの純収益を得た。J.P.Morgan Securities LLC、Cowen and Company、LLC、Piper Sandler&Co.が今回発行された共同簿記管理人と引受業者代表を務める。IPOに関連するいかなる費用も、取締役、上級管理者、10%以上の任意の種類の持分証券を所有する者、またはそれらの共同会社または当社の連属会社に支払われていない。
2021年5月27日に証券法第424(B)(4)条に基づいて米国証券取引委員会に提出された目論見書に記載されているように、我々が初めて公募して得られた資金の計画用途に実質的な変化はない。
発行者および関連購入者が株式証券を購入する
ない。
情報技術EM 6.保留
120
情報技術経営陣の財務状況と経営結果の検討と分析。
当社の財務状況と経営業績の検討と分析、本年度報告書でForm 10-K形式で提供されている連結財務諸表および関連付記をお読みください。本議論および分析に含まれるまたは本Form 10-K年次報告の他の部分に記載されている情報は、リスクおよび不確定要因に関する前向きな陳述を含む、我々の業務および関連融資の計画および戦略に関する情報を含む。本年度報告10-K表の“リスク要因”の部分に列挙された要素を含む多くの要因の影響により、我々の実際の結果は、以下の議論および分析に含まれる前向き陳述に記載または示唆された結果とは大きく異なる可能性がある。2021年12月31日までの財政年度と2020年12月31日までの財務業績の比較については、2022年3月7日に米国証券取引委員会に提出された2021年12月31日までの財政年度報告の第7項、すなわち経営陣の財務状況と経営成果の検討と分析を参照されたい。本報告で使用されるように、文脈が別に説明されていない限り、“私たち”、“当社”または“初日”は、初日のバイオ製薬会社を意味する。
概要
設立初日は重要な未満足の需要を解決するためだった:癌児童は抗癌薬開発革命で後ろに振られた。私たちの名前のインスピレーションは、医師と患者とその家族が初歩的な癌診断と治療計画について行った“初日トーク”に由来している。われわれの目標は,1日目から抗癌剤の開発を再想定し,年齢にかかわらずすべての癌患者の可能性を再定義することである。
我々は臨床段階の生物製薬会社であり,生命にかかわる疾患を有するすべての年齢の患者のための標的治療の開発と商業化に取り組んでいる。当初,癌を有する小児科患者に臨床開発努力を集中させたが,これは脆弱な集団であり,最近の標的治療や免疫腫瘍学革命では十分なサービスが得られていなかった。
私たちの主要な候補製品tovorafenib(DAY 101)は経口、脳透過、高選択性II型PAN急速加速型繊維肉腫、あるいはPAN-RAF、キナーゼ阻害剤である。Tovorafenib(DAY 101)は325人以上の患者で研究されており、単一療法として全体的な耐性が良好であることが証明されている。Tovorafenib(DAY 101)は特定の遺伝子変化を有する児童と成人群の中で鼓舞的な抗腫瘍活性を示し、このような変化はRAS/マイトジェン活性化プロテインキナーゼ(MAPK)経路の過剰活性化を招き、細胞成長の暴走を招く。
Tovorafenib(DAY 101)は2020年8月にFDAによってpLGG治療のための突破療法指定が承認され、これは第1段階試験の初歩的な結果に基づいており、この試験はpLGG患者が迅速な抗腫瘍活性と持続的な反応を有することを示している。児童低レベルグリオーマは児童の中で診断される最もよく見られる脳腫瘍であり、標準的な治療法もなく、大多数の患者に対して承認された治療方法もない。著者らは2020年9月にFDAから悪性グリオーマ治療の孤児薬物指定を獲得し、2021年5月にEUグリオーマ治療委員会から悪性グリオーマ治療の指定薬物を獲得した。さらに、FDAは2021年7月にtovorafenib(DAY 101)の活性化RAF変化が存在する低レベルグリオーマ(LGG)の存在する稀な小児科疾患の治療を許可した。
121
我々はすでにtovorafenib(DAY 101)の重要な2期試験あるいはFirefly-1を開始し、完全に募集し、児童再発或いは進行性低レベルグリオーマ患者の単一療法として、これらの患者は活性化BRAF変化を有する。最初の患者は2021年5月にFirefly−1の用量治療を受け,2022年5月にARM登録を完了した。Firefly-1試験はまた、(A)主要なキューが登録されているので、合格患者のアクセス範囲を拡大するために、他の2つの研究分枝に拡張された;および(B)6ケ月から25歳までの再発または進行性頭蓋外固形腫瘍を有し、RAF融合を活性化した患者におけるtovorafenib(DAY 101)の初歩的な治療効果を評価する。2022年6月のFirefly−1試験中期分析の予備データと2023年1月の全患者のトップデータを報告した。2023年1月のTOPLINEデータによると、69項目の神経腫瘍学的反応評価(RANO)評価可能な患者のうち、総応答率(OOR)は64%であり、3名の確認された完全応答(CR)および41名の部分応答(PR)(31名の確定診断された部分応答および10名の未確認部分応答(UPR))を含む。著者らは他の19名の安定期(SD)患者を観察し、臨床受益率は91%(CR+PR/UPR+SD)であった。77名の治療を受けた患者の安全性データによると,ボルラフェニの単一療法は全体的に耐性が良好であった。腫瘍の減少や安定はpLGG患者に臨床的意義があると考えられ,多くの患者が承認された治療法が乏しいことから,両者とも有益であると考えられている。2023年第2四半期の医学会議でFirefly−1試験からのより多くのデータを提出し、新薬申請前(NDA)にFDAとともに研究データの重要な部分を審査する予定です, 私たちは2023年上半期に秘密協定を提出する前に会議を開催する予定です。
我々はすでに2022年第2四半期にtovorafenib(DAY 101)のキー3期試験あるいはFirefly-2を開始し、pLGGの第一線の治療として、最初の試験は2022年6月にスタートした。2023年3月,1人目の患者はFirefly−2を服用した。
私たちの第二の候補製品pimasertibは経口的、高選択性のマイトジェン活性化プロテインキナーゼ1と2或いはMEKの小分子阻害剤であり、MEKはMAPK経路中の特徴が明確な重要なシグナルノードである。Pimasertibは850名を超える異なる腫瘍タイプの患者の10個以上の1/2期臨床試験で検討されており,単一療法としても併用標準看護療法としても検討されている。すでに発表された臨床前研究により、Pimasertibは他のMEK阻害剤と比較して高い中枢神経系(CNS)浸透率を有することが示唆された。
我々は、2つのサブ研究を含む開放ラベル、マルチセンター、1 b/2 a期の傘式主試験、または火-1、トレフラフィニ単一療法または併用療法を開始した。サブ研究1はトヴォラフェニ(DAY 101)の第2段階試験であり、12歳以上のRAF変化腫瘍を有する患者の単一治療に用いられ、1人目の患者は2021年11月に服用した。第二の研究はtovorafenib(DAY 101)とpimasertibの1 b/2期連合試験であり、12歳以上の各種MAPK変化を有する固形腫瘍の患者に用いられ、1人目の患者は2022年5月に治療を受けた。臨床前モデルでは,RAFとMEKの同時抑制は協同抗腫瘍活性を引き起こすことが証明されている。この組み合わせは、NRAS変異黒色腫および肺癌、クラスII BRAF変化によって駆動される腫瘍、BRAF野生型融合を有する腫瘍、およびKRAS変化によって駆動される腫瘍を含む、MAPK変化によって駆動される様々な成人固形腫瘍において増強された抗腫瘍活性を示す可能性がある。
著者らの業務開発能力は、腫瘍学薬物開発における著者らの豊富な経験と、研究と患者権益提唱コミュニティにおける深いつながりに加え、特に小児科環境において、すべての年齢の患者のための識別、獲得と治療方法の開発においてリードしていると信じている。私たちはすべての治療分野におけるtovorafenib(DAY 101)とpimasertibの世界的な独占権利を持っているが、いくつかのマイルストーンと特許権使用料を支払わなければならない。
122
次の表に私たちの候補製品ルートをまとめました。

2018年11月の設立以来、私たちは、私たちの候補製品を決定、取得、開発し、私たちのルートを確立するためにほとんどの資源を使用しています。私たちの会社を組織し、そのための人員を配置しています。業務計画、私たちの知的財産権の組み合わせを確立し、第三者と私たちの候補製品の生産計画を確立し、資金を調達し、商業発表の準備をし、これらの業務に一般的かつ行政的な支援を提供しています。私たちは商業販売に許可された製品もなく、製品販売や他の源から何の収入も得ておらず、私たちが運営を開始して以来純損失が発生した。2022年12月31日と2021年12月31日の年度までに、それぞれ1億422億ドルと7280万ドルの純損失を報告します。2022年12月31日と2021年12月31日までの累計赤字はそれぞれ2兆697億ドルと1兆275億ドルだった。予測可能な未来には、私たちが引き続き私たちの候補製品を開発し、規制部門の承認を求め、任意の承認された製品を商業化し、私たちの製品パイプラインを拡大し、私たちの組織に投資することを求め、費用と巨額の損失が大幅に増加すると予想される。また、重大な法律、監査、会計、規制、税務関連、取締役·役員保険、投資家関係、その他の民間会社として招かれていない費用など、上場企業の運営に関する追加コストが引き続き発生する見通しです。
私たちはこれまで、私たちの償還可能な優先株、変換可能手形、普通株を初回公募株とその後の公募株で売却することで、私たちの運営に資金を提供してきました。
2022年12月31日現在、現金と現金等価物および短期投資総額は3兆423億ドル。私たちの現在の運営計画によると、経営陣は2025年までの予想運営に資金を提供する十分な資本資源を持っていると信じています。製品開発に関連する多くのリスクや不確実性により、決して利益を実現しない可能性があり、それまで追加資本を調達し続ける必要があります。私たちが私たちの業務計画を支援するために十分な資金を得ることに成功するという保証はない。もし私たちが必要な時や魅力的な条項で資金を調達できない場合、私たちは候補製品の開発と商業化を大幅に延期、減少または停止したり、新しいライセンス内と買収の追求を縮小または終了しなければならないかもしれない。
123
私たちは所有したり経営したりしないし、現在は製造施設を設立する計画もない。私たちは依存し、引き続き第三者に依存して私たちの候補臨床試験製品を生産し、私たちの任意の候補製品が市場の承認を得た場合、商業製造に使用されることが予想される。私たちが開発を通じて私たちの候補製品を推進することに伴い、私たちは各候補製品のために活性薬物成分或いは原料薬、薬物製品、包装と調合の予備供給者を増加させ、任意の潜在的な供給中断を防止することを探索する。
新冠肺炎が大流行する
進行中の新冠肺炎大流行の全面的な影響は依然として高度に不確定であり,変化する可能性がある。新冠肺炎疫病をめぐって多くの不確定性が存在し、未来の事態の発展は予測できず、私たちの運営と財務状況に実質的な負の影響を与える可能性がある。私たちは、地方政府が息を吹き返し、新たな圧力を発生させるべきであるため、私たちの第三者サービスプロバイダが提供するサービスの変動を経験し、継続することが予想され、すべての新しい圧力は、保護措置の延長、延長、または強化をもたらす可能性がある。製造と供給において、私たちの薬品サプライチェーンは中断されないことが予想され、ロック措置や制限が実施されるかどうか、そしてもしあれば、私たちの施設や業務にどのような影響を与える可能性があります。
私たちの管理チームは引き続きこの絶えず変化する健康危機及び私たちの財務状況、流動性、運営、主要なサプライヤーと従業員チームへの影響を積極的に監視している。
“インフレ低減法案”
総裁·バイ登は2022年8月16日、会社調整後の財務諸表収入に基づく代替最低税率を含む2022年インフレ率低減法案に署名した。私たちの初歩的な評価によると、インフレ低減法案は私たちの所得税規定と現金税に実質的な影響を与えないと考えられる。私たちは私たちの業務に対するそれらの潜在的な影響を評価するために、税金法律法規の変化に注目し続けている。
重要な合意
武田資産協定
2019年12月16日、DOT Treateutics-1,Inc.または当社の子会社DOT-1は、武田製薬株式会社の関連先および関連会社ミレニアム製薬会社と資産購入契約または武田資産協定を締結した。武田資産プロトコルによれば、DOT-1は、TAK-580に関連するいくつかの技術的権利および独自技術(現在、TOVORAFenib(DAY 101))を購入し、原発性脳腫瘍または固形腫瘍脳転移患者の治療に新しい方法を提供する。DOT−1はまた,われわれのこのようなRAF−阻害剤の研究·開発活動のための臨床在庫用品を受け取り,指定された調査者臨床試験プロトコルを得た。武田はViracta治療会社(F/k/a Sunesis PharmPharmticals,Inc.)やViractaの独占ライセンスプロトコルやViractaライセンスプロトコルとDOT-1にも割り当てている.武田はまた、武田資産協定に基づいてDOT−1に指定特許項下のグローバル再許可独占許可と、武田資産協定に基づいて生成された他の特許及び独自技術項下の非独占許可とを付与した。DOT-1はまた、武田資産協定で定義されている帰還許可を武田に付与し、武田資産協定が規定する適用時間範囲内で指定された開発マイルストーンを実現できなければ、この許可を自動的に終了またはDOT−1で終了することができる。この武田に付与された帰還許可証は転換時に終了し、ミレニアム証券取引所協定と関係がある。
124
DOT-1は、資産の売却と譲渡および武田資産協定によるライセンス付与の代償として、100万ドルの現金を前払いし、2019年12月にDOT-1で9,857,143株のAシリーズ償還可能転換優先株を発行した。他の投資家がDOT-1のAシリーズ融資で発行済み株式に支払った価格によると、発行済み株の公正価値は990万ドルと推定される。ミレニアム証券取引所協定の条項によると、武田は転換発効後、2021年5月26日にDOT-1の9,857,143株Aシリーズ償還可能転換可能優先株を我々の普通株6,470,382株に交換した。
武田資産協定の有効期限は、各国のすべての譲渡特許権とすべてのライセンス特許権が満期になった後、国ごとに終了します。武田は我々が初めて商品を商業販売する前に武田資産協定を終了する可能性があり、一定期間連続して指定された時間内にいかなる開発活動も停止すれば、この停止は当事者の同意を得ておらず、規制当局の指導に応えるためでもない。また、武田は我々が破産した場合に武田資産協定を終了することができる。武田が我々の開発停止や破産により武田資産協定を終了すれば、譲渡されたすべての特許、ノウハウ、契約(Viractaライセンス契約を除く)が武田に譲渡され、武田は特許やノウハウに基づいて回復許可を得て、これ等で終了したすべての製品を利用する。
DOT-1は2021年12月31日から当社と合併して当社に組み込まれ、当社は既存の会社であり、武田資産購入協定に従ってDOT-1の義務を負う。
Viracta許可協定
2019年12月16日、DOT-1は、武田資産協定に従って譲渡されたViractaライセンス協定を修正し、再確認した。Viractaライセンスプロトコルによると,DOT−1は特定の特許権やノウハウにより世界的に独占的に許可されており,RAFタンパク質ファミリーに結合した化合物を含む製品を開発,使用,製造,商業化することができる。
DOT-1はViractaに現金200万ドルを前払いしており,このお金は2019年に研究開発費として記録されている.DOT−1は2021年2月にViractaに300万ドルの記念碑的支払いを支払い,2021年4月にマイルストーンを実現した際に研究開発費として記録された。DOT−1はまた,2つの適応の中で許可製品ごとに特定の開発と規制マイルストーンを実現する際に5400万ドルまでの追加マイルストーン支払いを要求されており,第2の適応は特定のマイルストーン事件を実現するために支払われるマイルストーンは第1の適応のマイルストーンよりも低い。また、DOT−1が許可製品に関する優先審査券を取得し、その優先審査券を第三者に販売するか、またはその優先審査券を使用する場合、DOT−1は、そのような販売のいずれかから受信したすべての純対価格または使用済み優先審査券の価値の特定のパーセンテージをViractaに支払う義務がある。DOT−1は,特許製品の1カ国/地域での初商業販売から,特許製品の純売上高の中央値から1桁パーセントまでの分級印税(あれば)の支払いを義務付けている。印税の支払い義務は、国/地域およびライセンス製品のライセンス製品をもとに終了し、1つの国/地域での最初の商業販売から、(I)当該国/地域で当該製品を使用または販売するViractaライセンス特許、共通所有の協力特許または指定特許に関する最後の有効主張が満了するまで続く。(Ii)当該国/地域での最後の法定独占経営権の満了、または(Iii)当該製品の国/地域での初商業販売の10周年である。以上のようにしない限り、他のマイルストーンはない, 達成され、2022年12月31日に満了される。
Viractaライセンス契約の期限は,会社がViractaにその製品の当該国/地域での印税を支払う義務期間が満了した後,ライセンス製品と国/地域で終了する.DOT-1は、指定された通知期間内に任意またはすべてのライセンス製品に関連するViractaライセンスプロトコルを任意に終了する権利がある。
DOT-1は2021年12月31日からわが社と合併して当社に合併し、わが社は生き残った会社であり、Viracta許可協定に従ってDOT-1の義務を負います。
125
メルクKGaA社とライセンス契約を結び、ドイツのダムシュタット
2021年2月10日、DOT Treateutics-2,Inc.または我々の子会社DOT-2は、ドイツのダムシュタットに位置するメルクKGaA製薬会社と、MRKDG許可協定であるライセンス契約を締結した。MRKDGライセンス協定によると、ドイツのダムシュタットのMerck KGaAは独占的なグローバルライセンスを付与し、特定の特許権利と技術ノウハウに基づいて、複数のレベルで再許可を付与する権利があり、PimasertibとMSC 2015103 Bの化合物を含む製品を研究、開発、製造し、商業化することができる。臨床在庫品も受け取り,その研究や開発活動に用いられている。著者らの独占許可はメルクKGaAが付与した非独占許可の制約を受けなければならず、メルクKGaAはドイツの癌研究組織の付属会社であり、ドイツDarmstadtメルクKGaAはいくつかのpimasertibに関連する臨床研究を直接或いは間接的に行う権利を保持する。
MRKDGライセンス協定によると、2029年までに商業的に合理的な努力を使用し、少なくとも2つの指定された主要市場国で少なくとも2つのライセンス製品を開発して商業化する義務がある
MRKDG許可協定により付与された権利や臨床用品を考慮すると,800万ドルが前払いされており,将来的には代替用途がなく,研究活動に用いられているため,研究開発費として記録されている。また,250万ドルの記念碑的支払いを行い,ライセンス契約の性質と,2022年12月31日までの1年間の初臨床試験における患者初投与に関するマイルストーン事件により,研究開発費として記録された。特定の開発、法規、ビジネスマイルストーンの実現状況、および将来のライセンス製品の純売上高の高ビット数印税パーセント(ある場合)に応じて、3億645億ドルの費用を追加的に支払うことも要求される可能性があります。マイルストーンと特許使用料は未来の事件に依存し、マイルストーンと満期支払いが達成されたときに記録される。
MRKDGライセンス契約の期限は,当該国/地域内の当該ライセンス製品についてライセンス側への印税支払い義務期間が満了した後,個々のライセンス製品と国/地域に基づいて満期となり,すべてのライセンス製品とMRKDGライセンス契約下のすべての国/地域に対する全支払義務期間が満了する。
2021年12月31日からDOT−2はわが社と合併し,わが社は生き残った会社であり,MRKDG許可協定によりDOT−2の義務を担っている。
経営成果の構成部分
運営費
研究開発費
研究開発費には、私たちの発見と許可内約束、および私たちの主要候補製品tovorafenib(DAY 101)と私たちの第2の候補製品pimasertibの開発を含む、私たちの研究活動によって生じる外部および内部費用が主に含まれています。
外部費用には、
126
内部費用には、
私たちは発生した金額に基づいて研究と開発費用を計上します。私たちは計画的に外部コストを追跡し、現在私たちのtovorafenib(DAY 101)計画とpimasertib計画の費用を含めている。我々は,プランに特定された間接コストを追跡しないが,これらのコストは複数のプランに配置されているため,単独では分類されない.
研究開発活動は私たちのビジネスモデルの核心だ。我々は,予見可能な将来において,我々の業務戦略を継続し,臨床試験によりtovorafenib(DAY 101)とpimasertibを普及させ,より大規模な臨床試験を行い,我々の研究開発努力を拡大し,より多くの候補製品を決定,獲得,開発するとともに,特に我々のより多くの候補製品が臨床開発と後期臨床開発段階に入るにつれて,我々の研究開発費が大幅に増加することを予想している。
Tovorafenib(DAY 101)、pimasertib、または私たちが開発または買収する可能性のある任意の他の候補製品の将来の臨床試験の持続時間およびコストを合理的に決定することはできず、任意の候補製品の商業化および販売からどの程度の収入を得るかを合理的に決定することはできない。私たちの候補製品の開発と商業化、そして必要な規制とマーケティング承認を得る能力は非常に不確定です。これは新薬の開発に関連する多くのリスクと不確実性が原因であり、その中の多くのリスクと不確定性は私たちの制御範囲内にないためである
127
これらの要因のいずれかの推定値の変化は、私たちの現在および将来の候補製品の開発に関連するコストおよび時間の大きな変化を意味するかもしれない。例えば、FDAや他の規制機関が現在予想されている候補製品の臨床開発を完了するために必要な試験ではなく、臨床試験を要求する場合、あるいは患者登録やその他の理由で、私たちが行っているおよび計画中の臨床試験に重大な遅延が生じた場合、私たちは臨床開発を完成するために多くの追加の財政資源と時間を費やす必要があるかもしれない。
一般と行政費用
一般と行政費用には,主に人事に関する費用,法律や専門サービス費用,保険料,施設に関する費用が含まれる。人事に関連するコストには、執行、財務、会社、業務発展および行政機能を有する者の賃金、ボーナス、福祉、株式ベースの給与、出張費用、およびその他の関連コストが含まれる。法律および専門サービス費用には、知的財産権および会社の事務に関連する法律費用、会計、監査、税務、人的資源、業務発展およびその他のコンサルティングサービスの専門費用、出張費および施設関連費用が含まれる。
私たちは、予測可能な未来に、私たちの一般的かつ行政的費用が大幅に増加することを予想しています。なぜなら、私たちの従業員数が増加し、私たちの候補製品のための研究開発努力の拡大を支持し、私たちのチームと商業市場のための準備に特化した資源を増加させ、私たちの全体的な運営を支援するからです。ナスダック世界ベスト市場(ナスダック)や米国証券取引委員会(米国証券取引委員会)要求に関連するコスト、追加の役員および役員保険コスト、投資家および広報コストなど、上場企業に関連する費用が増加することも予想される。
転換可能な非持株権益を償還するには純損失を占めなければならない
償還可能な転換可能な非持株権益による純損失はわが子会社DOT-1に割り当てられていない純損失の一部である。2021年5月26日、武田は“ミレニアム証券取引所合意”の条項により、武田が保有しているDOT-1株を私たちの普通株に交換した。当時、償還可能な転換可能な非持株権は消滅され、DOT-1は私たちの完全子会社となった。
償還可能な非持株権益株式を交換する−配当とする−
2021年12月31日までに武田が株式を交換したため,追加実収資本約1,000,000,000ドルの清算損失を確認し,転換時に武田に発行した普通株の公正価値と転換日に非持株権益を償還可能な帳簿価値との差額から計算した。普通株株主が純損失と1株当たり純損失を占めるべきであることを計算する際には、全株、非現金交換は配当とみなされる。
128
経営成果
2022年12月31日までと2021年12月31日までの年次比較
次の表は、2022年12月31日と2021年12月31日までの年間経営実績をまとめたものです
|
|
現在までの年度 |
|
|
|
|
|
|
|
|||||||
|
|
2022 |
|
|
2021 |
|
|
$Change |
|
|
変更率 |
|
||||
運営費用: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
研究開発 |
|
$ |
85,618 |
|
|
$ |
43,584 |
|
|
$ |
42,034 |
|
|
|
96.4 |
% |
一般と行政 |
|
|
61,291 |
|
|
|
29,159 |
|
|
|
32,132 |
|
|
|
110.2 |
% |
総運営費 |
|
|
146,909 |
|
|
|
72,743 |
|
|
|
74,166 |
|
|
|
102.0 |
% |
運営損失 |
|
|
(146,909 |
) |
|
|
(72,743 |
) |
|
|
(74,166 |
) |
|
|
102.0 |
% |
利子収入,純額 |
|
|
4,746 |
|
|
|
4 |
|
|
|
4,742 |
|
|
* |
|
|
その他の費用、純額 |
|
|
(18 |
) |
|
|
(15 |
) |
|
|
(3 |
) |
|
|
20.0 |
% |
純損失 |
|
|
(142,181 |
) |
|
|
(72,754 |
) |
|
|
(69,427 |
) |
|
|
95.4 |
% |
転換社債の償還は純損失を占めなければならない |
|
|
— |
|
|
|
(2,109 |
) |
|
|
2,109 |
|
|
* |
|
|
償還可能な非制御的権益を交換する |
|
|
— |
|
|
|
(99,994 |
) |
|
|
99,994 |
|
|
* |
|
|
普通株主/メンバーは純損失を占めるべきである |
|
$ |
(142,181 |
) |
|
$ |
(170,639 |
) |
|
$ |
28,458 |
|
|
|
-16.7 |
% |
研究開発費
研究開発費は4200万ドル増加し、2021年12月31日までの年度の4360万ドルから2022年12月31日までの年度の8560万ドルに増加した。2022年12月31日までの年間で、第三者費用は2021年12月31日と比較して2900万ドル増加したが、これは主に臨床試験、製造、その他の製品開発費用の増加、人員関連費用は追加従業員と株式ベースの報酬により1810万ドル増加し、前払いとマイルストーン費用はマイルストーン支払いのスケジュールにより850万ドル減少したためである。
次の表は、2022年12月31日と2021年12月31日までの年間私たちの外部·内部研究開発費をまとめています
|
|
現在までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
|
|
(単位:千) |
|
|||||
外部コスト: |
|
|
|
|
|
|
||
第三者CRO,CMO等第三者臨床試験費用(1) |
|
$ |
50,175 |
|
|
$ |
21,181 |
|
MRKDG許可協定に関するマイルストーン支払い |
|
|
2,500 |
|
|
|
— |
|
MRKDG許可協定に関する前金 |
|
|
— |
|
|
|
8,000 |
|
Viractaライセンス契約に関するマイルストーン支払い |
|
|
— |
|
|
|
3,000 |
|
実験室材料や用品を含む他の研究と開発費用 |
|
|
3,598 |
|
|
|
243 |
|
内部コスト: |
|
|
|
|
|
|
||
従業員関連費用 |
|
|
29,345 |
|
|
|
11,160 |
|
研究開発費総額 |
|
$ |
85,618 |
|
|
$ |
43,584 |
|
129
一般と行政費用
一般·行政費は3210万ドル増加し、2021年12月31日までの年度の2920万ドルから2022年12月31日までの年度の6130万ドルに増加した。一般·行政費の増加は、主に従業員数の増加による1,710万ドルの従業員報酬コスト、および商業能力の増強と業務運営の持続的な拡大に推進される1,060万ドルの専門サービス、法律および保険費用、440万ドルの施設コストおよびその他の費用である。
償還可能な非持株権益株式を交換する−配当とする−
2021年5月、DOT-1における武田の償還可能な非持株権益株式を初日のバイオ製薬会社の普通株に交換し、この取引を配当金として計上した。そこで,転換時に武田に発行された普通株の公正価値と転換日に非持株権益を償還可能な帳簿価値との差額から計算した追加実収資本の100,000,000ドルの非現金清算損失を確認した。これは総合経営報告書の純損失の中で配当金として開示され、2021年12月31日までの年度の1株当たり純損失計算に含まれる。2022年12月31日までの年度中には、同等の損失は確認されていない。
流動性と資本資源
流動資金源
2022年6月、私たちはPiper Sandler&Co.とJones Trading Institution Services LLCと販売エージェントとして株式分配協定を締結し、私たちの普通株の発行と販売に関連し、総発行価格は時々1.5億ドル、すなわち2022年のATMに達する。証券法によると、2022年にATM機がこれらの株を発行·売却する行為を市場で発行するとみなされている。2022年12月31日現在、2022年のATM下のいかなる普通株も販売していません。
2022年6月、私たちは後続発行を完了し、引受割引、手数料、発行コストを差し引いた後、1株15.00ドルで11,500,000株の普通株(引受業者が追加1,500,000株の普通株を購入する選択権を含む)を発行·売却し、純収益は約1.616億ドルだった。
2021年6月、私たちは初公募株を完成させ、2021年5月に引受業者が追加普通株を購入する選択権を全面的に行使して発行した150万株の普通株を含む合計1150万株の普通株を1株16.00ドルで一般に売却した。引受割引、手数料、発行コスト1700万ドルを差し引いた後、IPOから得られた純収益総額は1.67億ドルだった。初めて公募する前に、償還可能な転換可能な優先株と転換可能な手形を売ることで、私たちの運営に資金を提供します。私たちはこれまでAシリーズとBシリーズの償還可能な優先株と転換可能な手形を売却して発行することで約1.92億ドルの毛収入を集めた。
2022年12月31日までの累計赤字は2兆697億ドル、現金、現金等価物、短期投資は3兆423億ドルだった。私たちは私たちの現金、現金等価物、そして短期投資が今後12ヶ月と2025年の現金需要を満たすのに十分になると信じている。
私たちの現金の主な用途は運営費用に資金を提供することであり、その中には主に研究開発支出が含まれており、その中には私たちの免許、臨床試験と実験室コスト、及び私たちの給料とコンサルティング費用を含むより小さい一般と行政支出が含まれている。運営費に資金を提供するための現金は、これらの費用を支払う時間の影響を受けており、これは私たちの未払い売掛金や売掛金の変化に反映されている。私たちの物質的現金需要には以下の契約義務と他の義務が含まれている。
賃貸借証書
私たちはオフィス空間に対してレンタル経営義務があります。同社の固定賃貸支払い義務は2022年12月31日現在で約90万ドルであり、そのうち約50万ドルは12カ月以内に支払うべきである。
130
契約研究機関と契約製造機関
著者らは正常な業務過程においてCRO、CMOと他の第三者サプライヤーと臨床試験、製造、テストとその他の研究と開発活動の契約を締結した。これらの契約は一般的に通知後に終了することに規定されていますが、一部の費用はプロジェクト承認後にキャンセルできないため、サプライヤーの例外があります。2022年12月31日現在、中止やキャンセル費用に関する課税額はありません。これらは発生しにくいためです。
許可協定
私たちは具体的な活動の満足状況に応じてマイルストーン費用を支払うことを要求する許可協定を締結した。2022年12月31日までの1年間に,pimasertibを含む製品の第1回臨床試験での初回投与に関する250万ドルのマイルストーン支払いを行い,2021年12月31日までの年間にViractaライセンス契約に関する300万ドルを支払った。私たちはこれらの合意に基づいて開発された製品の販売に印税を支払うことを要求された。2022年12月31日まで、私たちのすべての製品は開発中で、このような印税を支払う必要はありません。2022年12月31日まで、このような支払いの金額、時間、可能性はまだ不明なので、支払い義務は何もありません。
キャッシュフロー
次の表は、私たちが列挙した期間の現金源と用途をまとめています
|
|
現在までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
経営活動のための現金純額 |
|
$ |
(109,874 |
) |
|
$ |
(48,539 |
) |
投資活動のための現金純額 |
|
|
(255,074 |
) |
|
|
(8,000 |
) |
融資活動が提供する現金純額 |
|
|
165,901 |
|
|
|
297,120 |
|
現金および現金等価物の純増加 |
|
$ |
(199,047 |
) |
|
$ |
240,581 |
|
経営活動
2022年12月31日現在、経営活動で使用されている現金純額は1.099億ドルで、1.422億ドルの純損失、約660万ドルの純営業資産と負債変動、2570万ドルの非現金費用が含まれており、主に2720万ドルの株式ベースの給与支出が含まれている。業務資産と負債の変化は、主に課税費用やその他の流動負債の920万ドルの増加と関係がある。売掛金は150万ドル減少し、経営リース負債は30万ドル増加し、預金やその他の長期資産は30万ドル増加し、前払い費用とその他の流動資産は50万ドル増加し、この減少額を部分的に相殺した。
2021年12月31日までの年度の経営活動に用いられた現金純額は4,850万ドルであり,このうち純損失は7,280万ドルであったが,純営業資産および負債270万ドルおよび非現金費用2,150万ドル分で相殺された。経営資産と負債の変化は、主に前払い費用および他の流動資産の370万ドルの増加によるものであり、その中には300万ドルのビラクタ許可証マイルストーン前払いが含まれているが、計算されなければならない費用は他の流動負債の510万ドルの増加と売掛金の150万ドルの増加によって部分的に相殺される。私たちの非現金費用には主に1330万ドルの株式ベースの給与支出と20万ドルの非現金レンタル支出が含まれています。MRKDG許可協定に関連した800万ドルも支払い,この費用は研究開発費として確認され,我々の総合キャッシュフロー表に投資活動を計上した。
投資活動
2022年12月31日までの1年間で、投資活動のための現金純額は2.551億ドルで、短期投資の購入および財産·設備支出40万ドルに起因することができる。これは10万ドルの短期投資満期収益部分によって相殺される。
2021年12月31日までの投資活動用現金純額は800万ドルであり,MRKDG許可協定による支払いによるものとすべきである。
131
融資活動
2022年12月31日までの1年間、融資活動が提供した現金純額は1兆659億ドルで、主に我々の後続の普通株発行に関連する普通株発行純収益によるものである。また,430万ドルの現金純額は融資活動から来ており,これらの活動は株式オプションを行使する際に普通株を発行する収益と,我々の2021年の従業員株式購入計画による購入に関係している.
2021年12月31日までの1年間に、融資活動が提供した現金純額は2.971億ドルで、うち1.67億ドルはIPO関連の普通株の純収益に関連し、1.298億ドルはBシリーズの償還可能な転換可能優先株の売却·発行の純収益に関連している。
資金需要
設立以来、私たちは重大な運営損失を受けた。予見可能な未来には,我々が行っている活動に関する費用は引き続き大幅に増加し,運営損失も増加していくと予想される。
私たちの既存の現金、現金等価物、短期投資は、2025年までの運営費用と資本支出需要に資金を提供できると信じている。私たちのこの推定は、不正確であることが証明される可能性があるという仮定に基づいており、私たちは現在予想されているよりも早く私たちが利用できる資本資源を使用することができる。
予想された支出の結果として、私たちは私たちの持続的な運営に関連した多くの追加資金を得る必要があるだろう。これまで、製品販売から相当な収入を得ることができなかった場合、株式発行、債務融資、協力、戦略連合とマーケティング、流通、または許可手配の組み合わせで現金需要を満たす予定です。受け入れ可能な条件で、私たちは十分な追加資金を得ることができないかもしれないし、全くないかもしれない。もし私たちが必要な時や魅力的な条件下で資金を調達できない場合、私たちは私たちの研究、製品開発計画、あるいは未来の商業化努力を延期、制限、減少または終了することを要求されるかもしれません。あるいは私たちが自分で開発し、マーケティングすることをより望んでいた候補製品の権利を与えることができます。
私たちが株式または転換可能な債務証券を売却することによって追加資本を調達する場合、あなたの所有権資本は希釈され、これらの証券の条項は清算または他の特典を含む可能性があり、普通株主としての権利に悪影響を及ぼす可能性がある。債務融資および優先株融資に関与する可能性のある協定は、追加債務を招く、買収または資本支出を行う、または配当を宣言するなど、私たちが具体的な行動をとる能力を制限または制限する契約を含む。
もし私たちが第三者との協力、戦略連合またはマーケティング、流通または許可手配を通じてより多くの資金を調達するならば、私たちは私たちの技術、将来の収入源、研究計画、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になる可能性のある条項でライセンスを付与しなければならないかもしれない。
我々のより多くの資金を調達する能力は、インフレと絶えず変化する金利、および米国と世界の信用と金融市場の中断と変動を含む潜在的なグローバル経済状況の悪化の悪影響を受ける可能性があり、原因は持続的な新冠肺炎の疫病またはその他の原因である。製品開発に関する多くのリスクや不確実性のため、費用の増加時間や金額を予測することもできませんし、利益が出るかどうかや経営活動から正のキャッシュフローが生じるかどうかを保証することもできません。
表外手配
本年報10-K表の他の部分に掲載された総合財務諸表付記6に記載された賠償協定以外に、提出期間内に、著者らはなく、現在アメリカ証券取引委員会規則と条例で定義されたいかなる表外手配もない。
132
重要な会計政策と試算の使用
私たちの経営陣は財務状況と経営結果の討論と分析は私たちの連結財務諸表に基づいています。これらの連結財務諸表はアメリカ公認会計原則あるいはGAAPに基づいて作成されています。公認会計基準に従って連結財務諸表を作成することは、連結財務諸表の日付の資産、負債及び又は有資産及び負債の報告金額及び報告期間内に報告された費用金額に影響を与えるために、管理層に推定及び仮定を要求する。我々は,歴史的経験,既知の傾向や事件,および当時の状況で合理的と考えられる様々な他の要因に基づいて推定し,これらの要因の結果は資産や負債の帳簿価値の判断の基礎を構成しているが,これらの資産や負債の帳簿価値は他の源から容易に見られるものではない.私たちは持続的な基礎の上で私たちの推定と仮定を評価する。異なる仮定や条件の下で、私たちの実際の結果はこれらの推定とは異なる可能性がある。
我々の主な会計政策は、本年報第8項内の総合財務諸表付記により詳細に記載されているが、以下の会計政策は、我々の総合財務諸表の作成に使用する判断と推定に最も重要であると信じている。
研究と開発費用を計算すべきである
臨床前と臨床研究および契約製造活動を含む第三者サービスプロバイダによる研究·開発活動の推定コストの計算すべき負債を記録した。我々は,提供されているが伝票が発行されていない見積りサービス金額に基づいて,研究と開発活動の見積りコストを記録する.我々は,完了した作業見積り数や,サービスプロトコルに基づいて我々の第三者サービスプロバイダとの合意などに基づいてこれらの費用を計算する.
われわれはCMOとCROとの契約に基づいて臨床試験に関する費用を支払い,これらの契約はわれわれの臨床試験を支援·管理している。これらの契約の財務条項は交渉する必要があり、これらの条項は契約によって異なり、材料やサービスを提供する期限と一致しない支払いを招く可能性がある。一般に、これらのプロトコルは、固定料金、単価、または時間および材料で実行される動作範囲を規定する。われわれは,各試験が完了した推定作業量からCROによる臨床試験活動のコストを計算した。臨床試験費用については,計算すべき費用を見積もる際に用いる重要な要素は,患者数の登録,患者1人あたりの活動,活発な臨床場所数,および患者登録試験参加時間である。我々は,内部審査,CROとの通信や契約条項の審査により,患者の登録レベルや関連活動を可能な限りモニタリングした。私たちの推定はその時得られる最高の情報に基づいている。漢方医のサービス料を受け取る時、サービスを提供する時間帯と、各期間の仕事量を見積もります。これらの用品の所有権が我々の手元に移転した場合,代替用途のない臨床用品在庫は研究開発費に計上される。
発生し始めたコストを決定しない場合、または提供されるサービスのレベルまたはこれらのサービスのコストを過小評価または過大評価した場合、実際の費用は私たちの推定とは異なる可能性がある。今まで、私たちは計算すべき費用と実際の費用の間のどんな大きな違いにも遭遇しなかった。しかし,推定の性質から,われわれの臨床試験や他の研究活動の状態や進行に関するより多くの情報が分かった場合には,将来的に我々の推定を変えることは保証されない。
133
株式ベースの報酬
初公募の前に、オプション定価モデルを用いて、すべての株式に基づく奨励、激励株、制限株奨励の推定公正価値に基づいて、付与日に株式による報酬支出を確認した。オプション定価モデルは主観的仮定を入力する必要があり、対象普通株の公正価値、奨励の期待期限、期待変動率、無リスク金利と配当率を含む。普通株式の公正価値を決定する際に、企業価値を推定するための方法は、発行された個人持株会社株式証券の推定値を補償するための米国公認会計士協会会計および推定ガイドラインと一致する方法、方法、および仮定を用いて行われる。参加敷居の金額は取締役会が付与された時に決定される。この間に付与された賠償金の期待期限は,クリーニング終了イベントまでの期待時間に基づいて決定される.付与時に発効した米国債収益率に基づく無リスク金利を採用しており、この金利は奨励の有効期限と一致している。予想変動率は、私たちの業務が予想される流動性時間と一致する業界内の同業者団体に基づいている。配当収益率はゼロに設定されており、対象証券がなくても配当金が支払われないことが予想される
IPO完了後、我々はBlack-Scholes推定モデルを用いて付与されたオプションの公正価値を推定し、内在価値を用いて制限株式報酬の公正価値を推定し、制限株式単位が付与された日に我々の普通株の公正価値を推定する。
ブラック·スコアーズオプション定価モデルは、株式オプション報酬の公正価値を推定するために使用され、以下の仮定を使用することが要求される
私たちは没収が発生した同じ時期に費用を減らすことで没収を確認します。業績条件のある報酬が条件を満たす可能性がある場合には、株式ベースの報酬支出を確認し、報酬が付与される。
134
これらの推定値に基づく仮定は、固有の不確実性と経営陣判断の応用に関連する管理職の最適な推定を表している。したがって、もし私たちが全く違う仮定や推定を使用すれば、私たちの普通株の公正価値と私たちの株式ベースの報酬支出は大きく異なるかもしれない。
新会計公告
最近発表·採択された会計声明の概要については、本年度報告書の他の場所のForm 10-Kに含まれる我々の総合財務諸表付記2を参照されたい。
新興成長型会社の地位
新興成長型会社、またはEGCとして、2012年にJumpStart Our Business Startups ActまたはJOBS Actによると、私たちは延長された過渡期間を利用して新しい会計基準や改訂された会計基準を遵守することができる。この規定は、これらの基準が民間会社に適用されるまで、企業会計基準委員会が特定の会計基準の採用を延期することを可能にする。私たちは、私たち(I)がもはや新興成長型企業または(Ii)が“雇用法案”に規定されている延長移行期間から撤退することを明確かつ撤回できないまで、この延長された移行期間を使用して、新しい会計基準または改正会計基準を遵守することを選択することを選択した。2023年12月31日から、新興成長型企業の地位を失い、各種報告要件の免除を利用することができなくなり、2024年に提出される2023年12月31日までの会計年度報告から開始します。
イットM 7 A。市場リスクに関する定量的で定性的な開示。
取引法第12 b-2条の定義によると、我々は小さな報告会社であり、本プロジェクトに要求される情報を提供する必要はない。
イットM 8.財務諸表および補足データ。
本プロジェクトに必要な資料は連結財務諸表とその付記に掲載し,本年度報告表格10−KのF−1ページから開始した。
情報技術EM 9.会計及び財務開示面の変更と会計士との相違。
ない。
135
ITEM 9 Aです。制御とプログラムです
情報開示制御とプログラムの評価
2022年12月31日現在、経営陣は、改正された1934年の証券取引法規則13 a-15(E)および15 d-15(E)または取引法の規定に適合している我々の最高経営責任者および財務会計官の参加の下、開示制御および手続の有効性を評価している。我々の開示制御および手続きは、取引所法案に基づいて提出または提出された報告書において開示を要求する情報が、米国証券取引委員会規則および表で指定された期間内に記録、処理、集約および報告され、これらの情報が、必要な開示に関する決定をタイムリーに行うために、最高経営者および最高財務会計官を含む私たちの管理層に蓄積されて伝達されることを目的としている。どのような制御やプログラムも,設計や操作がどんなに良好であっても,予想される制御目標を実現するために合理的な保証を提供することしかできないが,管理層は,可能な制御とプログラムのコスト-利得関係を評価する際にその判断を運用しなければならない.この評価に基づき、我々の最高経営責任者と財務·会計官は、2022年12月31日現在、我々の開示制御プログラムは、合理的な保証レベルで有効であると結論した。
財務報告の内部統制に関する経営陣の報告
私たちの経営陣は財務報告書の十分な内部統制を確立して維持する責任がある。取引所法案が公布された第13 a-15(F)及び15 d-15(F)条によれば、財務報告の内部統制は、米国公認会計基準に基づいて財務報告の信頼性及び外部目的の財務諸表の作成のための合理的な保証を提供するために、我々のCEO及び最高財務官が設計又はその監督の下で、我々の取締役会、管理層及び他の人員によって実施されるプログラムとして定義される。私たちの財務報告書に対する内部統制は以下の政策と手続きを含む
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。したがって,有効と判定されたシステムであっても,財務諸表の作成や列報に合理的な保証を提供することしかできない.将来的にどのような有効性評価を行うかの予測には,条件の変化により制御が不十分になる可能性がある,あるいは政策やプログラムを遵守する程度が悪化する可能性があるというリスクがある.また、開示制御およびプログラムを設計する際には、我々の管理層は、その判断を用いて、可能な開示制御とプログラムの費用対効果関係を評価しなければならない。任意の開示制御およびプログラムの設計もまた、将来のイベント可能性のいくつかの仮定に部分的に基づいており、任意の設計がすべての潜在的な将来の条件でその目標を成功的に達成することを保証することはできない。
我々の経営陣は、最高経営責任者と財務責任者の参加の下、2022年12月31日までの財務報告書の内部統制に対する有効性を評価した。この評価を行う際には,経営陣はトレデビル委員会後援組織委員会(COSO)がその内部統制−総合枠組み(2013年)で提案した基準を用いた。我々の評価によると、我々の経営陣は、2022年12月31日現在、財務報告に対する内部統制がこれらの基準に基づいて有効であると結論している。
136
この10-K表年次報告書には、財務報告の内部統制に関する当社の独立公認会計士事務所の認証報告は含まれていません。私たちがまだ“1933年証券法”(改正)第2(A)節または“証券法”(2012年“私たちの企業創業法案”改正を開始)で定義されている“新興成長型会社”である限り、この免除を利用して、独立公認会計士事務所に要求された財務報告の内部統制の有効性の証明を守らないつもりです。
財務報告の内部統制の変化
最近の四半期内に、財務報告の内部統制に大きな影響を与えていないか、または合理的に財務報告の内部統制に大きな影響を与える可能性のある変化が発生しています。
情報技術EM 9 B。他の情報。
ない。
情報技術EM 9 Cです。検査を妨害する外国司法管轄区域を開示する。
ない。
137
第三部
情報技術EM 10.取締役、行政者、会社管理。
本プロジェクトに要求される情報は、ここに組み込まれ、本10-K表年次報告書に含まれる財政年度終了後120日以内に米国証券取引委員会に提出される2023年株主総会の最終委託書における“我々の役員情報”、“取締役選挙”、“会社管理”、“特定の利益所有者及び管理層の保証所有権”に関する部分を参照する。
本条項が要求する取引所法案第16(A)条の遵守に関する情報については,吾等は,2023年株主総会に関する委託書に第16(A)条の報告書(ある場合)を開示し,当該等の開示(ある場合)が本明細書に組み込まれることを参考にする。
私たちは、私たちの最高経営責任者、最高財務官、最高会計官、財務総監、または同様の機能を実行する人を含む、私たちのすべての上級管理者、役員、および従業員に適用されるビジネス行動および道徳的基準を採択しました。この基準は、私たちのサイトに発表されます。私たちの商業行為と道徳基準は、S-Kルール406(B)項で定義された“道徳的基準”である。私たちは私たちの商業行為と道徳基準条項の改正または免除に関する任意の法律要求の情報を私たちのウェブサイトで開示するつもりだ。当社サイトに掲載されているか、当社サイトから取得可能な資料は、本10-K表年次報告の一部ではありません。
情報技術EM 11.役員報酬。
本プロジェクトに要求される情報は、本Form 10−K年度報告書に含まれる財政年度終了後120日以内に米国証券取引委員会に提出される2023年株主年次総会に関する最終依頼書の“役員報酬”、“役員選挙”、“コーポレートガバナンス”の一部を参照して本明細書に組み込まれる。
情報技術EM 12.特定の実益所有者の保証所有権および管理層および関連する株主事項。
本プロジェクトに要求される情報は、本Form 10−K年度報告書がカバーする財政年度終了後120日以内に米国証券取引委員会に提出される2023年株主年次総会に関する我々の最終委託書の“特定の利益所有者及び経営陣の担保所有権”および“持分補償計画情報”の一部を参照して統合される。
情報技術EM 13.何らかの関係や関連取引,および取締役の独立性.
本条項によって要求される情報は、ここに組み込まれ、本Form 10-K年次報告書に含まれる財政年度終了後120日以内に米国証券取引委員会に提出される2023年株主総会の最終委託書における“いくつかの関係および関連者取引”および“会社ガバナンス”に関する部分を参照する。
イットM 14.主な会計費用およびサービス。
本条項に要求される情報はここに組み込まれ、本10-K表年次報告に含まれる財政年度終了後120日以内に米国証券取引委員会に提出される2023年株主総会の最終委託書における“独立公認会計士事務所の承認”と題する章を参考にした。
138
第4部
情報技術EM 15.証拠物、財務諸表明細書
(1)連結財務諸表:
第15項(A)項に要求される連結財務諸表は、本年度報告書10-K表第8項“連結財務諸表及び補足データ”の一部として提出される
(2)連結財務諸表の付表
15(A)項に要求される財務諸表明細書は、適用されない、不要、または要求された資料が、本年度報告シートの第10-K第8項に記載されている連結財務諸表または付記に含まれているので省略される。
(3)展示品。
展示品 |
|
説明する |
|
表 |
|
書類番号. |
|
展示品 |
|
アーカイブ済み/搭載済み |
|
|
|
|
|
|
|
|
|
|
|
3.1 |
|
再記載された会社登録証明書は、2021年6月1日です。 |
|
10-Q |
|
001-40431 |
|
2021年8月10日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
3.2 |
|
付例を改訂して再制定し,日付は2023年2月17日である。 |
|
8-K |
|
001-40431 |
|
2023年2月23日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
3.3 |
|
所有権と合併証明書、日付は2021年12月23日です |
|
10-K |
|
001-40431 |
|
March 7, 2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
4.1 |
|
普通株の書式 |
|
S-1/A |
|
333-255754 |
|
May 24, 2021 |
|
|
|
|
|
|
|
|
|
|
|
|
|
4.2 |
|
2021年2月1日日の投資家権利協定が改訂され、再署名され、初日にバイオ製薬ホールディングスLLCとその特定の株主の間で合意された。 |
|
S-1 |
|
333-255754 |
|
May 4, 2021 |
|
|
|
|
|
|
|
|
|
|
|
|
|
4.3 |
|
登録者の証券説明 |
|
10-K |
|
001-40431 |
|
March 7, 2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.1^ |
|
役員及び上級者と締結した弁済協定の書式 |
|
S-1 |
|
333-255754 |
|
May 4, 2021 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.2^ |
|
支配権および離職契約のフォーマットを変更する |
|
10-K |
|
001-40431 |
|
March 7, 2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.3^ |
|
2021年株式インセンティブ計画と奨励協定の形式 |
|
S-1 |
|
333-255754 |
|
May 4, 2021 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.4^ |
|
2021年従業員株購入計画とその奨励協定のフォーマット |
|
S-1 |
|
333-255754 |
|
May 4, 2021 |
|
|
|
|
|
|
|
|
|
|
|
|
|
139
10.5 |
|
オフィスレンタルは,2022年4月1日で,カリフォルニア航空機技術出版社と初日バイオ製薬会社の間でレンタルされている。 |
|
10-Q |
|
001-40431 |
|
2022年8月4日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.6 |
|
DOT治療−1社とミレニアム製薬会社との間の資産譲渡と許可協定は,2019年12月16日から発効した。 |
|
S-1 |
|
333-255754 |
|
May 4, 2021 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.7 |
|
ロイヤル空軍の許可協定は、2019年12月16日から発効し、Sunesis製薬会社とDOT治療-1社が署名した。 |
|
S-1 |
|
333-255754 |
|
May 4, 2021 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.8 |
|
ライセンス契約は,2021年2月10日にドイツ·ダムシュタット·メルクKGaA社と初日バイオ製薬会社が署名した。 |
|
S-1 |
|
333-255754 |
|
May 4, 2021 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.9 |
|
株式交換協定は,期日は2021年5月4日であり,初日にバイオ製薬ホールディングスとミレニアル製薬会社が署名した。 |
|
S-1 |
|
333-255754 |
|
May 4, 2021 |
|
|
|
|
|
|
|
|
|
|
|
|
|
21.1 |
|
登録者の子会社 |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
23.1 |
|
独立公認会計士事務所が同意します。 |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
31.1 |
|
2002年のサバンズ·オキシリー法第302節で可決された1934年の証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて発行された首席執行幹事証明書。 |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
31.2 |
|
2002年サバンズ−オキシリー法第302節で可決された1934年証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて首席財務幹事が認証された。 |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
32.1** |
|
2002年の“サバンズ-オックススリー法案”906節で採択された“米国法典”第18編1350条による最高経営責任者と最高財務官の認証。 |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
101.INS |
|
XBRLインスタンスドキュメントを連結する |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
101.書院 |
|
イントラネットXBRL分類拡張アーキテクチャ文書 |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
140
101.カール |
|
インラインXBRL分類拡張はリンクベース文書を計算する. |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
101.def |
|
XBRLソート拡張を連結してLinkbase文書を定義する. |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
101.介護会 |
|
XBRL分類拡張ラベルLinkbase文書を連結する. |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
101.Pre
|
|
XBRL分類拡張プレゼンテーションLinkbaseドキュメントを内部接続する. |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
104 |
|
表紙対話データファイル(イントラネットXBRL文書に埋め込まれ、添付ファイル101に含まれる) |
|
|
|
|
|
|
|
X |
登録者が見落としたS−K条例第601(B)(10)項に許可された部分展示品。
^管理契約または補償計画を示します。
**本証明書は、取引法第18条の規定に基づいて提出されていないものとみなされ、証券法または取引法に規定されているいかなる文書にも引用されているものとみなされてはなりません。
情報技術EM 16.表格10-Kの概要
ない。
141
サイン
改正された1934年証券取引法第13又は15(D)節の要求に基づいて、登録者は、正式に許可された次の署名者がその代表を代表して本報告書に署名することを正式に手配した.
|
|
初日生物製薬会社です。 |
|
|
|
|
|
日付:2023年3月6日 |
|
差出人: |
ジェレミー·ベンデル博士商工管理修士 |
|
|
|
ジェレミー·ベンデル博士商工管理修士 |
|
|
|
CEO兼社長 (首席行政主任) |
|
|
初日生物製薬会社です。 |
|
|
|
|
|
日付:2023年3月6日 |
|
差出人: |
チャールズ·N·ヨーク2世M.B.A. |
|
|
|
チャールズ·ヨーク2世M.B.A. |
|
|
|
最高経営責任者と首席財務官 (首席財務会計官) |
142
授権依頼書
このような陳述を通じてすべての人を認識し、以下の署名のすべての人はここでJeremy Bender,Ph.D.,M.B.A.とCharles N.York II,M.B.A.を構成して任命し、彼らはそれぞれ完全な代替と再代替の権力を持ち、他の人なしに行動する権利があり、彼または彼女の真実と合法的な事実代理人と代理人として、彼または彼女の名義、場所、代替、そしてすべての人の名義と代表で各人を代表し、単独で以下に述べる身分で行動する。本年度報告の任意およびすべての修正をForm 10−Kの形態で提出し、本年度報告およびそのすべての証拠物および他の関連文書を米国証券取引委員会に提出し、上記の代理弁護士および代理人、ならびに彼ら一人一人に各行為および事柄の完全な権限および許可を行い、すべての上述の代理弁護士および代理人または彼らまたはそれらの代替者または代替者が合法的にまたはそれに至るすべてのことを和として行うことができることを承認および確認する。
本報告書は、改正された1934年の証券取引法の要求に基づき、以下の登録者によって登録者として指定日に署名された。
名前.名前 |
|
タイトル |
|
日取り |
|
|
|
|
|
ジェレミー·ベンデル博士商工管理修士 |
|
CEO兼社長 (首席行政主任) |
|
March 6, 2023 |
ジェレミー·ベンデル博士商工管理修士 |
|
|
|
|
|
|
|
|
|
チャールズ·N·ヨーク2世M.B.A. |
|
最高経営責任者と首席財務官 (首席財務会計官) |
|
March 6, 2023 |
チャールズ·ヨーク2世M.B.A. |
|
|
|
|
|
|
|
|
|
ゲイリー·ニコルソンM.B.A. |
|
椅子と役員 |
|
March 6, 2023 |
ゲイリー·ニコルソン |
|
|
|
|
|
|
|
|
|
ジュリー·グラントM.Phil M.B.A. |
|
役員.取締役 |
|
March 6, 2023 |
ジュリー·グラントM.Phil M.B.A. |
|
|
|
|
|
|
|
|
|
ダン·ベッカー医学博士博士 |
|
役員.取締役 |
|
March 6, 2023 |
ダン·ベッカー医学博士博士 |
|
|
|
|
|
|
|
|
|
/s/Scott Garland |
|
役員.取締役 |
|
March 6, 2023 |
スコット·ガランド |
|
|
|
|
|
|
|
|
|
/s/マイケル·グラストン |
|
役員.取締役 |
|
March 6, 2023 |
マイケル·グラストン |
|
|
|
|
|
|
|
|
|
/s/ナタリー·ホルス |
|
役員.取締役 |
|
March 6, 2023 |
ナタリー·ホルス |
|
|
|
|
|
|
|
|
|
ジョンA.ジョセフ博士M.B.A. |
|
役員.取締役 |
|
March 6, 2023 |
ジョン·A·ジョシー博士商工管理修士 |
|
|
|
|
|
|
|
|
|
タイトル/Saira Ramasastry,M.S.,M.Phil. |
|
役員.取締役 |
|
March 6, 2023 |
Saira Ramasastry M.S.,M.Phil. |
|
|
|
|
143
連結財務諸表索引
独立公認会計士事務所レポート(PCAOB ID: |
F-2 |
合併貸借対照表 |
F-3 |
連結業務報告書 |
F-4 |
合併全面損失表 |
F-5 |
償還可能な転換可能優先株、償還可能な非持株権益と株主権益/メンバー(損失)連結報告書 |
F-6 |
統合現金フロー表 |
F-7 |
連結財務諸表付記 |
F-8 |
F-1
独立公認会計士事務所報告
以下のメンバーと取締役会に
初日生物製薬会社です。
財務諸表のいくつかの見方
我々は,2022年12月31日と2021年12月31日までの初日バイオ製薬会社(当社)の合併貸借対照表,2022年12月31日までの3年度の関連総合経営表,全面赤字,償還可能な転換可能優先株,償還可能な非持株権益と株主権益/メンバー(赤字)と現金流量,および関連付記(総称して“総合財務諸表”と呼ぶ)を監査した。総合財務諸表は、すべての重要な面で、2022年12月31日、2022年12月31日、2021年12月31日までの会社の財務状況、および2022年12月31日までの3年度の経営結果とキャッシュフローを公平に反映しており、米国公認会計原則に適合していると考えられる。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査することを求められておらず、私たちも招聘されて監査を行っていません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性について意見を述べるためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
/s/
2021年以来、私たちは会社の監査役を務めてきた。
March 6, 2023
F-2
初日生物製薬会社です。
円錐体合併貸借対照表
(単位は千で、シェアは含まれていない)
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
短期投資 |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
流動資産総額 |
|
|
|
|
|
|
||
財産と設備、純額 |
|
|
|
|
|
|
||
経営的リース使用権資産 |
|
|
|
|
|
|
||
預金とその他の長期資産 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
負債と株主権益 |
|
|
|
|
|
|
||
流動負債: |
|
|
|
|
|
|
||
売掛金 |
|
$ |
|
|
$ |
|
||
費用とその他の流動負債を計算しなければならない |
|
|
|
|
|
|
||
賃貸負債の当期部分を経営する |
|
|
|
|
|
|
||
流動負債総額 |
|
|
|
|
|
|
||
賃貸負債の長期部分 |
|
|
|
|
|
|
||
総負債 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
株主権益: |
|
|
|
|
|
|
||
普通株、$ |
|
|
|
|
|
|
||
実収資本を追加する |
|
|
|
|
|
|
||
その他の総合損失を累計する |
|
|
( |
) |
|
|
|
|
赤字を累計する |
|
|
( |
) |
|
|
( |
) |
株主権益総額 |
|
|
|
|
|
|
||
総負債と株主権益 |
|
$ |
|
|
$ |
|
||
付記はこれらの連結財務諸表の構成要素である。
F-3
初日生物製薬会社です。
会社連結業務報告書
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
運営費用: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
一般と行政 |
|
|
|
|
|
|
|
|
|
|||
総運営費 |
|
|
|
|
|
|
|
|
|
|||
運営損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
利子収入,純額 |
|
|
|
|
|
|
|
|
( |
) |
||
その他の費用、純額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
派生ツール部分負債の公正価値変動 |
|
|
|
|
|
|
|
|
( |
) |
||
純損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
転換可能な非持株権益を償還するには純損失を占めなければならない |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
償還可能な非持株権益株式を交換する−配当とする− |
|
|
|
|
|
( |
) |
|
|
|
||
普通株主/メンバーは純損失を占めるべきである |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
1株当たり基本と希釈して純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
加重-計算に使用される普通株式平均 |
|
|
|
|
|
|
|
|
|
|||
付記はこれらの連結財務諸表の構成要素である。
F-4
初日生物製薬会社です。
合併全面損失表
(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
その他の全面的な損失: |
|
|
|
|
|
|
|
|
|
|||
証券売却可能な未実現損失 |
|
|
( |
) |
|
|
|
|
|
|
||
全面損失総額 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
付記はこれらの連結財務諸表の構成要素である。
F-5
初日生物製薬会社です。
短所償還可能な転換可能な優先株、償還可能な非持株権益と
株主権益·会員権益(赤字)
(単位は千で、シェアは含まれていない)
|
|
償還可能両替 |
|
|
償還可能である |
|
|
|
普通株 |
|
|
普通株 |
|
|
激励株 |
|
|
その他の内容 |
|
|
その他を累計する |
|
|
積算 |
|
|
株主の |
|
|||||||||||||||||||||||||
|
|
株 |
|
|
金額 |
|
|
利子 |
|
|
|
株 |
|
|
金額 |
|
|
株 |
|
|
金額 |
|
|
株 |
|
|
金額 |
|
|
資本 |
|
|
総合損失 |
|
|
赤字.赤字 |
|
|
(赤字) |
|
|||||||||||||
2019年12月31日の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
|
|
— |
|
|
$ |
— |
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
( |
) |
|||||||
Aシリーズを発行して転換可能な優先株を償還して現金と交換し、発行コストを差し引く$ |
|
|
|
|
|
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
||
激励株を発行する |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
奨励株を廃止する |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
決済時デリバティブ部分負債の再分類 |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
株式ベースの給与費用 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||
償還可能な非持株権益は純損失を占めなければならない |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
所有権変更に関連する償還可能な非持株権益に譲渡する |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
|
初日のバイオ製薬ホールディングスのメンバーの純損失に起因します |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2020年12月31日残高 |
|
|
|
|
$ |
|
|
$ |
|
|
|
|
— |
|
|
$ |
— |
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
( |
) |
|||||||
Bシリーズを発行して償還可能な優先株を発行して現金と交換し、発行コストを差し引くことができる$ |
|
|
|
|
|
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
||
激励株を発行する |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
奨励株を廃止する |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
償還可能な転換可能優先株、普通株、奨励株を普通株に変換する |
|
|
( |
) |
|
|
( |
) |
|
|
— |
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
償還可能な非制御的権益を普通株に変換する |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
IPOで発行された普通株は,発行コストを差し引いて$となる |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
従業員の株購入計画に基づいて株を発行する |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
株式ベースの給与費用 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
償還可能な非持株権益は純損失を占めなければならない |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
普通株主/メンバーは純損失を占めるべきである |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2021年12月31日の残高 |
|
|
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
|
|
|
|
$ |
|
|
|
— |
|
|
$ |
— |
|
|
|
— |
|
|
$ |
— |
|
|
$ |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
|
||||
後続発行により普通株を発行し、発行コスト#ドルを差し引く |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
株式オプション行使時に普通株を発行する |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
制限株式単位を解除して普通株を発行する |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
従業員の株式購入計画に基づいて普通株を発行する |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
普通株に帰属せず没収する |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
株式ベースの給与費用 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
証券売却可能な未実現損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
普通株主/会員は純損失を占めるべきである |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2022年12月31日の残高 |
|
|
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
|
|
|
|
$ |
|
|
|
— |
|
|
$ |
— |
|
|
|
— |
|
|
$ |
— |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
付記はこれらの連結財務諸表の構成要素である。
F-6
初日生物製薬会社です。
コンソール.コンソールキャッシュフロー表統合レポート
(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
経営活動のキャッシュフロー |
|
|
|
|
|
|
|
|
|
|||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
純損失と経営活動で使用される現金純額の調整: |
|
|
|
|
|
|
|
|
|
|||
買収している研究開発資産は |
|
|
|
|
|
|
|
|
|
|||
株式ベースの給与費用 |
|
|
|
|
|
|
|
|
|
|||
減価償却費用 |
|
|
|
|
|
|
|
|
|
|||
短期投資割引の増加、純額 |
|
|
( |
) |
|
|
|
|
|
|
||
経営的使用権資産の償却 |
|
|
|
|
|
|
|
|
|
|||
非現金利子支出 |
|
|
|
|
|
|
|
|
|
|||
デリバティブ部分負債の変動 |
|
|
|
|
|
|
|
|
|
|||
経営性資産と負債変動状況: |
|
|
|
|
|
|
|
|
|
|||
前払い費用と他の流動資産 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
預金とその他の長期資産 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
売掛金 |
|
|
( |
) |
|
|
|
|
|
|
||
費用とその他の流動負債を計算しなければならない |
|
|
|
|
|
|
|
|
|
|||
リース負債を経営する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
経営活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
投資活動によるキャッシュフロー |
|
|
|
|
|
|
|
|
|
|||
短期投資購入のための現金 |
|
|
( |
) |
|
|
|
|
|
|
||
短期投資満期収益 |
|
|
|
|
|
|
|
|
|
|||
財産と設備を購入して支払った現金 |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
買収している研究開発資産のために支払った現金 |
|
|
|
|
|
( |
) |
|
|
|
||
投資活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
融資活動によるキャッシュフロー |
|
|
|
|
|
|
|
|
|
|||
Aシリーズを発行して転換可能な優先株を償還して得られた金は,発行コストを差し引く |
|
|
|
|
|
|
|
|
|
|||
Bシリーズを発行して転換可能な優先株を償還して得た金は,発行コストを差し引くことができる |
|
|
|
|
|
|
|
|
|
|||
普通株発行で得られた金の純額 |
|
|
|
|
|
|
|
|
|
|||
株式オプション行使時に普通株で得られた金を発行する |
|
|
|
|
|
|
|
|
|
|||
ESPP購入時に普通株を発行する収益 |
|
|
|
|
|
|
|
|
|
|||
融資活動が提供する現金純額 |
|
|
|
|
|
|
|
|
|
|||
現金および現金等価物の純増加 |
|
|
( |
) |
|
|
|
|
|
|
||
期初現金及び現金等価物 |
|
|
|
|
|
|
|
|
|
|||
期末現金と現金等価物 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
非現金活動の補足開示 |
|
|
|
|
|
|
|
|
|
|||
使用権資産と引き換えに賃貸負債 |
|
|
|
|
$ |
|
|
$ |
|
|||
償還可能な転換可能な非持株権益に譲渡する |
|
$ |
|
|
$ |
|
|
$ |
|
|||
転換に関する45,331,483株優先株、普通株と奨励株の交換(注1) |
|
$ |
|
|
$ |
|
|
$ |
|
|||
償還可能な転換可能な非持株資本を6,470,382株普通株に交換する(付記11) |
|
$ |
|
|
$ |
|
|
$ |
|
|||
付記はこれらの連結財務諸表の構成要素である。
F-7
初日生物製薬会社です。
連結財務諸表付記
1.ビジネスおよび組織機関の説明
組織と業務
第一日バイオ製薬会社、又は同社は、全年齢の生命にかかわる疾患を有する患者のための目標療法の開発及び商業化に取り組んでいる臨床段階のバイオ製薬会社である。同社は2018年11月にデラウェア州法律に基づいて有限責任会社として設立され、名称は英雄治療持株会社。その後、同社は2018年12月に初日治療ホールディングスに改称し、2020年3月に初日バイオ製薬ホールディングスまたは初日ホールディングス有限責任会社に改称した。
2021年5月26日、同社は、デラウェア州国務長官に変換証明書を提出することにより変換を完了し、その名称をDay One BiopPharmticals,Inc.に変更した。2021年12月31日までに、DOT Treateutics-2,Inc.(前身はHero Treateutics Inc.およびDay One BiopPharmticals,Inc.)の2つの子会社があり、またはDOT-2は、2018年11月にデラウェア州に登録され、DOT Treateutics-1,Inc.,またはDOT-1は、2019年12月に華トラ州に登録される。DOT-2とDOT-1はここで総称して子会社と呼ばれる.
2021年12月、当社取締役会は、子会社が当社と合併または合併することを許可し、当社は既存の会社であり、2021年12月31日から発効する。連結が財務諸表に及ぼす影響に関するより多くの情報は、“列報基礎”の節を参照されたい
武田株の初公開、会社転換と交換
2021年6月1日、同社は初公募株式(IPO)を完了し、IPOで合計販売された
初公募株を行うため、会社は2021年5月26日に法人転換を完了し、初日にホールディングス有限責任会社(I)がデラウェア州州務卿に変換証明書を提出することでデラウェア州有限責任会社からデラウェア州会社に変換し、(Ii)を初日バイオ製薬会社に変更した。
変換の一部として:
F-8
“ミレニアム証券取引所協定”又は“ミレニアム証券取引所協定”の条項に基づいて、初公募株と転換、及び転換について、ミレニアム医薬株式会社
同社は初日ホールディングス有限公司のすべての財産と資産を保有し、初日持株有限責任会社のすべての債務と義務を負担する。転換日から、初日に持株有限公司の各取締役会メンバーと各上級管理者が取締役会メンバーと当社の上級管理者となる。変換は免税再編であり、株式の発行許可を含み、
初公募が終わった後、
株式分割
2021年5月23日、Day One Holding LLC取締役会は、会社の株式の長期分割を実現するために、その運営協定の改訂を承認した
新冠肺炎に関連するリスクと不確実性
進行中の新冠肺炎大流行の全面的な影響は依然として高度に不確定であり,変化する可能性がある。新冠肺炎疫病をめぐる多くの不確定性が存在し、将来の事態発展は予測できず、会社の運営と財務状況に実質的なマイナス影響を与える可能性がある。当社は、地方政府が新たな圧力の発生と発生に反応し、どの新たな圧力が保護措置の延長、延長、または強化をもたらす可能性があるため、その第三者サービスプロバイダが提供するサービスの変動を経験し、継続することが予想される。製造·供給において,同社はその薬品サプライチェーンが妨害されないことを予想しており,ロック措置や制限が実施されているかどうかも特定できず,もしあれば,その施設や運営にどのような影響を与える可能性がある。
同社の管理チームは引き続きこの変化しつつある健康危機及びその運営、主要なサプライヤーと従業員チームへの影響を積極的にモニタリングしている。
2.重要な会計政策の概要s
陳述の基礎
添付されている総合財務諸表は、会社子会社の勘定を含む米国公認の会計原則またはアメリカ公認会計原則に従って作成されている。すべての会社間取引と残高は合併で販売された。
F-9
統合(“組織および業務”の節を参照)は、会計基準編纂またはASC,250で定義された報告エンティティの変化を構成していないため、会計変更と誤り訂正当社は2021年12月31日より、子会社から移転した資産と負債を歴史的帳簿価値で報告している。
本説明における適用ガイドの任意の言及は、財務会計基準委員会(FASB)のASCおよび会計基準更新(ASU)における権威GAAPを意味する。
予算の使用
米国公認会計原則に基づいて連結財務諸表を作成することは、連結財務諸表の日付の資産、負債及び又は資産及び負債の報告金額、及び報告期間内に報告された費用金額に影響を与えるために、管理層に推定と仮定を要求する。添付されている総合財務諸表に作成された重大な推定および仮定には、株式報酬の推定値、繰延税金資産および所得税の不確実性の推定値、および研究·開発活動の課税項目が含まれているが、これらに限定されない。同社の見積もりは歴史的経験や様々な他の合理的な仮定に基づいている。実際の結果は,これらの推定や仮定とは異なる可能性がある.
細分化市場
同社はその最高経営責任者を最高経営決定者、CODMと略称することを決定した。同社は、全年齢層の遺伝子定義癌患者の識別と標的治療を推進する事業である報告と運営部門の形態で業務を経営·管理している。同社のCODMは、まとめた上で財務情報を審査し、資源を割り当て、財務業績を評価する。その会社のすべての資産はアメリカにあります。
信用リスクその他のリスクと不確定要因が集中している
会社を高度な集中信用リスクに直面させる金融商品は主に現金、現金等価物、短期投資を含む。同社の現金、現金等価物、短期投資は米国の複数の金融機関に保管されている。預金金額は連邦保険の限度額を超える場合があります。経営陣は、当社の現金、現金等価物、短期投資を持つ金融機関の財務状況が良好であるため、金融機関の信用リスクが最も低いと考えている。
当社はいくつかのリスクと不確定要素の影響を受け、以下のいずれの変化も当社の将来の財務状況または経営結果に重大な悪影響を及ぼす可能性があると考えられる:将来の融資を得る能力、候補製品の承認と市場受け入れおよび補償の規制要求;当社が依存する第三者臨床研究機関とメーカーの表現、販売ルートの発展、当社の知的財産権の保護、知的財産権、特許、製品、監督管理またはその他の要素による当社の訴訟またはクレーム;市場構造の変化;企業が成長を支援するために必要な従業員の能力を引き付けて維持することができます
同社は第三者メーカーに依存してその計画中の研究·開発活動に製品を提供している。特に,同社は少数のメーカーがこれらの計画に関連する活性医薬成分や処方薬の供給要求に依存していることに依存し続けると予想される。これらのプロジェクトは活性薬物成分や製剤薬物供給の深刻な中断の悪影響を受ける可能性がある。
F-10
現金と現金等価物
当社は購入時の原始満期日が三ヶ月以下のすべての高流動性投資を現金等価物と見なしています。同社の現金等価物には、通貨市場基金や米政府機関証券への投資が含まれている。現金等価物は公正価値に近い分担コストで確認した。
投資する
同社の投資には、米国政府機関証券と米国債が含まれる。投資は、購入時に管理職の意図に応じて、保有から満期まで、販売または取引可能に分類される。その会社のすべての投資は販売可能に分類されている。売却可能証券は推定公正価値に基づいて入金され、当該等投資の未実現保有量損益(税項影響を差し引く)は他の全面(損失)収益の中で総合全面損益表の単独構成部分として列報する。公正価値は、見積、金利、および収益率曲線のような観察可能な見積市場金利に基づいて、または観察可能なデータ点を利用することによって決定される。
販売可能な証券について、当社は、任意の減価が信用損失または非信用損失に関連しているかどうかを判断する。損失が信用或いはその他の要素から来ているかどうかを評価する時、管理層は公正価値が償却コストより低い程度、格付け機関の証券格付けに対するいかなる変化及び証券関連の不利な条件などの要素を考慮する。この評価が信用損失が存在することを示す場合、証券から受け取るキャッシュフローの現在値と証券の余剰コストベースを比較することが予想される。もし現金流量の現在値が余剰コスト基礎より低い場合、信用損失が存在し、そして減値準備を計上するが、公正価値が余剰コストベースより小さい金額を限度とする。解約または回収の形態で信用損失部分に関連する後続活動は、証券信用損失を売却するための準備可能な一部として確認された。
F-11
金融商品の公正価値
総合貸借対照表では,公正価値で常時記録されている資産と負債は,その公正価値を計測するための投入に関する判断レベルに基づいて分類される.公正価値は、計量日に市場参加者間の秩序ある取引において資産または負債の元本または最も有利な市場の負債を移動させるために、受信される交換価格または支払いされる退出価格として定義される。公正価値を計量するための推定技術は,観察可能な投入を最大限に利用し,観察不可能な投入を最大限に減少させなければならない。“公正価値計量に関する権威指針”は公正価値計量の開示のために三級公正価値等級を確立し、具体的には以下の通りである
第1レベル--観察可能な投入、例えば、計量日と同じ資産または負債のアクティブ市場における未調整オファー;
第2レベル-資産または負債の投入を直接または間接的に観察することができる(第1レベルに含まれる見積を除く)。これらのオファーは、アクティブ市場における同様の資産または負債の見積もりと、非アクティブ市場における同じまたは同様の資産または負債の見積もりとを含む
第三レベル-市場活動が少ないか、または市場活動支援の観察できない投入がなく、資産または負債の公正な価値に大きな意義を持っている。
ある程度、推定値は市場で観察または観察できないモデルや投入に基づいており、公正価値の決定にはより多くの判断が必要である。そのため、当社が価値を公平に判断する際に下した判断は、第3級に分類されたツールに対する判断度が最も高く、公正価値レベル内の金融商品レベルは、公正価値計量に大きな意味を持つ任意の投入の中で最低レベルに基づいている。
現金等価物,前払い費用,その他の流動資産,売掛金,売掛金およびその他の流動負債の短期的性質により,付随する総合貸借対照表に反映される帳簿金額はその公正価値に近い。
財産と設備、純額
財産と設備はコストから減価償却と償却を差し引いて入金される。減価償却は直線法で関連資産の推定耐用年数(3~5年)で確認され、リース収益は賃貸期間または資産の推定耐用年数の短いもので償却される。
賃貸借証書
リース定義に適合する契約スケジュールは、経営的または融資的賃貸に分類され、総合貸借対照表に使用権資産またはROU資産、およびリース負債として記録され、レンタル期間内の固定賃貸支払いをリースに暗黙的な金利または会社の増分借入金利(IBR)で割引する計算方法である。レンタルROU資産およびレンタル責任は、発効日レンタル期間内の将来の最低レンタル支払いの現在値で確認します。当社には現在融資リースは何もありません。
経営リースROU資産は、(I)開始日または以前に支払われた金、(Ii)による初歩的な直接コスト、および(Iii)リース下のテナント激励措置に基づいて調整される。運営リースの隠れた金利は特定できないため、当社はレンタル開始日を適用した資料に基づいて内部収益率を決定します。IBRは,当社が資産が存在する類似経済環境下で,類似期限に相当する賃貸金を抵当に借入して支払う金利に基づいて決定される.当社はリース期間を対象資産を使用する権利がある取消不可期間と見なし、当社が任意の選択権を行使して契約の任意の期間を延長することを決定する理由がある。
F-12
運営リースの最低賃貸支払いのリースコストは直線法でレンタル期間内に確認する。賃貸負債は毎期利息を増加させ、支払いを減少させ、ROU資産はレンタル期間内に償却する。可変レンタルコストは発生時に入金されます。ROU資産とリース負債を計量する際には、会社は合併リースと非レンタル組成物を選択する。当社では、リース開始時の初期期限が12ヶ月以下の短期賃貸は含まれておらず、会計政策選択として、このようなレンタルのレンタル期間内の賃貸料支出を直線的に確認しています。
長期資産減価準備
事件や状況変化が長期資産の帳簿価値が回収できない可能性があることを示した場合、当社は長期資産(物件や設備および使用権資産を含む)の減値を評価します。保有·使用する資産の回収可能性は,資産の帳簿価値と資産予想による将来の未割引現金流量を比較することで測定した。当該等の資産は減値とみなされ、確認すべき減値は、当該資産の帳簿価値が当該資産の公正価値を超える金額で計量される。これまで、連結財務諸表では減値は確認されていません。
研究と開発費
研究および開発費用には、会社を代表して臨床試験を行う第三者契約研究機関、契約製造機関、および他の第三者との合意によって生成された技術および知的財産許可証の取得に関連するコスト、実験室材料および用品を含む研究および開発プロジェクトに関連する他のコスト、会社の研究開発者の給料、福祉および株式ベースの報酬支出、および賃貸料および施設維持費用および償却費用を含む従業員に関するコストが含まれる。当社で発生した研究開発費。会社はある研究と開発協定に署名する際に前払い金を支払う義務がある。将来の研究開発活動のための商品またはサービスのための事前支払いは、払い戻し不可能な金額を含めて延期されます。この金額は、関連貨物が提供されたり、関連サービスが提供されたりする際に費用として確認されたり、会社が貨物またはサービスを提供しないことが予想される場合に費用として確認されたりします。
研究と開発費用を計算すべきである
同社は,臨床前と臨床研究および契約製造活動を含む第三者サービスプロバイダによる研究と開発活動の見積もりコストの計上負債を記録している。同社は、請求書が発行されていないサービスを提供する推定金額に基づいて研究·開発活動の推定コストを記録し、これらのコストを貸借対照表中の計算すべき負債と総合経営報告書における研究·開発費用に計上している。このような費用は私たちの研究開発費の重要な構成要素だ。当社は,完了した作業の見積り数などに基づいて,サービスプロトコルに基づいて第三者サービス提供者と締結したプロトコルに基づいて,これらの費用を計上する.当社は各報告期間の計上すべき負債残高を決定する際に判断と見積もりを行います。実際のコストが既知であるため、会社はその負債を調整した。当社の計算すべきコストと実際に発生したコストとの間には大きな差はありません。
同社は,臨床試験の実施·管理を支援する契約製造組織と契約研究組織との契約に基づき,臨床試験に関する費用を支払っている。これらの契約の財務条項は交渉する必要があり、これらの条項は契約によって異なり、材料やサービスを提供する期限と一致しない支払いを招く可能性がある。一般に、これらのプロトコルは、固定料金、単価、または時間および材料で実行される動作範囲を規定する。会社が将来の研究·開発活動のための商品やサービスを前払いする場合、支払いは延期され、前払い費用として資本化され、貨物を受け取ったり、関連サービスを提供したりする際に費用として確認される。このような支払いは、予期される達成された時間に基づいて、現在または長期的な分類評価を行う。
F-13
特許費用
特許出願の提出及び起訴に関するすべての特許に関する費用は、支出回収状況が不確定であることによる費用に基づいて計上される。発生した金額は,連結業務報告書では一般費用と行政費用に分類される。
転換可能優先株を償還する
当社は発行日の公正価値に基づいて発行コストを差し引いた償還可能転換優先株入金を行います。償還可能な転換可能優先株は株主損失以外に入金される。当該等の株式は強制償還可能ではないが、当社がコントロールできないものとみなされる清算事件が発生した場合、得られた金は償還可能転換優先株株主の清算優先順位に応じてまず償還可能な転換可能優先株株主に割り当てられるからである。当社は転換可能な優先株を償還可能な帳簿価値をその清算優先株に調整していません。いつ清算事件が発生するかどうかが確定していないので、当社には償還可能な転換可能な優先株保有者に清算優先株を支払う責任があります。2021年5月のIPO終了時に、償還可能な転換可能優先株はすべて普通株に変換することができる。
償還可能な非持株権益
償還可能な非持株権益とは、DOT-1の非直接または間接的に当社の株式(純資産)部分に帰属することである。償還可能な非持株権益は一時株式に分類され、所有者に発行される優先株には、自社の制御範囲内に完全にはない償還特徴が含まれているからである。
株式ベースの報酬
初公募前に、当社は株式に基づくすべての奨励、奨励株式、制限的株式奨励の推定公正価値に基づいて、オプション定価モデルを採用し、付与日に株式に基づく報酬支出を確認した。オプション定価モデルは主観的仮定を入力する必要があり、標的普通株の公正価値、奨励の期待期限、期待変動率、無リスク金利と配当収益率を含む。普通株式の公正価値を決定する際に、企業価値を推定するための方法は、米国公認会計士協会会計および推定ガイドラインと一致する方法、方法、および仮定を用いて実行される補償として発行された個人持株会社株式証券の推定値それは.参加敷居の金額は取締役会が付与された時に決定される。この間に付与された賠償金の期待期限は,クリーニング終了イベントまでの期待時間に基づいて決定される.同社は付与時に発効した米国債収益率をもとに、付与期限と一致した無リスク金利を採用している。予想変動率は、当社が業務を展開している業界同業グループが予想される流動資金時間と一致することに基づいている。配当収益率を
初めての公募終了後、会社はBlack-Scholes推定モデルを用いて従業員と非従業員のオプションを付与する公正価値を推定し、内在価値は制限性株式奨励の公正価値を推定し、及び制限性株式単位は日会社普通株の公正価値を授与した。
ブラック·スコアーズオプション定価モデルは、株式オプション報酬の公正価値を推定するために使用され、以下の仮定を使用することが要求される
F-14
当社は直線帰属法を用いて株式報酬費用を確認しています。当社は没収が発生した同時期に費用を減らすことで没収を確認します。業績条件のある報酬が条件を満たす可能性がある場合には、会社は株式ベースの報酬支出を確認し、報酬が付与される。当社は総合経営報告書において株式をもとに報酬支出を分類しており,受賞者の賃金コストや受賞者のサービス支払いを分類する方式と同様である。
所得税
初公募前、同社は国内税法に基づく“直通”実体であり、2社の子会社を持っていた。転換前に、会社のメンバー(前身は初日バイオ製薬ホールディングス、LLC)は、初日にバイオ製薬ホールディングスのそれぞれの所有権と活動について直接課税した。当社の連結会社子会社は、貸借対照法の下で所得税を計上し、以下のようになる。
初公募および株式交換が完了した後、当社の所得税は貸借対照法で入金されます。繰延税項資産及び負債は、既存資産及び負債の帳簿金額及びそのそれぞれの税ベースと営業損失純額及び税額相殺繰越との差額による将来の税項影響を確認することができる。繰延税項資産及び負債は、総合財務諸表帳簿額面と資産及び負債の課税基準との差額に基づいて決定され、予想差額に予想される年度に適用される課税収入の制定税率を用いて計量される。繰延税金資産の一部または全部が現金化できない可能性が高い場合、繰延税金資産減価準備。
関連税務機関の審査を経た後、不確定な所得税の頭寸は最大金額で確認され、その金額は持続しない可能性が高い。確認や計測の変化は判決が発生した期間に反映される.同社の政策は、所得税の減納に関する利息と罰金を所得税として確認するための構成要素である。これまで、未確認の税収割引に関する利息や罰金は記録されていない。
1株当たり純損失
当社は証券参加に必要な二段階法に基づいて1株当たりの基本純損失と希薄化純損失を計算します。2種類の法の下で、1株当たりの基本純損失の計算方法は、純損失を期間中に発行された普通株の加重平均数量で割ることであり、潜在的な希釈証券を考慮しない。1株当たり純損失の算出方法は、任意の償還可能な非持株権益の損失を差し引いた後、純損失を期内に発行された普通株の加重平均に期内の潜在的希薄化証券の希薄化影響の和を加えたものである。潜在的希釈証券は、初回公募前の激励株、帰属していない制限的な普通株、および償還可能な転換可能な優先株を含む。IPO後、希釈可能な証券は、未付与制限株式奨励、未付与制限株式単位、および株式オプションを含む。列報のすべての期間において、1株当たり純損失は1株当たりほぼ純損失と同じであり、これは潜在普通株計上の影響が逆薄であり、激励株の参加ハードルが達していないためである。
F-15
総合損失
総合損失とは株主権益のすべての変動であるが,株主による変動や株主に割り当てられた変動は含まれていない.同社が証券を売却できる未実現収益と損失は他の全面損失の唯一の構成部分であり、報告の純損失に含まれず、総合全面損失表に記載されている。
新興成長型会社の地位
同社は,2012年のJumpStart Our Business Startups ActやJOBS Actの定義に基づく新興成長型会社である。“雇用法案”によると、新興成長型企業は、これらの基準が民間企業に適用されるまで、“雇用法案”公布後に発表された新たなまたは改正された会計基準の採用を延期することができる。当社は、(I)新興成長型会社または(Ii)が“雇用法案”に規定されている延長移行期間から脱退することを明確かつ撤回できなくなるまで、この延長移行期間を使用することを選択した。したがって、これらの連結財務諸表は、上場企業の発効日までに新たなまたは改訂された会計声明を遵守している会社と比較できない可能性がある。
雇用法案は、新規または改正された会計基準が民間企業に適用される前に、新興成長型企業がこの基準を採用することを排除しない。同社は、まだ新興成長型企業である期間中に、延長された移行期間を任意の他の新たなまたは改正された会計基準に使用すると予想される。
最近採用された会計公告
2019年12月、FASBは、期間内の税収分配方法、中期所得税計算方法、および外部基礎差異の繰延税金負債確認に関連するいくつかの例外を除去したASU 2019-12、所得税(特別テーマ740):所得税会計を簡略化する、またはASU 2019-12を発表した。ASU 2019-12はまた、所得税会計の他の側面を明確にし、簡略化した。この基準は財政年度とこれらの財政年度内の過渡期に適用され,2020年12月15日以降から早期採用が許可されている。当社は2022年1月1日にASU 2019-12を採用しており、これは当社の財務諸表及び関連開示に大きな影響を与えていません。
3.経常的公正価値計測
以下の表は、公正価値システム内で公正価値を段階的に定期的に計量する同社の2022年12月31日と2021年12月31日までの金融商品を示している(千計)
|
|
2022年12月31日 |
|
|||||||||||||
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
|
合計する |
|
||||
金融資産: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
アメリカ国債 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ政府機関証券 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
公正価値に応じて計量された総資産 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
|
|
2021年12月31日 |
|
|||||||||||||
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
|
合計する |
|
||||
金融資産: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
公正価値に応じて計量された総資産 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
F-16
同社の通貨市場基金は、活発な市場からの同じ資産の観察可能な投入を用いて測定されているため、レベル1に分類されている。
同社の米国債および米国政府機関証券は、活発な市場における類似資産の見積もりと、非アクティブ市場における同じまたは類似した資産の見積もりを含む資産の直接的または間接的に観察可能な投入を用いて測定されるため、2段階に分類される。
2022年12月31日と2021年12月31日現在、レベル3に分類されている資産や負債はない。
いくつありますか
以下の表は、会社の現金等価物、短期投資に分類された売却可能証券、および関連する未実現損益の推定公正価値(単位:千):をまとめている
|
|
2022年12月31日 |
|
|||||||||||||
|
|
原価を償却する |
|
|
未実現収益 |
|
|
未実現損失 |
|
|
公正価値を見積もる |
|
||||
現金等価物: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
アメリカ政府機関証券 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
現金等価物合計 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
短期投資 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
アメリカ政府機関証券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
短期投資総額 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
|
|
2021年12月31日 |
|
|||||||||||||
|
|
原価を償却する |
|
|
未実現収益 |
|
|
未実現損失 |
|
|
公正価値を見積もる |
|
||||
現金等価物: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
現金等価物合計 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
次の表は我々の現金等価物と売却可能証券の満期日をまとめたものである2022年12月31日(千):
|
|
原価を償却する |
|
|
公正価値 |
|
||
1年以下で成熟しています |
|
$ |
|
|
$ |
|
||
合計する |
|
$ |
|
|
$ |
|
||
以下の表は、同社が未実現損失総額を有する売却可能証券の内訳と、2022年12月31日までのこれらの赤字未実現の持続時間(千単位)を示している
|
|
2022年12月31日 |
|
|||||||||||||||||||||
|
|
12ヶ月以下の未実現損失 |
|
|
12ヶ月以上の赤字を達成していません |
|
|
合計する |
|
|||||||||||||||
|
|
公正価値 |
|
|
未実現損失 |
|
|
公正価値 |
|
|
未実現損失 |
|
|
公正価値 |
|
|
未実現損失 |
|
||||||
金融資産: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
アメリカ国債 |
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
||||
アメリカ政府機関証券 |
|
|
|
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|
( |
) |
||||
金融資産総額 |
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
||||
F-17
“会社”ができた
当社は定期的にその証券格付けの変動を検討し、周囲の経済状況を監査し、期待される信用損失のリスクを評価する。2022年12月31日までに
4.貸借対照表項目
前払い費用と他の流動資産
前払い資産および他の流動資産には、以下が含まれる(千計)
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
研究開発費を前払いする |
|
$ |
|
|
$ |
|
||
前払い保険 |
|
|
|
|
|
|
||
他の前払い費用と他の資産 |
|
|
|
|
|
|
||
前払い費用とその他の流動資産総額 |
|
$ |
|
|
$ |
|
||
財産と設備、純額
財産と設備の純額は以下の部分からなる(千計)
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
賃借権改善 |
|
$ |
|
|
$ |
|
||
家具と固定装置 |
|
|
|
|
|
|
||
財産と設備、毛額 |
|
|
|
|
|
|
||
減算:減価償却累計 |
|
|
( |
) |
|
|
( |
) |
財産と設備、純額 |
|
$ |
|
|
$ |
|
||
2022年12月31日現在、2021年12月31日と2020年12月31日までの年間減価償却費用は約1億ドル
費用とその他の流動負債を計算しなければならない
計算すべき費用および他の流動負債には、以下の項目が含まれる(千で計算)
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
研究と開発費用を計算すべきである |
|
$ |
|
|
$ |
|
||
給与明細に関連する費用を計算しなければならない |
|
|
|
|
|
|
||
専門サービス費用を計算する |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
費用とその他の流動負債総額を計算しなければならない |
|
$ |
|
|
$ |
|
||
F-18
5.重要なプロトコル
メルクKGaA社とライセンス契約を結び、ドイツのダムシュタット
2021年2月10日、同社の子会社DOT-2は、ドイツのダムシュタットにあるメルクKGaA製薬会社とMRKDGライセンス契約を締結した。MRKDG許可協定によると、ドイツのダムシュタットのメルクKGaAは同社に独占グローバルライセンスを付与し、特定の特許権利と技術ノウハウに基づいて、複数のレベルで再許可を付与し、PimasertibとMSC 2015103 B化合物を含む製品を研究、開発、製造、商業化する権利がある。同社は臨床在庫用品も受け取り,その研究や開発活動に用いられている。同社が付与した独占許可は、メルクKGaAが付与した非独占許可によって制限されている。メルクKGaAはドイツの癌研究機関の付属会社であり、ドイツのダムシュタットメルクKGaAはいくつかのPimasertibに関する臨床研究を直接または間接的に行う権利を保持している。
MRKDGライセンス協定によると、同社はビジネス上合理的な努力を使用し、2029年までに少なくとも2つの指定された主要市場国で少なくとも2つのライセンス製品を開発して商業化する義務がある。
MRKDGライセンス契約により付与された権利と臨床用品の代償として,同社は#ドルを前払いした
MRKDGライセンス契約の期限は,会社がその国/地域のライセンス製品についてライセンス側に印税を支払う義務期間が満了した後,ライセンス製品と国/地域で満期となり,会社がMRKDGライセンス契約に基づいてすべてのライセンス製品とすべての国/地域のすべての支払い義務が満了したときにすべて満期となる。
DOT−2は2021年12月31日から会社と合併して会社に合併し,会社は既存の会社であり,MRKDG許可協定に基づいてDOT−2の義務を負う。
武田資産購入協定
2019年12月16日、同社の子会社DOT-1は、武田薬業有限公司の関連側と関連会社ミレニアム社と資産購入契約、すなわち武田資産協定を締結した。武田資産プロトコルによれば、DOT-1は、TAK-580に関連するいくつかの技術的権利および独自技術(現在、TOVORAFenib(DAY 101))を購入し、原発性脳腫瘍または固形腫瘍脳転移患者の治療に新しい方法を提供する。DOT−1は,同社のようなRAF−阻害剤の研究·開発活動のための臨床在庫供給と,指定された調査者臨床試験プロトコルも受け取っている。武田はViracta治療会社(F/k/a Sunesis PharmPharmticals,Inc.)やViractaの独占ライセンスプロトコルやViractaライセンスプロトコルとDOT-1にも割り当てている.武田はまた、武田資産協定に基づいてDOT−1に指定特許項下のグローバル再許可独占許可と、武田資産協定に基づいて生成された他の特許及び独自技術項下の非独占許可とを付与した。当社も武田資産協定の定義に基づいて、武田資産協定が規定する適用時間範囲内で指定された発展マイルストーンに到達できなかった場合に自動的に終了またはDOT-1で終了する許可を武田に付与します。この武田に付与された帰還許可証は転換時に終了し、ミレニアム証券取引所協定と関係がある。
F-19
DOT−1は,資産の売却·譲渡および武田資産協定によるライセンス付与の代償として#ドルを前払いした
武田資産協定の有効期限は、各国のすべての譲渡特許権とすべてのライセンス特許権が満期になった後、国ごとに終了します。武田は、同社が初めて商品を販売する前に武田資産協定を終了することができ、連続して指定された期間内にいかなる開発活動も停止すれば、この停止は当事者の同意を得られず、規制当局の指導に応えるためでもない。また、武田は会社倒産時に武田資産協定を終了することができる。武田が我々の開発停止や破産により武田資産協定を終了すれば、譲渡されたすべての特許、ノウハウ、契約(Viractaライセンス契約を除く)が武田に譲渡され、武田は特許やノウハウに基づいて回復許可を得て、これ等で終了したすべての製品を利用する。
DOT-1は2021年12月31日から会社と合併して会社に合併し、会社は既存の会社であり、武田資産購入協定に基づいてDOT-1の義務を負う。
Viracta許可協定
2019年12月16日、DOT-1は、武田資産協定に従って譲渡されたViractaライセンス協定を修正し、再確認した。Viractaライセンスプロトコルによると,DOT−1は特定の特許権やノウハウにより世界的に独占的に許可されており,RAFタンパク質ファミリーに結合した化合物を含む製品を開発,使用,製造,商業化することができる。
DOT-1支払いました$
Viractaライセンス契約の期限は,会社がViractaにその製品の当該国/地域での印税を支払う義務期間が満了した後,ライセンス製品と国/地域で終了する.DOT-1は、指定された通知期間内に任意またはすべてのライセンス製品に関連するViractaライセンスプロトコルを任意に終了する権利がある。
DOT-1は2021年12月31日から会社と合併して会社に合併し、会社は生き残った会社であり、Viracta許可協定に従ってDOT-1の義務を負う。
F-20
6.支払いの引受およびまたは事項
賃貸借証書
同社は2020年3月にカリフォルニア州サンフランシスコ南部にある会社のオフィス施設について賃貸契約を締結した。このようなプロトコルは、確定された資産使用を制御する権利が一定期間、価格と交換するために会社に譲渡されたので、レンタルとして決定される。当社はレンタル期間を一年間延長することができます
2022年4月、当社は賃貸契約を締結した
当社のリース契約は暗黙金利を提供していないため、当社はレンタル開始日を適用する利用可能情報に基づいてレンタル逓増借入金金利、またはIBRを決定しました。IBRは,当社が資産が存在する類似経済環境下で,類似期限に相当する賃貸金を抵当に借入して支払う金利に基づいて決定される.2022年12月31日現在の加重平均残存賃貸期間と加重平均割引率は
同社のレンタルは何の支払いや賃貸料も要求せず、財務制限を加えず、残額保証も含まれていない。
資産を使用する賃貸費用は、適用されるリース期間内に直線法で確認される。レンタル料金は$
2022年12月31日現在、将来の賃貸債務は以下の通り(千で計算)
|
|
|
|
2023 |
$ |
|
|
2024 |
|
|
|
将来の最低賃貸支払い総額 |
|
|
|
差し引く:推定利息 |
|
|
|
リース負債現在価値を経営する |
$ |
|
|
F-21
研究と開発協定
同社は正常な業務過程中に臨床研究機構、代理工組織とその他の第三者サプライヤーと臨床試験、製造、テストとその他の研究と開発活動について契約を締結した。これらの契約は一般的に通知後に終了することに規定されていますが、一部の費用はプロジェクト承認後にキャンセルできないため、サプライヤーの例外があります。2022年12月31日と2021年12月31日まで、それぞれ
許可協定
当社は、付記5に開示された者のように、当該合意に基づいて、特定事件の満足状況に応じてマイルストーン費用を支払うライセンス契約を締結しなければならない。Viractaライセンス協定に関する最初のマイルストーンが実現され,2021年12月31日までの年度に研究開発費が計上されている。第二のマイルストーンはMRKDG許可協定に関連しており,2022年12月31日までの年度に実現し開発として記録した。会社はこれらの合意に基づいて開発された製品の販売に印税を支払うように要求されるかもしれない。2022年12月31日と2021年12月31日まで、すべての製品が開発中であり、
法律訴訟
当社は時々正常な業務過程で発生する訴訟の当事者になる可能性があります。当社はいかなる重大な法律手続きの影響も受けておらず、その知る限り、現在のところ重大な法的手続きが待っていることや脅かされていることはない。
賠償協定
正常な業務過程において、会社は各種の陳述と保証を含む契約と協定を締結し、特定の責任に対して賠償を行うことを規定している。これらのプロトコルでのリスクの開口は未知であり,将来それに提起される可能性があるがまだクレームされていないことに関連しているからである.これまで、同社は何のクレームも支払っておらず、その賠償義務に関するいかなる訴訟も弁護されていない。しかし、このような賠償義務のため、会社は未来に費用を記録するかもしれない。会社はまた、その役員や上級管理者の特定の事件や事件における賠償義務を負うが、そのような身分でサービスを要求する場合には一定の制限がある。今までクレームはありませんでしたが、同社はこれらの賠償協定の公正価値が最も低いと考えています。そのため同社には
7.転換可能優先株式償還可能
2021年6月1日、会社は初公募株を完成し、全発売
2021年2月、当社は発表
8.普通株式
その会社の登録証明書によると,当社は発行する権利がある
F-22
2018年11月、当社は当社と普通株購入契約を締結しました
市場で製品を提供する
2022年6月、会社はPiper Sandler&Co.とJones Trading Institution Services LLCと販売エージェントとして株式分配協定を締結し、会社の普通株の発行と売却に関連し、総発行価格は最高$に達する
2022年6月に後続サービスを提供
2022年6月、同社は後続発行と発行·販売を完了した
同社は未来の発行のために普通株式を予約しており、具体的には以下の通りである
|
|
十二月三十一日 |
|
|
発行済みと未償還普通株式オプション |
|
|
|
|
将来の贈与に使える普通株 |
|
|
|
|
ESPPに使える普通株式 |
|
|
|
|
発行済みおよび未発行の限定株式単位 |
|
|
|
|
合計する |
|
|
|
|
9.株式の給与
転換前に、初日に持株有限責任会社は奨励株式計画に基づいて奨励株式を付与し、発行を許可した
奨励株の公正価値は、以下の仮定を有するオプション定価モデルを用いて推定される
|
|
十二月三十一日までの年度 |
||
|
|
2021 |
|
2020 |
普通株主公正価値 |
|
$ |
|
$ |
参加のハードル |
|
$ |
|
$ |
無リスク金利 |
|
|
||
波動率 |
|
|
||
流動性を実現する時間(年単位) |
|
|
||
付与日公正価値 |
|
$ |
|
$ |
F-23
転換期間中、会社はすべての奨励株式を既得株式および非帰属普通株に変換する。そのため、2022年12月31日までの年間奨励株活動はない。
当社はオプション定価モデルを用いて付与日の各奨励株の公正価値を試算しています。メンバーの権益価値は最近行われた企業推定分析と普通株公正価値に基づいている。参加敷居の金額は取締役会が付与された時に決定される。この間に付与された賠償金の期待期限は,クリーニング終了イベントまでの期待時間に基づいて決定される.同社は付与時に有効な米国債収益率に基づく無リスク金利を採用している。
普通株主公正価値
初めて公募する前に、経営陣が普通株式の公正価値を推定する方法は、米国公認会計士協会の業務援助で概説した方法と一致している補償として発行された個人持株会社株式証券の推定値あるいは、あるいは 実践援助は、独立第三者評価専門家の協力の下で普通株に対する評価;会社の発展段階と業務戦略、研究開発仕事の状況、及び商業と工業に関連する重大なリスクを含む;会社の経営結果と財務状況、利用可能な資本資源のレベルを含む、生命科学と生物技術領域の上場会社の評価、及び最近完成した同業者会社の合併と買収;普通株は市場性が不足している;いくつかの客観的と主観的要素を考慮した。公平な取引において投資家に販売される償還可能な転換可能な優先株の価格、および会社が普通株に対する転換可能な優先株を償還する権利、優先権、および特権;当時の市場条件の下で、普通株および償還可能な転換可能な優先株保有者が流動性イベントを実現する可能性、例えば、初回公募株または売却。普通株の公正価値は、会社株が既定の証券取引所または全国市場システムに上場するまで、取締役会によって承認される。
奨励株式は株式奨励に分類され、株式の報酬費用に基づいて付与日奨励の公正価値に基づいている。
以下の表は、株式奨励活動の概要を提供します
|
|
量 |
|
|
加重平均 |
|
||
2020年12月31日現在の未返済債務 |
|
|
|
|
$ |
|
||
授与する |
|
|
|
|
$ |
|
||
没収される |
|
|
( |
) |
|
$ |
|
|
普通株式に帰属していないものに転換する |
|
|
( |
) |
|
$ |
|
|
2021年12月31日現在の未返済債務 |
|
|
|
|
$ |
|
||
F-24
2022年株式インセンティブ計画
2022年10月、取締役会と株主は2022年株式インセンティブ計画、または2022年計画を承認した。2022年計画では、非法定株式オプションと制限株式単位の付与が規定されている。“2022年計画”によると予約発行された普通株式数は
次の表は,2022年12月31日までの年度における2022年計画における株式オプション活動の概要を提供している。
|
|
オプション |
|
|
加重平均 |
|
|
加重平均 |
|
|
骨材 |
|
||||
2021年12月31日現在の未返済債務 |
|
|
— |
|
|
$ |
— |
|
|
|
|
|
|
|
||
授与する |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
鍛えられた |
|
|
— |
|
|
$ |
— |
|
|
|
|
|
$ |
— |
|
|
没収する |
|
|
— |
|
|
$ |
— |
|
|
|
|
|
|
|
||
2022年12月31日に返済されていません |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
すでに帰属しており,2022年12月31日に帰属する予定である |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2022年12月31日に行使できます |
|
|
— |
|
|
$ |
— |
|
|
|
— |
|
|
$ |
— |
|
総内在価値は、関連する普通株の推定公正価値と発行された現金オプションの行権価格との差を表す。
あったことがある
2022年12月31日現在、株式オプションの未償却株式報酬は$
同社はブラック·スコアーズオプション定価モデルを用いて、付与された株式オプション報酬の公正価値を推定し、以下のように仮定した
|
|
現在までの年度 |
|
|
2022 |
予想期限(年単位) |
|
|
予想変動率 |
|
|
無リスク金利 |
|
|
期待配当収益率 |
|
— |
次の表は、2022年12月31日までの1年間、2022年計画での制限株式単位活動をまとめています
|
|
量 |
|
|
加重平均 |
|
||
2021年12月31日現在の未帰属制限株式単位 |
|
|
— |
|
|
$ |
— |
|
授与する |
|
|
|
|
$ |
|
||
既得 |
|
|
— |
|
|
$ |
— |
|
没収する |
|
|
— |
|
|
$ |
— |
|
2022年12月31日現在の未帰属制限株式単位 |
|
|
|
|
$ |
|
||
F-25
制限株式単位の未償却株報酬は2022年12月31日まで$である
2021年株式インセンティブ計画
IPOが完了する前に、発行されたすべての奨励株は普通株に変換される
|
|
量 |
|
|
加重平均 |
|
||
2021年12月31日現在の未帰属普通株 |
|
|
|
|
$ |
|
||
既得 |
|
|
( |
) |
|
$ |
|
|
没収する |
|
|
( |
) |
|
$ |
|
|
2022年12月31日現在の未帰属普通株 |
|
|
|
|
$ |
|
||
2021年5月、IPOについて、取締役会と株主は2021年株式インセンティブ計画、すなわち2021年計画を承認し、IPO発効日の前日に発効した。2021年計画では、奨励的株式オプション、非法定株式オプション、株式付加価値権、制限株式奨励、制限株式単位、その他の株式ベースの奨励を付与することが規定されている。“2021年計画”により発行保留のための普通株式数は:(X)
次の表は,2022年12月31日までの年間2021計画下の株式オプション活動の概要を提供している。
|
|
オプション |
|
|
加重平均 |
|
|
加重平均 |
|
|
骨材 |
|
||||
2021年12月31日現在の未返済債務 |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
授与する |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
鍛えられた |
|
|
( |
) |
|
$ |
|
|
|
|
|
$ |
|
|||
没収する |
|
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
2022年12月31日に返済されていません |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
すでに帰属しており,2022年12月31日に帰属する予定である |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2022年12月31日に行使できます |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
総内在価値は、関連する普通株の推定公正価値と発行された現金オプションの行権価格との差を表す。
2022年12月31日までおよび2021年12月31日まで年度内に帰属するオプションの総公平価値は$
F-26
2022年12月31日現在、株式オプションの未償却株式報酬は$
同社はブラック·スコアーズオプション定価モデルを用いて、付与された株式オプション報酬の公正価値を推定し、以下のように仮定した
|
|
現在までの年度 |
||
|
|
2022 |
|
2021 |
予想期限(年単位) |
|
|
||
予想変動率 |
|
|
||
無リスク金利 |
|
|
||
期待配当収益率 |
|
— |
|
— |
次の表は、2022年12月31日までの1年間の2021計画下の限定株式単位活動をまとめています
|
|
量 |
|
|
加重平均 |
|
||
2021年12月31日現在の未帰属制限株式単位 |
|
|
|
|
$ |
|
||
授与する |
|
|
|
|
$ |
|
||
既得 |
|
|
( |
) |
|
$ |
|
|
没収する |
|
|
( |
) |
|
$ |
|
|
2022年12月31日現在の未帰属制限株式単位 |
|
|
|
|
$ |
|
||
制限株式単位の未償却株報酬は2022年12月31日まで$である
演技賞
2022年6月、会社は2021年の株式激励計画に基づいて、非実行従業員に業績株式オプションと業績株式単位からなる業績奨励を授与した。各成績賞は、会社の取締役会報酬委員会が決定した業績期間内に業績に基づく指標を実現することによって獲得された。当社が業績条件を達成する可能性があると判断すると、業績条件を含む株式奨励の推定公正価値は奨励期間内に支出される。2022年12月31日までの年間で、当社は授与します
同社はブラック·スコアーズオプション定価モデルを用いてPSO報酬の公正価値を推定しており、以下の仮定を前提としている
|
|
現在までの年度 |
|
|
2022 |
予想期限(年単位) |
|
|
予想変動率 |
|
|
無リスク金利 |
|
|
期待配当収益率 |
|
— |
F-27
2021年従業員株購入計画
2021年5月、取締役会は2021年従業員株購入計画を採択し、株主は2021年5月26日に発効する計画を承認した。合計する
ESPPによって発行された普通株式の公正価値は、付与された日にブラック·スコアーズオプション定価モデルを使用して以下の仮定の下で推定される
|
|
現在までの年度 |
||
|
|
2022 |
|
2021 |
予想期限(年単位) |
|
|
||
予想変動率 |
|
|
||
無リスク金利 |
|
|
||
期待配当収益率 |
|
|
||
連結業務報告書に記録されている株式別給与費用は以下の通り(千単位)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
研究開発費 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
一般と行政費用 |
|
|
|
|
|
|
|
|
|
|||
株式に基づく報酬総支出 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
2022年12月31日までに
2022年12月31日までに
10.1株当たり純損失
転換後の普通株株主/株主は1株当たり基本と希釈後の純損失を以下のように計算する(株と1株当たりの金額を除く)
|
|
12ヶ月まで |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
転換可能な非持株権益を償還するには純損失を占めなければならない |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
償還可能な非持株権益株式を交換する−配当とする− |
|
|
|
|
|
( |
) |
|
|
|
||
普通株主/メンバーは純損失を占めるべきである |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
1株当たり基本と希釈して純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
加重-1株当たり純損失の普通株平均、基本損失および赤字を計算するために使用される |
|
|
|
|
|
|
|
|
|
|||
F-28
次のように発行された潜在的希薄化証券は、それらの影響が逆薄であるため、1株当たりの純損失の計算から除外されている
|
|
12月31日まで |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
株式オプション |
|
|
|
|
|
|
|
|
|
|||
普通株に属していない |
|
|
|
|
|
|
|
|
|
|||
制限株式単位 |
|
|
|
|
|
|
|
|
|
|||
ESPPによって約束された株式 |
|
|
|
|
|
|
|
|
|
|||
転換可能優先株を償還する |
|
|
|
|
|
|
|
|
|
|||
激励株 |
|
|
|
|
|
|
|
|
|
|||
合計する |
|
|
|
|
|
|
|
|
|
|||
11.償還可能な非持株権
合併前、会社子会社DOT-1は武田資産協定(付記5)により武田にAシリーズ償還可能転換優先株を発行する。同社の結論は、それは償還可能な非持株権益を代表するということだ。
会社は非制御権益を償還可能な帳簿価値を調整し、子会社の純損失を武田に分配した。償還可能非持株権益への移転と償還可能非持株権益からの移転は子会社から発行されたAシリーズ償還可能転換優先株発行コストの所有権と分配面の変化を代表する。
2021年5月26日、ミレニアム証券取引所協定の条項により、武田は交換した
F-29
12.所得税
最初の公募と転換の前に、同社は税務目的で共同企業とされていたため、所得税を納める必要はなかった。有限責任会社のメンバーは、初日にバイオ製薬ホールディングスが発生した任意の課税所得額または損失中のシェアを適切な税務当局に報告し、関連税金(あれば)を支払う責任がある。転換前に、当社の総合付属会社は税務目的で会社とみなされ、総合財務諸表に計上された所得税を払わなければなりません。転換後、税務目的のため、当社とその付属会社は総合企業グループとされています。2021年12月31日、免税国内税法第332条の清算により、当社は以前の子会社を当社として清算する。したがって、2022年12月31日までの年度の単一実体支出とその後の納税申告書が計算される。すべての税引前損失はアメリカで発生した。
会社所得税準備金(福祉)の有効税率は連邦法定税率と異なり、以下のようになる
|
|
十二月三十一日までの年度 |
|
||||||||
|
|
2022 |
|
|
2021 |
|
2020 |
|
|||
法定料率 |
|
|
% |
|
|
% |
|
% |
|||
州税 |
|
|
% |
|
|
( |
)% |
|
% |
||
恒久的差異 |
|
|
|
|
|
% |
|
( |
)% |
||
単位 |
|
|
% |
|
|
% |
|
% |
|||
評価免除額を変更する |
|
|
( |
)% |
|
|
( |
)% |
|
( |
)% |
株式ベースの報酬 |
|
|
( |
)% |
|
|
( |
)% |
|
( |
)% |
合計する |
|
|
— |
|
|
|
— |
|
|
— |
|
繰延所得税は、財務報告目的のための資産および負債の帳簿価値と所得税目的のための額との間の一時的な差異の純影響を反映する。同社の繰延税金資産と負債の主な構成要素は以下の通り(千計)
|
|
12月31日まで |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
繰延税金資産 |
|
|
|
|
|
|
|
|
|
|||
連邦と州の営業純損失が繰り越す |
|
$ |
|
|
$ |
|
|
$ |
|
|||
資本化R&D第174節費用 |
|
|
|
|
|
|
|
|
|
|||
単位 |
|
|
|
|
|
|
|
|
|
|||
無形資産基盤 |
|
|
|
|
|
|
|
|
|
|||
株式ベースの報酬 |
|
|
|
|
|
|
|
|
|
|||
費用を計算する |
|
|
|
|
|
|
|
|
|
|||
繰延税金資産総額 |
|
|
|
|
|
|
|
|
|
|||
繰延税金負債総額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
減算:推定免税額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
繰延税項目純資産 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
F-30
会社は設立以来毎年純営業損失を出しています。当社はこのような繰越経営損失純額の利益を総合財務諸表に反映していません。損失の歴史があり、他の肯定的な証拠が不足しているため、当社はその繰延税項目の純資産が現金化できない可能性が高いと考えているため、繰延税項の純額は2022年12月31日および2021年12月31日に推定値から完全に相殺される。会社は評価免税額を#ドル増加させるだろう
2022年12月31日現在、同社の連邦純営業損失繰越(NOL)は$
当社は、所有権変更が国税法第382条の規定及び同様の国の規定に基づいて発生したか否かを決定するための研究を完了していない。発生または将来発生する可能性のある所有権変更により、その純営業損失と所得税の繰越免除の利用は重大な年度制限を受ける可能性がある。これらの所有権変更は、将来の課税所得額を相殺するために毎年使用できる純営業損失と所得税控除控除額を制限する可能性がある。一般的に、国税法第382条で定義されている“所有権変更”とは、3年以内に行われる1回または一連の取引を意味し、ある株主の所有権変更が会社流通株の50ポイントを超えることを招く。
2017年の減税·雇用法案によると、2022年から国税法第174条下の研究·実験費またはR&E費用は資本化しなければならない。R&E料金は以下の期限での償却が要求されます
不確定税収状況
権威ある指針によると、不確定な所得税状況が所得税申告書に与える影響は、関連税務機関が監査した後に継続可能な最大金額で確認しなければならない。不確実な所得税状況が持続する可能性が50%未満であれば、それは確認されないだろう。
下表は、税収割引が確認されていない期初と期末金額(千単位)を調節した
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
年初残高 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
前年度に係る納税状況の増加に基づく |
|
|
|
|
|
|
|
|
|
|||
今年度に関連した納税額の増加に基づく |
|
|
|
|
|
|
|
|
|
|||
前年に関連した納税状況による減少 |
|
|
( |
) |
|
|
|
|
|
|
||
本年度に係る税収頭寸に基づく減税 |
|
|
|
|
|
|
|
|
|
|||
年末残高 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
確認すれば、未確認の税収割引の全金額が会社の実際の税率に影響を与えない。その会社は税金の構成要素として利息と罰金を選択した。同社は、2022年、2021年、2020年12月31日までの年間で
同社は米国連邦、カリフォルニア州、その他の州税務管轄区で所得税申告書を提出した。2018年12月31日から2022年12月31日までの連邦·州所得税申告書はまだ審査が必要です。
13.供給計画を定義する
当社は国税法第401(K)条の規定により、従業員貯蓄計画を維持している。この計画の要求に合致すれば、すべての従業員が参加する資格がある。2022年12月31日までに、当社は等額供与$を作成します
F-31